ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области к области фармацевтики и предполагает создание эффективного лекарственного средства для лечения ревматоидного артрита и других воспалительных заболеваний на основе карбоксамидного производного изоксазолина, обладающего свойством дезактивировать рецепторы PAR-2 in vivo. Изобретение включает в себя также способ получения активного компонента лекарственного средства, способ создания фармацевтической композиции и способ использования фармацевтической композиции для лечения ревматоидного артрита.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Хронические воспалительные заболевания суставно-связочного аппарата, и в частности, ревматоидный артрит, характеризуются высокой инвалидностью (около 70%) и, как правило, развиваются, начиная с возраста 30-35 лет, приводя к снижению доли трудоспособного населения. Стандартная терапия артритов включает применение нестероидных противовоспалительных препаратов, глюкокортикостероидов. Однако, эти лекарственные средства имеют побочные эффекты. Другие усилия сфокусированы на хирургической установке имплантатов суставов, таких как коленные или тазобедренные имплантаты. Эти способы включают длительные и дорогостоящие хирургические процедуры, а пациенту после операционного вмешательства требуется значительный восстановительный период. Принимая во внимание проблемы, связанные с текущими способами, относящимися к профилактике или лечению артритов и связанных с артритами воспалений, продолжает существовать потребность в новых способах и композициях, используемых для лечения артритов. Одним из эффективных подходом к лечению хронических воспалительных заболеваний, в том числе артрита, представляется регулирование процессов активации PAR-2 рецептора активируемого протеазами. В последние годы стало известно, что активация PAR-2 рецептора серин-протеазами, к которым относится фактор Ха, приводит к протеолитическому отщеплению внеклеточного N-концевого олигопептидного фрагмента. Образующийся в результате этого новый реакционноспособный N-конец способен к обратимому связыванию с поверхностным сайтом рецептора. Это связывание активирует возникновение воспаления, фиброза и разрастание плотных соединительных тканей. Ингибирование фактора Ха препятствует отрыву концевого фрагмента PAR-2 и, таким образом, предотвращает запуск механизма связывания и останавливает развитие патологии.
Карбоксамидные производные изоксазолина могут быть использованы в качестве селективных ингибиторов фактора Ха и могут принимать участие в дезактивации PAR-2 (трансмембранных рецепторов, активируемых протеазами) ин-виво, в этом плане они могут быть использованы в качестве эффективных противовоспалительных средств, в том числе, в качестве средств для лечения ревматоидного артрита.
Применение производных 4,5-дигидроизоксазол-5-карбоксамида в качестве противоартритных средств ранее не было описано. В ряде патентов приводится информация по синтезу соединений, имеющих в своем составе фрагмент 4,5-дигидроизоксазол-5-карбоксамида (US 2005/192302 A1, WO 2012/114223 A1, WO 2005/21516 A1, US 2015/245616 A1, US 2008/262032 A1, US 6747050 B1, WO 2014/160668 A1, US 6583141 B1, WO 2004/48392 A1, US 6747050 B1, WO 2015/121212 A1, WO 2008/111009 A1, WO 2007/29077 A1, US 2008/132542, WO 2010/94126 A1), проявляющих ингибирующую активность относительно фосфодиэстеразы, коэнзима - дельта-9 дезатуразы, каспазы, а также, проявляющие антибактериальную и противогрибковую активность, являющиеся модуляторами рецепторов хемокинов, применяемых для лечения гепатита С и фиброзы печени. В патентах (US 5939418 A, WO 2010/94126 A1, ЕР 874629 B1) производные 4,5-дигидроизоксазол-5-карбоксамида относятся к соединениям, и их пролекарствам, применяемые для профилактики и лечения тромботических состояний у млекопитающих. В данных патентах описано использование производных 4,5-дигидроизоксазол-5-карбоксамидов для ингибирования факторов коагуляции Ха, но при этом не установлена взаимосвязь с циклом дезактивации PAR-2, а основной механизм действия связан с антикоагулятивным действием. Как лекарства против ревматоидного артрита данные соединения не используются, а применяются в качестве антикоагулянтов. Также в качестве лекарственных средств, как ингибиторов образования факторов коагуляции Ха, IХа и тромбина, индуцированные фактором VIIa и тканевым фактором, могут быть использованы соединения, а также их физиологически приемлемые соли, описанные в изобретении WO 2005058868 А1. Изобретение относится к новым замещенным гетероциклическим производным миндальной кислоты, поэтому объект настоящего изобретения не попадает под действие данного патента, как и не попадает под действие четырех других изобретений (US 20050153965, WO 2002042272 А8, US 20050137168 A1, WO 2013098833 A2, US 6339099 B1), где объектом исследования являются соединения-ингибиторы факторов свертывания крови.
