Область техники, к которой относится изобретение
Настоящее изобретение относится к соединениям, имеющим потенциальное терапевтическое применение при лечении заболеваний, связанных с воздействием неправильной укладки белков, в особенности с накоплением неправильно свернутых белков. В частности, изобретением охватываются соединения, которые способны проявлять защитный эффект против цитотоксического воздействия на эндоплазматический ретикулум (ER).
Уровень техники
Соединение 2-(2,6-дихлорбензилиден)гидразинкарбоксимидамид, которое также именуется гуанабенз, является альфа-агонистом типа альфа-2 и применяется в качестве антигипертензивного средства:
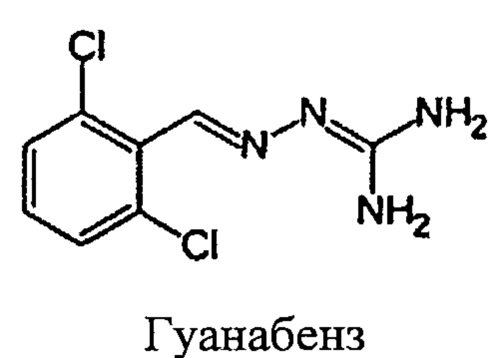
Известны и различные производные гуанабенза. Например, в US 3,982,020 (Sandoz, Inc.) описаны замещенные бензилиденгидразины и их применение в качестве гипо-гликемических-антигипергликемических средств, средств против ожирения и противовоспалительных средств. В US 2004/0068017 (Bausch & Lomb Inc.) раскрыты замещенные бензилиденгидразины, способные повышать активность желатиназы А в клетках глаза. Эти молекулы находят применение при лечении первичной открытоугольной глаукомы. В WO 2008/061647 (Acure Pharma АВ) раскрыто применение N-(2-хлор-3,4-диметокси-бензилиденамино)гуанидина в качестве ингибитора VEGFR и связанное с этим применение при лечении или профилактике нежелательного образования кровеносных сосудов при росте опухолей и/или при воспалительных заболеваниях. В WO 2005/031000 (Acadia Pharmaceuticals, Inc.) описаны замещенные бензилиденгидразины и их применение при лечении острой боли и хронической невропатической боли. Наконец, в ЕР 1908464 (CNRS) раскрыты гуанабенз и хлоргуанабенз и их применение при лечении связанных с накоплением полиглутамина заболеваний, включая болезнь Хантингтона.
Совсем недавно сообщалось, что гуанабенз обладает терапевтическим потенциалом и в ряде других областей. Недавно было отмечено, что гуанабенз обладает антиприоновой активностью (D. Tribouillard-Tanvier et al., 2008 PLoS One 3, e1981). Сообщалось, что его активность при защите от неправильной укладки белка на удивление оказалась гораздо шире и включает уменьшение накопления мутантного хантингтина при клеточных анализах (WO 2008/041133) и защиту от летальных эффектов экспрессии подверженного неправильной укладке мутантного инсулина Akita в эндоплазматическом ретикулуме (ER) панкреатических бета-клеток Min6 и INS-1 (Tsaytler et al., Science 2011 Vol. 332, 1 pp 91-94). В WO 2014/138298 и Way et al. (2015 Nature Communications 6:6532 DOI: 10.1038/ncomms7532) раскрыты гуанабенз и его применение при лечении демиелинизирующих заболеваний типа рассеянного склероза.
Также было показано, что гуанабенз способствует выживанию клеток HeLa, подвергнутых цитотоксическому ER-стрессу, вызванному ингибитором N-глико-зилирования туникамицином, зависимым от дозы образом (Tsaytler et al., Science 2011). Количественная оценка жизнеспособности клеток показала, что гуанабенз удвоил количество клеток, переживших ER-стресс, при средней эффективной концентрации ~0,4 мкМ. Ни агонист α2-адренергических рецепторов клонидин, ни антагонист α2-адренергических рецепторов эфароксан не защищали клетки от цитотоксического ER-стресса, и эфароксан не влиял на защитные эффекты гуанабенза (Tsaytler et al., Science 2011). Эти наблюдения показывают, что гуанабенз спасает клетки от летального ER-стресса по механизму, не зависящему от α2-адренергических рецепторов. Гуанабенз защищает клетки от летального накопления неправильно свернутых белков путем связывания с регуляторной субъединицей протеинфосфатазы 1, PPP1R15A (GADD34), избирательно нарушая индуцированное стрессом дефосфорилирование α-субъединицы фактора инициации трансляции 2 (eIF2α). Гуанабенз доводит скорость трансляции в подвергающихся стрессу клетках до уровня, управляемого имеющимися шаперонами, восстанавливая тем самым белковый гомеостаз. Сообщалось, что гуанабенз не связывается с конститутивным PPP1R15B (CReP), поэтому он не подавляет трансляцию в клетках, не подвергающихся стрессу (Tsaytler et al., Science 2011).
Неспособность поддерживать протеостаз в ER путем выработки адекватной реакции на развернутые белки (unfolded protein response, UPR) признано фактором, способствующим многим патологическим состояниям. Таким образом, описанные здесь молекулы, которые ингибируют фосфатазу eIF2α для тонкой настройки синтеза белка, могут приносить терапевтическую пользу при большом числе заболеваний, вызванных воздействием неправильной укладки белков, в частности при накоплении неправильно свернутых белков.
Настоящее изобретение направлено на получение альтернативных соединений на основе базовой структуры гуанабенза, имеющих возможное терапевтическое применение при лечении заболеваний, связанных с воздействием неправильной укладки белков, в особенности с накоплением неправильно свернутых белков.
Сущность изобретения
Первый аспект изобретения касается соединений формулы (I) либо их фармацевтически приемлемых солей:
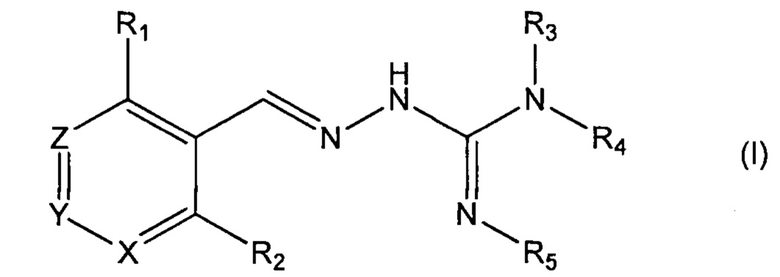
где: R1 означает алкил, О-алкил, Cl, F или Br;
R2 означает Н или F;
R3 выбран из Н и алкила;
R4 выбран из Н и C(O)R6;
R5 означает Н;
или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10;
R6 выбран из R7, OR7 и NR8R9;
R7, R8 и R9 каждый независимо выбран из алкила, циклоалкила, аралкила, циклоалкенила, гетероциклила и арила, каждый из которых необязательно замещен одной или несколькими группами R10;
каждый R10 независимо выбран из галогена, ОН,=O, CN, СОО-алкила, аралкила, SO2-алкила, SO2-арила, СООН, СО-алкила, СО-арила, NH2, NH-алкила, N(алкил)2, CF3, алкила и алкокси;
X и Z каждый независимо означает CR11, a Y выбран из CR11 и N;
R11 означает Н, алкил или F;
для применения при лечении протеинопатии и/или заболевания, связанного с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков.
Второй аспект изобретения касается соединений формулы (I) либо их фармацевтически приемлемых солей:
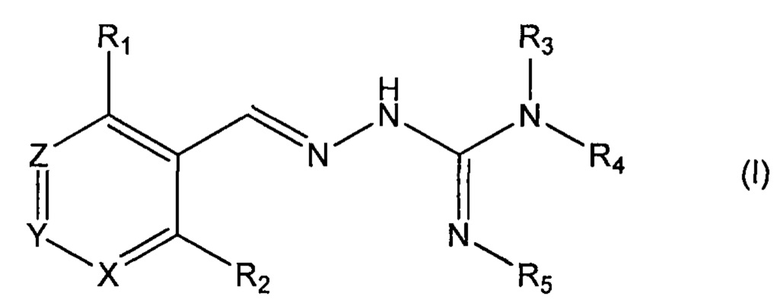
где: R1 означает алкил, О-алкил, Cl, F или Br;
R2 означает Н или F;
R3 выбран из Н и алкила;
R4 выбран из Н и C(O)R6;
R5 означает Н;
или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10;
R6 выбран из R7, OR7 и NR8R9;
R7, R8 и R9 каждый независимо выбран из алкила, циклоалкила, аралкила, циклоалкенила, гетероциклила и арила, каждый из которых необязательно замещен одной или несколькими группами R10;
каждый R10 независимо выбран из галогена, ОН,=O, CN, СОО-алкила, аралкила, SO2-алкила, SO2-арила, СООН, СО-алкила, СО-арила, NH2, NH-алкила, N(алкил)2, CF3, алкила и алкокси;
X, Y и Z каждый независимо означает CR11;
R11 означает Н, алкил или F;
для применения при лечении протеинопатии и/или заболевания, связанного с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков.
Третий аспект изобретения касается соединений формулы (I) либо их фармацевтически приемлемых солей:
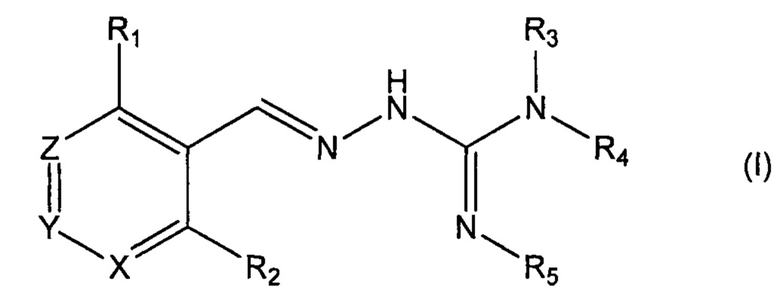
где: R1 означает Cl;
R2 означает Н;
R3 выбран из H и алкила;
R4 выбран из Н и C(O)R6;
R5 означает Н;
или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10;
R6 выбран из R7, OR7 и NR8R9;
R7, R8 и R9 каждый независимо выбран из алкила, циклоалкила, аралкила, цикло-алкенила, гетероциклила и арила, каждый из которых необязательно замещен одной или несколькими группами R10;
каждый R10 независимо выбран из галогена, ОН,=O, CN, СОО-алкила, аралкила, SO2-алкила, SO2-арила, СООН, СО-алкила, СО-арила, NH2, NH-алкила, N(алкил)2, CF3, алкила и алкокси;
X, Y и Z каждый независимо означает CR11;
R11 означает Н, алкил или F;
для применения при лечении протеинопатии и/или заболевания, связанного с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков.
Четвертый аспект изобретения касается соединений формулы (I) либо их фармацевтически приемлемых солей:
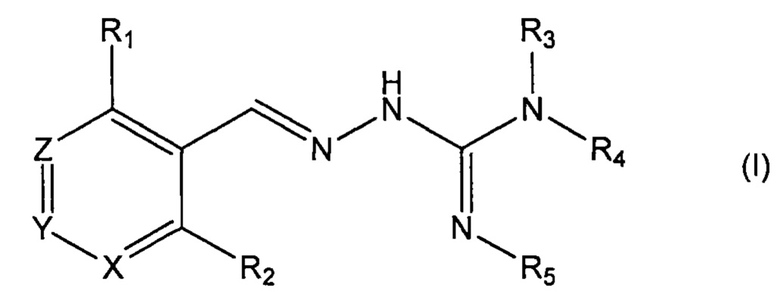
где: R1 означает алкил, О-алкил, Cl, F или Br;
R2 означает Н или F;
R3 выбран из Н и алкила;
R4 выбран из Н и C(O)R6;
R5 означает Н;
или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10;
R6 выбран из R7, OR7 и NR8R9;
R7, R8 и R9 каждый независимо выбран из алкила, циклоалкила, аралкила, цикло-алкенила, гетероциклила и арила, каждый из которых необязательно замещен одной или несколькими группами R10;
каждый R10 независимо выбран из галогена, ОН, =O, CN, СОО-алкила, аралкила, SO2-алкила, SO2-арила, СООН, СО-алкила, СО-арила, NH2, NH-алкила, N(алкил)2, CF3, алкила и алкокси;
X и Z каждый независимо означает CR11, a Y означает N;
R11 означает Н, алкил или F;
для применения при лечении протеинопатии и/или заболевания, связанного с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков.
Предыдущие исследования показали, что арильная группа должна быть по меньшей мере двузамещенной для того, чтобы соединения проявляли полезную фармакологическую активность (например, см. D. Tribouillard-Tanvier et al., PLoS One 3, e1981 (2008); и EP 1908464A, CNRS). Однако, в отличие от результатов предыдущих исследований, авторы настоящего изобретения неожиданно обнаружили, что однозамещенные производные арила тоже активны.
Кроме того, соединения формулы (I), как они определены выше, преимущественно не проявляют активности или проявляют низкую активность в отношении адренергических α2А-рецепторов по сравнению с соединениями предшествующего уровня техники типа гуанабенза. Эта потеря α2А-адренергической активности делает соединения терапевтически полезными при лечении протеопатий и/или расстройств, связанных с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков. Отсутствие α2А-адренергической активности означает, что соединения формулы (I) можно вводить в дозах, подходящих для лечения вышеуказанных заболеваний, без какого-либо существенного влияния на кровяное давление.
Еще один аспект изобретения касается фармацевтических композиций, содержащих соединения формулы (I), как описано выше, вместе с подходящим фармацевтически приемлемым разбавителем, наполнителем или носителем.
Раскрытие сущности изобретения
В настоящем изобретении термин "алкил" включает как насыщенные неразветвленные, так и насыщенные разветвленные алкильные группы. Предпочтительно алкильная группа представляет собой C1-20-алкил, более предпочтительно C1-15-алкил, еще более предпочтительно С1-12-алкил, еще более предпочтительно C1-6-алкил, более предпочтительно C1-3-алкил. Особенно предпочтительными алкильными группами являются, к примеру, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил.
В настоящем изобретении термин "циклоалкил" обозначает циклические алкильные группы. Предпочтительно циклоалкильная группа представляет собой С3-12-циклоалкил.
В настоящем изобретении термин "алкенил" обозначает группы, содержащие одну или несколько двойных связей углерод-углерод, которые могут быть разветвленными или неразветвленными. Предпочтительно алкенильная группа представляет собой С2-20-алкенил, более предпочтительно С2-15-алкенил, еще более предпочтительно С2-12-алкенил или же предпочтительно С2-6-алкенил, более предпочтительно С2-3-алкенил. Термин "циклический алкенил" следует понимать соответствующим образом.
В настоящем изобретении термин "арил" обозначает ароматические С6-12-группы. Типичные примеры включают фенил, нафтил и т.п.
В настоящем изобретении термин "гетероцикл" (также именуется здесь как "гетероциклил" и "гетероциклический") обозначает 4-12-членные, предпочтительно 4-12-членные насыщенные, ненасыщенные или частично ненасыщенные циклические группы, содержащие один или несколько гетероатомов, выбранных из N, О и S, которые необязательно также содержат одну или несколько групп СО. Термин "гетероцикл" охватывает как гетероарильные группы, так и гетероциклоалкильные группы, как определено ниже.
В настоящем изобретении термин "гетероарил" обозначает 4-12-членные ароматические группы, которые содержат один или несколько гетероатомов. Предпочтительно гетероарильная группа представляет собой 4-12-членную ароматическую группу, содержащую один или несколько гетероатомов, выбранных из N, О и S. Подходящими гетероарильными группами являются пиррол, пиразол, пиримидин, пиразин, пиридин, хинолин, тиофен, 1,2,3-триазол, 1,2,4-триазол, тиазол, оксазол, изотиазол, изооксазол, имидазол, фуран и др.
В настоящем изобретении термин "гетероциклоалкил" обозначает 4-12-членные циклические алифатические группы, которые содержат один или несколько гетероатомов. Предпочтительными гетероциклоалкильными группами являются пиперидинил, пирролидинил, пиперазинил, тиоморфолинил и морфолинил. Более предпочтительно гетероциклоалкильные группы выбирают из N-пиперидинила, N-пирролидинила, N-пиперазинила, N-тиоморфолинила и N-морфолинила.
В настоящем изобретении термин "аралкил" включает, без ограничения, группы, содержащие и арильные, и алкильные функциональные группы. В качестве примера, этот термин охватывает группы, в которых один из атомов водорода алкильный группы заменен арильной группой, например фенилом. Типичными аралкильными группами являются бензил, фенетил и т.п.
В одном предпочтительном воплощении R1 означает Cl, Br, Me или F, более предпочтительно Cl.
В одном предпочтительном воплощении R2 означает Н.
В одном предпочтительном воплощении Y означает CR11.
В другом предпочтительном воплощении Y означает N.
В одном предпочтительном воплощении R3 и R4 оба означают Н.
В одном предпочтительном воплощении R3 означает Н, a R4 означает C(O)R6.
В одном предпочтительном воплощении R6 означает алкил или алкокси, более предпочтительно Me или ОМе.
В одном предпочтительном воплощении R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетерциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10.
В одном предпочтительном воплощении данное соединение является соединением формулы (Ia) либо его фармацевтически приемлемой солью:
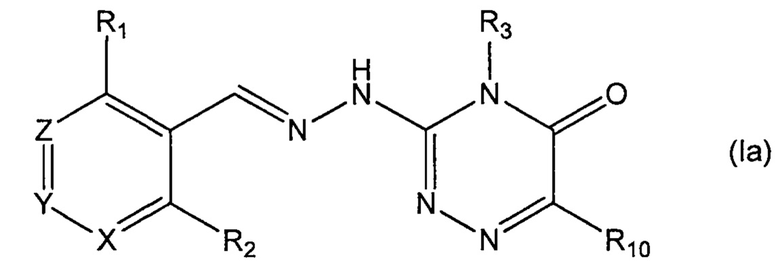
где R1, R2, R3 и R10 такие, как определено выше.
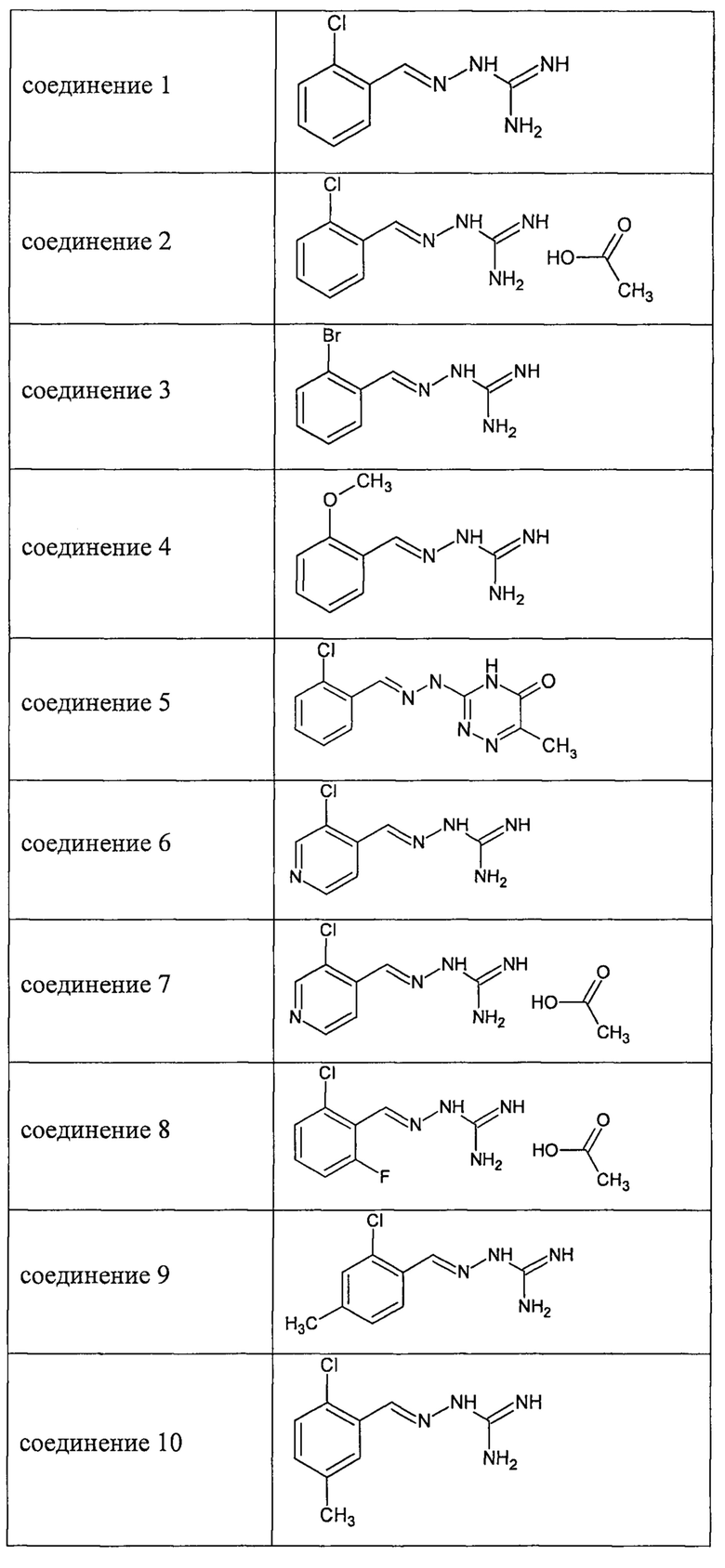
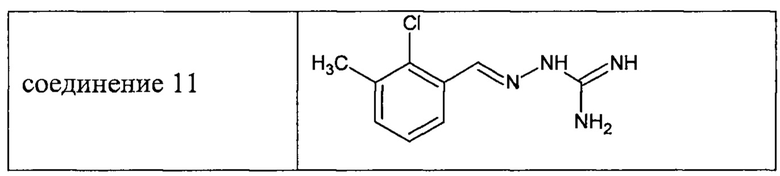
В одном особенно предпочтительном воплощении соединение формулы (I) выбирают из следующих соединений:
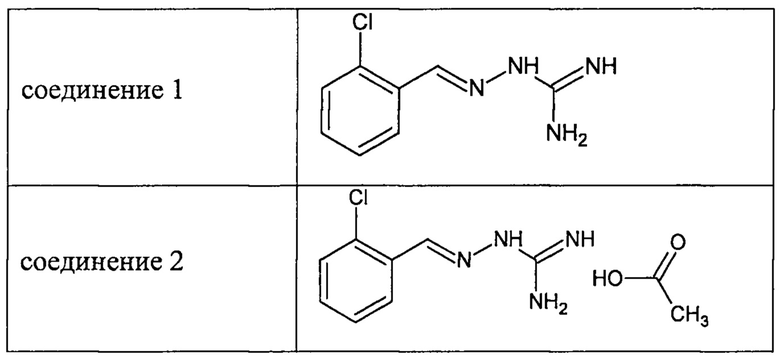
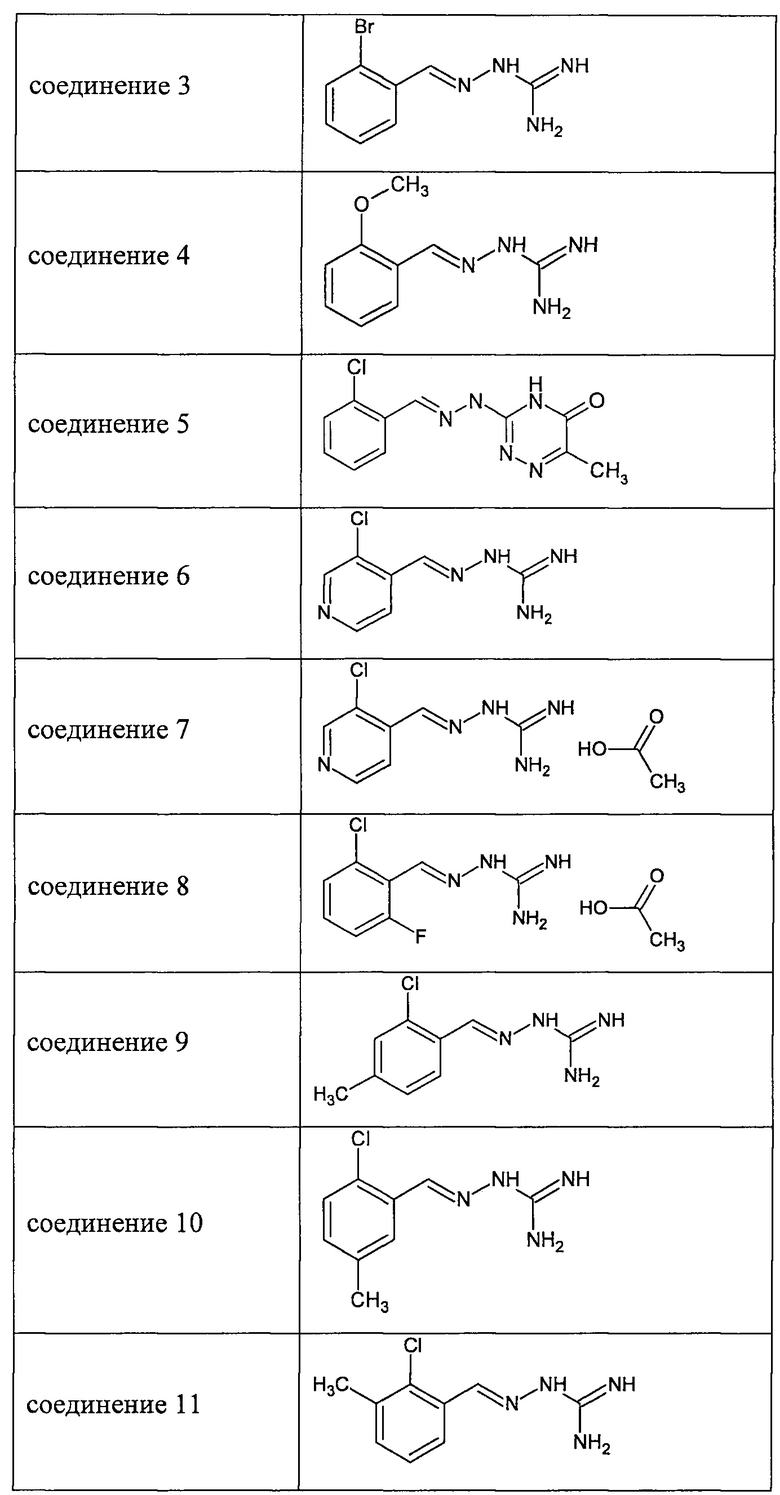
и их фармацевтически приемлемых солей.
