Область техники, к которой относится изобретение
Настоящее изобретение касается фармацевтических композиций для лечения рака, включающих в себя ингибиторы BRAF (например, вемурафениб) и/или ингибиторы MEK (например, траметиниб, RO5068760), в комбинации с антитубулиновыми соединениями по изобретению или другими известными ингибиторами тубулина, и применения таких композиций для лечения раковых заболеваний типа меланомы, лекарственно-устойчивого рака и раковых метастазов.
Уровень техники
Рак является второй по распространенности причиной смерти в США, уступая лишь сердечно-сосудистым заболеваниям. В США на рак приходится 1 из каждых 4 смертей. Относительная выживаемость на 5-летний срок у всех больных раком, которым поставлен диагноз в 1996-2003 гг., возросла до 66% по сравнению с 50% в 1975-1977 гг. (Cancer Facts & Figures, American Cancer Society: Atlanta, GA (2008)). Между 2000 и 2009 г. частота новых случаев рака снижалась в среднем на 0,6% в год среди мужчин и оставалась на том же уровне у женщин. С 2000 по 2009 г. смертность от всех видов рака в целом снижалась в среднем на 1,8% в год среди мужчин и 1,4% в год среди женщин. Такое улучшение выживаемости отражает прогресс в диагностике на ранней стадии и улучшения в лечении. Выявление высокоэффективных противораковых средств с низкой токсичностью является главной задачей раковых исследований.
Микротрубочки - это цитоскелетные филаменты, состоящие из αβ-гетеродимеров тубулина, которые участвуют в целом ряде клеточных функций, включая поддержание формы, везикулярный транспорт, подвижность и деление клеток. Тубулин является основным структурным компонентом микротрубочек и хорошо обоснован в качестве мишени для целого ряда очень успешных противораковых препаратов. Соединения, способные нарушать равновесие микротрубочки-тубулин в клетках, эффективны при лечении рака. Противораковые препараты типа таксола и винбластина, которые способны нарушить равновесие микротрубочки-тубулин в клетках, широко применяются при химиотерапии рака.
К сожалению, при клиническом применении противораковых препаратов, взаимодействующих с микротрубочками, возникают две основные проблемы: резистентность и нейротоксичность.
Злокачественная меланома является самой опасной формой рака кожи, которая составляет около 75% случаев смерти от рака кожи. Встречаемость меланомы неуклонно растет в западном мире. Число случаев удвоилось за последние 20 лет. Каждый год в мире диагностируется около 160000 новых случаев меланомы, причем она чаще встречается у мужчин и европеоидов. По данным отчета ВОЗ, за год во всем мире происходит около 48000 случаев смерти в связи с меланомой.
В настоящее время нет эффективного способа лечения запущенной/метастатической меланомы. Она очень устойчива к современной химиотерапии, лучевой терапии и иммунотерапии. Запущенная/метастатическая меланома имеет очень плохой прогноз, со средней выживаемостью в 6 месяцев и выживаемостью на 5-летний срок менее 5%.
В различных центрах применяются разные средства химиотерапии, в том числе дакарбазин (также называется DTIC), иммунотерапия (с помощью интерлейкина-2 (IL-2) или интерферона (IFN)), а также местная перфузия. В целом успешность при лечении метастатической меланомы довольно ограничена. IL-2 (пролейкин) является первым новым средством терапии, одобренным для лечения метастатической меланомы за 20 лет. Однако он дает всего лишь менее 5% случаев полной ремиссии у больных. В последние годы предпринимались большие усилия в борьбе с меланомой. Но ни комбинирование DTIC с другими химиотерапевтическими препаратами (например, цисплатином, винбластином и кармустином), ни добавление интерферона-α2b к DTIC не давали улучшения выживаемости по сравнению с лечением одним только DTIC. Совсем недавно клинические испытания с антителами и вакцинами для лечения поздних стадий меланомы также не смогли продемонстрировать удовлетворительную эффективность. Препарат ипилимумаб (Yervoy®) использует иммунную систему больного для борьбы с меланомой. Ипилимумаб применяется для лечения поздних стадий меланомы, которая вышла за пределы своей первоначальной локализации. При прицельной терапии применяются препараты, направленные на конкретных уязвимые места в раковых клетках.
Выявление мутации BRAF у ~60% больных меланомой и одобрение FDA ингибиторов BRAF (BRAFi; например, вемурафениб и дабрафениб (GSK2118436)) и ингибиторов MEK (MEKi; например, траметиниб (GSK1120212), RO5068760)) дало впечатляющие клинические результаты при лечении меланомы с мутацией BRAFV600. Применение авансом комбинации BRAFi + MEKi весьма эффективно при начальной терапии, но из-за неоднородности опухолей и активации альтернативных путей в пределах ~9 месяцев возникает устойчивость, приводящая к рецидиву заболевания и смерти пациентов.
Вемурафениб (Zelboraf®) является средством прицельной терапии, одобренным для лечения запущенной меланомы, которая не подлежит хирургии, или меланомы, распространившейся по всему организму. В отношении меланомы вемурафениб лечит только опухоли, имеющие определенную генетическую мутацию (BRAFV600). Кроме того, вемурафениб и другие ингибиторы BRAF могут быть активными при раке с различными мутациями BRAF. Примеры, при которых с высокой частотой происходят мутации В-RAF, включают меланому (30-60%), рак щитовидной железы (30-50%), колоректальный рак (5-20%), рак яичников (~30%) и другие виды рака (1-3%) (Wellbrock С, Karasarides М, Marais R. "The Raf protein takes centre stage". Nat. Rev. (2004) 5: 875-885).
Устойчивая клиническая активность вемурафениба у больных меланомой с мутацией BRAFV600 ограничивается быстрым развитием приобретенной устойчивости (Lee JT, Li L, Brafford PA, et al. "PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas". Pigment Cell Melanoma Res. (2010) 23: 820-827; Yang H, Higgins B, Kolinsky K, et al. "RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models". Cancer Res. (2010) 70: 5518-5527; Yang H, Higgins B, Kolinsky K, et al. "Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer". Cancer Res. (2012) 72: 779-789). Механизмы возникновения устойчивости широко изучались (Little AS, Smith PD, Cook SJ. "Mechanisms of acquired resistance to ERK1/2 pathway inhibitors". Oncogene (2013) 32(10): 1207-1215; Bollag G, HiRTh P, Tsai J, et al. "Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma". Nature (2010) 467: 596-599; Flaherty KT. "Targeting metastatic melanoma". Annu Rev Med. (2012) 63: 171-183; Su F, Bradley WD, Wang Q, et al. "Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation". Cancer Res. (2012) 72: 969-978). В литературе было предложено много различных механизмов, включая собственную устойчивость к BRAFi, амплификацию онкогена BRAF (Shi Н, Moriceau G, Kong X, et al. "Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance". Nat. Commun. (2012) 3: 724), повышающую регуляцию или активирующие мутации нижележащих киназ MEK, повышающую регуляцию экспрессии CRAF (Montagut С, Sharma SV, Shioda Т, et al. "Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma". Cancer Res. (2008) 68: 4853-4861), онкогенную активацию NRAS (Nazarian R, Shi H, Wang Q, et al. "Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation". Nature (2010) 468: 973-977), повышающую регуляцию пути EGFR-SFK-STAT3 (Girotti MR, Pedersen M, Sanchez-Laorden B, et al. "Inhibiting EFG receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma". Cancer Discov. (2013) 3(2): 158-167), "сторожевые" мутации (Whittaker S, Kirk R, Hayward R, et al. "Gatekeeper mutations mediate resistance to BRAF-targeted therapies". Sci. Transl. Med. (2010) 2: 35ra41; Balzano D, Santaguida S, Musacchio A, Villa F. "A general framework for inhibitor resistance in protein kinases". Chem. Biol. (2011) 18: 966-975; Sierra JR, Cepero V, Giordano S. "Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy". Mol. Cancer (2010) 9: 75), повышающую регуляцию таких рецепторов факторов роста, как рецептор инсулиноподобного фактора роста 1 (IFG1R) (Villanueva J, Vultur А, Lee JT, et al. "Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K". Cancer Cell (2010) 18: 683-695) или рецептор тромбоцитарного фактора роста (PDGFR), и ряд других механизмов устойчивости (Wilson TR, Fridlyand J, Yan Y, et al. "Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors". Nature (2012) 487: 505-509; Straussman R, Morikawa T, Shee K, et al. "Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion". Nature (2012) 487: 500-504). Было описано несколько способов поддержания уровня фосфорилирования связанной с внеклеточными сигналами киназы 1 и 2 (p-ERK1/2) в присутствии препаратов-ингибиторов BRAF, в том числе мутации ERK-киназы 1 (MEK1), рекрутмент альтернативных активаторов MEK1/2, мутации RAS или повышающая регуляция рецепторных тирозинкиназ (RTKs). Так, во многих случаях устойчивые к вемурафениб клетки перекрестно устойчивы к ингибиторам MEK (Little AS, Smith PD, Cook SJ. "Mechanisms of acquired resistance to ERK1/2 pathway inhibitors". Oncogene (2013) 32(10): 1207-1215; Atefi M, von Euw E, Attar N, et al. "Reversing melanoma cross-resistance to BRAF и MEK inhibitors by co-targeting the AKT/mTOR pathway". PLoS One (2011) 6: e28973; Poulikakos PI, Persaud Y, Janakiraman M, et al. "RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E)". Nature (2011) 480: 387-390). Поскольку одним из главных механизмов приобретенной устойчивости к вемурафениб является устойчивая активация нижележащей MEK/ERK, то наибольшее внимание привлекла комбинация BRAFi + MEKi, которая нацелена на элементы пути RAF-MEK-ERK, что привело к одобрению FDA комбинации дабрафениб + траметиниб в 2013 г. Тем не менее, из-за неоднородности опухолей и активации альтернативных путей при меланоме, устойчивость к этой комбинированной терапии развивается в среднем в течение 9,4 месяцев, а после возникновения устойчивости она почти не обладает клинической активностью.
Комбинирование препаратов с использованием средств с различными противораковыми механизмами может улучшить реакцию опухолей и выживаемость пациентов, особенно при лечении больных с запущенным раком (Carrick S, Parker S, Wilcken N, et al. "Single agent versus combination chemotherapy for metastatic breast cancer". Cochrane Database Syst. Rev. 2005: CD003372; Fassnacht M, Terzolo M, Allolio B, et al. "Combination chemotherapy in advanced adrenocortical carcinoma". N. Engl. J. Med. (2012) 366: 2189-2197; Pannu V, Kama P, Sajja HK, et al. "Synergistic antimicrotubule therapy for prostate cancer". Biochem. Pharmacol. (2011) 81: 478-487). Хотя комбинации вемурафениба со средствами, направленными на тот же путь митоген-активируемой протеинкиназы (MAPK), такими как ингибиторы MEK или ERK, широко изучались и проявляли клиническую эффективность (Greger JG, Eastman SD, Zhang V, et al. "Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations". Mol. Cancer Ther. (2012) 11: 909-920; Patel SP, Lazar AJ, Papadopoulos NE, et al. "Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma". Cancer (2013) 119(4): 799-805; Flaherty KT, Infante JR, Daud A, et al. "Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations". N. Engl. J. Med. (2012) 367: 1694-1703), но они лишь могут задержать клетки в фазе G0/G1. Такие стратегии комбинирования вряд ли будут эффективными в отношении резистентных клеток, способных избежать такого блокирования клеточного цикла.
Прошедшие хроническую селекцию устойчивые к вемурафениб клетки меланомы человека (например, A375RF21) не блокируются вемурафенибом в фазе G0/G1 при концентрации, эффективной для чувствительной исходной линии клеток (т.е. А375), а устойчивые к вемурафениб клетки легко переходят в фазу G2/M (фиг. 2А). Таким образом, комбинация вемурафениб с таким соединением, которое сильно индуцирует блокировку последующей фазы G2/M, должна успешно захватить устойчивые к вемурафениб клетки, избежавшие блокировки G0/G1, тем самым вызывая сильную синергию.
Недавно был открыт новый класс антимитотических средств, представленных каркасом 2-арил-4-бензоилимидазола (ABIs) (Chen J, Li CM, Wang J, et al. "Synthesis and antiproliferative activity of novel 2-aryl-4-benzoyl-imidazole derivatives targeting tubulin polymerization". Bioorg. Med. Chem. (2011) 19: 4782-4795; Chen J, Wang Z, Li CM, et al. "Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicine binding site in tubulin as potential anticancer agents". J. Med. Chem. (2010) 53: 7414-7427; Chen J, Ahn S, Wang J, et al. "Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J. Med. Chem. (2012) 55: 7285-7289; Li CM, Lu Y, Chen J, et al. "Orally bioavailable tubulin antagonists for paclitaxel-refractory cancer". Pharm. Res. (2012) 29: 3053-3063). Эти соединения проявляли антипролиферативные значения IC50 в низком наномолярном (нМ) диапазоне у нескольких линий клеток меланомы человека и мыши. Они связываются с тубулином по сайту связывания колхицина. По сравнению со многими существующими ингибиторами тубулина типа паклитакселя и винбластина соединения ABI могут эффективно обходить несколько клинически значимых механизмов множественной лекарственной устойчивости, в том числе лекарственной устойчивости, опосредованной Р-гликопротеином (Pgp), связанными со множественной лекарственной устойчивостью белками (MRPs) и белками устойчивости рака молочной железы (BCRP). Исследование in vivo показало, что они значительно ингибируют метастазирование клеток B16-F10 меланомы легких у мышей (Wang Z, Chen J, Wang J, et al. "Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma metastasis to the lung". Pharm. Res. (2012) 29: 3040-3052).
При быстро растущей заболеваемости раком и особенно меланомой и высокой устойчивости к современным терапевтических средствам, выявление более эффективных комбинаций препаратов, направленных на альтернативные пути для преодоления устойчивости к BRAFi при меланоме должно принести существенную пользу пациентам. Кроме того, поскольку мутации BRAF также распространены при многих других видах рака, включая рак яичников, колоректальный рак и папиллярный рак щитовидной железы, разработка новых стратегий комбинирования может оказать более широкое воздействие на те раковые заболевания, при которых существующие комбинации BRAFi + MEKi почти не проявляют клинической активности, причем это настоятельно необходимо.
Сущность изобретения
В одном воплощении настоящее изобретение касается фармацевтических композиций, содержащих ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; и фармацевтически приемлемый носитель.
В одном воплощении настоящее изобретение касается фармацевтических композиций, содержащих соединение, представленное структурной формулой II:
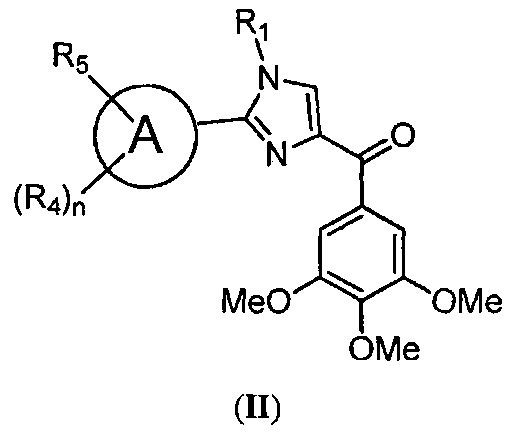
где: А означает одиночную или конденсированную ароматическую или гетероароматическую кольцевую систему;
R1 означает H, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=O)-арил, -(C=O)-фенил или OH;
R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
n означает целое число от 1 до 4;
либо его фармацевтически приемлемую соль, N-оксид, гидрат, таутомер или изомер; в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; и фармацевтически приемлемый носитель.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения (i) рака с мутацией BRAF, (ii) рака, устойчивого к ингибиторам BRAF, (iii) меланомы, (iv) лекарственно-устойчивой меланомы, (v) раковых метастазов у субъекта; или (vi) замедления или предотвращения устойчивого к ингибиторам BRAF рака у субъекта; включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или MEK; в комбинации с соединением, представленным структурной формулой II:
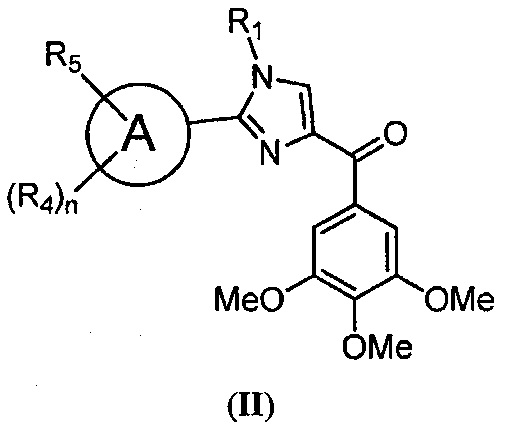
где: А означает одиночную или конденсированную ароматическую или гетероароматическую кольцевую систему;
R1 означает Н, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=O)-арил, -(C=O)-фенил или OH;
R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
n означает целое число от 1 до 4;
либо его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером;
субъекту, страдающему раком с мутацией BRAF, в условиях, эффективных для лечения данного рака.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения, торможения, устранения, замедления или предотвращения вторичной устойчивости рака к таксановым препаратам у субъекта, страдающего раком и ранее принимавшего таксановые препараты, включающего введение данному субъекту композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением, представленным структурной формулой II:
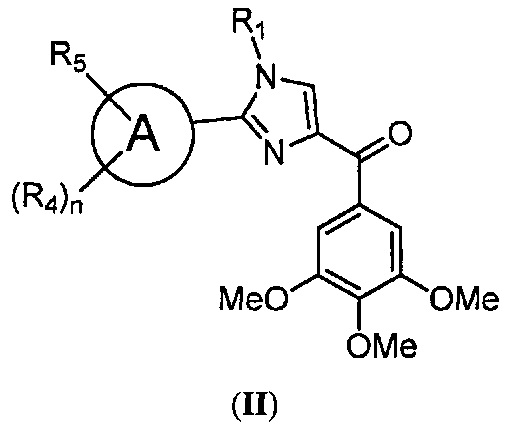
где: А означает одиночную или конденсированную ароматическую или гетероароматическую кольцевую систему;
R1 означает H, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=O)-арил, -(C=O)-фенил или OH;
R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
n означает целое число от 1 до 4;
либо его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером.
В одном воплощении настоящее изобретение касается способа: (i) лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака; (ii) подавления приобретенной устойчивости к ингибиторам BRAF; (iii) замедления или предотвращения возникновения устойчивости к ингибиторам BRAF; либо (iv) лечения, подавления, торможения, устранения, уменьшения, замедления или предотвращения метастазирования рака; включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с ингибитором тубулина, субъекту, страдающему лекарственно-устойчивым раком, в условиях, эффективных для лечения данного рака.
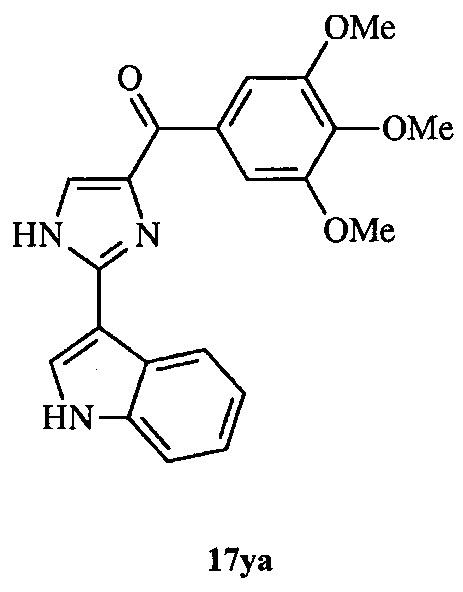
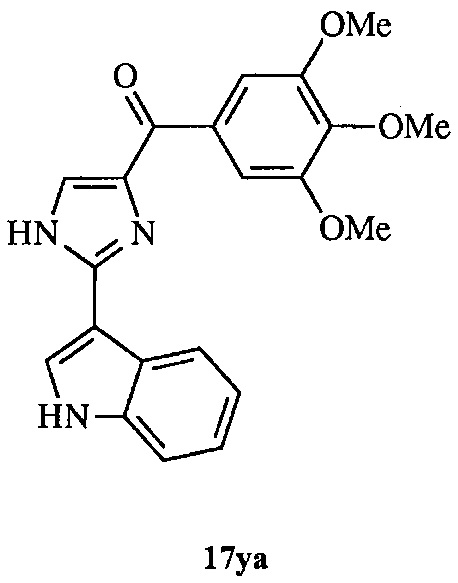
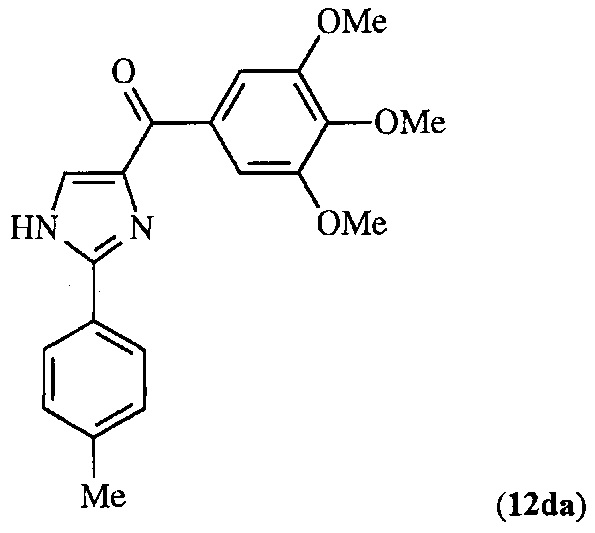
В другом воплощении соединение настоящего изобретения представлено соединением 12da. Еще в одном воплощении соединение настоящего изобретения представлено соединением 17уа.
В одном воплощении настоящее изобретение касается фармацевтических композиций, содержащих ингибитор тубулина, ингибитор BRAF и фармацевтически приемлемый носитель. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении ингибитором тубулина является соединение настоящего изобретению.
В одном воплощении настоящее изобретение касается фармацевтических композиций, содержащих соединение, представленное следующей формулой:
 или
или 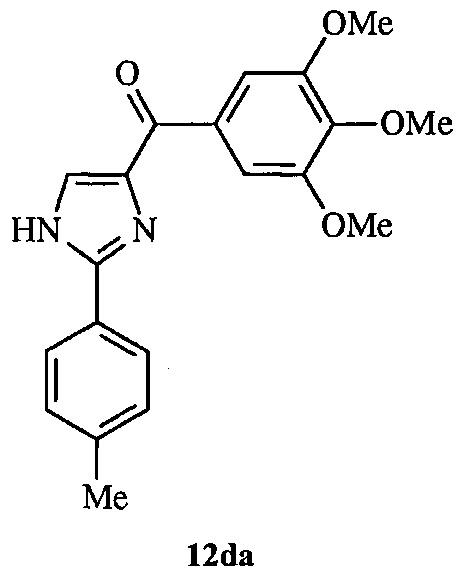
в комбинации с ингибитором BRAF и фармацевтически приемлемым носителем. В другом воплощении ингибитором BRAF является вемурафениб.
В одном воплощении настоящее изобретение касается фармацевтических композиций, содержащих соединение, представленное следующей структурой:
 или
или 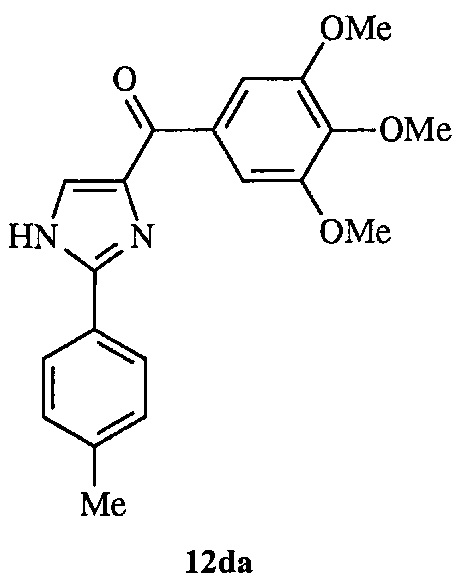
в комбинации с ингибитором MEK и фармацевтически приемлемым носителем. В другом воплощении ингибитором MEK является RO5068760.
В одном воплощении настоящее изобретение касается способа: (а) лечения, подавления, уменьшения тяжести, снижения риска или торможения рака с мутацией BRAF у субъекта; (b) лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивого к ингибиторам BRAF рака; (с) лечения, подавления, уменьшения тяжести, снижения риска или торможения меланомы; (d) лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивой меланомы; (е) лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака; (f) преодоления невосприимчивости к лечению ингибиторами BRAF у субъекта, страдающего лекарственно-устойчивым раком; или (g) предотвращения, устранения, уменьшения или замедления невосприимчивости к лечению рака у субъекта, страдающего раком; который включает введение композиции, содержащей соединение настоящего изобретения в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении рак представляет собой меланому, рак щитовидной железы, колоректальный рак или рак яичников. В другом воплощении рак представлен меланомой. В другом воплощении меланома является положительной по V600E. В другом воплощении рак представлен лекарственно-устойчивым раком. В другом воплощении меланома представлена лекарственно-устойчивой меланомой. В другом воплощении соединение по изобретению представлено соединением 12da. В другом воплощении соединение по изобретению представлено соединением 17уа.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака, который включает введение субъекту, страдающему лекарственно-устойчивым раком, композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, в условиях, эффективных для лечения данного рака. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любые их комбинации. В другом воплощении рак представляет собой меланому, рак щитовидной железы, колоректальный рак или рак яичников. В другом воплощении рак представлен меланомой.
Краткое описание фигур
Тематика настоящего изобретения подробно изложена и заявлена по пунктам в заключительной части спецификации. Однако изобретение, как в плане организации, так и способа применения, вместе с его задачами, признаками и преимуществами, лучше всего раскрывается с привлечением нижеследующего подробного описания вместе с прилагаемыми чертежами.
На фиг. 1 представлено получение линии устойчивых к вемурафениб клеток меланомы A375 (A375RF21) из исходной линии клеток A375 при хроническом отборе на протяжении 3 месяцев при возрастающих концентрациях вемурафениб. Анализ методом MTS показал, что значение IC50 для подавления пролиферации в исходных клетках меланомы А375 (0,57±0,03 мкМ) повысилось в 50 раз при измерении на устойчивых к вемурафениб клетках A375RF21 (28,9±0,6 мкМ). Напротив, значения IC50 у соединения 12da существенно не изменялись (10,7±1,5 нМ на клетках исходной линии А375 и 13,6±4,4 нМ на клетках A375RF21, соответственно). На рисунке приведены структуры соединений 12da и вемурафениб.
На фиг. 2 представлен анализ клеточного цикла (n=4). А. Клетки A375 или A375RF21 обрабатывали 1 мкМ вемурафениб в течение 24 ч и сравнивали с получавшей DMSO контрольной группой. Вемурафениб в дозе 1 мкМ эффективно блокировал клетки А375 в фазе G0/G1, но не блокировал устойчивые клетки A375RF21. В. Клетки A375RF21 обрабатывали DMSO, 30 мкМ вемурафениб, 20 нМ соединения 12da, 20 нМ доцетакселя и их комбинациями в течение 24 ч. Соединение 12da и доцетаксель индуцировали блокировку клеток A375RF21 в фазе G2/M, а их комбинации с вемурафениб блокировали клетки в фазах G1/G2/M.
На фиг. 3 показано, что комбинации ингибитора тубулина с вемурафениб синергически повышают степень апоптоза или гибели клеток у устойчивых клеток A375RF21. А. Репрезентативные диаграммы показывают распределение клеток в квадрантах Q1 (ранний апоптоз), Q2 (апоптоз), Q3 (живые) и Q4 (мертвые). Кластеры клеток с цитоморфологическим профилем типа высокое SSC (боковое рассеяние)/низкое FSC (прямое рассеяние) выделены черным цветом. Не отмечалось никаких отличий по обратному рассеянию между серой и черной популяцией. В. Степень апоптоза рассчитывали путем сложения процентов распределения в Q1, Q2 и Q4. В группах с комбинацией препаратов проявлялась значительно большая (*р<0,05) степень апоптоза по сравнению с простой суммой степени апоптоза в двух группах с монотерапией.
На фиг. 4 представлен эффект монотерапии и комбинированной терапии на полимеризацию тубулина на основе анализа очищенного белка (n=3). Вемурафениб при 20 мкМ практически не влияет на полимеризацию тубулина по сравнению с контрольной группой DMSO. Эффект ингибирования полимеризации тубулина в группе комбинированной терапии обусловлен исключительно соединением 12da.
На фиг. 5 представлен анализ методом вестерн-блот с указанными антителами на лизатах клеток A375RF21 (A), MDA-MB-435 и WM164 (В) через 48 ч после обработки. В качестве контроля на нанесение использовали GAPDH. А. Хотя указанные комбинированные терапии вызывали лишь умеренное снижение уровня p-ERK, они сильно ингибировали фосфорилирование AKT и повышали уровень маркеров апоптоза, в том числе расщепление PARP и расщепление каспазы-3. В. Соединение 12da также проявляло эффекты нокаута AKT на двух других линиях клеток меланомы человека с мутацией BRAFV600E, MDA-MB-435 и WM164.
На фиг. 6 представлено комбинирование in vivo вемурафениб и соединения 12da на модели устойчивых ксенотрансплантатов A375RF21 (n=7). А. Снимки выделенной опухолевой ткани. В. Кривые роста объема опухолей. С. График веса тела мышей от времени. D. Репрезентативные иммуногистохимические снимки при окрашивании срезов ткани опухолей Н&Е, Ki67, pAKT, pERK и S100 после 3-недельной монотерапии или комбинированной терапии. Синие отметки масштаба на снимках соответствуют 100 мкм.
На фиг. 7 представлено комбинирование in vivo высокой дозы вемурафениб (30 мг/кг) и соединения 12da (15 мг/кг) на модели устойчивых ксенотрансплантатов A375RF21 (n=5). А. Снимки выделенной опухолевой ткани. В. Кривые роста объема опухолей. С. График веса тела мышей от времени. Комбинация соединения 12da и вемурафениба при такой дозе вызывала регрессию опухолей на 44,9%.
На фиг. 8 представлена схема синтеза для получения арил-бензоил-имидазоловых (ABI) соединений настоящего изобретения. Реагенты и условия: (a) t-BuOH, I2, этилен-диамин, K2CO3, до кипения; (b) Phi (OAc)2, K2CO3, DMSO; (с) DBU, CBrCl3, DMF; (d) NaH, PhSO2Cl, THF, от 0°C до RT; (e) t-BuLi, замещенный бензоилхлорид, THF, -78°C; (f) Bu4NF, THF, RT.
На фиг. 9 представлена схема синтеза для получения арил-бензоил-имидазоловых (ABI) соединений настоящего изобретения. Реагенты и условия: (a) NH4OH, оксальдегид, этанол, RT; (b) NaH, PhSO2Cl, THF, от 0°C до RT; (с) t-BuLi, замещенный бензоилхлорид, THF, -78°C; (d) BU4NF, THF, RT; (е) BBr3, CH2Cl2; (f) c-HCl, AcOH, до кипения.
На фиг. 10 представлена схема синтеза для получения арил-бензоил-имидазоловых (ABI) соединений настоящего изобретения. Реагенты и условия: (a) NaH, замещенный бензоилхлорид, THF.
На фиг. 11 представлена схема синтеза соединений 12dc, 12fc, 12daa, 12dab, 12cba. (a) AlCl3, THF, до кипения; (b) NaH, CH3I для 12dab и 12cba и BnBr для 12daa, THF, до кипения.
На фиг. 12 представлена схема синтеза соединений 11gaa, 12la. (a) NH4OH, этанол, глиоксаль, RT; (b) NaH, замещенный PhSO2Cl, THF, от 0°C до RT; (с) t-BuLi (1,7 М в пентане), замещенный бензоилхлорид, THF, -78°C; (d) BU4NF, RT.
На фиг. 13 представлена схема синтеза соединения 12fa. (a) NH4OH, оксальдегид, этанол, RT; (b) NaH, PhSO2Cl, THF, от 0°C до RT; (с) t-BuLi, 3,4,5-триметоксибензоилхлорид, THF, -78°C; (d) Bu4NF, THF, RT.
На фиг. 14 представлена схема синтеза соединений 17уа, 17yab и 17уас. (а) 1. KOH, этанол, 2. PhSO2Cl, ацетон, RT; (b) NH4OH, глиоксаль, этанол, RT; (с) NaH, PhSO2Cl, THF, от 0°C до RT; (d) t-BuLi (1,7 М в пентане), 3,4,5-триметоксибензоилхлорид, THF, -78°C; (е) NaOH, этанол, H2O, до кипения; (f) TBAF, THF, RT; (g) NaH, CH3I, THF.
На фиг. 15 представлено распределение клеточного цикла клеток РС3 при 24-часовой обработке соединениями настоящего изобретения (12q, 70а, 70f и 70m).
На фиг. 16 представлены кривые "доза-эффект" 2-арил-4-бензоил-имидазоловых соединений (ABIs) в сравнении с другими противораковыми препаратами и соединениями на линию клеток меланомы со множественной лекарственной устойчивостью (клетки MDR) и соответствующую чувствительную исходную линию клеток (обычные клетки меланомы). Большое расстояние между двумя кривыми для паклитакселя, винбластина и колхицина указывает на то, что они являлись субстратами для Р-гликопротеина (P-gp). Перекрывание двух кривых у каждого ABI-соединения означает, что ABI-соединения не были субстратами для P-gp и подавляли множественную лекарственную устойчивость.
На фиг. 17 представлены эффекты ABI-соединений на полимеризацию тубулина in vitro. Тубулин (0,4 мг на пробу) обрабатывали ABI-соединениями при 10 мкМ (контроль на носитель: 5% DMSO). Отслеживали поглощение при 340 нм при 37°C каждую минуту на протяжении 15 мин и оказалось, что ABI-соединения 12da, 12db и 12cb ингибируют полимеризацию тубулина in vitro.
На фиг. 18 представлен анализ образования колоний меланомы B16-F1 в мягком агаре, который показал, что ABI-соединения ингибируют образование колоний в зависимости от концентрации. Фиг. 18А. Репрезентативные снимки у контроля и каждого исследуемого соединения (12cb, 12da и 12fb) при 100 нМ. Диаметр каждой лунки составлял 35 мм. Фиг. 18B. Количественное выражение результатов анализа для каждого исследуемого соединения (12cb, 12da и 12fb). Значения Р рассчитывали по сравнению с контролем по t-критерию Стьюдента с помощью программы GraphPad Prism. Столбцы - средние из трех повторов; планки - SD.
На фиг. 19 представлено исследование ABI-соединений in vivo. Фиг. 19А. Активность 12cb in vivo против опухолей меланомы B16-F1 у мышей C57/BL. Фиг. 19B. Активность 12fb in vivo против меланомы B16-F1 у мышей C57BL/6 и мышей SHO nude. Результаты показывают, что 12fb ингибирует рост опухолей меланомы дозо-зависимым образом. Мыши C57BL/6 несли аллотрансплантаты меланомы B16-F1 (n=5 на группу). Каждая мышь получала 0,5×106 клеток путем подкожной инъекции в бок. Когда размер опухоли достигал ~100 мм3, начинали ежедневную обработку по 30 мкл в/б. Фиг. 19C. Активность 12fb in vivo против ксенотрансплантатов меланомы A375 человека. Мыши SHO nude несли ксенотрансплантаты меланомы А375 человека (n=5 на группу). Каждая мышь получала 2,5×106 клеток путем подкожной инъекции в бок. Когда размер опухоли достигал ~150 мм3, начинали ежедневную обработку по 30 мкл в/б. Контроль: только носитель; точки: средние значения; планки: SD. DTIC=(5-(3,3-диметил-1-триазенил)-имидазол-4-карбоксамид, т.е. дакарбазин.
На фиг. 20 представлены эффекты 17уа и 55 на полимеризацию тубулина. Соединения 17уа и 55 связываются с сайтом связывания колхицина на тубулине и ингибируют полимеризацию тубулина. Фиг. 20А. Конкуренция за связывание. Тубулин (1 мг/мл) и колхицин (1,2 мкМ) инкубировали при различных концентрациях подофиллотоксина, винбластина, соединений 17уа и 55 (n=3; среднее ± SD). В качестве положительного и отрицательного контроля использовали подофиллотоксин и винбластин, соответственно. Фиг. 20B. Влияние на полимеризацию тубулина. Тубулин (0,4 мг) обрабатывали исследуемыми соединениями (5 мкМ). В качестве положительного контроля использовали колхицин. Фиг. 20C и 20D. Способность 17уа и 55 усиливать образование цитоплазматических комплексов ДНК-гистоны (апоптоз) через 24 ч на клетках РС-3 (С) и РС-3/TxR (D) (n=3; среднее ± SD). В качестве положительного контроля использовали доцетаксель.
На фиг. 21 представлено противораковое действие in vivo. Фиг. 21A. Мышей nude, несущих опухоли РС-3, обрабатывали доцетакселем (в/в, 10 или 20 мг/кг) в 1-й и 9-й день (n=5-6). Планки: SE. Фиг. 21B. Мышей nude, несущих опухоли РС-3/TxR, обрабатывали доцетакселем (в/в, 10 или 20 мг/кг) в 1-й и 9-й день и обрабатывали соединением 17уа (per os, 6,7 мг/кг) раз в день, пять дней в неделю (n=4-5). Планки: SE. Фиг. 21C. Мышей nude, несущих опухоли РС-3/TxR, обрабатывали соединением 17уа (per os, 3,3 мг/кг) два раза в день в течение 4 дней в первую неделю, а затем один раз в день, пять дней в неделю со 2-й по 4-ю неделю (n=7), а соединением 55 обрабатывали (per os, 10 или 30 мг/кг) два раза в день, пять дней в неделю в течение 4 недель (n=7). Планки: SE. Фиг. 21D. Мышей nude, несущих опухоли РС-3/TxR, обрабатывали соединением 17уа (per os, 10 мг/кг) три раза в неделю в течение 4 недель (n=5). Планки: SE.
На фиг. 22 представлено противораковое действие in vivo соединения 17уа на ксенотрансплантаты клеток лейкемии HL60.
На фиг. 23 представлен анализ клеточного цикла при двойном окрашивании антителом против фосфогистона Н3 и PI (йодидом пропидия) у устойчивых к вемурафениб клеток. Клетки A375RF21 (биологические пробы, n=4) обрабатывали средой для культуры клеток, содержащей либо 5% DMSO (контроль на носитель), либо один препарат или указанные комбинации в течение 24 ч, после чего проводили окрашивание с помощью антитела против фосфогистона H3-AlexaFluor® 488 и PI, а затем анализировали методом проточной цитометрии. А. Репрезентативные диаграммы иллюстрируют распределение клеток. Красные линии определяли вручную, чтобы показать, как по ним рассчитывали распределение фаз клеточного цикла. В. Количественные данные (среднее ± SD) по распределению фаз клеточного цикла. Ctrl: 5% DMSO; Vem: вемурафениб, 30 мкМ; ABI: соединение 12da, 20 нМ; Vem + ABI: вемурафениб, 30 мкМ + соединение 12da, 20 нМ; Doc: доцетаксель, 20 нМ; Vem + Doc: вемурафениб, 30 мкМ + доцетаксель, 20 нМ.
На фиг. 24 представлены кривые "доза-эффект" in vitro (n=5) по каждой комбинации на клетках А375 и A375RF21. По оси X: уровень дозы относительно концентрации IC50 препарата А или В на клетках А375 или A375RF21 при комбинированной терапии А + В. Фиг. 24A-24C - данные по клеткам А375, фиг. 24D-24F - данные по клеткам A375RF21.
На фиг. 25 показано, что основными механизмами устойчивости к вемурафениб у клеток A375RF21 являются гиперэкспрессия PDGFβ и активация пути PI3K-AKT. Оба механизма устойчивости были хорошо установлены на клинических опухолях, указывая, что механизмы устойчивости клеток A375RF21 могут представлять клинически значимые механизмы лекарственной устойчивости. Панель А. Анализ методом вестерн-блот для сравнения различных уровней белка у чувствительных исходных клеток A375 и устойчивых к вемурафениб клеток A375RF21, в присутствии или в отсутствие 2,5 мкМ вемурафениба (поддерживающая концентрация для культуры клеток A375RF21). Клетки инкубировали с контрольным носителем или 2,5 мкМ вемурафениб в течение 24 ч. Определяли уровень фосфо-PI3K после 30-минутной стимуляции с помощью 30 мкМ перекиси водорода. На графике представлены репрезентативные результаты из трех независимых экспериментов. Панель В. Эффективность ингибирования роста ингибиторами киназ при определении методом MTS (n=4) на клетках А375 и A375RF21. Траметиниб и селуменитиб являются ингибиторами MEK, а сунитиниб (малатная соль) является ингибитором RTK. Панель С. Сравнение расчетных значений IC50 (в виде среднего ± SD).
На фиг. 26 представлены кривые роста in vitro для клеток А375 и устойчивой к вемурафениб сублинии клеток A375RF21. В 96-луночные планшеты высеивали по 2000 клеток на лунку в 100 мкл культуральной среды (n=6) и инкубировали при 37°C, 5% CO2. Определяли общее количество белка методом SRB в каждой указанной точке времени, соответственно. Затем строили график значений поглощения при 564 нм в зависимости от времени роста клеток.
На фиг. 27 представлено схематическое описание стратегии исследований. Мышей NSG с привитыми вемурафениб-чувствительными или вемурафениб-устойчивыми PDX-опухолями Р2 приобретали у фирмы JAX. Опухоли Р2 размножали в дополнительных мышах NSG, чтобы получить достаточное количество опухолей Р3 для исследования. У полученных опухолей Р3 определяли генетические профили и гистологию и сверяли со стандартами из исходных опухолей Р0, поставленных на фирме JAX, чтобы обеспечить правильность опухолей. Кусочки опухолей Р3 имплантировали мышам для получения опухолей Р4 для исследований по эффективности, описанных в "Цели №1 и 2" (Пример 11). Кроме того, мышам вводили в хвостовую вену суспензии одиночных клеток из опухолей Р3 на экспериментальной модели метастазов меланомы в легких для оценки эффективности комбинаций против метастазирования меланомы в "Цели №3".
На фиг. 28 представлено ингибирование ABI-соединениями 12cb, 12da и 12fb метастазов меланомы в легких у мышей. Панель А. Репрезентативные снимки легких с узелками меланомы (черные точки, n=8 на группу). Обработка: в/б инъекции 5 дней в неделю в течение 2 недель. Панель В. Количество узелков меланомы на каждое легкое. Точки: индивидуальное количество узелков; длинная линия посредине: среднее значение; короткие линии наверху и внизу: 95% доверительные интервалы ** и ##: р<0,01. Панель С. Изменения веса тела мышей в ходе эксперимента. Точки: среднее; планки: SD. Контроль: только раствор носителя.
Следует иметь в виду, что для простоты и четкости изложения элементы, представленные на фигурах, не обязательно вычерчены в масштабе. Например, размеры некоторых элементов могут быть увеличены относительно других элементов для ясности. Кроме того, там, где это уместно, счетные номера могут повторяться между фигурами для обозначения соответствующих или аналогичных элементов.
Осуществление изобретения
В одном воплощении настоящее изобретение касается соединений, представленных структурной формулой I:
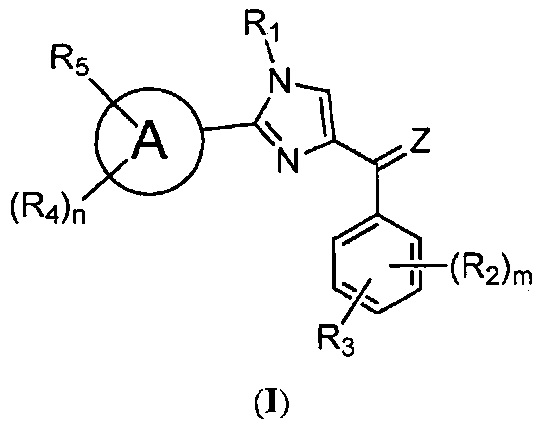
где: А означает одиночную или конденсированную ароматическую или гетероароматическую кольцевую систему;
Z означает O или S;
R1 означает H, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=O)-арил, -(C=O)-фенил или OH;
R2, R3, R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
m и n каждый независимо означает целое число от 0 до 4;
либо их фармацевтически приемлемых солей, N-оксидов, гидратов, таутомеров или изомеров.
В одном воплощении А означает арил. В другом воплощении А означает фенил. В другом воплощении А означает индолил. В другом воплощении А означает 3-индолил. В другом воплощении Z означает O.
В одном воплощении R2 означает OMe. В другом воплощении R3 означает H. В другом воплощении m равно 3. В другом воплощении R2 означает OMe, R3 означает H и m равно 3.
В одном воплощении R4 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R4 означает Me. В другом воплощении R4 означает H. В другом воплощении R5 означает H. В другом воплощении n равно 1. В другом воплощении R4 означает Me, R5 означает H и n равно 1. В другом воплощении R4 означает H, R5 означает H и n равно 1.
В другом воплощении R1 означает H. В другом воплощении R1 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R1 означает Me.
В одном воплощении настоящее изобретение касается соединений, представленных структурной формулой II:
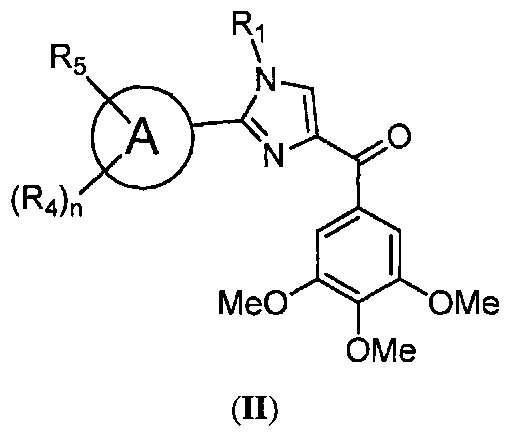
где: А означает одиночную или конденсированную ароматическую или гетероароматическую кольцевую систему;
R1 означает H, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=О)-арил, -(C=O)-фенил или OH;
R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
n означает целое число от 1 до 4;
либо их фармацевтически приемлемых солей, N-оксидов, гидратов, таутомеров или изомеров.