В ряде патентов (WO 2014020351 А1, WO 2014020350 А1, ЕР 1806141 A1, US 20090012263 A1, US 20060063930 A1, US 20020103138 A1) можно встретить информацию о том, что высокоактивные антагонисты PAR-2 предложены в качестве эффективного средства для лечения ревматоидного артрита, однако в качестве вторичных индикаций в патентах предложены заболевания дыхательных путей, невралгия, заболевания сердечнососудистой системы, инфаркт миокарда, атопический дерматит и рак. Кроме того, объект данного поиска не попадает под описание структуры Маркуша этих патентов. Авторы патента US 5888529 А предлагают способ лечения послеоперационной непроходимости кишечника. Лечение основано на открытии, что протеиназа-активированный рецептор-2 экспрессируется в мышечных клетках ободочной кишки, а также, что активация PAR-2 ингибирует моторику толстого кишечника. Рецептор PAR-2 активируется, по меньшей мере, частично, с помощью триптазы и химазы. Таким образом, использование рецепторов PAR-2 в качестве мишени для лечения ревматоидного артрита путем дезактивации производными 4,5-дигидроизоксазол-5-карбоксамида в настоящее время является свободной нишей и может быть использовано для создания изобретения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее йзобретение ставит своей целью создание новых веществ, способных дезактивировать трансмембранные рецепторы, активируемые протеазами (PAR-2) путем ингибирования протеазы фактора Ха, участвующей в цикле дезактивации PAR-2 в терапевтически эффективном количестве.
Поставленная цель достигается созданием новых соединений, представляющих собой карбоксамидные производные изоксазолина общей формулы 1,
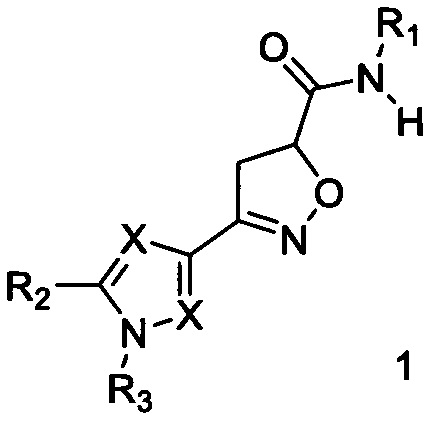
где X представляет атом азота, либо углерода: заместитель R1 представляет собой арил или гетероарил, содержащий атома азота 1 до 3; заместитель R2, R3 могут быть выбраны из следующего набора - водород, C1-С6 алкил, арил, алкокси, галоген, трифторметил.
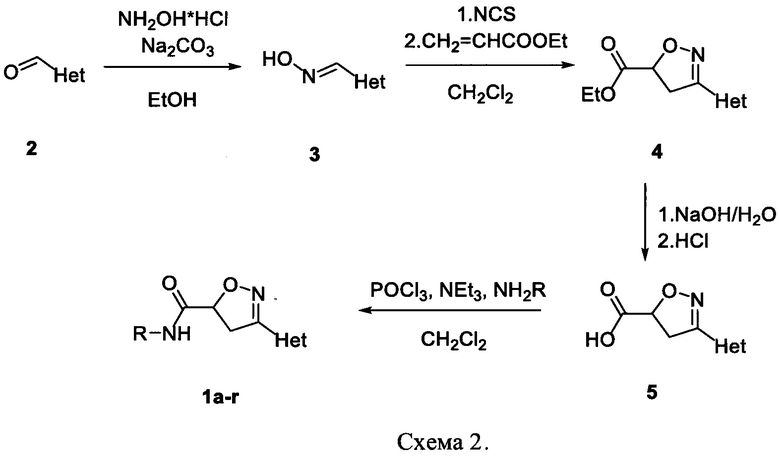
Изобретение также включает в себя фармацевтически приемлемые соли, сольваты и фармакологически функциональные производные соединений общей формулы 1, а также способ их получения. Метод синтеза соединений общей формулы 1 заключается в последовательном проведении 4-ех стадий (схема 1): 1) образования оксима из альдегида; 2) циклоприсоединения алкилакрилата к оксиму с образованием сложного эфира 5-изоксазолкарбоной кислоты, содержащей гетероциклический заместитель в положении 3; 3) гидролизе изоксазолкарбоксилата до изоказол-5-карбновой кислоты; 4) получении изоксазол-3-карбоксамида взаимодействием соответствующей кислоты с амином в присутствии хлорокиси фосфора и триэтиламина.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Для удобства приведены термины, использованные в описании, примерах и прилагаемой формуле изобретения.
«Активный компонент» (лекарственное вещество, лекарственная субстанция,) означает физиологически активное вещество синтетического или иного происхождения (биотехнологического, растительного, животного, микробного и прочего), обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и других готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы I и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, противомикробные агенты, любриканты, агенты пролонгированной доставки. Примерами суспендирующих агентов являются полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями.
Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные такие как мази и кремы, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения. «Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные.
Соли кислот также могут быть специально получены реакцией кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Фармацевтически приемлемый» включают молекулярные объекты и композиции, которые не вызывают неблагоприятную, аллергическую или другую вредную реакцию при введении животному или человеку соответственно. «Фармацевтически приемлемый носитель» включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, изотонические средства и средства, замедляющие всасывание, и тому подобное. Применение таких сред и средств для фармацевтических активных веществ известно в данной области техники. Рассматривается применение в терапевтических композициях любых стандартных сред или средств, за исключением тех, которые несовместимы с активным ингредиентом. В композиции также могут быть включены дополнительные активные ингредиенты.
