ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к фармацевтическим композициям, содержащим соединение пиррола. В частности, оно относится к фармацевтическим композициям, подходящим для перорального введения, к фармацевтическим композициям, которые подходят для парентерального введения и к применению таких композиций для предупреждения или лечения грибковых заболеваний. Оно также относится к способу получения фармацевтической композиции, описанной в данном документе.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Инвазивные грибковые инфекции общепризнаны заболеваниями хозяев с ослабленным иммунитетом. В течение последних двадцати лет произошло существенное увеличение количества зарегистрированных случаев грибковых инфекций. Отчасти это связано с повышением осведомленности и улучшением диагностики грибковых инфекций. Однако, главной причиной такой повышенной заболеваемости является существенное увеличение числа предрасположенных индивидуумов. Это обусловлено рядом факторов, включая новые и агрессивные иммуносуппрессивные терапии, увеличение выживаемости при интенсивной терапии, увеличение числа трансплантаций и более широкое применение антибиотиков во всем мире.
В некоторых группах пациентов частота возникновения грибковых инфекций высокая; у реципиентов трансплантатов легкого частота колонизации и инфицирования грибковыми организмами достигает 20%, а у реципиентов трансплантата гемопоэтических стволовых клеток грибковая инфекция достигает 15% (Ribaud et al., 1999, Clin Infect Dis. 28:322-30).
За последние годы улучшилось понимание вклада сенсибилизации к грибковой инфекции, колонизации, аллергии и локализованной инфекции в ухудшение существующих респираторных заболеваний. Так, грибок обнаружен при астме, ХОБЛ, бронхоэктазе и кистозном фиброзе. Аллергический бронхолегочный аспергиллез (АБЛА) представляет собой заболевание нижних дыхательных путей, вызываемое грибковой колонизацией, как правило, Apsergillus fumigatus. АБЛА встречается при астме с частотой 0,7-3,5%, а при кистозном фиброзе с частотой 7-9%.
В настоящее время существует четыре класса противогрибковых препаратов для лечения системных грибковых инфекций. Это полиены (например, амфотерицин В), азолы (например, кетоконазол или итраконазол), эхинокандины (например, каспофунгин) и флуцитозин.
Полиены представляют собой наиболее старый класс противогрибковых агентов, которые начали применять в 1950-х годах. Точный механизм действия остается невыясненным, однако полиены являются единственными эффективными препаратами против организмов, в наружных мембранах которых содержатся стеролы. Предполагают, что амфотерицин В взаимодействует с мембранными стеролами, образуя поры, через которые происходит утечка цитоплазматических компонентов и затем гибель клетки.
Азолы действуют путем ингибирования 14α-деметилазы по цитохром Р450-зависимому механизму. Это приводит к истощению мембранного стерола эргостерола и накоплению предшественников стерола, в результате чего нарушается текучесть и структура плазматической мембраны. Эхинокардины действуют путем ингибирования β-глюкансинтазы, фермента, синтезирующего клеточную стенку. Это приводит к аномалиям в образовании клеточной стенки, осмотической чувствительности и лизису клеток.
Флуцитозин представляет собой аналог пиримидина, нарушающего клеточный метаболизм пиримидинов, а также синтез ДНК, РНК и белков. Однако широкая устойчивость к флуцитозину ограничивает его терапевтическое применение.
Можно отметить, что в настоящее время существующие противогрибковые агенты действуют преимущественно на две клеточные мишени: мембранные стеролы (полиены и азолы) и β-глюкансинтазу (эхинокандины).
Часто отмечается устойчивость и к азолам, и к полиенам, и для борьбы с инвазивными грибковыми инекциями остаются только эхинокандины, которые начали применяться недавно. По мере нарастания применения эхинокандинов неизбежно возникнет резистентность к ним грибка.
Для обеспечения у пациентов положительных исходов терапии необходим поиск новых классов противогрибковых агентов.
Соединения пиррола также были отнесены к противогрибковым агентам. WO 2009/130481 описывает соединения пиррола, которые могут применяться для предупреждения или лечения грибковых заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы данного изобретения обнаружили, что соединение пиррола 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид (соединение Формулы I) является особенно эффективным противогрибковым агентом. Он имеет сильнодействующий характер при ингибировании ферментов, а также хорошую биодоступность и низкую токсичность в тестах с ингибированием грибковой инфекции. Тесты показали, что данное соединение пиррола ингибирует рост широкого спектра грибов, в частности, патогенного для человека гриба Aspergillus. Показано, что данное конкретное соединение обладает активностью против более широкого спектра видов рода Aspergillus по сравнению с другими соединениями пиррола. Кроме того, продемонстрирована повышенная эффективность соединения in vivo по сравнению с известным противогрибковым препаратом Вориконазол, в частности, повышенная эффективность против гриба Scedosporium. Таким образом, соединение 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид можно применять для эффективного лечения широкого спектра грибковых инфекций и заболеваний. Эти результаты описаны в международной заявке на патент с номером PCT/GB 2015/053546, содержание которой включено во всей полноте путем ссылки.
Авторы данного изобретения понимают необходимость эффективных композиций соединения пиррола 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамида. Для достижения оптимальных результатов при клиническом применении необходимы композиции соединения, обеспечивающие высокую биодоступность с минимальными побочными эффектами. Также существует необходимость в композициях 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамида, которые можно легко вводить пациенту, которому они необходимы.
Соответственно, в данном изобретении предложена фармацевтическая композиция, подходящая для перорального введения, которая содержит полученные посредством распылительной сушки частицы соединения формулы (I) или его фармацевтически приемлемой соли
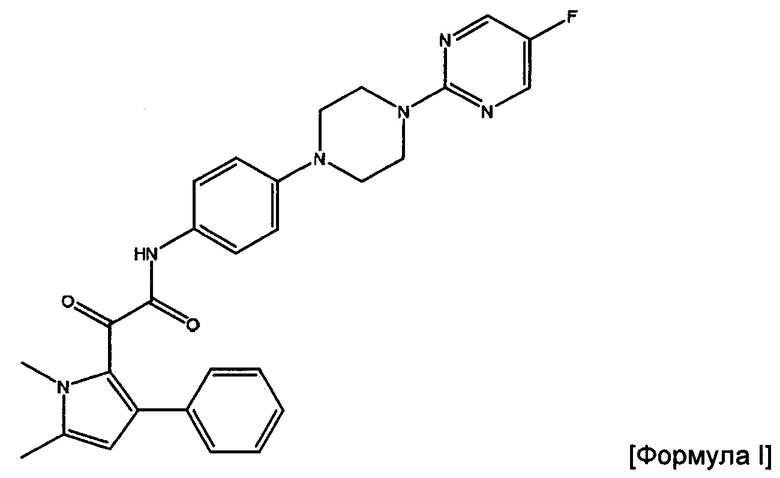
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид.
В одном аспекте композиция, подходящая для перорального введения, содержит полученные посредством распылительной сушки частицы соединения Формулы I. В другом аспекте соединение Формулы I является по существу аморфным. В одном аспекте фармацевтическая композиция дополнительно содержит один или более чем один эксципиент. В одном аспекте композиция содержит эксципиент гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS, от англ. hydroxypropyl methyl cellulose acetate succinate). В одном аспекте массовое соотношение соединения Формулы I и эксципиента составляет от 1:100 до 1:1, например, от 1:15 до 1:2. В одном аспекте фармацевтическая композиция содержит частицы, которые можно получать путем распылительной сушки из раствора, содержащего органический растворитель, выбранный из дихлорметана, метанола и их смесей. В одном аспекте фармацевтическая композиция находится в виде твердой пероральной лекарственной формы или в виде жидкой пероральной лекарственной формы. В одном аспекте жидкая пероральная лекарственная форма дополнительно содержит фармацевтически приемлемый буфер, имеющий pKa в диапазоне от 6,0 до 8,0. В одном аспекте буфер представляет собой фосфатный буфер с концентрацией от 1 мМ до 200 мМ, в котором композиция забуферена до рН приблизительно 7. В одном аспекте фармацевтическая композиция дополнительно содержит одно или более чем одно фармацевтически приемлемое связывающее вещество и/или один или более чем один фармацевтически приемлемый носитель и/или эксципиент и/или разбавитель и/или адъювант.
В данном изобретении также предложена фармацевтическая композиция, подходящая для парентерального введения, которая содержит (i) соединение формулы (I) или его фармацевтически приемлемую соль, (ii) циклодекстрин или модифицированный циклодекстрин и (iii) полиэтиленгликоль.
В одном аспекте композиция, подходящая для парентерального введения, содержит от 10 мас. % до 40 мас. % циклодекстрина или модифицированного циклодекстрина; и/или от 10 мас. % до 40 мас. % полиэтиленгликоля. В одном аспекте циклодекстрин или модифицированный циклодекстрин представляет собой гидроксипропил-бета-циклодекстрин. В одном аспекте полиэтиленгликоль представляет собой ПЭГ300 или ПЭГ400. В одном аспекте композиция дополнительно содержит поливинилпирролидон (Повидон). В одном аспекте соединение Формулы I или его фармацевтически приемлемая соль присутствует в концентрации от 1 мг/мл до 10 мг/мл. В одном аспекте композиция дополнительно содержит один или более чем один фармацевтически приемлемый носитель и/или эксципиент и/или разбавитель и/или адъювант.
В данном изобретении также предложена фармацевтическая композиция, описанная в данном документе, для применения в способе лечения человека или животного, которому это необходимо, в частности способ предупреждения или лечения грибковой инфекции у субъекта. Аналогично, в данном изобретении предложен способ предупреждения или лечения грибковой инфекции у человека или животного, которому это необходимо, указанный способ включает введение человеку или животному терапевтически эффективного количества фармацевтической композиции, описанной в данном документе; и применение фармацевтической композиции, описанной в данном документе, в изготовлении лекарственного средства для применения в предупреждении или лечении грибковой инфекции у человека или животного, которому это необходимо.
В изобретении также предложен способ получения фармацевтической композиции, содержащей соединение Формулы I или его фармацевтически приемлемую соль, где указанный способ включает распылительную сушку раствора соединения Формулы I или его соли. В одном аспекте способ включает: (i) растворение одного или более чем одного эксципиента в растворителе; (ii) добавление соединения Формулы I к раствору, полученному на стадии (i); и (iii) распылительную сушку раствора, полученного на стадии (ii).
В изобретении предложена фармацевтическая композиция, подходящая для перорального введения, которая содержит по существу аморфные частицы соединения Формулы (I) или его фармацевтически приемлемой соли.
Композиции по изобретению обладают преимуществами, поскольку обеспечивают чрезвычайно высокую биодоступность соединения Формулы I и обладают минимальными побочными эффектами.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 приведены фармакокинетические данные, полученные в клинических исследованиях на человеке композиции соединения Формулы I по изобретению, как описано в Примере 5. Ось у: Cmax в плазме (мкг/мл); ось х: время в часах.
На Фиг. 2 приведены фармакокинетические данные, полученные в клинических исследованиях на человеке композиции соединения Формулы I по изобретению, как описано в Примере 5. Ось у: Cmax в плазме (мкг/мл); ось х: доза соединения Формулы (I) (мг/кг).
На Фиг. 3 приведены фармакокинетические данные, полученные в клинических исследованиях на человеке композиции соединения Формулы I по изобретению, как описано в Примере 5. Ось у: AUC0-∞ в плазме (мкг*ч/мл); ось х: доза соединения Формулы (I) (мг/кг).
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В данном описании фармацевтически приемлемая соль представляет собой соль с фармацевтически приемлемой кислотой или основанием. Фармацевтически приемлемые кислоты включают как неорганические кислоты, например, соляную, серную, фосфорную, пирофосфорную, бромистоводородную, иодистоводородную или азотную кислоту, и органические кислоты, например, лимонную, фумаровую, малеиновую, яблочную, аскорбиновую, янтарную, винную, бензойную, уксусную, метансульфоновую, этансульфоновую, бензолсульфоновую, п-толуолсульфоновую кислоту, муравьиную, уксусную, пропионовую, гликолевую, молочную, пировиноградную, щавелевую, салициловую, трихлоруксусную, пикриновую, трифторуксусную, коричную, памовую, малоновую, миндальную, бисметиленсалициловую, этандисульфоновую, глюконовую, цитраконовую, аспарагиновую, стеариновую, пальмитиновую, этилендиаминтетрауксусную, п-аминобензойную или глутаминовую кислоту, сульфаты, нитраты, фосфаты, перхлораты, бораты, ацетаты, бензоаты, гидроксинафтоаты, глицерофосфаты или кетоглутараты. Другие примеры фармацевтически приемлемых неорганических или органических солей присоединения кислоты включают фармацевтически приемлемые соли, приведенные в Journal of Pharmaceutical Science, 66, 2 (1977), известные специалистам в области техники. Фармацевтически приемлемые основания включают гидроксиды щелочных металлов (например, натрия или калия) и щелочноземельных металлов (например, кальция или магния) и органические основания, например, алкиламины и гетероциклические амины, лизин, гуанидин, диэтаноламин и холин. К фармацевтически приемлемым солям присоединения кислоты также относятся гидраты, которые способно образовывать настоящее соединение. Соли присоединения кислоты можно получать непосредственно в виде продуктов синтеза соединений. В альтернативном варианте, свободное основание можно растворять в подходящем растворителе, содержащем соответствующую кислоту, и выделять соль путем выпаривания растворителя или разделять соль и растворитель иным образом. Соединение активного вещества может образовывать сольваты со стандартными низкомолекулярными растворителями при использовании способов, известных специалисту в области техники.
Если не указано иное, все значения в процентах, встречающиеся в данном описании, представляют собой массовые проценты (мас. %).
В изобретении предложена фармацевтическая композиция, подходящая для перорального введения, которая содержит полученные посредством распылительной сушки частицы соединения Формулы (I) или его фармацевтически приемлемой соли
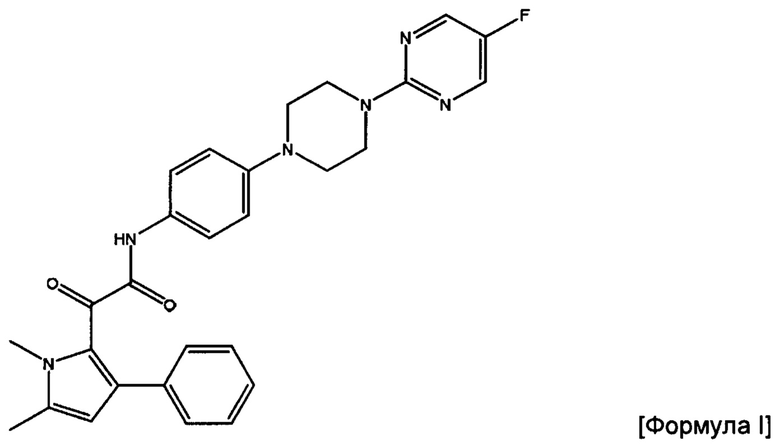
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид
Предпочтительно, композиция содержит полученные посредством распылительной сушки частицы соединения Формулы I. Более предпочтительно, соединение Формулы I находится в форме свободного основания.
Распылительная сушка представляет собой хорошо известный способ получения по существу аморфных частиц веществ, например, фармацевтически активных веществ, например, лекарств. Распылительная сушка особенно предпочтительна для получения твердых частиц температурочувствительных материалов и обладает преимуществом по сравнению с другими способами сушки, поскольку получаемые частицы имеют примерно одинаковый размер. Для сушки вещества можно применять любой подходящий аппарат для распылительной сушки, и имеются в продаже многочисленные распылительные сушилки. Например, распылительная сушилка может представлять собой распылительную сушилку одинарного или множественного действия. Зачастую предпочтительны распылительные сушилки множественного действия, поскольку с их применением можно получать частицы однородного размера. Распылительная сушка описана в стандартных руководствах, которые доступны специалистам, например, A.S. Mujumdar, Handbook of Industrial Drying, CRC Press 2014, где описана методика распылительной сушки и выбор оптимальных методик.
