В данном изобретении предложено новое соединение, его получение, содержащие его фармацевтические композиции и его применение в качестве терапевтически активного вещества. Соединение по изобретению является ингибитором BRAF и имеет свойства, нарушающие парадокс.
В данном изобретении предложено, в частности, новое соединение формулы (I)
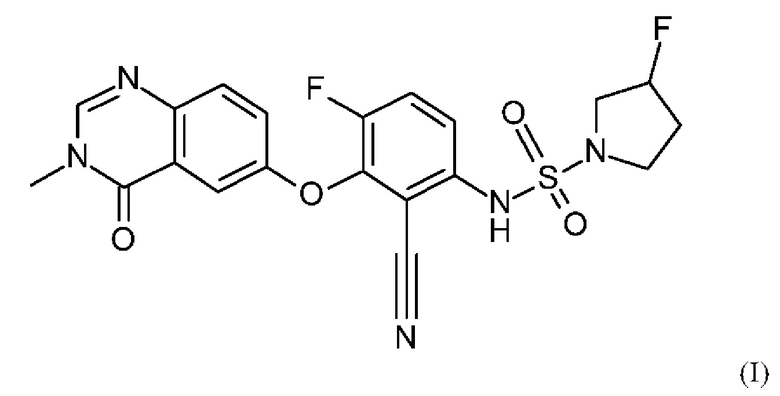
или его фармацевтически приемлемая соль.
Класс серин-треонинкиназ быстро развивающейся фибросаркомы (RAF, rapidly accelerated fibrosarcoma) состоит из трех элементов (ARAF, BRAF, RAF1), составляющих первый узел сигнального пути киназы MAP. Несмотря на очевидную избыточность трех изоформ RAF в передаче сигналов через фосфорилирование MEK1 и 2, частые онкогенные активирующие мутации обычно встречаются только для BRAF. В частности, замена V600 глутаминовой кислотой или лизином делает киназу высокоактивированной с последующей гиперстимуляцией пути MAPK независимо от внешних стимуляций (Cell. 2015 Jun 18; 161 (7): 1681-1696).
Мутантный BRAF является целевым онкогенным драйвером, и три ингибитора BRAF (вемурафениб, дабрафениб и энкорафениб), которые до этих пор вышли на рынок, демонстрируют эффективность при BRAFV600E-положительной меланоме. Однако почти повсеместно наблюдается быстрое приобретение устойчивости к лекарственному средству, а длительность терапевтического эффекта от целевой терапии остается ограниченной.
В дополнение, разработанные ингибиторы BRAF проявили неожиданную и «парадоксальную» способность подавлять передачу сигналов MAPK в опухолях, вызванных BRAFV600E, тогда как те же ингибиторы демонстрировали стимулирующую активность в отношении MAPK в моделях BRAF дикого типа (WT, wild type) (N Engl J Med 2012; 366: 271-273; и British Journal of Cancer volume 111, pages 640-645 (2014)).
Впоследствии механистические исследования парадокса RAF выяснили, что онкогенный BRAFV600E фосфорилирует MEK 1/2 в его мономерной цитозольной форме, тогда как активация WT BRAF и RAF1 требует сложного этапа событий, включающего транслокацию клеточной мембраны и гомо и/или гетеродимеризацию, чему способствует активированный RAS (KRAS, NRAS, HRAS) (Nature Reviews Cancer volume 14, pages 455-467 (2014)).
Связывание ингибиторов, таких как вемурафениб, дабрафениб или энкорафениб, с протомером WT BRAF или RAF1 быстро индуцирует гомо- и/или гетеродимеризацию RAF и мембранную ассоциацию новообразованного димера RAF. В димерной конформации один протомер RAF аллостерически индуцирует конформационные изменения второго, что приводит к активному состоянию киназы и, что важно, конформации, неблагоприятной для связывания ингибитора. В результате димер, индуцированный лечением лекарственным средством, способствует фосфорилированию MEK с помощью катализа, индуцируемого несвязанным протомером вместе с гиперактивацией пути.
Парадокс RAF приводит к двум клинически значимым последствиям: 1) ускоренный рост вторичных опухолей при монотерапии BRAFi (преимущественно кератохантомы и плоскоклеточного рака) (N Engl J Med 2012; 366: 271-273) и 2) приобретение устойчивости к лекарственному средству в условиях монотерапии BRAFi, а также при комбинации BRAFi+MEKi, представляющей собой активацию димер-опосредованной сигнализации RAF с помощью генетически обусловленных событий, включая мутации RAS, амплификации BRAF, экспрессию вариантов сплайсинга BRAF (Nature Reviews Cancer volume 14, pages 455-467 (2014)). Таким образом, существует потребность в ингибиторах RAF, способных нарушить этот парадокс.
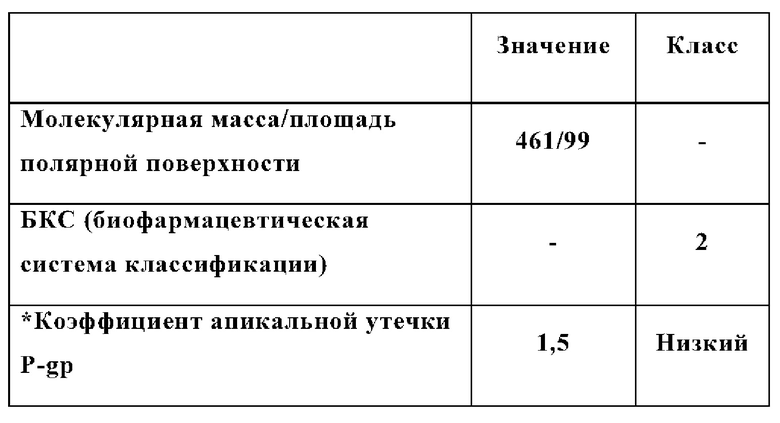
Кроме того, все одобренные до сих пор классические ингибиторы BRAF вемурафениб (Mol. Pharmaceutics 2012, 9, 11, 3236-3245), дабрафениб (J Pharmacol Ex Ther 2013, 344 (3) 655-664); и энкорафениб (Pharmacol Res. 2018; 129: 414-423) имеют очень плохую мозговую проницаемость. Это основное ограничение для использования этих классических ингибиторов BRAF для лечения рака головного мозга или метастазов в мозге. Таким образом, существует потребность в ингибиторах BRAF, улучшающих мозговую проницаемость.
Настоящее изобретение относится к неожиданному открытию, что ингибитор BRAF формулы (I) является более мощным и селективным ингибитором BRAF, демонстрирующим значительно меньшую парадоксальную активацию сигнального пути MAPK, сохраняя при этом высокую эффективность. Таким образом, это соединение можно назвать нарушителем парадокса или нарушителем парадокса RAF, в отличие от соединений, вызывающих парадокс RAF (и которые можно назвать индукторами парадокса или индукторами парадокса RAF). В дополнение к нарушению парадокса соединение формулы (I) также имеет очень мощные свойства проникновения в мозг, что обеспечивает срочно необходимую альтернативную терапию для лечения рака в мозге.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
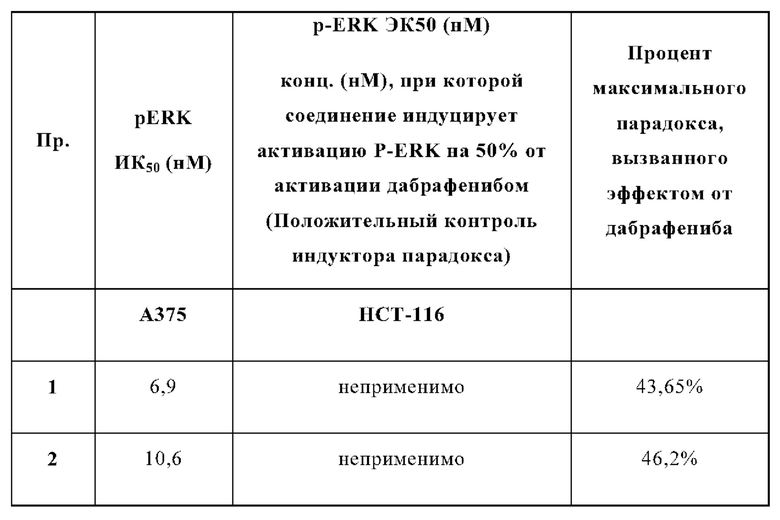
На Фиг. 1 показана кривая ингибирования P-ERK, индуцированная примером 1 в клеточной линии А375 мутантной по BRAF.
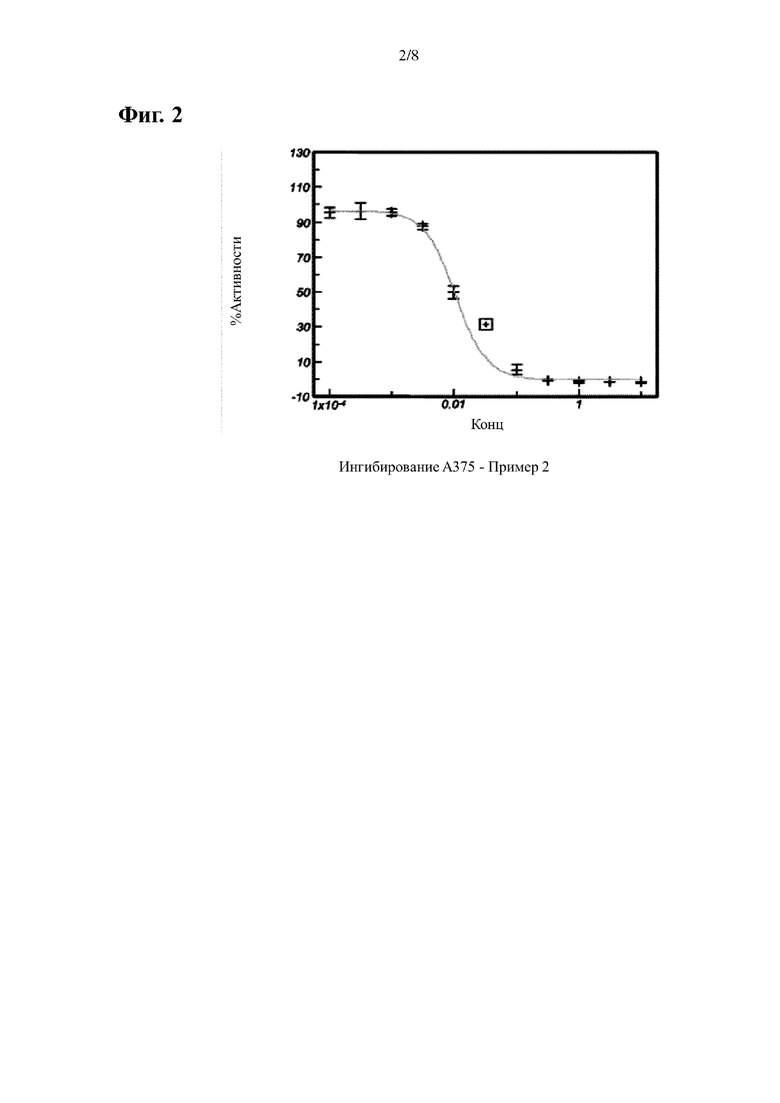
На Фиг. 2 показана кривая ингибирования P-ERK, индуцированная примером 2 в клеточной линии А375 мутантной по BRAF.
