ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к эластомерной полимерной композициии и ее получению. Полимерная композиция включает в сочетании односторонне терминально-модифицированный сопряженный диеновый (со)полимер и двусторонне терминально- модифицированный сопряженный диеновый (со)полимер. Полимерная композиция является полезной в приготовлении вулканизированной и, таким образом, сшитой эластомерной композиции, которая имеет относительно низкие гистерезисные потери, хорошее сцепление и низкие теплонакопительные свойства. Такие композиции полезны во многих изделиях, в том числе протекторах шин, которые обладают низким теплонакоплением и обеспечивают низкий расход топлива при езде, в сочетании с хорошим балансом других желательных физических и химических свойств, к примеру, хоршее сцепление с мокрой дорогой, сцепление на льду, стойкость к истиранию, прочность на растяжение и отличная пригодность для обработки.
УРОВЕНЬ ТЕХНИКИ
Гистерезисные энергитические потери вулканизированной резины, как правило, соотносятся с числом свободных концевых групп полимерной цепи. К примеру, полимер с высокой молекулярной массой обладает пониженным содержанием по массе концевых групп и имеет уменьшенные гистерезисные потери, но также имеет сниженную пригодность для обработки резиновых смесей. Функционализация концевых групп цепи с полярными группами, пригодными для взаимодействия с наполнителем, приводит к снижению количества свободных концевых групп цепи и, как правило, снижает гистерезис. Тем не менее, высокое количество взаимодействий концевых групп цепи и наполнителя приводит к заметному снижению пригодности для переработки соединения.
Эластомерные полимеры, полученные способами анионной полимеризации, в основном содержат линейные полимерные цепи. Высоко линейные эластомерные полимерные цепи показывают высокие вязкости раствора и низкотемпературные свойства. Чтобы преодолеть эти недостатки, были применены процессы связывания для образования, по меньшей мере частично, разветвленных полимерных цепей. Обычно используемые связующие вещества представляют собой дивинилбензол, галогенидные или алкоксидные соединения олова или кремния. Тем не менее, разветвленные или звездообразные полимеры, которые являются результатом таких реакций конденсации, часто демонстрируют повышенные гистерезисные потери или пониженную стойкость к истиранию.
WO 2007/047943 описывает применение силан сульфидного омега модификатора концевых групп цепи для получения эластомерного полимера с модифицированной концевой группой, который применяется в качестве компонента в вулканизированной композиции эластомерного полимера или протекторе шин.
Согласно WO 2007/047943, силан сульфидное соединение реагирует с анионно-инициированными живыми полимерами для получения полимеров "с модифицированными концевыми группами", которые впоследствии смешивают с наполнителями, вулканизирующими агентами, добавками-ускорителями или масляными мягчителями для получения вулканизированной композиции эластомерного полимера, имеющего низки гистерезисные потери.
Вулканизированные композиции эластомерных полимеров описаны как такие, которые показывают низкие значения тангенса δ при 60 °C, в частности, в сравнении с соединениями на основе соответствующих немодифицированных полимеров, без негативного влияния на значения тангенса δ при 0°C и технологических свойств, таких как значения соединения по Муни. Более низки значения тангенса δ при 60°C соответствуют более низкому сопротивлению качению, тогда как повышенный тангенс δ при 0°C соответствует улученому сцеплению шины с мокрой дорогой. Примерные отвержденные полимерные составы, как было показано, приводят к снижению тангенса δ при 60°C и значений теплонакопления, но эквивалентны значениям тангенса δ при 0 °C. Они описаны как имеющие пользу при изготовлении протекторов, имеющих более низкое сопротивление качению, в то же время с сохранением хороших свойств сцепления с влажным покрытием. Хотя гистерезисные свойства отвержденной резины могут быть значительно улучшены путем применения технологии, описанной в WO 2007/047943, воздействие технологии ограничено из-за того, что только один конец полимерной цепи может быть функционализирован с помощью описанного модифицирующего соединения. Соответственно, существует потребность в эффективной модификации второго конца полимерной цепи.
EП 2 518 104 описывает резиновую композицию для применения для шин, содержащую диеновый мономер на основе полимера и диоксида кремния. Диеновый полимер готовят с инициатором для получения живых полимерных цепей, имеющих две живые полимерные концевые группы.
Существует потребность в методах модификации и получающихся в результате полимеров, включая моифицированные полимеры, обеспечивающих превосходный баланс динамических свойств сшитым резиновым соединениям, содержащим диоксид кремния, таких как низкие гистерезисные потери и повышенная стойкость к истиранию, соответствующие повышенному сцеплению с мокрой поверхностью, низкое сопротивление качению и повышенная стойкость к истиранию в шинах, и поддержание приемлемой или улучшенной пригодности для обработки. Кроме того, желаемой задачей является обеспечить полимер, который обладает хорошей пригодностью для обработки (a) в процессе производства самого полимера, к примеру, в связи с низкой сцепляемостью во время процесса удаления растворителя и влаги, и (b) в ходе дальнейших этапов обработки, таких как подготовка и обработка насыщенных диоксидом кремния резиновых составов. Эти потребности были удовлетворены посредством следующего изобретения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В первом аспекте, настоящее изобретение предлагает полимерную композицию, которая, в ее общем варианте осуществления (также называемой в данном документе как "первая полимерная композиция"), содержит модифицированные полимеры согласно следующей Формуле 1 и Формуле 2:
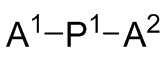 Формула 1
Формула 1
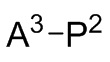 Формула 2
Формула 2
где
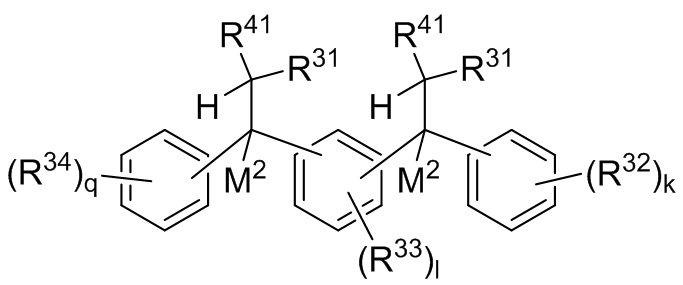
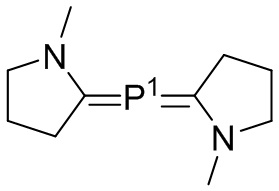
P1 и P2 каждый представляет собой независимо полимерную цепь, получаемую путем анионной полимеризации одного или более полимеризуемых мономеров, выбранных из сопряженных диенов и ароматических винильных соединений, причем каждая полимерная цепь P1 и P2 содержит по меньшей мере 40% по массе повторяющихся звеньев, полученных путем полимеризации указанных сопряженных диенов, и при этом по меньшей мере анионная полимеризация полимерной цепи P1 осуществляется в присутствии соединения следующей Формулы 9a:
 Формула 9a
Формула 9a
где каждый R31 независимо выбран из водорода, (C1-C10) алкила, (C6-C12) арила и (C7-C18) аралкила; каждый R32, R33 и R34 независимо выбран из водорода, (C1-C18) алкила и (C1-C18) алкокси; каждый R41 независимо выбран из (C1-C100) алкила и (C2-C100) алкенила, где каждый R41 необязательно замещен от одной до трех (C6-C12) арильными группами и необязательно связан со структурой Формулы 9a через олигомерную цепь, состоящую до 25 мономерных звеньев, выбранных из сопряженных диенов, особенно 1,3-бутадиена и изопрена, и ароматических винильных соединений, особенно стирола и дивинилбензола; каждый M2 независимо выбран из лития, натрия и калия; и k, l и q представляют собой целые числа, выбранные из 0, 1, 2 и 3;
A1, A2 и A3 независимо выбирают из следующих от Формулы 3 до Формулы 8:
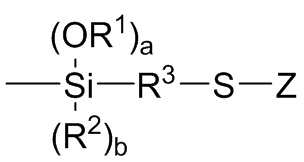 Формула 3
Формула 3
где
каждый R1 независимо выбран из (C1-C16) алкила;
каждый R2 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
a и b представляют собой целые числа, независимо выбранные из 0, 1 и 2, с a+b=2;
R3 независимо выбран из двухвалентного (C1-C16) алкила, двухвалентного (C6-C18) арила, двухвалентного (C7-C18) аралкила и -R4-O-R5-, где R4 и R5 независимо выбраны из двухвалентного (C1-C6) алкила; и
Z независимо выбран из (C1-C16) алкила, (C6-C18) арила, (C7-C18) аралкила, (C=S)-S-R6, где R6 выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила, и M1(R7)c(R8)d,
где
M1 представляет собой кремний или олово,
каждый R7 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R8 независимо выбран из -S-R3-Si(OR1)r(R2)s, где R1, R2 и R3 являются такими как определено для Формулы 3 выше, r представляет собой целое число, независимо выбранное из 1, 2 и 3 и s представляет собой целое число, независимо выбранное из 0, 1 и 2, с r+s=3;
c представляет собой целое число, независимо выбранное из 2 и 3; d представляет собой целое число, независимо выбранное из 0 и 1; и c+d=3;
 Формула 4
Формула 4
 Формула 5
Формула 5
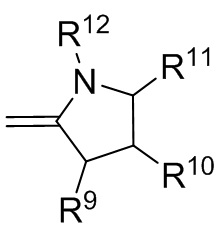
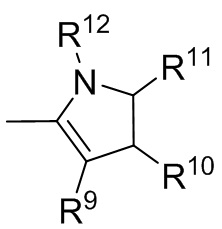
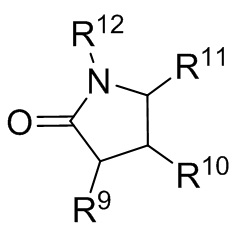
где R9, R10, R11 и R12 независимо выбранные из водорода, (C1-C16) алкила, (C6-C16) арила и (C7-C16) аралкила;
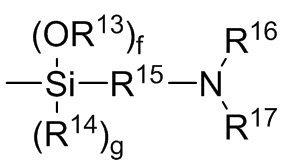 Формула 6
Формула 6
 Формула 7
Формула 7
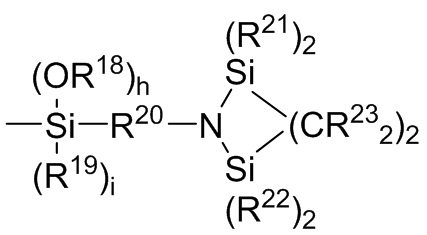
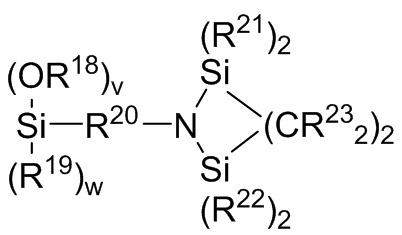
где каждый R13, R14, R18 и R19 независимо выбран из (C1-C16) алкила;
R15 и R20 независимо выбраны из двухвалентного (C1-C16) алкила, двухвалентного (C6-C18) арила, двухвалентного (C7-C18) аралкила и -R24-O-R25-, где R24 и R25 независимо выбраны из двухвалентного (C1-C6) алкила;
R16 и R17 независимо выбирают из (C1-C16) алкила и -SiR26R27R28, где R26, R27 и R28 независимо выбирают из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R21 и R22 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R23 независимо выбран из водорода и (C1-C6) алкила;
f, g, h и i представляют собой целые числа, независимо выбранные из 0, 1 и 2; f+g=2; и h+i=2;
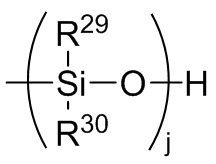 Формула 8
Формула 8
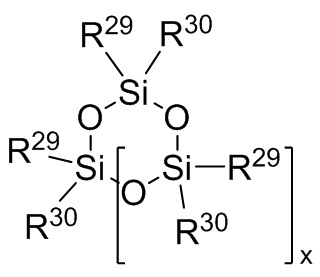
где каждый R29 и R30 независимо выбран из (C1-C16) алкила, (C6-C18) арила, (C7-C18) аралкила и винила; и
j представляет собой целое число, выбранное из от 1 до 200; и
где количество полимера Формулы 1 составляет от 15 до 85% в молях на основе общего количества полимера Формулы 1 и полимера Формулы 2.
Полимерная композиция в соответствии с данным изобретением может содержать один или более дополнительных компонентов, выбранных из (i) компонентов, которые добавляются или образованные в результате процесса полимеризации, применяемого для изготовления полимеров Формул 1 и 2 и (ii) компонентов, которые остаются после удаления растворителя из процесса полимеризации. Этот вариант осуществления первого аспекта изобретения также упоминается в данном документе как "вторая полимерная композиция". Компоненты, которые добавляют в процесс полимеризации включают, в частности, смягчители (масло для наполнения), стабилизаторы и полимеры, которые не являются полимерами Формулы 1 или Формулы 2. Компоненты, которые образуются в результате процесса полимеризации и которые остаются после удаления растворителя из процесса полимеризации, включают, в частности, полимеры, которые не являются полимерами Формулы 1 или Формулы 2.
Полимерная композиция в соответствии с данным изобретением (обе - первая и вторая полимерная композиция) может также содержать один или более дополнительных компонентов, которые добавляют после процесса полимеризации, включая один или более наполнителей, один или более дополнительных полимеров, которые не являются полимером Формулы 1 или Формулы 2, и один или более сшивающих агентов (вулканизирующих агентов). Этот вариант осуществления первого аспекта изобретения также упоминается в этом документе как "третья полимерная композиция". Компоненты, добавленные после полимеризации, как правило, добавляют к полимерной композиции после удаления растворителя из процесса полимеризации, и, как правило, добавляют с помощью механического перемешивания.
Во втором аспекте, настоящее изобретение предлагает способ получения полимерной композиции первого аспекта, причем указанный способ включает этапы
i) приведения в контакт смеси инициатора полимеризации, получаемой путем приведения в контакт соединения следующей Формулы 9:
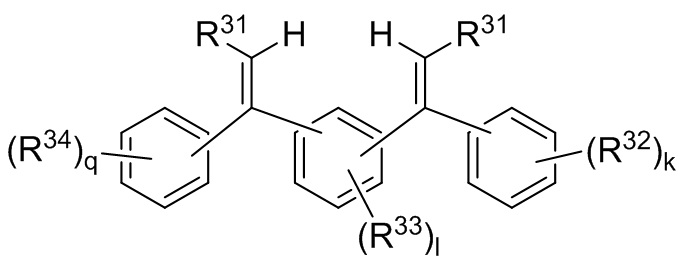 Формула 9
Формула 9
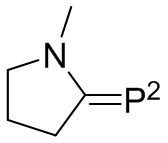
где каждый R31 независимо выбран из водорода, (C1-C10) алкила, (C6-C12) арила и (C7-C18) аралкила; каждый R32, R33 и R34 независимо выбран из водорода, (C1-C18) алкила и (C1-C18) алкокси; k, l и q представляют собой целые числа, независимо выбранные из 0, 1, 2 и 3,
с соединением следующей Формулы 10
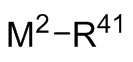 Формула 10
Формула 10
где M2 выбран из лития, натрия и калия и R41 выбран из (C1-C100) алкила и (C2-C100) алкенила, где каждый R41 необязательно замещен от одной до трех (C6-C12) арильными группами и необязательно связан с M2 через олигомерные цепи, состоящие из от 25 мономерных звеньев, выбранных из сопряженных диенов, особенно 1,3-бутадиена и изопрена, и ароматических винильных соединений, особенно стирола и дивинилбензола;
с одним или более полимеризуемых мономеров, выбранных из сопряженных диенов и ароматических винильных соединений для получения живих полимерных цепей, причем живые полимерные цепи содержат по меньшей мере 40% по массе повторяющихся звеньев, полученных путем полимеризации указанных сопряженных диенов, и
при этом молярное соотношение соединений Формулы 10 к соединениям Формулы 9 находится в диапазоне от 1:1 до 8:1, и
ii) приведения в контакте живых полимерных цепей этапа i) с одним или более агентами, модифицирующими концевую группу цепи, выбранными из соединений следующей Формулы 11 до Формулы 15:
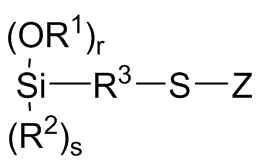 Формула 11
Формула 11
где R1, R2, R3, Z, r и s являются такими, как определено для Формулы 3 выше;
 Формула 12
Формула 12
где R9, R10, R11 и R12 являются такими, как определено для Формулы 4 выше;
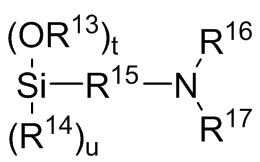 Формула 13
Формула 13
где R13, R14, R15, R16 и R17 являются такими, как определено для Формулы 6 выше; t представляет собой целое число, выбранное из 1, 2 и 3; u представляет собой целое число, выбранное из 0, 1 и 2; и t+u=3;
 Формула 14
Формула 14
где R18, R19, R20, R21, R22 и R23 являются такими, как определено для Формулы 7 выше; v представляет собой целое число, выбранное из 1, 2 и 3; w представляет собой целое число, выбранное из 0, 1 и 2; и v+w=3;
 Формула 15
Формула 15
где каждый R29 и R30 является таким, как определно для Формулы 8 выше; и x представляет собой целое число, выбранное из от 1 до 6; и
где агенты, модифицирующие концевую группу цепи Формулы 11 до Формулы 15 применяются в общем количестве
(i) от 0,15 до 0,85 молей на моль соединений Формулы 10, в то время, когда молярное соотношение соединений Формулы 10 к соединениям Формулы 9 находится в диапазоне от 1:1 до 2,1:1, и
(ii) от 0,5 до 3 молей на моль соединений Формулы 10 в то время, когда молярное соотношение соединений Формулы 10 к соединениям Формулы 9 находится в диапазоне от более чем 2,1:1 до 8:1.
В третьем аспекте, настоящее изобретение предлагает сшитую полимерную композицию, которую получают путем сшивания полимерной композиции в соответствии с данным изобретением, которая содержит один или более сшивающих (вулканизирующих) агентов.
В четвертом аспекте, настоящее изобретение предлагает способ получения сшитой полимерной композиции третьего аспекта, причем указанный способ включает этап сшивания полимерной композиции, которая содержит один или более сшивающих агентов.
В пятом аспекте, настоящее изобретение предлагает изделие, содержащее по меньшей мере один компонент, образованный из сшитой полимерной композиции в соответствии с данным изобретением. Изобретением может быть, например, шина, протектор шины, боковая стенка шины, автомобильная часть, компонент обуви, мяч для гольфа, ремень, уплотнитель, прокладка или шланг.
ГРАФИЧЕСКИЕ МАТЕРИАЛЫ
Чертеж: Коэффициенты трения при скольжении полимерных примеров.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
СМЕСЬ ИНИЦИАТОРА ПОЛИМЕРИЗАЦИИ И СПОСОБ ПРИГОТОВЛЕНИЯ
Инициатор полимеризации для получения полимерной композиции согласно первому аспекту данного изобретения представляет собой продукт реакции соединения Формулы 9 с соединением Формулы 10. Реакционный продукт представляет собой дианионное соединение Формулы 9a как определено выше, которое имеет по меньшей мере два карбаниона, каждый из которых связан с атомом металла M2. Типичные структуры и способы приготовления таких продуктов описаны, например, в EП 0 316 857, США 4172190, США 5521255, США 5561210 и EП 0 413 294.
В одном варианте реализации изобретения соединение Формулы 9a и лежащая в основе реакции между соединением Формулы 9 и соединением Формулы 10,
M2 представляет собой литий;
R41 выбран из (C1-C10) алкила;
каждый R31 независимо выбран из водорода и (C1-C10) алкила, предпочтительно водорода;
R32 и R34 идентичны и выбраны из водорода и (C1-C18) алкила;
каждый R33 независимо выбран из водорода и (C1-C18) алкила;
все другие заместители или группы, как правило, определены для Формулы 9 и Формулы 10.
Конкретные предпочтительные виды соединения Формулы 9 включают следующие соединения:
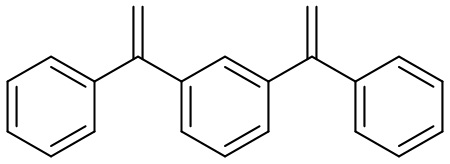
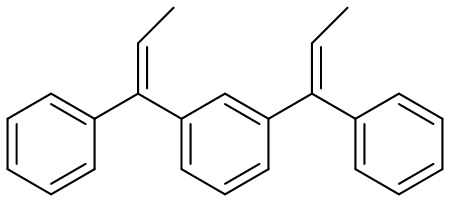
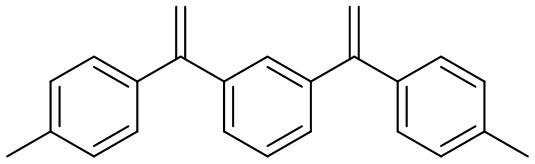
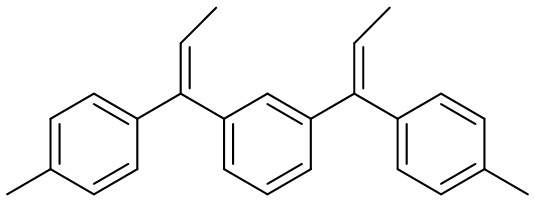
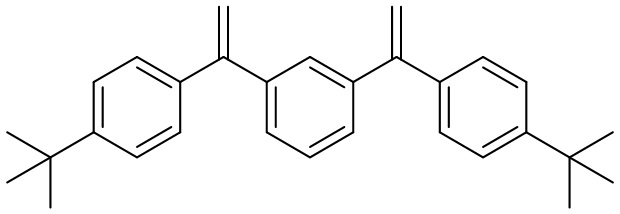
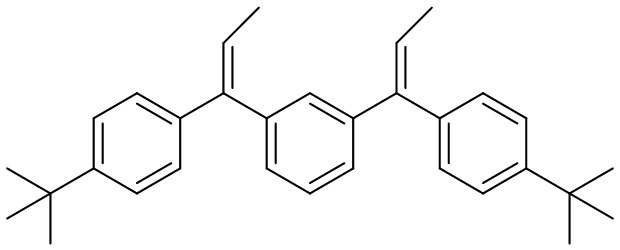
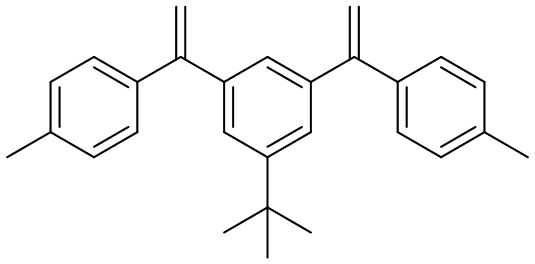
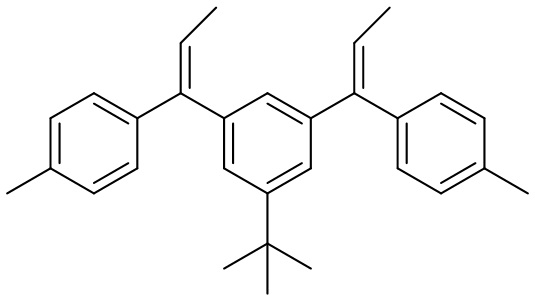
Конкретные предпочтительные виды соединения Формулы 10 включают следующие соединения:
метиллитий, этиллитий, пропиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий, фениллитий, гексиллитий, предпочтительно н-бутиллитий и втор-бутиллитий.
Конкретные предпочтительные виды соединения Формулы 9a включают те, которые получают путем приведения в контакт одного или более из указанных выше конкретных предпочтительных видов соединения Формулы 9 с одним или более из указанных выше конкретных предпочтительных видов соединения Формулы 10.
