Настоящее изобретение относится к новым соединениям, которые подходят для визуализации альфа-синуклеина и для диагностики заболеваний, связанных с агрегацией альфа-синуклеина. Соединения также применимы для лечения и профилактики заболеваний, которые связаны с агрегацией альфа-синуклеина.
УРОВЕНЬ ТЕХНИКИ
Известно большое количество неврологических и нейродегенеративных заболеваний, многие из которых в настоящее время неизлечимы и трудно поддаются диагностике. Все распространенные нейродегенеративные заболевания характеризуются неправильным сворачиванием, агрегацией и отложением специфических белков в головном мозге. Эти заболевания включают медицинские состояния, такие как болезнь Паркинсона (PD), деменция с тельцами Леви (DLB), множественная системная атрофия (MSA), болезнь Альцгеймера, прогрессирующий надъядерный паралич, кортикобазальная дегенерация, лобно-височная деменция, болезнь Крейтцфельдта-Якоба и многие другие.
Синуклеинопатии представляют собой группу заболеваний, характеризующихся накоплением и отложением агрегированного и неправильно свернутого белка альфа-синуклеина (αSYN). Синуклеинопатии включают болезнь Паркинсона (PD), деменцию с тельцами Леви (DLB) и множественную системную атрофию (MSA). Эти заболевания различаются в распределении накопления и отложения агрегированного и неправильно свернутого белка альфа-синуклеина в центральной и периферической нервной системе. Нейропатологически, их можно отличить от других нейродегенеративных заболеваний по патоспецифическому накоплению и отложению агрегированного и неправильно свернутого белка альфа-синуклеина, в то время как другие нейродегенеративные заболевания характеризуются накоплением и отложением других агрегированных и неправильно свернутых белков. Например, болезнь Альцгеймера характеризуется агрегированными и неправильно свернутыми белками A бета (Aβ) и тау, прогрессирующий надъядерный паралич и кортико-базальная дегенерация характеризуются агрегированным и неправильно свернутым белком тау, также некоторые случаи лобно-височной деменции характеризуются агрегированным и неправильно свернутым белком тау. Болезнь Крейтцфельдта-Якоба характеризуется агрегированным и неправильно свернутым прионным белком.
Патоспецифическое накопление и отложение агрегированных и неправильно свернутых белков является мишенью как для терапии, так и для диагностики с помощью соединений, которые связываются с этими агрегированными и неправильно свернутыми белковыми отложениями.
Одной из опций диагностического обнаружения патоспецифического накопления и отложения агрегированных и неправильно свернутых белков является использование обнаруживаемо меченых соединений, которые демонстрируют специфическое и селективное высокоаффинное связывание с указанными отложениями агрегированного белка. Это можно сделать, например, с помощью соединений, которые помечены подходящими радиоактивными изотопами и с помощью ПЭТ визуализации для обнаружения. Хотя соединения доступны для клинического применения для ПЭТ визуализации А бета и тау, до сих пор не изобретено соединений, которые можно было бы использовать для диагностической ПЭТ визуализации отложений альфа-синуклеина при синуклеинопатиях (Kotzbauer, P.T., Tu, Z. and Mach, R.H., Current status of the development of PET radiotracers for imaging alpha synuclein aggregates in Lewy bodies and Lewy neurites. Clin. Transl. Imaging, 2017. 5: p. 3-14). Соединения, разработанные и протестированные другими группами, до сих пор не обладали подходящей комбинацией свойств, включая высокую аффинность связывания с агрегированным и неправильно свернутым альфа-синуклеином, достаточную селективность аффинности связывания по сравнению с агрегированными и неправильно свернутыми другими белками, особенно А бета и тау (требуемой для дифференциальной диагностики различных заболеваний, характеризующихся патоспецифичным накоплением и отложением агрегированных и неправильно свернутых белков, и для способности точно обнаруживать присутствие агрегированных и неправильно свернутых альфа-синуклеинов также у пациентов, страдающих более чем одним заболеванием одновременно, то есть деменцией с тельцами Леви и болезнью Альцгеймера). Кроме того, требуемые свойства включают способность преодолевать гематоэнцефалический барьер и связываться с внутриклеточными отложениями, низкое неспецифическое связывание с тканью головного мозга и быстрое вымывание не связанного соединения из головного мозга.
WO 2009/146343 относится к некоторым пиразолам, 1,2,4-оксадиазолам и 1,3,4-оксадиазолам, которые являются мечеными веществами при визуализации позитронно-эмиссионной томографией (ПЭТ) для изучения отложений амилоида в головном мозге in vivo для диагностики болезни Альцгеймера. Болезнь Альцгеймера характеризуется агрегацией белков А бета и тау, и упомянутые меченые вещества ПЭТ связываются с агрегатами этих белков. Напротив, настоящее изобретение нацелено на заболевания, характеризующиеся агрегацией альфа-синуклеина. Для селективной диагностической визуализации агрегированного альфа-синуклеина требуется селективное связывание с альфа-синуклеином и отсутствие или слабое связывание с агрегированными А бета и тау.
В WO 00/66578 описаны специфические NPY антагонисты, которые можно использовать для лечения NPY-опосредованных заболеваний/состояний, таких как ожирение. Упоминается, что в дополнение к «прямому» действию соединений из WO 0066578 на подтип NPY5, существуют заболевания/состояния, которые могут выиграть при потере веса, такие как резистентность к инсулину, нарушение толерантности к глюкозе, диабет II типа, гипертония, гиперлипидемия, сердечно-сосудистые заболевания, камни в желчном пузыре, некоторые виды рака, апноэ во сне и т. д.
WO 2010/000372 относится к конкретному соединению, которое применяется для лечения или профилактики заболеваний, связанных с агрегацией белков, и/или нейродегенеративных заболеваний. Соединения WO 2010/000372 особенно подходят для лечения заболеваний, связанных с агрегацией белков, включая болезнь Паркинсона. Было показано, что они связываются с несколькими различными агрегированными белками, включая А бета и тау. Поэтому их можно использовать для диагностики нарушений, связанных с агрегацией белков, но невозможно надежно отличить нарушения, связанные с агрегацией альфа-синуклеина, от других нарушений, которые связаны с агрегацией амилоидных белков, таких как таупатии или болезнь Альцгеймера. Это, однако, было бы важно, потому что клинические проявления нарушений, связанных с агрегацией белков, очень похожи, и было бы очень желательно различать, например, между различными расстройствами для соответствующей адаптации лечения. Более того, молекулы, описанные в WO 2010/000372, обладают высоким неспецифическим связыванием с липидами и гидрофобными белками, что приводит к сильному неспецифическому связыванию в тканях мозга и других тканях. Таким образом, они не достигают требуемого специфического связывания и соотношения сигнала к шуму с альфа-синуклеином, как требуется для следовой ПЭТ визуализации.
