Область техники, к которой относится настоящее изобретение
Настоящее раскрытие относится к 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амину или его фармацевтически приемлемой соли для лечения врожденной гиперплазии надпочечников (САН). Настоящее раскрытие дополнительно относится к фармацевтическим составам и твердым формам 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли и их применению в лечении врожденной гиперплазии надпочечников (САН).
Предшествующий уровень техники настоящего изобретения
Классическая врожденная гиперплазия надпочечников (САН) представляет собой заболевание, которое включает группу болезней с аутосомно-рецессивным типом наследования, обуславливающих дефицит ферментов, при котором изменяется выработка стероидов надпочечников вследствие дефицита 21-гидроксилазы, состояния, которое приводит к недостаточному биосинтезу кортизола или его отсутствию. Одним из клинических проявлений отсутствия кортизола является отсутствие ингибирования по принципу обратной связи секреции адренокортикотропного гормона гипофиза (АСТН). Повышенные уровни АСТН вызывают гиперплазию надпочечников, а мутация фермента вызывает сдвиг в отношении превращения стероидов-предшественников кортизола в сторону альтернативных путей биосинтеза. Прежде всего сдвиг путей биосинтеза андрогенов приводит к вирилизации и другим нарушениям развития у лиц женского пола, а чрезмерное накопление АСТН ассоциировано с образованием эктопической ткани надпочечников в яичке у лиц мужского пола. Кроме того, поскольку этот же фермент (21-гидроксилаза) используется в пути биосинтеза минералокортикоидов, ряд таких пациентов страдают от дефицита альдостерона, что может приводить к дегидратации и смерти вследствие солевой недостаточности. Задокументированная распространенность в общей популяции США классической САН, обусловленной дефицитом 21-гидроксилазы, исходя из скрининга у новорожденных, составляла 1:10000-1:20800 (Trakakis et al., "An update to 21-hydroxylase deficient congenital adrenal hyperplasia," Gynecol. Endocrinol. (2010) 26(1):63-71; Hertzberg et al., "Birth prevalence rates of newborn screening disorders in relation to screening practices in the United States," J. Pediatr. (2011) 159(4):555-560).
Педиатрические пациенты от рождения до подросткового возраста и, в частности, лица женского пола, по всей видимости являются наиболее уязвимой популяцией пациентов с САН и представляют собой подгруппу пациентов с наибольшей неудовлетворенной медицинской потребностью (Cheng and Speiser, "Treatment outcomes in congenital adrenal hyperplasia," Adv. Pediatr. (2012) 59(1):269-281; Merke and Poppas, "Management of adolescents with congenital adrenal hyperplasia," Lancet Diabetes Endocrinol. (2013) 1(4):341-352). Избыточная выработка андрогенов у таких молодых пациентов приводит к преждевременному наступлению полового созревания и адренархе, изменениям в характере развития скелета, низкому росту, обусловленному преждевременным слиянием эпифизарных пластинок, а также значительному гирсутизму и акне. Хоть выживаемость должным образом обеспечивается за счет стратегий замещения стероидов с использованием физиологических доз глюкокортикоидов (например, гидрокортизона) и минералокортикоидов (например, флудрокортизона), такие дозы зачастую не в состоянии обеспечить подавление накопления АСТН и избыточной выработки прогестогена и андрогенов (например, 17-гидроксипрогестерона [17-ОНР], андростендиона и тестостерона). Неконтролируемые симптомы избытка андрогенов действительно оказывают существенное влияние на повседневную деятельность и развитие таких пациентов.
В настоящее время экзогенные кортикостероиды являются стандартным лечением для пациентов с классической САН. Такое лечение применяют для коррекции дефицита кортизола и снижения избыточных уровней АСТН и избытка андрогенов. Однако доза и продолжительность применения стероидов для подавления АСТН, как правило, намного превышают нормальный физиологический уровень, применяемый для замещения кортизола отдельно (как у пациентов с болезнью Аддисона.). Такое повышенное воздействие глюкокортикоидов может привести к развитию ятрогенного синдрома Кушинга, повышению частоты факторов риска развития сердечно-сосудистого заболевания, нарушению толерантности к глюкозе, снижению скорости роста и уменьшению минеральной плотности костной ткани у пациентов с САН (Elnecave et al., "Bone mineral density in girls with classical congenital adrenal hyperplasia due to CYP21 deficiency," J. Pediatr. Endocrinol. Metab. (2008) 21(12): 1155-1162; King et al., "Long-term corticosteroid replacement and bone mineral density in adult women with classical congenital adrenal hyperplasia,"./. Clin. Endocrinol. Metab. (2006) 91(3):865-869; Migeon and Wisniewski, "Congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Growth, development, and therapeutic considerations," Endocrinol. Metab. Clin. North Am. (2001) 30(1): 193-206).
Кортикотропин-рилизинг-гормон (CRF) представляет собой гормон гипоталамуса, высвобождаемый непосредственно в гипоталамо-гипофизарную портальную систему и воздействующий на специфические рецепторы кортикотропин-рилизинг-гормона 1 (CRF1) на кортикотрофах в передней доле гипофиза для стимуляции высвобождения АСТН. Было показано, что блокирование этих рецепторов уменьшает высвобождение АСТН как у животных, так и у людей. Следовательно, соединения, которые блокируют CRF1-рецепторы, обладают потенциалом в отношении прямого ингибирования избыточного высвобождения АСТН, которое происходит при САН и, следовательно, позволяют нормализовать выработку андрогенов, с применением при этом более низких физиологических доз гидрокортизона.
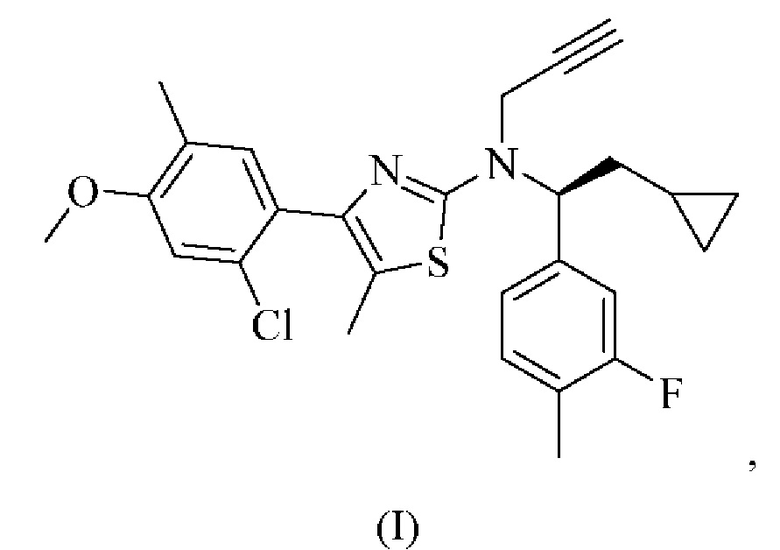
Соединение формулы (I)
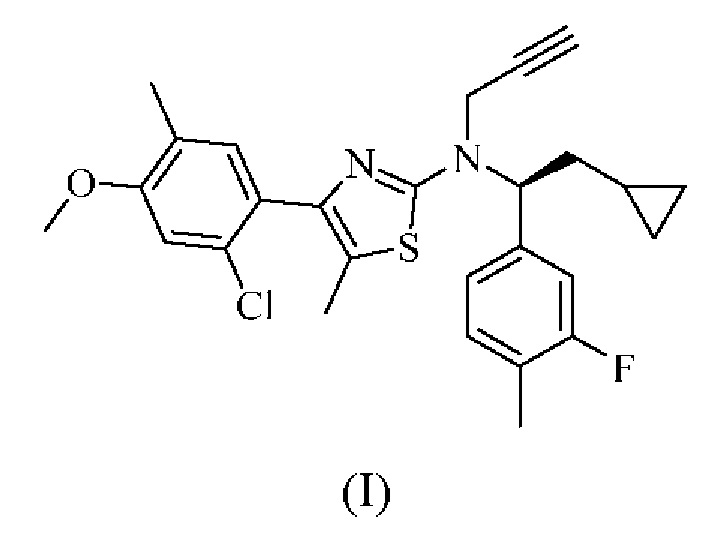
4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин представляет собой селективный антагонист рецептора CRF1. Соединение формулы (I) можно получать в соответствии со способами, описанными в заявках на выдачу патента США №№6586456 и 8314249, каждая из которых включена в настоящий документ посредством ссылки во всей своей полноте. Соединение формулы (I) представляет собой слаборастворимое соединение с низкой биодоступностью. Частично из-за его низкой растворимости, попытки получить состав на основе соединения формулы (I) оказались нелегкими, в частности, в случае составов, подходящих для введения педиатрическим пациентам.
Таким образом, существует потребность в методе лечения САН, который позволит избежать серьезных осложнений, связанных с терапией кортикостероидами. Также существует потребность в составе на основе соединения формулы (I) с повышенной биодоступностью и потребность в составе на основе соединения формулы (I), который подходит для введения педиатрическим пациентам. Составы и способы по настоящему изобретению удовлетворяют эти и другие потребности.
Краткое раскрытие настоящего изобретения
В настоящем документе представлена фармацевтическая композиция, содержащая: (а) соединение формулы (I):

или его фармацевтически приемлемую соль; и
(b) одно или несколько из основы с масляной фазой, эмульгирующего вещества, неионогенного поверхностно-активного вещества и солюбилизирующего вещества.
В настоящем документе представлена фармацевтическая композиция в лекарственной форме в виде перорального раствора, содержащая:
(a) соединение формулы (I):
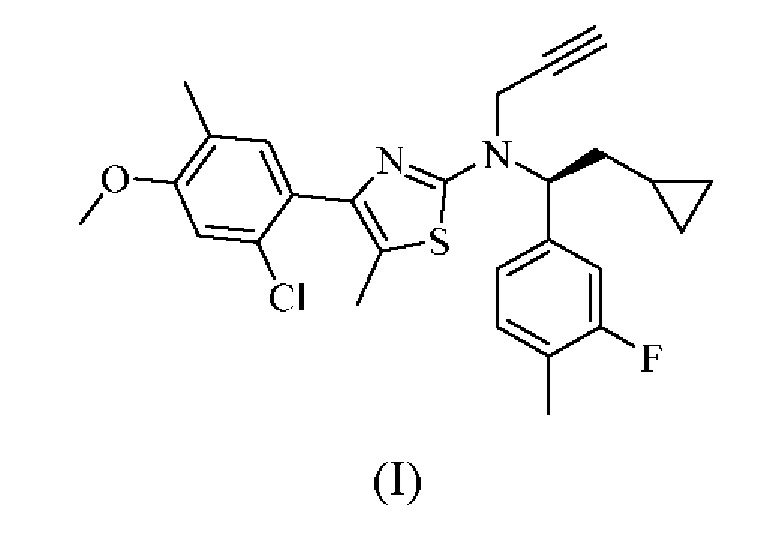
или его фармацевтически приемлемую соль;
(b) одно или несколько из подсластителя, антиоксиданта и вкусоароматической добавки; и
(c) жидкую основу.
Также в настоящем документе представлена фармацевтическая композиция по настоящему раскрытию (например, фармацевтическая композиция в лекарственной форме в виде перорального раствора по настоящему раскрытию) для применения в терапии, например, для применения в любом из раскрытых в настоящем документе способов.
В настоящем документе представлен способ лечения врожденной гиперплазии надпочечников (САН), предусматривающий введение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина, характеризующегося формулой (I):
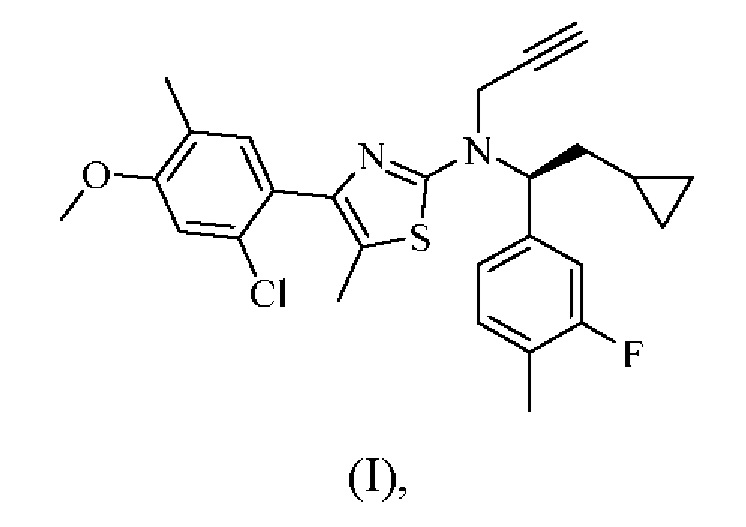
или его фармацевтически приемлемой соли.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе лечения врожденной гиперплазии надпочечников у субъекта.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе лечения врожденной гиперплазии надпочечников у субъекта.
В настоящем документе представлен способ лечения врожденной гиперплазии надпочечников у нуждающегося в этом субъекта, предусматривающий введение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли в количестве, достаточном для снижения у субъекта уровня одного или нескольких биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе лечения врожденной гиперплазии надпочечников у субъекта, причем соединение или его фармацевтически приемлемую соль вводят в количестве, достаточном для снижения у субъекта уровня одного или нескольких биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе лечения врожденной гиперплазии надпочечников у субъекта, причем соединение или его фармацевтически приемлемую соль вводят в количестве, достаточном для снижения у субъекта уровня одного или нескольких биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
Согласно некоторым вариантам осуществления снижение уровня любого из биомаркеров определяют путем сравнения уровня биомаркера, измеряемого во время циркадного высвобождения за сутки до введения 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, и уровня биомаркера, измеряемого во время циркадного высвобождения через сутки после введения 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления циркадное высвобождение происходит в период между 2 часами ночи и 10 часами утра.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят за три-восемь часов до циркадного высвобождения биомаркера.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 25%. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 50%.
Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 25%. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 40%. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 50%.
Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 25%. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 30%. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 50%.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 50% и уровень андростендиона снижается по меньшей мере на 50%.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят один раз в сутки в количестве, эквивалентном приблизительно 50 мг или приблизительно 100 мг 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина в виде свободного основания.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин вводят в форме свободного основания.
В настоящем документе представлен способ снижения тяжести одного или нескольких симптомов, выбранных из гирсутизма, преждевременного полового созревания, нарушений репродуктивной функции, акне и нарушения роста, у субъекта с классической врожденной гиперплазией надпочечников, предусматривающий введение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли в количестве, достаточном для снижения уровня андростендиона у субъекта. Согласно некоторым вариантам осуществления нарушение роста выбрано из одного или нескольких из увеличенной скорости роста, повышенной скорости увеличения массы тела или увеличенного костного возраста.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе снижения тяжести одного или нескольких симптомов, выбранных из гирсутизма, преждевременного полового созревания, нарушений репродуктивной функции, акне и нарушения роста, у субъекта с классической врожденной гиперплазией надпочечников, причем соединение или его фармацевтически приемлемую соль вводят в количестве, достаточном для снижения уровня андростендиона у субъекта.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-1Ч-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе снижения тяжести одного или нескольких симптомов, выбранных из гирсутизма, преждевременного полового созревания, нарушений репродуктивной функции, акне и нарушения роста, у субъекта с классической врожденной гиперплазией надпочечников,
причем соединение или его фармацевтически приемлемую соль вводят в количестве, достаточном для снижения уровня андростендиона у субъекта.
Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 25%. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 30%. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 50%.
В настоящем документе представлен способ снижения уровня одного или нескольких биомаркеров врожденной гиперплазии надпочечников у субъекта с врожденной гиперплазией надпочечников, предусматривающий введение субъекту 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-
метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе снижения уровня одного или нескольких биомаркеров врожденной гиперплазии надпочечников у субъекта с врожденной гиперплазией надпочечников.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе снижения уровня одного или нескольких биомаркеров врожденной гиперплазии надпочечников у субъекта с врожденной гиперплазией надпочечников.
Согласно некоторым вариантам осуществления один или несколько биомаркеров врожденной гиперплазии надпочечников выбраны из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
В настоящем документе представлен способ снижения дозировки кортикостероида, вводимого субъекту с врожденной гиперплазией надпочечников для контроля врожденной гиперплазии надпочечников, предусматривающий введение субъекту 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления кортикостероид представляет собой глюкокортикоид.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе снижения дозировки кортикостероида, вводимого субъекту с врожденной гиперплазией надпочечников.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе снижения дозировки кортикостероида, вводимого субъекту с врожденной гиперплазией надпочечников.
В настоящем документе представлен способ снижения тяжести одного или нескольких побочных эффектов лечения глюкокортикоидами у субъекта с врожденной гиперплазией надпочечников, предусматривающий введение субъекту 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, причем побочный эффект выбран из остеопороза, аваскулярного некроза кости, миопатии, гипергликемии, сахарного диабета, дислипидемии, набора лишнего веса, синдрома Кушинга, кушингоидных признаков, угнетения роста, угнетения функции надпочечников, гастрита, пептической язвы, желудочно-кишечного кровотечения, висцеральной перфорации, стеатоза печени, панкреатита, гипертензии, коронарной болезни сердца, ишемической болезни сердца, сердечной недостаточности, дерматопороза, атрофии кожи, экхимоза, пурпуры, эрозий, стрий, медленного заживления ран, склонности к образованию кровоизлияний, акне, гирсутизма, облысения, перепадов настроения, депрессии, эйфории, эмоциональной лабильности, раздражимости, акатизии, беспокойства, когнитивного нарушения, психоза, деменции, делирия, катаракты, глаукомы, птоза, мидриаза, оппортунистических инфекций глаз, центральной серозной хориоретинопатии, подавления клеточного иммунитета, предрасположенности к инфекциям и реактивации латентных инфекций.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе снижения тяжести одного или нескольких побочных эффектов лечения глюкокортикоидами у субъекта с врожденной гиперплазией надпочечников, причем побочный эффект выбран из остеопороза, аваскулярного некроза кости, миопатии, гипергликемии, сахарного диабета, дислипидемии, набора лишнего веса, синдрома Кушинга, кушингоидных признаков, угнетения роста, угнетения функции надпочечников, гастрита, пептической язвы, желудочно-кишечного кровотечения, висцеральной перфорации, стеатоза печени, панкреатита, гипертензии, коронарной болезни сердца, ишемической болезни сердца, сердечной недостаточности, дерматопороза, атрофии кожи, экхимоза, пурпуры, эрозий, стрий, медленного заживления ран, склонности к образованию кровоизлияний, акне, гирсутизма, облысения, перепадов настроения, депрессии, эйфории, эмоциональной лабильности, раздражимости, акатизии, беспокойства, когнитивного нарушения, психоза, деменции, делирия, катаракты, глаукомы, птоза, мидриаза, оппортунистических инфекций глаз, центральной серозной хориоретинопатии, подавления клеточного иммунитета, предрасположенности к инфекциям и реактивации латентных инфекций.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе снижения тяжести одного или нескольких побочных эффектов лечения глюкокортикоидами у субъекта с врожденной гиперплазией надпочечников, причем побочный эффект выбран из остеопороза, аваскулярного некроза кости, миопатии, гипергликемии, сахарного диабета, дислипидемии, набора лишнего веса, синдрома Кушинга, кушингоидных признаков, угнетения роста, угнетения функции надпочечников, гастрита, пептической язвы, желудочно-кишечного кровотечения, висцеральной перфорации, стеатоза печени, панкреатита, гипертензии, коронарной болезни сердца, ишемической болезни сердца, сердечной недостаточности, дерматопороза, атрофии кожи, экхимоза, пурпуры, эрозий, стрий, медленного заживления ран, склонности к образованию кровоизлияний, акне, гирсутизма, облысения, перепадов настроения, депрессии, эйфории, эмоциональной лабильности, раздражимости, акатизии, беспокойства, когнитивного нарушения, психоза, деменции, делирия, катаракты, глаукомы, птоза, мидриаза, оппортунистических инфекций глаз, центральной серозной хориоретинопатии, подавления клеточного иммунитета, предрасположенности к инфекциям и реактивации латентных инфекций.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят в количестве, достаточном для снижения уровня 17-гидроксипрогестерона (17-ОНР) по меньшей мере на 50% по сравнению с уровнем до введения.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят в количестве, достаточном для снижения уровня андростендиона по меньшей мере на 30% по сравнению с уровнем до введения.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят в количестве, достаточном для (а) снижения уровня 17-гидроксипрогестерона (17-ОНР) по меньшей мере на 50% по сравнению с уровнем до введения; и (b) снижения уровня андростендиона по меньшей мере на 30% по сравнению с уровнем до введения.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят один раз в сутки в количестве, эквивалентном от приблизительно 25 мг до приблизительно 150 мг 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина в виде свободного основания.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят один раз в сутки в количестве, эквивалентном приблизительно 50 мг или приблизительно 100 мг 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина в виде свободного основания.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин вводят в форме свободного основания.
В настоящем документе представлен способ лечения врожденной гиперплазии надпочечников у субъекта, предусматривающий
(i) измерение уровня одного или нескольких биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона, в биологическом образце, полученном от субъекта;
(ii) анализ уровня одного или нескольких биомаркеров для определения того, повышен ли уровень одного или нескольких биомаркеров в сравнении с таковым у здорового субъекта, у которого не наблюдается врожденной гиперплазии надпочечников; и
(iii) введение субъекту 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, если определено, что у субъекта наблюдаются повышенные уровни одного или нескольких биомаркеров.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе лечения врожденной гиперплазии надпочечников у субъекта, предусматривающем:
(i) измерение уровня одного или нескольких биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона, в биологическом образце, полученном от субъекта;
(ii) анализ уровня одного или нескольких биомаркеров для определения того, повышен ли уровень одного или нескольких биомаркеров в сравнении с таковым у здорового субъекта, у которого не наблюдается врожденной гиперплазии надпочечников; и
(iii) введение субъекту 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, если определено, что у субъекта наблюдаются повышенные уровни одного или нескольких биомаркеров.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе лечения врожденной гиперплазии надпочечников у субъекта, причем способ предусматривает:
(i) измерение уровня одного или нескольких биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона, в биологическом образце, полученном от субъекта;
(ii) анализ уровня одного или нескольких биомаркеров для определения того, повышен ли уровень одного или нескольких биомаркеров в сравнении с таковым у здорового субъекта, у которого не наблюдается врожденной гиперплазии надпочечников; и
(iii) введение субъекту 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, если определено, что у субъекта наблюдаются повышенные уровни одного или нескольких биомаркеров.
Согласно некоторым вариантам осуществления способ дополнительно предусматривает (iv) измерение уровня одного или несколько биомаркеров после введения 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли в биологическом образце, полученном от субъекта, для определения того, наблюдаются ли у субъекта сниженные уровни одного или нескольких биомаркеров по сравнению с таковыми при измерении на стадии (i). Согласно некоторым вариантам осуществления способ дополнительно предусматривает (ν) продолжение введения 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, если у субъекта наблюдаются сниженные уровни одного или нескольких биомаркеров.
Согласно некоторым вариантам осуществления стадии (i) и (iv) осуществляют в отношении биологических образцов, взятых у субъекта одинаковым способом и в пределах одного и того же окна времени суток. Согласно некоторым вариантам осуществления стадии (i) и (iv) осуществляют в отношении биологических образцов, взятых у субъекта в пределах окна времени суток с 2 ночи до 10 утра. Согласно некоторым вариантам осуществления стадии (i) и (iv) осуществляют в отношении биологических образцов, взятых у субъекта в пределах окна времени суток с 6 утра до 10 утра.
Согласно некоторым вариантам осуществления стадии (i) и (iv) предусматривают измерение уровней по меньшей мере двух биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
Согласно некоторым вариантам осуществления стадии (i) и (iv) предусматривают измерение уровней (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
Согласно некоторым вариантам осуществления стадия (i) предусматривает измерение уровня 17-гидроксипрогестерона (17-ОНР), причем уровень 17-гидроксипрогестерона (17-ОНР) повышен, если он превышает или равен 1000 нг/дл.
Согласно некоторым вариантам осуществления стадия (i) предусматривает измерение уровня андростендиона, причем уровень андростендиона повышен, если он превышает 200 нг/дл.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят один раз в сутки в количестве, эквивалентном от приблизительно 25 мг до приблизительно 150 мг 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина в виде свободного основания. Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят один раз в сутки в количестве, эквивалентном приблизительно 50 мг или приблизительно 100 мг 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина в виде свободного основания. Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин вводят в форме свободного основания.
В настоящем документе представлен способ лечения врожденной гиперплазии надпочечников (САН) у нуждающегося в этом субъекта, предусматривающий введение субъекту терапевтически эффективного количества 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, при этом субъект находится в сытом состоянии.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе лечения врожденной гиперплазии надпочечников (САН) у субъекта, при этом субъект находится в сытом состоянии.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе лечения врожденной гиперплазии надпочечников (САН) у субъекта, при этом субъект находится в сытом состоянии.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят субъекту с питательной композицией. Согласно некоторым вариантам осуществления питательная композиция представляет собой жидкую пищевую добавку, содержащую приблизительно 1500 калорий на литр, со следующим распределением калорий: приблизительно 14,7% белков, приблизительно 32% жиров и приблизительно 53,3% углеводов. Согласно некоторым вариантам осуществления питательную композицию принимают в количестве приблизительно 8 жидких унций. Согласно некоторым вариантам осуществления питательную композицию принимают в пределах 30 минут от введения 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят субъекту с питательной композицией. Согласно некоторым вариантам осуществления питательная композиция представляет собой жидкую пищевую добавку, содержащую 1500 калорий на литр, со следующим распределением калорий: 14,7% белков, 32% жиров и 53,3% углеводов. Согласно некоторым вариантам осуществления питательную композицию принимают в количестве приблизительно 8 жидких унций. Согласно некоторым вариантам осуществления питательную композицию принимают в пределах 30 минут от введения 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления введение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли характеризуется положительным влиянием пищи. Согласно некоторым вариантам осуществления положительное влияние пищи измеряют по Cmax, AUC или их комбинациям при сравнении перорального введения 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил- 1,3-тиазол-2-амина или его фармацевтически приемлемой соли в сытом состоянии и натощак.
Согласно некоторым вариантам осуществления соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение Cmax при введении в сытом состоянии и Cmax при введении натощак составляет от приблизительно 5 до приблизительно 10.
В настоящем документе представлен способ уменьшения глюкокортикоидной нагрузки у субъекта, как измерено после некоторого периода времени введения соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли, причем уменьшение глюкокортикоидной нагрузки является таковым относительно глюкокортикоидной нагрузки до введения соединения или его фармацевтически приемлемой соли.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе уменьшения глюкокортикоидной нагрузки у субъекта, причем уменьшение глюкокортикоидной нагрузки является таковым относительно глюкокортикоидной нагрузки до введения соединения или его фармацевтически приемлемой соли.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе уменьшения глюкокортикоидной нагрузки у субъекта, причем уменьшение глюкокортикоидной нагрузки является таковым относительно глюкокортикоидной нагрузки до введения соединения или его фармацевтически приемлемой соли.
В настоящем документе представлен способ улучшения в отношении одного или нескольких симптомов, выбранных из качества жизни, утомляемости, сна, инсулинорезистентности, толерантности к глюкозе, контроля уровня глюкозы, дислипидемии, гиперлипидемии, минеральной плотности костной ткани, обновления костной ткани, массы жировой ткани, веса, центрального ожирения, артериального давления, степени выраженности гирсутизма, цикличности менструации, контроля эктопической ткани надпочечников в яичке и репродуктивной функции, у субъекта с классической врожденной гиперплазией надпочечников, предусматривающий введение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, причем улучшение в отношении симптома происходит после некоторого периода времени введения соединения или его фармацевтически приемлемой соли, причем улучшение в отношении одного или нескольких симптомов является таковым относительно статуса одного или нескольких симптомов до введения соединения или его фармацевтически приемлемой соли.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в способе улучшения в отношении одного или нескольких симптомов, выбранных из качества жизни, утомляемости, сна, инсулинорезистентности, толерантности к глюкозе, контроля уровня глюкозы, дислипидемии, гиперлипидемии, минеральной плотности костной ткани, обновления костной ткани, массы жировой ткани, веса, центрального ожирения, артериального давления, степени выраженности гирсутизма, цикличности менструации, контроля эктопической ткани надпочечников в яичке и репродуктивной функции, у субъекта с классической врожденной гиперплазией надпочечников, причем улучшение в отношении симптома происходит после некоторого периода времени введения соединения или его фармацевтически приемлемой соли, причем улучшение в отношении одного или нескольких симптомов является таковым относительно статуса одного или нескольких симптомов до введения соединения или его фармацевтически приемлемой соли.
Также в настоящем документе представлено применение соединения, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил- 1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в способе улучшения в отношении одного или нескольких симптомов, выбранных из качества жизни, утомляемости, сна, инсулинорезистентности, толерантности к глюкозе, контроля уровня глюкозы, дислипидемии, гиперлипидемии, минеральной плотности костной ткани, обновления костной ткани, массы жировой ткани, веса, центрального ожирения, артериального давления, степени выраженности гирсутизма, цикличности менструации, контроля эктопической ткани надпочечников в яичке и репродуктивной функции, у субъекта с классической врожденной гиперплазией надпочечников, причем улучшение в отношении симптома происходит после некоторого периода времени введения соединения или его фармацевтически приемлемой соли, причем улучшение в отношении одного или нескольких симптомов является таковым относительно статуса одного или нескольких симптомов до введения соединения или его фармацевтически приемлемой соли.
Также в настоящем документе представлено соединение, которое представляет собой 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, или его фармацевтически приемлемая соль для применения в терапии, например, для применения в любом из раскрытых в настоящем документе способов.
Также в настоящем документе представлено применение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли для производства лекарственного препарата для применения в любом из раскрытых в настоящем документе способов.
Также в настоящем документе представлена высушенная распылением дисперсия, содержащая 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, характеризующийся формулой (I):
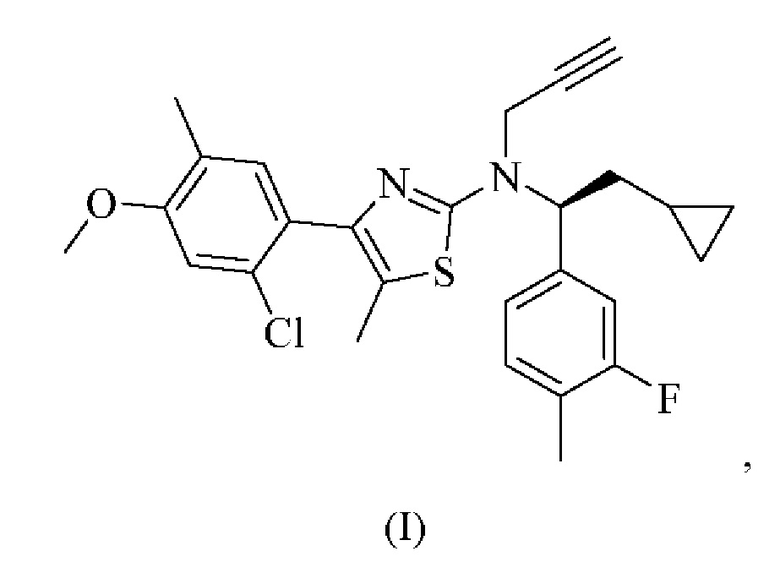
или его фармацевтически приемлемую соль и полимер. Согласно некоторым вариантам осуществления полимер выбран из нейтрального полимера, растворимого в кишечнике полимера и полимера пирролидона. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) и полимера составляет от приблизительно 1:9 до приблизительно 1:1.
Согласно некоторым вариантам осуществления полимер представляет собой нейтральный полимер. Согласно некоторым вариантам осуществления нейтральный полимер выбран из гидроксипропилметилцеллюлозы (НРМС) и гидроксиэтилцеллюлозы (НЕС).
Согласно некоторым вариантам осуществления полимер представляет собой растворимый в кишечнике полимер. Согласно некоторым вариантам осуществления растворимый в кишечнике полимер выбран из сукцината ацетата гидроксипропилметилцеллюлозы (HPMCAS), фталата ацетата целлюлозы (САР), фталата гидроксипропилметилцеллюлозы (НРМСР), сополимера аминометакрилата, сополимера аммониоалкилметакрилата и метакрилового сополимера.
Согласно некоторым вариантам осуществления полимер представляет собой полимер пирролидона. Согласно некоторым вариантам осуществления полимер пирролидона выбран из поливинилпирролидона (PVP) и сополимера винилпирролидона и винилацетата (PVP/VA). Согласно некоторым вариантам осуществления полимер пирролидона представляет собой PVP/VA. Согласно некоторым вариантам осуществления сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении от приблизительно 40:60 до приблизительно 60:40 по весу. Согласно некоторым вариантам осуществления сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении приблизительно 60:40 по весу. Согласно некоторым вариантам осуществления сополимер характеризуется структурой:
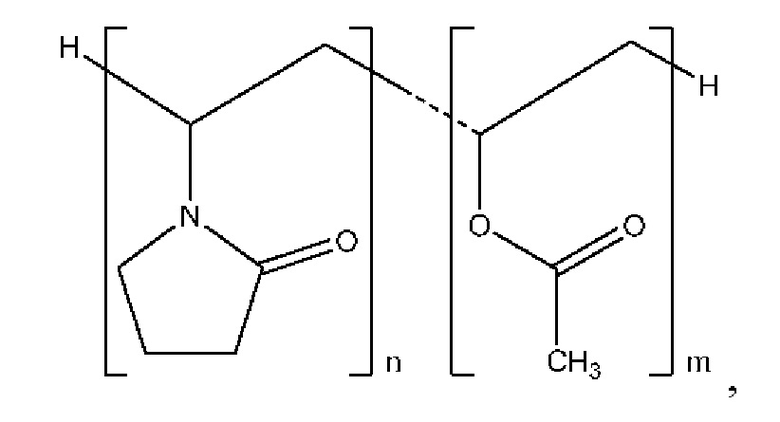
причем значение n превышает значение m с кратностью от приблизительно 1 до приблизительно 2 раз. Согласно некоторым вариантам осуществления сополимер представляет собой коповидон, причем значение n в приблизительно 1,16 раза превышает значение т.Согласно некоторым вариантам осуществления сополимер представляет собой коповидон со средней молекулярной массой от приблизительно 45000 до приблизительно 70000.
В настоящем документе представлена высушенная распылением дисперсия, содержащая 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, характеризующийся формулой (I):
или его фармацевтически приемлемую соль; и полимер, который представляет собой сополимер 1-винил-2-пирролидона и винилацетата, характеризующийся структурой:
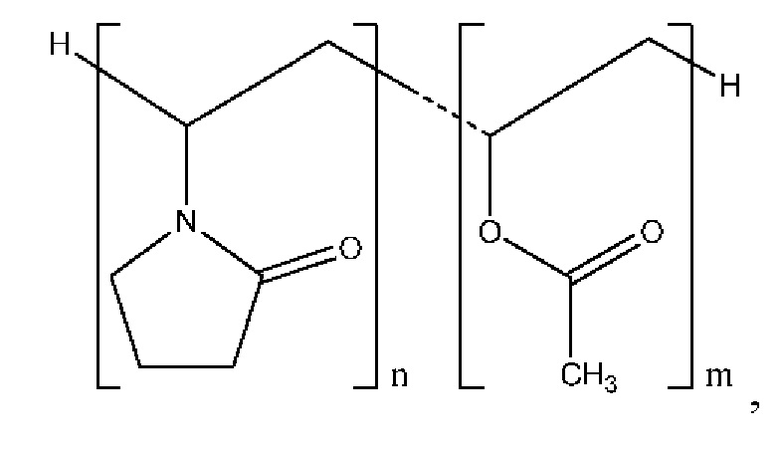
причем значение n превышает значение m с кратностью от приблизительно 1 до приблизительно 2 раз, и сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении приблизительно 60:40 по весу; и причем весовое соотношение соединения формулы (I) и сополимера составляет от приблизительно 1:1 до приблизительно 1:9.
Согласно некоторым вариантам осуществления высушенных распылением дисперсий по настоящему раскрытию, соединение формулы (I) и полимер вместе образуют однородные частицы. Согласно некоторым вариантам осуществления частицы характеризуются распределением частиц по размеру со значением D50 от приблизительно 5 мкм до приблизительно 100 мкм. Согласно некоторым вариантам осуществления частицы характеризуются распределением частиц по размеру со значением D50 от приблизительно 10 мкм до приблизительно 50 мкм. Согласно некоторым вариантам осуществления частицы характеризуются распределением частиц по размеру со значением D50 от приблизительно 15 мкм до приблизительно 30 мкм.
Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) и полимера составляет от приблизительно 1:1,5 до приблизительно 1:9. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) и полимера составляет от приблизительно 1:2,5 до приблизительно 1:4. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) и полимера составляет приблизительно 1:3.
Согласно некоторым вариантам осуществления частицы характеризуются содержанием остаточного растворителя менее чем приблизительно 2% по весу. Согласно некоторым вариантам осуществления частицы характеризуются содержанием остаточного растворителя менее чем приблизительно 1% вес. Согласно некоторым вариантам осуществления частицы характеризуются содержанием остаточного растворителя приблизительно 0,5% вес. или меньше.
Согласно некоторым вариантам осуществления соединение формулы (I) в дисперсии главным образом является аморфным.
Также в настоящем документе представлен способ получения высушенной распылением дисперсии по настоящему раскрытию, предусматривающий: растворение соединения формулы (I) или его фармацевтически приемлемой соли и полимера в органическом растворителе с образованием раствора; и высушивание распылением раствора с получением высушенной распылением дисперсии, причем высушивание распылением обеспечивает образование однородных частиц соединения формулы (I) и полимера.
Согласно некоторым вариантам осуществления способ предусматривает удаление органического растворителя после образования высушенной распылением дисперсии путем высушивания высушенной распылением дисперсии. Согласно некоторым вариантам осуществления высушенную распылением дисперсию сушат с помощью конвекционной полочной сушилки. Согласно некоторым вариантам осуществления органический растворитель представляет собой ацетон.
Согласно некоторым вариантам осуществления температура на входе распылительной сушилки составляет от приблизительно 60°С до приблизительно 80°С. Согласно некоторым вариантам осуществления температура на входе распылительной сушилки составляет приблизительно 72°С.
Согласно некоторым вариантам осуществления температура на выходе распылительной сушилки составляет от приблизительно 25°С до приблизительно 45°С. Согласно некоторым вариантам осуществления температура на выходе распылительной сушилки составляет приблизительно 35°С.
Согласно некоторым вариантам осуществления однородные частицы предусматривают насыпную плотность без утряски менее чем приблизительно 0,2 г/мл. Согласно некоторым вариантам осуществления однородные частицы предусматривают насыпную плотность без утряски менее чем приблизительно 0,15 г/мл.
Согласно некоторым вариантам осуществления однородные частицы предусматривают плотность после утряски менее чем приблизительно 0,3 г/мл. Согласно некоторым вариантам осуществления однородные частицы предусматривают плотность после утряски менее чем приблизительно 0,25 г/мл.
Также в настоящем документе представлена фармацевтическая композиция, содержащая высушенную распылением дисперсию по настоящему раскрытию и один или несколько фармацевтически приемлемых вспомогательных веществ. Согласно некоторым вариантам осуществления высушенная распылением дисперсия присутствует в количестве от приблизительно 20% до приблизительно 90% вес./вес. композиции. Согласно некоторым вариантам осуществления высушенная распылением дисперсия присутствует в количестве от приблизительно 40% до 80% вес/вес. композиции.
Согласно некоторым вариантам осуществления фармацевтические вспомогательные вещества выбраны из группы, состоящей из наполнителя, смазывающего вещества и их комбинаций. Согласно некоторым вариантам осуществления наполнитель выбран из группы, состоящей из связующего, разбавителя, разрыхлителя, вещества, способствующего скольжению, поверхностно-активного вещества и их комбинаций.
Согласно некоторым вариантам осуществления фармацевтическая композиция составлена в виде единичной лекарственной формы, причем соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от приблизительно 5 мг до приблизительно 200 мг. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от приблизительно 75 мг до приблизительно 150 мг. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве приблизительно 50 мг. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве приблизительно 100 мг.
Согласно некоторым вариантам осуществления фармацевтическая композиция составлена в виде таблетки, капсулы, саше, порошка, гранул, покрытой оболочкой частицы, покрытой оболочкой таблетки, покрытой кишечнорастворимой оболочкой таблетки, покрытой кишечнорастворимой оболочкой капсулы, пластинки, диспергируемой в полости рта, или пленки, диспергируемой в полости рта. Согласно некоторым вариантам осуществления фармацевтическая композиция находится в форме таблетки. Согласно некоторым вариантам осуществления фармацевтическая композиция находится в форме капсулы. Согласно некоторым вариантам осуществления фармацевтическая композиция покрыта оболочкой.
Согласно некоторым вариантам осуществления высушенная распылением дисперсия составлена для перорального введения и характеризуется положительным влиянием пищи при пероральном введении. Согласно некоторым вариантам осуществления высушенная распылением дисперсия характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления высушенная распылением дисперсия характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от приблизительно 5 до приблизительно 10.
Также в настоящем документе представлен способ получения фармацевтической композиции, предусматривающий объединение высушенной распылением дисперсии по настоящему раскрытию с одним или несколькими фармацевтически приемлемыми вспомогательными веществами.
Также в настоящем документе представлен способ лечения врожденной гиперплазии надпочечников (САН) у нуждающегося в этом субъекта, предусматривающий введение субъекту терапевтически эффективного количества высушенной распылением дисперсии по настоящему раскрытию или фармацевтической композиции, содержащей высушенную распылением дисперсию по настоящему раскрытию.
Также в настоящем документе представлены высушенная распылением дисперсия по настоящему раскрытию или фармацевтическая композиция, содержащая высушенную распылением дисперсию по настоящему раскрытию, для применения в способе лечения врожденной гиперплазии надпочечников (САН) у субъекта.
Также в настоящем документе представлено применение высушенной распылением дисперсии по настоящему раскрытию в производстве лекарственного препарата для применения в способе лечения врожденной гиперплазии надпочечников (САН) у субъекта.
Согласно некоторым вариантам осуществления высушенную распылением дисперсию или фармацевтическую композицию вводят субъекту в сытом состоянии. Согласно некоторым вариантам осуществления высушенную распылением дисперсию или фармацевтическую композицию вводят субъекту с питательной композицией. Согласно некоторым вариантам осуществления питательная композиция представляет собой жидкую пищевую добавку, содержащую от приблизительно 1000 до приблизительно 2000 калорий на литр с содержанием жиров более 30%. Согласно некоторым вариантам осуществления питательная композиция представляет собой жидкую пищевую добавку, содержащую 1500 калорий на литр, со следующим распределением калорий: 14,7% белков, 32% жиров и 53,3% углеводов. Согласно некоторым вариантам осуществления питательную композицию принимают в количестве приблизительно 8 жидких унций. Согласно некоторым вариантам осуществления питательную композицию принимают в пределах 30 минут от введения высушенной распылением дисперсии или фармацевтической композиции.
Согласно некоторым вариантам осуществления введение высушенной распылением дисперсии или фармацевтической композиции характеризуется положительным влиянием пищи. Согласно некоторым вариантам осуществления положительное влияние пищи измеряют по Cmax, AUC или их комбинациям при сравнении перорального введения высушенной распылением дисперсии или фармацевтической композиции в сытом состоянии и натощак. Согласно некоторым вариантам осуществления соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение Cmax при введении в сытом состоянии и Cmax при введении натощак составляет от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет от приблизительно 10 до приблизительно 20. Согласно некоторым вариантам осуществления соотношение Cmax при введении в сытом состоянии и Cmax при введении натощак составляет от приблизительно 10 до приблизительно 20.
Согласно некоторым вариантам осуществления раскрытых способов субъектом является педиатрический пациент.
Также в настоящем документе представлены высушенная распылением дисперсия по настоящему раскрытию или фармацевтическая композиция, содержащая высушенную распылением дисперсию по настоящему раскрытию, для применения в терапии, например, для применения в любом из раскрытых в настоящем документе способов.
Также в настоящем документе представлено применение высушенной распылением дисперсии по настоящему раскрытию для производства лекарственного препарата для применения в любом из раскрытых в настоящем документе способов.
Также в настоящем документе представлен способ лечения врожденной гиперплазии надпочечников (САН) у нуждающегося в этом субъекта, предусматривающий введение субъекту фармацевтической композиции по настоящему раскрытию, причем фармацевтическая композиция содержит терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли.
Также в настоящем документе представлена фармацевтическая композиция по настоящему раскрытию, причем фармацевтическая композиция содержит терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли для применения в способе лечения врожденной гиперплазии надпочечников (САН) у субъекта.
Также в настоящем документе представлено применение фармацевтической композиции по настоящему раскрытию, причем фармацевтическая композиция содержит терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, в производстве лекарственного препарата для применения в способе лечения врожденной гиперплазии надпочечников (САН) у субъекта.
Также в настоящем документе представлена фармацевтическая композиция по настоящему раскрытию, причем фармацевтическая композиция содержит терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли для применения в терапии, например, для применения в любом из раскрытых в настоящем документе способов.
Также в настоящем документе представлено применение фармацевтической композиции по настоящему раскрытию, причем фармацевтическая композиция содержит терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли для производства лекарственного препарата для применения в любом из раскрытых в настоящем документе способов.
Также в настоящем документе представлен способ улучшения абсорбции соединения формулы (I) в желудочно-кишечном тракте у субъекта, предусматривающий пероральное введение субъекту терапевтически эффективного количества высушенной распылением дисперсии по настоящему раскрытию или фармацевтической композиции, содержащей высушенную распылением дисперсию по настоящему раскрытию, причем улучшение является таковым относительно перорального введения соединения формулы (I), которое не было получено в виде высушенной распылением дисперсии.
Также в настоящем документе представлены высушенная распылением дисперсия по настоящему раскрытию или фармацевтическая композиция, содержащая высушенную распылением дисперсию по настоящему раскрытию, для применения в способе улучшения абсорбции соединения формулы (I) в желудочно-кишечном тракте у субъекта, причем улучшение является таковым относительно перорального введения соединения формулы (I), которое не было получено в виде высушенной распылением дисперсии.
Также в настоящем документе представлено применение высушенной распылением дисперсии по настоящему раскрытию в производстве лекарственного препарата для применения в способе улучшения абсорбции соединения формулы (I) в желудочно-кишечном тракте у субъекта, причем улучшение является таковым относительно перорального введения соединения формулы (I), которое не было получено в виде высушенной распылением дисперсии.
Согласно некоторым вариантам осуществления субъектом является педиатрический пациент.
Также в настоящем документе представлен способ улучшения биодоступности при пероральном введении соединения формулы (I) у субъекта, предусматривающий пероральное введение субъекту терапевтически эффективного количества высушенной распылением дисперсии по настоящему раскрытию или фармацевтической композиции, содержащей высушенную распылением дисперсию по настоящему раскрытию, причем улучшение является таковым относительно перорального введения соединения формулы (I), которое не было получено в виде высушенной распылением дисперсии.
Также в настоящем документе представлены высушенная распылением дисперсия по настоящему раскрытию или фармацевтическая композиция, содержащая высушенную распылением дисперсию по настоящему раскрытию, для применения в способе улучшения биодоступности при пероральном введении соединения формулы (I) у субъекта, причем улучшение является таковым относительно перорального введения соединения формулы (I), которое не было получено в виде высушенной распылением дисперсии.
Также в настоящем документе представлено применение высушенной распылением дисперсии по настоящему раскрытию в производстве лекарственного препарата для применения в способе улучшения биодоступности при пероральном введении соединения формулы (I) у субъекта, причем улучшение является таковым относительно перорального введения соединения формулы (I), которое не было получено в виде высушенной распылением дисперсии.
Согласно некоторым вариантам осуществления субъектом является педиатрический пациент.
Также в настоящем документе представлена кристаллическая соль, которая представляет собой соль 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил- 1,3-тиазол-2-амина и п-толуолсульфоновой кислоты.
Также в настоящем документе представлена кристаллическая соль, которая представляет собой соль 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил- 1,3-тиазол-2-амина и п-толуолсульфоновой кислоты для применения в терапии, например, для применения в любом из раскрытых в настоящем документе способов.
Также в настоящем документе представлено применение кристаллической соли, которая представляет собой соль 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина и п-толуолсульфоновой кислоты для производства лекарственного препарата для применения в любом из раскрытых в настоящем документе способов.
Другие признаки и преимущества представленных в настоящем документе способов, процессов, составов и применений будут очевидны из следующего подробного описания и фигур, а также из формулы изобретения.
Описание чертежей
На фиг. 1 показаны характеристики растворения нескольких составов на основе высушенной распылением дисперсии в 0,5% вес. искусственном кишечном соке (SIF) в забуференном фосфатом солевом растворе (PBS), рН 6,5.
На фиг. 2 показана ячейка для анализа вертикальной удельной проницаемости мембраны в составе μDiss Profiler™, применяемого для анализа удельной проницаемости мембраны.
На фиг. 3 показаны данные по растворению в насыщаемых условиях для нескольких составов на основе высушенной распылением дисперсии и соединения формулы (I) в 0,5% вес.SIF в PBS, рН 6,5.
Фиг. 4 представляет собой график, на котором показана удельная проницаемость мембраны для 1 мг/мл GB/IB 0,5% вес. SIF доз соединения формулы (I) и различных составов на основе высушенной распылением дисперсии с течением времени. Сплошные линии обозначают удельную проницаемость (мкг мин-1 см-2), а пунктирные линии обозначают концентрацию (мкг/мл) в 0,5% SIF.
Фиг. 5 представляет собой схему производственного процесса высушивания распылением, применяемого для получения 1000 г серии SDD, содержащей 25% соединения формулы (I) и 75% PVP/VA 64.
Фигуры 6А и 6В представляют собой линейные графики, на которых показаны результаты по фармакокинетике в рамках исследования биодоступности и влияния пищи у собак. На фиг. 6А показаны результаты для когорты 1, и на фиг. 6В показаны результаты для когорты 2.
Фиг. 7 представляет собой схему-алгоритм, на которой показан дизайн исследования 1 фазы фармакокинетических показателей и влияния пищи для соединения формулы (I) у здоровых взрослых субъектов.
Фигуры 8А и 8В представляют собой линейные графики, на которых показана средняя концентрация в плазме крови в зависимости от времени для соединения формулы (I) при введении соответственно натощак и в сытом состоянии у здоровых взрослых субъектов.
Фигуры 9А-9С представляют собой спагетти-графики фармакокинетических показателей соединения формулы (I) у здоровых взрослых субъектов при введении натощак и в сытом состоянии. На фиг. 9А показаны значения AUC0-tlast. На фиг. 9В показаны значения AUC0-∞. На фиг. 9С показаны значения Cmax.
Фиг. 10 представляет собой схему-алгоритм, на которой показан дизайн исследования 1 фазы биодоступности, фармакокинетических показателей и влияния пищи для соединения формулы (I) у здоровых взрослых субъектов.
На фиг. 11 показан дизайн исследования 2 фазы соединения формулы (I) с участием взрослых субъектов с врожденной гиперплазией надпочечников.
На фигурах 12А и 12В показаны средние арифметические значения для адренокортикотропного гормона (АСТН) (фиг. 12А) и 17-гидроксипрогестерона (17-ОНР) (фиг. 12В) для всех 8 субъектов когорты 1, отложенные на графике для каждого момента времени в случае исходного уровня до лечения (кружки), на 1-е сутки (квадраты) и на 14-е сутки (треугольники).
На фигурах 13А и 13В показаны средние арифметические значения для андростендиона (фиг. 13А) и тестостерона (фиг. 13В) для всех 8 субъектов когорты 1, отложенные на графике для каждого момента времени в случае исходного уровня до лечения (кружки), на 1-е сутки (квадраты) и на 14-е сутки (треугольники).
На фигурах 14А и 14В показано снижение АСТН в моменты времени через 8, 10 и 12 часов после введения дозы. На фиг. 14А показаны значения для каждого момента времени по сравнению с исходным уровнем. На фиг. 14В показаны средние значения по всем трем моментам времени.