Молярное соотношение в реакции между соединением Формулы 10 и соединением Формулы 9 находится в диапазоне от 1:1 до 8:1, предпочтительно от 1,5:1 до 8:1 и более предпочтительно от 2:1 до 6:1. При соотношении ниже 1:1, количество соединения Формулы 9, оставшегося после реакции с соединением Формулы 10 может вызвать значительное гелеобразование во время процесса полимеризации. При соотношении выше 8:1, количество соединения Формулы 10, оставшегося после реакции с соединением Формулы 9 приведет к менее чем 15% полимера Формулы 1 (на основе общего количества полимера Формулы 1 и полимера Формулы 2).
В одном варианте реализации изобретения, от 1 до 2,1 эквивалентов соединения Формулы 10 вступают в реакцию с одним эквивалентом соединения Формулы 9. В этом случае (учет погрешности эксперимента), полное количество соединения Формулы 10 вступает в реакцию с соединением Формулы 9. В последующем приготовлении полимерной композиции в соответствии с данным изобретением, необходимо использовать менее чем молярное (менее чем эквивалент) количество агентов, модифицирующих концевую группу цепи Формулы 11 до Формулы 15 относительно молярного количества соединения Формулы 9 с тем, чтобы получить композицию, содержащую полимеры как Формулы 1, так и Формулы 2.
В альтернативном варианте реализации изобретения, более чем 2,1 и до 8 эквивалентов соединения Формулы 10 вступают в реакцию с одним эквивалентом соединения Формулы 9, с получением таким образом смеси (композиции) дианионного инициатора (Формулы 9a) и моноанионного инициатора (оставшееся количество соединения Формулы 10). В таком случае, молярное количество агентов, модифицирующих концевую группу цепи Формулы 11 до Формулы 15, применяемого в приготовлении полимерной композиции в соответствии с данным изобретением может быть меньше, равно или более, чем молярное количество соединения Формулы 9 (соответственное к молярному количеству соединения Формулы 9a). Предпочтительное молярное соотношение соединения Формулы 10 к соединению Формулы 9 в этом варианте осуществления находится в диапазоне от более чем 2,1:1 до 7:1, более предпочтительно от более чем 2,1:1 до 6:1, даже более предпочтительно от 2,5:1 от 3,5:1, или приблизительно 3:1.
Способ получения смеси инициатора полимеризации включает этап приведения в контакт соединения-предшественника инициатора Формулы 9 с по меньшей мере одним соединением Формулы 10, необязательно в присутствии основания Льюиса. Реакцию предпочтительно проводят в неполярном растворителе, включая углеводородный растворитель, включая алифатический и ароматический растворитель, предпочтительно алифатический растворитель, такой как гексан, гептан, пентан, высокочистые изопарафины-растворители, циклогексан и метилциклогексан, и, как правило, проводят в течении периода времени от 2 секунд до 10 дней, предпочтительно от 5 секунд до 5 дней, даже более предпочтительно от 10 секунд до 2 дней, при температуре в диапазоне от -60°C до 130 °C, предпочтительно от 0°C до 100°C и даже более предпочтительно от 20°C до 70 °C.
В однома варианте реализации изобретения, соединение Формулы 10 может вступать в реакцию до реакции с соединением Формулы 9, с до 25 мономерными молекулами, выбранными из сопряженных диенов, особенно 1,3-бутадиена и изопрена, и ароматических винильных соединений, особенно стирола. Полученный в результате "олигомер", который находится в пределах Формулы 10, подвергают взаимодействию с соединением-предшественником инициатора Формулы 9, как описано в данном документе.
Необязательно может быть добавлена кислота Льюиса к соединению-предшественнику Формулы 9 перед добавлением и реакцией с соединением Формулы 10, с тем, чтобы она присутствовала в реакции с самого начала. Альтернативно, она может быть добавлена во время реакции или после завершения реакции. Любое из этих альтернативных добавлений приводит к образованию аддукта основания Льюиса с продуктом реакции, т.е. с соединением Формулы 9a. Когда присутствует основание Льюиса, его обычно применяют в соотношении соединения-предшественника инициатора Формулы 9 к основанию Льюиса в виде мольных эквивалентов от 0,01 до 20, предпочтительно от 0,05 до 5,0 и даже более предпочтительно от 0,1 до 3.0.
Для повышения стабильности свойств при хранении (предельный срок хранения) смеси инициатора полимеризации, возможна реакция полученной в результате реакции смеси, содержащей смесь инициатора полимеризации и включающей щелочной металл M2 с одним или более полимеризуемыми мономерами, выбранными из сопряженных диеновых мономеров, особенно выбранных из бутадиена и изопрена, и ароматических винильных соединений, особенно стирола. С этой целью удобно применть количество до 1000 эквивалентов, предпочтительно до 200 эквивалентов, более предпочтительно до 75 эквивалентов полимеризуемого мономера на эквивалент щелочного металла.
ПОЛИМЕРНАЯ КОМПОЗИЦИЯ
Полимерная композиция в соответствии с первым аспектом данного изобретения содержит модифицированные эластомерные полимеры согласно с Формулой 1 и Формулой 2 как определено в этом документе. В общем, количество полимера Формулы 1 представляет собой 15-85% в молях на основе общего количества полимера Формулы 1 и полимера Формулы 2. Если количество полимера Формулы 1 составляет более, чем 85% в молях, пригодность для обработки полимерной композиции, когда дополнительно содержится один или более наполнителей, может оказать негативное влияние. Если количество полимера Формулы 1 составляет меньше, чем 15% в молях, это может негативно повлиять на техническую характеристику сшитой полимерной композиции изобретения, в частности, в выражении сопротивления качению.
Конкретные предпочтительные варианты реализации полимера Формулы 1 включают, но не ограничиваются ими, следующие структуры (с таким P1 как определно в данном документе):
[Me3Si S (CH2)3 Si(OMe)(Me)]2P1, [Me3Si S (CH2)3 Si(OEt)(Me)]2P1, [Me3Si S (CH2)3 Si(OMe)2]2P1, [Me3Si S (CH2)3 Si(OEt)2]2P1, [Me3Si S (CH2)3 Si(OPr)(Me)]2P1, [Me3Si S (CH2)3 Si(OBu)(Me)]2P1, [Me3Si S (CH2)3 Si(OPr)2]2P1, [Me3Si S (CH2)3 Si(OBu)2]2P1, [Et3Si S (CH2)3 Si(OMe)(Me)]2P1, [Et3Si S (CH2)3 Si(OEt)(Me)]2P1, [Et3Si S (CH2)3 Si(OMe)2]2P1, [Et3Si S (CH2)3 Si(OEt)2]2P1, [Et3Si S (CH2)3 Si(OPr)(Me)]2P1, [Et3Si S (CH2)3 Si(OBu)(Me)]2P1, [Et3Si S (CH2)3 Si(OPr)2]2P1, [Et3Si S (CH2)3 Si(OBu)2]2P1, [(Bu)Me2Si S (CH2)3 Si(OMe)(Me)]2P1, [(Bu)Me2Si S (CH2)3 Si(OEt)(Me)]2P1, [(Bu)Me2Si S (CH2)3 Si(OMe)2]2P1, [(Bu)Me2Si S (CH2)3 Si(OEt)2]2P1, [(Bu)Me2Si S (CH2)3 Si(OPr)(Me)]2P1, [(Bu)Me2Si S (CH2)3 Si(OBu)(Me)]2P1, [(Bu)Me2Si S (CH2)3 Si(OPr)2]2P1, [(Bu)Me2Si S (CH2)3 Si(OBu)2]2P1, [Me3Si S (CH2) Si(OMe)2]2P1, [Me3Si S (CH2) Si(OEt)2]2P1, [Me3Si S (CH2)2 Si(OMe)2]2P1, [Me3Si S (CH2)2 Si(OEt)2]2P1, [Me3Si S (CH2) Si(OMe)(Me)]2P1, [Me3Si S (CH2) Si(OEt)(Me)]2P1, [Me3Si S (CH2)2 Si(OMe)(Me)]2P1, [Me3Si S (CH2)2 Si(OEt)(Me)]2P1, [Me3Si S (CH2) C(H)Me (CH2) Si(OMe)2]2P1, [Me3Si S (CH2) C(H)Me (CH2) Si(OEt)2]2P1, [Me3Si S (CH2) C(H)Me (CH2) Si(OMe)(Me)]2P1, [Me3Si S (CH2) C(H)Me (CH2) Si(OEt)(Me)]2P1, [(Bu)Me2Si S (CH2) Si(OMe)2]2P1, [(Bu)Me2Si S (CH2) Si(OEt)2]2P1, [(Bu)Me2Si S (CH2)2 Si(OMe)2]2P1, [(Bu)Me2Si S (CH2)2 Si(OEt)2]2P1, [(Bu)Me2Si S (CH2) Si(OMe)(Me)]2P1, [(Bu)Me2Si S (CH2) Si(OEt)(Me)]2P1, [(Bu)Me2Si S (CH2)2 Si(OMe)(Me)]2P1, [(Bu)Me2Si S (CH2)2 Si(OEt)(Me)]2P1, [(Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OMe)2]2P1, [(Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OEt)2]2P1, [(Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OMe)(Me)]2P1, [(Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OEt)(Me)]2P1, [Me3Si S C(H)Me (CH2)2 Si(OMe)2]2P1, [Me3Si S C(H)Me (CH2)2 Si(OEt)2]2P1, [Me3Si S C(H)Me (CH2)2 Si(OMe)(Me)]2P1, [Me3Si S C(H)Me (CH2)2 Si(OEt)(Me)]2P1, [(Bu)Me2Si S C(H)Me (CH2)2 Si(OMe)2]2P1, [(Bu)Me2Si S C(H)Me (CH2)2 Si(OEt)2]2P1, [(Bu)Me2Si S C(H)Me (CH2)2 Si(OMe)(Me)]2P1, [(Bu)Me2Si S C(H)Me (CH2)2 Si(OEt)(Me)]2P1, [Me3Si S (CH2)11 Si(OMe)2]2P1, [Me3Si S (CH2)11 Si(OEt)2]2P1, [Me3Si S (CH2)11 Si(OMe)(Me)]2P1, [Me3Si S (CH2)11 Si(OEt)(Me)]2P1, [(Bu)Me2Si S (CH2)11 Si(OMe)2]2P1, [(Bu)Me2Si S (CH2)11 Si(OEt)2]2P1, [(Bu)Me2Si S (CH2)11 Si(OMe)(Me)]2P1, [(Bu)Me2Si S (CH2)11 Si(OEt)(Me)]2P1, [(MeO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OMe)2]2P1, [(MeO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OMe)2]2P1, [(MeO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OMe)2]2P1, [(EtO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OEt)2]2P1, [(EtO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OEt)2]2P1, [(EtO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OEt)2]2P1, [(PrO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OPr)2]2P1, [(PrO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OPr)2]2P1, [(PrO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OPr)2]2P1, [(BuO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OBu)2]2P1, [(BuO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OBu)2]2P1, [(BuO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OBu)2]2P1, [(Me)(MeO)2Si (CH2)3 S Si(Me)2 S (CH2)3 Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2)3 S Si(Et)2 S (CH2)3 Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OMe)(Me)]2P1, [(Me)(EtO)2Si (CH2)3 S Si(Me)2 S (CH2)3 Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2)3 S Si(Et)2 S (CH2)3 Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OEt)(Me)]2P1, [(MeO)2(Me)Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OMe)(Me)]2P1, [(MeO)2(Me)Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OMe)(Me)]2P1, [(EtO)2(Me)Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OEt)(Me)]2P1, [(EtO)2(Me)Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OEt)(Me)]2P1, [(MeO)3Si (CH2) S Si(Me)2 S (CH2) Si(OMe)2]2P1, [(MeO)3Si (CH2) S Si(Et)2 S (CH2) Si(OMe)2]2P1, [(EtO)3Si (CH2) S Si(Me)2 S (CH2) Si(OEt)2]2P1, [(EtO)3Si (CH2) S Si(Et)2 S (CH2) Si(OEt)2]2P1, [(Me)(MeO)2Si (CH2) S Si(Me)2 S (CH2) Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2) S Si(Et)2 S (CH2) Si(Me)(OMe)]2P1, [(Me)(EtO)2Si (CH2) S Si(Me)2 S (CH2) Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2) S Si(Et)2 S (CH2) Si(Me)(OEt)]2P1, [(MeO)3Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OMe)2]2P1, [(MeO)3Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OMe)2]2P1, [(EtO)3Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OEt)2]2P1, [(EtO)3Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OEt)2]2P1, [(Me)(MeO)2Si (CH2)2 S Si(Me)2 S (CH2)2 Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2)2 S Si(Et)2 S (CH2)2 Si(Me)(OMe)]2P1, [(Me)(EtO)2Si (CH2)2 S Si(Me)2 S (CH2)2 Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2)2 S Si(Et)2 S (CH2)2 Si(Me)(OEt)]2P1, [(MeO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OMe)2]2P1, [(MeO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OMe)2]2P1, [(MeO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OMe)2]2P1, [(EtO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OEt)2]2P1, [(EtO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OEt)2]2P1, [(EtO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OEt)2]2P1, [(PrO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OPr)2]2P1, [(PrO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OPr)2]2P1, [(PrO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OPr)2]2P1, [(BuO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OBu)2]2P1, [(BuO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OBu)2]2P1, [(BuO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OBu)2]2P1, [(Me)(MeO)2Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OMe)(Me)]2P1, [(Me)(EtO)2Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OEt)(Me)]2P1, [(MeO)2(Me)Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OMe)(Me)]2P1, [(MeO)2(Me)Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OMe)(Me)]2P1, [(EtO)2(Me)Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OEt)(Me)]2P1, [(EtO)2(Me)Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OEt)(Me)]2P1, [(MeO)3Si (CH2) S Sn(Me)2 S (CH2) Si(OMe)2]2P1, [(MeO)3Si (CH2) S Sn(Et)2 S (CH2) Si(OMe)2]2P1, [(EtO)3Si (CH2) S Sn(Me)2 S (CH2) Si(OEt)2]2P1, [(EtO)3Si (CH2) S Sn(Et)2 S (CH2) Si(OEt)2]2P1, [(Me)(MeO)2Si (CH2) S Sn(Me)2 S (CH2) Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2) S Sn(Et)2 S (CH2) Si(Me)(OMe)]2P1, [(Me)(EtO)2Si (CH2) S Sn(Me)2 S (CH2) Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2) S Sn(Et)2 S (CH2) Si(Me)(OEt)]2P1, [(MeO)3Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OMe)2]2P1, [(MeO)3Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OMe)2]2P1, [(EtO)3Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OEt)2]2P1, [(EtO)3Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OEt)2]2P1, [(Me)(MeO)2Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(Me)(OMe)]2P1, [(Me)(MeO)2Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(Me)(OMe)]2P1, [(Me)(EtO)2Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(Me)(OEt)]2P1, [(Me)(EtO)2Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(Me)(OEt)]2P1,
 .
.
Конкретные предпочтительные варианты реализации полимера Формула 2 включают, но не ограничиваются ими, следующие структуры (с таким P2, как определено в этом документе):
Me3Si S (CH2)3 Si(OMe)(Me)P2, Me3Si S (CH2)3 Si(OEt)(Me)P2, Me3Si S (CH2)3 Si(OMe)2P2, Me3Si S (CH2)3 Si(OEt)2P2, Me3Si S (CH2)3 Si(OPr)(Me)P2, Me3Si S (CH2)3 Si(OBu)(Me)P2, Me3Si S (CH2)3 Si(OPr)2P2, Me3Si S (CH2)3 Si(OBu)2P2, Et3Si S (CH2)3 Si(OMe)(Me)P2, Et3Si S (CH2)3 Si(OEt)(Me)P2, Et3Si S (CH2)3 Si(OMe)2P2, Et3Si S (CH2)3 Si(OEt)2P2, Et3Si S (CH2)3 Si(OPr)(Me)P2, Et3Si S (CH2)3 Si(OBu)(Me)P2, Et3Si S (CH2)3 Si(OPr)2P2, Et3Si S (CH2)3 Si(OBu)2P2, (Bu)Me2Si S (CH2)3 Si(OMe)(Me)P2, (Bu)Me2Si S (CH2)3 Si(OEt)(Me)P2, (Bu)Me2Si S (CH2)3 Si(OMe)2P2, (Bu)Me2Si S (CH2)3 Si(OEt)2P2, (Bu)Me2Si S (CH2)3 Si(OPr)(Me)P2, (Bu)Me2Si S (CH2)3 Si(OBu)(Me)P2, (Bu)Me2Si S (CH2)3 Si(OPr)2P2, (Bu)Me2Si S (CH2)3 Si(OBu)2P2, Me3Si S (CH2) Si(OMe)2P2, Me3Si S (CH2) Si(OEt)2P2, Me3Si S (CH2)2 Si(OMe)2P2, Me3Si S (CH2)2 Si(OEt)2P2, Me3Si S (CH2) Si(OMe)(Me)P2, Me3Si S (CH2) Si(OEt)(Me)P2, Me3Si S (CH2)2 Si(OMe)(Me)P2, Me3Si S (CH2)2 Si(OEt)(Me)P2, Me3Si S (CH2) C(H)Me (CH2) Si(OMe)2P2, Me3Si S (CH2) C(H)Me (CH2) Si(OEt)2P2, Me3Si S (CH2) C(H)Me (CH2) Si(OMe)(Me)P2, Me3Si S (CH2) C(H)Me (CH2) Si(OEt)(Me)P2, (Bu)Me2Si S (CH2) Si(OMe)2P2, (Bu)Me2Si S (CH2) Si(OEt)2P2, (Bu)Me2Si S (CH2)2 Si(OMe)2P2, (Bu)Me2Si S (CH2)2 Si(OEt)2P2, (Bu)Me2Si S (CH2) Si(OMe)(Me)P2, (Bu)Me2Si S (CH2) Si(OEt)(Me)P2, (Bu)Me2Si S (CH2)2 Si(OMe)(Me)P2, (Bu)Me2Si S (CH2)2 Si(OEt)(Me)P2, (Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OMe)2P2, (Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OEt)2P2, (Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OMe)(Me)P2, (Bu)Me2Si S (CH2) C(H)Me (CH2) Si(OEt)(Me)P2, Me3Si S C(H)Me (CH2)2 Si(OMe)2P2, Me3Si S C(H)Me (CH2)2 Si(OEt)2P2, Me3Si S C(H)Me (CH2)2 Si(OMe)(Me)P2, Me3Si S C(H)Me (CH2)2 Si(OEt)(Me)P2, (Bu)Me2Si S C(H)Me (CH2)2 Si(OMe)2P2, (Bu)Me2Si S C(H)Me (CH2)2 Si(OEt)2P2, (Bu)Me2Si S C(H)Me (CH2)2 Si(OMe)(Me)P2, (Bu)Me2Si S C(H)Me (CH2)2 Si(OEt)(Me)P2, Me3Si S (CH2)11 Si(OMe)2P2, Me3Si S (CH2)11 Si(OEt)2P2, Me3Si S (CH2)11 Si(OMe)(Me)P2, Me3Si S (CH2)11 Si(OEt)(Me)P2, (Bu)Me2Si S (CH2)11 Si(OMe)2P2, (Bu)Me2Si S (CH2)11 Si(OEt)2P2, (Bu)Me2Si S (CH2)11 Si(OMe)(Me)P2, (Bu)Me2Si S (CH2)11 Si(OEt)(Me)P2, (MeO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OMe)2P2, (MeO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OMe)2P2, (MeO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OMe)2P2, (EtO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OEt)2P2, (EtO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OEt)2P2, (EtO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OEt)2P2, (PrO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OPr)2P2, (PrO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OPr)2P2, (PrO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OPr)2P2, (BuO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OBu)2P2, (BuO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OBu)2P2, (BuO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OBu)2P2, (Me)(MeO)2Si (CH2)3 S Si(Me)2 S (CH2)3 Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2)3 S Si(Et)2 S (CH2)3 Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OMe)(Me)P2, (Me)(EtO)2Si (CH2)3 S Si(Me)2 S (CH2)3 Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2)3 S Si(Et)2 S (CH2)3 Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OEt)(Me)P2, (MeO)2(Me)Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OMe)(Me)P2, (MeO)2(Me)Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OMe)(Me)P2, (EtO)2(Me)Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OEt)(Me)P2, (EtO)2(Me)Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OEt)(Me)P2, (MeO)3Si (CH2) S Si(Me)2 S (CH2) Si(OMe)2P2, (MeO)3Si (CH2) S Si(Et)2 S (CH2) Si(OMe)2P2, (EtO)3Si (CH2) S Si(Me)2 S (CH2) Si(OEt)2P2, (EtO)3Si (CH2) S Si(Et)2 S (CH2) Si(OEt)2P2, (Me)(MeO)2Si (CH2) S Si(Me)2 S (CH2) Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2) S Si(Et)2 S (CH2) Si(Me)(OMe)P2, (Me)(EtO)2Si (CH2) S Si(Me)2 S (CH2) Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2) S Si(Et)2 S (CH2) Si(Me)(OEt)P2, (MeO)3Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OMe)2P2, (MeO)3Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OMe)2P2, (EtO)3Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OEt)2P2, (EtO)3Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OEt)2P2, (Me)(MeO)2Si (CH2)2 S Si(Me)2 S (CH2)2 Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2)2 S Si(Et)2 S (CH2)2 Si(Me)(OMe)P2, (Me)(EtO)2Si (CH2)2 S Si(Me)2 S (CH2)2 Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2)2 S Si(Et)2 S (CH2)2 Si(Me)(OEt)P2, (MeO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OMe)2P2, (MeO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OMe)2P2, (MeO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OMe)2P2, (EtO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OEt)2P2, (EtO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OEt)2P2, (EtO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OEt)2P2, (PrO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OPr)2P2, (PrO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OPr)2P2, (PrO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OPr)2P2, (BuO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OBu)2P2, (BuO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OBu)2P2, (BuO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OBu)2P2, (Me)(MeO)2Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OMe)(Me)P2, (Me)(EtO)2Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OEt)(Me)P2, (MeO)2(Me)Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OMe)(Me)P2, (MeO)2(Me)Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OMe)(Me)P2, (EtO)2(Me)Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OEt)(Me)P2, (EtO)2(Me)Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OEt)(Me)P2, (MeO)3Si (CH2) S Sn(Me)2 S (CH2) Si(OMe)2P2, (MeO)3Si (CH2) S Sn(Et)2 S (CH2) Si(OMe)2P2, (EtO)3Si (CH2) S Sn(Me)2 S (CH2) Si(OEt)2P2, (EtO)3Si (CH2) S Sn(Et)2 S (CH2) Si(OEt)2P2, (Me)(MeO)2Si (CH2) S Sn(Me)2 S (CH2) Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2) S Sn(Et)2 S (CH2) Si(Me)(OMe)P2, (Me)(EtO)2Si (CH2) S Sn(Me)2 S (CH2) Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2) S Sn(Et)2 S (CH2) Si(Me)(OEt)P2, (MeO)3Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OMe)2P2, (MeO)3Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OMe)2P2, (EtO)3Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OEt)2P2, (EtO)3Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OEt)2P2, (Me)(MeO)2Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(Me)(OMe)P2, (Me)(MeO)2Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(Me)(OMe)P2, (Me)(EtO)2Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(Me)(OEt)P2, (Me)(EtO)2Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(Me)(OEt)P2,
 .
.
Фрагменты, полученные в процессе модификации концевых групп цепи применяют модифицирующие концевую группу цепи агенты и имеющие концевую тригидрокарбилсилильную группу, включая триалкилсилильную, диалкиларилсилильную и триарилсилильную, функционируют в качестве защитных групп, которые предотвращают непреднамеренную последующую реакцию полимерной цепи. Такие защитные группы могут быть удалены под воздействием соединения, содержащего реакционно-способную гидроксильную группу (-OH), такую как вода, спирты и органические или неорганические кислоты (к примеру, соляная кислота, серная кислота или карбоновые кислоты). Такие условия воздействия, как правило, присутствуют в процессе вулканизации. В тех случаях, когда концевая группа агента, модифицирующая концевую группу цепи является сульфидно-сшитой, воздействие реакционной гидроксильной группы и снятие защиты приведет к образованию незащищенной тиольной группы (SH) в качестве концевой группы полимерной цепи. В зависимости от рабочих условий для модифицированного полимера (к примеру, отгонка паром), может присутствовать и незащищенный модифицированный и защищенный модифицированный полимер.