В первом предпочтительном воплощении соединение формулы (I) выбирают из соединения 1, то есть 1-[[(2-хлорфенил)метилиден]амино]-гуанидина, и соединения 2, то есть 1-[[(2-хлорфенил)метилиден]амино]-гуанидинацетата, которые приведены выше.
Во втором предпочтительном воплощении соединение формулы (I) выбирают из соединения 8, которое приведено выше.
В третьем предпочтительном воплощении соединение формулы (I) выбирают из соединения 6 и соединения 7, которые приведены выше.
Терапевтическое применение
Соединения формулы (I) имеют потенциальное терапевтическое применение при лечении протеинопатий и/или расстройств, связанных с накоплением неправильно свернутых и/или развернутых белков. В частности, соединения формулы (I) обладают защитным действием против цитостатического воздействия на эндоплазматический ретикулум (ER) и возрастных расстройств.
Другой аспект изобретения касается применения соединений формулы (I), как они определены выше, при изготовлении лекарственных средств для лечения расстройств, связанных с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков.
Другой аспект изобретения касается применения соединений формулы (I), как они определены выше, при изготовлении лекарственных средств для лечения заболеваний, в которых накопление неправильно свернутых и/или развернутых белков вовлечено в механизм действия (Brown et al., 2012, Frontiers in Physiology, 3, Article 263).
Другой аспект изобретения касается применения соединений формулы (I), как они определены выше, при изготовлении лекарственных средств для лечения протеинопатий. Протеопатии представляют собой класс заболеваний, при которых некоторые белки становятся структурно аномальными и тем самым нарушают функцию клеток, тканей и органов в организме. Зачастую белки не сворачиваются в нормальную конформацию, и в таком неправильно свернутом и/или развернутом состоянии белки могут стать в некотором роде токсичными (приобретают токсическую функцию) или же могут потерять свою нормальную функцию или же у них может уменьшиться биологическая активность. Протеопатии, также известные как протеинопатии, нарушения конформации белков или заболевания с неправильной укладкой белков, охватывают многие заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, прионовые заболевания, диабет 2 типа, амилоидоз и целый ряд других заболеваний (см. неограничивающие примеры ниже).
В настоящем изобретении термины «протеинопатии», «протеопатии», «нарушения конформации белков», «заболевания с неправильной укладкой белков», «заболевания, связанные с воздействием неправильной укладки белков», «заболевания, связанные с цитотоксическим воздействием на ER (ER-стрессом)», «связанные с UPR заболевания" имеют одинаковые значения и относятся к заболеваниям, при которых некоторый белок становится структурно аномальным и тем самым нарушает клеточный гомеостаз.
В настоящем изобретении термины "неправильно свернутый белок" и "развернутый белок" имеют одинаковые значения и относятся к белкам, которые не укладываются в свою нормальную конформацию.
В настоящем изобретении выражение "приготовление лекарственного средства" включает применение одного или нескольких из вышеописанных соединений непосредственно в качестве лекарственного средства, наряду с его применением в программе скрининга для получения других активных средств или на любой стадии производства такого лекарственного средства.
Следующий аспект изобретения касается способа лечения протеинопатий и/или расстройств, связанных с воздействием неправильной укладки белков и/или с цитоток-сическим воздействием на ER (ER-стрессом), в частности с накоплением неправильно свернутых белков у нуждающихся в лечении субъектов, причем данный способ включает введение данному субъекту терапевтически эффективного количества соединения формулы (I), как оно определено выше.
Термин "способ" относится к способам, средствам, методам и процедурам для выполнения данной задачи, включая, без ограничения, способы, средства, методы и процедуры, которые либо известны, либо легко разрабатываются из известных способов, средств, методов и процедур практическими работниками в области химии, фармакологии, биологии, биохимии и медицины.
При этом термин "лечение" включает устранение, существенное торможение, замедление или нормализацию течения заболевания или расстройства, существенное ослабление клинических симптомов заболевания или расстройства либо существенное предотвращение появления клинических симптомов заболевания или расстройства.
В настоящем изобретении термины "болезнь", "расстройство", "заболевание" имеют одинаковое значение. Эти заболевания связаны с реакцией на ER-стресс и/или связаны с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков.
Термин "терапевтически эффективное количество" обозначает такое количество вводимого соединения, которое должно в некоторой степени устранить один или несколько симптомов заболевания или расстройства, подлежащего лечению.
В другом воплощении изобретение касается соединений формулы (I), как они определены выше, для применения при лечении UPR-заболеваний. Реакция на развернутые белки (UPR) является составной частью системы клеточной защиты от неправильно свернутых белков, которая адаптирует укладку в эндоплазматическом ретикулуме (ER) к изменяющимся условиям. UPR активируется в ответ на накопление развернутых или неправильно свернутых белков в просвете эндоплазматического ретикулума. В этом случае UPR имеет две главные цели: (i) восстановить нормальное функционирование клетки путем прекращения трансляции белков и (ii) активировать сигнальные пути, ведущие к увеличению продукции молекулярных шаперонов, участвующих в укладке белков. Если эти цели не достигаются за определенное время или же нарушение затягивается, то UPR нацеливается на апоптоз. Выше расположенными (upstream) компонентами UPR являются находящиеся в ER трансмембранные белки IRE1, ATF6 и PERK, которые детектируют дефекты укладки и перепрограммируют транскрипцию и трансляцию согласованным образом и восстанавливают протеостаз. Активированные IRE1 и ATF6 усиливают транскрипцию генов, участвующих в ER-укладке, типа тех, что кодируют шапероны BiP и GRP94. Активированная PERK ослабляет глобальный синтез белка путем фосфорилирования субъединицы фактора инициации трансляции 2 (eIF2α) по Ser51 и в то же время усиливает трансляцию фактора транскрипции ATF4. Последний контролирует экспрессию другого фактора транскрипции - CHOP, который в свою очередь усиливает экспрессию PPP1R15A/GADD34. PPP1R15A, эффектор отрицательной обратной связи, завершающей сигнализацию UPR, привлекает каталитическую субъединицу протеинфосфатазы 1 (РР1с) к дефосфорилированию eIF2α, что вызывает возобновление синтеза белка. Недостаточность UPR способствует многим патологическим состояниям, которые могли бы выправиться при адекватном запуске этой адаптивной реакции. Селективные ингибиторы индуцируемой стрессом eIF2α-фосфатазы, PPP1R15A-PP1, избирательно тормозят дефосфорилирование eIF2α и, тем самым, синтез белка в подвергнутых стрессу клетках, не влияя на синтез белка в не подвергающихся стрессу клетках. Это продлевает положительное воздействие UPR. Временное снижение синтеза белка полезно для подвергнутых стрессу клеток, так как уменьшение потока синтезируемых белков повышает доступность шаперонов и тем самым защищает от воздействия неправильной укладки (Tsaytler et al., Science 2011). Неселективные ингибиторы двух eIF2α-фосфатаз могут вызывать нежелательные эффекты, так как стойкое ингибирование трансляции является вредным. Так, генетическое устранение PPP1R15A и PPP1R15B приводит к ранней эмбриональной летальности у мышей, указывая на то, что ингибирование двух eIF2α-фосфатаз, PPP1R15A-PP1 и PPP1R15B-PP1, вредно для организма. Напротив, генетическое устранение PPP1R15A не имеет вредных последствий у мышей (Harding et al., 2009, Proc Natl Acad Sci USA, 106, 1832-1837). Кроме того, специфические ингибиторы PPP1R15A, как ожидается, должны быть инертными в отношении не подвергающихся стрессу клеток, так как PPP1R15A не экспрессируется в отсутствие стресса. Таким образом, селективные ингибиторы PPP1R15A должны быть безопасными. Неселективные ингибиторы двух eIF2α-фосфатаз тоже могут быть полезны для лечения заболеваний с неправильной укладкой белков при использовании их в дозах, вызывающих лишь частичное ингибирование фосфатаз.
Защита клеток (цитопротекция) от ER-стресса может быть измерена соответствующими методами. Например, цитопротекция может быть измерена на клетках HeLa, у которых ER-стресс вызывают добавлением сред, содержащих туникамицин, смесь гомологичных нуклеозидных антибиотиков, которые ингибируют ферменты семейства UDP-HexNAc:полипренол-P-HexNAc-1-P и применяются для индукции отклика на развернутые белки. Можно определять жизнеспособность клеток в присутствии и в отсутствие ингибиторных соединений через определенное время по измерению восстановления WST-8 в формазан с помощью стандартного набора для оценки жизнеспособности клеток (типа Cell Viability Counting Kit-8 фирмы Dojindo). Специалисты могут использовать и другие разновидности соединений тетразолия типа МТТ, MTS, ХТТ. Цитопротекция от ER-стресса измеряется в виде процентного увеличения числа жизнеспособных клеток (относительно контроля) после ER-стресса. Можно использовать альтернативные методы анализа жизнеспособности клеток типа люминогенного анализа АТФ. Более подробные сведения о таких методах приводятся в следующем разделе «Примеры».
В одном предпочтительном воплощении соединения формулы (I) способны продлевать защитное действие UPR относительно контроля (т.е. в отсутствие ингибиторного соединения) по меньшей мере на 10%, на 20%, более предпочтительно по меньшей мере на 30%, еще более предпочтительно по меньшей мере на 40%, на 50%, на 60%, на 70%, на 80%) и еще более предпочтительно по меньшей мере на 90%.
Соединения формулы (I) являются ингибиторами реакции PPP1R15A-PP1, что вызывает защитный эффект. Предпочтительно соединения проявляют защитное действие со значением ЕС50 менее 10 мкМ, более предпочтительно менее 5 мкМ и еще более предпочтительно менее 1 мкМ. Предпочтительно соединения должны быть лишены альфа-2-адренергической активности. Так, в одном предпочтительном воплощении соединения совсем не проявляют активности при функциональном анализе на альфа-2-адренергическую активность.
Некоторые соединения формулы (I) избирательно ингибируют PPP1R15A-PP1 и поэтому продлевают защитное действие UPR, тем самым, спасая клетки от воздействия неправильной укладки белков. Следовательно, ингибиторы PPP1R15A-PP1, описанные в настоящем изобретении, имеют терапевтическое применение при лечении различных заболеваний, связанных с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков, и/или протеинопатий.
В одном воплощении соединения формулы (I) способны ингибировать PPP1R15A и PPP1R15B. В наиболее предпочтительном воплощении соединения формулы (I) способны избирательно ингибировать PPP1R15A по сравнению с PPP1R15B.
В одном воплощении изобретение касается соединений формулы (I), как они определены выше, для применения при лечении заболеваний, связанных с путем фосфорилирования eIF2α, в механизме возникновения которых участвует накопление неправильно свернутых белков. Предпочтительно заболевание связано с PPP1R15A. Примеры таких заболеваний включают заболевания с неправильной укладкой белков и/или протеинопатий.
В другом воплощении изобретение касается соединений формулы (I), как они определены выше, для применения при лечении заболеваний, вызванных, связанных с или сопровождающихся фосфорилированием eIF2α и/или активностью PPP1R15A, в механизме возникновения которых участвует накопление неправильно свернутых белков.
В настоящем изобретении термин "заболевание, связанное с PPP1R15A" относится к заболеваниям, которые характеризуются аномальной активностью PPP1R15A, в механизме возникновения которых участвует накопление неправильно свернутых белков. Аномальная активность означает: (i) экспрессию PPP1R15A в таких клетках, которые в норме не экспрессируют PPP1R15A; (ii) повышение экспрессии PPP1R15A; или же (iii) повышение активности PPP1R15A.
В другом воплощении изобретение касается способа лечения млекопитающих с заболеваниями, которые облегчаются при ингибировании PP1R15A, в механизме возникновения которых участвует накопление неправильно свернутых белков, причем способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I), как оно определено выше.
В другом воплощении изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении заболеваний, связанных с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков, и/или UPR-заболеваний, причем данные соединения не обладают или обладают пониженной активностью адренергических альфа-2-агонистов по сравнению с гуанабензом.
В другом воплощении изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении заболеваний, связанных с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков, и/или UPR-заболеваний, причем данные соединения не ингибируют трансляцию белков в не подвергнутых стрессу клетках, экспрессирующих PPP1R15B.
В другом воплощении изобретение касается способа лечения заболеваний, которые характеризуются реакцией на ER-стресс с накоплением неправильно свернутых белков, причем способ включает введение пациенту терапевтически эффективного количества по меньшей мере одного соединение формулы (I), при этом данное соединение модулирует реакцию на ER-стресс.
В другом воплощении изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении протеопатий и/или заболеваний, связанных с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков, и/или UPR-заболеваний, причем данные соединения обладают селективностью в отношении голофосфатазы PPP1R15A-PP1, но не обладают или обладают пониженной активностью в отношении голофосфатазы PPP1R15B-PP1, причем соотношение (активность в отношении голофосфатазы PPP1R15B-РР1/активность в отношении PPP1R15B-PP1) у данных соединений по меньшей мере равно или превосходит соотношение (активность в отношении голофосфатазы PPP1R15A-РР1/активность в отношении PPP1R15B-PP1) для гуанабенза.
В другом воплощении изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении протеопатий и/или заболеваний, связанных с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков, и/или UPR-заболеваний, причем:
- данные соединения обладают активностью в отношении голофосфатазы PPP1R15A-PP1, но не обладают или обладают пониженной активностью в отношении голофосфатазы PPP1R15B-PP1;
- соотношение (активность в отношении голофосфатазы PPP1R15 В-РР1/активность в отношении PPP1R15B-PP1) у данных соединений по меньшей мере равно или превосходит соотношение (активность в отношении голофосфатазы PPPlR15A-PPl/активность в отношении PPP1R15B-PP1) для гуанабенза; и
- данные соединения не обладают или обладают пониженной активностью адренергических альфа-2-агонистов по сравнению с гуанабензом.
Заболевания и расстройства по изобретению представляют собой:
(i) связанные с реакцией на ER-стресс; и/или
(ii) с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых и/или развернутых белков; и/или
(iii) UPR-заболевания; и/или
(iv) связанные с PPP1R15A заболевания; и/или
(v) протеопатии.
Неограничивающие примеры заболеваний по изобретению включают следующие, но не ограничиваются ими.
Нейродегенеративные заболевания, такие как таупатии (типа болезни Альцгеймера, среди прочего), синуклеинопатии (типа болезни Паркинсона, среди прочего), болезнь Хантингтона и родственные полиглутаминовые заболевания, полиаланиновые заболевания (типа глазоглоточной мышечной дистрофии), прионовые заболевания (также известны как трансмиссивные губчатые энцефалопатии), демиелинизирующие заболевания типа болезни Шарко-Мари-Тута (также известна как наследственная моторная и сенсорная нейропатия), лейкодистрофии, боковой амиотрофический склероз (также известен как мотонейронная болезнь и как болезнь Лу Герига), сейпинопатии и рассеянный склероз.
Примеры таупатий включают, без ограничения, болезнь Альцгеймера, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию, лобно-височную лобарную дегенерацию или лобно-височную деменцию (FTD) (болезнь Пика). Нейродегенеративное заболевание FTD характеризуется прогрессирующей потерей нейронов, преимущественно затрагивающей лобные и/или височные доли; по распространенности оно уступает только болезни Альцгеймера (AD), причем на FTD приходится 20% случаев ранней деменции. Участие UPR в таупатиях хорошо установлено (см. Stoveken 2013, Journal of Neuroscience 33(36): 14285-14287). Не связывая себя какой-либо теорией, авторы полагают, что соединения по изобретению, которые являются ингибиторами PPP1R15A, будут ослаблять проявления таупатий. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении болезни Альцгеймера. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении заболеваний из числа лобно-височной деменции (FTD), надъядерного паралича и кортикобазальной дегенерации, предпочтительно FTD.
Примеры синуклеинопатий включают, без ограничения, болезнь Паркинсона, деменцию с тельцами Леви, чисто вегетативную недостаточность и множественную системную атрофию. Недавно Colla et al. (J. of Neuroscience 2012 Vol. 32 №10 pp. 3306-3320) показали, что небольшая молекула салубринал, которая усиливает фосфорилирование eIF2α путем ингибирования опосредованного PPP1R15A дефосфорилирования eIF2α (Воусе et al. 2005 Science Vol. 307 pp. 935-939), значительно ослабляет проявления болезни на двух животных моделях альфа-синуклеинопатии. Не связывая себя какой-либо теорией, авторы полагают, что соединения по изобретению, которые являются ингибиторами PPP1R15A, будут ослаблять проявления болезни при α-синуклеинопатиях. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении α-синуклеинопатий. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении болезни Паркинсона.
Примеры полиглутаминовых заболеваний включают, без ограничения, спинобульбарную мышечную атрофию (или болезнь Кеннеди), болезнь Хантингтона, дентаторубрально-паллидолюисовую атрофию, спиноцеребеллярную атаксию 1-го типа, спиноцеребеллярную атаксию 2-го типа, спиноцеребеллярную атаксию 3 типа (или болезнь Мачадо-Джозефа), спиноцеребеллярную атаксию 6 типа, спиноцеребеллярную атаксию 7-го типа и спиноцеребеллярную атаксию 17-го типа. Гуанабенз способен ослаблять накопление мутантного хантингтина при анализе на клетках (WO 2008/041133). Этот результат является неожиданным, так как хантингтин является либо цитозольным, либо ядерным. Однако существуют данные о том, что метаболизм мутантного хантингтина может быть связан с реакцией на ER-стресс (Nishitoh et al., 2002, Genes Dev, 16, 1345-55; Rousseau et al., 2004, Proc Natl Acad Sci USA, 101, 9648-53; Duennwald and Lindquist, 2008, Genes Dev, 22, 3308-19). To, что гуанабенз защищает клетки от цитотоксического ER-стресса и уменьшает накопление мутантного хантингтина дополнительно подтверждает идею о том, что могут быть такие аспекты реакции на ER-стресс, которые влияют на накопление мутантного хантингтина. Однако гуанабенз не применим для лечения заболеваний с неправильной укладкой белков у человека из-за его гипотензивного действия. Напротив, производные гуанабенза по изобретению, ингибиторы PPP1R15A, лишенные альфа2-адренергической активности, могут быть полезными для лечения полиглутаминовых заболеваний, в особенности из числа болезни Хантингтона, спинобульбарной мышечной атрофии (или болезни Кеннеди), дентаторубрально-паллидолюисовой атрофии, спиноцеребеллярной атаксии 1-го типа, спиноцеребеллярной атаксии 2-го типа, спиноцеребеллярной атаксии 3-го типа (или болезни Мачадо-Джозефа), спиноцеребеллярной атаксии 6 типа, спиноцеребеллярной атаксии 7 типа и спиноцеребеллярной атаксии 17-го типа. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении полиглутаминовых заболеваний. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении болезни Хантингтона.
Примеры полиаланиновых заболеваний включают глазоглоточную мышечную дистрофию, которая обусловлена полиаланиновой полосой в связывающем поли(А) ядерном белке 1 (PABPN1). Barbezier et al. (2011, EMBO Vol. 3 pp35-49) показали, что гуанабенз уменьшает агрегацию при глазоглоточной мышечной атрофии. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении полиаланиновых заболеваний. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении глазоглоточной мышечной атрофии.
Примеры прионовых заболеваний у человека включают, без ограничения, классическую болезнь Крейтцфельда-Якоба, новый вариант болезни Крейтцфельда-Якоба (nvCJD, заболевание у человека, родственное коровьей губчатой энцефалопатии), синдром Герстманна-Штреусслера-Шейнкера (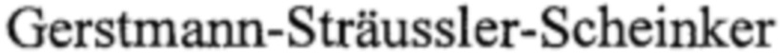 ), наследственную смертельную бессонницу и куру. Гуанабенз ослабляет симптомы у инфицированных прионами мышей (D. Tribouillard-Tanvier et al., 2008 PLoS One 3, e1981). Однако гуанабенз не применим для лечения заболеваний с неправильной укладкой белков у человека из-за его гипотензивного действия. Напротив, производные гуанабенза по изобретению, ингибиторы PPP1R15A, лишенные альфа2-адренергической активности, могут быть полезны для лечения прионовых заболеваний. В соответствии с предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении заболеваний из числа болезни Крейтцфельда-Якоба, нового варианта болезни Крейтцфельда-Якоба, синдрома Герстманна-Штреусслера-Шейнкера, смертельной семейной бессонницы и куру.
), наследственную смертельную бессонницу и куру. Гуанабенз ослабляет симптомы у инфицированных прионами мышей (D. Tribouillard-Tanvier et al., 2008 PLoS One 3, e1981). Однако гуанабенз не применим для лечения заболеваний с неправильной укладкой белков у человека из-за его гипотензивного действия. Напротив, производные гуанабенза по изобретению, ингибиторы PPP1R15A, лишенные альфа2-адренергической активности, могут быть полезны для лечения прионовых заболеваний. В соответствии с предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении заболеваний из числа болезни Крейтцфельда-Якоба, нового варианта болезни Крейтцфельда-Якоба, синдрома Герстманна-Штреусслера-Шейнкера, смертельной семейной бессонницы и куру.
Демиелинизирующие заболевания характеризуются потерей олигодендроцитов в центральной нервной системе или шванновских клеток в периферической нервной системе. Явлением, характерным для демиелинизирующих заболеваний, является снижение числа миелинизированных аксонов в центральной нервной системе или периферической нервной системе. Неограничивающие примеры белков, подвергающихся неправильной укладке в миелинизированных клетках (включая олигодендроциты и шванновские клетки), могут быть выбраны из группы, состоящей из СС1, основного белка миелина (МБР), церамид-галактозилтрансферазы (CGT), ассоциированного с миелином гликопротеина (MAG), миелинового гликопротеина олигодендроцитов (MOG), гликопротеина олигодендроцитов и миелина (OMG), фосфодиэстеразы циклических нуклеотидов (CNP), белка ноль миелина (MPZ), белка 22 периферического миелина (РМР22), коннексина 32 (Сх32), белка 2 (Р2), галактоцереброзида (GalC), сульфатида и протеолипидного белка (PLP). В случае шванновских клеток неправильной укладке преимущественно подвергаются белки MPZ, РМР22, Сх32 и Р2. У олигодендроцитов неправильной укладке предпочтительно подвергаются белки PLP, MBP, MAG.