В связи с вышеизложенным возникла потребность в соединениях, обладающих улучшенными диагностическими свойствами, в частности, необходимо улучшить специфичность связывания с альфа-синуклеином. Не специфическое связывание в тканях мозга и других тканях необходимо уменьшить, аффинность связывания необходимо увеличить, и физиологический период полужизни необходимо уменьшить.
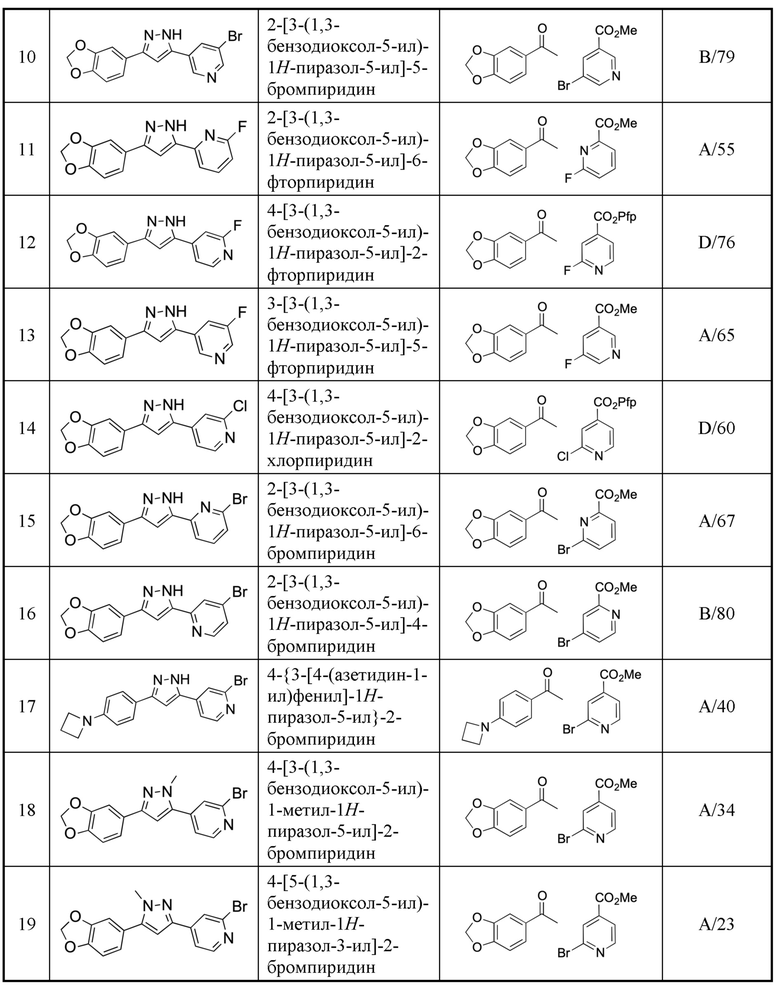
В US 2005/0075375 описаны специфические гетероциклические соединения для лечения вируса гепатита С.
краткое описание ЧЕРТЕЖА
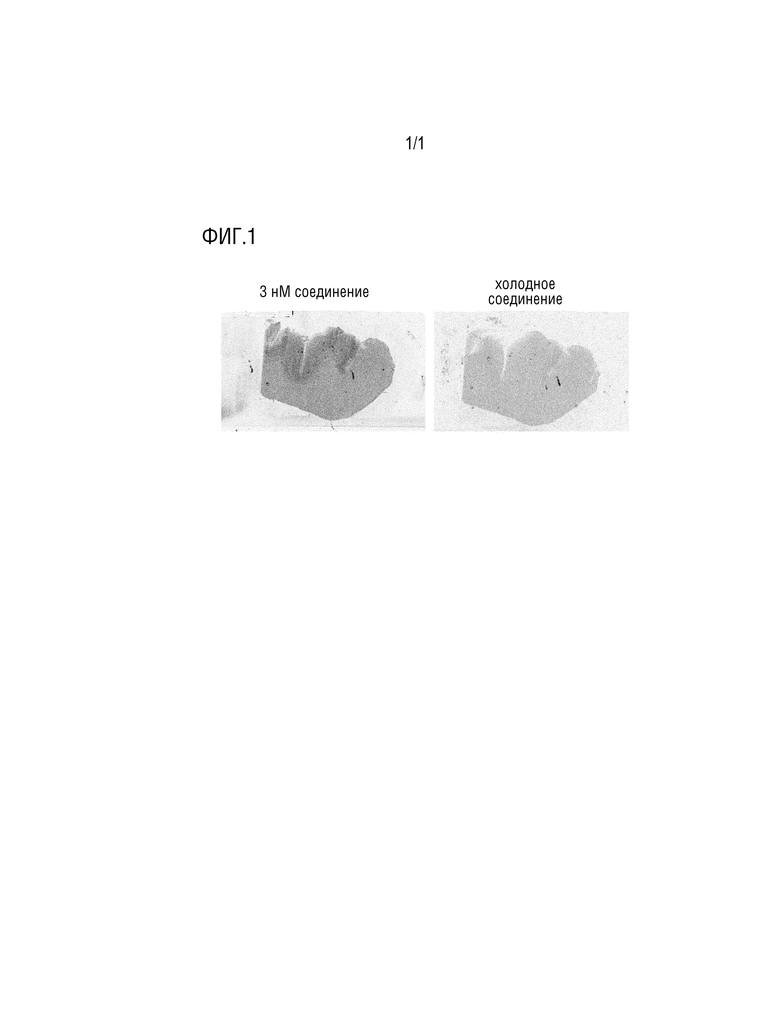
Фигура 1: Ауторадиография с использованием тканей головного мозга пациента, страдающего деменцией с тельцами Леви, с использованием [3H]-соединения 1.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
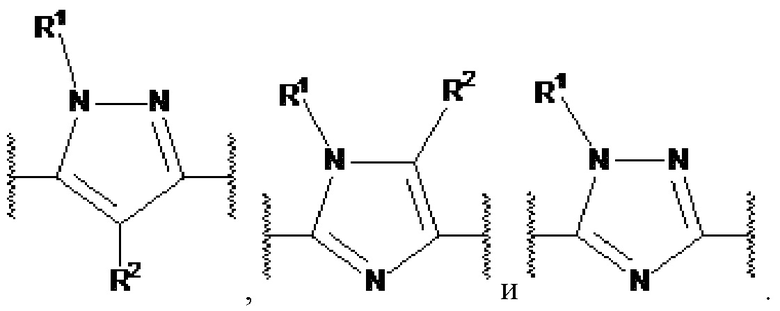
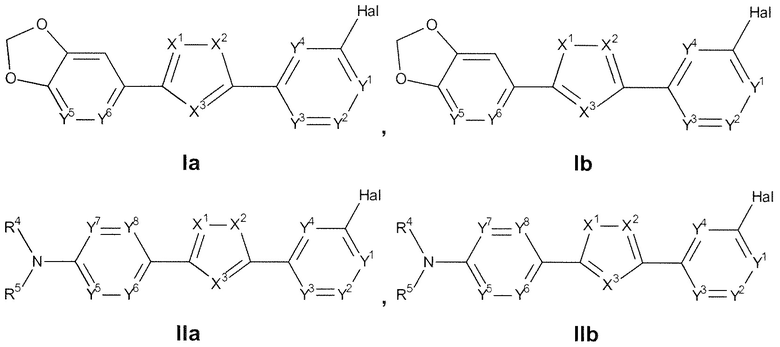
Настоящее изобретение относится к соединению, представленному общей формулой Ia, Ib, IIa или IIb.
