ОБЛАСТЬ ПРИМЕНЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение касается лечения и (или) профилактики атопического дерматита и родственных состояний. В частности, настоящее изобретение касается применения антагонистов рецептора интерлейкина-4 (IL-4R) для лечения или профилактики атопического дерматита у нуждающегося в этом пациента.
УРОВЕНЬ ТЕХНИКИ
Атопический дерматит (АД) представляет собой хроническое рецидивирующее воспалительное заболевание кожи, характеризующееся выраженным кожным зудом и наличием чешуйчатых и сухих экзематозных поражений. АД часто сочетается с другими атопическими заболеваниями, например аллергическим ринитом или бронхиальной астмой. Тяжелая форма атопического дерматита может стать причиной крайне выраженной инвалидизации вследствие серьезных психологических проблем, тяжелого нарушения сна и ухудшения качества жизни, приводящих к увеличению социально-экономического бремени заболевания.
В основе патофизиологии АД лежат сложные взаимодействия между опосредованной иммуноглобулином E (IgE) сенсибилизацией, иммунной системой и факторами окружающей среды. Первичное поражение кожи может являться иммунологическим нарушением, вызывающим IgE-опосредованную сенсибилизацию с дисфункцией эпителиального барьера, являющейся следствием генетических мутаций и местного воспаления. АД часто развивается у детей в возрасте до 5 лет и может сохраняться во взрослом возрасте.
Стандартное лечение АД включает местное применение лосьонов и увлажняющих средств, применение мазей и кремов, содержащих топические кортикостероиды, а также кортикостероидов в инъекционной форме. Однако большинство видов лечения обеспечивает временное и неполное уменьшение выраженности симптомов. Кроме того, у многих пациентов с умеренно тяжелым или тяжелым АД развивается резистентность к терапии топическими кортикостероидами или ингибиторами кальциневрина. В связи с этим, в данной области техники существует потребность в новых видах таргетной терапии для лечения и (или) профилактики АД.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Согласно определенным аспектам настоящего изобретения, представлены способы лечения и профилактики и (или) уменьшения выраженности симптомов атопического дерматита (АД), в том числе умеренно тяжелого или тяжелого АД. Определенные варианты осуществления настоящего изобретения касаются способов лечения, облегчения и профилактики умеренно тяжелого или тяжелого АД или уменьшения выраженности его симптомов у пациентов с устойчивостью заболевания к лечению топическим кортикостероидом или ингибитором кальциневрина. Согласно некоторым вариантам осуществления настоящего изобретения, настоящее изобретение включает способы лечения умеренно тяжелого или тяжелого АД при неэффективности топического кортикостероида или ингибитора кальциневрина. Способы настоящего изобретения включают введение нуждающемуся в этом испытуемому или пациенту фармацевтической композиции, содержащей антагонист рецептора интерлейкина-4 (IL-4R) в терапевтически эффективном количестве. Согласно определенным вариантам осуществления настоящего изобретения, антагонист IL-4R представляет собой антитело или его антигенсвязывающий фрагмент, которые специфически связываются с IL-4R. Примеры антител к IL-4R, которые могут использоваться в контексте способов настоящего изобретения, приведены в другом месте этого документа, в том числе в действующем примере 1. Согласно определенным вариантам осуществления настоящего изобретения, антагонист IL-4R является антителом к IL-4R, обладающим характеристиками связывания, присущими эталонному антигену, называемому в этом документе «mAb1» (например, антитело или его антигенсвязывающий фрагмент, включающий определяющие комплементарность участки mAb1). Согласно одному варианту осуществления настоящего изобретения, антитело или его антигенсвязывающий фрагмент, которые связываются с IL-4R, включает определяющие комплементарность участки (CDR) в паре последовательностей вариабельной области тяжелой цепи (HCVR) и вариабельной области легкой цепи (LCVR) со следующими идентификаторами SEQ ID NO: 162/164.
Некоторые варианты осуществления настоящего изобретения направлены на способы лечения, уменьшения выраженности, ослабления или профилактики кожного зуда у пациента и включают применение фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно одному варианту осуществления настоящего изобретения, пациент страдает умеренно тяжелым или тяжелым АД. Согласно некоторым вариантам осуществления настоящего изобретения, пациент страдает АД, устойчивым к лечению топическим кортикостероидом или ингибитором кальциневрина.
Согласно определенным вариантам осуществления настоящего изобретения, настоящее изобретение включают способы лечения умеренно тяжелого или тяжелого АД у пациента, включающие введение фармацевтической композиции, содержащей в терапевтически эффективном количестве антитело или его антигенсвязывающий фрагмент, которые связываются с IL-4R, и определение улучшения связанного с АД параметра. Улучшение можно устанавливать, определять или количественно оценивать способами, хорошо известными в данной области техники. Связанные с АД параметры обсуждаются в другом месте этого документа, в том числе, например, в действующем примере 7.
Согласно определенным примерам вариантов осуществления настоящего изобретения, настоящее изобретение предлагает способы улучшения одного или более связанных с АД параметров у нуждающегося в этом пациента. Улучшение связанных с АД параметров включает, например, уменьшение балла по шкале общей оценке исследователя (IGA - Investigator's Global Assessment); уменьшение площади пораженной кожи при атопическом дерматите (BSA - Body Surface Area); уменьшение индекса площади экзематозного поражения кожи и тяжести экземы (EASI - Eczema Area and Severity Index); уменьшение индекса SCORAD; уменьшение балла по опроснику из 5 вопросов для оценки кожного зуда; и (или) уменьшение балла по числовой шкале оценки интенсивности кожного зуда (NRS - Numeric Rating Scale). Согласно примерам вариантов осуществления настоящего изобретения, улучшение связанного с АД параметра выбирается из группы, включающей: (i) уменьшение от исходного значения балла по шкале IGA по меньшей мере на 25%; (ii) уменьшение от исходного значения показателя BSA по меньшей мере на 35%; (iii) уменьшение от исходного значения индекса EASI по меньшей мере на 45%; (iv) уменьшение от исходного значения индекса SCORAD по меньшей мере на 30%; (v) уменьшение от исходного значения балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 15%; (vi) уменьшение балла по числовой шкале оценки интенсивности кожного зуда (шкала NRS) по меньшей мере на 25%; и (vii) процентная доля пациентов с уменьшением индекса EASI ≥ 50% (EASI50).
Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения балла по шкале IGA по меньшей мере на 25% на день 22 и до по крайней мере дня 85 после введения фармацевтической композиции, содержащей антитело или его антигенсвязывающий фрагмент, которые связываются с IL-4R. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения показателя BSA по меньшей мере на 40% на день 29 и до по крайней мере дня 85 после введения фармацевтической композиции. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения индекса EASI по меньшей мере на 50% на день 29 и до по крайней мере дня 85 после введения фармацевтической композиции. Согласно определенным вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения индекса EASI по меньшей мере на 50% на день 29 по крайней мере у 70% пациентов после введения фармацевтической композиции. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения индекса SCORAD по меньшей мере на 30% на день 29 и до по крайней мере дня 85 после введения фармацевтической композиции. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 15% на день 15 и до по крайней мере дня 85 после введения фармацевтической композиции. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения балла по шкале NRS по меньшей мере на 25% к концу недели 2 и по крайней мере до конца недели 10 после введения фармацевтической композиции.
Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения балла по шкале IGA по меньшей мере на 45% на день 85 и до по крайней мере дня 197 после введения фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения показателя BSA по меньшей мере на 50% на день 85 и до по крайней мере дня 197 после введения фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения индекса EASI по меньшей мере на 60% на день 85 и до по крайней мере дня 197 после введения фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения индекса SCORAD по меньшей мере на 45% на день 85 и до по крайней мере дня 197 после введения фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 30% на день 85 и до по крайней мере дня 197 после введения фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра включает уменьшение от исходного значения балла по шкале NRS по меньшей мере на 50% на день 85 и до по крайней мере дня 197 после введения фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве.
Согласно другим примерам вариантов осуществления настоящего изобретения, настоящее изобретение предлагает способы лечения АД у пациента, и к этим способам относятся: (а) отбор пациента с повышенным уровнем по меньшей мере одного связанного с АД биомаркера; и (б) введение пациенту фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно определенным вариантам осуществления настоящего изобретения, антагонист IL-4R представляет собой антитело или его антигенсвязывающий фрагмент, которые связываются с IL-4R. К связанным с АД биомаркерам, которые можно определять, и (или) содержание которых можно измерять в контексте настоящего изобретения, относятся, помимо прочего, регулируемый тимусом и активацией хемокин (TARC, также известный как CCL17), иммуноглобулин E (IgE), эотаксин-3, лактатдегидрогеназа (ЛДГ), эозинофилы, антиген-специфический IgE (например, тест Phadiatop™) и периостин. Согласно некоторым вариантам осуществления настоящего изобретения, способы настоящего изобретения включают определение уровня связанного с АД биомаркера у нуждающегося в этом пациента, выбор пациента с повышенным уровнем связанного с АД биомаркера и введение в терапевтически эффективном количестве антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R. Согласно некоторым вариантам осуществления настоящего изобретения, пациента отбирают путем получения информации об уровне связанного с АД биомаркера у пациента. Согласно некоторым вариантам осуществления настоящего изобретения, уровень связанного с АД биомаркера определяют путем количественного анализа либо с помощью теста, известного в данной области техники, или как указано в другом месте этого документа. Согласно одному варианту осуществления настоящего изобретения, пациента отбирают на основании уровня IgE более чем приблизительно 1500 тыс. ед./л до или во время лечения. Согласно одному варианту осуществления настоящего изобретения, пациента отбирают на основании уровня TARC более чем приблизительно 1000 пг/мл до или во время лечения. Согласно одному соответствующему аспекту настоящего изобретения, предложены способы лечения АД, заключающиеся в введении пациенту фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве, где введение фармацевтической композиции пациенту приводит к уменьшению уровня по меньшей мере одного связанного с АД биомаркера в дни 4, 8, 15, 22, 25, 29, 36 или позже после введения фармацевтической композиции. Согласно определенным вариантам осуществления настоящего изобретения, у пациента после получения фармацевтической композиции наблюдается уменьшение уровня IgE от исходного значения на 5-20% на день 36 или позже. Согласно определенным вариантам осуществления настоящего изобретения, у пациента наблюдается уменьшение уровня TARC от исходного значения на 25-70% на день 4 или позже после введения фармацевтической композиции.
Настоящее изобретение также предлагает способы уменьшения уровня одного или более связанных с АД биомаркеров у пациента или улучшения одного или более связанных с АД параметров у пациента, которые включают последовательное однократное введение нуждающемуся в этому пациенту одной первичной дозы фармацевтической композиции, содержащей антагонист IL-4R, с последующим одно- или многократным введением вторичных доз фармацевтической композиции, содержащей антагонист IL-4R.
Согласно определенным вариантам осуществления настоящего изобретения, настоящее изобретение предлагает способы уменьшения уровня одного или более связанных с АД биомаркеров у пациента или улучшения одного или более связанных с АД параметров у пациента, которые включают введение пациенту приблизительно от 50 мг до приблизительно 600 мг фармацевтической композиции, содержащей антитело или его антигенсвязывающий фрагмент, которые специфически связываются с IL-4R. Согласно определенным вариантам осуществления настоящего изобретения, первичная доза и одна или более вторичных доз включают на каждую приблизительно от 75 мг до приблизительно 300 мг антитела или его антигенсвязывающего фрагмента. Согласно этому аспекту настоящего изобретения, фармацевтическую композицию можно вводить пациенту с частотой, например, один раз в неделю. Согласно некоторым вариантам осуществления настоящего изобретения, каждую вторичную дозу вводят через 1-8 недель после введения непосредственно предшествующей дозы. Согласно определенным вариантам осуществления настоящего изобретения, вводят по меньшей мере 4 дозы антитела или его антигенсвязывающего фрагмента. Согласно одному варианту осуществления настоящего изобретения, каждую вторичную дозу вводят через 1 неделю после введения непосредственно предшествующей дозы. Согласно определенным вариантам осуществления настоящего изобретения, первичная доза представляет собой первое количество антитела или его антигенсвязывающего фрагмента, a одна или более вторичных доз представляют собой второе количество антитела или его антигенсвязывающего фрагмента. Согласно некоторым вариантам осуществления настоящего изобретения, первое количество антитела или его антигенсвязывающего фрагмента представляет собой количество, кратное 1,5; 2; 2,5; 3; 3,5; 4 или 5 второго количества антитела или его антигенсвязывающего фрагмента. Согласно некоторым вариантам осуществления настоящего изобретения, фармацевтическую композицию вводят подкожно или внутривенно.
Согласно некоторым вариантам осуществления настоящего изобретения, настоящее изобретение предлагает способы лечения умеренно тяжелого или тяжелого АД, включающие совместное введение антагониста IL-4R и топического кортикостероида (ТКС). Согласно некоторым вариантам осуществления настоящего изобретения, эти способы далее включают количественную оценку улучшения связанного с АД параметра. Согласно определенным вариантам осуществления настоящего изобретения, настоящее изобретение предлагает способы улучшения одного или более связанных с АД параметров, и эти способы включают одновременное введение антагониста IL-4R и ТКС, где улучшение связанного с АД параметра выбирается из группы, включающей: (i) уменьшение от исходного значения балла по шкале IGA по меньшей мере на 45%; (ii) уменьшение от исходного значения показателя BSA по меньшей мере на 40%; (iii) уменьшение от исходного значения индекса EASI по меньшей мере на 65%; (iv) уменьшение от исходного значения индекса SCORAD по меньшей мере на 50%; (v) уменьшение от исходного значения балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 25%; и (vi) уменьшение балла по числовой шкале оценки интенсивности кожного зуда (шкала NRS) по меньшей мере на 60%. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра представляет собой уменьшение от исходного значения балла по шкале IGA по меньшей мере на 50% на день 29 после введения антитела или его антигенсвязывающего фрагмента, которые связываются с IL-4R. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра представляет собой уменьшение от исходного значения балла по числовой шкале оценки интенсивности кожного зуда (шкала NRS) по меньшей мере на 65% на день 29 после введения фармацевтической композиции. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра представляет собой уменьшение от исходного значения индекса EASI по меньшей мере на 70% на день 29 после введения фармацевтической композиции. Согласно некоторым вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра представляет собой уменьшение от исходного значения индекса SCORAD по меньшей мере на 60% на день 29 после введения фармацевтической композиции.
Согласно определенным вариантам осуществления настоящего изобретения, ТКС выбирают из группы, состоящей из ТКС группы I, ТКС группы II и ТКС группы III. Согласно некоторым вариантам осуществления настоящего изобретения, ТКС выбирают из группы, состоящей из метилпреднизолона ацепоната, мометазона фуроата, флутиказона пропионата, бетаметазона валерата и гидрокортизона бутирата.
Согласно соответствующим вариантам осуществления настоящего изобретения, настоящее изобретение предлагает способы уменьшения зависимости от ТКС у пациента с умеренно тяжелым или тяжелым АД, включающие совместное введение антагониста IL-4R и ТКС, где доза ТКС уменьшается на 50% от дозы для пациентов, не получающих антагонист IL-4R. Согласно одному варианту осуществления настоящего изобретения, настоящее изобретение предлагает способы уменьшения дозы ТКС при лечении умеренно тяжелого или тяжелого АД, включающие введение антагониста IL-4R совместно с ТКС в уменьшенной дозе. Доза ТКС может быть уменьшена, например, более чем на 10%, 20%, 30%, 40% или 50%. Согласно одному варианту осуществления настоящего изобретения, доза ТКС может быть уменьшена, например, более чем на 10%, 20%, 30%, 40% или 50% от дозы, которую пациент получал до лечения антагонистом IL-4R.
Настоящее изобретение также включает применение антагониста IL-4R, как рассматривается в этом документе, для лечения или профилактики АД для улучшения связанного с АД параметра, для уменьшения уровня по меньшей мере одного связанного с АД биомаркера и (или) для применения антагониста IL-4R по другим показаниям либо для лечения других состояний, рассматриваемых в этом документе.
Согласно определенным вариантам осуществления настоящего изобретения, антагонист IL-4R, данных способов, представляет собой антитело или антигенсвязывающий фрагмент, которые специфически связываются с IL-4R и которые включают последовательности CDR тяжелой и легкой цепей из пары последовательностей HCVR/LCVR, выбранной из группы, состоящей из последовательностей со следующими идентификаторами SEQ ID NO: 2/10, 18/20, 22/24, 26/34, 42/44, 46/48, 50/58, 66/68, 70/72, 74/82, 90/92, 94/96, 98/106, 114/116, 118/120, 122/130, 138/140, 142/144, 146/154, 162/164, 166/168, 170/178, 186/188, 190/192, 194/202, 210/212, 214/216, 218/226, 234/236, 238/240, 242/250, 258/260 и 262/264. Согласно одному варианту осуществления настоящего изобретения, антитело или антигенсвязывающий фрагмент, которые специфически связываются с IL-4R, содержат последовательности CDR тяжелой и легкой цепей из пары последовательностей HCVR/LCVR со следующими идентификаторами SEQ ID NO: 162/164. Согласно одному варианту осуществления настоящего изобретения, антитело или антигенсвязывающий фрагмент, которые специфически связываются с IL-4R, содержат три последовательности определяющего комплементарность участка тяжелой цепи (HCDR) со следующими идентификаторами SEQ ID NO: 148, 150, 152 соответственно; и три последовательности определяющего комплементарность участка легкой цепи (LCDR) со следующими идентификаторами SEQ ID NO: 156, 158 и 160 соответственно.
Согласно некоторым вариантам осуществления настоящего изобретения, фармацевтическую композицию вводят пациенту подкожно или внутривенно. Согласно некоторым вариантам осуществления настоящего изобретения, фармацевтическая композиция содержит приблизительно 50 мг до приблизительно 600 мг антитела или его антигенсвязывающего фрагмента, которые связываются с IL-4R. Согласно другим вариантам осуществления настоящего изобретения, фармацевтическая композиция содержит приблизительно 75 мг, приблизительно 100 мг, приблизительно 150 мг, приблизительно 200 мг, приблизительно 250 мг или приблизительно 300 мг антитела или его фрагмента, которые связываются с IL-4R.
Согласно определенным вариантам осуществления настоящего изобретения, фармацевтическую композицию вводят пациенту до, после применения второго терапевтического средства или одновременно с ним. Согласно некоторым вариантам осуществления настоящего изобретения, второе терапевтическое средство представляет собой топический кортикостероид (ТКС) или ингибиторы кальциневрина.
Согласно другому аспекту настоящего изобретения, настоящее изобретение предлагает мониторинг эффективности лечения антагонистом IL-4R умеренно тяжелого или тяжелого АД у пациента, и этот способ включает: (а) определение уровня экспрессии связанного с АД биомаркера, например TARC или сывороточного IgE в биологическом образце, взятом у пациента до лечения антагонистом IL-4R; (б) определение уровня экспрессии TARC или сывороточного IgE либо обоих этих биомаркеров в биологическом образце, взятом у пациента после лечения антагонистом IL-4R; (в) сравнение уровня, определенного на этапе (а), с уровнем, определенным на этапе (б); и (г) вынесение заключения об эффективности лечения, если уровень, определенный на этапе (б), ниже уровня, определенного на этапе (а), или вынесение заключения о том, что лечение не эффективно, если уровень, определенный на этапе (б), равен уровню, определенному на этапе (а) или выше данного уровня. Согласно одному варианту осуществления настоящего изобретения, уровень на этапе (б) определяют через 1, 2, 3, 4 или 5 недель после определения уровня на этапе (а). Согласно одному варианту осуществления настоящего изобретения, биомаркером является TARC, и если уровень TARC снижается после введения антагониста IL-4R, то лечение антагонистом IL-4R считается эффективным. Согласно одному варианту осуществления настоящего изобретения, антагонист IL-4R представляет собой антитело к IL-4R или его антигенсвязывающий фрагмент, содержащий последовательности CDR тяжелых и легких цепей пары последовательностей HCVR/LCVR, выбранной из группы, состоящей из последовательностей со следующими идентификаторами SEQ ID NO: 2/10, 18/20, 22/24, 26/34, 42/44, 46/48, 50/58, 66/68, 70/72, 74/82, 90/92, 94/96, 98/106, 114/116, 118/120, 122/130, 138/140, 142/144, 146/154, 162/164, 166/168, 170/178, 186/188, 190/192, 194/202, 210/212, 214/216, 218/226, 234/236, 238/240, 242/250, 258/260 и 262/264.
Уровень экспрессии биомаркера можно определять, например, через 1, 2, 3, 4, 5 недель или позже после введения антагониста IL-4R и можно сравнивать его с уровнем экспрессии до введения антагониста. Дозу или режим дозирования антагониста IL-4R (например, антитела к IL-4R) можно корректировать после определения уровня биомаркера. Например, если экспрессия биомаркера не повышается в течение недели 1, недели 2, недели 3, недели 4, недели 5 или позже после введения антагониста, то лечение антагонистом можно прекратить или увеличить его дозу. Если экспрессия биомаркера снижается после введения антагониста, дозу антагониста можно сохранить или уменьшить для определения минимальной эффективной дозы. Согласно некоторым вариантам осуществления настоящего изобретения, лечение продолжают с использованием минимальной эффективной дозы.
Согласно другому аспекту настоящего изобретения, настоящее изобретение предлагает способы мониторинга ответа на лечение антагонистом IL-4R, где пациент страдает умеренно тяжелым или тяжелым АД, и этот способ включает: (а) получение информации об уровне экспрессии TARC или IgE либо обоих этих биомаркеров в биологическом образце у пациента после введения антагониста IL-4R; и (б) подтверждение необходимости продолжения лечения, если уровень экспрессии TARC или IgE снизился относительно уровня экспрессии до лечения антагонистом IL-4R. Согласно одному варианту осуществления настоящего изобретения, биомаркером является TARC, и если уровни TARC снижаются после введения антагониста, то появляется основание для продолжения лечения антагонистом IL-4R.
Настоящее изобретение также включает применение антагониста IL-4R, как рассматривается в этом документе, для производства лекарственного средства для лечения и (или) профилактики атопического дерматита (АД) (например, умеренно тяжелого или тяжелого эозинофильного АД, экзогенного АД, эндогенного АД и др.) или для применения по другим показаниям или для лечения других состояний, рассматриваемых в этом документе.
Настоящее изобретение также включает применение антагониста IL-4R, как рассматривается в этом документе, для лечения и (или) профилактики АД (например, умеренно тяжелого или тяжелого эозинофильного АД и др.) или для применения по другим показаниям или для лечения других состояний, рассматриваемых в этом документе.
Согласно одному варианту осуществления настоящего изобретения, антагонист IL-4R представляет собой антитело к IL-4R или его антигенсвязывающий фрагмент.
Настоящее изобретение включает фармацевтическую композицию, содержащую антитело к IL-4R или его антигенсвязывающий фрагмент для применения с целью лечения и (или) профилактики АД и родственных состояний.
Настоящее изобретение также включает фармацевтическую композицию, содержащую антитело к IL-4R или его антигенсвязывающий фрагмент для применения с целью улучшения одного или более связанных с АД параметров у нуждающегося в этом пациента.
Кроме того, настоящее изобретение также включает фармацевтическую композицию, содержащую антитело к IL-4R или его антигенсвязывающий фрагмент для применения с целью снижения уровня одного или более связанных с АД биомаркеров у нуждающегося в этом пациента.
Настоящее изобретение включает фармацевтическую композицию, содержащую антитело к IL-4R или его антигенсвязывающий фрагмент, для применения с целью лечения АД у пациента с повышенным уровнем биомаркера, выбранного из группы, включающей регулируемый тимусом и активацией хемокин (TARC), IgE, эотаксин-3, лактатдегидрогеназу (ЛДГ) и периостин.
Настоящее изобретение также включает фармацевтическую композицию, содержащую антитело к IL-4R или его антигенсвязывающий фрагмент для применения с целью лечения АД у пациента, лечение которой приводит к снижению уровня связанного с АД биомаркера у пациента на день 4, 8, 15, 22, 25, 29 или 36 после лечения относительно уровня биомаркера у пациента до лечения. Согласно определенным вариантам осуществления настоящего изобретения, связанными с АД биомаркерами является TARC или IgE либо оба этих биомаркера.
Настоящее изобретение также включает фармацевтическую композицию, содержащую антитело к IL-4R или его антигенсвязывающий фрагмент для применения с целью улучшения связанного с АД параметра или снижения уровня связанного с АД биомаркера у нуждающегося в этом пациента, где данную фармацевтическую композицию вводят пациенту последовательно в виде одной первичной дозы с последующим введением одной или более вторичных доз.
Согласно одному варианту осуществления настоящего изобретения, одну или более вторичных доз вводят еженедельно.
Согласно некоторым вариантам осуществления настоящего изобретения, фармацевтическая композиция содержит 75-600 мг антитела к IL-4R или его антигенсвязывающего фрагмента. Согласно одному варианту осуществления настоящего изобретения, фармацевтическая композиция содержит приблизительно 300 мг антитела к IL-4R или его антигенсвязывающего фрагмента.
Согласно другому аспекту настоящего изобретения, настоящее изобретение предлагает способы мониторинга безопасности вводимой человеку терапевтической дозы антагониста рецептора интерлейкина-4 (IL-4R), и указанный способ включает: получение информации о безопасности антагониста после введения человеку, где эта информация включает сведения о возникновении одного или более явлений, выбранных из группы, состоящей из анафилактической реакции или острой аллергической реакции, требующей незамедлительного лечения, тяжелой реакции в месте введения длительностью более 24 часов, тяжелой инфекции, любой паразитарной инфекции, повышения уровня аланинаминотрансферазы (АЛТ) ≥ 2 раза выше верхней границы нормы (ВГН), интервала QTc ≥ 500 мс, беременности, передозировки и инфекции, вызванной вирусом простого герпеса типа II; определение возникновения одного или более из указанных явлений, определение небезопасности указанной терапевтической дозы и, в некоторых случаях, вынесение рекомендаций об отмене терапевтической дозы или ее снижении.
Согласно одному родственному аспекту настоящего изобретения, настоящее изобретение предлагает способы мониторинга безопасности вводимой человеку терапевтической дозы антагониста рецептора интерлейкина-4 (IL-4R), и указанный способ включает: получение информации о безопасности антагониста после введения человеку, где эта информация включает сведения о возникновении одного или более явлений, выбранных из группы, состоящей из анафилактической реакции или острой аллергической реакции, требующей незамедлительного лечения, тяжелой реакции в месте введения длительностью более 24 часов, тяжелой инфекции, любой паразитарной инфекции, повышения уровня аланинаминотрансферазы (АЛТ) ≥2 раза выше верхней границы нормы (ВГН), интервала QTc ≥ 500 мс, беременности, передозировки и инфекции, вызванной вирусом простого герпеса типа II; и определение отсутствия возникновения одного или более из указанных явлений и определение безопасности указанной терапевтической дозы.
Согласно одному варианту осуществления настоящего изобретения, инфекцией является инфекция верхних дыхательных путей, фарингит или синусит. Согласно одному варианту осуществления настоящего изобретения, реакцией в месте введения является эритема, боль, узелковое утолщение, гематома или кожный зуд. Согласно одному варианту осуществления настоящего изобретения, интенсивность боли превышает 2 мм по ВАШ, например составляет от 3 мм до 30 мм по ВАШ. Согласно одному варианту осуществления настоящего изобретения, диаметр эритемы составляет ≥9 мм.
Согласно одному варианту осуществления настоящего изобретения, безопасная терапевтическая доза составляет 500 мг или менее. Согласно одному варианту осуществления настоящего изобретения, безопасную терапевтическую дозу выбирают из группы, состоящей из 75 мг, 150 мг и 300 мг.
Согласно другому аспекту настоящего изобретения, настоящее изобретение предлагает способы количественного определения или мониторинга уровня антител к лекарственному веществу в сыворотке крови человека после введения препарата, где препаратом является антагонист рецептора интерлейкина-4 (IL-4R), и указанный способ включает: (а) получение образца сыворотки крови человека, получившего дозу указанного антагониста IL-4R; и (б) определение уровня антител к лекарственному веществу в указанном образце сыворотки крови.
Согласно другому аспекту настоящего изобретения, настоящее изобретение предлагает способы сравнения антагониста рецептора интерлейкина-4 (IL-4R), произведенного посредством первого процесса и предложенного эквивалентного второго процесса, и указанный способ включает: получение информации о безопасности антагониста, произведенного посредством первого процесса, после введения первому человеку, и безопасности антагониста, произведенного посредством второго процесса, после введения второму человеку, где эта информация включает сведения о возникновении одного или более явлений, выбранных из группы, состоящей из анафилактической реакции или острой аллергической реакции, требующей незамедлительного лечения, тяжелой реакции в месте введения длительностью более 24 часов, тяжелой инфекции, любой паразитарной инфекции, повышения уровня аланинаминотрансферазы (АЛТ) ≥ 2 раза выше верхней границы нормы (ВГН), интервала QTc ≥ 500 мс, беременности, передозировки и инфекции, вызванной вирусом простого герпеса типа II; и если данная информация не отличается существенно для антагониста, произведенного посредством первого процесса, и антагониста, произведенного посредством второго процесса, то эти два процесса считаются приемлемыми для производства эквивалентных антагонистов; а если данная информация отличается существенно для антагониста, произведенного посредством первого процесса, и антагониста, произведенного посредством второго процесса, то эти два процесса считаются неприемлемыми для производства эквивалентных антагонистов.
Согласно родственному аспекту настоящего изобретения, настоящее изобретение предлагает способы сравнения антагониста рецептора интерлейкина-4 (IL-4R), произведенного посредством первого процесса и посредством предложенного эквивалентного второго процесса, и указанный способ включает: получение информации о терапевтической дозе антагониста после введения дозы антагониста, произведенного посредством первого производственного процесса, первому человеку, и после введения дозы антагониста, произведенного посредством второго процесса, второму человеку, где эта информация включает одно или более из нижеследующего: (а) площадь под кривой зависимости концентрации лекарственного вещества в плазме крови от времени, рассчитанная с помощью метода трапеций, от момента времени «ноль» до момента определения (ППКпосл) от приблизительно 4 мг*ч/мл до приблизительно 20 мг*ч/мл; (б) максимальная определяемая концентрация в плазме крови (Смакс) от приблизительно 15 мкг/мл до приблизительно 42 мкг/мл; (в) время до первоначального достижения максимальной концентрации в плазме крови (tмакс) от приблизительно 40 ч до приблизительно 280 ч; (г) площадь под кривой зависимости концентрации лекарственного вещества в плазме крови от времени с экстраполяцией до бесконечности (AUC) от приблизительно 5 000 000 нг/ч*мл до приблизительно 25 000 000 нг/ч*мл; (д) длительность терминального периода полувыведения (t1/2z) от приблизительно 50 ч до приблизительно 200 ч; и если данная информация существенно не отличается для антагониста, произведенного посредством первого процесса, и антагониста, произведенного посредством второго процесса, то эти два процесса считаются приемлемыми для производства эквивалентных антагонистов, а если информация отличается существенно для антагониста, произведенного посредством первого процесса, и антагониста, произведенного посредством второго процесса, то эти два процесса считаются неприемлемыми для производства эквивалентных антагонистов.
Согласно другому аспекту настоящего изобретения, настоящее изобретение предлагает терапевтическую лекарственную форму фармацевтической композиции, содержащей антагонист рецептора интерлейкина-4 (IL-4R), где введение лекарственной формы человеку приводит к одному или более из нижеследующего: (а) площадь под кривой зависимости концентрации лекарственного вещества в плазме крови от времени, рассчитанная с помощью метода трапеций, от момента времени «ноль» до момента определения (ППКпосл) от приблизительно 4 мг*ч/мл до приблизительно 20 мг*/мл; (б) максимальная определяемая концентрация в плазме крови (Смакс) от приблизительно 15 мкг/мл до приблизительно 42 мкг/мл; (в) время до достижения первоначальной максимальной концентрации в плазме крови (tмакс) от приблизительно 40 ч до приблизительно 280 ч; (г) площадь под кривой зависимости концентрации лекарственного вещества в плазме крови от времени с экстраполяцией до бесконечности (AUC) от приблизительно 5 000 000 нг/ч*мл до приблизительно 25 000 000 нг/ч*мл; (д) длительность терминального периода полуэлиминации (t1/2z) от приблизительно 50 ч до приблизительно 200 ч.
Согласно одному варианту осуществления настоящего изобретения, безопасная терапевтическая доза составляет 500 мг или менее. Согласно одному варианту осуществления настоящего изобретения, безопасную терапевтическую дозу выбирают из группы, состоящей из дозы 75 мг, 150 мг и 300 мг.
Определенные аспекты настоящего изобретения имеют отношения к способам и композициям, которые могут применяться в вакцинировании. Настоящее изобретение предлагает способы усиления или потенцирования иммунного ответа на антиген у пациента. Согласно некоторым вариантам осуществления настоящего изобретения, способы усиления или потенцирования иммунного ответа на антиген у пациента включают введение фармацевтической композиции, содержащей антиген и антагонист IL-4R. Некоторые варианты осуществления настоящего изобретения касаются способов, включающих (а) введение пациенту вакцинной композиции, содержащей антиген; и (б) введение антагониста IL-4R до или после введения пациенту вакцинной композиции и (или) одновременное с ней. Настоящее изобретение также включает применение фармацевтических композиций для усиления или потенцирования иммунного ответа на антиген у пациента, и эти композиции содержат: (а) антиген; и (б) антагонист IL-4R. Согласно одному примеру варианта осуществления настоящего изобретения, антагонист IL-4R представляет собой антитело к IL-4R (как показано в примере 1 в этом документе). Согласно определенным вариантам осуществления настоящего изобретения, антагонист IL-4R является антителом к IL-4R, обладающим характеристиками связывания, присущими эталонному антигену, называемому в этом документе «mAb1» (например, антитело или его антигенсвязывающий фрагмент, включающие определяющие комплементарность участки mAb1).
Другие варианты осуществления настоящего изобретения будут очевидны по рассмотрении последующего подробного описания.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фиг. 1 показан график в декартовых координатах средних значений (СКО) профилей зависимости от времени концентрации в сыворотке крови функционального mAb1 после подкожного введения одной дозы.
На фиг. 2 схематически представлены процедура введения препарата и оценки боли, как указано в примере 5.
На фиг. 3 показано число пациентов с ответом на лечение по шкале IGA (балл от 0 до 1) - использование последних значений вместо недостающих (LOCF) для исследования в примере 6.
На фиг. 4 показано среднее изменение балла по шкале IGA от исходного значения - LOCF для исследования в примере 6.
На фиг. 5 показано среднее изменение балла по шкале IGA в процентах от исходного значения - LOCF для исследования в примере 6.
На фиг. 6 показано среднее изменение индекса EASI от исходного значения - LOCF для исследования в примере 6.
На фиг. 7 показано среднее изменение индекса EASI в процентах от исходного значения - LOCF для исследования в примере 6.
На фиг. 8 показано число пациентов с ответом на лечение в виде индекса EASI50 - LOCF для исследования в примере 6.
На фиг. 9 показано среднее изменение показателя BSA от исходного значения - LOCF для исследования в примере 6.
На фиг. 10 показано среднее изменение показателя BSA в процентах от исходного значения - LOCF для исследования в примере 6.
На фиг. 11 показано среднее изменение балла по опроснику из 5 вопросов для оценки кожного зуда от исходного значения - LOCF для исследования в примере 6.
На фиг. 12 показано среднее изменение балла по опроснику из 5 вопросов для оценки кожного зуда в процентах от исходного значения - LOCF для исследования в примере 6.
На фиг. 13 показано среднее изменение балла по шкале NRS от исходного значения - LOCF для исследования в примере 6.
На фиг. 14 показано среднее изменение балла по шкале NRS в процентах от исходного значения - LOCF для исследования в примере 6.
На фиг. 15 показано изменение показателя BSA в процентах от исходного значения у пациентов, получавших антитело к IL-4R в дозе 75 мг, 150 мг или 300 мг, в сравнении с плацебо для исследования в примере 8.
На фиг. 16 показано изменение балла по шкале IGA в процентах от исходного значения у пациентов, получавших антитело к IL-4R в дозе 75 мг, 150 мг или 300 мг, в сравнении с плацебо для исследования в примере 8.
На фиг. 17 показано изменение индекса EASI в процентах от исходного значения у пациентов, получавших антитело к IL-4R в дозе 75 мг, 150 мг или 300 мг, в сравнении с плацебо для исследования в примере 8.
На фиг. 18 показано изменение балла по шкале NRS в процентах от исходного значения у пациентов, получавших антитело к IL-4R в дозе 75 мг, 150 мг или 300 мг, в сравнении с плацебо для исследования в примере 8.
На фиг. 19 показано изменение с течением времени индекса EASI у пациентов с умеренно тяжелым или тяжелым АД, получавших антитело к IL-4R в дозе 300 мг, для исследования в примере 8.
На фиг. 20 показан процент пациентов с ответом на лечение в виде изменения индекса EASI у пациентов после получения антитела к IL-4R в дозе 75 мг, 150 мг или 300 мг в сравнении с плацебо для исследования в примере 8.
На фиг. 21 показан процент пациентов с ответом на лечение в виде изменения индекса EASI на неделе 4 (день 29) после получения антитела к IL-4R в дозе 75 мг, 150 мг или 300 мг в сравнении с плацебо для исследования в примере 8.
На фиг. 22 показан процент пациентов с баллом по шкале IGA ≤ 1 для исследования в примере 8.
На фиг. 23 показано среднее изменение индекса EASI в процентах от исходного значения до использованного вместо недостающего значения последнего значения (LOCF) для исследования в примере 10.
На фиг. 24 показан процент пациентов с ответом на лечение по шкале IGA (балл от 0 до 1) до использованного вместо недостающего значения последнего значения (LOCF) для исследования в примере 10.
На фиг. 25 показан процент пациентов с ответом на лечение по шкале IGA (уменьшение на 2 и более балла) до использованного вместо недостающего значения последнего значения (LOCF) для исследования в примере 10.
На фиг. 26 показан процент пациентов с ответом на лечение по индексу EASI (уменьшение индекса на 50% от исходного значения) до использованного вместо недостающего значения последнего значения (LOCF) для исследования в примере 10.
На фиг. 27 показано среднее изменение индекса EASI от исходного значения до LOCF для исследования в примере 10.
На фиг. 28 показано среднее изменение балла по шкале IGA от исходного значения до LOCF для исследования в примере 10.
На фиг. 29 показано среднее изменение балла по шкале IGA в процентах от исходного значения до LOCF для исследования в примере 10.
На фиг. 30 показано среднее изменение показателя BSA от исходного значения до LOCF для исследования в примере 10.
На фиг. 31 показано среднее изменение индекса SCORAD от исходного значения до LOCF для исследования в примере 10.
На фиг. 32 показано среднее изменение балла по шкале NRS от исходного значения до LOCF для исследования в примере 10.
На фиг. 33 показано среднее изменение балла по опроснику из 5 вопросов для оценки кожного зуда от исходного значения до LOCF для исследования в примере 10.
На фиг. 34 показано среднее изменение индекса EASI в процентах от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 35 показано среднее изменение индекса EASI от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 36 показан процент пациентов с ответом на лечение в виде показателя EASI50 - цензурированного LOCF для исследования в примере 11.
На фиг. 37 показан график Каплана - Мейера для времени до первоначального достижения ответа EASI50 - цензурированного LOCF для исследования в примере 11.
На фиг. 38 показано среднее изменение балла по шкале IGA в процентах от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 39 показано среднее изменение балла по шкале IGA от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 40 показан процент пациентов с ответом на лечение по шкале IGA (балл от 0 до 1) - цензурированного LOCF для исследования в примере 11.
На фиг. 41 показан график Каплана - Мейера для времени до первого балла по шкале IGA ≤ 1 - цензурированного LOCF для исследования в примере 11.
На фиг. 42 показан процент пациентов без рецидива заболевания с баллом по шкале IGA ≤ 1 на каждом визите - цензурированного LOCF для исследования в примере 11.
На фиг. 43 показан процент пациентов с уменьшением балла по шкале IGA ≥ 2 от исходного значения на каждом визите - цензурированного LOCF для исследования в примере 11.
На фиг. 44 показано среднее изменение индекса SCORAD в процентах от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 45 показано среднее изменение индекса SCORAD от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 46 показано среднее изменение балла по шкале NRS в процентах от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 47 показано среднее изменение балла по шкале NRS от исходного значения - цензурированного LOCF для исследования в примере 11.
На фиг. 48 показаны значения уровня сывороточного IgE на исходном уровне (А) и их медианное процентное изменение при введении различных доз mAb1 или плацебо (Б) для исследования в примере 12.
На фиг. 49 показаны значения уровня сывороточного TARC на исходном уровне (А) и их медианное процентное изменение при введении различных доз mAb1 или плацебо (Б) для исследования в примере 12.
На фиг. 50 показано изменение уровней TARC для объединенной группы mAb1 в сравнении с группой плацебо для исследования в примере 12.
На фиг. 51 показано распределение исходных значений уровня (А) TARC, (Б) уровня общего сывороточного IgE и (В) уровня лактатдегидрогеназы (ЛДГ) у пациентов в исследовании в разделе Б примера 12.
На фиг. 52 показано медианное изменение уровня IgE в процентах от исходного значения для исследования в разделе Б примера 12.
На фиг. 53 показано медианное изменение уровня ЛДГ в процентах от исходного значения для исследования в разделе Б примера 12.
На фиг. 54 показано медианное изменение уровня TARC в процентах от исходного значения для исследования в разделе Б примера 12.
На фиг. 55 показано медианное изменение уровня IgE в процентах от исходного значения для исследования в разделе В примера 12.
На фиг. 56 показано медианное изменение уровня TARC в процентах от исходного значения для исследования в разделе В примера 12.
ПОДРОБНОЕ ОПИСАНИЕ
Прежде чем приступить к описанию настоящего изобретения, необходимо понимать, что его применение не ограничено описанными конкретными способами и экспериментальными условиям, поскольку эти способы и условия могу различаться. Следует также понимать и то, что терминология, используемая в этом документе, предназначена для описания только конкретных вариантов осуществления настоящего изобретения и не является ограничительной, поскольку объем настоящего изобретения будет ограничен только прилагаемой формулой изобретения.
Если не указано иное, все технические и научные термины, используемые в этом документе, имеют то же значение, что и значение, которое обычно понимается средним специалистом в области, к которой принадлежит настоящее изобретение. В контексте этого документа термин «приблизительно», в случае применения к конкретному указанному числовому значению, означает, что это значение может отличаться от указанного значения не более чем на 1%. Например, в этом документе выражение «приблизительно 100» включает значения 99 и 101 и все промежуточные значения (например, 99,1, 99,2, 99,3, 99,4 и т.д.). В контексте этого документа термины «лечить», «лечение» и подобные им означают ослабление симптомов, временное или полное устранение причин симптомов, а также предупреждение либо замедление появления симптомов названного нарушения или состояния.
Настоящее изобретение включает способы, заключающиеся в введении нуждающемуся в этом пациенту терапевтической композиции, содержащей антагонист IL-4R. В контексте этого документа выражение «нуждающийся в этом пациент» означает человека или животного, у которого наблюдаются один или более симптомов или признаков атопического дерматита, и (или) у которых был диагностирован атопический дерматит. Согласно определенным вариантам осуществления настоящего изобретения, способы настоящего изобретения могут применяться для лечения пациентов с повышенным уровнем одного или более связанных с АД биомаркеров (описано в другом месте этого документа). Например, способы настоящего изобретения включают введение антагониста IL-4R пациентам с повышенным уровнем IgE или TARC либо периостина. Согласно некоторым вариантам осуществления настоящего изобретения, способы, указанные в этом документе, могут применяться для лечения АД у детей в возрасте ≤ 1 года. Например, настоящие способы могут применяться для лечения младенцев в возрасте до 1 месяца, в возрасте 1 месяца, 2 месяцев, 3 месяцев, 4 месяцев, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев или в возрасте до 12 месяцев. Согласно другим вариантам осуществления настоящего изобретения, настоящие способы могут применяться для лечения детей и (или) подростков в возрасте ≤ 18 лет. Например, настоящие способы могут применяться для лечения детей или подростков в возрасте до 17 лет, 16 лет, 15 лет, 14 лет, 13 лет, 12 лет, 11 лет, 10 лет, 9 лет, 8 лет, 7 лет, 6 лет, 5 лет, 4 лет, 3 лет или в возрасте до 2 лет.
В контексте настоящего изобретения к «нуждающемуся в этом пациенту» могут относиться, например, пациенты, у которых до лечения присутствуют (или присутствовали) один или более связанных с АД параметров, например увеличенные баллы по шкале IGA, показатель BSA, индекс EASI, индекс SCORAD, балл по опроснику из 5 вопросов для оценки кожного зуда и (или) балл по числовой шкале NRS, и (или) повышенный уровень одного или более связанных с АД биомаркеров, таких как IgE и (или) TARC (как указано в другом месте этого документа). Согласно определенным вариантам осуществления настоящего изобретения, к «нуждающемуся в этом пациенту» могут относиться пациенты из подгруппы популяции с большей предрасположенностью к АД, или с повышенным уровнем связанного с АД биомаркера. Например, к «нуждающемуся в этом пациенту» могут относиться пациенты из подгруппы популяции, сформированной в соответствии с расовым или этническим признаком этой популяции.
Термин «атопический дерматит» (АД), используемый в этом документе, представляет собой воспалительное заболевание кожи, характеризующееся выраженным кожным зудом и наличием чешуйчатых и сухих экзематозных поражений. Термин «атопический дерматит» включает, помимо прочего, АД, вызванный или связанный с нарушением барьерной функции эпидермиса, аллергией (например, аллергией на определенные продукты питания, пыльцу, плесень, пылевого клеща, животных и т.д.), воздействием ионизирующего излучения и (или) астмой. Настоящее изобретение охватывает способы лечения пациентов с легким, умеренно тяжелым или тяжелым АД. В контексте этого документа «умеренно тяжелый или тяжелый АД» характеризуется выраженным кожным зудом, распространенными поражениями кожи, которые часто осложняются персистирующими бактериальными, вирусными или грибковыми инфекциями. Умеренно тяжелый или тяжелый АД также включает хронический АД. Во многих случаях хронические поражения включат утолщенные бляшки на коже, лихенизацию и фиброзные папулы. У пациентов с умеренно тяжелым или тяжелым АД, как правило, поражено более 20% кожи или 10% кожи с поражением глаз, кистей и кожных складок. Умеренно тяжелый или тяжелый АД также присутствует у пациентов, нуждающихся в частом лечении топическими кортикостероидами. Об умеренно тяжелом или тяжелом АД также можно говорить и в том случае, если у пациента заболевание не поддается лечению топическим кортикостероидом или ингибитором кальциневрина, либо другим терапевтическим средством, известным в данной области техники.
Настоящее изобретение включает способы лечения как экзогенного, так и эндогенного АД. Для экзогенного АД характерно наличие IgE -опосредованной сенсибилизации и повышение уровня цитокинов Th2, и эта форма АД встречается у 70-80% пациентов. Эндогенный АД без IgE-опосредованной сенсибилизации встречается у 20-30% пациентов; у этих пациентов наблюдаются более низкие уровни IL-4 и IL -13, чем у пациентов с экзогенным АД.
Настоящее изобретение включает способы лечения АД у пациентов с устойчивостью, отсутствием ответа или недостаточным ответом заболевания на лечение топическим кортикостероидом (ТКС) или ингибитором кальциневрина. Термин «устойчивость, отсутствие ответа или недостаточный ответ заболевания на лечение ТКС или ингибитором кальциневрина», используемый в этом документе, относится к пациентам, получавшим лечение ТКС или ингибитором кальциневрина, где ТКС или ингибиторы кальциневрина не оказывают терапевтического эффекта. Согласно некоторым вариантам осуществления настоящего изобретения, данный термин относится к ухудшению соблюдения пациентом режима и схемы лечения, и (или) токсичности и побочным эффектам, и (или) неэффективности ТКС или ингибитора кальциневрина, применявшихся с целью снижения, ослабления или уменьшения выраженности симптомов АД. Согласно некоторым вариантам осуществления настоящего изобретения, данный термин относится к пациентам с умеренно тяжелым или тяжелым АД, устойчивым к лечению ТКС или ингибитором кальциневрина. Согласно некоторым вариантам осуществления настоящего изобретения, данный термин относится к пациентам с АД, течение которого не удается контролировать несмотря на лечение ТКС и (или) ингибитором кальциневрина. Согласно некоторым вариантам осуществления настоящего изобретения, у пациентов с «устойчивостью, отсутствием ответа или недостаточным ответом заболевания на лечение топическим ТКС или ингибитором кальциневрина» может не наблюдаться улучшение одного или более связанных с АД параметров. Примеры связанных с АД параметров приведены в другом месте этого документа. Например, лечение ТКС или ингибитором кальциневрина может не привести к уменьшению выраженности кожного зуда или к уменьшению индекса EASI, либо показателя BSA. Согласно некоторым вариантам осуществления настоящего изобретения, настоящее изобретение включает способы лечения умеренно тяжелого или тяжелого АД у пациентов, получавших ранее лечение ТКС или ингибитором кальциневрина в течение ≥ 1 месяца, у которых не наблюдалось улучшения одного или более связанных с АД параметров. Например, настоящие способы могут применяться для лечения пациентов с хроническим АД, получающих стабильную терапию ТКС или ингибитором кальциневрина, у которых показатель BSA составляет ≥ 10%, или балл по шкале IGA составляет ≥ 3.
Согласно другим вариантам осуществления настоящего изобретения, термин «нуждающийся в этом пациент» относится к пациентам с умеренно тяжелым или тяжелым АД, которые получали лечение одним или боле ТКС более 6 месяцев, более 1 года, более 2 лет, более приблизительно 5 лет, более приблизительно 7 лет или более приблизительно 10 лет. Пациенты могут желать минимизации выраженности нежелательных побочных эффектов ТКС или их отсутствия. Настоящее изобретение включает способы длительного более безопасного и эффективного лечения умеренно тяжелого или тяжелого АД у пациентов, и эти способы включают применение антагониста IL-4R совместно с ТКС, где дозу IL-4R корректируют для минимизации выраженности нежелательных побочных эффектов этого ТКС или предупреждения их возникновения. Согласно определенным вариантам осуществления настоящего изобретения, настоящее изобретение включает способы уменьшения зависимости от ТКС у пациента с умеренно тяжелым или тяжелым АД; эти способы включают применение антагониста IL-4R в терапевтически эффективном количестве совместно с сильным ТКС, где доза применяемого пациентом ТКС уменьшается приблизительно на 50% по сравнению с дозой пациента, не получающего антагонист IL-4R. Согласно определенным вариантам осуществления настоящего изобретения, настоящее изобретение включает способы уменьшения зависимости от ТКС у пациента с умеренно тяжелым или тяжелым АД; эти способы включают применение антагониста IL-4R в терапевтически эффективном количестве совместно с сильным ТКС, где доза применяемого пациентом ТКС уменьшается приблизительно на 50% по сравнению с дозой, получаемой пациентом до лечения антагонистом IL-4R. Согласно определенным вариантам осуществления настоящего изобретения, совместное применение антагониста IL-4R и ТКС приводит к аддитивному и синергическому эффекту этих препаратов при лечении АД в сравнении с монотерапией.
Термин «ТКС», используемый в этом документе, включает топические кортикостероиды группы I, группы II, группы III и группы IV. Согласно Анатомо-терапевтическо-химической классификации Всемирной организации здравоохранения, кортикостероиды делятся по своей активности на кортикостероиды с низкой активностью (группа I), умеренно активные (группы II), активные (группа III) и высокоактивные (группы IV), исходя из активности в сравнении с гидрокортизоном. ТКС группы IV (высокоактивные) более чем в 600 раз активнее гидрокортизона, и к ним относятся клобетазола пропионат и хальцинонид. ТКС группы III (активные) в 50-100 раз активнее гидрокортизона, и к ним относятся, помимо прочего, бетаметазона валерат, бетаметазона дипропионат, дифлукортолона валерат, гидрокортизона-17-бутират, мометазона фуроат и метилпреднизолона ацепонат. ТКС группы II (умеренно активные) более чем в 2-25 раз активнее гидрокортизона, и к ним относятся клобетазона бутират и триамцинолона ацетонид. К ТКС группы I (кортикостероиды с низкой активностью) относится гидрокортизон.
Хотя любые способы и материалы, схожие со способами и материалами, описанными в этом документе, или эквивалентные им, могут применяться при реализации на практике настоящего изобретения, ниже рассматриваются предпочтительные способы и материалы. Все публикации, упомянутые в этом документе, полностью включены в данный документ посредством ссылок.
Способы улучшения связанных с атопический дерматитом (АД) параметров
Настоящее изобретение включает способы улучшения одного или более связанных с атопическим дерматитом (АД) параметров у нуждающегося в этом пациента, которые включают введение пациенту фармацевтической композиции, содержащей антагонист рецептора интерлейкина-4 (IL-4R).
Примерами «связанных с АД параметров» являются: (а) балл по шкале общей оценки исследователя (IGA); (б) площадь пораженной кожи при атопическом дерматите (BSA); (в) индекс площади экзематозного поражения кожи и тяжести экземы (EASI); (г) индекс SCORAD; (д) балл по опроснику из 5 вопросов для оценки кожного зуда; и (е) балл по числовой шкале для оценки интенсивности кожного зуда (NRS). «Улучшение связанного с АД параметра» означает уменьшение исходного значения балла по шкале IGA, показателя BSA, индекса EASI, индекса SCORAD, балла по опроснику из 5 вопросов для оценки кожного зуда или балла по шкале NRS. В контексте этого документа термин «исходное значение» применительно к связанному с АД параметру означает числовое значение связанного с АД параметра у пациента до введения фармацевтической композиции, являющейся предметом настоящего изобретения.
Для определения возможного «улучшения» связанного с АД параметра этот параметр оценивают количественно до начала лечения и через один или более моментов времени после введения фармацевтической композиции, являющейся предметом настоящего изобретения. Например, величину связанного с АД параметра можно определять на день 1, день 2, день 3, день 4, день 5, день 6, день 7, день 8, день 9, день 10, день 11, день 12, день 14, день 15, день 22, день 25, день 29, день 36, день 43, день 50, день 57, день 64, день 71, день 85 или в конце недели 1, недели 2, недели 3, недели 4, недели 5, недели 6, недели 7, недели 8, недели 9, недели 10, недели 11, недели 12, недели 13, недели 14, недели 15, недели 16, недели 17, недели 18, недели 19, недели 20, недели 21, недели 22, недели 23, недели 24 или позже после первоначального лечения фармацевтической композицией, являющейся предметом настоящего изобретения. Различие между величиной параметра в конкретный момент времени после начала лечения и величиной этого параметра до начала лечения используют для определения возможного «улучшения» (например, уменьшения величины) связанного с АД параметра.
Шкала общей оценки исследователя (IGA). Шкала IGA представляет собой используемую в клинических условиях для оценки тяжести АД и клинического ответа на лечение 6-балльную шкалу со значениями от 0 (отсутствие воспалительных признаков атопического дерматита) до 5 (очень тяжелое заболевание). Согласно определенным вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале IGA. Например, настоящее изобретение включает терапевтические методы, которые приводят к уменьшению от исходного значения балла по шкале IGA по меньшей мере приблизительно на 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более на день 4, 8, 15, 22, 25, 29, 36, 43, 50, 57, 64, 71, 85 или позже после введения антагониста IL-4R (например, после подкожного введения антитела к IL-4R или его антигенсвязывающего фрагмента в дозе приблизительно 75 мг, 150 мг или 300 мг). Согласно определенным примерам вариантов осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале IGA по меньшей мере на 25% от исходного значения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале IGA по меньшей мере на 25% от исходного значения на день 15 после введения. Согласно определенным вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале IGA по меньшей мере на 35% от исходного значения на день 22 после введения. Согласно другим вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале IGA по меньшей мере на 40% или 45% от исходного значения на день 85 после введения.
Площадь пораженной кожи при атопическом дерматите (BSA). BSA определяется для участка пораженной кожи каждой крупной части тела (головы, туловища, рук и ног) и рассчитывается в виде процента от общей площади всех крупных частей тела. Согласно определенным вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению показателя BSA. Например, настоящее изобретение включает терапевтические методы, которые приводят к уменьшению от исходного значения показателя BSA по меньшей мере приблизительно на 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более на день 4, 8, 15, 22, 25, 29, 36, 43, 50, 57, 64, 71, 85 или позже после введения антагониста IL-4R (например, после подкожного введения антитела к IL-4R или его антигенсвязывающего фрагмента в дозе приблизительно 75 мг, 150 мг или 300 мг). Согласно определенным примерам вариантов осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению показателя BSA по меньшей мере на 35% от исходного значения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению показателя BSA по меньшей мере на 35% от исходного значения на день 29 после введения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению показателя BSA по меньшей мере на 40% от исходного значения на день 29 после введения. Согласно некоторым вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению показателя BSA по меньшей мере на 40% или 50% от исходного значения на день 85 после введения.
Индекс площади экзематозного поражения кожи и тяжести экземы (EASI). Индекс EASI представляет собой валидированный параметр, используемый в клинических условиях для оценки тяжести и степени АД (Hanifin et al. 2001, Exp. Dermatol. 10:11-18). Врачом или квалифицированным медицинским персоналом оценивается тяжесть четырех характеристик АД по шкале от 0 (отсутствие признаков заболевания) до 3 (тяжелое заболевание). Кроме того, оценивается в процентах площадь экзематозного поражения кожи головы, туловища, рук и ног, и полученное значение пересчитывается в баллы от 0 до 6. Согласно определенным вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса EASI. Например, настоящее изобретение включает терапевтические методы, которые приводят к уменьшению от исходного значения индекса EASI по меньшей мере приблизительно на 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более на день 4, 8, 15, 22, 25, 29, 36, 43, 50, 57, 64, 71, 85 или позже после введения антагониста IL-4R (например, после подкожного введения антитела к IL-4R или его антигенсвязывающего фрагмента в дозе приблизительно 75 мг, 150 мг или 300 мг). Согласно определенным примерам вариантов осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса EASI по меньшей мере на 45% от исходного значения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса EASI по меньшей мере на 45% от исходного значения на день 15 после введения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса EASI по меньшей мере на 50% от исходного значения на день 29 после введения. Согласно некоторым вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса EASI по меньшей мере на 55% или 60% от исходного значения на день 85 после введения.
Индекс SCORAD. Индекс «SCORing Atopic Dermatitis» (SCORAD) используется для клинической оценки тяжести (например, распространенности заболевания или тяжести проявлений) атопического дерматита и был разработан Европейской рабочей группой по атопическому дерматиту (консенсусный доклад Европейской рабочей группы по атопическому дерматиту, 1993 г., Dermatology (Basel) 186(1):23-31). Степень АД оценивается путем определения площади пораженной кожи каждой из определенных областей тела в процентах с последующим суммированием полученных значений, при этом максимальная величина составляет 100% (обозначается как параметр «А» при расчете общего индекса SCORAD). Тяжесть 6 специфических симптомов АД оценивается с использованием следующей шкалы: отсутствие симптомов (0), легкие симптомы (1), умеренные симптомы (2), тяжелые симптомы (3) (максимальное количество баллов составляет 18; обозначается как параметр «Б» при расчете общего индекса SCORAD). Осуществляется пациентивная оценка пациентом интенсивности кожного зуда и бессонницы с помощью визуально-аналоговой шкалы (ВАШ), согласно которой «0» означает отсутствие кожного зуда (или бессонницы), и «10» означает крайне сильный зуд (или бессонницу), при этом максимально возможное количество баллов составляет 20. Обозначается как параметр «В» при расчете общего индекса SCORAD. Индекс SCORAD рассчитывается следующим образом: А/5+7Б/2+В. Согласно определенным вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса SCORAD. Например, настоящее изобретение включает терапевтические методы, которые приводят к уменьшению от исходного значения индекса SCORAD по меньшей мере приблизительно на 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более на день 4, 8, 15, 22, 25, 29, 36, 43, 50, 57, 64, 71, 85 или позже после введения антагониста IL-4R (например, после подкожного введения антитела к IL-4R или его антигенсвязывающего фрагмента в дозе приблизительно 75 мг, 150 мг или 300 мг). Согласно определенным примерам вариантов осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса SCORAD по меньшей мере на 30% от исходного значения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса SCORAD по меньшей мере на 30% от исходного значения на день 29 после введения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса SCORAD по меньшей мере на 35% от исходного значения на день 29 после введения. Согласно некоторым вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению индекса SCORAD по меньшей мере на 40% или 45% от исходного значения на день 85 после введения.
Опросник из 5 вопросов для оценки кожного зуда. Опросник из 5 вопросов для оценки кожного зуда представляет собой одностраничный инструмент, состоящий из 5 вопросов, который используется в клинических условиях для оценки 5 параметров имеющегося кожного зуда: интенсивность зуда, длительность зуда, изменение интенсивности зуда, влияние зуда на жизнедеятельность пациента и локализация зуда (Elman and Hynan, 2010, Brit. J. Dermatol. 162:587-593). Каждый вопрос соответствует 1 из 5 параметров кожного зуда; пациенты оценивают интенсивность своих симптомов как «присутствуют» или по шкале от 1 до 5, где «5» является максимальным значением. Согласно определенным вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по опроснику из 5 вопросов для оценки кожного зуда. Например, настоящее изобретение включает терапевтические методы, которые приводят к уменьшению от исходного значения балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере приблизительно на 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более на день 4, 8, 15, 22, 25, 29, 36, 43, 50, 57, 64, 71, 85 или позже после введения антагониста IL-4R (например, после подкожного введения антитела к IL-4R или его антигенсвязывающего фрагмента в дозе приблизительно 75 мг, 150 мг или 300 мг). Согласно определенным примерам вариантов осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 15% от исходного значения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 15% от исходного значения на день 15 после введения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 20% от исходного значения на день 15 после введения. Согласно некоторым вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по опроснику из 5 вопросов для оценки кожного зуда по меньшей мере на 25% или 30% от исходного значения на день 85 после введения.
Числовая шкала для оценки интенсивности кожного зуда (шкала NRS). Числовая шкала для оценки интенсивности кожного зуда (шкала NRS) представляет собой инструмент оценки, включающий один вопрос и используемый для оценки у пациента самого сильного кожного зуда при АД за последние 12 часов по шкале от 1 до 10. Согласно определенным вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале NRS. Например, настоящее изобретение включает терапевтические методы, которые приводят к уменьшению от исходного значения балла по шкале NRS по меньшей мере приблизительно на 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более на день 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или позже после введения антагониста IL-4R (например, после подкожного введения антитела к IL-4R или его антигенсвязывающего фрагмента в дозе приблизительно 75 мг, 150 мг или 300 мг). Согласно определенным примерам вариантов осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале NRS по меньшей мере на 25% от исходного значения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале NRS по меньшей мере на 25% от исходного значения на день 2 после введения. Согласно одному варианту осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале NRS по меньшей мере на 30% от исходного значения на день 2 после введения. Согласно некоторым вариантам осуществления настоящего изобретения, введение пациенту антагониста IL-4R приводит к уменьшению балла по шкале NRS по меньшей мере на 45% или 50% от исходного значения на день 85 после введения.
Шкала общей оценки отдельных признаков (GISS). Отдельные компоненты поражения кожи при АД (эритема, инфильтрация/популяция, экскориация и лихенизация) оцениваются повсеместно (т.е. каждый параметр оценивается с учетом его распространенности по всему телу, а не по анатомической области) по шкале из 4 баллов (от 0 [отсутствие поражения] до 3 [тяжелое поражение]) с использованием критериев оценки тяжести индекса EASI.
Номинальная шкала для оценки кожного зуда. Номинальная шкала для оценки кожного зуда представляет собой 4-балльную шкалу, используемую для оценки симптомов, которая применяется в клинических исследованиях АД, и для которой характерна меньшая «усредненность» оценки (Kaufmann 2006). Оценка по шкале осуществляется следующим образом: 0 - отсутствие кожного зуда; 1 - легкий кожный зуд (эпизодический незначительный кожный зуд/почесывание); 2 - умеренный кожный зуд (постоянный или периодический кожный зуд/почесывание, не нарушающий сон); 3 - тяжелый кожный зуд (крайне беспокоящий кожный зуд/почесывание, нарушающий сон).
Опросник для пациента по оценке экземы (POEM). POEM представляет собой валидированный опросник из 7 вопросов, применяемый в клинической практике для оценки симптомов заболевания у детей и взрослых (Charman 2004). Опросник включает 7 вопросов (сухость кожи, кожный зуд, шелушение кожи, растрескивание кожи, потеря сна, кровоточивость и мокнутие кожи) с пятью вариантами ответов на каждый вопрос и дает общую балльную оценку от 0 до 28; высокий балл указывает на плохое качество жизни.
Дерматологический индекс качества жизни (DLQI). DLQI представляет собой валидированный опросник из 10 вопросов, применяемый в клинической практике и клинических исследованиях для оценки влияния симптомов АД и его лечения на качество жизни (Badia 1999). Опросник содержит 10 вопросов с четырьмя вариантами ответов на каждый вопрос, он предназначен для оценки качества жизни пациента в течение последней недели и позволяет получить общую балльную оценку от 0 до 30; высокий балл указывает на плохое качество жизни.
Опросник для оценки влияния кожного зуда на качество жизни. Опросник для оценки влияния кожного зуда на качество жизни представляет собой валидированный и специально разработанный для оценки кожного зуда инструмент, с помощью которого оцениваются симптоматический эмоциональный и функциональный эффект зуда. Для оценки трех названных параметров используется общий балл и отдельный балл по каждому параметру. Это надежный, достоверный и гибкий опросник (Desai 2008).
EQ-5D. Опросник EQ-5D представляет собой стандартизированный инструмент для оценки состояния здоровья, разработанным группой EuroQOl для простой и универсальной оценки состояния здоровья пациента для клинических и экономических целей. Опросник EQ-5D оценивает связанное со здоровьем качество жизни по 5 параметрам: подвижность, самообслуживание, повседневная деятельность, боль/дискомфорт и тревога/депрессия. Каждый параметр оценивается по 3-балльной шкале: «отсутствие трудностей» (1), «некоторые трудности» (2); «выраженные трудности» (3). Общее состояние здоровья выражается в виде 5-значного числа. Уровень здоровья, оцененный по 5 параметрам, можно трансформировать в соответствующий индекс для оценки состояния здоровья, где «0» означает «смерть» и «1» означает «отличное здоровье».
Опросник HADS. Опросник HADS представляет собой общую шкалу лайкертовского типа, используемую для выявления тревоги и депрессии (Bjelland 2002). Опросник состоит из 14 вопросов, из которых 7 вопросов предназначены для оценки тревоги, и 7 предназначены для оценки депрессии. Каждый вопрос оценивается по балльной шкале; пациент может набрать от 0 до 21 балла для тревоги или депрессии.
Общая оценка пациентом заболевания и эффекта лечения. Пациенты оценивают свое общее состояние здоровья по 5-балльной шкале лайкертовского типа от «плохо» до «отлично». Пациентам задают следующий вопрос: «Учитывая все, чем вас беспокоит экзема, оцените состояние своего здоровья». Имеются следующие варианты ответов: «Плохое», «Удовлетворительное», «Хорошее», «Очень хорошее», «Отличное».
Для оценки эффективности лечения пациенты оценивают свою удовлетворенность исследовательским лечением по 5-балльной шкале лайкертовского типа от «плохо» до «отлично». Пациентам задают следующий вопрос: «Как вы оцениваете эффективность исследуемого препарата в лечении экземы?» Имеются следующие варианты ответов: «Плохая», «Удовлетворительная», «Хорошая», «Очень хорошая», «Отличная».
Способы лечения хронического атопического дерматита
Настоящее изобретение включает способы длительного лечения умеренно тяжелого или тяжелого АД у пациента. Согласно определенным вариантам осуществления настоящего изобретения, эти способы включают применение антагониста IL-4R совместно с традиционным терапевтическим средством, например топическим кортикостероидом (ТКС). Согласно другим вариантам осуществления настоящего изобретения, антагонист IL-4R может быть антителом к IL-4R, как указано в этом документе.
Термин «традиционное терапевтическое средство», используемый в этом документе, относится к терапевтическим средствам и препаратам, часто или обычно применяемым для лечения у пациентов АД. К традиционным терапевтическим средствам относятся системные и топические средства. Например, наиболее часто применяемыми или назначаемыми препаратами являются топические кортикостероиды (ТКС). Другими примерами таких средств являются, помимо прочего, топические ингибиторы кальциневрина, антигистаминные препараты, пероральные иммуносупрессанты и глюкокортикоиды, системные иммуносупрессанты, такие как метотрексат, циклоспорин и азатиоприн. Традиционные терапевтические средства применяют для облегчения симптомов АД, однако они обладают многочисленными и существенными нежелательными побочными эффектами, в том числе способностью вызывать сахарный диабет, артериальную гипертензию, остеопороз, миелосупрессию, нефротоксичность, гепатотоксичность, лейкопению и способностью увеличивать риск микробных инфекций. Топические средства, например кортикостероиды и ингибиторы кальциневрина, не рекомендуются для длительного применения в связи с риском необратимой атрофии кожи, депигментации, угреподобной сыпи и рисков, связанных с системной абсорбцией, включая злокачественные новообразования кожи и лимфомы. Кроме того, повторное применение топических средств в течение длительного времени может привести к несоблюдению пациентом режима лечения.
Термин «длительное лечение АД», используемый в этом документе, относится к лечению или сдерживанию прогрессирования одного или более симптомов или болезненных состояний АД в течение длительного времени, обычно в течение приблизительно более 2 лет, более приблизительно 5 лет, более приблизительно 10 лет или более приблизительно 20 лет. Длительное лечение АД включает способы улучшения одного или более связанных с АД параметров в течение периода времени длительностью более 6 месяцев, более 1 года, более 2 лет или более приблизительно 5 лет, и эти способы включают применение антитела к IL-4R в комбинации с традиционным терапевтическим средством, например ТКС. Режим применения и доза антитела к IL-4R и ТКС корректируют или меняют таким образом, чтобы происходило существенное улучшение одного или более связанных с АД параметров, а также устранялось или минимизировалось токсическое действие традиционного средства. Согласно некоторым вариантам осуществления настоящего изобретения, антитело к IL-4R можно вводить в более высоких нагрузочных дозах с целью существенного улучшения связанного с АД параметра с последующим введением обычных доз для сохранения или поддержания улучшения. Совместно применяемый ТКС можно назначать в уменьшенной дозе, составляющей, обычно, приблизительно 20%, приблизительно 30%, приблизительно 40%, приблизительно 50% или приблизительно 60% от дозы для не получающего антитело к IL-4R пациента. Режимы применения и дозы описаны в другом месте этого документа. Согласно некоторым вариантам осуществления настоящего изобретения, настоящее изобретение включает способы уменьшения зависимости от ТКС пациента с умеренно тяжелым или тяжелым АД.
Согласно определенным вариантам осуществления настоящего изобретения, настоящее изобретение включает способы лечения пациентов, страдающих АД более 1 года, более приблизительно 5 лет, более приблизительно 10 лет или более приблизительно 15 лет, и эти способы включают применение антагониста IL-4R в терапевтически эффективном количестве в комбинации с традиционным терапевтическим средством, например ТКС.
Согласно другому аспекту настоящего изобретения, настоящее изобретение включает способы более безопасной и (или) более эффективной длительной терапии умеренно тяжелого или тяжелого АД. Термин «более безопасное и (или) более эффективное лечение», используемый в этом документе, относится к способам лечения, включающим применение антагониста IL-4R в комбинации с традиционным терапевтическим средством, например ТКС, с целью существенного улучшения одного или более связанных с АД параметров, а также устранения или минимизации побочных эффектов и токсичности традиционного средства. Согласно определенным вариантам осуществления настоящего изобретения, улучшение связанного с АД параметра выбирается из группы, включающей: (а) уменьшение от исходного значения балла по шкале оценке исследователя (IGA) по меньшей мере на 50%; (б) уменьшение от исходного значения балла по числовой шкале оценки интенсивности кожного зуда (шкала NRS) по меньшей мере на 65%; (в) уменьшение от исходного значения индекса площади экзематозного поражения кожи и тяжести экземы (EASI) по меньшей мере на 70%; и (г) уменьшение от исходного значения индекса SCORAD по меньшей мере на 60%. Согласно некоторым вариантам осуществления настоящего изобретения, доза традиционного средства снижается или уменьшается с целью минимизации нежелательных побочных эффектов. Согласно некоторым вариантам осуществления настоящего изобретения, способы лечения, описанные в этом документе, могут уменьшать или устранять риск рецидива заболевания после уменьшения дозы или отмены стероида.
Настоящее изобретение включает способы более эффективной и безопасной терапии при длительном лечении АД у пациентов, включая детей или молодежь, которые могут быть более восприимчивы или чувствительны к традиционным терапевтическим средствам.
Согласно другому аспекту настоящего изобретения, представлены способы уменьшения или устранения зависимости пациента с АД от традиционных терапевтических средств, например ТКС, во время лечения умеренно тяжелого или тяжелого АД. Согласно вариантам осуществления настоящего изобретения, эти способы включают: отбор пациентов с умеренно тяжелым или тяжелым АД, неконтролируемым или частично контролируемым с помощью базовой терапии; введение пациенту установленной дозы антагониста IL-4R, предпочтительно антитела к IL-4R, в начальном периоде лечения с продолжением базовой терапии; и постепенное уменьшение дозы одного или более компонентов базовой терапии в течение последующего периода лечения с продолжением терапии антагонистом IL-4R. Термин «базовая терапия», используемый в этом документе, относится к стандартным или традиционным терапевтическим средствам, известным в данной области техники и применяемым для лечения АД (описываются в другом месте этого документа). Согласно определенным вариантам осуществления настоящего изобретения, базовая терапия включает ТКС или топический ингибиторы кальциневрина. Согласно одному варианту осуществления настоящего изобретения, базовая терапия представляет собой терапию сильным ТКС группы III, например мометазона фуроатом, или метилпреднизолона ацепонатом. Согласно некоторым вариантам осуществления настоящего изобретения, традиционные терапевтические средства, например ТКС, полностью отменяются по прошествии начального периода лечения. Например, ТКС назначается в течение начального периода лечения и затем полностью отменяется в течение последующего периода лечения. Согласно определенным вариантам осуществления настоящего изобретения, доза ТКС уменьшается приблизительно на 10%, приблизительно на 20%, приблизительно на 30%, приблизительно на 40%, приблизительно на 50% или более от дозы, применявшейся в начальный период лечения.
Согласно одному примеру режима лечения для пациента с умеренно тяжелым или тяжелым АД, где пациенту с умеренно тяжелым или тяжелым АД вводят антагонист IL-4R, пациенту во время начального периода лечения (также называемого «стабильной фазой лечения») назначают в качестве базовой терапии традиционное терапевтическое средство, например ТКС. Во время последующего периода лечения (также называемого «фазой отмены лечения») доза ТКС постепенно уменьшается приблизительно на 5-60% от дозы начального периода лечения. Согласно одному варианту осуществления настоящего изобретения, лечение ТКС прекращают, т.е. дозу ТКС постепенно уменьшают в течение последующего периода лечения до его полной отмены.
Согласно родственному аспекту настоящего изобретения, представлены способы лечения АД, включающие применение определенной терапии в дополнение к базовой терапии с систематической отменой базовой терапии. Согласно определенным вариантам осуществления настоящего изобретения, антагонист IL-4R вводят в качестве дополнительной терапии пациенту с АД, получающему базовую терапию в течение определенного периода времени (например, в течение 1 недели, 2 недель, 3 недель, 1 месяца, 2 месяцев, 5 месяцев, 12 месяцев, 18 месяцев, 24 месяцев или более) (этот период времени также называется «стабильной фазой лечения»). Согласно некоторым вариантам осуществления настоящего изобретения, базовая терапия включат ТКС. За стабильной фазой лечения следует фаза отмены базовой терапии, где один или более компонентов базовой терапии отменяются, либо где уменьшается их доза, а дополнительная терапия продолжается. Согласно некоторым вариантам осуществления настоящего изобретения, доза препарата базовой терапии может быть уменьшена приблизительно на 5%, приблизительно на 10%, приблизительно на 20%, приблизительно на 30%, приблизительно на 40%, приблизительно на 50% или более в течение фазы отмены лечения. Фаза отмены лечения может длиться 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 7 недель, 8 недель, 9 недель, 10 недель, 11 недель, 12 недель или более.
Связанные с атопическим дерматитом биомаркеры
Настоящее изобретение также включает способы использования, количественного определения и анализа связанных с АД биомаркеров. В контексте этого документа термин «связанный с АД биомаркер» означает любого рода биологический ответ, тип клетки, параметр, белок, полипептид, фермент, ферментную активность, метаболит, нуклеиновую кислоту, углевод или другую биологическую молекулу, которые присутствуют или определяются у пациента с АД на уровне или в количестве, которое отличается (например, больше или меньше) от уровня или количества маркера, присутствующего или определяемого у пациента без АД. Согласно некоторым вариантам осуществления настоящего изобретения, термин «связанный с АД биомаркер» включает биомаркер, связанный с воспалением, которое опосредовано Т-хелперами типа 2 (Th2). К связанным с АД биомаркерам, которые можно определять и (или) измерять контексте настоящего изобретения, относятся, помимо прочего, регулируемый тимусом и активацией хемокин (TARC, также известный как CCL17), иммуноглобулин E (IgE), эотаксин-3 (также известный как CCL26), лактатдегидрогеназа (ЛДГ), эозинофилы, антиген-специфический IgE (например, тест Phadiatop™) и периостин. Термин «связанный с АД биомаркер» также включает ген или, генетический зонд, известный в данной области техники, который дифференциально экспрессируется у пациента с АД, в сравнении с пациентом без АД. Например, гены со значительно усиленной экспрессией у пациента с АД включают, помимо прочего, связанные с Т-хелперами 2-го типа (Th2) хемокины, например CCL13, CCL17, CCL18 и CCL26, маркеры эпидермальной пролиферации, например K16, Ki67, а также антигены T-клеток и дендритных клеток CD2, CD1b и CD1c (Tintle с соавт. 2011; J. Allergy Clin. Immunol. 128: 583-593). В качестве альтернативы, к «связанному с АД биомаркеру» также относятся гены с пониженной экспрессией вследствие АД, например белки терминальной дифференцировки (например, лорикрин, филаггрин и инволюкрин) (Tintle с соавт. 2011; J. Allergy Clin. Immunol. 128: 583-593). Определенные варианты осуществления настоящего изобретения касаются применения этих биомаркеров для мониторинга обратного развития заболевания при введении антагониста IL-4R. Способы обнаружения и (или) количественного определения таких связанных с АД биомаркеров известны в данной области техники; наборы для определения уровня таких связанных с АД биомаркеров доступны из разных коммерческих источников; и разные коммерческие диагностические лаборатории также предлагают услуги по определению этих биомаркеров.
Согласно определенным аспектам настоящего изобретения, представлены способы лечения АД, включающие: (а) отбор пациента с повышенным уровнем по меньшей мере одного связанного с АД биомаркера до или во время лечения, что означает наличие заболевания; и (б) введение пациенту фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве. Согласно определенным вариантам осуществления настоящего изобретения, отбирают тех пациентов, у которых повышен уровень связанного с АД биомаркера. Уровень связанного с АД биомаркера определяют или оценивают количественно путем взятия биологического образца у пациента для количественного определения биомаркера, известного в данной области техники. Согласно определенным другим вариантам осуществления настоящего изобретения, пациента отбирают путем получения информации о повышенном уровне связанного с АД биомаркера. Согласно определенным вариантам осуществления настоящего изобретения, пациента отбирают на основании повышенного уровня IgE или TARC либо периостина.
Для целей настоящего изобретения нормальным уровнем IgE у здорового пациента является уровень менее приблизительно 114 тыс. ед./л (например, при определении с помощью теста ImmunoCAP® [Phadia, Inc. Portage, MI]). Таким образом, настоящее изобретение включает способы, предполагающие отбор пациента с уровнем сывороточного IgE более приблизительно 114 тыс. ед./л, более приблизительно 150 тыс. ед./л, более приблизительно 500 тыс. ед./л, более приблизительно 1000 тыс. ед./л, более приблизительно 1500 тыс. ед./л, более приблизительно 2000 тыс. ед./л, более приблизительно 2500 тыс. ед./л, более приблизительно 3000 тыс. ед./л, более приблизительно 3500 тыс. ед./л, более приблизительно 4000 тыс. ед./л, более приблизительно 4500 тыс. ед./л или более приблизительно 5000 тыс. ед./л и введение пациенту фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве.
Уровни TARC у здорового пациента находятся в пределах от 106 нг/л до 431 нг/л, и средний уровень составляет приблизительно 239 нг/л. (Примером системы для измерения уровня TARC является комплект для количественного определения TARC методом иммуноферментного анализа, предлагаемый под каталожным номером DDN00 компанией R&D Systems, Minneapolis, MN.) Таким образом, настоящее изобретение включает способы, предполагающие отбор пациента с уровнем сывороточного TARC более приблизительно 431 нг/л, более приблизительно 500 нг/л, более приблизительно 1000 нг/л, более приблизительно 1500 нг/л, более приблизительно 2000 нг/л, более приблизительно 2500 нг/л, более приблизительно 3000 нг/л, более приблизительно 3500 нг/л, более приблизительно 4000 нг/л, более приблизительно 4500 нг/л или более приблизительно 5000 нг/л и введение пациенту фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве.
Другим связанным с АД биомаркером является антиген-специфический IgE. Тест Phadiatop™ представляет собой коммерчески доступный вариант теста для определения сывороточного специфического или антиген-специфического IgE, который используется для скринингового обследования на предмет аллергической сенсибилизации (Merrett с соавт. 1987, Allergy 17: 409-416). Данный тест позволяет одновременно определять сывороточные специфические IgE к ряду релевантных аллергенов, вызывающих распространенные ингаляционные аллергии. Данный тест дает качественный результат, положительный или отрицательный, в зависимости от степени флуоресценции. Если исследуемый биологический образец пациента дает флуоресценцию на уровне, который превышает или соответствует уровню флуоресценции контрольного образца, результат теста считается положительным. Если исследуемый биологический образец пациента дает флуоресценцию на уровне ниже уровня флуоресценции контрольного образца, результат теста считается отрицательным. Настоящее изобретение включает способы, предполагающие отбор пациента с положительным результатом теста и введение ему антагониста IL-4R в терапевтически эффективном количестве.
Периостин является белком внеклеточного матрикса, участвующим в воспалительном процессе, опосредованном Th2. Установлено, что у пациентов с АД уровни периостина повышены (Masuoka в соавт. 2012 J Clin Invest. 122(7):2590-2600. doi:10.1172/JCI58978). Настоящее изобретение включает способы, предполагающие введение антагониста IL-4R пациентам с целью снижения повышенного уровня периостина.
Лактатдегидрогеназа (ЛДГ) используется в качестве маркера повреждения тканей, и его уровень повышен у пациентов с АД (Kou с соавт. 2012; Arch. Dermatol. Res. 304: 305-312). Настоящее изобретение включает способы, предполагающие введение антагониста IL-4R пациентам с целью снижения повышенного уровня ЛДГ.
Согласно другим аспектам настоящего изобретения, предложены способы лечения АД, заключающиеся в введении пациенту фармацевтической композиции, содержащей антагонист IL-4R в терапевтически эффективном количестве, в которых введение фармацевтической композиции пациенту приводит к уменьшению уровня по меньшей мере одного связанного с АД биомаркера (например, IgE, TARC, эозинофилов, эотаксина-3, антиген-специфического IgE, ЛДГ и др.) во время введения фармацевтической композиции в сравнении с уровнем этого биомаркера у пациента до введения фармацевтической композиции.
Как будет очевидно среднему специалисту в данной области техники, повышение уровня связанного с АД биомаркера можно определить путем сравнения (i) уровня биомаркера, определенного у пациента в определенный момент времени после введения фармацевтической композиции, содержащей антагонист IL-4R, с (ii) уровнем биомаркера, определенного у пациента до введения фармацевтической композиции, содержащей антагонист IL-4R (т.е. «измерение на исходном уровне»). Определенными моментами времени, в которые определяют уровень биомаркера, могут быть, моменты времени приблизительно 4 часа, 8 часов, 12 часов, 1 день, 2 дня, 3 дня, 4 дня, 5 дней, 6 дней, 7 дней, 8 дней, 9 дней, 10 дней, 15 дней, 20 дней, 35 дней, 40 дней, 50 дней, 55 дней, 60 дней, 65 дней, 70 дней, 75 дней, 80 дней, 85 дней или более после введения фармацевтической композиции, содержащей антагонист IL-4R.
Согласно определенным конкретным вариантам осуществления настоящего изобретения, у пациента может наблюдаться повышенный уровень TARC и (или) IgE после введения фармацевтической композиции, содержащей антагонист IL-4R (например, антитела к IL-4R). Например, приблизительно на день 4, день 8, день 15, день 22, день 25, день 29, день 36, день 43, день 50, день 57, день 64, день 71 или день 85 после введения первой, второй или третьей дозы фармацевтической композиции, содержащей приблизительно 75, 150 или 300 мг антитела к IL-4R (например, mAb1), у пациента, согласно настоящему изобретению, может наблюдаться снижение уровня TARC приблизительно на 1%, 2%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% или более от исходного значения (где под «исходным значением» понимается уровень TARC у пациента до первого введения фармацевтической композиции). Аналогично, приблизительно на день 4, день 8, день 15, день 22, день 25, день 29, день 36, день 43, день 50, день 57, день 64, день 71 или день 85 после введения первой, второй или третьей дозы фармацевтической композиции, содержащей приблизительно 75, 150 или 300 мг антитела к IL-4R (например, mAb1), у пациента, согласно настоящему изобретению, может наблюдаться снижение уровня IgE приблизительно на 1%, 2%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% или более от исходного значения (где под «исходным значением» понимается уровень IgE у пациента до первого введения фармацевтической композиции).
Настоящее изобретение также включает способы выявления подходящего пациента, которому введение фармацевтической композиции, содержащей антагонист IL-4R, принесет пользу. Например, если у пациента до получения фармацевтической композиции, содержащей антагонист IL-4R, наблюдается уровень связанного с АД биомаркера, который указывает на наличие заболевания, считают, что этому пациенту введение фармацевтической композиции настоящего изобретения (композиция, содержащая антитело к IL-4R) принесет пользу. Согласно родственным вариантам осуществления настоящего изобретения, настоящее изобретение включает способы лечения подходящих пациентов, где у подходящего пациента может наблюдаться предрасположенность к более тяжелому течению АД, например в связи с расовой или этнической принадлежностью. Например, настоящее изобретение включает способы, предполагающие введение антагониста IL-4R пациентам афро-американского происхождения, у которых может наблюдаться предрасположенность к более тяжелому течению АД. У таких пациентов может наблюдаться повышенный уровень связанного с АД биомаркера.
Согласно определенным примерам вариантов осуществления настоящего изобретения, пациент может считаться подходящим кандидатом для терапии антителом к IL-4R, если у этого пациента наблюдается одно или более из следующего: (i) уровень IgE выше приблизительно 114 тыс. ед./л, выше приблизительно 150 тыс. ед./л, выше приблизительно 500 тыс. ед./л, выше приблизительно 1000 тыс. ед./л, выше приблизительно 1500 тыс. ед./л, выше приблизительно 2000 тыс. ед./л, выше приблизительно 2500 тыс. ед./л, выше приблизительно 3000 тыс. ед./л, выше приблизительно 3500 тыс. ед./л, выше приблизительно 4000 тыс. ед./л, выше приблизительно 4500 тыс. ед./л или выше приблизительно 5000 тыс. ед./л; или (ii) уровень TARC выше приблизительно 431 нг/л, выше приблизительно 500 нг/л, выше приблизительно 1000 нг/л, выше приблизительно 1500 нг/л, выше приблизительно 2000 нг/л, выше приблизительно 2500 нг/л, выше приблизительно 3000 нг/л, выше приблизительно 3500 нг/л, выше приблизительно 4000 нг/л, выше приблизительно 4500 нг/л, выше приблизительно 5000 нг/л; или (iii) положительный результат теста Phadiatop™. Дополнительные критерии, например другие клинические показатели (например, высокий балл по шкале IGA, увеличенный показатель BSA, увеличенный индекс EASI, увеличенный индекс SCORAD, высокий балл по опроснику из 5 вопросов для оценки кожного зуда и (или) высокий балл по шкале NRS, указывающий на АД), может применяться в комбинации с любым из вышеупомянутых биомаркеров для выявления пациентов, являющихся подходящими кандидатами для терапии антагонистом IL-4R, как указано в другом месте этого документа.
Антагонисты рецепторов IL-4
Как подробно описано выше, настоящее изобретение включает способы, заключающиеся в введении нуждающемуся в этом пациенту терапевтической композиции, содержащей антагонист рецептора интерлейкина-4 (IL-4R). В контексте этого документа, «антагонистом IL-4R» является любое вещество, которое связывается или взаимодействует с IL-4R и ингибирует нормальную биологическую сигнальную функцию IL-4R, где IL-4R экспрессируется клеткой in vitro или in vivo. Примерами групп антагонистов IL-4R являются, помимо прочего, низкомолекулярные антагонисты IL-4R, аптамеры к IL-4R, пептидные антагонисты IL-4R (например, молекулы пептид-ассоциированных антител) и антитела или антигенсвязывающие фрагменты антител, которые специфически связываются с человеческим IL-4R.
Термины «IL-4R», «hIL-4R» и другие, используемые в этом документе, относятся к альфа-цепи цитокинового рецептора человека, которая специфически связывается c интерлейкином-4 (IL-4), IL-4Rα (SEQ ID NO: 274). Если специально не указано то, что рецептор имеет отношение не к человеку, а к другим видам, термин «IL-4R», используемый в этом документе, означает альфа-цепь рецептора интерлейкина-4 человека.
Термин «антитело», используемый в этом документе, относится к молекулам иммуноглобулинов, состоящим из четырех полипептидных цепей: двух тяжелых (H) цепей и двух легких (L) цепей, соединенных дисульфидными связями, а также их мультимеры (например, IgM). Каждая тяжелая цепь состоят из вариабельной области (в этом документе используется сокращение HCVR или VH) и константной области. Константная область тяжелой цепи включает три домена: CH1, CH2 и CH3. Каждая легкая цепь состоит из вариабельной области (в этом документе используется сокращение LCVR или VL) и константной области. Константная область легкой цепи включает один домен (CL1). Области VH и VL в свою очередь включают гипервариабельные участки, называемые определяющими комплементарность участками (CDR), которые перемежаются с боле консервативными участками, называемыми каркасными (FR). Каждая область VH и VL включает три участка CDR и четыре участка FR, которые располагаются от N-конца до карбоксильного конца белковой цепи в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Согласно различным вариантам осуществления настоящего изобретения, участки FR антитела к IL-4R (или его антигенсвязывающего фрагмента) могут иметь последовательность, идентичную последовательностям зародышевой линии человека или могут иметь естественные или искусственные модификации. Аминокислотную консенсусную последовательность можно определить путем сопоставления двух или более участков CDR, расположенных «бок-о-бок».
Термин «антитело», используемый в этом документе, также включает антигенсвязывающие фрагменты или полные молекулы антител. Термины «антигенсвязывающая часть» антитела, «антигенсвязывающий фрагмент» антитела и им подобные, используемые в этом документе, включают любые естественные, получаемые ферментативным путем, синтетические или получаемые методом генной инженерии полипептиды или гликопротеины, которые специфически связываются с антигеном с образованием комплекса. Антигенсвязывающие фрагменты антитела можно получить, например, из полных молекул антител путем использования любых подходящих стандартных методик, например протеолитического расщепления или генной инженерии с применением рекомбинантных технологий с изменением и экспрессией ДНК, кодирующей вариабельные и, в некоторых случаях, константные домены антитела. Такая ДНК известна и (или) легко доступная из, например, коммерческих источников, библиотек ДНК (включая, например, библиотеки фаговых антител), либо ее можно синтезировать. ДНК можно секвенировать или изменять химически либо с помощью методик молекулярной биологии, например для придания одному или более вариабельным и (или) константным доменам подходящей конфигурации, или для введения кодонов, создания цистеиновых остатков, модификации, добавления или удаления аминокислот и т.д.
Примерами антигенсвязывающих фрагментов, помимо прочего, являются: (i) фрагменты Fab; (ii) фрагменты F(ab')2; (iii) фрагменты Fd; (iv) фрагменты Fv; (v) одноцепочечные молекулы Fv (scFv); (vi) фрагменты dAb; и (vii) единицы минимального распознавания аминокислотных остатков, имитирующие гипервариабельные участки антитела (например, отдельный определяющий комплементарность участок (CDR), такой как пептид CDR3), или пептиды c ограниченной конформационной свободой FR3-CDR3-FR4. Другие генно-инженерные молекулы, например домен-специфические антитела, антитела с одним доменом, антитела с удаленным доменом, химерные антитела, CDR-привитые антитела, диатела, триатела, тетратела, минитела, нанотела (например, моновалентные нанотела, бивалентные нанотела и др.), иммунофармацевтические вещества на основе модульного низкомолекулярного белка (SMIP) и вариабельные акульи домены IgNAR, также входят в понятие «антигенсвязывающий фрагмент», как указано в этом документе.
Антигенсвязывающий фрагмент антитела обычно включает по меньшей мере один вариабельный домен. Вариабельный домен может иметь любой размер или аминокислотную последовательность и обычно включает по меньшей мере один участок CDR, прилегающий к одной или более последовательностям каркасного участка или находящийся внутри каркаса. В антигенсвязывающих фрагментах с доменом VH, связанным с доменом VL домены VH и VL могут располагаться относительно друг друга любым соответствующим образом. Например, вариабельная область может быть димерной и содержать димеры VH-VH, VH-VL или VL-VL. В качестве альтернативы, антигенсвязывающий фрагмент антитела может содержать мономерный домен VH или VL.
Согласно определенным вариантам осуществления настоящего изобретения, антигенсвязывающий фрагмент антитела может содержать по меньшей мере один вариабельный домен, ковалентно связанный по меньшей мере с одним константным доменом. Примерами конфигураций вариабельных и константных доменов, которые можно обнаружить в антигенсвязывающем фрагменте антитела в рамках настоящего изобретения, помимо прочего, являются: (i) VH-CH1; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (v) VH-CH1-CH2-CH3; (vi) VH-CH2-CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (x) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1-CH2-CH3; (xiii) VL-CH2-CH3; и (xiv) VL-CL. При любой конфигурации вариабельных и константных доменов, включая любую из представленных выше конфигураций, вариабельные и константные домены могу либо быть напрямую связаны друг с другом, либо связаны полностью или частично посредством шарнирной области либо посредством линкерной области. Шарнирная область может включать по меньшей мере 2 (например, 5, 10, 15, 20, 40, 60 или более) аминокислоты, что приводит к возникновению гибкой или полугибкой связи между прилежащими вариабельными и (или) константными доменами в одной молекуле полипептида. Кроме того, антигенсвязывающий фрагмент антитела в рамках настоящего изобретения может включать гомодимер или гетеродимер (или другой мультимер) с любой из перечисленных вышей конфигураций вариабельных и константных доменов, связанных друг с другом нековалентной связью, и (или) с одним или более мономерными доменами VH или VL (например, дисульфидной(-ыми) связью(-ями)).
Как и в случае с полными молекулами антител, антигенсвязывающие фрагменты могут быть моноспецифическими или мультиспецифическими (например, биспецифическими). Мультиспецифический антигенсвязывающий фрагмент антитела обычно состоит по меньшей мере из двух разных вариабельных доменов, в которых каждый вариабельный домен способен специфически связываться с отдельным антигеном или с различными эпитопом этого же антигена. Любое мультиспецифическое антитело можно использовать как антигенсвязывающий фрагмент антитела в контексте настоящего изобретения с использованием стандартных методик, доступных в данной области техники.
Константная область антитела играет важную роль в способности антитела фиксировать комплемент и вызывать клеточно-опосредованную цитотоксичность. Таким образом, изотоп антитела можно выбирать с учетом желательности наличия для него способности опосредовать цитотоксичность.
Термин «человеческое антитело», используемый в этом документе, включает антитела с вариабельными и константными областями, полученными из последовательностей зародышевой линии иммуноглобулина человека. Человеческие антитела, являющиеся предметом настоящего изобретения, могут, тем не менее, включать аминокислотные остатки, не кодируемые последовательностями зародышевой линии иммуноглобулина человека (например, мутации, возникшие вследствие случайного сайт-специфического мутагенеза in vitro, или соматические мутации, возникшие in vivo), например в участках CDR и, в частности, в участке CDR3. Однако термин «человеческое антитело», используемый в этом документе, не включает антитела, в которых последовательности CDR, полученные из последовательностей зародышевой линии других видов млекопитающих, например мышей, были внесены в каркасную последовательность у человека.
Термин «рекомбинантное человеческое антитело», используемый в этом документе, включает все человеческие антитела, приготовленные, экспрессированные, созданные или выделенные с помощью рекомбинантных методик, например антитела, экспрессированные с применением вектора рекомбинантной экспрессии, трансфицированного в клетку-хозяина (описано ниже), антитела, выделенные из библиотеки рекомбинантных комбинаторных антител человека (описано ниже), антитела, выделенные от животного (например, мышей), являющегося трансгенным для генов иммуноглобулина человека (см., например, Taylor с соавт. (1992) Nucl. Acids Res. 20:6287-6295) или антитела, приготовленные, экспрессированные, созданные или выделенные любым другим способом, который предполагает сплайсинг гена иммуноглобулина человека до других последовательностей ДНК. Такие рекомбинантные человеческие антитела имеют вариабельные и константные области, полученные из последовательностей иммуноглобулина зародышевой линии человека. Согласно определенным вариантам осуществления настоящего изобретения, однако, такие рекомбинантные человеческие антитела подвержены мутагенезу in vitro (или, в котором используется животное с трансгенностью в отношении последовательностей человеческого Ig, соматическому мутагенезу in vivo), и, таким образом, аминокислотные последовательности областей VH и VL рекомбинантных антител представляют собой последовательности, которые, хотя и получены из родственных для человека последовательностей зародышевой линии VH и VL, могут отсутствовать в нормальных условиях в наборе антител зародышевой линии человека in vivo.
Человеческие антитела могут существовать в двух формах, связанных с гетерогенностью шарнирной области. В одной форме молекула иммуноглобулина содержит стабильную четырехцепочную конструкцию с молекулярной массой около 150-160 кДа, и ее димеры скреплены друг с другом межцепочечной дисульфидной связью тяжелой цепи. В другой форме димеры не соединены межцепочечными дисульфидными связями, и молекула массой приблизительно 75-80 кДа образуется состоящей из легкой и тяжелой цепей, соединенных ковалентной связью (полуантитело). Эти формы крайне тяжело отделить друг от друга даже после аффинной очистки.
Частота появления второй формы среди различных интактных изотипов IgG обусловлена, помимо прочего, структурными различиями, связанными с шарнирной областью изотипа антитела. Замена одной аминокислоты в шарнирной области человеческого IgG4 может существенно повлиять на частоту появление второй формы (Angal с соавт. (1993) Molecular Immunology 30:105) до уровней, обычно наблюдаемых при использовании шарнирной области человеческого IgG1. Настоящее изобретение охватывает антитела с одной или более мутациями в шарнирной области, область CH2 или CH3, что может быть желательно, например, при производстве, для увеличения процентной доли желаемой формы антитела.
Термин «выделенное антитело», используемый в этом документе, означает антитело, которое было идентифицировано и выделено и (или) получено по меньшей мере из одного компонента его естественного окружения. Например, антитело, которое было отделено от или получено из по меньшей мере одного компонента организма или из ткани либо клетки, в которой антитело существует или образуется в естественных условиях, является «выделенным антителом» для целей настоящего изобретения. Выделенным антителом также является антитело in situ в рекомбинантной клетке. Выделенными антителами являются антитела, которые прошли по меньшей мере одну стадию очистки или выделения. Согласно определенным вариантам осуществления настоящего изобретения, выделенное антитело может быть практически свободно от другого клеточного материала и (или) химических веществ.
Термин «специфически связывается» и подобные ему означают, что антитело или его антигенсвязывающий фрагмент образуют комплекс с антигеном, который относительно стабилен в физиологических условиях. Способы определения того, специфично ли связываются антитела с антигеном, хорошо известны в данной области техники и включают, например, равновесный диализ, поверхностный плазмонный резонанс, и тому подобные способы. Например, антитело, которое «специфически связывается» с IL-4R, в контексте настоящего изобретения, включает антитела, которые связываются с IL-4R или его частью с KD менее приблизительно 1000 нМ, менее приблизительно 500 нМ, менее приблизительно 300 нМ, менее приблизительно 200 нМ, менее приблизительно 100 нМ, менее приблизительно 90 нМ, менее приблизительно 80 нМ, менее приблизительно 70 нМ, менее приблизительно 60 нМ, менее приблизительно 50 нМ, менее приблизительно 40 нМ, менее приблизительно 30 нМ, менее приблизительно 20 нМ, менее приблизительно 10 нМ, менее приблизительно 5 нМ, менее приблизительно 4 нМ, менее приблизительно 3 нМ, менее приблизительно 2 нМ, менее приблизительно 1 нМ или менее приблизительно 0,5 нМ, при определении с помощью метода поверхностного плазмонного резонанса. Выделенное антитело, которые специфически связывается с IL-4R, может, однако, иметь перекрестную реактивность к другим антигенам, например молекулам IL-4R других видов (не человека).
Антитела к IL-4R, используемые для способов настоящего изобретения, могут включать одну или более аминокислотных замен, вставок и (или) удалений в каркасном участке и (или) участках CDR вариабельных доменов тяжелой и легкой цепей в сравнении с соответствующими последовательностями зародышевой линии, из которых были получены антитела. Такие мутации можно легко установить путем сравнения аминокислотных последовательностей, указанных в этом документе, с последовательностями зародышевой линии, представленными, например, в общедоступных базах последовательностей для антител. Настоящее изобретение включает способы, предполагающие применение антител и их антигенсвязывающих фрагментов, получаемых с использованием любых аминокислотных последовательностей, рассматриваемых в этом документе, где одна или более аминокислот в пределах одного или более каркасных участков и (или) участков CDR подвергаются мутации до соответствующего(-их) остатка(-ов) последовательности зародышевой линии, из которой было получено антитело, или до соответствующего(-их) остатка(-ов) другой последовательности зародышевой линии человека, или до консервативной замены аминокислоты соответствующего(-их) остатка(-ов) зародышевой линии (такие изменения последовательности называются в этом документе в целом «герминативные мутации»). Средний специалист в данной области техники, начиная с последовательностей вариабельных областей тяжелой и легкой цепей, рассматриваемых в этом документе, может легко получить разнообразные антитела и антигенсвязывающие фрагменты, включающие одну или более отдельных герминативных мутаций или их комбинаций. Согласно определенным вариантам осуществления настоящего изобретения, остатки участков CDR в пределах домена VH и (или) домена VL подвергаются обратной мутации до остатков, обнаруживаемых в исходной последовательности зародышевой линии, из которой были получены антитела. Согласно другим вариантам осуществления настоящего изобретения, только определенные остатки подвергаются обратным мутациям до исходной последовательности зародышевой линии, например только мутантные остатки обнаруживаемые в первых 8 аминокислотах фрагмента FR1 или в последних 8 аминокислотах фрагмента FR4, или только мутантные остатки, обнаруживаемые в участках CDR1, CDR2 или CDR3. Согласно другим вариантам осуществления настоящего изобретения, один или более остатков каркасных участков и (или) участков CDR подвергаются мутации до соответствующих остатков различных последовательностей зародышевой линии (т.е. последовательности зародышевой линии, которая отличается от последовательности зародышевой линии, из которой было изначально получено антитело). Кроме того, антитела настоящего изобретения могут содержать две или более герминативные мутации в каркасных участках и (или) участках CDR, например, где определенные отдельные остатки подвергаются мутации до соответствующего остатка конкретной последовательности зародышевой линии, а определенные другие остатки, которые отличаются от исходной последовательности зародышевой линии, сохраняются или подвергаются мутации до соответствующих остатков другой последовательности зародышевой линии. После получения, антитела и антигенсвязывающие фрагменты, содержащие одну или более герминативных мутаций, можно легко проверить на наличие одного или более желаемых свойств, например улучшенной специфичности связывания, улучшенных или усиленных антагонистических или агонистических биологических свойств (в зависимости от обстоятельств), сниженной иммуногенности и т.д. Применение антител и антигенсвязывающих фрагментов, полученных таким общим образом, охватывается настоящим изобретением.
В настоящее изобретение также входят способы, предполагающие использование антител к IL-4R, включающих варианты любых аминокислотных последовательностей HCVR, LCVR и (или) CDR, указанных в этом документе, имеющих одну или более консервативных замен. Например, настоящее изобретение включает использование антител к IL-4R, имеющих аминокислотные последовательности HCVR, LCVR и (или) CDR с, например, 10 или менее, 8 или менее, 6 или менее, 4 или менее и т.д. консервативными аминокислотными заменами относительно любых аминокислотных последовательностей HCVR, LCVR и (или) CDR, указанных в этом документе.
Термин «поверхностный плазмонный резонанс», используемый в этом документе, относится к оптическому феномену, который позволяет анализировать взаимодействия в реальном времени путем выявления изменений концентрации белка в биосенсорном матриксе, например с помощью системы BIAcore™ (подразделение Biacore Life Sciences компании GE Healthcare, Piscataway, NJ, США).
Термин «KD», используемый в этом документе, означает равновесную константу диссоциации для конкретного взаимодействия антитела и антигена.
Термин «эпитоп» означает антигенную детерминанту, которая взаимодействует со специфическим антигенсвязывающим участком в вариабельной области молекулы антитела, называемым паратопом. У одного антигена может быть несколько эпитопов. Таким образом, различные антитела могут связываться с различными участками антигена и могут обладать различными биологическими эффектами. Эпитопы могут быть конформационными и линейными. Конформационный эпитоп образуется за счет пространственного смежного расположения аминокислот из различных сегментов линейной полипептидной цепи. Линейный эпитоп образуется прилежащими аминокислотными остатками полипептидной цепи. В определенных случаях эпитопы антигена могут включать части сахаридов, фосфорильные группы или сульфонильные группы.
Получение человеческих антител
Способы получения человеческих антител у трансгенных мышей хорошо известны в данной области техники. Все эти способы можно использовать в контексте настоящего изобретения для получения человеческих антител, которые специфически связываются с человеческим IL-4R.
При использовании технологии VELOCIMMUNE™ (см., например, патент US 6,596,541, Regeneron Pharmaceuticals) или любого другого метода для получения моноклональных антител, изначально получают химерные антитела с высокой аффинностью к IL-4R, имеющие человеческую вариабельную область и мышиную константную область. Технология VELOCIMMUNE® предполагает получение трансгенной мыши с геномом, включающим вариабельные области человеческих тяжелой и легкой цепей, оперативно связанные с эндогенными мышиными константными областями, так что у мыши в ответ на антигенную стимуляцию образуются антитела, имеющие человеческую вариабельную область и мышиную константную область. ДНК, кодирующие вариабельные области тяжелой и легкой цепей антитела выделяют и оперативно связывают с ДНК, кодирующими константные области тяжелой и легкой цепей человека. ДНК затем экспрессируется в клетке, способной продуцировать полностью человеческое антитело.
Как правило, мышь с геномом, модифицированным по технологии VELOCIMMUNE®, сенсибилизируют исследуемым антигеном, и у нее забирают лимфоциты (например, В-лимфоциты), продуцирующие антитела. Полученные лимфоциты можно слить с миеломными клетками для создания бессмертных гибридомных клеточных линий, и такие гибридомные клеточные линии подвергаются скринингу и отбору для выявления линий, вырабатывающих антитела, специфичные по отношению к исследуемому антигену. ДНК, кодирующие вариабельные области тяжелой и легкой цепей можно выделить и соединить с ДНК, кодирующими требуемые изотипические константные области тяжелой и легкой цепей. Такой белок антитела может вырабатываться клеткой, например клеткой яичника китайского хомячка. В качестве альтернативы, ДНК, кодирующие антиген-специфические химерные антитела или вариабельные домены легкой и тяжелой цепей можно выделять непосредственно из антигенспецифических лимфоцитов.
Сначала выделяют химерные антитела с высокой аффинностью с человеческой вариабельной областью и мышиной константной областью. Определяют свойства этих антител и отбирают антитела с желаемыми свойствами, включая аффинность, селективность, характеристики эпитопа и т.д. с использованием стандартных методик, известных средним специалистам в данной области техники. Мышиные константные области заменяют желаемой человеческой константной областью для получения полностью человеческого антитела, являющегося предметом настоящего изобретения, например IgG1 или IgG4 дикого типа или модифицированного. Несмотря на то, что выбранная константная область может различаться в соответствии со специфичностью применения антитела, антигенсвязывающая способность с высокой аффинностью и мишень-специфические характеристики сохраняются в вариабельной области.
В целом, антитела, которые можно использовать для способов настоящего изобретения, обладают высокой аффинностью, как указано выше, при оценке их способности связываться с антигеном после иммобилизации на твердой фазе или в жидкой фазе. Мышиные константные области заменяют желаемыми человеческими константными областями для получения полностью человеческого антитела, настоящего изобретения. Несмотря на то, что выбранная константная область может различаться в соответствии со специфичностью применения антитела, антигенсвязывающая способность с высокой аффинностью и мишень-специфические характеристики сохраняются в вариабельной области.
