Перекрестные ссылки на родственные заявки
[1] По настоящей заявке испрашивается приоритет предварительной заявки США № 62/819487, поданной 15 марта 2019 г., предварительной заявки США № 62/819482, поданной 15 марта 2019 г., предварительной заявки США № 62/819472, поданной 15 марта 2019 г., предварительной заявки США № 62/819490, поданной 15 марта 2019 г., предварительной заявки США № 62/819476, поданной 15 марта 2019 г., предварительной заявки США № 62/821660, поданной 21 марта 2019 г., и Международной заявки № PCT/US 2019/039936, поданной 28 июня 2019 г., каждая из которых полностью включена в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
[2] Это изобретение относится к композициям и способам ингибирования CREB-связывающего белка (CBP). Композиции для ингибирования CBP полезны, например, в фармацевтических композициях для лечения определенных форм рака, зависимых от рецепторов андрогенов.
УРОВЕНЬ ТЕХНИКИ
[3] Рост и распространение опухолей, чувствительных к гормонам, зависят от онкогенных сигнальных программ, управляемых соответствующими ядерными рецепторами гормонов. Рецептор андрогенов (AR), ключевой фактор развития рака простаты и подмножеств рака молочной железы, контролирует экспрессию около 100 генов-мишеней, отвечающих за андроген. Экспрессия этих генов-мишеней AR важна для нормального развития тканей и клеточной активности, но может иметь патологические эффекты, которые лежат в основе инициации и прогрессирования опухоли. Прямое нацеливание на биосинтез андрогенов и взаимодействие андрогенов с AR может обеспечить клиническую ценность. Однако приобретенная устойчивость к этим методам лечения может обойти функцию AR, управляемую лигандами, при сохранении постоянной зависимости от управляемых AR транскрипционных программ.
[4] Ядерные рецепторы являются частью мультибелковых комплексов с участием коактиваторов и корепрессоров, которые контролируют влияние ядерного рецептора на гены-мишени, расположенные ниже по течению. В рамках AR-ассоциированного мультибелкового комплекса CBP/P300 являются критическими коактиваторами AR, модифицируя хроматиновое окружение, окружающее ядерный рецептор, для увеличения его внутренней транскрипционной активности и привлечения дополнительных кофакторов. Учитывая совместную регуляцию отношений между AR и CBP/P300, ингибирование активности CBP/P300 предлагает рациональный подход к подавлению AR-зависимых онкогенных программ в AR-зависимых опухолях, таких как рак молочной железы и простаты.
[5] Не существует одобренных терапевтических средств, специально нацеленных на пациентов с метастатической резистентностью к кастрату (mCRPC), для которых антагонисты андрогенов и терапия таксаном оказались неэффективными. К ним относятся пациенты с опухолями, несущими структурно измененные рецепторы андрогенов, включая форму сплайсинга AR-v7, которые продолжают стимулировать транскрипционную программу AR лиганд-независимым образом, на которые не влияют антагонисты андрогенов. Эта популяция представляет собой неудовлетворенную клиническую потребность.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[6] Настоящее изобретение обеспечивает способы и композиции для лечения рака AR+, включая пациентов с диагнозом определенных форм рака AR+, которые устойчивы к другим видам лечения, таких как пациенты, устойчивые к апалутамиду, даролутамиду или энзалутамиду или невосприимчивые к ним (например, пациенты с прогрессированием заболевания или с заболеванием, устойчивым к лечению энзалутамидом), путем введения соединения-ингибитора СВР (например, соединений формулы (I)) пациенту, нуждающемуся в этом. Композиции ингибитора CBP предпочтительно применяются в терапевтически эффективном количестве для ингибирования CBP и антагонизма передачи сигналов рецептора андрогена, что приводит к клиническому преимуществу при AR+ тройном отрицательном раке молочной железы (TNBC) и mCRPC, экспрессирующих форму сплайсинга AR-v7. AR является онкогенным фактором рака простаты, и прогрессирование заболевания в сторону резистентности к кастрации и к лекарствам связано с аберрациями AR, такими как амплификация AR, мутации в LBD и увеличении варианта сплайсинга AR без LBD (AR-v7). AR также экспрессируется в подмножестве TNBC, в котором он заменяет ER и управляет передачей сигналов ER посредством связывания с элементами ответа ER. Наконец, ER является основным онкогенным фактором при раке молочной железы ER+. Устойчивость к гормональной депривации при этом подтипе рака молочной железы может приводить к мутациям ER LBD, ведущим к лиганд-независимому росту. В одном аспекте ингибитор CBP полезен для лечения субъектов, положительных по рецепторам гормонов, с рецидивом рака или злокачественными опухолями, резистентными к гормональной терапии (например, путем антагонизма активности ER у этих субъектов).
[7] Настоящее изобретение частично основано на открытии того, что соединения формулы (I):
 (I)
(I)
и их фармацевтически приемлемые соли, где:
R1 представляет собой H или -OH;
каждый R2 независимо выбран из C1-C6 алкила (например, метила), галогена, -CN и -OR3 (например, метокси), где алкил необязательно замещен одним или более галогенами;
каждый R3 независимо представляет собой H или C1-C6 алкил (например, метил), где алкил необязательно замещен одним или более галогенами; и
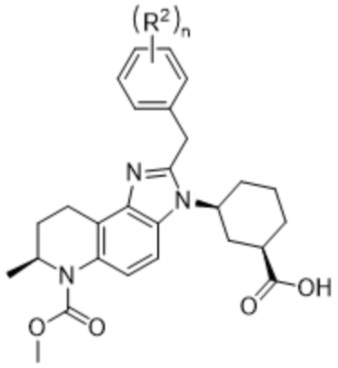
n равно целому числу, выбранному из 0, 1, 2, 3, 4 или 5, где n предпочтительно равно 0, 1, 2 или 3, обеспечивают активную составляющую, полезную для лечения AR-положительного рака, такого как определенные AR-положительные формы рака молочной железы (например, TNBC) и рака простаты (например, CRPC).
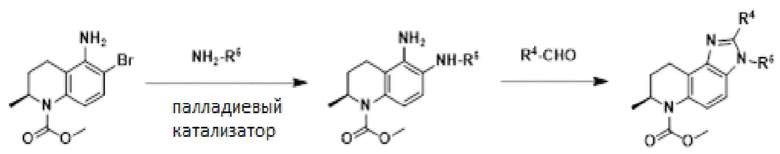
[8] Описание включает применение соединений формулы (I) и их фармацевтически приемлемых солей для лечения заболеваний или нарушений, связанных с ингибированием CBP, включая определенные AR-положительные формы рака, включая форму сплайсинга AR-v7 AR. CBP/P300 является важным кофактором в транскрипции, управляемой AR, включая андрогеннезависимые варианты AR, и является привлекательной мишенью для разработки новой терапии для удовлетворения потребностей этих пациентов.
[9] В некоторых вариантах осуществления, соединение формулы (I) представляет собой Соединение 1:
 (1)
(1)
или его стереоизомер и/или фармацевтически приемлемую соль.
[10] В некоторых вариантах осуществления изобретения Соединение 1 представляет собой первый элюируемый изомер при элюировании препаративной HPLC в условиях, определенных в Примере 1.2.
[11] В некоторых вариантах осуществления соединение формулы (I) представляет собой Соединение 2:
 (2)
(2)
или его стереоизомер и/или фармацевтически приемлемую соль.
[12] В некоторых вариантах осуществления изобретения Соединение 2 представляет собой первый элюируемый изомер при элюировании препаративной HPLC в условиях, определенных в Примере 1.3.
[13] В некоторых вариантах осуществления соединение формулы (I) представляет собой Соединение 3:
 (3)
(3)
или его стереоизомер и/или фармацевтически приемлемую соль.
[14] В некоторых вариантах осуществления изобретения Соединение 3 является вторым элюируемым изомером при элюировании препаративной HPLC в условиях, определенных в Примере 1.4.
[15] В некоторых вариантах осуществления соединение формулы (I) представляет собой Соединение 4:
 (4)
(4)
или его стереоизомер и/или фармацевтически приемлемую соль.
[16] Соединения формулы (I) активны в доклинических моделях, устойчивых к энзалутамиду, и, таким образом, представляют собой потенциальную новую терапию для этих пациентов с рефрактерным или резистентным заболеванием.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[17] Фиг. 1 представляет собой таблицу соединений в соответствии с различными вариантами осуществления настоящего изобретения.
[18] Фиг. 2 представляет собой серию схем реакций для химического синтеза соединений Формулы (I) и полезных промежуточных соединений при получении соединений Формулы (I).
[19] Фиг. 3 представляет собой иммуноблот, показывающий уровни экспрессии белков H3K27Ac, общего H3 и β-актина в клеточной линии рака молочной железы, подвергнутой воздействию возрастающих концентраций Соединения 1 в течение 24 часов.
[20] Фиг. 4 представляет собой график, показывающий активность Соединения 1 in vivo в модели ксенотрансплантата, полученной из клеточной линии, AR+ тройного отрицательного рака молочной железы.
[21] Фигура 5 представляет собой иммуноблот, показывающий уровни экспрессии AR и AR-v7 в линии клеток рака простаты AR-v7+ после 24-часового воздействия возрастающих концентраций Соединения 3.
[22] Фиг. 6 представляет собой график, показывающий активность Соединения 4 in vivo в модели ксенотрансплантата, полученной пациентом, при раке простаты, устойчивом к энзалутамиду.
ДЕТАЛЬНОЕ ОПИСАНИЕ
[23] Настоящее изобретение включает признание того, что соединения формулы (I) являются соединениями-ингибиторами CBP, определенными здесь как соединения, имеющие одну или более из следующих характеристик при тестировании в соответствии с протоколом биохимического анализа HTRF, приведенным ниже в Примере 2: (1) значение IC50 CBP менее 1 мкМ; и (2) значение IC50 CBP от 0,001 до 1 мкМ. Соединения-ингибиторы CBP могут иметь формулу (I):
(I)
или его фармацевтически приемлемая соль, где R1, R2 и n такие, как описано выше.
[24] Если здесь не указано иное, все изомерные формы указанных химических соединений представлены настоящим изобретением, включая их смеси (например, S, R и рацемическая ориентация в каждом хиральном центре). Если соединение содержит двойную связь, заместитель может иметь E или Z конфигурацию. Если соединение содержит дизамещенный циклоалкил, циклоалкильный заместитель может иметь цис- или транс-конфигурацию. Все таутомерные формы также предназначены для включения.
[25] Соединения формулы (I) и Группы A, если не указано иное, могут существовать в их таутомерной форме. Все такие таутомерные формы рассматриваются здесь как часть настоящего описания.
[26] Соединения формулы (I) и Группы A, если не указано иное, могут содержать один или более стереоцентров и, следовательно, существовать в различных стереоизомерных формах. Подразумевается, что, если не указано иное, все стереоизомерные формы соединений формулы (I) и Группы A, а также их смеси, включая рацемические смеси, составляют часть настоящего описания. Кроме того, настоящее описание охватывает все геометрические и позиционные изомеры. Например, если соединение формулы (I) или Группы A включает двойную связь или конденсированное кольцо, как цис-, так и транс-формы, а также смеси, охватываются объемом настоящего описания. Каждое раскрытое здесь соединение включает все энантиомеры, которые соответствуют общей структуре соединения. Соединения могут быть в рацемической или энантиомерно чистой формы или в любой другой форме с точки зрения стереохимии. Результаты анализа могут отражать данные, собранные для рацемической формы, энантиомерно чистой формы или любой другой формы с точки зрения стереохимии.
[27] Смеси диастереомеров можно разделить на их индивидуальные диастереомеры на основе их физико-химических различий методами, хорошо известными квалифицированным специалистам в данной области техники, такими как, например, хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить путем превращения энантиомерной смеси в диастереомерную смесь реакцией с подходящим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделением диастереомеров и превращением (например, гидролизом) отдельных диастереомеров в соответствующие чистые энантиомеры. Кроме того, некоторые из соединений формулы (I) или Группы A могут быть атропоизомерами (например, замещенными биарилами) и рассматриваются как часть этого описания. Энантиомеры также можно разделить с помощью хиральной колонки для HPLC.
[28] Соединения формулы (I) или Группы A могут образовывать кислотно-аддитивные соли, которые могут быть фармацевтически приемлемыми солями. Описание также включает фармацевтические композиции, содержащие одно или более соединений, как описано в данном документе, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В некоторых вариантах осуществления изобретения фармацевтические композиции, описанные в настоящем документе, могут быть предоставлены в виде стандартной лекарственной формы (например, капсулы, таблетки или тому подобное). В некоторых вариантах осуществления изобретения фармацевтические композиции, описанные в настоящем документе, могут быть представлены в пероральной лекарственной форме. В некоторых вариантах осуществления изобретения пероральная лекарственная форма соединения формулы (I) или Группы A может представлять собой капсулу. В некоторых вариантах осуществления изобретения пероральная лекарственная форма соединения формулы (I) или Группы A представляет собой таблетку. В некоторых вариантах осуществления изобретения пероральная лекарственная форма содержит один или более наполнителей, разрыхлителей, смазывающих веществ, скользящих веществ, антиадгезивов и/или антистатиков. В некоторых вариантах осуществления изобретения пероральную лекарственную форму получают путем сухого смешивания. В некоторых вариантах осуществления изобретения пероральная лекарственная форма представляет собой таблетку, которую получают путем сухого гранулирования.
[29] Соединение-ингибитор СВР настоящего изобретения можно дозировать на терапевтически эффективном уровне. Соединение селективного ингибитора СВР настоящего описания можно дозировать на терапевтически эффективном уровне.
Соединения описания
[30] В одном аспекте изобретение относится к соединениям формулы (I):
(I)
или их фармацевтически приемлемой соли, где:
R1 представляет собой H или -OH;
каждый R2 независимо выбран из C1-C6 алкила, галогена, -CN и -OR3, где алкил необязательно замещен одним или более галогенами;
каждый R3 независимо представляет собой H или C1-C6 алкил, где алкил необязательно замещен одним или более галогенами; и
n равно целому числу, выбранному из 0-5, где n предпочтительно равно 0, 1, 2 или 3.
[31] В некоторых вариантах реализации, R1 представляет собой H или -OH; каждый R2 независимо выбран из -F, -Cl, -CH3, -CHF2, -CN, и -OR3; каждый R3 независимо выбран из -CH3, -CHF2, и -CH(CH3)2, и n выбран из 0, 1, 2, и 3.
[32] В некоторых вариантах осуществления, соединение Формулы (I) обеспечиваются, когда R2 представляет собой -Cl, -CH3, -CHF2, -CN, -OCH3, -OCHF2, -OCH(CH3)2. В некоторых вариантах осуществления, R2 представляет собой -F, -CH3, -CHF2, -CN, или -OR3. В некоторых вариантах осуществления, R2 представляет собой -F, -Cl, -CHF2, -CN, или -OR3. В некоторых вариантах осуществления, R2 представляет собой -F, -Cl, -CH3, -CN, или -OR3. В некоторых вариантах осуществления, R2 представляет собой -F, -Cl, -CH3, -CHF2, или -OR3. В некоторых вариантах осуществления, R2 представляет собой -F, -Cl, -CH3, -CHF2, или -CN.
[33] В одном варианте осуществления изобретения в описании представлены соединения формулы (Ib):
 (Ib)
(Ib)
или их фармацевтически приемлемой соли, где:
R1 представляет собой H или -OH;
R2a выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами (например, CHF2);
R2b выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами (например, CHF2);
R2c выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами;
R2d выбран из H или галогена (например, F или Cl);
R2e выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), -CN, и -OR3, где алкил необязательно замещен одним или более галогенами (например, метил); и
R3 представляет собой H или C1-C6 алкил (например, метил), где алкил необязательно замещен одним или более галогенами (например, CHF2).
[34] В одном варианте осуществления изобретения в описании представлены соединения формулы (Ib):
(Ib)
или их фармацевтически приемлемой соли, где:
R1 представляет собой -OH;
R2a выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами (например, CHF2);
R2b выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами (например, CHF2);
R2c выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами;
R2d выбран из H или галогена (например, F или Cl);
R2e выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), -CN, и -OR3, где алкил необязательно замещен одним или более галогенами (например, метил); и
R3 представляет собой H или C1-C6 алкил (например, метил), где алкил необязательно замещен одним или более галогенами (например, CHF2).
[35] В одном варианте осуществления изобретения в описании представлены соединения формулы (Ib):
(Ib)
или их фармацевтически приемлемой соли, где:
R1 представляет собой H;
R2a выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами (например, CHF2);
R2b выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами (например, CHF2);
R2c выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами;
R2d выбран из H или галогена (например, F или Cl);
R2e выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), -CN, и -OR3, где алкил необязательно замещен одним или более галогенами (например, метил); и
R3 представляет собой H или C1-C6 алкил (например, метил), где алкил необязательно замещен одним или более галогенами (например, CHF2).
[36] В одном варианте осуществления изобретения в описании представлены соединения формулы (Ib):
(Ib)
или их фармацевтически приемлемой соли, где:
R1 представляет собой H или -OH;
R2a выбран из H, C1-C6 алкила (например, метил), галогена (например, F или Cl), и -OR3, где алкил необязательно замещен одним или более галогенами (например, CHF2);
R2b представляет собой H;
R2c представляет собой H;
R2d является независимо выбранным из H или галогена (например, F или Cl);
R2e представляет собой H; и
R3 представляет собой независимо H или C1-C6 алкил (например, метил), где алкил необязательно замещен одним или более галогенами (например, CHF2).
[37] В одном варианте осуществления изобретения в описании представлены соединения формулы (Ib):
(Ib)
или их фармацевтически приемлемой соли, где:
R1 представляет собой H или -OH;
R2a выбран из H, и -OR3;
R2b представляет собой H;
R2c представляет собой H;
R2d является независимо выбранным из H или галогена (например, F или Cl);
R2e представляет собой H; и
R3 представляет собой независимо H или C1-C6 алкил (например, метил), где алкил необязательно замещен одним или более галогенами (например, CHF2).
[38] В некоторых вариантах осуществления изобретения представлены Соединения селективного ингибитора СВР формулы (I). В некоторых вариантах осуществления изобретения представлены соединения селективного ингибитора CBP Формулы (Ib). Настоящее описание включает признание того, что соединения формулы (I) являются соединениями-ингибиторами CBP, определенными здесь как соединения, имеющие одну или более из следующих характеристик при тестировании в соответствии с протоколом биохимического анализа HTRF, приведенным ниже в Примере 2: (1) значение IC50 CBP менее 1 мкМ; и (2) значение IC50 CBP от 0,001 до 1 мкМ.
[39] В некоторых вариантах осуществления изобретения описание относится к соединениям формулы (I), которые имеют формулу, выбранную из Группы A:
 (A1),
(A1),  (A2),
(A2),
 (A3),
(A3),  (A4),
(A4),
 (A5),
(A5),  (A6),
(A6),
 (A7),
(A7),  (A8),
(A8),
 (A9),
(A9),  (A10),
(A10),  (A11) и
(A11) и  (A12),
(A12),
и их фармацевтически приемлемым солям.
[40] В некоторых вариантах реализации описание относится к соединению формулы (I), выбранному из Фиг. 1. Фигура 1 «Элюированный изомер» относится к порядку, в котором соединение элюируется препаративной HPLC.
[41] В некоторых вариантах осуществления, R1 представляет собой H или -OH. В некоторых вариантах осуществления, R1 представляет собой H. В некоторых вариантах осуществления, R1 представляет собой -OH.
[42] В некоторых вариантах осуществления, каждый R2 является независимо выбранным из C1-C6 алкила, галогена, -CN, и -OR3, где алкил необязательно замещен одним или более галогенами. В некоторых вариантах осуществления, R2 представляет собой C1-C6 алкил, где алкил необязательно замещен одним или более галогенами. В некоторых вариантах осуществления, R2 представляет собой C1-C6 алкил, где алкил замещен одним галогеном. В некоторых вариантах осуществления, R2 представляет собой C1-C6 алкил, где алкил замещен двумя галогенами. В некоторых вариантах осуществления, R2 выбран из -CH3 и -CHF2. В некоторых вариантах осуществления, R2 представляет собой -CH3. В некоторых вариантах осуществления, R2 представляет собой -CHF2. В некоторых вариантах осуществления R2 представляет собой галоген. В некоторых вариантах осуществления, R2 выбран из -F и -Cl. В некоторых вариантах осуществления, R2 представляет собой -F. В некоторых вариантах осуществления, R2 представляет собой -Cl. В некоторых вариантах осуществления, R2 представляет собой -CN. В некоторых вариантах осуществления, R2 представляет собой -OR3, где R3 является таким, как описано в данном документе.
[43] В некоторых вариантах осуществления, каждый R3 представляет собой независимо C1-C6 алкил, где алкил необязательно замещен одним или более галогенами. В некоторых вариантах осуществления, R3 представляет собой C1-C6 алкил, где алкил замещен одним галогеном. В некоторых вариантах осуществления, R3 представляет собой C1-C6 алкил, где алкил замещен двумя галогенами. В некоторых вариантах осуществления, R3 выбран из -CH3, -CHF2, и пропила. В некоторых вариантах осуществления, R3 представляет собой -CH3. В некоторых вариантах осуществления, R3 представляет собой -CHF2. В некоторых вариантах осуществления, R3 представляет собой пропил.
[44] В некоторых вариантах осуществления, n выбран из 0, 1, 2, и 3. В некоторых вариантах осуществления, n равно 0. В некоторых вариантах осуществления, n равно 1. В некоторых вариантах осуществления, n равно 2. В некоторых вариантах осуществления, n равно 3.
