Уровень техники
[0001] ATR(«ATM- и Rad3-родственная»)-киназа представляет собой протеинкиназу, участвующую в формировании клеточного ответа на повреждение ДНК. ATR-киназа действует совместно с АТМ («мутированной атаксия-телеангиэктазия») киназой и множеством других белков, регулируя клеточный ответ на повреждение ДНК, обычно называемый ответом на повреждение ДНК («DDR»). DDR стимулирует репарацию ДНК, способствует выживанию, вызывая задержку клеточного цикла путем активации контрольных точек клеточного цикла, что обеспечивает время для репарации. Без DDR, клетки являются гораздо более чувствительными к повреждению ДНК и быстро погибают из-за патологических изменений ДНК, вызванных эндогенными клеточными процессами, такими как репликация ДНК, или вызывающими повреждение ДНК экзогенными агентами, обычно используемыми при раковой терапии.
[0002] Здоровые клетки могут задействовать множество различных белков для репарации ДНК, включая DDR ATR киназу. В некоторых случаях, эти белки могут компенсировать друг друга путем активации функционально дублирующих процессов репарации ДНК. Напротив, многие раковые клетки «укрывают» дефекты в некоторых процессах репарации их ДНК, таких как АТМ-зависимая передача сигнала, и, следовательно, демонстрируют гораздо большую зависимость от оставшихся интактными белков, участвующих в репарации ДНК, которые включают ATR.
[0003] Кроме того, многие раковые клетки экспрессируют активированные онкогены или испытывают недостаток в ключевых генах-супрессорах опухолей, и это может привести к неуправляемым фазам репликации ДНК в раковых клетках, что, в свою очередь, вызывает повреждение ДНК. ATR считается ключевым компонентом DDR в ответе на репликацию поврежденной ДНК. В результате, выживание этих раковых клеток становится еще более зависимым от активности ATR по сравнению со здоровыми клетками. Соответственно, ингибиторы ATR можно использовать для лечения ракового заболевания либо отдельно, либо в комбинации с повреждающими ДНК агентами, так как они «выключают» механизм репарации ДНК, который является более важным для выживания раковых клеток, чем для нормальных здоровых клеток.
[0004] По сути, было показано, что нарушение функции ATR (например, в результате делеции гена) способствует гибели раковой клетки как в отсутствие, так и в присутствии повреждающих ДНК агентов. Это наводит на мысль, что ингибиторы ATR могут быть эффективными как в виде отдельных агентов, так и в виде сильных сенсибилизаторов к радиотерапии или генотоксичной терапии.
[0005] В силу этих причин существует необходимость в разработке эффективных и селективных ингибиторов ATR для лечения ракового заболевания, используемых либо в виде отдельных агентов, либо в виде комбинированной терапии с радиотерапией или генотоксичной терапией. Кроме того, имеется необходимость в методе синтеза ингибиторов ATR, который позволил бы осуществлять крупномасштабный синтез и улучшил бы используемые в настоящее время известные способы.
[0006] ATR-пептид можно экспрессировать и выделять, используя множество способов, известных из литературы (см., например, Ünsal-Kaçmaz и др., PNAS 99:10, сс. 6673-6678, 14 мая 2002 года; см. также Kumagai и др., Cell 124, сс. 943-955, 10 марта 2006 года; Ünsal-Kaçmaz и др., Molecular and Cellular Biology, февраль 2004 года, сс. 1292-1300; и Hall-Jackson и др., Oncogene 1999, 18, 6707-6713).
Краткое описание чертежей
Фиг.1а: XRPD Соединения I-1 в виде безводного свободного основания;
Фиг.2а: ТГА Соединения I-1 в виде безводного свободного основания;
Фиг.3а: ДСК Соединения I-1 в виде безводного свободного основания;
Фиг.1b: XRPD Соединения I-1 в виде гидрата;
Фиг.2b: ТГА Соединения I-1 в виде гидрата;
Фиг.3b: ДСК Соединения I-1 в виде гидрата;
Фиг.1с: XRPD Соединения I-1 в виде аддукта с винной кислотой;
Фиг.2с: ТГА Соединения I-1 в виде аддукта с винной кислотой;
Фиг.3с: ДСК Соединения I-1 в виде аддукта с винной кислотой.
Сущность изобретения
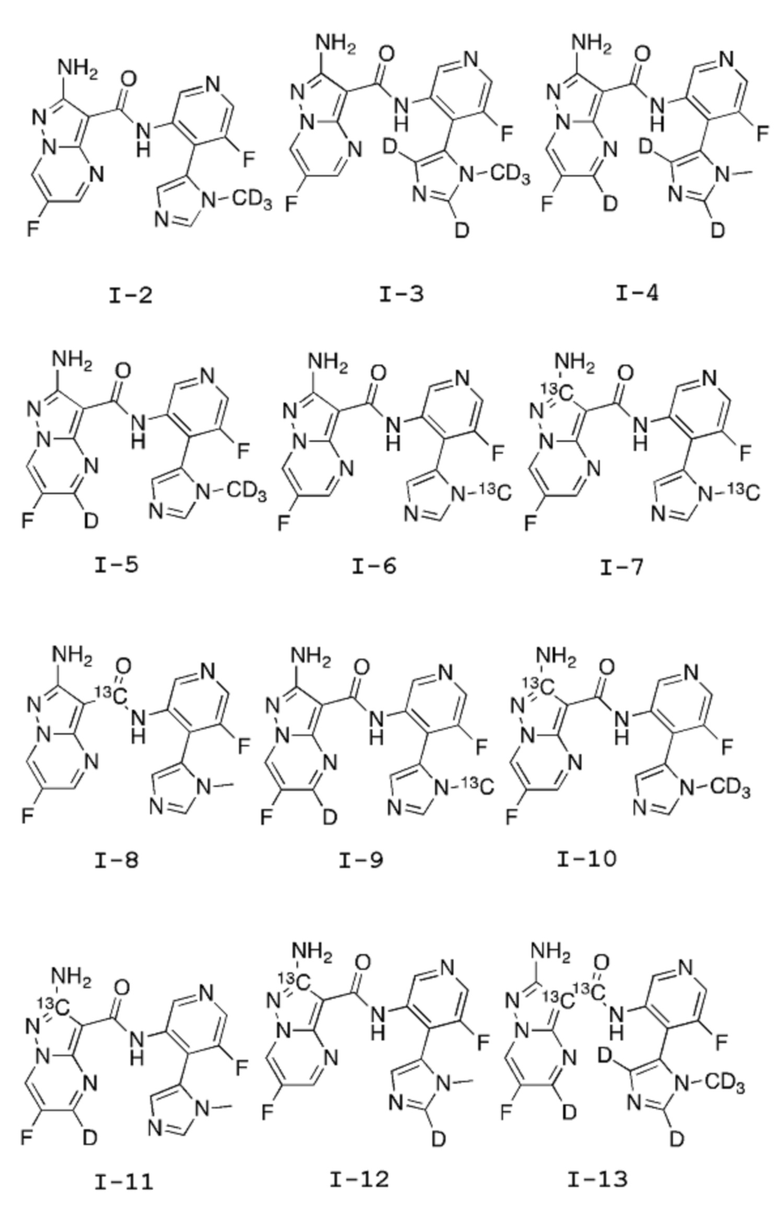
[0007] Настоящее изобретение относится к твердым формам ингибиторов ATR, а также к дейтерированным формам ингибиторов ATR. Настоящее изобретение также относится к способам и промежуточным продуктам для получения аминопиразолопиримидинового соединения, пригодного в качестве эффективного ингибитора ATR-киназы. Производные аминопиразолопиримидина являются пригодными в качестве ингибиторов ATR, а также для получения ингибиторов ATR.
[0008] Один из аспектов изобретения относится к способу получения соединения I-1:
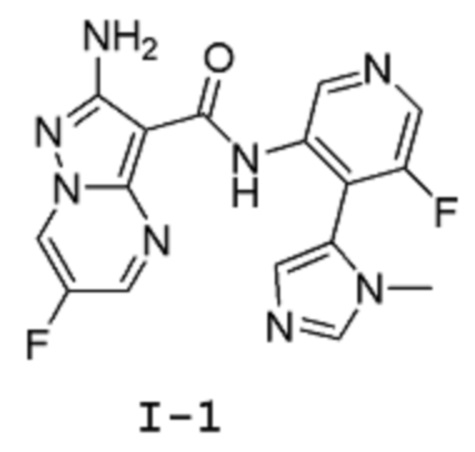
[0009] Другой аспект настоящего изобретения включает соединение формулы I-A:
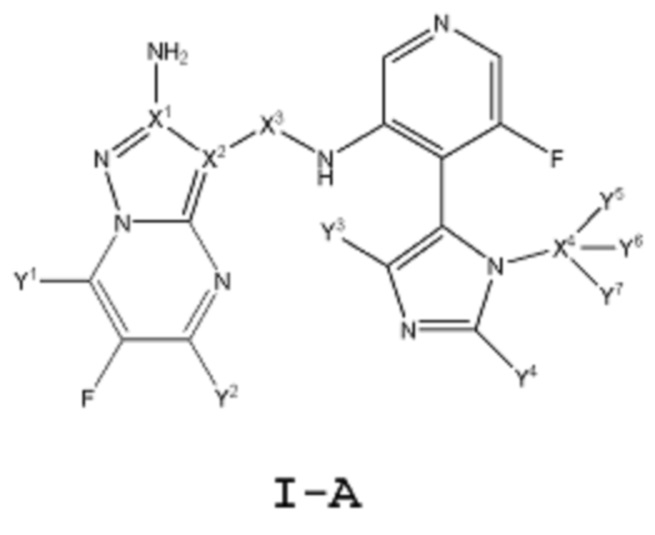
или его фармацевтически приемлемую соль или производное, где:
каждый из Y1, Y2, Y3, Y4, Y5, Y6 и Y7 независимо означает водород или дейтерий; при условии, что, по меньшей мере, один из Y1, Y2, Y3, Y4, Y5, Y6 и Y7 означает дейтерий;
каждый из Х1, Х2 и Х4 независимо выбирают из 12С или 13С; и
Х3 независимо выбирают из -12С(О)- или -13С(О)-.
[0010] Другой аспект изобретения относится к твердым формам соединения формулы I-1:
[0011] В настоящем описании изложены и другие аспекты изобретения.
Подробное описание изобретения
Способы
[0012] Другой аспект настоящего изобретения включает способ получения соединения формулы I-1:
включающий стадию взаимодействия соединения формулы 6b:
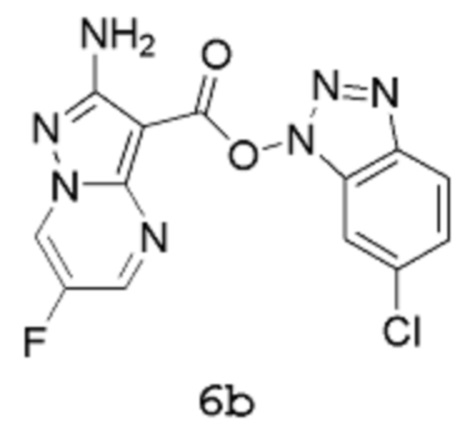
с соединением формулы 11:
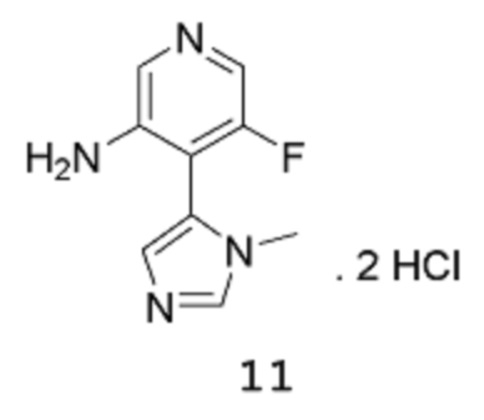
в подходящих условиях для образования амидной связи.
[0013] Подходящие условия для образования амидной связи включают взаимодействие соединения формулы 6b с замещенным 3-аминопиридином 11 в присутствии растворителя и органического основания. В одном из воплощений растворитель можно выбирать из NMP, ДМФА или анизола (предпочтительно). В другом воплощении органическое основание представляет собой алифатический амин, независимо выбираемый из триэтиламина или DIPEA (предпочтительно).
[0014] Другие воплощения настоящего изобретения включают способ получения соединения формулы 11:
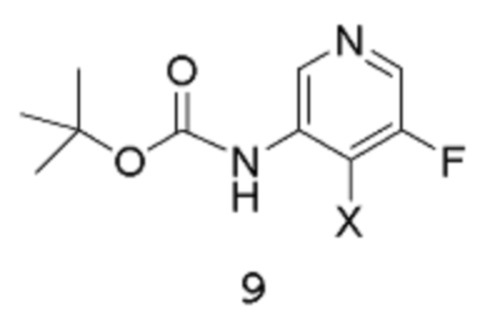
путем взаимодействия соединения формулы 9:
с соединением формулы 10:
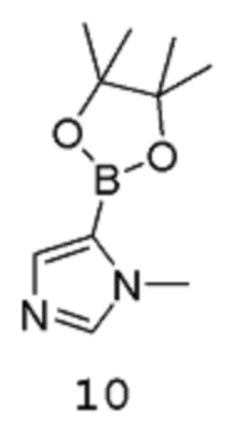
в подходящих условиях для катализируемой металлом реакции кросс-сочетания с образованием аддукта, содержащего защищенную аминогруппу; после чего полученный аддукт подвергают воздействию подходящих условий для снятия защиты.
[0015] Подходящие условия для катализируемой металлом реакции кросс-сочетания включают металлический катализатор, подходящий растворитель и подходящее основание. В некоторых вариантах осуществления металлический катализатор представляет собой палладиевый катализатор. Примеры подходящих палладиевых катализаторов включают, но не ограничиваются этим, PdCl2(PPh3)2, Pd(Ph3)4 и PdCl2(dppf) (где каждый Ph означает фенил, и dppf представляет собой 1,1-бис(дифенилфосфино)ферроцен). Подходящие основания включают, но не ограничиваются этим, фосфат калия, К2СО3, трет-BuOK и Na2CO3. Подходящие растворители включают, но не ограничиваются этим, DME, тетрагидрофуран, толуол и этанол.
[0016] Подходящие условия для снятия защиты для удаления защитной группы включают реакцию защищенного соединения в присутствии сильной кислоты, такой как HCl (предпочтительно), HBr, серная кислота или трифторуксусная кислота.
[0017] Другое воплощение относится к способу получения соединения формулы 9:
путем реакции соединения формулы 8:
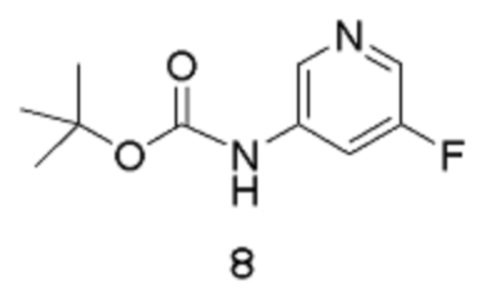
в подходящих условиях для галогенирования.
[0018] Подходящие условия для галогенирования включают реакцию соединения 8 в апротонном растворителе в присутствии сильного основания и электрофильного источника галогена. В одном из воплощений растворитель можно выбирать из DCM, диэтилового эфира или ТГФ (предпочтительно). В другом воплощении сильное основание выбирают из трет-BuLi, втор-BuLi или н-BuLi (предпочтительно). В другом воплощении электрофильное соединение, используемое для введения атома галогена, можно, например, выбирать из I2 (предпочтительно), CF3I, дииодэтана, Br2, CBr4.
