ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[001] Настоящее изобретение относится к лечению и/или предупреждению кожных инфекций, ассоциированных с IL-4R-связанными заболеваниями. В частности, настоящее изобретение относится к введению антагонистов рецепторов интерлейкина-4 (IL-4R) для снижения кожных инфекций у нуждающегося в этом пациента.
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
[002] Кожные инфекции, как правило, возникают в местах повреждения кожи, образующихся вследствие, например, атопического дерматита, ожогов, трещин кожи, порезов, волдырей, укусов насекомых, послеоперационных ран, внутривенных инъекций лекарственных средств или в местах введения внутривенных катетеров, или длительного применения местных стероидов. Кожные инфекции могут быть локализованными или диффузными с тяжелым воспалением слоев эпидермиса, дермы или подкожных слоев кожи. Они могут быть вызваны разными микроорганизмами, в том числе без ограничения Staphylococcus aureus, Streptococcus spp., вирусом простого герпеса, вирусом контагиозного моллюска, и грибами, такими как Microsporum spp. и Trichophyton spp.
[003] Атопический дерматит (AD) является хроническим/рецидивирующим воспалительным заболеванием кожи, которое характеризуется выраженным зудом (например, сильным раздражением) и шелушащимися или сухими экзематозными повреждениями. AD часто ассоциируется с другими атопическими нарушениями, такими как аллергический ринит и астма. Пациенты с атопическим дерматитом восприимчивы к тяжелым кожным инфекциям, вызываемым бактериями и вирусами, в том числе без исключением S. aureus и вирусом простого герпеса. S. aureus вызывает тяжелые локализованные и диффузные (например, импетиго) кожные инфекции. Колонизация и инфицирования повреждений S. aureus существенно влияет на активность и тяжесть заболевания AD.
[004] Типичные виды лечения включают лосьоны и увлажнители для местного применения, антибиотики, противовирусные и противогрибковые средства. Однако большинство методик лечения оказывает лишь временное, неполное, симптоматические облегчение. Кроме того, у пациентов с AD со степенью тяжести от средней до тяжелой длительное применение местных кортикостероидов или ингибиторов кальциневрина может приводить к повышенному риску инфекций кожи микроорганизмами. Таким образом, в данной области существует необходимость в новых целенаправленно воздействующих терапевтических препаратах для лечения и/или предупреждения кожных инфекций.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[005] Согласно определенным аспектам настоящего изобретения предложены способы лечения, предупреждения или уменьшения тяжести кожной инфекции у субъекта. Также включены способы уменьшения восприимчивости к кожной инфекции или уменьшения риска воспаления, возникающего в результате инфицирования микроорганизмами субъекта. Согласно определенным вариантам осуществления настоящее изобретение предлагает способы улучшения барьерной функции кожи с целью уменьшения колонизации микроорганизмами кожи субъекта. Способы по настоящему изобретению также предусматривают введение нуждающемуся в этом субъекту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста рецепторов интерлейкина-4 (IL-4R). Согласно определенным вариантам осуществления фармацевтическую композицию вводят подкожно в дозе 75-600 мг.
[006] Согласно определенным вариантам осуществления кожная инфекция может являться бактериальной инфекций или вирусной инфекцией. Согласно определенным вариантам осуществления кожная инфекция может быть вызвана микроорганизмом, выбранным из группы, состоящей из Staphylococcus aureus, Streptococcus spp., Pseudomonas aeruginosa, Bacteroides spp., вируса простого герпеса, вируса контагиозного моллюска, вируса Коксаки, вируса коровьей оспы, Candida albicans, Microsporum spp., Trichophyton spp., Penicillium spp., Cladosporium spp., Alternaria spp. и Aspergillus spp. Согласно определенным вариантам осуществления кожная инфекция выбрана из группы, состоящей из импетиго, целлюлита, инфекционного дерматита, герпетической экземы, фолликулита, инфицированного волдыря, микоза, отрубевидного лишая, инфекции, вызываемой Staphylococcus aureus, и инфекции, вызываемой Streptococcus.
[007] Согласно определенным вариантам осуществления кожная инфекция вызвана S. aureus. Согласно некоторым вариантам осуществления колонизация S. aureus на коже уменьшается при введении терапевтически эффективного количества антагониста IL-4R.
[008] Согласно определенным вариантам осуществления настоящее изобретение предлагает способы лечения или предупреждения кожной инфекции или уменьшения колонизации микроорганизмами кожи субъекта, где способы предусматривают последовательное введение субъекту от приблизительно 50 мг до приблизительно 600 мг фармацевтической композиции, содержащей антагонист IL-4R, в начальной дозе с последующим введением одной или нескольких вторичных доз. Согласно определенным вариантам осуществления каждая начальная доза и одна или несколько вторичных доз содержат от приблизительно 75 мг до приблизительно 300 мг антагониста IL-4R. Согласно определенным вариантам осуществления антагонист IL-4R вводят в начальной дозе 600 мг с последующим введением одной или нескольких вторичных доз, где каждая вторичная доза содержит 300 мг. Согласно данному аспекту изобретения фармацевтическую композицию можно вводить субъекту с частотой введения дозы, например, один раз в неделю, один раз в 2 недели, один раз в 3 недели или один раз в 4 недели. Согласно одному варианту осуществления каждую вторичную дозу вводят через 1 неделю сразу после непосредственно предшествующей дозы. Согласно одному варианту осуществления антагонист IL-4R вводят в начальной дозе 300 мг с последующим введением 3-15 вторичных доз, где каждая вторичная доза содержит 300 мг и ее вводят еженедельно.
[009] Согласно определенным вариантам осуществления настоящее изобретение предлагает способы лечения или предупреждения кожной инфекции у субъекта, где кожная инфекция связана с IL-4R-ассоциированным заболеванием или нарушением, например, атопическим дерматитом, астмой или аллергией. Согласно одному варианту осуществления у субъекта имеется атопический дерматит со степенью тяжести от средней до тяжелой.
[010] Согласно связанному аспекту настоящее изобретение предлагает способы улучшения барьерной функции кожи путем введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции, содержащей антагонист IL-4R. Согласно определенным вариантам осуществления барьерная функция кожи улучшается при поврежденной коже у пациента с атопическим дерматитом. Согласно определенным вариантам осуществления улучшение барьерной функции кожи при введении антитела к рецептору IL-4R выбрано из группы, состоящей из: (i) по меньшей мере 10% повышения от исходного уровня показателя увлажнения рогового слоя (SCH); (ii) по меньшей мере 20% снижения от исходной уровня показателя трансэпидермальной потери влаги (TEWL) и (iii) снижения значения pH кожной поверхности до значения кислого pH.
[011] Иллюстративные антагонисты IL-4R, которые можно использовать в контексте способов по настоящему изобретению, включают, например, низкомолекулярные химические ингибиторы IL-4R или его лигандов (IL-4 и/или IL-13), или биологические средства, которые целенаправленно воздействуют на IL-4R или его лиганды. Согласно определенным вариантам осуществления антагонист IL-4R представляет собой антигенсвязывающий белок (например, антитело или его антигенсвязывающий фрагмент), который связывается с IL-4Rα-цепь и блокирует передачу сигналов с участием IL-4, IL-13 или как IL-4, так IL-13. Согласно одному варианту осуществления антитело или его антигенсвязывающий фрагмент, которые специфически связываются с IL-4R, содержит участки, определяющие комплементарность (CDR), в паре последовательностей вариабельного участка тяжелой цепи (HCVR)/ вариабельного участка легкой цепи (LCVR) под SEQ ID NO: 1/2. Согласно определенным вариантам осуществления антитело или его антигенсвязывающий фрагмент содержит CDR тяжелой цепи (HCDR1) с аминокислотной последовательностью под SEQ ID NO: 3; HCDR2 с аминокислотной последовательностью под SEQ ID NO: 4; HCDR3 с аминокислотной последовательностью под SEQ ID NO: 5; CDR легкой цепи (LCDR1) с аминокислотной последовательностью под SEQ ID NO: 6; LCDR2 с аминокислотной последовательностью под SEQ ID NO: 7 и LCDR3 с аминокислотной последовательностью под SEQ ID NO: 8. Одним таким типом антигенсвязывающего белка, который можно использовать в контексте способов по настоящему изобретению, является антитело к IL-4Rα, такое как дупилумаб.
[012] Согласно некоторым вариантам осуществления фармацевтическую композицию вводят пациенту подкожно или внутривенно.
[013] Согласно определенным вариантам осуществления фармацевтическую композицию вводят пациенту до, после второго терапевтического средства или одновременно с ним. Согласно некоторым вариантам осуществления второе терапевтическое средство выбрано из группы, состоящей из антибактериального средства, противовирусного средства, противогрибкового средства, другого ингибитора IL-4R, ингибитора IgE, кортикостероида (например, местного кортикостероида), нестероидного противовоспалительного лекарственного средства (NSAID) и IFNγ.
[014] Согласно некоторым вариантам осуществления настоящее изобретение предлагает применение антагониста IL-4R по настоящему изобретению в изготовлении лекарственного препарата для лечения, или снижения, или предупреждения кожной инфекции у пациента.
[015] Другие варианты осуществления по настоящему изобретению будут очевидны из обзора следующего подробного описания.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[016] На фигуре 1 показано процентное изменение BSA от исходного уровня у пациентов, получавших 75 мг, 150 мг или 300 мг антитела к IL-4R, по сравнению с плацебо для исследования в примере 1.
[017] На фигуре 2 показано процентное изменение согласно IGA от исходного уровня у пациентов, получавших 75 мг, 150 мг или 300 мг антитела к IL-4R, по сравнению с плацебо для исследования в примере 1.
[018] На фигуре 3 показано процентное изменение EASI от исходного уровня у пациентов, получавших 75 мг, 150 мг или 300 мг антитела к IL-4R, по сравнению с плацебо для исследования в примере 1.
[019] На фигуре 4 показано процентное изменение показателя зуда согласно NRS от исходного уровня у пациентов, получавших 75 мг, 150 мг или 300 мг антитела к IL-4R, по сравнению с плацебо для исследования в примере 1.
[020] На фигуре 5 показа динамика ответа EASI у пациентов с AD со степенью тяжести от средней до тяжелой на 300 мг антитела к IL-4R для исследования в примере 1.
[021] На фигуре 6 показан процент респондеров по показателю EASI, получавших 75 мг, 150 мг или 300 мг антитела к IL-4R, по сравнению с плацебо для исследования в примере 1.
[022] На фигуре 7 показаны ответы EASI на 4 неделе (29 день) на антитела к IL-4R, вводимые в дозах 75 мг, 150 мг или 300 мг, по сравнению с плацебо для исследования в примере 1.
[023] На фигуре 8 показан процент пациентов, достигающих IGA ≤ 1, для исследования в примере 1.
[024] На фигуре 9 показано среднее процентное изменение показателя EASI от исходного уровня до переноса данных последнего наблюдения вперед (LOCF), для исследования в примере 2.
[025] На фигуре 10 показано количество респондеров по показателю IGA (балл 0 или 1) до LOCF для исследования в примере 2.
[026] На фигуре 11 показано количество респондеров по показателю IGA (снижение балла на 2 или более) до LOCF для исследования в примере 2.
[027] На фигуре 12 показано количество респондеров по показателю EASI (снижение балла на 50% от исходного уровня) до LOCF для исследования в примере 2.
[028] На фигуре 13 показано количество респондеров по показателю IGA (снижение балла на 75% от исходного уровня) до LOCF для исследования в примере 2.
[029] На фигуре 14 показано среднее изменение показателя EASI от исходного уровня до LOCF для исследования в примере 2.
[030] На фигуре 15 показано среднее процентное изменение составляющих компонентов EASI от исходного уровня на 12 неделе для исследования в примере 2.
[031] На фигуре 16 показано среднее изменение показателя IGA от исходного уровня до LOCF для исследования в примере 2.
[032] На фигуре 17 показано среднее процентное изменение показателя IGA от исходного уровня до LOCF для исследования в примере 2.
[033] На фигуре 18 показано среднее изменение BSA от исходного уровня до LOCF для исследования в примере 2.
[034] На фигуре 19 показано среднее изменение показателя SCORAD от исходного уровня до LOCF для исследования в примере 2.
[035] На фигуре 20 показано среднее изменение показателя NRS от исходного уровня до LOCF для исследования в примере 2.
[036] На фигуре 21 показано среднее процентное изменение показателя NRS от исходного уровня в течение 12 недель для исследования в примере 2.
[037] На фигуре 22 показано среднее изменение от исходного уровня показателя 5-D зуда до LOCF для исследования в примере 10.
[038] На фигуре 23 показано среднее процентное изменение от исходного уровня показателя зуда по шкале 5-D в течение 12 недель для исследования в примере 2.
[039] На фигуре 24 показано среднее процентное изменение составляющих компонентов показателя зуда по шкале 5-D от исходного уровня на 85 день для исследования в примере 2.
[040] На фигуре 25 показан процент пациентов, достигающих IGA 0-1 на 12 неделе, для исследования в примере 2.
[041] На фигуре 26 показан процент пациентов, достигающих средней величины процента BSA на 12 неделе, для исследования в примере 2.
[042] На фигуре 27 показано среднее изменение QoLIAD от исходного уровня к 12 неделе для исследования в примере 2.
[043] На фигуре 28 показана корреляция между изменением QoLIAD и изменением клинических результатов по EASI (A) и зуда по шкале 5-D (B) на 12 неделе для исследования в примере 2.
[044] На фигуре 29 показано медианное изменение SCH от исходного уровня в поврежденной коже для исследования в примере 3.
[045] На фигуре 30 показано медианное изменение SCH от исходного уровня в неповрежденной коже для исследования в примере 3.
[046] На фигуре 31 показано медианное изменение TEWL от исходного уровня в поврежденной коже для исследования в примере 3.
[047] На фигуре 32 показано медианное изменение TEWL от исходного уровня в неповрежденной коже для исследования в примере 3.
[048] На фигуре 33 показан процент образцов, положительных по S. aureus, в поврежденной коже для исследования в примере 4.
[049] На фигуре 34 показан процент образцов, положительных по S. aureus, в неповрежденной коже для исследования в примере 4.
[050] На фигуре 35 показано изменение процента образцов, положительных по S. aureus, от исходного уровня в поврежденной коже для исследования в примере 4.
[051] На фигуре 36 показано изменение процента образцов, положительных по S. aureus, от исходного уровня в неповрежденной коже для исследования в примере 4.
ПОДРОБНОЕ ОПИСАНИЕ
[052] Перед описанием настоящего изобретения необходимо понимать, что настоящее изобретение не ограничивается конкретными описанными способами и условиями экспериментов, поскольку такие способы и условия могут варьировать. Также необходимо понимать, что терминология, используемая в данном документе, предназначена лишь с целью описания конкретных вариантов осуществления, и не предполагает ограничительный характер, поскольку область настоящего изобретения будет ограничиваться лишь прилагаемой формулой изобретения.
[053] Если не определено иное, все технические и научные термины, используемые в данном документе, имеют такое же значение, которое обычно понимается специалистом в области, к которой принадлежит настоящее изобретение. Используемое в данном документе выражение «приблизительно», при использовании со ссылкой на конкретное описываемое числовое значение, означает, что значение может отличаться от описываемого значения не более чем на 1%. Например, используемое в данном документе выражение «приблизительно 100» включает 99 и 101 и все значения между ними (например, 99,1, 99,2, 99,3, 99,4 и т.д.). Используемые в данном документе выражения «лечить», «лечение» или им подобные означает облегчать симптомы, устранить причинность симптомов, либо на временной, либо на постоянной основе, или предупреждать или замедлять проявление симптомов указанного нарушения или состояния.
[054] Хотя любые способы и материалы, подобные или эквивалентные тем, которые описаны в данном документе, можно использовать в практике настоящего изобретения, ниже описаны предпочтительные способы и материалы. Все публикации, упомянутые в данном документе, включены в данный документ посредством ссылки во всей своей полноте.
Способы лечения, предупреждения или уменьшения тяжести кожных инфекций
[055] Настоящее изобретение включает способы, которые предусматривают введение нуждающемуся в этом субъекту терапевтической композиции, содержащей антагонист IL-4R. Используемое в данном документе выражение «нуждающийся в этом субъект», означает человека или любое животное за исключением человека, которые характеризуются одним или несколькими симптомами кожной инфекции, и/или которым был поставлен диагноз кожной инфекции.
[056] В контексте настоящего изобретения выражение «субъект» включает субъекта с кожной инфекцией, где кожная инфекция выбрана из группы, состоящей из импетиго, целлюлита, инфекционного дерматита, герпетической экземы, фолликулита, инфицированного волдыря, микоза, отрубевидного лишая, инфекции, вызываемой Staphylococcus aureus, и инфекции, вызываемой Streptococcus.
[057] Согласно определенным вариантам осуществления способы по настоящему изобретению можно применять для уменьшения воспаления и/или зуда, возникающих в результате инфекции кожи микроорганизмами.
[058] Согласно определенным вариантам осуществления выражение «субъект» включает субъектов с IL-4R-связанным заболеванием или нарушением, например, c атопическим дерматитом, астмой или аллергией.
[059] Настоящее изобретение предлагает способы уменьшения колонизации кожи микроорганизмами, предусматривающие введение терапевтически эффективного количества антагониста IL-4R. Используемое в данном документе выражение «субъект» включает субъекта, инфицированного микроорганизмом, в том числе без ограничения Staphylococcus aureus, Streptococcus spp., Pseudomonas aeruginosa, Bacteroides spp., вирусом простого герпеса, вирусом Коксаки, вирусом контагиозного моллюска, вирусом осповакцины, Candida albicans, Microsporum spp., Trichophyton spp., Penicillium spp., Cladosporium spp., Alternaria spp. и Aspergillus spp. Согласно определенным вариантам осуществления настоящее изобретение предлагает способы снижения колонизации S. aureus кожи пациентов с атопическим дерматитом. Согласно некоторым вариантам осуществления колонизация микроорганизмами уменьшается по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45%, по меньшей мере на 50%, по меньшей мере на 55%, по меньшей мере на 60%, по меньшей мере на 65%, по меньшей мере на 70%, по меньшей мере на 75% по сравнению с исходным уровнем при введении антагониста IL-4R.
[060] Колонизацию микроорганизмами можно измерять при помощи тестов и процедур, известных в данной области, например, при помощи ПЦР, культивирования микроорганизмов, микроскопии и окрашивания или иммунофлуоресценции. Согласно определенным вариантам осуществления колонизацию микроорганизмами можно измерять по присутствию белковых биомаркеров микроорганизмов, известных в данной области, например, токсинов микроорганизмов, таких как стафилококковый токсин-1, обуславливающий развитие синдрома токсического шока. Способы обнаружения и/или количественного определения таких биомаркеров известны в данной области.
[061] Согласно определенным вариантам осуществления выражение «субъект» включает субъекта, одновременного инфицированного одним или несколькими микроорганизмами, например, субъекта, одновременного инфицированного вирусом простого герпеса и S. aureus.
[062] Настоящее изобретение включает способы уменьшения восприимчивости субъекта к кожной инфекции. Используемое в данном документе выражение «субъект» относится к субъектам с повышенной восприимчивостью к кожной инфекции или с более высоким риском развития кожной инфекции, например, к субъектам с атопическим дерматитом. Согласно данному аспекту выражение «субъект» включает субъектов с тяжелым атопическим дерматитом, более высокой сенсибилизацией к аллергенам, а также субъектов с астмой или пищевой аллергией. Выражение «субъект» также включает субъектов с повышенными сывороточными уровнями общего и аллергенспецифического IgE или сывороточных хемокинов (например, CCL17 или CCL27).
Способы улучшения барьерной функции кожи
[063] Настоящее изобретение включает способы улучшения у субъекта барьерной функции кожи, предусматривающие введение нуждающемуся в этом субъекту терапевтически эффективного количества антагониста IL-4R. Выражение «барьерная функция кожи» относится к защитной функции кожи за счет проницаемости структурного барьера рогового слоя и секреции противомикробных пептидов. Проницаемость барьера, а также противомикробная защита исчезает или нарушается в том случае, если целостность кожи повреждается вследствие кожной инфекции или вследствие заболевания, такого как атопический дерматит. Используемое в данном документе выражение «субъект» может включать субъектов со сниженной барьерной функцией кожи или субъектов, у которых до лечения проявляются (или проявлялись) один или несколько параметров барьерной функции кожи. Например, выражение «субъект», используемое в данном документе, включает субъектов со сниженным образованием кожных противомикробных пептидов.
[064] Примеры параметров барьерной функции кожи включают: (a) увлажнение рогового слоя (SCH), (b) трансэпидермальную потерю воды (TEWL), (c) pH поверхности кожи и (d) профилометрию шероховатости кожи (Eberlein-Konig et al 2000, Acta Derm. Venereol. 80: 188-191). SCH и TEWL можно измерить при помощи методик корнеометрии и эвапориметрии, известных в данной области (например, Vergananini et al 2010, J. Dermatol. Treatment, 21: 126-129).
[065] Чтобы определить, «улучшился ли» параметр барьерной функции кожи, параметр измеряют количественно на исходном уровне и в одной или нескольких временных точках после введения фармацевтической композиции по настоящему изобретению. Например, параметр можно измерять в 1 день, 2 день, 3 день, 4 день, 5 день, 6 день, 7 день, 8 день, 9 день, 10 день, 11 день, 12 день, 14 день, 15 день, 22 день, 25 день, 29 день, 36 день, 43 день, 50 день, 57 день, 64 день, 71 день, 85 день; или в конце 1 недели, 2 недели, 3 недели, 4 недели, 5 недели, 6 недели, 7 недели, 8 недели, 9 недели, 10 недели, 11 недели, 12 недели, 13 недели, 14 недели, 15 недели, 16 недели, 17 недели, 18 недели, 19 недели, 20 недели, 21 недели, 22 недели, 23 недели, 24 недели или дольше после начального лечения фармацевтической композицией по настоящему изобретению. Различие между величиной параметра в определенной временной точке после начала лечения и величиной параметра на исходном уровне используют для установления того, произошло ли «улучшение» (например, снижение) параметра.
[066] Увлажнение рогового слоя (SCH): измерения SCH проводят корнеометром, который регистрирует электрическую емкость кожи в качестве показателя увлажнения кожи. Чем больше емкость, тем более увлажненной является кожа. Согласно определенным вариантам осуществления настоящего изобретения введение пациенту антагониста IL-4R приводит в результате к повышению показателя SCH. Согласно определенным вариантам осуществления повышение показателя SCH составляет по меньшей мере 10%, по меньшей мере 20%, по меньшей мере 30%, по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% или по меньшей мере 70% по сравнению с исходным уровнем.
[067] Трансэпидермальная потеря воды (TEWL): TEWL фиксируют при помощи эвапориметрии. Чем больше этот показатель, тем более высокой является потеря воды с кожи. Согласно определенным вариантам осуществления настоящего изобретения введение пациенту антагониста IL-4R приводит к уменьшению величины TEWL. Согласно определенным вариантам осуществления уменьшение составляет по меньшей мере 5%, по меньшей мере 15%, по меньшей мере 25%, по меньшей мере 35%, по меньшей мере 45%, по меньшей мере 55%, по меньшей мере 65% или по меньшей мере 75% по сравнению с исходным уровнем.
[068] Значение pН поверхности кожи: Значение pH поверхности кожи измеряют pH-метром. Значение pH поверхности кожи повышается вследствие нескольких видов кожного воспаления, в том числе инфекции. Согласно определенным вариантам осуществления настоящего изобретения введение пациенту антагониста IL-4R приводит к уменьшению значения pH поверхности кожи до значения кислого pH. Согласно определенным вариантам осуществления введение пациенту антагониста IL-4R приводит к снижению значения pH до pH 6,0, pH 5,9, pH 5,8, pH 5,7, pH 5,6, pH 5,5, pH 5,4, pH 5,3, pH 5,2, pH 5,1, pH 5,0, pH 4,9, pH 4,8, pH 4,7, pH 4,6 или pH 4,5.
[069] Шероховатость кожи: шероховатость кожи измеряют профилометром. Профили шероховатости кожи получают в виде электрических сигналов. Шероховатость кожи повышена при состояниях кожной инфекции и атопического дерматита. Согласно определенным вариантам осуществления настоящего изобретения введение пациенту антагониста IL-4R приводит к уменьшению шероховатости кожи.
[070] Согласно определенным вариантам осуществления выражение «субъект» включает субъектов с белком, или геном, или генетическим зондом («биомаркером»), ассоциированных с барьерной функцией кожи, которые могут дифференциально экспрессироваться вследствие сниженной барьерной функции кожи. Например, гены, которые положительно регулируются у субъекта с кожной инфекцией, могут включать гены маркеров эпидермальной пролиферации, такие как K16, Ki67; и гены, которые отрицательно регулируются, могут включать гены белков конечной дифференцировки, такие как филаггрин, лорикрин или инволюкрин. Согласно определенным вариантам осуществления выражение «субъект» относится к пациенту с заболеванием или нарушением, таким как атопический дерматит. Согласно определенным вариантам осуществления данное выражение может включать субъектов с тяжелым, активируемым аллергенами (или экзогенным) заболеванием AD.
[071] Согласно определенным аспектам настоящего изобретения предложены способы лечения кожной инфекции или улучшения барьерной функции кожи, которые предусматривают: (a) отбор субъекта, у которого проявляется уровень по меньшей мере одного параметра или биомаркера до или во время лечения, являющегося признаком состояния заболевания; и (b) введение субъекту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста IL-4R. Уровень биомаркера устанавливают или количественно определяют на основе получения образца от пациента для анализа биомаркера, известного в данной области. Согласно определенным другим вариантам осуществления пациента отбирают на основе получения информации, связанной с повышенным уровнем биомаркера у пациента.
