ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к катализатору окисления для дизельного двигателя и к системе выпуска выхлопных газов для дизельного двигателя, содержащей катализатор окисления. Данное изобретение также относится к способам и видам применения катализатора окисления для обработки выхлопных газов от дизельного двигателя.
УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ДАННОМУ ИЗОБРЕТЕНИЮ
Дизельные двигатели производят выброс выхлопных газов, которые обычно содержат по меньшей мере четыре класса загрязняющих веществ, которые запрещены в законодательном порядке межгосударственными организациями во всем мире: монооксид углерода (CO), несгоревшие углеводороды (HC), оксиды азота (NOx) и твердые частицы (PM). Нормы выпуска для дизельных двигателей, стационарных или подвижных (например, автомобильных дизельных двигателей), становятся все более жесткими. Поэтому имеется потребность в предложении улучшенных катализаторов и систем выпуска выхлопных газов, которые могут отвечать этим стандартам, и которые являются низкозатратными.
Системы выпуска выхлопных газов для дизельных двигателей могут включать несколько устройств для регулирования выбросов. Каждое устройство для регулирования выбросов имеет специализированное назначение и ответственно за обработку одного или более классов загрязняющих веществ в выхлопных газах. Эксплуатационные параметры устройства для регулирования выбросов выше по потоку могут влиять на эксплуатационные параметры устройства для регулирования выбросов ниже по потоку. Это обусловлено тем, что выхлопные газы от выпускного отверстия устройства для регулирования выбросов выше по потоку поступают в устройство для регулирования выбросов ниже по потоку. Взаимодействие между каждым устройством для регулирования выбросов в системе выпуска выхлопных газов важно для общей эффективности системы.
Катализаторы окисления, такие как катализаторы окисления дизельного топлива (DOC), обычно окисляют монооксид углерода (CO) и углеводороды (HC) в выхлопных газах, образованных дизельным двигателем. Катализаторы окисления дизельного топлива могут также окислять некоторую часть оксида азота (NO), который присутствует в выхлопных газах, до диоксида азота (NO2). Хотя диоксид азота (NO2) сам является загрязняющим веществом, превращение NO в NO2 может являться целесообразным. Произведенный NO2 может быть использован, чтобы регенерировать твердые частицы (PM), которые были захвачены, например, фильтром твердых частиц выхлопа дизельного двигателя (DPF) ниже по потоку или катализированным сажевым фильтром (CSF) ниже по потоку. Обычно NO2, генерируемый катализатором окисления, увеличивает отношение NO2:NO в выхлопных газах на выходе катализатора окисления по сравнению с выхлопными газами на входе. Это увеличенное отношение может быть выгодным для систем выпуска выхлопных газов, содержащих ниже по течению потока катализатор селективного каталитического восстановления (SCR) или фильтра с катализатором селективного каталитического восстановления (SCRFTM). Отношение NO2:NO в выхлопных газах, произведенных непосредственно дизельным двигателем, может быть слишком низким для оптимальных эксплуатационных параметров катализатора селективного каталитического восстановления (SCR) или фильтра с катализатором селективного каталитического восстановления (SCRF).
Несмотря на то, что обычно является выгодным включение в систему выпуска выхлопных газов катализатора окисления, такого как катализатор окисления дизельного топлива (DOC), который обладает хорошей активностью в отношении образования NO2, такое применение катализатора окисления может быть проблематичным в случае стремления к получению оптимальных эксплуатационных параметров от устройства для регулирования выбросов ниже по течению потока (например, катализатора селективного каталитического восстановления (SCR) или фильтра с катализатором селективного каталитического восстановления (SCRFTM)). Среднее количество NO2, образуемое катализатором окисления при заданной температуре выхлопных газов, может изменяться значительным образом на протяжении его срока службы. Это может приводить к затруднениям в калибровке дозирования азотсодержащего восстановителя для выполнения активного селективного каталитического восстановления (SCR).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы данного изобретения нашли, что превосходная активность в отношении окисления оксида азота (NO) в выхлопных газах от дизельного двигателя может быть получена, когда в катализаторе окисления марганец (Mn) объединен с платиной (Pt). Платина является дорогостоящей, и ее часто включают в катализаторы окисления в сравнительно больших количествах для, помимо прочего, их активности окисления в отношении NO. Включение марганца (Mn) в комбинации с платиной (Pt) может приводить к улучшению активности окисления NO или предоставлению возможности применения уменьшенного количества Pt для достижения заданного уровня окисления NO. Катализатор окисления также обладает высокой активностью в отношении окисления монооксида углерода (CO) и углеводородов (HC) в выхлопных газах, образованных дизельным двигателем.
Данное изобретение предлагает катализатор окисления для обработки выхлопных газов от дизельного двигателя, данный катализатор окисления содержит: область первого покрытия из пористого оксида «washcoat» для окисления монооксида углерода (CO) и углеводородов (HC), при этом область первого покрытия из пористого оксида «washcoat» содержит первый металл платиновой группы (МПГ) и первый материал-носитель; область второго покрытия из пористого оксида «washcoat» для окисления оксида азота (NO), при этом область второго покрытия из пористого оксида «washcoat» содержит платину (Pt), марганец (Mn) и второй материал-носитель; и основу (носитель), имеющую впускной конец и выпускной конец.
Для того, чтобы обеспечить высокую активность в отношении окисления монооксида углерода (CO) и углеводородов (HC), катализатор окисления по данному изобретению имеет две области покрытия из пористого оксида «washcoat»: (i) область первого покрытия из пористого оксида «washcoat» для окисления CO и HC, и (ii) область второго покрытия из пористого оксида «washcoat» для окисления NO, которая включает Pt и Mn.
Окислительная активность комбинации Pt и Mn в отношении оксида азота (NO) может быть особенно выгодной, когда определенные материалы на основе оксида алюминия используют в качестве материала-носителя.
Как правило, область второго покрытия из пористого оксида «washcoat» расположена таким образом, чтобы контактировать с выхлопными газами на выпускном конце основы и после контактирования выхлопных газов с областью первого покрытия из пористого оксида «washcoat».
В качестве дополнения или альтернативы, катализатор может проявлять выгодную окислительную активность (например, в отношении CO, HC и NO), и особенно в отношении NO, когда он имеет расположение, которое способствует контактированию выхлопных газов с областью покрытия из пористого оксида «washcoat», содержащей Pt и Mn, незадолго перед тем, как выхлопные газы выводятся из катализатора, и после того, как они находились в контакте с областью покрытия из пористого оксида «washcoat» для окисления CO и HC. При таком расположении катализатора окисления, когда выхлопные газы, поступают в катализатор, они первоначально приводятся в контакт с областью первого покрытия из пористого оксида «washcoat» для окисления CO и HC. После того, как выхлопные газы прошли через область первого покрытия из пористого оксида «washcoat» или поверх нее, они приводятся в контакт с областью второго покрытия из пористого оксида «washcoat» для окисления NO перед тем, как они в заключение проходят через выпускное отверстие катализатора окисления. Дополнительно было найдено, что, когда катализатор окисления расположен таким образом, Mn-содержащая область покрытия из пористого оксида «washcoat» является удивительно устойчивой к отравлению серой от дизельного топлива и может сохранять свою активность окисления NO.
Данное изобретение дополнительно предлагает систему выпуска выхлопных газов для дизельного двигателя. Система выпуска выхлопных газов содержит катализатор окисления по данному изобретению и устройство для регулирования выбросов.
Количество NO2 в выхлопных газах может влиять на эксплуатационные параметры устройства для регулирования выбросов ниже по течению потока для селективного каталитического восстановления оксидов азота (NOx). Катализаторы селективного каталитического восстановления ((SCR) и катализаторы фильтра селективного каталитического восстановления (SCRFTM) для обработки NOx (например, NO2+NO) часто требуют, чтобы отношение NO2 к NO во входном газе находилось в пределах определенного интервала для оптимальных каталитических эксплуатационных параметров. Оптимальная доля NO2 в NOx обычно зависит от вида композиции, используемой в катализаторе селективного каталитического восстановления (SCR) или катализаторе фильтра селективного каталитического восстановления (SCRFTM), однако отношение NO2 к NO в выхлопных газах непосредственно от дизельного двигателя обычно слишком низкое для оптимальных эксплуатационных параметров катализатора.
Катализатор окисления по данному изобретению может быть применен, чтобы преобразовывать NO в NO2 и тем самым увеличивать количество NO2 в выхлопных газах, которые выпускаются из катализатора, по сравнению с количеством NO2 в выхлопных газах на входе катализатора (т.е. отношения NO2:NOx и NO2:NO в выхлопных газах, которые выпускаются из катализатора окисления, являются более высокими, чем соответствующие отношения выхлопных газов на входе катализатора окисления). Катализатор окисления может изменять состав NOx в выхлопных газах для оптимальных рабочих характеристик селективного каталитического восстановления (SCR).
Проблема с применением катализатор окисления для увеличения содержания NO2 в выхлопных газах заключается в том, что активность окисления NO катализатора окисления обычно изменяется на протяжении его срока службы. Как правило, когда катализатор ʺстареетʺ (т.е. катализатор был использован в течение продолжительного периода времени), активность катализатора в отношении окисления NO уменьшается. Несмотря на то, что количество NO2 в выхлопных газах, которые выпускаются из «состаренного» катализатора окисления, может быть достаточным для оптимальных эксплуатационных параметров устройства для регулирования выбросов (например, катализатора селективного каталитического восстановления (SCR)) ниже по течению потока, это изменение в количестве образуемого NO2, является проблематичным для калибровки дозирования азотсодержащего восстановителя для выполнения активного селективного каталитического восстановления (SCR).
Было найдено, что катализатор окисления по данному изобретению может проявлять сравнительно стабильную активность окисления NO на протяжении своего срока службы. Соответственно, разница в активности окисления NO катализатора окисления в свежем состоянии (т.е. когда он является «новым» и не подвергался повторяющемуся неоднократному, длительному применению) и в состаренном состоянии является обычно небольшой.
Другой аспект данного изобретения относится к транспортному средству или устройству (например, стационарному или подвижному устройству). Транспортное средство или устройство содержит дизельный двигатель и катализатор окисления или систему выпуска выхлопных газов по данному изобретению.
Данное изобретение также относится к нескольким видам применения и способам.
Первый аспект данного изобретения в отношении способов предоставляет способ обработки выхлопных газов от дизельного двигателя. Данный способ включает приведение выхлопных газов в контактирование с катализатором окисления по данному изобретению или пропускание выхлопных газов через систему выпуска выхлопных газов по данному изобретению. Выражение «обработка выхлопных газов» в этом контексте относится к окислению монооксида углерода (CO), углеводородов (HC) и оксида азота (NO) в выхлопных газах от дизельного двигателя.
Второй аспект данного изобретения в отношении способов предоставляет способ регулирования содержания NOx в выхлопных газах от дизельного двигателя для устройства для регулирования выбросов. Данный способ включает: (a) регулирование содержания NOx в выхлопных газах посредством контактирования выхлопных газов с катализатором окисления по данному изобретению, чтобы образовывать обработанные выхлопные газы; и (b) пропускание обработанных выхлопных газов в устройство для регулирования выбросов.
Первый аспект данного изобретения в отношении видов применения относится к применению катализатора окисления для обработки выхлопных газов от дизельного двигателя, необязательно в комбинации с устройством для регулирования выбросов. Как правило, катализатор окисления применяют, чтобы обработать (например, окислить) монооксид углерода (CO) и углеводороды (HC) в выхлопных газах от дизельного двигателя.
Второй аспект данного изобретения в отношении видов применения относится к применению катализатора окисления для регулирования NOx в выхлопных газах от дизельного двигателя для устройства для регулирования выбросов (например, устройства для регулирования выбросов ниже по течению потока).
Третий аспект в отношении видов применения относится к применению катализатора окисления при регенерации устройства для регулирования выбросов, имеющего фильтрующую основу (например, устройства для регулирования выбросов ниже по течению потока, имеющего фильтрующую основу).
Четвертый аспект в отношении видов применения относится к применению марганца (Mn), предпочтительно в комбинации с платиной (Pt), в катализаторе окисления для дизельного двигателя, чтобы увеличить окисление оксида азота (NO) в выхлопных газах от дизельного двигателя.
Пятый аспект в отношении видов применения относится к применению марганца (Mn), предпочтительно в комбинации с платиной (Pt), в катализаторе окисления для дизельного двигателя, чтобы стабилизировать активность окисления NO катализатора окисления на протяжении его срока службы.
В аспектах с первого по пятый в отношении видов применения катализатор окисления является катализатором окисления в соответствии с данным изобретением.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1-10 являются схематическими представлениями катализаторов окисления по данному изобретению. На данных Фигурах, левая сторона катализатора окисления является впускным концом, и правая сторона является выпускным концом.
Фиг. 1 показывает катализатор окисления, содержащий область первого покрытия из пористого оксида «washcoat» (1) и область/зону второго покрытия из пористого оксида «washcoat» (2), расположенные на основе (3).
Фиг. 2 показывает катализатор окисления, содержащий область первого покрытия из пористого оксида «washcoat» (1) и область/зону второго покрытия из пористого оксида «washcoat» (2). Область первого покрытия из пористого оксида «washcoat» (1) расположена непосредственно на основе (3). Область/зона второго покрытия из пористого оксида «washcoat» (2) расположена на области первого покрытия из пористого оксида «washcoat» (1).
Фиг. 3 показывает катализатор окисления, содержащий область первого покрытия из пористого оксида «washcoat» (1) и область/зону второго покрытия из пористого оксида «washcoat» (2). Имеет место перекрывание между областью первого покрытия из пористого оксида «washcoat» (1) и областью/зоной второго покрытия из пористого оксида «washcoat» (2). Часть области первого покрытия из пористого оксида «washcoat» (1) расположена на области/зоне второго покрытия из пористого оксида «washcoat» (2). Как область первого покрытия из пористого оксида «washcoat» (1), так и область/зона второго покрытия из пористого оксида «washcoat» (2) расположены на основе (3).
Фиг. 4 показывает катализатор окисления, содержащий область первого покрытия из пористого оксида «washcoat» (1) и область/зону второго покрытия из пористого оксида «washcoat» (2). Имеет место перекрывание между областью первого покрытия из пористого оксида «washcoat» (1) и областью/зоной второго покрытия из пористого оксида «washcoat» (2). Часть области/зоны второго покрытия из пористого оксида «washcoat» (2) расположена на области первого покрытия из пористого оксида «washcoat» (1). Как область первого покрытия из пористого оксида «washcoat» (1), так и область/зона второго покрытия из пористого оксида «washcoat» (2) расположены на основе (3).
Фиг. 5 показывает катализатор окисления, содержащий слой первого покрытия из пористого оксида «washcoat» (1) и слой второго покрытия из пористого оксида «washcoat» (2), расположенные на основе (3). Слой второго покрытия из пористого оксида «washcoat» (2) расположен на слое первого покрытия из пористого оксида «washcoat» (1).
Фиг. 6 показывает катализатор окисления, содержащий зону первого покрытия из пористого оксида «washcoat» (1) и область/зону второго покрытия из пористого оксида «washcoat» (2). Как область/зона первого покрытия из пористого оксида «washcoat» (1), так и область/зона второго покрытия из пористого оксида «washcoat» (2) расположены на области/слое третьего покрытия из пористого оксида «washcoat» (4). Область/слой третьего покрытия из пористого оксида «washcoat» (4) находится на основе (3).
Фиг. 7 показывает катализатор окисления, содержащий зону первого покрытия из пористого оксида «washcoat» (1), зону второго покрытия из пористого оксида «washcoat» (2) и зону третьего покрытия из пористого оксида «washcoat» (4). Как зона первого покрытия из пористого оксида «washcoat» (1), так и зона второго покрытия из пористого оксида «washcoat» (2) расположены на основе (3). Зона третьего покрытия из пористого оксида «washcoat» (4) расположена на зоне второго покрытия из пористого оксида «washcoat» (2).
Фиг. 8 показывает катализатор окисления, содержащий зону первого покрытия из пористого оксида «washcoat» (1), зону второго покрытия из пористого оксида «washcoat» (2) и зону третьего покрытия из пористого оксида «washcoat» (4). Как зона первого покрытия из пористого оксида «washcoat» (1), так и зона третьего покрытия из пористого оксида «washcoat» (4) расположены на основе (3). Зона второго покрытия из пористого оксида «washcoat» (2) расположена на зоне третьего покрытия из пористого оксида «washcoat» (4).
Фиг. 9 показывает катализатор окисления, содержащий зону первого покрытия из пористого оксида «washcoat» (1), зону второго покрытия из пористого оксида «washcoat» (2) и зону третьего покрытия из пористого оксида «washcoat» (4). Как зона первого покрытия из пористого оксида «washcoat» (1), так и зона второго покрытия из пористого оксида «washcoat» (2) расположены на основе (3). Зона третьего покрытия из пористого оксида «washcoat» (4) расположена на зоне первого покрытия из пористого оксида «washcoat» (1).
Фиг. 10 показывает катализатор окисления, содержащий зону первого покрытия из пористого оксида «washcoat» (1), зону второго покрытия из пористого оксида «washcoat» (2), зону третьего покрытия из пористого оксида «washcoat» (4) и зону четвертого покрытия из пористого оксида «washcoat» (5). Как зона первого покрытия из пористого оксида «washcoat» (1), так и зона третьего покрытия из пористого оксида «washcoat» (4) расположены на основе (3). Зона второго покрытия из пористого оксида «washcoat» (2) расположена на зоне третьего покрытия из пористого оксида «washcoat» (4). Зона четвертого покрытия из пористого оксида «washcoat» (5) расположена на зоне первого покрытия из пористого оксида «washcoat» (1).
ПОДРОБНОЕ ОПИСАНИЕ ДАННОГО ИЗОБРЕТЕНИЯ
Катализатор окисления по данному изобретению содержит, или может состоять из них, область первого покрытия из пористого оксида «washcoat» для окисления монооксида углерода (CO) и углеводородов (HC), область второго покрытия из пористого оксида «washcoat» для окисления оксида азота (NO) и основу.
Обычно, область второго покрытия из пористого оксида «washcoat» расположена или ориентирована таким образом, чтобы контактировать с выхлопными газами после то, как они имели контакт с областью первого покрытия из пористого оксида «washcoat». Как правило, область первого покрытия из пористого оксида «washcoat» расположена или ориентирована, чтобы контактировать с выхлопными газами перед областью второго покрытия из пористого оксида «washcoat». Соответственно, область первого покрытия из пористого оксида «washcoat» может быть расположена таким образом, чтобы контактировать с выхлопными газами, когда они поступают в катализатор окисления, и область второго покрытия из пористого оксида «washcoat» может быть расположена таким образом, чтобы контактировать с выхлопными газами, когда они оставляют катализатор окисления. Примеры таких расположений описаны в данном документе.