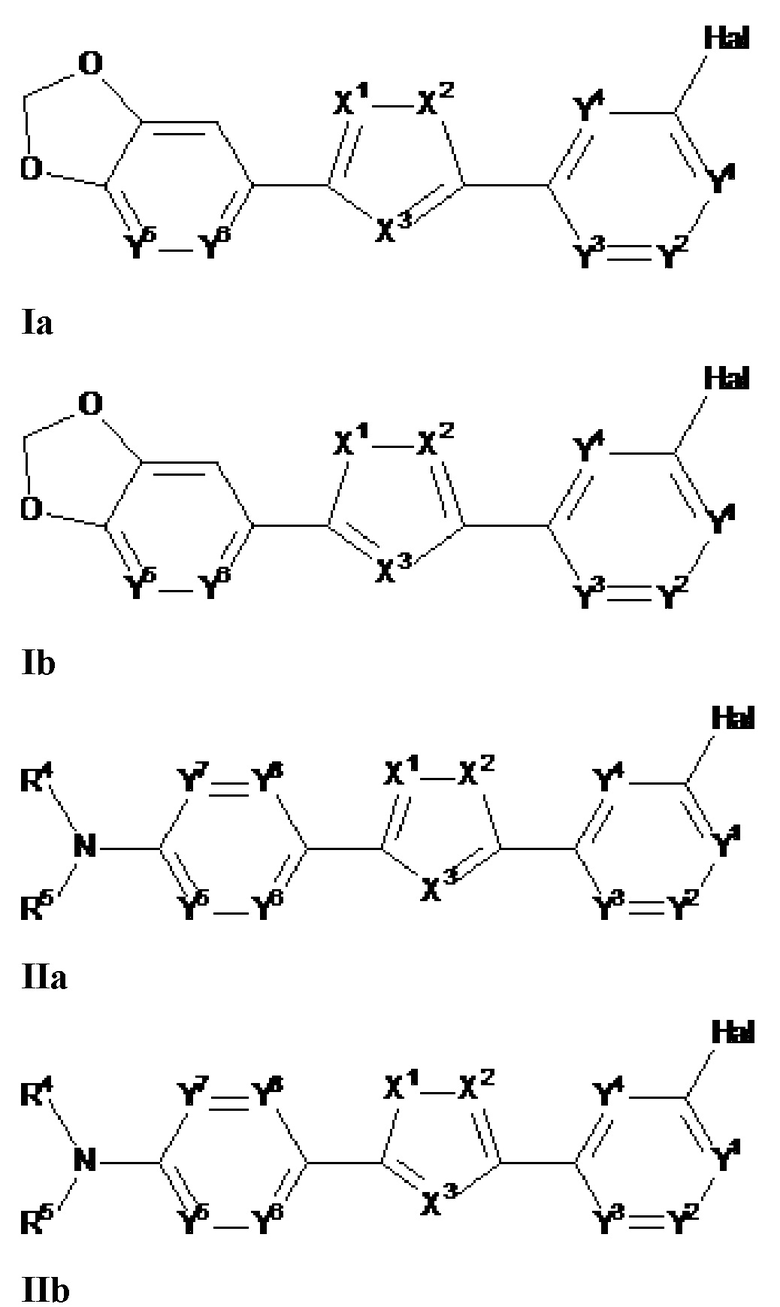
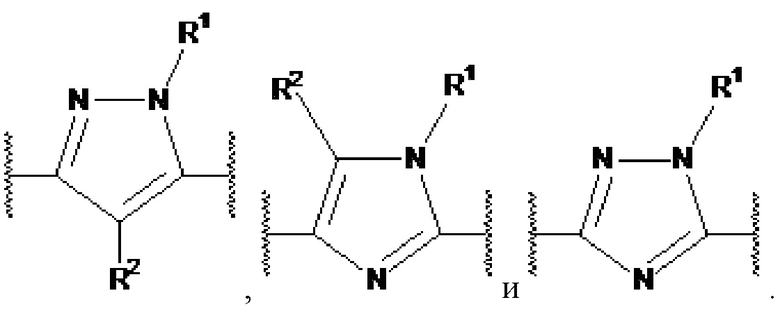
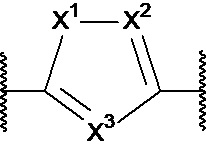
X1, X2 и X3 независимо выбраны из CR2, N и NR1, при условии, что, по меньшей мере, два из X1, X2 и X3 представляют собой N или NR1. Понятно, что N и NR1 присутствуют, настолько позволяет валентность, т.е., N может присутствовать только в положении =X1-, =X2- или =X3-, и NR1 может присутствовать только в положении -X1- или -X2-.
