[1] Настоящая заявка испрашивает приоритет следующих заявок:
CN202110472705.3, дата подачи: 29 апреля 2021 г.
CN202111112439.X, дата подачи: 18 сентября 2021 г.
ОБЛАСТЬ ТЕХНИКИ
[2] Настоящее изобретение относится к кристаллической форме производного тиофена и способу ее получения, в частности, к кристаллической форме формулы (I) и способу ее получения.
УРОВЕНЬ ТЕХНИКИ
[3] Подагрический артрит представляет собой распространенный и сложный тип артрита. Когда концентрация мочевой кислоты в крови человека превышает 7 мг/дл, мочевая кислота откладывается в суставах, хрящах и почках в виде мононатриевой соли, что приводит к сверхактивности (чувствительности) иммунной системы, тем самым вызывая болезненное воспаление. Типичными областями поражения являются плюснефаланговый сустав, голеностопный сустав, коленный сустав и т.д. Гиперурикемия является основной патологической причиной подагрического артрита. Гиперурикемия относится к нарушению метаболизма пуриновых веществ в организме человека, приводящему к повышению синтеза мочевой кислоты или понижению ее выведения, что приводит к аномально высокому уровню содержания мочевой кислоты в крови. На международном уровне стандарты диагностики гиперурикемии (HUA) определяются как: при нормальных в отношении пуринов условиях питания уровни мочевой кислоты в крови натощак, измеренные дважды в разные дни, превышают 400 мкмоль/л (6,8 мг/дл) у мужчин и 360 мкмоль/л (6 мг/дл) у женщин. Ее можно категоризировать на три типа, недостаточная экскреция мочевой кислоты, избыточная выработка мочевой кислоты или смешанный тип. Клиническое исследование указывает на то, что 90% первичной гиперурикемии подпадает под категорию недостаточной экскреции мочевой кислоты.
[4] Гиперурикемия неразрывно связана с подагрой и является независимым фактором риска метаболических заболеваний [таких как диабет, метаболический синдром (MS), гиперлипидемия], хронического заболевания почек, сердечно-сосудистого заболевания и инсульта. Следовательно, снижение уровня мочевой кислоты в организме человека может способствовать не только лечению или предупреждению гиперурикемии и подагры, но и снижению риска других осложнений, ассоциированных с гиперурикемией.
[5] В организме человека существует два источника пуринов: эндогенные пурины, возникающие в результате внутреннего синтеза или распада нуклеиновых кислот (примерно 600 мг/день), и экзогенные пурины, получаемые в результате поступления пуринов с пищей (примерно 100 мг/день). В нормальных условиях запас мочевой кислоты в организме составляет 1200 мг, при этом ежедневно вырабатывается приблизительно 700 мг мочевой кислоты. Из них 2/3 выводится через почки, 1/3 - через кишечник и очень небольшое количество выводится через потовые железы. Следовательно, обычно применяемые в клинической практике лекарственные средства, снижающие уровень мочевой кислоты, включают ингибиторы ксантиноксидазы (XO) (такие как аллопуринол и фебуксостат), которые подавляют выработку мочевой кислоты, и ингибиторы Urat1, которые способствуют экскреции мочевой кислоты (такие как бензбромарон и лесинурад).
[6] Ксантиноксидаза представляет собой фермент с низкой специфичностью; он может катализировать превращение гипоксантина в ксантин и впоследствии в мочевую кислоту, а также напрямую катализировать превращение ксантина в мочевую кислоту. Ингибиторы ксантиноксидазы представляют собой препараты первой линии для лечения гиперурикемии, при этом аллопуринол и фебуксостат являются основными лекарственными препаратами, представленными на рынке. Однако, такие лекарственные средства не отвечают клиническим потребностям всех пациентов и обладают заметными побочными эффектами. Аллопуринол является единственным доступным во всем мире терапевтическим средством, снижающим уровень мочевой кислоты, однако он может приводить к серьезным неблагоприятным явлениям со стороны кожи. Тяжелые реакции повышенной чувствительности, связанные с аллопуринолом, тесно ассоциированы с человеческим лейкоцитарным антигеном (HLA)-B*5801, при этом китайское население характеризуется более высокой частотой положительного результата в отношении HLA-B*5801 (6%-8%) по сравнению с европеоидами (приблизительно 2%), что увеличивает риск реакций повышенной чувствительности. Фебуксостат характеризуется превосходным эффектом снижения уровня мочевой кислоты по сравнению с аллопуринолом, но даже при высоких дозах, составляющих 80 мг в день, 40%-52% пациентов не достигают ожидаемого целевого уровня снижения мочевой кислоты, и это может увеличить частоту возникновения острых приступов подагры.
[7] На рынке все еще существует неудовлетворенная клиническая потребность в безопасных и эффективных лекарственных средствах, снижающих уровень мочевой кислоты.
СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
[8] В настоящем изобретении предусмотрена кристаллическая форма А соединения формулы (I), где кристаллическая форма A характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 12,35±0,20°, 15,05±0,20°, 18,19±0,20°, 20,10±0,20°, 23,05±0,20°, 25,05±0,20°, 25,87±0,20°, 27,16±0,20°,
 .
.
[9] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы A характеризуется характеристическими дифракционными пиками при следующих значениях угла 2θ: 10,89±0,20°, 12,35±0,20°, 13,42±0,20°, 15,05±0,20°, 18,19±0,20°, 20,10±0,20°, 21,82±0,20°, 23,05±0,20°, 25,05±0,20°, 25,87±0,20°, 27,16±0,20°, 30,28±0,20°.
[10] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы A характеризуется характеристическими дифракционными пиками при следующих значениях угла 2θ: 6,72°, 8,94°, 10,89°, 12,35°, 13,42°, 15,05°, 17,26°, 18,19°, 18,70°, 20,10°, 21,82°, 23,05°, 24,28°, 25,05°, 25,87°, 27,16°, 29,41°, 30,28°, 30,89°, 33,58°, 36,29°, 37,29°, 38,99°.
[11] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма А характеризуется дифрактограммой XRPD, по сути показанной на фигуре 1.
[12] В некоторых вариантах осуществления настоящего изобретения данные анализа из дифрактограммы XRPD кристаллической формы A являются такими, как показано в таблице 1.
[13]

[14] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма A характеризуется кривой термогравиметрического анализа с потерей веса 1,96% при 200°C±3°C.
[15] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма А характеризуется термограммой TGA, показанной на фигуре 2.
[16] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма А характеризуется кривой дифференциальной сканирующей калориметрии, содержащей эндотермический пик с началом при 244,3°C±2°C.
[17] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма А характеризуется термограммой DSC, показанной на фигуре 3.
[18] В настоящем изобретении предусмотрена кристаллическая форма B соединения формулы (I), где кристаллическая форма A характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 23,93±0,20°, 24,73±0,20°, 26,58±0,20°,
.
[19] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы B характеризуется характеристическими дифракционными пиками при следующих значениях угла 2θ: 13,02±0,20°, 14,68±0,20°, 16,44±0,20°, 19,50±0,20°, 22,69±0,20°, 23,93±0,20°, 24,73±0,20°, 26,58±0,20°.
[20] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы B характеризуется характеристическими дифракционными пиками при следующих значениях угла 2θ: 13,02±0,20°, 14,68±0,20°, 16,44±0,20°, 19,50±0,20°, 22,69±0,20°, 23,93±0,20°, 24,73±0,20°, 25,87±0,20°, 26,58±0,20°, 28,98±0,20°, 29,34±0,20°, 31,86±0,20°.
[21] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы B характеризуется характеристическими дифракционными пиками при следующих значениях угла 2θ: 5,37°, 11,72°, 13,02°, 14,68°, 15,44°, 16,05°, 16,44°, 16,94°, 18,68°, 19,50°, 20,69°, 21,13°, 21,32°, 21,70°, 22,41°, 22,69°, 23,46°, 23,93°, 24,73°, 25,87°, 26,58°, 27,78°, 28,98°, 29,34°, 29,66°, 30,07°, 31,26°, 31,38°, 31,86°, 32,73°, 33,71°, 34,02°, 34,68°, 35,41°, 36,64°, 37,30°, 37,86°, 38,30°.
[22] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма B характеризуется дифрактограммой XRPD, по сути показанной на фигуре 4.
[23] В некоторых вариантах осуществления настоящего изобретения данные анализа из дифрактограммы XRPD кристаллической формы B являются такими, как показано в таблице 2.
[24]