[45] В некоторых вариантах осуществления, n равно 2 и R2 представляет собой CH3 в одном случае и F во втором случае. В некоторых вариантах осуществления, n равно 2 и R2 представляет собой -OCH3 в одном случае и Cl во втором случае. В некоторых вариантах осуществления, n равно 2 и R2 представляет собой -OCH3 в одном случае и -OCH3 во втором случае. В некоторых вариантах осуществления, n равно 2 и R2 представляет собой -CH3 в одном случае и F во втором случае. В некоторых вариантах осуществления, n равно 2 и R2 представляет собой F в одном случае и F во втором случае. В некоторых вариантах осуществления, n равно 2 и R2 представляет собой -CHF2 в одном случае и F во втором случае. В некоторых вариантах осуществления, n равно 2 и R2 представляет собой -OCH(CH3)2 в одном случае и F во втором случае. В некоторых вариантах осуществления, n равно 2 и R2 представляет собой -OCHF2 в одном случае и F во втором случае. В некоторых вариантах осуществления, n равно 3 и R2 представляет собой -OCH3 в одном случае, F во втором случае, и F в третьем случае. В некоторых вариантах осуществления, n равно 3 и R2 представляет собой -OCH3 в одном случае, -OCH3 во втором случае, и F в третьем случае.
[46] В некоторых вариантах осуществления, R1 представляет собой H и n равно 0.
[47] В некоторых вариантах осуществления, R1 представляет собой -OH и n равно 0.
[48] В некоторых вариантах осуществления, R1 представляет собой -OH, n равно 2, и R2 представляет собой -F в одном случае и -OR3 во втором случае, где R3 представляет собой -CH3.
[49] В некоторых вариантах осуществления, R1 представляет собой -OH, n равно 2, и R2 представляет собой -F в одном случае и -OR3 во втором случае, где R3 представляет собой -CHF2.
[50] В некоторых вариантах осуществления, соединение формулы (I) представляет собой Соединение 1:
(1)
или его фармацевтически приемлемую соль.
[51] В некоторых вариантах осуществления, соединение формулы (I) представляет собой (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту.
[52] В некоторых вариантах осуществления, соединение формулы (I) представляет собой (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[53] В некоторых вариантах осуществления, соединение формулы (I) представляет собой Соединение 2:
(2)
или его фармацевтически приемлемую соль.
[54] В некоторых вариантах осуществления, соединение формулы (I) представляет собой 3-((7S)-2-(гидрокси(фенил)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[55] В некоторых вариантах осуществления, соединение формулы (I) является первым элюирующим изомером 3-((7S)-2-(гидрокси(фенил)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты при элюировании препаративной ВЭЖХ с использованием условий, определенных в Примере 1,3.
[56] В некоторых вариантах осуществления, соединение формулы (I) представляет собой (1R,3R)-3-((S)-2-((R)-гидрокси(фенил)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[57] В некоторых вариантах осуществления, соединение формулы (I) представляет собой Соединение 3:
 (3)
(3)
или его фармацевтически приемлемую соль.
[58] В некоторых вариантах осуществления, соединение формулы (I) представляет собой 3-((7S)-2-((2-(дифторметокси)-5-фторфенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[59] В некоторых вариантах осуществления, соединение формулы (I) является первым элюирующим изомером 3-((7S)-2-((2-(дифторметокси)-5-фторфенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты при элюировании из препаративной ВЭЖХ с использованием условий, определенных в Примере 1.5.
[60] В некоторых вариантах осуществления, соединение формулы (I) представляет собой (1R,3R)-3-((S)-2-((R)-(2-(дифторметокси)-5-фторфенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[61] В некоторых вариантах осуществления, соединение формулы (I) представляет собой Соединение 4:
(4)
или его фармацевтически приемлемую соль.
[62] В некоторых вариантах осуществления, соединение формулы (I) представляет собой 3-((S)-2-бензил-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[63] В некоторых вариантах осуществления, соединение формулы (I) представляет собой (1R,3R)-3-((S)-2-бензил-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[64] Описание также частично основано на признании того, что соединения формулы (I) являются соединениями селективных ингибиторов CBP, определяемыми здесь как ингибиторы CBP, имеющие значение BRD4 IC50 выше, чем их значение CBP IC50, предпочтительно, когда его значение BRD4 IC50 больше чем 1 мкМ (например, от 1 микромоль до 10 микромоль или больше), где значения IC50 определяют, как в процедурах, изложенных в анализе, описанном в Примере 2. В некоторых вариантах осуществления изобретения соединения формулы (I) могут быть соединениями-селективными ингибиторами CBP, где значение BRD4 IC50 больше 500 нМ (например, от 500 наномоль до 10 микромоль или больше), где значения IC50 определяют, как в процедурах, изложенных в анализе, описанном в Примере 2. Описание также частично основано на признании того, что Соединение 1 является соединением селективного ингибитора CBP, определенным здесь как ингибитор CBP, имеющий значение BRD4 IC50 больше, чем его значение CBP IC50, предпочтительно, где его значение BRD4 IC50 больше 1 мкМ (например, от 1 микромоль до 10 микромоль или более), где значения IC50 определяют, как в процедурах, изложенных в анализе, описанном в Примере 2.
[65] Открытие включает применение одного или более соединений формулы (I) и их фармацевтически приемлемых солей в фармацевтических препаратах для лечения пациентов, у которых диагностировано заболевание или расстройство, связанное с ингибированием CBP (например, некоторые формы рака). Композиции, содержащие одно или более соединений формулы (I) и их фармацевтически приемлемые соли, могут быть получены с помощью определенных способов, также предоставленных здесь. В некоторых вариантах осуществления изобретения соединение селективного ингибитора CBP формулы (I) применяется для лечения рака молочной железы (например, TNBC) или рака простаты. В некоторых вариантах осуществления изобретения соединение селективного ингибитора CBP формулы (Ib) применяется для лечения AR+ формы рака, включая AR+ рак молочной железы или рак простаты. Применение Соединения селективного ингибитора CBP формулы (I) предусмотрено для лечения пациента, у которого диагностирована форма рака AR+, такая как AR+ рак молочной железы (например, AR+ TNBC) или AR+ рак простаты (например, AR-v7+ форма рака простаты).
[66] В некоторых вариантах осуществления изобретения открытие включает применение (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновой кислоты (Соединение 1) и ее фармацевтически приемлемых солей в фармацевтических препаратах для лечения пациентов, у которых диагностировано заболевание или расстройство, связанное с ингибированием CBP (например, некоторые формы рака). Композиции, содержащие Соединение 1 и его фармацевтически приемлемые соли, могут быть получены определенными способами, также предусмотренными здесь.
[67] В некоторых вариантах осуществления изобретение включает применение Соединения 2 и его фармацевтически приемлемых солей в фармацевтических препаратах для лечения пациентов, у которых диагностировано заболевание или нарушение, связанное с ингибированием CBP (например, некоторых форм рака). Композиции, содержащие Соединение 2 и его фармацевтически приемлемые соли, могут быть получены определенными способами, также предусмотренными здесь.
[68] В некоторых вариантах осуществления изобретение включает применение Соединения 3 и его фармацевтически приемлемых солей в фармацевтических препаратах для лечения пациентов, у которых диагностировано заболевание или нарушение, связанное с ингибированием CBP (например, некоторых форм рака). Композиции, содержащие Соединение 3 и его фармацевтически приемлемые соли, могут быть получены определенными способами, также предусмотренными здесь.
[69] В некоторых вариантах осуществления изобретение включает применение Соединения 4 и его фармацевтически приемлемых солей в фармацевтических препаратах для лечения пациентов, у которых диагностировано заболевание или нарушение, связанное с ингибированием CBP (например, некоторых форм рака). Композиции, содержащие Соединение 4 и его фармацевтически приемлемые соли, могут быть получены определенными способами, также предусмотренными здесь.
Способы синтеза соединений
[70] Соединения настоящего описания могут быть получены различными способами, включая стандартные химические методы. Подходящие пути синтеза показаны в примерах, приведенных ниже.
[71] Соединения настоящего изобретения, то есть соединения Формулы (I), (II) или Группы A, или их фармацевтически приемлемые соли, могут быть получены способами, известными в области органического синтеза, которые частично изложены в схемах синтеза изображенных в примерах. В схемах, описанных ниже, хорошо понятно, что защитные группы для чувствительных или реакционноспособных групп используются там, где это необходимо, в соответствии с общими принципами или химическим составом. С защитными группами манипулируют согласно стандартным методам органического синтеза (T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999). Эти группы удаляют на удобной стадии синтеза соединения с использованием методов, которые очевидны для квалифицированных специалистов в данной области техники. Процессы выбора, а также условия реакции и порядок их выполнения должны соответствовать получению соединений Формулы (I), (II) или Группы A.
[72] Квалифицированные специалисты в данной области техники поймут, что стереоцентры существуют в соединениях Формулы (I), (II) или Группы A. Соответственно, настоящее описание включает оба возможных стереоизомера (если иное не указано и/или не определено в синтезе) и включает не только рацемические соединения, но также отдельные энантиомеры и/или диастереомеры. Если не указано иное, когда соединение желательно в виде единственного энантиомера или диастереомера, оно может быть получено стереоспецифическим синтезом или разделением конечного продукта или любого удобного промежуточного соединения. На разделение конечного продукта, промежуточного продукта или исходного материала можно повлиять любым подходящим способом, известным в данной области. См., например, «Stereochemistry of Organic Compounds» by E. L. Eliel, S. H. Wilen, and L. N. Mander (Wiley-lnterscience, 1994).
Способы применения соединений
[73] В некоторых вариантах осуществления изобретения соединения формулы (I) представляют собой инструментальные соединения, полезные для изучения эффектов ингибирования CBP/p300 in vitro или на модели in vivo. In vitro инструментальные соединения формулы (I) могут быть полезны для изучения эффектов ингибирования CBP/p300 на очищенные белки, клеточные экстракты, в интактных клетках и моделях клеточных линий и т.п. In vivo инструментальные соединения формулы (I) могут быть полезны для изучения эффектов ингибирования CBP/p300 в ксенотрансплантатах, полученных из клеточной линии, в ксенотрансплантатах, полученных от пациентов, на модели мышей с нокаутом, в моделях с нокаутом мышей и т.п.
[74] Предпочтительно в описании представлены фармацевтические препараты для лечения пациентов, у которых диагностирован рак AR+. В частности, соединения, представленные в настоящем документе, могут быть составлены в виде активной фармацевтической композиции, содержащей одно или более соединений формулы (I) (или их фармацевтически приемлемую соль и/или энантиомер), полезных для лечения рака простаты, включая метастатический устойчивый к кастрату рак простаты (CRPC) и/или AR+ рак молочной железы, включая местнораспространенный или метастатический AR+ рак молочной железы. Например, ингибирование CBP/P300 может нацеливаться на транскрипционную активность AR посредством H3K27Ac, снижение экспрессии гена-мишени AR или снижение экспрессии AR с, в конечном итоге, снижением пролиферации. Кроме того, ингибиторы CBP/P300 BRD представляют возможность подавления ER-управляемой передачи сигналов при гормон-рецептор-положительном раке молочной железы. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит Соединение 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит Соединение 2. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит Соединение 3. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит Соединение 4.
[75] Описанные здесь соединения и композиции являются ингибиторами CBP, имеющими более низкую концентрацию ингибирования по сравнению с его концентрацией ингибирования по отношению к BRD4.
[76] Способы лечения (например, путем ингибирования CBP) могут включать введение субъекту, нуждающемуся в этом, терапевтически эффективного количества (1) соединения формулы (I) или его фармацевтически приемлемой соли; (2) фармацевтической композиции, содержащей одно или более соединений формулы (I) или их фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[77] Способы лечения (например, путем ингибирования CBP) могут включать введение субъекту, нуждающемуся в этом, терапевтически эффективного количества (1) Соединения 1 или его фармацевтически приемлемой соли; (2) фармацевтической композиции, содержащей Соединение 1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[78] Способы лечения (например, путем ингибирования CBP) могут включать введение субъекту, нуждающемуся в этом, терапевтически эффективного количества (1) Соединения 2 или его фармацевтически приемлемой соли; (2) фармацевтической композиции, содержащей Соединение 2 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[79] Способы лечения (например, путем ингибирования CBP) могут включать введение субъекту, нуждающемуся в этом, терапевтически эффективного количества (1) Соединения 3 или его фармацевтически приемлемой соли; (2) фармацевтической композиции, содержащей Соединение 3 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[80] Способы лечения (например, путем ингибирования CBP) могут включать введение субъекту, нуждающемуся в этом, терапевтически эффективного количества (1) Соединения 4 или его фармацевтически приемлемой соли; (2) фармацевтической композиции, содержащей Соединение 4 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[81] Фармацевтические композиции можно вводить перорально в любой перорально приемлемой дозированной форме. Соответственно, пациент и/или субъект могут быть выбраны для лечения с применением соединения, описанного в данном документе, путем сначала оценки пациента и/или субъекта, чтобы определить, нуждается ли субъект в ингибировании CBP, и если субъект определен как нуждающийся в ингибировании CBP, затем введения субъекту композиции, описанной в данном документе.
[82] Соединения и композиции ингибиторов CBP полезны, например, для подавления программ транскрипции, управляемых AR, посредством ингибирования BRD CBP/P300, включая варианты AR-v7. Эффект ингибирования роста Соединений 1, 2 и 4 и энзалутамида определяли на панели линий клеток рака простаты, включая андроген-зависимые и независимые модели, а также AR-отрицательные клеточные линии. Соединение 1 индуцировало зависимое от концентрации снижение H3K27Ac, метки, специфичной для CBP/P300, в AR-положительной клеточной линии рака молочной железы (Фиг. 3). Соединения 1, 2 и 4 снижали экспрессию мРНК TMPRSS2 и XBP1 в AR-положительной клеточной линии рака молочной железы. Соединения 1, 2 и 4 ингибируют пролиферацию клеточных линий рака молочной железы после 10 дней непрерывного воздействия лекарственного средства, а клеточные линии с высокой экспрессией мРНК AR более чувствительны, чем линии с низкой экспрессией. Обработка Соединением 1 вызвала ингибирование роста опухоли в модели ксенотрансплантата, полученной из AR-положительной линии клеток рака молочной железы (Фиг. 4). В некоторых вариантах осуществления изобретения AR-положительная линия клеток рака молочной железы может представлять собой MDA-MB-453. Обработка клеток рака простаты Соединением 2 приводила к уменьшению как полноразмерных, так и вариантных форм AR, включая AR-v7 (Фиг. 5). Соединения 1, 2 и 4 снижали гены-мишени AR TMPRSS2 и KLK3, а также MYC зависимым от концентрации образом в линии клеток рака простаты AR-v7+. В некоторых вариантах осуществления изобретения линия клеток рака простаты AR-v7+ может представлять собой 22Rv1. Соединения 1, 2 и 4 обладали сильным и зависимым от концентрации эффектом ингибирования роста во всех линиях клеток AR+, включая линии клеток, несущие AR-v7. Обработка Соединением 4 в дозе 40 мг/кг/доза ежедневно с понедельника по четверг, повторенная еженедельно, или 80 мг/кг/доза в понедельник и четверг (дважды в неделю), повторенная еженедельно, привела к сильному противоопухолевому ответу на модели ксенотрансплантата, полученной от пациента, устойчивой к энзалутамиду (Фиг. 6).
[83] Энзалутамид является ингибитором рецепторов андрогенов, показанным для лечения пациентов с резистентным к кастрации раком простаты или метастатическим раком простаты, чувствительным к кастрации. Пациенты, получающие энзалутамид, также могут одновременно получать аналог гонадотропин-рилизинг-гормона (GnRH) или перенести двустороннюю орхиэктомию. Энзалутамид - это ингибитор рецептора андрогена, который действует на различных этапах пути передачи сигнала рецептора андрогена. Было показано, что энзалутамид конкурентно ингибирует связывание андрогенов с рецепторами андрогенов; и, следовательно, ингибирует ядерную транслокацию рецепторов андрогенов и их взаимодействие с ДНК. Основной метаболит, N-десметилэнзалутамид, проявлял in vitro активность, аналогичную энзалутамиду. Энзалутамид уменьшал пролиферацию и индуцировал гибель клеток рака простаты in vitro, а также уменьшал объем опухоли на модели ксенотрансплантата рака простаты у мышей.
[84] В некоторых вариантах осуществления изобретения соединения формулы (I) или их фармацевтически приемлемые соли показаны для лечения пациентов с устойчивым к кастрации раком простаты или метастатическим раком простаты, чувствительным к кастрации. Пациенты, получающие соединение формулы (I) или его фармацевтически приемлемую соль, также могут одновременно получать аналог гонадотропин-рилизинг гормона (GnRH) или перенести двустороннюю орхиэктомию.
[85] Энзалутамид был активен против андроген-зависимых клеточных линий, таких как VCaP, но был неактивен в AR-отрицательных клеточных линиях, а также в клетках AR-v7+. Напротив, Соединение 1 оказывало сильное и зависящее от концентрации ингибирующее действие на рост во всех линиях клеток AR+, включая линию клеток AR-v7+ линию клеток (IC50=0,6 мкМ). Соединение 1 также было неактивным в отношении линий AR-клеток. Это согласуется с предполагаемым механизмом действия Соединения 1.
[86] Не связываясь с теорией, считается, что CBP/P300 может взаимодействовать с AR напрямую через домен NRID в CBP/P300. Однако, в отличие от других ядерных рецепторов, это взаимодействие, как полагают, не зависит от связывания лиганда. Кроме того, CBP/P300 может взаимодействовать как с LBD AR, так и с его N-концевым доменом. Оба эти фактора имеют отношение к кастратурезистентному раку простаты, и ожидается, что CBP/P300 взаимодействует с AR-v7 очень аналогично, как и с AR. CBP/P300 также косвенно взаимодействует с AR через кофактор TIP60/SRC-1, который сам взаимодействует с AR. Этот подход может отличаться от прямых антагонистов рецепторов и может иметь то преимущество, что он не зависит от структурных изменений в домене связывания лиганда AR(LBD). Такие ингибиторы CBP/P300 BRD могут проявлять активность в ряде видов рака, зависящих от программ транскрипции ядерных гормонов рецепторов, таких как метастатический CRPC и местно-распространенный или метастатический AR+ рак молочной железы. Терапевтические преимущества ингибирования AR были продемонстрированы клинически при этих формах рака, но с ограничениями, которые подчеркивают необходимость альтернативных подходов с долгосрочным преимуществом и механизмами устойчивости, которые не совпадают с антиандрогенной терапией. Например, ингибирование CBP/P300 может нацеливаться на транскрипционную активность AR посредством снижения ацетилирования H3K27, снижения экспрессии гена-мишени AR и/или снижения экспрессии AR, что в конечном итоге приводит к снижению пролиферации. Ожидается, что опосредованное ингибитором CBP/P300 ингибирование активности AR будет нечувствительным к структурным вариациям LBD AR и, следовательно, нечувствительным к связанным с LBD механизмам устойчивости к антагонистам андрогенов. Наконец, ингибиторы CBP/P300 BRD могут быть полезны для подавления ER-управляемой передачи сигналов при раке молочной железы, положительном по рецепторам гормонов.
[87] Аберрации AR в LBD (вариант сплайсинга v7, мутации AR) могут сделать рецептор-лиганд независимым и нечувствительным к антагонистам AR. Идентифицировано около двадцати вариантов сплайсинга мРНК AR, причем подмножество является конститутивно активным. Примечательно, что все биологически активные формы AR сохраняют NTD; препараты, нацеленные на NTD, могут воздействовать на все формы AR, включая те, которые могут вызывать устойчивость к терапии, направленной на AR-LBD. Из вариантов AR только AR-v7 и ARv567 были обнаружены на уровне белка, и AR-v7 был наиболее изучен. Примечательно, что в случаях mCRPC, когда мужчины изначально лечились антагонистом AR, пациенты с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) демонстрировали более низкий ответ PSA и более короткие PFS и OS по сравнению с отрицательными для AR-v7 CTC. Кроме того, в образцах крови пациентов с mCRPC частота обнаружения белка AR-v7 в ядрах CTC увеличилась с 3% образцов от пациентов после первой линии терапии до 31% образцов для третьей или более линий терапии. Подобные результаты указывают на возможность использования AR-v7 в качестве биомаркера отбора пациентов и его вероятную полезность для определения того, каким мужчинам с mCRPC может быть выгодно лечение антагонистами AR по сравнению с химиотерапией.