Предпочтительно, фармацевтическая композиция, подходящая для перорального введения, содержит соединение Формулы I, которое является по существу аморфным. Аморфная частица представляет собой частицу, у которой отсутствует дальний кристаллографический порядок. Предпочтительно, соединение Формулы 1 является более чем на 50% аморфным, например, более чем на 70% аморфным, более предпочтительно более чем на 90% аморфным, еще более предпочтительно более чем на 95% аморфным, более предпочтительно более чем на 99% аморфным, например, более чем на 99,5% аморфным или более чем на 99,9% аморфным. Таким образом, по существу аморфные частицы представляют собой частицы, в которых соединение Формулы I является низкокристаллическим, например, менее чем на 50% кристаллическим, например, менее чем на 30% кристаллическим, предпочтительно, менее чем на 10% кристаллическим, особенно менее чем на 5%, например, менее чем на 1% кристаллическим, например, менее чем на 0,5% кристаллическим или менее чем на 0,1% кристаллическим. Кристалличность можно определить способами, известными специалистам в области техники. Существует множество способов тестирования аморфных частиц, которые известны специалистам в области техники и которые могут применяться для определения, является ли частица аморфной или кристаллической. Указанные способы включают, без ограничения, дифракцию рентгеновских лучей на порошке, дифференциальную сканирующую калориметрию, динамическую сорбцию паров, изотермическую микрокалориметрию, инверсионную газовую хроматографию, ближнюю инфракрасную спектроскопию и ЯМР в твердом состоянии.
Предпочтительно, фармацевтическая композиция, подходящая для перорального введения, содержит частицы соединения Формулы I, которые имеют средний размер частиц от приблизительно 0,5 мкм до приблизительно 1000 мкм, более предпочтительно от приблизительно 1 мкм до приблизительно 500 мкм, например, от 5 мкм до 100 мкм, например, от приблизительно 20 мкм до приблизительно 50 мкм. Термин «средний размер частиц» относится к значениям, известным как D50. Термин D50 означает, что 50 об. % частиц имеют диаметр, который меньше указанного значения, и 50 об. % частиц имеют диаметр, который больше указанного значения. Средний размер частиц можно измерять с помощью известных в области техники стандартных методов лазерной дифракции для определения распределения частиц по размерам. Одним из примеров устройств для определения размера частиц сухих порошков является Mastersizer 2000, изготавливаемый Malvern Instruments Ltd (Worcestershire, UK).
Предпочтительно, фармацевтическая композиция, подходящая для перорального введения, содержит частицы, которые можно получать путем распылительной сушки из раствора, содержащего органический растворитель. Предпочтительно, органический растворитель представляет собой один или более чем один растворитель, выбранный из дихлорметана, ацетона, метанола и этанола. Более предпочтительно, растворитель представляет собой смесь двух или более растворителей, выбранных из дихлорметана, ацетона, метанола и этанола. Еще более предпочтительно, растворитель представляет собой смесь дихлорметана и/или ацетона с метанолом и/или этанолом. Как правило, соотношение дихлорметана и/или ацетона и метанола и/или этанола составляет от 1:1 до 5:1, например, от 2:1 до 4:1, например, 3:1. Например, растворитель часто представляет собой смесь дихлорметана и метанола, где соотношение дихлорметана и метанола составляет от 2:1 до 4:1, например, 3:1. Более предпочтительно, растворитель представляет собой дихлорметан и метанол в соотношении 3:1. Соотношения растворителей можно определять по массе или по объему; предпочтительны объемные соотношения.
Предпочтительно, фармацевтическая композиция, подходящая для перорального введения, дополнительно содержит один или более чем один эксципиент. Фармацевтически приемлемые эксципиенты, известные специалистам в области техники, включают, например, связывающие агенты, например, сироп, гуммиарабик, желатин, сорбит, трагакант, поливинилпирролидон (Повидон), метилцеллюлозу, этил целлюлозу, натрий-карбоксиметилцеллюлозу, гидроксипропилметилцеллюлозу, сахарозу и крахмал; наполнители и носители, например, кукурузный крахмал, желатин, лактозу, сахарозу, микрокристаллическую целлюлозу, каолин, маннит, кальций фосфорнокислый двузамещенный, хлорид натрия и альгиновую кислоту; а также лубриканты, такие как стеарат магния, стеарат натрия и стеараты других металлов, глицеролстеарат, стеариновую кислоту, силиконовое масло, тальк, воска, масла и коллоидный диоксид кремния. Также можно применять корригенты, например, перечную мяту, масло винтергрена, ароматизирующие вещества вишни. Также может быть желательным добавление красителя для удобства распознавания лекарственной формы. Таблетки также могут быть покрыты оболочками при помощи хорошо известных в области техники способов.
Более предпочтительно, фармацевтическая композиция, подходящая для перорального введения, содержит эксципиент, выбранный из (i) целлюлозы или модифицированной целлюлозы, например, гипромеллозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы (НРМС, от англ. гидрокси propyl methyl cellulose), гидроксипропилметилцеллюлозы ацетата (НРМСА, от англ. гидрокси propyl methyl cellulose acetate) и гидроксипропилметилцеллюлозы ацетата сукцината (HPMCAS, от англ. гидрокси propyl methyl cellulose acetate succinate) и (ii) сополимера винилпирролидона и винилацетата, имеющего массовое соотношение винилпирролидона и винилацетата в составе сополимера от 10:1 до 1:10, например, от 5:1 до 1:5, например, от 3:1 до 1:1, например, от 2:1 до 1:1, например, 3:2 (что также может быть выражено как 6:4), например, Kollidon VA64 или Kollidon VA64 Fine, оба предоставляются компанией BASF; предпочтителен Kollidon VA64. Фармацевтическая композиция может содержать смесь (i) и (ii). Более предпочтительно, фармацевтическая композиция, подходящая для перорального введения, содержит эксципиент на основе целлюлозы согласно варианту (i). Наиболее предпочтительные эксципиенты включают НРМС, НРМСА и HPMCAS; еще более предпочтительным является HPMCAS.
HPMCAS представляет собой гидроксипропилметилцеллюлозы ацетат сукцинат. Содержание ацетильных и сукцинильных групп в составе полимера может определять свойства HPMCAS. HPMCAS типа L представлен полимерами с высоким соотношением сукцинильных заместителей к ацетильным заместителям; как правило, содержание сукцинильных групп составляет 14-18 мас. % и содержание ацетильных групп составляет 5-9%. HPMCAS типа М представлен полимерами с более низким соотношением; как правило, содержание сукцинильных групп составляет 10-14 мас. % и содержание ацетильных групп составляет 7-11%. HPMCAS типа Н как правило имеет содержание сукцинильных групп 4-8 мас. % и содержание ацетильных групп 10-14%. HPMCAS типа L как правило растворяется приблизительно при рН≥5,5; HPMCAS типа М как правило растворяется приблизительно при рН≥6,0, a HPMCAS типа Н как правило растворяется приблизительно при рН≥6.8. Как правило, HPMCAS содержит 12-28 мас. % метоксильных групп и 4-28% гидроксипропокси. HPMCAS имеется в продаже у таких поставщиков, как Shin-Etsu (продукт AQOAT) и Ashland ("AquaSolve"). Можно применять любые подходящие HPMCAS, известные специалистам в области техники.
Предпочтительно, когда эксципиент присутствует в фармацевтической композиции, подходящей для перорального введения, массовое соотношение соединения Формулы I и эксципиента составляет от 1:100 до 1:1. Более предпочтительно, массовое соотношение соединения Формулы I и эксципиента составляет от 1:50 до 1:1, например, от 1:25 до 1,5, например, от 1:15 до 1:2, например, 1:10, 1:7, 1:5, 1:4 или 1:3. Более предпочтительно, массовое соотношение соединения Формулы I и эксципиента составляет от 1:9 до 1:11, например, 1:10 или составляет от 1:3 до 1:5, например, 1:4. Массовое соотношение соединения Формулы 1 и эксципиента 1:4 может достигаться, например, при массовом соотношении соединения Формулы 1 и эксципиента 4:16.
Фармацевтическая композиция по изобретению, подходящая для перорального введения, обычно содержит от 1 до 50 мас. % соединения Формулы I, более конкретно, от 4 до 40 мас. %, например, от 7 до 30 мас. %. Например, фармацевтическая композиция может содержать от 5 до 20 мас. %, например, от 8 до 15 мас. %, например, от 9 до 11 мас. %, например, 10 мас. %. Композицию, содержащую приблизительно 10 мас. % соединения Формулы I, можно, например, изготавливать, используя композицию, содержащую только соединение Формулы I и эксципиент, например, HPMCAS, используя массовое соотношение 1:9 (соединение Формулы I: HPMCAS). В альтернативном варианте фармацевтическая композиция может содержать от 10 до 40 мас. % соединения Формулы I, например, от 15 до 30 мас. %, например, от 18 до 22 мас. %, например, приблизительно 20 мас. %. Композицию, содержащую приблизительно 20 мас. % соединения Формулы I, можно, например, изготавливать, используя композицию, содержащую только соединение Формулы I и эксципиент, например, HPMCAS, используя массовое соотношение 4:16 (соединение Формулы I: HPMCAS).
Фармацевтическая композиция, подходящая для перорального введения, может дополнительно содержать одно или более чем одно фармацевтически приемлемое связывающее вещество и/или один или более чем один фармацевтически приемлемый носитель и/или эксципиент (т.е. дополнительно к указанным выше эксципиентам) и/или разбавитель и/или адъювант.
Фармацевтическая композиция, подходящая для перорального введения, может предпочтительно быть представлена в виде: отдельных единиц дозы, таких как капсулы, саше или таблетки, каждая их которых содержит определенное количество активного агента; в виде порошков или гранул; в виде раствора или суспензии активного агента в водной жидкой среде или неводной жидкой среде; или в виде жидкой эмульсии масло-в-воде или вода-в-масле; или в виде болюса и т.д. Предпочтительно, фармацевтическая композиция, подходящая для перорального введения, находится в виде (i) твердой пероральной лекарственной формы или (i) в виде жидкой пероральной лекарственной формы. Более предпочтительно, фармацевтическая композиция, подходящая для перорального введения, находится в виде твердой пероральной лекарственной формы.
Твердые пероральные лекарственные формы включают, например, таблетки и капсулы. Твердые пероральные лекарственные формы могут содержать наряду с активным ингредиентом солюбилизирующие агенты, например, циклодекстрины или модифицированные циклодекстрины; разбавители, например, лактозу, декстрозу, сахарозу, целлюлозу, кукурузный крахмал или картофельный крахмал; лубриканты, например, диоксид кремния, тальк, стеариновую кислоту, магния или кальция стеарат и/или полиэтиленгликоли; связывающие агенты; например, крахмалы, гуммиарабик, желатин, метилцеллюлозу, карибоксиметилцеллюлозу или поливинилпирролидон; дезагрегирующие агенты, например, крахмал, альгиновую кислоту, альгинаты или карбоксиметилкрахмал натрия (натрия крахмала гликолят); шипучие смеси; красители; подсластители; смачивающие агенты, например, лецитин, полисорбаты, лаурилсульфаты; и в целом, нетоксичные и фармакологически неактивные вещества, применяемые в фармацевтических композициях. Такие фармацевтические препараты можно изготавливать любым известным способом, например, путем смешивания, гранулирования, таблетирования, покрытия сахарной оболочкой или покрытия пленочной оболочкой.
Таблетка может быть получена путем прессования или формования, возможно, с одним или более чем одним вспомогательным ингредиентом. Прессованные таблетки можно получать путем прессования в подходящей машине активного агента в свободнотекучем виде, таком как порошок или гранулы, возможно, смешивания со связывающим веществом, лубрикантом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим агентом. Формованные таблетки можно получать путем формования в подходящей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки можно покрывать оболочкой или наносить риски и можно составлять таким образом, чтобы обеспечивать замедленное или контролируемое высвобождение активного агента.
Другие твердые пероральные дозированные лекарственные формы включают леденцы, содержащие активный агент в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте, и пастилки, содержащие активный агент в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик.
Жидкие пероральные лекарственные формы включают растворы, сиропы, эмульсии и суспензии. Растворы могут содержать солюбилизирующие агенты, например, циклодекстрины или модифицированные циклодекстрины. Сиропы могут содержать в качестве носителей, например, сахарозу или сахарозу с глицерином и/или маннит и/или сорбит. Жидкие пероральные лекарственные формы включают растворы для полоскания рта, содержащие активный агент в подходящем жидком носителе.
Предпочтительно, когда фармацевтическая композиция, подходящая для перорального введения, находится в форме жидкой пероральной лекарственной формы, жидкая пероральная лекарственная форма дополнительно содержит фармацевтически приемлемый буфер, имеющий pKa в диапазоне от 6,0 до 8,0, предпочтительно около рН 7, например, от рН 6,5 до рН 7,5, например, от рН 7,0 до рН 7,5, предпочтительно от рН 7,1 до рН 7,3, например, около рН 7,2. Можно применять любой фармацевтически приемлемый буфер, способный поддерживать рН раствора в данном диапазоне. Например, подходящие буферные соли включают цитрат (например, цитрат натрия/лимонную кислоту), фосфат (например, Na2HPO4/NaH2PO4) и карбонат (например, карбонат натрия/бикарбонат натрия). Предпочтителен фосфатный буфер. Концентрация соли в буфере может быть любой концентрацией соли подходящей для получения желаемой жидкой пероральной композиции. Как правило, концентрацию соли в буферном растворе выбирают для поддержания рН раствора в желаемом диапазоне, например, около рН 7 (например, рН 7,2). Типичные концентрации соли составляют от 1 мМ до 200 мМ, например, от 5 мМ до 100 мМ, например, от 10 мМ до 50 мМ, например, от 20 мМ до 40 мМ, например, около 25 мМ, около 30 мМ или около 35 мМ.
Предпочтительные композиции по изобретению, подходящие для перорального введения, таким образом, содержат полученные посредством распылительной сушки частицы соединения Формулы I или его фармацевтически приемлемой соли, где соединение Формулы I является по существу аморфным и где композиция дополнительно содержит один или более чем один эксципиент.
Более предпочтительные композиции по изобретению, подходящие для перорального введения, содержат полученные посредством распылительной сушки частицы соединения Формулы I, где соединение Формулы I является по существу аморфным и где композиция дополнительно содержит эксципиент гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS).
Еще более предпочтительные композиции по изобретению, подходящие для перорального введения, содержат полученные посредством распылительной сушки частицы соединения Формулы I, где соединение Формулы I является по существу аморфным и где композиция дополнительно содержит эксципиент HPMCAS, и где массовое соотношение соединения Формулы I к эксципиенту составляет от 1:100 до 1:1, предпочтительно от 1:15 до 1:2.
Еще более предпочтительные композиции по изобретению, подходящие для перорального введения, содержат полученные посредством распылительной сушки частицы соединения Формулы I, где соединение Формулы I является по существу аморфным и может быть получено путем распылительной сушки из раствора, содержащего органический растворитель, выбранный из дихлорметана, метанола и их смесей и где композиция дополнительно содержит эксципиент HPMCAS, и где массовое соотношение соединения Формулы I к эксципиенту составляет от 1:15 до 1:2.
Наиболее предпочтительные композиции по изобретению, подходящие для перорального введения, содержат полученные посредством распылительной сушки частицы соединения Формулы I, где соединение Формулы I является по существу аморфным и где композиция содержит 10 мас. % соединения Формулы I и 90 мас. % HPMCAS (т.е. массовое соотношение соединения Формулы I к HPMCAS составляет 1:9). Соединение формулы I наиболее предпочтительно получают посредством распылительной сушки из смеси дихлорметан: метанол в соотношении 3:1 об./об.
Аналогично, наиболее предпочтительные композиции по изобретению подходящие для перорального введения, содержат полученные посредством распылительной сушки частицы соединения Формулы I, где соединение Формулы I является по существу аморфным и где композиция содержит 20 мас. % соединения Формулы I и 80 мас. % HPMCAS (т.е. массовое соотношение соединения Формулы I к HPMCAS составляет 1:4). Соединение формулы I наиболее предпочтительно получают посредством распылительной сушки из смеси дихлорметан: метанол в соотношении 3:1 об./об.