Считается, что некоторые концевые группы полимера, такие как незащищенная тиольная группа, являются ракционно-способными по отношению к наполнителям, таким как кремнезем и/или углеродная сажа, что может привести к более однородному распределению наполнителя в полимерной композиции.
Конкретные предпочтительные примеры полимеров, содержащих незащищенные концевые тиольные группы, включают один из следующих, включая их аддукты оснований по Льюису (при этом P1 и P2 будут такими, как определено в данном описании):
[HS (CH2)3 Si(OH)(Me)]2P1, [HS (CH2)3 Si(OH)2]2P1, [HS (CH2)3 Si(OH)(Et)]2P1, [HS (CH2) C(H)Me (CH2) Si(OH)2]2P1, [HS (CH2) C(H)Me (CH2) Si(OH)(Me)]2P1, [HS (CH2) C(H)Me (CH2) Si(OH)(Et)]2P1, [HS (CH2) Si(OH)2]2P1, [HS (CH2) Si(OH)(Me)]2P1, [HS (CH2) Si(OH)(Et)]2P1, [HS (CH2)2 Si(OH)2]2P1, [HS (CH2)2 Si(OH)(Me)]2P1, [HS (CH2)2 Si(OH)(Et)]2P1, [HS (CH2) C(Me)2 (CH2) Si(OH)2]2P1, [HS (CH2) C(Me)2 (CH2) Si(OH)(Me)]2P1, [HS (CH2) C(Me)2 (CH2) Si(OH)(Et)]2P1, [HS C(H)Me (CH2)2 Si(OH)2]2P1, [HS C(H)Me (CH2)2 Si(Me)(OH)]2P1, [HS C(H)Me (CH2)2 Si(Et)(OH)]2P1, HS (CH2)3 Si(OH)(Me)P2, HS (CH2)3 Si(OH)2P2, HS (CH2)3 Si(OH)(Et)P2, HS (CH2) C(H)Me (CH2) Si(OH)2P2, HS (CH2) C(H)Me (CH2) Si(OH)(Me)P2, HS (CH2) C(H)Me (CH2) Si(OH)(Et)P2, HS (CH2) Si(OH)2P2, HS (CH2) Si(OH)(Me)P2, HS (CH2) Si(OH)(Et)P2, HS (CH2)2 Si(OH)2P2, HS (CH2)2 Si(OH)(Me)P2, HS (CH2)2 Si(OH)(Et)P2, HS (CH2) C(Me)2 (CH2) Si(OH)2P2, HS (CH2) C(Me)2 (CH2) Si(OH)(Me)P2, HS (CH2) C(Me)2 (CH2) Si(OH)(Et)P2, HS C(H)Me (CH2)2 Si(OH)2P2, HS C(H)Me (CH2)2 Si(Me)(OH)P2, HS C(H)Me (CH2)2 Si(Et)(OH)P2.
Продукт реакции в виде полимера, модифицированного на конце цепи обычно содержит силанольные группы и алкоксильные группы в общем количестве от 0,01 до 3000 ммоль/кг полимера, предпочтительно от 0,05 до 1500 ммоль/кг, более предпочтительно от 0,1 до 500 ммоль/кг и даже более предпочтительно от 0,2 до 200 ммоль/кг.
Продукт реакции в виде полимера, модифицированного на конце цепи, предпочтительно содержит сульфидные группы (в виде тиоловых групп и/или сульфидно-сшитых защитных групп) в общем количестве от 0,01 до 1000 ммоль/кг полимера, предпочтительно от 0,05 до 500 ммоль/кг, более предпочтительно от 0,1 до 300 ммоль/кг и даже более предпочтительно от 0,2 до 200 ммоль/кг полимера.
Для большинства применений, полимер P1 и P2 представляет собой предпочтительно гомополимер, полученный из сопряженного диолефина, сополимер, полученный из сопряженного диолефинового мономера с ароматическим винильным мономером и/или терполимер одного или двух типов сопряженных диолефинов с одним или двумя типами ароматических винильных соединений. Примеры особенно полезных полимеров включают гомополимеры бутадиена или изопрена и случайные или блок со- и терполимеры бутадиена, изопрена и стирола, особенно случайный coполимер бутадиена с изопреном и случайный или блок coполимер бутадиена со стиролом.
В полимерах P1 и P2, ароматические виниловые мономеры составляют до 60%, предпочтительно от 2 до 55% и более предпочтительно от 5 до 50% по массе, основанном на общей массе полимера. Количество меньше, чем 2% по массе может привести к ухудшению баланса сопротивления качению, влажного заноса и стойкости к истиранию и к снижению прочности на растяжение, в то время как количество более, чем 60% по массе может привести к увеличению гистерезисных потерь. Полимер может быть блок или случайным coполимером ароматического винилового мономера, и предпочтительно 40% по массе или более ароматических винильных мономерных звеньев связаны однократно, и 10% по массе или меньше представляют собой полимерные "блоки" восьми или более ароматических винильных мономеров, связанных последовательно (длина последовательно связанных ароматических винильных звеньев может измеряться методом озонолиза гель-проникающей хроматографии, разработанной Tanaka и др. (Polymer, Vol. 22, pp. 1721-1723 (1981)). Сополимеры за пределами этого диапазона, как правило, имеют повышенные гистерезисные потери.
Хотя нет никаких конкретных ограничей, касающихся содержания 1,2-связей и/или 3,4-связей (далее называемых "содержание виниловой связи") сопряженной диолефиновой части полимера P1 и P2, для большинства применений содержание виниловой связи составляет предпочтительно от 2 до 90% по массе, особенно предпочтительно от 4 до 80% по массе (в расчете на общую массу диолефиновой части). Если содержание виниловой связи в полимере меньше, чем 2% по массе, полученный в результате продукт может иметь меньший баланс устойчивости к влажному заносу, сопротивлению качению и стойкости к истиранию. Если содержание винила в полимере превышает 90% по массе, полученный в результате продукт может проявлять ухудшенную прочность на растяжение и стойкость к истиранию, и относительно большие потери гистерезиса.
МОНЕМЕРЫ
Мономеры, применяемые в получении полимера в соответствии с данным изобретением, выбраны из сопряженных олефинов и необязательно из ароматических винильных соединений.
Подходящие сопряженные олефины включают сопряженные диены, такие как 1,3-бутадиен, 2-алкил-1,3-бутадиен, изопрен (2-метил-1,3-бутадиен), 2,3-диметил-1,3-бутадиен, 1,3-пентадиен, 2,4-гексадиен, 1,3-гексадиен, 1,3-гептадиен, 1,3-октадиен, 2-метил-2,4-пентадиен, циклопентадиен, 2,4-гексадиен и 1,3-циклооктадиен и их комбинацию из двух или более. 1,3-бутадиен и изопрен являются предпочтительными сопряженными олефинами, и 1,3-бутадиен является особенно предпочтительным.
Подходящие ароматические виниловые соединения включают стирол, C1-4 алкил-замещенный стирол, такой как 2-метилстирол, 3-метилстирол, 4-метилстирол, 2,4-диметилстирол, 2,4,6-триметилстирол, α-метилстирол и стильбен, 2,4-диизопропилстирол, 4-трет-бутилстирол, винилбензил диметиламин, (4-винилбензил)диметил аминоэтиловый эфир, N,N-диметиламиноэтил стирол, трет-бутоксистирол и винилпиридин и их кобинацию двух или более. Стирол является особенно предпочтительным ароматическим винильным соединением.
В дополнение к вышеуказанным сопряженным олефинам и ароматическим винильным соединениям, возможно применять один или более мономеров, выбранных из олефинов и несопряженных диолефинов, таких как C2-C20 α-олефины и не-сопряженные C4-C20 диолефины, особенно норборнадиен, этилидененорборнен, 1,4-гексадиен, 1,5-гексадиен, 1,7-октадиен, 4-винилциклогексен и дивинилбензол, включая 1,2-дивинилбензол, 1,3-дивинилбензол и 1,4-дивинилбензол и их смесь.
В одном варианте реализации изобретения, количество дивинилбензола, включая 1,2-дивинилбензол, 1,3-дивинилбензол и 1,4-дивинилбензол, составляет 1% в молях или менее (на основе общего молярного количества мономеров, которые применяются для изготовления полимера).
АГЕНТЫ, МОДИФИЦИРУЮЩИЕ КОНЦЕВУЮ ГРУППУ ЦЕПИ
Согласно второму аспекту изобретения, один или более агентов, модифицирующих концевую группу цепи (или коротко "модифицирующие агенты") применяются для реакции с концом полимерной цепи(ей), полученной из способа в соответствии со вторым изобретением. В общем, силан-сульфидные омега агенты, модифицирующие концевую группу цепи, такие как описано в WO 2007/047943, WO 2009/148932, США 6229036 и США 2013/0131263, каждый включен в данный документ в качестве ссылки в полном объеме, может быть использована для этой цели.
В предпочтительном варианте реализации изобретения, агент, модифицирующий концевую группу цепи, выбран из одного или более агентов, модифицирующих концевую группу цепи, представленных Формулой 11 до Формулы 15. В особенно предпочтительном варианте реализации изобретения, агент, модифицирующий концевую группу цепи, выбран из одного или более соединений, представленных Формулой 11. Конкретные предпочтительные виды агентов, модифицирующих концевую группу цепи Формулы 11, включают следующие соединения:
(MeO)3Si (CH2)3 S SiMe3, (EtO)3Si (CH2)3 S SiMe3, (PrO)3Si (CH2)3 S SiMe3, (BuO)3Si (CH2)3 S SiMe3, (MeO)3Si (CH2)2 S SiMe3, (EtO)3Si (CH2)2 S SiMe3, (PrO)3Si (CH2)2 S SiMe3, (BuO)3Si (CH2)2 S SiMe3, (MeO)3Si CH2 S SiMe3, (EtO)3Si CH2 S SiMe3, (PrO)3Si CH2 S SiMe3, (BuO)3Si CH2 S SiMe3, (MeO)3Si CH2 CMe2 CH2 S SiMe3, (EtO)3Si CH2 CMe2 CH2 S SiMe3, (PrO)3Si CH2 CMe2 CH2 S SiMe3, (BuO)3Si CH2 CMe2 CH2 S SiMe3, ((MeO)3Si CH2 C(H)Me CH2 S SiMe3, (EtO)3Si CH2 C(H)Me CH2 S SiMe3, (PrO)3Si CH2 C(H)Me CH2 S SiMe3, (BuO)3Si CH2 C(H)Me CH2 S SiMe3, (MeO)2(Me)Si (CH2)3 S SiMe3, (EtO)2(Me)Si (CH2)3 S SiMe3, (PrO)2(Me)Si (CH2)3 S SiMe3, (BuO)2(Me)Si (CH2)3 S SiMe3, (MeO)2(Me)Si (CH2)2 S SiMe3, (EtO)2(Me)Si (CH2)2 S SiMe3, (PrO)2(Me)Si (CH2)2 S SiMe3, (BuO)2(Me)Si (CH2)2 S SiMe3, (MeO)2(Me)Si CH2 S SiMe3, (EtO)2(Me)Si CH2 S SiMe3, (PrO)2(Me)Si CH2 S SiMe3, (BuO)2(Me)Si CH2 S SiMe3, (MeO)2(Me)Si CH2 CMe2 CH2 S SiMe3, (EtO)2(Me)Si CH2 CMe2 CH2 S SiMe3, (PrO)2(Me)Si CH2 CMe2 CH2 S SiMe3, (BuO)2(Me)Si CH2 CMe2 CH2 S SiMe3, ((MeO)2(Me)Si CH2 C(H)Me CH2 S SiMe3, (EtO)2(Me)Si CH2 C(H)Me CH2 S SiMe3, (PrO)2(Me)Si CH2 C(H)Me CH2 S SiMe3, (BuO)2(Me)Si CH2 C(H)Me CH2 S SiMe3, (MeO)(Me)2Si (CH2)3 S SiMe3, (EtO)(Me)2Si (CH2)3 S SiMe3, (PrO) Me)2Si (CH2)3 S SiMe3, (BuO)(Me)2Si (CH2)3 S SiMe3, (MeO)(Me)2Si (CH2)2 S SiMe3, (EtO)(Me)2Si (CH2)2 S SiMe3, (PrO)(Me)2Si (CH2)2 S SiMe3, (BuO)(Me)2Si (CH2)2 S SiMe3, (MeO)(Me)2Si CH2 S SiMe3, (EtO)(Me)2Si CH2 S SiMe3, (PrO)(Me)2Si CH2 S SiMe3, (BuO)(Me)2Si CH2 S SiMe3, (MeO)(Me)2Si CH2 CMe2 CH2 S SiMe3, (EtO)(Me)2Si CH2 CMe2 CH2 S SiMe3, (PrO)(Me)2Si CH2 CMe2 CH2 S SiMe3, (BuO)(Me)2Si CH2 CMe2 CH2 S SiMe3, ((MeO)(Me)2Si CH2 C(H)Me CH2 S SiMe3, (EtO)(Me)2Si CH2 C(H)Me CH2 S SiMe3, (PrO)(Me)2Si CH2 C(H)Me CH2 S SiMe3, (BuO)(Me)2Si CH2 C(H)Me CH2 S SiMe3, (MeO)3Si (CH2)3 S SiEt3, (EtO)3Si (CH2)3 S SiEt3, (PrO)3Si (CH2)3 S SiEt3, (BuO)3Si (CH2)3 S SiEt3, (MeO)3Si (CH2)2 S SiEt3, (EtO)3Si (CH2)2 S SiEt3, (PrO)3Si (CH2)2 S SiEt3, (BuO)3Si (CH2)2 S SiEt3, (MeO)3Si CH2 S SiEt3, (EtO)3Si CH2 S SiEt3, (PrO)3Si CH2 S SiEt3, (BuO)3Si CH2 S SiEt3, (MeO)3Si CH2 CMe2 CH2 S SiEt3, (EtO)3Si CH2 CMe2 CH2 S SiEt3, (PrO)3Si CH2 CMe2 CH2 S SiEt3, (BuO)3Si CH2 CMe2 CH2 S SiEt3, ((MeO)3Si CH2 C(H)Me CH2 S SiEt3, (EtO)3Si CH2 C(H)Me CH2 S SiEt3, (PrO)3Si CH2 C(H)Me CH2 S SiEt3, (BuO)3Si CH2 C(H)Me CH2 S SiEt3, (MeO)2(Me)Si (CH2)3 S SiEt3, (EtO)2(Me)Si (CH2)3 S SiEt3, (PrO)2(Me)Si (CH2)3 S SiEt3, (BuO)2(Me)Si (CH2)3 S SiEt3, (MeO)2(Me)Si (CH2)2 S SiEt3, (EtO)2(Me)Si (CH2)2 S SiEt3, (PrO)2(Me)Si (CH2)2 S SiEt3, (BuO)2(Me)Si (CH2)2 S SiEt3, (MeO)2(Me)Si CH2 S SiEt3, (EtO)2(Me)Si CH2 S SiEt3, (PrO)2(Me)Si CH2 S SiEt3, (BuO)2(Me)Si CH2 S SiEt3, (MeO)2(Me)Si CH2 CMe2 CH2 S SiEt3, (EtO)2(Me)Si CH2 CMe2 CH2 S SiEt3, (PrO)2(Me)Si CH2 CMe2 CH2 S SiEt3, (BuO)2(Me)Si CH2 CMe2 CH2 S SiEt3, ((MeO)2(Me)Si CH2 C(H)Me CH2 S SiEt3, (EtO)2(Me)Si CH2 C(H)Me CH2 S SiEt3, (PrO)2(Me)Si CH2 C(H)Me CH2 S SiEt3, (BuO)2(Me)Si CH2 C(H)Me CH2 S SiEt3, (MeO)(Me)2Si (CH2)3 S SiEt3, (EtO)(Me)2Si (CH2)3 S SiEt3, (PrO)Me)2Si (CH2)3 S SiEt3, (BuO)(Me)2Si (CH2)3 S SiEt3, (MeO)(Me)2Si (CH2)2 S SiEt3, (EtO)(Me)2Si (CH2)2 S SiEt3, (PrO)(Me)2Si (CH2)2 S SiEt3, (BuO)(Me)2Si (CH2)2 S SiEt3, (MeO)(Me)2Si CH2 S SiEt3, (EtO) (Me)2Si CH2 S SiEt3, (PrO)(Me)2Si CH2 S SiEt3, (BuO)(Me)2Si CH2 S SiEt3, (MeO)(Me)2Si CH2 CMe2 CH2 S SiEt3, (EtO)(Me)2Si CH2 CMe2 CH2 S SiEt3, (PrO)(Me)2Si CH2 CMe2 CH2 S SiEt3, (BuO)(Me)2Si CH2 CMe2 CH2 S SiEt3, ((MeO)(Me)2Si CH2 C(H)Me CH2 S SiEt3, (EtO)(Me)2Si CH2 C(H)Me CH2 S SiEt3, (PrO)(Me)2Si CH2 C(H)Me CH2 S SiEt3 и (BuO)(Me)2Si CH2 C(H)Me CH2 S SiEt3, (MeO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OMe)3, (MeO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OMe)3, (MeO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OMe)3, (EtO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OEt)3, (EtO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OEt)3, (EtO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OEt)3, (PrO)3Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OPr), (PrO)3Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OPr)3, (PrO)3Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OPr)3, (MeO)3Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OMe)3, (MeO)3Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OMe)3, (MeO)3Si (CH2)2 S Si(Bu)2 S (CH2)2 Si(OMe)3, (EtO)3Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OEt)3, (EtO)3Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OEt)3, (EtO)3Si (CH2)2 S Si(Bu)2 S (CH2)2 Si(OEt)3, (PrO)3Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OPr)3, (PrO)3Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OPr)3, (PrO)3Si (CH2)2 S Si(Bu)2 S (CH2)2 Si(OPr)3, (MeO)3Si CH2 S Si(Me)2 S CH2 Si(OMe)3, (MeO)3Si CH2 S Si(Et)2 S CH2 Si(OMe)3, (MeO)3Si CH2 S Si(Bu)2 S CH2 Si(OMe)3, (EtO)3Si CH2 S Si(Me)2 S CH2 Si(OEt)3, (EtO)3Si CH2 S Si(Et)2 S CH2 Si(OEt)3, (EtO)3Si CH2 S Si(Bu)2 S CH2 Si(OEt)3, (PrO)3Si CH2 S Si(Me)2 S CH2 Si(OPr)3, (PrO)3Si CH2 S Si(Et)2 S CH2 Si(OPr)3, (PrO)3Si CH2 S Si(Bu)2 S CH2 Si(OPr)3, (MeO)3Si CH2 CMe2 CH2 S Si(Me)2 S CH2 CMe2 CH2 Si(OMe)3, (MeO)3Si CH2 CMe2 CH2 S Si(Et)2 S CH2 CMe2 CH2 Si(OMe)3, (MeO)3Si CH2 CMe2 CH2 S Si(Bu)2 S CH2 CMe2 CH2 Si(OMe)3, (EtO)3Si CH2 CMe2 CH2 S Si(Me)2 S CH2 CMe2 CH2 Si(OEt)3, (EtO)3Si CH2 CMe2 CH2 S Si(Et)2 S CH2 CMe2 CH2 Si(OEt)3, (EtO)3Si CH2 CMe2 CH2 S Si(Bu)2 S CH2 CMe2 CH2 Si(OEt)3, (PrO)3Si CH2 CMe2 CH2 S Si(Me)2 S CH2 CMe2 CH2 Si(OPr)3, (PrO)3Si CH2 CMe2 CH2 S Si(Et)2 S CH2 CMe2 CH2 Si(OPr)3, (PrO)3Si CH2 CMe2 CH2 S Si(Bu)2 S CH2 CMe2 CH2 Si(OPr)3, (MeO)3Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OMe)3, (MeO)3Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OMe)3, (MeO)3Si CH2 C(H)Me CH2 S Si(Bu)2 S CH2 C(H)Me CH2 Si(OMe)3, (EtO)3Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OEt)3, (EtO)3Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OEt)3, (EtO)3Si CH2 C(H)Me CH2 S Si(Bu)2 S CH2 C(H)Me CH2 Si(OEt)3, (PrO)3Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OPr)3, (PrO)3Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OPr)3, (PrO)3Si CH2 C(H)Me CH2 S Si(Bu)2 S CH2 C(H)Me CH2 Si(OPr)3, (MeO)2(Me)Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OMe)2(Me), (EtO)2(Me)Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OEt)2(Me), (PrO)2(Me)Si (CH2)3 S Si(Me)2 S (CH2)3 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)3 S Si(Et)2 S (CH2)3 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)3 S Si(Bu)2 S (CH2)3 Si(OPr)2(Me), (MeO)2(Me)Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)2 S Si(Bu)2 S (CH2)2 Si(OMe)2(Me), (EtO)2(Me)Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)2 S Si(Bu)2 S (CH2)2 Si(OEt)2(Me), (PrO)2(Me)Si (CH2)2 S Si(Me)2 S (CH2)2 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)2 S Si(Et)2 S (CH2)2 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)2 S Si(Bu)2 S (CH2)2 Si(OPr)2(Me), (MeO)2(Me)Si CH2 S Si(Me)2 S CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 S Si(Et)2 S CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 S Si(Bu)2 S CH2 Si(OMe)2(Me), (EtO)2(Me)Si CH2 S Si(Me)2 S CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 S Si(Et)2 S CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 S Si(Bu)2 S CH2 Si(OEt)2(Me), (PrO)2(Me)Si CH2 S Si(Me)2 S CH2 Si(OPr) 2(Me), (PrO)2(Me)Si CH2 S Si(Et)2 S CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 S Si(Bu)2 S CH2 Si(OPr)2(Me), (MeO)2(Me)Si CH2 CMe2 CH2 S Si(Me)2 S CH2 CMe2 CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 CMe2 CH2 S Si(Et)2 S CH2 CMe2 CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 CMe2 CH2 S Si(Bu)2 S CH2 CMe2 CH2 Si(OMe)2(Me), (EtO)2(Me)Si CH2 CMe2 CH2 S Si(Me)2 S CH2 CMe2 CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 CMe2 CH2 S Si(Et)2 S CH2 CMe2 CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 CMe2 CH2 S Si(Bu)2 S CH2 CMe2 CH2 Si(OEt)2(Me), (PrO)2(Me)Si CH2 CMe2 CH2 S Si(Me)2 S CH2 CMe2 CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 CMe2 CH2 S Si(Et)2 S CH2 CMe2 CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 CMe2 CH2 S Si(Bu)2 S CH2 CMe2 CH2 Si(OPr)2(Me), (MeO)2(Me)Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 C(H)Me CH2 S Si(Bu)2 S CH2 C(H)Me CH2 Si(OMe)2(Me), (EtO)2(Me)Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 C(H)Me CH2 S Si(Bu)2 S CH2 C(H)Me CH2 Si(OEt)2(Me), (PrO)2(Me)Si CH2 C(H)Me CH2 S Si(Me)2 S CH2 C(H)Me CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 C(H)Me CH2 S Si(Et)2 S CH2 C(H)Me CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 C(H)Me CH2 S Si(Bu)2 S CH2 C(H)Me CH2 Si(OPr)2(Me), (MeO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OMe)3, (MeO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OMe)3, (MeO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OMe)3, (EtO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OEt)3, (EtO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OEt)3, (EtO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OEt)3, (PrO)3Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OPr), (PrO)3Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OPr)3, (PrO)3Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OPr)3, (MeO)3Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OMe)3, (MeO)3Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OMe)3, (MeO)3Si (CH2)2 S Sn(Bu)2 S (CH2)2 Si(OMe)3, (EtO)3Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OEt)3, (EtO)3Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OEt)3, (EtO)3Si (CH2)2 S Sn(Bu)2 S (CH2)2 Si(OEt)3, (PrO)3Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OPr)3, (PrO)3Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OPr)3, (PrO)3Si (CH2)2 S Sn(Bu)2 S (CH2)2 Si(OPr)3, (MeO)3Si CH2 S Sn(Me)2 S CH2 Si(OMe)3, (MeO)3Si CH2 S Sn(Et)2 S CH2 Si(OMe)3, (MeO)3Si CH2 S Sn(Bu)2 S CH2 Si(OMe)3, (EtO)3Si CH2 S Sn(Me)2 S CH2 Si(OEt)3, (EtO)3Si CH2 S Sn(Et)2 S CH2 Si(OEt)3, (EtO)3Si CH2 S Sn(Bu)2 S CH2 Si(OEt)3, (PrO)3Si CH2 S Sn(Me)2 S CH2 Si(OPr)3, (PrO)3Si CH2 S Sn(Et)2 S CH2 Si(OPr)3, (PrO)3Si CH2 S Sn(Bu)2 S CH2 Si(OPr)3, (MeO)3Si CH2 CMe2 CH2 S Sn(Me)2 S CH2 CMe2 CH2 Si(OMe)3, (MeO)3Si CH2 CMe2 CH2 S Sn(Et)2 S CH2 CMe2 CH2 Si(OMe)3, (MeO)3Si CH2 CMe2 CH2 S Sn(Bu)2 S CH2 CMe2 CH2 Si(OMe)3, (EtO)3Si CH2 CMe2 CH2 S Sn(Me)2 S CH2 CMe2 CH2 Si(OEt)3, (EtO)3Si CH2 CMe2 CH2 S Sn(Et)2 S CH2 CMe2 CH2 Si(OEt)3, (EtO)3Si CH2 CMe2 CH2 S Sn(Bu)2 S CH2 CMe2 CH2 Si(OEt)3, (PrO)3Si CH2 CMe2 CH2 S Sn(Me)2 S CH2 CMe2 CH2 Si(OPr)3, (PrO)3Si CH2 CMe2 CH2 S Sn(Et)2 S CH2 CMe2 CH2 Si(OPr)3, (PrO)3Si CH2 CMe2 CH2 S Sn(Bu)2 S CH2 CMe2 CH2 Si(OPr)3, (MeO)3Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OMe)3, (MeO)3Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OMe)3, (MeO)3Si CH2 C(H)Me CH2 S Sn(Bu)2 S CH2 C(H)Me CH2 Si(OMe)3, (EtO)3Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OEt)3, (EtO)3Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OEt)3, (EtO)3Si CH2 C(H)Me CH2 S Sn(Bu)2 S CH2 C(H)Me CH2 Si(OEt)3, (PrO)3Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OPr)3, (PrO)3Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OPr)3, (PrO)3Si CH2 C(H)Me CH2 S Sn(Bu)2 S CH2 C(H)Me CH2 Si(OPr)3, (MeO)2(Me)Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OMe)2(Me), (EtO)2(Me)Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OEt)2(Me), (PrO)2(Me)Si (CH2)3 S Sn(Me)2 S (CH2)3 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)3 S Sn(Et)2 S (CH2)3 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)3 S Sn(Bu)2 S (CH2)3 Si(OPr)2(Me), (MeO)2(Me)Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OMe)2(Me), (MeO)2(Me)Si (CH2)2 S Sn(Bu)2 S (CH2)2 Si(OMe)2(Me), (EtO)2(Me)Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OEt)2(Me), (EtO)2(Me)Si (CH2)2 S Sn(Bu)2 S (CH2)2 Si(OEt)2(Me), (PrO)2(Me)Si (CH2)2 S Sn(Me)2 S (CH2)2 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)2 S Sn(Et)2 S (CH2)2 Si(OPr)2(Me), (PrO)2(Me)Si (CH2)2 S Sn(Bu)2 S (CH2)2 Si(OPr)2(Me), (MeO)2(Me)Si CH2 S Sn(Me)2 S CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 S Sn(Et)2 S CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 S Sn(Bu)2 S CH2 Si(OMe)2(Me), (EtO)2(Me)Si CH2 S Sn(Me)2 S CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 S Sn(Et)2 S CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 S Sn(Bu)2 S CH2 Si(OEt)2(Me), (PrO)2(Me)Si CH2 S Sn(Me)2 S CH2 Si(OPr) 2(Me), (PrO)2(Me)Si CH2 S Sn(Et)2 S CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 S Sn(Bu)2 S CH2 Si(OPr)2(Me), (MeO)2(Me)Si CH2 CMe2 CH2 S Sn(Me)2 S CH2 CMe2 CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 CMe2 CH2 S Sn(Et)2 S CH2 CMe2 CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 CMe2 CH2 S Sn(Bu)2 S CH2 CMe2 CH2 Si(OMe)2(Me), (EtO)2(Me)Si CH2 CMe2 CH2 S Sn(Me)2 S CH2 CMe2 CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 CMe2 CH2 S Sn(Et)2 S CH2 CMe2 CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 CMe2 CH2 S Sn(Bu)2 S CH2 CMe2 CH2 Si(OEt)2(Me), (PrO)2(Me)Si CH2 CMe2 CH2 S Sn(Me)2 S CH2 CMe2 CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 CMe2 CH2 S Sn(Et)2 S CH2 CMe2 CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 CMe2 CH2 S Sn(Bu)2 S CH2 CMe2 CH2 Si(OPr)2(Me), (MeO)2(Me)Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OMe)2(Me), (MeO)2(Me)Si CH2 C(H)Me CH2 S Sn(Bu)2 S CH2 C(H)Me CH2 Si(OMe)2(Me), (EtO)2(Me)Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OEt)2(Me), (EtO)2(Me)Si CH2 C(H)Me CH2 S Sn(Bu)2 S CH2 C(H)Me CH2 Si(OEt)2(Me), (PrO)2(Me)Si CH2 C(H)Me CH2 S Sn(Me)2 S CH2 C(H)Me CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 C(H)Me CH2 S Sn(Et)2 S CH2 C(H)Me CH2 Si(OPr)2(Me), (PrO)2(Me)Si CH2 C(H)Me CH2 S Sn(Bu)2 S CH2 C(H)Me CH2 Si(OPr)2(Me).