[88] Различные методы измерения и определения AR-положительности могут быть использованы для отбора пациентов, получающих фармацевтическую композицию, содержащую одно или более соединений формулы (I). В случае рака простаты экспрессия AR в значительной степени сохраняется в целевой популяции пациентов с рецидивом. Кроме того, белок или мРНК AR и AR-v7 могут быть обнаружены в CTC, в то время как мутации AR и амплификация AR могут быть обнаружены из циркулирующей опухолевой ДНК (цоДНК). Современные методы IHC для измерения AR при раке молочной железы различаются по множеству факторов, включая используемые антитела, методологию IHC и критерий отсечения для положительности (Safarpour et al., Am. J. Cancer Res., 2014, 4:353-368). В большинстве исследований используется клон антитела AR441 от Dako (Agilent) и 1% или 10% положительных ядер в качестве порога положительности. В целом частота AR+ TNBC составляет от 20% до 30%.
[89] Рак предстательной железы, устойчивый к андрогенной депривации или антагонистам андрогенов, остается неудовлетворенной потребностью. В некоторых примерах ингибирование CBP/P300 BRD с его дифференцированным механизмом противодействия программе транскрипции, управляемой AR, используется в качестве лечения для этих пациентов с потенциальной полезностью в качестве более ранней линии терапии. Фармацевтические композиции, содержащие одно или более соединений формулы (I), можно использовать для лечения определенных форм рака простаты. Предпочтительно, композиции, содержащие одно или более соединений формулы (I), полезны для ингибирования CBP/P300 BRD с дифференцированным механизмом противодействия программе транскрипции, управляемой AR (например, в качестве ранней линии терапии). Фармацевтические композиции, содержащие одно или более соединений формулы (I), можно применять для лечения mCRPC или других видов рака, зависящих от транскрипции, управляемой AR.
[90] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей одно или более соединений формулы (I), пациентам с диагнозом mCRPC с прогрессирующим резистентным к кастрации заболеванием, которые потерпели неудачу или не переносили по меньшей мере два предшествующих системных лечения, включая по меньшей мере одну терапию на основе антагонистов андрогенов с поддающимся оценке заболеванием (антиандрогены+аналог LHRH, энзалутамид или абиратерон) и повышение уровня PSA с обнаруживаемым метастатическим заболеванием или без него в соответствии со стандартными определениями или нейроэндокринными признаками. Соединения формулы (I) применимы для лечения пациентов с диагнозом AR-v-7 вариантной формы AR. В некоторых способах пациентов с диагнозом mCRPC с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) можно лечить фармацевтической композицией, содержащей одно или более соединений формулы (I). В некоторых способах пациентов, у которых диагностировано прогрессирование заболевания после лечения энзалутамидом, можно лечить фармацевтической композицией, содержащей одно или более соединений формулы (I).
[91] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 1, пациентам с диагнозом mCRPC с прогрессирующей устойчивой к кастрации болезнью, которые не смогли или не переносили по меньшей мере два предшествующих системных лечения, включая по меньшей мере одну терапию на основе антагонистов андрогенов с оцениваемым заболеванием (антиандрогены+аналог LHRH, энзалутамид или абиратерон) и повышение уровня PSA с обнаруживаемым метастатическим заболеванием или без него в соответствии со стандартными определениями или нейроэндокринными признаками. Соединение 1 также полезно для лечения пациентов с диагнозом AR-v-7 вариантной формы AR. В некоторых способах пациентов с диагнозом mCRPC с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) можно лечить фармацевтической композицией, содержащей Соединение 1. В некоторых способах пациентов, у которых диагностировано прогрессирование заболевания после лечения энзалутамидом, можно лечить фармацевтической композицией, содержащей Соединение 1.
[92] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту пациентам с диагнозом mCRPC с прогрессирующим устойчивым к кастрации заболеванием, у которого не было эффекта или была непереносимость как минимум двух предшествующих системных терапий, включая как минимум одну терапию на основе антагонистов андрогенов с поддающимся оценке заболеванием (антиандрогены+аналог LHRH, энзалутамид или абиратерон), а также повышение уровня PSA с обнаруживаемым метастатическим заболеванием или без него в соответствии со стандартными определениями или нейроэндокринными признаками. (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновая кислота также полезна для лечения пациентов с диагнозом AR-v-7 вариантной формы AR. В некоторых методах пациентов с диагнозом mCRPC с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) можно лечить фармацевтической композицией, содержащей (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту. В некоторых методах пациентов, у которых диагностировано прогрессирование заболевания после лечения энзалутамидом, можно лечить фармацевтической композицией, содержащей (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[93] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 2, пациентам с диагнозом mCRPC с прогрессирующим резистентным к кастрации заболеванием, которые не смогли или не переносили по меньшей мере два предшествующих системных лечения, включая по меньшей мере одну терапию на основе антагонистов андрогенов с оцениваемым заболеванием (антиандрогены+аналог LHRH, энзалутамид или абиратерон) и повышение уровня PSA с обнаруживаемым метастатическим заболеванием или без него в соответствии со стандартными определениями или нейроэндокринными признаками. Соединение 2 также полезно для лечения пациентов с диагнозом AR-v-7 вариантной формы AR. В некоторых способах пациентов с диагнозом mCRPC с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) можно лечить фармацевтической композицией, содержащей Соединение 2. В некоторых способах пациентов, у которых диагностировано прогрессирование заболевания после лечения энзалутамидом, можно лечить фармацевтической композицией, содержащей Соединение 2.
[94] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 3, пациентам с диагнозом mCRPC с прогрессирующим резистентным к кастрации заболеванием, которые не смогли или не переносили по меньшей мере два предшествующих системных лечения, включая по меньшей мере одну терапию на основе антагонистов андрогенов с оцениваемым заболеванием (антиандрогены+аналог LHRH, энзалутамид или абиратерон) и повышение уровня PSA с обнаруживаемым метастатическим заболеванием или без него в соответствии со стандартными определениями или нейроэндокринными признаками. Соединение 3 также полезно для лечения пациентов с диагнозом AR-v-7 вариантной формы AR. В некоторых способах пациентов с диагнозом mCRPC с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) можно лечить фармацевтической композицией, содержащей Соединение 3. В некоторых способах пациентов, у которых диагностировано прогрессирование заболевания после лечения энзалутамидом, можно лечить фармацевтической композицией, содержащей Соединение 3.
[95] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 4, пациентам с диагнозом mCRPC с прогрессирующим резистентным к кастрации заболеванием, которые не смогли или не переносили по меньшей мере два предшествующих системных лечения, включая по меньшей мере одну терапию на основе антагонистов андрогенов с оцениваемым заболеванием (антиандрогены+аналог LHRH, энзалутамид или абиратерон) и повышение уровня PSA с обнаруживаемым метастатическим заболеванием или без него в соответствии со стандартными определениями или нейроэндокринными признаками. Соединение 4 также полезно для лечения пациентов с диагнозом AR-v-7 вариантной формы AR. В некоторых способах пациентов с диагнозом mCRPC с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) можно лечить фармацевтической композицией, содержащей Соединение 4. В некоторых способах пациентов, у которых диагностировано прогрессирование заболевания после лечения энзалутамидом, можно лечить фармацевтической композицией, содержащей Соединение 4.
[96] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей (1R,3R)-3-((S)-2-бензил-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту пациентам с диагнозом mCRPC с прогрессирующим резистентным к кастрации заболеванием, которые не смогли или не переносили по крайней мере два предшествующих системных лечения, включая по крайней мере одну терапию на основе антагонистов андрогенов с поддающимся оценке заболеванием (антиандрогены+аналог LHRH, энзалутамид или абиратерон) и повышением уровня PSA с обнаруживаемым метастатическим заболеванием или без него в соответствии со стандартными определениями или нейроэндокринными признаками. (1R,3R)-3-((S)-2-бензил-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновая кислота также полезна для лечения пациентов с диагнозом AR-v-7 вариантной формы AR. В некоторых методах пациентов с диагнозом mCRPC с AR-v7-положительными циркулирующими опухолевыми клетками (CTC) можно лечить фармацевтической композицией, содержащей (1R,3R)-3-((S)-2-бензил-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту. В некоторых способах пациентов с диагнозом прогрессирования заболевания после лечения энзалутамидом можно лечить фармацевтической композицией, содержащей (1R,3R)-3-((S)-2-бензил-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновую кислоту.
[97] Фармацевтические композиции, содержащие одно или более соединений формулы (I), полезны для лечения определенных форм рака молочной железы AR+. Например, подавление транскрипционной активности AR и/или ER с помощью соединения формулы (I) (или фармацевтической композиции, содержащей одно или более соединений формулы (I)) может быть полезно для лечения противоопухолевых эффектов при AR+ раке молочной железы, включая TNBC и метастатические ER+ опухоли за счет ингибирования CBP/P300 BRD. В некоторых вариантах осуществления изобретения подавление транскрипционной активности AR и/или ER с помощью соединения формулы (I) (или фармацевтической композиции, содержащей одно или более соединений формулы (I)) может быть полезно для лечения противоопухолевых эффектов при AR+ раке молочной железы, включая опухоли TNBC и ER+, посредством ингибирования CBP/P300 BRD. В некоторых вариантах осуществления изобретения подавление транскрипционной активности AR и/или ER с помощью Соединения 1 (или фармацевтической композиции, содержащей Соединение 1) может быть полезно для лечения противоопухолевых эффектов при AR+ раковых заболеваниях молочной железы, включая TNBC и метастатические ER+ опухоли, посредством его ингибирования CBP/P300 BRD. В некоторых вариантах осуществления изобретения подавление транскрипционной активности AR и/или ER с помощью Соединения 1 (или фармацевтической композиции, содержащей Соединение 1) может быть полезно для лечения противоопухолевых эффектов при раке молочной железы AR+, включая опухоли TNBC и ER+, посредством его ингибирования CBP/P300 BRD. В некоторых вариантах осуществления изобретения подавление транскрипционной активности AR и/или ER с помощью Соединения 2 (или фармацевтической композиции, содержащей Соединение 2) может быть полезно для лечения противоопухолевых эффектов при раке молочной железы AR+, включая опухоли TNBC и ER+, посредством его ингибирования CBP/P300 BRD. В некоторых вариантах осуществления изобретения подавление транскрипционной активности AR и/или ER с помощью Соединения 3 (или фармацевтической композиции, содержащей Соединение 3) может быть полезно для лечения противоопухолевых эффектов при раке молочной железы AR+, включая опухоли TNBC и ER+, посредством его ингибирования CBP/P300 BRD. В некоторых вариантах осуществления изобретения подавление транскрипционной активности AR и/или ER с помощью Соединения 4 (или фармацевтической композиции, содержащей Соединение 4) может быть полезно для лечения противоопухолевых эффектов при раке молочной железы AR+, включая опухоли TNBC и ER+, посредством его ингибирования CBP/P300 BRD.
[98] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей одно или более соединений формулы (I), пациентам с диагнозом инвазивная карцинома молочной железы с тройным отрицательным статусом (согласно рекомендациям Коллегии американских патологов [CAP]) и обнаруживаемой экспрессии AR в > 1% опухолевых клеток с прогрессирующим заболеванием, у которых не удалось как минимум два предшествующих системных лечения с поддающимся оценке заболеванием или инвазивной карциномой молочной железы, AR-положительный результат > 1% и ER, PR или HER-положительный (согласно руководящим принципам CAP) с прогрессирующим заболеванием, у которого не получилось по крайней мере, три предшествующих системных терапии. Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей одно или более соединений формулы (I), пациентам, у которых диагностирован рак молочной железы Her2. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей одно или более соединений формулы (I), пациентам, у которых диагностирован рак молочной железы ER-, PR- или ER-/PR.
[99] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 1, пациентам с диагнозом инвазивная карцинома молочной железы с тройным отрицательным статусом (в соответствии с рекомендациями Коллегии американских патологов [CAP]) и обнаруживаемой экспрессией AR в > 1% опухолевых клеток, с прогрессирующим заболеванием, не имевшем по крайней мере двух предшествующих системных терапий, с поддающимся оценке заболеванием или инвазивной карциномой молочной железы, AR-положительный результат > 1% и ER, PR или HER-положительный результат (согласно руководящим принципам CAP) с прогрессирующим заболеванием, при котором не удалось как минимум три предыдущих системных лечения. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 1, пациентам, у которых диагностирован рак молочной железы Her2. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 1, пациентам с диагнозом ER-, PR- или ER-/PR- рака молочной железы.
[100] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 2, пациентам с диагнозом инвазивная карцинома молочной железы с тройным отрицательным статусом (в соответствии с рекомендациями Коллегии американских патологов [CAP]) и обнаруживаемой экспрессией AR в > 1% опухолевых клеток, с прогрессирующим заболеванием, не имевшем по крайней мере двух предшествующих системных терапий, с поддающимся оценке заболеванием или инвазивной карциномой молочной железы, AR-положительный результат > 1% и ER, PR или HER-положительный результат (согласно руководящим принципам CAP) с прогрессирующим заболеванием, при котором не удалось как минимум три предыдущих системных лечения. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 2, пациентам, у которых диагностирован рак молочной железы Her2. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 2, пациентам с диагнозом ER-, PR- или ER-/PR- рака молочной железы.
[101] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 3, пациентам с диагнозом инвазивная карцинома молочной железы с тройным отрицательным статусом (в соответствии с рекомендациями Коллегии американских патологов [CAP]) и обнаруживаемой экспрессией AR в > 1% опухолевых клеток, с прогрессирующим заболеванием, не имевшем по крайней мере двух предшествующих системных терапий, с поддающимся оценке заболеванием или инвазивной карциномой молочной железы, AR-положительный результат > 1% и ER, PR или HER-положительный результат (согласно руководящим принципам CAP) с прогрессирующим заболеванием, при котором не удалось как минимум три предыдущих системных лечения. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 3, пациентам, у которых диагностирован рак молочной железы Her2. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 3, пациентам с диагнозом ER-, PR- или ER-/PR- рака молочной железы.
[102] Некоторые методы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 4, пациентам с диагнозом инвазивная карцинома молочной железы с тройным отрицательным статусом (в соответствии с рекомендациями Коллегии американских патологов [CAP]) и обнаруживаемой экспрессией AR в > 1% опухолевых клеток, с прогрессирующим заболеванием, не имевшем по крайней мере двух предшествующих системных терапий, с поддающимся оценке заболеванием или инвазивной карциномой молочной железы, AR-положительный результат > 1% и ER, PR или HER-положительный результат (согласно руководящим принципам CAP) с прогрессирующим заболеванием, при котором не удалось как минимум три предыдущих системных лечения. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 4, пациентам, у которых диагностирован рак молочной железы Her2. Некоторые способы включают введение терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 4, пациентам с диагнозом ER-, PR- или ER-/PR- рака молочной железы.
[103] Настоящее описание позволяет квалифицированному специалисту в соответствующей области техники создавать и применять изобретения, представленные в данном документе, в соответствии с многочисленными и разнообразными вариантами осуществления. Различные изменения, модификации и улучшения настоящего описания, которые легко приходят на ум квалифицированным специалистам в данной области техники, включая определенные изменения, модификации, замены и улучшения, также являются частью этого описания. Соответственно, вышеприведенное описание приведено в качестве примера для иллюстрации представленных здесь открытий. В настоящем описании представлены соединения, которые являются селективными ингибиторами CBP.
ПРИМЕРЫ
Определения, используемые в следующих Схемах и в других местах здесь:
ACN ацетонитрил
Ac2O уксусный ангидрид
(+)BINAP (±)-2,2′-бис(дифенилфосфино)-1,1′-бинафталин
Boc трет-бутоксикарбонил
n-BuOH бутанол
см сантиметр
DCE 1,2-дихлорэтан
DCM дихлорметан или хлористый метилен
DEA диэтиламин
DMC 2-Хлор-4,5-дигидро-1,3-диметил-1H-имидазолий хлорид
DMP Десс-Мартина периодинан
DMTMM 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид
DIEA N,N-диизопропилэтиламин
DMAP 4-(диметиламино)пиридин
DMF N,N-диметилформамид
DMSO диметилсульфоксид
DPPA дифенилфосфорилазид
dppf бис(дифенилфосфино)ферроцен
ES электрораспылительная ионизация
Et3N триэтиламин
EtOAc этилацетат
EtOH этанол
FA муравьиная кислота
FCC колоночная флэш-хроматография
ч часы
HATU 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизурония гексафторфосфат
HCl хлороводород
HOAc уксусная кислота
ВЭЖХ высокоэффективная жидкостная хроматография
(i-Pr)2NEt N,N-диизопропилэтиламин
L литр
ЖХ/МС жидкостная хроматография/масс-спектрометрия
LDA диизопропиламид лития
K2CO3 карбонат калия
MeOH метанол
мл миллилитр
ммоль миллимоль
мг миллиграмм
МГц мегагерц
МС масс-спектрометрия
m/z отношение масса/заряд
NBS N-бромсукцинимид
нм нанометр
NMM 4-метилморфолин
ЯМР ядерный магнитный резонанс
Pd2(dba)3 трис(дибензилиденацетон)дипалладий
Ph3P трифенилфосфин
PhCHO бензальдегид
PhMe толуол
м.д. частей на миллион
кт комнатная температура
RT время удерживания
SFC сверхкритическая жидкостная хроматография
STAB триацетоксиборгидрид натрия
p-TSA пара-толуолсульфоновый ангидрид
p-TsOH пара-толуолсульфоновая кислота
TFA трифторуксусная кислота
TFAA трифторуксусный ангидрид
THF тетрагидрофуран
УФ ультрафиолет
XPhos 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил
Материалы
[104] Если не указано иное, все материалы были получены от коммерческих поставщиков и использовались без дополнительной очистки. Безводные растворители были получены от Sigma-Aldrich (Милуоки, Висконсин) и использовались напрямую. Все реакции с участием реагентов, чувствительных к воздуху или влаге, проводились в атмосфере азота, а все реакции с использованием микроволнового облучения проводились на приборе Biotage Initiator EXP EU.
[105] Если не указано иное, масс-триггерная очистка HPLC и/или чистота и масс-спектральные данные с низким разрешением были измерены с использованием: (1) Системы ультраэффективной жидкостной хроматографии (UPLC) (Waters Acquity UPLC с устройством для пробоотбора и масс-спектрометром Waters Micromass ZQ) с УФ-детектированием при 220 нм и режимом низкорезонансных электрораспылительных положительных ионов (ESI) (Колонка: Acquity UPLC BEH C18 1,7 мкм 2,1×50 мм; градиент: 5-100% Растворитель B (95/5/0,09%: Ацетонитрил/вода/муравьиная кислота) в Растворителе A (95/5/0,1%: 10 мМ формиат аммония/ацетонитрил/муравьиная кислота) в течение 2,2 мин, затем 100-5% Растворитель B в Растворителе A в течение 0,01 мин, затем выдержка при 5% Растворителе B в Растворителе A в течение 0,29 мин) или (2) Системы высокоэффективной жидкостной хроматографии Waters HT2790 Alliance (ВЭЖХ) (одноквадратный масс-спектрометр Waters 996 PDA и Waters ZQ) с УФ-детектированием при 220 и 254 нм и режимом низкорезонансной ионизации электрораспылением (положительный/отрицательный) (ESI) (Колонка: XBridge Фенил или C18, 5 мкм 4,6×50 мм; градиент: 5-95% Растворителя B (95% метанола/5% воды с 0,1% муравьиной кислоты) в Растворителе A (95% воды/5% метанола с 0,1% муравьиной кислоты) в течение 2,5 минут, затем выдерживают при 95% Растворителе B в Растворителе A в течение 1 мин (только MS с низким разрешением).
Общие методы приготовления соединений
[106] Здесь описаны способы синтеза соединений настоящего изобретения. Соединения настоящего изобретения можно синтезировать в соответствии со схемами синтеза, представленными ниже. Приготовление исходного материала для Схем 1 и 2 («Промежуточное соединение 1») описано ниже. Приготовление исходного материала для Схем 3 и 4 можно найти в Примере 1, Часть A патента США № 4,404,207.
[107] Если не указано иное, заместители R4 и R5 в следующих схемах реакций определены следующим образом.