[25] В настоящем изобретении предусмотрен кристалл соединения формулы (I), где кристалл характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 13,28±0,30°, 15,34±0,30°, 25,14±0,30°,
.
[26] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристалла содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,30°, 13,28±0,30°, 15,34±0,30°, 18,16±0,30°, 22,06±0,30°, 25,14±0,30°, 26,75±0,30°, 27,25±0,30°.
[27] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристалла содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,30°, 11,21±0,30°, 13,28±0,30°, 15,34±0,30°, 18,16±0,30°, 22,06±0,30°, 23,15±0,30°, 25,14±0,30°, 25,97±0,30°, 26,75±0,30°, 27,25±0,30°, 30,82±0,30°.
[28] В настоящем изобретении предусмотрен кристалл соединения формулы (I), где кристалл характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 15,26±0,20°, 25,07±0,20°,
.
[29] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристалла содержит характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 26,66±0,20°, 28,38±0,20°, 30,70±0,20°.
[30] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристалла содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,03±0,20°, 11,13±0,20°, 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 26,66±0,20°, 28,38±0,20°, 29,41±0,20°, 30,70±0,20°, 38,53±0,20°.
[31] В настоящем изобретении предусмотрена кристаллическая форма C соединения формулы (I), где кристаллическая форма C характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 18,08±0,20°, 25,07±0,20°,
.
[32] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 26,66±0,20°, 28,38±0,20°, 30,70±0,20°.
[33] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 25,38±0,20°, 26,66±0,20°, 30,70±0,20°.
[34] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,03±0,20°, 11,13±0,20°, 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 26,66±0,20°, 28,38±0,20°, 29,41±0,20°, 30,70±0,20°, 38,53±0,20°.
[35] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,03±0,20°, 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 25,38±0,20°, 26,66±0,20°, 28,38±0,20°, 29,41±0,20°, 30,70±0,20°, 38,53±0,20°.
[36] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,03±0,20°, 11,13±0,20°, 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 24,09±0,20°, 25,07±0,20°, 25,38±0,20°, 26,66±0,20°, 27,17±0,20°, 28,38±0,20°, 29,41±0,20°, 30,70±0,20°, 31,02±0,20°, 38,53±0,20°.
[37] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 25,07±0,20° и в качестве альтернативы при 18,08±0,20°, и/или 9,03±0,20°, и/или 11,13±0,20°, и/или 15,26±0,20°, и/или 18,92±0,20°, и/или 21,99±0,20°, и/или 24,09±0,20°, и/или 25,38±0,20°, и/или 26,66±0,20°, и/или 27,17±0,20°, и/или 28,38±0,20°, и/или 29,41±0,20°, и/или 30,70±0,20°, и/или 31,02±0,20°, и/или 33,67±0,20°, и/или 35,40±0,20°, и/или 36,35±0,20°, и/или 37,26±0,20°, и/или 38,53±0,20°.
[38] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,03°, 11,13°, 13,20°, 15,26°, 18,08°, 18,92°, 21,99°, 24,09°, 25,07°, 25,38°, 26,66°, 27,17°, 28,38°, 29,41°, 30,70°, 31,02°, 33,67°, 35,40°, 36,35°, 37,26°, 38,53°.
[39] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при следующих значениях угла 2θ: 5,66°, 9,03°, 11,13°, 13,20°, 13,70°, 15,26°, 17,25°, 18,08°, 18,92°, 20,88°, 21,99°, 23,41°, 24,09°, 25,07°, 25,38°, 25,99°, 26,66°, 27,17°, 28,38°, 29,41°, 29,98°, 30,70°, 31,02°, 31,72°, 33,67°, 35,40°, 36,35°, 36,74°, 37,26°, 38,53°, 39,80°.
[40] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма C характеризуется дифрактограммой XRPD, по сути показанной на фигуре 5.
[41] В некоторых вариантах осуществления настоящего изобретения данные анализа из дифрактограммы XRPD кристаллической формы C являются такими, как показано в таблице 3.
[42]

[43] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма C характеризуется кривой термогравиметрического анализа с потерей веса 1,21% при 200°C±3°C.
[44] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма C характеризуется термограммой TGA, показанной на фигуре 6.
[45] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма C характеризуется кривой дифференциальной сканирующей калориметрии, содержащей эндотермический пик с началом при 250,0°C±2°C.
[46] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма C характеризуется термограммой DSC, как показано на фигуре 7.
[47] В настоящем изобретении предусмотрена кристаллическая форма D соединения формулы (I), где кристаллическая форма D характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,71±0,20°, 11,87±0,20°, 25,21±0,20°,
.
[48] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы D содержит характеристические дифракционные пики при следующих значениях угла 2θ: 6,71±0,20°, 11,87±0,20°, 13,39±0,20°, 15,44±0,20°, 20,77±0,20°, 22,16±0,20°, 25,21±0,20°, 27,05±0,20°.
[49] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы D содержит характеристические дифракционные пики при следующих значениях угла 2θ: 6,71±0,20°, 11,87±0,20°, 13,39±0,20°, 15,44±0,20°, 16,32±0,20°, 17,90±0,20°, 20,77±0,20°, 22,16±0,20°, 24,31±0,20°, 25,21±0,20°, 27,05±0,20°, 27,41±0,20°.
[50] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы D содержит характеристические дифракционные пики при следующих значениях угла 2θ: 6,45°, 6,71°, 9,22°, 10,40°, 11,61°, 11,87°, 12,53°, 13,39°, 13,82°, 15,44°, 16,32°, 17,37°, 17,90°, 18,27°, 19,07°, 19,67°, 19,90°, 20,77°, 22,16°, 24,31°, 25,21°, 26,10°, 27,05°, 27,41°, 28,50°, 29,59°, 30,10°, 30,89°, 31,17°, 32,81°, 33,77°, 34,17°, 35,52°, 36,57°, 38,20°, 38,68°.
[51] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма D характеризуется дифрактограммой XRPD, по сути показанной на фигуре 8.
[52] В некоторых вариантах осуществления настоящего изобретения данные анализа из дифрактограммы XRPD кристаллической формы D являются такими, как показано в таблице 4.
[53]

[54] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма D характеризуется кривой термогравиметрического анализа с потерей веса 1,14% при 200°C±3°C.
[55] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма D характеризуется термограммой TGA, показанной на фигуре 9.
[56] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма D характеризуется кривой дифференциальной сканирующей калориметрии, содержащей эндотермический пик с началом при 251,4°C±2°C.
[57] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма D характеризуется термограммой DSC, как показано на фигуре 10.
[58] В настоящем изобретении предусмотрена кристаллическая форма E соединения формулы (I), где кристаллическая форма E характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 13,28±0,20°, 15,34±0,20°, 25,14±0,20°,
.
[59] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 25,14±0,20°, 26,75±0,20°, 27,25±0,20°.
[60] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 12,43±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 23,15±0,20°, 25,14±0,20°.
[61] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 11,21±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 23,15±0,20°, 25,14±0,20°, 25,97±0,20°, 26,75±0,20°, 27,25±0,20°, 30,82±0,20°.
[62] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 11,21±0,20°, 12,43±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 23,15±0,20°, 25,14±0,20°, 25,97±0,20°, 26,75±0,20°, 27,25±0,20°.
[63] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при следующих значениях угла 2θ: 13,28±0,20°, 25,14±0,20° и в качестве альтернативы при 15,34±0,20°, и/или 9,11±0,20°, и/или 10,94±0,20°, и/или 11,21±0,20°, и/или 12,43±0,20°, и/или 18,16±0,20°, и/или 22,06±0,20°, и/или 23,15±0,20°, и/или 23,35±0,20°, и/или 24,19±0,20°, и/или 25,97±0,20°, и/или 26,75±0,20°, и/или 27,25±0,20°, и/или 28,45±0,20°, и/или 29,49±0,20°, и/или 30,82±0,20°, и/или 33,74±0,20°, и/или 36,39±0,20°, и/или 37,34±0,20°, и/или 38,57±0,20°.
[64] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 10,94±0,20°, 11,21±0,20°, 12,43±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 23,15±0,20°, 23,35±0,20°, 24,19±0,20°, 25,14±0,20°, 25,97±0,20°, 26,75±0,20°, 27,25±0,20°, 28,45±0,20°, 29,49±0,20°, 30,82±0,20°, 33,74±0,20°, 36,39±0,20°, 37,34±0,20°, 38,57±0,20°.
[65] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11°, 10,94°, 11,21°, 12,43°, 13,28°, 15,34°, 17,39°, 18,16°, 20,18°, 18,94°, 20,95°, 22,06°, 23,15°, 23,35°, 24,19°, 25,14°, 25,97°, 26,75°, 27,25°, 28,45°, 29,49°, 30,16°, 30,82°, 33,74°, 35,45°, 36,39°, 37,34°, 38,57°.
[66] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма E характеризуется дифрактограммой XRPD, по сути показанной на фигуре 11.
[67] В некоторых вариантах осуществления настоящего изобретения данные анализа из дифрактограммы XRPD кристаллической формы E являются такими, как показано в таблице 5.
[68]
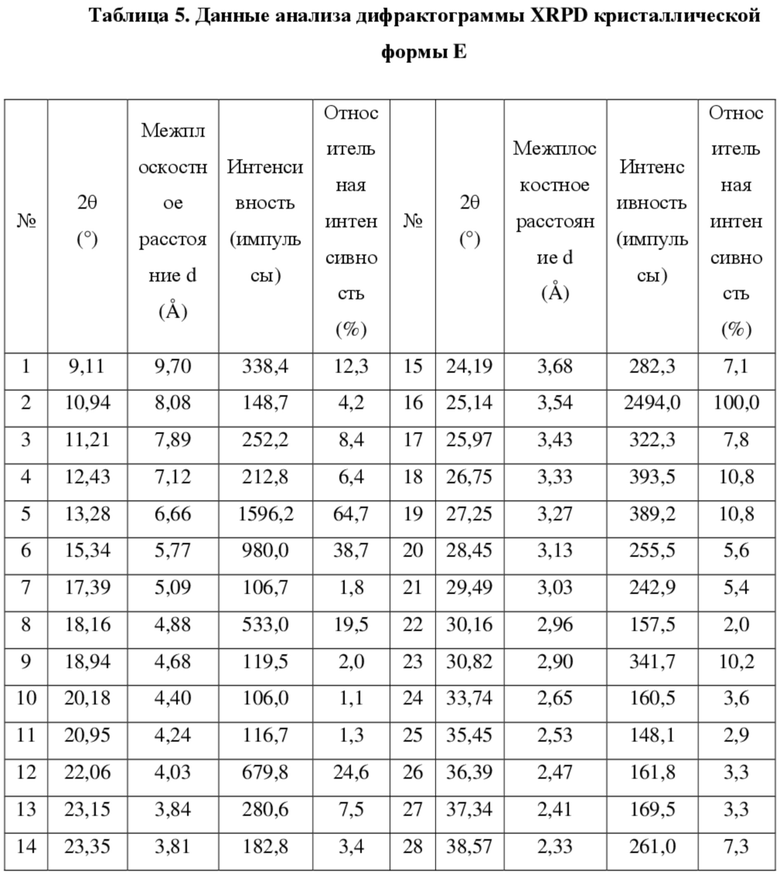
[69] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма E характеризуется кривой термогравиметрического анализа с потерей веса 0,79% при 200°C ± 3°C.
[70] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма E характеризуется термограммой TGA, показанной на фигуре 12.
[71] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма E характеризуется кривой дифференциальной сканирующей калориметрии, содержащей эндотермический пик с началом при 250,4°C ± 2°C.
[72] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма E характеризуется термограммой DSC, как показано на фигуре 13.
[73] В настоящем изобретении также предусмотрено применение кристаллических форм A, B, C, D и E соединения формулы (I) в изготовлении лекарственного препарата для лечения подагры и гиперурикемии.
[74] Технический эффект
[75] Соединение формулы (I) характеризуется устойчивыми кристаллическими свойствами и отсутствием гигроскопичности, демонстрируя хорошие перспективы для фармацевтической разработки.
[76] Определение и описание
[77] Если не указано иное, следующие термины и выражения, применяемые в данном документе, имеют следующие значения. Конкретное выражение или термин при отсутствии точного определения не следует считать неопределенными или неясными, а следует понимать в соответствии с общепринятым значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту.
[78] Промежуточные соединения по настоящему изобретению могут быть получены посредством различных способов синтеза, известных специалистам в данной области техники, в том числе посредством конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, образованных путем их объединения с другими способами химического синтеза, и эквивалентных альтернатив, известных специалистам в данной области техники, при этом предпочтительные варианты осуществления включают без ограничения примеры настоящего изобретения.
[79] Химические реакции конкретных вариантов осуществления настоящего изобретения завершаются в подходящем растворителе, при этом растворитель должен быть подходящим для химических изменений по настоящему изобретению и необходимых для этого реагентов и материалов. Для получения соединения по настоящему изобретению специалистам в данной области техники иногда необходимо модифицировать или выбирать способы синтеза или схемы реакций на основе существующих вариантов осуществления.
[80] Настоящее изобретение описано более подробно с помощью примеров ниже, но примеры не являются ограничительными в отношении настоящего изобретения.
[81] Все растворители, используемые в настоящем изобретении, являются коммерчески доступными и не требуют дополнительной очистки перед использованием.
[82] Соединения по настоящему изобретению названы в соответствии с традиционными принципами номенклатуры в данной области техники или с помощью программного обеспечения ChemDraw®, а для коммерчески доступных соединений используют названия согласно каталогу поставщика.
[83] Структура соединений по настоящему изобретению может быть подтверждена общепринятыми способами, известными специалистам в данной области техники, и если настоящее изобретение включает абсолютную конфигурацию соединения, то абсолютная конфигурация может быть подтверждена с использованием общепринятых методик из данной области техники. Например, в случае рентгеновской дифракции монокристаллов (SXRD) абсолютная конфигурация может быть подтверждена путем сбора данных об интенсивности дифракции выращенного монокристалла с применением дифрактометра Bruker D8 Venture с источником излучения CuKα в качестве источника света и следующим режимом сканирования: сканирование ϕ/ω, и после сбора соответствующих данных структура кристалла может быть дополнительно проанализирована прямым способом (Shelxs97).
[84] Применяемые в настоящем изобретении растворители являются коммерчески доступными. В настоящем изобретении используются следующие сокращения: -OMOM обозначает группу метоксиметилового эфира; HPE обозначает 100% активность ингибирования; ZPE обозначает 0% активность ингибирования; DPBS обозначает фосфатно-солевой буферный раствор Дульбекко.
[85] Рентгеновский порошковый дифрактометр (XRPD), используемый в настоящем изобретении
[86] Модель прибора: рентгеновский дифрактометр Bruker D2 PHASER
[87] Подробные параметры для XRPD указаны ниже.
[88] Источник излучения: Cu, k-альфа (λ = 1,54184Å).
[89] Напряжение на трубке: 30 кВ.
[90] Ток на трубке: 10 мА.
[91] Щель расходимости: 0,6 мм.
[92] Щель Соллера в основном оптическом приборе: 2,5°.
[93] Щель Соллера во вспомогательном оптическом приборе: 2,5°.
[94] Щель детектора: 5,827°.
[95] Противорассеивающая щель: 0 мм.
[96] Ось сканирования: θs-θd.
[97] Размер шага: 0,02 град.
[98] Время на шаг: 0,2 секунды.
[99] Область сканирования: 3-40 град.
[100] Дифференциальный сканирующий калориметр (DSC), используемый в настоящем изобретении
[101] Модель прибора: дифференциальный сканирующий калориметр TA Q2000.
[102] Способ тестирования: образец (приблизительно 1 мг) отбирают и помещают в алюминиевый тигель для DSC с целью проведения тестирования. В условиях 50 мл/мин и. N2 образец нагревают от 30°C (комнатная температура) до 250°C со скоростью 10°C/мин.
[103] Термогравиметрический анализатор (TGA), используемый в настоящем изобретении
[104] Модель прибора: термогравиметрический анализатор DISCOVERY 5500.
[105] Способ тестирования: образец (2-5 мг) отбирают и помещают в платиновый тигель для TGA с целью проведения тестирования. В условиях 25 мл/мин. и N2 образец нагревают от комнатной температуры до 300°C со скоростью 10°C/мин.
[106] Динамическая сорбция паров (DVS), используемая в настоящем изобретении
[107] Модель прибора: анализатор динамической сорбции паров Intrinsic.
Условия тестирования: образец (10-30 мг) отбирают и помещают в лоток для образцов DVS для проведения тестирования.
[108] Подробные параметры для DVS указаны ниже.
[109] Температура: 25°C.
[110] Равновесие: dm/dt = 0,002%/мин. (минимум: 10 минут, максимум: 180 минут).
[111] Тестовое приращение RH (%): 10 (90-0-90%), 5 (90-95%).
[112] Диапазон тестового приращения RH (%): 0%-95%-0%.
[113] Оценка и классификация гигроскопичности показаны в таблице 6.
[114]