Объектом данного изобретения являются новые карбоксамидные производные изоксазолина общей формулы 1 и их фармацевтически приемлимые соли,

где X представляет атом азота, либо углерода: заместитель R1 представляет собой арил или гетероарил, содержащий атома азота 1 до 3; заместитель R2, R3 могут быть выбраны из следующего набора - водород, C1-С6 алкил, арил, алкокси, галоген, трифторметил.
Более предпочтительно, соединения для лечения воспалительных заболеваний могут быть выбраны из следующего ряда:
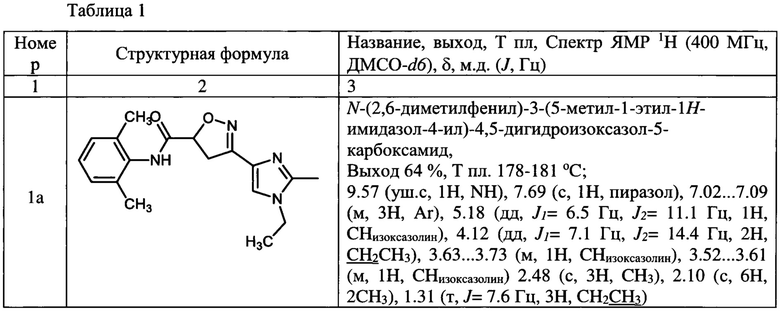
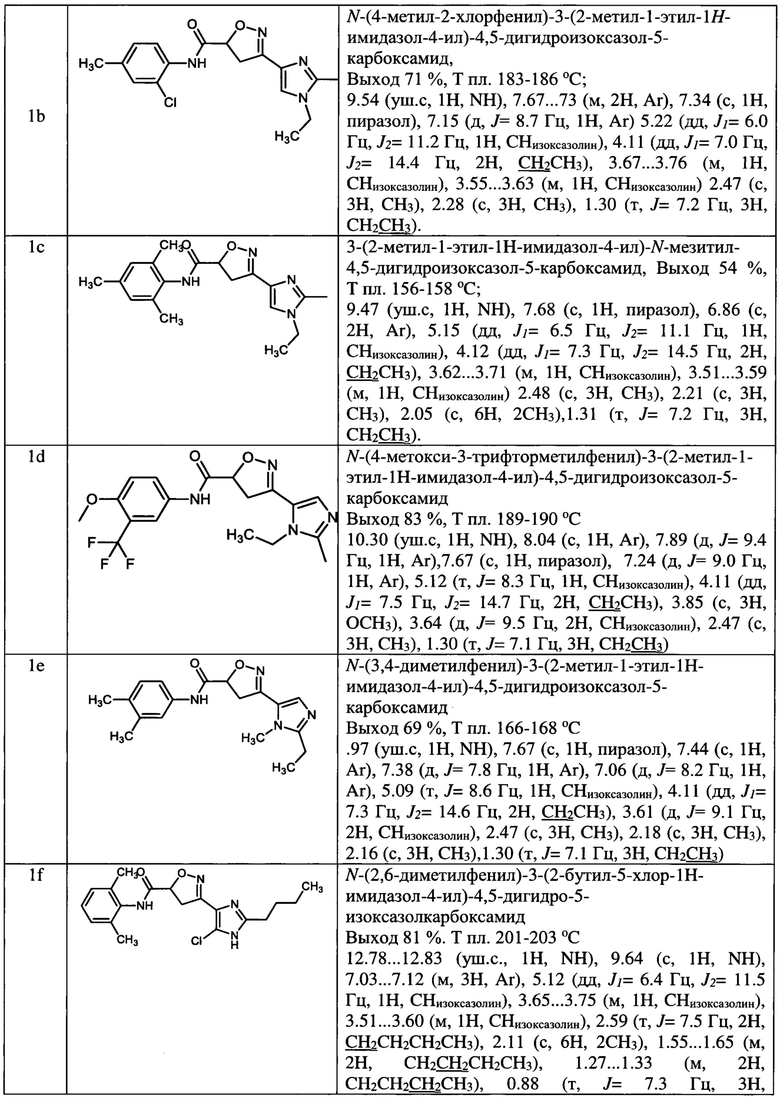
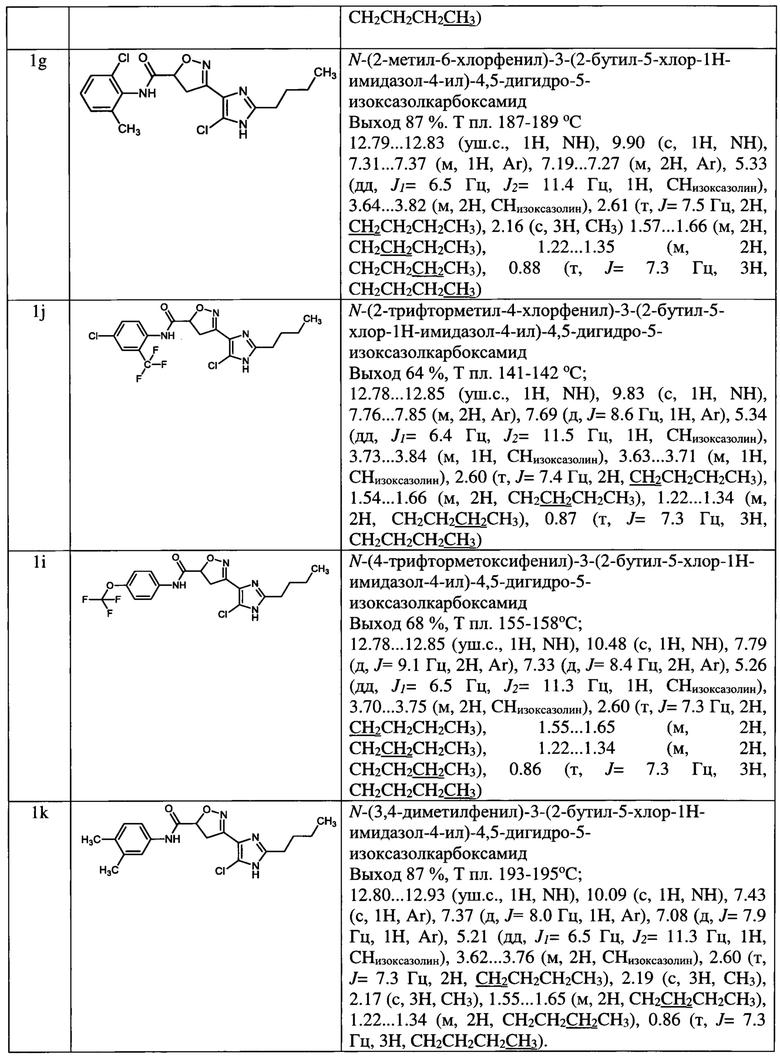
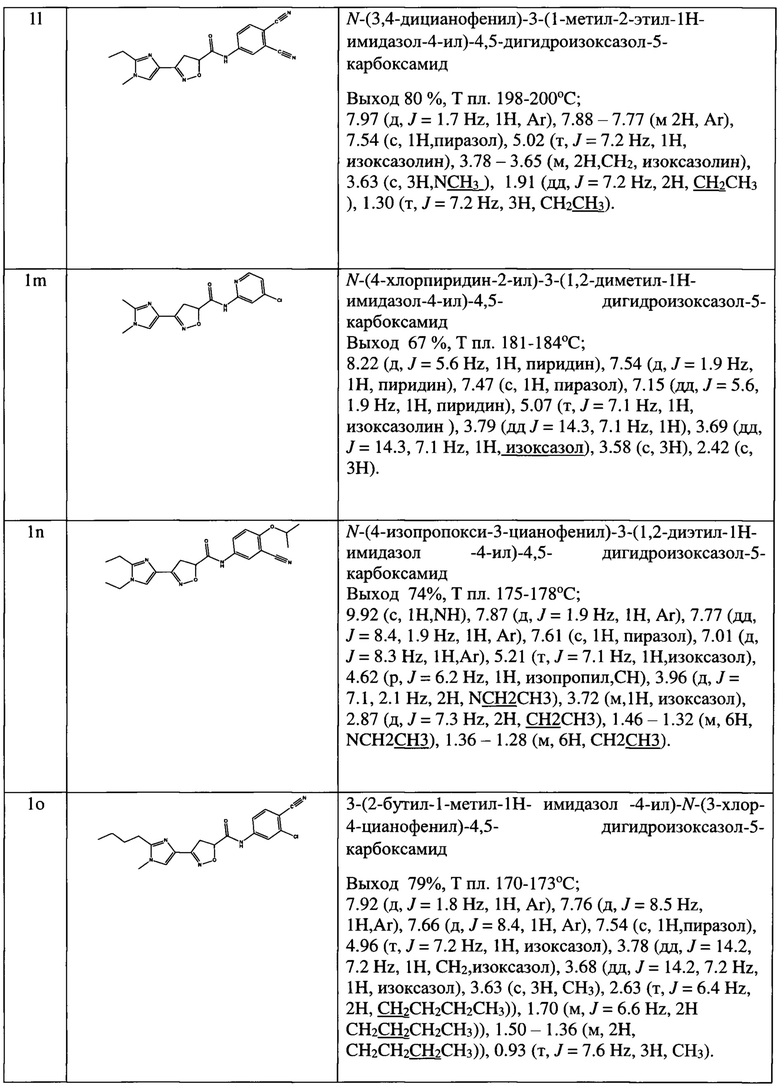
N-(2,6-диметилфенил)-3-(5-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид (1а);
N-(4-метил-2-хлорфенил)-3-(2-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид (1b);
3-(2-метил-1-этил-1Н-имидазол-4-ил)-К-мезитил-4,5-дигидроизоксазол-5-карбоксамид (1с);
N-(4-метокси-3-трифторметилфенил)-3-(2-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид (1d);
N-(3,4-диметилфенил)-3-(2-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид (1е);
N-(2,6-диметилфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид (1f);
N-(2-метил-6-хлорфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид (1g);
N-(2-трифторметил-4-хлорфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид (1h);
N-(4-трифторметоксифенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид (1i);
N-(3,4-диметилфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид (1j);
N-(3,4-дицианофенил)-3-(1-метил-2-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид (1k);
N-(4-хлорпиридин-2-ил)-3-(1,2-диметил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид (1l);
N-(4-изопропокси-3-цианофенил)-3-(1,2-диэтил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид (1m);
3-(2-бутил-1-метил-1Н-имидазол-4-ил)-N-(3-хлор-4-цианофенил)-4,5-дигидроизоксазол-5-карбоксамид (1n);
3-(1-метил-2-этил-1Н-имидазол-4-ил)-N-(4-(трифторметил)фенил)-4,5-дигидроизоксазол-5-карбоксамид (1о) и их фармацевтически приемлемых солей и сольватов.