В одном воплощении А означает арил. В другом воплощении А означает фенил. В другом воплощении А означает индолил. В другом воплощении А означает 3-индолил.
В одном воплощении R4 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R4 означает Me. В другом воплощении R4 означает H. В другом воплощении R5 означает H. В другом воплощении n равно 1. В другом воплощении R4 означает Me, R5 означает H и n равно 1. В другом воплощении R4 означает H, R5 означает H и n равно 1.
В другом воплощении R1 означает H. В другом воплощении R1 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R1 означает Me.
В одном воплощении настоящее изобретение касается соединений, представленных структурной формулой III:
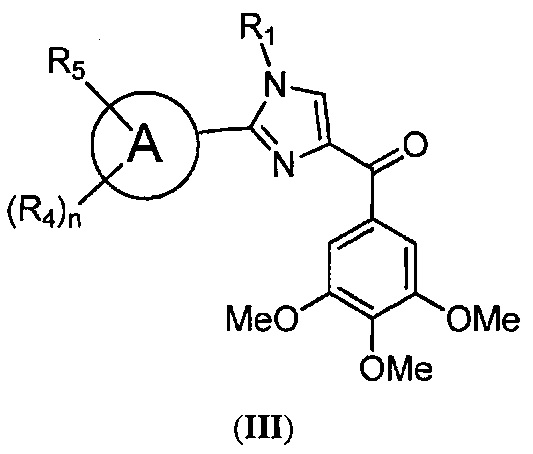
где: R1 и R9 каждый независимо означает H, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=O)-арил, -(C=O)-фенил или OH;
R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
n означает целое число от 1 до 4;
либо их фармацевтически приемлемых солей, N-оксидов, гидратов, таутомеров или изомеров.
В одном воплощении R4 означает H. В другом воплощении R5 означает H. В другом воплощении n равно 1. В другом воплощении R4 означает H, R5 означает H и n равно 1. В другом воплощении R9 означает H. В другом воплощении R9 означает Me.
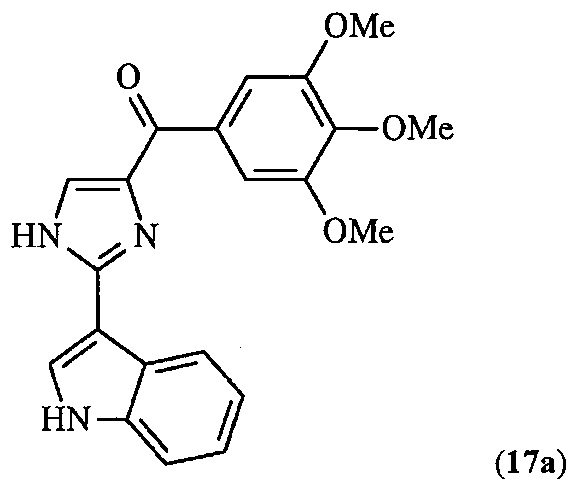
В другом воплощении R1 означает H. В другом воплощении R1 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R1 означает Me.
В другом воплощении соединение формулы III представлено структурой соединения 17уа:
В одном воплощении настоящее изобретение касается соединений, представленных структурной формулой IV:
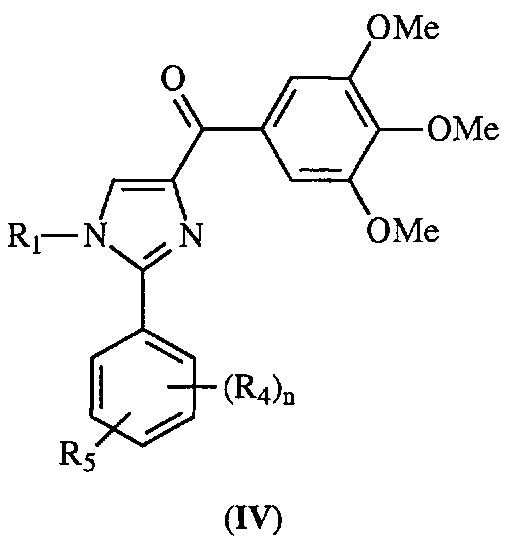
где: R1 означает H, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=O)-арил, -(C=O)-фенил или OH;
R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
n означает целое число от 1 до 4;
либо их фармацевтически приемлемых солей, N-оксидов, гидратов, таутомеров или изомеров.
В одном воплощении R4 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R4 означает Me. В другом воплощении R4 означает H. В другом воплощении R5 означает H. В другом воплощении n равно 1. В другом воплощении R4 означает Me, R5 означает H и n равно 1.
В другом воплощении R1 означает H. В другом воплощении R1 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R1 означает Me.
В другом воплощении соединение формулы IV представлено структурой соединения 12da:
В одном воплощении А у соединений формулы I и II означает Ph. В другом воплощении А у соединений формулы I и II означает индолил. В другом воплощении А у соединений формулы I и II означает 2-индолил. В другом воплощении А у соединений формулы I и II означает 3-индолил. В другом воплощении А у соединений формулы I и II означает 4-индолил. В другом воплощении А у соединений формулы I и II означает 5-индолил. В другом воплощении А у соединений формулы I и II означает 6-индолил. В другом воплощении А у соединений формулы I и II означает 7-индолил.
В другом воплощении R5 находится в пара-положении. В другом воплощении R5 находится в мета-положении. В другом воплощении R5 находится в орто-положении. В другом воплощении R5 означает 4-Ме. В другом воплощении R5 означает H. В другом воплощении R5 означает 4-F.
В другом воплощении R4 означает H.
В другом воплощении n равно 1.
В одном воплощении группа А в формулах I и II означает фуранил, бензофуранил, бензотиофенил, индолил, пиридинил, фенил, бифенил, трифенил, дифенилметан, адамантанил, флуоренил и другие гетероциклические аналоги, такие, например, как пирролил, пиразолил, пиридинил, имидазолил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, пирролизинил, индолил, изохинолинил, хинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хиналолинил, фталазинил, нафтиридинил, хиноксалинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, фуранил, пирилий, бензодиоксолил, тиранил, тиетанил, тетрагидротиофенил, дитиоланил, тетрагидротиопиранил, тиофенил, тиепинил, тианафтенил, оксатиоланил, морфолинил, тиоксанил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил.
В одном воплощении А означает фенил. В другом воплощении А означает группу индолила; наиболее предпочтительно 3-индолил или 5-индолил.
В одном воплощении Z в формуле I означает О. В другом воплощении Z означает S.
В одном воплощении R1 в формулах I, II, III и IV означает водород. В другом воплощении R1 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R1 означает Me. В другом воплощении R1 означает линейный или разветвленный C1-C6-галоалкил. В другом воплощении R1 означает CF3. В другом воплощении R1 означает фенил. В другом воплощении R1 означает бензил. В другом воплощении R1 означает SO2-арил. В другом воплощении R1 означает (C=O)-арил.
В одном воплощении R3 в формуле I находится в пара-положении. В другом воплощении R3 находится в мета-положении. В другом воплощении R3 находится в орто-положении.
В одном воплощении R2 и R3 в формуле I независимо означают водород. В другом воплощении R2 и R3 независимо означают линейный или разветвленный C1-C6-алкокси. В другом воплощении R2 и R3 независимо означают OCH3. В другом воплощении R2 и R3 независимо означают F. В другом воплощении R2 и R3 независимо означают 4-F. В другом воплощении R2 и R3 независимо означают Cl. В другом воплощении R2 и R3 независимо означают Br. В другом воплощении R2 и R3 независимо означают I. В другом воплощении R2 и R3 независимо означают линейный или разветвленный C1-C6-галоалкил. В другом воплощении R2 и R3 независимо означают CF3. В другом воплощении R2 и R3 независимо означают CN. В другом воплощении R2 и R3 независимо означают NH2. В другом воплощении R2 и R3 независимо означают OH. В другом воплощении R2 и R3 независимо означают линейный или разветвленный C1-C6-алкил. В другом воплощении R2 и R3 независимо означают CH3. В другом воплощении R2 и R3 независимо означают NO2. В другом воплощении R2 и R3 независимо означают алкиламино. В другом воплощении R2 и R3 независимо означают 4-N(Me)2.
В одном воплощении m в формуле I равно 0. В другом воплощении m равно 1. В другом воплощении m равно 2. В другом воплощении m равно 3. В другом воплощении m равно 4.
В одном воплощении R5 в формулах I, II, III и IV находится в пара-положении. В другом воплощении R5 находится в мета-положении. В другом воплощении R5 находится в орто-положении.
В одном воплощении R4 и R5 в формулах I, II, III и IV независимо означают водород. В другом воплощении R4 и R5 независимо означают линейный или разветвленный C1-C6-алкокси. В другом воплощении R4 и R5 независимо означают OMe. В другом воплощении R4 и R5 независимо означают F. В другом воплощении R4 и R5 независимо означают Cl. В другом воплощении R4 и R5 независимо означают Br. В другом воплощении R4 и R5 независимо означают I. В другом воплощении R4 и R5 независимо означают линейный или разветвленный C1-C6-галоалкил. В другом воплощении R4 и R5 независимо означают CF3. В другом воплощении R4 и R5 независимо означают CN. В другом воплощении R4 и R5 независимо означают NH2. В другом воплощении R4 и R5 независимо означают OH. В другом воплощении R4 и R5 независимо означают линейный или разветвленный C1-C6-алкил. В другом воплощении R4 и R5 независимо означают NO2. В другом воплощении R4 и R5 независимо означают алкиламино.
В одном воплощении n в формулах I, II, III и IV равно 0. В другом воплощении n равно 1. В другом воплощении n равно 2. В другом воплощении n равно 3. В другом воплощении n равно 4.
Следует иметь в виду, что у гетероциклических колец n ограничивается числом доступных для замены положений, то есть количеством групп CH минус один. Соответственно, если кольцом A, к примеру, является фуранил, тиофенил или пирролил, то n составляет от 0 до 2; а если кольцом А, к примеру, является оксазолил, имидазолил или тиазолил, то n равно 0 либо 1; а если кольцом А, к примеру, является оксадиазолил или тиадиазолил, то n равно 0.
В одном воплощении R9 в формуле III означает водород. В другом воплощении R9 означает линейный или разветвленный C1-C6-алкил. В другом воплощении R9 означает CH3. В другом воплощении R9 означает линейный или разветвленный C1-C6-галоалкил. В другом воплощении R9 означает CF3. В другом воплощении R9 означает фенил. В другом воплощении R9 означает -CH2Ph. В другом воплощении R9 означает SO2-арил. В другом воплощении R9 означает (C=O)-арил. В другом воплощении R9 означает (SO2)-Ph. В другом воплощении R9 означает (SO2)-Ph-OCH3.
В настоящем изобретении "одиночная или конденсированная ароматическая или гетероароматическая кольцевая система" может представлять собой любое такое кольцо, включая, без ограничения, фенил, индолил, 1Н-индол, изоиндолил, пиридинил, пиримидинил, пиридазинил, пиразинил, триазинил, тетразинил, тиазолил, изотиазолил, оксазолил, изоксазолил, имидазолил, пиразолил, пирролил, фуранил, тиофенил, изохинолинил, нафтил, антраценил, бензимидазолил, индазолил, 2Н-индазол, 4,5,6,7-тетрагидро-2Н-индазол, 3Н-индол-3-он, пуринил, бензоксазолил, 1,3-бензоксазолил, бензизоксазолил, бензотиазолил, 1,3-бензотиазол, 4,5,6,7-тетрагидро-1,3-бензотиазол, хиназолинил, хиноксалинил, циннолинил, фталазинил, хинолинил, изохинолинил, акридинил, бензофуранил, 1-бензофуран, изобензофуранил, бензотиофенил, бензо[с]-тиофенил, бензодиоксолил, тиадиазолил, [1,3]оксазоло[4,5-b]пиридин, оксадиазолил, имидазо[2,1-b][1,3]тиазол, 4Н,5Н,6Н-циклопента[d][1,3]тиазол, 5Н,6Н,7Н,8Н-имидазо[1,2-а]пиридин, 7-оксо-6Н,7Н-[1,3]тиазоло[4,5-d]пиримидин, [1,3]тиазоло[5,4-b]пиридин, 2Н,3Н-имидазо[2,1-b][1,3]тиазол, тиено[3,2-d]пиримидин-4(3Н)-он, 4-оксо-4Н-тиено[3,2-d][1,3]тиазин, имидазо[1,2-а]пиридин, пиразоло[1,5-а]пиридин, имидазо[1,2-а]пиразин, имидазо[1,2-а]пиримидин, 1Н-пирроло[2,3-b]пиридин, пиридо[2,3-b]пиразин, пиридо[2,3-b]пиразин-3(4Н)-он, 4Н-тиено[3,2-b]пиррол, хиноксалин-2(1Н)-он, 1Н-пирроло[3,2-b]-пиридин, 7Н-пирроло[2,3-d]пиримидин, оксазоло[5,4-b]пиридин, тиазоло[5,4-b]пиридин и др.
В настоящем изобретении "гетероциклическая кольцевая система" означает насыщенный или ненасыщенный N-гетероцикл, включая, без ограничения, такие аза- и диаза-циклоалкилы, как азиридинил, азетидинил, диазатидинил, пирролидинил, пиперидинил, пиперазинил и азоканил, пирролил, пиразолил, имидазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, пирролизинил, индолил, хинолинил, изохинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хинололинил, фталазинил, нафтиридинил, хиноксалинил и др.; насыщенные или ненасыщенные О-гетероциклы, включая, без ограничения, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, фуранил, бензофуранил, пирилий, бензодиоксолил и др.; насыщенные или ненасыщенные S-гетероциклы, включая, без ограничения, тиранил, тиетанил, тетрагидротиофенил, дитиоланил, тетрагидротиопиранил, тиофенил, бензотиофенил, тиепинил, тианафтенил и др.; насыщенные или ненасыщенные смешанные гетероциклы, которыми могут быть любые гетероциклы, содержащие два или несколько гетероатомов S, N или O, включая, без ограничения, оксатиоланил, морфолинил, тиоксанил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил и др.
В настоящем изобретении термин "алкил" может означать любой неразветвленный или разветвленный алкил, содержащий до 30 атомов углерода, если не указано иначе. В другом воплощении алкилы содержат 1-6 атомов углерода. В другом воплощении алкилы содержат 1-8 атомов углерода. В другом воплощении алкилы содержат 1-10 атомов углерода. В другом воплощении алкилы содержат 1-12 атомов углерода. В другом воплощении алкилы содержат 1-20 атомов углерода. В другом воплощении циклические алкилы содержат 3-8 атомов углерода. В другом воплощении разветвленным алкилом является алкил, замещенный боковой цепью алкила из 1-5 атомов углерода. В одном воплощении алкил может быть незамещенным. В другом воплощении алкил может быть замещен галогеном, галоалкилом, гидроксилом, алкокси, карбонилом, амидо, алкиламидо, диалкиламидо, циано, нитро, CO2H, амино, алкиламино, диалкиламино, карбоксилом, тио и/или тиоалкилом.
Алкильная группа может быть единственным заместителем или же она может быть компонентом более крупного заместителя, как-то алкокси, галоалкила, арилалкила, алкиламино, диалкиламино, алкиламидо, алкилкарбамида и т.д. Предпочтительными алкильными группами являются метил, этил и пропил, а также галометил, дигалометил, тригалометил, галоэтил, дигалоэтил, тригалоэтил, галопропил, дигалопропил, тригалопропил, метокси, этокси, пропокси, арилметил, арилэтил, арилпропил, метиламино, этиламино, пропиламино, диметиламино, диэтиламино, метиламидо, ацетамидо, пропиламидо, галометиламидо, галоэтиламидо, галопропиламидо, метилкарбамид, этилкарбамид, пропилкарбамид и др.
В настоящем изобретении термин "арил" относится к таким ароматическим кольцам, которые непосредственно связаны с другой группой. Арильная группа может быть единственным заместителем или же она может быть компонентом более крупного заместителя, как-то арилалкила, ариламино, ариламидо и т.д. Примеры арильных групп включают, без ограничения, фенил, толил, ксилил, фуранил, нафтил, пиридинил, пиримидинил, пиридазинил, пиразинил, триазинил, тиазолил, оксазолил, изоксазолил, пиразолил, имидазолил, тиофенил, пирролил, фенилметил, фенилэтил, фениламино, фениламидо и др. Заместители включают, без ограничения: F, Cl, Br, I, линейный или разветвленный C1-C5-алкил, линейный или разветвленный C1-C5-галоалкил, линейный или разветвленный C1-C5-алкокси, линейный или разветвленный C1-C5-галоалкокси, CF3, CN, NO2, -CH2CN, NH2, NH-алкил, N(алкил)2, гидроксил, -OC(O)CF3, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)Н или C(O)NH2.
В настоящем изобретении термин "алкокси" относится к эфирным группам, замещенным алкильной группой, как определено выше. К алкокси относятся как линейные, так и разветвленные алкоксигруппы. Неограничительные примеры алкоксигрупп представлены метокси, этокси, пропокси, изопропокси, трет-бутокси.
В настоящем изобретении термин "аминоалкил" относится к аминогруппам, замещенным алкильной группой, как определено выше. К аминоалкилам относятся моноалкиламины, диалкиламины или триалкиламины. Неограничительные примеры аминоалкильных групп представлены -N(Me)2, -NHMe, -NH3.
"Галоалкильные" группы относятся, в другом воплощении, к таким алкильным группам, как определено выше, которые замещены одним или несколькими атомами галогена, например, F, Cl, Br или I. Неограничительные примеры галоалкильных групп представлены CF3, CF2CF3, CH2CF3.
"Алкоксиалкильные" группы относятся, в другом воплощении, к таким алкильным группам, как определено выше, которые замещены алкоксигруппой, как определено выше, например, метокси, этокси, пропокси, изопропокси, трет-бутокси и т.д. Неограничительные примеры алкоксиалкильных групп представлены -CH2-О-CH3, -CH2-O-СН(CH3)2, -CH2-O-С(CH3)3, -CH2-CH2-О-CH3, -CH2-CH2-O-CH(CH3)2, -CH2-CH2-О-С(CH3)3.
"Циклоалкильные" или "карбоциклические" группы относятся, в одном воплощении, к кольцевым структурам, содержащим атомы углерода в качестве атомов кольца, которые могут быть насыщенными или ненасыщенными, замещенными или незамещенными. В другом воплощении циклоалкил представляет собой 3-12-членное кольцо. В другом воплощении циклоалкил представляет собой 6-членное кольцо. В другом воплощении циклоалкил представляет собой 5-7-членное кольцо. В другом воплощении циклоалкил представляет собой 3-8-членное кольцо. В другом воплощении циклоалкильная группа может быть незамещенной или замещенной галогеном, алкилом, галоалкилом, гидроксилом, алкокси, карбонилом, амидо, алкиламидо, диалкиламидо, циано, нитро, CO2H, амино, алкиламино, диалкиламино, карбоксилом, тио и/или тиоалкилом. В другом воплощении циклоалкильное кольцо может быть конденсировано с другим насыщенным или ненасыщенным циклоалкильным или гетероциклическим 3-8-членным кольцом. В другом воплощении циклоалкильное кольцо является насыщенным кольцом. В другом воплощении циклоалкильное кольцо является ненасыщенным. Неограничительные примеры циклоалкильных групп включают циклогексил, циклогексенил, циклопропил, циклопропенил, циклопентил, циклопентенил, циклобутил, циклобутенил, циклооктил, циклооктадиенил (COD), циклооктаен (COE) и др.
"Гетероциклы" или "гетероциклические" группы относятся, в одном воплощении, к кольцевым структурам, содержащим, наряду с атомами углерода, атомы серы, кислорода, азота или любые их комбинации в составе кольца. В другом воплощении гетероцикл представляет собой 3-12-членное кольцо. В другом воплощении гетероцикл представляет собой 6-членное кольцо. В другом воплощении гетероцикл представляет собой 5-7-членное кольцо. В другом воплощении гетероцикл представляет собой 3-8-членное кольцо. В другом воплощении гетероциклическая группа может быть незамещенной или замещенной галогеном, алкилом, галоалкилом, гидроксилом, алкокси, карбонилом, амидо, алкиламидо, диалкиламидо, циано, нитро, CO2H, амино, алкиламино, диалкиламино, карбоксилом, тио и/или тиоалкилом. В другом воплощении гетероциклическое кольцо может быть конденсировано с другим насыщенным или ненасыщенным циклоалкильным или гетероциклическим 3-8-членным кольцом. В другом воплощении гетероциклическое кольцо является насыщенным кольцом. В другом воплощении гетероциклическое кольцо является ненасыщенным. Неограничительные примеры гетероциклических колец включают пиридин, пиперидин, морфолин, пиперазин, тиофен, пиррол, бензодиоксол или индол.
В одном воплощении настоящим изобретением предусмотрены соединения по настоящему изобретению либо их изомеры, метаболиты, фармацевтически приемлемые соли, фармацевтические препараты, таутомеры, гидраты, N-оксиды, полиморфы или кристаллы либо их комбинации. В одном воплощении настоящим изобретением предусмотрены изомеры соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены метаболиты соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены фармацевтически приемлемые соли соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены фармацевтические препараты соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены таутомеры соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены гидраты соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены N-оксиды соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены полиморфы соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены кристаллы соединений по изобретению. В другом воплощении настоящим изобретением предусмотрены композиции, содержащие соединения по изобретению, как описано здесь, или, в другом воплощении, комбинации изомеров, метаболитов, фармацевтически приемлемых солей, фармацевтических препаратов, таутомеров, гидратов, N-оксидов, полиморфов или кристаллов соединений по изобретению.
В одном воплощении термин "изомер" включает, без ограничения, оптические изомеры и аналоги, структурные изомеры и аналоги, конформационные изомеры и аналоги и т.п. В другом воплощении изомеры являются оптическими изомерами.
В одном воплощении соединения по изобретению представляют собой чистые (E)-изомеры. В другом воплощении соединения по изобретению представляют собой чистые (Z)-изомеры. В другом воплощении соединения по изобретению представляют собой смесь (Е)- и (Z)-изомеров. В одном воплощении соединения по изобретению представляют собой чистые (R)-изомеры. В другом воплощении соединения по изобретению представляют собой чистые (S)-изомеры. В другом воплощении соединения по изобретению представляют собой смесь (R)- и (S)-изомеров.
Соединения настоящего изобретения также могут находиться в виде рацемической смеси, содержащей практически эквивалентные количества стереоизомеров. В другом воплощении соединения настоящего изобретения могут быть получены или иным образом выделены по известным методикам с получением стереоизомера, практически свободного от своего соответствующего стереоизомера (т.е. практически чистого). Под практически чистым имеется в виду то, что стереоизомер очищен по меньшей мере на 95%, более предпочтительно по меньшей мере на 98%, наиболее предпочтительно по меньшей мере на 99%.
Соединения настоящего изобретения также могут находиться в виде гидратов, что означает, что соединение дополнительно содержит стехиометрическое или нестехиометрическое количество воды, связанной нековалентными межмолекулярными силами.
Соединения настоящего изобретения могут существовать в виде одного или нескольких возможных таутомеров, и в зависимости от конкретных условий можно отделить некоторые или все таутомеры в виде отдельных индивидуальных и отдельных единиц. Следует иметь в виду, что при этом охватываются все возможные таутомеры, в том числе все дополнительные ено- и кето-таутомеры и/или изомеры. Например, к ним относятся следующие таутомеры, без ограничения:
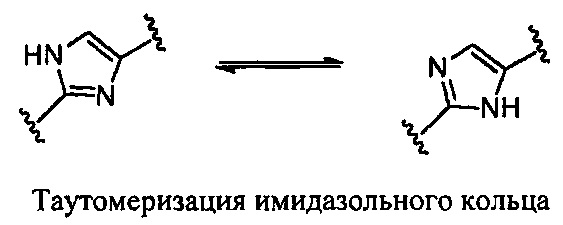
Таутомеры по изобретению представляют собой свободно превращающиеся друг в друга таутомеры, а не неразделенные смеси. Имидазолы и другие циклические системы по изобретению подвержены таутомеризации. Все таутомеры считаются частью изобретения.
Подразумевается, что в структурах, представленных в настоящем изобретении, если атом азота имеет меньше 3 связей, то для заполнения валентности азота присутствуют атомы H.
Изобретение включает и "фармацевтически приемлемые соли" соединений по изобретению, которые могут быть получены при реакции соединений по изобретению с кислотой или основанием. Некоторые соединения, особенно те, которые содержат кислотные или основные группы, могут находиться в виде соли, предпочтительно фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются биологически или иным образом нежелательными. Соли образуются с такими неорганическими кислотами, как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и др., и такими органическими кислотами, как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, n-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и др. Специалистам в данной области известны и другие соли, которые легко могут быть адаптированы для применения в соответствии с настоящим изобретением.
Подходящие фармацевтически приемлемые соли аминов соединений настоящего изобретения могут быть получены из неорганической кислоты или из органической кислоты. В одном воплощении примерами неорганических солей аминов являются бисульфаты, бораты, бромиды, хлориды, гемисульфаты, гидроброматы, гидрохлораты, 2-гидроксиэтилсульфонаты (гидроксиэтансульфонаты), йодаты, йодиды, изотионаты, нитраты, персульфаты, фосфаты, сульфаты, сульфаматы, сульфанилаты, соли сульфоновых кислот (алкилсульфонаты, арилсульфонаты, замещенные галогенами алкилсульфонаты, замещенные галогенами арилсульфонаты), сульфонаты и тиоцианаты.
В одном воплощении примеры органических солей аминов могут быть выбраны из алифатических, циклоалифатических, ароматических, арилалифатических, гетероциклических, карбоновых и сульфоновых классов органических кислот, примерами которых являются ацетаты, аргинины, аспартаты, аскорбаты, адипаты, антранилаты, алгинаты, алкановые карбоксилаты, замещенные алкановые карбоксилаты, альгинаты, бензолсульфонаты, бензоаты, бисульфаты, бутираты, бикарбонаты, битартраты, цитраты, камфораты, камфорсульфонаты, циклогексилсульфаматы, циклопентанпропионаты, эдетаты кальция, камсилаты, карбонаты, клавуланаты, циннаматы, дикарбоксилаты, диглюконаты, додецилсульфонаты, дигидроксихлориды, деканоаты, энантуаты, этансульфонаты, эдетаты, эдизилаты, эстолаты, эзилаты, фумараты, формиаты, фториды, галактуронаты, глюконаты, глутаматы, гликолаты, глюкораты, глюкогептаноаты, глицерофосфаты, глюцептаты, гликоллиларсанилаты, глютараты, глутаматы, гептаноаты, гексаноаты, гидроксималеаты, гидроксикарбоновые кислоты, гексилрезорцинаты, гидроксибензоаты, гидроксинафтоаты, гидрофтораты, лактаты, лактобионаты, лаураты, малаты, малеаты, метилен-бис(β-оксинафтоаты), малонаты, манделаты, мезилаты, метансульфонаты, метилбромиды, метилнитраты, метилсульфонаты, однокалиевые малеаты, мукаты, монокарбоксилаты, нафталинсульфонаты, 2-нафталинсульфонаты, никотинаты, нитраты, напсилаты, N-метилглюкамины, оксалаты, октаноаты, олеаты, памоаты, фенилацетаты, пикраты, фенилбензоаты, пивалаты, пропионаты, фталаты, фенилацетаты, пектинаты, фенилпропионаты, пальмитаты, пантотенаты, полигалактураты, пируваты, хинаты, салицилаты, сукцинаты, стеараты, сульфанилаты, себацетаты, тартраты, теофиллинацетаты, n-толуолсульфонаты (тозилаты), трифторацетаты, терефталаты, таннаты, теоклаты, тригалоацетаты, триэтиодиды, трикарбоксилаты, ундеканоаты и валераты.
В одном воплощении примеры неорганических солей карбоновых кислот или гидроксилов могут быть выбраны из солей аммония, щелочных металлов, включая литий, натрий, калий, цезий; щелочноземельных металлов, включая кальций, магний, алюминий; солей цинка, бария, холина, четвертичного аммония.
В другом воплощении примеры органических солей карбоновых кислот или гидроксилов могут быть выбраны из солей аргинина, органических аминов, включая алифатические органические амины, алициклические органические амины, ароматические органические амины, бензатины, трет-бутиламины, бенетамины (N-бензилфенэтиламин), дициклогексиламины, диметиламины, диэтаноламины, этаноламины, этилендиамины, гидрабамины, имидазолы, лизины, метиламины, мегламины, N-метил-D-глюкамины, N,N'-дибензилэтилендиамины, никотинамиды, органические амины, орнитины, пиридины, пиколины, пиперазины, прокаин, трис(гидроксиметил)метиламины, триэтиламины, триэтаноламины, триметиламины, трометамины и карбамиды.
В одном воплощении соли могут быть получены стандартными методами, как-то при реакции продукта в виде свободного основания или свободной кислоты с одним или несколькими эквивалентами соответствующей кислоты или основания в растворителе или в такой среде, в которой соль не растворяется, или в растворителе типа воды, который удаляется под вакуумом или при лиофилизации или при замене ионов существующей соли на другой ион на подходящей ионообменной смоле.
Фармацевтические композиции
Другой аспект настоящего изобретения касается фармацевтических композиций, включающих фармацевтически приемлемый носитель и по меньшей мере одно соединение в соответствии с аспектами настоящего изобретения. Фармацевтическая композиция может содержать одно или несколько из вышеприведенных соединений настоящего изобретения. Как правило, фармацевтическая композиция по настоящему изобретению включает в себя соединение настоящего изобретения типа соединения формулы I, II, III или IV либо 17уа или 12da либо его фармацевтически приемлемую соль в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; и фармацевтически приемлемый носитель. Фармацевтическая композиция настоящего изобретения также может включать ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; и фармацевтически приемлемый носитель. Термин "фармацевтически приемлемый носитель" относится к любым подходящим адъювантам, носителям, наполнителям или стабилизаторам, которые могут быть в твердом или жидком виде типа таблеток, капсул, порошков, растворов, суспензий или эмульсий. В некоторых воплощениях фармацевтическая композиция включает в себя комбинацию соединения настоящего изобретения типа соединения формулы I, II, III или IV либо 17уа или 12da либо его фармацевтически приемлемой соли с ингибитором BRAF; и фармацевтически приемлемый носитель. В некоторых воплощениях фармацевтическая композиция включает в себя комбинацию соединения настоящего изобретения типа соединения формулы I, II, III или IV либо 17уа или 12da либо его фармацевтически приемлемой соли с ингибитором MEK; и фармацевтически приемлемый носитель. В некоторых воплощениях фармацевтическая композиция включает в себя комбинацию соединения настоящего изобретения типа соединения формулы I, II, III или IV либо 17уа или 12da либо его фармацевтически приемлемой соли с ингибитором BRAF и ингибитором MEK; и фармацевтически приемлемый носитель. В некоторых воплощениях фармацевтическая композиция включает в себя комбинацию ингибитора тубулина с ингибитором BRAF; и фармацевтически приемлемый носитель. В некоторых воплощениях фармацевтическая композиция включает в себя комбинацию ингибитора тубулина с ингибитором MEK; и фармацевтически приемлемый носитель. В некоторых воплощениях фармацевтическая композиция включает в себя комбинацию ингибитора тубулина с ингибитором BRAF, ингибитор MEK и фармацевтически приемлемый носитель.
Как описано здесь, термин "BRAF" относится к гену человека, производящему белок, именуемый B-Raf. Белок B-Raf участвует в отправке внутриклеточных сигналов, которые участвуют в управлении клеточным ростом. В 2002 г. было показано, что он мутирован при раковых заболеваниях у человека. Были разработаны препараты для лечения раковых заболеваний, вызванных BRAF. Одним из ингибирующих BRAF препаратов является вемурафениб, который был одобрен FDA для лечения поздних стадий меланомы. Сейчас разрабатываются другие специфические ингибиторы мутантного белка B-Raf для применения против рака (которые здесь именуются "ингибиторы BRAF"). К ним относятся: GDC-0879, PLX-4720, сорафениб тозилат, дабрафениб и LGX818.
Как описано здесь, термин "MEK" относится к ферментам MEK1 и/или MEK2 - киназам активируемых митогенами протеинкиназ. Ферменты MEK представляют собой киназы, фосфорилирующие активируемые митогенами протеинкиназы. Киназы MEK являются представителями сигнального каскада MAPK, который активируется при меланоме. При ингибировании MEK блокируется пролиферация клеток и индуцируется апоптоз (контролируемая гибель клеток).
Термин "ингибитор MEK" относится к химическим веществам или препаратам, ингибирующим ферменты MEK1 и/или MEK2 - киназы активируемых митогенами протеинкиназ. Они могут применяться для воздействия на путь MAPK/ERK, который зачастую гиперактивен при некоторых раковых заболеваниях. Поэтому ингибиторы MEK обладают потенциалом для лечения некоторых раковых заболеваний, особенно меланомы с мутацией BRAF и колоректального рака с мутацией KRAS/BRAF. К ингибиторам MEK относятся, без ограничения: траметиниб (GSK1120212), селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб или XL518 и CI-1040 или PD035901.
В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие терапевтически эффективное количество двух соединений, обладающих противораковой активностью, и фармацевтически приемлемый носитель. В другом воплощении композиция включает ингибитор BRAF и соединение по изобретению типа соединения формулы I, II, III или IV либо 17уа или 12da. В другом воплощении композиция включает ингибитор MEK и соединение по изобретению типа соединения формулы I, II, III или IV либо 17уа или 12da. В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие терапевтически эффективное количество трех соединений, обладающих противораковой активностью, и фармацевтически приемлемый носитель. В другом воплощении композиция включает ингибитор BRAF, ингибитор MEK и соединение по изобретению типа соединения формулы I, II, III или IV либо 17уа или 12da. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб (GSK1120212), селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб или XL518, CI-1040 или PD035901 или же любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении соединением по изобретению является соединение формулы I, II, III или IV. В другом воплощении соединением по изобретению является соединение 17уа. В другом воплощении соединением по изобретению является соединение 12da. В другом воплощении соединение находится в виде своей фармацевтически приемлемой соли, N-оксида, гидрата, таутомера или изомера.
В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие терапевтически эффективное количество ингибитора тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; и фармацевтически приемлемый носитель. В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие комбинацию терапевтически эффективного количества ингибитора BRAF и ингибитора тубулина и фармацевтически приемлемый носитель. В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие комбинацию терапевтически эффективного количества ингибитора MEK и ингибитора тубулина и фармацевтически приемлемый носитель. В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие комбинацию терапевтически эффективного количества ингибитора BRAF, ингибитора MEK и ингибитора тубулина и фармацевтически приемлемый носитель. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб (GSK1120212), селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб или XL518, CI-1040 или PD035901 или же любая их комбинация. В другом воплощении ингибитором MEK является траметиниб или RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель.
Как правило, композиция должна содержать от 0,01 до 99%, предпочтительно от примерно 20 до 75% активных соединений, вместе со вспомогательными веществами, носителями и/или эксципиентами. Поскольку индивидуальные потребности могут различаться, то определение оптимальных интервалов эффективных количеств каждого компонента находится в компетенции специалистов. Типичные дозировки составляют от 0,01 до 100 мг/кг массы тела. Предпочтительные дозировки составляют от 0,1 до 100 мг/кг массы тела. Наиболее предпочтительными дозировки составляют от 1 до 100 мг/кг массы тела. Режим лечения для введения соединений настоящего изобретения также может быть легко определен рядовыми специалистами. То есть частоту введения и размер дозы можно установить по принципу оптимизации, предпочтительно сводя к минимуму побочные эффекты.
В одном воплощении способы настоящего изобретения могут включать введение соединения формулы I-IV по изобретению при различных дозах. В одном воплощении соединение формулы I-IV вводится в дозе 0,1-200 мг/кг. В одном воплощении соединение формулы I-IV вводится в дозе 0,01-1 мг/кг. В одном воплощении соединение формулы I-IV вводится в дозе 0,1-10 мг/кг, или в другом воплощении 0,1-25 мг/кг, или же в другом воплощении 10-50 мг/кг, или же в другом воплощении 10-25 мг/кг, или же в другом воплощении 0,3-30 мг/кг, или же в другом воплощении 0,5-25 мг/кг, или же в другом воплощении 0,5-50 мг/кг, или же в другом воплощении 0,75-15 мг/кг, или же в другом воплощении 0,75-60 мг/кг, или же в другом воплощении 1-5 мг/кг, или же в другом воплощении 1-20 мг/кг, или же в другом воплощении 3-15 мг/кг, или же в другом воплощении 30-50 мг/кг, или же в другом воплощении 30-75 мг/кг, или же в другом воплощении 100-2000 мг/кг. В другом воплощении соединение формулы I-IV вводится в дозе 5 мг/кг, 10 мг/кг, 15 мг/кг, 20 мг/кг, 25 мг/кг, 30 мг/кг или 35 мг/кг. В другом воплощении соединение формулы I-IV вводится в дозе 10 мг/кг. В другом воплощении соединение формулы I-IV вводится в дозе 15 мг/кг. В другом воплощении соединение формулы I-IV вводится в дозе 25 мг/кг.
В одном воплощении соединение формулы I-IV вводится в дозе 10 мг. В одном воплощении соединение формулы I-IV вводится в дозе 15 мг. В одном воплощении соединение формулы I-IV вводится в дозе 25 мг. В другом воплощении соединение формулы I-IV вводится в дозе 0,01 мг, 0,03 мг, 0,1 мг, 0,3 мг, 0,75 мг, 5 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг, 35 мг, 40 мг, 45 мг, 50 мг, 55 мг, 60 мг, 65 мг, 70 мг, 75 мг, 80 мг, 85 мг, 90 мг, 95 мг или 100 мг.
В одном воплощении способы настоящего изобретения могут включать введение ингибитора BRAF по изобретению при различных дозах. В одном воплощении ингибитор BRAF вводится в дозе 0,1-200 мг/кг. В одном воплощении ингибитор BRAF вводится в дозе 0,01-1 мг/кг. В одном воплощении ингибитор BRAF вводится в дозе 0,1-10 мг/кг, или в другом воплощении 0,1-25 мг/кг, или же в другом воплощении 10-50 мг/кг, или же в другом воплощении 10-25 мг/кг, или же в другом воплощении 0,3-30 мг/кг, или же в другом воплощении 0,5-25 мг/кг, или же в другом воплощении 0,5-50 мг/кг, или же в другом воплощении 0,75-15 мг/кг, или же в другом воплощении 0,75-60 мг/кг, или же в другом воплощении 1-5 мг/кг, или же в другом воплощении 1-20 мг/кг, или же в другом воплощении 3-15 мг/кг, или же в другом воплощении 30-50 мг/кг, или же в другом воплощении 30-75 мг/кг, или же в другом воплощении 100-2000 мг/кг. В другом воплощении ингибитор BRAF вводится в дозе 5 мг/кг, 10 мг/кг, 15 мг/кг, 20 мг/кг, 25 мг/кг, 30 мг/кг, 35 мг/кг, 40 мг/кг, 45 мг/кг или 50 мг/кг. В другом воплощении ингибитор BRAF вводится в дозе 10 мг/кг. В другом воплощении ингибитор BRAF вводится в дозе 20 мг/кг. В другом воплощении ингибитор BRAF вводится в дозе 30 мг/кг. В другом воплощении ингибитор BRAF вводится в дозе 40 мг/кг. В другом воплощении ингибитор BRAF вводится в дозе 45 мг/кг. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб.
В одном воплощении ингибитор BRAF вводится в дозе 10 мг. В одном воплощении ингибитор BRAF вводится в дозе 15 мг. В одном воплощении ингибитор BRAF вводится в дозе 25 мг. В одном воплощении ингибитор BRAF вводится в дозе 45 мг. В другом воплощении ингибитор BRAF вводится в дозе 5 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг, 35 мг, 40 мг, 45 мг или 50 мг.
В одном воплощении способы настоящего изобретения могут включать введение ингибитора MEK по изобретению при различных дозах. В одном воплощении ингибитор MEK вводится в дозе 0,1-200 мг/кг. В одном воплощении ингибитор MEK вводится в дозе 0,01-1 мг/кг. В одном воплощении ингибитор MEK вводится в дозе 0,1-1 мг/кг, или в другом воплощении 0,1-25 мг/кг, или же в другом воплощении 10-50 мг/кг, или же в другом воплощении 10-25 мг/кг, или же в другом воплощении 0,3-0,5 мг/кг, или же в другом воплощении 0,5-25 мг/кг, или же в другом воплощении 0,5-50 мг/кг, или же в другом воплощении 0,75-15 мг/кг, или же в другом воплощении 0,75-60 мг/кг, или же в другом воплощении 1-5 мг/кг, или же в другом воплощении 1-20 мг/кг, или же в другом воплощении 3-15 мг/кг, или же в другом воплощении 30-50 мг/кг, или же в другом воплощении 30-75 мг/кг, или же в другом воплощении 100-2000 мг/кг. В другом воплощении ингибитор MEK вводится в дозе 0,1 мг/кг, 0,2 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг, 0,6 мг/кг, 0,7 мг/кг, 0,8 мг/кг, 0,9 мг/кг или 1 мг/кг. В другом воплощении ингибитор MEK вводится в дозе 0,1 мг/кг. В другом воплощении ингибитор MEK вводится в дозе 0,3 мг/кг. В другом воплощении ингибитор MEK вводится в дозе 0,5 мг/кг. В другом воплощении ингибитор MEK вводится в дозе 0,7 мг/кг. В другом воплощении ингибитор MEK вводится в дозе 1 мг/кг. В другом воплощении ингибитором MEK является траметиниб или RO5068760.
Твердые стандартные дозовые формы могут быть обычного типа. Твердая форма может представлять собой капсулы типа обычных желатиновых капсул, содержащих соединения настоящего изобретения и носитель, к примеру, смазывающие вещества и инертные наполнители типа лактозы, сахарозы или кукурузного крахмала. В другом воплощении эти соединения заключены в таблетки со стандартными основами таблеток типа лактозы, сахарозы или кукурузного крахмала в сочетании со связующими веществами типа гуммиарабика, кукурузного крахмала или желатина, разрыхлителями типа кукурузного крахмала, картофельного крахмала или альгиновой кислоты и смазывающими веществами типа стеариновой кислоты или стеарата магния.
Таблетки, капсулы и т.п. могут также содержать связующее вещество типа трагакантовой камеди, гуммиарабика, кукурузного крахмала или желатина; наполнители типа дикальцийфосфата; дезинтегрирующее средство типа кукурузного крахмала, картофельного крахмала, альгиновой кислоты; смазывающее вещество типа стеарата магния; и подсластитель типа сахарозы, лактозы или сахарина. Если стандартная дозовая форма представляет собой капсулы, то, наряду с вышеприведенными материалами, она может содержать жидкий носитель типа жирного масла.
Различные другие материалы могут присутствовать в качестве покрытия или для модификации физической формы дозовой единицы. Например, таблетки могут быть покрыты шеллаком, сахаром или тем и другим. Сироп, наряду с активным ингредиентом, может содержать сахарозу в качестве подсластителя, метил- и пропилпарабены в качестве консервантов, краситель и ароматизатор типа аромата вишни или апельсина.
Для перорального терапевтического введения эти активные соединения могут быть заключены с наполнителями и применяться в виде таблеток, капсул, эликсиров, суспензий, сиропов и т.п. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процент соединения в этих композициях, конечно, может варьироваться и обычно составляет от 2% до 60% массы единицы. Количество активного соединения в таких терапевтически применимых композициях таково, чтобы получалась подходящая дозировка. Предпочтительные композиции по настоящему изобретению готовят таким образом, чтобы пероральная единица дозы содержала от 1 мг до 800 мг активного соединения.
Активные соединения настоящего изобретения могут вводиться перорально, к примеру, с инертным разбавителем или с усваиваемым съедобным носителем, или же они могут быть заключены в твердые или мягкие капсулы, или же они могут быть запрессованы в таблетки, или же они могут прямо входить в состав продуктов питания.
Фармацевтические формы, пригодные для применения в виде инъекций, включают стерильные водные растворы или дисперсии и стерильные порошки для приготовления ех temporo стерильных растворов или дисперсий. Во всех случаях эти формы должны быть стерильными и должны быть жидкими в такой степени, чтобы они легко вводились шприцем. Они должны быть стабильными в условиях производства и хранения и должны быть защищены от загрязняющего действия микроорганизмов типа бактерий и грибков. Носителем может быть растворитель или среда диспергирования, содержащая, к примеру, воду, этанол, полиол (например, глицерин, пропиленгликоль или жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Соединения или фармацевтические композиции настоящего изобретения также можно вводить в виде доз для инъекций путем растворения или суспендирования этих материалов в физиологически приемлемом разбавителе с фармацевтическим адъювантом, носителем или наполнителем. Такие адъюванты, носители и/или наполнители включают, без ограничения, стерильные жидкости, такие как вода и масло, с добавлением или без добавления поверхностно-активного вещества и других фармацевтически и физиологически приемлемых компонентов. Примеры масел представлены маслами из нефти, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло или минеральное масло. В общем, предпочтительными жидкими носителями, в особенности для растворов для инъекций, являются вода, физраствор, водные растворы декстрозы и родственных сахаров и такие гликоли, как пропиленгликоль или полиэтиленгликоль.
Эти активные соединения также можно вводить парентерально. Растворы или суспензии этих активных соединений могут быть приготовлены в воде в смеси с подходящим поверхностно-активным веществом типа гидроксипропилцеллюлозы. Дисперсии также могут быть приготовлены в глицерине, жидком полиэтиленгликоле и их смеси в маслах. Примеры масел представлены маслами из нефти, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло или минеральное масло. В общем, предпочтительными жидкими носителями, в особенности для растворов для инъекций, являются вода, физраствор, водные растворы декстрозы и родственных сахаров и такие гликоли, как пропиленгликоль или полиэтиленгликоль. При обычных условиях хранения и применения эти препараты содержат консервант для предотвращения роста микроорганизмов.