Если более, чем один агент, модифицирующий концевую группу цепи, применяется для цели модификации концевой группы цепи, агенты, модифицирующие концевую группу цепи, могут быть добавлены один за другим к раствору живого анионного полимера, или они могут быть смешаны вместе перед добавлением полученной смеси к раствору живого анионного полимера.
Агенты, модифицирующие концевую группу цепи, могут быть добавлены периодически (или с регулярными или нерегулярными интервалами) или непрерывно во время полимеризации, но их предпочтительно добавляют при скорости преобразования полимеризации более, чем 80 процентов и более предпочтительно при скорости преобразования более, чем 90 процентов. Предпочтительно, существенное количество концов полимерной цепи не заканчивается до реакции с агентом, модифицирующим концевую группу; то есть, концы живой полимерной цепи присутствуют и способны реагировать с модифицирующим агентом. Реакция модификации концевой группы цепи может происходить до, после или во время добавления любого связующего вещества. Предпочтительно, реакция модификации концевой группы цепи завершается после добавления любого связующего вещества. Смотри, например, WO 2009/148932, который включен сюда посредством ссылки.
ПРОЦЕСС МОДИФИКАЦИИ КОНЦЕВОЙ ГРУППЫ ЦЕПИ
Полимерная композиция в соответствии с первым аспектом изобретения представляет собой продукт реакции живых полимерных цепей, содержащих концы альфа- и омега,омега'-карбанионной полимерной цепи с по меньшей мере одним агентом, модифицирующим концевую группу цепи. Живые полимерные цепи, содержащие концы альфа- и омега,омега'-карбаниовоной полимерной цепи, представляют собой продукт реакции смеси инициатора, содержащей по меньшей мере один моноанионный полимеризационный инициатор и по меньшей мере один дианионный полимеризационный инициатор с мономерами. Моноанионные инициаторы формируют, после реакции с мономерами, концы живых моноанионных полимерных цепей (альфа-карбанионна концевая группа цепи), и дианионные инициаторы формируют, после реакции с мономерами, концы живых дианионных полимерных цепей (омега,омега'-концы карбанионной полимерной цепи). Каждый конец карбанионной полимерной цепи может реагировать с одним эквивалентом агента модифицирующего концевую группу цепи, давая в результате альфа- или омега,омега'-модифицированные концевые группы полимерной цепи. В соответствии с изобретением, реакция конца моноанионной полимерной цепи с модифицирующим агентом, приводит к альфа-модифицированной полимерной цепи Формулы 2, в то время как реакция обоих концов дианионной полимерной цепи с модифицированным агентом приводит к a) омега,омега'-модифицированной полимерной цепи Формулы 1 (когда модифицирующий агент, используемый в стехиометрическом отношении к концам анионной полимерной цепи), или b) смеси альфа-модифицированных полимерных цепей Формулы 2 и омега,омега'-модифицированных полимерных цепей Формулы 1 (когда модифицирующий агент применяется субстехиометрически в отношении к концам анионной полимерной цепи).
Агент, модифицирующий концевую группу цепи, может быть добавлен непосредственно к полимерному раствору без разбавления; тем не менее, это может быть выгодным добавлять модифицирующий агент в растворенной форме, такой как в инертном растворителе, (например, циклогексане). Количество агента, модифицирующего концевую группу цепи, добавленного к полимеризации, может изменяться в зависимости от вида мономеров, связующего вещества, агента, модифицирующего концевую группу цепи, реакционных условий и желаемых свойств полимера. В настоящем изобретении, количество агента, модифицирующего концевую группу цепи, специально корректируется на основе молярного соотношения соединений Формулы 9 и 10, как определено в данном документе. Реакция модификации концевой группы полимерной цепи может быть проведена при температуре в интервале от 0°C до 150 °C, предпочтительно от 15°C до 120°C и даже более предпочтительно от 30°C до 100 °C. Там нет ограничений в отношении продолжительности реакции модификации концевой группы цепи. Тем не менее, по отношению к экономике процесса полимеризации, например, в случае периодического процесса полимеризации, реакцию модификации концевой группы цепи обычно останавливают от около 5 до 60 минут после добавления модифицирующего агента.
Способ изготовления модифицированного полимера изобретения включает по меньшей мере следующие этапы A через C:
Этап A: приведение в контакт соединения Формулы 9 с соединением Формулы 10 для получения смеси инициатора моно- и дианионных инициаторов.
Этап B: приведение в контакт смеси инициатора с одним или более полимеризуемыми мономерами, выбранными из сопряженных олефинов, предпочтительно выбранных из бутадиена и изопрена, и необязательно ароматических винильных соединений, предпочтительно выбранных из стирола и альфа-метил стирола, полимеризация в растворителе и необязательно в присутствии мономера, который способен на реакцию с более чем одной растущей полимерной цепью, такой как дивинилбензол. Подходящие растворители для полимеризации включают не-полярные алифатические и не-полярные ароматические растворители, предпочтительно гексан, гептан, бутан, пентан, высокочистые изопарафины-растворители, циклогексан, толуол и бензол.
Этап C: приведение в контакт реакционного продукта этапа A с по меньшей мере одним агентом модифицирующим концевую группу цепи, выбранном из Формулы 11 до Формулы 15 (как описано в данном документе), предпочтительно из Формулы 11, для формирования полимера, модифицированного на конце цепи.
РАНДОМИЗИРУЮЩИЕ АГЕНТЫ
Полярные координационные соединения, называемые также рандомизирующие агенты, могут быть необязательно добавлены к полимеризации для подгонки микроструктуры сопряженной диеновой части (включая содержание винильных связей полибутадиеновой фракции), или для подгонки распределения состава ароматического винильного соединения, служащего таким образом в качестве рандомизирующего компонента. Два или более рандомизирующих агентов могут применяться в комбинации. Примерные рандомизирующие агенты представляют собой основания по Льюису и включают, но не ограничиваются такими, как эфирные соединения, такие как диетиловый эфир, ди-н-бутиловый эфир, этиленгликоль диетиловый эфир, этиленгликоль дибутиловый эфир, диэтиленгликоль диметиловый эфир, пропилен гликоль диметиловый эфир, пропиленгликоль диетиловый эфир, пропиленгликоль дибутиловый эфир, алкилтетрагидрофуриловые эфиры, такие как метилтетрагидрофуриловый эфир, этилтетрагидрофуриловый эфир, пропилтетрагидрофуриловый эфир, бутилтетрагидрофуриловый эфир, гексилтетрагидрофуриловый эфир, октилтетрагидрофуриловый эфир, тетрагидрофуран, 2,2-(бистетрагидрофурфурил)пропан, бистетрагидрофурфурилформил, метиловый эфир тетрагидрофурфурилового спирта, этиловый эфир тетрагидрофурфурилового спирта, бутиловый эфир тетрагидрофурфурилового спирта, α-метокситетрагидрофуран, диметоксибензол и диметоксиэтан, и третичные аминовые соединения, такие как триэтиламин, пиридин, N,N,N',N'-тетраметил этилендиамин, дипиперидиноэтан, метиловый эфир N,N-диэтилэтаноламина, этиловый эфир N,N-диэтилэтаноламина и N,N-диэтилэтаноламин. Примеры предпочтительных рандомизирующих соединений определены в WO 2009/148932, включены в даный документы посредством ссылки во всей своей полноте. Рандомизирующий агент(ы), как правило, добавляют в молярном соотношении рандомизированного соединения к инициаторному соединению от 0,012:1 до 10:1, предпочтительно от 0,1:1 до 8:1 и более предпочтительно от 0,25:1 до около 6:1.
СВЯЗУЮЩИЕ ВЕЩЕСТВА
Полимеризационные композиции изобретения могут необязательно быть подвергнуты приведению в контакт с одним или более связующими веществами для образования разветвленных полимеров.
Связующие вещества включают тетрахлорид олова, тетрабромид олова, тетрафторид олова, тетраиодид олова, тетрахлорид кремния, тетрабромид кремния, тетрафторид кремния, тетраиодид кремния, алкил олова и алкил кремния тригалогениды или диалкил олова и диалкил кремния дигалогениды. Полимеры, связанные с оловом или кремния тетрагалогенидами, имеют максимум четыре ответвления, полимеры, связанные с алкилом олова и алкил кремния тригалогенидами, имеют максимум три ответвления, и полимеры, связанные с диалкилом олова и диалкилом кремния дигалогенидами, имеют максимум два ответвления. Гексагалогенидные дисиланы или гексагалогенидные дисилоксаны могут также быть использованы в качестве связующих веществ, давая в результате полимеры с максимум шестью ответвлениями. Полезные связующие вещества олова и кремния галогенидов включают: SnCl4, (R1)3SnCl, (R1)2SnCl2, R1SnCl3, SiCl4, R1SiCl3, (R1)2SiCl2, (R1)3SiCl, Cl3Si-SiCl3, Cl3Si-O-SiCl3, Cl3Sn-SnCl3 и Cl3Sn-O-SnCl3, где R1 представляет собой группу гидрокарбила, предпочтительно алкильную группу. Примеры связующих веществ олова и кремния алкоксидов, кроме того включают: Sn(OMe)4, Si(OMe)4, Sn(OEt)4 и Si(OEt)4. Наиболее предпочтительные связующие вещества представляют собой: SnCl4, SiCl4, Sn(OMe)4 и Si(OMe)4.
Связующие вещества могут быть добавлены периодически (или с регулярными или нерегулярными интервалами) или непрерывно в ходе полимеризации, но предпочтительно добавляют при скорости преобразования полимеризации более чем 80 процентов и более предпочтительно при скорости преобразования более чем 90 процентов. Связующее вещество, как правило, добавляют только после того, как достигнута высокая степень преобразования.
Например, связующее вещество может быть непрерывно добавлено во время полимеризации, в том случае, когда желаемыми являются асимметричные связи. Такое непрерывное добавление обычно проводят в реакционной зоне, отделенной от зоны, где происходит основная часть полимеризации. Связующее вещество может быть добавлено в углеводородном растворе, например, в циклогексане, в смесь для полимеризации, с помощью подходящего перемешивания для распределения и реакции. Обычно, применяется от 0,01 до 2,0 моль, предпочтительно от 0,02 до 1,5 моль и более предпочтительно от 0,04 до 0,6 моль связующего вещества для каждых 4,0 моль концевой группы живой анионной полимерной цепи.
Предпочтительно, значительное количество концов полимерной цепи не заканчивается до реакции со связующим веществом; то есть, концевые группы живой полимерной цепи присутствуют и способны реагировать со связующим веществом в реакции конденсации полимерной цепи. Реакция конденсации происходит до, после или во время добавления любого агента модифицирующего концевую группу цепи. Реакция конденсации предпочтительно завершена до добавления агента, модифицирующего концевую группу цепи. В некоторых вариантах реализации изобретения, между 5 и 20 процентов концевых групп живой полимерной цепи, как определено с помощью GPC, прореагировали со связующим веществом до добавления агента, модифицирующего концевую группу цепи. В других вариантах реализации изобретения, между 20 и 35 процентов концевых групп живой полимерной цепи прореагировали со связующим веществом до добавления агента, модифицирующего концевую группу цепи. В еще другом варианте реализации изобретения, между 35 и 50 процентов концевых групп живой полимерной цепи прореагировали со связующим веществом до добавления агента, модифицирующего концевую группу цепи.
Сочетание различных связующих веществ, таких так Bu2SnCl2 и SnCl4; Me2SiCl2 и Si(OMe)4; Me2SiCl2 и SiCl4; SnCl4 и Si(OMe)4; SnCl4 и SiCl4, могут также применяться для связывания полимерных цепей. Особенно желательным является применение комбинации связующих веществ из олова и кремния в соединениях в протекторах покрышек, которые содержат как кремний, так и углеродную сажу. В таком случае, молярное соотношение олова к соединению кремния обычно находится в интервале от 20:80 до 95:5; более часто от 40:60 до 90:10 и предпочтительно от 60:40 до 85:15. Наиболее часто, в количестве от около 0,001 до 4,5 ммоль связующего вещества применяют на 100 г полимера. Как правило, предпочтительно применять от около 0,05 до около 0,5 ммоль связующего вещества на 100 г полимера для получения желаемой вязкости по Муни и для того, чтобы обеспечить функционализацию концевой группы цепи оставшейся фракции живого полимера. Большие количества, как правило, производят полимеры, содержащие концевые ракционно способные группы или недостаточное сопряжение и позволяет достичь только недостаточной модификации концевой группы цепи.
Реакцию конденсации полимера могут проводить при температурном диапазоне от 0°C до 150 °C, предпочтительно от 15°C до 120°C и даже более предпочтительно от 40°C до 100 °C. При этом нет ограничений для продолжительности реакции конденсации. Тем не менее, по отношению к экономике полимеризационного процесса, например, в случае периодической полимеризации, реакцию конденсации обычно останавливают при приблизительно от 5 до 60 минут после добавления связующего вещества.
В предпочтительном варианте реализации изобретения дивинилбензол применяют в качестве связующего вещества. Дивинилбензол, как правило, добавляют вместе с мономерами перед началом полимеризации путем добавления инициатора.
СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНОЙ КОМПОЗИЦИИ
Способ получения полимерной композиции в соответствии со вторым аспектом изобретения включает этапы
i) приведения в контакт смеси инициатора полимеризации, полученной путем приведения в контакт соединения Формулы 9 как определено в данном документе с соединением Формулы 10, с одним или более полимеризуемыми мономерами, выбранными из сопряженных диенов и ароматических винильных соединений для получения живых полимерных цепей, где живые полимерные цепи содержат, по меньшей мере, 40% по массе повторяющихся звеньев, полученных путем полимеризации указанных сопряженных диенов, и причем молярное соотношение соединений Формулы 10 к соединениям Формулы 9 находится в диапазоне от 1:1 до 8:1, и
ii) приведения в контакт живых полимерных цепей этапа i) с одним или более агентами, модифицирующими концевую группу цепи Формулы 11 до Формулы 15 как определено в данном документе, предпочтительно одним или более агентами, модифицирующими концевую группу цепи Формулы 11, и в условиях, как определено в данном документе.
В одном варианте реализации изобретения, способ получения полимерной композиции включает этапы:
во-первых, приведения в контакт соединения Формулы 9 с более чем от 2,1 и до 8 моль эквивалентами соединения Формулы 10 для образования смеси моно- и дианионных видов и дополнительно приведения в контакт его с по меньшей мере одним видом полимеризуемых мономеров, выбранных из сопряженных олефинов и ароматических винильных соединений, образуя таким образом смесь альфа-моноанионных живых полимерных цепей и омега,омега'-дианионных живых полимерных цепей, и
дальнейшего добавления и приведения в контакт по меньшей мере одного модификатора концевых групп цепи Формулы 11 до Формулы 15, предпочтительно Формулы 11, образуя таким образом полимерную композицию, содержащую a) полимеры Формулы 2, которые модифицированы на одном конце цепи (альфа-модифицированные) и b) полимеры Формулы 1, которые модифицированы на обоих концевых отрезках концов полимерной цепи (омега,омега'-модифицированные).
В другом варианте реализации изобретения, способ получения модифицированного полимера включает этапы:
во-первых, приведения в контакт соединения Формулы 9 с от 1 до 2,1 моль эквивалентами соединения Формулы 10 для образования дианионных видов и дополнительно приведения в контакт его с по меньшей мере одним типом полимеризуемых мономеров, выбранных из сопряженных олефинов и ароматических винильных соединений, формируя таким образом омега,омега'-дианионные живые полимерные цепи, и
дальнейшего добавления и приведения в контакт по меньшей мере одного модификатора концевых групп цепи Формулы 11 до Формулы 15, предпочтительно Формулы 11, причем отношение суммы модификатора концевых групп цепи к соединению Формулы 10 составляет от 0,15 до 0,85, формируя, таким образом, полимерную композицию, содержащую a) полимеры Формулы 2, которые модифицированы на одном конце цепи (альфа-модифицированные) и b) которые модифицированы на обоих концевых отрезках концов полимерной цепи (омега,омега'-модифицированные) и необязательно c) полимеры, которые не модифицированы.
Способ получения полимерной композиция обычно проводят в полимеризационном растворе как полимеризацию в растворе, причем образованный полимер является растворимым в реакционной смеси, или в виде полимеризации в суспензии/суспензионной полимеризации, причем образованный полимер является нерастворимым в реакционной смеси. Подходящие полимеризационные растворители включают не-полярные алифатические и не-полярные ароматические растворители, предпочтительно гексан, гептан, бутан, пентан, высокочистые изопарафины-растворители, циклогексан, толуол и бензол. Полимеризация в растворе обычно протекает при низких температурах, предпочтительно ниже 10 МПа, предпочтительно в температурном диапазоне от 0 до 120 °C. Полимеризацию обычно проводят при периодических, непрерывных или полунепрерывных условиях полимеризации.
В общем, применимая информация о технологиях полимеризации включает полярные координационные соединения и ускорители, каждый для увеличения реактивности инициатора, для случайного размещения ароматических винильных соединений, для случайного размещения 1,2-полибутадиеновых или 1,2-полиизопреновых или 3,4-полиизопреновых звеньев, введенных в полимер; количество каждого соединения; мономера (ов); и подходящих условий процесса описаны в WO 2009/148932, полностью включены в данном документе в качестве ссылки.
ПОЛИМЕРНАЯ КОМПОЗИЦИЯ- ДОПОЛНИТЕЛЬНЫЕ КОМПОНЕНТЫ
Полимерная композиция в соответствии с первым аспектом изобретения может дополнительно содержать один или более дополнительных компонентов, выбранных из (i) компонентов, которые добавлены или образуются в результате процесса полимеризации, применяемые для получения полимера и (ii) компоненты, которые остаются после удаления растворителя из процесса полимеризации. Как правило, эта "вторая полимерная композиция" представляет собой результат без растворителя способа получения полимерной композиции и может содержать компоненты, выбранные из масел (смягчители или масло для наполнения), стабилизаторов и дополнительных (не относящих к изобретению) полимеров. Подходящие масла являются такими, как определено в данном документе. Дополнительные полимеры могут быть изготовлены отдельно, например, в другом реакторе полимеризации, в растворе и могут быть добавлены в реактор до завершения процесса получения полимера.