Антагонисты рецепторов интерлейкина-4
[072] Способы по настоящему изобретению также включают введение нуждающемуся в этом субъекту терапевтической композиции, содержащей антагонист рецепторов интерлейкина-4 (IL-4R). Используемый в данном документе термин «антагонист IL-4R» (также обозначаемый в данной документе как «ингибитор IL-4R», «антагонист IL-4Rα», «блокатор IL-4R», «блокатор IL-4Rα» и т.п.) представляет собой любое средство, которые связывается или взаимодействует с IL-4Rα или лигандом IL-4R, и ингибирует или ослабляет нормальную биологическую сигнальную функцию рецептора IL-4 типа 1 и/или типа 2. Человеческий IL-4Rα имеет аминокислотную последовательность под SEQ ID NO: 9. Рецептор IL-4 типа 1 представляет собой димерный рецептор, содержащий IL-4Rα-цепь и γc-цепь. Рецептор IL-4 типа 2 представляет собой димерный рецептор, содержащий IL-4Rα-цепь и IL-13Rα1-цепь. Рецепторы IL-4 типа 1 взаимодействуют с IL-4 и стимулируются IL-4, в то время как рецепторы IL-4 типа 2 взаимодействуют как с IL-4, так и с IL-13, и стимулируются ими. Таким образом, антагонисты IL-4R, которые можно применять в способах по настоящему изобретению, могут выполнять функции путем блокирования IL-4-опосредованной передачи сигналов, IL-13-опосредованной передачи сигналов или как IL-4-опосредованной, так и IL-13-опосредованной передачи сигналов. Таким образом, антагонисты IL-4R по настоящему изобретению могут предупреждать взаимодействие IL-4 и/или IL-13 с рецептором типа 1 или типа 2.
[073] Неограничивающие примеры категорий антагонистов IL-4R включат низкомолекулярные ингибиторы IL-4R, аптамеры к IL-4R, ингибиторы IL-4R на основе пептидов (например, молекулы «пептидотел»), «рецепторные тела» (например, cконструированные молекулы, содержащие лиганд-связывающий домен компонента IL-4R) и антитела или антигенсвязывающие фрагменты антител, которые специфически связываются с человеческим IL-4Rα. Используемые в данном документе антагонисты IL-4R также включают антигенсвязывающие белки, которые специфически связываются с IL-4 и/или IL-13.
Антитела к IL-4Rα и их антигенсвязывающие фрагменты
[074] Согласно определенным иллюстративным вариантам осуществления настоящего изобретения антагонистом IL-4R является антитело к IL-4Rα или его антигенсвязывающий фрагмент. Термин «антитело», используемый в данном документе, включает молекулы иммуноглобулинов, содержащие четыре полипептидные цепи, две тяжелые (H) цепи и две легкие (L) цепи, соединенные между собой дисульфидными связями, а также их мультимеры (например, IgM). В типичном антителе каждая тяжелая цепь содержит вариабельный участок тяжелой цепи (в данном документе имеет аббревиатуру HCVR или VH) и константный участок тяжелой цепи. Константный участок тяжелой цепи содержит три домена: CH1, CH2 и CH3. Каждая легкая цепь содержит вариабельный участок легкой цепи (в данном документе имеет аббревиатуру LCVR или VL) и константный участок легкой цепи. Константный участок легкой цепи содержит один домен (CL1). Участки VH и VL можно дополнительно подразделять на участки гипервариабельности, называемые участками, определяющими комплементарность (CDR), чередующиеся с более консервативными участками, называемыми каркасными участками (FR). Каждый VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца до карбокси-конца в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Согласно разным вариантам осуществления настоящего изобретения FR антитела к IL-4R (или его антигенсвязывающего участка) могут быть идентичны последовательностям иммуноглобулина зародышевой линии человека или могут быть изменены естественным или искусственным путем. Аминокислотная консенсусная последовательность может быть определена на основании анализа «бок о бок» двух или более CDR.
[075] Термин «антитело», используемый в данном документе, также включает антигенсвязывающие фрагменты целых молекул антител. Термины «антигенсвязывающий участок» антитела, «антигенсвязывающий фрагмент» антитела и им подобные, используемые в данном документе, включают любой встречающийся в природе, получаемый ферментативным путем, синтетический или получаемый методиками генной инженерии полипептид или гликопротеин, специфически связывающийся с антигеном с образованием комплекса. Антигенсвязывающие фрагменты антитела могут быть получены, например, из целых молекул антител при помощи любых подходящих стандартных методик, таких как протеолитическое расщепление или рекомбинантные методики генной инженерии, включающие манипуляцию с ДНК, кодирующей вариабельные и необязательно константные домены антител, и ее экспрессию. Такая ДНК известна и/или легкодоступна, например, из коммерческих источников, библиотек ДНК (в том числе, например, библиотек «фаг-антитело») или ее можно синтезировать. ДНК можно секвенировать и с ней можно проводить химические манипуляции или манипуляции при помощи методик молекулярной биологии, например, для упорядочивания одного или нескольких вариабельных и/или константных доменов в подходящую конфигурацию, или для введения кодонов, создания цистеиновых остатков, модификации, присоединения или удаления аминокислот и т.д.
[076] Неограничивающие примеры антигенсвязывающих фрагментов включают: (i) Fab-фрагменты; (ii) F(ab')2-фрагменты; (iii) Fd-фрагменты; (iv) Fv-фрагменты; (v) одноцепочечные молекулы Fv (scFv); (vi) dAb-фрагменты и (vii) минимальные распознающие единицы, состоящие из аминокислотных остатков, имитирующих гипервариабельный участок антитела (например, выделенный участок, определяющий комплементарность (CDR), такой как пептид CDR3), или пептид c ограниченной конформационной свободой FR3-CDR3-FR4. Другие сконструированные молекулы, такие как домен-специфические антитела, однодоменные антитела, антитела с удаленным доменом, химерные антитела, CDR-привитые антитела, диатела, триатела, тетратела, минитела, нанотела (например, моновалентные антитела, бивалентные антитела и т.д.), иммунопрепараты на основе модульного белка с малым размером молекул (SMIP) и вариабельные домены IgNAR акулы, также включены в выражение «антигенсвязывающий фрагмент», используемый в данном документе.
[077] Антигенсвязывающий фрагмент антитела будет, как правило, содержать по меньшей мере один вариабельный домен. Вариабельный домен может быть любого размера или аминокислотного состава и будет, как правило, содержать по меньшей мере один CDR, который прилегает или находится в рамке считывания с одной или несколькими каркасными последовательностями. В антигенсвязывающих фрагментах, имеющих домен VH, связанный с доменом VL, домены VH и VL могут располагаться относительно другу друга в любом подходящем порядке. Например, вариабельный участок может быть димерным и содержать димеры VH-VH, VH-VL или VL-VL. Альтернативно, антигенсвязывающий фрагмент антитела может содержать мономерный домен VH или VL.
[078] Согласно определенным вариантам осуществления антигенсвязывающий фрагмент антитела может содержать по меньшей мере один вариабельный домен, ковалентно связанный с по меньшей мере одним константным доменом. Неограничивающие иллюстративные конфигурации вариабельных и константных доменов, которые можно выявить в антигенсвязывающем фрагменте антитела по настоящему изобретению, включают: (i) VH-CH1; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (v) VH-CH1-CH2-CH3; (vi) VH-CH2-CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (x) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1-CH2-CH3; (xiii) VL-CH2-CH3 и (xiv) VL-CL. В любой конфигурации вариабельных и константных доменов, в том числе каких-либо иллюстративных конфигурациях, изложенных выше, вариабельные и константные домены могут быть либо непосредственно связаны друг с другом, либо могут быть связаны при помощи всего или части шарнирного или линкерного участка. Шарнирный участок может состоять из по меньшей мере 2 (например, 5, 10, 15, 20, 40, 60 или более) аминокислот, которые приводят к образованию гибкой или полугибкой связи между прилегающими вариабельными и/или константными доменами в одной молекуле полипептида. Кроме того, антигенсвязывающий фрагмент антитела по настоящему изобретению может содержать гомодимер или гетеродимер (или другой мультимер) из любых конфигураций вариабельных и константных доменов, изложенных выше, в нековалентной ассоциации друг с другом и/или с одним или несколькими мономерными доменами VH или r VL (например, при помощи дисульфидной(дисульфидных) связи(связей)).
[079] Термин «антитело», используемый в данном документе, также включает мультиспецифические (например, биспецифические) антитела. Мультиспецифическое антитело или антигенсвязывающий фрагмент антитела будет обычно содержать по меньшей мере два различных вариабельных домена, где каждый вариабельный домен способен специфически связываться с отдельным антигеном или с другим эпитопом того же самого антигена. Любой формат мультиспецифических антител может быть адаптирован для применения в контексте антитела или антигенсвязывающего фрагмента антитела по настоящему изобретению при помощи стандартных методик, доступных в данной области. Например, настоящее изобретение включает способы, предусматривающие применение биспецифических антител, где один фрагмент иммуноглобулина является специфическим для IL-4Rα или его фрагмента, а другой фрагмент иммуноглобулина является специфическим для второй терапевтической мишени или конъюгирован с терапевтическим фрагментом. Иллюстративные биспецифические форматы, которые можно применять в контексте настоящего изобретения, включают без ограничения, например, биспецифические форматы на основе scFv или биспецифические форматы на основе диател, слияния IgG-scFv, двойной вариабельный домен (DVD)-Ig, квадрому, выступы-во-впадины, обычную легкую цепь (например, обычную легкую цепь с выступами-во-впадины и т.п.), CrossMab, CrossFab, (SEED)-тело, лейциновую застежку, DuoBody, IgG1/IgG2, Fab (DAF)-IgG двойного действия и Mab2 биспецифические форматы (см., например, Klein et al. 2012, mAbs 4:6, 1-11, и источники, упоминаемые в данном документе, для изучения вышеизложенных форматов). Биспецифические антитела также можно cконструировать при помощи конъюгации пептидов и нуклеиновых кислот, например, где не встречающиеся в природе аминокислоты с ортогональной химической реакционной способностью применяют для получения сайт-специфических конъюгатов антитело-нуклеотид, которые затем самособираются в мультимерные комплексы с определенными составом, валентностью и геометрической формой. (См., например, Kazane et al., J. Am. Chem. Soc. [Epub: Dec. 4, 2012]).
[080] Антитела, используемые в способах по настоящему изобретению, могут быть человеческими антителами. Термин «человеческое антитело», используемый в данном документе включает антитела, имеющие вариабельные и константные участки, происходящие из последовательностей иммуноглобулинов зародышевой линии человека. Человеческие антитела по настоящему изобретению при этом могут включать аминокислотные остатки, не кодируемые последовательностями иммуноглобулинов зародышевой линии человека (например, мутации, вводимые случайным или сайт-специфичным мутагенезом in vitro или соматической мутацией in vivo), например, в CDR и, в частности, CDR3. Однако термин «человеческое антитело», используемый в данном документе, не включает антитела, в которых последовательности CDR, полученные из зародышевой линии другого вида млекопитающего, такого как мышь, привиты на последовательности каркасных участков человека.
[081] Антитела, используемые в способах по настоящему изобретению, могут быть рекомбинантными человеческими антителами. Термин «рекомбинантное человеческое антитело», используемый в данном документе, включает все антитела, получаемые, экспрессируемые, создаваемые или выделяемые рекомбинантным способом, такие как антитела, экспрессируемые с помощью рекомбинантного вектора экспрессии, трансфицированного в клетку-хозяина (описанные далее), антитела, выделяемые из комбинаторной библиотеки рекомбинантных человеческих антител (описанные далее), антитела, выделяемые из животного (например, мыши), которое является трансгенным по генам человеческих иммуноглобулинов (см., например, Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295), или антитела, получаемые, экспрессируемые, создаваемые или выделяемые любым другим способом, включающим соединение последовательностей генов человеческих иммуноглобулинов с другими последовательностями ДНК. Такие рекомбинантные человеческие антитела имеют вариабельные и константные участки, происходящие из последовательностей иммуноглобулинов зародышевой линии человека. Однако согласно определенным вариантам осуществления такие рекомбинантные человеческие антитела подвергают мутагенезу in vitro (или в случае использования животного, трансгенного по последовательностям человеческого Ig, соматическому мутагенезу in vivo) и, таким образом, аминокислотные последовательности участков VH и VL рекомбинантных антител представляют собой последовательности, которые, будучи полученными из последовательностей VH и VL зародышевой линии человека и родственными последовательностям им, могут не встречаться в природе в репертуаре антител зародышевой линии человека in vivo.
[082] Согласно определенным вариантам осуществления антитела, используемые в способах по настоящему изобретению, специфически связываются с IL-4Rα. Выражение «специфически связывается» или подобное ему означает, что антитело или его антигенсвязывающий фрагмент образует комплекс с антигеном, который является относительно устойчивым в физиологических условиях. Способы определения наличия специфического связывания антитела с антигеном хорошо известны в данной области и включают, например, равновесный диализ, поверхностный плазмонный резонанс и им подобные. Например, антитело, которое «специфически связывается» с IL-4Rα, как используется в контексте настоящего изобретения, включает антитела, которые связываются с IL-4Rα или его частью с KD менее приблизительно 1000 нМ, менее приблизительно 500 нМ, менее приблизительно 300 нМ, менее приблизительно 200 нМ, менее приблизительно 100 нМ, менее приблизительно 90 нМ, менее приблизительно 80 нМ, менее приблизительно 70 нМ, менее приблизительно 60 нМ, менее приблизительно 50 нМ, менее приблизительно 40 нМ, менее приблизительно 30 нМ, менее приблизительно 20 нМ, менее приблизительно 10 нМ, менее приблизительно 5 нМ, менее приблизительно 4 нМ, менее приблизительно 3 нМ, менее приблизительно 2 нМ, менее приблизительно 1 нМ или менее приблизительно 0,5 нМ, измеренном в анализе поверхностного плазмонного резонанса. Однако выделенное антитело, которое специфически связывается с IL-4Rα человека, характеризуется перекрестной реактивностью по отношению к другим антигенам, таким как молекулы IL-4Rα из других (не относящихся к человеку) видов.
[083] Согласно определенным иллюстративным вариантам осуществления настоящего изобретения антагонистом IL-4R является антитело к IL-4Rα или его антигенсвязывающий фрагмент, содержащий вариабельный участок тяжелой цепи (HCVR), вариабельный участок легкой цепи (LCVR) и/или участки, определяющие комплементарность (CDR), содержащие аминокислотные последовательности антител к IL-4R, как изложено в патенте США №7608693. Согласно определенным иллюстративным вариантам осуществления антитело к IL-4Rα или его антигенсвязывающий фрагмент, который можно применять в контексте способов настоящего изобретения, содержит участки, определяющие комплементарность тяжелой цепи (HCDR) из вариабельного участка тяжелой цепи (HCVR), содержащие аминокислотную последовательность под SEQ ID NO: 1, и участки, определяющие комплементарность легкой цепи (LCDR), из вариабельного участка легкой цепи (LCVR), содержащие аминокислотную последовательность под SEQ ID NO: 2. Согласно определенным вариантам осуществления антитело к IL-4Rα или его антигенсвязывающий фрагмент содержит три HCDR (HCDR1, HCDR2 и HCDR3) и три LCDR (LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность под SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность под SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность под SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность под SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность под SEQ ID NO: 7 и LCDR3 содержит аминокислотную последовательность под SEQ ID NO: 8. Согласно определенным вариантам осуществления антитело к IL-4R или его антигенсвязывающий фрагмент содержит HCVR, содержащий SEQ ID NO: 1 и LCVR, содержащий SEQ ID NO: 2. Согласно определенным иллюстративным вариантам осуществления способы по настоящему изобретению предусматривают применение антитела к IL-4Rα, содержащего аминокислотные последовательности HCDR1-HCDR2-HCDR3-LCDR1-LCDR2-LCDR3 под SEQ ID NO: 3-4-5-6-7-8 (обозначаемые и известные в данной области как «дупилумаб»), или его биоэквивалента.
[084] Согласно определенным вариантам осуществления способы по настоящему изобретению включают применение антитела к IL-4R, где антитело содержит тяжелую цепь, содержащую аминокислотную последовательность под SEQ ID NO: 10. Согласно некоторым вариантам осуществления антитело к IL-4R содержит легкую цепь, содержащую аминокислотную последовательность под SEQ ID NO: 11. Иллюстративным антителом, содержащим тяжелую цепь, содержащую аминокислотную последовательность под SEQ ID NO: 10, и легкую цепь, содержащую аминокислотную последовательность под SEQ ID NO: 11, является полностью человеческое антитело к IL-4R, известное как дупилумаб. Согласно определенным иллюстративным вариантам осуществления способы по настоящему изобретению предусматривают применение дупилумаба или его биоэквивалента. Термин «биоэквивалент», используемый в данном документе, относится к антителам к IL-4R или IL-4R-связывающим белкам или их фрагментам, которые являются фармацевтическими эквивалентами или фармацевтическими альтернативами, скорость и/или степень всасывания которых значительно не отличается от таких показателей для дупилумаба при введении одинаковой молярной дозы в аналогичных экспериментальных условиях, либо однократной дозы, либо нескольких доз. В контексте настоящего изобретения данный термин относится к антигенсвязывающим белкам, которые связываются с IL-4R, которые в контексте клинической значимости не отличаются от дупилумаба по безопасности, чистоте и/или активности.
[085] Другие антитела к IL-4Rα, которые можно использовать в контексте способов по настоящему изобретению, включают, например, антитело, обозначаемое и известное в данной области как AMG317 (Corren et al., 2010, Am J Respir Crit Care Med., 181(8):788-796), или любое из антител к IL-4Rα, изложенных в патенте США № 7186809, патенте США № 7605237, патенте США № 7608693 или патенте США № 8092804.
[086] Антитела к IL-4Rα, используемые в контексте способов по настоящему изобретению, могут характеризоваться pH-зависимыми характеристиками связывания. Например, антитело к IL-4Rα для применения в способах по настоящему изобретению может характеризоваться ослабленным связыванием с IL-4Rα при значении кислого pH по сравнению с нейтральным значением pH. Альтернативно, антитело к IL-4Rα по настоящему изобретению может характеризоваться усиленным связыванием со своим антигеном при значении кислого pH по сравнению с значением нейтрального pH. Выражение «значение кислого pH» включает значения pH менее приблизительно 6,2, составляющие, например, менее приблизительно 6,0, 5,95, 5,9, 5,85, 5,8, 5,75, 5,7, 5,65, 5,6, 5,55, 5,5, 5,45, 5,4, 5,35, 5,3, 5,25, 5,2, 5,15, 5,1, 5,05, 5,0 или менее. Используемое в данном документе выражение «значение нейтрального pH» означает pH от приблизительно 7,0 до приблизительно 7,4. Выражение «значение нейтрального pH» включает значения pH, составляющие приблизительно 7,0, 7,05, 7,1, 7,15, 7,2, 7,25, 7,3, 7,35 и 7,4.
[087] В определенных примерах «ослабленное связывание с IL-4Rα при значении кислого pH по сравнению с значением нейтрального pH» выражают с точки зрения соотношения величины KD связывания антитела с IL-4Rα при значении кислого pH с величиной KD связывания антитела с IL-4Rα при значении нейтрального pH (или наоборот). Например, антитело или его антигенсвязывающий фрагмент можно рассматривать как характеризующиеся «ослабленным связыванием с IL-4Rα при значении кислого pH по сравнению со значением нейтрального pH» для целей настоящего изобретения, если антитело или его антигенсвязывающий фрагмент характеризуется соотношением KD в кислых/нейтральных условиях приблизительно 3,0 или выше. Согласно определенным иллюстративным вариантам осуществления соотношение KD в кислых/нейтральных условиях для антитела и антигенсвязывающего фрагмента по настоящему изобретению может составлять приблизительно 3,0, 3,5, 4,0, 4,5, 5,0, 5,5, 6,0, 6,5, 7,0, 7,5, 8,0, 8,5, 9,0, 9,5, 10,0, 10,5, 11,0, 11,5, 12,0, 12,5, 13,0, 13,5, 14,0, 14,5, 15,0, 20,0, 25,0, 30,0, 40,0, 50,0, 60,0, 70,0, 100,0 или более.
[088] Антитела с pH-зависимыми характеристиками связывания могут быть получены, например, при скрининге группы антител в отношении ослабленного (или усиленного) связывания с определенным антигеном при значении кислого pH по сравнению со значением нейтрального pH. Кроме того, модификации антигенсвязывающего домена на уровне аминокислот могут приводить к образованию антител с pH-зависимыми характеристиками. Например, при замене одной или нескольких аминокислот антигенсвязывающего домена (например, в CDR) остатком гистидина может быть получено антитело с ослабленным связыванием с антигеном при значении кислого pH по сравнению со значением нейтрального pH. Используемое в данном документе выражение «значение кислого pH» означает pH 6,0 или менее.
Фармацевтические композиции
[089] Настоящее изобретение включает способы, которые предусматривают введение пациенту антагониста IL-4R, где антагонист IL-4R содержится в фармацевтической композиции. Фармацевтические композиции по настоящему изобретению составляют с подходящими носителями, вспомогательными средствами и другими средствами, которые обеспечивают подходящий перенос, доставку, переносимость и им подобные. Множество подходящих составов можно найти в справочнике, известном всем химикам-фармацевтам: Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA. Эти составы включают, например, порошки, пасты, мази, желе, воска, масла, липиды, липидсодержащие (катионные или анионные) пузырьки (такие как LIPOFECTIN™), конъюгаты ДНК, безводные абсорбционные пасты, эмульсии типа «масло в воде» и «вода в масле», эмульсии карбовакс (полиэтиленгликоли с различной молекулярной массой), полужидкие гели и полужидкие смеси, содержащие карбовакс. См. также Powell et al. "Compendium of excipients for parenteral formulations" PDA (1998) J Pharm Sci Technol 52:238-311.
[090] Дозу антитела, вводимого пациенту в соответствии со способами по настоящему изобретению, можно варьировать в зависимости от возраста и размерных характеристик пациента, симптомов, состояний, пути введения и им подобных. Дозу обычно рассчитывают в соответствии с массой тела или площадью поверхности тела. В зависимости от тяжести состояния частоту и продолжительность лечения можно корригировать. Эффективные дозы и схемы введения фармацевтических композиций, содержащих антитела к IL-4R, можно определить эмпирически; например, динамику состояния пациента можно контролировать при помощи периодической оценки и, соответственно, корригировать дозу. Кроме того, межвидовое приведение доз можно осуществлять при помощи хорошо известных в данной области способов (например, Mordenti et al., 1991, Pharmaceut. Res. 8:1351). Конкретные иллюстративные дозы антител к IL4R и режимы введения, включающие то же самое, которые можно использовать в контексте настоящего изобретения, раскрыты в других частях данного документа.
[091] Известны различные системы доставки и их можно использовать для введения фармацевтической композиции по настоящему изобретению, например, инкапсулирование в липосомы, микрочастицы, микрокапсулы, рекомбинантные клетки, способные экспрессировать мутантные вирусы, опосредованный рецепторами эндоцитоз (см., например, Wu et al., 1987, J. Biol. Chem. 262:4429-4432). Способы введения включают без ограничения внутрикожные, внутримышечные, интраперитонеальные, внутривенные, подкожные, интраназальные, эпидуральные и пероральные пути. Композицию можно вводить любым удобным путем, например, инфузией или болюсной инъекцией, абсорбцией через эпителиальные или кожно-слизистые покровы (например, слизистую ротовой полости, слизистую прямой кишки и кишечника и др.), и можно вводить совместно с другими биологически активными средствами.
[092] Фармацевтическую композицию по настоящему изобретению можно доставить подкожно или внутривенно при помощи стандартной иглы и шприца. Кроме того, в отношении подкожной доставки при доставке фармацевтической композиции по настоящему изобретению широко используется шприц-ручка. Такая шприц-ручка может быть многоразовой или одноразовой. В многоразовой шприц-ручке, как правило, используется заменяемый картридж, который содержит фармацевтическую композицию. После того, как вся фармацевтическая композиция в картридже была введена и картридж опустел, пустой картридж можно легко утилизировать и заменить новым картриджем, который содержит фармацевтическую композицию. Затем шприц-ручку можно использовать повторно. В одноразовой шприц-ручке заменяемый картридж отсутствует. Вместо этого одноразовую шприц-ручку выпускают предварительно наполненной фармацевтической композицией, удерживаемой в резервуаре изделия. После того как резервуар освобождается от фармацевтической композиции, все изделие утилизируют.
[093] Многочисленные многоразовые шприц-ручки и изделия для автоинъекторной доставки используются при подкожной доставке фармацевтической композиции по настоящему изобретению. Примеры включают без ограничения AUTOPEN™ (Owen Mumford, Inc., Вудсток, Великобритания), шприц-ручку DISETRONIC™ (Disetronic Medical Systems, Бургдорф, Швейцария), шприц-ручку HUMALOG MIX 75/25™, шприц-ручку HUMALOG™, шприц-ручку HUMALIN 70/30™ (Eli Lilly and Co., Индианаполис, Индиана), NOVOPEN™ I, II и III (Novo Nordisk, Копенгаген, Дания), NOVOPEN JUNIOR™ (Novo Nordisk, Копенгаген, Дания), шприц-ручку BD™ (Becton Dickinson, Франклин-Лэйкс, Нью-Джерси), OPTIPEN™, OPTIPEN PRO™, OPTIPEN STARLET™ и OPTICLIK™ (Sanofi-Aventis, Франкфурт, Германия), при этом упомянуты лишь несколько из них. Примеры многоразовых шприц-ручек, используемых при подкожном введении фармацевтической композиции по настоящему изобретению, включают без ограничения шприц-ручку SOLOSTAR™ (Sanofi-Aventis), FLEXPEN™ (Novo Nordisk) и KWIKPEN™ (Eli Lilly), автоинъектор SURECLICK™ (Amgen, Таузанд-Окс, Калифорния), PENLET™ (Haselmeier, Штутгарт, Германия), EPIPEN (Dey, L.P.) и шприц-ручку HUMIRA™ (Abbott Labs, Эббот-Парк, Иллинойс), при этом упомянуты лишь несколько из них.
[094] В определенных ситуациях фармацевтическую композицию можно доставлять в системе с контролируемым высвобождением. Согласно одному варианту осуществления можно использовать насос (см. Langer, выше; Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201). Согласно другому варианту осуществления можно использовать полимерные материалы; см. Medical Applications of Controlled Release, Langer and Wise (eds.), 1974, CRC Pres., Boca Raton, Florida. Согласно еще одному варианту осуществления систему с контролируемым высвобождением можно разместить рядом с мишенью для композиции, при этом требуется лишь часть системной дозы (см., например, Goodson, 1984, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138). Другие системы с контролируемым высвобождением описаны в обзорной публикации Langer, 1990, Science 249:1527-1533.