Для применения в виде аэрозолей соединения настоящего изобретения в растворе или суспензии могут быть упакованы в контейнер для аэрозоля под давлением вместе с подходящими вытеснителями, к примеру, углеводородными вытеснителями типа пропана, бутана или изобутана, вместе с обычными вспомогательными веществами. Материалы настоящего изобретения также можно вводить и не в сжатом виде, как-то в распылителе или пульверизаторе.
В одном воплощении настоящим изобретением предусмотрены фармацевтические композиции, которые включают соединение формулы I-IV, как описано здесь, и/или его изомер, фармацевтически приемлемую соль, фармацевтический продукт, гидрат, N-оксид или любую их комбинацию, по отдельности или в комбинации с другим терапевтическим средством, таким, к примеру, как противораковое средство, включая, без ограничения: ингибиторы тубулина, ингибиторы BRAF, ингибиторы MEK или другие средства, подходящие для описанных здесь применений. В одном воплощении фармацевтические композиции соединений формулы I-IV, как описано здесь, включают соединение по изобретению в сочетании с ингибитором BRAF. В другом воплощении фармацевтические композиции включают соединение по изобретению в сочетании с ингибитором MEK. В другом воплощении фармацевтические композиции включают соединение по изобретению в сочетании с ингибитором BRAF и ингибитором MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб (GSK1120212), селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб или XL518, CI-1040 или PD035901 или же любая их комбинация. В другом воплощении ингибитором MEK является траметиниб или RO5068760.
В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; и фармацевтически приемлемый носитель. В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие ингибитор тубулина в комбинации с ингибитором BRAF и фармацевтически приемлемый носитель. В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие ингибитор тубулина в комбинации с ингибитором MEK и фармацевтически приемлемый носитель. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб (GSK1120212), селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб или XL518, CI-1040 или PD035901 или же любая их комбинация. В другом воплощении ингибитором MEK является траметиниб или RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель.
В одном воплощении соединения настоящего изобретения вводятся в комбинации с противораковым средством. В одном воплощении противораковым средством является ингибитор BRAF. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В одном воплощении противораковым средством является ингибитор MEK. В другом воплощении ингибитором MEK является траметиниб (GSK1120212), селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб или XL518, CI-1040 или PD035901 или же любая их комбинация. В другом воплощении ингибитором MEK является траметиниб или RO5068760.
В одном воплощении настоящее изобретение направлено на фармацевтические композиции, содержащие соединения, представленные структурной формулой II:
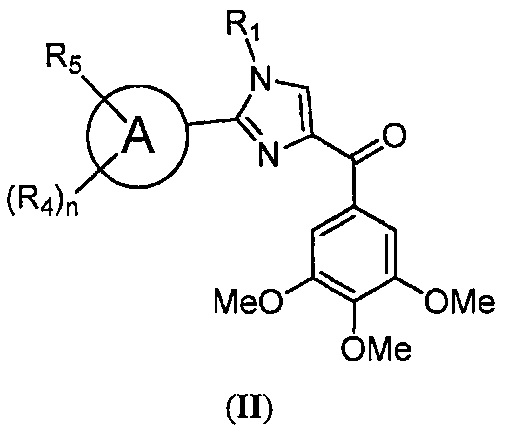
где: А означает одиночную или конденсированную ароматическую или гетероароматическую кольцевую систему;
R1 означает H, линейный или разветвленный C1-C6-алкил, арил, фенил, бензил, галоалкил, аминоалкил, -OCH2Ph, SO2-арил, SO2-фенил, -(C=O)-арил, -(C=O)-фенил или OH;
R4 и R5 каждый независимо означает водород, линейный или разветвленный C1-C6-алкил, линейный или разветвленный C1-C6-галоалкил, линейный или разветвленный C1-C6-алкокси, линейный или разветвленный C1-C6-галоалкокси, F, Cl, Br, I, CF3, CN, -CH2CN, NH2, OH, -OC(O)CF3, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, COOH, -C(O)Ph, C(O)O-алкил, C(O)H, -C(O)NH2 или NO2; и
n означает целое число от 1 до 4;
либо его фармацевтически приемлемую соль, N-оксид, гидрат, таутомер или изомер; в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; и фармацевтически приемлемый носитель.
В другом воплощении соединением настоящего изобретения является соединение 17уа. В другом воплощении соединением настоящего изобретения является соединение 12da.
Следующий аспект настоящего изобретения касается способа лечения рака, который включает выбор субъекта, нуждающегося в лечении рака, и введение субъекту фармацевтической композиции, содержащей терапевтически эффективные количества соединения формулы I, II, III или IV в комбинации по меньшей мере с одним из ингибиторов BRAF или MEK; и фармацевтически приемлемый носитель, в условиях, эффективных для лечения рака. В другом воплощении рак представлен лекарственно-устойчивым раком. В другом воплощении рак представляет собой меланому. В другом воплощении рак представлен меланомой с мутацией BRAF. В другом воплощении рак представлен устойчивым к вемурафенибу раком. В другом воплощении соединением по изобретению является соединение 17уа. В другом воплощении соединением по изобретению является соединение 12da.
При введении соединений настоящего изобретения их можно вводить системно или же непосредственно в то место, где находятся раковые или предраковые клетки. Таким образом, введение может осуществляться любым способом, эффективным для доставки соединений или фармацевтических композиций к раковым клеткам или предраковым клеткам. Типичные способы введения включают, без ограничения, введение соединений или композиций перорально, топически, трансдермально, парентерально, подкожно, внутривенно, внутримышечно, внутрибрюшинно, путем закапывания в нос, в полость или в пузырь, в глаза, интраартериально, интралезионально, либо путем нанесения на слизистые оболочки, к примеру, слизистые носа, горла и бронхов.
В некоторых воплощениях любая из композиций по изобретению содержит соединение формулы I-IV или ингибитор тубулина, в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, в любой форме или воплощении, как описано здесь. В некоторых воплощениях любая из композиций по изобретению состоит из соединения формулы I-IV или ингибитора тубулина, в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, в любой форме или воплощении, как описано здесь. В некоторых воплощениях любая из композиций по изобретению состоит в основном из соединения формулы I-IV или ингибитора тубулина, в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, в любой форме или воплощении, как описано здесь. В некоторых воплощениях термин "включает в себя" означает включение указанного активного средства типа соединения формулы I-IV или ингибитора тубулина, а также включение других активных средств и фармацевтически приемлемых носителей, наполнителей, мягчителей, стабилизаторов и пр., известных в фармацевтической промышленности. В некоторых воплощениях термин "состоит в основном из" относится к таким композициям, у которых единственным активным ингредиентом является указанный активный ингредиент, однако могут быть включены и другие соединения, которые служат для стабилизации, консервации и т.д. лекарственной формы, но не участвуют непосредственно в терапевтическом эффекте данного активного ингредиента. В некоторых воплощениях термин "состоит в основном из" может относиться к таким компонентам, которые способствуют высвобождению активного ингредиента. В некоторых воплощениях термин "состоит" относится к таким композициям, которые содержат активный ингредиент и фармацевтически приемлемый носитель или наполнитель.
В одном воплощении настоящим изобретением предусмотрены комбинированные препараты. В одном воплощении термин "комбинированный препарат" специально означает "комплект" в том смысле, что партнеры по комбинации, как определено выше, можно вводить самостоятельно или с помощью различных фиксированных комбинаций с различным количеством партнеров по комбинации, т.е. одновременно, параллельно, раздельно или последовательно. В некоторых воплощениях составные части комплекта могут вводиться, например, одновременно или хронологически смещенно, то есть в различные моменты времени и с одинаковыми или различными интервалами времени для любой части из комплекта. В некоторых воплощениях может вводиться лишь часть от общего количества партнеров по комбинации в комбинированном препарате. В одном воплощении комбинированный препарат может варьироваться, например, для того, чтобы учесть потребности субпопуляции пациентов при лечении или потребности одного пациента, причем различные потребности могут быть обусловлены конкретным заболеванием, тяжестью заболевания, возрастом, полом, массой тела, что может быть легко установлено специалистом.
Следует иметь в виду, что настоящее изобретение касается композиций и комбинированной терапии, как описано здесь, для любого заболевания, расстройства или состояния, по необходимости, как это должно быть известно специалистам. Некоторые применения таких композиций и комбинированной терапии уже были описаны выше, для определенных заболеваний, расстройств и состояний, представляющих воплощения настоящего изобретения, а способы лечения таких заболеваний, расстройств и состояний у субъектов путем введения описанных здесь соединений, самих по себе или в составе комбинированной терапии или с помощью композиций по изобретению, составляют дополнительные воплощения изобретения.
Биологическая активность
В одном воплощении изобретением предусмотрены соединения и композиции, в том числе любые описанные здесь воплощения, для применения в любом из способов по изобретению. В одном воплощении применение соединений по изобретению или содержащих их композиций должно иметь применимость при ингибировании, подавлении, усилении или стимулировании требуемой реакции у субъекта, как это должно быть понятно специалистам. В другом воплощении композиции также могут содержать и дополнительные активные ингредиенты, активность которых будет полезной для того применения, при котором вводится соединение по изобретению.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска развития или торможения рака, включающего введение соединения по изобретению страдающему раком субъекту в условиях, эффективных для лечения рака. В другом воплощении соединение вводится в комбинации с ингибитором BRAF. В другом воплощении соединение вводится в комбинации с ингибитором MEK. В другом воплощении соединение вводится в комбинации с ингибитором BRAF и ингибитором MEK. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак представляет собой меланому, рак щитовидной железы, колоректальный рак или рак яичников.
В одном воплощении настоящим изобретением предусмотрены способы: а) лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивых опухолей; b) лечения, подавления, уменьшения тяжести, снижения риска или торможения метастатического рака; с) лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака; d) лечения, подавления, уменьшения тяжести, снижения риска или торможения меланомы; е) лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака, причем рак представлен меланомой, раком щитовидной железы, раком желчных путей, немелкоклеточным раком легких (NSCLC), колоректальным раком или раком яичников; f) способ лечения, подавления, уменьшения тяжести, снижения риска или торможения метастатической меланомы; g) способ лечения, подавления, уменьшения тяжести, снижения риска или торможения рака с мутацией BRAF; h) способ лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивого к ингибиторам BRAF рака; i) способ лечения, подавления, уменьшения тяжести, снижения риска, торможения, устранения, замедления или профилактики устойчивого к ингибиторам BRAF рака; j) способ лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивой к ингибиторам BRAF меланомы; k) лечения, подавления, уменьшения тяжести, снижения риска или торможения рака у субъекта, причем субъект ранее получал химиотерапию, лучевую терапию или биологическую терапию; l) способ преодоления невосприимчивости к лечению ингибиторами BRAF у субъекта; m) лечения, подавления, уменьшения тяжести, снижения риска или торможения рака щитовидной железы; n) лечения, подавления, уменьшения тяжести, снижения риска или торможения колоректального рака; о) лечения, подавления, уменьшения тяжести, снижения риска или торможения рака яичников; р) лечения, подавления, уменьшения, торможения, устранения, замедления или профилактики метастазирования рака у субъекта, страдающего раком; или q) лечения, подавления, уменьшения, торможения, устранения, замедления или профилактики вторичного рака, устойчивого к таксановым препаратам, у пациента, страдающего раком и ранее получавшего таксановый препарат, включающего стадию введения данному субъекту соединения по изобретению и/или изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристаллов данного соединения или любой их комбинации, в сочетании по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; либо содержащей их композиции. В другом воплощении способы включают введение соединения по изобретению и/или изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристаллов данного соединения или любой их комбинации, в сочетании с ингибитором BRAF, либо содержащей их композиции. В другом воплощении способы включают введение соединения по изобретению и/или изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристаллов данного соединения или любой их комбинации, в сочетании с ингибитором MEK; либо содержащей их композиции. В другом воплощении способы включают введение соединения по изобретению и/или изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристаллов данного соединения или любой их комбинации, в сочетании по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; либо содержащей их композиции. В другом воплощении способы включают введение ингибитора тубулина в комбинации с ингибитором BRAF либо содержащей их композиции. В другом воплощении способы включают введение ингибитора тубулина в комбинации с ингибитором MEK либо содержащей их композиции. В другом воплощении способы включают введение ингибитора тубулина в комбинации с ингибитором BRAF и ингибитором MEK либо содержащей их композиции.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения рака с мутацией BRAF у субъекта, включающего введение композиции, содержащей ингибитор BRAF, ингибитор MEK или их комбинацию, в сочетании с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему раком с мутацией BRAF, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора BRAF. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора MEK. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак представляет собой меланому, рак щитовидной железы, колоректальный рак или рак яичников. В другом воплощении рак представлен меланомой. В другом воплощении меланома является положительной по V600E. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак представлен лекарственно-устойчивым раком. В другом воплощении рак устойчив к ингибиторам BRAF. В другом воплощении рак устойчив к таксанам. В другом воплощении рак устойчив к доцетакселю. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивого к ингибиторам BRAF рака у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему устойчивым к ингибиторам BRAF раком, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора BRAF. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора MEK. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен меланомой. В другом воплощении меланома является положительной по V600E. В другом воплощении рак представлен метастатическим раком. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивого к вемурафениб рака у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему устойчивым к вемурафениб раком, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора BRAF. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора MEK. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен меланомой. В другом воплощении меланома является положительной по V600E. В другом воплощении рак представлен метастатическим раком. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения меланомы у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему меланомой, в условиях, эффективных для лечения меланомы. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является Вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении меланома является лекарственно-устойчивой. В другом воплощении меланома является положительной по V600E. В другом воплощении меланома представлена метастатической меланомой. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения рака щитовидной железы у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему раком щитовидной железы, в условиях, эффективных для лечения рака щитовидной железы. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак щитовидной железы является лекарственно-устойчивым. В другом воплощении рак щитовидной железы является метастатическим. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения рака яичников у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему раком яичников, в условиях, эффективных для лечения рака яичников. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак яичников является лекарственно-устойчивым. В другом воплощении рак яичников является метастатическим. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения колоректального рака у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему колоректальным раком, в условиях, эффективных для лечения колоректального рака. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении колоректальный рак является лекарственно-устойчивым. В другом воплощении колоректальный рак является метастатическим. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивой меланомы у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему лекарственно-устойчивой меланомой, в условиях, эффективных для лечения меланомы. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении меланома является положительной по V600E. В другом воплощении меланома представлена метастатической меланомой. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака у субъекта, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему лекарственно-устойчивым раком, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак представлен меланомой. В другом воплощении рак представлен положительной по V600E меланомой. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа преодоления невосприимчивости к лечению ингибитором BRAF у субъекта, страдающего лекарственно-устойчивым раком, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему лекарственно-устойчивым раком. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора BRAF. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора MEK. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак представлен меланомой. В другом воплощении рак представлен положительной по V600E меланомой. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа предотвращения, устранения, уменьшения или замедления невосприимчивости к лечению рака у субъекта, страдающего раком, включающего введение композиции, содержащей по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; в комбинации с соединением по изобретению или его фармацевтически приемлемой солью, N-оксидом, гидратом, таутомером или изомером, субъекту, страдающему лекарственно-устойчивым раком. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора BRAF. В другом воплощении комбинация состоит в основном из соединения по изобретению и ингибитора MEK. В другом воплощении комбинация состоит в основном из соединения по изобретению, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак является метастатическим. В другом воплощении рак представлен меланомой. В другом воплощении рак представлен положительной по V600E меланомой. В другом воплощении соединение по изобретению представлено соединением формулы I-IV. В другом воплощении соединение по изобретению представлено соединением 17уа. В другом воплощении соединение по изобретению представлено соединением 12da.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения рака с мутацией BRAF у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему раком с мутацией BRAF, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK 162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен меланомой. В другом воплощении меланома является положительной по V600E. В другом воплощении рак представлен лекарственно-устойчивым раком. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак является устойчивым к ингибиторам BRAF.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивого к ингибиторам BRAF рака у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему устойчивым к ингибиторам BRAF раком, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак представлен меланомой. В другом воплощении меланома является положительной по V600E.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивого к вемурафениб рака у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему устойчивым к вемурафениб раком, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак представлен меланомой. В другом воплощении меланома является положительной по V600E.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения меланомы у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему меланомой, в условиях, эффективных для лечения меланомы. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении меланома является лекарственно-устойчивой. В другом воплощении меланома представлена метастатической меланомой. В другом воплощении меланома является положительной по V600E.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения рака щитовидной железы у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему раком щитовидной железы, в условиях, эффективных для лечения рака щитовидной железы. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении рак щитовидной железы является лекарственно-устойчивым. В другом воплощении рак щитовидной железы является метастатическим.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения колоректального рака у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему колоректальным раком, в условиях, эффективных для лечения колоректального рака. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении колоректальный рак является лекарственно-устойчивым. В другом воплощении колоректальный рак является метастатическим.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения рака яичников у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему раком яичников, в условиях, эффективных для лечения рака яичников. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении рак яичников является лекарственно-устойчивым. В другом воплощении рак яичников является метастатическим.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивой меланомы у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему лекарственно-устойчивой меланомой, в условиях, эффективных для лечения меланомы. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении меланома является положительной по V600E. В другом воплощении меланома является метастатической.
В одном воплощении настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему лекарственно-устойчивым раком, в условиях, эффективных для лечения рака. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак представлен меланомой. В другом воплощении рак представлен положительной по V600E меланомой.
В одном воплощении настоящее изобретение касается способа преодоления невосприимчивости к лечению ингибитором BRAF у субъекта, включающего введение композиции, содержащей ингибитор тубулина в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK, субъекту, страдающему лекарственно-устойчивым раком. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора BRAF. В другом воплощении комбинация состоит в основном из ингибитора тубулина и ингибитора MEK. В другом воплощении комбинация состоит в основном из ингибитора тубулина, ингибитора BRAF и ингибитора MEK. В другом воплощении ингибитором BRAF является вемурафениб, дабрафениб, GDC-0879, PLX-4720, сорафениб тозилат, LGX818 или любая их комбинация. В другом воплощении ингибитором BRAF является вемурафениб. В другом воплощении ингибитором BRAF является дабрафениб. В другом воплощении ингибитором MEK является траметиниб, селуметиниб, RO5068760, MEK162, PD-325901, кобиметиниб, CI-1040 или любая их комбинация. В другом воплощении ингибитором MEK является траметиниб. В другом воплощении ингибитором MEK является RO5068760. В другом воплощении ингибитором тубулина является паклитаксель, эпотилон, доцетаксель, дискодермолид, колхицин, комбрестатин, 2-метоксиэстрадиол, метоксибензолсульфонамид (Е7010), винбластин, винкристин, винорелбин, винфлуин, доластатин, галихондрин, гемиастерлин, криптофизин 52, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель, колхицин, винбластин, таксол или любая их комбинация. В другом воплощении ингибитором тубулина является доцетаксель. В другом воплощении рак представляет собой меланому, рак щитовидной железы, рак желчных путей, немелкоклеточный рак легких (NSCLC), колоректальный рак или рак яичников. В другом воплощении рак представлен метастатическим раком. В другом воплощении рак представлен меланомой. В другом воплощении рак представлен положительной по V600E меланомой.
Соединения настоящего изобретения применимы при лечении, уменьшении тяжести, снижении риска или торможении рака, метастатического рака, лекарственно-устойчивых опухолей, лекарственно-устойчивого рака и различных форм рака. В предпочтительном воплощении рак представляет собой рак кожи (например, меланому), рак щитовидной железы, колоректальный рак, рак яичников, рак простаты, рак молочной железы, рак легких, рак толстой кишки, рак желчных путей, немелкоклеточный рак легких (NSCLC), лейкемию, лимфому, рак головы и шеи, рак поджелудочной железы, пищевода, почек или рак ЦНС (например, глиому, глиобластому). Лечение этих различных раковых заболеваний подтверждается приведенными здесь Примерами. Кроме того, исходя из предполагаемого механизма действия их в качестве ингибиторов тубулина, полагаем, что другие формы рака тоже должны поддаваться лечению или профилактике при введении соединений или композиций настоящего изобретения пациентам. Предпочтительные соединения настоящего изобретения избирательно разрушают раковые клетки, вызывая абляцию раковых клеток, но предпочтительно не нормальных клеток. Важно отметить, что вред для нормальных клеток сводится к минимуму, так как раковые клетки подвержены разрушению при намного более низких концентрациях соединений настоящего изобретения.
В некоторых воплощениях настоящим изобретением предусмотрено применение описанных здесь соединений либо их изомеров, метаболитов, фармацевтически приемлемых солей, фармацевтических продуктов, таутомеров, полиморфов, кристаллов, N-оксидов, гидратов или любых их сочетаний, в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; для лечения, подавления, уменьшения тяжести, снижения риска или торможения рака у субъектов. В другом воплощении рак представляет собой рак кожи (например, меланому), рак щитовидной железы, колоректальный рак, рак яичников, адренокортикальную карциному, рак анального канала, рак мочевого пузыря, опухоль головного мозга, опухоль ствола мозга, рак молочной железы, глиому, астроцитому мозжечка, астроцитому головного мозга, эпендимому, медуллобластому, супратенториальную примитивную нейроэктодермальную опухоль, опухоль эпифиза, глиому гипоталамуса, карциноидную опухоль, карциному, рак шейки матки, рак толстой кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак внепеченочных желчных протоков, семейство опухолей Юинга (Pnet), внечерепную гоноцитарную опухоль, рак глаз, внутриглазную меланому, рак желчного пузыря, рак желудка, внегонадную гоноцитарную опухоль, гестационную трофобластную опухоль, рак головы и шеи, рак подглоточника, карциному островковых клеток, рак гортани, лейкемию, острую лимфобластную лейкемию, рак полости рта, рак печени, рак легких, немелкоклеточный рак легких, мелкоклеточную лимфому, связанную со СПИД лимфому, (первичную) лимфому центральной нервной системы, кожную Т-клеточную лимфому, болезнь Ходжкина, неходжкинскую лимфому, злокачественную мезотелиому, карциному диска Меркеля, метастатическую плоскоклеточную карциному, множественную миелому, миеломную болезнь, фунгоидную гранулему, миелодиспластический синдром, миелопролиферативные нарушения, рак носоглотки, нейробластому, рак ротоглотки, остеосаркому, рак эпителия яичников, гоноцитарную опухоль яичников, опухоль яичников с низким злокачественным потенциалом, рак поджелудочной железы, рак экзокринной части поджелудочной железы, карциному островковых клеток, рак придаточных пазух и носовой полости, рак паращитовидной железы, рак полового члена, феохромоцитому, рак гипофиза, плазмоцитарные новообразования, рак простаты, рабдомиосаркому, рак прямой кишки, рак почек, почечно-клеточный рак, рак слюнных желез, синдром Сезари, Т-клеточную кожную лимфому, рак кожи, саркому Калоши, меланому, рак тонкой кишки, саркому мягких тканей, рак яичек, злокачественную тимому, рак мочеиспускательного канала, рак матки, саркому, необычный детский рак, рак влагалища, рак вульвы, опухоль Вильмса или любую их комбинацию. В другом воплощении субъект ранее проходил химиотерапию, лучевую терапию или биологическую терапию.
В некоторых воплощениях настоящим изобретением предусмотрено применение описанных здесь соединений либо их изомеров, метаболитов, фармацевтически приемлемых солей, фармацевтических продуктов, таутомеров, полиморфов, кристаллов, N-оксидов, гидратов или любых их сочетаний, в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; для лечения, подавления, уменьшения тяжести, снижения риска или торможения метастатического рака у субъектов. В другом воплощении рак представляет собой рак кожи (например, меланому), рак щитовидной железы, колоректальный рак, рак яичников, адренокортикальную карциному, рак анального канала, рак мочевого пузыря, опухоль головного мозга, опухоль ствола мозга, рак молочной железы, глиому, астроцитому мозжечка, астроцитому головного мозга, эпендимому, медуллобластому, супратенториальную примитивную нейроэктодермальную опухоль, опухоль эпифиза, глиому гипоталамуса, карциноидную опухоль, карциному, рак шейки матки, рак толстой кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак внепеченочных желчных протоков, семейство опухолей Юинга (Pnet), внечерепную гоноцитарную опухоль, рак глаз, внутриглазную меланому, рак желчного пузыря, рак желудка, внегонадную гоноцитарную опухоль, гестационную трофобластную опухоль, рак головы и шеи, рак подглоточника, карциному островковых клеток, рак гортани, лейкемию, острую лимфобластную лейкемию, рак полости рта, рак печени, рак легких, немелкоклеточный рак легких, мелкоклеточную лимфому, связанную со СПИД лимфому, (первичную) лимфому центральной нервной системы, кожную Т-клеточную лимфому, болезнь Ходжкина, неходжкинскую лимфому, злокачественную мезотелиому, карциному диска Меркеля, метастатическую плоскоклеточную карциному, множественную миелому, миеломную болезнь, фунгоидную гранулему, миелодиспластический синдром, миелопролиферативные нарушения, рак носоглотки, нейробластому, рак ротоглотки, остеосаркому, рак эпителия яичников, гоноцитарную опухоль яичников, опухоль яичников с низким злокачественным потенциалом, рак поджелудочной железы, рак экзокринной части поджелудочной железы, карциному островковых клеток, рак придаточных пазух и носовой полости, рак паращитовидной железы, рак полового члена, феохромоцитому, рак гипофиза, плазмоцитарные новообразования, рак простаты, рабдомиосаркому, рак прямой кишки, рак почек, почечно-клеточный рак, рак слюнных желез, синдром Сезари, Т-клеточную кожную лимфому, рак кожи, саркому Калоши, меланому, рак тонкой кишки, саркому мягких тканей, рак яичек, злокачественную тимому, рак мочеиспускательного канала, рак матки, саркому, необычный детский рак, рак влагалища, рак вульвы, опухоль Вильмса или любую их комбинацию.
В некоторых воплощениях настоящим изобретением предусмотрено применение описанных здесь соединений либо их изомеров, метаболитов, фармацевтически приемлемых солей, фармацевтических продуктов, таутомеров, полиморфов, кристаллов, N-оксидов, гидратов или любых их сочетаний, в комбинации по меньшей мере с одним из ингибиторов BRAF или ингибиторов MEK; для лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого или невосприимчивого рака у субъектов. В другом воплощении рак представляет собой рак кожи (например, меланому), рак щитовидной железы, колоректальный рак, рак яичников, адренокортикальную карциному, рак анального канала, рак мочевого пузыря, опухоль головного мозга, опухоль ствола мозга, рак молочной железы, глиому, астроцитому мозжечка, астроцитому головного мозга, эпендимому, медуллобластому, супратенториальную примитивную нейроэктодермальную опухоль, опухоль эпифиза, глиому гипоталамуса, карциноидную опухоль, карциному, рак шейки матки, рак толстой кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак внепеченочных желчных протоков, семейство опухолей Юинга (Pnet), внечерепную гоноцитарную опухоль, рак глаз, внутриглазную меланому, рак желчного пузыря, рак желудка, внегонадную гоноцитарную опухоль, гестационную трофобластную опухоль, рак головы и шеи, рак подглоточника, карциному островковых клеток, рак гортани, лейкемию, острую лимфобластную лейкемию, рак полости рта, рак печени, рак легких, немелкоклеточный рак легких, мелкоклеточную лимфому, связанную со СПИД лимфому, (первичную) лимфому центральной нервной системы, кожную Т-клеточную лимфому, болезнь Ходжкина, неходжкинскую лимфому, злокачественную мезотелиому, карциному диска Меркеля, метастатическую плоскоклеточную карциному, множественную миелому, миеломную болезнь, фунгоидную гранулему, миелодиспластический синдром, миелопролиферативные нарушения, рак носоглотки, нейробластому, рак ротоглотки, остеосаркому, рак эпителия яичников, гоноцитарную опухоль яичников, опухоль яичников с низким злокачественным потенциалом, рак поджелудочной железы, рак экзокринной части поджелудочной железы, карциному островковых клеток, рак придаточных пазух и носовой полости, рак паращитовидной железы, рак полового члена, феохромоцитому, рак гипофиза, плазмоцитарные новообразования, рак простаты, рабдомиосаркому, рак прямой кишки, рак почек, почечно-клеточный рак, рак слюнных желез, синдром Сезари, Т-клеточную кожную лимфому, рак кожи, саркому Капоши, меланому, рак тонкой кишки, саркому мягких тканей, рак яичек, злокачественную тимому, рак мочеиспускательного канала, рак матки, саркому, необычный детский рак, рак влагалища, рак вульвы, опухоль Вильмса или любую их комбинацию.
В одном воплощении "метастатический рак" означает такой рак, который распространился (метастазировал) из своего первоначального сайта в другие области тела. Практически все раковые заболевания имеют потенциал к распространению. Будут ли возникать метастазы - это зависит от сложного взаимодействия многих факторов опухолевых клеток, в том числе от типа рака, степени зрелости (дифференцировки) опухолевых клеток, расположения и продолжительности наличия рака, а также других не совсем понятных факторов. Метастазы распространяются тремя способами - путем локального разрастания из опухоли в окружающие ткани, через кровоток в удаленные участки или через лимфатическую систему в соседние или удаленные лимфатические узлы. Каждый вид рака может иметь типичный способ распространения. Опухоли именуются по первичному участку (например, рак молочной железы, который распространился в мозг, называется "метастатический рак молочной железы в мозге").
В одном воплощении "лекарственно-устойчивый рак" означает то, что раковые клетки приобретают устойчивость к химиотерапии. Раковые клетки могут приобрести устойчивость к химиотерапии по ряду механизмов, включая мутацию или гиперэкспрессию мишени препарата, инактивацию препарата или выведение препарата из клетки. Опухоли, которые дают рецидивы после первоначальной реакции на химиотерапию, могут быть устойчивыми к нескольким препаратам (они обладают множественной лекарственной устойчивостью). По общепринятой точке зрения на лекарственную устойчивость, одна или несколько клеток в опухолевой популяции приобретают генетические изменения, которые придают лекарственную устойчивость. Соответственно, причины лекарственной устойчивости, среди прочего, состоят в том, что: а) некоторые из клеток, не погибших при химиотерапии, мутируют (изменяются) и становятся устойчивыми к препарату; после того, как они размножатся, устойчивых клеток может оказаться больше, чем клеток, чувствительных к химиотерапии; b) амплификация генов: раковая клетка может производить сотни копий определенного гена, который запускает перепроизводство белка, делающего противоопухолевый препарат неэффективным; с) раковые клетки могут выкачивать препарат из клетки столь быстро, как это происходит при помощи молекулы под названием Р-гликопротеин; d) раковые клетки могут прекратить захват препарата, потому что перестанет работать белок, транспортирующий препарат через клеточную стенку; е) раковые клетки могут научиться восстанавливать разрывы ДНК, вызванные некоторыми противораковыми препаратами; f) у раковых клеток может возникнуть механизм, инактивирующий препарат. Одним из главных механизмов множественной лекарственной устойчивости является избыточная экспрессия Р-гликопротеина (P-GP) или других насосов, откачивающих препараты (OAT, OCT, BCRP и др.). Этот белок является клинически важным транспортным белком, принадлежащим к семейству трансмембранных переносчиков типа АТФ-связывающей кассеты. Он может выкачивать субстраты, в том числе противораковые препараты, из опухолевых клеток по АТФ-зависимому механизму. Таким образом, устойчивость к противораковым средствам, используемым в химиотерапии, является основной причиной неэффективности лечения при злокачественных заболеваниях, вызывая невосприимчивость опухолей. Лекарственная устойчивость является главной причиной неблагоприятного исхода химиотерапии рака.
В одном воплощении "невосприимчивый рак" означает лекарственно-устойчивый рак, как описано выше. В другом воплощении "невосприимчивый рак" означает то, что раковые клетки приобретают устойчивость к любому лечению типа химиотерапии, лучевой терапии или биологической терапии.
В одном воплощении настоящее изобретение касается лечения, подавления, уменьшения тяжести, снижения риска или торможения рака у субъекта, причем субъект ранее проходил химиотерапию, лучевую терапию или биологическую терапию.
В одном воплощении "химиотерапия" означает химическую обработку при раке типа препаратов, непосредственно убивающих раковые клетки. Такие препараты именуются "противораковыми препаратами" или "противоопухолевыми средствами". В современной терапии применяется более 100 препаратов для лечения рака, чтобы излечить конкретный рак. Химиотерапия применяется для контролирования роста опухолей, когда невозможно излечение; для уменьшения опухолей перед операцией или перед лучевой терапией; для снятия симптомов (типа боли); и для разрушения микроскопических раковых клеток, которые могут остаться после удаления известной опухоли хирургическим путем (что называется адъювантной терапией). Адъювантная терапия проводится для предотвращения возможного рецидива рака.
В одном воплощении "лучевая терапия" означает применение рентгеновских лучей высокой энергии и аналогичных лучей (типа электронов) для лечения заболеваний. Многие люди с раковыми заболеваниями должны получать лучевую терапию как часть своего лечения. Она может проводиться в виде внешней радиотерапии с помощью рентгеновских лучей снаружи от организма либо изнутри в виде внутренней радиотерапии. Лучевая терапия работает путем уничтожения раковых клеток на обработанном участке. Хотя нормальные клетки также могут повреждаться при лучевой терапии, но обычно они восстанавливаются. Проведение лучевой терапии может излечить некоторые виды рака, а также может уменьшить вероятность рецидива рака после операции. Она может применяться для уменьшения симптомов рака.
В одном воплощении "биологическая терапия" означает то, что в природе встречаются вещества в организме, которые разрушают раковые клетки. Есть несколько видов терапии, включая: моноклональные антитела, ингибиторы роста раковых клеток, вакцины и генную терапию. Биологическая терапия также известна как иммунотерапия.
Следующий аспект настоящего изобретения касается способа лечения или профилактики раковых заболеваний, который включает: обеспечение композиции, содержащей соединение настоящего изобретения типа соединения формулы I, II, III или IV либо 17уа или 12da и по меньшей мере один из ингибиторов BRAF или ингибиторов MEK; а затем введение эффективного количества композиции пациенту способом, эффективным для лечения или профилактики ракового заболевания.
В соответствии с одним воплощением, подлежащий лечению пациент характеризуется наличием предракового заболевания, а введение композиции эффективно для предотвращения развития предракового заболевания в раковое заболевание. Это может происходить путем разрушении предраковых клеток до или одновременно с дальнейшим развитием их в раковое заболевание.
В соответствии с другим воплощением, подлежащий лечению пациент характеризуется наличием ракового заболевания, а введение композиции эффективно вызывает регрессию ракового заболевания или тормозит дальнейшее развитие рака, т.е. полностью прекращает его развитие или уменьшает скорость его развития. Предпочтительно это происходит путем уничтожения раковых клеток, независимо от их местонахождения в организме пациента, то есть будут ли раковые клетки находиться в первичном очаге опухоли или же раковые клетки уже метастазировали и образовали вторичные опухоли в организме пациента.
В настоящем изобретении субъект или пациент относится к любым больным млекопитающим, включая, без ограничения, людей и других приматов, собак, кошек, лошадей, коров, овец, свиней, крыс, мышей и других грызунов. В одном воплощении субъектом является мужчина. В другом воплощении субъектом является женщина. В некоторых воплощениях описанные здесь способы могут применяться для лечения как мужчин, так и женщин.
При введении соединений настоящего изобретения они могут вводиться системно или же непосредственно в то место, где находятся раковые клетки или предраковые клетки. Таким образом, введение может осуществляться любым способом, эффективным для доставки соединений или фармацевтических композиций к раковым клеткам или предраковым клеткам. Типичные способы введения включают, без ограничения, введение соединений или композиций перорально, топически, трансдермально, парентерально, подкожно, внутривенно, внутримышечно, внутрибрюшинно, путем закапывания в нос, в полость или в пузырь, интраокулярно, интраартериально, интралезионально, либо путем нанесения на слизистые оболочки, к примеру, слизистые носа, горла и бронхов.
При введении соединений или фармацевтических композиций настоящего изобретения для лечения, подавления, уменьшения тяжести, снижения риска или торможения раковых заболеваний, фармацевтическая композиция также может содержать или может вводиться в сочетании с другими терапевтическими средствами или схемами лечения, известными в настоящее время или разрабатываемыми в дальнейшем для лечения различных видов рака. Примеры других терапевтических средств или схем лечения включают, без ограничения, лучевую терапию, иммунотерапию, химиотерапию, хирургическое вмешательство и их комбинации.
Нижеследующие примеры приводятся для более полного раскрытия предпочтительных воплощений изобретения. Но они не должны никоим образом рассматриваться как ограничивающие полный объем изобретения.
ПРИМЕРЫ
Представленные ниже примеры приводятся только в целях иллюстрации и не предназначаются для ограничения, никоим образом, объема настоящего изобретения.
ПРИМЕР 1. Комбинация из соединения 12da или 17уа и вемурафениба для лечения меланомы с мутацией BRAF и устойчивого к вемурафениб рака
Вемурафениб - это новый препарат против меланомы, который одобрен для мутантов V600E, но в течение ~9 месяцев возникает устойчивость к нему. Проводили скрининг нескольких ингибиторов тубулина, включая соединения 12da и 17уа, чтобы оценить их антипролиферативные эффекты при комбинировании с вемурафенибом на исходные клетки А375 и MDA-MB-435, то есть линии клеток с мутацией BRAF V600E. Эти комбинации могут помочь преодолеть невосприимчивость.
Гипотезу о синергетической блокировке клеточного цикла при комбинировании вемурафениб с 12da или доцетакселем проверяли на панели исходных линий клеток меланомы с мутацией BRAFV600E и прошедшей хронический отбор устойчивой к вемурафенибу сублинии A375RF21 (Su F, Bradley WD, Wang Q, et al. "Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation". Cancer Res. (2012) 72: 969-978). Полученные устойчивые к вемурафениб клетки A375RF21 были использованы in vitro и in vivo в качестве модели рецидива заболевания, чтобы проверить, будет ли предложенная синергетическая комбинация препаратов потенциально приносить пользу для лечения при связанной с этим клинической устойчивости к вемурафенибу.
Материалы и методы
Реагенты и клеточные линии
Вемурафениб (также известный как PLX4032, RG7204 или R05185426), траметиниб, сунитиниб (малатная соль) и доцетаксель приобретали у LC Laboratories (Woburn, MA). Соединение 12da синтезировали сами, как описано ниже. Соединения растворяли в диметилсульфоксиде (DMSO, Sigma-Aldrich, St. Louis, МО), получая исходный раствор в 10 мМ. Линию клеток А375 меланомы человека получали из АТСС (Manassas, VA). Клетки WM164 и MDA-MB-435 получали от Dr. Meenhard Herlyn (Wistar Institute, Philadelphia, PA, и Dr. Robert Clarke (Georgetown University, Washington, DC), соответственно. Все клеточные линии проверяли перед использованием в этом исследовании. Клетки культивировали в среде DMEM (Mediatech, Inc., Manassas, VA) с добавлением 10% эмбриональной бычьей сыворотки (FBS, Atlanta Biologicals, Lawrenceville, GA), 1% смеси антибиотиков/противогрибковых средств (Sigma-Aldrich, St. Louis, МО) и 5 мкг/мл бычьего инсулина (Sigma-Aldrich, St. Louis, МО).
Приобретенная устойчивость к вемурафениб in vitro
Линию клеток меланомы с приобретенной устойчивостью к вемурафениб подвергали хроническому отбору путем культивирования исходных клеток A375 при возрастающих концентрациях вемурафениб, согласно опубликованному методу: Su F, Bradley WD, Wang Q, et al. "Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation". Cancer Res. (2012) 72: 969-978), на протяжении по меньшей мере трех месяцев. У выделенной линии устойчивых клеток A375RF21 значение IC50 для вемурафениба стойко повышалось более чем в 50 раз (28,9±0,6 мкМ на клетках A375RF21 по сравнению с 0,57±0,03 мкМ у исходной линии клеток А375 при определении методом MTS, фиг. 1). Устойчивая линия клеток A375RF21 поддерживалась в полной культуральной среде, содержащей 2,5 мкМ вемурафениба.
Пролиферация клеток и анализ комбинаций in vitro
Исследовали жизнеспособность клеток по пролиферации методом MTS или SRB, как описано ранее. Исследование in vitro комбинаций вемурафениба и ингибиторов тубулина разрабатывали и проводили с помощью программного обеспечения CalcuSyn (Biosoft, Ferguson, МО) при пяти повторах по каждой серии обработки. Концентрации препаратов выбирали, исходя из значений IC50 для каждого препарата при тестировании в пилотном исследовании. Синергизм, аддитивное действие или антагонизм определяли по методу Chou-Talalay (Chou ТС. "Drug combination studies and their synergy quantification using the Chou-Talalay method". Cancer Res. (2010) 70: 440-446), который дает индекс комбинирования (CI), рассчитанный на выходе программы.
Анализ клеточного цикла
Проводили анализ методом проточной цитометрии, как описано ранее (Wang Z, Chen J, Wang J, et al. "Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis". Pharm. Res. (2012) 29: 3040-3052). Для определения распределения клеток в фазах G2 и М клеточного цикла (фиг 23) их собирали с помощью трипсина, окрашивали с помощью антитела против фосфогистона H3-AlexaFluor® 488 на льду в течение 1 часа в темноте, после чего окрашивали раствором PI/РНКаза в течение 30 мин при комнатной температуре в темноте в соответствии с инструкциями изготовителя (#FCCH025103, EMD Millipore Corporation, Ballerica, MA). Затем данные обрабатывали и строили графики с помощью программы Modfit 2.0 (Verity Software House, Topsham, ME).
Анализ полимеризации тубулина
Анализ полимеризации HTS-тубулина проводили, как описано ранее, используя коммерческий набор согласно инструкциям изготовителя (#BK004P, Cytoskeleton, Inc., Denver, CO). Тубулин из бычьего мозга (0,4 мг) смешивали с 5 мкМ 12da, 20 мкМ вемурафениб или с комбинацией двух средств и инкубировали в 110 мкл общего буфера для тубулина (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EDTA, 1 мМ GTP) при pH 6,9. Проводили кинетическую запись поглощения при 340 нм через каждую 1 мин в течение 45 мин при 37°C на считывающем устройстве Synergy НТ (Bio-Tek Instruments, Winooski, VT). Данные по монотерапии или комбинированной терапии сравнивали с данными от положительной контрольной группы при 10 мкМ колхицина.
Вестерн-блоттинг
Через указанные промежутки времени собирали клетки меланомы человека A375RF21, MDA-MB-435 или WM164 для исследования соответствующих каскадных белков или маркеров апоптоза на вестерн-блотах. Экстрагировали общий белок путем лизиса клеток буфером RIPA (Sigma-Aldrich, St. Louis, МО), содержащим коктейль из ингибиторов фосфатаз и протеиназ (Sigma-Aldrich, St. Louis, МО). Затем определяли общую концентрацию белка методом ВСА с помощью набора (Sigma-Aldrich, St. Louis, МО). Лизаты клеток разбавляли до одинаковой общей концентрации белка с помощью стартового буфера Лэммли (Bio-Rad, Hercules, СА) и кипятили 5 мин для денатурации белка. Затем группу образцов, содержащих по 10 мкг общего белка, наносили в лунки готового 4-15% полиакриламидного геля (Bio-Rad, Hercules, СА) для электрофореза, после которого их переносили на нитроцеллюлозную мембрану в 0,2 мкм (Bio-Rad, Hercules, СА). После блокирования 5% бычьим сывороточным альбумином в 1×TBST в течение одного часа при комнатной температуре мембраны подвергали инкубации по отдельности с первичными кроличьими антителами (Cell Signaling Technology, Inc., Danvers, MA): против фосфо-ERK1/2 (Thr202/Tyr204; #9101), против p44/42 MAPK (ERK1/2; #9102), против фосфо-AKT (Ser473; #9271), против AKT (#9272), против циклина D1 (92G2; #2978); против расщепленного PARP (Asp214; #9185), против расщепленной каспазы-3 (Asp 175; #9664), против RAS (#3339), против ARAF (#4432), против BRAF (#9433), против CRAF (#9422), фосфо-PI3-киназы р85 (Tyr458)/р55 (Tyr199) (#4228), против PTEN (#9188), против β-рецептора PDGF (#3169) или против GAPDH (#3683) в течение ночи при 4°C. Затем мембраны инкубировали со вторичным антителом против IgG кролика, конъюгированным с HRP (Cell Signaling, #7071), в течение 1 ч при комнатной температуре. Искомые белки выявляли путем инкубации с реагентом 1×LumiGLO® (Cell Signaling, #7003) в течение 1 мин и экспонировали на рентгеновской пленке. Пленки сканировали по серой шкале и определяли интенсивность на дорожках с помощью программного обеспечения ImageJ (NIH, Bethesda, MD, USA).
Детектирование апоптоза
Клетки A375RF21 высеивали в 6-луночные планшеты (1×106 на лунку) и обрабатывали питательной средой, содержащей 5% DMSO, вемурафениб, 12da, доцетаксель или указанные комбинации. После 48-часовой инкубации проводили анализ апоптоза с помощью набора Annexin V-FITC Apoptosis Detection Kit (Abcam, Cambridge, MA) согласно инструкциям изготовителя и анализировали на цитометре BD LSR-II (BD Biosciences, San Jose, CA).