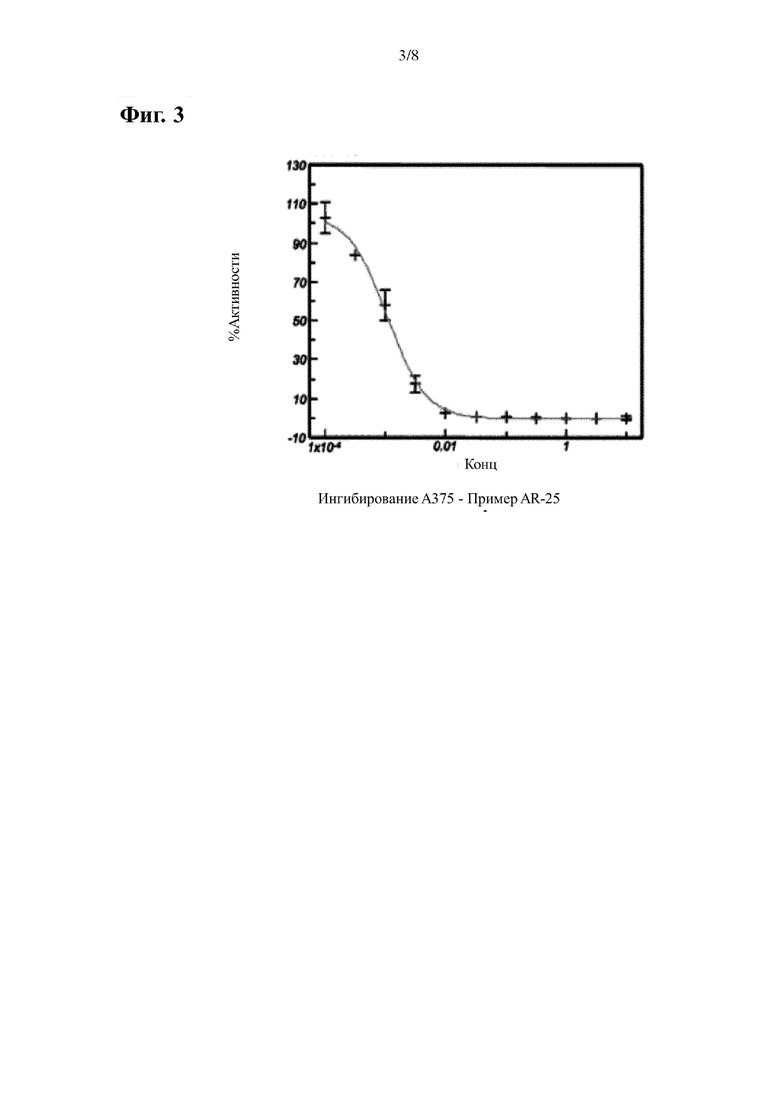
На Фиг. 3 показана кривая ингибирования P-ERK, индуцированная эталонным соединением AR-25 в клеточной линии А375 мутантной по BRAF.
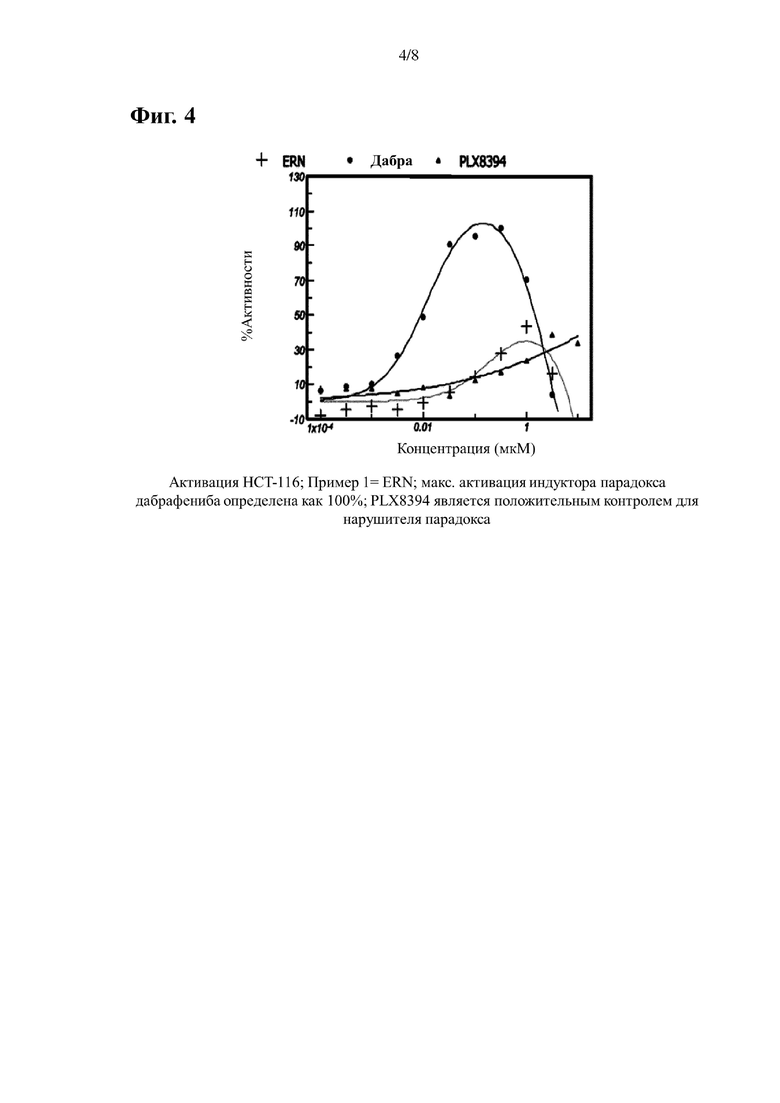
На Фиг. 4 показана кривая ингибирования P-ERK, индуцированная примером 1 в клеточной линии НСТ-116 WT BRAF. Для сравнения также приведены данные, полученные при лечении контрольными соединениями дабрафенибом (индуктор парадокса) и PLX-8394 (нарушитель парадокса).
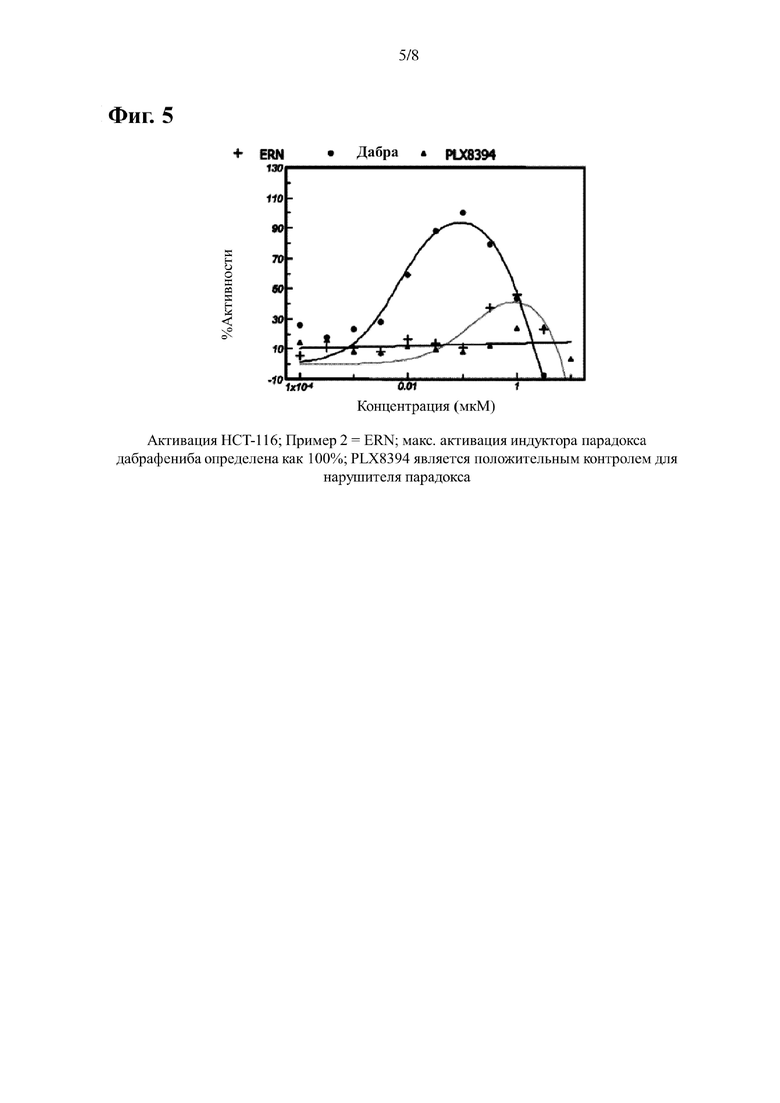
На Фиг. 5 показана кривая активации P-ERK, индуцированная примером 2 в клеточной линии НСТ-116 WT BRAF. Для сравнения также приведены данные, полученные при лечении контрольными соединениями дабрафенибом (индуктор парадокса) и PLX-8394 (нарушитель парадокса).