В некоторых воплощениях демиелинизирующие заболевания выбирают из группы, состоящей из болезней Шарко-Мари-Тута (СМТ). СМТ составляют группу наследственных нейропатических заболеваний, характеризующихся хронической двигательной и сенсорной полинейропатией. Идентифицированы различные типы СМТ, такие как СМТ1, СМТ2, СМТ4, СМТХ и болезнь Дежерина-Сотта (Dejerine-Sottas). Подтипы СМТ можно дополнительно подразделить главным образом по молекулярно-генетическим данным. Например, СМТ1 подразделяется на СМТ1А, 1В, 1С, 1D, 1E, 1F/2E. Свыше 100 мутаций гена, кодирующего белок ноль миелина (Р0), трансмембранный одноходовый белок, которая является главным белком, который вырабатывают миелинизирующие шванновские клетки, вызывают нейропатию Шарко-Мари-Тута (D'Antonio et al., 2009, J Neurosci Res, 87, 3241-9). Мутации наследуются доминантно и вызывают заболевания путем приобретения токсичной функции (D'Antonio et al., 2009, J Neurosci Res, 87, 3241-9). Делеция серина 63 из P0 (P0S63del) вызывает нейропатию Шарко-Мари-Тута 1В у людей и аналогичную демиелинизирующую нейропатию у трансгенных мышей. Мутантный белок накапливается в ER и индуцирует UPR (D'Antonio et al., 2009, J Neurosci Res, 87, 3241-9). Генетическое устранение проапоптотического гена CHOP при UPR восстанавливает двигательную функцию у мышей Шарко-Мари-Тут (Pennuto et al, 2008, Neuron, 57, 393-405). To, что ингибирование PPP1R15A в клетках почти устраняет экспрессию CHOP в подвергнутых ER-стрессу клетках, означает, что генетическое или фармакологическое ингибирование PPP1R15A должно уменьшить двигательные дисфункции у мышей Шарко-Мари-Тут. Недавно D'Antonio et al. (2013 J. Exp. Med. Vol. pp. 1-18) показали, что у мышей P0S63del, получавших салубринал, восстановилась почти нормальная двигательная активность при анализе методом вращающегося стержня (rotarod), причем это сопровождалось устранением морфологических и электро-физиологических нарушений. Накопление связанных с СМТ мутантных белков в ER не уникально для P0S63del; идентифицировано еще по меньшей мере пять других мутантов Р0, которые остаются в ER и вызывают UPR (Pennuto et al., 2008 Neuron Vol. 57 pp. 393-405; Saporta et al., 2012 Brain Vol. 135 pp. 2032-2047). Кроме того, оказалось, что неправильная укладка белков и накопление неправильно свернутых белков в ER задействовано в патогенезе и других нейропатий СМТ в результате мутаций в белках РМР22 и Сх32 (Colby et al, 2000 Neurobiol. Disease Vol. 7 pp. 561-573; Kleopa et al., 2002 J. Neurosci. Res. Vol. 68 pp. 522-534; Yum et al., 2002 Neurobiol. Dis. Vol. 11 pp. 43-52). Однако салубринал токсичен и не может применяться для лечения больных людей (D'Antonio et al., 2013 J. Exp. Med. Vol. pp. 1-18). Напротив, ингибиторы PPP1R15A формулы (I), как ожидают, являются безопасными и могли бы применяться для лечения заболеваний СМТ, предпочтительно СМТ-1А и 1В. В соответствии с предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении СМТ, более предпочтительно СМТ-1 и болезни Дежерина-Сотта. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении СМТ, связанных с накоплением неправильно свернутых белков в ER. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении СМТ-1А. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении СМТ-1В. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении СМТ-1Е.
В другом воплощении соединения формулы (I) предназначаются для применения при лечении СМТ, более предпочтительно для применения при лечении СМТ-1, в сочетании по меньшей мере с одним соединением, выбранным из группы D-сорбитола, баклофена, пилокарпина, налтрексона, метимазола, мифепристона, кетопрофена и их солей. В соответствии с другим воплощением изобретение касается гуанабенза или салубринала (т.е. ингибиторов PPP1R15A) либо их фармацевтически приемлемых солей для применения при лечении СМТ, предпочтительно СМТ-1, в сочетании с по меньшей мере одним соединением, выбранным из группы D-сорбита, баклофена, пилокарпина, налтрексона, метимазола, мифепристона, кетопрофена и их солей. Соединения комбинируются для совместного или раздельного введения, одновременно или последовательно.
Изобретение касается композиций, содержащих ингибитор PPP1R15A, выбранный из группы соединений формулы (I), гуанабенза и салубринала либо их фармацевтически приемлемых солей, и по меньшей мере одно коммерчески доступное соединение или его соль, для применения при лечении нейродегенеративных заболеваний, предпочтительно СМТ, более предпочтительно СМТ-1. Дозировка соединений в композициях должна находиться в пределах доз, не превышающих назначаемые обычно дозы для длительного поддерживающего лечения, или оказавшихся безопасными при клинических испытаниях 3 фазы; наиболее предпочтительно дозировка соединений в комбинации должна соответствовать количествам от 1% до 10% от тех, которые обычно назначаются для длительного поддерживающего лечения.
Итак, настоящее изобретение касается композиций, содержащих ингибитор PPP1R15A, выбранный из группы соединений формулы (I), гуанабенза и салубринала либо их фармацевтически приемлемых солей, и соединение, усиливающее экспрессию белка РМР22, выбранное из группы D-сорбита, баклофена, пилокарпина, налтрексона, метимазола, мифепристона, кетопрофена и их солей, для применения при лечении СМТ, предпочтительно СМТ-1, более предпочтительно СМТ-1А.
В других воплощениях демиелинизирующие заболевания выбирают из группы, состоящей из лейкодистрофий. Примеры лейкодистрофий включают, без ограничения, среди прочего, адренолейкодистрофию (ALD), болезнь Александера, болезнь Канавана, болезнь Краббе, метахроматическую лейкодистрофию (MLD), болезнь Пелицеуса-Мерцбахера (Pelizaeus-Merzbacher или PMD), детскую атаксию с гипомиелинизацией центральной нервной системы (также известна как болезнь исчезающего белого вещества), синдром CAMFAK, болезнь Рефсума (Refsum), синдром Коккейна (Cockayne), синдром Вер-дер-Кнаппа, синдром Зельвегер, синдром Гийена-Барре (GBS), хроническую воспалительную демиелинизирующую полинейропатию (CIDP), многоочаговую моторную нейропатию (MMN) и прогрессирующий надъядерный паралич, прогрессирующую многоочаговую лейкоэнцефалопатию (PML), энцефаломиелит, центральный понтильный миелиноз (СРМ), болезнь антител против MAG. Gow et al. (Neuron, 2002 Vol. 36, 585-596) показали, что при PMD активируется реакция на развернутые белки, и показали, что этот путь представлен дупликацией гена PLP1. В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении лейкодистрофий, предпочтительно болезни Пелицеуса-Мерцбахера (PMD).
Боковой амиотрофический склероз (ALS) известен как мотонейронная болезнь и как болезнь Лу Герига (Lou Gehrig). Теперь уже хорошо известно, что неправильная укладка белков играет главную роль при семейном и при спорадическом ALS (Matus et al. 2013 Int. J. Cell Biol. ID674751 http://dx.doi.org/10.1155/2013/674751). Saxena et al. (Nature Neuroscience 2009 Vol. 12 pp. 627-636) показали, что салубринал продлевает продолжительность жизни у трансгенных мышей G93A-SOD1 на модели мотонейронной болезни. Совсем недавно Jiang et al. (Neuroscience 2014) показали, что гуанабенз замедляет появление симптомов болезни, продлевает продолжительность жизни, улучшает двигательную активность и уменьшает потери двигательных нейронов на модели ALS у мышей G93A-SOD1. Das et al. (2015 Science 388, 239-242) показали, что производное гуанабенза предотвращает двигательные, морфологические и молекулярные дефекты ALS у мутантных мышей G93A-SOD1. В соответствии с предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении семейной и спорадической формы ALS.
Примеры сейпинопатий включают, без ограничения, связанную с врожденной липодистрофией Берардинелли-Сейпа (Berardinelli-Seip) 2-го типа (BSCL2) мотонейронную болезнь, врожденную генерализованную липодистрофию (CGL), синдром Сильвера (Silver), дистальную наследственную моторную нейропатию V-го типа (dHMN-V). Экспрессия мутантных форм сейпина в клеточных культурах активирует путь реакции на развернутые белки (UPR) и индуцирует опосредованную ER-стрессом гибель клеток (Ito & Suzuki, 2009 Brain 132: 87-15). В соответствии с предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении сейпинопатий.
В другом воплощении демиелинизирующее заболевание представлено рассеянным склерозом и родственными заболеваниями типа болезни Шильдера. В соответствии с предпочтительным воплощением, изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении рассеянного склероза.
Кистозный фиброз (CF)
Norez et al. (2008 Eur. J. Pharmacol. Vol. 592 pp. 33-40) показали, что гуанабенз активирует Са2+-зависимые хлорные токи в эпителиальных клетках дыхательных путей человека при кистозном фиброзе (муковисцидозе). Не связывая себя какой-либо теорией, авторы полагают, что соединения по изобретению, которые являются производными гуанабенза-ингибиторами PPP1R15A, будут ослаблять проявления болезни кистозного фиброза. В соответствии с предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении кистозного фиброза.
Заболевания сетчатки
В опубликованной недавно литературе представлены данные о том, что UPR участвует в развитии дегенерации сетчатки: наследственной дегенерации сетчатки типа цилиопатии сетчатки и пигментного ретинита, дегенерации желтого пятна, ретинопатии недоношенных, светоиндуцированной дегенерации сетчатки, отслоении сетчатки, диабетической ретинопатии и глаукомы (см. обзоры Gorbatyuk and Gorbatyuk, 2013, Molecular Vision Vol. 19 pp. 1985-1998; Jing et al., 2012, Exp Diabetes Res, 2012, 589-589).
Цилиопатии сетчатки представляют собой группу редких генетических заболеваний, возникающих из-за дефекта первичных ресничек фоторецепторов, который вызывает пигментный ретинит. Этот дефект, как сообщалось, индуцирует ER-стресс вследствие накопления белка во внутреннем сегменте фоторецепторов, что в свою очередь индуцирует UPR (WO 2013/124484). Дегенерация сетчатки очень часто встречается при цилиопатии и может наблюдаться при отдельных формах пигментного ретинита, таких как амавроз Лебера или Х-хромосомный пигментный ретинит, а также при таких синдромах, как синдром Барде-Бидля (BBS), синдром Альстрема (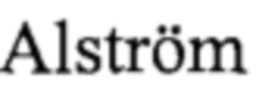 syndrome или ALMS) или синдром Ушера (Usher). Цилиопатии сетчатки входят в группу, состоящую из синдрома Барде-Бидля, синдрома Сеньора-Локена (Senior-Loken), синдрома Жубера (Joubert), синдрома Сальдино-Сайнцера (Salidono-Mainzer), синдрома Сенсенбреннера (Sensenbrenner), синдрома Жена (Jeune), синдрома Меккеля-Грубера (Meckel-Gruder), синдрома Альстрема (), синдрома MORM. В одном предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении заболеваний сетчатки, более предпочтительно наследственной дегенерации сетчатки, такие как цилиопатии сетчатки и пигментного ретинита, дегенерации желтого пятна, ретинопатии недоношенных, светоиндуцированной дегенерации сетчатки, отслоения сетчатки, диабетической ретинопатии и глаукомы.
syndrome или ALMS) или синдром Ушера (Usher). Цилиопатии сетчатки входят в группу, состоящую из синдрома Барде-Бидля, синдрома Сеньора-Локена (Senior-Loken), синдрома Жубера (Joubert), синдрома Сальдино-Сайнцера (Salidono-Mainzer), синдрома Сенсенбреннера (Sensenbrenner), синдрома Жена (Jeune), синдрома Меккеля-Грубера (Meckel-Gruder), синдрома Альстрема (), синдрома MORM. В одном предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении заболеваний сетчатки, более предпочтительно наследственной дегенерации сетчатки, такие как цилиопатии сетчатки и пигментного ретинита, дегенерации желтого пятна, ретинопатии недоношенных, светоиндуцированной дегенерации сетчатки, отслоения сетчатки, диабетической ретинопатии и глаукомы.
В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении пигментного ретинита. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении амавроза Лебера. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении синдрома Барде-Бидля. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении синдрома Альстрема. В соответствии с другим предпочтительным воплощением, изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении синдрома Ушера.
В предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении заболеваний сетчатки, более предпочтительно для применения при лечении заболеваний, входящих в группу наследственной дегенерации сетчатки, таких как цилиопатии сетчатки, пигментный ретинит, дегенерация желтого пятна, ретинопатия недоношенных, светоиндуцированная дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома, в сочетании с соединением, усиливающим экспрессию и/или активность белка BIP, типа вальпроевой кислоты или ее производных, трихостатина А, лития, 1-(3,4-дигидроксифенил)-2-тиоцианат-этанона и эксендина-4. Так, изобретение касается композиций, содержащих ингибитор PPP1R15A формулы (I) или его фармацевтически приемлемую соль и соединение, усиливающее экспрессию и/или активность белка BIP, предпочтительно вальпроевую кислоту, для применения при лечении заболеваний, входящих в группу наследственной дегенерации сетчатки, таких как цилиопатии сетчатки, пигментный ретинит, дегенерация желтого пятна, ретинопатия недоношенных, светоиндуцированная дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома.
В предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении заболеваний сетчатки, более предпочтительно для применения при лечении заболеваний, входящих в группу наследственной дегенерации сетчатки, таких как цилиопатии сетчатки, пигментный ретинит, дегенерация желтого пятна, ретинопатия недоношенных, светоиндуцированная дегенерация сетчатки, отслоение сетчатки, диабетическая ретинопатия и глаукома, в сочетании с векторами для генной терапии. Неограничивающие примеры векторов для генной терапии включают лентивирусные, аденовирусные и аденоассоциированные векторы (AAVs); эти векторы эффективны при доставке нужных генов в сетчатку и пигментный эпителий сетчатки для генной терапии глаз. Предполагают, что при внутриглазной генной терапии наследственной дегенерации сетчатки, связанной с накоплением неправильно свернутых мутантных белков, накопленные белки останутся в эндоплазматическом ретикулуме, тогда как нормальные белки будут экспрессироваться из вектора для генной терапии. При этом необходимо снижать накопление белка в клетке, предпочтительно в ER, с помощью ингибиторов PPP1R15A. Изобретение также касается композиций, содержащих ингибитор PPP1R15A, выбранный из группы соединений формулы (I), гуанабенза и салубринала либо их фармацевтически приемлемых солей, в сочетании с внутриглазной генной терапией.
Лизосомные болезни накопления
Лизосомные болезни накопления представляют собой группу из примерно 50 редких наследственных метаболических заболеваний, которые возникают из-за дефектов функции лизосом. Дисфункция лизосом обычно является следствием недостаточности одного из ферментов, необходимых для метаболизма липидов, гликопротеинов или так называемых мукополисахаридов. Примеры лизосомных болезней, которые можно лечить с помощью описанных здесь ингибиторов PPP1R15A формулы (I), включают, без ограничения, GM2-ганглиозидоз с дефицитом активатора, альфа-маннозидоз, аспартилглюкоз-аминурия, болезнь накопления сложных эфиров холестерина, цистиноз, болезнь Данона (Danon), болезнь Фабри, болезнь Фарбера, болезнь Ниманна-Пика, фукозидоз, галактосиалидоз, болезнь Гоше (I, II, III типа), GM1-ганглиозидоз (инфантильный, поздний инфантильный/ювенильный, взрослый/хронический), болезнь I-клеток/муколипидоз, детская болезнь накопления свободной сиаловой кислоты/ISSD, ювенильная недостаточность гексозаминидазы, болезнь Краббе (инфантильная форма, поздняя форма), недостаточность лизосомальной кислой липазы (ранняя форма/поздняя форма), метахроматическая лейкодистрофия, различные мукополисахаридозы (такие как псевдополидистрофия Гурлера (Hurler)/муколипидоз IIIA, мукополисахаридоз I (MPS I) с синдромом Гурлера, MPS I с синдромом Шейе (Scheie), MPS I с синдромом Гурлера-Шейе, MPS II с синдромом Гурлера, синдром Санфилиппо (Sanfilippo) типа A (MPS IIIA), синдром Санфилиппо типа В (MPS IIIB), синдром Санфилиппо типа С (MPS IIIC), синдром Санфилиппо типа D (MPS IIID), болезнь Моркио (Morquio) типа A/MPS IVA, болезнь Моркио типа B/MPS IVB, MPS IX с недостаточностью гиалуронидазы, MPS VI Марото-Лами (Maroteaux-Lamy), MPS VII с синдромом Слая (Sly), мукополилипидоз I/сиалидоз, муколипидоз IIIC, муколипидоз типа IV (множественная недостаточность сульфатазы), болезнь Ниманна-Пика (типа А, В, С), болезнь CLN6 (атипичная поздняя инфантильная, поздняя форма, ранняя ювенильная), болезнь/CLN3 Баттена-Шпильмайера-Вогта /ювенильная, финский вариант поздней инфантильной CLN5, болезнь Янского-Бильшовского (Jansky-Bielschosky)/поздняя инфантильная форма CLN2/TPP1, болезнь Куфса (Kufs/взрослая форма NCL/CLN4, вариант Northern с эпилепсией/поздняя инфантильная форма CLN8, болезнь Сантавуори-Халтия (Santavuori-Haltia)/инфантильная форма CLN1/PPT, бета-маннозидоз, болезнь Помпе/болезнь накопления гликогена типа II, пикнодизостоз, болезнь Сандхоффа (Sandhoff)/GM2-ганглиозидоз (взрослая форма, инфантильная форма, ювенильная форма), болезнь Шиндлера, болезнь Салла (Sall)/болезнь накопления сиаловой кислоты, болезнь Тея-Сакса/GM2-ганглиозидоз и болезнь Вольмана (Wolman). В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении лизосомных болезней накопления, которые являются следствием недостаточности по меньшей мере одного из ферментов, необходимых для метаболизма липидов, гликопротеинов или так называемых мукополисахаридов, причем данный фермент подвергается неправильной укладке в эндоплазматическом ретикулуме (ER). В соответствии с другим предпочтительным воплощением лизосомная болезнь накопления представлена болезнью Гоше.
Амилоидозы
Амилоидоз является неспецифическим термином, который относится к целому ряду различных заболеваний, которые собирательно именуют амилоидозом. Амилоиды - это белки, у которых изменяется вторичная структура, в результате чего они при укладке принимают характерную форму, β-складчатый лист. Когда растворимые в норме белки при укладке превращаются в амилоиды, то они становятся нерастворимыми, откладываются и накапливаются в органах или тканях, нарушая нормальную функцию. Различные типы амилоидоза имеют различные признаки и симптомы в зависимости от того, где и в каких органах агрегируют амилоидные белки. Примеры амилоидозов включают, без ограничения, среди прочего, амилоидоз AL, АН, ALH (амилоид происходит из легкой цепи, тяжелой цепи, тяжелой и легкой цепи антител, соответственно), амилоидоз АА (амилоид происходит из сывороточного белка А), амилоидоз ATTR (амилоид происходит из транстиретина), первичный системный амилоидоз, вторичный системный амилоидоз, старческий системный амилоидоз, семейная амилоидная полинейропатия 1, наследственная церебральная амилоидная ангиопатия, связанный с гемодиализом амилоидоз, семейная амилоидная полинейропатия III, финский наследственный системный амилоидоз, амилоидоз предсердий, наследственный безнейропатический системный амилоидоз, амилоидоз по месту инъекции, наследственный амилоидоз почек и болезнь Альцгеймера. В соответствии с одним предпочтительным воплощением амилоид представлен бета-амилоидом (Аβ или Abeta), а изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении болезни Альцгеймера.
В соответствии с другим предпочтительным воплощением амилоид представлен HLA-B27 (Colbert et al., 2009 Prion Vol. 3 (1), pp. 15-16), а изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении спондилоартропатии, более предпочтительно анкилозирующего спондилита.
Воспаление
PPP1R15A представляет собой перспективную мишень для контроля воспаления путем блокировки высвобождения воспалительных цитокинов и других секретируемых молекулярных медиаторов, вызывающих патогенные состояния. Неограничивающие примеры заболеваний или состояний, с которыми связано воспаление, которое можно лечить с помощью описанных здесь ингибиторов PPP1R15A формулы (I), включают, без ограничения, инфекционные или неинфекционные воспалительные заболевания легких (такие как сепсис, легочные инфекции, респираторный дистресс-синдром, бронхолегочная дисплазия и др.); инфекционные или неинфекционные воспалительные заболевания других органов, такие как колит, язвенный колит, воспалительная болезнь кишечника, диабетическая нефропатия, геморрагический шок, спондилоартропатия, панкреатит; вызванный воспалением рак (например, раковое перерождение у пациентов с колитом или воспалительной болезнью кишечника); и др.
Примеры таких патогенных воспалительных заболеваний включают аутоиммунные заболевания, наследственные заболевания, хронические заболевания и инфекционные заболевания, такие как аллергия, астма, гиперцитокинемия, в том числе заболевания типа реакции "трансплантат против хозяина" (GVHD), острый респираторный дистресс-синдром (ARDS), сепсис, синдром системной воспалительной реакции (SIRS) (см. WO 2011/061340). Предпочтительно инфекционные заболевания выбирают из инфекции вирусом гриппа, инфекции вирусом оспы, инфекции вирусом герпеса, тяжелого острого респираторного синдрома (SARS), инфекции вирусом чикунгунья, инфекции вирусом Западного Нила, инфекции вирусом денге, инфекции вирусом японского энцефалита, инфекции вирусом желтой лихорадки и инфекции вирусом гепатита С.
Предпочтительно аутоиммунные заболевания выбирают из синдрома Шегрена, системной красной волчанки, псориаза, герпетиформного дерматита, витилиго, грибовидного микоза, аллергического контактного дерматита, атопического дерматита, красного плоского лишая, острого лихеноидного и оспоподобного питириаза (PLEVA), артрита, катастрофического антифосфолипидного синдрома.
В соответствии с одним предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении заболеваний, входящих в группу колита, язвенного колита, воспалительной болезни кишечника, панкреатита, сепсиса. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении панкреатита. В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении сепсиса.
В соответствии с другим предпочтительным воплощением изобретение касается ингибиторов PPP1R15A формулы (I) либо их фармацевтически приемлемых солей для применения при лечении спондилоартропатии, более предпочтительно анкилозирующего спондилита.
Метаболические и/или сердечно-сосудистые заболевания, такие как липоматоз, гиперлипидемия, семейная гиперхолестеринемия, ожирение, атеросклероз, гипертония, сердечные заболевания, ишемия миокарда, инсульт, инфаркт миокарда, аортальный стеноз, ангиоспазм, диабет и родственные заболевания, в том числе гипергликемия, нарушение толерантности к глюкозе, гиперинсулинемия (преддиабет), гиперчувствительность к инсулину I типа и диабет II типа, резистентность к инсулину, синдром Уолкотта-Раллисона (Wolcott-Rallison), среди прочего.
В одном предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении атеросклероза. В другом предпочтительном воплощении, соединения формулы (I) предназначаются для применения при лечении заболеваний, входящих в группу гипертонии, сердечных заболеваний, сердечной ишемии, инсульта, инфаркта миокарда, аортального стеноза или ангиоспазма. В одном предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении ишемии миокарда. В другом предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении заболеваний, входящих в группу гипергликемии, нарушения толерантности к глюкозе, гиперинсулинемии (преддиабета), гиперчувствительности к инсулину I и II типа, резистентности к инсулину и синдрома Уолкотта-Раллисона. В другом предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении преддиабета или диабета, более предпочтительно диабета 2-го типа.
Остеопороз
Yokota et al. (ВМС Musculoskeletal disorders 2013, 14, 197) и He et al. (Cellular Signaling 2013, 25, 552-560) показали, что салубринал (Boyce et al., 2005) эффективно блокирует остеопороз на модели у мышей и стимулирует образование костной ткани. Однако салубринал токсичен и не может применяться для лечения больных людей. Напротив, ингибиторы PPP1R15A формулы (I) должны быть безопасными и могут быть полезными при лечении остеопороза. В одном предпочтительном воплощении соединения формулы (I) предназначаются для применения при лечении остеопороза.
Травмы нервной системы
Ohri et al. (Neurobiology of Disease, 2013 Vol. 58 pp. 29-37) показали, что салубринал значительно улучшает локомоцию задних конечностей, что соответствует лучшей сохранности белого вещества и уменьшению апоптоза олигодендроцитов, тем самым улучшая функциональное восстановление после повреждения спинного мозга.
Ингибиторы PPP1R15A формулы (I) по изобретению должны быть безопасными и могут быть полезными для уменьшения потери олигодендроцитов после травматического повреждения спинного мозга и для профилактического и/или терапевтического лечения травм спинного мозга. В одном предпочтительном воплощении соединения формулы (I) предназначаются для профилактического и/или терапевтического лечения травм спинного мозга.
Ишемия, ишемия головного мозга, апноэ во сне
Настоящим изобретением предусмотрены способы применения ингибиторов PPP1R15A формулы (I) по изобретению для предотвращения и/или лечения повреждения тканей в результате повреждения или гибели клеток вследствие некроза или апоптоза. Примеры повреждений нервной ткани включают ишемические и реперфузионные повреждения типа ишемического инсульта головного мозга и черепно-мозговой травмы. В одном предпочтительном воплощении соединения формулы (I) предназначаются для профилактического и/или терапевтического лечения ишемии головного мозга типа ишемического инсульта головного мозга и черепно-мозговой травмы.
Старение
Старение связано с дегенерацией клеток, тканей и органов, что приводит к таким заболеваниям, как рак, сердечно-сосудистая недостаточность, ожирение, сахарный диабет 2-го типа, неалкогольная жировая дистрофия печени и нейродегенеративные заболевания, а также к ухудшению большинства физиологических показателей.