Ксенотрансплантация опухолей и обработка
Самцов мышей nude в возрасте семь-восемь недель приобретали у Charles River Laboratories International, Inc. (Wilmington, MA). Клетки A375RF21 суспендировали в ледяной среде DMEM без FBS, лишенной фенолового красного и FBS, и смешивали с Matrigel при высокой концентрации (BD Biosciences, San Jose, СА) в соотношении 1:1 непосредственно перед использованием. Каждой мыши вводили подкожно (п/к) в левый бок со спины по 100 мкл этой смеси, содержащей 3×106 клеток. Через неделю после инокуляции мышей случайным образом делили на четыре группы (n=7 для комбинации препаратов с начальной низкой дозой и n=5 для последующей высокой дозы) и начинали обработку. Соединение 12da или вемурафениб разводили в PEG300 (Sigma-Aldrich, St. Louis, МО) и вводили посредством внутрибрюшинной (в/б) инъекции один раз в день, 5 дней в неделю на протяжении 3 недель. Контрольной группе носителя вводили в/б такой же объем (100 мкл) PEG300 при той же частоте дозирования. По окончании экспериментов мышей забивали, выделяли и взвешивали опухолевые ткани, а затем фиксировали в 10% растворе формалина с фосфатным буфером.
У каждой мыши 3 раза в неделю определяли объем опухоли и вес тела. Объем опухолей рассчитывали по формуле a×b×0,5, где а и b означают больший и меньший диаметр опухоли. Данные представляли в виде среднего ± SD для каждой группы и наносили на график зависимости от времени. Ингибирование роста опухолей (TGI) рассчитывали как 100-100×[(Т-Т0)/(С-С0)], а регрессию опухолей вычисляли как (Т-Т0)/Т0×100, где Т, Т0, С и С0 означают средний объем опухоли для данной группы в последний день обработки, средний объем опухоли у той же группы в первый день обработки, средний объем опухоли для контрольной группы носителя в последний день обработки и средний объем опухоли для контрольной группы носителя в первый день обработки, соответственно.
Патологический и иммуногистохимический анализ
Опухолевые ткани, фиксированные в формалиновом буфере свыше одной недели, окрашивали гематоксилином и эозином. Для иммуногистохимического анализа (IHC) использовали следующие первичные кроличьи антитела: против Ki67, против фосфо-AKT (Ser473) и против фосфо-ERK1/2 (Thr202/Tyr204) (#9027; #4060; #4376; Cell Signaling Technology, Inc., Danvers, MA). Первичное антитело против SI00 приобретали у Abcam (#ab868, Abcam Inc., Cambridge, MA). Анализы проводили согласно методикам производителя.
Статистический анализ
Данные анализированы с помощью программного обеспечения Prism 5.0 (GraphPad Software, Inc., San Diego, CA). Статистическую значимость (P<0,05) оценивали по непараметрическому критерию Mann-Whitney Rank Test и одностороннему ANOVA при изучении апоптоза in vitro и исследовании ксенотрансплантации. Получавшие обработку группы сравнивали с группой носителя, а группы комбинированной терапии сравнивали с группами, получавшими монотерапию, соответственно.
Результаты
Комбинация вемурафениба с ингибиторами тубулина 17уа и 12da проявляет сильную синергичность как на исходной, так и на устойчивой к вемурафениб линии клеток меланомы
Проводили скрининг нескольких ингибиторов тубулина, включая соединения 12da и 17уа, для оценки их антипролиферативного действия при комбинировании с вемурафенибом на клетках исходной линии А375 и линии MDA-MB-435, то есть клетках с мутацией BRAFV600E. Для сравнения включали доцетаксель и колхицин, два хорошо известных ингибитора тубулина (таблица 1 и фиг. 24). Как оказалось, расчетные значения CI для комбинации 12da и вемурафениб были столь низкие, как 0,32 (на линии клеток А375) и 0,10 (на линии клеток MDA-MB-435) при их ED50. Кроме того, оказалось, что ингибитор MEKi (траметиниб) и общий ингибитор рецепторных тирозинкиназ (RTKi; сунитиниб) проявляют лишь аддитивные значения CI (таблица 1).
Исходя из приведенных ниже результатов, эти комбинации могут способствовать преодолению невосприимчивости, наблюдающейся у пациентов, принимающих вемурафениб.
Соединения 17уа и 12da проявляли синергическое действие при комбинировании с доцетакселем или вемурафенибом. Работа начиналась с 17уа, но была перенесена на 12da для исследований in vivo. Вводится концепция индекса комбинирования (CI). По сути, значения CI <1,0 указывают на синергетическое действие, значение 1,0 указывает на аддитивное действие, а значения >1,0 указывают на менее чем аддитивное или антагонистическое действие.
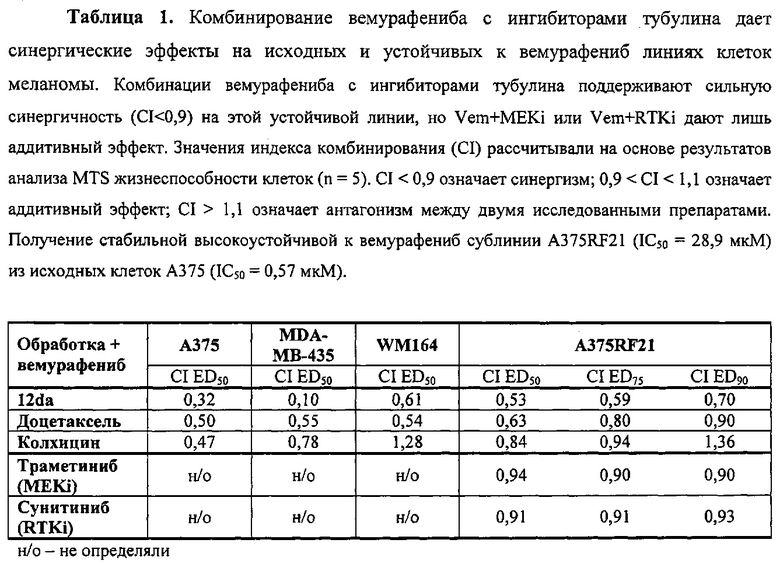
Клинически опухоли меланомы неизбежно дают рецидивы только через 3-6 месяцев после химиотерапии вемурафенибом, поэтому нужно было определить, будет ли наблюдаемая синергичность сохраняться у устойчивых к вемурафениб клеток. Для этого была создана устойчивая к вемурафениб сублиния A375RF21 из исходных клеток A375 меланомы человека посредством хронического отбора по приведенным в литературе методикам (Su F, Bradley WD, Wang Q, et al. "Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation". Cancer Res. (2012) 72: 969-978). Проводили анализ методом вестерн-блот на клетках A375 и клетках A375RF21, чтобы определить дифференциальную активацию белков, которая, как известно, приводит к устойчивости к вемурафениб. Как видно из фиг. 25А, у A375RF21 уровень pERK в присутствии 2,5 мкМ вемурафениба (поддерживающая концентрация его в культуральной среде) не изменялся, что подтверждает возникновение приобретенной устойчивости к вемурафениб. Экспрессия pMEK также оставалась активной в клетках A375RF21, что свидетельствует о потенциальной перекрестной устойчивости их к ингибиторам MEK1/2. Эта перекрестная устойчивость подтверждалась при инкубации клеток с двумя известными ингибиторами MEK (траметиниб и селуметиниб, фиг. 25B и 25C). В клетках A375RF21 путь PI3K/AKT был гиперактивирован, но не наблюдалось существенных изменений уровней RAS, BRAF, ARAF, CRAF. Эти результаты согласуются с работой Fei Su et al. Интересно, что в клетках A375RF21 также значительно возрастал уровень β-рецептора PDGF в присутствии или в отсутствие 2,5 мкМ вемурафениб в среде. Хорошо известно, что в опухолях у невосприимчивых к вемурафениб пациентов существуют оба механизма устойчивости (pAKT и PDGF-β), придающие лекарственную устойчивость клеткам A375RF21.
Как видно из таблицы 1, при повторении исследования по комбинированию препаратов на клетках A375RF21 расчетные значения CI для соединения 12da в комбинации с вемурафениб все оказались меньше 0,9 (диапазон: 0,53-0,70), что указывает на сильную синергичность при всех исследованных концентрациях. При ED50 все три ингибитора тубулина действовали синергетически с вемурафениб. При повышении концентрации препарата значения CI для групп доцетакселя или колхицина возрастали сравнительно быстро. При дозе ED90 комбинация доцетаксель с вемурафениб была почти аддитивной (значение CI=0,90), тогда как при комбинировании колхицина с вемурафениб эффект обращался в антагонизм (значение CI=1,36). По сравнению с двумя другими ингибиторами тубулина, соединение 12da проявляло большую синергичность в комбинации с вемурафениб на резистентных клетках A375RF21.
Вне ожидания, расчитанные значения CI для комбинации 12da и вемурафениба оказался столь низкими, как 0,1 и 0,3 при ЕС50. Поскольку клиническая регрессия опухолей сильно развивалась только через 3-6 месяцев после химиотерапии вемурафенибом, то исследование того, будет ли наблюдавшийся в первую очередь эффект синергии при комбинированной терапии сохраняться и на устойчивой к вемурафениб линии клеток A375RF21, было продолжено.
Из всех исследованных химиотерапевтических средств ингибитор мутантов B-RAF вемурафениб проявлял особенно синергетичную цитотоксичность на клетках MDA-MB-435, которые первоначально считались клетками рака молочной железы, но теперь известны как клетки меланомы (табл. 2), проявляя значение CI=0,10 для вемурафениб + 17уа, а также проявляя сильную, но меньшую синергичность при ЕС75 и ЕС90 (табл. 5). Такое действие можно объяснить как блокирование клеток ингибитором B-RAF и противотубулиновым средством (17уа) на различных стадиях клеточного цикла, G и G2/M (как обсуждалось здесь), соответственно. Соответственно, двойное воздействие на митоз должно более полно воздействовать на митотические клетки, вызывая гибель клеток. Синергические результаты по CI также видны из табл. 3 на клетках меланомы А375 не только у 17уа, но и других антитубулиновых средств, таких как колхицин, винбластин и таксол. Отмечалась ограниченная синергичность с ингибитором AKT и отсутствие синергичности с ингибитором MEK (табл. 4).
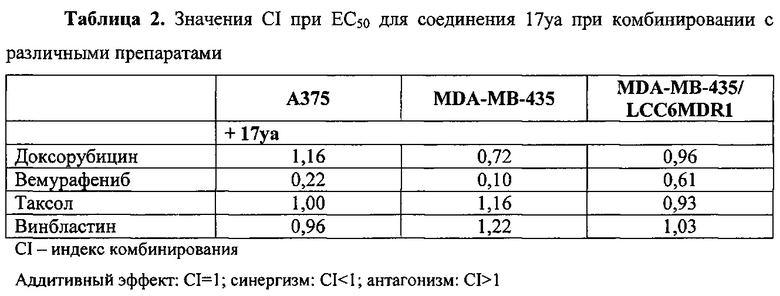
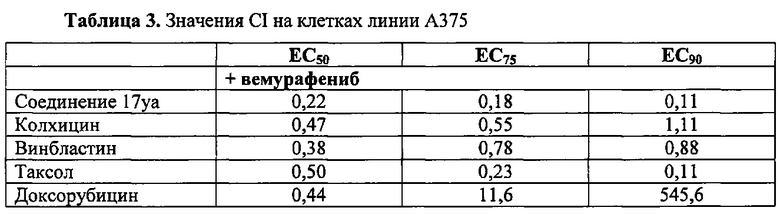


Комбинация 12da и вемурафениба вызывает синергетическую блокировку клеточного цикла на клетках A375RF21 (таблица 1)
Как ингибитор тубулина, связывающийся с колхициновым сайтом, соединение 12da эффективно блокирует исходную линию клеток A375 в фазе G2/M дозо-зависимым образом. Чтобы определить, будет ли комбинация соединения 12da и вемурафениба блокировать устойчивые к вемурафенибу клетки в различных фазах репликации, проводили анализ клеточного цикла на клетках A375RF21. После 24-часовой обработки раствором соединения при указанных концентрациях, приведенные на фиг. 2B данные четко указывают на синергетическую блокировку клеточного цикла. У контролей на DMSO 50% клеток A375RF21 находились в фазе G0/G1, а процент клеток в фазе S или G2/M составлял 12% или 32%, соответственно. В группе обработки одним лишь соединением 12da при концентрации 20 нМ содержание клеток в фазе G2/M возросло до 70%. При использовании вемурафениба в качестве единственного средства для получения такой же блокировки клеточного цикла G0/G1 на устойчивой линии клеток A375RF21 приходилось повысить концентрацию вемурафениба до 30 мкМ или выше в сравнении с менее чем 1 мкМ на исходных клетках A375. Как и ожидалось, комбинация вемурафениба и соединения 12da сильно блокировала клетки A375RF21 в обеих фазах G0/G1 (48%) и G2/M (43%). Кроме того, комбинированная обработка производила намного больше клеточных обломков, что указывает на усиление апоптоза раковых клеток. Обработка комбинацией вемурафениб и доцетаксель давала такие же синергетические эффекты.
Комбинированная обработка вызывает значительное усиление апоптотической гибели клеток на устойчивых к вемурафениб клетках
Для более четкого понимания возможного эффекта индукции апоптоза клеток при комбинированной обработке использовали проточную цитометрию при совместном окрашивании аннексином V и йодидом пропидия для дифференциации живых и апоптозных клеток A375RF21. Как и ожидалось, монотерапия давала лишь умеренные эффекты на индукцию апоптоза клеток при исследованных концентрациях; напротив, в группах комбинированной обработки апоптоз значительно усиливался (фиг. 3А). Как видно из фиг. 3В, где представлено процентное содержание клеток в Q1 (ранний апоптоз), Q2 (апоптоз) и Q4 (мертвые клетки), комбинация соединения 12da и вемурафениба приводила к апоптозу или гибели 50±7,6% от всех подсчитанных клеток, что гораздо выше, чем простая сумма процентов апоптоза в двух группах монотерапии (11,8±3,0% для соединения 12da и 11,9±3,5% для вемурафениба). Аналогичный эффект синергичности при индукции апоптоза наблюдался и в группе комбинированной обработки, содержащей доцетаксель (6,4±3,0% для группы самого доцетакселя, 38,1±2,6% для комбинации).
Комбинирование ослабляет приобретенную устойчивость к вемурафениб через понижающую регуляцию pAKT или общего AKT и активацию каскадов апоптоза
Установлено, что соединение 12da воздействует на полимеризацию тубулина (Chen J, Li CM, Wang J, et al. "Synthesis and antiproliferative activity of novel 2-aryl-4-benzoyl-imidazole derivatives targeting tubulin polymerization". Bioorg. Med. Chem. (2011) 19: 4782-4795; Chen J, Wang Z, Li CM, et al. "Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicine binding site in tubulin as potential anticancer agents". J. Med. Chem. (2010) 53: 7414-7427), а вемурафениб воздействует на BRAFV600E. В качестве первого подхода к пониманию ответственных за это молекулярных механизмов, ведущих к такой сильной синергетической комбинации, исследовали, опосредована ли синергия потенциированием прямой мишени соединения 12da или вемурафениба. Как видно из фиг. 4, вемурафениб сам по себе не оказывал никакого влияния на полимеризацию тубулина, даже при высокой концентрации в 20 мкМ. Добавление вемурафениба к соединению 12da вызывало лишь незначительно усиление ингибирования полимеризации тубулина по сравнению с одним только соединением 12da. Ингибирование полимеризации тубулина при комбинированной обработке обусловлено исключительно соединением 12da, свидетельствуя о том, что синергическая комбинация не опосредуется через усиление ингибирования непосредственной мишени соединения 12da. Затем исследовали, оказывает ли комбинация какие-либо эффекты на pERK, признак ингибирования BRAFV600E вемурафенибом, методом вестерн-блоттинга. Из фиг. 5А видно, что ни соединение 12da, ни комбинированная обработка не оказывала явного влияния на уровень pERK или общего ERK после 48-часовой инкубации с устойчивыми клетками A375RF21. Замена соединения 12da на другой ингибитор тубулина, доцетаксель, давала такие же результаты (фиг. 5А). Таким образом, синергическая комбинация вряд ли работает через усиление ингибирования BRAFV600E.
Комбинирование ослабляет приобретенную устойчивость к вемурафениб через понижающую регуляцию pAKT или общего AKT и активацию каскадов апоптоза
Недавно Fei Su et al. сообщали, что уровень pAKT у устойчивых к вемурафениб клонов A375 повышается по сравнению с исходными чувствительными к вемурафениб клетками (Su F, Bradley WD, Wang Q, et al. "Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation". Cancer Res. (2012) 72: 969-978). Поскольку клетки A375RF21 также проявляют сильную активацию pAKT (фиг. 25А), то было высказано предположение, что комбинированная обработка может вызывать его сильную синергичность через понижающую регуляцию пути AKT в устойчивых к вемурафениб клетках A375RF21. Как видно из фиг. 5А, и pAKT, и общий AKT (tAKT) сильно снижались при обработке только соединением 12da или при комбинированной обработке после 48-часовой инкубации, свидетельствуя о том, что синергетический антипролиферационный эффект может быть опосредован одновременным воздействием на фосфорилирование ERK и AKT. Доцетаксель тоже снижал экспрессию pAKT и tAKT и оказывал аналогичные эффекты при комбинировании его с вемурафениб. Например, наряду с явным снижением уровня tAKT, комбинация соединения 12da и вемурафениб снижала уровень pAKT до 61% относительно tAKT (расчитывали по относительному уровню плотности на дорожках: 0,08/0,13×100%), тогда как обработка только одним препаратом снижала уровень pAKT до 77% (12da, 0,6/0,77×10%) и 70% (вемурафениб, 0,34/0,48×100%) относительно соответствующего уровня tAKT, соответственно. Сильное дозо-зависимое ингибирование pAKT/tAKT соединением 12da далее было подтверждено на двух других линиях клеток с мутацией BRAFV600E, WM164 и MDA-MB-435 (фиг. 5B). В устойчивых к вемурафениб клетках (фиг. 5A) снижение уровня циклина D1 в группах вемурафениба и комбинированной обработки указывает на блокирование клеточного цикла на переходе G0/G1. Ингибиторы тубулина сильно индуцировали маркеры апоптоза, расщепленные белки PARP и каспазу-3, тогда как вемурафениб лишь слегка повышал их экспрессию. Этот результат согласуется с наблюдениями в эксперименте по выявлению апоптоза.
Комбинация вемурафениб и соединения 12da синергетически подавляет рост устойчивых к вемурафениб опухолей in vivo
Чтобы установить, можно ли сильный синергизм, наблюдаемый в отношении клеток A375RF21 in vitro, переносить на устойчивые к вемурафениб опухоли in vivo, сравнивали эффект комбинированного действия на рост опухолей с эффектом обработки только одним препаратом. Ранее было установлено, что соединение 12da эффективно подавляет рост опухолей исходной меланомы A375 in vivo в дозе 25 мг/кг (Wang Z, Chen J, Wang J, et al. "Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis". Pharm. Res. 29(11): 3040-3052). Пилотное исследование показало, что устойчивые к вемурафениб клетки A375RF21 проявляют такую же кинетику роста (фиг. 26), как и исходные клетки A375 в отсутствие обработки препаратами. Для того, чтобы избежать какой-либо потенциально неожиданной токсичности при комбинировании с вемурафенибом, дозу соединения 12da снижали до 10 мг/кг.

Как видно из фиг. 6 и табл. 6, обработка одним вемурафенибом (20 мг/кг) давала лишь минимальное (22,65%) TGI, а соединение 12da (10 мг/кг) само по себе давало лишь немного лучшее TGI в 38,12% на этой модели устойчивых к вемурафениб опухолей; напротив, при обработке их комбинацией ингибирование роста опухолей значительно повышалось до 88,56% после 3-недельной обработки (фиг. 6А, 6B и 6D, табл. 6). Три из семи мышей, получавших комбинированную обработку и содержавшихся еще 7 дней без обработки, не проявляли никаких существенных (Р=0,2857) рецидивов опухолей, сохраняя подавление опухолей на 81,27%. В течение всего эксперимента ни одна мышь в четырех группах не потеряла массы тела более чем на 10% (фиг. 6C), что указывает на отсутствие общей токсичности такой обработки. Когда мышей забивали, то выделяли основные органы, включая мозг, сердце, почки, печень, селезенку и легкие, и направляли на патологический анализ. Не наблюдалось никаких отклонений от нормы в этих органах. В совокупности, эти результаты убедительно свидетельствуют о том, что такая комбинированная обработка эффективно помогает преодолеть приобретенную устойчивость к вемурафениб на модели меланомы A375RF21 и дополнительно подтверждает синергетические антипролиферативные эффекты, наблюдавшиеся in vitro.
Чтобы установить, будет ли понижающая регуляция сигнализации AKT при комбинированной терапии, наблюдаемая in vitro, также функционировать in vivo, проводили иммуногистохимический анализ срезов опухолей из всех экспериментальных групп. Также определяли активность пути ERK и оценивали уровень пролиферации в срезах опухолей по маркеру Ki-67. Как видно из фиг. 6D, лучшее ингибирование пути и пролиферации в группе комбинированной обработки хорошо соответствовало общей реакции опухолей по данным TGI. Фосфорилирование ERK и AKT вместе с уровнем экспрессии Ki67 в ядре и в цитоплазме сильно снижалось в группе комбинированной обработки. Более того, широкая зона фоновой розовой окраски Matrigel у срезов опухолей при окрашивании Н&Е в группе комбинированной обработки свидетельствует, что после комбинированной обработки осталось очень немного опухолевых клеток, если вообще. Значительное сокращение клеток меланомы в группе комбинированной обработки также подтверждается снижением плотности при иммуноокрашивании на S100 (фиг. 6D).
Более высокая доза комбинации вемурафениб с соединением 12da приводит к значительной регрессии устойчивых к вемурафениб опухолей без заметной токсичности
Поскольку результаты, представленные на фиг. 6 и в табл. 6, были многообещающими, но не приводили к заметной регрессии опухолей, то эксперимент повторяли путем повышения дозы на 50% и для соединения 12da, и для вемурафениба. Результаты представлены на фиг. 7 и в табл. 7.

Отмечалось небольшое повышение эффективности для вемурафениба (TGI=22,65% при 20 мг/кг против 28,10% при 30 мг/кг) и значительное повышение эффективности для соединения 12da (TGI=38,12% при 10 мг/кг против 72,72% при 15 мг/кг). Отмечалась умеренная регрессия опухолей (44,9%) в группе комбинированной обработки при повышенной дозе препарата, как видно из фиг. 7B, тогда как при более низких дозах регрессия не наблюдалась. В совокупности эти данные дают первое убедительное свидетельство того, что комбинации новых ингибиторов тубулина типа соединения 12da с вемурафенибом могли бы преодолеть приобретенную устойчивость к вемурафениб у больных меланомой с мутацией BRAFV600E.
Итак, эти исследования убедительно свидетельствуют, что комбинации ингибитора BRAF (например, вемурафениба) и новых ингибиторов тубулина (например, соединения 12da) эффективно преодолевают приобретенную устойчивость к ингибиторам BRAF у меланом с мутацией BRAF по нескольким возможным механизмам, включая синергетическую блокировку клеточного цикла, усиление апоптоза и сильное ингибирование пути AKT. По крайней мере in vitro, такие комбинации кажутся более эффективным, чем комбинации вемурафениб с ингибиторами MEK или AKT либо существующими ингибиторами тубулина. При отсутствии устойчивой эффективности комбинации BRAFi + MEKi для меланомы, разработка такой стратегии комбинирования, нацеленной на альтернативные пути, могла бы иметь большое значение в этой области.
Обсуждение
Несмотря на то, что вемурафениб - первый препарат, одобренный для больных меланомой с мутацией BRAFV600E, показал замечательные ответы в начальной терапии, почти у всех пациентов, принимающих этот препарат, возникала устойчивость к нему в пределах нескольких месяцев (Bollag G, Tsai J, Zhang J, et al. "Vemurafenib: the first drug approved for BRAF-mutant cancer". Nat. Rev. Drug Discov. (2012) 11: 873-886). Понимание механизмов, лежащих в основе первичной или приобретенной устойчивости, и разработка подходящих стратегий комбинирования могли бы обеспечить более эффективные пути преодоления такой устойчивости. Существует обширная литература по доклиническим исследованиям и клиническим испытаниям для поиска эффективных комбинаций вемурафениба с другими средствами с целью устранения или уменьшения устойчивости опухолей меланомы к ингибиторам BRAF или MEK1/2 (Greger JG, Eastman SD, Zhang V, et al. "Combinations of BRAF, MEK, and PBK/mTOR inhibitors overcome acquired resistance to the BRAF ингибитор GSK2118436 dabrafenib, mediated by NRAS or MEK mutations". Mol. Cancer Ther. (2012) 11: 909-920; Patel SP, Lazar AJ, Papadopoulos NE, et al. "Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma". Cancer (2013) 119(4): 799-805; Flaherty KT, Infante JR, Daud A, et al. "Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations". N. Engl. J. Med. (2012) 367: 1694-1703; Niehr F, von Euw E, Attar N, et al. "Combination therapy with vemurafenib (PLX4032/RG7204) and metformin in melanoma cell lines with distinct driver mutations". J. Transl. Med. (2011) 9: 76; Paraiso KH, Haarberg HE, Wood E, et al. "The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms". Clin. Cancer Res. (2012) 18: 2502-2514; Koya RC, Mok S, Otte N, et al. "BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy". Cancer Res. (2012) 72: 3928-3937). Ингибиторы пути RAF/MEK/ERK в основном вызывают G1-блокировку клеточного цикла, но не гибель опухолевых клеток меланомы. Таким образом, комбинация средств, направленных на различные компоненты одного и того же пути (например, комбинация вемурафениба и ингибиторов MEK1/2), хотя и эффективна поначалу (Little AS, Smith PD, Cook SJ. "Mechanisms of acquired resistance to ERK1/2 pathway inhibitors". Oncogene (2012) 32(10): 1207-1215), не может поддерживать длительную синергию против устойчивых клеток, которые могут избежать этой G1-блокировки клеточного цикла. Поскольку одним из главных признаков ингибиторов тубулина является их способность сильно блокировать клетки в фазе G2/M, то считалось, что комбинация вемурафениба и ингибитора тубулина синергически блокирует клетки меланомы, вызывая усиление апоптоза, и преодолевает приобретенную устойчивость. В нашем исследовании для исследования комбинаций с вемурафенибом против опухолей меланомы был выбран новый ингибитор тубулина, соединение 12da. Была разработана устойчивая к вемурафениб линия клеток A375RF21 меланомы человека и было показано, что комбинация соединения 12da и вемурафениба обладает сильным синергизмом in vitro. Было подтверждено, что синергизм не может возникать путем усиления ингибирования полимеризации тубулина или уменьшения активации p-ERK. Напротив, экспериментальные результаты показали, что такая комбинированная обработка преодолевает приобретенную устойчивость к вемурафенибу через усиление индукции апоптоза посредством синергетической блокировки клеточного цикла в G1 и G2/M и существенного нарушения сигнального пути выживания, связанного с фосфорилированием AKT. Было показано, что сильный синергизм, наблюдавшийся in vitro, четко переводится в существенную эффективность in vivo при тестировании на модели устойчивых к вемурафенибу ксенотрансплантатов. Дальнейший иммуногистохимический анализ на срезах тканей подтвердил сильное ингибирование пролиферации опухолей и снижение активности pAKT.
Активация сигнального пути PI3K/AKT/MTOR, как было показано, способствует уменьшению чувствительности к ингибированию ERK1/2 в линиях клеток меланомы человека (Bartholomeusz С, Gonzalez-Angulo AM. "Targeting the PI3K signaling pathway in cancer therapy". Expert Opin. Ther. Targets (2012) 16: 121-130), а в нескольких недавних исследованиях четко продемонстрирована синергическая комбинация из ингибитора пути PI3K/AKT/mTOR и ингибитора BRAF или ингибитора MEK (Liu R, Liu D, Xing M. "The Akt inhibitor MK2206 synergizes, but perifosine antagonizes, the BRAF(V600E) inhibitor PLX4032 and the MEK1/2 inhibitor AZD6244 in the inhibition of thyroid cancer cells". J. Clin. Endocrinol. Metab. (2012) 97: E173-182). Недавно было представлено несколько новых классов соединений в качестве ингибиторов полимеризации тубулина, которые также проявляли сильное торможение пути AKT (Krishnegowda G, Prakasha Gowda AS, et al. "Synthesis and biological evaluation of a novel class of isatin analogs as dual inhibitors of tubulin polymerization and Akt pathway". Bioorg. Med. Chem. (2011) 19: 6006-6014; Viola G, Bortolozzi R, Hamel E, et al. "MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells". Biochem. Pharmacol. (2012) 83: 16-26; Zhang С, Yang N, Yang CH, et al. "S9, a novel anticancer agent, exerts its anti-proliferative activity by interfering with both PI3K-Akt-mTOR signaling and microtubule cytoskeleton". PLoS One (2009) 4: e4881). Кроме того, конститутивно активный путь PI3K/AKT, как оказалось, приводит к множественной лекарственной устойчивости к препаратам, направленным на полимеризацию тубулина микротрубочек (МТРА), а ингибирование опосредованного PDK/AKT сигнального пути, как оказалось, сенсибилизирует раковые клетки к индуцированному МТРА апоптозу (Bhalla KN. "Microtubule-targeted anticancer agents and apoptosis". Oncogene (2003) 22: 9075-9086). Эти исследования указывают на тесное взаимодействие между ингибиторами полимеризации тубулина и понижающей регуляцией AKT в раковых клетках. Кроме того, недавно сообщалось, что ингибитор MEK - AZD6244 индуцирет блокировку роста клеток меланомы и регрессию опухолей в комбинации с доцетакселем (Haass NK, Sproesser K, Nguyen TK, et al. "The mitogen activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel". Clin. Cancer Res. (2008) 14: 230-239). Интересно, что наши результаты согласуются с этими исследованиями. Исследования in vivo, представленные в данном изобретении, показывают эффективность комбинированной обработки на опухолевых клетках, уже обладающих устойчивостью к вемурафенибу, на модели ксенотрансплантатов A375RF21. Можно предположить, что если комбинацию использовать до того, как опухолевые клетки приобретут устойчивость к вемурафенибу, то регрессия опухолей будет более сильной, а появление устойчивых опухолевых клеток значительно замедлится или даже предотвратится. Это могло бы выражаться по крайней мере в более длительном времени без прогрессирования у пациентов и/или усилении регрессии заболевания. В совокупности, в данном исследовании представлено первое прямое свидетельство и обоснование для комбинирования сильного ингибитора тубулина с ингибитором, направленным на путь RAF/MEK/ERK, для значительного улучшения терапии больных меланомой.
ПРИМЕР 2. Синтез отдельных арил-бензоил-имидазоловых соединений
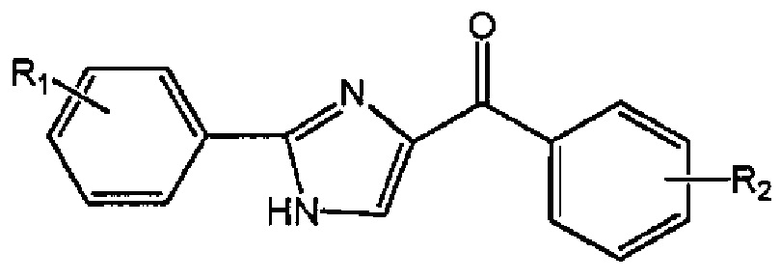
Получение 2-арил-4,5-дигидро-1Н-имидазолов 14b, 14c, 14x (фиг. 8)
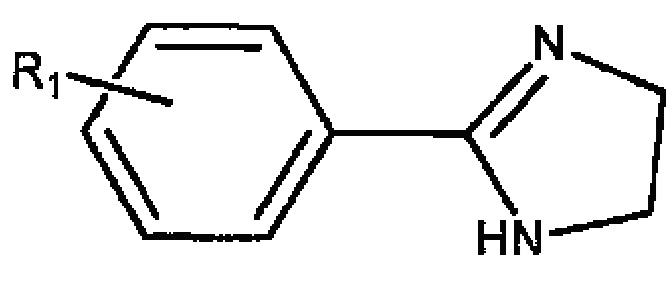
В раствор соответствующего бензальдегида 8(b, c, x) (60 ммоль) в t-BuOH (300 мл) добавляли этилендиамин (66 ммоль) и перемешивали 30 мин при комнатной температуре. В реакционную смесь последовательно добавляли карбонат калия (75 ммоль) и йод (180 ммоль), а затем перемешивали при 70°C в течение 3 ч. Добавляли сульфит натрия (Na2SO3) и экстрагировали смесь хлороформом. Органический слой сушили над сульфатом магния и упарировали. Остаток очищали методом колоночной флэш-хроматографии (хлороформ : метанол = 20:1), получая белое твердое вещество. Выход: 50-60%.
Получение 2-арил-1H-имидазолов 9a-j, 9р, 9х (фиг. 8 и 9)
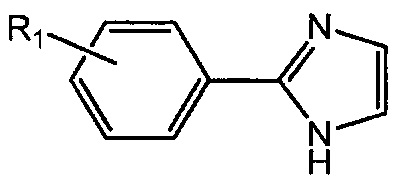
Метод A (нужен только для 9b, 9x; фиг. 8). В раствор 2-арил-4,5-дигидро-1Н-имидазола 14b, x (35 ммоль) в DMSO (100 мл) добавляли карбонат калия (38,5 ммоль) и диацетоксийодбензол (38,5 ммоль). Реакционную смесь перемешивали в течение ночи в темноте. Добавляли воду, после чего экстрагировали дихлорметаном. Органический слой сушили над сульфатом магния и упаривали. Остаток подвергали колоночной флэш-хроматографии (гексан : этилацетат = 3:2), получая белое твердое вещество. Выход: 30%-50%.
Метод В (нужен только для 9c; фиг. 8). В раствор 2-арил-4,5-дигидро-1Н-имидазола 14c в DMF (70 мл) добавляли DBU (55 ммоль) и CBrCl3 (55 ммоль). Реакционную смесь перемешивали в течение ночи и добавляли насыщенный раствор NaHCO3 (водный), после чего экстрагировали дихлорметаном. Органический слой сушили над сульфатом магния и упаривали. Остаток подвергали колоночной флэш-хроматографии (хлороформ : метанол = 50:1), получая белое твердое вещество. Выход: 7%.
Метод С (нужен только для 9а, 9d-j, 9р; фиг. 9). В раствор соответствующего бензальдегида (8а, 8d-j, 8р) (100 ммоль) в этаноле (350 мл) при 0°C добавляли раствор 40% оксальдегида в воде (12,8 мл, 110 ммоль) и 29% раствор гидроксида аммония в воде (1000 ммоль, 140 мл). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь упаривали, а остаток подвергали колоночной флэш-хроматографии с дихлорметаном в качестве элюента, получая искомое соединение в виде желтого порошка. Выход: 20%-40%.
Получение 2-арил-1-(фенилсульфонил)-1Н-имидазолов 10a-j, р, х (фиг. 8 и 9)
В раствор 2-арил-1Н-имидазола 9a-j, р, х (20 ммоль) в безводном THF (200 мл) при 0°C добавляли гидрид натрия (60% дисперсия в минеральном масле, 1,2 g, 30 ммоль) и перемешивали в течение 30 мин. Добавляли бензолсульфонилхлорид (2,82 мл, 22 ммоль) и перемешивали реакционную смесь в течение ночи. После разбавления 100 мл насыщенного раствора NaHCO3 (водного), реакционную смесь экстрагировали этилацетатом (500 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 2:1), получая бледное твердое вещество. Выход: 50%-70%.
Получение арил-(2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанонов 11aa-ai, ba, са, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa (фиг. 8 и 9)
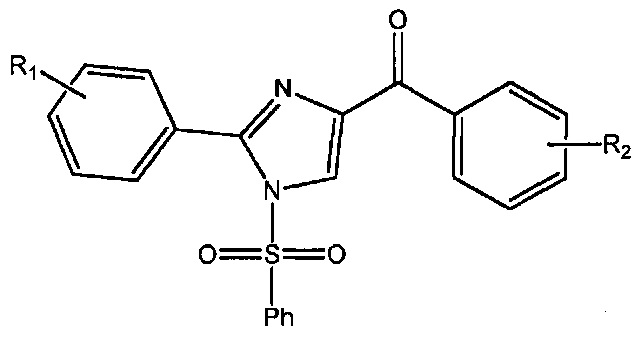
В раствор 2-арил-1-(фенилсульфонил)-1Н-имидазола (6,0 ммоль) 10а-j, р, х в безводном THF (30 мл) при -78°C добавляли 1,7 М трет-бутиллитий в пентане (5,3 мл, 9,0 ммоль) и перемешивали в течение 10 мин. Добавляли соответствующий замещенный бензоилхлорид (7,2 ммоль) при -78°C и перемешивали в течение ночи. Реакционную смесь разбавляли 100 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 4:1), получая белое твердое вещество. Выход: 15%-40%.
Общая методика получения арил-(2-арил-1Н-имидазол-4-ил)метанонов 12aa-ai, ba, са, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa (фиг. 8 и 9)
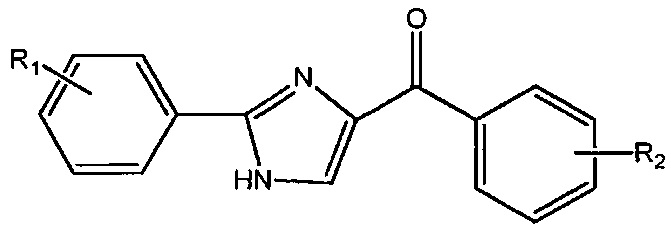
В раствор арил-(2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанона (2,0 ммоль) 11aa-ai, ba, са, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa в THF (20,0 мл) добавляли 1,0 M тетрабутиламмония фторид (4,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли 50 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 3:1) или перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 80-95%.
Получение (2-(4-гидроксифенил)-1Н-имидазол-4-ил)(арил)метанонов 12ka, 12kb (фиг. 9)
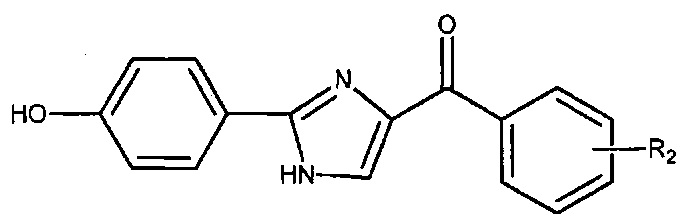
В раствор (2-(4-(бензилокси)фенил)-1Н-имидазол-4-ил)(арил)метанона 12ja или 12jb (1 ммоль) в AcOH (20 мл) добавляли конц. HCl (2 мл) и кипятили с обратным холодильником в течение ночи. После удаления растворителя остаток перекристаллизовывали из дихлорметана, получая искомое соединение в виде желтого твердого вещества. Выход: 70-85%.
Получение (2-арил-1Н-имидазол-4-ил)(3,4,5-тригидроксифенил)метанонов 13еа, 13fa, 13ha (фиг. 9)
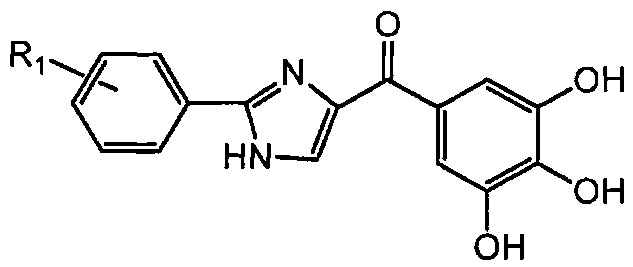
В раствор арил-(2-арил-1Н-имидазол-4-ил)метанона 12еа, 12fa или 12ha (0,5 ммоль) в CH2Cl2 (6,0 мл) добавляли 1,0 М BBr3 (2 ммоль) в CH2Cl2 и перемешивали 1 ч при комнатной температуре. Добавляли воду, чтобы разрушить излишки BBr3. Осажденное твердое вещество отфильтровывали и перекристаллизовывали из MeOH, получая желтое твердое вещество. Выход: 60-80%.
Получение арил-(2-арил-1Н-имидазол-4-ил)метанона в виде HCl-соли (12db-HCl)
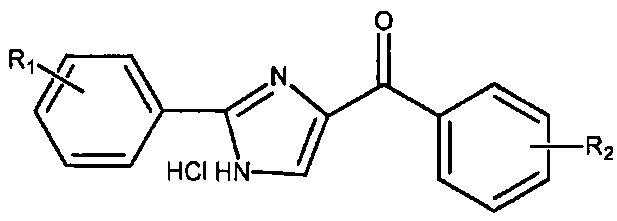
В раствор 12db (0,5 ммоль) в метаноле (20 мл) добавляли 2 М раствор HCl (5 ммоль) в этиловом эфире и перемешивали в течение ночи при комнатной температуре. Реакционную смесь упаривали, а остаток промывали CH2Cl2, получая искомое соединение. Выход: 95%.
Получение арил-(2-фенил-1Н-имидазол-1-ил)метанонов 12aba, 12ааа (фиг. 10).
В раствор 2-фенил-1Н-имидазола 9а (10 ммоль) в THF (20 мл) добавляли NaH (15 ммоль) и замещенный бензоилхлорид (12 ммоль) при 0°C.Реакционную смесь перемешивали в течение ночи и разбавляли насыщенным раствором NaHCO3, после чего экстрагировали этилацетатом. Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (хлороформ), получая белое твердое вещество. Выход: 12-16%.
Получение 1-замещенных (2-фенил-1Н-имидазол-1-ил)-арилметанонов 12dc, 12fc, 12daa, 12dab, 12cba, 11gaa, 12la (фиг. 11-12)
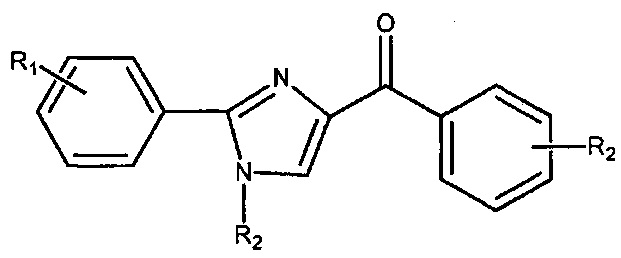
Синтез 12dc, 12fc и 12daa, 12dab и 12cba обобщен на фиг. 11. Соединения 12da, 12cb и 12fa синтезировали способом, описанным выше и на фиг. 8 и 9. Обработка 12da и 12fa хлоридом алюминия давала пара-деметилированные 12dc, 12fc с интактной 3,5-диметоксигруппой. Соединение 12daa получали бензилированием 12da в положении N-1. А метилирование 12da и 12cb в положении N-1 давало соединения 12dab и 12cba, соответственно.
Синтез 12dc, 12fc, 12daa, 12dab, 12cba: метод D (для 12dc и 12fc) [фиг. 11]
R1=CH3 (12dc)
R1=Cl (12fc)
В раствор 12da или 12fa (200 мг) в THF (20 мл) добавляли хлорид алюминия (10 экв.). Реакционную смесь перемешивали в течение ночи. Добавляли воду, после чего экстрагировали этилацетатом. Органический слой сушили над сульфатом магния и упаривали. Остаток подвергали колоночной флэш-хроматографии (гексан : этилацетат = 1:1), получая желтовато-белое твердое вещество. Выход: 60%-80%.
Синтез 12daa, 12dab, 12cba: метод Е [фиг. 11]
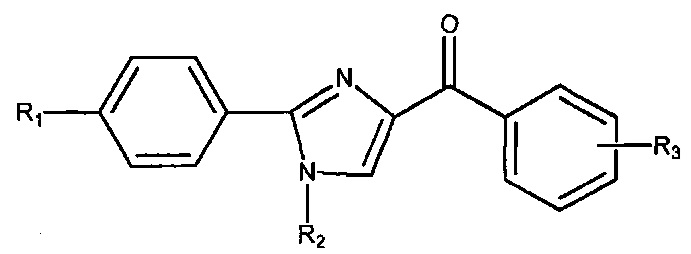
R1=Me; R2=Bn; R3=3,4,5-(OMe)3 (12daa)
R1=Me; R2=CH3; R3=3,4,5-(OMe)3 (12dab)
R1=OMe; R2=CH3; R3=F (12cba)
В раствор 12da или 12cb (100 мг) в THF (10 мл) на ледяной бане добавляли гидрид натрия (1,2 экв.), после чего добавляли метилйодид (для 12dab, 12cba) или бензилбромид (для 12daa) (2 экв.). Полученную реакционную смесь перемешивали 5 ч при кипячении с обратным холодильником. После разбавления 50 мл насыщенного раствора NaHCO3 (водного) реакционную смесь экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 2:1), получая белое твердое вещество. Выход: 50%-98%. 12daa: выход 92,8%; т.пл. 135-137°C. 1Н-ЯМР (CDCl3, 500 MHz) δ 7,81 (s, 1Н), 7,80 (d, J=6,5 Hz, 2H), 7,58 (d, J=8,0 Hz, 2H), 7,41-7,45 (m, 3H), 7,31-7,33 (m, 2H), 7,20 (d, J=7,0 Hz, 2H), 5,33 (s, 2H), 3,99 (s, 3H), 3,98 (s, 6H), 2,47 (s, 3H). MS (ESI): теор. 442,2 для C27H26N2O4, факт. 443,1 [М+Н]+. HPLC1: tR 4,28 мин, чистота >99%.
Синтез 11gaa и 12la (фиг. 12)
R1=N(Me)2; R2=(4-OMe)PhSO2 (11gaa)
R1=Br; R2=H(12la)
Замещенные бензальдегидные соединения 8(l, g) превращали в соединения 9(l, g) в присутствии гидроксида аммония и глиоксаля, получая имидазоловый каркас. Кольца имидазола у соединений 9(l, g) блокировали соответствующей фенилсульфонильной группой, после чего конъюгировали с 3,4,5-триметоксибензоилхлоридом, получая соединения 11(la, gaa). Обработка 11la фторидом трет-бутиламмония для удаления защитной группы давала 12la.
