Уровень техники
Область техники, к которой относится изобретение
В настоящей заявке описаны композиции и способы для лечения врожденной гиперплазии коры надпочечников.
Описание предшествующего уровня техники
Врожденная гиперплазия коры надпочечников (ВГКН) представляет собой группу аутосомно-рецессивных генетических нарушений, приводящих к снижению биосинтеза кортизола или к его отсутствию. Наиболее частой формой заболевания является дефицит 21-гидроксилазы, вызванный мутациями в гене CYP21A2, расположенном на хромосоме 6p21, который составляет приблизительно 95% всех случаев ВГКН (см., напр., обзор Speiser et al., Int. J. Pediatr. Endocrinol. 2010:494173 (2010)). Эти мутации могут варьировать от полной потери ферментативной активности, необходимой для синтеза кортизола в коре надпочечников до целого спектра частичной потери ферментативной активности, что обуславливает степень тяжести заболевания, которая является прямым следствием конкретной мутации. Классификация этого спектра недостаточности 21-гидроксилазы в широком смысле включает сольтеряющую и простую вирильную формы, относящиеся к классической ВГКН, и более умеренную форму, известную как неклассическая ВГКН или «поздняя» ВГКН, которая, как правило, диагностируется в позднем подростковом возрасте или у молодых взрослых. Пациенты с неклассической ВГКН являются либо гомозиготными, либо сложногетерозиготными, часто имеющими аллель классической ВГКН. Такие пациенты обладают достаточной ферментативной активностью (> 20-50% нормальной), чтобы не страдать от потери соли или от дефицита кортизола, при рождении обладают нормально развитыми наружными половыми органами и на протяжении всей жизни могут не иметь симптомов заболевания (Trapp et al., Steroids 77(4):342-46 (2012)). При более редкой форме заболевания, составляющей 5% случаев, к ВГКН приводит мутация в гене 11β-гидроксилазы CYP11B1 (11β-OH ВГКН).
Обе генетические мутации приводят к врожденной гиперплазии коры надпочечников, дефициту кортизола и избыточному образованию адренокортикотропного гормона (АКТГ) с повышенным образованием андрогенов. Такие пациенты нуждаются в пожизненном приеме глюкокортикоидов и страдают от сопутствующих проблем, связанных с таким лечением. Соответственно, существует острая потребность в схемах лечения, которые улучшали бы состояние здоровья, самочувствие, качество жизни и контролировали бы связанные заболевания у пациентов с ВГКН.
Краткое содержание изобретения
Кортиколиберин (CRF, corticotropin-releasing factor, кортикотропин-рилизинг фактор) активирует рецептор CRF1, относящийся к классу В рецепторов, сопряженных с G-белками (GPCR, G protein-coupled receptor). Антагонисты CRF1 могут напрямую ингибировать высвобождение АКТГ у пациентов с ВГКН, обеспечивая таким образом нормализацию образования андрогена при пониженной, более физиологичной дозе гидрокортизона и сниженных связанных с лечением побочных эффектах.
В одном варианте осуществления настоящего изобретения представлен способ лечения ВГКН посредством введения нуждающемуся в этом субъекту эффективного количества антагониста CRF1, включая (но не ограничиваясь) введение перед сном.
В более конкретном варианте осуществления настоящего изобретения антагонист CRF1 имеет период полудиссоциации (t1/2) свыше 30 минут, а в другом варианте осуществления настоящего изобретения - свыше 40 минут, а в другом варианте осуществления настоящего изобретения - свыше 50 минут.
Вариант осуществления настоящего изобретения 1. Способ лечения врожденной гиперплазии коры надпочечников (ВГКН) посредством введения нуждающемуся в этом субъекту антагониста рецептора CRF1, имеющего период полудиссоциации свыше 30 минут.
Вариант осуществления настоящего изобретения 2. Способ по варианту осуществления настоящего изобретения 1, где период полудиссоциации антагониста рецептора CRF1 составляет свыше 40 минут.
Вариант осуществления настоящего изобретения 3. Способ по варианту осуществления настоящего изобретения 1, где период полудиссоциации антагониста рецептора CRF1 составляет свыше 50 минут.
Вариант осуществления настоящего изобретения 4. Способ по любому из вариантов осуществления настоящего изобретения 1-3, где антагонист рецептора CRF1 является Соединением I (NBI-77860; 2,5-диметил-3-[2-метил-4-(метилокси)фенил]-N-[(1S-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил]пиразоло[1,5-a]пиримидин-7-амин).
Вариант осуществления настоящего изобретения 5. Способ по любому из вариантов осуществления настоящего изобретения 1-3, где антагонист рецептора CRF1 является NBI-30775, NBI-34041, SSR-126374, SSR-125543, анталармином (N-бутил-N-этил-2,5,6-триметил-7-(2,4,6-триметилфенил)пирроло[3,2-e]пиримидин-4-амином) или DMP904.
Вариант осуществления настоящего изобретения 6. Способ по любому из вариантов осуществления настоящего изобретения 1-5, где антагонист рецептора CRF1 вводят перед сном.
Вариант осуществления настоящего изобретения 7. Способ по любому из вариантов осуществления настоящего изобретения 1-6, где антагонист рецептора CRF1 вводят во время или перед расчетным временем высвобождения АКТГ в соответствии с его циркадными ритмом.
Вариант осуществления настоящего изобретения 8. Способ по варианту осуществления настоящего изобретения 7, где антагонист рецептора CRF1 вводят за 3-4 часа до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом.
Вариант осуществления настоящего изобретения 9. Способ снижения уровня 17-гидроксипрогестерона и АКТГ у субъекта с врожденной гиперплазией коры надпочечников (ВГКН), при этом указанный способ включает введение субъекту антагониста рецептора CRF1 перед сном.
Вариант осуществления настоящего изобретения 10. Способ по варианту осуществления настоящего изобретения 9, где антагонист рецептора CRF1 вводят во время или перед расчетным временем высвобождения АКТГ в соответствии с его циркадными ритмом.
Вариант осуществления настоящего изобретения 11. Способ по варианту осуществления настоящего изобретения 9 или по варианту осуществления настоящего изобретения 10, где антагонист рецептора CRF1 вводят за 3-4 часа до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом.
Вариант осуществления настоящего изобретения 12. Способ по любому из вариантов осуществления настоящего изобретения 9-11, где антагонист рецептора CRF1 является Соединением I (NBI-77860; 2,5-диметил-3-[2-метил-4-(метилокси)фенил]-N-[(1S-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил]пиразоло[1,5-a]пиримидин-7-амин).
Вариант осуществления настоящего изобретения 13. Способ по любому из вариантов осуществления настоящего изобретения 9-11, где антагонист рецептора CRF1 является NBI-30775, NBI-34041, SSR-126374, SSR-125543, анталармином (N-бутил-N-этил-2,5,6-триметил-7-(2,4,6-триметилфенил)пирроло[3,2-e]пиримидин-4-амином) или DMP904.
Вариант осуществления настоящего изобретения 14. Антагонист рецептора CRF1 для применения в лечении врожденной гиперплазии коры надпочечников (ВГКН), при этом период полудиссоциации антагониста рецептора CRF1 составляет свыше 30 минут.
Вариант осуществления настоящего изобретения 15. Антагонист рецептора CRF1 по варианту осуществления настоящего изобретения 14, где период полудиссоциации антагониста рецептора CRF1 составляет свыше 40 минут.
Вариант осуществления настоящего изобретения 16. Антагонист рецептора CRF1 по варианту осуществления настоящего изобретения 14, где период полудиссоциации антагониста рецептора CRF1 составляет свыше 50 минут.
Вариант осуществления настоящего изобретения 17. Антагонист рецептора CRF1 по любому из вариантов осуществления настоящего изобретения 14-16, где антагонист рецептора CRF1 является Соединением I (NBI-77860; 2,5-диметил-3-[2-метил-4-(метилокси)фенил]-N-[(1S-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил]пиразоло[1,5-a]пиримидин-7-амин).
Вариант осуществления настоящего изобретения 18. Антагонист рецептора CRF1 по любому из вариантов осуществления настоящего изобретения 14-16, где антагонист рецептора CRF1 является NBI-30775, NBI-34041, SSR-126374, SSR-125543, анталармином (N-бутил-N-этил-2,5,6-триметил-7-(2,4,6-триметилфенил)пирроло[3,2-e]пиримидин-4-амином) или DMP904.
Вариант осуществления настоящего изобретения 19. Антагонист рецептора CRF1 по любому из вариантов осуществления настоящего изобретения 14-18, где антагонист рецептора CRF1 подходит для введения перед сном.
Вариант осуществления настоящего изобретения 20. Антагонист рецептора CRF1 по любому из вариантов осуществления настоящего изобретения 14-19, где антагонист рецептора CRF1 подходит для введения во время или перед расчетным временем высвобождения АКТГ в соответствии с его циркадными ритмом.
Вариант осуществления настоящего изобретения 21. Антагонист рецептора CRF1 по любому из вариантов осуществления настоящего изобретения 14-20, где антагонист рецептора CRF1 подходит для введения за 3-4 часа до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом.
В других вариантах осуществления настоящего изобретения способы и применения, описанные в настоящей заявке выше и далее, включают уменьшение количества глюкокортикоида или минералокортикоида по меньшей мере на 10%, 15%, 20%, 30%, 40%, 50%, 60% от рекомендуемой суточной дозы глюкокортикоида (такого как гидрокортизона, преднизона, преднизолона, дексаметазона или флудрокортизона), вводимой взрослому субъекту (напр., человеку) с ВГКН. В других вариантах осуществления настоящего изобретения способы и применения, описанные в настоящей заявке выше и далее, включают уменьшение количества глюкокортикоида или минералокортикоида по меньшей мере на 10%, 15%, 20%, 30%, 40%, 50%, 60% от рекомендуемой суточной дозы глюкокортикоида (напр., гидрокортизона) или минералокортикоида (напр., флудрокортизона), вводимой взрослому субъекту (напр., человеку) с ВГКН.
Эти и другие варианты осуществления настоящего изобретения станут очевидны из ссылки на следующее подробное описание изобретения. С этой целью в настоящей заявке приведены различные ссылки, описывающие более подробно конкретную информацию об уровне техники, процедуры, соединения и композиции, каждая из которых в полном объеме включена в настоящую заявку посредством ссылки.
Терминам, специально не определенным в настоящей заявке, следует приписывать те значения, которые им давали бы специалисты в данной области техники в свете раскрытия и контекста изобретения. Однако, как применяют в настоящем описании изобретения, если иное не оговорено, термины имеют указанные значения. На протяжении всего описания настоящего изобретения ссылка на «один вариант осуществления настоящего изобретения» или на «вариант осуществления настоящего изобретения» означает, что конкретное свойство, структура или характеристика, описанная в связи с вариантом осуществления настоящего изобретения, включена по меньшей мере в один вариант осуществления настоящего изобретения. Таким образом, фразы «в одном варианте осуществления настоящего изобретения» или «во варианте осуществления настоящего изобретения» на протяжении всего описания настоящего изобретения необязательно относятся к одному и тому же варианту осуществления настоящего изобретения. Более того, конкретные свойства, структуры или характеристики можно комбинировать любым подходящим способом в одном или нескольких вариантах осуществления настоящего изобретения.
Также, как применяют в настоящем описании изобретения и в прилагаемой формуле изобретения, формы единственного числа включают объекты во множественном числе, если иное четко не определено контекстом. Таким образом, например, ссылка на «не относящееся к человеку животное» может относиться к одному или нескольким не относящимся к человеку животным или ко множеству таких животных, а ссылка на «клетку» или «определенную клетку» включает ссылку на одну или несколько клеток и их эквивалентов (напр., на множество клеток), известных специалистам в данной области техники, и т.п. При описании стадий способа или при указании стадий способа в формуле изобретения стадии описаны в определенном порядке, так что описание первой стадии, проводимой «перед» (т.е. до) второй стадией, имеет такое же значение, как при обратной формулировке, что вторая стадия проводится «после» первой стадии. Термин «приблизительно», относящийся к числу или числовому интервалу, обозначает, что это число или числовой интервал указаны с аппроксимацией в рамках экспериментальных отклонений (или в рамках статистической ошибки эксперимента), и, следовательно, число или числовой интервал может варьировать на величину от 1% до 15% от заявленного числа или числового интервала. Также следует отметить, что термин «или», как правило, применяют как включающий «и/или», если иное четко не определено контекстом. Термин «по меньшей мере один», например, при указании по меньшей мере одного соединения или по меньшей мере одной композиции, имеет то же значение, что и термин «один или несколько».
