ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение в целом относится к модифицированным эластомерным полимерам, характеризующимся низким содержанием виниловых связей, их применению при получении композиций на основе эластомерного полимера, а также вулканизированным изделиям, полученным из композиций на основе эластомерного полимера. Конкретно настоящее изобретение относится к модификации "живых" анионных эластомерных полимеров, характеризующихся низким содержанием включенного 1,2-бутадиена, также называемым в данном документе "1,2-связями" или "виниловыми связями", с помощью модифицирующего средства на основе силана-сульфида или с помощью последовательной комбинации по меньшей мере двух модифицирующих средств на основе силана с образованием модифицированного эластомерного полимера. Модифицированные эластомерные полимеры являются реакционноспособными по отношению к наполнителям или другим компонентам, присутствующим в композиции на основе эластомерного полимера. Соответствующие композиции на основе эластомерного полимера могут применяться для получения вулканизированных и, таким образом, сшитых эластомерных композиций, характеризующихся относительно низкими гистерезисными потерями. Такие вулканизированные композиции на основе эластомерного полимера применимы во многих изделиях, в том числе протекторах шины с низкой устойчивостью к качению в комбинации с хорошими балансировочными или другими необходимыми физическими и химическими свойствами, например, свойствами устойчивости к проскальзыванию на мокрой дороге, устойчивостью к истиранию, пределом прочности при растяжении и пригодностью для обработки.
УРОВЕНЬ ТЕХНИКИ
Повышение цен на нефть и национальное законодательство требует уменьшения автотранспортных выбросов углекислого газа и, таким образом, заставляет производителей шин и каучука получать "топливосберегающие" шины. Один общий подход получения топливосберегающих шин заключается в получении составов на основе каучука, которые характеризуются сниженными гистерезисными потерями.
Основной источник гистерезиса в вулканизированных эластомерных полимерах связан со свободными концевыми группами полимерной цепи, то есть той частью цепи эластомерного полимера, которая находится между последней поперечной связью и концом полимерной цепи. Данный свободный конец полимера не участвует в каком-либо процессе эффективного эластичного восстановления. Как результат, любая энергия, передаваемая к данной части полимера, теряется. Рассеянная энергия приводит к ярко выраженному гистерезису при динамической деформации.
Другой источник гистерезиса в вулканизированных эластомерных полимерах связан с недостаточным распределением частиц наполнителя в вулканизированной композиции на основе эластомерного полимера. Гистерезисные потери сшитой композиции на основе эластомерного полимера связаны с ее величиной тангенса δ при 60°C (см. ISO 4664-1:2005; Rubber, Vulcanized or thermoplastic; Determination of dynamic properties - part 1: General guidance). В общем, вулканизированные композиции на основе эластомерного полимера, характеризующиеся относительно небольшим тангенсом δ при 60°C, являются предпочтительными для достижения уменьшенных гистерезисных потерь. В конечном продукте, представляющем собой шину, они трансформируются в более низкую устойчивость к качению и улучшенную эффективность использования топлива.
Один в целом приемлемый подход для уменьшения гистерезисных потерь заключается в уменьшении числа свободных концевых групп цепи эластомерных полимеров. В литературе были описаны различные технологии, в том числе применение связующих средств, таких как четыреххлористое олово, с помощью которого можно функционализировать концевую группу полимерной цепи и привести в контакт, например, с наполнителем или ненасыщенными частями полимера (см. патенты США №№3281383, 3244664, 3692874, 3978103, 4048206, 4474908, 6777569, 3078254 и 4616069 и US №005/0124740). Применение связующих средств в качестве реагирующих веществ для живых полимеров обычно приводит к образованию полимерных смесей, содержащих одну фракцию линейных или несвязанных полимеров и одну или более фракций, содержащих более двух полимерных плеч в точке связывания. Тетрагалогенид кремния представляет собой типичный пример связующих средств на основе галогенида кремния.
В WO №007/047943 описывается применение модификатора на основе силана-сульфида для получения эластомерного полимера с модифицированными концевыми группами цепи, который применяют в вулканизированных композициях на основе эластомерного полимера, таких как протектор шины. В данном документе описывается реакция между силан-сульфидным соединением и анионно-инициируемыми живущими полимерами с получением полимеров с модифицированными концевыми группами цепи, которые затем смешивают и приводят в контакт с наполнителями, вулканизирующими средствами, ускорителями или нефтяными мягчителями с получением вулканизированной композиции на основе эластомерного полимера, характеризующейся низкими гистерезисными потерями. Для дополнительной регуляции молекулярного веса полимера и свойств полимера связующее средство ("связывающее средство") может применяться в способе в качестве необязательного компонента. Затем добавляют модификатор перед, после или во время добавления связующего средства и проводят реакцию модификации предпочтительно после добавления связующего средства.
В WO №009/148932 описывается комбинированное применение двух соединений-модификаторов, которые последовательно приводят в контакт живущим анионным эластомерным полимером с получением модифицированного эластомерного полимера с целью снижения связанных с шиной устойчивости к качению и связанного расхода топлива.
Несмотря на улучшение в отношении гистерезисных потерь с помощью решения, предусмотренного в WO №007/047943 и WO №009/148932, получают вулканизированные изделия с устойчивостью к проскальзыванию на мокрой дороге ("характеристикой сцепления шины с мокрым дорожным покрытием"), которая является недостаточной для некоторых применений.
Полимеры с низким содержанием виниловых связей, характеризующиеся низкой долей 1,2-полибутадиена в полибутадиеновой фракции бутадиенового сополимера или в полибутадиеновом гомополимере, применяют в соединениях для протекторов шины и боковой стенки шины вследствие их улучшенной устойчивости к истиранию. Напротив, их характеристика сцепления шины с мокрым дорожным покрытием является неудовлетворительной. Наилучшими полимерами на рынке с точки зрения их устойчивости к истиранию являются высокомолекулярные цис-полибутадиены, полученные с применением катализатора Циглера-Натта на основе неодима и характеризующиеся содержанием виниловых связей <2% и температурой стеклования (Tg) порядка около -105°C. Как результат, полимеры с низким содержанием виниловых связей преимущественно применялись в качестве добавок в полимерных смесях для улучшения устойчивости к истиранию.
Сцепление шины с мокрым дорожным покрытием и устойчивость к качению вулканизированного полимера также сильно зависит от его Tg, который снова находится под влиянием содержания виниловых связей в вулканизированном полимере, то есть низкое содержание виниловых связей связано с низким Tg. Для достижения полезных свойств вулканизированной композиции на основе эластомерного полимера для применения в протекторах шины, существует необходимость в сбалансировании устойчивости к истиранию и сцепления шины с мокрым дорожным покрытием при одновременном снижении как устойчивости к качению, так и тепловыделения насколько это возможно.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения неожиданно обнаружили, что соответствующее сцепление шины с мокрым дорожным покрытием в комбинации с улучшенной устойчивостью к истиранию может достигаться в вулканизированном эластомерном полимере посредством применения живого анионного эластомерного полимера, характеризующегося низким содержанием виниловых связей, и подвергания его определенной химической модификации. При применении в шине вулканизированный модифицированный эластомерный полимер имеет улучшенную (сниженную) устойчивость к качению при поддержании свойств сцепления шины с мокрым дорожным покрытием, известным для модифицированных эластомерных полимеров.
В первом аспекте в настоящем изобретении предлагается модифицированный эластомерный полимер на основе живого анионного эластомерного полимера, содержащий более чем 70 и менее чем 99 мас.% бутадиеновых элементарных звеньев и более чем 1 и менее чем 30 мас. мас.% стирольных элементарных звеньев и характеризующийся содержанием виниловых связей от 2 до менее чем 10% исходя из полибутадиеновой фракции живого анионного эластомерного полимера. Собственно говоря, с точки зрения его значений содержания как бутадиена, так и стирола живой анионный эластомерный полимер представляет собой "сополимер", но его называют в данном документе как "полимер" для удобства. Модифицированный эластомерный полимер получают посредством приведения в контакт живого анионного эластомерного полимера с одним или более модифицирующих соединений.
Во втором аспекте в настоящем изобретении дополнительно предлагается композиция на основе эластомерного полимера, содержащая модифицированный эластомерный полимер из первого аспекта настоящего изобретения и один или более дополнительных компонентов, выбранных из компонентов, которые добавляют в или образуют в результате способа полимеризации, применяемого для получения модифицированного эластомерного полимера из первого аспекта настоящего изобретения, и компонентов, которые остаются после удаления растворителя из способа полимеризации. Компоненты, которые добавляют в способ полимеризации, включают, в частности, масла (масла для наполнения), наполнители, стабилизаторы и дополнительные эластомерные полимеры.
В одном варианте реализации первого аспекта настоящего изобретения модифицированный эластомерный полимер получают посредством приведения в контакт живого анионного эластомерного полимера с модифицирующим соединением формулы (I)
где R независимо представляет собой C1-C16алкил или бензил; R'' представляет собой C1-C4алкил; R' выбран из C6-C18арила, C7-C50алкиларила, C1-C50алкила и C2-C50диалкилэфира (то есть -алкил-O-алкил-), где каждая группа необязательно замещена одной или более группами, выбранными из C1-C4алкила, C1-C4алкокси, C6-C12арила, C7-C16алкиларила, ди(C1-C7гидрокарбил)амино, бис(три(C1-C12алкил)силил)амино, трис(C1-C7гидрокарбил)силила и C1-C12тиоалкила; x представляет собой целое число, выбранное из 1, 2 и 3; y представляет собой целое число, выбранное из 0, 1 и 2; и x + y = 3.
В настоящем изобретении дополнительно предлагается способ получения модифицированного эластомерного полимера, включающий этап приведения в контакт живого анионного эластомерного полимера с модифицирующим соединением формулы (I).
В другом варианте реализации первого аспекта настоящего изобретения модифицированный эластомерный полимер получают посредством приведения в контакт живого анионного эластомерного полимера по меньшей мере с одним из модифицирующих соединений следующих формул (1) и (2) и по меньшей мере одним из модифицирующих соединений формул (3), (4), (5) и (6)
где R2, R3, R10, R11, R12, R14, R16, R17 и R18 независимо выбраны из C1-C16алкила и бензила, где алкильные группы для R10, R11 и R12, а также для R16, R17 и R18 могут быть связаны друг с другом таким образом, что образуют кольцо, содержащее два атома Si и N; R1 и R13 независимо выбраны из C1-C4алкила; R4, R9 и R15 независимо выбраны из C6-C18арила, C7-C50алкиларила, C1-C50алкила и C2-C50диалкилэфира (то есть -алкил-O-алкил-), где каждая группа необязательно замещена одной или более группами, выбранными из C1-C4алкила, C1-C4алкокси, C6-C12арила, C7-C16алкиларила, ди(C1-C7гидрокарбил)амино, бис(три(C1-C12алкил)силил)амино, трис(C1-C7гидрокарбил)силила и C1-C12тиоалкила; R5, R6 и R7 независимо выбраны из водорода, C1-C16алкила и C6-C12арила; R8 выбран из C1-C16алкила и C6-C12арила; и R19, R20 и R21 независимо выбраны из водорода и C1-C16алкила; каждый из x и p представляет собой целое число, выбранное из 1 и 2; каждый из y и q представляет собой целое число, выбранное из 1 и 2; x + y = 3; и p + q = 3.
В настоящем изобретении дополнительно предлагается способ получения модифицированного эластомерного полимера, включающий этапы (i) приведения в контакт живого анионного эластомерного полимера по меньшей мере с одним из модифицирующих соединений формул (1) и (2) с получением модифицированного полимера и (ii) приведения в контакт модифицированного полимера и по меньшей мере одного из модифицирующих соединений формул (3), (4), (5) и (6).
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Определения
Применяемый в данном документе включенный 1,2-бутадиен (или "виниловые связи", или "1,2-связи") относится к 1,3-бутадиеновым мономерам, включенным в полимерную цепь через первый и второй атом углерода мономерной молекулы. Содержание включенного 1,2-бутадиена (или содержание виниловых связей) выражается в процентах (или массовых процентах) относительно общего количества бутадиена в полимере.
Применяемый в данном документе термин "живой анионный эластомерный полимер" относится к полимеру, который содержит по меньшей мере одну реакционноспособную или "живую" анионную концевую группу.
Алкильные группы, как определяется в данном документе, либо как таковые, либо в связи с другими группами, такими как алкиларильная или алкокси, включают как алкильные группы с неразветвленной цепью, такие как метил (Me), этил (Et), н-пропил (Pr), н-бутил (Bu), н-пентил, н-гексил и т.д., так и разветвленные алкильные группы, такие как изопропил, трет-бутил и т.д., а также циклические алкильные группы, такие как циклогексил.
Алкоксигруппы, как определяется в данном документе, включают метокси (MeO), этокси (EtO), пропокси (PrO), бутокси (BuO), изопропокси, изобутокси, пентокси и т. д.
Арильные группы, как определяется в данном документе, включают фенил, бифенил и другие бензоидные соединения. Арильные группы предпочтительно содержат только одно ароматическое кольцо и наиболее предпочтительно содержат C6ароматическое кольцо.
Алкиларильные группы, как определяется в данном документе, относятся к комбинации одной или более арильных групп, связанных с одной или более алкильных групп, например, в форме алкил-арил, арил-алкил, алкил-арил-алкил и арил-алкил-арил. Алкиларильные группы предпочтительно содержат только одно ароматическое кольцо и наиболее предпочтительно содержат C6ароматическое кольцо.