Примеры  включают
включают
Примеры  включают
включают
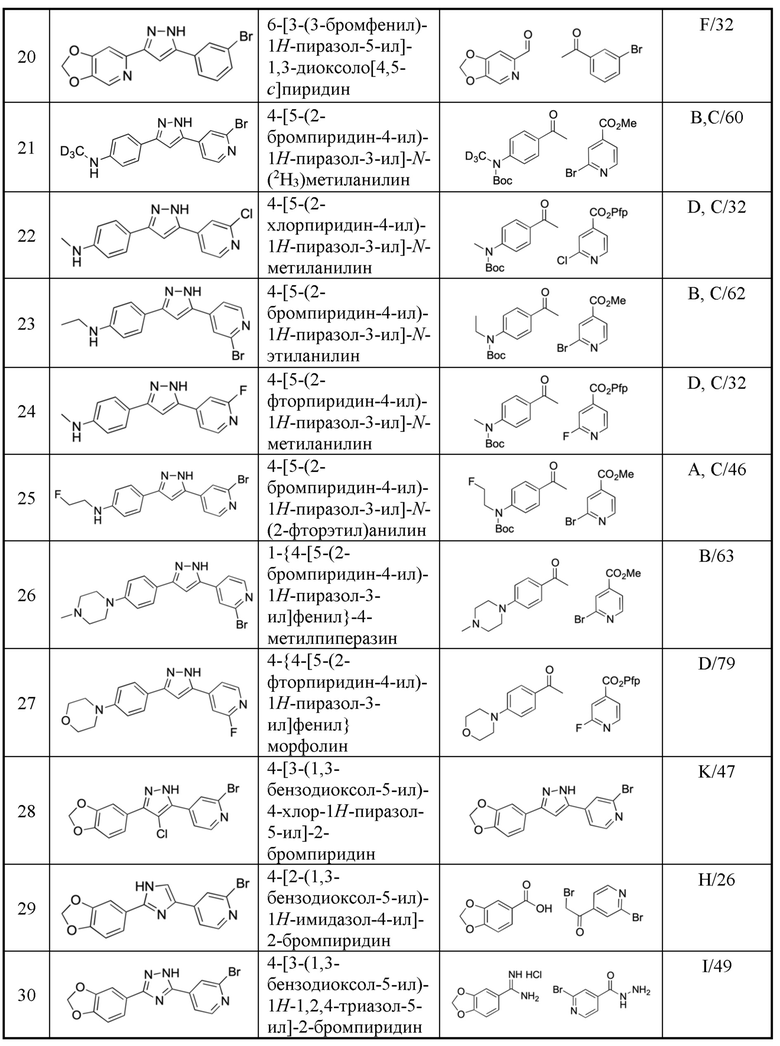
В предпочтительном варианте осуществления, 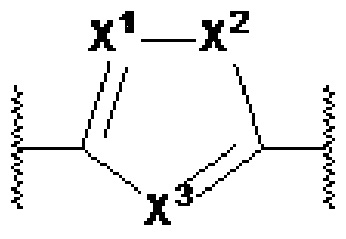 представляет собой
представляет собой 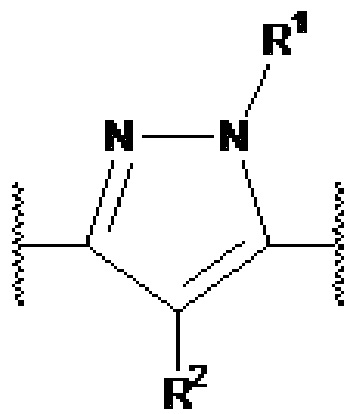 или
или 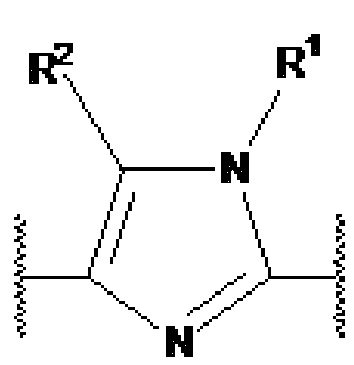 (более предпочтительно,
(более предпочтительно, 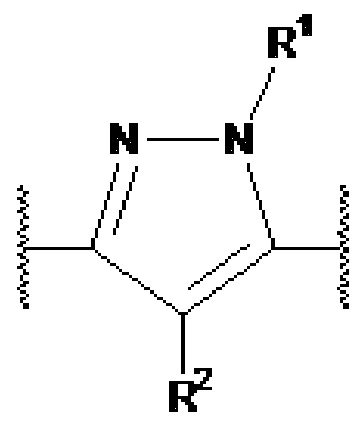 ) или
) или 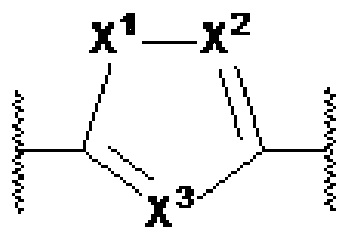 представляет собой
представляет собой 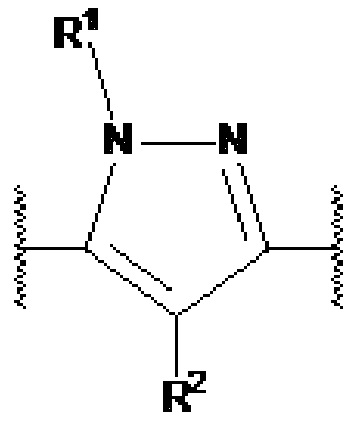 или
или 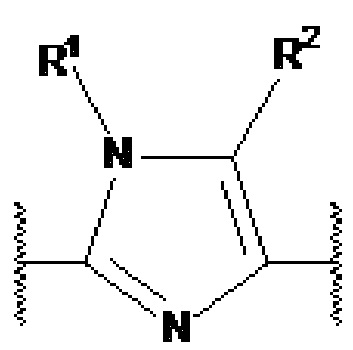 (более предпочтительно,
(более предпочтительно, 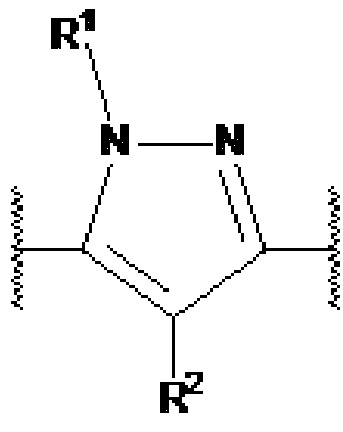 ).
).
Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 независимо выбраны из CR3 и N, при условии, что, по меньшей мере, один из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N.
В предпочтительном варианте осуществления, в формулах Ia или Ib, Y1, Y2, Y3, Y4, Y5 и Y6 независимо выбраны из CR3 и N, при условии, что, по меньшей мере, один из Y1, Y2, Y3, Y4, Y5 и Y6 представляет собой N. В более предпочтительном варианте осуществления, один или два или три из Y1, Y2, Y3, Y4, Y5 и Y6 представляет собой N, даже более предпочтительно, один или два из Y1, Y2, Y3, Y4, Y5 и Y6 представляет собой N, еще более предпочтительно, один из Y1, Y2, Y3, Y4, Y5 и Y6 представляет собой N. В предпочтительном варианте осуществления, Y1 представляет собой N.