На фигурах 15А и 15В показано снижение 17-ОНР в моменты времени через 8, 10 и 12 часов после введения дозы. На фиг. 15А показаны значения для каждого момента времени по сравнению с исходным уровнем. На фиг. 15В показаны средние значения по всем трем моментам времени.
На фигурах 16 А и 16В показано снижение андростендиона в моменты времени через 8, 10 и 12 часов после введения дозы. На фиг. 16А показаны значения для каждого момента времени по сравнению с исходным уровнем. На фиг. 16В показаны средние значения по всем трем моментам времени.
На фиг. 17А показаны средние концентрации АСТН в плазме крови после введения 50 мг дозы соединения формулы (I) qhs (когорта 1; n=8). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны АСТН: Лица женского пола, 6-58 пг/мл; лица мужского пола, 7-69 пг/мл.
На фиг. 17В показаны средние концентрации 17-ОНР в сыворотке крови после введения 50 мг дозы соединения формулы (I) qhs (когорта 1; n=8). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны 17-ОНР: Лица женского пола <207 нг/дл; лица мужского пола <139 нг/дл.
На фиг. 17С: средние концентрации андростендиона в сыворотке крови после введения 50 мг дозы соединения формулы (I) qhs (когорта 1; n=8). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны андростендиона: Лица женского пола, 26-214 нг/мл; лица мужского пола, 33-134 нг/мл.
На фиг. 18А показаны средние концентрации АСТН в плазме крови после введения 100 мг дозы соединения формулы (I) qhs (когорта 2; n=4). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны АСТН: Лица женского пола, 6-58 пг/мл; лица мужского пола, 7-69 пг/мл.
На фиг. 18В показаны средние концентрации 17-ОНР в сыворотке крови после введения 100 мг дозы соединения формулы (I) qhs (когорта 2; n=4). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны 17-ОНР: Лица женского пола <207 нг/дл; лица мужского пола <139 нг/дл.
На фиг. 18С концентрации андростендиона в сыворотке крови после введения 100 мг дозы соединения формулы (I) qhs (когорта 2; n=4). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны андростендиона: Лица женского пола, 26-214 нг/мл; лица мужского пола, 33-134 нг/мл.
На фиг. 19А показаны средние концентрации АСТН в плазме крови после введения 100 мг дозы соединения формулы (I) с ужином (когорта 3). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны АСТН: Лица женского пола, 6-58 пг/мл; лица мужского пола, 7-69 пг/мл.
На фиг. 19В показаны средние концентрации 17-ОНР в сыворотке крови после введения 100 мг дозы соединения формулы (I) с ужином (когорта 3). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны 17-ОНР: Лица женского пола <207 нг/дл; лица мужского пола <139 нг/дл.
На фиг. 19С показаны средние концентрации андростендиона в сыворотке крови после введения 100 мг дозы соединения формулы (I) с ужином (когорта 3). Планки погрешностей обозначают стандартную ошибку среднего для каждого момента времени в окне утреннего времени суток. Нормальные диапазоны андростендиона: Лица женского пола, 26-214 нг/мл; лица мужского пола, 33-134 нг/мл.
Фиг. 20 представляет собой схему производственного процесса для получения капсул с 50 мг соединения формулы (I).
Фиг. 21 представляет собой альтернативную схему производственного процесса для получения капсул с 50 мг соединения формулы (I).
На фигурах 22А и 22В показана схема производственного процесса для получения SDD-гранул соединения формулы (I).
Фиг. 23 представляет собой схему производственного процесса для получения жидкого состава 1 на основе соединения формулы (I) с концентрацией 50 мг/нл.
Фиг. 24 представляет собой схему производственного процесса для получения жидкого состава 2 на основе соединения формулы (I) с концентрацией 50 мг/нл.
Фиг. 25 представляет собой XRPD-спектр соединения формулы (I) в виде кристаллического свободного основания, форма I.
Фиг. 26 представляет собой DSC-спектр соединения формулы (I) в виде кристаллического свободного основания, форма I.
Фиг. 27 представляет собой XRPD-спектр соединения формулы (I) в виде кристаллической соли паратолуолсульфокислоты, форма 1.
Фиг. 28 представляет собой DSC- и TGA-спектр соединения формулы (I) в виде кристаллической соли паратолуолсульфокислоты, форма 1.
Подробное раскрытие настоящего изобретения
Как описано в настоящем документе, 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин, характеризующийся формулой (I):
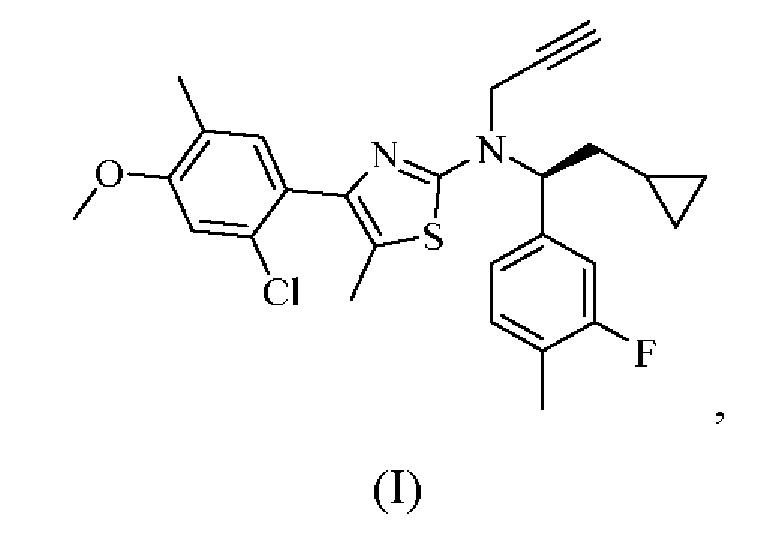
или его фармацевтически приемлемая соль представляют собой селективный антагонист рецептора CRF1, который, как было обнаружено, является эффективным в лечении врожденной гиперплазии надпочечников. В частности, соединение формулы (I), как было обнаружено, эффективно снижает уровень нескольких биомаркеров, ассоциированных с врожденной гиперплазией надпочечников.
Скрининг в отношении САН у новорожденных осуществляют с помощью иммунологического анализа для измерения уровней 17-ОНР в образцах капиллярной крови, взятой из пятки, полученных в пределах первых 72 часов жизни. Образец крови анализируют на предмет 17-ОНР с помощью коммерчески доступного иммунологического анализа на основе усиленной диссоциацией флуоресценции хелата лантаноида (DELFIA; PerkinElmer, Уолтем, Массачусетс) (White et al., J. Pediatr. 163:10-12 (2013)). Скрининговые тесты второго уровня, в которых используются биохимические и молекулярно-генетические способы тестирования, осуществляемые в промежутке времени с 8 по 14 дни жизни, используют в девяти штатах США и настоятельно рекомендуются еще 5 штатами. Биохимический способ включает иммунологический анализ с экстракцией органическими растворителями или жидкостную хроматографию с последующей тандемной масс-спектрометрией для измерения соотношений стероидов 17-ОНР, андростендиона и 21-дезоксикортизола и кортизола (см., например, Speiser et al., Int. J. Pediatr. Endocrinol. 2010:494173, 2010). Генетический скрининг направлен на поиск мутаций CYP21A2, которые ассоциированы с САН. Хотя это не так широко используется в США, добавление второго скрининга потенциально может улучшить чувствительность всего процесса скрининга, в котором чувствительность первого скрининга отдельно составляет примерно 72%.
При отсутствии результатов скрининга у новорожденных, классическую САН у младенцев-девочек обычно идентифицируют по наличию наружных половых органов промежуточного типа. У лиц мужского пола при рождении наблюдаются нормальные половые органы, поэтому диагноз не определяется, если не проводится скрининг у новорожденных или не обнаруживаются другие медицинские осложнения. Младенцы, у которых изначально не была диагностирована САН и не наблюдалась сольтеряющая форма заболевания, диагноз определяется позже по слабом наборе весе, рвоте, гиперкалиемии и гипонатриемии в пределах первых нескольких недель жизни.
Лечение САН основано на нормализации уровней гормонов и стероидов с применением ряда лекарственных препаратов с момента постановки диагноза в младенчестве и до наступления взрослой жизни. Глюкокортикоиды в настоящее время являются стандартным лечением при САН и применяются как для коррекции эндогенного дефицита кортизола, так и для снижения повышенных уровней АСТН, вырабатываемого гипофизом, который обуславливает повышенную выработку андрогенов. В отличие от лечения болезни Аддисона (надпочечниковая недостаточность), при котором замещение кортизола является достаточным, лечение САН также должно обеспечивать снижение выработки АСТН, для контроля последующего избытка андрогенов. Таким образом, цели лечения глюкокортикоидами включают замещение кортизола и подавление АСТН для предупреждения вирилизации и нарушений менструального цикла у лиц женского пола и для ингибирования эктопической ткани надпочечников в яичке у лиц мужского пола. Замещение минералокортикоидов необходимо для достижения нормальной активности ренина в плазме крови с целью поддержания нормального артериального давления, водно-солевого баланса и волемического статуса у таких пациентов с сольтеряющей формой САН.
Схема лечения глюкокортикоидами должна поддерживать нормальные физиологические процессы, а также обеспечивать доступность достаточного уровня кортизола во время событий, которые могут вызвать сильную стрессовую реакцию (например, сопутствующее заболевание, физическая нагрузка, гипотензия). Тщательный мониторинг также необходим для избежания развития ятрогенного синдрома Кушинга вследствие передозировки глюкокортикоидами с целью адекватного подавления выработки андрогенов или болезни Аддисона вследствие недостаточного лечения.
Передозировка минералокортикоидами может вызвать гипертензию, тогда как недостаточное лечение может привести к понижению артериального давления, потере солей, утомляемости и повышению потребности в глюкокортикоидах. Типичные лабораторные тесты для мониторинга эффективности лечения включают измерение концентраций в плазме крови 17-ОНР, андростендиона, тестостерона, активности ренина и электролитов.
У взрослых пациентов с САН наблюдается повышенная частота факторов риска развития сердечно-сосудистого заболевания, включая ожирение, гипертензию и инсулинорезистентность (см., например, Kim et al., Semin. Reprod. Med. 27(4):316-21 (2009)). В исследовании с участием крупной когорты педиатрических и взрослых пациентов с САН (n=244) было продемонстрировано, что пациентам назначают различные схемы лечения глюкокортикоидами, но у них часто наблюдается неудовлетворительный контроль гормонов и вышеупомянутые неблагоприятные исходы (см., например, Finkielstain et al., J. Clin. EndocrinolMetab. 97(12):4429-38 (2012)).
Лечение САН включает попытки нормализовать дефицит кортизола с помощью глюкокортикоидов (обычно гидрокортизона у детей, но часто более сильнодействующих препаратов с узкими терапевтическими индексами, таких как дексаметазон, у взрослых) и, если необходимо при солевой недостаточности, минералокортикоидов (обычно флудрокортизона). Однако, дозы глюкокортикоидов, необходимые для достижения достаточного подавления избыточной выработки андрогенов, обычно намного превышают нормальную физиологическую дозу, применяемую для замещения кортизола отдельно, как у пациентов с болезнью Аддисона. Такое повышенное воздействие глюкокортикоидов может привести к развитию ятрогенного синдрома Кушинга, повышению частоты факторов риска развития сердечно-сосудистого заболевания, нарушению толерантности к глюкозе и уменьшению минеральной плотности костной ткани у пациентов с САН (см., например, Elnecave et al., J. Pediatr. Endocrinol. Metab. 21:1155-62 (2008); King et al., J. Clin. Endocrinol. Metab. 91(3):8656-59 (2006); Migeon et al., Endocrinol. Metab. Clin. North Am. 30:193-206 (2001)). Недавно практические рекомендации по клинической тактике лечения врожденной гиперплазии надпочечников были опубликованы в Journal of Clinical Endocrinology and Metabolism (Speiser, P.W., et al. J. Clin. Endocrinol. Metab. November 2018, 103(11): 1-46). Эта статья включена посредством ссылки во всей своей полноте.
Кортикотропин-рилизинг-гормон (CRF) был выделен из гипоталамуса овцы и идентифицирован как пептид длиной 41 аминокислота. Было обнаружено, что CRF вызывает глубокие изменения в функции эндокринной, нервной и иммунной систем. Полагают, что CRF является основным физиологическим регулятором базального и индуцированного стрессом высвобождения адренокортикотропного гормона («АСТН»), β-эндорфина и других проопиомеланокортин-(«РОМС»)-производных пептидов, вырабатываемых передней долей гипофиза (см., например, Vale et al., Science 213:1394-1397, 1981). Секреция CRF вызывает высвобождение АСТН из кортикотрофов в передней доле гипофиза посредством связывания с рецептором CRF1, членом класса В семейства рецепторов, сопряженных с G-белком.
Ввиду физиологической значимости CRF, разработка биологически активных малых молекул, которые обладают значительной активностью в отношении связывания CRF1-рецептора и которые способны оказывать антагонистическое действие на CRF1-рецептор, остается желательной целью и является предметом текущих исследований и разработок для лечения беспокойства, депрессии, синдрома раздраженного кишечника, посттравматического стрессового расстройства и злоупотребления наркотических веществ.
Гормон гипофиза АСТН, под контролем кортикотропин-рилизинг-гормона (CRF) гипоталамуса, стимулирует поглощение холестерина и запускает синтез прегненолона, инициируя стероидогенез в надпочечниках. Кора надпочечников состоит из трех зон, в которых вырабатываются различные классы гормонов, многие из которых регулируются АСТН, мобилизующим холестерин через этот путь. Дефицит таких ферментов в результате мутации или делеции обуславливает повышение концентраций субстрата. При наиболее распространенной форме САН, возникающей в результате мутаций или делеций в гене 21-гидроксилазы (CYP21A2), сильные андрогены вырабатываются надпочечниками из-за накопления предшественников стероидов, прогестерона и 17-гидроксипрогестерона (17-ОНР). В таких случаях уровни 17-ОНР в плазме крови могут достигать 10-1000-кратного превышения относительно нормальной концентрации. Такое повышение приводит к избыточной выработке андрогенов, в частности, андростендиона, тестостерона и дигидрокситестостерона, обуславливая вирилизацию у лиц женского пола. Кроме того, дефицит 21-гидроксилазы при САН обуславливает недостаточный биосинтез глюкокортикоидов и минералокортикоидов, в частности, кортизола и альдостерона. Кортизол является критически важным работающим по принципу отрицательной обратной связи регулятором секреции CRF гипоталамуса и высвобождения АСТН гипофиза. Недостаточность синтеза и высвобождения глюкокортикоидов устраняет блокирующий механизм в отношении гипоталамуса и гипофиза, что обуславливает повышение уровней АСТН. Избыточная АСТН-стимуляция обуславливает гипертрофию пучковой зоны и сетчатой зоны, что приводит к гиперплазии надпочечников.
Определения
Если не указано иное, все применяемые в настоящем документе технические и научные термины имеют те же значения, которые обычно понятны рядовому специалисту в данной области, к которой относится настоящее раскрытие. В настоящем документе описаны способы и материалы для применения в настоящем раскрытии; другие подходящие способы и материалы, известные в данной области, также можно применять. Материалы, способы и примеры являются исключительно иллюстративными и не предназначены для ограничения. Все публикации, патентные заявки, патенты, последовательности, записи из баз данных и другие упоминаемые в настоящем документе источники включены посредством ссылки во всей своей полноте. В случае противоречий настоящее описание, включая определения, будет иметь преимущественную силу.
Термин «приблизительно» перед значением для DSC, TGA или Tg, показатели которых выражаются в градусах Цельсия, имеет допустимую вариабельность ±5°С. Во всех других случаях, если не указано иное, термин «приблизительно» перед указанным значением включает указанное значение, а также включает ±20% от указанного значения и, более конкретно, включает значения ±10%, ±5%, ±2% и ±1% от указанного значения.
Для более краткого описания, некоторые количественные выражения в настоящем документе приводятся в виде диапазона от приблизительного количества X до приблизительного количества Y. Следует понимать, что, когда приводится диапазон, такой диапазон не ограничивается приведенными верхней и нижней границами, а скорее включает полный диапазон от приблизительного количества X до приблизительного количества Υ или любой диапазон в его пределах.
«Комнатная температура» или «RT» относится к температуре окружающей среды в обычной лаборатории, которая, как правило, составляет около 25°С.
«Высушивания распылением» относится к способу получения сухого порошка из раствора или взвеси. Раствор или взвесь распыляют или подвергают быстрой сушке горячим газом, например, воздухом или азотом, что обуславливает быстрое и равномерное испарение растворителя. «Высушенная распылением дисперсия» относится к порошку, полученному в результате процесса высушивания распылением.
Термин «фармацевтически приемлемый носитель» или «фармацевтически приемлемое вспомогательное вещество» включает любые возможные растворители, сорастворители, комплексообразующие средства, диспергирующие среды, оболочки, противобактериальные и противогрибковые средства, изотонические средства и средства, замедляющие абсорбцию, и т.д., которые не являются биологически или иным образом неприемлемыми. Применение таких сред и средств для фармацевтически активных веществ хорошо известны в данной области. За исключением случаев, когда какие-либо общепринятые среды или средство несовместимы с активным ингредиентом, их применение в терапевтических составах предполагается. В составы также могут быть включены дополнительные активные ингредиенты. Кроме того, могут быть включены различные вспомогательные вещества, как, например, широко используемые в данной области. Эти и другие подобные соединения описаны в литературе, например, в Merck Index, Merck & Company, Рауэй, Нью-Джерси. Рекомендации по включению различных компонентов в фармацевтические композиции описаны, например, в Oilman et al. (Eds.) (2010); Goodman and Gilman's: The Pharmacological Basis of Therapeutics, 12th Ed., The McGraw-Hill Companies.
«Субъект» в контексте настоящего документа означает человека или отличного от человека млекопитающего, например, собаку, кошку, мышь, крысу, корову, овцу, свинью, козу, отличного от человека примата или птицу, например, курицу, а также любых других позвоночных или беспозвоночных животных. Согласно некоторым вариантам осуществления субъектом является человек.
Согласно некоторым вариантам осуществления субъект перенес и/или у него наблюдается по меньшей мере один симптом заболевания или нарушения, подлежащих лечению и/или предупреждению. Согласно некоторым вариантам осуществления у субъекта была идентифицирована или диагностирована врожденная гиперплазия надпочечников (САН). Согласно некоторым вариантам осуществления у субъекта имеется подозрение на наличие САН. Согласно некоторым вариантам осуществления у субъекта в медицинской карте указано, что у субъекта наблюдается САН (и необязательно в медицинской карте указано, что субъекта следует лечить с использованием любых из представленных в настоящем документе композиций). Согласно некоторым вариантам осуществления субъектом является педиатрический пациент.
Термин «педиатрический пациент» в контексте настоящего документа относится к субъекту в возрасте до 21 года на момент постановки диагноза или лечения. Термин «педиатрический» дополнительно может быть разделен на различные подгруппы, включая: новорожденных (с момента рождения и до первого месяца жизни); младенцев (в возрасте от 1 месяца до двух лет); детей (в возрасте от двух лет до 12 лет); и подростков (в возрасте от 12 лет до 21 года (до двадцать второго дня рождения, но не включая его)). Berhman et al., Textbook of Pediatrics, 15th Ed. Philadelphia: W.B. Saunders Company, 1996; Rudolph et al., Rudolph's Pediatrics, 21st Ed. New York: McGraw-Hill, 2002; и Avery et al., Pediatric Medicine, 2nd Ed. Baltimore: Williams & Wilkins; 1994. Согласно некоторым вариантам осуществления педиатрический пациент находится в возрасте от момента рождения и до первых 28 дней жизни, в возрасте от 29 дней до менее чем двух лет, в возрасте от двух лет до менее чем 12 лет или в возрасте от 12 лет до 21 года (до двадцать второго дня рождения, но не включая его). Согласно некоторым вариантам осуществления педиатрический пациент находится в возрасте от момента рождения и до первых 28 дней жизни, в возрасте от 29 дней до менее чем 1 года, в возрасте от одного месяца до менее чем четырех месяцев, в возрасте от трех месяцев до менее чем семи месяцев, в возрасте от шести месяцев до менее чем 1 года, в возрасте от 1 года до менее чем 2 лет, в возрасте от 2 лет до менее чем 3 лет, в возрасте от 2 лет до менее чем семи лет, в возрасте от 3 лет до менее чем 5 лет, в возрасте от 5 лет до менее чем 10 лет, в возрасте от 6 лет до менее чем 13 лет, в возрасте от 10 лет до менее чем 15 лет или в возрасте от 15 лет до менее чем 22 лет.
В контексте настоящего документа термины «лечить» или «лечение» относятся к терапевтическим или паллиативным мерам. Благоприятные или требуемые клинические результаты включают без ограничения ослабление, полностью или частично, симптомов, ассоциированных с заболеванием, или нарушением, или состоянием, уменьшение степени выраженности заболевания, стабилизацию (т.е. отсутствие ухудшения) течения заболевания, задержку или замедление прогрессирования заболевания, облегчение или смягчение течения болезненного состояния (например, одного или нескольких симптомов заболевания) и ремиссию (частичную или полную), не зависимо от того, является она выявляемой или невыявляемой. «Лечение» также может означать продление выживания по сравнению с ожидаемым выживанием в случае отсутствия получения лечения.
Термин «предупреждение» в контексте настоящего документа означает предупреждение проявления, рецидива или распространения, полностью или частично, описанных в настоящем документе заболевания или состояния или их симптома.
Термин «введение» или «осуществление введения» относится к способу предоставления дозировки соединения или фармацевтического состава позвоночному или беспозвоночному животному, включая млекопитающее, птицу, рыбу или амфибию. Предпочтительный способ введения может варьироваться в зависимости от различных факторов, например, компонентов фармацевтического состава, участка заболевания и тяжести заболевания.
В контексте настоящего документа «терапевтически эффективное количество» представляет собой количество соединения формулы (I) или его фармацевтически приемлемой соли или количество фармацевтической композиции, содержащей соединение формулы (I), которое является достаточным для достижения требуемого эффекта, и оно может варьироваться в соответствии с природой и тяжестью болезненного состояния и активностью соединения. Терапевтический эффект представляет собой облегчение, до некоторой степени, одного или нескольких из симптомов заболевания и может включать излечение заболевания. «Излечение» означает, что симптомы активного заболевания устранены. Однако, определенные отдаленные или постоянные последствия заболевания могут сохраняться даже после достижения излечения (такие как, например, обширное повреждение тканей).
Термин «аморфный» означает твердое вещество в твердом состоянии, которое является некристаллическим состоянием. Аморфные твердые вещества представляют собой неупорядоченно расположенные молекулы, и поэтому они не имеют различимой кристаллической решетки или элементарной ячейки и, следовательно, не характеризуются определяемым дальним порядком. Твердое состояние твердого вещества можно определять с помощью микроскопии в поляризованном свете, порошковой рентгеновской дифракции (XRPD), дифференциальной сканирующей калориметрии (DSC) или других стандартных методов, известных специалистам в данной области.
В контексте настоящего документа «окно времени суток» относится к периоду времени, определяемому временем начала окна и временем окончания окна. Все эти промежутки времени относятся к локальным промежуткам времени, в которые происходит забор образцов. Фраза «одно и тоже окно времени суток» при упоминании взятых у субъекта образцов означает, например, что образец, взятый в 8:15, и образец, взятый в 9:15 утра, считаются взятыми в одно и то же окно времени суток, например, с 2 ночи до 10 утра или с 6 утра до 10 утра.
Способы
Настоящее раскрытие относится к способам лечения врожденной гиперплазии надпочечников (САН). Способы включают введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления способ включает введение субъекту терапевтически эффективного количества SDD по настоящему раскрытию, которая включает полимер и соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления способ включает введение субъекту терапевтически эффективного количества фармацевтической композиции по настоящему раскрытию, содержащей SDD, которая включает полимер и соединение формулы (I) или его фармацевтически приемлемую соль.
В настоящем документе представлен способ лечения врожденной гиперплазии надпочечников (САН), предусматривающий введение соединения формулы (I) или его фармацевтически приемлемой соли для нормализации или частичной нормализации уровней биомаркеров, ассоциированных с врожденной гиперплазией надпочечников. Согласно некоторым вариантам осуществления нормализация или частичная нормализация уровней биомаркеров предусматривает снижение уровней повышенных биомаркеров или повышение уровней пониженных биомаркеров по сравнению с таковыми у субъекта без САН.
В настоящем документе представлен способ лечения врожденной гиперплазии надпочечников у нуждающегося в этом субъекта, предусматривающий введение соединения формулы (I) или его фармацевтически приемлемой соли в количестве, достаточном для снижения уровня одного или нескольких биомаркеров, ассоциированных с врожденной гиперплазией надпочечников. Согласно некоторым вариантам осуществления биомаркеры выбраны из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона у субъекта.
Согласно некоторым вариантам осуществления снижение уровня любых из биомаркеров (например, любого из 17-ОНР, АСТН и андростендиона) определяют путем сравнения уровня биомаркера, измеряемого во время циркадного высвобождения за сутки до введения соединения формулы (I) или его фармацевтически приемлемой соли, и уровня биомаркера, измеряемого во время циркадного высвобождения через сутки после введения соединения формулы (I) или его фармацевтически приемлемой соли. «За сутки до введения соединения формулы (I)» относится к субъекту, которому ранее не вводили соединение формулы (I) в пределах по меньшей мере последних 24 часов.
Согласно некоторым вариантам осуществления циркадное высвобождение биомаркеров, ассоциированных с САН, происходит в период между 2 часами ночи и 10 часами утра. Согласно другим вариантам осуществления циркадное высвобождение биомаркеров, ассоциированных с САН, происходит в период между 6 часами утра и 10 часами утра.
Согласно некоторым вариантам осуществления любых из раскрытых в настоящем документе способов соединение формулы (I) или фармацевтически приемлемую соль вводят субъекту в ночное время или перед сном (т.е. на ночь). Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят за три-восемь часов до циркадного высвобождения биомаркера. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят за шесть-восемь часов до циркадного высвобождения биомаркера. Введение до циркадного высвобождения может быть адаптировано для сменных работников (например, тех, кто работает ночью и спит днем), и в этом случае введение не обязательно будет происходить в ночное время. Следовательно, введение зависит от ожидаемого циркадного высвобождения биомаркера, и может варьироваться в зависимости от конкретных работы и режима сна индивидуума (т.е. субъекта, пациента).
Согласно некоторым вариантам осуществления представленных в настоящем документе способов уровень 17-гидроксипрогестерона снижается по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 50%, по меньшей мере на 55% или по меньшей мере на 60% от уровней до введения. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 25%. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 50%. Согласно некоторым вариантам осуществления представленных в настоящем документе способов уровень 17-гидроксипрогестерона снижается на величину от приблизительно 10% до приблизительно 90%, от приблизительно 15% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 25% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 35% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 55% до приблизительно 90% или от приблизительно 60% до приблизительно 90% от уровней до введения.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается до уровня, находящегося в пределах диапазона для 17-гидроксипрогестерона, ожидаемого у субъекта без САН, т.е. менее 1000 нг/дл или менее 200 нг/дл.
Согласно некоторым вариантам осуществления представленных в настоящем документе способов уровень адренокортикотропного гормона снижается по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 50%, по меньшей мере на 55% или по меньшей мере на 60% от уровней до введения. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 25%. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 40%. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 50%.
Согласно некоторым вариантам осуществления представленных в настоящем документе способов уровень адренокортикотропного гормона снижается на величину от приблизительно 10% до приблизительно 90%, от приблизительно 15% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 25% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 35% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 55% до приблизительно 90% или от приблизительно 60% до приблизительно 90% от уровней до введения.
Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается до уровня, находящегося в пределах диапазона для адренокортикотропного гормона, ожидаемого у субъекта без САН.
Согласно некоторым вариантам осуществления представленных в настоящем документе способов уровень андростендиона снижается по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 50%, по меньшей мере на 55% или по меньшей мере на 60% от уровней до введения. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 25%. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 30%. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 50%.
Согласно некоторым вариантам осуществления представленных в настоящем документе способов уровень андростендиона снижается на величину от приблизительно 10% до приблизительно 90%, от приблизительно 15% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 25% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 35% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 55% до приблизительно 90% или от приблизительно 60% до приблизительно 90% от уровней до введения.
Согласно некоторым вариантам осуществления уровень андростендиона снижается до уровня, находящегося в пределах диапазона для андростендиона, ожидаемого у субъекта без САН, т.е. менее 200 нг/дл.
Также в настоящем документе представлен способ снижения тяжести одного или нескольких симптомов, выбранных из гирсутизма, преждевременного полового созревания, нарушений репродуктивной функции, акне и нарушения роста, у субъекта с классической врожденной гиперплазией надпочечников, предусматривающий введение соединения формулы (I) или его фармацевтически приемлемой соли в количестве, достаточном для снижения одного или нескольких биомаркеров САН у субъекта, например, снижения андростендиона у субъекта. Нарушение роста может относиться, например, к увеличенной скорости роста, повышенной скорости увеличения массы тела и/или увеличенному костному возрасту.
В настоящем документе представлен способ снижения уровня одного или нескольких биомаркеров врожденной гиперплазии надпочечников у субъекта с врожденной гиперплазией надпочечников, предусматривающий введение субъекту соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления один или несколько биомаркеров врожденной гиперплазии надпочечников выбраны из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
В настоящем документе представлен способ снижения дозировки кортикостероида, вводимого субъекту с врожденной гиперплазией надпочечников для контроля врожденной гиперплазии надпочечников, предусматривающий введение субъекту соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления кортикостероид представляет собой глюкокортикоид.
Также в настоящем документе представлен способ снижения тяжести одного или нескольких побочных эффектов лечения глюкокортикоидами у субъекта с врожденной гиперплазией надпочечников, предусматривающий введение субъекту соединения формулы (I) или его фармацевтически приемлемой соли. Отдаленные последствия лечения глюкокортикоидами детально задокументированы в данной области (см., например, Oray, Μ. et al. (2016): Long-term effect of glucocorticoids, Expert Opinion on Drug Safety. DOI: 10.1517/14740338.2016.1140743). Такие побочные эффекты ассоциированы с каждой биологической системой, например, опорно-двигательным аппаратом (например, остеопороз, аваскулярный некроз кости и миопатия), эндокринной и метаболической системой (например, гипергликемия, сахарный диабет, дислипидемия, набор лишнего веса, синдром Кушинга, кушингоидные признаки, угнетение роста, угнетение функции надпочечников), желудочно-кишечным трактом (например, гастрит, пептическая язва, желудочно-кишечное кровотечение, висцеральная перфорация, стеатоз печени, панкреатит), сердечно-сосудистой системой (например, гипертензия, коронарная болезнь сердца, ишемическая болезнь сердца, сердечная недостаточность), кожей (например, дерматопороз, атрофия кожи, экхимоз, пурпура, эрозии, стрии, медленное заживление ран, склонность к образованию кровоизлияний, акне, гирсутизм и облысение), нейропсихиатрическими симптомами (например, перепады настроения, депрессия, эйфория, эмоциональная лабильность, раздражимость, акатизия, беспокойство, когнитивное нарушение, психоз, деменция и делирий), органами зрения (например, катаракта, глаукома, птоз, мидриаз, оппортунистические инфекции глаз и центральная серозная хориоретинопатия) и иммунной системой (например, подавление клеточного иммунитета, предрасположенность к инфекциям и реактивация латентных инфекций).
Соответственно, согласно некоторым вариантам осуществления побочные эффекты лечения глюкокортикоидами выбраны из остеопороза, аваскулярного некроза кости, миопатии, гипергликемии, сахарного диабета, дислипидемии, набора лишнего веса, синдрома Кушинга, кушингоидных признаков, угнетения роста, угнетения функции надпочечников, гастрита, пептической язвы, желудочно-кишечного кровотечения, висцеральной перфорации, стеатоза печени, панкреатита, гипертензии, коронарной болезни сердца, ишемической болезни сердца, сердечной недостаточности, дерматопороза, атрофии кожи, экхимоза, пурпуры, эрозий, стрий, медленного заживления ран, склонности к образованию кровоизлияний, акне, гирсутизма, облысения, перепадов настроения, депрессии, эйфории, эмоциональной лабильности, раздражимости, акатизии, беспокойства, когнитивного нарушения, психоза, деменции, делирия, катаракты, глаукомы, птоза, мидриаза, оппортунистических инфекций глаз, центральной серозной хориоретинопатии, подавления клеточного иммунитета, предрасположенности к инфекциям, реактивации латентных инфекций и любой их комбинации.
В настоящем документе представлен способ лечения врожденной гиперплазии надпочечников у субъекта, предусматривающий
(i) измерение уровня одного или нескольких биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона, в биологическом образце, полученном от субъекта;
(ii) анализ уровня одного или нескольких биомаркеров для определения того, повышен ли уровень одного или нескольких биомаркеров в сравнении с таковым у здорового субъекта, у которого не наблюдается врожденной гиперплазии надпочечников; и
(iii) введение субъекту соединения формулы (I) или его фармацевтически приемлемой соли, если определено, что у субъекта наблюдаются повышенные уровни одного или нескольких биомаркеров.
Согласно некоторым вариантам осуществления способ дополнительно предусматривает (iv) измерение уровня одного или несколько биомаркеров после введения соединения формулы (I) или его фармацевтически приемлемой соли в биологическом образце, полученном от субъекта, для определения того, наблюдаются ли у субъекта сниженные уровни одного или нескольких биомаркеров по сравнению с таковыми при измерении на стадии (i). Согласно некоторым вариантам осуществления способ дополнительно предусматривает (v) продолжение введения соединения формулы (I) или его фармацевтически приемлемой соли, если у субъекта наблюдаются сниженные уровни одного или нескольких биомаркеров.
Согласно некоторым вариантам осуществления стадии (i) и (iv) осуществляют в отношении биологических образцов, взятых у субъекта одинаковым способом и в пределах одного и того же окна времени суток. Согласно некоторым вариантам осуществления стадии (i) и (iv) осуществляют в отношении биологических образцов, взятых у субъекта в пределах окна времени суток с 2 ночи до 10 утра. Согласно некоторым вариантам осуществления стадии (i) и (iv) осуществляют в отношении биологических образцов, взятых у субъекта в пределах окна времени суток с 6 утра до 10 утра.
Согласно некоторым вариантам осуществления стадии (i) и (iv) предусматривают измерение уровней по меньшей мере двух биомаркеров, выбранных из (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
Согласно некоторым вариантам осуществления стадии (i) и (iv) предусматривают измерение уровней (а) 17-гидроксипрогестерона (17-ОНР); (b) адренокортикотропного гормона (АСТН); и (с) андростендиона.
Согласно некоторым вариантам осуществления стадия (i) предусматривает измерение уровня 17-гидроксипрогестерона (17-ОНР), причем уровень 17-гидроксипрогестерона (17-ОНР) повышен, если он превышает или равен 1000 нг/дл.
Согласно некоторым вариантам осуществления стадия (i) предусматривает измерение уровня андростендиона, причем уровень андростендиона повышен, если он превышает 200 нг/дл.
Согласно некоторым вариантам осуществления способов по настоящему раскрытию соединение формулы (I) вводят в количестве, эквивалентном от приблизительно 25 мг до приблизительно 150 мг соединения формулы (I) в виде свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве, эквивалентном приблизительно 50 мг или приблизительно 100 мг соединения формулы (I) в виде свободного основания.
Согласно некоторым вариантам осуществления раскрытых в настоящем документе способов соединение формулы (I) вводят в форме свободного основания.
Согласно некоторым вариантам осуществления раскрытых в настоящем документе способов соединение формулы (I) вводят один раз в сутки.
Также в настоящем документе представлен способ лечения САН у педиатрического пациента. Способы включают введение педиатрическому пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления способ включает введение педиатрическому пациенту терапевтически эффективного количества SDD по настоящему раскрытию, которая включает полимер и соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления способ включает введение педиатрическому пациенту терапевтически эффективного количества фармацевтической композиции по настоящему раскрытию, содержащей SDD, которая включает полимер и соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления педиатрическим пациентом является новорожденный. Согласно некоторым вариантам осуществления педиатрическим пациентом является младенец. Согласно некоторым вариантам осуществления педиатрическим пациентом является ребенок. Согласно некоторым вариантам осуществления педиатрическим пациентом является подросток.
Согласно некоторым вариантам осуществления способов по настоящему раскрытию соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту в сытом состоянии. Термин «в сытом состоянии» в контексте настоящего документа относится к введению соединения формулы (I) или его фармацевтически приемлемой соли в интервале времени от приблизительно 1 часа до принятия пищи или питательной композиции до приблизительно 1 часа после принятия пищи или питательной композиции. Термин «натощак» в контексте настоящего документа относится к промежутку по меньшей мере в два часа между приемом пищи или питательной композиции и введением соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту с пищей или питательной композицией, такой как биологически активная добавка или питательная смесь, заменяющий еду напиток, жидкая пищевая добавка или высококалорийная жидкая пища. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту в пределах приблизительно 1 часа до того, как субъект примет пищу или питательную композицию. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту в пределах приблизительно 1 часа после того, как субъект принял пищу или питательную композицию. Примеры подходящих питательных композиций включают без ограничения питательные смеси, пищевые добавки, заменители пищи и регидратационные композиции. Согласно некоторым вариантам осуществления пища представляет собой продукт, содержащий концентрированные калории и белок. Согласно некоторым вариантам осуществления питательная композиция представляет собой композицию, используемую для энтерального и парентерального дополнительного питания для младенцев, специальных питательных смесей, добавок для пожилых людей и добавок для лиц со сложностями пищеварения и/или мальабсорбцией. Питательные смеси для взрослых и детей хорошо известны в данной области и являются коммерчески доступными (например, Similac®, Ensure®, Jevity® и Alimentum® от Ross Products Division, Abbott Laboratories, Колумбус, Огайо).
Согласно некоторым вариантам осуществления питательная композиция находится в жидкой форме. Энергетическая ценность питательных композиций в жидкой форме может находиться в диапазоне от приблизительно 0,6 ккал до приблизительно 3 ккал на мл. Согласно некоторым вариантам осуществления питательная композиция находится в твердой форме или в форме порошка. В твердой форме или в форме порошка биологически активные добавки могут содержать от приблизительно 1,2 до более 9 ккал на грамм, как, например, от приблизительно 3 до 7 ккал на грамм.
Согласно некоторым вариантам осуществления питательная композиция представляет собой заменяющий питание батончик. Примеры включают PowerBar®, батончики Glucerna®, батончики Choice DM®, батончики Ensure® и батончики Boost®. Согласно некоторым вариантам осуществления питательная композиция представляет собой питательный коктейль или заменяющий еду напиток. Коммерчески доступные примеры включают фирменные продукты для взрослых Ensure® (такие как Ensure® Original, Ensure® Plus, Ensure® Enlive, Ensure® High Protein, Ensure® Clear и Ensure® Light), Glucerna®, Choice DM®, Slim Fast®, Pediasure®, Glytrol® и Resource®. Согласно некоторым вариантам осуществления питательная композиция представляет собой Ensure® Plus. Согласно некоторым вариантам осуществления питательная композиция представляет собой Ensure® Plus со вкусом ванили. Ensure Plus® представляет собой высококалорийную жидкую пищевую добавку, которая содержит 1500 калорий на литр, со следующим распределением калорий: 14,7% белков, 32% жиров и 53,3% углеводов.
Согласно некоторым вариантам осуществления раскрытых способов соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту с 8 жидкими унциями (237 мл) Ensure® Plus. Согласно некоторым вариантам осуществления Ensure® Plus имеет вкус ванили.
Согласно некоторым вариантам осуществления способов, соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту после приема питательной композиции. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту до приема питательной композиции. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту одновременно с приемом питательной композиции.
Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту с последующим приемом питательной композиции. Согласно некоторым вариантам осуществления питательную композицию принимают через приблизительно 1 минуту, приблизительно 5 минут, приблизительно 10 минут, приблизительно 15 минут, приблизительно 20 минут, приблизительно 25 минут, приблизительно 30 минут, приблизительно 35 минут, приблизительно 40 минут, приблизительно 45 минут или приблизительно 60 минут после введения соединения формулы (I) или его фармацевтически приемлемой соли, или в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления питательную композицию принимают через 1 минуту, 5 минут, 10 минут, 15 минут, 20 минут, 25 минут, 30 минут, 35 минут, 40 минут, 45 минут или 60 минут после введения соединения формулы (I) или его фармацевтически приемлемой соли, или в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления питательную композицию принимают в пределах 30 минут от введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления раскрытых способов соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту с 8 жидкими унциями (237 мл) Ensure® Plus. Согласно некоторым вариантам осуществления Ensure® Plus имеет вкус ванили.
Согласно некоторым вариантам осуществления питательная композиция принимается субъектом с последующим введением соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят через приблизительно 1 минуту, приблизительно 5 минут, приблизительно 10 минут, приблизительно 15 минут, приблизительно 20 минут, приблизительно 25 минут, приблизительно 30 минут, приблизительно 35 минут, приблизительно 40 минут, приблизительно 45 минут или приблизительно 60 минут после приема питательной композиции, или в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят через 1 минуту, 5 минут, 10 минут, 15 минут, 20 минут, 25 минут, 30 минут, 35 минут, 40 минут, 45 минут или 60 минут после приема питательной композиции, или в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в пределах 30 минут от введения питательной композиции. Согласно некоторым вариантам осуществления раскрытых способов соединение формулы (I) или его фармацевтически приемлемую соль вводят субъекту с 8 жидкими унциями (237 мл) Ensure® Plus. Согласно некоторым вариантам осуществления Ensure® Plus имеет вкус ванили.
Согласно некоторым вариантам осуществления способов, влияние пищи наблюдается при сравнении введения соединения формулы (I) или его фармацевтически приемлемой соли в сытом состоянии и натощак. Термин «влияние пищи» в контексте настоящего документа относится к относительной разнице в AUC (площадь под кривой AUC(0-t) и/или AUC(0-∞)) или Cmax (максимальная концентрация в плазме крови или пиковая концентрация в плазме крови) активного вещества при введении субъекту соединения формулы (I) или его фармацевтически приемлемой соли перорально, одновременно с пищей или в сытом состоянии, по сравнению с этими же показателями при введении субъекту этого же соединения формулы (I) или его фармацевтически приемлемой соли натощак. Влияние пищи (F) рассчитывают следующим образом:
F% = [(Хв сытом состоянии - Хв сытом состоянии)/Хнатощак × 100,
где Хв сытом состоянии и Хнатощак представляют собой значения AUC (AUC(0-t) и/или AUC(0-∞)) или Cmax при введении в сытом состоянии и натощак соответственно. Согласно некоторым вариантам осуществления повышенное или положительное влияние пищи наблюдается при введении субъекту соединения формулы (I) или его фармацевтически приемлемой соли в сытом состоянии. Согласно некоторым вариантам осуществления введение соединения формулы (I) или его фармацевтически приемлемой соли обеспечивает повышенное или положительное влияние пищи, при котором наблюдаются повышенные Cmax и/или AUC при пероральном введении в сытом состоянии по сравнению с введением натощак.
Согласно некоторым вариантам осуществления способов, соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет от приблизительно 5 до приблизительно 10, как, например, от приблизительно 5 до приблизительно 9, от приблизительно 5 до приблизительно 8, от приблизительно 5 до приблизительно 7, от приблизительно 5 до приблизительно 6, от приблизительно 6 до приблизительно 10, от приблизительно 6 до приблизительно 9, от приблизительно 6 до приблизительно 8, от приблизительно 6 до приблизительно 7, от приблизительно 7 до приблизительно 10, от приблизительно 7 до приблизительно 9, от приблизительно 7 до приблизительно 8, от приблизительно 8 до приблизительно 10, от приблизительно 8 до приблизительно 9 или от приблизительно 8 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет приблизительно 5, приблизительно 6, приблизительно 7, приблизительно 8, приблизительно 9 или приблизительно 10, или находится в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления способов, соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет от приблизительно 10 до приблизительно 20.
Согласно некоторым вариантам осуществления способов, соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет 5-10, как, например, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9 или 8-10. Согласно некоторым вариантам осуществления соотношение AUC при введении в сытом состоянии и AUC при введении натощак составляет 5, 6, 7, 8, 9 или 10, или находится в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления способов, соотношение L^max при введении в сытом состоянии и Cmax при введении натощак составляет от приблизительно 5 до приблизительно 10, как, например, от приблизительно 5 до приблизительно 9, от приблизительно 5 до приблизительно 8, от приблизительно 5 до приблизительно 7, от приблизительно 5 до приблизительно 6, от приблизительно 6 до приблизительно 10, от приблизительно 6 до приблизительно 9, от приблизительно 6 до приблизительно 8, от приблизительно 6 до приблизительно 7, от приблизительно 7 до приблизительно 10, от приблизительно 7 до приблизительно 9, от приблизительно 7 до приблизительно 8, от приблизительно 8 до приблизительно 10, от приблизительно 8 до приблизительно 9 или от приблизительно 8 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение Cmax при введении в сытом состоянии и Cmax при введении натощак составляет приблизительно 5, приблизительно 6, приблизительно 7, приблизительно 8, приблизительно 9 или приблизительно 10, или находится в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления средняя Cmax соединения формулы (I) или его фармацевтически приемлемой соли превышает таковую при введении в сытом состоянии в сравнении с введением натощак с кратностью от приблизительно 1,5 до приблизительно 3 раз. Согласно некоторым вариантам осуществления способов, соотношение Cmax при введении в сытом состоянии и Cmax при введении натощак составляет 5-10, как, например, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9 или 8-10. Согласно некоторым вариантам осуществления соотношение СШах при введении в сытом состоянии и Cmax при введении натощак составляет 5, 6, 7, 8, 9 или 10, или находится в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления средняя Cmax соединения формулы (I) или его фармацевтически приемлемой соли в 1,5-3 раза превышает таковую при введении в сытом состоянии в сравнении с введением натощак. Согласно некоторым вариантам осуществления средняя t_-max соединения формулы (I) или его фармацевтически приемлемой соли в приблизительно 2 раза превышает таковую при введении в сытом состоянии в сравнении с введением натощак. Согласно некоторым вариантам осуществления способов, соотношение Cmax при введении в сытом состоянии и Cmax при введении натощак составляет от приблизительно 10 до приблизительно 20.
Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят субъекту с пищей. Согласно некоторым вариантам осуществления пища представляет собой высококалорийную пищу с высоким содержанием жиров. Согласно некоторым вариантам осуществления пища представляет собой низкокалорийную пищу с низким содержанием жира. Согласно некоторым вариантам осуществления 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят в пределах примерно 5 минут после начала приема пищи. Согласно некоторым вариантам осуществления пища представляет собой ужин. Согласно некоторым вариантам осуществления пища представляет собой завтрак.
Согласно некоторым вариантам осуществления «в сытом состоянии» подразумевает прием пищи с высоким содержанием жиров. Согласно некоторым вариантам осуществления «в сытом состоянии» подразумевает прием пищи с низким содержанием жиров. FDA представило проект руководящих принципов, касающихся пищи с высоким и низким содержанием жиров («Оценка влияния пищевых продуктов на лекарственные средства при IND и NDA рекомендации по клинической фармакологии для промышленности» ("Assessing the Effects of Food on Drugs in INDs and NDAs - Clinical Pharmacology Considerations Guidance for Industry"), Министерство здравоохранения и социальных служб США, Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов, Центр по оценке и исследованию лекарственных средств (CDER), февраль 2019 года, Clinical Pharmacology). В таблице 1 показаны определения для пробного приема пищи, представленные в руководстве FDA.
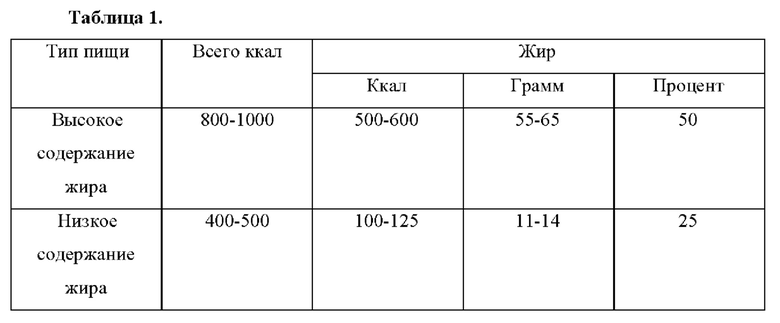
Композиция пищи с высоким содержанием жиров, представленная в руководстве FDA, показана в таблице 2.