Живой анионный эластомерный полимер
Живой анионный эластомерный полимер, применяемый в настоящем изобретении, представляет собой сополимер бутадиена и стирола, а также необязательно одного или более из дополнительных сомономеров. Количество бутадиена в живущем анионном эластомерном полимере составляет более чем 70 и менее чем 99 мас.% исходя из общей массы полимера. Количество бутадиена может составлять по меньшей мере 75, по меньшей мере 85 или по меньшей мере 90 мас.%. Количество стирола в живущем анионном эластомерном полимере составляет более чем 1 и менее чем 30 мас.% исходя из общей массы полимера. Количество стирола предпочтительно составляет по меньшей мере 2, более предпочтительно по меньшей мере 4, еще более предпочтительно по меньшей мере 6 мас.%. В одном варианте реализации изобретения живой анионный эластомерный полимер представляет собой сополимер только бутадиена и стирола с более чем 1 и менее чем 30 мас.% стирола, предпочтительно от 5 до 25 мас.%, более предпочтительно от 8 до 20 мас.% стирола. Стирол-бутадиеновый сополимер включает частично гидрированный стирол-бутадиеновый сополимер.
Сомономеры, отличные от бутадиена и стирола, которые могут применяться при получении живого анионного эластомерного полимера включают ненасыщенные мономеры, такие как α-олефины, внутренние олефины, диены с сопряженными двойными связями, диены с несопряженными двойными связями, ароматические виниловые мономеры и акриловые мономеры. Подходящие α-олефины включают C2-20-α-олефины, такие как этилен, пропилен и 1-бутен. Подходящие диены с сопряженными двойными связями включают 2-метил-1,3-бутадиен (изопрен), 2,3-диметил-1,3-бутадиен, 1,3-пентадиен, 2,4-гексадиен, 1,3-гексадиен, 1,3-гептадиен, 1,3-октадиен, 2-метил-2,4-пентадиен, циклопентадиен, 2,4-гексадиен и 1,3-циклооктадиен. Подходящие диены с несопряженными двойными связями включают норборнадиен, этилиденнорборнен, 1,4-гексадиен, 1,5-гексадиен, 1,7-октадиен и 4-винилциклогексен. Подходящие ароматические виниловые мономеры включают C1-4алкил-замещенный стирол, такой как 2-метилстирол, 3-метилстирол, α-метилстирол, 2,4-диметилстирол, 2,4,6-триметилстирол и стильбен, 2,4-диизопропилстирол, 4-трет-бутилстирол, винилбензилдиметиламин, (4-винилбензил)диметиламиноэтиловый эфир, N,N-диметиламиноэтилстирол, трет-бутоксистирол, винилпиридин, 1,2-дивинилбензол, 1,3-дивинилбензол и 1,4-дивинилбензол. Подходящие акриловые мономеры включают акрилонитрил, акрилаты, такие как акриловая кислота, метилакрилат, этилакрилат, пропилакрилат и бутилакрилат, и метакрилаты, такие как метилметакрилат, этилметакрилат, пропилметакрилат и бутилметакрилат.
Живой анионный эластомерный полимер может представлять собой статистический или блок-сополимер.
В соответствии с настоящим изобретением содержание включенного 1,2-бутадиена в живущем анионном эластомерном полимере составляет от 2 до менее 10% исходя из полибутадиеновой фракции полимера (исходя из либо массы, либо молей повторяющихся элементарных звеньев бутадиена). Включенный 1,2-бутадиен также называют в данном описании "1,2-связями" или "виниловыми связями". В предпочтительном варианте реализации изобретения содержание виниловых связей в полибутадиеновой фракции составляет от 4 до 9%.
Сополимеризация мономеров может быть достигнута с помощью известных из уровня техники реакций полимеризации анионного живущего типа. Температура полимеризация, как правило, составляет от -50 до 250°C, предпочтительно от 0 до 120°C при атмосферном давлении, давлении ниже атмосферного или повышенном давлении. Предпочтительно полимеризацию проводят при абсолютном давлении от 0,01 до 500 МПа, более предпочтительно от 0,01 до 10 МПа и наиболее предпочтительно от 0,1 до 2 МПа. В целом полимеризацию проводят партиями, непрерывно или полунепрерывно. Способ полимеризации может осуществляться в форме полимеризации в газовой фазе (например, в псевдоожиженном слое или в реакторе с перемешиванием в слое), полимеризации в растворе, где образованный полимер является практически растворимым в реакционной смеси, полимеризации в суспензии/взвеси, где образованный полимер является практически нерастворимым в реакционной среде, или полимеризации в объеме, при которой избыток мономера, подлежащий полимеризации, используют в качестве реакционной среды.
Полимеризация, как правило, инициируется с помощью анионного инициатора, такого как, но не ограничиваясь этим, органометаллическое соединение, содержащее по меньшей мере один атом лития, натрия или калия и содержащее от 1 до около 20 атомов углерода. Органометаллическое соединение предпочтительно содержит по меньшей мере один атом лития и представляет собой, например, этиллитий, пропиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий, фениллитий, гексиллитий, 1,4-дилитио-н-бутан или 1,3-ди(2-литио-2-гексил)бензол, предпочтительно н-бутиллитий или втор-бутиллитий. Органометаллический инициатор, в частности, органолитиевый инициатор, может применяться сам по себе или в комбинации двух или более из них. Общее количество органометаллического инициатора(-ов), в частности, органолитиевого инициатора(-ов), будет регулироваться в зависимости от мономера и целевого молекулярного веса. Его общее количество, как правило, составляет от 0,05 до 5 ммоль, предпочтительно от 0,2 до 3 ммоль на 100 грамм мономера.
Полярные координационные соединения традиционно добавляют в полимеризационную смесь в качестве рандомизирующего компонента, таким образом, обеспечивая регуляцию содержания виниловых связей в сопряженной диолефиновой части полимера и, например, оказывая влияние на распределение ароматических виниловых мономеров в сополимере бутадиена/изопрена и ароматического винилового соединения. Для уменьшения содержания виниловых связей в сопряженной диолефиновой части полимера подходящим является повышение температуры полимеризации и/или уменьшение или минимизация количества соединений, выполняющих функцию полярного координационного соединения или "рандомизирующего компонента", или полное исключение их применения. Полярные координационные соединения включают, например, эфирные соединения, такие как диэтиловый эфир, ди-н-бутиловый эфир, диэтиловый эфир этиленгликоля, дибутиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диметиловый эфир пропиленгликоля, диэтиловый эфир пропиленгликоля, дибутиловый эфир пропиленгликоля, алкилтетрагидрофуриловые эфиры, такие как, метилтетрагидрофуриловый эфир, этилтетрагидрофуриловый эфир, пропилтетрагидрофуриловый эфир, бутилтетрагидрофуриловый эфир, гексилтетрагидрофуриловый эфир, октилтетрагидрофуриловый эфир, тетрагидрофуран, 2,2-бис(тетрагидрофурфурил)пропан, бис(тетрагидрофурфурил)формаль, метиловый эфир тетрагидрофурфурилового спирта, этиловый эфир тетрагидрофурфурилового спирта, бутиловый эфир тетрагидрофурфурилового спирта, α-метокситетрагидрофуран, диметоксибензол и диметоксиэтан, и соединения третичного амина, такие как бутиловый эфир триэтиламина, пиридин, N,N,N',N'-тетраметилэтилендиамин, дипиперидиноэтан, метиловый эфир N,N-диэтилэтаноламина, этиловый эфир N,N-диэтилэтаноламина и N,N-диэтилэтаноламин. Другие полярные координационные соединения включают олигомерный оксоланилалкан, описанный в патентах США №№ 6790921 и 6664328. Полярное координационное соединение(-я) будет, как правило, добавляться при общем молярном отношении относительно соединения-инициатора не более 10:1 (включая случаи полностью неполярного координационного соединения), но, как правило, от 0,1: 1 до 8:1, предпочтительно от 0,25:1 до около 6:1 и более предпочтительно от 0,5: 1 до 4: 1.
Полимеризация необязательно может предусматривать ускорители для повышения реакционной способности инициатора со статистическим упорядочением ароматических виниловых соединений, введенных в полимер, или с получением одной цепи ароматических виниловых соединений, таким образом, оказывая влияние на распределение ароматических виниловых мономеров в живущем анионном эластомерном сополимере. Примеры ускорителей включают алкоксиды натрия или феноксиды натрия и алкоксиды калия или феноксиды калия, предпочтительно алкоксиды калия или феноксиды калия, такие как изопропоксид калия, т-бутоксид калия, т-амилоксид калия, н-гептилоксид калия, бензилоксид калия, феноксид калия; калиевые соли карбоновых кислот, таких как изовалериановая кислота, каприловая кислота, лауриновая кислота, пальмитиновая кислота, стеариновая кислота, олеиновая кислота, линоленовая кислота, бензойная кислота, фталевая кислота и 2-этилгексановая кислота; калиевые соли органических сульфоновых кислот, таких как додецилбензолсульфоновая кислота, тетрадецилбензолсульфоновая кислота, гексадецилбензолсульфоновая кислота и октадецилбензолсульфоновая кислота; и калиевые соли органических фосфористых кислот, таких как диэтилфосфит, диизопропилфосфит, дифенилфосфит, дибутилфосфит и дилаурилфосфит. Такие соединения-ускорители могут добавляться в общем количестве от 0,005 до 0,5 моль на 1,0 грамм-эквивалента атома литиевого инициатора. Если добавляется менее 0,005 моль, достаточный эффект, как правило, не достигается. С другой стороны, если количество соединения-ускорителя составляет более около 0,5 моль, производительность и эффективность реакции модификации концевой группы цепи может значительно снижаться.
Соединение, представляющее собой алкоксид щелочного металла, может также добавляться вместе с инициатором полимеризации для повышения полимеризационной реакционной способности. Соединение, представляющее собой алкоксид щелочного металла, может быть получено посредством приведения в контакт спирта с органическим соединением, содержащим щелочной металл. Приведение в контакт можно проводить в углеводородном растворителе в присутствии мономеров, таких как сопряженные диолефиновые мономеры и ароматические виниловые мономеры, перед сополимеризацией мономеров. Соединения, представляющее собой алкоксид щелочного металла, могут быть представлены алкоксидами щелочных металлов на основе тетрагидрофурфурилового спирта, N,N-диметилэтаноламина, N,N-диэтилэтаноламина, 1-пиперазинэтаноламина и т. п. Органическое соединение щелочного металла, предпочтительно органолитиевое соединение, такое как этиллитий, пропиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий, гексиллитий и смеси двух или более из них, может применяться в качестве реагирующего вещества вместе со спиртовым соединением для получения алкоксида щелочного металла. н-Бутиллитий и втор-бутиллитий являются предпочтительными органолитиевыми соединениями. Молярное отношение спиртового соединения к органолитиевому соединению должно составлять от 1:0,7 до 1:5,0, предпочтительно от 1:0,8 до 1:2,0 и более предпочтительно от 1:0,9 до 1: 1,2. Если молярное отношение органолитиевого соединения к спиртовому соединению составляет более 5,0, эффект в отношении улучшения предела прочности при растяжении, устойчивости к истиранию и в отношении гистерезиса может быть нарушен. Напротив, молярное отношение органолитиевого соединения к спиртовому соединению, составляющее менее 0,8, может снижать скорость полимеризации и значительно снижать производительность, таким образом, приводя к низкой эффективности модифицированной реакции сочетания, а также реакции модификации концевых групп цепи.
Для дополнительной регуляции молекулярного веса полимера и свойств полимера может применяться связующее средство ("связывающее средство"). Например, в реакцию полимеризации может добавляться галогенид олова, такой как тетрагалогенид олова, тригалогенид олова и дигалогенид олова, галогенид кремния, такой как тетрагалогенид кремния, тригалогенид кремния и дигалогенид кремния, алкоксид олова, алкоксид кремния или смесь двух или более из них. Связующие средства включают четыреххлористое олово, тетрабромид олова, тетрафторид олова, тетрайодид олова, тетрахлорид кремния, тетрабромид кремния, тетрафторид кремния и тетрайодид кремния. Полимеры, связанные с тетрагалогенидами олова или кремния, содержат максимум четыре плеча (или четыре связанные полимерные цепи), при этом тригалогениды олова и кремния обеспечивают максимум три плеча, и дигалогениды олова и кремния обеспечивают максимум два плеча связанного полимера. Связующие средства также включают гексагалогендисиланы и гексагалогендисилоксаны, приводящие в результате к связанному полимеру максимум с шестью плечами. Конкретные связующие средства, представляющие собой галогенид олова и галогенид кремния, включают SnCl4, R3SnCl, R2SnCl2, R-SnCl3, SiCl4, R3SiCl, R2SiCl2, R-SiCl3, Cl3Si-SiCl3, Cl3Si-O-SiCl3, Cl3Sn-SnCl3 и Cl3Sn-O-SnCl3, где R представляет собой C1-C18алкил, C7-C18алкиларил, C7-C18арилалкил и C6-C18арил. Конкретные связующие средства, представляющие собой алкоксид олова и алкоксид кремния, включают Sn(OMe)4, Si(OMe)4, Sn(OEt)4 и Si(OEt)4. Наиболее предпочтительные связующие средства представляют собой SnCl4, SiCl4, Sn(OMe)4 и Si(OMe)4.
Комбинация олово-содержащего связующего соединения и кремний-содержащего связующего соединения, как определено выше, необязательно может применяться для связывания полимера, который может приводить в результате улучшенным свойствам в отношении шинных каучуков, таких как уменьшенные гистерезисные потери, особенно для полимерных композиций протектора шины, которые содержат как диоксид кремния, так и углеродную сажу. В таких случаях молярное отношение олово-содержащего связующего соединения к кремний-содержащему связующему соединению обычно будет находиться в диапазоне от 20:80 до 95:5, чаще от 40:60 до 90:10 и предпочтительно от 60:40 до 85:15.
Общее количество применяемых связующих средств будет влиять на вязкость по Муни связанного полимера и, как правило, будет находиться в диапазоне от 0,01 до 4,5 миллиграмм-эквивалентов на 100 грамм эластомерного полимера, например, от 0,01 до около 1,5 миллиграмм-эквивалентов на 100 грамм полимера. При больших количествах связующих средств проявляется тенденция к получению полимеров, содержащих реакционноспособные концевые группы, или недостаточной степени связывания. Может применяться от нуля до менее одного эквивалента связывающей группы на основе олова и/или кремния на эквивалент литиевого инициатора для обеспечения последующей функционализации остальной фракции живущего полимера. Например, если тетрахлорид олова или кремния или их смесь применяют в качестве связующего средства, может применяться от 0 до менее 1,0 моля, предпочтительно от 0 до 0,8 моля и более предпочтительно от 0 до 0,6 моля связующего средства на каждые 4 моль живых содержащих литий концевых групп полимерной цепи.
