По настоящей заявке испрашивается приоритет на основании предварительной заявки на патент США № 62/377427, поданной 19 августа 2017 г. Содержание предварительной заявки включено в настоящую заявку в качестве ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение относится к композициям комбинации 5-[3-(4-бензилоксифенилтио)фур-2-ил]имидазолидин-2,4-диона или его аналогов с циклодекстрином и способам их применения. Композиции 5-[3-(4-бензилоксифенилтио)-фур-2-ил]имидазолидин-2,4-диона или его аналогов с циклодекстрином увеличивают растворимость в воде 5-[3-(4-бензилоксифенилтио)-фур-2-ил]-имидазолидин-2,4-диона или его аналогов, тем самым увеличивают биодоступность при пероральном введении. Кроме того, настоящее изобретение относится к применению фармацевтических композиций, включающих 5-[3-(4-бензилоксифенилтио)-фур-2-ил]-имидазолидин-2,4-дион или его аналоги с циклодекстрином, для лечения заболевания или состояния, которое является или считается чувствительным к ингибированию матриксной металлопротеиназы -12.
Уровень техники изобретения
Ряд соединений 5-[3-(4-бензилоксифенилтио)-фур-2-ил]-имидазолидин-2,4-диона или его аналогов был раскрыт в заявке на патент США 20060041000. Эти соединения были разработаны для использования в качестве ингибиторов макрофагальной эластазы. Все эти соединения являются производными гидантоина и исследованы in vitro для их использования в качестве ингибиторов матриксной металлопротеиназы (ММР).
Как показано в заявке на патент США 20060041000, все протестированные соединения показывают желаемую активность и благоприятный профиль селективности. Значения IC50 в отношении MMP-12 находятся в диапазоне 1-300 нМ, поэтому все они считаются активными. Большинство из приведенных выше соединений не проявляют ингибирования в отношении ММР-1 и ММР-7 при 10 мкМ. Их селективность в отношении ММР-12 по сравнению с MMP-2, MMP-3, MMP-9 и MMP-13 находится в интервале значений от 50- до 1000- кратных. Эти соединения, по-видимому, обладают некоторым потенциалом, который можно использовать при лечении заболеваний или состояний, опосредованных MMP-12, таких как астма, хронические обструктивные заболевания легких (ХОБЛ), артрит, рак, заболевание сердца и нефрит. Однако, за исключением данных IC50 для ограниченных ММР, дополнительные биологические данные не были предоставлены подробно. Кроме того, за исключением данных ЯМР и МС, никаких других данных о физических и химических свойствах представлено не было.
Следовательно, было бы желательно понять характеристики этих соединений и разработать подходящие составы для использования 5-[3-(4-бензилоксифенилтио)-фур-2-ил]-имидазолидин-2,4-диона или его аналогов для потенциального лечения различных заболеваний посредством ингибирования MMP.
Согласно заявке на патент США 20060041000, анализы ингибирования ММР проводили в водном буферном растворе (50 мМ Hepes, 10 мМ CaCl2, 0,05% Brij 35, pH 7,5), что указывает на то, что эти соединения должны быть достаточно растворимы в воде. Однако неожиданно было обнаружено, что эти соединения имеют очень низкую растворимость в воде. 5-[3-(4-Бензилоксифенилтио)-фур-2-ил]-имидазолидин-2,4-дион и его аналоги проявляют не только низкую растворимость в воде, но также в кислой среде. Следовательно, при пероральном введении в обычной твердой лекарственной форме можно ожидать низкую биодоступность.
Следовательно, остается потребность в разработке составов этих соединений, таких как составы, которые делают эти соединения пригодными для неинвазивного, такого как пероральное, интраназальное и/или сублингвальное введение.
Сущность изобретения
Настоящее изобретение относится к композициям или составам, включающим комбинацию соединения формулы (I) или его соли, или гидрата вышеуказанного, и циклодекстрина.
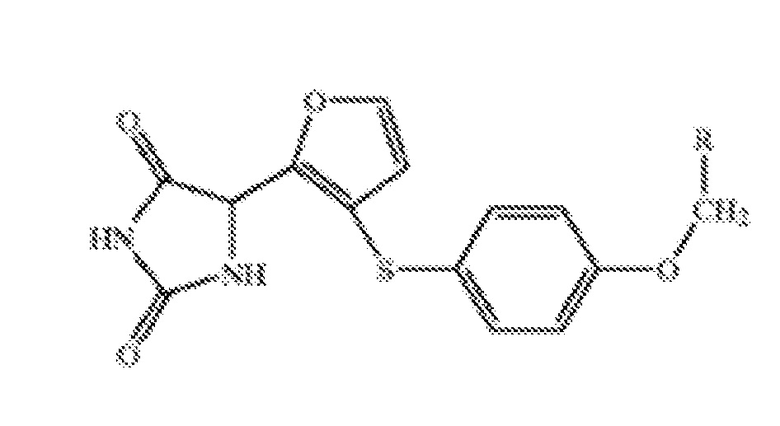
Формула (I)
Также предоставлены способы получения композиций, включающих соединение формулы (I) или его соль, и способы лечения заболевания или показания, которые чувствительны к соединению формулы (I) или его соли, включающие введение фармацевтической композиции, включающей соединение формулы (I) или его соль, животному или человеку.
Одной целью настоящего изобретения является повышение растворимости в воде соединения формулы (I). Таким образом, настоящее изобретение относится к способам улучшения растворимости соединения формулы (I), причем указанный способ включает фармацевтическую композицию, включающую комбинацию соединения формулы (I) и циклодекстрина.
Подробное описание изобретения
Настоящее изобретение относится к композиции, включающей соединение формулы (I) и циклодекстрин. Композиция значительно увеличивает растворимость соединения формулы (I) в воде.
В соответствии с настоящим изобретением, соединение формулы (I), где R выбран из группы, состоящей из фенила, 4-бензилоксифенила, 4-бифенила, 4-метоксифенила, 3-метоксифенила, 2-метоксифенила, 3,5-диметоксифенила, 4-хлорфенила, 3-хлорфенила, 2-хлорфенила, 4-метилфенила, 3-метилфенила, 2-метилфенила и 3-трифторметилфенила.
Следует понимать, что соли, такие как фармацевтически приемлемые соли и их сольваты, также подразумеваются описаниями, приведенными в данном документе. Таким образом, все солевые и несолевые формы соединения формулы (I) и сольваты вышеизложенного охватываются изобретением и описаниями соединения формулы (I), приведенными в настоящем документе.
В соответствии с настоящим изобретением, циклодекстрин для использования в композициях по настоящему изобретению представляет собой водорастворимый незамещенный или замещенный альфа-циклодекстрин (ACD), бета-циклодекстрин (BCD) или гамма-циклодекстрин (GCD). В некоторых вариантах осуществления бета-циклодекстрин выбран из группы, состоящей из метил-бета-циклодекстрина (MBCD), гидроксипропил-бета-циклодекстрина (HPBCD) и сульфобутилового эфира бета-циклодекстрина (SBEBCD). В некоторых вариантах осуществления бета-циклодекстрин представляет собой метил-бета-циклодекстрин или гидроксипропил-бета-циклодекстрин. В некоторых вариантах осуществления гамма-циклодекстрин представляет собой гидроксипропил-гамма-циклодекстрин (HPGCD). В одном предпочтительном варианте осуществления циклодекстрин представляет собой гидроксипропил-бета-циклодекстрин (HPBCD) или метил-бета-циклодекстрин (MBCD).
В соответствии с настоящим изобретением, предоставлены способы улучшения растворимости соединения формулы (I) в воде, включающие комбинацию соединения формулы (I) с циклодекстрином. В одном варианте осуществления предоставлен способ увеличения растворимости в воде соединения формулы (I), где способ включает образование комплекса включения соединения формулы (I) и циклодекстрина. В некоторых вариантах осуществления растворимость соединения формулы (I), когда оно присутствует в виде комплекса включения с циклодекстрином в деионизированной воде при комнатной температуре, увеличивается по меньшей мере в 2 раза по сравнению с растворимостью соединения формула (I) в незакомплексованной форме при тех же условиях. Термин «комнатная температура», как он определен в настоящем описании, составляет приблизительно 20 до 25 градусов Цельсия со средним значением 23°C. В других вариантах осуществления растворимость, такая как растворимость в воде, соединения формулы (I) в композиции увеличивается, по меньшей мере, в 5-2000 раз или более по сравнению с соединением формулы (I) в отдельности. Сравнения растворимости могут быть оценены способами, известными специалисту в данной области, такими как любой из конкретных способов и условий, подробно описанных в настоящем документе.