Композиция пищи с низким содержанием жиров, представленная в руководстве FDA, показана в таблице 3.

Согласно некоторым вариантам осуществления пища с высоким содержанием жиров в общей сложности содержит 800-1000 ккал и 500-600 ккал для жира. Согласно некоторым вариантам осуществления пища с низким содержанием жиров в общей сложности содержит 400-500 ккал и 100-125 ккал для жира.
Также в настоящем документе представлен способ улучшения абсорбции соединения формулы (I) или его фармацевтически приемлемой соли в желудочно-кишечном тракте у субъекта. Способ включает пероральное введение субъекту фармацевтической композиции по настоящему раскрытию, причем улучшение является таковым относительно перорального введения соединения формулы (I) или его фармацевтически приемлемой соли, которые не были получены в виде высушенной распылением дисперсии. Согласно некоторым вариантам осуществления субъектом является педиатрический пациент.
Также в настоящем документе представлен способ улучшения биодоступности при пероральном введении соединения формулы (I) или его фармацевтически приемлемой соли у субъекта. Способ включает пероральное введение субъекту фармацевтической композиции по настоящему раскрытию, причем улучшение является таковым относительно перорального введения соединения формулы (I) или его фармацевтически приемлемой соли, которые не были получены в виде высушенной распылением дисперсии.
Согласно некоторым вариантам осуществления представленных в настоящем документе способов, субъектом является педиатрический пациент.
Также в настоящем документе представлен способ лечения врожденной гиперплазии надпочечников (САН) у нуждающегося в этом субъекта, предусматривающий введение субъекту фармацевтической композиции по настоящему раскрытию, причем фармацевтическая композиция содержит терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления фармацевтическая композиция представляет собой липидный полутвердый состав. Согласно некоторым вариантам осуществления фармацевтическая композиция представляет собой жидкий состав. Согласно некоторым вариантам осуществления фармацевтическую композиция вводят субъекту в сытом состоянии.
Также в настоящем документе представлена фармацевтическая композиция по настоящему раскрытию для применения в способе лечения врожденной гиперплазии надпочечников (САН) у субъекта. Согласно некоторым вариантам осуществления субъект находится в сытом состоянии.
Согласно некоторым вариантам осуществления фармацевтическую композицию вводят субъекту с питательной композицией. Согласно некоторым вариантам осуществления питательная композиция представляет собой жидкую пищевую добавку, содержащую от приблизительно 1000 до приблизительно 2000 калорий на литр с содержанием жиров более чем приблизительно 30%. Согласно некоторым вариантам осуществления питательная композиция представляет собой жидкую пищевую добавку, содержащую 1500 калорий на литр, со следующим распределением калорий: 14,7% белков, 32% жиров и 53,3% углеводов. Согласно некоторым вариантам осуществления питательную композицию принимают в количестве от приблизительно 6 до приблизительно 12 жидких унций. Согласно некоторым вариантам осуществления питательную композицию принимают в количестве приблизительно 8 жидких унций. Согласно некоторым вариантам осуществления питательную композицию принимают в пределах 30 минут от введения фармацевтической композиции.
Согласно некоторым вариантам осуществления фармацевтическая композиция характеризуется положительным влиянием пищи. Согласно некоторым вариантам осуществления положительное влияние пищи измеряют по Cmax, AUC, или их комбинации, соединения формулы (I) при сравнении перорального введения фармацевтической композиции в сытом состоянии и натощак. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 10 до приблизительно 20. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 10 до приблизительно 20. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4 или от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4 или от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4. Согласно некоторым вариантам осуществления соотношение t^max соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 1,5 до приблизительно 3. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 1,5 до приблизительно 3. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет 1-4 или 5-10. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет 1-4 или 5-10. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет 1-4. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет 1-4. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет 1,5-3. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет 1,5-3.
Согласно некоторым вариантам осуществления субъектом является педиатрический пациент.
Согласно некоторым вариантам осуществления фармацевтическая композиция составлена для перорального введения и характеризуется положительным влиянием пищи при пероральном введении. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от приблизительно 10 до приблизительно 20. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от приблизительно 10 до приблизительно 20. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от приблизительно 1 до приблизительно 4 или от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соединение формулы
(I) Характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от приблизительно 1 до приблизительно 4 или от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от приблизительно 1 до приблизительно 4. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от приблизительно 1 до приблизительно 4. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от приблизительно 1,5 до приблизительно 3. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от приблизительно 1,5 до приблизительно 3. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак 1-4 или 5-10. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак 1-4 или 5-10. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак 1-4. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением max при введении в сытом состоянии и Cmax при введении натощак 1-4. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак 1,5-3. Согласно некоторым вариантам осуществления соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак 1,5-3.
Согласно некоторым вариантам осуществления фармацевтическую композицию вводят субъекту с пищей. Согласно некоторым вариантам осуществления пища представляет собой пищу с высоким содержанием жиров. Согласно некоторым вариантам осуществления пища представляет собой пищу с низким содержанием жиров. Согласно некоторым вариантам осуществления фармацевтическую композиция вводят в пределах приблизительно 5 минут после начала приема пищи. Согласно некоторым вариантам осуществления пища представляет собой ужин. Согласно некоторым вариантам осуществления пища представляет собой завтрак.
Согласно некоторым вариантам осуществления введение фармацевтической композиции характеризуется положительным влиянием пищи. Согласно некоторым вариантам осуществления положительное влияние пищи измеряют по Cmax, AUC, или их комбинациям, соединения формулы (I) при сравнении перорального введения фармацевтической композиции в сытом состоянии и натощак. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 10 до приблизительно 20. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 10 до приблизительно 20. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4 или от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4 или от приблизительно 5 до приблизительно 10. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 1 до приблизительно 4. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от приблизительно 1,5 до приблизительно 3. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от приблизительно 1,5 до приблизительно 3. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет 1-4 или 5-10. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет 1-4 или 5-10. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет 1-4. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет 1-4. Согласно некоторым вариантам осуществления соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет 1,5-3. Согласно некоторым вариантам осуществления соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет 1,5-3.
Во избежание неоднозначного толкования, также в настоящем документе представлено соответствующее соединение формулы (I) или его фармацевтически приемлемая соль или соответствующая фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, для применения в соответствующих способах, как описано в настоящем документе.
Во избежание неоднозначного толкования, также в настоящем документе представлено применение соответствующего соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в соответствующих способах, как описано в настоящем документе.
Во избежание неоднозначного толкования, также в настоящем документе представлено применение соответствующей фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, в производстве лекарственного препарата для применения в соответствующих способах, как описано в настоящем документе.
Снижение глюкокортикоидной нагрузки, вырабатываемые надпочечниками андрогены и предшественники
Глюкокортикоиды являются классом кортикостероидов, представляющих собой класс стероидных гормонов. Глюкокортикоиды представляют собой кортикостероиды, которые связываются с глюкокортикоидным рецептором, который присутствует почти в каждой клетке позвоночного животного. Согласно некоторым вариантам осуществления субъект одновременно получает дозу глюкокортикоида. Согласно некоторым вариантам осуществления глюкокортикоид выбран из кортизола (гидрокортизона), кортизона, преднизона, преднизолона, метилпреднизолона, дексаметазона, бетаметазона, триамцинолона, флудрокортизона ацетата и дезоксикортикостерона ацетата. Согласно некоторым вариантам осуществления глюкокортикоид представляет собой кортизол (гидрокортизон). Согласно некоторым вариантам осуществления глюкокортикоид представляет собой кортизон. Согласно некоторым вариантам осуществления глюкокортикоид представляет собой преднизон. Согласно некоторым вариантам осуществления глюкокортикоид представляет собой дексаметазон.
Согласно некоторым вариантам осуществления дозу глюкокортикоида измеряют в эквивалентах гидрокортизона. Согласно некоторым вариантам осуществления дозу глюкокортикоида измеряют как величину, кратную верхней границе нормы для физиологической дозы в эквивалентах гидрокортизона. Любой глюкокортикоид можно давать в дозе, которая обеспечивает примерно такие же эффекты глюкокортикоида, что и при нормальной выработке кортизола; это называется физиологической, замещающей или поддерживающей дозой.
Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу, как измерено после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу от приблизительно 4 до приблизительно 12 мг/м2/сутки, как измерено после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу от приблизительно 4 до приблизительно 9 мг/м2/сутки, как измерено после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу, которая составляет менее чем приблизительно 8 мг/м2/сутки, как измерено после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу, как измерено после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу от приблизительно 4 до приблизительно 12 мг/м2/сутки, как измерено после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу от приблизительно 4 до приблизительно 9 мг/м2/сутки, как измерено после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида представляет собой физиологическую дозу, которая составляет менее чем приблизительно 8 мг/м2/сутки, как измерено после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления доза глюкокортикоида, которую дают субъекту одновременно, представляет собой нормальную физиологическую дозу в эквивалентах гидрокортизона. Согласно некоторым вариантам осуществления дозу глюкокортикоида, которую дают субъекту одновременно, определяют после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления дозу глюкокортикоида, которую дают субъекту одновременно, определяют после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет от приблизительно 2 до приблизительно 16 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет от приблизительно 4 до приблизительно 12 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет от приблизительно 5 до приблизительно 11 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет от приблизительно 6 до приблизительно 10 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет от приблизительно 7 до приблизительно 9 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет от приблизительно 4 до приблизительно 9 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет приблизительно 8 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет приблизительно 12 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет менее чем приблизительно 8 мг/м2/сутки. Согласно некоторым вариантам осуществления нормальная физиологическая доза в эквивалентах гидрокортизона составляет приблизительно 2, приблизительно 3, приблизительно 4, приблизительно 5, приблизительно 6, приблизительно 7, приблизительно 8, приблизительно 9, приблизительно 10, приблизительно 11, приблизительно 12, приблизительно 13, приблизительно 14, приблизительно 15 или приблизительно 16 мг/м2/сутки, или находится в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления доза глюкокортикоида, которую дают субъекту одновременно, находится у верхней границы нормы для нормальной физиологической дозы в эквивалентах гидрокортизона. Согласно некоторым вариантам осуществления дозу глюкокортикоида, которую дают субъекту одновременно, определяют после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления дозу глюкокортикоида, которую дают субъекту одновременно, определяют после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления верхняя граница нормы в 1,5 раза превышает нормальную физиологическую дозу. Согласно некоторым вариантам осуществления верхняя граница нормы в приблизительно 1,5 раза превышает нормальную физиологическую дозу. Согласно некоторым вариантам осуществления верхняя граница нормы в приблизительно 1,5 раза превышает нормальную физиологическую дозу. Согласно некоторым вариантам осуществления верхняя граница нормы в приблизительно 2 раза превышает нормальную физиологическую дозу. Согласно некоторым вариантам осуществления верхняя граница нормы в приблизительно 2,5 раза превышает нормальную физиологическую дозу. Согласно некоторым вариантам осуществления верхняя граница нормы в приблизительно 1,0, приблизительно 1,1, приблизительно 1,2, приблизительно 1,3, приблизительно 1,4, приблизительно 1,5, приблизительно 1,6, приблизительно 1,7, приблизительно 1,8, приблизительно 1,9, приблизительно 2,0, приблизительно 2,1, приблизительно 2,2, приблизительно 2,3, приблизительно 2,4, приблизительно 2,5, приблизительно 2,6, приблизительно 2,7, приблизительно 2,8, приблизительно 2,9 или в приблизительно 3,0 раза превышает нормальную физиологическую дозу, или находится в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 10% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 20% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 30% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 40% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 50% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 60% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 70% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на менее чем приблизительно 20% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на величину, составляющую от приблизительно 20% до приблизительно 50%, после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоид а до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на более чем приблизительно 50% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 10% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 20% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 30% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 40% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 50% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 60% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на приблизительно 70% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на менее чем приблизительно 20% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на величину, составляющую от приблизительно 20% до приблизительно 50%, после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на более чем приблизительно 50% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления доза глюкокортикоида для субъекта снижается на величину, находящуюся в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 25% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня 17-гидроксипрогестерона является таковым относительно уровня 17-гидроксипрогестерона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 50% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня 17-гидроксипрогестерона является таковым относительно уровня 17-гидроксипрогестерона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона менее чем в 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона находится в пределах нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на приблизительно 25% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня 17-гидроксипрогестерона является таковым относительно уровня 17-гидроксипрогестерона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на приблизительно 50% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня 17-гидроксипрогестерона является таковым относительно уровня 17-гидроксипрогестерона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона в менее чем приблизительно 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона находится в пределах нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона у субъекта снижается на величину, находящуюся в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 25% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 40% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на 50% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона менее чем в 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона находится в пределах нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на приблизительно 25% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на приблизительно 40% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона снижается по меньшей мере на приблизительно 50% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона в менее чем приблизительно 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона находится в пределах нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления уровень адренокортикотропного гормона у субъекта снижается на величину, находящуюся в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 25% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 30% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на 50% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень андростендиона менее чем в 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень андростендиона находится в пределах нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на приблизительно 25% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на приблизительно 30% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень андростендиона снижается по меньшей мере на приблизительно 50% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень андростендиона в менее чем приблизительно 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень андростендиона находится в пределах нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления уровень андростендиона у субъекта снижается на величину, находящуюся в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления уровень тестостерона снижается по меньшей мере на 25% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень тестостерона снижается по меньшей мере на 30% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень тестостерона снижается по меньшей мере на 50% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень тестостерона менее чем в 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень тестостерона находится в пределах нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления уровень тестостерона снижается по меньшей мере на приблизительно 25% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень тестостерона снижается по меньшей мере на приблизительно 30% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень тестостерона снижается по меньшей мере на приблизительно 50% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень тестостерона в менее чем приблизительно 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень тестостерона находится в пределах нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления уровень тестостерона у субъекта снижается на величину, находящуюся в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на 50% и уровень андростендиона снижается по меньшей мере на 50% после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровня 17-гидроксипрогестерона и уровня андростендиона является таковым относительно уровня 17-гидроксипрогестерона и уровня андростендиона до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона менее чем в 1,5 раза превышает верхнюю границу нормы и уровень андростендиона менее чем в 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона находится в пределах нормы и уровень андростендиона находится в пределах нормы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона снижается по меньшей мере на приблизительно 50% и уровень андростендиона снижается по меньшей мере на приблизительно 50% после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем снижение уровня 17-гидроксипрогестерона и уровня андростендиона является таковым относительно уровня 17-гидроксипрогестерона и уровня андростендиона до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона в менее чем приблизительно 1,5 раза превышает верхнюю границу нормы и уровень андростендиона в менее чем приблизительно 1,5 раза превышает верхнюю границу нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона находится в пределах нормы и уровень андростендиона находится в пределах нормы после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления уровень 17-гидроксипрогестерона и андростендиона у субъекта снижается на величину, находящуюся в пределах диапазона, определенного любыми из предыдущих значений.
Согласно некоторым вариантам осуществления у субъекта наблюдается уменьшение глюкокортикоидной нагрузки после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем уменьшение глюкокортикоидной нагрузки является таковым относительно глюкокортикоидной нагрузки до введения соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления происходит улучшение в отношении одного или нескольких симптомов, выбранных из качества жизни, утомляемости, сна, инсулинорезистентности, толерантности к глюкозе, контроля уровня глюкозы, дислипидемии, гиперлипидемии, минеральной плотности костной ткани, обновления костной ткани, массы жировой ткани, веса, центрального ожирения, артериального давления, степени выраженности гирсутизма, цикличности менструации, контроля эктопической ткани надпочечников в яичке и репродуктивной функции, после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем улучшение в отношении одного или нескольких симптомов является таковым относительно статуса одного или нескольких симптомов до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта наблюдается уменьшение глюкокортикоидной нагрузки после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем уменьшение глюкокортикоидной нагрузки является таковым относительно глюкокортикоидной нагрузки до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления происходит улучшение в отношении одного или нескольких симптомов глюкокортикоидной нагрузки, выбранных из качества жизни, утомляемости, сна, инсулинорезистентности, толерантности к глюкозе, контроля уровня глюкозы, дислипидемии, гиперлипидемии, минеральной плотности костной ткани, обновления костной ткани, массы жировой ткани, веса, центрального ожирения, артериального давления, степени выраженности гирсутизма, цикличности менструации, контроля эктопической ткани надпочечников в яичке и репродуктивной функции, после некоторого периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, причем улучшение в отношении одного или нескольких симптомов является таковым относительно статуса одного или нескольких симптомов до введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления у субъекта улучшается качество жизни, как измерено с помощью опросника EuroQol в 5 категориях по 5 уровням (EQ-5D-5L), после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем улучшение в отношении результатов EuroQol в 5 категориях по 5 уровням (EQ-5D-5L) является таковым относительно результатов EuroQol в 5 категориях по 5 уровням (EQ-5D-5L) до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта снижается утомляемость после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение утомляемости является таковым относительно утомляемости до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается качество сна после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение качества сна является таковым относительно качества сна до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта снижается инсулинорезистентность после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение инсулинорезистентности является таковым относительно инсулинорезистентности до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта снижается толерантность к глюкозе после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение толерантности к глюкозе является таковым относительно толерантности к глюкозе до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается контроль уровня глюкозы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение контроля уровня глюкозы является таковым относительно контроля уровня глюкозы до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта снижаются уровни липидов, свидетельствующие о дислипидемии, после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровней липидов является таковым относительно уровней липидов до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта снижаются уровни липидов, свидетельствующие о гиперлипидемии, после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем снижение уровней липидов является таковым относительно уровней липидов до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается минеральная плотность костной ткани после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение минеральной плотности костной ткани является таковым относительно минеральной плотности костной ткани до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается обновление костной ткани после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение обновления костной ткани является таковым относительно обновления костной ткани до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта уменьшается масса жировой ткани после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем уменьшение массы жировой ткани является таковым относительно массы жировой ткани до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта уменьшается вес тела после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем уменьшение веса тела является таковым относительно веса тела до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта уменьшается центральное ожирение после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем уменьшение центрального ожирения является таковым относительно центрального ожирения до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается артериальное давление после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение артериального давления является таковым относительно артериального давления до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта уменьшается степень тяжести гирсутизма после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем уменьшение степени тяжести гирсутизма является таковым относительно степени тяжести гирсутизма до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается цикличность менструации после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение цикличности менструации является таковым относительно цикличности менструации до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается контроль эктопической ткани надпочечников в яичке после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение контроля эктопической ткани надпочечников в яичке является таковым относительно контроля эктопической ткани надпочечников в яичке до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышается репродуктивная функция после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение репродуктивной функции является таковым относительно репродуктивной функции до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышаются уровни гонадотропина после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение уровней гонадотропина является таковым относительно уровней гонадотропина до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышаются уровни прогестерона после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение уровней прогестерона является таковым относительно уровней прогестерона до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышаются уровни спермы после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение уровней спермы является таковым относительно уровней спермы до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления у субъекта повышаются уровни LH (лютеинизирующего гормона) после некоторого периода времени введения соединения формулы (I) или его фармацевтически приемлемой соли, причем повышение уровней LH является таковым относительно уровней LH до введения соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно некоторым вариантам осуществления период времени введения составляет по меньшей мере приблизительно 4 недели. Согласно некоторым вариантам осуществления период времени введения составляет по меньшей мере приблизительно 24 недели. Согласно некоторым вариантам осуществления период времени введения составляет по меньшей мере приблизительно один год. Согласно некоторым вариантам осуществления период времени введения составляет по меньшей мере 4 недели. Согласно некоторым вариантам осуществления период времени введения составляет по меньшей мере 24 недели. Согласно некоторым вариантам осуществления период времени введения составляет по меньшей мере один год. Согласно некоторым вариантам осуществления период времени введения составляет менее чем приблизительно 1 сутки. Согласно некоторым вариантам осуществления период времени введения составляет приблизительно 1, 2, 3, 4, 5, 6 или 7 суток, или находится в пределах диапазона любых из предыдущих значений. Согласно некоторым вариантам осуществления период времени введения составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 или 24 недели, или находится в пределах диапазона любых из предыдущих значений. Согласно некоторым вариантам осуществления период времени введения составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 месяцев, или находится в пределах диапазона любых из предыдущих значений. Следует понимать, что сравнительные измерения предпочтительно проводить утром.
Согласно некоторым вариантам осуществления субъектом является педиатрический пациент. Согласно некоторым вариантам осуществления возраст педиатрического пациента равен шести годам или меньше. Согласно некоторым вариантам осуществления возраст педиатрического пациента составляет более шести лет и менее одиннадцати лет. Согласно некоторым вариантам осуществления возраст педиатрического пациента составляет более десяти лет и менее пятнадцати лет. Согласно некоторым вариантам осуществления возраст педиатрического пациента составляет более четырнадцати лет и менее девятнадцати лет.
Согласно некоторым вариантам осуществления субъект является взрослым. Согласно некоторым вариантам осуществления возраст субъекта составляет более восемнадцати лет. Согласно некоторым вариантам осуществления субъектом является лицо женского пола.
Согласно некоторым вариантам осуществления субъектом является лицо мужского пола. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в виде описанной в настоящем документе фармацевтической композиции. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в виде описанной в примере 9 фармацевтической композиции. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в виде описанной в примере 11 фармацевтической композиции. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в виде описанной в примере 12 фармацевтической композиции. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в виде описанной в примере 13 фармацевтической композиции. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в виде соли соляной кислоты или соли п-толуолсульфоновой кислоты.
Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в виде описанной в настоящем документе соли п-толуолсульфоновой кислоты.
Во избежание неоднозначного толкования, также в настоящем документе представлено соответствующее соединение формулы (I) или его фармацевтически приемлемая соль или соответствующая фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, для применения в соответствующих способах, как описано в настоящем документе.
Во избежание неоднозначного толкования, также в настоящем документе представлено применение соответствующего соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в соответствующих способах, как описано в настоящем документе.
Во избежание неоднозначного толкования, также в настоящем документе представлено применение соответствующей фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, в производстве лекарственного препарата для применения в соответствующих способах, как описано в настоящем документе.
Соль п-толуолсульфоновой кислоты
Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль представляют собой соль 4-(2-хлор-4-метокси -5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина и п-толуолсульфоновой кислоты.
Согласно некоторым вариантам осуществления соль 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина и п-толуолсульфоновой кислоты представляет собой кристаллическую соль. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется формой 1.
Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется порошковой рентгенограммой, главным образом как показано на фиг. 27. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется DSC-термограммой, главным образом как показано на фиг. 28. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется термограммой термогравиметрического анализа (TGA), главным образом как показано на фиг. 28.
Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется наличием по меньшей мере одного пика порошковой рентгеновской дифракции (XRPD) для угла 2-тета (±0,2 градуса), выбранного из 9,1, 11,3, 13,2, 16,3 и 21,1 градусов. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется наличием по меньшей мере двух пиков порошковой рентгеновской дифракции (XRPD) для угла 2-тета (±0,2 градуса), выбранного из 9,1, 11,3, 13,2, 16,3 и 21,1 градусов. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется наличием по меньшей мере трех пиков порошковой рентгеновской дифракции (XRPD) для угла 2-тета (±0,2 градуса), выбранного из 9,1, 11,3, 13,2, 16,3 и 21,1 градусов. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется наличием по меньшей мере четырех пиков порошковой рентгеновской дифракции (XRPD) для угла 2-тета (±0,2 градуса), выбранного из 9,1, 11,3, 13,2, 16,3 и 21,1 градусов. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется наличием характеристических пиков порошковой рентгеновской дифракции (XRPD) для угла 2-тета (±0,2 градуса), на 9,1, 11,3, 13,2, 16,3 и 21,1 градусах. Согласно некоторым вариантам осуществления кристаллическая соль п-толуолсульфоновой кислоты характеризуется наличием эндотермического пика с началом плавления при приблизительно 156°С (22,2 Дж/г) на термограмме дифференциальной сканирующей калориметрии (DSC).
Липидный полутвердный состав
В настоящем документе представлен липидный полутвердый состав, который представляет собой фармацевтическую композицию, содержащую:
(a) соединение формулы (I):
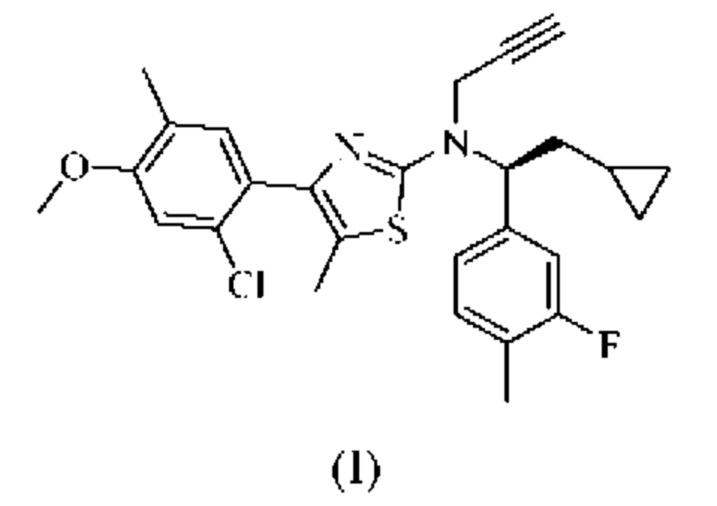
или его фармацевтически приемлемую соль; и
(b) одно или несколько из основы с масляной фазой, эмульгирующего вещества, неионогенного поверхностно-активного вещества и солюбилизирующего вещества.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес. до приблизительно 20% вес. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 5% вес. до приблизительно 15% вес. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 10% вес. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20% вес. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания, или их количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит основу с масляной фазой. Основа с масляной фазой представляет собой растворитель, который плохо смешивается с водой. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес. до приблизительно 50% вес. основы с масляной фазой. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 20% вес. до приблизительно 50% вес. основы с масляной фазой. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 35% вес. до приблизительно 45% вес. основы с масляной фазой. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 39% вес. основы с масляной фазой. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, или 45% вес. основы с масляной фазой, или ее количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления основа с масляной фазой выбрана из триглицеридов со средней длиной цепи, глицерина, пропиленгликоля, полиэтиленгликоля, оливкового масла, соевого масла, кукурузного масла и Transcutol. Согласно некоторым вариантам осуществления основа с масляной фазой представляет собой триглицериды со средней длиной цепи. Согласно некоторым вариантам осуществления триглицериды со средней длиной цепи представляют собой Labrafac ТМ Lipophile WL1349. Согласно некоторым вариантам осуществления триглицериды со средней длиной цепи представляют собой Miglyol 812N.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит эмульгирующее вещество. Эмульгирующее вещество представляет собой соединение или вещество, которое действует как стабилизатор для эмульсий. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 5% вес. до приблизительно 50% вес. эмульгирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 10% вес. до приблизительно 30% вес. эмульгирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 15% вес. до приблизительно 25% вес. эмульгирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 20% вес. эмульгирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30% вес. эмульгирующего вещества, или его количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления эмульгирующее вещество выбрано из триглицеридов со средней длиной цепи, пропиленгликольдикаприлата/дикапрата, глицерина, пропиленгликоля, полиэтиленгликоля, оливкового масла, соевого масла, кукурузного масла и Transcutol. Согласно некоторым вариантам осуществления эмульгирующее вещество представляет собой пропиленгликольдикаприлат/дикапрат. Согласно некоторым вариантам осуществления пропиленгликольдикаприлат/дикапрат представляет собой Labrafac ТМ PG.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит неионогенное поверхностно-активное вещество. Неионогенное поверхностно-активное вещество представляет собой вещество с гидрофильной «головкой» и гидрофобным «хвостом», которое не имеет заряда и которое является компонентом составов, добавляемым для улучшения растворимости или свойств эмульсии. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 5% вес. до приблизительно 50% вес. неионогенного поверхностно-активного вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 10% вес. до приблизительно 30% вес. неионогенного поверхностно-активного вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 15% вес. до приблизительно 25% вес. неионогенного поверхностно-активного вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 19% вес. неионогенного поверхностно-активного вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30% вес. неионогенного поверхностно-активного вещества, или его количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления неионогенное поверхностно-активное вещество выбрано из олеоилполиоксил-6-глицеридов, линолеоилполиоксил-6-глицеридов, полисорбата 80, полисорбата 20, Gelucire, лауроилполиоксил-32-глицеридов, полоксамера, стеарата PEG-32 и PEG-32 гидрогенизированных пальмовых глицеридов. Согласно некоторым вариантам осуществления неионогенное поверхностно-активное вещество представляет собой лауроилполиоксил-32-глицериды. Согласно некоторым вариантам осуществления лауроилполиоксил-32-глицериды представляют собой Gelucire® 44/14.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит солюбилизирующее вещество. Солюбилизирующее вещество представляет собой растворитель, который способствует солюбилизации соединения формулы (I) или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес. до приблизительно 50% вес. солюбилизирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес. до приблизительно 20% вес. солюбилизирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 5% вес. до приблизительно 15% вес. солюбилизирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 11% вес. солюбилизирующего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20% вес. солюбилизирующего вещества, или его количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления солюбилизирующее вещество выбрано из олеоилполиоксил-6-глицеридов, линолеоилполиоксил-6-глицеридов, полисорбата 80, полисорбата 20, витамина Е полиэтиленгликольсукцината, Gelucire, лауроилполиоксил-32-глицеридов и полоксамера. Согласно некоторым вариантам осуществления солюбилизирующее вещество представляет собой витамина Е полиэтиленгликольсукцинат. Согласно некоторым вариантам осуществления витамина Е полиэтиленгликольсукцинат представляет собой Kolliphor® TPGS. Согласно некоторым вариантам осуществления витамина Е полиэтиленгликольсукцинат представляет собой витамин E/TPGS 260.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль;
(b) основу с масляной фазой;
(c) эмульгирующее вещество;
(d) неионогенное поверхностно-активное вещество; и
(e) солюбилизирующее вещество.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 5% вес. до приблизительно 15% вес. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от приблизительно 35% вес. до приблизительно 45% вес. основы с масляной фазой;
(c) от приблизительно 15% вес. до приблизительно 25% вес. эмульгирующего вещества;
(d) от приблизительно 15% вес. до приблизительно 25% вес. неионогенного поверхностно-активного вещества; и
(e) от приблизительно 5% вес. до приблизительно 15% вес. солюбилизирующего вещества.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 10% вес. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) приблизительно 39% вес. основы с масляной фазой;
(c) приблизительно 20% вес. эмульгирующего вещества;
(d) приблизительно 19% вес. неионогенного поверхностно-активного вещества; и
(e) приблизительно 11% вес. солюбилизирующего вещества.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин;
(b) компонент в виде триглицеридов со средней длиной цепи;
(c) компонент в виде пропиленгликольдикаприлата/дикапрата;
(d) компонент в виде лауроилполиоксил-32-глицеридов; и
(e) компонент в виде витамина Е полиэтиленгликольсукцината.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 5% вес. до приблизительно 15% вес. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина;
(b) от приблизительно 35% вес. до приблизительно 45% вес. триглицеридов со средней длиной цепи;
(c) от приблизительно 15% вес. до приблизительно 25% вес. пропиленгликольдикаприлата/дикапрата;
(d) от приблизительно 15% вес. до приблизительно 25% вес. лауроилполиоксил-32-глицеридов; и
(e) от приблизительно 5% вес. до приблизительно 15% вес. витамина Е полиэтиленгликольсукцината.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 10% вес. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина;
(b) приблизительно 39% вес. триглицеридов со средней длиной цепи;
(c) приблизительно 20% вес. пропиленгликольдикаприлата/дикапрата;
(d) приблизительно 19% вес. лауроилполиоксил-32-глицеридов; и
(e) приблизительно 11% вес. витамина Е полиэтиленгликольсукцината.
Согласно некоторым вариантам осуществления липидная полутвердая фармацевтическая композиция характеризуется вязкостью от приблизительно 15 до приблизительно 40 сантипуаз при приблизительно 45°С. Согласно некоторым вариантам осуществления липидная полутвердая фармацевтическая композиция характеризуется вязкостью от приблизительно 26 до приблизительно 30 сантипуаз при приблизительно 45°С. Согласно некоторым вариантам осуществления липидная полутвердая фармацевтическая композиция характеризуется вязкостью от приблизительно 5 до приблизительно 25 сантипуаз при приблизительно 60°С. Согласно некоторым вариантам осуществления липидная полутвердая фармацевтическая композиция характеризуется вязкостью от приблизительно 14 до приблизительно 18 сантипуаз при приблизительно 60°С.
Согласно некоторым вариантам осуществления фармацевтическая композиция не содержит комбинацию маннита, кроскармеллозы натрия, маисового крахмала, гидроксипропилметилцеллюлозы и стеарата магния.
Согласно некоторым вариантам осуществления фармацевтическая композиция не содержит по меньшей мере одно из маннита, кроскармеллозы натрия, маисового крахмала, гидроксипропилметилцеллюлозы и стеарата магния.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит соединение формулы (I) или его фармацевтически приемлемую соль в кристаллической форме. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит соединение формулы (I) или его фармацевтически приемлемую соль в аморфной форме. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит соединение формулы (I) в виде свободного основания. Согласно некоторым вариантам осуществления кристаллическая форма соединения формулы (I) представляет собой форму I.
Согласно некоторым вариантам осуществления фармацевтическая композиция составлена в виде единичной лекарственной формы, причем соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от приблизительно 5 мг до приблизительно 200 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в единичной лекарственной форме в количестве от приблизительно 75 мг до приблизительно 150 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в единичной лекарственной форме в количестве приблизительно 50 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в единичной лекарственной форме в количестве приблизительно 100 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в единичной лекарственной форме в количестве приблизительно 25 мг, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления фармацевтическая композиция находится в форме таблетки, капсулы, саше, порошка, гранул, покрытой оболочкой частицы, покрытой оболочкой таблетки, покрытой кишечнорастворимой оболочкой таблетки, покрытой кишечнорастворимой оболочкой капсулы, пластинки, диспергируемой в полости рта, или пленки, диспергируемой в полости рта. Согласно некоторым вариантам осуществления фармацевтическая композиция находится в форме таблетки. Согласно некоторым вариантам осуществления фармацевтическая композиция находится в форме капсулы. Согласно некоторым вариантам осуществления лекарственная форма покрыта оболочкой.
Некоторые варианты осуществления относятся к способу получения фармацевтической композиции, предусматривающему:
(b) нагревание смеси основы с масляной фазой, эмульгирующего вещества, неионогенного поверхностно-активного вещества и солюбилизирующего вещества;
(b) перемешивание смеси из стадии (а) до получения однородной смеси; и
(c) перемешивание соединения формулы (I) или его фармацевтически приемлемой соли с однородной смесью из стадии (b) до тех пор, пока соединение формулы (I) или его фармацевтически приемлемая соль не растворятся, с образованием композиции.
Согласно некоторым вариантам осуществления способ дополнительно предусматривает:
(d) инкапсулирование композиции из стадии (с) в оболочку капсулы с образованием капсулы; и
(e) запайку капсулы из стадии (d) в смеси средства для запайки и растворителя для запайки.
Жидкие составы
В настоящем документе представлена фармацевтическая композиция в лекарственной форме в виде перорального раствора, содержащая:
(a) соединение формулы (I):
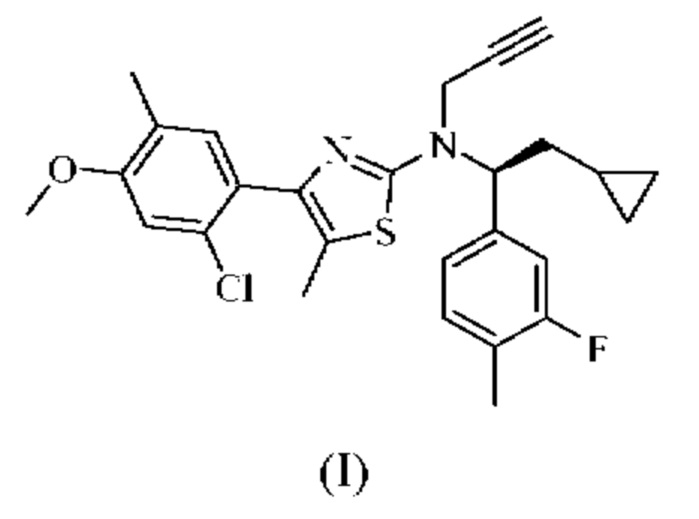
или его фармацевтически приемлемую соль;
(b) одно или несколько из подсластителя, антиоксиданта и вкусоароматической добавки; и
(c) жидкую основу.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес./об. до приблизительно 50% вес./об. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес./об. до приблизительно 10% вес./об. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 5% вес./об. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10% вес./об. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания, или их количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит подсластитель. Подсластитель представляет собой компонент состава, добавляемый для улучшения вкуса. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,01% вес./об. до приблизительно 1,5% вес./об. подсластителя. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,1% вес./об. до приблизительно 0,5% вес./об. подсластителя. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,15% вес./об. подсластителя. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,1, 0,2, 0,3, 0,4 или 0,5% вес./об. подсластителя, или его количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления подсластитель выбран из сахарина, сахарозы, сукралозы, аспартама, декстрозы, фруктозы, мальтита, маннита, сорбита и авантама. Согласно некоторым вариантам осуществления подсластитель представляет собой сахарин.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит антиоксидант. Антиоксидант представляет собой компонент состава, включаемый для улучшения стабильности путем предупреждения окисления. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,01% вес./об. до приблизительно 1,5% вес./об. антиоксиданта. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,1% вес./об. до приблизительно 0,5% вес./об. антиоксиданта. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,17% вес./об. антиоксиданта. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,1, 0,2, 0,3, 0,4 или 0,5% вес./об. антиоксиданта, или его количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления антиоксидант выбран из бутилированного гидрокситолуола, витамина Е TPGS, бутилированного гидроксианизола, аскорбиновой кислоты, лецитина, трет-бутилгидрохинона и лимонной кислоты. Согласно некоторым вариантам осуществления антиоксидант представляет собой бутилированный гидрокситолуол.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит вкусоароматическую добавку. Вкусоароматическая добавка представляет собой компонент состава, добавляемый для маскировки вкуса за счет ароматических веществ. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,01% вес./об. до приблизительно 0,5% вес./об. вкусоароматической добавки. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,05% вес./об. до приблизительно 0,2% вес./об. вкусоароматической добавки. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,10% вес./об. вкусоароматической добавки. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,05, 0,06, 0,07, 0,08, 0,09, 0,10, 0,11, 0,12, 0,13, 0,14, 0,15, 0,16, 0,17, 0,18, 0,19 или 0,2% вес./об. вкусоароматической добавки, или ее количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления вкусоароматическая добавка выбрана из вкусоароматической добавки FONA «Апельсин», вкусоароматической добавки FONA «Juicy», вкусоароматической добавки FONA «Виноград», вкусоароматической добавки Firmenich SA «Лимон», вкусоароматической добавки Firmenich Tetrarome «Апельсин», вкусоароматической добавки IFF «Вишня» и вкусоароматической добавки IFF «Виноград». Согласно некоторым вариантам осуществления вкусоароматическая добавка представляет собой вкусоароматическую добавку FONA «Апельсин».
Жидкая основа представляет собой растворитель, способный растворять или частично растворять соединение формулы (I) или его фармацевтически приемлемую соль с целью доставки в виде раствора для перорального приема. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 50% вес./об. до приблизительно 99,9% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 90% вес./об. до приблизительно 99% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 92% вес./об. до приблизительно 97% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 94,6% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99% вес./об. жидкой основы, или ее количество в пределах диапазона любых из предыдущих значений. Согласно некоторым вариантам осуществления жидкая основа выбрана из триглицеридов со средней длиной цепи, пропиленгликольдикаприлата/дикапрата, глицерина, пропиленгликоля, полиэтиленгликоля, оливкового масла, соевого масла, кукурузного масла и Transcutol. Согласно некоторым вариантам осуществления жидкая основа представляет собой триглицериды со средней длиной цепи. Согласно некоторым вариантам осуществления триглицериды со средней длиной цепи представляют собой Labrafac Lipophile WL1349.
Согласно некоторым вариантам осуществления фармацевтическая композиция дополнительно содержит поверхностно-активное вещество. Поверхностно-активное вещество представляет собой компонент состава, добавляемый для улучшения растворимости или свойств эмульсии. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес./об. до приблизительно 50% вес./об. поверхностно-активного вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 10% вес./об. до приблизительно 30% вес./об. поверхностно-активного вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 20% вес./об. поверхностно-активного вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30% вес./об. поверхностно-активного вещества, или его количество в пределах диапазона любых из предыдущих значений.
Согласно некоторым вариантам осуществления поверхностно-активное вещество выбрано из олеоилполиоксил-6-глицеридов, линолеоилполиоксил-6-глицеридов, полисорбата 80, полисорбата 20, витамина Е полиэтиленгликольсукцината, Gelucire, лауроилполиоксил-32-глицеридов, лаурилсульфата натрия, полоксамера, сложных эфиров PEG-6 и кукурузного масла и сложных эфиров PEG-6 и гидрогенизированного пальмового масла/пальмоядрового масла. Согласно некоторым вариантам осуществления поверхностно-активное вещество представляет собой олеоилполиоксил-6-глицериды. Согласно некоторым вариантам осуществления олеоилполиоксил-6-глицериды представляют собой LABRAFIL М 1944 CS.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 50% вес./об. до приблизительно 90% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 70% вес./об. до приблизительно 80% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 75% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 74,6% вес./об. жидкой основы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 или 80% вес./об. жидкой основы, или ее количество в пределах диапазона любых из предыдущих значений. Согласно некоторым вариантам осуществления жидкая основа выбрана из триглицеридов со средней длиной цепи, пропиленгликольдикаприлата/дикапрата, глицерина, пропиленгликоля, полиэтиленгликоля, оливкового масла, соевого масла, кукурузного масла и Transcutol. Согласно некоторым вариантам осуществления жидкая основа представляет собой триглицериды со средней длиной цепи. Согласно некоторым вариантам осуществления триглицериды со средней длиной цепи представляют собой Labrafac Lipophile WL1349.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль;
(b) подсластитель;
(c) антиоксидант;
(d) вкусоароматическую добавку; и
(e) жидкую основу.
Согласно некоторым вариантам осуществления фармацевтическая композиция дополнительно содержит поверхностно-активное вещество.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 4% вес./об. до приблизительно 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. подсластителя;
(c) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. антиоксиданта;
(d) от приблизительно 0,05% вес./об. до приблизительно 0,2% вес./об. вкусоароматической добавки; и
(e) от приблизительно 92% вес./об. до приблизительно 97% вес./об. жидкой основы.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) приблизительно 0,15% вес./об. подсластителя;
(c) приблизительно 0,17% вес./об. антиоксиданта;
(d) приблизительно 0,1% вес./об. вкусоароматической добавки; и
(e) приблизительно 94,6% вес./об. жидкой основы.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 4% вес./об. до приблизительно 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. подсластителя;
(c) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. антиоксиданта;
(d) от приблизительно 0,05% вес./об. до приблизительно 0,2% вес./об. вкусоароматической добавки;
(e) от приблизительно 15% вес./об. до приблизительно 25% вес./об. поверхностно-активного вещества; и
(f) от приблизительно 70% вес./об. до приблизительно 80% вес./об. жидкой основы.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) приблизительно 0,15% вес./об. подсластителя;
(c) приблизительно 0,17% вес./об. антиоксиданта;
(d) приблизительно 0,1% вес./об. вкусоароматической добавки;
(e) приблизительно 20% вес./об. поверхностно-активного вещества; и
(f) приблизительно 75% вес./об. жидкой основы.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль;
(b) сахарин;
(c) бутилированный гидрокситолуол;
(d) вкусоароматическую добавку FONA «Апельсин»; и
(e) триглицериды со средней длиной цепи.
Согласно некоторым вариантам осуществления фармацевтическая композиция дополнительно содержит олеоилполиоксил-6-глицериды.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 4% вес./об. до приблизительно 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. сахарина;
(c) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. бутилированного гидрокситолуола;
(d) от приблизительно 0,05% вес./об. до приблизительно 0,2% вес./об. вкусоароматической добавки FONA «Апельсин»; и
(e) от приблизительно 92% вес./об. до приблизительно 97% вес./об. триглицеридов со средней длиной цепи.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) приблизительно 0,15% вес./об. сахарина;
(c) приблизительно 0,17% вес./об. бутилированного гидрокситолуола;
(d) приблизительно 0,1% вес./об. вкусоароматической добавки FONA «Апельсин»; и
(e) приблизительно 94,6% вес./об. триглицеридов со средней длиной цепи.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 4% вес./об. до приблизительно 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. сахарина;
(c) от приблизительно 0,1% вес./об. до приблизительно 0,2% вес./об. бутилированного гидрокситолуола;
(d) от приблизительно 0,05% вес./об. до приблизительно 0,2% вес./об. вкусоароматической добавки FONA «Апельсин»;
(e) от приблизительно 15% вес./об. до приблизительно 25% вес./об. олеоилполиоксил-6-глицеридов; и
(f) от приблизительно 70% вес./об. до приблизительно 80% вес./об. триглицеридов со средней длиной цепи.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) приблизительно 0,15% вес./об. сахарина;
(c) приблизительно 0,17% вес./об. бутилированного гидрокситолуола;
(d) приблизительно 0,1% вес./об. вкусоароматической добавки FONA «Апельсин»;
(e) приблизительно 20% вес./об. олеоилполиоксил-6-глицеридов; и
(f) приблизительно 75% вес./об. триглицеридов со средней длиной цепи.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит соединение формулы (I) в виде свободного основания.
Согласно некоторым вариантам осуществления фармацевтическая композиция составлена в виде единичной лекарственной формы, причем соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от приблизительно 5 мг/мл до приблизительно 200 мг/мл, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в единичной лекарственной форме в количестве от приблизительно 75 мг/мл до приблизительно 150 мг/мл, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в единичной лекарственной форме в количестве приблизительно 50 мг/мл, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в единичной лекарственной форме в количестве приблизительно 100 мг/мл, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления жидкая фармацевтическая композиция характеризуется вязкостью от приблизительно 1 до приблизительно 50 сантипуаз при приблизительно 25°С.
Некоторые варианты осуществления относятся к способу получения фармацевтической композиции, предусматривающему:
(a) смешивание жидкой основы с подсластителем;
(b) смешивание смеси из стадии (а) с антиоксидантом и вкусоароматической добавкой;
(c) смешивание соединения формулы (I) или его фармацевтически приемлемой соли со смесью из стадии (b); и
(d) смешивание смеси из стадии (с) с дополнительной частью жидкой основы.
Согласно некоторым вариантам осуществления стадия (а) способа предусматривает смешивание жидкой основы с подсластителем и поверхностно-активным веществом.
Высушенные распылением дисперсии
Способы и применения по настоящему раскрытию могут предусматривать введение высушенной распылением дисперсии (SDD) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли и применение SDD в лечении врожденной гиперплазии надпочечников (САН).
Согласно некоторым вариантам осуществления повышение концентрации и биодоступности в водной среде лекарственного средства с низкой растворимостью в высушенной распылением дисперсии достигается, если SDD характеризуется одним или несколькими свойствами, включая, например, следующее: (1) твердая дисперсия является главным образом однородной; (2) лекарственное средство является главным образом аморфным; (3) SDD характеризуется относительно высоким содержанием лекарственного вещества; и (4) SDD характеризуется низким содержанием остаточного растворителя. Согласно некоторым вариантам осуществления дисперсия при введении в водную среду обеспечивает по меньшей мере временную концентрацию растворенного лекарственного средства в водной среде, которая превышает растворимость лекарственного средства в кристаллической форме в той же среде. Водная среда может представлять собой, например, среду in vitro, такую как среда для проверки растворимости (например, забуференный фосфатом солевой раствор (PBS)) или среду in vivo, такую как желудочно-кишечный тракт (GI) животного, например, человека. Согласно некоторым вариантам осуществления водная среда представляет собой нижний отдел GI-тракта, как, например, тонкий кишечник и толстый кишечник.
В настоящем раскрытии представлена высушенная распылением дисперсия, которая содержит полимер и соединение, характеризующееся структурой формулы (I):
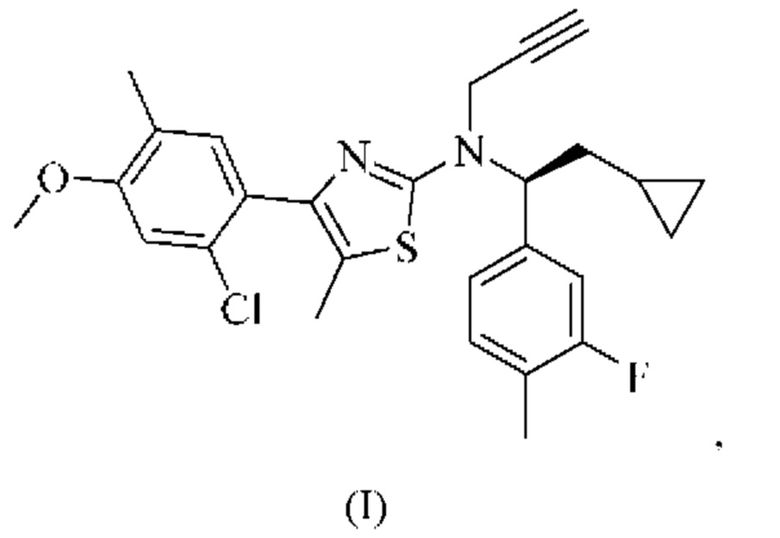
или его фармацевтически приемлемую соль. Согласно некоторым вариантам осуществления SDD включает полимер, выбранный из нейтрального полимера, растворимого в кишечнике полимера и полимера пирролидона. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимер составляет от приблизительно 1:1 до приблизительно 1:9.
Согласно некоторым вариантам осуществления полимер представляет собой нейтральный полимер. Например, полимер не содержит заряженных функциональных групп. Согласно некоторым вариантам осуществления нейтральный полимер представляет собой целлюлозный полимер. Например, целлюлозный полимер может представлять собой полимер по меньшей мере с одним заместителем, связанным сложноэфирной и/или простой эфирной связью, при этом полимер характеризуется степенью замещения по меньшей мере 0,05 для каждого заместителя. Примеры подходящих нейтральных полимеров включают без ограничения гидроксипропилметилцеллюлозу (НРМС) и гидроксиэтилцеллюлозу (НЕС).
Согласно некоторым вариантам осуществления полимер представляет собой растворимый в кишечнике полимер. «Растворимый в кишечнике полимер» в контексте настоящего документа представляет собой полимерное вещество, которое является главным образом нерастворимым и/или главным образом стабильным в кислой среде, характеризующейся рН менее чем приблизительно 7, и которое является главным образом растворимым или может разлагаться в среде, характеризующейся рН приблизительно 7 или больше. Примеры подходящих растворимых в кишечнике полимеров включают без ограничения карбоксиметилэтилцеллюлозу (СМЕС), фталат ацетата целлюлозы (САР), сукцинат ацетата гидроксипропилметилцеллюлозы (CAS), фталат метилцеллюлозы, фталат гидроксиэтилметилцеллюлозы, фталат гидроксипропилметилцеллюлозы (НРМСР), сукцинат ацетата гидроксипропилметилцеллюлозы (HPMCAS), фталат поливинилового спирта, фталат поливинилбутирата, фталат поливинилацетата, сополимер малеинового ангидрида и винилацетата, сополимер винилбутилового эфира и малеинового ангидрида, сополимер стирола и однозамещенного сложного эфира малеиновой кислоты, сополимер метилметакрилата и метакриловой кислоты, сополимер стирола и акриловой кислоты, сополимер метилакрилата, метакриловой кислоты и октилакрилата, сополимер метакриловой кислоты и метилметакрилата, сополимер аминометакрилата, сополимер аммониоалкилметакрилата, метакриловый сополимер и их смеси. Согласно некоторым вариантам осуществления растворимый в кишечнике полимер выбран из сукцината ацетата гидроксипропилметилцеллюлозы (HPMCAS), фталата ацетата целлюлозы (САР), фталата гидроксипропилметилцеллюлозы (НРМСР), сополимера аминометакрилата, сополимера аммониоалкилметакрилата и метакрилового сополимера. Согласно некоторым вариантам осуществления растворимый в кишечнике полимер представляет собой полимер Eudragit®, продаваемый Evonik Industries (Эссен, Германия). Согласно некоторым вариантам осуществления растворимый в кишечнике полимер представляет собой сополимер аминометакрилата. Согласно некоторым вариантам осуществления сополимер аминометакрилата представляет собой Eudragit® Е РО/100. Согласно некоторым вариантам осуществления растворимый в кишечнике полимер представляет собой сополимер аммониоалкилметакрилата. Согласно некоторым вариантам осуществления сополимер аммониоалкилметакрилата представляет собой Eudragit® RLPO. Согласно некоторым вариантам осуществления растворимый в кишечнике полимер представляет собой метакриловый сополимер. Согласно некоторым вариантам осуществления метакриловый сополимер представляет собой Eudragit® L100 или Eudragit® S100.
Согласно некоторым вариантам осуществления полимер представляет собой полимер пирролидона. Например, полимер пирролидона может представлять собой винилпирролидоновый полимер, такой как поливинилпирролидон (PVP) или сополимер винилпирролидона и винилацетата (PVP/VA), включая гомополимеры и сополимеры PVP и гомополимеры и сополимеры N-винилпирролидона. Согласно некоторым вариантам осуществления полимер пирролидона представляет собой PVP/VA. Согласно некоторым вариантам осуществления PVP/VA представляет собой сополимер 1-винил-2-пирролидона и винилацетата. Согласно некоторым вариантам осуществления сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении от приблизительно 30:70 до приблизительно 70:30 по весу, как, например, от приблизительно 40:60 до приблизительно 60:40 по весу или от приблизительно 45:55 до приблизительно 55:45 по весу. Согласно некоторым вариантам осуществления сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении приблизительно 30:70, приблизительно 35:65, приблизительно 40:60, приблизительно 45:55, приблизительно 50:50, приблизительно 55:45, приблизительно 60:40, приблизительно 65:35 или приблизительно 70:30 по весу. Согласно некоторым вариантам осуществления сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении приблизительно 60:40 по весу.
Согласно некоторым вариантам осуществления полимер пирролидона характеризуется структурой:
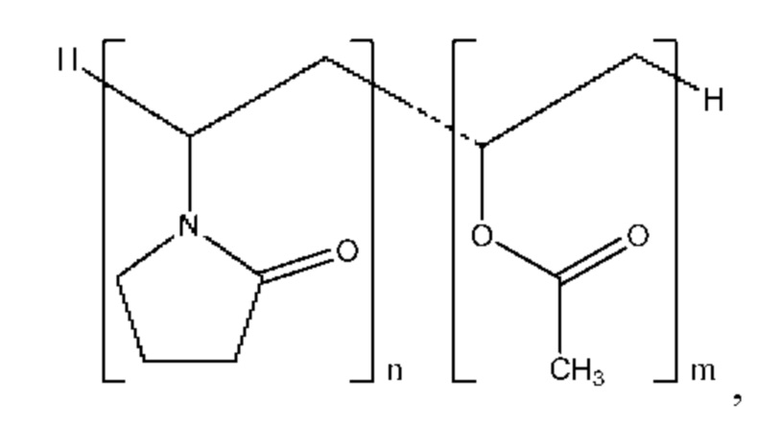
где значение n превышает значение m с кратностью от приблизительно 1 до приблизительно 2 раз. Например, значение n может в приблизительно 1, приблизительно 1,05, приблизительно 1,1, приблизительно 1,15, приблизительно 1,16, приблизительно 1,2, приблизительно 1,25, приблизительно 1,3, приблизительно 1,35, приблизительно 1,4, приблизительно 1,45, приблизительно 1,5, приблизительно 1,55, приблизительно 1,6, приблизительно 1,65, приблизительно 1,7, приблизительно 1,75, приблизительно 1,8, приблизительно 1,85, приблизительно 1,9, приблизительно 1,95 или приблизительно 2 раза превышать значение m, или значение кратности находится в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления значение n в приблизительно 1,16 раза превышает значение m. Согласно некоторым вариантам осуществления сополимер представляет собой коповидон, и значение n в приблизительно 1,16 раза превышает значение m.
Согласно некоторым вариантам осуществления полимер пирролидона характеризуется структурой:

где значение n превышает значение m с кратностью от 1 до 2 раз. Например, значение n может в приблизительно 1, 1,05, 1,1, 1,15, 1,16, 1,2, 1,25, 1,3, 1,35, 1,4, 1,45, 1,5, 1,55, 1,6, 1,65, 1,7, 1,75, 1,8, 1,85, 1,9, 1,95 или 2 раза превышать значение т, или значение кратности находится в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления значение n в 1,16 раза превышает значение m.
Согласно некоторым вариантам осуществления сополимер представляет собой коповидон, и значение n в 1,16 раза превышает значение т. Согласно некоторым вариантам осуществления применяемый в раскрытых SDD полимер присутствует в количестве, достаточном для повышения максимальной концентрации лекарственного средства в случае аморфной формы соединения формулы (I) или его фармацевтически приемлемой соли в среде применения (например, водной среде) по сравнению с контрольной композицией, которая содержит эквивалентное количество кристаллической формы соединения формулы (I) или его фармацевтически приемлемой соли, но без полимера. Согласно некоторым вариантам осуществления после введения SDD в среду применения (например, водную среду) полимер обеспечивает повышение концентрации в водной среде соединения формулы (I) или его фармацевтически приемлемой соли по сравнению с контрольной композицией. Следует понимать, что контрольная композиция не содержит солюбилизаторов или других компонентов, которые могут существенно повлиять на растворимость соединения формулы (I) или его фармацевтически приемлемой соли, и что соединение формулы (I) или его фармацевтически приемлемая соль находятся в твердой форме в контрольной композиции.
Согласно некоторым вариантам осуществления SDD весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимер составляет от приблизительно 1:1 до приблизительно 1:9. Например, весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера может составлять от приблизительно 1:1 до приблизительно 1:8, от приблизительно 1:1 до приблизительно 1:7, от приблизительно 1:1 до приблизительно 1:6, от приблизительно 1:1 до приблизительно 1:5, от приблизительно 1:1 до приблизительно 1:4, от приблизительно 1:1 до приблизительно 1:3, от приблизительно 1:1 до приблизительно 1:2, от приблизительно 1:1 до приблизительно 1:1,5, от приблизительно 1:1,5 до приблизительно 1:9 или от приблизительно 1:2,5 до приблизительно 1:4. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет приблизительно 1:9, приблизительно 1:8, приблизительно 1:7,5, приблизительно 1:7, приблизительно 1:6, приблизительно 1:5, приблизительно 1:4, приблизительно 1:3, приблизительно 1:2,5, приблизительно 1:2, приблизительно 1:1,5 или приблизительно 1:1, или весовое соотношение находится в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет приблизительно 1:9. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет приблизительно 1:1,5. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет приблизительно 1:1.
Согласно некоторым вариантам осуществления SDD весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимер составляет 1:1-1:9. Например, весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера может составлять 1:1-1:8, 1:1-1:7, 1:1-1:6, 1:1-1:5, 1:1-1:4, 1:1-1:3,1:1-1:2,1:1-1:1,5, 1:1,5-1:9 или 1:2,5-1:4. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет 1:9, 1:8, 1:7.5, 1:7, 1:6, 1:5, 1:4, 1:3, 1:2,5, 1:2, 1:1,5 или 1:1, или весовое соотношение находится в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет 1:9. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет 1:1,5. Согласно некоторым вариантам осуществления весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и полимера составляет 1:1.
Согласно некоторым вариантам осуществления высушенная распылением дисперсия включает соединение формулы (I) или его фармацевтически приемлемую соль и полимер, который представляет собой сополимер 1-винил-2-пирролидона и винилацетата, характеризующийся структурой:
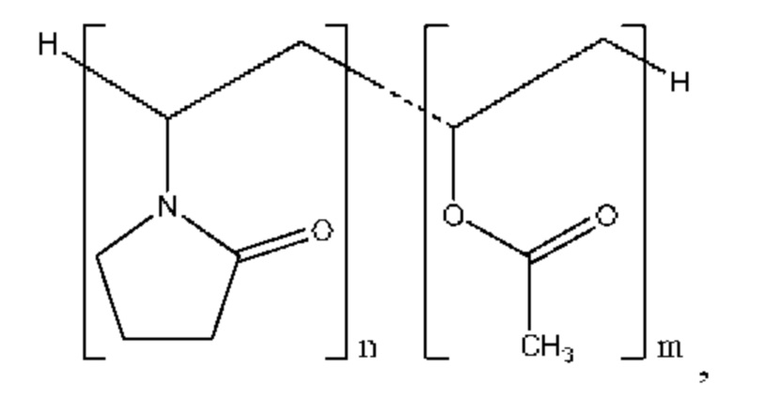
причем значение n превышает значение m с кратностью от приблизительно 1 до приблизительно 2 раз, и сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении приблизительно 60:40 по весу, и весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и сополимера составляет от приблизительно 1:1 до приблизительно 1:9.
Согласно некоторым вариантам осуществления высушенная распылением дисперсия включает соединение формулы (I) или его фармацевтически приемлемую соль и полимер, который представляет собой сополимер 1-винил-2-пирролидона и винилацетата, характеризующийся структурой:

причем значение n превышает значение m с кратностью 1-2 раза, и сополимер содержит 1-винил-2-пирролидон и винилацетат в соотношении 60:40 по весу, и весовое соотношение соединения формулы (I) или его фармацевтически приемлемой соли и сополимера составляет 1:1-1:9.
Согласно некоторым вариантам осуществления SDD, соединение формулы (I) или его фармацевтически приемлемая соль и полимер вместе образуют однородные частицы. Согласно некоторым вариантам осуществления частицы представляют собой главным образом однородную композицию, которая включает соединение формулы (I) или его фармацевтически приемлемую соль и полимер. В контексте настоящего документа «главным образом однородный» означает, что соединение формулы (I) или его фармацевтически приемлемая соль диспергированы как можно более однородно в полимере и могут считаться твердым раствором соединения формулы (I) или его фармацевтически приемлемой соли, диспергированным в полимере. Хотя дисперсия может характеризоваться некоторыми зонами повышенной концентрации соединения формулы (I) или его фармацевтически приемлемой соли, сама по себе дисперсия характеризуется единой температурой стеклования (Tg), которая демонстрирует, что дисперсия является главным образом однородной. Это контрастирует с простой механической смесью чистых аморфных частиц соединения формулы (I) или его фармацевтически приемлемой соли и чистых аморфных полимерных частиц, которые обычно демонстрируют два различных значения Tg, одно для соединения формулы (I) или его фармацевтически приемлемой соли и одно для полимера. «Tg» в контексте настоящего документа представляет собой характеристическую температуру, при которой стеклообразное вещество при постепенном нагревании претерпевает относительно быстрое (например, за 10-100 секунд) физическое изменение из стеклообразного состояния в резинообразное состояние.
Согласно некоторым вариантам осуществления частицы характеризуются распределением частиц по размеру (D-значение, D50) от приблизительно 5 мкм до приблизительно 100 мкм, как, например, от приблизительно 5 мкм до приблизительно 95 мкм, от приблизительно 5 мкм до приблизительно 90 мкм, от приблизительно 5 мкм до приблизительно 85 мкм, от приблизительно 5 мкм до приблизительно 80 мкм, от приблизительно 5 мкм до приблизительно 75 мкм, от приблизительно 5 мкм до приблизительно 70 мкм, от приблизительно 5 мкм до приблизительно 65 мкм, от приблизительно 5 мкм до приблизительно 60 мкм, от приблизительно 5 мкм до приблизительно 55 мкм, от приблизительно 5 мкм до приблизительно 50 мкм, от приблизительно 5 мкм до приблизительно 45 мкм, от приблизительно 5 мкм до приблизительно 40 мкм, от приблизительно 5 мкм до приблизительно 35 мкм, от приблизительно 5 мкм до приблизительно 30 мкм, от приблизительно 5 мкм до приблизительно 25 мкм, от приблизительно 5 мкм до приблизительно 20 мкм, от приблизительно 5 мкм до приблизительно 15 мкм, от приблизительно 5 мкм до приблизительно 10 мкм, от приблизительно 10 мкм до приблизительно 100 мкм, от приблизительно 10 мкм до приблизительно 95 мкм, от приблизительно 10 мкм до приблизительно 90 мкм, от приблизительно 10 мкм до приблизительно 85 мкм, от приблизительно 10 мкм до приблизительно 80 мкм, от приблизительно 10 мкм до приблизительно 75 мкм, от приблизительно 10 мкм до приблизительно 70 мкм, от приблизительно 10 мкм до приблизительно 65 мкм, от приблизительно 10 мкм до приблизительно 60 мкм, от приблизительно 10 мкм до приблизительно 55 мкм, от приблизительно 10 мкм до приблизительно 50 мкм, от приблизительно 10 мкм до приблизительно 45 мкм, от приблизительно 10 мкм до приблизительно 40 мкм, от приблизительно 10 мкм до приблизительно 35 мкм, от приблизительно 10 мкм до приблизительно 30 мкм, от приблизительно 10 мкм до приблизительно 25 мкм, от приблизительно 10 мкм до приблизительно 20 мкм, от приблизительно 10 мкм до приблизительно 15 мкм, от приблизительно 15 мкм до приблизительно 100 мкм, от приблизительно 15 мкм до приблизительно 95 мкм, от приблизительно 15 мкм до приблизительно 90 мкм, от приблизительно 15 мкм до приблизительно 85 мкм, от приблизительно 15 мкм до приблизительно 80 мкм, от приблизительно 15 мкм до приблизительно 75 мкм, от приблизительно 15 мкм до приблизительно 70 мкм, от приблизительно 15 мкм до приблизительно 65 мкм, от приблизительно 15 мкм до приблизительно 60 мкм, от приблизительно 15 мкм до приблизительно 55 мкм, от приблизительно 15 мкм до приблизительно 50 мкм, от приблизительно 15 мкм до приблизительно 45 мкм, от приблизительно 15 мкм до приблизительно 40 мкм, от приблизительно 15 мкм до приблизительно 35 мкм, от приблизительно 15 мкм до приблизительно 30 мкм, от приблизительно 15 мкм до приблизительно 25 мкм, от приблизительно 15 мкм до приблизительно 20 мкм, от приблизительно 20 мкм до приблизительно 100 мкм, от приблизительно 20 мкм до приблизительно 95 мкм, от приблизительно 20 мкм до приблизительно 90 мкм, от приблизительно 20 мкм до приблизительно 85 мкм, от приблизительно 20 мкм до приблизительно 80 мкм, от приблизительно 20 мкм до приблизительно 75 мкм, от приблизительно 20 мкм до приблизительно 70 мкм, от приблизительно 20 мкм до приблизительно 65 мкм, от приблизительно 20 мкм до приблизительно 60 мкм, от приблизительно 20 мкм до приблизительно 55 мкм, от приблизительно 20 мкм до приблизительно 50 мкм, от приблизительно 20 мкм до приблизительно 45 мкм, от приблизительно 20 мкм до приблизительно 40 мкм, от приблизительно 20 мкм до приблизительно 35 мкм, от приблизительно 20 мкм до приблизительно 30 мкм, от приблизительно 20 мкм до приблизительно 25 мкм, от приблизительно 25 мкм до приблизительно 100 мкм, от приблизительно 25 мкм до приблизительно 95 мкм, от приблизительно 25 мкм до приблизительно 90 мкм, от приблизительно 25 мкм до приблизительно 85 мкм, от приблизительно 25 мкм до приблизительно 80 мкм, от приблизительно 25 мкм до приблизительно 75 мкм, от приблизительно 25 мкм до приблизительно 70 мкм, от приблизительно 25 мкм до приблизительно 65 мкм, от приблизительно 25 мкм до приблизительно 60 мкм, от приблизительно 25 мкм до приблизительно 55 мкм, от приблизительно 25 мкм до приблизительно 50 мкм, от приблизительно 25 мкм до приблизительно 45 мкм, от приблизительно 25 мкм до приблизительно 40 мкм, от приблизительно 25 мкм до приблизительно 35 мкм, от приблизительно 25 мкм до приблизительно 30 мкм, от приблизительно 30 мкм до приблизительно 100 мкм, от приблизительно 30 мкм до приблизительно 95 мкм, от приблизительно 30 мкм до приблизительно 90 мкм, от приблизительно 30 мкм до приблизительно 85 мкм, от приблизительно 30 мкм до приблизительно 80 мкм, от приблизительно 30 мкм до приблизительно 75 мкм, от приблизительно 30 мкм до приблизительно 70 мкм, от приблизительно 30 мкм до приблизительно 65 мкм, от приблизительно 30 мкм до приблизительно 60 мкм, от приблизительно 30 мкм до приблизительно 55 мкм, от приблизительно 30 мкм до приблизительно 50 мкм, от приблизительно 30 мкм до приблизительно 45 мкм, от приблизительно 30 мкм до приблизительно 40 мкм, от приблизительно 30 мкм до приблизительно 35 мкм, от приблизительно 35 мкм до приблизительно 100 мкм, от приблизительно 35 мкм до приблизительно 95 мкм, от приблизительно 35 мкм до приблизительно 90 мкм, от приблизительно 35 мкм до приблизительно 85 мкм, от приблизительно 35 мкм до приблизительно 80 мкм, от приблизительно 35 мкм до приблизительно 75 мкм, от приблизительно 35 мкм до приблизительно 70 мкм, от приблизительно 35 мкм до приблизительно 65 мкм, от приблизительно 35 мкм до приблизительно 60 мкм, от приблизительно 35 мкм до приблизительно 55 мкм, от приблизительно 35 мкм до приблизительно 50 мкм, от приблизительно 35 мкм до приблизительно 45 мкм, от приблизительно 35 мкм до приблизительно 40 мкм, от приблизительно 40 мкм до приблизительно 100 мкм, от приблизительно 40 мкм до приблизительно 95 мкм, от приблизительно 40 мкм до приблизительно 90 мкм, от приблизительно 40 мкм до приблизительно 85 мкм, от приблизительно 40 мкм до приблизительно 80 мкм, от приблизительно 40 мкм до приблизительно 75 мкм, от приблизительно 40 мкм до приблизительно 70 мкм, от приблизительно 40 мкм до приблизительно 65 мкм, от приблизительно 40 мкм до приблизительно 60 мкм, от приблизительно 40 мкм до приблизительно 55 мкм, от приблизительно 40 мкм до приблизительно 50 мкм, от приблизительно 40 мкм до приблизительно 45 мкм, от приблизительно 45 мкм до приблизительно 100 мкм, от приблизительно 45 мкм до приблизительно 95 мкм, от приблизительно 45 мкм до приблизительно 90 мкм, от приблизительно 45 мкм до приблизительно 85 мкм, от приблизительно 45 мкм до приблизительно 80 мкм, от приблизительно 45 мкм до приблизительно 75 мкм, от приблизительно 45 мкм до приблизительно 70 мкм, от приблизительно 45 мкм до приблизительно 65 мкм, от приблизительно 45 мкм до приблизительно 60 мкм, от приблизительно 45 мкм до приблизительно 55 мкм, от приблизительно 45 мкм до приблизительно 50 мкм, от приблизительно 50 мкм до приблизительно 100 мкм, от приблизительно 50 мкм до приблизительно 95 мкм, от приблизительно 50 мкм до приблизительно 90 мкм, от приблизительно 50 мкм до приблизительно 85 мкм, от приблизительно 50 мкм до приблизительно 80 мкм, от приблизительно 50 мкм до приблизительно 75 мкм, от приблизительно 50 мкм до приблизительно 70 мкм, от приблизительно 50 мкм до приблизительно 65 мкм, от приблизительно 50 мкм до приблизительно 60 мкм, от приблизительно 50 мкм до приблизительно 55 мкм, от приблизительно 55 мкм до приблизительно 100 мкм, от приблизительно 55 мкм до приблизительно 95 мкм, от приблизительно 55 мкм до приблизительно 90 мкм, от приблизительно 55 мкм до приблизительно 85 мкм, от приблизительно 55 мкм до приблизительно 80 мкм, от приблизительно 55 мкм до приблизительно 75 мкм, от приблизительно 55 мкм до приблизительно 70 мкм, от приблизительно 55 мкм до приблизительно 65 мкм, от приблизительно 55 мкм до приблизительно 60 мкм, от приблизительно 60 мкм до приблизительно 100 мкм, от приблизительно 60 мкм до приблизительно 95 мкм, от приблизительно 60 мкм до приблизительно 90 мкм, от приблизительно 60 мкм до приблизительно 85 мкм, от приблизительно 60 мкм до приблизительно 80 мкм, от приблизительно 60 мкм до приблизительно 75 мкм, от приблизительно 60 мкм до приблизительно 70 мкм, от приблизительно 60 мкм до приблизительно 65 мкм, от приблизительно 65 мкм до приблизительно 100 мкм, от приблизительно 65 мкм до приблизительно 95 мкм, от приблизительно 65 мкм до приблизительно 90 мкм, от приблизительно 65 мкм до приблизительно 85 мкм, от приблизительно 65 мкм до приблизительно 80 мкм, от приблизительно 65 мкм до приблизительно 75 мкм, от приблизительно 65 мкм до приблизительно 70 мкм, от приблизительно 70 мкм до приблизительно 100 мкм, от приблизительно 70 мкм до приблизительно 95 мкм, от приблизительно 70 мкм до приблизительно 90 мкм, от приблизительно 70 мкм до приблизительно 85 мкм, от приблизительно 70 мкм до приблизительно 80 мкм, от приблизительно 70 мкм до приблизительно 75 мкм, от приблизительно 75 мкм до приблизительно 100 мкм, от приблизительно 75 мкм до приблизительно 95 мкм, от приблизительно 75 мкм до приблизительно 90 мкм, от приблизительно 75 мкм до приблизительно 85 мкм, от приблизительно 75 мкм до приблизительно 80 мкм, от приблизительно 80 мкм до приблизительно 100 мкм, от приблизительно 80 мкм до приблизительно 95 мкм, от приблизительно 80 мкм до приблизительно 90 мкм, от приблизительно 80 мкм до приблизительно 85 мкм, от приблизительно 85 мкм до приблизительно 100 мкм, от приблизительно 85 мкм до приблизительно 95 мкм, от приблизительно 85 мкм до приблизительно 90 мкм, от приблизительно 90 мкм до приблизительно 100 мкм, от приблизительно 90 мкм до приблизительно 95 мкм или от приблизительно 95 мкм до приблизительно 100 мкм. Согласно некоторым вариантам осуществления D50 составляет приблизительно 5 мкм, приблизительно 10 мкм, приблизительно 15 мкм, приблизительно 16 мкм, приблизительно 20 мкм, приблизительно 25 мкм, приблизительно 30 мкм, приблизительно 35 мкм, приблизительно 40 мкм, приблизительно 45 мкм, приблизительно 50 мкм, приблизительно 55 мкм, приблизительно 60 мкм, приблизительно 65 мкм, приблизительно 70 мкм, приблизительно 75 мкм, приблизительно 80 мкм, приблизительно 85 мкм, приблизительно 90 мкм, приблизительно 95 мкм или приблизительно 100 мкм. Согласно некоторым вариантам осуществления D50 составляет приблизительно 10 мкм, приблизительно 11 мкм, приблизительно 12 мкм, приблизительно 13 мкм, приблизительно 14 мкм, приблизительно 15 мкм, приблизительно 16 мкм, приблизительно 17 мкм, приблизительно 18 мкм, приблизительно 19 мкм или приблизительно 20 мкм или равняется значению в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления значение D50 составляет приблизительно 16 мкм. Значение D50 можно измерять с помощью стандартных методик измерения размера частиц, хорошо известных специалистам в данной области. Такие методики включают, например, проточное фракционирование в поперечном поле центробежных сил, фотонную корреляционную спектроскопию, светорассеяние, лазерную дифракцию и осаждение в тарельчатой центрифуге.
Согласно некоторым вариантам осуществления частицы характеризуются распределением частиц по размеру (D-значение, D50) от 5 мкм до 100 мкм, как, например, от 5 мкм до 95 мкм, от 5 мкм до 90 мкм, от 5 мкм до 85 мкм, от 5 мкм до 80 мкм, от 5 мкм до 75 мкм, от 5 мкм до 70 мкм, от 5 мкм до 65 мкм, от 5 мкм до 60 мкм, от 5 мкм до 55 мкм, от 5 мкм до 50 мкм, от 5 мкм до 45 мкм, от 5 мкм до 40 мкм, от 5 мкм до 35 мкм, от 5 мкм до 30 мкм, от 5 мкм до 25 мкм, от 5 мкм до 20 мкм, от 5 мкм до 15 мкм, от 5 мкм до 10 мкм, от 10 мкм до 100 мкм, от 10 мкм до 95 мкм, от 10 мкм до 90 мкм, от 10 мкм до 85 мкм, от 10 мкм до 80 мкм, от 10 мкм до 75 мкм, от 10 мкм до 70 мкм, от 10 мкм до 65 мкм, от 10 мкм до 60 мкм, от 10 мкм до 55 мкм, от 10 мкм до 50 мкм, от 10 мкм до 45 мкм, от 10 мкм до 40 мкм, от 10 мкм до 35 мкм, от 10 мкм до 30 мкм, от 10 мкм до 25 мкм, от 10 мкм до 20 мкм, от 10 мкм до 15 мкм, от 15 мкм до 100 мкм, от 15 мкм до 95 мкм, от 15 мкм до 90 мкм, от 15 мкм до 85 мкм, от 15 мкм до 80 мкм, от 15 мкм до 75 мкм, от 15 мкм до 70 мкм, от 15 мкм до 65 мкм, от 15 мкм до 60 мкм, от 15 мкм до 55 мкм, от 15 мкм до 50 мкм, от 15 мкм до 45 мкм, от 15 мкм до 40 мкм, от 15 мкм до 35 мкм, от 15 мкм до 30 мкм, от 15 мкм до 25 мкм, от 15 мкм до 20 мкм, от 20 мкм до 100 мкм, от 20 мкм до 95 мкм, от 20 мкм до 90 мкм, от 20 мкм до 85 мкм, от 20 мкм до 80 мкм, от 20 мкм до 75 мкм, от 20 мкм до 70 мкм, от 20 мкм до 65 мкм, от 20 мкм до 60 мкм, от 20 мкм до 55 мкм, от 20 мкм до 50 мкм, от 20 мкм до 45 мкм, от 20 мкм до 40 мкм, от 20 мкм до 35 мкм, от 20 мкм до 30 мкм, от 20 мкм до 25 мкм, от 25 мкм до 100 мкм, от 25 мкм до 95 мкм, от 25 мкм до 90 мкм, от 25 мкм до 85 мкм, от 25 мкм до 80 мкм, от 25 мкм до 75 мкм, от 25 мкм до 70 мкм, от 25 мкм до 65 мкм, от 25 мкм до 60 мкм, от 25 мкм до 55 мкм, от 25 мкм до 50 мкм, от 25 мкм до 45 мкм, от 25 мкм до 40 мкм, от 25 мкм до 35 мкм, от 25 мкм до 30 мкм, от 30 мкм до 100 мкм, от 30 мкм до 95 мкм, от 30 мкм до 90 мкм, от 30 мкм до 85 мкм, от 30 мкм до 80 мкм, от 30 мкм до 75 мкм, от 30 мкм до 70 мкм, от 30 мкм до 65 мкм, от 30 мкм до 60 мкм, от 30 мкм до 55 мкм, от 30 мкм до 50 мкм, от 30 мкм до 45 мкм, от 30 мкм до 40 мкм, от 30 мкм до 35 мкм, от 35 мкм до 100 мкм, от 35 мкм до 95 мкм, от 35 мкм до 90 мкм, от 35 мкм до 85 мкм, от 35 мкм до 80 мкм, от 35 мкм до 75 мкм, от 35 мкм до 70 мкм, от 35 мкм до 65 мкм, от 35 мкм до 60 мкм, от 35 мкм до 55 мкм, от 35 мкм до 50 мкм, от 35 мкм до 45 мкм, от 35 мкм до 40 мкм, от 40 мкм до 100 мкм, от 40 мкм до 95 мкм, от 40 мкм до 90 мкм, от 40 мкм до 85 мкм, от 40 мкм до 80 мкм, от 40 мкм до 75 мкм, от 40 мкм до 70 мкм, от 40 мкм до 65 мкм, от 40 мкм до 60 мкм, от 40 мкм до 55 мкм, от 40 мкм до 50 мкм, от 40 мкм до 45 мкм, от 45 мкм до 100 мкм, от 45 мкм до 95 мкм, от 45 мкм до 90 мкм, от 45 мкм до 85 мкм, от 45 мкм до 80 мкм, от 45 мкм до 75 мкм, от 45 мкм до 70 мкм, от 45 мкм до 65 мкм, от 45 мкм до 60 мкм, от 45 мкм до 55 мкм, от 45 мкм до 50 мкм, от 50 мкм до 100 мкм, от 50 мкм до 95 мкм, от 50 мкм до 90 мкм, от 50 мкм до 85 мкм, от 50 мкм до 80 мкм, от 50 мкм до 75 мкм, от 50 мкм до 70 мкм, от 50 мкм до 65 мкм, от 50 мкм до 60 мкм, от 50 мкм до 55 мкм, от 55 мкм до 100 мкм, от 55 мкм до 95 мкм, от 55 мкм до 90 мкм, от 55 мкм до 85 мкм, от 55 мкм до 80 мкм, от 55 мкм до 75 мкм, от 55 мкм до 70 мкм, от 55 мкм до 65 мкм, от 55 мкм до 60 мкм, от 60 мкм до 100 мкм, от 60 мкм до 95 мкм, от 60 мкм до 90 мкм, от 60 мкм до 85 мкм, от 60 мкм до 80 мкм, от 60 мкм до 75 мкм, от 60 мкм до 70 мкм, от 60 мкм до 65 мкм, от 65 мкм до 100 мкм, от 65 мкм до 95 мкм, от 65 мкм до 90 мкм, от 65 мкм до 85 мкм, от 65 мкм до 80 мкм, от 65 мкм до 75 мкм, от 65 мкм до 70 мкм, от 70 мкм до 100 мкм, от 70 мкм до 95 мкм, от 70 мкм до 90 мкм, от 70 мкм до 85 мкм, от 70 мкм до 80 мкм, от 70 мкм до 75 мкм, от 75 мкм до 100 мкм, от 75 мкм до 95 мкм, от 75 мкм до 90 мкм, от 75 мкм до 85 мкм, от 75 мкм до 80 мкм, от 80 мкм до 100 мкм, от 80 мкм до 95 мкм, от 80 мкм до 90 мкм, от 80 мкм до 85 мкм, от 85 мкм до 100 мкм, от 85 мкм до 95 мкм, от 85 мкм до 90 мкм, от 90 мкм до 100 мкм, от 90 мкм до 95 мкм или от 95 мкм до 100 мкм. Согласно некоторым вариантам осуществления D50 составляет 5 мкм, 10 мкм, 15 мкм, 16 мкм, 20 мкм, 25 мкм, 30 мкм, 35 мкм, 40 мкм, 45 мкм, 50 мкм, 55 мкм, 60 мкм, 65 мкм, 70 мкм, 75 мкм, 80 мкм, 85 мкм, 90 мкм, 95 мкм или 100 мкм. Согласно некоторым вариантам осуществления D50 составляет 10 мкм, 11 мкм, 12 мкм, 13 мкм, 14 мкм, 15 мкм, 16 мкм, 17 мкм, 18 мкм, 19 мкм или 20 мкм или равняется значению в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления значение D50 составляет 16 мкм. Значение D50 можно измерять с помощью стандартных методик измерения размера частиц, хорошо известных специалистам в данной области. Такие методики включают, например, проточное фракционирование в поперечном поле центробежных сил, фотонную корреляционную спектроскопию, светорассеяние, лазерную дифракцию и осаждение в тарельчатой центрифуге.
SDD по настоящему раскрытию характеризуются низким содержанием остаточного растворителя. «Содержание остаточного растворителя» в контексте настоящего документа относится к количеству растворителя, присутствующего в SDD после высушивания распылением непосредственно после выхода из распылительной сушилки. Присутствие растворителя в SDD снижает температуру стеклования (Tg) дисперсии. Согласно некоторым вариантам осуществления подвижность соединения формулы (I) или его фармацевтически приемлемой соли в SDD и их склонность к фазовому разделению и кристаллизации снижается по мере уменьшения количества остаточного растворителя в SDD. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя не более чем приблизительно 10% вес., как, например, не более чем приблизительно 5% вес. или не более чем приблизительно 1% вес. Например, SDD характеризуются содержанием остаточного растворителя приблизительно 2% вес., приблизительно 1,9% вес., приблизительно 1,8% вес., приблизительно 1,7% вес., приблизительно 1,6% вес., приблизительно 1,5% вес., приблизительно 1,4% вес., приблизительно 1,3% вес., приблизительно 1,2% вес., приблизительно 1,1% вес., приблизительно 1% вес., приблизительно 0,9% вес., приблизительно 0,8% вес., приблизительно 0,7% вес., приблизительно 0,6% вес., приблизительно 0,5% вес. или меньше. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя менее чем приблизительно 2% вес. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя менее чем приблизительно 1% вес. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя приблизительно 0,5% по весу или меньше. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя не более 10% вес., как, например, не более 5% вес. или не более 1% вес. Например, SDD характеризуются содержанием остаточного растворителя 2% вес., 1,9% вес., 1,8% вес., 1,7% вес., 1,6% вес., 1,5% вес., 1,4% вес., 1,3% вес., 1,2% вес., 1,1% вес., 1% вес., 0,9% вес., 0,8% вес., 0,7% вес., 0,6% вес., 0,5% вес. или меньше. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя менее 2% вес. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя менее 1% вес. Согласно некоторым вариантам осуществления SDD характеризуются содержанием остаточного растворителя 0,5% по весу или меньше.
Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль в высушенной распылением дисперсии являются главным образом аморфными. В контексте настоящего документа «главным образом аморфный» означает, что количество соединения формулы (I) или его фармацевтически приемлемой соли в аморфной форме составляет по меньшей мере 60% вес. и что количество присутствующей кристаллической формы не превышает 20% вес. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль в дисперсии являются «почти полностью аморфными» означает, что по меньшей мере 90% вес. лекарственного средства является аморфным или что количество соединения формулы (I) или его фармацевтически приемлемой соли в кристаллической форме не превышает 10% вес. Количества кристаллического лекарственного средства можно измерять с помощью порошковой рентгеновской дифракции (PXRD), анализа с использованием растрового электронного микроскопа (SEM), дифференциальной сканирующей калориметрии (DSC), микроскопии в поляризованном свете (PLM) или любого другого стандартного количественного или качественного измерения, применяемого для выявления кристаллического вещества. Не вдаваясь в конкретную теорию полагают, что аморфная или некристаллическая форма в сочетании с полимером обуславливает большее облегчение растворения и абсорбции в требуемом месте, например, кишечнике, что приводит к повышенной биодоступности по сравнению с кристаллической формой соединения формулы (I) без полимера.
Процесс получения высушенных распылением дисперсий
В настоящем раскрытии представлены способы получения высушенной распылением дисперсии, содержащей полимер и соединение формулы (I) или его фармацевтически приемлемую соль, такой как описанные в настоящем документе SDD. Согласно некоторым вариантам осуществления способ включает растворение соединения формулы (I) или его фармацевтически приемлемой соли и полимера в органическом растворителе с образованием раствора; и высушивание распылением раствора с получением высушенной распылением дисперсии, причем высушивание распылением обеспечивает образование однородных частиц соединения формулы (I) и полимера. Согласно некоторым вариантам осуществления продукт, получаемый путем высушивания распылением, сушат для удаления растворителя или смеси растворителей. Согласно некоторым вариантам осуществления органический растворитель представляет собой ацетон.
Раскрытые в настоящем документе SDD можно получать путем высушивания распылением смеси, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, один или несколько полимеров и соответствующий растворитель или смесь растворителей. Высушивание распылением включает распыление жидкой смеси, содержащей, например, твердое вещество и растворитель или смесь растворителей, и удаление растворителя или смеси растворителей. Распыление можно выполнять, например, через двухлоточную, или механическую, или ультразвуковую форсунку, или на вращающемся диске.
Высушивание распылением превращает подаваемую жидкость в форму высушенных частиц. Согласно некоторым вариантам осуществления высушивание распылением включает распыление подаваемого жидкого раствора в виде капельной струи и контактирование капель с горячим воздухом или газом в сушильной камере. Подлежащая высушиванию распылением композиция может представлять собой любой раствор, грубодисперсную суспензию, взвесь, коллоидную дисперсию или пасту, которые можно распылять с применением выбранного прибора для высушивания распылением. Согласно некоторым вариантам осуществления композиция представляет собой раствор, который является прозрачным и не содержит нерастворенных твердых веществ. Согласно некоторым вариантам осуществления спреи получают с помощью либо центробежного (вращающегося) распылителя, либо форсунки. Испарение влаги из капель и образование сухих частиц происходит в условиях контролируемой температуры и воздушного потока. Для проведения высушивания распылением можно применять коммерчески доступные типы приборов. Например, коммерческие распылительные сушилки производятся Buchi Ltd. и Niro (например, линейка PSD распылительных сушилок от Niro). Методики и способы высушивания распылением также можно найти в Perry's Chemical Engineering Handbook, 6th Ed., R. H. Perry, D. W. Green & J. O. Maloney, eds., McGraw-Hill Book Co. (1984); и Marshall, "Atomization and Spray-Drying" 50, Chem. Eng. Prog. Monogr. Series 2 (1954).
Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на входе от приблизительно 40°С до приблизительно 100°С, например, от приблизительно 60°С до приблизительно 80°С или от приблизительно 70°С до приблизительно 75°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на входе приблизительно 40°С, приблизительно 45°С, приблизительно 50°С, приблизительно 55°С, приблизительно 60°С, приблизительно 70°С, приблизительно 72°С, приблизительно 75°С, приблизительно 80°С, приблизительно 85°С, приблизительно 90°С, приблизительно 95°С, или приблизительно 100°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на входе составляет от приблизительно 60°С до приблизительно 80°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на входе приблизительно 61°С, приблизительно 63°С, приблизительно 65°С, приблизительно 67°С, приблизительно 69°С, приблизительно 71°С, приблизительно 73°С, приблизительно 75°С, приблизительно 77°С, приблизительно 79°С, приблизительно 81°С, приблизительно 83°С, или приблизительно 85°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на входе составляет приблизительно 72°С.
Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на входе от 40°С до 100°С, например, от 60°С до 80°С или от 70°С до 75°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на входе 40°С, 45°С, 50°С, 55°С, 60°С, 70°С, 72°С, 75°С, 80°С, 85°С, 90°С, 95°С или 100°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на входе составляет от 60°С до 80°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на входе 61°С, 63°С, 65°С, 67°С, 69°С, 71°С, 73°С, 75°С, 77°С, 79°С, 81°С, 83°С, или 85°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на входе составляет 72°С.
Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на выходе от приблизительно 20°С до приблизительно 75°С, например, от приблизительно 25°С до приблизительно 50°С или от приблизительно 30°С до приблизительно 40°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на выходе приблизительно 20°С, приблизительно 25°С, приблизительно 30°С, приблизительно 35°С, приблизительно 40°С, приблизительно 45°С, приблизительно 50°С, приблизительно 55°С, приблизительно 60°С, приблизительно 65°С, приблизительно 70°С или приблизительно 75°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на выходе составляет от приблизительно 25°С до приблизительно 45°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на выходе приблизительно 24°С, приблизительно 26°С, приблизительно 28°С, приблизительно 30°С, приблизительно 32°С, приблизительно 34°С, приблизительно 36°С, приблизительно 38°С, приблизительно 40°С, приблизительно 42°С, приблизительно 44°С или приблизительно 46°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на выходе составляет приблизительно 35°С.
Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на выходе от 20°С до 75°С, например, от 25°С до 50°С или от 30°С до 40°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на выходе 20°С, 25°С, 30°С, 35°С, 40°С, 45°С, 50°С, 55°С, 60°С, 65°С, 70°С или 75°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на выходе составляет от 25°С до 45°С. Согласно некоторым вариантам осуществления высушивание распылением проводят с температурой на выходе 24°С, 26°С, 28°С, 30°С, 32°С, 34°С, 36°С, 38°С, 40°С, 42°С, 44°С или 46°С, или значением температуры в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления температура на выходе составляет 35°С.
Согласно некоторым вариантам осуществления способ включает удаление органического растворителя после образования высушенной распылением дисперсии. Согласно некоторым вариантам осуществления органический растворитель удаляют путем высушивания SDD. Согласно некоторым вариантам осуществления с целью удаления или снижения количества остаточных растворителей, таких как органический растворитель, до фармацевтически приемлемых уровней, применяют процесс вторичной сушки, такой как сушка в псевдоожиженном слое, сушка в вакууме, сушка на полках, индукционная сушка, сушка в аппарате роторного типа или сушка в двухконусной вакуумной сушилке. Согласно некоторым вариантам осуществления SDD сушат с помощью конвекционной полочной сушилки.
Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют насыпную плотность без утряски менее чем приблизительно 0,2 г/мл или менее чем приблизительно 0,15 г/мл. Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют насыпную плотность без утряски приблизительно 0,19 г/мл, приблизительно 0,18 г/мл, приблизительно 0,17 г/мл, приблизительно 0,16 г/мл, приблизительно 0,15 г/мл, приблизительно 0,14 г/мл, приблизительно 0,13 г/мл, приблизительно 0,12 г/мл, приблизительно 0,11 г/мл, приблизительно 0,1 г/мл, приблизительно 0,09 г/мл, приблизительно 0,08 г/мл, приблизительно 0,07 г/мл, приблизительно 0,06 г/мл или приблизительно 0,05 г/мл, или насыпную плотность в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют насыпную плотность без утряски менее 0,2 г/мл или менее 0,15 г/мл. Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют насыпную плотность без утряски 0,19 г/мл, 0,18 г/мл, 0,17 г/мл, 0,16 г/мл, 0,15 г/мл, 0,14 г/мл, 0,13 г/мл, 0,12 г/мл, 0,11 г/мл, 0,1 г/мл, 0,09 г/мл, 0,08 г/мл, 0,07 г/мл, 0,06 г/мл или 0,05 г/мл, или насыпную плотность в пределах диапазона, определенного любыми из предыдущих значений. Термин «насыпная плотность без утряски» в контексте настоящего документа относится к свойству порошков и определяется как масса множества частиц материала, деленная на общий объем, который они занимают. Общий объем включает объем частиц, объем пустот между частицами и объем внутренних пор.
Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют плотность после утряски менее чем приблизительно 0,3 г/мл или менее чем приблизительно 0,25 г/мл. Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют плотность после утряски приблизительно 0,29 г/мл, приблизительно 0,28 г/мл, приблизительно 0,27 г/мл, приблизительно 0,26 г/мл, приблизительно 0,25 г/мл, приблизительно 0,24 г/мл, приблизительно 0,23 г/мл, приблизительно 0,22 г/мл, приблизительно 0,21 г/мл, приблизительно 0,2 г/мл или приблизительно 0,19 г/мл, или плотность после утряски в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют плотность после утряски менее 0,3 г/мл или менее 0,25 г/мл. Согласно некоторым вариантам осуществления однородные частицы, получаемые с помощью способов по настоящему раскрытию, имеют плотность после утряски 0,29 г/мл, 0,28 г/мл, 0,27 г/мл, 0,26 г/мл, 0,25 г/мл, 0,24 г/мл, 0,23 г/мл, 0,22 г/мл, 0,21 г/мл, 0,2 г/мл или 0,19 г/мл, или плотность после утряски в пределах диапазона, определенного любыми из предыдущих значений. Термин «плотность утряски» или «плотность после утряски» в контексте настоящего документа относится к мере плотности порошка. Насыпную плотность после утряски фармацевтического порошка определяют с применением прибора для измерения насыпной плотности после утряски, который настроен на утряску порошка с фиксированной ударной силой и частотой. Насыпную плотность после утряски с помощью способа USP определяют линейной прогрессией числа утрясок.
Дейтерированные соединения
Также в настоящем документе раскрыты соединения, характеризующиеся структурой следующей формулы (II):
или их фармацевтически приемлемая соль, где:
каждый R1 представляет собой независимо С(RA)3;
каждый RA представляет собой независимо водород или дейтерий;
каждый R2 представляет собой независимо водород или дейтерий;
каждый R3 представляет собой независимо водород или дейтерий;
R4 представляет собой 
R5 представляет собой водород или дейтерий;
R6 представляет собой C(RA)3; и
R7 представляет собой C(RB)3, причем по меньшей мере один из RA, RB, R2, R3 и R5 представляет собой дейтерий.
По отношению к представленным в настоящем документе соединениям, если конкретное положение атома обозначено как соответствующее дейтерию, или «D», или «d», следует понимать, что распространенность дейтерия в таком положении значительно превышает естественную распространенность дейтерия, которая составляет приблизительно 0,015%. Положение, обозначенное как соответствующее дейтерию, как правило, характеризуется минимальным коэффициентом изотопного обогащения, в определенных вариантах осуществления, по меньшей мере 3500 (52,5% включение дейтерия), по меньшей мере 4000 (60% включение дейтерия), по меньшей мере 4500 (67,5% включение дейтерия), по меньшей мере 5000 (75% включение дейтерия), по меньшей мере 5500 (82,5% включение дейтерия), по меньшей мере 6000 (90% включение дейтерия), по меньшей мере 6333,3 (95% включение дейтерия), по меньшей мере 6466,7 (97% включение дейтерия), по меньшей мере 6600 (99% включение дейтерия) или по меньшей мере 6633,3 (99,5% включение дейтерия), по каждому обозначенному положению дейтерия.
Согласно некоторым вариантам осуществления соединение формулы (II) может представлять собой одно из следующего, или его фармацевтически приемлемую соль:
Фармацевтические композиции
Раскрытые в настоящем документе способы и применения могут предусматривать введение соединения формулы (I) в виде фармацевтической композиции.
Согласно некоторым вариантам осуществления описанных в настоящем документе способов 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль вводят в фармацевтической композиции, дополнительно содержащей одно или несколько фармацевтически приемлемых вспомогательных веществ.
Также в настоящем документе представлена фармацевтическая композиция, содержащая 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-цикло пропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль для применения в любых из описанных в настоящем документе способов.
Согласно некоторым вариантам осуществления описанные в настоящем документе способы и применения предусматривают введение фармацевтической композиции, которая не содержит высушенной распылением дисперсии соединения формулы (I), как указано, например, в примере 1. Соответственно, согласно некоторым вариантам осуществления фармацевтическая композиция не содержит каких-либо из следующих полимеров: гидроксипропилметилцеллюлозы ацетата сукцинат-L (HPMCAS-L); сополимер винилпирролидона и винилацетата 64 (PVP/VA 64); HPMCAS-M; и метилметакрилатный сополимер (1:1) (Eudragit® L100).
Согласно некоторым вариантам осуществления описанные в настоящем документе способы и применения предусматривают введение фармацевтической композиции, которая не является эталонным составом, описанным в примере 9. Соответственно, согласно некоторым вариантам осуществления фармацевтическая композиция не содержит по меньшей мере трех из вспомогательных веществ, выбранных из каприлового/капринового триглицерида (Labrafac® Lipophile, Gattefosse, Франция); пропиленгликольдикаприлата/дикапрата (Labrafac® PG, Gattefosse, Франция); олеоилполиоксил-6-глицеридов (Labrafil® M 1944 CS, Gattefosse, Франция); полисорбата 20; полиоксил касторового масла (Kolliphor® RH 40, BASF, Германия); полиоксил 15 гидроксистеарата (Kolliphor® HS 15, BASF, Германия); лауроилполиоксил-32-глицеридов (Gelucire® 44/14, Gattefosse, Франция); d-a-токоферол полиэтиленгликоль 1000 сукцината (TPGS); и моноэтилового эфира диэтиленгликоля (Transcutol®, Gattefosse, Франция).
Согласно некоторым вариантам осуществления описанные в настоящем документе способы и применения предусматривают введение фармацевтической композиции, которая представляет собой состав, описанный в примере 9. Согласно некоторым вариантам осуществления описанные в настоящем документе способы и применения предусматривают введение фармацевтической композиции, которая представляет собой состав, описанный в примере 11. Согласно некоторым вариантам осуществления описанные в настоящем документе способы и применения предусматривают введение фармацевтической композиции, которая представляет собой состав, описанный в примере 12. Согласно некоторым вариантам осуществления описанные в настоящем документе способы и применения предусматривают введение фармацевтической композиции, которая представляет собой состав, описанный в примере 13.
Согласно некоторым вариантам осуществления фармацевтические композиции включают высушенную распылением дисперсию, содержащую полимер и соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно некоторым вариантам осуществления фармацевтическая композиция включает SDD, содержащую соединение формулы (I) и одно или несколько фармацевтически приемлемых вспомогательных веществ. Согласно некоторым вариантам осуществления SDD присутствует в фармацевтической композиции в количестве от приблизительно 20% до приблизительно 90% вес./вес. композиции, как, например, от приблизительно 20% до приблизительно 85%, от приблизительно 20% до приблизительно 80%, от приблизительно 20% до приблизительно 75%, от приблизительно 20% до приблизительно 70%, от приблизительно 20% до приблизительно 65%, от приблизительно 20% до приблизительно 60%, от приблизительно 20% до приблизительно 55%, от приблизительно 20% до приблизительно 50%, от приблизительно 20% до приблизительно 45%, от приблизительно 20% до приблизительно 40%, от приблизительно 20% до приблизительно 35%, от приблизительно 20% до приблизительно 30%, от приблизительно 20% до приблизительно 25%, от приблизительно 25% до приблизительно 90%, от приблизительно 25% до приблизительно 85%, от приблизительно 25% до приблизительно 80%, от приблизительно 25% до приблизительно 75%, от приблизительно 25% до приблизительно 70%, от приблизительно 25% до приблизительно 65%, от приблизительно 25% до приблизительно 60%, от приблизительно 25% до приблизительно 55%, от приблизительно 25% до приблизительно 50%, от приблизительно 25% до приблизительно 45%, от приблизительно 25% до приблизительно 40%, от приблизительно 25% до приблизительно 35%, от приблизительно 25% до приблизительно 30%, от приблизительно 30% до приблизительно 90%, от приблизительно 30% до приблизительно 85%, от приблизительно 30% до приблизительно 80%, от приблизительно 30% до приблизительно 75%, от приблизительно 30% до приблизительно 70%, от приблизительно 30% до приблизительно 65%, от приблизительно 30% до приблизительно 60%, от приблизительно 30% до приблизительно 55%, от приблизительно 30% до приблизительно 50%, от приблизительно 30% до приблизительно 45%, от приблизительно 30% до приблизительно 40%, от приблизительно 30% до приблизительно 35%, от приблизительно 35% до приблизительно 90%, от приблизительно 35% до приблизительно 85%, от приблизительно 35% до приблизительно 80%, от приблизительно 35% до приблизительно 75%, от приблизительно 35% до приблизительно 70%, от приблизительно 35% до приблизительно 65%, от приблизительно 35% до приблизительно 60%, от приблизительно 35% до приблизительно 55%, от приблизительно 35% до приблизительно 50%, от приблизительно 35% до приблизительно 45%, от приблизительно 35% до приблизительно 40%, от приблизительно 40% до приблизительно 90%, от приблизительно 40% до приблизительно 85%, от приблизительно 40% до приблизительно 80%, от приблизительно 40% до приблизительно 75%, от приблизительно 40% до приблизительно 70%, от приблизительно 40% до приблизительно 65%, от приблизительно 40% до приблизительно 60%, от приблизительно 40% до приблизительно 55%, от приблизительно 40% до приблизительно 50%, от приблизительно 40% до приблизительно 45%, от приблизительно 45% до приблизительно 90%, от приблизительно 45% до приблизительно 85%, от приблизительно 45% до приблизительно 80%, от приблизительно 45% до приблизительно 75%, от приблизительно 45% до приблизительно 70%, от приблизительно 45% до приблизительно 65%, от приблизительно 45% до приблизительно 60%, от приблизительно 45% до приблизительно 55%, от приблизительно 45% до приблизительно 50%, от приблизительно 50% до приблизительно 90%, от приблизительно 50% до приблизительно 85%, от приблизительно 50% до приблизительно 80%, от приблизительно 50% до приблизительно 75%, от приблизительно 50% до приблизительно 70%, от приблизительно 50% до приблизительно 65%, от приблизительно 50% до приблизительно 60%, от приблизительно 50% до приблизительно 55%, от приблизительно 55% до приблизительно 90%, от приблизительно 55% до приблизительно 85%, от приблизительно 55% до приблизительно 80%, от приблизительно 55% до приблизительно 75%, от приблизительно 55% до приблизительно 70%, от приблизительно 55% до приблизительно 65%, от приблизительно 55% до приблизительно 60%, от приблизительно 60% до приблизительно 90%, от приблизительно 60% до приблизительно 85%, от приблизительно 60% до приблизительно 80%, от приблизительно 60% до приблизительно 75%, от приблизительно 60% до приблизительно 70%, от приблизительно 60% до приблизительно 65%, от приблизительно 65% до приблизительно 90%, от приблизительно 65% до приблизительно 85%, от приблизительно 65% до приблизительно 80%, от приблизительно 65% до приблизительно 75%, от приблизительно 65% до приблизительно 70%, от приблизительно 70% до приблизительно 90%, от приблизительно 70% до приблизительно 85%, от приблизительно 70% до приблизительно 80%, от приблизительно 70% до приблизительно 75%, от приблизительно 75% до приблизительно 90%, от приблизительно 75% до приблизительно 85%, от приблизительно 75% до приблизительно 80%, от приблизительно 80% до приблизительно 90%, от приблизительно 80% до приблизительно 85% или от приблизительно 85% до приблизительно 90% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в количестве от приблизительно 40% до приблизительно 90% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в количестве от приблизительно 40% до приблизительно 80% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в фармацевтической композиции в количестве от приблизительно 60% до приблизительно 80% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в количестве приблизительно 80% вес./вес. композиции.
Согласно некоторым вариантам осуществления SDD присутствует в фармацевтической композиции в количестве от приблизительно 1% до приблизительно 20% вес./вес. композиции, как, например, приблизительно 13% вес./вес. композиции. Согласно некоторым вариантам осуществления фармацевтическая композиция включает SDD, содержащую соединение формулы (I) и одно или несколько фармацевтически приемлемых вспомогательных веществ. Согласно некоторым вариантам осуществления SDD присутствует в фармацевтической композиции в количестве от 20% до 90% вес./вес. композиции, как, например, от 20% до 85%, от 20% до 80%, от 20% до 75%, от 20% до 70%, от 20% до 65%, от 20% до 60%, от 20% до 55%, от 20% до 50%, от 20% до 45%, от 20% до 40%, от 20% до 35%, от 20% до 30%, от 20% до 25%, от 25% до 90%, от 25% до 85%, от 25% до 80%, от 25% до 75%, от 25% до 70%, от 25% до 65%, от 25% до 60%, от 25% до 55%, от 25% до 50%, от 25% до 45%, от 25% до 40%, от 25% до 35%, от 25% до 30%, от 30% до 90%, от 30% до 85%, от 30% до 80%, от 30% до 75%, от 30% до 70%, от 30% до 65%, от 30% до 60%, от 30% до 55%, от 30% до 50%, от 30% до 45%, от 30% до 40%, от 30% до 35%, от 35% до 90%, от 35% до 85%, от 35% до 80%, от 35% до 75%, от 35% до 70%, от 35% до 65%, от 35% до приблизительно 60%, от 35% до 55%, от 35% до 50%, от 35% до 45%, от 35% до 40%, от 40% до 90%, от 40% до 85%, от 40% до 80%, от 40% до 75%, от 40% до 70%, от 40% до 65%, от 40% до 60%, от 40% до 55%, от 40% до 50%, от 40% до 45%, от 45% до 90%, от 45% до 85%, от 45% до 80%, от 45% до 75%, от 45% до 70%, от 45% до 65%, от 45% до 60%, от 45% до 55%, от 45% до 50%, от 50% до 90%, от 50% до 85%, от 50% до 80%, от 50% до 75%, от 50% до 70%, от 50% до 65%, от 50% до 60%, от 50% до 55%, от 55% до 90%, от 55% до 85%, от 55% до 80%, от 55% до 75%, от 55% до 70%, от 55% до 65%, от 55% до 60%, от 60% до 90%, от 60% до 85%, от 60% до 80%, от 60% до 75%, от 60% до 70%, от 60% до 65%, от 65% до 90%, от 65% до 85%, от 65% до 80%, от 65% до 75%, от 65% до 70%, от 70% до 90%, от 70% до 85%, от 70% до 80%, от 70% до 75%, от 75% до 90%, от 75% до 85%, от 75% до 80%, от 80% до 90%, от 80% до 85% или от приблизительно 85% до 90% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в количестве от 40% до 90% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в количестве от 40% до 80% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в фармацевтической композиции в количестве от 60% до 80% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в количестве 80% вес./вес. композиции. Согласно некоторым вариантам осуществления SDD присутствует в фармацевтической композиции в количестве от приблизительно 1% до приблизительно 20% вес./вес. композиции, как, например, приблизительно 13% вес./вес. композиции.
Согласно некоторым вариантам осуществления раскрытых в настоящем документе фармацевтических композиций (например, композиции, включающей SDD), фармацевтически приемлемое вспомогательное вещество выбрано из группы, состоящей из наполнителя, смазывающего вещества и их комбинаций. Согласно некоторым вариантам осуществления фармацевтические вспомогательные вещества выбраны из группы, состоящей из вещества, способствующего скольжению, наполнителя, разрыхлителя, смазывающего вещества и их комбинации.
Согласно некоторым вариантам осуществления фармацевтическая композиция включает наполнитель. Согласно некоторым вариантам осуществления наполнитель выбран из группы, состоящей из связующих, разбавителей, разрыхлителей, веществ, способствующих скольжению, поверхностно-активных веществ и их комбинаций.
Согласно некоторым вариантам осуществления наполнитель включают сахариды (например, сахара, крахмал и целлюлозу), желатин, карбонат кальция и синтетические полимеры (например, поливинилпирролидон, полиэтиленгликоль и полоксамеры (например, полоксамер 188, сополимер полиоксиэтилена и полиоксипропилена)). Иллюстративные наполнители включают без ограничения глюкозу, сахарозу, лактозу, крахмал, включая виды модифицированного крахмала, такие как крахмалгликолят натрия (например, Explotab®), ксилит, декстрин, сахарозу, сорбит, маннит (например, Parteck® М 200 (маннит со средним размером частиц от приблизительно 50 мкм до приблизительно 500 мкм) или Parteck® М 100 (маннит со средним размером частиц менее 212 мкм)), целлюлозу, поливинилпирролидон, полиэтиленгликоль, поливиниловый спирт, полиметакрилат, двухзамещенный фосфат кальция, стеарат магния, стеарат кальция, стеарат натрия, стеариновую кислоту, гидрогенизированные растительные масла, минеральное масло, лаурилсульфат натрия, лаурилсульфат магния, глицерилпальмитостеарат, бензоат натрия, стеарилфумарат натрия, коллоидный диоксид кремния, бензоат натрия, олеат натрия, ацетат натрия, альгиновую кислоту, альгинаты (например, альгинат натрия), силикат кальция и ионообменные смолы. Иллюстративные целлюлозные наполнители включают микрокристаллическую целлюлозу (например, Avicel® PH-101 (микрокристаллическая целлюлоза со средним размером частиц примерно 50 мкм) или Avicel® РН 200 (микрокристаллическая целлюлоза со средним размером частиц примерно 180 мкм)), метилцеллюлозу, этилцеллюлозу, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Иллюстративные наполнители включают сшитый поливинилпирролидон (как, например, со средним размером частиц от 90 мкм до 130 мкм или со средним размером частиц от 10 мкм до 30 мкм). Другие наполнители, известные специалистам в данной области, также рассматриваются как пригодные при составлении в описанные в настоящем документе фармацевтические композиции.
Согласно некоторым вариантам осуществления наполнитель представляет собой связующее. Связующие включают средства, которые удерживают активный фармацевтический ингредиент (например, высушенную распылением дисперсию, содержащую полимер и соединение формулы (I) или его фармацевтически приемлемую соль) и неактивные ингредиенты вместе в связной смеси. Иллюстративные связующие включают без ограничения глюкозу, сахарозу, лактозу, крахмал, включая виды модифицированного крахмала, такие как крахмалгликолят натрия (Explotab®), ксилит, декстрин, сахарозу, сорбит, маннит (например, Parteck® М 200 (маннит со средним размером частиц от приблизительно 50 мкм до приблизительно 500 мкм), Parteck® М 100 (маннит со средним размером частиц менее 212 мкм)), желатин, трагакантовую камедь, гуммиарабик, целлюлозу, поливинилпирролидон, полиэтиленгликоль, поливиниловый спирт, полиметакрилат и крахмалгликолят натрия. Иллюстративные целлюлозные наполнители включают микрокристаллическую целлюлозу (например, Avicel® РН-101 (микрокристаллическая целлюлоза со средним размером частиц примерно 50 мкм) или Avicel® РН 200 (микрокристаллическая целлюлоза со средним размером частиц примерно 180 мкм)), простые эфиры целлюлозы, метилцеллюлозу, этилцеллюлозу, кроскармеллозу натрия, натрий карбоксиметилкрахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Иллюстративные поливинилпирролидоновые наполнители включают сшитый поливинилпирролидон, такой как Kollidon® CL (кросповидон со средним размером частиц от 90 мкм до 130 мкм) или Kollidon® CL-SF (кросповидон со средним размером частиц от 10 мкм до 30 мкм). Другие связующие, известные специалистам в данной области, также рассматриваются как пригодные при составлении в описанные в настоящем документе композиции.
Согласно некоторым вариантам осуществления наполнитель представляет собой разбавитель. Подходящие разбавители включают без ограничения лактозу, маннит, изомальт, сахарозу, декстрозу и сорбит.
Согласно некоторым вариантам осуществления наполнитель представляет собой разрыхлитель. Разрыхлители включают любое средство, которое содействует разрушению состава в водной среде, например, для содействия более быстрому высвобождению активного фармацевтического ингредиента (например, соединения формулы (I) или его фармацевтически приемлемой соли). Иллюстративные разрыхлители включают без ограничения крахмал и виды модифицированного крахмала, такие как кукурузный крахмал, картофельный крахмал, крахмалгликолят натрия или кроскармеллоза натрия, альгиновую кислоту, альгинаты, такие как альгинат натрия, поливинилпирролидон, бентонитовую глину, метилцеллюлозу, агар, карбоксиметилцеллюлозу, кросповидон, шипучие системы на основе гидрокарбонатов, такие как лимонная кислота с бикарбонатами, и ионообменные смолы. Другие разрыхлители, известные специалистам в данной области, также рассматриваются как пригодные при составлении в описанные в настоящем документе композиции.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит разрыхлитель. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес./вес. до приблизительно 30% вес./вес. разрыхлителя. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 5% вес./вес. до приблизительно 15% вес./вес. разрыхлителя. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 10% вес./вес. разрыхлителя. Согласно некоторым вариантам осуществления разрыхлитель выбран из кроскармеллозы натрия, крахмалгликолята натрия, кросповидона и бикарбоната натрия. Согласно некоторым вариантам осуществления разрыхлитель представляет собой кроскармеллозу натрия.
Согласно некоторым вариантам осуществления наполнитель представляет собой вещество, способствующее скольжению. Вещества, способствующие скольжению, можно применять для улучшения текучести порошка или гранул, или как одного, так и другого. Вещества, способствующие скольжению, включают без ограничения диоксид кремния, такой как коллоидный диоксид кремния или гидроокись кремния, силикат магния, алюмометасиликат магния, тальк, крахмал, силикат кальция, легкую безводную кремниевую кислоту и аэрогели на основе диоксида кремния.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит вещество, способствующее скольжению. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,1% вес./вес. до приблизительно 5% вес./вес. вещества, способствующего скольжению. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,1% вес./вес. до приблизительно 1% вес./вес. вещества, способствующего скольжению. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,67% вес./вес. вещества, способствующего скольжению. Согласно некоторым вариантам осуществления вещество, способствующее скольжению, выбрано из силиката кальция, диоксида кремния и талька. Согласно некоторым вариантам осуществления вещество, способствующее скольжению, представляет собой силикат кальция.
Согласно некоторым вариантам осуществления наполнитель представляет собой поверхностно-активное вещество, смачивающее вещество, солюбилизатор или их комбинацию. Примеры включают без ограничения моностеарат глицерина, цетостеариловый спирт, эмульгирующий воск с цетомакроголем, сложные эфиры сорбитана, полиоксиэтиленовые алкилэфиры (например, простые эфиры макрогола, такие как цетомакрогол 1000), полиоксиэтиленовые производные касторового масла, полиоксиэтиленовые сложные эфиры сорбитана и жирных кислот (например, Tween®), полиоксиэтиленстеараты, додецилсульфат натрия, тилоксапол (неионный жидкий полимер типа алкиларилполиэфира со спиртами, также известный как Superinone или Triton). Другие примеры включают без ограничения полоксамеры, такие как Pluronic® F68, F127 и F108, которые являются блок-сополимерами этиленоксида и пропиленоксида, и полоксамины, такие как Tetronic® 908 (также известный как Poloxamine® 908), который является тетрафункциональным блок-сополимером, полученным путем последовательного добавления пропиленоксида и этиленоксида к этилендиамину (доступен от BASF), декстран, лецитин, натриевые соли сложных диалкиловых эфиров сульфоянтарной кислоты, такие как Aerosol® ОТ, который представляет собой натриевую соль диоктилового эфира сульфоянтарной кислоты (доступный от American Cyanimid), Duponol® Р, который представляет собой лаурилсульфат натрия (доступный от DuPont), Triton® Х-200, который представляет собой алкиларилполиэфирсульфонат (доступный от Rohm and Haas), Tween® 20 и Tween® 80, которые представляют собой полиоксиэтиленовые сложные эфиры сорбитана и жирных кислот (доступные от ICI Specialty Chemicals), Carbowax™ 3550 и 934, которые представляют собой полиэтиленгликоли (доступные от Union Carbide), Crodesta™ F-110, который представляет собой смесь стеарата сахарозы и дистеарата сахарозы, и Crodesta™ SL-40 (оба доступны от Croda Inc.) и SA90HCO, который характеризуется химической формулой С18Н37-CH2(CON(CH3)CH2(CHOH)4CH2OH)2.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит наполнитель. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 30% вес./вес. до приблизительно 99% вес./вес. наполнителя. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 50% вес./вес. до приблизительно 90% вес./вес. наполнителя. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 75,5% вес./вес. наполнителя. Согласно некоторым вариантам осуществления наполнитель выбран из маннита, микрокристаллической целлюлозы, лактозы, крахмала, изомальта, силикатированной микрокристаллической целлюлозы, дикальцийфосфата, мальтодекстрина и их комбинации. Согласно некоторым вариантам осуществления наполнитель представляет собой комбинацию маннита и микрокристаллической целлюлозы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 30% вес./вес. до приблизительно 80% вес./вес. маннита. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 50% вес./вес. до приблизительно 60% вес./вес. маннита. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 56% вес./вес. маннита. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 1% вес./вес. до приблизительно 50% вес./вес. микрокристаллической целлюлозы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 10% вес./вес. до приблизительно 30% вес./вес. микрокристаллической целлюлозы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 20% вес./вес. микрокристаллической целлюлозы. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 56% вес./вес. маннита и приблизительно 20% вес./вес. микрокристаллической целлюлозы.
Согласно некоторым вариантам осуществления фармацевтическая композиция включает смазывающее вещество. Смазывающие вещества представляют собой средства, добавляемые в фармацевтические составы для снижения растирания в ходе обработки и предупреждения комкования ингредиентов между собой. Иллюстративные смазывающие вещества включают без ограничения тальк, крахмал, стеарат магния, стеарат кальция, стеарат натрия, стеарат цинка, стеариновую кислоту, растительный стеарин, адипиновую кислоту, жирные кислоты воска, такие как глицерилбегенат, гидрогенизированное растительное масло, минеральное масло, полиэтиленгликоль, ликоподий, лаурилсульфат натрия, лаурилсульфат магния, глицерилпальмитостеарат, бензоат натрия, хлорид натрия, Sterotex, моностеарат глицерина, стеарилфумарат натрия, коллоидный диоксид кремния, бензоат натрия, олеат натрия и ацетат натрия. Другие смазывающие вещества, известные специалистам в данной области, также рассматриваются как пригодные при составлении в описанные в настоящем документе композиции.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит смазывающее вещество. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,1% вес./вес. до приблизительно 10% вес./вес. смазывающего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит от приблизительно 0,1% вес./вес. до приблизительно 1% вес./вес. смазывающего вещества. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 0,5% вес./вес. смазывающего вещества. Согласно некоторым вариантам осуществления фармацевтическое смазывающее вещество выбрано из стеарилфумарата натрия, стеарата магния, стеариновой кислоты, лаурилсульфата натрия, олеата натрия, глицерилбегената и талька. Согласно некоторым вариантам осуществления смазывающее вещество представляет собой стеарилфумарат натрия.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) высушенную распылением дисперсию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль и полимер;
(b) вещество, способствующее скольжению;
(c) наполнитель; и
(d) разрыхлитель.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 1% вес./вес. до приблизительно 20% вес./вес. высушенной распылением дисперсии, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и полимер;
(b) от приблизительно 0,1% вес./вес. до приблизительно 1% вес./вес. вещества, способствующего скольжению;
(c) от приблизительно 50% вес./вес. до приблизительно 90% вес./вес. наполнителя; и
(d) от приблизительно 5% вес./вес. до приблизительно 0,2% вес./вес. разрыхлителя.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 13% вес./вес. высушенной распылением дисперсии, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и полимер;
(b) приблизительно 0,67% вес./вес. вещества, способствующего скольжению;
(c) приблизительно 75,5% вес./вес. наполнителя; и
(d) приблизительно 10% вес./вес. разрыхлителя.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) высушенную распылением дисперсию из примера 3;
(b) силикат кальция;
(c) комбинацию маннита и микрокристаллической целлюлозы; и
(d) кроскармеллозу натрия.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) от приблизительно 1% вес./вес. до приблизительно 20% вес./вес. высушенной распылением дисперсии из примера 3;
(b) от приблизительно 0,1% вес./вес. до приблизительно 1% вес./вес. силиката кальция;
(c) от приблизительно 50% вес./вес. до приблизительно 60% вес./вес. маннита и от приблизительно 10% вес./вес. до приблизительно 30% вес./вес. микрокристаллической целлюлозы; и
(d) от приблизительно 5% вес./вес. до приблизительно 0,2% вес./вес. кроскармеллозы натрия.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит:
(a) приблизительно 13% вес./вес. высушенной распылением дисперсии из примера 3;
(b) приблизительно 0,67% вес./вес. силиката кальция;
(c) приблизительно 56% вес./вес. маннита и приблизительно 20% вес./вес. микрокристаллической целлюлозы; и
(d) приблизительно 10% вес./вес. кроскармеллозы натрия.
В фармацевтические составы по настоящему раскрытию можно включать дополнительные вспомогательные вещества. Дополнительно примеры вспомогательных веществ включают без ограничения пигменты, красители, ароматизаторы, консерванты и подсластители. Вкусоароматические добавки и красители можно добавлять для улучшения вкуса или внешнего вида состава. Примеры консервантов, применяемых в фармацевтических композициях, представляют собой ароматические спирты, такие как бензиловый спирт или фенол, антиоксиданты, такие как витамин А, витамин Е, витамин С и селен, аминокислоты, такие как цистеин и метионин, лимонную кислоту и цитрат натрия или синтетические консерванты, такие как метилпарабен и пропилпарабен. Чтобы сделать ингредиенты более вкусными, можно добавлять подсластители, особенно в жевательные таблетки или жидкости, такие как сиропы.
Также представлены способы получения фармацевтической композиции, такой как фармацевтическая композиция по настоящему раскрытию. Согласно некоторым вариантам осуществления способ включает объединение описанной в настоящем документе высушенной распылением дисперсии с одним или несколькими фармацевтически приемлемыми вспомогательными веществами, например, описанным в настоящем документе фармацевтически приемлемым вспомогательным веществом.
Согласно некоторым вариантам осуществления способ получения фармацевтической композиции предусматривает:
(a) смешивание описанной в настоящем документе высушенной распылением дисперсии с веществом, способствующим скольжению;
(b) дальнейшее смешивание смеси из стадии (а) с наполнителем и разрыхлителем;
(c) просеивание смеси из стадии (b) для разрушения агрегатов и содействия однородности смеси;
(d) дополнительное перемешивание внутригранулярной смеси из стадии (с);
(e) прессование внутригранулярной смеси из стадии (d) посредством прессования на валковом прессе для образования гранул; и
(f) измельчение полученной в результате прессования на валковом прессе ленты с образованием гранул из стадии (е).
Согласно некоторым вариантам осуществления высушенная распылением дисперсия представляет собой высушенную распылением дисперсию, описанную в примере 3.
Фармацевтические композиции по настоящему раскрытию составляют для перорального введения. При получении композиций в виде лекарственной формы для перорального применения можно использовать любую из стандартных фармацевтических сред. В случае твердых препаратов для перорального применения, таких как, например, порошки, капсулы, каплеты, желатиновые капсулы и таблетки, подходящие носители и добавки включают виды крахмала, сахара, разбавители, гранулирующие средства, смазывающие вещества, связующие, средства для улучшения распадаемости таблеток и т.п. Подходящие связующие включают без ограничения крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, сахаристые вещества из кукурузы, натуральные и синтетические камеди, такие как гуммиарабик, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Разрыхлители включают без ограничения крахмал, метилцеллюлозу, агар, бентонитовую глину, ксантановую камедь и т.п.
Лекарственные формы для перорального применения могут представлять собой твердое вещество, гель или жидкость. Согласно некоторым вариантам осуществления лекарственная форма представляет собой твердую лекарственную форму. Согласно некоторым вариантам осуществления твердая лекарственная форма представляет собой пилюлю, таблетку, капсулу, каплет, желатиновые капсулы, гранулы, порошок, саше, пластинку, диспергируемую в полости рта, или пленку, диспергируемую в полости рта. Согласно некоторым вариантам осуществления твердая лекарственная форма покрыта оболочкой. Согласно некоторым вариантам осуществления оболочка представляет собой кишечнорастворимую оболочку, сахарную оболочку или пленочную оболочку. Согласно некоторым вариантам осуществления твердая лекарственная форма представляет собой покрытую оболочкой частицу, покрытую оболочкой таблетку, покрытую кишечнорастворимой оболочкой таблетку или покрытую кишечнорастворимой оболочкой капсулу. Согласно некоторым вариантам осуществления твердая лекарственная форма представляет собой пилюлю или таблетку. Типы таблеток для перорального приема включают полученные путем прессования жевательные пастилки и таблетки, которые могут быть покрыты кишечнорастворимой оболочкой, сахарной оболочкой или пленочной оболочкой. Согласно некоторым вариантам осуществления фармацевтическую композицию составляют в виде капсулы. Согласно некоторым вариантам осуществления фармацевтическую композицию составляют в виде порошка, раствора или суспензии (например, в пропиленкарбонате, растительных маслах, PEG, полоксамере 124 или триглицеридах) или инкапсулируют в капсулу (капсулу на основе желатина или целлюлозы). Капсулы могут представлять собой твердые или мягкие желатиновые капсулы, тогда как гранулы и порошки могут быть предоставлены в не шипучей или шипучей форме с комбинацией других ингредиентов, известных специалистам в данной области.
Фармацевтические композиции по настоящему раскрытию могут содержать, на единицу дозы, например, таблетки, капсулы, порошка и т.п., количество активного ингредиента, необходимого для доставки эффективной дозы, как описано выше.
Согласно некоторым вариантам осуществления фармацевтические композиции по настоящему раскрытию составляют в виде единичной лекарственной формы. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от приблизительно 5 мг до приблизительно 200 мг в единичной лекарственной форме. Например, они присутствуют в количестве от приблизительно 5 мг до приблизительно 175 мг, от приблизительно 5 мг до приблизительно 150 мг, от приблизительно 5 мг до приблизительно 125 мг, от приблизительно 5 мг до приблизительно 100 мг, от приблизительно 5 мг до приблизительно 75 мг, от приблизительно 5 мг до приблизительно 50 мг, от приблизительно 5 мг до приблизительно 25 мг, от приблизительно 25 мг до приблизительно 200 мг, от приблизительно 25 мг до приблизительно 175 мг, от приблизительно 25 мг до приблизительно 150 мг, от приблизительно 25 мг до приблизительно 125 мг, от приблизительно 25 мг до приблизительно 100 мг, от приблизительно 25 мг до приблизительно 75 мг, от приблизительно 25 мг до приблизительно 50 мг, от приблизительно 50 мг до приблизительно 200 мг, от приблизительно 50 мг до приблизительно 175 мг, от приблизительно 50 мг до приблизительно 150 мг, от приблизительно 50 мг до приблизительно 125 мг, от приблизительно 50 мг до приблизительно 100 мг, от приблизительно 50 мг до приблизительно 75 мг, от приблизительно 75 мг до приблизительно 200 мг, от приблизительно 75 мг до приблизительно 175 мг, от приблизительно 75 мг до приблизительно 150 мг, от приблизительно 75 мг до приблизительно 125 мг, от приблизительно 75 мг до приблизительно 100 мг, от приблизительно 100 мг до приблизительно 200 мг, от приблизительно 100 мг до приблизительно 175 мг, от приблизительно 100 мг до приблизительно 150 мг, от приблизительно 100 мг до приблизительно 125 мг, от приблизительно 125 мг до приблизительно 200 мг, от приблизительно 125 мг до приблизительно 175 мг, от приблизительно 125 мг до приблизительно 150 мг, от приблизительно 150 мг до приблизительно 200 мг, от приблизительно 150 мг до приблизительно 175 мг или от приблизительно 175 мг до приблизительно 200 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от приблизительно 25 мг до приблизительно 125 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от приблизительно 75 мг до приблизительно 150 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве приблизительно 5 мг, приблизительно 10 мг, приблизительно 25 мг, приблизительно 35 мг, приблизительно 50 мг, приблизительно 65 мг, приблизительно 75 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг или приблизительно 200 мг в единичной лекарственной форме, или в количестве в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве приблизительно 50 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве приблизительно 100 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве приблизительно 25 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от 5 мг до 250 мг в единичной лекарственной форме. Например, они присутствуют в количестве от 5 мг до 175 мг, от 5 мг до 150 мг, от 5 мг до 125 мг, от 5 мг до 100 мг, от 5 мг до 75 мг, от 5 мг до 50 мг, от 5 мг до 25 мг, от 25 мг до 200 мг, от 25 мг до 175 мг, от 25 мг до 150 мг, от 25 мг до 125 мг, от 25 мг до 100 мг, от 25 мг до 75 мг, от 25 мг до 50 мг, от 50 мг до 200 мг, от 50 мг до 175 мг, от 50 мг до 150 мг, от 50 мг до 125 мг, от 50 мг до 100 мг, от 50 мг до 75 мг, от 75 мг до 200 мг, от 75 мг до 175 мг, от 75 мг до 150 мг, от 75 мг до 125 мг, от 75 мг до 100 мг, от 100 мг до 200 мг, от 100 мг до 175 мг, от 100 мг до 150 мг, от 100 мг до 125 мг, от 125 мг до 200 мг, от 125 мг до 175 мг, от 125 мг до 150 мг, от 150 мг до 200 мг, от 150 мг до 175 мг или от 175 мг до 200 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от 25 мг до 125 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от 75 мг до 150 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве 5 мг, 10 мг, 25 мг, 35 мг, 50 мг, 65 мг, 75 мг, 90 мг, 100 мг, 125 мг, 150 мг, 175 мг или 200 мг в единичной лекарственной форме, или в количестве в пределах диапазона, определенного любыми из предыдущих значений. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве 50 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве 100 мг в единичной лекарственной форме. Согласно некоторым вариантам осуществления фармацевтические композиции по настоящему раскрытию составляют в виде таблетки. Согласно некоторым вариантам осуществления таблетка покрыта оболочкой. Согласно некоторым вариантам осуществления фармацевтические композиции по настоящему раскрытию составляют в виде капсул. Согласно некоторым вариантам осуществления фармацевтические композиции находятся в форме саше. Согласно некоторым вариантам осуществления фармацевтические композиции находятся в форме гранул.
Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в дозе приблизительно 25 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в дозе приблизительно 50 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в дозе приблизительно 75 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в дозе приблизительно 100 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическую композицию вводят в дозе приблизительно 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления фармацевтическую композицию вводят в дозе приблизительно 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическую композицию вводят в дозе приблизительно 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическую композицию вводят в дозе приблизительно 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления фармацевтическая композиция содержит приблизительно 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Суточная доза соединения формулы (I) или его фармацевтически приемлемой соли в фармацевтической композиции, как описано в настоящем раскрытии, может варьироваться в широком диапазоне от приблизительно 1,0 мг до приблизительно 10000 мг на взрослого человека в сутки, или выше, или в любом диапазоне в пределах указанных значений. В случае перорального введения, композиции могут быть представлены в форме таблеток, содержащих, например, приблизительно 0,01 мг, приблизительно 0,05 мг, приблизительно 0,1 мг, приблизительно 0,5 мг, приблизительно 1,0 мг, приблизительно 2,5 мг, приблизительно 5,0 мг, приблизительно 10,0 мг, приблизительно 15,0 мг, приблизительно 25,0 мг, приблизительно 50,0 мг, приблизительно 75,0 мг, приблизительно 100 мг, приблизительно 150 мг, приблизительно 200 мг, приблизительно 250 или приблизительно 500 миллиграммов соединения формулы (I) или его фармацевтически приемлемой соли, для симптоматической корректировки дозировки для подлежащего лечению пациента. Согласно некоторым вариантам осуществления эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли может быть доставлено при уровне дозы от приблизительно 0,1 мг/кг до приблизительно 1000 мг/кг веса тела в сутки, или при уровне дозы в любом диапазоне указанных значений, например, диапазон может составлять от приблизительно 0,5 мг/кг до приблизительно 500 мг/кг, от приблизительно 1,0 мг/кг до приблизительно 250 мг/кг, от приблизительно 0,1 мг/кг до приблизительно 100 мг/кг, от приблизительно 0,1 мг/кг до приблизительно 50,0 мг/кг веса тела в сутки, от приблизительно 0,1 мг/кг до приблизительно 15,0 мг/кг веса тела в сутки, от приблизительно 0,5 мг/кг до приблизительно 7,5 мг/кг веса тела в сутки, или любое количество в диапазоне указанных значений. Согласно некоторым вариантам осуществления эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли может быть доставлено при уровне дозы от 0,1 мг/кг до 1000 мг/кг веса тела в сутки, или при уровне дозы в любом диапазоне указанных значений, например, диапазон может составлять от 0,5 мг/кг до 500 мг/кг, от 1,0 мг/кг до 250 мг/кг, от 0,1 мг/кг до 100 мг/кг, от 0,1 мг/кг до 50,0 мг/кг веса тела в сутки, от 0,1 мг/кг до 15,0 мг/кг веса тела в сутки, от 0,5 мг/кг до 7,5 мг/кг веса тела в сутки, или любое количество в диапазоне указанных значений. Представленную в настоящем документе фармацевтическую композицию можно вводить по схеме 1-4 раза в сутки или в разовой суточной дозе.
Согласно некоторым вариантам осуществления суточная доза соединения формулы (I) или его фармацевтически приемлемой соли составляет приблизительно 25 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза соединения формулы (I) или его фармацевтически приемлемой соли составляет приблизительно 50 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза соединения формулы (I) или его фармацевтически приемлемой соли составляет приблизительно 75 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза соединения формулы (I) или его фармацевтически приемлемой соли составляет приблизительно 100 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза соединения формулы (I) или его фармацевтически приемлемой соли составляет приблизительно 150 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза соединения формулы (I) или его фармацевтически приемлемой соли составляет приблизительно 200 мг, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят два раза в сутки в дозе приблизительно 25 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят два раза в сутки в дозе приблизительно 50 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят два раза в сутки в дозе приблизительно 75 мг, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят два раза в сутки в дозе приблизительно 100 мг, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления суточная доза фармацевтической композиции составляет приблизительно 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза фармацевтической композиции составляет приблизительно 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза фармацевтической композиции составляет приблизительно 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза фармацевтической композиции составляет приблизительно 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза фармацевтической композиции составляет приблизительно 150 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления суточная доза фармацевтической композиции составляет приблизительно 200 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления фармацевтическую композицию вводят два раза в сутки в дозе приблизительно 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическую композицию вводят два раза в сутки в дозе приблизительно 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят два раза в сутки в дозе приблизительно 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления фармацевтическую композицию вводят два раза в сутки в дозе приблизительно 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления способ предусматривает введение суточной дозы фармацевтической композиции, содержащей приблизительно 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение суточной дозы фармацевтической композиции, содержащей приблизительно 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение суточной дозы фармацевтической композиции, содержащей приблизительно 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение суточной дозы фармацевтической композиции, содержащей приблизительно 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение суточной дозы фармацевтической композиции, содержащей приблизительно 150 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение суточной дозы фармацевтической композиции, содержащей приблизительно 200 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
Согласно некоторым вариантам осуществления способ предусматривает введение фармацевтической композиции два раза в сутки в дозе приблизительно 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение фармацевтической композиции два раза в сутки в дозе приблизительно 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение соединения формулы (I) или его фармацевтически приемлемой соли два раза в сутки в дозе приблизительно 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания. Согласно некоторым вариантам осуществления способ предусматривает введение фармацевтической композиции два раза в сутки в дозе приблизительно 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
Факторы, связанные с конкретным подлежащим лечению субъектом, включая возраст субъекта, вес., рацион и время введения, могут обуславливать необходимость корректировки дозировок. Согласно некоторым вариантам осуществления субъектом является взрослый человек. Согласно некоторым вариантам осуществления субъектом является педиатрический пациент.
Специалисту в данной области будет понятно, что клинические исследования, как in vivo, так и in vitro, с применением подходящих известных и общепринятых клеточных и/или животных моделей позволяют прогнозировать способность тестируемого соединения лечить или предупреждать данное нарушение. Специалисту в данной области также будет понятно, что клинические исследования с участием людей, включая испытания, предусматривающие первое применение препарата у людей, исследования с целью определения оптимальной дозы и исследования эффективности с участием здоровых субъектов и/или субъектов, страдающих от данного нарушения, можно проводить в соответствии со способами, хорошо известными в области клинических исследований и медицины. Например, надлежащие дозировки для педиатрических пациентов можно определять с применением известных способов, учитывающих вес., возраст и предусматривающих такие модели, как Simcyp® Pediatric Simulator (CERTARA, Принстон, Нью-Джерси), которую можно применять для создания фармакокинетического подхода к дозированию, в котором учитываются возраст субъекта, онтогенез путей выведения соединения формулы (I) или его фармацевтически приемлемой соли и площадь поверхности тела (BSA).
Согласно некоторым вариантам осуществления фармацевтические композиции по настоящему раскрытию остаются стабильными в течение по меньшей мере 3 месяцев. Согласно некоторым вариантам осуществления фармацевтические композиции остаются стабильными в течение по меньшей мере 6 месяцев. Согласно некоторым вариантам осуществления фармацевтические композиции остаются стабильными в течение по меньшей мере 9 месяцев. Согласно некоторым вариантам осуществления фармацевтические композиции остаются стабильными в течение по меньшей мере 12 месяцев. Например, композиции не проявляют изменений (например, более чем на 5%) в отношении внешнего вида, рН, процента примесей, активности (как измерено с помощью анализов in vitro) или осмолярности с течением времени, например, спустя по меньшей мере 3 месяца, 6 месяцев, 9 месяцев или по меньшей мере 12 месяцев по сравнению с исходной композицией после ее производства. Согласно некоторым вариантам осуществления фармацевтические композиции не проявляют значимых изменений, как определено Международной конференцией по гармонизации технических требований к регистрации лекарственных препаратов для человека (ICH), в отношении одного или нескольких из внешнего вида, рН, процента примесей, активности (как измерено с помощью анализов in vitro) или осмолярности с течением времени, например, спустя по меньшей мере 12 месяцев по сравнению с исходной композицией после ее производства.
Наборы
Также представлены наборы. Как правило, набор включает одну или несколько фармацевтических композиций, как описано в настоящем документе, например, фармацевтическую композицию, содержащую, например, высушенную распылением дисперсию, как описано в примерах 1-4, или состав, описанный в примере 9. Согласно определенным вариантам осуществления набор может включать одну или несколько систем доставки, например, для доставки или введения фармацевтической композиции, как представлено в настоящем документе, и указания для применения набора (например, инструкции для лечения субъекта). Согласно некоторым вариантам осуществления набор может включать фармацевтическую композицию, как описано в настоящем документе, и информацию о лекарственном препарате, в которой указаны количества, которые следует вводить субъекту с врожденной гиперплазией надпочечников. Фактическая доза соединения формулы (I) или его фармацевтически приемлемой соли, представленных в настоящем документе, зависит от конкретного состава, веса пациента и подлежащего лечению состояния.
Примеры
Пример 1. Составы на основе высушенной распылением дисперсии, содержащие соединение формулы (I) и различные полимеры.
Составы на основе высушенной распылением дисперсии
Получали серию составов на основе высушенной распылением дисперсии (SDD), содержащих соединение формулы (I) и полимер. SDD-составы включали: (1) 10% соединения формулы (1)/90% гидроксипропилметилцеллюлозы ацетата сукцината-L (HPMCAS-L); (2) 25% соединения формулы (I)/75% HPMCAS-L; (3) 40% соединения формулы (I)/60% HPMCAS-L; (4) 25% соединения формулы (1)/75% сополимера винилпирролидона и винилацетата 64 (PVP/VA 64); (5) 25% соединения формулы (Г/60% Cabosil (коллоидный диоксид кремния/15% HPMCAS-L; (6) 25% соединения формулы (1/75% HPMCAS-M; и (7) 25% соединения формулы (1/75% метилметакрилатного сополимера (1:1) (Eudragit® L100).
Полимер PVP/VA представлял собой сополимер 1-винил-2-пирролидона и винилацетата с соотношением 60:40 по весу 1-винил-2-пирролидон:винилацетат со средней молекулярной массой 45000-70000 (коповидон, продаваемый как Kollidon® VA 64, BASF, Флорхем Парк, Нью-Джерси). HPMCAS представлял собой смесь уксусной кислоты и сложных моноэфиров янтарной кислоты и гидроксипропилметилцеллюлозы, которая относилась либо к степени чистоты L (HPMCAS-L), с содержанием ацетила 5-9%, содержанием сукциноила 14-18%, содержанием метоксила 20-24% и содержанием гидроксипропокси 5-9% (продаваемый Shin-Etsu, Япония); либо к степени чистоты М (HPMCAS-M), с содержанием ацетила 7-11%, содержанием сукциноила 10-14%, содержанием метоксила 21-25% и содержанием гидроксипропокси 5-9% (продаваемый Shin-Etsu, Япония).
Характеристики растворения
Тестировали характеристики растворения нескольких из описанных выше SDD-составов (см. фиг. 1). 1000 мкгА/мл каждого SDD тестировали в 0,5% вес. искусственном кишечном соке (SIF) в PBS, рН 6,5. Образцы тестировали на 5, 10, 20, 45, 90 и 1200 минуте. Липидный состав, содержащий 10% соединения формулы (I), применяли в качестве контроля. Результаты показаны в таблице 4 ниже.

Растворение в насыщаемых условиях
Осуществляли анализ удельной проницаемости мембраны (см., например, Stewart et al., Mol. Pharm. (2017) 14:2032-2046), и данные по растворению в насыщаемых условиях получали для нескольких из описанных выше SDD-составов и сравнивали с данными для соединения формулы (I) и нескольких эталонных составов, включая полутвердый липидный состав (эталонный состав 1) и два состава на основе самоэмульгирующейся системы лекарственной доставки (SEDDS) (эталонные составы 2 и 3). Компоненты эталонных составов показаны в таблице 5 ниже и включают, помимо соединения формулы (I), каприловый/каприновый триглицерид (Labrafac® Lipophile, Gattefosse, Франция); пропиленгликольдикаприлат/дикапрат (Labrafac® PG, Gattefosse, Франция); олеоилполиоксил-6-глицериды (Labrafil® M 1944 CS, Gattefosse, Франция); полисорбат 20; полиоксил касторовое масло (Kolliphor® RH 40, BASF, Германия); полиоксил 15 гидроксистеарат (Kolliphor® HS 15, BASF, Германия); лауроилполиоксил-32-глицериды (Gelucire® 44/14, Gattefosse, Франция); d-α-токоферол полиэтиленгликоль 1000 сукцинат (TPGS); и моноэтиловый эфир диэтиленгликоля (Transcutol®, Gattefosse, Франция).
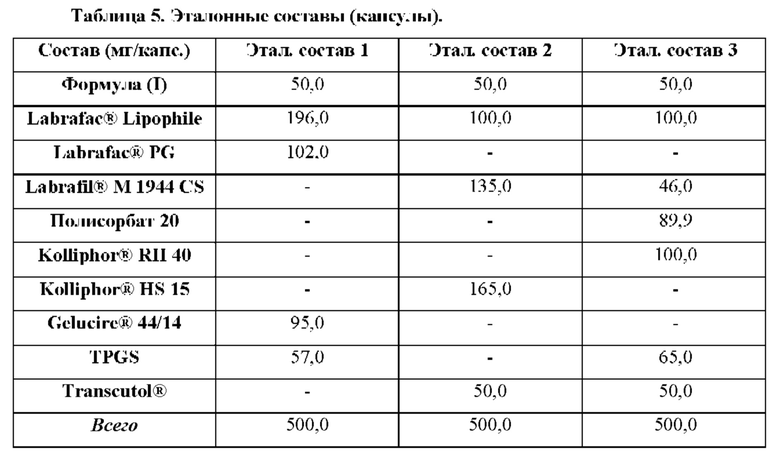
В анализе измеряли удельную проницаемость через искусственные стенки желудка и кишечника посредством УФ-спектроскопии (jiDiss ProfilerTM, Pion Inc., Биллерика, Массачусетс). Вкратце, анализ осуществляли следующим образом. Ячейку для анализа вертикальной удельной проницаемости мембраны, состоящую из компартмента-донора и компартмента-приемника и разделенную полипропиленовой мембраной Accurel РР 1Е (55% пористости, толщина 100 мкм) (3 М, Мейплвуд, Миннесота) (фиг. 2), наполняли 50 мкл липидного раствора Pion GIT-0, состоящего из 20% вес./вес. фосфол ипида, растворенного в додекане (Pion Inc., Биллерика, Массачусетс), и присоединяли к принимающему сосуду. Содержимое как компартмента-донора, так и компартмента-приемника перемешивали с помощью магнитной мешалки. Компартмент-приемник содержал пластиковый разделитель и решетку для подъема якоря мешалки над мембраной. Образцы вводили в донорный сосуд путем предварительного отвешивания непосредственно в донорный сосуд с последующим добавлением среды растворения. После добавления среды растворения в донорный сосуд, принимающий сосуд вставляли в донорный сосуд и суспендировали вертикально на 5 мм выше компартмента-донора с помощью полиэтиленовой перчатки. Для этого анализа искусственная желудочная среда (подаваемый материал) представляла собой 0,1 N HCl, рН 2, и включала 200 мкгА/мл каждого SDD, а искусственная кишечная среда (принимающий материал) представляла собой 0,5% вес. SIF в PBS, рН 6,5, и включала 100 мкгА/мл каждого SDD. Температура для анализа поддерживалась на уровне 44,5°С. УФ-зонды (длина оптического пути 10 мм), соединенные со спектрометром УФ Rainbow (Pion Inc.), применяли для определения эффективной концентрации лекарственного средства в принимающих сосудах. Образцы компартмента-донора отбирали с помощью пипетки одноразового пользования для центрифугирования с последующим HPLC- и DLS-анализом супернатанта. Результаты показаны на фиг. 3 и в таблице 6 ниже.
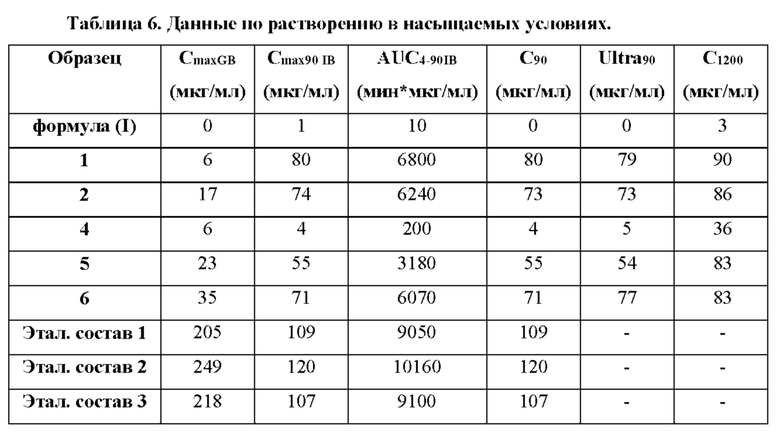
Также определяли удельную проницаемость мембраны в случае 1 мг/мл желудочного барьера/кишечного барьера (GB/IB) 0,5% вес. SIF доз соединения формулы (I) и высушенных распылением дисперсий (2) с 25% соединения формулы (I)/75% HPMCAS-L и (4) 25% соединения формулы (I)/75% PVP/VA 64. Результаты показаны на фиг. 4 в виде концентрации в приемнике в зависимости от времени и удельной проницаемости в зависимости от времени (сглаженная производная от концентрации в приемнике × объем/площадь поверхности).
Пример 2. Определение характеристик высушенной распылением дисперсии, содержащей 25% соединения формулы (I) и 75% сополимера винилпирролидона и винилацетата (PVP/VA).
Скрининг стабильности SDD
Несколько из описанных в примере 1 SDD тестировали на предмет химической и физической стабильности. Осуществляли исследования стабильности SDD с остаточным растворителем, при этом образцы хранили как при 5°С, так и при 25°С. Измерения проводили спустя 1 и 2 недели хранения. Результаты показаны в таблице 7 ниже. Столбец с временем удержания 32,36 мин соответствует соединению формулы (I).

Также осуществляли исследования стабильности раствора, при этом образцы хранили как при 5°С, так и при 25°С. Измерения проводили спустя 1 и 2 недели хранения. Результаты показаны в таблице 8 ниже. Столбец с временем удержания 32,36 мин соответствует соединению формулы (I).