Область второго покрытия из пористого оксида «washcoat», как правило, расположена таким образом, чтобы контактировать с выхлопными газами на выпускном конце основы и после контактирования выхлопных газов с областью первого покрытия из пористого оксида «washcoat».
Область первого покрытия из пористого оксида «washcoat» содержит первый металл платиновой группы (МПГ) и первый материал-носитель или может состоять по существу из них.
Как правило, первый МПГ выбирают из группы, состоящей из платины, палладия и комбинации платины и палладия. Первый МПГ может быть платиной. Первый МПГ может состоять по существу из платины (например, первый МПГ является лишь платиной). Первый МПГ может быть палладием. Первый МПГ может состоять по существу из палладия (например, первый МПГ является лишь палладием). Первый МПГ может быть комбинацией платины и палладия. Первый МПГ может состоять по существу из платины и палладия (например, первый МПГ является лишь платиной и палладием). Предпочтительно, первый МПГ выбирают из группы, состоящей из платины и комбинации платины и палладия.
Область первого покрытия из пористого оксида «washcoat» может содержать первый МПГ в виде лишь металла(ов) платиновой группы. Соответственно, только один или несколько МПГ, присутствующих в области первого покрытия из пористого оксида «washcoat», определяют как первый МПГ.
Когда первый МПГ является комбинацией платины и палладия, то первый МПГ может находиться в форме сплава, предпочтительно биметаллического сплава. Соответственно, первый МПГ может содержать сплав платины и палладия или состоять по существу из него.
Когда первый МПГ является палладием или комбинацией платины и палладия, то область первого покрытия из пористого оксида «washcoat» может дополнительно содержать золото. Область первого покрытия из пористого оксида «washcoat» может содержать сплав палладия и золота (например, палладий первого металла платиновой группы может присутствовать в виде сплава с золотом). Катализаторы, содержащие золото (Au), могут быть приготовлены при применении способа, описанного в заявке WO 2012/120292.
Когда область первого покрытия из пористого оксида «washcoat» содержит золото, например, сплав палладия и золота, то обычно область первого покрытия из пористого оксида «washcoat» имеет отношение общей массы палладия (Pd) к общей массе золота (Au) от 9:1 до 1:9, предпочтительно от 5:1 до 1:5 и более предпочтительно от 2:1 до 1:2.
Область первого покрытия из пористого оксида «washcoat» обычно имеет общую загрузку первого МПГ от 5 до 300 г/фут3 (0,177-10,594 кг/м3). Предпочтительно, чтобы область первого покрытия из пористого оксида «washcoat» имела общую загрузку первого МПГ от 10 до 250 г/фут3 (0,353-8,829 кг/м3) (например, от 75 до 175 г/фут3 (2,649-6,180 кг/м3)), более предпочтительно от 15 до 200 г/фут3 (0,530-7,063 кг/м3) (например, от 50 до 150 г/фут3 (1,766-5,297 кг/м3)), еще более предпочтительно от 20 до 150 г/фут3 (0,706-5,297 кг/м3).
Когда первый МПГ является комбинацией платины и палладия, то обычно область первого покрытия из пористого оксида «washcoat» имеет отношение по массе платины к палладию от 20:1 до 1:20 (например, от 15:1 до 1:15), предпочтительно от 10:1 до 1:10 (например, от 7,5:1 до 1:7,5), более предпочтительно от 5:1 до 1:5 (например, от 3:1 до 1:3) и еще более предпочтительно от 2,5:1 до 1:1.
Предпочтительно, чтобы, когда первый МПГ является комбинацией платины и палладия, то область первого покрытия из пористого оксида «washcoat» содержала общую массу платины, которая больше общей массы палладия или равна ей (например, отношение Pt:Pd по массе составляет≥1:1). Более предпочтительно, область первого покрытия из пористого оксида «washcoat» содержит общую массу платины, которая больше общей массы палладия (например, отношение Pt:Pd по массе составляет > 1:1). Выгодная активность «light-off» может быть получена, когда общая масса платины больше или равна общей массе палладия в области первого покрытия из пористого оксида «washcoat».
Обычно предпочтительно, чтобы область первого покрытия из пористого оксида «washcoat» имела отношение по массе платины к палладию от 20:1 до 1:1 (например, от 15,1:1 до 1,1:1), более предпочтительно от 10:1 до 1,25:1 (например, от 7,5:1 до 1,5:1), и еще более предпочтительно от 5:1 до 2:1.
Предполагается, что основной функцией области первого покрытия из пористого оксида «washcoat» является окисление монооксида углерода (CO) и углеводородов (HC). Тем не менее, следует принимать во внимание, что в некоторых вариантах осуществления катализатора окисления область первого покрытия из пористого оксида «washcoat» может также окислять некоторое количество NO до NO2, особенно когда значительная доля металла платиновой группы (МПГ) является платиной.
Как правило, первый МПГ размещают или поддерживают на первом материале-носителе. Первый МПГ может быть размещен непосредственно на первом материале-носителе или непосредственно поддерживаться первым материалом-носителем (например, без размещения промежуточного материала-носителя между первым МПГ и первым материалом-носителем). Например, платина и/или палладий могут быть диспергированы на первом материале-носителе.
Когда первый МПГ является комбинацией платины и палладия, то платина может быть размещена или поддерживаться на первом материале-носителе, и/или палладий может быть размещен или поддерживаться на первом материале-носителе. Предпочтительно, как платину, так и палладий размещают или поддерживают на первом материале-носителе (т.е. тот же самый материал-носитель используют как для платины, так и для палладия).
Когда первый МПГ является комбинацией платины и палладия, то область первого покрытия из пористого оксида «washcoat» может дополнительно содержать материал-носитель для палладия. Область первого покрытия из пористого оксида «washcoat» может поэтому содержать, или состоять по существу из них, первый МПГ, первый материал-носитель и материал-носитель для палладия. Платина может быть размещена или поддерживаться на первом материале-носителе, и палладий может быть размещен или поддерживаться на материале-носителе для палладия. Первый материал-носитель и материал-носитель для палладия являются предпочтительно разными (например, имеют разный состав).
Когда первый МПГ является комбинацией платины и палладия, то область первого покрытия из пористого оксида «washcoat» может дополнительно содержать материал-носитель для платины. Область первого покрытия из пористого оксида «washcoat» может поэтому содержать, или состоять по существу из них, первый МПГ, первый материал-носитель и материал-носитель для платины. Платина может быть размещена или поддерживаться на материале-носителе для платины, и палладий может быть размещен или поддерживаться на первом материале-носителе. Первый материал-носитель и материал-носитель для платины являются предпочтительно разными (например, имеют разный состав).
Как правило, первый материал-носитель содержит огнеупорный оксид металла или состоит по существу из него. Огнеупорные оксиды металлов, подходящие для применения в качестве каталитического компонента катализатора окисления для дизельного двигателя, хорошо известны в данной области техники.
Огнеупорный оксид металла, как правило, выбирают из группы, состоящей из оксида алюминия, кремнезема, диоксида титана, диоксида циркония, оксида церия и их смешанных или сложных оксидов, таких как смешанные или сложные оксиды двух или более этих оксидов. Например, огнеупорный оксид металла может быть выбран из группы, состоящей из оксида алюминия, кремнезема, диоксида титана, диоксида циркония, оксида церия, кремнезема-оксида алюминия, диоксида титана-оксида алюминия, диоксида циркония-оксида алюминия, оксида церия-оксида алюминия, диоксида титана-кремнезема, диоксида циркония-кремнезема, диоксида циркония-диоксида титана, оксида церия-диоксида циркония и оксида алюминия-оксида магния.
Первый материал-носитель или его огнеупорный оксид металла необязательно могут быть легированы (например, легирующей примесью). Легирующая примесь может быть выбрана из группы, состоящей из циркония (Zr), титана (Ti), кремния (Si), иттрия (Y), лантана (La), празеодима (Pr), самария (Sm), неодима (Nd) и их оксидов.
Включение легирующей примеси может термически стабилизировать огнеупорный оксид металла или материал-носитель. Следует иметь в виду, что любая ссылка на «легированный» в этом контексте относится к материалу, в котором объемная или основная кристаллическая решетка огнеупорного оксида металла легирована посредством замещения или внедрения легирующей примесью. В ряде случаев, небольшие количества легирующей примеси могут присутствовать на поверхности огнеупорного оксида металла. Однако большинство легирующей примеси будет обычно присутствовать в объеме огнеупорного оксида металла. Присутствие легирующей примеси часто влияет на химические и/или физические свойства огнеупорного оксида металла.
Когда первый материал-носитель или его огнеупорный оксид металла легирован, общее количество легирующей примеси составляет от 0,25 до 5% по массе, предпочтительно от 0,5 до 3% по массе (например, примерно 1% по массе).
Первый материал-носитель или его огнеупорный оксид металла могут содержать оксид алюминия, легированный легирующей примесью, или состоять по существу из него. Особенно предпочтительным является то, что первый материал-носитель или его огнеупорный оксид металла содержит, или состоит по существу из него, оксид алюминия, легированный легирующей примесью, когда область первого покрытия из пористого оксида «washcoat» содержит щелочноземельный металл.
Оксид алюминия может быть легирован легирующей примесью, содержащей кремний (Si), магний (Mg), барий (Ba), лантан (La), церий (Ce), титан (Ti) или цирконий (Zr) или комбинацию двух этих элементов или более. Легирующая примесь может содержать, или состоять по существу из него, оксид кремния, оксид магния, оксид бария, оксид лантана, оксид церия, оксид титана или оксид циркония. Предпочтительно, легирующая примесь содержит, или состоит по существу из него, кремний, магний, барий, церий или их оксид, особенно кремний или церий или их оксид. Более предпочтительно, легирующая примесь содержит, или состоит по существу из него, кремний, магний, барий или их оксид, особенно кремний, магний или их оксид, главным образом кремний или его оксид.
Примеры оксида алюминия, легированного легирующей примесью, включают оксид алюминия, легированный кремнеземом, оксид алюминия, легированный оксидом магния, оксид алюминия, легированный барием или оксидом бария, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированный оксидом церия, особенно оксид алюминия, легированный кремнеземом, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированный оксидом церия. Предпочтительно, чтобы оксидом алюминия, легированным легирующей примесью, являлся оксид алюминия, легированный кремнеземом, оксид алюминия, легированный барием или оксидом бария, или оксид алюминия, легированный оксидом магния. Более предпочтительно, чтобы оксидом алюминия, легированным легирующей примесью, являлся оксид алюминия, легированный кремнеземом, или оксид алюминия, легированный оксидом магния. Еще более предпочтительно, чтобы оксидом алюминия, легированным легирующей примесью, являлся оксид алюминия, легированный кремнеземом. Оксид алюминия, легированный легирующей примесью, может быть получен при применении способов, известных в данной области техники, или, например, способом, описанным в US 5045519.
Когда оксид алюминия является оксидом алюминия, легированным кремнеземом, то оксид алюминия легирован кремнеземом в общем количестве от 0,5 до 45% по массе (т.е. % по массе в расчете на оксид алюминия), предпочтительно от 1 до 40% по массе, более предпочтительно от 1,5 до 30% по массе (например, от 1,5 до 10% по массе), особенно предпочтительно от 2,5 до 25% по массе, еще более предпочтительно от 3,5 до 20% по массе (например, от 5 до 20% по массе), даже еще более предпочтительно от 4,5 до 15% по массе.
Когда оксид алюминия является оксидом алюминия, легированным оксидом магния, то оксид алюминия легирован магнием в количестве, как определено выше, или количестве от 1 до 30% по массе (т.е. % по массе в расчете на оксид алюминия), предпочтительно 5 до 25% по массе.
Предпочтительно, чтобы первый материал-носитель или его огнеупорный оксид металла не был легирован легирующей примесью, содержащей марганец или состоящей по существу из марганца. Соответственно, первый материал-носитель или его огнеупорный оксид металла не активирован активатором, таким как активатор, выбранный из группы, состоящей из олова, марганца, индия, металла группы VIII (например, Fe, Co, Ni, Ru, Rh, Pd, Os, Ir и Pt, особенно Ir) и их комбинаций.
В качестве альтернативы или дополнения, первый материал-носитель или его огнеупорный оксид металла может содержать алюминат щелочноземельного металла или состоять по существу из него. Термин «алюминат щелочноземельного металла» обычно относится к соединению формулы MAl2O4, где «M» представляет щелочноземельный металл, такой как Mg, Ca, Sr или Ba. Такие соединения обычно образуют структуру шпинели. Эти соединения могут быть получены при применении обычных способов, известных в данной области техники, или посредством способа, описанного в EP 0945165, US 6217837 или US 6517795.
Как правило, алюминат щелочноземельного металла является алюминатом магния (MgAl2O4), алюминатом кальция (CaAl2O4), алюминатом стронция (SrAl2O4), алюминатом бария (BaAl2O4) или смесью двух из них или более. Предпочтительно, алюминат щелочноземельного металла является алюминатом магния (MgAl2O4).
Обычно, когда первый материал-носитель или его огнеупорный оксид металла содержит, или состоит по существу из него, смешанный или сложный оксид с оксидом алюминия (например, кремнезем-оксид алюминия, оксид алюминия-оксид магния или смесь оксида алюминия и оксида церия), то предпочтительно смешанный или сложный оксид с оксидом алюминия содержит от по меньшей мере 50 до 99% по массе оксида алюминия, более предпочтительно от 70 до 95% по массе оксида алюминия, еще более предпочтительно от 75 до 90% по массе оксида алюминия.
Когда первый материал-носитель, или его огнеупорный оксид металла, содержит, или состоит по существу из него, оксид церия-диоксид циркония, то оксид церия-диоксида циркония может состоять по существу из 20-95% по массе оксида церия и 5-80% по массе диоксида циркония (например, 50-95% по массе оксида церия и 5-50% по массе диоксида циркония), предпочтительно 35-80% по массе оксида церия и 20-65% по массе диоксида циркония (например, 55-80% по массе оксида церия и 20-45% по массе диоксида циркония), даже еще более предпочтительно 45-75% по массе оксида церия и 25-55% по массе диоксида циркония.
Обычно, материал-носитель для палладия содержит огнеупорный оксид металла или состоит по существу из него. Материал-носитель для палладия, или его огнеупорный оксид металла, может быть таким материалом-носителем, который определен в данном документе выше в отношении первого материала-носителя. Когда область первого покрытия из пористого оксида «washcoat» содержит материал-носитель для палладия, предпочтительно, чтобы материал-носитель для палладия, или его огнеупорный оксид металла, содержал, или состоял по существу из него, оксид церия и/или оксид церия-диоксид циркония.
Как правило, материал-носитель для платины содержит огнеупорный оксид металла или состоит по существу из него. Материал-носитель для платины, или его огнеупорный оксид металла, может быть таким материалом-носителем, который определен в данном документе выше в отношении первого материала-носителя. Когда область первого покрытия из пористого оксида «washcoat» содержит материал-носитель для платины, предпочтительно, чтобы материал-носитель для платины или его огнеупорный оксид металла, содержал, или состоял по существу из него, оксид алюминия, при этом оксид алюминия необязательно легирован легирующей примесью, такой как описано выше. Когда материал-носитель для платины содержит оксид алюминия, легированный легирующей примесью, предпочтительно, чтобы легирующая примесь содержала, или состояла по существу из него, кремний, магний, церий, лантан или их оксид, более предпочтительно кремний или его оксид.
Материал-носитель для платины (или его огнеупорный оксид металла) и/или материал-носитель для палладия (или его огнеупорный оксид металла) не легирован легирующей примесью, содержащей марганец или состоящей по существу из марганца. Соответственно, материал-носитель для платины (или его огнеупорный оксид металла) и/или материал-носитель для палладия (или его огнеупорный оксид металла) не активирован активатором, таким как активатор, выбранный из группы, состоящей из олова, марганца, индия, металла группы VIII (например, Fe, Co, Ni, Ru, Rh, Pd, Os, Ir и Pt, особенно Ir) и их комбинаций.
Область первого покрытия из пористого оксида «washcoat» может содержать материал-носитель в количестве (например, общем количестве первого материала-носителя и, в случае присутствия, материала-носителя для платины и материала-носителя для палладия) от 0,1 до 4,5 г/дюйм3 (6-275 мг/см3) (например, от 0,25 до 4,2 г/дюйм3 (15-256 мг/см3)), предпочтительно от 0,3 до 3,8 г/дюйм3 (18-232 мг/см3), еще более предпочтительно от 0,5 до 3,0 г/дюйм3 (31-183 мг/см3) (от 1 до 2,75 г/дюйм3 (61-168 мг/см3) или от 0,75 до 1,5 г/дюйм3 (46-92 мг/см3)) и даже еще более предпочтительно от 0,6 до 2,5 г/дюйм3 (37-153 мг/см3) (например, от 0,75 до 2,3 г/дюйм3 (46-140 мг/см3)).
Область первого покрытия из пористого оксида «washcoat» может дополнительно содержать материал, адсорбирующий углеводороды. Материалом, адсорбирующим углеводороды, может быть цеолит.
Предпочтительно, чтобы цеолит являлся цеолитом со средним размером пор (например, цеолитом, имеющим максимальный размер кольца десяти тетраэдрически координированных атомов) или цеолитом с большим размером пор (например, цеолитом, имеющим максимальный размер кольца двенадцати тетраэдрически координированных атомов). Может являться предпочтительным, чтобы цеолит не являлся цеолитом с малым размером пор (например, цеолитом, имеющим максимальный размер кольца восьми тетраэдрически координированных атомов).
Примеры подходящих цеолитов или видов цеолита включают фожазит, клиноптилолит, морденит, силикалит, ферриерит, цеолит X, цеолит Y, ультрастабильный цеолит Y, цеолит AEI, цеолит ZSM-5, цеолит ZSM-12, цеолит ZSM-20, цеолит ZSM-34, цеолит CHA, цеолит SSZ-3, цеолит SAPO-5, оффретит, бета-цеолит или медьсодержащий цеолит CHA. Цеолит является предпочтительно цеолитом ZSM-5, бета-цеолитом или цеолитом Y.
Когда область первого покрытия из пористого оксида «washcoat» содержит адсорбент углеводородов, то общее количество адсорбента углеводородов составляет от 0,05 до 3,00 г/дюйм3 (3-183 мг/см3), предпочтительно от 0,10 до 2,00 г/дюйм3 (6-122 мг/см3), более предпочтительно от 0,2 до 1,0 г/дюйм3 (12-61 мг/см3). Например, общее количество адсорбента углеводородов может составлять от 0,8 до 1,75 г/дюйм3 (49-107 мг/см3), например, от 1,0 до 1,5 г/дюйм3 (61-92 мг/см3).
Однако может являться предпочтительным, чтобы область первого покрытия из пористого оксида «washcoat» не содержала материал, адсорбирующий углеводороды.