Синтез (2-(4-бромфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la) (фиг. 12)
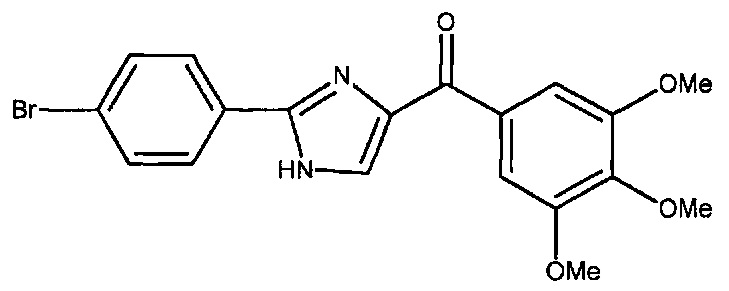
Синтез 9l, 9g. В раствор соответствующего бензальдегида (8l или 8g, 100 ммоль) в этаноле (400 мл) при 0°C добавляли 40% раствор оксалальдегида (глиоксаля) в воде (1,1 экв.) и 29% раствор гидроксида аммония в воде (10 экв.). После перемешивания 2-3 дня при комнатной температуре реакционную смесь упаривали, а остаток подвергали колоночной флэш-хроматографии с дихлорметаном в качестве элюента, получая искомое соединение в виде желтого порошка. Выход: 10%-30%.
Синтез 10la, 10gb. В раствор имидазола (9l, 9g) (10 ммоль) в безводном THF (200 мл) при 0°C добавляли гидрид натрия (60% дисперсия в минеральном масле, 1,2 экв.) и перемешивали в течение 20 мин. Добавляли 4-метоксибензолсульфонилхлорид (для 10gb) или бензолсульфонилхлорид (для других) (1,2 экв.) и перемешивали реакционную смесь в течение ночи. После разбавления 200 мл насыщенного раствора NaHCO3 (водного) реакционную смесь экстрагировали этилацетатом (600 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 2:1), получая бледное твердое вещество. Выход: 40%-95%.
Синтез 11la, 10gaa. В раствор 2-арил-1-(фенилсульфонил)-1Н-имидазола (10la, 10gb) (5,0 ммоль) в безводном THF (30 мл) при -78°C добавляли 1,7 М трет-бутиллитий в пентане (1,2 экв.) и перемешивали 10 мин. Добавляли 3,4,5-триметоксибензоилхлорид (1,2 экв.) при -78°C и перемешивали в течение ночи. Реакционную смесь разбавляли 100 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (300 мл). Органический слой сушили над сульфатом магния и упатровали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 3:1), получая белое твердое вещество. Выход: 5%-45%.
Синтез 12la. В раствор арил-(2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)-метанона (11la) (2,0 ммоль) в THF (25,0 мл) добавляли 1,0 М фторид тетрабутиламмония (2 экв.) и перемешивали в течение ночи. Реакционную смесь разбавляли 60 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (150 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 4:1) или перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 80-98%.
Синтез (4-фторфенил)(2-(4-метоксифенил)-1Н-имидазол-4-ил)метанона (12cb) (фиг. 8)
В раствор (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11cb, 872 мг, 2,0 ммоль) в THF (20,0 мл) добавляли 1,0 М фторид тетрабутиламмония (4,0 мл, 4,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли 50 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 90%; т.пл. 245-247°C.
Синтез (2-(n-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da) (фиг. 9)

В раствор (1-(фенилсульфонил)-2-(n-толил)-1Н-имидазол-4-ил)(3,4,5-триметокси-фенил)метанона (11da, 492 мг, 1,0 ммоль) в THF (15,0 мл) добавляли 1,0 М фторид тетрабутиламмония (2,0 мл, 2,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли 30 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (80 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 88,5%.
Синтез (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa) (фиг. 9 и 13)
2-(4-Хлорфенил)-1Н-имидазол (9f). В раствор 4-хлорбензальдегида (8f) (100 ммоль) в этаноле (350 мл) при 0°C добавляли 40% раствор оксалальдегида в воде (12,8 мл, 110 ммоль) и 29% раствор гидроксида аммония в воде (1000 ммоль, 140 мл). После перемешивания 2-3 дня при комнатной температуре, реакционную смесь упаривали, а остаток подвергали колоночной флэш-хроматографии с дихлорметаном в качестве элюента, получая искомое соединение в виде желтого порошка. Выход: 19,8%. 1H-ЯМР (500 MHz, DMSO-d6) δ 13,60 (br, 1Н), 7,94 (d, J=8,5 Hz, 2H), 7,51 (d, J=8,0 Hz, 2H), 7,27 (s, 1H), 7,03 (s, 1H). MS (ESI): теор. 178,0 для C9H7ClN2, факт. 178,9 [M+H]+.
2-(4-Хлорфенил)-1-(фенилсульфонил)-1Н-имидазол (10f). В раствор 2-(4-хлорфенил)-1Н-имидазола (9f) (20 ммоль) в безводном THF (200 мл) при 0°C добавляли гидрид натрия (60% дисперсия в минеральном масле, 1,2 г, 30 ммоль) и перемешивали в течение 30 мин. Добавляли бензолсульфонилхлорид (2,82 мл, 22 ммоль) и перемешивали реакционную смесь в течение ночи. После разбавления 100 мл насыщенного раствора NaHCO3 (водного) реакционную смесь экстрагировали этилацетатом (500 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 2:1), получая бледное твердое вещество. Выход: 54,9%. 1H-ЯМР (500 MHz, CDCl3) δ 7,65 (d, J=2,0 Hz, 1H), 7,58 (t, J=7,5 Hz, 1H), 7,43 (d, J=8,5 Hz, 2H), 7,38 (t, J=8,0 Hz, 2H), 7,34-7,36 (m, 4H), 7,12 (d, J=1,5 Hz, 1H). MS (ESI): теор. 318,0 для C15H11ClN2O2S, факт. 341,0 [M+Na]+.
(2-(4-Хлорфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11fa). В раствор 2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазола (10f) (6,0 ммоль) в безводном THF (30 мл) при -78°C добавляли 1,7 М трет-бутиллитий в пентане (5,3 мл, 9,0 ммоль) и перемешивали в течение 10 мин. Добавляли 3,4,5-триметоксибензоилхлорид (7,2 ммоль) при -78°C и перемешивали в течение ночи. Реакционную смесь разбавляли 100 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 4:1), получая белое твердое вещество. Выход: 36,8%. 1H-ЯМР (500 MHz, CDCl3) δ 8,05 (d, J=7,5 Hz, 2H), 7,77 (t, J=7,5 Hz, 1H), 7,62 (t, J=8,0 Hz, 2H), 7,48 (s, 1H), 7,44 (d, J=9,0 Hz, 2H), 7,39 (d, J=8,5 Hz, 2H), 7,37 (s, 2H). MS (ESI): теор. для C25H21ClN2O6S 512,1, факт. 513,1 [M+H]+.
(2-(4-Хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa). В раствор (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)-метанона (12fa) (2,0 ммоль) в THF (20,0 мл) добавляли 1,0 М фторида тетрабутиламмония (4,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли 50 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 3:1) или перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 80-95%. Выход: 36,9%; т.пл. 193-195°C. 1H-ЯМР (500 MHz, CDCl3) δ 10,75 (br, 1H), 7,96 (d, J=8,5 Hz, 2H), 7,83 (s, 1H), 7,47 (d, J=9,0 Hz, 2H), 7,23 (s, 2H), 3,97 (s, 3H), 3,94 (s, 6H), 2,43 (s, 3H). MS (ESI): теор. 372,1 для C19H17ClN2O4, факт. 395,1 [M+Na]+, 370,9 [M-H]-. HPLC с градиентом: растворитель A (вода) и растворитель B (метанол): 0-15 мин 40-100% В (линейный градиент), 15-25 мин 100% В; tR 16,36 мин, чистота >99%.
Синтез (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12fb) (фиг. 9)
В раствор (2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)-метанона (11fb, 440 мг, 1,0 ммоль) в THF (12,0 мл) добавляли 1,0 М фторид тетрабутиламмония (2,0 мл, 2,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли 20 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (60 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 83,7%.










ПРИМЕР 3. Синтез (индолил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенилметанонов 17уа, 17yab и 17уас (фиг. 14)
Синтез (2-(1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17уа)
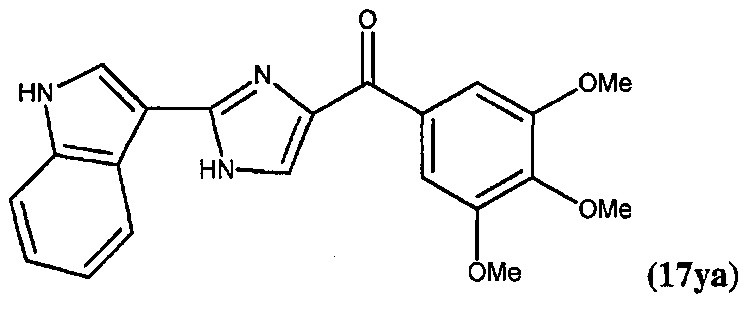
Синтез 1-(фенилсульфонил)-1Н-индол-3-карбоксальдегида (8уа). В раствор индол-3-карбоксальдегида (8у) (100 ммоль) в этаноле (500 мл) при комнатной температуре добавляли гидроокись калия (1,1 экв.). Смесь перемешивали до полного растворения. Этанол полностью удаляли под вакуумом, а остаток растворяли в ацетоне (250 мл), после чего добавляли бензолсульфонилхлорид (1,1 экв., 110 ммоль). Реакционную смесь перемешивали в течение получаса. Осадок отфильтровывали, а фильтрат упаривали и перекристаллизовывали из метанола, получая белое твердое вещество. Выход: 33%. 1H-NMR (500 MHz, CDCl3) δ 10,17 (s, 1Н), 8,25-8,39 (m, 2H), 7,97-8,09 (m, 3H), 7,69 (t, J=7,33 Hz, 1H), 7,59 (t, J=7,5 Hz, 2H), 7,39-7,54 (m, 2H). MS (ESI): теор. 285,1 для C15H11NO3S, факт. 286,0 [M+H]+.
Синтез 3-(1Н-имидазол-2-ил)-(фенилсульфонил)-1Н-индола (9уа). В раствор 1-(фенилсульфонил)-1H-индол-3-карбоксальдегида (8уа) (100 ммоль) в этаноле (400 мл) при 0°C добавляли 40% раствор оксалальдегида (глиоксаля) в воде (1,1 экв., 110 ммоль) и 29% раствор гидроксида аммония в воде (10 экв., 1000 ммоль). После перемешивания в течение 2-3 дней при комнатной температуре реакционную смесь гасили водой и экстрагировали дихлорметаном. Органический слой удаляли под вакуумом, а остаток подвергали колоночной флэш-хроматографии с гексан/этилацетатом (4:1-2:1) в качестве элюента, получая искомое соединение в виде желтого порошка. Выход: 12%. 1H-NMR (500 MHz, DMSO-d6) δ 8,33 (d, J=2,9 Hz, 2H), 8,13 (d, J=7,8 Hz, 2H), 7,98-8,04 (m, 1H), 7,62-7,67 (m, 1H), 7,55 (d, J=7,82 Hz, 2H), 7,22-7,34 (m, 4H). MS (ESI): теор. 323,1 для C17H13N3O2S, факт. 324,0 [M+H]+.
Синтез 1-(фенилсульфонил)-3-(1-(фенилсульфонил)-1H-имидазол-2-ил)-1H-индола (10уа). В раствор 3-(1Н-имидазол-2-ил)-1-(фенилсульфонил)-1H-индола (9уа) (20 ммоль) в безводном THF (300 мл) при 0°C добавляли гидрид натрия (60% дисперсия в минеральном масле, 1,2 экв., 24 ммоль) и перемешивали в течение 20 мин. Добавляли бензолсульфонилхлорид (1,2 экв., 24 ммоль) и перемешивали реакционную смесь в течение ночи. После разбавления 200 мл насыщенного раствора NaHCO3 (водного) реакционную смесь экстрагировали этилацетатом (600 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 5:1), получая белое твердое вещество. Выход: 40%. 1H-NMR (CDCl3, 300 MHz) δ 8,02-8,08 (m, 4Н), 7,72 (d, J=1,5 Hz, 1H), 7,35-7,60 (m, 8H), 7,23 (d, J=1,5 Hz, 1H), 7,10-7,16 (m, 3H). MS (ESI): теор. 463,1 для C23H17N3O4S2, факт. 486,0 [M+Na]+.
Синтез (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1H-индол-3-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17уаа). В раствор 1-(фенилсульфонил)-3-(1-(фенилсульфонил)-1Н-имидазол-2-ил)-1H-индола (10уа) (5,0 ммоль) в безводном THF (100 мл) при -78°C добавляли 1,7 М трет-бутиллитий в пентане (1,2 экв., 6,0 ммоль) и перемешивали в течение 10 мин. Добавляли раствор 3,4,5-триметоксибензоилхлорида (1,2 экв., 6,0 ммоль) в THF при -78°C и перемешивали в течение ночи. Реакционную смесь гасили 100 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (300 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 3:1), получая белое твердое вещество. Выход: 30%. 1H-NMR (500 MHz, CDCl3) δ 8,09 (d, J=10 Hz, 1H), 8,04 (d, J=10 Hz, 2H), 7,91 (s, 1H), 7,76 (d, J=5 Hz, 2H), 7,65 (t, J=10 Hz, 1H), 7,55-7,58 (m, 5H), 7,40 (s, 2H), 7,33-7,36 (m, 3H), 7,25 (t, J=10 Hz, 1H), 4,05 (s, 3H), 4,03 (s, 6H). MS (ESI): теор. 657,0 для C33H27N3O8, факт. 680,1 [M+Na]+.
Синтез (2-(1H-индол-3-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17уа). В раствор (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1H-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17уаа) (1 ммоль) в этаноле (40 мл) и воде (4 мл) добавляли гидроокись натрия (10 экв., 10 ммоль) и перемешивали в течение ночи при кипячении с обратным холодильником в темноте. Реакционную смесь разбавляли 50 мл воды и экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 1:1), получая желтое твердое вещество. Выход: 60%. 1H-NMR (500 MHz, CD3OD) δ 8,31 (d, J=6,5 Hz, 1H), 7,99 (s, 1H), 7,90 (s, 1H), 7,48-7,52 (m, 3Н), 7,24-7,28 (m, 2H), 4,00 (s, 6H), 3,93 (s, 3H). MS (ESI): теор. 377,1 для C21H19N3O4, факт. 400,1 [M+Na]+. Т.пл. 208-210°C.
Синтез (2-(1-(фенилсульфонил)-1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-три-метоксифенил)метанона (17yab)
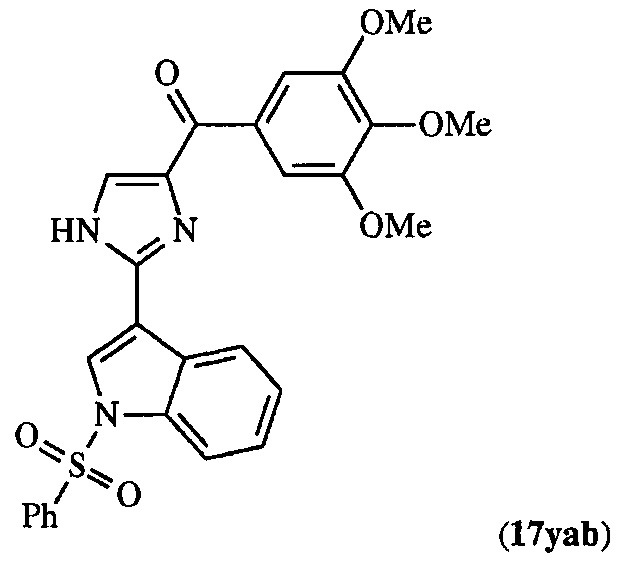
В раствор соединения 17уаа (66 мг) в THF (1,0 мл) добавляли 1,0 М фторид тетрабутиламмония (0,4 мл, 0,4 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли 20 мл насыщенного раствора NaHCO3 (водного) и экстрагировали этилацетатом (20 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан : этилацетат = 2:1), получая беловатое твердое вещество. Выход: 45%. Т.пл. 110-112°C. 1H-NMR (CDCl3, 500 MHz) δ 8,40-8,42 (m, 2Н), 8,09 (d, J=8,0 Hz, 1H), 7,93-7,98 (m, 4H), 7,59 (t, J=7,5 Hz, 1H), 7,41-7,49 (m, 5H), 4,01 (s, 3H), 3,97 (s, 6H). MS (ESI): теор. 517,1 для C27H23N3O6S, факт. 540,0 [M+Na]+. HPLC: tR 6,81 мин, чистота 96,3%.
Синтез (1-метил-2-(1-(метил)-1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-три-метоксифенил)метанона (17уас)
В раствор 17уа (75 мг, 0,2 ммоль) в безводном THF (20 мл) при 0°C добавляли гидрид натрия (60% дисперсия в минеральном масле, 20 мг, 0,5 ммоль) и перемешивали в течение 20 мин. Добавляли метилйодид (70 мг, 0,5 ммоль) и перемешивали реакционную смесь в течение 1 ч. После разбавления 20 мл насыщенного раствора NaHCO3 (водного) реакционную смесь экстрагировали этилацетатом (60 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 75%. Т.пл. 164-166°C. 1H-NMR (CDCl3, 500 MHz) δ 8,30 (d, J=7,5 Hz, 1H), 8,01 (s, 1H), 7,87 (s, 1H), 7,41 (t, J=8,5 Hz, 1H), 7,39 (s, 1H), 7,35 (t, J=7,0 Hz, 1H), 7,23 (t, J=7,0 Hz, 1H), 3,98 (s, 6H), 3,95 (s, 3H), 3,91 (s, 3H), 3,89 (s, 3H). MS (ESI): теор. 405,2 для C23H23N3O4, факт. 406,4 [M+H]+. HPLC: tR 4,80 мин, чистота >99%.
ПРИМЕР 4. Антипролиферативная активность отобранных ABI-соединений по изобретению
Анализ цитотоксичности на клеточных культурах
Материалы и методы
Исследовали антипролиферативную активность ABI-соединений на трех линиях клеток меланомы (A375 и WM-164, линии клеток меланомы человека; B16-F1, линия клеток меланомы мыши) и четырех линиях клеток рака простаты человека (LNCaP, DU 145, РС-3 и РРС-1). Все эти линии клеток были получены из АТСС (American Type Culture Collection, Manassas, VA), за исключением клеточной линии РРС-1. Клетки MDA-MB-435 и MDA-MB-435/LCCMDR1 были любезно предоставлены Dr. Robert Clarke из Georgetown University School of Medicine, Washington, DC. Клетки меланомы культивировали в среде DMEM (Cellgro Mediatech, Inc., Herndon, VA), а клетки рака простаты культивировали в среде RPMI 1640 (Cellgro Mediatech, Inc., Herndon, VA) с добавлением 10% FBS (Cellgro Mediatech). Культуры содержали при 37°C в увлажненной атмосфере, содержащей 5% CO2. В каждую лунку 96-луночных планшетов высеивали от 1000 до 5000 клеток в зависимости от скорости роста и подвергали воздействию различных концентраций исследуемых соединений в течение 48 ч (быстро растущие клетки меланомы) или 96 ч (медленно растущие клетки рака простаты) в 3-5 повторах. Количество клеток в конце обработки препаратом измеряли методом анализа с сульфородамином В (SRB). Вкратце, клетки фиксировали 10% трихлоруксусной кислотой и окрашивали 0,4% SRB, а затем измеряли оптическую плотность при 540 нм на считывающем устройстве (Dynex Technologies, Chantilly, VA). Строили графики выживаемости клеток в зависимости от концентрации препарата и получали значения IC50 (концентрации, при которой рост клеток ингибируется на 50% от необработанного контроля) методом нелинейной регрессии с помощью программы GraphPad Prism (GraphPad Software, San Diego, CA).
Результаты
Результаты по антипролиферативнй активности соединений по изобретению in vitro на трех линиях клеток меланомы (одна линия клеток меланомы мыши, B16-F1, и две линии клеток метастатической меланомы человека, A375 и WM-164) и четырех линиях клеток рака простаты человека (LNCaP, РС-3, Du 145 и РРС-1) приведены в табл. 8-10.
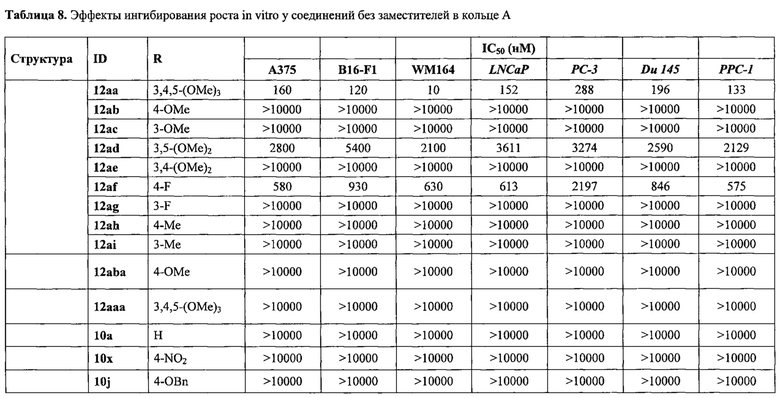
Как видно из табл. 8, соединения 12aa-12ai проявляли умеренную активность со значениями IC50 в мкМ диапазоне (средние из всех семи клеточных линий). Наиболее сильным из этого ряда было соединение 12аа со средним значением IC50 в 160 нМ. Удаление одной из метоксигрупп у 3,4,5-триметокси на кольце С (12ad, 12ае) приводило к значительной потере активности (IC50>10 мкМ для 12ае и среднее IC50 в 3,1 мкМ для 12ad). Соединение с 4-фтор на кольце С (12af) также проявляло сравнительно высокую активность (IC50=0,91 мкМ), что имеет важные последствия, поскольку замена группировки триметокси на группу 4-фтор может обеспечить хорошую активность и улучшение метаболической устойчивости. Положение фтора на кольце С имеет решающее значение для активности, поскольку переход от 4-фтор к 3-фтор приводит к полной потере активности (IC50>10 мкМ для 12ag по сравнению с 0,91 мкМ для 12af). Этот результат свидетельствует, что потенциальный донор водородной связи находится возле 4-го положения в этом кольце.
Из табл. 8 четко видно, что решающими являются положения в кольцах А и С. Простое перемещение С-кольцевой группировки из положения 4 в положение 1 в имидазольном кольце (кольцо В) приводит к полной потере активности (IC50>10 мкМ для 12aba, 12ааа, 10а, 10х, 10j).
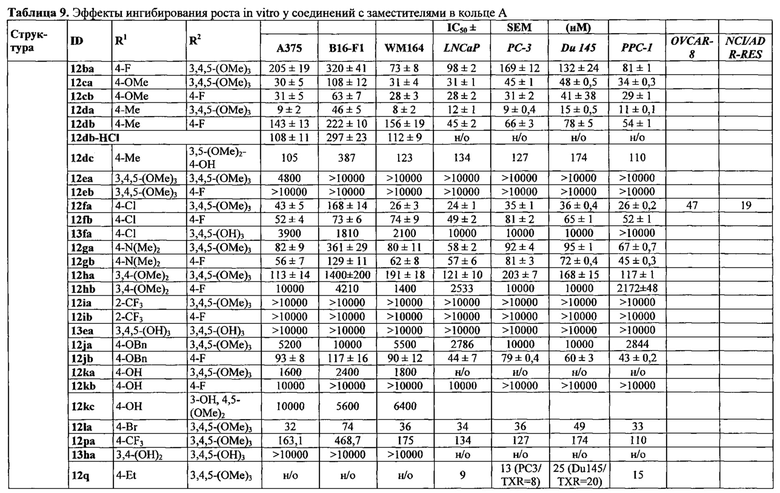

Как видно из табл. 9, соединения с заместителями 3,4,5-триметокси и 4-фтор у кольца C проявляли хорошую активность при различных заместителях у кольца А. Эти соединения проявляли отличную антипролиферативную активность с такими низкими значениями IC50, как 8,0 нМ, на клеточной линии WM164 (12da). В общем, соединения, включающие единственный заместитель в пара-положении кольца А, оказались более сильными, судя по активностям 12са, 12cb, 12da, 12db, 12fa, 12fb, 12ga и 12gb (IC50=7,9-110 нМ). HCl-соль 12db (IC50=172 нМ) проявляла немного меньшую активность, чем соответствующее свободное основание 12db (IC50=109 нМ). Соединение 12fb (IC50=63,7 нМ) с одним галогеновым заместителем в пара-положении у колец А и С оказалось сильным и без метоксигруппы. Соединения с 3,4,5-триметокси-заместителями в кольце А полностью теряли активность (IC50>10 мкМ для 12еа, 12eb), что свидетельствует об очень разном окружении для связывания возле кольца А и кольца С.Удаление 5-метокси-заместителя из кольца А значительно улучшало активность (IC50=330 нМ и >10 мкМ для 12ha, 12еа, соответственно). Деметилирование 3,4,5-триметоксигруппы резко снижало активность с 43 нМ (12fa) до 3,89 мкМ (13fa). Близкие результаты наблюдались для 13еа, 12ka, 12kb и 13ha при деметилировании заместителей у обоих колец A или C. Электроно-донорные группы (4-метокси, 4-диметиламино, 4-метил) и электроно-акцепторные группы (4-хлор, 2-трифторметил) у кольца А не проявляли существенных различий в активности. Введение трифторметильной группы в орто-положении кольца А вызывало полную потерю активности (IC50>10 мкМ для 12ia, 12ib). Наличие бензилоксигруппы в пара-положении кольца A (IC50=75 нМ для 12jb) приводило к 440-кратному возрастанию активности по сравнению с пара-гидрокси-соединением 12kb (IC50=33 мкм). Стоит отметить, что соединение 12jb с 4-фтор у кольца C обладает лучшей активностью, чем его аналог 12ja, содержащий 3,4,5-триметоксигруппу в кольце C (IC50=75 нМ для 12jb и 7,3 мкМ для 12ja).
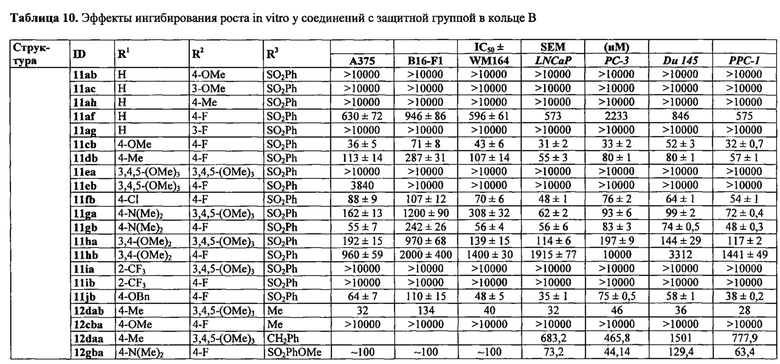
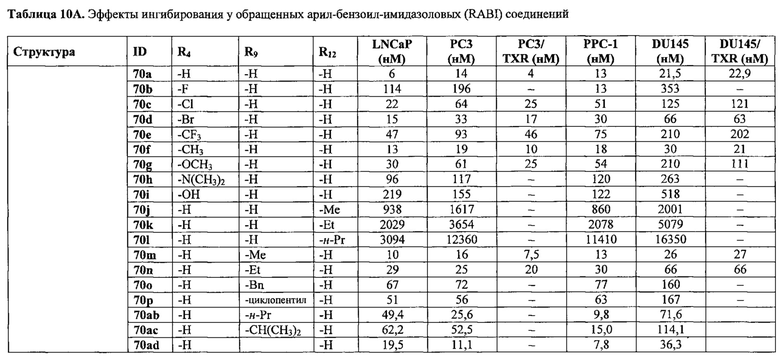
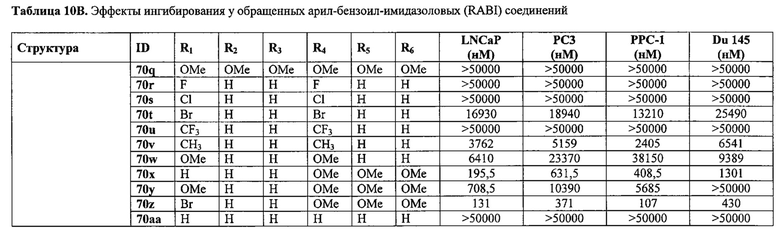
Как видно из табл. 10, соединения с защитной фенилсульфонильной группой, присоединенной к азоту в имидазольном кольце (11cb, 11db, 11fb, 11ga, 11gb, 11ha, 11jb), тоже были очень активными с IC50 в нМ диапазоне (табл. 10). Как правило, активность этих соединений сравнима с соответствующими незащищенными аналогами, о чем свидетельствует сравнение активности 11cb (43 нМ), 11db (111 нМ), 11fb (72 нМ), 11ga (285 нМ), 11gb (87 нМ), 11ha (268 нМ) и 11jb (61 нМ) с соответствующими незащищенными аналогами 12cb (36 нМ), 12db (109 нМ), 12fb (64 нМ), 12ga (131 нМ), 12gb (72 нМ), 12ha (330 нМ) и 12jb (75 нМ). Другие соединения (11ab-11ag, 11ea, 11eb, 11hb, 11ia и 11ib, 1-50 мкМ) в общем были гораздо менее активны, как и соответствующие их аналоги (12ab-12ag, 12еа, 12eb, 12hb, 12ia и 12ib, 1-50 мкМ).
Эффекты соединений по изобретению на распределение клеточного цикла у РС3 представлены на фиг. 15.
Анализ клеточного цикла
Распределение клеточного цикла определяли по окрашиванию йодидом пропидия (PI). Обработанные клетки промывали PBS и фиксировали 70% ледяным этанолом в течение ночи. Затем фиксированные клетки окрашивали 20 мкг/мл PI в присутствии РНКазы А (300 мкг/мл) при 37°C в течение 30 мин. Распределение клеточного цикла анализировали методом активированной флуоресценцией сортировки клеток (FACS) в Отделе базовых служб University of Tennessee Health Science Center, TN.
Результаты
Данные анализа клеточного цикла показывают, что обращенные ABIs (RABIs) блокируют клетки в фазе G2/M. Клетки РС3 обрабатывали соединениями 12q, 70а, 70f и 70m в течение 24 ч (фиг. 15) и исследовали распределение окрашенных PI клеток методом FACS. Для изучения эффекта дозы были выбраны четыре различные концентрации - 1, 10, 50 и 100 нМ каждого соединения. В группе, получавшей носитель, около 18% клеток РС3 оказались в фазе G2/M. Соединения RABIs повышали долю клеток в фазе G2/M вплоть до 70% примерно в зависимости от концентрации. Эффективность различных концентраций при блокировании клеток в фазе G2/M положительно коррелирует с ингибированием роста клеток in vitro. Антипролиферативная активность соединений RABIs представлена в табл. 10A и табл. 10B. Некоторые из RABIs проявляли довольно сильное антипролиферативное действие (например, соединение 70а).
ПРИМЕР 5. Действие арил-бензоил-имидазоловых (ABI) соединений на лекарственно-устойчивые клетки меланомы
Опосредованный Р-гликопротеином (Pgp) отток препаратов является главным механизмом у раковых клеток для предотвращения возрастания эффективной внутриклеточной концентрации противораковых препаратов. Сравнивали действие ABI-соединений на клетки меланомы (MDA-MB-435/LCCMDR1) со множественной лекарственной устойчивостью (MDR) и на исходные нерезистентные раковые клетки (MDA-MB-435). Хотя MDA-MB-435 была первоначально определена как линия раковых клеток молочной железы, однако было четко установлено, что она происходит из линии клеток меланомы М14. Соединения 12da, 12fb, 12cb, 11cb и 11fb вместе с другими антитубулиновыми препаратами, включая колхицин, паклитаксель и винбластин, тестировали и на MDR-линии клеток меланомы, и на исходной линии клеток меланомы (табл. 11A). Паклитаксель и винбластин клинически применяются как противораковые препараты, действующие на тубулин в клетках. Хотя колхицин и не является одобренным FDA препаратом для лечения рака, однако его пролекарство, ZD6126, проходит клинические испытания для твердых опухолей. Бортезомиб - первый терапевтический ингибитор протеасом, который в 2003 г. был одобрен FDA для применения при множественной миеломе. АВТ-751, как известно, нацелен на сайт связывания колхицина в тубулине. Он оказался перспективным препаратом-кандидатом при клинических испытаниях у детей с рецидивирующей или рефрактерной нейробластомой. Соединения 12da, 12fb, 12cb, 11cb, 11fb имели намного лучшие индексы устойчивости (3,0 для 12da, 0,9 для 12fb, 1,3 для 12cb, 0,8 для 11cb, 0,7 для 11fb), чем колхицин (65,8), паклитаксель (69,3) и винбластин (27,5). Хотя колхицин, паклитаксель и винбластин проявляли высокую активность на нерезистентной линии клеток меланомы (0,5-10 нМ), однако эти соединения были значительно менее сильными на MDR-линии клеток меланомы (277-658 нМ). Напротив, 12cb, 11cb, 11fb были практически одинаково эффективными как на MDR (15 нМ, 38 нМ, 30 нМ, 30 нМ, 35 нМ для 12da, 12fb, 12cb, 11cb и 11fb, соответственно), так и на нерезистентной линии клеток меланомы (5 нМ, 41 нМ, 24 нМ, 38 нМ, 50 нМ для 12da, 12fb, 12cb, 11cb и 11fb, соответственно). На клетках А375 и WM-164 соединение 12da было более активным, чем паклитаксель и колхицин.
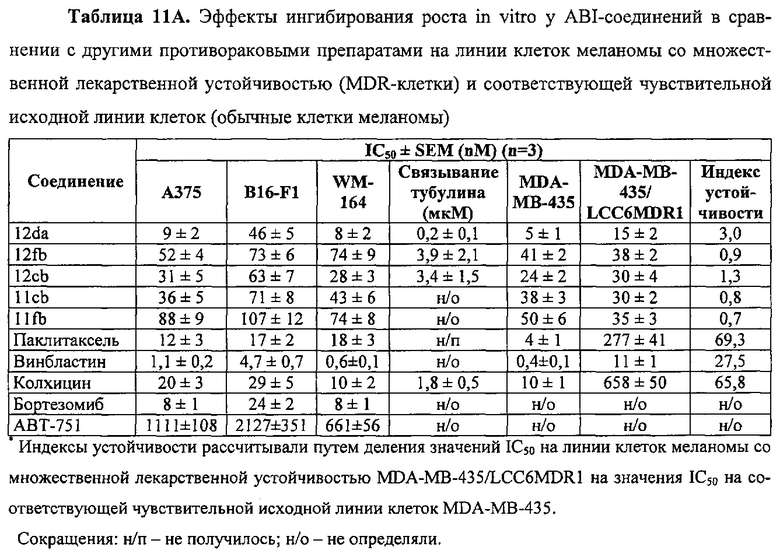
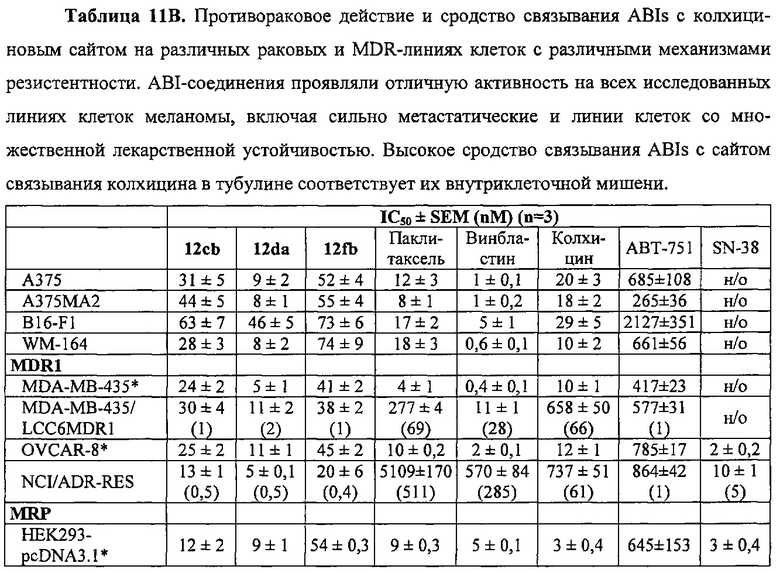
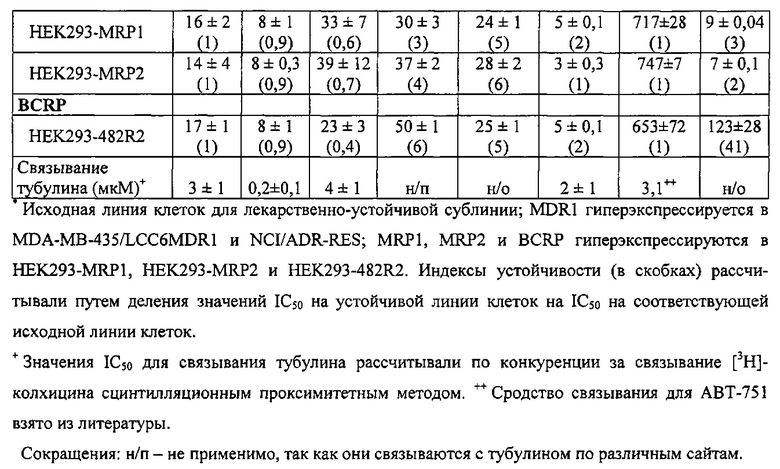
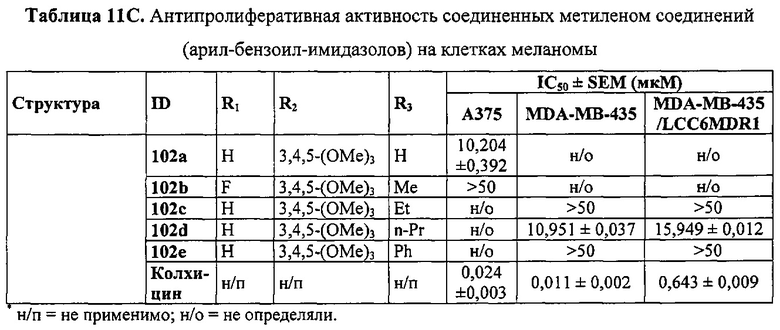
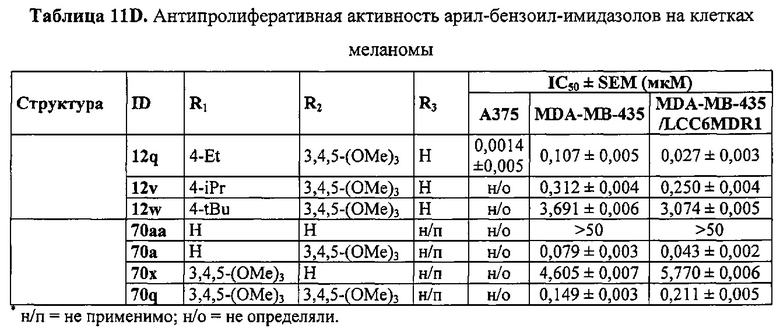
Из результатов в табл. 11A видно, что линия клеток MDA-MB-435/LCCMDR1 очень устойчива к колхицину, паклитакселю и винбластину. Но ABIs по изобретению проявляли одинаковую активность и на лекарственно-устойчивой линии клеток, и на чувствительной исходной линии клеток. Этот результат убедительно свидетельствует о том, что ABIs не являются субстратами для P-gp. Так, они преодолевают множественную лекарственную устойчивость у клеток MDA-MB-435/LCCMDR1. Кривые доза-ответ для 12fb, 12da, и 12cb представлены на фиг. 16. В табл. 11B более подробно исследованы механизмы устойчивости для паклитакселя, SN-38, винбластина и колхицина в сравнении ABI-соединениями 12cb, 12da и 12fb. MRP и BCRP придавали умеренную устойчивость к паклитакселю (индексы устойчивости 4 и 6, соответственно) и винбластину (индексы устойчивости 6 и 5, соответственно), a BCRP придавал значительную устойчивость к SN-38 (индекс устойчивости 41). Однако ни одно из ABI-соединений не было чувствительным к опосредованной MRP или BCRP устойчивости (индексы устойчивости от 0,4 до 1,0). АВТ-751, как и ABIs, не был чувствительным к MDR1, MRP или BCRP.
ПРИМЕР 6. Анализ полимеризации микротрубочек in vitro
Материалы и методы
Тубулин из мозга быка (0,4 мг) (Cytoskeleton, Denver, CO) смешивали с 10 мкМ исследуемого соединения и инкубировали в 110 мкл общего буфера для тубулина (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA и 1 мМ GTP) при pH 6,9. Измеряли поглощение при 340 нм через каждую 1 мин на протяжении 15 мин на считывающем устройстве Synergy 4 Microplate Reader (Bio-Tek Instruments, Winooski, VT). Для полимеризации тубулина спектрофотометр устанавливали на 37°C.
Результаты
Исследовали ингибирование полимеризации тубулина арил-бензоил-имидазоловыми (ABI) соединениями. Тубулин из мозга быка (чистота >97%) инкубировали с тремя сильнодействующими ABI-соединениями 12cb, 12da и 12db при концентрации 10 мкМ, чтобы определить влияние этих ABI-соединений на полимеризацию тубулина (фиг. 17). Полимеризация тубулина полностью ингибировалась соединением 12da, тогда как при инкубации с соединениями 12cb и 12db наблюдалось ингибирование на ~80%.
Эффект дестабилизации микротрубочек был аналогичен действию колхицина и винбластина, но противоположен эффекту паклитакселя. Результаты не только подтверждают, что ABIs могут непосредственно взаимодействовать с тубулином, но и свидетельствуют о том, что у них может быть общий с колхицином (или винбластином) сайт связывания.
ПРИМЕР 7. Ингибирование меланомы in vitro
Материалы и методы
Клетки меланомы B16-F1 высеивали при колониеобразующей плотности (2000 клеток на лунку в 6-луночных планшетах) на основу из 0,8% агара. Клетки культивировали в 0,4% агаре со средой DMEM с добавлением эмбриональной бычьей сыворотки и раствора антибиотиков и противогрибковых средств при 37°C в атмосфере из 95% воздуха и 5% CO2. Клетки обрабатывали соединениями 12da, 12cb и 12fb при различных концентрациях (20, 100 и 500 нМ). Соединения добавляли в среды из исходных 1 мМ растворов в DMSO, а в качестве контроля использовали соответствующее разбавление DMSO. Клетки культивировали на протяжении 14 дней. Планшеты фотографировали и измеряли количество колоний на автоматизированном счетчике Artek 880 Automated Colony Counter (Artek Systems Corporation, Farmingdale, NY).
Результаты
На фиг. 18 представлены четыре репрезентативные фотографии. После 14 дней инкубации в контролях (без обработки) обнаруживалось около 130 колоний (диаметром более 100 мкм).
Соединения 12cb и 12da эффективно ингибировали образование колоний меланомы B16-F1 даже при самой низкой исследуемой концентрации в 20 нМ (р<0,05 по сравнению с контролем). Соединение 12fb проявляло эффективное ингибирование при 100 нМ. Все три исследованных соединения проявляли полное ингибирование образования колоний при 0,5 мкМ, что дополнительно свидетельствует об эффективности ABIs против меланомы.
ПРИМЕР 8. Противоопухолевая активность in vivo
Материалы и методы
Животные. Самок мышей C57/BL в возрасте 4-6 недель приобретали у Harlan Laboratories (Harlan Laboratories Inc., Indianapolis, IN). Содержание животных отвечало требованием Association for Assessment and Accreditation and Laboratory Animal Care. Все процедуры проводились в соответствии с указаниями нашего Institutional Animal Care and Use Committee.
Оценка эффективности in vivo. Клетки меланомы B16-F1 мыши в лишенной FBS среде DMEM (Cellgro Mediatech) доводили до концентрации 5×106 жизнеспособных клеток/мл. Суспензию клеток (100 мкл) вводили подкожно в правый дорзальный бок каждой мыши. Когда опухоли достигали размера 100-150 мм3, примерно через 7 дней после инокуляции клеток, всех мышей, несущих опухоли, разделяли на группы обработки и контроля на основании размера опухолей (n=5 на группу). В каждой группе опухоли имели одинаковый средний размер. Мышам в контрольной группе (отрицательный контроль) вводили внутрибрюшинно 50 мкл раствора только носителя или DTIC в дозе 60 мг/кг (положительный контроль) один раз в день. Объем опухолей измеряли через каждые 2 дня с помощью регистрирующего электронного цифрового штангенциркуля (Fisher Scientific, Inc., Pittsburgh, PA) и рассчитывали по формуле а×b×0,5, где а и b означают больший и меньший диаметр, соответственно. Объем опухолей выражали в кубических миллиметрах. Данные выражали в виде среднего ± SE для каждой группы и наносили на график зависимости от времени. Снижение размера опухолей в процентах по завершении эксперимента (через 14 дней после начала обработки) рассчитывали по формуле 100-100×[(Т-Т0)/(С-C0)], где T означает средний объем опухолей в группе обработки в определенный день, T0 - средний объем опухолей в той же группе в первый день обработки, C - средний объем опухолей в контроле в определенный день, а C0 - средний объем опухолей в той же группе в первый день обработки. Для оценки токсичности соединений отслеживали активность животных и средний вес тела в каждой группе на протяжении всего эксперимента. По окончании обработки всех мышей забивали с помощью CO2 с последующим смещением шейных позвонков и извлекали опухоли для дальнейших исследований.
Результаты
Для оценки эффективности аналогов ABI in vivo исследовали противоопухолевую активность соединения 12cb на мышах с ксенотрансплантатами меланомы B16-F1. В качестве положительного контроля использовали DTIC, золотой стандарт при лечении злокачественной меланомы (фиг. 19А). Двадцать самок мышей C57/BL разделяли на 4 группы: группа контроля на носитель, группа обработки DTIC (60 мг/кг), группа обработки 12cb (10 мг/кг) и группа обработки 12cb (30 мг/кг). Каждой мыши подкожно вводили 0,5 млн. клеток меланомы B16-F1. Через 7 дней после прививки опухолей начинали обработку введением каждого соединения внутрибрюшинно раз в день (фиг. 19). Объем опухолей значительно (р<0,05) снижался на 47%, 51% и 73% для 12cb (10 мг/кг), DTIC (60 мг/кг) и 12cb (30 мг/кг), соответственно, после 14 дней обработки. Ни в одной из групп обработки не наблюдалось никакой существенной потери веса за время эксперимента.
Было выбрано два уровня доз 12fb: 15 и 45 мг/кг. В качестве положительного контроля использовали DTIC в дозе 60 мг/кг. Сначала для исследования была выбрана модель аллотрансплантатов меланомы B16-F1 на мышах C57BL/6. После 13 дней обработки (фиг. 19B) соединение 12fb тормозило рост опухоли меланомы (значение TGI) на 32% при 15 мг/кг и на 82% при 45 мг/кг. Значение p по t-критерию Стьюдента для 12fb при 45 мг/кг по сравнению с контролем составляло менее 0,001, что означает значимое отличие. Значение p по t-критерию для 12fb при 15 мг/кг по сравнению с контролем составляло 0,08, свидетельствуя, что эта доза была неэффективной. При сравнении 12fb в дозе 45 мг/кг с DTIC в дозе 60 мг/кг, у которого TGI было равно 51%, значение p по t-критерию составляло около 0,001, свидетельствуя, что 12fb обладало значительно лучшей активностью, чем DTIC. В контрольной группе и группе обработки 12fb в дозе 15 мг/кг средняя масса тела слегка увеличилась за время эксперимента.
Для дальнейшего подтверждения активности ABIs in vivo использовали модель ксенотрансплантатов меланомы А375 человека на мышах SHO и исследовали 12fb в дозе 25 мг/кг. В качестве положительного контроля опять использовали DTIC в дозе 60 мг/кг. После 31 дня обработки (фиг. 19C) соединение 12fb тормозило рост опухолей меланомы (значение TGI) на 69%, тогда как DTIC тормозил рост на 52%. Значение p t теста для 12fb в сравнении с контролем было меньше, чем 0,001, предполагая, что 12fb значительно ингибирует рост опухоли меланомы в дозе 25 мг/кг. Значение p по t-критерию для 12fb при сравнении с DTIC составляло менее 0,05, опять же свидетельствуя, что 12fb обладало лучшей активностью, чем DTIC. Средняя масса тела во всех группах слегка увеличилась за время эксперимента. Физическая активность у мышей также выглядела нормальной, свидетельствуя, что доза в 25 мг/кг хорошо переносится мышами SHO.
ПРИМЕР 9. Фармакология соединений 17уа, 12fa и 55 in vitro и in vivo
Материалы и методы
Культуры клеток рака простаты и анализ цитотоксичности. Все клеточные линии рака простаты (LNCaP, РС-3, DU145, РРС-1) получали из ATCC (American Type Culture Collection, Manassas, VA, USA). Линия PC-3_TxR человека была устойчива к паклитакселю и использовалась в качестве модели MDR для сравнения с РС-3. Материалы для клеточных культур приобретали у Cellgro Mediatech (Herndon, VA, USA). Все клеточные линии использовались для тестирования антипролиферативной активности соединений 17уа, 12fa и 55 методом анализа с сульфородамином В (SRB). Все линии раковых клеток поддерживали в среде RPMI 1640 с 2 мМ глутамина и 10% эмбриональной бычьей сыворотки (FBS).
Анализ полимеризации микротрубочек in vitro. Тубулин из мозга свиньи (0,4 мг) (Cytoskeleton, Denver, СО) смешивали с 1 и 5 мкМ исследуемого соединения или носителя (DMSO) и инкубировали в 100 мкл буфера (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA, pH 6,9, и 1 мМ GTP). Измеряли поглощение при 340 нм через каждую 1 мин на протяжении 15 мин (считывающее устройство Synergy 4 Microplate Reader, Bio-Tek Instruments, Winooski, VT). Для полимеризации тубулина спектрофотометр устанавливали на 37°C.
Метаболические инкубации. Исследования по метаболической стабильности проводили путем инкубации 0,5 мкМ исследуемого соединения в общем объеме реакции 1 мл, содержащем 1 мг/мл микросомального белка в реакционном буфере [0,2 М раствор фосфатного буфера (pH 7,4), 1,3 мМ NADP+, 3,3 мМ глюкозо-6-фосфата и 0,4 ед./мл глюкозо-6-фосфатдегидрогеназы] при 37°C на водяной бане с качалкой. Систему регенерации NADPH (растворы A и B) получали от BD Biosciences (Bedford, MA). Для исследования глюкуронизации инкубировали кофактор - 2 мМ UDP-глюкуроновой кислоты (Sigma, St. Louis, МО) в деионизированной воде с 8 мМ MgCl2, 25 мкг аламетицина (Sigma, St. Louis, МО) в деионизированной воде и регенерирующими NADPH растворами (BD Biosciences, Bedford, MA), как описано ранее. Общая концентрация ДМСО в реакционном растворе составляла примерно 0,5% (об/об). Из реакционной смеси отбирали аликвоты (100 мкл) для определения метаболической стабильности через 5, 10, 20, 30, 60 и 90 мин. Для гашения реакции и осаждения белков добавляли ацетонитрил (150 мкл), содержащий 200 нМ внутреннего стандарта. Затем образцы центрифугировали при 4000×g в течение 30 мин при комнатной температуре, а супернатант подвергали анализу непосредственно методом LC-MS/MS.
Аналитический метод. Раствор образца (10 мкл) вводили в систему HPLC серии Agilent (Agilent 1100 Series Agilent 1100 Chemstation, Agilent Technology Co, Ltd). Bee аналиты разделяли на узкоканальной колонке С18 (Alltech Alltima HP, 2,1×100 мм, 3 мкм, Fisher, Fair Lawn, NJ). Использовали режим из двух градиентов. Для исследований по метаболической стабильности использовали режим градиентов для разделения аналитов с использованием смесей подвижной фазы A [ACN/H2O=5%/95%, об/об, содержащей 0,1% муравьиной кислоты] и подвижной фазы B [ACN/H2O=95%/5%, об/об, содержащей 0,1% муравьиной кислоты] при скорости потока 300 мкл/мин. Использовали 10% подвижной фазы A от 0 до 1 мин, после чего следовал запрограммированный линейно градиент до 100% подвижной фазы B за 4 мин, поддерживая 100% подвижной фазы B в течение 0,5 мин, а затем быстрый откат до 10% подвижной фазы A. Подвижная фаза A продолжалась еще 10 мин до самого конца анализа.
Использовали трехквадрупольный масс-спектрометр API Qtrap 4000™ (Applied Biosystems/MDS Sciex, Concord, Ontario, Canada), работающий вместе с источником TurboIonSpray. На распылительной игле устанавливали напряжение в 5 кВ для положительного режима. Газовую завесу устанавливали на 10, газ 1 и газ 2 устанавливали на 50, газ для ударной диссоциации (CAD) - средний и температуру датчика у нагревателя источника на 500°C. Для получения наиболее чувствительных сигналов использовали режим множественного мониторинга реакций (MRM), m/z 378→210 (17уа), m/z 373→205 (12fa), m/z 410→242 (55) и m/z 309→171 (внутренний стандарт). Регистрацию и количественную обработку данных осуществляли с помощью программы Analyst™, версия 1.4.1 (Applied Biosystems).
Водорастворимость. Растворимость препаратов определяли с помощью Multiscreen Solubility Filter Plate (Millipore Corporate, Billerica, MA) в сочетании с LC-MS/MS. Вкратце, в 96-луночный планшет вносили по 198 мкл фосфатно-солевого буфера (PBS) (pH 7,4), добавляли по 2 мкл 10 мМ иследуемых соединений (в DMSO) и перемешивали при осторожном встряхивании (200-300 об/мин) в течение 1,5 ч при комнатной температуре (n=3). Планшет центрифугировали 10 мин при 800×g и использовали фильтраты для определения концентрации и растворимости иследуемых соединений методом LC-MS/MS, как описано выше.
Фармакокинетические исследования. Приобретали самцов мышей ICR (n=3 в группе) в возрасте от 6 до 8 недель у Harlan Inc. и использовали их для изучения фармакокинетики (PK) соединений 17уа, 12fa и 55. Все соединения (10 мг/кг) растворяли в DMSO/PEG300 (1:9) и вводили однократно путем внутривенной (i.v.) инъекции (50 мкл) в хвостовую вену. Через 5, 15, 30 мин и 1, 1,5, 2, 3, 4, 8, 12 и 24 ч после i.v. введения брали пробы крови. Для оценки пероральной биодоступности мышам вводили внутрь (р.о.) гаважем по 20 мг/кг (в Tween80/DMSO/H2O, 2:2:6) исследуемых соединений. Через 0,5, 1, 1,5, 2, 3, 4, 8, 12 и 24 ч после р.о. введения брали пробы крови.
Самок крыс Sprague-Dawley (n=3; 254±4 г) приобретали у Harlan Inc. (Indianapolis, IN). Катетеры для грудной яремной вены крыс приобретали у Braintree Scientific Inc. (Braintree, MA). По прибытии животных подвергали акклиматизации в течение 3 дней в помещении с контролируемой температурой (20-22°C) с 12-часовым циклом освещение/темнота перед какой-либо обработкой. Соединения 17уа, 12fa и 55 вводили i.v. в грудную яремную вену в дозе 5 мг/кг (в DMSO/PEG300, 1:9). Для замены удаляемой крови вводили равный объем гепаринизированного физраствора и через 10, 20, 30 мин и 1, 2, 4, 8, 12, 24 ч брали образцы крови (250 мкл) через катетер из яремной вены. Для оценки пероральной биодоступности крысам вводили внутрь (р.о.) гаважем по 10 мг/кг (в Tween80/DMSO/H2O, 2:2:6) исследуемых соединений. Через 30, 60, 90, 120, 150, 180, 210, 240 мин и 8, 12 24 ч после перорального введения брали пробы крови (250 мкл) через катетер из яремной вены. Перед взятием крови готовили гепаринизованные шприцы и флаконы. Образцы плазмы получали путем центрифугирования проб крови 5 мин при 8000×g. Все образцы плазмы немедленно хранили при -80°C до анализа.
Из 100 мкл плазмы экстрагировали анализируемые вещества с помощью 200 мкл ацетонитрила, содержащего 200 нМ внутреннего стандарта. Образцы тщательно перемешивали, центрифугировали и переносили органические экстракты в автоматический пробоотборник для анализа методом LC-MS/MS.
Исследования ксенотрансплантатов РС-3_TxR. Готовили клетки PC-3_TxR (10×107/мл) в культуральной среде RPMI 1640, содержащей 10% FBS, и смешивали с Matrigel (BD Biosciences, San Jose, СА) в соотношении 1:1. Опухоли получали путем введения 100 мкл смеси (5×106 клеток на 1 животное) подкожно (s.c.) в бок 6-8-недельным самцам бестимусных мышей nude. Измеряли длину и ширину опухолей и рассчитывали объем опухолей (мм3) по формуле π/6×L×W2, где длина (L) и ширина (W) определяется в мм. Когда объем опухолей достигал 300 мм3, животным, несущим опухоли PC-3_TxR, вводили перорально носитель [Tween80/DMSO/H2O, 2:2:6)] или 17уа (10 мг/кг). Схема дозировки: 3 раза в неделю на протяжении 4 недель.
Результаты
17уа и 55 проявляют широкую цитотоксичность на клетках, в том числе клетках со множественной лекарственной устойчивостью
Оценивали способность 17уа и 55 ингибировать рост клеток раковых линий методом анализа SRB (табл. 12). Оба соединения ингибировали рост клеток некоторых раковых линий человека, в том числе пяти линий клеток рака простаты и одной линии клеток глиомы, со значениями IC50 в низком наномолярном диапазоне. Соединение 17уа проявляло в 1,7-4,3 раз большую силу действия, чем 55 на этих линиях клеток. Для изучения влияния препаратов на устойчивость к 17уа и 55 и для сравнения с исходной линией клеток РС-3 использовали клетки устойчивой к паклитакселю линии РС-3 (РС-3/TxR), которая гиперэкспрессирует Р-гликопротеин (P-gp). Значения IC50 для доцетаксела составляли 1,2±0,1 нМ и 17,7±0,7 нМ для клеток РС-3 и РС-3/TxR, соответственно. Соединения 17уа и 55 были равносильными против исходной линии РС-3 и РС-3/TxR, тогда как паклитаксель и доцетаксель проявляли относительную 85- и 15-кратную устойчивость, соответственно. Эти данные означают, что соединения 17уа и 55 обходят опосредованную P-gp лекарственную устойчивость.
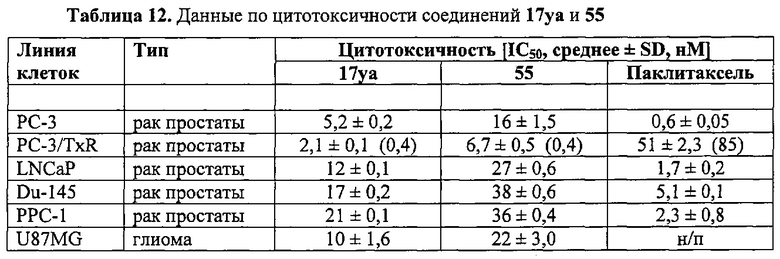

17уа и 55 связываются с сайтом связывания колхицина на тубулине, ингибируют полимеризацию тубулина и индуцируют апоптоз клеток (фиг. 20)
Для изучения взаимодействия небольших молекул ингибиторов с тубулином был разработан метод анализа конкурентного связывания. При этом используются различные концентрации 17уа или 55 для конкуренции за связывание колхицина с тубулином. Оба соединения эффективно конкурировали с колхицином за связывание с тубулином (фиг. 20A); однако кривые их конкурентного связывания существенно отклонялись от нуля при более высоких концентрациях по сравнению с подофиллотоксином, известным мощным лигандом сайта связывания с колхицином. Это свидетельствует о том, что соединения 17уа и 55 проявляют меньшее сродство, чем подофиллотоксин, или же они связываются с сайтом связывания колхицина лишь частично. Винбластин, отрицательный контроль, не ингибировал связывание колхицина с тубулином, что свидетельствует о специфичности этого метода анализа конкурентного связывания.
Тубулин из мозга свиньи (чистота >97%) инкубировали с 17уа или 55 (5 мкМ), чтобы проверить их влияние на полимеризацию тубулина (фиг. 20B). Соединения 17уа и 55 ингибировали полимеризацию тубулина на 47% и 40% за 15 мин, соответственно. В качестве положительного контроля использовали колхицин при 5 мкМ, который ингибировал полимеризацию тубулина на 32%. Эти данные свидетельствуют о том, что соединения 17уа и 55 несколько сильнее ингибируют полимеризацию тубулина, чем колхицин. Таким образом, молекулярный механизм этих соединений заключается в связывании с сайтом связывания колхицина, ингибировании полимеризации тубулина и индукции цитотоксичности.
Клетки РС-3 и РС-3/TxR подвергали воздействию от 0,8 до 600 нМ соединения 17уа или 55 либо доцетакселя в течение 24 ч. В качестве меры апоптоза клеток использовали уровень комплексов ДНК-гистон. Соединения 17уа и 55 были равносильными в индуцировании апоптоза клеток РС-3 (фиг. 20C) и РС-3/TxR (фиг. 20D) за 24 ч. Хотя доцетаксель был очень активен в индуцировании апоптоза клеток РС-3, однако он был слабее на клетках РС-3/TxR вследствие гиперэкспрессии P-gp.
17уа и 55 проявляют благоприятные фармакологические свойства
Исследовали такие фармакологические свойства соединений 17уа и 55, как метаболическая стабильность, проницаемость, водорастворимость и взаимодействия с другими лекарствами (табл. 13A). Соединение 17уа проявляло большую метаболическую стабильность и водорастворимость, чем 55. Оба препарата проявляли более чем адекватные значения проницаемости, что свидетельствует о возможности их приема внутрь. Кроме того, соединения 17уа и 55 проявляли высокие значения IC50 в микромолярном диапазоне при анализе ингибирования ферментов CYP, указывая на то, что оба соединения могут избежать взаимодействия с другими лекарствами через основные ферменты CYP печени. В целом, оба соединения проявляли благоприятные фармакологические свойства.
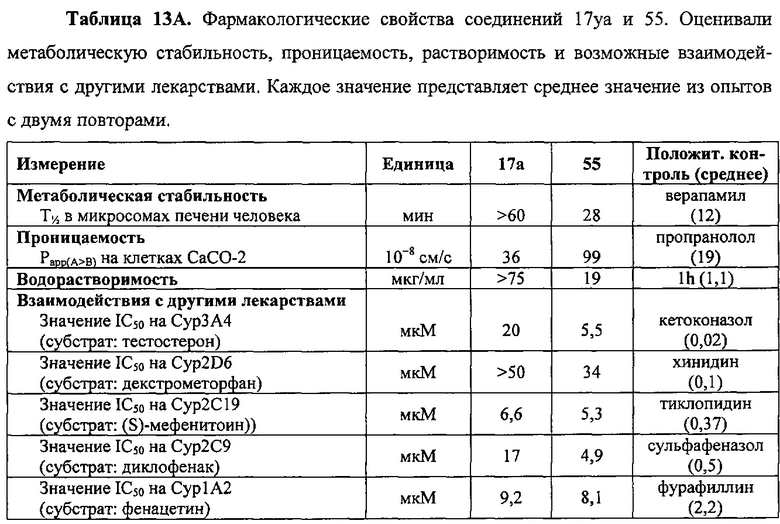
Как видно из табл. 13B, период полужизни для 17уа составлял 80 мин по реакции I фазы, свидетельствуя о том, что соединение 17уа было стабильным в метаболических процессах I фазы. Период полужизни (90 мин) в присутствии UDP-глюкуроновой кислоты был таким же, как и в ее отсутствие. Эти данные свидетельствуют, что соединение 17уа является стабильным в микросомах печени человека, и можно ожидать, что у человека будет низкий клиренс и длительный период полужизни. С другой стороны, соединение 55 проявляет период полужизни в 30 и 43 мин в присутствии или в отсутствие UDP-глюкуроновой кислоты, соответственно. Соединение 12fa проявляет период полужизни в 44 мин в I фазе. Эти данные говорят о том, что все три соединения проявляют приемлемую стабильность в микросомах печени человека, а 17уа более стабильно, чем 12fa и 55. При исследовании их метаболизма было обнаружено, что 12fa и 55 проявляют более высокий уровень восстановления кетонов (данные не приводятся), свидетельствуя, что 12fa и 55 более лабильны, чем 17уа.
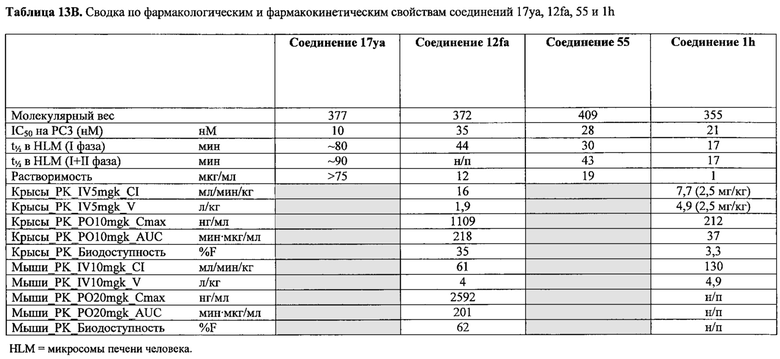
Соединение 17ya проявляет большую растворимость в воде, a 12fa и 55 проявляют приемлемую растворимость
Соединение 17уа содержит имидазольное кольцо, и это кольцо улучшает водорастворимость, которая составляет >75 мкг/мл (табл. 13A). Соединения 12fa и 55 проявляли меньшую водорастворимость, которая составляла 12 и 19 мкг/мл, соответственно. В целом, 17уа проявляло большую растворимость в воде, а соединения 12fa и 55 проявляли приемлемую растворимость в воде, которая была значительно лучше, чем у соединения 1h. Большая растворимость 12fa выражалась в значительном улучшении биодоступности по сравнению с 1h (35% против 3,3% у крыс). Аналогично для 17уа и 55, водорастворимость коррелировала со значительным улучшением биодоступности, как изложено ниже (табл. 14).
Фармакокинетические исследования 17уа и 55 на мышах, крысах и собаках
Фармакокинетические параметры 17уа и 55 при введении в виде однократной (i.v. или р.о.) дозы у мышей ICR, крыс Sprague-Dawley и беспородных собак приведены в табл. 14. Соединение 17уа проявляло низкий клиренс у мышей и крыс, свидетельствуя, что 17уа обладает метаболической стабильностью и минимальным пресистемным метаболизмом у этих видов. Кроме того, 17уа имело умеренный объем распределения у мышей и крыс, что указывает на то, что оно может правильно распределяться в тканях, включая опухоли. В отличие от мышей и крыс, общий клиренс 17уа у собак был на удивление высоким. Два распространенных метаболита в плазме крови собак, гидроксилированный метаболит и неизвестный метаболит с m/z +34 от исходного (данные не приводятся), соответствуют найденным в микросомах печени собак. Итак, у собак отмечается более высокий клиренс и меньшая перорального экспозиция для 17уа по сравнению с соединением 55, но не у мышей и крыс. Кроме того, обильные метаболиты 17уа отмечаются только в микросомах печени собак, но не в микросомах печени мышей, крыс или человека (данные не приводятся). Соединение 17уа проявляло приемлемую пероральную биодоступность в 21%, 36% и 50% у крыс, мышей и собак, соответственно. А соединение 55 проявляло низкий клиренс у крыс и умеренный клиренс у мышей и собак. Подобно 17уа, соединение 55 проявляло умеренный объем распределения у этих видов. Соединение 55 проявляло постоянную пероральную биодоступность у этих трех видов (24%-36%). Эти свойства означают, что соединения 17уа и 55 являются потенциальными перорально доступными ингибиторами тубулина.
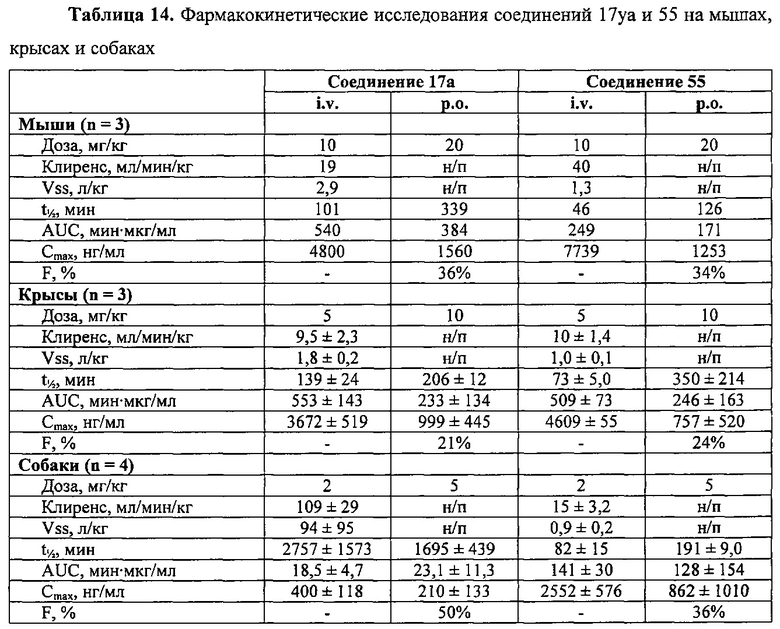
Соединения 17ya и 55 ингибируют рост устойчивых к паклитакселю ксенотрансплантатов рака простаты (РС-3/TxR)
Мышам nude инокулировали клетки РС-3 (фиг. 21A) и устойчивые к паклитакселю клетки рака простаты (РС-3/TxR) (фиг. 21B) и давали опухолям достичь объема 150-300 мм3. Доцетаксель (10 или 20 мг/кг), который применяется в клинике для лечения рака простаты, использовали для оценки его эффективности на моделях опосредованной P-gp лекарственной устойчивости in vivo. Опухоли РС-3/TxR оказались быстрорастущими, их объем достигал 1500-2500 мм3 по окончании исследования. Хотя при внутривенном введении 10 и 20 мг/кг доцетаксель проявлял зависимость от дозы на обеих моделях (фиг. 21A и 21B), эффект ингибирования роста опухоли (TGI) снижался с 84% TGI на опухолях РС-3 до 14% TGI на опухолях РС-3/TxR при внутривенной дозе в 10 мг/кг (табл. 15). Кроме того, при более высокой дозе (20 мг/кг) доцетаксель вызывал частичную регрессию (TGI>100%) опухолей РС-3, но всего лишь 56% TGI на опухолях РС-3/TxR. Эффективность доцетакселя на опухолях РС-3/TxR резко снижалась по сравнению с опухолями РС-3, свидетельствуя о том, что эффективность ухудшалась вследствие опосредованной P-gp лекарственной устойчивости, и эти результаты очень хорошо согласуются с нашими данными по цитотоксичности или апоптозу in vitro. В отличие от слабой эффективности доцетакселя на опухолях РС-3/TxR, соединение 17уа при пероральном введении (6,7 мг/кг) проявляло более чем 100% TGI, не влияя на массу тела (фиг. 21B и табл. 15). Кроме того, 2 из 4 мышей nude, несущих опухоли РС-3/TxR, оказались без опухолей на 19-й день (данные не приводятся).
Далее модель ксенотрансплантатов РС-3/TxR использовалась для оценки эффективности соединений 17уа (при других схемах введения) и 55. Предельно допустимая доза (потеря массы тела >20%) для 17уа оказалась равной 10 мг/кг при пероральном введении один раз в день на протяжении 4 дней; или 3,3 мг/кг при введении два раза в день (b.i.d.) на протяжении 5 дней (данные не приводятся). Как видно из фиг. 21C, соединение 17уа вводили по 3,3 мг/кг два раза в день на протяжении первых 4 дней подряд в первую неделю, а затем график был изменен на один раз в день со 2-й по 4-ю неделю. Результаты показывают, что происходила частичная регрессия с 4 по 19-й день, a TGI составил 97%, и 1 из 7 мышей оказалась без опухолей на 26-й день. Более высокая доза (10 мг/кг) 17уа при более низкой частоте дозирования (q2d) (фиг. 21D) вызывала частичную регрессию с 13 по 29-й день. Эти данные свидетельствуют о том, что при оптимизации дозы и схемы дозирования соединение 17уа будет успешно ингибировать опухоли РС-3/TxR. Соединение 55 вводили перорально мышам nude по 10 или 30 мг/кг два раза в день (b.i.d.) пять раз в неделю с 1-й по 4-ю неделю. Как видно из фиг. 21С, профили ингибирования проявляют зависимость от дозы на опухолях РС-3/TxR. Значение TGI составляло 59% в группе обработки при низкой дозе (10 мг/кг). Кроме того, при повышении дозы (30 мг/кг) начинала проявляться частичная регрессия (TGI>100%) с 19-го дня до окончания исследования (26-й день). Некоторые мыши в группе носителя теряли вес тела в конечной точке, отчасти из-за раковой кахексии. Напротив, мыши, получавшие 17уа (3,3 мг/кг) или 55 (30 мг/кг), набирали вес (табл. 15), свидетельствуя, что эти оптимизированные дозы 17уа или 55 хорошо переносятся и предотвращают раковую кахексию.
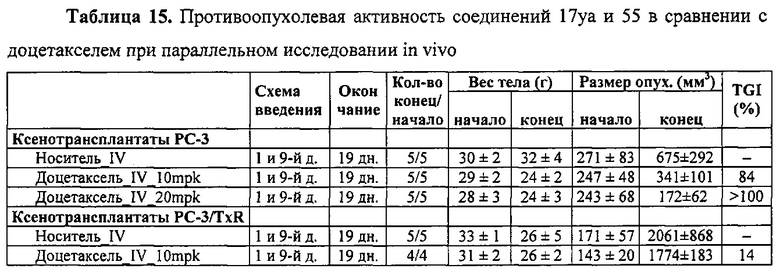

Проникновение соединений 17ya и 55 в мозг у мышей nude
Определяли концентрации соединений 17уа и 55 в целом мозге мышей nude через 1 ч и 4 ч после перорального введения в дозе 20 мг/кг (табл. 16). Определяли соотношение концентраций в мозге и плазме и сравнивали с доцетакселем у мышей nude. Соединение 55 проявляло большее проникновение в мозг, чем 17уа и доцетаксель. Соединение 17уа проявляло лишь немного большее соотношение концентраций мозг/плазма, чем доцетаксель через 1 ч и через 4 ч. Концентрации соединения 55 в мозге достигали от 14 до 19% его концентрации в плазме через 1 ч и 4 ч, соответственно, проявляя в 3,2 раза большее соотношение мозг/плазма через 1 ч и через 4 ч по сравнению с доцетакселем. Эти данные свидетельствуют, что соединение 55 обладает потенциально благоприятными свойствами для лечения глиомы, так как оно проявляет большее проникновение в мозг и большую мощность (22 нМ, табл. 12) на клетках глиомы.

ПРИМЕР 10. Эффективность на ксенотрансплантатах лейкемии (HL60) in vivo (фиг. 22)
Готовили клетки HL60 (10×107/мл) в культуральной среде RPMI 1640, содержащей 10% FBS, и смешивали с Matrigel (BD Biosciences, San Jose, CA) в соотношении 1:1. Опухоли получали путем введения 100 мкл смеси (5×106 клеток на 1 животное) подкожно в бок 6-8-недельным самцам бестимусных мышей nude. Измеряли длину и ширину опухолей и рассчитывали объем опухолей (мм3) по формуле π/6×L×W2, где длина (L) и ширина (W) определяется в мм. Когда объем опухолей достигал 200 мм3, животным, несущим опухоли HL60, вводили перорально носитель [Tween80/DMSO/H2O, 2:2:6)] или 17уа (10 мг/кг). Схема дозировки: 1 раз в неделю на протяжении 2 недель. Винкристин (1 мг/мл) вводили внутрибрюшинно раз в неделю.
Результаты
Мышам nude инокулировали клетки промиелоцитарной лейкемии человека HL60 и давали опухолям достичь объема около 200 мм3. Для оценки реакции этой модели in vivo в качестве положительного контрольного препарата использовали винкристин (1 мг/кг), который применяется в клинике для гематологических видов рака, включая лейкемию. Строили графики объема опухолей (мм3) в зависимости от времени в виде среднего + SD для 4-5 животных. Опухоли HL60 оказались быстрорастущими, их объем достигал 2000-3000 мм3 за две недели. Однако внутрибрюшинное введение 1 мг/кг винкристина давало очень мощный эффект игибирования роста опухолей (фиг. 22), причем торможение роста опухолей (TGI) составляло 84%. При пероральном введении 17уа (20 мг/кг) торможение роста опухолей составляло 40%. Размер опухолей HL60 сохранялся вплоть до 5 дней после обработки 17уа без заметного увеличения, но в течение следующих 2 дней размер опухолей значительно возрастал (60-100%). Это говорит о том, что эффект игибирования роста опухолей может повыситься при более частом введении 17уа.
ПРИМЕР 11. Комбинация BRAFi и ингибитора тубулина, нацеленная на альтернативные пути, может замедлить или предотвратить развитие устойчивости к вемурафенибу
Конкретные терапевтические цели этого исследования приведены на фиг. 27.
Комбинации доцетакселя (апробированный ингибитор тубулина) с BRAFi (вемурафениб или дабрафениб) либо MEKi (траметиниб) могут подавлять приобретенную устойчивость к вемурафенибу на моделях ксенотрансплантатов полученных от больных (PDX, или "xenopatient") меланомой опухолей
Полезность любой новой стратегии лечения в конечном счете зависит от ее способности проявлять устойчивую клиническую эффективность. Меланома хорошо известна как гетерогенная опухоль, проявляющая высокую фенотипическую и функциональную пластичность в ответ на микроокружение или эпигенетические факторы. Так, недавние работы показали, что устойчивые к вемурафенибу опухоли могут проявлять несколько механизмов устойчивости у одного пациента или даже в рамках одной и той же биопсии опухоли, взятой из одного метастатического сайта. В отличие от опухолей меланомы у пациентов, опухоли, выращенные из установленных клеточных линий типа A375, имеют два главных ограничения: (а) они потеряли неоднородность своих исходных опухолей, что может существенно повлиять на эффективность лекарственных препаратов; и (b) у них зачастую возникают необратимые генетические изменения вследствие адаптации к условиям клеточной культуры, которые отличаются от естественного микроокружения опухолей. Обширные исследования показали, что на ранних стадиях пассирования (≤5 пассажей) опухоли PDX сохраняют точное генетическое соответствие опухолям пациентов, сохраняют морфологию и неоднородность исходных опухолей, а реакция на терапию у них похожа на то, что наблюдается в клинике. Поэтому необходимо проверить, что сильная синергия и эффективность, наблюдаемая на опухолях A375RF21 (см. фиг. 6 и 7), остается в силе и на опухолях PDX. Также важно оценить возможность развития лекарственной устойчивости, используя опухоли PDX вместо опухолей, выращенных из установленных клеточных линий, для более прямого перехода от лабораторного стола к постели больного.
Поскольку существуют апробированные ингибиторы тубулина, то для быстрой разработки такой инновационной комбинации на пользу пациентам сначала проводили оценку эффективности комбинаций доцетакселя с тремя апробированными в настоящее время BRAFi/MEKi, чтобы определить оптимальное сочетание. Более того, новая стратегия комбинирования должна иметь большее воздействие, если она будет применяться для лечения двух групп больных меланомой с мутациями BRAFV600E. Первая группа - BRAFi-наивные пациенты, которые не принимали BRAFi и/или MEKi. Такие пациенты получат наибольшую пользу, если комбинация сможет значительно замедлить или даже предотвратить потенциальное развитие устойчивости к BRAFi. Вторая группа - BRAFi-устойчивые пациенты, которые получали лечение BRAFi и/или MEKi в клинических условиях. Эти пациенты выиграют, если комбинация сможет эффективно преодолеть приобретенную лекарственную устойчивость.
Получение опухолей PDX в ранней стадии из несущих PDX-опухоли мышей
Последние работы в данной области привели к стандартизации некоторых методик постановки успешных PDX-моделей. Различные PDX-модели теперь доступны из коммерческих источников. Использовали приобретенных мышей NSG (обычно 1-2 мыши), которым имплантировали две PDX-опухоли (P2) с небольшим количеством пассажей. Опухоли вырастали примерно до 1500 мм3, и их размножали примерно на 5 новых мышах NSG, чтобы получить достаточное количество опухолей Р3. Отбирали случайным образом пять опухолей Р3, изучали их генетический профиль и гистологию и сверяли с данными по исходным опухолям Р0, чтобы обеспечить общее генетическое и гистологическое соответствие, а затем кусочки опухолей имплантировали большому числу мышей для последующих исследований (фиг. 27). Для того, чтобы обеспечить однозначность при этих проверках, проводили гистологический анализ.
а. Определение эффективности in vivo на чувствительных к вемурафениб PDX-опухолях при обработке комбинацией доцетакселя с вемурафенибом, дабрафенибом или траметинибом
Тестировали три утвержденных в настоящее время BRAFi или MEKi, чтобы ранжировать их по эффективности комбинаций для клинической приоритетности в будущем. Хотя мыши NSG и являются самыми лучшими для размножения опухолей, но у них есть шерсть и они дороже (в 2~3 раза), чем "голые" мыши (nude). Многочисленные исследования показали, что окончательные экспериментальные результаты на мышах nude столь же верны, как и на более дорогих и трудных мышах NSG при исследованиях PDX. Поэтому в опытах по конечной эффективности использовали мышей nude. В качестве контрольной комбинированной обработки включали стандартную комбинацию дабрафениб + траметиниб. Кроме того, все препараты, используемые на этой стадии, являются апробированными и их фармакокинетические свойства известны. Поэтому в данном исследовании использовали принятые дозы и способы введения. В частности, вемурафениб (45 мг/кг), дабрафениб (30 мг/кг) и траметиниб (0,3 мг/кг) вводили мышам внутрь гаважем. Доцетаксель (10 мг/кг) вводили внутривенно через хвостовую вену, так как он недоступен перорально.
Использовали по 7 мышей в группе для оценки того, достигается ли стойкая регрессия опухолей для трех независимых вемурафениб-чувствительных PDX-опухолей. Для обеспечения статистической значимости в этих важных исследованиях на животных проводили статистический анализ и расчет численности выборки.
Вкратце, 6-недельных самцов бестимусных мышей nude приобретали у Charles River. Вемурафениб-чувствительные PDX-опухоли Р3 меланомы нарезали на мелкие кусочки (~3 мм), которые затем имплантировали подкожно в бок 63 анестезированным мышам nude по установленными методикам, описанным в литературе. Когда опухоли достигали примерно 100-200 мм3 через 2-3 недели после имплантации, мышей случайным образом разбивали на девять групп (n=7), сводя к минимуму различия в весе и размерах опухолей: группа отрицательного контроля только с носителем (группа 1); четыре группы монотерапии (группы 2-5) с беспрерывным ежедневным приемом внутрь вемурафениба, дабрафениба, траметиниба или доцетакселя (i.v.); и четыре группы комбинированной терапии (группы 6-9) с беспрерывным ежедневным введением комбинации дабрафениб + траметиниб (контрольная комбинация), доцетаксель + вемурафениб, доцетаксель + дабрафениб и доцетаксель + траметиниб при тех же дозах и способах введения, что и в виде индивидуальных препаратов.
Вследствие природы PDX, нецелесообразно прикреплять какие бы то ни было люминесцентные зонды in vivo для мониторинга опухолей. Поэтому опухоли измеряли каждые три дня штангенциркулем и рассчитывали их объемы по формуле: (ширина2 × длина)/2. Согласно предыдущим работам, чувствительные к вемурафенибу опухоли у мышей четко приобретали устойчивость к монотерапии вемурафенибом в пределах 60 дней. Поэтому опухоли отслеживали вплоть до 90 дней или до тех пор, пока опухоль не регрессировала полностью. Возникновение лекарственной устойчивости тщательно отслеживали по кинетике роста опухолей. Кроме того, из каждой опухоли раз в две недели брали серийные биоптаты с помощью тонкой иглы по известным методикам. В этих биоптатах определяли уровни BRAF и pERK/tERK методом вестерн-блот, чтобы отслеживать возникновение лекарственной устойчивости к BRAFi и MEKi. Внимательно следили за весом, активностью и внешним видом мышей на предмет потенциальной токсичности.
По окончании экспериментов брали терминальные пробы крови (0,8-1,0 мл/мышь) путем сердечной пункции для всестороннего анализа клинической патологии на фирме Charles River Laboratories. Всех животных забивали ингаляцией CO2 с последующим смещением шейных позвонков сразу после отбора крови и у каждой мыши извлекали основные органы (мозг, сердце, легкие, печень, селезенка, почки) и хранили отдельно в 10% растворе формалина с фосфатным буфером. Эти органы тщательно осматривали и анализировали на потенциальную токсичность лекарственных препаратов (например, гепатотоксичность) и признаки метастазирования. Осторожно извлекали опухоли, взвешивали и обрабатывали для определения эффектов препаратов на ключевые показатели клеточной пролиферации, антиангиогенеза и апоптоза, а также фиксировали и обрабатывали для гистопатологических исследований. Вышеописанный эксперимент повторяли на двух других моделях вемурафениб-чувствительных PDX-опухолей, чтобы подтвердить, что эффективность не связана с определенной моделью PDX-опухолей, так что общее количество мышей, использовавшихся на данной стадии, составляло 63×3=189.
b. Определение эффективности in vivo на устойчивых к вемурафениб PDX-опухолях при обработке комбинацией доцетакселя с вемурафенибом, дабрафенибом или траметинибом
Идеальной стратегией преодоления приобретенной устойчивости к вемурафенибу является применение авансом очень эффективных способов лечения, чтобы предотвратить ее развитие. К сожалению, это не реально при существующей терапии, и у большинства пациентов быстро вырабатывается устойчивость к вемурафенибу. Недавно Stuart et al. предположили, что схема прерывистой монотерапии с высокой дозировкой вемурафениба может ослабить развитие лекарственной устойчивости по сравнению со схемами беспрерывного дозирования. Однако психологическое воздействие на пациентов и долговременная клиническая эффективность такой стратегии "дней отдыха от препарата" остаются неясными. Поскольку у резистентных опухолей меланомы часто вырабатывается зависимость от активированного пути MEK-ERK, то подходящая комбинация ингибитора тубулина с BRAFi/MEKi должна быть очень эффективна в подавлении роста опухолей, тогда как монотерапия может оказаться недостаточной (фиг. 6 и 7). Поэтому важно подтвердить, что комбинация доцетакселя с BRAFi/MEKi вызывает значительную регрессию у вемурафениб-устойчивых опухолей. Такие результаты могут претворяться в продление срока жизни пациентов, даже если опухоли станут устойчивыми к вемурафенибу.
Аналогично описанным выше экспериментальным методикам, после получения опухолей PDX в ранней стадии из несущих вемурафениб-устойчивые PDX-опухоли мышей, 63 мышей разбивали на девять групп (одна группа только с носителем, четыре группы монотерапии и четыре группы комбинированной терапии), которые использовали для определения того, будет ли комбинация доцетакселя с апробированным препаратом BRAFi или MEKi эффективной на устойчивой к вемурафенибу PDX-модели. Измеряли размеры опухолей, брали серийные биоптаты опухолей, терминальные пробы крови и извлекали основные органы для оценки эффективности и потенциальной токсичности, аналогично описанным в предыдущем разделе. Тестирование комбинаций повторяли на двух дополнительных моделях вемурафениб-устойчивых PDX-опухолей; таким образом, на этой стадии использовали 63×3=189 мышей.
Даже если ожидается, что эти устойчивые к вемурафенибу PDX-опухоли будут устойчивы к монотерапии BRAFi/MEKi или комбинации дабрафениб + траметиниб, как это показано в клинических испытаниях и в приведенных выше результатах на модели ксенотрансплантатов A375RF21, полагаем, что они будут ценными стандартами для объективной оценки потенциальной эффективности комбинаций, содержащих доцетаксель. Аналогично экспериментам на чувствительных к вемурафенибу PDX-моделях, определяли размеры опухолей, потенциальную острую токсичность, клиническую патологию и уровни pERK/ERK в биоптатах опухолей для того, чтобы ранжировать эти комбинации по эффективности.
Определение потенциальной токсичности и целенаправленный вестерн-блоттинг для оценки эффективности лечения и идентификации потенциальных биомаркеров для клинического мониторинга заболевания
Определение потенциальной токсичности при комбинированной терапии на PDX-моделях
В дополнение к тщательному мониторингу веса и активности у несущих PDX-опухоли мышей во время лечения, пробы крови, взятые в конечных точках, отправляли на всесторонний анализ клинической патологии (химия крови) в пределах 24 ч после сбора. Определяли полный профиль патологии, химии и гематологии (общий анализ крови с дифференцированным) и представляли подробные результаты. Результаты анализировали на наличие потенциальных признаков токсичности, аналогично тому, что было описано ранее. Для патологического анализа готовили парафиновые блоки из фиксированных формалином тканей опухолей, а срезы окрашивали гематоксилином и эозином. Срезы сканировали, чтобы создать цифровую копию всех тканей на стеклянном предметном стекле с помощью ScanScope® XT (Aperio Technologies, Inc., CA) при 0,25 пиксель/мкм. Процесс сканирования позволяет рассматривать и анализировать изображения тканей при различном увеличении, что весьма соответствует традиционному рассматриванию тканей под обычным микроскопом.
Патологическая оценка пролиферации клеток и апоптоза на срезах опухолей PDX
Для оценки того, сохраняет ли комбинация препаратов повышенную эффективность, делали срезы собранных опухолей PDX и проводили иммуногистохимию.
Для антипролиферативной оценки срезы опухолей исследовали на снижение уровня pERK и окрашивали на Ki67, который является маркером пролиферации опухолевых клеток, по стандартным методикам иммуногистохимии. Для определения доли клеток меланомы на срезах опухолей использовали специфичные для меланомы маркеры, S100 и НМВ-45, в сочетании с окрашиванием Н&Е.
Для оценки апоптоза изучали морфологию ядер на предмет фрагментации ядер путем фиксации срезов опухолей, окрашивания срезов Hoechst 33342 и подсчета ядер с признаками фрагментации или нормальной морфологии. С другой стороны, изменения в ядрах оценивали методом TUNEL с последующим анализом апоптотических путей. Изменения трансмембранного потенциала митохондрий измеряли с помощью набора для проточной цитометрии MitoScreen. Концентрацию и субклеточное распределение (транслокацию из митохондрий в цитозоль) цитохрома с оценивали методом вестерн-блоттинга. Экспрессию антиапоптозных белков Bcl-2 и Bcl-xl; проапоптозных белков Вах и Noxa; и статус фосфорилирования проапоптозного белка Bad оценивали методом вестерн-блоттинга. Активацию инициаторной каспазы 9 и эффекторной каспазы 3 оценивали путем измерения специфического протеолитического расщепления флуорогенных субстратов Ac-LEHD-AFC (для каспазы 9) и Ac-DEVD-AMC (для каспазы 3).