[108] На Схеме 1 представлены методы, применимые для синтеза соединений Формулы I.
Схема 1

[109] На Схеме 2 представлены методы, применимые для синтеза соединений Формулы I.
Схема 2
[110] В качестве альтернативы на Схеме 3 представлены методы, применимые для синтеза некоторых соединений формулы I.
Схема 3

[111] В качестве альтернативы на Схеме 4 представлены методы, применимые для синтеза некоторых соединений формулы I.
Схема 4

Пример 1: Синтезы соединений изобретения
[112] Соединения, перечисленные на Фиг. 1, были получены с использованием стандартных химических манипуляций и процедур, аналогичных описанным здесь. Фигура 1 «Элюированный изомер» относится к порядку, в котором соединение элюируется препаративной HPLC.
Пример 1.1: Приготовление Промежуточного соединения 1: метил-(S)-5-амино-6-бромо-2-метил-3,4-дигидрохинолин-1(2H)-карбоксилата
[113] На Фигуре 2 (A) представлена схема синтеза получения Промежуточного соединения 1, как описано ниже.
Этап 1 . 8-хлор-5-метокси-2-метилхинолин гидрохлорид
[114] В 4-горлой круглодонной колбе объемом 5 л, продуваемую и поддерживаемую инертной атмосферой азота, растворяли 2-хлор-5-метоксианилин (250 г, 1,59 моль) в 1-бутаноле (1200 мл). Затем добавляли соляную кислоту (водн., 36,5%, 526,5 мл) и хлоранил (456,5 г, 1,86 моль). Полученную смесь перемешивали в течение 1 ч при 100°C в атмосфере азота. Затем по каплям добавляли раствор (E)-бут-2-еналя (169 мл, 2,06 моль) в 1-бутаноле (300 мл). Полученный раствор перемешивали в течение 1 ч при 100°C в атмосфере азота. Масляную баню охлаждали до 70°C и добавляли тетрагидрофуран (1500 мл). Затем полученную смесь перемешивали 1 ч при 70°C. Реакционную смесь охлаждали до 0°C и твердые вещества фильтровали. Твердые вещества промывали тетрагидрофураном (3 л) при 0°C, затем сушили в сушильном шкафу с получением гидрохлорида 8-хлор-5-метокси-2-метилхинолина (83,0 г, 74%) в виде желтого твердого вещества. MS (ES, m/z): 208 [M+H]+.
Этап 2. 5-метокси-2-метилхинолин
[115] В трехгорлой круглодонной колбе на 1000 мл растворяли 8-хлор-5-метокси-2-метилхинолин гидрохлорид (50 г, 204,82 ммоль) в метаноле (300 мл). Затем добавляли гидроксид натрия (3M, 205 мл) и 10% палладий на угле (25 г). В реакционную смесь загружали водород (г). Реакционную смесь перемешивали в атмосфере водорода в течение 3 ч при комнатной температуре. Реакционную смесь продували азотом и твердые вещества отфильтровывали через целит. Отфильтрованный раствор концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:5). Это давало указанное в заголовке соединение (28,5 г, 80%) в виде желтого масла. MS: (ES, m/z): 174 [M+H]+.
Этап 3. (2S)-5-метокси-2-метил-1,2,3,4-тетрагидрохинолин
[116] В реакторе под давлением на 30 мл (50 атм) растворяли 5-метокси-2-метилхинолин (4,0 г, 23,09 ммоль) в метаноле (10 мл). Тогда Ru(OTf)(η6-гексаметилбензол)((с, S)-TsDPEN) ([N-[(1S,2S)-2-(амино-κN)-1,2-дифенилэтил]-4-метилбензолсульфонамидато-κN] [(1,2,3,4,5,6-η)-1,2,3,4,5,6-гексаметилбензол](1,1,1-трифторметансульфонато-κO)-рутений, полученный в соответствии с процедурой в J. Am. Chem. Soc. 2011, 133, 9878-9891) (150 мг, 0,23 ммоль) был добавлен. К указанному выше водороду вводили. Полученный раствор перемешивали в течение 6 ч при комнатной температуре. Полученную смесь концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:4). Это давало указанное в заголовке соединение (3,0 г, 73%) в виде желтого масла. MS: (ES, m/z): 178 [M+H]+.
Этап 4. метил-(S)-5-метокси-2-метил-3,4-дигидрохинолин-1(2H)-карбоксилат
[117] В круглодонной колбе на 250 мл растворяли (2S)-5-метокси-2-метил-1,2,3,4-тетрагидрохинолин (18 г, 99,52 ммоль) в дихлорметане (100 мл). Затем добавляли пиридин (23,6 г, 298,36 ммоль), а затем метилкарбонохлоридат (9,4 г, 99,47 ммоль). Полученный раствор перемешивали 1 ч при комнатной температуре. Полученный раствор разбавляли в 100 мл дихлорметана и промывали 3×200 мл воды. Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:3). Это давало указанное в заголовке соединение (21 г, 89%) в виде желтого масла. MS: (ES, m/z): 236 [M+H]+.
Этап 5. метил-(S)-5-гидрокси-2-метил-3,4-дигидрохинолин-1(2H)-карбоксилат
[118] В трехгорлой круглодонной колбе на 500 мл растворяли метил-(2S)-5-метокси-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат (21 г, 89,36 ммоль) в дихлорметане (150 мл). Затем добавляли трибромид бора (150 мл, 0,15 моль, 1 М в CH2Cl2). Полученный раствор перемешивали 1 ч при комнатной температуре. Затем реакцию гасили добавлением 300 мл воды. Полученную смесь экстрагировали 3×300 мл дихлорметана. Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:2). Это давало указанное в заголовке соединение (13,5 г, 68%) в виде желтого твердого вещества. MS: (ES, m/z): 222 [M+H]+.
Этап 6. метил-(S)-2-метил-5-(((трифторметил)сульфонил)окси)-3,4-дигидрохинолин-1(2H)-карбоксилат
[119] В круглодонной колбе емкостью 250 мл растворяли метил-(2S)-5-гидрокси-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат (5 г, 18,08 ммоль) в дихлорметане (50 мл). Затем добавляли пиридин (14,3 г, 180,78 ммоль) и ангидрид трифторметансульфоновой кислоты (10,2 г, 36,15 ммоль). Полученный раствор перемешивали 1 ч при комнатной температуре. Полученную смесь промывали водой 3×100 мл. Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:3). Это давало указанное в заголовке соединение (5,5 г, 86%) в виде желтого масла. MS: (ES, m/z): 354 [M+H]+.
Этап 7. метил-(S)-5-((дифенилметилен)амино)-2-метил-3,4-дигидрохинолин-1(2H)-карбоксилат
[120] В круглодонную колбу на 500 мл, продуваемую и поддерживаемую в инертной атмосфере азота растворяли метил(2S)-2-метил-5-[(трифторметан)сульфонилокси]-1,2,3,4-тетрагидрохинолин-1-карбоксилат (23,5 г, 65,18 ммоль) в толуоле (100 мл). Затем добавляли дифенилметанимин (17,9 г, 97,78 ммоль), аддукт трис(дибензилиденацетон)дипалладий-хлороформа (1,19 г, 1,30 ммоль), (+/-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (2,43 г, 3,90 ммоль) и карбоната цезия (42,4 г, 130,13 ммоль). Полученный раствор перемешивали в течение ночи при 100°C в атмосфере азота. Реакционную смесь охлаждали и твердые вещества отфильтровывали. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:3). Это давало указанное в заголовке соединение (33 г, 80%) в виде желтого масла. MS: (ES, m/z): 385 [M+H]+.
Этап 8. метил-(S)-5-амино-2-метил-3,4-дигидрохинолин-1(2H)-карбоксилат
[121] В круглодонной колбе на 500 мл растворяли метил-(2S)-5-[(дифенилметилиден)амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат (33 г, 85,93 ммоль) в метаноле (200 мл). Затем добавляли ацетат натрия (17 г, 207,23 ммоль) и гидрохлорид гидроксиламина (12,3 г, 177,00 ммоль). Полученный раствор перемешивали 2 ч при комнатной температуре. Твердые вещества отфильтровывали. Полученную смесь концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:2). Это давало указанное в заголовке соединение (12,5 г, 66%) в виде желтого твердого вещества. MS: (ES, m/z): 221 [M+H]+.
Этап 9. метил-(S)-5-амино-6-бромо-2-метил-3,4-дигидрохинолин-1(2H)-карбоксилат (Промежуточное соединение 1)
[122] В трехгорлой круглодонной колбе объемом 100 мл растворяли метил-(2S)-5-амино-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат (1 г, 4,09 ммоль) в ацетонитриле (20 мл). Затем добавляли N-бромсукцинимид (730 мг, 4,10 ммоль). Полученный раствор перемешивали 30 мин при комнатной температуре. Полученную смесь концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (1:1). Это давало указанное в заголовке соединение (1,1 г, 90%) в виде желтого твердого вещества. MS: (ES, m/z): 299, 301 [M+H]+.
1H-ЯМР: (400 МГц, CD3OD, м.д.): 7,19(д, J=8,8 Гц, 1H), 6,84(д, J=8,8 Гц, 1H), 4,73-4,69 (м, 1H), 3,74 (с, 3H), 2,64-2,57 (м, 1H), 2,55-2,44 (м, 1H), 2,12-2,05 (м, 1H), 1,82-1,79 (м, 1H), 1,17 (д, J=6,9 Гц, 3H).
Пример 1.2: Синтез (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1карбоновой кислоты (1); (1R,3R)-3-[(7S)-2-[(S)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1карбоновой кислоты (1’)
[123] На Фигуре 2(B) представлена схема синтеза получения Соединения 1 и Соединения 1´, как описано ниже.
Синтез промежуточного соединения 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусной кислоты
Этап 1. 2-(5-фтор-2-метоксифенил)-2-[(триметилсилил)окси]ацетонитрил
[124] Раствор ZnI2 (1,6 мг, 0,01 ммоль), 5-фтор-2-метоксибензальдегида (1,54 г, 9,99 ммоль) в триметилсиланкарбонитриле (1,5 мл, 11,25 ммоль) перемешивали в течение 1 ч при комнатной температуре. Полученную смесь концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением 2-(5-фтор-2-метоксифенил)-2-[(триметилсилил)окси]ацетонитрила в виде белого твердого вещества ( 2,0 г, 79%).
Этап 2. 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусная кислота
[125] Раствор 2-(5-фтор-2-метоксифенил)-2-[(триметилсилил)окси]ацетонитрила (1,50 г, 5,92 ммоль) в соляной кислоте (10 мл, 12M) перемешивали в течение 1 ч при 25°C и затем перемешивали в течение 2 ч при 70°C. Реакционную смесь охлаждали и концентрировали под вакуумом. Неочищенный продукт очищали обращенно-фазовой хроматографией (Колонка: C18; Подвижная фаза, A: вода (содержащая 0,05% TFA) и B: ACN (от 5% до 20% за 30 мин); Детектор, УФ 254 нм) с получением 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусной кислоты в виде белого твердого вещества (1,10 г, 93%).
Этап 3. 6-фтор-2-метил-5-нитрохинолин
[126] Раствор трифторметансульфоновой кислоты (82,0 мл, 0,923 моль) в HNO3 (19,6 мл, 0,437 моль) перемешивали в течение 20 мин при 0°C. После этого добавляли 6-фтор-2-метилхинолин (50,0 г, 0,310 моль) в дихлорметане (300 мл) при 0°C. Полученную смесь перемешивали 15 ч при комнатной температуре (25°C). Реакционную смесь разбавляли водой (300 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали дихлорметаном (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (элюируя смесью 1:4 этилацетат/петролейный эфир) с получением 6-фтор-2-метил-5-нитрохинолина в виде светло-желтого твердого вещества (60,0 г, 94%). ЖХМС (ES, m/z): 207 [M+H]+.
Этап 4. (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин
[127] Раствор (S)-(-)-MeO-BIPHEP (1,03 г, 1,77 ммоль), димера хлор(1,5-циклооктадиен)иридия(I) (538 мг, 0,80 ммоль) в толуоле (100 мл) перемешивали в течение 30 мин при комнатной температуре (25°C) в атмосфере азота. После этого добавляли I2 (410 мг, 1,62 ммоль) и 6-фтор-2-метил-5-нитрохинолин (33,0 г, 0,160 моль) в толуоле (100 мл). Полученную смесь перемешивали в течение 20 ч при комнатной температуре (25°C) в атмосфере водорода (50 атм). Полученную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением неочищенного продукта (35,0 г). Неочищенный продукт растворяли в этилацетате (230 мл) с последующим добавлением D-камфорсульфоновой кислоты (36,9 г, 0,158 моль). Полученный раствор перемешивали в течение 1 ч при 60°C, а затем охлаждали до комнатной температуры. Твердые вещества собирали фильтрованием и промывали этилацетатом (120 мл). Твердые вещества растворяли в воде (50 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали этилацетатом (3×120 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина в виде красного твердого вещества (25,5 г, 76%). ЖХМС (ES, m/z): 211 [M+H]+
Этап 5. метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[128] Раствор (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина (25,3 г, 0,120 моль), пиридина (39,0 мл, 0,484 моль), метилкарбонохлоридата (18,7 мл, 0,242 моль) в дихлорметане (150 мл) перемешивали в течение 3 часов при комнатной температуре (25°C). Реакционную смесь промывали 1 М соляной кислотой (2×70 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде желтого твердого вещества (29,8 г, 92%). ЖХМС (ES, m/z): 269 [M+H]+.
Этап 6. метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро1,2,3,4-тетрагидрохинолин-1-карбоксилат
[129] Раствор метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (29,6 г, 0,110 моль), пиридина (29,6 мл, 0,368 моль), карбоната калия (30,5 г, 0,220 моль), метил-(1R,3R)-3-аминоциклогексан-1-карбоксилата (25,6 г, 162,84 ммоль) в ДМСО (270 мл) перемешивали в течение 15 ч при 90°C и затем охлаждали до комнатной температуры. Реакцию гасили добавлением воды (200 мл) и экстрагировали этилацетатом (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде красного масла (32 г, 72%). ЖХМС (ES, m/z): 406 [M+H]+.
Этап 7. метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[130] Раствор метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (31,0 г, 76,46 ммоль), NH4Cl (24,3 г, 454,28 ммоль), Fe (64,3 г, 1,15 моль) в тетрагидрофуране (300 мл), этаноле (300 мл) и воде (100 мл) перемешивали 1 ч при 80°C и затем охлаждали до комнатной температуры. Твердые вещества отделяли фильтрованием. Полученный раствор разбавляли водой (300 мл) и экстрагировали этилацетатом (3×400 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме, получая метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат в виде темно-зеленого твердого вещества (27,5 г, 92%). ЖХМС (ES, m/z): 376 [M+H]+.
Этап 8. метил-(2S)-5-[2-(5-фтор-2-метоксифенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[131] Раствор 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусной кислоты (240 мг, 1,20 ммоль), HATU (228 мг, 0,60 ммоль), метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (150 мг, 0,40 ммоль), DIEA (0,19 мл, 1,20 ммоль) в N,N-диметилформамиде (10 мл) перемешивали в течение 1 ч при 25°C. Полученный раствор разбавляли H2O (10 мл). Полученный раствор экстрагировали этилацетатом (3×15 мл) и органические слои объединяли. Полученную смесь промывали рассолом (2×20 мл). Смесь сушили над безводным сульфатом натрия и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 3:2 этилацетат/петролейный эфир) с получением метил-(2S)-5-[2-(5-фтор-2-метоксифенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде твердого вещества желтого цвета (180 мг, 81%). ЖХМС (ES, m/z): 558 [M+H]+.
Этап 9. метил-(7S)-2-[(5-фтор-2-метоксифенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилат .
[132] Раствор метил-(2S)-5-[2-(5-фтор-2-метоксифенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (180 мг, 0,32 ммоль) в AcOH (8 мл) перемешивали в течение ночи при 60°C. Реакционную смесь охлаждали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(7S)-2-[(5-фтор-2-метоксифенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H, 8H, 9H-имидазо[4,5-f]хинолин-6-карбоксилата в виде желтого твердого вещества (120 мг, 69%). ЖХМС (ES, m/z): 540 [M+H]+.
Этап 10. (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (1); (1R,3R)-3-[(7S)-2-[(S)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (1’)
[133] Раствор метил-(7S)-2-[(5-фтор-2-метоксифенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилата (120 мг, 0,22 ммоль), и LiOH (16 мг, 0,67 ммоль) в тетрагидрофуране (2,0 мл), метаноле (2,0 мл) и воде (2,0 мл) перемешивали в течение ночи при 25°C. Полученную смесь концентрировали под вакуумом. Неочищенный продукт очищали с помощью Prep-HPLC (колонка, колонка XBridge Prep C18 OBD, 19×150 мм, 5 мкм; Подвижная фаза, A: вода (содержащая 10 ммоль/л NH4HCO3) и B: ACN (от 15,0% до 29,0% за 14 минут); Детектор, УФ 220/254 нм). Продукт разделяли с помощью Chiral-Prep-HPLC (колонка, CHIRALPAK IE, 2×25 см, 5 мкм; Подвижная фаза, A: Hex (содержащий 0,1% FA) и B: этанол (выдержка 50,0% этанола в течение 12 мин); Детектор, УФ 220/254 нм). Фракции продукта концентрировали, чтобы получить (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту (1) в виде белого твердого вещества (23,6 мг, 20%); и (1R,3R)-3-[(7S)-2-[(S)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту (1’) в виде белого твердого вещества (23,8 мг, 20%). Стереоизомерную чистоту определяли с помощью ВЭЖХ: Колонка: CHIRALPAK IE-3, размер Колонки: 0,46×5 см; 3 мкм; Мобильная фаза: Hex (0,1% FA): EtOH=50:50, расход: 1,0 мл/мин.
Первый элюирующий изомер (1): 1H-ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,56-7,47 (м, 1H), 7,47-7,31 (м, 1H), 7,21-7,09 (м, 1H), 7,09-6,89 (м, 2H), 6,53 (с, 1H), 4,81-4,61 (м, 2H), 3,85 (с, 3H), 3,78 (с, 3H), 3,31-3,18 (м, 1H), 3,06-2,82 (м, 2H), 2,57-2,41 (м, 1H), 2,41-2,31 (м, 1H), 2,312,09 (м, 3H), 1,83-1,58 (м, 3H), 1,49-1,21 (м, 2H), 1,16 (д, J=6,8 Гц, 3H). ЖХМС (ES, m/z):
526 [M+H]+.
Второй элюирующий изомер (1’): 1H-ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,69-7,44 (м, 2H), 7,44-7,29 (м, 1H), 7,12-6,99 (м, 1H), 6,98-6,82 (м, 1H), 6,37 (с, 1H), 5,03-4,91 (м, 1H), 4,81-4,69 (м, 1H), 3,78 (с, 3H), 3,61 (с, 3H), 3,22-3,04 (м, 1H), 3,02-2,87 (м, 2H), 2,54-2,41 (м, 1H), 2,41-2,27 (м, 1H), 2,27-2,08 (м, 3H), 1,82-1,58 (м, 3H), 1,58-1,41 (м, 2H), 1,14 (д, J=6,4 Гц, 3H).
ЖХМС (ES, m/z): 526 [M+H]+.
[134] Композиция Формулы (I) может содержать соединение одной или более Формул (II-a), (II-b), (II-c), (II-d), (II-e), (II-f), (II-g), (II-h), (II-i), (II-j), (II-k), (II-l), (II-m), (II-n) и/или (II-o). Например, в некоторых вариантах реализации описание обеспечивает композицию, содержащую соединение 1 вышеуказанной структуры или его фармацевтически приемлемую соль с чистотой по меньшей мере 90%, где композиция содержит менее 10%, например менее 9%, менее 8%, менее 7%, менее 6%, менее 5%, менее 4%, менее 3%, менее 2% или менее 1% в совокупности из одного или более следующих стереоизомеров соединения 1, представленных Формулами (II-a) - (II-o) ниже:
 (II-a)
(II-a) (II-b)
(II-b) (II-c)
(II-c) (II-d)
(II-d) (II-e)
(II-e)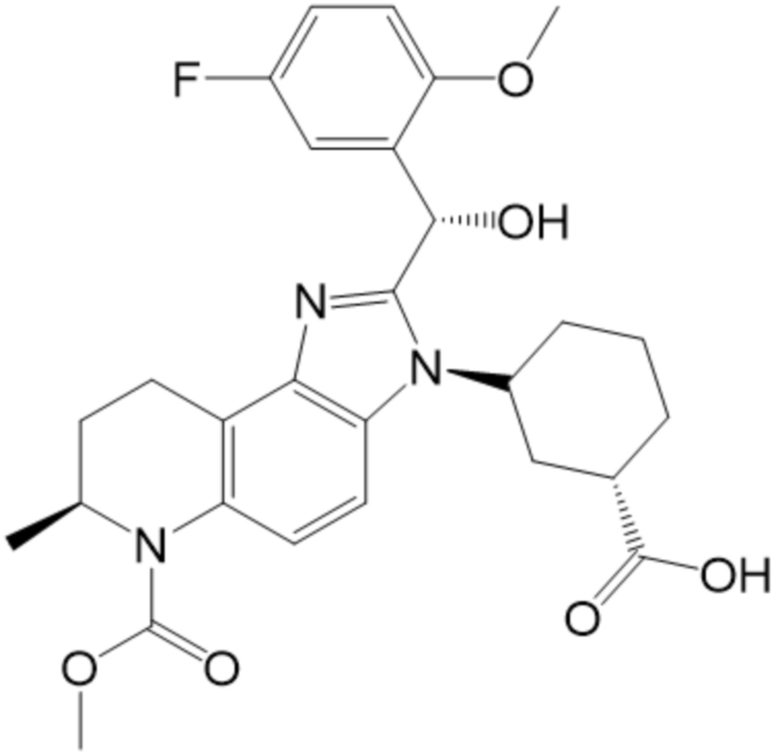 (II-f)
(II-f)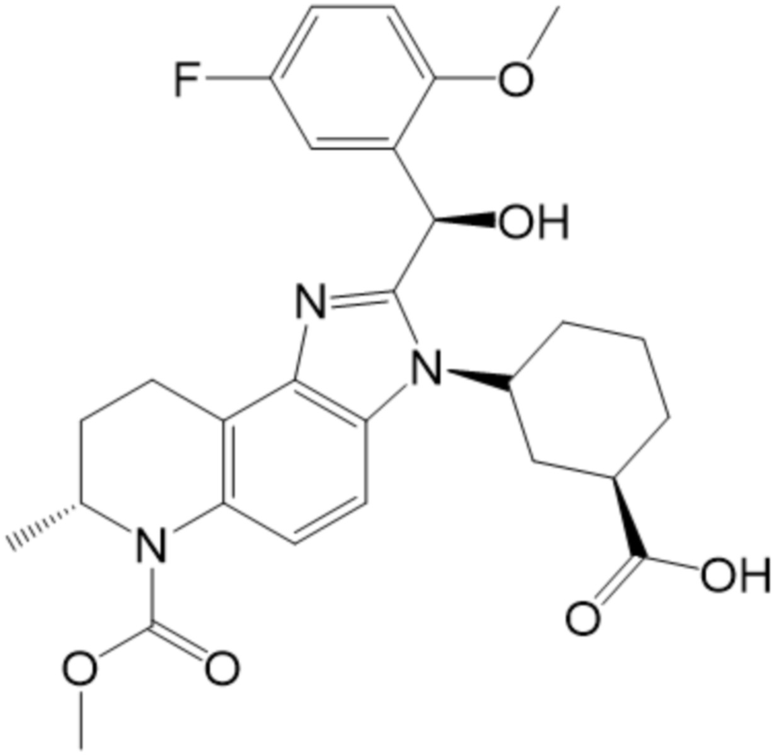 (II-g)
(II-g) (II-h)
(II-h) (II-i)
(II-i) (II-j)
(II-j) (II-k)
(II-k)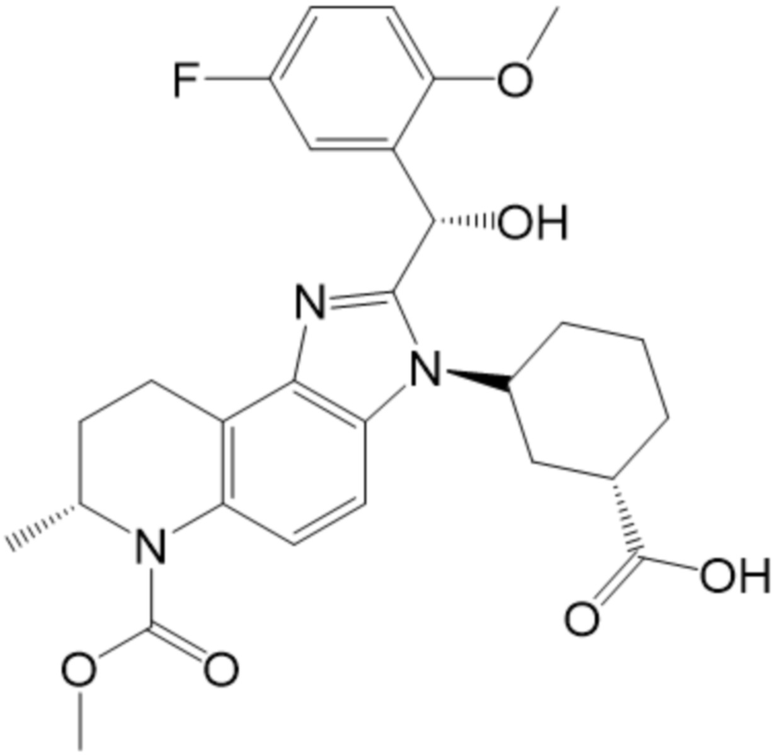
(II-l)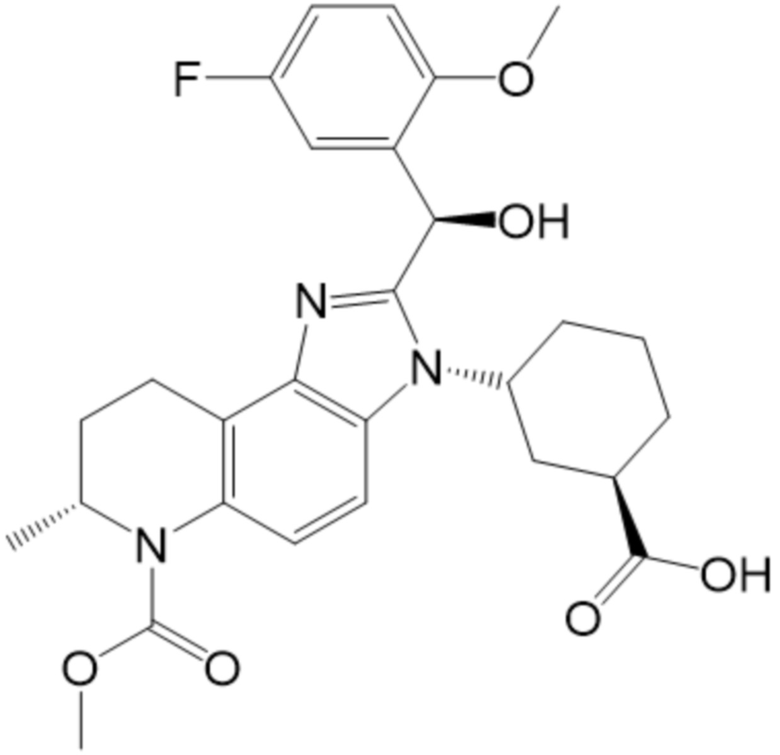 (II-m)
(II-m) (II-n)
(II-n) (II-o)/соединение 1’
(II-o)/соединение 1’
[135] Например, в описании предложена фармацевтическая композиция, содержащая соединение 1 или его фармацевтически приемлемую соль с чистотой по меньшей мере 95%, как определено указанным выше методом ВЭЖХ в Примере 1.7. В описании также представлена фармацевтическая композиция, содержащая соединение 1 с чистотой по меньшей мере 95%, как определено указанным выше методом ВЭЖХ.
[136] В описании предложено соединение Формулы II, полученное вышеуказанным способом, приведенным в Примере 1.2:
 (II)
(II)
или его фармацевтически приемлемая соль, энантиомер, гидрат, сольват, изомер или таутомер.
[137] Для квалифицированного специалиста будет очевидно, что каждый из стереоизомеров соединения Формулы (II) может быть получен путем изменения стереохимии соответствующих реагентов, используемых в способе Примера 1.2 выше. Например, регулируя реагент, используемый на стадии 4 Примера 1.2, можно синтезировать такие соединения, как соединения Формул (II-m) и (II-n). Точно так же на Этапе 6 Примера 1.2, реагент метил-(1S,3R)-3-аминоциклогексан-1-карбоксилат может использоваться вместо метил-(1R,3R)-3-аминоциклогексан-1-карбоксилата для получения соединения Формулы (II-b) и (II-e). Для квалифицированного специалиста будет очевидно, что путем совмещения этих типов модификаций процесса, изложенного в Примере 1,2, можно синтезировать каждое из соединений (II-a)-(II-o), изображенных выше.
Пример 1.3: (1R,3R)-3-[(7S)-2-[(R)-гидрокси(фенил)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (2)
[138] На Фигуре 2(C) представлена схема синтеза получения Соединения 2, как описано ниже.
Этап 1. 6-фтор-2-метил-5-нитрохинолин
[139] Раствор трифторметансульфоновой кислоты (82,0 мл, 0,923 моль) в HNO3 (19,6 мл, 0,437 моль) перемешивали в течение 20 мин при 0°C. После этого добавляли 6-фтор-2-метилхинолин (50,0 г, 0,310 моль) в дихлорметане (300 мл) при 0°C. Полученную смесь перемешивали 15 ч при комнатной температуре (25°C). Реакционную смесь разбавляли водой (300 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали дихлорметаном (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (элюируя смесью 1:4 этилацетат/петролейный эфир) с получением 6-фтор-2-метил-5-нитрохинолина в виде светло-желтого твердого вещества (60,0 г, 94%). ЖХМС (ES, m/z): 207 [M+H]+.
Этап 2. (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин
[140] Раствор (S)-(-)-MeO-BIPHEP (1,03 г, 1,77 ммоль), димера хлор(1,5-циклооктадиен)иридия(I) (538 мг, 0,80 ммоль) в толуоле (100 мл) перемешивали в течение 30 мин при комнатной температуре (25°C) в атмосфере азота. После этого добавляли I2 (410 мг, 1,62 ммоль), 6-фтор-2-метил-5-нитрохинолин (33,0 г, 0,160 моль) в толуоле (100 мл). Полученную смесь перемешивали в течение 20 ч при комнатной температуре (25°C) в атмосфере водорода (50 атм). Полученную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением неочищенного продукта (35,0 г). Неочищенный продукт растворяли в этилацетате (230 мл) с последующим добавлением D-камфорсульфоновой кислоты (36,9 г, 0,158 моль). Полученный раствор перемешивали в течение 1 ч при 60°C, а затем охлаждали до комнатной температуры. Твердые вещества собирали фильтрованием и промывали этилацетатом (120 мл). Твердые вещества растворяли в воде (50 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали этилацетатом (3×120 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина в виде красного твердого вещества (25,5 г, 76%). ЖХМС (ES, m/z): 211 [M+H]+.
Этап 3. метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[141] Раствор (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина (25,3 г, 0,120 моль), пиридина (39,0 мл, 0,484 моль), метилкарбонохлоридата (18,7 мл, 0,242 моль) в дихлорметане (150 мл) перемешивали в течение 3 часов при комнатной температуре (25°C). Реакционную смесь промывали 1 М соляной кислотой (2×70 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде желтого твердого вещества (29,8 г, 92%). ЖХМС (ES, m/z): 269 [M+H]+.
Этап 4. метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[142] Раствор метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (29,6 г, 0,110 моль), пиридина (29,6 мл, 0,368 моль), карбоната калия (30,5 г, 0,220 моль), метил-(1R,3R)-3-аминоциклогексан-1-карбоксилата (25,6 г, 162,84 ммоль) в ДМСО (270 мл) перемешивали в течение 15 ч при 90°C и затем охлаждали до комнатной температуры. Реакцию гасили добавлением воды (200 мл) и экстрагировали этилацетатом (3 х 300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде красного масла (32 г, 72%). ЖХМС (ES, m/z): 406 [M+H]+.
Этап 5. метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[143] Раствор (2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (31,0 г, 76,46 ммоль), NH4Cl (24,3 г, 454,28 ммоль), Fe (64,3 г, 1,15 моль) в тетрагидрофуране (300 мл), этаноле (300 мл), воде (100 мл) перемешивали 1 ч при 80°C и затем охлаждали до комнатной температуры. Твердые вещества отфильтровывали. Полученный раствор разбавляли водой (300 мл) и экстрагировали этилацетатом (3×400 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме, получая метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат в виде темно-зеленого твердого вещества (27,5 г, 92%). ЖХМС (ES, m/z): 376 [M+H]+.
Этап 6. метил-(2S)-5-((R)-2-гидрокси-2-фенилацетамидо)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[144] Раствор (R)-2-гидрокси-2-фенилуксусной кислоты (972 мг, 6,39 ммоль), HATU (1,20 г, 3,16 ммоль), метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (800 мг, 2,13 ммоль), DIEA (1,08 мл, 6,20 ммоль) в N,N-диметилформамиде (10 мл) перемешивали в течение 5 ч при комнатной температуре (25°C). Полученный раствор разбавляли водой (30 мл) и экстрагировали этилацетатом (3×50 мл). Органические слои объединяли и промывали рассолом (2×25 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(2S)-5-((R)-2-гидрокси-2-фенилацетамидо)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде бесцветного масла (600 мг, 55%). ЖХМС (ES, m/z): 510 [M+H]+.
Этап 7. метил-(7S)-2-[(R)-гидрокси(фенил)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилат
[145] Раствор метил-(2S)-5-((R)-2-гидрокси-2-фенилацетамидо)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (600 мг, 1,18 ммоль) в ледяной уксусной кислоте (5 мл, 98%) перемешивали в течение ночи при 40°C, а затем охлаждали до комнатной температуры. Реакционную смесь разбавляли водой (10 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали этилацетатом (3×15 мл). Органические слои объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(7S)-2-[(R)-гидрокси(фенил)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H, 8H, 9H-имидазо[4,5-f]хинолин-6-карбоксилата (400 мг, 69%) в виде бесцветного масла. ЖХМС (ES, m/z): 492 [M+H]+.
Этап 8. (1R,3R)-3-[(7S)-2-[(R)-гидрокси(фенил)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо-[4,5-f]-хинолин-3-ил]циклогексан-1-карбоновая кислота (2)
[146] Раствор метил-(7S)-2-[(R)-гидрокси(фенил)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилата (400 мг, 0,81 ммоль), LiOH (100 мг, 4,17 ммоль) в тетрагидрофуране (5 мл) и воде (2 мл) перемешивали в течение ночи при комнатной температуре (25°C). Полученную смесь концентрировали под вакуумом. Неочищенный продукт очищали препаративной ВЭЖХ (Колонка: Колонка XBridge Shield RP18 OBD, 5 мкм, 19×150 мм; Подвижная фаза, A: вода (содержащая 10 ммоль/л NH4HCO3) и B: ACN (от 3% до 30% за 21 мин); Детектор: УФ 254 нм). Фракции продукта лиофилизировали, чтобы получить (1R,3R)-3-[(7S)-2-[(R)-гидрокси(фенил)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо-[4,5-f]-хинолин-3-ил]циклогексан-1-карбоновую кислоту в виде белого твердого вещества (83,7 мг, 22%). Стереоизомерную чистоту определяли с помощью ВЭЖХ: Колонка: CHIRALPAK IE-3, размер Колонки: 0,46 х 5 см; 3 мкм; Мобильная фаза: Hex (0,1% FA): EtOH=85:15, Расход: 1,0 мл/мин.
1H-ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,47-7,28 (м, 7H), 6,12 (с, 1H), 4,84-4,74 (м, 2H), 3,79 (с, 3H), 3,33-3,25 (м, 1H), 3,03-2,96 (м, 1H), 2,86-2,82 (м, 1H), 2,38-2,25 (м, 2H), 2,25-2,07 (м, 3H), 1,79-1,72 (м, 1H), 1,64-1,57 (м, 2H), 1,40-1,29 (м, 2H), 1,16 (д, J=6,8 Гц, 3H). ЖХМС (ES, m/z): 478 [M+H]+; 99,13% ee.
[147] Композиция Формулы (I) может содержать соединение одной или более формул (III-a), (III-b), (III-c), (III-d), (III-e), (III-f), (III-g), (III-h), (III-i), (III-j), (III-k), (III-l), (III-m), (III-n), и/или (III-o). Например, в некоторых вариантах реализации описание обеспечивает композицию, содержащую соединение 2 вышеуказанной структуры или его фармацевтически приемлемую соль с чистотой по меньшей мере 90%, где композиция содержит менее 10%, например менее 9%, менее 8%, менее 7%, менее 6%, менее 5%, менее 4%, менее 3%, менее 2% или менее 1% в совокупности из одного или более следующих стереоизомеров соединения 2, представленных Формулами (III-a) - (III-o) ниже:
 (III-a)
(III-a) (III-b)
(III-b) (III-c)
(III-c)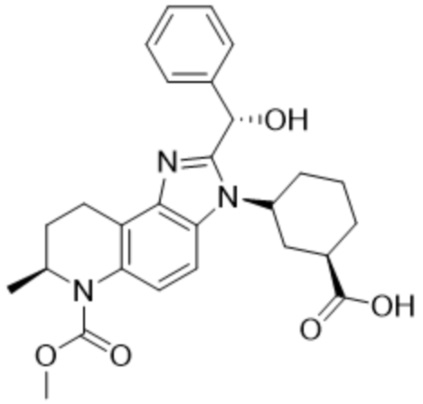 (III-d)
(III-d) (III-e)
(III-e) (III-f)
(III-f) (III-g)
(III-g) (III-h)
(III-h) (III-i)
(III-i) (III-j)
(III-j)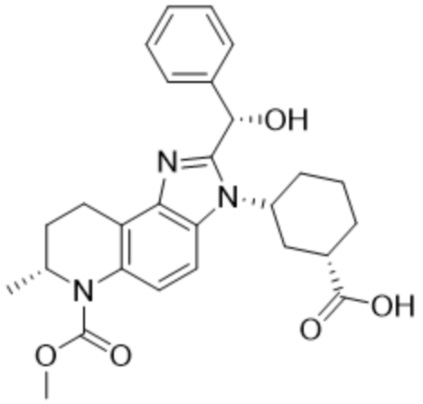 (III-k)
(III-k)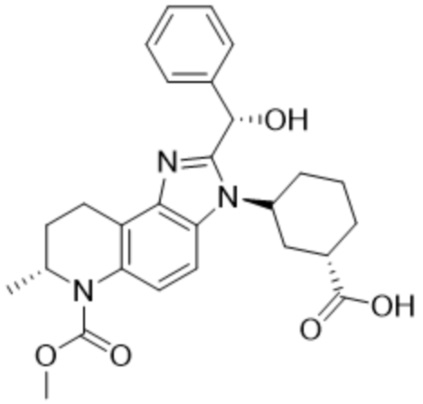 (III-l)
(III-l) (III-m)
(III-m) (III-n)
(III-n) (III-o)
(III-o)
[148] Например, в описании предложена фармацевтическая композиция, содержащая соединение 2 или его фармацевтически приемлемую соль с чистотой по меньшей мере 95%, как определено указанным выше методом ВЭЖХ в Примере 1.7. В описании также представлена фармацевтическая композиция, содержащая соединение 2 с чистотой по меньшей мере 95%, как определено указанным выше методом ВЭЖХ.
Пример 1.4: (1R,3R)-3-[(7S)-2-[(S)-[2-(дифторметокси)-5-фторфенил](гидрокси) метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (452), (1R,3R)-3-[(7S)-2-[(R)-[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо [4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (3)
[149] На Фигуре 2(D) представлена схема синтеза получения Соединения 3 и Соединения 452, как описано ниже.
Этап 1. 2-(дифторметокси)-5-фторбензальдегид
[150] Раствор 5-фтор-2-гидроксибензальдегида (2,0 г, 14,3 ммоль), диэтил(бромдифторметил)фосфоната (5,69 г, 21,3 ммоль), гидроксида калия (16,0 г, 285 ммоль) в MeCN (100 мл) и воде (50 мл) перемешивали в течение 1 ч при -30°C. Реакционную смесь разбавляли водой (20 мл). Полученный раствор экстрагировали этилацетатом (3×100 мл), органические слои объединяли и сушили над безводным сульфатом натрия. Твердые вещества отфильтровывали. Полученную смесь концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением 2-(дифторметокси)-5-фторбензальдегида в виде твердого вещества желтого цвета (1,46 г, 54%). ЖХМС (ES, m/z): 191 [M+H]+.
Этап 2. 2-[2-(дифторметокси)-5-фторфенил]-2-[(триметилсилил)окси]ацетонитрил
[151] Раствор 2-(дифторметокси)-5-фторбензальдегида (1,46 г, 7,68 ммоль), TMSCN (760 мг, 7,66 ммоль), ZnI2 (50 мг, 0,16 ммоль) в дихлорметане (3 мл) перемешивали в течение 2 ч при комнатной температуре (25°C). Полученную смесь концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением 2-[2-(дифторметокси)-5-фторфенил]-2-[(триметилсилил)окси]ацетонитрила в виде твердого вещества желтого цвета (800 мг, 36%). ЖХМС (ES, m/z):290 [M+H]+.
Этап 3. 2-[2-(дифторметокси)-5-фторфенил]-2-гидроксиуксусная кислота
[152] Раствор 2-[2-(дифторметокси)-5-фторфенил]-2-[(триметилсилил)окси]ацетонитрила (800 мг, 2,77 ммоль), 1,4-диоксана (2,0 мл), хлористого водорода (1,0 мл, 12M) в воде (2 мл) перемешивали в течение 12 ч при 70°C, а затем охлаждали до комнатной температуры. Полученный раствор концентрировали под вакуумом. Неочищенный продукт очищали колоночной хроматографией с обращенной фазой (вода (содержащая 0,05% TFA)/MeCN) с получением 2-[2-(дифторметокси)-5-фторфенил]-2-гидроксиуксусной кислоты (400 мг, 61%). ЖХМС (ES, m/z): 237 [M+H]+.
Этап 4. 6-фтор-2-метил-5-нитрохинолин
[153] Раствор трифторметансульфоновой кислоты (82,0 мл, 0,923 моль) в HNO3 (19,6 мл, 0,437 моль) перемешивали в течение 20 мин при 0°C. После этого добавляли 6-фтор-2-метилхинолин (50,0 г, 0,310 моль) в дихлорметане (300 мл) при 0°C. Полученную смесь перемешивали 15 ч при комнатной температуре (25°C). Реакционную смесь разбавляли водой (300 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали дихлорметаном (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (элюируя смесью 1:4 этилацетат/петролейный эфир) с получением 6-фтор-2-метил-5-нитрохинолина в виде светло-желтого твердого вещества (60,0 г, 94%). ЖХМС (ES, m/z): 207 [M+H]+.
Этап 5. (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин
[154] Раствор (S)-(-)-MeO-BIPHEP (1,03 г, 1,77 ммоль), димера хлор(1,5-циклооктадиен)иридия(I) (538 мг, 0,80 ммоль) в толуоле (100 мл) перемешивали в течение 30 мин при комнатной температуре (25°C) в атмосфере азота. После этого добавляли I2 (410 мг, 1,62 ммоль), 6-фтор-2-метил-5-нитрохинолин (33,0 г, 0,160 моль) в толуоле (100 мл). Полученную смесь перемешивали в течение 20 ч при комнатной температуре (25°C) в атмосфере водорода (50 атм). Полученную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением неочищенного продукта (35,0 г). Неочищенный продукт растворяли в этилацетате (230 мл) с последующим добавлением D-камфорсульфоновой кислоты (36,9 г, 0,158 моль). Полученный раствор перемешивали в течение 1 ч при 60°C, а затем охлаждали до комнатной температуры. Твердые вещества собирали фильтрованием и промывали этилацетатом (120 мл). Твердые вещества растворяли в воде (50 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали этилацетатом (3×120 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина в виде красного твердого вещества (25,5 г, 76%). ЖХМС (ES, m/z): 211 [M+H]+.
Этап 6. метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[155] Раствор (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина (25,3 г, 0,120 моль), пиридина (39,0 мл, 0,484 моль), метилкарбонохлоридата (18,7 мл, 0,242 моль) в дихлорметане (150 мл) перемешивали в течение 3 часов при комнатной температуре (25°C). Реакционную смесь промывали 1 М хлористым водородом (2×70 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде желтого твердого вещества (29,8 г, 92%). ЖХМС (ES, m/z): 269 [M+H]+.
Этап 7. метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[156] Раствор метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (29,6 г, 0,110 моль), пиридина (29,6 мл, 0,368 моль), карбоната калия (30,5 г, 0,220 моль), метил-(1R,3R)-3-аминоциклогексан-1-карбоксилата (25,6 г, 162,84 ммоль) в ДМСО (270 мл) перемешивали в течение 15 ч при 90°C и затем охлаждали до комнатной температуры. Реакцию гасили добавлением воды (200 мл) и экстрагировали этилацетатом (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде красного масла (32 г, 72%). ЖХМС (ES, m/z): 406 [M+H]+.
Этап 8. метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[157] Раствор метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (31,0 г, 76,46 ммоль), NH4Cl (24,3 г, 454,28 ммоль), Fe (64,3 г, 1,15 моль) в тетрагидрофурана (300 мл), этанола (300 мл), воды (100 мл) перемешивали 1 ч при 80°C и затем охлаждали до комнатной температуры. Твердые вещества отфильтровывали. Полученный раствор разбавляли водой (300 мл) и экстрагировали этилацетатом (3×400 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме, получая метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат в виде темно-зеленого твердого вещества (27,5 г, 92%). ЖХМС (ES, m/z): 376 [M+H]+.
Этап 9. метил-(2S)-5-[2-[2-(дифторметокси)-5-фторфенил]-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[158] Раствор метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (200 мг, 0,53 ммоль), 2-[2-(дифторметокси)-5-фторфенил]-2-гидроксиуксусной кислоты (220 мг, 0,93 ммоль), DMTMM (350 мг, 1,26 ммоль) в дихлорметане (5 мл) перемешивали в течение 1 ч при комнатной температуре (25°C). Полученный раствор концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(2S)-5-[2-[2-(дифторметокси)-5-фторфенил]-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде желтого твердого вещества (70,0 мг, 22%). ЖХМС (ES, m/z): 594 [M+H]+.
Этап 10. метил-(7S)-2-[[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилат
[159] Раствор метил-(2S)-5-[2-[2-(дифторметокси)-5-фторфенил]-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (70,0 мг, 0,12 ммоль) в ледяной уксусной кислоте (2,0 мл) перемешивали в течение ночи при 40°C, а затем охлаждали до комнатной температуры. Полученный раствор концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:2 этилацетат/петролейный эфир) с получением метил-(7S)-2-[[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилата в виде желтого твердого вещества (50,0 мг, 74%). ЖХМС (ES, m/z): 576 [M+H]+.
Этап 11. (1R,3R)-3-[(7S)-2-[(S)-[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (452); (1R,3R)-3-[(7S)-2-[(R)-[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (3)
[160] Раствор метил-(7S)-2-[[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилата (50,0 мг, 0,09 ммоль), LiOH (10,0 мг, 0,42 ммоль) в тетрагидрофуране (2,0 мл) и воды (2,0 мл) перемешивали в течение ночи при комнатной температуре (25°C). Полученную смесь концентрировали под вакуумом. Неочищенный продукт очищали с помощью Prep-HPLC (колонка, колонка XBridge Shield RP18 OBD, 30×150 мм, 5 мкм; Подвижная фаза, A: вода (содержащая 10 ммоль/л NH4HCO3) и B: ACN (от 25,0% до 35,0% за 8 мин); Детектор, УФ 254/220 нм). Фракции продукта концентрировали, чтобы получить (1R,3R)-3-[(7S)-2-[(S)-[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту (452) в виде белого твердого вещества (4,50 мг, 9%), и (1R,3R)-3-[(7S)-2-[(R)-[2-(дифторметокси)-5-фторфенил](гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту (515) в виде белого твердого вещества (4,30 мг, 9%). Энантиомерный избыток определяли с помощью ВЭЖХ: Колонка: CHIRALPAK IE-3, размер Колонки: 0,46×5 см; 3 мкм; Сорастворитель: IPA (20 мМ NH3) Градиент (B%): От 10% до 50% за 4,0 мин, удерживание 2,0 мин при 50%.
Первый элюирующий изомер (452): 1H-ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,63-7,61 (м, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,41 (д, J=9,2 Гц, 1H) 7,20-7,13 (м, 2H), 6,67-6,30 (м, 2H), 4,98-4,95 (м, 1H), 4,76-4,71 (м, 1H), 3,78 (с, 3H), 3,15-2,86 (м, 3H), 2,46-2,20 (м, 5H), 1,81-1,53 (м, 5H), 1,13 (д, J=6,8 Гц, 3H). ЖХМС (ES, m/z): 562 [M+H]+.
Второй элюирующий изомер (3): 1H-ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,55-7,53 (м, 1H), 7,47-7,42 (м, 2H), 7,40-7,12 (м, 2H), 6,85-6,44 (м, 2H), 4,94-4,91 (м, 1H), 4,76-4,71 (м, 1H), 3,78 (с, 3H), 3,22-2,84 (м, 3H), 2,46-2,23 (м, 5H), 1,84-1,61 (м, 5H), 1,14 (д, J=6,4 Гц, 3H). ЖХМС (ES, m/z): 562 [M+H]+ ; >99,99% ee.
[161] В некоторых вариантах осуществления описание обеспечивает первый элюируемый изомер, полученный на Этапе 11 процесса, описанного в Примере 1,4. В некоторых вариантах осуществления описание обеспечивает второй элюируемый изомер, полученный на Этапе 11 процесса, описанного в Примере 1,4.
[162] Композиция Формулы (I) может содержать соединение одной или более Формул (IV-a), (IV-b), (IV-c), (IV-d), (IV-e), (IV-f), (IV-g), (IV-h), (IV-i), (IV-j), (IV-k), (IV-l), (IV-m), (IV-n), и/или (IV-o). Например, в некоторых вариантах реализации описание обеспечивает композицию, содержащую соединение 3 вышеуказанной структуры или его фармацевтически приемлемую соль с чистотой по меньшей мере 90%, где композиция содержит менее 10%, например менее 9%, менее 8%, менее 7%, менее 6%, менее 5%, менее 4%, менее 3%, менее 2% или менее 1% в совокупности из одного или более следующих стереоизомеров соединения 3, представленных Формулами (IV-a) - (IV-o) ниже:
 (IV-a)
(IV-a) (IV-b)
(IV-b)
(IV-c) (IV-d)
(IV-d) (IV-e)
(IV-e) (IV-f)
(IV-f) (IV-g)
(IV-g) (IV-h)
(IV-h) (IV-i)
(IV-i) (IV-j)
(IV-j) (IV-k)
(IV-k) (IV-l)
(IV-l) (IV-m)
(IV-m) (IV-n)
(IV-n) (IV-o)
(IV-o)
[163] Например, в описании предложена фармацевтическая композиция, содержащая соединение 3 или его фармацевтически приемлемую соль с чистотой по меньшей мере 95%, как определено указанным выше методом ВЭЖХ в Примере 1.7. В описании также представлена фармацевтическая композиция, содержащая соединение 3 с чистотой по меньшей мере 95%, как определено указанным выше методом ВЭЖХ.
Пример 1.5: (1R,3R)-3-((S)-2-бензил-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновая кислота (4)
[164] На Фигуре 2(E) представлена схема синтеза получения Соединения 4, как описано ниже.
Этап 1. метил-(S)-5-амино-6-(((1R,3R)-3-(метоксикарбонил)циклогексил)амино)-2-метил-3,4-дигидрохинолин-1-(2H)-карбоксилат
[165] В 10-миллилитровой круглодонной колбе, продуваемой и поддерживаемой инертной атмосферой азота, растворяли метил-(1R,3R)-3-аминоциклогексан-1-карбоксилат гидрохлорида (130 мг, 0,67 ммоль) в диоксане (4 мл). Затем добавляли метил-(2S)-5-амино-6-бром-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (100 мг, 0,33 ммоль, Промежуточное соединение 1), Brettphos (72 мг, 0,13 ммоль) предварительный катализатор Brettphos 3-го поколения (61 мг, 0,07 ммоль) и трет-бутоксид натрия (97 мг, 1,01 ммоль). Полученный раствор перемешивали в течение 1 ч при 100°C в атмосфере азота. Реакционную смесь охлаждали и твердые вещества отфильтровывали. Полученную смесь концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (2:1). Это давало указанное в заголовке соединение (41,3 мг, 33%) в виде темно-зеленого твердого вещества. MS: (ES, m/z): 376 [M+H]+.
Этап 2. метил-(S)-2-бензил-3-((1R,3R)-3-(метоксикарбонил)циклогексил)-7-метил-3,7,8,9-тетрагидро-6H-имидазо[4,5-f]хинолин-6-карбоксилат
[166] В круглодонной колбе на 25 мл растворяли метилметил-(S)-5-амино-6-(((1R,3R)-3-(метоксикарбонил)циклогексил)амино)-2-метил-3,4-дигидрохинолин-1(2H)-карбоксилат (165,4 мг, 0,44 ммоль) в дихлорметане (5 мл). Затем добавляли 2-фенилацетальдегид (158,8 мг, 1,32 ммоль). Полученный раствор перемешивали 2 ч при комнатной температуре. Полученную смесь концентрировали под вакуумом. Остаток очищали FCC, элюируя смесью этилацетат/петролейный эфир (2:1). Это давало указанное в заголовке соединение (122,9 мг, 59%) в виде желтого твердого вещества. MS: (ES, m/z): 476 [M+H]+.
Этап 3. (1R,3R)-3-[(7S)-2-бензил-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (4)
[167] В круглодонной колбе на 25 мл растворяли метил-(S)-2-бензил-3-((1R,3R)-3-(метоксикарбонил)циклогексил)-7-метил-3,7,8,9-тетрагидро- 6H-имидазо[4,5-f]хинолин-6-карбоксилат (30 мг, 0,06 ммоль) в тетрагидрофуране (0,5 мл). Затем добавляли воду (0,5 мл), а затем гидроксид лития (7,0 мг, 0,29 ммоль). Полученный раствор перемешивали 3 ч при 85°C. Значение pH раствора доводили до 5-6 с помощью соляной кислоты (1 моль/л). Полученный раствор экстрагировали этилацетатом (3×20 мл). Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Неочищенный продукт очищали с помощью препаративной ВЭЖХ в следующих условиях: Колонка, XBridge C18 OBD Prep Column,  5 мкм, 19 мм X 250 мм; подвижная фаза, А: Вода (содержащая 10 ммоль/л NH4HCO3) и B: ACN (от 10,0% до 30,0% ACN за 10 мин); УФ-детектор: 254 нм. Это давало указанное в заголовке соединение (15,2 мг, 52%) в виде белого твердого вещества.
5 мкм, 19 мм X 250 мм; подвижная фаза, А: Вода (содержащая 10 ммоль/л NH4HCO3) и B: ACN (от 10,0% до 30,0% ACN за 10 мин); УФ-детектор: 254 нм. Это давало указанное в заголовке соединение (15,2 мг, 52%) в виде белого твердого вещества.