Если необходимо асимметричное связывание добавление связующего средства будут проводить непрерывным способом. Непрерывное добавление связующего средства обычно проводят в реакционной зоне, которая отделена от зоны, в которой происходит полимеризация основной массы. Связующее средство может добавляться в реакцию полимеризации в углеводородном растворителе, таком как циклогексан, и будет, как правило, добавляться уже после того, как произойдет превращение мономера в достаточно высокой степени. Например, связующее средство может добавляться при превращении мономера по меньшей мере около 85%, предпочтительно по меньшей мере около 90%.
Для способов полимеризации в растворе полимеризацию проводят в подходящем растворителе, диспергирующем средстве или разбавителе. Некоординирующие инертные жидкости предпочтительно включают, но не ограничиваясь этим, углеводороды с неразветвленными и разветвленными цепями, такие как пропан, бутан, изобутан, пентан, гексан, гептан, пентаметилгептан и октан, циклические и алициклические углеводороды, такие как циклогексан, циклогептан, метилциклогексан и метилциклогептан, ароматические и алкил-замещенные ароматические соединения, такие как бензол, толуол и ксилол, фракции минерального масла, такие как легкий или рядовой бензин, нафта, керосин и газойль, жидкости на основе фторированных углеводородов, такие как перфторированные C4-10алканы, и смеси двух или более из них. Дополнительные подходящие растворители включают жидкие олефины, которые могут выступать в качестве (со)мономеров в способе полимеризации, такие как пропилен, 1-бутен, 1-пентен, циклопентен, 1-гексен, 3-метил-1-пентен, 4-метил-1-пентен, бутадиен, изопрен, 1,4-гексадиен, 1,7-октадиен, 1-октен, 1-децен, стирол, дивинилбензол, этилиденнорборнен, аллилбензол, 2-метилстирол, 3-метилстирол, 4-метилстирол, 4-винилциклогексен, винилциклогексан и смеси двух или более из них.
Модифицирующее соединение (I)
Модифицирующее соединение формулы (I) предпочтительно определено следующим образом:
где R независимо представляет собой C1-C16алкил или бензил; R'' представляет собой C1-C4алкил; R' выбран из C6-C18арила, C7-C50алкиларила, C1-C50алкила и C2-C50диалкилэфира (то есть алкил-O-алкил), где каждая группа необязательно замещена одной или более из групп, выбранных из C1-C4алкила, C1-C4алкокси, C6-C12арила, C7-C16алкиларила, ди(C1-C7гидрокарбил)амино, бис(три(C1-C12алкил)силил)амино, трис(C1-C7гидрокарбил)силила и C1-C12тиоалкила; x представляет собой целое число, выбранное из 1, 2 и 3; y представляет собой целое число, выбранное из 0, 1 и 2; и x + y = 3.
В предпочтительном варианте реализации изобретения R независимо выбран из C1-C5алкила, и R' представляет собой C1-C5алкил.
Конкретные предпочтительные примеры модифицирующего соединения формулы (I) включают:
(MeO)3Si-(CH2)3-S-SiMe3, (EtO)3Si-(CH2)3-S-SiMe3, (PrO)3Si-(CH2)3-S-SiMe3, (BuO)3Si-(CH2)3-S-SiMe3, (MeO)3Si-(CH2)2-S-SiMe3, (EtO)3Si-(CH2)2-S-SiMe3, (PrO)3Si-(CH2)2-S-SiMe3, (BuO)3Si-(CH2)2-S-SiMe3, (MeO)3Si-CH2-S-SiMe3, (EtO)3Si-CH2-S-SiMe3, (PrO)3Si-CH2-S-SiMe3, (BuO)3Si-CH2-S-SiMe3, (MeO)3Si-CH2-CMe2-CH2-S-SiMe3, (EtO)3Si-CH2-CMe2-CH2-S-SiMe3, (PrO)3Si-CH2-CMe2-CH2-S-SiMe3, (BuO)3Si-CH2-CMe2-CH2-S-SiMe3, (MeO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (EtO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (PrO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (BuO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (MeO)2(Me)Si-(CH2)3-S-SiMe3, (EtO)2(Me)Si-(CH2)3-S-SiMe3, (PrO)2(Me)Si-(CH2)3-S-SiMe3, (BuO)2(Me)Si-(CH2)3-S-SiMe3, (MeO)2(Me)Si-(CH2)2-S-SiMe3, (EtO)2(Me)Si-(CH2)2-S-SiMe3, (PrO)2(Me)Si-(CH2)2-S-SiMe3, (BuO)2(Me)Si-(CH2)2-S-SiMe3, (MeO)2(Me)Si-CH2-S-SiMe3, (EtO)2(Me)Si-CH2-S-SiMe3, (PrO)2(Me)Si-CH2-S-SiMe3, (BuO)2(Me)Si-CH2-S-SiMe3, (MeO)2(Me)Si-CH2-CMe2-CH2-S-SiMe3, (EtO)2(Me)Si- CH2-CMe2-CH2-S-SiMe3, (PrO)2(Me)Si-CH2-CMe2-CH2-S-SiMe3, (BuO)2(Me)Si-CH2-CMe2-CH2-S-SiMe3, (MeO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (EtO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (PrO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (BuO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (MeO)(Me)2Si-(CH2)3-S-SiMe3, (EtO)(Me)2Si-(CH2)3-S-SiMe3, (PrO)Me)2Si-(CH2)3-S-SiMe3, (BuO) (Me)2Si-(CH2)3-S-SiMe3, (MeO)(Me)2Si-(CH2)2-S-SiMe3, (EtO) (Me)2Si-(CH2)2-S-SiMe3, (PrO)(Me)2Si-(CH2)2-S-SiMe3, (BuO)(Me)2Si-(CH2)2-S-SiMe3, (MeO)(Me)2Si-CH2-S-SiMe3, (EtO)(Me)2Si-CH2-S-SiMe3, (PrO)(Me)2Si-CH2-S-SiMe3, (BuO)(Me)2Si-CH2-S-SiMe3, (MeO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (EtO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (PrO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (BuO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (MeO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (EtO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (PrO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (BuO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (MeO)3Si-(CH2)3-S-SiEt3, (EtO)3Si-(CH2)3-S-SiEt3, (PrO)3Si-(CH2)3-S-SiEt3, (BuO)3Si-(CH2)3-S-SiEt3, (MeO)3Si-(CH2)2-S-SiEt3, (EtO)3Si-(CH2)2-S-SiEt3, (PrO)3Si-(CH2)2-S-SiEt3, (BuO)3Si-(CH2)2-S-SiEt3, (MeO)3Si-CH2-S-SiEt3, (EtO)3Si-CH2-S-SiEt3, (PrO)3Si-CH2-S-SiEt3, (BuO)3Si-CH2-S-SiEt3, (MeO)3Si-CH2-CMe2-CH2-S-SiEt3, (EtO)3Si-CH2-CMe2-CH2-S-SiEt3, (PrO)3Si-CH2-CMe2-CH2-S-SiEt3, (BuO)3Si-CH2-CMe2-CH2-S-SiEt3, (MeO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (EtO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (PrO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (BuO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (MeO)2(Me)Si-(CH2)3-S-SiEt3, (EtO)2(Me)Si-(CH2)3-S-SiEt3, (PrO)2(Me)Si-(CH2)3-S-SiEt3, (BuO)2(Me)Si-(CH2)3-S-SiEt3, (MeO)2(Me)Si-(CH2)2-S-SiEt3, (EtO)2(Me)Si-(CH2)2-S-SiEt3, (PrO)2(Me)Si-(CH2)2-S-SiEt3, (BuO)2(Me)Si-(CH2)2-S-SiEt3, (MeO)2(Me)Si-CH2-S-SiEt3, (EtO)2(Me)Si-CH2-S-SiEt3, (PrO)2(Me)Si-CH2-S-SiEt3, (BuO)2(Me)Si-CH2-S-SiEt3, (MeO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (EtO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (PrO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (BuO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (MeO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (EtO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (PrO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (BuO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (MeO)(Me)2Si-(CH2)3-S-SiEt3, (EtO)(Me)2Si-(CH2)3-S-SiEt3, (PrO)(Me)2Si-(CH2)3-S-SiEt3, (BuO)(Me)2Si-(CH2)3-S-SiEt3, (MeO)(Me)2Si-(CH2)2-S-SiEt3, (EtO)(Me)2Si-(CH2)2-S-SiEt3, (PrO)(Me)2Si-(CH2)2-S-SiEt3, (BuO)(Me)2Si-(CH2)2-S-SiEt3, (MeO)(Me)2Si-CH2-S-SiEt3, (EtO)(Me)2Si-CH2-S-SiEt3, (PrO)(Me)2Si-CH2-S-SiEt3, (BuO)(Me)2Si-CH2-S-SiEt3, (MeO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (EtO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (PrO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (BuO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (MeO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3, (EtO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3, (PrO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3, (BuO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3, (MeO)3Si-(CH2)3-S-SiMe2tBu, (EtO)3Si-(CH2)3-S-SiMe2tBu, (PrO)3Si-(CH2)S-S-SiMetBu, (BuO)3Si-(CH2-S-SiMe2tBu, (MeO)3Si-(CH2)2-S-SiMe2tBu, (EtO)3Si-(CH2)2-S-SiMe2tBu, (PrO)3Si-(CH2)2-S-SiMe2tBu, (BuO)3Si-(CH2)2-S-SiMe2tBu, (MeO)3Si-CH2-S-SiMe2tBu, (EtO)3Si-CH2-S-SiMe2tBu, (PrO)3Si-CH2-S-SiMe2tBu, (BuO)3Si-CH2-S-SiMe2tBu, (MeO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, (EtO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, (PrO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, (BuO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, (MeO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (EtO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (PrO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (BuO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (MeO)2(Me)Si-(CH2)3-S-SiMe2tBu, (EtO)2(Me)Si-(CH2)2-S-SiMe2tBu, (PrO)2(Me)Si-(CH2)3-S-SiMe2tBu, (BuO)2(Me)Si-(CH2)3-S-SiMe2tBu, (MeO)2(Me)Si-(CH2)2-S-SiMe2tBu, (EtO)2(Me)Si-(CH2)2-S-SiMe2tBu, (PrO)2(Me)Si-(CH2)2-S-SiMe2tBu, (BuO)2(Me)Si-(CH2)2-S-SiMe2tBu, (MeO)2(Me)Si-CH2-S-SiMe2tBu, (EtO)2(Me)Si-CH2-S-SiMe2tBu, (PrO)2(Me)Si-CH2-S-SiMe2tBu, (BuO)2(Me)Si-CH2-S-SiMe2tBu, (MeO)2(Me)Si-CH2-CMe2-CH2-S-SiMe2tBu, (EtO)2(Me)Si-CH2-CMe2-CH2-S-SiMe2tBu, (PrO)2(Me)Si-CH2-CMe2-CH2-S-SiMe2tBu, (BuO)2(Me)Si-CH2-CMe2-CH2-S-SiMe2tBu, (MeO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (EtO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (PrO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (BuO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (MeO) (Me)2Si-(CH2)3-S-SiMe2tBu, (EtO)(Me)2Si-(CH2)3-S-SiMe2tBu, (PrO)(Me)2Si-(CH2)3-S-SiMe2tBu, (BuO)(Me)2Si-(CH2)3-S-SiMe2tBu, (MeO)(Me)2Si-(CH2)2-S-SiMe2tBu, (EtO)(Me)2Si-(CH2)2-S-SiMe2tBu, (PrO)(Me)2Si-(CH2)2-S-SiMe2tBu, (BuO)(Me)2Si-(CH2)2-S-SiMe2tBu, (MeO)(Me)2Si-CH2-S-SiMe2tBu, (EtO)(Me)2Si-CH2-S-SiMe2tBu, (PrO)(Me)2Si-CH2-S-SiMe2tBu, (BuO)(Me)2Si-CH2-S-SiMe2tBu, (MeO)(Me)2Si-CH2-CMe2-CH2-S-SiMe2tBu, (EtO)(Me)2Si-CH2-CMe2-CH2-S-SiMe2tBu, (PrO)(Me)2Si-CH2-CMe2-CH2-S-SiMe2tBu, (BuO)(Me)2Si-CH2-CMe2-CH2-S-SiMe2tBu, (MeO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (EtO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (PrO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe2tBu и (BuO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe2tBu.
Модифицирующие соединения формулы (I) могут быть получены посредством приведения в контакт серосодержащего соединения формулы (II)
где R, R', R'', x и y имеют такие же значения, как определено в отношении формулы (I), с соединением формулы (C)
где Q представляет собой фтор, хлор или бром, и R имеет такое же значение, как определено в отношении формулы (I).
Модифицирующие соединения формулы (I) включают сульфанилсилановые соединения, описанные в патенте США № 6229036, содержание которого включено в данный документ посредством ссылки. Среди сульфанилсилановых соединений, описанных в нем, таковые без атомов галогена являются предпочтительными.
Модифицирующие соединения (1) и (2)
В одном варианте реализации модифицирующих соединений формул (1) и (2) R1 и R13 независимо выбраны из C1-C4алкила; R3, R10, R11 и R12 независимо выбраны из C1-C16алкила и бензила; и R4, и R9 независимо выбраны из C1-C16алкила, C6-C16арила и C7-C16алкиларила.