[0019] Другие воплощения настоящего изобретения относятся к способу получения соединения формулы 8:
путем реакции соединения формулы 7:
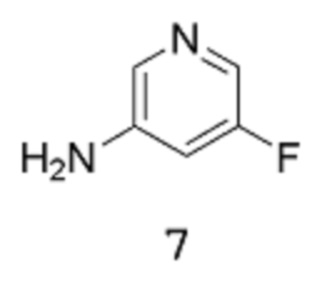
в подходящих условиях для получения защищенной аминогруппы.
[0020] Подходящие условия для введения защитной группы включают реакцию аминосоединения 7 в апротонном растворителе в присутствии Вос2О. Такая реакция может протекать в присутствии основания. В одном из воплощений растворитель можно выбирать из диэтилового эфира или ТГФ (предпочтительно). В другом воплощении сильное основание можно выбирать из DMAP, н-BuLi, LHMDS или NaHMDS (предпочтительно).
Дейтерированные соединения
[0021] Изотопы можно вводить в соединение I-1 посредством выбора элементов структуры, содержащих изотопные атомы (которые либо являются коммерчески доступными, либо могут быть получены в соответствии с литературой), и включения их в последовательность согласно новому способу по изобретению, описанному на примере немеченого изотопами вещества (см. выше).
[0022] Другой аспект настоящего изобретения относится к соединению формулы I-A:
или к его фармацевтически приемлемой соли или производному, где:
каждый из Y1, Y2, Y3, Y4, Y5, Y6 и Y7 независимо означает водород или дейтерий; при условии, что, по меньшей мере, один из Y1, Y2, Y3, Y4, Y5, Y6 и Y7 означает дейтерий;
каждый из Х1, Х2 и Х4 независимо выбирают из 12С или 13С; и
Х3 независимо выбирают из -12С(О)- или -13С(О)-.
[0023] Следующие меченые элементы структуры, которые можно использовать в методе синтеза для получения соединения формулы I-A, являются коммерчески доступными:
- 1,2-ди-13С-2-цианоуксусная кислота;
- этиловый эфир 1-13С-2-циано(13С)уксусной кислоты;
- этиловый эфир 2-13С-2-циано(13С)уксусной кислоты;
- 1-(тридейтерометил)-1Н-имидазол;
- 2,4,5-тридейтеро-1-(метил)-1Н-имидазол; и
- 2,4,5-тридейтеро-1-(тридейтерометил)-1Н-имидазол.
[0024] Другие меченые элементы структуры, которые можно использовать в методе синтеза для получения соединения формулы I-A, являются известными квалифицированному специалисту в данной области. Они могут включать, но не ограничиваться этим, следующие меченые элементы структуры:
- 2-циано(13С)уксусная кислота; Triplett и др., J Labelled Comp Radiopharm, 1978, 14(1), 35;
- 1-13С-2-цианоуксусная кислота; Matsumoto и др., Heterocycles, 1985, 23(8), 2041;
- 2-13С-2-цианоуксусная кислота; Baldwin и др., J Am Chem Soc, 1989, 111(9), 3319;
- 1-дейтеро-3-(диэтиламино)-2-фторакрилальдегид; Funabiki и др., Chem Lett, 1997, (8), 739;
- 2-дейтеро-1-(метил)-1Н-имидазол; Torregrosa и др., Tetrahedron, 2005, 61(47), 11148-11155;
- 4,5-дидейтеро-1-(метил)-1Н-имидазол; Pavlik и др., J. Org. Chem., 1991, 56(22), 6313-6320;
- 4,5-дидейтеро-1-(тридейтерометил)-1Н-имидазол; Mamer и др., Rapid Communications in Mass Spectrometry, 2005, 19(12), 1771-1774;
- 2-тритио-1-(метил)-1Н-имидазол; Buncel и др., Can. J. Chem., 1986, 64(6), 1240-1245;
- 2,4,5-тритритио-1-(метил)-1Н-имидазол; Grimmett, Scien of Synthesis, 2002, 325-528; и
- 1-(13С-метил)-1Н-имидазол; Van Thuijl и др., Organic Mass Spectrometry, 1973, 7(10), 1165-1172.
[0025] В одном или более воплощениях настоящего изобретения Y1, Y2, Y3 и Y4 независимо выбирают из дейтерия или водорода; и Y5, Y6 и Y7 означают дейтерий.
[0026] В некоторых воплощениях Y1 и Y2 независимо выбирают из дейтерия или водорода; и Y3, Y4, Y5, Y6 и Y7 означают дейтерий.
[0027] В другом воплощении Y1, Y2, Y5, Y6 и Y7 независимо выбирают из дейтерия или водорода; и Y3 и Y4 означают дейтерий.
[0028] В других воплощениях Y1, Y3 и Y4 независимо выбирают из дейтерия или водорода; и Y2, Y5, Y6 и Y7 означают дейтерий.
[0029] В других воплощениях Y1, Y2, Y3, Y4, Y5, Y6 и Y7 означают водород; и Х4 означает 13С.
[0030] В другом воплощении Y1, Y2, Y3, Y4, Y5, Y6 и Y7 означают водород; и Х1 и Х4 означают 13С.
[0031] В некоторых воплощениях Y1, Y2, Y3, Y4, Y5, Y6 и Y7 означают водород; и Х3 означает -13С(О)-.
[0032] В другом воплощении Y1, Y3, Y4, Y5, Y6 и Y7 означают водород; Y2 означает дейтерий; и Х4 означает 13С.
[0033] В других воплощениях Y1, Y2, Y3 и Y4 означают водород; Y5, Y6 и Y7 означают дейтерий; и Х1 означает 13С.
[0034] В других воплощениях Y1, Y3, Y4, Y5, Y6 и Y7 означают водород; Y2 означает дейтерий; и Х1 означает 13С.
[0035] В другом воплощении Y1, Y2, Y3, Y5, Y6 и Y7 означают водород; Y4 означает дейтерий; и Х1 означает 13С.
[0036] В другом воплощении Y1 означает водород; Y2, Y3, Y4, Y5, Y6 и Y7 означают дейтерий; Х2 означает 13С; и Х3 означает -13С(О)-.
[0037] В другом примере соединения формулы I-A согласно данному изобретению представлены в Таблице 1. Квалифицированному специалисту в данной области должно быть понятно, что соединения согласно настоящему изобретению могут находиться в различных таутомерных формах.
Таблица 1
Твердые формы
[0038] Другой аспект настоящего изобретения относится к твердой форме соединения формулы I-1:
где эту форму выбирают из группы, состоящей из соединения I-1 в виде безводного свободного основания, соединения I-1 в виде гидрата или соединения I-1 в виде аддукта с винной кислотой.
Соединение I-1 в виде безводного свободного основания
[0039] В некоторых аспектах настоящего изобретения твердая форма представляет собой соединение I-1 в виде безводного свободного основания. В другом аспекте настоящего изобретения твердая форма представляет собой кристаллическое соединение I-1 в виде безводного свободного основания. В некоторых воплощениях твердая форма характеризуется одним или более пиками, выраженными значениями 2-тета ± 0,2, равными примерно 9,9, 12,8, 15,4, 17,0, 23,1, 27,8, 29,0 и 30,1 градусов, на порошковой рентгеновской дифрактограмме, получаемой с использованием излучения Cu-K-альфа. В других воплощениях твердая форма характеризуется порошковой рентгеновской дифрактограммой по существу такой же, как представлена на Фиг.1а.
Соединение I-1 в виде гидрата
[0040] В некоторых аспектах настоящего изобретения твердая форма представляет собой соединение I-1 в виде гидрата. В другом аспекте настоящего изобретения твердая форма представляет собой кристаллическое соединение I-1 в виде гидрата. В других воплощениях кристаллическое соединение I-1 в виде гидрата имеет соотношение соединения I-1 к воде, равное 1:3. В других воплощениях соединение I-1 в виде гидрата характеризуется потерей массы от примерно 12,6% в диапазоне температуры от примерно 40°С до примерно 100°С. В некоторых воплощениях твердая форма характеризуется одним или более пиками, выраженными значениями 2-тета ± 0,2, равными примерно 27,5, 20,6 и 9,7 градусов, на порошковой рентгеновской дифрактограмме, получаемой с использованием излучения Cu-K-альфа. В других воплощениях твердая форма характеризуется порошковой рентгеновской дифрактограммой, по существу такой же, как представлена на Фиг.1b.
Соединение I-1 в виде аддукта с винной кислотой
[0041] В некоторых аспектах настоящего изобретения твердая форма представляет собой соединение I-1 в виде аддукта с винной кислотой. В другом аспекте настоящего изобретения твердая форма представляет собой кристаллическое соединение I-1 в виде аддукта с винной кислотой. В других воплощениях кристаллическое соединение I-1 в виде аддукта с винной кислотой имеет соотношение соединения I-1 к винной кислоте, равное 1:1. В некоторых воплощениях твердая форма характеризуется одним или более пиками, выраженными значениями 2-тета ± 0,2, равными примерно 7,1, 18,3 и 13,2 градусов, на порошковой рентгеновской дифрактограмме, получаемой с использованием излучения Cu-K-альфа. В других воплощениях твердая форма характеризуется порошковой рентгеновской дифрактограммой, по существу такой же, как представлена на Фиг.1с.
[0042] Следует отметить, что в настоящей заявке термины «воплощение», «пример» и «аспект» используются взаимозаменяемо.
[0043] Соединения согласно данному изобретению включают соединения, раскрытые в настоящем описании в общем виде, и далее проиллюстрированы посредством классов, подклассов и типов, раскрытых в настоящем описании. В настоящем описании используются приведенные ниже определения, если не указано иное. В данном изобретении химические элементы идентифицированы в соответствии с периодической таблицей элементов, версии CAS, Handbook of Chemistry and Physics, 75-ое изд. Дополнительно, общие принципы органической химии описаны в «Organic Chemistry», Thomas Sorrell, University Science Books, Sausalito: 1999, и «Marchʹs Advanced Organic Chemistry», 5-ое изд., ред.: Smith M.B. и March J., John Wiley & Sons, New York: 2001, полное содержание которых включено в данный контекст путем ссылки.
[0044] В настоящем описании указанный диапазон атомных чисел включает любое целое число. Например, группа, имеющая 1-4 атома, может иметь 1, 2, 3 или 4 атома.
[0045] В настоящем описании соединения согласно изобретению необязательно могут быть замещены одним или более заместителями, такими как проиллюстрировано в настоящем описании в общем виде или показано на примере конкретных классов, подклассов и типов согласно изобретению. Фраза «необязательно замещенный» используется взаимозаменяемо с фразой «замещенный или незамещенный». Вообще, термин «замещенный», независимо от того, находится или нет перед ним термин «необязательно», относится к замене радикалов водорода в данной структуре радикалами конкретного заместителя. Если не указано иное, необязательно замещенная группа может иметь заместитель в каждом замещаемом положении группы и, когда более чем одно положение в любой данной структуре может быть замещено более чем одним заместителем, выбираемым из конкретной группы, заместитель может быть либо одним и тем же, либо в каждом положении заместители могут быть разными. Комбинации заместителей, предусмотренные в данном изобретении, предпочтительно являются заместителями, которые приводят к образованию стабильных или химически подходящих соединений.
[0046] Если не указано иное, заместитель, соединенный связью, изображенной выходящей из центра кольца, означает, что заместитель может быть связан в любом положении кольца. В приведенном ниже примере i, например, Jw может быть связан в любом положении пиридильного кольца. В случае бициклических колец, связь, изображенная проходящей через оба цикла, указывает, что заместитель может быть связан в любом положении бициклического кольца. В приведенном ниже примере ii, например, Jw может быть связан с 5-членным кольцом (по атому азота, например) и с 6-членным кольцом.
[0047] Используемый в настоящем описании термин «стабильный» относится к соединениям, которые, по существу, не изменяются, когда подвергаются условиям, подходящим для их получения, обнаружения, рекуперации, очистки и применения согласно одному или более назначениям, раскрытым в настоящем описании. В некоторых воплощениях стабильное соединение или химически подходящее соединение представляет собой соединение, которое, по существу, не изменяется при хранении при температуре 40°С или менее в отсутствие влажности или при других химически активных условиях в течение, по крайней мере, недели.
[0048] Используемый в настоящем описании термин «алифатический» или «алифатическая группа» означает линейную (т.е., неразветвленную), разветвленную или циклическую, замещенную или незамещенную углеводородную цепь, полностью насыщенную или содержащую одну или более единиц ненасыщенности, которая имеет одну точку присоединения к остальной части молекулы.
[0049] Если не указано иное, алифатические группы содержат 1-20 алифатических атомов углерода. В некоторых воплощениях алифатические группы содержат 1-10 алифатических атомов углерода. В других воплощениях алифатические группы содержат 1-8 алифатических атомов углерода. В других воплощениях алифатические группы содержат 1-6 алифатических атомов углерода, тогда как в других воплощениях алифатические группы содержат 1-4 алифатических атомов углерода. Алифатические группы могут быть линейными или разветвленными, замещенными или незамещенными алкильными, алкенильными или алкинильными группами. Конкретные примеры включают, но не ограничиваются этим, метил, этил, изопропил, н-пропил, втор-бутил, винил, н-бутенил, этинил и трет-бутил. Алифатические группы также могут быть циклическими или иметь комбинацию линейных или разветвленных и циклических групп. Примеры таких типов алифатических групп включают, но не ограничиваются этим, циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, -СН2-циклопропил, СН2СН2СН(СН3)-циклогексил.
[0050] Термин «циклоалифатический» (или «карбоцикл» или «карбоциклил») относится к моноциклическому С3-С8-углеводороду или бициклическому С8-С12- углеводороду , который является полностью насыщенным или который содержит одну или более единиц ненасыщенности, но который не является ароматическим, и имеет одну точку присоединения к остатку молекулы, при этом любое отдельное кольцо в вышеуказанной бициклической системе имеет 3-7 членов. Примеры циклоалифатических групп включают, но не ограничиваются этим, циклоалкильные и циклоалкенильные группы. Конкретные примеры включают, но не ограничиваясь этим, циклогексил, циклопропил и циклобутил.
[0051] Термин «гетероцикл», «гетероциклил» или «гетероциклический», используемый в настоящем описании, означает неароматические, моноциклические, бициклические или трициклические кольцевые системы, в которых один или более членов кольца являются независимо выбранным гетероатомом. В некоторых воплощениях «гетероцикл», «гетероциклил» или «гетероциклическая» группа имеет три-четырнадцать членов кольца, причем один или более членов кольца представляют собой гетероатом, независимо выбираемый из кислорода, серы, азота или фосфора, и каждое кольцо в системе содержит 3-7 членов кольца.
[0052] Примеры гетероциклов включают, но не ограничиваются этим, 3-1Н-бензимидазол-2-он, 3-(1-алкил)бензимидазол-2-он, 2-тетрагидрофуранил, 3-тетрагидрофуранил, 2-тетрагидротиофенил, 3-тетрагидротиофенил, 2-морфолино, 3-морфолино, 4-морфолино, 2-тиоморфолино, 3-тиоморфолино, 4-тиоморфолино, 1-пирролидинил, 2-пирролидинил, 3-пирролидинил, 1-тетрагидропиперазинил, 2-тетрагидропиперазинил, 3-тетрагидропиперазинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 1-пиразолинил, 3-пиразолинил, 4-пиразолинил, 5-пиразолинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-пиперидинил, 2-тиазолидинил, 3-тиазолидинил, 4-тиазолидинил, 1-имидазолидинил, 2-имидазолидинил, 4-имидазолидинил, 5-имидазолидинил, индолинил, тетрагидрохинолинил, тетрагидроизохинолинил, бензотиолан, бензодитиан и 1,3-дигидроимидазол-2-он.
[0053] Циклические группы (например, циклоалифатические и гетероциклы) могут быть линейными, конденсированными, с мостиковой связью или спироциклическими.
[0054] Термин «гетероатом» означает один или более атомов кислорода, серы, азота, фосфора или кремния (включая любую окисленную форму азота, серы, фосфора или кремния; кватернизованную форму любого основного азота; или замещаемый азот гетероцикла, например, N (как в 3,4-дигидро-2Н-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле)).