В биологии старение является состоянием или процессом старения. На клеточном уровне старение представляет собой такое явление, когда выделенные клетки проявляют ограниченную способность к делению в культуре (ограничение Хейфлика, открытое Леонардом Хейфликом в 1961 г.), тогда как организменное старение представляет собой старение организма. Старение организма характеризуется снижением способности реагировать на стресс, возрастанием гомеостатического дисбаланса и повышенным риска заболеваний; в частности, с возрастом ухудшается UPR (Naidoo et al., 2008, J Neurosci, 28, 6539-48). Таким образом, усиление положительного эффекта UPR путем ингибирования фосфатазы eIF2α может облегчить связанные с возрастом заболевания. Следовательно, ингибиторы PPP1R15A формулы (I) по изобретению должны быть безопасными и могут быть полезными для предотвращения и/или лечения заболеваний, связанных с продолжительностью жизни или пролиферативной способностью клеток, а также заболеваний, вызываемых или усугубляемых старением клеток у животных, более конкретно у людей.
В соответствии с одним предпочтительным воплощением настоящее изобретение касается соединений из числа:
для применения при лечении и/или профилактике одного или нескольких заболеваний, входящих в группу кистозного фиброза (муковисцидоза), лизосомных болезней накопления, амилоидозов, раковых заболеваний, воспаления, предпочтительно сепсиса, колита и панкреатита, метаболических нарушений, диабета, сердечно-сосудистых заболеваний, остеопороза, травм центральной нервной системы, ишемии, заболеваний сетчатки, сейпинопатий, нейродегенеративных заболеваний, предпочтительно болезни Альцгеймера, болезни Паркинсона, бокового амиотрофическего склероза, болезни Хантингтона, полиглутаминовых и полиаланиновых заболеваний, болезней Шарко-Мари-Тута, лейкодистрофий и рассеянного склероза.
В соответствии с одним воплощением настоящее изобретение также касается соединений из числа вышеприведенных соединений 1-11, а также содержащих их фармацевтических композиций.
Фармацевтические композиции
Для применения в соответствии с настоящим изобретением соединения либо их физиологически приемлемые соли, сложные эфиры или другие физиологически функциональные производные, описанные здесь, могут быть представлены в виде лекарственных форм, содержащих соединения либо их физиологически приемлемые соли, сложные эфиры или другие физиологически функциональные производные, вместе с одним или несколькими фармацевтически приемлемыми носителями и необязательно также другими терапевтическими и/или профилактическими ингредиентами. Носители должны быть приемлемыми в смысле совместимости с другими ингредиентами состава и безвредными для получателей. Фармацевтические композиции могут предназначаться для применения на людях или животных в медицине и ветеринарии.
Примеры таких подходящих наполнителей для самых разных форм фармацевтических композиций, описанных здесь, приведены в "Handbook of Pharmaceutical Excipients", 2nd Edition, (1994), под редакцией A Wade и PJ Weller.
Приемлемые носители или разбавители для терапевтического применения хорошо известны в области фармацевтики и описаны, к примеру, в Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R. Gennaro, editor, 1985).
Примеры подходящих носителей включают лактозу, крахмал, глюкозу, метил-целлюлозу, стеарат магния, маннит, сорбит и др. Примеры подходящих разбавителей включают этанол, глицерин и воду.
Выбор фармацевтических носителей, наполнителей или разбавителей может быть сделан с учетом предполагаемого способа введения и стандартной фармацевтической практики. Фармацевтические композиции могут содержать в качестве или в дополнение к носителям, наполнителям или разбавителям любые подходящие связующие вещества, смазывающие вещества, суспендирующие средства, покрывающие вещества, солюбилизирующие средства, буферы, ароматизаторы, поверхностно-активные средства, загустители, консерванты (в том числе антиоксиданты) и др., а также вещества, включаемые с целью придания композиции изотоничности по отношению к крови предполагаемого получателя.
Примеры подходящих связующих веществ включают крахмал, желатин, такие природные сахара, как глюкоза, безводная лактоза, сыпучая лактоза, бета-лактоза, декстроза, природные и синтетические камеди, такие как аравийская камедь, трагакант или альгинат натрия, карбоксиметилцеллюлозу и полиэтиленгликоль.
Примеры подходящих смазывающих веществ включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и др.
В фармацевтические композиции могут входить консерванты, стабилизаторы, красители и даже ароматизаторы. Примеры консервантов включают бензоат натрия, сорбиновую кислоту и сложные эфиры п-гидроксибензойной кислоты. Также можно использовать антиоксиданты и суспендирующие вещества.
Фармацевтические составы включают формы, пригодные для перорального, местного (в том числе дермального, трансбуккального, глазного и подъязычного), ректального или парентерального (в том числе подкожного, внутрикожного, внутримышечного и внутривенного), интраназального, внутриглазного и легочного введения, например, путем ингаляции. Лекарственные формы, когда это целесообразно, для удобства могут быть представлены в виде дискретных дозовых единиц и могут быть получены любым из способов, хорошо известных в области фармацевтики. Все способы включают стадию приведения активного соединения в контакт с жидкими носителями или тонко измельченными твердыми носителями либо теми и другими, а затем, при необходимости, формование продукта в желательную форму.
Фармацевтические составы, пригодные для перорального введения, в которых носителем является твердое вещество, наиболее предпочтительно представлены в виде стандартных дозовых форм, таких как болюсы, капсулы или таблетки, содержащие заданное количество активного соединения. Таблетки получают прессованием или формованием, необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки могут быть получены путем таблетирования активного соединения в соответствующей машине в сыпучем виде типа порошка или гранул, необязательно в смеси со связующим веществом, скользящим веществом, инертным разбавителем, смазывающим веществом, поверхностно-активным средством или диспергирующим средством. Формованные таблетки могут быть получены путем формования активного соединения с инертным жидким разбавителем. Таблетки необязательно могут иметь покрытие, а если без оболочки, то необязательно с насечками. Капсулы могут быть получены путем заполнения активного соединения, по отдельности или в смеси с одним или несколькими вспомогательными ингредиентами, в капсульные оболочки, а затем запечатывания их обычным способом. Облатки аналогичны капсулам, в которых активное соединение вместе с любыми вспомогательными ингредиентами заделано в оболочку из рисовой бумаги. Активное соединение также может быть сформовано в виде диспергируемых гранул, которые, к примеру, можно суспендировать в воде перед введением или насыпать на пищевой продукт. Гранулы могут быть упакованы, например в пакетики-саше. Составы, пригодные для перорального введения, в которых носителем является жидкость, могут быть представлены в виде раствора или суспензии в водной или неводной жидкости или же в виде жидкой эмульсии типа масло-в-воде.
Составы для перорального введения включают дозовые формы с контролируемым высвобождением, например таблетки, в которых активное соединение заделано в соответствующий матрикс, контролирующий высвобождение, или покрыто соответствующей пленкой, контролирующей высвобождение. Такие формы особенно удобны для профилактического применения.
Фармацевтические составы, пригодные для ректального введения, в которых носителем является твердое вещество, наиболее предпочтительно представлены в виде стандартных дозовых форм типа свечей. Подходящие носители включают масло какао и другие материалы, обычно используемые в данной области. Свечи удобно получаются путем смешивания активного соединения с размягченным или расплавленным носителем с последующим охлаждением и заливанием в формочки.
Лекарственные формы, пригодные для парентерального введения, включают стерильные растворы или суспензии активного соединения в водных или маслянистых носителях.
Лекарственные формы по изобретению подходят для глазного введения, в частности, для внутриглазного, интравитреального, топического глазного или окологлазного введения, более предпочтительно для топического глазного или интравитреального введения.
Препараты для инъекций могут быть приспособлены для введения болюсом или непрерывного вливания. Такие препараты обычно представлены в виде стандартных дозовых форм или в многодозовых контейнерах, которые укупоривают после внесения препарата вплоть до использования. С другой стороны, активное соединение может быть в виде порошка, который восстанавливают с подходящим носителем типа стерильной апирогенной воды перед использованием.
Активное соединение также может быть сформировано в виде препаратов-депо пролонгированного действия, которые можно вводить путем внутримышечной инъекции или путем имплантации, например подкожно или внутримышечно. Препараты-депо могут включать в себя, к примеру, подходящие полимерные или гидрофобные материалы либо ионообменные смолы. Такие препараты пролонгированного действия особенно удобны для профилактического применения.
Составы, пригодные для легочного введения через ротовую полость, составляют таким образом, чтобы в бронхиальное дерево получателя попадали частицы, содержащие активное соединение, желательно диаметром от 0,5 до 7 микрон. В одном варианте такие формы имеют вид тонко измельченного порошка, который для удобства может находиться в специальной прокалываемой капсуле для ингалятора, к примеру, из желатина, или же в виде сжатой (self-propelling) формы, содержащей активное соединение, подходящий жидкий или газообразный вытеснитель и необязательно еще другие ингредиенты, такие как ПВА и/или твердый разбавитель. Подходящие жидкие вытеснители включают пропан и хлорфторуглеводороды, а подходящие газообразные вытеснители включают двуокись углерода. Можно использовать и такие сжатые формы, в которых активное соединение распыляется в виде капелек раствора или суспензии.
Такие сжатые формы аналогичны известным в этой области и могут быть получены существующими методами. Обычно они помещаются в контейнере, снабженном срабатывающим вручную или автоматически клапаном с желательными характеристиками распыления; предпочтительно это клапан дозировочного типа, выпускающий фиксированный объем, к примеру 25-100 мкл при каждом срабатывании.
В другом варианте активное соединение может находиться в виде раствора или суспензии для распылителя или пульверизатора, в котором для получения мелких капель аэрозоля для ингаляции используется воздушная струя или ультразвуковое воздействие.
Составы, пригодные для интраназального введения, включают препараты, которые в общем аналогичны описанным выше для легочного введения. При распылении такие формы должны давать частицы диаметром от 10 до 200 микрон, что позволит им оставаться в носовой полости; это осуществляется, по необходимости, при использовании порошка с частицами соответствующего размера или путем выбора надлежащего клапана. Другие подходящие формы включают крупные порошки с частицами диаметром от 20 до 500 микрон для введения путем быстрой ингаляции через носовой проход из контейнера, приставленного близко к носу, и капли в нос, содержащие от 0,2 до 5% мас. активного соединения в водном или маслообразном растворе или суспензии.
Фармацевтически приемлемые носители хорошо известны специалистам в этой области и включают, без ограничения, 0,1 М и предпочтительно 0,05 М фосфатный буфер или 0,8% солевой раствор. Кроме того, такие фармацевтически приемлемые носители могут представлять собой водные или неводные растворы, суспензии и эмульсии. Примеры неводных растворителей: пропиленгликоль, полиэтиленгликоль, растительные масла типа оливкового масла и органические сложные эфиры для инъекций типа этилолеата. Водные носители включают воду, спиртовые/водные растворы, эмульсии или суспензии, в том числе солевой раствор и буферные среды. Парентеральные носители включают раствор хлорида натрия, раствор Рингера с декстрозой, декстрозу и хлорид натрия, раствор Рингера с лактатом или нелетучие масла. Также могут присутствовать консерванты и другие добавки, такие, к примеру, как противомикробные средства, антиоксиданты, хелатирующие вещества, инертные газы и др.
Композиции, пригодные для местного применения, могут быть представлены, к примеру в виде гелей, кремов или мазей. Такие препараты можно наносить, например, на раны или язвы - просто намазать на поверхность раны или язвы либо нанести на подходящий носитель типа бинта, марли, сетки и т.п., которым можно будет покрыть участок, подлежащий обработке.
Также предусмотрены жидкие или порошкообразные формы, которые можно распылять или присыпать прямо на подлежащее обработке место, например на рану или язву. С другой стороны, состав можно распылить или насыпать на носитель типа бинта, марли, сетки и т.п., а затем этот носитель нанести на подлежащий обработке участок.
В соответствии с другим аспектом изобретения предусмотрен способ получения фармацевтических или ветеринарных композиций, как они описаны выше, причем способ включает приведение активного соединения в контакт с носителем, к примеру путем смешивания.
В общем, составы получают путем равномерного и тщательного перемешивания активного средства с жидкими носителями или мелкодисперсными твердыми носителями либо теми и другими, а затем, при необходимости, формования продукта. Изобретение распространяется на способы получения фармацевтических композиций, включающие приведение соединения общей формулы (I) в тесный контакт с фармацевтически или ветеринарно приемлемым носителем или жидким носителем.
Соли/эфиры
Соединения по изобретению могут присутствовать в виде солей или сложных эфиров, в частности фармацевтически и ветеринарно приемлемых солей или сложных эфиров.
Фармацевтически приемлемые соли соединений по изобретению включают в себя подходящие соли с кислотами или основаниями. Обзор подходящих фармацевтических солей приведен в Berge et al., J Pharm Sci, 66, 1-19 (1977). Соли образуются, к примеру, с сильными неорганическими кислотами типа минеральных кислот, например с галоидоводородными кислотами, такими как гидрохлориды, гидробромиды и гидроиодиды, серной кислотой - сульфаты, бисульфаты, гемисульфаты, тиоцианаты, персульфаты, с фосфорной кислотой и сульфокислотами; с сильными органическими карбоновыми кислотами, такими как алканкарбоновыми кислотами, содержащими от 1 до 4 атомов углерода, которые не замещены или замещены (например, галогенами), типа уксусной кислоты; с насыщенными или ненасыщенными дикарбоновыми кислотами, к примеру со щавелевой, малоновой, янтарной, малеиновой, фумаровой, фталевой или тетрафталиевой кислотой; с гидроксикарбоновыми кислотами, к примеру с аскорбиновой, гликолевой, молочной, яблочной, винной или лимонной кислотой; с аминокислотами, например с аспарагиновой или глутаминовой кислотой; с бензойной кислотой; или с органическими сульфоновыми кислотами, такими как (С1-С4)-алкил- или арилсульфоновыми кислотами, которые не замещены или замещены (к примеру, галогенами), типа метан- или п-толуолсульфоновой кислоты. Соли, которые не являются фармацевтически или ветеринарно приемлемыми, все-таки могут быть полезными в качестве промежуточных соединений.
Предпочтительными солями являются, к примеру, ацетат, трифторацетат, лактат, глюконат, цитрат, тартрат, малеат, малат, пантотенат, адипат, альгинат, аспартат, бензоат, бутират, диглюконат, циклопентанат, глюкогептанат, глицерофосфат, оксалат, гептаноат, гексаноат, фумарат, никотинат, пальмоат, пектинат, 3-фенилпропионат, пикрат, пивалат, пропионат, тартрат, лактобионат, пиволат, камфорат, ундеканоат и сукцинат, соли органических сульфокислот, таких как метансульфонат, этансульфонат, 2-гидроксиэтан-сульфонат, камфорсульфонат, 2-нафталинсульфонат, бензолсульфонат, п-хлорбензол-сульфонат и п-толуолсульфонат; и соли неорганических кислот, таких как гидрохлорид, гидробромид, гидроиодид, сульфат, бисульфат, гемисульфат, тиоцианат, персульфат, соли фосфорной и сульфоновой кислоты. В соответствии с предпочтительным воплощением соль представлена ацетатом.
Эфиры образуются из органических кислот либо спиртов/гидроксидов, в зависимости от того, какая функциональная группа будет этерифицирована. Органические кислоты включают карбоновые кислоты, такие как алканкарбоновые кислоты, содержащие от 1 до 12 атомов углерода, которые не замещены или замещены (например, галогенами), типа уксусной кислоты; насыщенные или ненасыщенные дикарбоновые кислоты, к примеру щавелевая, малоновая, янтарная, малеиновая, фумаровая, фталевая или тетрафталиевая кислота; гидроксикарбоновые кислоты, к примеру аскорбиновая, гликолевая, молочная, яблочная, винная или лимонная кислота; аминокислоты, например аспарагиновая или глутаминовая кислота; бензойную кислоту; или органические сульфоновые кислоты, такие как (С1-С4)-алкил- или арилсульфоновые кислоты, которые не замещены или замещены (например, галогенами), типа метан- или п-толуолсульфоновой кислоты. Подходящие гидроксиды включают неорганические гидроксиды, такие как гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия. Спирты включают алкановые спирты, содержащие 1-12 атомов углерода, которые могут быть не замещены или замещены, например, галогенами.
Энантиомеры/таутомеры
Во всех аспектах настоящего изобретения, изложенных выше, изобретение включает, где это целесообразно, все энантиомеры, диастереомеры и таутомеры соединений по изобретению. Специалисты в данной области смогут распознать соединения, которые обладают оптическими свойствами (одним или несколькими хиральными атомами углерода) или таутомерными характеристиками. Соответствующие энантиомеры и/или таутомеры могут быть выделены/получены методами, известными в данной области техники. Энантиомеры характеризуются абсолютной конфигурацией своих хиральных центров и описываются правилами R/S-старшинства Кана-Ингольда-Прелога. Такие правила хорошо известны в данной области (например, см. Advanced Organic Chemistry, 3rd edition, ed. March J., John Wiley and Sons, New York, 1985).
Таким образом, соединения формулы (I) также включают в себя таутомерные формы по формуле:
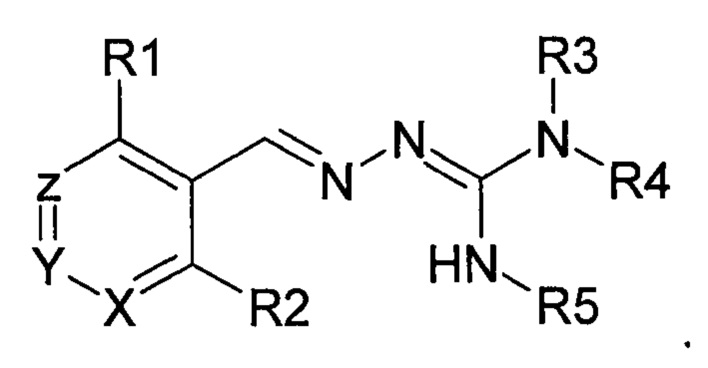
В качестве иллюстративного примера представлена таутомерная форма из примера 1:
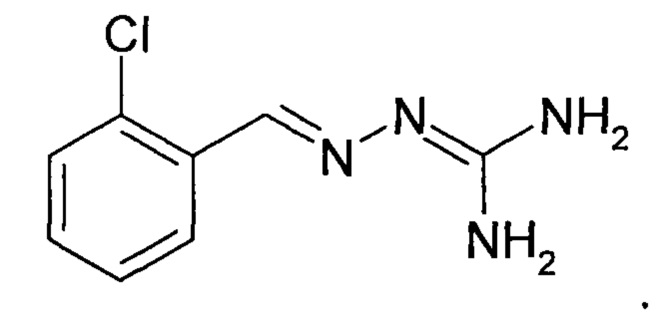
Соединения по изобретению, содержащие хиральные центры, могут применяться в виде рацемической смеси или обогащенной энантиомерами смеси, или же рацемическая смесь может быть разделена с помощью хорошо известных методов и индивидуальные энантиомеры могут применяться по отдельности.
Стереоизомеры и геометрические изомеры
Некоторые соединения по изобретению могут существовать в виде стереоизомеров и/или геометрических изомеров, например они могут обладать одним или несколькими асимметричными и/или геометрическими центрами и поэтому могут существовать в виде двух или нескольких стереоизомеров и/или геометрических форм. Настоящим изобретением предусматривается применение всех индивидуальных стереоизомеров и геометрических изомеров данных ингибиторных средств и их смесей. Термины, используемые в формуле изобретения, охватывают эти формы при условии, что данные формы сохраняют соответствующую функциональную активность (хотя и не обязательно в той же степени).
Настоящее изобретение также включает в себя все подходящие изотопные варианты этих средств либо их фармацевтически приемлемых солей. Изотопные варианты средств по настоящему изобретению либо их фармацевтически приемлемых солей определяется как такие, у которых по меньшей мере один атом заменен атомом, имеющим такой же атомный номер, но другую атомную массу, которая отличается от атомной массы, обычно встречающейся в природе. Примеры изотопов, которые можно вводить в средства и их фармацевтически приемлемые соли, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 17О, 18О, 31Р, 32Р, 35S, 18F и 36Cl, соответственно. Некоторые изотопные варианты средств и их фармацевтически приемлемых солей, например те, в которые включен радиоактивный изотоп типа 3Н или 14С, применимы при исследовании тканевого распределения лекарственных препаратов и/или субстратов. Особенно предпочтительны третированные, то есть 3Н, и изотопы углерод-14, то есть 14С, из-за легкости их получения и детектирования. Кроме того, замещение такими изотопами, как дейтерий, то есть 2Н, может давать определенные терапевтические преимущества вследствие большей метаболической стабильности получаемых веществ, например большего периода полураспада in vivo или потребности в меньших дозах, поэтому они могут быть предпочтительными в некоторых обстоятельствах. Например, изобретение включает в себя соединения общей формулы (I), в которых какой-либо атом водорода заменен атомом дейтерия. Изотопные варианты вещество по настоящему изобретению и их фармацевтически приемлемых солей обычно получают с помощью стандартных процедур, используя соответствующие изотопные варианты подходящих реагентов.
Пролекарства
Изобретение также включает в себя соединения по настоящему изобретению в виде пролекарственных форм, т.е. ковалентно связанных соединений, которые высвобождают исходное активное лекарственное средство общей формулы (I) in vivo. Такими пролекарствами обычно являются соединения по изобретению, у которых одна или несколько соответствующих групп подверглись модификации таким образом, чтобы эта модификация могла быть устранена при введении в организм человека или млекопитающего. Устранение обычно осуществляется ферментом, который естественным образом присутствует в таком организме, хотя возможно и введение второго вещества вместе с таким пролекарством, чтобы осуществлять устранение in vivo. Примеры таких модификаций включают сложные эфиры (например, любые из описанных выше), при этом устранение может осуществляться эстеразами и пр. Специалистам в данной области должны быть хорошо известны и другие такие системы.
Сольваты
Настоящее изобретение также включает в себя сольватные формы соединений по настоящему изобретению. Термины, используемые в формуле изобретения, охватывают эти формы.
Полиморфные формы
Изобретение также касается соединений по настоящему изобретению в виде различных кристаллических форм, полиморфных форм и безводных форм. В фармацевтической промышленности хорошо известно, что химические соединения могут быть выделены в любой из этих форм при небольшом изменении способа очистки и/или при выделении из растворителей, используемых при синтетическом получении таких соединений.
Способы введения
Фармацевтические композиции по настоящему изобретению могут быть приспособлены для ректального, интраназального, внутрибронхиального, местного (в том числе трансбуккального, подъязычного и глазного введения, в частности для внутриглазного, интравитреального, топического глазного или окологлазного введения), вагинального или парентерального (в том числе подкожного, внутримышечного, внутривенного, внутриартериального и внутрикожного), внутрибрюшинного или интратекального введения. Предпочтительно составы вводятся перорально. Составы для удобства могут быть представлены в виде стандартной дозовой формы, т.е. в виде дискретных порций, содержащих стандартную дозу или несколько доз или часть стандартной дозы. Для примера, состав может быть в виде таблеток и капсул с замедленным высвобождением, и могут быть получены любым способом, хорошо известным в области фармацевтики.
Составы для перорального введения в настоящем изобретении могут быть представлены в виде дискретных единиц типа капсул, желатиновых капсул, капель, облаток, пилюль или таблеток, содержащих заданное количество активного средства; в виде порошка или гранул; в виде раствора, эмульсии или суспензии активного средства в водной жидкости или неводной жидкости; или в виде жидкой эмульсии типа масло-в-воде или жидкой эмульсии типа вода-в-масле; или же в виде болюса и т.п. Предпочтительно эти композиции содержат от 1 до 250 мг, более предпочтительно от 10 до 100 мг и еще более предпочтительно от 1 до 100 мг активного ингредиента на 1 дозу.
В отношении композиций для перорального введения (например, таблеток и капсул) термин "приемлемый носитель" охватывает носители типа распространенных наполнителей, например связующие вещества, к примеру сироп, гуммиарабик, желатин, сорбит, трагакант, поливинилпирролидон (повидон), метилцеллюлоза, этилцеллюлоза, натриевая карбоксиметилцеллюлоза, гидроксипропилметилцеллюлоза, сахароза и крахмал; заполнители и носители, к примеру кукурузный крахмал, желатин, лактоза, сахароза, микрокристаллическая целлюлоза, каолин, маннит, дикальциевый фосфат, хлорид натрия и альгиновая кислота; и смазывающие вещества, такие как стеарат магния, стеарат натрия и стеараты других металлов, стеарат глицерина, стеариновая кислота, жидкий силикон, тальк, воск, масла и коллоидная двуокись кремния. Также можно использовать такие ароматизаторы, как мята перечная, винтергреневое масло, вишневый ароматизатор и др. Может потребоваться добавление красителя, чтобы дозовую форму было легко идентифицировать. Таблетки также могут быть покрыты оболочкой с помощью методов, хорошо известных в данной области.
Таблетки получают прессованием или формованием, необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки могут быть получены путем таблетирования активного соединения в соответствующей машине в сыпучем виде типа порошка или гранул, необязательно в смеси со связующим веществом, скользящим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим средством. Формованные таблетки могут быть получены путем формования в соответствующей машине порошкообразной смеси соединения, пропитанной инертным жидким разбавителем. Таблетки необязательно могут быть покрыты оболочкой или насечками и составлены таким образом, чтобы обеспечить медленное или контролируемое высвобождение активного средства.
