Изобретение относится к области медицины, позволяя проводить неинвазивный пренатальный скрининг плода на основе внеклеточной ДНК из плазмы крови матери. Изобретение позволяет обрабатывать результаты массового параллельного секвенирования для определения наиболее распространенных анеуплоидий плода, таких как синдром Дауна, синдром Патау и синдром Эдвардса, а также изменение количества половых хромосом, начиная с 9 недели, то есть в первом триместре беременности.
Пренатальная диагностика является важным этапом контроля состояния плода в процессе протекания беременности. Чтобы своевременно обнаружить патологические состояния и спрогнозировать вероятные риски, необходимо как можно раньше определить возможные отклонения в развитии плода. В современной клинической практике применяют набор методов, определяющих возможные осложнения при беременности по косвенным признакам, такие как биохимический анализ крови или ультразвуковое исследование плода. Однако эти методы не позволяют точно поставить диагноз и применяются скорее как первичные методы оценки состояния плода. Чтобы с уверенностью диагностировать хромосомные патологии плода, такие как синдром Дауна или синдром Патау, необходимо провести кариотипирование, то есть напрямую взять образцы тканей хориона или плаценты, что связано с риском развития осложнений для плода из-за проведенной инвазивной процедуры. С развитием технологий секвенирования ДНК было предложено несколько подходов к определению геномных аномалий плода неинвазивными методами. В 2011 году был предложен патент (RU 2599419, C12Q 1/68, опубл. 10.10.2016), описывающий методику определения таких аномалий путем анализа ДНК, выделенной из ядросодержащих эритроцитов, обнаруженных в крови матери. Считается, что такие эритроциты появляются в крови матери из плаценты и содержат ДНК плода. Однако такой подход имеет ряд ограничений, поскольку методика обнаружения и выделения таких клеток из крови матери сложна и неоднозначна в связи с тем, что в крови беременной женщины обнаруживается крайне мало клеток плода. Чтобы повысить вероятность нахождения таких клеток, авторы патента RU 2599419 предлагают такой скрининг, начиная с 12-й недели беременности, тогда как решение о прерывании беременности рекомендовано принимать до 12-й недели, что является пороговым безопасным сроком прерывания беременности по желанию женщины.
Помимо ядросодержащих эритроцитов, ДНК плода обнаруживается также в плазме крови матери в форме коротких фрагментов (свободноклеточная или внеклеточная ДНК). Этих фрагментов достаточно, чтобы провести скрининг крупных хромосомных аномалий плода, начиная с 9-й недели беременности. На данном уровне техники существует два основных подхода к анализу данных секвенирования плазмы крови матери с целью определения анеуплоидий у плода: таргетное секвенирование отдельных локусов и массовое параллельное секвенирование всей внеклеточной ДНК из плазмы матери. Последний подход основан на секвенировании всей доступной внеклеточной ДНК из плазмы крови матери, с последующим подсчетом количества чтений, картировавшихся на каждую из хромосом. Только некоторое относительно невысокое количество чтений будет происходить из тканей плаценты и содержать ДНК плода (фетальная фракция), тогда как основная масса чтений будет происходить из генома матери. Этот процент будет меняться в зависимости от срока беременности, индекса массы тела матери и некоторых других факторов, однако, как правило, фетальная фракция находится в пределах 10-15% от общего количества чтений. При наличии анеуплоидий у плода хромосомная фракция (то есть относительное количество чтений, нормированное на длину хромосомы) одной из хромосом будет меняться, что позволяет определить анеуплоидию. Однако чтобы получить статистически значимые отличия требуется более 7 миллионов чтений на образец (Bayindir et al., 2015, https://www.nature.com/articles/ejhg2014282), что повышает стоимость скрининга.
Необходимость высокого количества чтений обусловлена несколькими факторами. Первая сложность заключается в том, что процент фетальной фракции в секвенируемом материале может быть достаточно низкой, что затрудняет биоинформатический анализ. Улучшение методик выделения ДНК и создания геномных библиотек может заметно усилить точность определения анеуплоидий.
Еще одна сложность заключается в том, что существует феномен неравномерности процессов секвенирования в зависимости от GC-состава участка генома. Эта неравномерность имеет нелинейную природу, и может сильно варьировать в зависимости как от технологии секвенирования, так и от конкретного лабораторного протокола. Чтобы компенсировать эту неравномерность были предложены методы нормирования количества чтений в зависимости от GC-состава. Предыдущий патент (RU 2543155, С40В 20/00, G06F 19/10, G06F 19/20, C12Q 1/68, опубл. 27.02.2015) предлагал метод корректировки, основанный на ранжировании участков генома в зависимости от среднего GC-состава и делении уровня покрытия каждого участка на коэффициент, характеризующий превышение среднего покрытия всех участков с таким же GC-составом над средним покрытие по всем участкам. В настоящем изобретении для определения коэффициента поправки используется регрессионный метод, что дает возможность не отбрасывать участки генома с краевыми значениями GC-состава (Chandrananda D, Thorne NP, GanesamoorthyD, Bruno DL, Benjamini Y, Speed TP, Slater HR, Bahlo M. Investigating and Correcting Plasma DNA Sequencing Coverage Bias to Enhance Aneuploidy Discovery. PLoS One. 2014; 9(1): e86993. (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3906086/).
Стандартная схема оценки анеуплоидий после коррекции количества чтений на GC-состав предусматривает нормализацию чтений на каждом участке на общее количество чтений образца, и подсчет усредненной Z-метрики для таргетных хромосом. Однако данная методика все еще подвержена влиянию отдельных участков с выбивающимися значениями даже после нормализации, поэтому требуется дальнейшая компенсация на вариацию данных.
Известен способ определения геномных перестроек по фрагментам ДНК в плазме крови матери, состоящий в количественном анализе внеклеточной ДНК путем массового параллельного секвенирования при помощи полупроводниковой технологии (патент US 20130012399, C12Q 1/68; С40В 20/00; G01N 27/26). Длина чтений, полученных этим методом секвенирования, составляет в среднем 140-160 пн, что позволяет более специфично картировать эти чтения на референсный геном по сравнению с чтениями, полученными методом синтеза, длина которых, как правило, составляет 25-50 пн. Еще одним несомненным плюсом полупроводникового секвенирования является высокая скорость работы секвенатора. Это позволяет сократить время на обработку образца, что важно, если учитывать пороговый срок для относительно безопасной процедуры прерывания беременности до 12-й недели. Недостатками метода секвенирования синтезом применительно к неинвазивному пренатальному скринингу также является относительно высокая себестоимость каждого запуска секвенатора, использование же полупроводникового секвенирования позволяет снизить стоимость анализа, что позволит легче ввести метод в повседневную клиническую практику и сократить необходимость инвазивной диагностики до минимума.
Данное изобретение решает задачу создания способа неинвазивного пренатального скрининга анеуплоидий плода путем более точного определения наличия хромосомных аномалий у плода по данным массового параллельного секвенирования плазмы крови матери при низком покрытии секвенирования. В данном изобретении мы предлагаем ряд улучшений вышеописанной методики, позволяющих добиться более стабильных и точных результатов, а также улучшить разрешение метода, позволяя детектировать не только геномные аномалии, но крупные хромосомные перестройки, такие как частичная дупликация хромосом, которые тоже могут приводить к серьезным последствиям для плода.