Исследования стабильности также осуществляли для SDD, содержащей 25% соединения формулы (I) и 75% PVP/VA 64, при этом образцы хранили при 5°С (в закрытом виде, с влагопоглотителем), 25°С (60% RH, в закрытом виде, с влагопоглотителем) и 30°С (65% RH, в закрытом виде, с влагопоглотителем). Измерения проводили спустя 1 месяц, 2 месяца, 3 месяца, 6 месяцев и 12 месяцев хранения. Спустя 12 месяцев хранения изменений в чистоте не наблюдали. Результаты показаны в таблице 9 ниже. Столбец с временем удержания 30,2 мин соответствует соединению формулы (I).
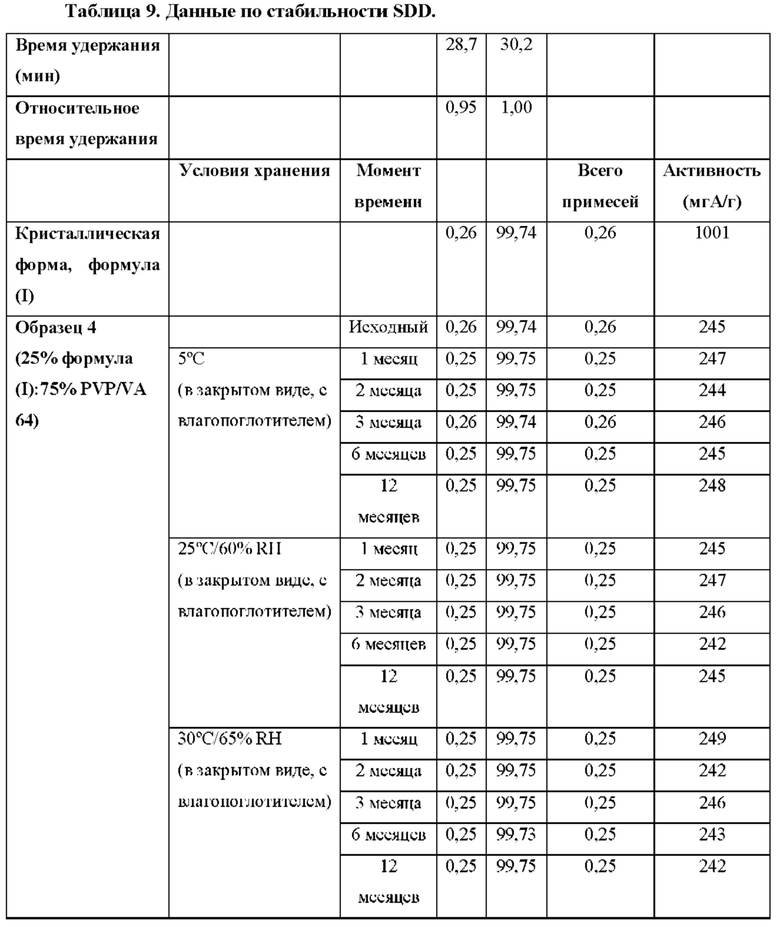
В то время как для образцов 1 и 2 наблюдали разрушение спустя приблизительно 2 недели хранения, было обнаружено, что SDD, содержащая 25% соединения формулы (I) и 75% PVP/VA 64 (образец 4), была как химически, так и физически стабильной, и ее дополнительно подвергали скринингу и определению характеристик, как описано ниже.
Параметры 1 раунда скрининга процесса производства SDD с 25% соединения формулы ф/75% PVP/VA 64
SDD с 25% соединения формулы (I)/75% PVP/VA 64 получали в распылительной сушилке для фармацевтических составов с газопроизводительностью для сушильного газа 100 кг/ч (PSD-1). Краткое описание производства показано в таблице 10 ниже.
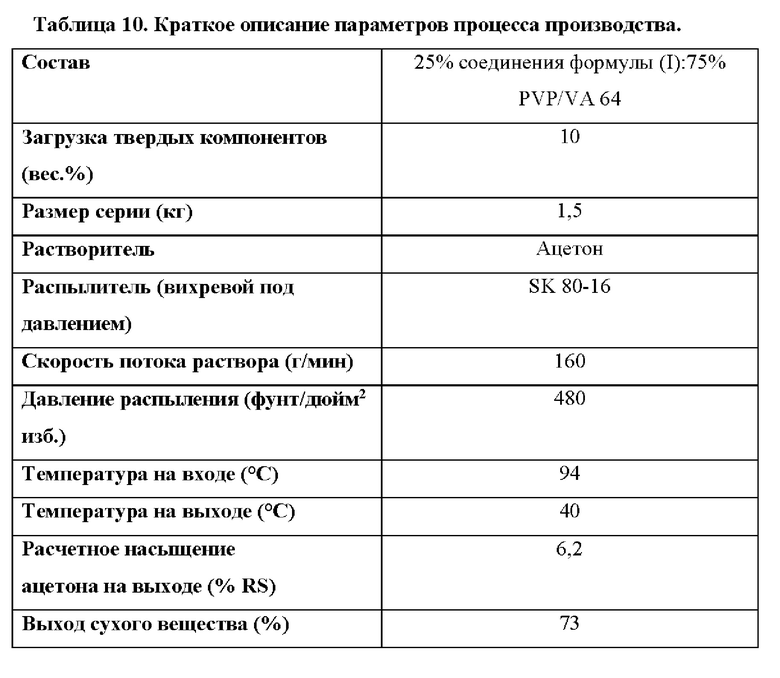
Исходя из 73% выхода, наблюдаемого в первом раунде скрининга процесса, было выполнено три распыления для исследования эффекта снижения производительности по раствору и температуры на выходе в отношении выхода продукта. Все процедуры распыления проводили при сниженной скорости потока 110 г/мин. Температура на выходе варьировалась со значениями 40°С (партия А), 35°С (партия В) и 30°С (партия С). Температуру на выходе уменьшали с поддержанием низкого насыщения ацетона на выходе для повышения разницы между температурой камеры на выходе и Tg SDD с остаточным растворителем, таким образом повышая выход продукта. Камеру распылительной сушилки и систему выходных каналов очищали после каждой производственной процедуры. Краткое описание производства показано в таблице 11.
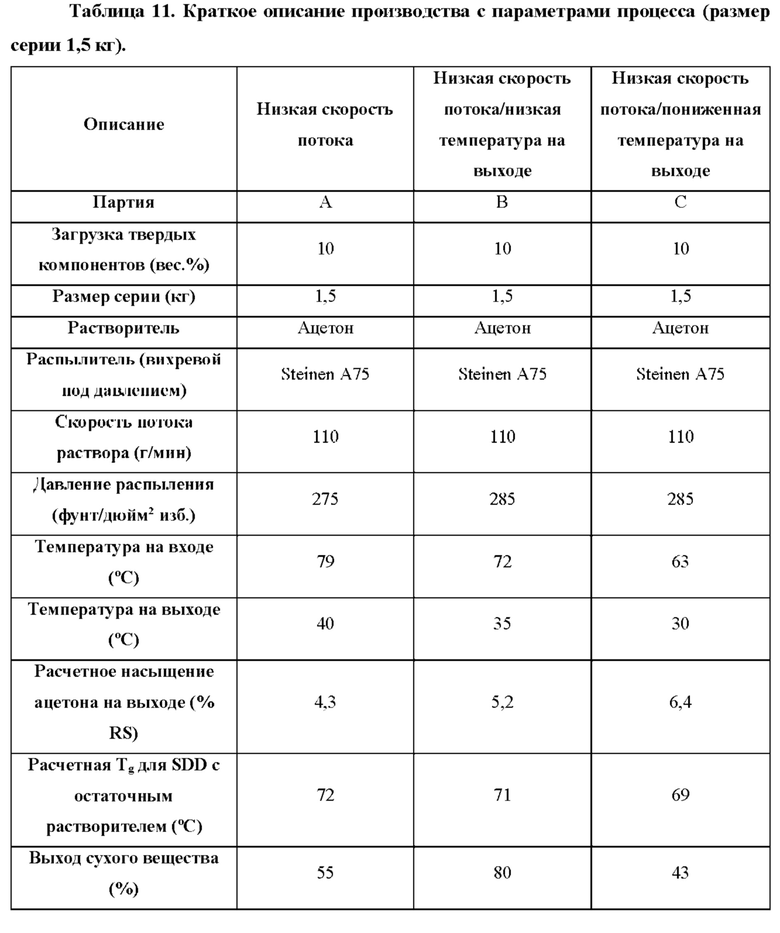
Было обнаружено, что условия, применяемые для партии В, обеспечивают наиболее высокий выход. Затем осуществляли одно дополнительное распыление при тех же условиях обработки, что и в случае партии В, с увеличением при этом размера серии с 1,5 кг до 3,5 кг для оценки согласованности процесса и для определения того, будет ли выход продукта повышаться с течением времени. Усредненные условия процесса для такой партии показаны в таблице 12.
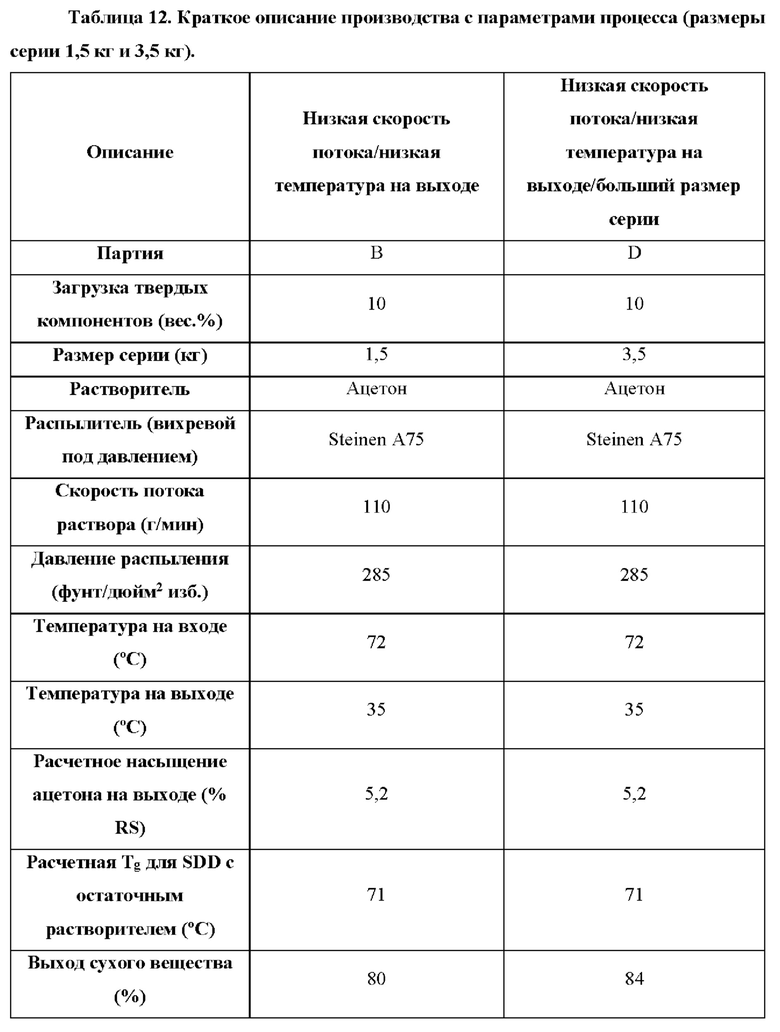
Размер серии 1,5 кг (партия D) распылялся с 84% выходом в сравнении с 80% выходом для 3,5 кг серии (партия В).
Определение характеристик скрининга процесса получения SDD с 25% соединения формулы (I)/75% PVP/VA 64 SDD с 25% соединения формулы (I)/75% PVP/VA 64, полученные для оценки параметров процесса, характеризовали в отношении свойств порошка, внешнего вида и физических и химических свойств. Тестирование включало оценку распределения частиц по размеру с помощью Malvern, определение насыпной плотности без утряски и после утряски, оценку растворения при использовании микроцентрифуги, модулированную дифференциальную сканирующую калориметрию (mDSC), порошковую рентгеновскую дифракцию (PXRD), сканирующую электронную микроскопию (SEM) и анализ посторонних примесей. Из результатов не видно какой-либо значимой разницы между партиями.
Распределение частиц по размеру (PSD) и табличные данные по свойствам порошка для SDD с 25% соединения формулы (I)/75% PVP/VA 64 показаны в таблице 13. Все SDD с 25% соединения формулы (I)/75% PVP/VA 64 характеризовались очень схожим PSD с D50 примерно 16 мкм. Все SDD с 25% соединения формулы (I)/75% PVP/VA 64 характеризовались низкими показателями насыпной плотности без утряски и после утряски.

Партию с размером загрузки 3,5 кг анализировали и сравнивали с партией А для оценки параметров процесса. Характеристики растворения были схожими для каждой из этих партий. Растворение происходило быстро с достижением Cmax и высокое содержание лекарственного средства в свободном виде сохранялось в течение 90 минут. Эти данные показаны в таблице 14.
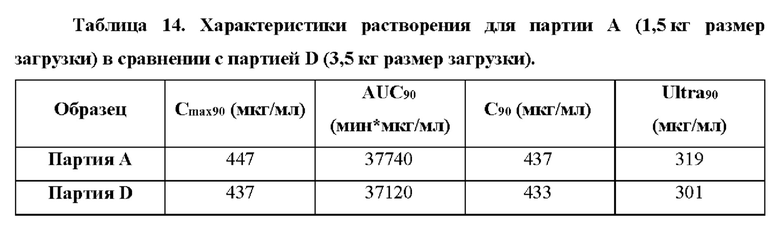
SDD с 25% соединения формулы (I)/75% PVP/VA 64 также оценивали с помощью DSC, PXRD и SEM. На DSC-термограммах показана единая Tg при 84°С, что указывает на однородность дисперсий. Из PXRD-дифрактограмм видно, что признаки наличия кристаллов в SDD отсутствуют. На SEM-изображениях представлена морфология раздутой сферы с некоторым количеством разрушенных частиц и некоторым количеством очень маленьких частиц.
Осуществляли дополнительное тестирование в отношении партии В, которое включало оценку химической/физической стабильности как распыляемого раствора, так и SDD до вторичного высушивания (SDD с остаточным растворителем) для установления максимального времени пребывания в процессе получения. Также оценивали концентрацию остаточного ацетона в зависимости от продолжительности вторичного высушивания в конвекционной полочной сушилке для назначения условий для сушки на полках, чтобы обеспечить сушку SDD в соответствии с рекомендациями Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для человека (ICH) для ацетона.
Содержание остаточного ацетона в зависимости от продолжительности сушки оценивали путем высушивания SDD с остаточным растворителем в полочной сушилке и сбора образцов в 24-часовом промежутке времени. SDD с остаточным растворителем сушили при 40°С/15% относительной влажности (RH), и высушивание с достижением показателей для ацетона согласно ЮН (0,5% вес, 5000 ppm) наблюдали через четыре часа.
Допустимое время хранения раствора для распыления определяли путем приготовления типичного раствора, который содержал 2,5% вес. соединения формулы (I), 7,5% вес. PVP/VA 64 и 90% вес. ацетона. Эти растворы изначально анализировали в отношении посторонних примесей, а затем выдерживали при 5°С и 25°С.Брали аликвоты и периодически анализировали в отношении посторонних примесей в течение 14 суток. Из результатов видно, что качественное и количественное содержание примесей не изменялось ни при каких условиях на протяжении 14 суток.
SDD с остаточным растворителем анализировали в отношении примесей после хранения при 5°С и 25°С в течение 1 и 2 недель и сравнивали с качественным и количественным содержанием примесей исходного соединения формулы (I) и SDD, которую подвергали вторичной сушке после высушивания распылением. Качественное и количественное содержание примесей было схожим с таковым изначального высушенного образца и исходного соединения формулы (I) на протяжении 2 недель хранения.
Образцы для проверки стабильности SDD с остаточным растворителем характеризовали в отношении физической стабильности с помощью DSC, PXRD и SEM. На DSC-термограммах показана единая Tg при 81°С, что указывает на однородность дисперсии без разделения на фазы. Из PXRD-дифрактограмм видно, что признаки наличия кристаллов после хранения в любых условиях отсутствуют. На SEM-изображениях представлена типичная морфология по большей части раздутой сферы с некоторым количеством разрушенных частиц.
Пример 3. Получение 1000 г серии высушенной распылением дисперсии, содержащей 25% соединения формулы (I) и 75% PVP/VA 64 1000 г серия высушенной распылением дисперсии, содержащей 25% соединения формулы (I) и 75% PVP/VA 64, получали, как описано в примере 2 для серий 1,5 кг и 3,5 кг. Вкратце, ацетон (90% (вес./вес.) от общей смеси) добавляли в емкость для смешивания с последующим добавлением 250,0 г соединения формулы (I) (2,5% (вес/вес.) от общей смеси). Смесь перемешивали в течение 30 минут в темноте при диапазоне температур от 15°С до 27°С. По окончании периода перемешивания раствор был прозрачным и не содержал нерастворенных твердых веществ. Затем добавляли PVP/VA 64 (750,0 г, 7,5% (вес./вес.) от общей смеси), и смесь перемешивали в течение еще 30 минут в темноте при диапазоне температур от 15°С до 27°С. По окончании периода перемешивания раствор был прозрачным и не содержал нерастворенных твердых веществ.
Раствор подавался под давлением и распылялся в сушильной камере. Высушенные распылением дисперсии получали в распылительной сушилке для фармацевтических составов с газопроизводительностью для сушильного газа 100 кг/ч (PSD-1). Температура на входе была установлена на 75°С (изменялась в диапазоне 60°С-90°С). Температура на выходе была установлена на 35°С (изменялась в диапазоне 32°С-38°С). Давление подачи было установлено на 280 фунт/дюйм2 изб. (изменялось в диапазоне 230-330 фунт/дюйм2 изб.). Скорость подачи была установлена на ПО г/мин (изменялась в диапазоне 90-130 г/мин). Высушенный распылением порошок затем сушили в конвекционной полочной сушилке с толщиной слоя ≤2,5 см при 40°С (±5°С) и 15% относительной влажности (±10%) в течение 24 часов под желтым светом. Остаточный ацетон после высушивания составлял<0,5% вес. (5000 ppm). Фиг. 5 представляет собой схему процесса производства.
Пример 4. Получение составов на основе соединения формулы (I) в виде высушенной распылением дисперсии для клинического применения.
Высушенную распылением дисперсию (SDD), содержащую 25% соединения формулы (I) и 75% PVP/VA 64, полученную, как описано выше, составляли в виде суспензии или капсулы для клинического применения.
Получение суспензии
Суспензию, которая содержала 50 мг SDD, получали следующим образом. Бутылку из темного стекла для дозирования на 30 мл взвешивали на весах. Затем в бутылку для дозирования отвешивали 200,0 мг SDD (50 мгА)±5%. С применением шприца на 10 мл, в бутылку для дозирования добавляли 5,0 мл воды (очищенной, USP) и закрывали бутылку, и аккуратно встряхивали в течение 30 секунд. SDD-суспензию хранили во флаконе из темного стекла при 2-8°С до применения и вводили в пределах 24 часов после получения.
Получение капсул
Пустую твердую желатиновую капсулу, размер 0 (Capsugel, Морристаун, Нью-Джерси), помещали на весы и регистрировали вес. Затем на бумаге для взвешивания или ее эквиваленте взвешивали 200,0 мг SDD (50 мгА)±5%. Все содержимое переносили в капсулу с применением прибора ProFunnel для капсул размера 0. Заполненную капсулу помещали на весы и регистрировали вес. Вес пустой капсулы вычитали из веса с загрузкой, обеспечивая, чтобы вес SDD внутри капсулы составлял 200,0 мг SDD±5% или от 190,0 мг до 210,0 мг. Капсулу плотно закрывали крышечкой, убедившись, что она встала на свое место. Капсулы хранили во флаконе из темного стекла при 2-8°С до применения и вводили в пределах 24 часов после получения.
Пример 5. Исследования относительной биодоступности и влияния пищи на собаках.
Получали четыре высушенные распылением дисперсии (SDD), составленные в 0,25% метилцеллюлозе в виде суспензии: (1) 25% соединения формулы (I)/75% HPMCAS-L; (2) 10% соединения формулы (I)/90% HPMCAS-L; (3) 25% соединения формулы (I)/75% метилметакрилатного сополимера (1:1) (Eudragit® L100); и (4) 25% соединения формулы (I)/75% PVP/VA 64. Состав для капсулы для клинического применения получали в виде эталонного состава (эталонный состав 1 из таблицы 5 выше).
Собакам (две когорты, шесть собак в каждой) вводили дозу за шесть сессий, включая сессии натощак и сессии в сытом состоянии (рацион с высоким содержанием жиров), с использованием 50 мг дозы одной из SDD или эталонного состава на собаку в рамках перекрестного дизайна с 3 факторами. Между каждой сессией имел место 3-суточный период вымывания. Все составы переносились хорошо. Дизайн исследования показан в таблице 15 ниже.
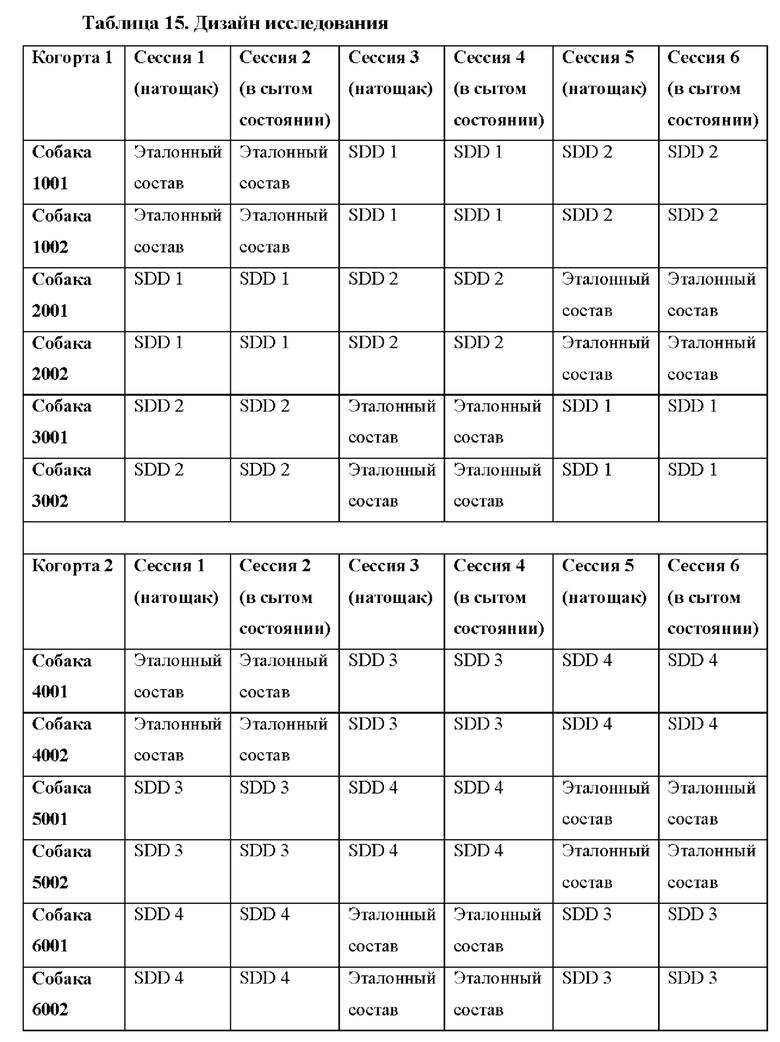
Рассчитывали площадь под кривой зависимости концентрации в плазме крови от времени для 0 часов, экстраполированную до бесконечности (AUC0-∞), максимальную концентрацию в плазме крови (Cmax), кажущийся период полувыведения в конечной фазе (t1/2) и время достижения максимальной концентрации в плазме крови (tmax). Результаты показаны в таблице 16 ниже и на фигурах 6А и 6В.

Из результатов видно, что t1/2 и tmax были схожими среди всех составов и сопоставимыми между введением в сытом состоянии и натощак. При введении в сытом состоянии, в случае пищи высоким содержанием жиров показатели экспозиции повышались, а вариабельность результатов между животными уменьшалась. Влияние пищи было более заметным в случае составов на основе высушенной распылением дисперсии, особенно для максимальной экспозиции (Cmax).
В сравнении с эталонной формой, SDD 4 (25% соединения формулы (I)/75% PVP/VA 64), по-видимому, характеризовалась более низкой вариабельность результатов между животными, более низкими показателями экспозиции при введении натощак и несколько более низкой Cmax, но относительно сопоставимой AUC при введении в сытом состоянии.
Пример 6. Исследование 1 фазы для оценки фармакокинетических показателей, влияния пищи на фармакокинетические показатели и безопасности соединения формулы (I) у здоровых взрослых субъектов.
Это исследование было разработано для оценки фармакокинетических показателей (РК) соединения формулы (I), а также для оценки влияния сытого состояния на РК соединения формулы (I). Для данного исследования была выбрана доза 50 мг доза, поскольку она находилась в пределах тестированного диапазона доз в завешенных испытаниях фазы 1 и фазы 2 и хорошо переносилась в этих исследованиях. Цели исследования были следующими: оценить РК 50 мг соединения формулы (I) у здоровых взрослых субъектов; оценить влияние пищи на РК 50 мг соединения формулы (I); и оценить безопасность и переносимость 50 мг соединения формулы (I).
Дизайн исследования
Исследование представляло собой открытое рандомизированное перекрестное исследование 1 фазы с 2 периодами для оценки РК и влияния пищи на РК соединения формулы (I) у 16 здоровых взрослых субъектов мужского и женского пола в возрасте от 18 до 55 лет.
После предоставления информированного согласия субъекты проходили скрининг в отношении соответствия критериям для участия в исследовании за 28 суток до 1-х суток периода лечения 1. Соответствующих критериям включения в исследование субъектов принимали в клинике на -1-е сутки и рандомизировали их в соответствии с 1 из 2 последовательностей лечения (16 субъектов [8 лиц мужского пола и 8 лиц женского пола]; см. таблицу 17 ниже). На 1-е сутки каждого периода лечения субъекты получали однократную дозу 50 мг соединения формулы (I) натощак или в сытом состоянии. Интервал между дозами составлял 21 сутки.
Субъектам необходимо было не принимать пищу по меньшей мере 4 часа до регистрации на -1-е сутки. При введении натощак, субъектам необходимо было не принимать пищу в течение ночи на протяжении по меньшей мере 10 часов до введения дозы и не принимать пищу в течение еще 4 часов после введения дозы. При введении в сытом состоянии, субъектам необходимо было не принимать пищу в течение ночи на протяжении по меньшей мере 10 часов, а затем принимать жидкую пищевую добавку с исследуемым лекарственным средством (жидкая пищевая добавка употреблялась в пределах 30 минут) и не употреблять никакой другой пищи в течение 4 часов после введения дозы. В ходе обоих периодов лечения употребление воды не допускалось в течение 1 часа до введения дозы и до истечения 2 часов после введения дозы, за исключением воды/жидкой пищевой добавки, предусмотренных для введения дозы исследуемого лекарственного средства. В качестве жидкой пищевой добавки применяли Ensure Plus® со вкусом ванили.
На 1-е сутки каждого периода лечения субъектам вводили 50 мг дозу соединения формулы (I). Образцы крови брали для РК-анализа в 36-часовом промежутке времени в ходе пребывания в стационаре. Субъекты оставались в клинике в день введения дозы и выписывались на 2-е сутки каждого периода лечения после завершения всех требуемых процедур. Утром 8-х и 15-х суток каждого периода лечения субъекты возвращались в клинику и пребывали там в амбулаторных условиях для взятия образца крови для оценок РК и безопасности. На 21-е сутки периода лечения 1 субъекты приходили в центр и к вечеру 21-х суток у них завершались оценки, и они оставались в центре на ночь и на следующий день начинали 1 сутки периода лечения 2. Заключительный предусмотренный исследованием визит последующего наблюдения имел место на 22-е сутки периода лечения 2 (21±2 суток после введения дозы в рамках периода лечения 2) или при преждевременном прекращении испытания.
В ходе каждого периода лечения брали образцы крови для РК-анализ в пределах 45 минут до введения дозы и через примерно 30 минут и 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 16, 24, 36, 168, 336 и 504 часов после введения дозы.
Оценки безопасности (включая лабораторные тесты по клинической безопасности, измерения физиологических показателей, физикальные обследования и электрокардиограммы (ECG)) проводили во время исследования по графику. Нежелательные явления (АЕ) и применение сопутствующих лекарственных препаратов оценивали в ходе всего исследования. На фиг. 7 проиллюстрирован дизайн исследования.
Исследуемый препарат, доза и способ введения
Соединение формулы (I) было предоставлено в виде инкапсулированного липидного полутвердого вещества, содержащего 50 мг соединения формулы (I) в виде эквивалента свободного основания для перорального введения. Субъекты проглатывали одну капсулу с примерно 240 мл воды натощак. Субъекты проглатывали одну капсулу с жидкой пищевой добавкой (Ensure Plus® [емкость 237 мл]) с дополнительной водой в объеме до 120 мл в сытом состоянии.
Продолжительность лечения
Продолжительность участия в исследовании для каждого взрослого субъект составляла примерно 10 недель, включая до 28 дней скрининга, 2 дня введения дозы с интервалом 21 день и заключительный предусмотренный исследованием визит последующего наблюдения через 21 день после получения последней дозы исследуемого лекарственного средства в ходе периода лечения 2.
Критерии оценки Фармакокинетические показатели
Для соединения формулы (I) были рассчитаны следующие РК-параметры в плазме крови:
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 часов до последней измеряемой концентрации (AUC0-tlast)
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 до 24 часов (AUC0-24)
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 часов, экстраполированная до бесконечности (AUC0-∞)
- Максимальная концентрация в плазме крови (Cmax)
- Время достижения максимальной концентрации в плазме крови (tmax)
- Время задержки между моментом введения дозы и появлением измеряемого тестируемого препарата (Tlag)
- Кажущийся период полувыведения в конечной фазе (t1/2)
- Кажущаяся константа скорости элиминации в конечной фазе (λz)
- Кажущееся среднее время удержания препарата (MRT)
- Молярное соотношение по AUC гидроксилированного метаболита соединения формулы (I) и исходного соединения формулы (I) в качестве лекарственного средства
Следующие РК-параметры в плазме крови были рассчитаны только для соединения формулы (I):
- Кажущийся системный клиренс после перорального введения (CL/F)
- Кажущийся объем распределения в конечной фазе после перорального введения (Vz/F)
Безопасность
Безопасность оценивали в ходе всего исследования, и были включены следующие оценки:
- Нежелательные явления (АЕ)
- Клинико-лабораторные исследования (анализ крови, анализ на свертываемость крови, клиническая биохимия и анализ мочи)
- Измерения физиологических показателей (включая ортостатическое артериальное давление и частоту пульса)
- Физикальные обследования
- Электрокардиограммы в 12 отведениях (ECG)
Статистические методы
Фармакокинетические параметры рассчитывали с применением некомпартментных методов и сводили по состоянию (в сытом состоянии или натощак) с применением описательной статистики. Двусторонние 90% доверительные интервалы рассчитывали для соотношения в сытом состоянии в сравнении с состоянием натощак по AUC0-∞, AUC0-tlast и Cmax для соединения формулы (I) и гидроксилированного метаболита соединения формулы (I)-
Данные по безопасности сводили с использованием описательной статистики.
Фармакокинетические показатели Оценки фармакокинетических показателей
Образцы плазмы крови для оценки РК для анализов соединения формулы (I) и гидроксилированного метаболита соединения формулы (I) брали в следующие моменты времени в ходе каждого периода лечения:
- 1-е сутки: в пределах 45 минут до введения дозы и через примерно 30 минут и 1, 2, 3, 4, 5,6, 7, 8, 9, 10, 12 и 16 часов после введения дозы.
- 2-е сутки: через примерно 24 и 36 часов после введения дозы.
- 8-е сутки: через примерно 168 часов после введения дозы.
- 15-е сутки: через примерно 336 часов после введения дозы.
- Через примерно 504 часа после введения дозы (в случае периода лечения 1 этот образец брали утром по меньшей мере за 30 минут до взятия образца, который берется до введения дозы, на 1-е сутки периода лечения 2).
- Заключительный предусмотренный исследованием визит для субъектов, которые преждевременно прекратили испытания: 1 образец.
Образцы крови на 1-е сутки брали в пределах 5 минут от предусмотренных графиком моментов времени отбора образцов (отличных от таковых для образца, который берется до введения дозы). Образцы крови на 2-е, 8-е и 15-е сутки брали в пределах 2 часов от предусмотренного графиком момента времени отбора образцов. Для взятого на 504 час образца крови для периода лечения 2 было предусмотрено ±2-дневное окно. Образец для РК должен был быть взят у субъектов, которые преждевременно прекратили участие в испытании. Регистрировали точное время отбора образцов в часах и минутах.
Биоаналитические методы
Образцы плазмы крови анализировали в отношении соединения формулы (I) и его гидроксилированного метаболита при помощи inVentiv Health, Принстон, Нью-Джерси, с соблюдением требований Надлежащей лабораторной практики (GLP) и соответствующих типовых рабочих инструкций (SOP).
Концентрации соединения формулы (I) и гидроксилированного метаболита соединения формулы (I) определяли количественно в образцах плазмы крови в соответствии с валидированными методами с применением тандемной масс-спектрометрии в режиме детекции положительно заряженных ионов. Этот метод был валидирован для анализа соединения формулы (I) и гидроксилированного метаболита соединения формулы (I) в 25,0 мкл образцов плазмы крови человека с дикалиевой солью этилендиаминтетрауксусной кислоты (K2-EDTA) в диапазонах концентраций от 5,00 до 2500 нг/мл и от 0,500 до 250 нг/мл соответственно. Все результаты анализов находились в пределах приемлемых границ. Повторный анализ активных испытанных образцов (ISR) был успешно выполнен в этом исследовании для обоих аналитов.
Результаты
Результаты по фармакокинетике.
В исследование были включены восемь субъектов мужского пола и восемь субъектов женского пола. Средний возраст составлял 37,1 года (диапазон 21-55 лет). Большинство субъектов были европеоидами (93,8%) и латиноамериканцами (81,3%). Средний вес при скрининге составлял 160,28 фунтов (диапазон 102,0-222,2 фунтов) и средний BMI составлял 25,50 кг/м2 (диапазон 20,7-30,5 кг/м2). Рандомизация была сбалансирована по демографическим данным и исходным характеристикам.
Все 16 субъектов были включены в выборку для анализа безопасности. Ни один субъект не был исключен из выборки для анализа безопасности и ни у одного субъекта не были исключены из анализа его или ее РК-данные.
Профили средней концентрации в плазме крови в зависимости от времени для соединения формулы (I) при введении натощак и в сытом состоянии представлены на фигурах 8А и 8В соответственно. Соединение формулы (I) медленно абсорбировалось после перорального введения как натощак, так и в сытом состоянии. Средние концентрации в плазме крови были ниже при введении натощак, чем при введении в сытом состоянии.
РК-параметры для соединения формулы (I) после лечения соединением формулы (I) с введением натощак и в сытом состоянии приведены в таблице 18 ниже, где AUC0-24=площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 до 24 часов, AUC0-tlast=AUC с момента времени 0 часов до последней измеряемой концентрации, AUC0-∞=AUC с момента времени 0 часов, экстраполированная до бесконечности, CL/F=кажущийся системный клиренс после перорального введения, CV = коэффициент вариации, Cmax = максимальная концентрация в плазме крови, CV(%) = коэффициент вариации, max = максимальный, мин = минимальный, MRT = кажущееся среднее время удержания препарата, РК = фармакокинетический, SD = стандартное отклонение, t1/2=кажущийся период полувыведения в конечной фазе, Tlag = время задержки между моментом введения дозы и появлением измеряемого тестируемого препарата, tmax = время достижения максимальной концентрации в плазме крови, VZ/Т = кажущийся объем распределения в конечной фазе после перорального введения.
РК-данные для tmax, Tlag, t1/2, MRT и Vz/F округляли до 2 значащих цифр, а все другие параметры (AUC0-24, AUC0-tlast, AUC0-∞, Cmax и CL/F) округляли до 3 значащих цифр. Последняя значащая цифра округлялась в большую сторону, если цифра справа от нее была >5, и округлялась в меньшую сторону, если цифра справа от нее была <4.

Как видно из таблицы 18, выше, введение 50 мг соединения формулы (I) в сытом состоянии в сравнении с введением натощак обуславливало более высокую среднюю Cmax соединения формулы (I) (примерно в 2 раза выше; 1550 в сравнении с 731 нг/мл), большее t1/2 (42 в сравнении с 33 часами), несколько меньшее медианное tmax (5,0 в сравнении с 6,0 часами) и более высокую среднюю AUC0-∞ (примерно в 2 раза выше; 17800 в сравнении с 9440 нг × ч/мл). Средние геометрические соотношения соединения формулы (I) для Cmax и AUC0-tlast для введения в сытом состоянии в сравнении с таковым натощак составляли 218,6% и 215,2% соответственно, что указывает на то, что абсорбция соединения формулы (I) была в примерно 2 раза выше при введении с едой. Верхние и нижние пределы 90% доверительного интервала (CI) как для Cmax (187,4% и 255,1% соответственно), так и для AUC0-tlast (182,9% и 253,1% соответственно) были за пределами диапазона «отсутствия эффекта» от 80,00% до 125,00%, что указывает на то, что имело место влияние пищи на экспозицию соединения формулы (I).
Плотность распределения значений Tlag и tmax представлена в таблице 19 и таблице 20 соответственно. Спагетти-графики для AUC0-tlast, AUC0-∞ по и Cmax соединения формулы (I) показаны на фигурах 9А, 9В и 9С соответственно.
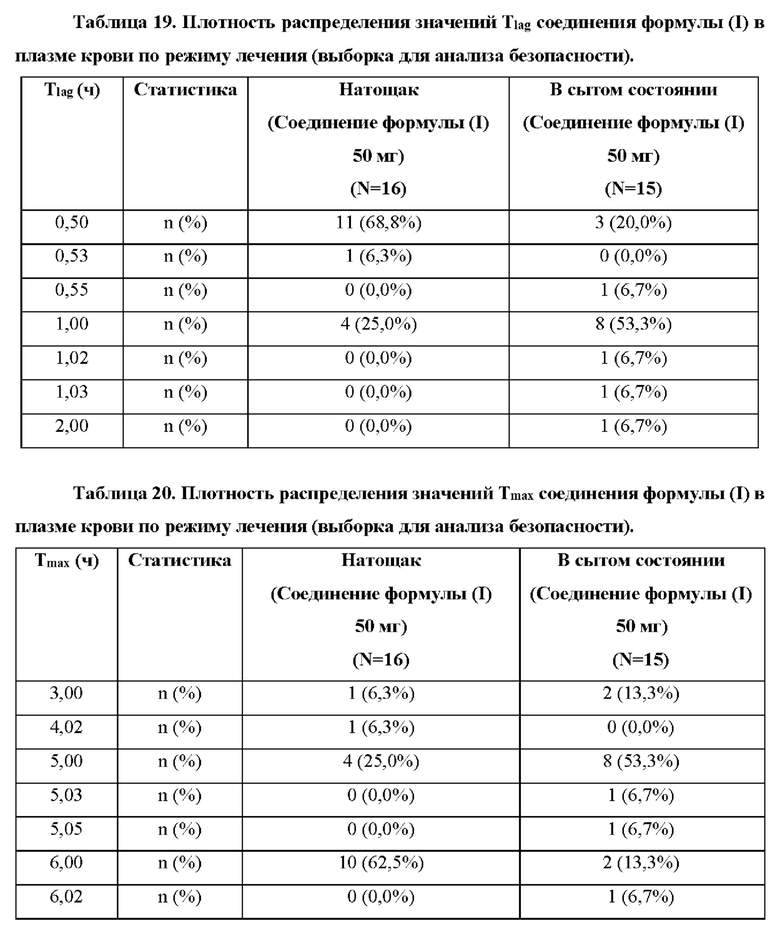
Соединение формулы (I) медленно абсорбировалось после перорального введения натощак и в сытом состоянии. При введении натощак, среднее Cmax соединения формулы (I) было на примерно 53% ниже такового при введении в сытом состоянии (731 в сравнении с 1550 нг/мл). Ввиду увеличенной фазы элиминации значения t1/2 и, следовательно, значения AUC0-∞ не могли быть определены для некоторых профилей зависимости концентрации соединения формулы (I) от времени. Среднее AUC0-∞ было на примерно 47% ниже при введении натощак, чем при введении в сытом состоянии (9440 в сравнении с 17800 нг × ч/мл) для тех субъектов, у которых можно было определить AUC0-∞. Среднее AUC0-tlast было на примерно 50% ниже при введении натощак, чем при введении в сытом состоянии (8020 в сравнении с 16200 нг × ч/мл). Медианное tmax было несколько больше при введении натощак, чем при введении в сытом состоянии (6,0 в сравнении с 5,0 часами), а среднее t1/2 было меньше при введении натощак, чем при введении в сытом состоянии (33 в сравнении с 42 часами), для тех субъектов, у которых можно было определить t1/2. Вариабельность РК соединения формулы (I) (геометрическое CV%) для AUC, Cmax, t1/2 и MRT была ниже при введении в сытом состоянии, в сравнении с введением натощак.
Средние геометрические соотношения и ассоциированные 90% CI для AUC0-tlast и Cmax соединения формулы (I) после лечения соединением формулы (I) для введения в сытом состоянии в сравнении с введением натощак представлены в таблице 21 ниже, где AUC0-tlast=AUC с момента времени 0 часов до последней измеряемой концентрации, Cmax = максимальная концентрация в плазме крови и РК = фармакокинетика.

Средние геометрические соотношения соединения формулы (I) для Cmax и AUC0-tlast для введения в сытом состоянии в сравнении с таковым натощак составляли 218,6% и 215,2% соответственно, что указывает на то, что абсорбция соединения формулы (I) была в примерно 2 раза выше при введении с едой. Верхние и нижние пределы 90% CI как для Cmax (187,4%, 255,1%), так и для AUC0-tlast (182,9%, 253,1%) были за пределами диапазона «отсутствия эффекта» от 80,00% до 125,00%, что указывает на то, что имело место влияние пищи на экспозицию соединения формулы (I). Из-за потери значений AUC0-∞ влияние пищи на общую экспозицию не оценивали с применением значений AUC0-∞.
Вывод:
Введение 50 мг соединения формулы (I) в сытом состоянии в сравнении с введением натощак обуславливало более высокую среднюю Cmax соединения формулы (I) (примерно в 2 раза выше; 1550 в сравнении с 731 нг/мл), большее t1/2 (42 в сравнении с 33 часами), несколько меньшее медианное tmax (5,0 в сравнении с 6,0 часами) и более высокую среднюю AUC0-∞ (примерно в 2 раза выше; 17800 в сравнении с 9440 нг × ч/мл). Средние геометрические соотношения соединения формулы (I) для Cmax и AUC0-tlast для введения в сытом состоянии в сравнении с таковым натощак составляли 218,6% и 215,2% соответственно, что указывает на то, что абсорбция соединения формулы (I) была в примерно 2 раза выше при введении с едой. Верхние и нижние пределы 90% CI как для Cmax (187,4%, 255,1%), так и для AUC0-tlast (182,9%, 253,1%) были за пределами диапазона «отсутствия эффекта» от 80,00% до 125,00%, что указывает на то, что имело место влияние пищи на экспозицию соединения формулы (I). Схожие результаты наблюдали в случае гидроксилированного метаболита соединения формулы (I). В совокупности эти результаты указывают на то, что 50 мг соединения формулы (I) хорошо переносилось у здоровых субъектов при введении натощак или в сытом состоянии, и что общие AUC и Cmax были повышены в том случае, когда соединение формулы (I) принималось с едой.
Пример 7. Исследование 1 фазы для оценки относительной биодоступности, влияния пищи на фармакокинетические показатели и безопасности составов на основе соединения формулы (I) у здоровых взрослых субъектов.
Было спланировано исследование 1 фазы для сравнения относительной биодоступности 50 мг дозы разных составов на основе соединения формулы (I), а также для оценки эффекта введения натощак и в сытом состоянии в отношении фармакокинетических показателей (РК) соединения формулы (I). Для данного исследования была выбрана доза 50 мг доза, поскольку она находится в пределах тестированного диапазона доз в завешенных испытаниях фазы 1 и фазы 2 и хорошо переносилась в этих исследованиях. Цели исследования являются следующими: оценить РК и сравнить относительную биодоступность составов с 50 мг соединения формулы (I) у здоровых взрослых субъектов; оценить влияние пищи на РК составов с 50 мг соединения формулы (I); и оценить безопасность и переносимость составов с 50 мг соединения формулы (I).
Дизайн исследования
Исследование представляет собой открытое рандомизированное перекрестное исследование 1 фазы с тремя периодами для оценки относительной биодоступности и влияния пищи на РК 50 мг соединения формулы (I) у здоровых взрослых субъектов. В ходе периода лечения 1 и периода лечения 2 субъекты будут получать однократную дозу 50 мг соединения формулы (I), вводимую в виде инкапсулированного липидного полутвердого вещества (эталон), и 1 из 2 разных тестируемых составов на основе высушенной распылением дисперсии (SDD) (суспензия или капсула) в сытом состоянии, и в ходе периода лечения три субъекта будут получать тот же тестируемый состав на основе SDD натощак.
Всего 36 здоровых взрослых субъектов будут рандомизированы в соответствии с 1 из 4 последовательностей лечения (9 субъектов на последовательность; приближенно равное распределение лиц мужского пола и лиц женского пола на последовательность; см. таблицу 22 ниже). Интервал между дозами будет составлять 21 сутки.

После предоставления информированного согласия субъекты будут проходить скрининг в отношении соответствия критериям для участия в исследовании. Скрининг начнется за 28 суток до 1-х суток периода лечения 1. Соответствующие критериям включения в исследование субъекты будут приняты в клинике на -1-е сутки и рандомизированы в соответствии с 1 из 4 последовательностей лечения на 1-е сутки периода лечения 1. В ходе периодов лечения 1 и 2 субъекты не будут принимать пищу в течение ночи на протяжении по меньшей мере 10 часов, а затем будут принимать жидкую пищевую добавку (Ensure Plus® со вкусом ванили, емкость 237 мл) с исследуемым лекарственным средством и не будут употреблять никакой другой пищи в течение 4 часов после введения дозы. В ходе периода лечения 3 субъекты не будут принимать пищу в течение ночи на протяжении по меньшей мере 10 часов до введения дозы и не будут принимать пищу в течение еще 4 часов после введения дозы. В ходе всех периодов лечения употребление воды не будет допускаться в течение 1 часа до введения дозы и до истечения 2 часов после введения дозы, за исключением воды/жидкой пищевой добавки, предусмотренных для введения дозы исследуемого лекарственного средства.
На 1-е сутки каждого периода лечения субъекты будут получать 50 мг дозу соединения формулы (I) и у них будут брать образцы крови для РК-анализа. Субъекты должны будут заполнить опросник удовлетворенности вкусовых качеств после приема исследуемого лекарственного средства на 1-е сутки периода лечения 3 (только для субъектов, которые получают SDD-суспензию натощак). Субъекты будут оставаться в клинике в день введения дозы и выписываться на 2-е сутки каждого периода лечения после завершения всех требуемых процедур, включая взятие образца для РК через 36 часов. Утром 8-х и 15-х суток каждого периода лечения субъекты будут возвращаться в клинику и пребывать там в амбулаторных условиях для взятия образца крови для оценок РК и безопасности. На 21-е сутки периода лечения 1 и периода лечения 2 субъекты будут приходить в центр и на 21-сутки у них уже завершится проведение оценок, и они будут оставаться в центре на ночь и на следующий день начнут 1-е сутки периода лечения 2 или периода лечения 3. Заключительный предусмотренный исследованием визит последующего наблюдения будет иметь место на 22-е сутки периода лечения 3 (21±2 суток после введения дозы в рамках периода лечения 3) или при преждевременном прекращении испытания.
Образцы крови для РК-анализа и оценки безопасности будут взяты/проведены в ходе всего исследования по графику. Дизайн исследования схематически показан на фиг. 10.
Продолжительность лечения
Ожидаемая продолжительность участия в исследовании для каждого здорового взрослого субъекта будет составлять примерно 13 недель, включая до 28 суток скрининга, 3 дозы, каждая с интервалом 21 сутки, и заключительный предусмотренный исследованием визит последующего наблюдения через 21 сутки после получения последней дозы исследуемого лекарственного средства в ходе периода лечения 3.
Исследуемый препарат, доза и способ введения
Соединение формулы (I) будет предоставлено в виде двух разных тестируемых составов для перорального введения: в виде порошка в бутылках для составления суспензии (20 мл) и в виде заполненных порошком капсул. Тестируемые составы на основе соединения формулы (I) будут содержать 50 мг соединения формулы (I) в виде эквивалента свободного основания. Субъекты должны проглотить исследуемое лекарственное средство с жидкой пищевой добавкой (Ensure Plus® [емкость 237 мл]) с дополнительными 100 мл воды (состав на основе SDD в виде капсулы) или с дополнительными 80 мл воды (состав на основе SDD в виде суспензии) в ходе периода лечения 1 или 2. Субъекты должны проглотить исследуемое лекарственное средство с 330 мл воды (состав на основе SDD в виде капсулы) или 310 мл воды (состав на основе SDD в виде суспензии) в ходе периода лечения 3.
Эталонный препарат, доза и способ введения
Эталонный состав на основе соединения формулы (I) (инкапсулированный липидный полутвердый состав) будет предоставлен в виде капсул для перорального введения. Эталонные капсулы с соединением формулы (I) будут содержать 50 мг соединения формулы (I) в виде эквивалента свободного основания. Субъекты должны проглотить одну капсулу с жидкой пищевой добавкой (Ensure Plus [емкость 237 мл]) с дополнительными 100 мл воды в ходе периода лечения 1 или 2.
Критерии оценки Фармакокинетические показатели
Образцы крови для оценки концентраций в плазме крови соединения формулы (I) и метаболитов будут брать в пределах 45 минут до введения дозы и через примерно 30 минут и 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 16, 24, 36, 168, 336 и 504 часов после введения дозы.
Для соединения формулы (I) и метаболитов будут рассчитаны следующие РК-параметры в плазме крови:
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 часов до последней измеряемой концентрации (AUC0-tlast)
- Площадь под кривой концентрации в плазме крови, экстраполированная с момента времени 0 часов до бесконечности (AUC0-∞)
- Максимальная концентрация в плазме крови (Cmax)
- Время достижения максимальной концентрации в плазме крови (tmax)
- Время задержки между моментом введения дозы и появлением измеряемого тестируемого препарата (Tlag)
- Кажущийся период полувыведения в конечной фазе (t1/2)
- Кажущаяся константа скорости элиминации в конечной фазе (λz)
- Кажущееся среднее время удержания препарата (MRT)
- Молярное соотношение по AUC первичного(-ых) метаболит(-ов) и исходного соединения формулы (I) в качестве лекарственного средства
Следующие РК-параметры в плазме крови будут рассчитаны только для соединения формулы (I):
- Кажущийся системный клиренс после перорального введения (CL/F)
- Кажущийся объем распределения в конечной фазе после перорального введения (Vz/F)
Другие оценки
Будет проведен опрос по опроснику удовлетворенности вкусовых качеств. Оценки безопасности
Безопасность будут оценивать в ходе всего исследования, и будут включены следующие оценки:
- АЕ
- Клинико-лабораторные исследования (анализ крови, анализ на свертываемость крови, клиническая биохимия и анализ мочи)
- Измерения физиологических показателей (включая ортостатическое артериальное давление и частоту пульса)
- Физикальные обследования
- Электрокардиограмма в 12 отведениях (ECG)
Статистические методы
Фармакокинетические параметры будут рассчитаны с применением некомпартментных методов и сведены по типу состава с применением описательной статистики. Двусторонние 90% доверительные интервалы будут рассчитаны для соотношения каждого тестируемого состава (SDD-суспензия и SDD-капсула) в сравнении с эталонным составом по AUC0-∞, AUC0-tlast и Cmax для соединения формулы (I) и метаболитов при введении в сытом состоянии. Кроме того, двусторонние 90% доверительные интервалы будут рассчитаны для соотношения каждого тестируемого состава в сытом состоянии в сравнении с состоянием натощак по AUC0-∞, AUC0-tlast и Cmax для соединения формулы (I) и метаболитов.
Данные по безопасности и опроснику удовлетворенности вкусовых качеств будут сведены с использованием описательной статистики.
Результаты
Результаты по фармакокинетике.
Результаты по фармакокинетике показаны в таблицах 23-26 ниже.