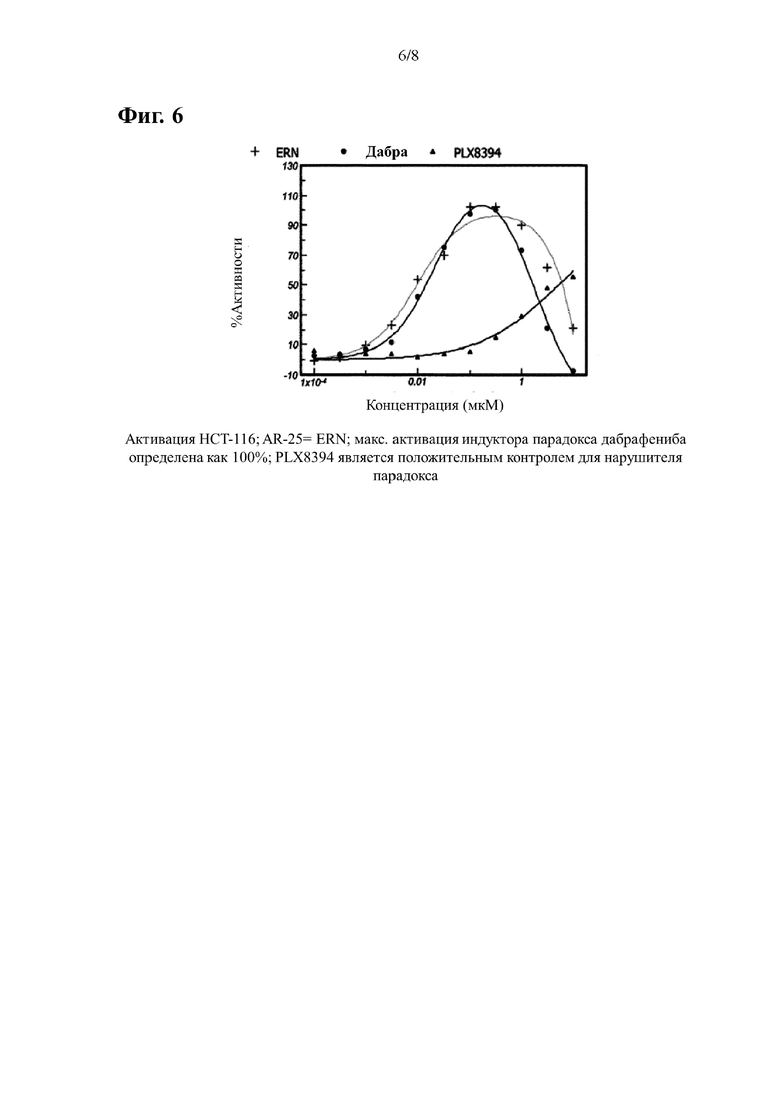
На Фиг. 6 показана кривая активации P-ERK, индуцированная эталонным соединением AR-25 в клеточной линии НСТ-116 WT BRAF. Для сравнения также приведены данные, полученные при лечении контрольными соединениями дабрафенибом (индуктор парадокса) и PLX-8394 (нарушитель парадокса).
На Фиг. 7 изображена парадоксальная активация пути МАР-киназы, индуцированная ингибиторами BRAF первого поколения. BRAF является частью первого узла сигнального пути киназы MAP, а мутантный BRAF является онкогенным драйвером (слева). В BRAF V600E/K-мутантных опухолях BRAF передает сигнал как мономер, в состоянии, при котором белок ингибируется ингибиторами BRAF первого поколения (средина). Ингибиторы BRAF первого поколения способствуют гомо- и/или гетеродимеризации BRAF WT (вверху справа). В этом контексте протомер, не занятый ингибитором BRAF, приобретает конформацию, неблагоприятную для связывания ингибитора (средина, справа). Результатом лечения ингибитором BRAF первого поколения в этом контексте является парадоксально повышенная активация MAPK и, следовательно, рост опухоли в клетках BRAF WT (внизу, справа).
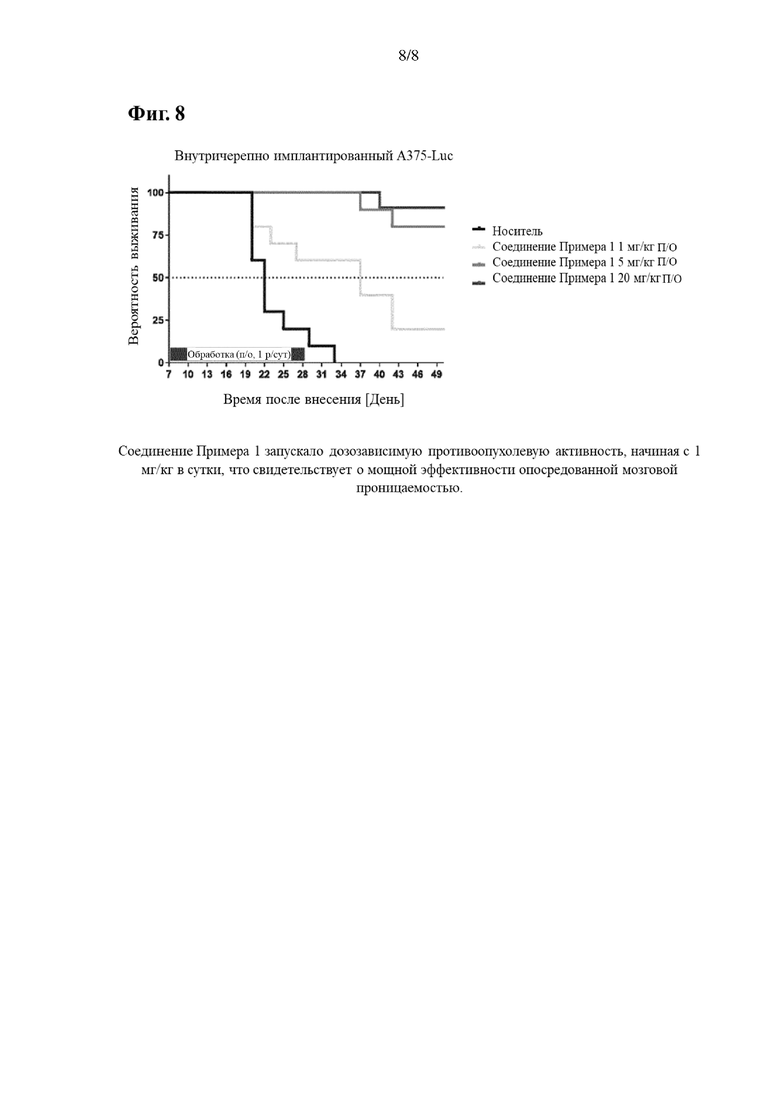
На Фиг. 8 показано, что соединение примера 1 вызывало дозозависимую противоопухолевую активность, начиная с 1 мг/кг в сутки, что свидетельствует о мощной эффективности опосредованной мозговой проницаемостью.
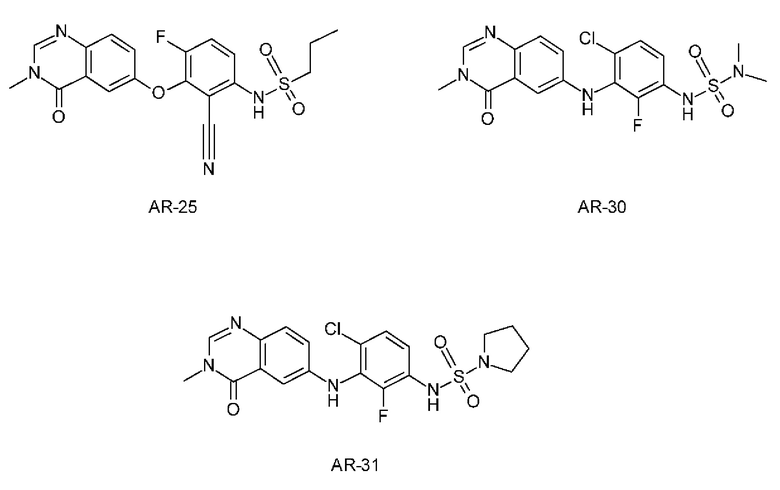
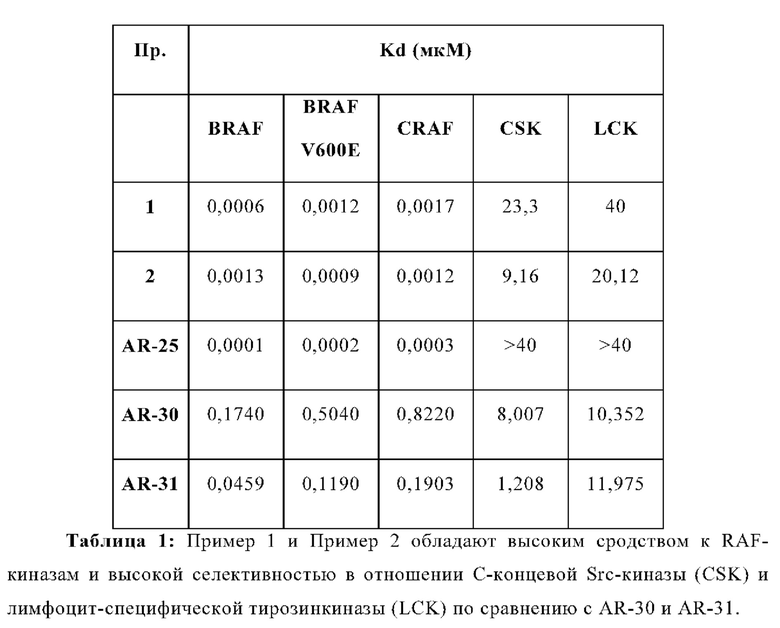
В WO 2012/118492 раскрыты эталонные соединения AR-25 как пример 25, AR-30 как Пример 30 и AR-31 как Пример 31.
Термин «фармацевтически приемлемая соль» относится к тем солям соединения формулы (I), которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот и которые не являются биологически или иным образом нежелательными. Соли образуются с такими неорганическими кислотами, как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., в частности хлористоводородная кислота, и такими органическими кислотами, как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и т.п. Кроме того, эти соли можно получать путем добавления неорганического основания или органического основания к свободной кислоте. Соли, полученные из неорганического основания, включают, но не ограничиваются этим, соли натрия, калия, лития, аммония, кальция, магния и т.п. Соли, полученные из органических оснований, включают, но не ограничиваются этим, соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе встречающихся в природе замещенных аминов, циклических аминов и основных ионообменных смол, например смол изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина, этаноламина, лизина, аргинина, N-этилпиперидина, пиперидина, полиимина и т.п. Конкретные фармацевтически приемлемые соли соединения формулы (I) представляют собой соли хлористоводородной кислоты, соли метансульфоновой кислоты и соли лимонной кислоты.
Соединение формулы (I) может содержать один асимметрический центр и может находиться в форме оптически чистых энантиомеров, смесей энантиомеров, таких как, например, рацематы, смеси диастереомеров, диастереомерные рацематы или смеси диастереомерных рацематов.
В соответствии с правилом Кана - Ингольда - Прелога асимметрический атом углерода может находиться в «R»- или «S»-конфигурации.