Другие составы, пригодные для перорального введения, включают леденцы, содержащие активное средство в ароматизированной основе, обычно в сахарозе и гуммиарабике или трагаканте; пастилки, содержащие активное средство в инертной основе типа желатина и глицерина либо сахарозы и гуммиарабика; и жидкости для полоскания рта, содержащие активное средство в подходящем жидком носителе.
Другие формы введения включают растворы или эмульсии, которые могут вводиться внутривенно, внутриартериально, интратекально, подкожно, внутрикожно, внутрибрюшинно, внутримышечно, в глаза, топически или около глаз, которые готовят из стерильных или стерилизуемых растворов.
Фармацевтические композиции по настоящему изобретению также могут быть в виде свечей, вагинальных свечей, суспензий, эмульсий, лосьонов, мазей, кремов, гелей, спреев, растворов или присыпок.
Альтернативным средством трансдермального введения является применение кожных пластырей. Например, активный ингредиент может быть включен в крем, состоящий из водной эмульсии полиэтиленгликоля или жидкого парафина. Активный ингредиент также может быть включен, в концентрации от 1 до 10% мас., в мазь, состоящую из белого воска или белого мягкого парафина в качестве основы, вместе с такими стабилизаторами и консервантами, которые могут потребоваться.
Дозировка
Рядовой специалист в данной области сможет легко определить надлежащую дозировку одной из композиций по изобретению для введения субъекту без излишних экспериментов. Как правило, врач должен определить такую дозировку, которая будет наиболее подходящей для данного пациента, которая будет зависеть от множества факторов, включая активность используемого соединения, продолжительность действия этого соединения, возраст, вес тела, общее состояние здоровья, пол, диету, способ и время введения, скорость выведения, сочетание лекарственных средств, тяжесть данного заболевания и того, кто проходит лечение. Приведенные здесь дозировки являются типичными для среднего случая. Конечно, бывают отдельные случаи, когда требуются более высокие или более низкие интервалы доз, причем эти случаи также находятся в рамках настоящего изобретения.
В соответствии с настоящим изобретением для лечения определенного заболевания нужно вводить эффективное количество соединения общей формулы (I). Конечно, такая дозировка будет затем модифицирована в соответствии со способом введения соединения. Например, для достижения "эффективного количества" при острой терапии предпочтительным является парентеральное введение соединения общей формулы (I). Наиболее эффективным является внутривенное вливание соединения в 5% растворе декстрозы в воде или обычном физрастворе либо аналогичной композиции с подходящими наполнителями, хотя применимо и внутримышечное введение болюсом. Как правило, парентеральная доза должна составлять от 0,01 до 100 мг/кг; предпочтительно от 0,1 до 20 мг/кг, таким образом, чтобы концентрация препарата в плазме поддерживалась на уровне эффективной концентрации. Соединения можно вводить от одного до четырех раз в день на таком уровне, чтобы полная суточная доза составляла от 0,4 до 400 мг/кг/день. Точное количество соединения по изобретению, которое будет терапевтически эффективным, и способ введения, которым такое соединение лучше всего вводится, легко определяется рядовым специалистом в данной области путем сравнения уровня препарата в крови с концентрацией, необходимой для достижения терапевтического эффекта.
Соединения по изобретению также можно вводить пациентам перорально, таким образом, чтобы концентрация препарата была достаточна для достижения одного или нескольких приведенных здесь терапевтических показаний. Как правило, фармацевтическая композиция, содержащая соединение, вводится перорально в дозе от 0,1 до 50 мг/кг, в зависимости от состояния пациента. Предпочтительно пероральная доза составляет от 0,1 до 20 мг/кг.
При введении соединений по настоящему изобретению в соответствии с настоящим изобретением не ожидается никаких неприемлемых токсикологических эффектов. Соединения по изобретению, которые могут обладать хорошей биодоступностью, можно протестировать одним из нескольких биологических методов, чтобы определить, какая концентрация соединения потребуется для достижения заданного фармакологического эффекта.
Комбинации
В одном особенно предпочтительном воплощении одно или несколько соединений по изобретению вводят в комбинации с одним или несколькими другими активными средствами, например существующими лекарствами, доступными на рынке. В таких случаях соединения по изобретению можно вводить последовательно, одновременно или поочередно с одним или несколькими другими активными средствами.
Лекарственных средства, в целом, более эффективны при их применении в комбинации. В частности, комбинированная терапия желательна для того, чтобы избежать наложения основных токсичностей, механизмов действия и механизмов резистентности. Кроме того, большинство лекарств также желательно вводить в максимально переносимых дозах с минимальными промежутками времени между такими дозами. Главное преимущество комбинирования лекарственных средств состоит в том, что оно может вызывать аддитивные или возможные синергические эффекты за счет биохимических взаимодействий, а также может уменьшить появление резистентности.
Благоприятные комбинации могут быть предложены путем изучения ингибиторной активности исследуемых соединений со средствами, известными или предполагаемыми как полезные для лечения определенного заболевания. Эта процедура также может применяться для определения порядка введения лекарств, то есть до, одновременно или после введения другого. Такая схема введения может относиться ко всем приведенным здесь активным средствам.
В соответствии с предпочтительным воплощением изобретение касается фармацевтических композиций, содержащих ингибитор PPP1R15A формулы (I) или его фармацевтически приемлемую соль и соединение, повышающее экспрессию и/или активность белка BiP, а также фармацевтически приемлемый носитель и/или наполнитель (см. WO 2013/124484). Предпочтительно соединение, усиливающее экспрессию и/или активность белка BiP, выбирают из группы, состоящей из вальпроевой кислоты или ее производных, трихостатина А, лития, 1-(3,4-дигидроксифенил)-2-тиоцианатэтанона и эксендина-4. В соответствии с одним предпочтительным воплощением для белка BiP предпочтительна вальпроевая кислота или ее производное типа 2-ен-вальпроевой кислоты.
В соответствии с одним предпочтительным воплощением изобретение касается фармацевтических композиций, содержащих ингибитор PPP1R15A формулы (I) или его фармацевтически приемлемую соль и соединение, повышающее экспрессию и/или активность белка BiP, а также фармацевтически приемлемый носитель и/или наполнитель, для лечения заболеваний, связанных с путем PPP1R15A и с воздействием неправильной укладки белков, в частности с накоплением неправильно свернутых белков. Предпочтительно заболевание выбирают из группы таупатий, синуклеинопатий, полиглутаминовых и полиаланиновых заболеваний, лейкодистрофий, болезней Шарко-Мари-Тута, сейпинопатий, кистозного фиброза (муковисцидоза), рассеянного склероза, лизосомных болезней накопления, амилоидозов, заболеваний сетчатки, воспаления, метаболических заболеваний, сердечно-сосудистых заболеваний, остеопороза, травм нервной системы, ишемии.
Методы анализа
Следующий аспект изобретения касается применения соединений, как описано выше, при анализе для идентификации дальнейших соединений-кандидатов, способных ингибировать PPP1R15А-РР1.
Предпочтительным методом является метод конкурентного связывания. Более предпочтительно метод конкурентного связывания включает приведение соединения по изобретению в контакт с PPP1R15A-PP1 и соединением-кандидатом и детектирование каких-либо изменений во взаимодействии между соединением по изобретению и PPP1R15A-PP1.
Предпочтительно соединение-кандидат получают путем стандартной модификации соединения по изобретению по принципу SAR (соотношения структура-активность). В настоящем изобретении термин "стандартная модификация по принципу SAR" относится к стандартным методам, известным в данной области для изменения соединений посредством химической дериватизации.
Так, в одном аспекте идентифицированное соединение может выступать в качестве модели (например, шаблона) для разработки других соединений. Используемые при таком тестировании соединения могут быть свободными в растворе, прикрепленными к твердой подложке, находиться на поверхности клеток или внутри клеток. Можно измерять устранение активности или образование комплексов при связывании между соединением и исследуемым веществом.
Анализ по настоящему изобретению может представлять собой скрининг, при котором тестируется целый ряд веществ. В одном аспекте метод анализа по настоящему изобретению представляет собой высокопроизводительный скрининг.
Изобретением также предусмотрено применение конкурентных методов скрининга лекарственных средств, в которых нейтрализующие антитела, способные специфически связываться с соединением, конкурируют с исследуемым соединением за связывание с данным соединением.
Другой метод скрининга предусматривает высокопроизводительный скрининг (HTS) веществ, обладающих подходящим сродством связывания с субстанциями, на основе метода, подробно описанного в WO 84/03564.
Ожидается, что методы анализа по настоящему изобретению подойдут и для мелкомасштабного, и для крупномасштабного скрининга исследуемых соединений, а также при количественном анализе.
Предпочтительно метод конкурентного связывания включает приведение соединения по изобретению в контакт с PPP1R15A-PP1 в присутствии известного субстрата PPP1R15A-PP1 и детектирование каких-либо изменений во взаимодействии между PPP1R15A-PP1 и данным известным субстратом.
В следующем аспекте изобретения предусмотрен способ детектирования связывания лиганда с PPP1R15A-PP1, который включает стадии:
(i) контактирование лиганда с PPP1R15A-PP1 в присутствии известного субстрата;
(ii) детектирование каких-либо изменений во взаимодействии между PPP1R15A-РР1 и данным известным субстратом;
причем данный лиганд представляет собой соединение по изобретению.
Один из аспектов изобретения касается способа, включающего стадии:
(a) проведение анализа методом, описанным выше;
(b) идентификацию одного или нескольких лигандов, способных связываться со связывающим лиганды доменом; и
(c) получение некоего количества данных одного или нескольких лигандов.
В другом аспекте изобретения предусмотрен способ, включающий стадии:
(a) проведение анализа методом, описанным выше;
(b) идентификацию одного или нескольких лигандов, способных связываться со связывающим лиганды доменом; и
(c) получение фармацевтической композиции, содержащей данные один или несколько лигандов.
В следующем аспекте изобретения предусмотрен способ, включающий стадии:
(a) проведение анализа методом, описанным выше;
(b) идентификацию одного или нескольких лигандов, способных связываться со связывающим лиганды доменом;
(c) модификацию данных одного или нескольких лигандов, способных связываться со связывающим лиганды доменом;
(d) проведение анализа методом, описанным выше;
(e) необязательно получение фармацевтической композиции, содержащей данные один или несколько лигандов.
Изобретение также касается лигандов, идентифицированных описанным выше способом.
Следующий аспект изобретения касается фармацевтических композиций, содержащих лиганды, идентифицированные описанным выше способом.
Другой аспект изобретения касается применения лигандов, идентифицированных описанным выше способом, при получении фармацевтических композиций, предназначенных для применения при лечении заболеваний, связанных с накоплением неправильно свернутых белков, как определено выше.
Вышеприведенные способы могут применяться для скрининга на предмет лигандов, применимых в качестве ингибиторов PPP1R15A-PP1.
Соединения общей формулы (I) могут применяться как в качестве лабораторных инструментов, так и в качестве терапевтических средств. В лаборатории некоторые соединения по изобретению применимы для выяснении того, выполняет ли известная или вновь обнаруженная мишень критическую или по крайней мере существенную биохимическую функцию при возникновении или прогрессировании заболевания, то есть в процессе, который обычно называется "проверкой мишени".
Далее настоящее изобретение будет описано с привлечением следующих фигур.
Краткое описание фигур
На фиг. 1 представлена зависимая от дозы защита клеток HeLa соединением 1 по изобретению от ER-стресса, вызванного 6-часовой обработкой туникамицином.
На фиг. 2 представлена зависимая от дозы защита поврежденных гамма-интерфероном олигодендроцитов крыс соединением 1 по изобретению.
На фиг. 3 представлена зависимая от дозы защита поврежденных ротеноном первичных мезенцефалических нейронов крыс соединением 2 по изобретению.
На фиг. 4 представлена зависимая от дозы защита поврежденных бета-амилоидом 1-42 первичных корковых нейронов крыс соединением 2 по изобретению.
На фиг. 5 представлена способность соединения 2 при различных концентрациях и схемах введения предотвращать двигательные дефекты при ALS у мутантных мышей SOD1.
IFB-B: соединение 2 вводили перорально.
BID: введение два раза в день.
QD: введение один раз в день.
На фиг. 6 представлена способность соединения 2 при различных концентрациях и схемах введения предотвращать двигательные дефекты при СМТ-1 А у трансгенных (TG) крыс с гиперэкспрессией РМР-22. Соединение 2 вводили перорально один раз в день.
На фиг. 7 представлена способность соединения 1 при 5 мкМ предотвращать накопление белка DM20 с мутацией Т181Р в клетках 293Т человека.
На фиг. 8 представлена способность соединения 6 предотвращать гибель клеток, связанную с накоплением склонного к неправильной укладке инсулина Akita, при экспрессировании в клетках Min6.
На фиг. 9 представлена способность соединения 1 и соединения 6 при различных концентрациях предотвращать гибель клеток инсулиномы Min6, связанную с накоплением неправильно свернутых белков при 6-часовой обработке туникамицином.
На фиг. 10 представлена способность соединения 1 и соединения 6 при различных концентрациях предотвращать гибель клеток инсулиномы INS1, связанную с накоплением неправильно свернутых белков при 6-часовой обработке туникамицином.
На фиг. 11 представлена способность соединения 2 защищать фоторецепторы от апоптоза и сохранять восприятие света у мышей BBS12-/-. Электроретинограмма (ERG). Средняя разность в процентах между глазом BBS12-/-, обработанным соединением 2 (2,5 мкМ) в сочетании с вальпроевой кислотой (0,2 мМ) либо одним лишь соединением 2 (2,5 мкМ), и глазом, обработанным носителем (PBS). Положительные значения означают увеличение ответа ERG, а отрицательные означают уменьшение ERG по сравнению с обработанным PBS глазом (n=10-14 на группу).
На фиг. 12 представлена способность соединения 2 уменьшать нагруженность белком в эндоплазматическом ретикулуме у мышей BBS12-/-. Трансмиссионная электронная микроскопия (ТЕМ) эндоплазматического ретикулума (ER) в фоторецепторах BBS12-/- в ответ на введение соединения 2 (2,5 мкМ) в сочетании с вальпроевой кислотой (VPA) (0,2 мМ) или PBS. Расширение цистерн ER наблюдается при введении одного лишь PBS (слева), тогда как соединение 2 в комбинации с VPA уменьшает такое расширение (справа) после однократной интравитреальной инъекции.
На фиг. 13 представлена способность соединений 1, 6 и 8 (при 25 мкМ) предотвращать продукцию интерферона 1-го типа в эмбриональных фибробластах мыши при липофекции с поли I:С.
На фиг. 14 показана способность соединения 2 защищать неонатальные кардиомиоциты крыс от вызванного гипоксией апоптоза. На графике представлено процентное содержание апоптотических клеток при измерении методом FACS. Кардиомиоциты подвергали гипоксии (0,3% О2) в течение 36 часов в отсутствие (0 мкМ) или в присутствии указанных концентраций соединения 2 (n=3).
Далее настоящее изобретение будет описано на следующих неограничивающих примерах.
ПРИМЕРЫ
1. Материалы и методы
Соединение 3 приобретали у фирмы Chembridge (ref: 5173161).
Соединение 4 приобретали у фирмы Chemdiv (ref: 0589-0012).
Соединение 5 приобретали у фирмы Chemdiv (ref: 1683-6502).
1.1. Получение соединений по настоящему изобретению
Реагенты и коммерческие соединения приобретали у фирмы Acros Organics, Sigma-Aldrich. Соединения по настоящему изобретению могут быть получены по следующей общей методике.
Общая методика А
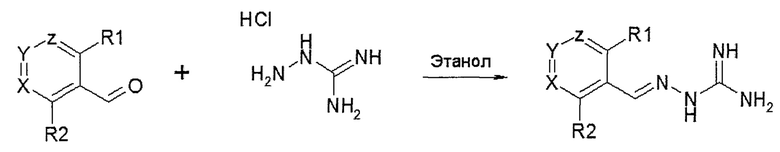
В раствор бензальдегида (1 экв.) в этаноле (300 мл) последовательно добавляли аминогуанидин гидрохлорид (1 экв.) и ацетат натрия (1 экв.) при 25°С. Полученную реакционную смесь нагревали при 80°С в течение следующих ~6 часов. Завершение реакции отслеживали по TLC со смесью дихлорметан/метанол (8/2) в качестве подвижной фазы. По завершении реакции реакционную смесь охлаждали до 25°С и выливали в насыщенный раствор NaHCO3 (700 мл). Образовавшийся осадок отфильтровывали под вакуумом и промывали водой (100 мл). Полученный твердый материал растирали с диэтиловым эфиром (2×25 мл) и высушивали под вакуумом, получая требуемое замещенное производное аминогуанидина.
Следующие соединения получали по общей методике А.
Соединение 1. 2-(2-Хлорбензилиден)гидразинкарбоксимидамид
Получали по общей методике А из 2-хлорбензальдегида (10 г), получая 11,1 г требуемого соединения (выход: 79,6%). 1H-NMR (DMSO-d6): δ (ppm) 5.66 (s, 2H), 6.05 (brs, 2H), 7.27 (m, 2H), 7.40 (m, 1H), 8.14 (dd, 1H), 8.27 (s, 1H); MS (ESI+): m/z=197,2 [M+H]+.
Соединение 2. 2-(2-Хлорбензилиден)гидразинкарбоксимидамид ацетат
В суспензию 2-хлорбензальдегида (30,0 г) и аминогуанидина бикарбоната (29,0 г) в метаноле (450 мл) добавляли уксусную кислоту (30 мл) при 25°С. Реакционную смесь перемешивали при 70°С в течение 30 минут. Завершение реакции отслеживали по TLC со смесью дихлорметан/метанол (8/2) в качестве подвижной фазы. По завершении реакции реакционную смесь охлаждали до 25°С и упаривали под вакуумом. Остаток суспендировали в метаноле (250 мл) и удаляли нерастворимый материал фильтрованием. Полученный фильтрат упаривали под вакуумом и описанный выше процесс (суспен-дирование в метаноле + фильтрование) повторяли еще три раза. Затем твердое вещество растирали с диэтиловым эфиром (3×100 мл) и высушивали под вакуумом, получая 46,0 г соли ацетата 2-(2-хлорбензилиден)гидразинкарбоксимидамида (выход: 84,2%). 1Н-NMR (DMSO-d6): δ (ppm) 1.81 (s, 3Н), 7.12 (m, 4H), 7.34 (m, 2H), 7.46 (m, 1H), 8.22 (m, 1H), 8.36 (s, 1H); LC-MS: m/z=197,2 [M+H]+.
Соединение 6. 2-[(3-Хлорпиридин-4-ил)метилиден]гидразинкарбоксимидамид
Получали по общей методике А из 2-хлорбензальдегида (0,5 г), получая 0,16 г требуемого соединения (выход: 23%). 1H-NMR (DMSO-d6): δ (ppm) 6.00 (brs, 2H), 6.32 (brs, 2Н), 8.10 (d, 1Н), 8.14 (s, 1H), 8.35 (dd, 1H), 8.52 (s, 1H); MS (ESI+): m/z=198,0 [M+H]+.
Соединение 7. 2-[(3-Хлорпиридин-4-ил)метилиден]гидразинкарбоксимидамид ацетат
В суспензию 3-хлоризоникотинальдегида (2,0 г) и аминогуанидина бикарбоната (2,12 г) в метаноле (28 мл) добавляли уксусную кислоту (2 мл) при 25°С. Реакционную смесь перемешивали при 70°С в течение ~2 часов. Завершение реакции отслеживали по TLC со смесью дихлорметан/метанол (8/2) в качестве подвижной фазы. По завершении реакции неочищенную смесь охлаждали до 25°С и упаривали под вакуумом. Твердое вещество растирали со смесью метанол:диэтиловый эфир (9:1) (4×50 мл) и высушивали под вакуумом, получая 2,0 г соли ацетата 2-[(3-хлорпиридин-4-ил)метилиден]гидразин-карбоксимидамида (выход: 55,1%). 1H-NMR (DMSO-d6): δ (ppm) 6.01 (brs, 2H), 6.48 (m, 4H), 8.12 (d, 1H), 8.16 (s, 1H), 8.38 (dd, 1H), 8.54 (s, 1H); MS (ESI+): m/z=198,1 [M+H]+.
Соединение 8. 2-(2-Хлор-6-фторбензилиден)гидразинкарбоксимидамид ацетат
В суспензию 2-хлор-6-фторбензальдегида (1,5 г) и аминогуанидина бикарбоната (1,29 г) в метаноле (22 мл) добавляли уксусную кислоту (1,5 мл) при 25°С. Реакционную смесь перемешивали при 70°С в течение ~1 часа. Завершение реакции отслеживали по TLC со смесью дихлорметан/метанол (8/2) в качестве подвижной фазы. По завершении реакции смесь охлаждали до 25°С и упаривали под вакуумом. Полученное твердое вещество растирали со смесью метанол:диэтиловый эфир (9:1) (3×50 мл) и высушивали под вакуумом, получая 2,2 г соли ацетата 2-(2-хлор-6-фторбензилиден)гидразинкарбо-ксимидамида (выход: 84,8%). 1H-NMR (DMSO-d6): δ (ppm) 1.89 (s, 3Н), 6.13 (brs, 4H), 7.24 (m, 1H), 7.33 (m, 2H), 8.17 (s, 1H); MS (ESI+): m/z=215,1 [M+H]+.
Соединение 9. 2-(2-Хлор-4-метилбензилиден)гидразинкарбоксимидамид
Получали по общей методике А из 2-хлор-4-метилбензальдегида (0,2 г), получая 255 мг требуемого соединения (выход: 93,8%). 1H-NMR (DMSO-d6): δ (ppm) 2.29 (s, 3Н), 5.60 (brs, 2H), 6.00 (brs, 2H), 7.10 (d, 2H), 7.27 (s, 1H), 8.02 (d, 1H), 8.24 (s, 1H); MS (ESI+): m/z=210,9 [M+H]+.
Соединение 10. 2-(2-Хлор-5-метилбензилиден)гидразинкарбоксимидамид
Получали по общей методике А из 2-хлор-5-метилбензальдегида (0,2 г), получая 156 мг требуемого соединения (выход: 57,4%). 1H-NMR (DMSO-d6): δ (ppm) 2.30 (s, 3Н), 5.64 (brs, 2H), 6.06 (brs, 2H), 7.07 (d, 2H), 7.27 (d, 1H), 7.97 (s, 1H), 8.24 (s, 1H); MS (ESI+): m/z=210,9 [M+H]+.
Соединение 11. 2-(2-Хлор-3-метилбензилиден)гидразинкарбоксимидамид
Получали по общей методике А из 2-хлор-3-метилбензальдегида (0,2 г), получая 226 мг требуемого соединения (выход: 83,1%). 1H-NMR (DMSO-d6): δ (ppm) 2.17 (s, 3Н), 5.64 (brs, 2H), 6.03 (brs, 2H), 7.18 (t, 2H), 7.24 (d, 1H), 7.99 (s, 1H), 8.37 (s, 1H); MS (ESI+): m/z=210,9 [M+H]+.
Выбранные соединения по изобретению приведены ниже в таблице
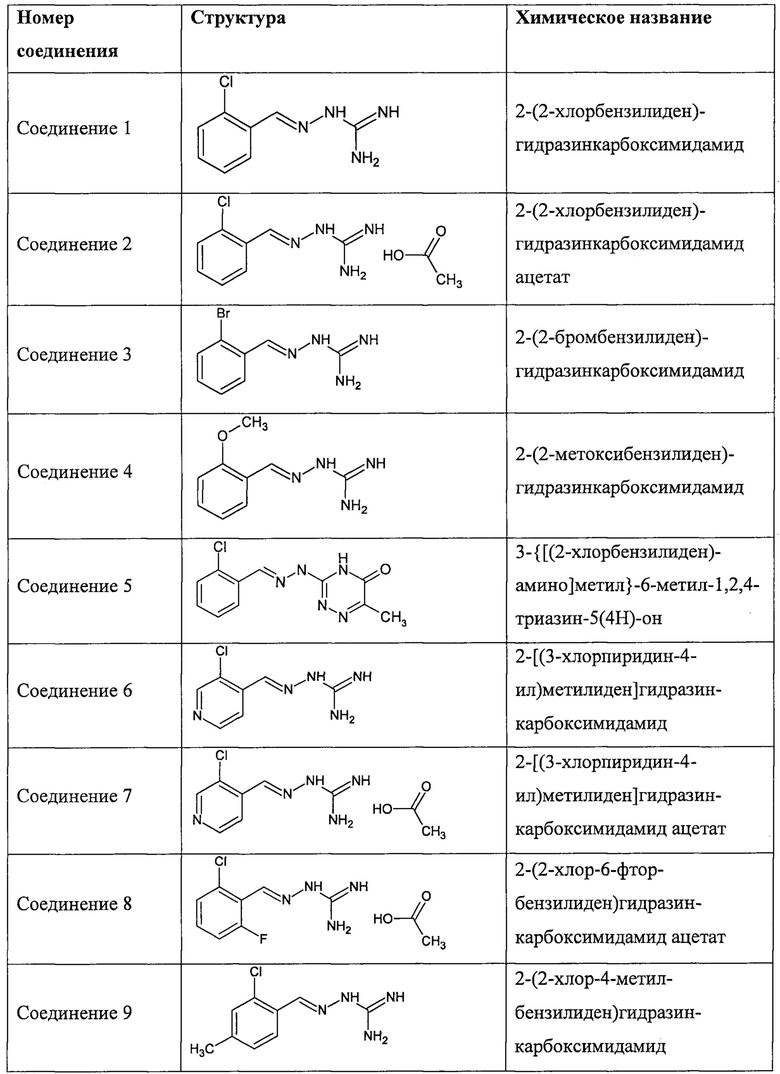
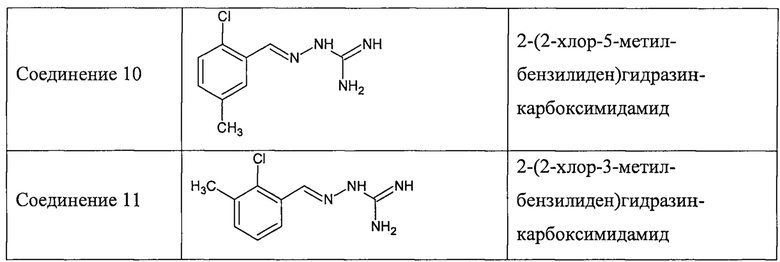
В некоторых из приведенных ниже экспериментов можно использовать соли этих соединений.
