Изобретение относится к области получения водных полиуретановых дисперсий, используемых в производстве покрытий и адгезивов.
Покрытия на основе водных полиуретановых дисперсий низкотоксичные, безопасны как в нанесении, так и в процессе эксплуатации, быстро высыхают после нанесения, а также обладают высокой эластичностью.
Данные преимущества водных дисперсий полиуретана способствуют все более широкому распространению данных материалов на рынке.
Водные полиуретановые дисперсии представляют собой стабильную коллоидную систему. Дисперсионной средой в такой системе является вода, а дисперсной фазой - полиуретан. Для повышения стабильности системы на стадии синтеза форполимера в цепочку полимера вводят гидрофильные группы.
Преимущество полиуретановых дисперсий состоит в том, что они однокомпонентны, однако для приведения водостойкости, химстойкости и стойкости к истиранию к оптимальному уровню требуется введение дополнительных добавок. Недостатком водных полиуретановых дисперсий является плохая адгезия к покрываемой поверхности и для устранения данного недостатка в композицию покрытия вводят адгезионные добавки.
В литературе описаны, в основном, два способа синтеза ПУД, а именно: предполимерный и ацетоновый. В этих способах дисперсию получают в две стадии: на первой стадии синтезируют полиуретановый предполимер по реакции полиизоцианатов с полиолами, на второй стадии производят диспергирование предполимера в воде с последующим его удлинением полиаминами в водной фазе. В предполимерном способе используют высококипящие растворители, такие как N-метилпирролидон и диметилформамид (в количестве до 30 масс. %), которые остаются в конечном продукте. В ацетоновом способе используют низкокипящие растворители, такие как ацетон или метилэтилкетон, которые удаляют из конечного продукта отгонкой при пониженном давлении. Этот способ позволяет широко варьировать сырьевые компоненты, но наличие энергоемкой стадии отгонки больших количеств растворителя, а также его регенерации приводит к значительному удорожанию процесса ( Samy A. Madbouly, Joshua U. Otaigbe // Recent advances in synthesis, characterization and rheological properties of polyurethanes and POSS / polyurethane nanocomposites dispersions and films. Progress in Polymer Science. 2009. V. 34 P. 1283-1332)
Известна сшиваемая полиуретановая дисперсия (патент на изобретение РФ № 2527946, опубл. 10.09.2014), получаемая следующим образом: осуществляют загрузку полиольного компонента, компонента с сульфонатными и/или карбоксилатными группами, при необходимости других реакционноспособных по отношению к изоцианатам соединений, после чего указанные компоненты нагревают до температуры от 20 до 100°С, и при перемешивании как можно быстро добавляют изоцианатный компонент. Используя экзотермический эффект, реакционную смесь перемешивают при температуре от 40 до 150°С до тех пор, пока не будет достигнуто теоретическое содержание изоцианатных групп. При этом при необходимости можно добавлять катализатор. Затем реакционную смесь разбавляют растворителем до содержания твердого вещества от 25 до 95% масс., предпочтительно от 40 до 80% масс., и при температуре от 30 до 120°С путем подачи разбавленного водой и/или растворителем аминокарбоновой кислоты и/или гидроксикарбоновой кислоты. По истечении времени реакции, составляющего от 2 до 60 минут, путем добавления дистиллированной воды, соответственно переведения реакционной смеси в добавленную дистиллированную воду, осуществляют диспергирование образовавшегося полимера, причем использованный растворитель частично или полностью отгоняют в процессе диспергирования или по его завершении.
Известен способ получения высококонцентрированной водной наноразмерной полиуретановой дисперсии, не содержащей органический растворитель, с концентрацией основного вещества 30-60%, представляющей собой продукт взаимодействия:
A) по меньшей мере одного полиизоцианата, содержащего по меньшей мере две изоцианатные группы;
B) одного или нескольких полиолов с молекулярной массой (ММ) от 1000 до 18000, имеющих по меньшей мере две гидроксильные группы;
C) одного или нескольких соединений по меньшей мере с двумя OH-функциональными группами, которые содержат по меньшей мере одну карбоксильную группу, которая может быть превращена полностью или частично в карбоксилатную группу в присутствии оснований;
D) возможно одного или нескольких полиолов и/или глицидиловых эфиров полиолов со средней молекулярной массой менее 500, содержащих 2 и более гидроксильные и/или эпоксидные группы;
E) одного или нескольких третичных аминов;
F) одного или нескольких полиаминов, содержащих по меньшей мере одну NH2-группу.
При этом подвергают одновременному взаимодействию компоненты (A), (B) и (C) до степени конверсии изоцианатных групп 70-98%, при необходимости вводят в реакционную массу компонент (D), затем полностью или частично нейтрализуют карбоксильные группы компонента (C) компонентом (E), диспергируют в воде, вводят компонент (F), нагревают дисперсию и выдерживают при температуре от 20 до 90°C в течение от одного до четырех часов.
Известен патент KR 11209295, в котором описан способ получения водной полиуретановой дисперсии с участием в качестве модификатора основной цепи амина с алкоксисилановыми группами. После гидролиза в водной среде алкоксисилановых групп, все образованные гидроксисилановые группы (-Si-ОН) вступают в реакцию поликонденсации, в результате чего силановый фрагмент выступает в качестве разветвляющего агента, что повышает физико-механические показатели полученной полиуретанмочевины. Описанная область применения такой дисперсии – основа красок против обрастания днищ морских судов микроорганизмами.