Решение поставленной задачи обеспечивается тем, что в способе неинвазивного пренатального скрининга анеуплоидий плода путем массового параллельного секвенирования при помощи полупроводниковой технологии получают плазму из образца крови беременной женщины при сроке беременности не менее 9-и недель, производят выделение внеклеточной ДНК из плазмы беременной женщины, создают полногеномные библиотеки с использованием внеклеточной ДНК, производят секвенирование библиотек при помощи ионного полупроводникового массового параллельного секвенирования, а также первичную подготовку и фильтрацию полученных чтений, затем производят картирование чтений на референсный геном, при этом используют алгоритм Super-maximal Exact Matching, затем производят вторичную фильтрацию чтений по качеству картирования, при этом используют для оценки качества картирования пороговое значение в 15 единиц, затем производят удаление копий чтений, возникших из-за ПЦР-амплификации; затем производят предварительную оценку фетальной фракции для образцов пакетом SeqFF, затем производят определение пола плода в образце по формуле
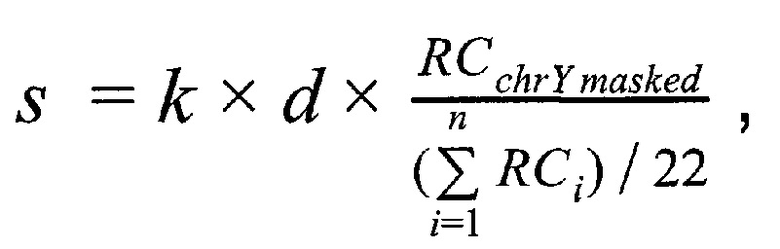
где 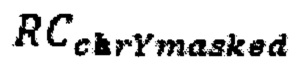 - количество чтений, которое выровнялось на хромосому Y после фильтрации неспецифичных участков,
- количество чтений, которое выровнялось на хромосому Y после фильтрации неспецифичных участков, 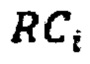 - количество чтений, которое выровнялось на аутосомную хромосому, n - количество аутосом (равное 22), k - эмпирически подобранный коэффициент (в диапазоне от 10000 до 1000000, подбирается в зависимости от представленности Y хромосомы относительно общего числа чтения так, чтобы s было не дробным), d - доля фетальной фракции образца, и если s≤1, значит, исследуемый образец считается женским полом, если s>1, то мужским.
- количество чтений, которое выровнялось на аутосомную хромосому, n - количество аутосом (равное 22), k - эмпирически подобранный коэффициент (в диапазоне от 10000 до 1000000, подбирается в зависимости от представленности Y хромосомы относительно общего числа чтения так, чтобы s было не дробным), d - доля фетальной фракции образца, и если s≤1, значит, исследуемый образец считается женским полом, если s>1, то мужским.
Коэффициент k необходим для того, что компенсировать разницу в порядке количества чтений, приходящихся на хромосому Y относительно порядка количества чтений, приходящихся на все остальные аутосомы, и позволяет получать не дробное значение s. На основе наших эмпирических данных, отношение суммарного количества чтений, картировавшихся на Y хромосому, относительно количества чтений, картировавшихся на аутосомы, получается менее одного к ста тысячам для образцов с плодом женского пола, и на порядок выше для образцов с плодом мужского пола. Однако это соотношение может варьировать в зависимости от использованной методики секвенирования, а также количества отфильтрованных из анализа неспецифичных участков Y хромосомы, поэтому этот коэффициент подбирается эмпирически в диапазоне от 10000 до 1000000 на основе измерений соотношения числа чтений, картировавшихся на Y хромосомы после фильтрации неспецифических участков, к количеству чтений, картировавшихся на аутосомы, чтобы получаемое значение s не было дробным.
Домножение на d добавляется в формулу для компенсации на колебаний в уровне фетальной ДНК от образца к образцу, т.к. чтения, картировавшиеся на хромосому Y, всегда приходят только от плодовой ДНК, тогда как чтения, картировавшиеся на остальные хромосомы, приходят как от ДНК плода, так и от ДНК матери.
Затем производят подсчет фетальной фракции для образцов методом DEFRAG и определяют фетальную фракцию для образцов по формуле:
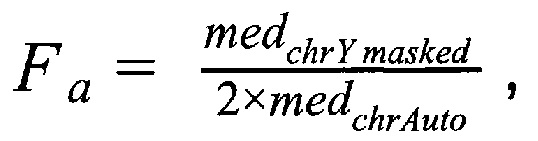
где 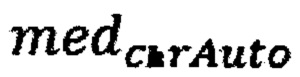 - медиана нормализованного количества чтений на аутосомных хромосомах,
- медиана нормализованного количества чтений на аутосомных хромосомах, 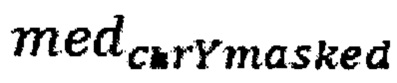 - медиана нормализованного количества чтений на Y хромосоме, где предварительно были удалены все чтения, которые выровнялись на неспецифичные регионы хромосомы; затем производят подсчет фетальной фракции для образцов по формуле для женского пола:
- медиана нормализованного количества чтений на Y хромосоме, где предварительно были удалены все чтения, которые выровнялись на неспецифичные регионы хромосомы; затем производят подсчет фетальной фракции для образцов по формуле для женского пола:
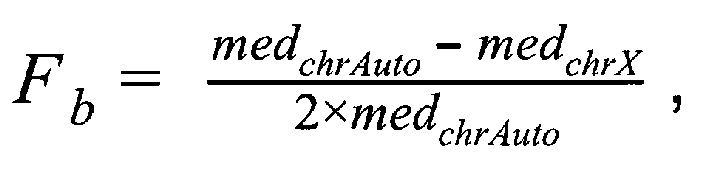
где 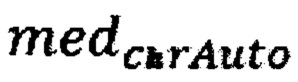 - медиана нормализованного количества чтений на аутосомных хромосомах,
- медиана нормализованного количества чтений на аутосомных хромосомах, 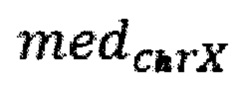 - медиана нормализованного количества чтений на X хромосоме образца; затем методом локальной регрессии LOESS производят нормализацию по GC-составу; затем производят коррекцию пиков и выбросов с помощью еще одного этапа нормализации количества чтений для каждого бина: количество чтений в бине умножают на коэффициент нормализации, который является средним количеством всех чтений во всех бинах на всех аутосомах контрольной группы образцов, поделенные на среднее количество чтений для всех чтений во всех бинах на всех аутосомах исследуемого образца по формуле:
- медиана нормализованного количества чтений на X хромосоме образца; затем методом локальной регрессии LOESS производят нормализацию по GC-составу; затем производят коррекцию пиков и выбросов с помощью еще одного этапа нормализации количества чтений для каждого бина: количество чтений в бине умножают на коэффициент нормализации, который является средним количеством всех чтений во всех бинах на всех аутосомах контрольной группы образцов, поделенные на среднее количество чтений для всех чтений во всех бинах на всех аутосомах исследуемого образца по формуле:
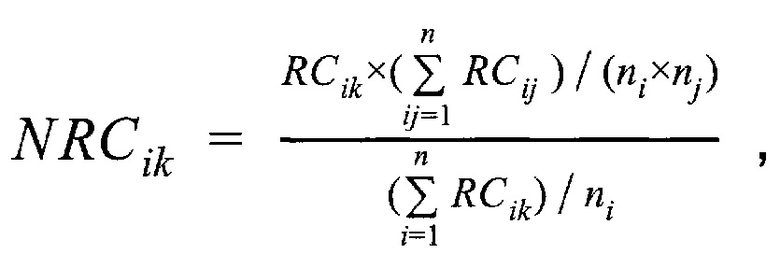
где 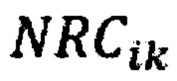 - нормализованное количество чтений конкретного бина,
- нормализованное количество чтений конкретного бина, 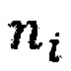 - количество бинов,
- количество бинов, 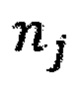 - количество образцов в контрольной группе,
- количество образцов в контрольной группе, 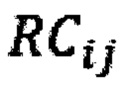 - количество чтений конкретного бина до нормализации; затем вычисляют
- количество чтений конкретного бина до нормализации; затем вычисляют 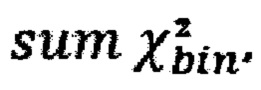 - сумму разности квадратов между нормализованным количеством чтений для бина для всех образцов из контрольной группы и нормализованным количеством чтений исследуемого образца по формуле:
- сумму разности квадратов между нормализованным количеством чтений для бина для всех образцов из контрольной группы и нормализованным количеством чтений исследуемого образца по формуле:
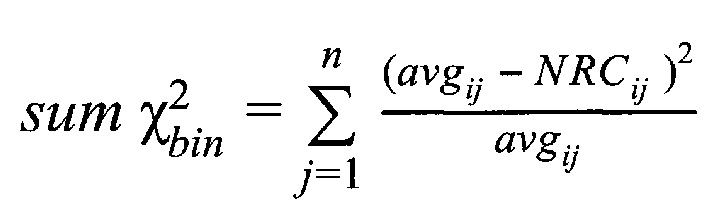
где 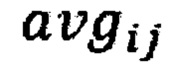 - среднее количество чтений для всех бинов после нормализации; затем из
- среднее количество чтений для всех бинов после нормализации; затем из 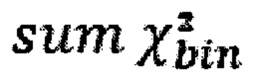 вычитают степень свободы
вычитают степень свободы 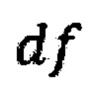 - количество образцов в контрольной группе минус один, и делят на квадратный корень из удвоенной степени свободы, таким образом, получая Z-метрику для всех бинов в образце, и, если значение метрики в бине превышает порог в 3.5, вклад такого бина в подсчет интегральной хромосомной z-метрики снижают путем деления на коэффициент
- количество образцов в контрольной группе минус один, и делят на квадратный корень из удвоенной степени свободы, таким образом, получая Z-метрику для всех бинов в образце, и, если значение метрики в бине превышает порог в 3.5, вклад такого бина в подсчет интегральной хромосомной z-метрики снижают путем деления на коэффициент 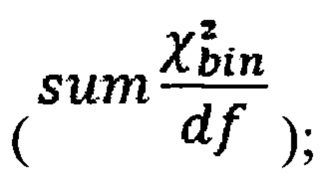 затем производят определение репрезентативности контрольной выборки путем сравнения паттерна покрытия бинов и хромосомных фракций образца с обобщенным паттерном группы контроля; затем производят методом линейной регрессии подсчет Z-метрики образца, для чего используют нормализованные показатели количества чтений для прямой и обратной нити ДНК каждой хромосомы, при этом выбирают четыре наиболее удачных набора предикторов на основе скорректированного значения квадрата R и определяют предсказанные значения Z-метрики на основании моделей с наиболее удачными предикторами; затем для каждой из тестируемых хромосом рассчитывают финальную Z-метрику на основе отношения наблюдаемой хромосомной фракции к предсказанной по формуле
затем производят определение репрезентативности контрольной выборки путем сравнения паттерна покрытия бинов и хромосомных фракций образца с обобщенным паттерном группы контроля; затем производят методом линейной регрессии подсчет Z-метрики образца, для чего используют нормализованные показатели количества чтений для прямой и обратной нити ДНК каждой хромосомы, при этом выбирают четыре наиболее удачных набора предикторов на основе скорректированного значения квадрата R и определяют предсказанные значения Z-метрики на основании моделей с наиболее удачными предикторами; затем для каждой из тестируемых хромосом рассчитывают финальную Z-метрику на основе отношения наблюдаемой хромосомной фракции к предсказанной по формуле
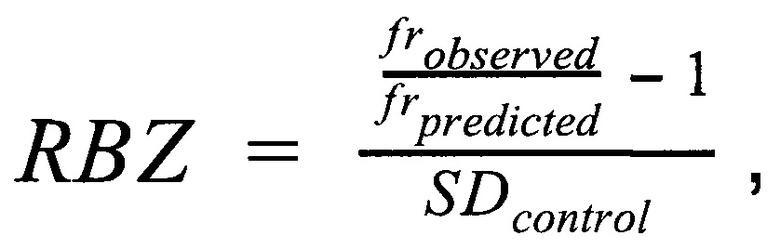
где 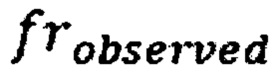 - наблюдаемая хромосомная фракция,
- наблюдаемая хромосомная фракция, 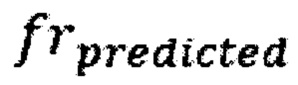 - предсказанная хромосомная фракция исследуемого образца,
- предсказанная хромосомная фракция исследуемого образца,  - среднеквадратичное отклонение относительной хромосомной фракции (наблюдаемой к предсказанной) в группе образцов контроля.
- среднеквадратичное отклонение относительной хромосомной фракции (наблюдаемой к предсказанной) в группе образцов контроля.
Полученное значение финальной Z-метрики позволяет классифицировать образец либо как не отличающийся от нормы, если значение Z-метрики для хромосомы находится в пределах значения 3х стандартных отклонений от среднего для контрольной выборки. Если значение Z-метрики для хромосомы меньше нижней границы указанного интервала, то образец считается аномальным, и является носителем моносомии, а если большей верхней границы указанного интервала, то образец является носителем трисомии по данной хромосоме. Образец считается не прошедшим контроль качества, если определяемый уровень фетальной фракции образца ниже порогового значения в 3,5%, или если после всех стадий предподготовки образца количество чтений стало меньше 1 млн.
Заявленное изобретение предлагает набор улучшений, позволяющий повысить точность и надежность метода по сравнению с ранее существующими способами.
На этапе предобработки данных секвенирования используется алгоритм выравнивания, адаптированный для данных с полупроводниковых секвенаторов (tmap). Технология полупроводникового секвенирования предусматривает большую длину чтений по сравнению с технологией секвенирования через синтез, что позволяет не дробить фетальные чтения, длина которых, как правило, составляет 125-150 пн (Chandrananda et al., 2014), на более мелкие части и, как следствие, точнее картировать эти чтения. На этапе анализа данных производится дополнительная коррекция избыточной вариации данных методом Хи квадрат, что позволяет не фильтровать выбивающиеся участки, а корректировать, и, таким образом, не терять информацию при подсчет общей Z-метрики, что позволяет получать достоверные результаты при более низкой глубине секвенирования.