[0055] Термин «ненасыщенный», используемый в настоящем описании, означает, что остаток имеет одну или более единиц ненасыщенности. Квалифицированному специалисту в данной области известно, что ненасыщенные группы могут быть частично ненасыщенными или полностью ненасыщенными. Примеры частично ненасыщенных групп включают, но не ограничиваются этим, бутен, циклогексен и тетрагидропиридин. Полностью ненасыщенные группы могут быть ароматическими, антиароматическими или неароматическими. Примеры полностью ненасыщенных групп включают, но не ограничиваются этим, фенил, циклооктатетраен, пиридил, тиенил и 1-метилпиридин-2(1Н)-он.
[0056] Термин «алкокси» или «тиоалкил», используемый в настоящем описании, относится к алкильной группе, как описано ранее, присоединенной через атом кислорода («алкокси») или атом серы («тиоалкил»).
[0057] Термины «галогеналкил», «галогеналкенил», «галогеналифатический» и «галогеналкокси» означают алкил, алкенил или алкокси, которые могут быть замещены одним или более атомами галогена. Этот термин включает перфторированные алкильные группы, такие как -СF3 и -СF2СF3.
[0058] Термины «галоген», «гало» и «гал» означают F, Cl, Br или I.
[0059] Термин «арил», используемый отдельно или как часть большего остатка, как в «арилалкиле», «арилалкокси» или «арилоксиалкиле», относится к моноциклическим, бициклическим и трициклическим кольцевым системам, имеющим в общей сложности от пяти до четырнадцати членов кольца, где, по меньшей мере, одно кольцо в этой системе является ароматическим, и где каждое кольцо в системе содержит 3-7 членов кольца. Термин «арил» можно использовать взаимозаменяемо с термином «арильное кольцо».
[0060] Термин «гетероарил», используемый отдельно или как часть большего остатка, как в «гетероарилалкиле» или «гетероарилалкокси», относится к моноциклическим, бициклическим и трициклическим кольцевым системам, имеющим в общей сложности от пяти до четырнадцати членов кольца, где, по меньшей мере, одно кольцо в этой системе является ароматическим, по меньшей мере, одно кольцо в системе содержит один или более гетероатом(ов), и где каждое кольцо в системе содержит 3-7 членов кольца. Термин «гетероарил» можно использовать взаимозаменяемо с термином «гетероарильное кольцо» или термином «гетероароматический». Примеры гетероарильных колец включают, но не ограничиваются этим, 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, бензимидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, N-пирролил, 2-пирролил, 3-пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил), 2-тиазолил, 4-тиазолил, 5-тиазолил, тетразолил (например, 5-тетразолил), триазолил (например, 2-триазолил и 5-триазолил), 2-тиенил, 3-тиенил, бензофурил, бензотиофенил, индолил (например, 2-индолил), пиразолил (например, 2-пиразолил), изотиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-триазолил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, пуринил, пиразинил, 1,3,5-триазинил, хинолинил (например, 2-хинолинил, 3-хинолинил, 4-хинолинил) и изохинолинил (например, 1-изохинолинил, 3-изохинолинил или 4-изохинолинил).
[0061] Должно быть понятно, что термин «гетероарил» включает определенные типы гетероарильных колец, две различные формы которых находятся в равновесном состоянии относительно друг друга. Более конкретно, например, такие соединения, как гидропиридин и пиридинон (и, аналогично, гидроксипиримидин и пиримидинон), охватываются определением «гетероарил».
[0062] Термины «защитная группа» и «защищающая группа», используемые в настоящем описании, являются взаимозаменяемыми и относятся к агенту, используемому для временного блокирования одной или более желательных функциональных групп в соединении, содержащем множество реакционноспособных участков. В некоторых вариантах осуществления, защитная группа имеет одну или более, или, предпочтительно, все из следующих характеристик: а) селективно добавляется к функциональной группе с хорошим выходом давая в результате защищенный субстрат, который b) является стабильным по отношению к реакциям, проходящим в одном или более других реакционноспособных участках; и с) является селективно удаляемой с хорошим выходом с помощью реагентов, которые не разрушают восстановленную функциональную группу со снятой защитой. Квалифицированному специалисту в данной области должно быть понятно, что в некоторых случаях реагенты не атакуют другие реакционноспособные группы в соединении. В других случаях реагенты также могут взаимодействовать с другими реакционноспособными группами в соединении. Примеры защитных групп подробно описаны у Greene T.W., Wuts P.G. в руководстве «Protective Groups in Organic Synthesis», третье издание, John Wiley & Sons, New York: 1999 (и другие издания руководства), полное содержание которого включено в данный контекст путем ссылки. Термин «защитная для азота группа», используемый в настоящем описании, относится к агенту, используемому для временного блокирования одного или более желательных реакционноспособных участков с атомом азота в многофункциональном соединении. Предпочтительные защитные группы для азота также имеют характеристики, проиллюстрированные выше для защитной группы, и некоторые примеры защитных групп для азота также подробно описаны в 7 главе у Greene T.W., Wuts P.G. в руководстве «Protective Groups in Organic Synthesis», третье издание, John Wiley & Sons, New York: 1999, полное содержание которого включено в данный контекст путем ссылки.
[0063] В некоторых вариантах осуществления метиленовый фрагмент алкила или алифатической цепи является необязательно замещенным другим атомом или группой. Примеры таких атомов или групп включают, но не ограничиваются этим, азот, кислород, серу, -С(О)-, -С(=N-CN)-, -C(=NR), -C(=NOR)-, -SO- или -SO2-. Эти атомы или группы можно комбинировать для образования более крупных групп. Примеры таких более крупных групп включают, но не ограничиваются этим, -ОС(О)-, -С(О)СО-, -СО2-, -C(O)NR-, -C(=N-CN), -NRCO-, -NRC(O)O-, -SO2NR-, -NRSO2-, -NRC(O)NR-, -OC(O)NR- и -NRSO2NR-, где R означает, например, Н или С1-6-алифатический радикал. Должно быть понятно, что эти группы могут быть связаны с метиленовыми фрагментами алифатической цепи посредством одинарной, двойной или тройной связей. Примером необязательного замещения (в этом случае, атом азота), где алифатическая цепь связана посредством двойной связи, может быть -CH2CH=N-CH3. В некоторых случаях, необязательное замещение, особенно на терминальном конце, может осуществляться посредством тройной связи с алифатической группой. Одним из таких примеров является CH2CH2CH2C≡N. Должно быть понятно, что в этой ситуации терминальный азот не связан с другим атомом.
[0064] Также должно быть понятно, что термин «метиленовый фрагмент» также может относиться к разветвленным или замещенным метиленовым фрагментам. Например, в изопропильном фрагменте [-CH(CH3)2] атом азота (например, NR), замещающий первый указанный «метиленовый фрагмент», приводит к образованию диметиламина [-N(CH3)2]. В таких случаях, квалифицированному специалисту в данной области должно быть понятно, что атом азота не будет связан ни с какими дополнительными атомами, и в этом случае «R» в «NR»будет отсутствовать.
[0065] Если не указано иное, необязательные замещения приводят к образованию химически стабильного соединения. Необязательные замещения могут происходить как в цепи, так и/или на любом конце цепи; т.е., как в точке присоединения, так и/или на терминальном конце. Два необязательных замещения также могут быть соседними по отношению друг к другу внутри цепи, если только они приводят к образованию химически стабильного соединения. Например, алифатический С3-радикал может быть необязательно замещен 2-мя атомами азота с образованием -C-N≡N. С помощью необязательных замещений также можно полностью заместить все атомы углерода в цепи. Например, алифатический С3-радикал может быть необязательно замещен -NR-, -C(O)- и -NR- с образованием -NRC(O)NR- (мочевины).
[0066] Если не указано иное, если замещение происходит на терминальном конце, атом замещения связан с атомом водорода на терминальном конце. Например, если метиленовый фрагмент -CH2CH2CH3 необязательно замещен -О-, то полученное соединение может представлять собой -OCH2CH3, -CH2OCH3 или -CH2CH2OH. Должно быть понятно, что если терминальный атом не содержит никаких свободных валентных электронов, тогда на терминальном конце не будет атома водорода (например, -CH2CH2CH=O или -CH2CH2C≡N).
[0067] Термин «реакция кросс-сочетания», используемый в настоящем описании, относится к реакции, при которой образуется связь углерод-углерод в присутствии металлического катализатора. Как правило, один из атомов углерода является связанным с функциональной группой («группой кросс-сочетания»), в то время как другой атом углерода связан с галогеном. Примеры реакций кросс-сочетания включают, но не ограничиваются этим, сочетание по Сузуки, сочетание по Стиллу и сочетание по Негиши.
[0068] Термин «группа кросс-сочетания», используемый в настоящем описании, относится к функциональной группе, способной взаимодействовать с другой функциональной группой (например, галогеном) в реакциях кросс-сочетания с образованием связи углерод-углерод («С-С»). В некоторых вариантах осуществления связь С-С образуется между двумя ароматическими группами.
[0069] Термин «условие кросс-сочетания», используемый в настоящем описании, относится к химическим условиям (например, температуре, продолжительности реакции, требуемому объему растворителя), необходимым для протекания реакции кросс-сочетания.
[0070] Примеры групп кросс-сочетания и соответствующих для них условий кросс-сочетания включают, но не ограничиваются этим, бороновые кислоты и эфиры бороновых кислот в условиях сочетания по Сузуки, SnBu3 (Bu: бутил) в условиях сочетания по Стиллу и ZnX (X: галоген) в условиях сочетания по Негиши.
[0071] Все три условия кросс-сочетания обычно включают использование катализатора, подходящего растворителя и, необязательно, основания. В условиях сочетания по Сузуки используют палладиевый катализатор и подходящий растворитель. Примеры подходящих палладиевых катализаторов включают, но не ограничиваются этим, PdCl2(PPh3)2, Pd(Ph3)4 и PdCl2(dppf) (где каждый Ph означает фенил, и dppf представляет собой 1,1-бис(дифенилфосфино)ферроцен). Подходящие основания включают, но не ограничиваются этим, К2СО3 и Na2CO3. Подходящие растворители включают, но не ограничиваются этим, тетрагидрофуран, толуол и этанол.
[0072] Условия сочетания по Стиллу включают использование катализатора (обычно, палладий, но иногда никель), подходящего растворителя и других необязательных реагентов. Примеры подходящих катализаторов включают, но не ограничиваются этим, PdCl2(PPh3)2, Pd(Ph3)4 и PdCl2(dppf). Подходящие растворители включают, но не ограничиваются этим, тетрагидрофуран, толуол и диметилформамид.
[0073] Условия сочетания по Негиши включают использование катализатора (палладий или никель) и подходящего растворителя. Примеры подходящих катализаторов включают, но не ограничиваются этим, Pd2(dba)3, Ni(PPh3)2Cl2, PdCl2(PPh3)2 и Pd(Ph3)4 (где «dba» представляет собой трис(дибензилиденацетон)дипалладий). Подходящие растворители включают, но не ограничиваются этим, тетрагидрофуран, толуол и диметилформамид.
[0074] Условия сочетаний по Сузуки, Стиллу и Негиши являются известными квалифицированному специалисту в данной области и описаны более подробно во множестве ссылочных материалов, включая «Marchʹs Advanced Organic Chemistry».
[0075] Квалифицированному специалисту в данной области должно быть понятно, что группы кросс-сочетания образуются из предшественников групп сочетания. Предшественник групп сочетания представляет собой реагент или группу реагентов, используемых для образования группы кросс-сочетания. Примеры включают, но не ограничиваются этим, бис(пинаколато)диборан с образованием боронатов, триметилбораты с образованием бороновых кислот, Bu3SnCl с образованием станнанов и ZnCl2 с образованием цинкатов в реакциях кросс-сочетания по Негиши. Примеры условий образования подходящей группы сочетания включают, но не ограничиваются этим, получение эфиров бороновых кислот путем опосредуемого палладием катализа; получение бороновых кислот путем гидролиза эфиров бороновых кислот; получение станнанов, используя двухстадийный способ: 1) обмен галогена на металл с последующим 2) трансметаллированием с помощью Bu3SnCl; и получение цинкатов, используя двухстадийный способ: 1) обмен галогена на металл с последующим 2) добавлением ZnCl2.
[0076] Если не указано иное, структуры, изображенные в настоящем описании, также включают все изомерные (например, энантиомерные, диастереомерные, геометрические, конформационные и ротационные) формы этих структур. Например, в данное изобретение включены R- и S-конфигурации каждого асимметрического центра, (Z)- и (E)-изомеры двойной связи и конформационные (Z)- и (E)-изомеры. Квалифицированному специалисту в данной области должно быть понятно, что заместитель может свободно вращаться вокруг любых связей, позволяющих вращение. Например, заместитель, изображенный как  , также представляет собой
, также представляет собой  .
.
[0077] Следовательно, отдельные стереохимические изомеры, а также смеси энантиомеров, диастереомеров, геометрических изомеров, конформационных изомеров и ротационных изомеров соединений согласно настоящему изобретению входят в объем изобретения.
[0078] Если не указано иное, все таутомерные формы соединений согласно изобретению входят в объем данного изобретения.
[0079] В соединениях согласно данному изобретению любой атом, не специально не указанный как являющийся конкретным изотопом, подразумевает любой стабильный изотоп этого атома. Если не указано иное, если положение специально обозначено «Н» или «водород», это означает, что в этом положении находится водород в его естественном изотопном составе. Также, если не указано иное, если положение специально обозначено «D» или «дейтерий», это означает, что в этом положении находится дейтерий в количестве, которое, по меньшей мере, в 3340 раз превышает естественное содержание дейтерия, равное 0,015% (т.е., по меньшей мере, 50,1% включение дейтерия).
[0080] Оба обозначения «D» и «d» относятся к дейтерию.
[0081] Дополнительно, если не указано иное, структуры, изображенные в настоящем описании, также включают соединения, которые отличаются только наличием одного или более изотопно-обогащенных атомов. Например, соединения, имеющие структуры согласно настоящему изобретению, за исключением замещения водорода дейтерием или тритием или замещения углерода обогащенным 13С- или 14С-углеродом, входят в объем изобретения. Такие соединения являются пригодными, например, в качестве аналитических «инструментов» или зондов в биологических тестах.
[0082] Используемый в настоящем описании термин «кристаллический» относится к твердому веществу, которое имеет определенное расположение и/или структуру молекул в кристаллической решетке.
[0083] Используемый в настоящем описании термин «аморфный» относится к твердым формам, которые состоят из беспорядочно расположенных молекул и не обладают различимой кристаллической решеткой.
[0084] Используемый в настоящем описании термин «сольват» относится к кристаллическому твердому аддукту, содержащему либо стехиометрические, либо нестехиометрические количества растворителя, включенного в кристаллическую структуру. Если включенный растворитель представляет собой воду, такой аддукт называется «гидратом».
Аббревиатуры
[0085] Использованы следующие аббревиатуры:
Способы
[0086] Способы и соединения, раскрытые в настоящем описании, являются пригодными для получения ингибиторов ATR, которые содержат аминопиразолопиримидиновое ядро. Общие способы синтеза, показанные на схемах в настоящем описании, являются пригодными для получения большого множества химических соединений, которые можно использовать при получении фармацевтических соединений.