Известные составы полиуретановых дисперсий не решают проблему низкой адгезии покрытия с поверхностью.
Технической задачей заявляемого изобретения является получение стабильной дисперсии полиуретана в воде, применяемой для производства покрытий и адгезивов, обладающей высоким уровнем адгезии к покрывной поверхности.
Технический результат заключается в повышении адгезионных свойств покрытия, полученного на основе водной дисперсии полиуретана.
Технический результат достигается тем, что способ получения водной полиуретановой дисперсии, представляющей собой продукт взаимодействия по меньшей мере одного диизоцианата и/или полиизоцианата с функциональностью от 2 до 3 по NCO-группе, по меньшей мере одного простого полиэфир-полиола с функциональностью по ОН-группам от 1,9 до 4 и гидроксильным числом от 14 до 250, растворителя, способного растворяться в органической и водной фазе, органической кислоты с функциональностью по СООН-группам от 1 до 3 и ОН-группам от 2 до 3, по меньшей мере одного третичного амина как нейтрализатора карбоксильной группы, по меньшей мере одного водорастворимого амина «А2» с функциональностью по NH2-группе от 2 до 4, по меньшей мере одного амина «А1» - продукта гидролиза и последующей поликонденсации аминоалкоксисилана H2N-R-Si-(OAlk)3 общей формулы
H2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2 (1)
где
R = -(СH2)k-, k от 1 до 5
Alk = CH3 или C2H5
содержит следующие стадии: полиольный компонент подвергают предварительному обезвоживанию в реакторе при температуре 93-95 °С под вакуумом до степени содержания остаточной влаги не более 0,03% с последующей подачей смеси органической кислоты и растворителя, перемешивания при температуре 50-80°С и введения изоцианатного компонента, осуществляют синтез форполимера при температуре 25-90°С в атмосфере азота до содержания расчетного количества остаточных изоцианатных групп, затем нейтрализуют карбоксильные группы третичным амином при температуре 40-80°С в течение 0,5-3ч, диспергируют форполимер в воде с добавлением пеногасителя, после чего вводят амин «А2», при рН в пределах 6.0 – 7.0 вводят амин «А1» и выдерживают до полного исчезновения NCO-групп.
В заявляемом изобретении в качестве одного из полиаминов (или единственного полиамина) используется продукт с алкоксисилановыми группами общей формулы
H2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2 (1)
где
R = -(СH2)k-, k от 1 до 5
Alk = CH3 или C2H5
Как известно, наиболее эффективными адгезивами на сегодняшний день являются бифункциональные соединения, способные к взаимодействию, как с полимерной основой материала, так и с поверхностью, на который этот материал наносится. Таким образом, адгезив выступает «мостиком», химически связывающим полимерную основу с поверхностью (стекло, металл, бетон и т.п.).
Функциональные алкоксисиланы – наиболее известные соединения для усиления адгезионных свойств:
F-R-Si(OAlk)3, (2)
где
Alk = CH3 или C2H5,
R = -(СH2)k- , k от 1 до 5
F – функциональная группа (эпоксидная, винильная, изоцианатная, сульфгидрильная, аминная, акрилатная).
Функциональной группой F адгезив взаимодействует с реакционными группами полимерной основы (связующего).
Алкоксисилановые группы на первой стадии подвергаются реакции с водой (гидролиз):
F-R-Si(OAlk)3 + 3Н2О = F-R-Si(OН)3 + 3AlkOH (3)
Полученные гидроксисилановые группы (-Si-OH) способны к реакции поликонденсации с ОН-группами, имеющимися на поверхности большинства материалов:
F-R-Si(OН)3 + НО-М = F-R-Si(OН)2 – М + Н2О, (4)
где М – материал поверхности.
В заявляемом изобретении предложено введение в состав водной полиуретановой дисперсии на этапе ее синтеза полиамина общей формулы:
H2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2 (1)
Полиамин данной формулы получается из двух молекул аминоалкилполиалкоксисилана путем их неполного гидролиза и последующей поликонденсации между собой:
Стадия гидролиза:
2 NH2-R-Si(OAlk)3 + 2Н2О = 2 NH2-R-Si(OН)(ОAlk)2 + 2AlkOH (5)
где
R = -(СH2)k-, k от 1 до 5;
Alk = CH3 или C2H5
Стадия поликонденсации:
2 NH2-R-Si(OН)(ОAlk)2 = H2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2 + H2O (6)
Указанный полиамин, взаимодействуя с диспергированным в воде уретановым форполимером, образует диспергированную в воде полиуретанмочевину с алкоксисилановыми группами:
2 ~R’-NCO + H2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2 =
~R’-N(H)C(O)N(H)-R - Si(OAlk)2 – O – Si(OAlk)2 – R – N(H)C(O)N(H)-R’~ (7)
Где R` - остаток уретанового форполимера
В водной среде алкоксисилановые группы полиуретанмочевины гидролизуются до гидроксисилановых:
~R’-N(H)C(O)N(H)-R - Si(OAlk)2 – O – Si(OAlk)2 – R – N(H)C(O)N(H)-R’~ + 4H2O =
~R’-N(H)C(O)N(H)-R - Si(OH)2 – O – Si(OH)2 – R – N(H)C(O)N(H)-R’~ + 4AlkOH (8)
Для обеспечения обратимости реакции поликонденсации полиуретанмочевины по гидроксильным группам в водной среде необходимо обеспечить нейтральность среды (рН в пределах 6.0 – 7.0), а также отсутствие катализаторов поликонденсации (солей металлов, свободных аминов и др.). В этих условиях гидроксильные группы сохранятся на полиуретанмочевине до нанесения дисперсии на поверхность обрабатываемого материала и испарения воды, соответственно, обеспечив необратимую реакцию по гидроксильным группам материала (см. реакцию 4) и, таким образом, обеспечив высокую адгезию.