1.2. Культивирование клеток млекопитающих, конструкции и трансфекция
Клетки HeLa культивировали в минимальной достаточной среде Игла (ЕМЕМ) с добавлением глутамина, пирувата натрия, заменимых аминокислот, пенициллина и стрептомицина (Lonza), содержащей 10% фетальной телячьей сыворотки (FBS) (Biowest).
Клетки 293Т культивировали в модифицированной Дульбекко среде Игла (DMEM) с добавлением пенициллина, стрептомицина, глутамина (Lonza) и 10% фетальной телячьей сыворотки (FBS) (Biowest).
Клетки Min6 культивировали в среде DMEM с добавлением пенициллина, стрептомицина, глутамина, пирувата натрия, 50 мкМ β-меркаптоэтанола и 15% фетальной телячьей сыворотки (FBS) (Biowest).
Клетки INS1 культивировали в среде RPMI с добавлением пенициллина, стрептомицина, глутамина, пирувата натрия, 50 мкМ β-МЕ и 10% фетальной телячьей сыворотки (FBS) (Biowest).
Каждую линию клеток содержали при 37°С в атмосфере с 5% СО2.
Последовательности открытых рамок считывания (ORF) для PLP1, DM20 и инсулина человека получали от фирмы Life Technologies (Invitrogen) (IOH41689, IOH5252 и IOH7334, соответственно). Клонирование конструкций в экспрессирующую плазмиду pDEST26 (Invitrogen) проводили с помощью смеси Gateway® LR Clonase™ II Enzyme Mix (Invitrogen). Мутации в ORF проводили с помощью набора QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) (мутация T181P для ORFs PLP1 и DM20, Akita (C96Y) для ORF инсулина).
Экспрессирование генов в клетках млекопитающих осуществляли посредством нуклеофекции с помощью системы Amaxa™ 4D-Nucleofector™ System (Lonza) или трансфекции с помощью липофектамина (Life Technologies).
1.3. Цитопротекция от ER-стресса
Этот метод описан в Tsaytler et al. (Science 2011).
Клетки HeLa культивировали в минимальной достаточной среде Игла (ЕМЕМ) с добавлением глутамина, пирувата натрия, заменимых аминокислот, пенициллина и стрептомицина, содержащей 10% фетальной телячьей сыворотки (FBS), при 37°С в атмосфере с 5% CO2.
Клетки высеивали в 96-луночные планшеты при плотности 17000 клеток/мл за день до обработки. ER-стресс вызывали добавлением 5 мкг/мл туникамицина (Sigma-Aldrich) вместе с ингибиторами фосфатаз (0,5-10 мкМ). Через 6 ч среду заменяли свежей средой и поддерживали цитопротекцию добавлением ингибиторов фосфатаз (0,5-10 мкМ). Через 48 или 72 ч после обработки туникамицином оценивали жизнеспособность клеток по восстановлению WST-8 в формазан с помощью набора Cell Counting Kit-8 (Sigma) в соответствии с рекомендациями поставщика.
Цитопротекцию от ER-стресса измеряли по величине цитопротекторного эффекта в сравнении с контрольным соединением гуанабензом (Tsaytler et al., Science 2011) после ER-стресса:
'-' нет цитопротекторного эффекта;
'+' меньший цитопротекторный эффект по сравнению с гуанабензом;
'++' аналогичный цитопротекторный эффект по сравнению с гуанабензом.
В таблице 1 суммированы результаты по цитопротекторному действию различных соединений по изобретению в сравнении с гуанабензом после стресса, вызванного 6-часовой обработкой туникамицином.
1.4. Оценка скорости трансляции в не подвергавшихся стрессу клетках
Клетки HeLa (100000 клеток/мл) высеивали в 6-луночные планшеты за 24 ч перед каждым экспериментом и либо оставляли без обработки, либо обрабатывали соединениями (50 мкМ) в течение 2,5, 5 и 9 ч. Культуральную среду заменяли средой DMEM без метионина (Invitrogen) за 30 мин до добавления соединений. За один час до каждой временной точки в культуральную среду добавляли 50 мкМ Click-iT® AHA (L-азидо-гомоаланин) (Invitrogen) для маркировки вновь синтезированного белка. По окончании каждой временной точки клетки промывали ледяным PBS и собирали путем диссоциации трипсином (Lonza), а затем лизировали в 50 мМ буфере трис-HCl, содержащем 1% SDS (Sigma) и ингибиторы протеаз и фосфатаз (Sigma). Образцы белка конъюгировали с алкинбиотином (Invitrogen) с помощью набора Click-iT® Protein Reaction Buffer Kit (Invitrogen). Образцы денатурировали при 70°C в течение 10 мин, разделяли на подготовленных гелях ECL 4-20% (GE Healthcare) и переносили на нитроцеллюлозные мембраны (GE Healthcare). Включенный во вновь синтезированные белки алкинбиотин, конъюги-рованный с Click-iT® AHA, детектировали с помощью стрептавидина с HRP (Gentex). Проявляли путем инкубации в ECL Prime (GE Healthcare) и считывали хемилюминесценцию на приборе Fusion Solo 3S (Vilber Lourmat).
1.5. Оценка скорости трансляции в подвергавшихся стрессу клетках
Проводили обработку так же, как при измерении трансляции в не подвергавшихся стрессу клетках, за исключением того, что вместе с соединениями добавляли туникамицин (5 мкг/мл).
1.6. Функциональный анализ GPCR для адренергических α2А-рецепторов (метод определения CellKey)
Агонистическую активность соединений оценивали на клетках СНО, эндогенно экспрессирующих рецептор альфа-2А человека, и определяли по измерению их влияния на модуляцию импеданса методом определения CellKey.
Клетки высеивали в 96-луночные планшеты при плотности 6×104 клеток/лунку в буфере HBSS (Invitrogen) + 20 мМ HEPES (Invitrogen) с 0,1% BSA и оставляли уравновешиваться на 60 мин при 28°С перед началом эксперимента. Планшеты помещали в установку и проводили измерения при температуре 28°С. Растворы добавляли одновременно во все 96 лунок с помощью встроенной струйной системы: HBSS (исходный контроль), контрольный агонист при 100 нМ (стимулированный контроль), контрольный агонист (определение ЕС50) или исследуемые соединения. Проводили измерения импеданса в течение 10 мин после добавления лиганда. Стандартным контрольным агонистом служил эпинефрин, который тестировали в каждом эксперименте при нескольких концентрациях для получения кривой концентрация-ответ, по которой вычисляли его значение ЕС50.
Данные типа доза-ответ по исследуемым соединениям анализировали с помощью программы Hill методом нелинейного регрессионного анализа кривых концентрация-ответ, составленных по средним значениям повторных измерений, используя аппроксимацию кривой по уравнению Хилла. Результаты представлены в табл. 1, причем соединения с ЕС50>33,3 мкМ считались не обладающими существенной альфа-2 адренергической активностью.
1.7. Модель болезни рассеянного склероза in vitro: повреждение олигодендроцитов крыс гамма-интерфероном при совместном культивировании с нейронами
Культивирование олигодендроцитов вместе с нейронами
Нейроны/ОРС культивировали, как описано ранее Yang et al. (2005 J Neurosci Methods, 149(1) pp. 50-6), с модификациями. Вкратце, извлекали целый мозг (без мозжечка) из 17-дневных эмбрионов крыс (Wistar, Janvier Labs). Целый мозг обрабатывали 20 мин при 37°С раствором трипсина-ЭДТА (PanBiotech) при конечной концентрации 0,05% трипсина и 0,02% ЭДТА. Диссоциацию останавливали добавлением модифицированной по Дульбекко среды Игла (DMEM) с 4,5 г/л глюкозы (PanBiotech), содержащей ДНКазу I категории II (конечная концентрация 0,5 мг/мл; PanBiotech, серия: Н140508) и 10% фетальной телячьей сыворотки (FCS, Invitrogen, серия: 41Q7218K). Клетки подвергали механической диссоциации пропусканием их три раза через наконечник пипетки на 10 мл. Затем клетки центрифугировали при 515 g в течение 10 мин при 4°С. Супернатант отбрасывали, а осадок ресуспендировали в определенной культуральной среде, состоящей из среды Neurobasal (Invitrogen, серия: 1636133) с добавлением 2% раствора В27 (Invitrogen, серия: 1660670), 2 ммоль/л L-глутамина (PanBiotech), 2% раствора PS, а также 1% FCS и 10 нг/мл тромбоцитарного фактора роста (PDGF-AA, серия: Н131205). Клетки высеивали при плотности 20000 клеток на лунку в 96-луночные планшеты, покрытые PLL (BD Corning, серия: 6614022) и ламинином (Sigma, серия: 083M4034V). Планшеты содержали при 37°С в инкубаторе с увлажнением, в атмосфере воздух (95%) - СО2 (5%). Через каждые 2 дня половину среды заменяли свежей средой. На 18-й день проводили преинкубацию исследуемых соединений за 1 час перед добавлением гамма-интерферона (70 ед./мл, 48 ч, R&D Systems, серия: AAL2214081).
Исследуемые соединения и обработка гамма-интерфероном
На 18-й день культивирования растворяли исследуемые соединения (4 концентрации) в культуральной среде, а затем преинкубировали с олигодендроцитами в совместной культуре с нейронами за 1 час перед добавлением гамма-интерферона (70 ед./мл). Через 1 час после инкубации с исследуемыми соединениями добавляли γ-интерферон в концентрации 70 ед./мл на 48 ч и все еще в присутствии исследуемых соединений. Затем клетки фиксировали холодным раствором этанола (95%, Sigma, серия: SZBD3080V) и уксусной кислоты (5%, Sigma, серия: SZBD1760V) в течение 5 мин при -20°С. После пермеабилизации 0,1% сапонином (Sigma, серия: BCBJ8417V) клетки инкубировали 2 часа с моноклональным антителом против O4, полученном на мышах (Sigma, серия: SLBF5997V),
в разведении 1/1000 в PBS (PAN, серия: 8410813), содержащем 1% FCS и 0,1% сапонина, в течение 2 ч при комнатной температуре. Это антитело выявляли с помощью козьего антитела с Alexa Fluor 488 против IgG мыши (Invitrogen, серия: 1664729) в разведении 1/400 в PBS, содержащем 1% FCS и 0,1% сапонина, в течение 1 ч при комнатной температуре.
Анализ общего числа клеток с O4
Для каждого условия делали по 30 снимков на лунку с помощью ImageXpress (Molecular Device) при 20-кратном увеличении. Все снимки делали в одних и тех же условиях. Анализ общего количества клеток с O4 проводили автоматически с помощью редактора модуля Custom (Molecular Device). Данные выражали в процентах от контроля (без интоксикации, без γ-интерферона = 100%) для отражения вызванного интерфероном-гамма повреждения. Все значения приведены в виде среднего ± SEM (s.e. среднего) (n=6 лунок на каждое условие).
1.8. Модель болезни Паркинсона in vitro: повреждение ротеноном первичных мезенцефалических нейронов крыс
Культивирование мезенцефалических дофаминергических нейронов
Дофаминергические нейроны крыс культивировали, как описано Schinelli et al. (1988 J. Neurochem 50, pp. 1900-07) и Visanji et al. (2008 FASEB J. 22(7) pp. 2488-97). Вкратце, средний мозг, взятый из 15-дневных эмбрионов крыс (Janvier Labs, Франция), препарировали под микроскопом. Извлекали эмбриональный средний мозг и помещали в ледяную среду Лейбовица (L15, PanBiotech, серия: 9310614), содержащую 2% пенициллина-стрептомицина (PS, PanBiotech, серия: 1451013) и 1% бычьего сывороточного альбумина (BSA, PanBiotech, серия: H140603). Для приготовления клеточных препаратов использовали вентральную часть мезенцефалического изгиба - область развивающегося мозга, богатую дофаминергическими нейронами.
Средний мозг диссоциировали трипсинизацией в течение 20 мин при 37°С (0,05% трипсина, 0,02% ЭДТА, PanBiotech, серия: 5890314). Реакцию останавливали добавлением модифицированной по Дульбекко среды Игла (DMEM, PanBiotech, серия: 1300714), содержащей ДНКазу I категории II (0,1 мг/мл, PanBiotech, серия: Н140508) и 10% фетальной телячьей сыворотки (FCS, Gibco, серия: 41Q7218K). Затем клетки подвергали механической диссоциации пропусканием их 3 раза через пипетку на 10 мл. Затем клетки центрифугировали при 180g в течение 10 мин при +4°С через слой BSA (3,5%) в среде L15. Супернатант отбрасывали, а клеточные осадки ресуспендировали в определенной культуральной среде, состоящей из среды Neurobasal (Invitrogen, серия: 1636133) с добавлением В27 (2%, Invitrogen, серия: 1660670), L-глутамина (2 мМ, PanBiotech, серия: 8150713) и 2% раствора PS, а также 10 нг/мл мозгового нейротрофического фактора (BDNF, PanBiotech, серия: H140108) и 1 нг/мл глиального нейротрофического фактора (GDNF, PanBiotech, серия: H130917). Подсчитывали жизнеспособные клетки в цитометре Нойбауэра по исключению трипанового синего. Клетки высеивали при плотности 40000 клеток на лунку в 96-луночные планшеты, покрытые поли-L-лизином (Corning Biocoat, серия: 6614022), и содержали в инкубаторе с увлажнением при 37°С, в атмосфере 5% СО2/95% воздуха. Через каждые 2 дня половину среды заменяли свежей средой.
На 6-й день культивирования среду удаляли и добавляли свежую среду, без или с ротеноном (Sigma, серия: 021M2227V), разведенным до 10 нМ в контрольной среде, по 3 лунки на каждое условие. Исследуемые соединения растворяли в культуральной среде, а затем преинкубировали с мезенцефалическими нейронами за 1 час перед добавлением ротенона.
После 24 часов интоксикации клетки фиксировали 4% раствором параформальдегида (Sigma, серия SLBF7274V) в PBS (PanBiotech, серия: 4831114) при рН 7,3 в течение 20 мин при комнатной температуре. Клетки дважды промывали в PBS, а затем пермеабилизовали и блокировали неспецифические сайты раствором PBS, содержащим 0,1% сапонина (Sigma, серия: BCBJ8417V) и 1% FCS, в течение 15 мин при комнатной температуре. Затем клетки инкубировали с мышиным моноклональным антителом против тирозингидроксилазы (ТН, Sigma, серия: 101М4796) в разведении 1/10000 в PBS, содержащем 1% FCS, 0,1% сапонина, в течение 2 ч при комнатной температуре. Это антитело выявляли с помощью козьего антитела с Alexa Fluor 488 против IgG мыши (Molecular Probes, серия: 1531668) в разведении 1/800 в PBS, содержащем 1% FCS, 0,1% сапонина, в течение 1 ч при комнатной температуре.
Анализ общего числа ТН-положительных нейронов
Помеченные иммунологически культуры автоматически исследовали с помощью ImageXpress (Molecular Device, США). Для каждого условия автоматически анализировали по 20 полей на лунку (что составляет ~80% от общей поверхности лунки) из 3-х лунок. Общее количество нейронов с ТН определяли автоматически с помощью редактора модуля Custom (Molecular Devices, США). Данные выражали в процентах от контроля (без интоксикации, без ротенона = 100%) для отражения вызванного ротеноном повреждения. Все значения приведены в виде среднего ± SEM (s.e. среднего) 1-й культуры (n=3 лунки на 1 условие на 1 культуру).
1.9. Модель болезни Альцгеймера in vitro: повреждение первичных корковых нейронов крыс бета-амилоидом 1-42
Культивирование корковых нейронов крыс
Корковые нейроны крыс культивировали, как описано Singer et al. (1999 J. Neuroscience 19 pp. 2455-63) и Callizot et al. (2013 J.Neurosci. Res. 91 pp. 706-16).
Беременных самок (Wistar; Janvier Labs) на 15-й день беременности забивали путем смещения шейных позвонков. Извлекали эмбрионы и сразу же помещали в ледяную среду L15 Лейбовица (PanBiotech, серия: 9310614) с 2% раствора пенициллина (10000 ед./мл) и стрептомицина (10 мг/мл) (PS; PanBiotech, серия: 1451013) и 1% бычьего сывороточного альбумина (BSA; PanBiotech, серия: Н140603). Кору головного мозга обрабатывали 20 мин при 37°С раствором трипсина-ЭДТА (PanBiotech, серия: 5890314) при конечной концентрации 0,05% трипсина и 0,02% ЭДТА. Диссоциацию останавливали добавлением модифицированной по Дульбекко среды Игла (DMEM) с 4,5 г/л глюкозы (PanBiotech, серия: 1300714), содержащей ДНКазу I категории II (конечная концентрация 0,5 мг/мл; PanBiotech, серия: Н140508) и 10% фетальной телячьей сыворотки (FCS; Invitrogen, серия: 41Q7218K). Клетки подвергали механической диссоциации пропусканием их три раза через наконечник пипетки на 10 мл. Затем клетки центрифугировали при 515 g в течение 10 мин при 4°С. Супернатант отбрасывали, а осадок ресуспендировали в определенной культуральной среде, состоящей из среды Neurobasal (Invitrogen, серия: 1636133) с добавлением 2% раствора В27 (Invitrogen, серия: 1660670), 2 ммоль/л L-глутамина (PanBiotech, серия: 8150713), 2% раствора PS и 10 нг/мл мозгового нейротрофического фактора (BDNF; PanBiotech, серия: Н140108). Подсчитывали жизнеспособные клетки в цитометре Нойбауэра по исключению трипанового синего. Клетки высеивали при плотности 30000 клеток на лунку в 96-луночные планшеты, покрытые поли-L-лизином (Corning Biocoat, серия: 6614022), и культивировали при 37°С в инкубаторе в атмосфере воздух (95%) - СО2 (5%). Через каждые 2 дня среду заменяли. Корковые нейроны подвергали интоксикации растворами А-бета (см. ниже) после 11 дней культивирования.
Исследуемые соединения и обработка бета-амилоидом 1-42
Препарат бета-амилоида 1-42 получали в соответствии с процедурой, описанной Callizot et al., 2013. Вкратце, растворяли пептид 1-42 бета-амилоида (Bachem, серия: 1014012) в приведенной выше определенной культуральной среде, лишенной сыворотки, при начальной концентрации 40 мкМ. Этот раствор перемешивали 3 дня при 37°С в темноте и использовали сразу же после разведения его должным образом в культуральной среде до используемых концентраций.
Исследуемые соединения растворяли в культуральной среде, а затем преинкуби-ровали с первичными корковыми нейронами за 1 час до добавления бета-амилоида 1-42. Препарат бета-амилоида 1-42 добавляли до конечной концентрации 20 мкМ (включая и ~2 мкМ токсичных олигомеров по данным WB) при разведении в контрольной среде в присутствии соединений. После 24-часовой интоксикации клетки фиксировали холодным раствором этанола (95%, Sigma, серия: SZBD3080V) и уксусной кислоты (5%, Sigma, серия: SZBD1760V) в течение 5 мин при -20°С. После пермеабилизации 0,1% сапонином (Sigma, серия: BCBJ8417V) клетки инкубировали 2 часа с мышиным моноклональным антителом против ассоциированного с микротрубочками белка-2 (МАР-2; Sigma, серия: 063М4802) в разведении 1/400 в PBS (PanBiotech, серия: 4831114), содержащем 1% фетальной телячьей сыворотки (Invitrogen, серия: 41Q7218K) и 0,1% сапонина. Это антитело выявляли с помощью козьего антитела с Alexa Fluor 488 против IgG мыши (Molecular Probe, серия: 1572559) в разведении 1/400 в PBS, содержащем 1% фетальной телячьей сыворотки и 0,1% сапонина, в течение 1 ч при комнатной температуре.
Анализ общего числа нейронов
Помеченные иммунологически культуры автоматически исследовали с помощью ImageXpress (Molecular Device, США) при 20-кратном увеличении. Для каждого условия автоматически анализировали по 20 полей на лунку (что составляет ~80% от общей поверхности лунки) из 3-х лунок. Общее количество нейронов определяли автоматически с помощью редактора модуля Custom (Molecular Devices, США). Данные выражали в процентах от контроля (без интоксикации, без бета-амилоида 1-42=100%) для отражения вызванного А-бета 1-42 повреждения. Все значения приведены в виде среднего ± SEM (s.e. среднего) (n=3 лунки на 1 условие на 1 культуру).
1.10. Модель бокового амиотрофического склероза (ALS) на мышах in vivo: трансгенные мыши SOD1-G93A
Для экспериментов использовали трансгенных (TG) мышей, экспрессирующих мутантный SOD1-G93A человека (гетерозиготный TgN-SOD1-G93A-1Gur; Gurney et al. (1994) Science 264, 1772-1775), и 5 однопометных особей дикого типа. Мышей G93A SOD1 разводили в Charles River Germany путем скрещивания гемизиготных TG-самцов (линия 002726М; B6SJL TG SOD1 × G93A 1GUR/J, JAX) с самками дикого типа (WT) (линия 10012, JAX), полученными из JAX Laboratories, США. Животных разбивали на группы следующим образом (самцов и самок распределяли поровну по опытным группам, т.е. каждая опытная группа должна была содержать равное число самцов и самок).
Трансгенные мыши G93A SOD1:
- 12 трансгенных мышей G93A SOD1, получавших носитель QD (т.е. 1 раз в день) гаважем, начиная с 60-дневного возраста и продолжая вплоть до конечной точки;
- 12 трансгенных мышей G93A SOD1, получавших соединение 2 (1,5 мг/кг) BID (т.е. 2 раза в день) гаважем, начиная с 60-дневного возраста и продолжая вплоть до конечной точки;
- 12 трансгенных мышей G93A SOD1, получавших соединение 2 (3 мг/кг) QD гаважем, начиная с 60-дневного возраста и продолжая вплоть до конечной точки;
- 12 трансгенных мышей G93A SOD1, получавших соединение 2 (3 мг/кг) QD в сочетании с рилузолом (20 мг/кг) гаважем, начиная с 60-дневного возраста и продолжая вплоть до конечной точки;
- 12 трансгенных мышей G93A SOD1, получавших соединение 2 (10 мг/кг) QD гаважем, начиная с 60-дневного возраста и продолжая вплоть до конечной точки.
Поведенческое тестирование
Испытание на вращающемся стержне (ротарод) проводили перед началом дозировки (исходный уровень, день 60) и примерно в дни 75, 90, 105 и 120. Для испытания на ротароде отбирали мышей, рожденных в пределах 2-4 дней. Одна дневная сессия включает одну тренировочную пробу на 5 мин при 4 об/мин на установке Rotarod (AccuScan Instruments, Columbus, США). Через 30 мин животные последовательно проходили 3 ускоряющиеся тест-пробы по 6 мин с изменением скорости от 0 до 40 об/мин за 360 секунд с промежутками между пробами по меньшей мере в 30 мин. Отмечали время до падения со стержня. Мышей, остававшихся на стержне более 360 сек, снимали, а их время засчитывали как 360 секунд.
1.11. Модель болезни Шарко-Мари-Тута 1А in vivo: трансгенные крысы с гиперэкспрессией РМР-22
Трансгенных крыс СМТ1А получали при скрещивании самцов крыс РМР-22 (Лаборатория проф. Nave, Max-Planck Institut fur experimentelle Medizin,  , Германия) и самок крыс Sprague-Dawley (Elevage Janvier, Франция). Животных размещали и содержали в Key-Obs (
, Германия) и самок крыс Sprague-Dawley (Elevage Janvier, Франция). Животных размещали и содержали в Key-Obs ( , Франция). Процедуры на животных проводили в строгом соответствии с директивой ЕС от 22 сентября 2010 г. (2010/63/UE).
, Франция). Процедуры на животных проводили в строгом соответствии с директивой ЕС от 22 сентября 2010 г. (2010/63/UE).