[095] Инъекционные формы могут включать лекарственные формы для внутривенных, подкожных, внутрикожных и внутримышечных инъекций, для капельного введения и т.п. Эти инъекционные препараты можно получать известными способами. Например, инъекционные формы можно получать, например, растворением, суспендированием или эмульгированием антитела или его соли, описанных выше, в стерильной водной среде или масляной среде, обычно используемой для инъекций. Водной средой для инъекций, является, например, физиологический солевой раствор, изотонический раствор, содержащий глюкозу и другие вспомогательные средства и т.п, которые можно использовать в комбинации с подходящим растворителем, таким как спирт (например, этанол), многоатомный спирт (например, пропиленгликоль, полиэтиленгликоль), неионогенное поверхностно-активное вещество [например, полисорбат 80, HCO-50 (полиоксиэтиленовый (50 моль) аддукт гидрогенизированного касторового масла)] и т.п. В качестве масляной среды используют, например, кунжутное масло, арахисовое масло и т.п., которые можно использовать в комбинации с растворителем, таким как бензилбензоат, бензиловый спирт и т.п. Приготовленным таким способом инъекционным составом можно наполнить подходящую ампулу.
[096] Преимущественно, фармацевтические композиции для перорального или парентерального применения, описанные выше, получают в лекарственных формах в единичной стандартной дозе, подходящей для подбора дозы активных ингредиентов. Такие лекарственные формы в стандартной дозе включают, например, таблетки, пилюли, капсулы, инъекции (ампулы), суппозитории и т.п.
[097] Иллюстративные фармацевтические композиции, содержащие антитело к IL-4R, которые можно использовать в контексте настоящего изобретения, раскрыты, например, в опубликованной патентной заявке США № 2012/0097565.
Доза
[098] Количество антагониста IL-4R (например, антитела к IL-4R), вводимого субъекту согласно способам по настоящему изобретению, как правило, представляет собой терапевтически эффективное количество. Используемое в данном документе выражение «терапевтически эффективное количество» означает количество антагониста IL-4R, которое в результате приводит к одному или нескольким из: (a) уменьшенной колонизации микроорганизмами, в том числе уменьшенной колонизации кожи S. aureus; (b) улучшенной барьерной функции кожи; (c) уменьшенному риску кожного воспаления вследствие инфекции микроорганизмами и/или (d) уменьшенной восприимчивости к инфицированию кожи микроорганизмами. Выражение «терапевтически эффективное количество» также включает количество антагониста IL-4R, которое ингибирует, предупреждает, снижает или задерживает прогрессирование кожной инфекции у субъекта. Согласно определенным вариантам осуществления выражение «терапевтически эффективное количество» означает количество антагониста IL-4R, которое приводит к поддающемуся обнаружению улучшению одного или нескольких симптомов или признаков, в том числе к уменьшенному числу рецидивов или обострений у субъекта с атопическим дерматитом.
[099] В случае антитела к IL-4R терапевтически эффективное количество может составлять от приблизительно 0,05 мг до приблизительно 600 мг, например, приблизительно 0,05 мг, приблизительно 0,1 мг, приблизительно 1,0 мг, приблизительно 1,5 мг, приблизительно 2,0 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 110 мг, приблизительно 120 мг, приблизительно 130 мг, приблизительно 140 мг, приблизительно 150 мг, приблизительно 160 мг, приблизительно 170 мг, приблизительно 180 мг, приблизительно 190 мг, приблизительно 200 мг, приблизительно 210 мг, приблизительно 220 мг, приблизительно 230 мг, приблизительно 240 мг, приблизительно 250 мг, приблизительно 260 мг, приблизительно 270 мг, приблизительно 280 мг, приблизительно 290 мг, приблизительно 300 мг, приблизительно 310 мг, приблизительно 320 мг, приблизительно 330 мг, приблизительно 340 мг, приблизительно 350 мг, приблизительно 360 мг, приблизительно 370 мг, приблизительно 380 мг, приблизительно 390 мг, приблизительно 400 мг, приблизительно 410 мг, приблизительно 420 мг, приблизительно 430 мг, приблизительно 440 мг, приблизительно 450 мг, приблизительно 460 мг, приблизительно 470 мг, приблизительно 480 мг, приблизительно 490 мг, приблизительно 500 мг, приблизительно 510 мг, приблизительно 520 мг, приблизительно 530 мг, приблизительно 540 мг, приблизительно 550 мг, приблизительно 560 мг, приблизительно 570 мг, приблизительно 580 мг, приблизительно 590 мг или приблизительно 600 мг антитела к IL-4R. Согласно определенным вариантам осуществления субъекту вводят 75 мг, 150 мг или 300 мг антитела к IL-4R.
[0100] Количество антагониста к IL-4R, содержащегося в отдельных дозах, можно выражать в миллиграммах антитела на килограмм массы тела пациента (т.е., мг/кг). Например, антагонист IL-4R можно вводить пациенту в дозе от приблизительно 0,0001 до приблизительно 10 мг/кг массы тела пациента.
Комбинированная терапия
[0101] Способы по настоящему изобретению согласно определенным вариантам осуществления предусматривают введение субъекту одного или нескольких терапевтических средств в комбинации с антагонистом IL-4R. Используемое в данном документе выражение «в комбинации с» означает, что дополнительные терапевтические средства вводят до, после содержащей антагонист IL-4R фармацевтической композиции или совместно с ней. Выражение «в комбинации с» также включает последовательное или одновременное введение антигониста IL-4R и второго терапевтического средства.
[0102] Например, при введении «до» фармацевтической композиции, содержащей антагонист IL-4R, дополнительное терапевтическое средство можно вводить за приблизительно 72 часа, приблизительно 60 часов, приблизительно 48 часов, приблизительно 36 часов, приблизительно 24 часа, приблизительно 12 часов, приблизительно 10 часов, приблизительно 8 часов, приблизительно 6 часов, приблизительно 4 часа, приблизительно 2 часа, приблизительно 1 час, приблизительно 30 минут, приблизительно 15 минут или приблизительно 10 минут до введения фармацевтической композиции, содержащей антагонист IL-4R. При введении «после» фармацевтической композиции, содержащей антагонист IL-4R, дополнительное терапевтическое средство можно вводить через приблизительно 10 минут, приблизительно 15 минут, приблизительно 30 минут, приблизительно 1 час, приблизительно 2 часа, приблизительно 4 часов, приблизительно 6 часов, приблизительно 8 часов, приблизительно 10 часов, приблизительно 12 часов, приблизительно 24 часа, приблизительно 36 часов, приблизительно 48 часов, приблизительно 60 часов или приблизительно 72 часа после введения фармацевтической композиции, содержащей антагонист IL-4R. Введение «одновременно» или вместе с фармацевтической композицией, содержащей антагонист IL-4R, означает, что дополнительное терапевтическое средство вводят субъекту в отдельной лекарственной форме в течение менее 5 минут (до, после или в то же самое время) от введения фармацевтической композиции, содержащей антагонист IL-4R, или вводят субъекту в одном комбинированном дозированном составе, содержащем как дополнительное терапевтическое средство, так и антагонист IL-4R.
[0103] Дополнительным терапевтическим средством может быть, например, антибактериальное средство (в том числе антибиотики для местного и системного применения, антибиотики узкого и широкого спектра действия), противовирусное средство (например, ацикловир или фоскарнет), противогрибковое средство (например, флуконазол и эконазола нитрат), другой антагонист IL-4R, антагонист IgE, интерферон-гамма (IFNγ), антибиотики, антисептический лосьон для местного применения или любой другой смягчающий препарат или их комбинация.
[0104] Способы по настоящему изобретению включают введение антагониста IL-4R в комбинации со вторым терапевтическим средством для дополнительной или синергетической активности с целью уменьшения риска возникновения кожных инфекций, например, у пациента с AD.
Режимы введения
[0105] Настоящее изобретение включает способы, предусматривающие введение субъекту фармацевтической композиции, содержащей антагонист IL-4R, с частотой введения дозы приблизительно четыре раза в неделю, два раза в неделю, один раз в неделю, один раз в две недели, один раз в три недели, один раз в четыре недели, один раз в пять недель, один раз в шесть недель, один раз в восемь недель, один раз в двенадцать недель или с меньшей частотой до тех пор, пока не будет достигнут терапевтический ответ. Согласно определенным вариантам осуществления, включающим введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 150 мг или 300 мг один раз в неделю.
[0106] Согласно определенным вариантам осуществления настоящего изобретения несколько доз антагониста IL-4R можно вводить субъекту в течение определенного периода времени. Способы по данному аспекту настоящего изобретения включают последовательное введение субъекту нескольких доз антагониста IL-4R. Используемое в данном документе выражение «последовательное введение» означает, что каждую дозу антагониста IL-4R вводят субъекту в разный момент времени, например, в разные дни, разделенные предварительно определенным интервалом (например, часами, днями, неделями или месяцами). Настоящее изобретение включает способы, которые предусматривают последовательное введение пациенту одной начальной дозы антагониста IL-4R, затем одной или нескольких вторичных доз антагониста IL-4R, а затем необязательно одной или нескольких третьих доз антагониста IL-4R.
[0107] Выражения «начальная доза», «вторичные дозы» и «третичные дозы» относятся к временной последовательности введения антагонистa IL-4R. Таким образом, «начальная доза» представляет собой дозу, которую вводят в начале режима терапии (также обозначается как «доза исходного уровня»); «вторичные дозы» представляют собой дозы, которые вводят после введения начальной дозы; и «третьи дозы» представляют собой дозы, которые вводят после вторичных доз. Все из начальных, вторичных и третичных доз могут содержать одинаковое количество антагониста IL-4R, но, как правило, могут отличаться друг от друга по частоте введения. Однако согласно определенным вариантам осуществления количество антагониcта IL-4R, содержащегося в начальных, вторичных и/или третичных дозах, отличается друг от друга (например, при необходимости корригируется в сторону повышения или снижения) в ходе курса лечения. Согласно определенным вариантам осуществления начальная доза содержит первое количество антитела или его антигенсвязывающего фрагмента, и каждая одна или несколько вторичных доз содержат второе количество антитела или его антигенсвязывающего фрагмента. Согласно некоторым вариантам осуществления первое количество антитела или его фрагмента составляет 1,5x, 2x, 2,5x, 3x, 3,5x, 4x или 5x второго количества антитела или его антигенсвязывающего фрагмента. Согласно определенным вариантам осуществления одну или несколько (например,1, 2, 3, 4 или 5) доз вводят в начале режима лечения в виде «ударных доз», затем последующие дозы вводят с меньшей частотой (например, «поддерживающие дозы»). Например, антагонист IL-4R можно вводить пациенту с кожной инфекцией в ударной дозе приблизительно 300 мг или приблизительно 600 мг, затем можно вводить одну или несколько поддерживающих доз от приблизительно 75 мг до приблизительно 300 мг. Согласно одному варианту осуществления каждая начальная доза и одна или нескольких вторичных доз включают от 50 мг до 600 мг антагониста IL-4R, например, от 100 мг до 400 мг антагониста IL-4R, например, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 400 мг или 500 мг антагониста IL-4R.
[0108] Согласно одному иллюстративному варианту осуществления настоящего изобретения каждую вторичную и/или третичную дозу вводят через 1-14 (например, 1, 1½, 2, 2½, 3, 3½, 4, 4½, 5, 5½, 6, 6½, 7, 7½, 8, 8½, 9, 9½, 10, 10½, 11, 11½, 12, 12½, 13, 13½, 14, 14½ или более) недель после непосредственно предшествующей дозы. Выражение «непосредственно предшествующая доза», используемое в данном документе, означает в последовательности нескольких введений дозу антагониста IL-4R, которую вводят пациенту до введения ближайшей следующей дозы в последовательности без промежуточных доз.
[0109] Способы по данному аспекту настоящего изобретения могут включать введение пациенту любого числа вторичных и/или третичных доз антагониста IL-4R. Например, согласно определенным вариантам осуществления пациенту вводят только одну вторичную дозу. Согласно другим вариантам осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или более) вторичных доз. Аналогичным образом, согласно определенным вариантам осуществления пациенту вводят только одну третью дозу. Согласно другим вариантам осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или более) третьих доз.
[0110] Согласно вариантам осуществления, включающим несколько вторичных доз, каждую вторичную дозу можно вводить с той же самой частотой, что и другие вторичные дозы. Например, каждую вторичную дозу можно вводить пациенту через 1-6 недель после непосредственно предшествующей дозы. Аналогичным образом, согласно вариантам осуществления, включающим несколько третьих доз, каждую третью дозу можно вводить с той же самой частотой, что и другие третьи дозы. Например, каждую третью дозу можно вводить пациенту через 2-4 недели после непосредственно предшествующей дозы. Альтернативно, частота, с которой вторичные и/или третичные дозы вводят пациенту, может отличаться в ходе режима лечения. Частоту введения может корректировать во время лечения врач в зависимости от потребностей отдельного пациента после клинического обследования.
[0111] Настоящее изобретение включает способы, предусматривающие последовательное введение пациенту антагониста IL-4R и второго терапевтического средства для лечения кожной инфекции. Согласно некоторым вариантам осуществления способы по настоящему изобретению предусматривают введение одной или нескольких доз антагониста IL-4R с последующим введением одной или нескольких доз второго терапевтического средства. Например, можно вводить одну или несколько доз от приблизительно 75 мг до приблизительно 300 мг антагониста IL-4R, после которых можно вводить одну или несколько доз второго терапевтического средства (например, антибиотика или любого другого терапевтического средства, описанного в других частях данного документа) для лечения, облегчения, снижения или уменьшения тяжести воспаления вследствие инфицирования кожи микроорганизмами. Согласно некоторым вариантам осуществления антагонист IL-4R вводят в одной или нескольких дозах, что приводит к улучшенной барьерной функции кожи, затем осуществляют введение второго терапевтического средства для уменьшения количества патогенной микрофлоры на коже. Альтернативные варианты осуществления настоящего изобретения относятся к сопутствующему введению антагониста IL-4R и второго терапевтического средства. Например, вводят одну или несколько доз антагониста IL-4R и вводят второе терапевтическое средство в отдельной дозе с одинаковой или разной частотой по отношению к антагонисту IL-4R. Согласно некоторым вариантам осуществления второе терапевтическое средство вводят до, после антагониста IL-4R или совместно с ним.
ПРИМЕРЫ
[0112] Следующие примеры изложены для того, чтобы обеспечить специалистов в данной области полным раскрытием и описанием получения и применения способов и композиций по настоящему изобретению, и они не предполагают ограничения объема того, что авторы настоящего изобретения рассматривают в качестве своего изобретения. Были приложены усилия для обеспечения точности по отношению к используемым числам (например, количеству, температуре и т.п.), однако необходимо учитывать некоторые ошибки и отклонения в экспериментах. Если не указано иное, то части являются частями по массе, молекулярная масса является средней молекулярной массой, температура представлена в градусах Цельсия, а давление является атмосферным или близким к атмосферному.
[0113] Иллюстративным антагонистом IL-4R, используемым в следующих примерах, является человеческое антитело к IL-4R, обозначаемое в данной области как дупилумаб, где антитело содержит три HCDR (HCDR1, HCDR2 и HCDR3) и три LCDR (LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность под SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность под SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность под SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность под SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность под SEQ ID NO: 7 и LCDR3 содержит аминокислотную последовательность под SEQ ID NO: 8 (также обозначаемое в данном документе как «mAb1»).
Пример 1. Лечение пациентов с атопическим дерматитом cо cтепенью тяжести от средней до тяжелой антителом к IL-4R: анализ объединенных исследований фазы 1b
[0114] Параметры эффективности лечения AD из двух отдельных клинических испытаний у пациентов с AD со степенью тяжести от средней до тяжелой измеряли и объединяли для анализа. «Исследование A» представляло собой 12-недельное двойное слепое рандомизированное плацебо-контролируемое исследование с последовательно нарастающими дозами для оценки безопасности и переносимости вводимого антитела к IL-4R (mAb1) у пациентов с атопическим дерматитом. Период лечения составлял 4 недели, затем пациентов наблюдали в течение 8 недель после окончания периода лечения. Пациентов рандомизировали в соотношении 4:1 для получения mAb1 или плацебо в каждой из трех когорт с нарастающими дозами (75 мг, 150 мг или 300 мг). Исследование состояло из периода отбора (с -14 дня до -3 дня), периода лечения (с 1 дня по 29 день) и периода последующего наблюдения (с 29 дня по 85 день). Во время периода лечения пациентов наблюдали в клинике один раз в неделю в целях безопасности, лабораторных оценок и оценок клинической эффективности на 1, 4, 8, 15, 22, 25 и 29 дни (4 неделя). Пациенты получали дозу mAb1 или плацебо на 1, 8, 15 и 22 дни. Окончание периода лечения в исследовании приходилось на 29 день (4 неделю). Контроль у пациентов осуществляли на базе исследовательского центра в течение 6 часов после инъекции (mAb1 или плацебо) на 1 день и в течение 3 часов после инъекции на 8, 15 и 22 дни. Во время периода последующего наблюдения пациентов наблюдали в клинике для оценок в рамках последующего наблюдения на 36, 43, 50, 57, 64, 71 и 85 дни (окончание визита в рамках исследования).
[0115] "Исследование B" представляло собой 12-недельное двойное слепое рандомизированное плацебо-контролируемое исследование с последовательно нарастающими повторными дозами у пациентов с AD со степенью тяжести от средней до тяжелой. Пациентам с AD вводили 150 мг или 300 мг mAb1, или плацебо на 1, 8, 15 и 22 дни исследования (четыре еженедельные дозы). Все введения в обоих исследованиях осуществляли подкожно.
[0116] Критерии включения пациента в исследования были следующими: (1) пациент должен быть мужчиной или женщиной в возрасте ≥18 лет; (2) иметь хронический атопический дерматит в течение 3 лет; (3) иметь EASI ≥12; (4) IGA ≥3; (5) ≥15% вовлечения BSA в AD (в США) или ≥10% вовлечения BSA в AD (за пределами США); и (6) иметь в анамнезе недостаточный ответ на постоянный режим лечения местными кортикостероидами (TCS) или ингибиторами кальциневрина.
[0117] Критерии исключения пациента из исследования были следующими: (1) WBC <3,5×103/мкл; (2) тромбоциты <125×103/мкл; (3) нейтрофилы <1,75×103/мкл; (4) AST/ALT >1,5x ULN; (5) положительный по гепатиту B или гепатиту C; и (6) лечение TCS или ингибиторами кальциневрина в течение 1 недели от исходного уровня.
[0118] Первичной конечной точкой исследований было осуществление контроля частоты возникновения нежелательных явлений, возникших на фоне лечения (TEAE) от исходного уровня до 12 недели. Исследовательскими конечными точками переменных эффективности были: (i) % достижения IGA 0 или 1 к 4 неделе; (ii) % снижение BSA и EASI от исходного уровня; и (iii) изменение от исходного уровня показателя по шкале NRS.
[0119] Исследовательские переменные эффективности, измеренные в данном исследовании, включали: (1) долю пациентов, у которых достигали показателя общего оценки исследователем (IGA) 0 или 1 к 4 неделе и к каждому визиту; (2) изменение и процентное изменение вовлечения площади поверхности тела в атопический дерматит (BSA), индекс распространенности и тяжести экземы (EASI), показатель SCORAD и 5-D зуда от исходного уровня к каждому визиту; (3) еженедельное изменение от исходного уровня оценки зуда по цифровой оценочной шкале (NRS); (4) изменение от исходного уровня эозинофилов крови, TARC, эотаксина-3 и общего уровня IgE к 4 недели; (5) изменение от исходного уровня эозинофилов, TARC, эотаксина-3 и общего уровня IgE к 12 неделе; и (6) изменение от исходного уровня эозинофилов, TARC, эотаксина-3, результатов Phadiatop™ и уровня общего IgE, связанных с ответом к 4 неделе.
[0120] Исходный уровень для переменных эффективности определяли как последнюю непропущенную величину в день или за день до рандомизации. Для пациента, у которого отсутствует величина в день или за день до его/ее рандомизации, в качестве исходного уровня будут использовать последнюю непропущенную величину в день или за день до инъекции первой дозы.
[0121] Общая оценка исследователем (IGA): IGA представляет собой шкалу оценки, используемую в клинических исследованиях для определения тяжести AD и клинического ответа на лечение на основании 6-балльной шкалы, варьирующей от 0 (отсутствует) до 5 (очень тяжелый). Показатель IGA оценивали при каждом визите в клинику.
[0122] Вовлечение площади поверхности тела в атопический дерматит (BSA): BSA, пораженную AD, оценивали для каждого основного участка тела (головы, туловища, верхних конечностей и нижних конечностей) и описывали в виде общего процента от каждого участка тела. Пациентов оценивали в отношении BSA в следующих визитах: отбор, 1 день/начальный момент (до введения препарата) и 15, 29, 36, 43, 57, 71 и 85 дни (окончание исследования) или досрочное завершение.
[0123] Индекс распространенности и тяжести экземы (EASI): EASI является валидированным показателем, используемым в клинической практике и клинических испытаниях для оценки тяжести и степени AD (Hanifin et al 2001, Exp. Dermatol. 10: 11-18). Расчет величины EASI основан на врачебной оценке отдельных признаков [эритемы (E), индурация/образование папул (I), экскориации (X) и лихенизации (L)], где каждый признак оценивается как 0=отсутствует, 1=слабый, 2=средний или 3=тяжелый, а также на основании показателя площади [исходя из пораженного % (BSA)], где 0=0% BSA, 1=1-9% BSA, 2=10-29% BSA, 3=30-49% BSA, 4=50-69% BSA, 5=70-89% BSA, 6=90-100% BSA.
[0124] Для каждого из основных участков тела (головы, верхних конечностей, туловища и нижних конечностей) показатель EASI=(E+I+X+L) x показатель поверхности. Общий показатель EASI представляет собой взвешенное общее EASI участка исходя из удельной массы 10%=голова, 20%=верхние конечности, 30%=туловище, 40%=нижние конечности. Минимальный возможный показатель EASI равняется 0 и максимальный возможный показатель EASI равняется 72, где более высокая величина указывает на повышенную тяжесть атопического дерматита. Достижение EASI 50 (50% или большее снижение показателя EASI считается дерматологами-исследователями клинически значимым уровнем снижения для использования в качестве конечной точки.
[0125] У пациентов оценивали показатель EASI в следующих визитах: отбор, 1 день/начальный момент (до введения препарата) и 15, 29, 36, 43, 57, 71 и 85 дни (окончание исследования) или досрочное завершение.
[0126] SCORAD: SCORAD является валидированным средством, используемым в клинических исследованиях и клинической практике, которое было разработано для стандартизации оценки степени и тяжести AD (Dermatology 1993, 186: 23-31). Степень AD оценивают в виде процента каждой определенной площади тела и описывают в виде суммы всех площадей, при этом максимальная величина составляет 100% (определяется как "A" в расчете всего показателя SCORAD). Тяжесть 6 определенных симптомов (эритемы, отека/образования папул, экскориации, лихенификации, просачивания/образования корок и сухости) AD оценивают при помощи следующей шкалы: отсутствует (0), слабая (1), средняя (2) или тяжелая (3) (для максимума из 18 баллов обозначается как "B" при расчете полного индекса SCORAD). Субъективную оценку зуда и бессонницы для каждого симптома дает пациент или родственник по визуальной аналоговой шкале (VAS), где 0 означает отсутствие зуда (или бессонницы) и 10 означает наиболее сильный воображаемый зуд (или бессонницу) при максимальном возможном показателе 20. Этот параметр определяется как "C" в расчете полного показателя SCORAD. Показатель SCORAD рассчитывают как A/5+7B/2+C. Максимальный показатель SCORAD составляет 103.
[0127] Пациенты подлежали оценке по шкале SCORAD в следующих визитах: отбор, 1 день/исходный уровень (до введения препарата) и 15, 29, 36, 43, 57, 71 и 85 дни (окончание исследования) или досрочное завершение.
[0128] Шкала 5-D зуда: Шкала 5-D для оценки зуда представляет собой средство из 5 вопросов, используемое в клинических испытаниях для оценки 5 параметров зуда на исходном уровне: степени, продолжительности, направленности, нарушения функций и распространенности (Elman et. al.2010, Brit. J. Dermatol. 162: 587-593). Пациенты ранжируют свои симптомы в течение предшествующего 2-недельного периода как «присутствуют» или шкале от 1 до 5, при этом 5 является максимальной степенью нарушения для каждого вопроса по степени, продолжительности, направленности и нарушению функций. Показатели для полей из одного пункта (продолжительность, степень или направленность) равны величине, указанной ниже варианта ответа (диапазон 1-5).
[0129] Поле нарушения функций включает четыре пункта, которые оценивают влияние зуда на повседневную деятельность: сон, отдых/социальную активность, домашнюю работу/поездки и работу/учебу. Показатель поля нарушения функций получают при учете наибольшей величины по любому из четырех пунктов.
[0130] Для поля распространенность суммируют число пораженных участков тела (возможная сумма 0-16) и сумму соотносят с пятью вариантами показателя: сумма 0-2=балл 1, сумма 3-5=балл 2, сумма 6-10=балл 3, сумма 11-13=балл 4 и сумма 14-16=балл 5.
[0131] Показатели по каждому из пяти полей получают отдельно и затем суммируют вместе для получения общего показателя 5-D. Показатели 5-D потенциально могут варьировать от 5 (отсутствие зуда) до 25 (наиболее тяжелый зуд).
[0132] Осуществляли оценку зуда у пациентов по шкале 5-D в следующих визитах: отбор, 1 день/исходный уровень (до введения препарата) и 15, 29, 43, 57, 71 и 85 дни (окончание исследования) или досрочное завершение.
[0133] Цифровая шкала для оценки зуда (NRS): NRS для оценки зуда представляет собой средство оценки из одного вопроса, который использовали для оценки наиболее сильного зуда у пациента в результате AD в предшествующие 12 часов. С пациентами связываются по IVRS два раза в сутки с вечера визита скринингового обследования и задают следующий вопрос, «по шкале от 0 до 10, при этом 0 означает 'отсутствие зуда' и 10 означает 'наиболее сильный воображаемый зуд', как вы бы оценили вашу самую сильную степень зуда, испытываемого в течение предшествующих 12 часов?» Пациентов обучают использовать IVRS для записи своего показателя зуда по шкале NRS при визите скринингового обследования и запрашивают информацию о соблюдении режима терапии при каждом следующем визите в клинику. Пациенты заполняют рейтинговую шкалу два раза в сутки до последнего визита в рамках исследования.
[0134] Исходную величину NRS определяют как среднее из зарегистрированных показателей NRS в течение времени от непосредственно после визита скринингового обследования до непосредственно перед визитом исходного уровня. Для показателя NRS после исходного уровня, средний еженедельный показатель NRS рассчитывают как среднее из описанных ежедневных показателей NRS в течение недели (пропорционально распределенное среднее).
[0135] Показатели IGA, BSA, EASI и SCORAD оценивали при каждом визите в клинику. Осуществляли оценку зуда у пациентов по шкале 5-D в следующих визитах: отбор, 1 день/исходный уровень (до введения препарата) и 15, 29, 43, 57, 71 и 85 дни (окончание исследования) или досрочное завершение. Пациенты использовали IVRS для записи своего показателя зуда по шкале NRS два раза в сутки ежедневно до последнего визита в рамках исследования.
[0136] Исходный уровень для переменных эффективности определяли как последнюю непропущенную величину в день или за день до рандомизации. Для пациента, у которого отсутствует величина в день или за день до его/ее рандомизации, в качестве исходного уровня будут использовать последнюю непропущенную величину в день или за день до инъекции первой дозы.
[0137] Исходные демографические характеристики для группы пациентов представлены ниже в таблице 1.
Основные демографические характеристики
[0138] Средние характеристики исходного уровня заболеваний представлены в таблице 2.
Средние исходные характеристики заболеваний
[0139] Результаты поисковых показателей эффективности, полученные из объединенных исследований, обобщены в таблицах 3-11 и фигурах 1-8.
Обобщенные результаты о субъектах, достигающих IGA ≤1 на 29 день и во все визиты в рамках исследования
Обобщенные результаты процента и абсолютного изменения от исходного уровня показателя BSA - все величины представлены в виде среднего (SD)
Обобщенные результаты процента и абсолютного изменении от исходного уровня показателя EASI - все величины представлены в виде среднего (SD)
Обобщенные результаты процента и абсолютного изменения от исходного уровня показателя 5-D зуда - все величины представлены в виде среднего (SD)
Обобщенные результаты процента и абсолютного изменения от исходного уровня среднего еженедельного показателя по шкале NRS - все величины представлены в виде среднего (SD)
Обобщенные результаты процента и абсолютного изменения от исходного уровня показателя IGA - все величины представлены в виде среднего (SD)
Число (%) субъектов, достигающих EASI-50 на 29 день и в каждый визит в рамках исследования - LOCF
Число (%) субъектов, достигающих EASI-25 на 29 день и в каждый визит в рамках исследования - LOCF
Число (%) субъектов, достигающих EASI-75 на 29 день и в каждый визит в рамках исследования - LOCF
[0140] mAb1 хорошо переносилось и было эффективным у взрослых с AD со степенью тяжести от средней до тяжелой. Введение mAb1 значительно снижало активность и тяжесть заболевания AD. На 4 неделе при введении 150 мг и 300 мг g mAb1 достигали значительного снижения по сравнению с плацебо по изменению от исходного уровня %BSA (p<0,05) (фигура 1), IGA (p<0,001) (фигура 2), EASI (p<0,001) (фигура 3) и зуда по шкале NRS (p<0,01, 300 мг) (фигура 4). У большего числа пациентов отмечали ≥50% снижение показателя EASI при 150 мг mAb1 (54,5%) и при 300 мг (71,4%) по сравнению с плацебо (18,8%; p<0,05 для обоих доз) (фигуры 5 и 6). Большее число пациентов достигали EASI-25, EASI-50 и EASI-75 при mAb1 по сравнению с плацебо на 4 неделе (фигура 7).
[0141] Что касается 300 мг mAb1, то значимое снижение наблюдали в течение 2 недель по %BSA (p<0,02), IGA (p<0,05) и EASI (p<0,0001). Снижение BSA, IGA и EASI (p<0,05 по сравнению с плацебо) сохранялось в течение 8 недель. Процент пациентов с IGA 0 или 1 на 4 неделе был выше, чем в группе плацебо, но был статистически не значим (фигура 8).
[0142] Наиболее распространенными нежелательными явлениями, возникшими на фоне лечения (AE), при введении mAb1 были назофарингит (19,6% по сравнению с 12,5% для плацебо) и головная боль (11,8% по сравнению с 6,3% для плацебо).
Пример 2. Клиническое испытание с повторными дозами подкожно вводимого антитела к IL-4R (mAb1) у взрослых пациентов с атопическим дерматитом со степенью тяжести от средней до тяжелой
А. План исследования
[0143] Данное исследование представляло собой 28-недельное рандомизированное двойное слепое плацебо-контролируемое исследование mAb к IL-4R, обозначаемого в данном документе как «mAb1», вводимого подкожно, у пациентов с атопическим дерматитом cо степенью тяжести от средней до тяжелой. Продолжительность периода лечения составляла 12 недель, затем пациентов наблюдали в течение дополнительных 16 недель после окончания лечения.
[0144] Для исследования включали и рандомизировали 109 пациентов в соотношении 1:1 (54 в группе плацебо и 55 в группе с 300 мг антитела). От участия в исследовании отказались 43 пациента (30 в группе плацебо и 13 в группе 300 мг). Рандомизацию проводили в соответствии с уровнями IgE (IgE<150 кЕ/л по сравнению с ≥150 кЕ/л в ходе визита скринингового обследования) для исследования эффективности mAb1 у пациентов с внешней или внутренней формой AD. Пациентов, которые соответствовали критериям отбора, оценивали на 1 день/исходный уровень, рандомизировали и затем они получали 300 мг mAb1 или плацебо подкожно. Каждую еженедельную дозу исследуемого лекарственного средства вводили в виде одной инъекции по 2 мл или разделяли на две инъекции по 1 мл. Для еженедельных визитов пациенты возвращались в клинику и получали инъекцию исследуемого лекарственного средства на 8, 15, 22, 29, 36, 43, 50, 57, 64, 71 и 78 дни. Осуществляли тщательный контроль состояния пациентов на базе исследовательского центра в течение минимум 2 часов после введения каждой дозы исследуемого лекарственного средства. Окончание периода лечения приходилось на 85 день. Визиты последующего наблюдения приходились на 92, 99, 106, 113, 120, 127, 134, 141, 148, 155, 162, 169, 176, 183, 190 дни и окончание визита в рамках исследования на 197 день.
[0145] Критерии включения в исследование были следующими: (1) мужчина или женщина в возрасте 18 лет или старше; (2) хронический AD, диагностированный по пересмотренному критерию Eichenfield по Hannifin and Rajka, который присутствовал в течение по меньшей мере 3 лет до визита скринингового обследования; (3) показатель EASI ≥16 в ходе визита скринингового обследования и визита исходного уровня; (4) величина IGA ≥3 в ходе визита скринингового обследования и визита исходного уровня; (5) ≥10% вовлечения BSA в AD в ходе визита скринингового обследования и визита исходного уровня; (6) наличие в анамнезе недостаточного ответа на постоянный (≥1 месяца) режим местными кортикостероидами или ингибиторами кальциневрина в качестве лечения AD в течение последних 3 месяцев до визита скринингового обследования; (7) пациенты должны были использовать постоянную дозу не содержащего примесей основного смягчающего средства два раза в сутки в течение по меньше мере 7 дней до визита исходного уровня; и (8) готовность, обязанность и возможность возвращаться для всех визитов в клинику и завершать все связанные с исследованием процедуры, а также готовность и возможность подписать форму информированного согласия (ICF).
[0146] Критерии исключения из исследования были следующими: (1) предшествующее лечение mAb1; (2) наличие любого из отклонений в следующих лабораторных показателях в ходе визита скринингового обследования: количество лейкоцитов <3,5×103/мкл; корличество тромбоцитов <125×103/мкл; количество нейтрофилов <1,75×103/мкл; количество аминотрасферазы (AST)/аланинтрансферазы (ALT) >1,5x ULN; и CPK >2x ULN; (3) положительные или промежуточные результаты в ходе визита скринингового обследования на поверхностный антиген вируса гепатита B, антитело к капсидному антигену вируса гепатита B или вируса гепатита C; (4) появление новой физической нагрузки или существенное изменение в предшествующей физической нагрузке в течение 4 недель до отбора (визит 1). Субъекты должны были изъявить готовность сохранять одинаковый уровень физической нагрузки в ходе исследования и воздерживаться от нетипичной интенсивной физической нагрузки в ходе испытания; (5) лечение экспериментальным лекарственным средством в течение 8 недель или в течение 5 периодов полужизни, если известно, в зависимости от того, что дольше, до исходного визита; (6) лечение живой (аттенуированной) вакциной в течение 12 недель до визита исходного уровня; (7) лечение при помощи аллерген-специфической иммунотерапии в течение 6 месяцев до визита исходного уровня; (8) лечение ингибиторами лейкотриенов в течение 4 недель до визита исходного уровня; (9) лечение системными кортикостероидами в течение 4 недель до визита исходного уровня; (10) лечение местными кортикостероидами, такролимусом и/или пимекролимусом в течение 1 недели до визита исходного уровня; (11) системное лечение AD иммуносупрессирующим/иммуномодулирующим соединением, например, циклоспорином, микофенолата мофетилом, IFN-γ, фототерапией (узкополосным УФ-B, УФ-A1, псораленом+УФ-A), азатиоприном, метотрексатом или биологическими препаратами в течение 4 недель до визита исходного уровня; (12) три или более отбеливающих ванны во время любой недели в течение 4 недель до визита исходного уровня; (13) лечение AD при помощи медицинского устройства (например, Atopiclair®, MimyX®, Epicerum®, Cerave® и т.п.) в течение 1 недели до визита исходного уровня; (14) хроническая или острая инфекция, требующая лечения антибиотиками для перорального или внутривенного введения, противовирусными препаратами, противопаразитарными средствами, антипротозойными средствами или противогрибковыми средствами в течение 4 недель до визита скринингового обследования, или поверхностные кожные инфекции в течение 1 недели до визита скринингового обследования; (15) ВИЧ-инфекция в анамнезе; (16) в анамнезе реакции гиперчувствительности к доксициклину или родственным соединениям; (17) в анамнезе клиническая паразитарная инфекция, отличная от вагинального трихомониаза; (18) в анамнезе злокачественная опухоль в течение 5 лет до визита исходного уровня со следующими исключениями; допускаются пациенты с полностью излеченной карциномой шейки матки in situ, и неметастатической сквамозно-клеточной или базально-клеточной карциномой кожи в анамнезе; (19) плановая хирургическая процедура во время участия пациента в исследовании; (20) применение вертикального солярия/солярия в течение 4 недель до визита скринингового обследования; (21) тяжелое сопутствующее заболевание или тяжелое заболевание в анамнезе, такое как психическое заболевание, заболевание сердца, заболевание почек, неврологическое, эндокринное, метаболическое или лимфатическое заболевание, или любое другое заболевание или состояние, которое бы нежелательным образом повлияло на участие субъекта в данном исследовании; (22) беременные женщины или женщины, кормящие грудью; и/или (23) нежелание использовать надлежащий контроль рождаемости. Надлежащий контроль рождаемости определяют как соглашение систематически применять эффективный и приемлемый способ контрацепции в ходе исследования и в течение 16 недель после последней дозы исследуемого лекарственного средства. Для женщин способы надлежащего контроля рождаемости определены использованием гормональных контрацептивов, внутриматочного устройства (IUD), или предполагают двойную барьерную контрацепцию (т.е., презерватив+диафрагма, презерватив или диафрагма+спермицидный гель или пена). Для мужчин способы надлежащего контроля рождаемости определены двойной барьерной контрацепцией (т.е., презерватив+диафрагма, презерватив или диафрагма+спермицидный гель или пена). Для женщин менопаузу определяют как отсутствие в течение 24 месяц менструаций; в сомнительных случаях необходимо подвтердить документально уровень фолликулостимулирующего гормона ≥25 ЕД/мл. Необходимо подтвердить докуменатально гистерэктомию, билатеральную оофорэктомию или билатеральное лигирование маточных труб, если это необходимости.
B. Переменные эффективности
[0147] Первичная конечная точка представляла собой процентное изменение показателя EASI от исходного уровня к 12 неделе. Вторичные конечные точки, измеряемые в данном исследовании, включали: (1) долю пациентов, которые достигали величины общей оценки исследователем (IGA) 0 или 1 на 12 неделе; (2) долю пациентов, которые достигали ≥50% общего снижения показателя EASI (также называемого EASI 50) от исходного уровня к 12 неделе; (3) изменение показателя EASI от исходного уровня к 12 неделе; (4) изменение и процентное изменение показателя IGA, вовлечения площади поверхности тела в атопический дерматит (BSA), индекса распространенности и тяжести экземы (EASI), SCORAD, показателя зуда согласно NRS и зуда по шкале 5-D от исходного уровня к 12 неделе; (5) частоту возникновения TEAE от исходного уровня к 28 неделе; (6) изменение от исходного уровня содержания эозинофилов, TARC, результаты Phadiatop™ и общий уровень IgE, связанный с ответом; (7) изменение QoLIAD от исходного уровня к 12 неделе; (8) процент пациентов, которые достигают снижения величины IGA ≥2 от исходного уровня к 12 неделе; (9) процент пациентов, которые достигают снижения величины IGA ≥3 от исходного уровня к 12 неделе; и (10) PD ответ эозинофилов крови, TARC и общий уровень IgE.
[0148] Исходный уровень для переменных эффективности определяли как последнюю непропущенную величину в день или за день до рандомизации. Для пациента, у которого отсутствует величина в день или за день до его/ее рандомизации, в качестве исходного уровня будут использовать последнюю непропущенную величину в день или за день до инъекции первой дозы.
Процедуры исследования
[0149] Переменные эффективности IGA, BSA, EASI, SCORAD, показатель зуда по шкале 5-D и показатель зуда согласно NRS описывали в других частях данного документа (см. пример 1).
[0150] Показатели IGA, BSA, EASI и SCORAD оценивали при каждом визите в клинику. У пациентов оценивали показатель зуда по шкале 5-D в следующие визиты: скрининг, 1 день/исходный уровень (до введения препарата) и 15, 29, 43, 57, 71, 85, 99, 113, 127, 141, 155, 169, 183 и 197 дни (окончание исследования) или досрочное завершение. Пациенты использовали IVRS для записи своего показателя зуда по шкале NRS два раза в сутки ежедневно до последнего визита в рамках исследования.
[0151] Индекс качества жизни при атопическом дерматите (QoLIAD): QoLIAD представляет собой валидированный опросник из 25 пунктов, используемый в клинической практике и клинических испытаниях для оценки влияния симптомов заболевания и лечения AD на QoL (Whalley et al 2004, Br. J. Dermatol. 150: 274-283; Meads et al 2005, Value Health 8: 331-332). Формат представлен в виде простого ответа да/нет на 25 пунктов с системой оценки от 0 до 25; высокий показатель указывает на плохое QoL. Анализ является чувствительным к изменению, при этом разниц на 2-3 пункта считается клинически значимой. Опросник предлагали подгруппе пациентов в ходе отбора и в 1 день/исходный уровень (до введения препарата), а также в 29, 57, 85 и 197 дни (окончание исследования) или в момент досрочного завершения. Разницу между средствами лечения сравнивали при помощи модели ковариационного анализа (ANCOVA) с соответствующим исходным уровнем в качестве ковариаты.
С. Экспериментальное средство лечения
[0152] Фармацевтический продукт на основе mAb1 предоставляли в виде лиофилизированного порошка в стеклянном флаконе на 5 мл для подкожного введения. При подкожном введении фармацевтический продукт на основе mAb1 восстанавливали 2,5 мл стерильной воды для инъекций, получая раствор, содержащий 150 мг/мл mAb1. Уровень исследуемой дозы mAb1 составлял 300 мг для подкожного введения. mAb1 или плацебо вводили в виде 1 (2 мл) или 2 (1 мл) подкожных инъекций в клинике в 1 день/исходный уровень и 8, 15, 22, 29, 36, 43, 50, 57, 64, 71 и 78 дни. Несмотря на то, что введение каждой еженедельной дозы исследуемого лекарственного средства в виде одной инъекции по 2 мл было предпочтительным, каждую еженедельную дозу могли разделять на две инъекции по 1 мл. Места для подкожных инъекций чередовали между следующими местами: тыльная сторона рук, живот (кроме пупка или области талии) и верхние части бедер. Введение в конечности не разрешалось вследствие возможности различий в абсорбции и биодоступности. Если требовалось введение нескольких инъекций в один и тот же день, то каждую инъекцию проводили в разных местах для инъекций (например, 1 инъекцию в правый нижний квадрант живота, а другую в левый нижний квадрант живота). Места для подкожных инъекций чередовали с тем, чтобы в одних и тех же местах не инъецировали в течение 2 последовательных недель.
[0153] Плацебо, соответствующее mAb1, готовили в том же составе, что и mAb1, но без добавления антитела.
[0154] Осуществляли контроль состояния пациентов на базе исследовательского центра в течение минимум 2 часов после введения каждой дозы исследуемого лекарственного средства.
[0155] Кроме того, от пациентов требовали применять в постоянных дозах основное смягчающее средство, не содержащее примесей, два раза в сутки в течение по меньшей мере 7 дней до визита исходного уровня и в ходе участия в исследовании. Пациенты отмечали соблюдение фонового лечения в ходе исследования при помощи IVRS или IWRS. Система запрашивала у пациентов ответ на следующий вопрос о применении смягчающего средства: «Вы пользуетесь увлажнителем, одобренным врачом-исследователем, на пораженных участках вашей кожи?».
D. Оценка безопасности
[0156] Безопасность оценивали в ходе исследования при помощи контроля нежелательных явлений и серьезных нежелательных явлений.
[0157] Нежелательным явлением (AE) является любое нежелательное медицинское явление у субъекта или субъекта, участвующего в клиническом исследовании, которому вводят фармацевтический продукт. Таким образом, AE может представлять собой любое неблагоприятный или непредусмотренный признак (в том числе не соответствующие норме данные лабораторного исследования), симптом или заболевание, временно ассоциированные с применением лекарственного препарата, который считается или не считается связанным с лекарственным (экспериментальным) препаратом. AE также включают: любое ухудшение (т.е. любое клинически значимое изменение частоты возникновения и/или интенсивности) ранее существующего состояния, которое временно ассоциировано с применением исследуемого лекарственного средства; не соответствующие норме данные лабораторного исследования, считающиеся исследователем клинически значимыми; и любое нежелательное медицинское явлением.
[0158] Серьезным нежелательным явлением (SAE) является любое нежелательное медицинское явление, которое при любой дозе приводит к смерти; угрожает жизни; требует стационарной госпитализации или продления существующей госпитализации; приводит к устойчивому или значительному нарушению функций/нетрудоспособности; является врожденной аномалией/пороком развития или является важным медицинским явлением.
[0159] Кроме того, в ходе исследования измеряли лабораторные переменные безопасности, переменные жизненно важных признаков, переменные электрокардиографии в 12 отведениях (ЭКГ) и переменные физического обследования.
[0160] Клинические лабораторные данные состоят из общего анализа крови, биохимического анализа крови и общего анализа мочи. Образцы крови для проведения общего анализа крови собирали во время каждого визита в рамках исследования; образцы крови для биохимического исследования сыворотки крови и образцы мочи для общего анализа мочи собирали для оценки общего состояния здоровья пациента в ходе скринингового обследования, в 1 день/исходный уровень (до приема препарата), 15 день, 29 день, 43 день, 57 день, 71 день, 85 день, 99 день, 113 день, 141 день, 169 день и 197 день (окончание исследования) или в момент досрочного завершения, если субъект прекращает участие в исследовании.
[0161] Параметры жизненно важных функций включают частоту дыхания (уд./мин.), частоту пульса (уд./мин.), систолическое и диастолическое кровяное давление (мм рт.ст.) и температуру тела (°C). Данные о жизненно важных функциях получали (до приема препарата, в дни введения дозы) в ходе скрининга и в 1 день/исходный уровень и 8, 15, 22, 29, 36, 43, 50, 57, 64, 71, 78, 85, 99, 113, 141, 169 и 197 дни (окончание исследования) или в момент досрочного завершения. Данные о жизненно важных функциях получали через 1 и 2 часа после инъекции, затем после введения доз исследуемого лекарственного средства в 1, 8, 15, 22, 29, 36, 43, 50, 57, 64, 71 и 78 дни.
[0162] Параметры ЭКГ в 12 отведениях включают: HR желудочков, интервал PR, интервал QRS, интервал QT с поправкой (QTcF=QT/[RR0,33] и QTcB=QT/[RR0,5]). Статус ЭКГ: нормальный, нарушенный клинически незначимый, или нарушенный клинически значимый. Стандартную ЭКГ в 12 отведениях выполняли при скрининге, в 141 день и 197 день (окончание исследования) или в момент досрочного завершения.
[0163] Образцы для исследования (сыворотку крови/РНК/плазму крови) собирали при скрининговом обследовании и в 1 день/начальный момент (до введения препарата) и 8, 15, 22, 29, 57, 85 и 197 дни (окончание исследования) или в момент досрочного завершения, и в ходе незапланированных визитов.
[0164] Тщательное и полное физическое обследование проводили при скрининговом обследовании, в 85 день и 197 день (окончание исследования) или в момент досрочного завершения.
E. Анализ данных
1. Анализ переменных поисковых показателей эффективности
[0165] Все качественные переменные анализировали с использованием точного критерия Фишера с номинальным p-значением и описанными доверительными интервалами. Все непрерывные переменные анализировали с помощью ковариационного анализа (ANCOVA) с использованием слоя исходного IgE (<150 кЕ/л по сравнению с ≥150 кЕ/л в ходе визита скринингового обследования). Если не указано иное, то оценки изменений от исходного уровня и построение доверительных интервалов для непрерывных показателей были основаны на модели ANCOVA, которая включает лечение в качестве основного фактора и исходную величину в качестве ковариат. Представлены точечное оценивание и 95% CI разницы в откорректированном среднем изменении от исходного уровня между двумя группами лечения. Отсутствующие величины будут замещать на основании подхода переноса данных последнего наблюдения вперед (LOCF). В случае, если предположения модели не подтверждены, то будут использовать ранговый ковариационный анализ.
2. Анализ данных по безопасности
[0166] Анализ безопасности основывается на описываемых AE, клинических лабораторных оценках, показателях жизненно важных функций и ЭКГ в 12 отведениях. Пороговые значения для потенциально клинически значимых величин (PCSV) лабораторных переменных, показателей жизненно важных функций и ЭКГ определяли в SAP. Временной интервал для обнаружения любого явления или нарушения находится между инфузией исследуемого терапевтического препарата и окончанием исследования. Данные, собранные за пределами этого интервала, исключают из расчета описательной статистики и выявления нарушений для лабораторных оценок, показателей жизненно важных функций и ЭКГ.
F. Безопасность Результаты
[0167] Как правило, mAb1 хорошо переносилось при благоприятном профиле безопасности. В ходе исследования не было зарегистрировано каких-либо клинически значимых результатов лабораторных тестов (биохимический анализ крови, общий анализ крови или общий анализ мочи). Ни в одном из лабораторных параметров не наблюдали динамики в среднем/медианном исходном уровне. В ходе исследования значимая динамика средних или медианных изменений от исходного уровня температуры или пульса отсутствовала. Клинически значимых нарушений в результатах физического обследования, ЭКГ или жизненно важных функций не наблюдали.
[0168] Общий профиль нежелательных явлений (AE) был характерен для выборки здоровых людей. Случаев смерти зарегистрировано не было. У 8 пациентов были SAE, из которых 1 был в группе mAb1 (перелом костей лицевого черепа) и 7 были в группе плацебо (стенокардия, целлюлит, герпетическая экзема, бактериальная инфекция кожи, почечная недостаточность, астматический кризис, легочное нарушение и атопический дерматит). У 8 пациентов были TEAE, которые в результате привели к прекращению приема исследуемого лекарственного средства, ьиз которых 1 находился в группе mAb1 и 7 в группе плацебо. В группе mAb1 были 87 пациентов с по меньшей мере одним TEAE (n=43 [78,2%] по сравнению с 44 [81,5%] в группе плацебо). Наиболее частыми TEAE были назофарингеальные инфекции у субъектов, которым вводили дозу mAb1 (n=22 [40%] по сравнению с 10 [18,5%] для плацебо). Другие TEAE в группе лечения включали глазные инфекции, нарушения со стороны нервной системы, а также общие нарушения и нарушения в месте введения. TEAE за период 28 недель обобщены в таблице 12.
Нежелательные явления, возникающие на фоне лечения, в течение 28 недель
[0169] Отмечалось заметно меньше число кожных инфекций, ассоциированное с лечением mAb1 (5,5%), по сравнению с плацебо (24,1%). Число и тип кожных инфекций, наблюдаемых у принимавших mAb1 пациентов по сравнению с плацебо показаны в таблице 13.
Кожные инфекции у принимавших mAb1 пациентов по сравнению с плацебо
[0170] Подкожное введение mAb1 взрослым пациентам с AD со степенью от средней до тяжелой, как правило, было безопасным и хорошо переносимым.
G. Эффективность Результаты
[0171] Результаты исходных показателей и поисковых показателей эффективности, полученных из исследования, обобщены на фигурах 9-28 и в таблицах 14-24. Как отмечали выше, пациентов лечили путем подкожного введения 300 мг mAb1 один раз в неделю в течение 12 недель или плацебо.
Обобщенные результаты исходных характеристик - все величины представлены в виде среднего (SD)
300 мг
Обобщенные результаты исходных характеристик подгруппы для оценки QoLIAD
300 мг
Обобщенные результаты о проценте и абсолютном изменении показателя EASI от исходного уровня к 12 неделе и каждому визиту в ходе последующего периода наблюдения - все величины представлены в виде среднего (SD)
Обобщенные результаты о проценте и абсолютном изменении показателя IGA от исходного уровня к 12 неделе и каждому визиту в ходе последующего периода наблюдения - все величины представлены в виде среднего (SD)
Обобщенные результаты абсолютного изменения показателя BSA от исходного уровня к 12 неделе и каждому визиту в ходе последующего периода наблюдения - все величины представлены в виде среднего (SD)
Обобщенные результаты абсолютного изменения показателя SCORAD от исходного уровня к 12 неделе и каждому визиту в ходе последующего периода наблюдения - все величины представлены в виде среднего (SD)
Обобщенные результаты абсолютного изменения показателя зуда по шкале 5-D от исходного уровня к 12 неделе и каждой неделе в ходе последующего периода наблюдения - все величины представлены в виде среднего (SD)
Обобщенные результаты абсолютного изменения средней величины NRS от исходного уровня к 12 неделе и каждой неделе в ходе последующего периода наблюдения - все величины представлены в виде среднего (SD)
Обобщенные результаты от субъектов, достигающих показателя IGA 0 или 1 к 12 неделе и каждому визиту в ходе периода последующего наблюдения
(N=54)
(N=55)
Обобщенные результаты от субъектов, достигающих EASI 50 к 12 неделе и каждому визиту в ходе периода последующего наблюдения
(N=54)
(N=55)
Корреляции QoLIAD на исходном уровне
H. Выводы
[0172] Подкожное введение антитела к IL-4R (mAb1) взрослым пациентам с атопическим дераматитом со степенью тяжести от средней до тяжелой, как правило, было безопасным и хорошо переносилось после введения доз 300 мг в течение 12 недель. Введение mAb1 по 300 мг приводило к значимому снижению показателей зуда IGA, EASI, BSA, SCORAD и NRS к 85 дню, как в среднем, так и в абсолютном и процентном изменении по сравнению с исходным уровнем (см. таблицы 14, 16-21). Процент пациентов, достигающих показателя по шкале IGA 0 или 1 на 85 день для группы 300 мг, составлял 40,0%, в то время как такая же величина для плацебо составляла 7,4% (таблица 22). На 85 день процент пациентов, которые достигали процентного снижения показателя EASI на 50% ("EASI-50"), составлял 85,5% для группы 300 мг, в то время как EASI-50 для пациентов, получавших плацебо, на 85 день составил 35,2% (таблица 23). Процентное изменение показателя EASI от исходного уровня к 12 неделе в группе mAb1 было статистически значимым по сравнению с группой плацебо (-74,0% по сравнению с -23,0%, p-значение <0,0001). Лечение mAb1 значимо снижало все четыре компонента заболевания согласно EASI по сравнению с плацебо: эритему, инфильтрацию/образование папул/отек, экскориацию и лихенификацию (p<0,0001 для всех 4 компонентов; фигура 15). Группа лечения значимо отличалась от группы плацебо по всем вторичным точкам эффективности. Ниже представлены p-значения для: статус респондеров IGA (0 или 1) (<0,0001), статус респондеров EASI (<0,0001), абсолютное изменение EASI исходного уровня (<0,0001), абсолютное изменение IGA от исходного уровня (<0,0001), процентное изменение IGA от исходного уровня (<0,0001), абсолютного изменения BSA (<0,0001), абсолютного изменения SCORAD (<0,0001), абсолютного изменения показателя зуда по шкале NRS (<0,0001) и абсолютного изменения от исходного уровня показателя зуда по шкале 5-D к 12 неделе (<0,0001), соответственно. Лечение mAb1 значимо снижало все пять компонентов показателя зуда по шкале 5-D: продолжительность, степень, направленность, нарушений функций и распространенность (p<0,0001 для продолжительности, степени, нарушения функций и распространности, p<0,001 для направленности; фигура 24). Введение 300 мг mAb1 значимо улучшало QoL по сравнению с плацебо в течение 12-недельного периода лечения (фигура 27). Улучшение от QoLIAD исходного уровня было значимо выше при применении mAb1 по сравнению с плацебо через 4 недели (различие средних значений -5,1; 95% CI -7,7, -2,4; P=0,0003), через 12 недель (различие средних значений -6,4; 95% CI -9,0, -3,8; P<0,0001) и по окончанию исследования (различие средних значений -6,3; 95% CI -9,0, -3,7; P<0,0001). Через 12 недель изменения QoLIAD при применении mAb1 умеренно коррелировали с изменениями активности заболевания и зудом, при этом коэффициенты корреляции Пирсона составляли 0,435 для показателя EASI, 0,406 для количественной шкалы оценки зуда и 0,494 для показателя зуда по шкале 5D (все P<0,05). Кроме того, лечение 300 мг mAb1 также приводило к меньшему количеству кожных инфекций у пациентов с AD (5,5%) по сравнению с плацебо (24,5%), включая бактериальные, вирусные и грибковые инфекции. Пациенты, получавшие mAb1, характеризовались сниженной восприимчивостью к инфекциям микроорганизмами, чем пациенты, получавшие плацебо.
Пример 3. Анализ барьерной функции кожи
[0173] Анализ барьерной функции кожи проводили с образцами, полученными от субъектов, которые принимали участие в клинических испытаниях mAb1.
Исследование A
[0174] В ʹисследовании Aʹ субъектам с AD вводили либо mAb1 (75, 150 или 300 мг), либо плацебо в 1, 8, 15 и 22 дни исследования (т.е., четыре еженедельные дозы). Увлажнение рогового слоя (SCH; корнеометрия) и трансэпидермальную потерю воды (TEWL; эвапориметрия) (Vergananini et al 2010, J. Dermatol. Treatment 21: 126-129) измеряли на исходном уровне, в 29 день (окончание исследования) и 85 день (окончание исследования) для подгруппы пациентов в данном исследовании.
[0175] Медианные величины SCH и TEWL исходного уровня обобщены в таблице 25.
Медианные SCH и TEWL исходного уровня
[0176] Что касается SCH, то чем больше емкость (ед. абсорб.), тем более увлажненной является кожа. Результаты медианной величины SCH как для поврежденной, так и для неповрежденной кожи были ниже таких результатов, описываемых для «нормальной» кожи (т.е., кожи человека без AD). Как и ожидалось, величины SCH при повреждении были ниже, чем для неповрежденных участков (таблица 25).
[0177] Что касается TEWL, то чем больше этот показатель, тем более высокой является потеря воды кожей. Медианные величины TEWL исходного уровня были высокими (то есть, слишком большое испарение воды с кожи) в исследованных поврежденных участках по сравнению с такими величинами, описанных для нормальной кожи. Медианные величины при повреждении были выше, чем такие величины для неповрежденных участков. Медианные результаты для неповрежденной кожи для групп плацебо и 150 мг были близкими к описанным нормальным величинам.
[0178] Лечение mAb1 демонстрировало повышение SCH поврежденных и неповрежденных участков в группах при всех дозах. Результаты показывают на то, что существует динамика улучшения SCH в поврежденных участках при лечении mAb1 по сравнению с плацебо на 29 день (фигура 29). Неповрежденные участки характеризовались лучшим SCH на исходном уровне, а наблюдаемые величины изменений после окончания лечения, как правило, были столь большими (фигура 30).
[0179] Что касается TEWL, то может наблюдаться динамика снижения в поврежденных участках, за исключением группы 150 мг (фигура 31). Динамику снижения TEWL наблюдали в ходе измерений, полученных для неповрежденной кожи, однако ее наблюдали преимущественно после 29 дня, когда разрешали местное лечение (фигура 32).
Исследование B
[0180] Исследование B представляло собой исследование фазы 2b для оценки безопасности и эффективности mAb1 у взрослых с AD со степенью тяжести от средней до тяжелой. Подробности протокола раскрыты в примере 9 опубликованной патентной заявки США № US20140072583, которая включена в данный документ по всей своей полноте.
[0181] После ударной дозы пациенты получали плацебо (PBO) или mAb1 (100 мг один раз в 4 недели, 300 мг один раз в 4 недели, 200 мг один раз в 2 недели, 300 мг один раз в 2 недели 300 мг раз в неделю) в течение 16 недель (нед.) с последующим дополнительным 16-недельным периодом последующего наблюдения для оценки безопасности. Наиболее распространенными TEAE (PBO по сравнению с mAb1) были назофарингит (26,2% против 28%), головная боль (3,3% против 10,7%) и реакция в месте инъекции (3,3% против 6,9%). Проводили дополнительное исследование для оценки TEWL и увлажнения рогового слоя (SCH). При наличии исходных повреждений кожи mAb1 значимо снижало TEWL к 4 неделе (-27%±4 все mAb1 [n=44] по сравнению с +696%±645 PBO [n=7], p<0,0001, среднее изменение±SE) и улучшенную или сохраненную величину TEWL к 16 неделе (-42%±5 все mAb1 [n=31] по сравнению с +62,3%±33 PBO [n=3], p<0,001). Устойчивой разницы по SCH между пациентами, получавшими mAb1 и PBO, не наблюдалось. Снижение TEWL коррелировало с подавлением тимуса и хемокина, регулируемого активацией (TARC) (среднее % изменение, 16 нед., r Спирмена=0,32, p <0,01). Средние % изменения TARC в группе биомаркеров Th2, периостина или эозинофилов оценивали для корреляции со средними % изменениями других клинических показателей. На 16 нед., наиболее высокими коэффициентами корреляции были средние % изменения TARC с клиническими результатами (зуд по шкале 5D, r=0,45; пациент-ориентированное измерение экземы (POEM), r=0,40; площадью поверхности тела, r=0,39; показателем атопического дерматита (SCORAD), r=0,36; общей оценкой исследователем (IGA), r=0,35; показателем распространенности и тяжести экземы (EASI), r=0,33; все p<0,0001). Эти корреляции были независимыми от лечения. Блокада IL-4Rα посредством mAb1 может нормализовать патогенез AD как вследствие воспаления, так и барьерного нарушения.
Пример 4. Колонизация кожи Staphylococcus aureus
[0182] Анализ колонизации кожи микроорганизмами проводили с образцами, полученными от субъектов, которые принимали участие в клинических исследованиях mAb1. В ʹисследовании Aʹ субъектам с AD вводили либо mAb1 (75, 150 или 300 мг), либо плацебо в 1, 8, 15 и 22 дни исследования (т.е., четыре еженедельные дозы). Смывы кожи собирали и культивировали на присутствие S. aureus. Оценивали процент положительных образцов, собранных до и после лечения. Более 50% всех исходных образцов из поврежденной кожи при исследовании были положительными в отношении колонизации S. aureus (фигура 33). Во всех группах лечения процент положительных образцов снижался после лечения на 29 день (окончание лечения; фигура 34). Наблюдали динамику ответа на введение дозы, которая проявлялась в более низкой колонизации при лечении. Результаты 85 дня объединяли, при этом происходило постоянное снижение в группах 75 мг и плацебо, а в двух других группах введения дозы mAb1 происходило повышение (но ниже исходного уровня). Подобное наблюдение было получено для процента положительных образцов из неповрежденной кожи, однако, что касается снижения процента, то зависимости от дозы не наблюдали (фигуры 35-36). Культивирование S. aureus из образцов смывов кожи, собранных в данном исследовании, указывало на то, что лечение mAb1 может снизить колонизацию кожи S. aureus у пациентов с AD.
[0183] Настоящее изобретение не ограничено по объему конкретными вариантами осуществления, описанными в данном документе. В действительности, разные модификации настоящего изобретения, кроме описанных в данном документе, будут очевидны специалистам в данной области из изложенного выше описания и сопровождающих графических материалов. Такие модификации находятся в пределах прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА С ПОМОЩЬЮ АНТАГОНИСТА IL-4R | 2013 |
|
RU2666630C2 |
| СПОСОБЫ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА С ПОМОЩЬЮ АНТАГОНИСТА IL-4R | 2013 |
|
RU2698907C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ТЯЖЕЛОГО АТОПИЧЕСКОГО ДЕРМАТИТА ПУТЕМ ВВЕДЕНИЯ ИНГИБИТОРА IL-4R | 2017 |
|
RU2759630C2 |
| СПОСОБЫ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2020 |
|
RU2828020C2 |
| СПОСОБ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА ПОСРЕДСТВОМ ВВЕДЕНИЯ ИНГИБИТОРА ИЛ-4R | 2019 |
|
RU2801204C2 |
| СПОСОБЫ ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ ВАКЦИНЫ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2753869C2 |
| СПОСОБЫ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ АЛЛЕРГИИ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2777328C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКОГО СИНУСИТА С ПОЛИПАМИ НОСА ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2734490C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2713406C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОЛИПОЗА НОСА ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2014 |
|
RU2674680C2 |






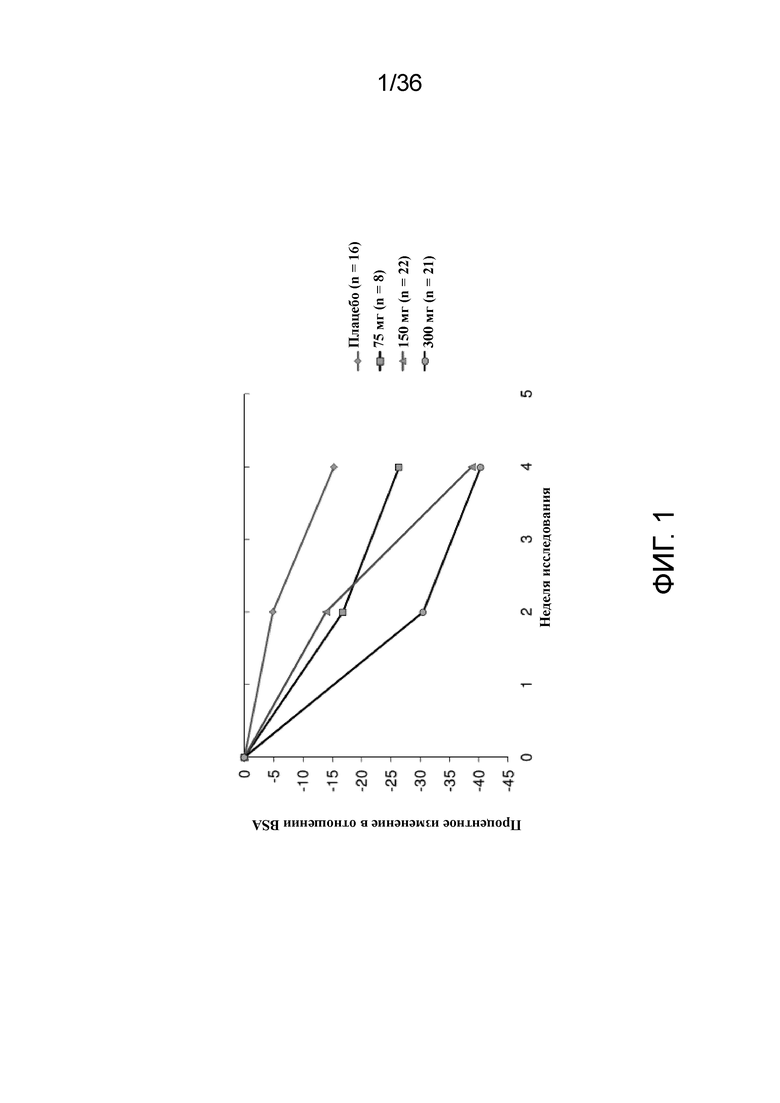
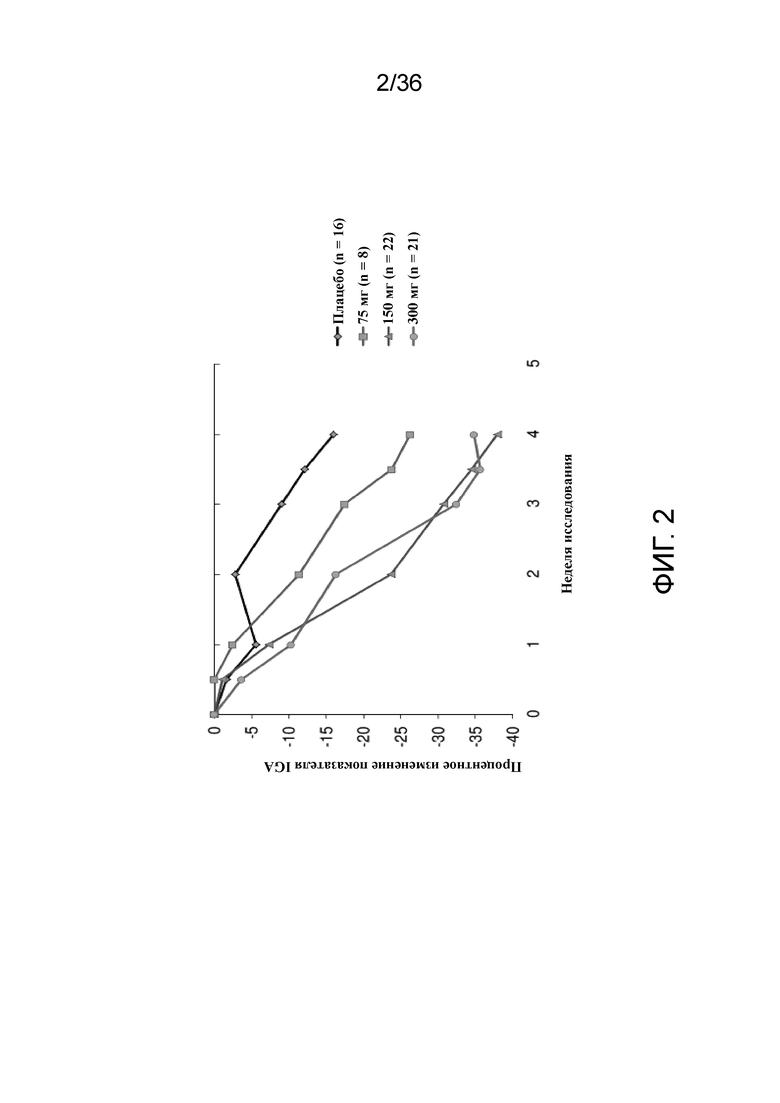

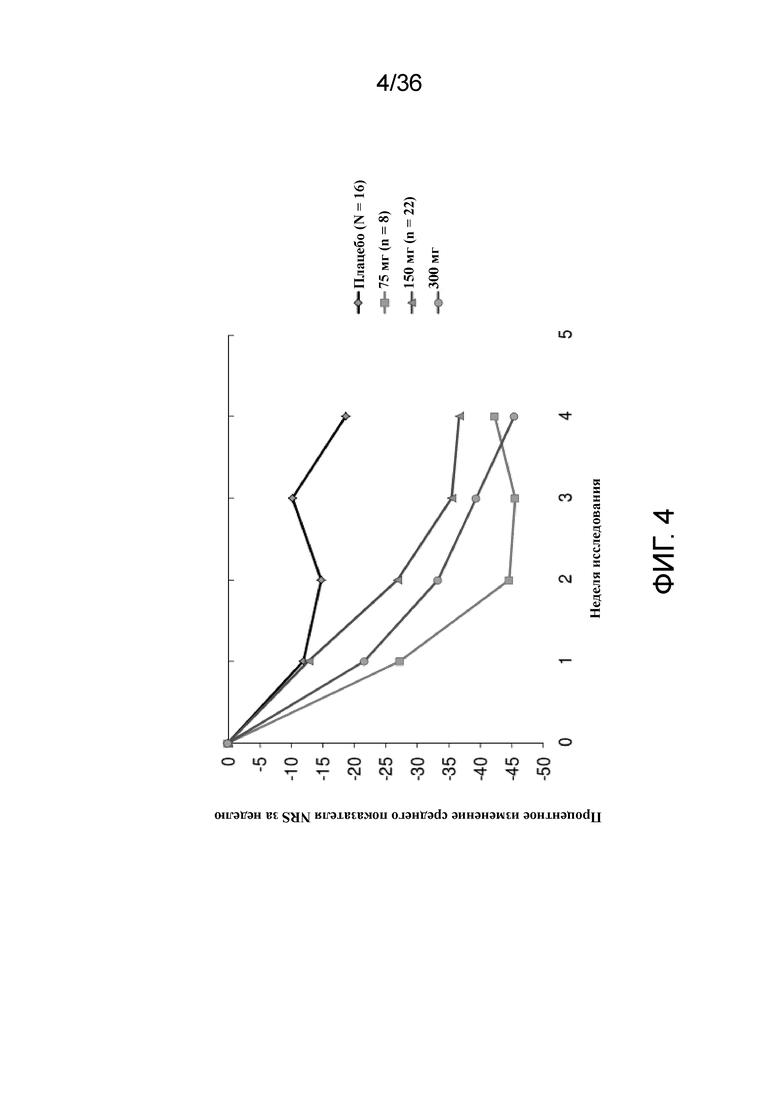
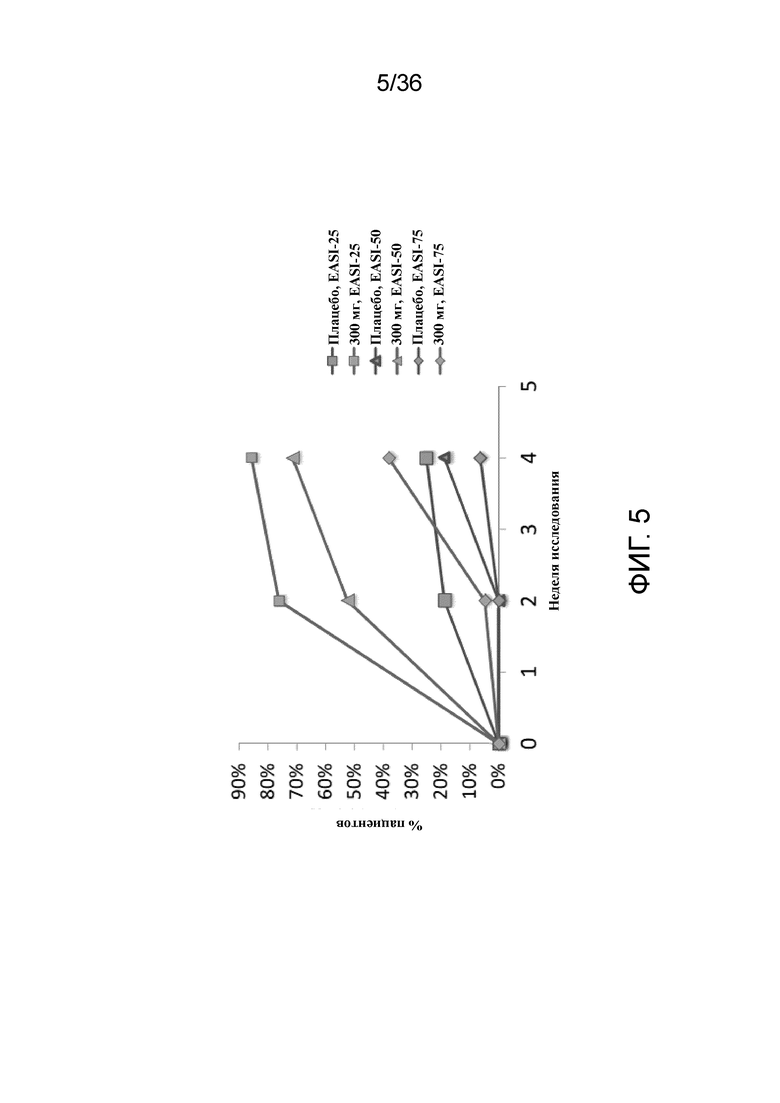
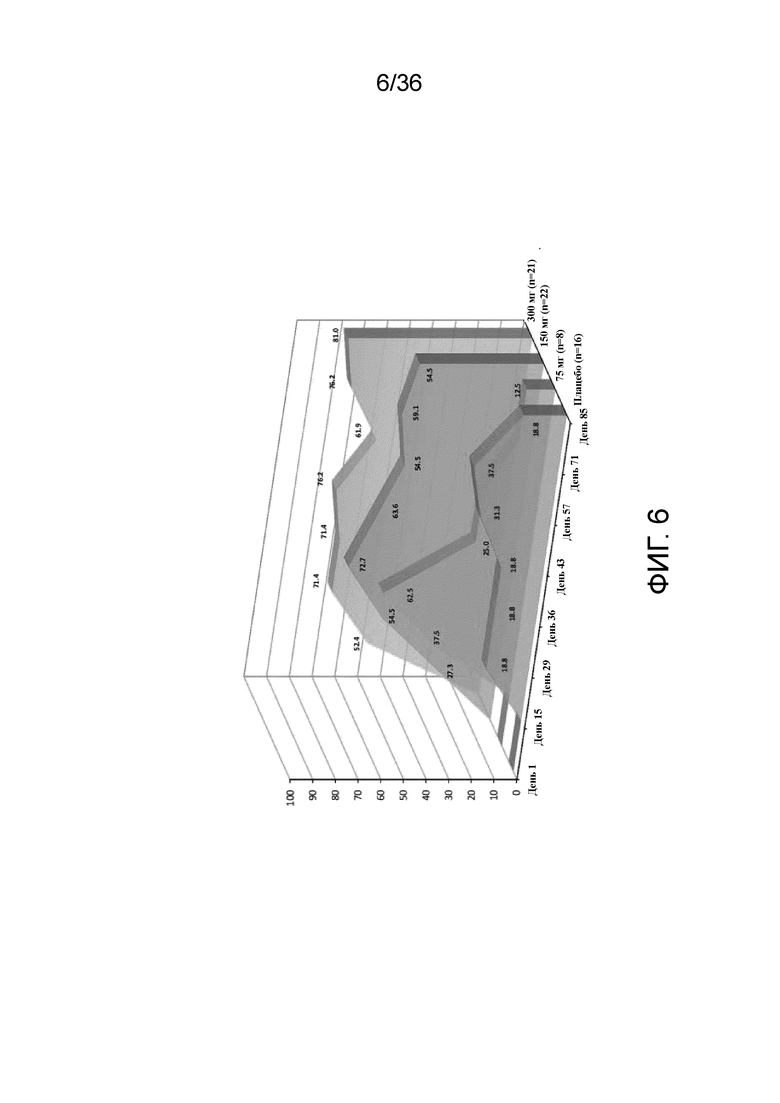
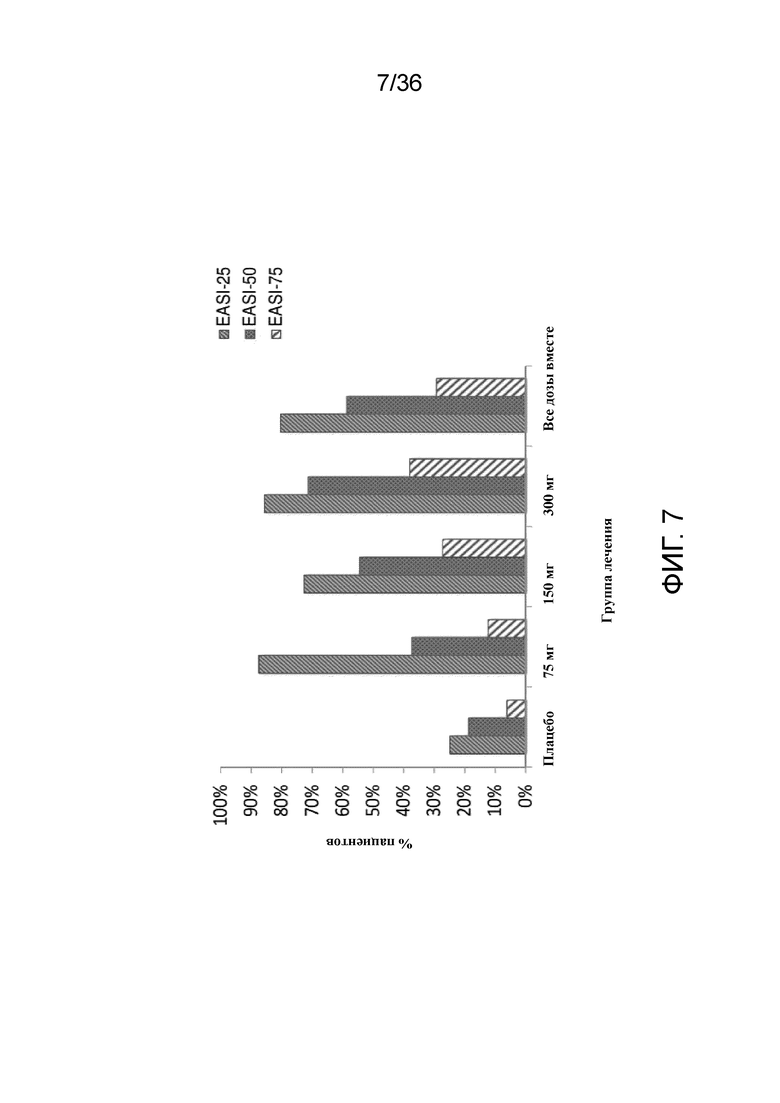

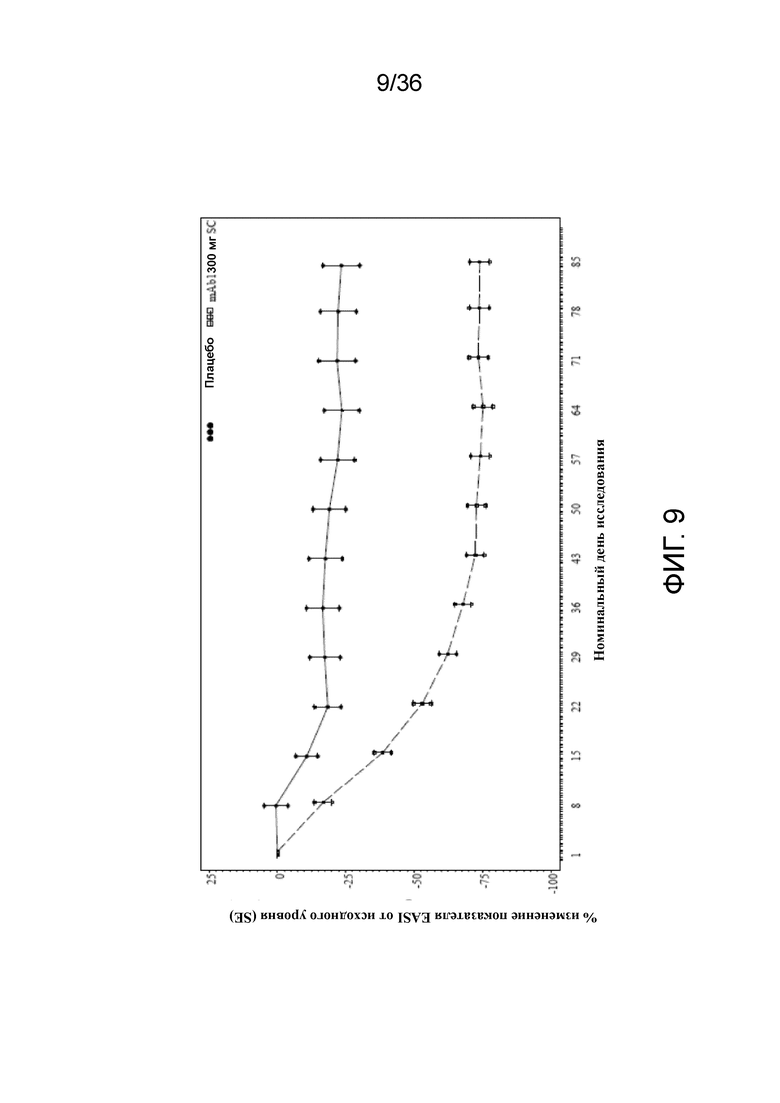
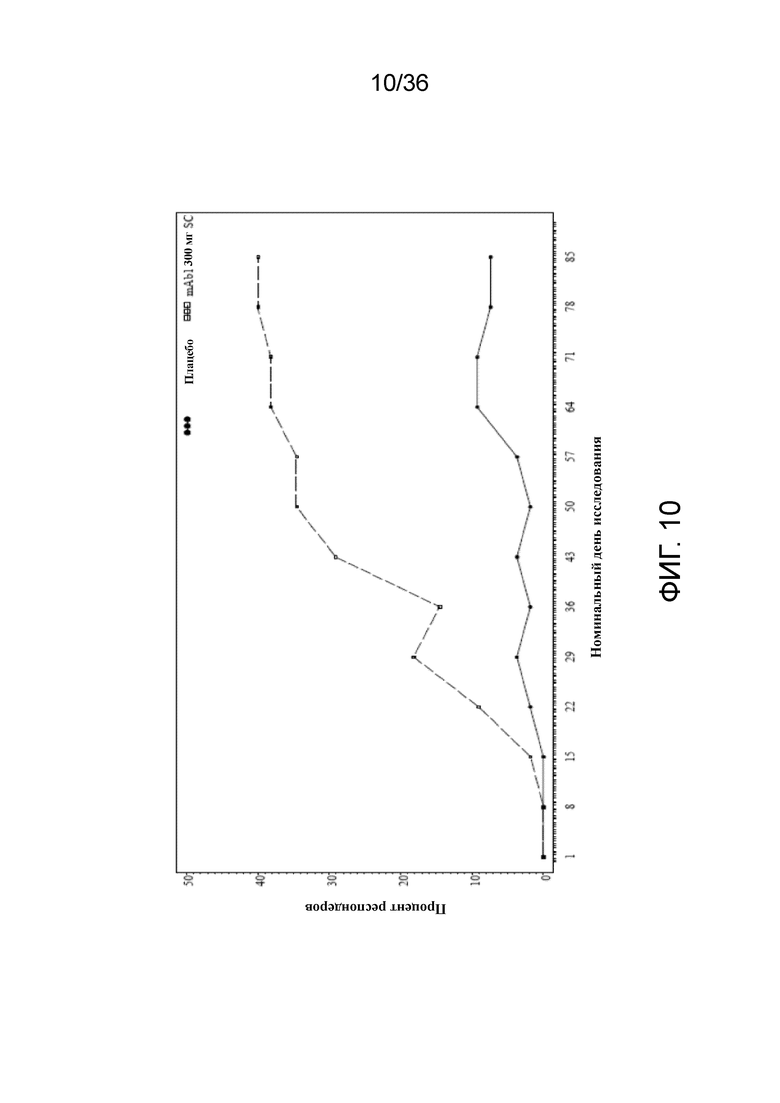
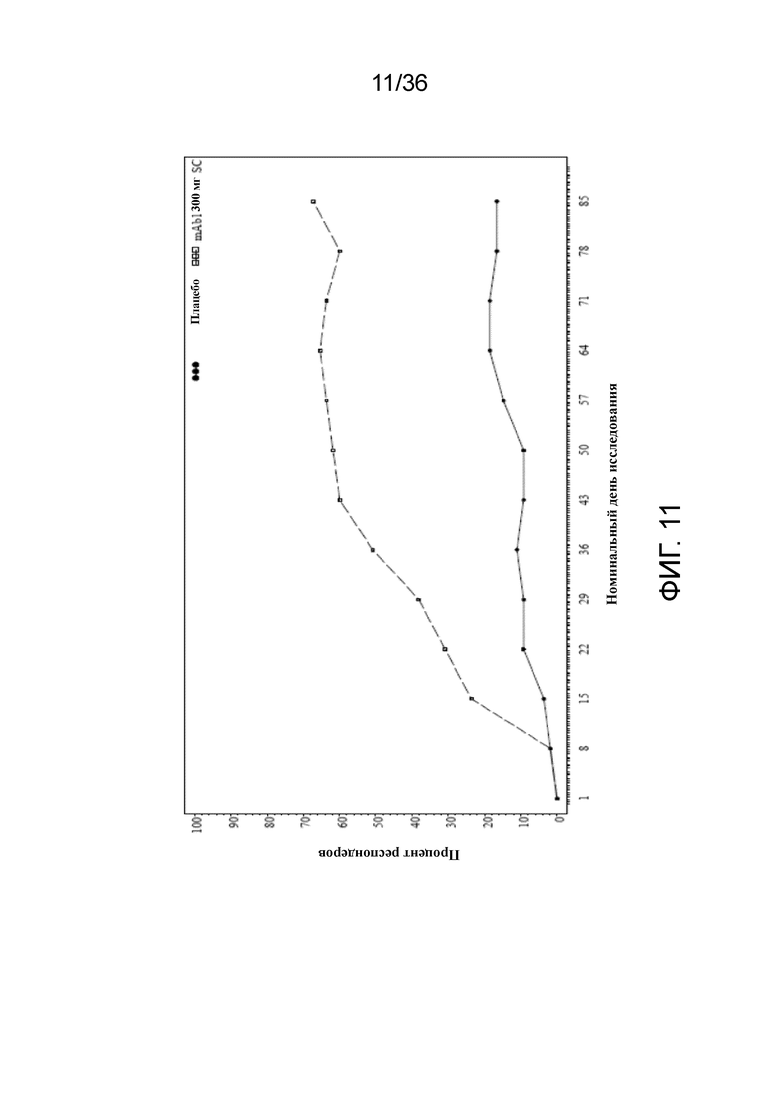
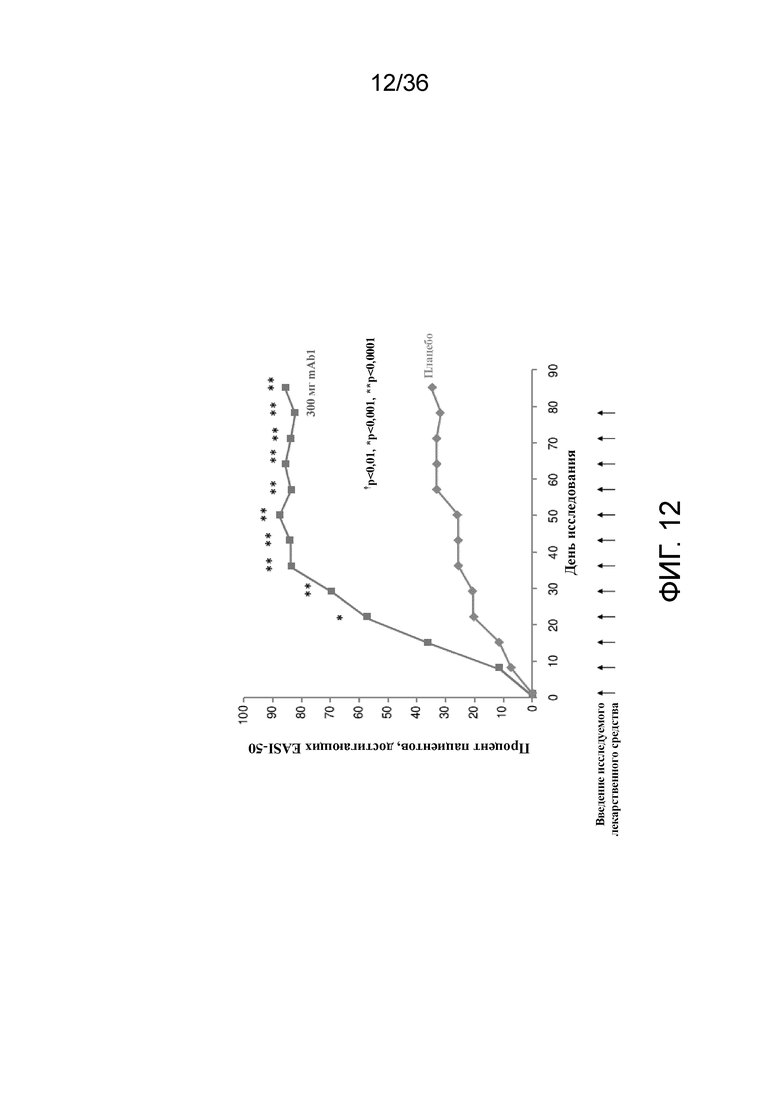
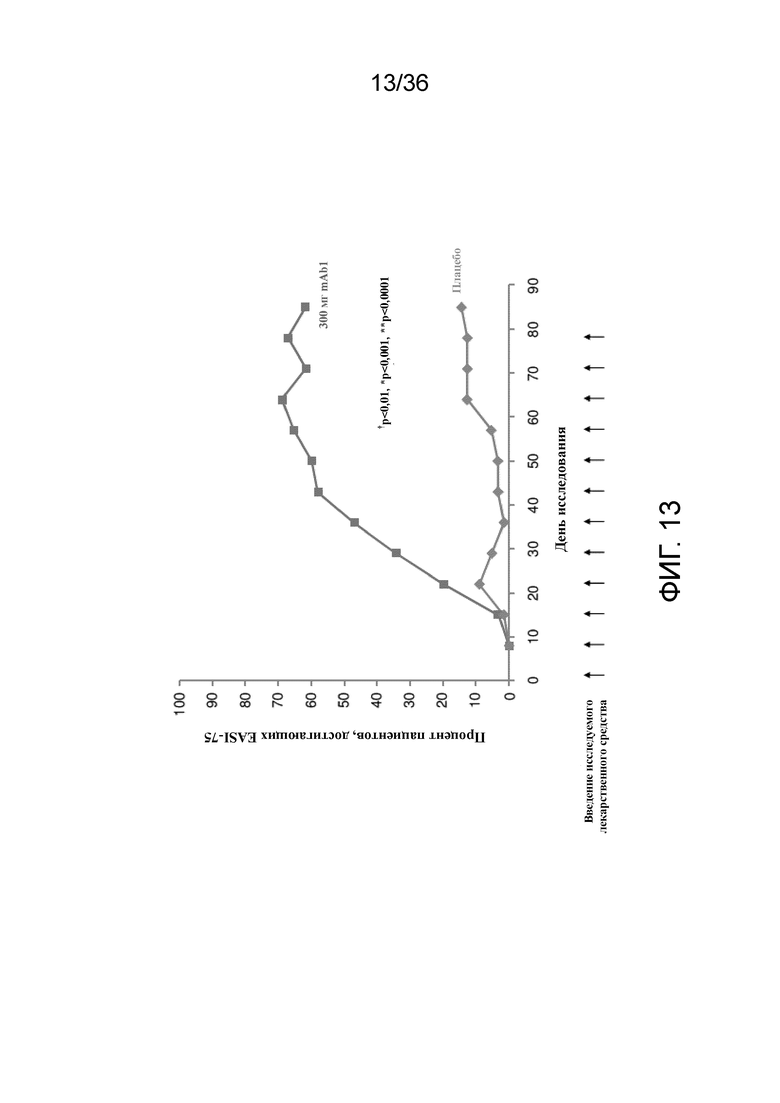
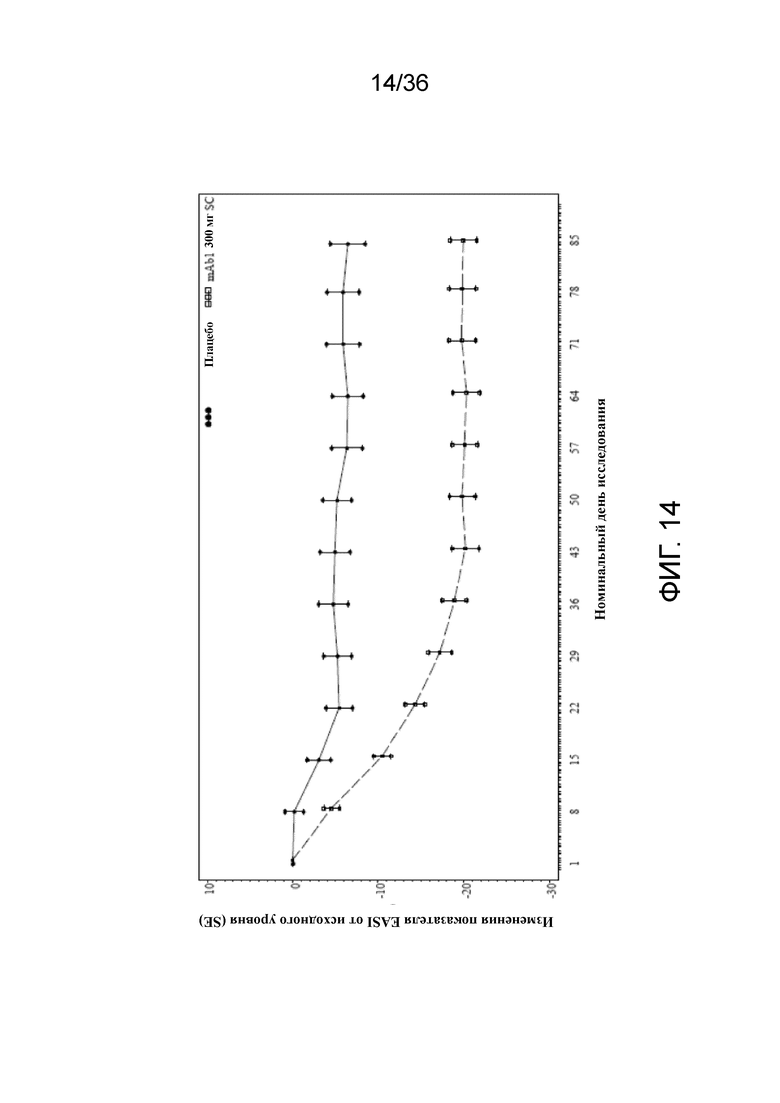
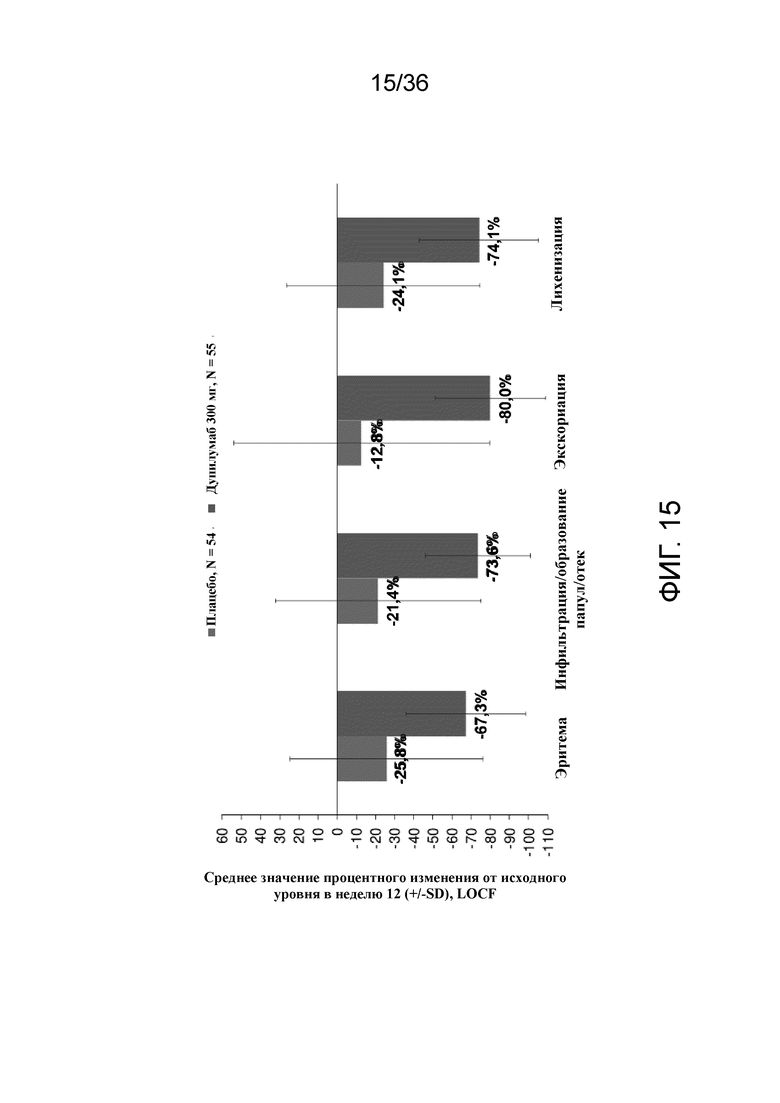
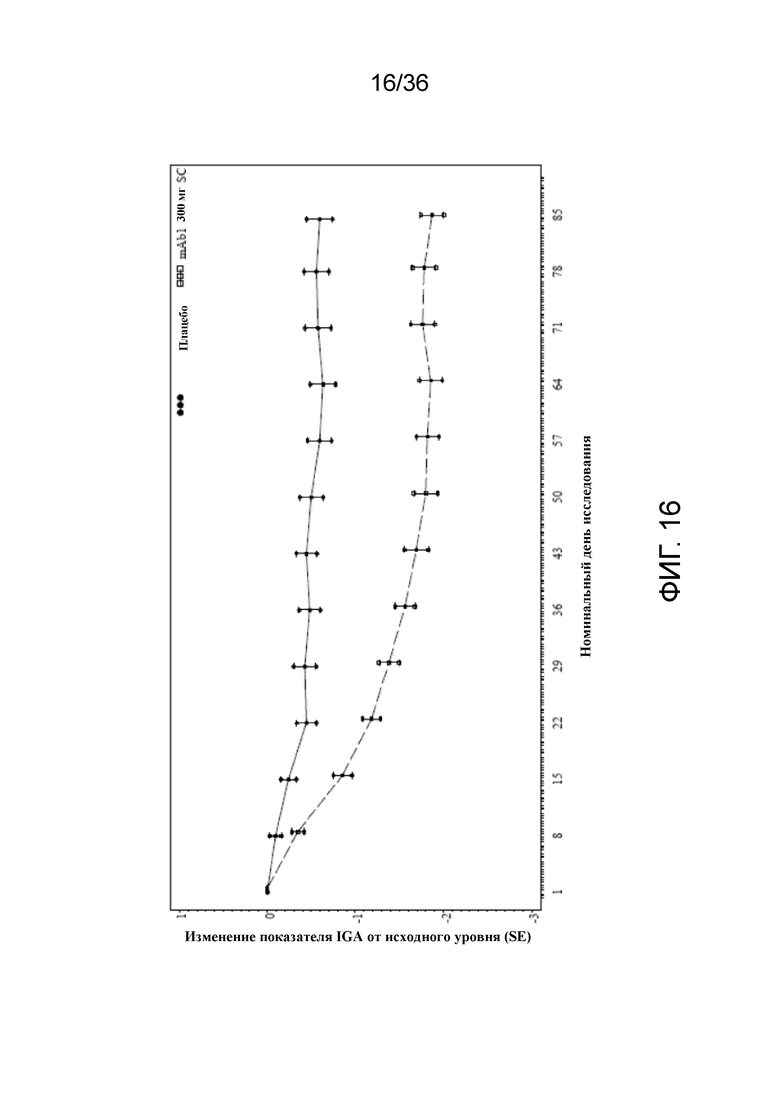
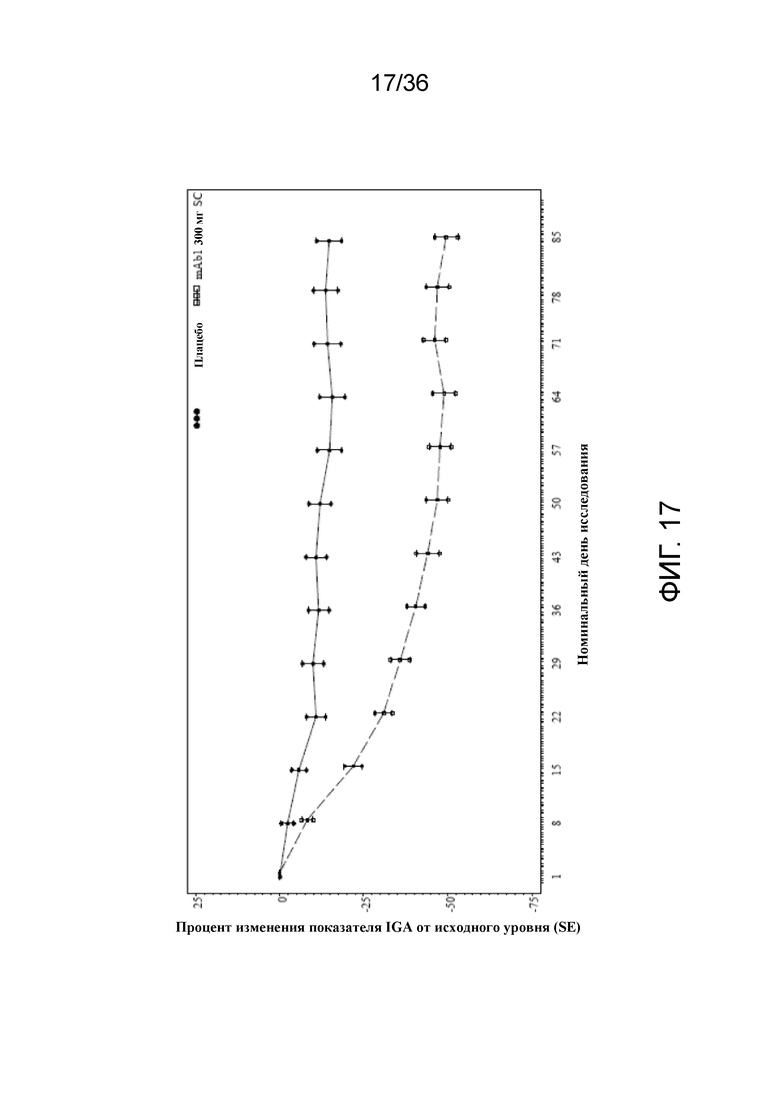
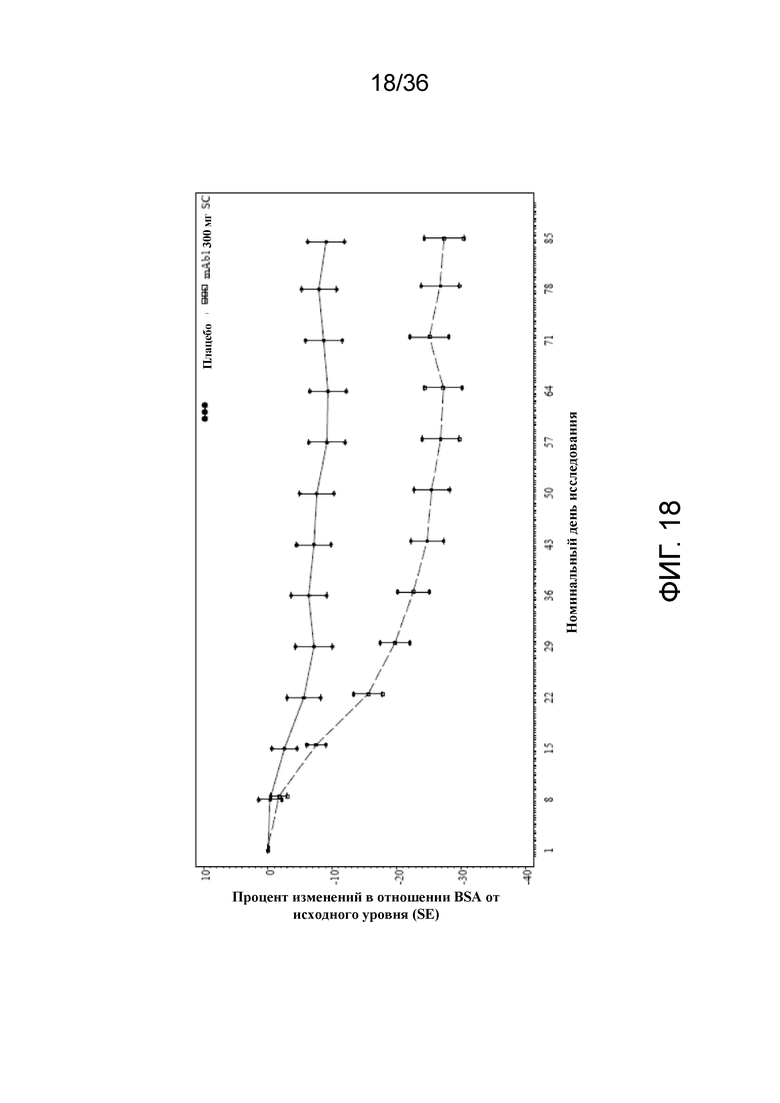
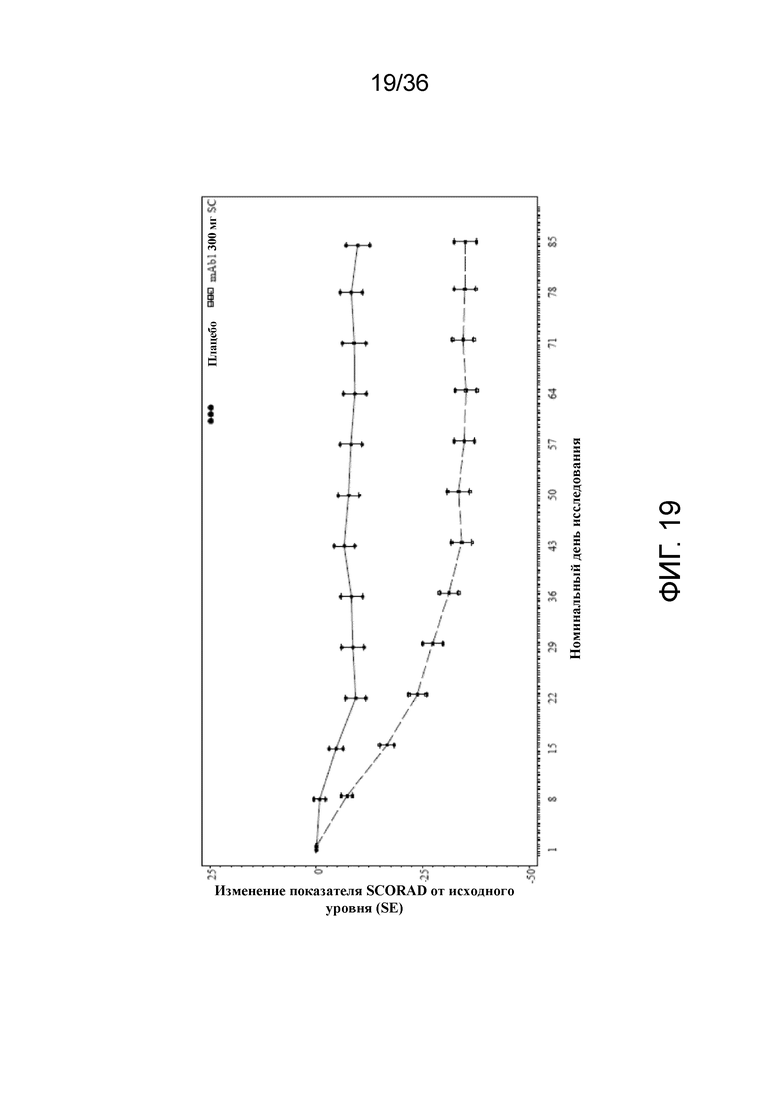
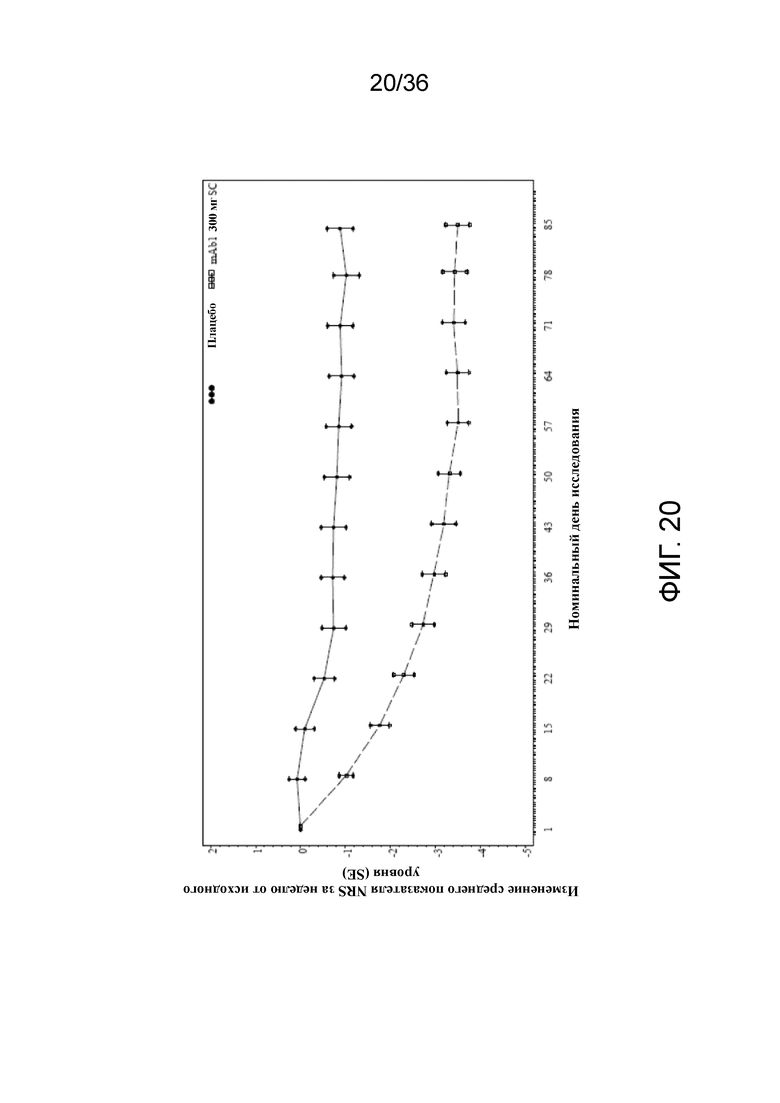

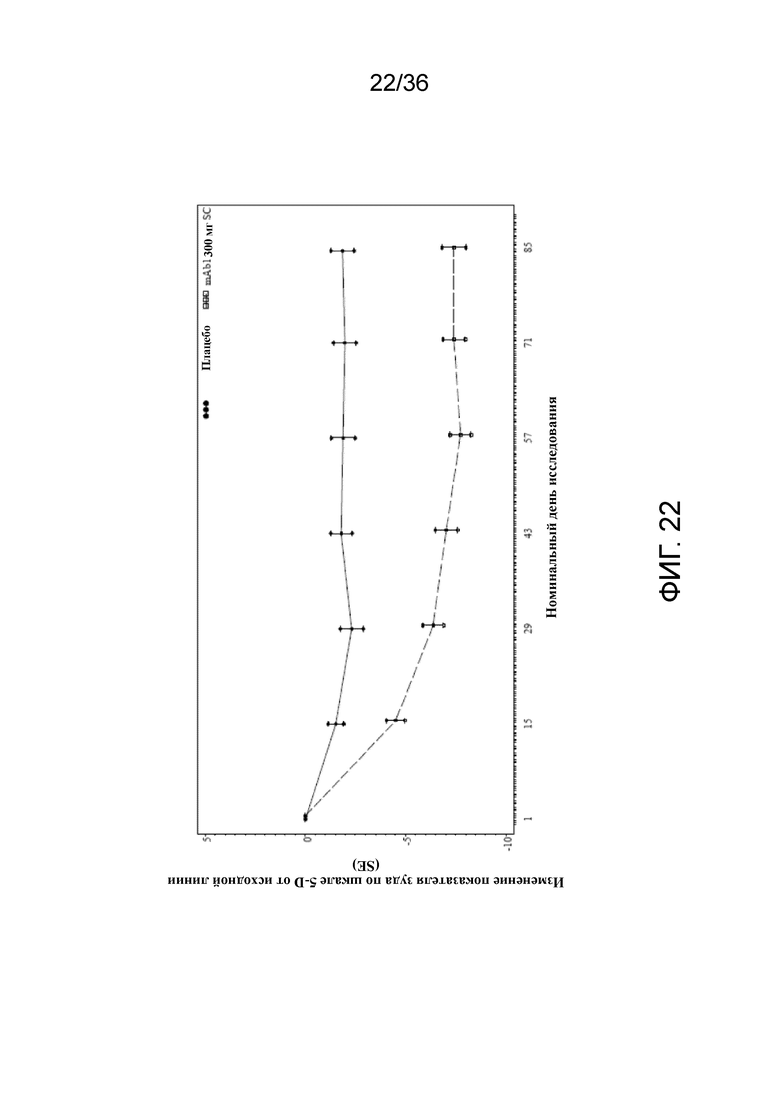
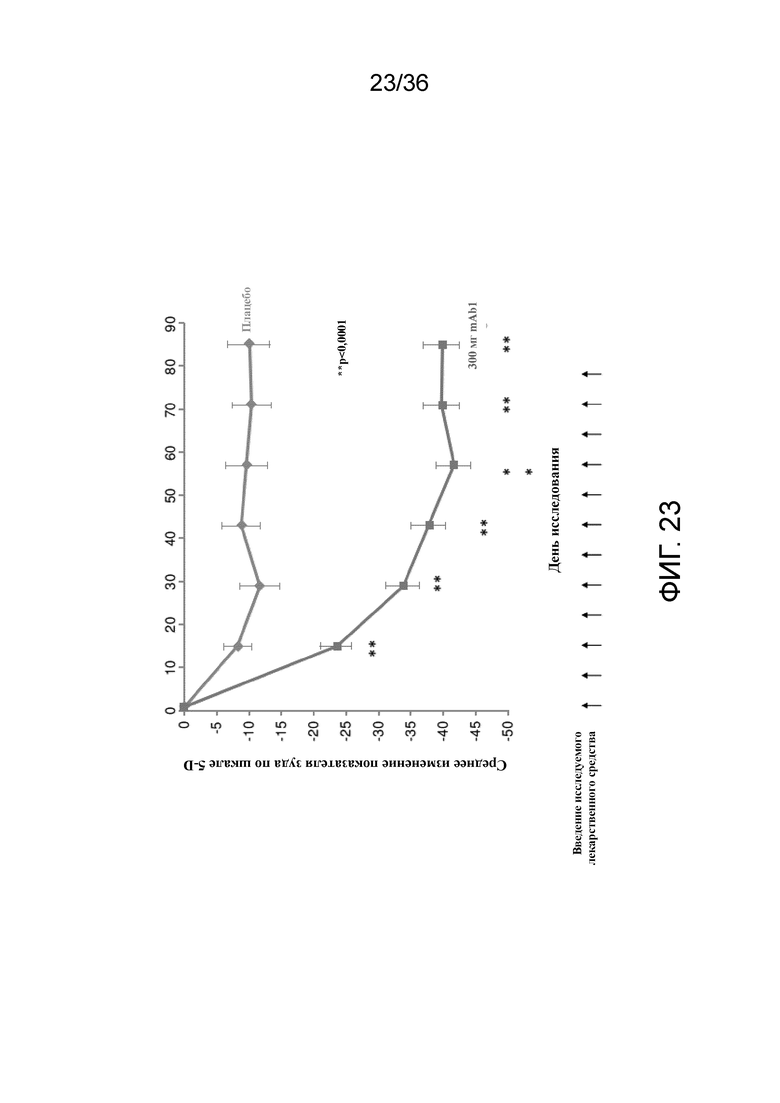
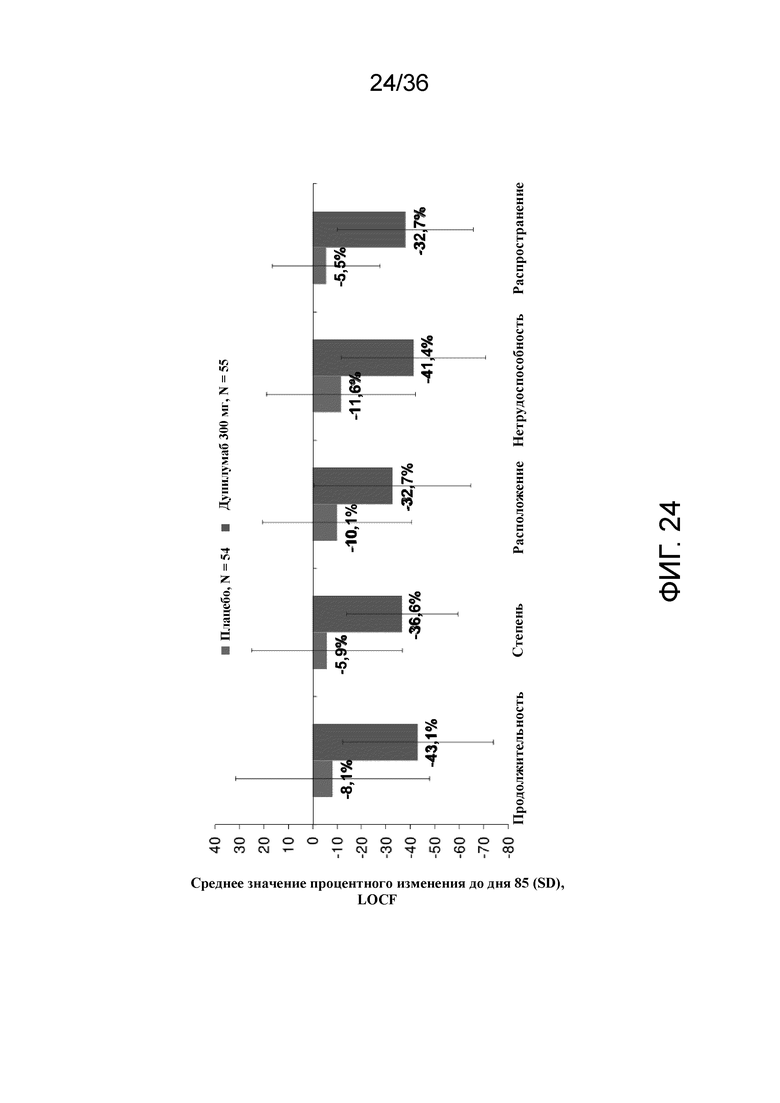
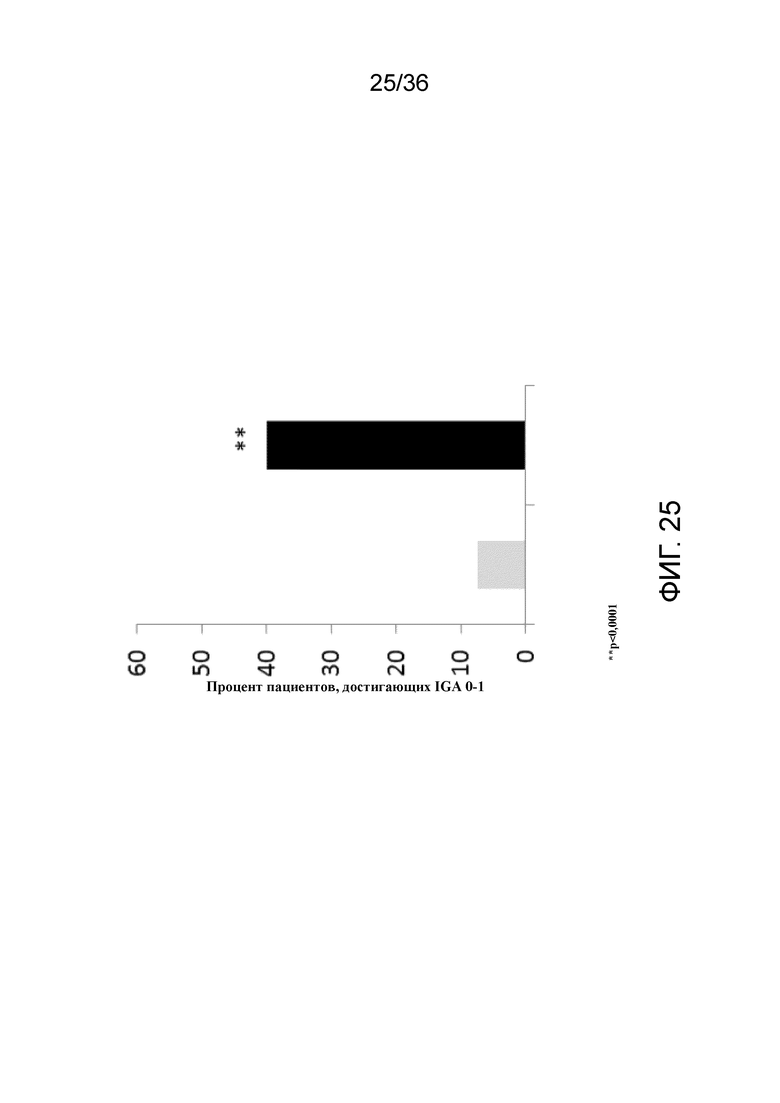
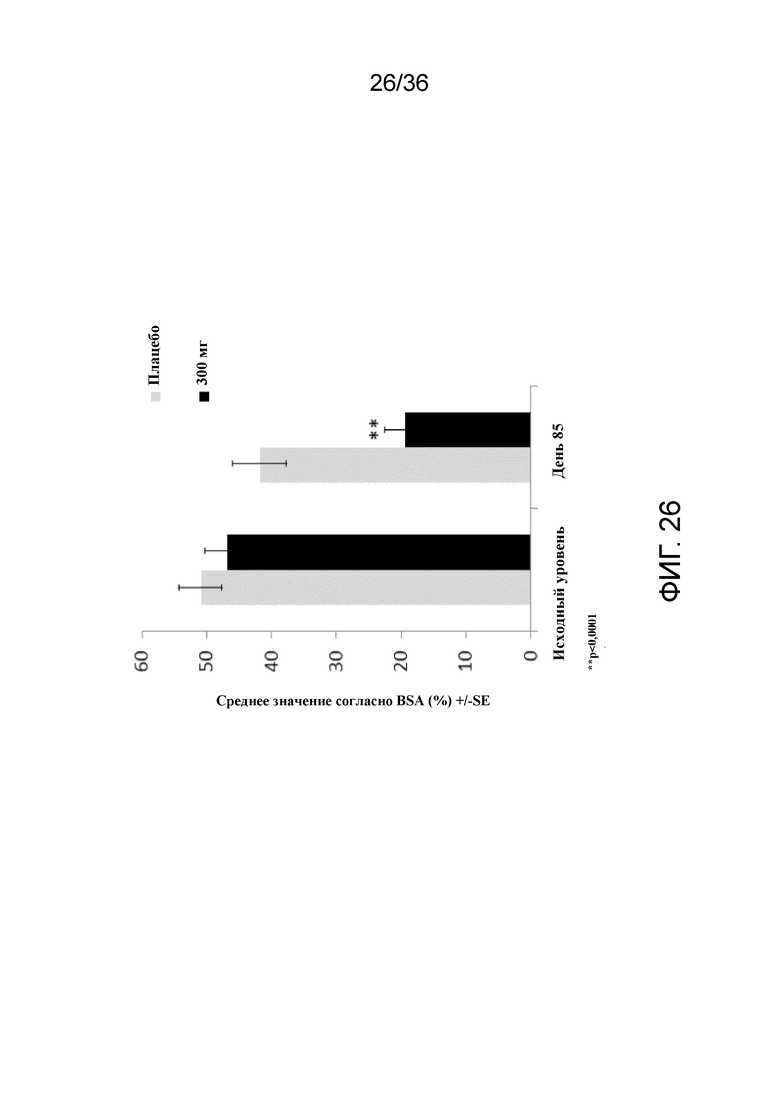
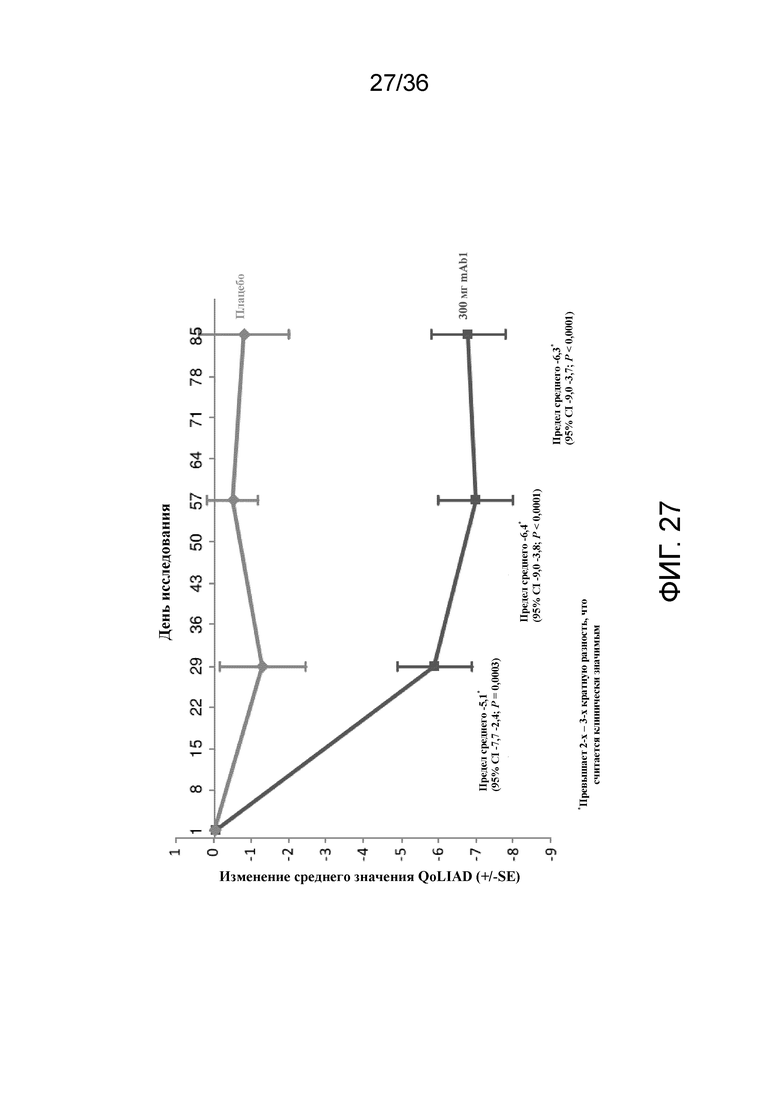
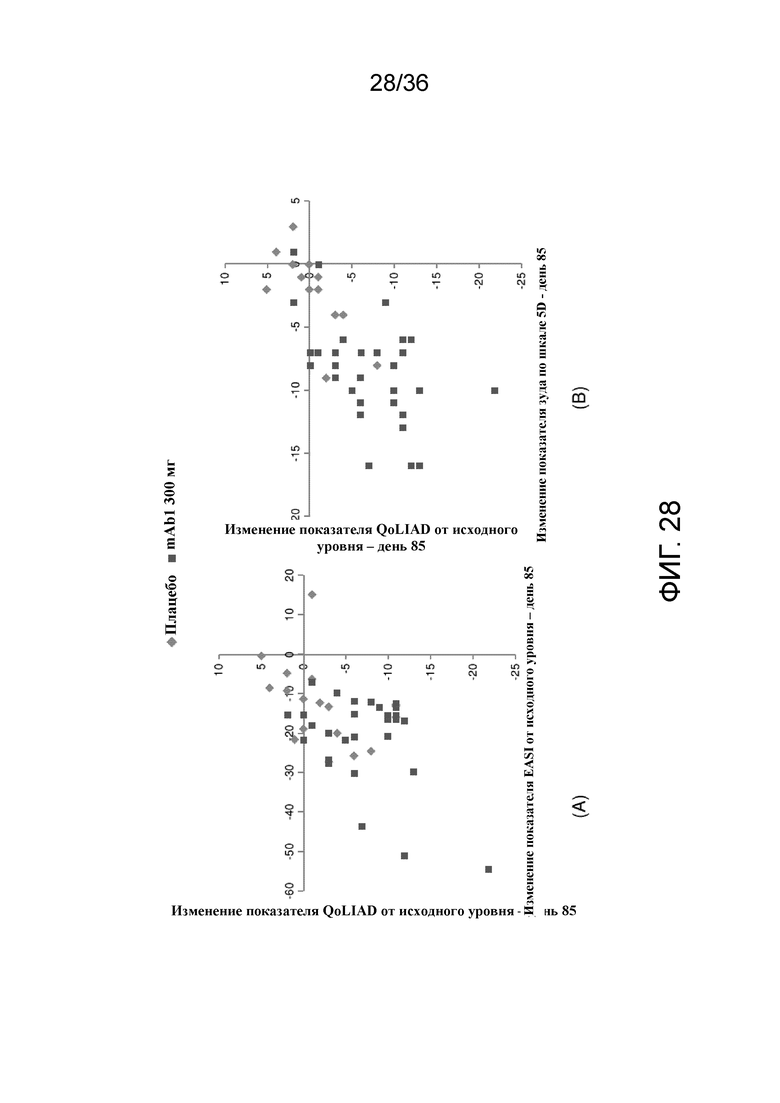
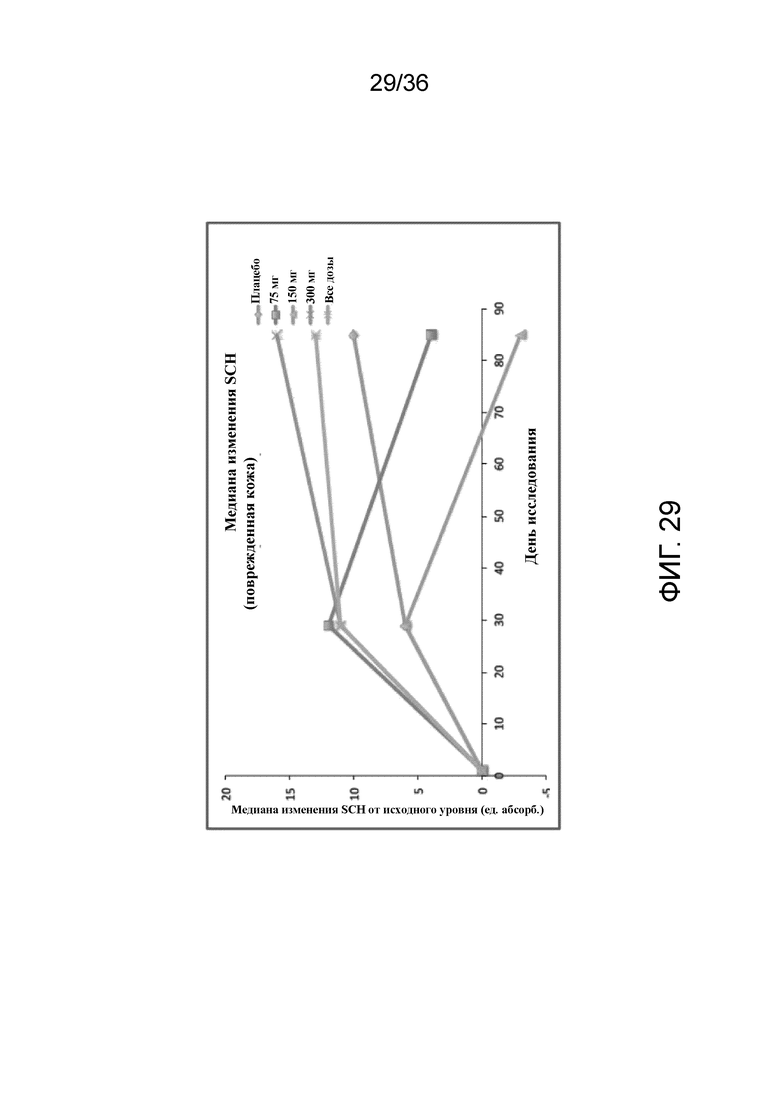
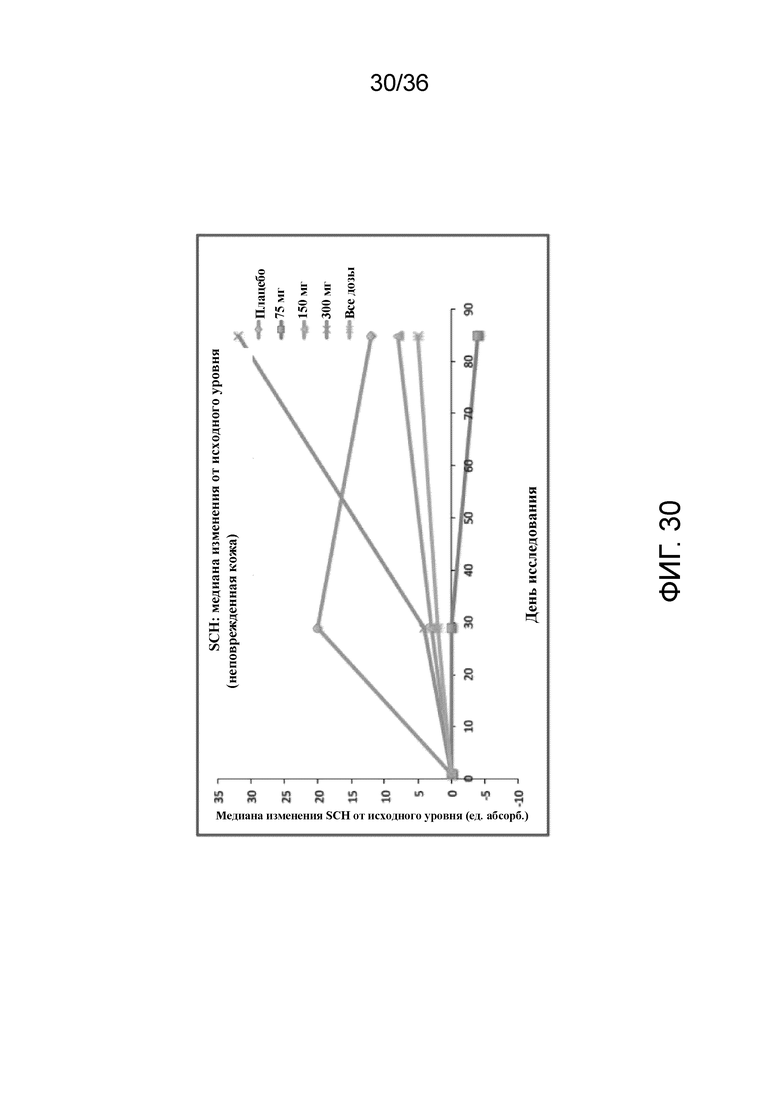
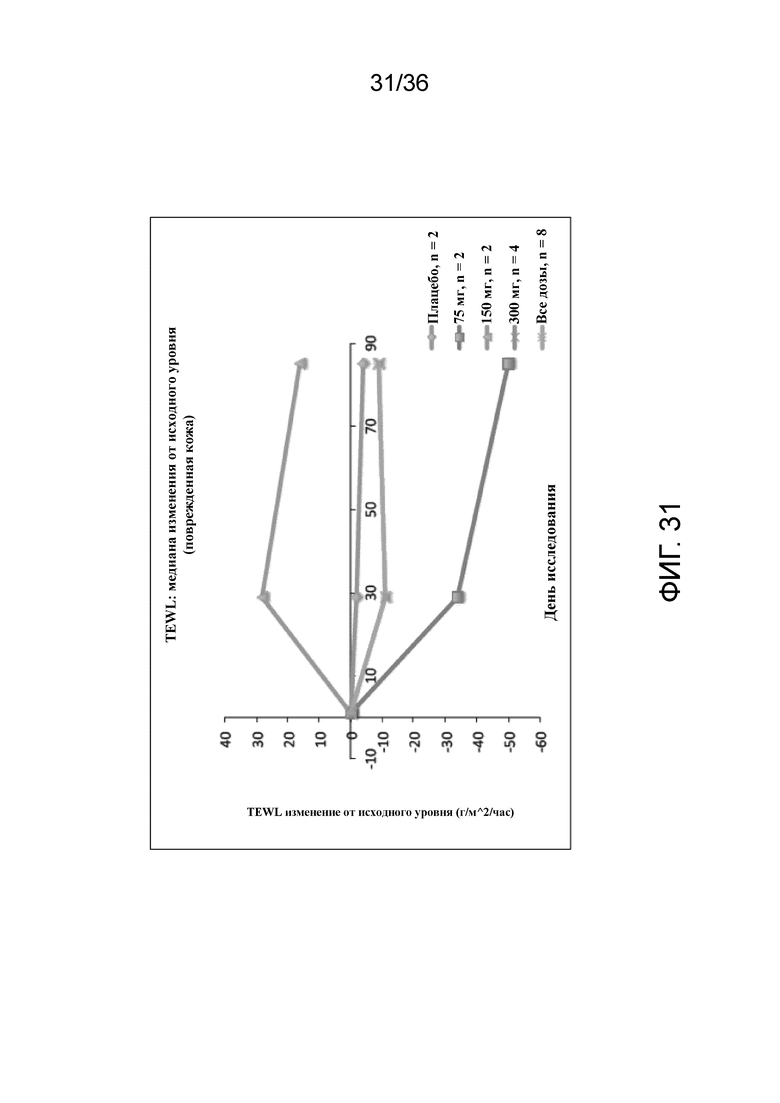
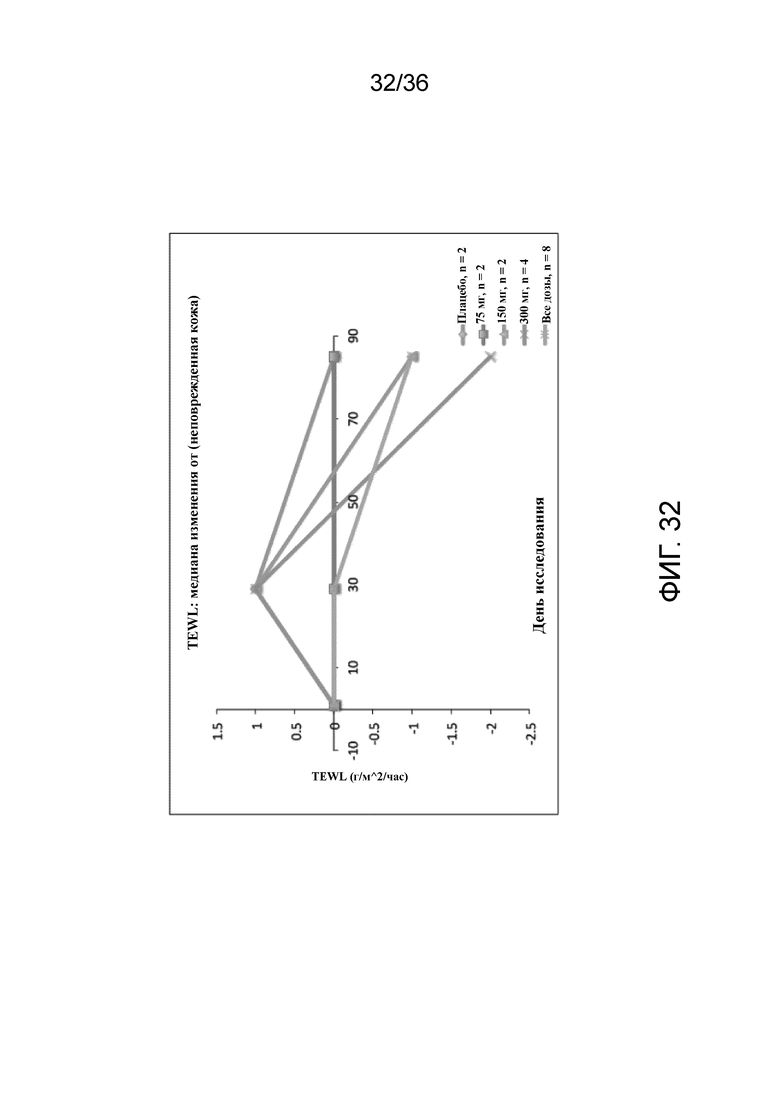

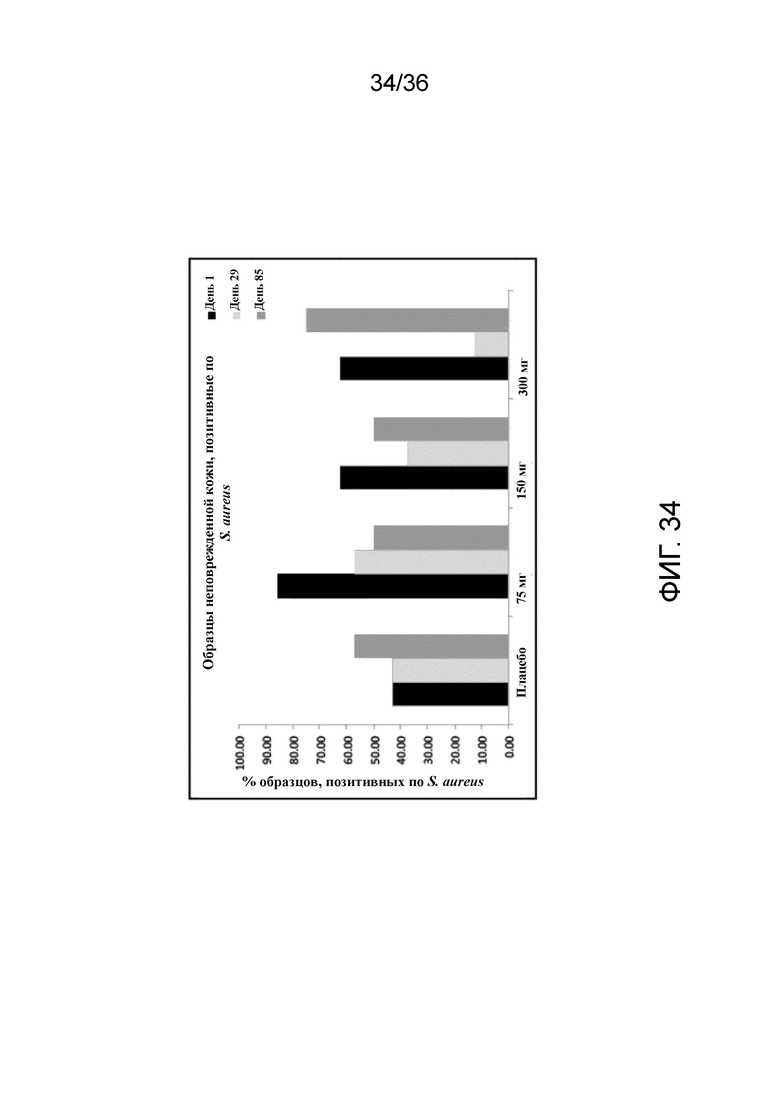
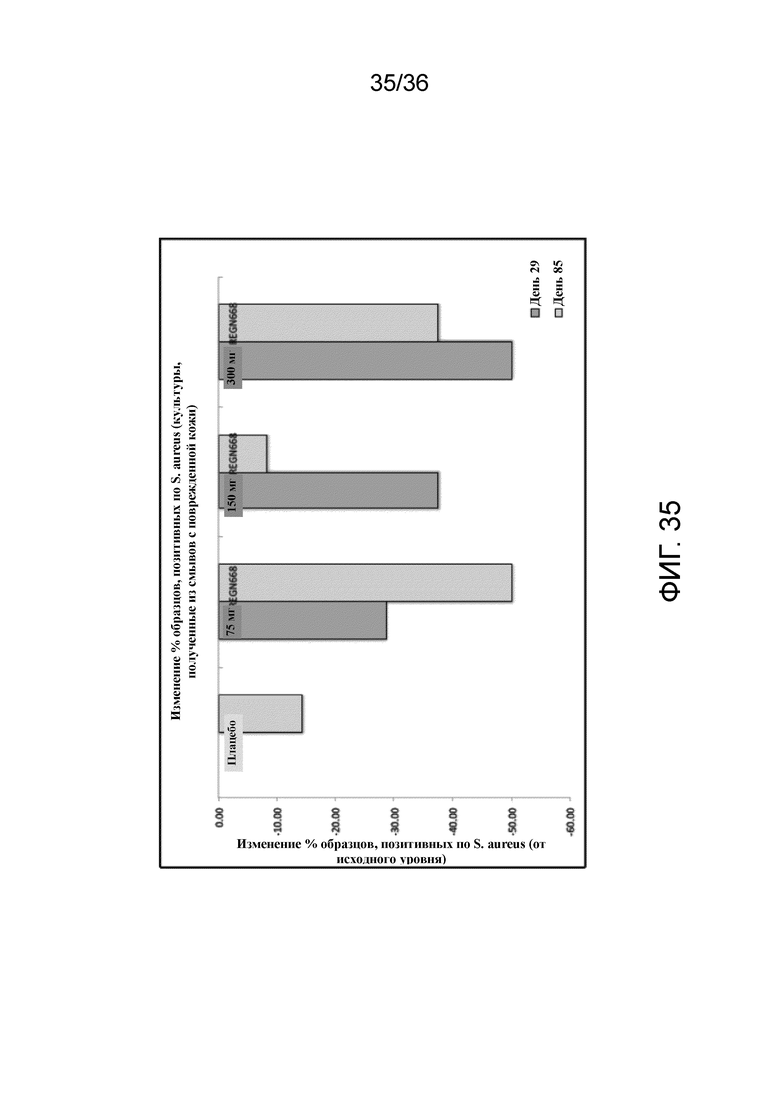
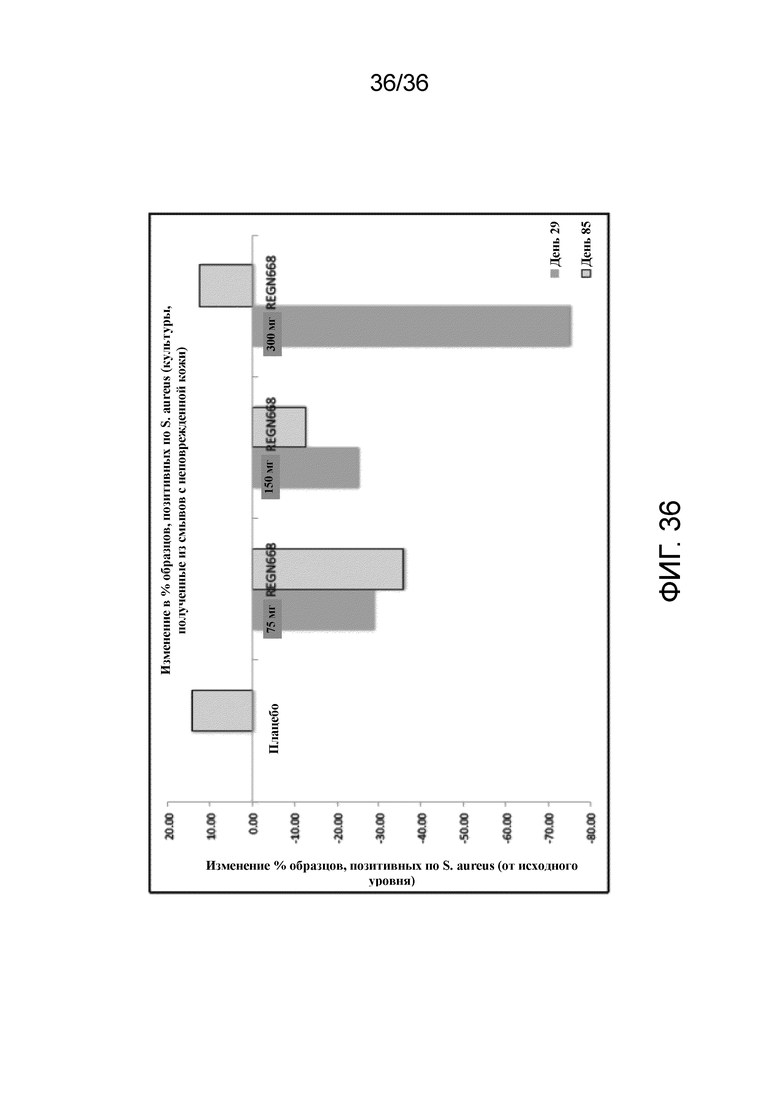
Изобретение относится к биотехнологии и медицине, а именно к способу лечения и/или предупреждения кожных инфекций, ассоциированных с IL-4R-связанными заболеваниями. Способ лечения или предупреждения тяжести кожной инфекции, выбранной из группы, состоящей из импетиго, целлюлита, инфекционного дерматита, герпетической экземы, фолликулита, инфицированного волдыря, микоза, отрубевидного лишая, инфекции, вызываемой Staphylococcus aureus, и инфекции, вызываемой Streptococcus, предусматривает введение нуждающемуся в этом субъекту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста IL-4R, где у субъекта атопический дерматит. Антагонист IL-4R представляет собой антитело или его антигенсвязывающий фрагмент, который специфически связывает IL-4Rα и содержит три CDR тяжелой цепи HCVR, содержащей SEQ ID NO: 1, и три CDR легкой цепи LCVR, содержащей SEQ ID NO: 2. Отмечалось заметно меньше число кожных инфекций, ассоциированное с лечением указанным антагонистом, его подкожное введение взрослым пациентам с АД со степенью от средней до тяжелой было безопасным и хорошо переносимым. Лечение также демонстрировало повышение значения SCH (увлажнение рогового слоя) поврежденных и неповрежденных участков при всех дозах. 14 з.п. ф-лы, 36 ил., 25 табл., 4 пр.