Полиуретановая дисперсия согласно заявляемому техническому решению является продуктом взаимодействия следующих компонентов:
1) по меньшей мере одного полиола;
2) по меньшей мере одного полиизоцианата;
3) растворителя;
4) органической кислоты с гидроксильными группами;
5) по меньшей мере одного третичного амина (нейтрализатора);
6) по меньшей мере одного амина «А1» - продукта гидролиза и последующей поликонденсации аминоалкоксисилана H2N-R-Si-(OAlk)3 общей формулы;
7) по меньшей мере одного водорастворимого амина «А2» с функциональностью по NH2-группе от 2 до 4.
В качестве полиола возможно использование простого полиэфир-полиола, с функциональностью по ОН-группам от 1.9 до 4, предпочтительно 1.9 – 2.5, и гидроксильным числом от 14 до 250, предпочтительно от 20 до 60.
В качестве органической кислоты могут быть использованы карбоновые кислоты с функциональностью по СООН-группам от 1 до 3 с гидроксильными группами (функциональность по ОН-группам от 2 до 3) (например, диметилопропионовая кислота (ДМПК)) в количестве, определяемым мольным соотношением между ОН группами полиола и кислоты (варьируется в пределах 0,5/1 до 4/1, предпочтительно от 1/1 до 2/1).
В процессе синтеза используется растворитель, способный растворяться как в органической, так и водной фазе (например, N-метилпирролидон, ацетон). Количество растворителя определяется в зависимости от требуемой вязкости форполимера при определенной температуре.
Полиизоцианатный компонент (с функциональностью по NCO-группе от 2 до 3, предпочтительно от 2 до 2.2) может быть использован как алифиатической (например, гексаметилендиизоцианат, изофорондиизоцианат и др.), так и ароматической (например, толуилендиизоцианат, дифенилметандиизоцианат и др.) природы. Полиизоцианатный компонент используется в количестве, соответствующему мольному соотношению между NCO-группами изоцианата и гидроксильными группами полиолов. Мольное соотношение между NCO-группами изоцианата и гидроксильными группами полиолов (в т.ч. гидроксилсодержащей кислоты) варьируется в пределах от 1.5 до 3, предпочтительно от 1.8 до 2.2.
В качестве нейтрализатора карбоксильной группы применяются третичные амины (возможно использование триэтиламина, диметилциклогексиламина или этилдиизопропиламина) в количестве, определяемым мольным соотношением между третичными аминами и карбоксильными группами (варьируется в пределах 0.5/1 до 1.5/1, предпочтительно 0.7/1 до 1/1).
В качестве полиамина с алкоксисилановыми группами «А1» используется продукт неполного гидролиза и последующей конденсации аминоалкоксилана (H2N-R-Si-(OAlk)3) общей формулы
H2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2 (1)
Амин «А1» служит для обеспечения адгезионной способности готового продукта.
Другой амин («А2») должен иметь функциональность по NH2-группе от 2 до 4 (предпочтительно от 2 до 2.2). В качестве «А2» могут быть использованы водорастворимые полиамины (этилендиамин, пропилендиамин, др.).
Мольное содержание NH2-групп продукта «А1» в смеси продуктов «А1» и «А2» составляет от 10 до 100% (предпочтительно от 20 до 30%).
Количество полиаминов определяется мольным соотношением NH2-групп полиаминов (суммарно «А1» и «А2») и NCO-групп форполимера (варьируется в пределах 0.5/1 до 1.5/1, предпочтительно в пределах 0.8/1 до 1/1).
Процесс получения водной полиуретановой дисперсии состоит из следующих основных стадий:
1. Обезвоживание полиола;
2. Синтез форполимера;
3. Нейтрализация форполимера;
4. Эмульгирование форполимера;
5. Получение полиуретановой дисперсии.
На первой стадии в реактор, выполненный из некорродирующего материала, снабженный мешалкой, системой обогрева и охлаждения, вакуумной и азотной линиями вводят полиол. При перемешивании полиол разогревается до 93-95°С. Далее в реакторе создается вакуум с остаточным давлением не более 20 мм рт.ст. Процесс обезвоживания ведут в течение 3 часов, после чего отбирается проба на содержание остаточной влаги. При содержании остаточной влаги более 0,03% ведут процесс обезвоживания еще 2 часа с повторным отбором пробы. По достижении содержания остаточной влаги не более 0,03% переходят к следующей стадии.