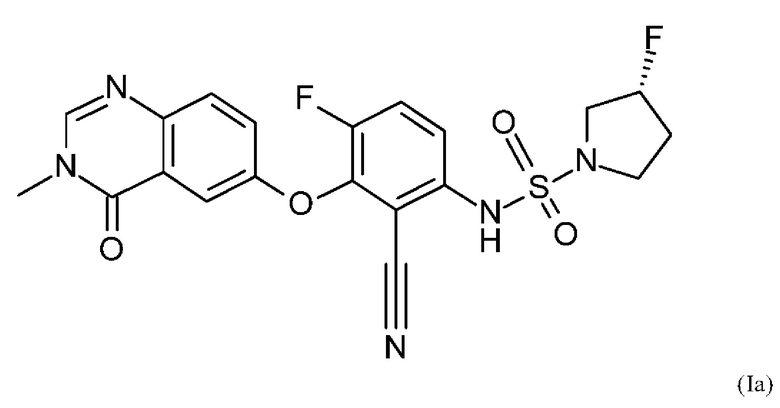
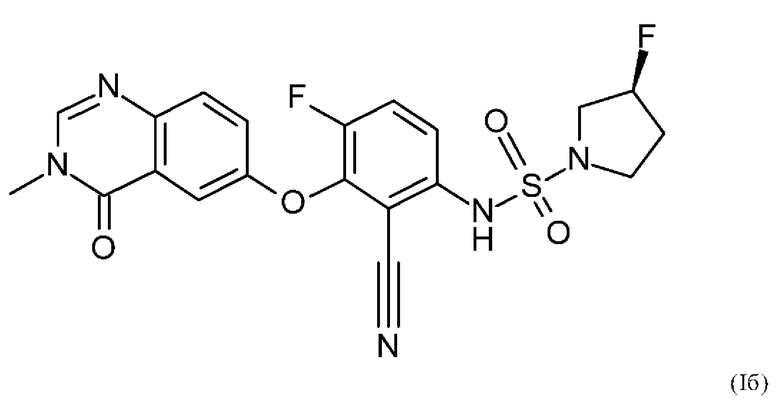
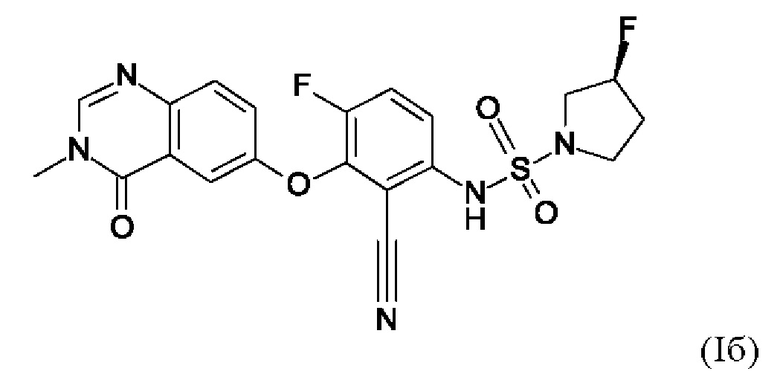
Также вариант реализации данного изобретения касается описанного в данном документе соединения формулы (I), как описано в данном документе, и его фармацевтически приемлемой соли, в частности соединения формулы (I), как описано в данном документе, более конкретно соединения формулы (Ia) или (Iб), как описано в данном документе.
Изобретение относится также к фармацевтически приемлемой соли соединения формулы (I), где фармацевтически приемлемая соль может быть выбрана из гидрохлоридных солей, солей метансульфоновой кислоты и солей лимонной кислоты.
Также вариантом реализации данного изобретения является соединение согласно формуле (Ia).
Также вариантом реализации данного изобретения является соединение согласно формуле (Iб).
Способы получения соединений формулы (Ia) или (Iб), как описано в данном документе, также является объектом изобретения.
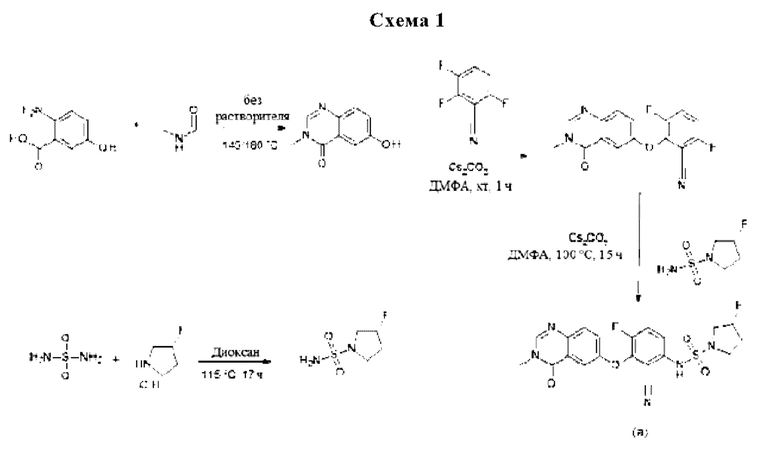
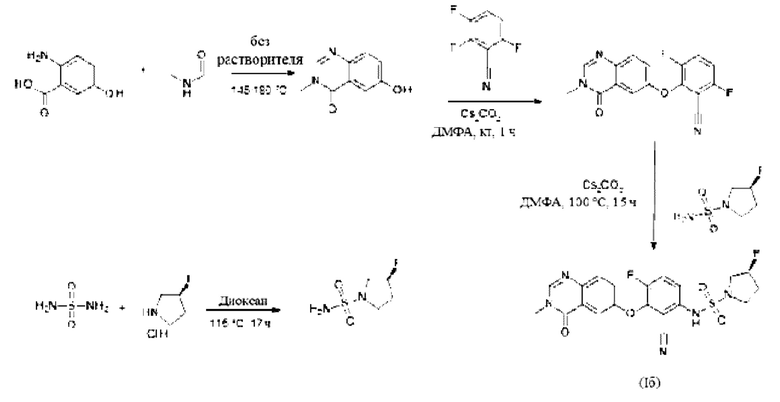
Получение соединения формулы (I) по данному изобретению можно осуществлять путем последовательного или конвергентного синтеза. Синтезы по данному изобретению проиллюстрированы на следующих общих схемах. Навыки, необходимые для проведения реакции и очистки получаемых в результате продуктов, известны специалистам в данной области техники.
Конкретнее, соединение формулы (I) можно получать способами, приведенными ниже, способами, приведенными в примерах, или аналогичными способами. Соответствующие условия реакций для отдельных шагов реакций известны специалисту в данной области техники. Последовательность реакции не ограничивается последовательностью, показанной на схеме 1, однако, в зависимости от исходных веществ и их соответствующей реакционной способности, последовательность шагов реакции может быть свободно изменена. Исходные материалы являются либо коммерчески доступными, либо их можно получить способами, аналогичными способам, приведенным ниже, способами, описанными в ссылках, приведенных в описании, или в примерах, или способами, известными в данной области техники.
Следует понимать, что соединение формулы (I) в данном изобретении может быть дериватизировано в функциональных группах, чтобы получить производные, которые способны к обратному превращению в исходное соединение in vivo.
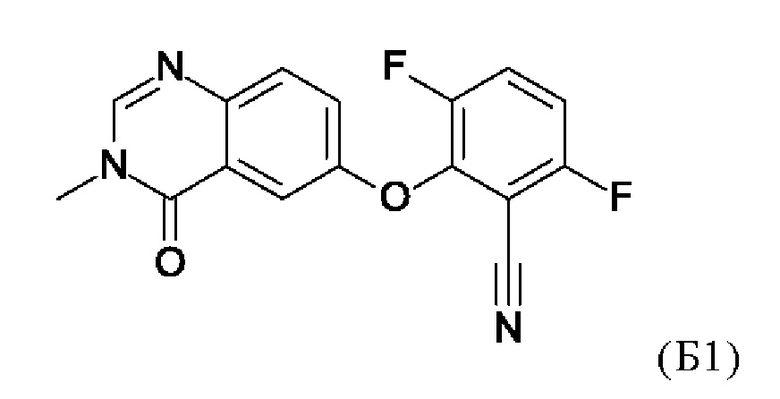
Таким образом, изобретение также относится к способу получения соединения по изобретению, включающему проведение реакции соединения формулы (Б1)
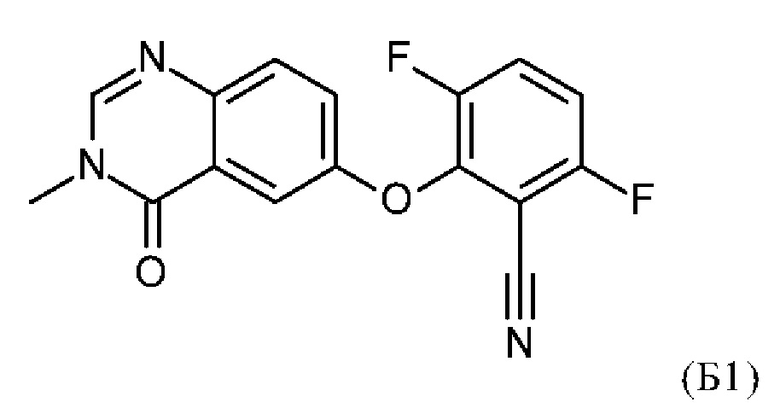
с соединением формулы (Б2)
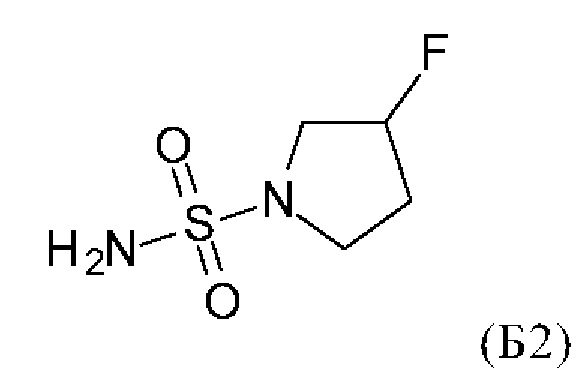
в присутствии основания.
Реакцию удобно проводить в растворителе. Растворителем может являться, например, ДМФА.
Реакцию удобно проводить в присутствии основания. Основой может служить, например, карбонат цезия.
Удобными условиями для реакции могут быть между примерно 30°С и примерно 150°С, в частности между примерно 50°С и примерно 130°С, более конкретно между примерно 70°С и примерно 120°С. Удобными условиями являются примерно 100°С в течение от примерно 1 часа до примерно 48 часов, в частности от примерно 2 часов до примерно 20 часов.
Изобретение также относится к соединению согласно изобретению, полученное способом согласно изобретению.
Изобретение, в частности, также относится к:
Соединению формулы (I) или его фармацевтически приемлемой соли для применения в качестве терапевтически активного вещества;
Фармацевтической композиции, содержащей соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемую соль и терапевтически инертный носитель;
Соединению формулы (I), описанной в данном документе, или его фармацевтически приемлемой соли для применения в лечении или профилактике рака;
Соединению формулы (I), как описано в данном документе, или его фармацевтически приемлемой соли для применения в лечении или профилактике рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или немелкоклеточного рака легкого (НМРЛ);
Применению соединения формулы (I), как описано в данном документе, или его фармацевтически приемлемой соли для лечения или профилактики рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ;
Применению соединения формулы (I), как описано в данном документе, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ;
Способу лечения рака, причем способ включает введение эффективного количества соединения формулы (I), как описано в данном документе, или его фармацевтически приемлемой соли нуждающемуся в этом пациенту; и
Способу лечения или профилактики рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ, причем способ включает введение пациенту эффективного количества соединения формулы (I), как описано в данном документе, или его фармацевтически приемлемой соли нуждающемуся в этом пациенту.