[115] Примечание: ΔW% указывает на увеличение веса образца за счет поглощения влаги при 25 ± 1°C и RH 80 ± 2%.
[116] КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[117] Фигура 1 представляет собой дифрактограмму XRPD Cu-Kα-излучения кристаллической формы А соединения формулы (I).
[118] Фигура 2 представляет собой термограмму TGA кристаллической формы А соединения формулы (I).
[119] Фигура 3 представляет собой термограмму DSC кристаллической формы А соединения формулы (I).
[120] Фигура 4 представляет собой дифрактограмму XRPD Cu-Kα-излучения кристаллической формы B соединения формулы (I).
[121 Фигура 5 представляет собой дифрактограмму XRPD Cu-Kα-излучения кристаллической формы C соединения формулы (I).
[122] Фигура 6 представляет собой термограмму TGA кристаллической формы C соединения формулы (I).
[123] Фигура 7 представляет собой термограмму DSC кристаллической формы C соединения формулы (I).
[124] Фигура 8 представляет собой дифрактограмму XRPD Cu-Kα-излучения кристаллической формы D соединения формулы (I).
[125] Фигура 9 представляет собой термограмму TGA кристаллической формы D соединения формулы (I).
[126] Фигура 10 представляет собой термограмму DSC кристаллической формы D соединения формулы (I).
[127] Фигура 11 представляет собой дифрактограмму XRPD Cu-Kα-излучения кристаллической формы E соединения формулы (I).
[128] Фигура 12 представляет собой термограмму TGA кристаллической формы E соединения формулы (I).
[129] Фигура 13 представляет собой термограмму DSC кристаллической формы E соединения формулы (I).
[130] Фигура 14 представляет собой термограмму DVS кристаллической формы C соединения формулы (I).
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
[131] Настоящее изобретение описано более подробно с помощью примеров ниже, но это не означает, что существуют какие-либо противоречащие ограничения в отношении настоящего изобретении. В данном документе подробно описано настоящее изобретение, а также раскрыты его конкретные варианты осуществления; для специалистов в данной области техники очевидно, что осуществление модификаций и улучшений по отношению к вариантам осуществления настоящего изобретения происходит без отступления от сущности и объема настоящего изобретения.
[132] Пример 1. Получение кристаллической формы А соединения формулы (I)
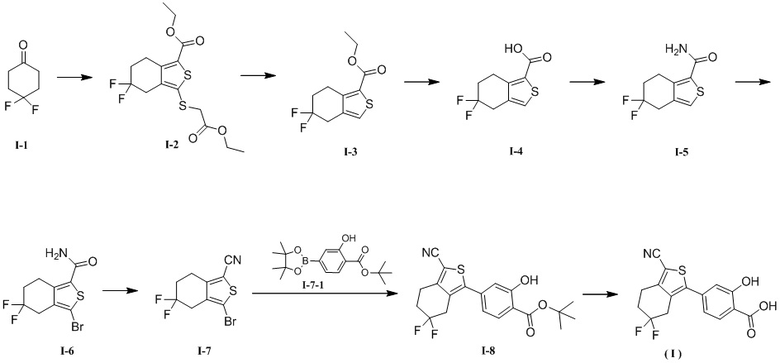
[133] Стадия 1. Синтез соединения I-2
[134] К диметилсульфоксиду (1200 мл) добавляли трет-бутоксид калия (234,26 г, 2,09 моль). Смесь перемешивали при комнатной температуре до прозрачности. К полученному по каплям добавляли раствор соединения I-1 (200 г, 1,49 моль) в диметилсульфоксиде (500 мл) при 15-20°C. После добавления смесь перемешивали в течение еще 40 минут. Затем к полученному по каплям добавляли сероуглерод (113,54 г, 1,49 моль, 90,11 мл) и поддерживали температуру реакционной смеси не выше 20°C. После добавления смесь перемешивали в течение еще 20 минут. В смесь медленно добавляли трет-бутоксид калия (100,40 г, 894,70 ммоль), поддерживали температуру между 15 и 20°C и смесь перемешивали в течение 30 минут. Затем к полученному по каплям добавляли этилбромацетат (498,05 г, 2,98 моль, 329,83 мл). Температуру смеси поддерживали при между 15 и 20°C и смесь перемешивали в течение 1,5 часа при той же температуре. К полученному добавляли карбонат калия (206,09 г, 1,49 моль), а реакционную смесь нагревали до 60°C и перемешивали в течение еще 1,5 часа. К реакционной смеси добавляли 1 л воды, добавляли 6 M водный раствор хлористоводородной кислоты для доведения pH до значения между 3 и 4 и смесь экстрагировали этилацетатом (1,5 л × 2). Объединенные органические фазы промывали насыщенным солевым раствором (200 мл × 3) и высушивали при пониженном давлении для удаления органического растворителя. К полученному в результате неочищенному продукту добавляли изопропанол (200 мл), смесь перемешивали до однородного состояния, оставляли отстаиваться в течение 15 часов, фильтровали и высушивали под вакуумом при 45°C в течение 1 часа с получением соединения I-2. 1H ЯМР (400 MГц, CDCl3) δ: 4,32 (q, J = 7,2 Гц, 2H), 4,19 (q, J = 7,2 Гц, 2H), 3,56 (s, 2H), 3,25 (t, J = 6,8 Гц, 2H), 3,19 (t, J = 14,4 Гц, 2H), 2,26 - 2,17 (m, 2H), 1,37 (t, J = 7,2 Гц, 3H), 1,27 (t, J = 7,2 Гц, 3H). MS масса/заряд = 364,8 [M+H]+.
[135] Стадия 2. Синтез соединения I-3
[136] Соединение I-2 (282 г, 773,82 ммоль) растворяли в этаноле (3,5 л) и к полученному добавляли никель Ренея (99,45 г, 1,16 моль). Реакционную систему трижды заменяли азотом, перемешивали и обеспечивали прохождение реакции при 85°C в течение 48 часов под давлением водорода, составляющим 2,5 МПа. Затем реакционную смесь охлаждали и фильтровали через диатомит в атмосфере азота. Фильтрат высушивали при пониженном давлении для удаления растворителя и получали соединение I-3. Полученное в результате соединение непосредственно применяли на следующей стадии без дополнительной очистки. 1H ЯМР (400 MГц, CDCl3) δ: 7,09 (s, 1H), 4,26 (q, J = 7,2 Гц, 2H), 3,20 (t, J = 6,8 Гц, 2H), 3,12 (t, J = 14,4 Гц, 2H), 2,20 - 2,10 (m, 2H), 1,30 (t, J = 6,8 Гц, 3H). MS масса/заряд = 247,0 [M+H]+.
[137] Стадия 3. Синтез соединения I-4
[138] Соединение I-3 (40,00 г, 162,42 ммоль) растворяли в метаноле (200 мл). К полученному добавляли 200 мл водного раствора гидроксида натрия (12,99 г, 324,84 ммоль), а реакционную смесь нагревали до 50°C и перемешивали в течение 2 часов. Реакционную смесь высушивали при пониженном давлении для удаления органического растворителя. К остатку добавляли 150 мл воды и добавляли 6 M водный раствор хлористоводородной кислоты, чтобы довести pH до значения между 2 и 3, что приводило в результате к осаждению большого количества белого твердого вещества. Смесь фильтровали. Осадок на фильтре промывали 100 мл воды и 50 мл петролейного эфира и высушивали при пониженном давлении при 50°C в течение 3 часов с получением соединения I-4. 1H ЯМР (400 MГц, CD3OD) δ: 7,38 (s, 1H), 3,33 - 3,17 (m, 4 H), 2,28 - 2,21 (m, 2H).
[139] Стадия 4. Синтез соединения I-5
[140] Соединение I-4 (35,0 г, 160,39 ммоль) растворяли в тетрагидрофуране (200 мл). К полученному добавляли карбонилдиимидазол (33,81 г, 208,51 ммоль). Реакционную смесь перемешивали в течение 2 часов в атмосфере азота, затем добавляли аммиачную воду (31,23 г, 240,58 ммоль, 34,32 мл) и смесь перемешивали в течение еще 15 часов. Смесь высушивали при пониженном давлении для удаления органического растворителя. К полученному в результате остатку добавляли 300 мл воды, остаток перемешивали в течение 10 минут и фильтровали. Осадок на фильтре промывали 100 мл воды, высушивали при пониженном давлении при 55°C в течение 2,5 часа с получением соединения I-5. 1H ЯМР (400 MГц, CDCl3) δ: 7,09 (s, 1H), 5,72 (brs, 2H), 3,30 - 3,18 (m, 4H), 2,29 - 2,19 (m, 2H).
[141] Стадия 5. Синтез соединения I-6
[142] Соединение I-5 (31 г, 142,70 ммоль) растворяли в N,N-диметилформамиде (200 мл). К полученному медленно порциями добавляли N-бромсукцинимид (27,94 г, 156,97 ммоль). Реакционную смесь перемешивали при 20°C в течение еще 2 часов. Реакционную смесь медленно выливали в 600 мл перемешанной воды, что приводило в результате к осаждению большого количества твердого вещества. После перемешивания в течение 10 минут смесь фильтровали. Осадок на фильтре промывали 200 мл воды и 100 мл петролейного эфира, затем высушивали под вакуумом при 50°C в течение 2 часов с получением соединения I-6. 1H ЯМР (400 MГц, CDCl3) δ: 5,62 (brs, 2H), 3,25 (t, J = 7,2 Гц, 2H), 3,04 (t, J = 14,0 Гц, 2H), 2,26 - 2,18 (m, 2H).
[143] Стадия 6. Синтез соединения I-7
[144] К этилацетату (250 мл) добавляли соединение I-6 (48 г, 162,09 ммоль) и триэтиламин (32,80 г, 324,18 ммоль, 45,12 мл). Смесь охлаждали до 0°C в атмосфере азота, а затем к полученному по каплям добавляли трифторуксусный ангидрид (44,26 г, 210,72 ммоль, 29,31 мл). Реакционную смесь перемешивали в течение 1 часа при той же температуре, затем нагревали до 20°C и перемешивали в течение еще 0,5 часа. Реакционную смесь разбавляли 250 мл этилацетата, последовательно промывали водой (100 мл × 2), насыщенным раствором бикарбоната натрия (150 мл) и насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и высушивали при пониженном давлении для удаления органического растворителя. Таким образом получали соединение I-7, которое непосредственно применяли на следующей стадии без дополнительной очистки. 1H ЯМР (400 MГц, CDCl3) δ: 3,13 - 2,97 (m, 4H), 2,30 - 2,20 (m, 2H).
[145] Стадия 7. Синтез соединения I-8
[146] К диметоксиэтану (60 мл) и воде (12 мл) добавляли соединение I-7 (6,0 г, 21,57 ммоль), соединение I-7-1 (7,60 г, 23,73 ммоль) и безводный фосфат калия (9,16 г, 43,15 ммоль). К полученному добавляли Pd(dppf)Cl2 (394,64 мг, 539,34 мкмоль) в атмосфере азота. Реакционную смесь нагревали до 85°C в атмосфере азота и перемешивали в течение еще 15 часов. Реакционную смесь охлаждали, добавляли 20 мл воды и 100 мл этилацетата, смесь перемешивали в течение 10 минут и фильтровали. Органическую фазу отделяли от фильтрата. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и высушивали при пониженном давлении для удаления органического растворителя. К полученному в результате неочищенному продукту добавляли этилацетат (80 мл) и последовательно добавляли активированный уголь (4 г) и диоксид кремния (4 г). Смесь нагревали до 80°C, перемешивали в течение 1 часа, затем охлаждали, фильтровали через диатомит и высушивали при пониженном давлении для удаления органического растворителя. Полученный в результате неочищенный продукт последовательно суспендировали трет-бутилметиловым эфиром (25 мл) при 25°C в течение 0,5 часа и фильтровали, а полученный в результате осадок на фильтре высушивали под вакуумом при 45°C в течение 1 часа с получением соединения I-8. 1H ЯМР (400 MГц, CDCl3) δ: 11,18 (s, 1H), 7,86 (d, J = 8,0 Гц, 1H), 7,01 (d, J = 1,6 Гц, 1H), 6,92 - 6,90 (m, 1H), 3,24 (t, J = 14,4 Гц, 2H), 3,12 (t, J = 6,8 Гц, 2H), 2,36 - 2,26 (m, 2H), 1,64 (s, 9H).
[147] Стадия 8. Синтез кристаллической формы A соединения формулы (I)
[148] Соединение I-8 (32 г, 81,75 ммоль) добавляли к трифторуксусной кислоте (250 мл) и реакционную смесь перемешивали при 20°C в течение 1 часа. Трифторуксусную кислоту удаляли при пониженном давлении и к полученному остатку добавляли воду (300 мл). Смесь суспендировали при комнатной температуре в течение 20 минут до полного диспергирования и фильтровали. Осадок на фильтре промывали водой (200 мл) и высушивали при пониженном давлении при 45°C в течение 1 часа с получением кристаллической формы A соединения формулы (I). 1H ЯМР (400 MГц, CD3OD) δ: 8,03 - 7,96 (m, 1H), 7,12 - 7,06 (m, 2H), 3,36 - 3,29 (m, 2H), 3,16 - 3,07 (m, 2H), 2,44 - 2,30 (m, 2H) Дифрактограмма XRPD кристаллической формы A показана на фигуре 1, ее термограмма TGA показана на фигуре 2 и ее термограмма DSC показана на фигуре 3.
[149] Пример 2. Получение соединения формулы (I)