На второй стадии синтеза форполимера в реактор подается смесь растворителя и органической кислоты с гидроксильными группами.
Перемешивание ведется до получения гомогенной смеси с охлаждением до 25-80°С, после чего в реактор вводят полиизоцианат. Синтез форполимера ведут при температуре от 25 до 95°С, предпочтительно от 5 до 80°С, в атмосфере азота до достижения расчетного значения остаточных изоцианатных групп.
На третьей стадии осуществляют нейтрализацию форполимера третичным амином. Процесс нейтрализации ведут при температуре в пределах 40 – 80°С в течение от 30 минут до 3 часов.
На четвертой стадии проводят эмульгирование форполимера в аппарате из некорродирующего материала, снабженного мешалкой, системой обогрева и охлаждения, азотной линией.
В аппарат-эмульгатор вводят дистиллированную воду в количестве от 30 до 80% от массы вводимого форполимера и пеногаситель в количестве от 0.01% до 1 % на готовый продукт.
Далее, при интенсивном перемешивании воды (скорость мешалки не менее 800 об/мин), в аппарат-эмульгатор вводится нейтрализованный форполимер. Время подачи форполимера – не более 10 минут. Температура воды перед подачей форполимера варьируется в пределах от 20 до 50°С, температура вводимого форполимера – от 20 до 60°С. Эмульгирование форполимера ведут от 5 минут до 1 часа при температуре от 20 до 60°С.
На пятой стадии осуществляют получение полиуретановой дисперсии, заключающееся в реакции между уретановым форполимером, диспергированным в воде, с удлинителями цепи – полиаминами - с образованием твердой полиуретанмочевины (дисперсной фазы) в воде (дисперсионная среда).
Расчетное количество амина «А2» вводится в аппарат с полиуретановой эмульсией сразу по окончании процесса эмульгирования. Температура эмульсии перед подачей амина «А2» – от 20 до 50°С. Скорость перемешивающего устройства при подаче амина «А2» и при дальнейшем перемешивании должна быть не ниже 800 об/мин.
Расчетное количество амина «А1» вводится в аппарат с полиуретановой эмульсией сразу при достижении рН эмульсии от 6.0 до 7.0. Скорость перемешивающего устройства при подаче амина «А1» и при дальнейшем перемешивании должна быть не ниже 800 об/мин. Перемешивание ведут при температуре 40-50°С до полного исчезновения NCO-групп.
Примеры осуществления изобретения
В приведенных примерах реализации в качестве полиолов использовали полиоксипропиленгликоли ППГ 2000 (функциональность 2, гидроксильное число 56 мгКОН/г) и ППГ 4000 (функциональность 2, гидроксильное число 28 мгКОН/г), ароматического изоцианата - смесь изомеров 2.4 и 2,6 - толуилендиизоцианат (ТДИ), алифатического изоцианата – изофорондиизоцианат (ИФДИ), органической кислоты (эмульгатора) – диметилолпропионовая кислота (ДМПК), растворителя – N-метилпирролидон (NMP), третичного амина – триэтиламин (ТЭА), в качестве полиамина «А1» - полиамин с алкоксисилановыми группами на основе гамма-аминопропилтриэтоксисилана (ОАМЕО), полиамина «А2» - этилендиамин (ЭДА), пеногаситель марки SN-Defoamer NXZ (на основе алифатических углеводородов и неионных ПАВ, производство San Nopco Limited (Япония)).
Пример 1
В реактор, снабженный мешалкой, системой обогрева и охлаждения, вакуумной и азотной линиями вводили 200гр полиола ППГ2000 и нагревали при непрерывном перемешивании до 93-95°С. Далее процесс обезвоживания полиола протекал в реакторе под вакуумом с остаточным давлением 20 мм рт.ст. до содержания остаточной влаги не более 0,03%.
Далее в реактор подавали смесь 30 гр NMP и 8,7 гр ДМПК с последующим перемешиванием до получения гомогенной смеси, смесь охлаждали до +50°С, после чего в реактор вводили 55гр ТДИ.
Синтез форполимера вели при температуре 60-65°С в атмосфере азота до достижения расчетного значения остаточных изоцианатных групп 4,22% (1 час).
Далее в реактор вводили 5,3гр ТЭА, далее вели нейтрализацию при температуре 60-65°С в течение 1 часа.
Эмульгирование форполимера осуществляли в аппарате-эмульгаторе, снабженном мешалкой, системой обогрева и охлаждения, азотной линией. Для этого вводили в смеситель дистиллированную воду в количестве 410,2 гр и пеногаситель NXZ в количестве 4 гр. Температуру водной фазы поддерживали при 20-25°С. Далее, при интенсивном перемешивании воды (1000 об/мин) в аппарат в течение 5 минут вводили охлажденный до 40°С форполимер в количестве 299,1гр. Эмульгирование форполимера в воде вели в течение 10 минут при температуре 25-30°С.
5,6гр ЭДА вводили в аппарат с полиуретановой эмульсией сразу по окончании процесса эмульгирования при непрерывном интенсивном перемешивании (1000 об/мин). Через 10 минут диспергирования проводится замер рН дисперсии. При рН от 6.0 до 7.0 в аппарат вводили 18гр ОАМЕО. Далее процесс получения полиуретановой дисперсии вели при температуре 40-50°С до полного исчезновения NCO-групп (около 45 минут).