В изобретении также предложена фармацевтическая композиция, подходящая для перорального введения, где композиция содержит по существу аморфные частицы соединения Формулы I или его фармацевтически приемлемой соли. Композиция является такой, как описано в данном документе.
Более предпочтительно, в изобретении предложена фармацевтическая композиция, подходящая для перорального введения, содержащая соединение Формулы I, где соединение Формулы I является по существу аморфным, и композиция дополнительно содержит HPMCAS, где массовое соотношение соединения Формулы I и HPMCAS составляет приблизительно от 1:3 до 1:5, например, 1:4, или приблизительно от 1:8 до 1:10, например, приблизительно 1:9. Таким образом, в изобретении предложена фармацевтическая композиция, подходящая для перорального введения, содержащая соединение Формулы I и HPMCAS, где соединение Формулы I является по существу аморфным и композиция содержит 10 мас. % соединения Формулы I и 90 мас. % HPMCAS. В изобретении также предложена фармацевтическая композиция, подходящая для перорального введения, содержащая соединение Формулы I и HPMCAS, где соединение Формулы I является по существу аморфным и композиция содержит 20 мас. % соединения Формулы I и 80 мас. % HPMCAS.
В изобретении также предложен способ получения фармацевтической композиции, содержащей соединение Формулы I или его фармацевтически приемлемую соль
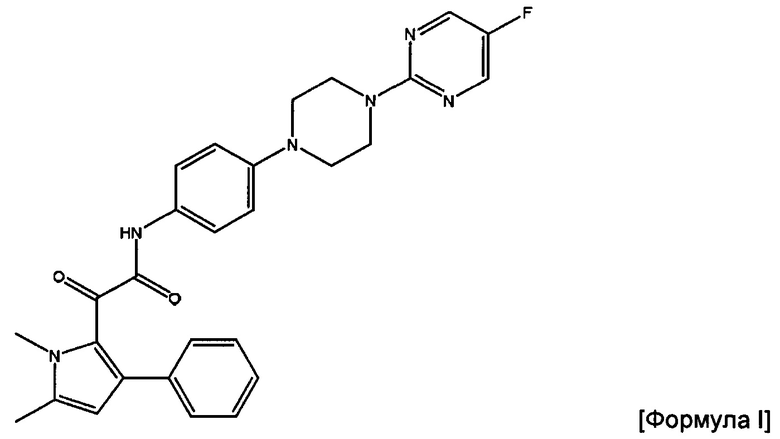
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид
где указанный способ включает распылительную сушку раствора соединения Формулы (I) или его соли.
Можно применять любой подходящий метод распылительной сушки. Распылительная сушка описана выше.
Предпочтительно, способ получения фармацевтической композиции, подходящей для перорального введения, включает стадии:
i) растворения одного или более чем одного эксципиента в растворителе;
ii) добавления соединения Формулы I к раствору, полученному на стадии (i); и
iii) распылительную сушку раствора, полученного на стадии (ii).
Предпочтительно, один или более чем один эксципиент представляют собой эксципиенты, описанные в данном документе. Более предпочтительно, один или более чем один эксципиент представляют собой (i) целлюлозу или модифицированную целлюлозу, описанные в данном документе, или (ii) сополимер винилпиролидона - винилацетата, описанный в данном документе, или смесь (i) и (ii). Более предпочтительно, эксципиент представляет собой HPMCAS, описанный в данном документе.
Предпочтительно, растворитель представляет собой органический растворитель, описанный в данном документе. Более предпочтительно, растворитель представляет собой смесь дихлорметана и/или ацетона с метанолом и/или этанолом. Еще более предпочтительно, растворитель представляет собой смесь дихлорметана и метанола, где соотношение дихлорметана и метанола составляет от 2:1 до 4:1, например, 3:1. Более предпочтительно, растворитель представляет собой дихлорметан и метанол в соотношении 3:1.
Так, например, в изобретении предложен способ получения фармацевтической композиции, подходящей для перорального введения, описанной в данном документе, в котором:
- эксципиент представляет собой гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS);
- растворитель представляет собой смесь дихлорметана и метанола, где соотношение дихлорметана и метанола составляет от 5:1 до 1:1;
- концентрация эксципиента в растворителе составляет от 5% до 20% мас./об.; и
- соединение Формулы I добавляют к раствору эксципиента в растворителе для получения концентрации от 0,5% до 10% по массе.
Более предпочтительно, в изобретении предложен способ получения фармацевтической композиции, подходящей для перорального введения, описанной в данном документе, в котором:
- эксципиент представляет собой гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS);
- растворитель представляет собой смесь дихлорметана и метанола, где объемное соотношение дихлорметана и метанола составляет от 4:1 до 2:1;
- концентрация эксципиента в растворителе составляет от 7% до 18% мас./об.; и
- соединение Формулы I добавляют к раствору эксципиента в растворителе для получения концентрации от 0,5% до 6% по массе.
Еще более предпочтительно, в изобретении предложен способ получения фармацевтической композиции, подходящей для перорального введения, описанной в данном документе, в котором:
- эксципиент представляет собой гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS);
- растворитель представляет собой смесь дихлорметана и метанола, где объемное соотношение дихлорметана и метанола составляет приблизительно 3:1; и
- (i) концентрация эксципиента в растворителе составляет от приблизительно 7 мас. % до приблизительно 11 мас. % и соединение Формулы I добавляют к раствору эксципиента в растворителе для получения концентрации от приблизительно 0,5% до приблизительно 2% по массе; или
(ii) концентрация эксципиента в растворителе составляет от приблизительно 12 мас. % до приблизительно 18 мас. % и соединение Формулы I добавляют к раствору эксципиента в растворителе для получения концентрации от приблизительно 3% до приблизительно 5% по массе.
Более предпочтительно, в изобретении предложен способ получения фармацевтической композиции, подходящей для перорального введения, включающий
i) растворение от приблизительно 7 мас. % до приблизительно 11 мас. % (например, приблизительно 9 мас. %) HPMCAS в растворителе, где растворитель представляет собой смесь дихлорметан : метанол в соотношении 3:1 об./об.;
ii) добавление соединения Формулы I к раствору, полученному на стадии (i) с получением раствора, в котором концентрация соединения Формулы I составляет от приблизительно 0,5% до приблизительно 2% по массе (например, приблизительно 1% по массе); и
iii) распылительную сушку раствора, полученного на стадии (ii).
Аналогично, в изобретении предложен способ получения фармацевтической композиции, подходящей для перорального введения, включающий
i) растворение от приблизительно 12 мас. % до приблизительно 18 мас. % (например, приблизительно 16 мас. %) HPMCAS в растворителе, где растворитель представляет собой смесь дихлорметан : метанол в соотношении 3:1 об./об.;
ii) добавление соединения Формулы I к раствору, полученному на стадии (i) с получением раствора, в котором концентрация соединения Формулы I составляет от приблизительно 3% до приблизительно 5% по массе (например, приблизительно 4% по массе); и
iii) распылительную сушку раствора, полученного на стадии (ii).
В изобретении также предложена фармацевтическая композиция, подходящая для парентерального введения, которая содержит (i) соединение Формулы I или его фармацевтически приемлемую соль,
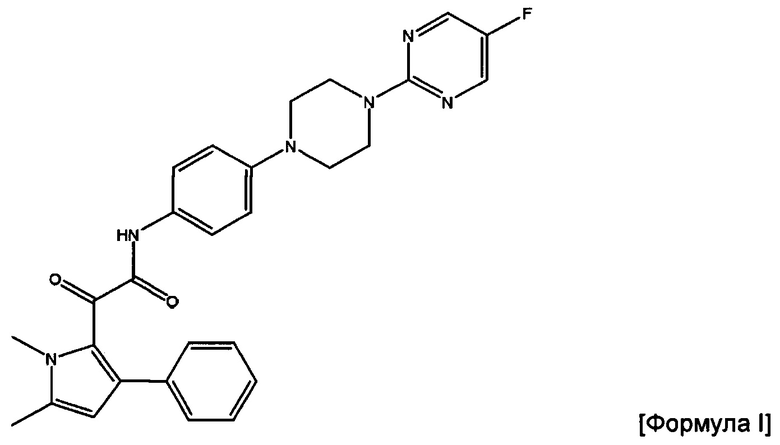
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид
(ii) циклодекстрин или модифицированный циклодекстрин и (iii) полиэтиленгликоль.
Фармацевтически приемлемые соли описаны в данном документе. Предпочтительно, фармацевтическая композиция, подходящая для парентерального введения, содержит соединение Формулы I. Более предпочтительно, фармацевтическая композиция, подходящая для парентерального введения, содержит соединение Формулы I в форме свободного основания.
Предпочтительно, циклодекстрин присутствует в количестве от приблизительно 10 мас. % до 40 мас. % по отношению к фармацевтической композиции. Типичное количество циклодекстрина, присутствующего в композиции, составляет от 20 мас. % до 30 мас. %, например, приблизительно 25 мас. %.
Предпочтительно, полиэтиленгликоль присутствует в количестве от приблизительно 10 мас. % до 40 мас. % по отношению к фармацевтической композиции. Типичное количество полиэтиленгликоля, присутствующего в композиции, составляет от 20 мас. % до 30 мас. %, например, приблизительно 25 мас. %.
Фармацевтическая композиция, подходящая для парентерального введения, может содержать любой подходящий циклодекстрин или его смесь. Типичные циклодекстрины содержат ряд мономеров глюкозы, количество которых в кольце варьирует от 6 до 8 единиц, образующих форму усеченного конуса. Циклодекстрины часто обозначают α (альфа)-циклодекстрины (содержащие 6-членную кольцевую молекулу сахара); β (бета)-циклодекстрины (содержащие 7-членную кольцевую молекулу сахара) или γ (гамма)-циклодекстрины (содержащие 8-членную кольцевую молекулу сахара). Поскольку циклодекстрины являются гидрофобными внутри и гидрофильными снаружи, они образуют комплексы с гидрофобными соединениями. Таким образом, они могут усиливать растворимость и биодоступность таких соединений. Альфа-, бета- и гамма-циклодекстрины общепризнаны безопасными Управлением по контролю качества пищевых продуктов и лекарственных средств (FDA). Циклодекстрины (CD) могут быть модифицированы различными способами, при этом оставаясь подходящими для применения в композициях по изобретению. Например, известные модифицированные циклодекстрины включают гидроксиэтил-β-CD (НЕ-β-CD), гидроксипропил-β-CD (HP-β-CD), сульфобутилэфир-β-CD (SBE-β-CD), метил-β-CD (М-β-CD), диметил-β-CD (DM-β-CD/DIMEB), произвольным образом диметилированный β-CD (RDM-β-CD), произвольным образом метилированный β-CD (RM-β-CD/RAMEB), карбоксиметил-β-CD (CM-β-CD), карбоксиметилэтил-β-CD (CME-β-CD), диэтил-β-CD (DE-β-CD), три-О-метил-β-CD (TRIMEB), три-О-этил-β-CD (TE-β-CD), три-О-бутирил-β-CD (TB-β-CD), три-О-валерил-β-CD (TV-β-CD), ди-О-гексаноил-β-CD (DH-β-CD), глюкозил-β-CD (G1-β-CD), мальтозил-β-CD (G2-β-CD) и 2-гидрокси-3-триметил-аммонийпропил-β-СО (HTMAPCD). Циклодекстрины и их применение в фармацевтических препаратах описано в стандартных руководствах, например,  and Szejtli, Cyclodextrins in Pharmacy, Springer, 1993, где описаны преимущества конкретных циклодекстринов в фармацевтических препаратах.
and Szejtli, Cyclodextrins in Pharmacy, Springer, 1993, где описаны преимущества конкретных циклодекстринов в фармацевтических препаратах.
Предпочтительно, циклодекстрины выбраны из гидроксипропилбетациклодекстрина и сульфобутилэфир бета-циклодекстрина (Captisol) и их смесей. Предпочтительным является гидроксипропил-бета-циклодекстрин.
Фармацевтическая композиция, подходящая для парентерального введения, может содержать любой подходящий полиэтиленгликоль или его смесь. Например, композиция может содержать любой полиэтиленгликоль, одобренный для внутривенного введения. Полиэтиленгликоли, которые могут применяться, включают ПЭГ200-ПЭГ500, например, ПЭГ300 и/или ПЭГ400. Предпочтительными являются ПЭГ300 и ПЭГ400, наиболее предпочтительным является ПЭГ400.
Специалистам в области техники понятно, что цифры после термина «ПЭГ» (например, 300 в «ПЭГ300») относятся к средней молекулярной массе молекулы ПЭГ. Так, ПЭГ400 обычно содержит приблизительно 9 единиц этиленгликоля в каждой молекуле полимера, а ПЭГ300 обычно содержит 7 единиц этиленгликоля в каждой молекуле полимера. При этом специалистам в области техники понятно, что множество имеющихся в продаже ПЭГов являются полидисперсными. В целом, распределение по молекулярной массе можно охарактеризовать статистически в терминах средневесовой молекулярной массы (Mw) и среднечисленной молекулярной массы (Mn), их соотношение часто обозначают индексом полидисперсности (Mw/Mn). Как Mw, так и Mn можно определять стандартными способами, например, при помощи масс-спектрометрии.
Предпочтительно, фармацевтическая композиция, подходящая для парентерального введения, содержит один или более чем один диспергирующий агент, например, низкомолекулярный повидон (поливинилпирролидон). Предпочтительно, повидон является свободным от эндотоксинов. Повидоны имеются в продаже у таких поставщиков, как например, Ashland (Plasdone). Предпочтительно, повидон имеет значение K от 5 до 20, например, от 10 до 18. Например, повидон может иметь номинальную молекулярную массу приблизительно 4000 и значение K от приблизительно 10 до приблизительно 14. В альтернативном варианте повидон может иметь номинальную молекулярную массу приблизительно 10000 и значение K от приблизительно 15 до приблизительно 18. Значение K является функцией от средней степени полимеризации и собственной вязкости полимера и может быть рассчитано по кинематической вязкости водного раствора полимера. Предпочтительно, повидон имеет Tg (температуру стеклования) от приблизительно 110°С до приблизительно 130°С, например, от приблизительно 120°С до приблизительно 126°С. Фармацевтическая композиция может содержать смесь двух или более повидонов.
Предпочтительно, когда фармацевтическая композиция, подходящая для парентерального введения, содержит повидон или их смесь, повидон или их смесь присутствуют в количестве от приблизительно 0,1 до 5 мас. %, более предпочтительно, от 0,5 до 2 мас. %, еще более предпочтительно приблизительно 1 мас. % по отношению к общей массе композиции.
Предпочтительно, соединение Формулы I присутствует в фармацевтической композиции, подходящей для парентерального введения, в концентрации от 1 мг/мл до 10 мг/мл. Более предпочтительно, концентрация соединения Формулы I в фармацевтической композиции, подходящей для парентерального введения, составляет от 2 до 7 мг/мл, например, от 3 до 5 мг/мл, например, 4 мг/мл.
Фармацевтическая композиция, подходящая для парентерального введения, может дополнительно содержать один или более чем один фармацевтически приемлемый носитель и/или эксципиент и/или разбавитель и/или адъювант. Например, композиция может содержать в качестве носителя, стерильную воду, или может быть в форме стерильных водных изотонических солевых растворов.
Предпочтительно, конечное значение рН фармацевтической композиции, подходящей для парентерального введения, доводят до рН от приблизительно 4 до рН приблизительно 8.
Более предпочтительно, конечное значение рН фармацевтической композиции, подходящей для парентерального введения, доводят до рН от приблизительно 4 до рН приблизительно 6, например, от рН приблизительно 4,5 до рН приблизительно 5,5, например, рН около 5, например, рН приблизительно 5,0. Можно доводить рН фармацевтической композиции при помощи любой фармацевтически приемлемой кислоты или основания. Предпочтительной является фосфорная кислота.
Предпочтительные фармацевтические композиции являются стерильными и свободными от пирогенов.
