Изобретение относится к области органической химии, в частности к усовершенствованию способа получения простых виниловых эфиров на основе реакции соответствующих спиртов и дихлорэтана, как синтетического эквивалента ацетилена.
Виниловые эфиры относятся к наиболее значимым классам органических соединений [Б.А. Трофимов, Гетероатомные производные ацетилена. Новые полифункциональные мономеры, реагенты и полупродукты, Москва, Наука, 1981, 319 с]. Функциональные виниловые эфиры являются стартовыми соединениями для тонкого органического синтеза, прогрессивными строительными блоками для новых технологий. На основе виниловых эфиров созданы синтетические волокна и пластмассы, лаки, клеи, флотореагенты и экстрагенты, препараты для медицины и сельского хозяйства и т.д.
В 1930-1940-е годы виниловые эфиры были получены М.Ф. Шостаковским под руководством академика А.Е. Фаворского [М.Ф. Шостаковский Академик Алексей Евграфович Фаворский. М.: ГНТИ химич. лит-ры, 1953, 157 с].
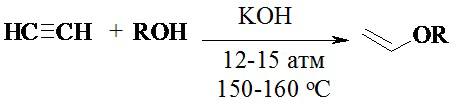
Было показано, что какой-либо спирт при нагревании в автоклаве (150-160°С), в присутствии КОН и ацетилена под давлением 12-15 атм образует виниловые эфиры с выходами около 96%. Уникальность метода состояла в том, что впервые для винилирования спиртов под давлением использовался чистый (не разбавленный инертным газом) ацетилен. Этот метод обеспечил более высокую производительность и был проще в технологическом оформлении [Б.А. Трофимов, Н.К. Гусарова, Успехи химии, 2007, 76, 550-570].
Позднее в Иркутском институте химии им. А.Е. Фаворского СО РАН были разработаны более общие подходы к синтезу простых виниловых эфиров на основе ацетилена с использованием суперосновных сред, состоящих из сильного основания и полярного негидроксильного комплексообразующего растворителя [Б.А. Трофимов, ЖОрХ, 1995, 31, 9, 1368-1387; B.A. Trofimov, Curr. Org. Chem., 2002, 6, 13, 1121-1162]. Они создали реальные возможности для направленного изучения этих высокореакционноспособных строительных блоков в органическом синтезе.
Безусловно, метод винилирования спиртов ацетиленом очень удобен, довольно недорогой и позволяет получать виниловые эфиры из дешевых реагентов, но использование газообразного ацетилена, особенно в промышленности, может быть сопряжено с рядом существенных ограничений:
• ацетилен–это газообразное вещество с высокой проникающей способностью;
• ацетилено-воздушные и ацетилено-кислородные смеси в очень широком диапазоне взрываются при наличии искры, открытого огня, нагретой поверхности или какого-либо другого источника воспламенения;
• ацетилен требует особой осторожности при хранении и эксплуатации.
Особенно серьезным недостатком для процессов с участием ацетилена является использование повышенного давления. В связи с этим во многих странах существует законодательное ограничение по этому параметру (обычно 0.4-0.5 атм) [Н.Ф. Кононов, С.А. Островский, А.А. Устынюк, Новая технология некоторых синтезов на основе ацетилена, Наука, Москва, 1977, 17 с].
Наиболее близкими по технической сущности к предлагаемому изобретению является следующий способ (прототип).
Показан синтез алкилвиниловых эфиров [Л.А. Опарина, С.И. Шайхудинова, Л.Н. Паршина, О.В. Высоцкая, Th. Preiss, J. Henkelmann, Б.А. Трофимов, ЖОрХ 2005, 41, 672-676] реакцией нуклеофильного присоединения первичных и вторичных спиртов к ацетилену с использованием каталитической системы CsF/MOH (M = Li, Na) при атмосферном (ДМСО, 100°С) или повышенном (без растворителя, 135–140°С) давлении ацетилена, который приводит к алкилвиниловым эфирам с выходами до 93%. Существенными недостатками первого способа (прототипа), несмотря на высокие выходы виниловых эфиров, является высокая взрывоопасность процесса, связанная с применением ацетилена в проточной системе, а также под давлением, при высокой температуре и с использованием фторида цезия, что требует специальных условий проведения процесса (инертная атмосфера, полное обезвоживание реагентов и растворителей). Перечисленные недостатки препятствуют практическому использованию способа-прототипа для получения виниловых эфиров в укрупненных количествах.