[150] Стадия 1. Синтез соединения I-4
[151] Соединение I-3 (2,5 г, 10,15 ммоль) растворяли в метаноле (10 мл), а затем к полученному добавляли воду (10 мл) и гидроксид натрия (1,62 г, 40,61 ммоль). Полученную в результате реакционную смесь помещали на масляную баню при 40°C и перемешивали в течение 2 часов. Затем реакционную смесь упаривали наполовину при пониженном давлении и к остатку добавляли воду (5 мл). После перемешивания к смеси добавляли 6 M хлористоводородную кислоту, чтобы довести pH до значения между 2 и 3, что приводило к осаждению большого количества белого твердого вещества. Твердое вещество посредством фильтрования и высушивали при пониженном давлении при 50°C в течение 3 часов с получением соединения I-4. 1H ЯМР (400 MГц, CDCl3) δ: 7,28 (s, 1H), 3,30 (t, J = 7,0 Гц, 2H), 3,22 (t, J = 14,3 Гц, 2H), 2,25 (tt, J = 6,8, 13,4 Гц, 2H).
[152] Стадия 2. Синтез соединения I-5
[153] Соединение I-4 (500 мг, 2,29 ммоль) растворяли в дихлорметане (5 мл), а затем к полученному добавляли карбонилдиимидазол (445,83 мг, 2,75 ммоль). Полученную в результате реакционную смесь перемешивали, обеспечивали прохождение реакции в течение 1 часа в атмосфере азота и выливали в энергично перемешиваемую аммиачную воду (2,87 г, 22,91 ммоль, 3,15 мл, содержание 28%) в тетрагидрофуране (5 мл). Реакционную смесь перемешивали и обеспечивали прохождение реакции в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении при 25°C. Остаток экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, а затем высушивали на роторном испарителе с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии (этилацетат/петролейный эфир = 0-45%) с получением соединения I-5. 1H ЯМР (400 MГц, CDCl3) δ: 7,10 (s, 1H), 5,58 (br s, 2H), 3,28 (t, J = 6,9 Гц, 2H), 3,21 (t, J = 14,4 Гц, 2H), 2,24 (tt, J = 6,9, 13,4 Гц, 2H).
[154] Стадия 3. Синтез соединения 2-4
[155] Соединение I-5 (320 мг, 1,47 ммоль) растворяли в DMF (3 мл) и полученный раствор охлаждали до 0°C, а затем к полученному добавляли цианурхлорид (298,81 мг, 1,62 ммоль). Конечную реакционную смесь перемешивали в течение 2 часов в атмосфере азота (в течение этого времени осаждалось большое количество белого твердого вещества). Реакционную смесь разбавляли этилацетатом (50 мл), а затем промывали водой (10 мл × 3) и насыщенным солевым раствором (10 мл). Органическую фазу высушивали над подходящим количеством безводного сульфата натрия и фильтровали, чтобы удалить любое высушивающее средство. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и получения неочищенного продукта 2-4, который непосредственно применяли на следующей стадии. 1H ЯМР: (400 MГц, CDCl3) δ: 7,25 (s, 1H), 3,21 (t, J = 14,3 Гц, 2H), 3,09 (t, J = 6,9 Гц, 2H), 2,28 (tt, J = 6,8, 13,2 Гц, 2H).
[156] Стадия 4. Синтез соединения 2-5
[157] Соединение 2-4 (290 мг, 1,46 ммоль) растворяли в уксусной кислоте (2 мл), а затем к полученному добавляли жидкий бром (348,94 мг, 2,18 ммоль, 112,56 мкл). Полученную реакционную смесь перемешивали при 25°C (комнатная температура) в течение 15 часов. Реакционную смесь затем высушивали на роторном испарителе и к остатку добавляли этилацетат (30 мл). К смеси добавляли насыщенный водный раствор карбоната натрия, чтобы довести pH до значения между 7 и 8, а органическую фазу отделяли. Водную фазу экстрагировали с помощью этилацетата (30 мл ). Органические фазы объединяли и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии (этилацетат/петролейный эфир = 0-5%) с получением соединения I-7. 1H ЯМР: (400 MГц, CDCL3) δ: 3,10-2,99 m, 4H), 2,32-2,19 (m, 2H).
[158] Стадия 5. Синтез соединения 2-6
[159] К диоксану (3 мл) и воде (0,6 мл) добавляли соединение I-7 (140 мг, 503,39 мкмоль), борат 2-5A (178,39 мг, 553,73 мкмоль) и карбонат калия (139,14 мг, 1,01 ммоль), а затем к полученному добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2) (36,83 мг, 50,34 мкмоль). Затем смесь перемешивали и обеспечивали прохождение реакции на масляной бане при температуре 105°C в течение 15 часов в атмосфере азота. Реакционную смесь высушивали на роторном испарителе с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (этилацетат/петролейный эфир = 0-25%) с получением соединения 2-6. 1H ЯМР: (400 MГц, CHCl3) δ: 7,87 (d, J = 8,0 Гц, 1H), 7,28 (d, J = 1,6 Гц, 1H), 7,10 (dd, J = 1,6, 8,0 Гц, 1H), 5,0 (s, 2H), 3,3 (s, 3H), 3,55(s, 3H), 3,23 (t, J = 14,4 Гц, 2H), 3,13 (t, J = 6,8 Гц, 2H), 2,39 - 2,24 (m, 2H).
[160] Стадия 6. Синтез соединения 2-7
[161] Соединение 2-6 (105 мг, 266,90 мкмоль) растворяли в тетрагидрофуране (2 мл), а затем к полученному добавляли водный раствор моногидрата гидроксида лития (2 M, 533,80 мкл). Полученную реакционную смесь перемешивали при 25°C (комнатная температура) в течение 15 часов. Реакционную смесь высушивали при 40°C на роторном испарителе для удаления тетрагидрофурана. К остатку добавляли 2 M хлористоводородную кислоту, чтобы довести pH до значения между 2 и 3, что приводило к осаждению большого количества твердого вещества. К полученному добавляли этилацетат (50 мл) и смесь перемешивали. Затем этилацетат отделяли и смесь высушивали на роторном испарителе с получением соединения 2-7, а неочищенный продукт непосредственно применяли на следующей стадии.
[162] Стадия 7. Синтез соединения формулы (I)
[163] Соединение 2-7 (105 мг, 276,77 мкмоль) растворяли в метаноле (1 мл) и к полученному добавляли хлористоводородную кислоту (60,55 мг, 1,66 ммоль, 59,36 мкл). Реакционная смесь становилась мутной, и ее перемешивали при 25°C в течение 3 часов. Реакционную смесь высушивали на роторном испарителе при 40°C и полученный остаток очищали посредством препаративной HPLC (хроматографическая колонка: Venusil ASB Phenyl 150 * 30 мм * 5 мкм; подвижная фаза: [вода(0,05%HCl)-ACN]; ACN%: 60%-90%, 9 минут) с получением соединения формулы (I). 1H ЯМР (400 MГц, CD3OD) δ: 8,00 (d, J = 8,0 Гц, 1H), 7,13 - 7,04 (m, 2H), 3,35-3,32 (m, 2H), 3,12 (t, J = 7,2 Гц, 2H), 2,45 - 2,30 (m, 2H); MS (ESI) масса/заряд: 334,02 [M-H]-.
[164] Пример 3. Получение кристаллической формы B соединения формулы (I)
[165] К дихлорметану (1 мл) добавляли кристаллическую форму A (20 мг, 0,06 ммоль) соединения формулы (I) и смесь перемешивали при 25°C в течение 120 часов. Смесь фильтровали. Осадок на фильтре высушивали при пониженном давлении при 50°C в течение 2-5 часов с получением кристаллической формы B соединения формулы (I). Дифрактограмма XRPD кристаллической формы B показана на фигуре 4.
[166] Пример 4. Получение кристаллической формы C соединения формулы (I)
[167] К смеси растворителей этилацетата (5 мл) и гептана (5 мл) добавляли кристаллическую форму A (1,0 г, 2,98 ммоль) соединения формулы (I), затем смесь перемешивали при 25°C в течение 72 часов. Смесь фильтровали. Осадок на фильтре высушивали при пониженном давлении при 45°C в течение 2 часов с получением кристаллической формы C соединения формулы (I). Дифрактограмма XRPD кристаллической формы C показана на фигуре 5, ее термограмма TGA является такой, как показано на фигуре 6, и ее термограмма DSC показана на фигуре 7.
[168] Пример 5. Получение кристаллической формы D соединения формулы (I)
[169] К смеси растворителей тетрагидрофурана (0,4 мл) и воды (0,4 мл) добавляли 20 мг навески кристаллической формы A соединения формулы (I). Смесь перемешивали при 50°C до растворения всех твердых веществ, охлаждали до 13°C и перемешивали в течение 72 часов. Смесь фильтровали. Осадок на фильтре высушивали при пониженном давлении при 45°C в течение 2 часов с получением кристаллической формы D соединения формулы (I). Дифрактограмма XRPD кристаллической формы D показана на фигуре 8, ее термограмма TGA показана на фигуре 9 и ее термограмма DSC показана на фигуре 10.
[170] Пример 6. Получение кристаллической формы E соединения формулы (I)
[171] К смеси растворителей тетрагидрофурана (3,3 мл) и воды (6,6 мл) добавляли кристаллическую форму A (1,0 г, 2,98 ммоль) соединения формулы (I). Полученную смесь перемешивали при 25°С в течение 72 часов. Смесь фильтровали. Осадок на фильтре высушивали при пониженном давлении при 45°C в течение 2 часов с получением кристаллической формы E соединения формулы (I). Дифрактограмма XRPD кристаллической формы E показана на фигуре 11, ее термограмма TGA показана на фигуре 12 и ее термограмма DSC показана на фигуре 13.
[172] Пример 7. Исследование гигроскопичности соединения формулы (I)
[173] Экспериментальные материалы:
[174] анализатор динамической сорбции паров DVS Intrinsic.
[175] Способы проведения эксперимента:
[176] 10-30 мг кристаллической формы C соединения формулы (I) отбирали и помещали в лоток для образцов DVS для проведения тестирования.
[177] Результаты эксперимента:
[178] спектр DVS для кристаллической формы C соединения формулы (I) является таким, как показано на фигуре 14, при этом ΔW равняется 0,196%.
[179] Заключение по итогам эксперимента:
[180] соединение формулы (I) в кристаллической форме C не проявляет гигроскопичности, при этом гигроскопический прирост веса составляет 0,196% при 25°C и 80% RH.
[181] Пример 8. Тест на стабильность в твердом состоянии соединения формулы (I) в кристаллической форме C
[182] На основании "Руководства по тестированию стабильности фармацевтических ингредиентов и составов" (Китайская фармакопея, издание 2015 г., общее правило 9001) стабильность соединения формулы (I) в кристаллической форме C изучали при высокой температуре (60°C, в открытом состоянии), высокой влажности (комнатная температура/относительная влажность 92,5%, в открытом состоянии) и сильном воздействии света (5000 люкс, в герметично закрытом состоянии).
[183] Двенадцать порций соединения формулы (I) в кристаллической форме C взвешивали параллельно, при этом каждая порция составляла примерно 1,5 г, и помещали в плоские сосуды для взвешивания (70 * 35 мм) или одноразовые чашки Петри, распределяя тонким слоем. Их по отдельности помещали в стабильные условия высокой температуры (60°C), высокой влажности (25°C/влажность 92,5%), комбинированной высокой температуры и высокой влажности (40°C/влажность 75%) и воздействия света. Для образца, помещенного в условия высокой температуры и высокой влажности, сосуд герметично закрывали алюминиевой фольгой, в которой пробивали небольшие отверстия, чтобы обеспечить полный контакт образцов с окружающим воздухом; образец, помещенный под сильное воздействие света, герметично закрывали крышкой из кварцевого стекла. Образцы, помещенные в условия высокой температуры (60°C) и высокой влажности (влажность 92,5%, комнатная температура), отбирали для тестирования на 5-е и 10-е сутки (внешний вид, сопутствующие вещества и содержание). Образцы, помещенные в условия комбинированной высокой температуры и высокой влажности (40°C/влажность 75%), отбирали для тестирования в 1-й, 2-й и 3-й месяцы (внешний вид, сопутствующие вещества и содержание). Образцы, помещенные в условия воздействия света, отбирали для тестирования, когда общее воздействие света достигало 1,2×106 люкс⋅ч. Результаты исследования сравнивали с исходными результатами тестирования в день 0 и результаты теста показаны в таблице 7 ниже.
[184]