Готовую полиуретановую дисперсию сливали в герметичную стеклянную тару и проводили анализ (см. таблицу 1).
Пример 2
В реактор, снабженный мешалкой, системой обогрева и охлаждения, вакуумной и азотной линиями вводили 200гр полиола ППГ2000 и нагревали при непрерывном перемешивании до 93-95°С. Далее процесс обезвоживания полиола протекал в реакторе под вакуумом с остаточным давлением 20 мм рт.ст. до содержания остаточной влаги не более 0,03%.
Далее в реактор подавали смесь 30 гр NMP и 8,6 гр ДМПК с последующим перемешиванием до получения гомогенной смеси, смесь охлаждали до +5°С, после чего в реактор вводили 55гр ТДИ.
Синтез форполимера вели при температуре 60-65°С в атмосфере азота до достижения расчетного значения остаточных изоцианатных групп 4,24% (1 час).
Далее в реактор вводили 5,2 гр ТЭА, далее вели нейтрализацию при температуре 60-650С в течение 1 часа.
Эмульгирование форполимера осуществляли в аппарате-эмульгаторе, снабженном мешалкой, системой обогрева и охлаждения, азотной линией. Для этого вводили в смеситель дистиллированную воду в количестве 396,6 гр и пеногаситель NXZ в количестве 4гр. Температуру водной фазы поддерживали при 20-25°С. Далее, при интенсивном перемешивании воды (1000 об/мин) в аппарат в течение 5 минут вводили охлажденный до 40°С форполимер в количестве 298,9 гр. Эмульгирование форполимера в воде вели в течение 10 минут при температуре 25-30°С.
6,8гр ЭДА вводили в аппарат с полиуретановой эмульсией сразу по окончании процесса эмульгирования при непрерывном интенсивном перемешивании (1000 об/мин). Через 10 минут диспергирования проводится замер рН дисперсии. При рН от 6.0 до 7.0 в аппарат вводили 8.8 гр ОАМЕО. Процесс получения полиуретановой дисперсии вели при температуре 40-50°С до полного исчезновения NCO-групп (около 45 минут).
Готовую полиуретановую дисперсию сливали в герметичную стеклянную тару и проводили анализ (см. таблицу 1).
Пример 3 (без применения ОАМЕО, сравнительный)
В реактор, снабженный мешалкой, системой обогрева и охлаждения, вакуумной и азотной линиями вводили 200 гр полиола ППГ2000 и нагревали при непрерывном перемешивании до 93-95°С. Далее процесс обезвоживания полиола протекал в реакторе под вакуумом с остаточным давлением 20 мм рт.ст. до содержания остаточной влаги не более 0,03%.
Далее в реактор подавали смесь 30гр NMP и 8,5 гр ДМПК с последующим перемешиванием до получения гомогенной смеси, смесь охлаждали до +50°С, после чего в реактор вводили 55 гр ТДИ.
Синтез форполимера вели при температуре 60-650С в атмосфере азота до достижения расчетного значения остаточных изоцианатных групп 4,27% (1 час).
Далее в реактор вводили 5,1 гр ТЭА, далее вели нейтрализацию при температуре 60-65°С в течение 1 часа.
Эмульгирование форполимера осуществляли в аппарате-эмульгаторе, снабженном мешалкой, системой обогрева и охлаждения, азотной линией. Для этого вводили в смеситель дистиллированную воду в количестве 385 гр и пеногаситель NXZ в количестве 4 гр. Температуру водной фазы поддерживали при 20-25°С. Далее, при интенсивном перемешивании воды (1000 об/мин) в аппарат в течение 5 минут вводили охлажденный до 40°С форполимер в количестве 298,7 гр. Эмульгирование форполимера в воде вели в течение 10 минут при температуре 25-30°С.
8,0гр ЭДА вводили в аппарат с полиуретановой эмульсией сразу по окончании процесса эмульгирования при непрерывном интенсивном перемешивании (1000 об/мин). Процесс получения полиуретановой дисперсии вели при температуре 40-50°С до полного исчезновения NCO-групп (около 45 минут).
Готовую полиуретановую дисперсию сливали в герметичную стеклянную тару и проводили анализ (см. таблицу 1).
Пример 4
В реактор, снабженный мешалкой, системой обогрева и охлаждения, вакуумной и азотной линиями вводили 200гр полиола ППГ2000 и нагревали при непрерывном перемешивании до 93-95°С. Далее процесс обезвоживания полиола протекал в реакторе под вакуумом с остаточным давлением 20 мм рт.ст. до содержания остаточной влаги не более 0,03%.
Далее в реактор подавали смесь 30гр NMP и 9,5 гр ДМПК с последующим перемешиванием до получения гомогенной смеси, смесь охлаждали до +50°С, после чего в реактор вводили 73 гр ИФДИ.
Синтез форполимера вели при температуре 80-85°С в атмосфере азота до достижения расчетного значения остаточных изоцианатных групп 4,15% (3 часа).
Далее в реактор вводили 5,8 гр ТЭА, далее вели нейтрализацию при температуре 60-650С в течение 1 часа.