Область первого покрытия из пористого оксида «washcoat» может дополнительно содержать щелочноземельный металл. Область первого покрытия из пористого оксида «washcoat» обычно содержит количество щелочноземельного металла, эффективное для промотирования окисления монооксида углерода (CO) и/или углеводородов (HC). Щелочноземельный металл может промотировать окисление CO и/или HC (например, низкотемпературная активность окисления CO и/или HC может быть улучшена), особенно когда щелочноземельный металл объединен с определенными материалами-носителями, такими как оксид алюминия, легированный кремния.
Щелочноземельный металл может быть выбран из магния (Mg), кальция (Ca), стронция (Sr), бария (Ba) и комбинации двух их или более. Щелочноземельный металл является предпочтительно кальцием (Ca), стронцием (Sr) или барием (Ba), более предпочтительно стронцием (Sr) или барием (Ba), и наиболее предпочтительно щелочноземельный металл является барием (Ba).
Обычно предпочтительно, чтобы щелочноземельный металл был расположен или поддерживался на материале-носителе, таком как первый материал-носитель. Когда область первого покрытия из пористого оксида «washcoat» содержит материал-носитель для платины, щелочноземельный металл может быть размещен или поддерживаться на материале-носителе для платины. Когда область первого покрытия из пористого оксида «washcoat» содержит материал-носитель для палладия, щелочноземельный металл может быть размещен или поддерживаться на материале-носителе для палладия.
Как правило, катализатор окисления или область первого покрытия из пористого оксида «washcoat» содержит щелочноземельный металл в общем количестве от 0,07 до 3,75 моль/фут3 (2,47-132 моль/м3), особенно от 0,1 до 3,0 моль/фут3 (3,53-106 моль/м3), более часто от 0,2 до 2,5 моль/фут3 (7,06-88,3 моль/м3) (например, от 0,25 до 1,0 моль/фут3 (8,83-35,3 моль/м3)), таком как от 0,3 до 2,25 моль/фут3 (10,6-79,5 моль/м3), особенно от 0,35 до 1,85 моль/фут3 (12,4-65,3 моль/м3), предпочтительно от 0,4 до 1,5 моль/фут3 (14,1-53,0 моль/м3), даже еще более предпочтительно от 0,5 до 1,25 моль/фут3 (17,7-44,1 моль/м3).
Обычно, катализатор окисления или область первого покрытия из пористого оксида «washcoat» содержит щелочноземельный металл в общем количестве от 10 до 500 г/фут3 (0,353-17,7 кг/м3) (например, от 60 до 400 г/фут3 (2,12-14,1 кг/м3) или от 10 до 450 г/фут3 (0,353-15,9 кг/м3)), особенно от 20 до 400 г/фут3 (0,706-14,1 кг/м3), более часто от 35 до 350 г/фут3 (1,24-12,4 кг/м3), таком как от 50 до 300 г/фут3 (1,77-10,6 кг/м3), главным образом от 75 до 250 г/фут3 (2,65-8,83 кг/м3).
Катализатор окисления обычно содержит щелочноземельный металл в количестве от 0,1 до 20% по массе, предпочтительно от 0,5 до 17,5% по массе, более предпочтительно от 1 до 15% по массе и даже еще более предпочтительно от 1,5 до 12,5% по массе. Количество щелочноземельного металла может составлять от 1,0 до 8,0% по массе, например, от 1,5 до 7,5% по массе, особенно от 2,0 до 7,0% по массе (например, от 2,5 до 6,5% по массе или от 2,0 до 5,0% по массе). Количество щелочноземельного металла может составлять от 5,0 до 17,5% по массе, например, от 7,5 до 15% по массе, особенно от 8,0 до 14% по массе (например, от 8,5 до 12,5% по массе или от 9,0 до 13,5% по массе).
Как правило, отношение по массе щелочноземельного металла к первому металлу платиновой группы (МПГ) составляет от 0,25:1 до 20:1 (например, от 0,3:1 до 20:1). Предпочтительно, чтобы отношение общей массы щелочноземельного металла к общей массе металла платиновой группы (МПГ) составляло от 0,5:1 до 17:1, более предпочтительно от 1:1 до 15:1, особенно от 1,5:1 до 10:1, еще более предпочтительно от 2:1 до 7,5:1 и даже еще более предпочтительно от 2,5:1 до 5:1.
Когда первый МПГ содержит платину (Pt) или состоит по существу из нее, то предпочтительно область первого покрытия из пористого оксида «washcoat» содержит щелочноземельный металл в общей массе больше, чем общая масса платины (Pt).
Область первого покрытия из пористого оксида «washcoat» может дополнительно содержать цеолитовый катализатор, такой как цеолитовый катализатор, определенный в данном документе ниже. Предпочтительно, чтобы область первого покрытия из пористого оксида «washcoat» содержала цеолитовый катализатор, когда область второго покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор. Таким образом, область первого покрытия из пористого оксида «washcoat» и область второго покрытия из пористого оксида «washcoat» содержат каждая цеолитовый катализатор.
Обычно, область первого покрытия из пористого оксида «washcoat» содержит щелочноземельный металл в количестве, эффективном, чтобы промотировать окисление CO и/или HC, например, при применении в комбинации с Pt или Pt и Pd в отношении по массе≥1:1 (например, с Pt и Pd в отношении по массе от 1:0 до 1:1).
Может, однако, являться предпочтительным, чтобы область первого покрытия из пористого оксида «washcoat» не содержала родий, щелочной металл и/или щелочноземельный металл, особенно щелочной металл и/или щелочноземельный металл, размещенный или поддерживаемый на материале-носителе (например, первом материале-носителе, материале-носителе для платины и/или материале-носителе для палладия). Соответственно, область первого покрытия из пористого оксида «washcoat» может не содержать родий, щелочной металл и/или щелочноземельный металл, особенно щелочной металл и/или щелочноземельный металл, размещенный или поддерживаемый на материале-носителе.
Во избежание неопределенности, общей особенностью области первого покрытия из пористого оксида «washcoat» (т.е. в любом или всех вариантах осуществления, указанных выше) является то, что в области первого покрытия из пористого оксида «washcoat» по существу отсутствует марганец или его оксид. Область первого покрытия из пористого оксида «washcoat» предпочтительно не содержит марганец или его оксид.
Катализатор окисления по данному изобретению содержит область второго покрытия из пористого оксида «washcoat» для окисления оксида азота (NO) до диоксида азота (NO2). Область второго покрытия из пористого оксида «washcoat» содержит или может состоять по существу из платины (Pt), марганца или его оксида, а также второй материал-носитель.
Область второго покрытия из пористого оксида «washcoat» может дополнительно содержать палладий, например, палладий, размещенный или поддерживаемый на втором материале-носителе. Когда область второго покрытия из пористого оксида «washcoat» содержит палладий, то отношение платины к палладию по общей массе составляет обычно≥2:1 (например, Pt:Pd составляет от 1:0 до 2:1), более предпочтительно≥4:1 (например, Pt:Pd составляет от 1:0 до 4:1).
Когда область второго покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор, такой как цеолитовый катализатор, определенный в данном документе ниже, то область второго покрытия из пористого оксида «washcoat» может иметь отношение по общей массе платины к палладию, поддерживаемому на втором материале-носителе, ≥2:1 (например, Pt:Pd составляет от 1:0 до 2:1), более предпочтительно ≥4:1 (например, Pt:Pd составляет от 1:0 до 4:1).
Обычно предпочтительно, чтобы область второго покрытия из пористого оксида «washcoat» по существу не содержала палладия, особенно по существу не содержала палладия (Pd), расположенного или поддерживаемого на втором материале-носителе. Более предпочтительно, область второго покрытия из пористого оксида «washcoat» не содержит палладий, особенно палладий, размещенный или поддерживаемый на втором материале-носителе. Присутствие палладия, особенно в большом количестве, в области второго покрытия из пористого оксида «washcoat» может быть вредным для активности окисления NO. Активность окисления NO для палладия обычно низкая при типичных условиях применения для катализатора окисления дизельного топлива. Также, любой присутствующий палладий может реагировать с некоторой частью присутствующей платины с образованием сплава. Это также может быть вредным для активности окисления NO в области второго покрытия из пористого оксида «washcoat», поскольку сплавы платины и палладия не являются не являются такими активными в отношении окисления NO как платина сама по себе.
Как правило, область второго покрытия из пористого оксида «washcoat» содержит платину (Pt) в качестве единственного металла платиновой группы. Область второго покрытия из пористого оксида «washcoat» предпочтительно не содержит один или несколько других металлов платиновой группы, таких как рутений (Ru), родий (Rh), палладий (Pd), осмий (Os) и/или иридий (Ir). Более предпочтительно, область второго покрытия из пористого оксида «washcoat» не содержит один или несколько других металлов платиновой группы, таких как рутений (Ru), родий (Rh), палладий (Pd), осмий (Os) и/или иридий (Ir), поддерживаемых на втором материале-носителе.
Платину (Pd) обычно размещают или поддерживают на втором материале-носителе. Платина может быть размещена непосредственно на втором материале-носителе или непосредственно поддерживаться вторым материалом-носителем (например, без размещения промежуточного материала-носителя между платиной и вторым материалом-носителем). Например, платина может быть диспергирована на втором материале-носителе.
Область второго покрытия из пористого оксида «washcoat» обычно имеет общую загрузку платины от 5 до 300 г/фут3 (0,177-10,594 кг/м3). Предпочтительно, чтобы область второго покрытия из пористого оксида «washcoat» имела общую загрузку платины от 10 до 250 г/фут3 (0,353-8,829 кг/м3) (например, от 75 до 175 г/фут3 (2,649-6,180 кг/м3)), более предпочтительно от 15 до 200 г/фут3 (0,530-7,063 кг/м3) (например, от 50 до 150 г/фут3 (1,766-5,297 кг/м3)), еще более предпочтительно от 20 до 150 г/фут3 (0,706-5,297 кг/м3).
Предполагается, что основной функцией области второго покрытия из пористого оксида «washcoat» является окисление оксида азота (NO) до диоксида азота (NO2). Однако, следует принимать во внимание, что в некоторых вариантах осуществления катализатора окисления, область второго покрытия из пористого оксида «washcoat» может также окислять некоторое количество монооксида углерода (CO) и/или некоторое количество углеводородов (HC) во время применения.
Область второго покрытия из пористого оксида «washcoat» также содержит марганец (Mn). Марганец может присутствовать в элементарной форме или в виде оксида. Область второго покрытия из пористого оксида «washcoat» обычно содержит марганец или его оксид.
Марганец (Mn) обычно размещают или поддерживают на втором материале-носителе. Марганец (Mn) может быть размещен непосредственно на втором материале-носителе или непосредственно поддерживаться вторым материалом-носителем (например, без размещения промежуточного материала-носителя между Mn и вторым материалом-носителем).
Область второго покрытия из пористого оксида «washcoat» обычно имеет общую загрузку марганца от 5 до 500 г/фут3 (0,177-17,7 кг/м3). Предпочтительно, чтобы область второго покрытия из пористого оксида «washcoat» имела общую загрузку марганца (Mn) от 10 до 250 г/фут3 (0,353-8,829 кг/м3) (например, от 75 до 175 г/фут3 (2,649-6,180 кг/м3)), более предпочтительно от 15 до 200 г/фут3 (0,530-7,063 кг/м3) (например, от 50 до 150 г/фут3 (1,766-5,297 кг/м3)), еще более предпочтительно от 20 до 150 г/фут3 (0,706-5,297 кг/м3).
Как правило, область второго покрытия из пористого оксида «washcoat» имеет отношение Mn:Pt по массе≤5:1, более предпочтительно < 5:1.
Обычно, область второго покрытия из пористого оксида «washcoat» имеет отношение Mn:Pt по массе ≥0,2:1 (например, ≥0,5:1), более предпочтительно > 0,2:1 (например, > 0,5:1).
Область второго покрытия из пористого оксида «washcoat» может иметь отношение по общей массе марганца (Mn) к платине от 5:1 до 0,2:1, такое как от 5:1 до 0,5:1 (например, от 5:1 до 2:3 или от 5:1 до 1:2), предпочтительно от 4.5:1 до 1:1 (например, от 4:1 до 1,1:1), более предпочтительно от 4:1 до 1,5:1. Отношение Mn:Pt по массе может быть важным для достижения активности в отношении NO, описанной в данном документе.
Как правило, второй материал-носитель содержит или состоит по существу из огнеупорного оксида металла. Огнеупорный оксид металла, как правило, выбирают из группы, состоящей из оксида алюминия, кремнезема, диоксида титана, диоксида циркония, оксида церия и их смешанных или сложных оксидов, таких как смешанные или сложные оксиды двух или более этих оксидов. Например, огнеупорный оксид металла может быть выбран из группы, состоящей из оксида алюминия, кремнезема, диоксида титана, диоксида циркония, оксида церия, кремнезема-оксида алюминия, диоксида титана-оксида алюминия, диоксида циркония-оксида алюминия, оксида церия-оксида алюминия, диоксида титана-кремнезема, диоксида циркония-кремнезема, диоксида циркония-диоксида титана, оксида церия-диоксида циркония и оксида алюминия-оксида магния.
Второй материал-носитель или его огнеупорный оксид металла необязательно могут быть легированы (например, легирующей примесью). Легирующая примесь может быть выбрана из группы, состоящей из циркония (Zr), титана (Ti), кремния (Si), иттрия (Y), лантана (La), празеодима (Pr), самария (Sm), неодима (Nd) и их оксидов.
Когда второй материал-носитель или его огнеупорный оксид металла легирован, общее количество легирующей примеси составляет от 0,25 до 5% по массе, предпочтительно от 0,5 до 3% по массе (например, примерно 1% по массе).
Второй материал-носитель или его огнеупорный оксид металла могут содержать оксид алюминия, легированный легирующей примесью, или состоять по существу из него. Особенно предпочтительным является то, что второй материал-носитель или его огнеупорный оксид металла, содержит оксид алюминия, легированный легирующей примесью, или состоит по существу из него. Было найдено, что комбинация марганца (Mn), платины (Pt) и материал-носитель на основе легированного оксида алюминия, особенно материал-носитель на основе оксида алюминия, легированного кремнеземом, предоставляет превосходную активность окисления NO и может стабилизировать активность окисления NO катализатора окисления на протяжении его срока службы.
Оксид алюминия может быть легирован легирующей примесью, содержащей кремний (Si), магний (Mg), барий (Ba), лантан (La), церий (Ce), титан (Ti) или цирконий (Zr) или комбинацию двух этих элементов или более. Легирующая примесь может содержать, или состоять по существу из него, оксид кремния, оксид магния, оксид бария, оксид лантана, оксид церия, оксид титана или оксид циркония. Предпочтительно, легирующая примесь содержит, или состоит по существу из него, кремний, магний, барий, церий или их оксид, особенно кремний или церий или их оксид. Более предпочтительно, легирующая примесь содержит, или состоит по существу из него, кремний, магний, барий или их оксид, особенно кремний, магний или их оксид, главным образом кремний или его оксид.
Примеры оксида алюминия, легированного легирующей примесью, включают оксид алюминия, легированный кремнеземом, оксид алюминия, легированный оксидом магния, оксид алюминия, легированный барием или оксидом бария, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированный оксидом церия, особенно оксид алюминия, легированный кремнеземом, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированный оксидом церия. Предпочтительно, чтобы оксидом алюминия, легированным легирующей примесью, являлся оксид алюминия, легированный кремнеземом, оксид алюминия, легированный барием или оксидом бария, или оксид алюминия, легированный оксидом магния. Более предпочтительно, чтобы оксидом алюминия, легированным легирующей примесью, являлся оксид алюминия, легированный кремнеземом, или оксид алюминия, легированный оксидом магния. Еще более предпочтительно, чтобы оксидом алюминия, легированным легирующей примесью, являлся оксид алюминия, легированный кремнеземом.
Когда оксид алюминия является оксидом алюминия, легированным кремнеземом, то оксид алюминия легирован кремнеземом в общем количестве от 0,5 до 45% по массе (т.е. % по массе в расчете на оксид алюминия), предпочтительно от 1 до 40% по массе, более предпочтительно от 1,5 до 30% по массе (например, от 1,5 до 10% по массе), особенно предпочтительно от 2,5 до 25% по массе, еще более предпочтительно от 3,5 до 20% по массе (например, от 5 до 20% по массе), даже еще более предпочтительно от 4,5 до 15% по массе.
Когда оксид алюминия является оксидом алюминия, легированным оксидом магния, то оксид алюминия легирован магнием в количестве, как определено выше, или количестве от 1 до 30% по массе (т.е. % по массе в расчете на оксид алюминия), предпочтительно 5 до 25% по массе.
Предпочтительно, чтобы второй материал-носитель или его огнеупорный оксид металла не был легирован легирующей примесью, содержащей марганец или состоящей по существу из марганца. Соответственно, второй материал-носитель или его огнеупорный оксид металла не активирован активатором, таким как активатор, выбранный из группы, состоящей из олова, марганца, индия, металла группы VIII (например, Fe, Co, Ni, Ru, Rh, Pd, Os, Ir и Pt, особенно Ir) и их комбинаций.
В качестве альтернативы или дополнения, второй материал-носитель или его огнеупорный оксид металла может содержать алюминат щелочноземельного металла, такой как алюминат щелочноземельного металла, имеющий структуру шпинели, или состоять по существу из него.
Как правило, алюминат щелочноземельного металла является алюминатом магния (MgAl2O4), алюминатом кальция (CaAl2O4), алюминатом стронция (SrAl2O4) или алюминатом бария (BaAl2O4) или же смесью двух из них или более. Предпочтительно, алюминат щелочноземельного металла является алюминатом магния (MgAl2O4).
Обычно, когда второй материал-носитель или его огнеупорный оксид металла содержит, или состоит по существу из него, смешанный или сложный оксид с оксидом алюминия (например, кремнезем-оксид алюминия, оксид алюминия-оксид магния или смесь оксида алюминия и оксида церия), то предпочтительно смешанный или сложный оксид с оксидом алюминия содержит от по меньшей мере 50 до 99% по массе оксида алюминия, более предпочтительно от 70 до 95% по массе оксида алюминия, еще более предпочтительно от 75 до 90% по массе оксида алюминия.
Когда второй материал-носитель, или его огнеупорный оксид металла, содержит, или состоит по существу из него, оксид церия-диоксид циркония, то оксид церия-диоксида циркония может состоять по существу из 20-95% по массе оксида церия и 5-80% по массе диоксида циркония (например, 50-95% по массе оксида церия и 5-50% по массе диоксида циркония), предпочтительно 35-80% по массе оксида церия и 20-65% по массе диоксида циркония (например, 55-80% по массе оксида церия и 20-45% по массе диоксида циркония), даже еще более предпочтительно 45-75% по массе оксида церия и 25-55% по массе диоксида циркония.