Объектом данного изобретения является способ получения карбоксамидных производных изоксазолинов общей формулы 1, заключающийся в последовательном проведении 4-ех стадий (схема 2): а) образования оксима из альдегида, б) циклоприсоединения алкилакрилата с образованием сложного эфира 5-изоксазолкарбоной кислоты, содержащей гетероциклический заместитель в положении 3 в) гидролизе изоксазолкарбооксилата г) получении изоксазолкарбоксамида общей взаимодействием соответствующей кислоты с аминов в присутствии хлорокиси фосфора и триэтиламина.
Предметом настоящего изобретения является способ дезактивации рецептора PAR-2, заключающийся во введении в контакт указанного рецептора и соединения общей формулы 1, либо его фармацевтически приемлемые соли, сольваты.
Предметом настоящего изобретения является активный компонент, обладающий свойствами, позволяющими ему дезактивировать рецептор PAR-2, представляющий собой соединение общей формулы 1 или его фармацевтические-приемлемые соли, сольваты.
Предметом настоящего изобретения является фармацевтическая композиция, содержащая соединение общей формулы 1 и (или) их фармацевтически приемлемую соль или активный компонент в терапевтически приемлемом количестве, пригодная для лечения воспалительных заболевания, в том числе ревматоидного артрита. Клиническая дозировка фармацевтической композиции или лекарственного средства, содержащих в качестве активного начала соединения общей формулы 1 у пациентов может корректироваться в зависимости от: терапевтической эффективности и биодоступностиактивных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента.
Предметом настоящего изобретения является средство для лечения и (или) профилактики ревматоидного артрита и (или) другого воспалительного процесса в виде мази, таблетки, капсулы, помещенное в фармацевтически приемлемую упаковку, содержащее соединение общей формулы 1 или его фармацевтически приемлемую соль в качестве активного компонента или фармацевтическую композиции по п.6 в терапевтически эффективно количестве, предназначенную для лечения воспалительных заболеваний, в том числе ревматоидного артрита и других заболеваний, связанных с повышенной активностью рецепторов PAR-2.
Предметом настоящего изобретения является способ лечения заболевания, связанного с повышенной активностью рецепторов PAR-2, заключающийся в местном введении (на очаг воспаления), в терапевтическом количестве активного компонента или фармацевтической композиции или лекарственного средства.
Предметом настоящего изобретения является способ лечения заболевания, связанного с повышенной активностью рецепторов PAR-2, заключающийся в пероральном введении в терапевтическом количестве активного компонента или фармацевтической композиции по или лекарственного средства.
Ниже перечислены экспериментальные процедуры, которые служат не для ограничения данного патента, а лишь иллюстрируют настоящее изобретение, в частности способ получения соединений, среди которых может выбран активный компонент для лечения воспалительных заболеванием посредством дезактивации рецептора PAR-2, метод.
Пример 1. Общая методика получения оксимов 3.
В 100 мл этилового спирта растворяли 0,11 моль карбальдегида 2, к полученному раствору прибавляли смесь 30,21 г (0,43 моль) гидроксиламина гидрохлорида и 34,50 г (0,32 моль) карбоната натрия в 100 мл дистиллированной воды. Реакционную смесь перемешивали при кипении в течение 16 часов. После истечения времени этиловый спирт упаривали в вакууме на ротационном испарителе, выпавший осадок отфильтровывали, промывали 2×20 мл дистиллированной водой, сушили при 50°С в течение 24 ч.
Получили 10,01 г оксима 2-метил-1-этил-1H-имидазол-4-карбальдегида 3а в виде порошка белого цвета. Выход продукта составил 60%.
Т.пл. 135-137°С. Спектр ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д. (J, Гц): 8.04 (с, 1H, СН), 7.99 (с, 1H, СН), 7,52 (с, 1H, пиразол), 7.28 (с, 1H, пиразол), 4.06 (дд, J1=7.3 Гц, J2=14.5 Гц, 2Н, СH2СН3), 2.34 (с, 3Н, СН3), 1.28 (т, J=7.2 Гц, ЗН, СH2СН3).
Получили 8,71 г оксима 2-бутил-5-хлор-1H-имидазол-4-карбальдегида 3b в виде порошка белого цвета. Выход продукта составил 75%.
Т.пл. 143-145°С. Спектр ЯМР 1Н (400 МГц, ДМСО-d6), δ, м.д. (J, Гц): 11.73…12.55 (уш. с, 1Н, NH), 7.85 (с, 1Н, СН), 7.24 (с, 1Н, CН), 2.66 (т, J=7.4 Гц, 1H, СН2СН2СН2СН3), 2.57 (т, J=7.4 Гц, 2Н, СН2СН2СН2СН3), 1.52…1.65 (м, 2Н, СН2СН2СН2СН3), 1.20…1.36 (м, 2Н, СН2СН2СН2СН3), 0.87 (т, J=7.4 Гц, 3Н, СН2СН2СН2СН3).