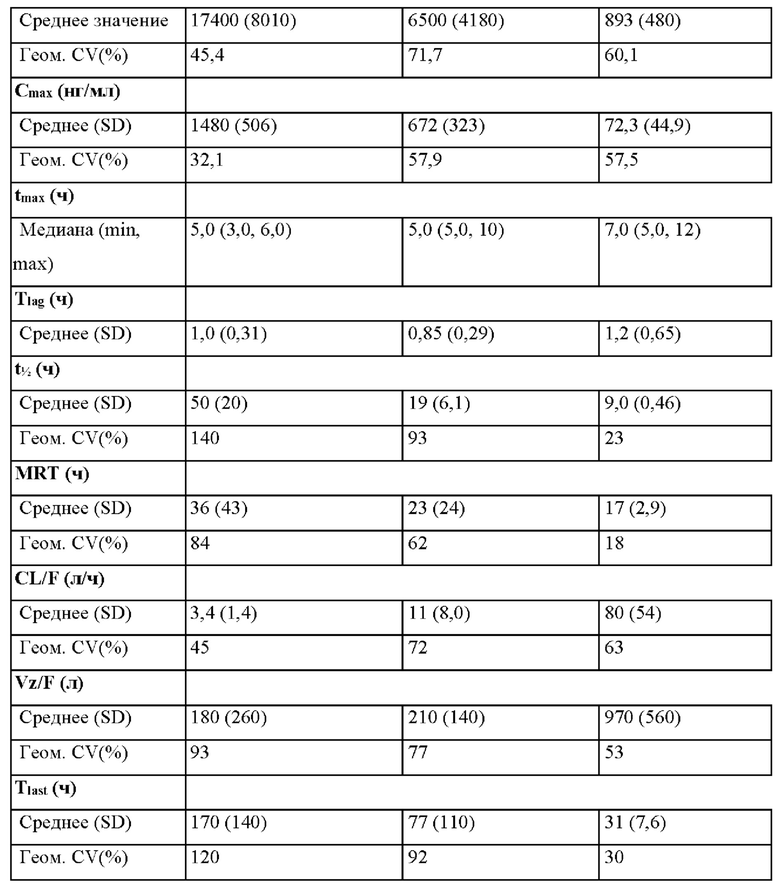
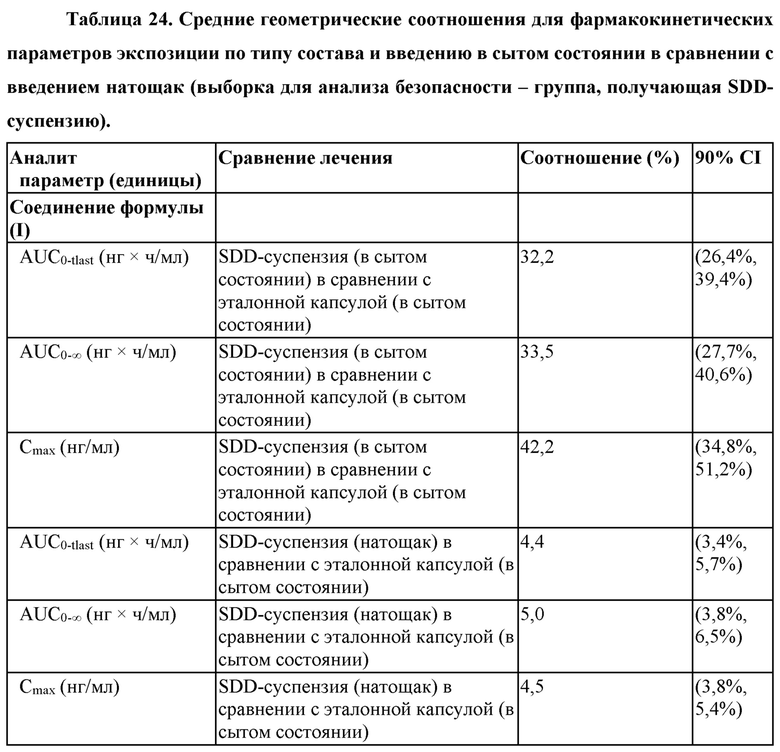
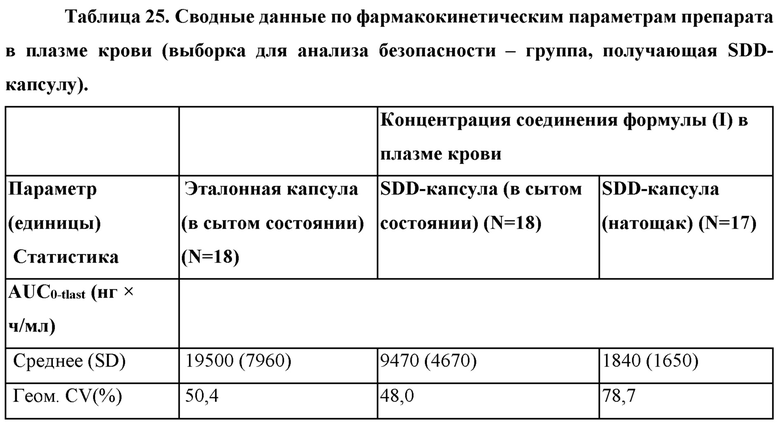
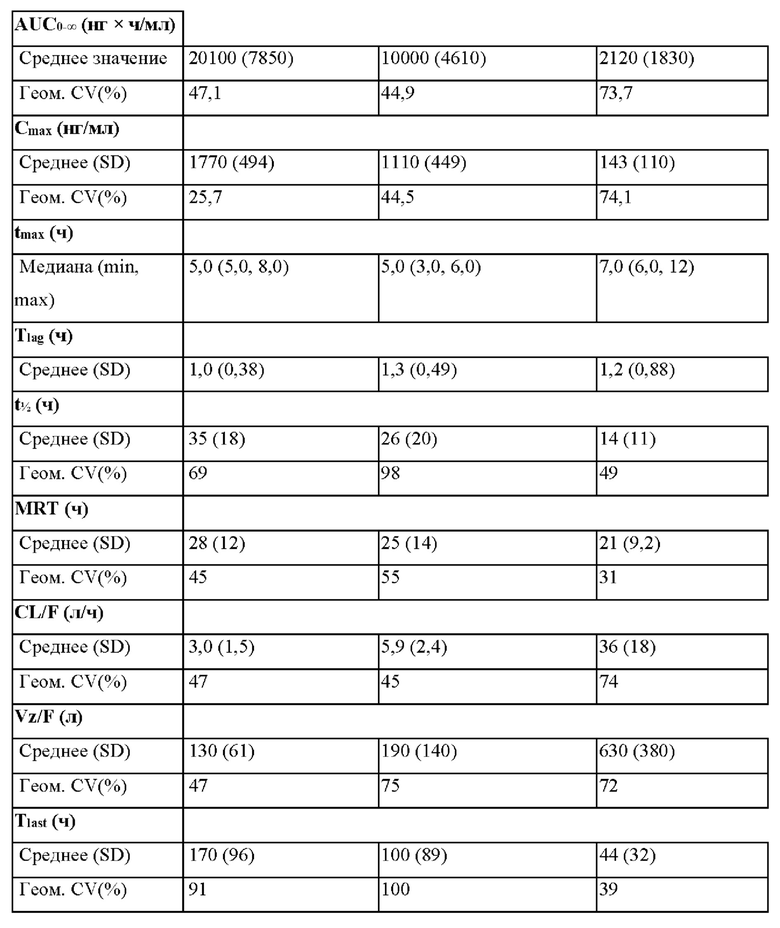
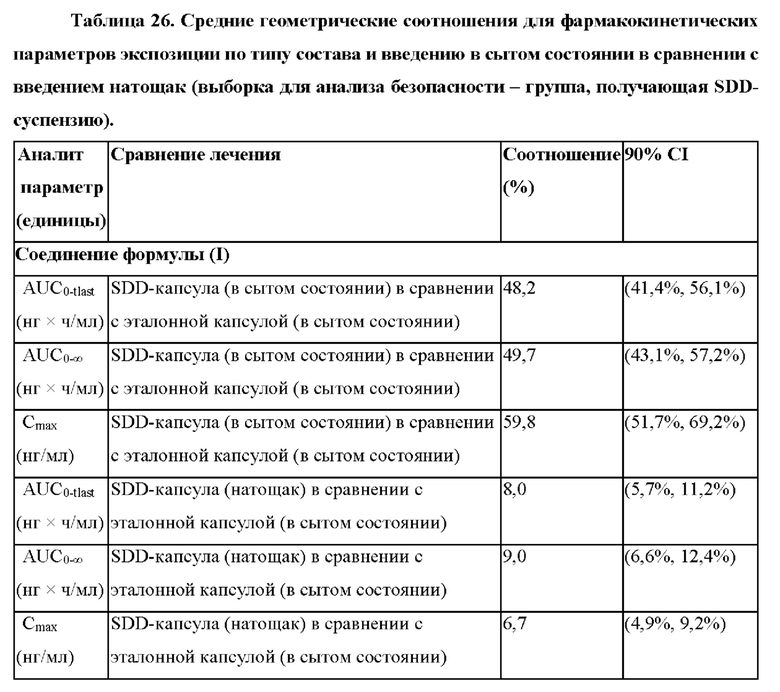
Пример 8. Исследование 2 фазы соединения формулы (I) с участием взрослых субъектов с врожденной гиперплазией надпочечников.
Было спланировано исследование 2 фазы для оценки безопасности, переносимости, фармакокинетических показателей (РК) и фармакодинамических показателей (PD) соединения формулы (I) у взрослых субъектов с классической врожденной гиперплазией надпочечников (САН). Цели исследования являются следующими: оценить безопасность и переносимость двух нарастающих доз соединения формулы (I) у взрослых субъектов с САН; оценить эффект повторных доз соединения формулы (I) в отношении эндогенных уровней PD-биомаркеров у взрослых субъектов с САН; и оценить показатели экспозиции в плазме крови после повторных доз соединения формулы (I), вводимых каждый вечер.
Более низкая доза, выбранная для этого исследования, 50 мг соединения формулы (I), хорошо переносилась у здоровых добровольцев в исследованиях безопасности и РК как с одной дозой, так и с повторными дозами. Дозы вплоть до 100 мг также хорошо переносились у здоровых добровольцев в исследованиях 1 фазы как с одной дозой, так и с повторными дозами, и, что важно, в крупном исследовании 2 фазы у не пожилых субъектов женского и мужского пола с большим депрессивным расстройством, получающих соединение формулы (I) в ходе 8-недельного периода лечения в двойном слепом режиме. Кроме того, ожидаемые показатели экспозиции в равновесном состоянии при использовании выбранных доз соединения формулы (I), с применением предсказанных Cmax и AUC, находятся в пределах приемлемых границ безопасности, определенных в доклинических токсикологических исследованиях, которые были проведены до настоящего времени.
Дизайн исследования
Было спланировано открытое исследование 2 фазы с использованием возрастающих многократных доз для оценки безопасности, переносимости, РК и PD соединения формулы (I) у примерно 30 взрослых субъектов женского и мужского пола (в возрасте 18-50 лет) с задокументированным медицинским диагнозом, представляющим собой классическую САН, обусловленную дефицитом 21-гидроксилазы. Исследование будет включать дизайн, предусматривающий последовательные когорты, с четырьмя когортами, получающими дозу соединения формулы (I): 50 мг и 100 мг, при этом каждая доза вводится в течение 14 суток подряд:
- Когорта 1: 50 мг соединения формулы (I) один раз в сутки с бутылкой Ensure Plus® со вкусом ванили (-237 мл) примерно в 22:00.
- Когорта 2: 100 мг соединения формулы (I) один раз в сутки с бутылкой Ensure Plus® со вкусом ванили (-237 мл) примерно в 22:00.
- Когорта 3:100 мг соединения формулы (I) один раз в сутки с ужином примерно в 19:00.
- Когорта 4: 100 мг соединения формулы (I) два раза в сутки с завтраком примерно в 07:00 и с ужином примерно в 19:00.
Прежде чем перейти от когорты 1 к когорте 2, потребуется приблизительно 2 недели для оценки данных по безопасности и переносимости. Субъекты, которые ранее завершили текущее исследование в когорте 1 или когорте 2, могут быть повторно включены в когорты 3 или 4 (в дополнение к новым субъектам). В таблице 27 ниже приведены когорты по величине дозы, дозы и число субъектов на когорту.
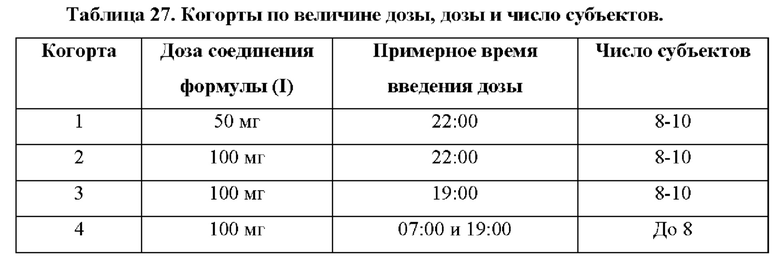
Субъекты будут проходить скрининг в отношении соответствия критериям для участия в исследовании на протяжении периода вплоть до примерно 3 недель (сутки с -28 по -8). Субъекты, которые будут повторно включены в исследование и у которых имела место стабильная схема приема лекарственных препаратов от САН с момента их последнего визита в этом исследовании, не должны проходить скрининг; субъекты, которые будут повторно включены в исследование и у которых имело место изменение схемы приема лекарственных препаратов от САН должны пройти второй скрининговый визит. В ходе скрининга субъекты предоставят один образец крови утром с 07:00 до 10:00 (до первой утренней дозы гидрокортизона) для определения у них уровней 17-гидроксипрогестерона (17-ОНР) для включения в исследование.
Соответствующие критериям включения в исследование субъекты, у которых по результатам скрининга уровень 17-ОНР≥1000 нг/дл, будут приняты в исследовательский центр на 1 ночь и у них будут взяты серийные образцы для оценки PD на исходном уровне в пределах 24-часового периода, начиная с -7-х суток. Серийные образцы для оценки PD на исходном уровне будут взяты примерно в 21:45, 23:00, 00:00, 1:00, 2:00, 4:00, 6:00, 8:00, 10:00, 12:00, 14:00, 16:00 и 22:00. На -6-е сутки свою обычную утреннюю дозу стероидных препаратов субъекты примут после взятия в 10:00 образца для оценки PD. Субъекты будут выписываться на -6-сутки после взятия последнего образца для оценки PD.
Субъекты в каждой когорте по величине дозы будут приняты в исследовательский центр на 1-е и 14-е сутки (первые и последние сутки введения дозы). На 1-е сутки у субъектов будут брать образец крови для генотипирования в отношении CYP21A2. Образцы для исходных оценок безопасности будут брать на 1-е сутки до введения первой дозы исследуемого лекарственного средства. Исследуемое лекарственное средство (50 или 100 мг соединения формулы (I)) будут вводить примерно в 22:00 для когорт 1 и 2 и примерно в 18:45, 20:00, 21:00, 22:00, 23:00, 00:00, 1:00, 2:00, 4:00, 6:00, 8:00, 10:00, 12:00, 14:00, 18:00, 19:00 и 22:00 для когорт 3 и 4. На -6-е сутки свою обычную утреннюю дозу стероидных препаратов субъекты примут после взятия в 10:00 образца для оценки PD. Субъекты будут выписываться на -6-сутки после взятия последнего образца для оценки PD.
Субъекты в когортах 1 и 2 будут приняты в исследовательский центр на 1-е и 14-е сутки (первые и последние сутки введения дозы). На 1-е сутки у субъектов будут брать образец крови для генотипирования в отношении цитохрома Р450 (CYP) 21А2. Образцы для исходных оценок безопасности будут брать на 1-е сутки до введения первой дозы исследуемого лекарственного средства. Исследуемое лекарственное средство (50 мг или 100 мг соединения формулы(1)) будут вводить примерно в 22:00. Свою обычную утреннюю дозу сопутствующих стероидных препаратов субъекты примут после взятия образцов для оценки PK/PD, которые берутся в пределах 12 часов после введения дозы (примерно в 10:00), на 2-е сутки и после взятия образцов для оценки PK/PD, которые берутся в пределах 16 часов после введения дозы (примерно в 14:00), на 15-е сутки. Субъекты выпишутся из исследовательского центра вечером на 2-е и 15-е сутки после завершения всех связанных с исследованием процедур для этих дней. Перед выпиской на 2-е сутки исследуемое лекарственное средство будет введено в исследовательском центре примерно в 22:00. Исследуемое лекарственное средство будет вводиться самостоятельно каждый вечер дома примерно в 22:00 с 3-е по 13-е сутки. Субъекты будут принимать их обычную утреннюю дозу сопутствующих стероидных препаратов примерно в 10:00 с 3-е по 14-е сутки. На 7-е сутки периода лечения оценки РК, PD и оценки безопасности будут выполняться в исследовательском центре в амбулаторных условиях.
На 1-е сутки у субъектов в когортах 3 и 4 будут брать образец крови для генотипирования в отношении CYP21A2 (только для субъектов, которые ранее не участвовали в когортах 1 или 2). Образцы для исходных оценок безопасности будут брать на 1-е сутки до введения первой дозы исследуемого лекарственного средства. В случае когорты 3 исследуемое лекарственное средство (100 мг соединения формулы(1)) будет вводиться субъектом дома с 1-е по 13-е сутки примерно в 19:00 с каждым ужином. В случае когорты 4 исследуемое лекарственное средство (100 мг соединения формулы(1)) будет вводиться субъектом дома с 2-е по 14-е сутки примерно в 7:00 с каждым завтраком и с 1-е по 13-е сутки примерно в 19:00 с каждым ужином. В случае обеих когорт вечерняя доза на 14-е сутки будет вводится в исследовательском центре. Субъекты будут принимать их обычную утреннюю дозу сопутствующих стероидных препаратов примерно в 10:00 с 1-е по 14-е сутки. На 7-е сутки периода лечения оценки РК, PD и оценки безопасности будут выполняться в исследовательском центре в амбулаторных условиях. Субъекты будут приняты в исследовательский центр на 14-е сутки (последние сутки введения дозы). На 14-е сутки субъекты будут получать исследуемое лекарственное средство в исследовательском центре примерно в 19:00 со стандартным (с умеренным содержанием жира/умеренным содержанием калорий) ужином. Свою обычную утреннюю дозу сопутствующих стероидных препаратов субъекты примут после взятия образцов для оценки PK/PD примерно в 14:00 на 15-е сутки. Субъекты выпишутся из исследовательского центра вечером на 15-е сутки после завершения всех связанных с исследованием процедур.
Для всех когорт визиты последующего наблюдения на 21-е, 28-е и 35-е сутки будут осуществляться в исследовательском центре или дома у субъекта квалифицированным медицинским работником на дому (в зависимости от предпочтения субъекта). Заключительный предусмотренный исследованием визит будет иметь место в исследовательском центре через примерно 5 недель после последней дозы исследуемого лекарственного средства (на 49-е сутки или при преждевременном прекращении испытания). Будет иметь место интервал для проведения визита -8 часов для 7-х суток, -8 часов/+3 суток для 21-х, 28-х и 35-х суток и+7 суток для заключительного предусмотренного исследованием визита. Безопасность, переносимость, РК и PD будут оценивать по графику в ходе всего исследования. Дизайн исследования схематически показан на фиг. 11.
Процедура повышения дозы
Когорта 1 будет состоять из примерно 8-10 субъектов, которые будут получать суточную дозу 50 мг соединения формулы (I) примерно в 22:00 в течение 14 суток (субъекты будут получать исследуемое лекарственное средство в центре на 1-е, 2-е и 14-е сутки и самостоятельно вводить исследуемое лекарственное средство дома с 3-е по 13-е сутки). После завершения оценок на 15-е сутки для всех субъектов в когорте 1, медицинский наблюдатель изучит накопленные результаты по безопасности и переносимости чтобы убедиться, что при переходе к 100 мг дозе (когорты 2 и 3) отсутствуют какие-либо опасения по поводу безопасности, и для определения того, достигалась ли максимальная переносимая доза (MTD). Если MTD достигается, повышение дозы проводить не будут. Между когортами 1 и 2 будет примерно 2-недельный период для проведения таких процедур контроля безопасности. Аналогичную процедуру будут применять перед переходом к дозе 100 мг два раза в сутки (когорта 4).
Если медицинский наблюдатель определит, что можно безопасно переходить к 100 мг соединения формулы (I), субъектам в когорте 2 будут вводить 100 мг соединения формулы (I) ежесуточно в течение 14 суток. Введение дозы для когорт 3 и 4 можно начинать одновременно с когортой 2.
В ходе 14-суточного периода введения дозы для любой когорты введение дозы может быть отсрочено или приостановлено, если у одного или нескольких субъектов наблюдается тяжелое или серьезное нежелательное явление (АЕ) или если тип, частота или тяжесть АЕ становятся неприемлемыми. Если введение дозы отсрочено, медицинский наблюдатель изучит все доступные данные по безопасности, переносимости и РК, прежде чем будет допущено получение исследуемого лекарственного средства для других субъектов.
Исследуемая популяция
В исследование будут включены примерно 30 взрослых субъектов женского и мужского пола (в возрасте 18-50 лет) с задокументированным медицинским диагнозом, представляющим собой классическую САН, обусловленную дефицитом 21-гидроксилазы, которые соответствуют всем протокольным требованиям, необходимым для участия. Субъекты, которые ранее завершили текущее исследование в когорте 1 или когорте 2, могут быть повторно включены в когорты 3 или 4 (в дополнение к новым субъектам).
Продолжительность лечения
Ожидаемая продолжительность участия в исследовании для каждого субъекта составляет примерно 11 недель, включая скрининговый период вплоть до примерно 3 недель, 24-часовой период определения PD на исходном уровне (примерно 7 суток до первых суток введения дозы), 14 суток введения дозы и период последующего наблюдения примерно 5 недель. Общая продолжительность исследования будет составлять 8-11 недель для субъектов, повторно включенных в исследование.
Исследуемый препарат, доза и способ введения
Соединение формулы (I) будет предоставлено в виде капсул, содержащих 50 мг соединения формулы (I) в виде свободного основания для перорального введения (см., например, эталонный состав 1, как описано в примере 9). Дозы соединения формулы (I) составляют 50 мг и 100 мг, вводимые в форме капсул для перорального применения. Каждая доза исследуемого лекарственного средства для когорт 1 и 2 будет вводиться с бутылкой Ensure Plus® со вкусом ванили (-237 мл). Каждая доза исследуемого лекарственного средства для когорты 3 будет вводиться субъектом с каждым ужином примерно в 19:00. Каждая доза исследуемого лекарственного средства для когорты 4 будет вводиться субъектом с каждым завтраком примерно в 19:00 (т.е. общая суточная доза 200 мг).
Критерии оценки
Когорты 1 и 2
Образцы крови для оценки PD на исходном уровне в пределах 24-часового периода будут взяты в период с -7-е по -6-е сутки примерно в 21:45, 23:00, 00:00, 1:00, 2:00, 4:00, 6:00, 8:00, 10:00, 12:00, 14:00, 16:00 и 22:00. Образцы крови для оценки РК- и PD-параметров соединения формулы (I) будут взяты в период с 1-е по 2-е сутки и с 14-е по 15-е сутки: за 15 минут до введения дозы и через 1, 2, 3, 4, 6, 8, 10, 12, 14, 16, 20 и24 часа после введения дозы; 7-е сутки (через примерно 24 часа после введения дозы); 21-е, 28-е и 35-е сутки (через примерно 168, 336 и 504 часа после введения дозы); и во время заключительного предусмотренного исследованием визита (49-е сутки или при преждевременном прекращении испытания). Когорты 3 и 4
Образцы крови для оценки PD на исходном уровне в пределах 24-часового периода будут взяты в период с -7-е по -6-е сутки примерно в 18:45, 20:00, 21:00, 22:00, 23:00, 24:00, 1:00, 2:00, 4:00, 6:00, 8:00, 10:00, 12:00, 14:00, 18:00, 19:00 и 22:00. Образцы крови для оценки РК- и PD-параметров соединения формулы (I) будут взяты в период с 14-е по 15-е сутки в следующие моменты времени (для когорты 4, все моменты времени относятся к вечернему введению дозы, если не указано иное): за 15 минут до введения дозы и через 1, 2, 3, 4, 5, 6, 7, 9, 11, 13, 15, 17, 19, 23, 24 и 27 часов после введения дозы; 7-е сутки (через 24 часа после введения дозы для когорты 3 или через 12 часов после введения утренней дозы, но перед вечерней дозой для когорты 4); 21-е, 28-е и 35-е сутки (через примерно 168, 336 и 504 часа после введения дозы); и во время заключительного предусмотренного исследованием визита (49-е сутки или при преждевременном прекращении испытания).
Фармакокинетические показатели
Для соединения формулы (I) и метаболитов будут рассчитаны следующие РК-параметры в плазме крови:
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 часов до последней измеряемой концентрации (AUC0-tlast)
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 до 24 часов (AUC0-24)
- Максимальная концентрация в плазме крови (Cmax)
- Время достижения максимальной концентрации в плазме крови (tmax)
- Время задержки между моментом введения дозы и появлением измеряемого тестируемого препарата (Tlag)
- Период полувыведения в конечной фазе (t1/2)
- Кажущаяся константа скорости элиминации в конечной фазе (λz)
- Кажущееся среднее время удержания препарата (MRT) Дополнительные РК-параметры, только для 14-х суток:
- Средняя концентрация в плазме крови в равновесном состоянии (CaVg)
- Процентное отклонение в равновесном состоянии (% отклонение)
- Индекс накопления в равновесном состоянии
- Кажущийся системный клиренс после перорального введения (CL/F) (только соединение формулы (I))
Форм акодин амика
Первичные параметры: утренний 17-ОНР (сыворотка крови; нг/дл) в образцах, взятых в 6:00, 8:00 и 10:00 (образцы, взятые через 8, 10 и 12 часов после введения дозы, для когорт 1 и 2, и образцы, взятые через 11, 13 и 15 часов после введения дозы, для когорт 3 и 4.
Вторичные параметры: 17-ОНР во все другие моменты времени, андростендион (сыворотка крови; нг/дл), тестостерон (сыворотка крови; нг/дл), кортизол (сыворотка крови; мкг/дл) и адренокортикотропный гормон (АСТН в плазме крови; пг/мл).
Безопасность
Безопасность и переносимость будут оценивать в ходе всего исследования, и будут включены следующие оценки:
- Нежелательные явления
- Клинико-лабораторные исследования - клиническая биохимия (включая креатинкиназу, миоглобин, общий и конъюгированный билирубин), анализ крови, анализ на свертываемость крови (протромбиновое время, аРТТ, d-димер, фибриноген) и анализ мочи (включая количественный миоглобин, цилиндры и кристаллы)
- Основные физиологические показатели
- Физикальные обследования (включая обследование опорно-двигательного аппарата)
- Электрокардиограммы в 12 отведениях (ECG)
- Шкала Колумбийского университета для оценки степени тяжести суицидальных проявлений (C-SSRS)
- Краткая психиатрическая оценочная шкала (BPRS)
Статистические методы
Переменные безопасность, РК и PD будут сведены в каждой когорте по величине дозы (50 мг и 100 мг соединения формулы (I)) с применением описательной статистики. Сводные данные по PD-показателям будут включать как наблюдаемые значения, так и изменения по сравнению со значениями до введения дозы.
Результаты
Результаты по фармакокинетике.
В это исследование были включены восемь субъектов (четверо лиц женского пола и четверо лиц мужского пола), и они завершили участие в исследовании в составе когорты 1. Информация касательно возраста, пола и BMI для участников исследования показана в таблице 28 ниже.

Субъекты получали суточную дозу соединения формулы (I) при 50 мг примерно в 22:00 (-10 вечера или перед сном) в течение 14 суток. Средние арифметические значения для АСТН (фиг. 12А) и 17-ОНР (фиг. 12 В) всех 8 субъектов когорты 1 откладывали на графике для каждого момента времени для результатов исходного уровня до лечения, 1-е сутки, и 14-х суток. Из профилей концентрации как АСТН, так и 170НР на 1-е сутки и 14-е сутки видно явное снижение относительно профилей средних концентраций на исходном уровне. Средние значения РК-Параметров Tmax, Cmax и AUC24 в когорте 1 для соединения формулы (I) показаны в таблице 29 ниже. Эти РК-параметры согласуются с результатами наблюдений из исследований 1 фазы с участием здоровых добровольцев.
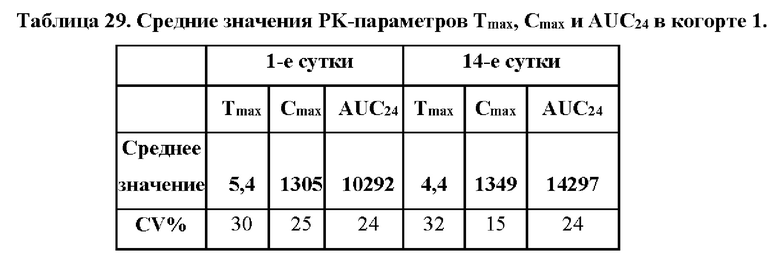
Дополнительные средние значения РК-параметров Tmax, Cmax и AUC24 в когорте 1 и когорте 2 для соединения формулы (I) на 1-е сутки после введения дозы показаны в таблице 30 ниже.

Дополнительные средние значения РК-параметров Tmax, Cmax и AUC24 в когорте 1, когорте 2 и когорте 3 для соединения формулы (I) на 14-е сутки после введения дозы показаны в таблице 31 ниже.
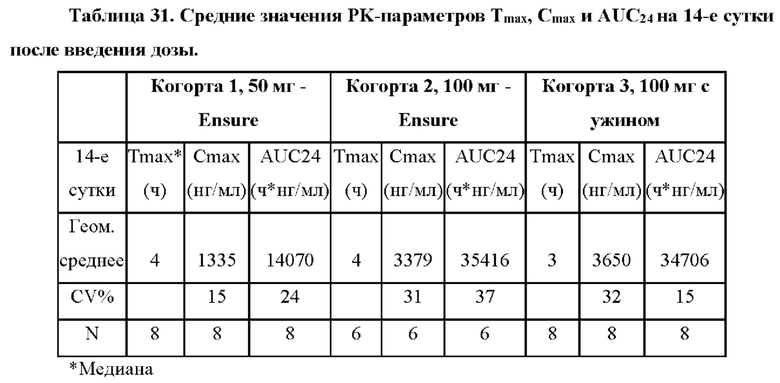
Средние арифметические значения для андростендиона (фиг. 13А) и тестостерона (фиг. 13В) всех 8 субъектов когорты 1 откладывали на графике для каждого момента времени для результатов исходного уровня до лечения, 1-е сутки, и 14-х суток. Из профиля концентрации андростендиона на 1-е сутки и 14-е сутки видно явное снижение относительно профиля средних концентраций на исходном уровне.
Если обратить внимание исключительно на критическое окно в утреннее время суток (моменты времени через 8, 10 и 12 часов после введения дозы) с 6:00 утра до 10:00 утра, можно наблюдать заметное снижение уровней АСТН относительно исходного уровня по каждому из 3 моментов времени на 1-е и 14-е сутки (фиг. 14А). Средние арифметические значения по всем трем моментам времени демонстрируют >=50% снижение относительно исходного уровня на 1-е сутки и на 14-е сутки (фиг. 14В).
Если обратить внимание исключительно на критическое окно в утреннее время суток (моменты времени через 8, 10 и 12 часов после введения дозы) с 6:00 утра до 10:00 утра, можно наблюдать заметное снижение уровней 17-ОНР относительно исходного уровня по каждому из 3 моментов времени на 1-е и 14-е сутки (фиг. 15А). Средние арифметические значения по всем трем моментам времени демонстрируют>=40% снижение относительно исходного уровня на 1-е сутки и на 14-е сутки (фиг. 15В).
Если обратить внимание исключительно на критическое окно в утреннее время суток (моменты времени через 8, 10 и 12 часов после введения дозы) с 6:00 утра до 10:00 утра, можно наблюдать заметное снижение уровней андростендиона относительно исходного уровня по каждому из 3 моментов времени на 1-е и 14-е сутки (фиг. 16А). Средние арифметические значения по всем трем моментам времени демонстрируют >=30% снижение относительно исходного уровня на 1-е сутки и на 14-е сутки (фиг. 16В).
Сводные данные по снижению 17-ОНР и андростендиона в когорте 1 показаны в таблице 32. Кроме того, уровни андростендиона у трех субъектов были нормализованы по типу лечения к значениям у трех субъектов с ID №№001, 002 и 006).


Спустя 14 суток после введения соединения формулы (I) один раз в сутки у большей части участников в когортах 1-3 наблюдали сниженные концентрации в сыворотке крови вырабатываемых надпочечниками андрогенов и предшественников. Изменения средних значений относительно исходного уровня (±стандартное отклонение) в когорте 1 были следующими: 17-ОНР, -2341,0±1535,0 нг/дл; андростендион, -98,4±98,7 нг/дл; и АСТН, -157,0±194,9 пг/мл. Снижения средних значений были существеннее в когорте 2 (17-ОНР, -4406,0±5516,1; андростендион, -362,8±354,0; АСТН, -180,9±155,2) и когорте 3 (17-ОНР, -4760,1±4018,2; андростендион, -210,9±188,6; АСТН, -358,9±177,6), что свидетельствует о возможном наличии дозозависимого эффекта. На фигурах 17А-17С, 18А-18С и 19А-19С показаны результаты для когорт 1, 2 и 3 соответственно.
Краткий итог
Результаты этого продолжающегося открытого исследования фазы II продемонстрировали снижение по меньшей мере на 50 процентов уровней 17-гидроксипрогестерона (17-ОНР) и адренокортикотропного гормона (АСТН) относительно исходного уровня у более 50 процентов пациентов с САН в когорте 1, которых лечили соединением формулы (I) в течение 14 суток (т.е. у 6 из 8 пациентов в когорте 1 наблюдали снижение на>50% уровней 17-ОНР относительно исходного уровня по меньшей мере для одного момента времени в окне утреннего времени суток, см., например, таблицу 32). Значимые снижения также наблюдали в отношении других биомаркеров, включая андростендион (т.е. у 4 из этих пациентов также наблюдали снижение на >50% уровней андро стенд иона относительно исходного уровня по меньшей мере для одного момента времени в окне утреннего времени суток, см., например, таблицу 32). Большее снижение уровней биомаркеров в когортах 2 и 3, в которых лечение проводилось двойной дозой соединения формулы (I), в сравнении с когортой 1, свидетельствует о возможном наличии дозозависимого эффекта. Кроме того, соединение формулы (I) хорошо переносилось, при этом сообщалось об относительно малом числе легких нежелательных явлений (АЕ) (например, головная боль, болевые ощущения при овуляции, утомляемость, местная инфекция (палец стопы), головокружение, тошнота, URI, кровоподтек, причем чаще всего имела место головная боль). Клинически значимых результатов стандартных лабораторных исследований, основных физиологических показателей или электрокардиограммы не наблюдали.
Пример 9. Эталонный состав 1 на основе соединения формулы (I).
В таблицах 34А и 34В показан эталонный состав 1 на основе соединения формулы (I), применяемый в клинических исследованиях, описанных в примерах 6 и 8 выше. Иллюстративный производственный процесс показан на фиг. 20. Другой иллюстративный производственный процесс показан на фиг. 21.

Пример 10. Исследование для оценки эффекта Ensure Plus, пудинга Ensure, молока и пищи с высоким содержанием жиров с использованием эталонной капсулы.
Дизайн исследования
Исследование представляет собой открытое рандомизированное перекрестное исследование 1 фазы с четырьмя периодами для оценки влияния пищи с разными уровнями содержания жира и калорий на РК, безопасность и переносимость соединения формулы (I) у здоровых взрослых субъектов.
Всего 16 здоровых взрослых субъектов (8 лиц мужского пола и 8 лиц женского пола) будут случайным образом распределены в соответствии с 1 из 4 последовательностей лечения (4 субъекта на последовательность [2 лиц мужского пола и 2 лиц женского пола на последовательность]; см. таблицу 35 ниже). В ходе каждого периода лечения субъекты будут получать однократную дозу 100 мг соединения формулы (I), вводимую с соответствующей пищей, согласно схеме рандомизации. Между каждой дозой будет иметь место период вымывания длительностью по меньшей мере 21 сутки.
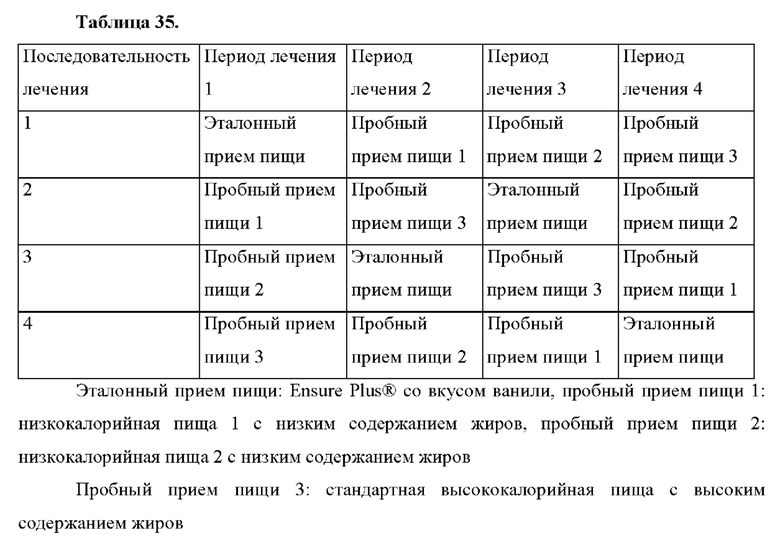
После предоставления информированного согласия субъекты будут проходить скрининг в отношении соответствия критериям для участия в исследовании. Скрининг начнется за 28 суток до 1-х суток периода лечения 1. Соответствующие критериям включения в исследование субъекты будут приняты в клинике на -1-е сутки и рандомизированы в соответствии с 1 из 4 последовательностей лечения на 1-е сутки периода лечения 1. В ходе каждого периода лечения субъекты не будут принимать пищу в течение ночи на протяжении по меньшей мере 10 часов до начала приема назначенной пищи, в соответствии со схемой рандомизации, и будут принимать исследуемое лекарственное средство примерно в 8:00. Субъекты должны полностью завершить прием пищи в течение указанного периода времени и не должны употреблять какую-либо другую пищу в течение 4 часов после введения дозы. Для всех периодов лечения употребление воды не будет допускаться в течение 1 часа до введения дозы и до истечения 2 часов после введения дозы, за исключением воды, предусмотренной при введении дозы исследуемого лекарственного средства и запланированных приемах пищи.
На 1-е сутки каждого периода лечения субъектам будут вводить 100 мг дозу соединения формулы (I). Образцы крови будут брать для РК-анализа в 36-часовом промежутке времени в ходе пребывания в стационаре. В ходе каждого периода лечения субъекты будут оставаться в клинике в день введения дозы и будут выписываться на 2-е сутки после завершения всех требуемых процедур. Утром 8-х и 15-х суток каждого периода лечения субъекты будут возвращаться в клинику для осуществления амбулаторного визита с целью взятия образца крови для оценки РК и оценок безопасности. На 21-е сутки периодов лечения 1-3 субъекты будут приходить в центр и на 21-сутки у них уже завершится проведение оценок и они будут оставаться в центре на ночь и на следующий день начнут 1-е сутки последующего периода лечения. Заключительный предусмотренный исследованием визит последующего наблюдения будет иметь место на 22-е сутки периода лечения 4 (21±2 суток после введения дозы в рамках периода лечения 4) или при преждевременном прекращении испытания.
Образцы крови для оценки РК будут взяты в ходе всего исследования по графику.
Исследуемая популяция
В исследование будут включены шестнадцать здоровых взрослых субъектов (8 лиц мужского пола и 8 лиц женского пола) в возрасте 18-55 лет включительно, которые соответствуют всем протокольным требованиям, необходимым для участия.
Продолжительность лечения
Ожидаемая продолжительность участия в исследовании для каждого здорового взрослого субъекта будет составлять примерно 16 недель, включая до 28 суток скрининга, 4 суток введения дозы с интервалом длительностью по меньшей мере 21 сутки между последовательными дозами и заключительный предусмотренный исследованием визит последующего наблюдения через 21 сутки (±2 суток) после получения последней дозы исследуемого лекарственного средства в ходе периода лечения 4.
Исследуемый препарат, доза и способ введения
Соединение формулы (I) будет предоставлено в виде капсул для перорального введения (инкапсулированный липидный полутвердый состав, например, пример 9). Капсулы с соединением формулы (I) будут содержать 50 мг соединения формулы (I) в виде эквивалента свободного основания. В ходе каждого периода лечения субъекты будут получать две капсулы по 50 мг (100 мг) исследуемого лекарственного средства вместе с пищей и водой, как определено схемой рандомизации. Прием пищи, воды и исследуемого лекарственного средства осуществляется следующим образом:
Эталонный прием пищи: Две капсулы исследуемого лекарственного средства будут вводиться через примерно 5 минут после начала приема жидкой пищевой добавки (т.е. Ensure Plus® [емкость 237 мл]) и с дополнительными 120 мл воды для введения дозы исследуемого лекарственного средства.
Пробный прием пищи 1: Две капсулы исследуемого лекарственного средства будут вводиться через примерно 5 минут после начала приема низкокалорийной пищи 1 с низким содержанием жиров с 120 мл воды для введения дозы исследуемого лекарственного средства.
Пробный прием пищи 2: Две капсулы исследуемого лекарственного средства будут вводиться через примерно 5 минут после начала приема низкокалорийной пищи 2 с низким содержанием жиров с 120 мл воды для введения дозы исследуемого лекарственного средства.
Пробный прием пищи 3: Две капсулы исследуемого лекарственного средства будут вводиться через примерно 30 минут после начала приема высококалорийной пищи с высоким содержанием жиров с 120 мл воды для введения дозы исследуемого лекарственного средства.
КРИТЕРИИ ОЦЕНКИ
Фармакокинетические показатели
Образцы крови для оценки концентраций в плазме крови соединения формулы (I) и метаболитов будут брать в пределах 45 минут до введения дозы и через примерно 30 минут и 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 16, 24, 36, 168, 336 и 504 часов после введения дозы.
Для соединения формулы (I) и метаболитов будут рассчитаны следующие РК-параметры в плазме крови:
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 часов до последней измеряемой концентрации (AUC0-tlast)
- Площадь под кривой зависимости концентрации в плазме крови от времени с момента времени 0 часов, экстраполированная до бесконечности (AUC0-∞)
- Максимальная концентрация в плазме крови (Cmax)
- Время достижения максимальной концентрации в плазме крови (tmax)
- Время задержки между моментом введения дозы и появлением измеряемого тестируемого препарата (Tlag)
- Кажущийся период полувыведения в конечной фазе (t1/2)
- Кажущаяся константа скорости элиминации в конечной фазе (λz)
- Кажущееся среднее время удержания препарата (MRT)
- Молярное соотношение по AUC первичного(-ых) метаболит(-ов) и исходного соединения формулы (I) в качестве лекарственного средства. Следующие РК-параметры в плазме крови будут рассчитаны только для соединения формулы (I):
- Кажущийся системный клиренс после перорального введения (CL/F)
- Кажущийся объем распределения в конечной фазе после перорального введения (Vz/F)
Оценки безопасности
Безопасность будут оценивать в ходе всего исследования, и будут включены следующие оценки:
- Нежелательные явления (АЕ)
- Клинико-лабораторные исследования (анализ крови, анализ на свертываемость крови, клиническая биохимия и анализ мочи)
- Измерения физиологических показателей (включая ортостатическое артериальное давление и частоту пульса)
- Физикальные обследования
- Электрокардиограммы в 12 отведениях (ECG)
Статистические методы
Фармакокинетические параметры будут рассчитаны с применением некомпартментных методов и сведены по типу пищи (пробный прием пищи/эталонный прием пищи) с применением описательной статистики. Двусторонние 90% доверительные интервалы будут рассчитаны для соотношения каждого пробного приема пищи в сравнении с эталонным приемом пищи по AUC0-∞, AUC0-tlast и Cmax для соединения формулы (I) и метаболитов.
Данные по безопасности будут сведены с использованием описательной статистики. Результаты
Результаты по фармакокинвтике
Результаты по фармакокинетике показаны в таблице 36 ниже.


Пример 11. Гранулярный состав на основе соединения формулы (I) с использованием высушенной распылением дисперсии (SDD-G).
В таблице 37 показан гранулярный состав на основе соединения формулы (I) с применением SDD, полученной в соответствии с примером 3 выше. Иллюстративный производственный процесс показан на фиг. 22А и фиг. 22В.
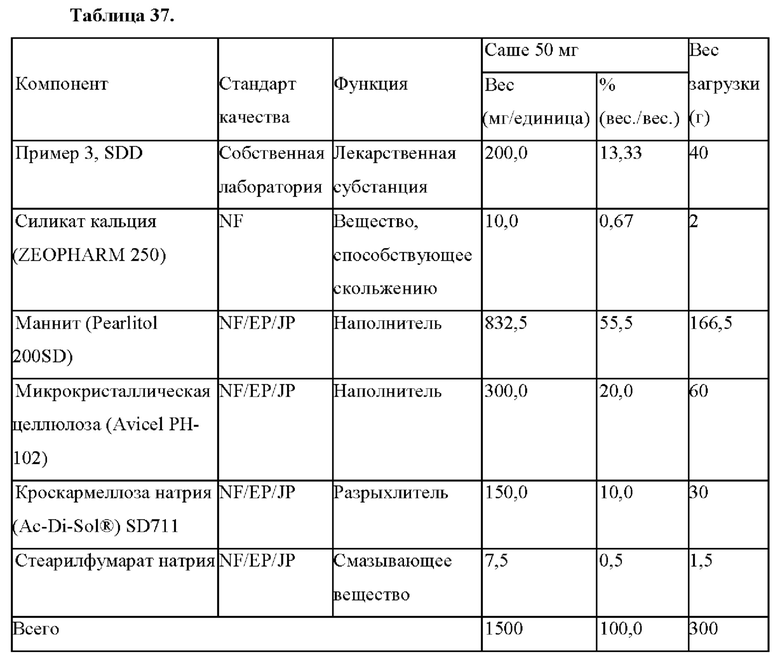
Пример 12. Жидкий состав 1 на основе соединения формулы (I)
В таблице 38 показан жидкий состав 1 на основе соединения формулы (I) в виде свободного основания. Иллюстративный производственный процесс показан на фиг. 23.
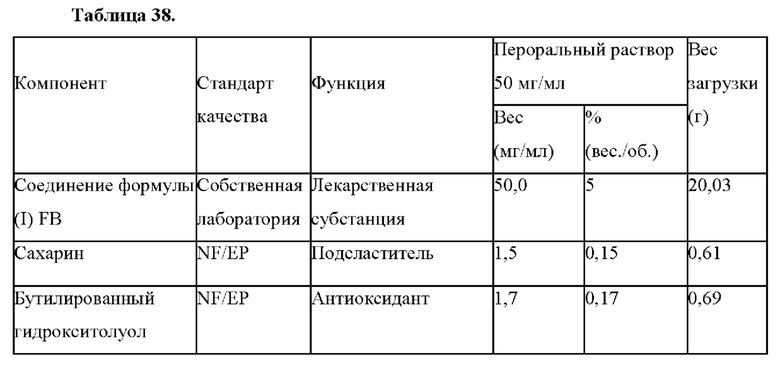
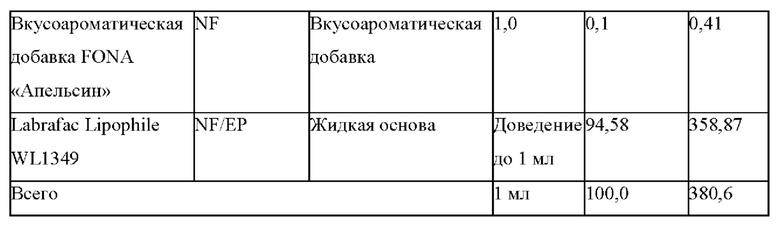
Пример 13. Жидкий состав 2 на основе соединения формулы (I)
В таблице 39 показан жидкий состав 2 на основе соединения формулы (I) в виде свободного основания. Иллюстративный производственный процесс показан на фиг. 24.
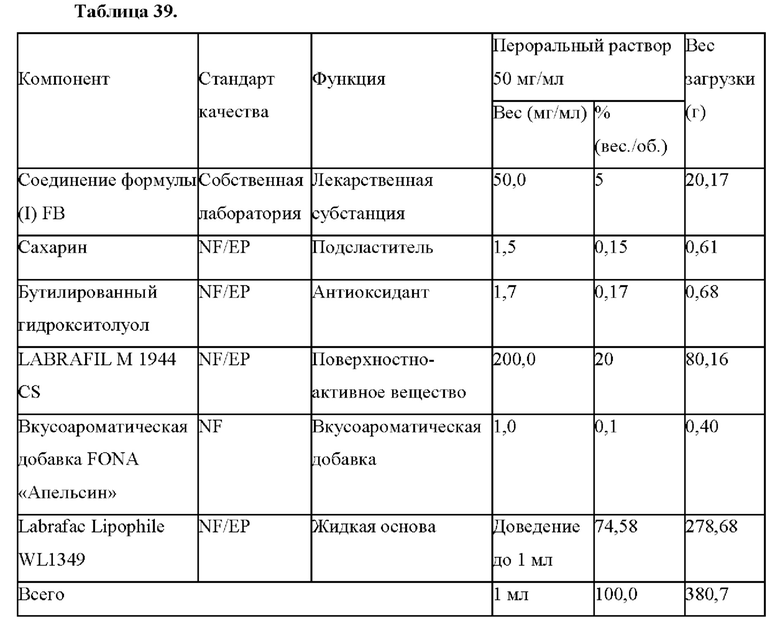
Пример 14. Открытое исследование фазы I для оценки фармакокинетических показателей, относительной биодоступности, влияния пищи, безопасности и переносимости разных составов-прототипов на основе соединения формулы (I) у здоровых взрослых субъектов.
Методология
Речь пойдет об одноцентровом, открытом, рандомизированном, перекрестном исследовании с 4 периодами и применением однократной дозы у здоровых взрослых субъектов, спланированном для изучения фармакокинетики (РК) и безопасности до 4 жидких липидных составов-прототипов на основе соединения формулы (I) (пероральный раствор соединения формулы (I), составы-прототипы, 50 мг/мл), состава на основе соединения формулы (I) в виде высушенной распылением дисперсии (гранулы соединения формулы (I) для вскрываемых капсул, 25-50 мг) и соединения формулы (I), капсулы с 50 мг (эталон). Планируется включение 36 субъектов, которые будут распределены в 3 когорты по 12 субъектов на когорту, с 6 субкогортами по 6 субъектов на субкогорту. В каждой из этих 6 субкогорт, 6 субъектов будут определены в одну из 3 субкогорт, в которой однократную пероральную дозу исследуемого лекарственного средства (IMP) вводят в рамках 4 периодов введения дозы (периоды 1-4) в сытом состоянии или натощак, или в одну из 3 субкогорт, в которой однократную пероральную дозу IMP вводят в рамках 2 периодов введения дозы (периоды 1 и 2) только в сытом состоянии. В пределах каждой субкогорты субъекты будут также рандомизированы до введения первой дозы IMP в рамках периода 1 в соответствии с одной из следующих последовательностей лечения (таблица 40):
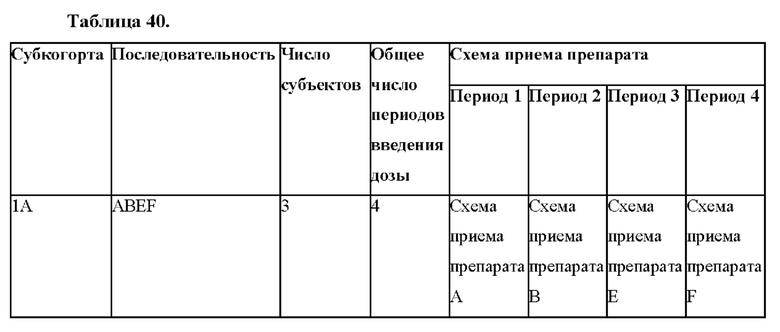
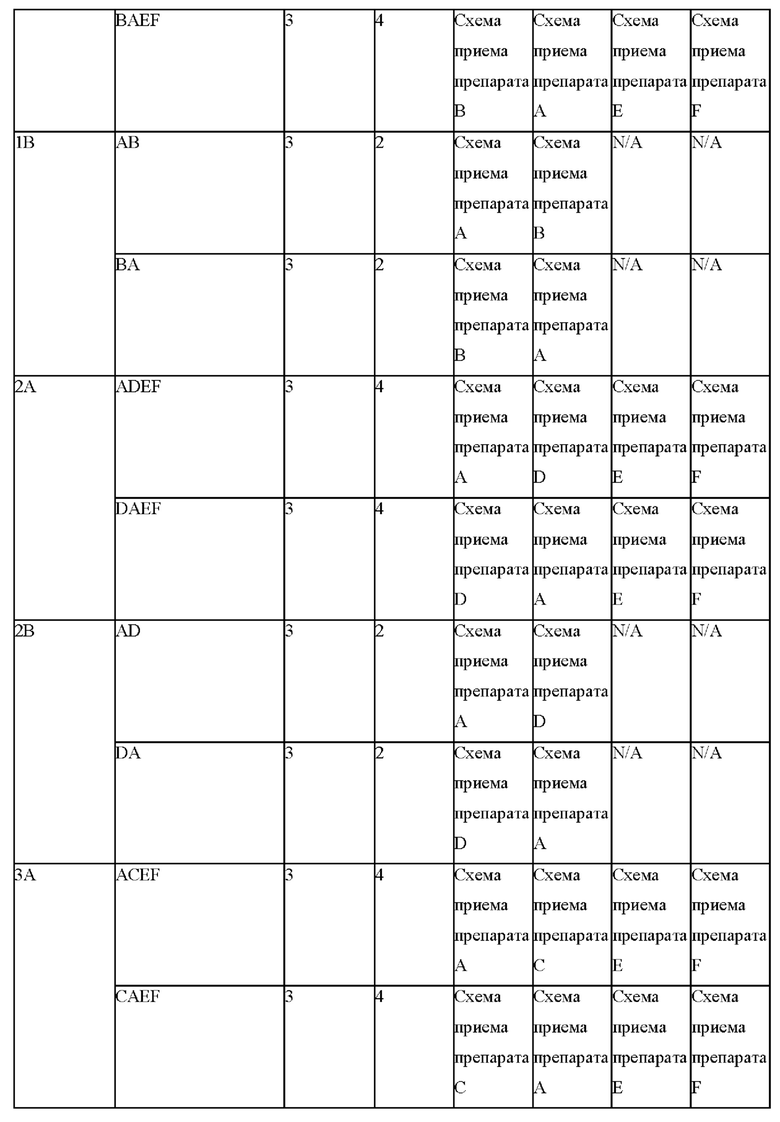
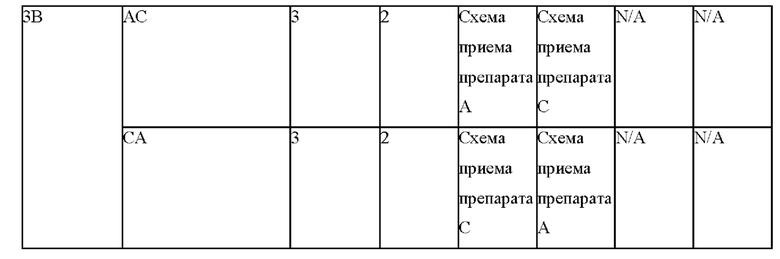
При заполнении информированного согласия субъекты будут соглашаться принимать участие в 2 периодах исследования или 4 периодах исследования. После определения в субкогорту порядок, в котором субъекты будут получать исследуемые препараты, будет рандомизирован в соответствии с приведенным выше графиком.
Субъекты будут проходить до 4 схем приема препарата лечения в рамках до 4 периодов в определенном схемой рандомизации порядке в пределах каждой субкогорты.
Эффект разных относящихся к приему пищи состояний в отношении РК соединения формулы (I) можно изучать в рамках периодов 3 и 4 путем введения натощак или после приема альтернативной пищи (например, завтрака с высоким содержанием жиров, стандартного или легкого завтрака и т.д.).
Предлагаемые схемы приема препарата представлены в таблице 41 ниже:
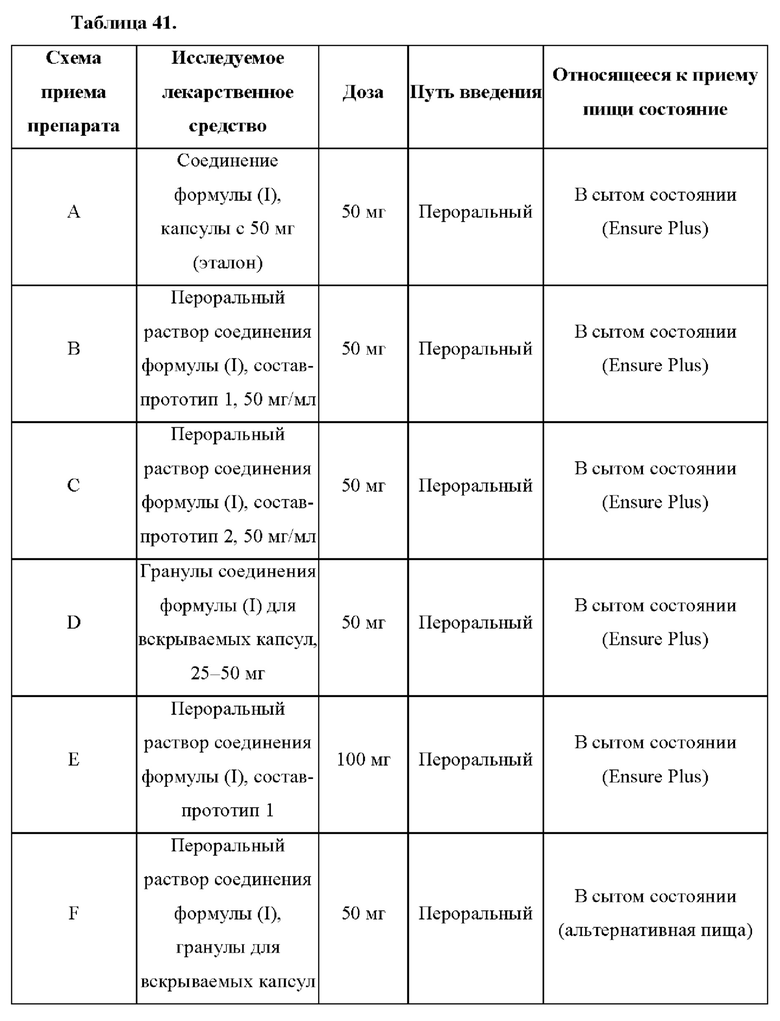
Дизайн исследования:
Субъекты будут проходить скрининг в отношении соответствия критериям для участия в исследовании на протяжении периода вплоть до 28 суток до введения первой дозы IMP в рамках периода 1. Каждый период исследования будет проходить по одному и тому же дизайну исследования. Субъекты будут приняты в клинике вечером до введения IMP (-1-е сутки). В случае периодов 1 и 2 (схема приема препарата А и одна из схем приема препарата В, С или D), все субъекты будут получать составы на основе соединения формулы (I) утром в соответствии со схемой рандомизации в сытом состоянии с жидкой пищевой добавкой (Ensure Plus). В случае периодов 3 и 4 (схемы приема препарата Е и F), субъекты будут получать составы на основе соединения формулы (I) утром в соответствии со схемой рандомизации в сытом состоянии с жидкой пищевой добавкой или в альтернативном относящемся к приему пищи состоянии (натощак или альтернативная пища). Введение IMP будет осуществляться на 1-е сутки с соответствующим интервалом между субъектами в зависимости от материально-технического обеспечения (приблизительно 10 мин). В рамках периодов приемы пищи будут стандартизированы для каждой схемы лечения.
Субъекты будут оставаться в клинике до истечения 36 ч после введения дозы, после чего они будут выписаны. Субъекты будут возвращаться в клинику через 168 ч (7 суток) и 336 ч (14 суток) после введения дозы для взятия образца крови для оценки РК и оценок безопасности. Минимальный период вымывания между процедурами введения дозы IMP будет составлять 14 суток между периодами 1 и 2 и 21 сутки или более для накопления промежуточных данных между периодами 2 и 3 и между периодами 3 и 4.
Через 18-24 суток после заключительного визита будет осуществляться телефонный звонок в рамках последующего наблюдения, чтобы убедиться, что субъекты все еще хорошее себя чувствуют.
После завершения периода 2 для всех когорт, будет проводиться обзор промежуточных данных, в ходе которого будут оценивать данные по РК и безопасности, а также любые возникшие релевантные данные о стабильности в контексте химического состава, процесса производства и контроля качества (CMC) для определения состава, уровня дозы и относящегося к приему пищи состояния, в котором следует вводить IMP, в периоде 3 (схема приема препарата Е). Аналогичный промежуточный обзор будет проводиться после завершения периода 3 (введение по схеме приема препарата Е) для определения состава, уровня дозы и относящегося к приему пищи состояния, в котором следует вводить IMP, в периоде 4 (схема приема препарата F). Критерии для принятия решений в рамках промежуточного анализа будут основываться на доступных РК-данных для соединения формулы (I): например, Cmax, Tmax, AUC(0-36), Frei и данных по безопасности.
Число субъектов согласно плану:
Планируется включение 36 здоровых субъектов мужского и женского (небеременных и не кормящих) пола в 6 субкогорт по 6 субъектов на субкогорту. Эти субкогорты будут объединены в группы по 2 для создания 3 когорт с n=12 для периодов 1 и 2 с целью получения данных у 10 подходящих для оценки субъектов в каждой когорте в контексте первичных целей на вариант состава. Всего 18 субъектов, 6 из каждой из субкогорт 1А, 2А и ЗА, участвующих в периодах 1 и 2, дополнительно будут участвовать в периодах 3 и 4, с целью получения данных минимум для 6 подходящих для оценки субъектов. Субъект будет считаться подходящим для оценки в рамках конкретной схемы приема препарата, если он/она получали IMP и прошли достаточно запланированных оценок РК за период времени вплоть до 336 ч (14 суток) после введения дозы для такой схемы приема препарата, чтобы можно было оценить конечные точки исследования. Субъект будет считаться подходящим для оценки с целью конкретного сравнения (например, влияния пищи, относительной биодоступности), если он/она получали оба сравниваемых IMP и для них имеется достаточно РК-данных за период времени вплоть до 14 суток после прохождения каждой схемы приема препарата, чтобы можно было оценить конечные точки исследования.
Субъекты, исключенные из исследования из-за связанного с IMP нежелательного явления (АЕ), не будут замещены. Субъекты, исключенные из исследования по другим причинам, могут быть замещены по соглашению между главным исследователем (PI) и спонсором для обеспечения достаточного числа подходящих для оценки субъектов к концу каждого периода исследования. Для замещающих субъектов может потребоваться введение дозы определенных составов из предыдущих схем приема препарата с целью получения минимального числа подходящих для оценки субъектов, необходимых для промежуточных решений и для получения данных для любой другой схемы приема препарата, которая требуется для выполнения сравнений в рамках целей исследования, за исключением того, что любая предыдущая доза IMP, которая считалась субоптимальной, вводиться не будет. В общей сложности в исследование могу быть включены до 8 замещающих субъектов. Максимальное число субъектов, которым можно вводить дозы, в общей сложности составляет 44.
Если субъект исключен из субкогорты 1А, 2А или 3А после периода 2, допускается его/ее замещение субъектом из субкогорты 1B, 2В или 3В, при условии, что субъект подпишет обновленную форму согласия, давая согласие на участие в четырех периодах лечения. По усмотрению исследователя, такой субъект может не проходить повторные процедуры скрининга.
Продолжительность исследования:
В случае субъектов, включенных для получения однократной дозы в 4 отдельных случаях в периоды с 1 по 4 (субкогорты 1А, 2А и 3А), расчетное время с момента скрининга до осуществления телефонного звонка в рамках последующего наблюдения составляет примерно 15-16 недель.
В случае субъектов, включенных для получения однократной дозы в 2 отдельных случаях только в периоды с 1 по 2 (субкогорты 1B, 2В и 3В), расчетное время с момента скрининга до осуществления телефонного звонка в рамках последующего наблюдения составляет примерно 8-9 недель.
Оценки фармакокинетических показателей
Данные по концентрации в плазме крови соединения формулы (I) будут проанализированы для финального отчета Quotient Sciences и промежуточных обзоров Neurocrine Biosciences, Inc. (NBI) с применением Phoenix WinNonlin v8.0 или ее более новой версии (Certara USA, Inc., США). NBI будет ответственна за РК-анализ для промежуточного обзора.
РК-анализ полученных данных по зависимости концентрации от времени будут осуществлять с применением соответствующих некомпартментных методик для получения показателей следующих РК-параметров (таблица 42), где это возможно и целесообразно:
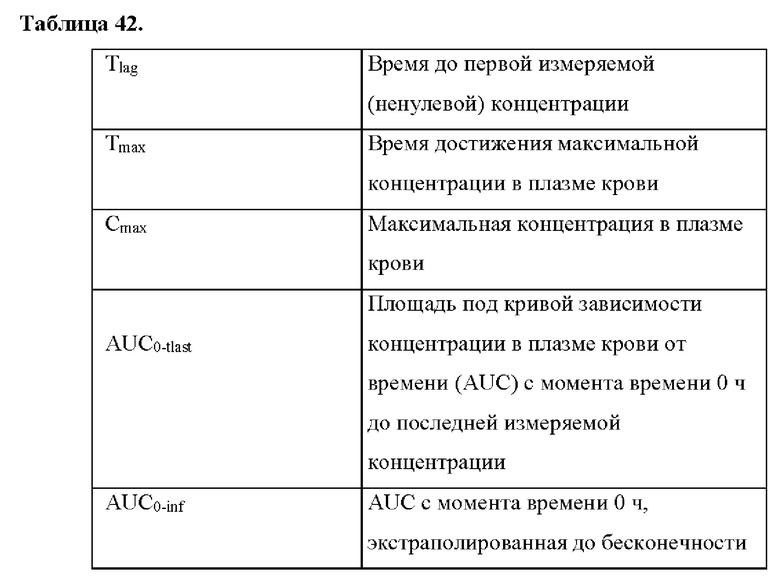

Оценки вкусовых качеств:
Вкусовые качества будут оцениваться для каждого IMP-состава и основы (например, Ensure Plus, легкая пища) с применением опросника, разработанного с этой целью и адаптированного по мере необходимости для данного конкретного исследования.
В опроснике субъектам будет предложено оценить приемлемость запаха, сладости, горечи, вкуса, привкуса и послевкусия по 6-балльной шкале, а общие ощущения - по 5-балльной шкале для каждого IMP-состава независимо от любых предыдущих составов.
Статистическая методология:
Будут предоставлены описательные сводки для всех данных по безопасности, оценок РК и данных опросника удовлетворенности вкусовых качеств. Проверка гипотез в отношении данных по безопасности или опросника удовлетворенности вкусовых качеств проводиться не будет.
Периоды 1 и 2 - для каждой когорты отдельно
Относительная биодоступность
Статистическое моделирование будет осуществляться по РК-параметрам (AUC(0-tlast), AUC(0-inf) и Cmax) для соединения формулы (I), преобразованным на основе натурального логарифма, для оценки относительной биодоступности (Frel) с применением модели со смешанными эффектами с параметрами для схемы приема препарата, периода и последовательности в качестве фиксированных эффектов и субъекта в рамках определенной последовательности в качестве случайного эффекта. Будут представлены средние геометрические соотношения (GMR) и 90% доверительный интервал (CI) для релевантного представляющего интерес сравнения, т.е. между каждым из составов-прототипов (пероральный раствор соединения формулы (I), состав-прототип 1, 50 мг/мл, например, пример 12; пероральный раствор соединения формулы (I), состав-прототип 2, 50 мг/мл, например, пример 13; и гранулы соединения формулы (I) для вскрываемых капсул, 25-50 мг, например, пример 11 [схемы приема препарата В, С и D соответственно]) и соединение формулы (I), капсулы с 50 мг, например, пример 9 (эталон; схема приема препарата А).
Все периоды (периоды 1-4)
Влияние пищи
Статистическое моделирование будет осуществляться по РК-параметрам AUC(0-tlast), AUC(0-inf) и Cmax для соединения формулы (I) для оценки влияния пищи, в соответствующем случае. Преобразованные на основе натурального логарифма РК-параметры будут проанализированы в отношении биодоступности с применением модели со смешанными эффектами с параметрами для относящегося к приему пищи состояния (и типа пищи, если применимо) в качестве фиксированного эффекта и субъекта в качестве случайного эффекта. Будут представлены средние геометрические соотношения и 90% CI для релевантных представляющих интерес сравнений, где соотношение определяется как натощак/в сытом состоянии или пробный прием пищи/эталонный прием пищи (если применимо).
Относительная биодоступность
Статистическое моделирование будет осуществляться по РК-параметрам (AUC(0-tlast), AUC(0-inf) и Cmax) для соединения формулы (I), преобразованным на основе натурального логарифма, для оценки относительной биодоступности с применением модели со смешанными эффектами с параметрами для схемы приема препарата в качестве фиксированного эффекта и субъекта в качестве случайного эффекта. Будут представлены средние геометрические соотношения и 90% CI для релевантного представляющего интерес сравнения, т.е. между каждым из составов-прототипов (схемы приема препарата Е и F, IMP должны быть определены в рамках промежуточных обзоров после завершения периодов 2 и 3) и соединением формулы (I), капсулы с 50 мг (эталон; схема приема препарата А).
Результаты
Сводные предварительные РК-данные представлены в таблице 43 ниже:

Пример 15. Соединение формулы (I) в виде кристаллического свободного основания, форма I.
Пример 15А
Схема 1: Получение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(15)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропин-1-ил)-2-тиазоламина (соединение формулы (I), форма I)
Схема 1

Стадия 1: Получение (S)-2-циклопропил-1-(3-фтор-4-метилфенил)-N-(1-фенилэтил)этан-1-имина (соединение 3-А)
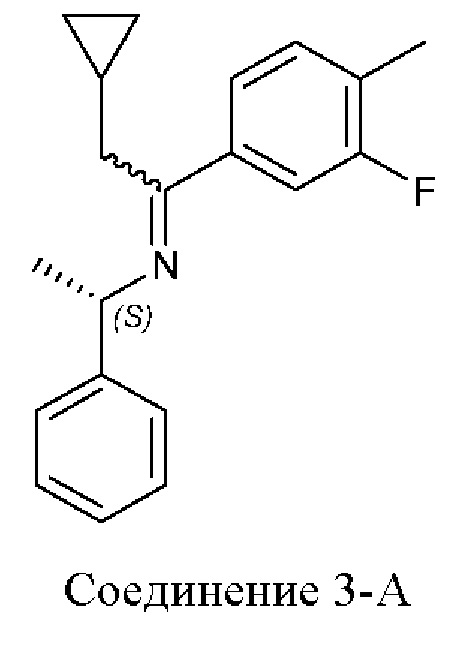
Смесь 2-циклопропил-1-(3-фтор-4-метилфенил)этан-1-она (1-А, 150,7 кг, 1 экв., в виде 27,6% вес./вес. раствора в толуоле, пример 15С), (&)-(-)-1-фенилэтиламина (2-А, 112,9 кг, 1,19 экв.) и п-толуолсульфоновой кислоты (7,4 кг, 0,05 экв.) нагревали с обратным холодильником при 110-120°С в течение 23-25 ч в реакторе, имеющем конфигурацию по типу аппарата Дина-Старка. Затем растворитель удаляли при 125 135°С при атмосферном давлении до прекращения дистилляции и добавляли часть толуола (275 кг, 2,24 вес./вес.) с получением суспензии. Суспензию нагревали с обратным холодильником при П0-120°С в течение 23-25 ч. Смесь охлаждали до 22°С и промывали дважды водным NH4CI (10%, 301,2 кг, 0,72 экв.) и один раз водным NaHCO3 (5%, 301,2 кг, 0,23 экв., проверка рН 8-9). Растворитель удаляли при 125-135°С и атмосферном давлении до достижения целевого объема 256 л, смесь фильтровали через целит, осадок на фильтре промывали толуолом (25 кг). Полученную в результате смесь, содержащую соединение 3-А, применяли непосредственно в следующей стадии без выделения. Выход определяли с поправкой на чистоту LOD и GC-FID образца (208,4 кг, 90,0% с поправкой, 0,89% соединения 2-A). EI-MS: 294,1 [М-Н]+, 190,1 [М-С6Н5СН(СН3)]+, 105,1 [С6Н5СН(СН3)]+.
Стадия 2: Получение гидрохлорида (5г)-2-циклопропил-1-(3-фтор-4-метилфенил)-N-((S)-1-фенилэтил)этан-1-амина (соединение 4-А)
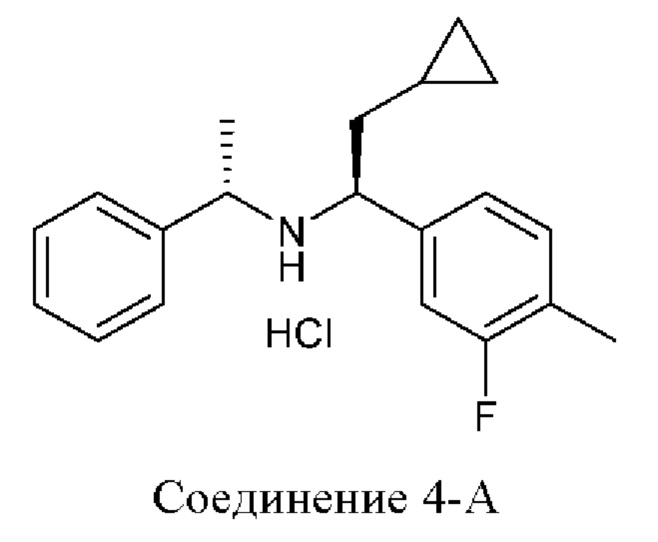
Никелевый губчатый катализатор (144 кг, 0,70 вес/вес, поставляемый в виде 50% вес/вес.суспензии в воде) добавляли в реактор гидрогенизации, оснащенный погружной трубой, способной удалять материал из верхней части массы внутри, сводя к минимуму количество введенной воды. Супернатант выбрасывали, добавляли этанол (329,3 кг, 1,58 вес./вес, безводный), суспензию перемешивали, а затем давали ей осесть. Этот процесс повторяли еще четыре раза и проверяли супернатант по Карлу Фишеру (KF) (≤1% H2O вес/вес). Соединение 3-А (208,4 кг, 1 экв., в виде 62,6% раствора в толуоле) добавляли к смеси в реактор гидрогенизации, и применяли этанол (387,6 кг, 1,86 вес./вес.) для промывки дополнительной колбы в реакторе гидрогенизации. В реакторе гидрогенизации два раза повышали/сбрасывали давление с использованием азота (2 бара) и два раза с использованием водорода (5 бар), затем повышали давление с использованием водорода (9,8-10,2 бара) и нагревали до 33-37°С и перемешивали в течение 17-19 ч. В системе три раза повышали/сбрасывали давление с использованием азота (1 бар) и суспензию фильтровали и три раза промывали этанолом (493,8 кг, 2,37 вес/вес). Добавляли НС1 (концентрированную, 83,4 кг, 1,07 экв.) и перемешивали смесь 25-35 мин при 20-24°С. Смесь концентрировали с помощью дистилляции при 78-80°С и атмосферном давлении с целью удаления воды с заданным объемом дистиллята 1167 л (5,6 л/кг, соединение 3-А) и проверяли раствор по KF (≤1,5% H2O вес./вес). Смесь перемешивали при 48-52°С в течение 55-65 мин, затем при 68-72°С в течение 55-65 мин, затем охлаждали до 20-24°С при скорости 12°С/ч и перемешивали в течение 25-35 мин, затем охлаждали до 0-4°С при скорости 10°С/ч и перемешивали в течение 55-65 мин. Суспензию фильтровали, осадок на фильтре дважды промывали предварительно охлажденным этанолом (329,2 кг, 1,58 вес./вес, 0°С) и собранное твердое вещество сушили при 40°С с получением соединения 4-А (156,5 кг, 66,4% без поправки). 1H ЯМР (400 МГц, DMSO-d6) δ ppm -0,33-0,06 (m, 2Н) 0,11-0,31 (m, 3Н) 1,57 (d, J=6,57 Гц, 3Н) 1,95 (br X, J=1,01 Гц, 2Н) 2,26 (d, J=1,26 Гц, 3Н) 3,68 (br d, J=7,83 Гц, 1H) 3,92 (br t, J=6,44 Гц, 1H) 6,98 (dd, J=7,71, 1,14 Гц, 1H) 7,28-7,36 (m, 2Н) 7,37-7,50 (m, 5Н). ESI-MS: 298,2 m/z [М+Н]+.
Стадия 3: Получение гидрохлорида (5)-2-циклопропил-1-(3-фтор-4-метилфенил)этан-1-амина (соединение 5-А)
Соединение 4-А (156,5 кг, 1,00 экв.) и Pd/C (7,8 кг, 10% по Pd) добавляли в реактор гидрогенизации в инертной атмосфере. Затем в реакторе два раза повышали/сбрасывали давление с использованием азота (2 бара), а затем добавляли метанол (494,5 кг, 3,16 вес./вес). В реакторе три раза повышали/сбрасывали давление с использованием азота (2 бара), затем три раза с использованием водорода (5 бар), повышали давление с использованием водорода (9,8-10,2 бар), нагревали до 58-62°С и перемешивали в течение 7-9 ч. Реакционную смесь охлаждали до 20-24°С. В реакторе три раза повышали/сбрасывали давление с использованием азота (1 бар) и суспензию фильтровали и три раза промывали метанолом (492,9 кг, 3,15 вес./вес). Раствор концентрировали при 63-67°С и атмосферном давлении до достижения заданного объема дистиллята 1408 л (9,0 л/кг соединения 4-А). Добавляли н-гептан (1173,8 кг, 7,5 вес/вес) и нагревали смесь с обратным холодильником при 65-80°С и атмосферном давлении в реакторе, имеющем конфигурацию по типу аппарата Дина-Старка, с целью удаления метанола. Суспензию охлаждали до 31-35°С и фильтровали, осадок на фильтре промывали н-гептаном (147,1 кг, 0,94 вес./вес.) и твердое вещество сушили при 40°С (101,0 кг, 93,8% без поправки, 99,2% ее). 1Н ЯМР (400 МГц, DMSO-d6) δ ppm -0,12-0,14 (m, 2Н) 0,26-0,42 (m, 2Н) 0,44-0,55 (m, 1H) 1,70-1,83 (m, 2Н) 2,23 (d, J=1,52 Гц, 3Н) 4,24 (t, J=7,33 Гц, 1Н) 7,22-7,29 (m, 1H) 7,29-7,36 (m, 1H) 7,40 (dd, J=10,99, 1,39 Гц, 1H). ESI-MS: 194,2 [М+Н]+, 177,0 [M-NH2]+.
Стадия 4: Получение (5)-4-(2-хлор-4-метокси-5-метилфенил)-N-(2-циклопропил-1-(3-фтор-4-метилфенил)этил)-5-метилтиазол-2-амина (соединение 7-А)
Перемешивали смесь н-гептана (146 кг), воды (142 кг), соединения 5-А (57,4 кг) и водного раствора гидроксида натрия (30% вес/вес, 41,0 кг) перемешивали. Разделяли слои и удаляли водный слой. Органический слой промывали водой (170 кг) и разделяли слои. Органический слой выделяли с применением н-гептана (40 кг) для промывки, и в реактор добавляли н-гептан (145 кг) и 1-(2-хлор-4-метокси-5-метилфенил)-2-тиоцианатопропан-1-он (6-А, 66,1 кг) и нагревали до 85°С. При 84-85°С в реактор добавляли выделенный ранее органический слой, содержащий свободное основание соединения 5-А, и промывали его н-гептаном (20 кг). Полученную в результате смесь перемешивали в течение 2 ч при 83°С.Далее растворитель заменяли на метанол посредством четырех добавлений по принципу «положи и возьми»/вакуумной дистилляции метанола (180 кг) при 55°С, с заданным остающимся в реакторе объемом, составляющим 287 л. Суспензию охлаждали до 5°С и добавляли воду (570 кг) в течение 4 ч при 5-10°С, при этом первые 60 кг добавляли очень медленно. Суспензию выдерживали 2 ч при °С, а затем выделяли путем фильтрации и промывали смесью метанол/вода (91/115 кг), а затем смесью метанол/вода (134/57 кг). Желтое твердое вещество сушили при 25°С и 1 мбар в течение 17 ч, затем 40°С и 1 мбар в течение 22 ч с получением соединения 7-А (97,4 кг, выход 87,5%). 1Н ЯМР (400 МГц, DMSO-d6) δ ppm -0,01-0,14 (m, 2Н) 0,29-0,42 (m, 2Н) 0,61-0,73 (m, 1H) 1,47 (dt, J=13,83, 6,85 Гц, 1H) 1,76 (dt, J=13,89, 7,20 Гц, 1Н) 2,00 (s, 3Н) 2,11 (s, 3Н)2,19 (d, J=1,01 Гц, 3Н) 3,82 (s, 3Н) 4,54 (q, J=7,58 Гц, 1Н) 7,00 (s, 1H) 7,06 (d, J=0,76 Гц, 1H) 7,08-7,14 (m, 2Н) 7,18-7,23 (m, 1H) 7,89 (d, J=8,08 Гц, 1Н). ESI-MS: 445,3 m/z [М+Н]+.
Стадия 5: Получение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропин-1-ил)-2-тиазоламина (соединение формулы (I))
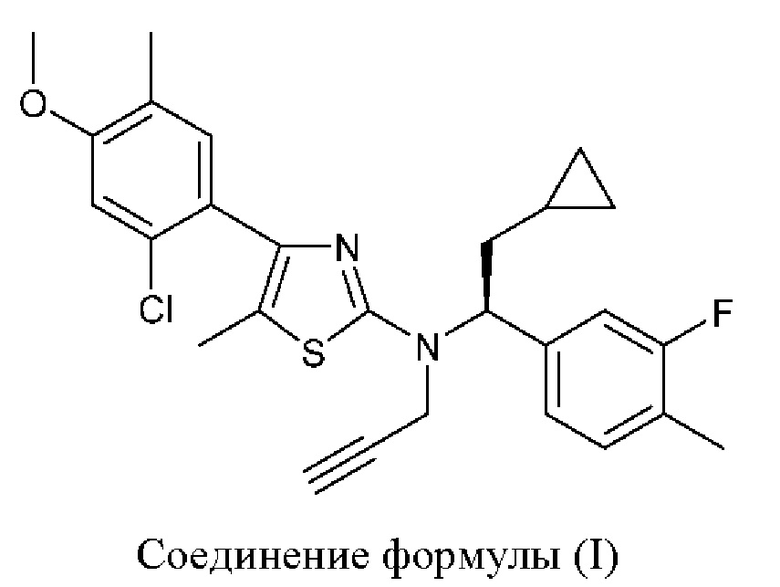
Смесь МТВЕ (279 кг), тетра-н-бутиламмония бромида (10,5 кг) и соединения 7-А (95,4 кг) нагревали при наружной температуре 60°С в течение 30 мин, а затем охлаждали до 0°С. Добавляли водный раствор гидроксида калия (52,4% вес/вес, 364 кг) и пропаргилбромид (39,4 кг в виде 80% вес./вес. раствора в толуоле, 1,19 экв.) при 0-5°С и двухфазную смесь выдерживали 14,5 ч при 4-6°С с энергичным перемешиванием. Далее добавляли воду (191 кг) и сливали водный слой. Органический слой два раза промывали водой (382 кг) и один раз водным раствором уксусной кислоты (5,26% вес./вес, 190 кг) при 20°С.Смесь окончательно фильтровали, промывали этанолом (11 кг), а затем растворитель заменяли на этанол посредством 3 добавлений по принципу «положи и возьми»/вакуумной дистилляции этанола (300 кг) при 25-30°С для первого цикла, а затем при 35-40°С, с заданным остающимся в реакторе объемом для каждого цикла, составляющим 250 л. Добавляли этанол (164 кг) и нагревали смесь при наружной температуре 60°С в течение 0,5 ч перед охлаждением до 25°С за 1 ч и вносили соединение формулы (I) в качестве затравки в исходном виде (0,340 кг), которое можно получать, как описано ниже в примере 15 В. Суспензию выдерживали в течение 5 ч, охлаждали до 0°С за 2 ч, выдерживали 12 ч, фильтровали и два раза промывали этанолом (24 кг каждый раз), предварительно охлажденным до 0°С. Белое твердое вещество сушили при 40°С и 1 мбар в течение 19 ч с получением 80,15 кг соединения формулы (I), форма I (выход 77,2%). 1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,14 (qt, J=8,59, 4,42 Гц, 2H) 0,29-0,48 (m, 2Н) 0,61-0,82 (m, 1H) 1,89 (dt, J=14,08, 6,98 Гц, 1H), 2,07 (br d, J=7,83 Гц, 1H) 2,10 (s, 3Н) 2,14 (s, 3H) 2,20 (d, J=1,01 Гц, 3H) 3,11 (t, J=2,27 Гц, 1H) 3,83 (s, 3H) 3,94-4,22 (m, 2H) 5,26 (t, J=7,58 Гц, 1H) 7,05 (s, 1H) 7,10-7,36 (m, 4H). ESI-MS: 483,2 m/z [M+H]+.
Кристалличность кристаллического соединения формулы (I) в виде свободного основания, форма I, подтверждали с помощью XRPD (фиг. 25, таблица 44) и дополнительно подтверждали с помощью DSC (фиг. 26), результаты которой указывают на наличие кристаллического соединения с началом плавления при приблизительно 84,4°С (71,9 Дж/г). С помощью TGA кристаллического свободного основания была показана приблизительно 0,6% потеря веса за счет растворителя/H2O.
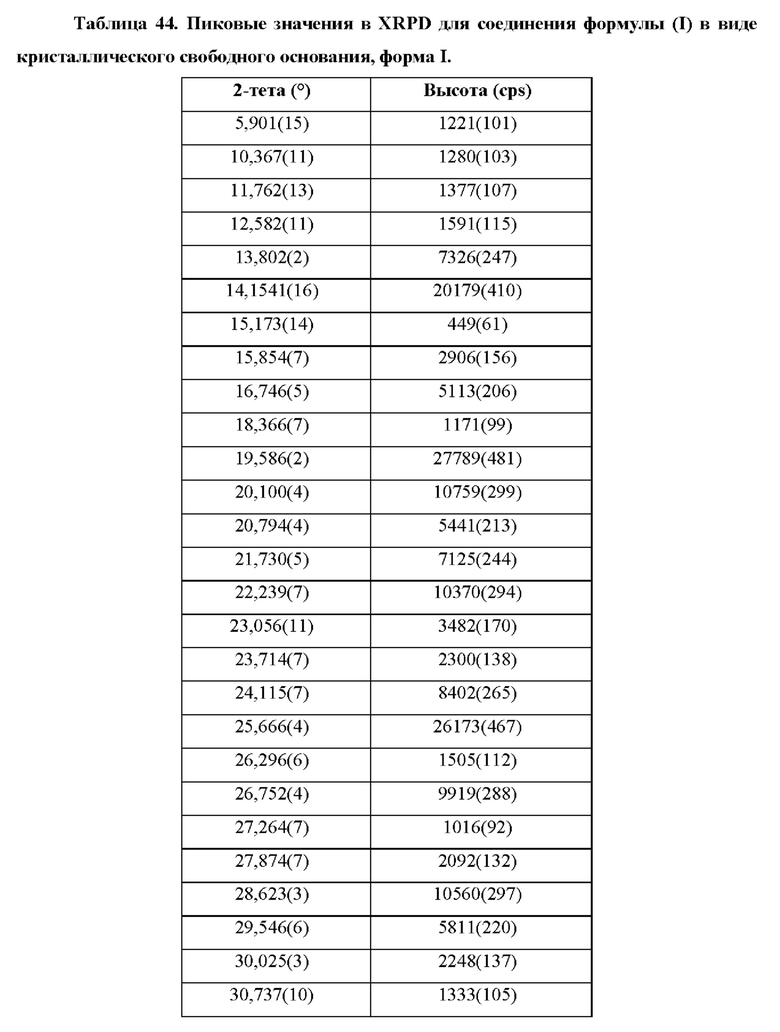
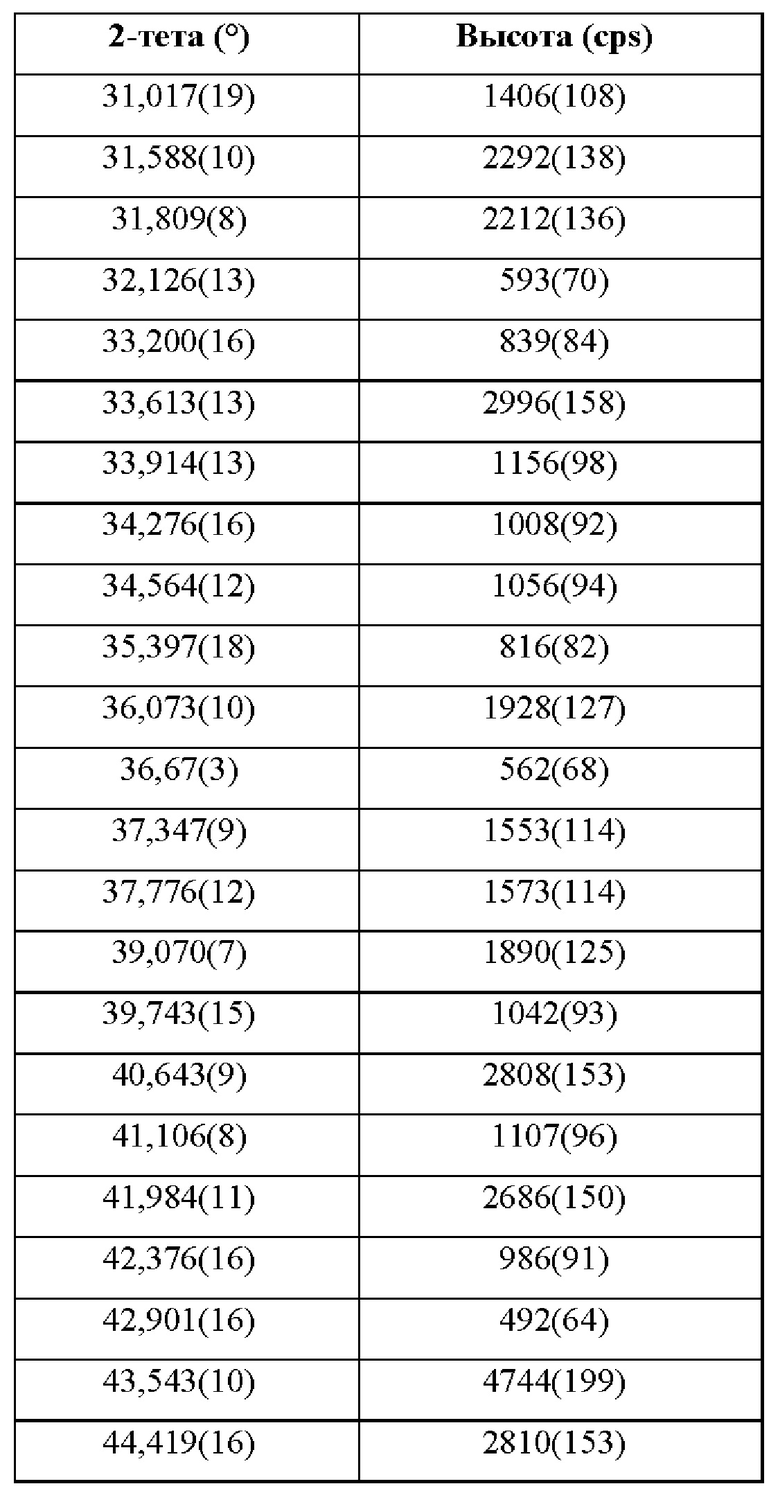
Пример 15В
Получение 4-(2-хлор-4-метокси-5-метилфенил)-N-[(15r)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропин-1-ил)-2-тиазоламина (соединение формулы (I), затравочная загрузка)
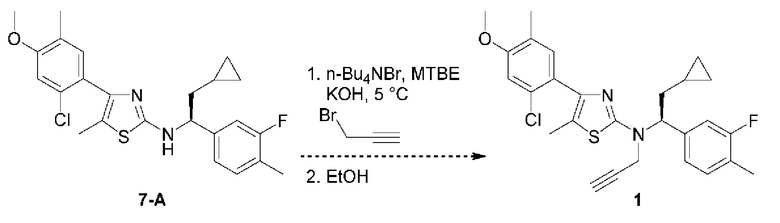
Смесь МТВЕ, тетра-н-бутиламмония бромида и соединения 7-А, охлажденную до 0°С, обрабатывали водным раствором гидроксида калия и пропаргилбромидом, поддерживая температуру на 0-5°С. Полученную в результате двухфазную смесь выдерживали 23 ч при 4-6°С. Далее добавляли воду и МТВЕ и сливали водный слой. Органический слой два раза промывали водой и один раз водным раствором уксусной кислоты при 20°С.Добавляли этанол, а затем растворитель заменяли на этанол посредством 3 добавлений по принципу «положи и возьми»/вакуумной дистилляции этанола при 35-40°С с заданным остающимся в сосуде объемом для каждого цикла, за исключением третьего цикла, в котором смесь концентрировали досуха. В сосуд добавляли этанол и нагревали смесь при наружной температуре 60°С в течение 0,5 ч перед ее охлаждением до 20°С за 1 ч и выдерживали 18 ч с получением суспензии. Суспензию охлаждали до 0°С, выдерживали 6 ч, фильтровали и два раза промывали этанолом, предварительно охлажденным до 0°С с получением твердого вещества. Твердое вещество сушили при 40°С в вакууме с получением соединения формулы (I).
Пример 15С
Схема 2: Получение 2-циклопропил-1-(3-фтор-4-метилфенил)этан-1-она (соединение 1-А)
Схема 2
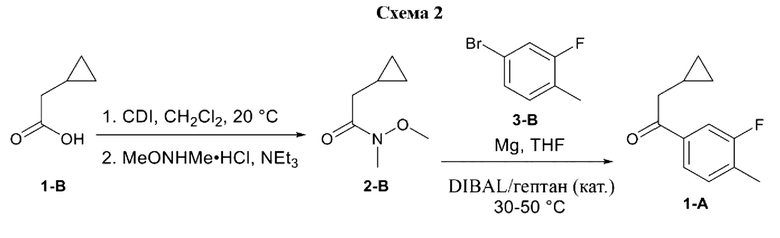
Стадия 1: Получение 2-циклопропил-N-метокси-N-метилацетамида (соединение 2-В)

Суспензию 1,1'-карбонилдиимидазола (152,6 кг, 1,01 экв.) в DCM (682 кг, 513 л, 7,3 вес/вес. относительно 2-циклопропилуксусной кислоты) обрабатывали раствором 2-циклопропилуксусной кислоты (1-В, 93,6 кг, 1 экв.) в DCM (248 кг, 186 л, 2,65 вес./вес.) в течение по меньшей мере 1 ч, поддерживая температуру ≤25°С и компенсируя значительное вскипание. Полученную в результате смесь перемешивали в течение 15 мин при 22°С, а затем частями добавляли N,O-диметилгидроксиламин*НС1 (93,3 кг, 1,03 экв.), поддерживая температуру ≤30°С. Далее при 20-25°С к перемешиваемой смеси добавляли триэтиламин (46,4 кг, 0,49 экв.). Полученную в результате смесь перемешивали при 22°С по меньшей мере 1 ч. Смесь один раз промывали раствором KHSO4 (0,24 М, 357,1 кг, 0,09 экв.), один раз раствором KHSO4 (0,40 М, 365,4 кг, 0,15 экв.), один раз раствором KHSO4 (0,80 М, 384,5 кг, 0,30 экв.) и один раз раствором NaHCO3 (0,60 М, 393,1 кг, 0,24 экв.). Остаточный DCM удаляли посредством трех процедур по принципу «положи и возьми» с использованием THF (166,6 кг, 1,78 вес./вес.) и вакуумной дистилляции (50-60°С, до достижения минимального объема/до остановки дистилляции). Добавляли THF (333,2 кг, 3,56 вес/вес.) и выход определяли с поправкой на чистоту LOD и GC-FID образца (131,5 кг, 98,2% с поправкой). 1H-ЯМР (400 МГц, DMSO-d6) δ: -0,01-0,03 (m, 2Н) 0,32-0,36 (m, 2Н) 0,81-0,90 (br m, 1Н) 2,18 (d, J=6,80 Гц, 2H) 2,97 (s, 3H) 3,53 (s, 3H). ESI-MS: 144,0 [M+H]+.
Стадия 2: Получение 2-циклопропил-1-(3-фтор-4-метилфенил)этан-1-она (соединение 1-А)

Mg (стружки, 28,6 кг, 1,37 экв.) суспендировали в THF (244,7 кг, 2,0 вес/вес.) и по каплям при 30°С добавляли DIBAL-H (1М в н-гептане, 18,9 кг, 0,03 экв.). Полученную в результате смесь перемешивали при 30°С в течение по меньшей мере 10 мин, а затем в течение по меньшей мере 30 мин при 30-50°С добавляли 4-бром-2-фтор-1-метилбензол (3-В, неразбавленное, 21,1 кг, 0,13 экв.). Далее смесь обрабатывали раствором 4-бром-2-фтор-1-метилбензола (3-В, 191,6 кг, 1,18 экв.) в THF (414,5 кг, 3,37 вес/вес.) при 30-50°С в течение 3 ч или меньше. Смесь перемешивали при 30°С в течение по меньшей мере 1 ч. Далее смесь обрабатывали 2-циклопропил-N-метокси-N-метилацетамидом (2-В, 123,0 кг, 1 экв., 25,9% вес./вес. раствор в THF) в течение по меньшей мере 1 ч при 15-25°С. Полученную в результате смесь перемешивали при 20-25°С в течение по меньшей мере 1 ч. Перемешиваемую смесь затем обрабатывали водным НС1 (3 М, 10,3% вес/вес, 668,9 кг, 2,24 экв.) при 10-25°С и полученную в результате смесь перемешивали по меньшей мере 2 ч (проверка рН 3,0-3,5). Слои разделяли и водный слой объединяли с гептаном (290,3 кг, 2,36 вес./вес). Слои разделяли и органический слой один раз промывали раствором NaHCO3 (0,63 М, 211,6 кг, 0,15 экв.) и один раз раствором NaCl (2,57 М, 213,0 кг, 0,55 экв.). Остаточные растворители удаляли посредством вакуумной дистилляции при 58-62°С до остановки дистилляции, а затем посредством одной процедуры по принципу «положи и возьми» с использованием толуола (275,5 кг, 2,24 вес/вес.) при 107-117°С до остановки дистилляции. Добавляли толуол (275,5 кг, 2,24 вес/вес.) и выход определяли с поправкой на чистоту LOD и GC-FID образца (150,7 кг, 91,3% с поправкой). 1Н ЯМР (400 МГц, DMSO-d6) δ ppm 0,07-0,21 (m, 2Н) 0,40-0,54 (m, 2Н) 1,02 (ttt, J=8,16, 8,16, 6,68, 6,68, 4,86, 4,86 Гц, 1H) 2,30 (d, J=1,77 Гц, 3Н) 2,91 (d, J=6,57 Гц, 2H) 7,44 (t, J=7,83 Гц, 1H) 7,57-7,78 (m, 2H). ESI-MS: 193,1 [M+H]+.
Пример 16. Соединение формулы (I) в виде кристаллической соли пар атолуолсульфокислоты, форма 1.
Примерно 20 мг соединения формулы (I) отвешивали во флакон. С применением пипетки позитивного вытеснения добавляли 250 мкл растворителя (IPA) во флакон вместе с якорем мешалки. Флакон помещали в алюминиевый блок на мешалке Reacti-Therm и нагревали до 60°С в течение ~1 часа. Во флакон добавляли молярный эквивалент пара-толуолсульфоновой кислоты (20 мкл 2М раствора в воде) и обеспечивали перемешивание. Образец медленно охлаждали до комнатной температуры вместе с мягким газообразным азотом для испарения. Осадок собирали, оставляли сушиться на ночь, а затем анализировали с помощью XRPD, DSC и TGA.
Кристалличность кристаллической соли паратолуолсульфокислоты, форма 1, подтверждали с помощью XRPD (фиг. 27, таблица 45) и дополнительно подтверждали с помощью DSC (фиг. 28), результаты которой указывают на наличие кристаллического соединения с началом плавления при приблизительно 156°С (22,2 Дж/г). Результаты TGA кристаллического соединения представлены на фиг. 28, и они демонстрируют приблизительно 0,5% потерю веса за счет растворителя/Н2О.
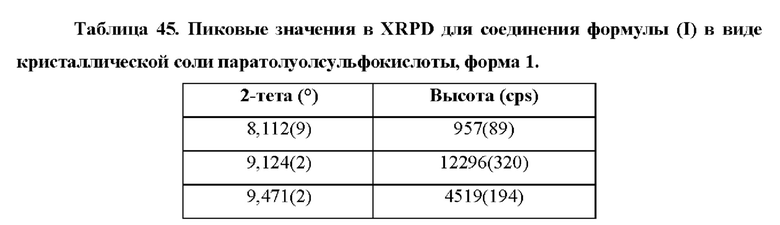
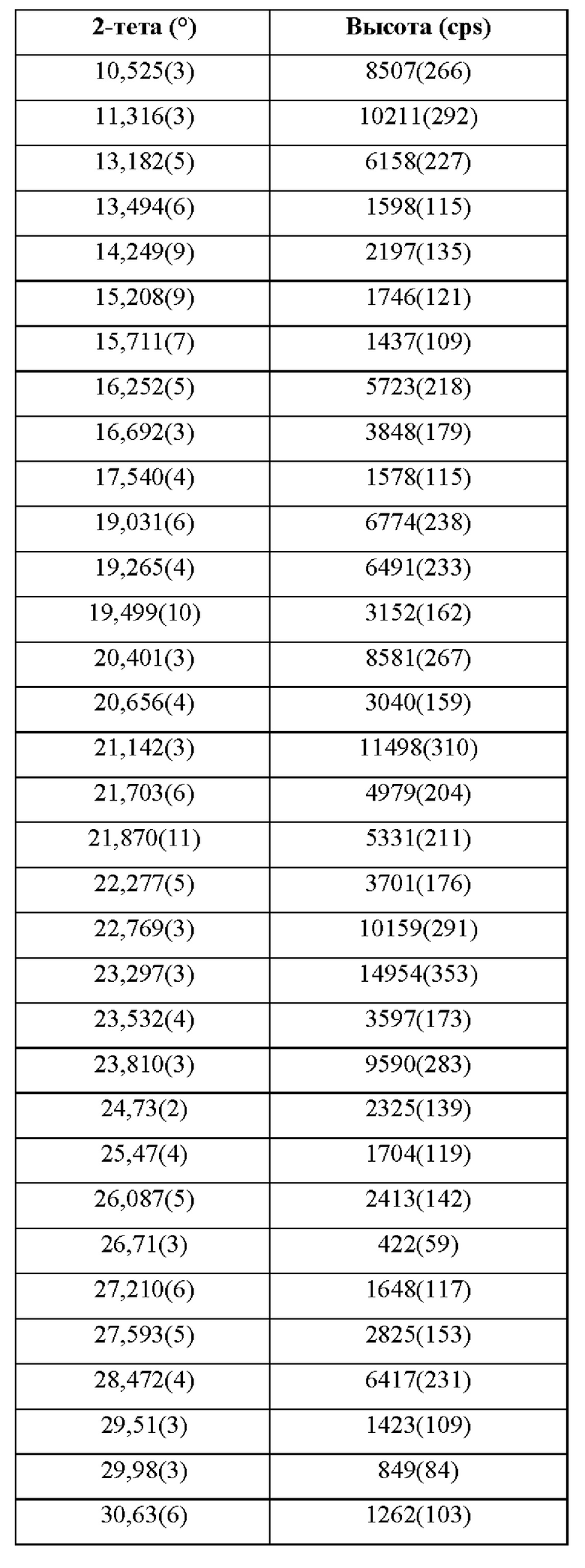
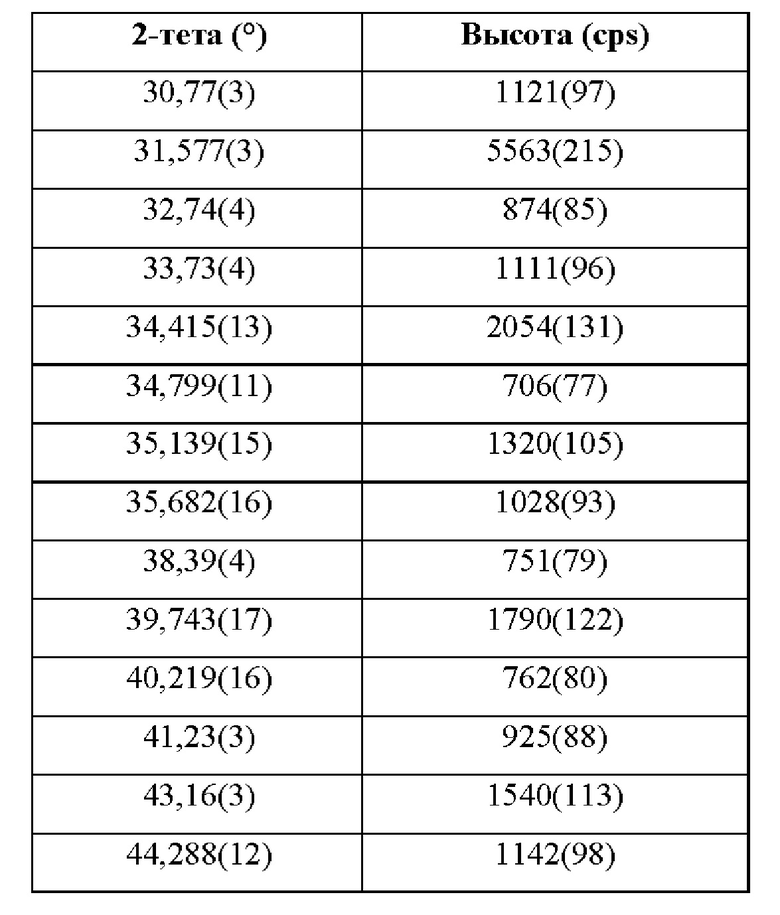
Другие варианты осуществления
Следует понимать, что вышеприведенное описание предназначено для иллюстрации, а не ограничения объема настоящего раскрытия, которое определяется объемом прилагаемой формулы изобретения. Другие аспекты, преимущества и модификации находятся в пределах объема следующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТАГОНИСТЫ РЕЦЕПТОРА CFR ДЛЯ ЛЕЧЕНИЯ ВРОЖДЕННОЙ ГИПЕРПЛАЗИИ КОРЫ НАДПОЧЕЧНИКОВ | 2015 |
|
RU2812318C2 |
| СПОСОБЫ ЛЕЧЕНИЯ СИНДРОМА ШЕГРЕНА С ПРИМЕНЕНИЕМ ИНГИБИТОРА ТИРОЗИНКИНАЗЫ БРУТОНА | 2020 |
|
RU2824354C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКОЙ СПОНТАННОЙ КРАПИВНИЦЫ С ПРИМЕНЕНИЕМ ИНГИБИТОРА ТИРОЗИНКИНАЗЫ БРУТОНА | 2020 |
|
RU2818678C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА CFR1 ДЛЯ ЛЕЧЕНИЯ ВРОЖДЕННОЙ ГИПЕРПЛАЗИИ КОРЫ НАДПОЧЕЧНИКОВ | 2015 |
|
RU2718918C2 |
| ПРОЛЕКАРСТВА АБИРАТЕРОНА | 2020 |
|
RU2822219C2 |
| Твёрдые лекарственные формы палбоциклиба | 2016 |
|
RU2686840C1 |
| ПОВЫШЕНИЕ БИОДОСТУПНОСТИ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ТЕРАПИИ НАЛТРЕКСОНОМ | 2011 |
|
RU2640561C2 |
| ПОВЫШЕНИЕ БИОДОСТУПНОСТИ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ТЕРАПИИ НАЛТРЕКСОНОМ | 2011 |
|
RU2781141C2 |
| КИШЕЧНОРАСТВОРИМЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2018 |
|
RU2812901C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ АГОНИСТОВ FXR | 2017 |
|
RU2737324C2 |

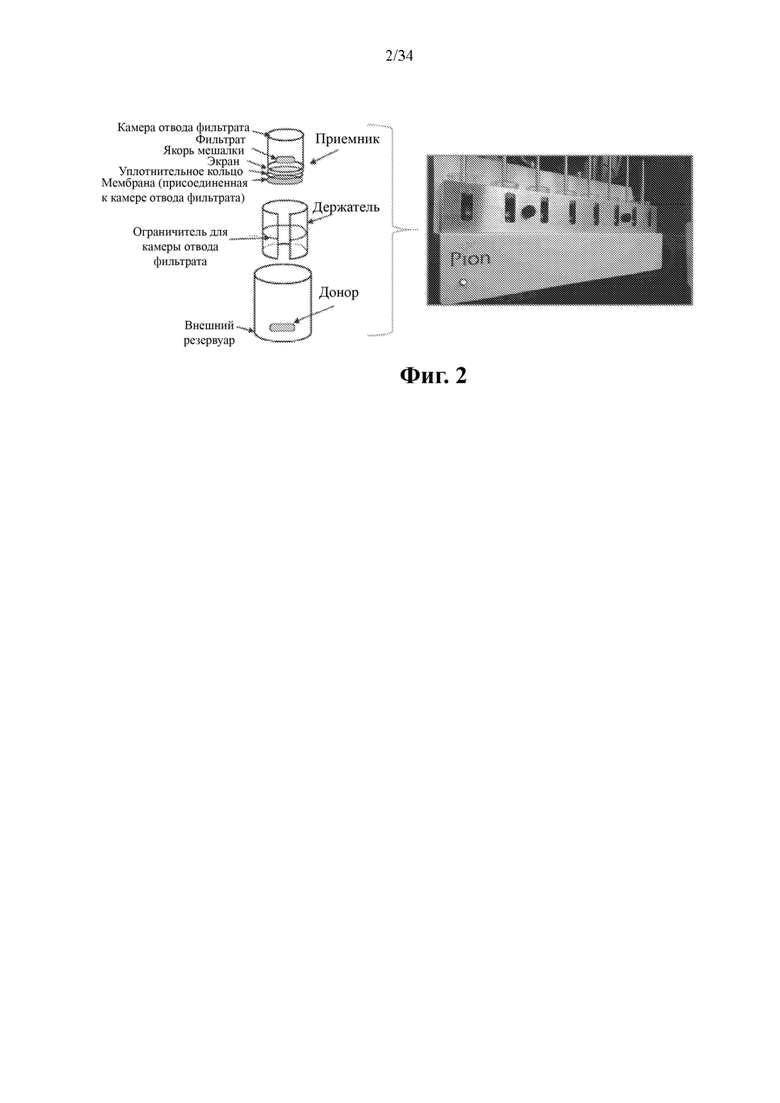
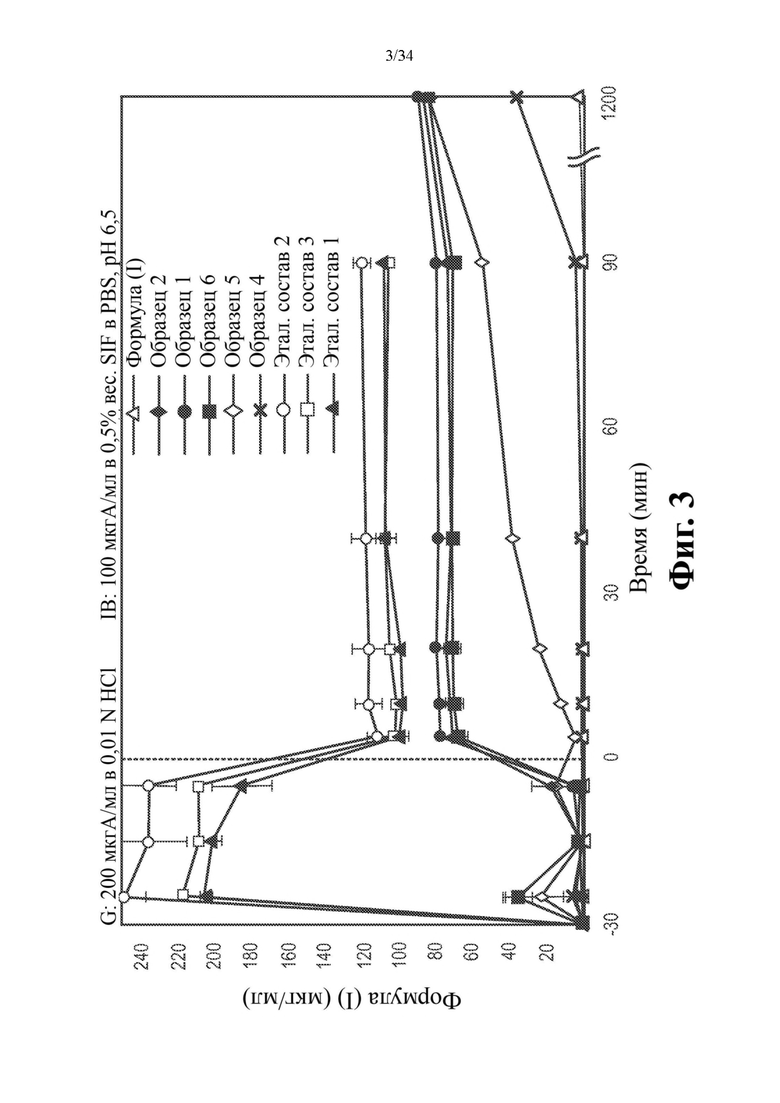
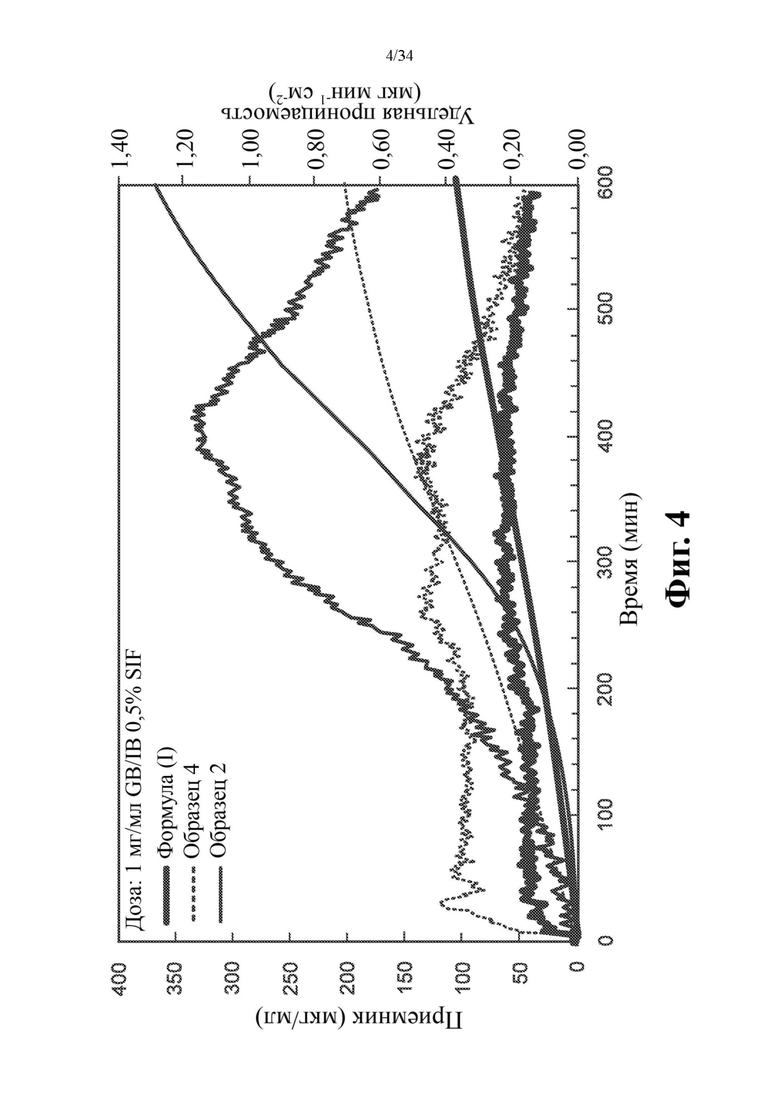
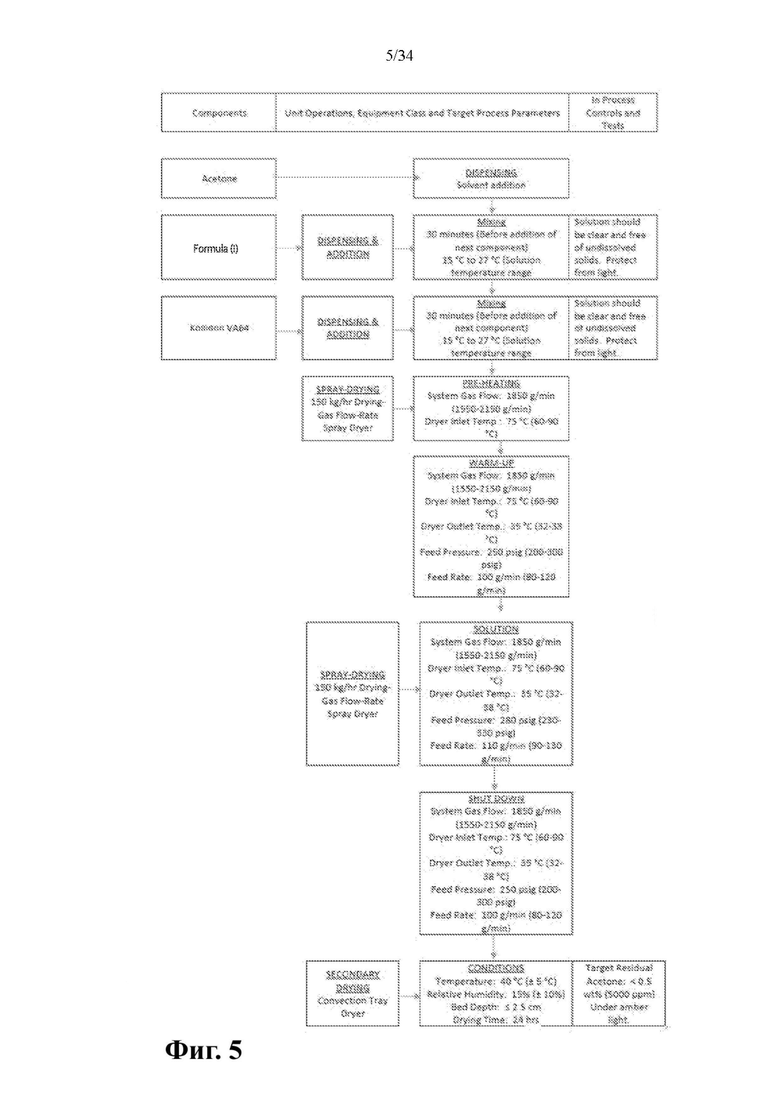
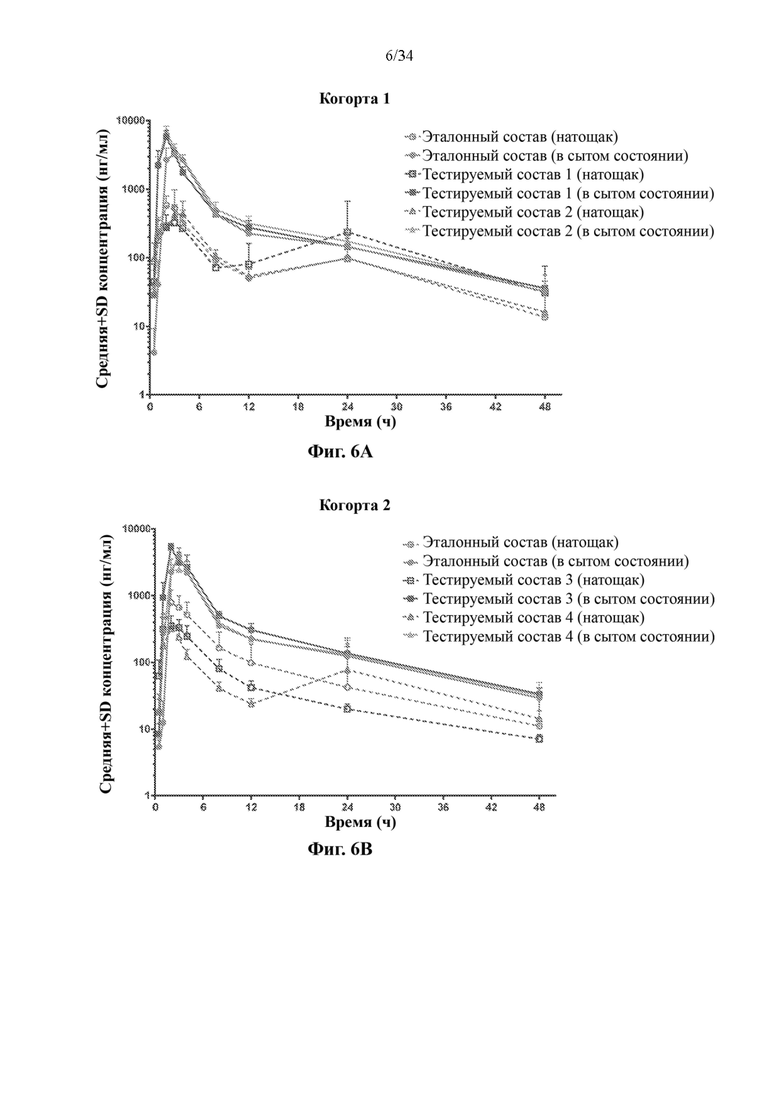
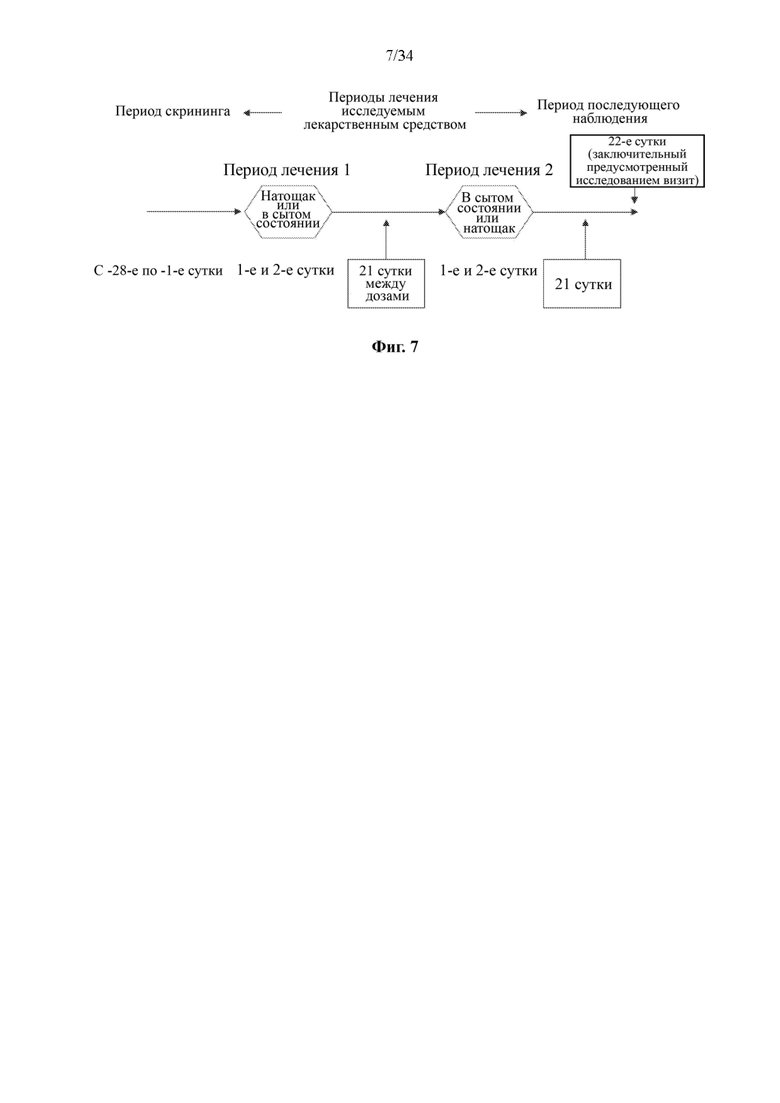


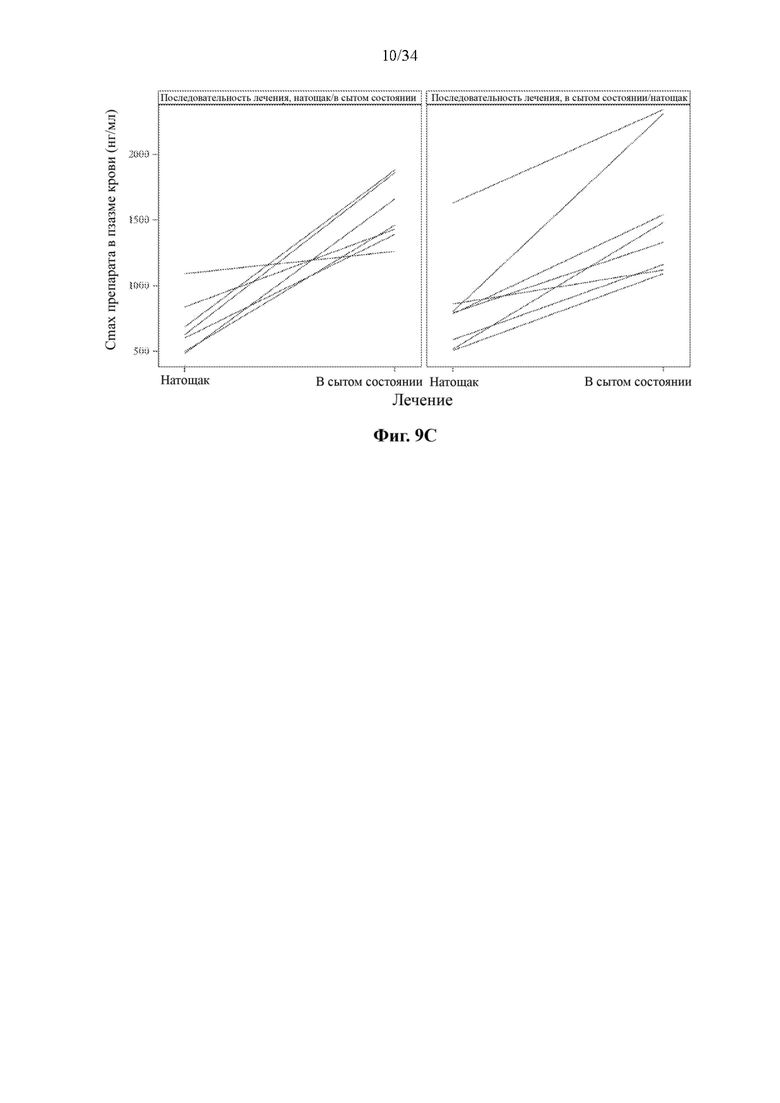


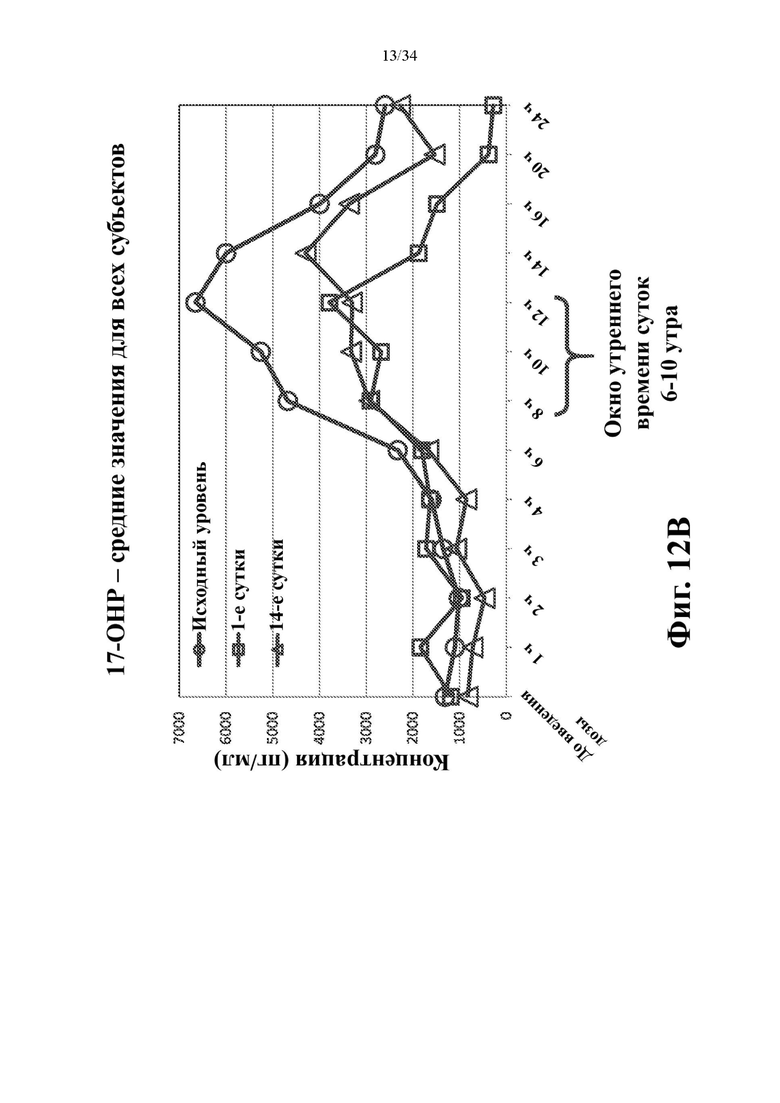

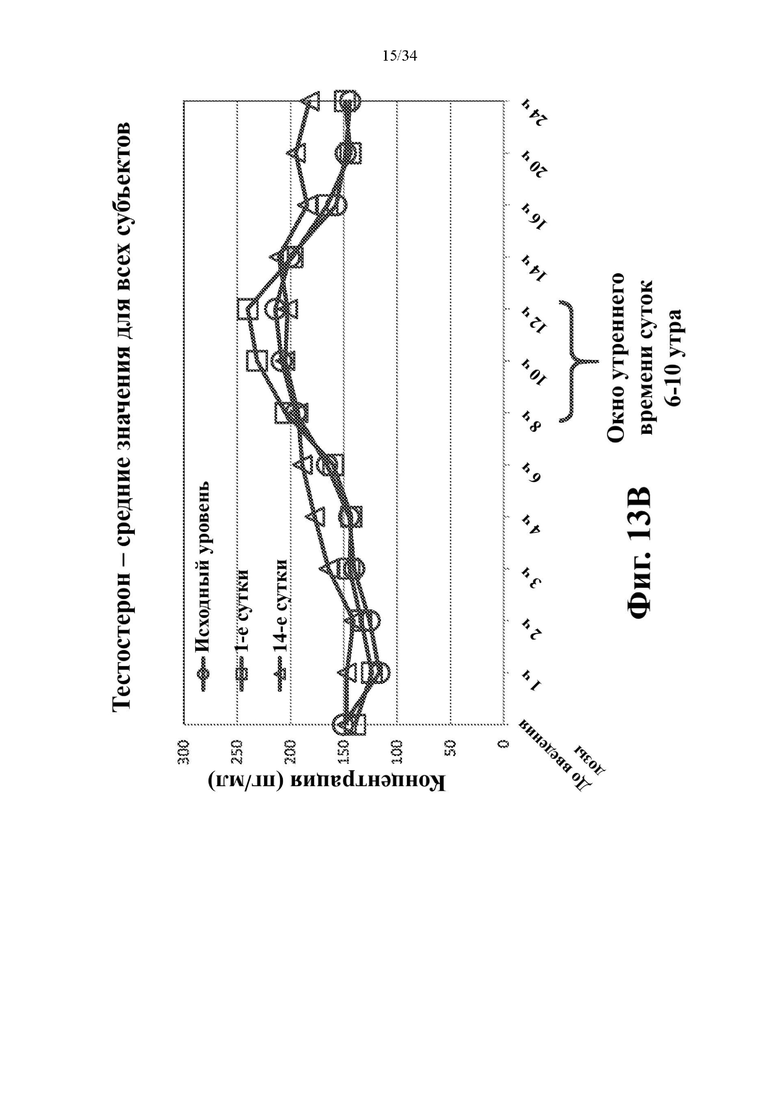
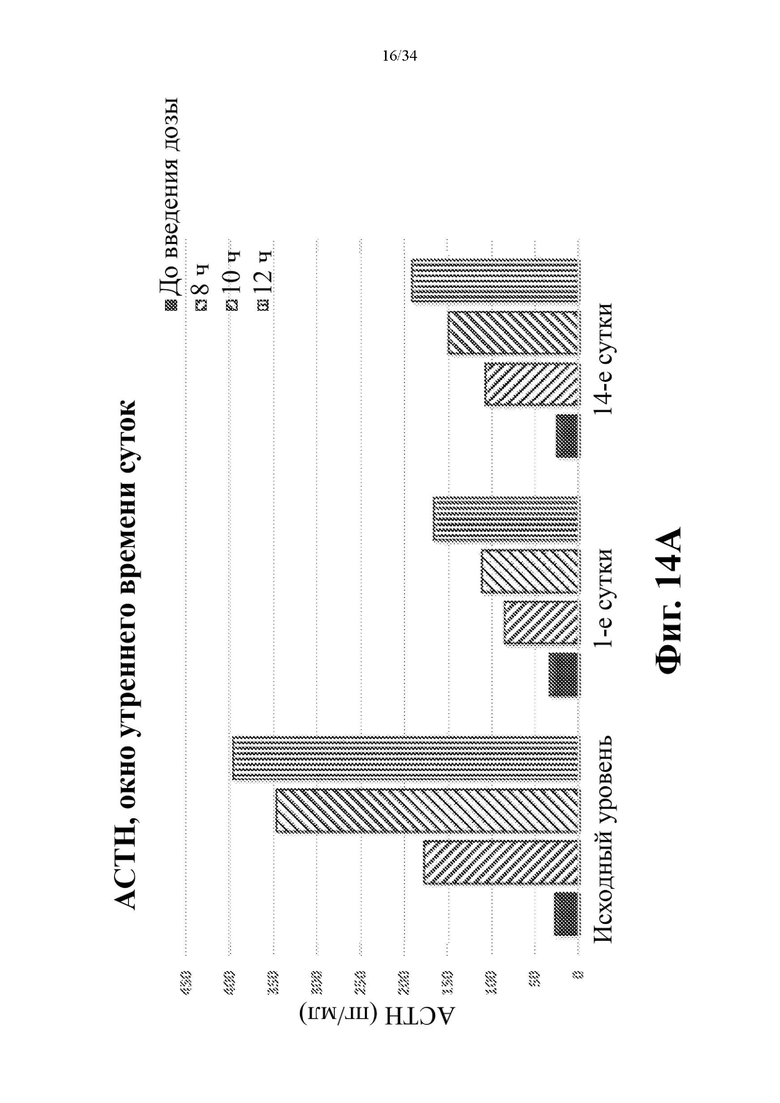
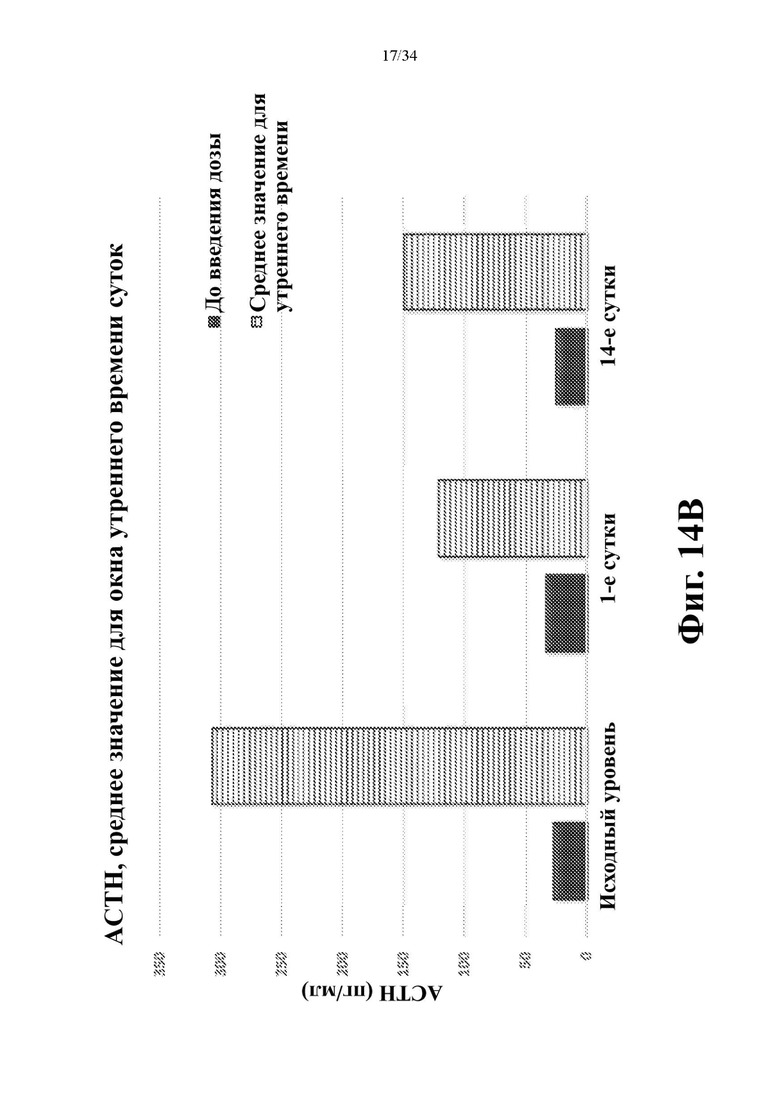
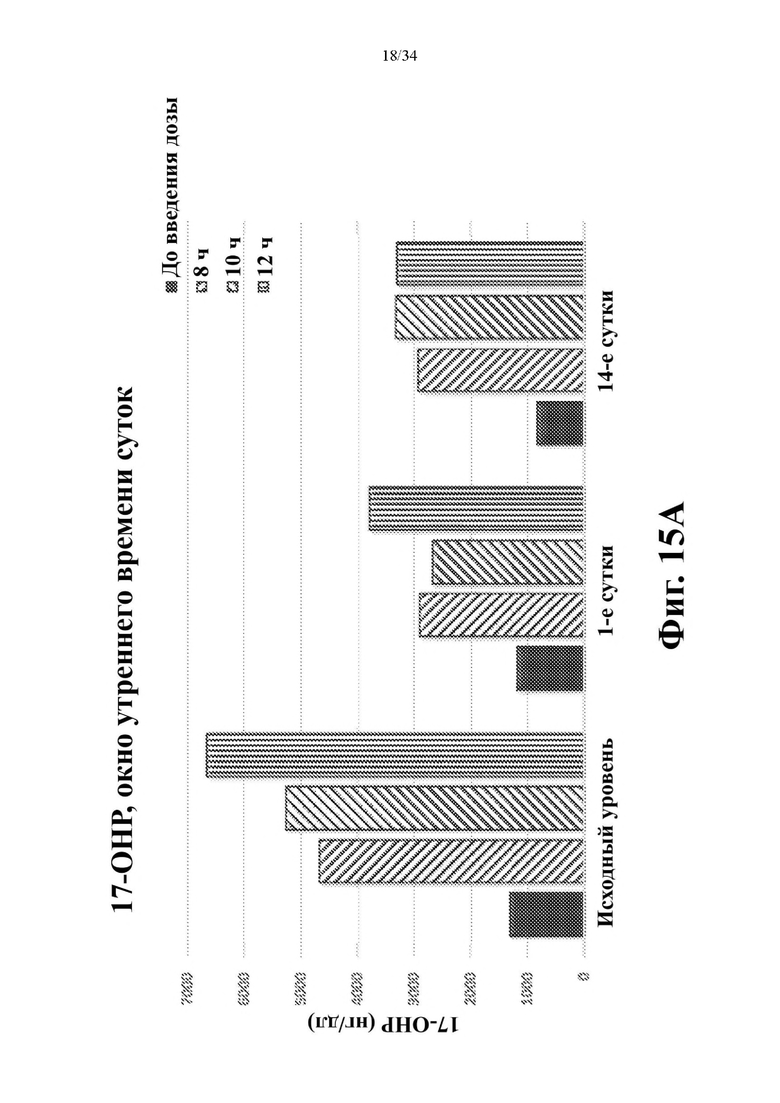