1. Способ лечения или предупреждения тяжести кожной инфекции, предусматривающий введение нуждающемуся в этом субъекту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста рецептора интерлейкина-4 (IL-4R), где у субъекта атопический дерматит, где антагонист IL-4R представляет собой антитело или его антигенсвязывающий фрагмент, который специфически связывает IL-4Rα, где антитело или его антигенсвязывающий фрагмент содержит три CDR тяжелой цепи (HCDR1, HCDR2 и HCDR3) HCVR, содержащей SEQ ID NO: 1, и три CDR легкой цепи (LCDR1, LCDR2 и LCDR3) LCVR, содержащей SEQ ID NO: 2, где кожная инфекция выбрана из группы, состоящей из импетиго, целлюлита, инфекционного дерматита, герпетической экземы, фолликулита, инфицированного волдыря, микоза, отрубевидного лишая, инфекции, вызываемой Staphylococcus aureus, и инфекции, вызываемой Streptococcus.
2. Способ по п. 1, где кожной инфекцией является инфекция, вызываемая Staphylococcus aureus.
3. Способ по п.1 или 2, где фармацевтическую композицию вводят подкожно.
4. Способ по п. 3, где фармацевтическую композицию вводят подкожно в дозе 75-600 мг.
5. Способ по п. 4, где фармацевтическую композицию вводят в дозе 300 мг.
6. Способ по любому из пп.1-5, где фармацевтическая композиция содержится в контейнере, выбранном из группы, состоящей из шприца, шприц-ручки, стеклянного флакона и автоинъектора.
7. Способ по п.6, где фармацевтическая композиция содержится в шприце.
8. Способ по п.6, где фармацевтическая композиция содержится в шприц-ручке.
9. Способ по п.8, где шприц-ручка предварительно заполнена.
10. Способ по п.6, где фармацевтическая композиция содержится в стеклянном флаконе.
11. Способ по п.6, где фармацевтическая композиция содержится в автоинъекторе.
12. Способ по любому из пп. 1-6, где у субъекта имеется атопический дерматит со степенью тяжести от средней до тяжелой.
13. Способ по п. 1, где второе терапевтическое средство вводят субъекту до, после фармацевтической композиции или одновременно с ней, где второе терапевтическое средство выбрано из группы, состоящей из антибактериального средства, противовирусного средства, противогрибкового средства, другого антагониста IL-4R, ингибитора IgE, кортикостероида, нестероидного противовоспалительного лекарственного средства (NSAID) и IFNγ.
14. Способ по п. 11, где антитело или его антигенсвязывающий фрагмент содержит CDR тяжелой цепи (HCDR1) с аминокислотной последовательностью SEQ ID NO: 3; HCDR2 с аминокислотной последовательностью SEQ ID NO: 4; HCDR3 с аминокислотной последовательностью SEQ ID NO: 5; CDR легкой цепи (LCDR1) с аминокислотной последовательностью SEQ ID NO: 6; LCDR2 с аминокислотной последовательностью SEQ ID NO: 7 и LCDR3 с аминокислотной последовательностью SEQ ID NO: 8.
15. Способ по п. 1, где антителом или его антигенсвязывающим фрагментом является дупилумаб.
| УСТРОЙСТВО ДЛЯ ПОВОРАЧИВАНИЯ ВАЛА МЕСИЛЬНО-ФОРМУЮЩИХ ТОРФЯНЫХ МАШИН | 1927 |
|
SU11302A1 |
| ONG P | |||
| Y | |||
| "Editorial update on emerging treatments of atopic dermatitis" | |||
| Expert Opinion on Emerging Drugs, 2012, 17(2), p.129-133 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| US 7608693 B2, 27.10.2009 | |||
| "Sanofi and Regeneron Report Positive Proof-of-Concept Data for Dupilumab, an IL-4R alpha Antibody, in Atopic Dermatitis", 71 ST ANNUAL MEETING OF THE AMERICAN ACADEMY OF DERMATOLOGY, 02.03.2013, pages 1-3, Найдено в Интернет [12/11/2018] https://investor.regeneron.com/news-releases/news-release-details/sanofi-and-regeneron-report-positive-proof-concept-data/ | |||
| WO 2001092340 A2, 06.12.2001 | |||
| US 2003124121 A1, 03.07.2003 | |||
| RING J | |||
| et al | |||
| "Guidelines for treatment of atopic eczema (atopic dermatitis) Part I" | |||
| Journal of the European Academy of Dermatology and Venereology, 2012, 26(8), 1045-1060 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| PETRY V | |||
| ET AL | |||
| "Bacterial skin colonization and infections in patients with atopic dermatitis", ANAIS BRASILEIROS DE DERMATOLOGIA, 2012, v.87, no.5, p.729-734 | |||
| LIN YU-TSAN ET AL | |||
| "Role of bacterial pathogens in atopic dermatitis", CLINICAL REVIEWS IN ALLERGY & IMMUNOLOGY, 2007, v.33, no.3, p.167-177 | |||
| NAVARINI A | |||
| A | |||
| et al | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| J ALLERGY CLIN IMMUNOL, NOVEMBER 2011, 128(5):1128-30, doi:10.1016/j.jaci.2011.09.009. | |||