Пример 2. Общая методика получения этилового эфира 3-(гетерил)-4,5-дигидро-5-изоксазолкарбоновой кислоты 4,
К суспензии N-хлорсукцинимида (1, 1 экв.) в хлористом метилене (1 г - 10,6 мл) добавляют порциями оксим 2 (1 экв.), затем реакционную массу перемешивают при комнатной температуре в течение 12 часов. Далее к смеси прибавляют этиловый эфир акриловой кислоты (2 экв.) и триэтиламин (1,2 экв.), поддерживают температуру не выше 25°С. Далее реакционную массу перемешивают при комнатной температуре в течение 90 мин. Органический раствор промывают дистиллированной водой, насыщенным раствором NaCl, сушат над безводным сульфатом натрия, пропускают через слой силикагеля и упаривают в вакууме на ротационном испарителе. Получают соединения формул 4 в виде масла.
Пример 3 Общая методика получения 3-(гетерил)-4,5-дигидро-5-изоксазолкарбоновых кислот 5.
Соответствующие соединения эфира 3 (1 экв.) прибавляют порциями в 10%-ный раствор NaOH в 50% метаноле. Реакционную массу охлаждают, так чтобы температура смеси не превышала 25°С. Далее смесь перемешивают при комнатной температуре в течение 12 часов. Затем частично упаривают массу на ротационном испарителе. Далее примеси экстрагируют толуолом, водный слой подкисляют концентрированной соляной кислотой (36%) до рН=3.0, при охлаждении. Выпавший осадок отфильтровывают, промывают охлажденной водой, сушат при 40°С в течение 24 ч. Получают аналитически чистые соответствующие карбоновые кислоты 5 в виде порошка.
Пример 4. Общая методика синтеза амида карбоновой кислоты соединений формул 6.
Растворяют соответствующей кислоты 5 (1 экв.) в дихлорметане (20 мл на 1 г вещества), к раствору прибавляют триэтиламин (2 экв.) и соответствующий амин (1 экв.), полученную смесь перемешивают 1 час при комнатной температуре. Затем к охлажденной реакционной массе прибавляют 1,5 экв. хлорокиси фосфора, при температуре не выше 10°С, далее смесь перемешивают при комнатной температуре в течение 12 часов.
После этого органический слой последовательно промывают: дистиллированной водой, 7%-ным раствором K2СО3, насыщенным раствором NaCl; сушат над безводным сульфатом натрия, упаривают под вакуумом на ротационном испарителе, сушат при 40°С в течение 24 ч.
Амид дважды перекристаллизовывают: 1) в 10 объемах метанола; 2) отфильтрованный и высушенный осадок далее растворяют в 2-х объемах этилацетата при нагревании и прибавляют 8 объемов изопропилового спирта, раствор охлаждают, выпавший осадок фильтруют, промывают изопропиловым спиртом. Получают аналитически чистые соединений формул 6 в виде порошка белого цвета.
Выходы, данные идентификации амидов представлены в таблице 1

Пример 5. Исследование противоартритной и противовоспалительной активности на животных моделях.
Исследование в модели адьювант-индуцированного артрита выполнено на самцах линии Wistar. Соединения 1а-1р исследованил в дозах 5-500 мг/кг. Раствор препаратов готовили с использованием 0,5% Твин 80 и вводили внутрижелудочно. Контроьным животным вводили растворитель в эквивалентном объеме.
Исследование в модели коллаген-индуцированного артрита выполнено на самцах линии Lewis. Соединения 1а-1р исследованил в дозах 5-500 мг/кг. Раствор препаратов готовили с использованием 0,5% Твин 80 и вводили внутрижелудочно. Контрольным животным вводили растворитель в эквивалентном объеме.
В качестве препарат сравнения использовали Индометацин в дозе 20 мг/кг, который вводили перорально в объеме 10 мл/кг Тест на гипотензивное действие (понижение внутриглазного давления). Препараты вводили 14 дней.
В модели адьювант-индуцированного артрита введение начинали на следующий день после введения адъюванта. В модели коллаген-индуцированного артрита - после рандомизации группы. Рандомизацию проводили по баллам проявления признаков артрита так, чтобы средний счет в группе был 0,25-0,75.
Терапевтическую активность препаратов оценивали визуально по шкале выраженности признаков артрита, в модели адьювант-индуцированного артрита кроме этого также проводили измерение объема лапок. Также следили за изменением массы и смертностью по сравнению с контрольными группами.
Адъювант-индуцированный артрит
Полнй адъювант Фрейнда вводили крысам субплантарно в правую заднюю лапку в обьеме 0,1 мл. В связи с тем, что в клинической практике планируется пероральное ввденеие человеку, исследдуемые препараты вводили внутрижелудочно при помощи зонда в дозах 0-500 мг/кг.