Схема А
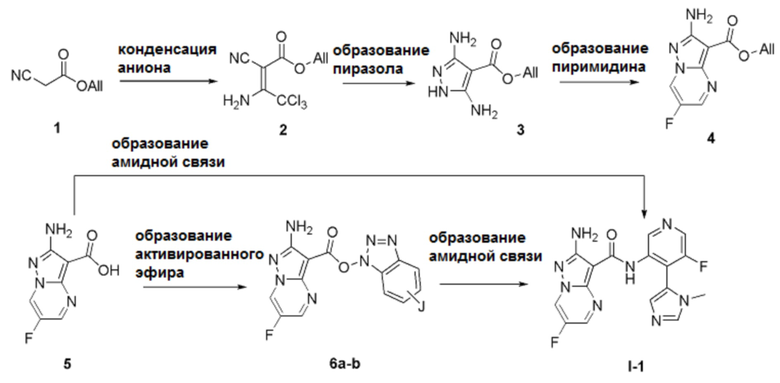
[0087] Соединения согласно данному изобретению можно синтезировать способами, аналогичными изображенному на Схеме А.
Стадия 1
[0088] Анион коммерчески доступного аллилцианоацетата 1 можно вводить во взаимодействие, например, с трихлорацетонитрилом с получением промежуточного продукта 2. На стадии конденсации аниона анион коммерчески доступного аллилцианоацетата 1 можно получать с помощью основания, такого как ацетат калия в соответствующем растворителе, таком как спирт (например, изопропиловый спирт). Анион затем вводят во взаимодействие с трихлорацетонитрилом при комнатной температуре.
Стадия 2
[0089] Промежуточный продукт 2 затем вводят во взаимодействие с гидразином с образованием диаминопиразола 3. На стадии образования пиразола промежуточный продукт 2 вводят во взаимодействие с гидразином (или его гидратом) в апротонном растворителе, таком как ДМФА, с получением диаминопиразола 3. Взаимодействие протекает в основных условиях (например, в присутствии ацетата калия или AcONa) при нагревании (например, при температуре ≥ 110°С) для обеспечения полной циклизации.
Стадия 3
[0090] Промежуточный продукт 3 далее можно конденсировать с диэлектрофильным партнером сочетания с образованием пиримидина 4. На стадии образования пиримидина промежуточный продукт 3 взаимодействует с 1,3-диэлектрофильным соединением (например, 1,3-диальдегидом или 3-(диалкиламино)проп-2-еналем) в различных типах растворителей (например, ДМФА или ДМСО/вода) с получением бициклических ядер 4. Когда один или два электрофильных центра являются защищенными/замаскированными (например, альдегид, замаскированный как кеталь), для высвобождения реакционноспособной функциональной группы необходимо введение сульфоновой кислоты (например, ПТСК).
Стадия 4
[0091] Снятие защиты аллильного сложного эфира, например, посредством гидролиза, приводит к получению карбоновых кислот 5. Стадия снятия защиты соединения 4 происходит в гидролитических условиях, известных квалифицированному специалисту в данной области. Например, обработка соединения 4 фенилсиланом или 4-метилбензолсульфинатом в присутствии каталитического количества палладия (например, Pd(PPh3)4) приводит к образованию соответствующей карбоновой кислоты 5. Альтернативно, соединения 4 можно обрабатывать водным раствором щелочи (например, NaOH, LiOH или KOH) с получением кислот 5.
Стадия 5
[0092] На стадии образования активированного сложного эфира карбоновые кислоты 5 вводят во взаимодействие с агентами сочетания амидов, известными квалифицированному специалисту в данной области. Подходящие партнеры для сочетания амидов включают, но не ограничиваются этим, TBTU, TCTU, HATU, T3P и COMU. Когда агент сочетания выбран подходящим образом, реакции могут проходить быстро (~1 ч) при комнатной температуре в присутствии органического основания, такого как алифатический амин (например, триэтиламин, DIPEA), с получением активированных сложных эфиров 6а-b. Например, когда используются агенты сочетания амидов - TBTU [J=H] или TCTU [J=Cl], соединения 6а-b получают просто путем фильтрации реакционной смеси.
[0093] Образование активированных сложных эфиров 6а-b до образования амидной связи для получения соединения I-A, как правило, является предпочтительным, несмотря на возможность прямого превращения соединения 5 в соединение формулы I-A согласно данному изобретению. Также могут быть использованы альтернативные активированные сложные эфиры (выделенные или образованные in situ), которые должны быть известны квалифицированному специалисту в данной области (например, агенты сочетания - TBTU, TCTU, HATU, T3P, COMU).
Стадия 6
[0094] На стадии образования амидной связи активированные сложные эфиры 6а-b можно вводить во взаимодействие с замещенным 3-аминопиридином 11 с получением соединения I-1 согласно данному изобретению. Реакцию сочетания амидов проводят в растворителе (например, анизоле, NMP, пиридине, ДМФА и т.д.) при нагревании (например, при температуре ≥ 90°С).
[0095] Альтернативно, две описанные выше стадии можно комбинировать: карбоновую кислоту 5 можно использовать в качестве исходных «точек» для образования амидной связи, генерируя активированные сложные эфиры in situ с помощью таких же агентов для сочетания амидов, как описано выше. Соединения согласно данному изобретению выделяют способом, аналогичным описанному выше.
Получение и примеры
[0096] Все коммерчески доступные растворители и реагенты использовали без какой-либо предварительной обработки. Реакции под воздействием микроволнового излучения осуществляли, используя микроволновую печь CEM Discovery. Флэш-хроматографию, например, осуществляли в системе ISCO© CombiflashR Companion™, элюируя в градиенте 0-100% EtOAc/петролейный эфир. Для осуществления флэш-хроматографии также использовали другие способы, известные в данной области. Образцы использовали предварительно абсорбированными на диоксиде кремния. Там, где указано, сверхкритическую флюидную хроматографию (SFC) осуществляли на установке Berger Minigram SFC. Все 1Н-ЯМР-спектры регистрировали, используя прибор Bruker Avance III 500, при 500 МГц. МС-образцы анализировали на масс-спектрометре Waters SQD, работающем в режиме регистрации положительных и отрицательных ионов. Образцы вводили в масс-спектрометр, используя хроматографию. Все конечные продукты имели чистоту ≥ 95%, если в подробно описанных экспериментах не указано иное. Чистоту определяли методом ВЭЖХ, используя систему Waters Acquity UPLC, на приборе Waters SQD MS, оборудованном колонкой Waters UPLC BEH C8, 1,7 мкм, размером 2,1 × 50 мм и защитной колонкой Vanguard BEH C8, 1,7 мкм, размером 2,1 × 5 мм.
[0097] Используемый в настоящем описании термин «Rt(мин)» относится к выраженному в минутах времени удерживания соединения в ВЭЖХ. Если не указано иное, для получения представленных времен удерживания использовали способы ВЭЖХ, как описано ниже:
ВЭЖХ, Способ В
Прибор: Waters Acquity UPLC-MS;
Колонка: Waters UPLC BEH C8, 1,7 мкм, размером 2,1 × 50 мм, с защитной колонкой Vanguard BEH C8, 1,7 мкм, размером 2,1 × 5 мм;
Температура колонки: 45°С;
Подвижная фаза А: 10 мМ формиат аммония в смеси вода:ацетонитрил (95:5), рН=9;
Подвижная фаза В: ацетонитрил;
Детектирование: 210-400 нм;
Градиент: 0-0,40 мин: 2% В; 0,40-4,85 мин: 2%-98% В; 4,85-4,90 мин: 98%-2% В; 4,90-5,00 мин: удерживание при 2% В;
Объемная скорость потока: 0,6 мл/мин.
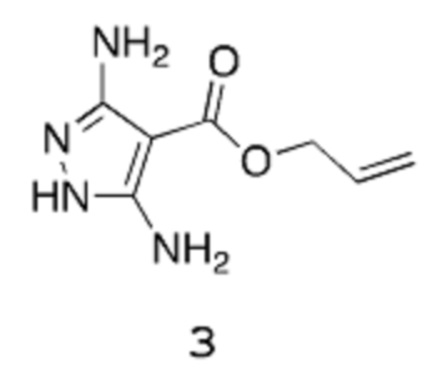
Получение 1: Аллил-3,5-диамино-1Н-пиразол-4-карбоксилат
Стадия 1: Аллил-3-амино-4,4,4-трихлор-2-цианобут-2-еноат 2
[0098] К раствору КОАс (589,4 г, 6,006 моль) в изопропаноле (3 л) добавляют аллилцианоацетат (429,4 г, 403,2 мл, 3,432 моль), и реакционную смесь охлаждают до температуры 5°С. Порциями по 50 мл добавляют трихлорацетонитрил (495,5 г, 3,432 моль), поддерживая температуру ниже 15°С. Реакционную смесь затем оставляют нагреваться до температуры 20°С, перемешивая в течение 3 часов. Добавляют воду (~4 л) для растворения неорганических веществ, при этом происходит осаждение желательного продукта. Смесь перемешивают в течение 20 мин, и твердое вещество выделяют путем фильтрации под вакуумом. Это твердое вещество отфильтровывают, промывают водой (2 раза по 0,5 л) и высушивают в вакууме в течение ночи при температуре 40°С, получая аллил-3-амино-4,4,4-трихлор-2-цианобут-2-еноат 2 в виде не совсем белого порошкообразного вещества (787 г, выход 85%).
Стадия 2: Аллил-3,5-диамино-1Н-пиразол-4-карбоксилат 3
[0099] К суспензии аллил-3-амино-4,4,4-трихлор-2-цианобут-2-еноата 2 (619 г, 2,297 моль) и КОАс (676,3 г, 6,891 моль) в ДМФА (2,476 л) при температуре 0°С медленно добавляют гидразингидрат (172,5 г, 167,6 мл, 3,446 моль) в течение 15 мин. Реакционную смесь затем перемешивают при температуре окружающей среды в течение 2 часов, на стадии которой 1Н-ЯМР показывает, что исходное вещество прореагировало полностью. Реакционную смесь затем нагревают при температуре 110°С в течение ночи, после чего охлаждают до температуры окружающей среды, перемешивая в течение следующих 48 часов. Смесь отфильтровывают через воронку из пористого стекла для удаления осажденного твердого вещества, и фильтрат выпаривают при пониженном давлении, получая густую жидкость. Добавляют ДХМ (приблизительно 2 л), и смесь снова отфильтровывают для удаления дополнительных твердых веществ, выпавших в осадок. Фильтрат очищают, пропуская через слой силикагеля массой 1 кг (градиент ДХМ/МеОН в качестве элюента), и растворитель удаляют, получая твердое вещество оранжевого цвета, которое суспендируют в ацетонитриле и нагревают при температуре примерно 70°С до тех пор, пока все твердые вещества не перейдут в раствор, после чего раствор оставляют охлаждаться до температуры окружающей среды, затем охлаждают до температуры 2°С. Образовавшийся осадок выделяют вакуумной фильтрацией, промывают охлажденным MeCN (~50 мл) и высушивают до постоянной массы в вакуумном шкафу, получая указанное в заголовке соединение в виде не совсем белого порошкообразного вещества (171,2 г, выход 41%).
Получение 2а: 1Н-Бензо[d][1,2,3]триазол-1-ил-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилат
Стадия 1: Аллил-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилат 4
[00100] К суспензии аллил-3,5-диамино-1Н-пиразол-4-карбоксилата 3 (42,72 г, 234,5 ммоль) в смеси ДМСО (270,8 мл)/вода (270,8 мл) добавляют гидрат п-TsOH (46,72 г, 245,6 ммоль) и 3-(диизопропиламино)-2-фторпроп-2-еналь (описанный в Tetrahedron Letters, 33(3), 357-60, 1992) (38,69 г, 223,3 ммоль). Реакционную смесь нагревают до температуры 100°С в течение 3 часов, в течение которых твердое вещество медленно осаждается из раствора. Суспензию оранжевого цвета оставляют на ночь охлаждаться до комнатной температуры. Твердое вещество отфильтровывают, промывают водой и высушивают в вакууме, получая аллил-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилат 4 в виде твердого вещества песочного цвета (45,05 г, выход 85%).
Стадия 2: 2-Амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоновая кислота 5
[00101] К суспензии аллил-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилата 4 (45 г, 190,5 ммоль) в ДХМ (1,35 л) добавляют фенилсилан (41,23 г, 46,96 мл, 381,0 ммоль), затем Pd(PPh3)4 (8,805 г, 7,620 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов 30 мин. Реакционную смесь отфильтровывают, и твердое вещество промывают ДМХ, получая твердое вещество светло-желтого цвета (43,2 г). Это твердое вещество далее растирают в ДХМ (225 мл) при комнатной температуре в течение 45 мин, потом отфильтровывают и высушивают в течение ночи в вакууме, получая 2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоновую кислоту 5 в виде твердого вещества светло-желтого цвета (37,77 г, выход 100%).
[00102] В альтернативном способе, 4-метилбензолсульфинат (безводный, 1,2 экв., 22,6 г, 127 ммоль) суспендируют в безводном ДМСО (20 об., 500 мл). Перемешиваемую смесь нагревают до температуры 30°С в атмосфере азота. После завершения растворения добавляют Pd(PPh3)4 (2% мол., 2,4 г, 2,1 ммоль). Смесь перемешивают в течение 10 мин при температуре 25-30°С, после чего образуется мутный раствор желтого цвета. Порциями добавляют аллил-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилат 4 (25 г, 105,8 ммоль), при этом поддерживая температуру в пределах 25-30°С. После завершения добавления мутный раствор перемешивают до тех пор, пока, согласно данным ВЭЖХ, не завершится реакция (2-3 часа). Спустя 15 мин после добавления субстрата образуется обильный осадок. Смесь становится более густой, так как продолжается реакция. Реакционную смесь разбавляют водой (125 мл), и медленно добавляют 2 М раствор HCl (66 мл), все время поддерживая температуру в пределах 25-30°С. Суспензию перемешивают в течение 30 мин, затем отфильтровывают. Процесс фильтрования проводят медленно (2 часа). Полученное твердое вещество промывают водой, потом высушивают в печи для спекания. Это твердое вещество суспендируют в ДХМ (8 об.) в течение 1 часа. Твердое вещество отфильтровывают (быстрая фильтрация) и промывают ДХМ. Твердое вещество повторно суспендируют в хлороформе (8 об.) в течение 1 часа. Кислоту отфильтровывают и высушивают в печи для спекания. Этот продукт далее высушивают в вакуумном сушильном шкафу при температуре 50°С в течение 24 часов. Продукт 5 получают в виде не совсем белого твердого вещества (18,6 г, выход 85%); 1Н-ЯМР (500 МГц, ДМСО-d6): δ 12,14 (1Н, уш.с), 9,31 (1Н, дд), 8,69 (1Н, м), 6,47 (2Н, уш.с); 19F-ЯМР (500 МГц, ДМСО-d6): δ -153,65; MS (ES+): 197,1.
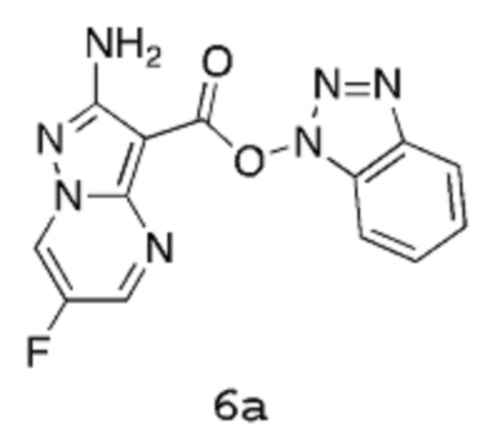
Стадия 3: 1Н-Бензо[d][1,2,3]триазол-1-ил-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилат 6а
[00103] К суспензии 2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоновой кислоты 5 (20 г, 102,0 ммоль) в хлороформе (300 мл) добавляют Et3N (11,35 г, 15,63 мл, 112,2 ммоль). Суспензию перемешивают в течение ~5 мин, и затем добавляют (бензотриазол-1-илоксидиметиламинометилен)диметиламмоний бортетрафторид (32,75 г, 102,0 ммоль). Суспензию нагревают до температуры 60°С в течение 1 часа, а затем густую суспензию оставляют охлаждаться до комнатной температуры. Полученную суспензию отфильтровывают, промывают хлороформом (200 мл) и высушивают в вакууме в течение ночи, получая указанное в заголовке соединение 6а в виде порошкообразного вещества светло-желтого цвета (32,5 г, выход 88%).