Вторая полимерная композиция, которая получена после удаления растворителя и технической воды из процесса полимеризации предпочтительно имеет вязкость по Муни (ML 1+4, 100 °C, как измеряно в соответствии с ASTM D 1646 (2004) с применением прибора Монсанто MV2000) до 150, предпочтительно от 20 до 120 и более предпочтительно от 30 до 100. Если вязкость по Муни полимерной композиции более чем 150, пригодность для обработки, что отражается введением наполнителя и теплонакоплением в закрытом резиносмесителе, объединением на вальцовой мельнице, скоростью экструзии, продольной усадкой экструдируемого потока, гладкостью, и т.д., скорее всего, будет негативно влиять, потому что машинное сцепление используемое производителями шин не предназначено для работы с такими высокими профилями значений по Муни резины, и стоимость обработки увеличивается. В некоторых случаях вязкость по Муни менее чем 20 может быть не предпочтительной из-за увеличенной липкости и текучести на холоде несшитого полимера, что в результате приведет к сложности в обработке, слабой прочности формовочного материала в сыром состоянии и плохой стабильности размеров во время хранения. В других случаях, когда полимерная композиция применяется как умягчитель, компатибилизатор или вещество для улучшения технологических свойств в полимерных составах, вязкость по Муни менее чем 20 может быть предпочтительной.
Предпочтительное молекулярно-массовое распределение полимерной композиции, полученной после удаления растворителя из процесса полимеризации, отражается соотношением средневесовой молекулярной массы к среднечисловой молекулярной массе (Mw/Mn), которое находится от 1,0 до 10,0, предпочтительно от 1,1 до 8,0 и более предпочтительно от 1,2 до 4,5.
В полимерной композиции первого аспекта изобретения, полимеры Формулы 1 и 2 содержатся по меньшей мере в количестве 15% по массе настоящего полимера, более предпочтительно по меньшей мере 30% по массе и даже более предпочтительно по меньшей мере 45% по массе. Оставшееся количество полимера состоит из дополнительных полимеров, упомянутых выше. Примерами подходящих дополнительных полимеров являются такие, которые определены в WO 2009/148932 и предпочтительно включают стирол-бутадиеновый сополимер, натуральные резины, полиизопрен и полибутадиен. Желательно, чтобы такие дополнительные полимеры имели вязкость по Муни (ML 1+4, 100°C как измеряно в соответствии с ASTM D 1646 (2004)) в диапазоне от 20 до 150, предпочтительно от 30 до 100.
Полимерная композиция в соответствии с данным изобретением может также содержать один или более дополнительных компонентов, которые добавляют после процесса полимеризации, в том числе один или более наполнителей, один или более дополнительных полимеров, которые не являются полимерами Формулы 1 или Формулы 2, и один или более сшивающих агентов (вулканизирующих агентов) ("третья полимерная композиция"). Содержащая наполнитель полимерная композиция обычно является результатом механического процесса смешивания с применением первой или второй композиции изобретения, одного или больше наполнителей и других необязательных компонентов.
Полимерная композиция изобретения может дополнительно содержать один или более сшивающий агент как определено ниже. Полимерная композиция, содержащая сшивающий агент может быть приготовлена путем смешивания полимерной композиции, необязательно содержащей один или более компонентов, выбранных из масел, стабилизаторов, наполнителей и дополнительных полимеров, и одного или более сшивающих агентов в смесителе при от 140 до 180 °C. Кроме того, такая полимерная композиция может быть получена путем смешивания полимерной композиции изобретения в смесителе при от 140 до 180°C для формирования композиции "первого этапа". Получение композиции "первого этапа" может включать один или более этапов смешивания, предпочтительно от 2 до 7 этапов смешивания. После охлаждения, сшивающие (вулканизирующие) агенты, такие как сера, сшивающие (вулканизирующие) ускорители, необязательно оксид цинка и тому подобные добавляют в композицию "первого этапа" и полученную композицию "второго этапа" смешивают с применением смесителя Брабендера, смесителя Банбери или мельницы с открытыми валами с перфорированной рубашкой для формирования желаемой формы.
МАСЛА
Одно или более масел могут быть использованы в комбинации с несшитой полимерной композицией, необязательно содержащей компоненты (i) и/или (ii), наполнитель и/или сшивающий агент, для уменьшения вязкости или значений Муни или для улучшения пригодности для обработки полимерной композиции и различных эксплуатационных свойств полимерных композиций раз сшитых.
Масла могут быть добавлены к полимерной композиции до конца процесса приготовления полимера и в качестве отдельного компонента после процесса приготовления. Для образцовых примеров и классификации масел смотри WO 2009/148932 и США 2005/0159513, каждый из которых включен в настоящее описание посредством ссылки во всей своей полноте.
Образцовые масла включают MES (сольват мягкой экстракции), RAE (остаточный ароматический экстракт, включая T-RAE (обработанный остаточный ароматический экстракт) и S-RAE), DAE (дистиллированный ароматический экстракт, включая TDAE (обработанный дистиллированный ароматический экстракт)) и NAP (легкое и тяжелое нафтеновые масла, включая Nytex 4700, Nytex 8450, Nytex 5450, Nytex 832, Tufflo 2000 и Tufflo 1200). Кроме того, натуральные масла, включая растительные масла, могут быть использованы в качестве масла для наполнения. Образцовые масла также включают функционализированные вариации этих масел, в частности эпоксидированные масла или гидроксилированные масла. Масла могут содержать различные концентрации полициклических ароматических соединений, парафинов, нафтенов и ароматических соединений и могут иметь различные температуры стеклования.
ТЕХНОЛОГИЧЕСКИЕ ДОБАВКИ
Технологические добавки могут быть необязательно добавлены к полимерной композиции. Их обычно добавляют для уменьшения вязкости. В результате, период перемешивания уменьшается и/или уменьшается количество этапов смешивания и, следовательно, уменьшается энергия потребления и/или достигается повышение пропускной способности в ходе процесса экструзии резиновой смеси. Образцовые технологические добавки описаны в Rubber Handbook, SGF, The Swedish Institution of Rubber Technology 2000 и в Werner Kleemann, Kurt Weber, Elastverarbeitung-Kennwerte und Berechnungsmethoden, Deutscher Verlag für Grundstoffindustrie (Лейпциг, 1990), каждый из которых включен в настоящее описание посредством ссылки во всей полноте. Примеры образцовых технологических добавок включают в частности:
(A) жирные кислоты, включая олеиновую кислоту, приолен, пристерен и стеариновую кислоту;
(B) соли жирных кислот, включая Aktiplast GT, PP, ST, T, T-60, 8, F; Deoflow S; Kettlitz Dispergator FL, FL Plus; Dispergum 18, C, E, K, L, N, T, R; Polyplastol 6, 15, 19, 21, 23; Struktol A50P, A60, EF44, EF66, EM16, EM50, WA48, WB16, WB42, WS180, WS280 и ZEHDL;
(C) диспергирующие агенты, включая Aflux 12, 16, 42, 54, 25; Deoflow A, D; Deogum 80; Deosol H; Kettlitz Dispergator DS, KB, OX; Kettlitz-Mediaplast 40, 50, Pertac/GR; Kettlitz-Dispergator SI; Struktol FL и WB 212; и
(D) диспергирующие агенты или технологические добавки для высокой активности белых наполнителей, включая Struktol W33, WB42, HT207, HT254, HT276; Ultra-Flow 440 и.700S (Активные добавки).
НАПОЛНИТЕЛИ
В одном варианте реализации изобретения, полимерная композиция в соответствии с данным изобретением содержит один или более наполнителей, выступающих в качестве армирующих агентов. Примеры подходящих наполнителей включают углеродную сажу (в том числе электропроводящую углеродную сажу), углеродные нанотрубки (CNT) (в том числе дискретные CNT, полые углеродные волокна (HCF) и модифицированный CNT, несущий одну или более функциональных групп, таких как гидроксильные, карбоксильные и карбонильные группы) графит, графен (в том числе дискретные графеновые пластинки), кремнезем, углеродно-кремнеземный двухфазный наполнитель, глины (слоистые силикаты, в том числе расслоенная наноглина и органоглина), карбонат кальция, карбонат магния, лигнин, аморфные наполнители, такие как наполнители на основе стеклянных частичек, наполнители на основе крахмала и их комбинацию. Дополнительные примеры подходящих наполнителей описаны в WO 2009/148932, которые полностью включены в настоящее описание посредством ссылки.
Примеры подходящей углеродной сажи включают одну, традиционно изготовленную путем обжига в печи, например, такую, которая имеет удельную поверхность адсорбции азота 50-200 м2/г и абсорбцию DBP масла 80-200 мл/100 грамм, такую как углеродная сажа FEF, HAF, ISAF или SAF класса, и электропроводящую углеродную сажу. В некоторых вариантах реализации изобретения применяют углеродную сажу высокоагломерированного типа. Углеродную сажу обычно применяют в количестве от 2 до 100 частей по весу, или от 5 до 100 частей по весу, или от 10 до 100 частей по весу, или от 10 до 95 частей по весу на 100 частей по весу общего полимера.
Примеры подходящего кремнеземного наполнителя включают кремнезем, приготовленный технологией жидкостной обработки, кремнезем, приготовленный сухим способом производства и кремнезем типа синтетических силикатов. Кремнезем с частичками малого диаметра и высокой удельной поверхностью проявляет повышенный усиливающий эффект. Маленький диаметр, высокоагломерированного типа кремнезем (т.е. имеющий большую удельную поверхность и высокую абсорбционную способность масла) демонстрирует превосходную диспергируемость в полимерной композиции, приводящей в результате к отличной пригодности для обработки. Средний диаметр частиц кремнезема в выражении диаметра первичных частиц может быть от 5 до 60 нм, или от 10 до 35 нм. Удельная площадь поверхности кремнеземных частичек (измерянных по методу БЭТ) может быть от 35 до 300 м2/г. Кремнезем обычно применяется в количестве от 10 до 150 частей по весу, или от 30 до 130 частей по весу, или от 50 до 130 частей по весу на 100 частей по весу общего полимера.
Кремнеземные наполнители могут применяться в комбинации с другими наполнителями, в том числе углеродной сажей, углеродными нанотрубками, углерод-кремнеземным двухфазным наполнителем, графеном, графитом, глиной, карбонатом кальция, карбонатом магния и их комбинация.
Углеродная сажа и кремнезем могут быть добавлены вместе, в этом случае общее количество углеродной сажи и кремнезема составляет от 30 до 150 частей по весу или от 50 до 150 частей по весу на 100 частей по весу общего полимера.
Углерод-кремнеземный двухфазный наполнитель, также называемый, как покрытая кремнеземом углеродная сажа, изготовлена путем нанесения кремнезема на поверхность углеродной сажи и коммерчески доступна под торговой маркой CRX2000, CRX2002 или CRX2006 (продукты Cabot Co.). Углерод-кремнеземный двухфазный наполнитель добавляют в том же количестве, как описано выше в отношении кремнезема.
СИЛАНОВЫЕ СВЯЗУЮЩИЕ ВЕЩЕСТВА
В некоторых вариантах реализации изобретения, силановое связующее вещество (применяется для улучшения совместимости полимера и наполнителей) может быть добавлено к полимерной композиции изобретения, когда дополнительно содержится один или более кремнезем, слоистый кремнезем (такой как магадиит) и углерод-кремнеземный двухфазный наполнитель. Обычное количество добавленного силанового связующего вещества составляет от около 1 до около 20 частей по весу и, в некоторых вариантах реализации изобретения, от около 5 до около 15 частей по весу на 100 частей по весу общего количества кремнезема и/или углерод-кремнеземного двухфазного наполнителя.
Силановые связующие вещества могут быть классифицированы согласно с Fritz Röthemeyer, Franz Sommer: Kautschuk Technologie, (Carl Hanser Verlag 2006):
(A) бифункциональные силаны, включая Si230 ((EtO)3Si(CH2)3Cl), Si225 ((EtO)3SiCH=CH2), Si263 ((EtO)3Si(CH2)3SH), [(EtO)3Si(CH2)3Sx(CH2)3Si(OEt)3] с x=3.75 (Si69), 2.35 (Si75) или 2.15 (Si266), Si264 ((EtO)3Si-(CH2)3SCN) и Si363 ((EtO)Si((CH2-CH2-O)5(CH2)12CH3)2(CH2)3SH)) (Evonic Industries AG); NXT (3-октаноилтио-1-пропилтриэтоксисилан), NXT-Z45, NXT-Z100 (Momentive Performance Materials Inc.); Xiameter® ofs-6030 силан (метакрилоксипропилтриметоксисилан), Xiameter® ofs-6300 силан ((MeO)3SiCH=CH2), и
(B) монофункциональные силаны, включая Si203 ((EtO)3-Si-C3H7) и Si208 ((EtO)3-Si-C8H17).
Дополнительные подходящие примеры силановых связующих веществ даны в WO 2009/148932 и включают бис-(3-гидрокси-диметилсилил-пропил)тетрасульфид, бис-(3-гидрокси-диметилсилил-пропил) дисульфид, бис-(2-гидрокси-диметилсилил-этил) тетрасульфид, бис-(2-гидрокси-диметилсилил-этил)дисульфид, 3-гидрокси-диметилсилил-пропил-N,N-диметилтиокарбамоил тетрасульфид и 3-гидрокси-диметилсилил-пропилбензотиазол тетрасульфид.
СШИВАЮЩИЕ АГЕНТЫ (ВУЛКАНИЗИРУЮЩИЕ АГЕНТЫ )
Любой сшивающий агент, который обычно применяется в производстве резиновых изделий, может применяться в изобретении, и может применяться комбинация одного или более сшивающих агентов.
Сера, серо-содержащие соединения, действующие в качестве доноров серы, ускоряющие системы на основе серы и пероксиды являются наиболее распространенными сшивающими агентами. Примеры серо-содержащих соединений, действующих в качестве доноров серы, включают дитиодиморфолин (DTDM), тетраметилтиурам дисульфид (TMTD), тетраэтилтиурам дисульфид (TETD) и дипентаметилентиурам тетрасульфид (DPTT). Примеры ускоряющих систем на основе серы включают аминопроизводные, производные гуанидина, конденсационные продукты альдегидамина, тиазолы, сульфиды тиурама, дитиокарбаматы и тиофосфаты. Примеры пероксидов включают ди-терт-бутил-пероксиды, ди-(терт-бутил-перокси-триметил-циклогексан), ди-(терт-бутил-перокси-изопропил-)бензол, дихлоро-бензоилпероксид, дикумилпероксиды, терт-бутил-кумил-пероксид, диметил-ди(терт-бутил-перокси)гексан, диметил-ди(терт-бутил-перокси)гексин и бутил-ди(терт-бутил-перокси)валерат (Rubber Handbook, SGF, The Swedish Institution of Rubber Technology 2000).
Дополнительные примеры и дополнительная информация, касающаяся сшивающих агентов может быть найдена в Kirk-Othmer, Encyclopedia of Chemical technology 3rd, Ed., (Wiley Interscience, N.Y. 1982), volume 20, pp. 365-468, (в частности " Vulcanizing Agents and Auxiliary Materials " pp. 390-402).
Сшивающие ускорители сульфен амидного типа, гуанидинового типа или тиурамового типа могут применяться вместе со сшивающим агентом, как требуется. Другие добавки, такие как цинковые белила, сшивающие вспомогательные вещества, вещества, предотвращающие старение, вещества, вспомогающие в переаботке и тому подобные, могут быть необязательно добавлены. Сшивающий агент обычно добавляют в полимерную композицию в количестве от 0,5 до 10 частей по весу или, в некоторых вариантах реализации изобретения, от 1 до 6 частей по весу на 100 частей по весу общего веса полимера. Примерами ускорителей сшивания и их добавленное количество по отношению к общей массе полимера приведены в WO 2009/148932.
Ускоряющие системы на основе серы могут или могут не содержать оксид цинка. Оксид цинка предпочтительно применяется как компонент ускоряющей системы на основе серы.
СШИТАЯ ПОЛИМЕРНАЯ КОМПОЗИЦИЯ
Сшитую полимерную композицию в соответствии с м третьим аспектом изобретения получают путем сшивания полимерной композиции изобретения, которая содержит по меньшей мере один сшивающий агент. Поскольку сшитые эластомерные полимерные композиции изобретения проявляют низкое сопротивление качению, низкое динамическое теплонакопление и отличное произведение влажного заноса, они хорошо подходят для применения в производстве шин, протекторов шин, боковых стенок и несущих конструкций шин, а также других промышленных продуктов, таких как ремни, шланги, гасители вибраций и компоненты обуви.
Сшитая полимерная композиция является результатом реактивного полимер-полимер сшивающего процесса формовки, который выполняют из полимерной композиции изобретения, содержащей один или более сшивающих агентов. Реактивная обработка преобразует, по существу, несшитую полимерную композицию в сшитую (или вулканизированную) полимерную композицию.
Данное изобретение также относится к изделию, содержащему по меньшей мере один компонент, сформированный из сшитой полимерной композиции изобретения. Изделием может быть шина, протектор шины, боковая стенка шины, автомобильная часть, компонент обуви, мяч для гольфа, пояс, уплотнитель, прокладка или шланг.
Для изготовления шины транспортного средства, следующие дополнительные полимеры представляют особый интерес для применения в сочетании с полимером изобретения: натуральный каучук; низкий цис-полибутадиен (LCBR) содержащий менее чем 20 процентов по массе 1,2-полибутадиена, эмульсия SBR (ESBR) и раствор SBR (SSBR) резин с температурой стеклования выше -50 °C; полибутадиеновая резина с высоким содержанием цис-1,4-звеньев (>90%), таких которые получены путем применения катализатора на основе никеля, кобальта, титана, ванадия, гадолиния или неодимия; и полибутадиеновая резина с содержанием винила от 0 до 75%; и их комбинацию; полибутадиеновая резина с содержанием высоких транс-1,4-звеньев (>75%) или содержащих SBR, например, между 5 и 45% масс. стирола и имеет содержание высокого транс-1,4-полибутадиена (>75%) в полибутадиеновой фракции coполимера (каждый тип полимера, SBR или BR, может быть получена с одним или больше соединениями инициатора, содержащими соединения щелочноземельных металлов, таких, которые описаны в Патентах США №№. 6693160; 6627715; 6489415; 6103842; 5753579; 5086136; и 3629213, каждый из которых включен в настоящее описание посредством сслыки во всей полноте; или с применением катализаторов на основе кобальта, например, как описано в Патенте США №№ 6310152; 5834573; 5753761; 5448002 и 5089574 и публикации заявки патента США No. 2003/0065114, каждый из которых включен в настоящее описание посредством сслыки во всей полноте; или с применением катализаторов на основе ванадия, например, как описано в EП 1 367 069; JP 11301794 и США 3,951,936, каждый из которых включен в настоящее описание посредством сслыки во всей полноте; или с применением катализаторов на основе неодимия, как описано в EП 0 964 008, EП 0 924 214 и Патент США №№ 6184168; 6018007; 4931376; 5134199 и 4689368, каждый из которых включен в настоящее описание посредством сслыки во всей полноте).
Композицию изобретения могут также применять для производства ударопрочного полистирола (HIPS) и бутадиен-модифицированного акронитрил-бутадиен-стирольного сополимера (ABS) (смотри, например, WO 2009/148932, включены в данное описание в качестве ссылки).
ОПРЕДЕЛЕНИЯ
Алкильные группы как определено в данном документе, будь то сами по себе или в сочетании с другими группами, такими как алкарил или алкокси, включают как прямые цепи алкильных групп, таких как метил (Me), этил (Et), н-пропил (Pr), н-бутил (Bu), н-пентил, н-гексил, и т.д., разветвленные алкильные группы, такие как изопропил, трет-бутил, и т.д., и циклические алкильные группы, такие как циклогексил.
Алкокси группы, как определено в этом документе включают метокси (MeO), этокси (EtO), пропокси (PrO), бутокси (BuO), изопропокси, изобутокси, пентокси, и пр.
Арильные группы как определено в этом документе включают фенил, бифенил и другие бензоидные соединения. Арильные группы предпочтительно содержат только одно ароматическое кольцо и более предпочтительно содержат C6 ароматическое кольцо.
Аралкильные группы, как определено в этом документе относятся к комбинации одной или более арильных групп, связанных с одной или более алкильными группами, например, в форме алкил-арил, арил-алкил, алкил-арил-алкил и арил-алкил-арил. Аралкильные группы предпочтительно содержат только одно ароматическое кольцо и более предпочтительно содержат C6 ароматическое кольцо.
ПРИМЕРЫ
Следующие примеры приведены с целью дополнительной иллюстрации изобретения, и не должны толковаться как ограничивающие изобретение. Примеры включают приготовление и тестирование модифицированных эластомерных полимеров; и приготовление и тестирование несшитых полимерных композиций, а также поперечно сшитых или отвержденных полимерных композиций, также упоминаемых как вулканизованная полимерная композиция. Если не указано иное, все части и процентные выражения выражены по массе. "Комнатная температура" означает температуру 20 °C. Все полимеризации производятся в атмосфере азота при удалении влаги и кислорода.
Содержание винильных групп в полибутадиеновой фракции измеряли с помощью ФУРЬЕ-ИК-спектроскопии на Nicolet AVATAR 360 ФУРЬЕ-ИК-спектроскопии или Nicolet iS 10 ФУРЬЕ-ИК- спектроскопии, на основе определения калибровки 1H-ЯМР методом как описаны выше. ИК образцы были приготовлены с применением пресса.
Содержание связанного стирола: калибровочная кривая были получена с помощью ФУРЬЕ-ИК-спектроскопии (Nicolet AVATAR 360 ФУРЬЕ-ИК- спектроскопия или Nicolet iS 10 ФУРЬЕ-ИК-спектроскопия). ИК образцы были приготовлены с применением пресса. Для ИК определения связанного стирола в стирол-бутадиеновых сополимерах, проверялись четыре полоски: a) полоска для транс-1,4-полибутадиеновых звеньев при 966 см-1, b) полоска для цис-1,4-полибутадиеновых звеньев при 730 см-1, c) полоска для 1,2-полибутадиеновых звеньев при 910 см-1 и d) полоска для связанного стирола (стирольная ароматическая связь) при 700 см 1. Вершины связей нормализованы в соответствии с соответствующими коэффициентами ослабления и обобщены в общей сложности до 100%. Нормализация осуществляется через 1H- и 13C-ЯМР (Avance 400 от Bruker Analytik GmbH, 1H=400 МГц; 13C=l00 МГц).
1D ЯМР спектры были собраны на BRUKER Avance 400 ЯМР спектрометре (BRUKER Corp.), с применением ʺ5 мм двойного детекторного зонда.ʺ Однородность поля была оптимизирована за счет максимизации дейтериевого сигнального замыкателя. Образцы расклинивали путем оптимизации дейтериевого сигнального замыкателя. Образцы вводили в действие при комнатной температуре (298 K). Были использованы следующие дейтериевые растворители: C6D6 (7,16 частей на миллион для 1H; 128,06 частей на миллион для 13C-ЯМР), CDCl3 (7,26 частей на миллион для 1H; 77,16 частей на миллион для 13C-ЯМР), сигналы оставшихся протонов дейтериевых растворителей каждый был использован в качестве внутреннего стандарта.
Для спектральной обработки, было использовано програмное обеспечение BRUKER TopSpin. Коррекция фазировки и базисной линии и спектральная интеграция полученных спектров были сделаны в ручном режиме. Для получения параметров см. Таблицу 1.