Определенный вариант реализации изобретения относится к описанному в данном документе соединению формулы (I) или его фармацевтически приемлемой соли для применения в терапевтическом и/или профилактическом лечении рака, в частности рака, вызванного мутантами BRAF, в частности рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
Конкретный вариант реализации изобретения относится к описанному в данном документе соединению формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для применения в терапевтическом и/или профилактическом лечении рака, вызванного мутантами BRAF, в частности рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
Определенный вариант реализации изобретения относится к фармацевтической композиции, содержащей описанное в данном документе соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемое вспомогательное вещество.
Определенный вариант реализации изобретения касается способа терапевтического и/или профилактического лечения рака, в частности рака, вызванного мутантами BRAF, в частности рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или немелкоклеточного рака легкого (НМРЛ) путем введения эффективного количества соединения формулы (I), как описано в данном документе, или ее фармацевтически приемлемой соли нуждающемуся в этом пациенту.
Определенный вариант реализации изобретения относится к описанному в данном документе соединению формулы (I) или его фармацевтически приемлемой соли для применения в качестве лекарственного средства в терапевтическом и/или профилактическом лечении пациента с раком, вызванным мутантами BRAF, в частности раком щитовидной железы, колоректальным раком, раком мозга, меланомой или НМРЛ, включающем определение статуса мутации BRAF у указанного пациента, а затем введение соединения формулы (I), как описано в данном документе, или его фармацевтически приемлемой соли указанному пациенту.
Определенный вариант реализации изобретения относится к описанному в данном документе соединению формулы (I) или его фармацевтически приемлемой соли для применения в терапевтическом и/или профилактическом лечении метастазов в мозге.
Кроме того, в тех случаях, когда это применимо, данное изобретение включает все заместители соединения формулы (I) в их соответствующей дейтерированной форме.
Кроме того, в тех случаях, когда это применимо, данное изобретение включает все заместители соединения формулы (I) в их соответствующей третированной форме.
Определенный вариант реализации изобретения относится к описанному в данном документе соединению формулы (I) или его фармацевтически приемлемой соли, где по меньшей мере один заместитель содержит по меньшей мере один радиоизотоп. Конкретные примеры радиоизотопов представляют собой 2Н, 3Н, 13С, 14С и 18F.
Кроме того, изобретение включает все оптические изомеры, т.е. диастереомеры, диастереомерные смеси, рацемические смеси, все их соответствующие энантиомеры и/или таутомеры, а также их сольваты, соединений формулы (I), где это применимо.
При необходимости, рацемические смеси соединения по изобретению можно разделять так, чтобы выделить отдельные энантиомеры. Разделение можно осуществлять способами, хорошо известными в данной области техники, например сочетанием рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси, с последующим разделением отдельных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография.
В вариантах реализации, в которых предложены оптически чистые энантиомеры, выражение «оптически чистый энантиомер» означает, что соединение содержит >90% необходимого изомера по массе, в частности >95% необходимого изомера по массе или, конкретнее, >99% необходимого изомера по массе, при этом указанное процентное содержание приведено с учетом общей массы изомера соединения. Хирально чистое или хирально обогащенное соединение можно получать посредством хирально-селективного синтеза или посредством разделения энантиомеров. Разделение энантиомеров можно проводить для конечного продукта или, в альтернативном варианте, для подходящего промежуточного соединения.
Другой вариант реализации изобретения предусматривает фармацевтическую композицию или лекарственное средство, содержащее соединение по изобретению и терапевтически инертный носитель, разбавитель или наполнитель, а также способ применения соединений по изобретению для получения такой композиции и лекарственного средства. В одном примере соединение формулы (I) может быть составлено путем смешивания при температуре окружающей среды, при соответствующем рН и при необходимой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, которые являются нетоксичными для реципиентов, при дозировках и концентрациях, используемых в галеновой форме для введения. рН состава зависит главным образом от конкретного применения и концентрации соединения, но предпочтительно составляет в диапазоне от приблизительно 3 до приблизительно 8. В одном примере соединение формулы (I) составлено в ацетатном буфере при рН 5. В другом варианте реализации соединение формулы (I) является стерильным. Соединение можно хранить, например, в виде твердой или аморфной композиции, в виде лиофилизированого состава или в виде водного раствора.
Композиции составляют, дозируют и вводят способом, соответствующим надлежащей медицинской практике. Факторы, которые необходимо учитывать в этом контексте, включают конкретное нарушение, подлежащее лечению, конкретное млекопитающее, подлежащее лечению, клиническое состояние отдельного пациента, причину нарушения, место доставки средства, способ введения, график введения и другие факторы, известные практикующим врачам.
Также одним из вариантов реализации данного изобретения является соединение формулы (I), как описано в данном документе, полученное в соответствии с любым из описанных процессов.
Аналитические процедуры
Материалы
Среда DMEM без красного фенола, дополненная L-глутамином, была приобретена у (Thermo Fisher Scientific). Фетальная бычья сыворотка (FBS) была приобретена у VWR. Расширенный набор ERK phospho-T202 /Y204 - 10000 тестов был приобретен у Cisbio каталожный номер # 64AERPEH. Клетки А375 и НСТ116 были первоначально получены из АТСС и сохранены в репозитории Roche. 384-луночные микропланшеты были приобретены у Greiner Bio-One, 384-луночные (с крышкой, HiBase, малый объем, кат. 784-080).
Анализ HTRF для определения P-ERK в клетках А375 или НСТ116
А375 представляет собой модель клеточного рака, экспрессирующую BRAF с мутацией V600E, и НСТ116, модель клеточного рака, экспрессирующую BRAF WT. Ингибиторы BRAF первого поколения, такие как, например, дабрафениб оказывает парадоксальное действие на опухолевые клетки, поскольку они ингибируют рост клеток BRAF с мутацией V600E (такой как, например, А375), в то время как они активируют рост клеток с BRAF дикого типа (таких как, например, НСТ116). Фосфорилирование ERK 1,2 (конечный элемент каскада фосфорилирования пути MAPK) в дальнейшем считается основным показателем статуса активации пути MAPK. Перед анализом клеточные линии А375 и НСТ116 культивируют в среде DMEM без красного фенола с добавлением 10% фетальной бычьей сыворотки (FBS). После обработки соединением уровни P-ERK определяют путем измерения сигнала флуоресценции FRET, индуцированного селективным связыванием 2 антител, предоставленных в упомянутом наборе (Cisbio каталожный номер # 64AERPEH), на белке ERK при фосфорилировании по Thr202/Tyr204. Коротко, 8000 клеток/лунка в 12 мкл среды/лунка высевают в 384-луночный планшет и оставляют на ночь в инкубаторе (при 37°С с увлажненной 5% атмосферой CO2), на следующий день обрабатывают планшет в двух повторах исследуемыми соединениями, дабрафенибом и PLX8394 (последние два как контрольные) в следующих конечных концентрациях лекарственного препарата: 10 мкМ - 3 мкМ - 1 мкМ - 0,3 мкМ - 0,1 мкМ - 0,03 мкМ - 0,01 мкМ - 0,003 мкМ - 0,001 мкМ, все лунки подвергают нормализации по ДМСО и проводят инкубацию с лекарственным препаратом в течение 1 часа. Затем в лунки добавляют 4 мкл 4Х буфера для лизиса, поставляемого с набором, затем центрифугируют планшет в течение 30 секунд (300 rcf) и инкубируют на шейкере для планшетов в течение 1 ч при комнатной температуре.
В конце инкубации добавляют 4 мкл/лунка улучшенного раствора антител Р-ERK (приготовленного согласно инструкции производителя), а затем 4 мкл/лунка криптатного раствора антитела P-ERK (подготовленного согласно инструкции производителя) (Cisbio каталожный номер # 64AERPEH) к тестовым лункам.
Чтобы обеспечить надлежащую нормализацию данных, контрольные лунки, которые не обрабатывались лекарственными средствами, указанные в следующей таблице, всегда включены в каждый планшет (согласно инструкции производителя):
Композиции лунок p-ERK HTRF (мкл):
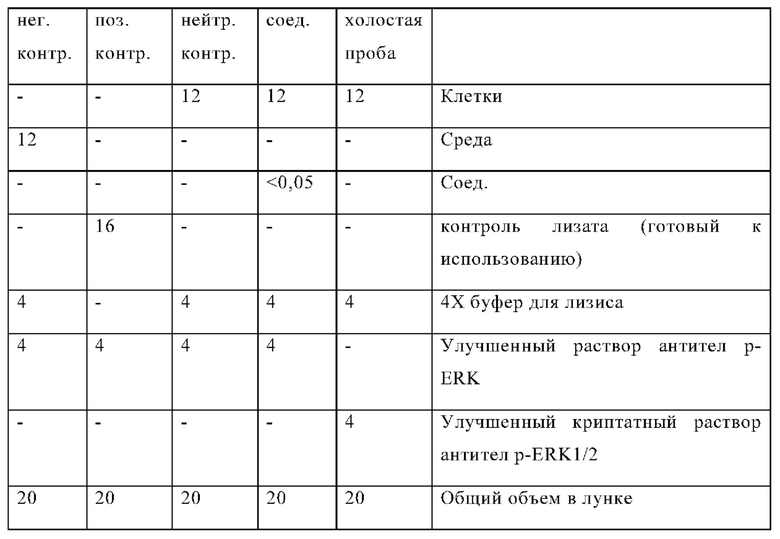
Затем центрифугируют планшет при 300 rcf в течение 30 секунд, герметизируют для предотвращения испарения и инкубируют в течение ночи в темноте при комнатной температуре.
Затем планшет анализируют и считывают значение флуоресценции с помощью аппарата Pherastast FSX (BMG Labtech) при 665 и 620 нм.
Полученные значения флуоресценции обрабатывают по формуле Соотношение = Сигнал(620 нм)/Сигнал(625 нм)*10000, после чего среднее соотношение холостой пробы вычитается из всех значений.
В случае клеток А375 (ингибирование BRAF) данные нормализуют учитывая среднее соотношение (вычитается холостая проба), полученного из клеток обработанных только ДМСО, как 100%, и с учетом среднего соотношения (вычитается холостая проба), полученного из клеток обработанных 10 мкМ, как 0%. Среднее значение нормализованных точек наносят на сигмовидную кривую и определяют ИК50. Результаты приведены в Таблицах 1-2 и Фигурах 1-3.