[185] Заключение: соединение формулы (I) в кристаллической форме C демонстрирует хорошую стабильность под воздействием таких факторов, как высокая температура, высокая влажность, сильное воздействие света и условия ускоренного хранения.
[186] Данные биологического анализа
[187] Экспериментальный пример 1. Тест на ингибирующую активность в отношении ксантиноксидазы
[188] 1. Цель эксперимента
[189] Оценить уровень ингибирования активности ксантиноксидазы соединением.
[190] 2. Реагенты
[191] В число основных реагентов, используемых в данном исследовании, входит ксантин (Sigma, номер по каталогу: X4002-1G, номер партии: SLBB5664V) и ксантиноксидаза (Sigma, номер по каталогу: X4376-5UN, номер партии: SLBQ1518V).
[192] 3. Приборы
[193] Основным прибором, применяемым в данном исследовании является многомодельный микропланшетный ридер.
[194] 4. Способы проведения эксперимента
[195] 1) В лунку с фоновым контролем соединения и лунку HPE (со 100% активностью ингибирования) с положительным контролем добавляли 50 мкл фосфатно-солевого буферного раствора Дульбекко (DPBS).
[196] 2) 2 Ед./мл ксантиноксидазы разбавляли DPBS, получая в результате концентрацию 0,04 Ед./мл, а в лунку для тестирования активности соединения и лунку для отрицательного контроля ZPE (с активностью ингибирования 0%) добавляли 50 мкл ксантиноксидазы.
[197] 3) Проводили серийное разведение соединения с использованием 3-кратного градиента по 8 точкам данных DMSO, а затем дополнительно разбавляли DPBS, добавляя по 50 мкл в каждую лунку в трех повторностях. В каждую лунку положительного контроля HPE (степень ингибирования 100%) и лунку отрицательного контроля ZPE (степень ингибирования 0%) добавляли по 50 мкл DPBS.
[198] 4) 200 мМ ксантина разбавляли DPBS до 300 мкМ. В каждую лунку добавляли 100 мкл ксантина и обеспечивали протекание реакции при комнатной температуре в течение 30 минут, что приводило в результате к получению конечной концентрации 0,01 Ед./мл ксантиноксидазы и конечной концентрации 0,5% DMSO в каждой лунке. Лунка положительного контроля HPE (активность ингибирования 100%) содержала ксантин, но не содержала ксантиноксидазу. Лунка отрицательного контроля ZPE (активность ингибирования 0%) содержала как ксантин, так и ксантиноксидазу. Лунка фонового контроля соединения содержала различные концентрации соединения и ксантина, но не содержала ксантиноксидазы.
[199] 5) Значения поглощения измеряли при 290 нм с применением спектрофотометра.
[200] 6) Анализ данных. Скорость ингибирования ксантиноксидазы для каждой лунки рассчитывали по следующей формуле:

[201] * ODтестовый образец относится к значению оптической плотности в отношении лунок для тестирования активности соединения, которые содержали соединение, ксантин и ксантиноксидазу;
[202] ODконтроль соединения относится к значению фоновой оптической плотности для различных концентраций лунок тестируемого соединения, которые содержали соединение и ксантин, но не ксантиноксидазу;
[203] ODZPE представляет собой среднее значение оптической плотности в отношении контрольной лунки с нулевой ингибирующей активностью, которая содержала 0,5% DMSO, ксантин и ксантиноксидазу;
[204] ODHPE представляет собой среднее значение оптической плотности контрольной лунки со 100% ингибирующей активностью, которая содержала 0,5% DMSO и ксантин, но не ксантиноксидазу.
[205] 7) С применением программного обеспечения GraphPad Prism данные в отношении степени ингибирования (степень ингибирования в %) соединений подвергали анализу нелинейной аппроксимации кривой с использованием способа "логарифм(агонист) против ответа -- переменный угловой коэффициент", позволяющий определить значение IC50 для соединения, при этом эмпирическое уравнение выглядит следующим образом:
[206] Y = нижнее значение + (верхнее значение - нижнее значение)/(1 + 10^((LogIC50 - X) * угловой коэффициент Хилла)).
[207] 5. Результаты эксперимента
[208] Результаты экспериментов показаны в таблице 8.
[209]

[210] Заключение: соединение по настоящему изобретению проявляет подходящую ингибирующую активность в отношении ксантиноксидазы.
[211] Экспериментальный пример 2. Тестирование ингибирующей активности соединения в отношении поглощения мочевой кислоты
[212] 1. Цель эксперимента
[213] В настоящем исследовании использовали стабильную трансфицированную линию клеток Urat1 человека для оценки ингибирующей активности тестируемого соединения в отношении поглощения мочевой кислоты.
[214] 2. Материалы для эксперимента
[215] 2.1 Клеточная линия
[216] Стабильная трансфицированная линия клеток Urat1 человека была сконструирована компанией Wuxi APPTEC (Shanghai) Co., Ltd. Стабильная трансфицированная линия клеток Urat1 человека (Urat1-MDCK) была получена путем трансфекции клеток MDCK геном Urat1 человека с последующей селекцией с помощью G418. Линию клеток культивировали в MEM, содержащей 10% фетальной бычьей сыворотки (FBS), 100 Ед./мл пенициллина, 100 г/мл стрептомицина, 2 мМ L-глутамина, 1% заменимых аминокислот и 250 г/мл G418.
[217] 2.2 Реагенты
[218] В число основных реагентов, используемых в настоящем исследовании, входила 14C-мочевая кислота (ARC, номер по каталогу: ARC-0513, номер по каталогу: 200122).
[219] 2.3 Приборы
[220] Основным прибором, применяемым в настоящем исследовании, являлся жидкостный сцинтилляционный анализатор (Perkin Elmer, Tri-Carb 4910TR).
[221] 3. Способы проведения эксперимента
[222] 3.1 Посев клеток
[223] 3.1.1 Клетки Urat1-MDCK, культивированные во флаконах T150 для культивирования клеток, расщепляли 0,25% трипсином, а затем разбавляли свежей культуральной средой, что в результате приводило к получению суспензии с концентрацией 200000 клеток/мл.
[224] 3.1.2 Клетки высевали в 48-луночный планшет для культивирования клеток по 0,5 мл на лунку, в результате чего конечная плотность составляла 100000 клеток/лунку.
[225] 3.1.3 Планшет для культивирования клеток помещали в инкубатор при 37°C и 5% CO2 и культивировали в течение ночи.
[226] 3.2 Обработка и тестирование соединения
[227] 3.2.1 Соединение разводили в DMSO с 5-кратным градиентом для 4 точек, при этом разбавленные концентрации в 200 раз превышали конечную концентрацию для тестирования. Затем соединение разбавляли в 10 раз буфером HBSS.
[228] 3.2.2 10 мМ концентрированный исходный раствор 14C-мочевой кислоты разбавляли до 1 мМ с использованием буфера HBSS.
[229] 3.2.3 После инкубации планшета для культивирования клеток в течение ночи среду для культивирования клеток удаляли из планшета, и клетки трижды промывали буфером HBSS, после чего в каждую лунку добавляли по 90 мкл буфера HBSS.
[230] 3.2.4 В каждую лунку добавляли 5 мкл разбавленного соединения, и клетки помещали в инкубатор при 37°C, 5% CO2 в течение 20 минут. Каждая лунка содержала 0,5% DMSO. Тестируемое соединение (10 мкМ) применяли в качестве контроля 100% степени ингибирования, а 0,5% DMSO применяли в качестве контроля 0% степени ингибирования.
[231] 3.2.5 В каждую лунку планшета для клеток добавляли 5 мкл разбавленной 14С-мочевой кислоты с конечной концентрацией мочевой кислоты, составляющей 50 мкM в каждой лунке. Клетки помещали в инкубатор при 37°C, 5% CO2 в течение 15 минут. Затем клетки трижды промывали предварительно охлажденным буфером HBSS.
[232] 3.2.6 В каждую лунку добавляли 150 мкл 0,1 M NaOH для лизирования клеток в течение 10 минут.
[233] 3.2.7 Клеточный лизат собирали во флаконы для жидкостного сцинтилляционного тестирования, при этом в каждый флакон для тестирования добавляли дополнительно по 2 мл сцинтилляционной жидкости.
[234] 3.2.8 Содержание 14C в каждом образце определяли с применением жидкостного сцинтилляционного анализатора.
[235] 3.2.9 Анализ данных.
Степень ингибирования, % = (HC-CPD)/(HC-LC) × 100% *
[236] * CPD представляет собой значение радиоактивного сигнала для лунки с соединением;
[237] HC представляет собой среднее значение радиоактивного сигнала для контрольной лунки с 0% ингибированием;
[238] LC представляет собой среднее значение радиоактивного сигнала для контрольной лунки с 100% ингибированием.
[239] 3.2.10 В программном обеспечении GraphPad Prism нелинейная регрессия выполняется с использованием способа "логарифм(ингибитор) против ответа -- переменный угловой коэффициент". Кривую зависимости "доза-эффект" аппроксимируют в соответствии со следующей формулой и определяют значения IC50 и IC90 для соединения.
[240] Y = нижнее значение + (верхнее значение - нижнее значение)/(1 + 10^((LogIC50 - X) * угловой коэффициент Хилла))
[241] 4. Результаты эксперимента
[242] Результаты экспериментов показаны в таблице 9.
[243]