Для оцентки противоспалительного действия препарата оценивали первичную реакцию (отек на правой лапке) онкометрически на 3-день после инъекции адъюванта. Для оценки терапевтического действия препарата оценивали вторичную иммунологическую реакцию (отек на левой лапке) на 14 день после ввдениея адъюванта. Следили за развитием воспалительной реакции и массой тела. Дополнительно учитываои симптомы генерализированной реакии организма на ведение адъюванта: отек ушей, полиартрит, ухудшение общего состояния, снижение массы тела, гибель.
Коллаген индуцированный артрит
Животным вводили внтурикожно воснование хвоста раствор коллалена второго типа(2 мг/кг) в полном адъюванте Фрейнда, соддержащем H37Ra (2,5 мг/мл) в объеме 100 мкл. После проявления признаков артрита (на 21 день) животных рандомизировани по группам.
Эффективность исследуемых препаратов оценивали на основе измерения площади кривой (AUC), построенной на основе баллов по шкале. Оценивают, как потенциальную терапевтическую эффективность по индивидуальным лапам с значением баллов >0 в момент рандомизации). Так и потенциальнуюпрофилактическую эффективность (по индивидуальным лапам со значением баллов больше 0 в момент рандомизации.
Пример 6. Получение фармацевтической композиции в форме капсул с гранулятом препарата.
Примерный состав компонентов для приготовления 5, 725 кг гранулята с содержанием основного вещества 30%:
1) Активный компонент 1а-1р, просеяный- 2,813 кг
2) МКЦ (микрокристаллическая целлюлоза) просеяная - 1,831 кг
3) Лактоза просеяная 1,745 кг
4) Аэросил просеяный 0,075 кг
5) Коллидон просеяный 0,367 кг, Методика приготовления гранулята
В реактор для получения загружают 0,344 кг коллидона, 3,088 кг воды очищеной, перемешивают реакционную массу 5 минут.В лабораторный миксер-гранулятор загружают 2,640 кг препарата, 1,716 МКЦ, 1,636 кг лактозы 0,201 кг крахмала, 0,069 кг аэросила. Перемешивают смесь 30 минут, со скоростью 200 оборотов в винуту. Далее задают вращение миксера 500 оборотов в минуту и вносят раствор коллидона. Перемешивают смесь 30 минут со скоростью 1500 оборотов в минуту. Полученную смесь выгружают из миксера. Перед началом операции гранулирвоания в емкость влажного гранулятора устанавливают сито с размером 1 мм.
Устанавливают значения вращения вала 120 об/минуту и перемешивают смесь в течение 25 минут. Выгружают влажный гранулят и переносят его в устройство для сушки гарнулята. На панели устройства для сушки гранулята задают следующие параметры - Т приточного воздуха - 50 С, расход воздуха 78 м3, время фазы 22 минуты. Далее готовый гранулят калибруют на сите с размером отверстий 1 мм. На выходе получают 5,725 кг гранул препарата (86%).
Получение капсул препарата
В бункер для капсул загружают 0,17 кг тверых желатиновых капсул (1700 шт). В бункер для наполнителя загружают 0,400 кг гранулята препарата. Включают устройство лдя капсулирования. На выходе получают 1700 капсул препарата с содержанием препарата 90 мг.
Приведенное выше описание говорит о принципах настоящего изобретения с примерами, приводимыми для целей иллюстрации, однако, будет понятно, что осуществление настоящего изобретения охватывает все обычные варианты, адаптации и/или модификации, которые входят в объем следующей далее формулы изобретения и ее эквивалентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2-АРИЛЗАМЕЩЕННЫЕ N-АРИЛИМИДАЗОЛИНЫ СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ЦИКЛООКСИГЕНАЗЫ-2, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2014 |
|
RU2565667C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИНА, ОБЛАДАЮЩИЕ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2357958C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АГОНИСТЫ РЕЦЕПТОРОВ ЖЕЛЧНЫХ КИСЛОТ TGR5, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2543485C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ МАКУЛЯРНОЙ ДЕГЕНЕРАЦИИ | 2012 |
|
RU2576380C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2233272C2 |
| БЕНЗОПИРАНОВЫЕ И БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ РI3K И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2506267C2 |
| ТИЕНОПИРИДОН КАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2005 |
|
RU2374250C2 |
| ПРОИЗВОДНЫЕ 4,5-ДИГИДРО-1H-ПИРАЗОЛА, ОБЛАДАЮЩИЕ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2281941C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 4,5-ДИГИДРО-1H-ПИРАЗОЛА, ИМЕЮЩИЕ CB-АНТАГОНИСТИЧЕСКУЮ АКТИВНОСТЬ | 2002 |
|
RU2299199C2 |
| НОВЫЕ АНТАГОНИСТЫ 5-НТ2 | 2016 |
|
RU2706229C2 |
Изобретение относится к карбоксамидным производным изоксазолинового ряда общей формулы 1, обладающим свойствами, позволяющими ему дезактивировать рецепторы PAR-2 (рецепторы, активируемых протеазами), а также к способу дезактивации рецептора PAR-2, а также к активному компоненту формулы 1, обладающему противовоспалительным действием. В общей формуле 1 X представляет атом азота, либо С-Н, либо С-Cl; заместитель R1 представляет собой 2,6-диметилфенил, 4-метил-2-хлорфенил, мезитил, 4-метокси-3-трифторметилфенил, 3,4-диметилфенил, 2-метил-6-хлорфенил, 2-трифторметил-4-хлорфенил, 4-трифторметоксифенил, 3,4-дицианофенил, 4-хлорпиридин-2-ил, 4-изопрпопокси-3-цианофенил, 3-хлор-4-цианофенил, 4-(трифторметилфенил); заместитель R2, R3 могут быть выбраны из следующего набора - водород, C1-С6 алкил. Соединения могут быть использованы для лечения воспалительных заболевания, в частности ревматоидного артрита, а также других заболеваний, лечение которых может быть связано с дезактивацией рецепторов PAR2. 3 н. и 1 з.п. ф-лы, 1 табл., 6 пр.