Краткое описание чертежей
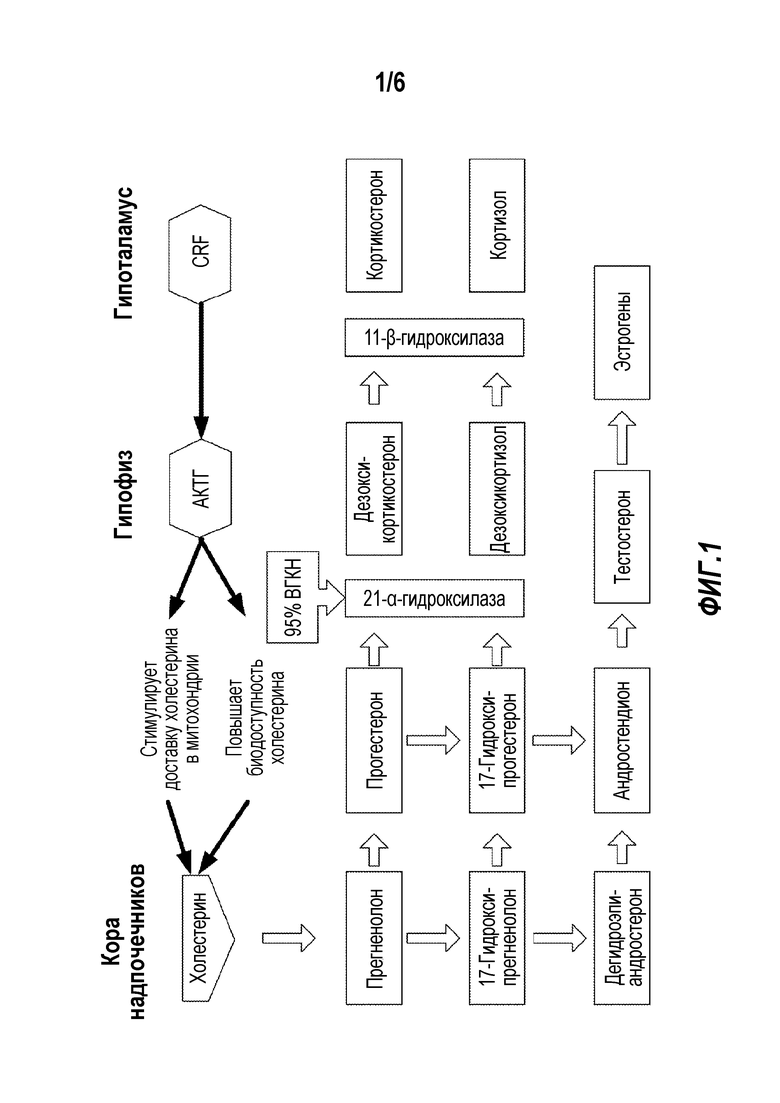
Фигура 1 показывает схему путей синтеза стероидов в коре надпочечников. Наиболее частая форма ВГКН вызвана дефицитом 21-гидроксилазы (также называемой 21-α-гидроксилазой), приводящей к снижению уровня кортизола и росту уровня андрогенов, таких как тестостерон и эстроген. Более редкий тип классической ВГКН является дефицитом 11β-гидроксилазы.
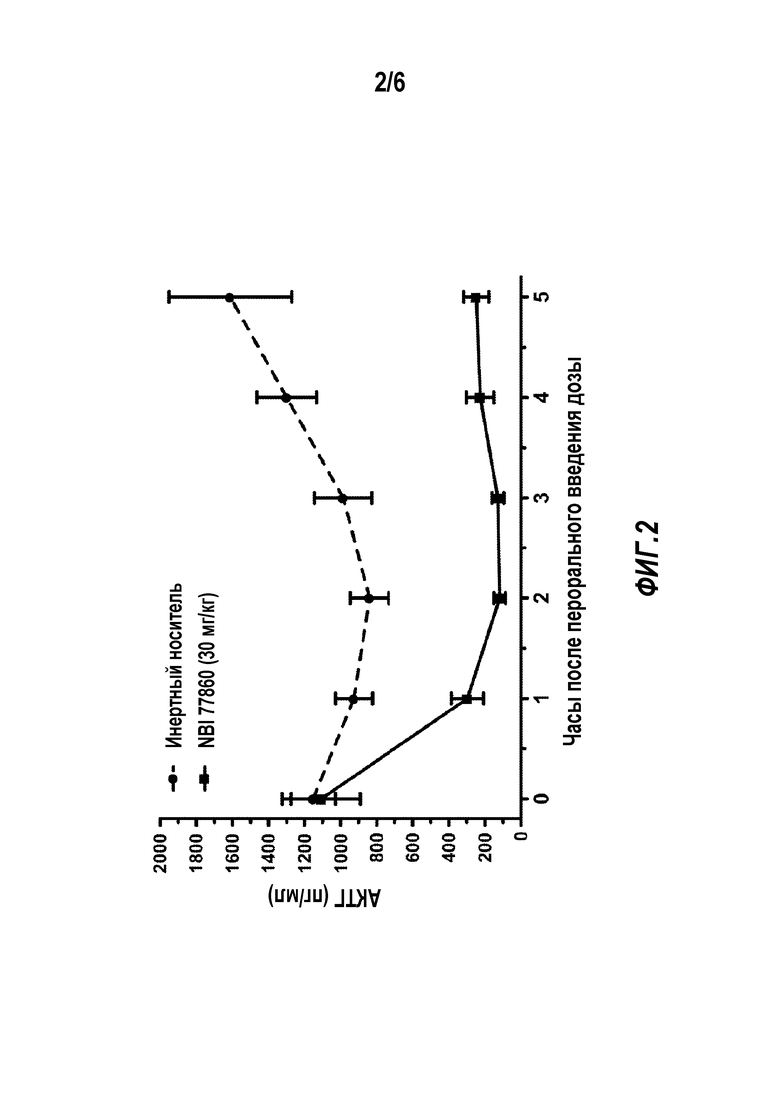
Фигура 2 представляет график, иллюстрирующий действие Соединения I (NBI 77860) на концентрацию АКТГ у адреналэктомированных крыс. Крысы получали 30 мг/кг Соединения I (NBI 77860) перорально. Данные представлены как средняя концентрация АКТГ в плазме (±стандартная ошибка среднего).
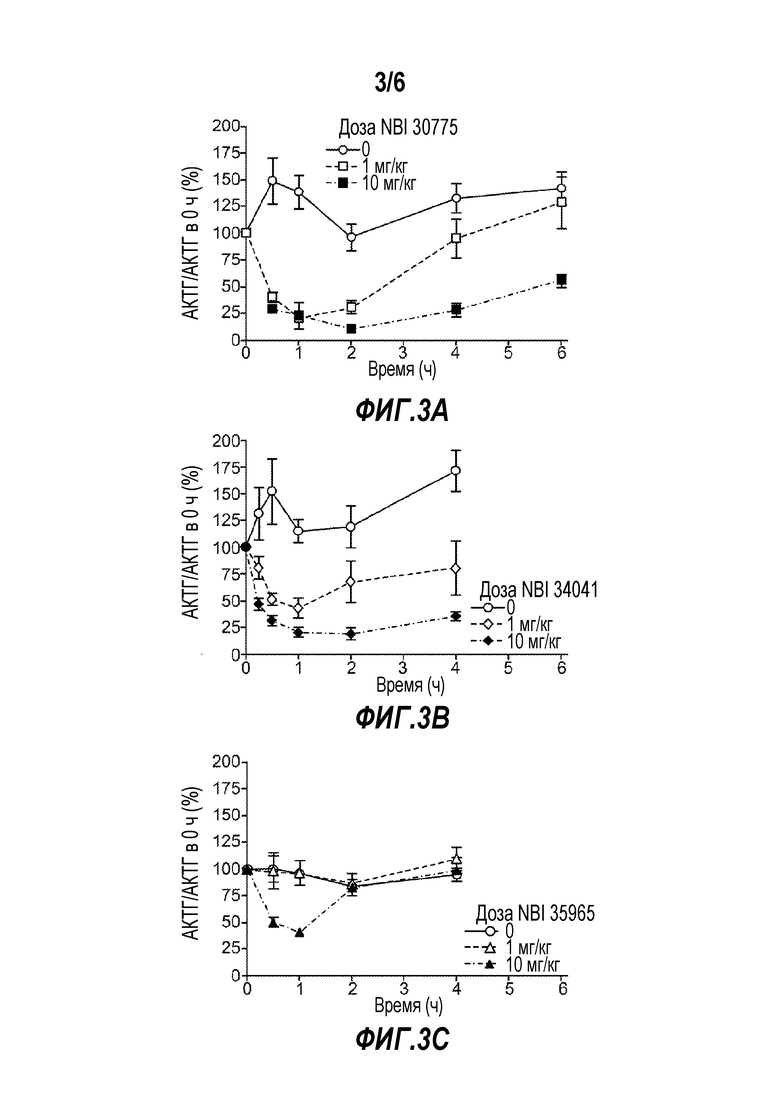
Фигуры 3A-C представляют графики, иллюстрирующие эффект антагонистов рецептора CRF1, различающихся по периоду полудиссоциации, на концентрацию АКТГ у адреналэктомированных крыс.
Фигура 4 представляет схему плана клинического исследования, описанного в Примере 6, спланированного для оценки безопасности, переносимости и суммарного количества в плазме NBI-77860, а также действия этого соединения на уровни эндогенных гормонов гипоталамо-гипофизарно-надпочечниковой системы.
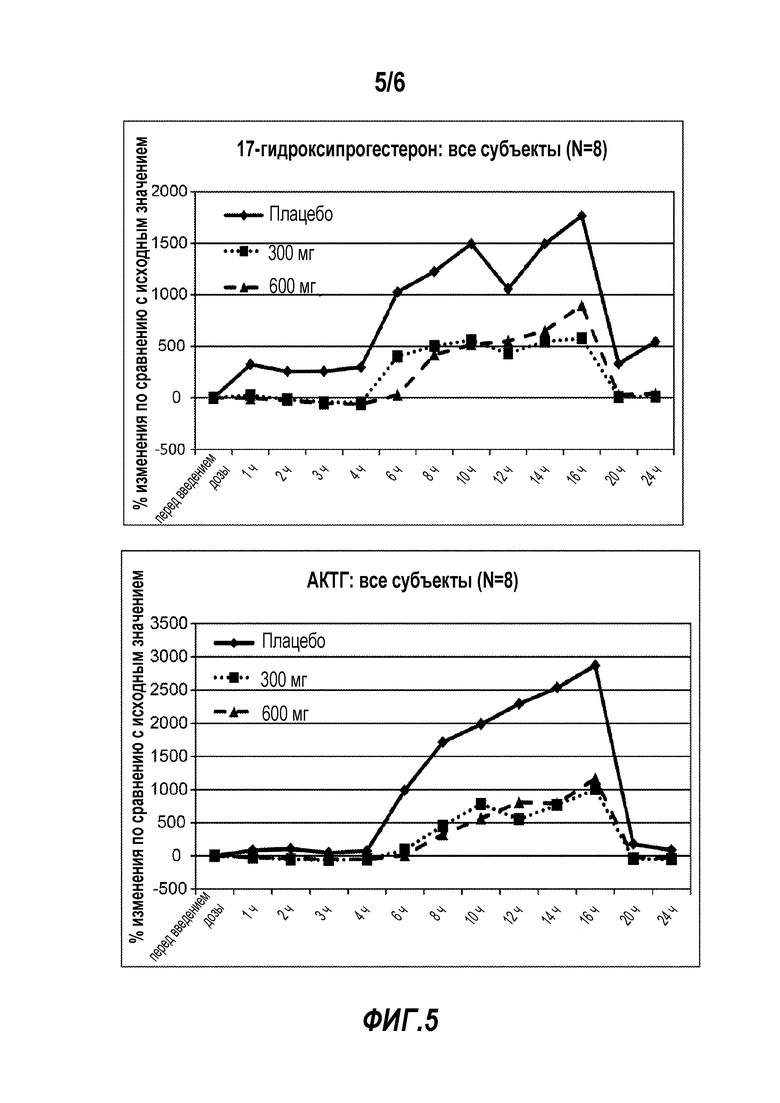
Фигура 5 представляет данные по среднему уровню 17-гидроксипрогестерона (верхняя панель) и АКТГ (нижняя панель) на протяжении 24-часового периода после введения дозы в клиническом исследовании, описанном в Примере 6.