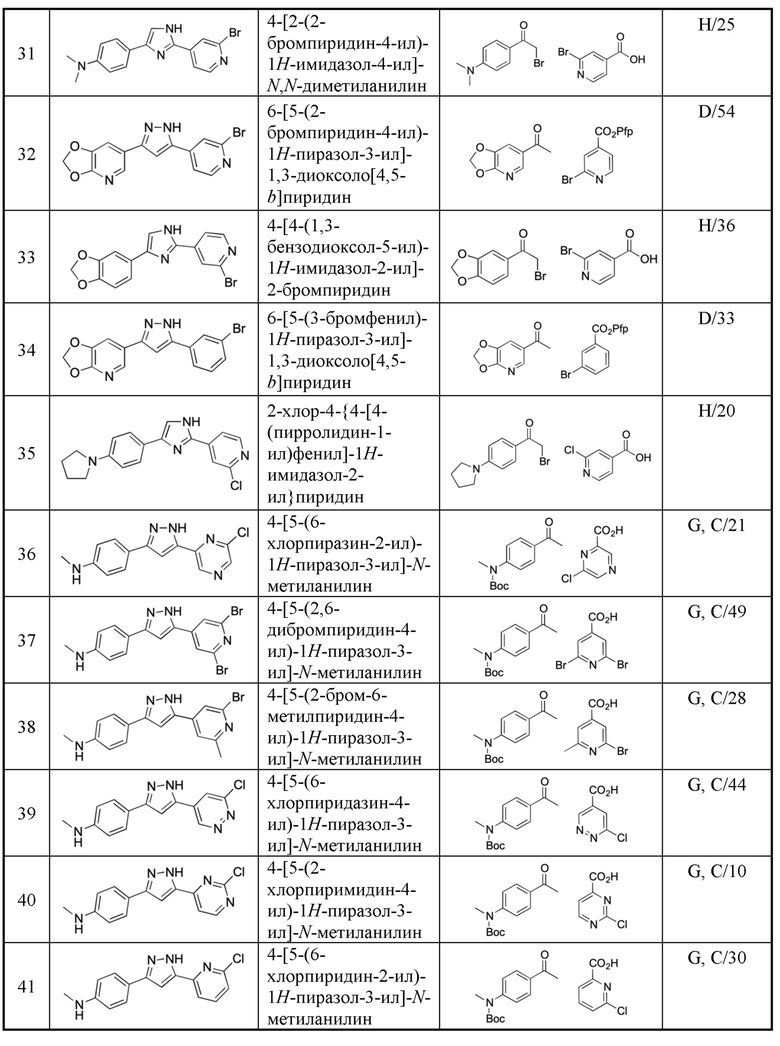
В предпочтительном варианте осуществления, в формулах Ia или Ib, по меньшей мере, один из Y1, Y3, Y4 и Y6 представляет собой N. В другом предпочтительном варианте осуществления, в формулах Ia или Ib, Y1 представляет собой N и, по меньшей мере, один из Y3, Y4 и Y6 представляет собой N.
В предпочтительном варианте осуществления, в формулах Ia или Ib, по меньшей мере, один из Y1, Y3, Y4 и Y6 представляет собой N и другие из Y1, Y2, Y3, Y4, Y5 и Y6 представляют собой CR3 (такой как CH). В другом предпочтительном варианте осуществления, в формулах Ia или Ib, Y1 представляет собой N, по меньшей мере, один из Y3, Y4 и Y6 представляет собой N, и другие из Y2, Y3, Y4, Y5 и Y6 представляют собой CR3 (такой как CH).
В предпочтительном варианте осуществления, в формулах Ia или Ib, Y1 представляет собой N и другие из Y2, Y3, Y4, Y5 и Y6 представляют собой CR3, более предпочтительно, Y1 представляет собой N и Y2, Y3, Y4, Y5 и Y6 представляют собой CH.
В предпочтительном варианте осуществления, в формулах IIa или IIb, Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 независимо выбраны из CR3 и N, при условии, что по меньшей мере, один из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N. В более предпочтительном варианте осуществления, один или два или три из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N, даже более предпочтительно, один или два из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N, еще более предпочтительно, один из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N. В предпочтительном варианте осуществления, Y1 представляет собой N.
В предпочтительном варианте осуществления, в формулах IIa или IIb, по меньшей мере, один из Y1, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N. В предпочтительном варианте осуществления, в формулах IIa или IIb, по меньшей мере, один из Y1, Y3, Y4, Y5 и Y7 представляет собой N. В другом предпочтительном варианте осуществления, в формулах IIa или IIb, Y1 представляет собой N и, по меньшей мере, один из Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N.
В предпочтительном варианте осуществления, в формулах IIa или IIb, по меньшей мере, один из Y1, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N, и другие из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой CR3 (такой как СН). В предпочтительном варианте осуществления, в формулах IIa или IIb, по меньшей мере, один из Y1, Y3, Y4, Y5 и Y7 представляет собой N и другие из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой CR3 (такой как СН). В другом предпочтительном варианте осуществления, в формулах IIa или IIb, Y1 представляет собой N, по меньшей мере, один из Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N, и другие из Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой CR3 (такой как СН). В предпочтительном варианте осуществления, в формулах IIa или IIb, Y1 представляет собой N, по меньшей мере, один из Y3, Y4, Y5 и Y7 представляет собой N и другие из Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой CR3 (такой как СН).
В предпочтительном варианте осуществления, в формулах IIa или IIb, Y1 представляет собой N и другие из Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой CR3, более предпочтительно, Y1 представляет собой N и Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой СН.
Неожиданно было обнаружено, что введение одного или нескольких атомов азота в качестве Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 сильно увеличивает селективность связывания с альфа-синуклеином, таким образом, соответствующие нарушения, связанные с агрегацией альфа-синуклеина, такие как болезнь Паркинсона, деменция с тельцами Леви и множественная системная атрофия, можно отличить от нарушений с агрегацией других белков, таких как болезнь Альцгеймера, при которой агрегируются преимущественно А бета и тау. Соотношение сигнала к шуму улучшается.
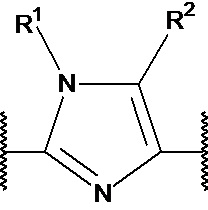
R1 выбран из водорода, C1-4 алкила и -(CH2)-O-P(=O)(OR)(OR), где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами. В предпочтительном варианте осуществления, R1 представляет собой водород, метил или 2-фторэтил. Если он присутствует, предпочтительно, чтобы -(CH2)-O-P(=O)(OR)(OR) был присоединен к X1 или X2.
R представляет собой водород или катион. Катион может быть любым фармацевтически приемлемым катионом. Предпочтительно, катион представляет собой одновалентный катион. Примерами являются натрий, литий, калий, аммоний и протонированные формы этаноламина, холина, лизина, меглумина, пиперазина, и трометамина. Предпочтительно, катион представляет собой натрий. В соединениях по настоящему изобретению, оба R могут быть водородом, оба R могут быть катионами (одинаковыми или разными катионами), или один R может быть водородом, и другой может быть катионом. Предпочтительно, оба R представляют собой натрий. Двухвалентные катионы, такие как Ca2+, Mg2+ и Zn2+, или трехвалентные катионы, такие как Al3+, возможны, но не предпочтительны, так как образующиеся соли менее растворимы в воде. Соединения, в которых R1 представляет собой -(CH2)-O-P(=O)(OR)(OR), и их синтез описан в WO 2017/102893, которая включена в настоящий документ посредством ссылки.
R2 независимо выбран из водорода, галогена и С1-4 алкила, где С1-4 алкил может быть необязательно замещен одним или несколькими галогенами. В предпочтительном варианте осуществления, R2 представляет собой водород.