Также в алгоритм входит новый этап контроля качества образца и репрезентативности контрольной выборки путем сравнения паттернов хромосомных фракций образца и контрольной группы методом суммирования среднеквадратичных отклонений хромосомных фракций. Определение анеуплоидий производится при помощи модифицированной, ранее не использованной, Z-метрики, в основе которой лежит регрессионный анализ, а для вычисления Z-метрики применяется отношение наблюдаемой хромосомной фракции к предсказанной, что позволяет определить оптимальный набор хромосом-предикторов и построить свою предсказательную модель для определения финальной Z-метрики образца. Уникальность такого подхода заключается в том, что за счет построения индивидуальной модели с подбором уникальных предикторов можно выборочно скорректировать неравномерность секвенирования. Таким образом, расчет Z-метрики адаптирован к конкретному инструменту и лаборатории, где выполняется неинвазивный пренатальный скрининг. Заявленное изобретение предлагает набор улучшений, позволяющий повысить точность и надежность метода по сравнению с ранее существующими способами.
На этапе предобработки данных секвенирования используется алгоритм выравнивания, адаптированный для данных с полупроводниковых секвенаторов (tmap). Технология полупроводникового секвенирования предусматривает большую длину чтений по сравнению с технологией секвенирования через синтез, что позволяет не дробить фетальные чтения, длина которых, как правило, составляет 125-150 пн (Chandrananda et al., 2014), на более мелкие части и, как следствие, точнее картировать эти чтения. На этапе анализа данных производится дополнительная коррекция избыточной вариации данных методом Хи квадрат, что позволяет не фильтровать выбивающиеся участки, а корректировать, и, таким образом, не терять информацию при подсчет общей Z-метрики, что позволяет получать достоверные результаты при более низкой глубине секвенирования.
Также в алгоритм входит новый этап контроля качества образца и репрезентативности контрольной выборки путем сравнения паттернов хромосомных фракций образца и контрольной группы методом суммирования среднеквадратичных отклонений хромосомных фракций. Определение анеуплоидий производится при помощи модифицированной, ранее не использованной, Z-метрики, в основе которой лежит регрессионный анализ, а для вычисления Z-метрики применяется отношение наблюдаемой хромосомной фракции к предсказанной, что позволяет определить оптимальный набор хромосом-предикторов и построить свою предсказательную модель для определения финальной Z-метрики образца. Уникальность такого подхода заключается в том, что за счет построения индивидуальной модели с подбором уникальных предикторов можно выборочно скорректировать неравномерность секвенирования. Таким образом, расчет Z-метрики адаптирован к конкретному инструменту и лаборатории, где выполняется неинвазивный пренатальный скрининг.
Изобретение осуществляется следующим образом.
Процесс подготовки образцов и секвенирования включает следующие стадии.
А) Получение плазмы из образца крови беременной женщины (срок беременности не менее 9-и недель).
Образцы венозной крови собирают в пробирки, содержащие 0,5 М раствор ЭДТА (рН=8,0). Минимальный объем пробирок 9 мл. Содержимое пробирок перемешивают переворачиванием вверх - вниз 10 раз. Не позже чем через 4 часа после забора крови проводят центрифугирование образцов в течение 10 минут при 2000g при температуре +4°С Далее переносят супернатант (плазму) в чистую пробирку и проводят центрифугирование в течение 10 минут при 16000g при температуре +4°С. Собранный супернатант (плазму) используют для выделения внеклеточной ДНК.
Б) Выделение внеклеточной ДНК из плазмы беременной женщины.
Для выделения ДНК используют 2 мл плазмы. Выделение ДНК проводят с применением магнитных частиц с помощью набора MagMAX Cell-Free DNA Isolation Kit (Thermo Fisher Scientific Inc., USA) (Catalog no. A29319) согласно рекомендациям производителя. Определяют концентрацию ДНК и качество.
В) Создание полногеномных библиотек с использованием внеклеточной ДНК.
Для каждого образца при создании библиотек используют весь объем выделенной ДНК. Подготовку библиотек проводят по протоколу набора "Ion Plus Fragment Library" ("Thermo Fisher Scientific Inc.", USA) (Catalog no. 4471252) с некоторыми модификациями. Библиотеки конструируют в несколько этапов. На первом этапе проводят достройку и затупление концов молекул ДНК согласно протоколу. Далее фрагменты ДНК очищают при помощи магнитных частиц Agencourt AMPure ХР (Beckman Coulter, Inc., USA) (Catalog no. A63881). На следующем этапе к молекулам ДНК пришивают адаптеры Ion Xpress Barcode Adapters 1-16 Kit ("Thermo Fisher Scientific Inc.", USA) (Catalog no. 4471250) согласно протоколу. Далее проводили двухстадийную очистку молекул ДНК при помощи магнитных частиц Agencourt AMPure ХР (Beckman Coulter, Inc., USA) (Catalog no. A63881) с целью отбора по длине. Полученные молекулы ДНК используют в качестве матрицы для последующей амплификации. Определяют концентрацию ДНК и качество. На заключительном этапе проводят пулирование полученных библиотек с последующим разведением. Для секвенирования используют 0,3 нг образца. В процесс подготовки библиотек внесены изменения:
исключен этап фрагментирования (поскольку внеклеточная ДНК представлена короткими молекулами ДНК);
внесены изменения в этап очистки ДНК после пришивки адаптеров с целью удаления больших фрагментов ДНК (поскольку после выделения образец содержит фрагменты длиной более 200 нт (вероятно, геномная ДНК беременной), то необходимо исключить эти фрагменты из исследования);
увеличено число циклов амплификации (в связи с низкой концентрацией внеклеточной ДНК);
увеличено количество образца, используемого для секвенирования (в связи с низкой концентрацией внеклеточной ДНК).
Г) Секвенирование библиотек при помощи ионного полупроводникового массового параллельного секвенирования.
Подготовку образцов к секвенированию и загрузку их на чип осуществляют с помощью системы «Ion Chef System» ("Thermo Fisher Scientific Inc.", USA), используя набор Ion 540 Kit-Chef (Catalog no. A30011) и микрочипы "Ion 540 Chip" ("Thermo Fisher Scientific Inc.", USA) (Catalog no. A27766) согласно инструкции производителя. Секвенирование проводили на приборе "Ion Torrent S5" ("Thermo Fisher Scientific Inc.", USA).
Процесс биоинформатического анализа результатов секвенирования включает следующие стадии.
А) Первичная подготовка и фильтрация полученных чтений.
Производится обрезания концов чтений с качеством чтения ниже 17 по шкале Phred, и фильтрация чтений длиннее 200 пн или короче 25 пн. Известно, что фрагменты ДНК фетального происхождения в среднем имеют длину 140-150 пн, поэтому фрагменты длиннее 200 пн отбрасываются как заведомо происходящие из генома матери, чтобы повысить процент фетальной фракции, то есть содержание чтений, происходящих от плода, а значит, и статистическую силу анализа.
Б) Картирование чтений на референсный геном.
После первичной фильтрации и оценки качества производится картирование чтений на референсный геном (GRCh37), то есть поиск места локализации каждого чтения на референсном геноме и определение координат чтения.
Каждая из технологий секвенирования имеет свои особенности и недостатки, что следует учитывать при картировании, поэтому данное изобретение использует алгоритмический подход картирования, созданный специально для работы с чтениями, полученными после полупроводникового секвенирования (этот алгоритм разработан ранее и находится за рамками данного изобретения). Большинство инструментов картирования разработаны для данных, полученных при секвенировании синтезом, и рассчитаны на фиксированную короткую длину чтений, тогда как технология полупроводникового секвенирования позволяет получать чтения разной длины, что и учитывает при этом алгоритм Super-maximal Exact Matching (Li, Heng. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. 2012. Bioinformatics, 28, 14: 1838-1844), используемый в инструменте tmap, применяемом для картирования чтений в данном изобретении.