Одним из возможных решений проблем способа-прототипа может стать замена газообразного ацетилена на его предшественник, выделяющий ацетилен в ходе реакции. Ранее был предложен синтез виниловых эфиров с использованием карбида кальция, как синтетического предшественника ацетилена, и различных ароматических спиртов в суперосновной системе KOH/ДМСО [R. Matake, A. Yusuke, H. Matsubara, Green Chem., 2016, 18, 2614-2618]. В пробирку из термостойкого стекла помещают ароматический спирт, карбид кальция, воду, гидроксид калия и диметилсульфоксид, после чего смесь нагревают до 120°С и перемешивают в течение 15 часов. Существенным недостатком этого способа, несмотря на высокие выходы виниловых эфиров, является длительное время реакции (до 15 часов), а также использование карбида кальция, который не позволит провести укрупнение синтеза до промышленных масштабов.
Во-первых, с технологической точки зрения использование карбида кальция связано с энергозатратностью процесса, так как карбид кальция получают прокаливанием смеси оксида кальция с коксом в электрических печах при температуре 1900-1950°С. Таким образом, это может быть невыгодно экономически. Во-вторых, при генерации ацетилена из воды и карбида кальция образуется так называемая пушонка – это рыхлая структура гидроксида кальция (гашеная известь), которая всегда мешает технологизации процесса ввиду «забивания» технологичных узлов.
Таким образом, несмотря на некоторые литературные данные о способах винилирования различных спиртов, большинство из них не воплощены в укрупненном масштабе или являются опасными.
Технической задачей настоящего изобретения является разработка способа получения функционализированных алкилвиниловых эфиров, обладающего существенной новизной и не имеющего недостатков указанных способов-прототипов.
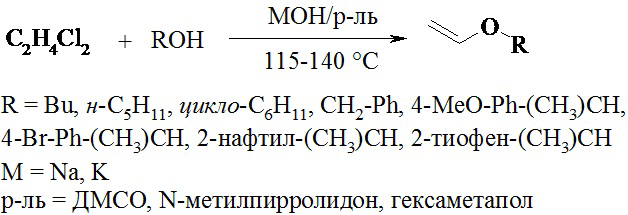
Целью настоящего изобретения является усовершенствование способа получения виниловых эфиров с использованием в качестве синтетического эквивалента ацетилена дихлорэтана (ДХЭ); процесс приводит к сравнимым и устойчивым выходам, что позволяет применить предлагаемый способ для крупномасштабного производства. Дихлорэтан является полупродуктом в производстве винилхлорида, а также отходом производств ПВХ (до 30% масс. в общей массе хлорорганических отходов по данным центральной лаборатории ОАО «Саянскхимпром» 1998-2006 гг.) [Б.М. Ласкин, М.З. Вдовец, Д.А. Мухортов, О.Н. Вознюк, А.И. Тугай, Химия и химическая технология. Технология неорганических веществ, 2013, 20, 35-40], при этом является жидким удобно дозируемым веществом. Использование дихлорэтана в качестве синтетического эквивалента ацетилена более перспективно, так как ДХЭ не взрывоопасен и прост в обращении. Данная технология позволит создать гибкие технологические схемы и сателлитные производства на базе промплощадок заводов, занимающихся синтезом ПВХ [В.Г. Данилкина, Т.М. Филиппова, Сборник научных трудов Ангарской государственной технической академии, 2013, 1, 277-283].
Нами была изучена реакция дегидрохлорирования ДХЭ в суперосновной системе KOH/ДМСО.