Животных разбивали на группы следующим образом (в эксперименты включали только самцов):
- 8 трансгенных крыс, получавших носитель гаважем QD (т.е. 1 раз в день), начиная с 5-недельного возраста и продолжая вплоть до конечной точки;
- 8 трансгенных крыс, получавших соединение 2 (1 мг/кг) гаважем QD, начиная с 5-недельного возраста и продолжая вплоть до конечной точки;
- 8 трансгенных крыс, получавших соединение 2 (3 мг/кг) гаважем QD, начиная с 5-недельного возраста и продолжая вплоть до конечной точки.
Поведенческое тестирование
Животных тестировали случайным образом и вслепую для оценки лечения и исхода лечения. Поведенческие эксперименты и показатели при испытании на перекладине проводились и проверялись на объекте Key-Obs исследователями, проводившими обработку слепым методом. Испытание на перекладине проводили на крысах СМТ1А после 3 недель и 5 недель лечения. В испытании на перекладине оценивается мышечная сила у четырех лап и показатели равновесия на неподвижном стержне. Крысу ставили на четырех лапах на середину деревянного стержня (диаметр: 2,5 см; длина: 50 см). Отмечали время, проведенное на перекладине (латентность до падения) при каждой пробе, и количество падений. Проводили пять последовательных проб (макс. 60 сек).
1.12. Модель лейкодистрофий (PMD) in vitro: гиперэкспрессия мутантного PLP1 и DM20 в линии клеток человека
За день до трансфекции высеивали клетки 293Т при плотности 300000 клеток/мл. Клетки 293Т трансфецировали конструкциями с мутантными PLP1 и DM20 с помощью липофектамина 2000 в соответствии с методикой производителя. После трансфекции. клетки обрабатывали соединениями или оставляли без обработки. В качестве контроля клетки трансфецировали нативными формами белков. Через 48 ч получали клеточные лизаты. Оценивали накопление белков методом вестерн-блоттинга.
1.13. Модель диабета 2-го типа in vitro: линии клеток Min6 и INS1
Цитопротекция от ER-стресса
Клетки высеивали в 96-луночные планшеты при плотности 0,5×106 клеток/мл для линии клеток Min6 и 0,4×106 клеток/мл для линии клеток INS1 за день до обработки. ER-стресс вызывали добавлением 2,5 мкг/мл туникамицина (Sigma-Aldrich) вместе с ингибиторами фосфатаз. Через 6 ч среду заменяли свежей средой и поддерживали цитопротекцию добавлением ингибиторов фосфатаз. Через 72 ч после обработки туникамицином оценивали жизнеспособность клеток по восстановлению WST-8 в формазан с помощью набора Cell Counting Kit-8 (Sigma) в соответствии с рекомендациями поставщика.
Защита от накопления склонного к неправильной укладке инсулина Akita
Клетки Min6 подвергали нуклеофекции конструкциями с мутантным инсулином Akita и высеивали в 96-луночные планшеты при плотности 300000 клеток/мл, а через 24 ч обрабатывали клетки соединениями или оставляли без обработки. В качестве контроля клетки подвергали нуклеофекции посторонней плазмидой. Через 6 дней добавляли селективный реагент (G418).
Через 9 дней после обработки оценивали жизнеспособность клеток по восстановлению WST-8 в формазан с помощью набора Cell Counting Kit-8 (Sigma) в соответствии с рекомендациями поставщика.
1.14. Модель воспаления/инфекционного заболевания in vitro: индуцированные поли-I:С эмбриональные фибробласты мыши
Экспериментальные методики
Эмбриональные фибробласты мыши (MEFs) подвергали липофекции с поли-I:С и обрабатывали соединениями по изобретению при двух концентрациях (25 мкМ) в течение 6 ч. После 6-часового культивирования определяли фосфорилирование eIF2α (eIF2α-P) и экспрессию PPP1R15A (GADD34) методом вестерн-блоттинга, а в супернатантах культур определяли продукцию β-интерферона (IFN) I-го типа методом ELISA. В качестве отрицательного и положительного контролей служили контроль без обработки (nt) и поли-LC/DMSO, соответственно.
Поли-I:С (полиинозиновая:полицитидиловая кислота либо натриевая соль полиинозиновой-полицитидиловой кислоты) является иммуностимулятором, который применяется для моделирования вирусных инфекций. Поли-I:С, которая по структуре подобна двухцепочечной РНК, взаимодействует с toll-подобным рецептором 3, который экспрессируется во внутриклеточных компартментах В-клеток и дендритных клеток.
В качестве контрольного ингибиторного соединения использовали гуанабенз (25 мкМ).
Культивирование клеток
MEFs культивировали в среде DMEM с 10% FCS (HyClone, Perbio), 100 ед./мл пенициллина, 100 мкг/мл стрептомицина, 2 мМ глутамина, заменимых аминокислот 1×MEM и 50 мкМ 2-меркаптоэтанола. MEFs обрабатывали в течение указанного времени 10 мкг/мл поли-I:С (InvivoGen) в сочетании с липофектамином 2000 (Invitrogen).
Иммуноблоттинг
Клетки лизировали в 1% Triton Х-100, 50 мМ Hepes, 10 мМ NaCl, 2,5 мМ MgCl2, 2 мМ ЭДТА, 10%) глицерина с добавлением мини-таблеток полного коктейля ингибиторов протеаз (Roche). Определение белка проводили с помощью набора BCA Protein Assay (Pierce). По 25-50 мкг растворимого в Triton Х-100 материала наносили на градиентный 2%-12%) или 8%-й гель для SDS-PAGE, а затем проводили иммуноблоттинг и хемилюми-несцентное детектирование (SuperSignal West Pico Chemi-luminescent Substrate, Pierce). Кроличьи поликлональные антитела, распознающие GADD34 (С-19), получали от Santa Cruz Biotechnology, а антитела против eIF2α[pS52] - от Invitrogen.
ELISA
Определение бета-IFN в супернатантах культур проводили с помощью набора Mouse Interferon Beta ELISA Kit (PBL Interferon Source) в соответствии с инструкциями производителя.
1.15. Симптомы цилиопатии сетчатки и пигментного ретинита in vivo: нокаутные мыши BBS 12
Получение и разведение нокаутных мышей
Мышей Bbs12-/-/J поддерживали на генетическом фоне C57BL/6 (Mockel et al., 2012, J. Biol. Chem. 287 pp. 37483-494). Мышей содержали и разводили в помещениях с контролируемой влажностью и температурой с 12-часовым циклом освещение/темнота со свободным доступом к нормальному корму и воде. Полностью нокаутных мышей Bbs12-/-выявляли посредством генотипирования методом полимеразной цепной реакции (ПЦР) с помощью набора КАРА Mouse Genotyping Kit (кат. №КК7302, Кара Biosystems, Woburn, Massachusetts, США).
Реагенты для интравитреального введения
Раствор для интравитреального введения готовили в стерильных условиях. Исходные растворы: 1,25 мМ соединения 2 и 100 мМ вальпроевой кислоты (VPA), затем разбавляли в PBS, рН 6, получая раствор соединения 2 (2,5 мкМ) + VPA (0,2 мМ) и раствор соединения 2 (2,5 мкМ). Вальпроевую кислоту получали от Sigma-Aldrich (кат. №4543).
Интравитреальное введение
Фенотипы сетчатки глаз у мышей Bbs12-/- и их механизм уже были опубликованы (Mockel et al., 2012). Мышам в постнатальном возрасте 14-16 дней делали интравитреальные инъекции (в стекловидное тело). Операцию проводили под операционным микроскопом. Мышей анестезировали изофлураном. Зрачки у мышей расширяли глазными каплями с 0,3% атропина (Alcon). В полость стекловидного тела со стороны лимба роговицы вставляли иглу 33-го калибра, соединенную с автоматическим дозатором (Hamilton Bonaduz AG, Bonaduz, Швейцария). Положение иглы контролировали под микроскопом. Вводили 1 мкл раствора для обработки в левый глаз мышей и 1 мкл PBS, рН 6, в правый глаз в качестве контроля. Мышей с кровоизлияниями в стекловидном теле или с повреждением сетчатки исключали из анализа.
Электроретинограммы
Электроретинограммы (ERGs) снимали через две недели после интравитреальной инъекции на установке HMsERG (Ocuscience®, Kansas City, Missouri, США). Мышей адаптировали к темноте в течение ночи, а затем анестезировали путем внутрибрюшинной инъекции домитора (7,6 мкг/г массы тела) и кетамина (760 мкг/г массы тела). Зрачки расширяли, как описано выше. Эксперименты проводили при тусклом красном освещении (кат. №R125IRR, Philips, Suresnes, Франция). Использовали стандартную процедуру ERG в соответствии с методикой производителя (Ocuscience®, Kansas City, Missouri, США). Вкратце, методика заключалась в регистрации адаптированной к темноте ERG (скотопической ERG) после фотонных стимулов с интенсивностью от 0,1 до 25 кд⋅с/м2. Результаты ERG усиливали и записывали в цифровом виде с помощью ERG View system 4.3 (Xenotec, Ocuscience®, Kansas City, Missouri, США). Затем измеряли а-волны и b-волны скотопических реакций.
Трансмиссионная электронная микроскопия
Образцы фиксировали погружением в 2,5% глутаральдегид и 2,5% параформальдегид в какодилатном буфере (0,1 М, рН 7,4) со вторичной фиксацией в 1% тетроксиде осмия в 0,1 М какодилатном буфере в течение 1 часа при 4°С и обезвоживали путем проводки через набор спиртов (50, 70, 90, 100%) и окись пропилена по 30 минут каждый раз. Образцы заключали в Epon™ 812 (Sigma-Aldrich, Saint-Louis, Missouri, США). Делали полутонкие срезы толщиной в 2 мкм с помощью ультрамикротома (Leica Ultracut UCT, Leica Biosystems, Wetzlar, Германия), окрашивали их толуидиновым синим и проводили гистологический анализ методом световой микроскопии. Нарезали ультратонкие срезы толщиной в 70 нм, контрастировали уранилацетатом и цитратом свинца и исследовали при 70 кВ под электронным микроскопом Morgagni 268D. Получали снимки в цифровом виде с помощью камеры Mega View III (System Soft Imaging).
1.16. Индуцированный гипоксией апоптоз в культуре неонатальных кардиомиоцитов крыс
Культивирование клеток
Первичные культуры неонатальных кардиомиоцитов крыс получали из желудочков 1-дневных крысят Sprague Dawley (Janvier, Франция). Крыс подвергали эвтаназии и извлекали у них сердце. Сердце нарезали мелкими кусочками (1-2 мм3) и подвергали ферментативному расщеплению с помощью набора Neonatal Heart Dissociation Kit Rat и GentleMACS™ Dissociator (MiltenyiBiotec, Германия). После диссоциации гомогенаты фильтровали (70 мкм), получая суспензию одиночных клеток. Выделенные клетки собирали центрифугированием и ресуспендировали в модифицированной по Дульбекко среде Игла (DMEM), содержащей 10% лошадиной сыворотки (HS), 5% фетальной телячьей сыворотки (FBS) и 1%) пенициллина/стрептомицина. Культуры обогащали миоцитами путем предварительного посева на 90 мин, чтобы истощить популяцию не-миоцитов. Не прикрепившиеся клетки высеивали в 6- или 96-луночные планшеты при соответствующей плотности клеток. Клетки культивировали при 37°С в атмосфере 95% воздуха/5% CO2 в течение 24 ч. Затем культуральную среду заменяли свежей средой DMEM, содержащей 1% FBS и различные концентрации исследуемого соединения, за 30 мин до инкубации в нормальной или гипоксической (N2/CO2, 95%/5%; 0,3% O2) камере для культивирования.
Обработка исследуемым соединением
Очищенные неонатальные кардиомиоциты крыс высеивали в 96-луночный планшет при 106 клеток на 2 мл для экспериментов по проточной цитометрии. Через 24 часа кардиомиоциты обрабатывали при различных концентрациях исследуемого соединения в культуральной среде с 0,1% DMSO. Клетки положительного контроля обрабатывали только культуральной средой (0,1% DMSO). Через 30 минут после начала обработки клетки инкубировали в гипоксической камере для культивирования (N2/CO2, 95%/5%; конечное значение O2: 0,3%) в течение 36 часов. Клетки отрицательного контроля оставляли в нормоксических условиях при 37°С в культуральной среде (1% FBS, 0,1% DMSO) на такое же время.
Измерение апоптотических клеток
По истечении времени обработки проводили проточную цитометрию для измерения содержания апоптотических клеток. Использовали набор для выявления апоптоза Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit фирмы Miltenyi. Клетки дважды промывали PBS и ресуспендировали в буфере для связывания. Добавляли FITC-аннексин V и пропидия йодид в соответствии с методикой производителя. Смесь инкубировали 15 мин в темноте при комнатной температуре, а затем измеряли клеточную флуоресценцию методом проточной цитометрии FACScan.
РЕЗУЛЬТАТЫ
2.1. Цитопротекция и избирательность соединений
Результаты различных анализов с выбранными соединениями по изобретению приведены ниже в табл. 1. В качестве примера, на фиг. 1 представлен цитопротективный эффект соединения 1 после стресса, вызванного воздействием туникамицина.
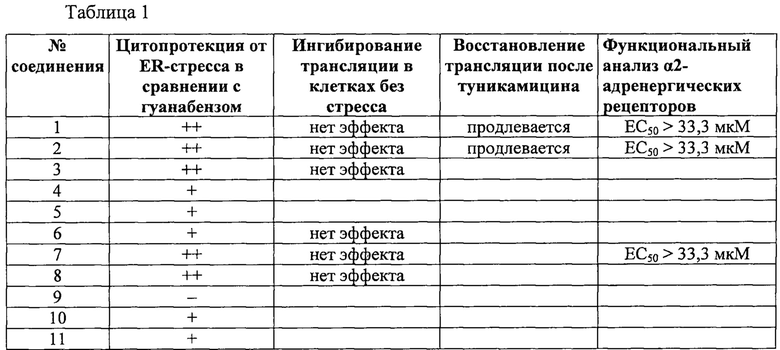
2.2. Рассеянный склероз
На фиг. 2 представлена зависимая от дозы защита поврежденных гамма-интерфероном олигодендроцитов крыс соединением 1 по изобретению.
Эти данные свидетельствуют, что соединения по изобретению являются перспективным эффективным средством лечения рассеянного склероза.
2.3. Болезнь Паркинсона (PD)
На фиг. 3 представлена зависимая от дозы защита поврежденных ротеноном первичных мезенцефалических нейронов крыс соединением 2 по изобретению.
Эти данные свидетельствуют, что соединения по изобретению являются перспективным эффективным средством лечения синуклеопатий и в особенности болезни Паркинсона.
2.4. Болезнь Альцгеймера (AD) и амилоидоз
На фиг. 4 представлена зависимая от дозы защита поврежденных бета-амилоидом 1-42 первичных корковых нейронов крыс соединением 2 по изобретению.
Эти данные свидетельствуют, что соединения по изобретению являются перспективным эффективным средством лечения амилоидоза и в особенности болезни Альцгеймера.
2.5. Боковой амиотрофический склероз (ALS)
На фиг. 5 представлены результаты испытания на ротароде трансгенных мышей SOD1 G93A после 90-дневной обработки соединением 2 по изобретению.
Эти данные свидетельствуют, что соединения по изобретению, а именно соединения 1 и 2, устраняют двигательные дефекты у трансгенных мышей и являются перспективным эффективным средством лечения ALS.
2.6. Болезнь Шарко-Мари-Тута 1А (СМТ-1А)
На фиг. 6 представлены результаты испытания на перекладине у трансгенных крыс с гиперэкспрессией РМР22 после 3-й 5-недельной обработки соединением 2 по изобретению.
Эти данные свидетельствуют, что соединения по изобретению, а именно соединения 1 и 2, устраняют двигательные дефекты у трансгенных крыс с гиперэкспрессией РМР22 и являются перспективным эффективным средством лечения демиелинизирующих заболеваний типа СМТ, более конкретно СМТ1А и СМТ1В.
2.7. Лейкодистрофия: болезнь Пелицеуса-Мерцбахера (PMD)
Были описаны мутации Т181Р и L223P в белках PLP1 и DM20, вызывающие тяжелый фенотип болезни Пелицеуса-Мерцбахера (Strautnieks et al., 1992, Am. J. Hum. Genet. 51(4): 871-878; Gow and Lazzarini, 1996, Nat Genet. 13(4):422-8).
Соединение 1 по изобретению (5 мкМ) предотвращает накопление белка DM20 с мутацией Т181Р, экспрессирующегося в клетках 293Т человека (фиг. 7).
Эти данные свидетельствуют, что соединения по изобретению, а именно соединения 1 и 2, являются перспективным эффективным средством лечения демиелинизирующих заболеваний типа лейкодистрофий, более конкретно PMD.
2.8. Диабет 2-го типа
На фиг. 8 представлены результаты гиперэкспрессии несущего мутацию Akita пре-проинсулина в клетках Min6 при обработке соединением 6 по изобретению.
Соединение 1 и соединение 6 в различных концентрациях предотвращают гибель клеток инсулиномы Min6, связанную с накоплением неправильно свернутых белков, вызванным 6-часовым воздействием туникамицина (фиг. 9).
Соединение 1 и соединение 6 в различных концентрациях предотвращают гибель клеток инсулиномы INS1, связанную с накоплением неправильно свернутых белков, вызванным 6-часовым воздействием туникамицина (фиг. 10).
Эти данные свидетельствуют, что соединения по изобретению являются перспективным эффективным средством лечения преддиабета и диабета, предпочтительно преддиабета 2-го типа и диабета 2-го типа.
2.9. Цилиопатия сетчатки/синдром Барде-Бидля
Соединение 2 защищает фоторецепторы от апоптоза и предохраняет восприятие света (фиг. 11) и уменьшает нагруженность белком в эндоплазматическом ретикулуме (фиг. 12) у мышей ВВS12-/-. По данным ЭРГ, у мышей BBS12-/- проявляется усиление реакции при обработке комбинацией соединения 2 и вальпроевой кислоты (VPA) либо одним лишь соединением 2 (фиг. 11). На фиг. 12 представлен полученный методом трансмиссионной электронной микроскопии репрезентативный снимок ER в фоторецепторах у мышей BBS12-/- в ответ на указанную проведенную обработку или генотип. При введении только PBS наблюдается дилатация, тогда как соединение 2 (2,5 мкМ) в сочетании с VPA (вальпроевая кислота) (0,2 мМ) уменьшают эту дилатацию после одной единственной интравитреальной инъекции.
Эти данные свидетельствуют, что соединения по изобретению в сочетании с соединением, повышающим экспрессию и/или активность белка BIP типа вальпроевой кислоты, являются перспективным эффективным средством лечения цилиопатии сетчатки типа синдрома Барде-Бидля и пигментного ретинита.
Хотя это и не проверяли, авторы предполагают, что такое лечение может способствовать снижению и других форм клеточного стресса типа фотонного стресса.
2.10. Инфекционные и неинфекционные воспалительные заболевания
Нормальная реакция эмбриональных фибробластов мыши (MEFs) на поли-I:С характеризуется экспрессией PPP1R15A, возрастанием eIF2α-P (вариабельным по времени и связанным с уровнем экспрессии PPP1R15A), опосредованным активацией PKR, и выработкой IFN типа I (от 500 до 700 пг/мл). MEFs с нокаутом РРР1R15А-/- не способны вырабатывать этот цитокин в ответ на поли-I:С.
Способность соединений по изобретению ингибировать PPP1R15A оценивали по возрастанию фосфорилирования eIF2α, снижению экспрессии PPP1R15A вследствие его собственного фармакологического ингибирования, что приводит к торможению общего синтеза белка и выработки IFN типа I.
Исследуемые соединения по изобретению оказались эффективными при 25 мкМ, усиливая фосфорилирование eIF2α, снижая экспрессию PPP1R15A и предотвращая продукцию IFN типа I. Для примера, на фиг. 13 представлена способность соединений 1, 6 и 8 (при 25 мкМ) предотвращать продукцию IFN типа I при липофекции MEFs с поли-I:С.
Эти данные свидетельствуют, что соединения по изобретению являются перспективным эффективным средством лечения инфекционных и неинфекционных воспалительных заболеваний.
2.11. Ишемия миокарда
Соединение 2 по изобретению защищает культуры неонатальных кардиомиоцитов крыс от вызванного гипоксией апоптоза (фиг. 14). Эти данные свидетельствуют, что соединения по изобретению являются перспективным эффективным средством лечения ишемии, а именно ишемии миокарда.
Специалистам в данной области должны быть очевидны различные модификации и варианты изобретения, не выходящие за рамки и не отходящие от сущности изобретения. Несмотря на то, что изобретение было описано со ссылкой на конкретные предпочтительные воплощения, следует понимать, что заявленное изобретение не должно неоправданно ограничиваться такими конкретными воплощениями. Так, настоящее изобретение охватывает различные модификации описанных способов осуществления изобретения, которые очевидны специалистам в соответствующих областях.