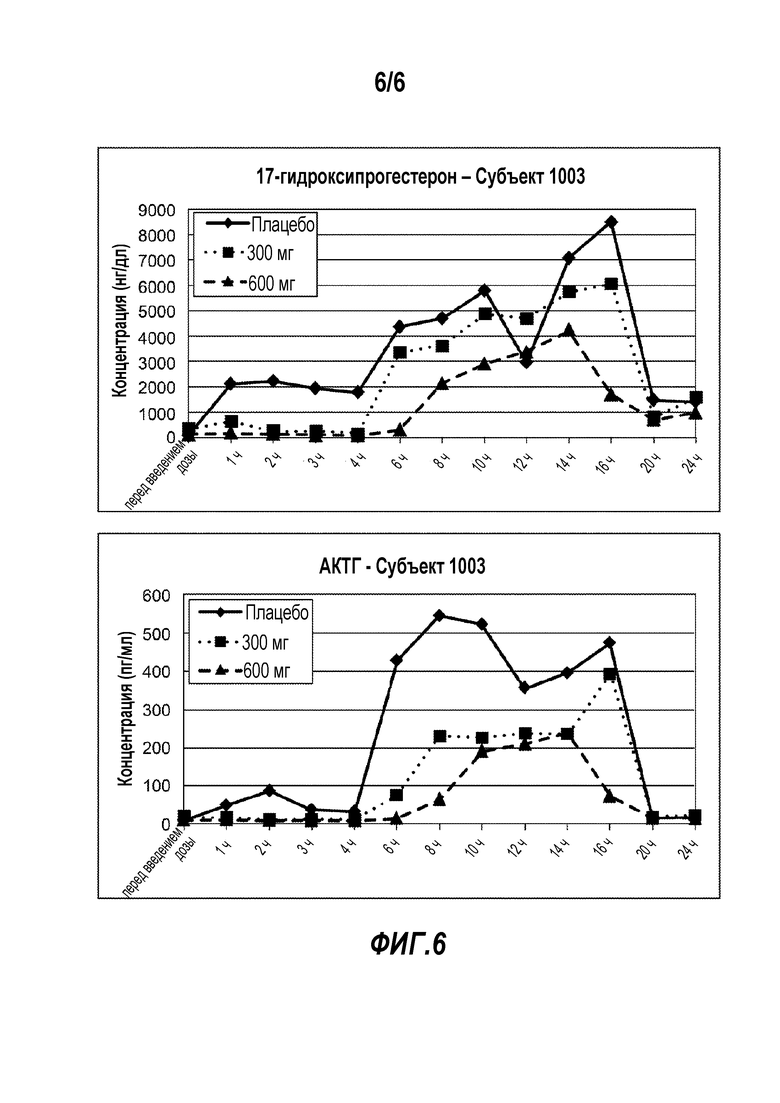
Фигура 6 представляет ответ конкретного индивидуального субъекта в отношении уровней 17-гидроксипрогестерона (верхняя панель) и АКТГ (нижняя панель) после введения 300 мг и 600 мг NBI-77860 и плацебо в течение времени.
Подробное описание изобретения
Как описано в настоящей заявке, было обнаружено, что антагонисты рецептора CRF1 напрямую ингибируют высвобождение АКТГ у пациентов с ВГКН и таким образом нормализует образование андрогена. Введение антагониста рецептора CRF1 позволяет применять пониженные, более физиологичные дозы гидрокортизона для субъектов с ВГКН и таким образом снижает связанные с лечением побочные эффекты.
Скрининг новорожденных на предмет ВГКН проводят с помощью способа иммунологического анализа для измерения уровня 17- гидроксипрогестерона в образцах капиллярной крови, полученных с помощью пяточной пункции в течение первых 72 часов жизни. Образцы крови анализируют на предмет 17-гидроксипрогестерона с помощью коммерчески доступного усиленного диссоциацией лантанидного флуоресцентного иммуноанализа (DELFIA, dissociation-enhanced lanthanide fluoroimmunoassay) (White et al., J. Pediatr. 163:10-12 (2013)).
Скрининговые тесты второго уровня с применением биохимических и молекулярно-генетических способов тестирования, проводимые между 8 и 14 днями жизни, проводят в девяти штатах США, и их настоятельно рекомендуют проводить в дополнительных 5 штатах. Биохимический способ включает иммунологическое исследование с экстракцией органическим растворителем или жидкостную хроматографию с последующей тандемной масс-спектрометрией для измерения отношения стероидов 17-гидроксипрогестерона, андростендиона и 21-дезоксикортизола к кортизолу (см., напр., Speiser et al., Int. J. Pediatr. Endocrinol. 2010:494173, 2010). Генетический скрининг выявляет мутации CYP21A2, ассоциированные с ВГКН. Дополнение вторым скринингом, хотя он и не является широко применимым в США, может улучшить чувствительность всего процесса скрининга, в котором чувствительность первого скрининга составляет 72%.
В отсутствие результатов скрининга новорожденных младенцев женского пола с классической ВГКН, как правило, идентифицируют по наличию наружных половых органов промежуточного типа. У мальчиков при рождении наружные половые органы нормальные, и поэтому ВГКН у них не диагностируется, если не проводится скрининг новорожденных или если нет других заметных медицинских осложнений. Младенцам, у которых исходно не диагностировали ВГКН и которые страдают от сольтеряющей формы заболевания, ставят диагноз позднее на фоне плохого набора веса, рвоты, гиперкалиемии и гипонатриемии в течение первых недель жизни.
Лечение ВГКН основано на нормализации уровней гормонов и стероидов с помощью ряда лекарственных средств с момента диагностики в младенчестве и на протяжении всего взрослого возраста. Глюкокортикоиды являются принятым в настоящее время стандартным лечением при ВГКН и применяются как для коррекции дефицита эндогенного кортизола, так и для снижения повышенного уровня АКТГ, образованного в гипофизе, что обусловливает повышенное образование андрогенов. В отличие от лечения болезни Аддисона (недостаточности коры надпочечников), при которой достаточно провести замещение кортизолом, лечение ВГКН также должно снижать образование АКТГ, чтобы также контролировать последующий избыток андрогена. Таким образом, цели глюкокортикоидного лечения включают кортизолозамещение и подавление АКТГ для предотвращения вирилизации и менструальных нарушений у женщин и для ингибирования образования тестикулярных опухолей добавочного надпочечника у мужчин. Замещение минералокортикоидами необходимо для достижения нормального уровня активности ренина в плазме для поддержания нормального кровяного давления, баланса электролитов и объема крови у пациентов с сольтеряющей формой ВГКН.
Схема лечения глюкокортикоидами должна поддерживать нормальную физиологию и также обеспечивать доступность достаточного уровня кортизола во время явлений, которые могут вызвать развитие сильного стрессового ответа (напр., сопутствующего заболевания, физических упражнений, гипотензии). Тщательный мониторинг также необходим для предотвращения развития ятрогенного синдрома Кушинга из-за передозировки глюкокортикоидов в попытке подавить образование андрогенов на соответствующем уровне, или болезни Аддисона из-за недостаточного лечения. Передозировка минералокортикоидов может вызвать гипертензию, в то время как недостаточное лечение может привести к низкому кровяному давлению, потери соли, слабости и повышенной потребности в глюкокортикоидах. Стандартные лабораторные исследования для наблюдения за эффективностью лечения включают измерение плазменной концентрации 17-гидроксипрогестерона, андростендиона, тестостерона, активности ренина и содержания электролитов.
Взрослые пациенты с ВГКН имеют больше факторов риска развития сердечно-сосудистых заболеваний, включая ожирение, гипертензию и инсулинорезистентность (см., напр., Kim et al., Semin. Reprod. Med. 27(4):316-21 (2009)). Исследование большой когорты детей и взрослых с ВГКН (n=244) показало, что пациентам назначают ряд схем лечения глюкокортикоидами, хотя они часто страдают от плохого контроля гормонального уровня и вышеупомянутых нежелательных исходов (см., напр., Finkielstain et al., J. Clin. Endocrinol Metab. 97(12):4429-38 (2012)).
Лечение ВГКН включает попытки нормализовать дефицит кортизола с помощью глюкокортикоидов (обычно с помощью гидрокортизона у детей, но часто с помощью более сильных агентов с узкими терапевтическими показателями, такими как дексаметазон, у взрослых) и при необходимости при потере соли с помощью минералокортикоидов (обычно с помощью флудрокортизона). Однако, как правило, дозы глюкокортикоидов, необходимые для достижения эффективного подавления избытка андрогенов, сильно превышают нормальную физиологическую дозу, применяемую только для кортизолозамещения, как у пациентов с болезнью Аддисона. Такое увеличенное действие глюкокортикоидов может привести к ятрогенному синдрому Кушинга, повышению факторов риска развития сердечно-сосудистых заболеваний, непереносимости глюкозы и пониженной минеральной плотности костей у пациентов с ВГКН (см., напр., Elnecave et al., J. Pediatr. Endocrinol. Metab. 21:1155-62 (2008); King et al., J. Clin. Endocrinol. Metab. 91(3):8656-59 (2006); Migeon et al., Endocrinol. Metab. Clin. North Am. 30:193-206 (2001)).
Кортиколиберин (CRF, corticotropin-releasing factor, кортикотропин-рилизинг фактор) был выделен из гипоталамуса овцы и идентифицирован как пептид, состоящий из 41 аминокислоты. Было обнаружено, что CRF вызывает сильные изменения функций эндокринной, нервной и иммунной систем. CRF считается основным физиологическим регулятором базального и индуцированного стрессом высвобождения адренокортикотропного гормона (АКТГ), β-эндорфина и других пропиомеланокортиновых пептидов из передней доли гипофиза (см., напр., Vale et al., Science 213:1394-1397, 1981). Секреция CRF вызывает высвобождение АКТГ из кортикотрофов в аденогипофизе посредством связывания рецептора CRF1 члена класса В семейства рецепторов, сопряженных с G-белками (GPCR, G protein-coupled receptor).
Из-за физиологического значения CRF, разработка биологически активных малых молекул, обладающих выраженной связывающей рецептор CRF1 активностью и способных быть антагонистами рецептора CRF1, остается актуальной задачей и является предметом проводимых исследований и разработок для лечения тревоги, депрессии, синдрома раздраженного кишечника, посттравматических стрессовых расстройств и злоупотребления психоактивными веществами.
Гормон гипофиза АКТГ под контролем кортиколиберина CRF, corticotropin-releasing factor, кортикотропин-рилизинг фактора) гипоталамуса стимулирует захват холестерина и запускает синтез прегненолона, инициируя биосинтез стероидов в надпочечниках (см. Фигуру 1). Кора надпочечников состоит из трех зон, образующих определенные классы гормонов, многие из которых образуются под действием АКТГ, мобилизующего холестерин по этому пути. Дефицит этих ферментов в результате мутации или делеции вызывает увеличение концентрации субстрата. При наиболее распространенной форме ВГКН, вызванной мутациями или делециями в гене 21-гидроксилазы (CYP21A2), сильные андрогены образуются в надпочечниках из-за скопления предшественников стероидов, прогестерона и 17-гидроксипрогестерона. В этих случаях уровень 17-гидроксипрогестерона в плазме может превышать нормальную концентрацию в 10-1000 раз. Такое повышение приводит к повышенному образованию андрогенов, а именно, андростендиона, тестостерона и дигидрокситестостерона, вызывая вирилизацию у женщин. Дополнительно, дефицит 21-гидроксилазы в случае ВГКН вызывает недостаточный биосинтез глюкокортикоидов и минералокортикоидов, а именно кортизола и альдостерона. Кортизол является важным регулятором отрицательной обратной связи при секреции CRF в гипоталамусе и при высвобождении гипофизарного АКТГ. Отсутствие синтеза и высвобождения глюкокортикоидов снимает ограничение с гипоталамуса и гипофиза, что вызывает повышение уровня АКТГ. Избыточная стимуляция АКТГ вызывает гипертрофию пучковой зоны и сетчатой зоны, что приводит к гиперплазии коры надпочечников.
В одном варианте осуществления настоящего изобретения антагонист рецептора CRF1, применимый для лечения ВГКН, является NBI-77860, 2,5-диметил-3-[2-метил-4-(метилокси)фенил]-N-[(1S)-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил]пиразоло[1,5-a]пиримидин-7-амином (также называемым в настоящей заявке «Соединением I»), имеющим следующую структуру.
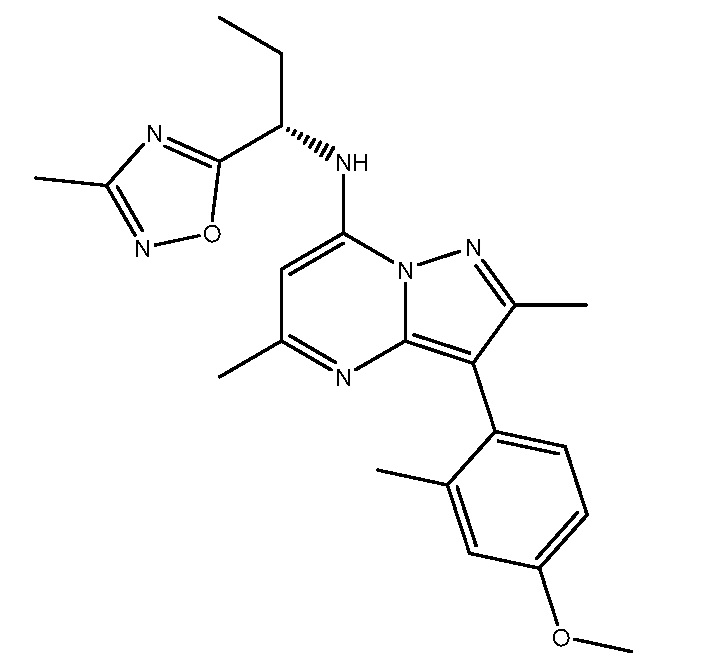
Соединение I
Соединение I является сильным антагонистом CRF1, обладающим pKi связывания=8.2 (см., напр., Tellew et al., Bioorg. Med. Chem. Lett. 20:7259, 2010 и Публикацию международной патентной заявки № WO 2006/044958; обе эти ссылки включены в настоящую заявку в полном объеме посредством ссылки). Как описано в настоящей заявке, Соединение I обладает сильным понижающим АКТГ действием, как показано на адреналэктомированных крысах.
В другом варианте осуществления настоящего изобретения антагонист рецептора CRF1, применимый для лечения ВГКН, является низкомолекулярным антагонистом, как описано в US 6586456, US 6806282, US 6531475, US 6664261, US 6610678, WO 98/08846, WO 98/11075, WO 99/10350, WO 2000/059888, WO 2006/044821, WO 2006/102194, WO 2006/107784, WO 2006/116412, WO 2006/126718, WO 2007/069565, WO 2007/069671, WO 2008/036541, WO 2008/036579, WO 2008/051533, WO 2008/082003, WO 2008/083070, WO 2008/136377, WO 2009/008552, WO 2009/144632, WO 2010/014280, WO 2010/014687, WO 2010/015628, WO 2010/015655, WO 2010/062718, WO 2010/096426, WO 2011/043387, WO 2011/092293, WO 2011/095450, WO 2011/092290 и WO 2011/043381.
В еще одном варианте осуществления настоящего изобретения антагонист рецептора CRF1 является NBI-30775, CP-316,311, пексацерфонтом, эмицерфонтом, SSR-125543 [4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропин-1-ил)-2-тиазоламином], SSR-126374, ONO-2333, NBI-34041, JNJ-19567470, GSK586529, PF-00572778, CP-376395, анталармином (N-бутил-N-этил-2,5,6-триметил-7-(2,4,6-триметилфенил)пирроло[3,2-e]пиримидин-4-амином) и DMP904.
В другом варианте осуществления настоящего изобретения антагонист рецептора CRF1 имеет период полудиссоциации (t1/2) свыше 30 минут, а в другом варианте осуществления настоящего изобретения - свыше 40 минут, а в другом варианте осуществления настоящего изобретения - свыше 50 минут. Период полудиссоциации конкретного антагониста рецептора CRF1 определяют посредством техники, раскрытой в Примере 3. Репрезентативные антагонисты рецептора CRF1 из этих вариантов осуществления настоящего изобретения включают Соединение I, NBI-30775, NBI-34041, SSR-125543A, анталармин (N-бутил-N-этил-2,5,6-триметил-7-(2,4,6-триметилфенил)пирроло[3,2-e]пиримидин-4-амин) и DMP904.
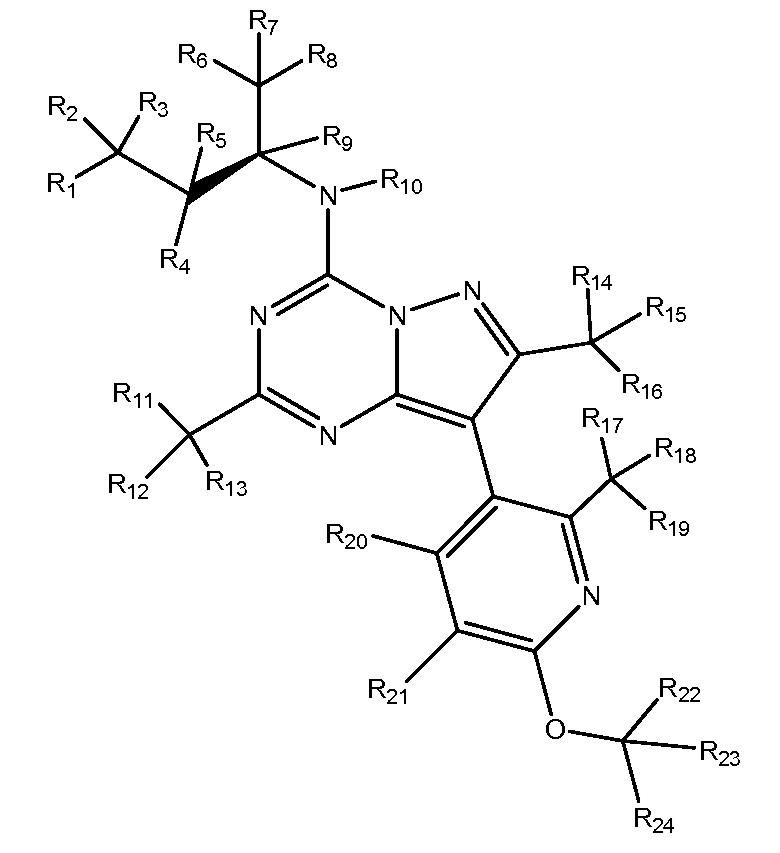
Применительно к описанным в настоящей заявке соединениям следует понимать, что обозначение водорода в конкретном положении подразумевает, что этот водород (H) может быть заменен дейтерием (D). Включение дейтерия вместо водорода, как известно, оказывает выраженные эффекты на физиологическую и фармакологическую активность замещенного соединения. В связи с этим следует понимать, что замена водорода дейтерием обозначает, что количество дейтерия в данном положении существенно превышает природное количество дейтерия. Соответственно, в одном варианте осуществления настоящего изобретения репрезентативные соединения включают следующие:
NBI-77860
NBI-77860
NBI-77860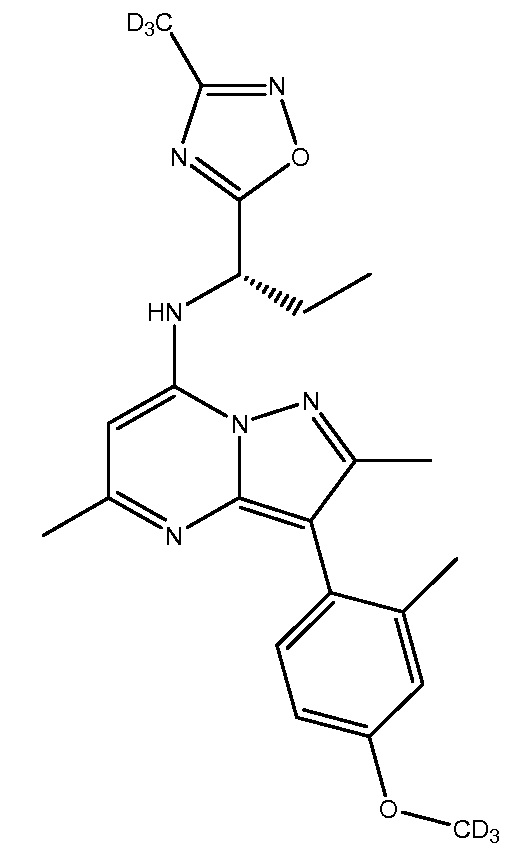
NBI-77860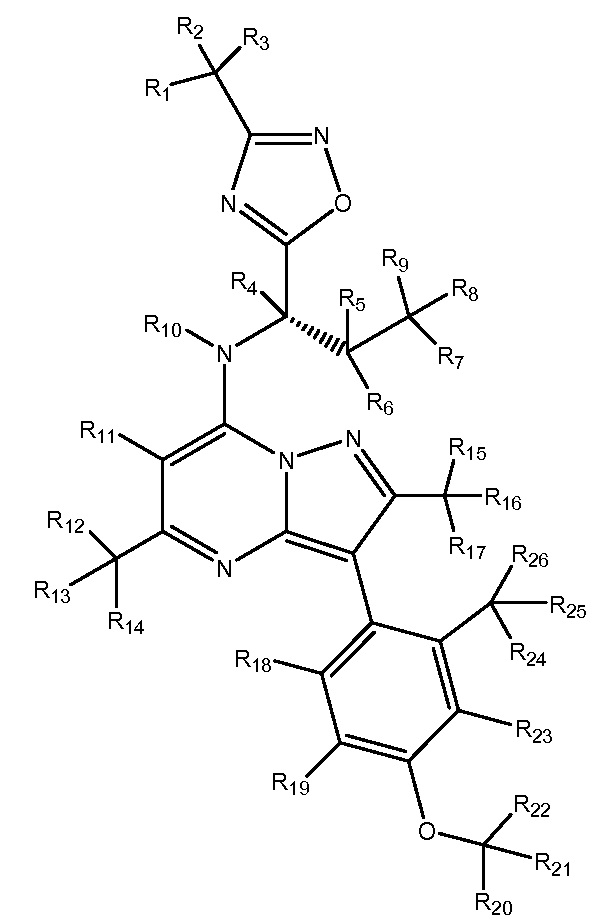
R1-R26 независимо являются H или D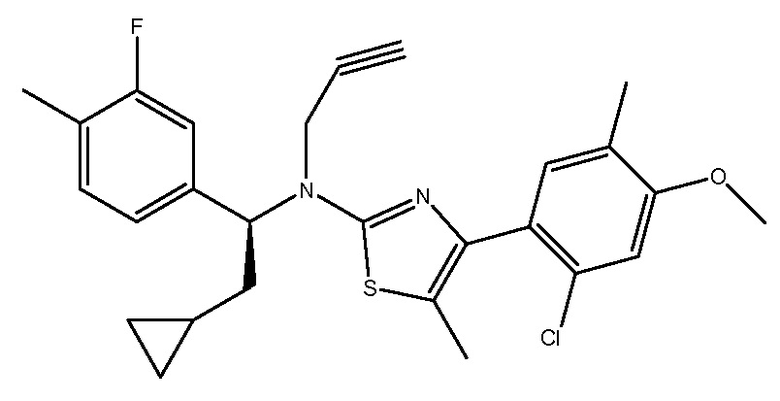
SSR-125543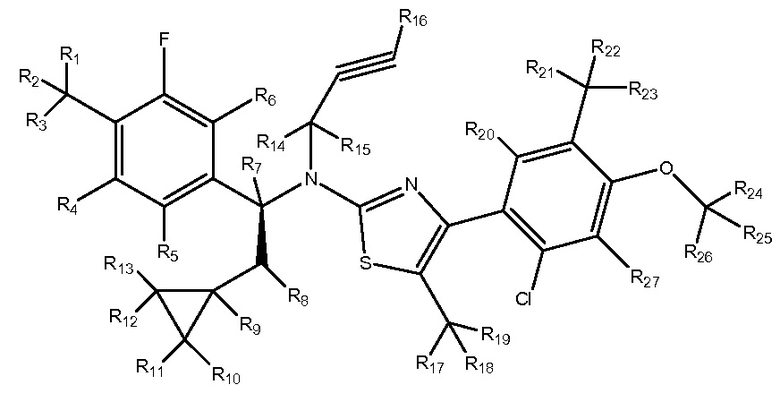
R1-R27 независимо являются H или D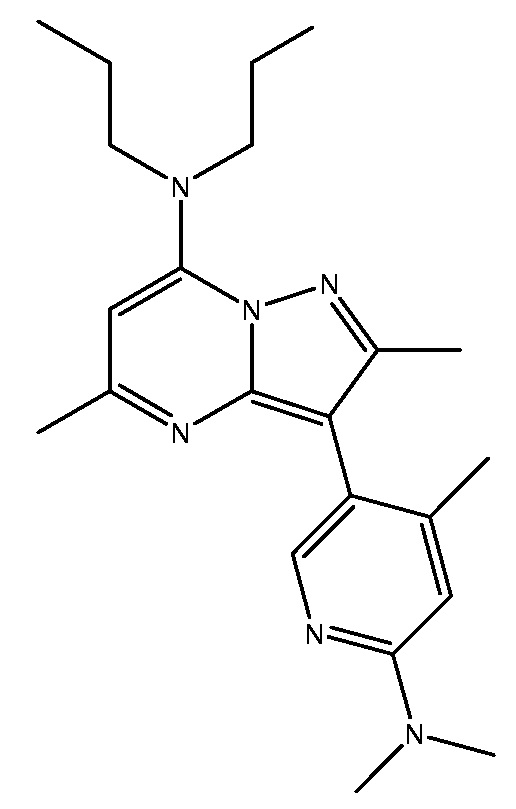
NBI-30775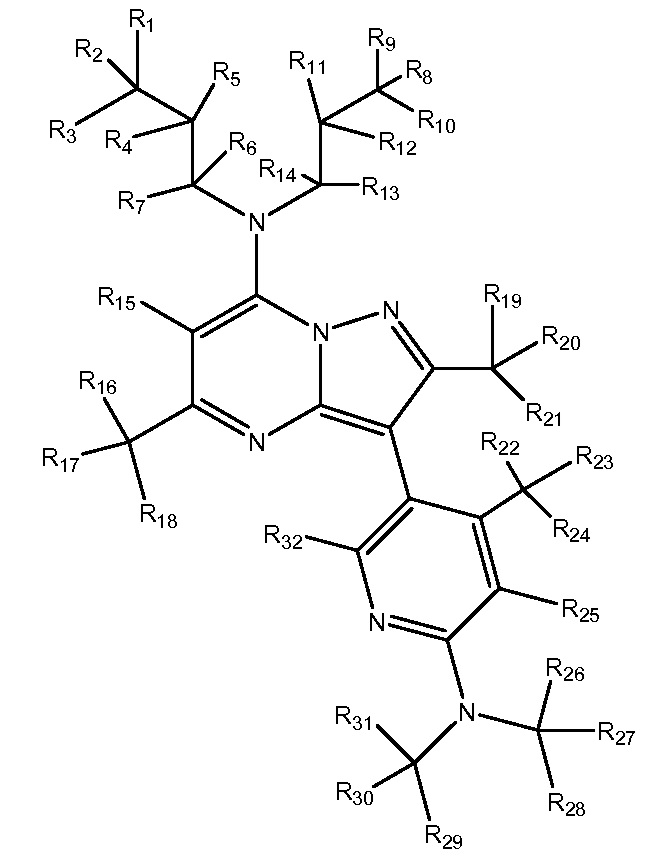
R1-R32 независимо являются H или D
NBI-34041
R1-R26 независимо являются H или D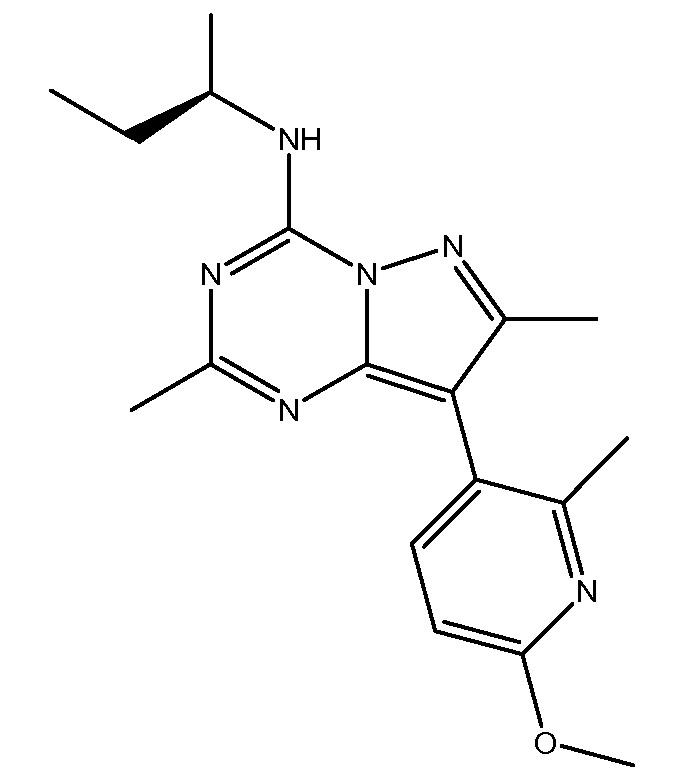
R1-R24 независимо являются H или D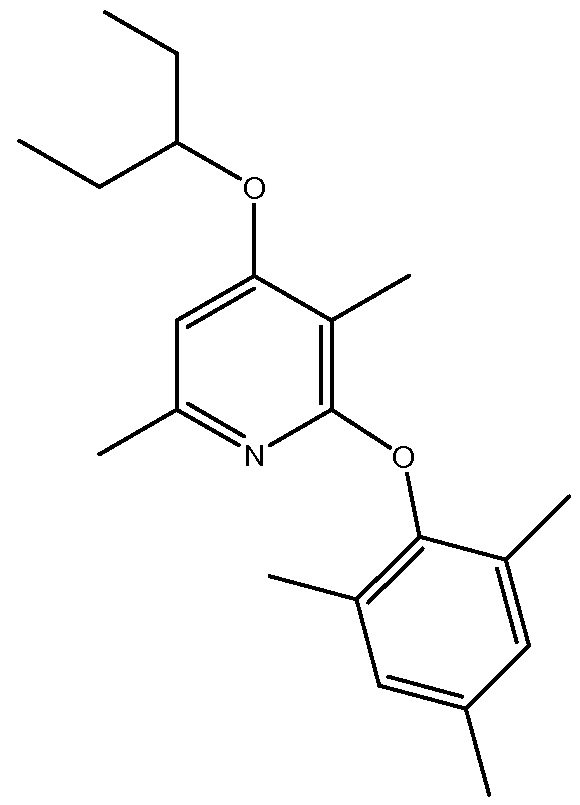
CP-316,311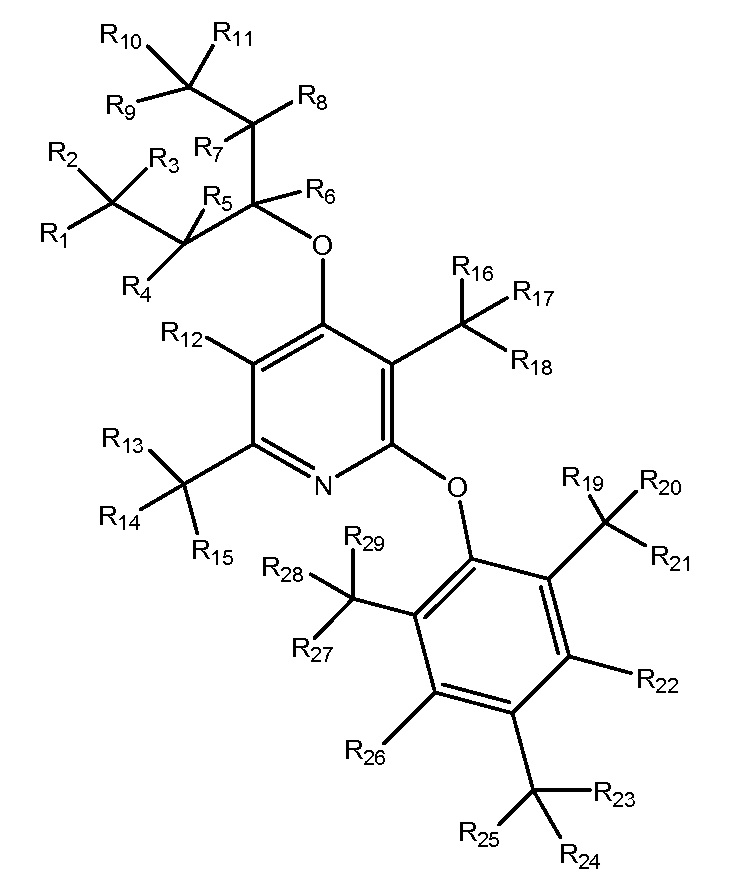
R1-R29 независимо являются H или D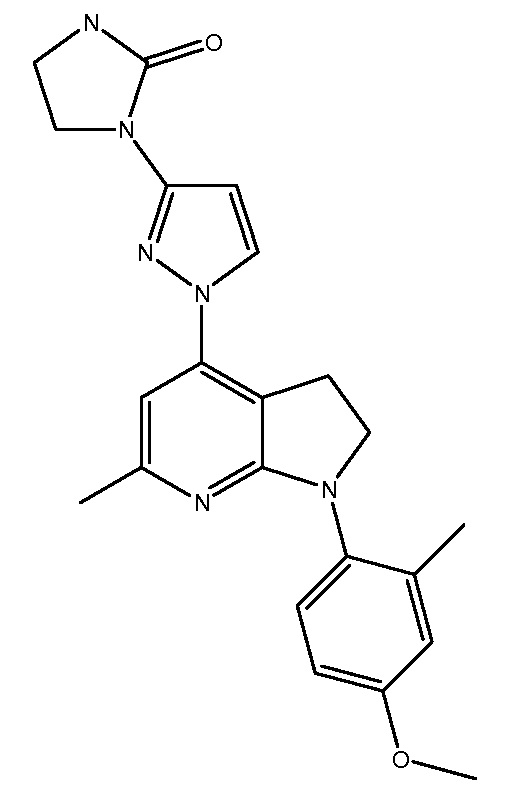
GSK876008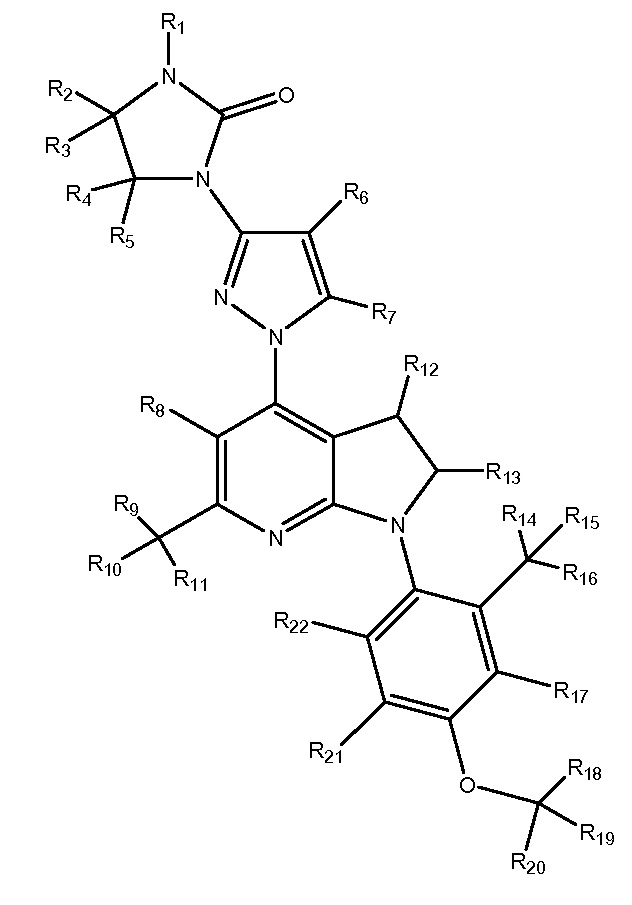
R1-R22 независимо являются H или D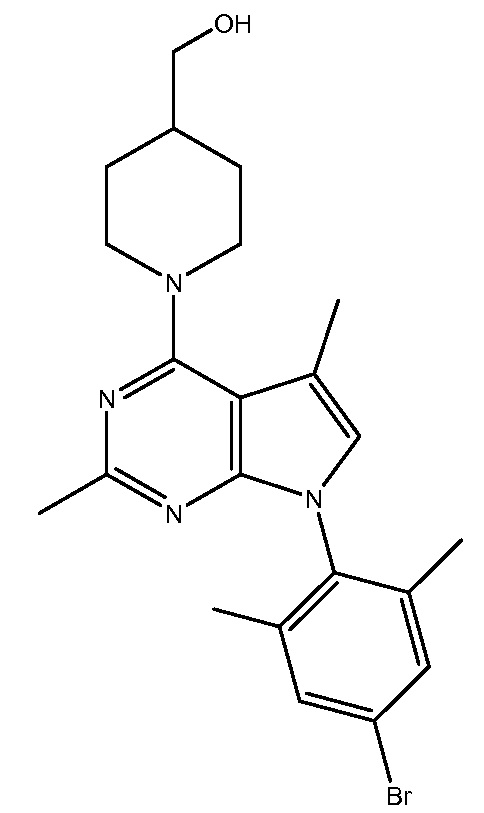
CRA5626/R317573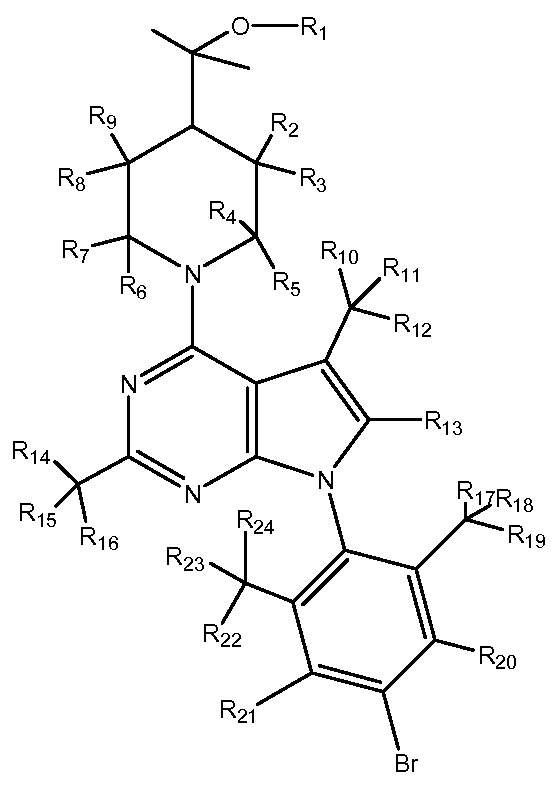
R1-R24 независимо являются H или D
ONO2333
R1-R27 независимо являются H или D
NBI76169
R1-R22 независимо являются H или D
В другом варианте осуществления настоящего изобретения любое из перечисленных выше соединений может включать стабильные или радиоактивные изотопы. Соответственно, в настоящее изобретение также включено применение меченных изотопами соединений. Идентичных описанным в настоящей заявке, где один или несколько атомов заменены атомом с атомной массой или с массовым числом, отличным от атомной массы или массового числа, обычно обнаруживаемых в природе. Примеры изотопов, которые можно включать в эти соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как, но не ограничиваясь дейтерием, как описано выше (2H), а также 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно. Определенные меченные изотопами соединения, например, соединения, в которые включены радиоактивные изотопы, такие как 3H и 14C, также применимы в исследованиях распределения лекарственного средства или вещества в тканях. Изотопы трития (3H) и углерода-14 (14C) являются особенно предпочтительными из-за простоты получения и детектирования. Замена более тяжелыми изотопами, такими как дейтерий (2H), может предоставить определенные терапевтические преимущества, обусловленные повышенной метаболической стабильностью, например, увеличенным временем полужизни или пониженной необходимой дозой, и, следовательно, могут быть предпочтительными в определенных обстоятельствах. Меченные изотопами соединения, как правило, можно получить, проводя рутинные процедуры, применяемые в данной области техники.
Действие на содержание АКТГ в плазме у адреналэктомированных крыс
Адренэктомия уничтожает кортикостерон (первичный глюкокортикоид) в кровотоке у крыс и снимает контроль по принципу отрицательной обратной связи гипоталамо-гипофизарно-надпочечниковой системы как на гипоталамусном, так и на гипофизарном (на уровне кортикотрофных клеток) уровне и таким образом хронически поднимает уровень АКТГ в плазме (см., напр., Mims et al., J. Natl. Med. Assoc. 69:145-47 (1977)). Было показано, что внутривенная инъекция пептидных антагонистов рецептора CRF1 снижает высокие уровни АКТГ в плазме у адреналэктомированных крыс (см., напр., Rivier et al., J. Med. Chem. 12:42:3175-82 (1999)). Эти наблюдения повторили с применением низкомолекулярного NBI-77860 (Соединения I). У адреналэктомированных крыс Соединение I обладает выраженной способностью снижать АКТГ. Максимальное снижение АКТГ коррелировало с пиком концентрации NBI-77860 в плазме; однако продолжительность эффекта снижения АКТГ превышала продолжительность присутствия лекарственного средства в плазме. Таким образом, у адреналэктомированных крыс существует прогнозируемая связь между суммарным содержанием NBI 77860 в плазме и in vivo эффективностью после перорального введения.
Эффективность соединения в качестве антагониста рецептора CRF можно определить различными способами. Антагонисты CRF, описанные в настоящей заявке, могут ингибировать специфичное связывание CRF со своим рецептором и, как следствие, проявлять антагонизирующую активность, ассоциированную с CRF. Активность соединения как антагониста CRF можно оценить с помощью одного или нескольких общепринятых исследований, включая исследование, описанное в Примерах. Антагонисты CRF, применимые в способах, описанных в настоящей заявке, включают соединения, обладающие аффинностью к рецептору CRF.
Не желая быть связанными какой-либо теорией, авторы настоящего изобретения полагают, что при лечении ВГКН антагонисты рецептора CRF будут предпочтительно блокировать высвобождение АКТГ из гипофизарных кортикотрофов, снижая тем самым образование андрогенов, и будут обеспечивать более оптимальный терапевтический подход к замещению кортизола. Исследования, проведенные на животных и человеке, показали фармакологический эффект Соединения I (NBI-77860) на высвобождение АКТГ. Для наблюдения за эффектами этого антагониста рецептора CRF1 можно применять оценки стандартных биомаркеров, применяемые эндокринологами для наблюдения за эффективностью лечения. Уровни 17-гидроксипрогестерона, андростендиона, тестостерона, кортизола и АКТГ в плазме, а также уровни метаболитов этих стероидных соединений в моче легко можно измерять как у детей, так и у взрослых, быстро получая значащие данные об эффектах лечения.
Фармацевтические композиции и способы лечения
Настоящее раскрытие также представляет фармацевтические композиции, включающие любое из соединений-антагонистов CRF, описанных в настоящей заявке, и фармацевтически приемлемый эксципиент для применения в способах для лечения ВГКН. Фармацевтически приемлемый эксципиент является физиологически и фармацевтически подходящим нетоксичным и неактивным материалом или ингредиентом, которые не влияет на активность активного ингредиента; эксципиент также можно назвать носителем. Соединения-антагонисты CRF можно составлять в форме фармацевтической композиции для применения в лечении или в предотвращении (или в профилактике) заболевания (напр., в снижении обострения ВГКН или проявления или рецидива одного или нескольких симптомов заболевания). Способы и эксципиенты, описанные в настоящей заявке, приведены в качестве примеров и не являются ограничивающими каким-либо образом. Фармацевтически приемлемые эксципиенты хорошо известны в фармацевтической области техники и описаны, например, в Rowe et al., Handbook of Pharmaceutical Excipients: A Comprehensive Guide to Uses, Properties, and Safety, 5th Ed., 2006, и в Remington: The Science and Practice of Pharmacy (Gennaro, 21st Ed. Mack Pub. Co., Easton, PA (2005)). Примеры фармацевтически приемлемых эксципиентов включают стерильный солевой раствор и фосфатно-солевой буфер при физиологическом значении pH. В фармацевтической композиции могут быть консерванты, стабилизаторы, красители, буферы и подобное. Дополнительно, также можно применять антиоксиданты и суспендирующие агенты.
Терапевтическое и/или профилактическое действие включает, например, улучшенный клинический исход, как терапевтическую обработку, так и профилактические или превентивные меры, где целью является предотвращение или замедление или задержка (уменьшение) нежелательного физиологического изменения или нарушения или предотвращение или замедление (уменьшение) распространения или степени тяжести такого нарушения. Как обсуждают в настоящей заявке, благоприятные или желаемые клинические результаты лечения субъекта включают, но не ограничиваются ослаблением, уменьшением или смягчением симптомов, вызванных или ассоциированных с подлежащим лечению заболеванием, состоянием или нарушением; уменьшением частоты развития симптомов; улучшением качества жизни; увеличением продолжительности периода ремиссии (т.е. уменьшением вероятности или предрасположенности к развитию у субъекта симптомов, на основании которых диагностируют заболевание); уменьшением степени заболевания; стабилизацией (т.е. отсутствием ухудшения) течения заболевания; отсрочкой или замедлением прогрессии заболевания; уменьшением или временным облегчением течения заболевания; и ремиссией (частичной или полной), детектируемой или недетектируемой; и/или общей выживаемостью. «Лечение» также может обозначать продление выживания по сравнению с ожидаемой продолжительностью выживания, если субъект на получает лечение. Нуждающиеся в лечении субъекты включают субъектов с уже имеющимся состоянием или нарушением, а также субъектов, предрасположенных к заболеванию или имеющих риск развития заболевания, состояния или нарушения, и субъектов, нуждающихся в предотвращении заболевания, нарушения или состояния (т.е. снижении вероятности развития заболевания, нарушения или состояния). Субъект может быть человеком или не являющимся человеком млекопитающим (напр., крысой, мышью, собакой, кошкой, сельскохозяйственным животным, содержащимся в зоопарке животным).
Оптимальные дозы, как правило, можно определить с помощью экспериментальных моделей и/или клинических исследований. Оптимальная доза может зависеть от массы тела, веса или объема крови субъекта. Как правило, количество описанного в настоящей заявке соединения в дозе варьирует от приблизительно 0,1 мг до приблизительно 30 мг на кг веса субъекта. В определенных вариантах осуществления настоящего изобретения однократная доза составляет приблизительно 50-1000 мг. Как правило, предпочтительным является применение минимальной дозы, достаточной для проведения эффективной терапии. Обычно можно наблюдать за пациентами на предмет терапевтической эффективности, проводя клиническую оценку и применяя исследования, подходящие для проходящего лечение или профилактику состояния, при этом данные исследования известны специалистам в данной области техники и описаны в настоящей заявке. За уровнем соединения, введенного субъекту, можно наблюдать, определяя уровень соединения в биологической жидкости, например, в крови, во фракции крови (напр., в сыворотке) и/или в моче, и/или в другом биологическом образце, отобранном у субъекта. Любой способ, применяемый в данной области техники для детектирования соединения, можно применять для измерения уровня соединения на протяжении схемы лечения.
Доза композиции, включающей по меньшей мере одно из описанных в настоящей заявке соединений для лечения ВГКН или связанного заболевания или нарушения может зависеть от состояния субъекта, то есть от стадии заболевания, степени тяжести симптомов, вызванных заболеванием, от общего состояния здоровья, а также от возраста, пола и веса и от других факторов, очевидных для специалиста в области медицины. Аналогично, дозу соединения можно определить в соответствии с параметрами, известными специалистам в области медицине.
Описанные в настоящей заявке фармацевтические композиции, содержащие по меньшей мере один из описанных в настоящей заявке антагонистов CRF1, можно вводить нуждающемуся в этом субъекту одним или несколькими способами, которые обеспечивают эффективную доставку эффективного количества соединения. Такие способы введения включают, например, пероральный, парентеральный, энтеральный, ректальный, интраназальный, трансбуккальный, подъязычный, внутримышечный и трансдермальный. Композиции, вводимые с помощью этих способов введения и другие, более подробно описаны в настоящей заявке.
Введение раскрытых соединений или композиций включает введение ночью или введение на ночь (т.е. введение перед сном). Как применяют в настоящей заявке, введение перед сном обозначает введение дозы, предназначенное для доставки клинически значимых концентраций антагониста CRF1 во время или до (например, за 2-5 часов до) расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом. Поскольку высвобождение АКТГ, как правило, происходит в 1:00-2:00 ночи и поскольку у перорально вводимых лекарственных средств время достижения максимальной концентрации (Tmax) составляет несколько часов, желательно вводить дозу, например, в 22:00, т.е. за 3-4 часа до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом. Такое же преимпульсное введение дозы можно адаптировать для людей, работающих посменно (напр., для работающих в ночную смену и спящих днем); в этом случае введение не обязательно проводить ночью. Таким образом, введение зависит от расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом и может варьировать в зависимости от конкретной работы и режима сна индивида (т.е. субъекта, пациента). В определенных вариантах осуществления настоящего изобретения описанный в настоящей заявке антагонист рецептора CRF1 (например, NBI-77860 или любой из NBI-30775, NBI-34041, SSR-126374, SSR-125543, анталармина (N-бутил-N-этил-2,5,6-триметил-7-(2,4,6-триметилфенил)пирроло[3,2-e]пиримидин-4-амина) или DMP904) вводят за 2-5 часов до (т.е. перед, заранее) расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом. В других вариантах осуществления настоящего изобретения антагонист рецептора CRF1 вводят субъекту приблизительно за 2-4 часа или приблизительно за 3-5 часов до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом. В более конкретном варианте осуществления настоящего изобретения антагонист рецептора CRF1 вводят субъекту приблизительно за 3-4 часа до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом.
Также в настоящей заявке представлен способ снижения уровней 17-гидроксипрогестерона и/или АКТГ у субъекта (т.е. у пациента, индивида) с ВГКН посредством введения антагониста рецептора CRF1. В определенных вариантах осуществления настоящего изобретения антагонист вводят во время или до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом. В других определенных вариантах осуществления настоящего изобретения антагонист рецептора CRF1 вводят приблизительно за 2-4 часа или приблизительно за 3-5 часов до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом. В более конкретном варианте осуществления настоящего изобретения антагонист рецептора CRF1 вводят субъекту приблизительно за 3-4 часа до расчетного времени высвобождения АКТГ в соответствии с его циркадными ритмом.
Описанные в настоящей заявке способы, включающие введение антагониста рецептора CRF1 нуждающемуся в этом субъекту женского пола описанным в настоящей заявке способом, который вызывает снижение уровня АКТГ и 17-гидроксипрогестерона у субъекта, может привести к уменьшению высвобождения андрогенов, таких как тестостерон и андростендион. Параллельно можно снижать дозу глюкокортикоидов на клинически значимое количество, что в свою очередь приводит к уменьшению побочных эффектов.
Количество глюкокортикоидов и минералокортикоидов для поддерживающей терапии молодых и растущих пациентов с ВГКН известно специалистам в данной области техники. Например, указания описаны в Speiser et al. (J. Clin. Endocrinol. Metab. 95:4133-60 (2010), которая включена в полном объеме посредством ссылки) и показаны в Таблицах 1 и 2 там же. В частности, для молодых, растущих пациентов с ВГКН специалисты в данной области техники считают, что введение высоких доз глюкокортикоидов может задерживать рост тела пациента. Соответственно, описанные в настоящей заявке способы лечения пациента с помощью антагониста рецептора CRF1 могут включать снижение дозы глюкокортикоидов клинически значимым способом.
В определенных вариантах осуществления настоящего изобретения способы лечения ВГКН с помощью введения антагониста рецептора CRF1 могут также включать введение глюкокортикоидов в дозе ниже дозы глюкокортикоидов, рекомендуемой в настоящее время для лечения субъектов с ВГКН. После окончания роста субъекта дозу глюкокортикоидов, такую как дозу гидрокортизона, преднизона, преднизолона, дексаметазона или флудрокортизона, рекомендуемую для поддерживающей терапии у взрослого пациента, можно снизить приблизительно на 10%, 15%, 20%, 30%, 40%, 50%, 60% или более от рекомендуемой дозы равной 15-25 мг/день гидрокортизона; 5-7,5 мг/день преднизона, 4-6 мг/день преднизолона; 0,25-0.5 мг/день дексаметазона или 0,05-0,2 мг/день флудрокортизона. У растущего субъекта с ВГКН, рекомендуемую дозу глюкокортикоидов, например, общую рекомендуемую дозу гидрокортизона равную 10-15 мг/м2 в день и/или общую дозу флудрокортизона равную 0,05-0,2 мг/день, можно уменьшить приблизительно на 10%, 15%, 20%, 30%, 40%, 50%, 60% или более у субъекта, получающего антагонист рецептора CRF1, как описано в настоящей заявке. В одном варианте осуществления настоящего изобретения описанные в настоящей заявке способы, включающие введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывают клинически значимое снижение уровня АКТГ относительно плацебо. В конкретном варианте осуществления настоящего изобретения введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывает клинически значимое снижение уровня АКТГ относительно плацебо, где снижение составляет по меньшей мере 25%. В другом варианте осуществления настоящего изобретения введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывает клинически значимое снижение уровня АКТГ относительно плацебо, где снижение составляет по меньшей мере 50%. См. руководство по введению глюкокортикоидов и минералокортикоидов в Speiser et al., указанной выше.
В другом варианте осуществления настоящего изобретения описанные в настоящей заявке способы, включающие введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывают клинически значимое снижение уровня 17-гидроксипрогестерона относительно плацебо. В другом конкретном варианте осуществления настоящего изобретения введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывает клинически значимое снижение уровня 17-гидроксипрогестерона относительно плацебо, где снижение составляет по меньшей мере 25%. В другом конкретном варианте осуществления настоящего изобретения введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывает клинически значимое снижение уровня 17-гидроксипрогестерона относительно плацебо, где снижение составляет по меньшей мере 50%.
В одном варианте осуществления настоящего изобретения описанные в настоящей заявке способы, включающие введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывают клинически значимое снижение как уровня АКТГ, так и уровня 17-гидроксипрогестерона относительно плацебо. В определенном варианте осуществления настоящего изобретения введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывает клинически значимое снижение как уровня АКТГ, так и уровня 17-гидроксипрогестерона относительно плацебо, где снижение составляет по меньшей мере 25%. В другом конкретном варианте осуществления настоящего изобретения введение нуждающемуся в этом субъекту эффективного количества антагониста CRF1, вызывает клинически значимое снижение как уровня АКТГ, так и уровня 17-гидроксипрогестерона относительно плацебо, где снижение составляет по меньшей мере 50%.
Фармацевтические композиции могут быть в форме раствора. Альтернативно, они могут быть в твердой форме, такой как порошок, таблетки или подобное. Композицию, включающую любое из описанных в настоящей заявке соединений можно составлять в форме для пролонгированного или замедленного высвобождения. Такие композиции, как правило, можно получить с помощью хорошо известной технологии и ввести, например, посредством пероральной, ректальной или подкожной имплантации, или посредством имплантации в заданное место. Формы для пролонгированного высвобождения могут содержать соединение, диспергированное в матриксе носителя и/или находящееся внутри резервуара, окруженного мембраной, которая контролирует скорость высвобождения. Эксципиенты для применения в таких формах являются биосовместимыми и также могут быть биодеградируемыми; предпочтительно, чтобы форма обеспечивала относительно постоянный уровень высвобождения активного компонента. Количество активного соединения, содержащееся в форме для пролонгированного высвобождения, зависит от места имплантации, скорости и ожидаемой продолжительности высвобождения, и от природы состояния, подлежащего лечению или предотвращению.
Для пероральных форм по меньшей мере одно из описанных в настоящей заявке соединений можно применять в индивидуальном виде или в комбинации с соответствующими добавками для получения таблеток, порошков, гранул или капсул, например, со стандартными добавками, такими как лактоза, маннит, кукурузный крахмал или картофельный крахмал; со связывающими веществами; с разрыхлителями; с лубрикантами; и, при желании, с растворителями, буферными агентами, увлажняющими агентами, консервантами, красителями и ароматизаторами. Соединения можно составлять с буферным агентом, чтобы обеспечить защиту соединения от низкого pH в желудке, и/или с кишечнорастворимой оболочкой. Соединение, включенное в композиции, можно составлять в форме для пероральной доставки с ароматизатором, напр., в виде жидкой, твердой или полутвердой формы, и/или с кишечнорастворимой оболочкой. Формы для перорального введения можно получать в виде желатиновых капсул, которые могут содержать активное соединение наряду с порошкообразными носителями, такими как лактоза, крахмал, производные целлюлозы, стеарат магния, стеариновая кислота и подобное. Похожие носители и растворители можно применять для получения прессованных таблеток.
Примеры
Пример 1
Связывающая рецептор CRF активность
Антагонисты CRF, как применяют в описанных в настоящей заявке способах, можно оценить на предмет их связывающей активности в отношении рецептора CRF посредством стандартного исследования связывания радиоактивно меченного лиганда, как в общих чертах описано Grigoriadis et al. (см., напр., Mol. Pharmacol vol 50, pp 679-686, 1996) и Hoare et al. (см., напр., Mol. Pharmacol 63: 751-765, 2003.) Применяя радиоактивно меченные лиганды CRF, можно использовать исследование для оценки связывающей активности описанных в настоящей заявке соединений с любым подтипом рецептора CRF.
Вкратце, исследование связывания включает замещение радиоактивно меченного лиганда CRF на рецепторе CRF. В частности, исследование связывания проводят в 96-луночных планшетах с применением 1-10 мкг клеточных мембран из клеток, стабильно трансфицированных человеческими рецепторами CRF. В каждую лунку добавляют приблизительно 0,05 мл буфера для исследования (напр., фосфатно-солевого буфера Дульбекко, 10 мМ хлорида магния, 2 мМ ЭГТА), содержащего заданное соединение или лиганд сравнения (например, саувагин, урокортин I или CRF), 0,05 мл [125I] тирозин-саувагина (конечная концентрация ~150 пМ или приблизительно KD, по данным анализа Скетчарда (Scatchard)) и 0,1 мл суспензии клеточных мембран, содержащих рецептор CRF. Смесь инкубируют в течение 2 часов при 22°C, затем разделяют связавшиеся и свободные радиоактивно меченные лиганды посредством скоростной фильтрации через стекловолоконные фильтры. После трех промываний фильтры высушивают и определяют радиоактивность (электроны Оже из 125I) с помощью сцинтилляционного счетчика. Все данные по связыванию радиоактивно меченного лиганда можно анализировать с помощью программ нелинейной аппроксимации методом наименьших квадратов Prism (GraphPad Software Inc) или XLfit (ID Business Solutions Ltd).
Пример 2
Активность агониста рецептора CRF1
В соответствии с Fleck et al. (J. Pharmacology and Experimental Therapeutics, 341(2):518-531, 2012) (здесь и далее «Fleck et al.», включенная в полном объеме посредством ссылки) представлена активность идентифицированных ранее антагонистов рецептора CRF1. Такая активность представлена как кинетически полученная аффинность (Ki), рассчитанная по константам скорости ассоциации (k1) и диссоциации (k-1) в соответствии со следующим уравнением:
Ki=k-1/k1
Также в соответствии с Fleck et al. представлены кинетические Ki антагонистов рецептора CRF1, перечисленные в Таблице 1 ниже:
Таблица 1
Репрезентативные антагонисты рецептора CRF1
С помощью такой же техники определяли следующую кинетическую Ki Соединения I (NBI-77860):
Таблица 1 (продолжение)
Пример 3
Период полудиссоциации ( t 1/2) антагонистов рецептора CRF1
Период полудиссоциации (t1/2) антагониста рецептора CRF1, как применяют в описанных в настоящей заявке способах, оценивают с помощью техники, описанной Fleck et al. Согласно этому описанию, константа скорости диссоциации меченых и немеченых лигандов обозначают k-1, в то время как период полудиссоциации лекарственного средства от рецептора (t1/2), равный среднему времени удержания, рассчитывают по константе скорости диссоциации (k-1) в соответствии со следующим уравнением:
t 1/2=0,693/k-1
Также в соответствии с Fleck et al. представлены периоды полудиссоциации (t1/2) антагонистов рецептора CRF1, перечисленные в Таблице 2 ниже.
Таблица 2
Период полудиссоциации репрезентативных соединений
С помощью такой же техники определили следующий период полудиссоциации Соединения I (NBI-77860):
Таблица 2 (продолжение)
Соответственно, антагонисты рецептора CRF1 с периодом полудиссоциации (t1/2) свыше 30 минут включают (но не ограничиваются указанными соединениями) анталармин, NBI-34041, DMP904, NBI-30775, SSR125543A и NBI-77860 (Соединение I). Эти же соединения также являются репрезентативными антагонистами рецептора CRF1 с периодом полудиссоциации (t1/2) свыше 40 минут и с периодом полудиссоциации (t1/2) свыше 50 минут.
Пример 4
Снижение АКТГ у адреналэктомированных крыс
Соединение I (NBI-77860) (см., напр., Tellew et al., Bioorg. Med. Chem. Lett. 2010, 20:7259; WO2006044958) является сильным антагонистом CRF1 с pKi связывания равной 8,2, с кинетической Ki, равной 49 нМ (Таблица 1 выше) и с t1/2 диссоциации равным 58 минут (Таблица 2 выше).
Было показано, что внутривенная инъекция пептидных антагонистов рецептора CRF1 снижает высокий уровень АКТГ в плазме адреналэктомированных крыс (см., напр., Rivier et al., J. Med. Chem. 12:42:3175-82 (1999)). Эти наблюдения повторили с применением низкомолекулярного NBI-77860. При пероральном введении адреналэктомированным крысам (n=6/группа) однократная доза NBI 77860 равная 30 мг/кг существенно снижала уровень АКТГ в плазме на протяжении периода, составляющего до 5 часов (см. Фигуру 2). Продолжительность эффекта коррелировала с пиком концентрации соединения в плазме; при этом продолжительность эффекта превышала продолжительность присутствия лекарственного средства в плазме. У адреналэктомированных крыс существует прогнозируемая связь между суммарным содержанием NBI 77860 в плазме и in vivo эффективностью после перорального введения.
Fleck et al. также сообщали об эффектах антагонистов рецептора CRF1, различающихся по времени полудиссоциации, на концентрацию АКТГ в плазме адреналэктомированных крыс, а именно NBI 30775, NBI 34041 и NBI 35965. При максимальной дозе (10 мг/кг), все три лиганда резко снижали уровень АКТГ (1 час после инъекции, Фигура 3A-C). После более продолжительного периода появлялось четкое различие между NBI 35965 и двумя другими лигандами. Уровень АКТГ возвращался к уровню при применении инертного носителя за 2 часа при применении NBI 35965 (Фиг. 3C), в то время как при применении NBI 30775 и NBI 34041 ответ поддерживался на протяжении 4-6 ч (Фиг. 3A и 3B).
Пример 5
Фармакодинамические эффекты Соединения I у человека
В этом исследовании фармакодинамические эффекты NBI-77860 (Соединения I) на человеческих субъектах оценивали, наблюдая эффект однократных пероральных доз на ответ гипоталамо-гипофизарно-надпочечниковой системы, развившийся после приема метирапона (0,04 г/кг) относительно плацебо и относительно однократной дозы алпразолама (0,75 мг). Метирапон блокирует синтез кортизола в коре надпочечников, имитируя таким образом дефицит кортизола при ВГКН, и метирапон связан с острым рефлекторным ростом уровня АКТГ.
Первичный анализ проводили, определяя значения АКТГ в период времени с 30 минут и до 4 часов после введения дозы (AUC (area under curve, площадь под кривой) (30 мин - 4 ч)). В этом исследовании при сравнении с плацебо делали следующие наблюдения. Существенное снижение AUC АКТГ (30 мин - 4 ч) наблюдали при применении алпразолама и NBI-77860, обработка 400 мг. Недостоверное снижение AUC АКТГ (30 мин - 4 ч) наблюдали при применении NBI-77860, обработка 50 мг. Увеличение AUC (30 мин - 4 ч) набдюдали при применении NBI-77860, обработка 10 мг.
При анализе значения АКТГ между 2 ч и 4 ч после введения дозы (AUC (2 ч - 4 ч)) (период, когда суммарное количество соединения в крови достигало Cmax значения) наблюдали существенное снижение уровня АКТГ при применении NBI-77860, периоды обработки 400 мг и 50 мг сравнивали с периодом обработки плацебо; однако снижение не наблюдали при применении NBI-77860 в дозе 10 мг.
Пример 6
Клиническое исследование
В данном исследовании NBI-77860 (Соединение I) оценивали в рамках клинического исследования «Простое слепое плацебо-контролируемое клиническое исследование фазы 1 с фиксированной последовательностью действий с однократным введением исследуемого соединения, проводимое с целью оценки безопасности и переносимости NBI-77860 у взрослых женщин с врожденной гиперплазией коры надпочечников» (Исследование нового препарата 117388). Исследование было простым слепым, плацебо-контролируемым, одноцентровым клиническим исследованием с фиксированной последовательностью действий с однократным введением исследуемого соединения взрослым пациенткам женского пола с классической ВГКН. Исследование было спланировано для оценки безопасности, переносимости и суммарного содержания NBI-77860 в плазме, а также для оценки эффекта этого соединения на уровень эндогенных гормонов гипоталамо-гипофизарно-надпочечниковой системы.
Всего 8 субъектам женского пола в возрасте от 19 до 58 лет с медицинским диагнозом классической ВГКН с дефицитом 21-гидролазы перед сном вводили однократные дозы (ночные дозы) NBI-77860 300 мг, 600 мг и плацебо на протяжении трех отдельных периодов лечения (см. схему плана исследования на Фигуре 4). Введение обычной утренней дозы сопутствующей стероидной терапии у этих субъектов откладывали до момента взятия образцов крови по истечении 16-часового периода после введения дозы (т.е. приблизительно до 14:00).
Фармакодинамические конечные точки для данного исследования включали заданные биомаркеры гипоталамо-гипофизарно-надпочечниковой системы в данной популяции пациентов, а именно, 17-гидроксипрогестерон (17-гидроксипрогестерон; в качесвте первичной фармакодинамической конечной точки), адренокортикотропный гормон (АКТГ), андростендион, тестостерон и уровень кортизола в сыворотке. Исходный анализ фармакодинамических переменных представлял собой анализ сгруппированных данных субъектов для каждого биомаркера, выраженных как среднее изменение процента относительно уровня перед введением дозы для условий введения двух активных доз относительно условия введения плацебо. Данные по среднему уровню 17-гидроксипрогестерона и АКТГ на протяжении 24-часового периода после введения дозы представлены на Фигуре 5.
У этих пациентов с ВГКН наблюдали схожее и клинически значимое снижение как 17-гидроксипрогестерона, так и АКТГ относительно уровня перед введением дозы на протяжении периода после введения NBI-77860 по сравнению с плацебо. В дополнение к данным по среднему значению в группе оценивали индивидуальный ответ, и «отвечающих на лечение» осторожно определяли как субъектов, у которых уровни 17-гидроксипрогестерона и АКТГ снижались по меньшей мере на 50% под действием активного NBI-77860 относительно плацебо во время пикового утреннего периода. Этот анализ отвечающих на лечение выявил значительный уровень ответа в исследовании равный 50% (ни один из субъектов не отвечал на лечение во время исходного периода обработки плацебо). Более того, доза в 300 мг оказывала почти такое же действие на 17-гидроксипрогестерон и АКТГ, как и доза в 600 мг. Пример индивидуального ответа субъекта для этих биомаркеров представлен на Фигуре 6.
Разные описанные выше варианты осуществления настоящего изобретения можно комбинировать с образованием других вариантов осуществления настоящего изобретения. Все патенты США, публикации заявок на патенты США, заявки на патенты США, иностранные патенты, иностранные заявки на патенты и публикации, не являющиеся патентами, на которые приведены ссылки в данном описании изобретения и/или которые перечислены в Информационном листке настоящей заявки, включая предварительные заявки на патент США с серийными номерами 61929941, поданную 21 января 2014 г.; 61981033, поданную 17 апреля 2014 г.; и 62/069155, поданную 27 октября 2014 г., включены в настоящую заявку в полном объеме посредством ссылки. Аспекты вариантов осуществления настоящего изобретения можно изменять при необходимости для включения сущности различных патентов, заявок и публикаций с образованием других вариантов осуществления настоящего изобретения.
Эти и другие изменения можно вносить во варианты осуществления настоящего изобретения в свете приведенного выше подробного описания изобретения. Как правило, в следующей формуле изобретения приведенные термины не предназначены для ограничения формулы изобретения конкретными вариантами осуществления настоящего изобретения, раскрытыми в описании изобретения и в формуле изобретения, но предназначены для включения всех возможных вариантов осуществления настоящего изобретения наряду с полным объемом эквивалентов, к которым относится формула изобретения. Соответственно, формула изобретения не ограничивается раскрытием.

Настоящая группа изобретений относится к медицине, а именно к терапии и эндокринологии, и касается лечения врожденной гиперплазии надпочечников (ВГКН). Для этого вводят эффективное количество антагониста рецептора CRF1 с периодом полудиссоциации более 50 минут, а именно соединение 2,5-диметил-3-[2-метил-4-(метилокси)фенил]-N-[(1S)-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил]пиразоло[1,5-a]пиримидин-7-амина (NBI-77860) или 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропин-1-ил)-2-тиазоламина (SSR-125543). Это позволяет корректировать уровни гормонов, входящих в состав «гипоталамо-гипофизарной оси», в частности 17-гидроксипрогестерона и АКТГ, и таким образом обеспечивать нормализацию образования андрогенов при применении пониженных, более физиологичных доз гидрокортизона, снижая таким образом связанные с лечением побочные эффекты. 2 н. и 4 з.п. ф-лы, 6 пр., 6 ил., 2 табл.
1. Способ лечения врожденной гиперплазии коры надпочечников (ВГКН) посредством введения нуждающемуся в этом субъекту антагониста рецептора CRF1 с периодом полудиссоциации свыше 50 минут, при этом антагонист рецептора CRF1 выбран из группы, состоящей из 2,5-диметил-3-[2-метил-4-(метилокси)фенил]-N-[(1S)-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил]пиразоло[1,5-a]пиримидин-7-амина (NBI-77860) и 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропин-1-ил)-2-тиазоламина (SSR-125543).
2. Способ по п.1, где антагонистом рецептора CRF1 является NBI-77860.
3. Способ по п.1, где антагонистом рецептора CRF1 является SSR-125543.
4. Способ снижения уровней 17-гидроксипрогестерона и АКТГ у субъекта с врожденной гиперплазией коры надпочечников (ВГКН), при этом указанный способ включает введение антагониста рецептора CRF1, где антагонист рецептора CRF1 выбран из группы, состоящей из 2,5-диметил-3-[2-метил-4-(метилокси)фенил]-N-[(1S)-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил]пиразоло[1,5-a]пиримидин-7-амина (NBI-77860) и 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропин-1-ил)-2-тиазоламина (SSR-125543).
5. Способ по п.4, где антагонистом рецептора CRF1 является NBI-77860.
6. Способ по п.4, где антагонистом рецептора CRF1 является SSR-125543.
| US 2010222339 A1 02.09.2010 | |||
| Tellew J.E | |||
| et al | |||
| ПЕРЕНОСНЫЙ СТАНОК ДЛЯ ОБТОЧКИ ШЕЕК ПАРОВОЗНЫХ ОСЕЙ | 1948 |
|
SU77860A1 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| М.Ф.ЛОГАЧЕВА и др | |||
| "Врожденная гиперплазия надпочечников: современные проблемы терминологии и | |||