Получение 2b: (6-Хлорбензотриазол-1-ил)-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилат 6b
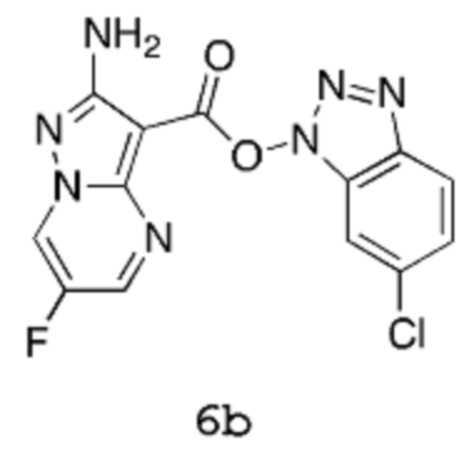
[00104] В трехгорлую колбу емкостью 2,5 л, оснащенную мешалкой, конденсатором, линией подачи азота и датчиком температуры Hanna, вводят 2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоновую кислоту 5 (60 г, 305,9 ммоль), хлороформ (900,0 мл) и триэтиламин (32,44 г, 44,68 мл, 320,6 ммоль). Порциями в течение 5 мин добавляют [(6-хлорбензотриазол-1-ил)окси(диметиламино)метилен]диметиламмоний (бортетрафторид-ион (1)) (87,00 г, 244,7 ммоль) (в процессе добавления внутренняя температура снижается от 22,7°С до 21,5°С). Смесь нагревают при температуре 60°С (внутренняя температура) в течение 2 часов, которая все еще сохраняется в виде суспензии кремового цвета. Смесь охлаждают до комнатной температуры, затем твердое вещество собирают методом фильтрации, хорошо промывают хлороформом (пока фильтрат не станет по существу бесцветным) и сушат путем отсасывания, получая продукт 6b в виде твердого вещества кремового цвета (82,2 г, выход 77%). 1Н-ЯМР (500 МГц, ДМСО-d6): δ 9,55 (дд, 1Н), 8,91 (д, 1Н), 8,22 (дд, 1Н), 8,09 (дд, 1Н), 7,57 (дд, 1Н) и 6,87 (с, 2Н). MS (ES+): 348,1.
[00105] В альтернативном способе 2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоновую кислоту 5 (30 г, 153 ммоль) суспендируют в ацетонитриле (540 мл). Добавляют триэтиламин (22,5 мл, 153 ммоль), затем [(6-хлорбензотриазол-1-ил)окси(диметиламино)метилен]диметиламмоний тетрафторборат (TCTU, 54,4 г, 153 ммоль). Смесь перемешивают при комнатной температуре в течение 2 часов. Продукт выделяют путем фильтрации, фильтровальный осадок промывают ацетонитрилом (2 раза по 60 мл). Продукт получают в виде твердого вещества коричневого цвета (49,3 г, выход 93%). 1Н-ЯМР (500 МГц, ДМСО-d6): δ 9,55 (дд, 1Н), 8,91 (д, 1Н), 8,22 (дд, 1Н), 8,09 (дд, 1Н), 7,57 (дд, 1Н) и 6,87 (с, 2Н); 19F-ЯМР (500 МГц, ДМСО-d6): δ -150,1; MS (ES+): 348,1.
Пример 1: Синтез 2-амино-6-фтор-N-(5-фтор-4-(1-метил-1Н-имидазол-5-ил)пиридин-3-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (Соединение I-1 )
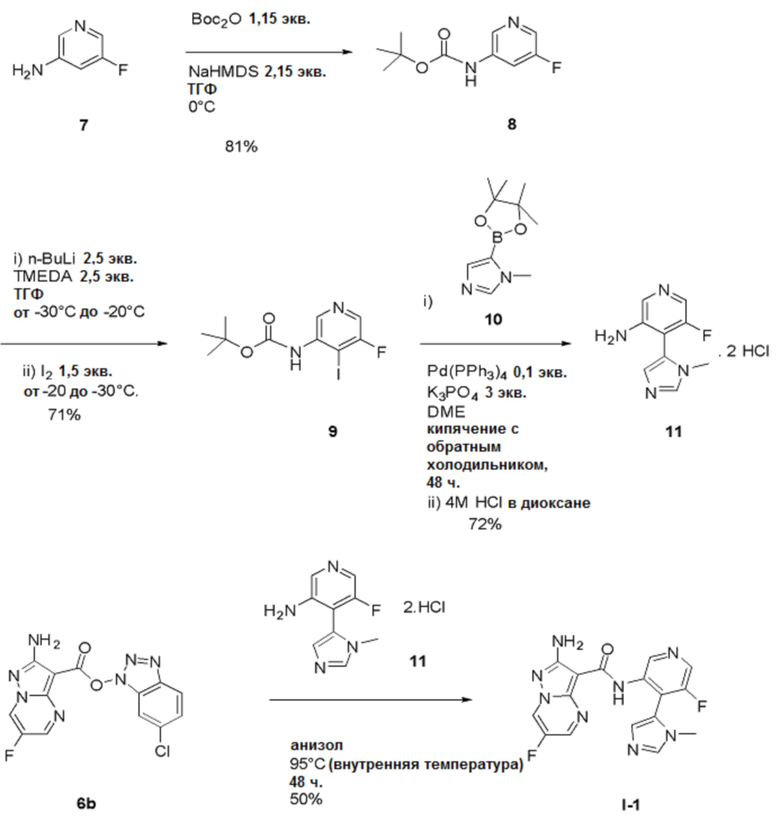
Стадия 1: трет-Бутил(5-фторпиридин-3-ил)карбамат
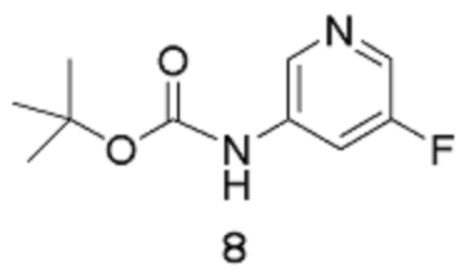
[00106] В заключенный в кожух сосуд емкостью 50 л вводят ТГФ (2,5 л), 5-фторпиридин-3-амин 1 (500 г, 4,460 моль), затем дополнительное количество ТГФ (5 л). К этой перемешиваемой смеси добавляют раствор трет-бутоксикарбонил-трет-бутилкарбоната (1,119 кг, 5,129 моль) в ТГФ (2,5 л), подаваемый насосом через вакуумную линию. Линию потом промывают ТГФ (1 л) до реакционного сосуда. Температуру реакции снижают до 0°С перед добавлением NaHMDS (4,794 л 2 М раствора в ТГФ, 9,589 моль) в виде 12 порций по 400 мл (после каждого добавления температура повышается приблизительно на 5°С, дозирование продолжают после охлаждения до внутренней температуры 0°С). Добавление завершается спустя 1 час. Внутреннюю температуру повышают до 5°С, и смесь перемешивают при этой же температуре в течение 1 часа. Реакцию осторожно останавливают путем медленного добавления насыщенного водного раствора хлорида аммония (1 л, экзотермическая реакция). Внутреннюю температуру повышают до 10°С, и добавляют дополнительное количество насыщенного водного раствора хлорида аммония (3 л). Внутреннюю температуру повышают до 25°С, и реакционную смесь экстрагируют EtOAc (1 х 5 л, затем 1 х 2,5 л). Объединенные органические слои промывают водой (1 х 5,5 л, затем 1 х 3 л), потом насыщенным раствором хлорида натрия (3 л).
[00107] Органическую фазу концентрируют в вакууме до общего объема, равного приблизительно 6 л, сушат (MgSO4), отфильтровывают через фильтровальную бумагу и концентрируют в вакууме (на роторном испарителе, температура бани составляет 40°С) до тех пор, пока продукт не выкристаллизовывается (остается приблизительно 2 л растворителя). Добавляют гептан (2,5 л), и смесь вращают на роторном испарителе при температуре 40°С. Раствор концентрируют в вакууме (на роторном испарителе, температура бани составляет 40°С) для удаления большего количества EtOAc до тех пор, пока продукт не выкристаллизовывается из раствора. Смесь затем оставляют охлаждаться и отстаиваться при температуре окружающей среды в течение ночи. Твердое вещество собирают путем фильтрации через фильтровальную бумагу «Ватман №1», промывают гептаном до тех пор, пока фильтрат, по существу, не станет бесцветным. Твердое вещество высушивают в течение приблизительно 5 часов, получая выход 1 продукта в виде не совсем белого твердого вещества, равный 382,51 г.
[00108] Маточный раствор медленно концентрируют в вакууме (на роторном испарителе, температура бани составляет 40°С) до тех пор, пока твердое вещество не выкристаллизовывается. Смесь оставляют отстаиваться при температуре окружающей среды в течение ночи, и твердое вещество собирают путем фильтрации, промывают гептаном и высушивают путем отсасывания, получая выход 2 продукта 8 в виде не совсем белого твердого вещества, равный 203,5 г. Процесс с использованием маточного раствора повторяют, получая выход 3 продукта в виде не совсем белого твердого вещества, равный 178,7 г. Общий выход продукта составляет 764,71 г, 81%. 1Н-ЯМР (500 МГц, ДМСО-d6): δ 9,86 (с, 1Н), 8,44 (с, 1Н), 8,17 (д, J=2,6 Гц, 1Н), 7,83 (д, J=11,6 Гц, 1Н), 3,30 (с, 1Н). MS (ES+): 213,0.
Стадия 2: трет-Бутил(5-фтор-4-иодпиридин-3-ил)карбамат
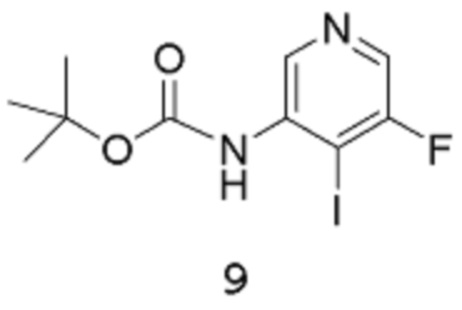
[00109] В заключенный в кожух сосуд емкостью 50 л вводят ТГФ (2,5 л), трет-бутил-N-(5-фтор-3-пиридил)карбамат 8 (400 г, 1,885 моль) в ТГФ (2,5 л), дополнительное количество ТГФ (3 л) и N,N,Nʹ,Nʹ-тетраметилэтан-1,2-диамин (547,6 г, 711,2 мл, 4,712 моль). Реакционную смесь охлаждают до температуры -28°С (внутренняя температура), затем добавляют n-BuLi (1,885 л 2,5 М раствора в гексане, 4,712 моль) посредством канюли с такой скоростью, чтобы внутренняя температура сохранялась ниже -20°С (т.е., в течение 2 часов). После завершения добавления реакционную смесь перемешивают при температуре между -30°С и -20°С (внутренняя температура) в течение следующих 50 мин. Медленно примерно 12 эквивалентными порциями добавляют твердый молекулярный йод (765,5 г, 3,016 моль) в течение 1 часа (после каждой добавленной порции температура через некоторое время повышается приблизительно на 2/3°С), поддерживая внутреннюю температуру ниже -20°С. После завершения добавления йода реакционную смесь перемешивают при температуре -30°С (внутренняя температура) в течение следующих 45 мин.
[00110] Реакцию останавливают путем медленного добавления насыщенного водного раствора хлорида аммония (2 л) (экзотермическая реакция). Потом добавляют воду (2 л), и реакционную смесь нагревают до температуры 20°С (внутренняя температура) и оставляют отстаиваться в течение ночи. К реакционной смеси добавляют EtOAc (5 л) и продолжают перемешивать в течение 10 мин. Водную фазу удаляют, затем к органической фазе добавляют насыщенный водный раствор тиосульфата натрия (2 л), энергично перемешивают в течение 10 мин. Добавляют дополнительное количество EtOAc (2,5 л) и воды (2 л), и продолжают перемешивать в течение 10 мин. Водную фазу удаляют, а органическую фазу далее промывают насыщенным водным раствором тиосульфата натрия (2 л) и водой (1 х 2 л, затем 1 х 2,5 л) и затем - насыщенным раствором хлорида натрия (2 л). Органическую фазу концентрируют в вакууме (роторный испаритель) до объема, при котором происходит кристаллизация продукта с получением густой суспензии. Смесь оставляют отстаиваться при комнатной температуре в течение ночи.
[00111] Твердое вещество собирают путем фильтрации, промывают минимальным количеством EtOAc (несколько сот мл), затем хорошо промывают гептаном, высушивают путем отсасывания в течение 3 часов, получая выход 1 продукта 9 в виде твердого вещества белого цвета, равный 311,99 г. Маточный раствор концентрируют в вакууме (роторный испаритель) досуха, получая твердое вещество темно-зеленого цвета (приблизительно 200 г), которое растворяют в EtOAc (750 мл) при кипячении с обратным холодильником. Затем добавляют активированный уголь (20 г), и смесь перемешивают при кипячении с обратным холодильником в течение 10 мин. Смесь отфильтровывают через фильтровальную бумагу, потом медленно концентрируют на роторном испарителе до тех пор, пока не образуется густая суспензия. Полученное твердое вещество собирают путем фильтрации, промывают минимальным количеством EtOAc, затем гептаном, высушивают путем отсасывания, затем в вакуумном шкафу при температуре 40°С в течение 2 часов, получая выход 2 продукта в виде твердого вещества белого цвета, равный 103,9 г. Маточный раствор снова концентрируют до тех пор, пока не образуется густая суспензия. Твердое вещество собирают путем фильтрации, промывают гептаном и высушивают путем отсасывания в вакууме (роторный испаритель), затем в вакуумном шкафу при температуре 40°С в течение нескольких часов, получая выход 3 продукта в виде твердого вещества белого цвета, равный 39,4 г. Общий выход составляет 455,29 г, 71%. 1Н-ЯМР (500 МГц, ДМСО-d6): δ 8,98 (с, 1Н), 8,27 (дд, J=1,2, 0,6 Гц, 2Н), 1,47 (с, 9Н). MS (ES+): 338,9.
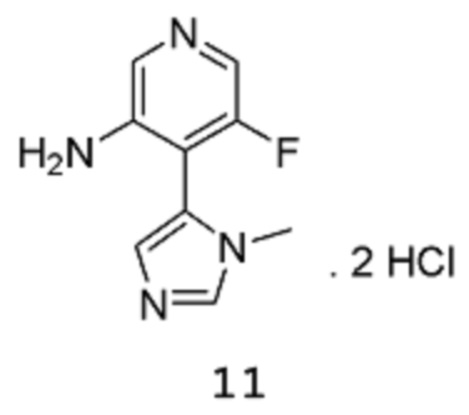
Стадия 3: 5-Фтор-4-(1-метил-1Н-имидазол-5-ил)пиридин-3-аминдигидрохлорид
[00112] К дегазированной (3 цикла вакуум/азот) смеси трет-бутил-N-(5-фтор-4-иод-3-пиридил)карбамата 9 (190 г, 561,9 ммоль), 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)имидазола 10 (175,4 г, 842,8 ммоль) и фосфата калия (226,0 г, 1,686 ммоль) в DME (2,28 л) добавляют Pd(PPh3)4 (64,93 г, 56,19 ммоль). Реакционный сосуд снова продувают азотом циклами вакуум/азот (3 раза). Смесь кипятят с обратным холодильником в атмосфере азота в течение 48 часов. Смесь охлаждают до комнатной температуры, затем пропускают через слой целита, промывают EtOAc до тех пор, пока фильтрат не станет практически бесцветным (приблизительно 1,5 л). Фильтрат концентрируют в вакууме, получая 339,7 г вязкого твердого вещества коричневого цвета.