Как правило, область второго покрытия из пористого оксида «washcoat» содержит второй материал-носитель в количестве от 0,1 до 4,5 г/дюйм3 (6-275 мг/см3) (например, 0,25 до 4,0 г/дюйм3 (15-244 мг/см3)), предпочтительно от 0,5 до 3,0 г/дюйм3 (31-183 мг/см3), более предпочтительно от 0,6 до 2,5 г·дюйм-3 (37-153 мг/см3) (например, от 0,75 до 1,5 г/дюйм3 (46-92 мг/см3).
Область второго покрытия из пористого оксида «washcoat» может дополнительно содержать цеолитовый катализатор, такой как цеолитовый катализатор, определенный в данном документе ниже.
В некоторых видах применения может обычно являться предпочтительным, чтобы область второго покрытия из пористого оксида «washcoat» по существу не содержала материал, адсорбирующий углеводороды, особенно цеолит. Соответственно, область второго покрытия из пористого оксида «washcoat» может не содержать материал, адсорбирующий углеводороды.
Может, кроме того, являться предпочтительным, чтобы область второго покрытия из пористого оксида «washcoat» по существу не содержала цеолитовый катализатор, такой как цеолитовый катализатор, описанный в данном документе ниже. Соответственно, область второго покрытия из пористого оксида «washcoat» может не содержать цеолитовый катализатор.
Область второго покрытия из пористого оксида «washcoat» обычно не содержит индий и/или иридий. Более предпочтительно, область второго покрытия из пористого оксида «washcoat» не содержит индий, иридий и/или магний.
Может являться предпочтительным, чтобы область второго покрытия из пористого оксида «washcoat» не содержала оксид церия или смешанный или сложный его оксид, такой как (i) смешанный или сложный оксид из оксида церия и оксида алюминия и/или (ii) смешанный или сложный оксид из оксида церия и диоксида циркония.
В качестве дополнения или альтернативы, область второго покрытия из пористого оксида «washcoat» может по существу не содержать родий, щелочной металл и/или щелочноземельный металл, особенно щелочной металл и/или щелочноземельный металл, размещенный или поддерживаемый на втором материале-носителе. Соответственно, область второго покрытия из пористого оксида «washcoat» может не содержать родий, щелочной металл и/или щелочноземельный металл, особенно щелочной металл и/или щелочноземельный металл, размещенный или поддерживаемый на втором материале-носителе.
Как правило, область первого покрытия из пористого оксида «washcoat» содержит >25% от общей концентрации металла платиновой группы (т.е. катализатора окисления). Предпочтительно, чтобы область первого покрытия из пористого оксида «washcoat» содержала >30%, более предпочтительно≥40%, от общей концентрации металла платиновой группы.
Обычно, общая концентрация первого МПГ больше, чем общая концентрация платины в области второго покрытия из пористого оксида «washcoat».
Как правило, катализатор окисления содержит общее количество материала-носителя (например, первого материала-носителя, второго материала-носителя и любого материала-носителя для платины и материала-носителя для палладия) в количестве от 0,1 до 4,5 г/дюйм3 (6-275 мг/см3) (например, от 0,25 до 4,2 г/дюйм3 (15-256 мг/см3)), предпочтительно 0,2 до 3,8 г/дюйм3 (12-232 мг/см3), таком как от 0,3 до 3,0 г/дюйм3 (18-183 мг/см3), особенно 0,5 до 2,5 г/дюйм3 (30-153 мг/см3) (например, 0,75 до 2,3 г/дюйм3) (46-140 мг/см3), еще более предпочтительно от 0,6 до 2,0 г/дюйм3 (37-122 мг/см3) и даже еще более предпочтительно от 0,75 до 1,75 г/дюйм3 (46-107 мг/см3).
Область первого покрытия из пористого оксида «washcoat» и/или область второго покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на основе.
Область первого покрытия из пористого оксида «washcoat» может быть размещена непосредственно на основе (т.е. область первого покрытия из пористого оксида «washcoat» находится в контакте с поверхностью основы; см. Фиг. 1-5). Область второго покрытия из пористого оксида «washcoat» может быть:
(a) размещена или поддерживаться на области первого покрытия из пористого оксида «washcoat» (например, см. Фиг. 2, 4 и 5); и/или
(b) размещена непосредственно на основе [т.е. область второго покрытия из пористого оксида «washcoat» находится в контакте с поверхностью основы] (например, см. Фиг. 1, 3, 4); и/или
(c) в контакте с областью первого покрытия из пористого оксида «washcoat» [т.е. область второго покрытия из пористого оксида «washcoat» расположена рядом с областью первого покрытия из пористого оксида «washcoat» или примыкает к ней].
Когда область второго покрытия из пористого оксида «washcoat» размещают непосредственно на основе, то часть или участок области второго покрытия из пористого оксида «washcoat» может находиться в контакте с областью первого покрытия из пористого оксида «washcoat», или область первого покрытия из пористого оксида «washcoat» и область второго покрытия из пористого оксида «washcoat» могут быть разделены (например, посредством зазора).
Когда область второго покрытия из пористого оксида «washcoat» располагают или поддерживают на области первого покрытия из пористого оксида «washcoat», вся область второго покрытия из пористого оксида «washcoat» или ее часть предпочтительно расположена непосредственно на области первого покрытия из пористого оксида «washcoat» (т.е. область второго покрытия из пористого оксида «washcoat» находится в контакте с поверхностью области первого покрытия из пористого оксида «washcoat»). Область второго покрытия из пористого оксида «washcoat» может быть слоем второго покрытия из пористого оксида «washcoat», и область первого покрытия из пористого оксида «washcoat» может быть слоем первого покрытия из пористого оксида «washcoat».
Область второго покрытия из пористого оксида «washcoat» может быть размещена непосредственно на основе (т.е. область второго покрытия из пористого оксида «washcoat» находится в контакте с поверхностью основы; см. Фиг. 1, 3 и 4). Область первого покрытия из пористого оксида «washcoat» может быть:
(i) размещена или поддерживаться на области второго покрытия из пористого оксида «washcoat» (например, см. Фиг. 3 и 4); и/или
(ii) размещена непосредственно на основе [т.е. область первого покрытия из пористого оксида «washcoat» находится в контакте с поверхностью основы] (например, см. Фиг. 3 и 4); и/или
(iii) в контакте с областью второго покрытия из пористого оксида «washcoat» [т.е. область первого покрытия из пористого оксида «washcoat» расположена рядом с областью второго покрытия из пористого оксида «washcoat» или примыкает к ней].
Область первого покрытия из пористого оксида «washcoat» может быть размещена непосредственно на области второго покрытия из пористого оксида «washcoat» (т.е. область первого покрытия из пористого оксида «washcoat» находится в контакте с поверхностью области второго покрытия из пористого оксида «washcoat»).
Предпочтительно, чтобы лишь участок или часть области первого покрытия из пористого оксида «washcoat» была расположена или поддерживалась на области второго покрытия из пористого оксида «washcoat». Соответственно, область первого покрытия из пористого оксида «washcoat» не полностью перекрывает или закрывает область второго покрытия из пористого оксида «washcoat».
Как правило, возможно, чтобы как область первого покрытия из пористого оксида «washcoat», так и область второго покрытия из пористого оксида «washcoat» не были расположены непосредственно на основе (т.е., чтобы ни область первого покрытия из пористого оксида «washcoat», ни область второго покрытия из пористого оксида «washcoat» не находились в контакте с поверхностью основы).
Катализатор окисления может дополнительно содержать область третьего покрытия из пористого оксида «washcoat». Соответственно, по меньшей мере одна из области первого покрытия из пористого оксида «washcoat» и области второго покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на области третьего покрытия из пористого оксида «washcoat». Область третьего покрытия из пористого оксида «washcoat» может содержать или может не содержать металл платиновой группы.
Область третьего покрытия из пористого оксида «washcoat» может быть размещена непосредственно на основе (т.е. область третьего покрытия из пористого оксида «washcoat» находится в контакте с поверхностью основы; см. Фиг. 6, 8 и 10). Область второго покрытия из пористого оксида «washcoat» может быть:
(a) размещена или поддерживаться на области третьего покрытия из пористого оксида «washcoat» (например, см. Фиг. 6, 8 и 10); и/или
(b) размещена непосредственно на основе [т.е. область второго покрытия из пористого оксида «washcoat» находится в контакте с поверхностью основы]; и/или
(c) в контакте с областью третьего покрытия из пористого оксида «washcoat» [т.е. область второго покрытия из пористого оксида «washcoat» расположена рядом с областью третьего покрытия из пористого оксида «washcoat» или примыкает к ней].
В качестве дополнения или альтернативы, область третьего покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на области первого покрытия из пористого оксида «washcoat» и/или области второго покрытия из пористого оксида «washcoat» (например, см. Фиг. 7 и 9).
Когда область третьего покрытия из пористого оксида «washcoat» расположена непосредственно на основе, то область первого покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на области третьего покрытия из пористого оксида «washcoat» (например, см. Фиг. 6). Область второго покрытия из пористого оксида «washcoat» может также быть размещена или поддерживаться на области третьего покрытия из пористого оксида «washcoat». Область третьего покрытия из пористого оксида «washcoat» может быть слоем третьего покрытия из пористого оксида, область первого покрытия из пористого оксида «washcoat» может быть зоной первого покрытия из пористого оксида «washcoat», и область второго покрытия из пористого оксида «washcoat» может быть зоной второго покрытия из пористого оксида «washcoat». Область/зона второго покрытия из пористого оксида «washcoat» может находиться в контакте с областью/зоной первого покрытия из пористого оксида «washcoat» [т.е. область/зона второго покрытия из пористого оксида «washcoat» расположена рядом с областью/зоной первого покрытия из пористого оксида «washcoat» или примыкает к ней]. В качестве альтернативы, область/зона первого покрытия из пористого оксида «washcoat» и область/зона второго покрытия из пористого оксида «washcoat» могут быть разделены (например, посредством зазора).
Когда область третьего покрытия из пористого оксида «washcoat» расположена непосредственно на основе, то область первого покрытия из пористого оксида «washcoat» может быть размещена непосредственно на основе (например, см. Фиг. 8 и 10). Область второго покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на области третьего покрытия из пористого оксида «washcoat» и/или области первого покрытия из пористого оксида «washcoat», предпочтительно область второго покрытия из пористого оксида «washcoat» размещена или поддерживается на области третьего покрытия из пористого оксида «washcoat». Область третьего покрытия из пористого оксида «washcoat» может быть зоной третьего покрытия из пористого оксида «washcoat», и область первого покрытия из пористого оксида «washcoat» может быть зоной первого покрытия из пористого оксида «washcoat». Область/зона третьего покрытия из пористого оксида «washcoat» может находиться в контакте с областью/зоной первого покрытия из пористого оксида «washcoat» [т.е. область/зона третьего покрытия из пористого оксида «washcoat» расположена рядом с областью/зоной первого покрытия из пористого оксида «washcoat» или примыкает к ней]. В качестве альтернативы, область/зона первого покрытия из пористого оксида «washcoat» и область/зона третьего покрытия из пористого оксида «washcoat» могут быть разделены (например, посредством зазора).
Область второго покрытия из пористого оксида «washcoat» может быть слоем второго покрытия из пористого оксида «washcoat» или зоной второго покрытия из пористого оксида «washcoat», предпочтительно зоной второго покрытия из пористого оксида «washcoat». Когда область второго покрытия из пористого оксида «washcoat» является зоной второго покрытия из пористого оксида «washcoat», катализатор окисления может дополнительно содержать область четвертого покрытия из пористого оксида «washcoat» (например, см. Фиг. 10). Область четвертого покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на зоне первого покрытия из пористого оксида «washcoat». Область четвертого покрытия из пористого оксида «washcoat» может находиться в контакте с зоной второго покрытия из пористого оксида «washcoat» [т.е. область четвертого покрытия из пористого оксида «washcoat» расположена рядом с зоной второго покрытия из пористого оксида «washcoat» или примыкает к ней]. В качестве альтернативы, область четвертого покрытия из пористого оксида «washcoat» и зона второго покрытия из пористого оксида «washcoat» могут быть разделены (например, посредством зазора).
Область четвертого покрытия из пористого оксида «washcoat» может быть зоной четвертого покрытия из пористого оксида «washcoat».
Область третьего покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на области второго покрытия из пористого оксида «washcoat» (например, см. Фиг. 9). Область второго покрытия из пористого оксида «washcoat» может быть зоной второго покрытия из пористого оксида «washcoat», и область первого покрытия из пористого оксида «washcoat» может быть зоной первого покрытия из пористого оксида «washcoat». Область/зона второго покрытия из пористого оксида «washcoat» может находиться в контакте с областью/зоной первого покрытия из пористого оксида «washcoat» [т.е. область/зона второго покрытия из пористого оксида «washcoat» расположена рядом с областью/зоной первого покрытия из пористого оксида «washcoat» или примыкает к ней]. В качестве альтернативы, область/зона первого покрытия из пористого оксида «washcoat» и область/зона второго покрытия из пористого оксида «washcoat» могут быть разделены (например, посредством зазора).
Область третьего покрытия из пористого оксида «washcoat» может быть слоем третьего покрытия из пористого оксида «washcoat» или зоной третьего покрытия из пористого оксида «washcoat».
Когда область третьего покрытия из пористого оксида «washcoat» является зоной третьего покрытия из пористого оксида «washcoat», то зона третьего покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на зоне второго покрытия из пористого оксида «washcoat» (например, см. Фиг. 7). Область первого покрытия из пористого оксида «washcoat» может быть зоной первого покрытия из пористого оксида «washcoat». Зона третьего покрытия из пористого оксида «washcoat» может находиться в контакте с зоной первого покрытия из пористого оксида «washcoat» [т.е. зона третьего покрытия из пористого оксида «washcoat» расположена рядом с зоной первого покрытия из пористого оксида «washcoat» или примыкает к ней]. В качестве альтернативы, зона первого покрытия из пористого оксида «washcoat» и зона третьего покрытия из пористого оксида «washcoat» могут быть разделены (например, посредством зазора).
Когда область третьего покрытия из пористого оксида «washcoat» является зоной третьего покрытия из пористого оксида «washcoat», то зона третьего покрытия из пористого оксида «washcoat» может быть размещена или поддерживаться на зоне первого покрытия из пористого оксида «washcoat» (например, см. Фиг. 9). Область второго покрытия из пористого оксида «washcoat» может быть зоной второго покрытия из пористого оксида «washcoat». Зона третьего покрытия из пористого оксида «washcoat» может находиться в контакте с зоной второго покрытия из пористого оксида «washcoat» [т.е. зона третьего покрытия из пористого оксида «washcoat» расположена рядом с зоной второго покрытия из пористого оксида «washcoat» или примыкает к ней]. В качестве альтернативы, зона второго покрытия из пористого оксида «washcoat» и зона третьего покрытия из пористого оксида «washcoat» могут быть разделены (например, посредством зазора) и/или не находиться в контакте (например, зона второго покрытия из пористого оксида «washcoat» не находится в физическом контакте с зоной третьего покрытия из пористого оксида «washcoat»).
Когда область третьего покрытия из пористого оксида «washcoat» является слоем третьего покрытия из пористого оксида «washcoat», то слой третьего покрытия из пористого оксида «washcoat» может быть размещен или поддерживаться как на области/зоне первого покрытия из пористого оксида «washcoat», так и на области/зоне второго покрытия из пористого оксида «washcoat».
В катализаторе окисления по данному изобретению, область второго покрытия из пористого оксида «washcoat» обычно расположена таким образом, чтобы контактировать с выхлопными газами на выпускном конце основы. Это достигается посредством расположения области/слоя/зоны второго покрытия из пористого оксида «washcoat» на выпускном конце основы.
Область второго покрытия из пористого оксида «washcoat» обычно расположена или ориентирована таким образом, чтобы контактировать с выхлопными газами после то, как они имели контакт с областью первого покрытия из пористого оксида «washcoat». Это может быть достигнуто посредством расположения области второго покрытия из пористого оксида «washcoat» различным образом на основе по отношению к области первого покрытия из пористого оксида «washcoat».
Соответственно, область второго покрытия из пористого оксида «washcoat» расположена или ориентирована таким образом, чтобы контактировать с выхлопными газами после то, как они имели контакт с областью первого покрытия из пористого оксида «washcoat», когда:
(a) область второго покрытия из пористого оксида «washcoat» является зоной второго покрытия из пористого оксида «washcoat», размещенной на выпускном конце основы, и необязательно область первого покрытия из пористого оксида «washcoat» является зоной первого покрытия из пористого оксида «washcoat», размещенной на впускном конце основы;
(b) область первого покрытия из пористого оксида «washcoat» является первым слоем покрытия из пористого оксида «washcoat» и область второго покрытия из пористого оксида «washcoat» является зоной второго покрытия из пористого оксида «washcoat», при этом зона второго покрытия из пористого оксида «washcoat» расположена на слое первого покрытия из пористого оксида «washcoat» на выпускном конце основы; или
(c) область первого покрытия из пористого оксида «washcoat» является первым слоем покрытия из пористого оксида «washcoat», и область второго покрытия из пористого оксида «washcoat» является слоем второго покрытия из пористого оксида «washcoat», и где слой второго покрытия из пористого оксида «washcoat» расположен на слое первого покрытия из пористого оксида «washcoat».
Как правило, область первого покрытия из пористого оксида «washcoat» расположена или ориентирована, чтобы контактировать с выхлопными газами перед областью второго покрытия из пористого оксида «washcoat». Соответственно, область первого покрытия из пористого оксида «washcoat» может быть расположена таким образом, чтобы контактировать с выхлопными газами, когда они поступают в катализатор окисления, и область второго покрытия из пористого оксида «washcoat» может быть расположена таким образом, чтобы контактировать с выхлопными газами, когда они оставляют катализатор окисления. Зональное расположение областей первого второго покрытия из пористого оксида «washcoat», показанное на Фиг. 1 и 6-10, является особенно выгодным в этом отношении.
Область второго покрытия из пористого оксида «washcoat» размещена таким образом, чтобы контактировать с выхлопными газами на выпускном конце основы и после контактирования выхлопных газов с областью первого покрытия из пористого оксида «washcoat» при любом с первого по третье расположении катализатора окисления, которые описаны в данном документе ниже.
Предпочтительно, чтобы область второго покрытия из пористого оксида «washcoat» являлась зоной второго покрытия из пористого оксида «washcoat». Более предпочтительно, зону второго покрытия из пористого оксида «washcoat» размещают или поддерживают на выпускном конце основы или вблизи него.