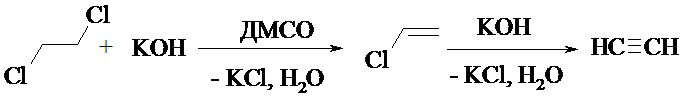
Выяснено, что при контроле температурного интервала от 25°С до 140°С, выделение газообразных продуктов начинается от 115 до 120°С. Для доказательства выделения ацетилена нами были проведены качественные реакции:
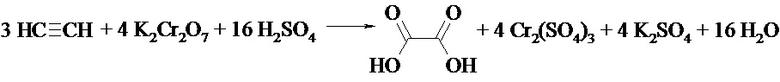
1) С обесцвечиванием водного раствора KMnO4
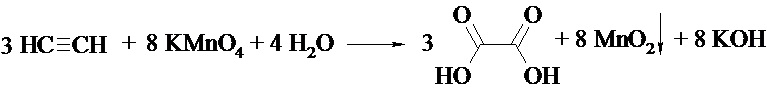
2) С [Сu(NH3)2]Cl (изменение цвета: из голубого раствора [Сu(NH3)2]Cl образуется красно-кирпичный осадок)

3) С K2Cr2O7 (изменение цвета: из оранжевого цвета раствора в зеленый)
Таким образом, для реакции винилирования различных алифатических спиртов оптимальной температурой является диапазон от 115-140°С. Далее нами была проведена оптимизация по варьированию количества щелочи и ДХЭ. Опытным путем выяснено, что при соотношении реагентов спирт : ДХЭ : щелочь - 1:3:8 выход н-бутилвинилового эфира составил 65%.
Винилирование н-бутанола дихлорэтаном в среде KOH/ДМСО при температуре 115-120°С
При замене полярного негидроксильного комплексообразующего растворителя в реакции винилирования н-бутанола ДМСО на N-метилпирролидон и гексаметапол, происходит снижение выхода винилового эфира на 33 и 28 % соответственно. А также при замене щелочи с КОН на NaOH происходит также снижение выхода до 42%.
Реакция винилирования различных алифатических спиртов проводится в интервале температур 115-140°С, в течение 1,5-2 часов в каталитической системе: гидроксид щелочного металла/полярный негидроксильный комплексообразующий растворитель (МOH/растворитель).
Синтетический эквивалент ацетилена - дихлорэтан - подается в реакцию постепенно, что обеспечивает примерно постоянную его концентрацию в реакционной смеси в течение всего времени реакции. Целевые виниловые эфиры выделяются вакуумной разгонкой из реакционной смеси. При этом из реакционной смеси выгоняется чистый растворитель, который можно повторно использовать в дальнейшем для получения виниловых эфиров или других целей. После вакуумной разгонки в колбе остается водорастворимый твердый остаток, представляющий из себя смесь хлорида и гидроксида калия. В промышленности хлорид калия является ценным удобрением, а также его можно использовать в электролизе с образованием водорода, хлора и гидроксида калия. Таким образом, предлагаемый нами способ получения виниловых эфиров станет практически безотходным и экологически безопасным. Выходы чистых виниловых эфиров варьируются в диапазоне 51-78% и представляют собой жидкости от прозрачного до желтого цвета. Следует отметить, что в процессе используются коммерчески доступные растворители без предварительной очистки и осушки.
Процесс винилирования спиртов, дихлорэтаном в системе МOH/растворитель (M=Na, K) сопровождается предварительной подготовкой: смесь необходимых для проведения синтеза количеств щелочи в полярном негидроксильном комплексообразующем растворителе греют при интенсивном перемешивании при температуре 70°С в течение 30-40 мин, после чего добавляют спирт и поднимают температуру реакции до 115-140°С, затем начинают прикапывать дихлорэтан. Такая подготовка обеспечивает приготовление активной формы сверхосновного катализатора.