[244] Заключение: соединение по настоящему изобретению проявляет хорошую ингибирующую активность в отношении поглощения мочевой кислоты.
[245] Экспериментальный пример 3. Исследование печеночной метаболической стабильности (HMS) в клетках печени
[246] 1. Цель эксперимента
[247] Проверить метаболическую стабильность тестируемого соединения в клетках печени человека и крысы.
[248] 2. Материалы для эксперимента
[249] 2.1 Тестируемое соединение (10 мМ), контрольные соединения: 7-этоксикумарин (30 мМ), 7-гидроксикумарин (контроль, 30 мМ).
[250] 2.2 Клетки
[251] Информация о клетке показана в таблице 10.
[252] Таблица 10. Информация о клетке

[253] 2.3 Буферная система
[254] Среда для размораживания: среда Вильямса Е, которая содержала 5% фетальной бычьей сыворотки и 30% раствора перколла наряду с другими вспомогательными средствами.
[255] Инкубационная среда: среда Вильямса Е (без фенолового красного), которая содержала 2 мМ L-глутамина и 25 мМ HEPES.
[256] Раствор для остановки реакции: ацетонитрил, который содержал 200 нг/мл толбутамида и лабеталола в качестве внутренних стандартов.
[257] Растворитель для разбавления: сверхчистая вода.
[258] 3. Способы проведения эксперимента
[259] 1) 30 мМ раствор получали путем растворения точного количества соединения положительного контроля в диметилсульфоксиде (DMSO).
[260] 2) В 96-луночном планшете 10 мМ тестируемое соединение и 30 мМ соединение положительного контроля разбавляли до 1 мМ и 3 мМ соответственно, используя DMSO.
[261] 3) Ацетонитрил использовали для дальнейшего разведения 1 мМ тестируемого соединения и 3 мМ соединенияположительного контроля до получения растворов для количественного анализа с конечными концентрациями 100 мкМ и 300 мкМ соответственно.
[262] 4) Хранившиеся клетки размораживали, отделяли и суспендировали в культуральных средах. Затем эти клетки разбавляли до концентрации 0,5×10^6 клеток/мл с использованием предварительно нагретых культуральных сред.
[263] 5) В 96-луночный планшет добавляли 198 мкл предварительно нагретой клеточной суспензии.
[264] 6) В предварительно помеченный набор 96-луночных планшетов переносили 100 мкл раствора для остановки реакции (ацетонитрил, содержащий 200 нг/мл толбутамида и 200 нг/мл лабеталола в качестве внутренних стандартов).
[265] 7) В двух повторностях в каждую лунку 96-луночного планшета добавляли по 2 мкл 100 мкМ тестируемого соединения или 300 мкМ раствора положительного контроля.
[266] 8) Образцы Т0 перемешивали примерно в течение одной минуты для получения гомогенной суспензии, после чего немедленно переносили по 20 мкл каждого образца в лунки, содержащие 100 мкл охлажденного на льду раствора для остановки реакции, и их перемешивали.
[267] 9) Все планшеты инкубировали при 37°C в инкубаторе с 95% увлажненной атмосферой при 5% CO2, и реакцию инициировали постоянным встряхиванием со скоростью примерно 600 об/мин.
[268] 10) Через 15, 30, 60 и 90 минут образцы смешивали, а затем по 20 мкл каждого образца в каждый момент времени переносили в лунки, содержащие 100 мкл охлажденного на льду раствора для остановки реакции, и их перемешивали.
[269] 11) Планшеты для образцов контроля со средой (MC) для T0 и T90 получали путем добавления тех же компонентов, что и в другие лунки, за исключением клеточной суспензии, и помечали как T0-MC и T90-MC. Была составлена итоговая таблица концентраций.
[270] 12) В каждый соответствующий момент времени реакцию останавливали, вынимая планшеты из инкубатора и смешивая их содержимое со 100 мкл охлажденного на льду раствора для остановки реакции.
[271] 13) Немедленное встряхивание планшетов проводили при 500 об/мин. на мешалке с платформой в течение 10 минут. Впоследствии все планшеты с образцами центрифугировали при 3220 x g в течение 20 минут при 4°C.
[272] 14) После центрифугирования 35 мкл супернатанта на лунку из планшетов для образцов переносили в другой набор предварительно помеченных 96-луночных планшетов, содержащих 70 мкл сверхчистой воды.
[273] 15) Аналитические планшеты запечатывали и хранили при 4°C до анализа LC-MS-MS.
[274] По следующей формуле рассчитывали значения остаточного содержания тестируемого соединения и контрольного соединения.

[275] Константу скорости элиминации k для тестируемого соединения и контрольного соединения в гепатоцитах рассчитывали путем построения графика зависимости логарифма остаточного содержания от времени. Период полувыведения (T1/2) и скорость собственного клиренса (CLint) определяли на основе константы скорости выведения k. Формула выглядит следующим образом.
[276] T1/2 = 0,693 / k
[277] CLint (hep) = k / количество клеток на миллилитр (миллион клеток/мл)
[278] CLint (liver) = CLint (hep) × соотношение веса печени к весу тела × количество гепатоцитов на грамм печени
[279] Параметры для каждого вида в формуле показаны в таблице 11 ниже.
[280] Таблица 11. Параметры для каждого вида
[281] 4. Результаты эксперимента
[282] Результаты показаны в таблице 12.
[283]

[284] Заключение: соединение по настоящему изобретению проявляет умеренную степень клиренса в клетках печени человека и высокую степень клиренса в клетках печени крысы.
[285] Экспериментальный пример 4. Тест на проницаемость мембраны MDR1
[286] 1. Цель эксперимента
[287] Клетки MDR1-MDCK II представляют собой клетки почек собаки Мадин-Дарби, трансфицированные геном MDR1 человека, обеспечивающим стабильно высокую экспрессию P-gp. Целью настоящего исследования является проверка двунаправленной проницаемости соединения в модели клеток MDR1-MDCK II и оценка его потенциала в отношении эффлюксного переноса.
[288] 2. Культура клеток
[289] Клетки MDR1-MDCK II (полученные от Piet Borst из Нидерландского института рака) высевали на мембраны из полиэтилентерефталата (PET) в 96-луночную систему с вкладышем с плотностью 2,5×105 клеток/мл. Клетки культивировали до тех пор, пока они не образовывали сливающийся монослой в течение 4-7 дней.
[290] 3. Способы проведения эксперимента
[291] Тестируемые соединения разбавляли в буфере для переноса (HBSS, содержащем 10 мМ Hepes с DMSO, pH 7,4) до концентрации 2 мкМ (DMSO <1%) и наносили либо на апикальную, либо на базолатеральную сторону клеточного монослоя. Повторные измерения тестируемого соединения проводили как в направлениях A-B, так и B-A. Дигоксин также тестировали в обоих направлениях при концентрации 10 мкМ, тогда как надолол и метопролол тестировали при 2 мкМ в направлении А-В. Планшет инкубировали в инкубаторе с CO2 при 37±1°C и 5% CO2 в среде с насыщенной влажностью в течение 2,5 часа без встряхивания. Кроме того, измеряли значения коэффициента оттока для каждого соединения и количественно определяли как тестируемые, так и эталонные соединения на основе отношения площадей пиков аналита к IS с помощью анализа LC/MS/MS. После анализа переноса целостность клеточного монослоя определяли с использованием анализа исключения Люцифера желтого. Буфер удаляли как из апикальной, так и из базолатеральной камер, после чего добавляли 75 мкл 100 мкМ флуоресцеина в буфере для переноса в апикальную камеру и 250 мкл буфера для переноса как в апикальную, так и базолатеральную камеры. Планшет инкубировали при 37°C и 5% CO2 и при насыщенной влажности в течение 30 минут без встряхивания. Через 30 минут инкубации 20 мкл образца флуоресцеина экстрагировали из апикальной камеры и к полученному добавляли 60 мкл буфера для переноса. Затем 80 мкл образца флуоресцеина собирали с базолатеральной стороны клетки. Относительные единицы флуоресценции (RFU) для флуоресцеина измеряли при 425/528 нм (возбуждение/испускание) с использованием микропланшет-ридера Envision.
[292] 4. Расчет данных
[293] Коэффициент кажущейся проницаемости (Papp, см/с.), коэффициент оттока и степень высвобождения рассчитывали по следующим формулам.
[294] Коэффициент кажущейся проницаемости (Papp, см/с.) рассчитывают следующим образом.
[295] Papp = (dCr/dt) × Vr / (A × C0),
[296] где dCr/dt представляет собой кумулятивную концентрацию соединения на приемном конце в единицу времени (М/с.); Vr представляет собой объем принимающего раствора (объемы апикального и базолатерального растворов составляют 0,075 мл и 0,250 мл соответственно); A представляет собой относительную площадь поверхности клеточного монослоя (0,0804 см2); и C0 представляет собой исходную концентрацию тестируемого соединения (нМ) или отношение площадей пиков контроля.
[297] Коэффициент оттока рассчитывают по следующей формуле.
[298] Коэффициент оттока = Papp (BA) / Papp (AB)
[299] Степень высвобождения рассчитывают по следующей формуле.
[300] Степень высвобождения, % = 100 × [(Vr × Cr) + (Vd × Cd)] / (Vd × C0),
[301] где C0 представляет собой исходную концентрацию (нМ) тестового соединения или отношение площадей пиков контроля; Vd представляет собой объем на дозирующем конце (апикальная сторона - 0,075 мл, а базолатеральная сторона - 0,250 мл); Cd и Cr представляют собой конечные концентрации (нМ) тестового соединения на дозирующей и принимающей сторонах соответственно или отношение площадей пиков контроля.
[302] Долю в процентах флуоресцеина в базолатеральных лунках рассчитывают по следующей формуле.