Таблица 1: 1D-ЯМР параметры сбора, с применением последовательностей стандартных импульсов BRUKER
GPC-метод: SEC откалибровано узко-распределенным полистиролным стандартом.
Подготовка образцов:
a) около 9-11 мг высушенного полимерного образца (содержание влаги < 0,6%) растворяли в 10 мл тетрагидрофурана, с применением коричневой пробирки 10 мл размера. Полимер растворяли путем встряхивания пробирки в течение 20 мин при 200 оборотов в минуту.
b) Полимерный раствор перемещали в 2 мл пробирку с применением 0,45 мкм съемного фильтра.
c) 2 мл пробирка была помещена на пробоотборник для GPC-анализа.
Скорость элюирования: 1,00 мл/мин
Вводимый объем: 100,00 мкл
Полидисперсность (Mw/Mn) использовали в качестве меры для ширины молекулярно-массового распределения. Значения Mw и Mn (средневесовая молекулярная масса (Mw) и среднечисловая молекулярная масса (Mn)) были измеряны с помощью гель-проникающей хроматографии на SEC с определением рефракционного индекса (универсальная калибровка). Измерение проводили в ТГФ при 40 °C. Аппарат: Agilent Serie 1100 /1200; настройка модуля: дегазатор, Исо насос, автопробоотборник, термостат, UV - детектор, RI - детектор.
В каждом GPC-устройстве, 4 колонки использовали в соединенном режиме: 1x Agilent Plgel 10 мкм Guard 50×7,5 мм ( Часть № PL1110-1120 ) плюс 3 x Agilent PLgel 10 мкм Mixed-B 300×7,5 мм ( Часть № PL1110-6100 ).
GPC стандартные растворы: EasiCal PS-1 Полистирольные стандартные растворы, Spatula A+B (Agilent Technologies, Часть № PL2010-0505).
Mp1, Mp2 и Mp3 соответствует (пик максимума) молекулярной массе, измерянной при третьем, втором и первом пике GPC кривой, соответственно (пик Mp1 (самая низкаямолекулярная масса) находиться на правой части кривой, и пик Mp3 (наивысшая молекулярная масса) находится на левой стороне кривой). Пик максимума молекулярной массы означает молекулярную массу пика в точке максимума интенсивности. Mp2 и Mp3 представляют собой две или три полимерные цепи связанные с одной макромолекулой. Mp1 представляет собой одну полимерную цепь (на основе молекулярной массы - без соединения двух или более полимерных цепей с одной макромолекулой).
Третья полимерная композиция была получена путем совмещения компонентов перечисленных в Таблице 4, Таблице 5 или Таблице 6 в 380 мл смесителе Бенбери (Labstation 350S от Brabender GmbH & Co KG), с последующим двух-стадийным процессом смешивания. Стадия 1 - смешивание всех компонентов вместе, за исключением компонентов вулканизационного комплекса, с образованием состава стадии 1. Стадия 2 - компоненты вулканизационного комплекса смешивают в составе стадии 1 с образованием состава стадии 2.
Вязкость по Муни измеряли в соответствии с ASTM D 1646 (2004), с временем предварительного разогрева одна минута и временем работы ротора 4 минуты, при температуре 100°C [ML1+4(100 °C)], на MV 2000E от Alfa Technologies UK. Измерения вязкости по Муни резины выполняли на сухом (без растворителя) сыром полимере (несшытая резина). Значения по Муни сырых полимеров приведены в Таблице 2.
Измерения несшитых реологических свойств проводили в соответствии с ASTM D 5289-95 (переодобрено 2001), с применением безроторного сдвигового реометра (MDR 2000 E от Alfa Technologies UK), чтобы измерить время отверждения (TC). Измерения реометром проводили при постоянной температуре 160°C на несшитом полимерном составе второй стадии, в соответствии с Таблицей 4, Таблицей 5 или Таблицей 6. Количество полимерного образца составляет около 4,5 г. Форма образца и приготовление образца стандартизированы и определяются измерительным устройством (MDR 2000 E от Alfa Technologies UK). TC 50, TC 90 и TC 95 значения представляют собой соответствующие периоды времени, необходимые для достижения 50%, 90% и 95% конверсии реакции сшивания. Крутящий момент измеряют как функцию времени реакции. Конверсию сшивания автоматически рассчитывают из сгенерированнорго крутящего момента по сравнению с кривой времени. TS 1 и TS 2 значения представляют собой соответствующие периоды времени, требуемые для увеличения крутящего момента 1 dNm и 2 dNm выше минимума соответствующего крутящего момента (ML) во время сшивания.
Прочность на растяжение, удлинение при разрыве и модуль при 300% удлинении (Модуль 300) были измеряны в соответствии с ASTM D 412-98A (перодобрено 2002), с сприменением C-образного образца для испытаний на Zwick Z010. Использовались стандартизированные C-образные образцы для испытаний 2 мм толщины. Измерения прочности на растяжение проводили при комнатной температуре на отвержденном полимерном образце второй стадии, приготовленном в соответствии с Таблицей 4, Таблицей 5 или Таблицей 6. Составы стадии 2 сшивали в течение 16-25 минут при 160°C до TC 95 (95% конверсия сшивания) (см. данные отверждения в Таблице 7).
Теплонакопление было измеряно в соответствии с ASTM D 623, метод A, на Doli 'Goodrich'-флексометре. Измерение теплонакопления было произведено на сшитом полимерном образце второй стадии в соответствии с Таблицей 4, Таблицей 5 или Таблицей 6. Составы Стадии 2 были сшиты при 160°C до TC 95 (95% конверсия сшивания) (см. данные отверждения в Таблице 7).
Эластичность на отскок была измеряна в соответствии с DIN 53512 при 0°C и 60°C на Zwick 5109. Измерения проводили на отвержденном полимерном образце второй стадии, приготовленной в соответствии с Таблицей 4, Таблицей 5 или Таблицей 6. Составы стадии 2 были сшиты при 160°C до TC 95 (95% конверсия сшивания) плюс 5 минут дополнительного времени (см. данные отверждения в Таблице 7). Чем меньше индекс при 0 °C, тем лучше сопротивление влажного заноса (ниже=лучше). Чем больше индекс при 60 °C, тем ниже гистерезисные потери и тем ниже сопротивление качению (выше=лучше).
DIN истирание измеряли в соответстиви с DIN 53516 (1987-06-01). Чем выше данный индекс, тем ниже стойкость к истиранию (ниже=лучше). Измерение истирания проводили на сшитом полимерном составе второй стадии в соответствии с Таблицей 4, Табицей 5 или Таблицей 6.
Измерения тангенса δ при 60°C и тангенса δ при 0 °C, а также тангенса δ при 10°C проводились на прямоугольном образце с толщиной 2 мм и шириной 10 мм, с применением динамического механического термального спектрометра "Eplexor 150N," производства Gabo Qualimeter Testanlagen GmbH (Германия), с применением динамической деформации 2%, при частоте 2 Гц, при соответствующих температурах. Начальная длина образца, которая равна зажимному расстоянию оборудования, составляет 10 мм. Чем меньше индекс при температуре 60 °C, тем ниже сопротивление качению (ниже=лучше). Тангенс δ при 0°C и тангенс δ при -10°C измеряли с применением такого же оборудования и условий загрузки при 0°C и -10 °C, соответственно. Чем больше индекс при 0 °C, тем лучше сопротивление влажного заноса и чем больше индекс при -10 °C, тем лучше свойства сцепления на льду (выше=лучше). Тангенс δ при 60°C и тангенс δ при 0 °C, а также тангенс δ при 10°C были измеряны (см. Талицу 8). Составы стадии 2 были сшиты при 160°C до TC 95 (95% конверсия сшивания) (см. данные отверждения в Таблице 7). Процесс приводит к образованию визуально безпузырьковых однородных отвержденных резиновых пластин толщиной 2 мм. Образец был вырезан с помощью железной резки образцов с соответствующей шириной 10 мм.
В целом, чем выше значения удлинения при разрыве, прочности на растяжение, Модуль 300, и тангенс δ при 0 °C, эластичности на отскок при 60 °C, тем лучше функциональные характеристики образца; в то же время чем ниже тангенс δ при 60 °C, теплонакопление, DIN истриание и эластичность на отскок при 0 °C, тем лучше функциональные характеристики образца.
Коэффициенты трения скольжения были определены с помощью метода в соответствии с EN ISO 13287. Измерения проводятся машиной для испытания сопротивления скольжению (PFI Germany: GSP 3034), с точностью измерении более, чем 2%. Резиновый образец отпрессовали на металлическую поверхность с определенной нормальной перпендикулярной силой к горизонтальной поверхности, установленной на уровне 30Н±2%. Образец устанавливают под углом 7° к горизонтальной линии, чтобы уменьшить поверхность контакта между металлом и резиной и силу прикладывали стационарно в течение 0,2 секунд. Образец двигали с определенной скоростью 0,3±0,03 м/с по металлической поверхности, в это время измеряли силу трения, которая представляет собой необходимую силу, параллельную поверхности и против направления движения образца. Коэффициент трения определяется как соотношение между силой трения и нормальной силой. На протяжении измерений были взяты около 60 точек измерений, рассчитано среднее значение этих измерений. Нижняя пластина выполнена из стали Номер 14371, соответствующей EN10088-2:2005. Образцы готовили путем прессования резинового образца в форме размерами 15×80×115 мм при 100°C с 200 бар в течение 7 минут. Для прессования применяются хромированные листы глянцевой поверхности, со слоем фольги с полиэстерной пленкой между резиновым образцом и стальным листом, чтобы получить очень гладкую и ровную поверхность образцов. Для каждого материала готовятся и измеряются четыре образца.
Соединения P1 и P2 были синтезированы согласно со способом, который описан в патенте США 4982029 и показаны и охарактеризованы ниже:
 P1
P1
1H-ЯМР (300 МГц, 23°C, CDCl3): δ=7,42 (с, 1H, Ar-H), 7,30 (с, 7H, Ar-H), 6,73 (д, 4H, Ar-H), 5,40 (с, 2H, =CH2), 5,28 (с, 2H, =CH2), 2,98 (с, 12H, NCH3) м.д.; 13C-ЯМР (75 МГц, 23°C, CDCl3) δ=149,94, 149,70, 141,70, 128,97, 128,46, 127,78, 127,64, 112,20, 111,49, 40,65 м.д.
 P2
P2
1H ЯМР (400 МГц, 23°C, CDCl3): δ=7,39-7,40 (м, 1H, Ar-H), 7,35-7,26 (м, 11H, Ar-H), 5,47 (д, J=1,2 Hz, 2 H, HCH=C), 5,40 (д, J=1,2 Hz, 2 H, HCH=C), 1,33 (с, 18H, C(CH3)3) м.д.; 13C ЯМР (100 МГц, 23°C, CDCl3) δ=150,74, 149,60, 141,53, 138,18, 128,20, 127,85, 127,75, 125,04, 113,85, 34,54, 31,32 м.д.
Приготовление смеси инициатора I1
Соединения P1 (34,9 г, 94,9 ммоль) и TMEDA (38,7 г, 333 ммоль) были растворены в циклогексане (286 г) и затем добавляли н-бутиллития (60,3 г, 190 ммоль, 20 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI1. GC анализ образца I1, который гидролизуют метанолом, показал полное превращение P1 в соответствующий дианион DI1.
Из-за очень сжатого воздуха и чувствительности к влаге DI1, соединение было определено как его гидролизованный продукт DI1a. Таким образом, смесь инициатора была гидролизована избытком метанола и охарактеризована путем ЯМР и GC-MS.
1H-ЯМР (400 МГц, 23°C, C6D6): δ=6,94-6,86 (м, 8H, Ar-H), 6,33-6,31 (м, 4H, Ar-H), 3,63-3,58 (м, 2H, 2x Ar-CH-Ar), 2,22 (с, 12H, NCH3), 1,82-1,76 (м, 2H, Ar2CH-CH2), 1,07-0,88 (м, 12H, (CH2)3CH3), 0,54 (т, 6H, (CH2)3CH3) м.д.; 13C-ЯМР (101 МГц, 23°C, C6D6) δ=149,43, 146,72, 134,07, 128,86, 128,65, 128,16, 128,15, 127,91, 113,35, 50,99, 40,50, 36,63, 32,34, 28,28, 22,99 м.д.
GC-MS (EI, 70 eV): m/z (%)=485 (M+, 22), 413 (M+ -CH3 -C4H9, 100), 327 (4), 207 (4), 171 (51), 134 (4).
 DI1
DI1  DI1a
DI1a
Приготовление смеси инициатора I2
Соединения P2 (22,1 г, 56,1 ммоль) и TMEDA (22,9 г, 196 ммоль) были растворены в циклогексане (500 г) и затем был добавлен н-бутиллития (36,0 г, 112 ммоль, 20 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2. GC анализ I2 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Из-за очень сжатого воздуха и чувствительности к влаге DI2, соединение было определено как его гидролизованный продукт DI2a. Таким образом, смесь инициатора была гидролизована избытком метанола и охарактеризована путем ЯМР и GC-MS.
 DI2
DI2  DI2a
DI2a
1H-ЯМР (400 МГц, 23°C, CDCl3): δ=7,28-7,27 (м, 2H, Ar-H), 7,26-7,24 (м, 2H, Ar-H), 7,16-7,11 (м, 6H, Ar-H), 7,02-6,99 (м, 2H, Ar-H), 3,81 (т, 2H, 2x Ar-CH-Ar), 2,01-1,95 (м, 4H, Ar2CH-CH2), 1,28 (с, 18H, 2x C(CH3)3), 1,27-1,17 (м, 12H, 2x (CH2)3CH3), 0,83 (т, 6H, 2x (CH2)3CH3) м.д.; 13C-ЯМР (101 МГц, 23°C, CDCl3) δ=148,59, 145,29, 142,67, 128,40, 127,81, 127,46, 125,69, 125,24, 51,00, 36,02, 34,41, 32,00, 31,49, 27,86, 22,66, 14,20 м.д.
GC-MS (EI, 70 eV): m/z (%)=511 (M+, 5), 439 (M+ -CH3 -C4H9, 100), 240 (12), 212 (13), 147 (5), 57 (11).
Приготовление смеси инициатора I3
Соединения P2 (31,5 г, 79,8 ммоль) и TMEDA (7,43 г, 63,9 ммоль) были растворены в циклогексане (254 г) и затем был добавлен н-бутиллития (68,2 г, 214 ммоль, 20 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2. A GC анализ I3 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Приготовление смеси инициатора I4
Соединение P2 (86,7 г, 109 ммоль, 50 мас.% раствор в циклогексане) и TMEDA (10,0 г, 87,2 ммоль) были растворены в циклогексане (91,7 г) и затем был добавлен н-бутиллития (95,5 г, 301 ммоль, 20 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2, A GC анализ I4 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Приготовление смеси инициатора I5
Соединение P2 (34,2 г, 86,7 ммоль), TMEDA (10,4 г, 89,2 ммоль), циклогексан (75,0 г) и н-бутиллития (60,5 г, 186 ммоль, 20 мас.% раствор в циклогексане) были добавлены в следующем порядке в двухстенный, 0,5 литровый стальной реактор при 40 °C. Смесь сразу же достигает 70°C после введения н-бутиллития. Смесь перемешивали на протяжении 100 мин, позволяя температуре остыть до 40 °C. GC анализ смеси, которую гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2. Кроме того, добавляли 1,3-бутадиен (45,7 г, 0,84 моль) в течении 10 мин. Полученную смесь применяют для полимеризации полимерного образца 28.
Приготовление смеси инициатора I6
Соединения P2 (6,80 г, 8,60 ммоль) и TMEDA (0,80 г, 6,90 ммоль) были растворены в циклогексане (30 г) и затем был добавлен н-бутиллитий (7,34 г, 16,7 ммоль, 15 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2. A GC анализ I6 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Приготовление смеси инициатора I7
Соединение P2 (10,0 г, 25,3 ммоль) и TMEDA (9,81 г, 88,7 ммоль) были растворены в циклогексане (150 г) и затем был добавлен н-бутиллитий (16,0 г, 50,7 ммоль, 20 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2. A GC анализ I7 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Приготовление смеси инициатора I8
Соединение P2 (4,0 г, 10,1 ммоль) и TMEDA (4,16 г, 35,0 ммоль) были растворены в циклогексане (80 г) и затем был добавлен н-бутиллитий (6,39 г, 20,3 ммоль, 20 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2. A GC анализ I8 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Приготовление смеси инициатора I9
Соединения P2 (16,7 г, 42,3 ммоль) и TMEDA (17,2 г, 148 ммоль) были растворены в циклогексане (520 г) и затем был добавлен н-бутиллитий (27,0 г, 84,7 ммоль, 20% мас. раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2. A GC анализ I9 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Приготовление смеси инициатора I10
Соединения P2 (16,7 г, 42,3 ммоль) и TMEDA (17,2 г, 148 ммоль) были растворены в циклогексане (520 г) и затем был добавлен н-бутиллитий (27,0 г, 84,7 ммоль, 20 мас.% раствор в циклогексане). Цвет раствора сразу же стал темно-красным, указывая на образование дианиона DI2. A GC анализ I10 который гидролизуют метанолом, показал полное превращениеP2 в соответствующий дианион DI2.
Дивинилбензол (DVB) был приобретен у Bowden Chemicals ltd. и применялся в качестве 0,16 M раствора в циклогексане. Соотношение изомеров 1,3-DVB/1,4-DVB составляет 70/30.
Агенты, модифицирующие концевую группу цепи
Агент, модифицирующий концевую группу цепи E1 представленный Формулой E1 ниже, и был синтезирован в соответствии со способом, описанным в WO 2009/148932.
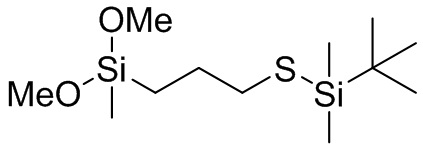
Формула E1
Агент, модифицирующий концевую группу цепи E2 был приобретен как 1-метил-2-пирролидон от Merck Millipore.
Формула E2
Агент, модифицирующий концевую группу цепи E3 представлен Формулой E3 ниже, и был синтезирован следующим образом:
Соединение E1 (114 г, 388 ммоль) растворяли в 400 мл трет-бутил метиловом эфире (MTBE) и добавляли метилмагний хлорид через капельную воронку (140 мл, 420 ммоль, 3M раствор в тетрагидрофуране). После завершения добавления реактива Гриньяра, смесь перемешивали на протяжении ночи. Метанол (2 мл) добавляли для гидролизации небольшого избытка реактива Гриньяра с последующей фильтрацией для удаления осажденных солей магния. Все летучие компоненты удаляли при пониженном давлении и остаток частично дистиллировали с получением 170 г (79%) безсцветной жидкости.
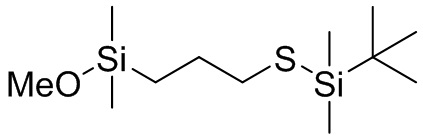
Формула E3
1H-ЯМР (400 МГц, 23°C, C6D6): δ=3,25 (с, 3H, OCH3), 2,50 (т, 2H, S-CH2), 1,75-1,68 (м, 2H, CH2-CH2-CH2), 1,00 (с, 9H, C(CH3)3), 0,69-0,65 (м, 2H, Si-CH2), 0,23 (с, 6H, Si(t-Bu)(CH3)2), 0,04 (с, 6H, Si(OMe)(CH3)2) м.д.; 13C-ЯМР (101 МГц, 23°C, C6D6) δ=50,02, 30,27, 27,61, 26,55, 19,14, 15,84, -2,54, -3,39 м.д.
Агент, модифицирующий концевую группу цепи E4 представлен Формулой E4 ниже, и был синтезирован в соответствии с процедурой описанной в WO 2009/148932.
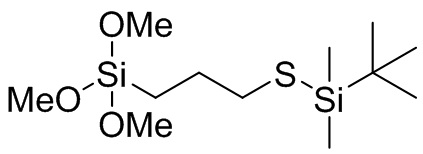
Формула E4
Сополимеризация 1,3-Бутадиена со Стиролом
Производство образцов 1-11, 14-18, 20-21, 24, 28-34 и 36-39
Сополимеризацию проводили в двухстенном, 10 литровом стальном реакторе, который был сначала продут азотом перед добавлением органического растворителя, мономеров, полярного координационного соединения, соединения инициатора или других компонентов. Затем были добавлены следующие компоненты в таком порядке: растворитель циклогексан (4600 грамм); бутадиеновый мономер, стирольный монмоер, тетраметилэтилендиамин (TMEDA) и необязательно дивинилбензол (DVB), и смесь нагревали до 40 °C, с последующим титрованием н-бутиллитием до удаления следов влаги или других примесей. Желаемое соединение инициатора полимеризации добавляли в реактор полимеризации для инициации реакции полимеризации. Если применяется смесь соединений (например, I3 и н-бутиллитий в Примере 16 в Таблице 2), если не указано иное, то они были добавлены в то же смое время. Полимеризацию проводили в течении, по меньшей мере, 80 минут, не позволяя температуре полимеризации превышать 60 °C, до тех пор, пока конверсия мономера не достигнет, по меньшей мере 97%. После этого было добавлено 2,3% от общего количества бутадиенового мономера, с последующим добавлением модификатора концевых групп цепи, если не указано иное. После 20 минут полимеризацию останавливали добавлением метанола (2 эквивалента, основанных на количестве инициатора). К полимерному раствору добавляли 2,20 г IRGANOX 1520 в качестве стабилизатора. Эту смесь перемешивали на протяжении 10 минут. Полученный в результате полимерный раствор необязательно смешивали с 27,3% мас TDAE масла (VivaTec 500, Hansen & Rosenthal KG), затем разделили паром в течение одного часа для удаления растворителя и других летучих веществ, и сушили в сушильном шкафу при 70°C в течение 30 минут и затем дополнительно в течение одного или трех дней, при комнатной температуре.
Количество реагентов, полученная в результате полимерная композиция и некоторые ее свойства приведены в Таблице 2 и Таблице3.
Производство Примеров 12, 13, 19 и 22
Сo-полимеризацию проводили в двухстенном, 40 литровом стальном реакторе, который был сначал продут азотом, перед добавлением органического растворителя, мономеров, полярного координирующего соединения, соединения инициатора или других компонентов. Следующие компоненты были затем добавлены в следующем порядке: циклогексановый растворитель (20200 грамм); бутадиеновый мономер, стирольный мономер, тетраметилэтилендиамин (TMEDA) и дивинилбензол (DVB), и смесь была нагрета до 40 °C, затем протитрована н-бутиллитием для удаления следов влаги или других примесей. Желаемые инциирующие полимеризацию соединения были добавлены в полимеризационный реактор для инциирования реакции полимеризации. Если применяется смесь инициаторных соединений (к примеру, I3 и н-бутиллитий в Примере 13 в Таблице 2), если не указано иное, они были добавлены в то же время. Полимеризацию проводили в течение по меньшей мере 80 минут, не позволяя температуре полимеризации превышать 60°C до тех пор, пока конверсия мономера будет по меньше мере 97%. После этого, было добавлено 2.3% от общего количества мономеров бутадиена, с последующим добавлением модификатора концевых групп цепи, если не указано иное. Спустя 20 минут полимеризацию останавливали добавлением метанола (2 эквивалента на основе количества инициатора). К полимерному раствору, 2.20 г IRGANOX 1520 было добавлено в качестве стабилизатора. Эту смесь перемешивали в течение 10 минут. Полученный в результате полимерный раствор смешивали с 27.3 мас % TDAE масла (VivaTec 500, Hansen & Rosenthal KG), затем десорбировали паром в течение одного часа для удаления растворителя и других летучих веществ, и сушили в сушильном шкафу при 70°C в течение 30 минут и затем дополнительно в течение от одного до трех дней, при комнатной температуре.