В случае клеток НСТ116 (активация BRAF) данные нормализуют учитывая среднее отношение (вычитается холостая проба), полученного из клеток обработанных только ДМСО, как 0%, и с учетом среднего соотношения (вычитается холостая проба), полученного из клеток обработанных дабрафенибом при концентрации, которая обеспечивает самый высокий сигнал, как 100%. Отдельные точки наносят на кривую сигмовидной или куполообразной формы, и определяют процент активации по сравнению с максимальной активацией, опосредованной дабрафенибом. ЭК50 представляет собой концентрацию, при которой достигается активация, равная 50% от максимально достигнутой дабрафенибом. Результаты приведены в Таблице 2 и Фигурах 4-6.
Если активация не достигает 50% от максимально достигнутого дабрафенибом, расчет ЭК50 не применяется.
Процент максимального эффекта, вызывающий парадокс, вызванный дабрафенибом, определяется путем расчета процента, при котором исследуемое соединение индуцирует свой максимальный сигнал P-ERK в процентах от наивысшего сигнала, вызванный дабрафенибом в пределах исследуемого диапазона доз.

Измерение СМЖ Kp,uu для оценки потенциала мозговой проницаемости
СМЖ Kp,uu представляет собой соотношение концентрации в спинномозговой жидкости (СМЖ): экспозиция несвязанной плазмы и значение Kp,uu≥1 указывают на хорошую мозговую проницаемость. Для соединения Примера 1 исследования однократной пероральной дозы на мышах и крысах, последовательные концентрации в плазме и спинномозговой жидкости (до 24 ч после введения дозы) измеряли с помощью РХ-МС/МС для вычисления СМЖ Kp,uu. Для исследований многократных пероральных доз на крысах и карликовых свинках концентрации в плазме и спинномозговой жидкости, приближающиеся к Tmax (через 3 ч после последней дозы), измеряли с помощью ЖХ-МС/МС и использовали для расчета СМЖ Kp,uu.

Внутричерепно имплантированный A375-Luc
Раковые клетки А375 BRAF V600E, конститутивно экспрессирующие люциферазу, вводили внутричерепно мышам с ослабленным иммунитетом. Обработку соединением Примера 1 начинали на 7-й день от внутричерепной инъекции и продолжали в течение 2 недель. Различные группы подвергались ежедневному пероральному введению 1 мг/кг, 5 мг/кг и 20 мг/кг Примера 1 соответственно. Результаты приведены на Фиг. 8.
Соединение формулы (I) и его фармацевтически приемлемая соль может быть использована как лекарственное средство (например, в форме фармацевтического препарата). Фармацевтические препараты можно вводить внутрь, например, перорально (например, в форме таблеток, покрытых оболочкой таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий), назально (например, в форме назальных спреев), ректально (например, в форме суппозиториев) или местно через глаза (например, в форме растворов, мазей, гелей или водорастворимых полимерных вкладок). При этом введение также можно осуществлять парентерально, а именно, внутримышечно, внутривенно или внутриокулярно (например, в форме стерильных инъекционных растворов).
Соединение формулы (I) и его фармацевтически приемлемую соль можно обрабатывать фармацевтически инертными, неорганическими или органическими адъювантами для производства таблеток, покрытых оболочкой таблеток, драже, твердых желатиновых капсул, инъекционных растворов или составов для местного применения. В качестве таких адъювантов для таблеток, драже и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.д.
Подходящими адъювантами для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые вещества и жидкие полиолы и т.д.
Подходящими адъювантами для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар, глюкоза и т.д.
Подходящими адъювантами для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.д.
Подходящими адъювантами для суппозиториев являются, например, натуральные или отвержденные масла, воски, жиры, полутвердые или жидкие полиолы и т.д.
Подходящими адъювантами для местных глазных составов являются, например, циклодекстрины, манит или множество других носителей и эксципиентов, известных в данной области техники.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, вещества, повышающие вязкость, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Также они могут содержать другие имеющие терапевтическую ценность вещества.
Дозировка может варьироваться в широком диапазоне и, конечно, ее следует подбирать под индивидуальные требования в каждом конкретном случае. В целом, в случае перорального введения суточная доза составляет от около 0,1 мг до 20 мг на кг массы тела, предпочтительно от около 0,5 мг до 4 мг на кг массы тела (например, около 300 мг на человека), предпочтительно разделенная на 1-3 отдельные дозы, которые могут состоять, например, из одинаковых количеств, если это уместно. В случае местного применения состав может содержать от 0,001% до 15% по массе лекарственного средства, а необходимую дозу, которая может составлять от 0,1 до 25 мг, можно вводить один раз в день или в неделю, или в виде нескольких доз (от 2 до 4) в день, или в виде нескольких доз в неделю. При этом следует понимать, что в случае соответствующего показания верхняя или нижняя граница, приведенная в данном документе, может быть превышена.
Фармацевтические композиции
Соединение формулы (I) и его фармацевтически приемлемую соль можно использовать в качестве терапевтически активного вещества, например в форме фармацевтического препарата. Фармацевтический препарат можно вводить перорально, например в форме таблеток, таблеток с оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако введение можно осуществлять ректально, например в форме суппозиториев, или парентерально, например в форме инъекционных растворов.
Соединение формулы (I) и его фармацевтически приемлемые соли можно подвергать обработке с фармацевтически инертными неорганическими или органическими носителями для получения фармацевтических препаратов. Лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и тому подобное можно использовать, например, в качестве таких носителей для таблеток, таблеток с оболочкой, драже и твердых желатиновых капсул. Пригодные носители для мягких желатиновых капсул представляют собой, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное. Однако в зависимости от природы активного вещества обычно нет необходимости в носителях в случае мягких желатиновых капсул. Пригодные носители для получения растворов и сиропов представляют собой, например, воду, полиолы, глицерин, растительные масла и тому подобное. Пригодные носители для суппозиториев представляют собой, например, природные или гидрогенизированные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное.
Кроме того, фармацевтический препарат может содержать фармацевтически приемлемые вспомогательные вещества, такие как консерванты, солюбилизаторы, стабилизаторы, увлажнители, эмульгаторы, подсластители, красители, ароматизаторы, соли для смены осмотического давления, буферы, маскирующие агенты или антиоксиданты. Также они могут содержать другие имеющие терапевтическую ценность вещества.
Лекарственные средства, содержащие соединение формулы (I) или его фармацевтически приемлемую соль и терапевтически инертный носитель также представлены в данном изобретении, как и способ их получения, который включает придание одному или более соединениям формулы (I) и/или их фармацевтически приемлемым солям и, если необходимо, одному или более другим терапевтически ценным веществам галеновой формы для введения вместе с одним или более терапевтически инертными носителями.
Дозировка может варьироваться в широком диапазоне и, конечно, будет подбираться под индивидуальные требования в каждом конкретном случае. В случае перорального введения дозировка для взрослых может изменяться от около 0,01 мг до около 1000 мг в сутки соединения общей формулы (I) или соответствующего количества его фармацевтически приемлемой соли. Суточную дозировку можно вводить в виде однократной дозы или несколькими дозами, а кроме того, можно превышать верхнюю границу, когда установлено, что существуют показания.
Следующие примеры иллюстрируют данное изобретение, не ограничивая его, а могут служить только в качестве иллюстрации. Фармацевтические препараты обычно содержат примерно 1-500 мг, в том числе 1-100 мг соединения формулы (I). Примеры композиций по изобретению:
Пример А
Таблетки на основе следующей композиции изготавливают традиционным образом:
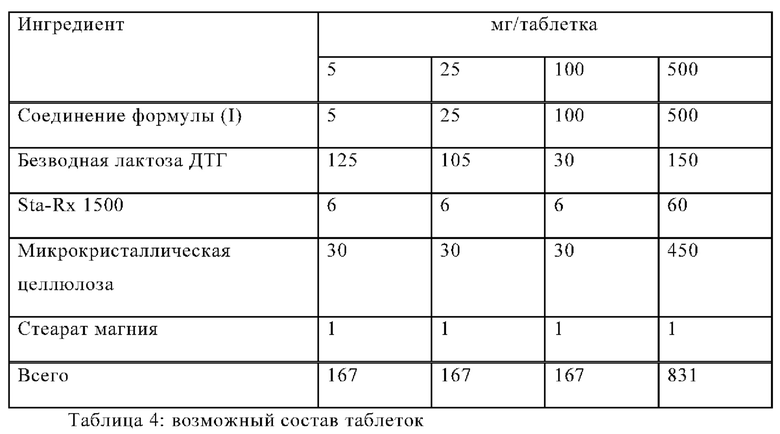
Процедура изготовления
1. Смешать ингредиенты 1, 2, 3 и 4 и гранулировать с очищенной водой.
2. Высушить гранулы при 50°С.
3. Пропустить гранулы через подходящее помольное оборудование.
4. Добавить ингредиент 5 и перемешивать в течение трех минут; спрессовать на подходящем прессе.
Пример Б-1
Капсулы на основе следующей композиции изготавливают таким образом:
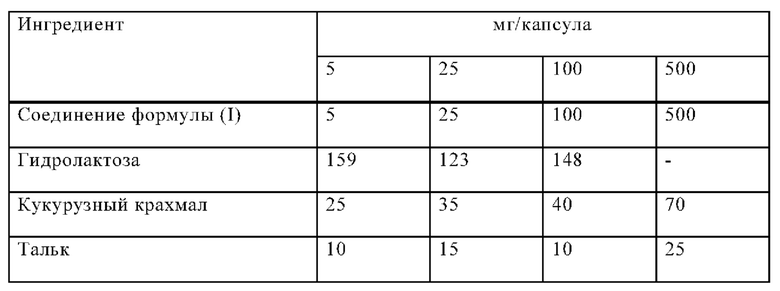

Процедура изготовления
1. Смешать ингредиенты 1, 2 и 3 в подходящем смесителе в течение 30 минут.
2. Добавить ингредиенты 4 и 5 и смешивать в течение 3 минут.
3. Наполнить соответствующие капсулы.
Соединение формулы (I), лактозу и кукурузный крахмал сначала смешивают в смесителе, а потом в измельчителе. Смесь возвращают в смеситель; к ней добавляют тальк и тщательно перемешивают. Машина наполняет смесью соответствующие капсулы, например, твердые желатиновые капсулы.
Пример Б-2
Изготавливают мягкие желатиновые капсулы со следующим составом:



Процедура изготовления
Соединение формулы (I) растворяют в теплом расплаве других ингредиентов, и смесью заполняют мягкие желатиновые капсулы подходящего размера. Заполненные мягкие желатиновые капсулы обрабатывают в соответствии с обычными методиками.