[303] в которой RFUApical и RFUBasolateral представляют собой относительные единицы флуоресценции флуоресцеина в апикальных и базолатеральных лунках соответственно; VApical и VBasolateral представляют собой объемы апикальных и базолатеральных лунок соответственно (0,075 мл и 0,25 мл). Доля в процентах флуоресцеина должна составлять менее 2%.
[304] 5. Результаты эксперимента
[305] Результаты показаны в таблице 13.
[306]

[307] Заключение: соединение по настоящему изобретению характеризуется высокой проницаемостью.
[308] Экспериментальный пример 5. Тестирование ингибирующей активности в отношении изофермента цитохрома P450
[309] 1. Цель эксперимента
[310] Определить ингибирующую активность тестируемого соединения в отношении различных подтипов изоферментов цитохрома Р450 человека.
[311] 2. Способы проведения эксперимента
[312] Получали тестируемые соединения, стандартные ингибиторы (при 100× конечной концентрации) и рабочие растворы смешанных субстратов; микросомы (приобретенные у Corning Inc.), хранившиеся при -80°C, вынимали и размораживали. В соответствующие лунки добавляли по 20 мкл тестируемого соединения и растворов стандартного ингибитора. Тем временем 20 мкл соответствующего растворителя добавляли в контрольные лунки без ингибитора (NIC) и лунки холостого контроля. Затем в соответствующие лунки добавляли по 20 мкл раствора смешанного субстрата за исключением пустых лунок, в которые добавляли 20 мкл фосфатного буфера (PB). Получали раствор микросом печени человека (возвращали в холодильник сразу после отметки о дате использования). Затем во все лунки добавляли по 158 мкл этого раствора. Планшет с образцами помещали на водяную баню с температурой 37°C для предварительной инкубации. Затем сразу же получали раствор фактора кофермента (NADPH). Через 10 минут во все лунки добавляли по 20 мкл раствора NADPH. Планшет с образцами встряхивали для перемешивания и помещали обратно на водяную баню при 37°C для инкубации еще в течение 10 минут. В соответствующие моменты времени реакцию останавливали посредством добавления 400 мкл холодного раствора ацетонитрила (внутренний стандарт в концентрации 200 нг/мл толбутамида и лабеталола). Планшет с образцами тщательно перемешивали, а затем центрифугировали при 4000 об/мин. в течение 20 минут для осаждения белков. Отбирали 200 мкл супернатанта и смешивали со 100 мкл воды, после чего его отправляли на анализ LC/MS/MS.
[313] 3. Результаты эксперимента
[314] Результаты показаны в таблице 14.
[315] Таблица 14. Значения IC50 соединения для ингибирования изоферментов P450

[316] Заключение: соединение по настоящему изобретению проявляет чрезвычайно низкую ингибирующую активность в отношении CYP1A2, CYP2C19, CYP2D6 и CYP3A4-M и умеренную ингибирующую активность в отношении CYP2C9.
[317] Пример 6. Фармакокинетика у крыс SD in vivo
[318] 1. Цель эксперимента
[319] Проверить фармакокинетику соединения на крысах SD in vivo.
[320] 2. Экспериментальные материалы
[321] Крысы Sprague Dawley (самцы, 180-350 г, возраст 6-10 недель, Beijing Vital River).
[322] 3. Способы проведения эксперимента
[323] Соединение смешивали с 5% DMSO/10% солютола/85% воды, перемешивали и встряхивали с получением прозрачного раствора 0,6 мг/мл для введения в группе, получавшей инъекцию. Затем раствор фильтровали через микропористую мембрану для последующего использования. Соединение смешивали с 5% DMSO/10% солютола/85% воды, перемешивали и встряхивали с получением прозрачного раствора 1 мг/мл для перорального введения. Шесть самцов крыс SD разделяли на две группы. В первой группе животным однократно внутривенно вводили инъекцию в дозе 3 мг/кг, используя в качестве растворителя смесь 5% DMSO/10% солютола/85% воды, при этом объем дозирования составлял 5 мл/кг. Во второй группе животные получали однократную пероральную дозу, вводимую через желудочный зонд, тестируемого соединения в дозе 10 мг/кг. Растворитель для перорального применения представлял собой 5% DMSO/10% солютола/85% воды, при этом объем дозирования составлял 10 мл/кг. Образцы цельной крови собирали через 0 (только для группы приема через желудочный зонд), 0,083 (только для группы приема путем внутривенной инъекции), 0,25, 0,5, 1, 2, 4, 8 и 24 часа после введения. Цельную кровь центрифугировали при 3200 g в течение 10 минут при 4°C с получением плазмы крови. Значения концентрации соединения и мочевой кислоты (только для группы приема через желудочный зонд) в плазме крови измеряли с применением способа LC/MS/MS. Фармакокинетические параметры, такие как пиковая концентрация, время достижения пиковой концентрации, скорость клиренса, период полувыведения, площадь под кривой и биологическая доступность, рассчитывали с применением программного обеспечения Phoenix WinNonlin.
[324] Результаты показаны в таблице 15 ниже.
[325]

[326] Заключение: соединения по настоящему описанию характеризуются предпочтительными фармакокинетическими свойствами и высокой биологической доступностью при пероральном приеме, где C0 представляет собой исходную концентрацию, T1/2 представляет собой период полувыведения, Vdss представляет собой кажущийся объем распределения в установившемся состоянии, Cl представляет собой общую скорость клиренса, AUC0-last представляет собой площадь под кривой зависимости концентрации в плазме крови от времени от момента времени 0 до последней поддающейся количественному измерению точки времени, AUC0-inf представляет собой площадь под кривой зависимости концентрации в плазме крови от момента времени 0, экстраполированной до бесконечности, Cmax представляет собой пиковую концентрацию, а Tmax представляет собой время достижения пиковой концентрации.






Изобретение относится к кристаллической форме производного тиофена, выбранной из формы С и Е. Кристаллическая форма C соединения формулы (I) характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 25,38±0,20°, 26,66±0,20°, 30,70±0,20°. Кристаллическая форма E соединения формулы (I) характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 12,43±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 23,15±0,20°, 25,14±0,20°. Технический результат - кристаллические формы C и E производного тиофена формулы (I) для изготовления лекарственного препарата для лечения подагры и гиперурикемии. 2 н. и 16 з.п. ф-лы, 14 ил., 15 табл., 14 пр.

1. Кристаллическая форма C соединения формулы (I), где кристаллическая форма C характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 25,38±0,20°, 26,66±0,20°, 30,70±0,20°,
 .
.
2. Кристаллическая форма C по п. 1, где ее порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,03±0,20°, 13,20±0,20°, 15,26±0,20°, 18,08±0,20°, 21,99±0,20°, 25,07±0,20°, 25,38±0,20°, 26,66±0,20°, 28,38±0,20°, 29,41±0,20°, 30,70±0,20°, 38,53±0,20°.
3. Кристаллическая форма C по п. 2, где ее порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих значениях угла 2θ: 13,20°, 15,26°, 18,08°, 21,99°, 25,07°, 25,38°, 26,66°, 30,70°.
4. Кристаллическая форма C по п. 3, где ее порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих значениях угла 2θ: 5,66°, 9,03°, 11,13°, 13,20°, 13,70°, 15,26°, 17,25°, 18,08°, 18,92°, 20,88°, 21,99°, 23,41°, 24,09°, 25,07°, 25,38°, 25,99°, 26,66°, 27,17°, 28,38°, 29,41°, 29,98°, 30,70°, 31,02°, 31,72°, 33,67°, 35,40°, 36,35°, 36,74°, 37,26°, 38,53°, 39,80°.
5. Кристаллическая форма C по любому из пп. 1-4, где кристаллическая форма C характеризуется дифрактограммой XRPD, показанной на фигуре 5.
6. Кристаллическая форма C по любому из пп. 1-4, где кристаллическая форма C характеризуется кривой термогравиметрического анализа с потерей веса 1,21% при 200°C±3°C.
7. Кристаллическая форма C по п. 6, где кристаллическая форма C характеризуется термограммой TGA, показанной на фигуре 6.
8. Кристаллическая форма C по любому из пп. 1-4, где кристаллическая форма C характеризуется кривой дифференциальной сканирующей калориметрии, содержащей эндотермический пик с началом при 250,0°C±2°C.
9. Кристаллическая форма C по п. 8, где кристаллическая форма C характеризуется термограммой DSC, показанной на фигуре 7.
10. Кристаллическая форма E соединения формулы (I), где кристаллическая форма E характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 12,43±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 23,15±0,20°, 25,14±0,20°,
.
11. Кристаллическая форма E по п. 10, где ее порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11±0,20°, 11,21±0,20°, 12,43±0,20°, 13,28±0,20°, 15,34±0,20°, 18,16±0,20°, 22,06±0,20°, 23,15±0,20°, 25,14±0,20°, 25,97±0,20°, 26,75±0,20°, 27,25±0,20°.
12. Кристаллическая форма E по п. 11, где ее порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11°, 12,43°, 13,28°, 15,34°, 18,16°, 22,06°, 23,15°, 25,14°.
13. Кристаллическая форма E по п. 12, где ее порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,11°, 10,94°, 11,21°, 12,43°, 13,28°, 15,34°, 17,39°, 18,16°, 18,94°, 20,18°, 20,95°, 22,06°, 23,15°, 23,35°, 24,19°, 25,14°, 25,97°, 26,75°, 27,25°, 28,45°, 29,49°, 30,16°, 30,82°, 33,74°, 35,45°, 36,39°, 37,34°, 38,57°.
14. Кристаллическая форма E по любому из пп. 10-13, где кристаллическая форма E характеризуется дифрактограммой XRPD, показанной на фигуре 11.
15. Кристаллическая форма E по любому из пп. 10-13, где кристаллическая форма E характеризуется кривой термогравиметрического анализа с потерей веса 0,79% при 200°C±3°C.
16. Кристаллическая форма E по п. 15, где кристаллическая форма E характеризуется термограммой TGA, показанной на фигуре 12.
17. Кристаллическая форма E по любому из пп. 10-13, где кристаллическая форма E характеризуется кривой дифференциальной сканирующей калориметрии, содержащей эндотермический пик с началом при 250,4°C±2°C.
18. Кристаллическая форма E по п. 17, где кристаллическая форма E характеризуется термограммой DSC, показанной на фигуре 13.
| WO 2010044403 A1, 22.04.2010 | |||
| ПРОИЗВОДНЫЕ КАРБОКСИЗАМЕЩЕННЫХ (ГЕТЕРО)АРОМАТИЧЕСКИХ КОЛЕЦ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2733750C2 |
| Sarma B | |||
| et al | |||
| Solid formation of pharmaceuticals: Polymorphs, salt and cocrystals | |||
| Korean J.Chem.Eng., 2011, 28(2), p.315-322; | |||
| Narayan Variankaval; et al | |||
| From form to function: Crystallization of active pharmaceutical ingredients, AlChE, 2008, vol.54(7), p.1682-1688 | |||
| Дж | |||
| Бернштейн | |||

