Область техники, к которой относится изобретение
Настоящее изобретение имеет отношение к соединениям, обладающим потенциальной возможностью применения в терапевтических целях для лечения заболеваний, связанных со стрессом, вызванным неправильным сворачиванием (мисфолдингом) белков и, в частности, с накоплением неправильно свернутых белков. В частности, изобретение предоставляет соединения, способные оказывать защитное действие от цитотоксического стресса эндоплазматического ретикулума (ER).
Уровень техники
Соединение 2-(2,6-дихлорбензилиден)гидразинкарбоксимидамин, также имеющее название гуанабенз, является альфа-агонистом альфа-2 типа, который используется как антигипертензивное средство.
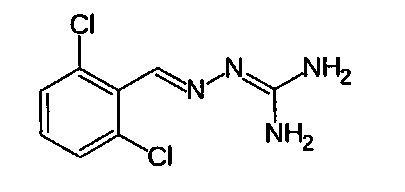
Гуанабенз
Известны также различные производные гуанабенза. Например, США 3,982,020 (Sandoz, Inc.) раскрывает замещенные бензилиден-гидразины и их использование в качестве гипогликемических средств, средств от ожирения и противовоспалительных средств. США 2004/0068017 (Bausch & Lomb Inc.) раскрывает замещенные бензилиден-гидразины, способные увеличивать активность желатиназы А в зрительных клетках. Эти молекулы находят применение при лечении первичной открытоугольной глаукомы. WO 2008/061647 (Acure Pharma АВ) раскрывает использование N-(2-хлор-3,4,-диметокибензилиденамино)гуанидина в качестве ингибитора VEGFR и его применение, связанное с этим, при лечении или предотвращении нежелательного образования кровеносных сосудов в ходе роста опухолей и/или в условиях воспаления. WO 2005/031000 (Acadia Pharmaceuticals, Inc.) раскрывает замещенные бензилиден-гидразины и их использование при лечении острой боли и хронической нейропатической боли. Наконец, ЕР 1908464 (CNRS) раскрывает гуанабенз и хлоргуанабенз и их применение при лечении болезней, связанных с удлинением полиглутаминового фрагмента, включая болезнь Хантингтона.
В последние годы сообщалось, что гуанабенз обладает терапевтическим потенциалом в ряде других областей. Недавно стало известно, что гуанабенз обладает антиприонной активностью (Tribouillard-Tanvier et al., 2008 PLoS One 3, e1981). Сообщалось, что его активность в отношении защиты от нарушения сворачивания белка неожиданно оказалась намного более широкой и включает ослабление накопления мутантного белка хантингтина в исследованиях на клетках (WO 2008/041133) и защиту от летальных эффектов экспрессии предрасположенного к неправильному сворачиванию мутантного инсулина в эндоплазматическом ретикулуме (ER) Min6 и INS-1 панкреатических бета-клеток у мышей Akita (Tsaytler et al., 2011 Science 332 pp 91-94). WO 2014/138298 и Way et al. (2015 Nature Communications 6:6532 DOI: 10.1038/ncomms7532) раскрывает гуанабенз и его использование при лечении демиелинизирующего заболевания, такого как рассеянный склероз.
Также было показано, что гуанабенз способствует выживаемости клеток HeLa, подвергнутых другому цитотоксическому ER-стрессу, индуцированному ингибитором N-гликозилирования туникамицином, дозо-зависимым образом (Tsaytler, et al., Science, 2011). Количественная оценка выживаемости клеток показала, что гуанабенз удваивал количество клеток, выживших после ER-стресса при использовании средней эффективной концентрации ~ 0.4 μМ. Ни агонист α2-адренергического рецептора клонидин, ни антагонист α2-адренергического рецептора эфароксан не защищали клетки от цитотоксического ER-стресса, причем эфароксан не препятствовал защитному действию гуанабенза (Tsaytler, et al., Science, 2011). Эти наблюдения показывают, что гуанабенз спасает клетки от летального ER-стресса с помощью механизма, независимого от α2-адренергического рецептора. Гуанабенз иным образом защищает клетки от летального накопления неправильно свернутых белков путем связывания с регуляторной субъединицей протеинфосфатазы 1, PPP1R15A (GADD34), выборочно нарушая индуцированное стрессом дефосфорилирование α-субъединицы фактора 2 инициации трансляции 2 (eIF2α). Гуанабенз регулирует скорость трансляции в подвергнутых стрессу клетках до уровня, осуществимого имеющимися шаперонами, тем самым восстанавливая гомеостаз белков. Сообщалось, что гуанабенз не связывается с конститутивной PPP1R15B (CReP) и, следовательно, не ингибирует трансляцию в неподвергнутых стрессу клетках (Tsaytler, et al., Science, 2011).
Неспособность поддерживать протеостаз в ER путем формирования соответствующей реакции несвернутых белков (UPR) считается фактором, сопутствующим многим патологическим состояниям. Таким образом, описанные в этом документе молекулы, ингибирующие eIF2α фосфатазу с целью тонкой регулировки синтеза белков, могут быть терапевтически полезными в отношении большого числа болезней, вызванных стрессом неправильного сворачивания белков и в частности, накоплением неправильно свернутых белков.
Tribouillard-Tanvier et al., PLoS One 3, e1981 (2008) и EP 1908464A раскрывают производные бензилиденгуанидина, содержащие гуанидин в виде концевой группы. Однако заявитель обнаружил, что концевая группа подлежит метаболизированию, при котором меняется биодоступность соединений. Кроме того, предшествующие исследования также показали, что (гетеро)арильная группа должна быть, по меньшей мере, дигалогенирована, для того, чтобы соединения проявляли подходящую фармакологическую активность (смотри, например, Tribouillard-Tanvier et al., PLoS One 3, e1981 (2008) и ЕР 1908464A, CNRS). Однако, в отличие от результатов предшествующих исследований, настоящий заявитель неожиданно обнаружил, что моногалогенированные (гетеро)арильные производные, содержащие модифицированную концевую группу, также могут быть активными. Таким образом, желательно предоставить альтернативу с улучшенным профилем активности и/или биодоступности.
Настоящее изобретение собирается предоставить альтернативные соединения, основанные на основной структуре гуанабенза, обладающие потенциальной возможностью применения в терапевтических целях для лечения заболеваний, связанных со стрессом, вызванным неправильным сворачиванием белков, и, в частности, с накоплением неправильно свернутых белков.
Сущность изобретения
Первый аспект изобретения имеет отношение к соединению формулы (I) или его фармацевтически приемлемой соли,
в которой: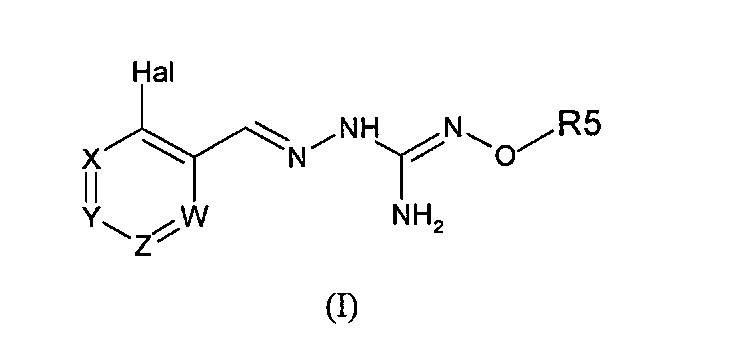
Hal = F, Cl, Br, I
X представляет собой или -CR1= или -N=,
Y представляет собой или -CR2= или -N=,
Z представляет собой или -CR3= или -N=,
W представляет собой или -CR4= или -N=,
R1 выбирают из Н, Hal, алкила и О-алкила;
R2 выбирают из Н, Hal, алкила, О-алкила и C(O)R6;
R3 выбирают из Н, Hal, алкила и О-алкила;
R4 представляет собой Н, Cl, F, I или Br;
R5 представляет собой Н или алкил, циклоалкил, аралкил, алкенил, циклоалкенил, гетероциклил, арил, С(O)-алкил, и С(O)-арил, каждый из которых необязательно является замещенным одной или более R7 группами;
R6 выбирают из ОН, О-алкила, О-арила, аралкила, NH2, NH-алкила, N(алкил)2, NH-арила, CF3, алкила и алкоксигруппы;
каждый R7 независимо выбирают из галогена, ОН, CN, СОО-алкила, аралкила, гетероциклила, S-алкила, SO-алкила, SO2-алкила, SO2-арила, СООН, СО-алкила, СО-арила, NH2, NH-алкила, N(алкил)2, CF3, алкила и алкоксигруппы.
И в которой, если Hal является Cl, и R4 является Cl, тогда R5 не является Н.
Второй аспект изобретения имеет отношение к фармацевтической композиции, содержащей соединение формулы (II):
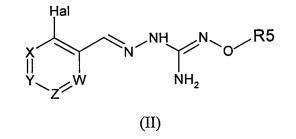
в которой:
Hal=F, Cl, Br, I
X представляет собой или -CR1= или -N=,
Y представляет собой или -CR2= или -N=,
Z представляет собой или -CR3= или -N=,
W представляет собой или -CR4= или -N=,
R1 выбирают из Н, Hal, алкила и О-алкила;
R2 выбирают из Н, Hal, алкила, О-алкила и C(O)R6;
R3 выбирают из Н, Hal, алкила и О-алкила;
R4 представляет собой Н, Cl, F, I или Br;
R5 представляет собой Н или алкил, циклоалкил, аралкил, алкенил, циклоалкенил, гетероциклил, арил, С(O)-алкил, и С(O)-арил, каждый из которых необязательно является замещенным одной или более R7 группами;
R6 выбирают из ОН, О-алкила, О-арила, аралкила, NH2, NH-алкила, N(алкил)2, NH-арила, CF3, алкила и алкоксигруппы;
каждый R7 независимо выбирают из галогена, ОН, CN, СОО-алкила, аралкила, гетероциклила, S-алкила, SO-алкила, SO2-алкила, SO2-арила, СООН, СО-алкила, СО-арила, NH2, NH-алкила, N(алкил)2, CF3, алкила и алкоксигруппы;
в сочетании с подходящим фармацевтически приемлемым разбавителем, эксципиентом или носителем.
Третий аспект изобретения имеет отношение к соединению формулы (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении заболевания, связанного со стрессом, вызванным неправильным сворачиванием белка, в частности, с накоплением неправильно свернутых белков, конкретнее, протеопатии:
в которой:
Hal=F, Cl, Br, I
X представляет собой или -CR1= или -N=,
Y представляет собой или -CR2= или -N=,
Z представляет собой или -CR3= или -N=,
W представляет собой или -CR4= или -N=,
R1 выбирают из Н, Hal, алкила и О-алкила;
R2 выбирают из Н, Hal, алкила, О-алкила и C(O)R6;
R3 выбирают из Н, Hal, алкила и О-алкила;
R4 представляет собой Н, Cl, F, I или Br;
R5 представляет собой Н или алкил, циклоалкил, аралкил, алкенил, циклоалкенил, гетероциклил, арил, С(O)-алкил и С(O)-арил, каждый из которых необязательно является замещенным одной или более R7 группами;
R6 выбирают из ОН, О-алкила, О-арила, аралкила, NH2, NH-алкила, N(алкил)2, NH-арила, CF3, алкила и алкоксигруппы;
каждый R7 независимо выбирают из галогена, ОН, CN, СОО-алкила, аралкила, гетероциклила, S-алкила, SO-алкила, SO2-алкила, SO2-арила, СООН, СО-алкила, СО-арила, NH2, NH-алкила, N(алкил)2, CF3, алкила и алкоксигруппы.
И фармацевтически приемлемый эксципиент.
Формула (I) представляет собой отдельный вариант осуществления формулы (II).
В предпочтительном варианте осуществления соединения формулы (I) или (II), определенные выше, преимущественно не демонстрируют активности в отношении адренергического α2А рецептора по срвнению с соединениями предшествующего уровня техники, такими как гуанабенз. Это отсутствие альфа-2-адренергической активности делает соединения терапевтически пригодными при лечении заболеваний, связанных со стрессом, вызванным неправильным сворачиванием белков, и в частности, с накоплением неправильно свернутых белков. Отсутствие альфа-2-адренергической активности означает, что соединения формулы (I) или (II) могут вводиться в дозировках, подходящих для лечения вышеупомянутых болезней, без какого-либо значительного воздействия на кровяное давление.
Осуществление изобретения
Использованный в описании термин "алкил" включает насыщенные алкильные группы с неразветвленной и разветвленной цепью. Предпочтительно, алкильная группа представляет собой С1-20 алкильную группу, более предпочтительно C1-15, еще более предпочтительно С1-12 алкильную группу, еще более предпочтительно C1-6 алкильную группу, более предпочтительно С1-3 алкильную группу. В частности, предпочтительные алкильные группы включают, например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил.
Использованный в описании термин "циклоалкил" относится к циклической алкильной группе. Предпочтительно, циклоалкильная группа является С3-12 циклоалкильной группой.
Использованный в описании термин "алкенил" относится к группе, содержащей одну или более углерод-углеродных двойных связей, которая может быть разветвленной или неразветвленной. Предпочтительно алкенильная группа является С2-20 алкенильной группой, более предпочтительно С2-15 алкенильной группой, еще более предпочтительно С2-12 алкенильной группой или предпочтительно С2-6 алкенильной группой, более предпочтительно С2-3 алкенильной группой. Термин "циклический алкенил" следует истолковывать соответственно.
Использованный в описании термин "арил" относится к С6-12 ароматической группе. Типичные примеры включают фенил и нафтил и т.д.
Использованный в описании термин "гетероцикл" (также упоминающийся в настоящем документе как "гетероциклил" и "гетероциклический") относится к 4-12, предпочтительно 4-6-членной насыщенной, ненасыщенной или частично ненасыщенной циклической группе, содержащей один или более гетероатомов, выбранных из N, О и S, и которая необязательно дополнительно содержит одну или более СО групп. Термин "гетероцикл" включает как гетероарильные группы, так и гетероциклоалкильные группы, как определно ниже.
Использованный в описании термин "гетероарил" относится к 4-12-членной ароматической группе, содержащей один или более гетероатомов. Предпочтительно, гетероарильная группа представляет собой 4-6-членную ароматическую группу, состоящую из одного или более гетероатомов, выбранных из N, О и S. Подходящие гетероарильные группы включают пиррол, пиразол, пиримидин, пиразин, пиридин, хинолин, тиофен, 1,2,3-триазол, 1,2,4-триазол, тиазол, оксазол, изо-тиазол, изо-оксазол, имидазол, фуран и тому подобное.
Использованный в описании термин "гетероциклоалкил" относится к 3-12-членной, предпочтительно 4-6-членной циклической алифатической группе, которая содержит один или более гетероатомов, выбранных из N, О и S. Предпочтительными являются N-содержащие 5-6-членные гетероциклоалкилы. Предпочтительные гетероциклоалкильные группы включают пиперидинил, пирролидинил, пиперазинил, тиоморфолинил и морфолинил. Более предпочтительно, гетероциклоалкильную группу выбирают из N-пиперидинила, N-пирролидинила, N-пиперазинила, N-тиоморфолинила и N-морфолинила.
Использованный в описании термин "аралкил" включает, но не ограничивается этим, группу, имеющую как арильную, так и алкильную функциональность. В качестве примера, термин включает группы, в которых один из атомов водорода алкильной группы заменен арильной группой, например, фенильной группой. Типичные аралкильные группы включают бензильную, фенетильную и тому подобные.
Ниже приведены конкретные варианты осуществления формулы (I) или (II):
В одном предпочтительном варианте осуществления Hal является Cl.
В одном предпочтительном варианте осуществления X представляет собой -CR1=.
В одном предпочтительном варианте осуществления Y представляет собой -CR2=.
В другом предпочтительном варианте осуществления Y представляет собой N.
В одном предпочтительном варианте осуществления Z=-CR3=.
В одном предпочтительном варианте осуществления W=-CR4=.
В одном предпочтительном варианте осуществления R1 представляет собой Н или F, более предпочтительно Н.
В одном предпочтительном варианте осуществления R2 представляет собой Н или F, более предпочтительно Н.
В одном предпочтительном варианте осуществления R3 представляет собой Н или F, более предпочтительно Н.
В одном предпочтительном варианте осуществления R4 представляет собой Н, Cl или F, более предпочтительно Н или F, более предпочтительно Н.
В одном предпочтительном варианте осуществления R3 и R4 оба являются Н.
В одном варианте осуществления R5 представляет собой Н, алкенил или алкил, каждый из алкенила или алкила является необязательно замещенным одной или более R7 группами.
В одном варианте осуществления R7 группы выбирают из галогена, ОН, гетероциклила, SO-алкила, SO2-алкила, О-алкила.
В одном особенно предпочтительном варианте осуществления соединение формулы (I) или (II) выбирают из следующих соединений:
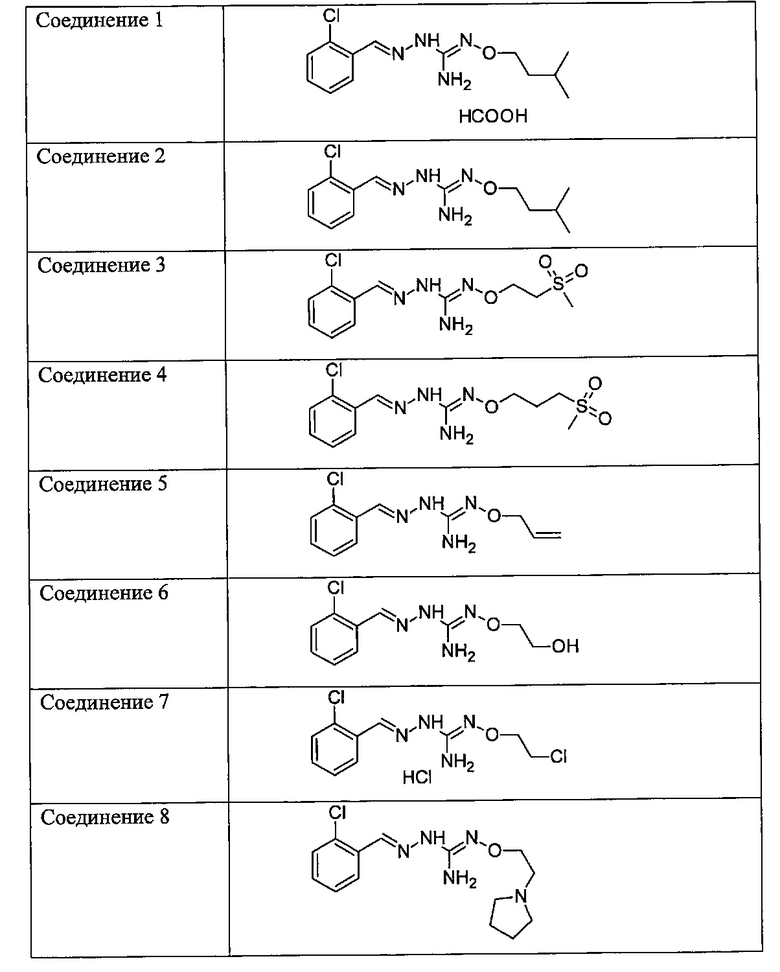
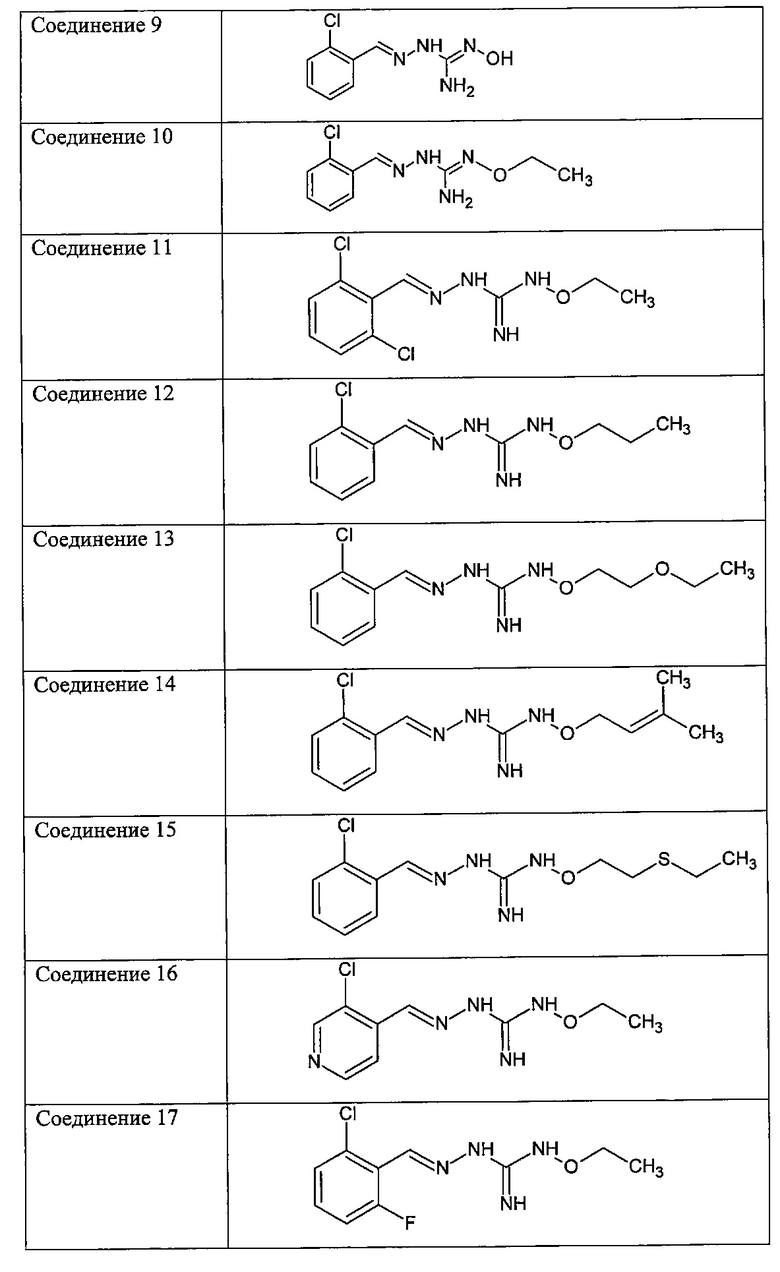
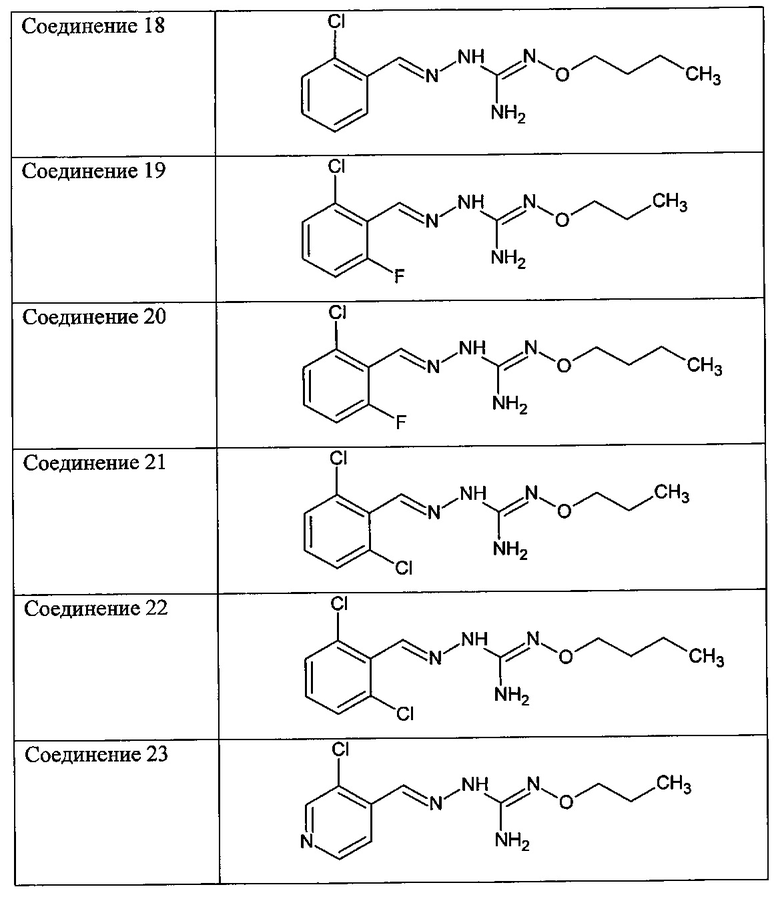
и их фармацевтически приемлемых солей.
В предпочтительном варианте осуществления соединение формулы (I) или (II) выбирают из Соединений 4, 6, 10, 11, 12, 14, 17, 18, указанных выше, более предпочтительно выбирают из Соединений 4, 11, 17, 18, указанных выше.
СОЕДИНЕНИЯ
Один аспект изобретения имеет отношение к соединениям формулы (I) или их фармацевтически приемлемым солям, как определено выше. Предпочтительные аспекты изобретения применяются mutatis mutandis (с необходимыми изменениями). В частности, предпочтительные соединения для этого аспекта изобретения включают Соединения 1, 2, 5 и 10, описанные в этом документе.
Способ получения
Следующий аспект изобретения имеет отношение к способу получения соединения формулы (I) или (II) или его фармацевтически приемлемых солей, как описано выше, включающему стадию взаимодействия соединения формулы (А) или его таутомерной формы:
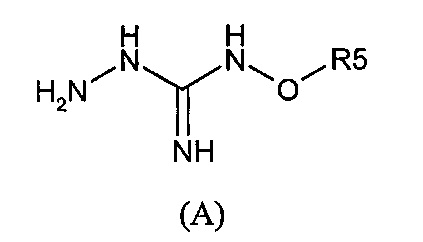
в которой R5 соответствует приведенному выше определению, с соединением формулы (В):
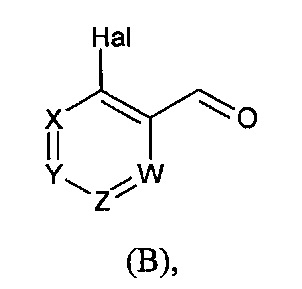
в которой X, Y, Z, W и Hal соответствуют приведенному выше определению,
необязательно с последующей стадией модификации R5 группы соединения, возникающей вследствие взаимодействия между соединениями формул (А) и (В), как описано выше, в другую R5 группу.
Предпочтительно, данный способ также может включать дополнительную стадию очистки соединения (I) или (II), полученного выше.
Реакция соединения между соединениями (А) и (В) может быть проведена в органическом растворителе, таком как спирт, например, этанол. Она может быть проведена при температуре, находящейся в диапазоне между комнатной температурой и температурой кипения реакционной смеси.
Реакция модификации R5 группы может быть проведена с помощью применения или адаптации известных методов. Например, в соединении, полученном после соединения (А) и (В), R5 может быть алкильной группой, замещенной группами R7: таким образом, может быть желательна замена R7 группы. Такие реакции замещения общеизвестны. В качестве характерного примера, может быть желательно замещение R7=OH R7=галогеном в соединении формулы (I) или (II). Такая реакция может быть проведена в присутствии галогенирующего агента, такого как хлорирующий агент, например, SOCl2. В большинстве случаев такая реакция может проводиться в органическом растворителе, таком как дихлорметан. Другим характерным примером является замещение R7=галогена R7=N-содержащим гетероциклом, таким как пирролидин. Такая реакция может быть проведена в присутствии основания, такого как TEA. В большинстве случаев такая реакция может быть проведена в органическом растворителе, таком как THF.
Согласно одному варианту осуществления способ может дополнительно включать стадию получения соединения формулы (А), как определено выше, путем взаимодействия соединения формулы (С):
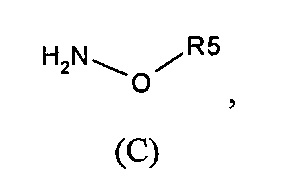
или одной из его солей,
в которой R5 является, как определено выше,
с S-метилизотиосемикарбазид иодгидратом (D):
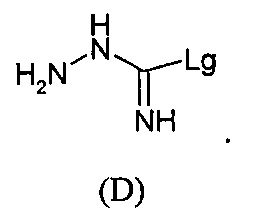
где Lg представляет собой уходящую группу, такую как -S-Алкил, например, -S-Метил.
или одной из его солей.
В большинстве случаев реакция между соединениями формул (С) и (D) может проводиться в водном растворе основания, например, в водном растворе, содержащем гидроксид натрия.
Реакция соединения между соединениями формул (С) и (D) может сопровождаться дополнительной стадией очистки.
В одном варианте осуществления способ необязательно може включать дополнительную стадию получения соединения формулы (С) путем взаимодействия соединения формулы (Е):
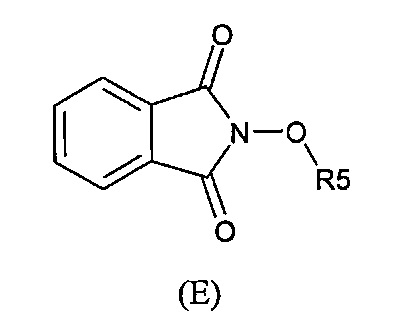
с соединением производным гидразина, например гидразингидратом или метилгидразином.
Способ изобретения необязательно может содержать стадию получения соединения формулы (Е) из соединения формулы (Е'):
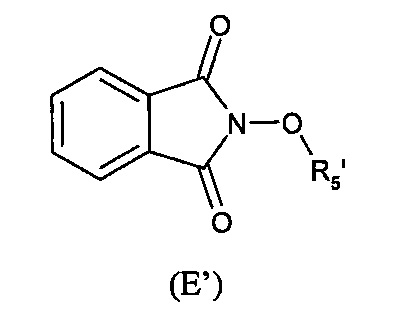
где (R5') представляет группу-предшественницу группы R5.
Эта реакция может быть желательна, в том случае, когда (Е) не является коммерчески доступным и невозможно получить (Е) из (F) и (Г), как раскрывается далее.
Таким образом, может быть желательно использование предшественника (Е'), который может быть трансформирован в (Е).
Предшественник представляет собой группу или соединение, которое может быть модифицировано в желательное соединение путем замены, устранения или иной разновидности химической реакции.
В качестве иллюстративного варианта осуществления реакция модификации R5' в желательную R5 группу может быть проведена путем применения или адаптации известных способов. Например, в (Е) R5 может быть алкильной группой, замещенной R7 группами: таким образом, может быть желательно модифицировать R7' группы в (Е') в желательные группы R7' в (Е). Такие реакции модификации общеизвестны. В качестве характерного примера, может быть желательно заменить предшественник R5', содержащий группу R7'=S(Алкил), группой R7=SO2(Алкил). Такая реакция может быть проведена в присутствии МСРВА. В большинстве случаев такая реакция может быть проведена в органическом растворителе, таком как дихлорметан.
Способ изобретения может включать стадию получения (Е) или (Е'), в случае необходимости, путем взаимодействия соединения (F)
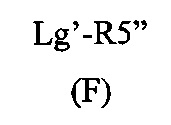
где R5'' представляет собой или R5 или R5', как определено выше, и Lg' представляет уходящую группу, такую как атом галогена или гидроксильная (ОН) группа,
с N-гидроксифталимидом (G):
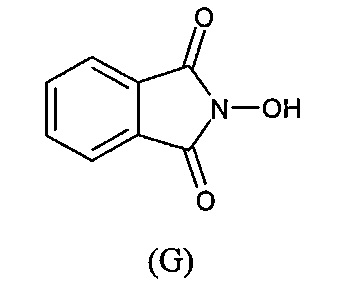
В общем, соединение (F) и (G) может быть проведено в соответствии с условиями синтеза Габриэля.
Согласно иллюстративному варианту осуществления эта реакция может быть проведена в присутствии основания, например органического или неорганического основания, как правило, TEA или K2CO3, или NaOAc, в частности, когда Lg содержит галоген(ы).
Согласно другому иллюстративному варианту осуществления первая стадия может проводиться в присутствии диизопропил азодикарбоксилата и PPh3, в частности, когда Lg=OH.
Соединения (F), (G), (В) обычно являются коммерчески доступными.
Соединения формулы (D):
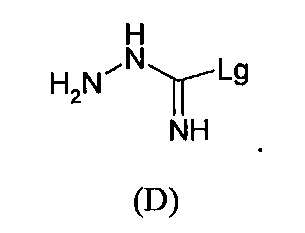
где Lg представляет собой -S-алкил, например, -S-метил, также являются частью изобретения.
В дополнение к способу, раскрытому выше, соединения настоящего изобретения могут быть приготовлены, а способ настоящего изобретения может быть осуществлен различными путями, хорошо известными специалистам в данной области техники. Специалистам должно быть понятно, что соединения могут быть синтезированы, например, путем применения или адаптации способов, описанных ниже, или их вариантов. Для специалистов данной области соответствующие модификации и замены являются очевидными и хорошо известными или их легко можно найти в научной литературе.