В соответствии с настоящим изобретением, пероральная биодоступность соединения формулы (I), в присутствии циклодекстрина, по меньшей мере на 50% больше, чем пероральная биодоступность соединения формулы (I) в отсутствие циклодекстрина. Пероральная биодоступность и ее сравнения могут быть оценены способами, известными в данной области, включая любой из конкретных способов, описанных в настоящем документе.
В соответствии с настоящим изобретением, предоставлена композиция соединения формулы (I) и циклодекстрина, где композиция системно индуцирует более высокую максимальную концентрацию (Cmax) соединения, чем это достигается, когда соединение вводят отдельно в том же количестве и при тех же условиях. В одном варианте осуществления композиция соединения формулы (I) системно индуцирует, по меньшей мере, в 1,5 раза или выше Cmax для соединения, чем это достигается, когда соединение вводят отдельно в том же количестве и при тех же условиях. В некоторых вариантах осуществления Cmax соединения формулы (I) при введении животному или человеку с циклодекстрином, по меньшей мере, в 2 раза выше, чем Cmax соединения формулы (I), вводимого отдельно при тех же самым условия.
В соответствии с настоящим изобретением, предоставлена композиция соединения формулы (I) и циклодекстрина, где композиция индуцирует большую площадь под кривой зависимости концентрации в плазме от времени (AUC) соединения, чем это достигается, когда соединение вводят в отсутствие циклодекстрина в том же количестве и при тех же условиях. В одном варианте осуществления композиция соединения формулы (I) с циклодекстрином индуцирует по меньшей мере в 2 или более раз больше AUC соединения, чем это достигается, когда соединение вводят в отсутствие циклодекстрина в том же количестве и при тех же условиях. В некоторых вариантах осуществления AUC соединения формулы (I) при введении животному с циклодекстрином, по меньшей мере, в 2 раза больше, чем AUC соединения формулы (I), вводимого в отсутствие циклодекстрина в том же количестве и при тех же условиях.
В соответствии с настоящим изобретением, предоставлена композиция соединения формулы (I) и циклодекстрина, где композиция индуцирует изменение времени достижения максимального уровня в плазме (Tmax) соединения, чем это достигается, когда соединение вводят в отсутствие циклодекстрина в том же количестве и при тех же условиях. В другом варианте осуществления композиция соединения формулы (I) с циклодекстрином снижает максимальный уровень в плазме (Tmax) соединения в 1 или 2 раза по сравнению с тем, что достигается, когда соединение вводят в отсутствие циклодекстрина в таком же количестве и при тех же условиях. В некоторых вариантах осуществления Tmax соединения формулы (I) при введении индивидууму с циклодекстрином по меньшей мере в 2 раза короче, чем у соединения формулы (I), вводимого в отсутствие циклодекстрина в таком же количестве и при тех же условиях. В некоторых вариантах осуществления соединение формулы (I) с циклодекстрином снижает Tmax по меньшей мере на 1, 2 и 3 часа или более.
В соответствии с настоящим изобретением, предоставлена композиция, включающая соединение формулы (I) и циклодекстрин, где молярное отношение соединения формулы (I) к циклодекстрину составляет от 1:1 до 1:300, предпочтительно от 1:1 до 1:50 и более предпочтительно от 1:1 до 1:10. В одном варианте осуществления композиция включает комплекс соединения формулы (I) и циклодекстрина, где, по меньшей мере, часть соединения формулы (I) встроена, по меньшей мере, частично в полость циклодекстрина с образованием комплекса включения. В другом варианте осуществления композиция включает физическую смесь циклодекстрина и соединения формулы (I), где физическая смесь не включает или по существу не включает, по меньшей мере, часть соединения формулы (I), вставленную, по меньшей мере, частично, в полость циклодекстрина. В другом варианте осуществления предоставлена композиция, включающая а) соединение формулы (I) или его соль, или сольват вышеупомянутого; b) циклодекстрин; и с) добавку. В одном варианте осуществления добавка представляет собой фармацевтически приемлемый эуксципиент. В другом варианте осуществления добавка будет дополнительно увеличивать растворимость соединения формулы (I) в водном растворе Добавка может быть в жидкой, твердой или полутвердой форме. В некоторых вариантах осуществления добавка выбрана из группы, но не ограничивается ими, состоящей из лимонной кислоты, PEG-4000, PVP K40, PVP K10, NaCMC, L-аргинина, лизина и D-маннитиола. В одном предпочтительном варианте осуществления добавка представляет собой L-аргинин, и в другом предпочтительном варианте осуществления добавка представляет собой лизин.
Композиции, включающие соединение формулы (I) и циклодекстрин, могут дополнительно включать дополнительные компоненты состава, также называемые в настоящем документе дополнительными агентами. В некоторых вариантах осуществления составов, описанных в настоящем документе, состав дополнительно включает носитель; в некоторых других вариантах осуществления составов, описанных в настоящем документе, состав дополнительно включает антиоксидант.
В соответствии с настоящим изобретением, композиция, включающая (а) соединение формулы (I) или его соль, или сольват вышеуказанного; (b) циклодекстрин; и (с) носитель представляет собой твердый состав. В некоторых вариантах состав является полутвердым. В некоторых вариантах осуществления состав представляет собой жидкость.
ПРИМЕРЫ
Нижеследующие примеры иллюстрируют композиции и способы по настоящему изобретению. Примеры не ограничивают изобретение, но предоставлены для объяснения метода изготовления полезных композиций для доставки лекарственного средства с контролируемым высвобождением.
Пример 1: Синтез соединения формулы (I)
Синтез соединения формулы (I), то есть 5-[3-(4-бензилоксифенилтио)-фур-2-ил]-имидазолидин-2,4-диона и его аналогов, проводили согласно способу, раскрытому в заявка на патент США 20060041000. Следующие соединения были синтезированы и охарактеризованы.
IVE: 5-{3-[4-(3-Метоксибензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-дион
IVH: 5-{3-[4-(4-Хлорбензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-дион
IVO: 5-{3-[4-(3-Метил-бензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-дион
IVP: 5-{3-[4-(2-Метил-бензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-дион
IVQ: 5-{3-[4-(3-трифторметил-бензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-дион
Пример 2: Влияние различных циклодекстринов на растворимость в воде 5-{3-[4-(3-метоксибензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-диона (IVE)
В таблице 1 перечислены различные коммерчески доступные циклодекстрины (CD). Для анализа влияния различных CD на растворимость в воде соединения IVE, готовили 1 мл каждого из следующих водных растворов, описанных в таблице 2. Избыточный IVE добавляли к каждому из этих растворов, и образцы встряхивали при комнатной температуре в течение 24 часов на орбитальном шейкере при 200 об/мин. Избыточный IVE присутствовал во всех образцах во всех случаях для того чтобы получить растворимость. Через 24 часа образцы центрифугировали. Аликвоту супернатанта разбавляли, при необходимости, и анализировали с помощью ВЭЖХ. Таблица 2 показывает, что как ACD, так и HPGCD значительно не улучшают растворимость в воде соединения IVE, в то время как MBCD, SBEBCD и HPBCD могут значительно повысить растворимость в воде соединения IVE. Эти результаты показывают, что только бета-циклодекстрины могут улучшить растворимость соединения IVE. Размер соединения может позволить ему образовывать комплекс включения, что приводит к более высокой растворимости в воде. Эффект усиления на растворимость в воде соединения IVO имеет порядок MBCD> SBEBCD >HPBCD> HPGCD >ACD. MBCD является наиболее эффективным агентом, повышающим растворимость, в этом эксперименте.