В одном варианте реализации изобретения R4 и R9 независимо выбраны из линейного C1-C10алкила, циклического C6-C12алкила, C6-C15арила и C7-C12алкиларила, таких как -CH2- (метилен), -(CH2)2- (этилиден), -(CH2)3- (пропилиден), -(CH2)4- (бутилиден), -(CH2)-C(CH3)2-CH2- (изо-пентилиден), -CH2-C6H4-CH2- (ксилиден) и -C6H4-C(CH3)2-C6H4-.
В одном варианте реализации изобретения R1, R3, R10, R11, R12 и R13 независимо выбраны из CH3- (метила), CH3-CH2- (этила), CH3-(CH2)2- (пропила), (CH3)2-(CH)- (пропила), CH3-(CH2)3- (н-бутила) и CH3-C(CH3)2- (трет-бутила).
В одном варианте реализации изобретения R1 и R13 независимо выбраны из C1-C3алкила, предпочтительно метила, этила, н-пропила и изо-пропила, и R3, R10, R11 и R12 независимо выбраны из линейного C1-C6алкила и циклического C6-C12алкила.
Конкретные примеры модифицирующих соединений формулы (1) включают (MeO)3Si-(CH2)S-S-SiMe3, (EtO)3Si-(CH2)3-S-SiMe3, (PrO)3Si-(CH2)3-S-SiMe3, (BuO)3Si-(CH2)3-S-SiMe3, (MeO)3Si-(CH2)2-S-SiMe3, (EtO)3Si-(CH2)2-S-SiMe3, (PrO)3Si-(CH2)2-S-SiMe3, (BuO)3Si-(CH2)2-S-SiMe3, (MeO)3Si-CH2-S-SiMe3, (EtO)3Si-CH2-S-SiMe3, (PrO)3Si-CH2-S-SiMe3, (BuO)3Si-CH2-S-SiMe3, (MeO)3Si-CH2-CMe2-CH2-S-SiMe3, (EtO)3Si-CH2-CMe2-CH2-S-SiMe3, (PrO)3Si-CH2-CMe2-CH2-S-SiMe3, (BuO)3Si-CH2-CMe2-CH2-S-SiMe3, (MeO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (EtO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (PrO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (BuO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (MeO)3Si-(CH2)3-S-SiEt3, (EtO)3Si-(CH2)3-S-SiEt3, (PrO)3Si-(CH2)3-S-SiEt3, (BuO)3Si-(CH2)3-S-SiEt3, (MeO)3Si-(CH2)2-S-SiEt3, (EtO)3Si-(CH2)2-S-SiEt3, (PrO)3Si-(CH2)2-S-SiEt3, (BuO)3Si-(CH2)2-S-SiEt3, (MeO)3Si-CH2-S-SiEt3, (EtO)3Si-CH2-S-SiEt3, (PrO)3Si-CH2-S-SiEt3, (BuO)3Si-CH2-S-SiEt3, (MeO)3Si-CH2-CMe2-CH2-S-SiEt3, (EtO)3Si-CH2-CMe2-CH2-S-SiEt3, (PrO)3Si-CH2-CMe2-CH2-S-SiEt3, (BuO)3Si-CH2-CMe2-CH2-S-SiEt3, (MeO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (EtO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (PrO)3Si-CH2-C(H)Me-CH2-S-SiEt3 и (BuO)3Si-CH2-C(H)Me-CH2-S-SiEt3.
Конкретные примеры модифицирующих соединений формулы (2) включают (MeO)3Si-(CH2)-N(SiMe3)2, (EtO)3Si-(CH2)3-N(SiMe3)2, (PrO)3Si-(CH2)3-N(SiMe3)2, (BuO)3Si-(CH2)3-N(SiMe3)2, (MeO)3Si-(CH2)2-N(SiMe3)2, (EtO)3Si-(CH2)2-N(SiMe3)2, (PrO)3Si-(CH2)2-N(SiMe3)2, (BuO)3Si-(CH2)2-N(SiMe3)2, (MeO)3Si-CH2-N(SiMe3)2, (EtO)3Si-CH2-N(SiMe3)2, (PrO)3Si-CH2-N(SiMe3)2, (BuO)3Si-CH2-N(SiMe3)2, (MeO)3Si-CH2-CMe2-CH2-N(SiMe3)2, (EtO)3Si-CH2-CMe2-CH2-N(SiMe3)2, (PrO)3Si-CH2-CMe2-CH2-N(SiMe3)2, (BuO)3Si-CH2-CMe2-CH2-N(SiMe3)2, (MeO)3Si-CH2-C(H)Me-CH2-N(SiMe3)2, (EtO)3Si-CH2-C(H)Me-CH2-N(SiMe3)2, (PrO)3Si-CH2-C(H)Me-CH2-N(SiMe3)2, (BuO)3Si-CH2-C(H)Me-CH2-N(SiMe3)2,
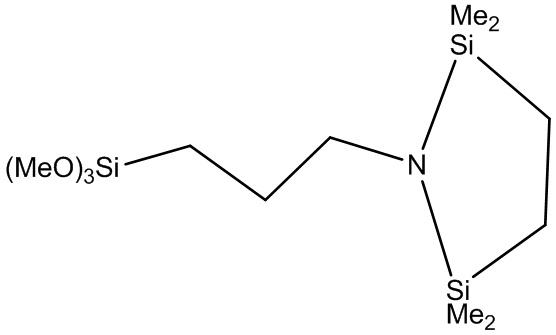 ,
, 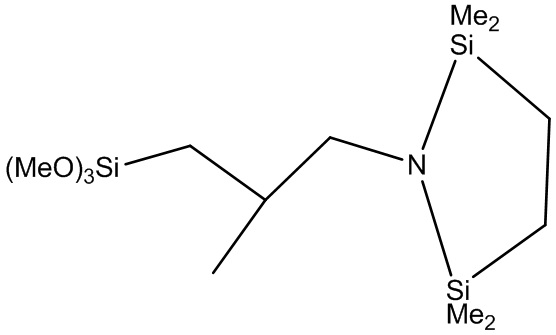 и
и
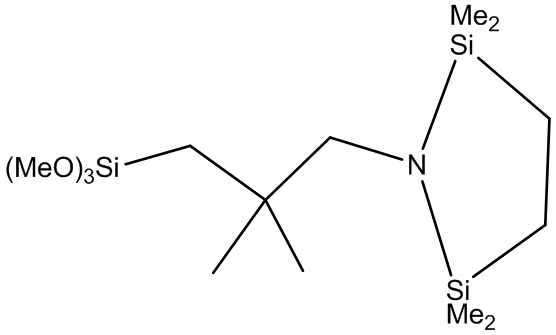 .
.
Модифицирующее соединение формулы (1) может быть получено посредством приведения в контакт серосодержащего соединения формулы (7)
где R1 и R4 имеют такие же значения, как определено в отношении формулы (1), с соединением формулы (8)
где Q представляет собой фтор, хлор или бром. Предпочтительно основание, такое как триэтиламин, применяют в реакции для гашения образованной галогенводородной кислоты формулы HQ.
Модифицирующее соединение формулы (1) также может быть получено посредством приведения в контакт серосодержащего соединения формулы (9)
где M представляет собой литий, натрий или калий, и R1 и R4 имеют такие же значения, как определено в отношении формулы (1), с соединением формулы (8).
Получение модифицирующих соединений формулы (2) является традиционным в уровне техники и может проводится, как описано, например, в WO №2003/029299 (соответствующей US №2004/0254301 и EP №1457501).
Реакция модификации с применением модифицирующего соединения формулы (1), как полагают, приводит в результате к модифицированному эластомерному полимеру, представленному формулой (P1)
где D представляет собой цепь эластомерного полимера; z представляет собой целое число, выбранное из 1, 2 и 3; и R1, R3 и R4 определены в отношении формулы (1).
Контакт с влагой, как полагают, приводит в результате к модифицированному эластомерному полимеру, представленному формулой (P3)
где D представляет собой цепь эластомерного полимера; z представляет собой целое число, выбранное из 1, 2 и 3; и R3 и R4 определены в отношении формулы (1).
Применение модифицирующих соединений (I), (1) и (2)
Модифицирующие соединения формул (I), (1) и (2) могут добавляться периодически (через равные или случайные промежутки времени) или непрерывно во время полимеризации, но предпочтительно добавляются, как только степень превращения при полимеризации достигает более 80%, и более предпочтительно при степени превращения более 90%. Предпочтительно значительное количество концевых групп полимерной цепи не закрывают перед реакцией с модифицирующим соединением; то есть концевые группы цепи живущего полимера присутствуют и являются способными к приведению в контакт с модифицирующим соединением в реакции модификации полимера. Реакция модификации может проводиться перед, после или во время добавления связующего средства (если применяется). Предпочтительно реакцию модификации завершают после добавления связующего средства (если применяется). В некоторых вариантах реализации изобретения более 10% концевых групп полимерной цепи приводится в контакт со связующим средством перед добавлением модифицирующего соединения. В некоторых вариантах реализации изобретения не применяют связующее средство, и цепи живущего полимера вступают в контакт с модифицирующим соединением. В ходе реакции модификации с модифицирующим соединением может приводиться в контакт одна или более полимерных цепей. Как результат, одна или более полимерных цепей связывается с функциональной группой из модифицирующего соединения.
Модифицирующее соединение может добавляться прямо в раствор полимера без разбавления; однако может быть полезным добавление модифицирующего соединения в растворе, например, в инертном растворителе, таком как циклогексан. Количество модифицирующего соединения, добавляемого в полимеризацию, варьируется в зависимости от мономерных веществ, модифицирующего соединения, условий реакции и необходимых свойств концевых групп модифицированного эластомерного полимера, но в целом составляет от 0,05 до 5 моль-эквивалентов, предпочтительно от 0,1 до 2,0 моль-эквивалентов и наиболее предпочтительно от 0,2 до 1,5 моль-эквивалентов на моль-эквивалент щелочного металла в органическом соединении щелочного металла, применяемом в качестве инициатора полимеризации. Реакция модификации может проводиться в температурном диапазоне от 0 до 150°C, предпочтительно от 15 до 100°C, еще более предпочтительно от 25 до 80°C. Отсутствуют ограничения в отношении продолжительности реакции функционализации, однако, что касается экономичного способа полимеризации, реакцию модификации обычно останавливают через от около 10 до 60 мин после добавления модифицирующего соединения.
Модифицирующие соединения (3)-(6)
После реакции по меньшей мере с одним из модифицирующих соединений формул (1) и (2) полимер дополнительно модифицируют посредством приведения в контакт по меньшей мере с одним из модифицирующих соединений формул (3), (4), (5) и (6).
В одном варианте реализации модифицирующих соединений формул (3), (4), (5) и (6) R2, R3, R8, R10, R11, R12, R14, R16, R17 и R18 независимо выбраны из C1-C16алкила, такого как CH3- (метил), CH3-CH2- (этил), CH3- (CH2)2- (н-пропил), (CH3)2-(CH)- (изо-пропил), CH3-(CH2)3- (н-бутил) и CH3-C(CH3)2- (трет-бутил), R1 и R13 независимо выбраны из C1-C4алкила, R5, R6, R7, R19, R20 и R21 независимо выбраны из водорода и C1-C16алкила, и R4, R9 и R15 независимо выбраны из C1-C16алкила, в частности, линейного C1-C10алкила или циклического C6-C12алкила, такого как -CH2- (метилен), -(CH2)2- (этилиден), -(CH2)3- (пропилиден), -(CH2)4- (бутилиден) и -(CH2)-C(CH3)2-CH2- (изо-пентилиден), C6-C15арила и C7-C16алкиларила, такого как -CH2-C6H4-CH2- (ксилиден) и -C6H4-C(CH3)2-C6H4-.
В одном варианте реализации изобретения R1 и R13 независимо выбраны из метила, этила и пропила (в том числе изомеров), R2, R3, R10, R11, R12, R14, R16, R17 и R18 независимо выбраны из линейного C1-C6алкила, R5, R6, R7, R8, R19, R20 и R21 независимо выбраны из водорода и линейного C1-C15алкила, и R4, R9 и R15 независимо выбраны из C1-C10алкила.