Таким образом, предпочтительные композиции по изобретению, подходящие для парентерального введения, содержат:
- от 10 мас. % до 40 мас. % циклодекстрина или модифицированного циклодекстрина;
- от 10 мас. % до 40 мас. % полиэтиленгликоля; и
- диспергирующий агент, например, повидон.
Например, композиции по изобретению, подходящие для парентерального введения, могут содержать:
- от 1 до 10 мг/мл соединения формулы (I) или его фармацевтически приемлемой соли;
- от 10 мас. % до 40 мас. % гидроксипропил бета-циклодекстрина;
- от 10 мас. % до 40 мас. % ПЭГ300 или ПЭГ400; и
- диспергирующий агент, например, повидон, где повидон является таким, как описано в данном документе; и
где рН композиции доводят до рН от приблизительно 4 до рН приблизительно 8.
Более предпочтительные композиции по изобретению, подходящие для парентерального введения, содержат:
- от 1 до 10 мг/мл соединения Формулы I или его фармацевтически приемлемой соли;
- от 10 мас. % до 40 мас. % гидроксипропил бета-циклодекстрина;
- от 10 мас. % до 40 мас. % ПЭГ300 или ПЭГ400; и
- диспергирующий агент, например, повидон, где повидон является таким, как описано в данном документе; и
где рН композиции доводят до рН от приблизительно 4 до рН приблизительно 6.
Еще более предпочтительные композиции по изобретению, подходящие для парентерального введения, содержат:
- от 3 до 5 мг/мл соединения Формулы I;
- от 20 мас. % до 30 мас. % гидроксипропил бета-циклодекстрина;
- от 20 мас. % до 30 мас. % ПЭГ300 или ПЭГ400, предпочтительно, ПЭГ400; и
- от 0,1 до 5 мас. % повидона; и
где рН композиции доводят до рН от приблизительно 4,5 до рН приблизительно 5,5.
Наиболее предпочтительные композиции по изобретению, подходящие для парентерального введения, содержат:
- 4 мг/мл (относительно конечного объема композиции) соединения Формулы I
- 25 мас. % гидроксипропил-бета-циклодекстрина;
- 25 мас. % ПЭГ400;
- 1 мас. % поливинилпирролидона (Повидона);
- фосфорную кислоту в количестве, достаточном для доведения рН фармацевтической композиции до рН 5,0; и
- воду до 100%.
Как описано в данном документе, фармацевтическая композиция по изобретению может дополнительно содержать один или более чем один адъювант, например, местный анестетик, консервант или буферизующий агент.
Фармацевтически приемлемые связывающие вещества включают растворы связывающих веществ и сухие связывающие вещества. Растворы связывающих веществ растворяют в растворителе (например, в способах влажной грануляции можно использовать воду или спирт). Примеры включают желатин, целлюлозу, производные целлюлозы, поливинилпирролидон, крахмал, сахарозу и полиэтиленгликоль. Сухие связывающие вещества добавляют в порошковую смесь либо после стадии влажной грануляции, либо как часть самого состава для прессования порошков. Примеры включают целлюлозу, метилцеллюлозу, поливинилпирролидон и полиэтиленгликоль.
Фармацевтические носители включают липосомы, наносферы, мицеллы, комплексы ДНК-белок, наногели и природные растворители, например, водные и неводные растворы.
Другие вещества, которые можно применять в качестве эксципиентов, разбавителей или носителей, включают гуммиарабик, альгинат, альгиновую кислоту, ацетат алюминия, бензиловый спирт, бутилпарабен, бутилированный гидрокситолуол, лимонную кислоту, карбонат кальция, канделильский воск, кроскармеллозу натрия, кондитерский сахар, коллоидный диоксид кремния, целлюлозу, фосфат кальция, карнаубский воск, кукурузный крахмал, карбоксиметилцеллюлозу кальция, стеарат кальция, кальциево-динатриевую соль этилендиаминтетрауксусной кислоты, кополивидон, гидрогенизированное касторовое масло, кальция гидрофосфата дигидрат, цетилпиридинхлорид, цистеин HCl, кросповидон, натрий фосфорнокислый двузамещенный, диметикон, натрия эритрозин, этилцеллюлозу, желатин, глицерилмоноолеат, глицерин, глицин, глицерилмоностеарат, глицерилбегенат, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, гипромеллозу, гидроксипропилметилцеллюлозы фталат, лактозу, стеарат магния, маннит, метилцеллюлозу, магния карбонат, минеральное масло, оксид магния, метилпарабен, повидон, полисорбат 80, полиэтиленоксид, полоксамер 407 или 188, бикарбонат калия, сорбат калия, картофельный крахмал, фосфорную кислоту, полиоксил-стеарат, карбоксиметилкрахмал натрия (натрия крахмала гликолят), кроскармеллозу натрия, натрия лаурилсульфат, крахмал, диоксид кремния, бензоат натрия, стеариновую кислоту, сахарозу, сорбиновую кислоту, карбонат натрия, сахарин натрия, альгинат натрия, силикагель, сорбитан моноолеат, натрия стеарилфумарат, натрия хлорид, натрия метабисульфит, натрия цитрата дигидрат, карбоксиметилцеллюлозу натрия, янтарную кислоту, пропионат натрия, диоксид титана, тальк, триацетин и триэтилцитрат.
Фармацевтическая композиция, описанная в данном документе, может содержать частицы соединения Формулы I, где средний размер частиц (по данному описанию) уменьшился благодаря технологиям микронизации или нанонизации.
Композиции по изобретению обладают особыми преимуществами, поскольку обеспечивают повышенную биодоступность соединения Формулы I. В данном описании биодоступность определяют таким образом, что, например, соединение Формулы I при внутривенном введении имеет биодоступность 100%. Фармацевтические композиции по изобретению, подходящие для перорального введения, особенно эффективны, поскольку обеспечивают повышенную биодоступность соединения Формулы I. Предпочтительно, композиция по изобретению обеспечивает биодоступность соединения Формулы I по меньшей мере 50%, более предпочтительно, по меньшей мере 70%, например, по меньшей мере 80%, еще более предпочтительно, по меньшей мере 90%, например, по меньшей мере 95%. Специалисту в области техники понятно, что биодоступность может определяться различными факторами, включая характеристики субъекта, которому вводят композицию (возраст, вес, пол и т.д.). Таким образом, отрицательный результат в одной группе субъектов не является решающим.
Биодоступность можно определять в фармакокинетических (ФК) исследованиях, в которых концентрация лекарства в плазме определяется как функция времени после внутривенного (в/в) и внесосудистого (например, перорального) введения. Абсолютная биодоступность (Fabs) представляет собой скорректированную на дозу (D) площадь под кривой «концентрация-время» (AUC) (не-внутривенное введение), поделенную на AUC (внутривенное введение). В данном описании Fabs для лекарства, вводимого пероральным способом (РО) рассчитывают согласно формуле:
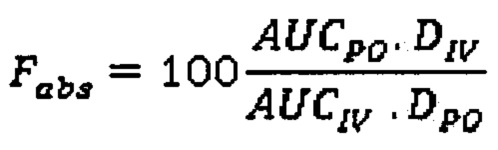
Композиции по изобретению могут найти применение в лечении патологических состояний человека или животного, которому это необходимо.
Соответственно, в изобретении предложена фармацевтическая композиция, описанная в данном документе, для применения в способе лечения человека или животного, которому это необходимо. Предпочтительно, в изобретении предложена фармацевтическая композиция, описанная в данном документе, для применения в лечении человека или животного, которому это необходимо, где лечение включает предупреждение или лечение грибковой инфекции у субъекта.
В изобретении также предложен способ предупреждения или лечения грибковой инфекции у человека или животного, которому это необходимо, указанный способ включает введение человеку или животному терапевтически эффективного количества фармацевтической композиции по данному описанию.
В изобретении также предложено применение фармацевтической композиции по данному описанию в изготовлении лекарственного средства для применения в предупреждении или лечении грибковой инфекции у человека или животного, которому это необходимо.
Фармацевтическую композицию по изобретению можно применять в способе лечения человека или животного, где лечение включает введение композиции в комбинации с дополнительным противогрибковым агентом по данному описанию.
Терапевтически эффективное количество композиции по изобретению можно вводить пациенту, которому это необходимо. Например, композицию обычно вводят в таком количестве, чтобы обеспечить субъекту суточную дозу соединения Формулы I вплоть до 200 мг, например, до 100 мг или до 50 мг на кг массы тела, например, от 0,001 до 200 или от 0,001 до 50 мг на кг массы тела, в соответствии (например) с возрастом, массой тела и состоянием субъекта, подлежащего лечению, характера и тяжести заболевания и частоты и способа введения. Предпочтительно, суточные дозы находятся на уровне до 200 мг, например, до 150 мг, до 100 мг, до 50 мг или до 40 мг на кг массы тела. Суточные дозы находятся на уровне, например, по меньшей мере 1 мг, по меньшей мере 2 мг или по меньшей мере 5 мг на кг массы тела. В одном воплощении суточные дозы находятся на уровне от 0,05 мг до 2 г, предпочтительно от 0,1 мг до 10 мг. Уровни соответствующих доз может легко установить квалифицированный специалист-медик.
Когда композицию по изобретению вводят вместе со вторым противогрибковым агентом, второй противогрибковый агент обычно вводят в дозе, которая равна или меньше стандартной дозы, используемой для этого лекарства. Таким образом, известные противогрибковые агенты можно вводить в более низких дозах по сравнению с используемыми в настоящее время, благодаря чему токсические эффекты будут снижаться.
Композиция по изобретению может найти применение в лечении или предупреждении грибковых заболеваний. Предпочтительно, грибковое заболевание включает грибковые инфекции, например, инфекции, вызываемые аскомицетами. Предпочтительно, грибковое заболевание включает инфицирование организмом, выбранным из рода Absidia; Acremonium; Alternaria; Aspergillus; Bipolaris; Blastomyces; Blumeria;; Cladosporium; Coccidioides; Colletotrichium; Curvularia; Encephalitozoon; Epicoccum; Epidermophyton; Exophiala; Exserohilum; Fusarium; Histoplasma; Leptosphaeria; Microsporum; Mycosphaerella; Neurospora, Paecilomyces; Penicillium; Phytophthora; Plasmopara; Pneumocystis; Pyricularia; Pythium; Puccinia; Rhizoctonia; Rhizomucor; Scedospohum; Scopulahopsis; Trichophyton; Trichosporon; и Ustilago.
Предпочтительно, грибковое заболевание включает инфицирование организмом рода Aspergillus, Scedospohum или Fusarium, например, грибковое заболевание включает инфицирование организмом родом Aspergillus или Scedosporium, в частности Aspergillus. В одном воплощении грибковое заболевание включает инфицирование организмом рода Aspergillus. В другом воплощении грибковое заболевание включает инфицирование организмом рода Scedosporium.
Предпочтительно, грибковое заболевание включает инфицирование организмом, выбранным из видов Absidia corymbifera; Acremonium spp; Alternaria alternata; Aspergillus flavus; Aspergillus fumigatus; Aspergillus nidulans; Aspergillus niger; Aspergillus parasiticus; Aspergillus terreus; Bipolaris spp; Blastomyces dermatitidis; Blumeria graminis;; Cladosporium cladosporoides; Cladosporium herbarium; Coccidioides immitis; Coccidioides posadasii; Curvularia lunata; Colletotrichium trifolii;; Encephalitozoon cuniculi; Epicoccum nigrum; Epidermophyton floccosum; Exophiala spp; Exserohilum rostratum; Fusarium graminarium; Fusarium solani; Fusarium sporotrichoides; Histoplasma capsulatum; Leptosphaeha nodorum; Microsporum canis; Mycosphaerella graminicola; Paecilomyces lilanicus; Paecilomyces varioti; Penicillium chrysogenum; Phytophthora capsici; Phytophthora infestans; Plasmopara viticola; Pneumocystis jiroveci; Puccinia coronata; Puccinia graminis; Pyricularia oryzae; Pythium ultimum; Rhizoctonia solani; Rhizomucor spp; Rhizopus spp; Scedospohum apiospermum; Scedospohum prolificans; Scedospohum species d; Scopulariopsis brevicaulis; Trichophyton mentagrophytes; Trichophyton interdigitale; Trichophyton rubrum; Trichosporon asahii; Trichosporon beigelii и Ustilago maydis.
Предпочтительно, грибковое заболевание включает инфицирование А. fumigatus, A. flavus, A. terreus, A. niger, A, lentulus, S. apiospermum, S. prolificans или S. species d. В частности, грибковое заболевание включает инфицирование А. fumigatus, A. flavus, A. terreus или A. niger. В одном воплощении грибковое заболевание включает инфицирование S. prolificans.
Примеры грибковых заболеваний, которые можно предупреждать или лечить с использованием композиций по изобретению, включают как системные, так и поверхностные инфекции. Грибковые заболевания включают инвазивные грибковые заболевания, вызванные видами Aspergillus, например, аспергиллез, но также и местные формы таких инфекций. Например, грибковые заболевания включают инвазивные грибковые заболевания, вызванные видами Aspergillus, например, аспергиллез, но также и местные формы таких инфекций. Композиции по изобретению особенно полезны при заболеваниях, вызванных видами Aspergillus, при которых необходимо фунгицидное лекарственное средство, обладающее меньшей токсичностью по сравнению с амфотерицином. В изобретении также предложено лечение дерматологических инфекций.
В одном воплощении фармацевтическая композиция по изобретению предназначена для применения в предупреждении или лечении заболевания, вызванного видами Aspergillus. Заболевание, вызванное видами Aspergillus, включает заболевания, вызванные A. fumigatus, A. flavus, A. terreus и A. niger.
Примеры системных инфекций, которые можно предупреждать или лечить с применением фармацевтической композиции по изобретению, включают: аспергилез легких, например, у пациентов с ослабленным иммунитетом, например, у реципиентов костного мозга или пациентов со СПИД; системный аспергилез, риноцеребральный мукомикоз, бластомикоз, гистоплазмоз, кокцидиомикоз, паракокцидиомикоз, лобомикоз, споротрихоз, хромобластомикоз, феогифомикоз и диссеминированный споротрихоз.
Примеры поверхностных инфекций, которые можно предупреждать или лечить с применением фармацевтической композиции по изобретению, включают: дерматомикоз, стопу атлета и онихомикоз (инфекцию ногтей).
Примеры заболеваний или состояний, которые вызываются грибком или при которых грибок усиливает аллергическую реакцию, и которые можно предупреждать или лечить с применением фармацевтической композиции по изобретению, включают аллергический бронхолегочный аспергиллез (АБЛА), астму, тяжелую астму с грибковой сенсибилизацией, кистозный фиброз с грибковой колонизацией, риносинусит и синусит. Например, заболевание может быть вызвано грибковой сенсибилизацией или заболевание может представлять собой аллергический бронхолегочный аспергиллез (АБЛА) или астму.
Фармацевтические композиции, описанные в данном документе, можно вводить в комбинации со вторым противогрибковым агентом. Предпочтительно, фармацевтическую композицию вводят отдельно от или последовательно со вторым противогрибковым агентом. Например, композицию по изобретению и второй противогрибковый агент могут быть представлены в виде набора. Так, набор может содержать композицию по изобретению и второй противогрибковый агент.
Второй противогрибковый агент может представлять собой любой подходящий противогрибковый агент, который специалист в области техники сочтет полезным в данных обстоятельствах. Например, таким образом можно осуществлять лечение любого из описанных в данном документе состояний.
Наиболее подходящие классы противогрибковых агентов включают азолы, полиены, ингибиторы пуриновых нуклеотидов, ингибиторы пиримидиновых нуклеотидов, ингибиторы маннана, ингибиторы белкового фактора элонгации, ингибиторы синтетазы хитина, ингибиторы синтетазы бета-глюканов, эхинокандины, аллиламины, антитела к HSP90, бактерицидные/увеличивающие проницаемость белковые продукты и полиоксины. Другие подходящие противогрибковые агенты, которые не относятся к указанным выше классам, включают соединения 5-фтор-1,3-дигидро-1-гидрокси-2,1-бензоксаборал (AN269), 5-хлор-1,3-дигидро-1-гидрокси-2,1-бензоксаборал (AN2718) и икофунгипен.