Таблица 1. Коммерчески доступные циклодекстрины
циклодекстрин
W7 M Pharma
HPB Pharma
гамма-циклодекстрин
W8 HP Pharma
Таблица 2. Растворимость соединения IVE в различных растворах CD
Пример 3: Влияние различных циклодекстринов на растворимость в воде 5-{3-[4-(2-метил-бензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-диона (IVP)
Для анализа влияния различных CD на растворимость в воде соединения IVP, готовили 1 мл каждого из следующих водных растворов, описанных в таблице 3. Избыточный IVP добавляли к каждому из этих растворов, и образцы встряхивали при комнатной температуре в течение 24 часов на орбитальном шейкере при 200 об/мин. Избыточный IVP присутствовал во всех образцах во всех случаях. Через 24 часа образцы центрифугировали. Аликвоту супернатанта разбавляли, при необходимости, и анализировали с помощью ВЭЖХ. Таблица 3 показывает, что как ACD, так и HPGCD значительно не улучшают растворимость в воде соединения IVP, в то время как MBCD, SBEBCD и HPBCD могут значительно повысить растворимость в воде соединения IVP. Эти результаты показывают, что только бета-циклодекстрины могут улучшить растворимость соединения IVP. Размер соединения может позволить ему образовывать комплекс включения, что приводит к более высокой растворимости в воде. Эффект усиления на растворимость в воде соединения IVP имеет порядок MBCD> SBEBCD >HPBCD> HPGCD >ACD. MBCD является наиболее эффективным агентом, повышающим растворимость, в этом эксперименте.
Таблица 3. Растворимость соединения IVP в различных растворах CD
Пример 4: Влияние различных циклодекстринов на растворимость в воде 5-{3-[4-(3-метил-бензилокси)фенилтио]-фур-2-ил}-имидазолидин-2,4-диона (IVO)
Для анализа влияния различных CD на растворимость в воде соединения IVO, готовили 1 мл каждого из следующих водных растворов, описанных в таблице 4. Избыточное количество IVO добавляли к каждому из этих растворов, и образцы встряхивали при комнатной температуре в течение 24 часов на орбитальном шейкере при 200 об/мин. Избыточное количество IVO присутствовало во всех образцах во всех случаях. Через 24 часа образцы центрифугировали. Аликвоту супернатанта разбавляли, при необходимости, и анализировали с помощью ВЭЖХ. Таблица 4 показывает, что как ACD, так и HPGCD не улучшают растворимость в воде соединения IVO, в то время как MBCD, SBEBCD и HPBCD могут значительно повысить растворимость в воде соединения IVO. Эти результаты показывают, что только бета-циклодекстрины могут улучшить растворимость соединения IVO. Размер соединения может позволить ему образовывать комплекс включения, что приводит к более высокой растворимости в воде. Эффект усиления на растворимость в воде соединения IVO имеет порядок MBCD> SBEBCD >HPBCD> HPGCD >ACD. MBCD является наиболее эффективным агентом, повышающим растворимость, в этом эксперименте.
Таблица 4. Растворимость соединения IVO в различных растворах CD
Пример 5: Влияние различных циклодекстринов на растворимость в воде 5-{3-[4-(3-трифторметил-бензилокси) фенилтио]фур-2-ил}имидазолидин-2,4-диона (IVQ)
Для анализа влияния различных CD на растворимость в воде соединения IVQ, готовили 1 мл каждого из следующих водных растворов, описанных в таблице 5. Избыточное количество IVQ добавляли к каждому из этих растворов, и образцы встряхивали при комнатной температуре в течение 24 часов на орбитальном шейкере при 200 об/мин. Избыточное количество IVQ присутствовало во всех образцах во всех случаях. Через 24 часа образцы центрифугировали. Аликвоту супернатанта разбавляли, при необходимости, и анализировали с помощью ВЭЖХ. Таблица 5 показывает, что как ACD, так и HPGCD не улучшают значительно растворимость в воде соединения IVQ, в то время как MBCD, SBEBCD и HPBCD могут значительно повысить растворимость в воде соединения IVQ. Эти результаты показывают, что только бета-циклодекстрины могут улучшить растворимость соединения IVQ. Размер соединения может позволить ему образовывать комплекс включения, что приводит к более высокой растворимости в воде. Эффект усиления на растворимость в воде соединения IVQ имеет порядок MBCD> SBEBCD >HPBCD> HPGCD >ACD. MBCD является наиболее эффективным агентом, повышающим растворимость, в этом эксперименте.
Таблица 5. Растворимость соединения IVQ в различных растворах CD
Пример 6: Получение комплекса соединения IVO с HPBCD методом выпаривания растворителя.
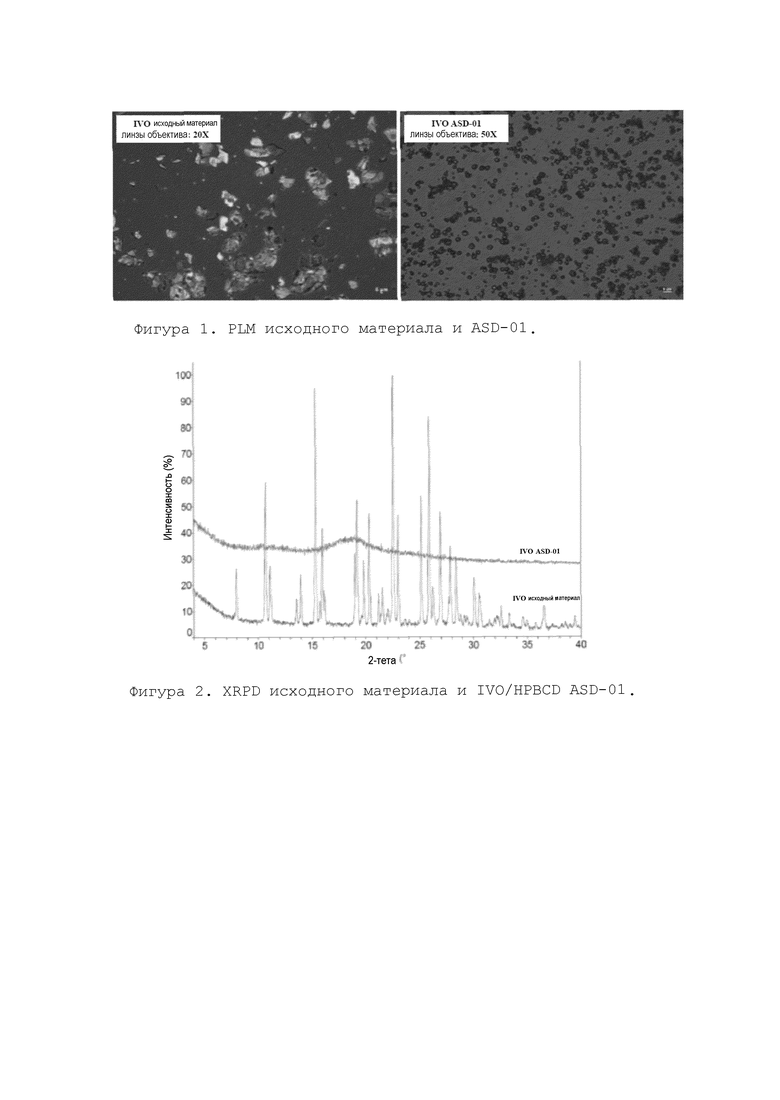
Приблизительно 10,0 г соединения IVO отвешивали в мерную колбу и полностью растворяли в 660 мл метанола (MeOH) с помощью ультразвука (15,2 мг/мл) с получением прозрачного раствора. После фильтрации на мембранном фильтре 0,45 мкм для удаления возможных остатков кристаллического твердого вещества в раствор добавляли приблизительно 30,05 г HPBCD (Ashland) в соотношении 25:75 (масс./масс.) IVO:HPBCD. Образец перемешивали в течение 60 минут до образования прозрачного раствора, а затем высушивали распылением с получением твердой дисперсии. Образец, полученный распылительной сушкой, дополнительно высушивали в условиях вакуума при 30°С в течение 24 часов. На основании результатов поляризационной микроскопии (PLM) (фиг. 1) и рентгеновской порошковой дифрактометрии (XRPD) (фиг. 2), полученный комплекс соединения IVO:HPBCD (1:3, масс./масс.) методом распылительной сушки был аморфным и был назван как соединение IVO/HPBCD ASD-01. Фиг.1 (слева) показывает, что только IVO является двоякопреломляющимся, что указывает на то, что IVO находится в кристаллической форме. Фиг.1 (справа) показывает, что комплекс IVO/HPBCD не показывает двойного лучепреломления. На фиг. 2 показано, что комплекс IVO/HPBCD не проявляет никаких сигналов дифракции на кристалле. IVO/HPBCD, скорее всего, находится в форме комплекса включения, то есть, по меньшей мере, часть соединения формулы (I) встроена, по меньшей мере, частично, в полость циклодекстрина. Образование комплекса включения изменяет IVO от кристаллической формы до аморфного статуса.
Таблица 6. Детали IVO/HPBCD ASD-01
Таблица 7. Установка параметров распылительной сушки и результаты
Пример 7: Сравнимый анализ растворимости для IVO ASD-01
Избыток IVO, 450 мг HPBCD, 150 мг IVO+450 мг HPBCD физическая смесь и 600 мг ASD-1 были приготовлены в 1,5 мл деионизированной воды, затем эти образцы встряхивали при комнатной температуре в течение 24 часов на орбитальном шейкере при 200 об/мин. Образцы центрифугировали в течение 3 и 24 часов. Аликвоту супернатанта разбавляли, при необходимости, и анализировали с помощью ВЭЖХ. При тех же условиях анализа, IVO ASD-01 показал гораздо более высокую растворимость в воде, чем физическая смесь (Таблица 8).
Таблица 8. Результат сравнимого анализа растворимости
Пример 8: Эффект дополнительного эксципиента на растворимость IVO
Это исследование должно было изучить, есть ли какой-либо синергетический эффект путем добавления дополнительных эксципиентов. Как показано в таблице 9, был приготовлен каждый из следующих водных растворов, содержащий 0; 0,125% 0,25%; 0,5%; 0,75%, 1,5% и 2% дополнительного эксципиента в 1 мл 30 мг/мл водного раствора MBCD. Избыток соединения IVO добавляли в каждый из этих растворов и образцы встряхивали в течение 24 часов на орбитальном шейкере при 200 об/мин и при комнатной температуре. Избыток соединения IVO присутствовал во всех образцах во всех случаях. Через 24 часа образцы центрифугировали. Аликвоту супернатанта разбавляли, при необходимости, и анализировали с помощью ВЭЖХ. Как показано в таблице 9, только L-аргинин (L-ARG) демонстрирует значительное улучшение растворимости IVO в растворе MBCD 30 мг/мл. Растворимость IVO уменьшается с увеличением концентрации лимонной кислоты и PEG-400, в то время как для PVP K40, PVP K10, NaCMC и D-маннитола эффект не наблюдался.
Таблица 9. Эффект эксципиента на растворимость соединения IVO в растворе MBCD 30 мг/мл
Пример 9: Эффект L-аргинина на растворимость в воде соединения IVO
Получали 1 мл каждого из нижеследующих водных растворов, содержащих от 0,15 до 2,5% L-ARG в деионизированной воде по массе. Избыточное соединение IVO добавляли к каждому из этих растворов, и образцы встряхивали при комнатной температуре в течение 24 часов на орбитальном шейкере при 200 об/мин. Избыточное соединение IVO присутствовало во всех образцах во всех случаях. Через 24 часа образцы центрифугировали. Аликвоту супернатанта разбавляли, при необходимости, и анализировали с помощью ВЭЖХ. Таблица 10 показывает, что только L-ARG может незначительно улучшить растворимость в воде соединения IVO, намного меньше, чем наблюдается в примере 8. Это указывает на то, что существует синергетический эффект при комбинации MBCD и L-аргинина. Один из возможных вариантов состоит в том, что L-аргинин может способствовать образованию комплекса включения между IVO и MBCD.
Таблица 10. Растворимость соединения IVO в растворах, содержащих L-аргинин
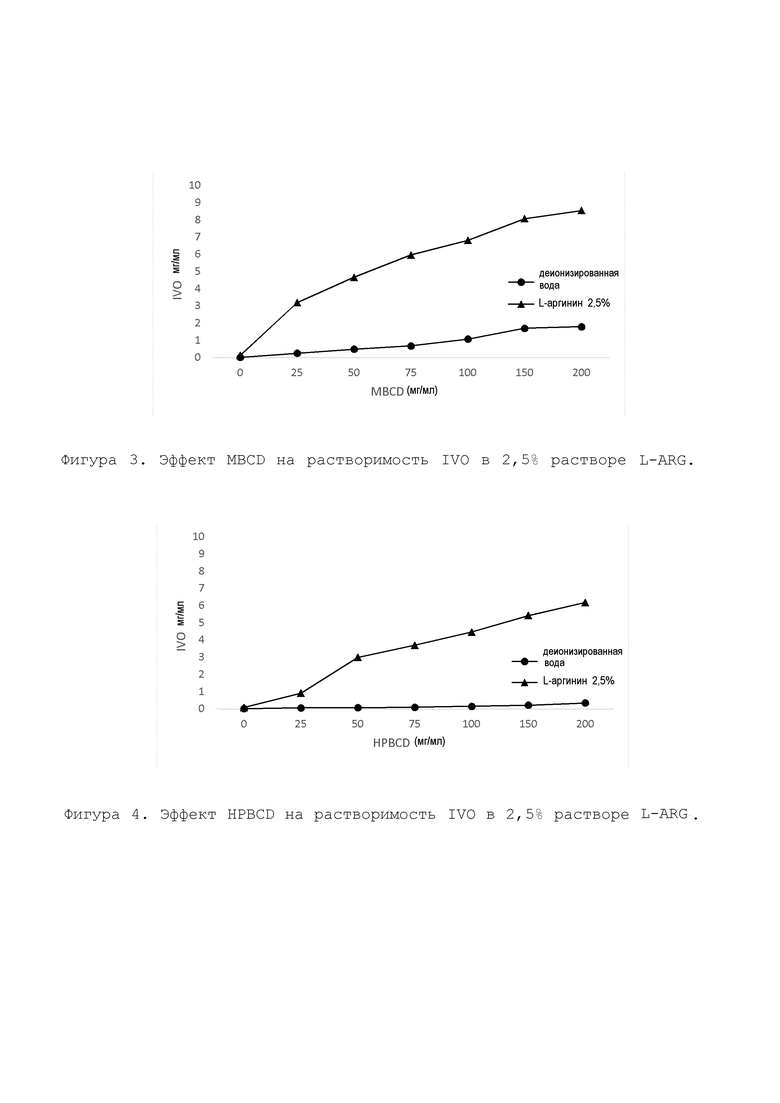
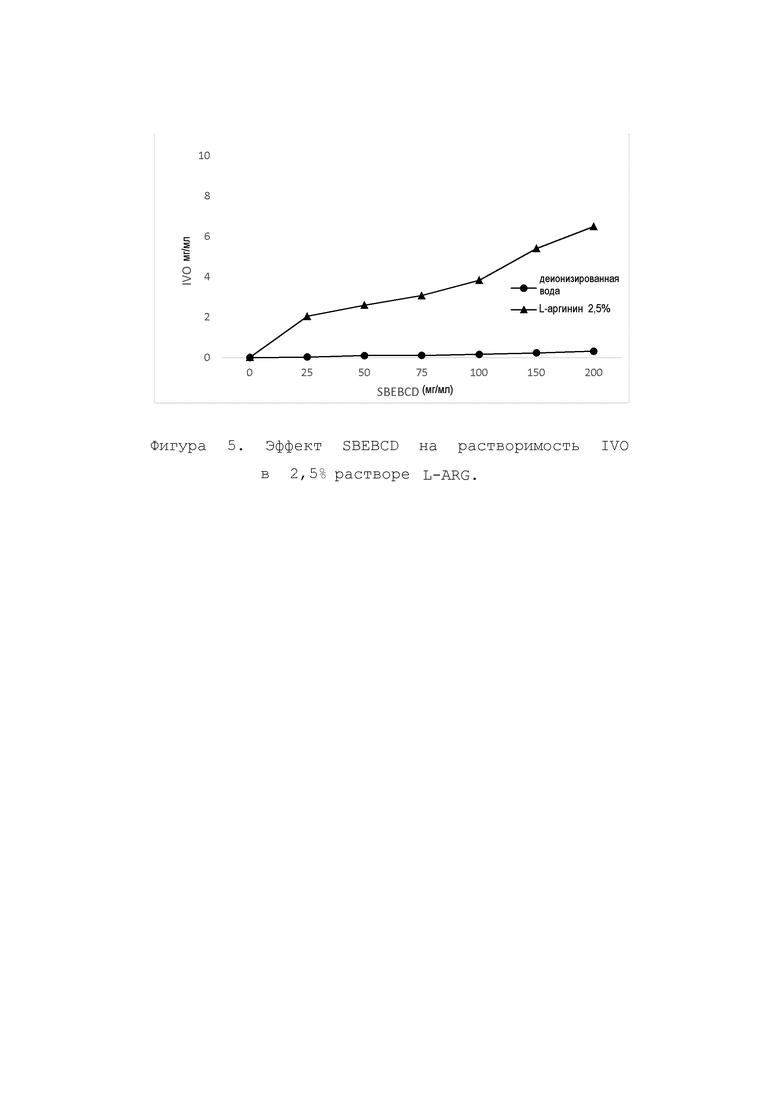
Пример 10: Эффект CD на растворимость соединения IVO в 2,5% растворе L-ARG
Получали 1 мл каждого из следующих водных растворов, содержащих 0; 25; 50; 75; 100; 150 и 200 мг/мл CD в 2,5% растворе L-ARG. Избыток соединения IVO добавляли к каждому из этих растворов, и образцы встряхивали при комнатной температуре в течение 24 часов на орбитальном шейкере при 200 об/мин. Избыточное соединение IVO присутствовало во всех образцах во всех случаях. Через 24 часа образцы центрифугировали. Аликвоту супернатанта при необходимости разбавляли и анализировали с помощью ВЭЖХ. На фиг. 3, 4 и 5 показано, что L-ARG проявляет сингенный эффект на улучшение растворимости со всеми 3 производными β-CD. Добавление L-аргинина улучшило растворимость IVO в водном растворе MBCD, HPBCD и SBEBCD более чем в 5, 18 и 20 раз, соответственно.
Пример 11: Фармакокинетическое исследование составов IVO у собак породы бигль
Введение
Этот пример описывает фармакокинетику (PK) IVO после однократного перорального введения (PO) или внутривенного введения (IV) различных составов, содержащих IVO, кобелям собак породы бигль.
Материалы
Подопытные животные: Виды: собака; порода: бигль; пол: кобель; общее количество: всего 3 кобеля были отобраны для исследования; масса тела: от 8 до 13 кг; Поставщик:: Marshalls BioResources (New York, NY, USA) Все истории жизни хранятся в CTPS, QPS Taiwan.
Материалы исследования
Немикронизированная суспензия IVO
Немикронизированный IVO (PT-C12071028-F13001) вводили животным посредством перорального введения в виде суспензии. Суспензию получали в 10% (масс./об.) гидроксипропил-β-циклодекстрине (HPBCD). Дозу 20 мг/кг использовали для 1-й группы дозирования.
Микронизированная суспензия IVO
Микронизированный IVO (D-1405FP1321-01) вводили животным посредством перорального введения в виде суспензии. Суспензию получали в 10% (масс./об) HPBCD. Дозу 20 мг/кг использовали для 2-ой группы дозирования.
Немикронизированный IVO в капсуле
Немикронизированный IVO (PT-C12071028-F13001) вводили животным путем перорального введения в виде капсулы. Капсулами заполняли желатиновые капсулы при тестировании средства в каждый день дозирования. Дозу 20 мг/кг использовали для 3-й группы дозирования.
Микронизированный IVO в капсуле
Микронизированный IVO (D-1405FP1321-01) вводили животным путем перорального введения в виде капсулы. Капсулами заполняли желатиновые капсулы при тестировании средства в каждый день дозирования. Дозу 20 мг/кг использовали для 4-ой группы дозирования.
Внутривенное введение (IV) немикронизированного IVO
Немикронизированный IVO (PT-C12071028-F13001) растворяли в DMSO и вводили животным посредством внутривенного введения. Дозу 4 мг/кг использовали для 5-ой группы дозирования.
Способы
План исследования
Дозирование и взятие крови
Это исследование было разработано в перекрестном способе. Три кобеля последовательно получали однократную дозу немикронизированной суспензии IVO (20 мг/кг IVO, 1-я дозировка), одну дозу микронизированной суспензии IVO (20 мг/кг IVO, 2-я дозировка), одну дозу немикронизированного IVO в капсуле (20 мг/кг IVO, 3-я дозировка), одну дозу микронизированного IVO в капсуле (20 мг/кг IVO, 4-я дозировка) и одну дозу ВВ немикронизированного IVO (4 мг/кг IVO, 5-я дозировка) с периодом выведения (≥3 дня) между каждым лечением в этом фармакокинетическом исследовании. Кровь брали у собак-кобелей до введения дозы и через 0,25, 0,5, 1, 2, 4, 6, 8, 12 и 24 ч после введения дозы у животных в 1-й группе дозирования, 2-й группе дозирования, 3-й группе дозирования и 4-й группе дозирования, и до введения дозы, и через 5 мин после введения дозы и через 0,25, 0,5, 1, 2, 4, 6, 8, 12 и 24 ч после введения дозы у животных из 5-й группы дозирования в пробирки, содержащие K2EDTA в качестве антикоагулянта. Образцы крови немедленно помещали на лед и центрифугировали (1500×g в течение 10 минут при 4°C) в течение 60 минут после взятия крови. Образцы плазмы хранили при -60°C или ниже в CTPS, QPS Taiwan, до передачи в QPS Taiwan. Сводные показатели о дозировке и времени взятия крови показана в таблице 11.
Таблица 11. Дозирование и взятие крови
1, 2, 4, 6, 8, 12 и
24 ч после введения
после введения и 0,25,
0,5, 1, 2, 4, 6, 8, 12
и 24 ч после введения
a Одним и тем же собакам применяли различные виды лечения перекрестным способом с периодом выведения более 3 дней между каждым введением дозы.
bИдентификаторы животных были 13M00001, 13M00007 и 13M00012.
сАнтикоагулянт был K2EDTA.
Биоаналитический метод анализа плазмы
Образцы плазмы анализировали в QPS Taiwan с использованием валидированного метода LC-MS/MS с LLOQ 5,000 нг/мл для IVO. Концентрации в плазме ниже самого низкого стандарта были зарегистрированы как ниже предела количественного определения (BQL).
АНАЛИЗЫ ДАННЫХ
Параметры PK
Параметры PK определяли в QPS Taiwan с использованием некомпартментного анализа индивидуального профиля с использованием Phoenix® WinNonlin® 6.3 (Pharsight Corporation, Mountain View, CA, USA). Наблюдаемая максимальная концентрация в плазме (Cmax) и время Cmax (Tmax) были определены непосредственно из данных. Площадь под кривой концентрация в плазме-время от времени-0 до 24 часов после введения дозы (AUC0-24ч) и площадь под кривой концентрация в плазме-время от времени-0, экстраполированной на бесконечность (AUC0-∞), были определены с помощью линейного правила трапеций:
AUC0-24ч=(t2-t1)×(C1+C2)/2
с экстраполяцией на бесконечность с использованием:
AUC0-∞=AUClast+Clast/λ
Там, где это возможно, кажущийся конечный период полувыведения (t1/2) рассчитывали по следующей формуле, где λ представляет собой константу скорости терминальной элиминации:
t1/2=ln(2)/λ
Критерии отбора для включения точек данных в расчет λ требовали, чтобы по крайней мере три точки данных, представляющие конечную фазу, были регрессированы и чтобы r2 ≥ 0,85 при округлении. Период полувыведения был определен как не определено (ND), если эти критерии не были выполнены. Общий клиренс (CL), среднее время удержания, экстраполированное на бесконечность (MRT0-∞), объем распределения (Vz) и биодоступность при пероральном введении препарата (F) определяли по следующим формулам:
CL=Доза/AUC0-∞, IV
MRT0-∞=AUMC0-∞/AUC0-∞
Vz=Доза/(λ•AUC0-∞)
F=(AUC 0-∞,РО/ДозаРО)/(AUC0-∞,IV/ДозаIV)
Номинальное время сбора образцов использовали для расчетов AUC, CL и t1/2. Номинальные дозы использовали для нормализованные по дозе AUC0-∞ во всех группах, получавших испытуемое изделие.
Соглашения о предоставлении данных
Анализ данных PK плазмы
Отдельные или средние концентрации в плазме указывали с точностью до трех знаков после запятой. Средние концентрации в плазме были рассчитаны с использованием SAS® и представлены с точностью до трех знаков после запятой. Отдельные концентрации, которые были BQL были установлены на ноль для расчета параметров PK.
Отдельные концентрации в плазме вводили в WinNonlinТМ с использованием значений с точностью до трех знаков после запятой. Параметры PK со значениями до 999 были представлены в виде до трех значащих цифр и значения ≥ 1000 были представлены как целые числа со следующими исключениями:
Значения Tmax указываются с одним десятичным знаком, если значение составляет один час или более, и с двумя десятичными знаками, если значение составляет менее одного часа.
Значения λ указываются с точностью до трех знаков после запятой.
CV% значения указываются с одним десятичным знаком.
Округление
Генерируемые компьютером данные, показанные в таблицах, были соответствующим образом округлены для включения в этот пример. В результате вычисление значений из данных в этом примере в некоторых случаях приводит к незначительным отклонениям.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Группы дозирования и график взятия крови для этого исследования приведены в таблице 11. Параметры PK для всех групп, получавших лечение IVO, приведены в таблице 12.
Таблица 12. Параметры PK IVO у кобелей собак после однократного перорального введения или внутривенного введения различных составов IVO
в капсуле (20 мг/кг)
в капсуле (20 мг/кг)
(4 мг/кг)
(ч•нг/мл)
(ч•нг/мл)
(ч)
(л/ч/кг)
--: не определено.
a средняя величина (диапазон).
bF(%)рассчитывали как (AUC0-∞,РО/ДозаРО)/(AUC0-∞,IV/Dos)
В общей сложности 3 кобелям породы бигль было дозировано в перекрестном способе пять составов IVO с периодом вымывания по меньшей мере 3 дня между каждой дозировкой в соответствии с протоколом. Не было отмечено ни одного инцидента с потенциально значительным влиянием на исход исследования.
Никаких количественных уровней IVO не было найдено во всех образцах до введения дозы. После перорального введения немикронизированной суспензии IVO (1-я группа дозирования), микронизированной суспензии IVO (2-я группа дозирования), немикронизированного IVO в капсуле (3-я группа дозирования) и микронизированного IVO в капсуле (4-я группа дозирования) Cmax IVO наблюдали в течение 2,0- 4,0 ч, 2,0-4,0 ч, все 4 ч и 0,5-2,0 ч, соответственно. Средние значения Cmax составляли 1037, 2524, 200 и 272 нг/мл в 1-й, 2-й, 3-й и 4-й группах дозирования, соответственно.
Средние значения AUC0-24ч IVO составляли 13105, 29085, 1807 и 1922 ч•нг/мл, и средние значения AUC0-∞ IVO составляли 15876, 35021, 1808 и 1993 после перорального введения в 1-й, 2-й, 3-й и 4-й группах дозирования, соответственно. Уровни в плазме снизились с t1/2 7,6, 8,2, 3,3 и 4,8 ч в 1-й, 2-й, 3-й и 4-й группах дозирования, соответственно (Таблица 12).
После внутривенного болюсного введения немикронизированного IVO при уровнях дозы 4 мг/кг Cmax IVO наблюдали при первом времени отбора проб, 0,08 ч после введения дозы, и в среднем составляли 6818 нг/мл. Средние значения AUC0-24ч и AUC0-∞ для IVO составляли 26959 и 27856 ч•нг/мл, соответственно. Уровни в плазме снизились с t1/2 4,7 ч. Средний системный клиренс составил 0,148 л/ч/кг, и средний Vz составил 0,972 л/кг. Среднее значение MRT0-∞ составляло 6,6 ч (таблица 12). После перорального введения немикронизированной суспензии IVO, микронизированной суспензии IVO, немикронизированного IVO в капсуле и микронизированного IVO в капсуле, биодоступность при пероральном введении препарата (F) IVO составила 11,1%, 25,1%, 1,43% и 1,46%, соответственно.
Выводы
Животным вводили IVO в виде внутривенного или перорального состава путем однократного введения. Результаты исследования показали, что скорость абсорбции и воздействие лекарственного средства IVO в группах, получавших суспензию, были выше, чем в группах, получавших капсулы. После перорального введения немикронизированной суспензии IVO, микронизированной суспензии IVO, немикронизированного IVO в капсуле и микронизированного IVO в капсуле, биодоступность при пероральном введении препарата (F) составила 11,1%, 25,1%, 1,43% и 1,46%, соответственно.
Пример 12: Параллельное исследование относительной биодоступности при пероральном введении препарата пероральных составов IVO после однократного перорального (PO) введения ʺне-наивнымʺ кобелям собак породы Бигль
ЦЕЛЬ ИССЛЕДОВАНИЯ
Цель этого исследования состояла в определении относительной биодоступности при пероральном введении двух составов IVO (заполненных в капсулы и дозированных в дозе 150 мг/собаку) по сравнению с эталонным составом немикронизированной суспензии IVO в дозе 20 мг/кг после однократного перорального введения ʺне-наивнымʺ кобелям породы бигль. Образцы плазмы брали у всех животных в течение 24 часов после введения дозы, и концентрацию IVO определяли с помощью метода LC-MS/MS.
Соответствие требованиям содержания животных
Все применимые части исследования соответствовали следующим инструкциям и рекомендациям относительно ухода за животными и санитарно-бытовых условий:
Рекомендации AAALAC International and NIH, изложенные в ʺGuide for the Care and Use of Laboratory Animals,ʺ National Research Council - ILAR, Revised 2011. People's Republic of China, Ministry of Science & Technology, ʺRegulations for the Administration of Affairs Concerning Experimental Animalsʺ, 1988.
МАТЕРИАЛЫ И СПОСОБЫ
Информация об исследуемом препарате и эталонном образце
Исследуемые составы и эталонный стандартный порошок были предоставлены Спонсором. Детали и протокол анализа (CoA) перечислены ниже:
(Внутренний стандарт)
(Биоаналитический Стандарт)
немикронизированный API)
*Солевой фактор=Молекулярная масса в солевой форме/Молекулярная масса в свободной форме
(группа 2)
Поправочный коэффициент=(Молекулярная масса в солевой форме /Молекулярная масса в свободной форме)/Чистота/Активность
Тест-система, план исследования и уход за животными
Тест-система
В этом исследовании использовали девять ʺне-наивныхʺ кобелей собак породы бигль (7,40-9,78 кг). Животные были получены от утвержденного поставщика (Marshall Bioresources, Beijing, China), и у каждого животного был уникальный номер татуировки кожи на ухе в качестве идентификации.
План исследования
суспензия
Носитель: 10% (масс/об) Гидроксипропил-β-циклодекстрин (HPBCD) в воде для инъекций
мг/собаку
гранулы
мг/собаку
Уход за животными
Комнату для животных контролировали и мониторировали на влажность (целевой средний диапазон от 40% до 70%) и температуру (целевой средний диапазон от 18°C до 26°C) с 10-20 проветривание помещения/час. В комнате был 12-часовой цикл свет/темнота, за исключением случаев, когда перерывы были необходимы в ходе процесса исследований.
Животных содержали индивидуально в сетчатых клетках из нержавеющей стали в условиях жизни, что соответствовало National Research Council ʺGuide for the Care and Use of Laboratory Animalsʺ
Животных кормили два раза в день. Группу собак ежедневно кормили приблизительно 220 граммами сертифицированного корма для собак (Beijing Vital Keao Feed Co., Ltd. Beijing, P. R. China). Эти количества были скорректированы по мере необходимости на основе потребления пищи в группе или индивидуальных изменений массы тела группы или индивидуума и/или изменений в сертифицированном корме.
Для группы натощак (группы с пероральным введением (PO) дозы) животных кормили днем (с 3:30 до 4:00 после полудня) до дня перорального приема, а оставшуюся пищу убирали примерно в 7:00 после полудня. Пищу не давали до 4 часов после введения дозы, если это не указано в этом протоколе. Животных группы натощак кормили один раз в день дозирования в количестве приблизительно 220 г.
Животным давали очищенную обратным осмосом и хлорированную воду неограниченно с помощью автоматической системы поения.
Пищевые компоненты и примеси окружающей среды в корме регулярно анализировались ежедневно сторонней или независимой лабораторией, соответственно. Отчеты об анализе и номера партий хранятся в испытательной лаборатории.
Питьевую воду для животных анализировали на предмет загрязнения каждый четверть часа независимой лабораторией. Отчеты по анализу воды хранятся в отделе ветеринарной операции в испытательной лаборатории.
Получение лекарственной формы
Для получения состава суспензии:
Суспензию IVO для группы 1 в концентрации 5 мг/мл в 10% (мас./об.) гидроксипропил-β-циклодекстрина в воде для инъекций получали в день введения. Подробная информация об используемом носителе и процедуре приготовления дозового состава была записана в папке исследования.
Для получения состава капсулы:
Желатиновые капсулы (размер: 0#) использовали в этом исследовании для групп 2 и 3.
a. Животных взвешивали в день введения дозы и отбирали с массой тела в пределах 10±1 кг, за исключением 7,91 кг животного D303 отбирали.
b. 150 мг API состава заполняли в капсулы.
c. 3 капсулы всего дозировали на собаку.
Подробное описание массы капсул представлены в таблице 13.
Таблица 13. Массы капулы
Введение
Лекарственные формы IVO вводили перорально в соответствии с СОП:
Пероральное введение лекарственной формы в виде суспензии:
Дозы для введения через зонд вливали с использованием 6 мл носителя (приблизительно в 3 раза больше объема трубки для зондирования). Все трубки были одинакового размера и нарезаны на одинаковую длину, чтобы объем промывки был сопоставим.
Пероральное введение лекарственной формы в виде капсулы:
a. Оттягивали нижнюю челюсть собаки вниз и помещали капсулу глубоко в горло. Затем капсулу проталкивали в глотку большим или указательным пальцем.
b. Для облегчения проглатывания капсулы животному давали 6 мл воды после каждого введения капсулы.
c. После введения капсулы глотание стимулировали легким поглаживанием собаки по горлу.
d. После введения рот собаки проверяли на предмет проглатывания капсулы.
Животных взвешивали перед каждым введением дозы и массы тела отдельных животных представлены в таблице 14.
Таблица 14. Масса тела животного
Сбор и подготовка образцов
Серийные образцы крови (приблизительно 0,8 мл в 10 мкл 0,5 М K2-EDTA) собирали с помощью венопункции из головной вены у животных без использования седативного средства до введения дозы (0), через 0,25 (15 мин), 0,5 (30 минут), 1, 2, 4, 6, 8, 12 и 24 часа после введения дозы. Фактическое время сбора образцов было записано в папке исследования. Для образцов, отобранных в течение первого часа с момента введения, допустима ± 1 минута запланированного времени. Для остальных точек времени образцы, которые были взяты в течение 5% от запланированного времени, были приемлемыми и не рассматривались как отклонение от протокола.
После сбора образцы крови осторожно переворачивали несколько раз и сразу же помещали на влажный лед перед центрифугированием при 2-8°C и 3000×g в течение 10 минут в течение 1 часа после сбора крови. Затем образцы плазмы переносили в полипропиленовые микроцентрифужные пробирки с маркировкой, быстро замораживали и переносили в отдел биоанализа на сухом льду и хранили замороженными в морозильной камере, в которой поддерживалась температура -60°C или ниже, до проведения биоанализа.
Клиническое наблюдение
Дважды в день (примерно в 9:30 до полудня и 3:30 после полудня) животных наблюдали на предмет смертности и признаков боли и дистресса. Наблюдения у клетки за общим состоянием здоровья и внешним видом проводили один раз в день. В день введения дозы животных наблюдали до и после каждого момента сбора крови. Любые необычные данные наблюдения, отмеченные на протяжении всего исследования, были записаны в папке исследования.
Анализ образцов
Образцы плазмы анализировали с использованием метода LC/MS-MS. Нижний предел количественного определения (LLOQ) для IVO в плазме составлял 2,00 нг/мл и верхний предел количественного определения (ULOQ) составлял 3000 нг/мл.
Анализ фармакокинетических данных
Профили концентрация в плазме-время IVO подвергали некомпартментному фармакокинетическому анализу с использованием программного обеспечения WinNonlin (версия 6.2.1).
Среднее время удержания (MRT), площадь под кривой «концентрация в плазме-время» (AUC) от нуля до времени последнего измерения (AUC0-last) и AUC от нуля до бесконечности (AUC0-inf) рассчитывались с использованием линейно-логарифмического правила трапеции (см.: Gabrielsson J. and Weiner D. Non-compartmental analysis in ʺPharmacokinetic and Pharmacodynamic Data Analysis: Concepts & Applicationsʺ, 3rd edition, Chapter 3.7.2., page 141-146. Swedish Pharmaceutical Press; 2002).
Все значения, кроме значений времени, были представлены в трех значащих цифрах. Значения времени указывались с точностью до двух знаков после запятой.
Номинальное время отбора проб использовали для расчета всех фармакокинетических параметров, поскольку не было отклонения между фактическим и номинальным временем отбора проб.
РЕЗУЛЬТАТЫ
Клинические наблюдения
Приблизительно 10 мл светло-желтой слизистой рвоты с пустой оболочкой капсулы наблюдали у животного D203 группы 2 через 1 час после введения дозы. Никакого ненормального эффекта не наблюдалось у других исследуемых животных во время этого исследования.
Верификация концентрации дозы
Верификация концентрации дозы в группе 1 показала точность 101%.
Фармакокинетика
Фармакокинетические параметры IVO представлены в Таблице 15.
Таблица 15. Фармакокинетические параметры лекарственных форм IVO
суспензия (20 мг/кг)
(капсула, 150 мг/собака)
Выводы
После однократного перорального введения немикронизированной суспензии IVO в дозе 20 мг/кг ʺне-наивнымʺ кобелям собак породы бигль натощак, максимальная концентрация в плазме (Cmax) составила 1453±90,7 нг/мл через 4,00±3,46 часов после введения дозы (Tmax). Содержание вещества в плазме, AUC0-inf и AUC0-last составило 12963±4191 и 12630±3948 нг·ч/мл, соответственно.
После однократного перорального введения IVO ASD-01 в капсуле в дозе 150 мг/собаку ʺне-наивнымʺ кобелям собак породы бигль натощак, максимальная концентрация в плазме (Cmax) составила 3947±1740 нг/мл через 1,67±0,58 часов после введения дозы (Tmax). Содержание вещества в плазме, AUC0-inf и AUC0-last составило 20233±8545 и 20100±8391 нг·ч/мл, соответственно.
После однократного перорального введения гранул IVO в капсуле в дозе 150 мг/собаку ʺне-наивнымʺ кобелям собак породы бигль натощак, максимальная концентрация в плазме (Cmax) составила 1071±597 нг/мл через 3,33±2,31 часа после введения дозы (Tmax). Содержание вещества в плазме, AUC0-inf и AUC0-last составило 6215±NC и 7077±3749 нг·ч/мл, соответственно.
По сравнению с контрольным составом IVO ASD-01 в капсуле абсорбировалось быстрее (Tmax=1,67), в то время как гранулы IVO в капсулах демонстрировали сходную Tmax через 3,33 часа по сравнению с эталонным составом. Количество в системном кровотоке (AUC) и максимальные концентрации в плазме (Cmax) IVO были намного выше для IVO ASD-01 в капсуле, чем для эталонного состава. Гранулы IVO в капсуле показали более низкую пероральную абсорбцию по сравнению с эталонным составом.
Относительная биодоступность IVO ASD-01 и гранул IVO составила 200% и 65,5% по сравнению с эталонным составом, соответственно.
Пример 13: Сравнение IVO/HPBCD ASD-в-капсуле с IVO API-в-капсуле у собак породы бигль
Состав пероральной суспензии немикронизированного IVO (20 мг/кг) использовали в качестве эталона. Суспензию получали в 10% (масс./об.) гидроксипропил-β-циклодекстрине (HPBCD). Использовали дозу 20 мг/кг (суспензия IVO), как показано в примерах 11 и 12.
IVO API-в-капсуле исследовали в Примере 11 с эталонным составом. Комплекс IVO:HPBCD в соотношении 25:75 (масс./масс.) получали в соответствии с процедурой, описанной в примере 6. IVO/HPBCD ASD-в-капсуле исследовали в примере 12 вместе с эталонным составом.
В таблице 16 показаны параметры PK трех различных составов, вводимых перорально собакам породы бигль. IVO API является высококристаллическим и обладает очень низкой растворимостью в воде. Состав IVO API-в-капсуле (чистый API, заполненный в пероральные капсулы) показал более низкие значения AUC и Cmax, чем у эталонного состава. Напротив, улучшенный состав для перорального введения, IVO/HPBCD ASD-в-капсуле, показывает значительно повышенную растворимость в воде. В результате пероральная абсорбция (AUC) и Cmax IVO были значительно увеличены с использованием IVO/HPBCD комплекса ASD-в-капсуле, примерно в 17 и 18 раз, соответственно, у собак по сравнению с IVO API-в-капсуле, как показано в таблице 16.
Таблица 16: Сравнение PK IVO/HPBCD ASD-в-капсуле с IVO API-в-капсуле у собак породы бигль
(ASD/API)a
(ASD/API)a
a Отношения рассчитывали после нормализации с параметрами эталонного состава.
По сравнению с эталонными составами IVO/HPBCD ASD-01 в капсуле абсорбировался быстрее (Tmax=1,7), в то время как IVO API-в-капсуле показывал то же значение Tmax через 4 часа по сравнению с эталонным составом. В общем и целом, HPBCD может значительно улучшить растворимость IVO в воде и увеличить системную абсорбцию.
Группа изобретений относится к области медицины и фармацевтики и представляет собой фармацевтические композиции (4 варианта), включающие соединение формулы I:
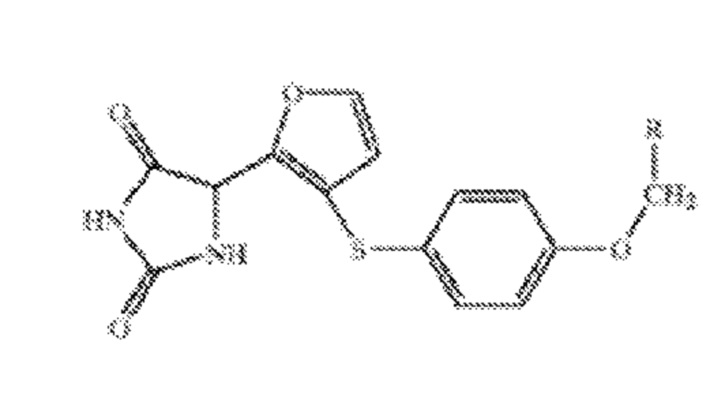 ,
,
где R выбран из группы, состоящей из фенила, 4-бензилоксифенила, 4-бифенила, 4-метоксифенила, 3-метоксифенила, 2-метоксифенила, 3,5-диметоксифенила, 4-хлорфенила, 3-хлорфенила, 2-хлорфенила, 4-метилфенила, 3-метилфенила, 2-метилфенила и 3-трифторметилфенила, или его соль; и водорастворимый бета-циклодекстрин. Группа изобретений обеспечивает повышение растворимости указанных соединений, тем самым увеличивая биодоступность при пероральном введении. 4 н. и 17 з.п. ф-лы, 5 ил., 19 табл., 13 пр.
1. Фармацевтическая композиция, включающая соединение формулы (I) или его соль; и водорастворимый бета-циклодекстрин, где R выбран из группы, состоящей из фенила, 4-бензилоксифенила, 4-бифенила, 4-метоксифенила, 3-метоксифенила, 2-метоксифенила, 3,5-диметоксифенила, 4-хлорфенила, 3-хлорфенила, 2-хлорфенила, 4-метилфенила, 3-метилфенила, 2-метилфенила и 3-трифторметилфенила.

Формула (I)
2. Фармацевтическая композиция по п. 1, где молярное отношение соединения формулы (I) к бета-циклодекстрину составляет от 1:1 до 1:50.
3. Фармацевтическая композиция по п. 1, где фармацевтическая композиция увеличивает растворимость соединения формулы (I) в деионизированной воде при комнатной температуре, по меньшей мере, в 1,5 раза по сравнению с растворимостью соединения формулы (I) без бета-циклодекстрина в тех же условиях.
4. Фармацевтическая композиция по п. 1, где фармацевтическая композиция увеличивает биодоступность при пероральном введении соединения формулы (I) у млекопитающего по меньшей мере на 50% по сравнению с биодоступностью при пероральном введении соединения формулы (I) без бета-циклодекстрина.
5. Фармацевтическая композиция по п. 1, где фармацевтическая композиция индуцирует, по меньшей мере, в 2 раза выше Cmax соединения формулы (I), чем это достигается, когда соединение вводят отдельно в том же количестве и при тех же условиях.
6. Фармацевтическая композиция по п. 1, где бета-циклодекстрин выбран из группы, состоящей из метил-бета-циклодекстрина, гидроксипропил-бета-циклодекстрина и сульфобутилового эфира бета-циклодекстрина.
7. Фармацевтическая композиция по п. 1, где, по меньшей мере, часть соединения формулы (I) встроена, по меньшей мере, частично в полость бета-циклодекстрина.
8. Фармацевтическая композиция по п. 1, где композиция включает физическую смесь соединения формулы (I) и бета-циклодекстрина.
9. Фармацевтическая композиция, включающая соединение формулы (I) или его соль; водорастворимый бета-циклодекстрин; и эксципиент, где R выбран из группы, состоящей из фенила, 4-бензилоксифенила, 4-бифенила, 4-метоксифенила, 3-метоксифенила, 2-метоксифенила, 3,5-диметоксифенила, 4-хлорфенила, 3-хлорфенила, 2-хлорфенила, 4-метилфенила, 3-метилфенила, 2-метилфенила и 3-трифторметилфенила.
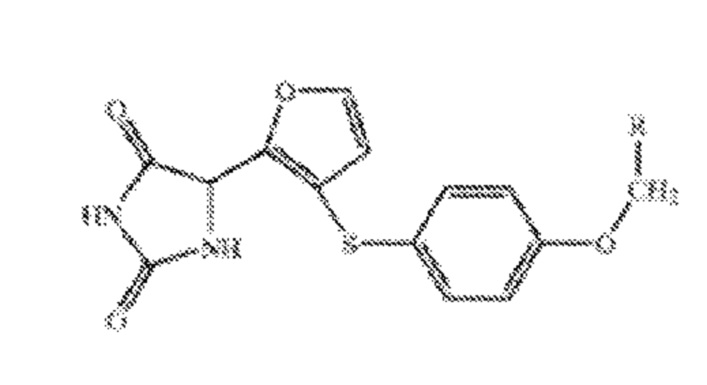
Формула (I)
10. Фармацевтическая композиция по п. 9, где эксципиент представляет собой L-аргинин.
11. Фармацевтическая композиция по п. 9, где молярное соотношение соединения формулы (I) к бета-циклодекстрину составляет от 1:1 до 1:50.
12. Фармацевтическая композиция по п. 9, где фармацевтическая композиция увеличивает растворимость соединения формулы (I) в деионизированной воде при комнатной температуре, по меньшей мере, в 1,5 раза по сравнению с растворимостью соединения формулы (I) без бета-циклодекстрина в тех же условиях.
13. Фармацевтическая композиция по п. 9, где фармацевтическая композиция увеличивает биодоступность при пероральном введении соединения формулы (I) у млекопитающего по меньшей мере на 50% по сравнению с биодоступностью при пероральном введении соединения формулы (I) без бета-циклодекстрина.
14. Фармацевтическая композиция по п. 9, где фармацевтическая композиция индуцирует, по меньшей мере, в 2 раза выше Cmax соединения формулы (I), чем это достигается, когда соединение вводят отдельно в том же количестве и при тех же условиях.
15. Фармацевтическая композиция по п. 9, где бета-циклодекстрин выбран из группы, состоящей из метил-бета-циклодекстрина, гидроксипропил-бета-циклодекстрина и сульфобутилового эфира бета-циклодекстрина.
16. Фармацевтическая композиция по п. 9, где эксципиент выбран из группы, состоящей из L-аргинина, лимонной кислоты, PEG-4000, PVP K40, PVP K10, NaCMC и D-маннитола.
17. Фармацевтическая композиция по п. 9, где, по меньшей мере, часть соединения формулы (I) встроена, по меньшей мере, частично в полость циклодекстрина.
18. Фармацевтическая композиция по п. 9, где композиция включает физическую смесь соединения формулы (I), бета-циклодекстрина и эксципиента.
19. Фармацевтическая композиция, включающая соединение 5-{3-[4-(3-метил-бензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-диона (IVO); и циклодекстрин, где циклодекстрин представляет собой гидроксипропил-бета-циклодекстрин.
20. Фармацевтическая композиция по п. 19, где массовое отношение соединения формулы (I) к циклодекстрину составляет 1:3.
21. Фармацевтическая композиция, включающая соединение 5-{3-[4-(3-метил-бензилокси)фенилтио]фур-2-ил}имидазолидин-2,4-диона (IVO); циклодекстрин и эксципиент, где циклодекстрин представляет собой гидроксипропил-бета-циклодекстрин и эксципиент представляет собой L-аргинин.
| US 2006041000 A1, 23.02.2006 | |||
| US 4727064 A, 23.02.1988 | |||
| WO 2010126818 A1, 04.11.2010. |