Пример В
Изготавливают суппозитории со следующим составом:
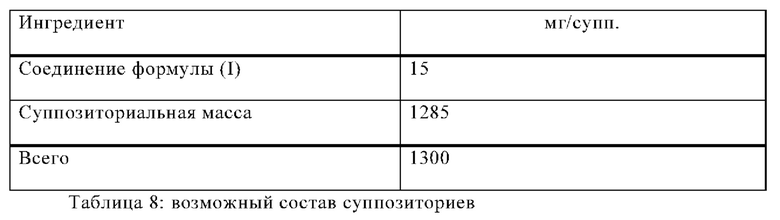
Процедура изготовления
Массу для суппозитория плавят в стеклянном или стальном сосуде, тщательно перемешивают и охлаждают до 45°С. После этого в нее добавляют мелко измельченное соединение формулы (I) и перемешивают до полного исчезновения. Смесь выливают в формы для суппозиториев пригодного размера, оставляют охлаждаться; затем суппозитории вынимают из форм и отдельно упаковывают в вощеную бумагу или металлическую фольгу.
Пример Г
Инъекционные растворы на основе следующей композиции изготавливают таким образом:
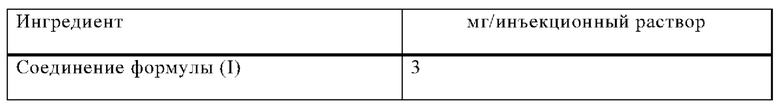

Процедура изготовления
Соединение формулы (I) растворяют в смеси полиэтиленгликоля 400 и воды для инъекций (часть). рН доводят до 5,0 уксусной кислотой. Объем доводят до 1,0 мл путем добавления остаточного количества воды. Раствор фильтруют, наливают во флаконы с необходимым избытком и стерилизуют.
Пример Д
Изготавливают саше со следующим составом:
Процедура изготовления
Соединение формулы (I) смешивают с лактозой, микрокристаллической целлюлозой и карбоксиметилцеллюлозой натрия и гранулируют со смесью поливинилпирролидона в воде. Гранулят смешивают со стеаратом магния и ароматизирующими добавками и помещают в саше.
Примеры
Сокращения
ДХМ = дихлорметан; ДМФА = диметилформамид; ДМСО = диметилсульфоксид; DRF = определение диапазона доз; ПЭР = ионизация электрораспылением; EtOAc = этилацетат; ЖХ-МС/МС = жидкостная хроматография-МС/МС; МеОН = метанол; МС = масс-спектрометрия; к.т. = комнатная температура; P-gp = Р-гликопротеин; СКЖХ = сверхкритическая жидкостная хроматография.
Эталонные соединения AR-25, AR-30 и AR-31 получали согласно синтезу, раскрытому в WO 2012/118492 в примере 25, примере 30 и примере 31 соответственно.
6-гидрокси-3-метилхиназолин-4-он
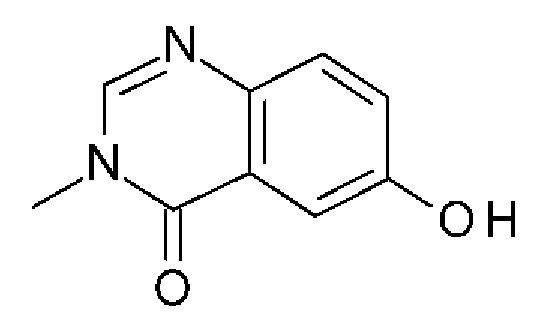
2-Амино-5-гидроксибензойную кислоту (10 г, 65,3 ммоль, Экв: 1,0) и N-метилформамид (30 г, 29,9 мл, 503 ммоль, Экв: 7,7) нагревали при 145°С в течение 21 ч 45 мин, затем охлаждали до к.т. Реакционную смесь разбавляли 50 мл Н2О и перемешивали при к.т.в течение 20 мин Образовавшийся осадок собирали фильтрованием. Светло-коричневое твердое вещество промывали 3×20 мл воды. Твердое вещество переносили в толуол и выпаривали досуха (3×). Твердое вещество сушили в вакууме при 40°С в течение ночи под высоким вакуумом, получая указанное в заголовке соединение в виде светло-коричневого твердого вещества (10,3 г, выход 89%). МС (ИЭР) м/з: 177,1 [М+Н]+.
3,6-дифтор-2-(3-метил-4-оксохиназолин-6-ил)оксибензонитрил
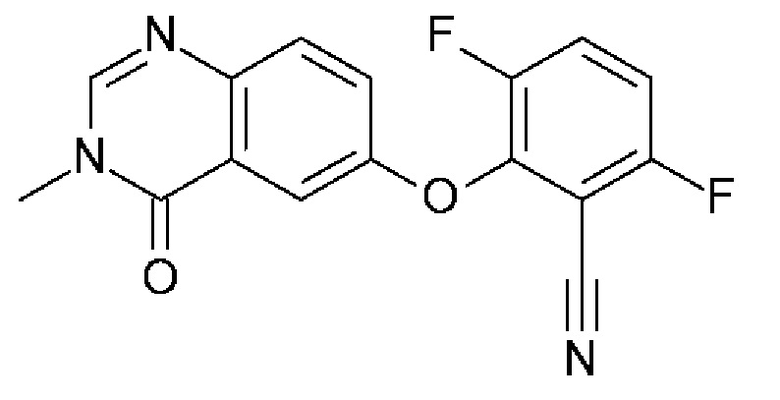
Карбонат цезия (3,22 г, 9,79 ммоль, Экв: 1,15) добавляли при к.т. к раствору 6-гидрокси-3-метилхиназолин-4-она (1500 мг, 8,51 ммоль, Экв: 1,0) в N,N-диметилформамиде (35 мл). Смесь перемешивали в течение 30 мин. при к.т., затем добавляли 2,3,6-трифторбензонитрил (1,47 г, 1,08 мл, 9,37 ммоль, Экв: 1,1). Через 1 ч реакционную смесь охлаждали на льду и разбавляли водой (120 мл). Полученное твердое вещество собирали фильтрованием, промывали ледяной водой (100 мл) и гептаном (100 мл) и сушили отсасыванием. Твердое вещество переносили в толуол и выпаривали досуха (3×), затем сушили в течение ночи в вакууме с получением указанного в заголовке соединения в виде твердого вещества светло-коричневого цвета (2,58 г, выход 97%). МС (ИЭР) м/з: 314,1 [М+Н]+.
(3R)-3-фторпирролидин-1-сульфонамид
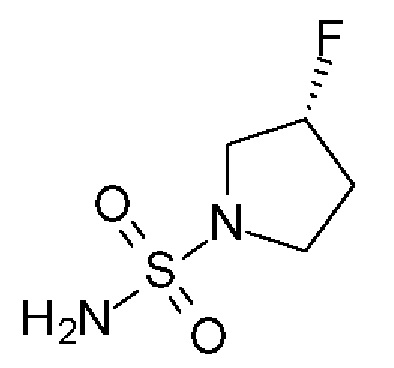
(R)-3-фторпирролидина гидрохлорид (1,8 г, 14,3 ммоль, Экв: 1,2) добавляли к раствору диамида серной кислоты (1,148 г, 11,9 ммоль, Экв: 1,0) и триэтиламина (2,42 г, 3,33 мл, 23,9 ммоль, Экв: 2) в диоксане (10 мл). Реакционную смесь перемешивали при 115°С в течение 15,5 ч, охлаждали до к.т.и концентрировали в вакууме. Остаток разбавляли ДХМ, выпаривали с силикагелем досуха и переносили на колонку. Очистка флэш-хроматографией (40 г диоксида кремния, 80% EtOAc) давала указанное в заголовке соединение в виде белого кристаллического твердого вещества (1,82 г, выход 91%). МС (ИЭР) м/з: 169,1 [М+Н]+.
(3S)-3-фторпирролидин-1-сульфонамид
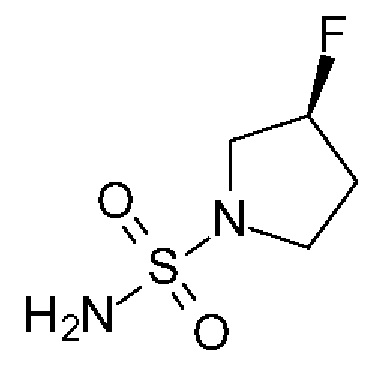
Триэтиламин (304 мг, 419 мкл, 3,01 ммоль, Экв: 2.0) добавляли к суспензии диамида серной кислоты (146 мг, 1,5 ммоль, Экв: 1,0) и (S)-3-фторпирролидина гидрохлорид (234 мг, 1,8 ммоль, Экв: 1,2) в диоксане (1,3 мл). Реакционную смесь перемешивали в запаянной пробирке при 115°С в течение 16 ч 35 мин, затем концентрировали в вакууме. Остаток разбавляли МеОН, выпаривали с силикагелем досуха и переносили на колонку. Очистка флэш-хроматографией (40 г диоксида кремния, 0-8% МеОН/ДХМ) давала указанное в заголовке соединение в виде твердого вещества светло-желтого цвета (193 мг, выход 75%). МС (ИЭР) м/з: 169,1 [М+Н]+.
(3R)-N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид (Пример 1)
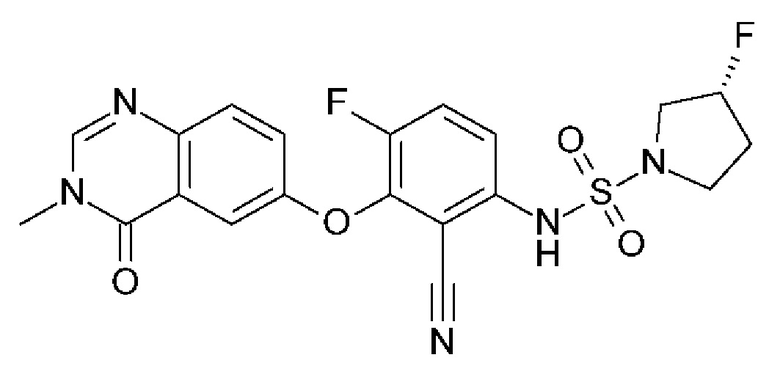
(R)-3-фторпирролидин-1-сульфонамид (1,26 г, 7,51 ммоль, Экв: 2,1) и карбонат цезия (2,56 г, 7,87 ммоль, Экв: 2.2) суспендировали в сухом ДМФА (10,2 мл) в атмосфере аргона. Реакционную смесь перемешивали при 50°С в течение 30 мин. Реакционную смесь охлаждали до к.т. и добавляли раствор 3,6-дифтор-2-((3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)бензонитрила (1,12 г, 3,58 ммоль, Экв: 1,0) в ДМФА (25,5 мл). Реакционную смесь перемешивали при 100°С в течение 15 ч, затем концентрировали в вакууме. Остаток растворяли в насыщ. водном NH4Cl (100 мл) и EtOAc (100 мл). Фазы разделяли и водный слой дополнительно экстрагировали 2×100 мл EtOAc. Объединенные органические слои промывали водой (200 мл) и солевым раствором (200 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Водный слой экстрагировали обратно EtOAc (3×100 мл). Объединенные органические экстракты промывали солевым раствором (200 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток разбавляли ДХМ и МеОН и концентрировали на силикагеле. Очистка флэш-хроматографией (120 г, 0,5-2% МеОН/ДХМ) давало грязно-белое твердое вещество, которое тритурировали с 1:1 гептан/ДХМ (20 мл) с помощью ультразвука, затем сушили в вакууме с получением указанного в заголовка соединения в виде твердого бесцветного вещества (1,087 г, выход 66%). МС (ИЭР) м/з: 426,2 [М+Н]+. Хиральная СЖХ: RT=4,594 мин [колонка Chiralpak IC, 4,6 х 250 мм, размер частиц 5 мкм (Daicel); градиент 20-40% МеОН, содержащий 0,2% NHEt2, в течение 8 мин.; поток: 2,5 мл/мин.; противодавление 140 бар].