Примеры модифицирующих соединений формулы (3) включают (MeO)2(Me)Si-(CH2)2-S-SiMe3, (EtO)2(Me)Si-(CH2)3-S-SiMe3, (PrO)2(Me)Si-(CH2)3-S-SiMe3, (BuO)2(Me)Si-(CH2)3-S-SiMe3, (MeO)2(Me)Si-(CH2)2-S-SiMe3, (EtO)2(Me)Si-(CH2)2-S-SiMe3, (PrO)2(Me)Si-(CH2)2-S-SiMe3, (BuO)2(Me)Si-(CH2)2-S-SiMe3, (MeO)2(Me)Si-CH2-S-SiMe3, (EtO)2(Me)Si-CH2-S-SiMe3, (PrO)2(Me)Si-CH2-S-SiMe3, (BuO)2(Me)Si-CH2-S-SiMe3, (MeO)2(Me)Si-CH2-CMe2-CH2-S-SiMe3, (EtO)2(Me)Si-CH2-CMe2-CH2-S-SiMe3, (PrO)2(Me)Si-CH2-CMe2-CH2-S-SiMe3, (BuO)2(Me)Si-CH2-CMe2-CH2-S-SiMe3, ((MeO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (EtO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (PrO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (BuO)2(Me)Si-CH2-C(H)Me-CH2-S-SiMe3, (MeO)(Me)2Si-(CH2)3-S-SiMe3, (EtO)(Me)2Si-(CH2)3-S-SiMe3, (PrO)(Me)2Si-(CH2)3-S-SiMe3, (BuO)(Me)2Si-(CH2)3-S-SiMe3, (MeO)(Me)2Si-(CH2)2-S-SiMe3, (EtO)(Me)2Si-(CH2)2-S-SiMe3, (PrO)(Me)2Si-(CH2)2-S-SiMe3, (BuO)(Me)2Si-(CH2)2-S-SiMe3, (MeO)(Me)2Si-CH2-S-SiMe3, (EtO)(Me)2Si-CH2-S-SiMe3, (PrO)(Me)2Si-CH2-S-SiMe3, (BuO)(Me)2Si-CH2-S-SiMe3, (MeO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (EtO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (PrO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (BuO)(Me)2Si-CH2-CMe2-CH2-S-SiMe3, (MeO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (EtO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (PrO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (BuO)(Me)2Si-CH2-C(H)Me-CH2-S-SiMe3, (MeO)2(Me)Si-(CH2)3-S-SiEt3, (EtO)2(Me)Si-(CH2)3-S-SiEt3, (PrO)2(Me)Si-(CH2)3-S-SiEt3, (BuO)2(Me)Si-(CH2)3-S-SiEt3, (MeO)2(Me)Si-(CH2)2-S-SiEt3, (EtO)2(Me)Si-(CH2)2-S-SiEt3, (PrO)2(Me)Si-(CH2)2-S-SiEt3, (BuO)2(Me)Si-(CH2)2-S-SiEt3, (MeO)2(Me)Si-CH2-S-SiEt3, (EtO)2(Me)Si-CH2-S-SiEt3, (PrO)2(Me)Si-CH2-S-SiEt3, (BuO)2(Me)Si-CH2-S-SiEt3, (MeO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (EtO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (PrO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (BuO)2(Me)Si-CH2-CMe2-CH2-S-SiEt3, (MeO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (EtO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (PrO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (BuO)2(Me)Si-CH2-C(H)Me-CH2-S-SiEt3, (MeO)(Me)2Si-(CH2)3-S-SiEt3, (EtO)(Me)2Si-(CH2)3-S-SiEt3, (PrO)(Me)2Si-(CH2)3-S-SiEt3, (BuO)(Me)2Si-(CH2)3-S-SiEt3, (MeO)(Me)2Si-(CH2)2-S-SiEt3, (EtO)(Me)2Si-(CH2)2-S-SiEt3, (PrO)(Me)2Si-(CH2)2-S-SiEt3, (BuO)(Me)2Si-(CH2)2-S-SiEt3, (MeO)(Me)2Si-CH2-S-SiEt3, (EtO)(Me)2Si-CH2-S-SiEt3, (PrO)(Me)2Si-CH2-S-SiEt3, (BuO)(Me)2Si-CH2-S-SiEt3, (MeO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (EtO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (PrO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (BuO)(Me)2Si-CH2-CMe2-CH2-S-SiEt3, (MeO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3, (EtO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3, (PrO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3 и (BuO)(Me)2Si-CH2-C(H)Me-CH2-S-SiEt3.
Модифицирующее соединение формулы (3) может быть получено посредством приведения в контакт серосодержащего соединения формулы (10)
где R1, R2, R4, x и y имеют такие же значения, как определено в отношении формулы (3), с соединением формулы (8). Предпочтительно основание, такое как триэтиламин, применяют в реакции для гашения образованной галогенводородной кислоты формулы HQ.
Модифицирующие соединения формулы (3) также могут быть получены посредством приведения в контакт серосодержащего соединения формулы (11)
где M представляет собой литий, натрий или калий, и R1, R2, R4, x и y имеют такие же значения, как определено в отношении формулы (3), с соединением формулы (8).
Применение модифицирующих соединений (3)-(6)
Модифицирующие соединения формул (3)-(6) могут добавляться периодически (через равные или случайные промежутки времени) или непрерывно во время полимеризации, но предпочтительно добавляются, когда степень превращения при полимеризации достигает более 80%, и более предпочтительно при степени превращения более 90%. Предпочтительно значительное количество концевых групп полимерной цепи не закрывают перед реакцией с такими модифицирующими соединениями; то есть концевые группы цепи живущего полимера присутствуют и являются способными к приведению в контакт с модифицирующим соединением в реакции модификации концевой группы полимерной цепи. Реакция модификации с применением модифицирующих соединений формул (3)-(6) может проводиться перед, после или во время добавления модифицирующего соединения формулы (1) или (2). Однако реакцию модификации с применением модифицирующих соединений формул (3)-(6) предпочтительно проводят после добавления модифицирующих соединений формулы (1) или (2). В одном варианте реализации изобретения по меньшей мере 10% концевых групп полимерной цепи, например от 10 до 20%, предпочтительно по меньшей мере 20%, например от 20 до 40% или от 40 до 70%, как определено GPC, приводится контакт с модифицирующим соединением формулы (1) или (2) перед добавлением модифицирующего соединения формул (3)-(6). В одном варианте реализации изобретения более 10%, предпочтительно более 20%, полимерной цепи, как определено GPC, образованной в ходе осуществления способа полимеризации, связывается с модифицирующим соединением формулы (3)-(6) в способе модификации концевой группы полимера.
Модифицирующие соединения формул (3)-(6) могут добавляться прямо в раствор полимера без разбавления; однако может быть полезным добавление модифицирующего соединения(-ий) в растворе, например, в инертном растворителе (например, циклогексане). Общее количество модифицирующего соединения формул (3)-(6), добавляемого в полимеризацию, варьируется в зависимости от мономерных веществ, модифицирующего соединения формулы (1) или (2), модифицирующего соединения формулы (3)-(6), условий реакции и необходимых свойств концевых групп модифицированного эластомерного полимера, но в целом составляет от 0,05 до 5,00 моль-эквивалентов, предпочтительно от 0,10 до 2,00 моль-эквивалентов и наиболее предпочтительно от 0,15 до 1,50 моль-эквивалентов на моль-эквивалент щелочного металла в органическом соединении щелочного металла, применяемом в качестве инициатора полимеризации. Реакция модификации может проводиться в температурном диапазоне от 0 до 150°C, предпочтительно от 15 до 100°C, еще более предпочтительно от 25 до 80°C. Отсутствуют ограничения в отношении продолжительности реакции модификации, однако, что касается экономичного способа полимеризации, реакцию модификации обычно останавливают через от около 5 до 60 мин после добавления модифицирующего соединения.
Модифицированный эластомерный полимер
Эластомерный полимер из первого аспекта настоящего изобретения получают посредством приведения в контакт живого анионного эластомерного полимера, как определено выше, с одним или более модифицирующих соединений, как определено выше, а именно посредством приведения в контакт с модифицирующим соединением формулы (I) или посредством приведения в контакт с (i) модифицирующим соединением формулы (1) или (2), затем (ii) модифицирующим соединением формулы (3), (4), (5) или (6). В частности, приведения в контакт модифицирующих соединений с живущим анионным эластомерным полимером могут проводится, как описано в WO №2007/047943 и WO №2009/148932, содержание и описание каждой из которых включены в данный документ в полном объеме. WO №2007/047943 и WO №2009/148932 относятся к модифицирующим соединениям как "силан-сульфидный модификатор", "силановое модифицирующее соединение" или "модифицирующее соединение", каждое из которых является модифицирующим соединением в соответствии с определением в настоящей заявки.
Реакция модификации с применением модифицирующего соединения формулы (I), как полагают, приводит в результате к модифицированному эластомерному полимеру, представленному формулой (III)
где D представляет собой эластомерный полимер; x представляет собой целое число, выбранное из 0, 1 и 2; y представляет собой целое число, выбранное из 0, 1 и 2; z представляет собой целое число, выбранное из 1, 2 и 3; x + y + z = 3; и R, R'' и R' определены в отношении формулы (I).
Контакт с влагой, как полагают, по меньшей мере частично приводит в результате к модифицированному эластомерному полимеру, представленному формулой (P4)
где D представляет собой эластомерный полимер; x представляет собой целое число, выбранное из 0, 1 и 2; y представляет собой целое число, выбранное из 0, 1 и 2; z представляет собой целое число, выбранное из 1, 2 и 3; x + y + z = 3; и R и R' определены в отношении формулы (I).
Реакция модификации с применением модифицирующего соединения формулы (3) в комбинации с модифицирующим соединением формулы (1), как полагают, приводит в результате к модифицированному эластомерному полимеру, представленному следующей формулой (P2)
где D представляет собой цепь эластомерного полимера; x представляет собой целое число, выбранное из 0 и 1; y представляет собой целое число, выбранное из 1 и 2; z равняется 1; и x + y + z = 3; и R1, R2, R3 и R4 определены в отношении формулы (3).
Контакт с влагой, как полагают, по меньшей мере частично приводит в результате к модифицированному эластомерному полимеру, представленному следующей формулой (P5)
где D представляет собой цепь эластомерного полимера; x представляет собой целое число, выбранное из 0 и 1; y представляет собой целое число, выбранное из 1 и 2; z равняется 1; и x + y + z = 3; и R2, R3 и R4 определены в отношении формулы (3).
Триалкилсилильная (-SiR3) группа в формуле (III) и триалкилсилильная, три(алкиларил)силильная или триарилсилильная (-SiR33) группы в формулах (P1) и (P2), как полагают, выполняют функцию защитной группы, которая предотвращает непредусмотренную последующую реакцию. Данная защитная группа может быть удалена посредством воздействия соединения, содержащего группы -OH, такого как вода, спирты, неорганические и органические кислоты (в том числе хлористоводородная кислота, серная кислота и карбоновые кислоты), таким образом, с образованием "незащищенной" тиольной (-SH) группы. Такие условия, как правило, имеют место во время вулканизации. В зависимости от выделения полимера могут присутствовать как незащищенные, так и/или защищенные модифицированные эластомерные полимеры. Например, с помощью отгонки с водяным паром раствора полимера, содержащего модифицированный эластомерный полимер в соответствии с данным изобретением, будут удалять некоторую часть защитных групп, приводя в результате к незащищенной форме с открытой тиольной группой. В альтернативном варианте с помощью процедуры безводного выделения можно обеспечить получение модифицированного эластомерного полимера в соответствии с формулой (III), (P1) или (P2).
Полагают, что незащищенная тиольная группа модифицированного эластомерного полимера является реакционноспособной в отношении наполнителей, таких как диоксид кремния и/или углеродная сажа, если имеются. Такое взаимодействие, как полагают, приводит к электростатическим взаимодействиям, которые обеспечивают более однородное распределение наполнителя в композиции на основе эластомерного полимера.
Полагают что силанольная группа модифицированного эластомерного полимера, в частности, полимеров согласно формулам (P3), (P4) и (P5), является реакционноспособной в отношении наполнителей, таких как, например, диоксид кремния, если имеется. Такое взаимодействие, как полагают, в результате приводит к образованию связей в случае некоторых наполнителей, например, связей Si-O-Si в случае диоксида кремния, или электростатическим взаимодействиям, которые в результате приводят к более однородному распределению наполнителя в композиции на основе эластомерного полимера.
Полученный модифицированный эластомерный полимер предпочтительно содержит сульфидные группы (например, тиольные) в количестве от 0,0005 до 0,20 или от 0,0010 до 0,20 ммоль/грамм эластомерного полимера, или от 0,0010 до 0,10 ммоль/грамм, более предпочтительно от 0,0025 до 0,1 ммоль/грамм и еще более предпочтительно от 0,0025 до 0,05 или от 0,0030 до 0,05 ммоль/грамм.
В зависимости от желаемого применения модифицированный эластомерный полимер в соответствии с данным изобретением предпочтительно характеризуется вязкостью по Муни в диапазоне от 20 до 150, более предпочтительно от 30 до 100 (ML 1+4, 100°C, измеренная в соответствии с ASTM D 1646 (2004) с применением прибора Monsanto MV2000). Если вязкость по Муни составляет менее 20, свойства устойчивости к истиранию и свойства в отношении гистерезисных потерь могут быть нарушены. Более того, липкость и текучесть в естественных условиях несшитого эластомерного полимера может повышаться, приводя в результате к сложности обработки, плохой когезионной прочности в невулканизированном состоянии и плохой формоустойчивости при хранении. Если вязкость по Муни составляет более 150, пригодность для обработки, отражаемая во включении наполнителя и тепловыделении во внутреннем смесителе, полосности на вальцовой мельнице, степени вытягивания, разбухании экструдируемого потока, гладкости и т. д., может быть нарушена с увеличением затрат на обработку.
Предпочтительное распределение молекулярного веса рассматриваемого модифицированного полимера относительно отношения средневесового молекулярного веса к среднечисловому молекулярному весу, Mw/Mn, предпочтительно находится в диапазоне от 1,2 до 3,0. Пригодность для обработки полимера может быть нарушена, если Mw/Mn составляет менее 1,2. Плохая пригодность для обработки не только повышает затраты на производство, но также ухудшает характеристики смешивания компонентов в полимерной композиции, такие как недостаточная степень дисперсности наполнителей и других добавок, которые могут привести к плохим физическим свойствам. Если Mw/Mn составляет более 3,0, содержание компонентов с низким молекулярным весом повышается, и могут увеличиться гистерезисные потери.
Композиция на основе эластомерного полимера
Композиция на основе эластомерного полимера в соответствии с данным изобретением содержит модифицированный эластомерный полимер в соответствии с данным изобретением и один или более дополнительных компонентов, выбранных из компонентов, которые добавляют в или образуют в результате способа полимеризации, применяемого для получения модифицированного эластомерного полимера, и компонентов, которые остаются после удаления растворителя из способа полимеризации. Компоненты, которые добавляют в способ полимеризации включают, в частности, масла (масла для наполнения), наполнители, стабилизаторы и дополнительные полимеры.
В одном варианте реализации изобретения композиция на основе эластомерного полимера в соответствии с данным изобретением содержит модифицированный эластомерный полимер в соответствии с данным изобретением в комбинации с одним или более масел для наполнения.