Целенаправленный вестерн-блоттинг для оценки эффективности комбинированной терапии и идентификации потенциальных биомаркеров для клинического мониторинга заболевания
Для тех PDX-опухолей, которые изначально чувствительны к вемурафенибу, проводили целенаправленный вестерн-блоттинг, чтобы определить изменения уровней ключевых белков, известных как придающие устойчивость к BRAFi или MEKi, и идентифицировать потенциальные биомаркеры, применимые для мониторинга терапевтической эффективности в будущем, используя серийные биоптаты опухолей, собранные во время экспериментов. В частности, исходя из представленных выше данных, усилия были сосредоточены на изучении тех элементов путей MAPK, PDGFβ, PI3K/AKT и апоптозных путей, которые, как известно, задействованы в устойчивости к вемурафенибу. Анализировали уровни экспрессии белков RAS, RAF, MEK1/2, фосфо-MEK1/2 (Ser217/221), ERK1/2, фосфо-ERK1/2 (Thr202/Tyr204), AKT, фосфо-AKT (Ser473), PDGFβ, расщепленного PARP или каспазы-3, аналогично работе на модели A375RF21. Для тех PDX-опухолей, которые устойчивы к вемурафенибу, определяли профили тех генов у опухолей РО, которые определяют базовые механизмы устойчивости, и отслеживали терапевтическую эффективность методом целенаправленного вестерн-блоттинга тех белков, которые отвечают за изначальную базовую устойчивость.
Кроме того, применение таксанов, в том числе доцетакселя, дает лишь небольшое преимущество по выживаемости, а большинство пациентов в конечном счете прогрессирует из-за присущей или приобретенной лекарственной устойчивости. Один из основных механизмов устойчивости опосредован ABC-переносчиками, которые могут уменьшать внутриклеточную концентрацию доцетакселя. Поэтому, в дополнение к мониторингу уровня белков, ответственных за устойчивость к BRAFi/MEKi, отслеживали возможное развитие устойчивости к доцетакселю путем изучения экспрессии ключевых ABC-переносчиков, включают Р-гликопротеин (Pgp), белок множественной лекарственной устойчивости (MRP) и белки устойчивости рака молочной железы (BCRP), используя методики, аналогичные описанным в предыдущих исследованиях. Экстракцию белков, вестерн-блоттинг и выявление антигенов проводили по стандартным методикам, с модификациями в зависимости от антигена-мишени. Уровень белков количественно определяли с помощью денситометрии (средние значения из экспериментов в трех повторах).
Ожидаемые результаты, подводные камни и альтернативные подходы
Комбинации доцетакселя с BRAFi или MEKi, как ожидается, будут эффективными и на чувствительных к вемурафенибу PDX-моделях, и на устойчивых к вемурафенибу PDX-моделях. Поскольку все препараты, используемые в данном исследовании, являются апробированными, то результаты данного исследования, если они окажутся эффективными, легко применимы на практике и могут быть быстро проверены в клинике, чтобы служить в качестве комбинированной терапии первого ряда и приносить немедленную пользу пациентам.
Комбинации ABI (новые ингибиторы тубулина) с BRAFi или MEKi подавляют приобретенную устойчивость к BRAFi и вторичную устойчивость к таксанам у PDX-опухолей
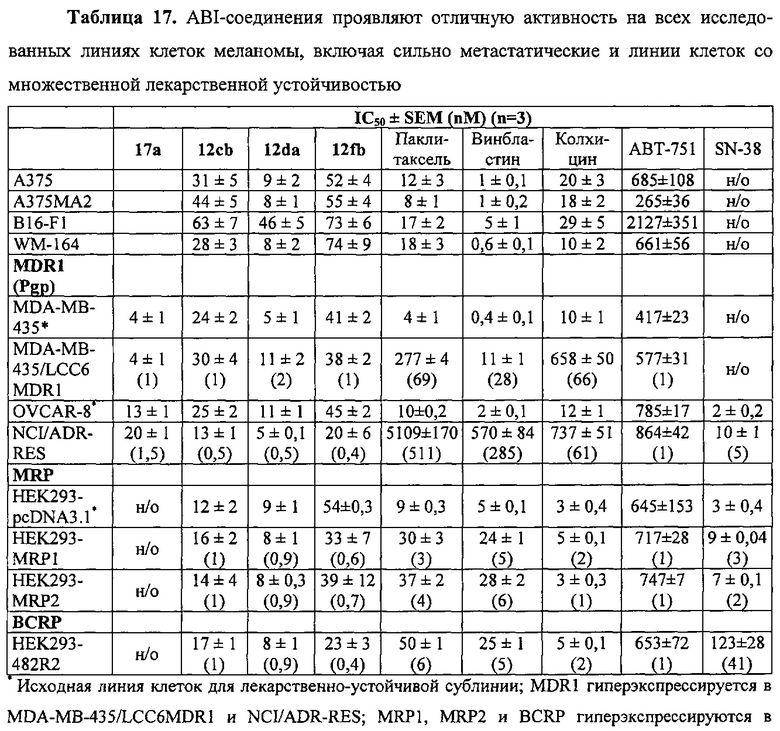

При клиническом применении доцетакселя может возникать вторичная устойчивость к таксанам при комбинировании доцетакселя с BRAFi/MEKi. По сравнению с апробированными ингибиторами тубулина, ABI-соединения связываются с другим сайтом в тубулине и имеют четкие преимущества, включая высокую активность, приемлемую пероральную биодоступность, отличные фармакокинетические свойства и эффективное преодоление опосредованной ABC-переносчиками множественной лекарственной устойчивости (табл. 17). Предварительные исследования токсичности показали, что ABIs обладают значительно более низкой токсичностью, чем существующие ингибиторы тубулина, такие как доцетаксель или винбластин. Поэтому для разработки нового поколения комбинированной терапии, чтобы преодолеть потенциальную вторичную устойчивость к комбинациям первой очереди, содержащим доцетаксель, определяли эффективность комбинаций BRAFi/MEKi с двумя перспективными ABIs (соединение 12da и соединение 17уа) на моделях PDX. Как ABIs, так и апробированные BRAFi/MEKi являются перорально активными средствами. Никаких неблагоприятных лекарственных взаимодействий между ингибиторами тубулина и BRAFi/MEKi не отмечено. Поэтому, исходя из модели ксенотрансплантатов A375RF21, при совместном приеме внутрь ABI с вемурафенибом, дабрафенибом или траметинибом, вероятно, сильная синергичность в подавлении роста опухолей меланомы сохранится и на опухолях PDX.
Придерживались тех же методик, что описаны выше, которые вкратце описаны ниже.
c. Определение эффективности in vivo на чувствительных к вемурафениб PDX-опухолях при обработке комбинацией ABI (соединения 12da и 17уа) с вемурафенибом, дабрафенибом или траметинибом
Для этого исследования использовали мышей NSG (обычно 1-2 мыши), которым имплантировали две PDX-опухоли (P2) с небольшим количеством пассажей. Опухоли вырастали примерно до 1500 мм3, и их размножали примерно на 5 новых мышах NSG, чтобы получить достаточное количество опухолей Р3. Отбирали случайным образом пять опухолей Р3, изучали их генетический профиль и гистологию и сверяли с данными по исходным опухолям Р0, чтобы обеспечить точное общее генетическое и гистологическое соответствие, а затем кусочки опухолей имплантировали большому числу мышей для последующих исследований (фиг. 27).
Поскольку фармакокинетические свойства ABI-соединений и BRAFi/MEKi уже установлены, то аналогично тому, что описано выше, определяли эффективность комбинаций, потенциальную токсичность и возможные биомаркеры для мониторинга заболевания на трех независимых чувствительных к вемурафенибу моделях PDX. Так как результаты по стандартной монотерапии вемурафенибом, дабрафенибом или траметинибом уже получены, как описано выше, то, когда закончилась оценка комбинаций, содержащих доцетаксель, исследование комбинированной терапии продолжали путем включения наиболее эффективной из приведенных выше комбинаций с доцетакселем, а также контрольной комбинации (дабрафениб + траметиниб). Итак, для соединения 12da, небольшие кусочки (~3 мм3) чувствительных к вемурафенибу опухолей Р3 имплантировали подкожно в бок 49 анестезированным мышам nude (6-недельным самцам бестимусных мышей nude). Когда опухоли достигали примерно 100-200 мм3 через 2-3 недели после имплантации, мышей случайным образом разбивали на семь групп (n=7), сводя к минимуму различия в весе и размерах опухолей: группа отрицательного контроля только с носителем (группа 1); одна группа монотерапии соединением 12da (15 мг/кг, группа 2); и пять групп комбинированной терапии с беспрерывным ежедневным введением комбинации дабрафениб + траметиниб (контрольная комбинация, группа 3), наиболее эффективной из приведенных выше комбинаций, содержащих доцетаксель (группа 4), и соединения 12da в комбинации с вемурафенибом, дабрафенибом или траметинибом (группы 5-7). Каждые три дня измеряли опухоли штангенциркулем и рассчитывали их объем по формуле: (ширина2 × длина)/2. Опухоли отслеживали вплоть до 90 дней или до тех пор, пока опухоль не регрессирует полностью. Возникновение лекарственной устойчивости тщательно отслеживали по кинетике роста опухолей. Внимательно следили за весом, активностью и внешним видом мышей на предмет потенциальной токсичности. Терминальные пробы крови отправляли на химический анализ крови, а срезы опухолей обрабатывали для анализа клинической патологии. Кроме того, из каждой опухоли раз в неделю брали серийные биоптаты с помощью тонкой иглы по известным методикам. Проводили целенаправленный вестерн-блоттинг для изучения элементов путей MAPK, PDGFβ, PI3K/AKT и апоптозных путей, чтобы отслеживать приобретенную лекарственную устойчивость и потенциальные биомаркеры для оценки терапевтической эффективности. Эксперименты повторяли на двух других вемурафениб-чувствительных PDX-опухолях, так что для тестирования соединения 12da использовали 49×3=147 мышей. Наконец, вместе с тестированием соединения 17уа использовали 147×2=294 мыши.
d. Определение эффективности in vivo на устойчивых к вемурафениб опухолях PDX при обработке комбинацией ABI (соединение 12da или 17уа) с вемурафенибом, дабрафенибом или траметинибом
Аналогично экспериментальным методикам, описанным вкратце в предыдущих разделах, эксперименты повторяли на трех моделях устойчивых к вемурафенибу PDX-опухолей с использованием 294 мышей для определения эффективности комбинаций ABI (соединение 12da или 17уа) с вемурафенибом, дабрафенибом или траметинибом. Измеряли размеры опухолей, брали серийные биоптаты опухолей, терминальные пробы крови и извлекали основные органы для оценки эффективности и потенциальной токсичности, аналогично описанным в предыдущем разделе. Потенциальную токсичность определяли путем мониторинга потери веса во время экспериментов и последующих анализов на предмет патологии. Проводили целенаправленный вестерн-блоттинг для оценки эффективности лечения и выявления потенциальных биомаркеров, как изложено выше.
Ожидаемые результаты, подводные камни и альтернативные подходы
Комбинации доцетакселя с BRAFi или MEKi, как ожидается, будут эффективными и на чувствительных, и на устойчивых к вемурафенибу моделях PDX. Следует ожидать, что такие комбинации будут сопоставимы или более эффективны, чем те, что содержат доцетаксель, но будут обладать преимуществом преодоления потенциальной вторичной лекарственной устойчивости, связанной с применением доцетакселя.
Комбинации ингибиторов тубулина с BRAFi или MEKi эффективны на моделях метастазов меланомы
Основная задача в обеспечении длительной выживаемости у больных меланомой состоит в том, чтобы найти эффективное лечение от метастазов меланомы. Вследствие их подкожной природы, PDX-модели редко дают метастазы. Поэтому, в дополнение к оценке эффективности системного введения комбинаций препаратов против опухолей меланомы на этой подкожной модели PDX, как описано выше, определяли эффективность комбинаций на экспериментальных моделях метастазирования в легких на мышах nude. Модель метастазирования в легких выбрана потому, что это один из главных участков метастазирования злокачественной меланомы, который имеет наихудшую выживаемость на 5-летний срок среди всех метастазов меланомы. В нашей лаборатории есть хорошо себя зарекомендовавшая методика для оценки метастазов меланомы в легких (фиг. 28), которая показала, что ABIs в виде монотерапии могут эффективно подавлять метастазирование меланомы в легких (в качестве примера приведено соединение 17уа). Аналогичные модели широко применяются в литературе. Кроме того, в отличие от клеток, культивируемых из давно устоявшихся клеточных линий типа A375, одноклеточные суспензии, выделенные непосредственно из опухолей PDX, никогда не растут на пластике. Таким образом, вероятно, у них сохраняется неоднородность опухолей, точное генетическое соответствие и реакции на лекарственные препараты, которые ожидаются для клеток в исходных опухолях пациентов.
Выделение одноклеточных суспензий из опухолей PDX с небольшим количеством пассажей
Одноклеточные суспензии получали из опухолей PDX по стандартной методике. Вкратце, после приобретения мышей NSG (обычно 1-2 мыши) им имплантировали две PDX-опухоли (Р2) с небольшим количеством пассажей и опухоли вырастали примерно до 2000 мм3. В пределах 1 часа после хирургического извлечения из мышей NSG, свежие опухоли промывали 3 раза средой DMEM, чтобы избежать загрязнения (по 5 минут на льду). Опухоли PDX, лишенные некротической и соединительной ткани, нарезали на мелкие кусочки (1×1 мм или меньше) с помощью стерильных скрещенных лезвий в стерильных условиях. Кусочки опухолей смешивали с раствором коллагеназы IV типа ultra-pure (Gibco 17104-019) при концентрации 1 мг/мл в среде DMEM и инкубировали 1 час при 37°C с легким встряхиванием. После гидролиза полученную смесь фильтровали через нейлоновый сетчатый фильтр (BD Biosciences, поры в 70 мкм, 352340), получая одноклеточные суспензии. Определяли число клеток с помощью счетчика клеток Auto Т4 Cell Counter, осаждали и ресуспендировали их в среде DMEM для инъекции в хвостовую вену. Жизнеспособность полученных этим методом клеток при определении с трипановым синим обычно составляет более 95%.
Подавление метастазирования в легких с помощью вемурафениб-чувствительных или вемурафениб-устойчивых одноклеточных суспензий
Поскольку наилучшие комбинации уже были классифицированы в предыдущем исследовании, описанном выше, то тестировали оптимальные комбинации. В качестве контрольной комбинации всегда включали апробированную комбинацию дабрафениб + траметиниб. Вкратце, каждой из 50 мышей nude (6-недельные, самцы и самки) через хвостовую вену вводили 75000 одиночных клеток, суспендированых в 100 мкл DMEM, полученных из вемурафениб-чувствительной или вемурафениб-устойчивой модели PDX-опухолей, и разбивали мышей на пять групп (n=10): группа отрицательного контроля только с носителем (группа 1); группа, получавшая контрольную комбинацию дабрафениб + траметиниб (группа 2); оптимальную комбинацию доцетакселя с BRAFi/MEKi, как определено выше (группа 3); оптимальную комбинацию соединения 12da или 17уа с BRAFi/MEKi, как определено выше (группа 4 и 5). Количество мышей в каждой группе увеличили с 7 до 10, чтобы обеспечить статистическую значимость, из-за более высокой ожидаемой вариабельности при этой экспериментальной модели метастазов в легких. Через 7-10 дней после инъекции опухолевых клеток мышам вводили носитель или комбинацию препаратов перорально (кроме доцетакселя, который не доступен перорально и вводится внутривенно) ежедневно на протяжении 4 недель. По окончании обработки всех мышей забивали путем ингаляции CO2 с последующим смещением шейных позвонков. Мышей вскрывали и извлекали легкие. Точно подсчитывали количество опухолевых узелков в легких и оценивали эффективность лечения на основании отсутствия или уменьшения количества опухолевых узелков в легких. Также проверяли меланомный характер узелков и морфологию опухолей после фиксации и обработки для получения заключенных в парафин срезов с последующим окрашиванием Н&Е. Для выполнения этого эксперимента с использованием клеток из трех вемурафениб-чувствительных и трех вемурафениб-устойчивых моделей PDX нужно использовать до 50×3×2=300 мышей nude.
Ожидаемые результаты, подводные камни и альтернативные подходы
Ожидается, что эти комбинации будут эффективно снижать количество метастазов в легких при вемурафениб-чувствительных опухолях и в меньшей степени вемурафениб-устойчивых опухолях. Комбинации, содержащие ингибитор тубулина (доцетаксель, соединение 12da или 17уа), должны быть более эффективными, чем стандартная комбинация дабрафениб + траметиниб, особенно при метастазировании вемурафениб-устойчивых опухолей. В том маловероятном случае, когда одноклеточные суспензии, выделенные из опухолей PDX, не будут образовывать метастазы в легких, в качестве альтернативного подхода следует использовать признанные линии меланомы человека BRAFV600E (YUGEN8, YUSAC2, YUKOLI и YUSIK) на ранних стадиях пассирования.
Все описанные здесь признаки (включая прилагаемую формулу изобретения, реферат и чертежи) и/или все раскрытые при этом стадии любого способа или процесса могут комбинироваться с любым из вышеприведенных аспектов в любом сочетании, за исключением тех сочетаний, в которых по крайней мере некоторые из таких признаков и/или стадий являются взаимоисключающими. Хотя здесь были представлены и подробно описаны предпочтительные воплощения, однако специалистам в соответствующей области должно быть ясно, что могут производиться различные модификации, дополнения, замены и пр., не отходящие от сущности изобретения, которые при этом считаются в рамках объема изобретения, как определено в нижеследующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ PAC-1 | 2016 |
|
RU2720509C2 |
| КОМПОЗИЦИИ АПИЛИМОДА И СПОСОБЫ ЕГО ИСПОЛЬЗОВАНИЯ ПРИ ЛЕЧЕНИИИ МЕЛАНОМЫ | 2015 |
|
RU2731908C2 |
| Способ адъювантного лечения рака | 2013 |
|
RU2640180C2 |
| КОМПОЗИЦИИ АПИЛИМОДА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ КОЛОРЕКТАЛЬНОГО РАКА | 2015 |
|
RU2739992C2 |
| ИНГИБИТОР BRAF КИНАЗЫ N-(3-(5-(4-ХЛОРОФЕНИЛ)-1H-ПИРАЗОЛО[3,4-B]ПИРИДИН-3-КАРБОНИЛ)-2,4-ДИФТОРОФЕНИЛ) ПРОПАН-1-СУЛЬФОНАМИД | 2018 |
|
RU2687107C1 |
| КОМПОЗИЦИИ НА ОСНОВЕ ИНГИБИТОРА SHP2 И СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2805355C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МЕТИЛХИНАЗОЛИНОНА | 2020 |
|
RU2802968C1 |
| ТЕРАПЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР RAF И ИНГИБИТОР ERK | 2017 |
|
RU2774612C2 |
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ С ПРИМЕНЕНИЕМ КОМБИНАЦИЙ ИНГИБИТОРОВ ERK И RAF | 2014 |
|
RU2722784C2 |
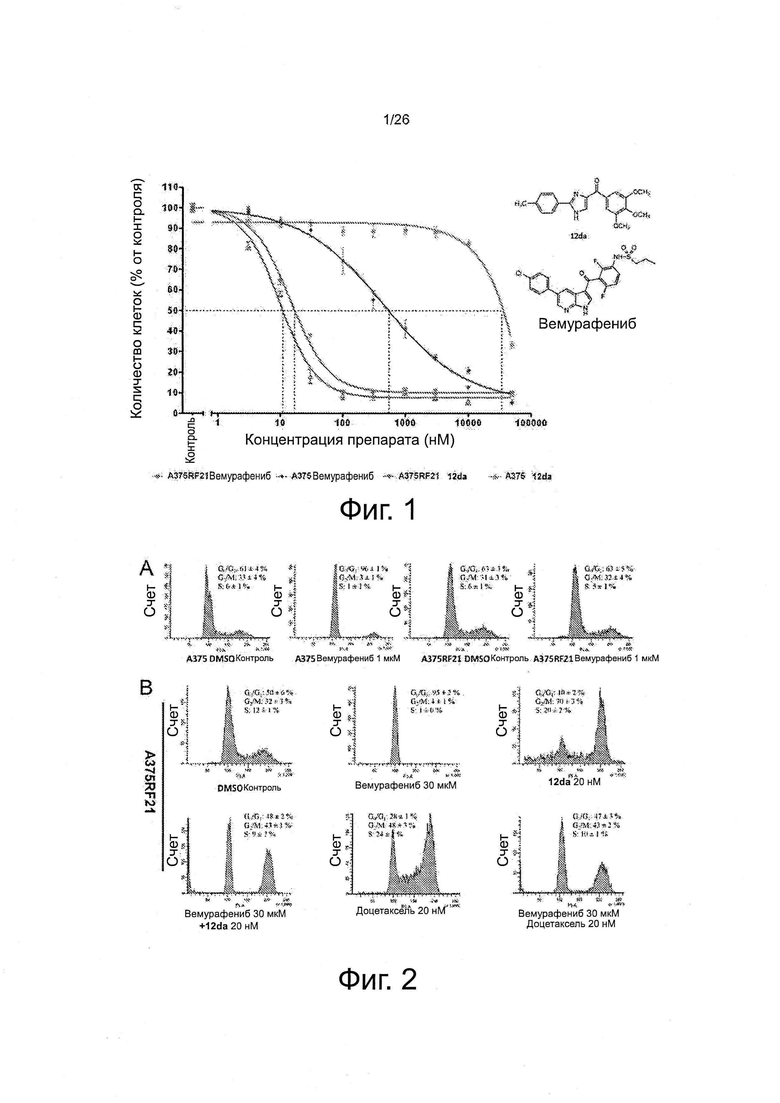
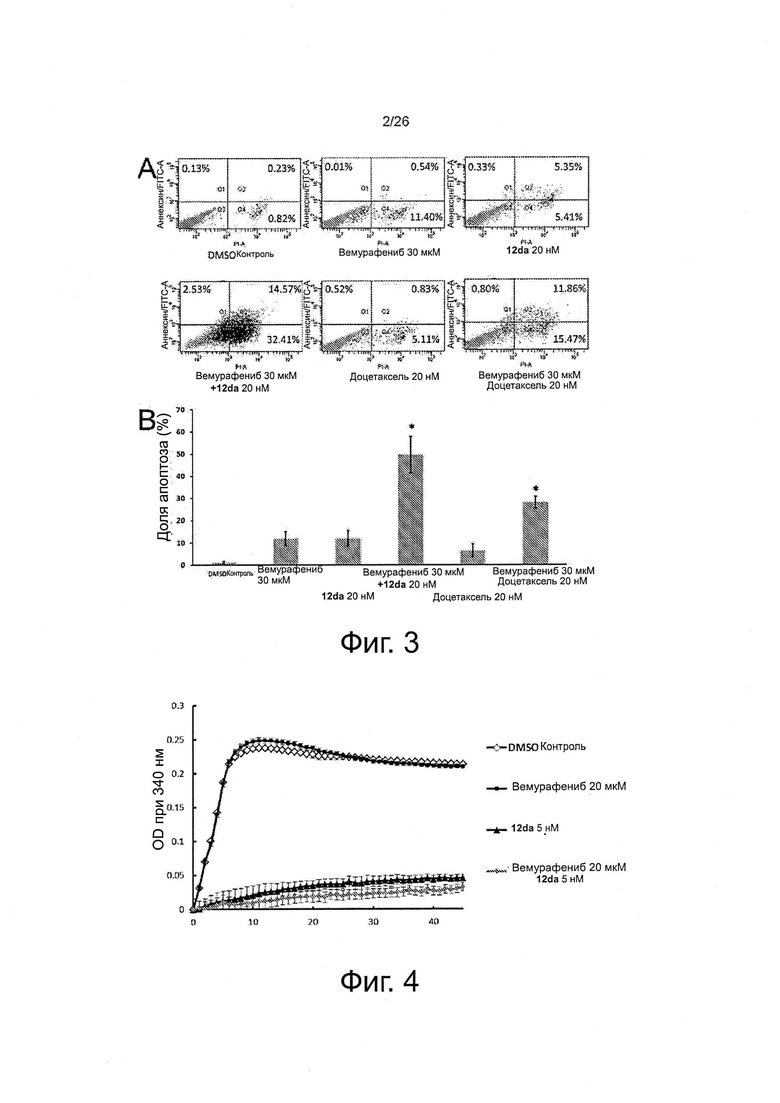
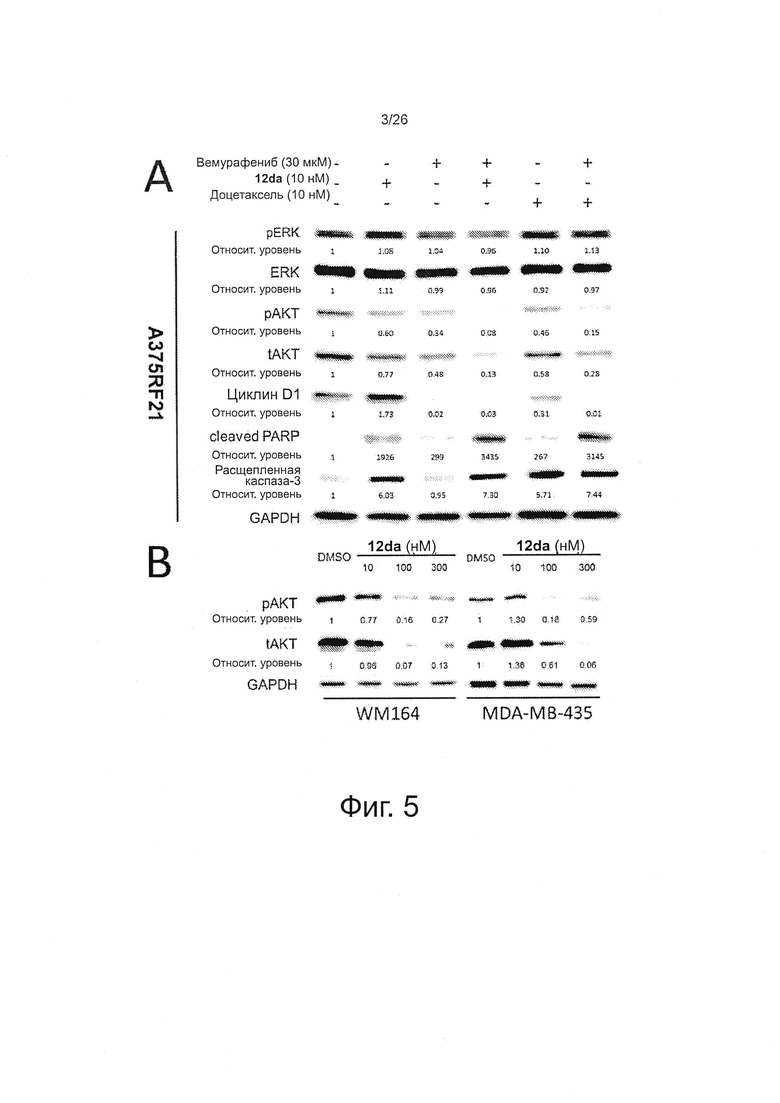
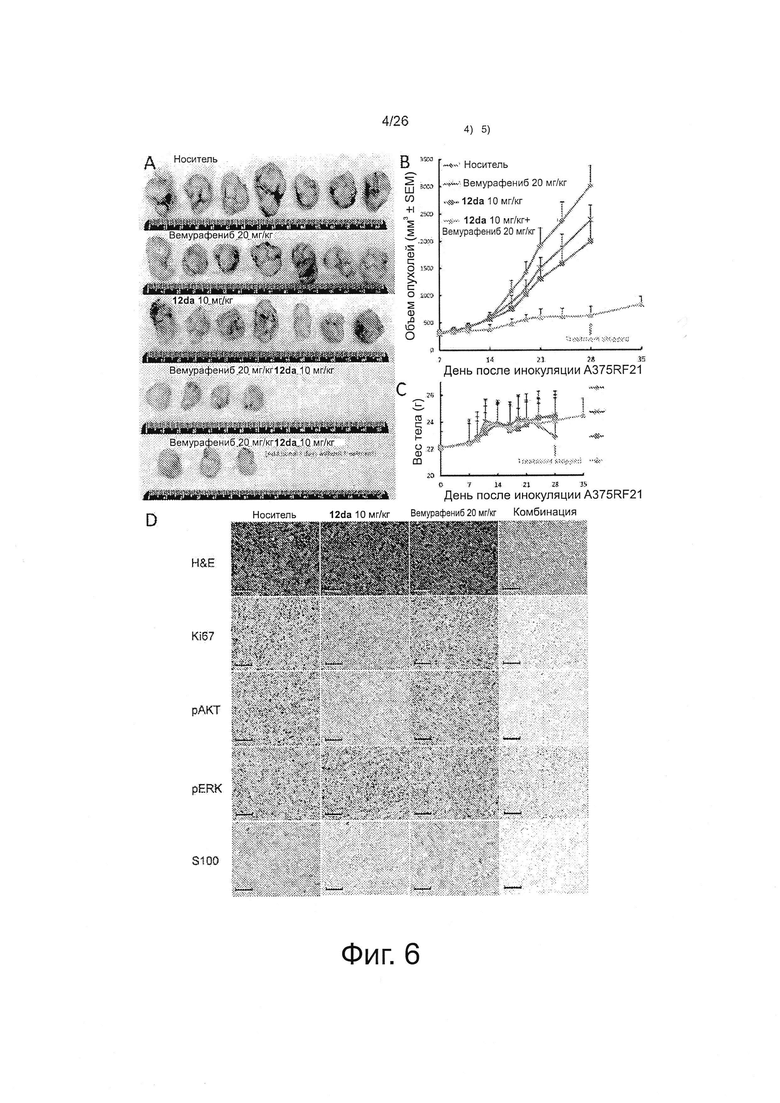
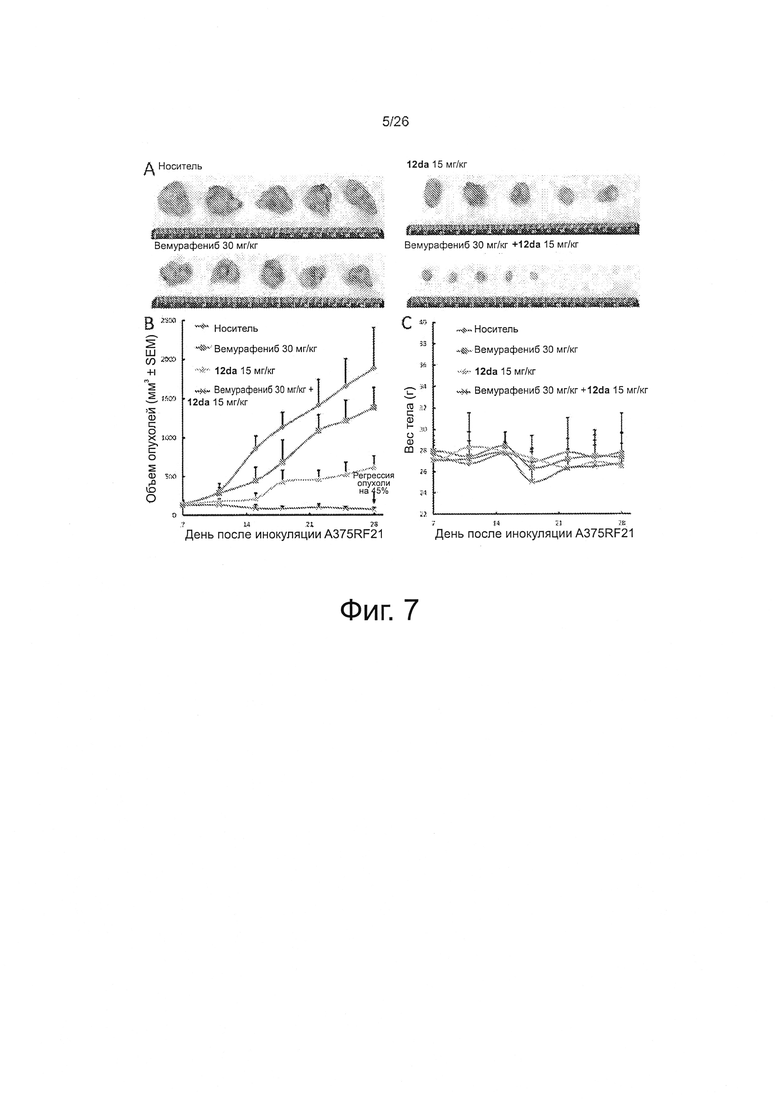
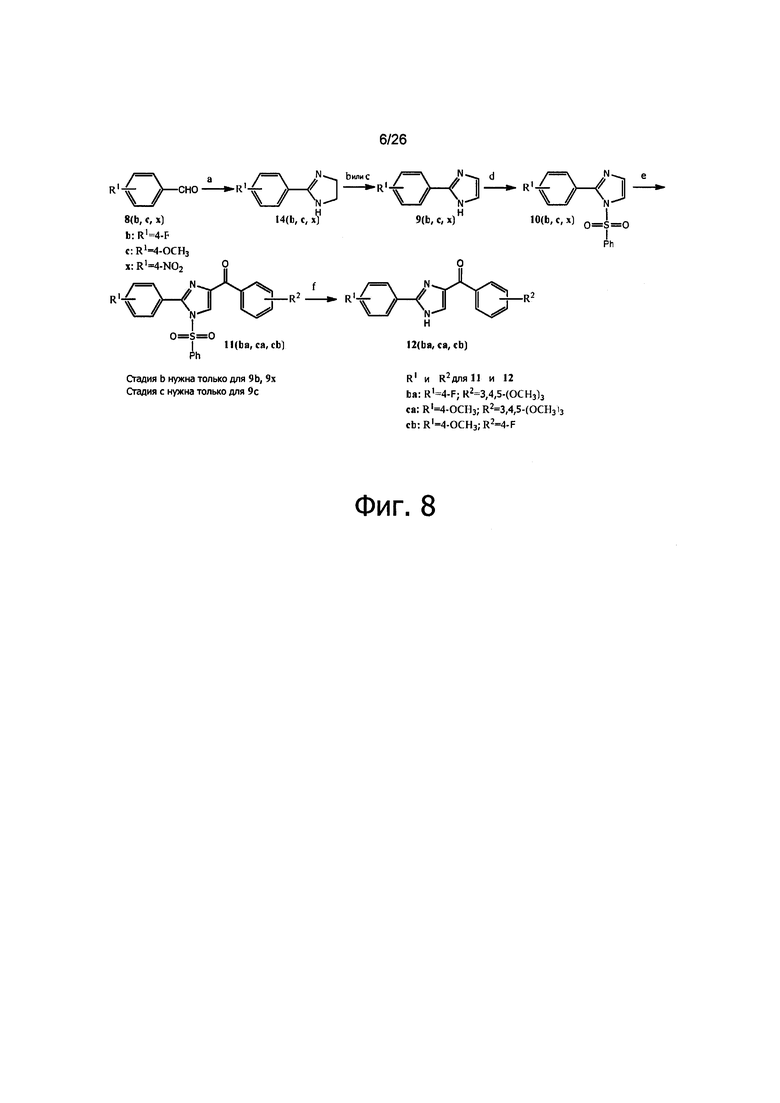
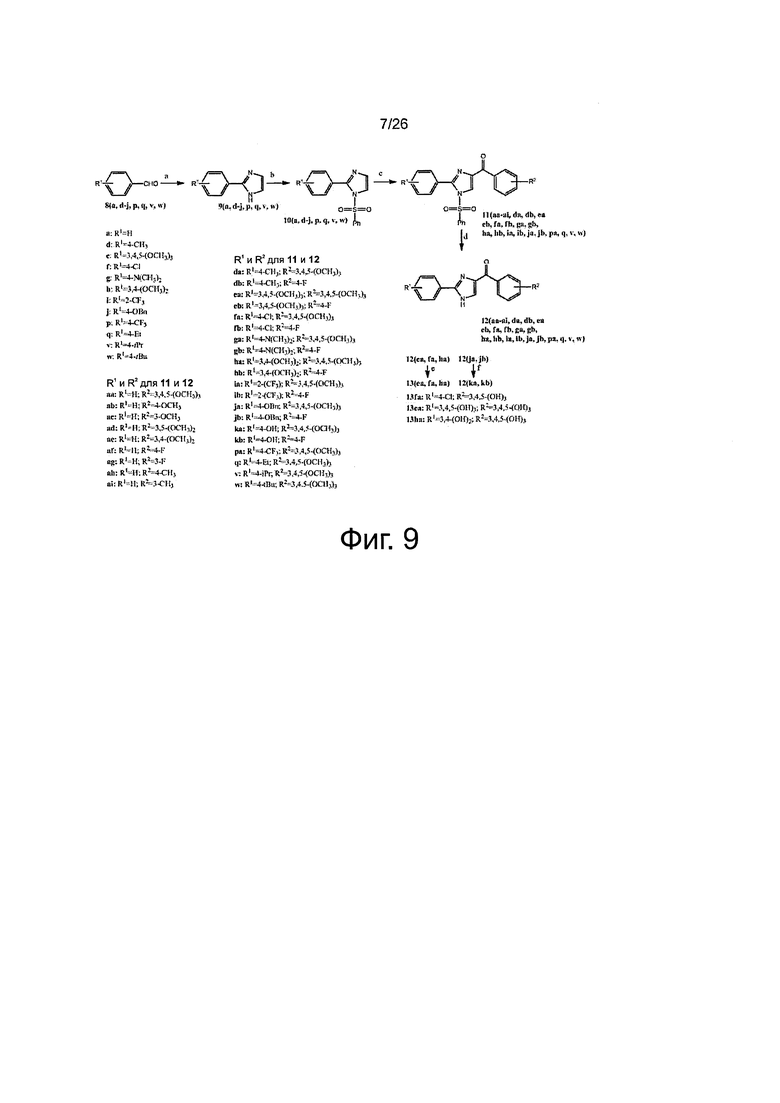
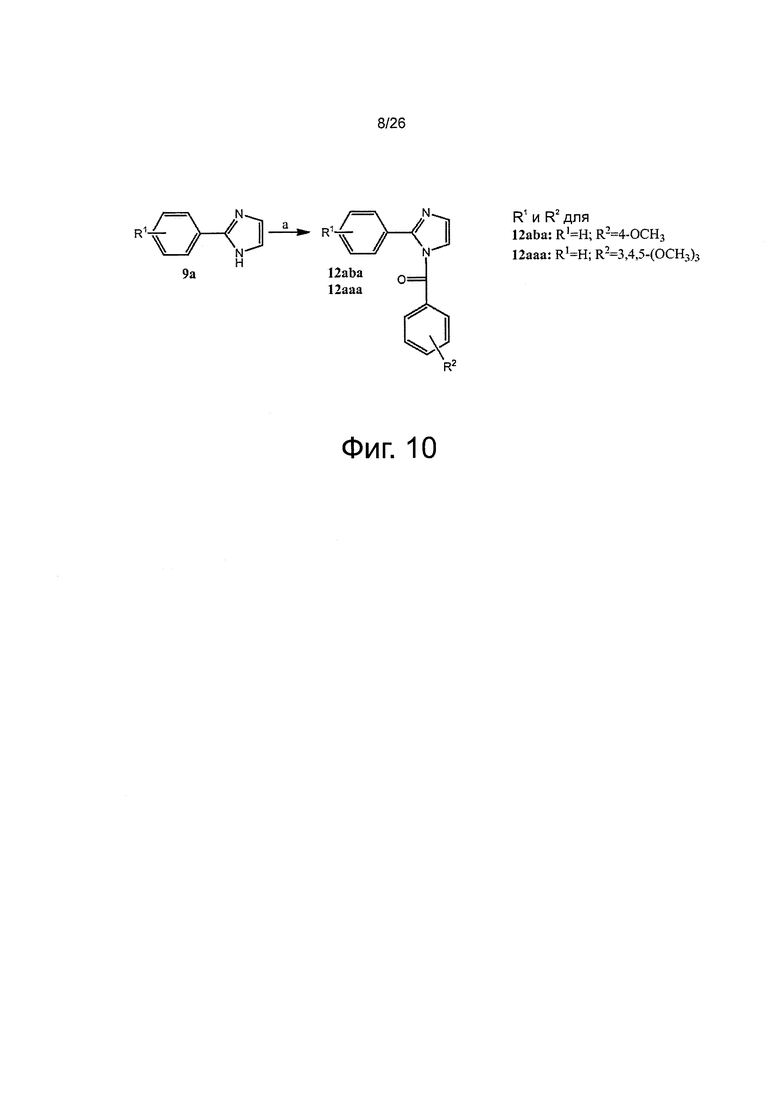
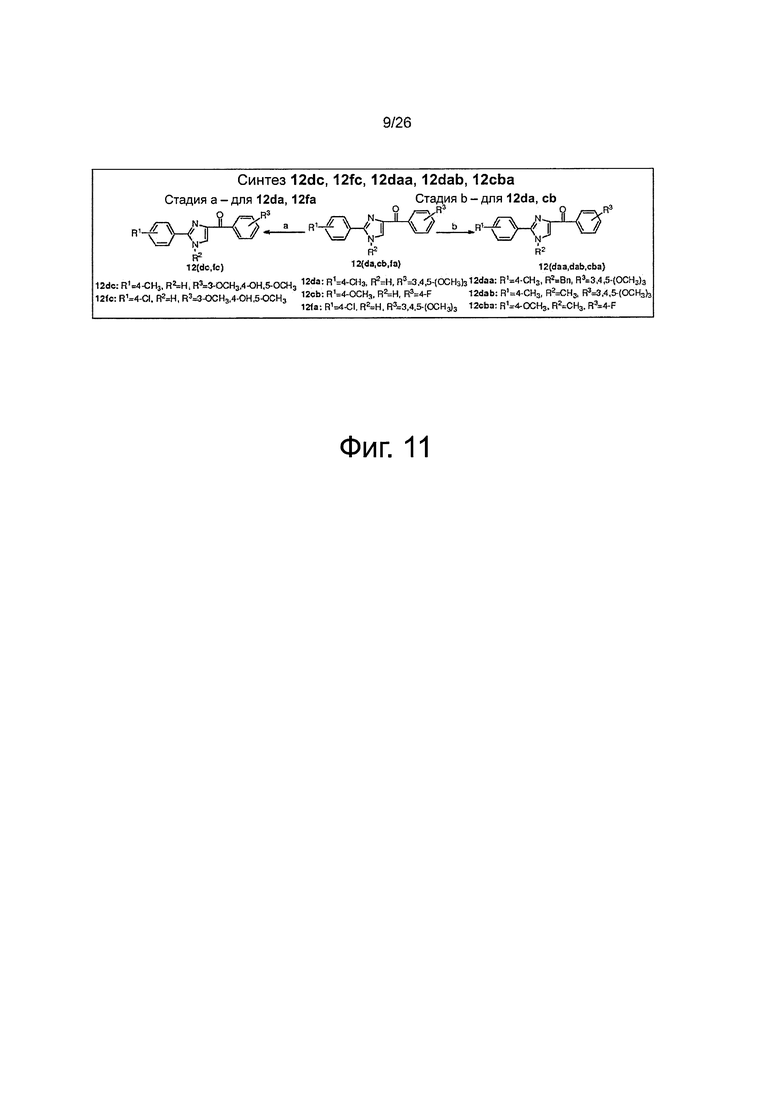
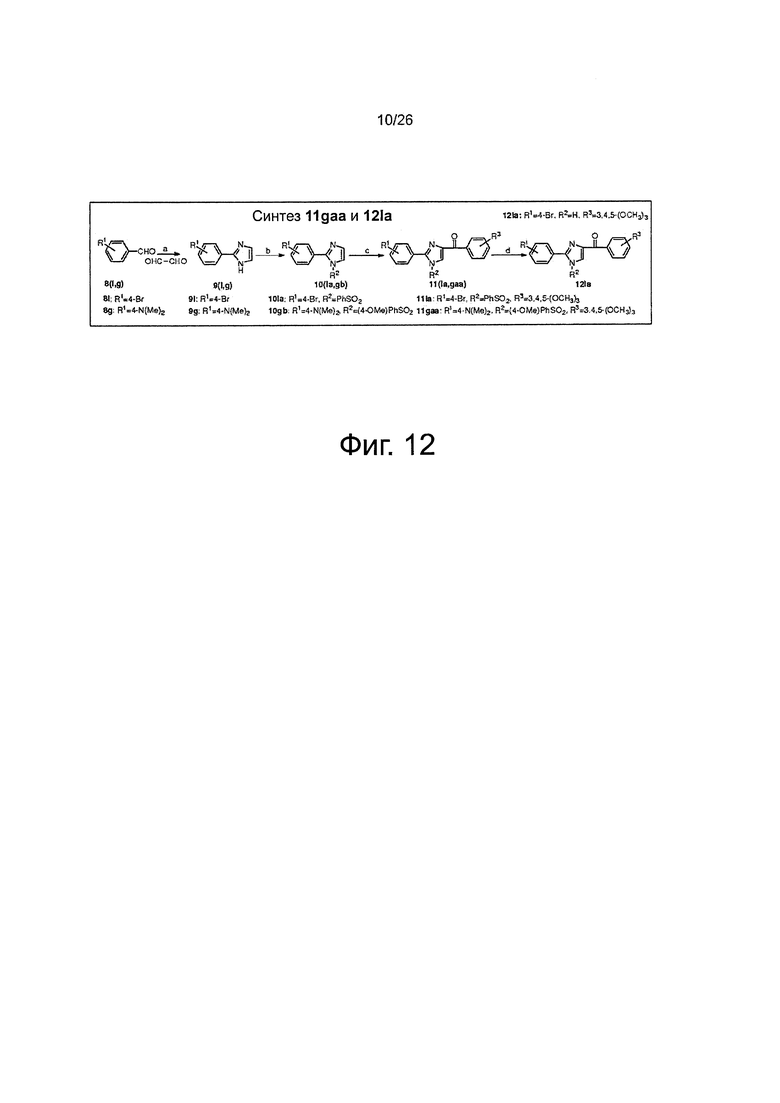
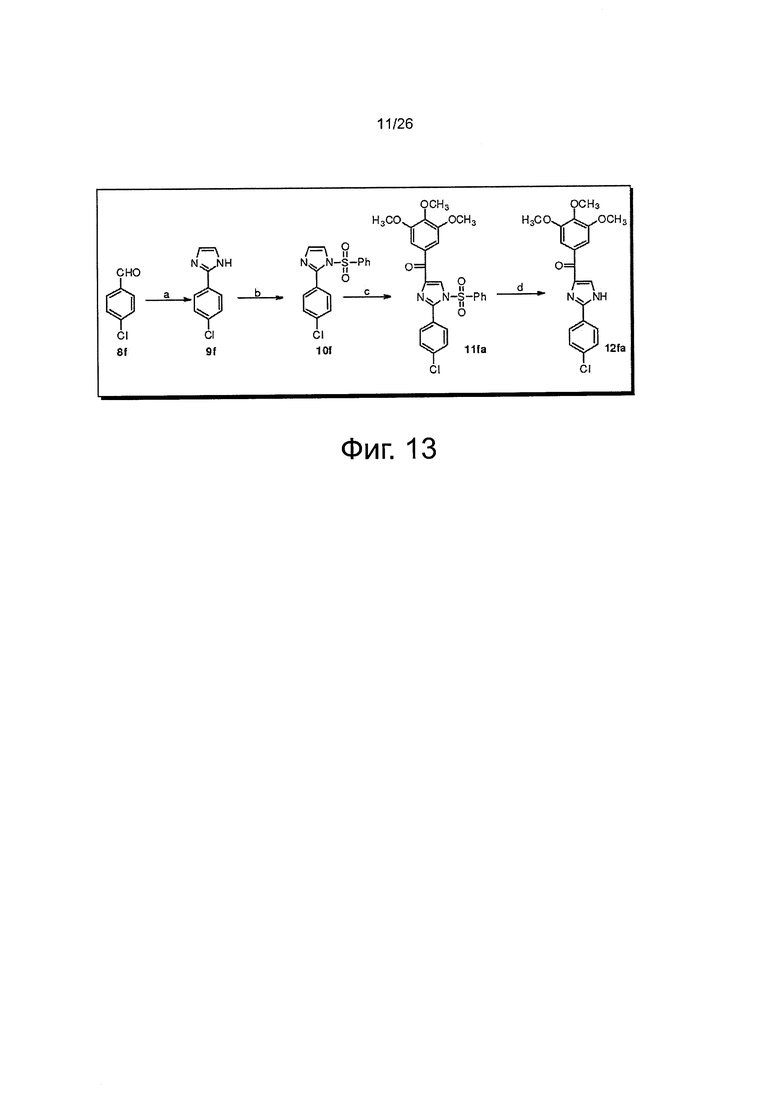
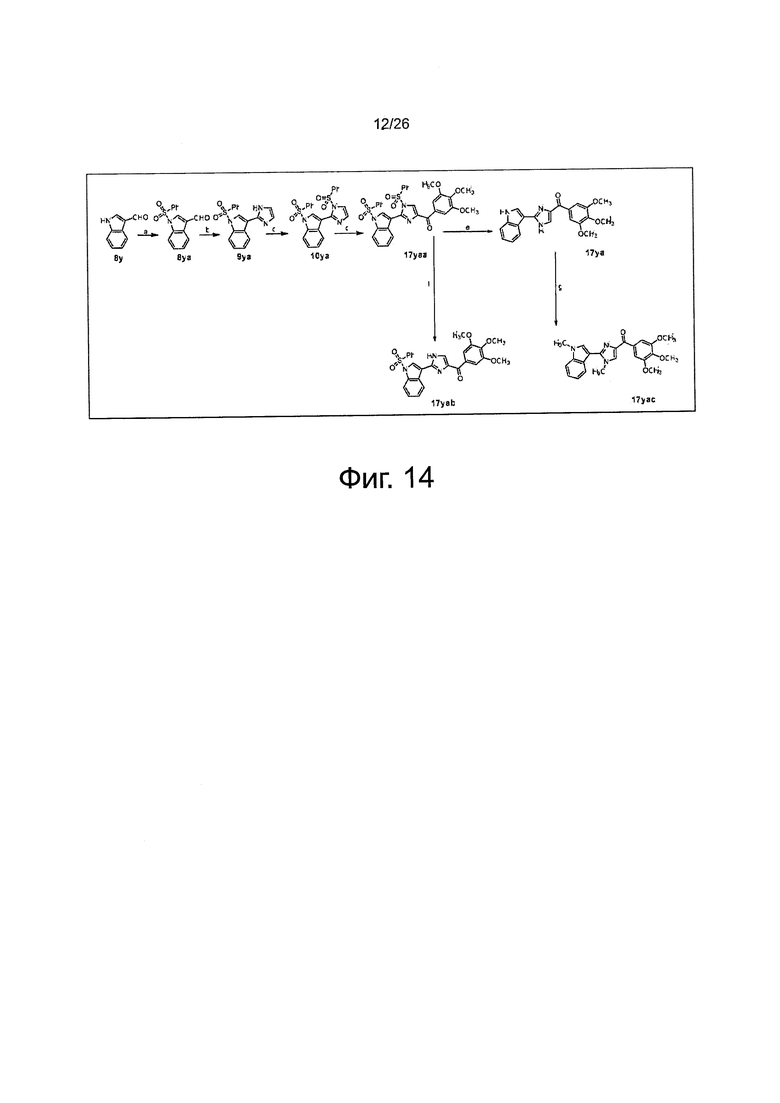

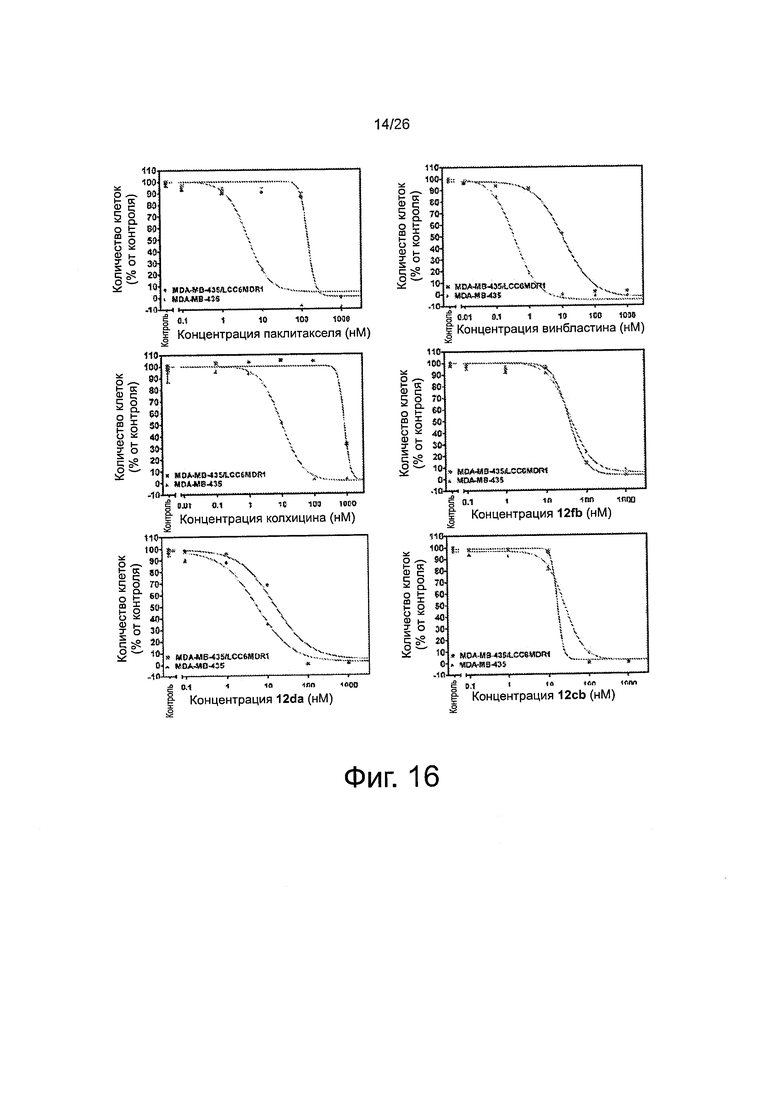
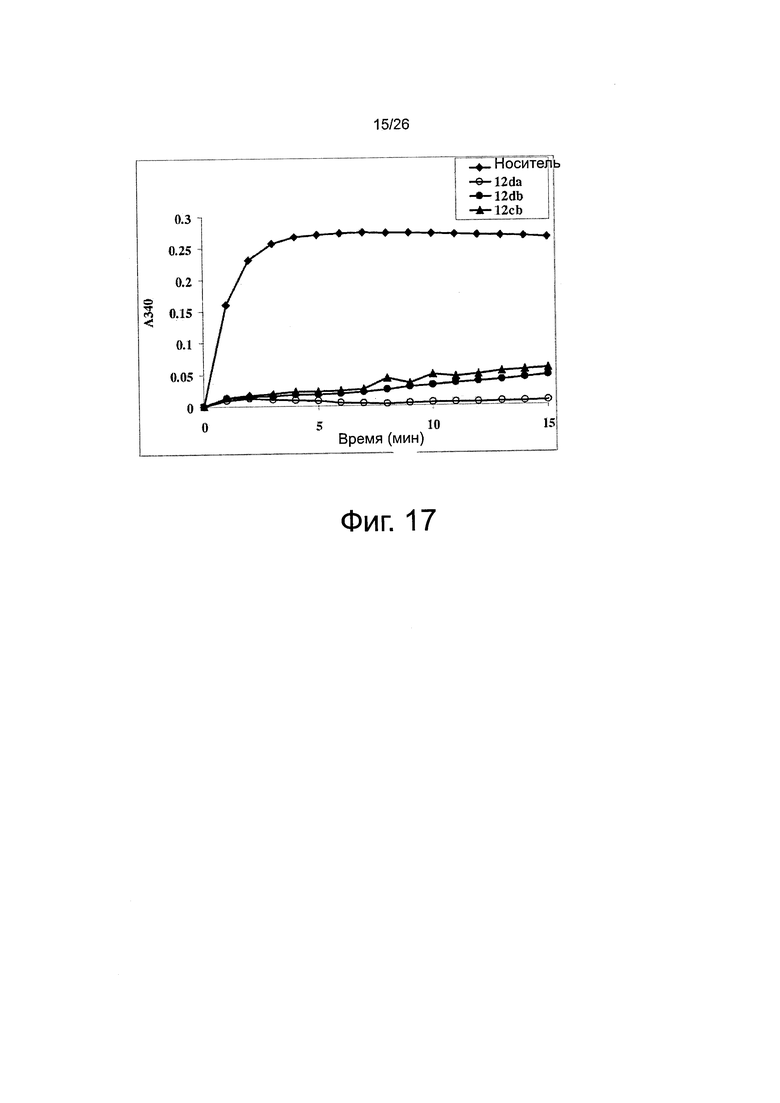
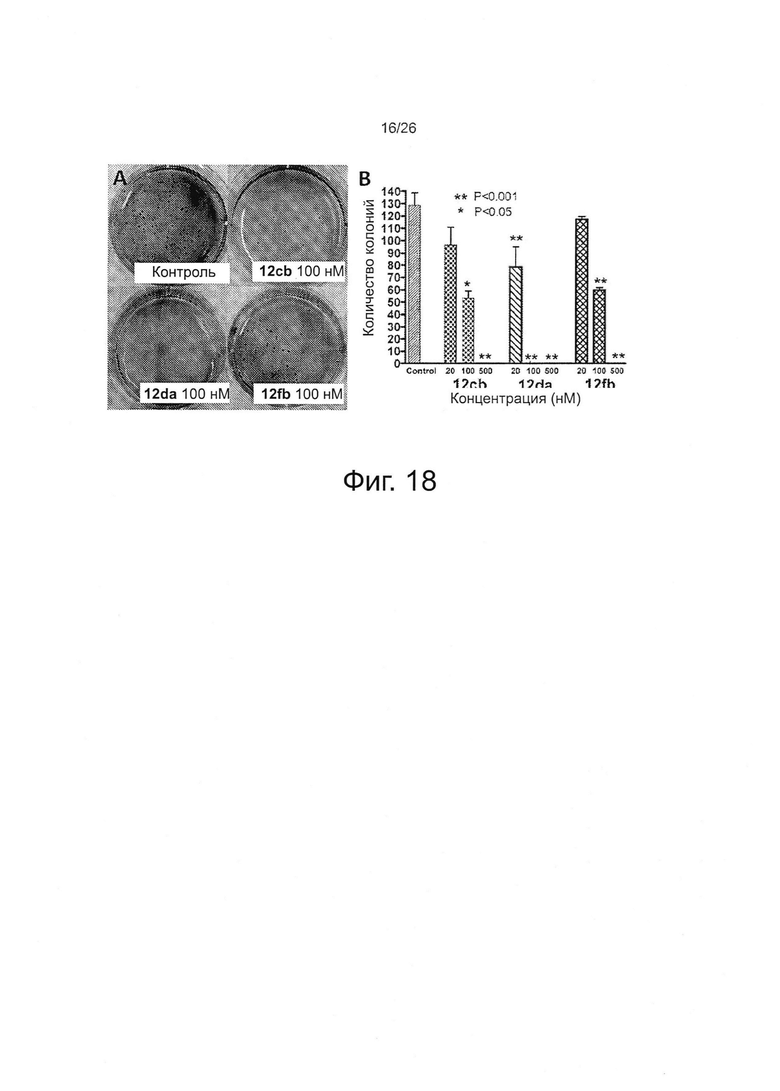
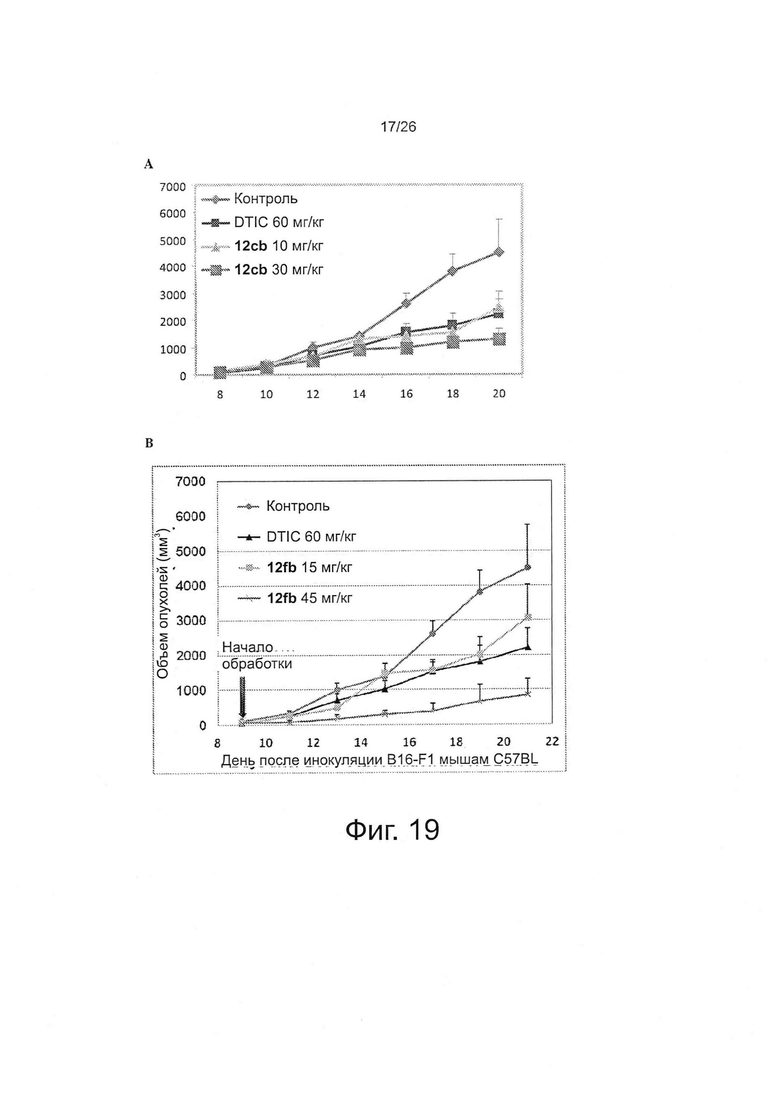
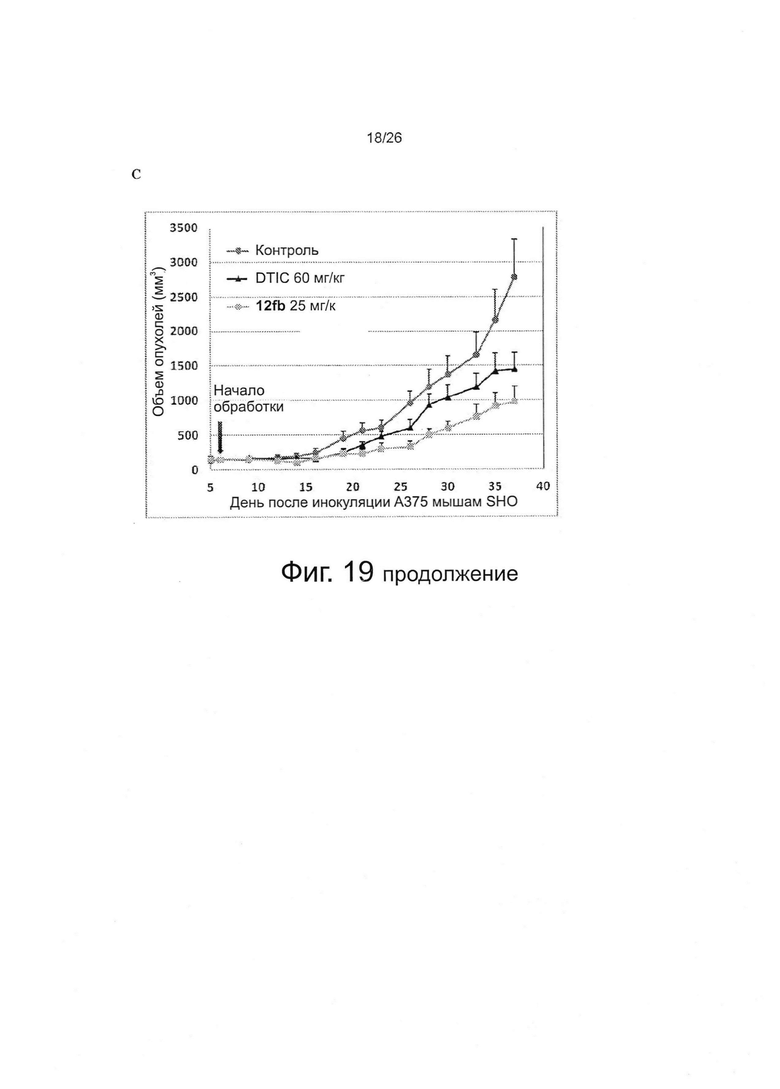
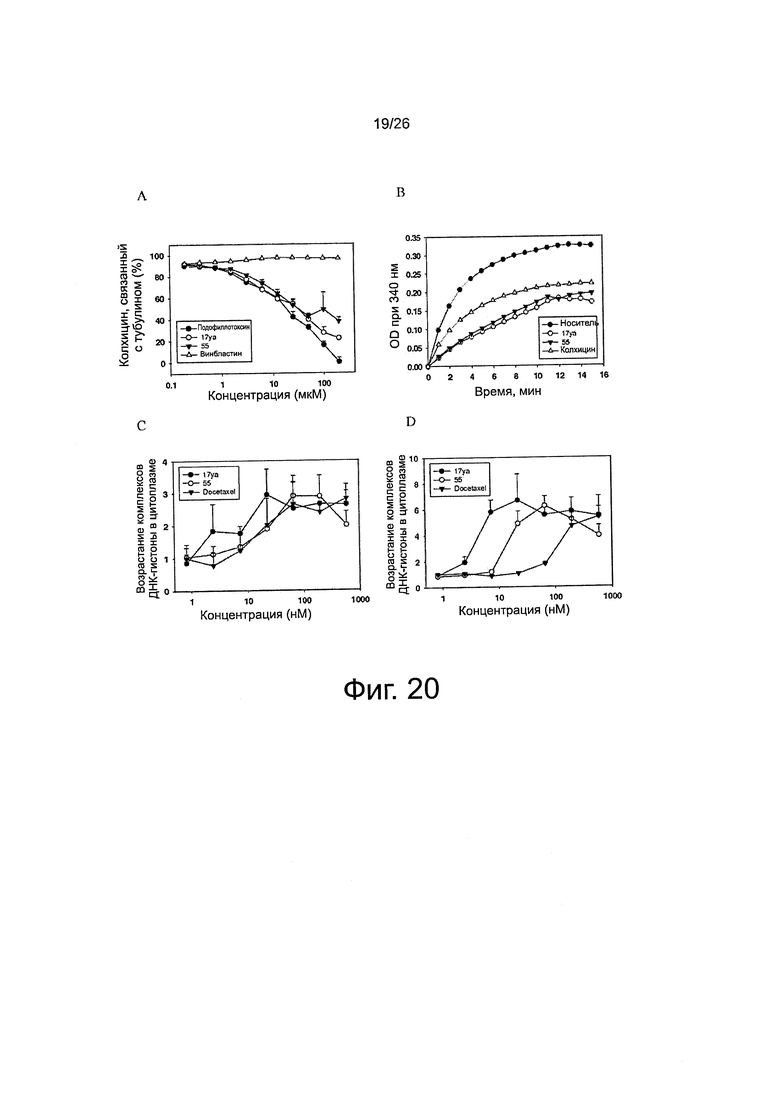
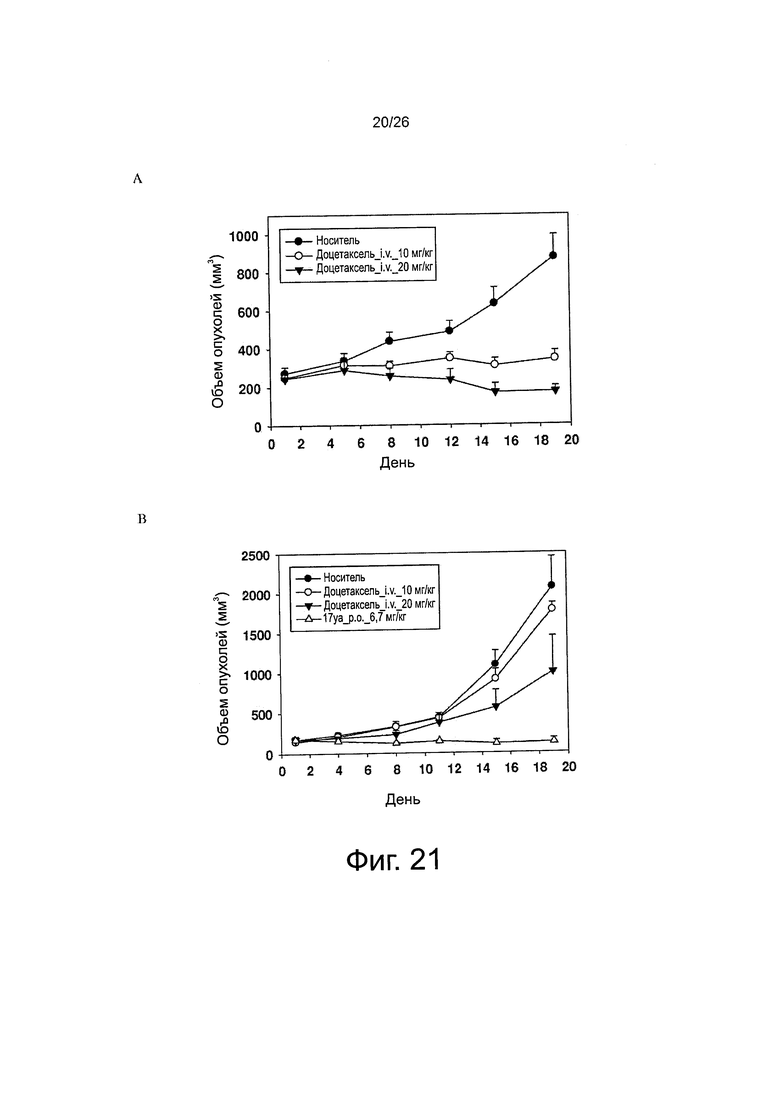
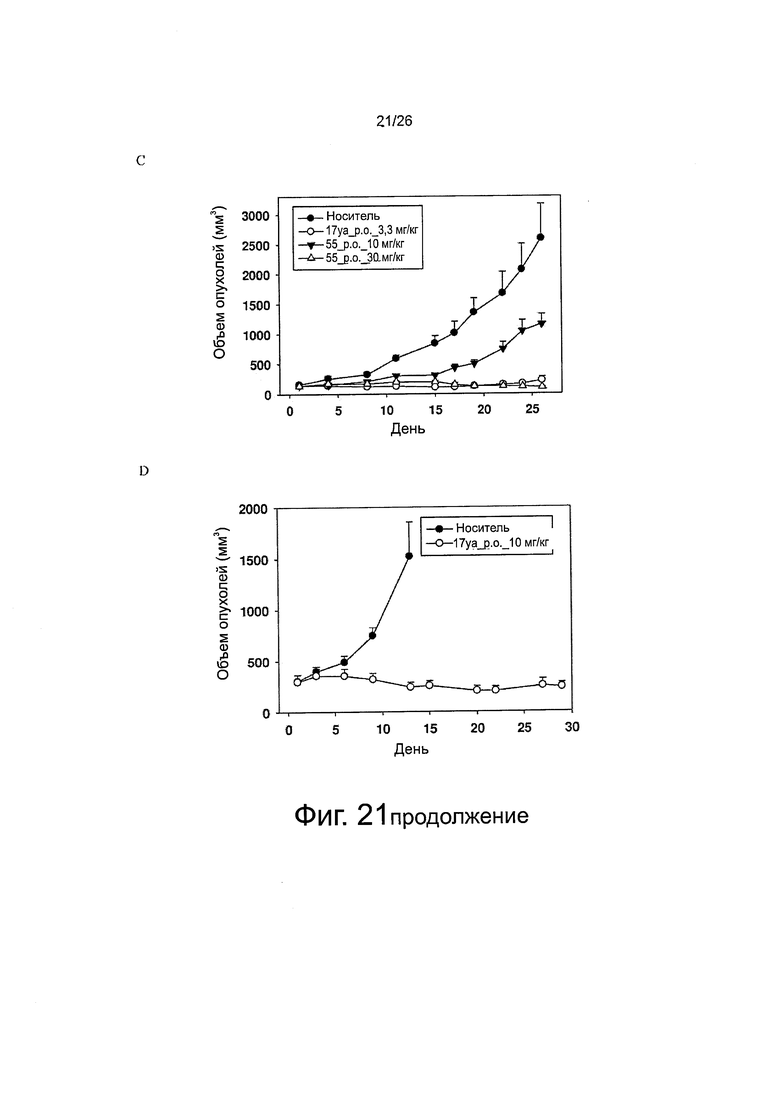
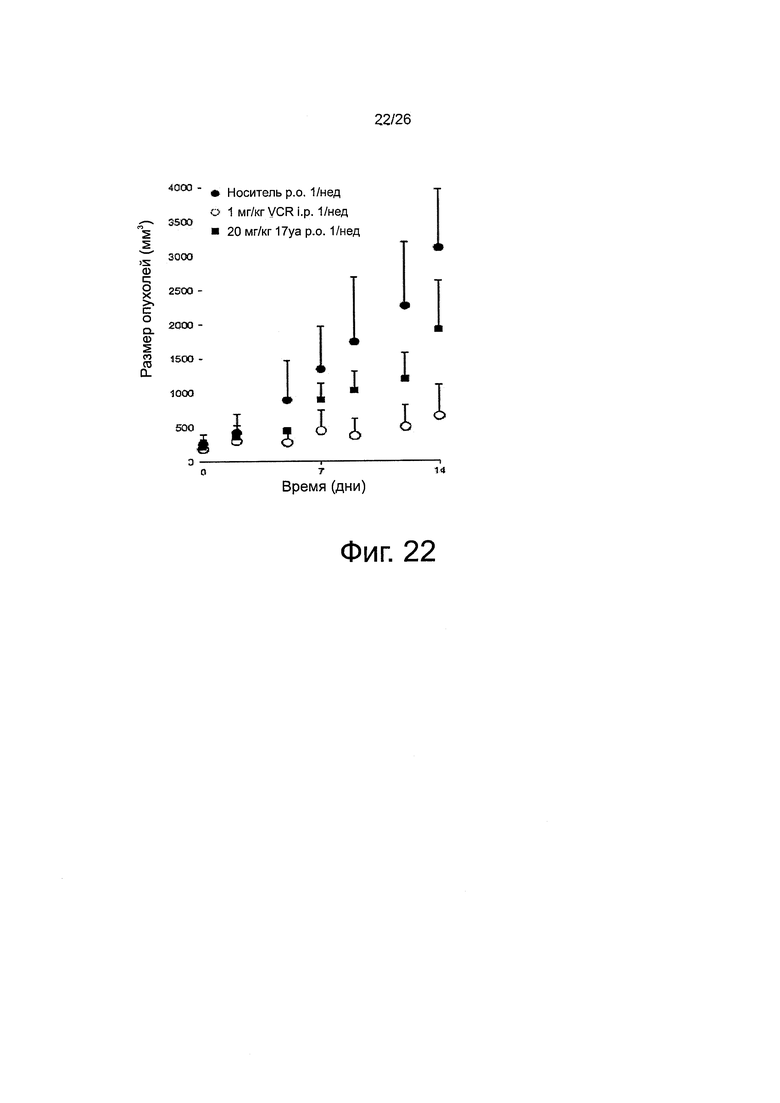
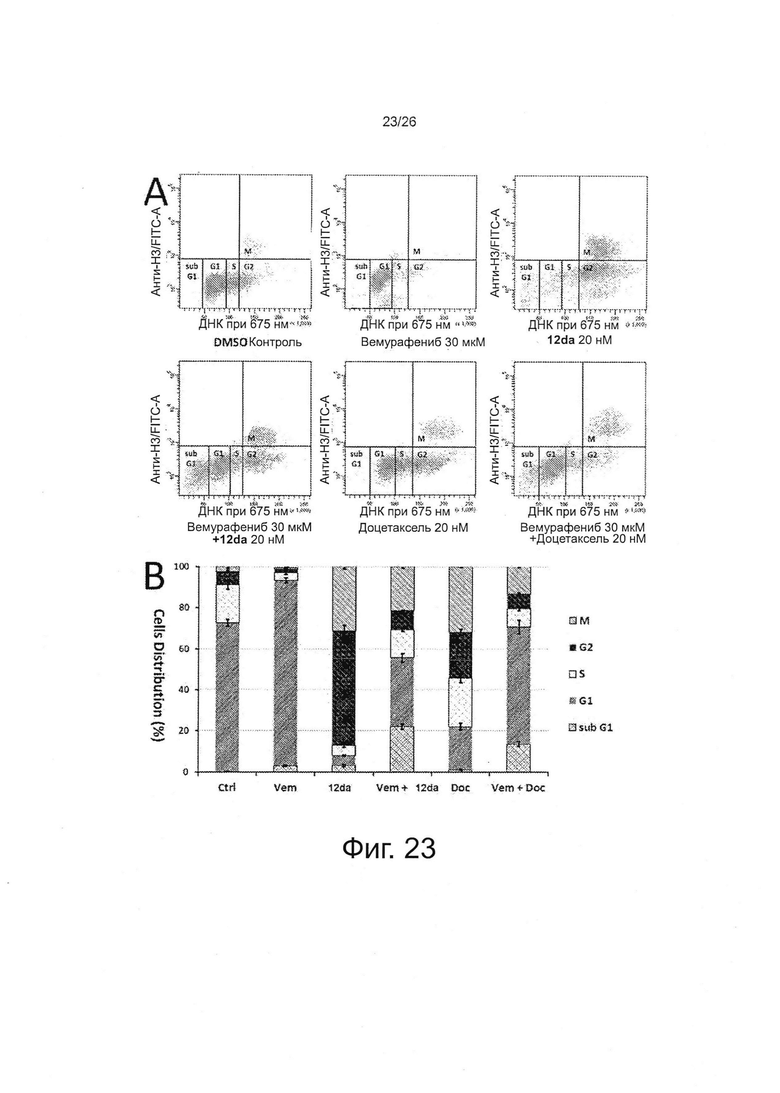
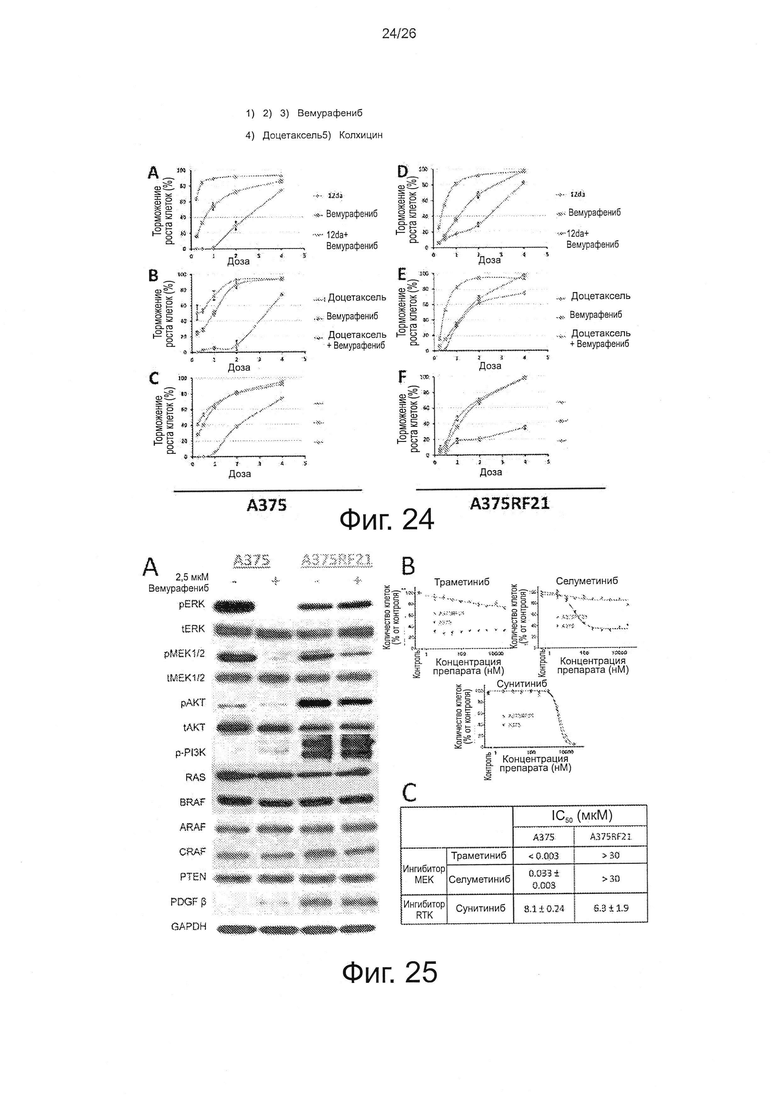
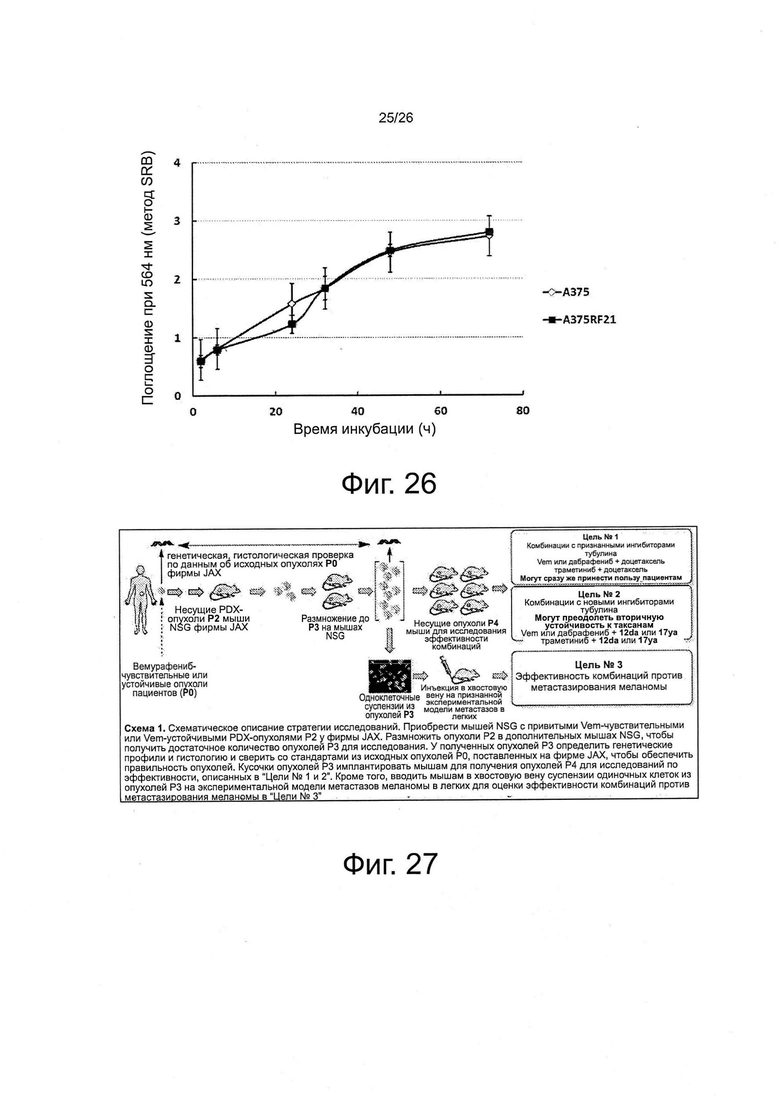
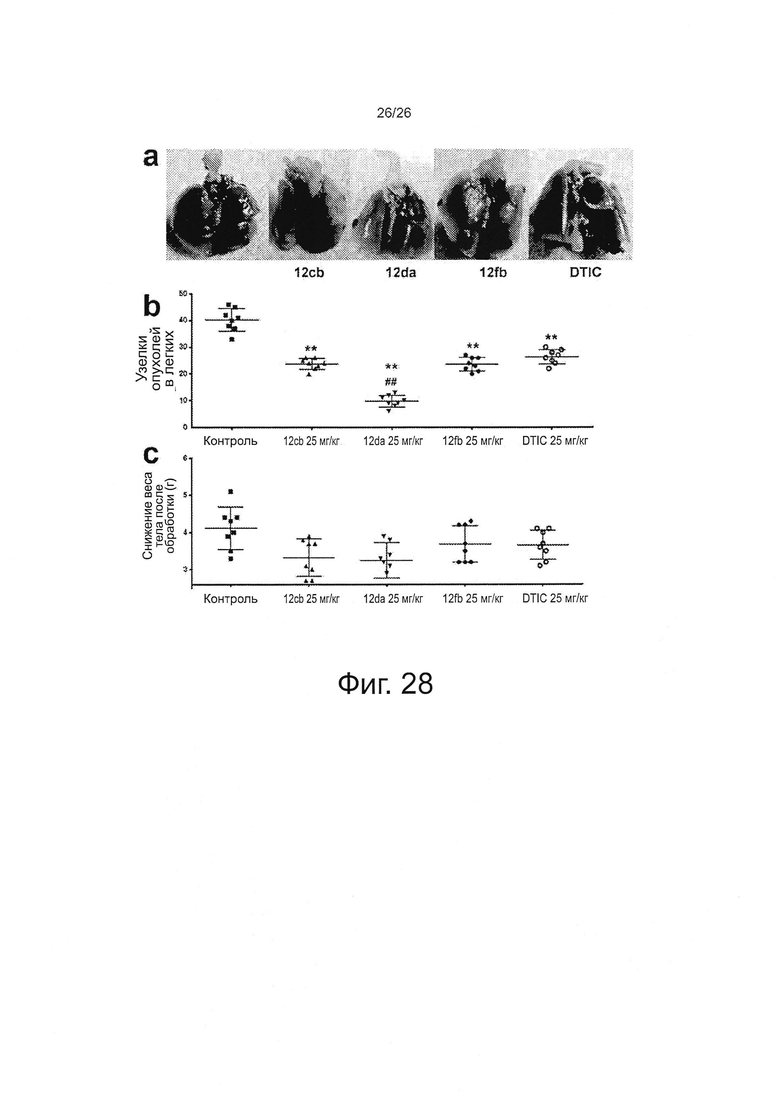
Группа изобретений относится к медицине, а именно к онкологии, и может быть использована при лечении рака. Фармацевтическая композиция по изобретению содержит ингибитор BRAF вемурафениб и ингибитор тубулина, представляющий собой соединение формулы 17ya или 12da
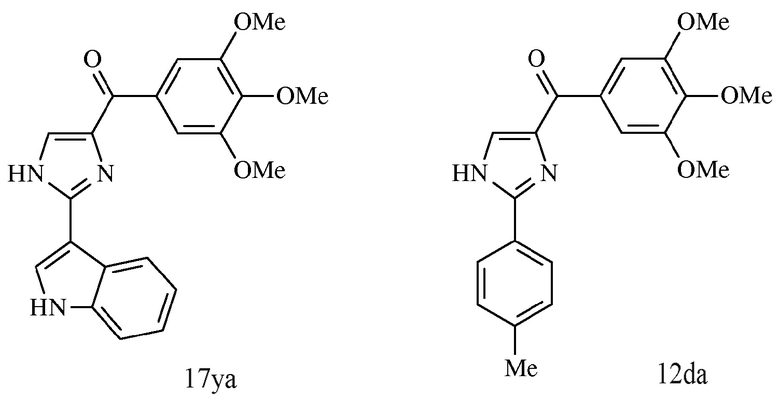
Способы по изобретению включают введение композиции. Использование изобретений позволяет подавлять рост опухолей за счет синергетического действия комбинации, снизить число побочных эффектов. 11 н. и 1 з.п. ф-лы, 17 табл., 28 ил., 11 пр.
1. Фармацевтическая композиция, обладающая антипролиферативным эффектом, содержащая (а) ингибитор тубулина, (b) ингибитор BRAF вемурафениб и (c) фармацевтически приемлемый носитель, где ингибитор тубулина представляет собой соединение формулы 17ya или 12da:
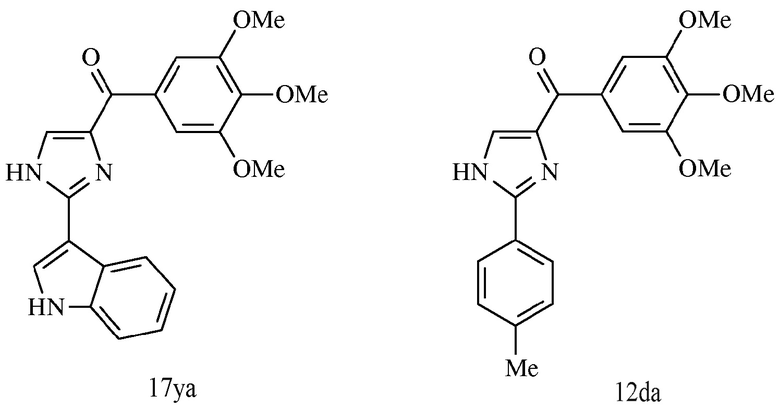
или их комбинацию,
либо фармацевтически приемлемую соль соединения, его N-оксид, гидрат, таутомер или изомер.
2. Способ лечения, подавления, уменьшения тяжести, снижения риска или торможения рака с мутацией BRAF у субъекта, включающий введение композиции по п. 1 субъекту, страдающему раком с мутацией BRAF.
3. Способ лечения, подавления, уменьшения тяжести, снижения риска или торможения устойчивого к ингибиторам BRAF рака у субъекта, страдающего раком, включающий введение данному субъекту композиции по п. 1 субъекту, страдающему устойчивым к ингибиторам BRAF раком.
4. Способ лечения, подавления, уменьшения тяжести, снижения риска или торможения меланомы, включающий введение композиции по п. 1 субъекту, страдающему меланомой.
5. Способ лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивой меланомы, включающий введение композиции по п. 1 субъекту, страдающему лекарственно-устойчивой меланомой.
6. Способ замедления или предотвращения устойчивого к ингибиторам BRAF рака у субъекта, страдающего раком, включающий введение данному субъекту композиции по п. 1.
7. Способ лечения, подавления, уменьшения, торможения, устранения, замедления или предотвращения раковых метастазов у субъекта, страдающего раком, включающий введение данному субъекту композиции по п. 1.
8. Способ лечения, подавления, уменьшения, торможения, устранения, замедления или предотвращения вторичной устойчивости рака к таксановым препаратам у субъекта, страдающего раком и ранее принимавшего таксановые препараты, включающий введение данному субъекту композиции по п. 1.
9. Способ по п. 8, при этом таксан представлен доцетакселем.
10. Способ лечения, подавления, уменьшения тяжести, снижения риска или торможения лекарственно-устойчивого рака, включающий введение композиции по п. 1 субъекту, страдающему лекарственно-устойчивым раком.
11. Способ подавления приобретенной устойчивости к ингибиторам BRAF у субъекта, страдающего раком, включающий введение композиции по п. 1 данному субъекту, страдающему раком.
12. Способ замедления или предотвращения развития устойчивости к ингибиторам BRAF у субъекта, страдающего раком, включающий введение композиции по п. 1 данному субъекту, страдающему раком.
| WO 2012136776, 11.10.2012 | |||
| US 20120071524, 22.03.2012 | |||
| US 20120053185 A1, 01.03.2012 | |||
| US 20080146555 A1, 19.06.2008 | |||
| WATANABE K | |||
| et al | |||
| Blockade of the extracellular signal-regulated kinase pathway enhances the therapeutic efficacy of microtubule-destabilizing agents in human tumor xenograft models//Clin Cancer Res | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| doi: | |||