Зона второго покрытия из пористого оксида «washcoat» обычно имеет длину от 10 до 90% длины основы (например, от 10 до 45%), предпочтительно от 15 до 75% длины основы (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 30 до 65%, такую как от 25 до 45%) длины основы, еще более предпочтительно от 25 до 65% (например, от 35 до 50%).
Когда область третьего покрытия из пористого оксида «washcoat» является зоной третьего покрытия из пористого оксида «washcoat», то зона третьего покрытия из пористого оксида «washcoat» обычно имеет длину от 10 до 90% длины основы (например, от 10 до 45%), предпочтительно от 15 до 75% длины основы (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 30 до 65%, такую как от 25 до 45%) длины основы, еще более предпочтительно от 25 до 65% (например, от 35 до 50%).
В первом расположении катализатора окисления, область первого покрытия из пористого оксида «washcoat» размещена или поддерживается выше по течению потока по отношению к зоне второго покрытия из пористого оксида «washcoat». Предпочтительно, область первого покрытия из пористого оксида «washcoat» является зоной первого покрытия из пористого оксида «washcoat». Более предпочтительно, зону первого покрытия из пористого оксида «washcoat» размещают или поддерживают на впускном конце основы или вблизи него. Mn-содержащая зона второго покрытия из пористого оксида «washcoat» может проявлять высокую устойчивость к сере, когда катализатор окисления имеет такое «зональное» расположение.
Зона первого покрытия из пористого оксида «washcoat» может прилегать к зоне второго покрытия из пористого оксида «washcoat». Предпочтительно, зона первого покрытия из пористого оксида «washcoat» контактирует с зоной второго покрытия из пористого оксида «washcoat». Когда зона первого покрытия из пористого оксида «washcoat» прилегает к зоне второго покрытия из пористого оксида «washcoat», или зона первого покрытия из пористого оксида «washcoat» находится в контакте с зоной второго покрытия из пористого оксида «washcoat», то зона первого покрытия из пористого оксида «washcoat» и зона второго покрытия из пористого оксида «washcoat» могут быть размещены или поддерживаться на основе в виде слоя (например, в виде одиночного слоя). Соответственно, слой (например, одиночный слой) может быть сформирован на основе, когда зоны первого и второго покрытий из пористого оксида «washcoat» прилегают друг к другу или находятся в контакте одна с другой. Такое расположение может способствовать избежанию проблем с противодавлением.
Зона первого покрытия из пористого оксида «washcoat» может быть отделена от зоны второго покрытия из пористого оксида «washcoat». Может иметься зазор (например, пространство) между зоной первого покрытия из пористого оксида «washcoat» и зоной второго покрытия из пористого оксида «washcoat».
Зона первого покрытия из пористого оксида «washcoat» может частично накладываться на зону второго покрытия из пористого оксида «washcoat». Соответственно, конечный участок или часть зоны первого покрытия из пористого оксида «washcoat» могут быть размещены или поддерживаться на зоне второго покрытия из пористого оксида «washcoat». Зона первого покрытия из пористого оксида «washcoat» может быть полностью или частично наложена на зону второго покрытия из пористого оксида «washcoat». Когда зона первого покрытия из пористого оксида «washcoat» наложена на зону второго покрытия из пористого оксида «washcoat», предпочтительно, чтобы зона первого покрытия из пористого оксида «washcoat» лишь частично перекрывала зону второго покрытия из пористого оксида «washcoat» (т.е. чтобы верхняя, внешняя поверхность зоны второго покрытия из пористого оксида «washcoat» не была полностью покрыта зоной первого покрытия из пористого оксида «washcoat»).
В качестве альтернативы, зона второго покрытия из пористого оксида «washcoat» может частично накладываться на зону первого покрытия из пористого оксида «washcoat». Соответственно, конечный участок или часть зоны второго покрытия из пористого оксида «washcoat» могут быть размещены или поддерживаться на зоне первого покрытия из пористого оксида «washcoat». Зона второго покрытия из пористого оксида «washcoat» обычно лишь частично покрывает зону первого покрытия из пористого оксида «washcoat».
Предпочтительно, чтобы зона первого покрытия из пористого оксида «washcoat» и зона второго покрытия из пористого оксида «washcoat» не были существенным образом наложены одна на другую.
Как правило, зона первого покрытия из пористого оксида «washcoat» имеет длину от 10 до 90% длины основы (например, от 10 до 45%), предпочтительно от 15 до 75% длины основы (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 30 до 65%, такую как от 25 до 45%) от длины основы, еще более предпочтительно от 25 до 65% (например, от 35 до 50%). Предпочтительно, чтобы длина зоны первого покрытия из пористого оксида «washcoat» была больше, чем длина зоны второго покрытия из пористого оксида «washcoat».
Во втором расположении катализатора окисления область первого покрытия из пористого оксида «washcoat» является первым слоем покрытия из пористого оксида «washcoat». Предпочтительно, чтобы слой первого покрытия из пористого оксида «washcoat» был протянут на полную длину (т.е. по существу по всей длине) основы, особенно на полную длину каналов монолитной основы.
Зона второго покрытия из пористого оксида «washcoat» типично размещена или поддерживается на слое первого покрытия из пористого оксида «washcoat». Предпочтительно, зону второго покрытия из пористого оксида «washcoat» размещают непосредственно на слое первого покрытия из пористого оксида «washcoat» (т.е. зона второго покрытия из пористого оксида «washcoat» находится в контакте с поверхностью слоя первого покрытия из пористого оксида «washcoat»).
Когда зона второго покрытия из пористого оксида «washcoat» размещена или поддерживается на слое первого покрытия из пористого оксида «washcoat», предпочтительно, чтобы зона второго покрытия из пористого оксида «washcoat» на полную длину была размещена или поддерживалась на слое первого покрытия из пористого оксида «washcoat». Длина зоны второго покрытия из пористого оксида «washcoat» меньше, чем длина слоя первого покрытия из пористого оксида «washcoat».
В третьем расположении катализатора окисления область первого покрытия из пористого оксида «washcoat» является первым слоем покрытия из пористого оксида «washcoat». Предпочтительно, чтобы слой первого покрытия из пористого оксида «washcoat» был протянут на полную длину (т.е. по существу по всей длине) основы, особенно на полную длину каналов монолитной основы
Область второго покрытия из пористого оксида «washcoat» является слоем второго покрытия из пористого оксида «washcoat». Предпочтительно, чтобы слой второго покрытия из пористого оксида «washcoat» был протянут на полную длину (т.е. по существу по всей длине) основы, особенно на полную длину каналов монолитной основы
Слой второго покрытия из пористого оксида «washcoat» размещают или поддерживают на на слое первого покрытия из пористого оксида «washcoat». Предпочтительно, слой второго покрытия из пористого оксида «washcoat» размещают непосредственно на слое первого покрытия из пористого оксида «washcoat» (т.е. слой второго покрытия из пористого оксида «washcoat» находится в контакте с поверхностью слоя первого покрытия из пористого оксида «washcoat»).
Катализатор окисления по данному изобретению может содержать цеолитовый катализатор. Область первого покрытия из пористого оксида «washcoat», область второго покрытия из пористого оксида «washcoat», область третьего покрытия из пористого оксида «washcoat» и/или область четвертого покрытия из пористого оксида «washcoat» могут содержать цеолитовый катализатор.
Может являться предпочтительным, чтобы область второго покрытия из пористого оксида «washcoat» содержала цеолитовый катализатор. Более предпочтительно, область первого покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор, и область второго покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор (например, как область первого покрытия из пористого оксида «washcoat», так и область второго покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор).
В качестве дополнения или альтернативы, область третьего покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор или состоит по существу из него. Катализатор окисления может содержать область четвертого покрытия из пористого оксида «washcoat». Область четвертого покрытия из пористого оксида «washcoat» может содержать цеолитовый катализатор или состоять по существу из него. Когда каждая из области третьего покрытия из пористого оксида «washcoat» и области четвертого покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор, то область третьего покрытия из пористого оксида «washcoat» может содержать первый цеолитовый катализатор или состоять по существу из него, и область четвертого покрытия из пористого оксида «washcoat» может содержать второй цеолитовый катализатор или состоять по существу из него, при этом первый цеолитовый катализатор отличается (т.е. имеет другой состав) от второго цеолитового катализатора. Первый цеолитовый катализатор может быть цеолитовым катализатором, описанным в данном документе ниже. Второй цеолитовый катализатор может быть цеолитовым катализатором, описанным в данном документе ниже.
Когда область третьего покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор или состоит по существу из него, то предпочтительно область первого покрытия из пористого оксида «washcoat» и/или область второго покрытия из пористого оксида «washcoat» не содержит цеолитовый катализатор.
Как правило, цеолитовый катализатор содержит, или состоит по существу из них, благородный металл и цеолит. Целитовые катализаторы могут быть получены способом, описанным в WO 2012/166868.
Благородный металл обычно выбирают из группы, состоящей из палладия (Pd), платины (Pt), родия (Rh), золота (Au), серебра (Ag), иридия (Ir), рутения (Ru), осмия (Os) и смесей двух их или более. Предпочтительно, благородный металл выбирают из группы, состоящей из палладия (Pd), платины (Pt), родия (Rh), золота (Au), серебра (Ag), иридия (Ir), рутения (Ru) и смесей двух их или более. Более предпочтительно, благородный металл выбирают из группы, состоящей из палладия (Pd), платины (Pt) и родия (Rh). Еще более предпочтительно, благородным металлом является палладий (Pd). Даже еще более предпочтительно, чтобы цеолитовый катализатор содержал палладий в качестве единственного благородного металла.
Цеолитовый катализатор может дополнительно содержать неблагородный металл. Соответственно, цеолитовый катализатор может содержать, или состоять по существу из них, благородный металл, цеолит и необязательно неблагородный металл.
Неблагородный металл может быть выбран из группы, состоящей из железа (Fe), меди (Cu), марганца (Mn), хрома (Cr), кобальта (Co), никеля (Ni) и олова (Sn), а также смесей двух их или более. Предпочтительно, чтобы неблагородный металл был выбран из группы, состоящей из железа, меди и кобальта, более предпочтительно из железа и меди. Еще более предпочтительно, неблагородным металлом является железо.
В качестве альтернативы, цеолитовый катализатор может по существу не содержать неблагородный металл. Соответственно, цеолитовый катализатор не содержит неблагородный металл.
Как правило, предпочтительно, чтобы цеолитовый катализатор не содержал неблагородный металл.
Цеолит обычно выбирают из алюмосиликатного цеолита и алюмофосфатного цеолит.
Обычно, цеолит может быть цеолитом с замененным металлом (например, алюмосиликатным цеолитом с замененным металлом или алюмофосфатным цеолитом с замененным металлом). Металл цеолита с замененным металлом может быть благородный металл (например, цеолит является цеолит с замещением благородным металлом). Когда цеолитовый катализатор содержит неблагородный металл, то цеолит может быть цеолитом с замещением благородным и неблагородным металлом.
Цеолитовый катализатор обычно содержит по меньшей мере 1% по массе (т.е. количества благородного металла цеолитового катализатора) благородного металла, расположенного внутри пор цеолита, предпочтительно по меньшей мере 5% по массе, более предпочтительно по меньшей мере 10% по массе, например, по меньшей мере 25% по массе, еще более предпочтительно по меньшей мере 50% по массе.
Цеолит может быть выбран из цеолита с малым размером пор (т.е. цеолита, имеющего максимальный размер кольца восьми тетраэдрически координированных атомов), цеолита со средним размером пор (т.е. цеолита, имеющего максимальный размер кольца десяти тетраэдрически координированных атомов) и цеолита с большим размером пор (т.е. цеолита, имеющего максимальный размер кольца двенадцати тетраэдрически координированных атомов). Более предпочтительно, цеолит выбирают из цеолита с малым размером пор и цеолита со средним размером пор.
Как правило, цеолит состоит из алюминия, кремния и/или фосфора. Цеолит обычно имеет трехмерное расположение SiO4, AlO4 и/или PO4, которые соединены посредством совместного использования атомов кислорода. Цеолит может иметь анионную структуру. Заряд анионной структуры может быть уравновешен катионами, например, катионами щелочных и/или щелочноземельных элементов (например, Na, K, Mg, Ca, Sr и Ba), катионами аммония и/или протонами.
В первом варианте осуществления цеолитового катализатора, цеолит является цеолитом с малым размером пор. Цеолит с малым размером пор предпочтительно имеет тип каркасной структуры, выбранный из группы, состоящей из ACO, AEI, AEN, AFN, AFT, AFX, ANA, APC, APD, ATT, CDO, CHA, DDR, DFT, EAB, EDI, EPI, ERI, GIS, GOO, IHW, ITE, ITW, LEV, KFI, MER, MON, NSI, OWE, PAU, PHI, RHO, RTH, SAT, SAV, SIV, THO, TSC, UEI, UFI, VNI, YUG и ZON, а также смесей или сростков любых двух или более этих типов. Сростки предпочтительно выбирают из KFI-SIV, ITE-RTH, AEW-UEI, AEI-CHA и AEI-SAV. Более предпочтительно, цеолит с малым размером пор имеет тип каркасной структуры, который является AEI, CHA или сростком AEI-CHA. Еще более предпочтительно, цеолит с малым размером пор имеет тип каркасной структуры, который является AEI или CHA.
Во втором варианте осуществления цеолитового катализатора, цеолит имеет тип каркасной структуры, выбранный из группы, состоящей из AEI, MFI, EMT, ERI, MOR, FER, BEA, FAU, CHA, LEV, MWW, CON и EUO, а также смесей любых двух или более этих типов.
В третьем варианте осуществления цеолитового катализатора, цеолит является цеолитом со средним размером пор. Цеолит со средним размером пор предпочтительно имеет тип каркасной структуры, выбранный из группы, состоящей из MFI, FER, MWW и EUO, более предпочтительно MFI.
В четвертом варианте осуществления цеолитового катализатора, цеолит является цеолитом с большим размером пор. Цеолит с большим размером пор предпочтительно имеет тип каркасной структуры, выбранный из группы, состоящей из CON, BEA, FAU, MOR и EMT, более предпочтительно BEA.
Цеолит обычно имеет молярное отношение кремнезема к оксиду алюминия (SAR) от 10 до 200 (например, от 10 до 40), более предпочтительно от 15 до 80 (например, от 15 до 30).
Цеолитовый катализатор первого, третьего и четвертого вариантов осуществления цеолитового катализатора (и также для некоторых типов каркасной структуры второго варианта осуществления цеолитового катализатора) могут иметь спектр инфракрасного излучения, имеющий характеристический пик поглощения в интервале от 750 см-1 до 1050 см-1 (в дополнение к пикам поглощения для самого цеолита). Предпочтительно, характеристический пик поглощения находится в интервале от 800 см-1 до 1000 см-1, более предпочтительно в интервале от 850 см-1 до 975 см-1.
Неожиданно было найдено, что область покрытия из пористого оксида «washcoat», содержащая цеолитовый катализатор первого варианта осуществления цеолитового катализатора, обладает активностью пассивного адсорбера NOx (PNA). Область покрытия из пористого оксида «washcoat» с активностью пассивного абсорбера NOx (PNA) может быть использована, чтобы сохранять NOx, когда температуры выхлопных газов являются сравнительно низкими, такими как непосредственно после пуска дизельного двигателя. Сохранение NOx цеолитовым катализатором происходит при температурах (например, ниже чем 200°С), которые ниже температуры, при которой платина и марганец, являющиеся компонентами области второго покрытия из пористого оксида «washcoat», выполняют значительное окисление оксида азота (NO) до диоксида азота (NO2).
Когда дизельный двигатель разогревается, температура выхлопных газов увеличивается, и температура цеолитового катализатора (и области покрытия из пористого оксида «washcoat», обладающей активностью пассивного абсорбера NOx (PNA)) будет также увеличиваться. Цеолитовый катализатор будет высвобождать адсорбированные NOx при этих более высоких температурах (например, 200°С или выше). Цеолитовый катализатор может высвобождать адсорбированные NOx, когда платина и марганец, являющиеся компонентами области второго покрытия из пористого оксида «washcoat», достигли своей эффективной температуры для окисления NO, или цеолитовый катализатор может высвобождать адсорбированные NOx при температуре несколько ниже этой эффективной температуры. Цеолитовый катализатор, как было неожиданно найдено, имеет высокую температуру высвобождения для адсорбированных NOx.
Также неожиданно было найдено, что область покрытия из пористого оксида «washcoat», содержащая цеолитовый катализатор второго варианта осуществления цеолитового катализатора, обладает активностью катализатора для холодного пуска (CSCTM). Такая активность может уменьшать выбросы во время периода холодного пуска посредством адсорбирования NOx и углеводородов (HC) при сравнительно низких температурах выхлопных газов (например, менее чем 200°С). Адсорбированные NOx и/или HC могут быть высвобождены областью покрытия из пористого оксида «washcoat», когда температура цеолитового катализатора близка или выше эффективной температуры других компонентов катализатора для окисления NO и/или HC. Активность в качестве катализатора для холодного пуска для цеолитового катализатора является особенно целесообразной, когда он объединен с областью второго покрытия из пористого оксида «washcoat», содержащей марганец.
Когда область третьего покрытия из пористого оксида «washcoat» содержит, или состоит по существу из него, цеолитовый катализатор, особенно цеолитовый катализатор первого или второго варианта осуществления цеолитового катализатора, температура, при которой адсорбированные NOx высвобождаются из цеолитового катализатора, может быть изменена (а именно, увеличена), когда катализатор окисления имеет определенное расположение. Выгодное изменение (а именно, увеличение) температуры цеолитового катализатора может получено, когда область второго покрытия из пористого оксида «washcoat» размещена или поддерживается на области третьего покрытия из пористого оксида «washcoat» (см., например, Фиг. 6, 8 и 10).
Когда область покрытия из пористого оксида «washcoat» содержит цеолитовый катализатор, главным образом область третьего покрытия из пористого оксида «washcoat» и/или область четвертого покрытия из пористого оксида «washcoat», то общая загрузка благородного металла (т.е. цеолитового катализатора в каждой области покрытия из пористого оксида «washcoat») составляет обычно от 1 до 250 г/фут3 (0,035-8,829 кг/м3) (например, от 1 до 75 г/фут3 (0,035-2,649 кг/м3)), предпочтительно от 5 до 150 г/фут3 (0,177-5,297 кг/м3) (например, от 5 до 50 г/фут3 (0,177-1,766 кг/м3)), более предпочтительно от 10 до 100 г/фут3 (0,353-3,531 кг/м3) (например, от 10 до 40 г/фут3 (0,353-1,413 кг/м3)).
Основы для поддержки катализаторов окисления для обработки выхлопных газов от дизельного двигателя хорошо известны в данной области техники. Способы изготовления покрытий из пористого оксида «washcoat» и нанесения покрытий из пористого оксида «washcoat» на основу также известны в данной области техники, (см., например, наши заявки WO 99/47260, WO 2007/077462 и WO 2011/080525).
Основа обычно имеет множество каналов (например, для протекания через нее выхлопных газов). Как правило, основа является керамическим материалом или металлическим материалом.
Предпочтительным является, чтобы основа была сделана или состояла из кордиерита (SiO2-Al2O3-MgO), карбида кремния (SiC), сплава Fe-Cr-Al, сплава Ni-Cr-Al или нержавеющей стали.
Как правило, основа является монолитом (также называемой в данном документе монолитной основой). Такие монолиты хорошо известны в данной области техники. Монолитная основа может быть проточным монолитом или фильтрующим монолитом.
Проточный монолит обычно выполнен в виде сотового монолита (например, металлического или керамического сотового монолита), имеющего множество каналов, вытянутых через него, данные каналы открыты на обоих концах. Когда основа является проточным монолитом, то катализатор окисления по данному изобретению является обычно катализатором окисления дизельного топлива (DOC) или применяется в качестве катализатора окисления дизельного топлива (DOC).
Фильтрующий монолит обычно содержит множество впускных каналов и множество выпускных каналов, при этом впускные каналы открыты на конце выше по течению потока (т.е. на стороне впускного отверстия для выхлопных газов) и закупорены или закрыты на конце ниже по течению потока (т.е. на стороне выпускного отверстия для выхлопных газов), выпускные каналы закупорены или закрыты на конце выше по течению потока и открыты на конце ниже по течению потока, и при этом каждый впускной канал отделен от выпускного канала пористой структурой. Когда основа является фильтрующим монолитом, то катализатор окисления по данному изобретению является обычно катализированным сажевым фильтром (CSF) или применяется в качестве катализированного сажевого фильтра (CSF).
Когда монолит является фильтрующим монолитом, предпочтительно, чтобы фильтрующий монолит являлся фильтром с протеканием через стенки. В фильтре с протеканием через стенки каждый впускной канал поочередным образом отделен от выпускного канала стенкой пористой структуры и наоборот. Предпочтительным является, чтобы впускные каналы и выпускные каналы были расположены в виде сотовой структуры. При расположении в виде сотовой структуры предпочтительным является, чтобы каналы, смежные в вертикальном и в боковом направлении с впускным каналом были закупорены на конце выше по течению потока и наоборот (т.е. каналы, смежные в вертикальном и в боковом направлении с выпускным каналом закупорены на конце ниже по течению потока). Если смотреть со стороны любого из двух концов, поочередно закрытые и открытые концы каналов имеют вид шахматной доски.
В принципе, основа может иметь любую форму или размер. Однако форму и размер основы обычно выбирают, чтобы оптимизировать воздействие каталитически активных материалов в катализаторе на выхлопные газы. Основа может, например, иметь трубчатую форму, волокнистую форму или находиться в виде твердых частиц. Примеры подходящих поддерживающих основ включают монолитную кордиеритовую основу с сотовой структурой, монолитную основу из SiC с сотовой структурой, слоистую волокнистую основу или основу из тканого материала, основу из пеноматериала, основу с перекрестным потоком, сетчатую основу из металлической проволоки, основу из пористого металлического материала и основу из керамических частиц.
Обычно, катализатор окисления по данному изобретению применяют в качестве катализатора окисления дизельного топлива (DOC) или катализированного сажевого фильтра (CSF). На практике, композиции катализаторов, применяемые в катализаторах окисления дизельного топлива (DOC) и катализированных сажевых фильтрах (CSF), являются сходными. Обычно, принципиальным различием между катализатором окисления дизельного топлива (DOC) и катализированным сажевым фильтром (CSF) является основа, которую покрывает композиция катализатора, и общее количество платины, палладия и любых других каталитически активных металлов, которые нанесены на основу.
Следует понимать, что любая ссылка на катализатор окисления по данному изобретению для применения в качестве катализатора окисления дизельного топлива (DOC) может включать активность пассивного адсорбера NOx (DOC-PNA) или активность катализатора для холодного пуска (DOC-CSC).
Данное изобретение также предоставляет систему выпуска выхлопных газов, содержащую катализатор окисления и устройство для регулирования выбросов. Примеры устройства для регулирования выбросов включают фильтр твердых частиц выхлопа дизельного двигателя (DPF), ловушку для обедненных NOx (LNT), катализатор для обедненных NOx (LNC), катализатор селективного каталитического восстановления (SCR), катализатор окисления дизельного топлива (DOC), катализированный сажевый фильтр (CSF), катализатор фильтра селективного каталитического восстановления (SCRFTM), катализатор, предотвращающий проскакивание аммиака, (ASC) и комбинации двух их или более. Такие устройства для регулирования выбросов все хорошо известны в данной области техники.
Некоторые из вышеуказанных устройств для регулирования выбросов имеют фильтрующие основы. Устройство для регулирования выбросов, имеющее фильтрующую основу, может быть выбрано из группы, состоящей из фильтра твердых частиц выхлопа дизельного двигателя (DPF), катализированного сажевого фильтра (CSF) и катализатора фильтра селективного каталитического восстановления (SCRFTM).
Предпочтительно, чтобы система выпуска выхлопных газов содержала устройство для регулирования выбросов, выбранное из группы, состоящей из ловушки для обедненных NOx (LNT), катализатора, предотвращающего проскакивание аммиака, (ASC), фильтра твердых частиц выхлопа дизельного двигателя (DPF), катализатора селективного каталитического восстановления (SCR), катализированного сажевого фильтра (CSF), катализатора фильтра селективного каталитического восстановления (SCRF™) и комбинаций двух их или более. Более предпочтительно, устройство для регулирования выбросов выбирают из группы, состоящей из фильтра твердых частиц выхлопа дизельного двигателя (DPF), катализатора селективного каталитического восстановления (SCR), катализированного сажевого фильтра (CSF), катализатора фильтра селективного каталитического восстановления (SCRF™) и комбинаций двух их или более. Еще более предпочтительно, устройство для регулирования выбросов является катализатором селективного каталитического восстановления (SCR) или катализатором фильтра селективного каталитического восстановления (SCRF™).
Когда система выпуска выхлопных газов по данному изобретению содержит катализатор селективного каталитического восстановления (SCR) фильтра селективного каталитического восстановления (SCRF™), то система выпуска выхлопных газов может дополнительно содержать инжектор для инжекции азотсодержащего восстановителя, такого как аммиак, или предшественника аммиака, такого как мочевина или формиат аммония, предпочтительно мочевины, в выхлопные газы ниже по течению потока по отношению к катализатору окисления и выше по течению потока по отношению к катализатору селективного каталитического восстановления (SCR) или катализатору фильтра селективного каталитического восстановления (SCRF™). Такой инжектор может быть соединен с возможностью протекания текучей среды с источником (например, резервуаром) предшественника азотсодержащего восстановителя. Дозирование с дроссельным регулированием предшественника в поток выхлопных газов может быть отрегулировано посредством средства управления работой двигателя, запрограммированного подходящим образом, и закрытого контура или открытого контура обратной связи, снабженного датчиками, контролирующими состав выхлопных газов. Аммиак может также быть образован нагреванием карбамата аммония (твердотельного), и образованный аммиак может быть инжектирован в выхлопные газы.
В качестве альтернативы или в дополнение к инжектору, аммиак может быть образован на месте (например, во время регенерации ловушки для обедненных NOx (LNT), расположенной выше по течению потока по отношению к катализатору селективного каталитического восстановления (SCR) или катализатору фильтра селективного каталитического восстановления (SCRF™). Соответственно, система выпуска выхлопных газов может дополнительно содержать средство управления работой двигателя для обогащения выхлопных газов углеводородами.
Катализатор селективного каталитического восстановления (SCR) или катализатор фильтра селективного каталитического восстановления (SCRFTM) может содержать по меньшей мере один металл, выбранный из группы, состоящей из Cu, Hf, La, Au, In, V, лантаноидов и переходных металлов Группы VIII (например, Fe), при этом данный металл поддерживается на тугоплавком оксиде или молекулярном сите. Металл предпочтительно выбирают из Ce, Fe, Cu и комбинаций любых двух или более этих металлов, более предпочтительно металл является Fe или Cu.
Огнеупорный оксид для катализатора селективного каталитического восстановления (SCR) или катализатора фильтра селективного каталитического восстановления (SCRFTM) может быть выбран из группы, состоящей из Al2O3, TiO2, CeO2, SiO2, ZrO2 и смешанных оксидов, содержащих два их или более. Нецеолитовый катализатор может также включать оксид вольфрама (например, V2O5/WO3/TiO2, WOx/CeZrO2, WOx/ZrO2 или Fe/WOx/ZrO2).
Является особенно предпочтительным, когда катализатор селективного каталитического восстановления (SCR), катализатор фильтра селективного каталитического восстановления (SCRFTM) или покрытие из пористого оксида «washcoat» с ним содержит по меньшей мере одно молекулярное сито, такое как алюмосиликатный цеолит или цеолит SAPO. По меньшей мере одно молекулярное сито может быть молекулярным ситом с малым, средним или большим размером пор. Под «молекулярным ситом с малым размером пор» в данном документе мы подразумеваем молекулярные сита, содержащие максимальный размер кольца из 8 атомов, такие как CHA; под «молекулярным ситом со средним размером пор» в данном документе мы подразумеваем молекулярное сито, содержащее максимальный размер кольца из 10 атомов, такое как ZSM-5; и под «молекулярным ситом с большим размером пор» в данном документе мы подразумеваем молекулярное сито имеющее максимальный размер кольца из 12 атомов, такое как бета. Молекулярные сита с малым размером пор являются потенциально выгодными для применения в катализаторах селективного каталитического восстановления (SCR).
В системе выпуска выхлопных газов по данному изобретению, предпочтительными молекулярными ситами для катализатора селективного каталитического восстановления (SCR) или катализатора фильтра селективного каталитического восстановления (SCRF™) являются синтетические алюмосиликатные цеолитовые молекулярные сита, выбранные из группы, состоящей из AEI, ZSM-5, ZSM-20, ERI, включая ZSM-34, морденита, ферриерита, BEA, включая бета, Y, CHA, LEV, включая Nu-3, MCM-22 и EU-1, предпочтительно AEI или CHA, и имеющие отношение кремнезема к глинозему от примерно 10 до примерно 50, например, от примерно 15 до примерно 40.
В первом варианте осуществления системы выпуска выхлопных газов, система выпуска выхлопных газов содержит катализатор окисления по данному изобретению, предпочтительно такой как катализатор окисления дизельного топлива (DOC), и катализированный сажевый фильтр (CSF). Такое расположение может быть названо как катализатор окисления дизельного топлива (DOC)/катализированный сажевый фильтр (CSF). За катализатором окисления (например, расположенным выше по течению потока) обычно следует катализированный сажевый фильтр (CSF). Соответственно, например, выпускное отверстие катализатора окисления соединено с впускным отверстием катализированного сажевого фильтра.
Во втором варианте осуществления системы выпуска выхлопных газов, система выпуска выхлопных газов содержит катализатор окисления дизельного топлива и катализатор окисления по данному изобретению, предпочтительно такой как катализированный сажевый фильтр (CSF). Это расположение может также называться расположением катализатор окисления дизельного топлива (DOC)/катализированный сажевый фильтр (CSF). Как правило, за катализатором окисления дизельного топлива (DOC) (например, расположенным выше по течению потока) следует катализатор окисления по данному изобретению. Соответственно, выпускное отверстие катализатора окисления дизельного топлива соединено с впускным отверстием катализатора окисления по данному изобретению.
Третий вариант осуществления системы выпуска выхлопных газов относится к системе выпуска выхлопных газов, содержащей катализатор окисления по данному изобретению, предпочтительно такой как катализатор окисления дизельного топлива (DOC), катализированный сажевый фильтр (CSF) и катализатор селективного каталитического восстановления (SCR). Такое расположение может быть названо как катализатор окисления дизельного топлива (DOC)/катализированный сажевый фильтр (CSF)/катализатор селективного каталитического восстановления (SCR), и оно является предпочтительной системой выпуска выхлопных газов для малотоннажных транспортных средств с дизельным двигателем. За катализатором окисления (например, расположенным выше по течению потока) обычно следует катализированный сажевый фильтр (CSF). За катализированным сажевым фильтром (например, расположенным выше по течению потока) обычно следует катализатор селективного каталитического восстановления (SCR). Инжектор азотсодержащего восстановителя может быть расположен между катализированным сажевым фильтром (CSF) и катализатором селективного каталитического восстановления (SCR). Соответственно, за катализированным сажевым фильтром (CSF) (например, расположенным выше по течению потока) может следовать инжектор азотсодержащего восстановителя, и за данным инжектором азотсодержащего восстановителя (например, расположенным выше по течению потока) может следовать катализатор селективного каталитического восстановления (SCR).
Четвертый вариант осуществления системы выпуска выхлопных газов относится к системе выпуска выхлопных газов, содержащей катализатор окисления дизельного топлива (DOC), катализатор окисления по данному изобретению, предпочтительно такой как катализированный сажевый фильтр (CSF), и катализатор селективного каталитического восстановления (SCR). Это является также расположением катализатор окисления дизельного топлива (DOC)/катализированный сажевый фильтр (CSF)/катализатор селективного каталитического восстановления (SCR). Как правило, за катализатором окисления дизельного топлива (DOC) (например, расположенным выше по течению потока) следует катализатор окисления по данному изобретению. За катализатором окисления по данному изобретению (например, расположенным выше по течению потока) обычно следует катализатор селективного каталитического восстановления (SCR). Инжектор азотсодержащего восстановителя может быть расположен между катализатором окисления и катализатором селективного каталитического восстановления (SCR). Соответственно, за катализатором окисления (например, расположенным выше по течению потока) может следовать инжектор азотсодержащего восстановителя, и за данным инжектором азотсодержащего восстановителя (например, расположенным выше по течению потока) может следовать катализатор селективного каталитического восстановления (SCR).
В пятом варианте осуществления системы выпуска выхлопных газов, система выпуска выхлопных газов содержит катализатор окисления по данному изобретению, предпочтительно такой как катализатор окисления дизельного топлива (DOC), катализатор селективного каталитического восстановления (SCR) и катализированный сажевый фильтр (CSF) или фильтр твердых частиц выхлопа дизельного двигателя (DPF). Данное расположение является расположением катализатор окисления дизельного топлива (DOC)/катализатор селективного каталитического восстановления (SCR)/катализированный сажевый фильтр (CSF) или расположением катализатор окисления дизельного топлива (DOC)/катализатор селективного каталитического восстановления (SCR)/фильтр твердых частиц выхлопа дизельного двигателя (DPF).
В пятом варианте осуществления системы выпуска выхлопных газов, за катализатором окисления по данному изобретению (например, расположенным выше по течению потока) обычно следует катализатор селективного каталитического восстановления (SCR). Инжектор азотсодержащего восстановителя может быть расположен между катализатором окисления и катализатором селективного каталитического восстановления (SCR). Соответственно, за катализатором окисления (например, расположенным выше по течению потока) может следовать инжектор азотсодержащего восстановителя, и за данным инжектором азотсодержащего восстановителя (например, расположенным выше по течению потока) может следовать катализатор селективного каталитического восстановления (SCR). За катализатором селективного каталитического восстановления (SCR) (например, расположенным выше по течению потока) следует катализированный сажевый фильтр (CSF) или фильтр твердых частиц выхлопа дизельного двигателя (DPF).
Шестой вариант осуществления системы выпуска отработавших газов содержит катализатор окисления по данному изобретению, предпочтительно такой как катализатор окисления дизельного топлива (DOC), и катализатор фильтра селективного каталитического восстановления (SCRF™). Такое расположение может быть названо как катализатор окисления дизельного топлива (DOC)/катализатор фильтра селективного каталитического восстановления (SCRF™). За катализатором окисления по данному изобретению (например, расположенным выше по течению потока) обычно следует катализатор фильтра селективного каталитического восстановления (SCRFTM). Инжектор азотсодержащего восстановителя может быть расположен между катализатором окисления и катализатором фильтра селективного каталитического восстановления (SCRF™). Соответственно, за катализатором окисления (например, расположенным выше по течению потока) может следовать инжектор азотсодержащего восстановителя, и за данным инжектором азотсодержащего восстановителя (например, расположенным выше по течению потока) может следовать катализатор фильтра селективного каталитического восстановления (SCRFTM).
В каждом из с третьего по шестой вариантов осуществления системы выпуска отработавших газов, описанных в данном документе выше, катализатор, предотвращающий проскакивание аммиака, (ASC) может быть расположен ниже по течению потока по отношению к катализатору селективного каталитического восстановления (SCR) или катализатору фильтра селективного каталитического восстановления (SCRF™) (т.е. в качестве отдельной монолитной основы), или, более предпочтительно, зона ниже по течению потока или примыкающая к концу монолитной основы, содержащей катализатор селективного каталитического восстановления (SCR), может быть использована в качестве носителя для катализатора, предотвращающего проскакивание аммиака, (ASC).
Другой аспект данного изобретения относится к транспортному средству или устройству. Транспортное средство или устройство содержит дизельный двигатель. Дизельный двигатель может быть двигателем с воспламенением от сжатия однородной смеси (HCCI), двигателем с воспламенением от сжатия предварительно перемешанной смеси (PCCI) или двигателем со сгоранием при низкой температуре (LTC). Предпочтительным является, чтобы дизельный двигатель являлся обычным (т.е. традиционным) дизельным двигателем.
Транспортное средство может являться малотоннажным транспортным средством с дизельным двигателем (LDV), таким как определено в законодательстве США или Европы. Малотоннажное транспортное средство с дизельным двигателем обычно имеет массу <2840 кг, более предпочтительно массу <2610 кг.
В США к малотоннажному транспортному средству с дизельным двигателем (LDV) относится дизельное транспортное средство, имеющее полный вес≤8500 фунтов (≤3856 кг). В Европе термин «малотоннажное транспортное средство с дизельным двигателем (LDV)» относится к (i) пассажирским транспортным средствам, имеющим не более чем восемь сидений в дополнение к сиденью водителя и имеющим максимальную массу, не превышающую 5 тонн, и (ii) транспортным средствам для перевозки грузов, имеющим максимальную массу, не превышающую 12 тонн.
В качестве альтернативы, транспортное средство может являться большегрузным транспортным средством с дизельным двигателем (HDV), таким как дизельное транспортное средство, имеющее полный вес >8500 фунтов (>3856 кг), как определено в законодательстве США.
Данное изобретение также предоставляет способ регулирования содержания NOx в выхлопных газах от дизельного двигателя для устройства для регулирования выбросов. Стадия (b) пропускания обработанных выхлопных газов в устройство для регулирования выбросов обычно включает пропускание непосредственным образом обработанных выхлопных газов в устройство для регулирования выбросов. Соответственно, выпускное отверстие катализатора окисления соединено непосредственно (например, без промежуточных узлов) с впускным отверстием устройства для регулирования выбросов.
Как правило, устройство для регулирования выбросов является катализатором селективного каталитического восстановления (SCR), катализатором фильтра селективного каталитического восстановления (SCRF™), фильтром твердых частиц выхлопа дизельного двигателя (DPF) или катализированным сажевым фильтром (CSF). Предпочтительно, устройство для регулирования выбросов является катализатором селективного каталитического восстановления (SCR) или катализатором фильтра селективного каталитического восстановления (SCRF™).
Любая ссылка на «регулирование содержания NOx», как использовано в данном документе, особенно в отношении аспектов способа или применения данного изобретения, относится к изменению (т.е. регулированию) или поддержанию, предпочтительно изменению, отношения (в млн-1 или об.%, обычно при температуре и давлении выхлопных газов) NO:NO2 в пределах предписанного интервала отношения при определенной температуре или в определенном температурном интервале выхлопных газов. Предписанное отношение обычно составляет менее чем 17:3, предпочтительно составляет от 5:1 до 1:5, более предпочтительно от 2,5:1 до 1:2,5 и даже еще более предпочтительно от 2:1 до 1:2 (например, от 1,5:1 до 1:1,5 или примерно 1:1).
Данное изобретение также относится к применению катализатора окисления при регенерации устройства для регулирования выбросов, имеющего фильтрующую основу (например, устройства для регулирования выбросов ниже по течению потока, имеющего фильтрующую основу).
Устройство для регулирования выбросов, имеющее фильтрующую основу, может быть выбрано из группы, состоящей из фильтра твердых частиц выхлопа дизельного двигателя (DPF), катализированного сажевого фильтра (CSF), катализатора фильтра селективного каталитического восстановления (SCRF™) и комбинации двух их или более.
Когда катализатор окисления по данному изобретению применяют при регенерации устройства для регулирования выбросов, имеющего фильтрующую основу, он может быть применен для активной или пассивной регенерации устройства для регулирования выбросов, предпочтительно для активной регенерации.
Катализатор окисления может быть использован, чтобы регенерировать устройство для регулирования выбросов, имеющее фильтрующую основу, при температуре по меньшей мере 220°С, предпочтительно по меньшей мере 240°С, более предпочтительно по меньшей мере 260°С, еще более предпочтительно по меньшей мере 280°С, посредством окисления оксида азота (NO) до диоксида азота (NO2).
ОПРЕДЕЛЕНИЯ
Термин «покрытие из пористого оксида «washcoat»» хорошо известен в данной области техники и относится к присоединенному покрытию, которое нанесено на основу обычно во время изготовления катализатора.
Термин «область покрытия из пористого оксида «washcoat»», как использовано в данном документе, относится к области, занимаемой покрытием из пористого оксида «washcoat» на основе. «Область покрытия из пористого оксида «washcoat»» может, например, быть размещена или поддерживаться на основе в виде «слоя» или «зоны». Площадь или расположение покрытия из пористого оксида «washcoat» на основе обычно регулируют во время процесса нанесения покрытия из пористого оксида «washcoat» на основу. «Область покрытия из пористого оксида «washcoat»» обычно имеет отчетливые границы или края (т.е. возможно отличить одну область покрытия из пористого оксида «washcoat» от другой области покрытия из пористого оксида «washcoat» при применении обычных аналитических методов).
Как правило, «область покрытия из пористого оксида «washcoat»» имеет по существу одинаковую длину. Ссылка на «по существу одинаковую длину» в этом контексте относится к длине, которая не отклоняется (например, в отношении разницы между максимальной и минимальной длиной) на более чем 10%, предпочтительно не отклоняется на более чем на 5%, более предпочтительно не отклоняется более чем на 1%, от ее средней величины.
Предпочтительным является, чтобы каждая «область покрытия из пористого оксида «washcoat»» имела по существу однородный состав (т.е. отсутствовала существенная разница в составе покрытия из пористого оксида «washcoat» при сравнении одной части области покрытия из пористого оксида «washcoat» с другой частью данной области покрытия из пористого оксида «washcoat»). «По существу однородный состав» в этом контексте относится к материалу (например, области покрытия из пористого оксида «washcoat»), в котором разница в составе при сравнении одной части области покрытия из пористого оксида «washcoat» с другой частью области покрытия из пористого оксида «washcoat» составляет 5% или менее, обычно 2,5% или менее, и наиболее обычно 1% или менее.
Термин «зона покрытия из пористого оксида «washcoat»», как использовано в данном документе, относится к области покрытия из пористого оксида «washcoat», имеющей длину, которая меньше, чем полная длина основы, например, ≤75% от полной длины основы. «Зона покрытия из пористого оксида «washcoat»» обычно имеет длину (т.е. по существу одинаковую длину) по меньшей мере 5% (например, ≥5%) от полной длины основы.
Общая длина основы представляет собой расстояние между ее впускным концом и ее выпускным концом (например, между противоположными концами основы).
Любая ссылка на «зону покрытия из пористого оксида «washcoat», расположенную на впускном конце основы», использованная в данном документе, относится к зоне покрытия из пористого оксида «washcoat», размещенной или поддерживаемой на основе, когда зона покрытия из пористого оксида «washcoat» ближе к впускному концу основы, чем к выпускному концу основы. Соответственно, средняя точка зоны покрытия из пористого оксида «washcoat» (т.е. на половине ее длины) находится ближе к впускному концу основы, чем к выпускному концу основы. Аналогичным образом, любая ссылка на «зону покрытия из пористого оксида «washcoat», расположенную на выпускном конце основы», использованная в данном документе, относится к зоне покрытия из пористого оксида «washcoat», размещенной или поддерживаемой на основе, когда зона покрытия из пористого оксида «washcoat» ближе к выпускному концу основы, чем к впускному концу основы. Соответственно, средняя точка зоны покрытия из пористого оксида «washcoat» (т.е. на половине ее длины) находится ближе к выпускному концу основы, чем к впускному концу основы.
Когда основа является фильтром с протеканием через стенки, то обычно любая ссылка на «зону покрытия из пористого оксида «washcoat», размещенную на впускном конце основы» относится к зоне покрытия из пористого оксида «washcoat», размещенной или поддерживаемой на основе, которая:
(a) ближе к впускному концу (например, открытому концу) впускного канала основы, чем данная зона покрытия из пористого оксида «washcoat» расположена по отношению к закрытому концу (например, блокированному или закупоренному концу) данного впускного канала, и/или
(b) ближе к закрытому концу (например, блокированному или закупоренному концу) выпускного канала основы, чем данная зона покрытия из пористого оксида «washcoat» расположена по отношению к выпускному концу (например, открытому концу) данного выпускного канала.
Соответственно, средняя точка зоны покрытия из пористого оксида «washcoat» (т.е. на половине ее длины) (a) ближе к впускному концу впускного канала основы, чем к закрытому концу впускного канала, и/или (b) ближе к закрытому концу выпускного канала основы, чем к выпускному концу выпускного канала.
Аналогичным образом, любая ссылка на «зону покрытия из пористого оксида «washcoat», размещенную на выпускном конце основы», когда основа является фильтром с протеканием через стенки, относится к зоне покрытия из пористого оксида «washcoat», размещенной или поддерживаемой на основе, которая:
(a) ближе к выпускному концу (например, открытому концу) выпускного канала основы, чем данная зона покрытия из пористого оксида «washcoat» расположена по отношению к закрытому концу (например, блокированному или закупоренному концу) данного выпускного канала, и/или
(b) ближе к закрытому концу (например, блокированному или закупоренному концу) впускного канала основы, чем данная зона покрытия из пористого оксида «washcoat» расположена по отношению к впускному концу (например, открытому концу) данного впускного канала.
Соответственно, средняя точка зоны покрытия из пористого оксида «washcoat» (т.е. на половине ее длины) (a) ближе к выпускному концу выпускного канала основы, чем к закрытому концу выпускного канала, и/или (b) ближе к закрытому концу впускного канала основы, чем к впускному концу впускного канала.
Зона покрытия из пористого оксида «washcoat» может удовлетворять как (a), так и (b), когда покрытие из пористого оксида «washcoat» присутствует в виде стенки фильтра с протеканием через стенки (т.е. зона покрытия из пористого оксида «washcoat» является внутренней стенкой).
Сокращение «МПГ», как оно используется в данном документе, относится к «металлу платиновой группы». Термин «металл платиновой группы» обычно относится к металлу, выбранному из группы, состоящей из Ru, Rh, Pd, Os, Ir и Pt, предпочтительно металлу, выбранному из группы, состоящей из Ru, Rh, Pd, Ir и Pt. Как правило, термин «МПГ» предпочтительно относится к металлу, выбранному из группы, состоящей из Rh, Pt и Pd.
Термин «смешанный оксид», как он используется в данном документе, обычно относится к смеси оксидов в одной фазе, как это обычно известно в данной области техники. Термин «сложный оксид», как использовано в данном документе, обычно относится к композиции оксидов, имеющей более чем одну фазу, как это обычно известно в данной области техники.
Любая ссылка на зоны покрытия из пористого оксида «washcoat», которые не «наложены существенным образом одна на другую», как использовано в данном документе, относится к частичному наложению (т.е. расстоянию между концами соседних зон на основе) менее чем 10% от длины основы, предпочтительно менее 7,5% от длины основы, более предпочтительно менее чем 5% от длины основы, особенно менее чем 2,5% от длины основы, даже еще более предпочтительно менее чем 1% от длины основы, и наиболее предпочтительно они вообще не перекрываются.
Выражение «состоять по существу из», как оно используется в данном документе, ограничивает объем признака, означая включение конкретных материалов или стадий и любых других материалов или стадий, которые по существу не влияют на базовые характеристики этого признака, таких как, например, минорные примеси. Выражение «состоять по существу из» охватывает выражение «состоять из».
Выражение «по существу не содержит», как использовано в данном документе, при ссылке на материал, обычно применительно к содержанию компонентов области покрытия из пористого оксида «washcoat», слоя покрытия из пористого оксида «washcoat» или зоны покрытия из пористого оксида «washcoat», означает, что данный материал содержится в незначительном количестве, таком как≤5% по массе, предпочтительно≤2% по массе, более предпочтительно≤1% по массе. Выражение «по существу не содержит» охватывает выражение «не содержит».
Любая ссылка на количество легирующей примеси, особенно общее количество, выраженное как % по массе, как использовано в данном документе, относится к массе материала-носителя или его огнеупорного оксида металла.
ПРИМЕРЫ
Изобретение будет теперь проиллюстрировано представленными ниже неограничивающими примерами.
Пример 1
Порошок кремнезема-оксида алюминия суспендировали в воде и измельчали до d90<20 микрон. Затем добавляли растворимую соль платины и смесь перемешивали, чтобы гомогенизировать ее. Суспензию наносили на кордиеритовый проточной монолит, имеющий 400 ячеек на квадратный дюйм, при применении общепринятых способов нанесения покрытия. Опытный образец сушили и обжигали при 500°С. Загрузка Pt на образец составляла 30 г/фут3 (1,059 кг/м3).
Пример 2
Способ Примера 1 повторяли, за исключением того, что после добавления растворимой соли платины добавляли нитрат марганца перед перемешиванием смеси для ее гомогенизации. Загрузка Pt на образец составляла 30 г/фут3 (1,059 кг/м3). Загрузка марганца на образец составляла 40 г/фут3 (1,413 кг/м3). Массовое отношение Pt к марганцу составляло 3:4.
Пример 3
Использовали способ Примера 2, за тем исключением, что порошок оксида алюминия суспендировали в воде и измельчали до d90<20 микрон вместо порошка кремнезема-оксида алюминия. Загрузка Pt на образец составляла 30 г/фут3 (1,059 кг/м3). Загрузка марганца на образец составляла 40 г/фут3 (1,413 кг/м3). Массовое отношение Pt к марганцу составляло 3:4.
Пример 4
Порошок оксида алюминия, легированного кремнеземом, суспендировали в воде и измельчали до d90<20 микрон. Ацетат бария добавляли к суспензии с последующим добавлением соответствующих количеств растворимых солей платины и палладия. Бета-цеолит добавляли таким образом, чтобы суспензия содержала 78% оксида алюминия, легированного кремнеземом, и 22% цеолита по массе. Суспензию затем перемешивали для ее гомогенизации. Полученный материал покрытия из пористого оксида «washcoat» наносили на впускные каналы кордиеритового проточного монолита, имеющего 400 ячеек на квадратный дюйм, при применении общепринятых способов нанесения покрытия, и затем сушили.
Вторую суспензию приготавливали посредством применения порошка оксида алюминия, легированного кремнеземом, и измельчения его до d90<20 микрон. Растворимую соль платины добавляли к суспензии с последующим добавлением раствора нитрата марганца. Суспензию перемешивали для ее гомогенизации. Эту вторую суспензию наносили на выпускные каналы проточного монолита при применении обычных способов нанесения покрытия. Опытный образец сушили и затем обжигали при 500°С. Конечный катализатор имел общую загрузку МПГ 75 г/фут3 (2,649 кг/м3) и загрузку марганца на покрытии выпускных каналов 100 г/фут3 (3,531 кг/м3). Массовое отношение платины к марганцу составляло 3:5.
Пример 5
Порошок оксида алюминия, легированного кремнеземом, суспендировали в воде и измельчали до d90<20 микрон. Ацетат бария добавляли к суспензии с последующим добавлением соответствующих количеств растворимых солей платины и палладия. Бета-цеолит добавляли таким образом, чтобы суспензия содержала 78% оксида алюминия, легированного кремнеземом, и 22% цеолита по массе. Суспензию затем перемешивали для ее гомогенизации. Полученный материал покрытия из пористого оксида «washcoat» наносили на впускные каналы кордиеритового проточного монолита, имеющего 400 ячеек на квадратный дюйм, при применении общепринятых способов нанесения покрытия, и затем сушили.
Вторую суспензию приготавливали посредством применения порошка оксида алюминия, легированного кремнеземом, и измельчения его до d90<20 микрон. Надлежащее количество растворимой соли платины добавляли к суспензии и перемешивали ее для гомогенизации. Эту вторую суспензию наносили на выпускные каналы проточного монолита при применении обычных способов нанесения покрытия. Опытный образец сушили и затем обжигали при 500°С. Конечный катализатор имел общую загрузку МПГ 75 г/фут3 (2,649 кг/м3).
Экспериментальные результаты
Измерение окисления NO
Керновые образцы извлекали из каждого из катализаторов Примеров 1-5. Все керны гидротермически «состаривали» в печи при 750°С в течение 15 часов. Дополнительные керны извлекали для Примеров 4 и 5 и поддерживали в «свежем» состоянии (т.е. их поддерживали без какой-либо термической обработки в печи).
Каталитическую активность определяли при применении стендового испытания на активность катализатора в отношении синтетического газа (SCAT). Свежие и состаренные керны испытывали в газовом устройстве для смоделированного испытания на активность катализатора (SCAT) при применении вводимых газовых смесей, представленных в Таблице 1. В каждом случае остатком являлся азот.
Таблица 1
Результаты
Результаты смоделированных испытаний на активность катализатора в отношении синтетического газа (SCAT) представлены в Таблицах 2-4 ниже.
Таблица 2
(Состаренное состояние)
Результаты в Таблице 2 показывают выполнение окисления NO для Примеров 1, 2 и 3 при 250°С. Пример 1 (не содержит марганец) имеет наиболее низкую активность окисления NO. Пример 2 (содержит марганец и носитель из кремнезема-оксида алюминия) имеет наиболее высокую активность окисления NO. Пример 3 (содержит марганец и носитель из оксида алюминия) имеет среднюю активность окисления NO. Включение марганца приводит к улучшенному выполнению окисления NO. Результаты для Примера 2 показывают, что комбинация марганца с материалом-носителем, содержащим легированный оксид алюминия, такой как оксид алюминия, легированный кремнеземом, является особенно выгодной.
Результаты для Примеров 4 и 5 представлены в Таблицах 3 и 4.
Таблица 3
(Свежее состояние)
(Состаренное состояние)
Результаты в Таблице 3 показывают, что катализатор Примера 4 (содержит марганец в зоне Pt) в состаренном состоянии при 200°С имеет более высокую степень окисления NO, чем катализатор Примера 5.
Таблица 4
(Свежее состояние)
(Состаренное состояние)
Результаты в Таблице 4 показывают, что катализатор Примера 4 в состаренном состоянии при 220°С показывает более высокую активность окисления NO, чем катализатор Примера 5.
Таблицы 3 и 4 также показывают, что разница в активности окисления NO между свежим и состаренным состояниями катализатора меньше для Примера 4, чем для Примера 5. Соответственно, катализатор Примера 4 показывает более стабильное выполнение окисления NO, чем Пример 5. Стабильное выполнение окисления NO между свежим и состаренным состояниями является выгодным для калибровок дозирования катализатора селективного каталитического восстановления (SCR) ниже по течению потока.
Пример 6
Нитрат Pd добавляли к суспензии цеолита с малым размером пор со структурой CHA и перемешивали. Суспензию наносили на кордиеритовый проточный монолит, имеющий структуру с 400 ячейками на квадратный дюйм, при применении общепринятых способов нанесения покрытия. Покрытие сушили и обжигали при 500°С. При этом получали покрытие, содержащее Pd-замещенный цеолит. Загрузка Pd в этом покрытии составляла 40 г/фут3 (1,413 кг/м3).
Вторую суспензию приготавливали при применении порошка кремнезема-оксида алюминия, измельченного до d90<20 микрон. Затем добавляли растворимую соль платины и смесь перемешивали, чтобы гомогенизировать ее. Суспензию наносили на выпускной конец проточного монолита при применении общепринятых способов нанесения покрытия. Покрытие затем сушили.
Третью суспензию приготавливали при применении порошка кремнезема-оксида алюминия, измельченного до d90<20 микрон. После добавления соответствующих количеств растворимых солей платины и палладия добавляли бета-цеолит таким образом, что суспензия содержала 75% оксида алюминия, легированного кремнеземом, и 25% цеолит по массе. Суспензию затем перемешивали для ее гомогенизации. Полученное покрытие из пористого оксида «washcoat» наносили на впускные каналы проточного монолита при применении общепринятых способов нанесения покрытия. Опытный образец затем сушили и обжигали при 500°С. Загрузка Pt в конечном образце составляла 51 г/фут3 (1,801 кг/м3) и загрузка Pd составляла 47,5 г/фут3 (1,677 кг/м3).
Пример 7
Нитрат Pd добавляли к суспензии цеолита с малым размером пор со структурой CHA и перемешивали. Суспензию наносили на кордиеритовый проточный монолит, имеющий структуру с 400 ячейками на квадратный дюйм, при применении общепринятых способов нанесения покрытия. Покрытие сушили и обжигали при 500°С. При этом получали покрытие, содержащее Pd-замещенный цеолит. Загрузка Pd в этом покрытии составляла 40 г/фут3 (1,413 кг/м3).
Вторую суспензию приготавливали при применении 5% марганца, поддерживаемого на порошке кремнезема-оксида алюминия, измельченном до d90<20 микрон. Затем добавляли растворимую соль платины и смесь перемешивали, чтобы гомогенизировать ее. Суспензию наносили на выпускной конец проточного монолита при применении общепринятых способов нанесения покрытия. Покрытие затем сушили.
Третью суспензию приготавливали при применении порошка кремнезема-оксида алюминия, измельченного до d90<20 микрон. После добавления соответствующих количеств растворимых солей платины и палладия добавляли бета-цеолит таким образом, что суспензия содержала 75% оксида алюминия, легированного кремнеземом, и 25% цеолит по массе. Суспензию затем перемешивали для ее гомогенизации. Полученное покрытие из пористого оксида «washcoat» наносили на впускные каналы проточного монолита при применении общепринятых способов нанесения покрытия. Опытный образец затем сушили и обжигали при 500°С. Загрузка Pt в конечном образце составляла 51 г/фут3 (1,801 кг/м3) и загрузка Pd составляла 47,5 г/фут3 (1,677 кг/м3).
Экспериментальные результаты
Измерение окисления NO
Керновые образцы извлекали из катализаторов Примеров 6 и 7. Оба керновых образца гидротермически состаривали в печи при 800°С в течение 16 часов в 10% воды.
Каталитическую активность определяли при применении стендового испытания на активность катализатора в отношении синтетического газа (SCAT). Состаренные керны испытывали в газовом устройстве для смоделированного испытания на активность катализатора (SCAT) при применении вводимых газовых смесей, представленных в Таблице 5. В каждом случае остатком являлся азот.
Таблица 5
Результаты
Результаты смоделированного испытания на активность катализатора (SCAT) представлены в Табл. 6 ниже.
Таблица 6
Результаты в Таблице 6 показывают степень окисления NO для Примеров 6 и 7 при 250°С. Пример 7, который содержит марганец, показывает более высокий уровень окисления NO при 250°С, чем Пример 6.
Во избежание каких-либо сомнений, полное содержание всех без исключения документов, цитированных в данном документе, включено посредством ссылки в представленную заявку.
| название | год | авторы | номер документа |
|---|---|---|---|
| АДСОРБЕР-КАТАЛИЗАТОР NO | 2017 |
|
RU2759725C2 |
| КАТАЛИЗАТОР ДЛЯ ХОЛОДНОГО ПУСКА И ЕГО ПРИМЕНЕНИЕ В ВЫХЛОПНЫХ СИСТЕМАХ | 2012 |
|
RU2612136C2 |
| СИСТЕМА КАТАЛИЗАТОРА ДЛЯ БЕНЗИНОВЫХ ДВИГАТЕЛЕЙ С ПРЯМЫМ ВПРЫСКОМ, РАБОТАЮЩИХ НА ОБЕДНЕННОЙ ТОПЛИВНОЙ СМЕСИ | 2016 |
|
RU2729060C2 |
| КАТАЛИЗАТОР ДЛЯ ОБРАБОТКИ ВЫХЛОПНЫХ ГАЗОВ | 2014 |
|
RU2675821C2 |
| КАТАЛИЗАТОР ОКИСЛЕНИЯ ДЛЯ ДВИГАТЕЛЯ С ВОСПЛАМЕНЕНИЕМ ОТ СЖАТИЯ | 2014 |
|
RU2721563C2 |
| КАТАЛИЗАТОР НА ОСНОВЕ МЕТАЛЛА ПЛАТИНОВОЙ ГРУППЫ (МПГ) ДЛЯ ОБРАБОТКИ ВЫХЛОПНЫХ ГАЗОВ | 2012 |
|
RU2665464C2 |
| КАТАЛИЗАТОР ДЛЯ ОБРАБОТКИ ВЫХЛОПНЫХ ГАЗОВ | 2014 |
|
RU2771714C2 |
| КАТАЛИЗАТОР, ПРЕДОТВРАЩАЮЩИЙ ПРОСКОК АММИАКА, СПРОЕКТИРОВАННЫЙ В КАЧЕСТВЕ ПЕРВОГО В СИСТЕМЕ СЕЛЕКТИВНОГО КАТАЛИТИЧЕСКОГО ВОССТАНОВЛЕНИЯ (SCR) | 2016 |
|
RU2715653C2 |
| ОКИСЛИТЕЛЬНЫЙ КАТАЛИЗАТОР И СИСТЕМА ВЫПУСКА ВЫХЛОПНЫХ ГАЗОВ ДЛЯ ДИЗЕЛЬНОГО ДВИГАТЕЛЯ | 2015 |
|
RU2733407C2 |
| КОМПОЗИТ КАТАЛИЗАТОРА ОКИСЛЕНИЯ, СПОСОБ ОБРАБОТКИ ПОТОКА ВЫХЛОПНЫХ ГАЗОВ И СИСТЕМА ОБРАБОТКИ ПОТОКА ВЫХЛОПНЫХ ГАЗОВ | 2014 |
|
RU2685426C1 |
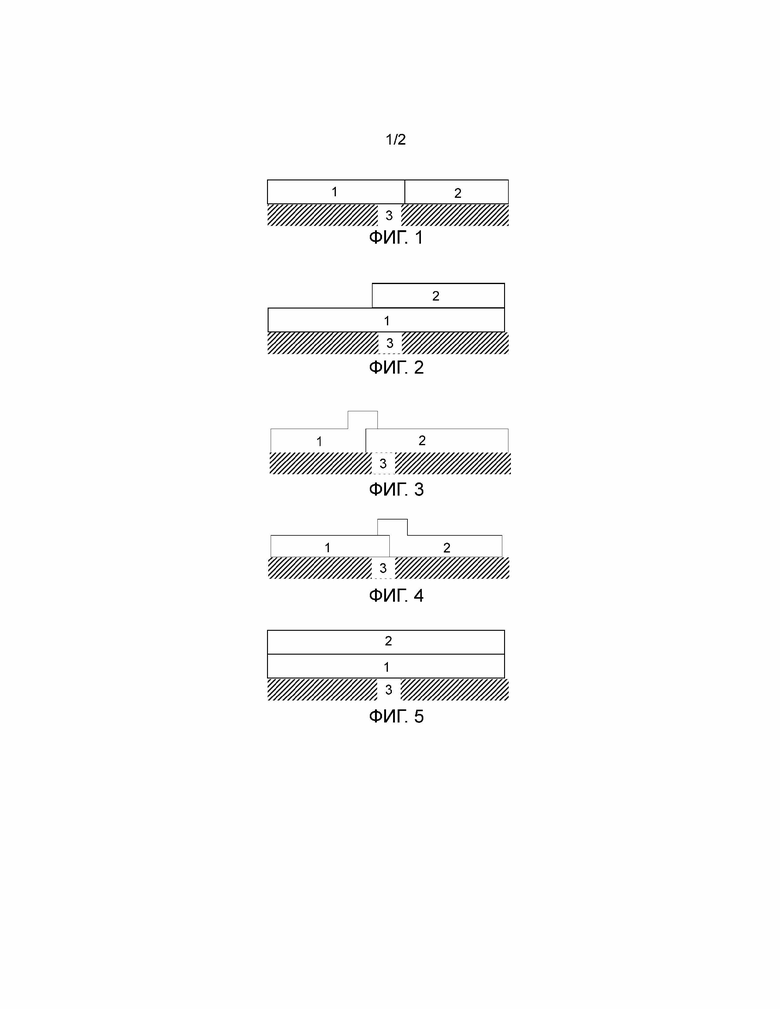
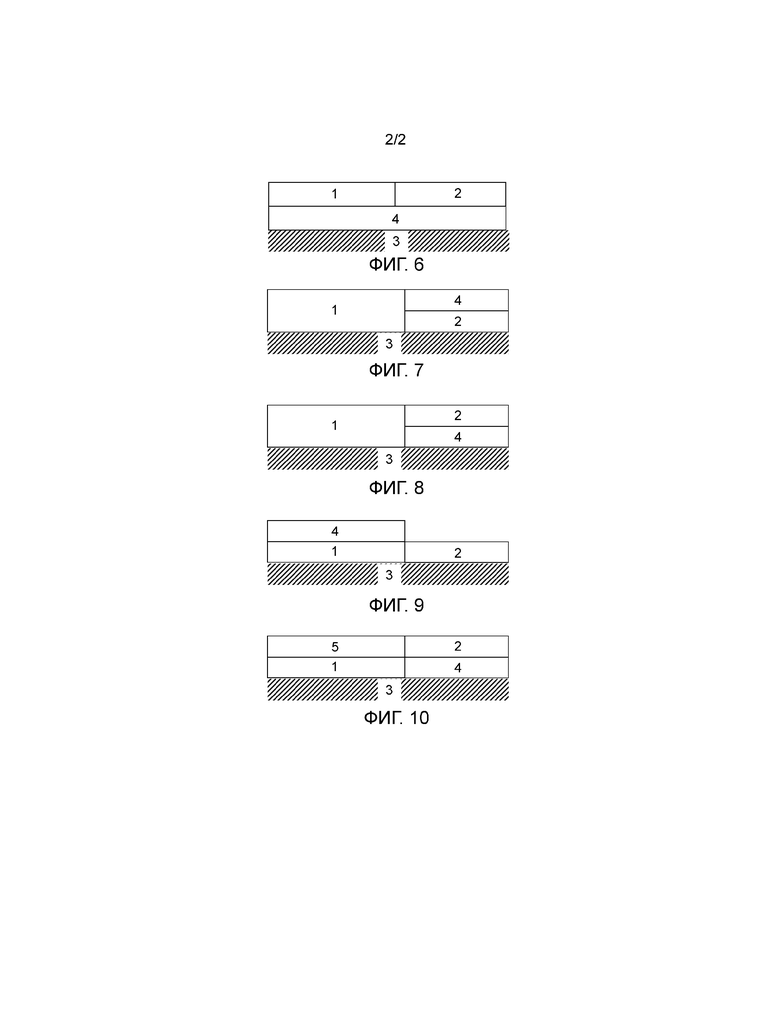
Изобретение относится к выхлопной системе для дизельного двигателя, включающей катализатор окисления для обработки выхлопных газов из дизельного двигателя и устройство контроля токсичности выхлопных газов, причем данный катализатор окисления содержит: область первого покрытия из пористого оксида для окисления монооксида углерода (CO) и углеводородов (HC), где область первого покрытия из пористого оксида содержит первый металл платиновой группы (МПГ) и первый материал-носитель, и где область первого покрытия не содержит марганца или его оксида; область второго покрытия из пористого оксида для окисления оксида азота (NO), где область второго покрытия из пористого оксида содержит платину (Pt), марганец (Mn) и второй материал-носитель, содержащий тугоплавкий оксид металла, который представляет собой диоксид кремния-оксид алюминия или оксид алюминия, легированный диоксидом кремния, где платина (Pt) размещена на втором материале-носителе или нанесена на второй материал-носитель и марганец (Mn) размещен на втором материале-носителе или нанесен на второй материал-носитель; и основу, имеющую впускной конец и выпускной конец, и где область первого покрытия из пористого оксида представляет собой зону первого покрытия из пористого оксида, размещенную на впускном конце основы, а область второго покрытия из пористого оксида представляет собой зону второго покрытия из пористого оксида, размещенную на выпускном конце основы, так что область второго покрытия из пористого оксида размещена таким образом, чтобы контактировать с выхлопными газами на выпускном конце основы и после контактирования выхлопных газов с областью первого покрытия из пористого оксида. Изобретение также относится к транспортному средству и способу обработки выхлопных газов из дизельного двигателя. Технический результат заключается в улучшении активности окисления NO или предоставления возможности применения уменьшенного количества Pt для достижения заданного уровня окисления NO. 3 н. и 25 з.п. ф-лы, 10 ил., 6 табл., 7 пр.
1. Выхлопная система для дизельного двигателя, включающая катализатор окисления для обработки выхлопных газов из дизельного двигателя и устройство контроля токсичности выхлопных газов, причем данный катализатор окисления содержит:
область первого покрытия из пористого оксида для окисления монооксида углерода (CO) и углеводородов (HC), где область первого покрытия из пористого оксида содержит первый металл платиновой группы (МПГ) и первый материал-носитель, и где область первого покрытия не содержит марганца или его оксида;
область второго покрытия из пористого оксида для окисления оксида азота (NO), где область второго покрытия из пористого оксида содержит платину (Pt), марганец (Mn) и второй материал-носитель, содержащий тугоплавкий оксид металла, который представляет собой диоксид кремния-оксид алюминия или оксид алюминия, легированный диоксидом кремния, где платина (Pt) размещена на втором материале-носителе или нанесена на второй материал-носитель и марганец (Mn) размещен на втором материале-носителе или нанесен на второй материал-носитель; и
основу, имеющую впускной конец и выпускной конец, и
где область первого покрытия из пористого оксида представляет собой зону первого покрытия из пористого оксида, размещенную на впускном конце основы, а область второго покрытия из пористого оксида представляет собой зону второго покрытия из пористого оксида, размещенную на выпускном конце основы, так что область второго покрытия из пористого оксида размещена таким образом, чтобы контактировать с выхлопными газами на выпускном конце основы и после контактирования выхлопных газов с областью первого покрытия из пористого оксида.
2. Выхлопная система по п. 1, в которой зона первого покрытия из пористого оксида и зона второго покрытия из пористого оксида расположены на основе в виде одиночного слоя.
3. Выхлопная система по п. 1, в которой область первого покрытия из пористого оксида является первым слоем покрытия из пористого оксида, и где зона второго покрытия из пористого оксида расположена на слое первого покрытия из пористого оксида.
4. Выхлопная система по п. 1, в которой область первого покрытия из пористого оксида является первым слоем покрытия из пористого оксида, а область второго покрытия из пористого оксида является слоем второго покрытия из пористого оксида, и где слой второго покрытия из пористого оксида расположен на слое первого покрытия из пористого оксида.
5. Выхлопная система по п. 4, в которой слой второго покрытия из пористого оксида распространяется на всю длину основы.
6. Выхлопная система по любому из пп. 1-4, в которой область второго покрытия из пористого оксида содержит платину (Pt) в качестве единственного металла платиновой группы.
7. Выхлопная система по любому из пп. 1-4, в которой область второго покрытия из пористого оксида дополнительно содержит палладий (Pd) и имеет отношение платины к палладию по общей массе ≥ 2:1.
8. Выхлопная система по любому из пп. 1-4, в которой область второго покрытия из пористого оксида имеет общую загрузку платины от 150 до 300 г/фут3 (5,297-10,59 кг/м3).
9. Выхлопная система по любому из пп. 1-4, в которой область второго покрытия из пористого оксида по существу не содержит цеолита.
10. Выхлопная система по любому из пп. 1-4, в которой область второго покрытия из пористого оксида не содержит родия, щелочного металла и/или щелочноземельного металла.
11. Выхлопная система по любому из пп. 1-4, в которой область первого покрытия из пористого оксида содержит первый металл платиновой группы (PGM) как единственный металл платиновой группы.
12. Выхлопная система по любому из пп. 1-4, в которой первый металл платиновой группы (МПГ) выбирают из группы, состоящей из платины (Pt), палладия (Pd) и их комбинации.
13. Выхлопная система по любому из пп. 1-4, в которой область первого покрытия из пористого оксида имеет общую загрузку первого металла платиновой группы (МПГ) от 150 до 300 г/фут3 (5,30-10,59 кг/м3).
14. Выхлопная система по любому из пп. 1-4, в которой первый материал-носитель содержит огнеупорный оксид металла, выбранный из группы, состоящей из оксида алюминия, диоксида кремния, диоксида титана, диоксида циркония, оксида церия и смешанного или сложного оксида двух или более этих оксидов.
15. Выхлопная система по п. 14, в которой первый материал-носитель содержит оксид алюминия, легированный легирующей примесью.
16. Выхлопная система по п. 15, в которой первый материал-носитель содержит оксид алюминия, легированный диоксидом кремния.
17. Выхлопная система по любому из пп. 1-4, в которой область первого покрытия из пористого оксида дополнительно содержит материал, адсорбирующий углеводороды, который представляет собой цеолит.
18. Выхлопная система по любому из пп. 1-4, в которой область первого покрытия из пористого оксида дополнительно содержит щелочноземельный металл.
19. Выхлопная система по любому из пп. 1-4, в которой катализатор окисления дополнительно содержит область третьего покрытия из пористого оксида.
20. Выхлопная система по п. 19, в которой область третьего покрытия из пористого оксида расположена непосредственно на основе.
21. Выхлопная система по п. 19, в которой область третьего покрытия из пористого оксида расположена на области первого покрытия из пористого оксида и/или области второго покрытия из пористого оксида.
22. Выхлопная система по п. 19, в которой область третьего покрытия из пористого оксида содержит цеолитовый катализатор, при этом цеолитовый катализатор содержит благородный металл, цеолит и необязательно неблагородный металл.
23. Выхлопная система по п. 22, в которой благородным металлом является палладий.
24. Выхлопная система по п. 22, в которой цеолит является цеолитом с малым размером пор, имеющим тип каркасной структуры, которым является AEI, CHA или сросток AEI-CHA.
25. Выхлопная система по любому из пп. 1-4, в которой основа является проточной основой.
26. Выхлопная система по любому из пп. 1-4, в которой устройство для регулирования выбросов выбрано из группы, состоящей из устройства для регулирования выбросов, выбранного из группы, состоящей из ловушки для обедненных NOx (LNT), катализатора, предотвращающего проскакивание аммиака (ASC), фильтра твердых частиц выхлопа дизельного двигателя (DPF), катализатора селективного каталитического восстановления (SCR), катализированного сажевого фильтра (CSF), фильтра с катализатором селективного каталитического восстановления (SCRF™) и комбинаций двух или более данных устройств.
27. Транспортное средство, содержащее дизельный двигатель и выхлопную систему по любому из пп. 1-26.
28. Способ обработки выхлопных газов из дизельного двигателя, включающий пропускание выхлопных газов через выхлопную систему по любому из пп. 1-4.
| WO2006044974 А1, 27.04.2006 | |||
| Бетоноукладчик,например,для подводного бетонирования | 1981 |
|
SU1046423A2 |
| US 8449852 В1, 28.05.2013 | |||
| EP 1264629 В1, 27.10.2004 | |||
| US 6221804 В1, 24.04.2001 | |||
| СПОСОБ СЖИГАНИЯ УГЛЕВОДОРОДНЫХ ТОПЛИВ (ВАРИАНТЫ) И КАТАЛИЗАТОРЫ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2008 |
|
RU2372556C2 |