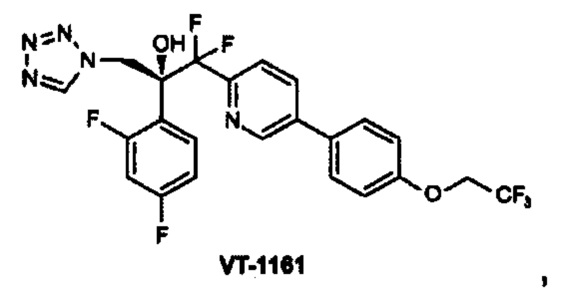
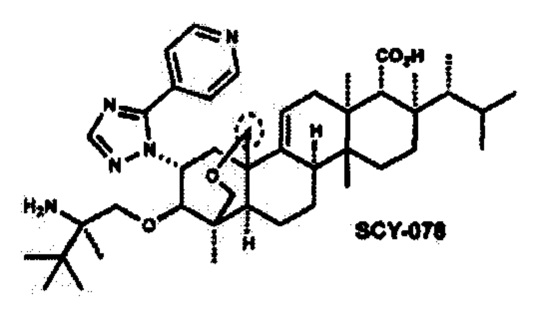
Например, второй противогрибковый агент может быть выбран из группы, состоящей из азолов, полиенов, ингибиторов пуриновых нуклеотидов, ингибиторов пиримидиновых нуклеотидов, ингибиторов маннана, ингибиторов белкового фактора элонгации, эхинокандинов, аллиламинов, антител к HSP90, бактерицидных/увеличивающих проницаемость белковых продуктов или полиоксинов или одного из соединений 5-фтор-1,3-дигидро-1-гидрокси-2,1-бензоксаборала (AN269), 5-chloro-1,3-дигидро-1-гидрокси-2,1-бензоксаборала (AN2718), икофунгипена, VT116 или SCY078.
VT116 представляет собой 2-пиридинэтанол, α-(2,4-дифторфенил)-β,β-дифтор-α-(1H-тетразол-1-илметил)-5-[4-(2,2,2-трифторэтокси)фенил]-, (αR)-,
a SCY078 078 (также обозначаемый MK-3118) представляет собой полусинтетическое производное энфумафунгина, 4Н-1,4а-пропано-2Н-фенантро[1,2-с]пиран-7-карбоновую кислоту, 15-[(2R)-2-амино-2,3,3-триметилбутокси]-8-[(1R)-1,2-диметилпропил]-1,6,6а,7,8,9,10,10а,10b,11,12,12а-додекагидро-1,6а,8,10а-тетраметил-14-[5-(4-пиридинил)-1H-1,2,4-триазол-1-ил]-, (1S,4aR,6aS,7R,8R,10aR,10bR,12aR,14R,15R):
Предпочтительными азолами являются клотримазол, эконазол, бифоназол, бутоконазол, фентиконазол, флуконазол, изоконазол, итраконазол, кетоконазол, миконазол, оксиконазол, сертаконазол, сулконазол, тиоконазол, изавуконазол, равуконазол, позаконазол, терконазол и вориконазол, луликоназол. Предпочтительными эхинокандинами являются анидулафунгин, каспофунгин, микафунгин и биафунгин. Предпочтительными аллиламинами являются тербинафин, бутенафин, аморолфин и нафтифин. Предпочтительными полиенами являются амфотерицин В и нистатин. Предпочтительным примером ингибитора пуриновых или пиримидиновых нуклеотидов является флуцитозин. Предпочтительным ингибитором маннана является прадамицин. Предпочтительным ингибитором белкового фактора элонгации является сордарин и его аналоги. Предпочтительным полиоксином является никкомицин Z.
Наиболее предпочтительными вторыми противогрибковыми агентами являются каспофунгин, микафунгин, анидулофунгин, амфотерицин В, вориконазол, позаконазол, изавуконазол, флуконазол и итраконазол.
Синтез
Соединение Формулы I представляет собой 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид или его фармацевтически приемлемую соль.
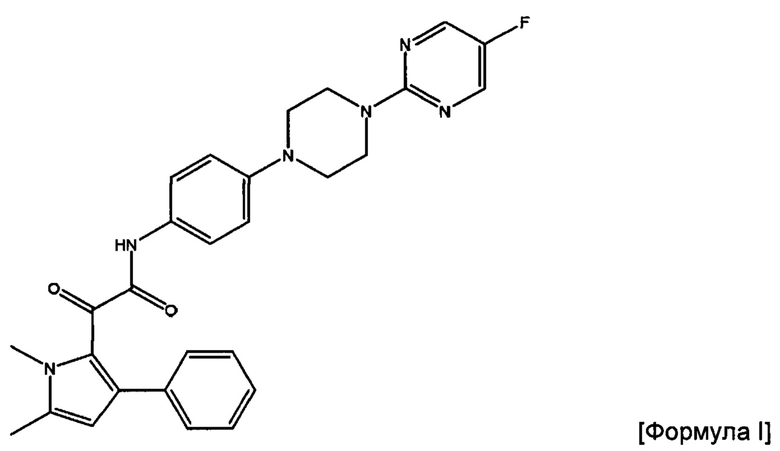
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид
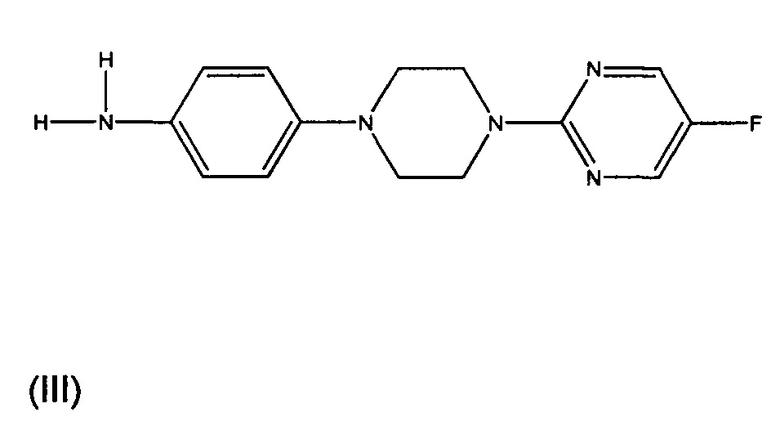
Один из синтетических путей соединения Формулы I описан в данном документе. В целом, соединение Формулы I может быть синтезировано в реакции между соединением формулы (II) с соединением формулы (III). Как правило, реакция протекает в присутствии органического растворителя и основания. Предпочтительно, растворитель представляет собой дихлорметан или тетрагидрофуран, а основание представляет собой триэтиламин или пиридин. Как правило, реакцию вначале проводят при 0°С, пока добавляют реагенты, а затем перемешивают при комнатной температуре до завершения реакции. Соединение формулы (III), как правило, может быть приобретено в коммерческих источниках или может быть получено известными способами.
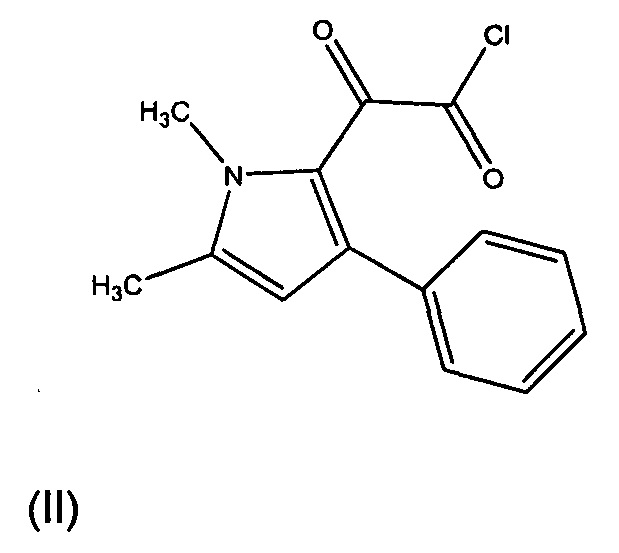
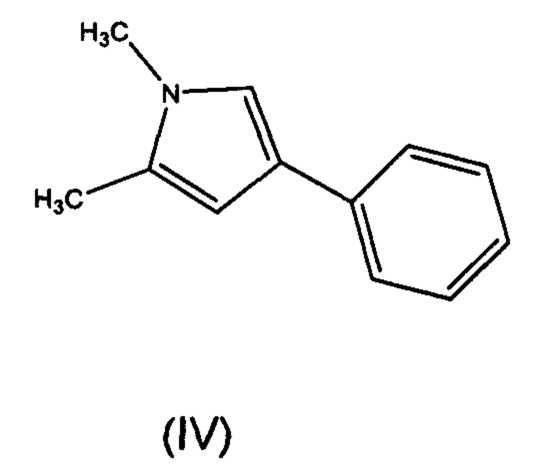
Соединение формулы (II) может быть получено в результате реакции соединения формулы (IV), предпочтительно, с оксалила хлоридом. Как правило, реакция протекает в органическом растворителе. Предпочтительно, растворитель представляет собой дихлорметан. Как правило, реакцию вначале проводят при 0°С, пока добавляют реагенты, а затем перемешивают при комнатной температуре до завершения реакции.
Все исходные материалы, упомянутые в реакциях, описанных выше, могут быть приобретены в коммерческих источниках или могут быть получены по аналогии с известными способами.
Приведенные ниже Примеры иллюстрируют изобретение и не должны рассматриваться как ограничивающие объем изобретения. В этой связи важно понимать, что конкретные тесты, использованные в разделе Примеры, служат исключительно для подтверждения противогрибковой активности. Для определения такой активности существует множество тестов и поэтому отрицательный результат в каком-либо одном конкретном тесте не является определяющим.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Пример 1. Синтез соединений Формулы I (2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамида) Синтез соединения Формулы I описан в международной заявке на патент с номером PCT/GB 2015/053546. Информация, относящаяся к синтезу соединения Формулы I, включена путем ссылки. Приведенный ниже Пример воспроизведен из указанной заявки на патент.
На схеме синтеза, приведенной ниже, показан способ синтеза:
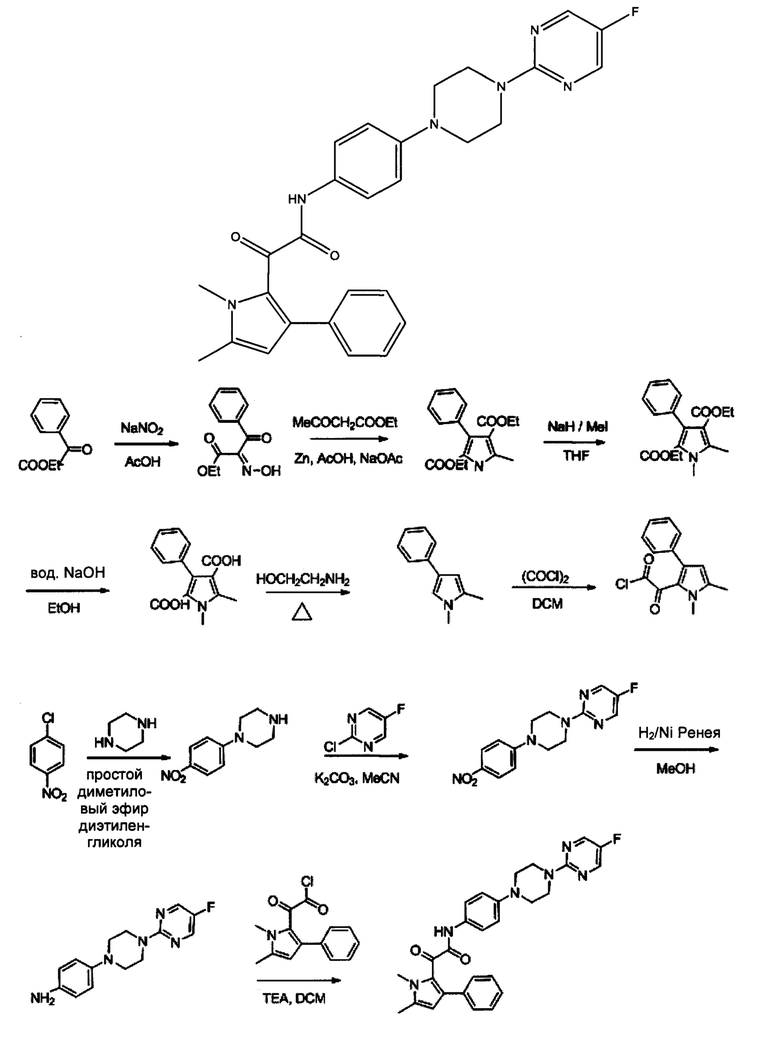
Этиловый эфир 2-гидроксиимино-3-оксо-3-фенилпропионовой кислоты (А)
Раствор нитрита натрия 1,07 кг, 45,62 моль) в воде (4 л) медленно добавляли к раствору этилбензоилацетата (2 кг, 10,41 моль) в ледяной уксусной кислоте (6 л), при 0-10°С в течение 2 ч. Продукт начинал преципитировать в ходе добавления. Реакционную смесь нагревали до комнатной температуры и перемешивали дополнительно в течение 1 ч. Добавляли воду (2,5 л) и перемешивали смесь дополнительно в течение 1 ч. Фильтровали с применением отсоса, промывали водой (2 л). Твердое вещество растворяли в хлороформе (8 л) и промывали водой (2×500 мл), рассолом (2×500 мл), высушивали над безводным сульфатом натрия и концентрировали под вакуумом насухо с получением 2,0 кг (86%) этилового эфира 2-гидроксиимино-3-оксо-3-фенилпропионовой кислоты А в виде белого твердого вещества. [система ТСХ: Этилацетат: Петролейный эфир (3:7); значение Rf: 0,28].
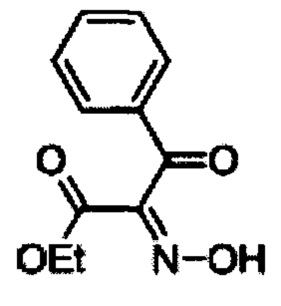
Диэтиловый эфир 5-метил-3-фенил-1Н-пиррол-2,4 дикарбоновой кислоты 1В)
Смесь этилацетоацетата (329 г, 2,53 моль), цинковой пыли (443 г, 6,78 моль) и безводного ацетата натрия (463 г, 5,65 моль) в ледяной уксусной кислоте (800 мл) нагревали до 60°С. Раствор А (500 г, 2,26 моль) в ледяной уксусной кислоте (1,5 л) добавляли тремя порциями при интенсивном перемешивании в течение ~1 ч. В ходе добавления температура поднималась до приблизительно 93°С. Температуру реакционной смеси поддерживали при 60-75°С в течение 3 ч. К реакционной смеси добавляли еще цинковую пыль (221 г, 3,39 моль) в течение 15 мин и перемешивали смесь при 60-75°С в течение 1 ч, охлаждали до комнатной температуры и отфильтровывали твердые вещества. Фильтрат выпаривали под вакуумом и осадок перегоняли совместно с толуолом (2×500 мл). К осадку добавляли воду (5 л) и этилацетат (1 л) и перемешивали до получения двух прозрачных слоев. Органический слой последовательно промывали водой (2×500 мл), насыщенным раствором бикарбоната (2×500 мл), рассолом (2×500 мл), высушивали над безводным сульфатом натрия и концентрировали с получением 360 г неочищенного смолистого продукта. Его перемешивали со смесью дихлорметана в петролейном эфире (200 мл: 1200 мл; 1:6) при комнатной температуре в течение 15 мин, фильтровали и промывали петролейным эфиром (100 мл) с получением 250 г (36%) диэтилового эфира 5-метил-3-фенил-1Н-пиррол-2,4 дикарбоновой кислоты B в виде почти белого твердого вещества. [система ТСХ: этилацетат: Петролейный эфир (3:7); значение Rf: 0,45]. Аналогично 1,5 кг (500 г × 3) А превращали в 500 г [245 г (36%) + 255 г (37%) + 250 г (36%)] В тремя порциями.