1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,47 (д, J=9,0 Гц, 1H), 7,39 (д, J=8,9 Гц, 1H), 7,35-7,19 (м, 5H), 4,84-4,68 (м, 2H), 4,45-4,25 (м, 2H), 3,79 (с, 3H), 3,22-3,14 (м, 1H), 2,98-2,85 (м, 2H), 2,40-2,02 (м, 5H), 1,83-1,70 (м, 1H), 1,64-1,54 (м, 2H), 1,33-1,13 (м, 5H). MS: (ES, m/z): 462 [M+H]+.
[168] Соединение 17, Соединение 18 и Соединение 19 получали с использованием стандартных химических манипуляций и процедур, аналогичных тем, которые использовались для получения Примера 16.
Пример 1.6: 3-((7S)-2-((4-хлорофенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновая кислота (413); и (1R,3R)-3-((S)-2-((S)-(4-хлорофенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновая кислота (501)
[169] На Фигуре 2(F) представлена схема синтеза получения Соединения 413 и Соединения 501, как описано ниже.
Этап 1. 6-фтор-2-метил-5-нитрохинолин
[170] Раствор трифторметансульфоновой кислоты (82,0 мл, 0,923 моль) в HNO3 (19,6 мл, 0,437 моль) перемешивали в течение 20 мин при 0°C. После этого добавляли 6-фтор-2-метилхинолин (50,0 г, 0,310 моль) в дихлорметане (300 мл) при 0°C. Полученную смесь перемешивали 15 ч при комнатной температуре (25°C). Реакционную смесь разбавляли водой (300 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали дихлорметаном (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (элюируя смесью 1:4 этилацетат/петролейный эфир) с получением 6-фтор-2-метил-5-нитрохинолина в виде светло-желтого твердого вещества (60,0 г, 94%). ЖХМС (ES, m/z): 207 [M+H]+.
Этап 2. (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин
[171] Раствор (S)-(-)-MeO-BIPHEP (1,03 г, 1,77 ммоль), димера хлор(1,5-циклооктадиен)иридия(I) (538 мг, 0,80 ммоль) в толуоле (100 мл) перемешивали в течение 30 мин при комнатной температуре (25°C) в атмосфере азота. После этого добавляли I2 (410 мг, 1,62 ммоль) и 6-фтор-2-метил-5-нитрохинолин (33,0 г, 0,160 моль) в толуоле (100 мл). Полученную смесь перемешивали в течение 20 ч при комнатной температуре (25°C) в атмосфере водорода (50 атм). Полученную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением неочищенного продукта (35,0 г). Неочищенный продукт растворяли в этилацетате (230 мл) с последующим добавлением D-камфорсульфоновой кислоты (36,9 г, 0,158 моль). Полученный раствор перемешивали в течение 1 ч при 60°C, а затем охлаждали до комнатной температуры. Твердые вещества собирали фильтрованием и промывали этилацетатом (120 мл). Твердые вещества растворяли в воде (50 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали этилацетатом (3×120 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина в виде красного твердого вещества (25,5 г, 76%). ЖХМС (ES, m/z): 211 [M+H]+.
Этап 3. метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[172] Раствор (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина (25,3 г, 0,120 моль), пиридина (39,0 мл, 0,484 моль), и метилкарбонохлоридата (18,7 мл, 0,242 моль) в дихлорметане (150 мл) перемешивали в течение 3 часов при комнатной температуре (25°C). Реакционную смесь промывали 1 н. хлористым водородом (водн., 2 х 70 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде желтого твердого вещества (29,8 г, 92%). ЖХМС (ES, m/z): 269 [M+H]+.
Этап 4. метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[173] Раствор метил-(2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (29,6 г, 0,110 моль), пиридина (29,6 мл, 0,368 моль), карбоната калия (30,5 г, 0,220 моль), и метил-(1R,3R)-3-аминоциклогексан-1-карбоксилата (25,6 г, 162,84 ммоль) в DMSO (270 мл) перемешивали в течение 15 ч при 90°C, а затем охлаждали до комнатной температуры. Реакцию гасили добавлением воды (200 мл) и экстрагировали этилацетатом (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде красного масла (32 г, 72%). ЖХМС (ES, m/z): 406 [M+H]+.
Этап 5. метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[174] Раствор метил-(2S)-2-метил-5-нитро-6-[[(1R,3R)-4-(метоксикарбонил)циклогексил]амино]-1,2,3,4-тетрагидрохинолин-1-карбоксилата (31,0 г, 76,46 ммоль), NH4Cl (24,3 г, 454,28 ммоль), и Fe (порошок, 64,3 г, 1,15 моль) в тетрагидрофуране (300 мл), этаноле (300 мл), воде (100 мл) перемешивали в течение 1 ч при 80°C, а затем охлаждали до комнатной температуры. Твердые вещества отфильтровывали. Полученный раствор разбавляли водой (300 мл) и экстрагировали этилацетатом (3×400 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением метил-(2S)-5-((R)-2-гидрокси-2-фенилацетамидо)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил] амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде темно-зеленого твердого вещества (27,5 г, 92%). ЖХМС (ES, m/z): 376 [M+H]+.
Этап 6. метил-(2S)-5-[2-(4-хлорофенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[175] Раствор 2-(4-хлорофенил)-2-гидроксиуксусной кислоты (112 мг, 0,60 ммоль), HATU (304 мг, 0,80 ммоль), метил-(2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (150 мг, 0,40 ммоль), и DIEA (155 мг, 1,20 ммоль) в N,N-диметилформамиде (2 мл) перемешивали 15 ч при комнатной температуре (25°C). Полученный раствор разбавляли водой (30 мл) и экстрагировали этилацетатом (3×50 мл). Органические слои объединяли и промывали рассолом (2×25 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(2S)-5-[2-(4-хлорофенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(мет-ксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде желтого масла (70,0 мг, 32%). ЖХМС (ES, m/z): 544 [M+H]+.
Этап 7. метил-(7S)-2-[(4-хлорофенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилат
[176] Раствор метил-(2S)-5-[2-(4-хлорофенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (60,0 мг, 0,11 ммоль) в AcOH (2 мл) перемешивали 15 ч при 40°C и затем охлаждали до комнатной температуры. Реакционную смесь разбавляли водой (10 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали этилацетатом (3×15 мл). Органические слои объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил-(7S)-2-[(4-хлорофенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилата в виде желтого масла (46,0 мг, 79%). ЖХМС (ES, m/z): 526 [M+H]+.
Этап 8. (1R,3R)-3-[(7S)-2-[(R)-(4-хлорофенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (413); (1R,3R)-3-[(7S)-2-[(S)-(4-хлорофенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (501)
[177] Раствор метил-(7S)-2-[(4-хлорофенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилата (50,0 мг, 0,10 ммоль), и LiOH (11,4 мг, 0,48 ммоль) в тетрагидрофуране (1 мл) и воде (1 мл) перемешивали 15 ч при 25°C. Полученную смесь концентрировали под вакуумом. Неочищенный продукт очищали препаративной ВЭЖХ (Колонка: Колонка XBridge Shield RP18 OBD, 5 мкм, 19×150 мм; Подвижная фаза, A: вода (содержащая 10 ммоль/л NH4HCO3) и B: ACN (от 10% до 37% за 12 мин); Детектор: УФ 254 нм). Фракции продукта лиофилизировали, чтобы получить (1R,3R)-3-[(7S)-2-[(R)-(4-хлорофенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту (413) в виде белого твердого вещества (10,5 мг, 43%); и (1R,3R)-3-[(7S)-2-[(S)-(4-хлорофенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту (501) в виде белого твердого вещества (7,0 мг, 29%).
Первый элюирующий изомер (413): 1H-ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,49 (д, J=9,0 Гц, 1H), 7,42-7,33 (м, 5H), 6,19 (с, 1H), 4,92-4,90 (м, 1H), 4,82-4,72 (м, 1H), 3,79 (с, 3H), 3,34-3,20 (м, 1H), 3,02-2,94 (м, 1H), 2,90-2,87 (м, 1H), 2,36-2,09 (м, 4H), 1,99-1,96 (м, 1H), 1,80-1,42 (м, 5H), 1,16 (д, J=6,6 Гц, 3H). ЖХМС (ES, m/z): 512 [M+H]+.
Второй элюирующий изомер (501): 1H-ЯМР (CD3OD, 400 МГц) δ (м.д.): 7,52-7,33 (м, 6H), 6,22 (с, 1H), 4,84-4,73 (м, 2H), 3,78 (с, 3H), 3,27-3,16 (м, 1H), 3,04-2,92 (м, 1H), 2,90-2,88 (м, 1H), 2,46-2,35 (м, 2H), 2,30-2,22 (м, 1H), 2,15-2,02 (м, 2H), 1,82-1,71 (м, 1H), 1,63-1,55 (м, 2H), 1,40-1,28 (м, 1H), 1,15 (д, J=6,6 Гц, 4H). ЖХМС (ES, m/z): 512 [M+H]+.
Пример 1.7: Условия ВЭЖХ
[178] В любом из вышеупомянутых вариантов осуществления указанная процентная чистота может быть определена с помощью ВЭЖХ. В некоторых вариантах реализации процентную чистоту определяют с использованием следующего метода ВЭЖХ:
5 мин 30% B
15 мин 45% B
21 мин 80% B
22 мин 80% B
22,1 мин 10% B
Пример 2: Биохимический анализ HTRF на активность CBP и BRD4
[179] Способность соединений формулы (I) селективно ингибировать CBP определяли с использованием следующего биохимического анализа HTRF на активность CBP и BRD4. Анализ проводили в конечном объеме 6 мкл в буфере для анализа, содержащем 50 мМ Hepes pH 7,5, 0,5 мМ GSH, 0,01% BGG, 0,005% BSA и 0,01% Triton X-100. Нанолитровые количества 10-точечного 3-кратного серийного разведения в ДМСО предварительно разливали в 1536 аналитических планшетов с максимальной концентрацией 33 мкМ и половинными логарифмическими разведениями. 3 мкл 2х Белка и 3 мкл 2х пептидного лиганда добавляли в аналитические планшеты (предварительно проштампованные соединением). Планшеты инкубировали в течение различного времени до 4 часов при комнатной температуре перед измерением сигнала TR-FRET. Значения IC50 показаны на Фигуре 1. Как показано на Фиг. 1, значение IC50, большее или равное 0,001 мкМ и меньше или равное 0,01 мкМ, обозначено как «++++»; значение больше 0,01 мкМ и меньше или равное 0,1 мкМ помечено как «+++»; значение больше 0,1 мкМ и меньше или равное 1 мкМ обозначается «++»; и значение больше 1 мкМ и меньше 1000 мкМ отмечено знаком «+».
Пример 3: Соединения 1 и Соединение 1 продемонстрировали in vitro активность против CBP.
[180] Эффективность и селективность соединений-ингибиторов CBP/P300, включая Соединение 1, определяли в биохимических анализах флуоресценции с временным разрешением с использованием GST-слияния бромодоменов CBP и BRD4. Вкратце, ингибиторы CBP предварительно разливали в 1536 аналитических планшетов для конечной тестовой концентрации от 33 мкМ до 1,7 нМ. Планшеты и инкубируют в течение 4 ч. Данные были представлены как процент ингибирования по сравнению с контрольными лунками. Значения IC50 определяли путем аппроксимации кривой стандартного 4-параметрического алгоритма логистической аппроксимации. В этих условиях было определено, что Соединение 1 является мощным ингибитором CBP с IC50 <2 нМ (N=16). В аналогичном анализе определяли активность BRD4, и Соединение 1 показало IC50 <500 нМ (N=15), что указывает на >200-кратную селективность.
[181] Селективность Соединения 1 оценивали в скрининговых анализах на ингибирование киназы и связывание BRD. Соединение 1 показало аффинность связывания от нулевой до низкой для человеческих киназ и соответствующих заболеванию мутантных вариантов, оцененных с помощью скрининга KINOMEscan™. Панель из 10 BRD, представляющих различные ветви или дерево бромодоменов, тестировали с использованием AlphaScreen. Из 10 исследованных бромодоменов Соединение 1 было неактивным по сравнению с 8. Значения IC50 Соединения 1 для бромодоменов CREBBP и BRD4 (тандемный BD1/BD2) составляли 0,1 и >10 мкМ, соответственно, подтверждая высокую селективность Соединения 1 в отношении CBP.
[182] Способность Соединения 1 и Соединения 1 селективно ингибировать CBP определяли с использованием биохимического анализа из Примера 2 на активность CBP и BRD4. Результаты показаны в Таблице 1 ниже:
Таблица 1
[183] Как Соединение 1, так и Соединение 1´ сильно ингибировали (например, IC50<100 нМ) CBP в биохимическом анализе HTRF из Примера 2, в то время как Соединение 1 было примерно в 3,5 раза более селективным в отношении ингибирования CBP по сравнению с BRD4 при использовании этого анализа.
Пример 4: Соединение 1 демонстрирует модуляцию H3K27Ac in vitro в клетках рака молочной железы.
[184] AR-положительные клетки рака молочной железы подвергали воздействию возрастающих концентраций Соединения 1 в течение 24 часов. Лизаты готовили в загрузочном буфере E-PAGE (Invitrogen) и анализировали вестерн-блоттингом с антителами, разведенными 1:1000 для анти-H3K27Ac, 1:2000 для антител к общему H3 и 1:10 000 для анти-β-актина. Блоты сканировали и анализировали на анализаторе изображений LI-COR Odyssey. Уровни H3K27Ac были нормализованы по β-актину.
[185] Соединение 1 индуцировало зависимое от концентрации снижение H3K27Ac, метки, специфичной для CBP/P300, в AR-положительной клеточной линии рака молочной железы. (Фиг. 3). Снижение на 50% наблюдалось между 0,03 и 0,1 μM с максимальным уменьшением приблизительно 60%, наблюдаемым при 0,3 μM.
Пример 5: Соединения 1, 2 и 4 уменьшают AR-целевой ген TMPRSS2 и ER-целевой ген XBP1 в AR-положительных клетках рака молочной железы
[186] AR-положительные клетки рака молочной железы подвергались воздействию увеличивающихся концентраций соединений в течение 24 часов. RNA экстрагировали и экспрессию гена измеряли с использованием тестов Taqman® для TMPRSS2 и XBP1.
[187] Все соединения снижали экспрессию мРНК TMPRSS2 и XBP1 в AR-положительной линии клеток рака молочной железы.
[188] Как показано в Таблице 2 ниже, значение IC50 менее 100 нМ обозначено как «+++»; значение больше 100 нМ и меньше 500 нМ обозначается «++»; и значение больше 500 нМ отмечено знаком «+».
Таблица 2
Пример 6: Соединения 1, 2 и 4 обладают антипролиферативной активностью в отношении линий клеток рака молочной железы AR+ in vitro.
[189] Линии клеток молочной железы культивировали в соответствии с рекомендациями дистрибьютора. На следующий день клетки подвергали воздействию соединений непрерывно в течение 10 дней. Жизнеспособность клеток оценивали с помощью теста CellTiter-Glo® (Promega) в конце периода инкубации. Эффект ингибирования роста оценивали по концентрации, ингибирующей рост на 50%, с использованием уравнения нелинейной регрессии и переменного наклона (Graphpad Prism).
[190] Соединения 1, 2 и 4 ингибируют пролиферацию клеточных линий рака молочной железы после 10 дней непрерывного воздействия лекарственного средства. Линии клеток с высокой экспрессией мРНК AR более чувствительны, чем линии с низкой экспрессией.
[191] Как показано в Таблице 3 ниже, значение IC50 менее 0,2 мкМ обозначено как «++++»; значение более 0,2 мкМ и менее 0,5 мкМ обозначается «+++»; значение больше 0,5 мкМ и меньше 1 мкМ отмечено «++»; и значение более 1 мкМ отмечено знаком «+».
Таблица 3
Пример 7: Соединение 1 продемонстрировало эффективность in vivo в AR-положительных ксенотрансплантатах рака молочной железы человека (TNBC).
[192] Противоопухолевую активность Соединения 1 тестировали на модели ксенотрансплантата, полученной из AR-положительной клеточной линии, для AR+ тройного отрицательного рака молочной железы (Robinson et al., «Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1», The EMBO Journal (2011) 30, 3019-3027 (2011), которая полностью включена в настоящий документ посредством ссылки). Вкратце, AR-положительные опухолевые клетки рака молочной железы (1×107) имплантировали подкожно в бок 6-8-недельных мышей NOD SCID. Мышей рандомизировали, и лечение начинали, когда средний размер опухоли достигал 160 мм3 (8 мышей на когорту). 50 мг/кг Соединения 1 вводили перорально ежедневно в течение эксперимента. Объем опухоли (TV) измеряли дважды в неделю штангенциркулем, и объем опухоли (мм3) рассчитывали следующим образом: TV=a×b×b/2, где «a» и «b» - длинный и короткий диаметры опухоли соответственно.
[193] Результаты показаны на Фигуре 4. В конце периода лечения обработка Соединением 1 вызвала ингибирование роста опухоли (TGI) на 104% (p<0,001) по сравнению с контролем с носителем (TGI=[1-(TreatedTVfinal-TreatedTVinitial)/(VehicleTVfinal-VehicleTVinitial)]*100, где «TVfinal» и «TVinitial» - средние объемы опухоли в последний день и начальный день дозирования). Средняя потеря массы тела составила 3,7%.
Пример 8: Соединение 2 модулирует уровень AR и вариантов на уровне белка.
[194] Клетки рака предстательной железы AR-v7+ подвергали действию Соединения 2 в течение 24 часов, в это время готовили лизаты и оценивали влияние соединения на уровень белка AR с помощью вестерн-блоттинга. Результаты показаны на Фигуре 5. Обработка клеток рака простаты Соединением 2 приводила к уменьшению как полноразмерных, так и вариантных форм AR, включая AR-v7.
Пример 9: Соединения 1, 2 и 4 демонстрируют модуляцию генов-мишеней AR TMPRSS2 и KLK3, а также MYC в линии клеток рака предстательной железы AR-v7+.
[195] Клетки рака простаты AR-v7+ подвергали воздействию соединений 1, 2 и 4 в течение 24 часов. RNA экстрагировали с помощью Qiacube RNAeasy Mini (Qiagen). Для всех протестированных генов реакции qPCR проводили в трех экземплярах с 250 нг РНК на реакцию и праймер-зондами Taqman® GAPDH использовался как ген «домашнего хозяйства».
[196] В испытанных условиях все 3 соединения снижали гены-мишени AR TMPRSS2 и KLK3, а также MYC в зависимости от концентрации в клетках рака простаты AR-v7+.
[197] Как показано в Таблице 4 ниже, значение IC50 менее 10 нМ обозначено как «++++»; значение больше 10 нМ и меньше 50 нМ помечено как «+++»; значение больше 50 нМ и меньше 100 нМ обозначается «++»; а значение больше 100 нМ отмечено знаком «+».
Таблица 4
Пример 10: Соединения 1, 2 и 4 демонстрируют антипролиферативную активность в клеточных линиях рака простаты.
[198] Клетки рака простаты, андроген-зависимые (LnCaP и VCaP) и андроген-независимые (22Rv1), высевали и инкубировали в течение ночи. На следующий день клетки непрерывно обрабатывали соединениями 1, 2, 4 и энзалутамидом (конечная максимальная концентрация 10 мкМ, полулогарифмические разведения). Жизнеспособность клеток оценивали с помощью теста CellTiter-Glo® (Promega) после 10-дневного воздействия лекарственного средства. Эффект ингибирования роста оценивали по концентрации, ингибирующей рост на 50%, с использованием уравнения нелинейной регрессии и переменного наклона (Graphpad Prism).
[199] Энзалутамид был активен в отношении андроген-зависимых клеточных линий LnCaP и VCaP, но был неактивен в отношении AR-отрицательных (PC3 и DU145) и AR-v7, экспрессирующих клеточную линию 22Rv1. Напротив, все соединения обладали сильным и зависимым от концентрации эффектом ингибирования роста во всех линиях клеток AR+, включая клетки 22Rv1. Все соединения были неактивны в клеточных линиях AR.
[200] Как указано в Таблице 5 ниже, значение IC50 менее 0,5 мкМ обозначено как «+++»; значение больше 0,5 мкМ и меньше 1 мкМ отмечено «++»; и значение более 1 мкМ отмечено знаком «+».
Таблица 5
AR-v7+
Пример 11: Соединение 4 демонстрирует противоопухолевую активность на полученной от пациента модели ксенотрансплантата рака простаты, устойчивого к энзалутамиду.
[201] Самцам мышей NOG (возраст 6-8 недель) имплантировали фрагменты опухоли PDX простаты. Когда опухоли достигли среднего объема опухоли 100-300 мм3, животных рандомизировали в разные когортные группы и начали дозирование в тот же день (День 0). Соединение 4 готовили в (0,5 CMC/0,5% Tween 80) pH8 и вводили в виде раствора. Животных взвешивали дважды в неделю. Опухоли измеряли дважды в неделю. Максимальный объем опухоли у контрольных животных составлял 1500 мм3.
[202] Соединение 4 вводили через желудочный зонд в дозе и графике 40 мг/кг/доза ежедневно с понедельника по четверг, повторяя еженедельно, или 80 мг/кг/дозу в понедельник и четверг (дважды в неделю), повторяя еженедельно.
[203] Обработка Соединением 4 в дозе 40 мг/кг/доза ежедневно с понедельника по четверг, повторенная еженедельно, или 80 мг/кг/доза в понедельник и четверг (дважды в неделю), повторяемая еженедельно, привела к сильному противоопухолевому ответу (Фиг. 6) со значениями ингибирования роста опухоли 84% и 82% соответственно. Энзалутамид обладал умеренной активностью (TGI=21%).
Пример 12: Фармацевтическая композиция, содержащая Соединение 1 и Соединение 1´
[204] Фармацевтическая композиция может содержать одно или более соединений формулы (I), как предусмотрено здесь, включая Соединение 1 и/или Соединение 1´.
[205] В одном примере активный фармацевтический ингредиент (API) может содержать примерно 90% или более Соединения 1 и до около 10% (предпочтительно до около 5%, наиболее предпочтительно до около 2,5%, включая около 1,5%) Соединения 1´.
[206] Пероральные лекарственные формы, содержащие Соединение 1, могут быть приготовлены в виде лекарственного средства в капсуле (DiC), инкапсулированного простого гранулированного сухого смешения и раствора на основе липидов в капсуле с твердой оболочкой. Капсулы могут содержать фармацевтически приемлемые наполнители, а инкапсулированные капсулы могут быть упакованы в бутылки из полиэтилена высокой плотности, запечатанные индукционным способом.
[207] Дополнительные варианты осуществления изобретения изложены в следующих пронумерованных пунктах:
1. Способ лечения пациента с диагнозом энзалутамид-устойчивой формы рака AR+, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей Соединение 1 или его фармацевтически приемлемую соль:

2. Способ лечения пациента с диагнозом энзалутамид-резистентная форма рака AR+, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту или ее фармацевтически приемлемую соль.
3. Способ по п. 1 или 2, отличающийся тем, что Соединение 1 вводят пациенту в пероральной стандартной лекарственной форме.
4. Способ по любому из пп. 1-3, отличающийся тем, что фармацевтическая композиция дополнительно содержит Соединение 1' или его фармацевтически приемлемую соль:

5. Способ по п. 4, отличающийся тем, что фармацевтическая композиция содержит примерно 95% или более (по данным HPLC) Соединения 1 и до 5% по весу Соединения 1´.
6. Способ по одному из пп. 1-5, в котором рак предстательной железы AR+ представляет собой кастрационно-резистентный рак предстательной железы (AR+) (CRPC).
7. Способ по любому из пп. 1-5, отличающийся тем, что рак AR+ представляет собой рак молочной железы AR+.
8. Способ по п. 7, в котором пациенту поставлен диагноз TNBC.
9. Способ по любому из пп. 1-8, отличающийся тем, что у пациента диагностирован рак, несущий сплайсинговую форму AR-v7 рецептора андрогенов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРОВАНИЕ ЦИКЛИЧЕСКОГО AMP-ЧУВСТВИТЕЛЬНОГО ЭЛЕМЕНТ-СВЯЗЫВАЮЩЕГО БЕЛКА (CREB) | 2020 |
|
RU2831142C2 |
| ИНГИБИРОВАНИЕ CREB-СВЯЗЫВАЮЩЕГО БЕЛКА (CBP) | 2019 |
|
RU2803290C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2019 |
|
RU2809763C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| ЛИГАНДЫ ЦЕРЕБЛОНА И БИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ | 2018 |
|
RU2795146C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2561109C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811403C2 |