Количество реагентов, полученная в результате полимерная композиция и некоторые ее свойства приведены в Таблице 2 и Таблице3.
Производство Примера 23
Сo-полимеризацию проводили в двухстенном, 10 литровом стальном реакторе, который был сначала продут азотом, перед добавлением органического растворителя, мономеров, полярного координирующего соединения, соединения инициатора или других компонентов. Следующие компоненты были затем добавлены в следующем порядке: циклогексановый растворитель (4600 грамм); бутадиеновый мономер, стирольный мономер, тетраметилэтилендиамин (TMEDA) и смесь была нагрета до 40 °C, затем протитрована н-бутиллитием для удаления следов влаги или других примесей. Инциатор полимеризации I1 был добавлен в полимеризационный реактор для инциирования реакции полимеризации. Полимеризацию проводили в течение 75 минут, не позволяя температуре полимеризации превышать 60 °C. После этого, было добавлено 0,5% от общего количества мономеров бутадиена, с последующим добавлением хлорида олова (IV) в качастве связующего вещества. Спустя 10 мин, было добавлено 1,8% от общего количества бутадиенового мономера, с последующим добавлением модификатора концевых групп цепи. Спустя 20 минут полимеризацию останавливали добавлением метанола (2 эквивалента на основе количества инициатора). После удаления полимерного раствора из реактора, большое количество гелеобразного остатка остается вокруг мешалки. Количество реагентов, полученная в результате полимерная композиция и некоторые ее свойства приведены в Таблице 2 и Таблице3.
Производство Примера 26
Сo-полимеризацию проводили в двухстенном, 5 литровом стальном реакторе, который был сначал продут азотом, перед добавлением органического растворителя, мономеров, полярного координирующего соединения, соединения инициатора или других компонентов. Следующие компоненты были затем добавлены в следующем порядке: циклогексановый растворитель (2260 грамм); бутадиеновый мономер, стирольный мономер, тетраметилэтилендиамин (TMEDA) и дивинилбензол (DVB), и смесь была нагрета до 40 °C, затем протитрована н-бутиллитием для удаления следов влаги или других примесей. Инциатор полимеризации I2 был добавлен в полимеризационный реактор для инциирования реакции полимеризации. Полимеризацию проводили в течение 80 минут, не позволяя температуре полимеризации превышать 60 °C. После этого, было добавлено 2,3% от общего количества мономеров бутадиена, с последующим добавлением модификатора концевых групп цепи, если не указано иное. Спустя 20 минут полимеризацию останавливали добавлением метанола (2 эквивалента на основе количества инициатора). К полимерному раствору, 2,20 г IRGANOX 1520 было добавлено в качестве стабилизатора. Эту смесь перемешивали в течение 10 минут. Полученный в результате полимерный раствор затем десорбировали паром в течение одного часа для удаления растворителя и других летучих веществ, и сушили в сушильном шкафу при 70°C в течение 30 минут и затем дополнительно в течение от одного до трех дней, при комнатной температуре.
Количество реагентов, полученная в результате полимерная композиция и некоторые ее свойства приведены в Таблице 2 и Таблице3.
Производство Примеров 25 и 35
Сo-полимеризацию проводили в двухстенном, 10 литровом стальном реакторе, который был сначала продут азотом, перед добавлением органического растворителя, мономеров, полярного координирующего соединения, соединения инициатора или других компонентов. Следующие компоненты были затем добавлены в следующем порядке: циклогексановый растворитель (4600 грамм); бутадиеновый мономер, стирольный мономер, тетраметилэтилендиамин (TMEDA) и смесь была нагрета до 40 °C, затем протитрована н-бутиллитием для удаления следов влаги или других примесей. Инциирующий полимеризацию н-бутиллитий был добавлен в полимеризационный реактор для инциирования реакции полимеризации. Полимеризацию проводили в течение, по меньшей мере, 80 минут, не позволяя температуре полимеризации превышать 60 °C, до тех пор, пока конверсия мономера будет по меньше мере 97%. После этого, было добавлено 0.5% от общего количества мономеров бутадиена, с последующим добавлением связующего вещества. Смесь перемешивали в течение 20 минут. Впоследствии, было добавлено 1,8% от общего количества модификатора концевых групп цепи, если не указано иное. Спустя 20 минут полимеризацию останавливали добавлением метанола (2 эквивалента на основе количества инициатора). К полимерному раствору, 2,20 г IRGANOX 1520 было добавлено в качестве стабилизатора. Эту смесь перемешивали в течение 10 минут. Полученный в результате полимерный раствор необязательно смешивали с 27,3% мас TDAE масла (VivaTec 500, Hansen & Rosenthal KG), затем десорбировали паром в течение одного часа для удаления растворителя и других летучих веществ, и сушили в сушильном шкафу при 70°C в течение 30 минут и затем дополнительно в течение от одного до трех дней, при комнатной температуре.
Количество реагентов, полученная в результате полимерная композиция и некоторые ее свойства приведены в Таблице 2 и Таблице3.
Производство Примера 39
Сo-полимеризацию проводили в двухстенном, 10 литровом стальном реакторе, который был сначала продут азотом, перед добавлением органического растворителя, мономеров, полярного координирующего соединения, соединения инициатора или других компонентов. Следующие компоненты были затем добавлены в следующем порядке: циклогексановый растворитель (4600 грам); бутадиеновый мономер (83% от общего количества бутадиена), стирольный мономер, тетраметилэтилендиамин (TMEDA) и дивинилбензол (DVB), и смесь была нагрета до 40 °C, затем протитрована н-бутиллитием для удаления следов влаги или других примесей. Инциатор полимеризации I3 и н-бутиллитий были добавлены в полимеризационный реактор для инциирования реакции полимеризации. Полимеризацию проводили в течение 100 минут, не позволяя температуре полимеризации превышать 60 °C. После этого, было добавлено 17% от общего количества мономеров бутадиена и смесь была дополнительно полимеризована в течение 50 мин, с последующим добавлением модификатора концевых групп цепи. Спустя 20 минут полимеризацию останавливали добавлением метанола (2 эквивалента на основе количества инициатора). К полимерному раствору, 2,20 г IRGANOX 1520 было добавлено в качестве стабилизатора. Эту смесь перемешивали в течение 10 минут. Полученный в результате полимерный раствор смешивали с 27.3 мас % TDAE масла (VivaTec 500, Hansen & Rosenthal KG), затем десорбировали паром в течение одного часа для удаления растворителя и других летучих веществ, и сушили в сушильном шкафу при 70°C в течение 30 минут и затем дополнительно в течение от одного до трех дней, при комнатной температуре.
Количество реагентов, полученная в результате полимерная композиция и некоторые ее свойства приведены в Таблице 2 и Таблице 3.
Производство Примера 40
Сo-полимеризацию проводили в двухстенном, 10 литровом стальном реакторе, который был сначала продут азотом, перед добавлением органического растворителя, мономеров, полярного координирующего соединения, соединения инициатора или других компонентов. Следующие компоненты были затем добавлены в следующем порядке: циклогексановый растворитель (4600 грамм); бутадиеновый мономер (83% от общего количества бутадиена), стирольный мономер (83% от общего количества стирола), тетраметилэтилендиамин (TMEDA) и дивинилбензол (DVB), и смесь была нагрета до 40 °C, затем протитрована н-бутиллитием для удаления следов влаги или других примесей. Инциатор полимеризации I3 и н-бутиллитий были добавлены в то же время в полимеризационный реактор для инциирования реакции полимеризации. Полимеризацию проводили в течение 120 минут, не позволяя температуре полимеризации превышать 60 °C. После этого, было добавлено 14,7% от общего количества мономеров бутадиена и 17% от общего количества мономера стирола, и смесь была дополнительно полимеризована в течение 60 мин, с последующим добавлением 2.3% от общего количества мономеров бутадиена и спустя 5 минут после этого добавлено модификатор концевых групп цепи. Спустя 20 минут полимеризацию останавливали добавлением метанола (2 эквивалента на основе количества инициатора). К полимерному раствору, 2,20 г IRGANOX 1520 было добавлено в качестве стабилизатора. Эту смесь перемешивали в течение 10 минут. Полученный в результате полимерный раствор смешивали с 27,3% мас TDAE масла (VivaTec 500, Hansen & Rosenthal KG), затем десорбировали паром в течение одного часа для удаления растворителя и других летучих веществ, и сушили в сушильном шкафу при 70°C в течение 30 минут и затем дополнительно в течение от одного до трех дней, при комнатной температуре.
Количество реагентов, полученная в результате полимерная композиция и некоторые ее свойства приведены в Таблице 2 и Таблице 3.
Использование тире ʺ-ʺ в Таблицах ниже указывает, что компонент не был добавлен. "б.м." указывает, что измерение не было принято, или что соответствующие данные были недоступны. Если не указано иное н-бутиллитий (BuLi) использовался как 20% мас. раствор в циклогексане.
Таблица 2: Количество реагентов, используемых для содания примеров полимерных композиций
(ммоль)
(моль)
(моль)
(E2) 4,513
(E1) 4,553
16,22/0,381
3,325/2,19 a
3,320/2,13b
3,330/0,110
12,04/0,370
2,708/0,081
3,531/0,105
14,36/0,435
16,95/0,321
15,57/0,513
3,336/0,102
3,545/0,102
a BuLi был добавлен сначала в реактор полимеризации и спустя 5 мин I3 был добавлен в реактор, концентрация раствора н-бутиллития была 0,159 моль/кг
b BuLi был добавлен сначала в реактор полимеризации и спустя 10 мин I3 был добавлен в реактор, концентрация раствора н-бутиллития была 0,159 моль/кг
c Концентрация раствора н-бутиллития была 0,179 моль/кг
d Концентрация раствора н-бутиллития была 0,384 моль/кг
Таблица 3: Характеристики первых полимерных композиций
(% площадь)
(% площадь)
(% площадь)
[MU]
[% масс.]
[% масс.]
[% масс.]
(87,6)
(12,4)
(85,8)
(14,2)
(6,7)
(72,1)
(16,5)
(11,4)
(71,4)
(17,7)
(10,9)
(63,7)
(18,1)
(18,2)
(63,3)
(18,2)
(18,5)
(65,1)
(18,0)
(16,9)
(66,3)
(18,9)
(14,8)
(76,9)
(14,1)
(9,0)
(76,8)
(23,2)
(75,7)
(24,3)
(46,9)
(53,1)
(45,2)
(39,5)
(15,3)
(55,6)
(44,4)
(46,3)
(39,0)
(14,7)
(48,7)
(34,5)
(16,8)
(47,7)
(52,3)
(52,0)
(48,0)
(55,0)
(45,0)
(47,4)
(52,6)
(42,3)
(31,4)
(26,3)
(95,1)
(4,9)
(74,8)
(7,7)
(17,5)
(82,3)
(17,7)
(35,9)
(64,1)
(75,1)
(24,9)
(71,0)
(29,0)
(76,3)
(16,3)
(7,4)
(71,3)
(12,0)
(31,7)
(54,3)
(45,7)
(46,2)
(53,8)
(24,0)
(55,9)
(20,1)
(22,6)
(59,1)
(18,3)
(18,9)
(62,6)
(18,5)
a определено с помощью SEC
b содержание винила означает сожержание 1,2-полибутадиеновых звеньев конечного coполимера, и определено с помощью ИК Спектроскопии
c содержание стирола конечного coполимера, и определено с помощью ИК Спектроскопии
d содержание стирола и винила содержание 1,2-полибутадиеновых звеньев конечного coполимера было определено с помощью ЯМР спектроскопии
e данные измерянные из полимерного раствора, который не был желатинизирован
Полимерные композиции
Третьи полимерные композиции были приготовлены смешиванием и соединением компонентов приведенных ниже в Таблице 4 в 370 мл смесителе Бенбери и сшиты в вулканизационном прессе при 200 бар и при 160 °C. Данные процесса сшивания и физические свойства для каждой примерной композиции приведены в Таблице 7 и Таблице 8.
Сшитые полимерные композиции (как перечислено в Таблице 4, Таблице 5 и Таблице 6), которые получены при одинаковых условиях в тот же день, тем же оператором, определены с большой буквы, к примеру, A, B, и т.д. Первая полимерная композиция, содержащаяся в сшитой полимерной композиции показана номером первой полимерной композиции, к примеру, 1, 2, и т.д. В результате, имеются серии сшитых полимерных композиций, такие как 6A, 8A и 11A, которые могут быть непосредственно сравнены друг с другом.
Таблица 4: Рецептура производства третьих полимерных композиций 6A, 8A, 11A, 33D, 34D
(част)a
(раствор стирол бутадиенового coполимера)
Dusantox 6PPDg
2,0
a част=частей на сто частей каучука, на основе суммы веса стирол бутадиенового coполимера и высокого цис 1,4-полибутадиена
b Styron Deutschland GmbH
c Evonic Industries AG
d Бис(триэтоксисилилпропил)дисульфан, эквиваленты серы на молекулу: 2.35
e Cognis GmbH
f Свет & озон защитный воск, Rhein Chemie Rheinau GmbH
g N-(1,3-диметилбутил)-N'-фенил-1,4-бензолдиамин, Duslo, a.s.
h Grillo-Zinkoxid GmbH
i VivaTec 500, Hansen & Rosenthal KG
j Сольвей AG
k N-трет-бутил-2-бензотиазил -сульфенамид; Rhein Chemie Rheinau GmbH
l дифенилгуанидин, Vulkacit D, Lanxess AG
Таблица 5: Рецептура производства третьих полимерных композиций 12C, 13C, 36C, 37C
(раствор стирол бутадиенового coполимера)
или 37
Dusantox 6PPDg
2,0
a част=частей на сто частей каучука, на основе суммы веса стирол бутадиенового coполимера (без содержания масла) и высокого цис 1,4-полибутадиена
b Styron Deutschland GmbH
c Evonic Industries AG
d Бис(триэтоксисилилпропил)дисульфан, серы эквиваленты на молекулу: 2,35
e Cognis GmbH
f Свет & озон защитный воск, Rhein Chemie Rheinau GmbH
g N-(1,3-диметилбутил)-N'-фенил-1,4-бензолдиамин, Duslo, a.s.
h Grillo-Zinkoxid GmbH
i Сольвей AG
j N-трет-бутил-2-бензотиазил -сульфенамид; Rhein Chemie Rheinau GmbH
k дифенилгуанидин, Vulkacit D, Lanxess AG
Таблица 6: Рецептура для производства третьих полимерных композиций 6B, 8B, 11B
(раствор стирол бутадиенового coполимера)
a част=частей на сто частей каучука, на основе суммы веса стирол бутадиенового coполимера
b IRB 8, международная ref. углеродная сажа, Sid Richardson
c Cognis GmbH
d Grillo-Zinkoxid GmbH
e Сольвей AG
f N-трет-бутил-2-бензотиазил -сульфенамид; Rhein Chemie Rheinau GmbH
Таблица 7: Данные процесса сшивания & кремнезем или углеродная сажа, имеющие свойства сшитой полимерной композиции
[MU]
a модификация содержащей SSBR полимерной композиции, где 0=без модифицированных концевых групп полимерные макромолекулы, 1=модификация концевой группы цепи на одной концевой группе полимерной цепи (в соответствии с Формулой 2), 2=модификация концевой группы цепи на обоих концевых группах полимерной цепи (в соответствии с Формулой 1); полимерные композиции изобретения представлены 1/2 или 0/1/2.
Таблица 8: Кремнезем или углеродная сажа, имеющая свойства сшитой полимерной композиции
Таблица 9: Сравнение коэффициентов трения при скольжении для линейных и частично разветвленных первых полимерных композиций
(через DVB)
a модификация содержащей SSBR полимерной композиции, где 0=без модифицированных концевых групп полимерные макромолекулы, 1=модификация концевой группы цепи на одной концевой группе полимерной цепи (в соответствии с Формулой 2), 2=модификация концевой группы цепи на обоих концевых группах полимерной цепи (в соответствии с Формулой 1); полимерные композиции изобретения представлены 1/2.
Было обнаружено, что полимерные композиции изобретения при применении для приготовления сшитых (усиленный кремнеземом или углеродной сажей) полимерных композициях (см., к примеру, 6A в Таблице 7 и Таблице 8) имеют относительно сниженный тангенс δ при 60 °C, относительно сниженное теплонакопление и относительно увеличенную эластичность на отскок при 60°C по сравнению со сшитыми резиновыми композициями, сделанными из немодифицированной полимерной композиции, сделанной не в соответствии с изобретением (см., к примеру, 11A в Таблице 7 и Таблице 8). Неожиданно также было обнаружено, что полимерные композиции изобретения показывают улучшенный баланс переработки при механическом смешивании с наполнителем и другими ингредиентами, если сравнивать с модифицированными полимерными композициями, произведенными не в соответствии с изобретением. Кроме того, эксплуатационные характеристики, в особенности на мокром покрытии, измерянные как тангенс дельта @ 0 °C, прочность на растяжение и удлинение при разрыве сшитых полимерных композиций изобретения также улучшились, в то время как сопротивление качению, измерянное как тангенс дельта @ 60 °C, теплонакопление и DIN стойкость к истиранию сшитых полимерных композиций изобретения являются сопоставимыми по сравнению с модифицированными полимерами не в соответствии с изобретением.
Полимерные композиции изобретения могут быть преобразованы в сшитые полимерные композиции путем первой стадии смешивания (этап смешивания, в котором кремнезем или углеродная сажа и другие ингредиенты добавляются к полимеру) и второй стадии смешивания (этап смешивания, в котором сшивающий агент добавляют к полимерной композиции) в соответствии с Таблицами 4-6, с последующим сшиванием в соответствии с Таблицами 4-6 при 160°C в течение 20 мин как описано в данном документе. Полимерные композиции и сшитые полимерные композиции (как приведено в Таблицах 4-6), приготовленные при одинаковых условиях в тот же день тем же оператором, определены с большой буквы, к примеру, A, B, и т.д. Полимер, который содержится в сшитой полимерной композиции определяетя по номеру полимера, к примеру, 6, 8, и т.д. В результате имеются серии сшитых полимерных композиций, такие как 6A, 8A, 11A, которые могут быть непосредственно сравнены друг с другом.
Полимерные композиции изобретения имеют улучшенную пригодность для обработки и хорошо сбалансированную производительность при гистерезисных потерях, сцеплении с мокрым покрытием, свойства теплонакопления и стойкости к истиранию, при сравнении с полимерными композициями не в соответствии с изобретением, такие как полимерные композиции, содержащие существенным образом только макромолекулы, которые модифицированы в обоих концевых группах полимерной цепи, при этом "существенным образом" означает, что более чем 85% всех полимерных цепей модифицированы в обоих концевых группах цепи (ω,ω'-модифицированный, в соответствии с Формулой 1), котороый не в соответствии с изобретением.
Как показано в Таблице 7 и Таблице 8, значения соединений по Муни несшитых, содержащих наполнители полимерных композиций изобретения уменьшены по сравнению с не-сшитыми полимерными композициями, содержащими существенным образом эластомерные макромолекулы, которые модифицированы на обоих концевых группах полимерной цепи. ʺПрочность на растяжениеʺ, ʺудлинение при разрывеʺ и ʺтангенс δ при 0°Cʺ значения сшитых полимерных композиций, содержащих первые полимерные композиции изобретения увеличены, по сравнению с сшитыми полимерными композициями, сделанными не в соответствии с изобретением. Сниженные значения по Муни несшитых, содержащих наполнители полимерных композиций указывает на снижение вязкости, что в результате приводит к более экономичным технологическим свойствам в процессе механического смешивания полимерной композиции с наполнителями и другими опциональными ингредиентами. Увеличенные значения ʺпрочности на растяжениеʺ и ʺудлинения при разрывеʺ приводят в результате к повышению прочности на разрыв сшитой полимерной композиции. Значения ʺтангенса δ при 0 °Cʺ коррелируют со свойствами сцепления на влажной поверхности, повышенные значения соответствуют повышонному сцеплению с влажным покрытием. Теплонакопление, значения ʺэластичности на отскок при 60 °Cʺ и ʺтангенса δ при 60 °Cʺ сшитых резиновых композиций в соответствии с изобретением не ухудшились или незначительно ухудшились по сравнению со сшитыми полимерными композициями, в которых макромолекулы полимера существенным образом модифицированы в обоих концевых группах цепи. Считается, что снижение "теплонакопления" полимера улучшит прочность получающейся в результате сшитой полимерной композиции и соответствует снижению гистерезисных энергетических потерь сшитой полимерной композиции. Кроме того, снижение "теплонакопления", как правило, сочетается с уменьшенным сопротивлением качению, с повышенной общей эластичностью.
Для демонстрации преимуществ изобретения в деталях приведены некоторые дополнителные примеры.
Полимерная композиция примера 6 (Таблица 2 и Таблица 3) была произведена в результате реакции соединения P2 с 2 эквивалентами н-бутиллития с образованием дианионного инициатора DI2, который впоследствии был использован в полимеризации 1,3-бутадиена и стирола с получением дианионных цепей живого стирол-бутадиенового полимера, которые впоследствии были модифицированы реакцией с модифиактором E1 в соотношении модификатора к литию 1:2 с выходом 1:1 смеси ω- и ω,ω'-модифицированных полимерных цепей. Это было использовано для приготовления содержащей наполнители полимерной композиции изобретения 6A (Таблица 7). Пример 6A имеет значительно меньшее теплонакопление, тангенс δ при 60°C и повышенную эластичность на отскок при 60 °C, прочность на растяжение и удлинение при разрыве, по сравнению с неизобретательской полимерной композицией 11A, содержащей неизобретательский немодифицированный полимер, который был сделан при той же процедуре, но без применения модификатора концевой группы цепи E1 (Таблица 7). Кроме того, содержащие наполнители полимерные композиции содержат полимеры, которые созданы путем a) coполимеризации 1,3-бутадиена и стирола с применением смешанной инициаторной системы, содержащей дианионный DI2 и н-бутиллитий и b) модификации полученных в результате ω,ω'-дианионной и ω-моноанионной coполимерных цепей с модификатором E1 для получения смеси ω- (в соответствии с Формулой 2) и ω,ω'- (в соответствии с Формулой 1) модифицированных макромолекул (пример 37 в Таблице 2 и Таблице 3). В соответствии с примером 37C, несшитая содержащая наполнитель полимерная композиция (в соответствии с Таблицей 5) имеет значительно ниже вязкость соединения по Муни 126.6 по сравнению с 145,2 примером неизобретательской несшитой полимерной композиции 12C (в соответствии с Таблицей 5). Неизобретательская полимерная композиция 12C основана на примере неизобретательского полимера 12 созданного путем полимеризации 1,3-бутадиена и стирола с диинициатором DI2 и модификации полученных в результате дианионных полимерных макромолекул с модификатором E1. Таким образом, неизобретательский полимер 12 содержит существенным образом ω,ω'-модифицированные полимерные цепи (в соответствии с Формулой 1). Баланс сцепления с мокрым покрытием /сопротивления качению примера 37C все еще находится в аналогичом порядке по сравнению с упомянутым 12C, с учетом слегка ухудшенного тангенса δ при 60°C и слегка улучшенных значений тангенса δ при 0°C (0,536 примера 37C против 0,466 примера 12C). Кроме того, более высокие значения прочности на растяжение и значения удлинения при разрыве примера 37C указывают на слегка улучшенные механические свойства для изобретательского примера.