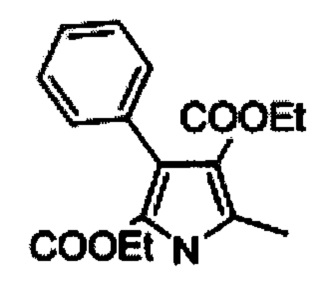
Диэтиловый эфир 1,5-диметил-3-фенил-1Н-пиррол-2,4-дикарбоновой кислоты (С)
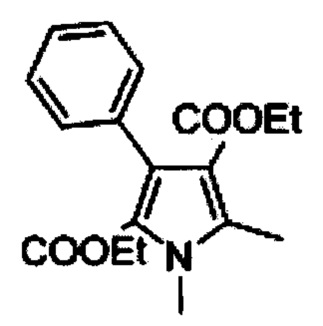
Раствор В (1 кг, 3,322 моль) в сухом тетрагирофуране (4 л) добавляли к суспензии гидрида натрия (60% мас./мас.; 254 г, 6,644 моль) в сухом тетрагидрофуране (4 л) при 0°С в течение 1 ч. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч и снова охлаждали до 0°С. Метил иодид (517 мл; 8,305 моль) добавляли в течение 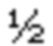 ч и перемешивали реакционную смесь при комнатной температуре в течение 18 ч. Гасили ледяной водой (100 мл) и добавляли 1N соляную кислоту (2 л). Отделяли оганические слои и экстрагировали водный слой дихлорметаном (2×500 мл). Объединенные органические слои последовательно промывали рассолом (2×200 мл), высушивали над безводным сульфатом натрия и концентрировали насухо с получением 950 г (91%) диэтилового эфира 1,5-диметил-3-фенил-1Н-пиррол-2,4-дикарбоновой кислоты С в виде желтого твердого вещества [система ТСХ: этилацетат: Петролейный эфир (3:7); значение Rf: 0,56].
ч и перемешивали реакционную смесь при комнатной температуре в течение 18 ч. Гасили ледяной водой (100 мл) и добавляли 1N соляную кислоту (2 л). Отделяли оганические слои и экстрагировали водный слой дихлорметаном (2×500 мл). Объединенные органические слои последовательно промывали рассолом (2×200 мл), высушивали над безводным сульфатом натрия и концентрировали насухо с получением 950 г (91%) диэтилового эфира 1,5-диметил-3-фенил-1Н-пиррол-2,4-дикарбоновой кислоты С в виде желтого твердого вещества [система ТСХ: этилацетат: Петролейный эфир (3:7); значение Rf: 0,56].
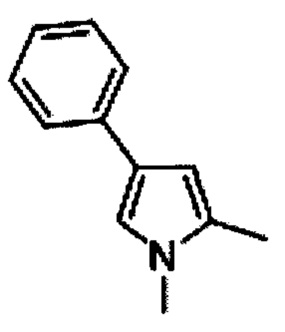
1,5-Диметил-3-фенил-1Н-пиррол-2,4-дикарбоновая кислота (D)
Раствор гидроксида натрия (1,21 кг, 30,25 моль) в воде (3,6 л) добавляли к раствору С (950 г, 3,025 моль) в этаноле (5 л) и нагревали с обратным холодильником в течение 15 ч. Этанол выпаривали под пониженным давлением, осадок разводили водой (1 л) и охлаждали до 0°С. Медленно добавляли концентрированную соляную кислоту (2 л) для доведения рН до ~2, поддерживая температуру ниже 10°С и перемешивали в течение 1 ч. Отфильтровывали преципитировавшее твердое вещество, промывали водой (1 л) и петролейным эфиром (1 л) и высушивали под вакуумом при 60°C с получением 550 г (70%) 1,5-диметил-3-фенил-1Н-пиррол-2,4-дикарбоновой кислоты D в виде белого твердого вещества. [система ТСХ: этилацетат: Петролейный эфир (3:7); значение Rf: 0,15].
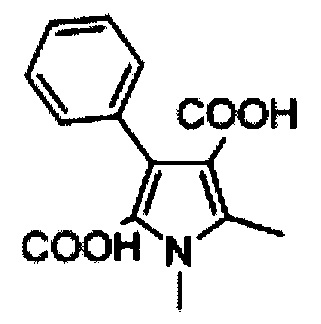
1,2-Диметил-4-фенил-1Н-пиррол (Е)
Суспензию Е (550 г, 2,123 моль) в этаноламине (1,5 л) нагревали до 175°С (в атмосфере N2) и поддерживали в течение 1 ч. Охлаждали реакционную смесь до комнатной температуры, разводили водой (500 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои последовательно промывали водой (2×100 мл) и рассолом (2×100 мл), высушивали над безводным сульфатом натрия и концентрировали под вакуумом при температуре ниже 40°C с получением неочищенного продукта. Флэш-хроматография на нейтральном оксиде алюминия с использованием 5% этилацетата в петролейном эфире в качестве элюента позволила получить 280 г (77%) 1,2-диметил-4-фенил-1Н-пиррола Е, в виде белого твердого вещества. [система ТСХ: этилацетат: Петролейный эфир (3:7); значение Rf: 0.75].
(1,5-Диметил-3-фенил-1Н-пиррол-2-ил)-оксоацетилхлорид (F)
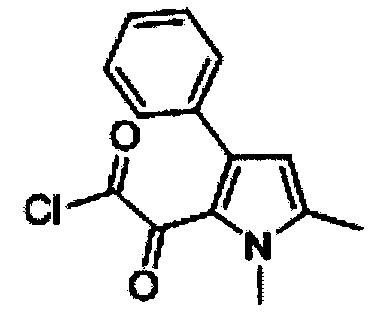
Оксалилхлорид (116 мл, 1,286 моль) медленно добавляли к охлажденному раствору Е (250 г, 1,169 моль) в сухом дихлорметане (3×200 мл) при 0°С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Выпаривали растворитель насухо под вакуумом с получением 340 г (89%) 1,5-диметил-3-фенил-1Н-пиррол-2-ил)-оксоацетилхлорида F в виде коричневой маслянистой жидкости. [система ТСХ: этилацетат: Петролейный эфир (3:7); значение Rf: 0,65].
4-нитрофенилпиперазин (G)
Раствор 1-хлор-4-нитробензола (650 г, 4,140 моль) в простом диметиловом эфире диэтиленгликоля (1 л) добавляли к раствору пиперазина (2,84 кг, 33,12 моль) в простом диметиловом эфире диэтиленгликоля (500 мл) при 100°С и полученную массу перемешивали при 100°С в течение 6 ч. Смесь охлаждали до 40-45°С, добавляли воду (5 л); нагревали до комнатной температуры и перемешивали в течение 1 ч. Отфильтровывали преципитировавшее твердое вещество, промывали водой (1 л), петролейным эфиром (500 мл) и высушивали с получением 700 г (81%) 4-нитрофенилпиперазина G в виде твердого вещества желтой окраски. [система ТСХ: Этилацетат: Петролейный эфир (3:7); значение Rf: 0,70].
5-Фтор-2-[4-(4-нитрофенил)-пиперазин-1-ил]-пиримидин (Н)
2-хлор-5-фторпиримидин (281 г, 2,12 моль) добавляли к суспензии 4-нитрофенилпиперазина G (400 г, 1,93 моль) и карбоната калия (532 г, 3,85 моль) в простом диметиловом эфире диэтиленгликоля (2,5 л), полученную смесь перемешивали при 100°С в течение 6 ч. После завершения реакции смесь охлаждали до 0°С и фильтровали, твердое вещество переносили в воду (5 л) и перемешивали в течение 30 мин. Суспензию фильтровали, твердый осадок промывали водой (1 л) и петролейным эфиром (1 л) и высушивали под вакуумом с получением 500 г (85%) 5-фтор-2-[4-(4-нитрофенил)-пиперазин-1-ил]-пиримидина Н в виде твердого вещества желтой окраски. [система ТСХ: Этилацетат: Петролейный эфир (3:7); значение Rf: 0,70].
4-[4-(5-Фтор-пиримидин-2-ил)-пиперазин-1-ил]-фениламин (I)
Раствор дитионита натрия (1,27 кг, 7,32 моль) в воде (6 л) добавляли к суспензии Н (500 г, 1,83 моль) и бикарбоната натрия (614 г, 7,32 моль) в метаноле (6 л) при 65°С. Полученную смесь перемешивали при 65°С в течение 2 ч. Реакционную смесь охлаждали до 10-15°С и фильтровали. Осадок распределялся между водой (2 л) и этилацетатом (5 л), органический слой промывали водой (2 л), рассолом (2 л) и высушивали над безводным сульфатом натрия. Концентрировали под вакуумом с получением 290 г (64%) 4-[4-(5-фтор-пиримидин-2-ил)-пиперазин-1-ил]-фенил амина I в виде твердого вещества. [система ТСХ: Метанол: Хлороформ (1:9); значение Rf: 0,50].
2-(1,5-Диметил-3-фенил-1Н-пирро-2-ил)-N-{4-[4-(5-фтор-пиримидин-2-ил)-пиперазин-1-ил]-фенил)-2-оксацетамид
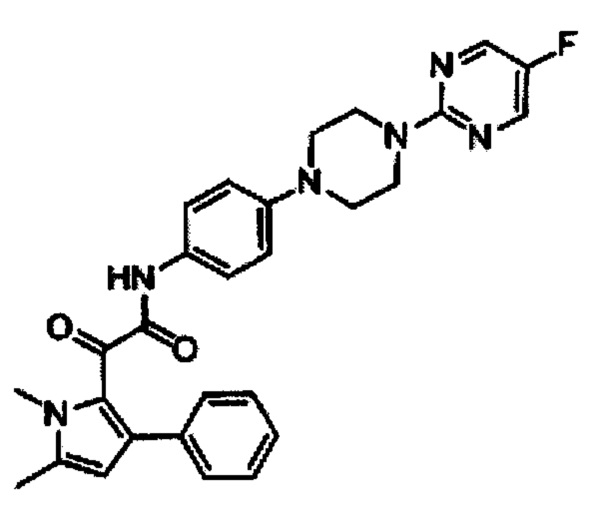
Раствор F (332 г, 1,27 моль) в дихлорметане (3 л) добавляли к перемешиваемому раствору I (290 г, 1,06 моль) и триэтиламина (294 мл, 2,12 моль) в дихлорметане (3 л) при 0°С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 мин. Реакционную смесь гасили водой и экстрагировали дихлорметаном (6×500 мл). Объединенные органические слои последовательно промывали насыщенным раствором бикарбоната (1,5 л), водой (1 л), рассолом (1,5 л) и, наконец, высушивали над безводным сульфатом натрия. Органический слой перемешивали с нейтральным оксидом алюминия (1 кг) при комнатной температуре в течение 30 мин и фильтровали. Фильтрат концентрировали под вакуумом с получением неочищенного соединения, которое после промывания диэтиловым эфиром (300 мл) и последующего растирания в этаноле (3 л) при 80°С в течение 1 ч и охлаждения до комнатной температуры, фильтровали, промывали этанолом (500 мл) и затем гексаном (200 мл) и высушивали с получением 340 г (64%) 2-(1,5-диметил-3-фенил-1Н-пирро-2-ил)-N-{4-[4-(5-фтор-пиримидин-2-ил-пиперазин-1ил]-фенил}-2-оксацетамида в виде твердого вещества желтой окраски. [система ТСХ: Этилацетат: Петролейный эфир (1:1); значение Rf: 0,65].
Данные ЯМР для 2-(1,5-диметил-3-фенил-1Н-пирро-2-ил)-N-{4-[4-(5-фтор-пиримидин-2-ил)-пиперазин-1-ил]-фенил}-2-оксацетамида (1Н ЯМР (400 МГц, CDCl3)) представлены на Фиг. 1. Сигнал регистрировали в масс-спектре при 499,1 [М+Н]+.
Пример 2. Противогрибковая активность соединения Формулы I
Данные свидетельствующие, что 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид подавляет рост грибов широкого спектра, представлены в заявке на международный патент с номером PCT/GB 2015/053546. Информация, относящаяся к биологической активности соединения Формулы I, включена путем ссылки.
PCT/GB 2015/053546 описывает эксперименты, в которых сравнивают противогрибковую активность соединения формулы I с различными референтными соединениями. Эксперименты, описанные в PCT/GB 2015/053546, показывают, что соединение Формулы I ингибирует рост грибковых организмов с минимальной ингибирующей концентрацией (MIC, от англ. minimum inhibitory concentration), т.е. наименьшей концентрацией лекарства, ингибирующей рост организма более чем на 80% по сравнению с контролем без лекарства, указанной ниже:

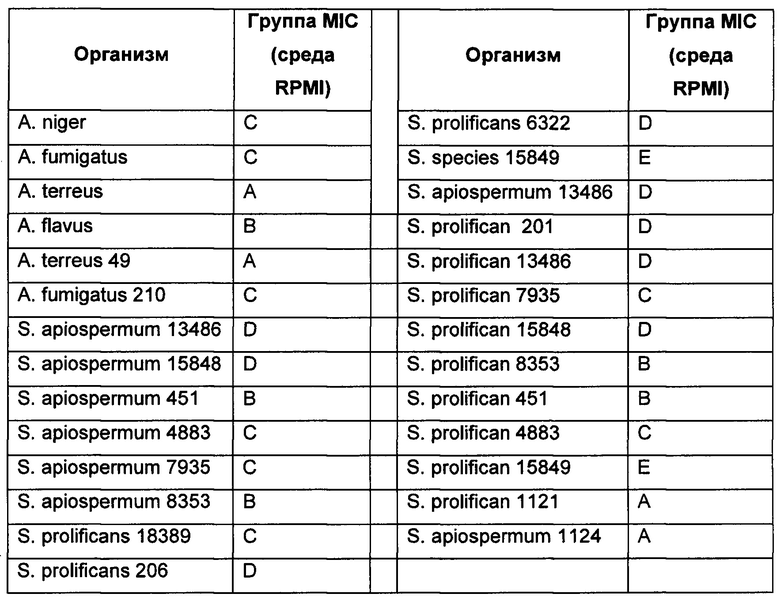
Соединение Формулы I также демонстрировало хорошую противогрибковую активность против S. dehoogii, S. boydii и S. aurantiacum.
PCT/GB 2015/053546 также описывает эксперименты, в которых соединение Формулы I также исследовали in vivo на модели на мышах. Данные в этой заявке свидетельствуют, что соединение Формулы I обладает превосходной эффективностью в мышиных моделях инвазивного аспергиллеза и что соединение Формулы I способно уменьшать показатели галактоманнана у мышей, инфицированных А. fumigatus. Соединение Формулы I также способно увеличивать выживаемость мышей, инфицированных Lomentospora prolificans FMR 3569, при сравнении с контрольными экспериментами, в которых применяли противогрибковый агент вориконазол.
В экспериментах in vivo, описанных в PCT/GB 2015/053546, соединение Формулы I вводили перорально через желудочный зонд. В PCT/GB 2015/053546 не осуществляли введение соединения Формулы I с применением композиций по данному изобретению.
Пример 3. ФК эксперименты - эксперименты на грызунах in vivo для определения предпочтительных пероральных композиций соединения Формулы I
Различные композиции исследовали в ФК исследованиях in vivo на крысах для определения оптимальной пероральной композиции для введения животным и человеку.
Соединение Формулы I вводили крысам в/в в дозе 10 мг/кг. Носитель содержал 15% гидроксипрол-бета-циклодекстрина (Kelptose НРВ для парентерального введения), 5% ДМСО и воду для инъекций. Перед использованием композицию фильтровали с использованием полиэфирсульфоновой мембраны (PES) ≤0,22 мкм. Объем вводимой дозы составлял 5 мл/кг. Образцы крови 0,2 мл из подъязычной вены брали под изофлурановой анестезией через 5 минут после в/в введения и хранили в ЭДТА. Плазму отделяли путем центрифугирования (1500 × g, 10 мин, прибл. 4°С) и замораживали при -70°С до исследования. Анализ для рассчета концентрации соединения Формулы I в плазме проводили посредством ЖХ-МС/МС. Регистрировали типичные значения AUC (AUCIV), составлявшие 13500 нг*ч/мл, соответствующие 100% биодоступности (по определению). Затем рассчитывали значение AUCIV для рассчета биодоступности приведенных в качестве примера пероральных композиций по данному описанию.
Композиция 1
Кристаллический образец соединения Формулы I находился в ПЭГ300. Композицию вводили перорально (через желудочный зонд) крысам в дозе 10 мг/кг (объем дозы = 5 мл/кг). Образцы крови 0,2 мл из подъязычной вены брали под изофлурановой анестезией через 15 минут после перорального введения и хранили в ЭДТА. Образцы брали через 0,5, 1, 2, 4, 8, 12 и 24 часов после введения. ФК данные (которые определяли как для в/в композиции) показали AUCPO (0-24 часа) 6610 нг*ч/мл, что соответствовало биодоступности (F) 49%.
Композиция 2
Кристаллический образец соединения Формулы I находился в 90% растворе ПЭГ300: 10% TPGS (d-α-токоферил полиэтиленгликоль 1000 сукцинат). TPGS известен как усилитель биодоступности, который действует как растворитель лекарства и ингибитор преципитации и был включен, поскольку был продемонстрирован полезный эффект его присутствия в композициях других нерастворимых соединений. При пероральном введении в виде раствора в носителе, представляющем собой PEG300/TPGS в дозе 150 мг/кг (концентрация композиции 15 мг/мл), значения AUCPO (0-24 часа) у самцов и самок крыс натощак составляли 34271 нг*ч/мл и 76963 нг*ч/мл, соответственно, что эквивалентно значениям биодоступности 17% и 38%, соответственно. Однако, когда указанные композиции вводили в течение длительного периода, наблюдался побочный эффект в виде диареи, что указывало на то, что композиция не подходит для клинического применения.
Композиции 3 и 4.
Кристаллический образец соединения Формулы I микронизировали посредством размола на струйной мельнице до конечного размера частиц D(v0,9) = 6,7 мкм, а затем суспендировали в смеси НРМС (75%) и SDS (додецилсульфата натрия 0,05%). Эту композицию (композицию 3) вводили перорально самцам и самкам крыс пл 150 мг/кг. Значения AUCPO (0-24 часа) в ФК экспериментах составили 9004 нг*ч/мл и 26286 нг*ч/мл для самцов и самок крыс, соответственно, что эквивалентно биодоступности 4,5% и 13%, соответственно.
Соединение Формулы I нанонизировали аналогичным образом и готовили в HPMC/SDS в виде композиции 3 (для получения композиции 4), и вводили самцам и самкам крыс перорально. Значения AUCPO (0-24 часа) в ФК экспериментах составили 17785 нг*ч/мл и 40272 нг*ч/мл для самцов и самок крыс, соответственно, что эквивалентно биодоступности 9% и 20%, соответственно.
Ни композиция 3, ни композиция 4 не продемонстрировали адекватной биодоступности соединения Формулы I. Более того, ФК эксперименты обнаружили, что время достижения максимальной концентрации соединения Формулы I в плазме (Tmax) существенно замедлялось, что позволяло предположить замедленную/неполную абсорбцию.
Композиция 5
Соединение Формулы I находилось в гидроксипропилметилцеллюлозы ацетате сукцинате (HPMCAS) (10% мас./мас. соединения Формулы I: 90% HPMCAS). Композицию подвергали распылительной сушке из раствора дихлорметан : метанол в соотншении 3:1 об./об. Композицию вводили перорально самцам и самкам крыс в дозе 10 мг/кг в 20 мМ фосфатном буфере (суспензия). Значения AUCPO (0-24 часа) в ФК экспериментах составили 13202 нг*ч/мл и 27774 нг*ч/мл для самцов и самок крыс, соответственно, что указывало на биодоступность >95% у обоих полов.
В отличие от композиции 2, композиция 5 хорошо переносилась как самцами, так и самками крыс без побочных эффектов со стороны ЖКТ. Это позволило провести токсикологические исследования на протяжение 1-3 месяцев. Композиция 5 хорошо переносилась как самцами, так и самками крыс в течение указанного периода.
Сравнение Композиции 2 и Композиции 5.
Сравнивали результаты ФК экспериментов, проводившихся с использованием композиций 2 и 5 в эквивалентных дозах (50 мг/кг) при пероральном введении дважды в сутки (BID) в течение 28 дней. Эти данные показали, что воздействие при применении композиции 5 было лучше, чем при применении композиции 2, дополнительным преимуществом было отсутствие негативного влияния на ЖКТ, ассоциированного с композицией 2.
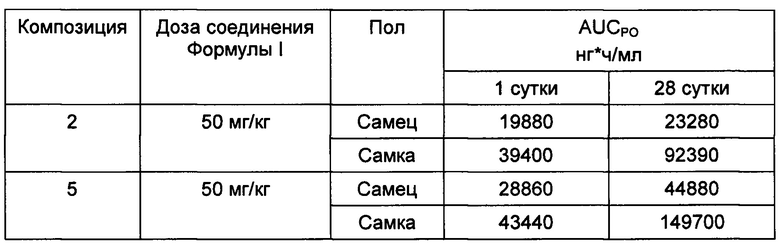
Пример 4. ФК эксперименты - эксперименты на яванских макаках in vivo для определения предпочтительных пероральных композиций соединения формулы I
Различные композиции исследовали в ФК исследованиях in vivo на яванских макаках для определения оптимальной пероральной композиции для введения животным и человеку.
Соединение Формулы I вводили самцам и самкам яванского макака в/в в дозе 10 мг/кг. В/в композиция была такой, как описано в Примере 3. Типичные значения AUC (AUCIV), зарегистрированные в ФК экспериментах, составляли 18000 нг*ч/мл. Затем значение AUCIV использовали для рассчета биодоступности приведенных в качестве примера пероральных композиций по данному описанию.
Композиция 2
Исследования проводили с использованием Композиции 2, как описано выше. Соединение Формулы I вводили самцам и самкам яванского макака перорально в дозе 10 мг/кг (объем дозы 5 мл/кг). Образцы крови 2 мл получали в течение 5 минут путем бедренной пункции (вена/аретрия) в имеющиеся в продаже пробирки с K2EDTA с применением игл 23G, присоединенных к соответствующему шприцу. Образцы брали через 0,25, 0,5, 1, 2, 4, 8, 12 и 24 часа после введения композиции. Образцы крови держали на льду до центрифугирования при 1600 × g (10 мин, прибл. 5°С) для отделения плазмы. Анализ для расчета концентрации соединения Формулы I в плазме проводили посредством ЖХ-МС/МС. Значения AUCPO (0-24 часа) в ФК экспериментах составили 14915 нг*ч/мл и 8192 нг*ч/мл для самцов и самок, соответственно, что соответствовало биодоступности 83% и 46%, у самцов и самок, соответственно.
К сожалению, проблемы с ЖКТ (в частности, диарея), наблюдавшиеся в экспериментах с крысами (см. Пример 3 выше) также отмечались у яванских макак. Исследования на протяжение 1 месяца с носителем PEG/TPGS показали, что диарея была основной проблемой несмотря на применение противодиарейного препарата (Диосмектит; "Смекта", 1 пакетик (3 г) на животного через 4 часа после введения). Эти исследования показали, что, несмотря на перспективную биодоступность соединения Формулы I при пероральном приеме, композиция 2 была неподходящей для клинического применения.
Композиция 5
Исследования проводили с использованием Композиции 5, как описано в Примере 3. Соединение Формулы I вводили самцам и самкам яванского макака перорально в дозе 10 мг/кг (объем дозы: 5 мл/кг; концентрация композиции 2 мг/мл соединения Формулы I, соответствующий 20 мг/мл полученных посредством распылительной сушки частиц).
Частицы, полученные, как описано для Композиции 5, суспендировали в различных буферных композициях/объемах. Значения AUCPO (0-24 часа) в ФК экспериментах были следующими:

Как можно заметить, при использовании низких концентраций буферных солей (20 мМ) и больших объемов буферов (10 мл) или при использовании малых объемов буферов (5 мл) с высокой буферной силой (40 мМ) наблюдали высокие значения AUCPO (0-24 часа). При таких условиях наблюдалась биодоступность >90% при использовании 10 мл 20 мМ фосфатного буфера и биодоступность >70% при использовании 5 мл 40 мМ фосфатного буфера. (Наблюдалась небольшая вариация в результатах, полученных у самок, получавших 5 мл 40 мМ буфера, что объясняет очевидно худшую биодоступность в данной группе субъектов).
Исследования, проведенные с композицией 5 в различных дозах у яванского макака обнаужило полное отсутствие побочных эффектов со стороны желудочно-кишечного тракта; таким образом, композиция существенно лучше переносится млекопитающими по сравнению с композицией 2, описанной выше. После повторного введения достигались высокие уровни лекарственного средства и наблюдалась лучшая пропорциональная зависимость от дозы.
Сравнение Композиции 2 и Композиции 5.
Сравнивали результаты ФК экспериментов, проводившихся с использованием композиций 2 и 5 в эквивалентных дозах (50 мг/кг) при пероральном введении дважды в сутки (BID) в течение 14 дней. Эти данные показали, что воздействие при применении композиции 5 было лучше, чем при применении композиции 2, дополнительным преимуществом было отсутствие негативного влияния на ЖКТ, ассоциированного с композицией 2.
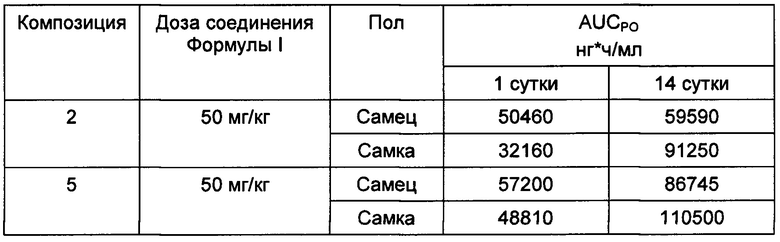
Приведенные выше данные, полученные как на крысах, так и на яванских макаках, показали, что, когда соединение Формулы I было представлено в Композиции 5, описанной выше, наблюдалась превосходная фармакокинетика как у крыс, так и у яванских макак. Композиция хорошо переносилась и оказывала особенно благотворное влияние по сравнению с наполнителями на основе растворителей, такими как PEG/TPGS, из-за отсутствия симптомов со стороны ЖКТ, которые бы потребовали вмешательства с применением противодиарейных препаратов при клиническом применении. При использовании данной композиции также наблюдалась лучшая пропорциональная зависимость параметров AUC и Cmax от дозы по сравнению с композициями на основе растворителя.
Композиция 6
Соединение Формулы I готовили в растворе 25% ДМСО: 75% ПЭГ300 (% об./об.) (концентрация соединения в носителе: 2 мг/мл). Композицию вводили самцам и самкам яванского макака перорально в дозе 10 мг/кг (объем вводимой дозы 5 мл/кг). Образцы крови 2 мл получали в течение 5 минут путем бедренной пункции (вена/артерия) в имеющиеся в продаже пробирки с K2EDTA с применением игл 23G, присоединенных к соответствующему шприцу. Образцы брали через 0,25, 0,5, 1, 2, 4, 8, 12 и 24 часа после введения композиции. Образцы крови держали на льду до центрифугирования при 1600 × g (10 мин, прибл. 5°С) для отделения плазмы. Анализ для расчета плазменной концентрации соединения Формулы I проводили посредством ЖХ-МС/МС. Значения AUCPO (0-24 часа) в ФК экспериментах составили 2900 нг*ч/мл и 1480 нг*ч/мл для самцов и самок, соответственно, что соответствовало биодоступности 16% и 8%, у самцов и самок, соответственно.
Композиция 7
Разработали композицию на основе липидов (Catalent), предназначенную для улучшения солюбилизации (и, следовательно, увеличения биодоступности) соединения Формулы I и протестировали на аванских макаках, как описано выше. Указанную композицию вводили самцам и самкам яванского макака перорально в дозе 10 мг/кг. Значения AUCPO (0-24 часа) в ФК экспериментах составили 3282 нг*ч/мл и 1924 нг*ч/мл для самцов и самок, соответственно, что соответствовало низкой биодоступности 18% и 11%, соответственно.
Пример 5. ФК эксперименты - исследования на человеке in vivo
Проводили исследования с использованием полученной посредством распылительной сушки композиции, соответствующей композициям по изобретению. Композиция включала в себя 20% соединения Формулы I / 80% HPMCAS, суспендированного в 20 мМ фосфатном буфере, как описано для композиции 5 в Примере 4. Образцы крови брали после введения композиции и определяли концентрации соединения Формулы I в плазме, как описано выше.
Первоначальные эксперименты проводили для определения эффекта варьирования дозы композиции. Результаты представлены в следующей Таблице.
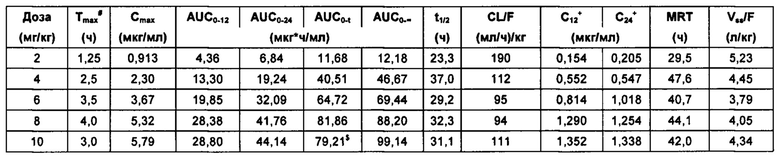
# Медианные значения
+ Концентрации через 12 и 24 ч после введения (для целей возможного повторного введения дозы).
$ в настоящее время представлены только значения AUC0-t от 0-72 ч для предупреждения демаскирования.
В приведенной выше таблице: Tmax = время, когда наблюдалась максимальная концентрация соединения Формулы I; Cmax = наблюдавшаяся максимальная концентрация; AUCA-B = площадь под фармакокинетической кривой «концентрация - время», где А представляет собой момент времени = 0 часов и В = время, до которого производили построение кривой; t1/2 = кажущийся конечный период полувыведения; CL = клиренс; Cn (n=12, 24) = концентрация соединения Формулы I в указанный момент времени; MRT = среднее время удержания; VSS = объем распределения в равновесном состоянии.
Графики зависимости Cmax и AUC0-∞ от вводимой дозы обнаруживали хорошую пропорциональную зависимость от дозы (Cmax: пропорциональность = 1,18; AUC: пропорциональность = 1,30), как показано на Фиг. 1-3. Наблюдалась превосходная биодоступность приблизительно 95%+.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВОГРИБКОВЫЕ АГЕНТЫ | 2015 |
|
RU2727520C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ СОЕДИНЕНИЯ ПИРАЗОЛА, ДИСПЕРГИРОВАННОГО В МАТРИЦЕ ПОЛИМЕРА | 2020 |
|
RU2826604C1 |
| КОМБИНАЦИЯ ВОРИКОНАЗОЛА И ПРОТИВОГРИБКОВОГО ИНГИБИТОРА CYP2C19 | 2005 |
|
RU2345769C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 2019 |
|
RU2820457C2 |
| ПРОИЗВОДНЫЕ (1,10b-ДИГИДРО-2-(АМИНОАЛКИЛФЕНИЛ)-5Н-ПИРАЗОЛО[1,5-c][1,3]БЕНЗОКСАЗИН-5-ИЛ)ФЕНИЛМЕТАНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСНОЙ РЕПЛИКАЦИИ ВИЧ | 2006 |
|
RU2416615C2 |
| ТОЗИЛАТНАЯ СОЛЬ ТЕРАПЕВТИЧЕСКОГО СОЕДИНЕНИЯ И ЕЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2008 |
|
RU2430099C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2159107C2 |
| ИНГИБИРУЮЩИЕ ВИЧ ПРОИЗВОДНЫЕ 2-(4-ЦИАНОФЕНИЛАМИНО)-ПИРИМИДИН-ОКСИДА | 2006 |
|
RU2398768C2 |
| СЛОЖНЫЕ ЭФИРЫ АМИНОКИСЛОТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1998 |
|
RU2197489C2 |
| ГРАНУЛА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2125445C1 |
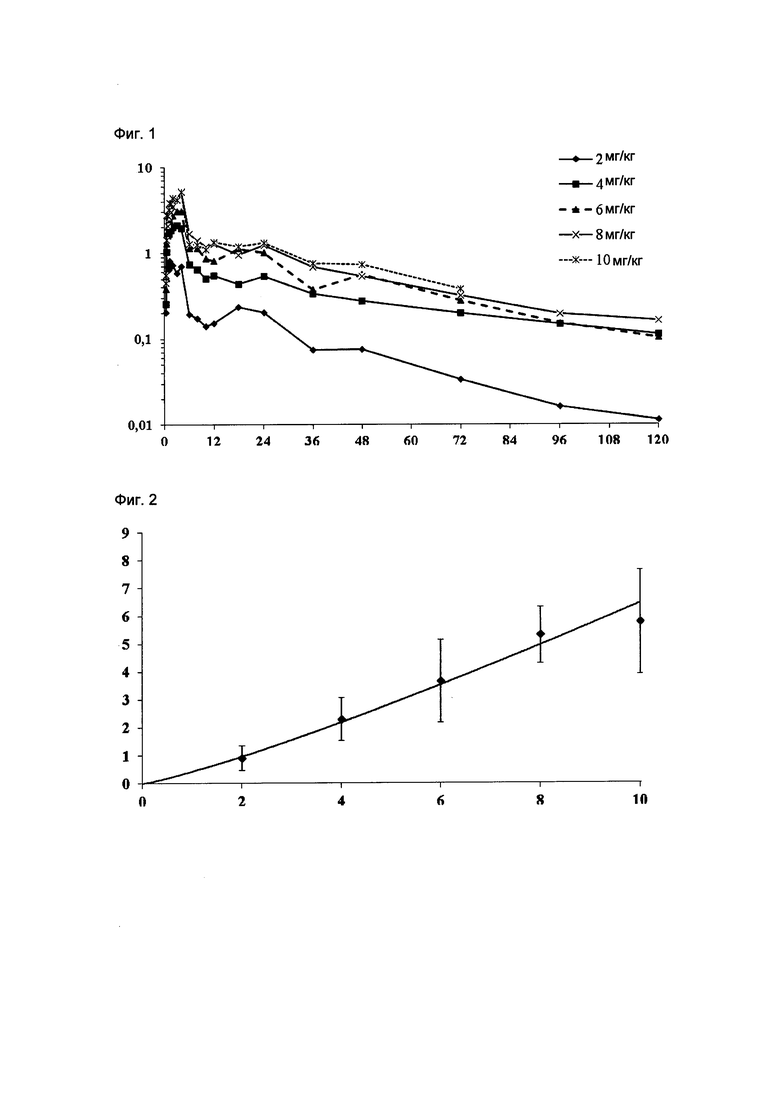
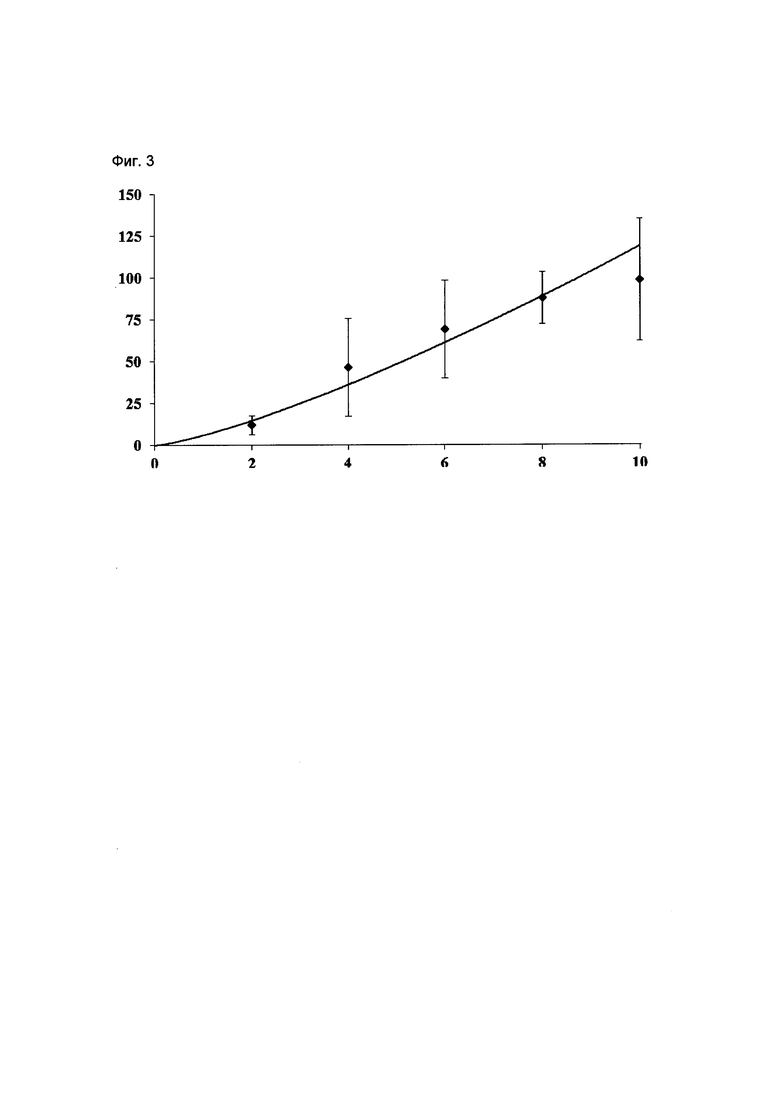
Изобретение относится к фармацевтическим композициям, содержащим частицы, полученные посредством распылительной сушки частицы 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамида, а также применению фармацевтических композиций для предупреждения или лечения грибковой инфекции. Технический результат: получены фармацевтические композиции на основе 2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамида, обладающие противогрибковыми свойствами. 7 н. и 23 з.п. ф-лы, 3 ил., 1 табл., 5 пр.
1. Фармацевтическая композиция, обладающая противогрибковой активностью, которая содержит полученные посредством распылительной сушки частицы соединения Формулы (I)
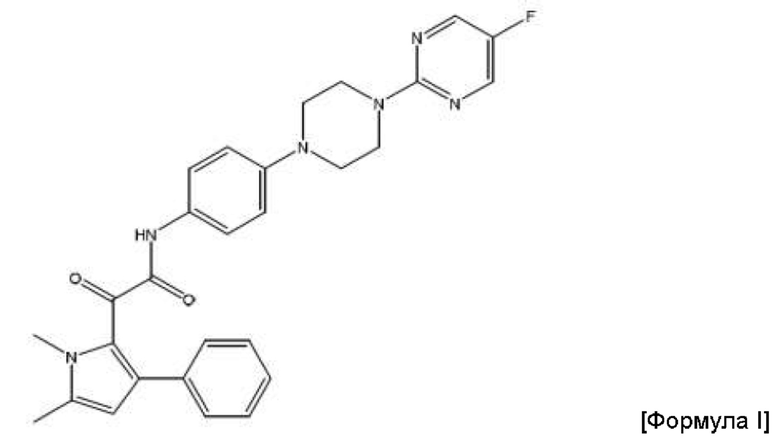
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид
в эффективном количестве и один или более чем один фармацевтически приемлемый эксципиент.
2. Фармацевтическая композиция по п. 1, в которой соединение Формулы (I) является по меньшей мере на 95% аморфным.
3. Фармацевтическая композиция по п. 1, подходящая для перорального введения.
4. Фармацевтическая композиция по п. 1, где композиция содержит эксципиент гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS).
5. Фармацевтическая композиция по п. 1, в которой массовое соотношение соединения Формулы (I) и эксципиента составляет от 1:100 до 1:1.
6. Фармацевтическая композиция по п. 1, в которой массовое соотношение соединения Формулы (I) и эксципиента составляет от 1:15 до 1:2.
7. Фармацевтическая композиция по п. 1, в которой частицы можно получать посредством распылительной сушки из раствора, содержащего органический растворитель, выбранный из дихлорметана, метанола и их смесей.
8. Фармацевтическая композиция по п. 1, в которой частицы можно получать посредством распылительной сушки из раствора, содержащего этанол.
9. Фармацевтическая композиция по п. 1, дополнительно содержащая одно или более чем одно фармацевтически приемлемое связывающее вещество и/или один или более чем один фармацевтически приемлемый носитель и/или разбавитель и/или адъювант.
10. Фармацевтическая композиция по п. 1, которая находится в виде твердой пероральной лекарственной формы.
11. Фармацевтическая композиция по п. 10, где твердая пероральная лекарственная форма представляет собой гранулы.
12. Фармацевтическая композиция по п. 10, где твердая пероральная лекарственная форма представляет собой таблетки.
13. Фармацевтическая композиция по п. 1, которая находится в виде жидкой пероральной лекарственной формы.
14. Фармацевтическая композиция по п. 1, которая находится в виде суспензии.
15. Фармацевтическая композиция по п. 13, где жидкая пероральная лекарственная форма дополнительно содержит фармацевтически приемлемый буфер, имеющий pKa в диапазоне от 6,0 до 8,0.
16. Фармацевтическая композиция по п. 15, в которой буфер представляет собой фосфатный буфер с концентрацией от 1 мМ до 200 мМ, в котором композиция забуферена до рН приблизительно 7.
17. Фармацевтическая композиция, обладающая противогрибковой активностью, подходящая для парентерального введения, которая содержит (i) соединение формулы (I)
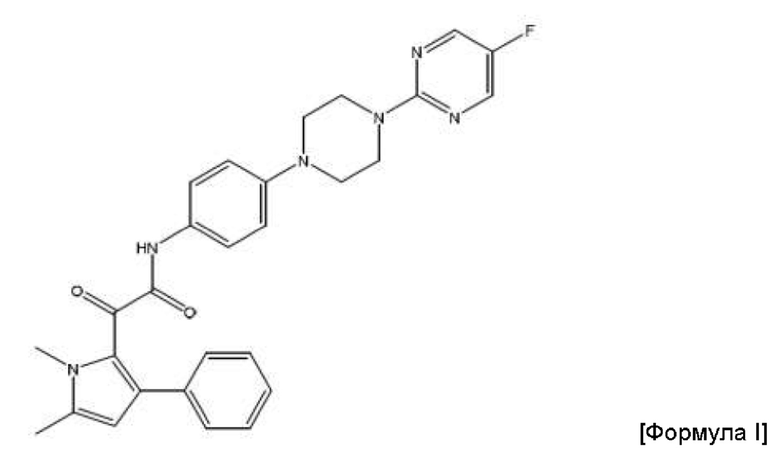
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид
в эффективном количестве,
(ii) циклодекстрин или модифицированный циклодекстрин и (iii) полиэтиленгликоль.
18. Фармацевтическая композиция по п. 17, содержащая:
- от 10 мас. % до 40 мас. % циклодекстрина или модифицированного циклодекстрина; и/или
- от 10 мас. % до 40 мас. % полиэтиленгликоля.
19. Фармацевтическая композиция по п. 17, в которой циклодекстрин или модифицированный циклодекстрин представляет собой гидроксипропил-бета-циклодекстрин.
20. Фармацевтическая композиция по п. 17, в которой полиэтиленгликоль представляет собой ПЭГ300 или ПЭГ400.
21. Фармацевтическая композиция по п. 17, дополнительно содержащая поливинилпирролидон (Повидон).
22. Фармацевтическая композиция по п. 17, в которой соединение Формулы (I) присутствует в концентрации от 1 мг/мл до 10 мг/мл.
23. Фармацевтическая композиция по п. 17, дополнительно содержащая один или более чем один фармацевтически приемлемый носитель, и/или эксципиент, и/или разбавитель, и/или адъювант.
24. Фармацевтическая композиция по п. 17, содержащая:
- 4 мг/мл (относительно конечного объема композиции) соединения Формулы (I),
- 25 мас. % гидроксипропил-бета-циклодекстрина;
- 25 мас. % ПЭГ400;
- 1 мас. % поливинилпирролидона (Повидона);
- фосфорную кислоту в количестве, достаточном для доведения рН фармацевтической композиции до рН 5,0; и
- воду до 100%.
25. Применение фармацевтической композиции по любому из пп. 1-24 для предупреждения или лечения грибковой инфекции у субъекта.
26. Способ предупреждения или лечения грибковой инфекции у субъекта, которому это необходимо, при котором указанному субъекту вводят терапевтически эффективное количество фармацевтической композиции по любому из пп. 1-24.
27. Применение фармацевтической композиции по любому из пп. 1-24 в изготовлении лекарственного средства для предупреждения или лечения грибковой инфекции.
28. Способ получения фармацевтической композиции, содержащей соединение Формулы (I)
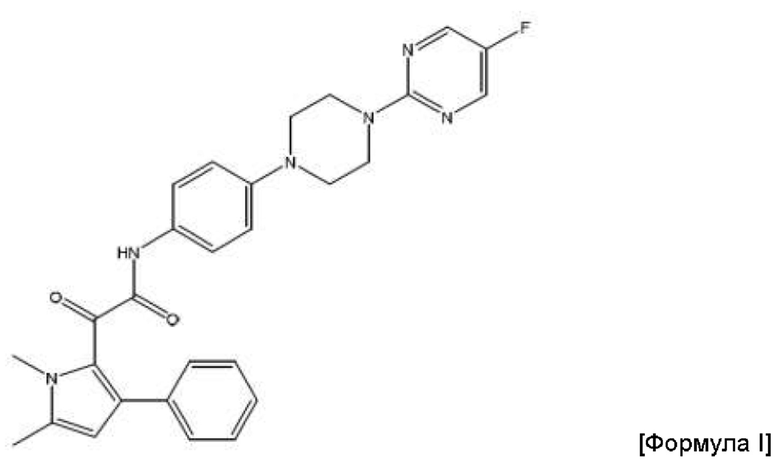
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид,
включающий распылительную сушку раствора соединения Формулы (I), при этом указанный способ включает:
i) растворение одного или более чем одного эксципиента в растворителе;
ii) добавление соединения Формулы (I) к раствору, полученному на стадии (i); и
iii) распылительную сушку раствора, полученного на стадии (ii).
29. Способ по п. 28, в котором:
- эксципиент представляет собой гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS);
- растворитель представляет собой смесь дихлорметана и метанола, где соотношение дихлорметана и метанола составляет от 5:1 до 1:1;
- концентрация эксципиента в растворителе составляет от 5% до 20% мас./об.; и
- соединение Формулы (I) добавляют к раствору эксципиента в растворителе для получения концентрации от 0,5% до 10% по массе.
30. Фармацевтическая композиция, обладающая противогрибковой активностью, подходящая для перорального введения, где композиция содержит частицы соединения Формулы (I)
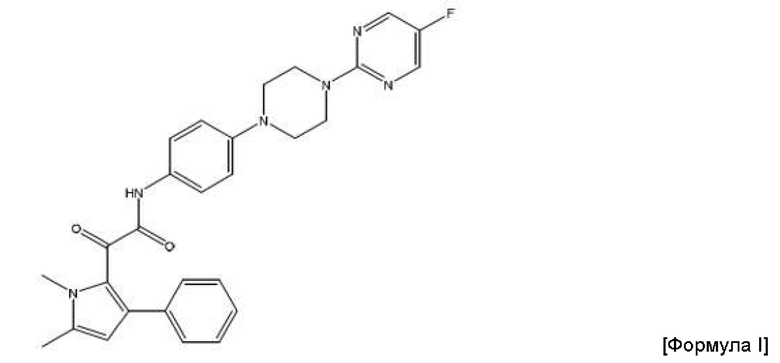
2-(1,5-диметил-3-фенил-1Н-пиррол-2-ил)-N-(4-(4-(5-фторпиримидин-2-ил)пиперазин-1-ил)фенил)-2-оксацетамид
в эффективном количестве и один или более чем один фармацевтически приемлемый эксципиент, при этом указанное соединение Формулы (I) является по меньшей мере на 95% аморфным.
| WO2009130481A1, 29.10.2009 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ переработки окисленных цинк-и железосодержащих материалов | 1980 |
|
SU901318A1 |