Эмульгирование форполимера осуществляли в аппарате-эмульгаторе, снабженном мешалкой, системой обогрева и охлаждения, азотной линией. Для этого вводили в смеситель дистиллированную воду в количестве 439,1гр и пеногаситель NXZ в количестве 4гр. Температуру водной фазы поддерживали при 20-25°С. Далее, при интенсивном перемешивании воды (1000 об/мин) в аппарат в течение 5 минут вводили охлажденный до 40°С форполимер в количестве 318,4гр. Эмульгирование форполимера в воде вели в течение 10 минут при температуре 25-30°С.
5,9гр ЭДА вводили в аппарат с полиуретановой эмульсией сразу по окончании процесса эмульгирования при непрерывном интенсивном перемешивании (1000 об/мин). Через 10 минут диспергирования проводится замер рН дисперсии. При рН от 6.0 до 7.0 в аппарат вводили 18.5 гр ОАМЕО. Процесс получения полиуретановой дисперсии вели при температуре 40-50°С до полного исчезновения NCO-групп (около 45 минут).
Готовую полиуретановую дисперсию сливали в герметичную стеклянную тару и проводили анализ (см. таблицу 1).
Пример 5
В реактор, снабженный мешалкой, системой обогрева и охлаждения, вакуумной и азотной линиями вводили 200 гр полиола ППГ4000 и нагревали при непрерывном перемешивании до 93-95°С. Далее процесс обезвоживания полиола протекал в реакторе под вакуумом с остаточным давлением 20 мм рт.ст. до содержания остаточной влаги не более 0,03%.
Далее в реактор подавали смесь 30гр NMP и 8,1 гр ДМПК с последующим перемешиванием до получения гомогенной смеси, смесь охлаждали до +50°С, после чего в реактор вводили 37гр ТДИ.
Синтез форполимера вели при температуре 80-85°С в атмосфере азота до достижения расчетного значения остаточных изоцианатных групп 3,01% (2 часа).
Далее в реактор вводили 4,9гр ТЭА, далее вели нейтрализацию при температуре 60-650С в течение 1 часа.
Эмульгирование форполимера осуществляли в аппарате-эмульгаторе, снабженном мешалкой, системой обогрева и охлаждения, азотной линией. Для этого вводили в смеситель дистиллированную воду в количестве 368 гр и пеногаситель NXZ в количестве 4гр. Температуру водной фазы поддерживали при 20-25°С. Далее, при интенсивном перемешивании воды (1000 об/мин) в аппарат в течение 5 минут вводили охлажденный до 40°С форполимер в количестве 280,1 гр. Эмульгирование форполимера в воде вели в течение 10 минут при температуре 25-30°С.
4,0 гр ЭДА вводили в аппарат с полиуретановой эмульсией сразу по окончании процесса эмульгирования при непрерывном интенсивном перемешивании (1000 об/мин). Через 10 минут диспергирования проводится замер рН дисперсии. При рН от 6.0 до 7.0 в аппарат вводили 12 гр ОАМЕО. Процесс получения полиуретановой дисперсии вели при температуре 40-50°С до полного исчезновения NCO-групп (около 45 минут).
Готовую полиуретановую дисперсию сливали в герметичную стеклянную тару и проводили анализ (см. таблицу 1).
Пример 6
В реактор, снабженный мешалкой, системой обогрева и охлаждения, вакуумной и азотной линиями вводили 200 гр полиола ППГ2000 и нагревали при непрерывном перемешивании до 93-95°С. Далее процесс обезвоживания полиола протекал в реакторе под вакуумом с остаточным давлением 20 мм рт.ст. до содержания остаточной влаги не более 0,03%.
Далее в реактор подавали смесь 45 гр NMP и 15гр ДМПК с последующим перемешиванием до получения гомогенной смеси, смесь охлаждали до +5°С, после чего в реактор вводили 71 гр ТДИ.
Синтез форполимера вели при температуре 80-85°С в атмосфере азота до достижения расчетного значения остаточных изоцианатных групп 4,88% (1 час).
Далее в реактор вводили 9гр ТЭА, далее вели нейтрализацию при температуре 60-650С в течение 1 часа.
Эмульгирование форполимера осуществляли в аппарате-эмульгаторе, снабженном мешалкой, системой обогрева и охлаждения, азотной линией. Для этого вводили в смеситель дистиллированную воду в количестве 444,6гр и пеногаситель NXZ в количестве 4 гр. Температуру водной фазы поддерживали при 20-25°С. Далее, при интенсивном перемешивании воды (1000 об/мин) в аппарат в течение 5 минут вводили охлажденный до 40°С форполимер в количестве 340,1 гр. Эмульгирование форполимера в воде вели в течение 10 минут при температуре 25-30°С.
7,4 гр ЭДА вводили в аппарат с полиуретановой эмульсией сразу по окончании процесса эмульгирования при непрерывном интенсивном перемешивании (1000 об/мин). Через 10 минут диспергирования проводится замер рН дисперсии. При рН от 6.0 до 7.0 в аппарат вводили 24 гр ОАМЕО. Процесс получения полиуретановой дисперсии вели при температуре 40-50°С до полного исчезновения NCO-групп (около 45 минут).
Готовую полиуретановую дисперсию сливали в герметичную стеклянную тару и проводили анализ (см. таблицу 1).