Как правило, принято считать, что линейные полимерные цепи имеют ухудшенную стабильность свойств при хранении из-за повышенной текучести на холоде и повышенной сцепляемости. Обычно линейные полимерные цепи, по меньшей мере, частично находятся в сочетании с галогенидами металлов или алкоксидами (к примеру, SnCl4, Si(OMe)4, SiCl4) для получения звездообразной формы полимеров. Пример полимерной композиции 23 показывает, что применение SnCl4 в качестве связующего вещества приводит к значительному застыванию, которое происходит из-за реакции с полимерными цепями, имеющими две реакционно-способные группы цепи (ω,ω'-анионные концевые группы полимерной цепи). Неожиданно, кроме того, было обнаружено, что полимерные композиции в соответствии с изобретением имеют пониженную сцепляемость, что соответствует отосительно сниженному коэффициенту трения при скольжении. Примеры для коэффициентов трения при скольжении неизобретательских и изобретательских полимерных композиций приведены в Таблице 9 и чертеже. Неизобретательская полимерная композиция 38 имеет относительно высокий коэффициент трения при скольжении 1,93, в то время как изобретательская полимерная композиция 15 имеет относительно низкое значение 1,76. Дальнейшее снижение коэффициента трения при скольжении до 1,60 было обнаружено для изобретательской полимерной композиции 19, которая дополнительно содержит связанные полимерные цепи, по сравнению с полимерными композициями 15 и 38.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНИЦИАТОРЫ ПОЛИМЕРИЗАЦИИ | 2013 |
|
RU2666359C2 |
| ШИНА ДЛЯ КОЛЕС ТРАНСПОРТНЫХ СРЕДСТВ | 2017 |
|
RU2731944C2 |
| ПОЛИМЕРЫ, МОДИФИЦИРОВАННЫЕ АМИНОСИЛАНОМ | 2012 |
|
RU2609166C2 |
| СМЕСЬ ФУНКЦИОНАЛИЗИРОВАННЫХ ПОЛИМЕРОВ ДЛЯ ШИН | 2016 |
|
RU2716689C2 |
| СИЛАНСУЛЬФИДНЫЕ МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2012 |
|
RU2617403C2 |
| МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ СОПОЛИМЕРЫ С НИЗКИМ СОДЕРЖАНИЕМ ВИНИЛОВЫХ СВЯЗЕЙ | 2013 |
|
RU2661220C2 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2558597C2 |
| МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2009 |
|
RU2504555C2 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2012 |
|
RU2599723C2 |
| РЕЗИНОВЫЕ СМЕСИ | 2012 |
|
RU2612148C2 |

Данное изобретение относится к эластомерной полимерной композиции и ее приготовлению. Описана полимерная композиция для получения сшитых резиновых соединений, содержащая модифицированные полимеры согласно следующей Формуле 1 и Формуле 2:
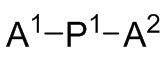 Формула 1
Формула 1
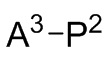 Формула 2,
Формула 2,
где
P1 и P2 каждый независимо представляет собой полимерную цепь, полученную анионной полимеризацией одного или более полимеризуемых мономеров, выбранных из сопряженных диенов и ароматических винильных соединений, причем каждая полимерная цепь P1 и P2 содержит по меньшей мере 40% по массе повторяющихся звеньев, полученных путем полимеризации указанных сопряженных диенов, и при этом по меньшей мере анионная полимеризация полимерной цепи P1 осуществляется в присутствии соединения следующей Формулы 9a:
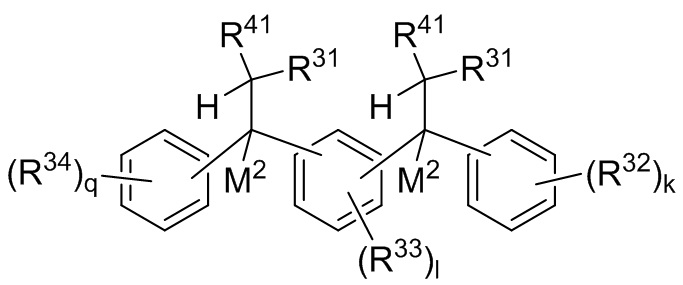 Формула 9a,
Формула 9a,
где каждый R31 независимо выбран из водорода, (C1-C10) алкила, (C6-C12) арила и (C7-C18) аралкила; каждый R32, R33 и R34 независимо выбран из водорода, (C1-C18) алкила и (C1-C18) алкокси; каждый R41 независимо выбран из (C1-C100) алкила и (C2-C100) алкенила, где каждый R41 необязательно замещен от одной до трех (C6-C12) арильными группами и необязательно связан с структурой Формулы 9a через олигомерную цепь, состоящую из до 25 мономерных звеньев, выбранных из сопряженных диенов, особенно 1,3-бутадиена и изопрена, и ароматических винильных соединений, особенно стирола и дивинилбензола; каждый M2 независимо выбран из лития, натрия и калия; и k, l и q представляют собой целые числа, выбранные из 0, 1, 2 и 3;
A1, A2 и A3 независимо выбирают из следующих Формулы 3 до Формулы 8:
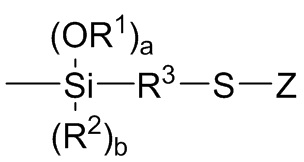 Формула 3,
Формула 3,
где
каждый R1 независимо выбран из (C1-C16) алкила;
каждый R2 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
a и b представляют собой целые числа, независимо выбранные из 0, 1 и 2, причем a+b=2;
R3 независимо выбран из двухвалентного (C1-C16) алкила, двухвалентного (C6-C18) арила, двухвалентного (C7 C18) аралкила и -R4-O-R5-, где R4 и R5 независимо выбраны из двухвалентного (C1-C6) алкила; и
Z независимо выбран из (C1-C16) алкила, (C6-C18) арила, (C7-C18) аралкила, (C=S)-S-R6, где R6 выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила, и M1(R7)c(R8)d, где
M1 представляет собой кремний или олово,
каждый R7 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R8 независимо выбран из -S-R3-Si(OR1)r(R2)s, где R1, R2 и R3 являются такими как определено для Формулы 3 выше, r представляет собой целое число, независимо выбранное из 1, 2 и 3 и s представляет собой целое число, независимо выбранное из 0, 1 и 2, причем r+s=3;
c представляет собой целое число, независимо выбранное из 2 и 3; d представляет собой целое число, независимо выбранное из 0 и 1; и c+d=3;
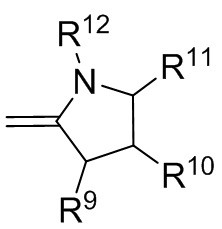 Формула 4
Формула 4
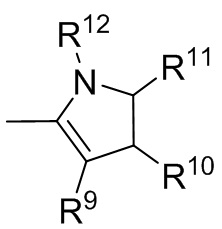 Формула 5,
Формула 5,
где R9, R10, R11 и R12 независимо выбирают из водорода, (C1-C16) алкила, (C6-C16) арила и (C7-C16) аралкила;
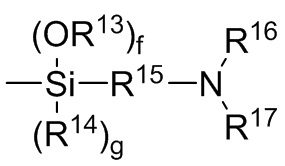 Формула 6
Формула 6
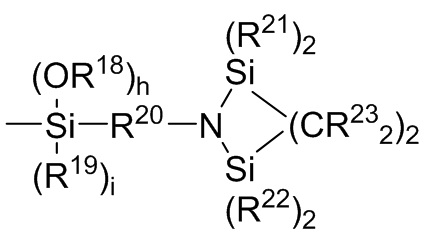 Формула 7,
Формула 7,
где каждый R13, R14, R18 и R19 независимо выбран из (C1-C16) алкила;
R15 и R20 независимо выбраны из двухвалентного (C1-C16) алкила, двухвалентного (C6-C18) арила, двухвалентного (C7-C18) аралкила и -R24-O-R25-, где R24 и R25 независимо выбраны из двухвалентного (C1-C6) алкила;
R16 и R17 независимо выбирают из (C1-C16) алкила и -SiR26R27R28, где R26, R27 и R28 независимо выбирают из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R21 и R22 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R23 независимо выбран из водорода и (C1-C6) алкила;
f, g, h и i представляют собой целые числа, независимо выбранные из 0, 1 и 2; f+g=2; и h+i=2;
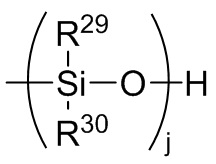 Формула 8,
Формула 8,
где каждый R29 и R30 независимо выбран из (C1-C16) алкила, (C6-C18) арила, (C7-C18) аралкила и винила; и
j представляет собой целое число, выбранное из от 1 до 200; и
при этом количество полимера Формулы 1 составляет от 15 до 85% в молях на основе общего количества полимера Формулы 1 и полимера Формулы 2. Также описаны способ получения полимерной композиции и сшитая полимерная композиция. Технический результат: повышение динамических свойств сшитых резиновых соединений. 4 н. и 17 з.п. ф-лы, 1 ил., 9 табл., 43 пр.
1. Полимерная композиция для получения сшитых резиновых соединений, содержащая модифицированные полимеры согласно следующей Формуле 1 и Формуле 2:
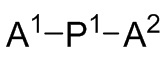 Формула 1
Формула 1
 Формула 2,
Формула 2,
где
P1 и P2 каждый независимо представляет собой полимерную цепь, полученную анионной полимеризацией одного или более полимеризуемых мономеров, выбранных из сопряженных диенов и ароматических винильных соединений, причем каждая полимерная цепь P1 и P2 содержит по меньшей мере 40% по массе повторяющихся звеньев, полученных путем полимеризации указанных сопряженных диенов, и при этом по меньшей мере анионная полимеризация полимерной цепи P1 осуществляется в присутствии соединения следующей Формулы 9a:
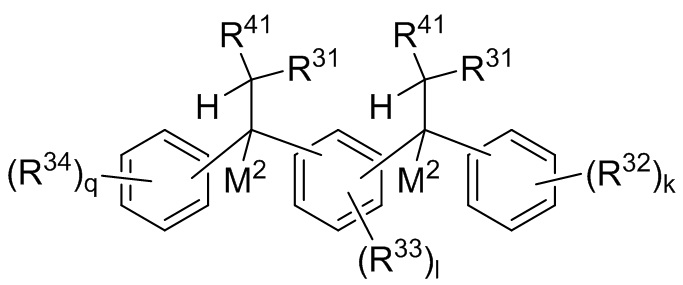 Формула 9a,
Формула 9a,
где каждый R31 независимо выбран из водорода, (C1-C10) алкила, (C6-C12) арила и (C7-C18) аралкила; каждый R32, R33 и R34 независимо выбран из водорода, (C1-C18) алкила и (C1-C18) алкокси; каждый R41 независимо выбран из (C1-C100) алкила и (C2-C100) алкенила, где каждый R41 необязательно замещен от одной до трех (C6-C12) арильными группами и необязательно связан с структурой Формулы 9a через олигомерную цепь, состоящую до 25 мономерных звеньев, выбранных из сопряженных диенов, особенно 1,3-бутадиена и изопрена, и ароматических винильных соединений, особенно стирола и дивинилбензола; каждый M2 независимо выбран из лития, натрия и калия; и k, l и q представляют собой целые числа, выбранные из 0, 1, 2 и 3;
A1, A2 и A3 независимо выбирают из следующих Формулы 3 до Формулы 8:
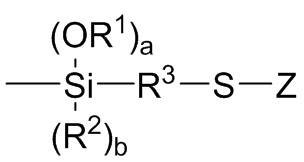 Формула 3,
Формула 3,
где
каждый R1 независимо выбран из (C1-C16) алкила;
каждый R2 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
a и b представляют собой целые числа, независимо выбранные из 0, 1 и 2, причем a+b=2;
R3 независимо выбран из двухвалентного (C1-C16) алкила, двухвалентного (C6-C18) арила, двухвалентного (C7 C18) аралкила и -R4-O-R5-, где R4 и R5 независимо выбраны из двухвалентного (C1-C6) алкила; и
Z независимо выбран из (C1-C16) алкила, (C6-C18) арила, (C7-C18) аралкила, (C=S)-S-R6, где R6 выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила, и M1(R7)c(R8)d, где
M1 представляет собой кремний или олово,
каждый R7 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R8 независимо выбран из -S-R3-Si(OR1)r(R2)s, где R1, R2 и R3 являются такими, как определено для Формулы 3 выше, r представляет собой целое число, независимо выбранное из 1, 2 и 3, и s представляет собой целое число, независимо выбранное из 0, 1 и 2, причем r+s=3;
c представляет собой целое число, независимо выбранное из 2 и 3; d представляет собой целое число, независимо выбранное из 0 и 1; и c+d=3;
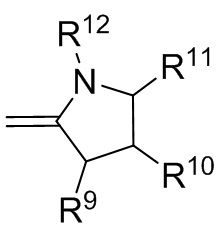 Формула 4
Формула 4
 Формула 5,
Формула 5,
где R9, R10, R11 и R12 независимо выбирают из водорода, (C1-C16) алкила, (C6-C16) арила и (C7-C16) аралкила;
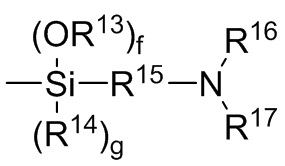 Формула 6
Формула 6
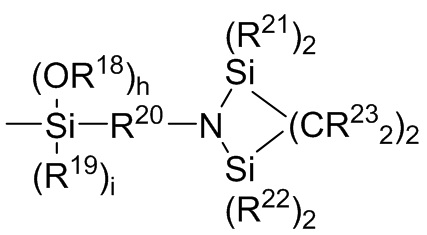 Формула 7,
Формула 7,
где каждый R13, R14, R18 и R19 независимо выбран из (C1-C16) алкила;
R15 и R20 независимо выбраны из двухвалентного (C1-C16) алкила, двухвалентного (C6-C18) арила, двухвалентного (C7-C18) аралкила и -R24-O-R25-, где R24 и R25 независимо выбраны из двухвалентного (C1-C6) алкила;
R16 и R17 независимо выбирают из (C1-C16) алкила и -SiR26R27R28, где R26, R27 и R28 независимо выбирают из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R21 и R22 независимо выбран из (C1-C16) алкила, (C6-C18) арила и (C7-C18) аралкила;
каждый R23 независимо выбран из водорода и (C1-C6) алкила;
f, g, h и i представляют собой целые числа, независимо выбранные из 0, 1 и 2; f+g=2; и h+i=2;
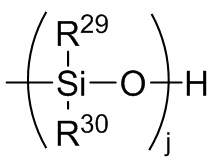 Формула 8,
Формула 8,
где каждый R29 и R30 независимо выбран из (C1-C16) алкила, (C6-C18) арила, (C7-C18) аралкила и винила; и
j представляет собой целое число, выбранное из от 1 до 200; и
при этом количество полимера Формулы 1 составляет от 15 до 85% в молях на основе общего количества полимера Формулы 1 и полимера Формулы 2.
2. Полимерная композиция по п. 1, отличающаяся тем, что содержит один или более дополнительных компонентов, выбранных из (i) компонентов, которые добавлены или образованы в результате процесса полимеризации, применяемого для получения полимеров Формул 1 и 2, и (ii) компонентов, которые остаются после удаления растворителя из процесса полимеризации.
3. Полимерная композиция по п. 2, отличающаяся тем, что один или более дополнительных компонентов выбраны из масла для наполнения, стабилизаторов и дополнительных полимеров, которые не являются полимерами Формулы 1 или Формулы 2.
4. Полимерная композиция по любому из пп. 1-3, отличающаяся тем, что содержит один или более компонентов, которые добавлены после процесса полимеризации, которые выбраны из одного или более наполнителей, одного или более дополнительных полимеров, которые не являются полимерами Формулы 1 или Формулы 2, и одного или более сшивающих агентов.
5. Полимерная композиция по п. 4, отличающаяся тем, что содержит один или более сшивающих агентов.
6. Полимерная композиция по любому из пп. 1-5, отличающаяся тем, что полимеры Формулы 1 и Формулы 2 составляют по меньшей мере 15% по массе присутствующего полимера.
7. Полимерная композиция по п. 6, отличающаяся тем, что полимеры Формулы 1 и Формулы 2 составляют по меньшей мере 30% по массе, более предпочтительно по меньшей мере 45% по массе присутствующего полимера.
8. Полимерная композиция по любому из пп. 1-7, отличающаяся тем, что A1, A2 и A3 независимо выбраны из Формулы 3, как определено в п. 1.
9. Полимерная композиция по любому из пп. 1-8, отличающаяся тем, что в соединении Формулы 9a
M2 представляет собой литий;
R41 выбран из (C1-C10) алкила;
каждый R31 независимо выбран из водорода и (C1-C10) алкила, предпочтительно водорода;
R32 и R34 идентичны и выбраны из водорода и (C1-C18) алкила;
каждый R33 независимо выбран из водорода и (C1-C18) алкила.
10. Полимерная композиция по любому из пп. 1-9, отличающаяся тем, что P1 и P2 каждый независимо выбран из гомополимеров бутадиена и изопрена, случайных или блок сополимеров бутадиена и изопрена, бутадиена и стирола, и изопрена и стирола, и случайных или блок терполимеров бутадиена, изопрена и стирола.
11. Полимерная композиция по любому из пп. 1-10, отличающаяся тем, что ароматические виниловые мономеры составляют от 2 до 55% по массе полимеров P1 и P2.
12. Способ получения полимерной композиции по любому из пп. 1-11, в котором указанный способ включает этапы
i) приведения в контакт смеси инициатора полимеризации, полученной путем приведения в контакт соединения следующей Формулы 9:
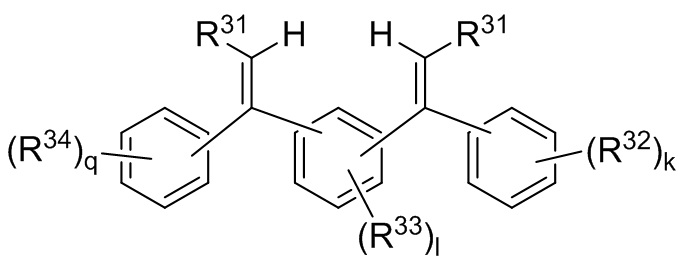 Формула 9,
Формула 9,
где каждый R31 независимо выбран из водорода, (C1-C10) алкила, (C6-C12) арила и (C7-C18) аралкила; каждый R32, R33 и R34 независимо выбран из водорода, (C1-C18) алкила и (C1-C18) алкокси; k, l и q представляют собой целые числа, независимо выбранные из 0, 1, 2 и 3,
с соединением следующей Формулы 10:
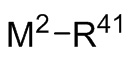 Формула 10,
Формула 10,
где M2 выбран из лития, натрия и калия и R41 выбран из (C1-C100) алкила и (C2-C100) алкенила, где каждый R41 необязательно замещен от одной до трех (C6-C12) арильными группами и необязательно связан с M2 через олигомерную цепь, состоящую из до 25 мономерных звеньев, выбранных из сопряженных диенов, особенно 1,3-бутадиена и изопрена, и ароматических винильных соединений, особенно стирола и дивинилбензола;
с одним или более полимеризуемыми мономерами, выбранными из сопряженных диенов и ароматических винильных соединений для получения полимерных цепей, причем живые полимерные цепи содержат по меньшей мере 40% по массе повторяющихся звеньев, полученных путем полимеризации указанных сопряженных диенов, и
при этом молярное соотношение соединений Формулы 10 к соединениям Формулы 9 находится в диапазоне от 1:1 до 8:1, и
ii) приведения в контакт живых полимерных цепей этапа i) с одним или более агентами, модифицирующими концевую группу цепи, выбранными из соединений следующей Формулы 11 до Формула 15:
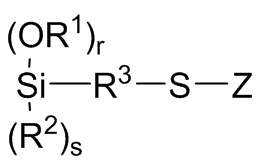 Формула 11,
Формула 11,
где R1, R2, R3, Z, r и s являются такими, как определено для Формулы 3 в п. 1;
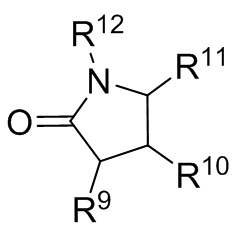 Формула 12,
Формула 12,
где R9, R10, R11 и R12 являются такими, как определено для Формулы 4 в п. 1;
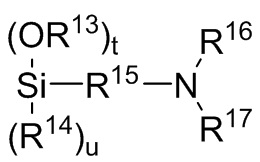 Формула 13,
Формула 13,
где R13, R14, R15, R16 и R17 являются такими, как определено для Формулы 6 в п. 1; t представляет собой целое число, выбранное из 1, 2 и 3; u представляет собой целое число, выбранное из 0, 1 и 2; и t+u=3;
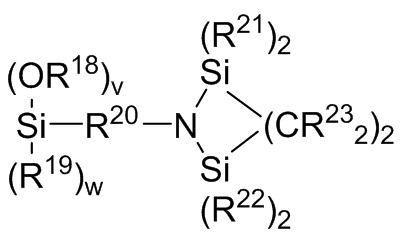 Формула 14,
Формула 14,
где R18, R19, R20, R21, R22 и R23 являются такими, как определено для Формулы 7 в п. 1; v представляет собой целое число, выбранное из 1, 2 и 3; w представляет собой целое число, выбранное из 0, 1 и 2; и v+w=3;
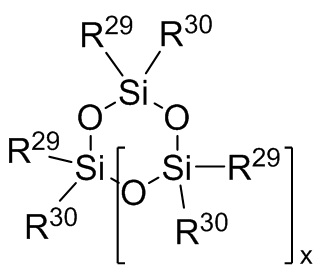 Формула 15,
Формула 15,
где каждый R29 и R30 является таким, как определено для Формулы 8 в п. 1; и x представляет собой целое число, выбранное из от 1 до 6; и
где агенты, модифицирующие концевую группу цепи Формулы 11 до Формулы 15, применяются в общем количестве
(i) от 0,15 до 0,85 молей на моль соединений Формулы 10 в случае, если молярное соотношение соединений Формулы 10 к соединениям Формулы 9 находится в диапазоне от 1:1 до 2,1:1, и
(ii) от 0,5 до 3 молей на моль соединений Формулы 10 в случае, если молярное соотношение соединений Формулы 10 к соединениям Формулы 9 находится в диапазоне от более чем от 2,1:1 до 8:1.
13. Способ по п. 12, отличающийся тем, что в соединении Формулы 9
каждый R31 независимо выбран из водорода и (C1-C10) алкила, предпочтительно водорода;
R32 и R34 идентичны и выбраны из водорода и (C1-C18) алкила;
каждый R33 независимо выбран из водорода и (C1-C18) алкила; и
в соединении Формулы 10
M2 представляет собой литий; и
R41 выбран из (C1-C10) алкила.
14. Способ по п. 12 или 13, отличающийся тем, что молярное соотношение между соединением Формулы 10 и соединением Формулы 9 находится в диапазоне от 1,5:1 до 8:1, предпочтительно от 2:1 до 6:1.
15. Способ по пп. 12-14, отличающийся тем, что молярное соотношение соединения Формулы 10 к соединению Формулы 9 находится в диапазоне от более чем 2,1:1 до 7:1, предпочтительно от 2,1:1 до 6:1, более предпочтительно от 2,5:1 до 3,5:1, даже более предпочтительно приблизительно 3:1.
16. Способ по пп. 12-15, отличающийся тем, что один или более агентов, модифицирующих концевую группу цепи, выбраны из соединений Формулы 11, как определено в п. 11.
17. Способ по пп. 12-16, отличающийся тем, что добавляют один или более агентов, выбранных из рандомизирующего агента и связующего вещества.
18. Способ по п. 17, отличающийся тем, что связующее вещество представляет собой дивинилбензол.
19. Сшитая полимерная композиция для применения в производстве шин и других промышленных продуктов, полученная путем сшивания полимерной композиции, как определено в п. 5.
20. Изделие, содержащее по меньшей мере один компонент, образованный из сшитой полимерной композиции, как определено в п. 19.
21. Изделие по п. 20, отличающееся тем, что представляет собой шину, протектор шины, боковую стенку шины, автомобильную часть, компонент обуви, мяч для гольфа, пояс, уплотнитель, прокладку или шланг.
| МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2009 |
|
RU2504555C2 |
| US 5521255 A, 28.05.1996 | |||
| JP 2009256566 A, 5.11.2009 | |||
| US 4172100 A, 23.10.1979. | |||