В) Вторичная фильтрация чтений по качеству картирования.
В дальнейшем анализе учитываются только те чтения, качество картирования которых, определяемое алгоритмом картирования tmap, было выше порогового значения в 15 единиц. Качество картирования определяется на основе количества несовпадений между референсной последовательностью и последовательностью чтения. Фильтрация по качеству картирования позволяет отсеять все неоднозначно картировавшиеся чтения или чтения, в которых было много ошибок секвенирования, из-за чего они могли картироваться неправильно.
Г) Удаление копий чтений, возникших из-за ПЦР-амплификации.
Удаление копий чтений производилось при помощи утилиты MarkDuplicates из пакета picard tools. Это важный этап в предподготовке данных, поскольку полимеразная цепная реакция (ПЦР), которая проводится на этапе подготовки геномных библиотек, может породить дисбаланс в количестве копий, непропорционально увеличив количество копий чтений на каком-то участке генома, что скажется на дальнейшем анализе.
Д) Определение пола плода в образце.
Аккуратное и точное определение пола важно при дальнейшем анализе на геномные перестройки, связанные с половыми хромосомами. Так, если плод является носителем синдрома Кляйнфельтера (двух X хромосом и одной Y хромосомы), то может ошибочно определяться как девочка, что, в свою очередь, приведет к ошибочному определению образца как нормального. Также стоит учитывать то, что существуют неспецифичные участки Y хромосомы, на которые могут картироваться чтения из образца с плодом женского пола. Чтобы снизить вероятность ошибки при определении пола, данное изобретение использует улучшенный метод определения пола, учитывающий фактор неспецифичности картирования на некоторые участки, фильтруя чтения, картирующиеся на такие участки.
Таким образов, в данном изобретении пол образца определяется по следующей формуле:
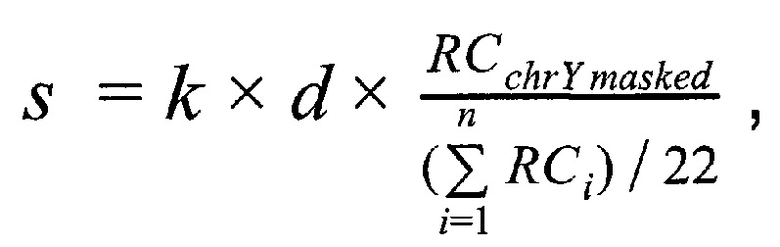
где 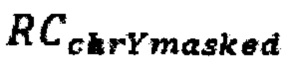 - количество чтений, которое выровнялось на хромосому Y после фильтрации неспецифичных участков, RCi - количество чтений, которое выровнялось на аутосомную хромосому, n - количество аутосом (равное 22), k - эмпирически подобранный коэффициент (в диапазоне от 100000 до 1000000, подбирается в зависимости от представленности Y хромосомы относительно общего числа чтения так, чтобы s было не дробным), d - доля фетальной фракции образца, и если s≤1, значит, исследуемый образец считается женским полом, если s>1, то мужским.
- количество чтений, которое выровнялось на хромосому Y после фильтрации неспецифичных участков, RCi - количество чтений, которое выровнялось на аутосомную хромосому, n - количество аутосом (равное 22), k - эмпирически подобранный коэффициент (в диапазоне от 100000 до 1000000, подбирается в зависимости от представленности Y хромосомы относительно общего числа чтения так, чтобы s было не дробным), d - доля фетальной фракции образца, и если s≤1, значит, исследуемый образец считается женским полом, если s>1, то мужским.
Коэффициент k необходим для того, что компенсировать разницу в порядке количества чтений, приходящихся на хромосому Y относительно порядка количества чтений, приходящихся на все остальные аутосомы, и позволяет получать не дробное значение s. На основе наших эмпирических данных, отношение суммарного количества чтений, картировавшихся на Y хромосому, относительно количества чтений, картировавшихся на аутосомы, получается менее одного к ста тысячам для образцов с плодом женского пола, и на порядок выше для образцов с плодом мужского пола. Однако это соотношение может варьировать в зависимости от использованной методики секвенирования, а также количества отфильтрованных из анализа неспецифичных участков Y хромосомы, поэтому этот коэффициент подбирается эмпирически в диапазоне от 10000 до 1000000 на основе измерений соотношения числа чтений, картировавшихся на Y хромосомы после фильтрации неспецифических участков, к количеству чтений, картировавшихся на аутосомы, чтобы получаемое значение s не было дробным.
Домножение на d добавляется в формулу для компенсации на колебаний в уровне фетальной ДНК от образца к образцу, т.к. чтения, картировавшиеся на хромосому Y, всегда приходят только от плодовой ДНК, тогда как чтения, картировавшиеся на остальные хромосомы, приходят как от ДНК плода, так и от ДНК матери.
Е) Подсчет фетальной фракции образца.
Это необходимый этап для дальнейшего корректного определения риска хромосомных аномалий образца. При фетальной фракции ниже порогового значения в 4% (Canick JA, Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE. The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat Diagn. 2013) результаты определения анеуплоидий считаются недостоверными, и требуется повторный забор крови у матери для получения нового образца.
В связи с важностью этого этапа данное изобретение использует сразу несколько разных методик определения фетальной фракции:
1.1.1. Подсчет фетальной фракции для образцов методом DEFRAG
1.1.2. Подсчет фетальной фракции для образцов пакетом SeqFF
1.1.3. Подсчет фетальной фракции для образцов по формуле:
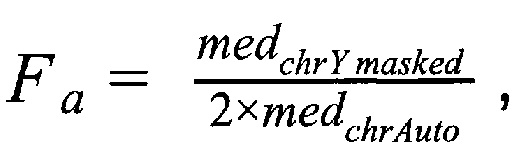
где 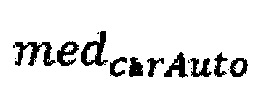 - медиана нормализованного количества чтений на аутосомных хромосомах,
- медиана нормализованного количества чтений на аутосомных хромосомах, 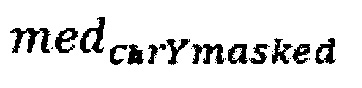 - медиана нормализованного количества чтений на Y хромосоме, где наше предварительно были удалены все чтения, которые выровнялись на неспецифичные регионы хромосомы.
- медиана нормализованного количества чтений на Y хромосоме, где наше предварительно были удалены все чтения, которые выровнялись на неспецифичные регионы хромосомы.
1.1.4. Подсчет фетальной фракции для образцов по формуле:
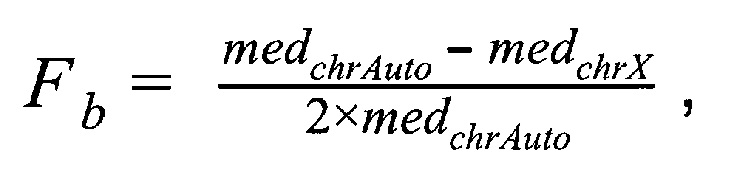
где 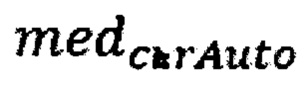 - медиана нормализованного количества чтений на аутосомных хромосомах,
- медиана нормализованного количества чтений на аутосомных хромосомах, 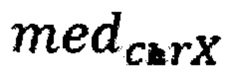 - медиана нормализованного количества чтений на X хромосоме образца.
- медиана нормализованного количества чтений на X хромосоме образца.
Наиболее точные результаты дает подход, основанный на оценке представленности Y хромосомы, однако такой метод применим только для образцов с плодом мужского пола, поэтому для девочек используются более сложные модели, включающие машинное обучение (этот алгоритм разработан ранее и находится за рамками данного изобретения).
Далее вычисляется среднее фетальной фракции образца по полученным разными методами значениям.
Ж) Нормализация по GC-составу.
Чтобы выровнять представленность хромосом при подсчете финальной Z-метрики, необходимо нормализовать количество чтений на GC-состав референсной последовательности. Для этого чтения группируются по непересекающимся участкам длиной 50 кб (далее бины). Нормализация количества чтений на бин с учетом GC-состава референсного генома производится методом локальной регрессии LOESS. Неравномерность секвенирования хорошо коррелирует с GC-составом фрагментов, однако зависимость нелинейная и значительно меняется как от технологии секвенирования, так и от варианта пробоподготовки, принятого в лаборатории. Локальная регрессия LOESS позволяет по данным контрольной выборки построить модель и подобрать компенсирующие коэффициенты для среднего значения GC-состава в каждом бине, при этом бины с нулевым количеством чтений удаляются из анализа.
З) Коррекция пиков и выбросов.
Однако коррекция по GC-составу не способна полностью выровнять покрытие, поэтому в данном изобретении применяет дополнительное снижение избыточной вариабельности данных методом Хи-квадрат, которое уменьшает вклад в подсчет Z-метрики бинов с вариабельностью выше, чем ожидается случайно. Сперва производится еще один этап нормализации количества чтений для каждого бина: количество чтений в бине умножается на коэффициент нормализации, который является средним количеством всех чтений во всех бинах на всех аутосомах контрольной группы образцов, поделенные на среднее количество чтений для всех чтений во всех бинах на всех аутосомах исследуемого образца. Далее вычисляется 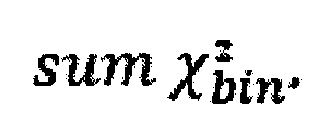 то есть сумма разности квадратов между нормализованным количеством чтений для бина для всех образцов из контрольной группы и нормализованным количеством чтений исследуемого образца. Из вычитается степень свободы
то есть сумма разности квадратов между нормализованным количеством чтений для бина для всех образцов из контрольной группы и нормализованным количеством чтений исследуемого образца. Из вычитается степень свободы 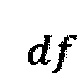 (количество образцов в контрольной группе минус один), и делится на квадратный корень из удвоенной степени свободы. Таким образом, мы получаем Z-метрику для всех бинов в образце, и если значение метрики в бине превышает порог в 3.5, вклад такого бина в подсчет интегральной хромосомной z-метрики снижается путем деления на коэффициент
(количество образцов в контрольной группе минус один), и делится на квадратный корень из удвоенной степени свободы. Таким образом, мы получаем Z-метрику для всех бинов в образце, и если значение метрики в бине превышает порог в 3.5, вклад такого бина в подсчет интегральной хромосомной z-метрики снижается путем деления на коэффициент 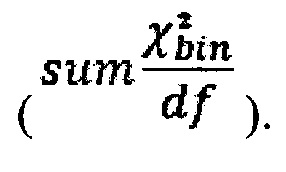
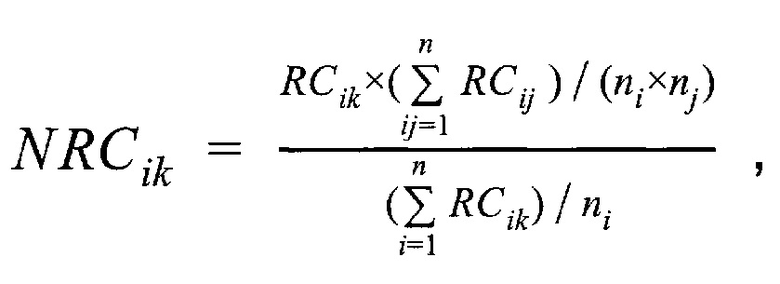
где 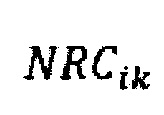 - нормализованное количество чтений конкретного бина,
- нормализованное количество чтений конкретного бина, 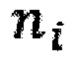 - количество бинов,
- количество бинов, 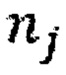 - количество образцов в контрольной группе,
- количество образцов в контрольной группе, 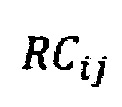 - количество чтений конкретного бина до нормализации.
- количество чтений конкретного бина до нормализации.
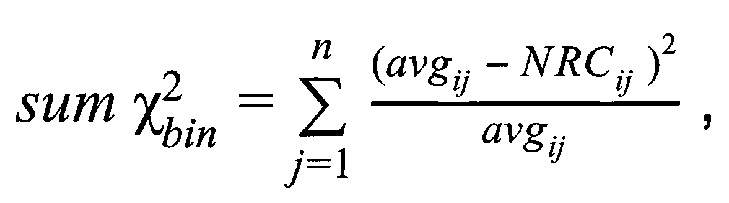
где 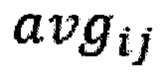 - среднее количество чтений для всех бинов после нормализации.
- среднее количество чтений для всех бинов после нормализации.
Такой подход позволяет не отбрасывать бины с выпадающими значениями, как делалось в методе, предложенном патентом RU 2529784, а использовать их, что сохраняет мощность теста при меньшей глубине секвенирования.
И) Определение репрезентативности контрольной выборки.
Очень важным этапом контроля качества является определение репрезентативности контрольной группы по отношению к исследуемому образцу. Для этого данное изобретение проводит сравнение паттерна покрытия бинов и хромосомных фракций образца с обобщенным паттерном группы контроля. Схожие паттерны покрытия означают то, что образец входит в ту же генеральную совокупность, что и контрольная выборка, и что проводить подсчет финальной Z-метрики на основе данных контрольной группы корректно. Такой подход является улучшением данного изобретения, позволяющим снижать вероятность получить некорректные значения оценки риска хромосомных аномалий.
К) Подсчет Z-метрики образца.
Финальным этапом анализа данных в данном изобретении является определение анеуплоидий методом регрессионной Z-метрики. Для получения финального значения Z-метрики для каждой хромосомы используется метод линейной регрессии. В качестве предикторов для построения модели и предсказания Z-метрики используются нормализованные показатели количества чтений для прямой и обратной нити ДНК каждой хромосомы. Алгоритм выбирает четыре наиболее удачных набора предикторов на основе скорректированного значения квадрата R, и выдает предсказанные значения Z-метрики на основании моделей с наиболее удачными предикторами. Финальная Z-метрика рассчитывается для каждой из тестируемых хромосом на основе отношения наблюдаемой хромосомной фракции к предсказанной по представленной формуле.