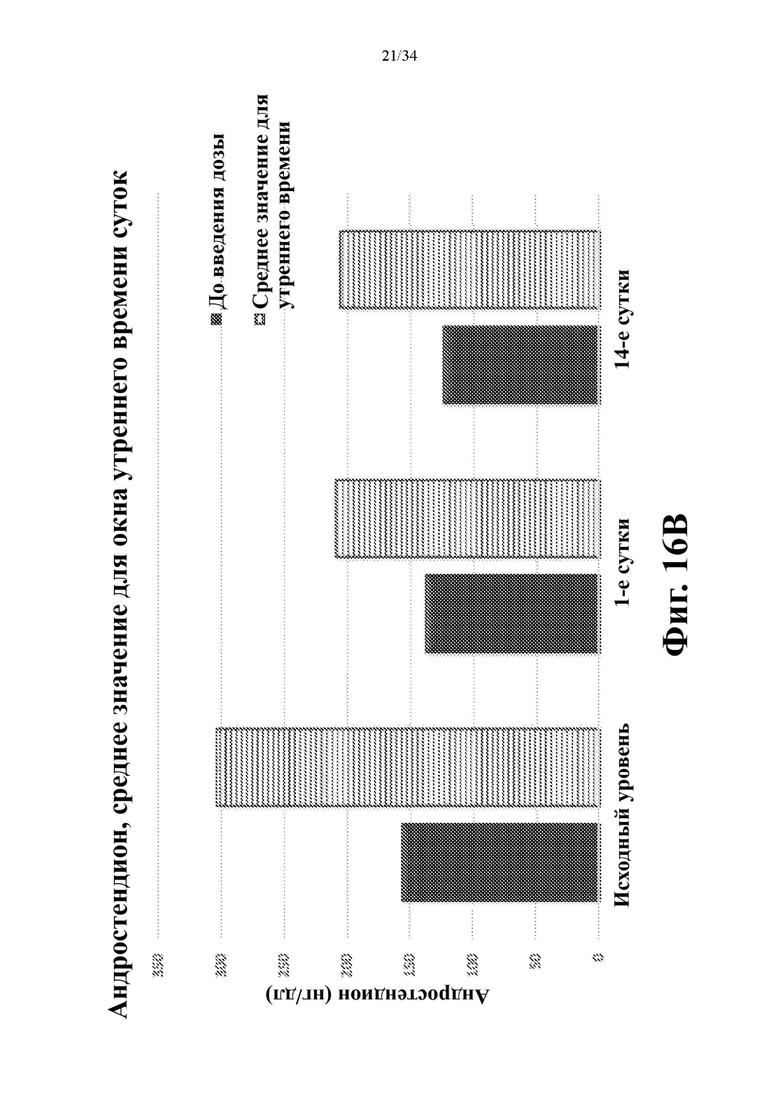
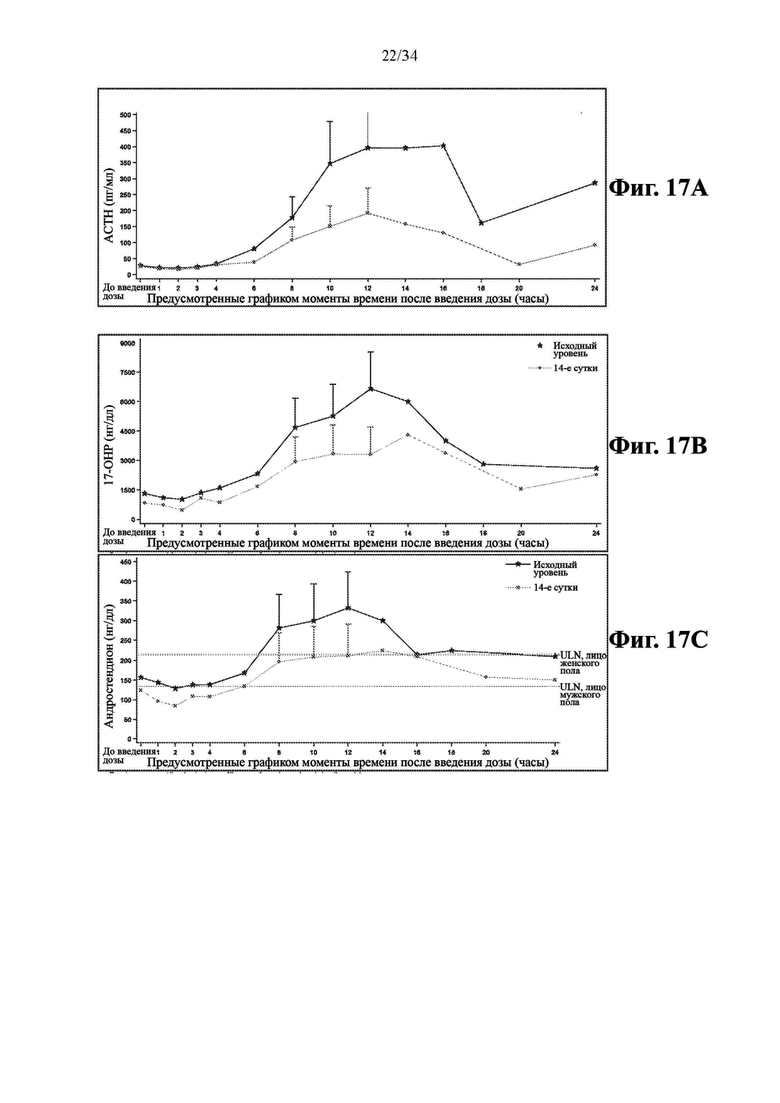


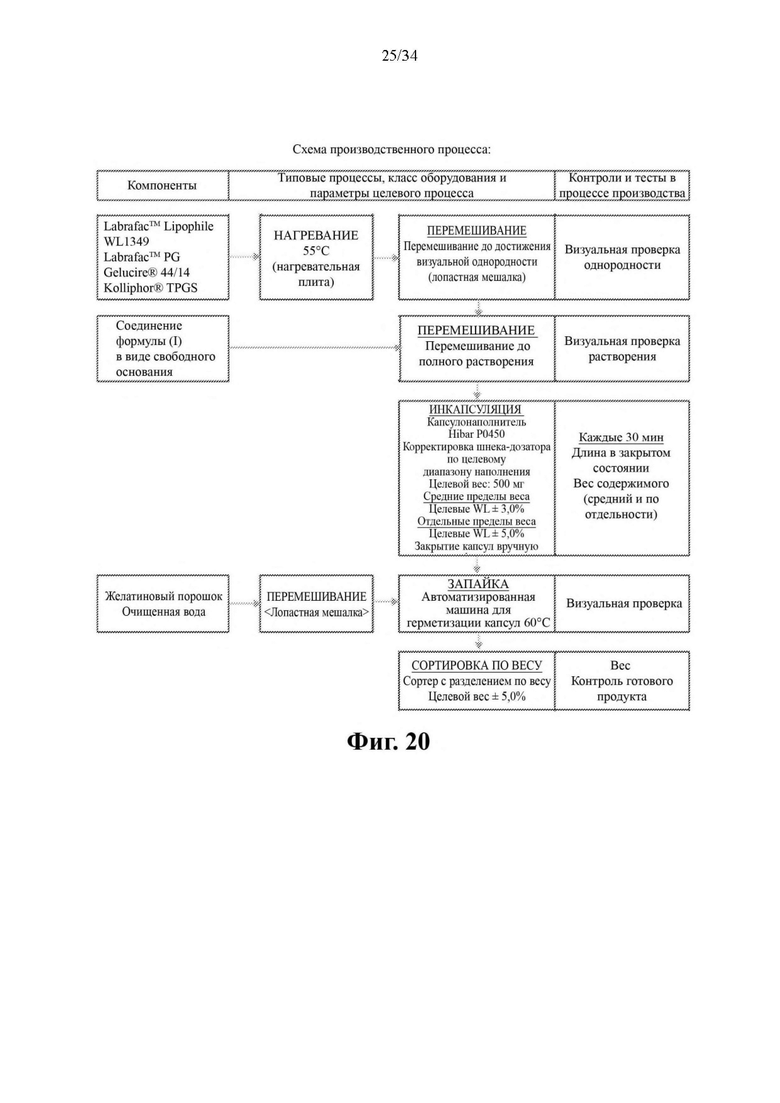
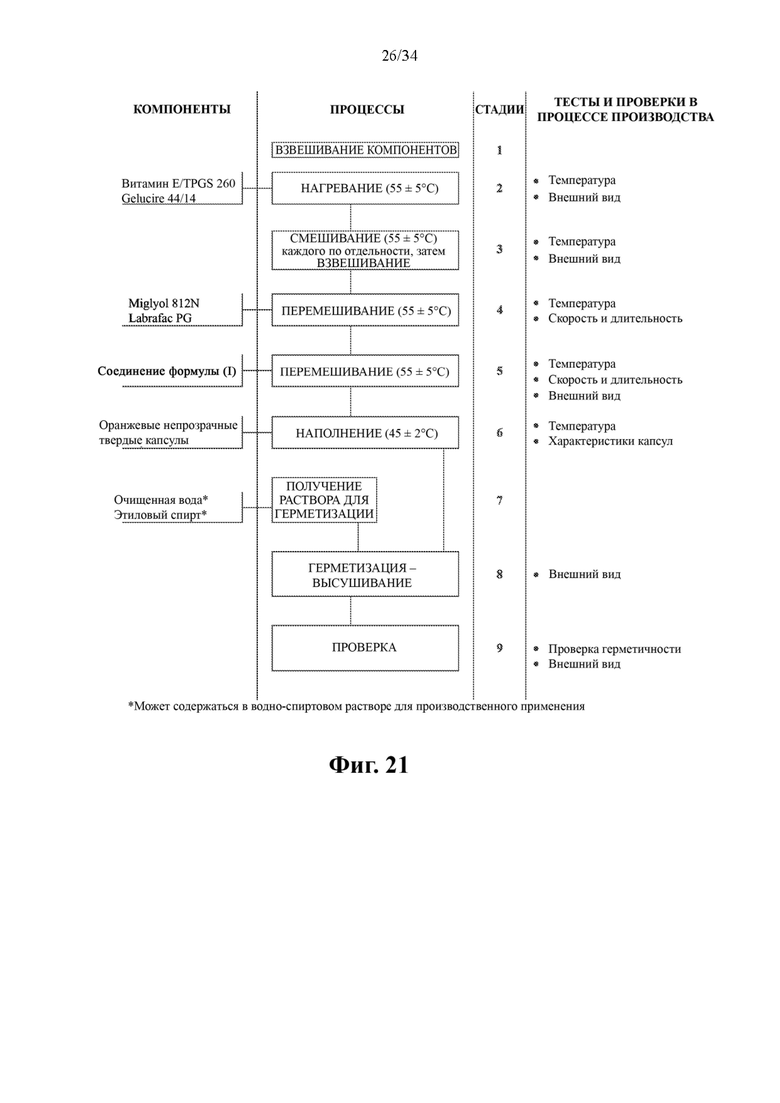
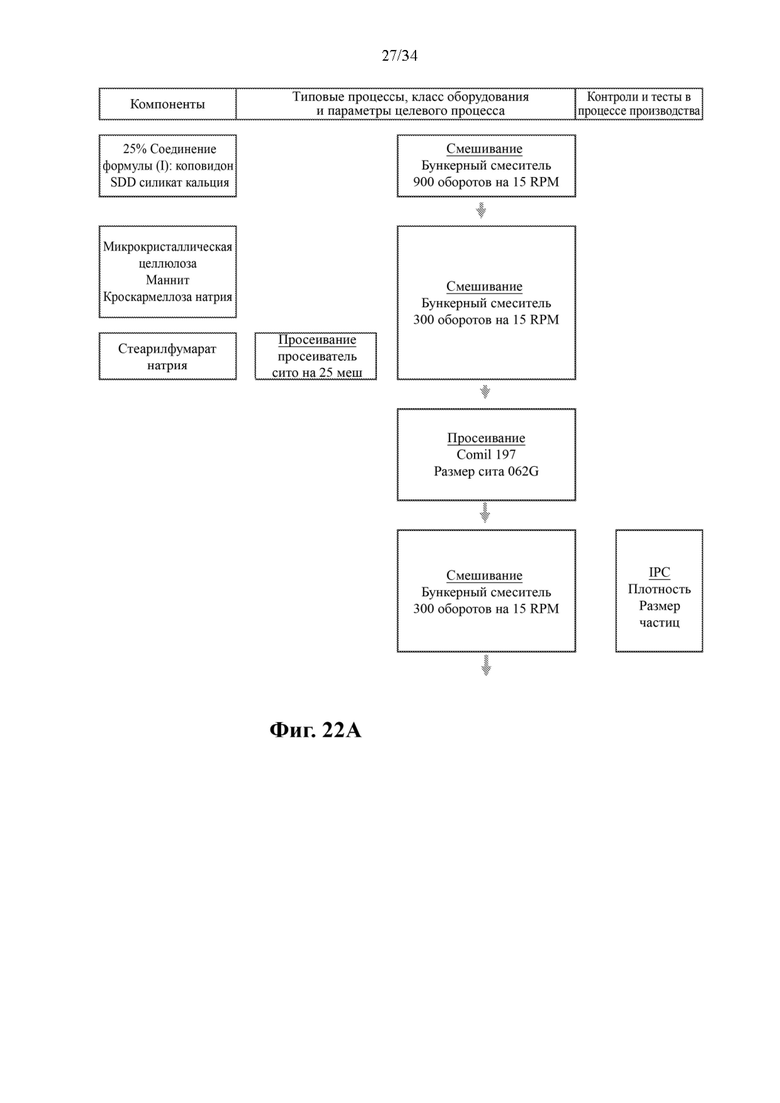
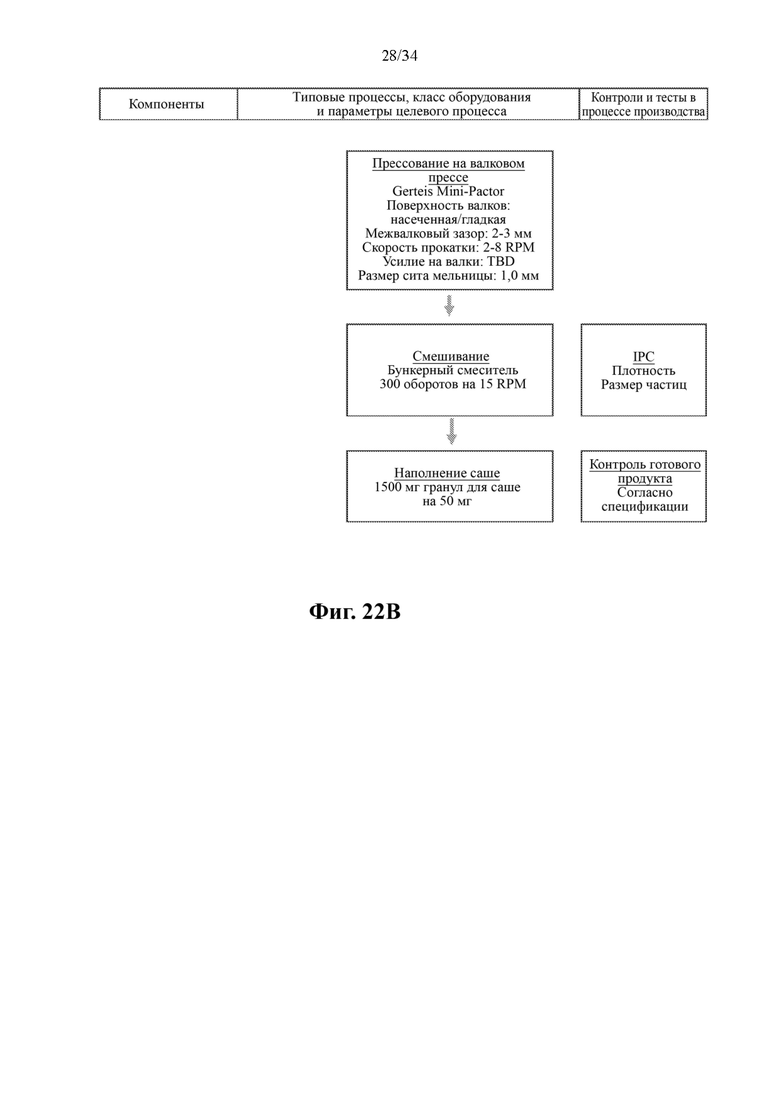
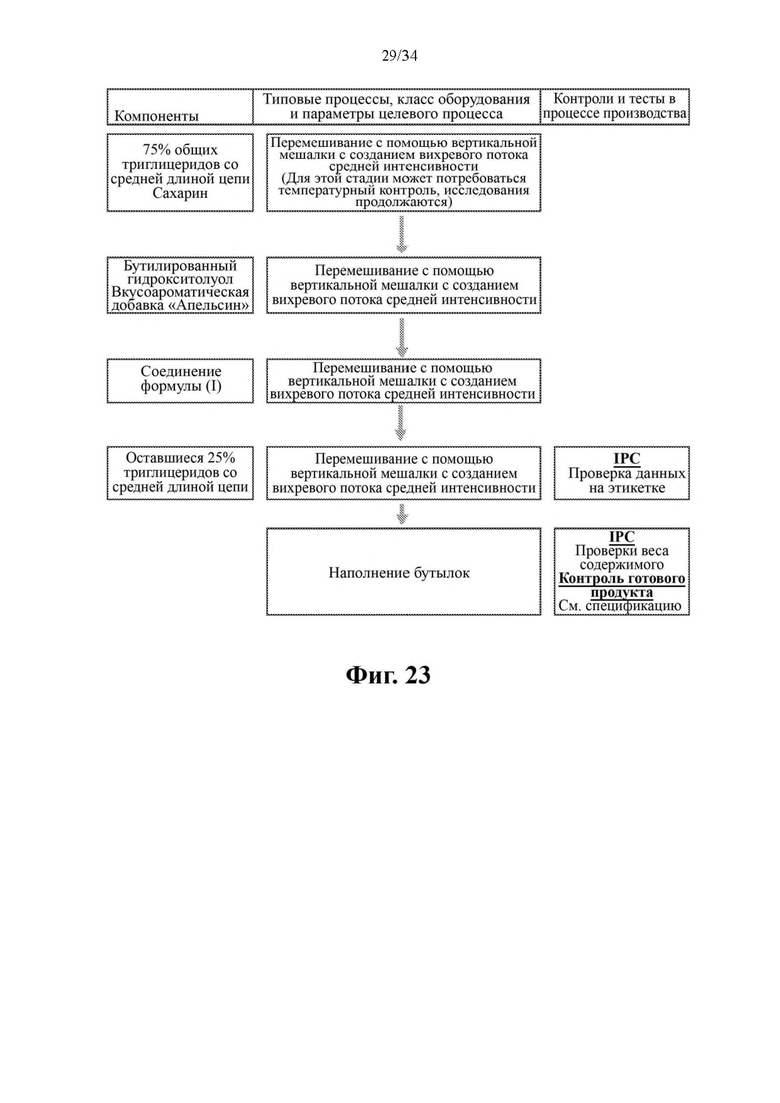
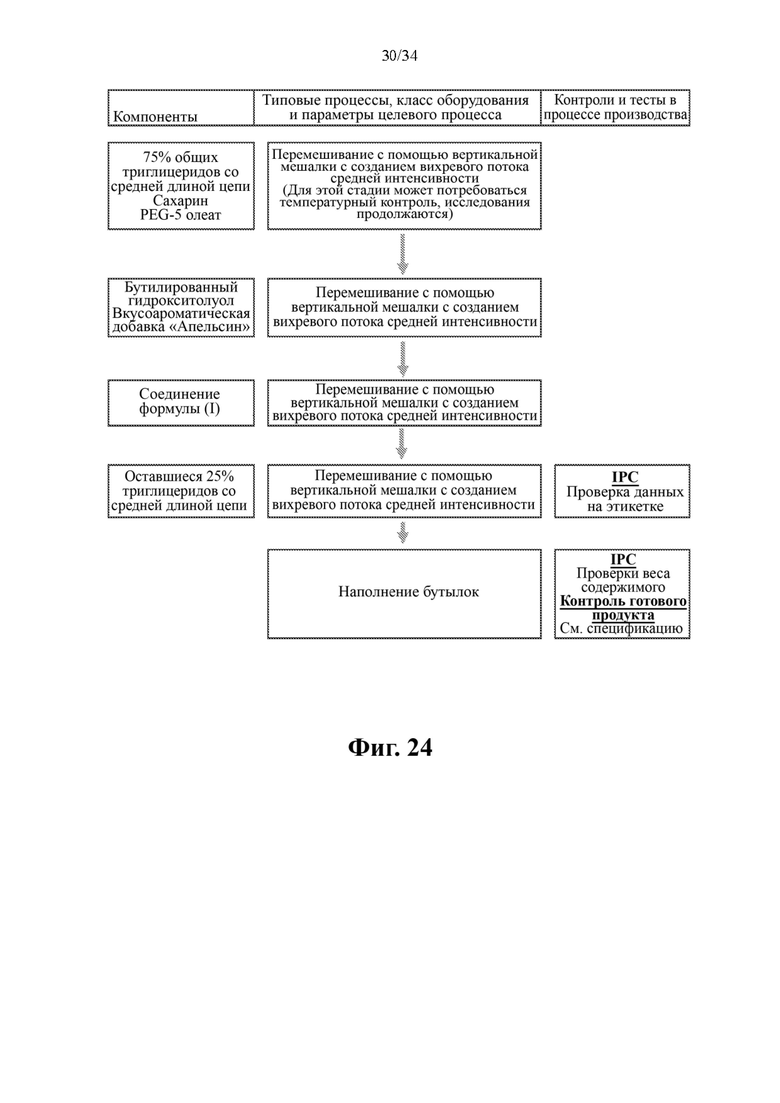
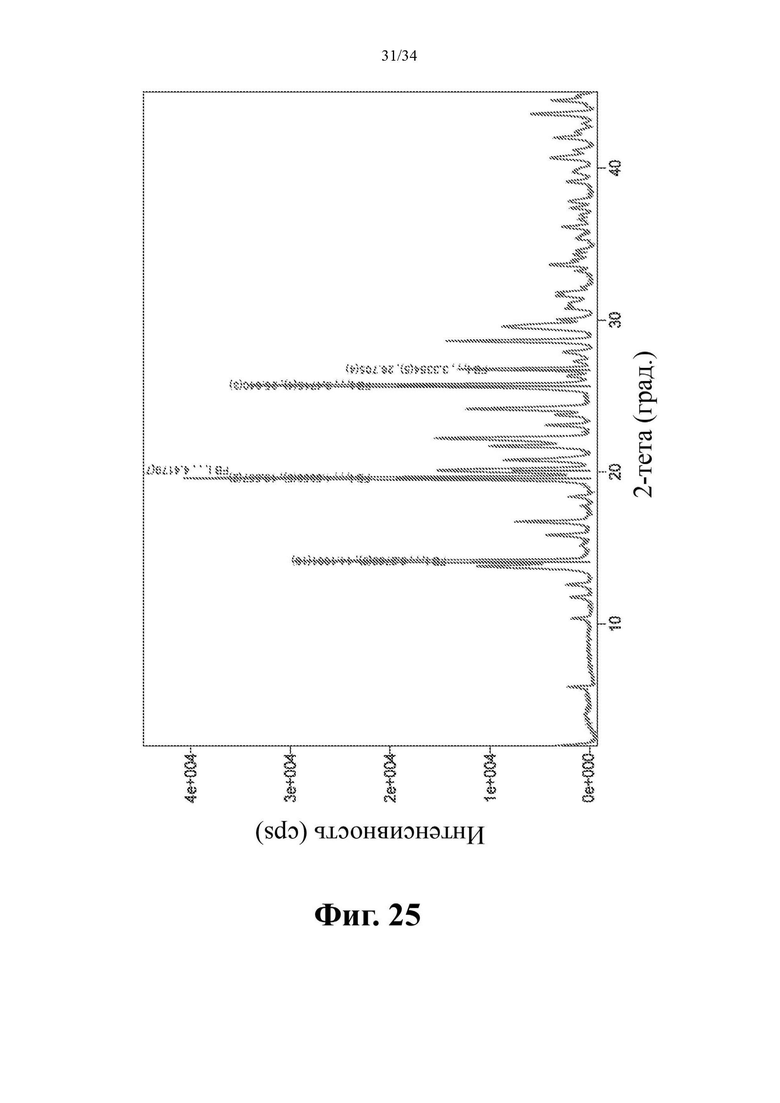

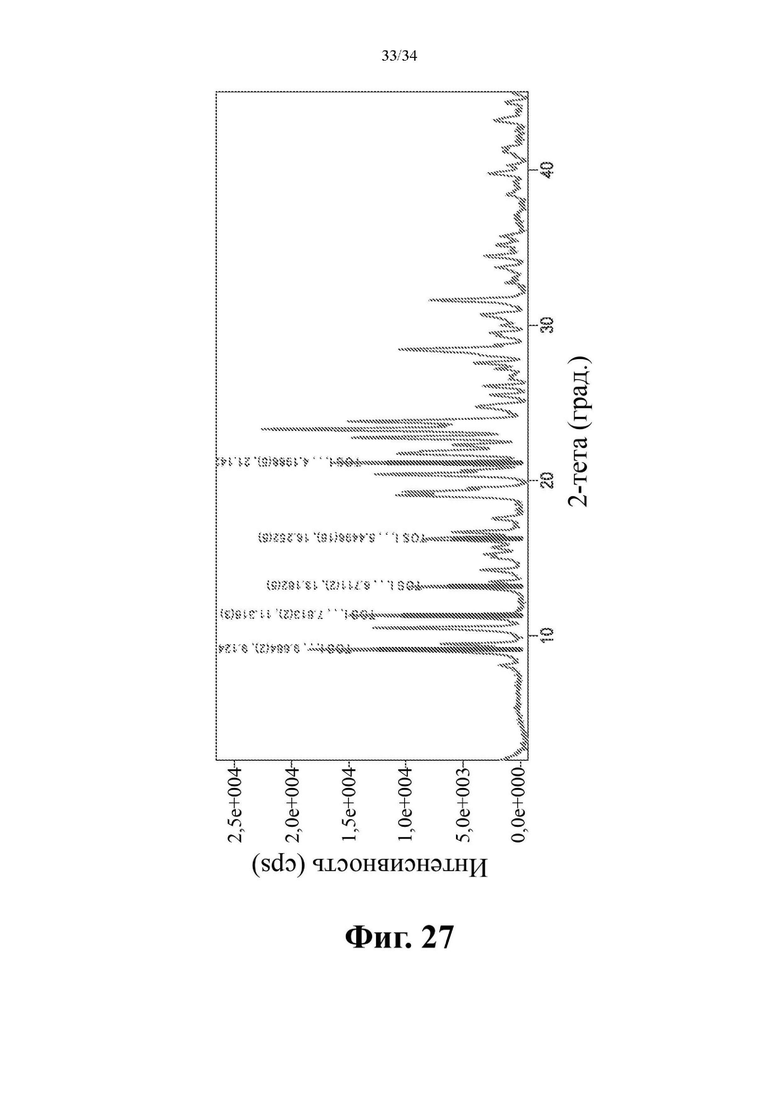
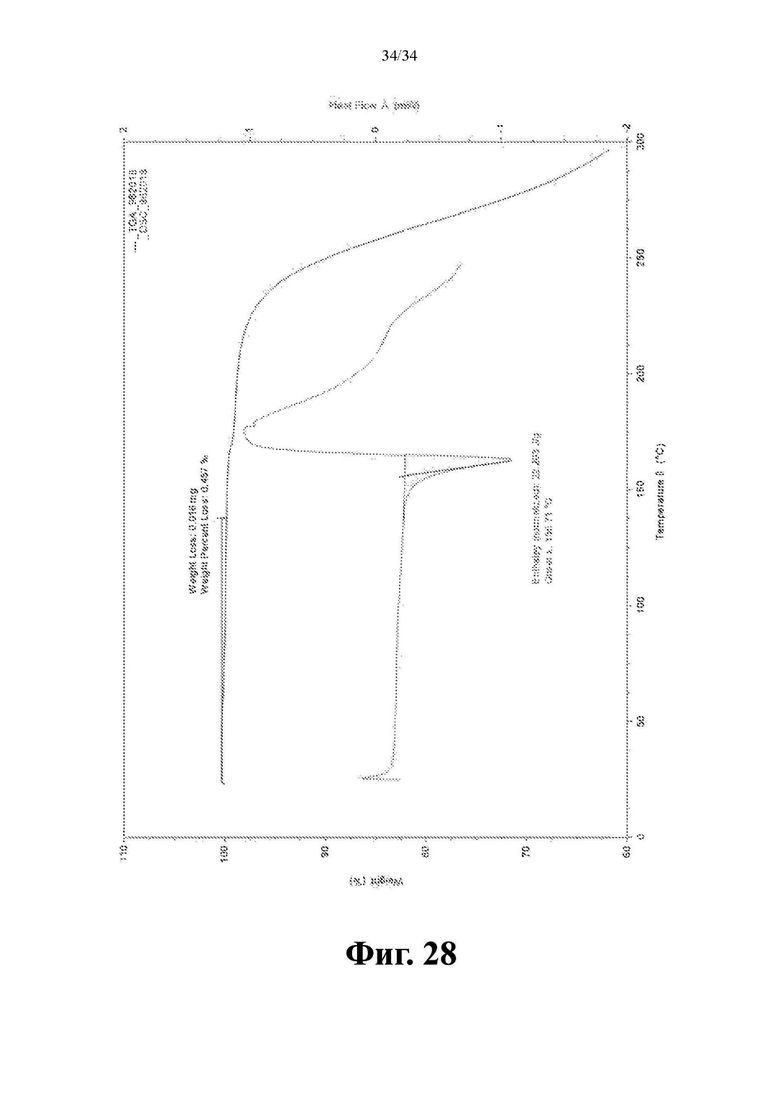
Группа изобретений относится к химии и фармацевтической промышленности, в частности к фармацевтической композиции для лечения врожденной гиперплазии надпочечников у субъекта в лекарственной форме в виде перорального раствора, содержащей: (а) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин [соединение формулы (I)]
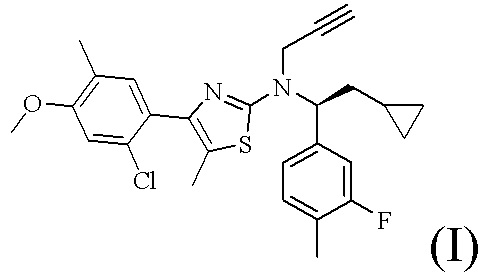
или его фармацевтически приемлемую соль; (b) одно или несколько из подсластителя, антиоксиданта и вкусоароматической добавки; и (c) триглицериды со средней длиной цепи. Также предложены способ получения указанной фармацевтической композиции и ее применение для лечения врожденной гиперплазии надпочечников у субъекта. Группа изобретений обеспечивает создание лекарственной формы соединения формулы (I) в виде перорального раствора, подходящего для введения, например, педиатрическим пациентам. 3 н. и 157 з.п. ф-лы, 44 ил., 45 табл., 16 пр.
1. Фармацевтическая композиция в лекарственной форме в виде перорального раствора для лечения врожденной гиперплазии надпочечников (САН) у субъекта, содержащая:
(a) соединение формулы (I)
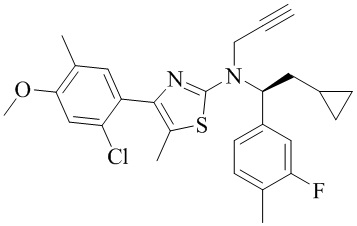
(I)
или его фармацевтически приемлемую соль;
(b) одно или несколько из подсластителя, антиоксиданта и вкусоароматической добавки; и
(c) триглицериды со средней длиной цепи.
2. Фармацевтическая композиция по п. 1, содержащая от 1 до 50% вес./об. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
3. Фармацевтическая композиция по п. 1, содержащая от 1 до 10% вес./об. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
4. Фармацевтическая композиция по п. 1, содержащая 5% вес./об. соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
5. Фармацевтическая композиция по любому из пп. 1-4, содержащая подсластитель.
6. Фармацевтическая композиция по любому из пп. 1-5, содержащая от 0,01 до 1,5% вес./об. подсластителя.
7. Фармацевтическая композиция по любому из пп. 1-5, содержащая от 0,1 до 0,5% вес./об. подсластителя.
8. Фармацевтическая композиция по любому из пп. 1-5, содержащая 0,15% вес./об. подсластителя.
9. Фармацевтическая композиция по любому из пп. 1-8, где подсластитель выбран из сахарина, сахарозы, сукралозы, аспартама, декстрозы, фруктозы, мальтита, маннита, сорбита и адвантама.
10. Фармацевтическая композиция по любому из пп. 1-9, где подсластитель представляет собой сахарин.
11. Фармацевтическая композиция по любому из пп. 1-10, содержащая антиоксидант.
12. Фармацевтическая композиция по любому из пп. 1-11, содержащая от 0,01 до 1,5% вес./об. антиоксиданта.
13. Фармацевтическая композиция по любому из пп. 1-11, содержащая от 0,1 до 0,5% вес./об. антиоксиданта.
14. Фармацевтическая композиция по любому из пп. 1-11, содержащая 0,17% вес./об. антиоксиданта.
15. Фармацевтическая композиция по любому из пп. 1-14, где антиоксидант выбран из бутилированного гидрокситолуола, витамина E полиэтиленгликольсукцината, бутилированного гидроксианизола, аскорбиновой кислоты, лецитина, трет-бутилгидрохинона и лимонной кислоты.
16. Фармацевтическая композиция по любому из пп. 1-15, где антиоксидант представляет собой бутилированный гидрокситолуол.
17. Фармацевтическая композиция по любому из пп. 1-16, содержащая вкусоароматическую добавку.
18. Фармацевтическая композиция по любому из пп. 1-17, содержащая от 0,01 до 0,5% вес./об. вкусоароматической добавки.
19. Фармацевтическая композиция по любому из пп. 1-17, содержащая от 0,05 до 0,2% вес./об. вкусоароматической добавки.
20. Фармацевтическая композиция по любому из пп. 1-17, содержащая 0,10% вес./об. вкусоароматической добавки.
21. Фармацевтическая композиция по любому из пп. 1-20, где вкусоароматическая добавка выбрана из вкусоароматической добавки FONA «Апельсин», вкусоароматической добавки FONA «Juicy», вкусоароматической добавки FONA «Виноград», вкусоароматической добавки Firmenich SA «Лимон», вкусоароматической добавки Firmenich Tetrarome «Апельсин», вкусоароматической добавки IFF «Вишня» и вкусоароматической добавки IFF «Виноград».
22. Фармацевтическая композиция по любому из пп. 1-21, где вкусоароматическая добавка представляет собой вкусоароматическую добавку FONA «Апельсин».
23. Фармацевтическая композиция по любому из пп. 1-22, содержащая от 50 до 99,9% вес./об. триглицеридов со средней длиной цепи.
24. Фармацевтическая композиция по любому из пп. 1-22, содержащая от 90 до 99% вес./об. триглицеридов со средней длиной цепи.
25. Фармацевтическая композиция по любому из пп. 1-22, содержащая от 92 до 97% вес./об. триглицеридов со средней длиной цепи.
26. Фармацевтическая композиция по любому из пп. 1-22, содержащая 94,6% вес./об. триглицеридов со средней длиной цепи.
27. Фармацевтическая композиция по любому из пп. 1-26, где триглицериды со средней длиной цепи представляют собой Labrafac Lipophile WL1349.
28. Фармацевтическая композиция по любому из пп. 1-27, дополнительно содержащая поверхностно-активное вещество.
29. Фармацевтическая композиция по п. 28, содержащая от 1 до 50% вес./об. поверхностно-активного вещества.
30. Фармацевтическая композиция по п. 28, содержащая от 10 до 30% вес./об. поверхностно-активного вещества.
31. Фармацевтическая композиция по п. 28, содержащая 20% вес./об. поверхностно-активного вещества.
32. Фармацевтическая композиция по любому из пп. 28-31, где поверхностно-активное вещество выбрано из олеоилполиоксил-6-глицеридов, линолеоилполиоксил-6-глицеридов, полисорбата 80, полисорбата 20, витамина Е полиэтиленгликольсукцината, Gelucire, лауроилполиоксил-32-глицеридов, лаурилсульфата натрия, полоксамера, сложных эфиров PEG-6 и кукурузного масла и сложных эфиров PEG-6 гидрогенизированного пальмового масла/пальмоядрового масла.
33. Фармацевтическая композиция по любому из пп. 28-32, где поверхностно-активное вещество представляет собой олеоилполиоксил-6-глицериды.
34. Фармацевтическая композиция по любому из пп. 32, 33, где олеоилполиоксил-6-глицериды представляют собой LABRAFIL M 1944 CS.
35. Фармацевтическая композиция по любому из пп. 1-34, содержащая от 70 до 80% вес./об. триглицеридов со средней длиной цепи.
36. Фармацевтическая композиция по любому из пп. 1-34, содержащая 75% вес./об. триглицеридов со средней длиной цепи.
37. Фармацевтическая композиция по любому из пп. 35, 36, где триглицериды со средней длиной цепи представляют собой Labrafac Lipophile WL1349.
38. Фармацевтическая композиция по п. 1, содержащая:
(a) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль;
(b) подсластитель;
(c) антиоксидант;
(d) вкусоароматическую добавку; и
(e) триглицериды со средней длиной цепи.
39. Фармацевтическая композиция по п. 38, дополнительно содержащая поверхностно-активное вещество.
40. Фармацевтическая композиция по п. 1, содержащая:
(a) от 4 до 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от 0,1 до 0,2% вес./об. подсластителя;
(c) от 0,1 до 0,2% вес./об. антиоксиданта;
(d) от 0,05 до 0,2% вес./об. вкусоароматической добавки; и
(e) от 92 до 97% вес./об. триглицеридов со средней длиной цепи.
41. Фармацевтическая композиция по п. 1, содержащая:
(a) 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) 0,15% вес./об. подсластителя;
(c) 0,17% вес./об. антиоксиданта;
(d) 0,1% вес./об. вкусоароматической добавки; и
(e) 94,6% вес./об. триглицеридов со средней длиной цепи.
42. Фармацевтическая композиция по п. 1, содержащая:
(a) от 4 до 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от 0,1 до 0,2% вес./об. подсластителя;
(c) от 0,1 до 0,2% вес./об. антиоксиданта;
(d) от 0,05 до 0,2% вес./об. вкусоароматической добавки;
(e) от 15 до 25% вес./об. поверхностно-активного вещества; и
(f) от 70 до 80% вес./об. триглицеридов со средней длиной цепи.
43. Фармацевтическая композиция по п. 1, содержащая:
(a) 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) 0,15% вес./об. подсластителя;
(c) 0,17% вес./об. антиоксиданта;
(d) 0,1% вес./об. вкусоароматической добавки;
(e) 20% вес./об. поверхностно-активного вещества; и
(f) 75% вес./об. триглицеридов со средней длиной цепи.
44. Фармацевтическая композиция по п. 1, содержащая:
(a) 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амин или его фармацевтически приемлемую соль;
(b) сахарин;
(c) бутилированный гидрокситолуол;
(d) вкусоароматическую добавку FONA «Апельсин»; и
(e) триглицериды со средней длиной цепи.
45. Фармацевтическая композиция по п. 44, дополнительно содержащая олеоилполиоксил-6-глицериды.
46. Фармацевтическая композиция по п. 45, содержащая:
(a) от 4 до 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от 0,1 до 0,2% вес./об. сахарина;
(c) от 0,1 до 0,2% вес./об. бутилированного гидрокситолуола;
(d) от 0,05 до 0,2% вес./об. вкусоароматической добавки FONA «Апельсин»; и
(e) от 92 до 97% вес./об. триглицеридов со средней длиной цепи.
47. Фармацевтическая композиция по п. 1, содержащая:
(a) 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) 0,15% вес./об. сахарина;
(c) 0,17% вес./об. бутилированного гидрокситолуола;
(d) 0,1% вес./об. вкусоароматической добавки FONA «Апельсин»; и
(e) 94,6% вес./об. триглицеридов со средней длиной цепи.
48. Фармацевтическая композиция по п. 1, содержащая:
(a) от 4 до 6% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) от 0,1 до 0,2% вес./об. сахарина;
(c) от 0,1 до 0,2% вес./об. бутилированного гидрокситолуола;
(d) от 0,05 до 0,2% вес./об. вкусоароматической добавки FONA «Апельсин»;
(e) от 15 до 25% вес./об. олеоилполиоксил-6-глицеридов; и
(f) от 70 до 80% вес./об. триглицеридов со средней длиной цепи.
49. Фармацевтическая композиция по п. 1, содержащая:
(a) 5% вес./об. 4-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-проп-2-инил-1,3-тиазол-2-амина или его фармацевтически приемлемой соли, в пересчете на вес свободного основания;
(b) 0,15% вес./об. сахарина;
(c) 0,17% вес./об. бутилированного гидрокситолуола;
(d) 0,1% вес./об. вкусоароматической добавки FONA «Апельсин»;
(e) 20% вес./об. олеоилполиоксил-6-глицеридов; и
(f) 75% вес./об. триглицеридов со средней длиной цепи.
50. Фармацевтическая композиция по любому из пп. 1-49, содержащая соединение формулы (I) в виде свободного основания.
51. Фармацевтическая композиция по любому из пп. 1-50, составленная в виде единичной лекарственной формы, причем соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от 5 до 200 мг/мл, в пересчете на вес свободного основания.
52. Фармацевтическая композиция по п. 51, где соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве от 75 до 150 мг/мл, в пересчете на вес свободного основания.
53. Фармацевтическая композиция по п. 51, где соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве 50 мг/мл, в пересчете на вес свободного основания.
54. Фармацевтическая композиция по п. 51, где соединение формулы (I) или его фармацевтически приемлемая соль присутствуют в количестве 100 мг/мл, в пересчете на вес свободного основания.
55. Способ получения фармацевтической композиции по любому из пп. 1-54, предусматривающий следующие стадии:
(a) смешивание триглицеридов со средней длиной цепи с подсластителем;
(b) смешивание смеси из стадии (a) с антиоксидантом и вкусоароматической добавкой;
(c) смешивание соединения формулы (I) или его фармацевтически приемлемой соли со смесью из стадии (b); и
(d) смешивание смеси из стадии (c) с дополнительной частью триглицеридов со средней длиной цепи.
56. Способ по п. 55, в котором стадия (a) предусматривает смешивание триглицеридов со средней длиной цепи с подсластителем и поверхностно-активным веществом.
57. Применение фармацевтической композиция по любому из пп. 1-54 для лечения врожденной гиперплазии надпочечников (CAH) у субъекта.
58. Применение по п. 57, где субъект находится в сытом состоянии.
59. Применение по п. 57 или 58, где фармацевтическая композиция вводится субъекту с питательной композицией.
60. Применение по п. 59, где питательная композиция представляет собой жидкую пищевую добавку, содержащую от 1000 до 2000 калорий на литр с содержанием жиров более чем 30%.
61. Применение по п. 59, где питательная композиция представляет собой жидкую пищевую добавку, содержащую 1500 калорий на литр, со следующим распределением калорий: 14,7% белков, 32% жиров и 53,3% углеводов.
62. Применение по любому из пп. 59-61, где питательная композиция принимается в количестве от 6 до 12 жидких унций.
63. Применение по любому из пп. 59-62, где питательная композиция принимается в количестве 8 жидких унций.
64. Применение по любому из пп. 59-63, где питательная композиция принимается в пределах 30 минут от введения фармацевтической композиции.
65. Применение по любому из пп. 58-64, где введение фармацевтической композиции характеризуется положительным влиянием пищи.
66. Применение по п. 65, где положительное влияние пищи измеряется по Cmax, AUC, или их комбинациям, соединения формулы (I) при сравнении перорального введения фармацевтической композиции в сытом состоянии и натощак.
67. Применение по любому из пп. 58-64, где соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от 1 до 4 или от 5 до 10.
68. Применение по любому из пп. 58-64, где соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от 1 до 4 или от 5 до 10.
69. Применение по любому из пп. 58-64, где соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от 1,5 до 3.
70. Применение по любому из пп. 58-64, где соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от 1,5 до 3.
71. Применение по любому из пп. 57-70, где субъектом является педиатрический пациент.
72. Применение по любому из пп. 58-64, где фармацевтическая композиция характеризуется положительным влиянием пищи при пероральном введении.
73. Применение по п. 72, где соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от 1 до 4 или от 5 до 10.
74. Применение по п. 72, где соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от 1 до 4 или от 5 до 10.
75. Применение по п. 72, где соединение формулы (I) характеризуется соотношением AUC при введении в сытом состоянии и AUC при введении натощак от 1,5 до 3.
76. Применение по п. 72, где соединение формулы (I) характеризуется соотношением Cmax при введении в сытом состоянии и Cmax при введении натощак от 1,5 до 3.
77. Применение по любому из пп. 57-76, где фармацевтическая композиция вводится субъекту с пищей.
78. Применение по п. 77, где пища представляет собой пищу с высоким содержанием жиров.
79. Применение по п. 77, где пища представляет собой пищу с низким содержанием жиров.
80. Применение по любому из пп. 77-79, где фармацевтическая композиция вводится в пределах 5 минут после начала приема пищи.
81. Применение по любому из пп. 77-80, где пища представляет собой ужин.
82. Применение по любому из пп. 77-80, где пища представляет собой завтрак.
83. Применение по любому из пп.77-82, где введение фармацевтической композиции характеризуется положительным влиянием пищи.
84. Применение по п. 83, где положительное влияние пищи измеряется по Cmax, AUC, или их комбинациям, соединения формулы (I) при сравнении перорального введения фармацевтической композиции в сытом состоянии и натощак.
85. Применение по любому из пп. 77-84, где соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от 1 до 4 или от 5 до 10.
86. Применение по любому из пп. 77-85, где соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от 1 до 4 или от 5 до 10.
87. Применение по любому из пп. 77-84, где соотношение AUC соединения формулы (I) при введении в сытом состоянии и AUC соединения формулы (I) при введении натощак составляет от 1,5 до 3.
88. Применение по любому из пп. 77-84, где соотношение Cmax соединения формулы (I) при введении в сытом состоянии и Cmax соединения формулы (I) при введении натощак составляет от 1,5 до 3.
89. Применение по любому из пп. 57-88, где фармацевтическая композиция содержит 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
90. Применение по любому из пп. 57-88, где фармацевтическая композиция содержит 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
91. Применение по любому из пп. 57-88, где фармацевтическая композиция содержит 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
92. Применение по любому из пп. 57-88, где фармацевтическая композиция содержит 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
93. Применение по любому из пп. 57-88, где способ предусматривает введение суточной дозы фармацевтической композиции, содержащей 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
94. Применение по любому из пп. 57-88, где способ предусматривает введение суточной дозы фармацевтической композиции, содержащей 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
95. Применение по любому из пп. 57-88, где способ предусматривает введение суточной дозы фармацевтической композиции, содержащей 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
96. Применение по любому из пп. 57-88, где способ предусматривает введение суточной дозы фармацевтической композиции, содержащей 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
97. Применение по любому из пп. 57-88, где способ предусматривает введение суточной дозы фармацевтической композиции, содержащей 150 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
98. Применение по любому из пп. 57-88, где способ предусматривает введение суточной дозы фармацевтической композиции, содержащей 200 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
99. Применение по любому из пп. 57-88, где способ предусматривает введение фармацевтической композиции два раза в сутки в дозе 25 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
100. Применение по любому из пп. 57-88, где способ предусматривает введение фармацевтической композиции два раза в сутки в дозе 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
101. Применение по любому из пп. 57-88, где способ предусматривает введение фармацевтической композиции два раза в сутки в дозе 75 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
102. Применение по любому из пп. 57-88, где способ предусматривает введение фармацевтической композиции два раза в сутки в дозе 100 мг соединения формулы (I) или его фармацевтически приемлемой соли, в пересчете на вес свободного основания.
103. Применение по любому из пп. 57-102, где субъект одновременно получает дозу глюкокортикоида.
104. Применение по п. 103, где глюкокортикоид выбран из кортизола (гидрокортизона), кортизона, преднизона, преднизолона, метилпреднизолона, дексаметазона, бетаметазона, триамцинолона, флудрокортизона ацетата и дезоксикортикостерона ацетата.
105. Применение по п. 103 или 104, где глюкокортикоид представляет собой кортизол (гидрокортизон).
106. Применение по п. 103 или 104, где глюкокортикоид представляет собой кортизон.
107. Применение по п. 103 или 104, где глюкокортикоид представляет собой преднизон.
108. Применение по любому из пп. 103-107, где доза глюкокортикоида измеряется в эквивалентах гидрокортизона.
109. Применение по любому из пп. 103-108, где доза глюкокортикоида измеряется как величина, кратная верхней границе нормы для физиологической дозы в эквивалентах гидрокортизона.
110. Применение по любому из пп. 103-109, где доза глюкокортикоида представляет собой физиологическую дозу, как измерено после периода времени введения фармацевтической композиции.
111. Применение по любому из пп. 103-109, где доза глюкокортикоида представляет собой физиологическую дозу от 4 до 12 мг/м2/сутки, как измерено после периода времени введения фармацевтической композиции.
112. Применение по любому из пп. 103-109, где доза глюкокортикоида представляет собой физиологическую дозу от 4 до 9 мг/м2/сутки, как измерено после периода времени введения фармацевтической композиции.
113. Применение по любому из пп. 103-109, где доза глюкокортикоида представляет собой физиологическую дозу, которая составляет менее чем 8 мг/м2/сутки, как измерено после периода времени введения фармацевтической композиции.
114. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на 10% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
115. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на 20% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
116. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на 30% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
117. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на 40% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
118. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на 50% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
119. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на 60% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
120. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на 70% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
121. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на менее чем 20% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
122. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на величину, составляющую от 20 до 50%, после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
123. Применение по любому из пп. 103-113, где доза глюкокортикоида для субъекта снижается на более чем 50% после периода времени введения фармацевтической композиции, причем снижение дозы глюкокортикоида является таковым относительно дозы глюкокортикоида до введения фармацевтической композиции.
124. Применение по любому из пп. 57-123, где уровень 17-гидроксипрогестерона снижается по меньшей мере на 25% после периода времени введения фармацевтической композиции, причем снижение уровня 17-гидроксипрогестерона является таковым относительно уровня 17-гидроксипрогестерона до введения фармацевтической композиции.
125. Применение по любому из пп. 57-123, где уровень 17-гидроксипрогестерона снижается по меньшей мере на 50% после периода времени введения фармацевтической композиции, причем снижение уровня 17-гидроксипрогестерона является таковым относительно уровня 17-гидроксипрогестерона до введения фармацевтической композиции.
126. Применение по любому из пп. 57-123, где уровень 17-гидроксипрогестерона в менее чем 1,5 раза превышает верхнюю границу нормы после периода времени введения фармацевтической композиции.
127. Применение по любому из пп. 57-123, где уровень 17-гидроксипрогестерона находится в пределах нормы после периода времени введения фармацевтической композиции.
128. Применение по любому из пп. 57-127, где уровень адренокортикотропного гормона снижается по меньшей мере на 25% после периода времени введения фармацевтической композиции, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения фармацевтической композиции.
129. Применение по любому из пп. 57-127, где уровень адренокортикотропного гормона снижается по меньшей мере на 40% после периода времени введения фармацевтической композиции, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения фармацевтической композиции.
130. Применение по любому из пп. 57-127, где уровень адренокортикотропного гормона снижается по меньшей мере на 50% после периода времени введения фармацевтической композиции, причем снижение уровня адренокортикотропного гормона является таковым относительно уровня адренокортикотропного гормона до введения фармацевтической композиции.
131. Применение по любому из пп. 57-127, где уровень адренокортикотропного гормона в менее чем приблизительно 1,5 раза превышает верхнюю границу нормы после периода времени введения фармацевтической композиции.
132. Применение по любому из пп. 57-123, где уровень адренокортикотропного гормона находится в пределах нормы после периода времени введения фармацевтической композиции.
133. Применение по любому из пп. 57-132, где уровень андростендиона снижается по меньшей мере на 25% после периода времени введения фармацевтической композиции, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения фармацевтической композиции.
134. Применение по любому из пп. 57-132, где уровень андростендиона снижается по меньшей мере на 30% после периода времени введения фармацевтической композиции, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения фармацевтической композиции.
135. Применение по любому из пп. 57-132, где уровень андростендиона снижается по меньшей мере на 50% после периода времени введения фармацевтической композиции, причем снижение уровня андростендиона является таковым относительно уровня андростендиона до введения фармацевтической композиции.
136. Применение по любому из пп. 57-132, где уровень андростендиона в менее чем 1,5 раза превышает верхнюю границу нормы после периода времени введения фармацевтической композиции.
137. Применение по любому из пп. 57-132, где уровень андростендиона находится в пределах нормы после периода времени введения фармацевтической композиции.
138. Применение по любому из пп. 57-137, где уровень тестостерона снижается по меньшей мере на 25% после периода времени введения фармацевтической композиции, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения фармацевтической композиции.
139. Применение по любому из пп. 57-137, где уровень тестостерона снижается по меньшей мере на 30% после периода времени введения фармацевтической композиции, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения фармацевтической композиции.
140. Применение по любому из пп. 57-137, где уровень тестостерона снижается по меньшей мере на 50% после периода времени введения фармацевтической композиции, причем снижение уровня тестостерона является таковым относительно уровня тестостерона до введения фармацевтической композиции.
141. Применение по любому из пп. 57-137, где уровень тестостерона в менее чем 1,5 раза превышает верхнюю границу нормы после периода времени введения фармацевтической композиции.
142. Применение по любому из пп. 57-137, где уровень тестостерона находится в пределах нормы после периода времени введения фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
143. Применение по любому из пп. 57-137, где уровень 17-гидроксипрогестерона снижается по меньшей мере на 50% и уровень андростендиона снижается по меньшей мере на 50% после периода времени введения фармацевтической композиции, причем снижение уровня 17-гидроксипрогестерона и уровня андростендиона является таковым относительно уровня 17-гидроксипрогестерона и уровня андростендиона до введения фармацевтической композиции.
144. Применение по любому из пп. 57-123, где уровень 17-гидроксипрогестерона в менее чем 1,5 раза превышает верхнюю границу нормы и уровень андростендиона в менее чем 1,5 раза превышает верхнюю границу нормы после периода времени введения фармацевтической композиции.
145. Применение по любому из пп. 57-123, где уровень 17-гидроксипрогестерона находится в пределах нормы и уровень андростендиона находится в пределах нормы после периода времени введения фармацевтической композиции.
146. Применение по любому из пп. 103-145, где у субъекта наблюдается уменьшение глюкокортикоидной нагрузки после периода времени введения фармацевтической композиции, причем уменьшение глюкокортикоидной нагрузки является таковым относительно глюкокортикоидной нагрузки до введения фармацевтической композиции.
147. Применение по п. 146, где происходит улучшение в отношении одного или нескольких симптомов глюкокортикоидной нагрузки, выбранных из качества жизни, утомляемости, сна, инсулинорезистентности, толерантности к глюкозе, контроля уровня глюкозы, дислипидемии, гиперлипидемии, минеральной плотности костной ткани, обновления костной ткани, массы жировой ткани, веса, центрального ожирения, артериального давления, степени выраженности гирсутизма, цикличности менструации, контроля эктопической ткани надпочечников в яичке и репродуктивной функции, после периода времени введения фармацевтической композиции, причем улучшение в отношении одного или нескольких симптомов является таковым относительно статуса одного или нескольких симптомов до введения фармацевтической композиции.
148. Применение по любому из пп. 110-147, где период времени введения составляет по меньшей мере 4 недели.
149. Применение по любому из пп. 110-147, где период времени введения составляет по меньшей мере 24 недели.
150. Применение по любому из пп. 110-147, где период времени введения составляет по меньшей мере один год.
151. Применение по любому из пп. 57-150, где субъектом является педиатрический пациент.
152. Применение по п. 151, где возраст педиатрического пациента равен шести годам или меньше.
153. Применение по п. 151, где возраст педиатрического пациента составляет более шести лет и менее одиннадцати лет.
154. Применение по п. 151, где возраст педиатрического пациента составляет более десяти лет и менее пятнадцати лет.
155. Применение по п. 151, где возраст педиатрического пациента составляет более четырнадцати лет и менее девятнадцати лет.
156. Применение по любому из пп. 57-150, где субъект является взрослым субъектом.
157. Применение по п. 156, где возраст взрослого субъекта составляет более восемнадцати лет.
158. Применение по любому из пп. 57-157, где субъектом является лицо женского пола.
159. Применение по любому из пп. 57-157, где субъектом является лицо мужского пола.
160. Применение по любому из пп. 57-159, где соединение формулы (I) или его фармацевтически приемлемая соль вводятся в виде соли соляной кислоты или соли п-толуолсульфоновой кислоты.
| WO 2015112642 A1, 30.07.2015 | |||
| СПОСОБ ПОЛУЧЕНИЯ [4-(2- ХЛОР-4- МЕТОКСИ-5- МЕТИЛФЕНИЛ)-5- МЕТИЛТИАЗОЛО-2- ИЛ] [2-ЦИКЛОПРОПИЛ-1- (3- ФТОР-4- МЕТИЛФЕНИЛ) - ЭТИЛ ]- АМИНА | 2010 |
|
RU2523793C2 |
| КОМПОЗИЦИИ ТВЕРДЫХ РАСТВОРОВ И ИХ ПРИМЕНЕНИЕ ПРИ СИЛЬНОЙ БОЛИ | 2014 |
|
RU2667977C2 |
| WO 2018219804 A1, 06.12.2018. | |||