[00113] Сырой продукт растворяют в диоксане (950 мл) и метаноле (431,1 мл), и раствор охлаждают на бане со льдом (внутренняя температура составляет 10°С), потом 8 примерно эквивалентными порциями добавляют HCl (4 М раствор в 1,4-диоксане) (842,8 мл 4 М раствора, 3,371 моль) в течение 20 мин (после каждого добавления происходит повышение температуры приблизительно на 3-4°С). После завершения добавления смесь нагревают до температуры 40°С и перемешивают при этой же температуре в течение 3 часов, затем оставляют охлаждаться до комнатной температуры в течение ночи при перемешивании. Твердое вещество собирают путем фильтрации, промывают 1,4-диоксаном и высушивают в вакууме в течение 1 часа, получая продукт 11 в виде твердого вещества песочного/коричневого цвета (107,9 г, выход 72%). 1Н-ЯМР (500 МГц, оксид дейтерия): δ 9,09 (с, 1Н), 8,24 (с, 1Н), 8,15 (уш.с, 1Н), 7,91-7,90 (1Н, уш.с), 7,88 (м, 1Н), 3,85 (с, 3Н). MS (ES+): 193,1.
Стадия 4: 2-Амино-6-фтор-N-(5-фтор-4-(1-метил-1Н-имидазол-5-ил)пиридин-3-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
(Соединение I-1)
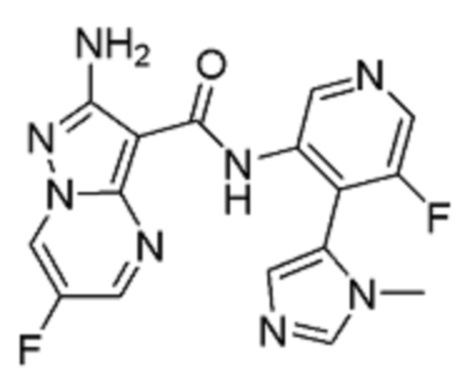
[00114] Смесь 5-фтор-4-(3-метилимидазол-4-ил)пиридин-3-аминдигидрохлорида 11 (8,006 г, 30,2 ммоль) и (6-хлорбензотриазол-1-ил)-2-амино-6-фторпиразоло[1,5-a]пиримидин-3-карбоксилата 6b (10 г, 28,76 ммоль) суспендируют в анизоле (100 мл). К этой суспензии добавляют DIPEA (8,177 г, 11,02 мл, 63,27 ммоль), и смесь нагревают при температуре 95°С (внутренняя температура) в течение 44 часов, затем оставляют охлаждаться до комнатной температуры в течение ночи. Твердое вещество собирают путем фильтрации, промывают минимальным количеством анизола (приблизительно 20 мл), высушивают в вакууме в течение 1 часа, затем твердое вещество высушивают в вакуумном шкафу при температуре 45°С (внутренняя температура) в течение 2 часов, получая 7,8 г продукта в виде твердого вещества светло-желтого цвета. Это твердое вещество суспендируют в воде (78 мл) и MeCN (117 мл) и добавляют ТФУ (2,4 г, 1,62 мл, 1 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 10 мин, затем отфильтровывают через фильтровальную бумагу, промывают небольшим количеством воды. Фильтрат подщелачивают до рН=8 добавлением 2 М раствора карбоната натрия при постоянном перемешивании. Твердое вещество собирают путем фильтрации, промывают водой, затем высушивают в вакууме в течение 1 часа. Твердое вещество потом высушивают в вакуумном шкафу при температуре 45°С (внутренняя температура) в течение ночи, получая 5,29 г продукта I-1 в виде твердого вещества бледно-желтого цвета. 1Н-ЯМР (500 МГц, ДМСО-d6): δ 9,68 (с, 2Н), 9,42 (дд, J=4,8, 2,5 Гц, 1Н), 8,46 (с, 1Н), 8,31 (д, J=2,5 Гц, 1Н), 8,07 (с, 1Н), 7,25 (д, J=1,0 Гц, 1Н), 6,71 (с, 2Н), 3,46 (с, 3Н). MS (ES+): 371,0.
Аналитические данные соединения
Твердые формы Соединения I-1
[00115] Соединение I-1 получают в различных твердых формах, включая безводные формы. Твердые формы согласно настоящему изобретению являются пригодными в производстве лекарственных средств для лечения ракового заболевания. Одно из воплощений относится к применению твердой формы, раскрытой в настоящем описании, для лечения ракового заболевания. В некоторых воплощениях раковое заболевание представляет собой тройной негативный рак молочной железы, рак поджелудочной железы, мелкоклеточный рак легких, колоректальный рак, рак яичников или немелкоклеточный рак легких. Другое воплощение относится к фармацевтической композиции, содержащей твердую форму, раскрытую в настоящем описании, и фармацевтически приемлемый носитель.
[00116] В настоящем описании раскрыта новая твердая форма Соединения I-1. Название и стехиометрия твердой формы представлены в Таблице 2, ниже.
Таблица 2
Пример 2: Соединение I-1 (безводное свободное основание)
[00117] Соединение I-1 (безводное свободное основание) можно получать согласно способам, описанным в Примере 1, стадия 4.
XRPD соединения I-1 (безводное свободное основание)
[00118] Порошковую рентгеновскую дифрактограмму соединения I-1 в виде безводного свободного основания регистрируют при комнатной температуре в режиме отражения, используя дифрактометр PANalytical, снабженный источником - трубкой Empyrean и детектором PIXcel 1D (PANalytical, Нидерланды). Источник рентгеновских лучей работает при напряжении 45 кВ и силе тока 40 мА. Порошкообразный образец помещают на силиконовый держатель. Данные регистрируют в диапазоне 3°-39° 2-тета с размером шага 0,013° и временем измерения 0,5 с на шаг. На Фиг.1а представлена порошковая рентгеновская дифрактограмма образца, характерная для кристаллического лекарственного вещества.
[00119] XRPD характеристические пики Соединения I-1 в виде безводного свободного основания:
Термоанализ соединения I-1 (безводное свободное основание)
[00120] Термогравиметрический анализ (ТГА) соединения I-1 в виде безводного свободного основания осуществляют для определения потери массы в процентах в виде функции от температуры, используя прибор Discovery TGA (TA Instruments Trios). Образец (2,84 мг) добавляют в предварительно взвешенную алюминиевую кювету и нагревают от комнатной температуры до температуры 400°С со скоростью 10°С/мин. Результаты ТГА, представленные на Фиг.2а, показывают очень незначительную потерю массы, наблюдаемую до начала плавления или термического разложения. При нагревании от комнатной температуры до температуры 261°С потеря массы составляет 0,60%. Начальная температура плавления/деструкции составляет 299°С.
Дифференциальная сканирующая калориметрия соединения I-1 (безводное свободное основание)
[00121] Дифференциальную сканирующую калориметрию (ДСК) соединения I-1 в виде безводного свободного основания осуществляют, используя устройство TA Instrument DSC Q2000. Образец (1,71 мг) взвешивают в точечно проколотой герметической алюминиевой кювете и нагревают от комнатной температуры до температуры 400°С со скоростью 10°С/мин. Результаты ДСК, представленные на Фиг.3а, показывают одиночную эндотерму плавления при температуре 302°С (начало).
Пример 3: Соединение I-1 (гидрат)
[00122] Соединение I-1 в виде безводного свободного основания, полученное способом, описанным в Примере 1, стадия 4, суспендируют в воде или смесях органического растворителя с водой с получением гидрата соединения I-1.
XRPD Соединения I-1 (гидрат)
[00123] Порошковую дифрактограмму соединения I-1 в виде гидрата регистрируют при комнатной температуре в режиме отражения, используя дифрактометр PANalytical, снабженный источником - трубкой Empyrean и детектором PIXcel 1D (PANalytical, Нидерланды). Источник рентгеновских лучей работает при напряжении 45 кВ и силе тока 40 мА. Порошкообразный образец помещают на силиконовый держатель. Данные регистрируют в диапазоне 3°-39° 2-тета с размером шага 0,013° и временем измерения 0,5 с на шаг. На Фиг.1b представлена порошковая рентгеновская дифрактограмма образца, которая характерна для кристаллического лекарственного вещества.
[00124] XRPD характеристические пики Соединения I-1 в виде гидрата:
Термоанализ соединения I-1 (гидрат)
[00125] Термогравиметрический анализ (ТГА) соединения I-1 в виде гидрата осуществляют для определения потери массы в процентах в виде функции от температуры, используя прибор Discovery TGA (TA Instruments Trios). Образец (4,74 мг) добавляют в предварительно взвешенную алюминиевую кювету и нагревают от комнатной температуры до температуры 400°С со скоростью 10°С/мин. Согласно данным ТГА, представленным на Фиг.2b, ниже 100°С происходит большая потеря массы, равная 12,6%. Эта потеря массы соответствует приблизительно 3 молярным эквивалентам воды. Последующая потеря массы при температуре выше 250°С является результатом плавления и разложения.
Дифференциальная сканирующая калориметрия соединения I-1 (гидрат)
[00126] Дифференциальную сканирующую калориметрию (ДСК) соединения I-1 в виде гидрата осуществляют, используя TA Instrument DSC Q2000. Образец (2,78 мг) взвешивают в точечно проколотой герметической алюминиевой кювете и нагревают от комнатной температуры до температуры 370°С со скоростью 10°С/мин. Результаты ДСК, представленные на Фиг.3b, показывают широкую эндотерму десольватации ниже температуры 100°С с последующей экзотермической рекристаллизацией до соединения I-1 в виде безводного свободного основания при температуре между 100-150°С. Пик эндотермы при температуре между 300-305°С указывает на плавление соединения I-1 в виде безводного свободного основания.
Пример 4: Соединение I-1 (винная кислота)
[00127] Соединение I-1 в виде безводного свободного основания, полученное способом, описанным в Примере 1, стадия 1, суспендируют с винной кислотой и этанолом с получением соединения I-1 в виде аддукта с винной кислотой.
XRPD соединения I-1 (винная кислота)
[00128] Порошковую рентгеновскую дифрактограмму соединения I-1 в виде аддукта с винной кислотой регистрируют при комнатной температуре в режиме отражения, используя дифрактометр PANalytical, снабженный источником - трубкой Empyrean и детектором PIXcel 1D (PANalytical, Нидерланды). Источник рентгеновских лучей работает при напряжении 45 кВ и силе тока 40 мА. Порошкообразный образец помещают на силиконовый держатель. Данные регистрируют в диапазоне 4,5°-39° 2-тета, с размером шага 0,013° и временем измерения 299,6 с на шаг. На Фиг.1с представлена порошковая рентгеновская дифрактограмма, характерная для кристаллического лекарственного вещества.
[00129] XRPD характеристические пики Соединения I-1 в виде аддукта с винной кислотой:
Термоанализ соединения I-1 (винная кислота)
[00130] Термогравиметрический анализ (TGA) соединения I-1 в виде аддукта с винной кислотой осуществляют для определения потери массы в процентах в виде функции от температуры, используя прибор Discovery TGA (TA Instruments Trios). Образец (3,35 мг) добавляют в предварительно взвешенную алюминиевую кювету и нагревают от комнатной температуры до температуры 300°С со скоростью 10°С/мин. Согласно данным ТГА, представленным на Фиг.2с, потеря массы происходит в три этапа - 12,4%, 12,6% и 8,55%, при температуре между 150-330°С.
Дифференциальная сканирующая калориметрия соединения I-1 (винная кислота)
[00131] Дифференциальную сканирующую калориметрию (ДСК) соединения I-1 в виде аддукта с винной кислотой осуществляют, используя TA Instrument DSC Q2000. Образец (1,08 мг) взвешивают в точечно проколотой герметической алюминиевой кювете и нагревают от комнатной температуры до температуры 350°С со скоростью 10°С/мин. Результаты ДСК, представленные на Фиг.3с, показывают первые 2 экзотермических пика при температуре между 200°С и 275°С, соответствующие первым 2 этапам потери массы при ТГА, и последний эндотермический пик при температуре выше 275°С, соответствующий потере массы последнего этапа при ТГА.
Пример 5: Анализ ингибирования клеточного ATR
[00132] Соединения могут быть подвергнуты скринингу в отношении их способности ингибировать внутриклеточный ATR, используя метод иммунофлуоресцентной микроскопии, для детектирования фосфорилирования гистона Н2АХ субстрата ATR в клетках, обработанных гидроксимочевиной. Клетки НТ29 высевали по 14000 клеток на лунку в 96-луночные планшеты черного цвета для визуализации (BD 353219) на среды McCoyʹs 5А (Sigma M8403), дополненные 10% фетальной бычьей сывороткой (JRH Biosciences 12003), раствором пенициллин/стрептомицин, разведенным в соотношении 1:100 (Sigma P7539), и 2 мМ L-глутамином (Sigma G7513), и оставляли на ночь для адгезии при температуре 37°С в 5% СО2. Соединения затем добавляли к клеточным средам с конечной концентрацией 25 мкМ в трехкратных последовательных разведениях, и клетки инкубировали при температуре 37°С в 5% СО2. Спустя 15 минут добавляли гидроксимочевину (Sigma H8627) до конечной концентрации 2 мМ.
[00133] После обработки гидроксимочевиной в течение 45 минут клетки промывали в PBS, фиксировали в течение 10 минут в 4% формальдегиде, разведенном в PBS (Polysciences Inc 18814), промывали в 0,2% Tween-20 в PBS (буфер для промывки) и пермеабилизировали в течение 10 минут в 0,5% Triton X-100 в PBS; все процессы осуществляли при комнатной температуре. Клетки затем промывали один раз в буфере для промывки и блокировали в течение 30 минут при комнатной температуре в 10%-ной козьей сыворотке (Sigma G9023), разведенной в буфере для промывки (буфер для блокирования). Для детектирования уровней фосфорилирования Н2АХ клетки затем инкубировали в течение 1 часа при комнатной температуре с первичным антителом (мышиное моноклональное антитело против фосфорилированного по Ser139 гистона Н2АХ ; Upstate 05-636), разведенным в соотношении 1:250, в буфере для блокирования. Клетки затем промывали пять раз в буфере для промывки перед инкубацией в течение 1 часа при комнатной температуре в темноте в смеси вторичного антитела (конъюгированное с Alexa Fluor 488 козье антитело к мышиным антигенам ; Invitrogen A11029) и красящего вещества Hoechst (Invitrogen H3570), разведенной в соотношении 1:500 и 1:5000, соответственно, в буфере для промывки. Клетки затем промывали пять раз в буфере для промывки и, в заключение, перед визуализацией в каждую лунку добавляли по 100 мкл PBS.
[00134] Клетки визуализировали по интенсивности Alexa Fluor 488 и Hoechst, используя программное обеспечение BD Pathway 855 и Attovision (BD Biosciences, версия 1.6/855), для определения количества фосфорилированного Р2АХ и окрашивания ДНК, соответственно. Затем для каждой лунки рассчитывали процент фосфорилированных Р2АХ-положительных ядер в монтаже 9-ти изображений при 20-кратном увеличении, используя программное обеспечение BD Image Data Explorer (BD Biosciences, версия 2.2.15). Фосфорилированные Н2АХ-положительные ядра определяли как представляющие интерес Hoechst-положительные области, содержащие Alexa Fluor 488 с интенсивностью в 1,75 раз, превышающей среднюю интенсивность Alexa Fluor 488 в клетках, не подвергнутых обработке гидроксимочевиной. В заключение, строили график процента Н2АХ-положительных ядер по отношению к концентрации для каждого соединения, и значения IC50 для ингибирования внутриклеточного ATR определяли, используя программное обеспечение Prism (GraphPad Prism, версия 3.0сх для Macintosh, программное обеспечение GraphPad, San Diego, California, USA).
[00135] Соединения, раскрытые в настоящем описании, также могут быть протестированы другими способами, известными в уровне техники (см. Sarkaria и др., «Inhibition of ATM and ATR Kinase Activities by the Radiosensitizing Agent, Caffeine»: Cancer Research 59: 4375-5382 (1999); Hickson и др., «Identification und Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM»: Cancer Research 64: 9152-9159 (2004); Kim и др., «Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members»: The Journal of Biological Chemistry, 274(53): 37538-37543 (1999); и Chiang и др., «Determination of the catalytic activities of mTOR and other members of the phosphoinositide-3-kinase-related kinase family»: Methods Mol. Biol. 281:125-41 (2004)).
Пример 6: Анализ ингибирования ATR
[00136] Соединения могут быть подвергнуты скринингу в отношении их способности ингибировать ATR-киназу, используя анализ включения радиоактивного фосфата. Анализ осуществляли в смеси 50 мМ Tris/HCl (рН 7,5), 10 мМ MgCl2 и 1 мМ DTT. Конечные концентрации субстрата составляли 10 мкМ [γ-33Р]АТФ (3 мКи 33Р АТФ/ммоль АТФ, Perkin Elmer) и 800 мкМ пептида-мишени (ASELPASQPQPFSAKKK).
[00137] Анализ проводили при температуре 25°С в присутствии 5 нМ полноразмерного ATR. Готовили раствор исходного буфера для анализа, содержащий все из вышеперечисленных реагентов, за исключением АТФ и представляющего интерес тестируемого соединения. 13,5 мкл исходного раствора помещали в 96-луночный планшет с последующим добавлением 2 мкл исходного ДМСО, содержащего последовательные разведения тестируемого соединения (как правило, исходя из конечной концентрации 15 мкМ с 3-кратными последовательными разведениями) с тройным повтором (конечная концентрация ДМСО составляет 7%). Планшет предварительно инкубировали в течение 10 минут при температуре 25°С, и реакцию инициировали путем добавления 15 мкл [γ-33Р]АТФ (конечная концентрация составляет 10 мкМ).
[00138] Реакцию останавливали через 24 часа путем добавления 30 мкл 0,1 М фосфорной кислоты, содержащей 2 мМ АТФ. 96-Луночный планшет с фосфоцеллюлозным фильтром для мультискрининга (Millipore, номер по каталогу MAPHN0B50) предварительно, перед добавлением 45 мкл смеси для остановки анализа, обрабатывали 100 мкл 0,2 М фосфорной кислотой. Планшет промывали пятикратно по 200 мкл с помощью 0,2 М фосфорной кислоты. После высушивания, перед подсчетом сцинтилляций (1450 Microbeta Liquid Scintillation Counter, Wallac), в лунку добавляли 100 мкл коктейля для жидкостной сцинтилляции Optiphase «SuperMix» (Perkin Elmer).
[00139] После удаления средних фоновых значений для всех точек данных вычисляли данные Кi(app) методом нелинейного регрессионного анализа начальной скорости, используя пакет программного обеспечения Prism (GraphPad Prism, версия 3.0сх для Macintosh, GraphPad Software, San Diego, California, USA).
[00140] Соединения согласно настоящему изобретению являются эффективными в основном в отношении ингибирования ATR. Соединение I-1 ингибирует ATR при значениях Кi ниже 1 мкМ.
Пример 7: Анализ сенсибилизации к цисплатину
[00141] Соединения могут быть подвергнуты скринингу в отношении их способности сенсибилизировать клетки НСТ116 колоректального рака по отношению к цисплатину, используя 96-часовой тест на жизнеспособность клеток (MTS). Клетки НСТ116 с нарушенной АТМ-зависимой передачей сигнала к цисплатину (см. Kim и др.; Oncogene 21:3864 (2002); см. также, Takemura и др.; JBC 281:30814 (2006)) высевали по 470 клеток на лунку в 96-луночные полистирольные планшеты (Costar 3596) в 150 мл среды McCoyʹs 5A (Sigma M8403), дополненной 10% фетальной бычьей сывороткой (JRH Biosciences 12003), раствором пенициллин/стрептомицин, разведенным в соотношении 1:100 (Sigma P7539), и 2 мМ L-глутамином (Sigma G7513), и оставляли на ночь для адгезии при температуре 37°С в 5% СО2. Затем к клеточным средам одновременно добавляли соединения и цисплатин в 2-кратных последовательных разведениях начиная от верхней конечной концентрации 10 мМ, в виде полной матрицы концентраций в конечном клеточном объеме, равном 200 мл, и клетки затем инкубировали при температуре 37°С в 5% СО2. Спустя 96 часов в каждую лунку добавляли 40 мл реагента MTS (Promega G358a), и клетки инкубировали в течение 1 часа при температуре 37°С в 5% СО2. И наконец, определяли абсорбцию при 490 нм, используя планшет-ридер SpectraMax Plus 384 (Моlecular Devices), при этом может быть зарегистрирована концентрация соединения, необходимая для снижения значения IC50 цисплатина, по меньшей мере, в три раза (с точностью до 1 знака после запятой).
[00142] Соединения согласно настоящему изобретению являются эффективными в отношении сенсибилизации раковых клеток в основном по отношению к цисплатину. Значения сенсибилизации цисплатина, полученные для Соединения I-1, < 0,2 мкМ.
Пример 8: Активность одного агента в отношении НСТ116
[00143] Соединения могут быть подвергнуты скринингу для выявления активности одного агента в отношении клеток НСТ116 колоректального рака, используя 96-часовой тест на жизнеспособность клеток (MTS). Клетки НСТ116 высевали по 470 клеток на лунку в 96-луночные полистирольные планшеты (Costar 3596) в 150 мл среды McCoyʹs 5A (Sigma M8403), дополненной 10% фетальной бычьей сывороткой (JRH Biosciences 12003), раствором пенициллин/стрептомицин, разведенным в соотношении 1:100 (Sigma P7539), и 2 мМ L-глутамином (Sigma G7513), и оставляли на ночь для адгезии при температуре 37°С в 5% СО2. Затем к клеточным средам добавляли соединения в 2-кратных последовательных разведениях, начиная от верхней конечной концентрации 10 мМ, в виде полной матрицы концентраций в конечном клеточном объеме, равном 200 мл, и клетки затем инкубировали при температуре 37°С в 5% СО2. Спустя 96 часов в каждую лунку добавляли 40 мл реагента MTS (Promega G358a), и клетки инкубировали в течение 1 часа при температуре 37°С в 5% СО2. И наконец, определяли абсорбцию при 490 нм, используя планшет-ридер SpectraMax Plus 384 (Моlecular Devices), что позволяло рассчитать значения IC50.
Пример 9: Анализ ингибирования ATR-комплекса
[00144] Соединения подвергали скринингу в отношении их способности ингибировать ATR-киназу в присутствии белков-партнеров ATRIP, CLK2 и TopBP1, используя анализ включения радиоактивного фосфата. Анализ выполняли в смеси 50 мМ Tris/HCl (рН 7,5), 10 мМ МgCl2 и 1 мМ DTT. Конечные концентрации субстрата составляли 10 мкМ [g-33P]АТФ (3,5 мкКи 33Р АТФ/нмоль АТР, Perkin Elmer, Massachusetts, USA) и 800 мкМ пептида-мишени (ASELPASQPQPFSAKKK, Isca Biochemicals, Cambridgeshire, UK).
[00145] Анализ выполняли при температуре 25°С в присутствии 4 нМ полноразмерного ATR, 40 нм полноразмерного ATRIP, 40 нМ полноразмерного CLK2 и 600 нМ TopBP1 (A891-S1105). Готовили ферментный маточный буферный раствор, содержащий все из вышеперечисленных реагентов, за исключением пептида-мишени, АТФ и представляющего интерес тестируемого соединения. Этот ферментный маточный раствор предварительно инкубировали в течение 30 минут при температуре 25°С. 8,5 мкл ферментного маточного раствора помещали в 96-луночный планшет с последующим добавлением 5 мкл пептида-мишени и 2 мкл исходного раствора ДМСО, содержащего последовательные разведения тестируемого соединения (как правило, исходя из конечной концентрации 1,5 мкМ с 2,5-кратными последовательными разведениями), с двойным повтором (конечная концентрация ДМСО составляет 7%). Планшет предварительно инкубировали в течение 10 минут при температуре 25°С, и реакцию инициировали путем добавления 15 мкл [g-33P]АТФ (конечная концентрация 10 мкМ).
[00146] Реакцию останавливали через 20 часов путем добавления 30 мкл 0,3 М фосфорной кислоты, содержащей 2 мМ АТФ. 96-Луночный планшет с фосфоцеллюлозным фильтром (Multiscreen HTS MAPHNOB50, Merck-Millipore, Massachusetts, USA) предварительно, перед добавлением 45 мкл смеси для остановки анализа, обрабатывали 100 мкл 0,1 М фосфорной кислоты. Планшет пятикратно промывали 200 мкл 0,1 М фосфорной кислоты. После высушивания, перед подсчетом сцинтилляций (Wallac 1450 Microbeta Liquid Scintillation Counter, Perkin Elmer, Massachusetts, USA), в лунку добавляли 50 мкл коктейля для жидкостной сцинтилляции Optiphase «SuperMix» (Perkin Elmer, Massachusetts, USA)).
[00147] После удаления средних фоновых значений для всех точек данных, вычисляли данные Кi(app) методом нелинейного регрессионного анализа начальной скорости, используя пакет программного обеспечения Prism (GraphPad Prism, версия 6.0с для Macintosh, GraphPad Software Inc., SanDiego, USA).
[00148] Хотя авторами настоящего изобретения описано большое число вариантов осуществления данного изобретения, несомненно, что приведенные в описании настоящего изобретения основные примеры могут быть изменены для возможности получения других вариантов осуществления, в которых используются соединения, способы и процессы настоящего изобретения. Следовательно, должно быть понятно, что объем настоящего изобретения определяется прилагаемыми пунктами фоpмулы изобpетения, а не его конкpетными воплощениями, котоpые пpедставлены в настоящем описании в качестве пpимеpa.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2014 |
|
RU2687276C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ATR КИНАЗЫ (ВАРИАНТЫ) | 2014 |
|
RU2720408C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2020 |
|
RU2750148C1 |
| СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR | 2013 |
|
RU2689996C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR | 2012 |
|
RU2677292C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2632203C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ДИАГНОСТИКИ, ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АГРЕГАЦИЕЙ АЛЬФА-СИНУКЛЕИНА | 2020 |
|
RU2822486C1 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ КАК МОДУЛЯТОРЫ ФОСФОИНОЗИТИД-3-КИНАЗЫ | 2014 |
|
RU2672910C9 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-3-ХЛОР-5-ФТОР-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2545021C1 |
| ПРОИЗВОДНЫЕ N-(ЗАМЕЩЕННОГО)-5-ФТОР-4-ИМИНО-3-МЕТИЛ-2-ОКСО-3,4-ДИГИДРОПИРИМИДИН-1(2Н)-КАРБОКСИЛАТА | 2013 |
|
RU2638556C2 |
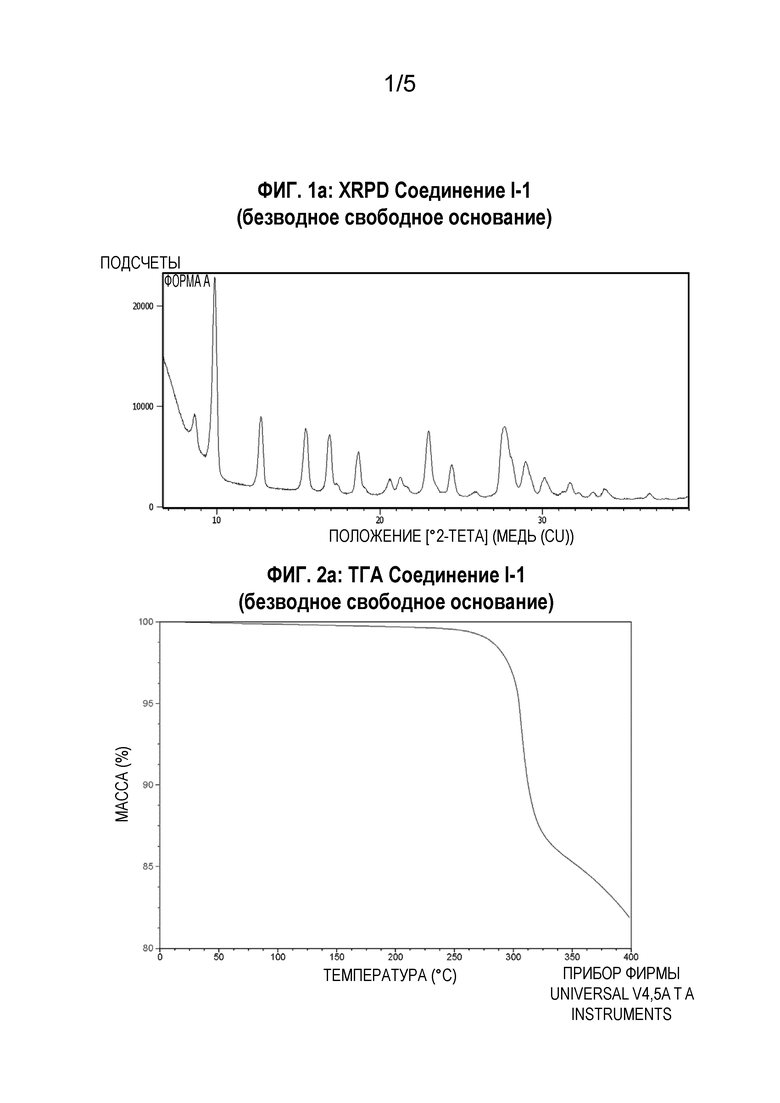
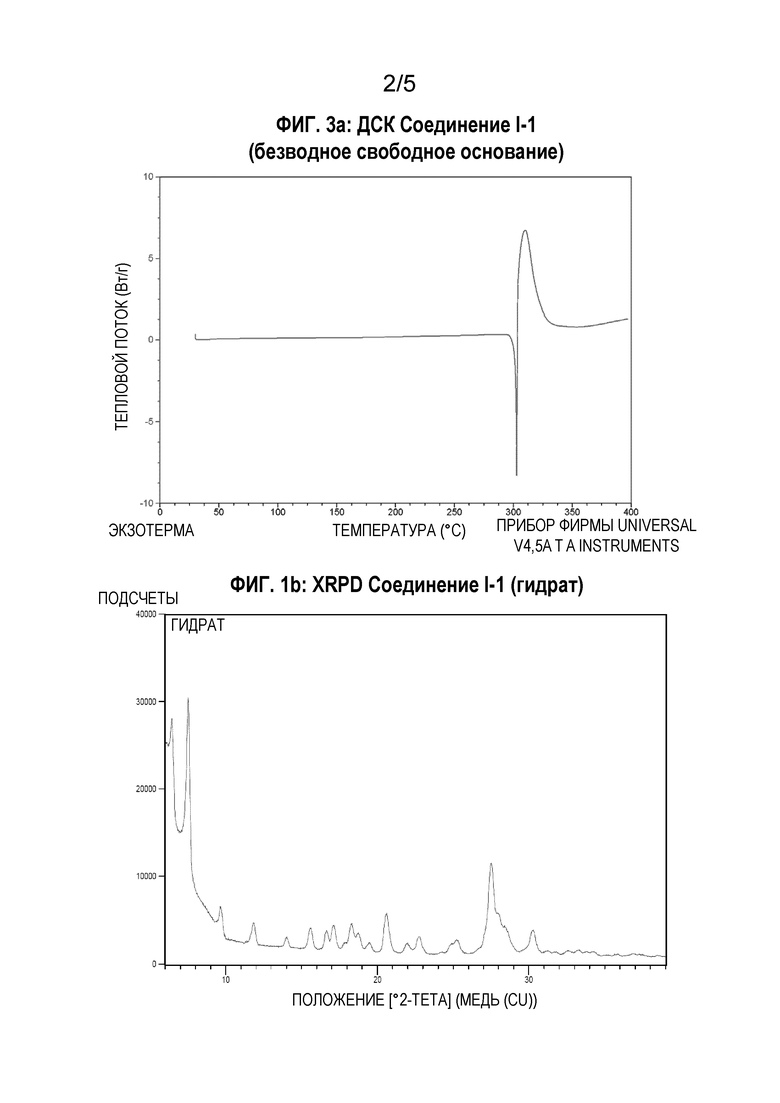
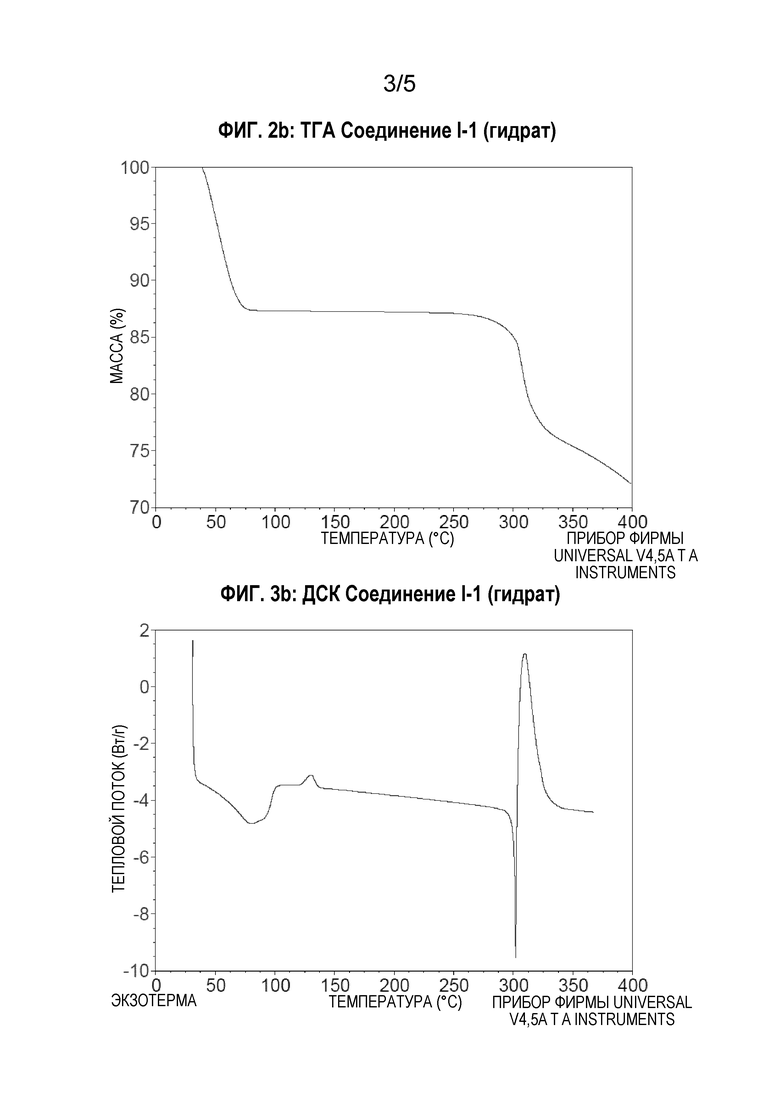
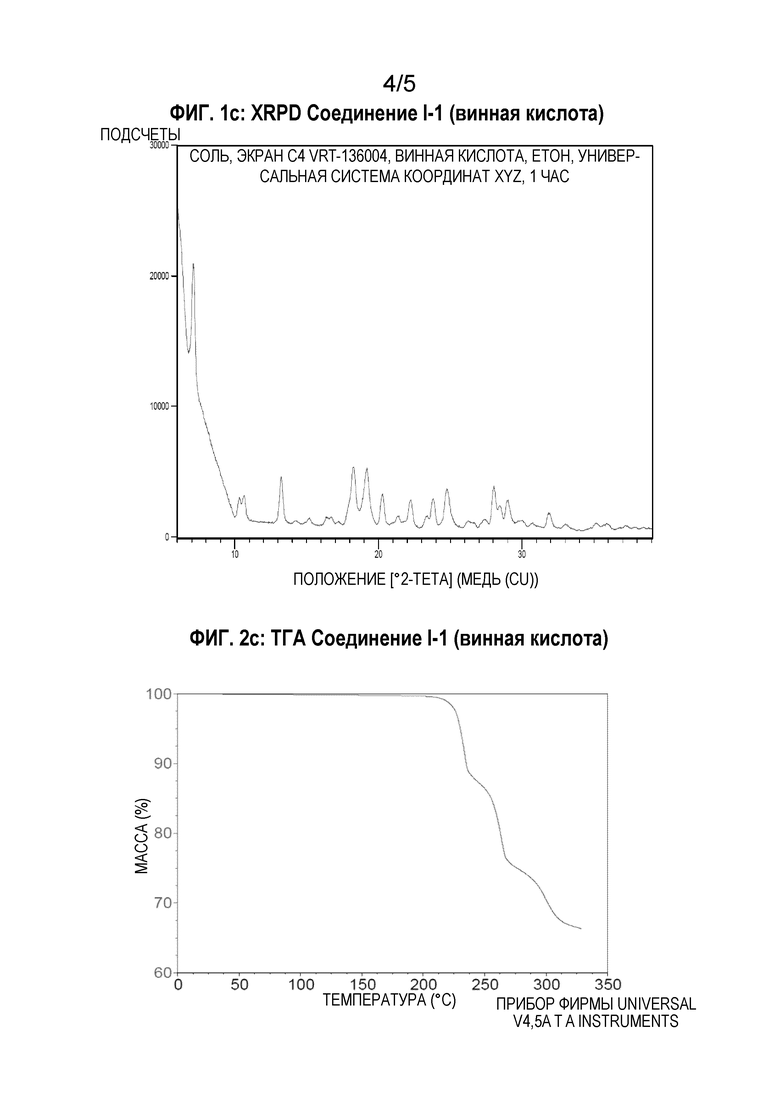
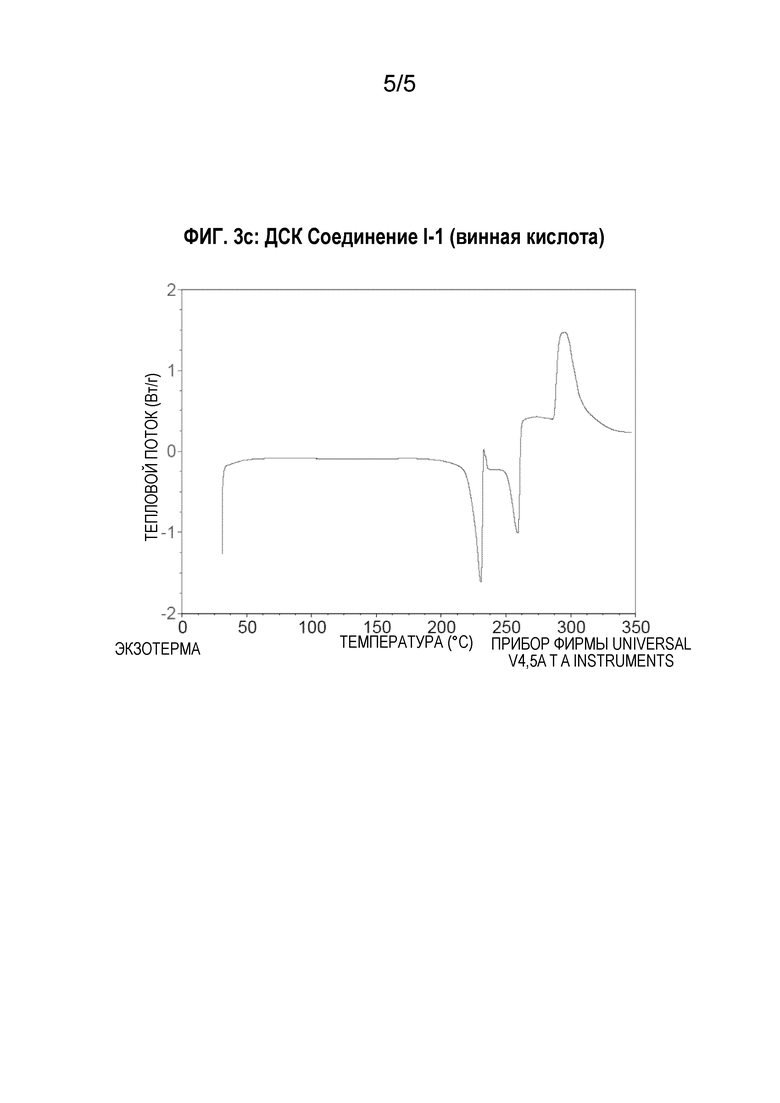
Настоящее изобретение относится к способу получения соединения формулы I-1, который включает стадию взаимодействия соединения формулы 6b с соединением формулы 11. Данное соединение пригодно в качестве ингибитора ATR протеинкиназы. 9 з.п. ф-лы, 9 ил., 6 табл., 8 пр.
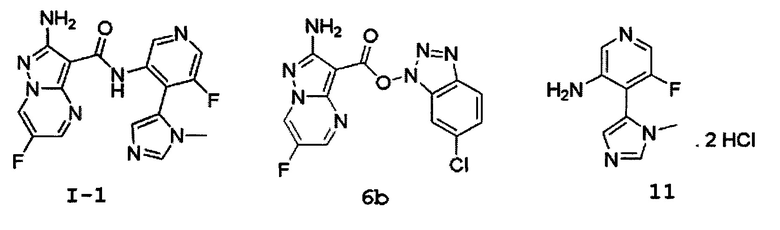
1. Способ получения соединения формулы I-1:
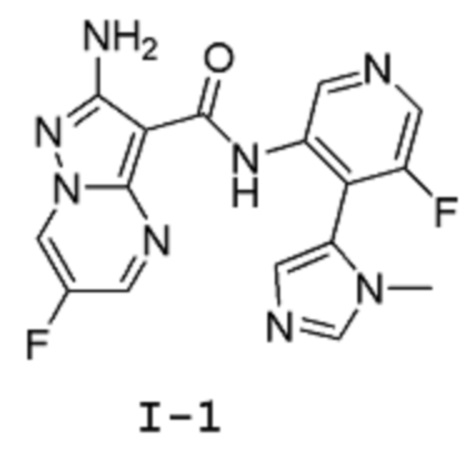 ,
,
включающий стадию взаимодействия соединения формулы 6b:
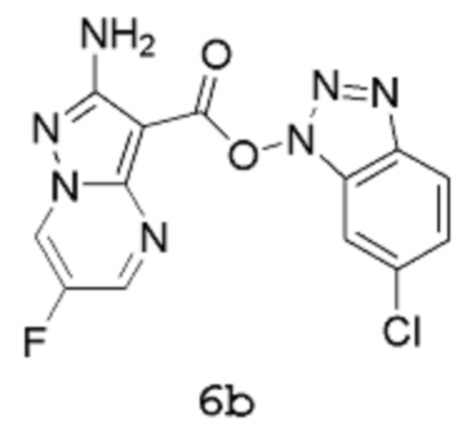
с соединением формулы 11:
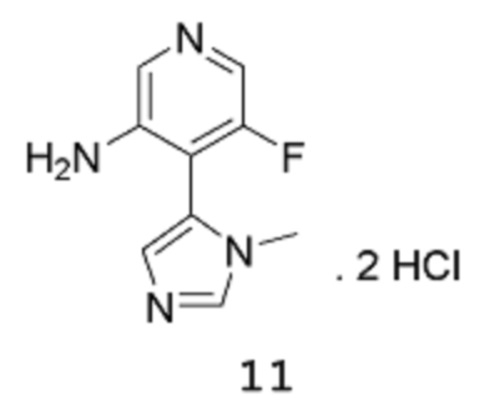
в подходящих условиях для образования амидной связи;
где подходящие условия для образования амидной связи включают взаимодействие соединения формулы 6b с соединением формулы 11 в присутствии растворителя и органического основания.
2. Способ по п.1, дополнительно включающий стадию получения соединения формулы 11:
путем взаимодействия соединения формулы 9:
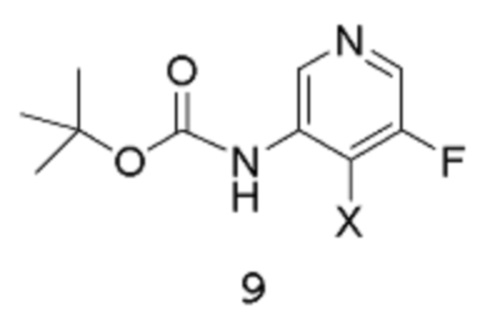 ,
,
где Х представляет собой галоген,
с соединением формулы 10:
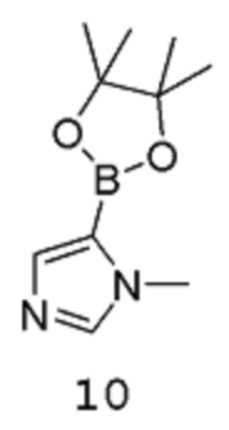
в подходящих условиях для катализируемой палладием реакции кросс-сочетания с образованием аддукта, содержащего защищенную аминогруппу;
где подходящие условия для катализируемой палладием реакции кросс-сочетания включают палладиевый катализатор, подходящий растворитель и подходящее основание;
после чего полученный аддукт подвергают воздействию подходящих условий для снятия защиты;
где подходящие условия для снятия защиты включают воздействие на полученный аддукт сильной кислотой.
3. Способ по п.2, дополнительно включающий стадию получения соединения формулы 9:
,
где Х представляет собой галоген;
путем реакции соединения формулы 8:
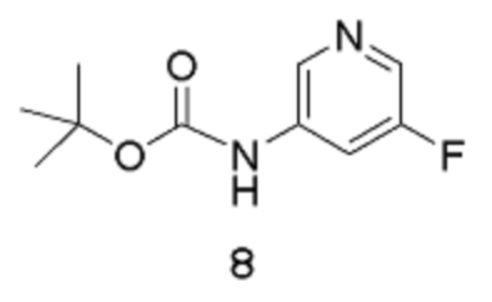
в подходящих условиях для галогенирования;
где подходящие условия для галогенирования включают реакцию соединения 8 в апротонном растворителе в присутствии сильного основания электрофильного источника галогена.
4. Способ по п.3, дополнительно включающий стадию получения соединения формулы 8:
путем реакции соединения формулы 7:
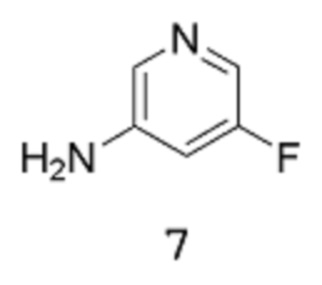
в подходящих условиях для получения защищенной аминогруппы;
где подходящие условия для получения защищенной аминогруппы включают реакцию соединения формулы 7 в апротонном растворителе в присутствии Boc2O.
5. Способ по п.1, где растворитель выбирают из NMP, ДМФА или анизола.
6. Способ по п.1, где органическое основание представляет собой алифатический амин.
7. Способ по п.6, где алифатический амин выбирают из триэтиламина и DIPEA.
8. Способ по п.2, где палладиевый катализатор выбирают из PdCl2(PPh3)2, Pd(Ph3)4 и PdCl2(dppf).
9. Способ по п.2, где подходящее основание включает одно или более из фосфата калия, K2CO3, tBuOK и Na2CO3.
10. Способ по п.2, где подходящий растворитель включает один или более из DME, тетрагидрофурана, толуола и этанола.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| RU 2012103487 A1, 10.08.2013, формула | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |