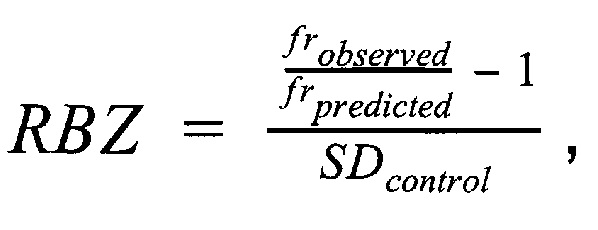
где 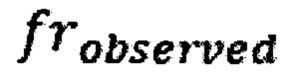 - наблюдаемая хромосомная фракция,
- наблюдаемая хромосомная фракция,  - предсказанная хромосомная фракция исследуемого образца,
- предсказанная хромосомная фракция исследуемого образца,  - среднеквадратичное отклонение относительной хромосомной фракции (наблюдаемой к предсказанной) в группе образцов контроля.
- среднеквадратичное отклонение относительной хромосомной фракции (наблюдаемой к предсказанной) в группе образцов контроля.
Полученное значение финальной Z-метрики позволяет классифицировать образец либо как не отличающийся от нормы, если значение Z-метрики для хромосомы находится в пределах значения 3х стандартных отклонений от среднего для контрольной выборки. Если значение Z-метрики для хромосомы меньше нижней границы указанного интервала, то образец считается аномальным, и является носителем моносомии, а если большей верхней границы указанного интервала, то образец является носителем трисомии по данной хромосоме. Образец считается не прошедшим контроль качества, если определяемый уровень фетальной фракции образца ниже порогового значения в 3,5%, или если после всех стадий биоинформатической подготовки образца количество чтений стало меньше 1 млн.
Данное изобретение позволяет быстрее и точнее определять хромосомные аномалии плода. Показана 100% чувствительность и специфичность теста на валидирующей выборке, собранной на базе Научно-исследовательском институте акушерства, гинекологии и репродуктологии им. Отта. Метод успешно определяет не только анеуплоидий, но и крупные хромосомные дупликации. Так, была успешно обнаружена частичная дупликация 13-й хромосомы в образце с генотипом 47,XX,der(13;13)(q10;q10),+13+21[15]/47,ХХ,+21[14], позже подтвержденном при помощи инвазивной диагностики.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения кариотипа плода беременной женщины на основании секвенирования гибридных прочтений, состоящих из коротких фрагментов внеклеточной ДНК | 2019 |
|
RU2717023C1 |
| СПОСОБ НЕИНВАЗИВНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА МЕТОДОМ СЕКВЕНИРОВАНИЯ | 2014 |
|
RU2543155C1 |
| СПОСОБ НЕИНВАЗИВНОЙ ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА | 2014 |
|
RU2583830C2 |
| НЕИНВАЗИВНОЕ ОБНАРУЖЕНИЕ ГЕНЕТИЧЕСКОЙ АНОМАЛИИ ПЛОДА | 2011 |
|
RU2589681C2 |
| Способ неинвазивного пренатального выявления эмбриональной хромосомной анеуплоидии по материнской крови | 2016 |
|
RU2744604C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ АНЕУПЛОИДИИ ПЛОДА В ОБРАЗЦЕ КРОВИ БЕРЕМЕННОЙ ЖЕНЩИНЫ | 2021 |
|
RU2777072C1 |
| СПОСОБ НЕИНВАЗИВНОЙ ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА | 2015 |
|
RU2627673C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИСТОЧНИКА АНЕУПЛОИДНЫХ КЛЕТОК ПО КРОВИ БЕРЕМЕННОЙ ЖЕНЩИНЫ | 2016 |
|
RU2674700C2 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ОПРЕДЕЛЕНИЯ ФРАКЦИИ ВНЕКЛЕТОЧНЫХ НУКЛЕИНОВЫХ КИСЛОТ В БИОЛОГИЧЕСКОМ ОБРАЗЦЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2699728C2 |
| НЕИНВАЗИВНЫЙ ДИАГНОСТИЧЕСКИЙ ТЕСТ ДНК ДЛЯ ОБНАРУЖЕНИЯ АНЕУПЛОИДИИ | 2012 |
|
RU2638456C2 |
Изобретение относится к области медицины. Предложен способ неинвазивного пренатального скрининга анеуплоидий плода путем массового параллельного секвенирования при помощи полупроводниковой технологии. Производят выделение внеклеточной ДНК из плазмы беременной женщины, создают полногеномные библиотеки с использованием внеклеточной ДНК. Производят подсчет фетальной фракции. Методом линейной регрессии производят подсчет Z-метрики образца, для чего используют нормализованные показатели количества чтений для прямой и обратной нити ДНК каждой хромосомы. Классифицируют образец либо как не отличающийся от нормы, либо как носитель моносомии или трисомии по данной хромосоме. Изобретение обеспечивает точный способ неинвазивного пренатального скрининга анеуплоидий плода.
Способ неинвазивного пренатального скрининга анеуплоидий плода путем массового параллельного секвенирования при помощи полупроводниковой технологии, отличающийся тем, что получают плазму из образца крови беременной женщины при сроке беременности не менее 9-и недель, производят выделение внеклеточной ДНК из плазмы беременной женщины, создают полногеномные библиотеки с использованием внеклеточной ДНК, производят секвенирование библиотек при помощи ионного полупроводникового массового параллельного секвенирования, а также первичную подготовку и фильтрацию полученных чтений, затем производят картирование чтений на референсный геном, при этом используют алгоритм Super-maximal Exact Matching, затем производят вторичную фильтрацию чтений по качеству картирования, при этом используют для оценки качества картирования пороговое значение в 15 единиц, затем производят удаление копий чтений, возникших из-за ПЦР-амплификации; затем производят предварительную оценку фетальной фракции для образцов пакетом SeqFF, затем производят определение пола плода в образце по формуле
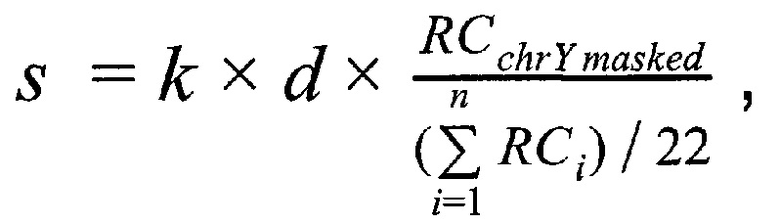
где RCchrYmasked - количество чтений, которое выровнялось на хромосому Y после фильтрации неспецифичных участков, RCi - количество чтений, которое выровнялось на аутосомную хромосому, n - количество аутосом, равное 22, k - эмпирически подобранный коэффициент, располагающийся в диапазоне от 100000 до 1000000, подбирается в зависимости от представленности Y хромосомы относительно общего числа чтения так, чтобы s было не дробным, d - доля фетальной фракции образца, и если s≤1, значит, исследуемый образец считается женским полом, если s>1, то мужским; затем производят подсчет фетальной фракции для образцов методом DEFRAG и определяют фетальную фракцию для образцов по формуле:
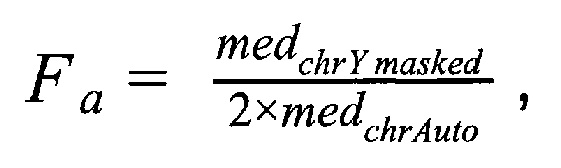
где medchrAuto - медиана нормализованного количества чтений на аутосомных хромосомах, medchrYmasked - медиана нормализованного количества чтений на Y хромосоме, где предварительно были удалены все чтения, которые выровнялись на неспецифичные регионы хромосомы; затем производят подсчет фетальной фракции для образцов по формуле:
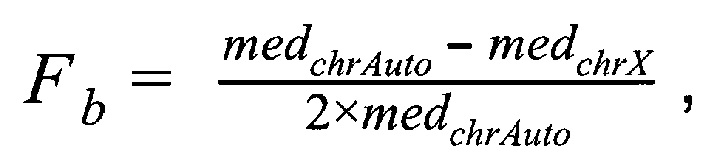
где medchrAuto - медиана нормализованного количества чтений на аутосомных хромосомах, medchrХ - медиана нормализованного количества чтений на X хромосоме образца; затем методом локальной регрессии LOESS производят нормализацию по GC-составу; затем производят коррекцию пиков и выбросов с помощью еще одного этапа нормализации количества чтений для каждого бина: количество чтений в бине умножают на коэффициент нормализации, который является средним количеством всех чтений во всех бинах на всех аутосомах контрольной группы образцов, поделенные на среднее количество чтений для всех чтений во всех бинах на всех аутосомах исследуемого образца по формуле:
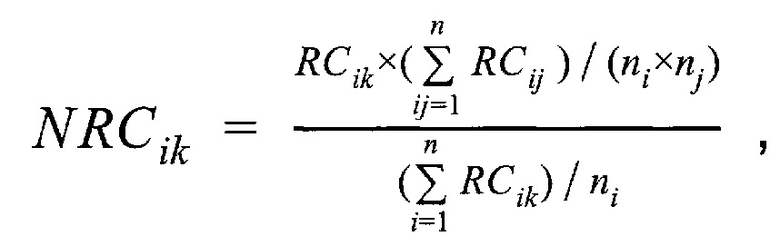
где NRCik - нормализованное количество чтений конкретного бина, ni - количество бинов, nj - количество образцов в контрольной группе, RCij - количество чтений конкретного бина до нормализации; затем вычисляют 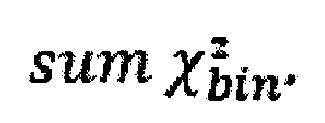 - сумму разности квадратов между нормализованным количеством чтений для бина для всех образцов из контрольной группы и нормализованным количеством чтений исследуемого образца по формуле:
- сумму разности квадратов между нормализованным количеством чтений для бина для всех образцов из контрольной группы и нормализованным количеством чтений исследуемого образца по формуле:
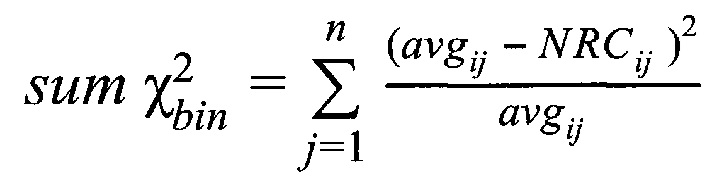 ,
,
где avgij - среднее количество чтений для всех бинов после нормализации; затем из 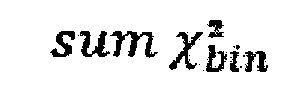 вычитают степень свободы df - количество образцов в контрольной группе минус один, и делят на квадратный корень из удвоенной степени свободы, таким образом, получая Z-метрику для всех бинов в образце, и, если значение метрики в бине превышает порог в 3.5, вклад такого бина в подсчет интегральной хромосомной Z-метрики снижают путем деления на коэффициент
вычитают степень свободы df - количество образцов в контрольной группе минус один, и делят на квадратный корень из удвоенной степени свободы, таким образом, получая Z-метрику для всех бинов в образце, и, если значение метрики в бине превышает порог в 3.5, вклад такого бина в подсчет интегральной хромосомной Z-метрики снижают путем деления на коэффициент 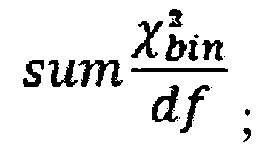 затем производят определение репрезентативности контрольной выборки путем сравнения паттерна покрытия бинов и хромосомных фракций образца с обобщенным паттерном группы контроля; затем производят методом линейной регрессии подсчет Z-метрики образца, для чего используют нормализованные показатели количества чтений для прямой и обратной нити ДНК каждой хромосомы, при этом выбирают четыре наиболее удачных набора предикторов на основе скорректированного значения квадрата R и определяют предсказанные значения Z-метрики на основании моделей с наиболее удачными предикторами; затем для каждой из тестируемых хромосом рассчитывают финальную Z-метрику на основе отношения наблюдаемой хромосомной фракции к предсказанной по формуле:
затем производят определение репрезентативности контрольной выборки путем сравнения паттерна покрытия бинов и хромосомных фракций образца с обобщенным паттерном группы контроля; затем производят методом линейной регрессии подсчет Z-метрики образца, для чего используют нормализованные показатели количества чтений для прямой и обратной нити ДНК каждой хромосомы, при этом выбирают четыре наиболее удачных набора предикторов на основе скорректированного значения квадрата R и определяют предсказанные значения Z-метрики на основании моделей с наиболее удачными предикторами; затем для каждой из тестируемых хромосом рассчитывают финальную Z-метрику на основе отношения наблюдаемой хромосомной фракции к предсказанной по формуле:
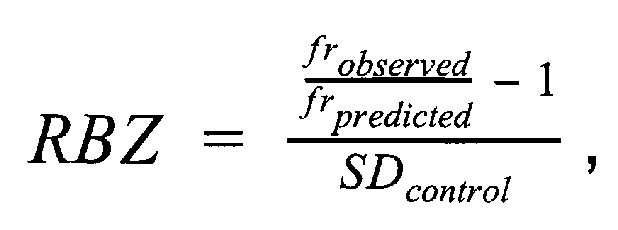
где frobserved - наблюдаемая хромосомная фракция, frpredicted - предсказанная хромосомная фракция исследуемого образца, SDcontrol - среднеквадратичное отклонение относительной хромосомной фракции (наблюдаемой к предсказанной) в группе образцов контроля; затем на основе финальной Z-метрики классифицируют образец либо как не отличающийся от нормы, если значение Z-метрики для хромосомы находится в пределах значения плюс и минус 3х стандартных отклонений от среднего, если значение Z-метрики для хромосомы меньше нижней границы указанного интервала, то образец считается аномальным, и является носителем моносомии, а если больше верхней границы указанного интервала, то образец является носителем трисомии по данной хромосоме; образец считается не прошедшим контроль качества, если определяемый уровень фетальной фракции образца ниже порогового значения в 3,5%, или если после всех стадий биоинформатической подготовки образца количество чтений стало меньше 1 млн.
| СПОСОБ НЕИНВАЗИВНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА МЕТОДОМ СЕКВЕНИРОВАНИЯ | 2014 |
|
RU2543155C1 |
| ТЕХНОЛОГИЯ ОПРЕДЕЛЕНИЯ ТРИСОМИИ ХРОМОСОМ МЕТОДОМ СЕКВЕНИРОВАНИЯ | 2012 |
|
RU2507269C2 |
| СПОСОБ НЕИНВАЗИВНОЙ ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА | 2015 |
|
RU2627673C2 |
| СПОСОБ НЕИНВАЗИВНОЙ ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА | 2014 |
|
RU2583830C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИСТОЧНИКА АНЕУПЛОИДНЫХ КЛЕТОК ПО КРОВИ БЕРЕМЕННОЙ ЖЕНЩИНЫ | 2016 |
|
RU2674700C2 |
| WO 2018132400 A1, 19.07.2018. | |||