В частности, такие методы можно найти в работе R.C. Larock, Comprehensive Organic Transformations, VCH publishers, 1989
Следует понимать, что соединения настоящего изобретения могут содержать один или более асимметрично замещенных атомов углерода и могут быть выделены в оптически активных или рацемических формах. Таким образом, имеются в виду все хиральные, диастереомерные, рацемические формы и все геометрические изомерные формы структуры, если конкретно не указана специфическая стереохимическая или изомерная форма. В данной области техники хорошо известно, как получить и выделить такие оптически активные формы. Например, смеси стереоизомеров можно разделить с помощью стандартных методов, включая, но не ограничиваясь этим, разделение рацемических форм, нормальную, обращенно-фазную и хиральную хроматографию, предпочтительное солеобразование, перекристаллизацию и тому подобное, или с помощью хирального синтеза из хиральных исходных материалов или путем преднамеренного синтеза целевых хиральных центров.
Соединения настоящего изобретения можно получить, используя целый ряд синтетических подходов. Реагенты и исходные материалы имеются в продаже на рынке, в другом случае средний специалист в данной области может их легко синтезировать с помощью хорошо известных в данной области техники методов. Все заместители, если не указано иное, определены ранее.
В описанных здесь реакциях может потребоваться защитить реакционноспособные функциональные группы, например гидрокси, амино, имино, тио или карбоксигруппы, в том случае, если такие группы желательны в конечном продукте, чтобы избежать их нежелательного участия в реакциях. Могут использоваться обычные защитные группы в соответствии со стандартной практикой, например, смотри T.W. Greene и P.G.М. Wuts in Protective Groups in Organic Synthesis, John Wiley и Sons, 1991; J.F.W. McOmie in Protective Groups in Organic Chemistry, Plenum Press, 1973.
Некоторые реакции могут проводиться в присутствии основания. Не существует особого ограничения относительно природы основания, которое должно использоваться в этой реакции, и любое основание, обычно используемое в реакциях этого типа, может равным образом, использоваться в этом случае, при условии, что оно не оказывает отрицательного воздействия на другие части молекулы. Примеры подходящих оснований включают гидроксид натрия, карбонат калия, триэтиламин, гидриды щелочных металлов, такие как гидрид натрия и гидрид калия; соединения алкиллития, такие как метиллитий и бутиллитий; и алкоксиды щелочных металлов, такие как метоксид натрия и этоксид натрия.
В большинстве случаев реакции проводятся в подходящем растворителе. Можно использовать целый ряд растворителей, при условии, что он не оказывает отрицательного действия на реакцию или на участвующие в реакции реагенты. Примеры подходящих растворителей включают углеводороды, которые могут быть ароматическими, алифатическими или циклоалифатическими углеводородами, такими как гексан, циклогексан, бензол, толуол и ксилен; амиды, такие как диметил-формамид; спирты, такие как этанол, метанол и простые эфиры, такие как диэтиловый эфир и тетрагидрофуран.
Реакции могут проходить в широком диапазоне температур. В общем, мы обнаружили, что удобно проводить реакцию при температуре от 0°С до 150°С (более предпочтительно примерно от комнатной температуры до 100°С). Необходимое для реакции время также может широко варьировать в зависимости от многих факторов, а именно, температуры реакции и природы реагентов. Однако, при условии, что реакция осуществляется при предпочтительных условиях, изложенных выше, периода от 3 часов до 20 часов обычно будет достаточно.
Полученное таким образом соединение может быть восстановлено из реакционной смеси с помощью обычных способов. Например, соединение может быть восстановлено путем отгонки растворителя из реакционной смеси или, если необходимо, после отгонки растворителя из реакционной смеси, можно вылить остаток в воду, а затем экстрагировать с несмешивающимся с водой органическим растворителем и отогнать растворитель из экстракта. Кроме того, продукт при желании может быть дополнительно очищен с помощью различных хорошо известных методов, таких как перекристаллизация, повторное осаждение или с помощью различных хроматографических методов, а именно колоночной хроматографии или препаративной тонкослойной хроматографии.
Способ изобретения также может включать дополнительную стадию выделения полученного продукта формулы (I).
Исходные продукты и/или реагенты могут быть коммерчески доступными, либо специалист в данной области может их легко получить путем применения или адаптации методов, раскрытых далее в экспериментальной части.
Применение в терапевтических целях
Соединения формулы (I) или (II) обладают потенциальной возможностью терапевтического применения при лечении заболеваний, связанных с накоплением неправильно свернутых и/или несвернутых белков. В частности, соединения формулы (I) или (II) могут оказывать защитное действие от цитотоксического стресса эндоплазматического ретикулума (ER) и возрастных болезней.
Другой аспект изобретения имеет отношение к использованию соединения формулы (I) или (II), как определено выше, для изготовления лекарственного средства для лечения заболевания, связанного со стрессом, вызванным неправильным сворачиванием белка, и в частности, с накоплением неправильно свернутых белков.
В одном предпочтительном варианте осуществления изобретения соединение формулы (I) или (II) предназначается для лечения заболеваний, в которых накопление неправильно свернутых и/или несвернутых белков имеет отношение к механизму действия (Brown et al, 2012, Frontiers in Physiology, 3, Article 263).
Другой аспект изобретения имеет отношение к использованию соединения формулы (I) или (II), как определено выше, для изготовления лекарственного средства для лечения протеопатий. Протеопатии относятся к классу болезней, при которых некоторые белки становятся структурно аномальными и вследствие этого нарушают функцию клеток, тканей и органов в организме. Часто такие белки не в состоянии принимать свою нормальную конформацию, и в таком неправильно свернутом и/или несвернутом состоянии белки в некоторых случаях могут становиться токсичными (приобретать токсическую функцию) или они могут утратить свою нормальную функцию или может наблюдаться снижение их биологической активности. Протеопатии, известные также как протеинопатии, связанные с конформационными нарушениями белков, или болезни, связанные с мисфолдингом (неправильным сворачиванием) белков, включают множество болезней, таких как болезнь Альцгеймера, болезнь Паркинсона, прионная болезнь, диабет 2 типа, амилоидоз и ряд других заболеваний (смотри неограничивающие примеры далее).
Использованные в описании термины "протеинопатии, протеопатии, заболевания, связанные с конформационными нарушениями белков, болезни, связанные с мисфолдингом белков, болезни, связанные со стрессом, вызванным мисфолдингом белков, болезни, связанные с накоплением неправильно свернутых белков, болезни, связанные с цитотоксическим ER-стрессом, болезни, связанные с UPR, имеют одинаковое значение и относятся к заболеваниям, при которых определенный белок становится структурно аномальным и тем самым нарушает клеточный гомеостаз.
Использованные в описании термины "неправильно свернутый белок" и "несвернутый белок" имеют одинаковое значение и относятся к белку, утратившему способность принимать свою нормальную конформацию.
Использованная в описании фраза "изготовление лекарственного средства" включает использование одного или более из описанных выше соединений непосредственно в качестве лекарственного средства наряду с его использованием в программе скрининга дополнительных активных веществ или в любой стадии производства такого лекарственного средства.
Еще один аспект изобретения имеет отношение к способу лечения протеинопатии и/или заболевания, связанного со стрессом, вызванным неправильным сворачиванием белков, и в частности, с накоплением неправильно свернутых белков у субъекта, нуждающегося в этом, указанный способ включает введение терапевтически эффективного количества соединения формулы (I) или (II), как определено выше, указанному субъекту.
Термин "способ" имеет отношение к методам, средствам, приемам и процедурам, предназначенным для выполнения определенной задачи, включая, но не ограничиваясь этим, или уже известные методы, средства, приемы и процедуры или разработанные на основе известных методов, средств, приемов и процедур специалистами-практиками в области химии, фармакологии, биологии, биохимии и медицины.
В описании термин "лечение" включает нейтрализацию, в значительной степени ингибирование, замедление или обратное развитие прогрессирования болезни или нарушения, уменьшение интенсивности клинических симптомов болезни или нарушения или предотвращение появления клинических симптомов болезни или нарушения.
Использованные в описании термины «болезнь», «нарушение», «состояние» имеют одинаковое значение. Болезнь является болезнью, связанной с активной реакцией на ER-стресс и/или связанной со стрессом, вызванным мисфолдингом белков, и в частности, с накоплением неправильно свернутых белков.
Термин "терапевтически эффективное количество" относится к тому количеству соединения, которое будучи введенным, до некоторой степени будет ослаблять один или более из симптомов болезни или нарушения, которое нуждается в лечении.
В другом варианте осуществления изобретение имеет отношение к соединению формулы (I) или (II), как определено выше, предназначенному для лечения UPR нарушений. Термин "реакция несвернутых белков" или UPR имеет отношение к компоненту системы клеточной защиты от неправильно свернутых белков, которая приводит фолдинг в эндоплазматическом ретикулуме (ER) в соответствие с изменяющимися условиями. UPR активируется в ответ на накопление несвернутых или неправильно свернутых белков в просвете эндоплазматического ретикулума. В такой ситуации UPR имеет две основные цели: (i) сохранение нормальной функции клетки путем остановки трансляции белка и (ii) активирование сигнальных путей, приводящих к повышенной выработке молекулярных шаперонов, вовлеченных в сворачивание белков. В том случае, если эти цели не достигаются в пределах определенного времени, или нарушение продолжается, UPR «ставит своей целью» апоптоз. Вышерасположенные компоненты UPR являются резидентными трансмембранными белками эндоплазматического ретикулума IRE1, ATF6 и PERK, которые определяют дефекты фолдинга, чтобы согласованно перепрограммировать транскрипцию и трансляцию и восстановить протеостаз. Активированные IRE1 и ATF6 повышают транскрипцию генов, вовлеченных в ER-фолдинг, например, кодирующих шапероны BiP и GRP94.
Активированный PERK ослабляет общий синтез белков путем фосфорилирования субъединицы фактора 2 инициации трансляции (eIF2α) на Ser51, при этом способствуя трансляции фактора транскрипции ATF4. Последний контролирует экспрессию CHOP, другого транскрипционного фактора, который в свою очередь способствует экспрессии PPP1R15A/GADD34. PPP1R15A, эффектор петли отрицательной обратной связи, которая завершает сигнальный путь UPR, мобилизирует каталитическую субъединицу протеинфосфатазы 1 (РР1с), чтобы дефосфорилировать eIF2α, обеспечивая возможность возобновления синтеза белков. Отсутствие UPR способствует развитию многих патологических состояний, которые могут быть скорректированы эффективным увеличением этого адаптивного ответа. Селективные ингибиторы индуцированной стрессом eIF2α фосфатазы PPP1R15A-PP1 задерживают eIF2α дефосфорилирование и, следовательно, синтез белков, избирательно в клетках, подвергнутых стрессу, не затрагивая синтез белков в неподвергнутых стрессу клетках, которые конститутивно экспрессируют eIF2α фосфатазу PPP1R15B-PP1. Это продлевает благоприятные эффекты UPR. Временное уменьшение синтеза белков полезно для подвергнутых стрессу клеток, потому что уменьшение поступления синтезированных белков увеличивает жизнеспособность шаперонов и таким образом защищает от тресса, вызванного мисфолдингом (Tsaytler et al., 2011 Science, 332, 91-94). Неселективные ингибиторы 2 eIF2α фосфатаз PPP1R15A-PP1 и PPP1R15B-PP1 могут оказывать нежелательные воздействия, так как постоянное ингибирование трансляции является вредным. Действительно, генетическое устранение и PPP1R15A и PPP1R15B приводит к ранней эмбриональной смертности у мышей, что показывает, что ингибирование двух eIF2α фосфатаз PPP1R15A-PP1 и PPP1R15B-PP1 является вредоносным в отношении всего организма. В отличие от этого генетическое удаление PPP1R15A не имеет пагубных последствий у мышей (Harding et al., 2009, Proc. Natl. Acad. Sci. USA, 106, 1832-1837). Более того, прогнозируется, что специфические ингибиторы PPP1R15A будут неактивными в неподвергнутых стрессу клетках, поскольку PPP1R15A не экспрессируется в отсутствие стресса. Таким образом, предполагается, что селективные PPP1R15A ингибиторы будут безопасными. Неселективные ингибиторы двух eIF2α фосфатаз также можно использовать для лечения болезней, связанных с мисфолдингом белков, при использовании в дозах, которые приводят только к частичному ингибированию фосфатаз.
Цитопротекция от ER-стресса может быть измерена с помощью подходящего метода анализа. Например, цитопротекция может быть измерена в клетках HeLa, в которых ER-стресс вызывают добавлением среды, содержащей туникамицин, смесь гомологичных нуклеозидных антибиотиков, которые ингибируют семейство ферментов UDP-HexNAc: полипренол-Р HexNAc-1-P и используются для того, чтобы вызывать реакцию несвернутых белков. Жизнеспособность клеток может быть определена в присутствии и в отсутствие ингибирующего соединения после заданного периода времени, путем измерения восстановления WST-8 в формазан с использованием стандартного набора для определения жизнеспособности клеток (такого как Cell Viability Counting Kit-8 от компании Dojindo). Цитопротекцию от ER-стресса определяют по величине процента увеличения жизнеспособных клеток (относительно контроля) после ER-стресса. Дополнительные подробности подходящего метода анализа предоставлены в разделе Примеры.
В одном предпочтительном варианте осуществления соединение формулы (I) или (II) способно пролонгировать защитный эффект UPR по сравнению с контролем (т.е. в отсутствие соединения-ингибитора), по меньшей мере, на 10%, по меньшей мере, на 20%, более предпочтительно, по меньшей мере, на 30%, даже более предпочтительно, по меньшей мере, на 40%, по меньшей мере, на 50%, по меньшей мере, на 60%, по меньшей мере, на 70%, по меньшей мере, на 80%, еще более предпочтительно, по меньшей мере, на 90%.
Соединения формулы (I) или (II) являются ингибиторами PPP1R15A-PP1 взаимодействия, которое индуцирует защитный эффект. Предпочтительно, соединение демонстрирует защитный эффект при показателе ЕС50 менее, чем около 5μM, даже более предпочтительно, менее чем около 2μМ, еще более предпочтительно менее, чем около 1μM. Предпочтительно соединение не должно иметь альфа-2-адренергической активности. Таким образом, в одном предпочтительном варианте осуществления соединение не проявляет какой-либо активности в функциональном анализе альфа-2-адренергической активности.
Некоторые соединения формулы (I) или (II) селективно ингибируют PPP1R15A-РР1, и таким образом, пролонгируют защитный эффект UPR, тем самым спасая клетки от стресса, вызванного неправильным сворачиванием белков. Следовательно, ингибиторы PPP1R15A-PP1, описанные в настоящем изобретении, могут иметь терапевтическое применение при лечении целого ряда болезней, связанных со стрессом, вызванным неправильным сворачиванием белков, и в частности, с накоплением неправильно свернутых белков, конкретнее при лечении протеинопатий.
В одном варианте осуществления соединение формулы (I) или (II) способно ингибировать PPP1R15A и PPP1R15B. В одном предпочтительном варианте осуществления соединение формулы (I) или (II) способно селективно ингибировать PPP1R15A по сравнению с PPP1R15B.
В одном варианте осуществления изобретение имеет отношение к соединению формулы (I) или (II), как определено выше, предназначенному для использования при лечении заболевания, связанного с путем фосфорилирования eIF2α, когда накопление неправильно свернутых белков вовлечено в механизм действия. Предпочтительно, заболевание является заболеванием или нарушением, связанным с PPP1R15A.
В другом варианте осуществления изобретение имеет отношение к соединению формулы (I) или (II), как определено выше, предназначенному для использования при лечении заболевания, вызванного, связанного с или сопровождающегося eIF2α фосфорилированием и/или PPP1R15A активностью, когда накопление неправильно свернутых белков вовлечено в механизм действия.
В другом варианте осуществления изобретение имеет отношение к соединению формулы (I) или (II), как определено выше, предназначенному для использования при лечении UPR нарушения, такого как, но без ограничения, старение (Naidoo et al., 2008, J Neurosci, 28, 6539-48).
При использовании в описании термин "болезнь или нарушение, связанное с PPP1R15A," имеет отношение к болезни или нарушению, характеризующемуся ненормальной PPP1R15A активностью, когда накопление неправильно свернутых белков вовлечено в механизм действия. Ненормальная активность относится к: (i) PPP1R15A экспрессии в клетках, которые не экспрессируют PPP1R15A; (ii) повышенной PPP1R15A экспрессии; или (iii) повышенной PPP1R15A активности.
В другом варианте осуществления изобретение имеет отношение к способу лечения млекопитающего, имеющего болезненное состояние, которое облегчается ингибированием PP1R15A, когда накопление неправильно свернутых белков вовлечено в механизм действия, при этом способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I) или (II), как определено выше.
В другом варианте осуществления изобретение имеет отношение к ингибитору PPP1R15A формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении нарушений, связанных со стрессом, вызванным неправильным сворачиванием белков, и в частности, с накоплением неправильно свернутых белков и/или нарушениями UPR, при этом указанное соединение не обладает или обладает уменьшенной активностью в качестве альфа-2-адренергического агониста по сравнению с гуанабензом.
В другом варианте осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении заболеваний, связанных со стрессом, вызванным неправильным сворачиванием белков, и в частности, с накоплением неправильно свернутых белков и/или нарушениями UPR, при этом указанное соединение не ингибирует трансляцию белков в неподвергнутых стрессу клетках, экспрессирующих PPP1R15B.
В другом варианте осуществления изобретение имеет отношение к способу лечения заболевания, характеризующегося активной реакцией на ER-стресс в сочетании с накоплением неправильно свернутых белков, данный способ включает введение пациенту терапевтически эффективного количества, по меньшей мере, одного соединения формулы (I) или (II), при этом указанное соединение модулирует ответ на ER-стресс.
В другом варианте осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для лечения заболеваний, связанных со стрессом, вызванным неправильным сворачиванием белков, и в частности, с накоплением неправильно свернутых белков и/или нарушениями UPR, при этом указанное соединение обладает селективностью в отношении PPP1R15A-PP1 голофосфатазы, не обладая или обладая уменьшенной активностью в отношении PPP1R15B-PP1 голофосфатазы, и при этом отношение (активность в отношении PPP1R15A-PP1 голофосфатазы / активность в отношении PPP1R15B-PP1) для указанного соединения, по меньшей мере, является равным или превышает отношение (активность в отношении PPP1R15A-PP1 голофосфатазы / активность в отношении PPP1R15B-PP1) для гуанабенза.
В другом варианте осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении нарушений, связанных со стрессом, вызванным мисфолдингом белков, и в частности, с накоплением неправильно свернутых белков и/или UPR нарушениями, при этом:
- указанное соединение обладает активностью в отношении PPP1R15A-PP1 голофосфатазы, но не обладает или обладает уменьшенной активностью в отношении PPP1R15B-PP1 голофосфатазы, и;
- при этом отношение (активность в отношении PPP1R15A-PP1 голофосфатазы / активность в отношении PPP1R15B-PP1) для указанного соединения, по меньшей мере, является равным или превосходит отношение (активность в отношении PPP1R15A-PP1 голофосфатазы / активность в отношении PPP1R15B-PP1) для гуанабенза; и
- при этом указанное соединение не обладает или обладает уменьшенной активностью в качестве альфа-2-адренергического агониста в сравнении с гуанабензом.
В другом варианте осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенной для использования при лечении заболевания или состояния, характеризующегося, по меньшей мере, одним из числа (1) стресса ER, (2) накопления в клетке несвернутого или неправильно свернутого белка и (3) UPR.
В другом варианте осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении болезни у субъекта, характеризующейся или связанной, по меньшей мере, с одним из числа (1) стресса эндоплазматического ретикулума (ER), (2) накопления в клетке несвернутых или неправильно свернутых белков и (3) реакции несвернутых белков.
Указанная болезнь связана с активной реакцией на ER-стресс и/или связана со стрессом, вызванным неправильным сворачиванием белков, и в частности, с накоплением несвернутых или неправильно свернутых белков; конкретнее болезнь является протеинопатией. Неограничивающие примеры болезни согласно изобретению включают, но не ограничиваются этим:
- Нейродегенеративные болезни, такие как таупатия (например, болезнь Альцгеймера в числе прочих), синуклеопатия (например, болезнь Паркинсона в числе прочих), болезнь Хантингтона и родственные полиглутаминные болезни, полиаланиновые заболевания (такие как окулофарингеальная мышечная дистрофия), прионные болезни (также называемые трансмиссивными губчатообразными энцефалопатиями), болезни, связанные с демиелинизацией, такие как болезнь Шарко-Мари-Тута (также называемая наследственной двигательной и сенсорной невропатией), лейкодистрофия, боковой амиотрофический склероз (также называемый заболеванием двигательных нейронов и болезнью Лу Герига) и рассеянный склероз.
Примеры таупатии включают, но не ограничиваются этим, болезнь Альцгеймера, прогрессирующий надъядерный паралич, кортико-базальную дегенерацию, лобновисочную лобарную дегенерацию (болезнь Пика). Лобно-височная деменция (FTD) является нейродегенеративным заболеванием, характеризующимся прогрессирующей потерей нейронов, преимущественно затрагивающим лобную и/или височную доли; уступающим по распространению только болезни Альцгеймера (AD), FTD обусловливает 20% случаев раннего начала деменции. Участие UPR в таупатии хорошо подтверждено (смотри Stoveken 2013, The Journal of Neuroscience 33(36): 14285-14287). Без связи с какой-либо теорией, ожидается, что соединения изобретения, которые являются PPP1R15A ингибиторами, будут уменьшать болезненные проявления таупатии. В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения болезни Альцгеймера. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении болезни, выбранной из числа лобно-височной деменции (FTD), надъядерного паралича и кортико-базальной дегенерации, предпочтительно FTD.
Примеры синуклеопатий включают, но не ограничиваются этим, болезнь Паркинсона, деменцию с тельцами Леви, первичную вегетативную недостаточность и множественную системную атрофию. Недавно Colla et al. (J. of Neuroscience 2012 Vol. 32 №10 рр 3306-3320) продемонстрироовали, что салубринал, небольшая молекула, которая увеличивает фосфорилирование eIF2 альфа путем ингибирования опосредованного PPP1R15A дефосфорилирования eIF2 альфа (Воусе et al. 2005 Science Vol. 307 pp 935-939), значительно ослабляет проявления болезни на двух животных моделях альфа-синуклеопатии. Соединения изобретения, которые являются ингибиторами PPP1R15A, будут ослаблять болезненные проявления альфа-синуклеопатии, такой как болезнь Паркинсона. В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения альфа-синуклеопатии, такой как болезнь Паркинсона.
Примеры полиглутаминных заболеваний включают, но не ограничиваются этим, спинобульбарную мышечную атрофию (или болезнь Кеннеди), болезнь Хантингтона, дентато-рубро-паллидо-льюисову атрофию, спинально-церебеллярную атаксию 1 типа, спинально-церебеллярную атаксию 2 типа, спинально-церебеллярную атаксию 3 типа (или болезнь Мачадо-Джозефа), спинально-церебеллярную атаксию 6 типа, спинально-церебеллярную атаксию 7 типа и спинально-церебеллярную атаксию 17 типа. Гуанабенз способен уменьшать накопление мутантного хантингтина в исследованиях на клетках (WO 2008/041133). Это открытие является неожиданным, поскольку мутантный белок хантингтин является либо цитозольным, либо ядерным. Однако, существует доказательство того, что метаболизм мутантного хантингтина ранее был связан с ответом на стресс ER (Nishitoh et al., 2002, Genes Dev, 16, 1345-55; Rousseau et al., 2004, Proc Natl Acad Sci USA, 101, 9648-53; Duennwald и Lindquist, 2008, Genes Dev, 22, 3308-19). Обнаружение того, что гуанабенз защищает клетки от цитотоксического стресса ER и уменьшает накопление мутантного хантингтина, дополнительно поддерживает идею о том, что возможно есть аспекты ответа на ER-стресс, которые влияют на накопление мутантного хантингтина. Тем не менее, гуанабенз не используется для лечения болезней человека, связанных с неправильным сворачиванием белков, вследствие его гипотензивной активности. В противоположность этому производные гуанабенза ингибиторы PPP1R15A, лишенные альфа-2-адренергической активности, могут использоваться для лечения полиглутаминных заболеваний и, конкретнее, болезни Хантингтона. В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения болезни Хантингтона.
Примеры полиаланиновых заболеваний включают окулофарингеальную мышечную дистрофию, которая вызывается поли-аланиновым участком в ядерном поли(А) связывающем белке 1 (PABPN1). Barbezier et al. (2011, EMBO Vol. 3 pp 35-49) показал, что гуанабенз уменьшает агрегацию при окулофарингеальной мышечной дистрофии. Согласно предпочтительному варианту осуществления изобретение имеет отношение к ингибитору PPP1R15A формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенной для использования при лечении полиаланиновых заболеваний, конкретнее окулофарингеальной мышечной дистрофии.
Примеры прионных болезней человека включают, но не ограничиваются этим, классическую болезнь Крейтцфельда-Якоба, новый вариант болезни Крейтцфельда-Якоба (nvCJD, заболевание человека, связанное с губчатой энцефалопатией крупного рогатого скота), синдром Гертсмана-Штраусслера-Шейнкера, спорадическую фатальную инсомнию и куру. Гуанабенз уменьшает симптомы у мышей, инфицированных прионами (D. Tribouillard-Tanvier et al., 2008 PLoS One 3, e1981). Тем не менее, гуанабенз не применяется для лечения болезней человека, связанных с нарушением правильного сворачивания (фолдинга) белков, вследствие его гипотензивной активности. В то же время производные гуанабенза ингибиторы PPP1R15A, лишенные альфа-2-адренергической активности, могут использоваться для лечения прионных болезней. Согласно предпочтительному варианту осуществления изобретение имеет отношение к ингибитору PPP1R15A формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении болезни, выбранной из болезни Крейтцфельда-Якоба, нового варианта болезни Крейтцфельда-Якоба, синдрома Гертсмана-Штраусслера-Шейнкера, смертельной семейной бессонницы и куру.
Нарушения, связанные с демиелинизацией, характеризуются потерей олигодендроцитов в центральной нервной системе или шванновских клеток в периферической нервной системе. Явление, связанное с нарушением демиелинизации, характеризуется уменьшением миелиновых аксонов в центральной нервной системе или периферической нервной системе. Неограничивающие примеры неправильно свернутых белков миелинизирующей клетки (включая олигодендроцит и шванновскую клетку) выбирают из группы, состоящей из СС1, основного миелинового белка (МВР), церамид галактозилтрансферазы (CGT), миелин-ассоциированного гликопротеина (МАГ), миелинового олигодендроцитарного гликопротеина (МОГ), олигодендроцит-миелинового гликопротеина (ОМГ), фосфодиэстеразы циклических нуклеотидов (CNP), белка миелина zero (MPZ), периферического миелинового белка 22 (РМР22), коннексина 32 (Сх32), белка 2 (Р2), галактоцереброзида (Ga1C), сульфатида и протеолипидного белка (PLP). MPZ, РМР22, Сх32 и Р2 являются преимущественными неправильно свернутыми белками для шванновских клеток. PLP, МБР, MAG являются преимущественными неправильно свернутыми белками для олигодендроцитов.
В некоторых вариантах осуществления нарушение, связанное с демиелинизацией, выбирают из группы, состоящей из болезней Шарко-Мари-Тута (СМТ). Болезни СМТ относятся к группе наследственных невропатий, отличающихся хронической двигательной и сенсорной полиневропатией. Были установлены различные типы СМТ, такие как СМТ1, СМТ2, СМТ4, СМТХ и болезнь Дежерина-Сотта. Подтипы СМТ могут быть дополнительно подразделены на основе молекулярно-генетических открытий. Например, СМТ1 подразделяется на СМТ1А, 1В, 1С, 1D, 1E, 1F/2E, 1X. Свыше 100 мутаций в гене, кодирующем миелиновый белок zero (Р0), трансмембранный белок с однократным пересечением мембраны, который является основным белком, вырабатываемым миелинизирующими шванновскими клетками, вызывают невропатию Шарко-Мари-Тута (D'Antonio et al., 2009, J Neurosci Res, 87, 3241-9). Эти мутации являются в основном наследственными и вызывают болезнь в результате увеличения токсической функции (D'Antonio et al., 2009, J Neurosci Res, 87, 3241-9). Делеция серина 63 из P0 (P0S63del) вызывает невропатию Шарко-Мари-Тута 1В у людей и аналогичную демиелинизирующую нейропатию у трансгенных мышей. Мутантный белок накапливается в ER и индуцирует UPR (D'Antonio et al., 2009). Генетическое удаление CHOP, проапоптотического гена в UPR, восстанавливает двигательную функцию у мышей с болезнью Шарко-Мари-Тута (Pennuto et al., 2008, Neuron, 57, 393-405). Открытие того, что PPP1R15A ингибирование в клетках почти прекращает CHOP экспрессию в клетках, подвергнутых ER-стрессу, показывает, что генетическое или фармакологическое ингибирование PPP1R15A должно уменьшать двигательную дисфункцию у мышей с болезнью Шарко-Мари-Тута. Недавно D'Antonio et al (2013 J. Exp. Med Vol. pp 1-18) продемонстрировали, что у P0S63del-мышей, которых лечили салубриналом, восстановилась почти нормальная двигательная способность (тест на вращающемся барабане), что сопровождалось избавлением от морфологических и электрофизиологических нарушений. Накопление связанного с СМТ мутанта в белках ER характерно не только для P0S63del; было установлено, по меньшей мере, пять других Р0 мутантов, которые хранятся в ER и вызывают UPR (Pennuto et al., 2008; Saporta et al., 2012 Brain Vol. 135 pp 2032-2047). Кроме того, неправильное сворачивание белков и накопление неправильно свернутых белков в ER участвует в патогенезе других СМТ невропатий вследствие мутаций в РМР22 и Сх32 (Colby et al., 2000 Neurobiol. Disease Vol. 7 pp 561-573; Kleopa et al., 2002 J. Neurosci. Res. Vol. 68 pp 522-534; Yum et al., 2002 Neurobiol. Dis. Vol. 11 pp 43-52). Однако, салубринал является токсичным и не может использоваться для лечения людей (D'Antonio et al. (2013)). Напротив, предполагается, что PPP1R15A ингибиторы формулы (I) или (II) являются безопасными и могут использоваться для лечения СМТ, предпочтительно СМТ-1, и более предпочтительно СМТ-1А, СМТ-1В, СМТ-1Е, СМТ-1X. В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения болезней Шарко-Мари-Тута, предпочтительно СМТ-1, более предпочтительно СМТ-1А, СМТ-1В, СМТ-1Е и СМТ-1X. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении СМТ, более предпочтительно СМТ-1 и болезни Дежерина-Сотта. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для лечения СМТ, связанной с накоплением неправильно свернутого белка в ER. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для лечения СМТ-1А. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или его фармацевтически приемлемой соли, предназначенному для лечения СМТ-1В. Согласно предпочтительному варианту осуществления изобретение имеет отношение к ингибитору PPP1R15A формулы (I) или его фармацевтически приемлемой соли, предназначенному для лечения СМТ-1Е. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или его фармацевтически приемлемой соли, предназначенному для лечения СМТ-1X.
В другом варианте осуществления соединение формулы (I) или (II) предназначается для лечения СМТ, более предпочтительно для применения при лечении СМТ-1, в сочетании, по меньшей мере, с одним соединением, выбранным из группы, включающей D-сорбитол, баклофен, пилокарпин, налтрексон, метимазол, мифепристон, кетопрофен и их соли. Соединения объединяют для введения группами или по отдельности, одновременно или последовательно.
Изобретение имеет отношение к композиции, содержащей PPP1R15A ингибитор, выбранный из группы соединения формулы (I) или (II), гуанабенза и салубринала или его фармацевтически приемлемой соли, и, по меньшей мере, одного имеющегося на рынке соединения и его солей, предназначенных для использования при лечении нейродегенеративных болезней, предпочтительно СМТ, более предпочтительно СМТ-1. Дозировка соединения в композиции должна находиться внутри диапазона доз, не превышающих дозы, обычно назначаемые в течение длительного поддерживающего лечения, или являющихся безопасными, что доказано на 3 фазе клинических испытаний; большинство предпочтительных дозировок соединения в комбинации должно соответствовать количеству от 1% до 10% от дозировок, которые обычно назначаются в течение длительного поддерживающего лечения.
Таким образом, изобретение имеет отношение к композиции, содержащей PPP1R15A ингибитор, выбранный из группы, включающей соединение формулы (I) или (II), гуанабенз и салубринал или его фармацевтически приемлемую соль, и соединение, увеличивающее экспрессию РМР22 белка, выбранное из группы, включающей D-сорбитол, баклофен, пилокарпин, налтрексон, метимазол, мифепристон, кетопрофен и их соли, предназначенной для использования при лечении СМТ, предпочтительно СМТ-1, более предпочтительно СМТ-1А, более предпочтительно СМТ-1А, СМТ-1В, СМТ-1Е и СМТ-1X.
В других вариантах осуществления нарушения, связанные с демиелинизацией, выбирают из группы, состоящей из лейкодистрофий. Примеры лейкодистрофий включают, но не ограничиваются этим, адренолейкодистрофию (ALD), болезнь Александера, болезнь Канавана, болезнь Краббе, метахроматическую лейкодистрофию (MLD), болезнь Пелицеуса-Мерцбахера (PMD), детскую атаксию с гипомиелинизацией центральной нервной системы (также известную как болезнь исчезающего белого вещества головного мозга), синдром CAMFAK, болезнь Рефсума, синдром Коккейна, синдром Ван дер Кнаппа, синдром Зельвегера, синдром Гийена-Барре (GBS), хроническую воспалительную демиелинизирующую полинейропатию (CIDP), мультифокальную двигательную нейропатию (MMN) и прогрессирующий надъядерный паралич, прогрессирующую мультифокальную лейкоэнцефалопатию (РМЛ), энцефаломиелит, центральный миелиноз моста (СРМ), болезнь, связанную с выработкой антител к миелин-ассоциированному гликопротеину (Anti-MAG) в числе прочих. Gow et al. (Neuron, 2002 Vol. 36, 585-596) продемонстрировали, что реакция несвернутых белков активируется при PMD, и показали, что этот путь представляет собой удвоение PLP1 гена. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении лейкодистрофий, и предпочтительно болезни Пелицеуса-Мерцбахера (PMD).
Амиотрофический боковой склероз (ALS) известен как заболевание двигательных нейронов и как болезнь Лу Герига. В настоящее время широко известно, что нарушенный фолдинг белка играет центральную роль как в семейном, так и в спорадическом ALS (Matus et al. 2013 Int. J. Cell Biol. ID674751 http://dx.doi.org/10.1155/2013/674751). Saxena et al. (Nature Neuroscience 2009 Vol.12 pp627-636) продемонстрировали, что салубринал увеличиает продолжительность жизни на G93A-SOD1 трансгенной мышиной модели болезни двигательных нейронов. Совсем недавно, Jiang et al. (Neuroscience 2014) продемонстрировали, что гуанабенз задерживает начало появления симптомов болезни, увеличиает продолжительность жизни, улучшает двигательную активность и уменьшает потерю двигательных нейронов на SOD1 G93A мышиной модели ALS. Без связи с какой-либо теорией, ожидается, что соединения изобретения, являющиеся производными гуанабенза и PPP1R15A ингибиторами, будут облегчать болезненные проявления ALS в сочетании с SOD1 мутацией G93A. Поэтому соединения формулы (I) и (II) могут использоваться для лечения как семейной, так и спорадической форм ALS.
Примеры сейпинопатий включают, но не ограничиваются этим, врожденную липодистрофию Берардинелли-Сейпа 2 типа, связанную с мутацией гена BSCL2, генерализованную врожденную липодистрофию (CGL), синдром Сильвера, дистальную наследственную двигательную невропатию V типа (dHMN-V). Экспрессия мутантных форм сейпина в культивируемых клетках активирует путь реакции несвернутых белков (UPR) и вызывает гибель клеток, индуцированную стрессом ER (Ito & Suzuki, 2009 Brain 132: 87-15). Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении сейпинопатии.
В другом варианте осуществления упоминаемое в описании заболевание, связанное с демиелинизацией, представляет собой рассеянный склероз и связанную с ним болезнь, такую как болезнь Шильдера. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении рассеянного склероза.
- Муковисцидоз (CF)
Norez et al. (2008 Eur. J. Pharmacol. Vol. 592 рр 33-40) продемонстрировали, что гуанабенз активирует Са2+ зависимые токи хлора в эпителиальных клетках дыхательных путей человека с муковисцидозом. Без связи с какой-либо теорией, предполагается, что соединения изобретения, являющиеся производными гуанабенза и PPP1R15A ингибиторами, будут облегчать болезненные проявления муковисцидоза. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении муковисцидоза.
- Ретинальные болезни (болезни сетчатки глаза).
В недавно опубликованных работах были предоставлены доказательства того, что UPR участвует в развитии дегенерации сетчатки: наследственной дегенерации сетчатки, такой как ретинальная цилиопатия и пигментная дегенерация сетчатки, дегенерация желтого пятна, ретинопатия недоношенных, индуцированная светом дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома (смотри обзор Gorbatyuk et Gorbatyuk 2013 - Retinal degeneration: Focus on the unfolded protein response, Molecular Vision Vol. 19 pp 1985-1998). Новые данные подтверждают роль ER-стресса в ретинальном апоптозе и клеточной смерти (Jing et al., 2012, Exp Диабет Res, 2012, 589589).
Ретинальные цилиопатии представляют собой группу редких наследственных заболеваний, которые возникают в результате дефекта в первичных ресничках фоторецепторов, вызывающего пигментную дегенерацию сетчатки (пигментный ретинит). Сообщалось, что этот дефект индуцирует стресс ER вследствие накопления белка во внутреннем сегменте фоторецептора, который в свою очередь индуцирует UPR (WO 2013/124484). Дегенерация сетчатки является очень часто встречающимся признаком цилиопатии, который может наблюдаться как при изолированном пигментном ретините, таком как врожденный амавроз Лебера или связанная с Х-хромосомой пигментная дегенерация сетчатки, или также при синдромных состояниях, таких как синдром Барде-Бидля (BBS), синдром Альстрема (ALMS) или синдром Ушера. Ретинальные цилиопатии выбирают из группы, состоящей из синдрома Барде-Бидля, синдрома Сениора-Локена, синдрома Жубера, синдрома Сальдино-Майнзера, синдрома Сенсенбреннера, синдрома Жене, синдрома Меккеля-Грубера, синдрома Альстрёма, синдрома MORM, врожденного амавроза Лебера, вызванного мутацией в цилиарном гене, и связанной с Х-хромосомой пигментной дегенерации сетчатки, вызванной мутацией в RPGR гене.
Пигментная дегенерация сетчатки является наследственной, дегенеративной болезнью глаз, которая вызывает сильное нарушение зрения и часто слепоту. Это наиболее частая причина генетически обусловленной слепоты. У пациентов наблюдается один или более из следующих симптомов, включая куриную (ночную) слепоту; туннельное зрение (отсутствие периферического зрения); боковое зрение (отсутствие центрального зрения); сетчатое (решетчатое) зрение; неприятие яркого света; медленную адаптацию от темноты к свету и наоборот; неясность зрения; плохое разделение по цвету и чрезмерную усталость. Пигментный ретинит (RP) возникает в результате свыше 100 мутаций в гене родопсина (Dryja et al, 1991, Proc Natl Acad Sci USA, 88, 9370-4). Родопсин является светочувствительным рецептором клеток-палочек, связанным с G-белком, и представляет собой ковалентный комплекс между трансмембранным белком опсином, состоящим из 348 аминокислот, и 11-цис ретиналем (Palczewski, 2006, Annu Rev Biochem, 75, 743-67). Мутации родопсина, вызывающие RP, представляют собой в основном миссенс-мутации, распределенные по всему белку (Dryja et al., 1991), подобно ALS-вызванным мутациям SOD1 (Valentine et al, 2005, Annu Rev Biochem, 74, 563-93). Мутации родопсина, вызывающие RP, были исследованы в различных системах и происходят в результате гетерологичной экспрессии белков в клетках млекопитающих, у трансгенных мышей и дрозофилы are consistent (Griciuc et al., 2011, Trends Mol Med, 17, 442-51). Наиболее распространенные мутанты родопсина, вызывающие RP, не способны сворачиваться, не связываются с 11-цис-ретиналем, не доходят до клеточной поверхности, а сохраняются в ER (Griciuc et al., 2011, Trends Mol Med, 17, 442-51). Мисфолдинг мутантов родопсина вызывает стресс ER и смерть палочковидных зрительных клеток (Griciuc et al., 2011). Это убедительно указывает на то, что PPP1R15A ингибиторы, подобные гуанабензу, но преимущественно не проявляющие активности в отношении адренергического альфа-2А рецептора, такие как соединения изобретения, будут улучшать RP.
В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения ретинальных болезней, более предпочтительно, наследственной дегенерации сетчатки, такой как ретинальная цилиопатия и пигментная дегенерация сетчатки, дегенерация желтого пятна, ретинопатия недоношенных, индуцированная светом дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении синдромной пигментной дегенерации сетчатки и/или внесиндромного пигментного ретинита. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении врожденного амавроза Лебера. Согласно другому предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или его фармацевтически приемлемой соли, предназначенному для использования при лечении синдрома Барде-Бидля. Согласно другому предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или его фармацевтически приемлемой соли, предназначенному для использования при лечении синдрома Альстрёма. Согласно другому предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или его фармацевтически приемлемой соли, предназначенному для использования при лечении синдрома Ушера.
Возрастная макулярная дегенерация (AMD) является главной причиной гражданской (практической) слепоты в Соединенных Штатах среди людей в возрасте свыше 65 лет.Shen et al. (2011 Effect of Guanabenz on Rat AMD Models и Rabbit Choroidal Blood - Vol. 5 pp 27-31) показали, что гуанабенз в значительной степени защищал пигментный эпителий сетчатки (RPE) от NaIO3-индуцированной дегенерации, препятствовал развитию хориоидальной неоваскуляризации (CNV) на индуцированной лазером модели AMD у крыс и заметно увеличивал хориоидальный ток крови in vivo. Соединения изобретения производные гуанабенза, которые являются PPP1R15A ингибиторами, подобно гуанабензу, которые, однако, не демонструруют активности в отношении адренергического альфа-2А рецептора, будут пригодны для лечения дегенерации сетчатки или макулярной дегенерации.
В предпочтительном варианте осуществления соединение формулы (I) предназначается для лечения ретинальных болезней, более предпочтительно для использования при лечении болезней, выбранных из группы наследственной дегенерации сетчатки, такой как ретинальная цилиопатия, пигментная дегенерация сетчатки, макулярная дегенерация, ретинопатия недоношенных, индуцированная светом дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома в сочетании с соединением, увеличивающим экспрессию и/или активность BIP белка, таким как вальпроевая кислота или ее производное, трихостатин А, литий, 1-(3,4-дигидрокси-фенил-2-тиоцианат-этанон и эксендин-4. Таким образом, изобретение имеет отношение к композиции, содержащей PPP1R15A ингибитор формулы (I) или его фармацевтически приемлемую соль и соединение, увеличивающее экспрессию и/или активность BIP белка, предпочтительно вальпроевую кислоту, предназначенной для использования при лечении болезней, выбранных из группы наследственной дегенерации сетчатки, такой как ретинальная цилиопатия, пигментная дегенерация сетчатки, макулярная дегенерация, ретинопатия недоношенных, индуцированная светом дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома. В предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения болезней сетчатки, более предпочтительно для использования при лечении болезней, выбранных из группы наследственной дегенерации сетчатки, такой как ретинальная цилиопатия, пигментная дегенерация сетчатки, макулярная дегенерация, ретинопатия недоношенных, индуцированная светом дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома, в сочетании с генотерапевтическими векторами. Неограничивающие примеры генотерапевтических векторов включают лентивирусные, аденовирусные и аденоассоциированные векторы (AAVs); эти векторы являются эффективными при доставке представляющих интерес генов в сетчатку и пигментный эпителий сетчатки с целью офтальмологической генотерапии. Ожидается, что при офтальмологической генной терапии наследственной дегенерации сетчатки, связанной с накоплением мутированных неправильно свернутых белков, будет сохраняться накопление белка в эндоплазматическом ретикулуме, вместе с тем нормальный белок будет экспрессироваться из генотерапевтического вектора. Таким образом, сохраняется необходимость уменьшения накопления белка/«нагрузки» в клетке, преимущественно в ER, с помощью PPP1R15A ингибиторов. Изобретение также имеет отношение к композиции, содержащей PPP1R15A ингибитор, выбранный из группы, включающей соединение формулы (I) или (II), гуанабенз и салубринал или его фармацевтически приемлемую соль в сочетании с офтальмологической генотерапией.
- Лизосомные болезни накопления;
Лизосомные болезни накопления представляют собой группу приблизительно из 50 редких наследственных метаболических нарушений, возникающих в результате дефектов в функции лизосом. Лизосомальная дисфункция обычно является следствием недостатка отдельного фермента, необходимого для метаболизма липидов, гликопротеинов или так называемых мукополисахаридов. Примеры лизосомных болезней накопления, которые можно лечить с помощью PPP1R15A ингибиторов формулы (I) или (II), описанных в данном документе, включают, но не ограничиваются этим, недостаток активатора/GM2 ганглиозидоз, альфа-маннозидоз, аспартиглюкозаминурию, болезнь нкопления эфиров холестерола, цистиноз, болезнь Данона, болезнь Фабри, болезнь Фарбера, болезнь Ниманна-Пика, фукозидоз, галактосиалидоз, болезнь Гоше (типы I, II, II), GM1 ганглиозидоз (детский, поздний детский/ювенильный, взрослый/хронический), болезнь «I-клеток»/муколипидоз, детскую болезнь накопления свободной сиаловой кислоты/ISSD, ювенильный дефицит гексозаминидазы А, болезнь Краббе (с началом в детстве, с поздним началом), дефицит лизосомной кислой липазы (раннее начало/позднее начало), метахроматическую лейкодистрофию, мукополисахаридозы (такие как псевдополидистрофия Гурлера/муколипидоз IIIA, мукополисахаридоз I (MPS I) синдром Гурлера, MPS I синдром Шейе, MPS I синдром Гурлера-Шейе, MPS II синдром Хантера, синдром Санфилиппо типа A (MPS IIIA), синдром Санфилиппо типа В (MPS IIIB), синдром Санфилиппо типа С (MPS IIIC), синдром Санфилиппо типа D (MPS IIID), болезнь Моркио типа A/MPS IVA, болезнь Моркио типа B/MPS IVB, MPS IX дефицит гиалуронидазы, MPS VI Марото-Лами, MPS VII синдром Слая, мукополилипидоз I/сиалидоз, муколипидоз IIIC, муколипидоз типа IV (множественную сульфатазную недостаточность, болезнь Ниманна-Пика (типы А, В, С), CLN6 болезнь (атипичная поздняя детская, вариант с поздним началом, ранняя юношеская), болезнь Шпильмайера-Фогта-Баттена/ювенильную NCL/CLN3 болезнь, Финский вариант поздней детской формы CLN5 болезни, болезнь Бильшовского-Янского/позднюю детскую CLN2/TPP1 болезнь, болезнь Куфса/болезнь NCL/CLN4 с началом в зрелом возрасте, северную эпилепсию/вариант поздней детской CLN8 болезни, болезнь Сантавуори-Халтиа/детскую CLN1/PPT болезнь, бета-маннозидоз, болезнь Помпе/болезнь накопления гликогена II типа, пикнодизостоз, болезнь Сандгоффа /GM2 ганглиозидоз (с началом в зрелом возрасте, с началом в детском возрасте, с началом в юношеском возрасте), болезнь Шиндлера, болезнь Салла/болезнь накопления сиаловой кислоты, болезнь Тея-Сакса/GM2 ганглиозидоз и болезнь Вольмана. Согласно предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении лизосомных болезней накопления, которые являются следствием недостатка, по меньшей мере, одного отдельного фермента, необходимого для метаболизма липидов, гликопротеинов или так называемых мукополисахаридов, при этом указанный фермент является неправильно свернутым в эндоплазматическом ретикулуме (ER). Согласно предпочтительному варианту осуществления лизосомная болезнь накопления является болезнью Гоше.
- Амилоидоз:
Амилоидоз - это неспецифический термин, относящийся к целому ряду различных болезней, собирательно называемых амилоидозом. Амилоиды представляют собой белки, вторичная структура которых изменена, что приводит к сворачиванию белков в характерную форму, бетаскладчатую конформацию. Когда нормально растворимые белки сворачиваются, становясь амилоидными, они становятся нерастворимыми, откладываются и накапливаются в органах или тканях, мешая нормальному функционированию.
Различные типы амилоидозов имеют разные признаки и симптомы в зависимости от того, где и в каких органах скапливаются амилоидные белки. Примеры амилоидоза включают, но не ограничиваются этим, AL, АН, ALH амилоидозы (амилоид, полученный из легкой цепи, тяжелой цепи, тяжелой и легкой цепи антител, соответственно), АА амилоидоз (амилоид, полученный из сывороточного А белка), ATTR амилоидоз (амилоид, полученный из транстиретина), первичный системный амилоидоз, вторичный системный амилоидоз, старческий системный амилоидоз, семейную амилоидную полинейропатию 1, наследственную церебральную амилоидную ангиопатию, амилоидоз, связанный с гемодиализом, семейную амилоидную полинейропатию III, Финский наследственный системный амилоидоз, атриальный амилоидоз, наследственный не невропатический системный амилоидоз, амилоидоз, расположенный в месте инъекции, наследственный амилоидоз почек и болезнь Альцгеймера, в числе прочих.
Согласно другому предпочтительному варианту осуществления амилоид является амилоидом бета (Аβ или Абета) и изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении болезни Альцгеймера.
Согласно другому предпочтительному варианту осуществления амилоид представляет собой HLA-B27 (Colbert et al. 2009 Prion Vol. 3 (1) pp 15-16) и изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для использования при лечении спондилоартропатии, более предпочтительно анкилозирующего спондилоартрита.
- Злокачественные заболевания (рак)
Раковые клетки имеют высокие метаболические потребности, и их пролиферация зависит от эффективного синтеза белков. Инициация трансляции играет решающую роль в контролировании гомеостаза белков, дифференцировки, пролиферации и злокачественной трансформации. Увеличение инициации трансляции способствует инициированию развития рака, наоборот, уменьшение инициации трансляции может уменьшить опухолевый рост (Donze et al., 1995, EMBO J, 14, 3828-34; Pervin et al., 2008, Cancer Res, 68, 4862-74; Chen et al., 2011, Nat Chem Biol, 7, 610-6). Без связи с какой-либо теорией, предполагается, что ингибирование PPP1R15A способно селективно уменьшать трансляцию в опухолевых клетках и таким образом уменьшать опухолевый рост. Примеры типов рака, которые можно лечить с помощью PPP1R15A ингибиторов формулы (I) или (II), описанных в данном документе, включают, но не ограничиваются этим, карциному, лимфому, бластому, саркому и лейкемию. Более конкретные примеры таких видов рака включают плоскоклеточный рак, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак желудочно-кишечного тракта, рак поджелудочной железы, нейробластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак толстого кишечника, колоректальный рак, карциному эндометрия, карциному слюнной железы, рак почки, рак предстательной железы, рак влагалища, рак щитовидной железы, гепатокациному, остеосаркому, рак желудка, меланому, множественную миелому, медуллярную кациному щитовидной железы и рак головы и шеи.
- Воспаление
PPP1R15A представляет собой перспективную мишень для контролирования воспаления путем блокирования высвобождения воспалительных цитокинов и других секретируемых молекулярных медиаторов, приводящих к патогенным условиям. Неограничивающие примеры болезней или состояний, сопровождающихся воспалением, которые можно лечить с помощью PPP1R15A ингибиторов формулы (I) или (II), описанных в данном документе, включают, но не ограничиваются этим, связанные с инфекцией или неинфекционные воспалительные состояния легких (т.е., сепсис, легочные инфекции, синдром острой дыхательной недостаточности, бронхолегочную дисплазию и т.д.); связанные с инфекцией или неинфекционные воспалительные состояния других органов, такие как колит, язвенный колит, воспалительное заболевание кишечника, диабетическая нефропатия, геморрагический шок, спондилоартропатия, панкреатит; вызванный воспалением рак (т.е., развитие рака у пациентов, связанного с колитом или воспалительным заболеванием кишечника); и тому подобные. Примеры таких патогенных воспалительных состояний включают аутоиммунные болезни, наследственные заболевания, хронические болезни и инфекционные болезни, такие как аллергия, астма, гиперцитокинемия, включая реакцию «трансплантат против хозяина» (GVHD), синдром острой дыхательной недостаточности (ARDS), сепсис, синдром системной воспалительной реакции (SIRS) (смотри WO 2011/061340). Предпочтительно, инфекционные болезни выбирают из инфекции вирусом гриппа, инфекции вирусом натуральной оспы, инфекции вирусом герпеса, тяжелого острого респираторного синдрома (SARS), инфекции вирусом чикунгунья, инфекции вирусом Западного Нила, инфекции вирусом денге, инфекции вирусом японского энцефалита, инфекции вирусом желтой лихорадки и инфекции вирусом гепатита С.
Предпочтительно аутоиммунное заболевание выбирают из синдрома Шёгрена, системной красной волчанки, псориаза, герпетиформного дерматита, витилиго, грибовидного микоза, аллергического контактного дерматита, атопического дерматита, красного плоского лишая, острого лихеоидного и вариолиформного парапсориаза (PLEVA), артрита, катастрофического антифосфолипидного синдрома.
Согласно другому предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II), или его фармацевтически приемлемой соли, предназначенному для применения при лечении болезни, выбранной из группы, включающей колит, язвенный колит, воспалительное заболевание кишечника, панкреатит, сепсис. Согласно другому предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для применения при лечении панкреатита. Согласно другому предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II) или его фармацевтически приемлемой соли, предназначенному для применения при лечении сепсиса.
Согласно другому предпочтительному варианту осуществления изобретение имеет отношение к PPP1R15A ингибитору формулы (I) или (II), или его фармацевтически приемлемой соли, предназначенному для применения при лечении спондилоартропатии, более предпочтительно анкилозирующего спондилита.
- Метаболические и/или сердечно-сосудистые нарушения, такие как ожирение, гиперлипидемия, семейная гиперхолестеринемия, атеросклероз, гипертензия, сердечнососудистые заболевания, ишемия сердца, инсульт, инфаркт миокарда, транс-аортальный стеноз и диабет, а также родственные заболевания, включают гипергликемию, нарушение толерантности к глюкозе, гиперинсулинемию (предиабет), гиперчувствительность к инсулину I и II типа, диабет, резистентность к инсулину, синдром Уолкотта-Раллисона в числе прочих.
В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения предиабета или диабета, более предпочтительно предиабета 2 типа или диабета 2 типа. В другом предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения болезни, выбранной из группы, включающей гипергликемию, нарушение толерантности к глюкозе, гиперинсулинемию (предиабет), гиперчувствительность к инсулину I и II типа, резистентность к инсулину и синдром Уолкотта-Раллисона. Действительно, секретирующие инсулин β-клетки поджелудочной железы имеют строго и четко регулируемую биосинтетическую «обязанность», заключающуюся в секреции инсулина. Таким образом, важной потребностью этих клеток является сохранение гомеостаза ER (Back и Kaufman, 2012, Annu Rev Biochem, 81, 767-93). Диабет 2 типа проявляется повышенными уровнями глюкозы в крови вследствие резистентности к инсулину в жировой ткани, мышцах и печени и/или уменьшенной секрецией инсулина из β-клеток поджелудочной железы. В ответ масса β-клеток увеличивается и усиливается их функционирование. В результате нагрузка на β-клетки становится чрезмерной, приводя к их прогрессирующей деградации и гибели. Увеличивается количество данных, показывающих, что гибель β-клеток является результатом стресса ER (Back и Kaufman, 2012, Annu Rev Biochem, 81, 767-93). Важно отметить, что делеция Chop улучшает функционирование β-клеток в различных моделях диабета (Song et al., 2008, J Clin Invest, 118, 3378-89). Без связи с какой-либо теорией, предполагается, что ингибиторы PPP1R15A-PP1 будут улучшать функционирование β-клеток при диабете 2 типа, поскольку ингибирование PPP1R15A-PP1 уменьшает уровни проапоптотического белка CHOP в ходе ER-стресса (Tsaytler et al, 2011, Science).
В другом варианте осуществления соединение формулы (I) или (II) предназначается для лечения болезни, выбранной из группы, включающей гипертензию, сердечнососудистые заболевания, ишемию сердца, инсульт, инфаркт миокарда, трансаортальное сужение или сосудистый инсульт. В другом предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения ишемии сердца. В другом предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для лечения атеросклероза.
- Остеопороз:
Yokota et al. (ВМС Musculoskeletal disorders 2013, 14, 197) и He et al (Cellular Signaling 2013, 25 552-560) продемонтрировали, что салубринал (Boyce et al. 2005) эффективно блокирует остеопороз и стимулирует образование кости на мышиной модели. Однако салубринал является токсичным и не используется для лечения людей. В противоположность этому, предсказывается, что PPP1R15A ингибиторы формулы (I) или (II) будут безопасными и могут использоваться для лечения остеопороза. Соединение формулы (I) или (II) предназначается для лечения остеопороза.
- Травма центральной нервной системы
Ohri et al. (Neurobiology of disease, 2013 Vol. 58 pp 29-37) продемонстрировали, что салубринал значительно улучшает способность задних конечностей двигаться, что соответствует лучшему сохранению белого вещества и пониженному апоптозу олигодендроцитов, таким образом улучшая функциональное восстановление после повреждения спинного мозга. Следовательно, предполагается, что PPP1R15A ингибиторы формулы (I) или (II) изобретения будут безопасными и могут использоваться для уменьшения потери олигодендроцитов после травматического повреждения синного мозга и для лечения повреждения спинного мозга. В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для профилактического и/или терапевтического лечения повреждения спинного мозга.
- Ишемия, ишемия головного мозга и апноэ во сне
Настоящее изобретение предоставляет способы применения PPP1R15A ингибиторов формулы (I) или (II) изобретения для предотвращения и/или лечения повреждения ткани, являющегося результатом повреждения или смерти клетки вследствие некроза или апоптоза. Примеры повреждения нервной ткани включают ишемическое и реперфузионное повреждение, такое как мозговой ишемический инсульт и травма головы. В одном предпочтительном варианте осуществления соединение формулы (I) или (II) предназначается для профилактического и/или терапевтического лечения ишемии головного мозга, такой как мозговой ишемический инсульт и травма головы.
- Старение
Старение связано с дегенерацией клеток, тканей и органов, приводящей к таким заболеваниям, как рак, сердечнососудистая недостаточность, ожирение, сахарный диабет 2 типа, неалкогольная жировая дистрофия печени и нейродегенеративные болезни, а также снижению большинства показателей физиологической работоспособности.
В биологии под старением имеется в виду состояние или процесс старения. Клеточное старение представляет собой явление, когда изолированные клетки демонстрируют ограниченную способность к делению в культуре (предел Хейфлика, открытый Леонардом Хейфликом в 1961), тогда как организменное старение означает старение организмов. Организменное старение характеризуется снижением способности отвечать на стресс, увеличением гомеостатического дисбаланса и повышением риска заболевания; в частности, UPR ухудшается с возрастом (Naidoo et al., 2008, J Neurosci, 28, 6539-48). Таким образом, продление положительного эффекта UPR путем ингибирования eIF2α фосфатазы может улучшить нарушения, связанные с возрастом. Следовательно, предсказывается, что PPP1R15A ингибиторы формулы (I) или (II) изобретения будут безопасными и могут ипользоваться для предотвращения и/или лечения болезней или нарушений, связанных с продолжительностью жизни или пролиферативной способностью клеток, и болезней или болезненных состояний, вызванных или усугубленных клеточным старением у животных, конкретнее у людей.
Согласно одному варианту осуществления настоящее изобретение также касается соединения формулы (I) или (II), предназначенного для применения при лечении и/или предотвращении заболевания, выбранного из группы таупатий, выбранных из числа болезни Альцгеймера, прогрессирующего надъядерного паралича, кортико-базальной дегенерации, лобно-височной лобарной дегенерации или лобно-височной деменции (FTD) (болезнь Пика); синуклеопатий, выбранных из болезни Паркинсона, деменции с тельцами Леви, первичной вегетативной недостаточности и множественной системной атрофии; полиглутаминных и полиаланиновых заболеваний, выбранных из болезни Хантингтона, спинобульбарной мышечной атрофии (или болезни Кеннеди), дентато-рубро-паллидо-льюисовой атрофии, спинально-церебеллярной атаксии 1 типа, спинально-церебеллярной атаксии 2 типа, спинально-церебеллярной атаксии 3 типа (или болезни Мачадо-Джозефа), спинально-церебеллярной атаксии 6 типа, спинально-церебеллярной атаксии 7 типа и спинально-церебеллярной атаксии 17 типа, окулофарингеальной мышечной дистрофии; демиелинизирующих заболеваний, таких как лейкодистрофия, болезнь Шарко-Мари-Тута и рассеянный склероз, муковисцидоза, сейпинопатии, лизосомной болезни накопления, амилоидоза, воспаления, метаболических нарушений и сердечно-сосудистых нарушений, выбранных из ожирения, гиперлипидемии, семейной гиперхолестеринемии, атеросклероза, гипертензии, сердечно-сосудистых заболеваний, ишемии сердца, инсульта, инфаркта миокарда, трансаортального сужения, сосудистого инсульта; остеопороза, травмы нервной системы, ишемии, остеопороза, ретинальных болезней, подобных пигментной дистрофии сетчатки, ретинальной цилиопатии, глаукоме, макулярной дегенерации и старению.
Согласно одному варианту осуществления заболевание выбирают, в частности, из числа рассеянного склероза; лейкодистрофии, предпочтительно болезни Пелицеуса-Мерцбахера; демиелинизирующего заболевания, такого как болезнь Шарко-Мари-Тута, предпочтительно СМТ-1А; сердечно-сосудистого заболевания, такого как гипертензия, ишемия сердца, инсульт, инфаркт миокарда, трансаортальное сужение; колита, язвенного колита, воспалительного заболевания кишечника, панкреатита, сепсиса; амилоидоза, такого как болезнь Альцгеймера и анкилозирующий спондилит; предиабета и диабета, такого как диабет 2 типа.
Согласно дополнительному варианту осуществления настоящее изобретение также имеет отношение к соединению формулы (I) или (II) в сочетании с соединением, увеличивающим экспрессию и/или активность белка BIP, предназначенному для применения при лечении ретинальных болезней, выбранных из группы, включающей наследственную дегенерацию сетчатки, такую как ретинальная цилиопатия, пигментная дистрофия сетчатки, макулярная дегенерация, ретинопатия недоношенных, индуцированная светом дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома.
Фармацевтические композиции
Для использования согласно настоящему изобретению соединения или физиологически приемлемые соли, сложные эфиры или другие физиологически функциональные производные, описанные в данном документе, могут быть предоставлены в виде фармацевтической композиции, содержащей указанные соединения или их физиологически приемлемую соль, сложный эфир или другое физиологически функциональное производное в сочетании с одним или более фармацевтически приемлемыми носителями и необязательно другими терапевтическими и/или профилактическими ингердиентами. Носитель(и) должен быть приемлемым в том смысле, что он должен быть совместим с другими ингредиентами композиции и не вредным для реципиента. Фармацевтические композиции могут предназначаться для применения в медицине и ветеринарии для лечения человека и животных. Примеры подходящих эксципиентов для различных форм фармацевтических композиций, описанных в данном документе, можно найти в "Handbook of Pharmaceutical Excipients, 2nd Edition, (1994), Edited by A Wade и PJ Weller.
Приемлемые для терапевтического применения носители или разбавители хорошо известны в фармацевтике и описаны, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R. Gennaro edit. 1985). Примеры подходящих носителей включают лактозу, крахмал, глюкозу, метилцеллюлозу, стеарат магния, маннитол, сорбитол и тому подобное. Примеры подходящих разбавителей включают этанол, глицерин и воду. Выбор фармацевтического носителя, эксципиента или разбавителя может быть сделан с учетом предполагаемого способа введения и общепринятой фармацевтической практики. Фармацевтические композиции могут содержать в качестве или в дополнение к носителю, эксципиенту или разбавителю любое подходящее связывающее вещество(а), смазывающее вещество(а), суспендирующее вещество(а), покрывающее вещество(а), солюбилизирующее вещество(а), буфер(ы), вкусовое вещество(а), поверхностно-активное вещество(а), загуститель(и), консервирующее вещество(а) (включая антиоксиданты) и тому подобное, и вещества, включенные с целью приведения композиции в изотоническое состояние с кровью предполагаемого реципиента.
Примеры подходящих связывающих веществ включают крахмал, желатин, естественные натуральные сахара, такие как глюкоза, безводная лактоза, свободная лактоза, бета-лактоза, сахаристые вещества из кукурузы, природные и искусственные камеди, такие как гуммиарабик, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлоза и полиэтиленгликоль.
Примеры подходящих смазывающих веществ включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное.
Консервирующие вещества, стабилизирующие вещества, красящие вещества и даже вкусовые вещества могут использоваться в фармацевтической композиции.
Примеры консервирующих веществ включают бензоат натрия, сорбиновую кислоту и сложные эфиры р-гидроксибензойной кислоты. Также могут использоваться антиоксиданты и суспендирующие вещества.
Фармацевтические композиции включают композиции, подходящие для перорального, местного (включая кожное, щечное, окулярное и подъязычное), ректального или парентерального (включая подкожное, внутрикожное, внутримышечное и внутривенное), назальное, внутриглазное и легочное введение, например, путем ингаляции. При необходимости композиция может быть удобно предоставлена в отдельных единицах дозирования и может быть приготовлена с помощью какого-либо из способов, хорошо известных в области фармацевтики. Все способы включают стадию соединения активного вещества с жидкими носителями или мелкоизмельченными твердыми носителями или и тем и другим, а затем, если необходимо, стадию придания продукту формы желательной композиции.
Подходящие для перорального приема фармацевтические композиции, в которых носитель является твердым веществом, наиболее предпочтительно предоставляются в виде композиций с однократной дозой, таких как пилюли, капсулы или таблетки, каждая из которых содержит заранее определенное количество активного соединения. Таблетка может быть получена с помощью прессования или формования, необязательно с одним или более дополнительными ингредиентами. Прессованные таблетки могут быть изготовлены путем прессования в подходящей машине активного соединения в сыпучей форме, такой как порошок или гранулы, и необязательно смешивания со связывающим веществом, смазывающим веществом, инертным разбавителем, поверхностно-активным веществом или диспергирующим веществом. Формованные таблетки могут быть изготовлены путем формования активного соединения с инертным жидким разбавителем. Таблетки необязательно могут быть покрытыми и, если являются непокрытыми, необязательно могут иметь риску. Капсулы можно изготовить, наполнив оболочки капсул активным соединением в отдельности или в смеси с одним или более дополнительными ингредиентами, а и затем закупорив их обычным способом. Крахмальные облатки являются аналогами капсул, в которых активное соединение вместе с любым дополнительным ингредиентом(ами) «впаивается» в конверт из рисовой бумаги. Активное соединение также может иметь форму диспергируемых гранул, которые например, можно развести в воде перед употреблением или разбрызгать на пищу. Гранулы могут быть упакованы, например, в пакет-саше. Подходящие для перорального приема композиции, в которых носитель является жидкостью, могут иметь форму раствора или суспензии в водосодержащей или неводной жидкости или форму жидкой эмульсии масло-в-воде.
Композиции для перорального приема включают лекарственные формы с контролируемым высвобождением, например, таблетки, в которых активное соединение заключено в состав подходящей матрицы, обеспечивающей контролируемое высобождение, или покрыто соотетствующей пленкой, обеспечивающей контролируемое высобождение. Такие композиции, в частности, могут быть удобны для профилактического применения.
Подходящие для ректального введения фармацевтические композиции, в которых носитель является твердым веществом, наиболее предпочтительно предоставляются в виде суппозиториев с однократной дозой. Подходящие носители включают масло какао и другие вещества, обычно используемые в данной области техники. Суппозитории можно удобно изготовить путем смешивания активного соединения с размягченным или расплавленным носителем(ями) с последующим охлаждением и формованием в формах.
Фармацевтические композиции, подходящие для парентерального введения, включают стерильные растворы или суспензии активного соединения в водном или маслянистом разбавителе.
Фармацевтические композиции изобретения пригодны для глазного введения, в частности, внутриокулярного, местного глазного или периокулярного (окологлазного) введения, более предпочтительно местного глазного или периокулярного введения.
Инъекционные препараты могут быть приспособлены для болюсной инъекции или длительной инфузии. Такие препараты удобно предоставляются в контейнерах с одной дозой или в многодозных контейнерах, которые закупориваются после заполнения композицией и остаются закупоренными до употребления. Альтернативно, активное соединение может иметь порошкообразную форму, которая перед употреблением перемешивается с подходящим разбавителем, таким как, например, стерильная, апирогенная вода.
Активное соединение также может быть включено в состав длительно действующих депо препаратов, которые могут вводиться с помощью внутримышечной инъекции или имплантации, например, подкожно или внутримышечно. Депо препаратов могут включать, например, подходящие полимерные или гидрофобные материалы, или ионообменные смолы. Такие длительно действующие композиции удобны, в частности, для профилактического применения.
Композиции, подходящие для ингаляционного введения через ротовую полость, содержат частицы, включающие активное соединение и желательно имеющие диаметр в пределах от 0.5 до 7 микрон, которые доставляются в бронхиальное дерево реципиента. Как один из вариантов, такие композиции имеют форму тонко измельченных порошков, которые могут удобно предоставляться или в прокалываемой капсуле, например, из желатина, подходящей для использования в устройстве для ингаляции, или альтернативно как самораспыляющаяся композиция, содержащая активное соединение, подходящий жидкий или газообразный пропеллент (распыляющее вещество) и необязательно другие ингредиенты, такие как поверхностно-активное вещество и/или твердый разбавитель. Подходящие жидкие пропелленты включают пропан и хлорфторуглероды, и подходящие газообразые пропелленты включают двуокись углерода. Также могут использоваться самораспыляющиеся композиции, в которых активное соединение дозируется в виде капель раствора или суспензии.
Такие самораспыляющиеся композиции являются аналогичными композициям, известным специалистам в данной области техники, и могут быть приготовлены с помощью общепринятых методов. Такие композиции удобно предоставляются в контейнере с ручным управлением или с автоматически функционирующим клапаном, имеющим желательные характеристики брызгообразования; преимущественно клапан обеспечивает доставку отмеренного фиксированного объема, например, от 25 до 100 микролитров, после каждого его срабатывания.
В качестве дополнительной возможности активное соединение может иметь форму раствора или суспензии, предназначенной для использования в аэрозольном ингаляторе или небулайзере, при этом для получения мелких капель в виде тумана для ингаляции применяется ускоренный воздушный поток или перемешивание ультразвуком.
Композиции, подходящие для назального применения, включают препараты, в основном сходные с описанными выше, для ингаляционного введения. Желательно, чтобы такие композиции содержали частицы с диаметром в пределах от 10 до 200 микрон, обеспечивая удерживание в носовой полости; это может достигаться в зависимости от требований использованием порошка с подходящим размером частиц или выбором соответствующего клапана. Другие подходящие композиции включают крупные порошки, имеющие частицы с диаметром в пределах от 20 до 500 микрон, для введения путем резкого вдоха через носовой канал из контейнера, поднесенного близко к носу, и капли в нос, содержащие от 0.2 до 5% вес/об активного соединения в водном или масляном растворе или суспензии.
Фармацевтически приемлемые носители хорошо известны специалистам в данной области техники и включают, но не ограничиваются этим, 0.1 М и предпочтительно 0.05 М фосфатный буфер или 0.8% солевой раствор. В дополнение к этому, такие фармацевтически приемлемые носители могут быть водными или неводными растворами, суспензиями и эмульсиями. Примерами неводных растворителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и подходящие для инъекций сложные эфиры, такие как этилолеат. Водные носители включают воду, спиртовые/водные растворы, эмульсии или суспензии, включая солевые растворы и забуференные среды. Парентерально приемлемые разбавители включают раствор хлорида натрия, раствор Рингера с декстрозой, декстрозу, раствор Рингера с лактатом или жирные масла. Также могут присутствовать консервирующие вещества и другие дополнительные компоненты, такие как, например, противомикробные препараты, антиоксиданты, хелатирущие агенты, инертные газы и тому подобное.
Композиции, подходящие для местного применения, могут предоставляться, например, в виде гелей, кремов или мазей. Такие препараты можно наносить на рану или язву, например, непосредственно распределять на поверхности раны или язвы или наносить на подходящую подложку, такую как марлевый бинт, марлевая салфетка, сетка или тому подобное, что может быть помещено в или сверху участка, который необходимо обраотать.
Также могут предоставляться жидкие или порошкообразные композиции, которые могут быть распылены или разбрызганы непосредственно на участок, который должен быть обработан, например, рану или язву. Альтернативно, композиция может быть напылена или набрызгана на носитель, такой как бинт, марля, сетка и тому подобное, после чего носитель может быть наложен на участок, который необходимо лечить.
Согласно дополнительному аспекту изобретения предоставляется процесс получения фармацевтической или ветеринарной композиции, как описано выше, процесс включает соединение активного соединения(й) с носителем, например, путем смешивания.
Как правило, композиции приготавливают путем равномерного и тщательного смешивания активного вещества с жидкими носителями или мелкоизмельченными твердыми носителями, или и тем и другим, а затем, по необходимости придания формы продукту. Изобретение распространяется на способы приготовления фармацевтической композиции, включающие объединение соединения общей формулы (I) с фармацевтически или с точки зреия ветеринарии приемлемым носителем или разбавителем.
Соли
Соединения изобретения могут предоставляться в виде солей, в частности, солей, приемлемых с точки зрения фармацевтики и ветеринарии.
Фармацевтически приемлемые соли соединений изобретения включают подходящие соли присоединения кислоты или основания. Обзор фармацевтически подходящих солей можно найти в Berge et al, J Pharm Sci, 66, 1-19 (1977). Соли образуются, например, при взаимодействии с сильными неорганическими кислотами, такими как минеральные кислоты, например, галогенводородными кислотами, такими как хлористоводородная, бромистоводородная и йодистоводородная, серная кислота и фосфорная кислота с образованием сульфата, бисульфата, гемисульфата, тиоцианата, персульфата, и сульфоновыми кислотами; с сильными органическими карбоновыми кислотами, такими как алканкарбоновые кислоты, в которых от 1 до 4 атомов углерода являются незамещенными или замещенными (например, галогеном), например, уксусной кислотой; с насыщенными или ненасыщенными дикарбоновыми кислотами, например щавелевой, малоновой, янтарной, малеиновой, фумарой, фталевой или тетрафталевой; с оксикарбоновыми кислотами, например, аскорбиновой, гликолевой, молочной, яблочной, винной или лимонной кислотой; с аминокислотами, например, аспарагиновой или глутаминовой кислотой; с бензойной кислотой; или с органическими сульфоновыми кислотами, такими как (С1-С4)-алкил- или арил-сульфоновые кислоты, которые являются незамещенными или замещенными (например, галогеном), такими как метан- или р-толуол-сульфоновая кислота. Неприемлемые фармацевтически или с точки зрения ветеринарии соли также могут иметь ценность как промежуточные соединения.
Предпочтительные соли включают, например, ацетат, трифторацетат, лактат, глюконат, цитрат, тартрат, малеат, малат, пантотенат, адипат, альгинат, аспартат, бензоат, бутират, диглюконат, циклопентанат, глюкогептанат, глицерофосфат, оксалат, гептаноат, гексаноат, фумарат, никотинат, пальмоат, пектинат, 3-фенилпропионат, пикрат, пивалат, пропионат, тартрат, лактобионат, пиволат, камфорат, ундеканоат и сукцинат, соли органических сульфоновых кислот, такие как метансульфонат, этансульфонат, 2-гидроксиэтан-сульфонат, камфорсульфонат, 2-нафталенсульфонат, бензолсульфонат, р-хлорбензолсульфонат и р-толуенсульфонат; и соли неорганических кислот, такие как гидрохлорид, гидробромид, иодгидрат, сульфат, бисульфат, гемисульфат, тиоцианат, персульфат, соли фосфорной и сульфоновой кислот. Согласно предпочтительному варианту осуществления соль представляет собой ацетат.
Энантиомеры/таутомеры
Во всех обсужденных ранее аспектах настоящее изобретение включает, если потребуется, все энантиомеры, диастереоизомеры и таутомеры соединения изобретения. Специалисту в данной области техники известны соединения, обладающие оптическими свойствами (одним или более хиральными атомами углерода) и/или таутомерными свойствами. Соответствующие энантиомеры и/или таутомеры могут быть выделены/получены известными в данной области техники методами. Энантиомеры характеризуются абсолютной конфигурацией их хиральньгх центров и описываются правилами R- и S-последовательностей Кана-Ингольда-Прелога. Такие правила хорошо известны в данной бласти техники (смотри, например, 'Advanced Organic Chemistry', 3rd edition, ed. March, J., John Wiley и Sons, New York, 1985).
Таким образом, соединения формулы (I) или (II) также включают таутомерные формы формулы:
 или
или 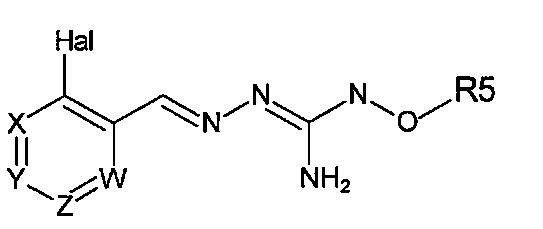
В качестве иллюстративного примера, таутомерная форма соединения 2 представляет собой:
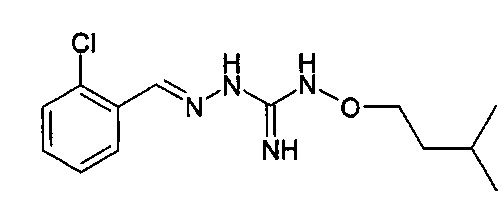 или
или 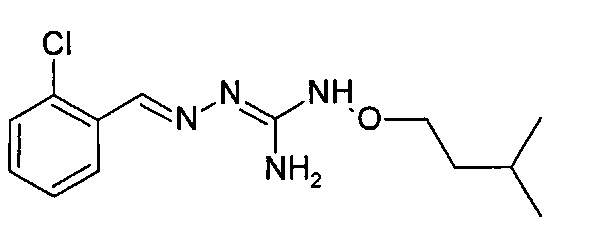
Соединения изобретения, содержащие хиральный центр, могут использоваться в виде рацемической смеси, энантиомерно обогащенной смеси, или рацемическая смесь может быть разделена с помощью хорошо известных методов, а отдельные энантиомеры могут использоваться по отдельности.
Стерео и геометрические изомеры
Некоторые из соединений изобретения могут существовать как стереоизомеры и/или геометрические изомеры - например, они могут обладать одним или более асимметричными и/или геометрическими центрами и потому могут существовать в двух или более стереоизомерных и/или геометрических формах как E/Z (Entgegen/Zusammen) изомеры. Настоящее изобретение рассматривает применение всех отдельных стереоизомеров и геометрических изомеров веществ-ингибиторов и их смесей. Термины, используемые в формуле изобретения, рассматривают эти формы, при условии, что указанные формы сохраняют соответствующую функциональную активность (хотя необязательно в той же степени).
Таким образом, соединения формулы (I) или (II) также включают Е и/или Z изомерные формы формулы:
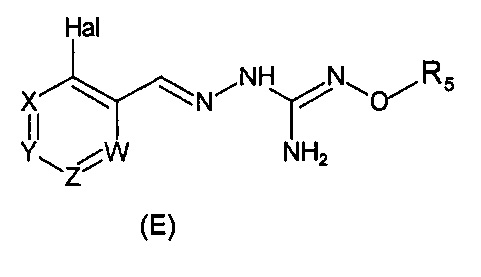 или
или 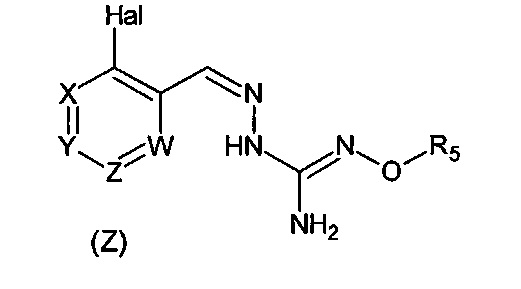
Настоящее изобретение также включают все подходящие изотопные варианты средства или его фармацевтически приемлемых солей. Изотопный вариант средства настоящего изобретения или его фармацевтически приемлемой соли определяется как вариант, в котором, по меньшей мере, один атом заменяется атомом, имеющим то же самое атомное число, но имеющий атомную массу, отличную от атомной массы, которая обычно обнаруживается в природе. Примеры изотопов, которые могут быть введены в состав вещества и его фармацевтически приемлемых солей, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 17O, 18O,31Р, 32Р, 35S, 18F и 36Cl, соответственно. Некоторые изотопные варианты вещества и его фармацевтически приемлемых солей, например, такие, в которые введен радиоактивный изотоп, такой как 3Н или 14С, используются в исследованиях распределения в тканях лекарственного средства и/или субстрата. Особенно предпочтительными являются изотопы, меченные тритием, т.е., 3Н, и углеродом-14, т.е., 14С, вследствие удобства и простоты в получении и обнаружении. Кроме того, замена такими изотопами как дейтерий, т.е., 2Н, может предоставить некоторые терапевтические преимущества, являющиеся результатом  метаболической стабильности, например, увеличения периода полувыведения in vivo или уменьшения необходимой дозировки, и, следовательно, может быть предпочтительной в ряде случаев. Например, изобретение включает соединения общей формулы (I), в которых какой-либо атом водорода заменяется атомом дейтерия. Изотопные варианты средства настоящего изобретения и его фармацевтически приемлемых солей в большинстве случаев могут быть получены с помощью общепринятых методов с использованием соответствующих изотопных вариантов подходящих реагентов.
метаболической стабильности, например, увеличения периода полувыведения in vivo или уменьшения необходимой дозировки, и, следовательно, может быть предпочтительной в ряде случаев. Например, изобретение включает соединения общей формулы (I), в которых какой-либо атом водорода заменяется атомом дейтерия. Изотопные варианты средства настоящего изобретения и его фармацевтически приемлемых солей в большинстве случаев могут быть получены с помощью общепринятых методов с использованием соответствующих изотопных вариантов подходящих реагентов.
Пролекарства
Изобретение дополнительно включает соединения настоящего изобретения в форме пролекарств, т.е. ковалентно связанных соединений, которые in vivo высвобождают активное исходное лекарственное средство согласно общей формуле (I). В большинстве случаев такие пролекарства представляют собой соединения изобретения, в которых одна или более соответствующих групп модифицирована так, что модификация может быть «отменена» после введения человеку или млекопитающему. Возвращение к прежнему состоянию, как правило, осуществляется с помощью фермента, естественным образом присутствующего у такого субъекта, хотя возможно введение вместе с таким пролекарством второго вещества, предназначенного для осуществления «возвращения» к прежнему состоянию in vivo. Примеры таких модификаций включают сложный эфир (например, какой-либо из описанных выше), в котором возвращение к прежнему состоянию может осуществляться эстеразой и т.д. Другие такие системы хорошо известны специалистам в данной области техники.
Сольваты
Настоящее изобретение также включает сольватные формы соединений настоящего изобретения. Термины, использованные в формуле изобретения, включают эти формы.
Полиморфы
Кроме того, изобретение имеет отношение к соединениям настоящего изобретения в их различных кристаллических формах, полиморфных формах и (без)водных формах. В фармацевтической индустрии хорошо установлено, что химические соединения могут быть выделены в любой из таких форм путем небольшого изменения метода очистки и/или выделения из растворителей, использованных при синтетическом получении таких соединений.
Введение
Фармацевтические композиции настоящего изобретения могут быть приспособлены для ректального, назального, внутрибронхиального, местного (включая щечное, подъязычное и офтальмологическое введение, в частности, для интраокулярного, интраветриального, местного глазного или периокулярного введения), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное, внутриартериальное и внутрикожное), внутрибрюшинного или подоболочечного введения. Предпочтительно композиция предназначается для перорального введения. Композиции могут удобно предоставляться в стандартой лекарственной форме, т.е. в форме отдельных частей, содержащих однократную дозу, или множество доз или субдозу однократной дозы. В качестве примера, композиции могут иметь форму таблеток и капсул с замедленным высвобождением, и могут быть изготовлены любым из способов, хорошо известных в области фармацевтики.
Композиции для перорального применения в настоящем изобретении могут предоставляться в виде отдельных единиц, таких как капсулы, желатинозные капсулы, пилюли, драже, облатки, крахмальные капсулы или таблетки, каждая из которых содержит заранее определенное количество активного вещества; порошки или гранулы; раствор, эмульсия или суспензия активного вещества в водосодержащей жидкости или неводной жидкости; или в виде жидкой эмульсии масло-в-воде или жидкой эмульсии вода-в-масле; или в виде болюса и т.д. Предпочтительно, эти композиции содержат от 1 до 250 мг, более предпочтительно от 10 до 100 мг, и даже более предпочтительно от 1 до 100 мг, активного ингредиента на дозу.
В отношении композиций, предназначенных для перорального введения (например, таблеток и капсул), термин "приемлемый носитель" включает разбавители, такие как обычные эксципиенты, например, связывающие вещества, например сироп, гуммиарабик, желатин, сорбитол, трагакантовая камедь, поливинилпирролидон (повидон), метилцеллюлозу, этилцеллюлозу, натрий карбоксиметилцеллюлозу,
гидроксипропилметилцеллюлозу, сахарозу и крахмал; наполнители и носители, например кукурузный крахмал, желатин, лактозу, сахарозу, микрокристаллическую целлюлозу, каолин, маннитол, дикальция фосфат, хлорид натрия и альгиновую кислоту; и смазывающие вещества, такие как стеарат магния, стеарат натрия и другие стеараты металлов, глицерил стеарат, стеариновую кислоту, силиконовую жидкость, тальк, воски, масла и коллоидную двуоксиь кремния. Также могут использоваться вкусоароматические вещества, такие как масло мяты перечной, масло гаультерии, вишневый ароматизатор и тому подобное. Чтобы сделать лекарственную форму легко узнаваемой, при необходимости может быть добавлено красящее вещество. Таблетки также могут иметь покрытие, нанесенное с помощью известных в данной области техники методов.
Таблетка может быть изготовлена путем прессования или формования необязательно в сочетании с одним или более дополнительными ингредиентами. Прессованные таблетки могут изготавливаться путем прессования в подходящей машине активного вещества в сыпучей форме, такой как порошок или гранулы, необязательно смешанного со связывающим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим средством. Формованные таблетки могут изготавливаться путем формования в подходящей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки могут быть необязательно покрыты оболочкой, могут иметь риску и могут быть изготовлены таким образом, чтобы обеспечить медленное или контролируемое высвобождение активного вещества.
Другие композиции, подходящие для перорального введения включают таблетки для рассасывания, содержащие активное вещество в ароматизированной основе, обычно сахарозе, и гуммиарабик или трагакантовую камедь; пастилки, содержащие активное вещество в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик; и жидкости для полоскания рта, содержащие активное вещество в подходящем жидком носителе.
Другие формы для введения включают растворы или эмульсии, которые могут быть введены с помощью инъекции внутривенно, внутриартериально, в полость позвоночного канала, подкожно, внутрикожно, внутрибрюшинно, интраокулярно, местно, периокулярно или внутримышечно, и которые приготавливают из стерильных или стерилизуемых растворов.
Фармацевтические композиции настоящего изобретения также могут быть в форме суппозиториев, пессариев, суспензий, эмульсий, лосьонов, мазей, кремов, гелей, спреев, растворов или порошков для присыпки.
Альтернативным способом чрескожного введения является использование кожного пластыря. Например, активный ингредиент может быть введен в состав крема, состоящего из водной эмульсии полиэтиленгликолей или жидкого парафина. Активный ингредиент также может быть включен в состав мази в концентрации от 1 до 10% по весу, состоящей из основы из белого воска или белого мягкого парафина при необходимости вместе с стабилизирующими и консервирующими веществами.
Дозировки
Средний специалист в данной области техники может легко определить подходящую для введения субъекту дозировку одной из рассматриваемых композиций без излишнего экспериментирования. В большинстве случаев реальную дозировку, наиболее подходящую для отдельного пациента, будет определять врач, причем дозировка будет зависеть от целого ряда факторов, включая активность применяемого конкретного соединения, метаболическую стабильность и продолжительность действия данного соединения, возраст, вес тела, общее состояние здоровья, пол, диету, способ и время введения, скорость выведения, комбинацию лекарственных средств, тяжесть определенного состояния и лечение, которое проводится индивидууму. Раскрытые в описании дозировки являются типичными средними дозировками. Конечно, могут быть отдельные случаи, при которых используются более высокие или более низкие пределы дозировок, такие случаи включаются в рамки этого изобретения.
В соответствии с данным изобретением эффективное количество соединения общей формулы (I) может вводиться с ориентировкой на конкретное состояние или болезнь. Разумеется, этот размер дозировки будет дополнительно изменяться в соответствии со способом введения соединения. Например, для достижения "эффективного количества" в ходе интенсивной терапии предпочтительным является парентеральное введение соединения общей формулы (I). Внутривенное вливание соединения в 5% декстрозе в воде или нормальном физиологическом растворе или сходной композиции с подходящими эксципиентами является наиболее эффективным, хотя также используется внутримышечная болюсная инъекция. В большинстве случаев парентеральная дозировка будет составлять примерно от 0.01 до 100 мг/кг; предпочтительно между 0.1 и 20 мг/кг, для того, чтобы сохранить эффективную концентрацию лекарственного средства в плазме. Соединения могут вводиться от одного до четырех раз в день, до достижения общей суточной дозы примерно от 0.4 до 400 мг/кг/день. Точное количество соединения, являющееся терапевтически эффективным, и лучший способ введения такого соединения легко определит средний специалист в данной области техники путем сравнения уровня средства в крови с концентрацией, необходимой для получения терапевтического эффекта.
Соединения этого изобретения также могут быть введены пациенту перорально и таким образом, чтобы концентрация лекарственного средства являлась достаточной для достижения одного или более из терапевтических показаний, раскрытых в описании. Как правило, фармацевтическая композиция, содержащая соединение, вводится в пероральной дозировке примерно от 0.1 до 50 мг/кг в соответствии с состоянием пациента. Предпочтительно пероральная дозировка будет составлять примерно от 0.1 до 20 мг/кг.
Не ожидается недопустимых токсикологических воздействий при введении соединений настоящего изобретения в соответствии с настоящим изобретением. Соединения этого изобретения, которые могут обладать хорошей биодоступностью, могут быть протестированы в одном из нескольких биологических тестов с целью определения концентрации соединения, необходимой для получения определенного фармакологического эффекта.
Комбинации
В особенно предпочтительном варианте осуществления одно или более соединений изобретения вводятся в комбинации с одним или более другими активными веществами, например, современными лекарственными средствами, имеющимися в продаже. В таких случаях соединения изобретения могут быть введены последовательно или одновременно с одним или более другими активными веществами.
В большинстве случаев лекарственные средства являются более эффективными при использовании в комбинации. В частности, комбинированная терапия желательна для того, чтобы избежать большой токсичности, перекрывания механизма действия и развития механизма(ов) резистентности. Более того, также желательным является введение большинства лекарственных средств в их максимально переносимых дозах с минимальными промежутками времени между такими дозами. Основные преимущества комбинированного применения лекарственных средств заключаются в том, что это может способствовать появлению аддитивных или синергетических эффектов в результате биохимических взаимодействий, а также может уменьшить возможность развития резистентности.
Эффективные комбинации могут быть обнаружены при исследовании ингибиторной активности тестируемых соединений в сочетании с известными препаратами или с препаратами, которые предположительно будут полезными при лечении конкретного заболевания. Эта процедура также может использоваться для определения порядка введения средств, т.е. до, одновременно или после доставки. Такое планирование может быть отличительной чертой всех активных веществ, установленных в данном документе.
Согласно предпочтительному варианту осуществления изобретение имеет отношение к фармацевтической композиции, содержащей PPP1R15A ингибитор формулы (I) или (II), или его фармацевтически приемлемой соли, и соединение, увеличивающее экспрессию и/или активность белка BiP, и фармацевтически приемлемый носитель и/или эксципиент (смотри WO 2013/124484). Предпочтительно, соединение, увеличивающее экспрессию и/или активность белка BiP, выбирают из группы, состоящей из вальпроевой кислоты или ее производного, трихостатина А, лития, 1-(3,4-дигидрокси-фенил)-2-тиоцианат-этанона и эксендина-4. Согласно предпочтительному варианту осуществления белок BiP представляет собой вальпроевую кислоту или ее производное, такое как 2-ен-вальпроевая кислота.
Согласно предпочтительному варианту осуществления изобретение имеет отношение к фармацевтической композиции, содержащей PPP1R15A ингибитор формулы (I) или (II) или его фармацевтически приемлемую соль и соединение, увеличивающее экспрессию и/или активность белка BiP, и фармацевтически приемлемый носитель и/или эксципиент для лечения заболевания, связанного с путем PPP1R15A и связанного с стрессом, вызванным неправильным сворачиванием белка, и, в частности, с накоплением неправильно свернутых белков. Предпочтительно, болезнь выбирают из группы, включающей муковисцидоз, лизосомную болезнь накопления, амилоидоз, рак, воспаления, метаболические нарушения, сердечнососудистые заболевания, остеопороз, травму центральной нервной системы, ишемию, ретинальные болезни, сейпинопатии, таупатии, синуклеопатии, полиглутаминные и полиаланиновые заболевания, нейродегенеративные болезни, предпочтительно болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склерлоз, болезнь Хантингтона, болезни Шарко-Мари-Тута, лейкодистрофии, рассеянный склероз.
Метод анализа
Дополнительный аспект изобретения имеет отношение к использованию соединений, описанных выше, в методах анализа по идентификации дополнительных соединений-кандидатов, способных ингибировать PPP1R15A-PP1. Предпочтительно, метод анализа является конкурентно-связывающим анализом.
Более предпочтительно, конкурентно-связывающий анализ включает контактирование соединения изобретения с PPP1R15A-PP1 и соединением-кандидатом и обнаружение какого-либо изменения во взаимодействии между соединением согласно изобретению и PPP1R15A-PP1.
Предпочтительно, соединение-кандидат образуется посредством обычной SAR модификации соединения изобретения. Использованный в описании термин "обычная SAR модификация" относится к стандартным методам, известным в данной области техники, имеющим целью изменение данного соединения посредством химического получения производных.
Таким образом, в одном аспекте идентифицированное соединение может действовать как модель (например, шаблон) для создания других соединений. Соединения, используемые в таком тесте, могут быть свободными в растворе, прикрепленными к твердой подложке, связанными с клеточной поверхностью или расположенными внутриклеточно. Аннулирование активности или образование связывающих комплексов между соединением и тестируемым соединением может быть измерено.
Метод анализа настоящего изобретения может быть скринингом, с помощью которого тестируется ряд веществ. В одном аспекте метод анализа настоящего изобретения является методом скрининга с высокой пропускной способностью.
Данное изобретение также рассматривает использование конкурентного метода скрининга лекарственных веществ, в котором нейтрализующие антитела, способные к связыванию с соединением, специфически конкурируют с тестируемым соединением за связывание.
Другая методика скрининга предоставляет способ скрининга с высокой поропускной способностью (HTS) веществ, обладающих подходящей аффинностью связывания с данными веществами, основанный на методе, подробно описанном в WO 84/03564.
Предполагается, что методы анализа настоящего изобретения будут пригодны и для небольшого и для крупномасштабного скрининга тестируемых соединений, а также в количественных методах анализа.
Предпочтительно, метод анализа на основе конкурентного связывания включает контактирование соединения изобретения с PPP1R15A-PP1 в присутствии известного субстрата PPP1R15A-PP1 и обнаружение какого-либо изменения во взаимодействии между указанным PPP1R15A-PP1 и указанным известным субстратом.
Дополнительный аспект изобретения предоставляет способ обнаружения связывания лиганда с PPP1R15A-PP1, указанный способ включает стадии:
(i) контактирование лиганда с PPP1R15A-PP1 в присутствии известного субстрата;
(ii) обнаружение какого-либо изменения во взаимодействии между PPP1R15A-РР1 и указанным известным субстратом;
при этом указанный лиганд является соединением изобретения.
Один аспект изобретения имеет отношение к процессу, включающему стадии:
(a) проведение метода анализа, описанного выше;
(b) идентификацию одного или более лигандов, способных связываться с лиганд-связывающим доменом; и
(c) получение некоторого количества указанного одного или более лигандов.
Другой аспект изобретения предоставляет процесс, содержащий стадии:
(a) проведение метода анализа, описанного выше;
(b) идентификацию одного или более лигандов, способных связываться с лиганд-связывающим доменом; и
(c) приготовление фармацевтической композиции, содержащей указанный один или более лигандов.
Другой аспект изобретения предоставляет процесс, содержащий стадии:
(a) проведение метода анализа, описанного выше;
(b) идентификацию одного или более лигандов, способных связываться с лиганд-связывающим доменом;
(c) модификацию указанного одного или более лигандов, способных к связыванию с лиганд-связывающим доменом;
(d) проведение метода анализа, описанного выше;
(e) необязательно приготовление фармацевтической композиции, содержащей указанный один или более лигандов.
Кроме того, изобретение имеет отношение к лиганду, идентифицированному с помощью описанного выше способа. Еще один аспект изобретения имеет отношение к фармацевтической композиции, содержащей лиганд, идентифицированный с помощью описанного выше способа. Другой аспект изобретения имеет отношение к применению лиганда, установленного способом, описанным выше, при получении фармацевтической композиции, предназначенной для использования при лечении заболевания, связанного с накоплением неправильно свернутых и/или несвернутых белков, как описано выше.
Приведенные выше способы могут использоваться для отбора лиганда, пригодного в качестве ингибитора PPP1R15 А-РР1.
Соединения общей формулы (I) используются как в качестве лабораторного инструмента, так и в качестве терапевтических средств. В условиях лаборатории отдельные соединения изобретения используются при установлении, вносит ли известная или вновь открытая мишень решающий или, по меньшей мере, существенный биохимический вклад во время формирования или развития болезненного состояния, процесс обычно называется 'оценкой правильности (валидацией) мишени'.
Настоящее изобретение дополнительно описывается со ссылкой на следующие фигуры, на которых:
Фигура 1 демонстрирует дозозависимую защиту клеток Hela с помощью соединения 12 от стресса ER, вызванного 6-часовой экспозицией с туникамицином.
Фигура 2 демонстрирует дозозависимую защиту поврежденных интерфероном-гамма крысиных олигодендроцитов при использовании соединения 11, соединения 12 и соединения 17.
Фигура 3 демонстрирует дозозависимую защиту поврежденных ротеноном первичных мезэнцефальных крысиных нейронов соединением 5, соединением 12 и соединением 17 изобретения.
Фигура 4 демонстрирует дозозависимую защиту поврежденных амилоидом-бета 1-42 первичных кортикальных нейронов крысы соединением 12 изобретения.
Фигура 5 демонстрирует способность соединения 12 и соединения 17 в концентрациях 5 микроМ и 10 микроМ, соответственно, предотвращать накопление Т181Р мутированного DM20 белка в клетках 293Т человека.
Фигура 6 демонстрирует способность соединения 16 предотвращать клеточную гибель, связанную с накоплением склонного к неправильному сворачиванию инсулина Akita, экспрессированного в клетках Min6.
Фигура 7 демонстрирует способность соединения 12, соединения 16 и соединения 17 в разных концентрациях предотвращать гибель клеток инсулиномы Min6, связанную с накоплением неправильно свернутого белка, вызванного 6-часовой экспозицией с туникамицином.
Фигура 8 демонстрирует способность соединения 11, соединения 12, соединения 16 и соединения 17 в различных концентрациях предотвращать гибель клеток инсулиномы INS1, связанную с накоплением неправильно свернутого белка, вызванного 6-часовой экспозицией с туникамицином.
Фигура 9 демонстрирует способность соединений 6, 10, 11, 12, 15, 16 и 17 (в концентрации 25 микроМ) предотвращать продукцию интерферона I типа фибробластами эмбриона мыши, подвергнутыми липофекции поли I:С.
Фигура 10 демонстрирует способность соединения 10 защищать кардиомиоциты новорожденных крыс от апоптоза, индуцированного гипоксией. График показывает процент апоптотических клеток, измеренный с помощью FACS-анализа. Кардиомиоциты подвергались гипоксии (0.3% О2) в течение 36 часов в отсутствие (0 мкМ) или в присутствии указанных концентраций Соединения 2 (n=3).
Настоящее изобретение дополнительно описывается со ссылкой на следующие неограничивающие примеры.
Примеры
1. Материалы и методы
1.1 Получение соединений согласно настоящему изобретению
Реактивы и имеющиеся на рынке соединения были куплены у компании Acros Organics, Sigma-Aldrich. Соединения согласно настоящему изобретению могут быть получены в соответствии со следующей общей методикой:
Соединения 1 и 2: получение 2-(2-xnop6emun)-N'-(3-метилбутокси)гидразинкарбоксимидамин формиата (соединение 1) и 2-(2-хлорбензил)-N'-(3-метилбутокси)гидразинкарбоксимидамина (соединение 2)
2-(3-метилбутокси)-1Н-изоиндол-1,3 (2Н)-дион (I-1)

Триэтиламин (49.58 г) добавили по каплям к перемешиваемому раствору N-гидроксифталимида (40 г) и 1-бром-3-метил бутана (37.4 г) в DMF (600 мл) при комнатной температуре. Реакционную смесь перемешивали при 70°С в течение 18 часов. Позволили реакционной смеси охладиться до комнатной температуры. Смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток суспендировали в холодной воде (1000 мл). Полученную суспензию хорошо перемешивали в течение некоторого времени, а твердое вещество отфильтровали при уменьшенном давлении. Твердое вещество дополнительно промыли деминерализованной водой (200 мл) и гексаном (100 мл). Полученное в результате твердое вещество высушивали при уменьшенном давлении с получением сырого вещества, которое очистили с помощью колоночной хроматографии на силикагеле. Целевой продукт элюировали приблизительно в 2% метаноле в дихлорметане. Выпаривание чистой продуктовой фракции давало 50.0 г 2-(3-метилбутокси)-1Н-изоиндол-1,3 (2Н)-диона (выход: 87.4%). 1Н-ЯМР (DMSO-d6): δ (ppm) 0.93 (d, 6Н), 1.57 (q, 2Н), 1.82 (m, 1Н), 4.16 (t, 2H), 7.86 (s, 4H); LC-MS: m/z=234.25 (M+H).
1-(амино-окси)-3-метилбутан гидрохлорид (I-2)
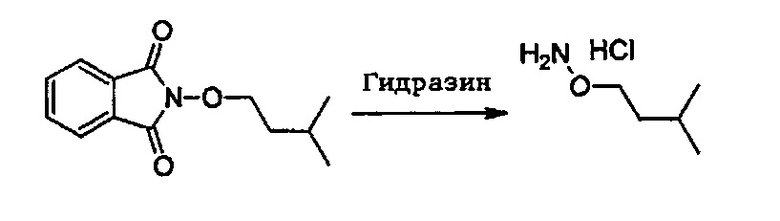
Гидразин гидрат (12.8 г) добавили по каплям к перемешиваемому раствору 2-(3-метилбутокси)-1Н-изоиндол-1,3(2Н)-диона (45 г) в метаноле (600 мл) при комнатной температуре. Реакционную смесь перемешивали при той же самой температуре в течение 24 часов. Реакционную смесь профильтровали, чтобы удалить нерастворимый побочный продукт, полученный фильтрат концентрировали при уменьшенном давлении с получением сырого вещества, которое очистили с помощью колоночной хроматографии на силикагеле. Целевой продукт элюировали приблизительно в 1% метаноле в дихлорметане. Выпаривание чистой продуктовой фракции давало желаемое промежуточное соединение в виде свободного основания, которое было превращено в хлористоводородную соль с использованием 4М HCl в 1,4-диоксане, что давало в результате 3.3 г 1-(аминокси)-3-метилбутан гидрохлорида. 1Н-ЯМР (DMSO-d6): δ (ppm) 0.89 (d, 6Н), 1.46 (q, 2Н), 1.65 (m, 1H), 4.01 (t, 2H), 10.84 (s, 3H).
N'-(3-метилбутокси)гидразинкарбоксимидамин (I-3)
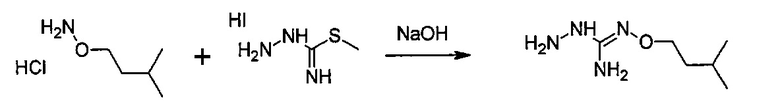
Раствор 2N NaOH (3.6 мл) добавили по каплям к перемешиваемому раствору 1-(амино-окси)-3-метилбутан гидрохлорида (1.2 г) и s-метилизотиосемикарбазид гидроиодида (2.02 г) в воде (3.6 мл) при комнатной температуре и перемешивали в течение 48 часов. Затем, реакционные смеси концентрировали при уменьшенном давлении и остаток остаток азеотропировали метанолом (5 мл). Полученный остаток суспендировали в этаноле (10 мл), а нерастворимые неорганические соли удалили фильтрованием. Фильтрат непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. Получение N'-(3-метилбутокси)гидразинкарбоксимидамина подтвердили с помощью LCMS-анализа. LC-MS: m/z=161.5 (М+Н).
2-(2-хлорбензил)-N'-(3-метилбутокси)гидразинкарбоксимидамин формиат (соединение 1)
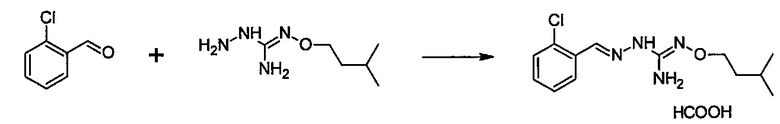
2-хлорбензальдегид (1.81 г) по каплям добавили к фильтрату, который содержит N'-(3-метилбутокси)гидразинкарбоксимидамин при комнатной температуре, и перемешивали в течение 2 часов. Затем, реакционную смесь концентрировали при уменьшенном давлении, и полученный таким образом остаток дополнительно очистили с помощью препаративной ВЭЖХ с использрованием 0.1% НСООН/вода/MeCN с получением 0.27 г 2-(2-хлорбензил)-N'-(3-метилбутокси)гидразинкарбоксимидамин формиата (выход: 13.1%). 1Н-ЯМР (DMSO-d6): δ (ppm) 0.88 (d, 6Н), 1.48 (q, 2Н), 1.68 (m, 1Н), 3.75 (t, 2H), 7.32 (m, 2H), 7.44 (m, 2H), 8,10 (m, 1H), 8.14 (m, 1H), 8.25 (m, 1H), 11.80 (s broad, 2H). LC-MS: m/z=282.88 (M+H).
2-(2-хлорбензил)-N'-(3-метилбутокси)гидразинкарбоксимидамин (соединение 2)
2-(2-хлорбензил)-N'-(3-метилбутокси)гидразинкарбоксимидамин формиат (220 мг) растворили в воде и превратили в основание с помощью насыщенного водного раствора NaHCO3. Основной водный раствор экстрагировали дихлорметаном, органический слой промыли водой, высушили над сульфатом натрия и выпарили при уменьшенном давлении с получением 180 мг 2-(2-хлорбензил)-N'-(3-метилбутокси)гидразинкарбоксимидамина в виде свободного основания (выход: 95%). 1H-ЯМР (DMSO-d6): δ (ppm) 0.89 (d, 6Н), 1.49 (q, 2Н), 1.69 (m, 1H), 3.75 (t, 2Н), 5.73 (s broad, 2H), 7.30 (m, 2H), 7.44 (m, 1H), 8,11 (m, 1H), 8.15 (m, 1H), 10.48 (s broad, 1H). LC-MS: m/z=282.82 (M+H).
Соединение 3: Получение 2-(2-хлорбензилиден)-N'-[2-(метилсулъфонил)этокси]гидразинкарбоксимидамина
2-[2-(метилсульфанил)этокси]-1H-изоиндол-1,3(2H)-дион (I-4)
2-хлорэтил-метилсульфид (10.1 г) по каплям добавили к перемешиваемому раствору N-гидроксифталимида (12.5 г), иодида калия (2.5 г) и карбоната калия (21.1 г) в DMF (150 мл) при комнатной температуре и перемешивали при 80°С в течение 18 часов. Дали реакционной смеси возможность охладиться до комнатной температуры и загрузили в 500 мл холодной воды. Затем, полученное таким образом твердое вещество отфильтровали при уменьшенном давлении. Полученное твердое вещество высушили при уменьшенном давлении с получением 9.7 г 2-[2-(метилсульфанил)этокси]-1H-изоиндол-1,3(2H)-диона (Выход: 52.8%) и использовали для следующей стадии без какой-либо дополнительной обработки. 'Н-ЯМР (DMSO-d6): δ (ppm) 2.16 (s, 3Н), 2.84 (t, 2Н), 4.29 (t, 2Н), 7.87 (s, 4Н). LC-MS: m/z=238.4 (M+H).
2-[2-(метилсульфонил)этокси] -1H-изоиндол-1,3 (2H)-дион (I-5)
m-СРВА (11 г) добавили порциями к перемешиваемому раствору 2-[2-(метилсульфанил)этокси]-1H-изоиндол-1,3(2H)-диона (9.6 г) в дихлорметане (100 мл) при комнатной температуре и перемешивали при комнатной температуре в течение 6 часов. Сырое непереработанное вещество концентрировали при уменьшенном давлении и полученный остаток суспендировали в насыщенном растворе NaHCO3 (100 мл) и перемешивали в течение 30 минут.Полученное твердое вещество профильтровали при уменьшенном давлении, промыли водой (50 мл) и высушили при уменьшенном давлении с получением 9.0 г 2-[2-(метилсульфонил)этокси]-1H-изоиндол-1,3(2H)-диона (выход: 82.6%) и использовали для следующей стадии без какой-либо дополнительной обработки. 1H-ЯМР (DMSO-d6): δ (ppm) 3.15 (s, 3Н), 3.66 (t, 2Н), 4.54 (t, 2Н), 7.88 (s, 4Н). LC-MS: m/z=270.3 (M+H).
1-(аминокси)-2-(метилсульфонил)этан гидрохлорид (I-6)
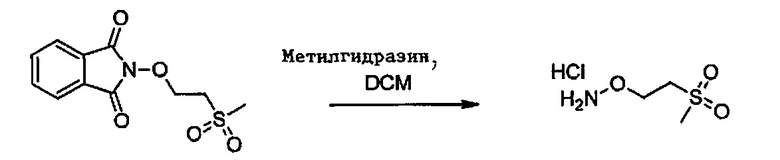
85% метилгидразина (2.0 г) добавили по каплям к перемешанной суспензии 2-[2-(метилсульфонил)этокси]-1H-изоиндол-1,3(2H)-диона (9.0 г) в дихлорметане (100 мл) при комнатной температуре и перемешивали в течение 6 часов. Затем реакционную смесь отфильтровали при уменьшенном давлении, чтобы удалить нерастворимый побочный продукт. Полученный фильтрат концентрировали при уменьшенном давлении при низкой температуре. Осадок суспендировали в 1N HCl (100 мл) и экстрагировали этилацетатом (3 × 250 мл). Полученный в результате водный раствор, содержащий требуемый продукт, концентрировали при уменьшенном давлении с получением белого твердого вещества, которое дополнительно истирали в порошок с диэтиловым эфиром и высушивали при уменьшенном давлении с получением 4.0 г 1-(аминокси)-2-(метилсульфонил)этан гидрохлорид (выход: 68.3%). 1Н-ЯМР (DMSO-d6): δ (ppm) 3.04 (s, 3Н), 3.60 (t, 2Н), 4.38 (t, 2Н), 10.09 (s broad, 2H). LC-MS: m/z=270.3 (M+H).
N'-[2-(метилсульфонил)этокси]гидразинкарбоксимидамин (I-7)
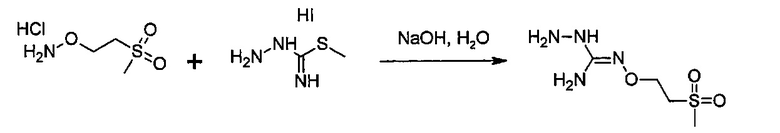
2N раствор NaOH (4.28 мл) добавили по каплям к перемешиваемому раствору 1-(аминокси)-2-(метилсульфонил)этан гидрохлорида (1.5 г) и s-метилизотиосемикарбазид иодгидрата (1.99 г) в воде (4.5 мл) при комнатной температуре и перемешивали в течение 48 часов. Затем реакционную смесь концентрировали при уменьшенном давлении, а остаток азеотропировали метанолом (5 мл). Полученное вещество суспендировали в этаноле (10 мл) и нерастворимые неорганические соли удалили фильтрованием. Полученный фильтрат, содержащий N'-[2-(метилсульфонил)этокси]гидразинкарбоксимидамин, непосредственно использовали для следующей стадии без какой-либо дополнительной обработки.
2-(2-хлорбензилиден)-N'-[2-(метилсульфонил)этокси]гидразинкарбоксимидамин (Соединение 3)
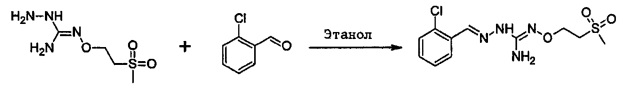
2-хлорбензальдегид (1.32 г) по каплям добавили к фильтрату, содержащему N'-[2-(метилсульфонил)этокси]гидразинкарбоксимидамин, при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при уменьшенном давлении и полученный таким образом остаток дополнительно очистили с помощью препаративной ВЭЖХ с использованием 0.1% NH3/вода/MeCN с получением 20 мг 2-(2-хлорбензилиден)-N'-[2-(метилсульфонил)этокси]гидразинкарбоксимидамина (выход: 0.7% в течение 2 стадий). 1Н-ЯМР (DMSO-d6): δ (ppm) 3.03 (s, 3Н), 3.45 (m, 2Н), 4.12 (m, 2Н), 6.11 (s broad, 2H), 7.40 (m, 2H), 7.44 (m, 1H), 8.15 (m, 1H), 8.26 (s broad, 1H), 10.48 (s, 1H). LC-MS: m/z=318.83 (M+H).
Соединение 4: 2-(2-хлорбензилиден)-N'-[3-(метилсулъфонил)пропокси]гидразинкарбоксимидамин
2-[3-(метилсульфанил)пропокси]-1H-изоиндол-1,3(2H)-дион (I-8)
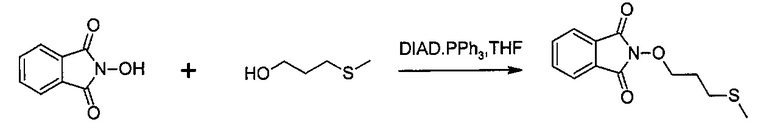
Диизопропил азодикарбоксилат (77.92 мл) по каплям добавили к перемешиваемому раствору N-гидроксифталимида (36.8 г), 3-(метилсульфанил)-1-пропанола (30 г) и трифенилфосфина (37.1 г) в безводном THF (600 мл) в атмосфере азота при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 минут, а затем дали ей нагреться до комнатной температуры и перемешивали в течение 18 часов. Затем реакционную смесь концентрировали при уменьшенном давлении с получением сырого материала, который очистили с помощью колоночной хроматографии на силикагеле. Целевой продукт элюировали в 4% этилацетате в гексане. Выпаривание чистой продуктовой фракции давало 30 г 2-[3-(метилсульфанил)пропокси]-1H-изоиндол-1,3(2H)-диона (выход: 42.2%). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.94 (q, 2Н), 2.07 (s, 3Н), 2.67 (t, 2Н), 4.23 (t, 2Н), 7.87 (s, 4Н). LC-MS: m/z=252.4 (M+H).
2-[3-(метилсульфонил)пропокси]-1H-изоиндол-1,3(2H)-дион (I-9)
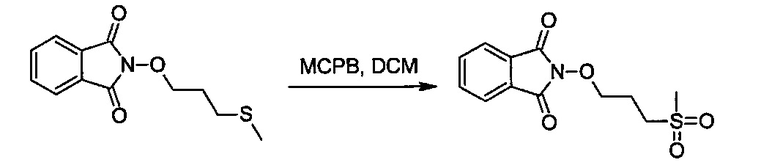
m-CPBA (61.89 г) добавили порциями к перемешиваемому раствору 2-[3-(метилсульфанил)пропокси]-1H-изоиндол-1,3(2H)-диона (30.0 г) в дихлорметане (550 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 5 часов. Затем реакционные смеси концентрировали при уменьшенном давлении с получением сырого материала, который суспендировали в насыщенном растворе NaHCO3 (250 мл) и хорошо перемешивали в течение 30 минут.Полученное твердое вещество отфильтровали при уменьшенном давлении и промыли водой (100 мл). Твердое вещество высушили при уменьшенном давлении с получением 22 г 2-[3-(метилсульфонил)пропокси]-1H-изоиндол-1,3(2H)-диона (выход: 65%). 1Н-ЯМР (CDCl3): δ (ppm) 2.32 (m, 2Н), 3.00 (s, 3Н), 3.50 (t, 2Н), 4.39 (t, 2Н), 7.83 (m, 4Н). LC-MS: m/z=283.9 (M+H).
1-(аминокси)-3-(метилсульфонил)пропан гадрохлорид (I-10)
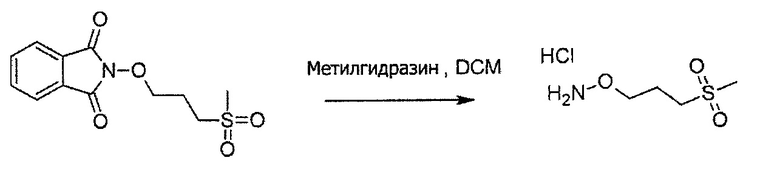
85% метилгидразин (4.2 г) добавили по каплям к перемешанной суспензии 2-[3-(метилсульфонил)пропокси]-1H-изоиндол-1,3(2H)-диона (20 г) в дихлорметане (300 мл) при комнатной температуре и перемешивали в течение 6 часов. Затем раствор профильтровали при уменьшенном давлении, чтобы удалить нерастворимый побочный продукт.Полученный в результате фильтрат концентрировали при уменьшенном давлении при низкой температуре. Остаток суспендировали в 1N HCl (200 мл) и экстрагировали этилацетатом (3 × 500 мл), чтобы удалить нежелательные примеси. Полученный водный раствор концентрировали при уменьшенном давлении с получением белого твердого вещества, которое дополнительно истирвали в порошок с диэтиловым эфиром и высушивали при уменьшенном давлении с получением 8.0 г 1-(аминокси)-3-(метилсульфонил)пропан гидрохлорида (выход: 59.8%). 1Н-ЯМР (DMSO-d6): δ (ppm) 2.04 (m, 2Н), 3.02 (s, 3Н), 3.19 (t, 2Н), 4.12 (t, 2Н), 11.06 (s broad, 3Н).
N'-[3-(метилсульфонил)пропокси]гидразинкарбоксимидамин (I-11)
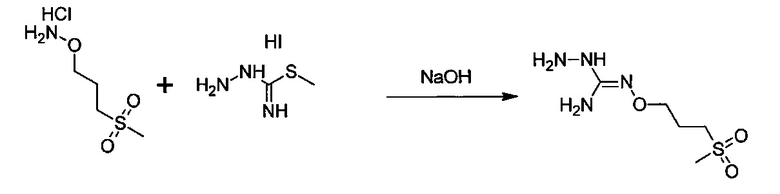
2N раствор NaOH (5.28 мл) добавили по каплям к перемешиваемому раствору 1-(аминокси)-3-(метилсульфонил)пропан гидрохлорид (2.0 г) и s-метилизотиосемикарбазид иодгидрата (2.46 г) в воде (6.0 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Образование N'-[3-(метилсульфонил)пропокси]гидразинкарбоксимидамина подтвердили с помощью LCMS-анализа. Затем смесь концентрировали при уменьшенном давлении и остаток азеотропировали метанолом (15 мл). Полученный материал суспендировали в этаноле (15 мл), а нерастворимые неорганические соли удалили фильтрованием. Фильтрат непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. LC-MS: m/z=210.8 (М+Н).
2-(2-хлорбензилиден)-N'-[3-(метилсульфонил)пропокси]гидразинкарбоксимидамин (соединение 4)
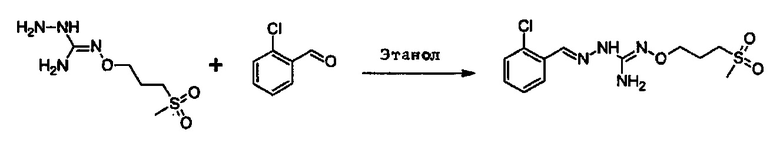
2-хлорбензальдегид (1.62 г) добавили по каплям к фильтрату, содержащему N'-[3-(метилсульфонил)пропокси]гидразинкарбоксимидамин, при комнатной температуре. Полученную реакционную смесь перемешивали при той же самой температуре в течение 2 часов. Сырое вещество концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью препаративной ВЭЖХ с использованием 0.1% NH3/вода/MeCN. После очистки вещество перемешивали в насыщенном растворе NaHCO3, и полученное твердое вещество профильтровали при уменьшенном давлении, промыли водой и высушили с получением 0.14 г чистого 2-(2-хлорбензилиден)-N'-[3-(метил сульфонил)пропокси]гидразинкарбоксимидамина (выход: 4% в течение 2 стадий). 1Н-ЯМР (DMSO-d6): δ (ppm) 2.01 (m, 2Н), 2.98 (s, 3Н), 3.24 (t, 2Н), 3.82 (t, 2Н), 5.90 (s, 2Н), 7.31 (m, 2Н), 7.43 (d, 1Н), 8.13 (m, 2H), 10.48 (s, 1H). LC-MS: m/z=333.5 (M+H).
Соединение 5: 2-(2-хлорбензилиден)-N'-(проп-2-ен-1-илокси) гидразин карбоксимидамин
N'-(проп-2-ен-1-илокси)гидразинкарбоксимидамин (I-12)
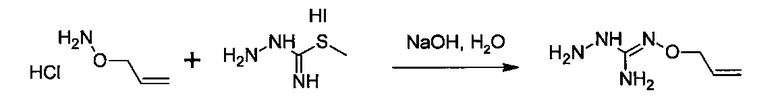
2N раствор NaOH (6.8 мл) добавили по каплям к перемешиваемому раствору О-аллилгидроксиламин гидрохлорида (1.5 г) и s-метилизотиосемикарбазид иодгидрата (3.22 г) в воде (4.2 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Образование промежуточного продукта I-12 N'-(проп-2-ен-1-илокси)гидразинкарбоксимидамина подтвердили с помощью LCMS-анализа. Затем смесь концентрировали при уменьшенном давлении и остаток азеотропировали метанолом (5 мл). Полученный материал суспендировали в этаноле (10 мл) и нерастворимые неорганические соли удалили фильтрованием. Фильтрат непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. LC-MS: m/z=130.6 (М+Н).
2-(2-хлорбензилиден)-N'-(проп-2-ен-1-илокси)гидразинкарбоксимидамин (Соединение 5)

2-хлорбензальдегид (1.9 г) добавили по каплям к фильтрату, содержащему N'-(проп-2-ен-1-илокси)гидразинкарбоксимидамин при комнатной температуре и перемешивали в течение 2 часов. Реакционную смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью препаративной ВЭЖХ с использованием 0.1% НСООН/вода/MeCN с получением 0.25 г 2-(2-хлорбензилиден)-N'-(проп-2-ен-1-илокси)гидразинкарбоксимидамина (выход: 6.1% за 2 стадии. 1Н-ЯМР (DMSO-d6): δ (ppm) 3.17 (s, 1Н), 4.23 (m, 2H), 5.82 (s broad, 2H), 5.98 (m, 1H), 7.37 (m, 2H), 8.15 (m, 3H). LC-MS: m/z=252.8 (M+H).
Соединение 6: 2-(2-хлорбензилиден)-N'-(2-гидроксиэтокси) гидразин карбоксимидамин
2-(2-гидроксиэтокси)-1H-изоиндол-1,3 (2H)-дион (I-13)
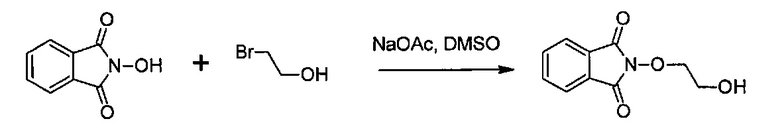
2-бромэтанол (13.26 мл) добавили по каплям к перемешиваемому раствору N-гидроксифталимида (10.0 г) и ацетата натрия (25.14 г) в DMF (50 мл) при комнатной температуре. Полученную реакционную смесь перемешивали при 80°С в течение 1.5 часов. Реакционной смеси дали возможность охладиться до комнатной температуры, загрузили в 500 мл холодной воды и экстрагировали продукт этилацетатом (2 × 400 мл). Полученные органические слои объединили и перегнали в вакууме. Остаток перемешали в холодной воде и полученное в результате твердое вещество отфильтровали под вакуумом. Твердое вещество высушили при уменьшенном давлении с получением 6.0 г 2-(2-гидроксиэтокси)-1H-изоиндол-1,3(2H)-диона (выход: 47.3%), который использовали для следующей стадии без какой-либо дополнительной обработки. 1Н-ЯМР (DMSO-d6): δ (ppm) 3.70 (q, 2Н), 4.18 (t, 2Н), 4.83 (t, 1Н), 7.87 (s, 4H). LC-MS: m/z=208.34 (M+H).
2-(аминокси)этанол гидрохлорид (I-14)

85% метилгидразина (1.25 г) добавили по каплям к перемешиваемой суспензии 2-(2-гидроксиэтокси)-1H-изоиндол-1,3(2H)-диона (6.0 г) в дихлорметане (25 мл) при комнатной температуре и перемешивали в течение 2 часов. Затем реакционную смесь отфильтровали при уменьшенном давлении, чтобы удалить нерастворимый побочный продукт.Фильтрат концентрировали при уменьшенном давлении при низкой температуре. Остаток суспендировали в 2N HCl в этилацетате (20 мл) и концентрировали при уменьшенном давлении при низкой температуре. Полученное твердое вещество истирали в порошок с дихлорметаном (2 × 15 мл) и высушивали при уменьшенном давлении с получением 2.8 г 2-(аминокси)этанол гидрохлорида (выход: 85.5% в виде моногидрохлорид salt). 1Н-ЯМР (DMSO-d6): δ (ppm) 3.61 (m, 2Н), 4.04 (t, 2Н), 4.73 (m, 1Н), 11.02 (s broad, 2H).
N'-(2-гидроксиэтокси)гидразинкарбоксимидамин (I-15)
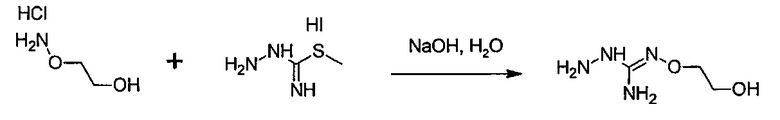
2N раствор NaOH (10.6 мл) добавили по каплям к перемешиваемому раствору 2-(аминокси)этанол хлористоводоодной соли (2.4 г) и s-метил изотиосемикарбазид иодгидрата (4.98 г) в воде (8.4 мл) при комнатной температуре и перемешивали в течение 24 часов. Образование N'-(2-гидроксиэтокси)гидразин карбоксимидамина подтвердили с помощью LCMS-анализа. Смеси концентрировали при уменьшенном давлении и полученный остаток азеотропировали метанолом (15 мл). Полученное вещество суспендировали в этаноле (10 мл), а нерастворимые неорганические соли удалили фильтрованием. Фильтрат, содержащий N'-(2-гидроксиэтокси)гидразинкарбоксимидамин, непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. LC-MS: m/z=134.6 (М+Н)
2-(2-хлорбензилиден)-N'-(2-гидроксиэтокси)гидразинкарбоксимидамин
(Соединение 6)
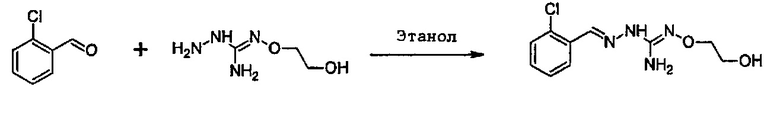
2-хлорбензальдегид (3.28 г) добавили по каплям к фильтрату, содержащему N'-(2-гидроксиэтокси)гидразинкарбоксимидамин, при комнатной температуре и перемешивали в течение 2 часов. Реакционную смесь концентрировали при уменьшенном давлении и полученный таким образом остаток дополнительно очистили с помощью препаративной ВЭЖХ с использованием 0.1% NH3/вода/MeCN с получением 0.24 г 2-(2-хлорбензилиден)-N'-(2-гидроксиэтокси)гидразинкарбоксимидамина (выход: 4.4% за 2 стадии). 1Н-ЯМР (DMSO-d6): δ (ppm) 3.68 (m, 2Н), 3.97 (m, 2Н), 5.82 (s broad, 2H), 5.07 (m, 1H), 7.50 (m, 2H), 7.55 (m, 1H), 8.34 (m, 1H) 8.47 (s, 1H), 8.67 (s, 1H), 11.78 (m, 1H), 12.09 (m, 1H). LC-MS: m/z=256.73 (M+H).
Соединение 7: 2-(2-хлорбензилиден)-N'-(2-хлорэтокси) гидразин карбоксимидамин гидрохлорид
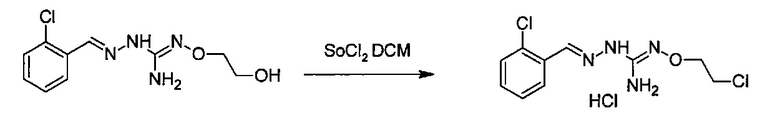
SoCl2 (0.26 мл) добавили по каплям к перемешиваемому раствору 2-(2-хлорбензилиден)-N'-(2-гидроксиэтокси)гидразин карбоксимидамина (0.22 г) в дихлорметане (10 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Затем реакционные смеси концентрировали при уменьшенном давлении. Полученный остаток истирали в порошок с н-пентаном (2 × 5 мл) и высушивали при уменьшенном давлении с получением 0.26 г 2-(2-хлорбензилиден)-N'-(2-хлорэтокси)гидразинкарбоксимидамина гидрохлорида (выход: 99.5%). LC-MS: m/z=274.8 (М+Н).
Соединение 8: 2-(2-хлорбензилиден)-N'-[2-(пирролидин-1-ил) этокси] гидразин карбоксимидамин
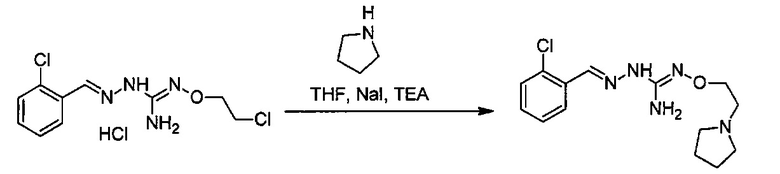
Пирролидин (0.23 г) добавили к перемешиваемому раствору 2-(2-хлорбензилиден)-N'-(2-хлорэтокси)гидразин карбоксимидамин гидрохлорида (0.27 г), триэтиламина (0.35 г) и иодида натрия (0.04 г) в THF (10 мл) при комнатной температуре. Полученную смесь перемешивали при 50°С в течение 24 часов. Затем реакционной смеси дали возможность охладиться до комнатной температуры, и сырое вещество загрузили в 50 мл холодной воды. Продукт экстрагировали этилацетатом (2 × 50 мл). Затем органические слои объединили и перегоняли в вакууме, полученный таким образом остаток дополнительно очистили с помощью препаративной ВЭЖХ с использованием 0.1% NH3/вода/MeCN с получением 14 мг 2-(2-хлорбензилиден)-N'-[2-(пирролидин-1-ил)этокси]-гидразинкарбоксимидамина (выход: 5.3%).). 1H-ЯМР (MeOD): δ (ppm) 1.91 (m, 4Н), 2.75 (m, 4Н), 2.88 (t, 2Н), 3.97 (t, 2Н), 7.32 (m, 2Н), 7.41 (m, 1H), 8.07 (m, 1H), 8.32 (s, 1H). LC-MS: m/z=310.33 (M+H).
Соединение 10: 2-(2-хлорбензилиден)-N'-этоксигидразинкарбоксимидамин
N'-(2-этокси)гидразинкарбоксимидамин (I-16)
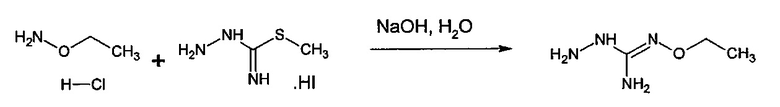
1N раствор NaOH (5.12 мл) добавили по каплям к перемешиваемому раствору этоксиамин хлористоводородной соли (0.5 г) и s-метил изотиосемикарбазид иодгидрата (1.19 г) в воде (5.0 мл) при комнатной температуре и перемешивали в течение 48 часов. Образование N'-(2-этокси)гидразинкарбоксимидамина подтвердили с помощью LCMS-анализа. Смеси концентрировали при уменьшенном давлении, а полученный остаток растворили в этаноле (15 мл). Нерастворимые твердые вещества удалили фильтрованием. Фильтрат концентрировали и N'-(2-этокси)гидразинкарбоксимидамин непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. LC-MS: m/z=118.8 (М+Н).
2-(2-хлорбензилиден)-N'-этоксигидразинкарбоксимидамин (соединение 10)
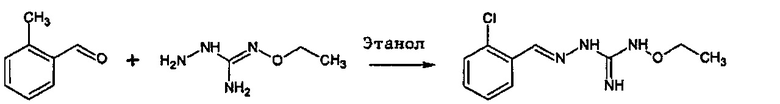
2-хлорбензальдегид (0.717 г) по каплям добавили к N'-(2-этокси)гидразинкарбоксимидамину в растворе этанола (10 мл) и ацетата натрия (0.42 г) при комнатной температуре и перемешивали в течение 2 часов при 90°С. Реакционную смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 21.4 мг 2-(2-хлорбензилиден)-N'-(2-этокси)гидразинкарбоксимидамина (выход: 1.7% за 2 стадии). 1Н- ЯМР (DMSO-d6): δ (ppm) 1.18 (t, 3Н), 3.77 (q, 2H), 5.77 (s broad, 2H), 7.31 (m, 2H), 7.43 (m, 1H), 8.11 (m, 1H), 8.15 (s, 1H), 10.45 (s broad, 1H). LC-MS: m/z=240.9 (M+H).
Соединение 11: 2-(2,6-дихлорбензилиден)-N-этоксигидразинкарбоксимидамин
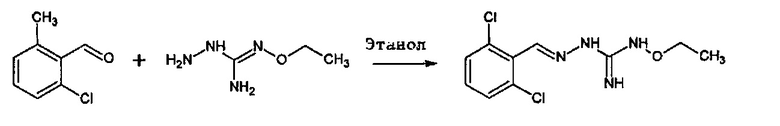
2,6-дихлорбензальдегид (0.896 г) по каплям добавили к 1 эквиваленту N'-(2-этокси)гидразинкарбоксимидамина (I-16) в раствор этанола (10 мл) и ацетата натрия (0.42 г) при комнатной температуре и перемешивали в течение 2 часов при 90°С. Реакционную смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 57 мг 2-(2,6-дихлорбензилиден)-N'-(2-этокси)гидразинкарбоксимидамина (выход: 4.1% за 2 стадии). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.77 (t, 3Н), 3.78 (q, 2Н), 5.48 (s broad, 2H), 7.33 (t, 1H), 7.52 (m, 2H), 8.04 (s, 1H), 8.16 (m, 1H). LC-MS: m/z=277.1 (M+H).
Соединение 12: 2-(2-хлорбензилиден)-N-пропоксигидразинкарбоксимидамин
N'-пропоксигидразинкарбоксимидамин (I-17)
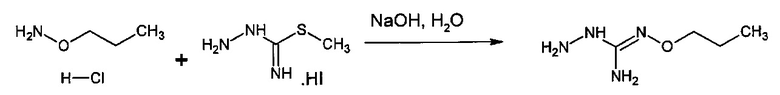
2N раствор NaOH (1.23 мл) по каплям добавили к перемешиваемому раствору О-пропилгидроксиамин хлористоводородной соли (0.28 г) и s-метил изотиосемикарбазид иодгидрата (0.58 г) в воде (2.0 мл) при комнатной температуре и перемешивали в течение 24 часов. Образование N'-(пропокси)гидразинкарбоксимидамин подтвердили с помощью LCMS-анализа. Смеси концентрировали при уменьшенном давлении, а полученный остаток растворили в этаноле (15 мл). Нерастворимые твердые вещества удалили фильтрованием. Фильтрат концентрировали и N'-(пропокси)гидразинкарбоксимидамин непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. LC-MS: m/z=132.9 (М+Н)
2-(2-хлорбензилиден)-N-пропоксигадразинкарбоксимидамин (соединение 12)
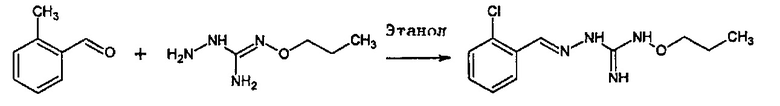
2-хлорбензальдегид (0.35 г) по каплям добавили к N'-(2-пропокси)гидразинкарбоксимидамину в растворе этанола (10 мл) и перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали при уменьшенном давлении и полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 25 мг 2-(2-хлорбензилиден)-N'-(2-пропокси)гидразинкарбоксимидамина (выход: 3.9% за 2 стадии). 1Н-ЯМР (DMSO-d6): δ (ppm) 0.88 (t, 3Н), 1.58 (m, 2Н), 3.66 (t, 2Н), 5.75 (s broad, 1H), 7.29 (m, 2H), 7.41 (m, 1H), 8.10 (m, 2H), 10.45 (s broad, 2H). LC-MS: m/z=255.1 (M+H).
Соединение 13: 2-(2-хлорбензилиден)-N-(2-этоксиэтокси) гидразинкарбоксимидамин
2-(2-этоксиэтокси)-1,3-диметилиден-2,3-дигидро-1H-изоиндол (I-18)
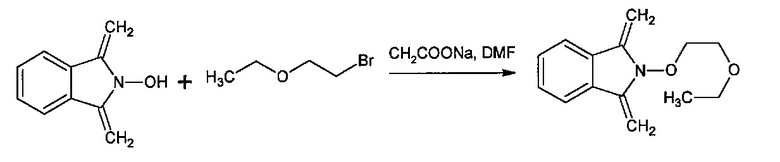
N-гидроксифталимид (4.0 г) и 1-бром-2-этоксиэтан (11.25 г) растворили в DMF (40.0 мл) и добавили CH3COONa (10.0 г) к раствору при комнатной температуре. Реакционную смесь перемешивали при 70°С в течение 12 часов. Реакционной смеси дали возможность охладиться до комнатной температуры и вылили в воду, а затем экстрагировали два раза этилацетатом. Органический слой концентрировали при уменьшенном давлении и очистили с помощью колоночной хроматографии на силикагеле. Целевой продукт элюировали 0-30% этилацетатом в гексане. Выпаривание чистых продуктовых фракций давало 4.8 г 2-(2-этоксиэтокси)-1,3-диметилиден-2,3-дигидро-1H-изоиндол (I-18) (выход: 83.3%). 1Н-ЯМР (DMSO-d6): δ (ppm) 0.98 (t, 3Н),3.39 (q, 2Н), 3.73 (t, 2Н), 4.27 (t, 2Н), 7.87 (s, 4Н). LC-MS: m/z=236.2 (M+H).
1-(аминокси)-2-этоксиэтан гидрохлорид (I-19)
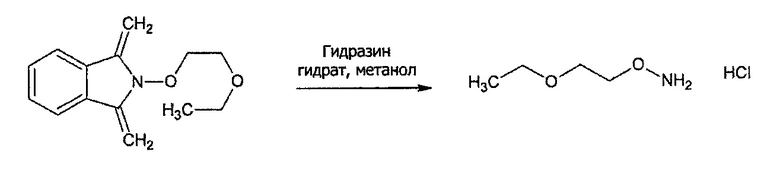
Гидразин гидрат (1.32 г) по каплям добавили к перемешиваемому раствору 2-(2-этоксиэтокси)-1,3-диметилиден-2,3-дигидро-1H-изоиндола (4.8 г) в метаноле (10 мл) при комнатной температуре и перемешивали в течение 30 минут. Затем реакционную смесь отфильтровали при уменьшенном давлении, чтобы удалить нерастворимые побочные продукты. Фильтрат концентрировали при уменьшенном давлении при низкой температуре, истирали в порошок с эфиром, а нерастворимые вещества удалили фильтрованием. Затем, к фильтрату по каплям добавили 4N HCl в диоксане (10.2 мл), а осажденную соль собрали фильтрованием и высушили с получением 2.0 г 1-(аминокси)-2-этоксиэтан гидрохлорида (выход: 69.4 в виде хлористоводородной соли). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.11 (t, 3Н), 3.44 (q, 2Н), 3.59 (m, 2Н), 4.14 (m, 2Н), 11.02 (s broad, 2Н). LC-MS: m/z=106.1 (М+Н).
N'-(2-этоксиэтокси)гидразинкарбоксимидамин (I-20)
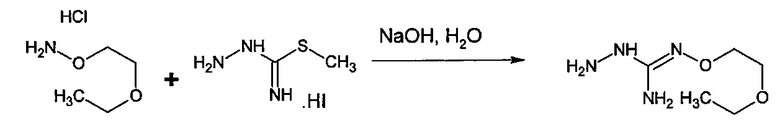
1N раствор NaOH (4.23 мл) по каплям добавили к перемешиваемому раствору 1-(аминокси)-2-этоксиэтан хлористоводородной соли (0.6 г) и s-метил изотиосемикарбазид иодгидрата (0.99 г) в воде (2.1 мл) при комнатной температуре и перемешивали в течение 48 часов. Образование N'-(2-этоксиэтокси)гидразинкарбоксимидамина подтвердили с помощью LCMS-анализа. Смеси концентрировали при уменьшенном давлении, а полученный остаток растворили в этаноле (10 мл). Нерастворимые твердые вещества удалили фильтрованием. Фильтрат концентрировали и N'-(2-этоксиэтокси)гидразинкарбоксимидамин непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. LC-MS: m/z=163.0 (М+Н).
2-(2-хлорбензилиден)-N-(2-этоксиэтокси)гидразинкарбоксимидамин (соединение 13)
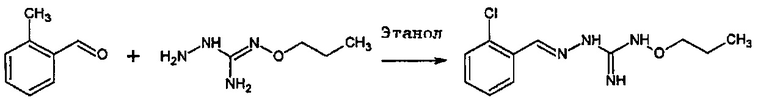
2-хлорбензальдегид (0.59 г) по каплям добавили к N'-(2-этоксиэтокси) гидразинкарбоксимидамину в растворе этанола (5 мл) и перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 19 мг 2-(2-хлорбензилиден)-N'-(2-пропокси)гидразинкарбоксимидамина (выход: 1.8% за 2 стадии). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.24 (t, 3Н), 3.48 (q, 2Н), 3.56 (m, 2Н), 3.83 (m, 2Н), 5.80 (s broad, 1H), 7.43 (m, 1H), 8.12 (m, 1H), 8.17 (s, 1H), 10.50 (s broad, 2H). LC-MS: m/z=285.0 (M+H).
Соединение 14: 2-(2-хлорбензилиден)-N'-[(3-метилбут-2-ен-1-ил)окси] гидразинкарбоксимидамин
2-[(3-метилбут-2-ен-1-ил)окси]- 1H-изоиндол-1,3(2H)-дион (I-21)
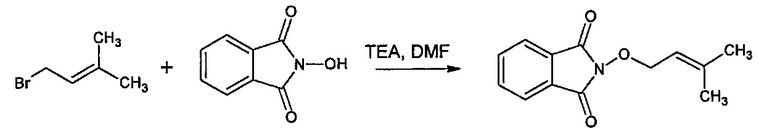
Триэтиламин (12.13 г) по каплям добавили к перемешиваемому раствору N-гидроксифталимида (9.85 г) и 1-бром-3-метил бутена (9.0 г) в DMF (30 мл) при комнатной температуре. Реакционную смесь перемешивали при 70°С в течение 2 часов. Реакционной смеси дали возможность охладиться до комнатной температуры. Смесь концентрировали при уменьшенном давлении и таким образом полученный остаток суспендировали в холодной воде. Полученную суспензию хорошо перемешали в течение некоторого времени, потом твердое вещество отфильтровали при уменьшенном давлении. Твердое вещество дополнительно промыли деминерализованной водой (200 мл) и гексаном (100 мл). Полученное твердое вещество высушивали при уменьшенном давлении с получением сырого вещества, которое было очищено с помощью колоночной хроматографии на силикагеле с получением 9.0 г -[(3-метилбут-2-ен-1-ил)окси]-1H-изоиндол-1,3(2H)-диона (выход: 64.5%). 1H-ЯМР (DMSO-d6): δ (ppm) 1.70 (d, 6Н), 4.63 (m, 2Н), 5.45 (m, 1Н), 7.87 (s, 4H). LC-MS: m/z=232.1 (M+H).
1-(аминокси)-3-метилбут-2-ен гидрохлорид (I-22)

Гидразин гидрат (2.52 г) по каплям добавили к перемешиваемому раствору 2-[(3-метилбут-2-ен-1-ил)окси]-1H-изоиндол-1,3(2H)-диона (9.0 г) в метаноле (120 мл) при комнатной температуре. Реакционную смесь перемешивали при той же самой температуре в течение 30 минут.Реакционную смесь профильтровали, чтобы удалить нерастворимые побочные продукты, а полученный фильтрат концентрировали при уменьшенном давлении с получением сырого вещества, которое очистили с помощью колоночной хроматографии на силикагеле. Сырое вещество истирали с эфиром, а нерастворимую массу удаляли фильтрованием. Фильтрат обработали 4 М HCl в диоксане (19 мл) по каплям, осадок профильтровали, собрали и высушили под вакуумом с получением 2.9 г 1-(аминокси)-3-метилбут-2-ен гидрохлорида (выход: 73.6%). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.70 (s, 3Н), 1.75 (s, 3Н), 1.65 (m, 1H), 4.50 (d, 2H), 5.30 (t, 1H), 10.89 (s, 3H).
N'-[(3-метилбут-2-ен-1-ил)окси] гидразинкарбоксимидамин (I-23)
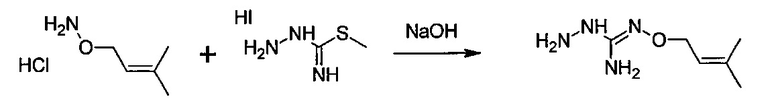
1N раствор NaOH (3.63 мл) по каплям добавили к перемешиваемому раствору 1-(аминокси)-3-метилбут-2-ен гидрохлорида (0.5 г) и s-метилизотиосемикарбазид иодгидрата (0.85 г) в воде (3 мл) при комнатной температуре и перемешивали в течение 48 часов. Затем реакционную смесь концентрировали при уменьшенном давлении. Полученный остаток суспендировали в этаноле (15 мл) и нерастворимые неорганические соли удалили фильтрованием. Фильтрат концентрировали и непосредственно использовали для следующей стадии без какой-либо дополнительной обработки. Получение N'-[(3-метилбут-2-ен-1-ил)окси] гидразинкарбоксимидамина было подтверждено с помощью LCMS-анализа. LC-MS: m/z=159.15 (М+Н).
2-(2-хлорбензилиден)-N'-[(3-метилбут-2-ен-1-ил)окси]гидразинкарбоксимидамин (соединение 14)

2-хлорбензальдегид (0.5 г) по каплям добавили к раствору N'-[(3-метилбут-2-ен-1-ил)окси] гидразинкарбоксимидамина в этаноле (3 мл) при комнатной температуре и перемешивали в течение 2 часов при 90°С. Реакционную смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 139 мг 2-(2-хлорбензилиден)-N'-[(3-метилбут-2-ен-1-ил)окси]гидразинкарбоксимидамина (выход: 13.5% за 2 стадии). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.64 (s, 3Н), 1.71 (s, 3Н), 3.17 (s, 1Н), 4.25 (d, 2H), 5.39 (t, 1H), 5.75 (s broad, 2H), 7.32 (m, 1H), 7.43 (m, 1H), 8.10 (m, 1H), 8.15 (m, 1H), 8.17 (s broad, 1H). LC-MS: m/z=281.2 (M+H).
Соединение 15: 2-(2-хлорбензилиден)-N-[2-(этилсульфанил)этокси] гидразинкарбоксимидамин
2-бромэтил этил сульфид (I-24)
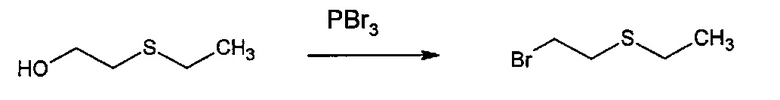
PBr3 (10 мл) по каплям добавили к 2-(этилсульфанил)этанолу в растворе дихлорметана (100 мл) при 0°С и перемешивали в течение 2 часов. Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Реакционную смесь охладили при 0°С и добавили 10 мл воды. Затем реакционную смесь нейтрализовали насыщенным раствором Na2CO3 (до рН 7) и экстрагировали дихлорметаном (3 × 250 мл). Органические слои отделили, объединили, высушили (Na2SO4) и концентрировали с получением 13.0 г 2-бромэтил этил сульфида (выход: 72.7%).1Н-ЯМР (CDCl3): δ (ppm) 1.30 (t, 3Н), 2.62 (q, 2Н), 2.97 (m, 2Н), 3.50 (m, 2Н).
2-[2-(этилсульфанил)этокси]-1H-изоиндол-1,3(2H)-дион (I-25)
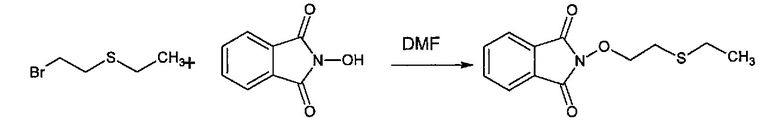
The N-гидроксифталимид (3.9 г) и 2-бромэтил-этил-сульфид (12.1 г) растворили в DMF (40.0 мл) и добавили частями CH3COONa (9.7 г) к раствору при комнатной температуре. Реакционную смесь перемешивали при 70°С в течение 2 часов. Реакционной смеси позволили охладиться до комнатной температуры и вылили в холодную воду, а затем два раза экстрагировали этилацетатом. Органический слой концентрировали при уменьшенном давлении и очистили с помощью колоночной хроматографии на силикагеле с получением 6.0 г 2-[2-(этилсульфанил)этокси]-1H-изоиндол-1,3(2H)-диона (I-25) (выход: 98%). 1Н-ЯМР (CDCl3): δ (ppm) 1.29 (t, 3Н), 2.63 (q, 2Н), 2.94 (t, 2Н), 4.36 (t, 2Н), 7.77 (m, 2Н), 7.86 (m, 2Н).
1-(аминокси)-2-(этилсульфанил)этан гидрохлорид (I-26)
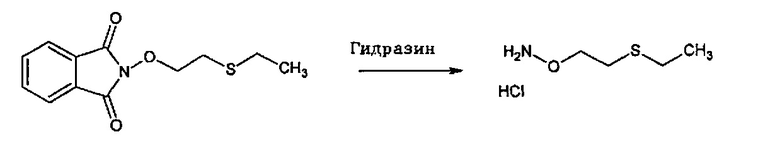
Гидразин гидрат (0.25 г) по каплям добавили к перемешиваемому раствору 2-[2-(этилсульфанил)этокси]-1H-изоиндол-1,3(2H)-диона (1.0 г) в метаноле (10 мл) при комнатной температуре. Реакционную смесь перемешивали при той же самой температуре в течение 30 минут.Реакционную смесь профильтровали, чтобы удалить нерастворимый побочный продукт, полученный фильтрат концентрировали при уменьшенном давлении, затем растворили в DCM и нерастворимое вещество удалили фильтрованием. Фильтрат концентрировали при уменьшенном давлении, затем сырое вещество истирали в порошок с эфиром, а нерастворимую массу удалили фильтрованием. Фильтрат обработали 4 М HCl в диоксане (2 мл) каплями. Затем удалили растворитель выпариванием и остаток истирали в порошок с диэтиловым эфиром с получением 454 мг 1-(аминокси)-2-(этилсульфанил)этан гидрохлорида (выход: 72.5%). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.18 (s, 3Н), 2.53 (m, 2Н), 2.79 (t, 2Н), 4.16 (t, 2Н), 11.14 (s broad, 3Н).
N'-[2-(этилсульфанил)этокси]гидразинкарбоксимидамин (I-27)
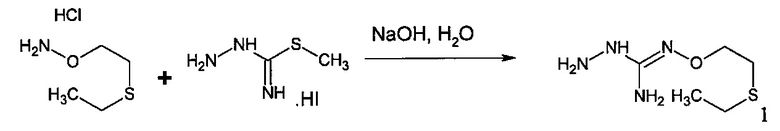
N Раствор NaOH (2.88 мл) по каплям добавили к перемешиваемому раствору 1-(аминокси)-2-(этилсульфанил)этан гидрохлорида (0.5 г) и s-метил изотиосемикарбазид иодгидрата (0.7 г) в воде (5 мл) при комнатной температуре и перемешивали в течение 48 часов. Смеси концентрировали при уменьшенном давлении, а полученный остаток растворили в этаноле (15 мл). Нерастворимые твердые вещества удалили фильтрованием. Фильтрат концентрировали и N'-[2-(этилсульфанил)этокси]гидразинкарбоксимидамин непосредственно использовали в следующей стадии без какой-либо дополнительной обработки.
2-(2-хлорбензилиден)-N-[2-(этилсульфанил)этокси]гидразинкарбоксимидамин (соединение 15)
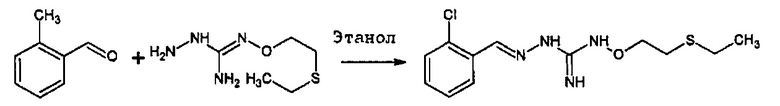
2-хлорбензальдегид (0.4 г) по каплям добавили к раствору N'-[2-(этилсульфанил)этокси]гидразинкарбоксимидамина в этаноле (5 мл) и перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали при уменьшенном давлении, полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 15 мг 2-(2-хлорбензилиден)-N-[2-(этилсульфанил)этокси]гидразинкарбоксимидамина (выход: 1.5% за 2 стадии). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.90 (t, 3Н), 2.54 (q, 2Н), 2.75 (t, 2Н), 3.85 (t, 2Н), 5.84 (s broad, 2H), 7.30 (m, 2H), 7.44 (m, 1H), 8.12 (m, 1H), 8.16 (s, 1H), 10.50 (s broad, 1H). LC-MS: m/z=301.9 (M+H).
Соединение 16: 2-[(3-хлорпиридин-4-ил)метилиден]-N-этоксигидразинкарбоксимидамин
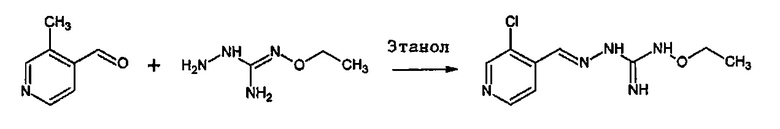
3-хлоризоникотинальдегид (0.72 г) по каплям добавили к 1 эквиваленту раствора N'-(2-этокси)гидразинкарбоксимидамина (I-16) в этаноле (5 мл) и ацетата натрия (0.42 г) при комнатной температуре и перемешивали в течение 2 часов при 80°С. Реакционную смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 184 мг 2-[(3-хлорпиридин-4-ил)метилиден]-N-этоксигидразинкарбоксимидамина (выход: 15% за 2 стадии). 1Н-ЯМР (DMSO-d6): δ (ppm) 1.19 (t, 3Н), 3.79 (q, 2Н), 5.96 (s broad, 2H), 8.05 (s, 1H), 8.11 (d, 1H), 8.41 (s, 1H), 10.89 (s broad, 1H). LC-MS: m/z=242.0 (M+H).
Соединение 17: 2-(2-хлор-6-фторбензилиден)-N-этоксигидразинкарбоксимидамин
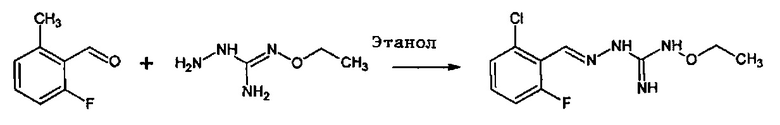
2-хлор-6-фторбензальдегид (0.81 г) по каплям добавили к 1 эквиваленту раствора N'-(2-этокси)гидразинкарбоксимидамина (I-16) в этаноле (5 мл) и ацетата натрия (0.42 г) при комнатной температуре и перемешивали в течение 2 часов при 80°С. Реакционную смесь концентрировали при уменьшенном давлении, а полученный таким образом остаток дополнительно очистили с помощью хроматографии с получением 215 мг 2-(2-хлор-6-фторбензилиден)-N-этоксигидразинкарбоксимидамина (выход: 17.2% за 2 стадии). 1H-ЯМР (DMSO-d6): δ (ppm) 1.17 (m, 3Н), 3.78 (q, 2Н), 5.48 (s broad, 2H), 7.30 (m, 3Н), 8.01 (s, 1H), 10.54 (s broad, 1H). LC-MS: m/z=258.9 (M+H).
Соединение 18: N'-бутокси-2-(2-хлорбензилиден)гидразинкарбоксимидамин
Соединение 18 получают тем же самым методом, как и соединение 12 из 2-хлорбензальдегида и N'-(2-бутокси)гидразинкарбоксимидамина.
Соединение 19: 2-(2-хлор-6-фторбензилиден)-N'-пропоксигидразинкарбоксимидамин
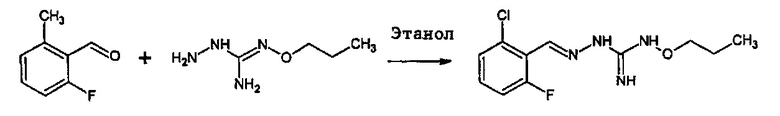
Соединение 19 получают тем же самым методом, как и соединение 17 из 2-хлор-6-фторбензальдегида и N'-(2-пропокси)гидразин карбоксимидамина (I-17) с получением 2-(2-хлор-6-фторбензилиден)-N'-пропоксигидразинкарбоксимидамина LC-MS: m/z=273.0 (М+Н).
Соединение 20: 2-(2-хлор-6-фторбензилиден)-N'-бутоксигидразинкарбоксимидамин
Соединение 20 получают тем же самым методом, как и соединение 17 из 2-хлор-6-фторбензальдегида и N'-(2-бутокси)гидразинкарбоксимидамина.
Соединение 21: 2-(2,6-дихлорбензилиден)-N-пропоксигидразинкарбоксимидамин
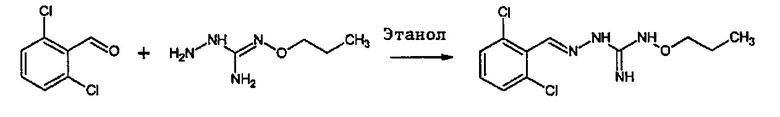
Соединение 21 получают тем же самым методом, как и соединение 11 из 2,6-дихлорбензальдегида и N'-(2-пропокси)гидразинкарбоксимидамина (I-17) с получением 2-(2-хлор-6-фторбензилиден)-N'-пропоксигидразинкарбоксимидамина LC-MS: m/z=290.9 (М+Н).
Соединение 22: 2-(2,6-дихлорбензилиден)-N-бутоксигидразинкарбоксимидамин
Соединение 22 получают тем же самым методом, как и соединение 17 из 2,6-дихлорбензальдегида и N'-(2-бутокси)гидразинкарбоксимидамина.
Соединение 23: 2-[(3-хлорпиридин-4-ил)метилиден]-N-пропоксигидразинкарбоксимидамин
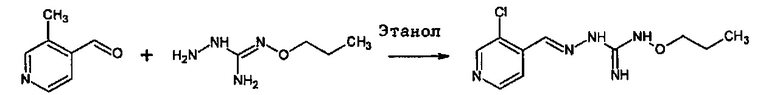
Соединение 23 получают тем же самым методом, как и соединение 16 из 3-хлоризоникотинальдегида и N'-(2-пропокси)гидразинкарбоксимидамина (I-17) с получением 2-(2-хлор-6-фторбензилиден)-N'-пропоксигидразинкарбоксимидамина LC-MS: m/z=255.9 (М+Н).
Выбранные соединения согласно изобретению представлены в Таблице ниже:
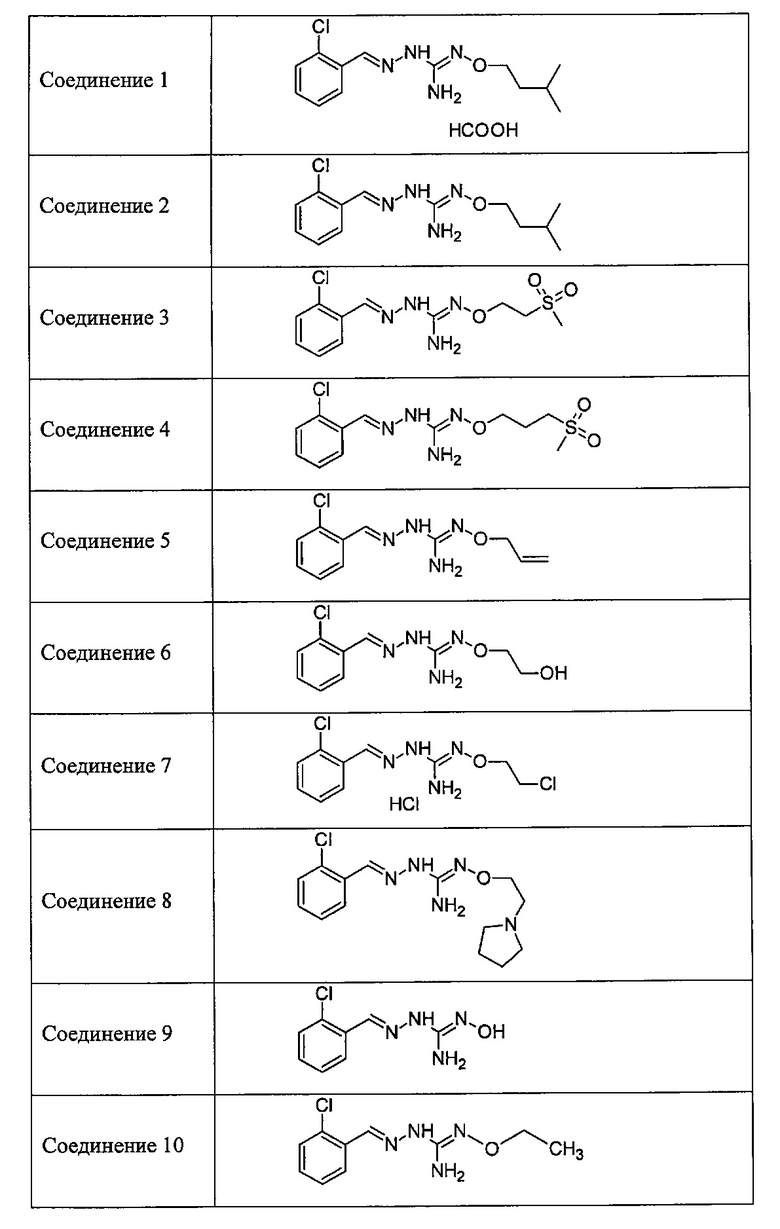
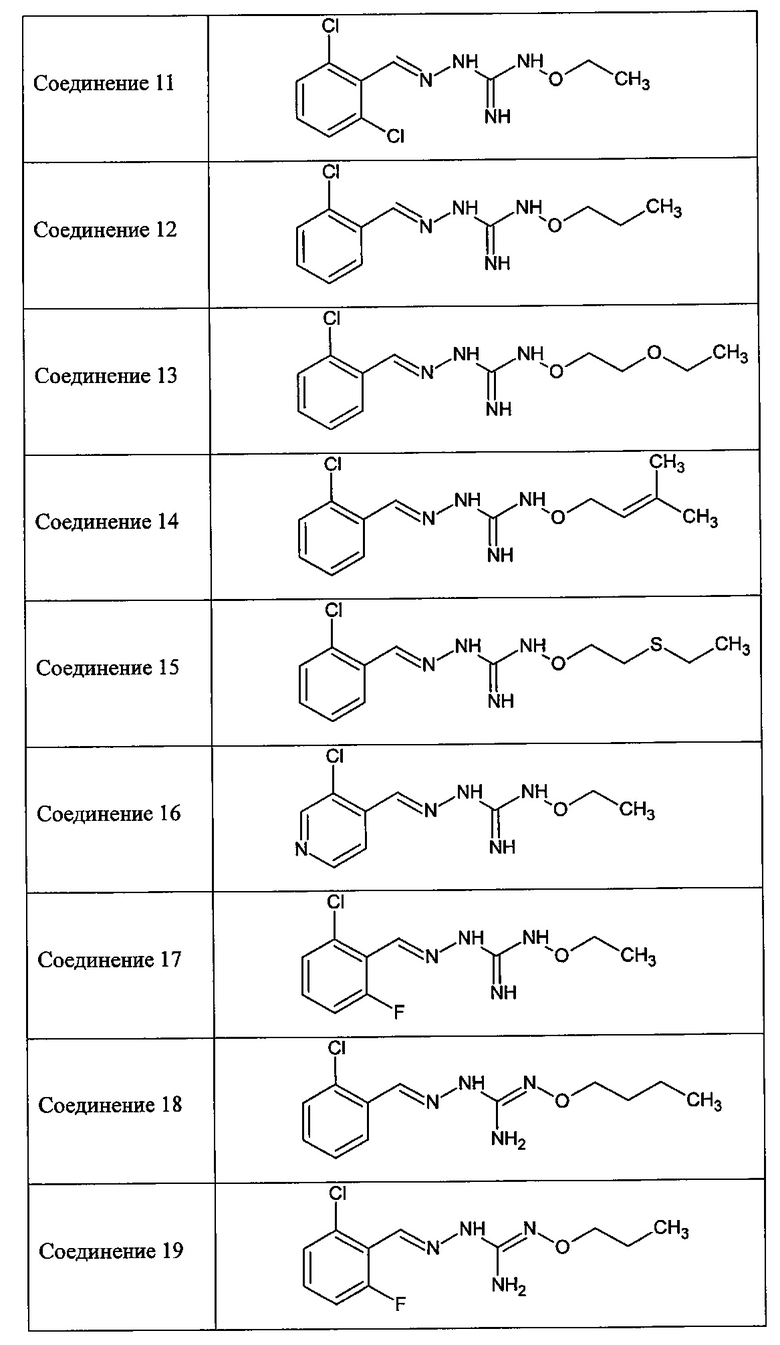
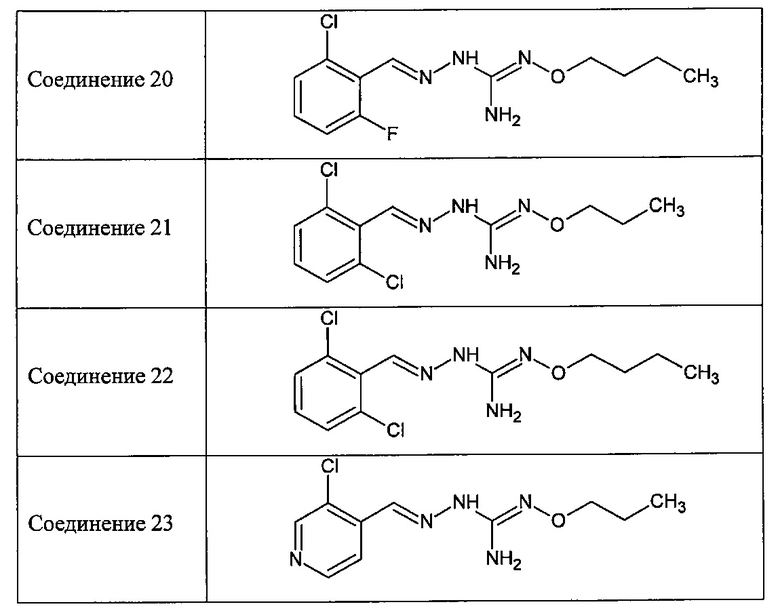
В некоторых из экспериментов, представленных ниже, может использоваться соль этих соединений.
1.2. Культивирование клеток млекопитающих, конструкты и трансфекция.
Клетки HeLa культивировали в минимальной питательной среде Игла (ЕМЕМ) с добавлением глутамина, пирувата натрия, не относящихся к незаменимым аминокислот, пенициллина и стрептомицина (Lonza), содержащей 10% эмбриональной бычьей сыворотки (FBS) (Biowest). Клетки 293Т культивировали в среде Игла, модифицированной по способу Дульбекко (DMEM), с добавлением пенициллина, стрептомицина, глутамина (Lonza) и 10% эмбриональной бычьей сыворотки (FBS) (Biowest).
Клетки Min6 культивировали в среде DMEM с добавлением пенициллина, стрептомицина, глутамина, пирувата натрия, 50 мкМ β-меркаптоэтанола и 15% эмбриональной бычьей сыворотки (FBS) (Biowest).
Клетки INS1 культивировали в среде RPMI с добавлением пенициллина, стрептомицина, глутамина, пирувата натрия (Lonza), 50 мкМ β-меркаптоэтанола и 10% эмбриональной бычьей сыворотки (FBS) (Biowest).
Каждую линию клеток поддерживали при 37°С в атмосфере, содержащей 5% СО2.
Последовательности открытых рамок считывания (ORF) человека для PLP1, DM20 и инсулина были получены от компании Life Technologies (Invitrogen) (IOH41689, IOH5252 и IOH7334, соответственно). Конструкт, помещенный в экспрессионную плазмиду pDEST26 (Invitrogen), был сделан Gateway® LR Clonase™ II Enzyme Mix (Invitrogen). ORF мутации проводили с использованием набора QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) (T181P мутация для PLP1 и DM20 ORFs, Akita (C96Y) для ORF инсулина).
Экспрессию гена в клетах млекопитающих осуществляли с помощью нуклеофекции с использованием системы Amaxa™ 4D-Nucleofector™ (Lonza) или путем трансфекции с использованием липофектамина (Life technologies).
1.3. Цитопротекция от стресса ER
Этот метод анализа описан в работе Tsaytler et al. (Science 2011). Клетки HeLa культивировали в минимальной питательной среде Игла (ЕМЕМ) с добавлением глутамина, пирувата натрия, не относящихся к незаменимым аминокислот, пенициллина и стрептомицина, содержащей 10% эмбриональной бычьей сыворотки (FBS), при 37°С в атмосфере с содержанием 5% СО2. Клетки высевали в 96-луночные планшеты при плотности 17,000 клеток/мл за день до обработки. Стресс ER вызывали добавлением 5 мкг/мл туникамицина (Sigma-Aldrich) вместе с PPP1R15A ингибиторами (0.5-10 мкМ). Через 6 часов заменяли среду свежей средой, а цитопротекцию обеспечивали добавлением PPP1R15A ингибиторов (0.5-10 мкМ). Жизнеспособность клеток оценивали путем измерения восстановления WST-8 в формазан с использованием набора Cell Counting Kit-8 (Sigma) в соответствии с инструкциями производителя через 48 часов или 72 часа после обработки туникамицином. Цитопротекция от стресса ER измеряется с точки зрения цитопротекторной эффективности по сравнению с эталонным соединением гуанабензом (Tsaytler et al., Science 2011) полсе ER стресса:
- ‘-‘ цитопротекторный эффект отсутствует;
- ‘+’ более низкий цитопротекторный эффект по сравнению с гуанабензом;
- ‘++’ сходный цитопротекторный эффект при сравнении с гуанабензом;
- ‘+++’ более высокий цитопротекторный эффект по сравнению с гуанабензом.
Таблица 1 суммирует результаты цитопротекторного эффекта различных соединений изобретения по сравнению с гуанабензом после стресса, индуцированного 6-часовым воздействием туникамицина.
1.4. Оценка уровней трансляции в клетках, не подвергнутых стрессу.
Клетки HeLa (100,000 клеток/мл) высевали в 6-луночные планшеты за 24 часа до каждого эксперимента и оставляли необработанными или обрабатывали соединениями (50 мкМ) в течение 2.5, 5 и 9 часов. Культуральную среду заменяли средой DMEM, не содержащей метионин (Invitrogen) за 30 минут перед добавлением соединений. За один час перед каждой временной точкой добавляли 50 мкМ Click-iT® AHA (L-азидогомоаланин) (Invitrogen) в культуральную среду, для того, чтобы пометить вновь синтезированные белки. В конце каждой временной точки клетки промывали очень холодным PBS и собирали с помощью диссоциации Трипсином (Lonza), затем лизировали в 50 мМ Tris-HCl буфере, содержащем 1% SDS (Sigma) и ингибиторы протеазы и фосфатазы (Sigma). Образцы белков связывали с алкин-биотином (Invitrogen) с использованием набора Click-iT® Protein Reaction Buffer Kit (Invitrogen). Образцы подвергали денатурации при 70°С в течение 10 минут, разлагали на ECL 4-20% преформированных гелях (GE Healthcare) и переносили на нитроцеллюлозные мембраны (GE Healthcare). Алкин-биотин, соединенный с Click-iT® AHA, включенный во вновь синтезированные белки, обнаруживали с помощью стрептавидин-HRP (Gentex). Обнаружение осуществляли путем инкубации с ECL Prime (GE Healthcare), показатель хемолюминесценции регистрировали с помощью прибора Fusion Solo 3S (Vilber Lourmat).
1.5. Оценка уровней трансляции в клетках, подвергнутых стрессу.
Обработку проводили так же, как для измерения трансляции в клетках, неподвергнутых стрессу, за исключением того, что туникамицин (5 мкг/мл) добавляли вместе с соединениями.
1.6. Функциональный GPCR-анализ в отношении адренергического α2А рецептора (CellKey-метод обнаружения)
Агонистическую активность соединений оценивали на клетках СНО, эндогенно экспрессирующих человеческий альфа2А рецептор, путем измерения их эффектов на изменение сопротивления (impedance modulation) с помощью CellKey метода обнаружения. Клетки высевали в 96-луночные планшеты при плотности 6×104 клеток/лунку в HBSS буфере (Invitrogen) + 20 mM HEPES (Invitrogen) с добавлением 0.1% BSA и давали им возможность прийти в равновесие в течение 60 минут при 28°С до начала эксперимента. Планшеты помещали в систему и проводили измерения при температуре 28°С. Растворы были добавлены одновременно во все 96 лунок с помощью интегрированной струйной системы: HBSS (основной контроль), эталонный агонист при 100 нМ (стимулированный контроль), эталонный агонист (определение ЕС50) или тестируемые соединения. Измерения сопротивления проводили в течение 10 минут после добавления лиганда. Стандартным эталонным агонистом является эпинефрин, который тестируется в каждом эксперименте при нескольких концентрациях с целью получения кривой концентрация-эффект, из которой вычисляется значение ЕС50.
Данные о зависимости дозы и эффекта, полученные в результате тестирования
соединений, анализировали с помощью программного обеспечения Hill с использованием нелинейного регрессионного анализа кривых концентрация-эффект, полученных с помощью среднего из повторных значений с использованием уравнения Хилла при обработке кривых. Результаты представлены в Таблице 1, считается, что соединения со значением ЕС50 > 33.3 мкМ не обладают значительной альфа-2 адренергической активностью.
1.7. In vitro модель рассеянного склероза: поврежденные интерфероном-гамма крысиные олигодендроциты, культивируемые совместно с нейронами
Культура олигодендроцитов, культивируемая совместно с нейронами
Нейроны / ОРС культивировали, как описано ранее Yang et al. (2005 J Neurosci Methods; 149(1) pp 50-6), с некоторыми модификациями. Коротко, у крысиных эмбрионов 17-дневного возраста (Wistar, Janvier labs) брали весь мозг (без мозжечка). Мозг обрабатывали в течение 20 минут при 37°С раствором трипсин-EDTA (Pan Biotech) в окончательной концентрации 0.05% трипсина и 0.02% EDTA. Диссоциацию останавливали добавлением среды Игла, модифицированной по способу Дульбекко (DMEM), с содержанием глюкозы 4.5 г/литр (Pan Biotech), содержащей ДНКазу I степени II (конечная концентрация 0.5 мг/мл; Pan Biotech, партия: h140508) и 10% эмбриональной телячьей сыворотки (FCS; Invitrogen, партия: 41Q7218K). Клетки механически диссоциировали, пропустив три раза через наконечник 10-мл пипетки. Затем клетки центрифугировали при 515g в течение 10 минут при 4°С. Супернатант отобрали, а осадок ресуспендировали в указанной культуральной среде, включающей нейробазальную среду (Invitrogen, партия: 1636133) с добавлением 2% раствора В27 (Invitrogen, партия: 1660670), 2 ммоль/литр L-глутамина (Pan Biotech), 2% раствора PS и 1% FCS и 10 нг/мл тромбоцитарного фактора роста (PDGF-AA, партия: H131205). Клетки высевали при плотности 20000 клеток на лунку в 96-луночные планшеты, предварительно покрытые PLL (BD гранулирование, партия: 6614022) и ламинином (Sigma, партия: 083M4034V). Планшеты держали при 37°С в инкубаторе с увлажненной атмосферой с содержанием 5% CO2 в воздухе (95%). Половину среды заменяли новой средой каждые 2 дня. На 18 день тестируемые соединения были предварительно проинкубированы в течение 1 часа до нанесения интерферона-гамма (70 U/мл, 48Н, R&D system, партия: AAL2214081).
Тестируемые соединения и интерферон-гамма interferon-gamma exposure
На 18 день культивирования тестируемые соединения (4 концентрации) растворили в культуральной среде и затем предварительно инкубировали с олигодендроцитами совместно культивированными с нейронами в течение 1 часа до нанесения интерферона-гамма (70 U/мл, 48Н). Через один час после инкубации с тестируемым соединением добавляли интерферон-гамма в концентрации 70 U/мл на 48 часов по-прежнему в присутствии тестируемого соединения. Затем клетки фиксировали холодным раствором этанола (95%, Sigma, партия: SZBD3080V) и уксусной кислоты (5%, Sigma, партия: SZBD1760V) в течение 5 минут при -20°С. После пермеабилизации 0.1% сапонином (Sigma, партия: BCBJ8417V) клетки инкубировали в течение 2 часов моноклональным анти-04 антителом, выработанным в мыши (Sigma, партия: SLBF5997V), при разведении 1/1000 в PBS (PAN, партия: 8410813), содержащем 1% FCS, 0.1% сапонина в течение 2 часов при комнатной температуре. Это антитело обнаруживали с помощью Alexa Fluor 488 козьего анти-мышь IgG (Invitrogen, партия: 1664729) в разведении 1/400 в PBS, содержащем 1% FCS, 0.1% сапонина в течение 1 часа при комнатной температуре.
Анализ общего количества клеток O4
Для каждого условия было сделано 30 снимков на 1 лунку с помощью ImageXpress (молекулярное устройство) при увеличении 20х. Все снимки были сделаны в одинаковых условиях. Анализ общего количества 04 клеток проводился автоматически с использованием программы пользовательского модуля (Molecular Device). Результаты выражали в процентах от контрольных условий (без интоксикации, без интерферона-гамма = 100%) для того, чтобы выразить повреждение интерфероном-гамма. Все значения были выражены как среднее +/- SEM (стандартная ошибка среднего) (n=6 лунок на условие).
1.8. In vitro модель болезни Паркинсона: поврежденные ротеноном первичные мезэнцефальные нейроны крыс
Культура мезэнцефальных допаминергических нейронов
Допаминэргические нейроны крыс культивировали, как описано в Schinelli et al., (1988 J. Neurochem 50 pp 1900-07) и Visanji et al., (2008 FASEB J. 22(7) pp 2488-97). Коротко, средний мозг, полученный из крысиных эмбрионов 15-дневного возраста (Janvier Labs, Франция) разрезали под микроскопом. Извлекали средний мозг эмбрионов и помещали в охлажденную до температуры льда среду Лейбовица (L15, Pan Biotech, партия: 9310614), содержащую 2% пенициллина-стрептомицина (PS, Pan Biotech, партия: 1451013) и 1% эмбрионального сывороточного альбумина (BSA, Pan Biotech, партия: h140603). Вентральный участок среднемозгового изгиба, участок развивающегося мозга, богатый допаминергическими нейронами, использовали для приготовления клеточных препаратов.
Средний мозг диссоциировали трипсинизацией в течение 20 минут при 37°С (Трипсин 0.05% EDTA 0.02%, PanBiotech, партия: 5890314). Реакцию останавливали добавлением среды Игла, модифицированной по способу Дульбекко (DMEM, PanBiotech, партия: 1300714), содержащей ДНКазу I степени II (0.1 мг/мл, PanBiotech, партия: Н140508) и 10% эмбриональной бычьей сыворотки (FCS, Gibco, партия: 41Q7218K). Затем клетки механически диссоциировали, три раза пропустив через наконечник 10 мл пипетки. Затем клетки центрифугировали при 180 × g в течение 10 минут при +4°С на слое BSA (3.5%) в среде L15. Супернатант удалили, а клеточные осадки ресуспендировали в определенной культуральной среде, состоящей из нейробазальной среды (Invitrogen, партия: 1636133) с добавлением В27 (2%, Invitrogen, партия: 1660670), L-глутамина (2 мМ, PanBiotech, партия: 8150713) и 2% раствора PS и 10 нг/мл нейротрофического фактора головного мозга (BDNF, PanBiotech, партия: Н140108) и 1 нг/мл глиального нейротрофического фактора (GDNF, Pan Biotech, партия: Н130917). Жизнеспособные клетки подсчитывали в цитометре Neubauer, используя тест вытеснения трипанового синего. Клетки высевали при плотности 40000 клеток/лунку в 96-луночные планшеты, предварительно покрытые поли-L-лизином (Corning Biocoat, партия: 6614022) и культивировали в инкубаторе при 37°С во влажной атмосфере с содержанием 5% СО2 и 95% воздуха. Половину среды заменяли свежей каждые 2 дня. На 6 день культивирования среду удаляли и добавляли свежую среду с или без ротенона (Sigma, партия: 021M2227V) при разведении 10 нМ в контрольной среде, оценивали 3 лунки на условие. Тестируемые соединения растворяли в культуральной среде и затем предварительно инкубировали с мезэнцефальными нейронами в течение 1 часа до нанесения ротенона.
Через 24 часа интоксикации клетки фиксировали раствором 4% параформальдегида (Sigma, партия SLBF7274V) в PBS (Pan Biotech, партия: 4831114), рН=7.3 в течение 20 минут при комнатной температуре. Клетки вновь дважды промыли PBS, затем пермеабилизировали и неспецифические сайты блокировали раствором PBS, содержащим 0.1% сапонина (Sigma, партия: BCBJ8417V) и 1% FCS в течение 15 минут при комнатной температуре. Затем, клетки инкубировали с моноклональными анти-тирозингидроксилазными антителами, полученными в мыши (ТН, Sigma, партия: 101М4796) при разведении 1/10000 в PBS, содержащем 1% FCS, 0.1% сапонина, в течение 2 часов при комнатной температуре. Это антитело обнаруживали с помощью Alexa Fluor 488 козьего анти-мышь IgG (Molecular Probes, партия: 1531668) при разведении 1/800 в PBS, содержащем 1% FCS, 0.1% сапонина, в течение 1 часа при комнатной температуре.
Анализ общего числа ТН- положительных нейронов
Иммуномеченные культуры автоматически исследовали с помощью ImageXpress (Молекулярное устройство, США). Для каждого условия автоматически анализировали 20 полей на лунку (представляющих ~80% общей поверхности лунки) из 3 лунок. Общее количество ТН нейронов автоматически анализировали с помощью пользовательского модуля (Молекулярное устройство, США). Результаты выражались в процентах от контрольных условий (без интоксикации, без ротенона = 100%) для того, чтобы выразить повреждение ротеноном. Все значения были выражены как среднее +/- SEM 1 культуры (n=3 лунки на условие на культуру).
1.9. In Vitro модель болезни Альцгеймера: поврежденные амилоидом-бета 1-42 первичные кортикальные нейроны крысы.
Культура кортикальных нейронов крысы
Кортикальные нейроны крыс культивировали, как описано в Singer et al., (1999 J. Neuroscience 19 pp 2455-63) и Callizot et al., (2013 J. Neurosci. Res. 91 pp 706-16). Беременных самок (Wistar; Janvier Labs) на 15 день беременности забивали смещением шейных позвонков. Зародышей собирали и немедленно помещали в охлажденную до температуры льда среду L15 Лейбовица (Pan Biotech, партия: 9310614), содержащую 2% раствор пенициллина (10,000 U/мл) и стрептомицина (10 мг/мл) (PS; Pan Biotech, партия: 1451013) и 1% бычьего сывороточного альбумина (BSA; Pan Biotech, партия: h140603). Кору головного мозга обрабатывали в течение 20 минут при 37°С раствором трипсин-EDTA (Pan Biotech, партия: 5890314) в окончательной концентрации 0.05% трипсина и 0.02% EDTA. Диссоциацию останавливали добавлением среды Игла, модифицированной по способу Дульбекко (DMEM), с 4.5 г/литр глюкозы (Pan Biotech, партия: 1300714), соеджащей ДНКазу I степени II (окончательная концентрация 0.5 мг/мл; Pan Biotech, партия: h140508) и 10% эмбриональной бычьей сыворотки (FCS; Invitrogen, партия: 41Q7218K). Клетки механически диссоциировали с помощью 10-мл пипетки, пропустив три раза через наконечник. Затем клетки центрифугировали при 515g в течение 10 минут при 4°С. Супернатант удаляли, а осадок ресуспендировали в определенной культуральной среде, состоящей из необазальной среды (Invitrogen, партия: 1636133) с добавленим 2% раствора В27 (Invitrogen, партия: 1660670), 2 ммоль/литр L-глутамина (Pan Biotech, партия: 8150713), 2% раствора PS, и 10 нг/мл нейротрофического фактора головного мозга (BDNF; Pan Biotech, партия: Н140108). Живые клетки подсчитывали в цитометре Neubauer, используя тест вытеснения трипанового синего. Клетки высевали при плотности 30000 на лунку в 96-луночные планшеты, предварительно покрытые поли-L-лизином (Corning Biocoat, партия: 6614022) и культивировали при 37°С в инкубаторе в атмосфере с содержание 95% воздуха и 5% CO2. Среду заменяли каждые два дня. Кортикальные нейроны подвергали интоксикации растворами А-бета (смотри ниже) через 11 дней культивирования.
Воздействие тестируемыми соединениями и амилоидом-бета 1-42
Приготовление амилоида-бета 1-42 было проведено в соответствии с методом, описанным Callizot et al., 2013. Коротко, пептид амилоида-бета 1-42 (Bachem, партия: 1014012) растворили в определенной культуральной среде, упомянутой выше, не содержащей серы, в исходной концентрации 40 мкмоль/литр. Этот раствор перемешивали в течение 3 дней при 37°С в темноте и немедленно использовали после тщательного разбавления в культуральной среде до используемых концентраций. Тестируемые соединения растворили в культуральной среде, а затем предварительно инкубировали с первичными кортикальными нейронами в течение 1 часа до нанесения амилоида-бета 1-42. Препарат амилоида-бета 1-42 добавили до конечной концентрации 20 мкМ (включая ~2 мкМ токсичных олигомеров, измеренных WB) при разведении в контрольной среде в присутствии лекарственных средств. Через 24 часа интоксикации клетки фиксировали холодным раствором этанола (95%, Sigma, партия: SZBD3080V) и уксусной кислоты (5%, Sigma, партия: SZBD1760V) в течение 5 минут при -20°С. После пермеабилизации 0.1% сапонином (Sigma, партия: BCBJ8417V) клетки инкубировали в течение 2 часов с мышиными моноклональными антителами к белку 2, ассоциированному с микротрубочками (МАР-2; Sigma, партия: 063М4802) при разведении 1/400 в PBS (Pan biotech, партия: 4831114), содержащими 1% эмбриональной бычьей сыворотки (Invitrogen, партия: 41Q7218K) и 0.1% сапонина. Это антитело обнаруживали с помощью Alexa Fluor 488 козьим анти-мышь IgG (Molecular probe, партия: 1572559) при разведении 1/400 в PBS, содержащем 1% эмбриональной бычьей сыворотки и 0.1% сапонина в течение 1 часа при комнатной температуре.
Анализ общего числа нейронов
Иммуномеченные культуры автоматически исследовали с помощью ImageXpress (Молекулярное устройство, США). Для каждого условия автоматически анализировали 30 полей на лунку (представляющих ~80% общей поверхности лунки) из 3 лунок. Общее количество нейронов автоматически анализировали с помощью пользовательского модуля (Молекулярное устройство, США). Результаты выражали в процентах от контрольных условий (без интоксикации, без амилоида-бета 1-42=100%) для того, чтобы выразить повреждение А-бета 1-42. Все значения были выражены как среднее +/- SEM (n=3 лунки на условие на культуру).
1.10. In vitro модель лейкодистрофии (PMD): сверхэкспрессия мутированных PLP1 и DM20 в линии клеток человека
За один день до трансфекции клетки 293Т высевали при плотности 300,000 клеток/мл. Клетки 293Т трансфицировали PLP1 и DM20 мутантными конструктами с использованием липофектамина 2000 согласно методу производителя. После трансфекции клетки обрабатывали молекулами или оставляли необработанными. В качестве контроля клетки трансфицировали нативными формами белков. Через 48 часов собирали клеточные лизаты. Накопление белка оценивали вестерн-блоттингом.
1.11. In vitro модель диабета 2 типа: линии клеток Min6 и INS1
Цитопротекция от ER-cmpecca
Клетки высевали в 96-луночные планшеты при плотности 0.5.106 клеток/мл для линии клетк Min6 и 0,4.106 клеток/мл для линии клеток INS1 за один день до обработки. ER стресс вызывали добавлением 2.5 мкг/мл туникамицин (Sigma Aldrich) вместе с ингибиторами фосфатазы. Среду заменили через 6 часов свежей средой, и цитопротекцию поддерживали добавлением ингибиторов фосфатазы. Жизнеспособность клеток оценивали путем измерения восстановления WST-8 в формазан с помощью набора Cell Counting Kit-8 (Sigma) в соответствии с рекомендациями поставщика, через 72 часа после обработки туникамицином.
Защита от накопления склонного к неправильному сворачиванию инсулинаАkita
Клетки Min6 подвергали нуклеофекции конструктами мутантного инсулинаАkita и высевали в 96-луночные планшеты при концентрации 300,000 клеток/мл, через 24 часа клетки или обрабатывали молекулами или оставляли необработанными. В качестве контроля клетки подвергали нуклеофекции нерелевантной плазмидой. Через 6 дней добавляли селективный агент (G418). Жизнеспособность клеток оценивали через 9 дней после обработки путем измерения восстановления WST-8 в формазан с использованием набора Cell Counting Kit-8 (Sigma) в соответствии с инструкциями поставщика.
1.12. In vitro модель воспаления/инфекционной болезни: индуцированные поли I:С фибробласты эмбриона мыши
Протоколы экспериментов
Фибробласты эмбриона мыши (MEFs) подвергали липофекции поли I:С и обрабатывали двумя концентрациями соединения изобретения (25 мкМ) в течение 6 часов. Через 6 часов культивирования еIF2альфа-фосфорилирование (eIF2a-P) и PPP1R15A (GADD34) экспрессию определяли с помощью вестерн-блоттинга, при этом выработку интерферона(IFN)-бета I типа определяли количественно в культуральных супернатантах методом ELISA. Контроль (nt) и поли I:C/DMSO соответственно являются отрицательным и положительным контролями. Поли I:С (полиинозиновая:полицитидиловая кислота или натриевая соль полиинозиновой:полицитидиловой кислоты) является иммуностимулирующим средством, используемым для моделирования вирусных инфекций. Известно, что поли I:С, которая структурно сходна с двухцепочечной РНК, взаимодействует с toll-подобным рецептором 3, который экспрессируется во внутриклеточных компартментах В-клеток и дендритных клеток. Гуанабенз (25 мкМ) использовали как эталонное ингибиторующее соединение.
Культура клеток
Клетки MEF культивировали в среде DMEM, содержащей 10% FCS (HyClone, Perbio), 100 единиц/мл пенициллина, 100 мкг/мл стрептомицина, 2 мМ глутамина, 1 × MEM не относящихся к незаменимым аминокислот и 50 мкМ 2-меркаптоэтанола. MEFS обрабатывали в течение указанного времени 10 мкг/мл поли I:C (InvivoGen) в комбинации с липофектамином 2000 (Invitrogen).
Иммуноблоттинг
Клетки лизировали в 1% Тритоне Х-100, 50 мМ Hepes, 10 мМ NaCl, 2.5 мМ MgCl2, 2 мМ EDTA, 10% глицерина с добавлением таблеток полной смеси ингибиторов протеаз (Roche). Количественное определение белка проводили методом ВСА-анализа содержания белков (Pierce). 25-50 мкг Тритон Х-100-растворимого материала загрузили на 2%-12% градиентный или 8% SDS-PAGE до иммуноблоттинга и обнаружения хемилюминесценции (SuperSignal West Pico Chemi-luminescent Substrate, Pierce). Использовали кроличьи поликлональные антитела, распознающие GADD34 (С-19), от компании Santa Cruz Biotechnology и анти-еIF2альфа[рS52] от компании Invitrogen.
Анализ методом Elisa
Количественное определение IFN-бета в культуральном супернатанте проводили с помощью набора Mouse Interferon Beta ELISA (PBL Interferon Source) согласно инструкциям производителя.
1.13. Апоптоз, индуцированный гипоксией, в культивируемых кардиомиоцитах новорожденных крыс
Культура клеток
Первичные культуры кардиомиоцитов новорожденных крыс получали из желудочков крыс Sprague Dawley 1-дневного возраста (Janvier, Франция). Крыс подвергали эвтаназии и извлекали сердца. Сердца разрезали на небольшие кусочки (1-2 мм3) и ферментативно обработали с использованием набора для диссоциации сердца новорожденных крыс и диссоциатора gentleMACS™ Dissociator (MiltenyiBiotec, Германия). После диссоциации гомогенаты профильтровали (70 мкм), чтобы получить одноклеточную суспензию. Изолированные клетки собирали центрифугированием и ресуспендировали в среде Игла, модифицированной по способу Дульбекко (DMEM), содержащей 10% лошадиной сыворотки (HS), 5% эмбриональной бычьей сыворотки (FBS) и 1% пенициллина/стрептомицина. Культуры обогатили миоцитами путем предварительного посева на 90 минут, чтобы истощить популяцию не миоцитов. Незакрепленные клетки высевали в 6- или 96-луночные планшеты с соответствующей плотностью клеток. Клетки культивировали при 37°С в атмосфере 95% воздух/5% СО2 в течение 24 часов. Затем культуральную среду заменяли свежей средой DMEM, содержащей 1% FBS и различные концентрации тестируемого соединения за тридцать минут до инкубации в нормальной или гипоксической (N2/CO2, 95%/5%; 0.3% О2) культуральной камере.
Обработка тестируемым соединением
Очищенные кардиомиоциты новорожденных крыс высевали в 96-луночные планшеты при 106 клеток/ 2 мл для проведения проточной цитометрии.
Через 24 часа кардиомиоциты обрабатывали разными концентрациями тестируемого соединения в культуральной среде с 0.1% DMSO. Положительные клеточные контроли обрабатывали культуральной средой (0.1% DMSO). Через тридцать минут после начала обработки клетки инкубировали в гипоксической культуральной камере (N2/CO2, 95%/5%; окончательно измеренное содержание О2: 0.3%) в течение 36 часов.
Отрицательные клеточные контроли оставляли в условиях с нормальным содержанием кислорода при 37°С в культуральной среде (1% FBS, 0.1% DMSO) в течение тех же самых периодов времени.
Определение количества апоптотических клеток
В конце периода обработки проводили проточную цитометрию для измерения количества апоптотических клеток. Использовали набор для определения апоптоза с помощью аннексин V-флуоресцеин изотиоцианата (FITC) от компании Miltenyi. Клетки дважды промывали PBS и ресуспендировали в связывающем буфере. FITC-аннексин V и пропидий иодид добавляли согласно протоколам производителя. Смесь инкубировали в течение 15 минут в темноте при комнатной температуре, а затем измеряли клеточную флуоресценцию с помощью FACS scan проточной цитометрии.
2. Результаты
2.1. Цитопротекция и селективность соединения
Результаты разных анализов с выбранными соединениями изобретения показаны ниже в Таблице 1.
В качестве примера, Фигура 1 демонстрирует цитопротекторное действие соединения 12 после стресса, индуцированного воздействием туникамицина.
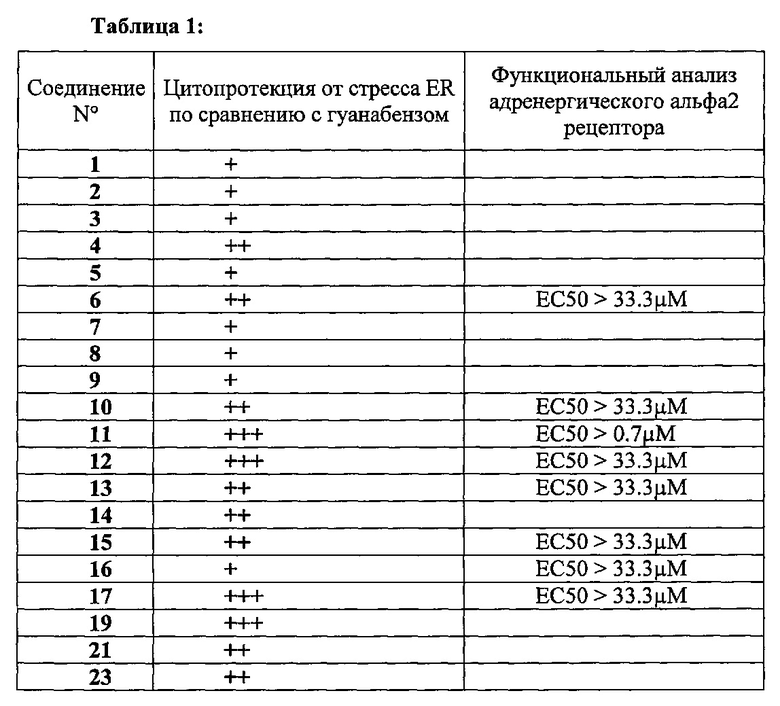
2.2. Рассеянный склероз
Фигура 2 показывает дозо-зависимую защиту от повреждения интерфероном-гамма крысиных олигодендроцитов соединениями 11, 12 и 17 изобретения.
Эти данные показывают, что соединения этого изобретения являются перспективными для эффективного лечения рассеянного склероза.
2.3. Болезнь Паркинсона (PD)
Фигура 3 показывает дозо-зависимую защиту от повреждения ротеноном первичных мезэнцефальных нейронов крыс соединениями 5, 11 и 12 изобретения.
Эти результаты показывают, что соединения этого изобретения являются перспективными для эффективного лечения синуклеопатии, и конкретнее, болезни Паркинсона.
2.4. Болезнь Альцгеймера (AD) и амилоидоз
Фигура 4 показывает дозо-зависимую защиту поврежденных амилоидом-бета 1-42 первичных кортикальных нейронов соединением 12 изобретения.
Эти результаты показывают, что соединения этого изобретения являются перспективными для эффективного лечения амилоидоза и, конкретнее, болезни Альцгеймера.
2.5. Лейкодистрофия: болезнь Пелицеуса-Мерцбахера (PMD).
Описано, что мутации Т181Р и L223P в белках PLP1 и DM20 вызывают тяжелый фенотип болезни Пелицеуса-Мерцбахера (Strautnieks et al. 1992, Am. J. Hum. Genet. 51 (4): 871-878; Gow и Lazzarini, 1996 Nat Genet. 13(4):422-8).
Соединение 12 и 17 изобретения (5 микроМ) способно предотвратить накопление Т181Р мутированного DM20 белка, экспрессированного в человеческих клетках 293Т (Фигура 5).
Эти результаты показывают, что соединения этого изобретения, в частности, соединения 12 и 17, являются перспективными для эффективного лечения заболеваний, связанных с демиелинизацией, подобных лейкодистрофии, конкретнее PMD.
2.6. Диабет 2 типа
Фигура 6 представляет результаты сверхэкспрессии препроинсулина, несущего мутацию Akita, в клетках Min6 с соединением 16 изобретения.
Соединения 12, 16 и 17 в различных концентрациях предотвращают смерть клеток инсулиномы Min6, связанную с накоплением неправильно свернутого белка, вызванного 6-часовой экспозицией с туникамицином (Фигура 7)
Соединения 11, 12, 16 и 17 в различных концентрациях предотвращают смерть клеток INS 1 инсулиномы, связанную с накоплением неправильно свернутого белка, вызванного 6-часовой экспозицией с туникамицином (Фигура 8).
Эти результаты показывают, что соединения изобретения являются перспективными для эффективного лечения предиабета и диабета, предпочтительно предиабета 2 типа и диабета 2 типа.
2.7. Связанные с инфекцией или неинфекционные воспалительные состояния.
Нормальный ответ MEF (эмбриональныхе мышиных фибробластов) на поли I:С характеризуется экспрессией PPP1R15A, увеличением еIF2альфа-Р (изменяющийся с течением времени и свзязанный с уровнями PPP1R15A экспрессии), опосредованного активацией PKR и выработкой IFN типа I (в пределах от 500 до 700 пг/мл). Нокаутные PPP1R15A -/- MEFs не способны продуцировать этот цитокин в ответ на поли I:С.
Потенциальная возможность соединений изобретения ингибировать PPP1R15A была оценена путем измерения увеличения еIF2альфа фосфорилирования, уменьшения PPP1R15A экспрессии вследствие его собственного фармакологического инигибирования, приводящего к ингибированию общего синтеза белка и продуцированию IFN I типа.
Было обнаружено, что оцененные соединения изобретения являются эффективными при 25 мкМ в отношении увеличения еIF2альфа фосфорилирования, уменьшения экспрессии PPP1R15A и предотвращения выработки IFN I типа. В качестве примера, Фигура 9 показывает способность соединения 6, 10, 11, 12, 15, 16 и 17 (при 25 микроМ) предотвращать продуцирование IFN типа-I мышиными эмбриональными фибробластами, подвергнутыми липофекции поли I:С.
Эти результаты показывают, что соединения этого изобретения являются перспективными для эффективного лечения связанных с инфекцией или неинфекционных воспалительных состояний.
2.8. Ишемия сердца
Соединение 10 изобретения защищает культивируемые кардиомиоциты новорожденных крыс от апоптоза, вызванного гипоксией (Фигура 10). Эти результаты показывают, что соединения этого изобретения являются перспективными для эффективного лечения ишемии, в частности, ишемии сердца.
Различные модификации и варианты изобретения будут очевидны специалистам в данной области техники без отступления от существа и объема изобретения. Несмотря на то, что изобретение описано в связи с конкретными предпочтительными вариантами осуществления, следует понимать, что заявленное изобретение не должно неоправданно ограничиваться такими конкретными вариантами осуществления. Безусловно, различные модификации описанных способов осуществления изобретения, которые являются очевидными специалистам в соответствующих областях, охватываются настоящим изобретением.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ БЕНЗИЛИДЕНГУАНИДИНА ДЛЯ ЛЕЧЕНИЯ ПРОТЕИНОПАТИЙ | 2015 |
|
RU2712452C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛИДЕНГУАНИДИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С НЕПРАВИЛЬНЫМ СВОРАЧИВАНИЕМ БЕЛКОВ | 2014 |
|
RU2654910C2 |
| СВЯЗЫВАЮЩИЕ АГЕНТЫ ЯДЕРНЫХ РЕЦЕПТОРОВ | 2011 |
|
RU2604666C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АЛКИЛАМИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ МЕЖДУ MDM2 И Р53 | 2007 |
|
RU2434863C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ КАССЕТНЫХ ТРАНСПОРТЕРОВ | 2008 |
|
RU2512682C2 |
| ТРИАЗОЛЫ ДЛЯ РЕГУЛЯЦИИ ГОМЕОСТАЗА ВНУТРИКЛЕТОЧНОГО КАЛЬЦИЯ | 2017 |
|
RU2753509C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ИЗОИНДОЛОВ, КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ, В ЧАСТНОСТИ В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВНОСТИ БЕЛКА-ШАПЕРОНА Hsp90 | 2006 |
|
RU2375361C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ АНДРОГЕН-РЕЦЕПТОР-ПОЛОЖИТЕЛЬНЫХ ФОРМ РАКА | 2020 |
|
RU2817802C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АМИЛОИДОМ ИЛИ АМИЛОИДПОДОБНЫМИ БЕЛКАМИ | 2007 |
|
RU2469026C2 |
| ИНДАЗОЛЬНЫЕ ИНГИБИТОРЫ СИГНАЛЬНОГО ПУТИ WNT И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2017 |
|
RU2682245C1 |
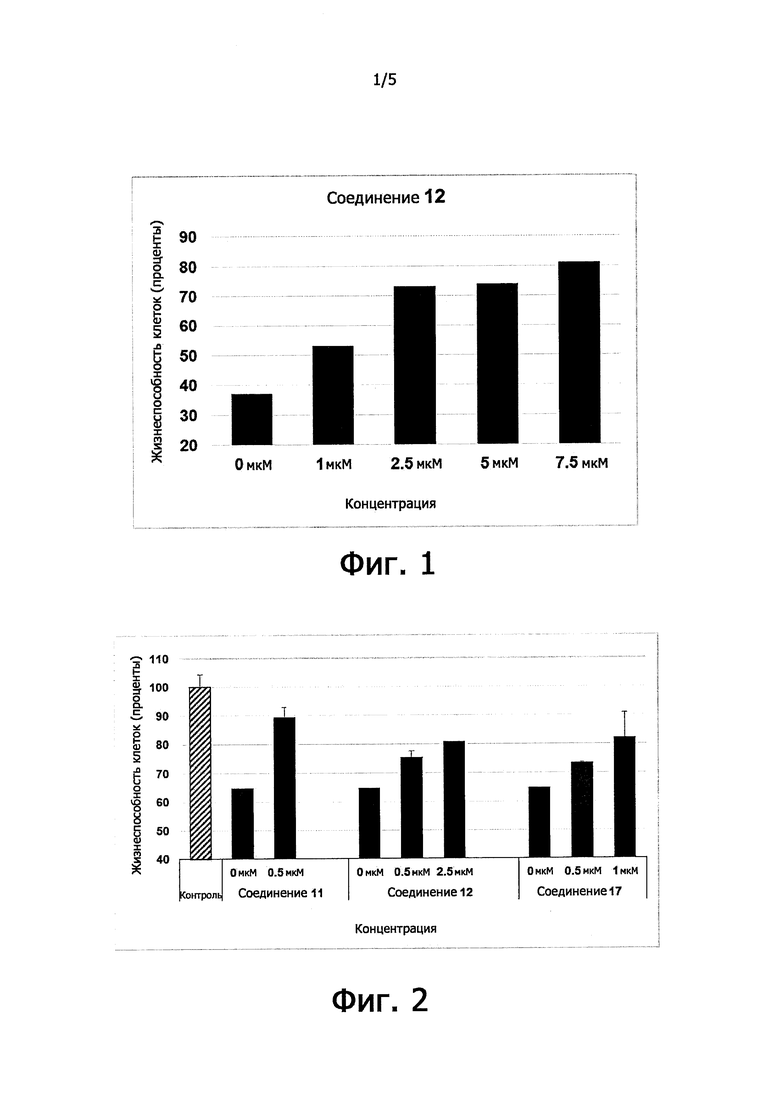
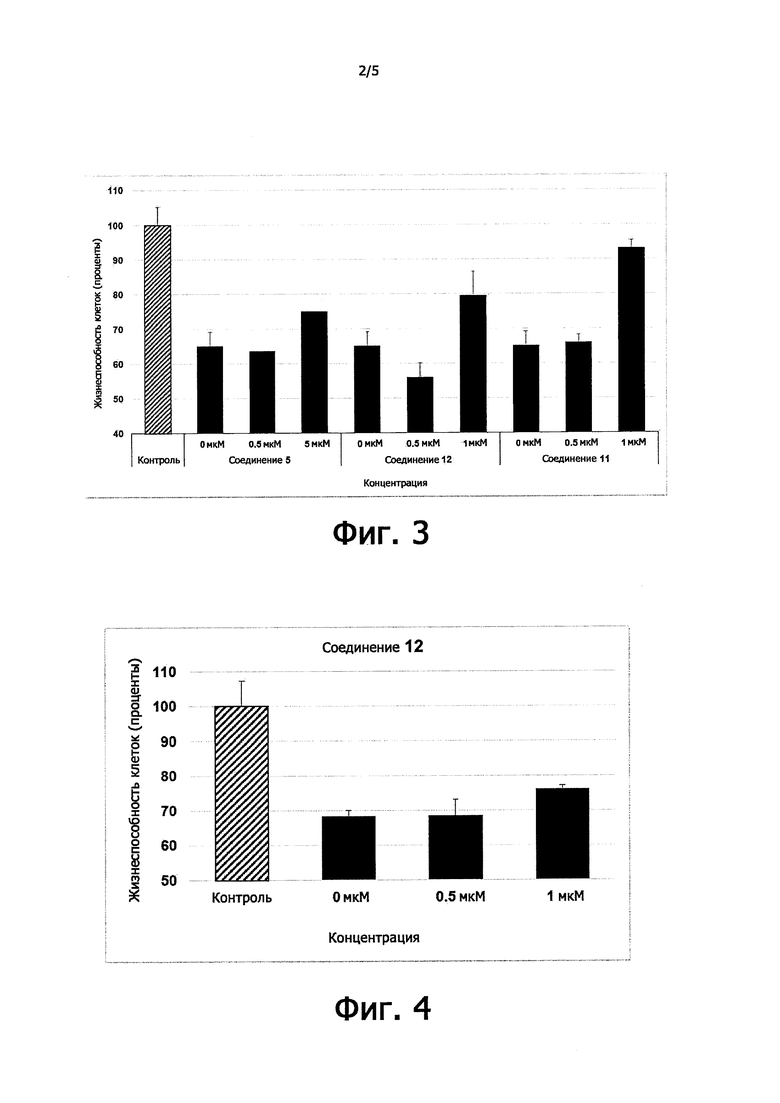
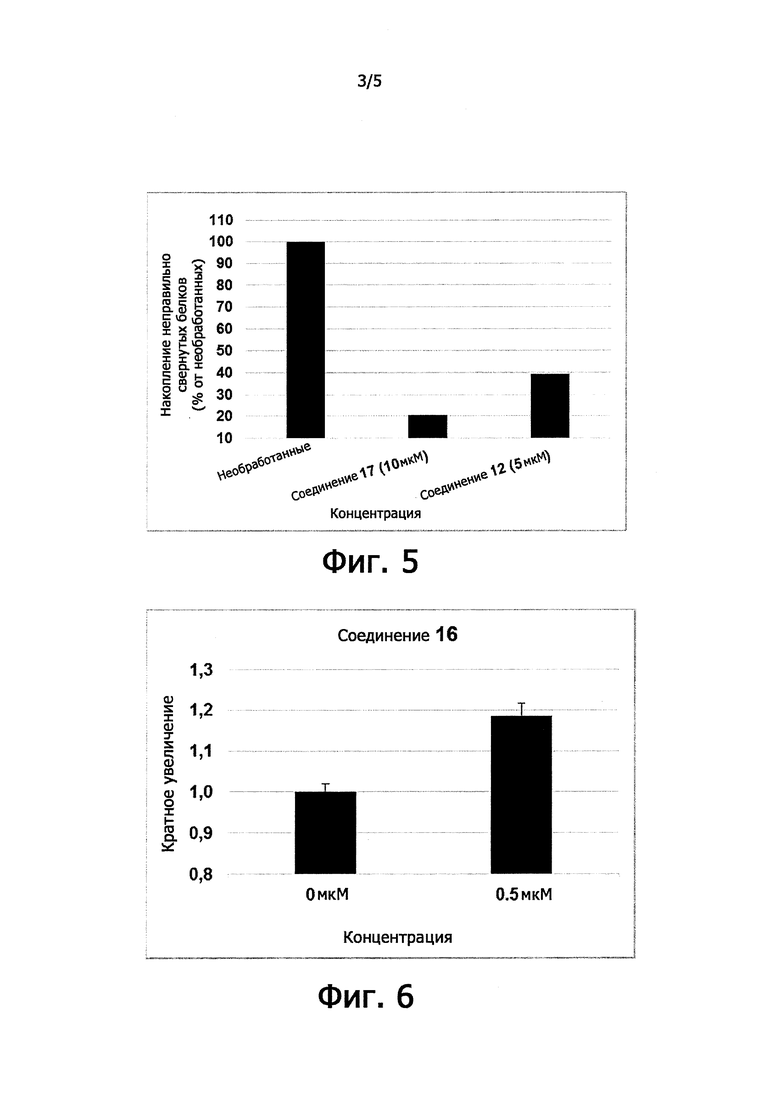
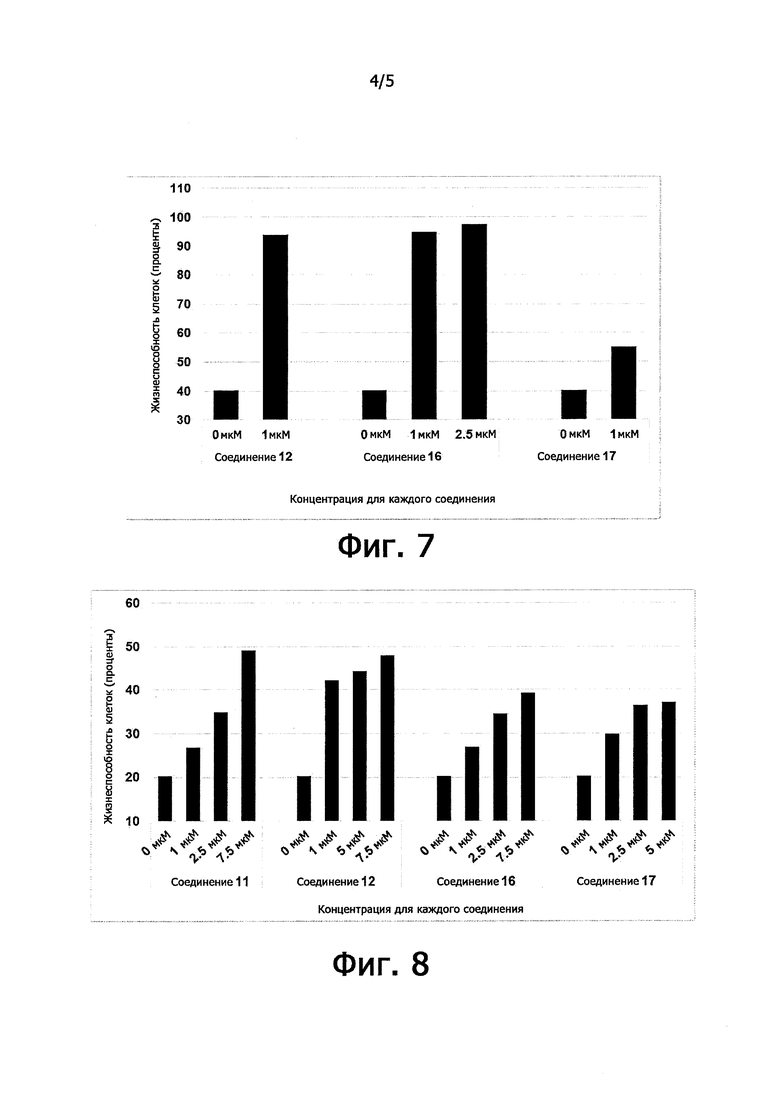
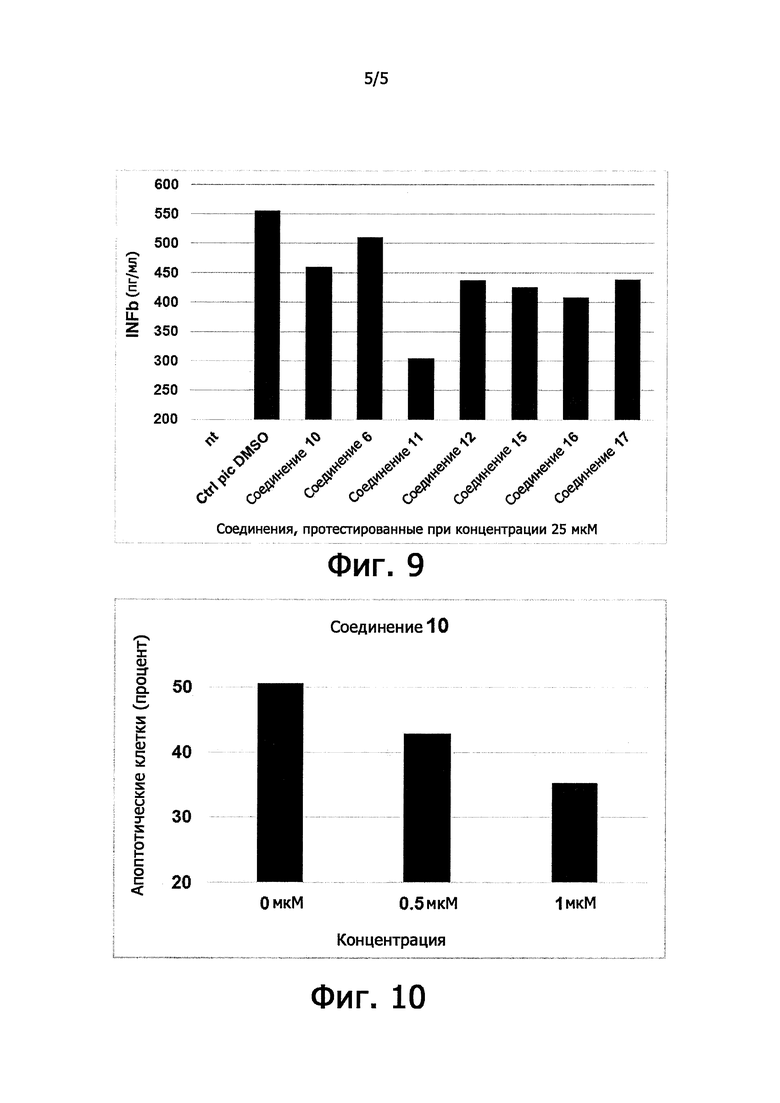
Изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или таутомерам, которые могут найти применение при лечении заболевания, связанного со стрессом, вызванным мисфолдингом белков, и в частности с накоплением неправильно свернутых белков. В формуле (I) Hal представляет собой F, Cl, Br, I; X представляет собой –CR1=; Y представляет собой –CR2= или –N=; Z представляет собой –CR3=; W представляет собой –CR4=; R1 представляет собой H; R2 представляет собой H; R3 представляет собой H; R4 представляет собой H, Cl, F, I или Br; R5 представляет собой алкил, алкенил, каждый из которых необязательно является замещенным одной или более R7 группами; каждый R7 независимо выбирают из галогена, OH, гетероциклила, S-алкила, SO-алкила, SO2-алкила и алкоксигруппы, где алкил представляет собой С1-С12 алкил; алкенил представляет собой С2-С12 алкенил; гетероцикл представляет собой 4-12-членную циклическую группу, содержащую один или более гетероатомов, выбранных из N, О и S. Изобретение относится также к способу получения соединений формулы (I), фармацевтической композиции и их применению при лечении заболевания, связанного со стрессом, вызванным мисфолдингом белков. 4 н. и 8 з.п. ф-лы, 10 ил., 1 табл., 13 пр.
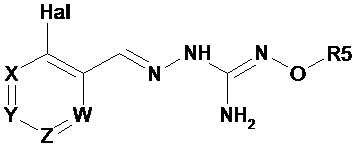 (I)
(I)
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
 (I),
(I),
или его таутомер, в которой
Hal = F, Cl, Br, I;
X представляет собой –CR1=;
Y представляет собой –CR2= или –N=;
Z представляет собой –CR3=;
W представляет собой –CR4=;
R1 представляет собой H;
R2 представляет собой H;
R3 представляет собой H;
R4 представляет собой H, Cl, F, I или Br;
R5 представляет собой алкил, алкенил, каждый из которых необязательно является замещенным одной или более R7 группами;
каждый R7 независимо выбирают из галогена, OH, гетероциклила, S-алкила, SO-алкила, SO2-алкила и алкоксигруппы,
где алкил представляет собой С1-С12 алкил; алкенил представляет собой С2-С12 алкенил; гетероцикл представляет собой 4-12-членную циклическую группу, содержащую один или более гетероатомов, выбранных из N, О и S.
2. Соединение по п. 1, в котором Hal представляет собой Cl.
3. Соединение по п. 1, в котором Y представляет собой –CR2= и R2 представляет собой H.
4. Соединение по п. 1, в котором W представляет собой –CR4= и R4 представляет собой H, Cl или F.
5. Соединение по п. 1, в котором R5 выбирают из алкенила или алкила, каждый из которых необязательно является замещенным одной или более R7 группами, выбранными из галогена, OH, гетероциклила, S-алкила, SO-алкила, SO2-алкила, O-алкила.
6. Соединение по п. 1, которое выбирают из следующих:
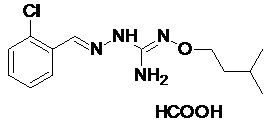
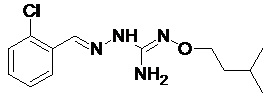
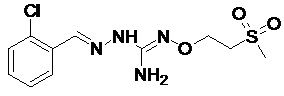
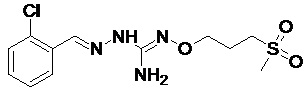
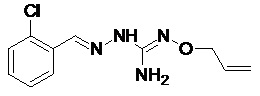
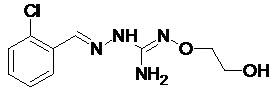

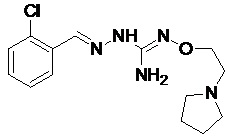
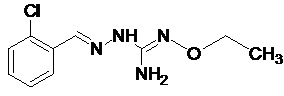
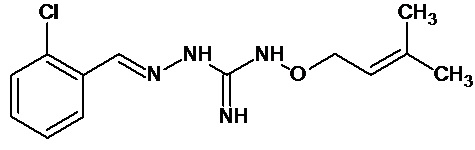

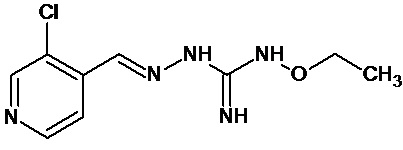
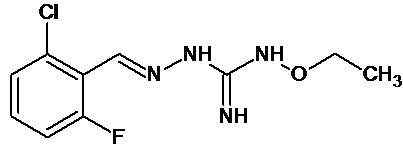
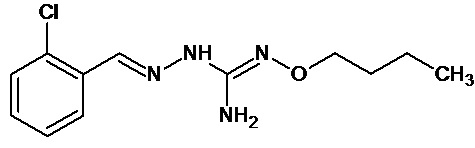
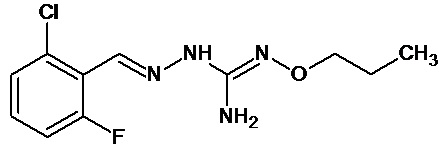

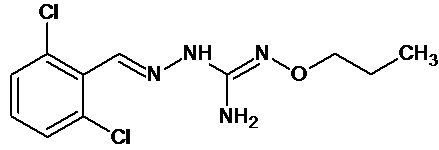
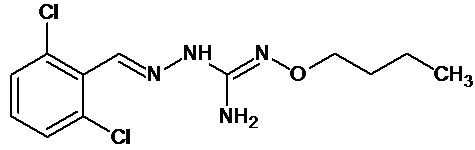
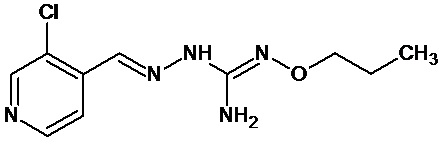
или его таутомер, или его фармацевтически приемлемая соль.
7. Способ получения соединения формулы (I) или его фармацевтически приемлемых солей по любому из пп. 1-6, включающий стадию взаимодействия соединения формулы (A):
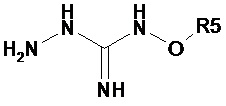 (A),
(A),
в которой R5 определяется как в любом из пп. 1-6, с соединением формулы (B):
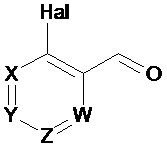 (B),
(B),
в которой X, Y, Z, W и Hal определяются как в любом из пп. 1-6.
8. Способ по п. 7, который включает дополнительную стадию очистки.
9. Фармацевтическая композиция для лечения заболевания, связанного со стрессом, вызванным мисфолдингом белков, выбранного из болезни Альцгеймера, болезни Паркинсона, полиглутаминных и полиаланиновых заболеваний, выбранных из болезни Хантингтона, спинобульбарной мышечной атрофии (или болезни Кеннеди), дентато-рубро-паллидо-льюисовой атрофии, спинально-церебеллярной атаксии 1 типа, спинально-церебеллярной атаксии 2 типа, спинально-церебеллярной атаксии 3 типа (или болезни Мачадо-Джозефа), спинально-церебеллярной атаксии 6 типа, спинально-церебеллярной атаксии 7 типа и спинально-церебеллярной атаксии 17 типа, окулофарингеальной мышечной дистрофии; демиелинизирующих заболеваний, таких как лейкодистрофия, болезнь Шарко-Мари-Тута и рассеянный склероз, воспаления, и связанных с воспалением заболеваний, выбранных из системной красной волчанки, панкреатита, колита, язвенного колита, воспалительного заболевания кишечника, сепсиса, синдрома системной воспалительной реакции (SIRS), вызванного воспалением рака, рака, диабета 2 типа, амиотрофического бокового склероза, ишемии сердца и старения, содержащая эффективное количество соединения формулы (I), как определено в любом из пп. 1-6, вместе с подходящим фармацевтически приемлемым разбавителем, эксципиентом или носителем.
10. Соединение формулы (I) для применения при лечении заболевания, связанного со стрессом, вызванным мисфолдингом белков, и в частности с накоплением неправильно свернутых белков, где соединение (I) определяется по любому из пп. 1-6.
11. Соединение по п. 10, отличающееся тем, что болезнь связана с путем PPP1R15A.
12. Соединение по п. 11, отличающееся тем, что болезнь выбирают из болезни Альцгеймера, болезни Паркинсона, полиглутаминных и полиаланиновых заболеваний, выбранных из болезни Хантингтона, спинобульбарной мышечной атрофии (или болезни Кеннеди), дентато-рубро-паллидо-льюисовой атрофии, спинально-церебеллярной атаксии 1 типа, спинально-церебеллярной атаксии 2 типа, спинально-церебеллярной атаксии 3 типа (или болезни Мачадо-Джозефа), спинально-церебеллярной атаксии 6 типа, спинально-церебеллярной атаксии 7 типа и спинально-церебеллярной атаксии 17 типа, окулофарингеальной мышечной дистрофии; демиелинизирующих заболеваний, таких как лейкодистрофия, болезнь Шарко-Мари-Тута и рассеянный склероз, воспаления, и связанных с воспалением заболеваний, выбранных из системной красной волчанки, панкреатита, колита, язвенного колита, воспалительного заболевания кишечника, сепсиса, синдрома системной воспалительной реакции (SIRS), вызванного воспалением рака, рака, диабета 2 типа, амиотрофического бокового склероза, ишемии сердца и старения.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| P | |||
| PRUSIS ET AL., Synthesis and Quantitative Structure-Activity Relationship of Hydrazones of N-Amino-N-hydroxyguanidine as Electron Acceptors for Xanthine Oxidase, J | |||
| MED | |||
| CHEM., 2004, Vol | |||
| Способ очищения сернокислого глинозема от железа | 1920 |
|
SU47A1 |
| ЭЛЕКТРОЛИТИЧЕСКИЙ АППАРАТ ДЛЯ ПОЛУЧЕНИЯ СВИНЦОВЫХ БЕЛИЛ | 1924 |
|
SU3105A1 |
| US 6599943 B1, 29.07.2003 | |||
| P | |||
| TSAYTLER ET AL., Selective Inhibition of a Regulatory Subunit of Protein | |||