Нами показано, что генерирование ацетилена из дихлорэтана в суперосновной системе начинается лишь при температуре выше 115°С. При более низкой температуре дегидрохлорирование не идет. Однако многие спирты по литературным данным винилируются при более низкой температуре. В этом случае мы применили другую модификацию реакции: в колбу, содержащую суперосновную суспензию MOH/растворитель и нагретую до 115-140°С, по каплям прикапываем дихлорэтан; выделяющийся ацетилен по газоотводной трубке поступает в другую колбу, содержащую раствор спирта в суперосновной системе и нагретую до температуры винилирования спирта. Такая модификация процесса позволяет существенно расширить количество вовлекаемых в реакцию спиртов и не ведет к значительному снижению технологичности процесса.
За счет замены газообразного ацетилена на его синтетический аналог - дихлорэтан - обеспечивается усовершенствование процесса винилирования функционализированных алифатических спиртов, а также упрощение выделения чистых целевых виниловых эфиров. Наглядной демонстрацией преимуществ предлагаемого способа является безотходное и экологически безопасное производство.
Предлагаемый способ позволяет также значительно упростить стадию выделения целевого продукта путем его вакуумной разгонки непосредственно из реакционной смеси без предварительных подготовительных операций. Одновременно это упрощает проблему регенерации и очистки растворителя. Однако следует отметить, что традиционная методика выделения виниловых эфиров с разбавлением реакционной смеси водой и дальнейшей экстракцией диэтиловым эфиром тоже может быть применена, что продемонстрировано на примерах.
Необходимо подчеркнуть, что предлагаемый способ обеспечивает практически полную утилизацию хлорорганических отходов на производстве ПВХ и созданию дополнительных производственных мощностей.
Следующие неограничивающие примеры иллюстрируют изобретение и показывают, что процессом можно легко управлять, меняя его параметры, такие как соотношение реагентов, растворитель, температура и время. Спектральные характеристики полученных продуктов полностью соответствуют описанным в литературе [Л.А. Опарина, С.И. Шайхудинова, Л.Н. Паршина, О.В. Высоцкая, Th. Preiss, J. Henkelmann, Б.А. Трофимов, ЖОрХ, 2005, 41, 672-676].
Пример 1
В 3-х горловую колбу объемом 100 мл, снабженную обратным эффективным холодильником, термометром, магнитной мешалкой и склянкой Дрекселя для фиксации газообразных продуктов, помещают 35 мл ДМСО, 20.8 г (0.32 моль) КОН⋅0.5Н2О. Перемешивают при температуре 70°С 30 минут (время отсчитывают от точки выхода на режим 70°С), затем добавляют 2.96 г (0.040 моль) н-бутанола, смывая 5 мл ДМСО (общее количество ДМСО 40 мл), и повышают температуру до 115°С. По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,2-дихлорэтана (ДХЭ) в течение 1.5 часов, при этом после добавления последних капель дихлорэтана продолжают перемешивание в течение 30 минут до полного превращения дихлорэтана. Соотношение реагентов: 8 (KOH) : 3 (ДХЭ) : 1 (спирт). После охлаждения реакционную смесь разбавляют 100 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×30 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, остаток перегоняют по фракциям. Получают 2.60 г н-бутилвинилового эфира с т. кип. 93-94°С (выход 65%).
Пример 2.
В условиях примера 1, проводим реакцию при повышенной температуре (130-140°С) в течение 1,5-2 ч, по окончании прикапывания 1,2-дихлорэтана реакционную смесь охлаждают, проводят экстракцию и сушат К2СО3. Получают, как описано в примере 1, 2.19 г н-бутилвинилового эфира с т. кип. 93-94°С (выход 54%).
Пример 3.
В условиях примера 1, используют те же загрузки исходных реагентов. Проводят реакцию при той же температуре и том же времени реакции. По окончанию прикапывания 1,2-дихлорэтана реакционную смесь охлаждают, переливают в колбу Кляйзена и перегоняют при пониженном давлении. В охлажденной ловушке собирают азеотроп, который представляет собой н-бутилвиниловый эфир с водой. Далее отгоняют ДМСО (до 80°С). По окончании перегонки полученный азеотроп переливают в делительную воронку и разделяют слои. Получают, как описано в примере 1, 2.55 г н-бутилвинилового эфира (выход 64%).
Пример 4.
В условиях примера 1, в той же колбе, используя те же загрузки КОН⋅0.5Н2О и ДМСО, а также 2.96 г (0.040 моль) н-бутанола, повышая температуру до 115-120°С (температура реакции). По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,1-дихлорэтана в течение 1.5-2 часов. Далее реакционную смесь экстрагируют диэтиловым эфиром (3×10 мл). Получают, как описано в примере 1, 1.99 г н-бутил винилового эфира (выход 50%).
Пример 5.
В условиях примера 1, в двух разных колбах. В первой колбе при 115-120°С происходит генерация ацетилена из ДХЭ с отводом во вторую колбу, в которой происходит винилирование спирта при 90°С. Загрузки в первой колбе 20 мл ДМСО, 7.8 г (0.12 моль) КОН⋅0.5Н2О, 5.94 г (0.06 моль) 1,2-дихлорэтана. Соотношение реагентов: 6 (KOH) : 3 (ДХЭ). Загрузки во второй колбе 20 мл ДМСО, 1.48 г (0.020 моль) н-бутанола и 1.3 г (0.020 моль) КОН⋅0.5Н2О. Соотношение реагентов: 1 (спирт) : 1 (КОН). По достижению температуры начинают прикапывать 1,2-дихлорэтан (ДХЭ) в течение 2 часов. После охлаждения реакционную смесь второй колбы разбавляют 50 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×15 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, остаток перегоняют по фракциям. Получают 1.19 г н-бутилвинилового эфира с т. кип. 93-94°С (выход 60%).
Пример 6.
В условиях примера 1, изменяют исходный спирт. В 3-х горловую колбу объемом 100 мл, помещают 35 мл ДМСО, 20.8 г (0.32 моль) КОН⋅0.5Н2О. Перемешивают при температуре 70°С 30 минут (время отсчитывают от точки выхода на режим 70°С), затем добавляют 3.53 г (0.040 моль) н-пентанола смывая 5 мл ДМСО (общее количество ДМСО 40 мл), и повышают температуру до 115°С. По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,2-дихлорэтана (ДХЭ) в течение 1.5 часов, при этом после добавления последних капель дихлорэтана продолжают перемешивание в течение 30 минут до полного превращения дихлорэтана. Соотношение реагентов: 8 (KOH) : 3 (ДХЭ) : 1 (спирт). После охлаждения реакционную смесь разбавляют 100 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×30 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, остаток перегоняют по фракциям. Получают 3.57 г амилвинилового эфира с т. кип. 110-111°С (выход 78%).
Пример 7.
В условиях примера 1, изменяют исходный спирт. В 3-х горловую колбу объемом 100 мл, помещают 35 мл ДМСО, 20.8 г (0.32 моль) КОН⋅0.5Н2О. Перемешивают при температуре 70°С 30 минут (время отсчитывают от точки выхода на режим 70°С), затем добавляют 4 г (0.040 моль) циклогексанола смывая 5 мл ДМСО (общее количество ДМСО 40 мл), и повышают температуру до 115°С. По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,2-дихлорэтана (ДХЭ) в течение 1.5 часов, при этом после добавления последних капель дихлорэтана продолжают перемешивание в течение 30 минут до полного превращения дихлорэтана. Соотношение реагентов: 8 (KOH) : 3 (ДХЭ) : 1 (спирт). После охлаждения реакционную смесь разбавляют 100 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×30 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, остаток перегоняют по фракциям. Получают 3.18 г винилциклогексанового эфира с т. кип. 147-148°С (выход 63%).
Пример 8.
В условиях примера 1, изменяют исходный спирт. В 3-х горловую колбу объемом 100 мл, помещают 35 мл ДМСО, 20.8 г (0.32 моль) КОН⋅0.5Н2О. Перемешивают при температуре 70°С 30 минут (время отсчитывают от точки выхода на режим 70°С), затем добавляют 4.32 г (0.04 моль) бензилового спирта смывая 5 мл ДМСО (общее количество ДМСО 40 мл), и повышают температуру до 115°С. По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,2-дихлорэтана (ДХЭ) в течение 1.5 часов, при этом после добавления последних капель дихлорэтана продолжают перемешивание в течение 30 минут до полного превращения дихлорэтана. Соотношение реагентов: 8 (KOH) : 3 (ДХЭ) : 1 (спирт). После охлаждения реакционную смесь разбавляют 100 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×30 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, остаток перегоняют по фракциям. Получают 3.65 г винилбензилового эфира (выход 68%).
Пример 9.
В условиях примера 1, изменяют исходный спирт. В 3-х горловую колбу объемом 100 мл, помещают 35 мл ДМСО, 20.8 г (0.32 моль) КОН⋅0.5Н2О. Перемешивают при температуре 70°С 30 минут (время отсчитывают от точки выхода на режим 70°С), затем добавляют 6.09 г (0.04 моль) 1-(4-метоксифенил)-этанола смывая 5 мл ДМСО (общее количество ДМСО 40 мл), и повышают температуру до 115°С. По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,2-дихлорэтана (ДХЭ) в течение 1.5 часов, при этом после добавления последних капель дихлорэтана продолжают перемешивание в течение 30 минут до полного превращения дихлорэтана. Соотношение реагентов: 8 (KOH) : 3 (ДХЭ) : 1 (спирт). После охлаждения реакционную смесь разбавляют 100 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×30 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, очищают колоночной хроматографией на силикагеле, элюент - гексан. Получают 4.99 г виниловый эфир 1-(4-метоксифенил)-этанола (выход 70%).
Пример 10.
В условиях примера 1, изменяют исходный спирт. В 3-х горловую колбу объемом 100 мл, помещают 35 мл ДМСО, 20.8 г (0.32 моль) КОН⋅0.5Н2О. Перемешивают при температуре 70°С 30 минут (время отсчитывают от точки выхода на режим 70°С), затем добавляют 8.04 г (0.04 моль) 1-(4-бромфенил)-этанола смывая 5 мл ДМСО (общее количество ДМСО 40 мл), и повышают температуру до 115°С. По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,2-дихлорэтана (ДХЭ) в течение 1.5 часов, при этом после добавления последних капель дихлорэтана продолжают перемешивание в течение 30 минут до полного превращения дихлорэтана. Соотношение реагентов: 8 (KOH) : 3 (ДХЭ) : 1 (спирт). После охлаждения реакционную смесь разбавляют 100 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×30 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, очищают колоночной хроматографией на силикагеле, элюент - гексан. Получают 4.63 г виниловый эфир 1-(4-бромфенил)этанола (выход 51%).
Пример 11.
В условиях примера 1, изменяют исходный спирт. В 3-х горловую колбу объемом 100 мл, помещают 35 мл ДМСО, 20.8 г (0.32 моль) КОН⋅0.5Н2О. Перемешивают при температуре 70°С 30 минут (время отсчитывают от точки выхода на режим 70°С), затем добавляют 6.89 г (0.04 моль) 1-(нафтил)-этанола смывая 5 мл ДМСО (общее количество ДМСО 40 мл), и повышают температуру до 115°С. По достижению температуры начинают прикапывать 11.88 г (0.12 моль) 1,2-дихлорэтана (ДХЭ) в течение 1.5 часов, при этом после добавления последних капель дихлорэтана продолжают перемешивание в течение 30 минут до полного превращения дихлорэтана. Соотношение реагентов: 8 (KOH) : 3 (ДХЭ) : 1 (спирт). После охлаждения реакционную смесь разбавляют 100 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×30 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, очищают колоночной хроматографией на силикагеле, элюент - гексан. Получают 6.03 г виниловый эфир 1-(нафтил)-этанола (выход 76%).
Пример 12.
В условиях примера 1, в двух разных колбах. В первой колбе при 115-120°С происходит генерация ацетилена из ДХЭ с отводом во вторую колбу, в которой происходит винилирование спирта при 90°С. Загрузки в первой колбе 20 мл ДМСО, 7.8 г (0.12 моль) КОН⋅0.5Н2О, 5.94 г (0.06 моль) 1,2-дихлорэтана. Соотношение реагентов: 6 (KOH) : 3 (ДХЭ). Загрузки во второй колбе 20 мл ДМСО, 3 г (0.02 моль) 1-(тиофен-2-ил)-этанола и 1.52 г (0.02 моль) КОН⋅0.5Н2О. Соотношение реагентов: 1 (спирт) : 1 (КОН). По достижению температуры начинают прикапывать 1,2-дихлорэтана (ДХЭ) в течение 2 часов. После охлаждения реакционную смесь второй колбы разбавляют 50 мл воды, проводят экстракцию диэтиловым эфиром (4×30 мл), эфирный экстракт промывают водой (3×15 мл) для удаления ДМСО и сушат К2СО3. Удаляют растворитель на роторном испарителе, очищают колоночной хроматографией на силикагеле, элюент - гексан. Получают 2.02 г винилового эфира 1-(тиофен-2-ил)этанола (выход 56%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛОВЫХ ЭФИРОВ НИЗШИХ СПИРТОВ | 1986 |
|
SU1504969A1 |
| Способ получения винилового эфира циклогексанола | 1990 |
|
SU1796613A1 |
| Способ получения виниловых эфиров аминофенолов | 2016 |
|
RU2640808C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛФУРФУРИЛОВЫХ ЭФИРОВ | 2008 |
|
RU2359965C1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕТУЛИНОВОЙ КИСЛОТЫ | 2014 |
|
RU2565773C1 |
| Атом-экономный безотходный способ получения 2-(4-фторфенил)-бут-3-ин-2-ола | 2016 |
|
RU2629024C1 |
| Способ получения водорастворимых пропаргиловых эфиров полисахарида арабиногалактана | 2016 |
|
RU2650544C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРИЛ (ГЕТАРИЛ) ЭТИНИЛКАРБИНОЛОВ | 2012 |
|
RU2479565C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИДИВИНИЛФОСФИНОВОЙ КИСЛОТЫ ИЗ КРАСНОГО ФОСФОРА И АЦЕТИЛЕНА | 2015 |
|
RU2632816C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛСУЛЬФИДОВ | 2005 |
|
RU2284320C1 |
Изобретение относится к способу получения простых виниловых эфиров, который заключается во взаимодействии различных алифатических спиртов и дихлорэтана, как синтетического эквивалента ацетилена, в суперосновной системе гидроксид щелочного Ме/высокополярный негидроксильный растворитель, например диметилсульфоксид. Процесс проводят при температуре 115-140°С в течение 1,5-2 часов. Мольное соотношение реагентов спирт : дихлорэтан : щелочь 1:3:8. Технический результат - усовершенствование процесса винилирования алифатических спиртов, а также упрощение выделения чистых целевых виниловых эфиров (простота и экологическая безопасность). 12 пр.
Способ получения простых виниловых эфиров взаимодействием различных алифатических спиртов, щелочей и синтетического эквивалента ацетилена, отличающийся тем, что в качестве синтетического эквивалента ацетилена используют 1,1- или 1,2-дихлорэтан в суперосновной системе гидроксид щелочного Ме/высокополярный негидроксильный растворитель и процесс проводят при температуре 115-140°С в течение 1,5-2 часов при мольном соотношении реагентов спирт : дихлорэтан : щелочь 1:3:8.
| Л.А | |||
| Опарина и др | |||
| Нуклеофильное присоединение к ацетиленам в суперосновных каталитических системах | |||
| XIII | |||
| Фторид-цезийсодержащие системы, эффективные катализаторы винилирования алканолов, Российский журнал органической химии, 2005, том 41, выпуск 5, с | |||
| Прибор для механического определения проекций линий данной длины и данного направления | 1923 |
|
SU656A1 |
| Способ получения винилового эфира циклогексанола | 1990 |
|
SU1796613A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛОВЫХ ЭФИРОВ НИЗШИХ СПИРТОВ | 1986 |
|
SU1504969A1 |
| Matake, R | |||
| et al., Synthesis of | |||