Масла для наполнения, предназначенные для применения в композиции на основе эластомерного полимера, включают минеральные масла. Минеральные масла могут подразделяться на категории как масла для наполнения ароматического типа, масла для наполнения алициклического типа и масла для наполнения алифатического типа. Среди таких масел для наполнения минеральное масло ароматического типа, характеризующееся гравитационно-вязкостной постоянной (V.G.C.) 0,900-1,049 (ароматическое масло), и минеральное масло алициклического типа, характеризующееся V.G.C. 0,850-0,899 (нафтеновое масло), являются особенно предпочтительными для обеспечения улучшенных свойств в отношении низкотемпературных гистерезисных потерь и связанной улучшенной устойчивости к проскальзыванию на мокрой дороге. Подходящие масла для наполнения включают MES (сольват мягкой экстракции), TDAE (ароматический экстракт из обработанного дистиллята), RAE (остаточный ароматический экстракт), DAE и NAP (нафтеновые). Природные масла также могут применяться в качестве масел для наполнения. Вышеуказанные масла для наполнения содержат различные концентрации полициклических ароматических соединений, парафинистых, нафтеновых и ароматических соединений и характеризуются разными температурами стеклования (для описания см. "Kautschuk Gummi Kunststoffe", vol. 52, с. 799-805). MES и TDAE являются особенно предпочтительными маслами для наполнения, предназначенными для каучука.
Наполнение модифицированного эластомерного полимера в соответствии с данным изобретением маслом для наполнения обеспечивает однородную степень дисперсности наполнителей, таких как углеродная сажа и диоксид кремния, в полимере и улучшает пригодность для обработки и другие свойства вулканизированных изделий, полученных из композиции на основе полимера. Количество масла для наполнения, применяемого в настоящем изобретении, обычно составляет от 0 до 100 м.ч., предпочтительно от 0 до 80 м.ч. и более предпочтительно от 0 до 70 м.ч., для 100 м.ч. модифицированного эластомерного полимера в форме конечного продукта реакции, представляющего собой блок-полимер, перед составлением каучука и процессом вулканизации. Если масло для наполнения добавляют в раствор полимера, временной интервал добавления должен находиться после модификации полимера или окончания полимеризации, например, после добавления модифицирующего средства или средства прекращения полимеризации. После добавления масла для наполнения маслонаполненную композицию на основе полимера получают посредством отделения любого растворителя для полимеризации от полимера посредством способа конвективной сушки или отгонки с водяным паром, сушки каучука с применением вакуумной сушилки, сушилки с обогревом горячим воздухом, вальцовой сушилки и т. п.
В другом варианте реализации изобретения композиция на основе эластомерного полимера, которая необязательно содержит одно или более масел для наполнения, как определено выше, дополнительно содержит один или более наполнителей. Наполнители выступают в качестве армирующих наполнителей и включают углеродные нанотрубки (CNT) (в том числе дискретные CNT, полые углеродные волокна (HCF) и модифицированные CNT, несущие одну или более функциональных групп, таких как гидроксильные, карбоксильные и карбонильные группы), углеродную сажу (в том числе электропроводную углеродную сажу), графит, графен (в том числе дискретные графеновые пластинки), диоксид кремния, углерод-кремнеземный двухфазный наполнитель, глины (слоистые силикаты, в том числе расслаивающуюся наноглину и органоглину), карбонат кальция, карбонат магния, лигнин, аморфные наполнители, такие как наполнители на основе частиц стекла, наполнители на основе крахмала, и их комбинации. Является предпочтительным комбинированное применение углеродной сажи с диоксидом кремния, применение углерод-кремнеземного двухфазного наполнителя, комбинированное применение углерод-кремнеземного двухфазного наполнителя с углеродной сажей и/или диоксидом кремния и комбинированное применение углеродных нанотрубок с одним или более из углерод-кремнеземного двухфазного наполнителя, углеродной сажи и диоксида кремния и необязательно других нанонаполнителей.
Подходящая углеродная сажа, которая может быть изготовлена посредством способа с применением печи, таким образом, имеет удельную площадь поверхности поглощения азота 50-200 м2/г и поглощения DBP-масла 80-200 мл/100 грамм. Примеры предпочтительных типов углеродной сажи включают классы углеродной сажи FEF, HAF, ISAF и SAF и электропроводную углеродную сажу. Углеродная сажа высоко агломерированного типа является особенно предпочтительной. Углеродную сажу, как правило, добавляют в количестве от 2 до 100 м.ч., предпочтительно от 5 до 100 м.ч., более предпочтительно от 10 до 100 м.ч., еще более предпочтительно от 10 до 95 м.ч. на 100 м.ч. от общего количества эластомерного полимера в композиции на основе полимера.
Примеры наполнителей на основе диоксида кремния включают диоксид кремния, полученный мокрым способом, диоксид кремния, полученный сухим способом, и диоксид кремния синтетического типа. Диоксид кремния, имеющий небольшой диаметр частиц, проявляет высокий армирующий эффект. Диоксид кремния высоко агломерированного типа с небольшим диаметром частиц (то есть с большой площадью поверхности и высокой поглотительной способностью масла) проявляет превосходную степень дисперсности в композиции на основе эластомерного полимера, таким образом, способствуя необходимым свойствам и исключительной пригодности для обработки. Средний диаметр частиц диоксида кремния относительно первичного диаметра частиц предпочтительно составляет от 5 до 60 нм, более предпочтительно от 10 до 35 нм. Удельная площадь поверхности частиц диоксида кремния (измеренная посредством способа BET с применением азота) предпочтительно составляет от 45 до 280 м2/г. Диоксид кремния, как правило, добавляют в количестве от 10 до 100 м.ч., предпочтительно от 30 до 100 м.ч., еще более предпочтительно от 30 до 95 м.ч. на 100 м.ч. от общего количества эластомерного полимера в композиции на основе полимера. Углеродная сажа и диоксид кремния могут добавляться вместе, в этом случае добавляемое общее количество углеродной сажи и диоксида кремния составляет от 30 до 100 м.ч., предпочтительно от 30 до 95 м.ч. на 100 м.ч. общего количества эластомерного полимера. Поскольку наполнители диспергируются в эластомерной композиции однородным образом, повышение количества (в пределах вышеуказанных диапазонов) приводит в результате к композиции с превосходной пригодностью для обработки с помощью прокатки и экструзии, и при этом вулканизированные продукты проявляют подходящие свойства в отношении гистерезисных потерь, устойчивость к качению, улучшенную устойчивость к проскальзыванию на мокрой дороге, устойчивость к истиранию и предел прочности при растяжении.
Углерод-кремнеземный двухфазный наполнитель может проявлять эффекты, аналогичные таковым, полученным посредством комбинированного применения углеродной сажи и диоксида кремния, даже в том случае, когда добавляют только его. Углерод-кремнеземный двухфазный наполнитель представляет собой так называемую покрытую диоксидом кремния углеродную сажу, полученную посредством нанесения покрытия из диоксида кремния на поверхность углеродной сажи, и является коммерчески доступным под торговой маркой CRX2000, CRX2002 или CRX2006 (продукты от Cabot Co.). Углерод-кремнеземный двухфазный наполнитель добавляют в тех же количествах, как описано ранее для диоксида кремния. Углерод-кремнеземный двухфазный наполнитель может применяться в комбинациях с другими наполнителями, например, углеродными нанотрубками, углеродной сажей, диоксидом кремния, глиной, карбонатом кальция и карбонатом магния. Среди таких наполнителей применение с углеродной сажей и/или диоксидом кремния является предпочтительным.
Помимо модифицированного эластомерного полимера и масла для наполнения и/или наполнителя композиция на основе эластомерного полимера в соответствии с данным изобретением может дополнительно содержать один или более дополнительных компонентов, в том числе, но не ограничиваясь этим, связующие средства и дополнительные эластомерные полимеры.
Дополнительные эластомерные полимеры, называемые в данном документе, представляют собой эластомерные полимеры, которые не соответствуют модифицированному полимеру в соответствии с данным изобретением. Они включают дополнительные немодифицированные эластомерные полимеры и дополнительные модифицированные эластомерные полимеры. Дополнительные немодифицированные эластомерные полимеры относятся к несшитым эластомерным полимерам, которые не приводились в контакт с модифицирующими соединениями в соответствии с настоящим изобретением, но были получены и закрыты традиционным способом. Дополнительные модифицированные эластомерные полимеры получают из несшитых немодифицированных эластомерных полимеров, которые были приведены в контакт с модифицирующими соединениями, не соответствующими настоящему изобретению. Модифицированный эластомерный полимер в соответствии с данным изобретением предпочтительно составляет по меньшей мере 30 мас.% от общего количества эластомерного полимера, присутствующего в композиции на основе эластомерного полимера, более предпочтительно по меньшей мере 50 мас.%. Остальная часть эластомерного полимера образована дополнительными эластомерными полимерами, включающими модифицированные и немодифицированные их варианты реализации. Предпочтительные дополнительные эластомерные полимеры включают цис-1,4-изопреновый полимер, натуральный каучук, 1,2- и 3,4-изопреновый полимер, стирол/бутадиеновый сополимер, стирол/изопрен/бутадиеновый тройной сополимер, полибутадиен, в том числе (i) полибутадиен, содержащий по меньшей мере 90 % масс. цис-1,4-полибутадиеновых элементарных звеньев, и (ii) низкомолекулярный цис-полибутадиен, содержащий менее 40 мас.% цис-полибутадиеновых и более 5 мас.% 1,2-полибутадиеновых элементарных звеньев, и (iii) полибутадиен, содержащий более 75 мас. транс-1,4-полибутадиеновых элементарных звеньев, акрилонитрил/бутадиеновый сополимер и хлоропреновый полимер, в том числе комбинации двух или более из них. Стирол-бутадиеновый сополимер, натуральные каучуки, полиизопрен и полибутадиен являются предпочтительными. Необходимо, чтобы дополнительные эластомерные полимеры (модифицированные или немодифицированные) имели вязкость по Муни (ML 1 +4, 100°C согласно ASTM D 1646 (2004)) в диапазоне от 20 до 200, предпочтительно от 25 до 150. Добавление дополнительных эластомерных полимеров в вышеуказанных диапазонах обеспечивает изготовление композиции на основе эластомерного полимера в соответствии с данным изобретением при низких затратах практически без ухудшения их характеристик.
Предпочтительно добавление силанового связующего средства в композицию на основе полимера, если применяют диоксид кремния или углерод-кремнеземный двухфазный наполнитель. Типичное количество добавляемого силанового связующего средства составляет от около 1 до около 20 м.ч., предпочтительно от 5 до 15 м.ч. на 100 м.ч. общего количества диоксида кремния и/или углерод-кремнеземного двухфазного наполнителя. Предпочтительным силановым связующим средством является то, которое содержит в своей молекуле как функциональную группу, реакционноспособную по отношению к поверхности диоксида кремния, такую как алкоксисилильная группа, так и функциональную группу, реакционноспособную по отношению к углерод-углеродной двойной связи полимера, такую как полисульфидная группа, меркаптогруппа или эпоксигруппа. Примеры включают бис-(3-триэтоксисилилпропил)тетрасульфид, бис-(3-триэтоксисилилпропил)дисульфид, бис-(2-триэтоксисилилэтил)тетрасульфид, бис-(2-триэтоксисилилэтил)дисульфид, 3-меркаптопропилтриметоксисилан, 3-триэтоксисилилпропил-N,N-диметилтиокарбамоила тетрасульфид, 3-триэтоксисилилпропилбензотиазола тетрасульфид и 3-октаноилтио-1-пропилтриэтоксисилан (NXT силан, Crompton Corporation). Применение такого силанового связующего средства повышает армирующий эффект, обусловленный комбинированным применением углеродной сажи и диоксида кремния или применением углерод-кремнеземного двухфазного наполнителя.
Композиция на основе эластомерного полимера в соответствии с данным изобретением предпочтительно не содержит вулканизирующее средство.
Настоящее изобретение будет описано более подробно с помощью примеров, которые не предназначены для ограничения настоящего изобретения.
ПРИМЕРЫ
Модифицирующее соединение (A) представляет собой пример формулы (1) и имеет следующую химическую структуру: (MeO)3Si-(CH2)2-S-SiMe2C(Me)3.
Модифицирующее соединение (B) представляет собой пример формулы (3) и имеет следующую химическую структуру: (MeO)2(Me)Si-(CH2)2-S-SiMe2C(Me)3.
Сравнительный пример C1 (SSBR с высоким содержанием виниловых связей, не модифицированный (A) или (B))
Загружали 18742,3 г циклогексана, 1824,6 г бутадиена, 208,26 г стирола и 14,750 ммоль TMEDA в деаэрированный 40-л реактор и перемешиваемую смесь нагревали до 40°C. Затем по каплям загружали н-бутиллитий для приведения в контакт с загрязняющими примесями, содержащимися в реакционной смеси, пока цвет реакционной смеси не стал желтоватым (этап титрования). После этого загружали количество по составу 17,036 ммоль н-бутиллития, соответствующее целевому молекулярному весу полимера, непосредственно через насос для инициации полимеризации. Время начала загрузки основного количества н-бутиллития применяли в качестве времени начала полимеризации. Первый этап полимеризации характеризовался обычной периодической полимеризацией в течение 10 мин без загрузки каких-либо дополнительных реагирующих веществ. Параллельно повышали температуру посредством нагревания или охлаждения горячей водой в стенке реакторов начиная с загрузки основного количества н-бутиллития до конечной температуры полимеризации Tpm 60°C со скоростью 2°C/мин. После этих 10 мин начинался этап первой загрузки 1328,4 г бутадиена и 151,04 г стирола с применением постоянной скорости подачи в течение 60 мин. Добавляли 20 мин к продолжительности реакции для завершения превращения. В течение одной мины с помощью насоса добавляли 13,94 г бутадиена с последующим добавлением 1,3534 ммоля четыреххлористого олова и 50 г циклогексана с помощью цилиндра. Второе добавление 51,36 г бутадиена выполняли через 20 мин в течение одной мины. Реакцию оставляли до завершения в течение 15 мин. После этого загружали 71,8 г метанола для завершения реакции. Раствор полимера стабилизировали с помощью 7,13 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 52,0%. Содержание стирола составляло 10,6%. Температура стеклования полимера составляла -46,2°C. Полный набор данных для образца приведен в таблице 1.