(3S)-N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид (Пример 2)
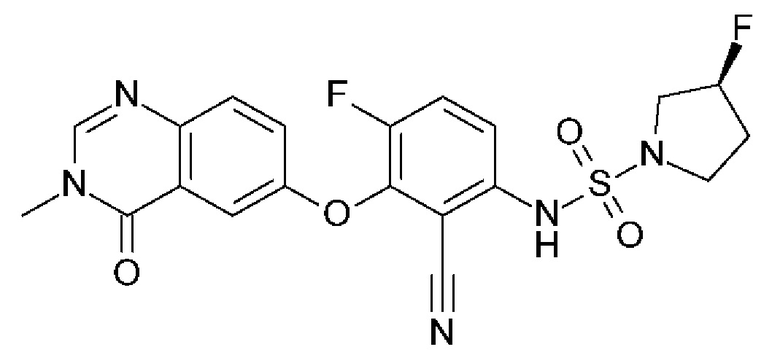
(S)-3-фторпирролидин-1-сульфонамид (181 мг, 1,08 ммоль, Экв: 2,1) растворяли в ДМФА (1,6 мл). Добавляли карбонат цезия при к.т.(368 мг, 1,13 ммоль, Экв: 2,2) и реакционную смесь перемешивали при 50°С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и добавляли раствор 3,6-дифтор-2-((3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)бензонитрила (160,8 мг, 513 мкмоль, Экв: 1,0) в ДХМ (4 мл). Реакционную смесь перемешивали при 105°С в течение 2 ч 50 мин, затем концентрировали в вакууме. Остаток растворяли в ДХМ и промывали насыщ. водным NH4Cl. Водный слой дважды экстрагировали ДХМ. Объединенные органические слои сушили над Na2SO4, фильтровали и выпаривали. Остаток (коричневое масло) разбавляли ДХМ и переносили на колонку. Очистка флэш-хроматографией (80 г, 0-100% EtOAc в ДХМ) давала твердое вещество, которое затем очищали с помощью СРХ с получением указанного в заголовке соединения в виде твердого вещества светло-желтого цвета (119 мг, выход 50%). МС (ИЭР) м/з: 426,2 [М+Н]+. Хиральная СЖХ: КТ=4,411 мин [колонка Chiralpak IC, 4,6 × 250 мм, размер частиц 5 мкм (Daicel); градиент 20-40% МеОН, содержащий 0,2% NHEt2, в течение 8 мин; поток: 2,5 мл/мин; противодавление 140 бар].
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ИНГИБИТОРЫ BRAF КАК «РАЗРУШИТЕЛИ ПАРАДОКСА» | 2020 |
|
RU2825870C1 |
| СОЕДИНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2708247C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
| ТЕРАПЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР RAF И ИНГИБИТОР ERK | 2017 |
|
RU2774612C2 |
| ИНГИБИТОР BRAF КИНАЗЫ N-(3-(5-(4-ХЛОРОФЕНИЛ)-1H-ПИРАЗОЛО[3,4-B]ПИРИДИН-3-КАРБОНИЛ)-2,4-ДИФТОРОФЕНИЛ) ПРОПАН-1-СУЛЬФОНАМИД | 2018 |
|
RU2687107C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА N-(3-(5-(4-ХЛОРОФЕНИЛ)-1Н-ПИРАЗОЛО[3,4-В]ПИРИДИН-3-КАРБОНИЛ)-2,4-ДИФТОРОФЕНИЛ) ПРОПАН-1-СУЛЬФОНАМИДА, АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2018 |
|
RU2678455C1 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ PAC-1 | 2016 |
|
RU2720509C2 |
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| НОВЫЕ ИНГИБИТОРЫ BRAF И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ КОЖНЫХ РЕАКЦИЙ | 2018 |
|
RU2779185C2 |
| Способ адъювантного лечения рака | 2013 |
|
RU2640180C2 |

Изобретение относится к новому соединению, которое имеет общую формулу (I), или его фармацевтически приемлемой соли. Соединение формулы (I) может быть использовано в качестве лекарственного средства для применения в лечении или профилактике рака, вызванного мутантами BRAF. 6 н. и 7 з.п. ф-лы, 8 ил., 10 табл., 2 пр.
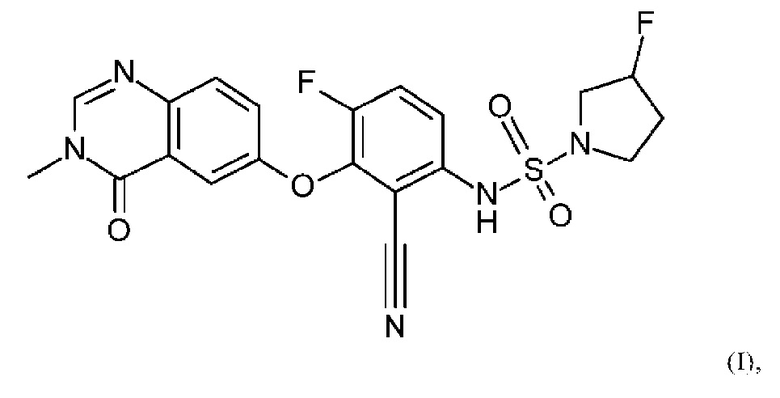
1. Соединение формулы (I)
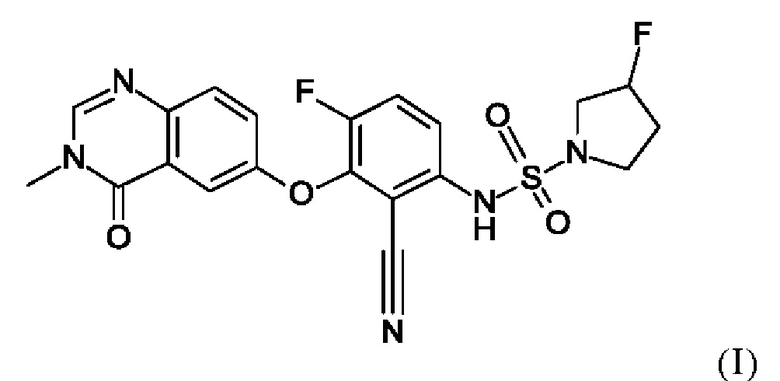
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где соединение представляет собой соединение формулы (I).
3. Соединение по п. 1 или 2, где соединение представляет собой соединение формулы (Ia).
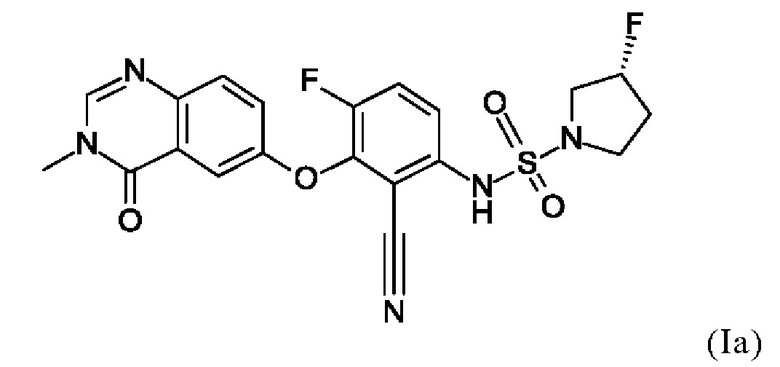
4. Соединение по п. 1 или 2, где соединение представляет собой соединение формулы (Iб).
5. Способ получения соединения по любому из пп. 1-4, включающий проведение реакции соединения формулы (Б1)
с соединением формулы (Б2)
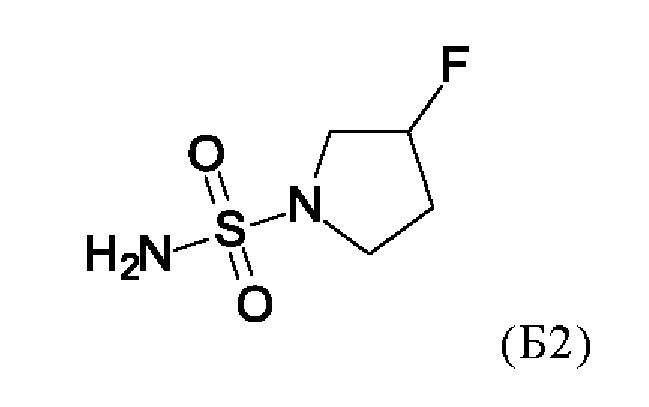
в присутствии основания.
6. Соединение по любому из пп. 1-4 для применения в качестве терапевтически активного вещества.
7. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении BRAF, содержащая эффективное количество соединения по любому из пп. 1-4 и терапевтически инертный носитель.
8. Соединение по любому из пп. 1-4 для применения в лечении или профилактике рака, вызванного мутантами BRAF.
9. Соединение по любому из пп. 1-4 для применения в лечении или профилактике рака, вызванного мутантами BRAF, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
10. Применение соединения по любому из пп. 1-4 для лечения или профилактики рака, вызванного мутантами BRAF, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
11. Применение соединения по любому из пп. 1-4 для получения лекарственного средства для лечения или профилактики рака, вызванного мутантами BRAF, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
12. Способ лечения рака, вызванного мутантами BRAF, причем указанный способ включает введение эффективного количества соединения, охарактеризованного в любом из пп. 1-4, нуждающемуся в этом пациенту.
13. Способ по п. 12, где рак, вызванный мутантами BRAF, выбран из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
| WO 2012118492 A1, 07.09.2012 | |||
| WENGLOWSKY STEVE et al., "Highly potent and selective 3-N-methylquinazoline-4(3H)-one based inhibitors of B-RafV600Ekinase", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, 2014, vol | |||
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Раздвижной золотник-байпас | 1925 |
|
SU1923A1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ ИНГИБИТОРА CDK4/6 И ИНГИБИТОРА В-Raf | 2013 |
|
RU2685250C2 |