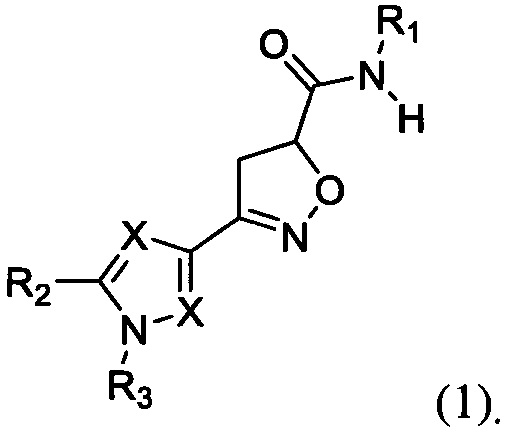
1. Карбоксамидные производные изоксазолина общей формулы 1,
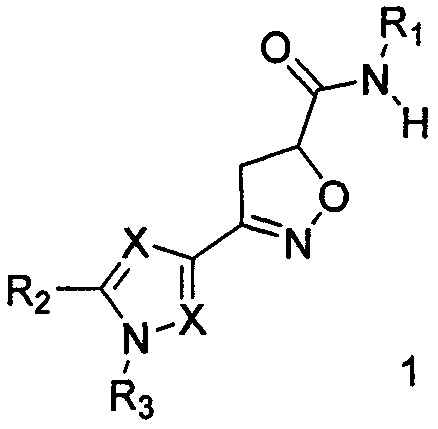
где X представляет атом азота, либо С-Н, либо С-Cl; заместитель R1 представляет собой 2,6-диметилфенил, 4-метил-2-хлорфенил, мезитил, 4-метокси-3-трифторметилфенил, 3,4-диметилфенил, 2-метил-6-хлорфенил, 2-трифторметил-4-хлорфенил, 4-трифторметоксифенил, 3,4-дицианофенил, 4-хлорпиридин-2-ил, 4-изопрпопокси-3-цианофенил, 3-хлор-4-цианофенил, 4-(трифторметилфенил); заместитель R2, R3 могут быть выбраны из следующего набора - водород, C1-С6 алкил.
2. Соединения по п. 1, выбранные из ряда соединений
N-(2,6-диметилфенил)-3-(5-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид;
N-(4-метил-2-хлорфенил)-3-(2-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид;
3-(2-метил-1-этил-1Н-имидазол-4-ил)-N-мезитил-4,5-дигидроизоксазол-5-карбоксамид;
N-(4-метокси-3-трифторметилфенил)-3-(2-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид;
N-(3,4-диметилфенил)-3-(2-метил-1-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид;
N-(2,6-диметилфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид;
N-(2-метил-6-хлорфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид;
N-(2-трифторметил-4-хлорфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид;
N-(4-трифторметоксифенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид;
N-(3,4-диметилфенил)-3-(2-бутил-5-хлор-1Н-имидазол-4-ил)-4,5-дигидро-5-изоксазолкарбоксамид;
N-(3,4-дицианофенил)-3-(1-метил-2-этил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид;
N-(4-хлорпиридин-2-ил)-3-(1,2-диметил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид;
N-(4-изопропокси-3-цианофенил)-3-(1,2-диэтил-1Н-имидазол-4-ил)-4,5-дигидроизоксазол-5-карбоксамид;
3-(2-бутил-1-метил-1Н-имидазол-4-ил)-N-(3-хлор-4-цианофенил)-4,5-дигидроизоксазол-5-карбоксамид;
3-(1-метил-2-этил-1Н-имидазол-4-ил)-N-(4-(трифторметил)фенил)-4,5-дигидроизоксазол-5-карбоксамид.
3. Способ дезактивации рецептора PAR-2 (рецептора, активируемого протеазами), заключающийся во введении в контакт указанного рецептора и соединения общей формулы 1 по п.1-2 в терапевтически эффективном количестве.
4. Активный компонент, представляющий собой соединение общей формулы 1 по п. 1-2, обладающий свойствами, позволяющими ему оказывать противовоспалительное действие на организм.
| WO 2010094126 A1, 26.08.2010 | |||
| Кабель с масляным наполнением | 1931 |
|
SU26361A1 |
| WO 2012101453 A1, 02.08.2012 | |||
| Способ концентрирования золота | 1979 |
|
SU874629A1 |