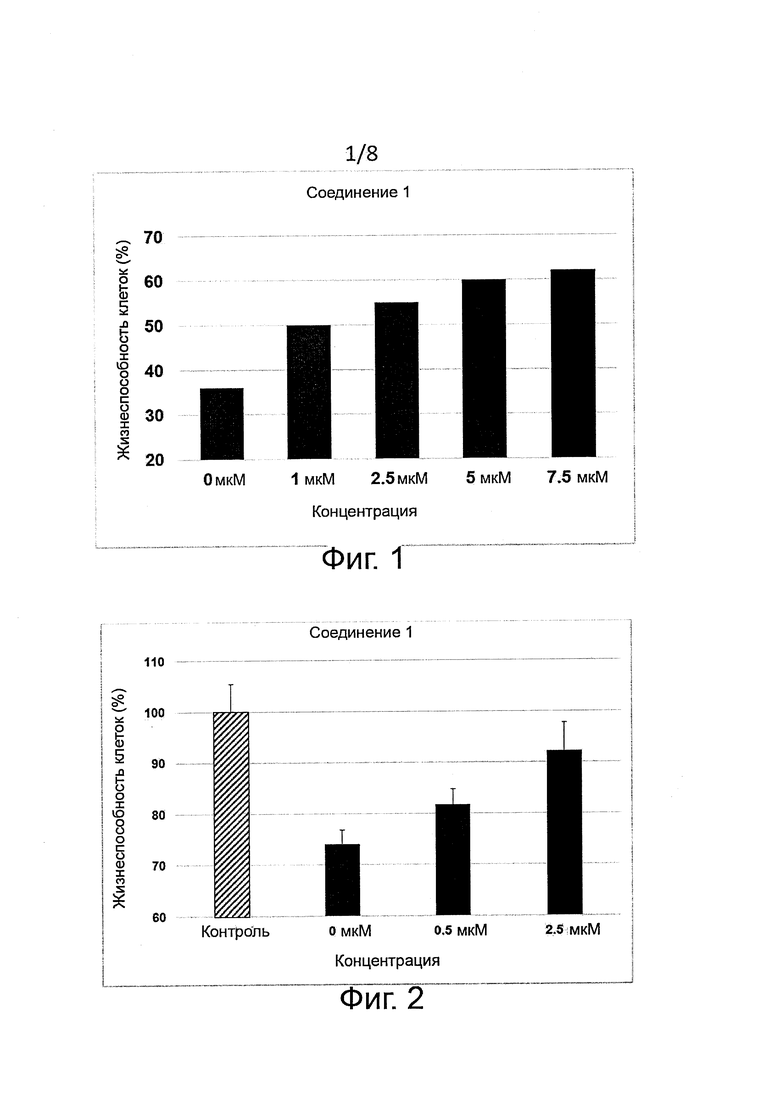
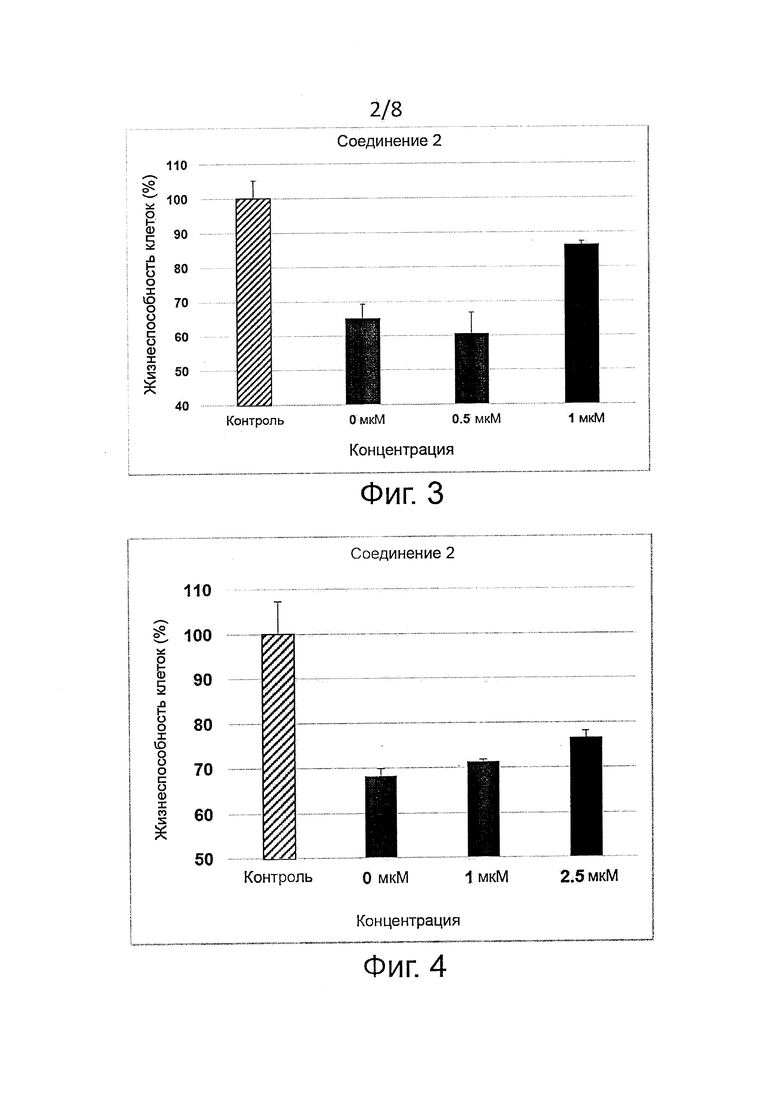
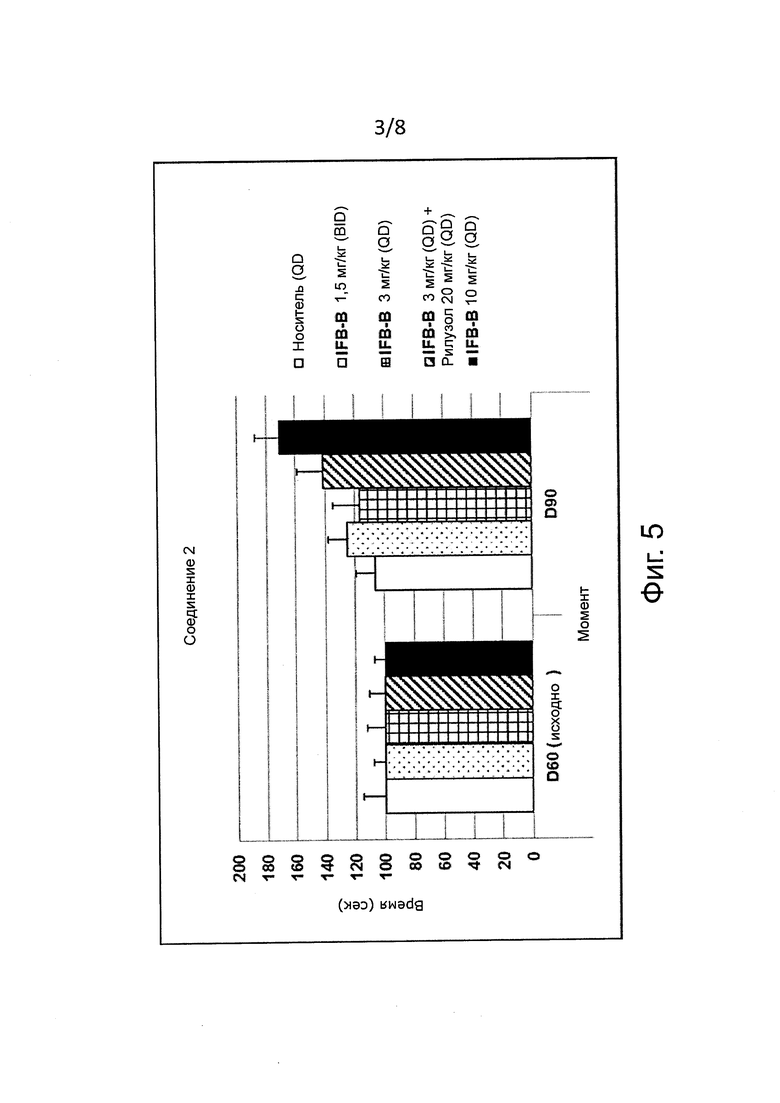
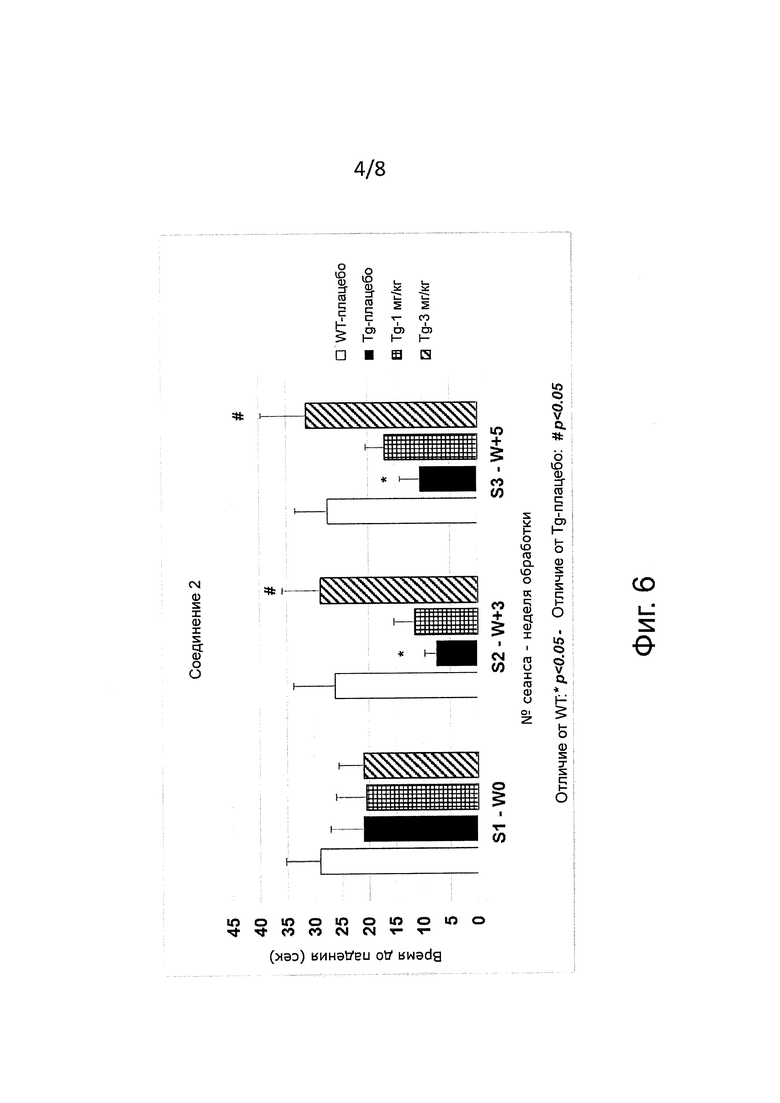
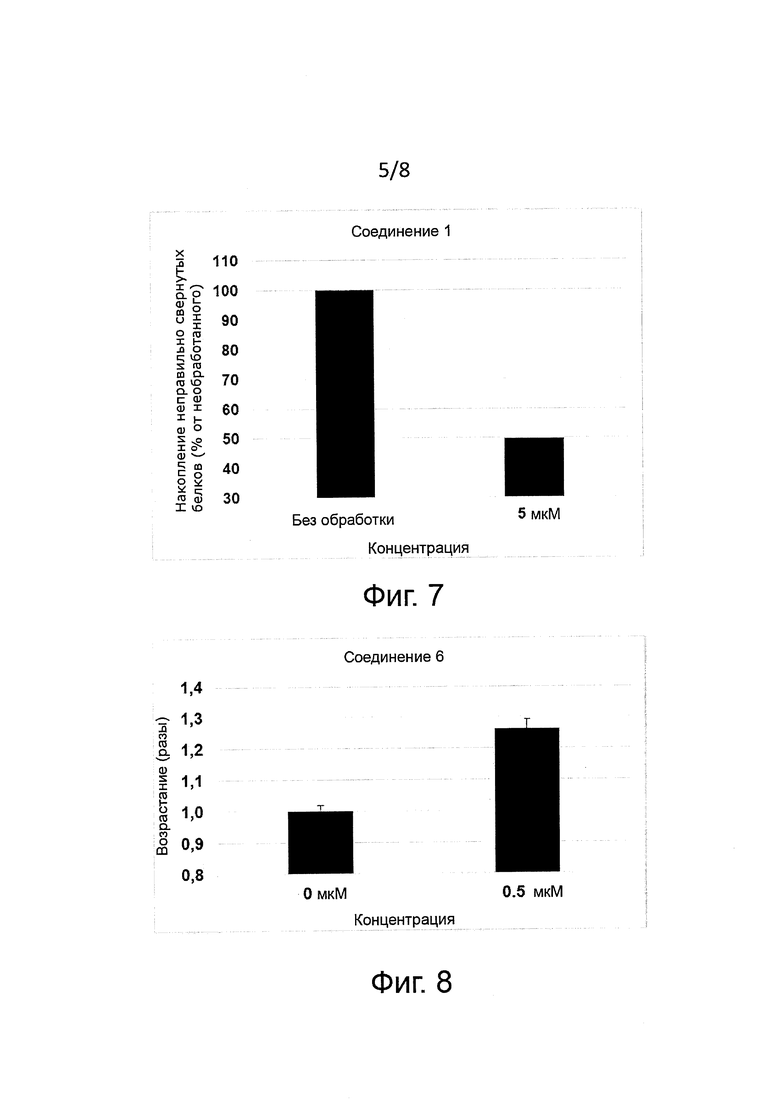
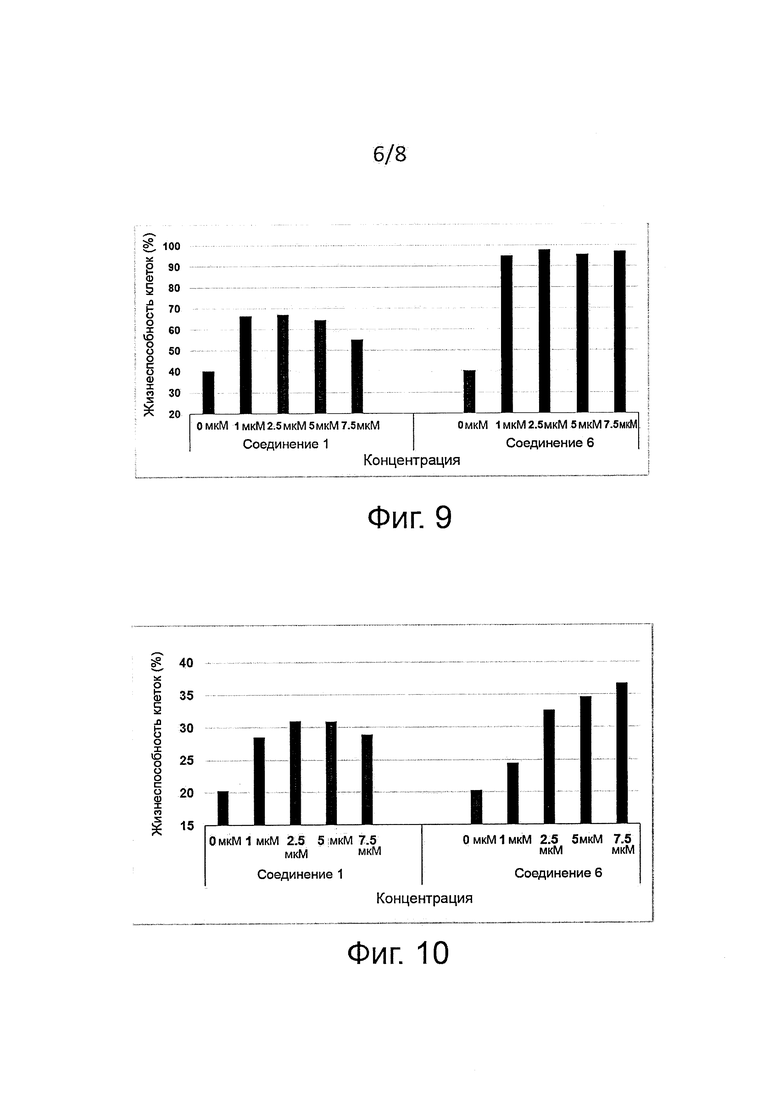
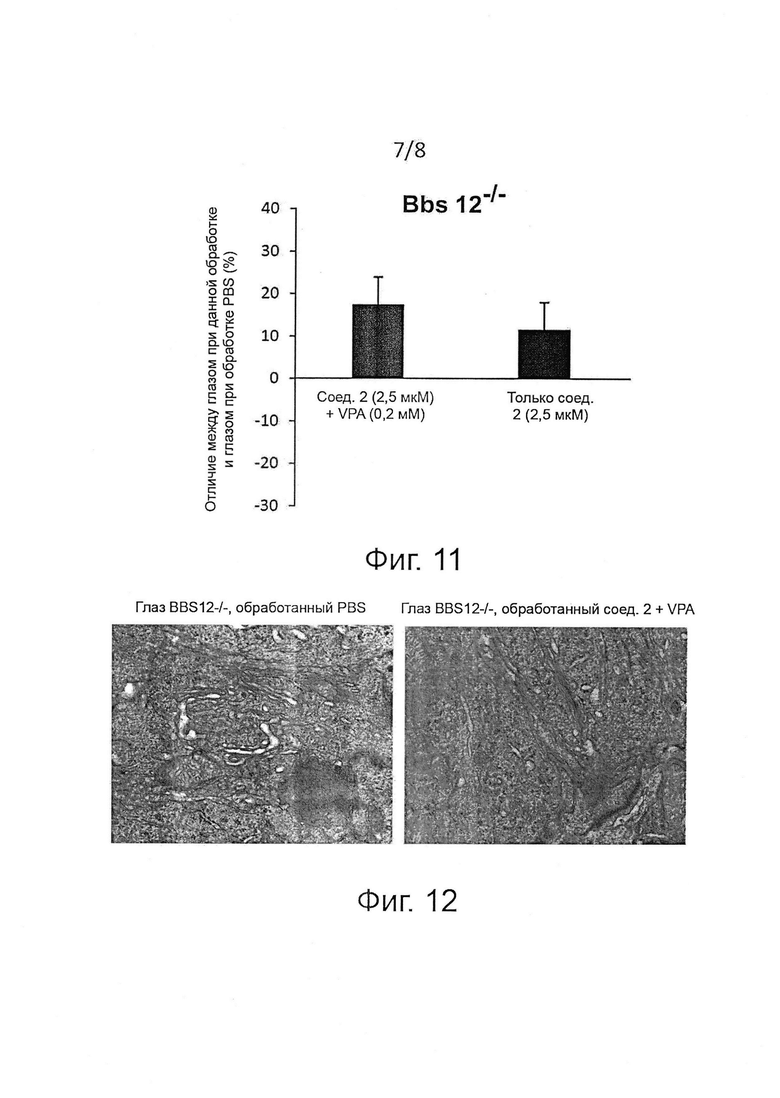
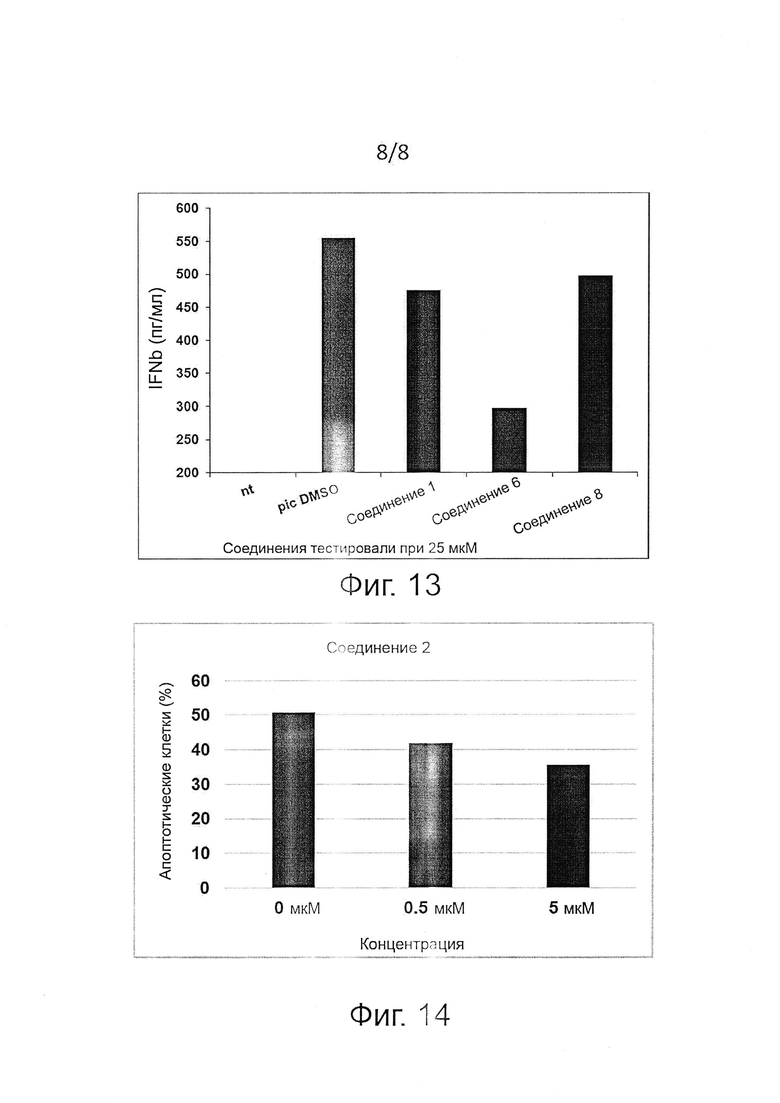
Изобретение относится к применению соединения формулы (I) либо его фармацевтически приемлемой соли или таутомера в качестве ингибитора PPP1R15A. В формуле (I) R1 означает O-C1-12алкил, Cl, F или Br; R2 означает H или F; R3 представляет собой H; R4 представляет собой H; R5 означает H; или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10; каждый R10 независимо выбран из =O и С1-12алкила; X и Z каждый независимо означает CR11, а Y выбран из CR11 и N; R11 означает H или C1-12алкил. Изобретение относится также к применению соединения формулы (I) либо его фармацевтически приемлемой соли или таутомера, где R1 означает Cl, F или Br; R2 означает H или F; R3 представляет собой H; R4 представляет собой H; R5 означает H; или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10; каждый R10 независимо выбран из C1-12алкила; X и Z каждый независимо означает CR11, а Y выбран из CR11 и N; R11 означает H, для лечения заболевания, связанного с воздействием неправильной укладки белков, в особенности с накоплением неправильно свернутых белков, и выбранного из группы лейкодистрофий. 2 н. и 11 з.п. ф-лы, 14 ил., 1 табл., 16 пр.
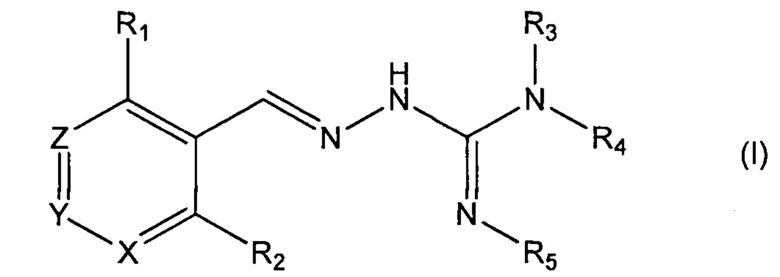
1. Применение соединения формулы (I)
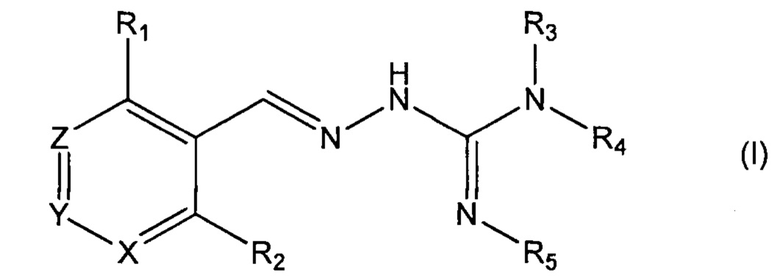
либо его фармацевтически приемлемой соли или таутомера, где
R1 означает O-C1-12алкил, Cl, F или Br;
R2 означает H или F;
R3 представляет собой H;
R4 представляет собой H;
R5 означает H;
или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10;
каждый R10 независимо выбран из =O и С1-12алкила;
X и Z каждый независимо означает CR11, а Y выбран из CR11 и N;
R11 означает H или C1-12алкил;
в качестве ингибитора PPP1R15A.
2. Применение по п. 1, в котором ингибирование PPP1R15A используется для лечения заболевания, связанного с воздействием неправильной укладки белков, в особенности с накоплением неправильно свернутых белков, и выбранного из группы, состоящей из воспалительных состояний; вызванного воспалением рака; муковисцидоза, полиглутаминовых заболеваний (таких как болезнь Мачадо-Джозефа); полиаланиновых заболеваний (таких как глазоглоточная мышечная дистрофия); лейкодистрофий и рассеянного склероза.
3. Применение по п. 2, в котором воспалительное состояние выбрано из сепсиса, синдрома системной воспалительной реакции (SIRS), колита, язвенного колита, воспалительной болезни кишечника, диабетической нефропатии, геморрагического шока, спондилоартропатии и панкреатита.
4. Применение соединения формулы (I)
либо его фармацевтически приемлемой соли или таутомера, где
R1 означает Cl, F или Br;
R2 означает H или F;
R3 представляет собой H;
R4 представляет собой H;
R5 означает H;
или же R4 и R5 соединяются с образованием 5-6-членной насыщенной или ненасыщенной гетероциклической группы, необязательно содержащей 1 или 2 гетероатома, таких как N, в дополнение к тем атомам N, с которыми связаны R4 и R5, причем данная гетероциклическая группа необязательно замещена одной или несколькими группами R10;
каждый R10 независимо выбран из C1-12алкила;
X и Z каждый независимо означает CR11, а Y выбран из CR11 и N;
R11 означает H;
для лечения заболевания, связанного с воздействием неправильной укладки белков, в особенности с накоплением неправильно свернутых белков, и выбранного из группы лейкодистрофий.
5. Применение по любому из пп. 1-4, в котором R1 означает Cl, Br или F, более предпочтительно Cl.
6. Применение по любому из пп. 1-5, в котором R2 означает H.
7. Применение по любому из предыдущих пунктов, в котором Y означает CR11.
8. Применение по любому из предыдущих пунктов, в котором соединение выбрано из следующих:
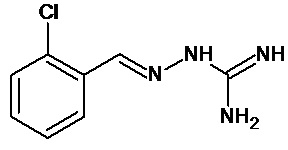
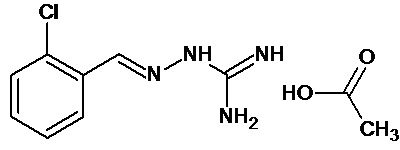
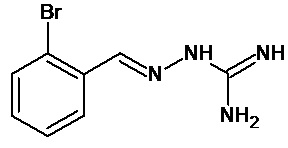
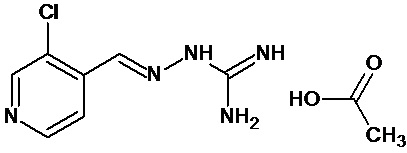
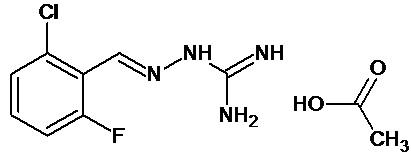
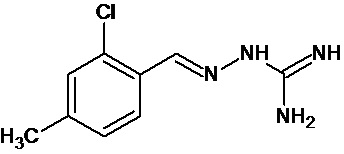
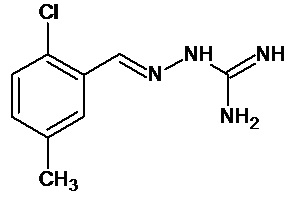
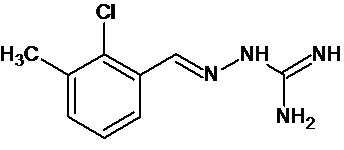
либо их таутомеров или фармацевтически приемлемых солей.
9. Применение по любому из предыдущих пунктов, в котором соединение выбрано из следующих:
либо их таутомеров или фармацевтически приемлемых солей.
10. Применение по п. 8, в котором соединение является соединением 1 либо его фармацевтически приемлемой солью.
11. Применение по любому из пп. 1, 2, 5-10, в котором заболеванием является рассеянный склероз.
12. Применение по любому из пп. 1, 2, 5-10, в котором заболеванием является лейкодистрофия.
13. Применение по любому из пп. 1, 2, 5-10, в котором заболеванием является болезнь Пелицеуса-Мерцбахера.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 3982020 А, 21.09.1976 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| V.N | |||
| SONAR ET AL., (E)-1-[(2-Chlorophenyl)methyleneamino]guanidinium chloride, ACTA CRYST., 2007, E63, o974-o975 | |||
| US 3975533 А, 17.08.1976 | |||
| D | |||
| TRIBOUILLARD-TANVIER ET AL., Antihypertensive drug guanabenz is active in vivo against both | |||