Конкретными примерами человеческих антител или антигенсвязывающих фрагментов, которые специфически связываются с IL-4R и которые можно применять в контексте способов настоящего изобретения, являются любые антитела или антигенсвязывающие фрагменты, включающие три участка CDR тяжелой цепи (HCDR1, HCDR2 и HCDR3), входящие в вариабельную область тяжелой цепи (HCVR) с аминокислотной последовательностью, выбранной из группы, состоящей из последовательностей со следующими идентификаторами SEQ ID NO: 2, 18, 22, 26, 42, 46, 50, 66, 70, 74, 90, 94, 98, 114, 118, 122, 138, 142, 146, 162, 166, 170, 186, 190, 194, 210, 214, 218, 234, 238, 242, 258 и 262. Антитело или антигенсвязывающий фрагмент может включать три участка CDR легкой цепи (LCVR1, LCVR2, LCVR3), входящие в вариабельную область легкой цепи (LCVR) с аминокислотной последовательностью, выбранной из группы, состоящей из последовательностей со следующими идентификаторами SEQ ID NO: 10, 20, 24, 34, 44, 48, 58, 68, 72, 82, 92, 96, 106, 116, 120, 130, 140, 144, 154, 164, 168, 178, 188, 192, 202, 212, 216, 226, 236, 240, 250, 260 и 264. Методы и технология выявления участков CDR в аминокислотных последовательностях HCVR и LCVR хорошо известны в данной области техники и могут применяться для выявления участков CDR в определенных аминокислотных последовательностях HCVR и (или) LCVR, указанных в этом документе. Примерами определений, которые можно использовать для выявления границ CDR, включают определение Kabat, определение Chothia и определение AbM. В общих чертах, определение Kabat основывается на вариабельности последовательности, определение Chothia основывается на расположении структурных петлевых фрагментов, и определение AbM представляет собой компромисс между определением Kabat и определением Chothia. См., например, статью Kabat «Sequences of Proteins of Immunological Interest», National Institutes of Health, Bethesda, Md, США. (1991); Al-Lazikani et al., J. Mol. Biol. 273:927-948 (1997); and Martin et al., Proc. Natl. Acad. Sci. USA 86:9268-9272 (1989). С целью выявления последовательностей CDR антитела можно также использовать общедоступные базы данных.
Согласно определенным вариантам осуществления настоящего изобретения, антитело или его антигенсвязывающий фрагмент включает шесть участков CDR (HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 и LCDR3) из пар аминокислотных последовательностей вариабельных областей тяжелой и легкой цепей (HCVR/LCVR), выбранных из группы, состоящей из последовательностей со следующими идентификаторами SEQ ID NO: 2/10, 18/20, 22/24, 26/34, 42/44, 46/48, 50/58, 66/68, 70/72, 74/82, 90/92, 94/96, 98/106, 114/116, 118/120, 122/130, 138/140, 142/144, 146/154, 162/164, 166/168, 170/178, 186/188, 190/192, 194/202, 210/212, 214/216, 218/226, 234/236, 238/240, 242/250, 258/260 и 262/264.
Согласно определенным вариантам осуществления настоящего изобретения, антитело или его антигенсвязывающий фрагмент включает шесть участков CDR (HCDR1/HCDR2/HCDR3/LCDR1/LCDR2/LCDR3) с аминокислотными последовательностями, выбранными из группы, состоящей из последовательностей со следующими идентификаторами SEQ ID NO: 4/6/8/12/14/16; 28/30/32/36/38/40; 52/54/56/60/62/64; 76/78/80/84/86/88; 100/102/104/108/110/112; 124/126/128/132/134/136; 148/150/152/156/158/160; 172/174/176/180/182/184; 196/198/200/204/206/208; 220/222/224/228/230/232; и 244/246/248/252/254/256.
Согласно определенным вариантам осуществления настоящего изобретения, антитело или его антигенсвязывающий фрагмент включает пары аминокислотных последовательностей HCVR/LCVR, выбранные из группы, состоящей из последовательностей со следующими идентификаторами SEQ ID NO: 2/10, 18/20, 22/24, 26/34, 42/44, 46/48, 50/58, 66/68, 70/72, 74/82, 90/92, 94/96, 98/106, 114/116, 118/120, 122/130, 138/140, 142/144, 146/154, 162/164, 166/168, 170/178, 186/188, 190/192, 194/202, 210/212, 214/216, 218/226, 234/236, 238/240, 242/250, 258/260 и 262/264.
Фармацевтические композиции
Настоящее изобретение включает способы, заключающиеся в введении пациенту антагониста IL-4R, где антагонист IL-4R входит в состав фармацевтической композиции. В состав фармацевтических композиций настоящего изобретения входят подходящие носители, вспомогательные вещества и другие вещества, обеспечивающие надлежащие перенос, доставку и переносимость композиции, а также прочие ее свойства. Множество подходящих лекарственных форм можно найти в справочниках, известных всем химикам-фармацевтам: Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, США. К этим лекарственным формам относятся, например: порошки, пасты, мази, желе, парафины, масла, липиды, липидосодержащие везикулы (катионные или анионные) (например, LIPOFECTIN™), конъюгаты ДНК, безводные абсорбционные пасты, эмульсия масла в воде и воды в масле, эмульсия карбовакса (полиэтиленгликоль различной молекулярной массы), полутвердые гели и полутвердые смеси, содержащие карбовакс. См. также Powell с соавт. «Compendium of excipients for parenteral formulations» PDA (1998) J Pharm Sci Technol 52:238-311.
Доза антитела, вводимая пациенту согласно способам настоящего изобретения, может различаться в зависимости от возраста и массы пациента, симптомов, состояний, способа введения и тому подобного. Дозу обычно рассчитывают согласно массе тела или площади поверхности тела. В зависимости от тяжести состояния, частоту и длительность введения можно корректировать. Эффективные дозы и режимы введения фармацевтических композиций, содержащих антитела к IL-4R, можно определять эмпирически; например улучшение состояния пациента можно определять путем его периодического обследования и корректировать дозу препарата согласно результатам этого обследования. Кроме того, увеличение дозы для различных видов можно выполнять с использованием способов, хорошо известных в данной области техники (например, Mordenti с соавт., 1991, Pharmaceut. Res. 8:1351). Конкретные примеры доз антител к IL-4R и режимов введения фармацевтической композиции, включающие дозы и режимы введения, которые могут использоваться в контексте настоящего изобретения, указаны в другом месте этого документа.
Известны и могут использоваться различные системы для введения фармацевтической композиции, являющейся предметом настоящего изобретения, например инкапсуляция в липосомах, микрочастицах, микрокапсулах, рекомбинантных клетках, способных экспрессировать мутантные вирусы, опосредованный рецепторами эндоцитоз (см., например, Wu с соавт., 1987, J. Biol. Chem. 262:4429-4432). Способы введения включают, помимо прочих, внутрикожное, внутримышечное, внутрибрюшное, внутривенное, подкожное, интраназальное, эпидуральное и пероральное введение. Настоящая композиция может быть доставлена в организм любым удобным способом, например путем инфузии или болюсной инъекции, либо за счет всасывания через эпителиальные и слизисто-кожные выстилки (например, слизистая оболочка полости рта, прямой кишки и кишечника и т.д.); также ее можно вводить с другими биологически активными средствами.
Фармацевтическую композицию, являющуюся предметом настоящего изобретения, можно вводить подкожно или внутривенно с помощью стандартных иглы и шприца. Кроме того, что касается подкожного введения, то фармацевтическую композицию, являющуюся предметом настоящего изобретения, можно вводить с помощью шприц-ручки. Такая шприц-ручка может быть предназначена для многократного или однократного пользования. Шприц-ручка многократного пользования обычно имеет заменяемый картридж, содержащий фармацевтическую композицию. После введения фармацевтической композиции, содержащейся в картридже, пустой картридж можно выбросить и заменить на новый, содержащий фармацевтическую композицию. Шприц-ручку затем можно использовать заново. Шприц-ручка однократного пользования не имеет заменяемого картриджа. Шприц-ручка однократного пользования имеет емкость, заполненную фармацевтической композицией. Шприц-ручку после введения фармацевтической композиции выбрасывают.
Для подкожного введения фармацевтической композиции, являющейся предметом настоящего изобретения, можно использовать различные шприц-ручки и им подобные устройства многократного пользования. Примерами многочисленных видов, помимо прочего, являются шприц-ручка AUTOPEN™ (Owen Mumford, Inc., г. Вудсток, Великобритания), шприц-ручка DISETRONIC™ (Disetronic Medical Systems, г. Бергдорф, Швейцария), шприц-ручка HUMALOG MIX 75/25™, шприц-ручка HUMALOG™, шприц-ручка HUMALIN 70/30™ (Eli Lilly and Co., г. Индианаполис, штат Индианаполис, США), NOVOPEN™ I, II и III (Novo Nordisk, г. Копенгаген, Дания), NOVOPEN JUNIOR™ (Novo Nordisk, г. Копенгаген, Дания), шприц-ручка BD™ (Becton Dickinson, г. Франклин Лейкс, штат Ною-Джерси, США), OPTIPEN™, OPTIPEN PRO™, OPTIPEN STARLET™ и OPTICLIK™ (sanofi-aventis, г. Франкфурт, Германия). Примерами многочисленных видов, шприц-ручек однократного пользования, которые могут быть использованы для подкожного введения фармацевтической композиции, являющейся предметом настоящего изобретения, являются, помимо прочего, шприцы-ручки SOLOSTAR™ (sanofi-aventis), FLEXPEN™ (Novo Nordisk), KWIKPEN™ (Eli Lilly), SURECLICK™ (Amgen, г. Саузенд Оукс, штат Калифорния, США), PENLET™ (Haselmeier, г. Штутгарт, Германия), EPIPEN (Dey, L.P.) и HUMIRA™ Pen (Abbott Labs, Abbott Park IL).
В определенных случаях фармацевтическую композицию можно вводить с помощью системы с контролируемым высвобождением лекарственного вещества. Согласно одному варианту осуществления настоящего изобретения, можно использовать помпу (см. Langer, supra; Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201). Согласно другому варианту осуществления настоящего изобретения, можно использовать полимерные материалы; см. Medical Applications of Controlled Release, Langer and Wise (eds.), 1974, CRC Pres., Boca Raton, Florida, США. Согласно другому варианту осуществления настоящего изобретения, систему с контролируемой доставкой лекарственного вещества можно размещать вблизи целевого органа, что требует введения лишь части системной дозы (см., например, Goodson, 1984, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138). Другие системы с контролируемой доставкой лекарственного вещества рассматриваются в обзорной статье Langer, 1990, Science 249:1527-1533.
К инъекционным препаратам могут относиться лекарственные формы для внутривенного, подкожного, внутрикожного и внутримышечного инъекционного введения, формы для внутривенного капельного введения и др. Эти инъекционные препараты можно готовить с помощью известных способов. Например, инъекционные препараты можно готовить путем растворения, суспендирования или эмульсифицирования антитела или его соли, описанной выше, в стерильной водной среде или масляной среде, обычно используемой для приготовления инъекционных препаратов. В качестве водных сред для приготовления инъекционных препаратов используются изотонический раствора хлорида натрия, изотонический раствор глюкозы и других вспомогательных веществ, например веществ, которые могут быть использованы в комбинации с подходящими солюбилизирующими веществами, например спиртом (например, этиловым спиртом), полиспиртом (например, пропиленгликолем), неионными сурфактантами (например, полисорбатом 80, аддуктом гидрогенизированного касторового масла HCO-50 (полиоксиэтилен (50 моль)) и др. В качестве масляной среды используются, например, кунжутное масло, соевое масло и др., которые можно использовать в комбинации с солюбилизирующим веществом, например бензилбензоатом, бензиловым спиртом и др. Приготовленный таким образом инъекционный препарат можно помещать в подходящую ампулу.
Предпочтительно, фармацевтические композиции для перорального или парентерального применения, рассмотренные выше, готовят в виде лекарственных форм с унифицированной дозой, соответствующей дозе действующих веществ. К таким лекарственным формам с унифицированной дозой относятся, например, таблетки, пилюли, капсулы, инъекционные лекарственные формы (раствор в ампулах), свечи и др.
Примеры фармацевтических композиций, содержащих антитело к IL-4R, которые можно использовать в контексте настоящего изобретения, указаны, например, в опубликованной патентной заявке для США № 2012/0097565.
Дозировка
Количество антагониста IL-4R (например, антитело к IL-4R), вводимое пациенту согласно способам настоящего изобретения, обычно является терапевтически эффективным количеством. В контексте этого документа, фраза «терапевтически эффективное количество» означает количество антагониста IL-4R, вызывающие одно или более из нижеследующих: (а) улучшение одного или более связанных с АД параметров (как указано в другом месте этого документа); и (или) (б) определяемое улучшение одного или более симптомов или признаков атопического дерматита. «Терапевтически эффективное количество» также означает количество антагониста IL-4R, подавляющее, предотвращающее, уменьшающее или замедляющее прогрессирование АД у пациента.
В случае антитела к IL-4R, терапевтически эффективное количество может составлять от приблизительно 0,05 мг до приблизительно 600 мг, например приблизительно 0,05 мг, приблизительно 0,1 мг, приблизительно 1,0 мг, приблизительно 1,5 мг, приблизительно 2,0 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 110 мг, приблизительно 120 мг, приблизительно 130 мг, приблизительно 140 мг, приблизительно 150 мг, приблизительно 160 мг, приблизительно 170 мг, приблизительно 180 мг, приблизительно 190 мг, приблизительно 200 мг, приблизительно 210 мг, приблизительно 220 мг, приблизительно 230 мг, приблизительно 240 мг, приблизительно 250 мг, приблизительно 260 мг, приблизительно 270 мг, приблизительно 280 мг, приблизительно 290 мг, приблизительно 300 мг, приблизительно 310 мг, приблизительно 320 мг, приблизительно 330 мг, приблизительно 340 мг, приблизительно 350 мг, приблизительно 360 мг, приблизительно 370 мг, приблизительно 380 мг, приблизительно 390 мг, приблизительно 400 мг, приблизительно 410 мг, приблизительно 420 мг, приблизительно 430 мг, приблизительно 440 мг, приблизительно 450 мг, приблизительно 460 мг, приблизительно 470 мг, приблизительно 480 мг, приблизительно 490 мг, приблизительно 500 мг, приблизительно 510 мг, приблизительно 520 мг, приблизительно 530 мг, приблизительно 540 мг, приблизительно 550 мг, приблизительно 560 мг, приблизительно 570 мг, приблизительно 580 мг, приблизительно 590 мг или приблизительно 600 мг антитела к IL-4R. Согласно определенным вариантам осуществления настоящего изобретения, пациенту вводят 75 мг, 150 мг или 300 мг антитела к IL-4R.
Количество антагониста IL-4R, содержащееся в отдельной дозе, можно выражать в миллиграммах антитела на килограмм массы тела пациента (т.е. мг/кг). Например, антагонист IL-4R можно вводить пациенту в дозе приблизительно от 0,0001 до приблизительно 10 мг/кг массы тела.
Комбинированная терапия
Согласно определенным вариантам осуществления настоящего изобретения, способы настоящего изобретения включают применение у пациента одного или более дополнительных терапевтических средств в комбинации с антагонистом IL-4R. В контексте этого документа, выражение «в комбинации с» означает, что дополнительные терапевтические средства применяются до, после фармацевтической композиции или одновременно с ней, содержащей антагонист IL-4R. Термин «в комбинации с» также включает последовательное или одновременное применение антагониста IL-4R и второго терапевтического средства.
Например, при применении дополнительного терапевтического средства «перед» введением фармацевтической композиции, содержащей антагонист IL-4R, его можно применять приблизительно за 72 часа, приблизительно за 60 часов, приблизительно за 48 часов, приблизительно за 36 часов, приблизительно за 24 часа, приблизительно за 12 часов, приблизительно за 10 часов, приблизительно за 8 часов, приблизительно за 6 часов, приблизительно за 4 часа, приблизительно за 2 часа, приблизительно за 1 час, приблизительно за 30 минут, приблизительно за 15 минут или приблизительно за 10 минут до введения фармацевтической композиции, содержащей антагонист IL-4R. При применении дополнительного терапевтического средства «после» введения фармацевтической композиции, содержащей антагонист IL-4R, его можно применять приблизительно через 10 минут, приблизительно через 15 минут, приблизительно через 30 минут, приблизительно через 1 час, приблизительно через 2 часа, приблизительно через 4 часа, приблизительно через 6 часов, приблизительно через 8 часов, приблизительно через 10 часов, приблизительно через 12 часов, приблизительно через 24 часа, приблизительно через 36 часов, приблизительно через 48 часов, приблизительно через 60 часов или приблизительно через 72 часа после введения фармацевтической композиции, содержащей антагонист IL-4R. Применение дополнительного терапевтического средства «одновременно» или вместе с фармацевтической композицией, содержащей антагонист IL-4R, означает, что это средство применяется у пациента в виде отдельной лекарственной формы не более чем 5 минут (до, после или в одно время) от применения фармацевтической композиции, содержащей антагонист IL-4R, или что это средство применяется у пациента в виде одной комбинированной лекарственной формы, содержащей как дополнительное терапевтическое средство, так и антагонист IL-4R.
Дополнительным терапевтическим средством может быть, например, антагонист IL-4R, антагонист IL -1 (включая, например, IL -1 по патенту US 6,927,044), антагонист IL -6, антагонист IL -6Р (включая, например, антитело к IL -6Р по патенту US 7,582,298), антагонист IL -13, антагонист ФНО, антагонист IL -8, антагонист IL -9, антагонист IL -17, антагонист IL -5, антагонист IgE, антагонист CD48, антагонист IL -31 (включая, например, IL -31 по патенту US 7,531,637), тимический стромальный лимфопротеин (TSLP) (включая, например, TSLP по патенту US 2011/027468), гамма-интерферон (IFNγ), антибиотики, топические кортикостероиды, такролимус, пимекролимус, циклоспорин, азатиоприн, метотрексат, кромолин-натрий, ингибиторы протеиназы или их комбинации. Согласно определенным вариантам осуществления настоящего изобретения, фармацевтическая композиция, содержащая антагонист IL-4R, применяется у пациента в сочетании с немедикаментозной терапией, например терапией ультрафиолетовым (УФ) излучением.
Способы настоящего изобретения включают применение антагониста IL-4R в комбинации со вторым дополнительным терапевтическим средством для получения аддитивного или синергического эффекта при лечении АД. Согласно одному варианту осуществления настоящего изобретения, настоящее изобретение включает способы лечения умеренно тяжелого или тяжелого АД. Определенные варианты осуществления настоящего изобретения включают способы лечения умеренно тяжелого или тяжелого АД путем применения антагониста IL-4R вместе с ТКС. ТКС может быть сильным по своей активности, например принадлежать к группе III. Примерами ТКС группы II являются метилпреднизолона ацепонат, мометазона фуроат, флутиказона пропионат и бетаметазона валерат. Согласно некоторым вариантам осуществления настоящего изобретения, ТКС может быть умеренным по своей активности, например из группы II, или слабым по своей активности, например из группы I.
Режимы введения
Настоящее изобретение включает способы, предполагающие введение пациенту фармацевтической композиции, содержащей антагонист IL-4R, с частотой приблизительно четыре раза в неделю, два раза в неделю, один раз в неделю, один раз каждые две недели, один раз каждые три недели, один раз каждые четыре недели, один раз каждые пять недель, один раз каждые восемь недель, один раз каждые двенадцать недель или реже до получения терапевтического ответа. Согласно определенным вариантам осуществления настоящего изобретения, включающим введение фармацевтической композиции, содержащей антитело к IL-4R, может использоваться режим дозирования с введением препарат один раз в неделю в дозе приблизительно 75 мг, 150 мг или 300 мг.
Согласно определенным вариантам осуществления настоящего изобретения, пациенту можно многократно вводить антагонист IL-4R в течение определенного периода времени. Согласно этому аспекту настоящего изобретения, способы включают последовательное многократное введение пациенту доз антагониста IL-4R. В контексте этого документа «последовательное введение» означает, что каждую дозу антагониста IL-4R вводят пациенту в разные моменты времени, например в разные дни через заранее определенные интервалы (например, часы, дни, недели или месяцы). Настоящее изобретение включает способы, заключающиеся в ведении пациенту одной первичной дозы антагониста IL-4R с последующим введением одной или более вторичных доз антагониста IL-4R и, необязательно, с последующим введением одной или более третичных доз антагониста IL-4R.
Термины «первичные дозы», «вторичные дозы» и «третичные дозы» относятся к временной последовательности введения антагониста IL-4R. Таким образом, «первичная доза» это доза, которую вводят в начале лечения (также называется «исходная доза»); «вторичные дозы» это дозы, которые вводят после введения первичной дозы; и «третичные дозы» это дозы, которые вводят после введения вторичных доз. Все первичные, вторичные и третичные дозы могут содержать одинаковое количество антагониста IL-4R, однако обычно они отличаются друг от друга в плане частоты введения. Согласно определенным вариантам осуществления настоящего изобретения, однако, количество антагониста IL-4R в первичных, вторичных и (или) третичных дозах может различаться (например, увеличиваться или уменьшаться согласно необходимости) в ходе лечения. Согласно определенным вариантам осуществления настоящего изобретения, вводят одну или более доз (например, 1, 2, 3, 4 или 5) в начале лечения в качестве «нагрузочных доз» с последующим более редким введением доз (например, «поддерживающих доз»). Например, антагонист IL-4R можно вводить пациенту с АД в нагрузочной дозе приблизительно 300 мг или приблизительно 600 мг с последующим введением поддерживающих доз приблизительно 75 мг или приблизительно 300 мг. Согласно одному варианту осуществления настоящего изобретения, первичная доза и одна или более вторичных доз составляют на каждую от 50 мг до 600 мг антагониста IL-4R, например от 100 мг до 400 мг антагониста IL-4R, например 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 400 мг или 500 мг антагониста IL-4R. Согласно некоторым вариантам осуществления настоящего изобретения, первичная доза или одна или более вторичных доз представляют собой на каждую такое же количество антагониста IL-4R. Согласно другим вариантам осуществления настоящего изобретения, первичная доза включает первое количество антагониста IL-4R, и одна или более вторичных доз включают на каждую второе количество антагониста IL-4R. Например, первое количество антагониста IL-4R может превышать в 1,5, 2, 2,5, 3, 3,5, 4, 5 или более раз второе количество антагониста IL-4R.
Согласно одному примеру варианта осуществления настоящего изобретения, каждую вторичную и (или) третичную дозу вводят через 1-14 (например, 1, 1½, 2, 2½, 3, 3½, 4, 4½, 5, 5½, 6, 6½, 7, 7½, 8, 8½, 9, 9½, 10, 10½, 11, 11½, 12, 12½, 13, 13½, 14, 14½ или более) недель после непосредственно предшествующей дозы. Фраза «непосредственно предшествующая доза», используемая в этом документе, означает, что в ряде последовательных введений нескольких доз пациенту вводят дозу антагониста IL-4R сразу же перед введением следующей дозы без введения других промежуточных доз.
Согласно этому аспекту настоящего изобретения, способы могут включать введение пациенту любого количества вторичных и (или) третичных доз антагониста IL-4R. Например, согласно определенным вариантам осуществления настоящего изобретения, пациенту вводят только одну вторичную дозу. Согласно другим вариантам осуществления настоящего изобретения, пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или более) вторичных доз. Аналогично, согласно определенным вариантам осуществления настоящего изобретения, пациенту вводят только одну третичную дозу. Согласно другим вариантам осуществления настоящего изобретения, пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или более) третичных доз.
Согласно вариантам осуществления настоящего изобретения с введением нескольких вторичных доз, каждую вторичную дозу можно вводить с той же частотой, что и другие вторичные дозы. Например, каждую вторичную дозу можно вводить пациенту через 1-2 недели после введения непосредственно предшествующей дозы. Аналогично, согласно вариантам осуществления настоящего изобретения с введением нескольких третичных доз, каждую третичную дозу можно вводить с той же частотой, что и другие третичные дозы. Например, каждую третичную дозу можно вводить пациенту через 2-4 недели после введения непосредственно предшествующей дозы. В качестве альтернативы, частота введения вторичных и (или) третичных доз пациенту может различаться в течение периода лечения. Частота введения также может корректироваться в течение периода лечения врачом в зависимости от потребностей отдельного пациента после клинического обследования.
Настоящее изобретение включает способы, предполагающие последовательное введение пациенту антагониста IL-4R и второго терапевтического средства для лечения АД. Согласно некоторым вариантам осуществления настоящего изобретения, данные способы включают введение одной или более доз антагониста IL-4R с последующим применением одной или более доз второго терапевтического средства. Например, можно вводить одну или более доз приблизительно от 75 мг до приблизительно 300 мг антагониста IL-4R с последующим применением одной или более доз второго терапевтического средства (например, топического кортикостероида или ингибитора кальциневрина или любого другого терапевтического средства, как указано в другом месте этого документа) с целью ослабления, уменьшения или устранения одного или более симптомов АД. Согласно некоторым вариантам осуществления настоящего изобретения, вводят одну или более доз антагониста IL-4R, что приводит к улучшению одного или более связанных с АД параметров, и затем назначают второе терапевтическое средство для предотвращения повторного возникновения по меньшей мере одного симптома АД. Альтернативные варианты осуществления настоящего изобретения касаются одновременного применения антагониста IL-4R и второго терапевтического средства. Например, вводят одну или более доз антагониста IL-4R и применяют второе терапевтическое средство в отдельной дозе с частотой применения схожей с частотой применения антагониста IL-4R или отличающейся от нее. Согласно некоторым вариантам осуществления настоящего изобретения, второе терапевтическое средство применяют до, после антагониста IL-4R или одновременно с ним.
Антагонисты IL-4R в качестве вакцинных адъювантов
Настоящее изобретение также включает композиции и способы, которые могут применять в вакцинировании. Например, антагонист IL-4R (например, антитело к IL-4R, рассматриваемое в этом документе) можно вводить пациенту в сочетании с вакциной для улучшения или усиления иммунного ответа (включая гуморальный и клеточный иммунный ответы) на вакцину, т.е. в качестве вакцинного адъюванта. Согласно определенным вариантам осуществления настоящего изобретения, антагонист IL-4R вводят непосредственно перед, после введения вакциной композиции пациенту и (или) одновременно с ней. Например, настоящее изобретение включает способы вызывания или усиления иммунного ответа на антиген у пациента путем введения ему фармацевтической композиции, содержащей антагонист IL-4R, с последующим введением вакцинной композиции, содержащей антиген (отдельно или в комбинации с антагонистом IL-4R), и, по желанию, введение дополнительных доз антагониста IL-4R в течение определенного периода времени после введения пациенту вакцинного антигена.
Антагонисты IL-4R, являющиеся предметом настоящего изобретения, можно вводить в качестве адъювантов с вакцинами любого типа, например с живыми вакцинами, живыми ослабленными вакцинами, убитыми вакцинами, субъединичными вакцинами, ДНК-вакцинами и иммунотерапевтическими противоопухолевыми вакцинами. К вакцинам, которые можно использовать в сочетании с антагонистами IL-4R, являющимися предметом настоящего изобретения, относятся вакцины против бактериальных патогенов, вирусов, паразитов и других инфекционных возбудителей. Примерами инфекционных возбудителей и заболеваний против которых могут применяться вакцинные композиции и способы настоящего изобретения являются, помимо прочего, ВИЧ, ВГB, РСВ, Neisseria meningitides, стрептококки, туберкулез, малярия, натуральная оспа, дифтерия, коклюш, столбняк, полиомиелит, корь, краснуха, эпидемический паротит, грипп, сибирская язва, ТОРС, вирус Эбола, хантавирус, вирус Денге и др.
Настоящее изобретение также включает фармацевтические композиции, содержащие антагонист IL-4R и один или более вакцинных антигенов. Согласно этому аспекту настоящего изобретения, фармацевтические композиции могут содержать один или более дополнительных иммуностимуляторов, например MPL, MDP, олигонуклеотиды CpG, липопептиды, сапонины, дцРНК, низкомолекулярные иммуностимуляторы и др.
Кроме антагонистов IL-4R в контексте вакцинных методов и композиций, рассматриваемых в этом документе, могут использоваться другие ингибиторы сигнального пути IL-4/IL-13 (например, антитела к IL-4, антитела к IL-13, биспецифические антитела к IL-4 и IL -13 и др.).
ПРИМЕРЫ
Следующие примеры приведены с целью предоставления средним специалистам в данной области техники полной информации о выполнении и применении способов и композиций, являющихся предметом настоящего изобретения, и не имеют своей целью ограничить объем того, что изобретатели считают своим изобретением. Был приложены все усилия для обеспечения точности приведенных числовых значений (например, количество, температура и т.д.), однако следует учитывать вероятность некоторых экспериментальных ошибок и отклонений. Если не указано иное, части - это части по массе, молекулярная масса - это средняя молекулярная масса, температура - это температура в градусах Цельсия и давление - это давление на уровне атмосферного давления или близкое к нему.
Пример 1. Получение человеческих антител к человеческому IL-4R
Человеческие антитела к IL-4R получали согласно патенту США № 7,608,693. В таблице 1 приведены идентификаторы пар аминокислотных последовательностей вариабельных областей тяжелой и легкой цепей и идентификаторы аминокислотных последовательностей участков CDR отдельных антител к IL-4R и названия соответствующих антител.
Антагонист IL-4R, используемый в приведенных ниже примерах, является человеческим антителом к IL-4R, указанный в таблице 1 как H1H098-b (также упоминается в этом документе как «mAb1»).
Пример 2: Клиническое исследование с применением однократных возрастающих доз антител к IL-4Р (mAb1), вводимых внутривенно и подкожно здоровым пациентам
А. Дизайн исследования
Данное исследование было рандомизированным двойным слепым плацебо-контролируемым исследованием с применением однократных последовательно возрастающих доз mAb1, вводимых внутривенно (в/в) и подкожно (п/к) здоровым пациентам. Основной целью данного исследования было изучение безопасности и переносимости здоровыми пациентами препарата mAb1, вводимого внутривенно и подкожно.
Скрининг выполнялся в период с -21-го по -3-й день. В 1-й день (исходная точка) пациенты были рандомизированы по группам, в которых исследуемый препарат (mAb1 или плацебо) вводился либо внутривенно, либо подкожно посредством двухчасовой инфузии. Пациенты посещали клинику в дни 4, 8, 11, 15, 22, 29, 43, 57 и 85 (конец исследования) для проверки безопасности и взятия образцов крови для выполнения клинических лабораторных анализов.
Всего в исследовании приняло участие 48 пациентов. В группе внутривенного введения было предусмотрено четыре когорты с последовательно возрастающими дозами (1,0, 3,0, 8,0 и 12,0 мг/кг), а в группе подкожного введения - две когорты с последовательно возрастающими дозами (150 и 300 мг). В каждой когорте находилось по 8 пациентов (если когорта не расширялась): путем рандомизации 6 пациентам был назначен препарат mAb1, а 2 пациентам - плацебо. В целях оптимальной безопасности первые 3 пациента в 1-й когорте внутривенного введения (1,0 мг/кг) получали препарат с интервалом в 24 часа друг между другом, а остальные 5 пациентов получали препарат спустя 5-7 дней. В последующих когортах внутривенного введения 3 из 8 пациентов получали препарат в день 1, а остальные 5 пациентов - спустя 5-7 дней. Все 8 пациентов в 1-й когорте с подкожным применением препарата (150 мг) получали препарат в один и тот же день. Все 8 пациентов в последующей когорте с подкожным применением препарата (300 мг) тоже получали препарат в один и тот же день. В когортах с подкожным применением препарат вводили пациентам после того, как он был введен пациентам когорт с внутривенным введением.
Критерии включения в исследование были следующими: (1) мужчины и женщины в возрасте 18-65 лет; (2) масса тела >50 кг и <120 кг; (3) в случае женщин, способных к деторождению - отрицательный результат теста сыворотки крови на беременность на скрининговый визит (визит 1) и отрицательный результат теста мочи на беременность на день -1; (4) готовность воздерживаться от употребления более чем 2 стандартных алкогольных напитков в любой 24-часовой промежуток времени на протяжении исследования. Стандартный алкогольный напиток считался эквивалентным 350 мл пива, 150 мл вина или 50 мл крепкого алкоголя; (5) готовность воздерживаться от употребления алкоголя на протяжении 24 часов, предшествующих каждом визиту в рамках исследования; (6) в случае мужчин, способных к зачатию, и женщин, способных к деторождению - готовность пользоваться надлежащими средствами контрацепции и не беременеть (или не приводить к беременности партнерши (партнерш)) на всем протяжении исследования. В надлежащие средства контрацепции входят внутриматочное устройство (ВМУ), двусторонняя перевязка маточных труб, вазэктомия, презерватив или диафрагма плюс контрацептивная губка или пена или гель; и (7) готовность, обязательство и способность приходить в клинику на все визиты и проходить все связанные с исследованием процедуры.
Критерии исключения из исследования были следующими: (1) Инициация нового комплекса физических упражнений или значительное изменение предыдущего комплекса физических упражнений в период 4 недель, предшествующих скринингу (визит 1). Пациенты должны были быть готовы на протяжении исследования придерживаться аналогичного уровня физических нагрузок и воздерживаться от необычайно сильных физических нагрузок; (2) беременные или кормящие грудью женщины; (3) значительное сопутствующее заболевание или его наличие в анамнезе, например сердечные, почечные, неврологические, эндокринные, метаболические, лимфатические заболевания, любое иное заболевание или состояние, которое бы оказало нежелательное влияние на участие пациента в этом исследовании; (4) какие-либо клинически значительные отклонения, наблюдаемые в ходе скринингового визита; (5) госпитализация по какой-либо причине в период 60 дней, следующих за скринингом (визит 1); (6) подтвержденное наличие в анамнезе заболевания вирусом иммунодефицита человека (ВИЧ), гепатитом В или гепатитом С и (или) положительный результат выполненных на скрининговом визите анализов на поверхностный антиген вируса гепатита В, на антитела к вирусу гепатита С или на ВИЧ; (7) злоупотребление наркотиками или спиртными напитками в течение года, предшествующего скрининговому визиту - на основании сведений в анамнезе или по результатам анализа на наркотики; (8) гиперчувствительность к доксициклину или схожим веществам на основании сведений в анамнезе; (9) участие в каком-либо клиническом исследовании иного экспериментального препарата или терапии в период 30 дней, предшествующих скрининговому визиту, или в предшествующий скрининговому визиту период, равный пяти периодам полувыведения экспериментального препарата, в зависимости от того, что дольше; (10) в прошлом воздействие на организм пациента какого-либо терапевтического или экспериментального биологического средства; (11) какое-либо медицинское или психическое нарушение, которое, по мнению исследователя, создало бы риск для пациента, помешало бы его участию в исследовании или помешало бы толкованию результатов исследования; (12) пациенты с положительным результатом анализа на туберкулез QuantiFERON; (13) паразитарная инфекция в анамнезе или недавние (имевшие место в предшествующие 6 месяцев) поездки в места с паразитарными эндемиями; (14) злоупотребление алкоголем или наркотическими веществами в последние 5 лет; (15) положительный результат анализа мочи на наркотики при скрининге (визит 1) или в исходной точке (визит 2); и (или) (16) применение живой/аттенуированной вакцины в период 12 недель, следующих за скринингом, или на протяжении исследования.
B. Исследуемое лечение
Лекарственный препарат mAb1 поставлялся в виде лиофилизированного порошка в стеклянных ампулах по 20 мл для в/в или п/к введения. При в/в введении лекарственный препарат mAb1 смешивался в одноразовой ампуле с 7,8 мл стерильного инъекционного раствора, в результате чего получался раствор, содержащий 50 мг mAb1 на 1 мл. Фармацевт или уполномоченное лицо набирали в шприц необходимое количество разведенного mAb1 (в зависимости от дозы и массы тела пациента) или плацебо и вводили его в инфузионный мешок с физиологическим раствором для в/в введения. Инфузия длилась 2 часа.
При п/к введении лекарственный препарат mAb1 смешивали с 2,3 мл стерильного инъекционного раствора, в результате чего получался раствор, содержащий 150 мг mAb1 на 1 мл. Фармацевт или уполномоченное лицо вводили инъекцию в области живота; введение в области конечностей не допускалось по причине возможного отклонения параметров абсорбции и биодоступности. Если в один и тот же день требовалось введение нескольких инъекций, то инъекции выполнялись в разных местах.
Были испытаны следующие дозы mAb1: 1,0, 3,0, 8,0 и 12,0 мг/кг внутривенно, а также 150 и 300 мг подкожно.
Приготовление плацебо выполнялось в тех же пропорциях, что и в случае mAb1, но при этом антитела не добавлялись.
C. Результаты и выводы
В целом, mAb1 хорошо переносился и продемонстрировал благоприятный профиль безопасности. Общий профиль нежелательных явлений (НЯ) был характерен для популяции здоровых пациентов. Менее чем у 55% пациентов, получавших mAb1 (19 из 36), наблюдалось как минимум одно возникающее во время лечения нежелательное явление (ВЛНЯ), а в группе приема плацебо этот показатель составил менее 59% (7 из 12). Наиболее часто сообщалось о следующих ВЛНЯ: повышение уровня креатининфосфокиназы (КФК) в крови, повышение артериального давления, назофарингит и зубная боль. У большинства пациентов ВЛНЯ были легкой или умеренной степени; только у 3 пациентов сообщалось о ВЛНЯ, степень которых считалась тяжелой. Исследователь счел, что только одно ВЛНЯ тяжелой степени (повышение уровня КФК в крови) было связано с лечением. В ходе исследования сообщалось об одном серьезном нежелательном явлении (СНЯ), и исследователь счел его не связанным с исследуемым препаратом. Ни один пациент не прекратил участие в исследовании по причине НЯ. Не сообщалось о смертельных исходах. В ходе исследования не сообщалось ни о каких иных клинически значительных отклонениях в результатах лабораторных анализов (общий и биохимический анализы крови, анализ мочи). В средних и медианных значениях исходных показателей лабораторных анализов не было выявлено никаких тенденций. В ходе исследования не наблюдалось каких-либо значительных тенденций в средних и медианных значениях изменений показателей температуры тела и пульса относительно исходного уровня. При физикальном осмотре, на ЭКГ и в показателях жизнедеятельности организма не наблюдалось клинически значительных отклонений.
Исследование характерно тем, что в популяции пациентов была велика доля чернокожих/афроамериканцев (таблица 2).
Демографические характеристики групп лечения
(n=12)
(n=6)
(n=6)
(n=6)
(n=6)
(n=6)
(n=6)
Хотя пациенты были здоровыми волонтерами, афроамериканцы как группа могут быть более подвержены атопическим заболеваниям (Caggana и др., 1999 г.; Genet. Med. 1: 267-271), и соответственно эту популяцию можно считать приемлемой для доказывания механизма действия на основании поискового анализа биомаркеров.
Фармакокинетический (ФК) анализ показал нелинейность кинетических характеристик. Насыщение мишень-опосредованного пути выведения наблюдалось при в/в дозах 8 и 12 мг/кг, которые приводили к концентрации функционального mAb1 свыше 30 мг/л. У 9 пациентов наблюдались низкие титры антител к препарату. Не наблюдалось внезапного и устойчивого снижения концентрации функционального mAb1, что свидетельствует о том, что антитела к препарату не имели значительного воздействия на ФК.
Пример 3: Клиническое исследование двух разных лекарственных препаратов с содержанием антител к IL-4R (mAb1) после подкожного введения антител к IL-4R (mAb1) здоровым пациентам
A. Дизайн исследования
Данное исследование было одноцентровым, двойным слепым, рандомизированным исследованием с применением однократных доз и без плацебо-контроля для изучения безопасности и фармакокинетических характеристик подкожного применения двух разных лекарственных препаратов с содержанием антител к IL-4R (mAb1), произведенных с использованием разных линий клеток и разных производственных процессов. Лекарственные препараты предоставлялись в дозировке 2 мл в концентрации 150 мг/мл, и 30 здоровым взрослым пациентам в двух параллельных группах (15 пациентов на группу) вводил 300 мг препарата (2 мл) подкожно. Из 30 пациентов 22 пациента были мужского пола (73,3%), 8 - женского (26,7%), возраст пациентов был от 19 до 45 лет, масса тела - от 54,8 до 94,3 кг.
Использовались показатели концентрации mAb1 в сыворотке крови для определения следующих параметров ФК: максимальная концентрация в сыворотке (Смакс), площадь под кривой [концентрация в сыворотке относительно времени] от нулевой временной точки до реальной временной точки, в которой в последний раз наблюдался показатель концентрации свыше нижнего предела количественного определения (tпосл (ППКпосл), а также площадь под кривой графика концентрации в сыворотке относительно времени от нулевой временной точки с экстраполяцией до бесконечности (ППК). Также измерялось время до достижения максимальной концентрации (tмакс) и период полувыведения (t1/2z).
В. Критерии оценки и методы
Безопасность оценивалась путем измерения нежелательных явлений (включая возникшие во время лечения нежелательные явления (ВЛНЯ), наступившие в период до двух месяцев после введения дозы), выполнения клинических лабораторных анализов (биохимический и общий анализы крови, анализ мочи), измерения основных показателей жизнедеятельности организма, снятия электрокардиограммы (ЭКГ) с автоматической расшифровкой данных, измерения уровня антител к mAb1 (отрицательный результат или титр антител), а также выполнения анализов местной переносимости (включая измерение боли в месте введения с помощью визуальной аналоговой шкалы [ВАШ; 100-миллиметровая линия без градации], эритемы [диаметр в месте введения в мм] и отека [диаметр в месте введения в мм]).
Нежелательными явлениями, представляющими особый интерес (НЯПОИ), были те НЯ (как серьезные, так и нет), которые вызывают озабоченность с научной и медицинской точек зрения и требуют индивидуального отслеживания, протоколирования и лечения, как указано в протоколе. Следующие НЯ считались НЯПОИ: гиперчувствительность/анафилаксия: анафилактическая реакция или острая аллергическая реакция, требующая проведения немедленного лечения; тяжелая реакция в месте введения препарата, продолжающаяся более 24 часов; тяжелая инфекция; любая паразитарная инфекция; повышение уровня аланинаминотрансферазы (АЛТ) ≥ 2 ВПН; QTc ≥ 500 мс; беременность; передозировка.
Забор крови для выполнения общего и биохимического анализов крови осуществлялся до введения препарата в день -1 и в дни 2 (через 24 часа после введения препарата) и 57, и биохимический анализ крови ограничивался изучением параметров деятельности печени в дни 8, 15, 22, 29, 36, 43 и 50.
В день 1 и в дни 15, 29 и 57 выполнялся забор крови для выявления антител к mAb1 в сыворотке.
Анализы местной переносимости выполнялись до введения препарата в день 1, а также через 2 минуты, 2 часа, 6 часов и 12 часов после введения препарата, а также в дни 2 (через 24 часа после введения препарата), 3, 4 и 8 после введения mAb1.
Забор крови для выполнения фармакокинетического и фармакогенетического анализов осуществлялся до введения препарата в день 1 и через 12 часов после введения препарата, а также в дни 2, 3, 4, 8, 11, 15, 22, 29, 36, 43, 50 и 57 после введения mAb1. Концентрация функционального mAb1 в сыворотке крови определялась с помощью иммуноферментного анализа с нижним пределом количественного определения (НПКО) в 78 нг/мл.
Для оптимального выполнения фармакогенетических анализов образцы брали в исходной точке (в день 1 до введения препарата).
Фармакокинетические показатели функционального mAb1 в сыворотке крови были сведены по группам лечения с помощью описательной статистики (среднее, геометрическое среднее, медиана, стандартное отклонение (СО), коэффициент вариации (КВ), минимум и максимум). На основании логарифмически преобразованных показателей Cмакс, ППКпосл и ППК были изучено отношение исследуемого и эталонного лечения с помощью линейной модели с фиксированными эффектами, в которой пол и лечение - фиксированные эффекты, а масса тела - независимая переменная. По показателям Смакс, ППКпосл и ППК были рассчитаны оценки и 90%-ные доверительные интервалы (ДИ) отношения исследуемого и эталонного лечения.
Оценка безопасности выполнялась путем изучения отдельных значений и описательных статистических показателей. Все НЯ кодировались согласно ред. 15.0 Медицинского словаря для регуляторной деятельности (MedDRA), а частота возникающих во время лечения нежелательных явлений (ВЛНЯ) подсчитывалась (количество и процентная доля) по основным системно-органным классам, предпочтительным терминам и группам лечения, данные о потенциально клинических значительных отклонениях (ПКЗО; определения согласно ред. 2.0 от 14 сентября 2009 г.) клинических лабораторных показателей, основных показателей жизнедеятельности организма, показателей ЭКГ, а также выходящие за пределы нормы показатели клинических лабораторных анализов помечали и указывали в виде сводных данных по группам лечения.
Результаты анализов на антитела к mAb1 приводили по группам лечения, пациентам и визитам либо как отрицательные, либо указывался титр в случае положительных результатов. Данные были сведены в форме количества пациентов (количество и процентная доля) с отрицательным и положительным результатом анализа на антитела к препарату в каждой группе лечения.
По каждой группе лечения и каждой запланированной временной точке предоставлялись описательные статистические показатели (среднее, СО, минимум, медиана и максимум) боли (согласно ВАШ), диаметра эритемы и диаметра отека. Также по группам лечения приводились сводные данные о средних значениях этих показателей на временном отрезке (от введения исследуемого препарата до оценки в день 8 включительно) и о пиковых значениях этих показателях (по результатам оценки после введения препарата).
C. Результаты в части фармакокинетики
Среднее ±СО (среднее геометрическое) [КВ %] фармакокинетических показателей функционального mAb1 в сыворотке крови
(27300) [31,6]
(25300) [36,6]
(72,00-240,00)
(48,00-240,00)
(10500000) [41,0]
(11200000) [35,7]c
(12800000) [29,4]b
(11600000) [38,2]c
(129) [33,8]b
(124) [36,2]c
b N=13, т.к. поскольку для 2 пациентов невозможно было представить конечную фазу элиминации/выведения в линейно-логарифмических координатах;
с N=11, т.к. 2 пациента прекратили участие в исследовании.
Точечные оценки величины отношения показателей сравниваемых лекарственных препаратов с указанием 90%-ного доверительного интервала
Лекарственный препарат А: Группа А (испытываемый препарат), 150 мг/мл (лекарственный препарат 1)×2,0 мл (300 мг mAb1);
Лекарственный препарат Б: Группа Б (эталонный препарат), 150 мг/мл (лекарственный препарат 2)×2,0 мл (300 мг mAb1).
Оценки выполнены на основе линейной модели с фиксированными эффектами, в которой пол, масса тела и лечение - фиксированные эффекты.
D. Результаты в части безопасности
ВЛНЯ наблюдались в группе лекарственного препарата А (испытываемый лекарственный препарат) у 12 из 15 пациентов (80,0%), а в группе лекарственного препарата Б (эталонный лекарственный препарат) - у 8 из 15 пациентов (53,3%). Очевидный дисбаланс лечения наблюдался лишь по некоторым основным системно-органным классам и, похоже, был связан с явлениями, которые не имели отношения к экспериментальному медицинскому продукту, т.к. зачастую выявлялась другая причина. На протяжении исследования наблюдалось четыре СНЯ у двух пациентов.
У 23-летнего пациента мужского пола в группе лечения лекарственным препаратом 1 наблюдалась «инфекция вирусом простого герпеса II типа» с такими симптомами, как нечеткость зрения, повышенное потоотделение, повышенная температура тела и головная боль начиная с 4-го дня после введения дозы, а также отек языка (с 6-го дня после введения дозы), а также кашель, заложенность в груди и мышечные судороги в нижних конечностях (с 7-го дня после введения дозы). В ходе этих явлений пациент несколько раз обращался в больницу по скорой помощи, и ему проводилось различное лечение, включая Solumedrol® (три дозы в/в), преднизон (9 дней), Rocephin® (одна доза в/в), Zithromax® (5 дней). Стоит отметить, что за 3 месяца до введения препарата пациент сделал косметический прокол языка. Через девятнадцать дней после введения препарата все симптомы разрешились. Результат выполненного через 10 дней после введения препарата теста на титры иммуноглобулина G, вызванного вирусом простого герпеса II типа, был отрицательным, а результат повторного теста, выполненного через 7 недель после введения препарата, был положительным. Исследователь и компания сочли это СНЯ связанным с экспериментальным медицинским продуктом. Более чем через 4 недели после введения препарата пациенту был поставлен диагноз «неврит лицевого нерва» с левой стороны, и Исследователь счел данное явление последствием инфекции вирусом простого герпеса II типа. По поводу данного явления проводилось лечение преднизоном (6 дней) и ацикловиром (10 дней). Исследователь счел, что данное СНЯ не связано с экспериментальным медицинским препаратом, т.к. в острой фазе инфекции вирусом простого герпеса II типа имело место многократное и повторное применение стероидных препаратов, что было принято за альтернативное объяснение явления. Компания сочла, что нельзя исключать причинно-следственную связь с экспериментальным медицинским препаратом. По состоянию на конец исследования оба явления были в процессе разрешения. На протяжении исследования у этого пациента не развились антитела к препарату
У 22-летнего пациента мужского пола из группы лечения лекарственным препаратом А наблюдался «повышенный уровень АЛТ» (уровень АЛТ до 11,4 ВПН), сопровождаемый «рабдомиолизом» (уровень креатининфосфокиназы до 392 ВПН), и оба явления были обнаружены в ходе запланированных лабораторных анализов через 7 недель после введения препарата. Данные явления наступили после физической нагрузки (состоявшей из плавания, отжиманий, подтягиваний, подъемов туловища из положения лежа и других упражнений на выносливость; пациент работает спасателем на воде), а также травмы трицепсов (нежелательное явление, не являющееся серьезным). Пациент был госпитализирован для восполнения потери жидкости. Была исключена связь повышенных показателей печеночной деятельности с какими-либо проблемами печени, а повышенный уровень АЛТ (а также повышенный уровень аспартатаминотрансферазы [АСТ] до 50,5 ВПН) был отнесен на счет рабдомиолиза. В ходе этих явлений уровень креатинина и громерулярная функция оставались в пределах нормы. Оба явления разрешились через 3 недели. Исследователь счел эти два СНЯ не связанными с экспериментальным медицинским препаратом. Также у пациента развились антитела к препарату - положительные значения титров были обнаружены в день 29 (значение титра = 120) и на визите в связи с окончанием лечения в день 58 (значение титра = 30).
Помимо повышенного уровня АЛТ иных НЯПОИ в ходе исследования не наблюдалось.
В ходе исследования у 4 (четырех) пациентов развились инфекции. Помимо описанной выше инфекции простого герпеса, наблюдались легкие случаи инфекции верхних дыхательных путей, фарингита и синусита (один случай на каждое заболевание) через 54, 7 и 1 день после введения препарата соответственно. Эти три явления наблюдались у пациентов, получавших C1P2.
У 12 пациентов наблюдалось 15 (пятнадцать) случаев реакций в месте введения препарата: эритема (8 случаев у 8 пациентов: по 4 на каждую группу), боль (3 случая: 2 - в группе C2P1, 1 - в группе C1P2), образование «узелков» (2 случая: по одному в каждой группе), гематома (1 случай в группе C1P2) и кожный зуд (1 случай в группе C2P1). Все случаи были легкими и разрешились в течение 24 часов с момента введения препарата.
Иные ВЛНЯ наблюдались в каждой из групп не более чем у 1 пациента, за исключением 2 случаев кожного зуда (не в месте введения препарата) у 2 пациентов, получавших C2P1, и 3 случаев головной боли (у 1 пациента, получавшего C2P1, и 2 пациентов, получавших C1P2).
Помимо ранее описанных отклонений показателей лабораторных анализов от нормы, не наблюдалось иного повышения показателей сверх пределов, установленных для ПКЗО.
У 6 из 27 (22,22%) пациентов, полностью прошедших исследование, был положительный результат теста на антитела к mAb1 (ни у одного пациента из тех, что досрочно выбыли из исследования, не были обнаружены антитела к препарату). Из 6 пациентов с положительными значениями титров антител к препарату, четверо получали C2P1, двое - C1P2. Между выработкой антител к препарату и ВЛНЯ не наблюдалось связи.
E. Отдельные анализы местной переносимости
Средние пиковые значения по 100-миллиметровой шкале боли ВАШ составляли 4,4 и 4,2 мм в группах приема C2P1 и C1P2 соответственно, а медиана составила 2,0 мм в обеих группах. В каждой группе у 5 (пяти) из 15 пациентов наблюдалось «отсутствие боли» (пиковое значение 0 мм). Самые высокие значения составили 17 и 18 мм в группах C2P1 и C1P2 соответственно и обычно регистрировались через 2 минуты после введения препарата (диапазон: от 2 минут до 12 часов после введения препарата).
Средние пиковые значения диаметра эритемы составили 12,5 и 10,9 мм в группах приема C2P1 и C1P2 соответственно. В каждой группе у 9 (девяти) из 15 пациентов эритемы не было ни в какой временной точке. Максимальные значения составили 40 мм в обеих группах, и все они регистрировались через 2 минуты после введения препарата, за исключением одного пациента, у которого максимальное значение (3 мм) наблюдалось через 48 часов после введения препарата.
Средние пиковые значения диаметра отека составили 1,1 и 0 мм в группах приема C2P1 и C1P2 соответственно. У 13 из 15 пациентов в группе приема C2P1 и у 15 из 15 пациентов в группе приема C1P2 отека не было ни в какой временной точке. Максимальные значения составили 15 и 1 мм у 2 пациентов в группе приема C2P1, и они наблюдались через 2 часа после введения препарата.
F. Выводы
После подкожного введения здоровым пациентам однократной дозы mAb1 в 300 мг, экспозиция функционального mAb1 в сыворотке крови была в группах двух испытываемых лекарственных препаратов схожей. Среднее геометрическое величины отношения показателей сравниваемых лекарственных препаратов (ЛП1/ЛП2) с 90%-ным ДИ составляло: Смакс: 1,10 (0,89-1,35); ППКпосл: 0,90 (0,71-1,16); ППК: 1,05 (0,86-1,29).
В целом, mAb1 хорошо переносился. У одного пациента, получавшего ЛП1, наблюдалось серьезное нежелательное явление «инфекция вирусом простого герпеса II типа», после чего последовал «неврит лицевого нерва».
Не наблюдалось клинически важных проблем по части местной переносимости, и между группами лечения не наблюдалось явных различий по параметрам местной переносимости (т.е. боль, эритема и отек).
Наиболее распространенным ВЛНЯ была эритема в месте введения (у 8 из 30 пациентов), и оно наблюдалось с одинаковой частотой в обеих группах лечения (у 4 из 15 пациентов [26,7%] в каждой из групп).
В заключение: при однократном п/к введении 300 мг mAb1 здоровым пациентам каких-либо клинически важных различий между двумя лекарственными препаратами по части их фармакокинетических характеристик, безопасности и местной переносимости не наблюдалось.
Пример 4: Клиническое исследование безопасности, переносимости и фармакокинетики при подкожном введении возрастающих однократных доз антител к IL-4Р здоровым взрослым пациентам мужского пола из Японии
A. Дизайн исследования
Данное исследование было рандомизированным, двойным слепым, плацебо-контролируемым исследованием с подкожным введением возрастающих однократных доз антител к IL-4Р (mAb1) здоровым взрослым пациентам мужского пола из Японии. Основной целью было изучение безопасности и переносимости mAb1 при подкожном введении возрастающих однократных доз здоровым пациентам мужского пола из Японии. Второстепенными целями было изучение фармакокинетики, иммуногенности, а также поисковое исследование фармакодинамики возрастающих однократных подкожных доз mAb1 при введении здоровым пациентам мужского пола из Японии.
Препарат mAb1 был получен из линии клеток 2 и предоставлен в жидком виде в концентрации либо 75 мг/мл, либо 150 мг/мл в ампулах. Однократные возрастающие дозы mAb1 в 75, 150, 300 и 600 мг вводили подкожно в день 1 (1 инъекция 75 мг и 150 мг, 2 инъекции 300 мг, 4 инъекции 600 мг). Продолжительность наблюдения каждого пациента составила приблизительно 11 недель (включая скрининговый период на протяжении 2-21 дней до введения препарата, 5 дней в клинике [от дня -1 до дня 4, из которых один день был днем лечения] и последующее амбулаторное наблюдение продолжительностью до 57 дней после введения препарата в форме визитов).
В. Критерии оценки
Безопасность: Нежелательные явления (НЯ), физикальный осмотр, клинические лабораторные анализы (общий и биохимические анализы крови, анализ мочи), основные показатели жизнедеятельности (артериальное давление и частота сердечных сокращений в положении лежа и стоя, температура тела), электрокардиограмма (ЭКГ) в 12-ти отведениях и антитела к mAb1
Фармакокинетика: Были рассчитаны следующие фармакокинетические показатели функционального mAb1 в сыворотке крови с использованием анализа без камерной модели: максимальная наблюдаемая концентрация в сыворотке крови (Смакс), время до наступления максимальной концентрации в сыворотке крови (tмакс), нормализованная по дозе Cмакс (Смакс/доза), площадь под кривой графика концентрации в сыворотке крови относительно времени от нулевой временной точки до реальной временной точки наступления последней концентрации свыше нижнего предела количественного определения tпосл (ППКпосл), нормализованная по дозе ППКпосл (ППКпосл/доза), площадь под кривой графика концентрации в сыворотке относительно времени от нулевой временной точки с экстраполяцией до бесконечности (ППК), очевидный объем распределения в условиях равновесного состояния (Vss/F), очевидный общий клиренс (CL/F), среднее время нахождения (СВН) и период полувыведения (t½z)
Фармакодинамика (ФД): фармакодинамические эффекты mAb1 на общий уровень IgE и TARC
Забор крови для выполнения фармакокинетических исследований осуществлялся до введения препарата (день 1) и в дни 1, 2, 4, 8, 11, 15, 18, 22, 25, 29, 36, 43, 50 и 57 (±1 день в случае дней 15-25; ±2 дня в случае дней 29-57) после введения mAb1. Концентрация mAb1 в сыворотке крови определялась с помощью валидированного иммуноферментного анализа с нижним пределом количественного определения (НПКО) в 78 нг/мл (0,078 мг/мл). Забор крови для выполнения фармакодинамических исследований осуществлялся до введения препарата в день -1 и в день 1, а затем в дни 8, 15, 22, 29, 43 и 57 (±1 день в случае дней 15-25; ±2 дня в случае дней 29-57) после введения mAb1. Измерение общего уровня IgE и TARC в сыворотке крови выполнялось с использованием валидированных методов.
C. Статистические методы
Оценка безопасности выполнялась путем изучения отдельных значений и описательных статистических показателей. Все нежелательные явления кодировались согласно ред. 15.1 MedDRA, а частота возникающих во время лечения нежелательных явлений (ВЛНЯ) подсчитывалась (количество и процентная доля) по основным системно-органным классам, предпочтительным терминам и группам лечения. Данные о потенциально клинических значительных отклонениях клинических лабораторных показателей, основных показателей жизнедеятельности организма, показателей ЭКГ, а также выходящие за пределы нормы показатели клинических лабораторных анализов помечали и указывали в виде сводных данных по группам лечения. Также с помощью описательной статистики были сведены исходные данные и изменения основных показателей жизнедеятельности, показателей ЭКГ и отдельных показателей лабораторных анализов относительно исходных значений.
По каждой группе лечения были сведены фармакокинетические показатели функционального mAb1 в сыворотке с применением описательной статистики (среднее, среднее геометрическое, стандартная ошибка среднего [СОС], медиана, стандартное отклонение [СО], коэффициент вариации [КВ], минимум и максимум). Пропорциональность доз изучали с помощью показателей Cмакс, ППКпосл и ППК с применением модели мощности. Влияние дозы на t½z изучали с применением линейной модели с фиксированными эффектами. Распределение значений tмакс проиллюстрировали с помощью гистограммы. Данные о ФД биомаркерах mAb1 (общий IgE и TARC: CCL17) были сведены по каждой группе лечения с применением описательной статистики.
D. Результаты в части безопасности
Однократные подкожные дозы mAb1 в 600 мг хорошо переносились здоровыми пациентами мужского пола из Японии с медианным показателем массы тела в 65,1 кг. В ходе этого исследования не отмечались ни серьезные ВЛНЯ, ни досрочное прекращение участия. В ходе 57-дневного периода наблюдения, следовавшего за введением препарата, среди 32 пациентов исследования наблюдалось 3 ВЛНЯ: у 1 из 8 пациентов группы приема плацебо (грипп), у 1 из 6 пациентов группы приема 150 мг (грипп) и у 1 из 6 пациентов группы приема 600 мг (ортостатическая гипотензия).
При применении объемов до 2,0 мл в 4 местах введения (600 мг) не наблюдалось местных кожных реакций и дискомфорта в месте введения препарата.
У 5 из 32 пациентов наблюдался положительный результат теста на антитела к mAb1, и значения титров были низкими (1 пациент в группе приема 75 мг, 2 - в группе приема 150 мг, 1 - в группе приема 300 мг, 1 - в группе приема 600 мг). Антитела к препарату не определялись ни у одного пациента в исходной точке и ни у одного пациента в группе приема плацебо. Ни у одного пациента с положительным результатом теста на антитела к препарату не наблюдалось никакого ВЛНЯ.
В группе приема mAb1 отмечалось очень низкое количество ПКЗО показателей общих и биохимических анализов крови, и при этом связи между дозой и частотой возникновения отклонений не наблюдалось. Особо стоит отметить, что не наблюдалось изменений по части печеночных ферментов. Наблюдалось малое количество ПКЗО основных показателей жизнедеятельности и показателей ЭКГ, и связи с дозой не отмечалось. В ходе исследования ни у одного из пациентов не наблюдалось удлинение интервала QTcB (>450 мс), и не наблюдалось изменений относительно исходного уровня более чем на 60 мс.
E. Результаты в части фармакокинетики
На фиг. 1 приведены средние показатели концентрации функционального mAb1 в сыворотке крови относительно времени после применения однократных подкожных доз. В таблице 5 сведены фармакокинетические показатели функционального mAb1 в сыворотке крови всех пациентов, получавших mAb1.
Среднее ±СО (среднее геометрическое) [КВ %] фармакокинетических показателей mAb1 в сыворотке крови
75 мг
150 мг
300 мг
600 мг
(3,00-7,03)
(3,00-7,03)
(6,99-10,00)
(3,00-7,02)
(14,01-21,05)
(21,01-24,04)
(35,00-42,02)
(42,00-56,02)
b N=4 т.к. поскольку для 2 пациентов невозможно было представить конечную фазу элиминации/выведения в линейно-логарифмических координатах
Медиана показателя tмакс mAb1 при любой дозе составляла 7 дней. Средний показатель периода полувыведения (t½z) был дозозависимым (p<0,01) и варьировался от 2,77 дней при дозе 75 мг до 8,77 дней при дозе 600 мг. Восьмикратное увеличение дозы от 75 до 600 мг приводило к 13,1-кратному, 30,4-кратному и 24,2-кратному увеличению среднего геометрического Смакс, ППКпосл и ППК соответственно.
F. Результаты в части фармакодинамики
Между различными группами лечения наблюдалась сильная разница по показателям сывороточного IgE и TARC. При однократном подкожном введении доз в 75 и 150 мг не наблюдалось какого-либо связанного с препаратом воздействия на сывороточный IgE на протяжении отрезка времени (изменение в процентах относительно исходной точки). При дозах в 300 и 600 мг наблюдалась тенденция к снижению сывороточного IgE после введения препарата. Лечение оказывало воздействие на TARC. При введении однократных подкожных доз от 75 до 600 мг - в отличие от плацебо - наблюдалось снижение сывороточного TARC. При повышении дозы наблюдалось более устойчивое снижение.
G. Выводы
Однократные подкожные дозы mAb1 до 600 мг хорошо переносились здоровыми пациентами мужского пола из Японии. В ходе этого исследования не отмечались ни серьезные ВЛНЯ, ни досрочное прекращение участия. У 32 пациентов исследования было зарегистрировано 3 ВЛНЯ. При применении объемов до 2,0 мл в 4 местах введения (600 мг) не наблюдалось местных кожных реакций и дискомфорта в месте введения препарата. В целом, наблюдавшиеся ВЛНЯ, а также показатели лабораторных анализов, основные показатели жизнедеятельности и показатели ЭКГ не дают основания предполагать наличие дозозависимых эффектов.
После введения однократных доз здоровым пациентам мужского пола из Японии медианный показатель абсорбции mAb1 tмакс составлял 7 дней, а средний дозозависимый показатель периода полувыведения (t½z) варьировался от 2,77 дней при дозе в 75 мг до 8,77 дней при дозе в 600 мг. Средний показатель экспозиции функционального mAb1 в сыворотке крови увеличивался быстрее чем дозопропорционально: восьмикратное увеличение дозы от 75 до 600 мг приводило к 13,1-кратному, 30,4-кратному и 24,2-кратному увеличению среднего геометрического Смакс, ППКпосл и ППК соответственно.
Наблюдался фармакодинамический эффект. После введения mAb1 уровень сывороточного TARC снижался. При повышении дозы наблюдалось более устойчивое снижение. У 5 из 32 пациентов наблюдались низкие значения титров антител к препарату. Антитела к препарату не определялись ни у одного пациента в исходной точке и ни у одного пациента в группе приема плацебо. Ни у одного пациента с положительным результатом теста на антитела к препарату не наблюдалось никаких ВЛНЯ.
Пример 5: Клиническое исследование для изучения воздействия скорости инъекции на безопасность и переносимость mAb1 при подкожном введении здоровым волонтерам
A. Обзор и дизайн исследования
Данное исследование проводилось для обоснования разработки инъекционного устройства большого объема, предназначенного для введения mAb1. В исследовании с помощью сравнения двух разных скоростей инъекции прогнозировались соответствующие характеристики двух разных устройств для подкожного введения: быстрая инъекция соответствовала автоинъектору, медленная - инфузионной микро-помпе. Основной целью исследования было сравнить безопасность и переносимость однократной дозы mAb1 в 300 мг (2 мл) при подкожном введении с двумя разными скоростями здоровым волонтерам. Второстепенными целями исследования были: сравнить фармакокинетические (ФК) характеристики однократной дозы mAb1 в 300 мг (2 мл) при подкожном введении с двумя разными скоростями здоровым волонтерам, распределенным по двум раздельным когортам, а также сравнить характеристики иммуногенности однократной дозы mAb1 в 300 мг (2 мл) при подкожном введении с двумя разными скоростями здоровым волонтерам.
Исследование было открытым, рандомизированным, проводилось в параллельных группах с применением однократных доз для изучения безопасности, переносимости, ФК и иммуногенности mAb1 при п/к введении с двумя разными скоростями. Исследование было рандомизированным для того, чтобы избежать какой-либо возможной предвзятости при распределении пациентов по группам лечения и минимизировать систематические различия между группами лечения по части исходных переменных во избежание искажения результатов. Маскировка метода и продолжительности инъекции не была возможной, поэтому исследование было открытым. В рамках одного центра в США был осуществлен набор и рандомизация 36 пациентов (по 18 в каждую группу лечения). Объем выборки для данного исследования был определен эмпирическим путем. Формальный объем выборки или расчет мощности на основе первичной конечной точки не использовались. Однако было рассчитано, что набор 18 пациентов в каждую группу обеспечит 80%-ную мощность для определения разницы показателя боли по шкале ВАШ в двух группах лечения в 20 очков, если исходить из того, что обычное стандартное отклонение показателя боли по ВАШ составляет 20,8 при двустороннем уровне значимости 0,05.
Пациенты проходили скрининг между днем -14 и днем -2. В день -1 пациенты ложились в больницу для тренинга и ознакомления с процедурой введения препарата, и они были рандомизированы либо в группу 1 (быстрая инъекция), либо в группу 2 (медленная инъекция):
Группа 1 (быстрая инъекция): пациенты получали исследуемый препарат путем мануальной подкожной инъекции продолжительностью 30 секунд.
Группа 2 (медленная инъекция): пациенты получали исследуемый препарат путем применения набора для подкожных инфузий, подсоединенного к шприцевому насосу, запрограммированному на введение 2 мл на протяжении 10 минут.
В день -1 пациентам была сделана «холостая» инъекция, в ходе которой набор для п/к инфузий на непродолжительное время присоединялся к поверхности кожи. Процесс подразумевал введение 6-миллиметровой иглы 27G на 10-15 секунд, после чего она изымалась. Пациенты оценивали свою боль/дискомфорт от каждого этапа «холостой» инъекции следующим образом:
Сразу же (в течение 10 секунд) после введения иглы и изъятия иглы пациенты оценивали свою боль/дискомфорт от каждого этапа процедуры.
Общая оценка (ОО): Через 1 минуту после изымания иглы пациентов просили дать ОО, вспомнив и оценив боль/дискомфорт, испытанный в ходе всей процедуры.
Сравнительная оценка (СО): приблизительно через 1 минуту после изымания иглы (сразу же после ОО), в дополнение к оценке по визуальной аналоговой шкале (ВАШ), пациентов просили дать СО путем соотнесения их общей боли/дискомфорта со знакомыми ощущениями, например укусом пчелы или прививкой от гриппа.
В день 1 всем пациентам вводили 300 мг mAb1 в объеме 2 мл, и они дали оценку по ВАШ согласно графику на фиг. 2.
По всем пациентам (групп 1 и 2) отслеживались данные о частоте возникновения, объеме и степени реакций в месте инъекции (РМИ), таких как эритема, отек, уплотнение, чувствительность и зуд. Объем (наибольший диаметр в мм) эритемы, отека и уплотнения, а также степень эритемы и отека оценивались через 1, 2, 4 и 8 часов после инъекции, а также в ходе визитов последующего наблюдения вплоть до окончания исследования или до того, как место инъекции имело нормальное состояние при 2 последовательных оценках согласно всем отслеживаемым параметрам. Также пациентов просили оценить степень кожного зуда и чувствительности (боли при нажатии) с использованием ВАШ.
В день 2 пациентов выписывали из клиники домой. Пациенты приходили в клинику на амбулаторные визиты для последующего наблюдения в дни 4, 8, 11, 15, 22, 29, 36, 43, 50, 57 и 64 (окончание исследования). Визиты в клинику в дни 8, 11 и 15 допускались с временным отклонением в +/- 1 день. Визиты в клинику в дни 22-64 допускались с временным отклонением в +/- 2 дня. Период наблюдения каждого пациента итого составлял 9 недель от введения препарата в день 1.
В. Анализ данных и статистические методы
Были сведены следующие демографические и исходные характеристики: возраст при скрининге (год), пол, этническая принадлежность, расовая принадлежность, исходная масса тела (кг), рост (м) и ИМТ (кг/м2), боль/дискомфорт согласно ВАШ. Основные переменные включали в себя следующие показатели безопасности и переносимости: (i) частота возникновения и степень связанных возникающих во время лечения нежелательных явлений (ВЛНЯ) до дня 64 (окончание исследования); частота возникновения, объем, степень и продолжительность РМИ до дня 64; (ii) общая боль/дискомфорт в связи с инъекцией (ОО); (iii) отдельные составляющие боли/дискомфорта при введении иглы, при введении исследуемого препарата и при изымании иглы; и (iv) остаточная боль/дискомфорт на протяжении временного отрезка: текущая боль/дискомфорт через 5, 10, 15 и 30 минут и через 1, 2, 4 и 8 часов после введения препарата и на последующих визитах в рамках исследования.
По всем пациентам (групп 1 и 2) отслеживались данные о частоте, объеме и степени тяжести РМИ, таких как эритема, отек, уплотнение, чувствительность и зуд. Оценивался объем (наибольший диаметр в мм) эритемы, отека и уплотнения. Также степень тяжести эритемы и отека оценивалась количественно с помощью стандартной шкалы оценки переносимости явлений на коже (Draize, от 0 до 4) через 1, 2, 4 и 8 часов после инъекции, а также в ходе визитов последующего наблюдения вплоть до окончания исследования или до того, как место инъекции имело нормальное состояние при 2 последовательных оценках согласно всем отслеживаемым параметрам.
Для оценки степени тяжести эритемы и отека использовались следующие баллы:
Эритема:
0 = отсутствие эритемы
1 = очень легкая эритема (почти незаметная)
2 = хорошо выраженная эритема
3 = эритема в степени от умеренной до тяжелой
4 = тяжелая эритема («свекольная» краснота) с образованием струпов (глубокие поражения)
Отек:
0 = отсутствие отека
1 = очень легкий отек (почти незаметный)
2 = легкий отек (края хорошо выражены)
3 = умеренный отек (приподнятость > 1 мм)
4 = тяжелый отек (приподнятость > 1 мм, распространяется за границы подверженного воздействию участка)
ВАШ - это непрерывная шкала (0-100 мм), с помощью которой пациенты оценивают свою боль/дискомфорт от инъекции исследуемого препарата. Левый край ВАШ обозначен как «отсутствие боли», а правый край - как «самая(ый) сильная(ый) боль/дискомфорт, которая(ый) возможна(ен)». Эта же шкала использовалась для количественной оценки зуда и чувствительности - параметров, которые оценивались в рамках РМИ.
Безопасность и переносимость mAb1 оценивали посредством физикальных осмотров, измерения основных показателей жизнедеятельности, снятия электрокардиограмм (ЭКГ) и выполнения лабораторных анализов. Пациентов просили отслеживать и сообщать о всех нежелательных явлениях (НЯ), наблюдавшихся с момента подписания формы информированного согласия до визита в связи с окончанием исследования в день 64. Определение нежелательных явлений, серьезных нежелательных явлений и возникших во время лечения нежелательных явлений дано в иных местах этого документа.
Образцы крови для фармакокинетических анализов брали на каждом визите в рамках исследования, начиная с исходной точки (день 1, до введения препарата и после введения препарата [в конце инъекции и через 1, 2, 4, 8 и 12 часов после инъекции]). Образцы крови для определения уровня антител к mAb1 брали в день 1 (до введения препарата) и в дни 29 и 64 (окончание исследования).
Непрерывные переменные анализировали с использованием следующих параметров описательной статистики: количество включенных в расчет пациентов (n), среднее, медиана, стандартное отклонение, минимум и максимум. В случае категориальных и порядковых данных указывали показатели частоты возникновения и процентные показатели по каждой категории.
C. Результаты
Боль в месте инъекции и остаточная боль: Оба инъекционных метода - быстрой и медленной инъекции - хорошо переносились, и наблюдался относительно низкий уровень боли при введении препарата. В обоих методах максимальная боль наблюдалась через 15-30 секунд после начала инъекции. Средние пиковые значения боли по шкале ВАШ (0-100 мм) составляли менее 15 мм. Показатели среднего балла по результатам оценки боли, включая общую оценку (впечатления об общей боли через 1 минуту после инъекции), а также оценку остаточной боли относительно времени, были схожими при быстрых и медленных инъекциях; наблюдавшиеся различия не имели клинически релевантный характер (т.е. Δ < 10 по шкале ВАШ (0-100 мм)). При медленных инъекциях большее количество пациентов, в сравнении с быстрыми инъекциями, сообщало об отсутствии или низком уровне боли (ВАШ < 5 мм). В целом при медленных инъекциях наблюдались незначительно более благоприятные характеристики по части боли при инъекции, но это не позволяет однозначно дифференцировать два инъекционных метода.
Реакции в месте инъекции: Показатели частоты возникновения РМИ в двух группах исследования в целом были схожими (89% в группе быстрых инъекций и 94% в группе медленных инъекций). Однако частота возникновения объективных РМИ (эритема и (или) уплотнение) была выше в группе медленных инъекций (83%; для сравнения: в группе быстрых инъекций - 44%), особенно что касается эритемы в месте инъекции (61% и 11% соответственно). Субъективные РМИ включали в себя чувствительность и кожный зуд в месте инъекции, и частота их возникновения была немного выше в группе быстрых инъекций (72%; для сравнения: в группе медленных инъекций - 56%), особенно что касается чувствительности в месте инъекции (72% и 39% соответственно). Начало наступления РМИ приходилось на период от 1 часа до нескольких дней после инъекции. Для разрешения РМИ был необходим период от 1 часа до нескольких дней после начала РМИ. В целом характеристики по части РМИ были немного более благоприятными в группе быстрых инъекций, но это не позволяет однозначно дифференцировать два инъекционных метода.
Нежелательные явления: Количество и частота возникновения возникающих во время лечения нежелательных явлений были выше в группе медленных инъекций (35 ВЛНЯ у 15 пациентов), чем в группе быстрых инъекций (19 ВЛНЯ у 11 пациентов). Большинство ВЛНЯ представляли собой РМИ, которые регистрировались как нежелательные явления по результатам оценки клинической релевантности исследователем. ВЛНЯ не приводили к прекращению пациентами участия в исследовании. Наблюдалось 3 серьезных ВЛНЯ, связанных с одним и тем же случаем, и они не были связаны с исследуемым препаратом или инъекционным методом. В целом, немного более благоприятные характеристики по части нежелательных явлений наблюдались в группе быстрых инъекций, и это по большей части обуславливалось тем, что РМИ регистрировались как нежелательные явления.
D. Выводы
Первичные и вторичные цели, указанные в протоколе исследования, были достигнуты. Препарат mAb1 был безопасным и хорошо переносился и при быстрых, и при медленных инъекциях. По результатам исследования не следует однозначной дифференциации двух инъекционных методов.
Пример 6: Клиническое исследование с применением последовательно возрастающих повторных подкожных доз антител к IL-4Р (mAb1) с участием пациентов c атопическим дерматитом в степени от умеренной до тяжелой
A. Дизайн исследования
Это исследование было рандомизированным, двойным слепым, плацебо-контролируемым исследованием фазы 1b с применением последовательно возрастающих повторных подкожных доз mAb1 с участием пациентов с экзогенным атопическим дерматитом (АД) в степени от умеренной до тяжелой. В исследовании было рандомизировано 30 пациентов (6 - в группу плацебо, по 8 - в группы приема 75, 150 и 300 мг). Лечение получили 28 пациентов. Продолжительность периода лечения составила 4 недели; последующее наблюдение пациентов длилось 8 недель после окончания периода лечения. Методом рандомизации пациенты были распределены в пропорции 4:1 по группам приема mAb1 и плацебо. Были сформированы 3 когорты с возрастающими дозами mAb1 (75, 150 и 300 мг). Основной целью исследования было изучение безопасности и переносимости. Второстепенной целью было изучение ФК. Поисковыми целями было изучение эффективности и биомаркеров. Поисковые переменные эффективности включали в себя: (i) доля пациентов, у которых балл по шкале IGA составлял от 0 до 1 очка к неделе 4 и на каждом визите в рамках исследования; (ii) изменение и процентное изменение - к каждому визиту относительно исходного уровня - показателя BSA, индекса EASI и балла по опроснику из 5 вопросов для оценки кожного зуда; и (iii) еженедельное изменение балла по шкале NRS.
В. Переменные эффективности
Переменные эффективности - балл по шкале IGA, показатель BSA, индекс EASI, индекс SCORAD, балл по опроснику из 5 вопросов для оценки кожного зуда - описаны в иных частях настоящего документа (например, см. пример 7).
Балл по шкале IGA, показатель BSA, индекс EASI и индекс SCORAD измеряли на каждом визите в клинику. Опросник из 5 вопросов для оценки кожного зуда использовался на следующих визитах: скрининг, день 1/исходная точка (до введения препарата) и дни 15, 29, 43, 57, 71 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании. Пациенты протоколировали свой балл по шкале оценки кожного зуда NRS два раза в день до последнего визита в рамках исследования.
Исходным показателем переменной эффективности является последнее имеющееся в наличии значение на дату рандомизации или до такой даты. Если по пациенту на дату рандомизации или до такой даты не имеется значений, то исходной точкой считается последний имеющийся в наличии показатель на день первой дозы или до такого дня.
C. Статистические методы
Показатели безопасности и поисковые переменные эффективности сводились по группам лечения и в совокупности. Сведение показателей безопасности и переносимости осуществлялось на основе популяции для анализа безопасности. Анализы безопасности были основаны на зафиксированных нежелательных явлениях (НЯ), результатах клинических лабораторных анализов, основных показателях жизнедеятельности организма и показателях ЭКГ в 12-ти отведениях.
Все категориальные переменные анализировали с помощью точного критерия Фишера с указанием номинального значения p и доверительных интервалов.
Все непрерывные переменные анализировали с помощью ковариационного анализа (ANCOVA). Если не указано иное, оценка изменений относительно исходного уровня и построение доверительных интервалов непрерывных параметров основывались на модели ANCOVA, в которой лечение было основным фактором, а исходные показатели - независимыми переменными. Указывалась точечная оценка и 95%-ный ДИ разницы наблюдавшихся в двух группах лечения скорректированных средних показателей изменений относительно исходного уровня. По причине небольшого размера популяции исследования в описательных целях указывались значения p по результатам анализов поисковых переменных эффективности. Отсутствующие значения были подставлены методом переноса данных последнего наблюдения (LOCF).
D. Распределение пациентов
Пациенты в группе приема плацебо были самыми молодыми. Тридцать три процента пациентов в группе приема плацебо были испаноговорящими или латиноамериканцами. В других группах лечения таких пациентов не было. В таблице 6 сведены демографические характеристики пациентов.
Обзор демографических характеристик
В таблице 7 сведены исходные характеристики заболевания среди популяции пациентов.
Исходные характеристики заболевания
Участники исследования демонстрировали следующие средние исходные значения: балл по шкале IGA - 3,8, индекс EASI 28,2, показатель BSA - 48,5, балл по шкале для оценки кожного зуда NRS - 6,4.
E. Результаты
Подкожное введение mAb1 пациентам с АД в степени от умеренной до тяжелой было безопасным и хорошо переносилось в рамках этого исследования. Было зарегистрировано одно серьезное нежелательное явление у пациента в группе приема 150 мг: у него было зафиксировано связанное с физической нагрузкой повышение уровня КФК. Смертельных исходов не было. У 25 из получивших лечение пациентов (83%) наблюдалось хотя бы одно возникшее во время лечения нежелательное явление (ВЛНЯ). Наиболее частыми ВЛНЯ в группах лечения mAb1 были инфекции и инвазии (n=7 [29%] относительно 1 [17%] в группе плацебо), а также головные боли (n=3 [13%] относительно 1 [17%] в группе плацебо).
На фиг. 3-14 сведены исходные показатели и поисковые показатели эффективности по результатам исследования. Введение mAb1 не индуцировало статистически значительное улучшение параметров АД в каких-либо поисковых конечных точках. Это может быть обусловлено малым размером популяции или тем фактом, что в группе плацебо пациенты имели более легкую степень заболевания и были моложе, чем в группе активного лечения.
Пример 7: Клиническое исследование с подкожным введением антител к IL-4Р (mAb1) пациентам с атопическим дерматитом в степени от умеренной до тяжелой
A. Дизайн исследования
Это исследование было 12-недельным, двойным слепым, рандомизированным, плацебо-контролируемым исследованием с применением последовательно возрастающих повторных доз для изучения безопасности и фармакокинетических характеристик подкожных доз антител к IL-4Р (далее - «mAb1»), вводимых взрослым пациентам с атопическим дерматитом в степени от умеренной до тяжелой. У пациентов с АД в степени от умеренной до тяжелой наблюдался индекс распространенности и тяжести экземы (Eczema Area and Severity Index (EASI)) ≥ 12, и было поражено не менее 10% поверхности кожи. Продолжительность периода лечения составила 4 недели; последующее наблюдение пациентов длилось 8 недель после окончания периода лечения. На протяжении периода не менее 1 недели, предшествующей исходной точке, пациенты не должны были применять топические средства (например, пимекролимус, такролимус, топические кортикостероиды). Также в период ≥ 4 недель, предшествующих исходной точке, не допускалось применение пероральных кортикостероидов и иммуносупрессивных средств (например, циклоспорина, микофенолат-мофетила, ИФН-гамма).
Пациенты были распределены методом рандомизации в пропорции 3:1 по группам приема mAb1 и плацебо. Были сформированы 2 когорты с возрастающими дозами препарата (150 и 300 мг). Исследование состояло из периода скрининга (от дня -14 до дня -3), периода лечения (от дня 1 до дня 29) (применение топических стероидных препаратов не допускалось) и периода последующего наблюдения (от дня 29 до дня 85) (применение топических стероидных препаратов допускалось). В ходе периода лечения пациенты приходили в клинику не реже одного раза в неделю для проверки параметров безопасности, клинических эффектов и выполнения лабораторных анализов - в дни 1, 4, 8, 15, 22, 25 и 29 (неделя 4). Пациенты получали дозу исследуемого препарата в дни 1, 8, 15 и 22. После приема каждой дозы исследуемого препарата пациентов наблюдали в исследовательском центре на протяжении 2 часов. Визит в связи с окончанием периода лечения приходился на день 29 (неделя 4). В ходе периода последующего наблюдения пациенты приходили в клинику на визиты для последующего наблюдения - в дни 36, 43, 50, 57, 64, 71 и 85 (визит в связи с окончанием исследования).
В. Переменные эффективности
В этом исследовании измерялись следующие поисковые переменные эффективности: (1) доля пациентов, которые к неделе 4 и на каждом визите в рамках исследования демонстрировали балл по итогам общей оценки исследователем (IGA) от 0 до 1 очка; (2) изменение и процентное изменение, между исходной точкой и каждым визитом, показателя площади кожи, пораженной атопическим дерматитом (BSA), индекса площади экзематозного поражения кожи и тяжести экземы (EASI), индекса SCORAD и балла по опроснику из 5 вопросов для оценки кожного зуда; (3) еженедельное изменение относительно исходной точки балла по числовой шкале для оценки кожного зуда (NRS); (4) изменение, между исходной точкой и неделей 4, показателей циркулирующих эозинофилов, TARC, эотаксина-3 и общего IgE; (5) изменение, между исходной точкой и неделей 12, показателей циркулирующих эозинофилов, TARC, эотаксина-3 и общего IgE; и (6) связываемое с ответом на препарат изменение, между исходной точкой и неделей 4, показателей циркулирующих эозинофилов, TARC, эотаксина-3, результатов теста Phadiatop™ и общего IgE.
Исходным показателем переменной эффективности является последнее имеющееся в наличии значение на дату рандомизации или до такой даты. Если по пациенту на дату рандомизации или до такой даты не имеется значений, то исходной точкой считается последний имеющийся в наличии показатель на день первой дозы или до такого дня.
Общая оценка исследователем (IGA): Шкала IGA представляет собой используемую в клинических условиях для оценки тяжести АД и клинического ответа на лечение 6-балльную шкалу со значениями от 0 (отсутствие воспалительных признаков атопического дерматита) до 5 (очень тяжелое заболевание). Балл по шкале IGA измерялся на каждом визите в клинику.
Площадь поверхности кожи, пораженной атопическим дерматитом (BSA): Определялась площадь пораженной атопическим дерматитом поверхности кожи каждой крупной части тела (голова, туловище, верхние и нижние конечности), и показатель BSA рассчитывался как сумма процентных значений по всем частям тела. Показатель BSA у пациентов измерялся на следующих визитах: скрининг, день 1/исходная точка (до введения препарата) и дни 15, 29, 36, 43, 57, 71 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании.
Индекс площади экзематозного поражения кожи и тяжести экземы (EASI): Индекс EASI представляет собой валидированный параметр, используемый в клинической практике и клинических исследованиях для оценки тяжести и распространенности АД (Hanifin и др., 2001 г., Exp. Dermetol. 10: 11-18). Индекс EASI рассчитывается на основании «оценки врачом отдельных симптомов» (Physician's Assessment of Individual Signs) [эритема (E), уплотнение/папула (I), экскориация (X) и лихенизация (L)]: по каждому симптому выставляется балл: 0 (отсутствует), 1 (в слабой степени), 2 (в умеренной степени) или 3 (в тяжелой степени), - а также на основании показателя площади [на основании показателя площади пораженной поверхности кожи в процентах]: 0=0% BSA, 1=1-9% BSA, 2=10-29% BSA, 3=30-49% BSA, 4=50-69% BSA, 5=70-89% BSA, 6=90-100% BSA.
По каждой крупной части тела (голова, туловище, верхние конечности и нижние конечности) индекс EASI рассчитывается как сумма (E+I+X+L), умноженная на показатель площади. Итоговый индекс EASI равняется взвешенной сумме индексов EASI по частям тела, где голова = 10%, верхние конечности = 20%, туловище = 30%, нижние конечности = 40%. Минимальный возможный индекс EASI составляет ноль, максимальный возможный - 72. Более высокий балл означает более тяжелую степень атопического дерматита. Если пациент достигает индекса EASI 50 (улучшение индекса EASI на 50 или более процентов), то дерматологическими исследователями это считается настолько клинически значительным улучшением, что индекс EASI 50 можно использовать в качестве конечной точки.
Оценка пациентов по индексу EASI выполнялась на следующих визитах: скрининг, день 1/исходная точка (до введения препарата) и дни 15, 29, 36, 43, 57, 71 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании.
SCORAD: SCORAD - это применяемый в клинических исследованиях и клинической практике валидированный параметр, разработанный в целях стандартизации оценки распространенности и тяжести АД (Dermatology 1993, 186: 23-31). Распространенность АД устанавливается путем оценки того, какая процентная доля каждой определенной части тела поражена заболеванием. Соответствующий балл представляет собой сумму показателей по всем частям тела. Максимально возможный балл - 100% (эквивалентно баллу «А» в общем подсчете по SCORAD). Степень тяжести 6 конкретных симптомов АД (эритема, отек/уплотнение, экскориация, лихенизация, выделения/корочки и сухость) оценивается по следующей шкале: отсутствие (0), в легкой степени (1), в умеренной степени (2) или в тяжелой степени (3) (максимально возможный балл - 18, что эквивалентно баллу «B» в общем подсчете по SCORAD). Пациент или родственник с помощью визуальной аналоговой шкалы (ВАШ) выполняет субъективную оценку степени кожного зуда и бессонницы в связи с каждым симптомом: 0 - нет зуда (или бессонницы), 10 - самый сильный зуд (бессонница), который(ую) можно представить. Максимально возможный балл - 20. Он эквивалентен баллу «С» в общем подсчете по SCORAD. Индекс SCORAD рассчитывается как А/5+7В/2+С. Максимально возможный индекс SCORAD - 103.
Оценка пациентов по индексу SCORAD выполнялась на следующих визитах: скрининг, день 1/исходная точка (до введения препарата) и дни 15, 29, 36, 43, 57, 71 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании.
Опросник из 5 вопросов для оценки кожного зуда: Опросник из 5 вопросов для оценки кожного зуда используется в клинических исследованиях для оценки 5 параметров имеющегося кожного зуда: интенсивность зуда, длительность зуда, изменение интенсивности зуда, влияние зуда на жизнедеятельность пациента и локализация зуда (Elman и др., 2010 г., Brit. J. Dermatol. 162: 587-593). Пациенты оценивают свои симптомы за предшествующие 2 недели как «присутствующие» или по шкале от 1 до 5, где «5» означает наибольшую степень относительно каждого вопроса по части интенсивности, длительности, изменения интенсивности и влияния на жизнедеятельность. Балл по односложным параметрам опросника (длительность, интенсивность и изменение интенсивности) равняется числовому значению, выбранному пациентом (в диапазоне от 1 до 5).
Параметр опросника «влияние на жизнедеятельность» состоит из четырех пунктов, с помощью которых оценивается влияние кожного зуда на повседневную деятельность: сон, досуг/общение, домашние/хозяйственные дела и работу/учебу. Балл по параметру «влияние на жизнедеятельность» - это самый высокий балл из тех, что были выставлены по четырем пунктам параметра.
Балл по параметру «изменение интенсивности» рассчитывается путем подсчета количества пораженных заболеванием частей тела (сумма может составлять от 0 до 16), и затем полученная сумма приравнивается к определенному баллу в зависимости от того, в каком диапазоне она находится: сумма от 0 до 2=1 очко, сумма от 3 до 5=2 очка, сумма от 6 до 10=3 очка, сумма от 11 до 13=4 очка, сумма от 14 до 16=5 очков.
Балл по каждому из пяти параметров выставляется отдельно, а затем все баллы суммируются в итоговый балл. Итоговый балл может составлять от 5 (нет кожного зуда) до 25 (самый сильный кожный зуд).
Опросник из 5 вопросов для оценки кожного зуда использовался на следующих визитах: скрининг, день 1/исходная точка (до введения препарата) и дни 15, 29, 43, 57, 71 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании.
Числовая шкала для оценки кожного зуда NRS: Шкала NRS для оценки кожного зуда включает в себя один вопрос и используется для оценки самого сильного кожного зуда, который наблюдался у пациента в связи с АД в предыдущие 12 часов. Начиная с вечера дня, в который состоялся скрининговый визит, пациенты дважды в день звонят в интерактивную систему голосового ответа и отвечают на следующий вопрос: «Как вы оцениваете момент, в который вы испытывали самый сильный кожный зуд за предыдущие 12 часов, по шкале от 0 до 10, где 0 означает отсутствие зуда, а 10 - самый сильный суд, который можно себе представить?» На скрининговом визите пациентам объясняют, как протоколировать балл по шкале NRS для оценки кожного зуда посредством интерактивной системы голосового ответа, а на всех последующих визитах в клинику пациентов спрашивают, соблюдали ли они указания. Пациенты протоколируют балл по данной шкале два раза в день до последнего визита в рамках исследования.
Исходный балл по шкале NRS рассчитывается как среднее число баллов по шкале NRS, зарегистрированных за период, начиная сразу после скринингового визита и заканчивая временной точкой непосредственно перед исходным визитом. После исходной точки средний балл по шкале IRS за неделю рассчитывается как среднее число баллов по шкале NRS, регистрируемых ежедневно в течение недели (пропорционально распределенное среднее).
В. Оценка безопасности
Безопасность в ходе исследования отслеживалась посредством оценки нежелательных явлений и серьезных нежелательных явлений.
Нежелательным явлением (НЯ) является любое неблагоприятное с медицинской точки зрения явление, наблюдающееся у принимающего фармацевтический препарат пациента или участника клинического исследования. Таким образом, НЯ может представлять собой любой неблагоприятный или непредвиденный признак (включая отклонение показателя лабораторного анализа от нормы), симптом или заболевание, время возникновения которого приходится на время применения лекарственного препарата, вне зависимости от наличия или отсутствия причинно-следственной связи с лекарственным (экспериментальным) препаратом. НЯ также включают: любое ухудшение (то есть клинически значительное изменение частоты и (или) интенсивности) ранее существовавшего заболевания, если время возникновения такого ухудшения приходится на время применения исследуемого препарата; отклонения показателей лабораторных анализов от нормы, если исследователь считает такие отклонения клинически значительными; а также любые неблагоприятные с медицинской точки зрения явления.
Серьезным нежелательным явлением (СНЯ) является любое неблагоприятное с медицинской точки зрения явление, которое, вне зависимости от дозировки, приводит к смертельному исходу или угрожает жизни или приводит к госпитализации или продлении ранее начатой госпитализации или приводит к устойчивой или значительной утрате трудоспособности/инвалидности или является врожденной аномалией/пороком или является важным с медицинской точки зрения явлением.
Также в ходе исследования оценивались лабораторные показатели безопасности, основные показатели жизнедеятельности, показатели электрокардиограммы (ЭКГ) в 12-ти отведениях и результаты физикального осмотра.
Клинические лабораторные данные включают в себя показатели общего и биохимических анализов крови и анализа мочи. Образцы крови для выполнения общего анализа крови брались на всех визитах в рамках исследования; образцы крови для выполнения биохимического анализа сыворотки крови и образцы мочи для выполнения анализа мочи брались для оценки общего состояния здоровья пациентов при скрининге, в день 1/исходной точке (до введения препарата), в дни 8, 15, 29, 36, 57, 85 (окончание исследования) или при досрочном прекращении участия в исследовании.
Основные показатели жизнедеятельности включают частоту дыхательных движений (циклов в минуту), частоту пульса (ударов в минуту), систолическое и диастолическое артериальное давление (мм рт.ст.) и температуру тела (°C). Основные показатели жизнедеятельности измерялись (до введения препарата, в дни приема препарата) при скрининге, в день 1/исходной точке и в дни 4, 8, 15, 22, 25, 29, 36 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании. В дни 1, 8, 15 и 22 основные показатели жизнедеятельности измерялись через 1 и 2 часа после введения исследуемого препарата.
Показатели ЭКГ в 12-ти отведениях включают: желудочковая ЧСС, интервал PR, интервал QRS, скорректированный интервал QT (QTcF=QT/[RR0,33] и QTcB=QT/[RR0,5]) Статус ЭКГ: норма, клинически незначительная патология, клинически значительная патология. Стандартная ЭКГ в 12-ти отведениях выполнялась при скрининге, в дни 29 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании.
Тщательный, полный физикальный осмотр выполнялся при скрининге, в дни 29 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании.
D. Анализ данных
1. Анализы поисковых показателей эффективности
Все категориальные переменные анализировались с помощью точного критерия Фишера с указанием номинального значения p и доверительных интервалов. Все непрерывные переменные были проанализированы с помощью ковариационного анализа (ANCOVA). Если не указано иное, оценка изменений относительно исходного уровня и построение доверительных интервалов непрерывных параметров основывались на модели ANCOVA, в которой лечение было основным фактором, а исходные показатели - независимыми переменными. Указывалась точечная оценка и 95%-ный ДИ разницы наблюдавшихся в двух группах лечения скорректированных средних показателей изменений относительно исходного уровня. Отсутствующие значения были подставлены методом переноса данных последнего наблюдения (LOCF). Если использование предположений по результатам моделирования не являлось оправданным, использовался ранговый ковариационный анализ. Корреляционный анализ выполнялся с использованием коэффициента корреляции Спирмена.
2. Анализ данных по безопасности
Анализ безопасности был основан на зафиксированных НЯ, результатах клинических лабораторных анализов, основных показателях жизнедеятельности организма и показателях ЭКГ в 12-ти отведениях. Пределы потенциально клинически значительных значений (ПКЗЗ) результатов лабораторных анализов, основных показателей жизнедеятельности и ЭКГ установлены в плане статистического анализа. Временной интервал, в который отслеживается наступление каких-либо событий или патологии, приходится на период времени между инфузией исследуемого препарата и окончанием исследования. Данные, поступившие за пределами этого периода времени, не учитываются при расчете описательных статистических показателей и выявлении патологий в результатах лабораторных анализов, основных показателях жизнедеятельности и ЭКГ.
E. Результаты
Как отмечалось выше, пациенты получали подкожно раз в неделю на протяжении четырех недель либо 150 мг mAb1, либо 300 мг mAb1, либо плацебо. Демографические и клинические характеристики участников различных групп лечения были в целом схожими, за исключением того, что участники группы приема 300 мг, в которой диагностировано заболевание, были более старшего возраста (Таблица 8). Популяция исследования в основном состояла из мужчин (62,2%), представителей европеоидной расы (94,6%). Средний возраст участников - 43,6 (15,4) лет. Из 37 пациентов 31 пациент (83,8%) окончил лечение, а 25 пациентов (67,6%) дошли до самого конца исследования. Наиболее частая причина прекращения участия - недостаток эффективности (4 пациента в группе приема плацебо и по одному пациенту в каждой из групп приема исследуемого препарата). Не наблюдалось случаев досрочного прекращения участия в исследовании по причине нежелательных явлений в связи с приемом mAb1.
В таблицах 9-14 сведены исходные показатели и поисковые показатели эффективности по результатам исследования.
Обзор исходных показателей - все значения приводятся как среднее (СО)
Обзор изменений балла по IGA относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений индекса EASI относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений показателя BSA относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений индекса SCORAD относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений (относительно исходного уровня, в процентах и в абсолютном выражении) балла по опроснику из 5 вопросов для оценки кожного зуда - все значения приводятся в виде средних (СО)
Обзор средних еженедельных изменений балла по шкале NRS относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
F. Выводы
Антитела к IL-4Р (mAb1), вводимые подкожно взрослым пациентам с атопическим дерматитом в степени от умеренной до тяжелой, в целом были безопасными и хорошо переносились по результатам применения 4 еженедельных доз в 150 и 300 мг; уровень нежелательных явлений (НЯ) в группах лечения препаратом и в группе лечения плацебо был схожим; не наблюдалось каких-либо дозолимитирующих токсических явлений или серьезных НЯ. Наиболее распространенными НЯ при приеме mAb1 были назофарингит и головная боль. Препарат mAb1 быстро (к 8-му дню) и дозозависимо приводил к уменьшению кожного зуда и улучшению симптомов кожного заболевания. Применение 150 и 300 мг mAb1 приводило к значительному улучшению баллов по шкале IGA, индекса EASI, показателя BSA, индекса SCORAD и балла по шкале NRS для оценки зуда, что выражалось в средних показателях и изменениях относительно исходной точки в процентном и абсолютном выражении (см. таблицы 9-14); и такое улучшение наблюдалось уже к 8-му дню и имело место до 85-го дня. На 29-й день в группе приема 300 мг доля пациентов, достигших ответа согласно EASI50, составляла 71,4%, а в группе приема плацебо - 18,8% (p=0,0025). Балл по шкале NRS для оценки кожного зуда снизился в этих группах на 45,4% и 18,6% соответственно (p=0,0016). Этот эффект продолжался до 85-го дня по наблюдениям согласно EASI50 и до 75-го дня по наблюдениям согласно шкале NRS для оценки кожного зуда. Положительный эффект в группе приема 300 мг, в сравнении с группой приема плацебо, был значительным на протяжении еще 6 недель после окончания периода лечения. Прием mAb к 29-му дню значительно улучшал прочие показатели клинического исхода, такие как среднее изменение (в процентах) балла по шкале IGA (p=0,0002), индекса EASI (p<0,0001), показателя BSA (p=0,0037) и балла по опроснику из 5 вопросов для оценки кожного зуда (p<0,0001). Эти улучшения обычно проявлялись к 8-му дню и сохранялись после окончания лечения. После окончания лечения синдрома отмены не наблюдалось.
Соответственно, результаты этого исследования доказывают, что mAb1 безопасен и эффективен при лечении атопического дерматита.
Пример 8: Лечение пациентов с атопическим дерматитом в степени от умеренной до тяжелой с помощью антител к IL-4Р: объединенный анализ результатов исследований фазы 1б
Результаты двух раздельных клинических исследований в части эффективности лечения пациентов с АД в степени от умеренной до тяжелой были объединены для выполнения анализа. Исследование «А» было 12-недельным, двойным слепым, рандомизированным, плацебо-контролируемым исследованием с применением последовательно возрастающих повторных доз для изучения безопасности и переносимости антител к IL-4Р (mAb1) при применении пациентами с атопическим дерматитом. Продолжительность периода лечения составила 4 недели; последующее наблюдение пациентов длилось 8 недель после окончания периода лечения. Пациентов методом рандомизации распределяли в пропорции 4:1 по группам приема mAb1 и плацебо. Были сформированы 3 когорты с возрастающими дозами mAb1 (75, 150 и 300 мг). Исследование состояло из периода скрининга (со дня -14 по день -3), периода лечения (со дня 1 по день 29) и периода последующего наблюдения (со дня 29 по день 85). В ходе периода лечения пациенты один раз в неделю приходили в клинику для проверки параметров безопасности, клинических эффектов и выполнения лабораторных анализов - в дни 1, 4, 8, 15, 22, 25 и 29 (неделя 4). Пациенты получали дозу mAb1 или плацебо в дни 1, 8, 15 и 22. Визит в связи с окончанием периода лечения приходился на день 29 (неделя 4). В день 1 пациентов наблюдали в исследовательском центре на протяжении 6 часов после инъекции (mAb1 или плацебо), а в дни 8, 15 и 22 - на протяжении 3 часов. В ходе периода последующего наблюдения пациенты приходили в клинику на визиты для последующего наблюдения - в дни 36, 43, 50, 57, 64, 71 и 85 (визит в связи с окончанием исследования).
Исследование «Б» было 12-недельным, двойным слепым, рандомизированным, плацебо-контролируемым исследованием с применением последовательно возрастающих повторных доз для лечения пациентов с АД в степени от умеренной до тяжелой. Пациентам с АД вводилось 150 или 300 мг mAb1 или плацебо в дни 1, 8, 15 и 22 (четыре еженедельных дозы) (см. пример 3 в этом документе). Во всех исследованиях препарат и плацебо вводились подкожно.
Критерии включения в исследования были следующими: (1) пациенты мужского и женского полов в возрасте ≥18 лет; (2) страдают хроническим атопическим дерматитом на протяжении 3 лет; (3) индекс EASI ≥ 12; (4) балл по шкале IGA ≥ 3; (5) ≥ 15% площади поверхности кожи (BSA) подвержено АД - в США; ≥ 10% - за пределами США; и (6) наличие в анамнезе ненадлежащего ответа на стабильный режим лечения топическими кортикостероидами (ТКС) или ингибиторами кальциневрина.
Критерии исключения из исследования были следующими: (1) количество лейкоцитов < 3,5×103/μл; (2) количество тромбоцитов < 125×103/μл; (3) количество нейтрофилов < 1,75×103/μл; (4) АСТ/АЛТ > 1,5×ВПН; (5) положительный результат анализов на гепатит В или гепатит С; и (6) лечение ТКС или ингибиторами кальциневрина в течение 1 недели, предшествующей исходной точке.
Основной конечной точкой исследований была частота возникновения возникших во время лечения нежелательных явлений (ВЛНЯ) за период между исходной точкой и неделей 12. Поисковыми конечными точками по части эффективности были: (i) процент пациентов, у которых к неделе 4 балл по IGA составил 0 или 1 очко; (ii) процент пациентов, у которых наблюдалось улучшение показателя BSA и индекса EASI относительно исходного уровня; и (iii) изменение балла по шкале NRS относительно исходного уровня.
Переменные эффективности - балл по IGA, показатель BSA, индекс EASI, индекс SCORAD, балл по опроснику из 5 вопросов для оценки кожного зуда и балл по шкале для оценки кожного зуда NRS - описаны в иных частях настоящего документа (например, см. пример 4).
Балл по шкале IGA, показатель BSA, индекс EASI и индекс SCORAD измерялись на каждом визите в клинику. Опросник из 5 вопросов для оценки кожного зуда использовался на следующих визитах: скрининг, день 1/исходная точка (до введения препарата) и дни 15, 29, 43, 57, 71 и 85 (окончание исследования) или при досрочном прекращении участия в исследовании. Пациенты протоколировали свой балл по шкале оценки кожного зуда NRS два раза в день до последнего визита в рамках исследования.
Исходным показателем переменной эффективности является последнее имеющееся в наличии значение на дату рандомизации или до такой даты. Если по пациенту на дату рандомизации или до такой даты не имеется значений, то исходной точкой считается последний имеющийся в наличии показатель на день первой дозы или до такого дня.
В таблице 15 ниже приведены исходные демографические характеристики популяции пациентов.
Исходные демографические характеристики
В таблице 16 приводятся средние исходные демографические показатели.
Средние исходные демографические показатели
В таблицах 17-25 и на фигурах 15-22 сведены объединенные данные исследований по части поисковых показателей эффективности.
Обзор данных пациентов, у которых ко дню 29 и ко всем визитам в рамках исследования балл по IGA составлял ≤ 1
Обзор изменений показателя BSA относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений индекса EASI относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений (относительно исходного уровня, в процентах и в абсолютном выражении) балла по опроснику из 5 вопросов для оценки кожного зуда - все значения приводятся в виде средних (СО)
Обзор средних еженедельных изменений балла по шкале NRS относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений балла по IGA относительно исходного уровня в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Количество пациентов (в %), достигших индекса EASI-50 к 29-му дню и по состоянию на каждый визит в рамках исследования - ПДПН
Количество пациентов (в %), достигших индекса EASI-25 к 29-му дню и по состоянию на каждый визит в рамках исследования - ПДПН
Количество пациентов (в %), достигших индекса EASI-75 к 29-му дню и по состоянию на каждый визит в рамках исследования - ПДПН
Препарат mAb1 хорошо переносился и был эффективен при применении взрослыми пациентами с АД в степени от умеренной до тяжелой. Применение mAb1 значительно улучшило показатели активности и степени заболевания. К 4-й неделе у пациентов в группах приема 150 и 300 мг mAb1, в сравнении с пациентами группы приема плацебо, наблюдались значительные улучшения, выразившиеся в изменениях в процентах, относительно исходного уровня, показателя BSA (р<0,05) (фиг. 15), балла по шкале IGA (р<0,001) (фиг. 16), индекса EASI (р<0,001) (фиг. 17) и балла по шкале оценки кожного зуда NRS (р<0,01, 300 мг) (фиг. 18). Количество пациентов, у которых индекс EASI снижался на ≥ 50%, было больше в группах приема 150 и 300 мг mAb1, чем в группе приема плацебо: 54,5%, 71,4% и 18,8% соответственно (р<0,05 для всех групп) (фиг. 19 и 20). На 4-й неделе большее количество пациентов достигли индексов EASI-25, EASI-50 и EASI-75 в группах приема mAb1, чем в группе приема плацебо (фиг. 21).
В группе приема 300 мг mAb1 через две недели проявлялись значительные улучшения (в процентах) показателей BSA (p<0,02), баллов по шкале IGA (р<0,05) и индекса EASI (p<0,0001). Улучшение показателей BSA, баллов по шкале IGA и индекса EASI (р<0,05 в сравнении с плацебо) сохранялось на протяжении 8 недель. В группах приема препарата пропорция пациентов, у которых на 4-й неделе балл по шкале IGA составлял 0 или 1 очко, была выше, чем в группе приема плацебо, но разница не была статистически значительной (фиг. 22).
При приеме mAb1 наиболее распространенными возникающими при лечении нежелательными явлениями (НЯ) были назофарингит (19,6% в сравнении с 12,5% в группе приема плацебо) и головная боль (11,8% в сравнении с 6,3% в группе приема плацебо).
Пример 9: Клиническое исследование с подкожным введением антител к IL-4Р (mAb1) в различных дозировках взрослым пациентам с атопическим дерматитом в степени от умеренной до тяжелой в параллельных группах
A. Дизайн исследования
Данное исследование было 32-недельным, рандомизированным, двойным слепым, плацебо-контролируемым исследованием для изучения зависимости ответа на лечение от дозы при применении еженедельных доз mAb1 взрослыми пациентами с АД в степени от умеренной до тяжелой в параллельных группах. Основной целью исследования было оценить эффективность различных режимов дозирования mAb1 в сравнении с плацебо при применении взрослыми пациентами с АД в степени от умеренной до тяжелой. Вторичными целями были: (1) оценка безопасности различных режимов дозирования mAb1 в сравнении с плацебо при применении взрослыми пациентами с АД в степени от умеренной до тяжелой; (2) оценка фармакокинетики (ФК) различных режимов дозирования mAb1 при применении взрослыми пациентами с АД в степени от умеренной до тяжелой; и (3) оценка потенциального ответа иммунной системы при использовании различных режимов дозирования mAb1 и сравнение с плацебо при применении взрослыми пациентами с АД в степени от умеренной до тяжелой.
Целевая популяция включала взрослых пациентов с АД в степени от умеренной до тяжелой, у которых заболевание не могло быть надлежащим образом сдержано с помощью топических препаратов или их применение не было рекомендовано по другой причине (например, побочные эффекты, связанные с безопасностью риски). В исследование было набрано приблизительно 240-288 пациентов. Соответствующие критериям набора пациенты были методом рандомизации распределены в пропорции 1:1:1:1:1:1 по 6 режимам еженедельного лечения (5 групп - прием активного препарата, 1 группа - прием плацебо). Стратификация при рандомизации выполнялась по признаку степени заболевания (умеренная или тяжелая) и географического региона (Япония и остальные страны). Применявшийся режим дозирования приводится в таблице 26
С 1-й по 15-ю недели дозы состояли из 1 инъекции по 2 мл
300 мг
300 мг
300 мг
200 мг
200 мг
Плацебо
Все пациенты получали в 1-й день 2 инъекции (насыщающая доза), после чего они получали еженедельные инъекции. При режимах дозирования «раз в 2 недели» и «раз в 4 недели» следующая доза исследуемого препарата вводилась на 2-й и 4-й неделе соответственно. Пациенты, распределенные в группы режимов дозирования «раз в 2 недели» и «раз в 4 недели», получали плацебо в идентичном объеме во все недели, в которые препарат mAb1 не применялся. После предоставления информированного согласия пациенты в ходе скринингового визита проходили оценку соответствия критериям набора. Пациенты, соответствовавшие критериям набора, проходили обследования в рамках дня 1 (исходный визит), рандомизацию, а затем получали еженедельные инъекции исследуемого препарата со дня 1 по неделю 15. На протяжении этого времени пациенты приходили в клинику на еженедельные визиты, а в некоторые недели осуществлялся лишь контакт по телефону. Пациентам (и (или) ухаживающим за ним лицам) на визитах 2, 3, 4, 5 и 6 давались указания по введению исследуемого препарата. На последующих визитах, в рамках которых требовался лишь контакт по телефону, введение исследуемого препарата осуществлялось самостоятельно. После каждой из 5 изначальных еженедельных инъекций пациентов тщательно наблюдали в исследовательском центре в течение как минимум 1 часа. В рамках отдельных визитов в клинику выполнялись обследования по части безопасности, лабораторные анализы и оценка клинических эффектов. Визит в связи с окончанием лечения выполнялся на 16-й неделю, т.е. через 1 неделю после введения последней дозы исследуемого препарата, для изучения показателей основной конечной точки. Визиты для последующего наблюдения осуществлялись каждые 2 недели с недели 18 по неделю 32. Визит в связи с окончанием лечения осуществлялся на 32-й неделе. По необходимости участникам исследования, по усмотрению исследователя, предоставлялось лечение для экстренного купирования симптомов АД (лекарственные препараты и (или) светотерапия). В случае если пациент испытывал(а) необходимость в лечении для экстренного купирования симптомов, он(а) немедленно прекращал(а) прием исследуемого препарата, и его (ее) просили по возможности продолжать прохождение запланированных в рамках исследования обследований. Перед применением лечения для экстренного купирования симптомов снимались показатели эффективности (балл по шкале IGA (общая оценка исследователем), индекс EASI (площадь экзематозного поражения кожи и тяжести экземы) и проч.). Если пациент соглашался участвовать в необязательной геномной части исследования, у него (нее) брался один образец для анализа ДНК и несколько образцов для анализа РНК.
Исследуемое лечение: mAb1 подкожно: 300 мг раз в неделю, 300 мг раз в 2 недели, 300 мг раз в 4 недели, 200 мг раз в 2 недели и 100 мг раз в 4 недели начиная со дня 1 оканчивая неделей 15 ИЛИ подкожная доза плацебо раз в неделю начиная со дня 1 оканчивая неделей 15. Со дня -7 по день 8 два раза в день применялось стандартное топическое смягчающее средство без добавок.
Конечные точки исследования: Основной конечной точкой исследования было изменение в процентах индекса EASI за период между исходной точкой и неделей 16. Вторичные конечные точки исследования включали: (1) доля пациентов, у которых к неделе 16 балл по IGA достиг 0 (воспалительные признаки отсутствуют) или 1 очка (воспалительные признаки почти отсутствуют); (2) доля пациентов, у которых к неделе 16 балл по IGA снизился ≥ 2 очка; (3) изменение индекса EASI в абсолютном выражении за период между исходной точкой и неделей 16; (4) доля пациентов, которые к неделе 16 достигли индексов EASI-50, EASI-75 или EASI-90 (50%-ное, 75%-ное и 90%-ное уменьшение индекса EASI в сравнении с исходным уровнем); (6) доля пациентов, которые к неделе 16 достигли индексов SCORAD-50, SCORAD-75 и SCORAD-90 (50%-ное, 75%-ное и 90%-ное уменьшение индекса SCORAD в сравнении с исходным уровнем); (7) изменение баллов по шкалам оценки зуда в абсолютном и процентном выражениях в сравнении с исходным уровнем (шкала NRS и номинальная 4-балльная шкала); (8) изменение баллов по шкале POEM в абсолютном и процентном выражениях в сравнении с исходным уровнем; (9) изменение компонентов шкалы GISS (эритема, уплотнение/папула, экскориация и лихенизация) в сравнении с исходным уровнем; (10) изменение суммарного балла по шкале GISS в сравнении с исходным уровнем; (11) частота возникновения возникающих при лечении нежелательных явлений (ВЛНЯ) за период между исходной точкой и неделей 32; и (12) фармакокинетические характеристики различных режимов дозирования препарата mAb1.
Прочие поисковые конечные точки включали: (1) распределение баллов, отражающих степень заболевания (например, баллы по шкале IGA, индексы EASI, SCORAD) и изменение за период между исходной точкой и различными временными точками до недели 16; (2) изменения баллов по шкале оценки кожного зуда NRS, номинальной 4-балльной шкале оценки кожного зуда, индекса SCORAD (ВАШ для оценки кожного зуда и ВАШ для оценки помех для сна), результатов общей оценки пациентом своего заболевания, общей оценки пациентом эффекта от лечения, баллов по шкалам DLQI, POEM, EQ-5D, опроснику для оценки влияния кожного зуда на качество жизни, опроснику HADS - за периоды времени между исходной точкой и различными временными точками вплоть до недели 16; (3) изменение, в абсолютном и процентом выражениях, процентного показателя BSA, индексов SCORAD, EASI, балла по шкале оценки кожного зуда NRS- за периоды времени между исходной точкой и различными временными точками вплоть до недели 16; (4) доля пациентов, у которых балл по IGA уменьшился на ≥ 2 очка - за периоды времени между исходной точкой и различными временными точками вплоть до недели 16; (5) пропорция пациентов, у которых балл по IGA уменьшился на ≥ 3 очка - за периоды времени между исходной точкой и различными временными точками вплоть до недели 16; (6) изменения показателей эффективности за период между неделей 16 и неделей 32; (7) частота возникновения и характеристики (титры относительно времени) антител к mAb1; (8) влияние концентрации mAb1 в плазме на выработку и устойчивость антител к препарату; (9) влияние антител к препарату на концентрацию mAb1 в плазме; (14) влияния антител к препарату на показатели клинического исхода (безопасность и эффективность); (10) влияние ФК параметров (Смакс и ППК) на показатели клинического исхода; и (11) влияние показателя массы тела на воздействие препарата и показатели клинического исхода.
Обоснование дизайна исследования: Целью этого исследования является нахождение оптимального режима дозирования, который затем подлежит дальнейшему изучению в рамках подтверждающих исследований фазы 3. Дизайн этого исследования фазы 2б был основан на результатах предыдущего исследования препарата mAb1, в котором изучалась безопасность и эффективность mAb1 при введении 300 мг раз в неделю на протяжении 12 последовательных недель пациентам с АД в степени от умеренной до тяжелой. Выбор режимов дозирования для фазы 2б также был продиктован наблюдаемыми и моделируемыми связями между фармакокинетическими (ФК) и фармакодинамическими (ФД) показателями (модели ФК/ФД) по результатам предыдущих клинических исследований. Доза 300 мг раз в неделю (т.е. изучаемый в рамках фазы 2б режим дозирования) была установлена в качестве верхнего предела диапазона доз. Целью исследования было установить режим дозирования с наименьшим содержанием препарата и максимальной (или близкой к максимальной) эффективностью и (или) найти режим дозирования, в котором оптимальное соотносятся преимущества и риски, в зависимости от того, какие характеристики продемонстрирует mAb1 по части безопасности. Соответственно, было выбрано 5 режимов дозирования, которые бы надлежащим образом покрыли необходимый диапазон - от потенциально сверхтерапевтического режима (т.е. верхний предел) до режима с явно субоптимальной эффективностью (т.е. нижний предел). Согласно протоколу также была предусмотрена группа приема плацебо в обеспечение возможности сравнить все режимы дозирования активного препарата с контрольным режимом.
Использование насыщающих доз: В день 1 большинство пациентов получали насыщающую дозу, которая является удвоением обычной дозы, вводимой на последующих визитах. Это позволяет быстрее достигнуть равновесного состояния и целевого показателя системной концентрации mAb1, а также потенциально уменьшить срок времени до наступления клинической пользы. Исследуемое лечение осуществлялось на протяжении 16 недель в обеспечение стабилизации системной концентрации функционального mAb1 при всех исследуемых режимах дозирования. По результатам фармакокинетического моделирования можно предположить, что режим дозирования «раз в 4 недели» может привести к уменьшению остаточной концентрации после введения изначальной насыщающей дозы. Соответственно, иммуногенный потенциал этих режимов дозирования может не полностью проявиться в ходе сокращенного курса лечения. После введения последней дозы исследуемого препарата осуществлялось последующее наблюдение всех пациентов на протяжении еще 16 недель, чтобы удостовериться, что клиренс mAb1 почти завершился (концентрация в плазме ниже нижнего предела количественного определения) до визита в связи с окончанием исследования.
Обоснование выбора дозы: В этом исследовании режимом с самой высокой дозой mAb1 был режим «300 мг раз в неделю». При коротком курсе лечения (4 недели) этот режим дозирования был безопасным и продемонстрировал себя как наиболее эффективный в рамках предыдущих клинических исследований фазы 1б, где он исследовался совместно с режимами с более низкими дозами (150 мг раз в неделю и 75 мг раз в неделю). По результатам фармакокинетического моделирования можно было предположить, что в долгосрочной перспективе режим «300 мг раз в неделю» может стать сверхтерапевтическим: концентрация mAb1 в плазме не достигала равновесного состояния к неделе 4, а стабилизация ожидалась на уровне, который бы был значительно выше уровня, необходимого для насыщения мишени, т.е. мембраносвязанной альфа-субъединицы рецептора IL-4. Однако это нужно было подтвердить путем сравнения режима «300 мг раз в неделю» с режимами с более низкими дозами в контексте более длительного лечения (12 или более недель), чтобы концентрация в плазме могла достичь равновесного состояния при всех исследуемых режимах дозирования. Хотя режим дозирования «300 мг раз в неделю» применялся на протяжении 12 недель в рамках более раннего исследования (на фазе 2а - проверка концепции), этот режим дозирования был вновь исследован на фазе 2б для подтверждения результатов фазы 2а и выполнения прямого сравнения с режимами с более низкими дозами в рамках одного и того же исследования. Таким образом, режим дозирования «300 мг раз в неделю» был верхним пределом дозового диапазона этого исследования.
Нижним пределом дозового диапазона был режим дозирования «100 мг раз в 4 недели». Основываясь на ФК/ФД моделировании ожидалось, что концентрация mAb1 в плазме в равновесном состоянии будет стабильно ниже показателей мишень-опосредованного клиренса (т.е. на уровне, который настолько низок, чтобы обеспечить выведение mAb1 в основном посредством связывания с рецептором IL-4), что позволяло предположить, что клинический ответ на этот режим дозирования является неполным. В диапазоне между нижним и верхним пределом были выбраны еще три режима дозирования. Ниже приводится обзор этих режимов дозирования и обоснование их использования:
300 мг раз в неделю: верхний предел. Тот же режим дозирования изучался на фазе 2а.
300 мг каждые 2 недели: высокая вероятность успешного применения согласно ФК/ФД данным и моделированию. Дозировка может оказаться достаточной для поддержания терапевтического уровня препарата в организме в интервалах между дозами.
300 мг раз в 4 недели: В ходе ФК-моделирования было продемонстрировано, что после введения насыщающей дозы уровень mAb1 в плазме быстро поднимался сверх уровня 60 мг/л, и это связывалось с быстрым наступлением эффекта. Дозировка раз в 4 недели может оказаться достаточной для закрепления терапевтического эффекта на протяжении определенного временного отрезка. Так как доза в 300 мг являлась самой высокой дозой из всех возможных, ее использование в режиме дозирования раз в 4 недели давало наибольшие шансы достигнуть эффективности.
200 мг раз в 2 недели: ожидалась некоторая степень эффективности без достижения максимального терапевтического эффекта. Режим дозирования представляет полезность для изучения ответа на различные дозы и выполнения дальнейшего ФК/ФД-моделирования. Также помогает оценить полный диапазон режимов дозирования раз в 2 недели.
100 мг раз в 4 недели: нижний предел. Скорей всего дает неоптимальную эффективность.
Плацебо: представляет собой надежное сравнительное средство для оценки каких-либо явных эффектов препарата.
Критерии включения и исключения: Для набора в исследование пациент должен был соответствовать следующим критериям: (1) мужчина или женщина в возрасте 18 лет или старше; (2) хронический АД (согласно Единым критериям Американской академии дерматологии, [Eichenfeld 2004]) присутствует не менее 3 лет до скринингового визита; (3) индекс EASI ≥ 16 при скрининге и на исходном визите; (4) балл по шкале IGA ≥ 3 (по шкале от 0 до 4) при скрининге и на исходном визите; (5) ≥ 10% площади поверхности кожи поражено АД (BSA) согласно наблюдениям при скрининге и на исходном визите; (6) у пациента в анамнезе запротоколирован недавний (в течение 3 месяцев, предшествующих скрининговому визиту) ненадлежащий ответ на амбулаторное лечение топическими препаратами, или по другим причинам нежелательно применение пациентом топического лечения (например, серьезные побочные эффекты или связанные с безопасностью риски)*; (7) пациент должен был не менее 7 дней до исходного визита два раза в сутки применять в стабильной дозе стандартное смягчающее средство без добавок; (8) готовность и способность осуществлять все визиты в клинику и проходить все связанные с исследованием процедуры; (9) способность понимать и заполнять предусмотренные исследованием анкеты; и (10) предоставление информированного согласия в письменной форме. *ПРИМЕЧАНИЕ: С позиций данного протокола, ненадлежащим ответом является недостижение стабильной ремиссии или низкой активности заболевания (например, балл по шкале IGA от 0 (воспалительные признаки отсутствуют) до 2 (умеренная степень)) несмотря на применение топических кортикостероидов умеренной до высокой активности (с применением или без применения топических ингибиторов кальциневрина по необходимости) ежедневно на протяжении не менее 28 дней или на протяжении максимального срока, рекомендуемого согласно рецепту (например, в случае сильнодействующих топических кортикостероидов - 14 дней) в зависимости от того, что дольше. Серьезные побочные эффекты и связанные с безопасностью риски - это те, которые по мнению исследователя или лечащего врача пациента перевешивают потенциальную пользу от лечения (например, гиперчувствительные реакции, существенная атрофия кожи, системные эффекты и проч. или неминуемая опасность таковых).
Пациенты, отвечающие какому-либо из нижеследующих критериев, не подлежали набору в это исследование: (1) лечение препаратом mAb1 в прошлом; (2) лечение каким-либо экспериментальным препаратом в период 8 недель, предшествующих исходному визиту, или в предшествующий исходному визиту период, равный 5 периодам полувыведения такого препарата (если эти данные известны) в зависимости от того, что дольше; (3) применение какой-либо перечисленной ниже терапии в период 4 недель, предшествующих исходному визиту (или наличие какого-либо заболевания, которое с вероятностью потребует применение такой терапии в первые 4 недели исследуемого лечения: системные кортикостероиды, иммуносупрессивные/иммуномодулирующие препараты (например, циклоспорин, микофенолат-мофетил, ИФН-гамма, азатиоприн или метотрексат) или светотерапия по поводу АД; (4) лечение топическими кортикостероидами, такролимусом и (или) пимекролимусом в течение 1 недели, предшествующей исходному визиту; (5) следующее лечение биологическими препаратами: какими-либо источающими клетки средствами, включая, помимо прочего, ритуксимаб (в период 6 месяцев, предшествующих исходному визиту, или до тех пор, пока уровень лимфоцитов и CD 19+ не вернется к норме, в зависимости от того, что дольше; инфликсимаб, адалимумаб, голимумаб, цертолизумаб пегол, абатацепт, этанерцепт, анакинра (в период 16 недель, предшествующих исходному визиту - по любым показаниям; или в период 5 лет, предшествующих исходному визиту - по дерматологическим показаниям) или иные биопрепараты (в период 16 предшествующих недель или в предшествующий период, равный 5 периодам полувыведения (если эти данные известны) зависимости от того, что дольше; (6) лечение АД отпускаемыми по рецепту увлажняющими средствами, считающимися медицинскими средствами (например, Atopiclair®, MimyX®, Epicerum®, Cerave® и проч.), в течение 1 недели, предшествующей исходному визиту; (7) регулярное посещение (более 2 раз в неделю) солярия в период 4 недель, предшествующих исходному визиту; (8) планируемое/ожидаемое использование каких-либо неразрешенных лекарств или процедур (включая, помимо прочего, топический такролимус и пимекролимус; кортикостероиды; отпускаемые по рецепту увлажняющие средства, считающиеся медицинскими средствами, такие как Atopiclair®, MimyX®, Epicerum®, Cerave® и проч.; аллергенная иммуннотерапия; системное лечение АД иммунностимулирующим/иммунномодулирующим средством; применение «живой» (аттенуированной) вакцины или экспериментального препарата (помимо mAb1), обширное плановое оперативное вмешательство в ходе исследуемого лечения; (9) применение «живой» (аттенуированной) вакцины в период 12 недель, предшествующих исходному визиту; (10) хроническая или острая инфекция, требующая лечение антибиотиками, противовирусными, противопаразитарными, противопротозойными или противогрибковыми препаратами в период 4 недель, предшествующих скрининговому визиту, или наличие поверхностных кожных инфекций в течение 1 недели, предшествующей исходному визиту; (11) наличие или подозрение на угнетение иммунитета, включая инвазивные оппортунистические инфекции в анамнезе (например, гистоплазмоз, листериоз, кокцидиоидомикоз, пневмоцистоз, аспергиллез) даже при разрешении инфекции, или прочие инфекции, рецидивирующие с патологической частотой, или длительные инфекции, дающие основания предполагать сниженный иммунитет - по мнению исследователя; (12) достоверное наличие в анамнезе заболевания вирусом иммунодефицита человека (ВИЧ) или положительный результат теста на ВИЧ при скрининговом визите; (13) положительный или неопределенный результат теста на поверхностный антиген вируса гепатита В (HBsAg), на антитела к ядерному антигену вируса гепатита В (HBcAb) или на антитела к вирусу гепатита С при скрининговом визите; (14) повышение уровня трансаминаз (АЛТ и (или) АСТ) более чем в три раза в сравнении с верхним пределом нормы (> 3 х ВПН) при скрининговом визите; (15) наличие в анамнезе клинического эндопаразитоза в период 12 месяцев, предшествующих исходному визиту, за исключением вылеченного вагинального трихомоноза; (16) наличие сопутствующих заболеваний кожи, которые могут помешать проведению предусмотренных исследованием обследований; (17) наличие в анамнезе злокачественных образований в период 5 лет, предшествующих исходному визиту, за исключением полностью вылеченной карциномы in situ шейки матки, полностью удаленной неметастатическая плоскоклеточной или базальной карциномы кожи; (18) наличие в анамнезе доброкачественных лимфопролиферативных нарушений; (19) высокий риск паразитарной инфекции, например проживание в районах с эндопаразитарными эндемиями или недавние поездки в такие районы (в период 12 месяцев, предшествующих исходному визиту), если обстоятельства свидетельствуют о контакте с паразитами (например, продолжительная поездка, нахождение в сельскохозяйственной местности или трущобах, отсутствие водопровода, потребление сырой, слабо приготовленной или по иной причине возможно зараженной еды, тесный контакт с носителями и переносчиками болезней и проч.), за исключением тех случаев, когда по результатам последующих медицинских обследований (например, анализ стула, крови и проч.) паразитарные инфекции/инвазии исключаются; (20) наличие в анамнезе злоупотребления алкоголем или наркотиками в период 2 лет, предшествующих скрининговому визиту; (21) тяжелое(ые) сопутствующее(ие) заболевание(я), которое(ые) по мнению исследователя будет(ут) иметь отрицательное воздействие на участие пациента в исследовании. Примеры включают, помимо прочего, пациентов с короткой ожидаемой продолжительностью жизни, неконтролируемым диабетом (HbA1c ≥ 9%), сердечно-сосудистыми заболеваниями (например, сердечная недостаточность III или IV степени по классификации Нью-Йоркской кардиологической ассоциации), серьезными почечными проблемами (например, пациент проходил диализ), гепатобилиарными проблемами (например, класс В или С по система Child-Puig), неврологическими проблемами (например, демиелинизирующее заболевание), серьезными аутоиммунными заболеваниями в активной стадии (например, волчанка, воспалительные заболевания кишечника, ревматоидный артрит), прочими серьезными эндокринными, желудочно-кишечными, метаболическими, легочными и лимфатическими заболеваниями. По пациентам, исключенным по данному критерию, в документации исследования (медицинских карточках, индивидуальных регистрационных картах (ИРК) и проч.) будет указано конкретное обоснование; (22) любые другие медицинские и психиатрические проблемы, включая актуальные отклонения от нормы показателей лабораторных анализов, выполненных при скрининге, которые по мнению исследователя дают основание предполагать наличие нового и (или) недостаточно изученного заболевания, имеют потенциал создания неоправданного риска для пациента при его (ее) участии в этом клиническом исследовании, дают основания для того, чтобы не считать участие пациента в исследовании надежным, или могут создавать помехи для проведения предусмотренных исследованием обследований. В документации исследования (медицинской карточке, ИРК и проч.) будет приведено конкретное обоснование исключения пациентов согласно данному критерию; (23) на период участия пациента в исследовании запланирована обширное оперативное вмешательство; (24) пациент является членом исследовательской группы и (или) близким родственником члена исследовательской группы; (25) беременные и кормящие грудью женщины; и (26) нежелание использовать надлежащие методы контрацепции в случае наличия репродуктивного потенциала и сексуальной активности. Требование по части надлежащих методов контрацепции подразумевают обязанность постоянно пользоваться эффективными и допустимыми средствами контрацепции на протяжении исследования и 16 недель после введения последней дозы исследуемого препарата.
В. Безопасность
Безопасность в ходе исследования отслеживалась посредством оценки нежелательных явлений и серьезных нежелательных явлений.
Нежелательным явлением (НЯ) является любое неблагоприятное с медицинской точки зрения явление, наблюдающееся у принимающего фармацевтический препарат пациента или участника клинического исследования. Таким образом, НЯ может представлять собой любой неблагоприятный или непредвиденный признак (включая отклонение показателя лабораторного анализа от нормы), симптом или заболевание, время возникновения которого приходится на время применения лекарственного препарата, вне зависимости от наличия или отсутствия причинно-следственной связи с лекарственным (экспериментальным) препаратом. НЯ также включают: любое ухудшение (то есть клинически значительное изменение частоты и (или) интенсивности) ранее существовавшего заболевания, если время возникновения такого ухудшения приходится на время применения исследуемого препарата; отклонения показателей лабораторных анализов от нормы, если исследователь считает такие отклонения клинически значительными; а также любые неблагоприятные с медицинской точки зрения явления.
Серьезным нежелательным явлением (СНЯ) является любое неблагоприятное с медицинской точки зрения явление, которое, вне зависимости от дозировки, приводит к смертельному исходу, или угрожает жизни, или приводит к госпитализации, или продлении ранее начатой госпитализации, или приводит к устойчивой или значительной утрате трудоспособности/инвалидности, или является врожденной аномалией/пороком или является важным с медицинской точки зрения явлением.
Также в ходе исследования оценивались лабораторные показатели безопасности, основные показатели жизнедеятельности, показатели электрокардиограммы (ЭКГ) в 12-ти отведениях и результаты физикального осмотра.
Клинические лабораторные данные включают в себя показатели общего и биохимических анализов крови и анализа мочи. Образцы крови для выполнения общего анализа крови брались на всех визитах в рамках исследования; образцы крови для выполнения биохимического анализа сыворотки крови и образцы мочи для выполнения анализа мочи брались для оценки общего состояния здоровья пациентов при скрининге, в день 1/исходной точке (до введения препарата), в дни 15, 29, 43, 57, 71, 85, 99, 113, 141, 169 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании.
Основные показатели жизнедеятельности включают частоту дыхательных движений (циклов в минуту), частоту пульса (ударов в минуту), систолическое и диастолическое артериальное давление (мм рт.ст.) и температуру тела (°C). Основные показатели жизнедеятельности измерялись (до введения препарата, в дни приема препарата) при скрининге, в день 1/исходной точке и в дни 4, 8, 15, 22, 25, 29, 43, 64, 71, 85, 99, 113, 127, 141, 155, 169, 183, 197 и 211 (окончание исследования) или при досрочном прекращении участия в исследовании. В дни 1, 8, 15 и 22 основные показатели жизнедеятельности измерялись через 1 и 2 часа после введения исследуемого препарата.
Показатели ЭКГ в 12-ти отведениях включают: желудочковая ЧСС, интервал PR, интервал QRS, скорректированный интервал QT (QTcF=QT/[RR0,33] и QTcB=QT/[RR0,5]) Статус ЭКГ: норма, клинически незначительная патология, клинически значительная патология. Стандартная ЭКГ в 12-ти отведениях выполнялась при скрининге, в дни 29 и 113 (окончание лечения) или при досрочном прекращении участия в исследовании.
Тщательный, полный физикальный осмотр выполнялся при скрининге, в дни 29 и 113 (окончание лечения) или при досрочном прекращении участия в исследовании.
C. Переменные эффективности
Переменные эффективности - балл по шкале IGA, показатель BSA, индекс EASI, индекс SCORAD, балл по опроснику из 5 вопросов для оценки кожного зуда и балл по шкале оценки кожного зуда NRS - описаны в иных частях настоящего документа (например, см. пример 7).
Балл по шкале IGA, показатель BSA, индекс EASI и индекс SCORAD измерялись на каждом визите в клинику. Оценка кожного зуда с помощью опросника из 5 вопросов выполнялась на следующих визитах: скрининг, день 1/исходная точка (до введения препарата), день 113 (окончание лечения) и день 211 (окончание исследования) или при досрочном прекращении участия в исследовании. Пациенты протоколировали свой балл по шкале оценки кожного зуда NRS два раза в день до последнего визита в рамках исследования.
Также изучались прочие показатели, такие как баллы по шкале общей оценки отдельных признаков (GISS), номинальной шкале оценки кожного зуда, опроснику для пациента для оценки экземы (POEM), дерматологическому индексу качества жизни (DLQI), опроснику для оценки влияния кожного зуда на качество жизни, опроснику EQ-50, опроснику HADS и общей оценке пациентом заболевания и эффекта лечения.
Исходной точкой для переменных эффективности было последнее имеющееся значение на дату рандомизации или до такой даты. Если по пациенту на дату рандомизации или до такой даты не имелось значений, то исходной точкой считалось последнее имеющееся значение на день первой дозы или до такого дня.
Пример 10: Клиническое исследование с применением повторных подкожных доз антител к IL-4Р (mAb1) с участием взрослых пациентов с атопическим дерматитом в степени от умеренной до тяжелой
A. Дизайн исследования
Данное исследование было 28-недельным, рандомизированным, двойным слепым, плацебо-контролируемым исследованием антител к IL-4Р (далее - «mAb1»), вводимых подкожно пациентам с атопическим дерматитом в степени от умеренной до тяжелой. Продолжительность периода лечения составила 12 недель; последующее наблюдение пациентов длилось 16 недель после окончания периода лечения.
В исследование было набрано 109 пациентов, которые были рандомизированы в пропорции 1:1 (54 - в группу приема плацебо, 55 - в группу приема 300 мг препарата с антителами). Сорок три пациента прекратили участие в исследовании (30 - в группе приема плацебо, 13 - в группе приема 300 мг). При рандомизации стратификация выполнялась согласно уровню IgE (IgE < 150 тыс. ед./л или ≥ 150 тыс. ед./л при скрининговом визите) для проверки эффективности mAb1 при применении пациентами с экзогенной и эндогенной формами АД. Пациенты, соответствовавшие критериям набора, проходили обследования в рамках дня 1 (исходный визит), рандомизацию, а затем получали 300 мг mAb1 или плацебо подкожно. Все еженедельные дозы исследуемого препарата применялись в форме одной 2-миллилитровой инъекции или двух 1-миллилитровых инъекций. Пациенты совершали еженедельные визиты в клинику и получали инъекции исследуемого препарата в дни 8, 15, 22, 29, 36, 43, 50, 57, 64, 71 и 78. После приема каждой дозы исследуемого препарата пациентов тщательно наблюдали в исследовательском центре на протяжении не менее 2 часов. Окончание периода лечения приходилось на день 85. Визиты для последующего наблюдения приходились на дни 92, 99, 106, 113, 120, 127, 134, 141, 148, 155, 162, 169, 176, 183, 190, а визит в связи с окончанием исследования - на день 197.
Критерии включения в исследование были следующими: (1) мужчина или женщина в возрасте 18 лет или старше; (2) хронический АД (диагноз поставлен согласно критериям Hannifin и Rajka в редакции Eichefield) присутствует не менее 3 лет до скринингового визита; (3) индекс EASI ≥ 16 при скрининге и на исходном визите; (4) балл по IGA ≥ 3 при скрининге и на исходном визите; (5) ≥ 10% площади поверхности кожи (BSA) поражено АД согласно наблюдениям при скрининге и на исходном визите; (6) у пациента в анамнезе запротоколирован ненадлежащий ответ на стабильный (≥ 1 месяца) прием топических кортикостероидов или ингибиторов кальциневрина (по поводу АД) в период 3 месяцев, предшествующих скрининговому визиту; (7) пациент должен был не менее 7 дней до исходного визита два раза в сутки применять в стабильной дозе стандартное смягчающее средство без добавок; и (8) готовность, желание и способность осуществлять все визиты в клинику и проходить все связанные с исследованием процедуры, а также готовность и способность подписать форму информированного согласия (ФИС).
Критерии исключения из исследования были следующими: (1) лечение препаратом mAb1 в прошлом; (2) наличие каких-либо следующих отклонений от нормы в показателях лабораторных анализов, выполненных во время скринингового визита: количество лейкоцитов < 3,5×103/μл; количество тромбоцитов < 125×103/μл; количество нейтрофилов < 1,75×103/μл; аспартатаминотрансфераза (АСТ)/аланинаминотрансфераза (АЛТ) > 1,5 × ВПН; КФК > 2 × ВПН; (3) положительный или неопределенный результат выполненного во время скринингового визита анализа на поверхностный антиген вируса гепатита В, на антитела к ядерному антигену вируса гепатита В или на антитела к вирусу гепатита С; (4) инициация нового комплекса физических упражнений или значительное изменение ранее практиковавшегося комплекса физических упражнений в период 4 недель, предшествующих скринингу (визит 1). Пациенты должны были быть готовы не изменять уровень физических нагрузок на протяжении исследования и воздерживаться от необычайно сильных физических нагрузок на протяжении исследования; (5) лечение экспериментальным препаратом в период 8 недель, предшествующих исходному визиту, или в предшествующий исходному визиту период, равный 5 периодам полувыведения такого препарата, если эти данные известны, в зависимости от того, что дольше; (6) применение живой (аттенуированной) вакцины в период 12 недель, предшествующих исходному визиту; (7) применение противоаллергической иммунотерапии в период 6 месяцев, предшествующих исходному визиту; (8) лечение ингибиторами лейкотриена в период 4 недель, предшествующих исходному визиту; (9) лечение системными кортикостероидами в период 4 недель, предшествующих исходному визиту; (10) лечение топическими кортикостероидами, такролимусом и (или) пимекролимусом в течение 1 недели, предшествующей исходному визиту; (11) системное лечение АД иммуносупрессивными/иммуномодулирующими веществами, например циклоспорином, микофенолат-мофетилом, ИФН-гамма, светотерапией (узкополосный УФВ, УФВ, УФА, псорален+УФА), азатиоприном, метотрексатом или биологиками в период 4 недель, предшествующих исходному визиту; (12) принятие трех или более «отбеливающих ванн» в течение какой-либо одной недели в рамках 4 недель, предшествующих исходному визиту; (13) лечение АД медицинским средством (например, Atopiclair®, MimyX®, Epicerum®, Cerave® и др.) в течение 1 недели, предшествующей исходному визиту; (14) хроническая или острая инфекция, требующая применения пероральных или внутривенных антибиотиков, противовирусных, противопаразитарных или противогрибковых препаратов в период 4 недель, предшествующих скрининговому визиту, или поверхностная инфекция кожи в течение 1 недели, предшествующей скрининговому визиту; (15) наличие ВИЧ-инфекции в анамнезе; (16) наличие в анамнезе гиперчувствительных реакций на доксициклин или схожие вещества; (17) наличие в анамнезе клинической паразитарной инфекции, за исключением вагинального трихомоноза; (18) наличие в анамнезе злокачественных образований в период 5 лет, предшествующих исходному визиту, за исключением случаев полностью вылеченной карциномы шейки матки in situ и неметастатической плоскоклеточной или базальной карциномы кожи; (19) запланированная на период участия пациента в исследовании хирургическая процедура; (20) пользование солярием в период 4 недель, предшествующих скрининговому визиту; (21) значительное сопутствующее заболевание или наличие в анамнезе значительных заболеваний, например психиатрических, сердечных, почечных, неврологических, эндокринных, метаболических или лимфатических заболеваний или любого иного заболевания или состояния, которое бы имело нежелательное влияние на участие пациента в этом исследовании; (22) беременные и кормящие грудью женщины; и (или) (23) нежелание пользоваться надлежащими методами контрацепции. Требование о надлежащих методах контрацепции подразумевает обязанность постоянно пользоваться эффективными и допустимыми средствами контрацепции на протяжении исследования и 16 недель после введения последней дозы исследуемого препарата. Для женщин надлежащими методами контрацепции являются: гормональные контрацептивы, внутриматочное устройство (ВМУ), двойная барьерная контрацепция (т.е. презерватив+диафрагма; презерватив или диафрагма+спермицидный гель или пена). Для мужчин надлежащими методами контрацепции являются: двойная барьерная контрацепция (т.е. презерватив+диафрагма; презерватив или диафрагма+спермицидный гель или пена). У женщин менопаузой считается период в 24 месяца без наступления месячных; в случае наличия сомнений требуется документальное подтверждение уровня фолликулостимулирующего гормона ≥25 ед/мл. В случае гистерэктомии, двусторонней оофорэктомии или двусторонней перевязки маточных труб требуется документальное подтверждение.
B. Переменные эффективности
Основной конечной точкой было изменение в процентах индекса EASI за период между исходной точкой и неделей 12. В этом исследовании оценивались следующие вторичные конечные точки: (1) доля пациентов, у которых балл по IGA в неделю 12 составлял от 0 до 1 очка; (2) доля пациентов, у которых за период между исходной точкой и неделей 12 наступило ≥ 50%-ное улучшение индекса EASI (такое улучшение также именуется «EASI 50»); (3) изменение индекса EASI за период между исходной точкой и неделей 12; (4) изменение и изменение в процентах балла по IGA, показателя площади поверхности кожи, пораженной атопическим дерматитом (BSA), индекса площади экзематозного поражения кожи и тяжести экземы (EASI), индекса SCORAD, балла по шкале оценки кожного зуда NRS и балла по опроснику из 5 вопросов для оценки кожного зуда за период между исходной точкой и неделей 12; (5) частота возникновения ВЛНЯ за период между исходной точкой и неделей 28; (6) связываемое с ответом на лечение изменение, относительно исходной точки, показателей эозинофилов, TARC, результатов теста Phadiatop™ и общего IgE; (7) изменение индекса QoLIAD за период между исходной точкой и неделей 12; (8) доля пациентов, у которых за период между исходной точкой и неделей 12 балл по IGA снизился на ≥ 2 очка; (9) доля пациентов, у которых за период между исходной точкой и неделей 12 снижение балла по IGA составляет ≥ 3 очка; и (10) ФД показатели ответа циркулирующих эозинофилов, TARC и общего IgE.
Исходным показателем переменной эффективности является последнее имеющееся в наличии значение на дату рандомизации или до такой даты. Если по пациенту на дату рандомизации или до такой даты не имеется значений, то исходной точкой считается последний имеющийся в наличии показатель на день первой дозы или до такого дня.
Предусмотренные исследованием процедуры
Переменные эффективности - балл по шкале IGA, показатель BSA, индекс EASI, индекс SCORAD, балл по опроснику из 5 вопросов для оценки кожного зуда и балл по шкале оценки кожного зуда NRS - описаны в иных частях настоящего документа (например, см. пример 7).
Балл по шкале IGA, показатель BSA, индекс EASI и индекс SCORAD измерялись на каждом визите в клинику. Опросник из 5 вопросов для оценки кожного зуда применялся на следующих визитах: скрининг, день 1/исходная точка (до введения препарата) и дни 15, 29, 43, 57, 71, 85, 99, 113, 127, 141, 155, 169, 183 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании. Пациенты протоколировали свой балл по шкале оценки кожного зуда NRS два раза в день до последнего визита в рамках исследования.
Индекс качества жизни пациента с атопическим дерматитом (QoLIAD): QoLIAD является валидированным опросником из 25 вопросов и используется в клинической практике и клинических исследованиях для оценки воздействия симптомов АД и лечения АД на качество жизни. Формат анкеты предусматривает простые ответы «да» или «нет» на 25 вопросов. Выставляется балл от 0 до 25. Более высокий балл означает более низкое качество жизни. Пациенты заполняли анкету при скрининге, в день 1/исходной точке и в дни 29, 57, 85, 99, 113, 127, 141, 155, 169, 183 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании.
C. Исследуемое лечение
Лекарственный препарат mAb1 поставлялся в виде лиофилизированного порошка в стеклянных ампулах по 5 мл для п/к введения. При п/к введении лекарственный препарат mAb1 смешивался с 2,5 мл стерильного инъекционного раствора, в результате чего получался раствор, содержащий 150 мг mAb1 на 1 мл. Испытывалось подкожное применение mAb1 в дозе 300 мг. Препарат mAb1 или плацебо вводился в клинике посредством одной (2 мл) или двух (1 мл) п/к инъекций в день 1/исходной точке и в дни 8, 15, 22, 29, 36, 43, 50, 57, 64, 71 и 78. Хотя предпочтительно было, чтобы все еженедельные дозы исследуемого препарата вводились посредством одной 2-миллилитровой инъекции, все еженедельные дозы можно было разделить на две 1-миллилитровых инъекции. Места подкожного введения препарата чередовались, он вводился в заднюю часть рук, область живота (за исключением пупка и области талии), верхнюю часть бедер. Введение препарата в конечности не допускалось в силу того, что это могло привести к иной абсорбции и биодоступности. Если в один день требовалось выполнение нескольких инъекций, то они выполнялись в разных местах (например, одна инъекция выполнялась в нижнем правом квадрате области живота, а другая - в левом нижнем квадрате). Места подкожного введения препарата чередовались таким образом, чтобы в одно и то же место инъекции не вводились на протяжении 2 последовательных недель.
Приготовление плацебо выполнялось в тех же пропорциях, что и в случае mAb1, но при этом антитела не добавлялись.
После приема каждой дозы исследуемого препарата пациентов наблюдали в исследовательском центре на протяжении не менее 2 часов.
Также пациенты должны были применять стандартное смягчающее средство без добавок два раза в сутки в стабильной дозировке на протяжении не менее 7 дней до исходного визита и на протяжении участия в исследовании. В ходе исследования пациенты протоколировали применение ими базовой терапии с помощью интерактивной системы голосового ответа и интерактивной системы веб-ответа. Система задавала пациентам следующий вопрос насчет применения ими смягчающего средства: «Наносили ли вы утвержденное вашим врачом-исследователем увлажняющее средство на пораженные участки кожи?»
D. Оценка безопасности
Безопасность в ходе исследования отслеживалась посредством оценки нежелательных явлений и серьезных нежелательных явлений.
Нежелательным явлением (НЯ) является любое неблагоприятное с медицинской точки зрения явление, наблюдающееся у принимающего фармацевтический препарат пациента или участника клинического исследования. Таким образом, НЯ может представлять собой любой неблагоприятный или непредвиденный признак (включая отклонение показателя лабораторного анализа от нормы), симптом или заболевание, время возникновения которого приходится на время применения лекарственного препарата, вне зависимости от наличия или отсутствия причинно-следственной связи с лекарственным (экспериментальным) препаратом. НЯ также включают: любое ухудшение (то есть клинически значительное изменение частоты и (или) интенсивности) ранее существовавшего заболевания, если время возникновения такого ухудшения приходится на время применения исследуемого препарата; отклонения показателей лабораторных анализов от нормы, если исследователь считает такие отклонения клинически значительными; а также любые неблагоприятные с медицинской точки зрения явления.
Серьезным нежелательным явлением (СНЯ) является любое неблагоприятное с медицинской точки зрения явление, которое, вне зависимости от дозировки, приводит к смертельному исходу или угрожает жизни или приводит к госпитализации или продлении ранее начатой госпитализации или приводит к устойчивой или значительной утрате трудоспособности/инвалидности или является врожденной аномалией/ пороком или является важным с медицинской точки зрения явлением.
Также в ходе исследования оценивались лабораторные показатели безопасности, основные показатели жизнедеятельности, показатели электрокардиограммы (ЭКГ) в 12-ти отведениях и результаты физикального осмотра.
Клинические лабораторные данные включают в себя показатели общего и биохимических анализов крови и анализа мочи. Образцы крови для выполнения общего анализа крови брались на всех визитах в рамках исследования; образцы крови для выполнения биохимического анализа сыворотки крови и образцы мочи для выполнения анализа мочи брались для оценки общего состояния здоровья пациентов при скрининге, в день 1/исходной точке (до введения препарата), в дни 15, 29, 43, 57, 71, 85, 99, 113, 141, 169 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании.
Основные показатели жизнедеятельности включают частоту дыхательных движений (циклов в минуту), частоту пульса (ударов в минуту), систолическое и диастолическое артериальное давление (мм рт.ст.) и температуру тела (°C). Основные показатели жизнедеятельности измерялись (до введения препарата, в дни приема препарата) при скрининге, в день 1/исходной точке и в дни 8, 15, 22, 29, 36, 43, 50, 57, 64, 71, 78, 85, 99, 113, 141, 169 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании. В дни 1, 8, 15, 22, 29, 36, 43, 50, 57, 64, 71 и 78 основные показатели жизнедеятельности измерялись через 1 и 2 часа после введения исследуемого препарата.
Показатели ЭКГ в 12-ти отведениях включают: желудочковая ЧСС, интервал PR, интервал QRS, скорректированный интервал QT (QTcF=QT/[RR0,33] и QTcB=QT/[RR0,5]) Статус ЭКГ: норма, клинически не значительная патология, клинически значительная патология. Стандартная ЭКГ в 12-ти отведениях выполнялась при скрининге, в дни 141 и 197 (окончание исследования) исследования или при досрочном прекращении участия в исследовании.
Исследовательские образцы (сыворотка/РНК/плазма) брались при скрининге, в день 1/исходная точка (до введения препарата) и дни 8, 15, 22, 29, 57, 85 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании, а также во время внеплановых визитов.
Тщательный, полный физикальный осмотр выполнялся при скрининге, в дни 85 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании.
E. Анализ данных
1. Анализы поисковых показателей эффективности
Все категориальные переменные анализировались с помощью точного критерия Фишера с указанием номинального значения p и доверительных интервалов. Все непрерывные переменные были проанализированы с помощью ковариационного анализа (ANCOVA) со стратификацией по исходному уровню IgE (< 150 тыс. ед./л или ≥ 150 тыс. ед./л на скрининговом визите). Если не указано иное, оценка изменений относительно исходного уровня и построение доверительных интервалов непрерывных параметров основывались на модели ANCOVA, в которой лечение было основным фактором, а исходные показатели - независимыми переменными. Указывалась точечная оценка и 95%-ный ДИ разницы наблюдавшихся в двух группах лечения скорректированных средних показателей изменений относительно исходного уровня. Отсутствующие значения будут подставлены методом переноса данных последнего наблюдения (LOCF). Если использование предположений по результатам моделирования не является оправданным, будет использован ранговый ковариационный анализ.
2. Анализ данных по безопасности
Анализ безопасности был основан на зафиксированных НЯ, результатах клинических лабораторных анализов, основных показателях жизнедеятельности организма и показателях ЭКГ в 12-ти отведениях. Пределы потенциально клинически значительных значений (ПКЗЗ) результатов лабораторных анализов, основных показателей жизнедеятельности и ЭКГ установлены в плане статистического анализа. Временной интервал, в который отслеживается наступление каких-либо событий или патологии, приходится на период времени между инфузией исследуемого препарата и окончанием исследования. Данные, поступившие за пределами этого периода времени, не учитываются при расчете описательных статистических показателей и определении наличия патологий в результатах лабораторных анализов, основных показателей жизнедеятельности и ЭКГ.
F. Безопасность: Результаты
В целом, mAb1 хорошо переносился и продемонстрировал благоприятный профиль безопасности. Общий профиль нежелательных явлений (НЯ) был характерен для здоровой популяции. Смертельных исходов не было. У 8 пациентов наблюдались СНЯ: один пациент был из группы приема mAb1 (перелом лицевой кости черепа), семь - из группы приема плацебо (стенокардия, целлюлит, герпетическая экзема, бактериальная инфекция кожи, почечная недостаточность, приступ астмы, нарушение функции легких и атопический дерматит). У 8 пациентов наблюдались ВЛНЯ, что привело к прекращению участия этих пациентов в исследовании: один пациент был из группы приема mAb1, семь - из группы приема плацебо. У 87 пациентов наблюдалось как минимум одно ВЛНЯ (n=43 [78,2%] в группе приема mAb1; n=44 [81,5%] в группе приема плацебо). Наиболее частым ВЛНЯ у пациентов, получавших mAb1, был назофарингит (n=22 [40%] относительно 10 [18,5%] в группе приема плацебо). Прочие ВЛНЯ, наблюдавшиеся в группе лечения, включали глазные инфекции, расстройства нервной системы, расстройства общего характера и реакции в месте введения препарата. В ходе исследования не сообщалось ни о каких иных клинически значительных отклонениях в результатах лабораторных анализов (общий и биохимический анализы крови, анализ мочи). В средних и медианных значениях исходных показателей лабораторных анализов не было выявлено никаких тенденций. В ходе исследования не наблюдалось каких-либо значительных тенденций в средних и медианных значениях изменений показателей температуры тела и пульса относительно исходного уровня. При физикальном осмотре, на ЭКГ и в показателях жизнедеятельности организма не наблюдалось клинически значительных отклонений.
Подкожное введение mAb1 пациентам с АД в степени от умеренной до тяжелой было в целом безопасным и хорошо переносилось.
G. Эффективность: Результаты
На рисунках 23-33 и в таблицах 27-35 сведены исходные показатели и поисковые показатели эффективности по результатам исследования. Как отмечалось выше, пациенты получали либо 300 мг mAb1 подкожно раз в неделю на протяжении 12 недель, либо плацебо.
Обзор исходных показателей - все значения приводятся как среднее (СО)
300 мг
Обзор изменений индекса EASI за период между исходной точкой и неделей 12/каждым визитом периода последующего наблюдения - в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений балла по шкале IGA за период между исходной точкой и неделей 12/каждым визитом периода последующего наблюдения - в процентах и в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений показателя BSA за период между исходной точкой и неделей 12/каждым визитом периода последующего наблюдения - в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений индекса SCORAD за период между исходной точкой и неделей 12/каждым визитом периода последующего наблюдения - в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений балла по опроснику из 5 вопросов D для оценки кожного зуда за период между исходной точкой и неделей 12/каждой неделей периода последующего наблюдения - в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор изменений среднего балла по шкале NRS за период между исходной точкой и неделей 12/каждой неделей периода последующего наблюдения - в абсолютном выражении - все значения приводятся в виде средних (СО)
Обзор пациентов, у которых балл по шкале IGA достиг нуля или 1 очка по состоянию на неделю 12 и все визиты периода последующего наблюдения
(N=54)
(N=55)
Обзор пациентов, достигших индекса EASI 50 по состоянию на неделю 12 и все визиты периода последующего наблюдения
(N=54)
(N=55)
H. Выводы
Антитела к IL-4Р (mAb1), вводимые подкожно взрослым пациентам с атопическим дерматитом в степени от умеренной до тяжелой, в целом были безопасными и хорошо переносились по результатам применения 12 еженедельных доз в 300 мг. Применение 300 мг mAb1 приводило к значительному улучшению балла по шкале IGA, индекса EASI, показателя BSA, индекса SCORAD и балла по шкале оценки зуда NRS, что выражалось в средних показателях, изменениях в процентах и изменениях в абсолютном выражении относительно исходной точки; и такое улучшение наблюдалось вплоть до 85-го дня (см. таблицы 27-33). Показатель доли пациентов из группы приема 300 мг препарата, у которых балл по IGA достиг нуля или 1 очка ко дню 85, составил 40,0%. В группе плацебо этот показатель составлял 7,4% (таблица 34). Доля пациентов в группе приема 300 мг, которые ко дню 85 достигли 50%-ного снижения индекса EASI («EASI-50»), составила 85,5%. В группе приема плацебо ко дню 85 индекс EASI-50 наблюдался у 35,2% пациентов (таблица 35). Разница между группой приема mAb1 и группой приема плацебо по части изменения индекса EASI за период между исходной точкой и неделей 12 в процентном выражении была статистически значительной (-74,0% относительно 23,0%, p<0,0001). По всем вторичным конечным точкам эффективности разница в показателях между группой приема препарата и группой приема плацебо была статистически значительной. Далее приводятся значения p: доля пациентов с изменением балла по шкале IGA (0 или 1) (<0,0001), доля пациентов с изменением индекса EASI (<0,0001), изменение индекса EASI в абсолютном выражении относительно исходной точки (<0,0001), изменение балла по шкале IGA в абсолютном выражении относительно исходной точки (<0,0001), изменение балла по шкале IGA в процентном выражении относительно исходной точки (<0,0001), изменение показателя BSA в абсолютном выражении (<0,0001), изменение индекса SCORAD в абсолютном выражении (<0,0001), изменение балла по шкале оценки кожного зуда NRS в абсолютном выражении (<0,0001) и изменение балла по опроснику из 5 вопросов для оценки кожного зуда в абсолютном выражении за период между исходной точкой и неделей 12 (<0,0001) соответственно.
Пример 11: Клиническое исследование для изучения безопасности mAb1, применяемого совместно с топическими кортикостероидами пациентами с АД в степени от умеренной до тяжелой
A. Дизайн исследования
Это исследование было рандомизированным, двойным слепым, плацебо-контролируемым исследованием, проводимым в параллельных группах для изучения безопасности и эффективности повторных подкожных доз mAb1, применяемых совместно с топическими кортикостероидами (ТКС) для лечения пациентов с АД в степени от умеренной до тяжелой. Пациенты были рандомизированы в пропорции 2:1 в группы приема 300 мг mAb1 и плацебо в форме подкожной инъекции один раз в неделю на протяжении 4 последовательных недель (в дни 1, 8, 15 и 22). Все пациенты проходили сопутствующее открытое ежедневное лечение сильнодействующим ТКС (в 50-100 раз сильнее гидрокортизона), таким как метилпреднизолона ацепонат 0,1%, мометазона фуроат 0,1% или бетаметазона валерат 0,1%, сроком до 28 дней. Для лечения вызванных АД поражений на лице, в складках и в области гениталий прочие топические препараты, такие как менее активные ТКС или топические ингибиторы кальциневрина (ТИК).
Начиная со скринингового визита пациенты применяли стандартное смягчающее средство без добавок два раза в сутки на протяжении не менее 7 дней до исходного визита и далее на протяжении исследования (один раз в день в дни лечения в местах нанесения ТКС). По усмотрению исследователя допускались мероприятия по контролю окружающей пациента среды и нефармакологические формы лечения, такие как избежание воздействия аллергенов и «отбеливающие ванны».
Скрининг выполнялся в период с -21-го по -1-й день. В день 1 пациентам вводилась первая инъекция препарата (300 мг mAb1 или плацебо), а затем в дни 8, 15 и 22 (+/-1 день) пациенты приходили в клинику для выполнения дополнительных инъекций препарата - всего вводилось 4 еженедельных дозы. Начиная с дня 1 пациенты применяли указанный(ые) выше топический(е) препарат(ы) один раз в сутки вечером и продолжали наносить его (их) на все подверженные АД участки тела (т.е. участки с активными поражениями, вызванными АД) до тех пор, пока не был достигнут их контроль - сроком до 28 дней. После того, как был достигнут контроль, нанесение ТКС на подверженные АД участки тела без активных поражений (т.е. участки, на которых поражения прошли) допускалось на чаще 2 дней в неделю до дня 28. После дня 28 применение топических препаратов для лечения остаточных поражений, вызванных АД, могло быть продолжено в необходимой степени. На протяжении исследования пациенты продолжали применять стандартное смягчающее средство без добавок два раза в сутки (один раз в сутки в дни, когда применялись топические препараты, на участках тела, на которые наносились топические препараты). Пациенты приходили в клинику на визиты в дни 29, 36, 50, 64 и 78 (окончание исследования).
Критерии включения в исследование были следующими: (1) мужчина или женщина в возрасте 18 лет или старше; (2) хронический АД (диагноз поставлен согласно критериям Hannifin и Rajka в редакции Eichefield) присутствует не менее 2 лет до скрининга; (3) активность АД согласно баллу по шкале IGA ≥ 3 и индексу SCORAD > 20 при скрининге и на исходном визите, и при этом имеется одно вызванное АД поражение или более, по поводу чего показано лечение сильнодействующим ТКС; (4) АД подтверждено не менее 10% площади поверхности кожи (BSA) согласно наблюдениям при скрининге и на исходном визите; (5) пациент должен был не менее 7 дней до исходного визита два раза в сутки применять стандартное смягчающее средство без добавок; (6) готовность и способность осуществлять все визиты в клинику и проходить все связанные с исследованием процедуры; и (7) способность прочитать, понять и подписать форму информированного согласия.
Критерии исключения из исследования были следующими: (1) лечение препаратом mAb1 в прошлом; (2) гиперчувствительные реакции на кортикостероиды или какие-либо иные ингредиенты, содержащиеся в применяемом в рамках исследования ТКС; (3) вызванные АД поражения в основном (≥ 50% общей площади пораженной кожи) расположены на лице, в складках и в области гениталий; (4) наличие сопутствующих кожных патологий, которые могут помешать выполнению предусмотренных исследованием оценок; (5) проведение какого-либо перечисленного далее лечения в период 4 недель, предшествующих исходному визиту, или какие-либо состояния, которые могут потребовать проведения такого лечения в ходе исследования: системные кортикостероиды, иммуносупрессивные и иммуномодулирующие препараты, например циклоспорин, микофенолат-мофетил, ИФН-гамма, азатиоприн или метотрексат; (6) лечение следующими биологиками: (а) какие-либо средства, истощающие клетки, включая, помимо прочего, ритуксимаб (в период 6 месяцев, предшествующих исходному визиту, или до тех пор, пока уровень лимфоцитов и CD 19+ не вернется к норме, в зависимости от того, что дольше;, (б) инфликсимаб, адалимумаб, голимумаб, цертолизумаб пегол, абатацепт, этанерцепт, анакинра (в период 8 недель, предшествующих исходному визиту); и (в) иные биологики (в период 8 предшествующих недель или в предшествующий период, равный 5 периодам полувыведения (если эти данные известны)); (7) какая-либо светотерапия по поводу заболевания кожи (например, узкополосный УФВ, УФВ, УФА1, псоларен+УФА) - в период 4 недель, предшествующих исходному визиту; (8) регулярное посещение (более 2 раз в неделю) солярия в период 4 недель, предшествующих исходному визиту; (9) лечение живой аттенуированной вакциной в период 12 недель, предшествующих исходному визиту; (10) лечение экспериментальным препаратом в течение предшествующих исходному визиту 8 недель или предшествующему исходному визиту периода, равному 5 периодам полувыведения, в зависимости от того, что дольше; (11) хроническая или острая инфекция, требующая лечения пероральными или внутривенными антибиотиками, противовирусными, противопаразитарными, противопротозойными или противогрибковыми препаратам и в период 4 недель, предшествующих скринингу, или поверхностная инфекция кожи в течение 1 недели, предшествующей скрининговому визиту; (12) наличие в анамнезе инвазивных оппортунистских инфекций, таких как гистоплазмоз, листериоз, кокцидиоидомикоз, кандидоз, пневмоцистная пневмония, аспергиллез, вне зависимости от того, разрешилось ли заболевание, а также вирус Джона Каннингема (прогрессивная мультифокальная лейкоэнцефалопатия); (13) достоверное наличие в анамнезе ВИЧ-инфекции; (14) положительный или неопределенный результат теста на поверхностный антиген вируса гепатита В, на на антитела к ядерному антигену вируса гепатита В или на антитела к вирусу гепатита С при скрининговом визите; (15) наличие каких-либо следующих отклонений от нормы показателей лабораторных анализов, выполненных во время скринингового визита: креатининфосфокиназа выше верхнего предела нормы (ВПН) более чем в 2 раза; аспартатаминотрансфераза (АСТ) и (или) аланинаминотрансфераза (АЛТ) > 2×ВПН; количество нейтрофилов < 1,75×103/μл; количество тромбоцитов < 100×103/μл; (16) инициация нового комплекса физических упражнений или значительное изменение предыдущего комплекса физических упражнений в период 2 недель, предшествующих рандомизации, или нежелание придерживаться текущего уровня физических нагрузок (без его увеличения) на протяжении участия в исследовании; (17) наличие в анамнезе гиперчувствительных реакций к доксициклину или прочим тетрациклинам; (18) наличие клинической эндопаразитарной инфекции в период 12 месяцев, предшествующих исходному визиту, за исключением вылеченного вагинального трихомоноза; (19) наличие в анамнезе злокачественной опухоли в период 5 лет, предшествующих исходному визиту, за исключением полностью вылеченной карциномы шейки матки in situ, полностью удаленной неметастатической плоскоклеточной или базальной карциномы кожи; (20) наличие в анамнезе доброкачественных лимфопролиферативных нарушений; (21) беременные и кормящие грудью женщины; (22) мужчины, способные к зачатию, и женщины, способные к деторождению, если они не желают пользоваться контрацептивными средствами; (23) наличие в анамнезе злоупотребления алкоголем и наркотическими веществами в период 2 лет, предшествующих скрининговому визиту; (24) недавняя поездка (в период 12 месяцев, предшествующих рандомизации) в районы с паразитарными эндемиями, например развивающиеся африканские страны, тропические/субтропические регионы Азии; (25) наличие в настоящем или прошлом серьезного(ых) сопутствующего(их) заболевания(й), которое(ые) бы неблагоприятным образом сказалось(ись) на участии пациента в исследовании, например, сердечная недостаточность III или IV степени, серьезные почечные, неврологические, эндокринные, желудочно-кишечные, гепатобилиарные, метаболические, легочные или лимфатические заболевания; и (26) любые иные состояния, которые могут создавать неоправданный риск для пациента или давать основания считать, что участие пациента в исследовании ненадежно, или мешать выполнению предусмотренных исследованием оценок.
Первичными конечными точками исследования была частота возникновения и степень неблагоприятных явлений. Вторичные конечные точки имели поисковый характер и включали: (1) индекс EASI50 - бинарная переменная, демонстрирующая наступило ли у пациента ≥50%-ное снижение индекса EASI за период между исходной точкой и днем 29 или иными временными точками, следующими за исходной; (2) достижение балла по шкале IGA ≤ 1 очка (воспалительные признаки отсутствуют или почти отсутствуют) ко дню 29 или иным временным точкам, следующим за исходной; (3) период времени до достижения балла по шкале IGA ≤ 1 и до достижения индекса EASI50; (4) изменение баллов по IGA, индексов EASI и SCORAD за период между исходной точкой и днем 29 или иными временными точками, следующими за исходной; и (5) доля пациентов, у которых на неделе 4 балл по IGA составлял ≤ 1 очка и у которых до конца периода наблюдений не наблюдалось рецидивов заболевания.
В. Переменные эффективности
Переменные эффективности - балл по IGA, показатель BSA, индекс EASI, индекс SCORAD и балл по шкале оценки кожного зуда NRS - описаны в иных частях настоящего документа (например, см. пример 7). Баллы по шкалам ООИ, BSA, EASI, NRS (оценка зуда) и SCORAD измерялись на каждом визите в клинику.
C. Процедуры и обследования
Характеристики безопасности изучались путем измерения частоты возникновения нежелательных явлений (описаны в иных частях настоящего документа) (НЯ) в период со дня 1 по день 78, а также путем изучения подробностей анамнеза, тщательного физикального осмотра, измерения основных показателей жизнедеятельности организма, снятия электрокардиограмм (ЭКГ) и выполнения клинических лабораторных анализов. На постоянной основе изучались маскированные данные о безопасности. Данные о сопутствующих препаратах и процедурах собирались начиная со скрининга и оканчивая днем 78 (окончание исследование) или днем досрочного прекращения участия (если таковое имело место). Во время каждого визита в клинику измерялись параметры безопасности, выполнялись лабораторные анализы и изучалась эффективность. Во время каждого визита, предусмотренного исследованием в период между исходной точкой (день 1) и началом лечения, брались образцы крови для определения системной остаточной концентрации функционального mAb1. Образцы крови брались заранее определенные временные точки для изучения уровня антител к mAb1. Также брались образцы для использования в исследовательских целях и для выполнения поискового анализа биомаркеров. Эффективность mAb1 измерялась путем изучения индексов EASI, SCORAD, баллов по шкалам IGA, NRS (оценка кожного зуда), показателя BSA (площадь поверхности кожи, пораженной АД). В заранее определенные временные точки брались образцы крови для фармакокинетических (ФК) анализов и измерения уровня антител к mAb1. Также брались образцы для использования в исследовательских целях и для выполнения поискового анализа биомаркеров.
D. Статистические методы
Так как все упоминаемые в этом разделе статистические анализы имели поисковой характер, учет множественности сравнений для ошибки первого рода не выполнялся. Во всех анализах уровень значимости был установлен на уровне 5%. Все категориальные переменные (доля пациентов, достигших индекса EASI-50 и снижения балла по шкале IGA по состоянию на каждый следующий за исходной точкой визит; доля пациентов, достигших снижения балла по шкале IGA по состоянию на день 29 и не имевших дальнейших рецидивов) были проанализированы с помощью точного критерия Фишера с использованием номинального значения p, рассчитанного путем сравнения групп приема mAb1 и плацебо. Были указаны точечные оценки и доверительные интервалы показателей долей пациентов. Отношение показателей долей пациентов ко времени было представлено на графиках. Все непрерывные переменные (изменение балла по шкале IGA, индексов EASI, SCORAD, балла по шкале NRS в абсолютном и процентном выражении за период между исходной точкой и каждым следующих за исходной точкой визитом) изучались методом ковариационного анализа (ANCOVA). Если не указано иное, оценка изменений относительно исходного уровня и построение доверительных интервалов непрерывных параметров основывались на модели ANCOVA, в которой лечение было основным фактором, а исходные показатели - независимыми переменными. Указывались точечные оценки и 95%-ный ДИ разницы между двумя группами лечения по скорректированным средним значениям изменений относительно исходной точки. Будет указано номинальное значение p, рассчитанное путем сравнения групп приема mAb1 и плацебо. Если использование предположений по результатам моделирования не являлось оправданным, использовался ранговый ковариационный анализ. Средние изменения относительно исходной точки были проиллюстрированы с помощью графиков. Для сравнения mAb1 с плацебо переменные, определяемые временем до наступления события (время до достижения индекса EASI50 и время до ответа согласно баллу по шкале IGA) анализировались с помощью логрангового критерия. По двум группам лечения были выполнены кривые выживаемости Каплана-Майера. В этом исследовании применялись следующие методы анализа: (а) цензурированный ПДПН (LOCF): Если пациент применял запрещенный препарат или выбывал из исследования, то значения по данным эффективности считались отсутствующими. Затем все отсутствующие значения были подставлены методом простого ПДПН. (b) Простой метод наблюдаемых случаев (observed case): Анализировались только наблюдаемые случаи.
E. Безопасность
В целом в этом исследовании mAb1 был безопасным и хорошо переносился. Смертельных исходов не было. Было зарегистрировано одно серьезное нежелательное явление (СНЯ) у пациента в группе приема плацебо - потеря сознания; в результате пациент выбыл из исследования. У других пациентов не было нежелательных явлений, которые бы приводили к прекращению лечения. Девятнадцать из 31 пациента, набранных в исследование, сообщали как минимум об одном возникшем во время лечения нежелательном явлении (ВЛНЯ) - 7 пациентов (70%) были из группы приема плацебо, 12 (57%) - из группы приема mAb1. По системно-органным классам (СОК) наиболее частым ВЛНЯ в группе приема mAb1 были инфекции и инвазии: они наблюдались у 12 пациентов (57%) (в группе приема плацебо - у 3 пациентов (30%)). Наиболее частой инфекцией был назофарингит: наблюдался у 5 пациентов (24%) в группе приема mAb1 и у 2 пациентов (20%) в группе приема плацебо. Тяжелые или оппортунистские инфекции не наблюдались. Среди прочих ВЛНЯ, возникавших более чем у одного пациента, были неспецифические симптомы, такие как головная боль (у 3 пациентов (14%) в группе приема mAb1 и у 1 пациента (10%) в группе приема плацебо), сонливость (у 2 пациентов (9,5%) в группе приема mAb1 и у 0% в группе приема плацебо), боль в ротоглотке (у 3 пациентов (14%) в группе приема mAb1 и у 1 пациента (10%) в группе приема плацебо) и кашель (у 2 пациентов (9,5%) в группе приема mAb1 и у 0% в группе приема плацебо). Большинство НЯ имели степень от легкой до умеренной и проходили в течении 2 недель. В группе приема mAb1 наблюдалось одно серьезное НЯ - бактериальный бронхит, начавшийся в день 63 (последняя доза исследуемого препарата была введена в день 22) и считавшийся не связанным с исследуемым лечением. В группе приема mAb1 не наблюдалось нежелательных явлений, которые бы свидетельствовали о неблагоприятном взаимодействии препаратов (mAb1 с ТКС) на кожном уровне. По результатам изучения полученных в ходе лечения потенциально клинически значительных значений (ПКЗЗ) показателей лабораторных анализов, основных показателей жизнедеятельности организма и показателей ЭКГ был установлено, что между двумя группами лечения имеется баланс ПКЗЗ, и не наблюдалось систематического распределения значений или иных четких тенденций, что позволяет предположить, что ПКЗЗ возникали случайно и не были связаны с исследуемым лечением.
F. Результаты
В этом исследовании mAb1 применялся совместно с ТКС пациентами с АД в степени от умеренной до тяжелой. Согласно текущим стандартам лечения АД на протяжении первых 4 недель требовалось контролируемое применение ТКС (т.е. совместно с исследуемым препаратом), что описывается в иных частях настоящего документа. В таблице 36 перечислены ТКС, применявшиеся пациентами, участвующими в исследовании. Пациенты должны были наносить ТКС на все активные поражения кожи один раз в день каждый день до исчезновения поражений, после чего ТКС наносились на участки, предрасположенные к возникновению поражений (т.е. участки, на которых раньше были поражения), один раз в день два дня в неделю. Не менее чем на 50% поражений необходимо было наносить сильнодействующий ТКС (класса III). На поражения, находящиеся на лице, в складках кожи или области гениталий - участках, на которые обычно не показано нанесение сильнодействующих ТКС, - можно было наносить более слабые ТКС (класса I или II). Количество ТКС, использованного за каждую неделю, измерялось путем взвешивания емкостей с ТКС во время их выдачи пациентам и во время следующего визита в клинику. В таблицах 37 и 38 сведены данные об использовании ТКС в период между днем 1 и днем 29.
Препараты ТКС
ТКС, используемые в период между днем 1 и днем 29 - все значения приводятся как средние (СО)
ТКС класса III, используемые в период между днем 1 и днем 29
Демографические характеристики и характеристики заболевания у пациентов обеих групп были по большей части однородными (таблицы 39 и 40). Средние исходные показатели степени АД (согласно шкале IGA, индексам EASI, SCORAD, показателю BSA и шкале оценки зуда NRS) также были достаточно сбалансированными.
Обзор демографических характеристик
Обзор исходных показателей - все значения приводятся как среднее (СО)
300 мг
На фиг. 34-47 и в таблицах 41-44 сведены поисковые показатели эффективности по результатам исследования. Несмотря на относительно небольшое количество пациентов и ограниченный период лечения, по результатам анализа были установлены статистически значительные и клинически релевантные эффекты mAb1 в сравнении с плацебо по основным поисковым конечным точкам эффективности, включая долю пациентов, достигших индекса EASI-50, а также изменение индексов EASI, SCORAD, баллов по шкале IGA и шкале оценки кожного зуда NRS в абсолютном и процентном выражении относительно исходной точки. Некоторые показатели продолжали улучшаться на протяжении нескольких недель после прекращения исследуемого лечения. Сто процентов пациентов в группе приема mAb1 плюс ТКС к 29-му дню достигли индекса EASI-50. В группе приема плацебо плюс ТКС этот показатель составлял 50% (p = 0,0015). По прочим конечным точкам (таким как доля пациентов, у которых балл по шкале IGA достиг 0-1 очка) в группе приема mAb1 в сравнении с группой приема плацебо наблюдалось превосходство числовых показателей, но оно не было статистически значительным (47,6% и 30,0% соответственно). Следует отметить, что пациенты в группе приема mAb1 в среднем использовали приблизительно на 50% меньше ТКС, что могло привести к недооценке эффектов лечения mAb1 относительно группы приема плацебо (прием только ТКС).
Обзор изменений индекса EASI в процентах и в абсолютном выражении за период между исходной точкой и днем 29
Обзор изменений балла по шкале IGA в процентах и в абсолютном выражении за период между исходной точкой и днем 29
Обзор изменений индекса SCORAD в процентах и в абсолютном выражении за период между исходной точкой и днем 29
Обзор изменений балла по шкале оценки кожного зуда NRS в процентах и в абсолютном выражении за период между исходной точкой и днем 29
Препарат mAb1, вводимый подкожно взрослым пациентам с АД в степени от умеренной до тяжелой при попутном применении ТКС, в целом был безопасным и хорошо переносился. Совместное лечение препаратом mAb1 и ТКС приводило к значительно более благоприятному исходу в сравнении с лечением лишь ТКС. Числовые показатели доли пациентов, достигших индекса EASI-50, значительно превосходили результаты недавних исследований, в которых mAb1 применялся в качестве монотерапии (до этого самый высокий показатель доли пациентов, достигших индекса EASI-50, составлял 75%), что дает основания полагать, что эффект mAb1 и ТКС имеет кумулятивный, синергетический характер. Однако, возможно, что такие результаты частично обуславливаются небольшим количеством пациентов и незначительной разницей в характеристиках популяций пациентов различных исследований.
По результатам исследования также установлена дополнительная эффективность mAb1 для пациентов с АД в степени от умеренной до тяжелой при применении ими ТКС. Эти результаты дают основание полагать, что комбинационная терапия может принести дополнительную клиническую пользу пациентам с АД в степени от умеренной до тяжелой, которая бы не наблюдалась при применении лишь mAb1 или лишь ТКС в качестве монотерапии. По результатам исследования также можно предположить о возможной способности mAb1 снижать потребность в ТКС, что потенциально может обеспечить более безопасное долгосрочное лечение пациентов с АД.
Пример 12: Анализ биомаркеров
Анализ биомаркеров выполнялся с использованием образцов, взятых у участников клинических исследований препарата mAb1. В частности, на основе образцов, взятых у пациентов в исходной точке и в различные временные точки после начала исследуемого лечения, измерялся уровень IgE и регулируемый тимусом и активацией хемокин (TARC). Для определения уровня антиген-специфического IgE выполнялся анализ Phadiatop™. Также определялся молекулярный профиль поражений на коже пациентов, участвовавших в клинических исследованиях препарата mAb1.
A. Введение mAb1 здоровым пациентам
В первом клиническом исследовании пациентам вводились однократные дозы mAb внутривенно (в/в) (1,0, 3,0, 8,0 и 12,0 мг/кг) или подкожно (п/к) (150 и 300 мг), либо вводилось плацебо (см. пример 2). Образцы для анализа биомаркеров брались у пациентов, принимавших антитела и плацебо, в дни 1 (исходная точка), 8, 29 и 85 (или при досрочном прекращении участия). В каждом образце измерялись уровни IgE и TARC. Значение p менее 0,10 считалось статистически значительным и допускало небольшое количество участников. Для анализа средних значений использовалась смешанная модель повторных измерений, а для анализа медианных значений - непараметрический анализ. Данные о средних изменениях в процентах уровней IgE и TARC в образцах пациентов сведены в таблицах 45 и 46 соответственно.
Медианное изменение уровня IgE в процентах после введения mAb1 или плацебо, относительно исходного уровня
Медианное изменение уровня TARC в процентах после введения mAb1 или плацебо, относительно исходного уровня
Исходные значения IgE сильно варьировались, что видно из результатов сравнения групп лечения по средним и медианным исходным значениям IgE (фиг. 48А). Границы нормы IgE согласно использованному методу анализа составляют 0-114 тыс. ед./л, и у 15 из 40 пациентов общий уровень IgE в исходной точке превышал 114 тыс. ед./л. Как следует из таблицы 43 и фиг. 48В, в целом уровень IgE снижался пропорционально дозе и времени воздействия mAb1. К 85-му дню после введения препарата у пациентов, получавших mAb1 подкожно, наблюдалось медианное снижение уровня IgE на 16,5% (150 мг) и 17,2% (300 мг). У пациентов, получавших mAb1 внутривенно, также наблюдалось значительное снижение IgE: на 10,7% в группе приема 8 мг/кг и на 25,6% в группе приема 12 мг/кг. В противоположность этому у пациентов, принимавших плацебо, уровень IgE с течением времени возрастал.
Медианные значения уровня TARC в сыворотке крови в исходной точке были в целом схожими во всех группах лечения (фиг. 49А) и превышали значения, о которых сообщается в литературе. Средний уровень TARC в исходной точке составлял 616 пг/мл (границы нормы 134-1327 пг/мл). У здоровых пациентов описывается диапазон уровня TARC в 106-431 нг/л (Hijnen и др., 2004 г., J. Allergy Clin. Immunol. 113: 334-340). В образцах, взятых у пациентов, получавших mAb1 в обеих дозах, наблюдалось значительное снижение уровня TARC в сравнении с образцами пациентов группы плацебо (p=0,044 в группе приема 150 мг; p=0,047 в группе приема 300 мг) (фиг. 49В и таблица 44). Например, введение 300 мг mAb1 подкожно приводило к медианному снижению уровня TARC почти на 35% к 8-му дню (p=0,052), в то время как уровень TARC у пациентов, получавших плацебо, возрастал на 7,7%. Значительное снижение уровня TARC у пациентов, получавших mAb1 как подкожно, так и внутривенно, продолжалось от 29-го дня до окончания исследования (85-й день). По результатам совокупного анализа данных всех пациентов, получавших mAb1, было установлено, что общая разница между группой приема mAb1 и группой приема плацебо по показателям изменения уровня TARC в процентах относительно исходной точки была значительной (p=0,004) (фиг. 50). Значительная разница также наблюдалась в дни 8 (р=0,012) и 29 (р=0,022).
B. Введение mAb1 пациентам с атопическим дерматитом
Уровень биомаркеров также измерялся в образцах, взятых в рамках двух раздельных клинических исследований у пациентов с атопическим дерматитом (АД). В исследовании «А» пациентам с АД вводился либо препарат mAb1 (75, 150 или 300 мг), либо плацебо в дни 1, 8, 15 и 22 (т.е. четыре еженедельных дозы). В исследовании «Б» пациентам с АД вводилось либо 150 или 300 мг mAb1, либо плацебо в дни 1, 8, 15 и 22 (т.е. четыре еженедельных дозы) (см. Пример 7 в этом документе). В обоих исследованиях препарат и плацебо вводились подкожно (п/к). Образцы для анализа биомаркеров брались у участников обоих исследований, принимавших антитела и плацебо, в дни 1 (исходная точка), 4, 8, 15, 22, 25, 29, 36, 43, 50, 57, 64, 71 и 85 (или при досрочном прекращении участия в исследовании). В каждом образце измерялись уровни IgE, TARC, лактатдегидрогеназы (ЛДГ) и антиген-специфического IgE (Phadiatop).
Уровень TARC в сыворотке измерялся с помощью валидированного анализа (набор Quantikine ELISA kit для измерения CCL17/TARC у человека, компания R&D Systems; валидация и анализы выполнялись компанией Quest Diagnostics). Общий уровень сывороточного IgE определялся с помощью анализа ImmunoCAP® (компания Thermo Scientific, утвержден Управлением по контролю качества продуктов питания и медикаментов (FDA); выполнялся компанией Quest Diagnostics). Уровень лактатдегидрогеназы (ЛДГ) измерялся с помощью модульного анализа Roche (утвержден FDA; выполнялся компанией Covance Central Laboratories). Анализ Phadiatop® (компания Thermo Scientific, утвержден FDA) выполнялся компанией Viracor-IBT. Для сравнения изменений уровня биомаркеров относительно исходной точки между группой приема mAb1 и группой приема плацебо применялся двухвыборочный медианный критерий.
У всех пациентов с АД в исследовании «Б» средние исходные уровни TARC, общего IgE и ЛДГ в сыворотке были выше принятого верхнего предела нормы (ВПН) (таблица 47 и фиг. 51).
Обзор исходных показателей биомаркеров
Все пациенты (n=37)
Плацебо (n=10)
Средний исходный уровень эозинофилов находился в верхней части диапазона нормы (таблица 47). У всех пациентов, по которым имеются данные - за исключением двух, - наблюдался положительный результат теста Phadiatop. У обоих этих пациентов также наблюдался нормальный общий уровень сывороточного IgE. По одному пациенту данные теста Phadiatop отсутствовали.
У набранных в исследование пациентов с АД в степени от умеренной до тяжелой наблюдался широкий диапазон значений исходного уровня TARC и IgE. У 27 из 36 пациентов уровень сывороточного TARC превышал 1000 пг/мл (что приблизительно в два раза больше среднего уровня, наблюдаемого у здоровых волонтеров) (фиг. 51А). У 32 из 36 пациентов уровень IgE составлял ≥ 150 тыс. ед./л (зачастую это значение используется для разграничения экзогенного и эндогенного АД) (фиг. 51В). У 17 из 37 пациентов уровень ЛДГ превышал 234 ед./л (фиг. 51С). Ни у одного пациента уровень ЛДГ не был ниже 100 ед./л.
С помощью линейной регрессии в обеих группах лечения препаратом было зафиксировано общее терапевтическое преимущество mAb1 в сравнении с плацебо по части воздействия на общий уровень сывороточного IgE (p<0,0001) (фиг. 52). Общий уровень сывороточного IgE снижался по мере лечения препаратом mAb1, в то время как в группе приема плацебо к концу исследования наблюдалось увеличение этого параметра.
В таблице 48 сведены результаты исследований «А» и «Б» по части медианного изменения уровня IgE в процентах относительно исходной точки в каждой из групп лечения (комбинированные данные).
Медианное изменение уровня IgE в процентах относительно исходного уровня (комбинированные данные исследований «А» и «Б»)
Как следует из таблицы 48 и фиг. 52, в образцах пациентов, получавших mAb1, наблюдалось статистически значительное снижение уровня IgE в сравнении с образцами пациентов группы плацебо. По состоянию на день 85 в группе приема 300 мг mAb1 медианное изменение уровня IgE в процентах составило -23,9%. В группе приема плацебо этот показатель вырос и составлял 41,7% (p<0,0001). В группе приема 150 мг медианное изменение в процентах за период между исходной точкой и всеми временными точками периода между днями 29 и 85 было значительным в сравнении с группой приема плацебо (р<0,03). В группе приема 300 мг медианное изменение в процентах за период между исходной точкой и всеми временными точками периода между днями 15 и 85 было значительным в сравнении с группой приема плацебо (р<0,04).
С помощью линейной регрессии было зафиксировано общее терапевтическое воздействие на уровень ЛДГ. В группе приема 300 мг наблюдалось статистически значительное снижение ЛДГ (р=0,0051) (фиг. 53). Медианное изменение в процентах ни в одной отдельной временной точке не было статистически значительным, но при этом наблюдалась тенденция во времени (p=0,008).
У пациентов с АД лечение препаратом mAb1 приводило к быстрому подавлению уровня сывороточного TARC (фиг. 54). В таблице 49 сведены результаты обоих исследований по части медианного изменения уровня TARC в процентах относительно исходной точки в каждой из групп лечения (комбинированные данные).
Медианное изменение уровня TARC в процентах относительно исходного уровня (комбинированные данные исследований «А» и «Б»)
У пациентов, принимавших 300 мг mAb1, наблюдалось статистически значительное снижение уровня сывороточного TARC в сравнении с пациентами, принимавшими плацебо (p<0,0001; линейный регрессионный анализ). У пациентов, принимавших 300 мг mAb1, статистически значительное подавление продолжалось до 50-го дня, т.е. приблизительно на протяжении одного месяца после введения последней дозы (вводилась в 21-й день исследования). В группе приема 150 мг наблюдалась сравнимая степень эффекта подавления, однако показатели начинали повышаться быстрее, чем в группе приема 300 мг. Статистически значительное подавление (медианное изменение уровня TARC в процентах относительно исходного уровня в сравнении с плацебо) наблюдалось в дни 36 и 43 в группе приема 150 мг (р<0,03), а также в дни 22, 25, 29, 36 и 50 в группе приема 300 мг (р<0,04).
Между пациентами, принимавшими плацебо, на протяжении исследования наблюдались различия по части уровня TARC. В силу активного выбывания пациентов из группы приема плацебо к концу исследования имелись данные только по 4 пациентам этой группы.
В заключение, лечение пациентов с АД препаратом mAb1 приводило к подавлению уровней TARC, IgE и ЛДГ - биомаркеров, связанных с воспалением Th2 и (или) активностью заболевания. У пациентов с АД применение mAb1, в сравнении с плацебо, приводило к быстрому снижению уровня сывороточного TARC. Продолжительность подавляющего эффекта, похоже, зависела от дозы, и результаты показывают, что эффект может продолжаться даже после прекращения приема препарата. У пациентов, принимавших mAb1, наблюдалось значительное снижение уровня общего сывороточного IgE. В группе приема 300 мг снижение уровня IgE (медианное изменение в процентах) продолжалось после фазы лечения, что показывает, что максимальный эффект подавления IgE не был достигнут. У пациентов, принимавших mAb1, наблюдалось устойчивое снижение уровня ЛДГ в сравнении с исходным уровнем. О наличии прямой связи между ЛДГ и IL-4 и IL -13 неизвестно, но связь ЛДГ со степенью заболевания дает основания предполагать, что ЛДГ может быть показателем степени повреждения кожи у пациентов с АД. Подавление TARC и IgE показывает, что mAb1 является мощным ингибитором воспаления Th2.
Взаимосвязь между биомаркерами и связанными с АД параметрами
В исследовании «Б» (см. пример 7) пациентам с сильной степенью АД вводилось 150 или 300 мг mAb1 или плацебо еженедельно на протяжении четырех недель. Для оценки зуда два раза в день применялась числовая рейтинговая шкала оценки кожного зуда (NRS, от 0 до 10). Рассчитывался средний балл по NRS за неделю. Раз в две недели применялся опросник из 5 вопросов для оценки кожного зуда. Предусмотренные опросником 5 вопросов оценивают различные аспектов кожного зуда: интенсивность, длительность, изменение интенсивности, влияние на жизнедеятельность пациента и локализация. Средние исходные баллы составляли 5,5 очков по шкале NRS и 19 очков по опроснику из 5 вопросов. В группе приема 300 мг средний балл по шкале NRS за неделю быстро снижался (среднее изменение в процентах относительно исходного уровня): ко 2-й неделе - на 31,9% (р<0,02), к 7-й неделе - на 55,2% (р<0,01). В группе приема плацебо эти показатели составляли +1,3% и -17,3% соответственно. У пациентов в группе приема 300 мг mAb1 также наблюдалось быстрое снижение балла по опроснику из 5 вопросов (среднее изменение в процентах составило -28,2% ко дню 15 (р=0,0009), -37,1% ко дню 29 (р=0,0007), -42,5% ко дню 43 (р=0,012); в группе приема плацебо эти показатели составляли +3,6%, +8,1% и -9,4% соответственно). В группе лечения препаратом также быстро снижался уровень CCL17 в сыворотке - маркера активности IL 4/ IL 13. На протяжении нескольких недель после окончания лечения наблюдалось сохранение эффекта подавления как CCL17, так и кожного зуда. В таблице 50 продемонстрирована взаимосвязь кожного зуда (согласно опроснику из 5 вопросов и шкале NRS) с показателями исхода дерматита (индекс EASI) и CCL17.
В общем, в этом исследовании во всех группах лечения балл по опроснику из 5 вопросов был значительно связан со значениями CCL17 (r=0,46, p=0,004 в исходной точке; r=0,55, p=0,002 в день 29) и индексом EASI (r=0,41, p=0,011 в исходной точке; 0,62, p<0,0001 в день 29). Изменение балла по опроснику из 5 вопросов в процентах было значительно связано с изменением индекса EASI относительно исходного уровня (r=0,65, p<0,0001 в день 15; r=0,61, p<0,0001 в день 29) и CCL17 (r=0,46, p=0,0089 в день 15; r=0,48, p=0,0105 в день 29) по совокупным данным групп лечения в дни 15 и 29. Данные групп лечения также отдельно изучались на предмет взаимосвязи балла по опроснику из 5 вопросов для оценки кожного зуда с индексом EASI и значениями CCL17. По состоянию на день 15 только в группе приема 150 мг наблюдалась сильная и значительная взаимосвязь между изменением индекса EASI в процентах и изменением балла по опроснику из 5 вопросов в процентах (r=0,81, p=0,0005). По аналогии, по состоянию на день 29 значительная взаимосвязь наблюдалась только в группе приема 150 мг (r=0,57, p=0,0036). Хотя и в день 15, и в день 29 наблюдалась значительная общая взаимосвязь между изменением уровня CCL17 в процентах и изменением балла по опроснику из 5 вопросов в процентах, в группах лечения по отдельности такой взаимосвязи ни в один из таких дней не наблюдалось.
Была продемонстрирована значительная взаимосвязь (в степени от умеренной до сильной) между силой зуда, измеряемой с помощью шкалы NRS, и индексом EASI. Однако взаимосвязь балла по шкале NRS со значениями CCL17 наблюдалась только в исходной точке, а по показателям изменений в процентах относительно исходного уровня значительной взаимосвязи установлено не было. Так как лечение взрослых пациентов с АД препаратом mAb1 приводило к быстрому и стабильному снижению кожного зуда, это дает основания полагать, что сигнальная функция IL-4/ IL -13 является основным механизмом в основе связанного с АД кожного зуда. Взаимосвязь между кожным зудом и CCL17 подчеркивает взаимосвязанность IL-4/ IL -13-опосредованного воспаления, активности АД и кожного зуда при сильном АД.
C. Многократное введение mAb1 пациентам с атопическим дерматитом в степени от умеренной до сильной
Уровни IgE и TARC измерялись в образцах, взятых в рамках клинического исследования у пациентов с атопическим дерматитом (АД) в степени от умеренной до сильной. Пациентам с АД вводилось 300 мг mAb1 либо плацебо в дни 1, 8, 15, 22, 29, 36, 43, 50, 57, 64, 71 и 78 (т.е. 12 еженедельных доз) (см. пример 10 в этом документе). В обоих исследованиях препарат и плацебо вводились подкожно (п/к). Образцы сыворотки крови для анализа биомаркеров брались у участников обоих исследований, принимавших антитела и плацебо, в дни 1 (исходная точка), 8, 15, 22, 25, 29, 36, 43, 50, 57, 64, 71, 85, 99, 113, 127, 141, 155, 169, 183 и 197 (окончание исследования) или при досрочном прекращении участия в исследовании. В каждом образце измерялись уровни IgE, TARC и антиген-специфического IgE (тест Phadiatop™).
TARC - это хемокин, индуцируемый IL-4/ IL -13, сильно влияющий на степень АД. Возможно, что он задействован в патогенезе заболевания. Измерялся исходный уровень TARC для прогноза потенциальной пользы лечения. В образцах, взятых после лечения, был измерен фармакодинамический эффект mAb1 на TARC.
У пациентов с АД зачастую наблюдается повышенный уровень IgE. Была установлена взаимосвязь общего уровня IgE со степенью АД. Возможно, что IgE задействован в патогенезе заболевания. Измерялся исходный уровень IgE для прогноза потенциальной пользы лечения. В образцах, взятых после лечения, был измерен фармакодинамический эффект mAb1 на общий уровень IgE.
Тест Phadiatop™ - это диагностический скрининговый инструмент, применяемый in vitro для определения антиген-специфического IgE, вырабатываемого в ответ на распространенные вдыхаемые аллергены. Исходные результаты теста Phadiatop™ анализировались для прогноза потенциальной пользы лечения. В образцах, взятых после лечения, был измерен фармакодинамический эффект mAb1 на антигенные показатели теста Phadiatop™.
По аналогии с результатами предыдущих клинических исследований (см. пп. А и В выше), уровни TARC и IgE снижались и оставались ниже исходных значений на протяжении 16-недельного периода последующего наблюдения, следующего за лечением (фиг. 55-56).
В сравнении с 4-недельным лечением препаратом mAb1, 12-недельное лечение 300 мг mAb1 приводило к более сильному подавлению IgE в 16-недельный период последующего наблюдения (медиана -57%). Степень подавления TARC по состоянию на конец лечения была схожей после 12 недель и недель лечения препаратом mAb1 (медиана -83% и -76% соответственно).
D. Совместное применение mAb1 и топических кортикостероидов пациентами с атопическим дерматитом в степени от умеренной до сильной
Модуляция TARC и IgE изучалась в рамках исследования безопасности и эффективности mAb1 при применении взрослыми пациентами с АД в степени от умеренной до сильной в комбинации с топическими кортикостероидами (ТКС). Между собой сравнивались две группы лечения, в которых применялись еженедельные дозы на протяжении 4 недель: (300 мг mAb1+ТКС) и (плацебо+ТКС). ТКС применялись со дня 1 вплоть до дня 28 (если поражения кожи проходили, применение ТКС прекращалось) (см. пример 11 в этом документе). Измерение показателей у пациентов выполнялось при скрининге, в исходной точке (день 1), затем еженедельно до недели 5, затем раз в две недели до недели 11. У обеих группах лечения уровень TARC снижался. В группе применения mAb1+ТКС подавление TARC было сильнее, чем в группе приема плацебо+ТКС. В дни 22, 29 и 50 наблюдалась статистически значительная разница. В обеих группах лечения также снижался уровень IgE. Между группами лечения не наблюдалось статистически значительной разницы в подавлении IgE.
Исходный уровень TARC по группам лечения
Уровень TARC измерялся с помощью набора Quantikine ELISA kit компании R&D Systems. В таблице 51 сведены показатели исходного уровня TARC по группам лечения. В обеих группах лечения средние и медианные исходные показатели уровня TARC находились за границами нормы в 106-431 пг/мл (Weihrauch и др., 2005 г., Cancer Res. 65: 13), а также были выше исходных показателей, описанных в п. А выше.
Среднее изменение уровня TARC в процентах относительно исходного уровня с указанием стандартного отклонения (СО)
В обеих группах лечения уровень TARC по сравнению с исходным уровнем снижался. Соответственно ТКС сами по себе имеют способность снижать уровень сывороточного TARC у пациентов с АД. Хотя в группе приема mAb1+ТКС, в сравнении с группой приема плацебо+ТКС, степень медианных изменений уровня TARC в процентах относительно исходного уровня была стабильно больше, разница являлась статистически значительной только в дни 22, 29 и 50 (средняя разность наименьших квадратов по результатам ковариационного анализа) (таблица 52).
В таблице 53 сведены средние и медианные исходные показатели IgE.
Исходный уровень IgE по группам лечения
В обеих группах лечения уровень IgE снижался. После дня 29 наблюдалась тенденция к более сильной степени медианного изменения уровня IgE в процентах в группе приема mAb1+ТКС. Ни в одной временной точке исследования разница не была статистически значительной (средняя разность наименьших квадратов по результатам ковариационного анализа, таблица 54).
среднее изменение в процентах относительно исходного уровня с указанием СО
В группе приема плацебо+ТКС ко дню 29 приблизительно 50% пациентов достигли индекса не менее EASI50, и показатели подавления TARC и IgE в этой группе соответствовали наблюдавшимся клиническим улучшениям. Ко дню 29 все пациенты в группе приема mAb1+ТКС достигли индекса не менее EASI50 (см. пример 11). В группе приема mAb1+ТКС, в сравнении с группой приема плацебо+ТКС, наблюдалось более сильное подавление TARC и IgE. Однако разница в подавлении TARC была статистически значительной только в дни 22, 29 и 50.
Применение настоящего изобретения не ограничивается конкретными вариантами осуществления настоящего изобретения, описанными в этом документе. Более того, различные модификации данного изобретения, помимо модификаций, описанных в этом документе, будут очевидны средним специалистам в данной области, исходя из вышеупомянутого описания и сопровождающих текст фигур. Такие модификации предполагаются входить в объем, охватываемый формулой данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА С ПОМОЩЬЮ АНТАГОНИСТА IL-4R | 2013 |
|
RU2698907C2 |
| СПОСОБ ЛЕЧЕНИЯ КОЖНОЙ ИНФЕКЦИИ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2704999C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ТЯЖЕЛОГО АТОПИЧЕСКОГО ДЕРМАТИТА ПУТЕМ ВВЕДЕНИЯ ИНГИБИТОРА IL-4R | 2017 |
|
RU2759630C2 |
| СПОСОБЫ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2020 |
|
RU2828020C2 |
| СПОСОБ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА ПОСРЕДСТВОМ ВВЕДЕНИЯ ИНГИБИТОРА ИЛ-4R | 2019 |
|
RU2801204C2 |
| СПОСОБЫ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ АЛЛЕРГИИ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2777328C2 |
| ЛЕЧЕНИЕ АТОПИЧЕСКОГО ДЕРМАТИТА С ПОМОЩЬЮ ТРАДИПИТАНТА | 2021 |
|
RU2818164C1 |
| СПОСОБЫ ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ ВАКЦИНЫ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2753869C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОЛИПОЗА НОСА ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2014 |
|
RU2674680C2 |
| ПРИМЕНЕНИЕ АНТАГОНИСТОВ IL-13 ДЛЯ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА | 2017 |
|
RU2752785C2 |
















































































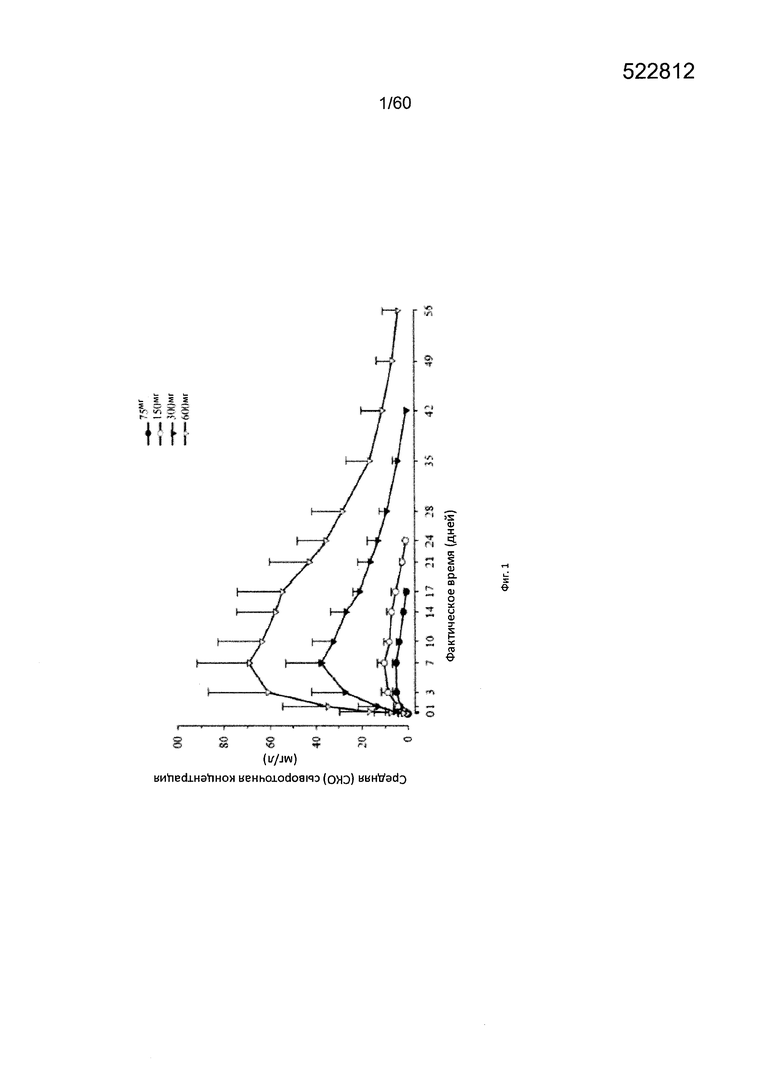

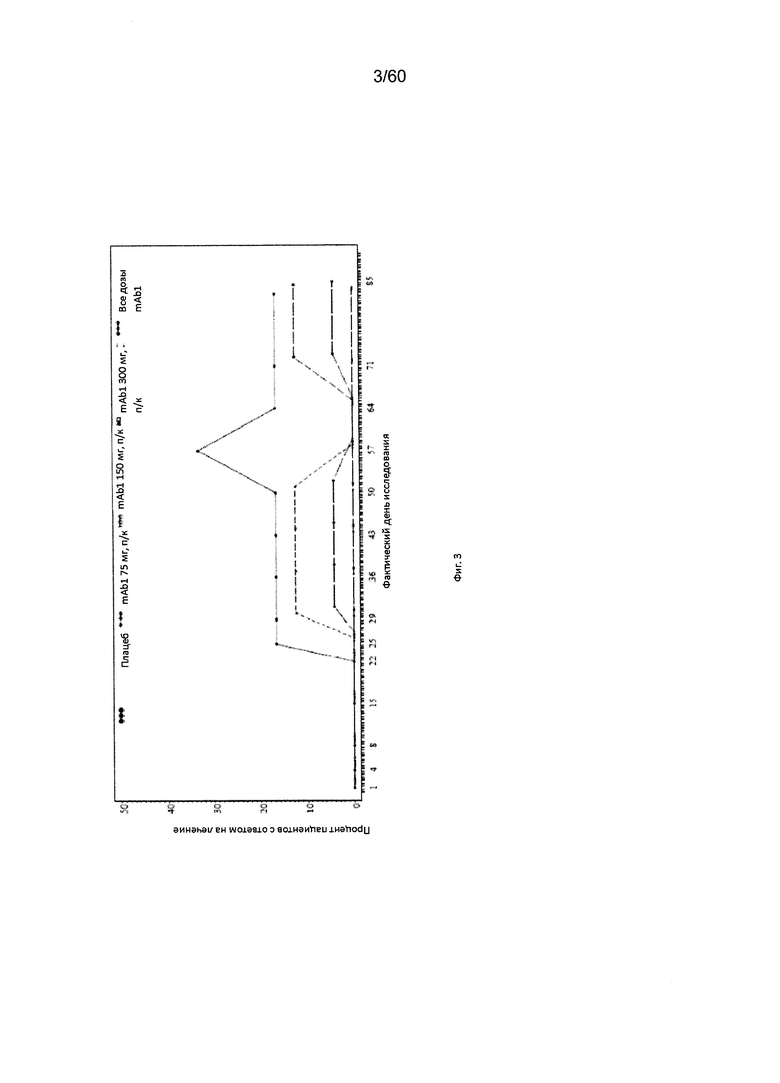
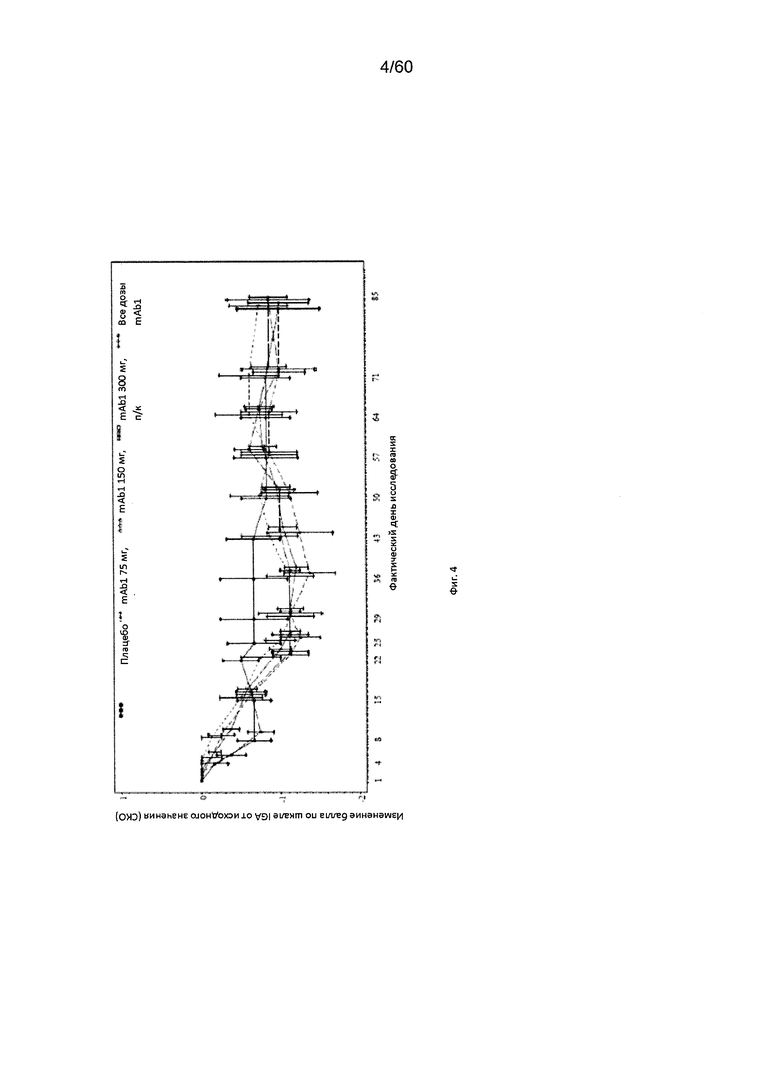
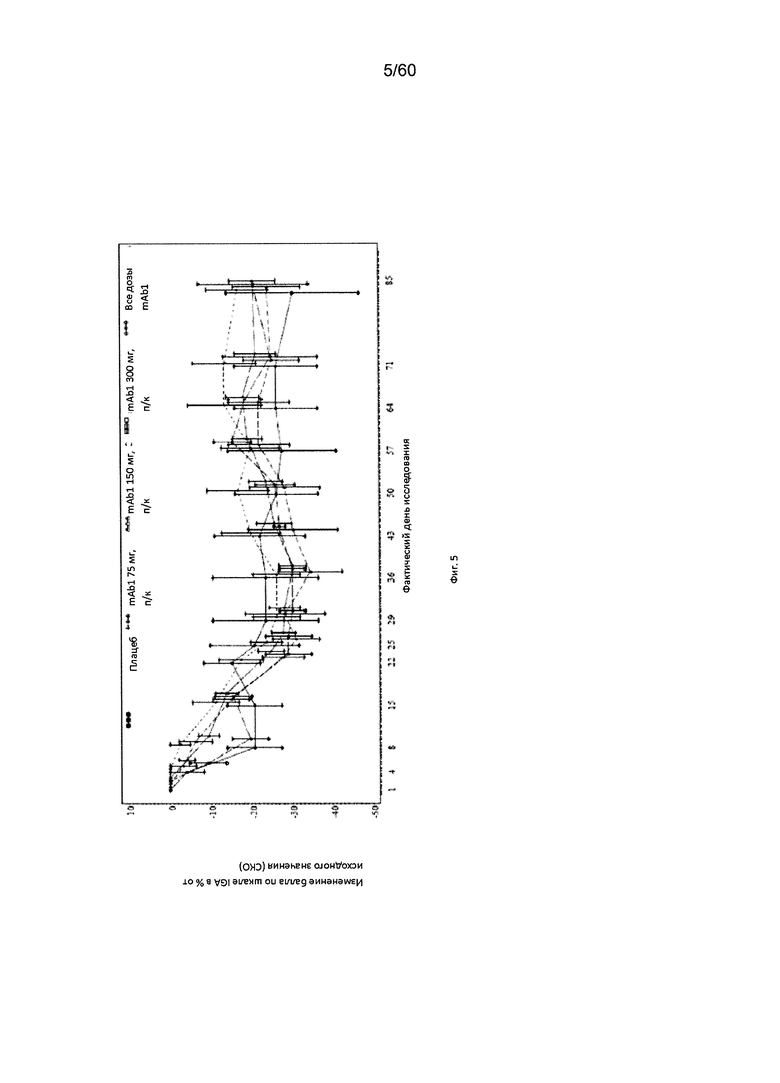
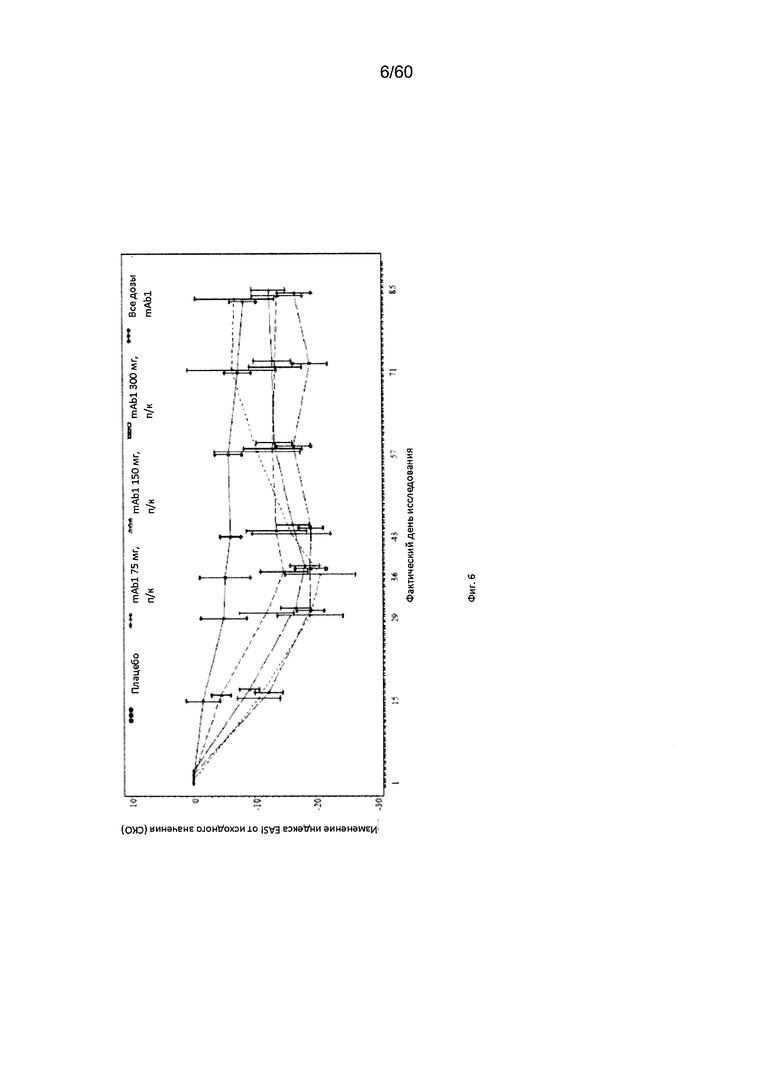
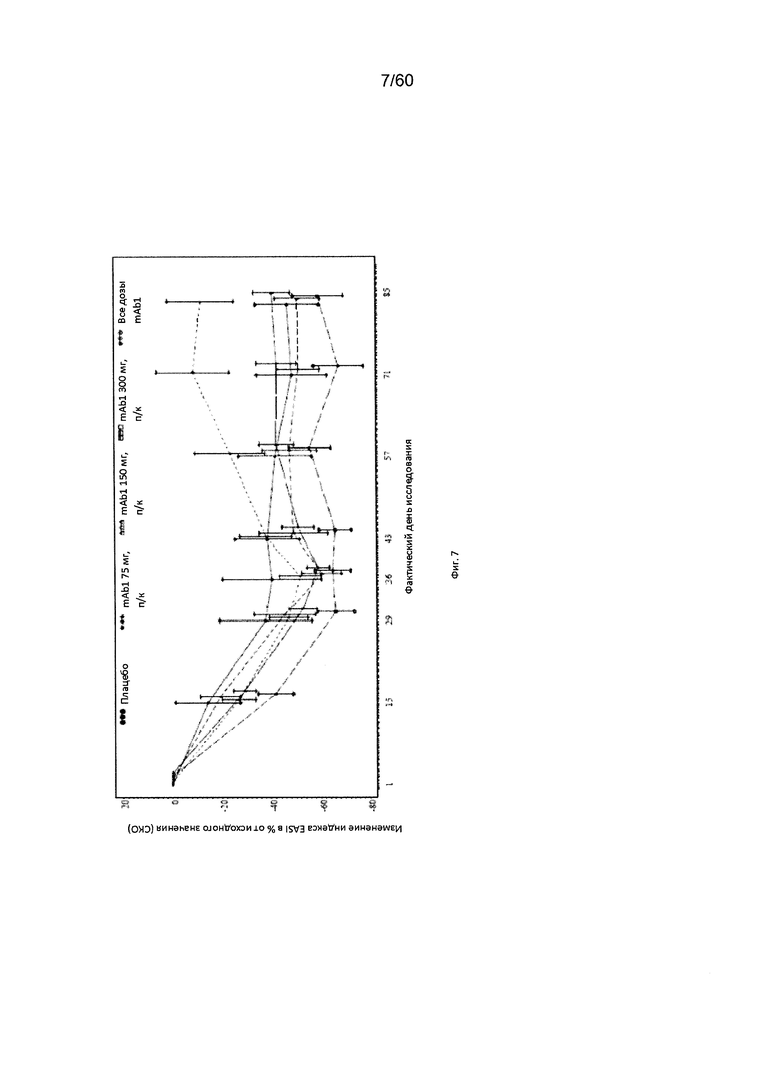
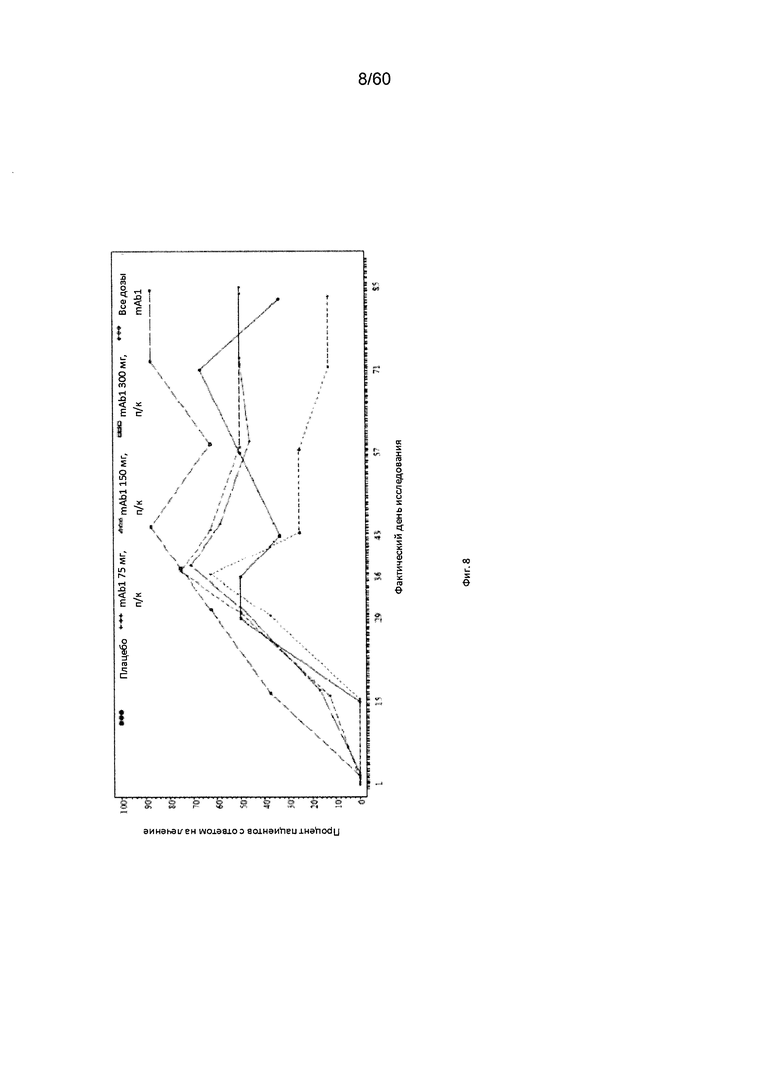
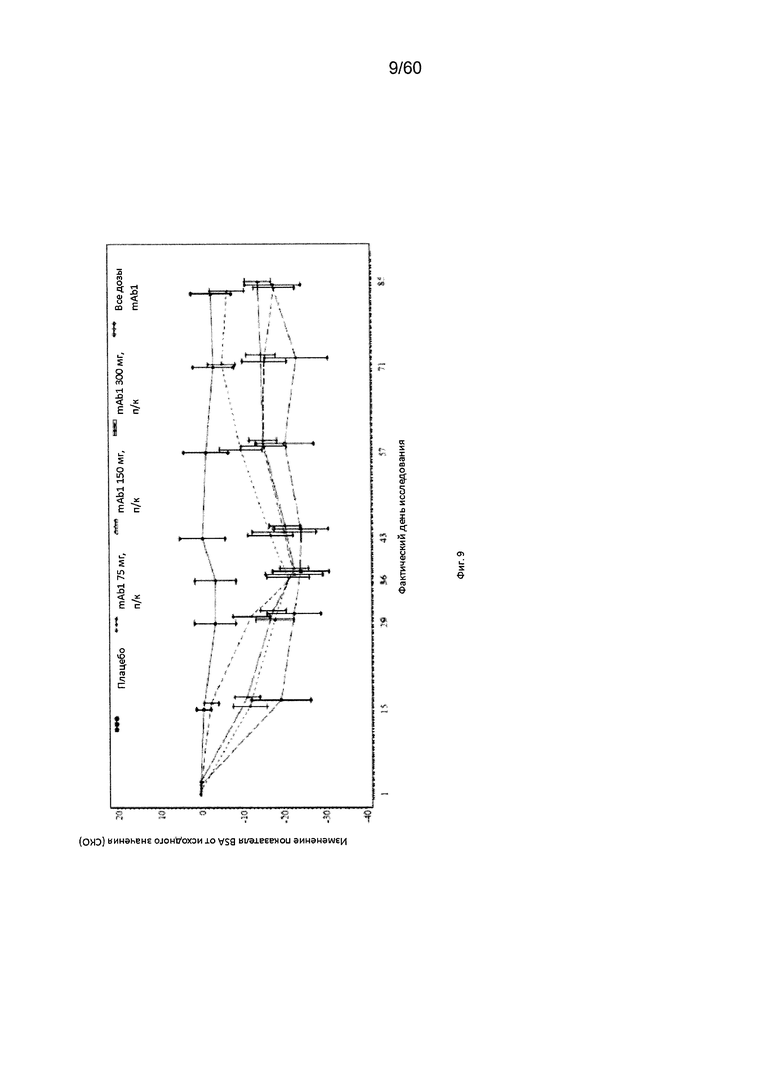
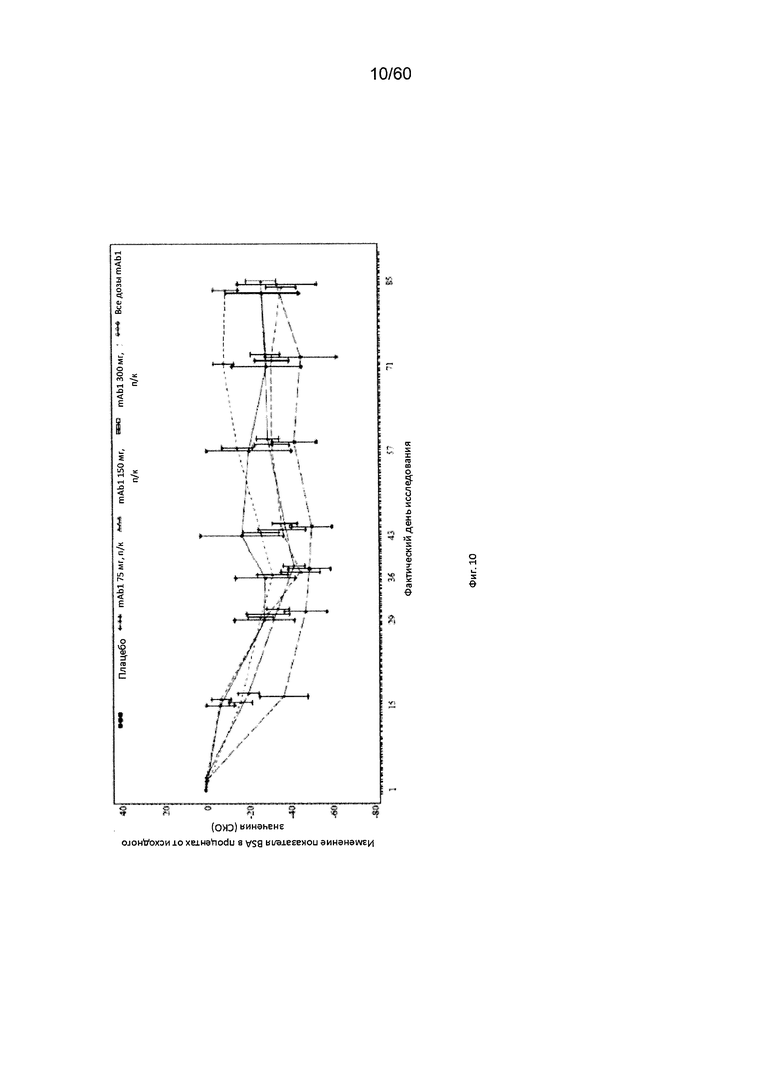
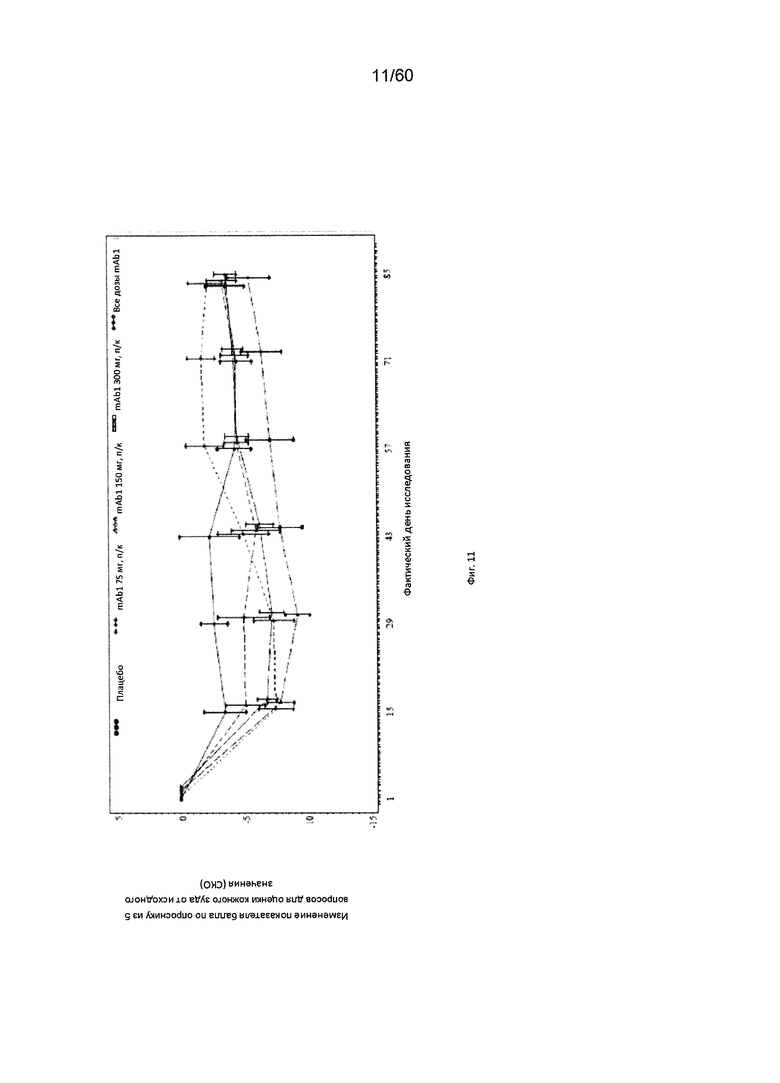
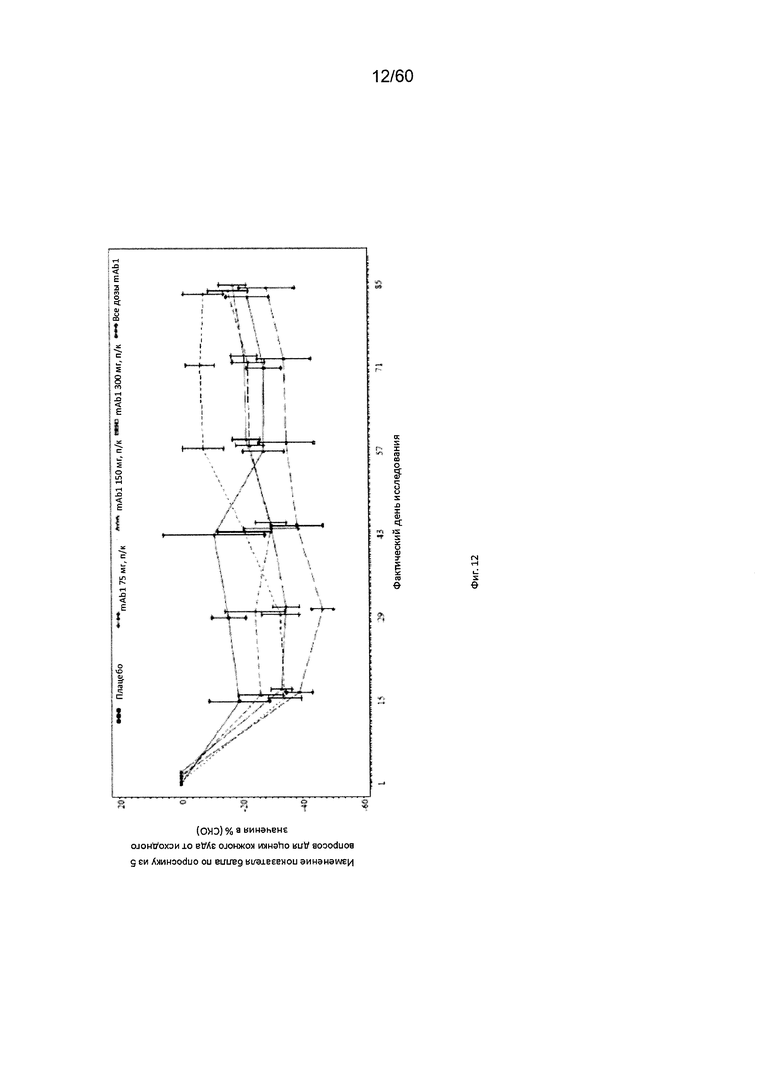
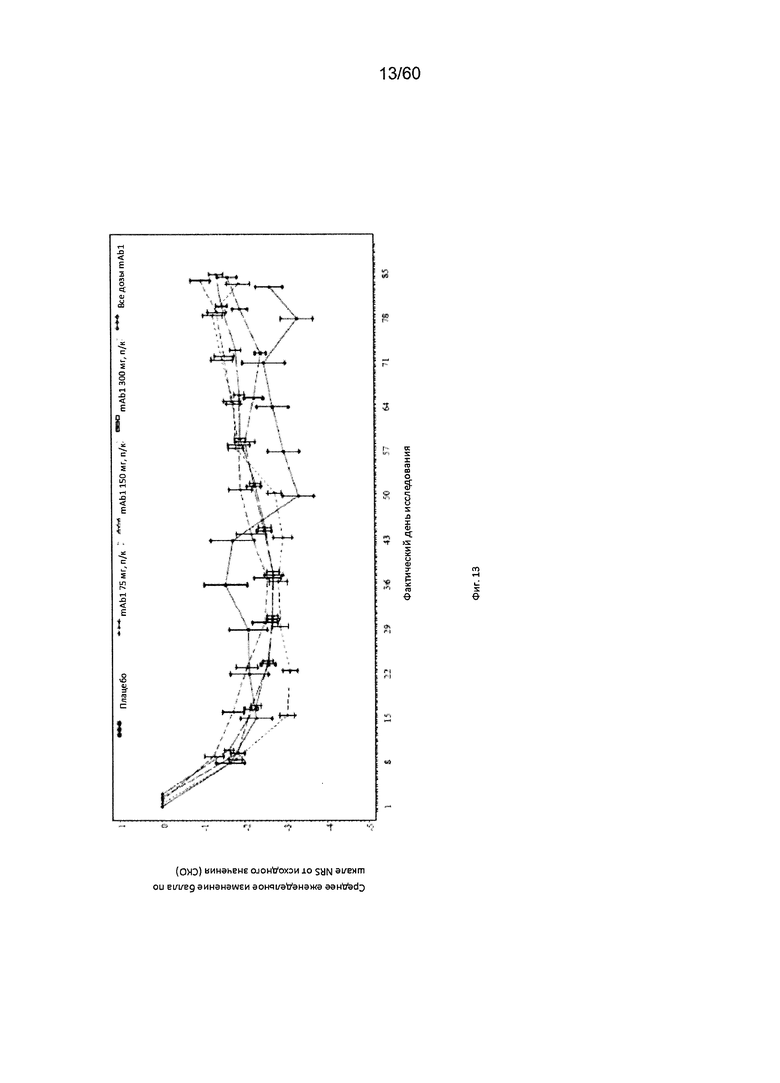
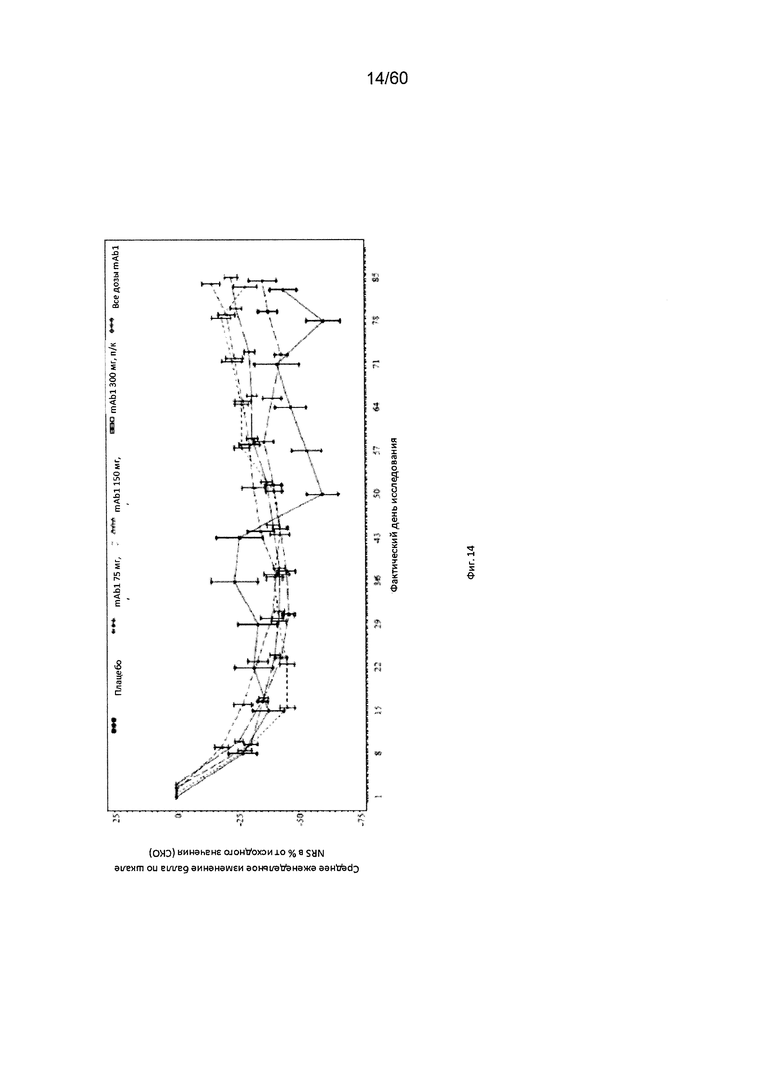
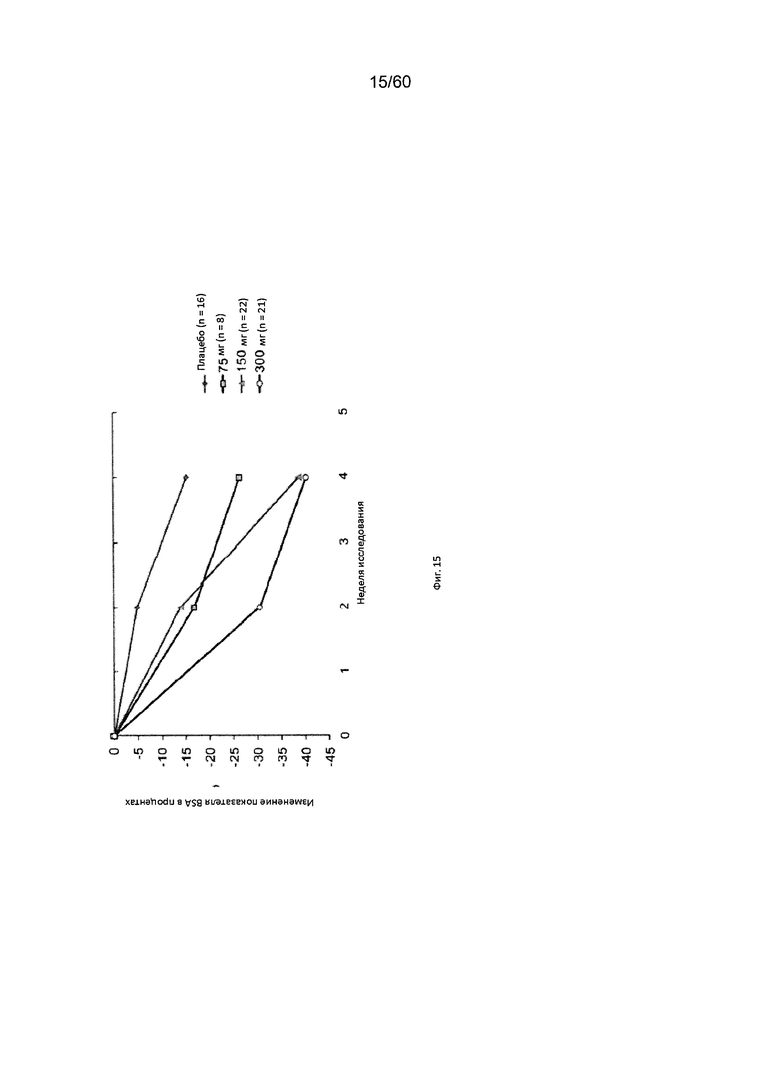
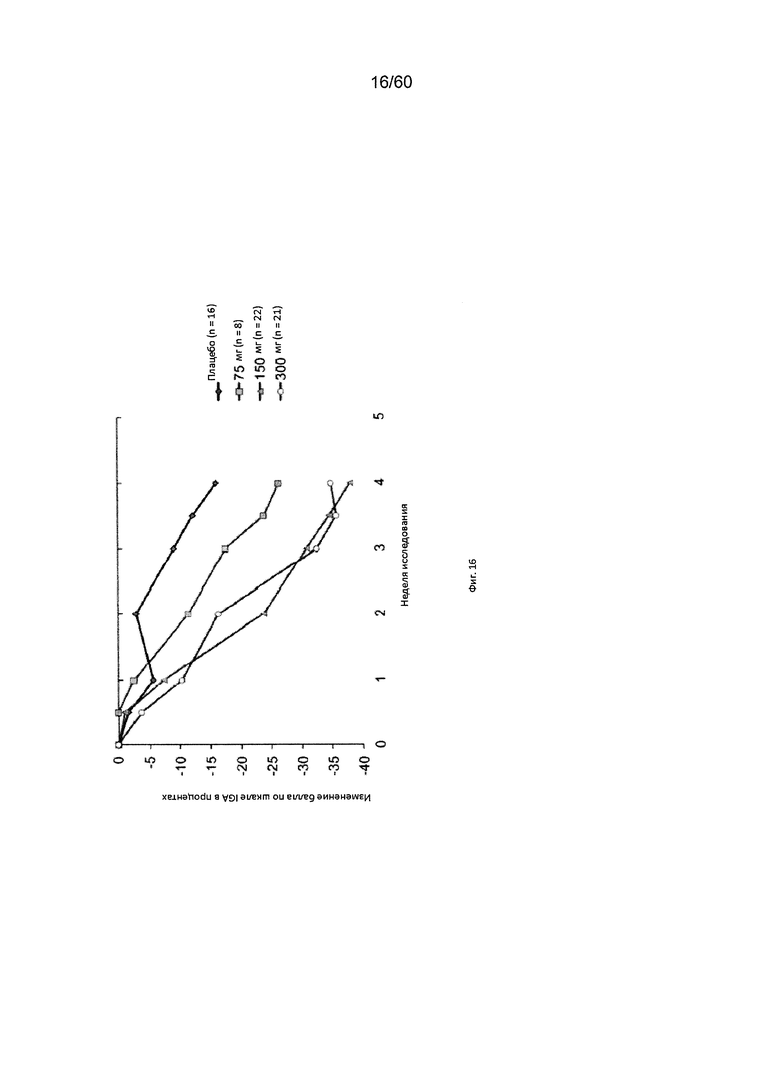
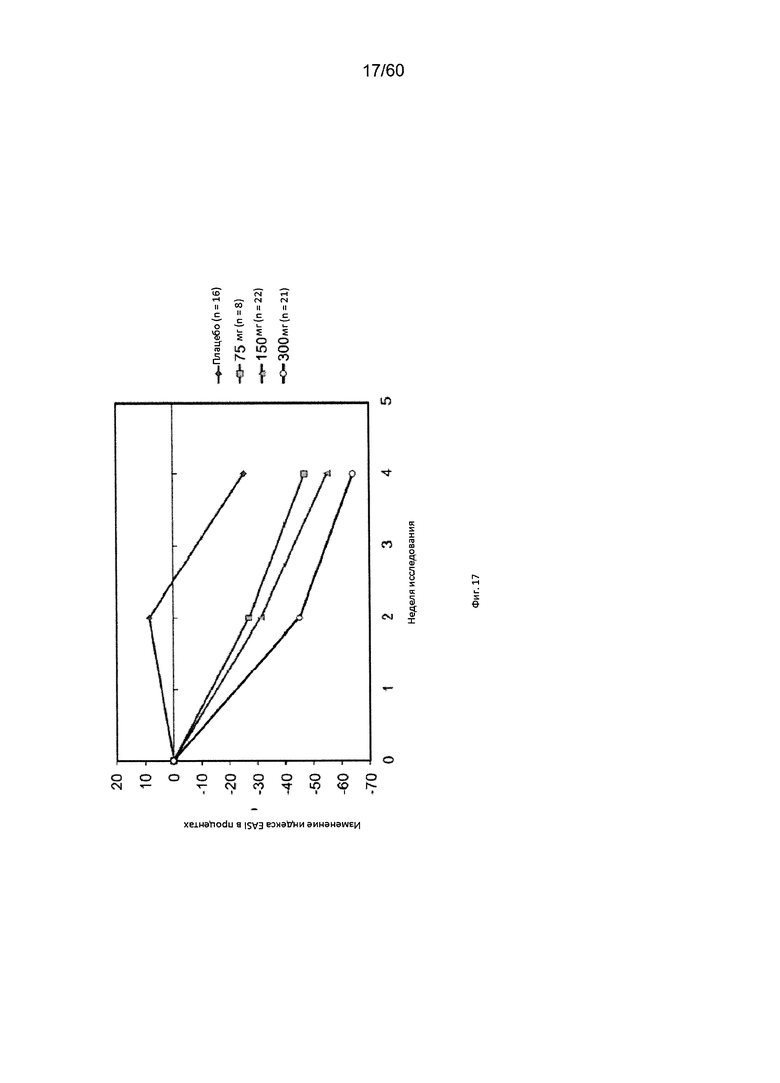
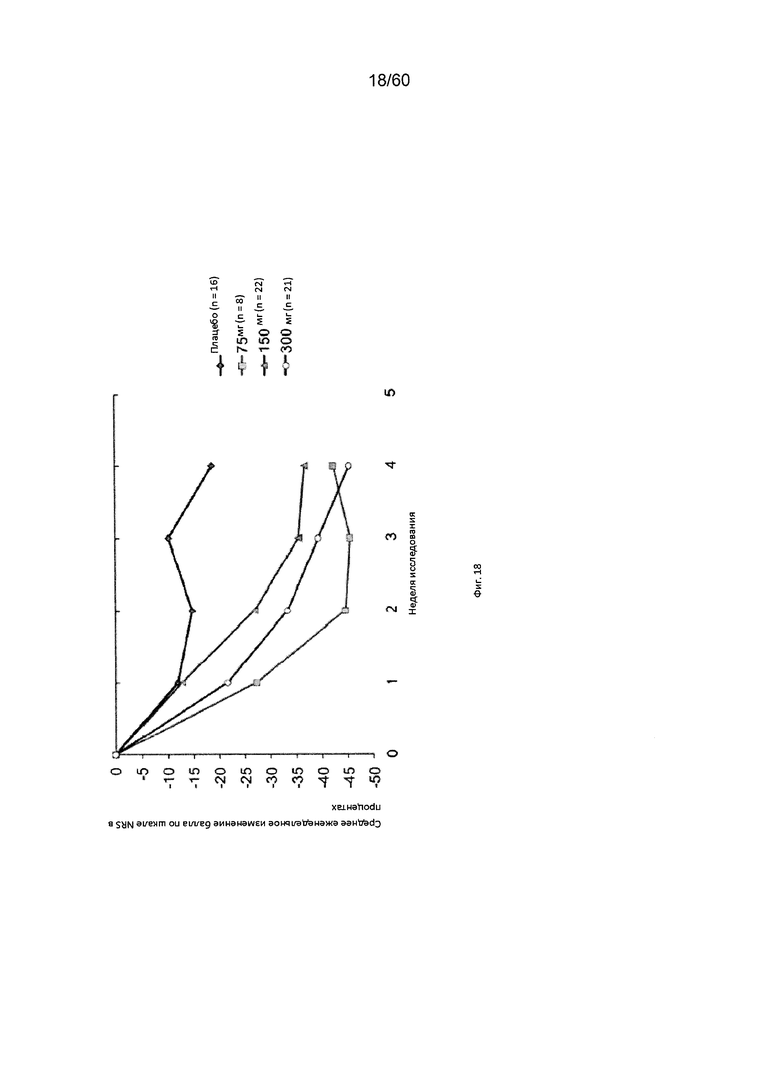
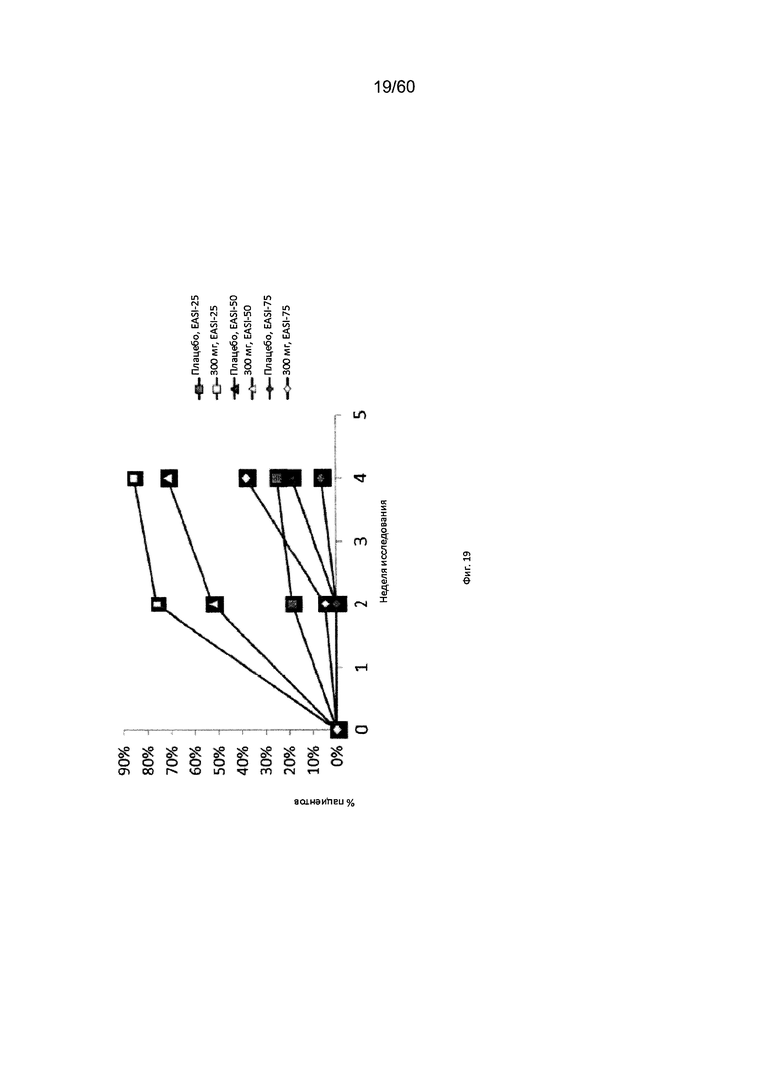
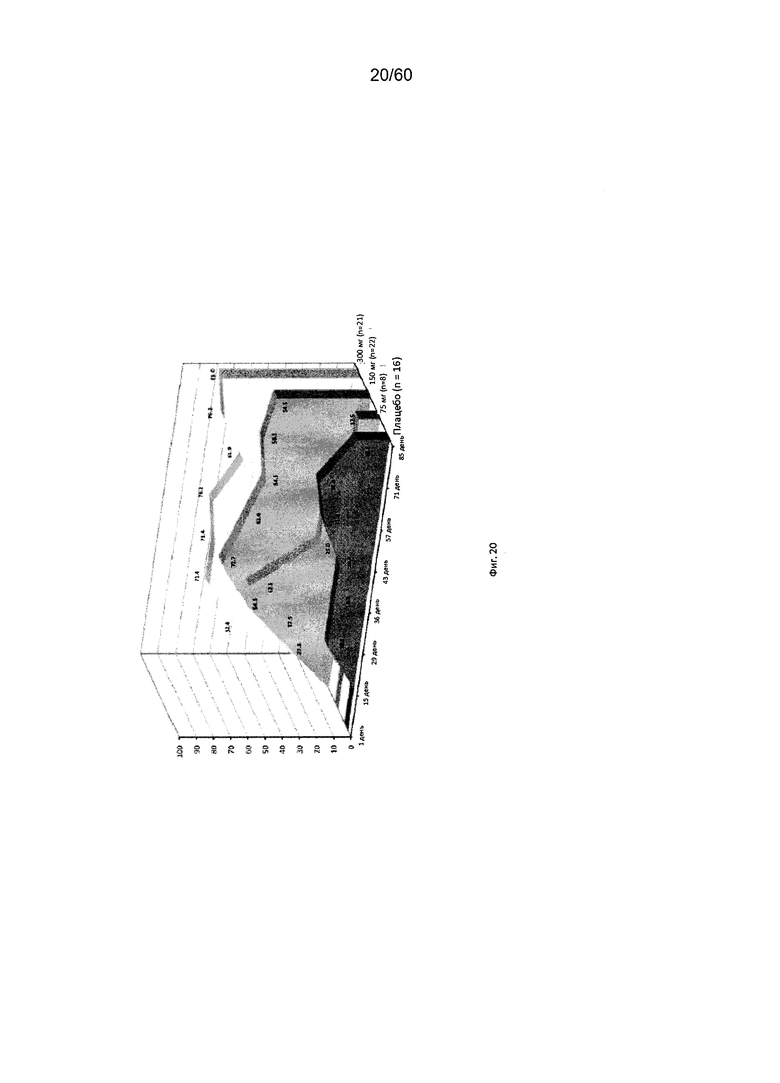



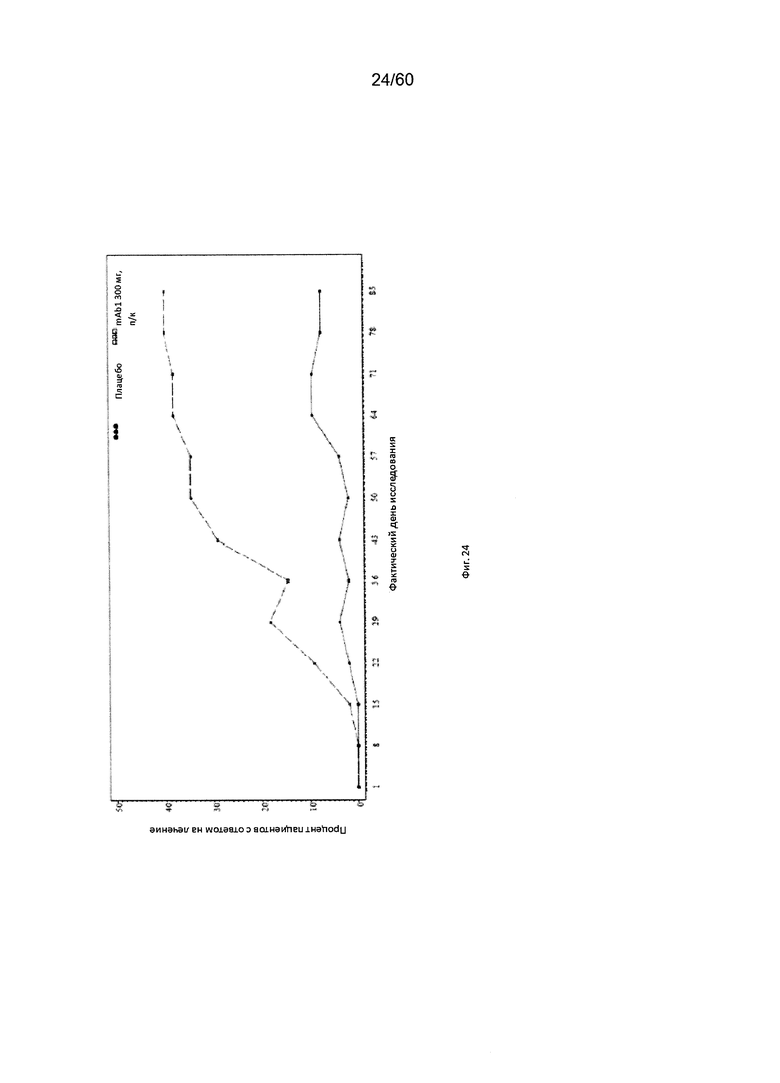
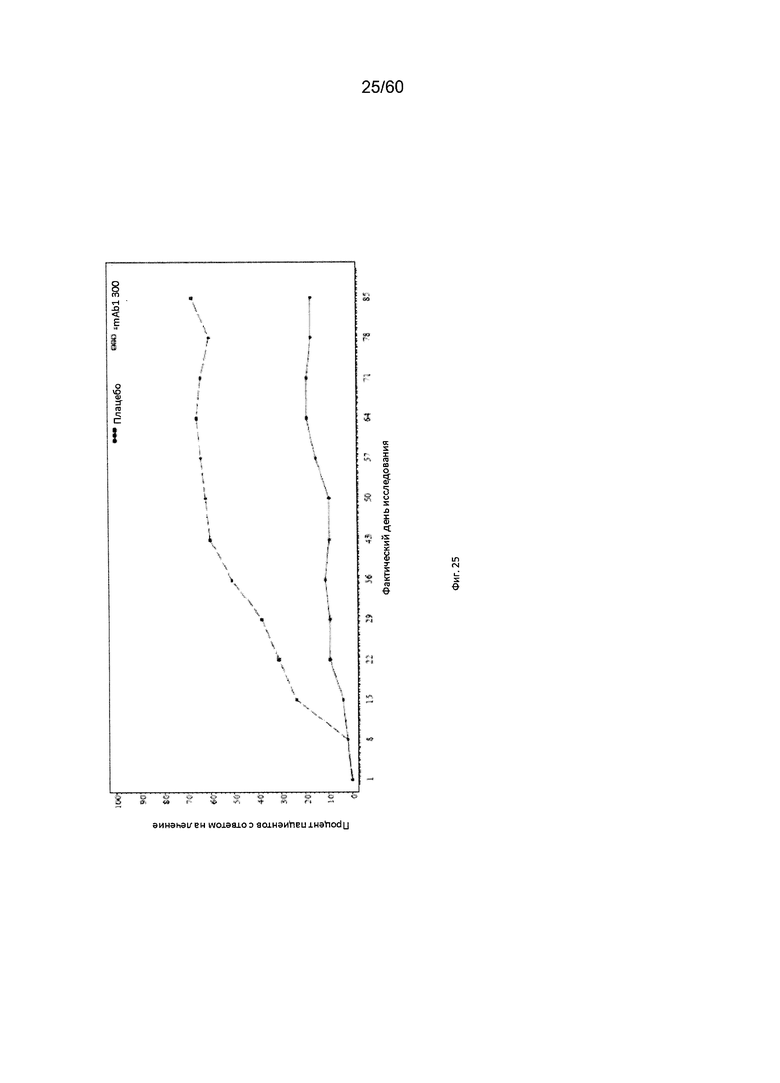
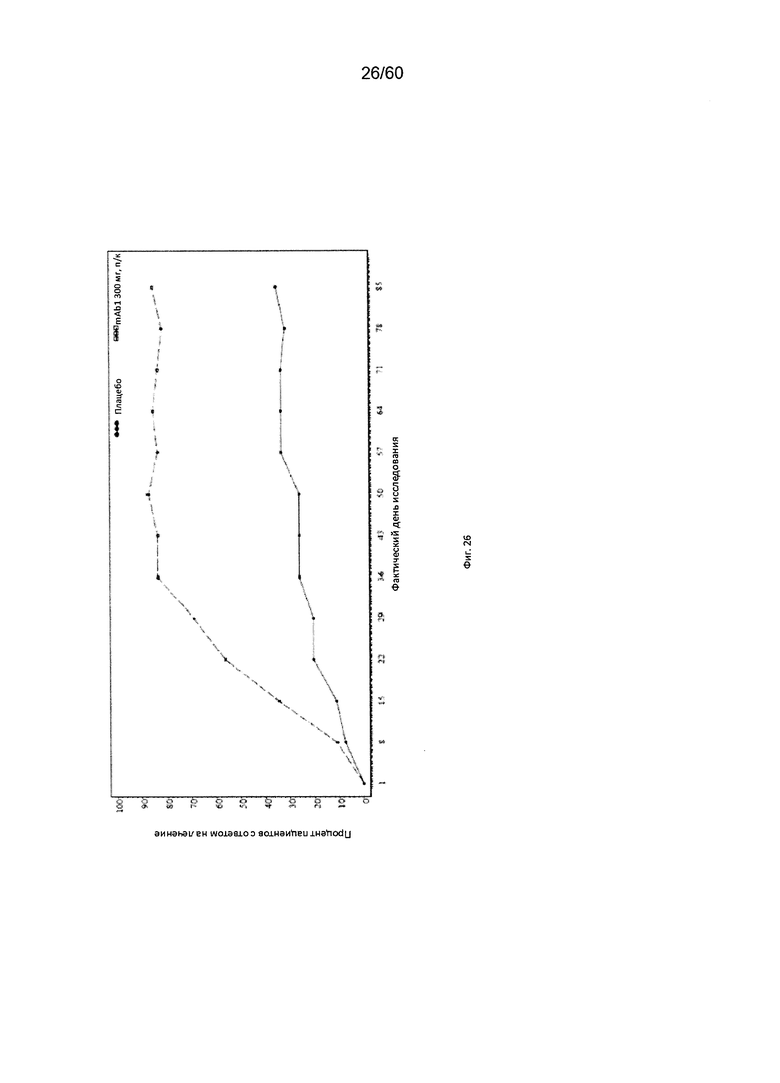
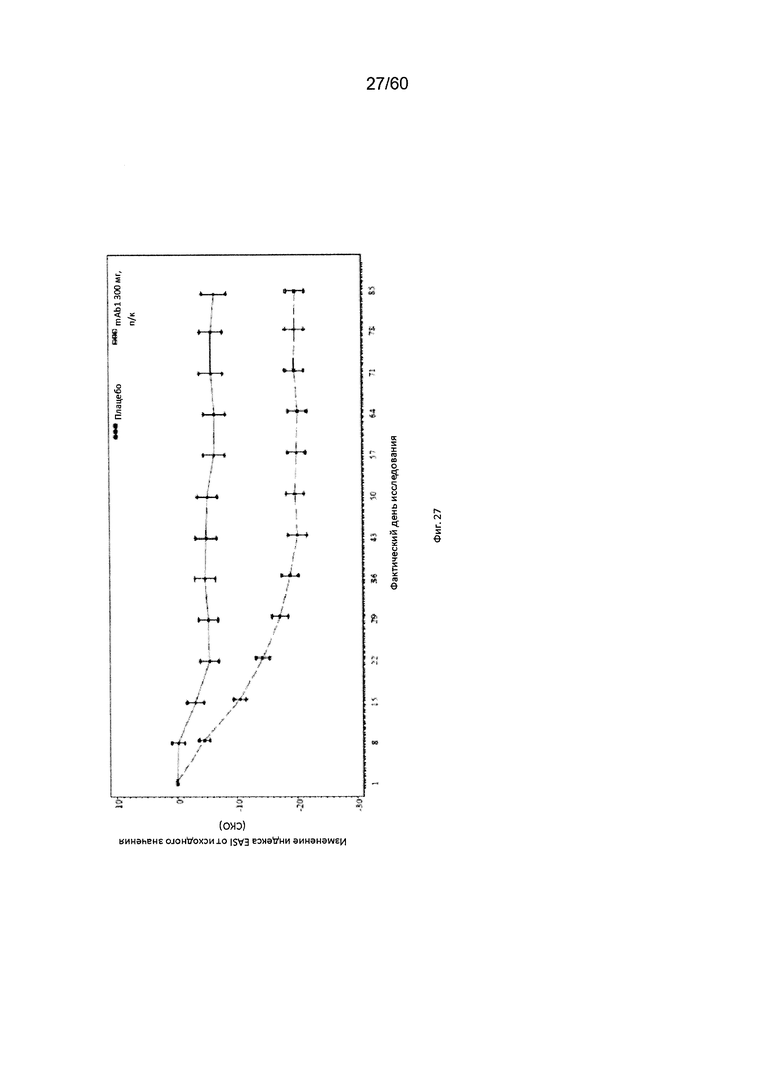


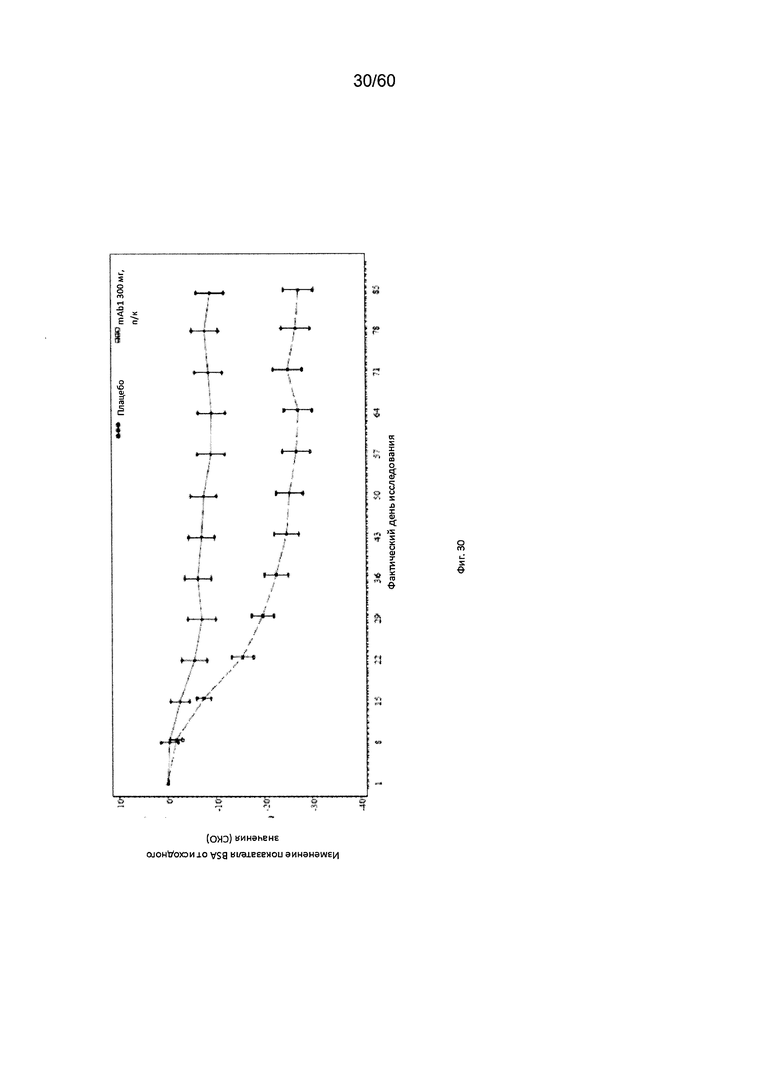
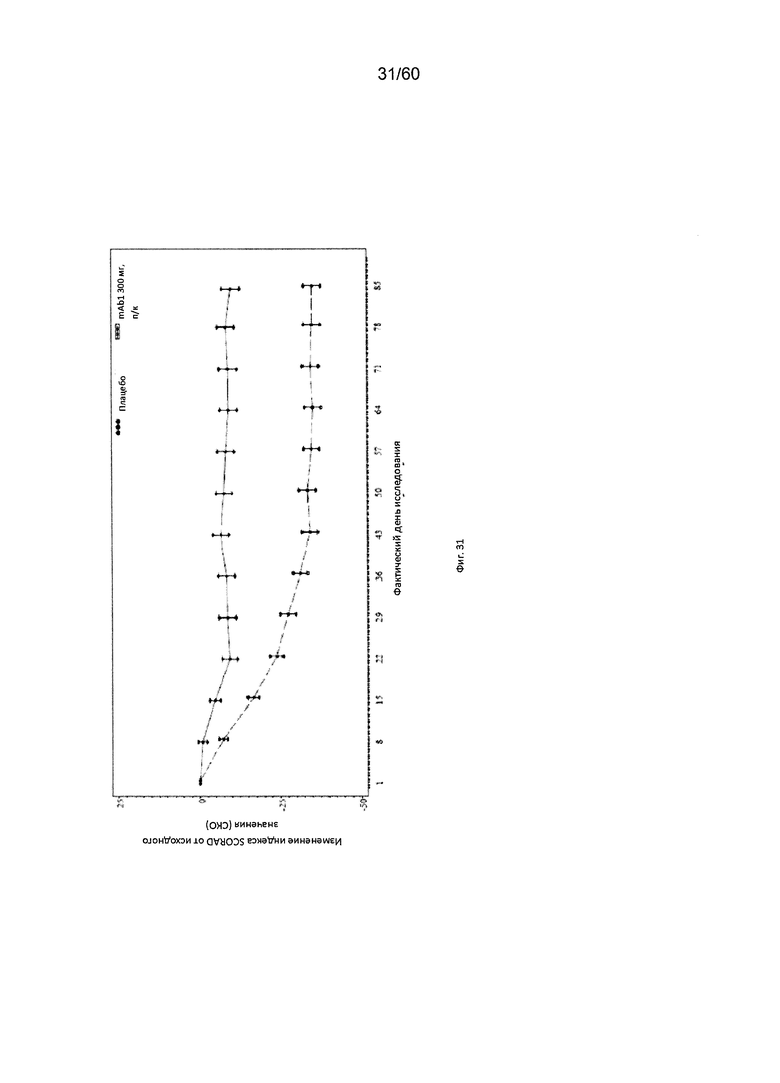

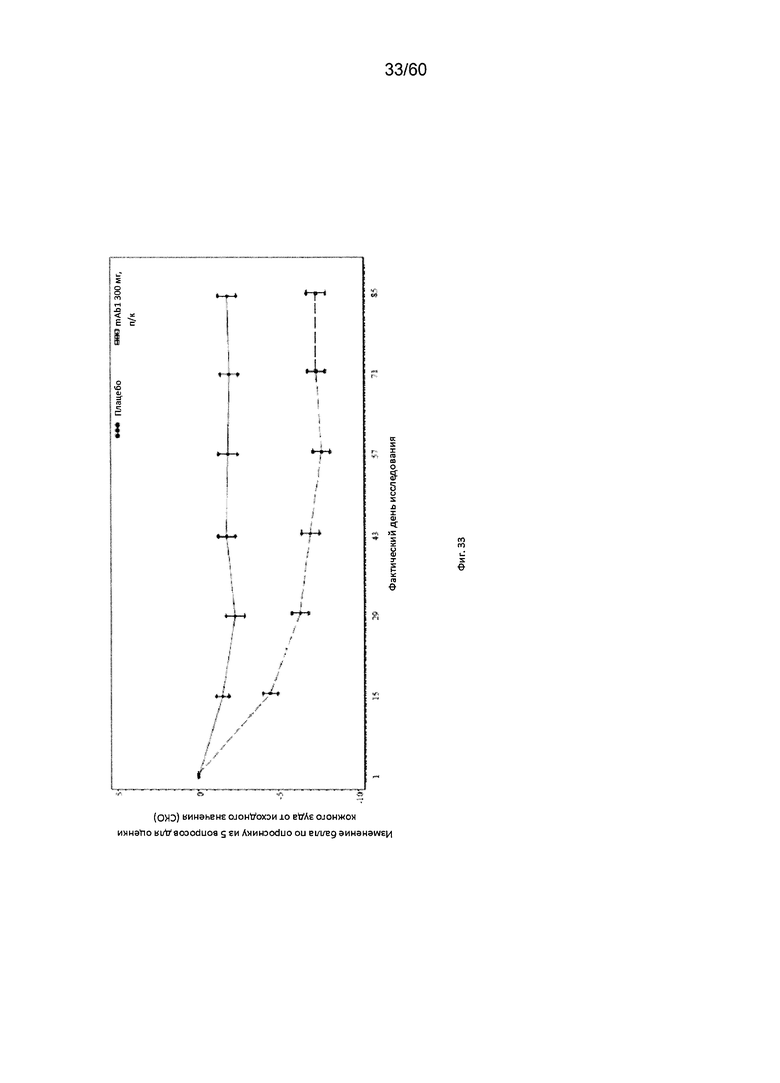

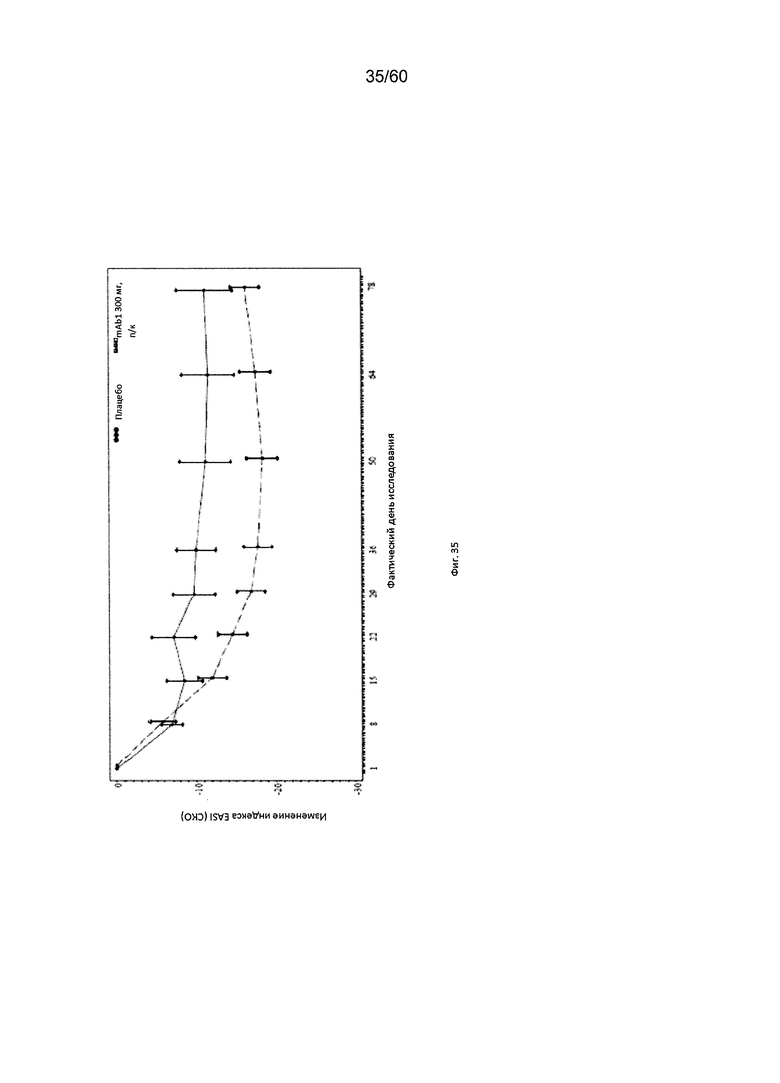
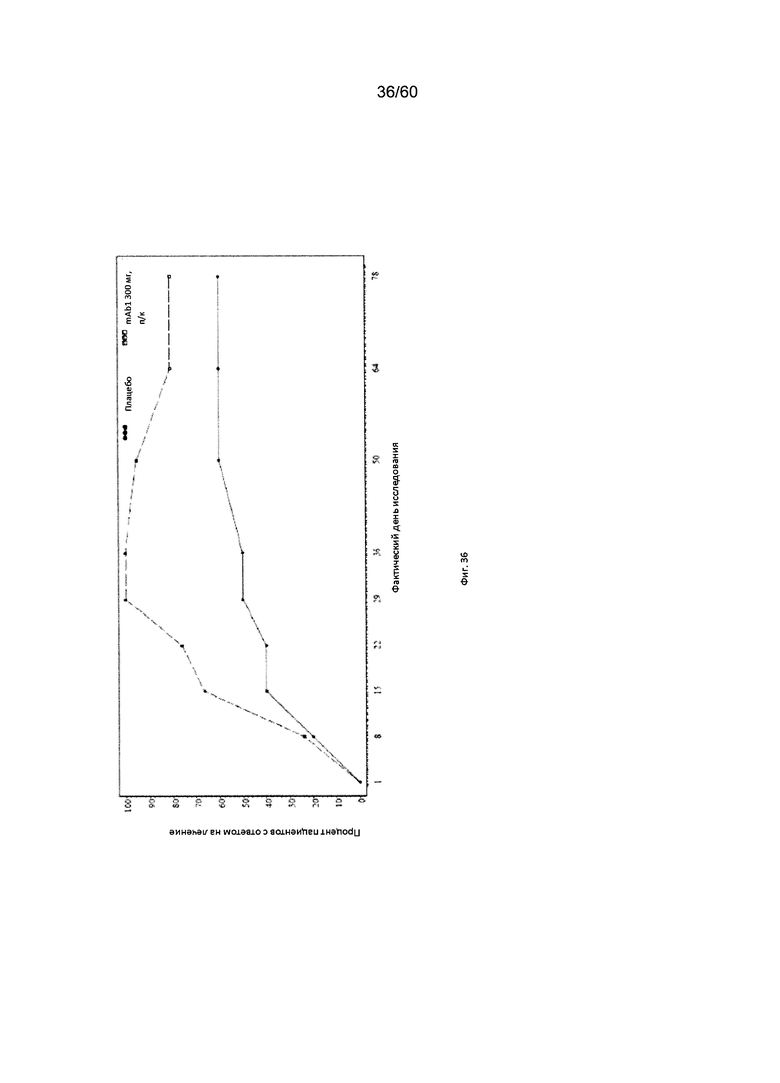
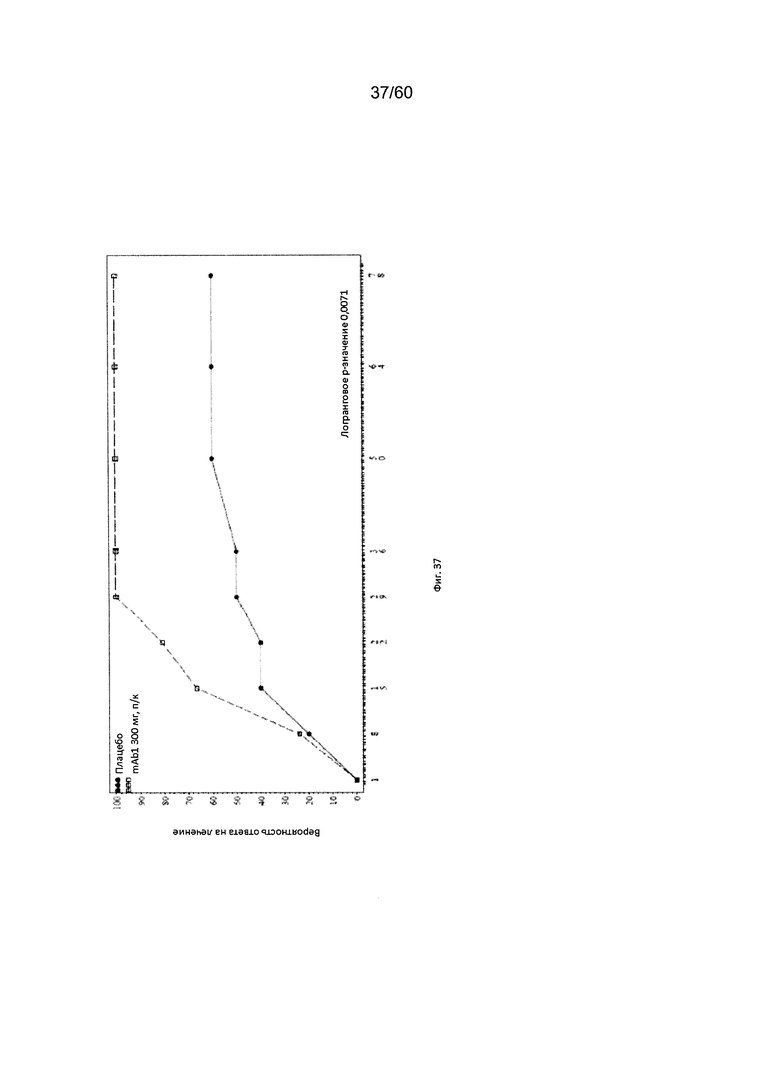
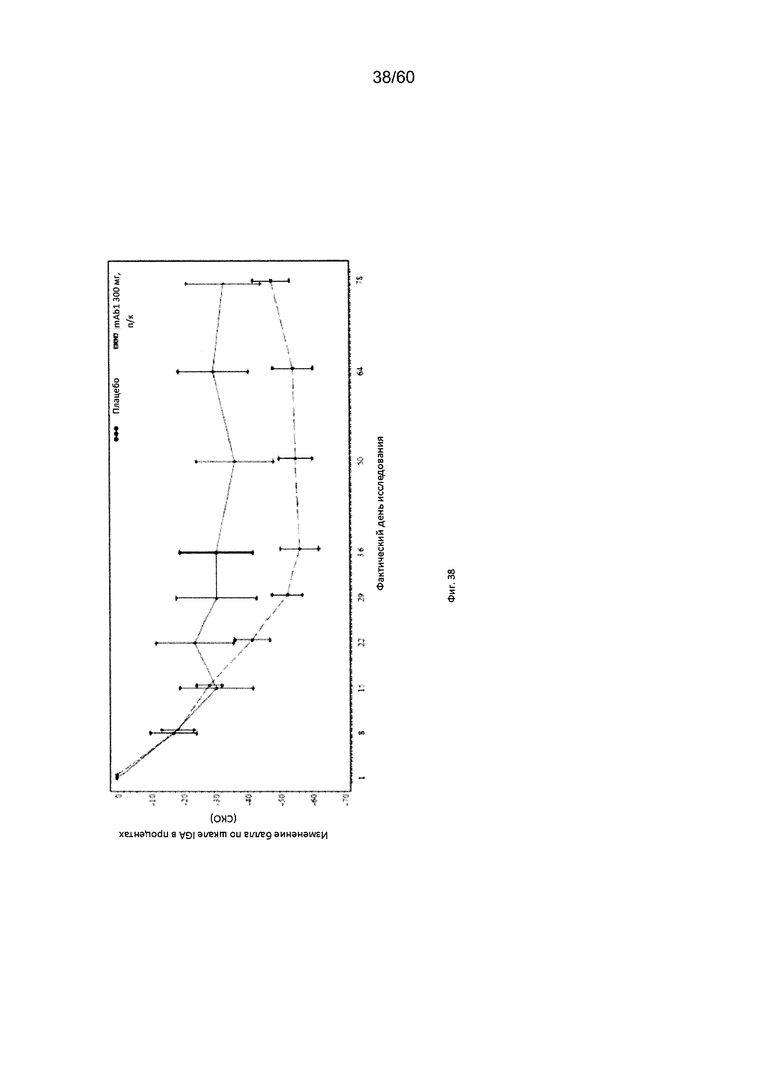
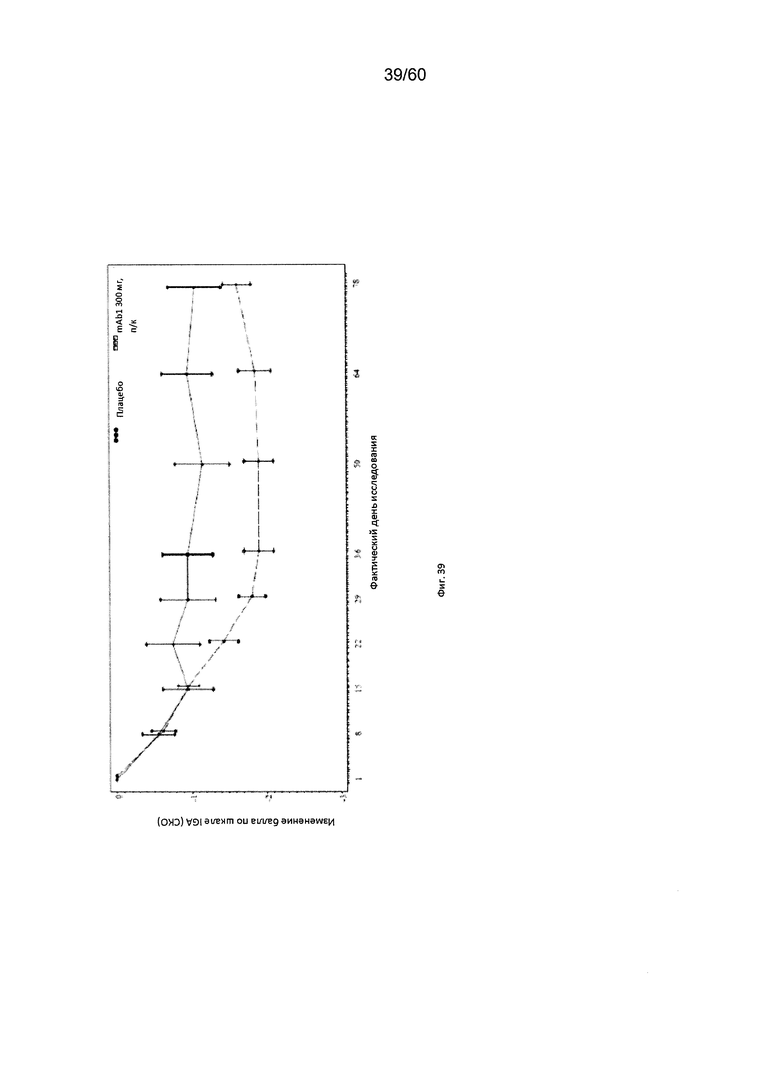
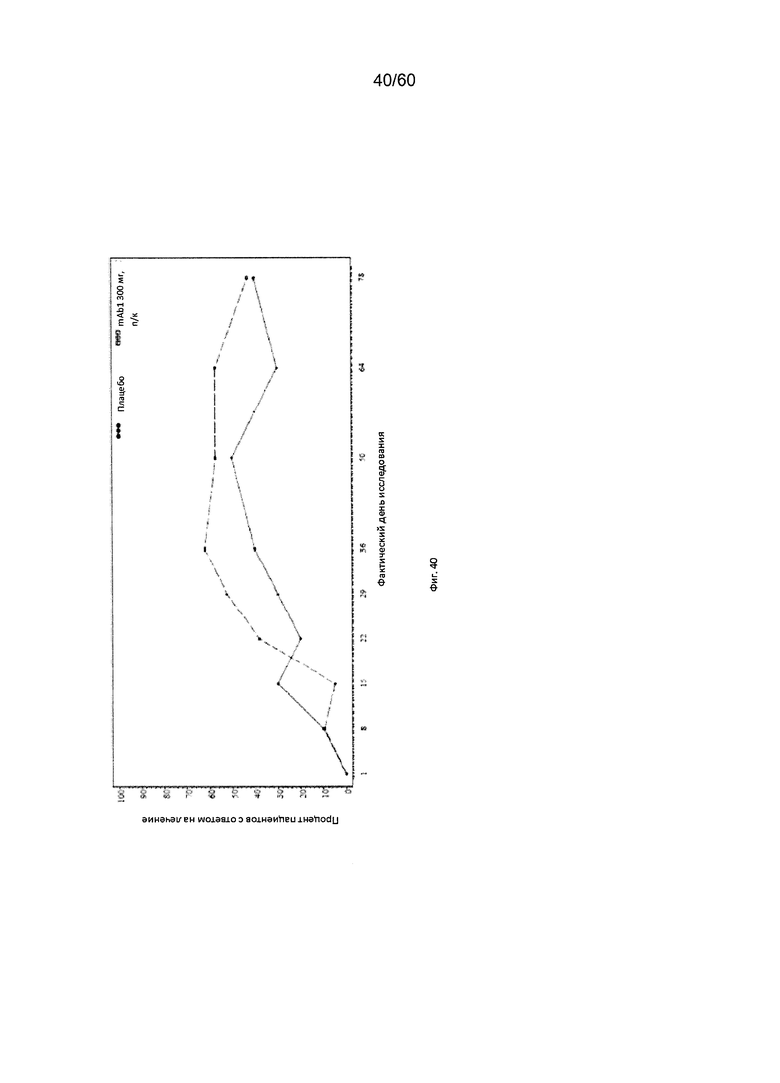
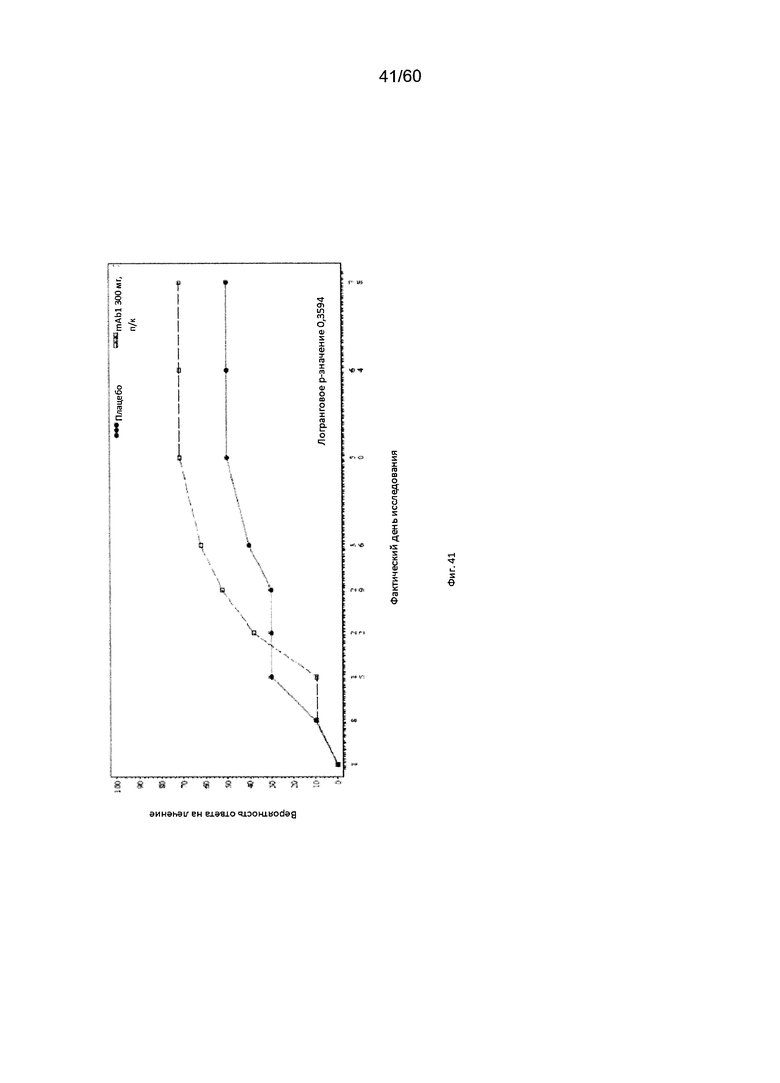
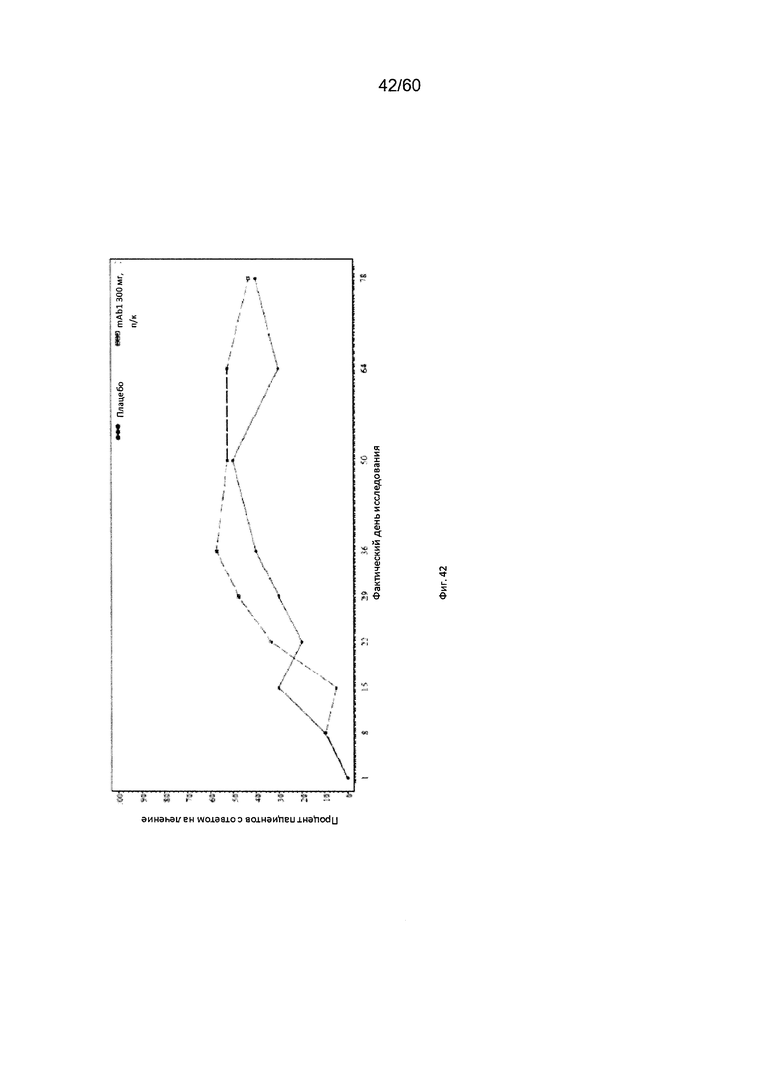
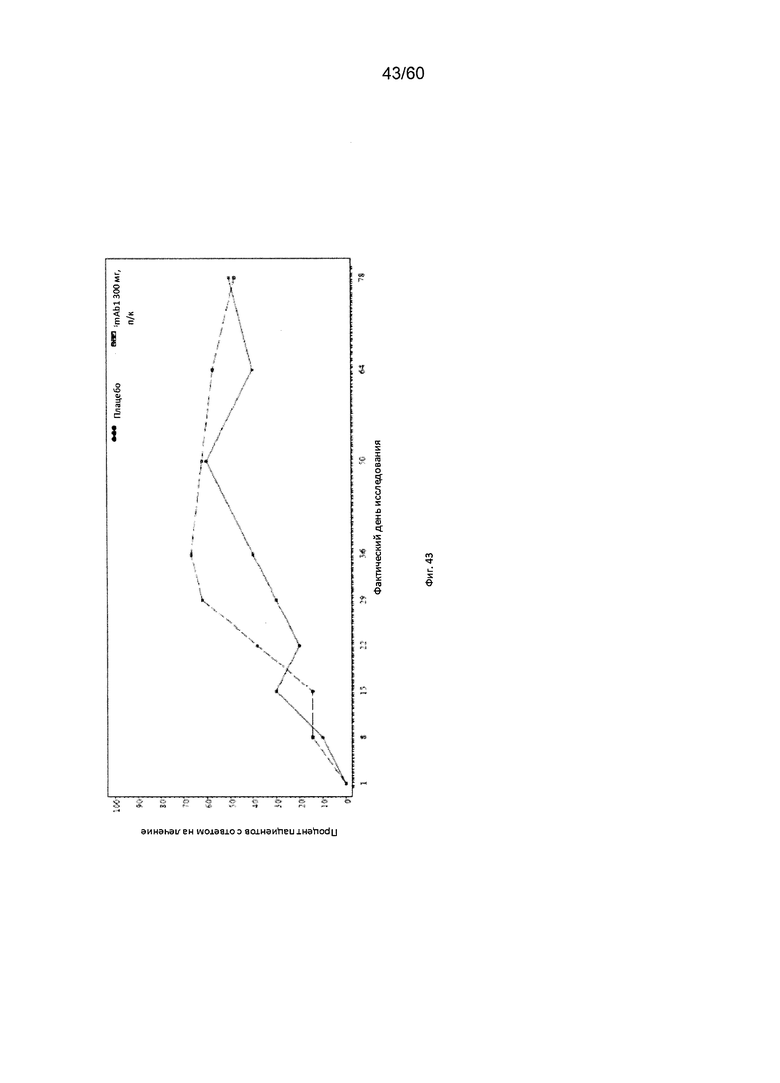
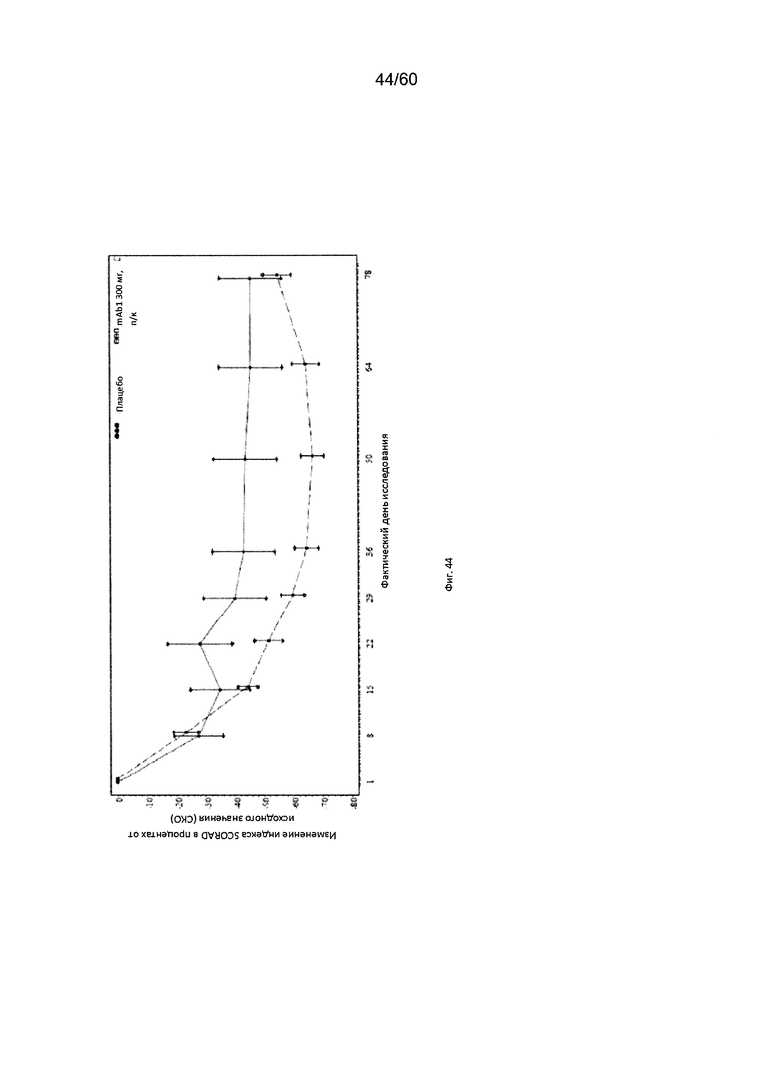
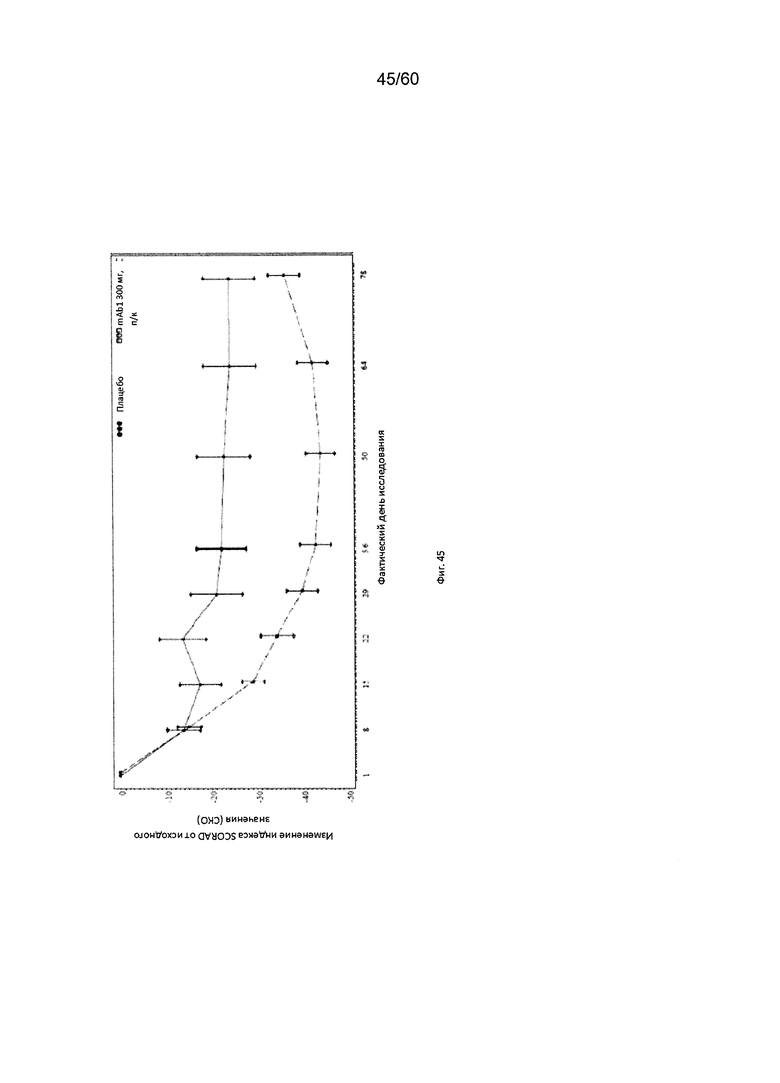
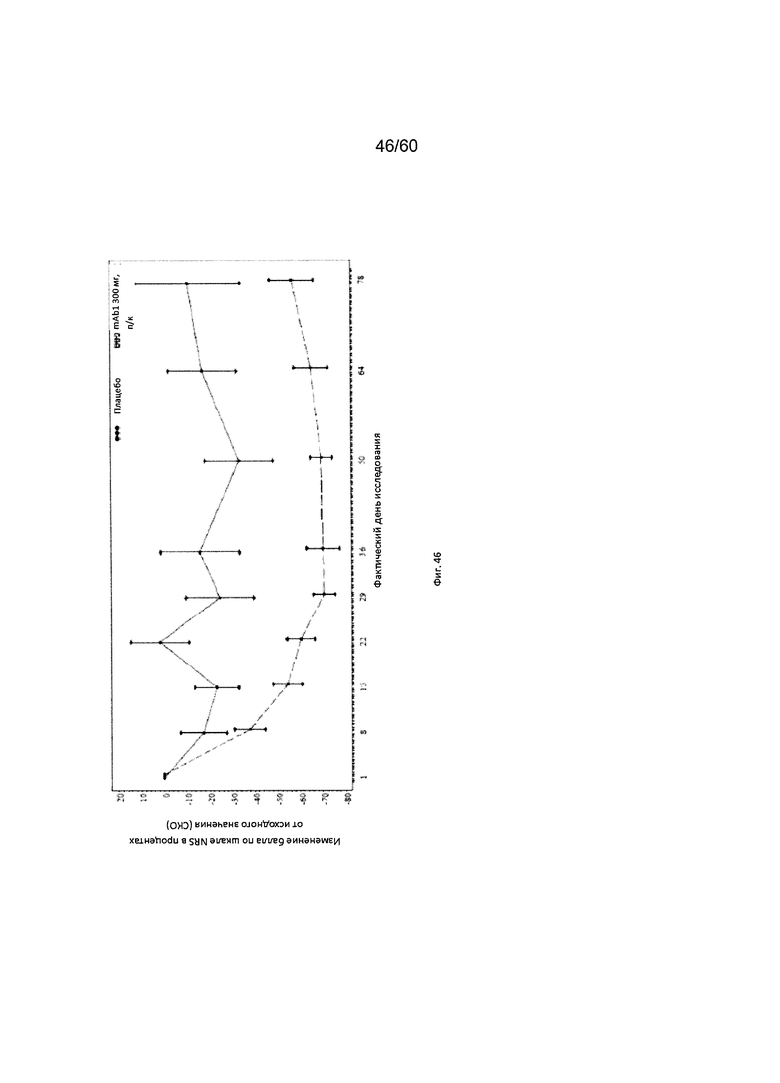
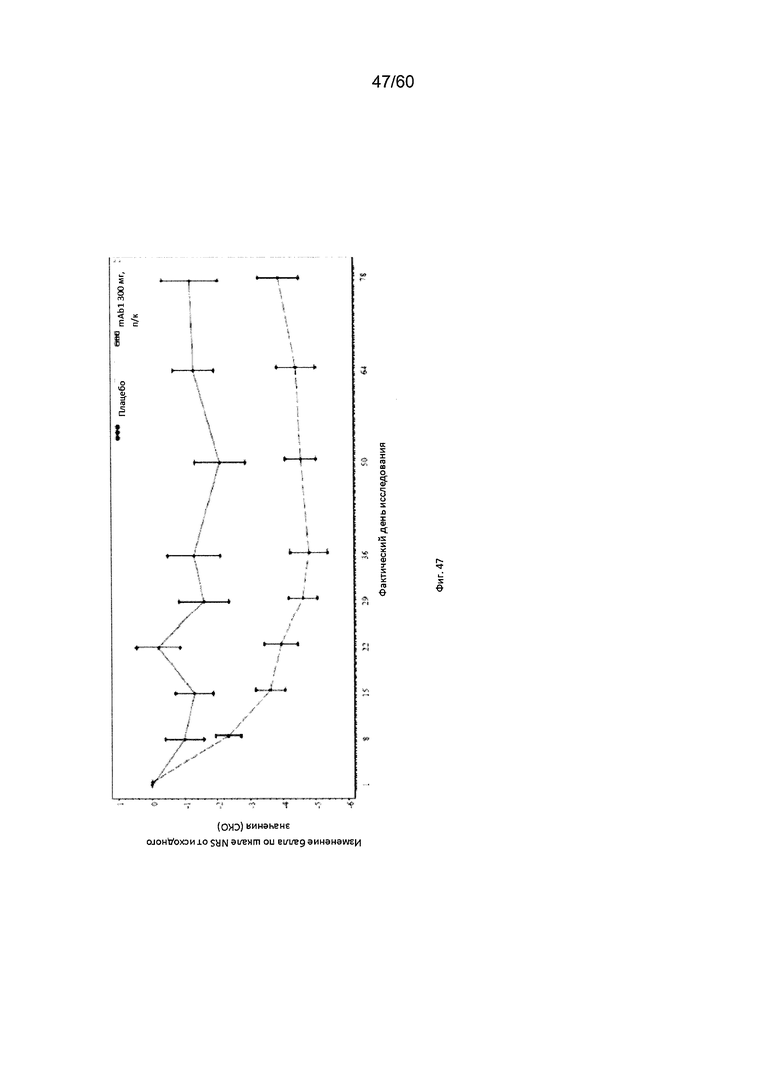

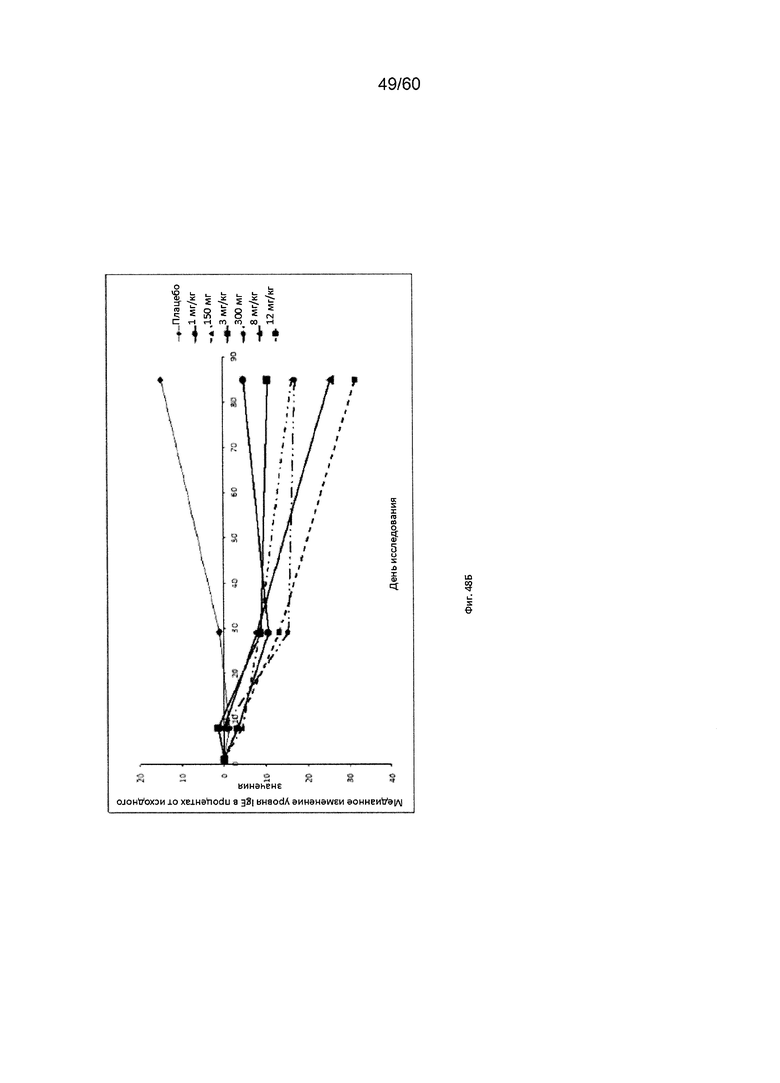

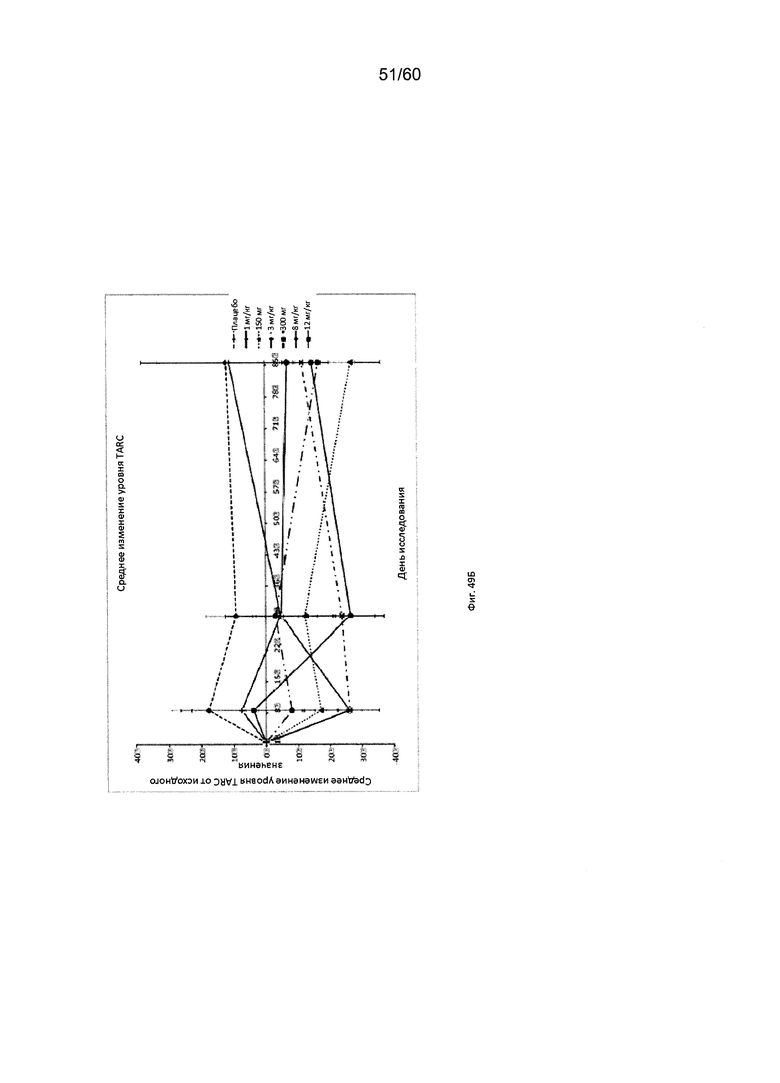
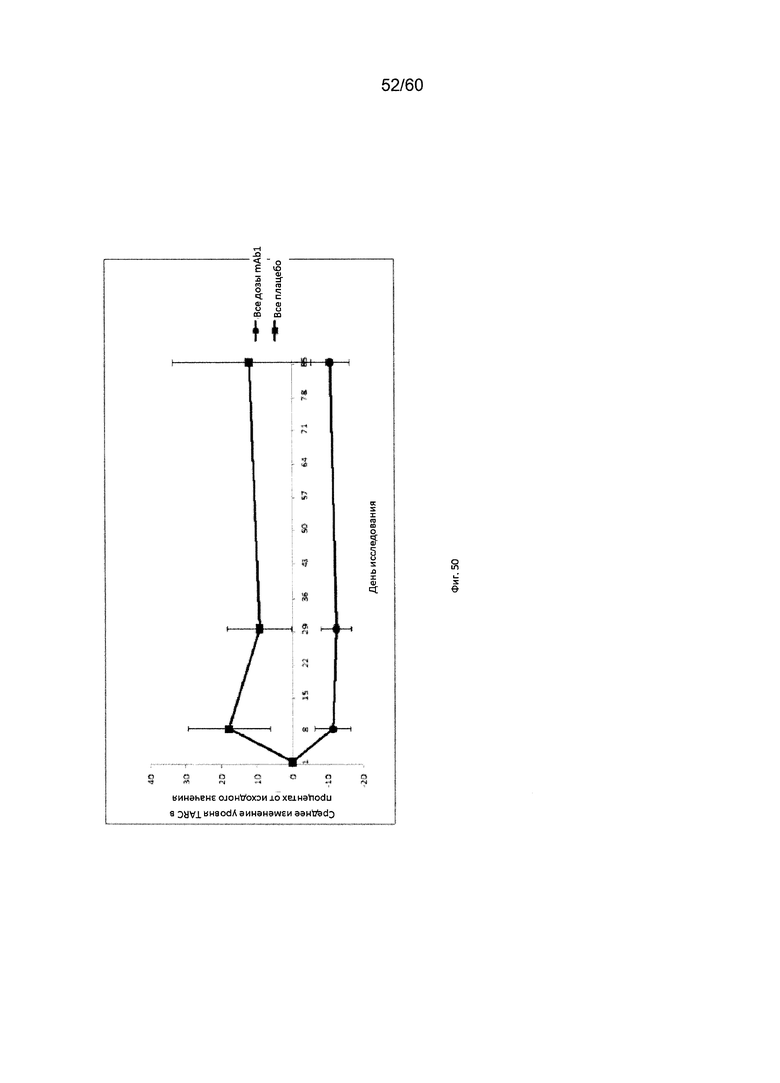




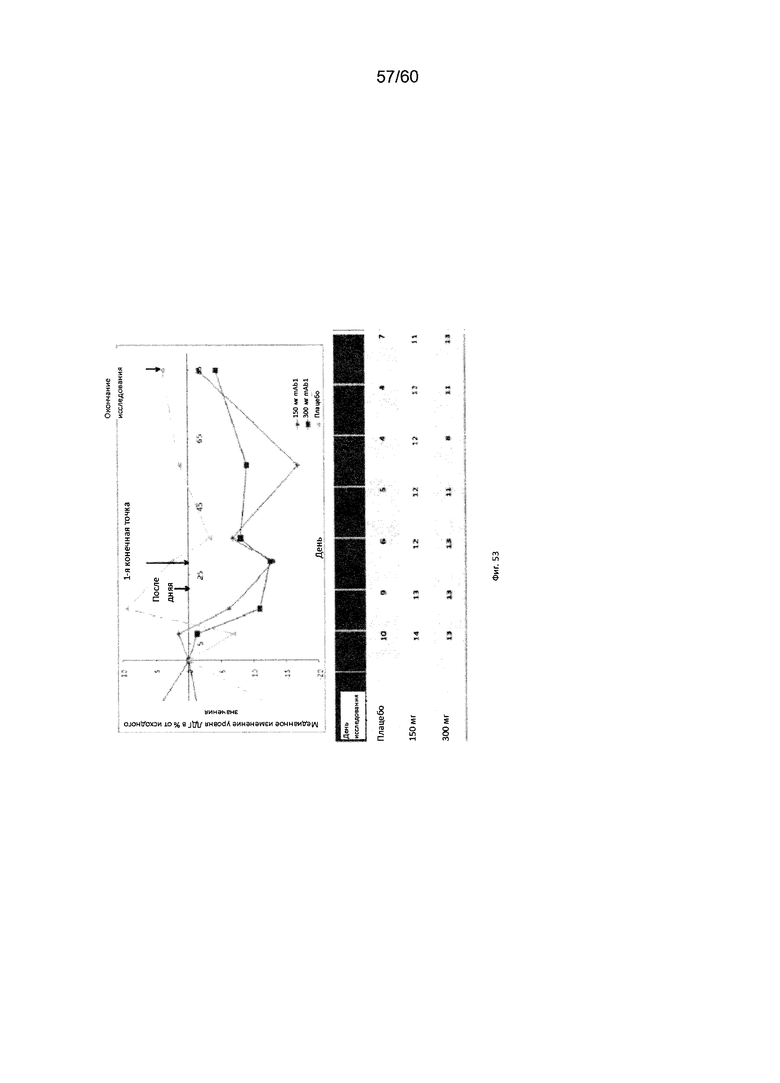

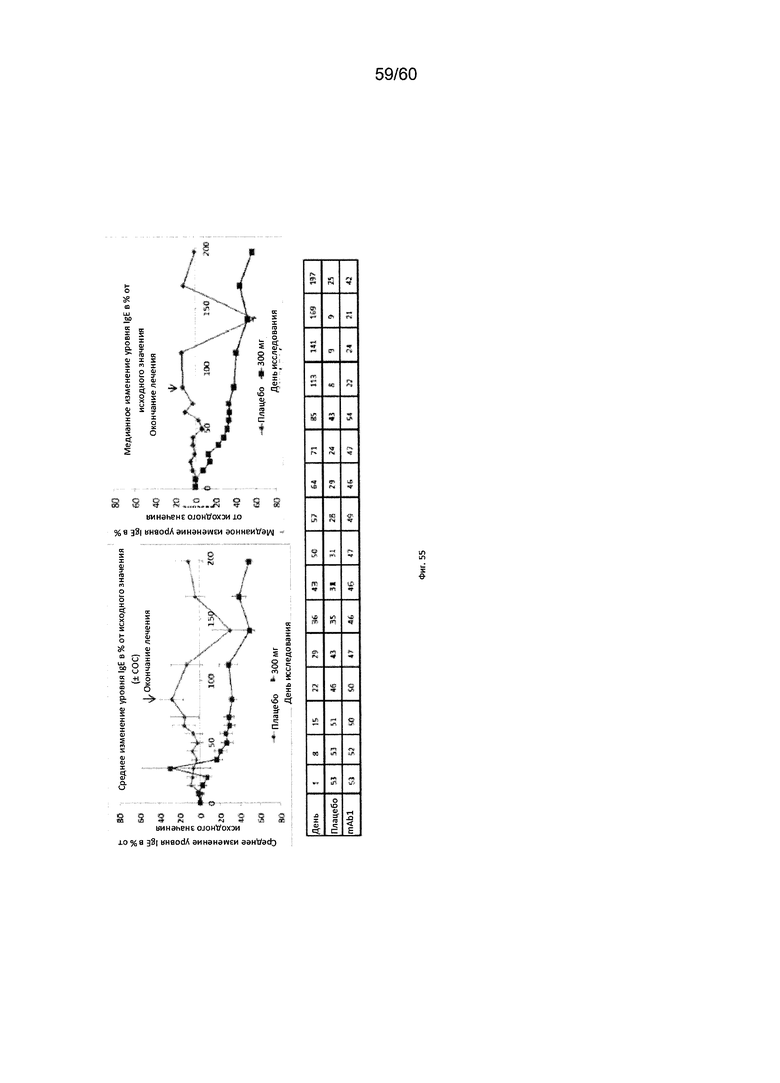
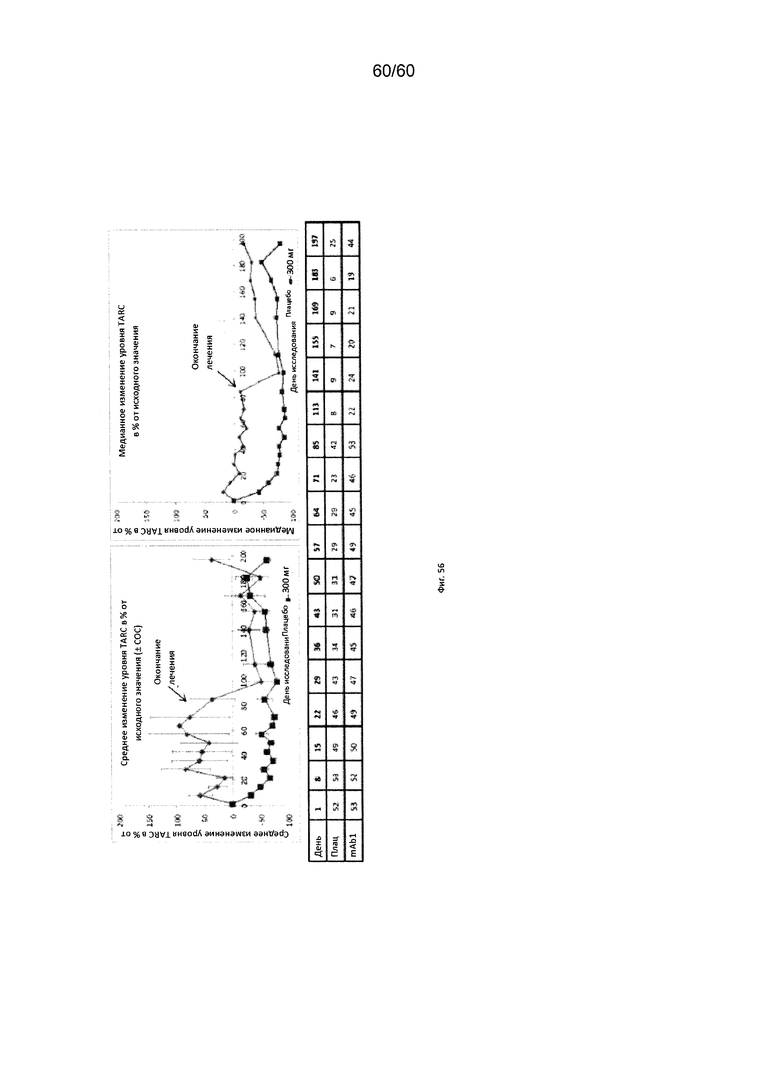
Изобретение относится к медицине, а именно к аллергологии и дерматологии, и касается фармацевтической композиции для лечения от умеренно тяжелого до тяжелого атопического дерматита (АД) у пациента с устойчивостью, отсутствием ответа или неадекватным ответом на топические кортикостероиды (ТКС) или ингибиторы кальциневрина. Фармацевтическая композиция содержит терапевтически эффективное количество антитела к рецептору интерлейкина-4 (IL-4R) человека или его антигенсвязывающего фрагмента. Изобретение обеспечивает эффективное лечение указанной патологии у пациента. 15 з.п. ф-лы, 60 ил., 54 табл.
1. Фармацевтическая композиция, содержащая терапевтически эффективное количество антитела к рецептору интерлейкина-4 (IL-4R) человека или его антигенсвязывающего фрагмента для лечения от умеренно тяжелого до тяжелого атопического дерматита (АД) у пациента с устойчивостью, отсутствием ответа или неадекватным ответом на топические кортикостероиды (ТКС) или ингибиторы кальциневрина.
2. Фармацевтическая композиция по п. 1, где пациент имеет в запротоколированном недавнем анамнезе, в течение 3 месяцев, предшествующих скрининговому визиту, ненадлежащий ответ на амбулаторное лечение топическими кортикостероидами или топическими ингибиторами кальциневрина, который включает одно или несколько из: (i) отсутствие достижения балла по общей оценке исследователя (IGA) от 0 (воспалительные признаки отсутствуют) до 2 (умеренная степень) несмотря на лечение топическими кортикостероидами от умеренной до высокой активности, применяемыми ежедневно на протяжении по меньшей мере 28 дней; или (ii) отсутствие достижения балла по IGA от 0 (воспалительные признаки отсутствуют) до 2 (умеренная степень) несмотря на лечение сильнодействующими топическими кортикостероидами, применяемыми ежедневно на протяжении по меньшей мере 14 дней.
3. Фармацевтическая композиция по п. 1 или 2, где каждая доза фармацевтической композиции, вводимая пациенту, содержит от 50 до 600 мг антитела к IL-4R человека или его связывающего фрагмента.
4. Фармацевтическая композиция по п. 1 или 2, где каждая доза фармацевтической композиции, вводимая пациенту, содержит 300 мг антитела к IL-4R человека или его антигенсвязывающего фрагмента.
5. Фармацевтическая композиция по п. 1 или 2, где лечение включает введение первоначальной дозы фармацевтической композиции пациенту с последующим введением одной или более последующих доз фармацевтической композиции пациенту, где каждую последующую дозу вводят пациенту через одну или две недели после непосредственно предшествующей дозы.
6. Фармацевтическая композиция по п. 5, где первоначальная доза содержит 600 мг антитела к IL-4R человека или его антигенсвязывающего фрагмента, и где каждая последующая доза содержит от 75 до 300 мг антитела к IL-4R человека или его антигенсвязывающего фрагмента.
7. Фармацевтическая композиция по п. 5, где первоначальная доза представляет собой двойное количество последующей доз(ы).
8. Фармацевтическая композиция по любому из пп. 1, 2, 6 или 7, где антитело или его антигенсвязывающий фрагмент содержат определяющие комплементарность участки (CDR) тяжелой и легкой цепей в паре аминокислотных последовательностей вариабельной области тяжелой цепи/вариабельной области легкой цепи (HCVR/LCVR), содержащей SEQ ID NO: 162/164.
9. Фармацевтическая композиция по п. 8, где антитело или его антигенсвязывающий фрагмент содержат три CDR тяжелой цепи (HCDR), содержащие SEQ ID NO: 148, 150 и 152 соответственно; и три последовательности CDR легкой цепи (LCDR), содержащие SEQ ID NO: 156, 158 и 160 соответственно.
10. Фармацевтическая композиция по любому из пп. 1, 2, 6, 7 или 9, где пациент после введения фармацевтической композиции показывает улучшение связанного с атопическим дерматитом (АД) параметра, где улучшение связанного с АД параметра представляет собой:
(I) уменьшение балла по шкале общей оценки исследователя (IGA) по меньшей мере на 25% от исходного значения;
(II) уменьшение балла по числовой шкале оценки интенсивности кожного зуда (шкала NRS) по меньшей мере на 25% от исходного значения;
(III) уменьшение индекса площади экзематозного поражения кожи и тяжести экземы (EASI) по меньшей мере на 45% от исходного значения;
(IV) уменьшение индекса оценки атопического дерматита (SCORAD) по меньшей мере на 30% от исходного значения;
(V) уменьшение балла согласно 5-D Pruritus по меньшей мере на 15% от исходного значения; или
(VI) уменьшение балла площади пораженной поверхности кожи при атопическом дерматите (BSA) по меньшей мере на 35% от исходного значения.
11. Фармацевтическая композиция по п. 10, где улучшение связанного с АД параметра представляет собой:
(a) уменьшение IGA по меньшей мере на 35% от исходного значения к 22 дню после введения первой дозы фармацевтической композиции, содержащей антитело к IL-4R человека или его антигенсвязывающий фрагмент;
(b) уменьшения NRS по меньшей мере на 30% от исходного значения к концу недели 2 после введения первой дозы фармацевтической композиции, содержащей антитело к IL-4R человека или его антигенсвязывающий фрагмент;
(c) уменьшение EASI по меньшей мере на 50% от исходного значения к 29 дню после введения первой дозы фармацевтической композиции, содержащей антитело к IL-4R человека или его антигенсвязывающий фрагмент;
(d) уменьшение SCORAD по меньшей мере на 35% от исходного значения к 29 дню после введения первой дозы фармацевтической композиции, содержащей антитело к IL-4R человека или его антигенсвязывающий фрагмент; или
(e) уменьшение балла согласно 5-D Pruritus по меньшей мере на 25% или по меньшей мере 30% от исходного значения до 85 дня после введения первой дозы фармацевтической композиции, содержащей антитело к IL-4R человека или его антигенсвязывающий фрагмент.
12. Фармацевтическая композиция по п. 10, где улучшение связанного с АД параметра представляет собой уменьшение балла BSA по меньшей мере на 35% от исходного значения к 29 дню после введения первой дозы фармацевтической композиции, содержащей антитело к IL-4R человека или его антигенсвязывающий фрагмент.
13. Фармацевтическая композиция по любому из пп. 1, 2, 6, 7, 9, 11 или 12, где лечение дополнительно включает совместное введение топических кортикостероидов.
14. Фармацевтическая композиция по п. 13, где лечение включает начальный период, когда вводят антитело к IL-R4 человека или его антигенсвязывающий фрагмент, затем последующий период, когда дозу топических кортикостероидов постепенно уменьшают, тогда как введение антитела к IL-4R человека или его антигенсвязывающего фрагмента продолжают.
15. Фармацевтическая композиция по п. 14, где количество топических кортикостероидов снижается на 10%, 20%, 30%, 40%, 50% или более по сравнению с дозой в начальный период лечения.
16. Фармацевтическая композиция по любому из пп. 1, 2, 6, 7, 9, 11, 12, 14 или 15, где фармацевтическую композицию вводят подкожной инъекцией.
| WO2010053751 A1, 14.05.2010 | |||
| ONG PECK Y, Editorial update on emerging treatments of atopic dermatitis// EXPERT OPINION ON EMERGING DRUGS ENGLAND, INFORMA HEALTHCARE, UK, (20120601), vol | |||
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| K | |||
| JAHNZ-ROZYK ET AL, Serum thymus and activation-regulated chemokine, macrophage-derived chemokine and eotaxin as markers of severity of atopic dermatitis // ALLERGY, (20050501), vol | |||
| Способ получения молочной кислоты | 1922 |
|
SU60A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| J | |||
| CORREN ET AL, "A Randomized, Controlled, Phase 2 Study of AMG 317, an IL-4R Antagonist, in Patients with Asthma", AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE, (20100415), vol | |||
| Водяные лыжи | 1919 |
|
SU181A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |