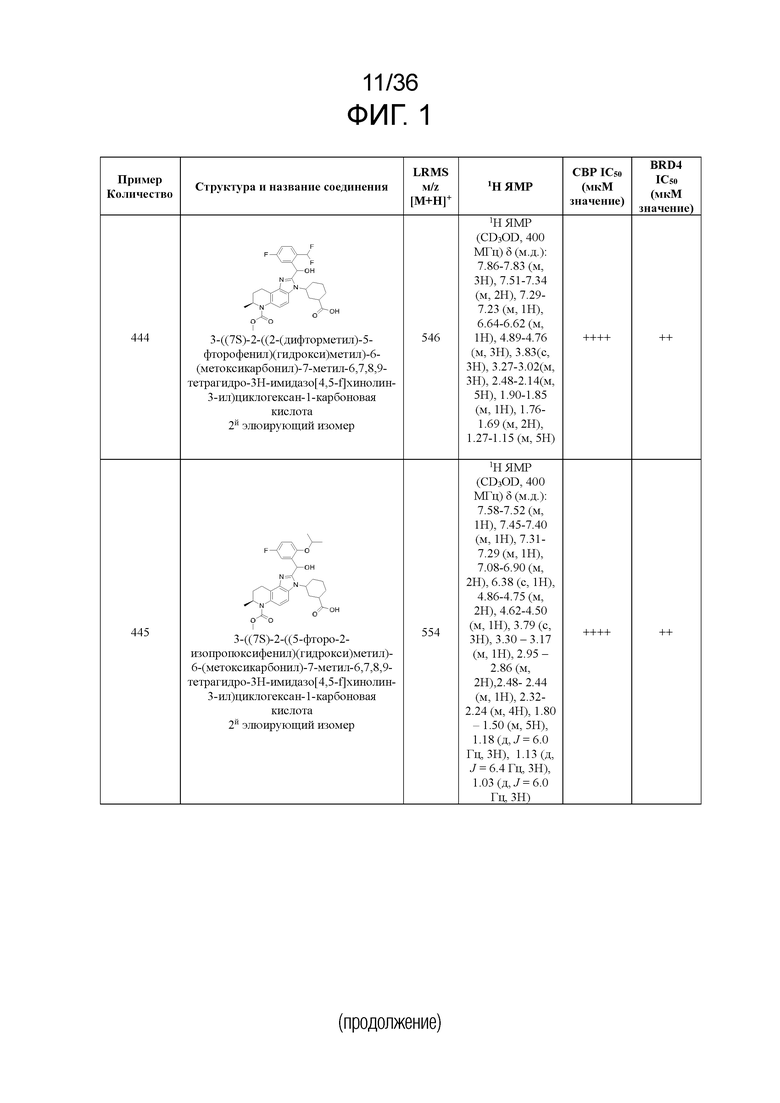

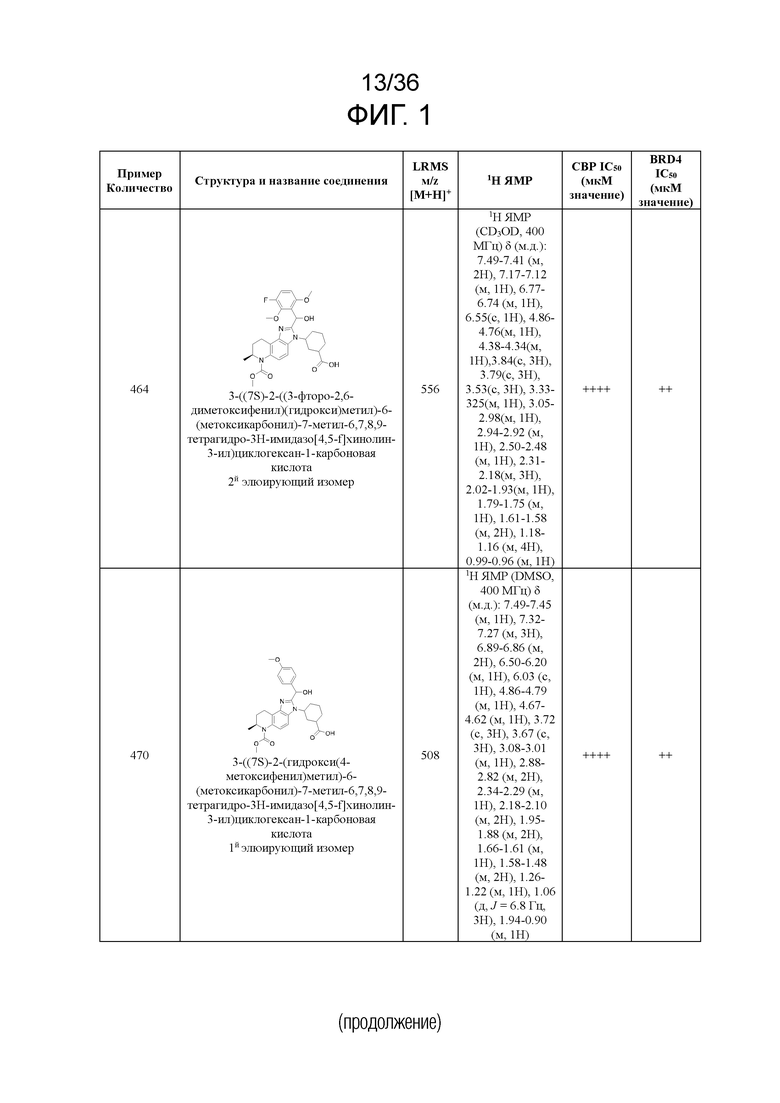

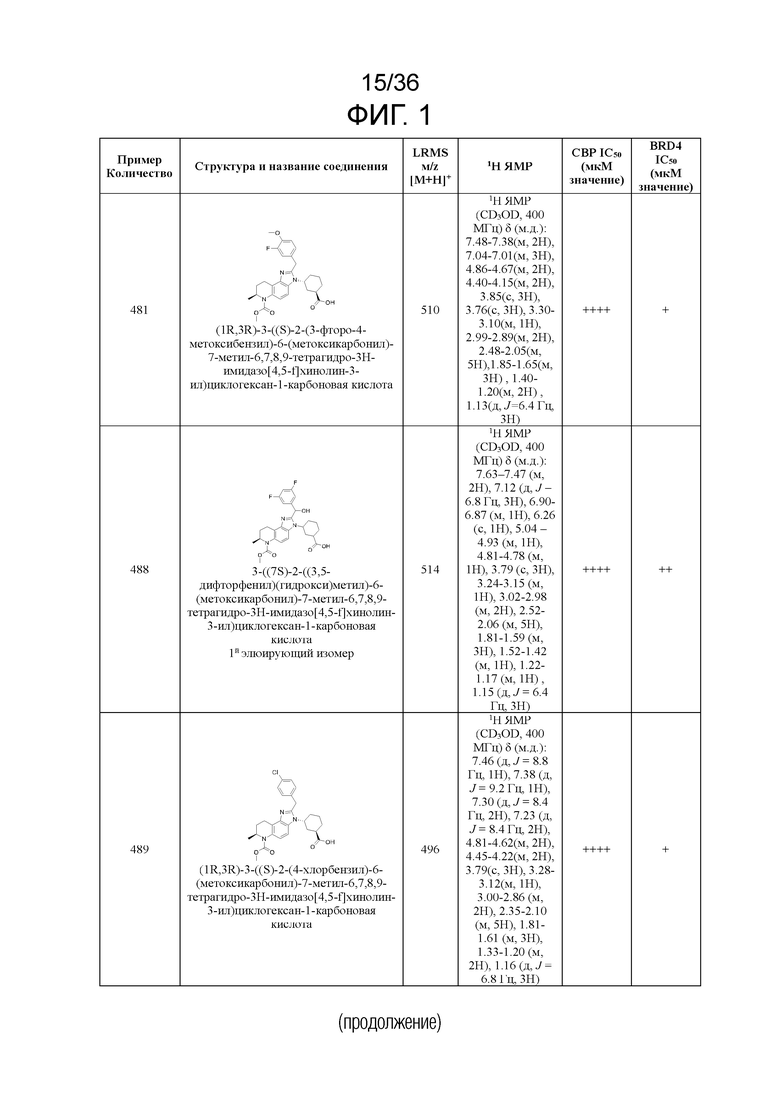


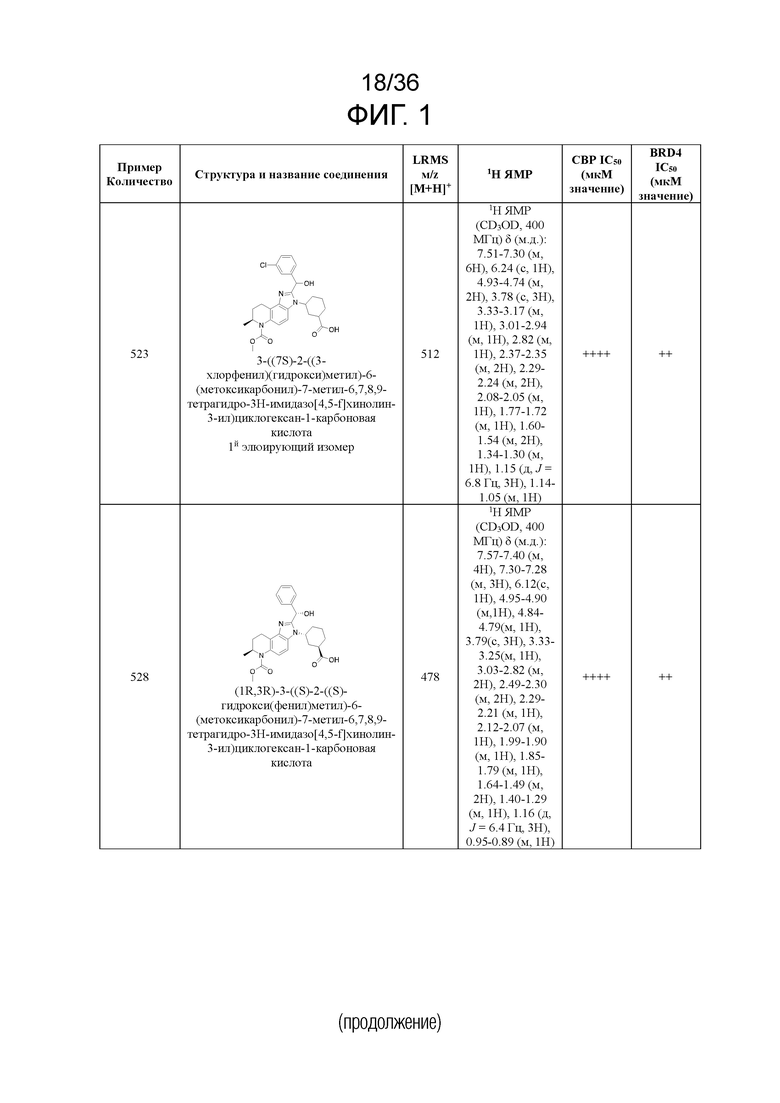







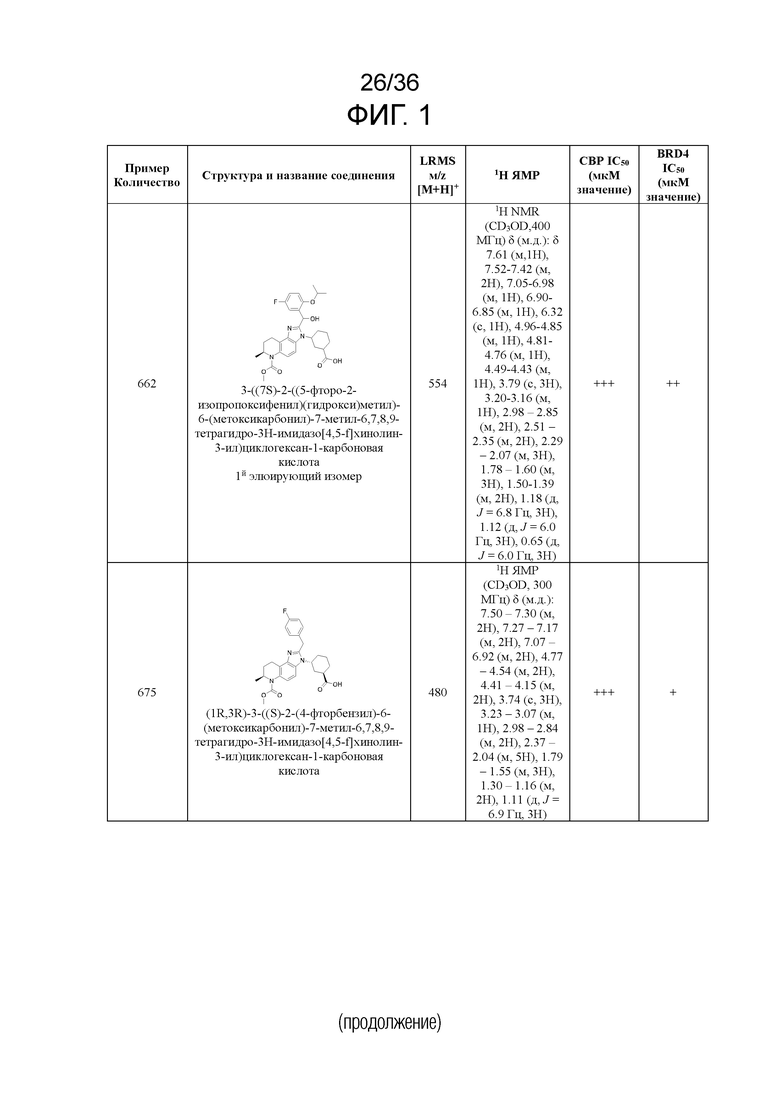


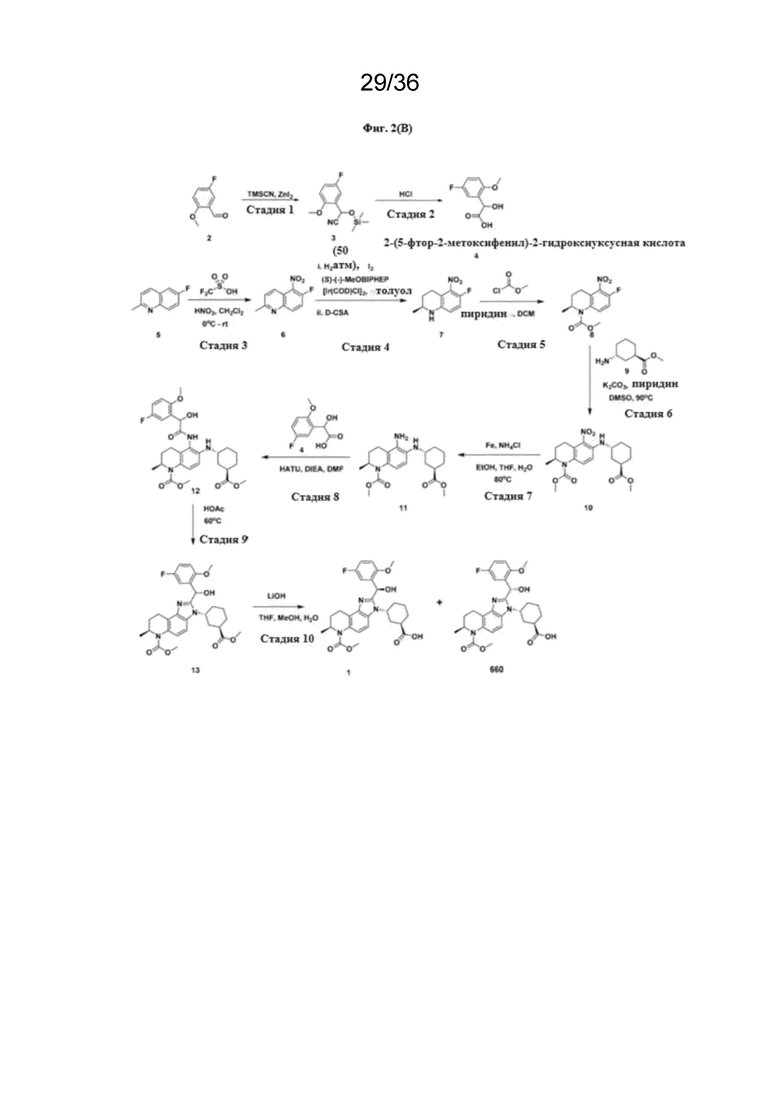
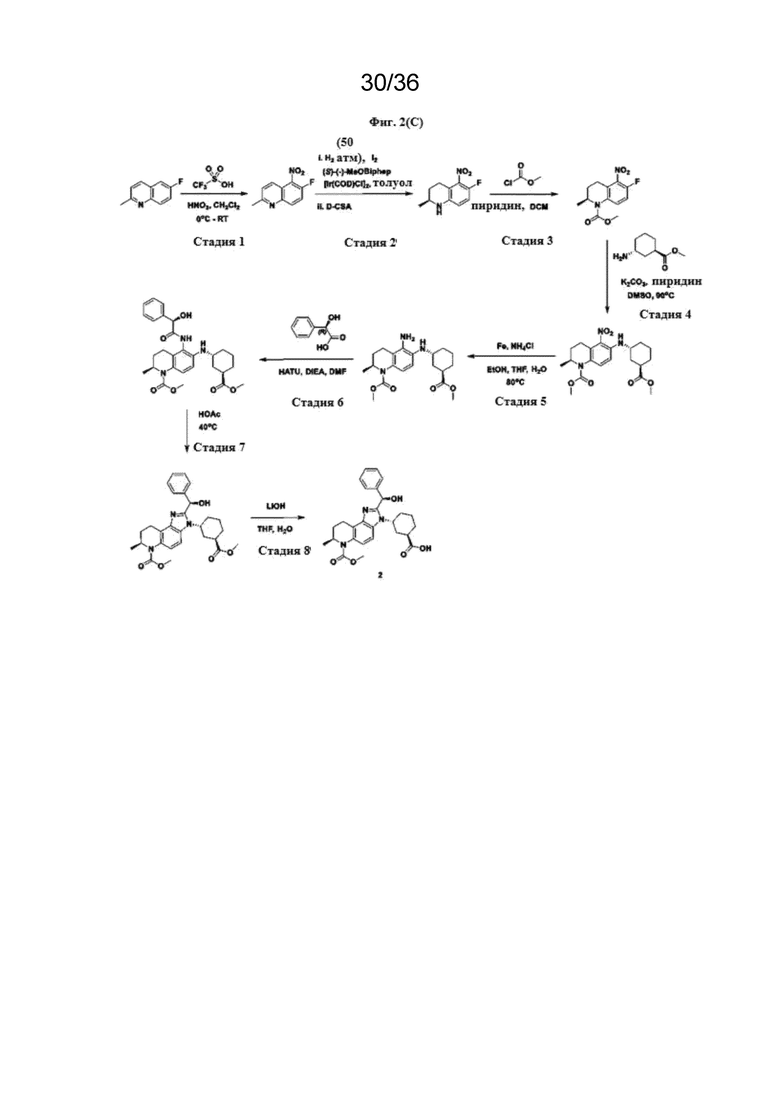
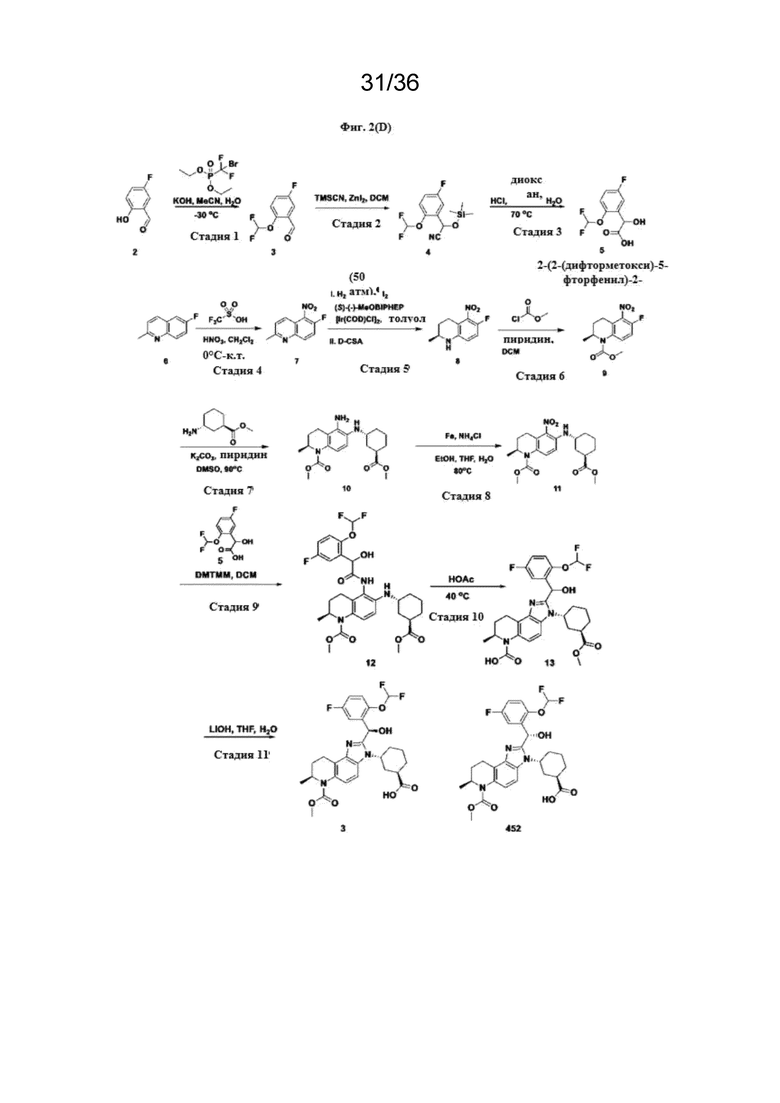



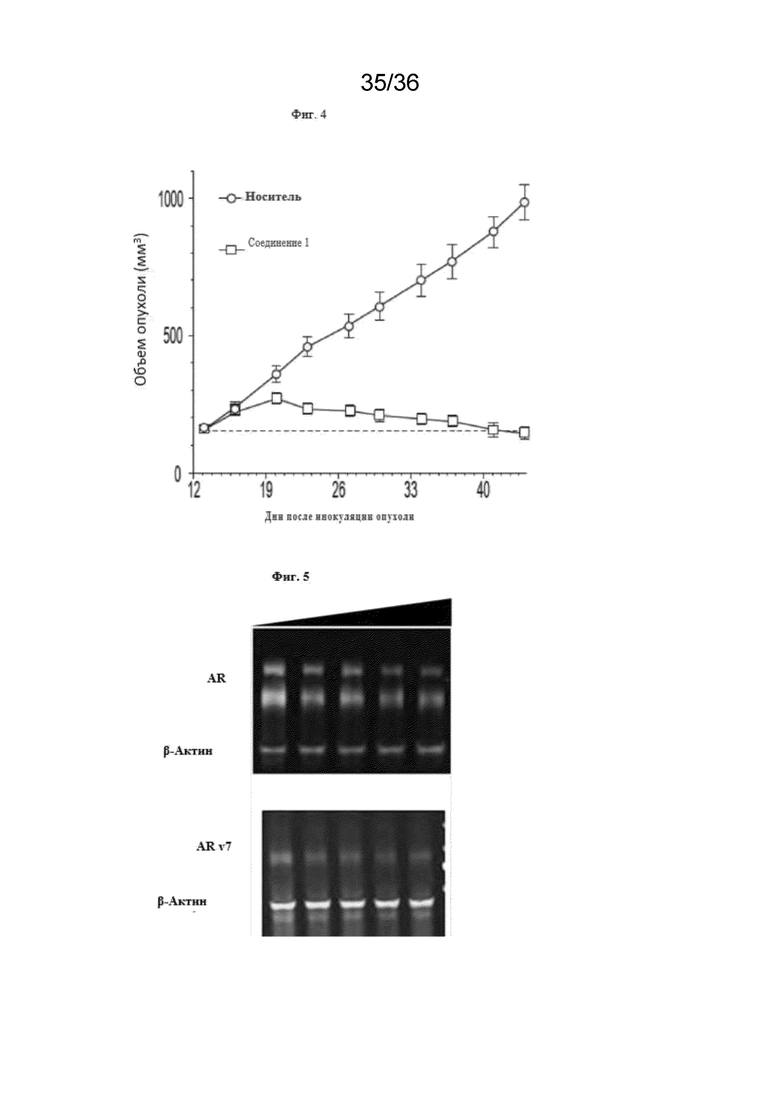

Группа изобретений относится в области медицины, а именно к способам лечения рака молочной железы, простаты и к способам лечения рака, экспрессирующего рецептор андрогенов. Способ лечения рака молочной железы у пациента и способ лечения рака простаты включают введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (I)

или его фармацевтически приемлемую соль, где R1 представляет собой H или -OH; каждый R2 независимо выбран из галогена и -OR3; каждый R3 независимо представляет собой C1-C6 алкил, где алкил необязательно замещен одним или более галогенами; и n равно 0, 1 или 2. Способ лечения рака, экспрессирующего рецептор андрогенов, у пациента включает введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту или ее фармацевтически приемлемую соль. Способ лечения рака, экспрессирующего рецептор андрогенов, у пациента включает введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей
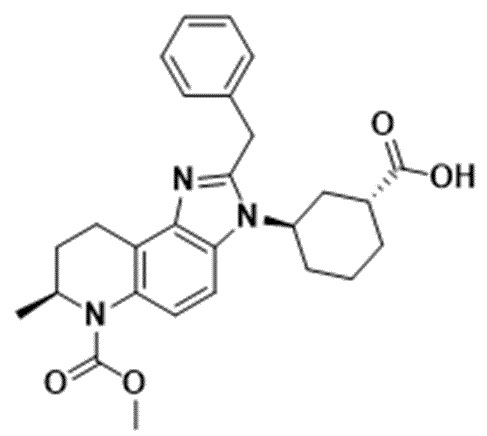
или его фармацевтически приемлемую соль. Вышеописанная группа изобретений позволяет проводить лечение рака молочной железы, рака простаты и рака, экспрессирующего андрогеновый рецептор. 4 н. и 32 з.п. ф-лы, 6 ил., 5 табл., 12 пр.
1. Способ лечения рака молочной железы у пациента, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (I)
 (I)
(I)
или его фармацевтически приемлемую соль,
где R1 представляет собой H или -OH;
каждый R2 независимо выбран из галогена и -OR3;
каждый R3 независимо представляет собой C1-C6 алкил, где алкил необязательно замещен одним или более галогенами; и
n равно 0, 1 или 2.
2. Способ по п. 1, отличающийся тем, что рак молочной железы представляет собой тройной отрицательный рак молочной железы.
3. Способ по п. 1, отличающийся тем, что рак молочной железы представляет собой AR+.
4. Способ по п. 1, отличающийся тем, что соединение выбрано из:

5. Способ по п. 4, отличающийся тем, что рак молочной железы представляет собой тройной отрицательный рак молочной железы.
6. Способ по п. 4, отличающийся тем, что рак молочной железы представляет собой AR+.
7. Способ по п. 1, отличающийся тем, что соединение представляет собой

8. Способ по п. 1, отличающийся тем, что соединение представляет собой

9. Способ по п. 1, отличающийся тем, что соединение представляет собой

10. Способ по п. 1, отличающийся тем, что соединение представляет собой

11. Способ по п. 1, отличающийся тем, что рак молочной железы представляет собой AR+ и где соединение выбрано из:

12. Способ по п. 11, отличающийся тем, что рак молочной железы представляет собой тройной отрицательный рак молочной железы.
13. Способ лечения рака простаты у пациента, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (I)
 (I)
(I)
или его фармацевтически приемлемую соль,
где R1 представляет собой H или -OH;
каждый R2 независимо выбран из галогена и -OR3;
каждый R3 независимо представляет собой C1-C6 алкил, где алкил необязательно замещен одним или более галогенами; и
n равно 0, 1 или 2.
14. Способ по п. 13, отличающийся тем, что рак простаты представляет собой AR+.
15. Способ по п. 13, отличающийся тем, что рак простаты представляет собой AR-v7+.
16. Способ по п. 13, отличающийся тем, что у пациента диагностирован устойчивый к кастрации рак простаты или метастатический рак простаты, чувствительный к кастрации.
17. Способ по п. 13, отличающийся тем, что соединение выбрано из:

18. Способ по п. 17, отличающийся тем, что у пациента диагностировано прогрессирование заболевания после лечения энзалутамидом.
19. Способ по п. 13, отличающийся тем, что соединение выбрано из:

20. Способ по п. 19, отличающийся тем, что рак простаты представляет собой AR+.
21. Способ по п. 20, отличающийся тем, что рак простаты представляет собой AR-v7+.
22. Способ по п. 19, отличающийся тем, что у пациента диагностирован устойчивый к кастрации рак простаты или метастатический рак простаты, чувствительный к кастрации.
23. Способ лечения рака, экспрессирующего рецептор андрогенов, у пациента, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту или ее фармацевтически приемлемую соль.
24. Способ по п. 23, отличающийся тем, что рак, экспрессирующий андрогенный рецептор, представляет собой рак молочной железы AR+.
25. Способ по п. 24, отличающийся тем, что рак молочной железы AR+ представляет собой тройной отрицательный рак молочной железы.
26. Способ по п. 24, отличающийся тем, что рак молочной железы AR+ представляет собой рак молочной железы Her2.
27. Способ по п. 24, отличающийся тем, что AR+ рак молочной железы выбран из ER-рака молочной железы, PR-рака молочной железы и ER-/PR-рака молочной железы.
28. Способ по п. 23, отличающийся тем, что рак, экспрессирующий рецептор андрогенов, представляет собой рак простаты AR+.
29. Способ по п. 28, отличающийся тем, что рак простаты AR+ представляет собой AR-v7+.
30. Способ по п. 23, отличающийся тем, что рак, экспрессирующий рецептор андрогенов, представляет собой устойчивый к кастрации рак простаты или метастатический рак простаты, чувствительный к кастрации.
31. Способ по п. 29, отличающийся тем, что у пациента диагностировано прогрессирование заболевания после лечения энзалутамидом.
32. Способ лечения рака, экспрессирующего рецептор андрогенов, у пациента, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей
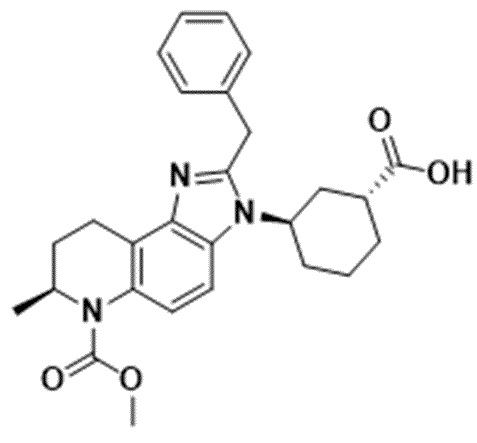
или его фармацевтически приемлемую соль.
33. Способ по п. 32, отличающийся тем, что рак, экспрессирующий андрогенный рецептор, представляет собой рак молочной железы AR+.
34. Способ по п. 22, отличающийся тем, что рак молочной железы AR+ представляет собой тройной отрицательный рак молочной железы.
35. Способ по п. 32, отличающийся тем, что рак, экспрессирующий рецептор андрогенов, представляет собой рак простаты AR+.
36. Способ по п. 35, отличающийся тем, что рак простаты AR+ представляет собой AR-v7+.
| US 2016257692 A1, 08.09.2016 | |||
| WO 2014113260 A1, 24.07.2014 | |||
| WO 2016086200 A1, 02.06.2016 | |||
| СПОСОБ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2010 |
|
RU2435596C1 |