Сравнительный пример C2 (SSBR с высоким содержанием виниловых связей, модифицированный (B))
Загружали 18714,5 г циклогексана, 1821,7 г бутадиена, 207,66 г стирола и 14,555 ммоль TMEDA в деаэрированный 40-л реактор и перемешиваемую смесь нагревали до 40°C. Затем по каплям загружали н-бутиллитий для приведения в контакт с загрязняющими примесями пока цвет реакционной смеси не изменился на желтоватый (этап титрования). После этого загружали количество по составу 16,591 ммоль н-бутиллития, соответствующее целевому молекулярному весу полимера, непосредственно через насос для инициации полимеризации. Время начала загрузки основного количества н-бутиллития применяли в качестве времени начала полимеризации. Первый этап полимеризации характеризовался обычной периодической полимеризацией в течение 10 мин без загрузки каких-либо дополнительных реагирующих веществ. Параллельно повышали температуру посредством нагревания или охлаждения горячей водой в стенке реакторов начиная с загрузки основного количества н-бутиллития до конечной температуры полимеризации Tpm 60°C со скоростью 2°C/мин. После этих 10 мин начинался этап первой загрузки 1326,4 г бутадиена и 150,74 г стирола с применением постоянной скорости подачи в течение 60 мин. Добавляли 20 мин к продолжительности реакции для завершения превращения. В течение одной мины с помощью насоса добавляли 13,94 г бутадиена с последующим добавлением 1,3467 ммоля четыреххлористого олова и 50 г циклогексана с помощью цилиндра. Второе добавление 51,36 г бутадиена выполняли через 20 мин в течение одной мины с последующим добавлением 15,6295 ммоль (B). Реакцию оставляли до завершения в течение 15 мин. После этого загружали 71,8 г метанола для завершения реакции. Раствор полимера стабилизировали с помощью 7,13 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 52,8%. Содержание стирола составляло 10,5%. Температура стеклования полимера составляла -45,4°C. Полный набор данных для образца приведен в таблице 1.
Сравнительный пример C3 (SSBR с высоким содержанием виниловых связей, модифицированный (A) и (B))
Загружали 18724,8 г циклогексана, 1823,1 г бутадиена, 207,76 г стирола и 13,9647 ммоль TMEDA в деаэрированный 40-л реактор и перемешиваемую смесь нагревали до 40°C. Затем по каплям загружали н-бутиллитий для приведения в контакт с загрязняющими примесями пока цвет реакционной смеси не изменился на желтоватый (этап титрования). После этого загружали количество по составу 18,3452 ммоль н-бутиллития, соответствующее целевому молекулярному весу полимера, непосредственно через насос для инициации полимеризации. Время начала загрузки основного количества н-бутиллития применяли в качестве времени начала полимеризации. Первый этап полимеризации характеризовался обычной периодической полимеризацией в течение 10 мин без загрузки каких-либо дополнительных реагирующих веществ. Параллельно повышали температуру посредством нагревания или охлаждения горячей водой в стенке реакторов начиная с загрузки основного количества н-бутиллития до конечной температуры полимеризации Tpm 60°C со скоростью 2°C/мин. После этих 10 мин начинался этап первой загрузки 1326,6 г бутадиена и 150,74 г стирола с применением постоянной скорости подачи в течение 60 мин. Добавляли 20 мин к продолжительности реакции для завершения превращения. В течение одной мины с помощью насоса добавляли 14,04 г бутадиена с последующим добавлением 2,9766 ммоля (A) и 50 г циклогексана с помощью цилиндра. Второе добавление 51,46 г бутадиена выполняли через 20 мин в течение одной мины с последующим добавлением 15,748 ммоль (B). Реакцию оставляли до завершения в течение 15 мин. После этого загружали 71,8 г метанола для завершения реакции. Раствор полимера стабилизировали с помощью 7,13 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 49,3%. Содержание стирола составляло 10,2%. Температура стеклования полимера составляла -49,4°C. Полный набор данных для образца приведен в таблице 1.
Сравнительный пример C4 (SSBR с низким содержанием виниловых связей, не модифицированный (A) или (B))
Загружали 19788,6 г циклогексана и 270,54 г стирола в деаэрированный 40-л реактор и перемешиваемую смесь нагревали до 60°C. Затем по каплям загружали н-бутиллитий для приведения в контакт с загрязняющими примесями пока цвет реакционной смеси не изменился на желтоватый (этап титрования). После этого загружали количество по составу 13,5484 ммоль н-бутиллития, соответствующее целевому молекулярному весу полимера, непосредственно через насос для инициации полимеризации. Время начала загрузки основного количества н-бутиллития применяли в качестве времени начала полимеризации. Непосредственно после начала полимеризации загружали 1581,78 г бутадиена с постоянной скоростью подачи в течение 75 мин. Параллельно повышали температуру посредством нагревания или охлаждения горячей водой в стенке реакторов начиная с загрузки основного количества н-бутиллития до конечной температуры полимеризации Tpm 85°C со скоростью 2,08°C/мин. После первой загрузки дополнительно загружали 791,16 г бутадиена с применением постоянной скорости подачи в течение дополнительных 75 мин. Добавляли 30 мин к продолжительности реакции для завершения превращения. В течение одной мины с помощью насоса добавляли 11,94 г бутадиена с последующим добавлением 1,3159 ммоля четыреххлористого олова и 50 г циклогексана с помощью цилиндра. Второе добавление 44,26 г бутадиена выполняли через 20 мин в течение одной мины. Реакцию оставляли до завершения в течение 15 мин. Реакцию завершали с помощью загрузки 65,5 г метанола. Раствор полимера стабилизировали с помощью 5,40 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 8,8%. Содержание стирола составляло 10,5%. Температура стеклования полимера составляла -82,4°C. Полный набор данных для образца приведен в таблице 1.
Пример E1
Загружали 19748 г циклогексана и 270,13 г стирола в деаэрированный 40-л реактор и перемешиваемую смесь нагревали до 60°C. Затем по каплям загружали н-бутиллитий для приведения в контакт с загрязняющими примесями пока цвет реакционной смеси не изменился на желтоватый (этап титрования). После этого загружали количество по составу 13,5119 ммоль н-бутиллития, соответствующее целевому молекулярному весу полимера, непосредственно через насос для инициации полимеризации. Время начала загрузки основного количества н-бутиллития применяли в качестве времени начала полимеризации. Непосредственно после начала полимеризации загружали 1578,48 г бутадиена с постоянной скоростью подачи в течение 75 мин. Параллельно повышали температуру посредством нагревания или охлаждения горячей водой в стенке реакторов начиная с загрузки основного количества н-бутиллития до конечной температуры полимеризации Tpm 85°C со скоростью 2,08°C/мин. После первой загрузки дополнительно загружали 789,46 г бутадиена с применением постоянной скорости подачи в течение дополнительных 75 мин. Добавляли 30 мин к продолжительности реакции для завершения превращения. В течение одной мины с помощью насоса добавляли 11,94 г бутадиена с последующим добавлением 1,3169 ммоля четыреххлористого олова и 50 г циклогексана с помощью цилиндра. Второе добавление 44,16 г бутадиена выполняли через 20 мин в течение одной мины с последующим добавлением 14,28 ммоль (B). Реакцию оставляли до завершения в течение 15 мин. Реакцию завершали с помощью загрузки 65,2 г метанола. Раствор полимера стабилизировали с помощью 5,40 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 8,6%. Содержание стирола составляло 10,4%. Температура стеклования полимера составляла -82,9°C. Полный набор данных для образца приведен в таблице 2.
Пример E2
Загружали 19781,7 г циклогексана и 270,74 г стирола в деаэрированный 40-л реактор и перемешиваемую смесь нагревали до 60°C. Затем по каплям загружали н-бутиллитий для приведения в контакт с загрязняющими примесями пока цвет реакционной смеси не изменился на желтоватый (этап титрования). После этого загружали количество по составу 13,5119 ммоль н-бутиллития, соответствующее целевому молекулярному весу полимера, непосредственно через насос для инициации полимеризации. Время начала загрузки основного количества н-бутиллития применяли в качестве времени начала полимеризации. Непосредственно после начала полимеризации загружали 1581,08 г бутадиена с постоянной скоростью подачи в течение 75 мин. Параллельно повышали температуру посредством нагревания или охлаждения горячей водой в стенке реакторов начиная с загрузки основного количества н-бутиллития до конечной температуры полимеризации Tpm 85°C со скоростью 2,08°C/мин. После первой загрузки дополнительно загружали 790,76 г бутадиена с применением постоянной скорости подачи в течение дополнительных 75 мин. Добавляли 30 мин к продолжительности реакции для завершения превращения. В течение одной мины с помощью насоса добавляли 11,94 г бутадиена с последующим добавлением 2,2581 ммоля (A) и 50 г циклогексана с помощью цилиндра. Второе добавление 44,16 г бутадиена выполняли через 20 мин в течение одной мины с последующим добавлением 14,327 ммоль (B). Реакцию оставляли до завершения в течение 15 мин. Реакцию завершали с помощью загрузки 65,2 г метанола. Раствор полимера стабилизировали с помощью 5,4 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 8,8%. Содержание стирола составляло 10,5%. Температура стеклования полимера составляла -83,2°C. Полный набор данных для образца приведен в таблице 2.
Сравнительный пример C5 (SSBR с низким содержанием виниловых элементарных звеньев, не модифицированный (A) или (B))
Загружали 5007,7 г циклогексана и 410,98 г стирола в деаэрированный 20-л реактор и перемешиваемую смесь нагревали до 60°C. Затем по каплям загружали н-бутиллитий для приведения в контакт с загрязняющими примесями пока цвет реакционной смеси не изменился на желтоватый (этап титрования). После этого загружали количество по составу 5,9485 ммоль н-бутиллития, соответствующее целевому молекулярному весу полимера, непосредственно через насос для инициации полимеризации. Время начала загрузки основного количества н-бутиллития применяли в качестве времени начала полимеризации. Повышали температуру посредством нагревания или охлаждения горячей водой в стенке реакторов начиная с загрузки основного количества н-бутиллития до конечной температуры полимеризации Tpm 85°C со скоростью 2,08°C/мин. Параллельно начинали этап первой загрузки 335,62 г бутадиена с применением постоянной скорости подачи в течение 45 мин. После чего продолжительность реакции не продолжали. Затем загружали 253,35 г бутадиена с постоянной скоростью подачи в течение дополнительных 105 мин. Реакцию оставляли до завершения в течение 15 мин. Затем в течение одной мины с помощью насоса добавляли 12,67 г бутадиена с последующим добавлением 0,5625 ммоля четыреххлористого олова и 50 г циклогексана с помощью цилиндра. Второе добавление 12,67 г бутадиена выполняли через 20 мин в течение одной мины. Реакцию оставляли до завершения в течение 15 мин. Реакцию завершали с помощью загрузки 12,3 г метанола. Раствор полимера стабилизировали с помощью 2,04 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 8,1%. Содержание стирола составляло 40,5%. Температура стеклования полимера составляла -42,9°C. Полный набор данных для образца приведен в таблице 1.
Сравнительный пример C6 (SSBR с высоким содержанием виниловых элементарных звеньев, не модифицированный (A) или (B))
Загружали 18895,8 г циклогексана, 521,09 г стирола, 1532,84 г бутадиена и 11,6145 ммоль TMEDA в 40-литровый реактор в атмосфере азота и нагревали смесь до 55ºC при перемешивании. Затем по каплям загружали н-бутиллитий в смесь (для приведения в контакт с загрязняющими примесями), пока цвет реакционной смеси не изменялся на желтоватый (этап титрования). Затем 19,1583 ммоль н-бутиллития, соответствующие целевому молекулярному весу полимера, загружали непосредственно через насос для инициации полимеризации. Время начала загрузки количества по составу н-бутиллития применяли в качестве времени начала первой реакции полимеризации. Температуру реакции регулировали с применением циркуляции горячей воды в стенке реакторов после начала загрузки н-бутиллития до конечной температуры полимеризации Tpm 75ºC при скорости 2ºC/мин для ускорения реакции полимеризации и сокращения продолжительности реакции. Первый этап полимеризации характеризовался обычной периодической полимеризацией в течение 10 мин без загрузки каких-либо дополнительных реагирующих веществ. После этого загружали 1116,31 г бутадиена и 379,71 г стирола с применением постоянной скорости подачи в течение 60 мин. После данного добавления добавляли 20 мин к продолжительности реакции для завершения реакции. В течение одной мины с помощью насоса добавляли 11,64 г бутадиена с последующим добавлением 2,786 ммоля (A) и 50 г циклогексана с помощью цилиндра. Второе добавление 42,96 г бутадиена выполняли через 20 мин в течение одной мины с последующим добавлением 16,781 ммоль (B). Реакцию оставляли до завершения в течение 15 мин. Реакцию завершали с помощью загрузки 76,8 г метанола. Раствор полимера стабилизировали с помощью 7,2 г Irganox 1520, извлекали полимер посредством отгонки с водяным паром и высушивали до достижения содержания остаточных летучих веществ <0,7%. Содержание виниловых связей в полимере составляло 28,9%. Содержание стирола составляло 24,9%. Температура стеклования полимера составляла -50,7°C. Полный набор данных для образца приведен в таблице 1.
Таблица 1: Данные сравнительных примеров C1-C6 полимера
Таблица 2: Данные примеров E1-E2 полимера в соответствии с данным изобретением
Введение модификации в случае полимеров с низким содержанием виниловых связей E1, E2 по сравнению с C4 в таблице 3 приводит в результате к значительному улучшению динамических характеристик. В частности, имеет место относительно значительное улучшение в отношении HBU (тепловыделения) (13-28%) по сравнению с эффектом в случае среды или высокомолекулярных виниловых полимеров C2, C3 по сравнению с C1 (0-10%). Применение модифицированных полимеров с низким содержанием виниловых связей в соответствии с данным изобретением может приводить к значительно более низкому истиранию, улучшенной устойчивости к качению, но, в дополнение, также к уменьшенному HBU.
Таблица 3: Характеристики вулканизированной заполненной композиции на основе модифицированного полимера
Состав 1 соединения
Tg [°C]
[°C]
[мм3]
Применяемые композиции на основе эластомерного полимера
1 применяют сравнительные примеры полимера или примеры полимера в соответствии с настоящим изобретением
Способы
Анализы молекулярного веса проводили посредством SEC/RI с применением HEWLETT PACKARD HP 1100. Элюируемый THF дегазировали на линии. Расход растворителя составлял 1,0 мл/мин. Вводили 100 мкл раствора полимера на анализ. Анализы проводили при 40°C. Значения молекулярного веса сначала рассчитывали на основе калибровки по полистиролу и приводили в таблицах как полистирол. Фактические значения молекулярного веса (значения молекулярного веса SSBR) определяли посредством деления на коэффициент, полученный из более раннего сравнения значений молекулярного веса SEC/RI и SEC/MALLS. Значения и коэффициенты зависят от композиции на основе полимера (содержания стирола и бутадиен). Коэффициент 1,64 применяли для SSBR, содержащего 10% стирола, коэффициент 1,52 применяли для SSBR с 25% стирола и коэффициент 1,47 применяли для SSBR с 40% стирола. Mp (как SSBR) применяли для расчета молярных отношений TMEDA, (A), (B)/активные участки (I*).
1H-ЯМР-спектроскопию применяли для определения содержания виниловых связей и содержания стирола. Образцы растворяли в дейтерированном хлороформе и получали спектры с применением спектрометра Bruker 200 MГц. Длинный блок относится к стирольным блокам с более чем 6 последовательными стирольными элементарными звеньями. Содержание виниловых связей VC относится к 1,2-полибутадиену, содержащемуся в полибутадиеновой фракции сополимера.
Температуру стеклования определяли с применением DSC Q2000 при следующих условиях:
Измерения для каждого образца проводили по меньшей мере один раз. Измерения включали два цикла нагревания. Для определения температуры стеклования применяли 2-ой цикл нагревания.
Измерения реологических свойств невулканизированных образцов согласно ASTM D 5289-95 проводили с применением безроторного сдвигового реометра (MDR 2000 E) для определения вулканизационных характеристик. Образцы для испытания вулканизировали за t95 при 160°C, в частности, для испытания на твердость и упругий отскок образцы для испытания вулканизировали за t95+5 мин при 160°C. Предел прочности при растяжении и модуль измеряли согласно ASTM D 412 на Zwick Z010. Истирание по DIN измеряли согласно DIN 53516 (1987-06-01). Испытания на твердость по Шору по шкале A (ASTM D 2240) и упругий отскок (ISO 4662) измеряли при 0°C, RT и 60°C. Динамические свойства, то есть тангенс δ при 0°C и 60°C, измеряли с применением динамического спектрометра Eplexor 150N/500N, изготовленного Gabo Qualimeter Testanlagen GmbH (Германия), с прикладыванием сжимающего динамического напряжения 0,2% при частоте 2 Гц. Тепловыделение измеряли согласно ASTM D 623, способ A, на устройстве для испытания на многократный изгиб Doli 'Goodrich'.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ СШИТЫЕ РЕЗИНОВЫЕ КОМПОЗИЦИИ | 2015 |
|
RU2701830C2 |
| ИНИЦИАТОРЫ ПОЛИМЕРИЗАЦИИ | 2013 |
|
RU2666359C2 |
| ШИНА ДЛЯ КОЛЕС ТРАНСПОРТНЫХ СРЕДСТВ | 2017 |
|
RU2731944C2 |
| СМЕСЬ ФУНКЦИОНАЛИЗИРОВАННЫХ ПОЛИМЕРОВ ДЛЯ ШИН | 2016 |
|
RU2716689C2 |
| ПОЛИМЕРЫ, МОДИФИЦИРОВАННЫЕ АМИНОСИЛАНОМ | 2012 |
|
RU2609166C2 |
| МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2009 |
|
RU2504555C2 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2012 |
|
RU2599723C2 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2558597C2 |
| СИЛАНСУЛЬФИДНЫЕ МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2012 |
|
RU2617403C2 |
| ПРОЦЕСС ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО ИНТЕРПОЛИМЕРА ИЛИ МОДИФИЦИРОВАННОГО ПОЛИМЕРА | 2009 |
|
RU2531821C2 |
Изобретение относится к модифицированному эластомерному полимеру. Модифицированный эластомерный полимер получают посредством приведения в контакт живого анионного эластомерного полимера с конкретным модифицирующим концевую группу цепи соединением, причем живой анионный эластомерный полимер содержит по меньшей мере 75 и менее чем 99 мас.% бутадиеновых элементарных звеньев и более чем 1 и менее чем 25 мас.% стирольных элементарных звеньев и характеризуется содержанием виниловых связей от 2 до менее чем 10% исходя из полибутадиеновой фракции живого анионного эластомерного полимера. Изобретение позволяет улучшить сцепление шины при применении вулканизированного модифицированного полимера с мокрым дорожным покрытием, повысить устойчивость к истиранию, улучшить устойчивость к качению. 4 н. и 21 з.п. ф-лы, 4 табл.
1. Модифицированный эластомерный полимер, полученный посредством приведения в контакт живого анионного эластомерного полимера и модифицирующего соединения формулы (I)
где R независимо представляет собой C1-C16алкил или бензил; R'' представляет собой C1-C4алкил; R' выбран из C6-C18арила, C7-C50алкиларила, C1-C50алкила и C2-C50диалкилэфира, где каждая группа необязательно замещена одной или более группами, выбранными из C1-C4алкила, C1-C4алкокси, C6-C12арила, C7-C16алкиларила, ди(C1-7гидрокарбил)амино, бис(три(C1-12алкил)силил)амино, трис(C1-C7гидрокарбил)силила и C1-C12тиоалкила; x представляет собой целое число, выбранное из 1, 2 и 3; y представляет собой целое число, выбранное из 0, 1 и 2; и x+y=3, и
где живой анионный эластомерный полимер содержит по меньшей мере 75 и менее чем 99 мас.% бутадиеновых элементарных звеньев и более чем 1 и до 25 мас.% стирольных элементарных звеньев и характеризуется содержанием виниловых связей от 2 до менее чем 10% исходя из полибутадиеновой фракции живого анионного эластомерного полимера.
2. Модифицированный эластомерный полимер по п. 1, где R независимо выбран из C1-C5алкила, и R' представляет собой C1-C5алкил.
3. Способ получения модифицированного эластомерного полимера по п. 1 или 2, включающий этап приведения в контакт живого анионного эластомерного полимера и модифицирующего соединения формулы (I).
4. Модифицированный эластомерный полимер по п.1, полученный посредством приведения в контакт живого анионного эластомерного полимера с модифицирующим соединением формул (1) и по меньшей мере одним из модифицирующих соединений формул (3), (4), (5) и (6)
где R2, R3, R10, R11, R12, R14, R16, R17 и R18 независимо выбраны из C1-C16алкила и бензила, где алкильные группы для R10, R11 и R12, а также для R16, R17 и R18 могут быть связаны друг с другом таким образом, что образуют кольцо, содержащее два атома Si и N; R1 и R13 независимо выбраны из C1-C4алкила; R4, R9 и R15 независимо выбраны из C6-C18арила, C7 C50алкиларила, C1-C50алкила и C2-C50диалкилэфира, где каждая группа необязательно замещена одной или более группами, выбранными из C1-C4алкила, C1-C4алкокси, C6-C12арила, C7-C16алкиларила, ди(C1-C7гидрокарбил)амино, бис(три(C1-C12алкил)силил)амино, трис(C1-C7гидрокарбил)силила и C1-C12тиоалкила; R5, R6 и R7 независимо выбраны из водорода, C1-C16алкила и C6-C12арила; R8 выбран из C1-C16алкила и C6-C12арила; и R19, R20 и R21 независимо выбраны из водорода и C1-C16алкила; каждый из x и p представляет собой целое число, выбранное из 1 и 2; каждый из y и q представляет собой целое число, выбранное из 1 и 2; x+y=3; и p+q=3, и
где живой анионный эластомерный полимер содержит более чем 75 и менее чем 99 мас.% бутадиеновых элементарных звеньев и более чем 1 и менее чем 25 мас.% стирольных элементарных звеньев и характеризуется содержанием виниловых связей от 2 до менее чем 10% исходя из полибутадиеновой фракции живого анионного эластомерного полимера.
5. Модифицированный эластомерный полимер по п. 4, где в формуле (1) R1 выбран из C1-C4алкила; R3 выбран из C1-C16алкила и бензила; и R4 выбран из C1-C16алкила, C6-C16арила и C7-C16алкиларила.
6. Модифицированный эластомерный полимер по п. 4, где в формулах (3), (4), (5) и (6) R2, R3, R8, R10, R11, R12, R14, R16, R17 и R18 независимо выбраны из C1-C16алкила, R1 и R13 независимо выбраны из C1-C4алкила, R5, R6, R7, R19, R20 и R21 независимо выбраны из водорода и C1-C16алкила, и R4, R9 и R15 независимо выбраны из C1-C16алкила, C6-C15арила и C7-C16алкиларила.
7. Модифицированный эластомерный полимер по п. 4, который получен посредством приведения в контакт живого анионного эластомерного полимера с модифицирующим соединением формулы (1) с получением модифицированного полимера и затем приведения в контакт модифицированного полимера с по меньшей мере одним из модифицирующих соединений формул (3), (4), (5) и (6).
8. Модифицированный эластомерный полимер по п. 4, который получают посредством приведения в контакт живого анионного эластомерного полимера с модифицирующим соединением формулы (1) и модифицирующим соединением формулы (3).
9. Модифицированный эластомерный полимер по любому из пп. 1, 2 и 4-8, отличающийся тем, что количество стирола в живом анионном эластомерном полимере составляет по меньшей мере 2 мас.%.
10. Модифицированный эластомерный полимер по п. 9, отличающийся тем, что живой анионный эластомерный полимер представляет собой сополимер бутадиена и стирола.
11. Модифицированный эластомерный полимер по любому из пп. 1, 2 или 4-8, отличающийся тем, что содержание виниловых связей в полибутадиеновой фракции живого анионного эластомерного полимера составляет от 4 до 9%.
12. Способ получения модифицированного эластомерного полимера по п. 7 или 8, включающий этапы (i) приведения в контакт живого анионного эластомерного полимера по меньшей мере с одним из модифицирующих соединений формул (1) и (2) с получением модифицированного полимера и (ii) приведения в контакт модифицированного полимера с по меньшей мере одним из модифицирующих соединений формул (3), (4), (5) и (6).
13. Способ по п. 12, включающий этапы (i) приведения в контакт живого анионного эластомерного полимера с модифицирующим соединением формулы (1) с получением модифицированного полимера и (ii) приведения в контакт модифицированного полимера с модифицирующим соединением формулы (3).
14. Способ получения модифицированного эластомерного полимера по п.13, отличающийся тем, количество стирола в живом анионном эластомерном полимере составляет по меньшей мере 2 мас.%.
15. Способ получения модифицированного эластомерного полимера по п. 14, отличающийся тем, что живой анионный эластомерный полимер представляет собой сополимер бутадиена и стирола.
16. Способ получения модифицированного эластомерного полимера по любому из пп. 12-15, отличающийся тем, что содержание виниловых связей в полибутадиеновой фракции живого анионного эластомерного полимера составляет от 4 до 9%.
17. Композиция на основе эластомерного полимера, содержащая модифицированный эластомерный полимер по любому из пп. 1, 2, 4-8 и один или более дополнительных компонентов, выбранных из компонентов, которые добавляют в или образуют в результате способа полимеризации, применяемого для получения модифицированного эластомерного полимера, и компонентов, которые остаются после удаления растворителя из способа полимеризации.
18. Композиция на основе эластомерного полимера по п. 17, содержащая одно или более масел для наполнения.
19. Композиция на основе эластомерного полимера по п. 17, содержащая один или более наполнителей.
20. Композиция на основе эластомерного полимера по п. 18, содержащая один или более наполнителей.
21. Композиция на основе эластомерного полимера по п. 19 или 20, отличающаяся тем, что один или более наполнителей выбраны из углеродных нанотрубок, углеродной сажи, диоксида кремния, углерод-кремнеземного двухфазного наполнителя, глины, карбоната кальция и карбоната магния.
22. Композиция на основе эластомерного полимера по п. 19 или 20, отличающаяся тем, что наполнитель выбран из комбинации углеродной сажи с диоксидом кремния, углерод-кремнеземного двухфазного наполнителя, комбинации углерод-кремнеземного двухфазного наполнителя с углеродной сажей и/или диоксидом кремния, и комбинации углеродных нанотрубок с одним или более из углерод-кремнеземного двухфазного наполнителя, углеродной сажи и диоксида кремния.
23. Композиция на основе эластомерного полимера по любому из пп. 17-20, содержащая один или более дополнительных эластомерных полимеров.
24. Композиция на основе эластомерного полимера по п. 21, содержащая один или более дополнительных эластомерных полимеров.
25. Композиция на основе эластомерного полимера по п. 22, содержащая один или более дополнительных эластомерных полимеров.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| ТЕРМОСТОЙКИЙ КАТИОННЫЙ БУРОВОЙ РАСТВОР | 2015 |
|
RU2602262C1 |
| JP 2012172077 A, 10.09.2012 | |||
| JP 2007270020 A, 18.10.2007 | |||
| Способ приготовления белкового кормового продукта на основе соево-мясных композиций | 2015 |
|
RU2607103C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО СОПРЯЖЕННОГО ДИЕНОВОГО ПОЛИМЕРА, МОДИФИЦИРОВАННЫЙ СОПРЯЖЕННЫЙ ДИЕНОВЫЙ ПОЛИМЕР И РЕЗИНОВАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2481361C2 |
| RU 2011141342 A, 10.06.2013. | |||