Образцы полученных дисперсий анализировали по следующим методикам:
рН: ГОСТ 33776-2016 «Методы испытаний химической продукции, представляющей опасность для окружающей среды. Определение рН, кислотности и щелочности».
Средний размер частиц: ГОСТ Р 8.777-2011 «Дисперсный состав аэрозолей и взвесей. Определение размеров частиц по дифракции лазерного излучения».
Вязкость: ГОСТ 25271-93 «Пластмассы. Смолы жидкие, эмульсии и дисперсии. Определение кажущейся вязкости по Брукфильду».
Прочность при растяжении: ГОСТ 18299-72 «Материалы лакокрасочные. Метод определения предела прочности при растяжении, относительного удлинения при разрыве и модуля упругости».
Адгезия: ГОСТ 15140-78 «Материалы лакокрасочные. Методы определения адгезии».
В таблице 1 представлены массовые и основные мольные соотношения используемых видов сырья, результаты испытаний.
Таблица 1
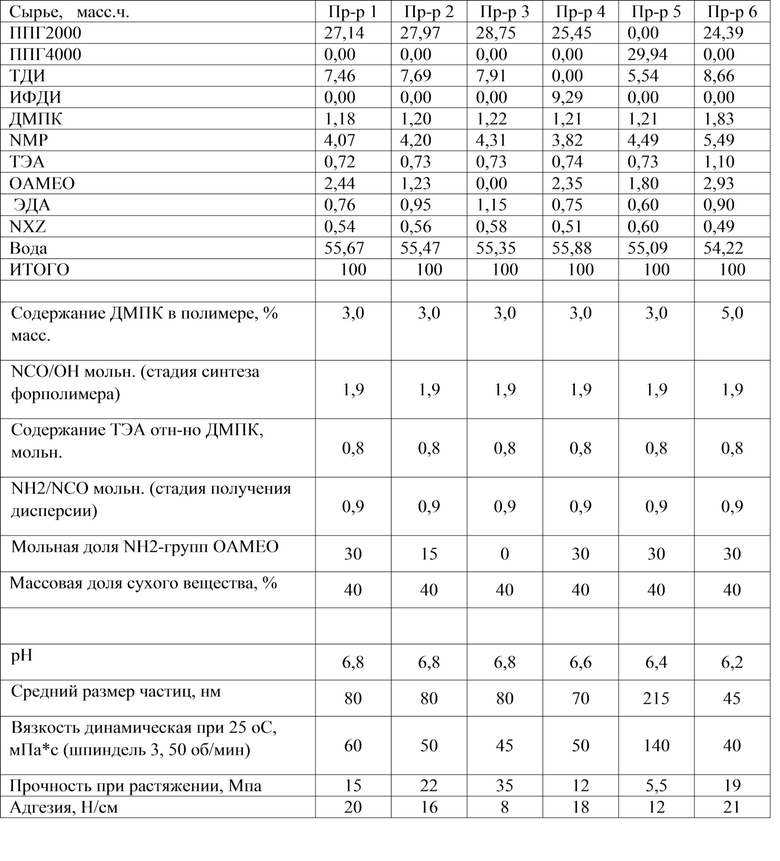
Как видно из приведенных примеров, дисперсии, содержащие в структуре полиуретанмочевины алкоксисилановые группы, обладают более высокой адгезией. Клеевые составы на основе таких продуктов обладают хорошей адгезией к различным материалам без термоактивации.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕХНОЛОГИЯ (СПОСОБ) ПОЛУЧЕНИЯ ВОДНОЙ ПОЛИУРЕТАНОВОЙ ДИСПЕРСИИ | 2021 |
|
RU2791545C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИЭФИРУРЕТАНТИОЛОВ С АЛКОКСИСИЛАНОВЫМИ ГРУППАМИ | 2018 |
|
RU2669567C1 |
| ВЫСОКОКОНЦЕНТРИРОВАННАЯ ВОДНАЯ НАНОРАЗМЕРНАЯ ПУ-ДИСПЕРСИЯ, НЕ СОДЕРЖАЩАЯ РАСТВОРИТЕЛЬ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2014 |
|
RU2554882C1 |
| НЕИОННО-ГИДРОФИЛИЗИРОВАННЫЕ СВЯЗУЮЩИЕ ДИСПЕРСИИ | 2008 |
|
RU2479600C2 |
| ОДНОКОМПОНЕНТНЫЕ СИСТЕМЫ ДЛЯ ПОКРЫТИЙ | 2004 |
|
RU2353628C2 |
| КОСМЕТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ СМОЛЫ И ЕЕ ПРИМЕНЕНИЕ В КОСМЕТИКЕ | 2000 |
|
RU2234912C2 |
| ПОЛИУРЕТАНОВАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПОЛИАЛЬДИМИН | 2003 |
|
RU2291162C2 |
| ВОДНЫЕ ПОЛИУРЕТАНОВЫЕ ДИСПЕРСИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ КЛЕЯ | 2005 |
|
RU2385331C2 |
| МИКРОПОРИСТОЕ ПОКРЫТИЕ НА ОСНОВЕ ПОЛИУРЕТАН-ПОЛИМОЧЕВИНЫ | 2007 |
|
RU2443722C2 |
| СПОСОБ ПОЛУЧЕНИЯ РЕАКЦИОННО-СПОСОБНОЙ ПОЛИУРЕТАНОВОЙ ЭМУЛЬСИИ | 2010 |
|
RU2496799C2 |
Настоящее изобретение относится к способу получения водной полиуретановой дисперсии, используемой в производстве покрытий и адгезивов. Указанный способ содержит следующие стадии: предварительное обезвоживание полиольного компонента в реакторе при 93-95°С под вакуумом до степени содержания остаточной влаги не более 0,03% с последующей подачей смеси органической кислоты и растворителя, перемешивание при 50-80°С и введение изоцианатного компонента, осуществление синтеза форполимера при 25-90°С в атмосфере азота до содержания расчетного количества остаточных изоцианатных групп, нейтрализация карбоксильных групп третичным амином при 40-80°С в течение 0,5-3 ч, диспергирование форполимера в воде с добавлением пеногасителя, после чего осуществляется введение амина «А2», далее при рН в пределах 6,0-7,0 осуществляют введение амина «А1» и выдерживание до полного исчезновения NCO-групп. Амин «А1» представляет собой продукт гидролиза и последующей поликонденсации аминоалкоксисилана H2N-R-Si-(OAlk)3 общей формулы
Н2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2. Амин «А2» является водорастворимым амином с функциональностью по NH2-группе от 2 до 4. Изоцианатный компонент является диизоцианатом и/или полиизоцианатом с функциональностью от 2 до 3 по NCO-группе. Полиольный компонент представляет собой, по меньшей мере, один простой полиэфир-полиол с функциональностью по ОН-группам от 1,9 до 4 и гидроксильным числом от 14 до 250. Растворитель способен растворяться в органической и водной фазе. Органическая кислота имеет функциональность по СООН-группам от 1 до 3 и ОН-группам от 2 до 3. Покрытия и адгезивы, полученные на основе данной водной полиуретановой дисперсии, обладают повышенными адгезионными свойствами. 3 з.п. ф-лы, 1 табл., 6 пр.
1. Способ получения водной полиуретановой дисперсии, представляющей собой продукт взаимодействия по меньшей мере одного диизоцианата и/или полиизоцианата с функциональностью от 2 до 3 по NCO-группе, по меньшей мере одного простого полиэфир-полиола с функциональностью по ОН-группам от 1,9 до 4 и гидроксильным числом от 14 до 250, растворителя, способного растворяться в органической и водной фазе, органической кислоты с функциональностью по СООН-группам от 1 до 3 и ОН-группам от 2 до 3, по меньшей мере одного третичного амина как нейтрализатора карбоксильной группы, по меньшей мере одного водорастворимого амина «А2» с функциональностью по NH2-группе от 2 до 4, по меньшей мере одного амина «А1» - продукта гидролиза и последующей поликонденсации аминоалкоксисилана H2N-R-Si-(OAlk)3 общей формулы
H2N- R - Si(OAlk)2 – O – Si(OAlk)2 – R – NH2 (1)
где
R = -(СH2)k-, k от 1 до 5
Alk = CH3 или C2H5,
содержит следующие стадии: полиольный компонент подвергают предварительному обезвоживанию в реакторе при температуре 93-95°С под вакуумом до степени содержания остаточной влаги не более 0,03% с последующей подачей смеси органической кислоты и растворителя, перемешивания при температуре 50-80°С и введения изоцианатного компонента, осуществляют синтез форполимера при температуре 25-90°С в атмосфере азота до содержания расчетного количества остаточных изоцианатных групп, затем нейтрализуют карбоксильные группы третичным амином при температуре 40-80°С в течение 0,5-3 ч, диспергируют форполимер в воде с добавлением пеногасителя, после чего вводят амин «А2», при рН в пределах 6.0-7.0 вводят амин «А1» и выдерживают до полного исчезновения NCO-групп.
2. Способ получения водной полиуретановой дисперсии по п.1, отличающийся тем, что мольное соотношение между ОН-группами полиольного компонента и органической кислоты находится в пределах 0,5/1 до 4/1, предпочтительно от 1/1 до 2/1.
3. Способ получения водной полиуретановой дисперсии по п.1, отличающийся тем, что мольное соотношение NH2/NCO варьируется в пределах от 0,5/1 до 1.5/1, предпочтительно от 0.8/1 до 1/1.
4. Способ получения водной полиуретановой дисперсии по п.1, отличающийся тем, что мольное содержание NH2-групп в смеси аминов «А1» и «А2» составляет от 10 до 100%, предпочтительно от 20 до 30%.
| KR 101209295 B1, 06.12.2012 | |||
| СПОСОБ ДЛЯ НАНЕСЕНИЯ ПОКРЫТИЙ, СКЛЕИВАНИЯ И СОЕДИНЕНИЯ МИНЕРАЛЬНЫХ ПОВЕРХНОСТЕЙ | 2009 |
|
RU2533126C2 |
| ПОЛИМЕРНАЯ ДИСПЕРСИЯ И КЛЕЙ, ИЗГОТОВЛЕННЫЙ С ЕЕ ПРИМЕНЕНИЕМ | 1999 |
|
RU2241728C2 |