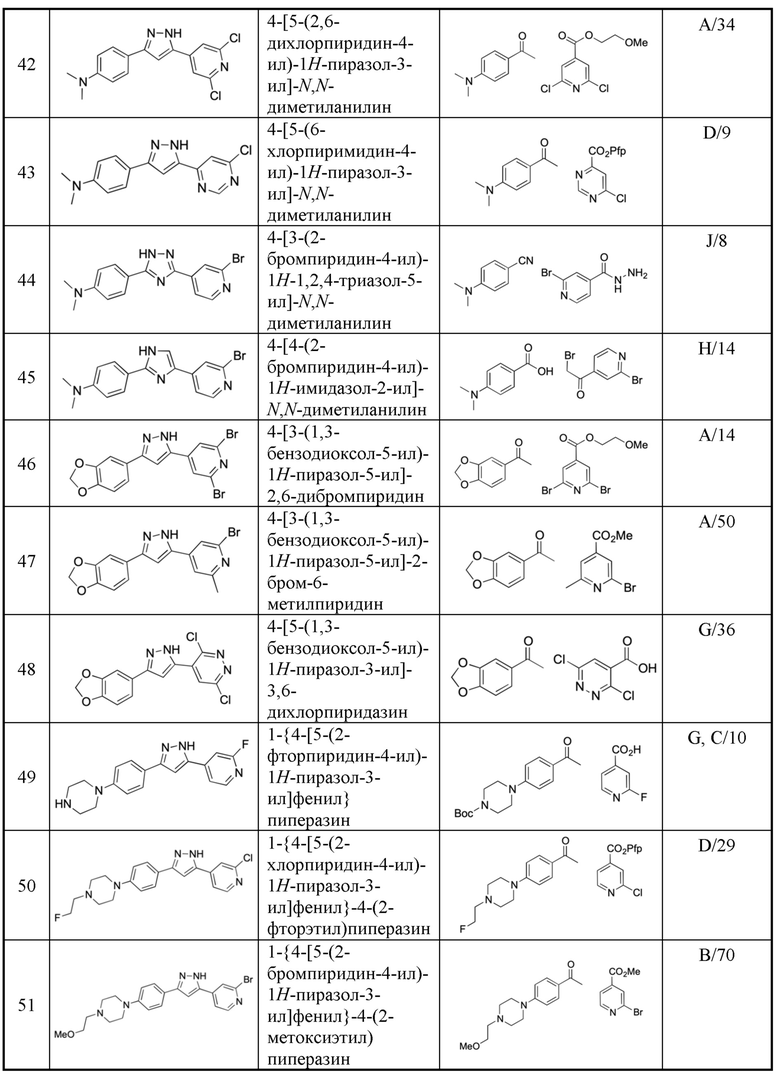
R3 представляет собой водород, галоген, С1-4 алкил, ОН и С1-4 алкокси, где С1-4 алкил и С1-4 алкокси могут быть необязательно замещены одним или несколькими галогенами. В предпочтительном варианте осуществления, R3 представляет собой водород или фтор. Еще более предпочтительно, R3 представляет собой водород.
R4 и R5 независимо выбраны из H и C1-4 алкила, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами, или где R4 и R5 вместе с атомом азота, с которым они связаны, образуют 4-6-членное насыщенное гетероциклическое кольцо, которое необязательно содержит один или несколько гетероатомов, выбранных из O и N, в дополнение к атому азота, с которым связаны R4 и R5, где 4-6-членное насыщенное гетероциклическое кольцо необязательно может быть замещено одним или несколькими R6.
В одном варианте осуществления, R4 и R5 независимо выбраны из H и C1-4 алкила, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами. Предпочтительно, R4 и R5 независимо выбраны из H и C1-4 алкила. В предпочтительном варианте, по меньшей мере, один из R4 и R5 представляет собой C1-4 алкил, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами. В более предпочтительном варианте осуществления, R4 представляет собой водород и R5 представляет собой C1-4 алкил, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами. В более предпочтительном варианте, по меньшей мере, один из R4 и R5 представляет собой C1-4 алкил. В более предпочтительном варианте, R4 представляет собой водород и R5 представляет собой C1-4 алкил.
В другом варианте осуществления R4 и R5 вместе с атомом азота, с которым они связаны, образуют 4-6-членное насыщенное гетероциклическое кольцо, которое необязательно содержит один или несколько (например, один) гетероатомов, выбранных из О и N, в добавление к атому азота, с которым связаны R4 и R5, где 4-6-членное насыщенное гетероциклическое кольцо может быть необязательно замещено одним или несколькими R6. В предпочтительном варианте осуществления, 4-6-членное гетероциклическое кольцо выбрано из азетидинила, пирролидинила, пиперидинила, морфолинила и пиперазинила, где азетидинил, пирролидинил, пиперидинил, морфолинил и пиперазинил могут быть необязательно замещены одним или несколькими R6. Более предпочтительно, 4-6-членное насыщенное гетероциклическое кольцо выбрано из азетидинила, пирролидинила, пиперидинила, морфолинила и пиперазинила, где N атом пиперазинила может быть необязательно замещен R6.
R6 независимо выбран из галогена, C1-4 алкила, ОН и C1-4 алкокси, где C1-4 алкил и C1-4 алкокси могут быть необязательно замещены одним или несколькими галогенами, предпочтительно, R6 представляет собой C1-4 алкил или фтор.
Hal представляет собой галоген, такой как Br, Cl и F, предпочтительно, Hal представляет собой Br или F.
m представляет собой количество R3 групп, отличных от водорода, в кольце, которое содержит Y1, Y2, Y3 и Y4. m представляет собой целое число от 0 до mmax, где mmax представляет собой число атомов углерода в кольце, которое содержит Y1, Y2, Y3 и Y4. Предпочтительно, m равно 0.
n представляет собой количество групп R3, отличных от водорода, в кольце, которое содержит Y5, Y6, Y7 и Y8 (формулы IIa и IIb). n представляет собой целое число от 0 до nmax, где nmax представляет собой количество атомов углерода в кольце, которое содержит Y5, Y6, Y7 и Y8. Предпочтительно, n равно 0.
p представляет собой количество групп R3, отличных от водорода, в кольце, которое содержит Y5 и Y6 (формулы Ia и Ib). p представляет собой целое число от 0 до nmax, где nmax представляет собой число атомов углерода в кольце, которое содержит Y5 и Y6. Предпочтительно, р равно 0.
Соединения по настоящему изобретению могут также присутствовать в форме пролекарств, их сольватов или солей.
Соединения по настоящему изобретению образуют соли, которые также входят в объем настоящего изобретения. Понятно, что ссылка на соединение по настоящему изобретению в настоящем документе включает ссылку на его соли, если не указано иное. Фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли являются предпочтительными, хотя другие соли также могут быть использованы, например, на стадиях выделения или очистки, которые могут быть использованы во время получения или в способах in vitro. Соли соединений по настоящему изобретению могут быть получены, например, путем взаимодействия соединения с количеством кислоты, таким как эквивалентное количество, в среде, такой как среда, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
Соединения, которые содержат основную группу, могут образовывать соли с различными органическими и неорганическими кислотами. Примеры кислотно-аддитивных солей включают ацетаты (например, образованные с уксусной кислотой или тригалогенуксусной кислотой, например, трифторуксусной кислотой), адипаты, альгинаты, аскорбаты, аспартаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, фумараты, глюкогептаноаты, глицерофосфаты, гемисульфаты, гептаноаты, гексаноаты, гидрохлориды, гидробромиды, гидроиодиды, гидроксиэтансульфонаты (например, 2-гидроксиэтансульфонаты), лактаты, малеаты, метансульфонаты, нафталинсульфонаты (например, 2-нафталинсульфонаты), никотинаты, нитраты, оксалаты, пектинаты, персульфаты, фенилпропионаты (например, 3-фенилпропионаты), фосфаты, пикраты, пивалаты, пропионаты, салицилаты, сукцинаты, сульфаты (например, те, которые образуются с серной кислотой), сульфонаты (например, те, которые упомянуты в настоящем документе), тартраты, тиоцианаты, толуолсульфонаты, такие как тозилаты, ундеканоаты и подобные.
В настоящем документе также рассматриваются пролекарства и сольваты соединений по настоящему изобретению. Термин «пролекарство», используемый в настоящем документе, обозначает соединение, которое при введении субъекту подвергается химическому превращению посредством метаболических или химических процессов с получением соединения по настоящему изобретению или их соли и/или сольваты.
Сольваты соединений по настоящему изобретению включают, например, гидраты.
Все стереоизомеры соединений по настоящему изобретению (например, те, которые могут существовать благодаря асимметрии атомов углерода в различных заместителях), включая энантиомерные формы и диастереомерные формы, входят в объем настоящего изобретения. Индивидуальные стереоизомеры соединений по изобретению могут, например, по существу не содержать другие изомеры (например, в виде чистого или по существу чистого оптического изомера, обладающего определенной активностью), или могут быть смешаны, например, в виде рацематов или со всеми другими, или другими выбранными, стереоизомерами. Хиральные центры соединений по настоящему изобретению могут иметь S- или R-конфигурацию, как определено в Рекомендациях IUPAC 1974.
Рацемические формы могут быть разделены физическими способами, такими как фракционная кристаллизация, разделение или кристаллизация диастереомерных производных или разделение с помощью хиральной колоночной хроматографии. Индивидуальные оптические изомеры могут быть получены из рацематов любым подходящим способом, включая, без ограничения, образование соли с оптически активной кислоты с последующей кристаллизацией.
Рассматриваются все конфигурационные изомеры соединений по настоящему изобретению, либо в смеси, либо в чистой или по существу чистой форме. Определение соединений по настоящему изобретению охватывают и цис (Z) и транс (Е) алкеновые изомеры, а также цис и транс изомеры циклических углеводородных или гетероциклических колец.
Также могут быть представлены дейтерированные варианты заявленных соединений. Положение, в котором присутствует дейтерирование, особо не ограничено, но оно может находиться, например, в С1-4 алкиле R4 и R5.
По всему описанию, группы и их компоненты могут быть выбраны для получения стабильных групп и соединений.
Соединения по настоящему изобретению могут быть представлены в форме диагностической или фармацевтической композиции, которая необязательно включает фармацевтически приемлемый носитель или эксципиент.
В соответствии с настоящим изобретением, термин «диагностическая композиция» относится к композиции для определения присутствия агрегированного альфа-синуклеина, лежащего в основе заболевания, связанного с агрегацией альфа-синуклеина.
В предпочтительном варианте осуществления, соединение по настоящему изобретению является обнаруживаемом или обнаруживаемо меченным. В соответствии с настоящим изобретением понимается, что соединение является обнаруживаемом или обнаруживаемо меченным, если его присутствие можно контролировать с помощью обычных методов, таких как ЯМР спектроскопия, однофотонная эмиссионная компьютерная томография (ОФЭКТ), оптическое обнаружение, позитронно-эмиссионная томография (ПЭТ), электронная микроскопия, магнитно-резонансная томография (МРТ), спектрометрия, хроматография, анализ ELISA, обнаружение радиоактивного излучения, предпочтительно с помощью ПЭТ, сцинтилляционный счет или гамма-счет, более предпочтительно, ПЭТ.
Когда соединения по настоящему изобретению предназначены для применения в качестве зондов для визуализации агрегированного альфа-синуклеина, они должны быть помечены. Конкретная природа метки будет зависеть от способа, который будет использоваться для визуализации. Обычно буду полезны радиоактивные метки, испускающие позитроны (ПЭТ) и имеющие короткий период полужизни, такие как 18F, 11C, 125I, 123I, 131I, 77Br и 76Br, в частности 18F и 11C. Из-за их коротких периодов полужизни, меченые соединения по настоящему изобретению следует готовить незадолго до их использования для тестирования. Следовательно, диагностическая композиция по настоящему изобретению также может быть представлена в виде набора, состоящего, по меньшей мере, из двух предшественников соединения по настоящему изобретению, которые вступают в реакцию с образованием желаемого соединения по настоящему изобретению.
Специалист в данной области техники сможет разработать способы, с помощью которых обнаруживаемая метка может быть присоединена к соединению по настоящему изобретению. Следующие схемы могут служить иллюстративными примерами.
Мечение 18F:
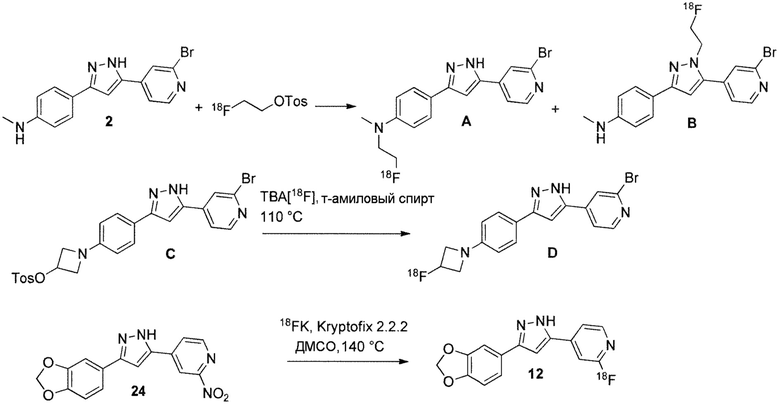
2-[18F]фторэтил тозилат является полезным предшественником для включения 18F посредством фторэтилирования соединений, содержащих нуклеофилы азота, кислорода и серы, как описано в WO 2010/000372, стр. 32, схема A для получения соединения 21 и соединения 22, начиная с соединения 23. Соединения A и B могут быть получены таким же образом. Другой способ включает прямое нуклеофильное замещение подходящей уходящей группы, как показано для превращения C в D (см. J. Med. Chem. 2013, 56, 4568-4579, схема 2) или превращения соединения 24 в соединение 12 на схеме 1.
Схема 1
Мечение 11C:
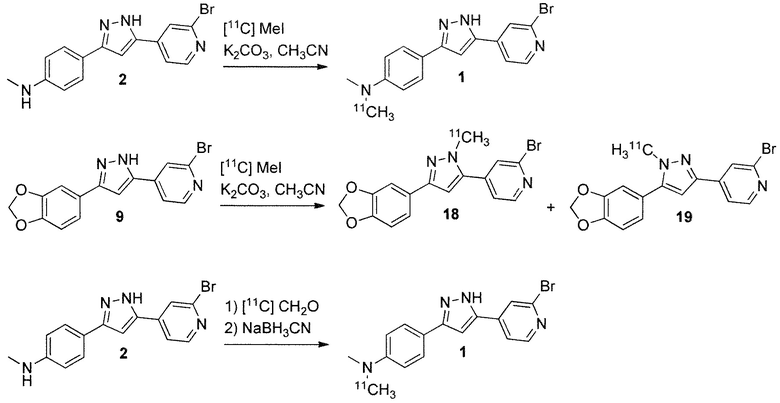
Соединения по изобретению, меченные 11C, могут быть получены прямым нуклеофильным алкилированием подходящего предшественника [11C] MeI, как описано в WO 2010/000372, стр. 32, схема B, для получения соединения 27 и соединение 28, начиная с соединения 23. Соединение 1 по настоящему изобретению можно синтезировать аналогичным образом, начиная с соединения 2. Таким же образом можно синтезировать соединение 18 и соединение 19, начиная с соединения 9, как показано на схеме 2. Альтернативно, простой и доступный способ (J.M. Hooker et al., Angew. Chem. Int. Ed. 2008, 47, 5989-5992) получения [11C]формальдегида в виде раствора в диметилформамиде путем превращения [11C]метилйодида в [11C]формальдегид в мягких условиях без потери удельной активности, может применяться для превращения соединения 2 в соединение 1 через восстановительное аминирование.
Схема 2:
Настоящее изобретение представляет способ визуализации отложений агрегированного альфа-синуклеина, который включает следующие стадии:
(i) введение субъекту обнаруживаемого количества композиции, содержащей обнаруживаемое меченое соединение по настоящему изобретению;
(ii) предоставление достаточного количества времени для связывания соединения с агрегированным альфа-синуклеином; и
(iii) обнаружение соединения, связанного с агрегированным альфа-синуклеином.
Композицию, содержащую обнаруживаемо меченое соединение, можно вводить субъекту любым путем введения, описанным ниже, таким как, например, пероральный или парентеральный. Меченое соединение может быть введено пациенту и через некоторое количество времени, достаточное для связывания соединения с агрегированным альфа-синуклеином, меченое соединение обнаруживают неинвазивно внутри пациента. Альтернативно, меченое соединение может быть введено пациенту, дается достаточное время для связывания соединения с агрегированным альфа-синуклеином, и затем берется образец ткани у пациента и меченого соединения обнаруживают в ткани отдельно от пациента. Образец ткани также может быть взят у пациента перед введением меченого соединения в образец ткани. После достаточного количества времени для связывания соединения с агрегированным альфа-синуклеином, соединение может быть обнаружено.
Визуализация агрегированного альфа-синуклеина также может быть выполнена количественно, чтобы можно было определить количество агрегированного альфа-синуклеина.
Настоящее изобретение также относится к соединению по настоящему изобретению, а также к его пролекарству, сольвату или соли, для применения при лечении или профилактике заболевания, связанного с агрегацией альфа-синуклеина.
Другими вариантами осуществления являются применение соединения по настоящему изобретению для приготовления фармацевтической композиции для лечения или профилактики заболевания, связанного с агрегацией альфа-синуклеина, а также способ лечения или профилактики заболевания, связанного с агрегацией альфа-синуклеина, включающий введение терапевтически эффективного количества соединения по настоящему изобретению пациенту, нуждающемуся в этом. Он включает применение соединения в виде «фармацевтической композиции», как описано ниже.
Термин «агрегация» в соответствии с настоящим изобретением относится к образованию олигомерных или мультимерных комплексов альфа-синуклеина, которое может сопровождаться интеграцией дополнительных биомолекул, таких как углеводы, нуклеиновые кислоты, липиды и/или ионы металлов, в комплексы.
Термин «заболевание, связанное с агрегацией альфа-синуклеина», как он используется в настоящем документе, относится к тем заболеваниям, которые характеризуются наличием агрегированного альфа-синуклеина. Такой агрегированный альфа-синуклеин может образовывать отложения в определенной ткани, более предпочтительно, в нервной ткани или ткани головного мозга. Степень агрегации зависит от конкретного заболевания.
В соответствии с настоящим изобретением, термин «фармацевтическая композиция» относится к композиции для введения пациенту, предпочтительно млекопитающему, более предпочтительно, пациенту-человеку. Фармацевтическая композиция по изобретению содержит соединения, перечисленные выше, и, необязательно, другие молекулы, способные изменять характеристики соединений по изобретению, таким образом, например, стабилизируя, модулируя и/или активируя их функции. Композиция может быть в твердой, жидкой или газообразной форме, и может быть, среди прочего, в форме порошка(ов), таблетка(ок), раствора(ов) или аэрозоля(ей). Фармацевтическая композиция по настоящему изобретению может, необязательно и дополнительно, содержать фармацевтически приемлемый носитель или эксципиент. Примеры подходящих фармацевтических носителей и эксципиентов хорошо известны в данной области техники и включают забуференные фосфатом солевые растворы, воду, эмульсии, такие как масляно-водные эмульсии, различные типы смачивающих агентов, стерильные растворы, органические растворители, в том числе ДМСО и т.д. Композиции, содержащие такие носители, могут быть составлены хорошо известными традиционными способами.
Фармацевтическая композиция будет составлена и дозирована в соответствии с надлежащей медицинской практикой, принимая во внимание клиническое состояние отдельного пациента, место доставки фармацевтической композиции, способ введения, схему введения и другие факторы, известные практикующим специалистам. «Эффективное количество» фармацевтической композиции для целей настоящего изобретения, таким образом, определяется такими соображениями. Специалисту в данной области техники известно, что эффективное количество фармацевтической композиции, вводимой индивидууму, будет, среди прочего, зависеть от природы соединения.
Фармацевтические композиции по изобретению можно вводить перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей, капель или чрескожных пластырей), буккально или в виде перорального или назального спрея. Предпочтительно, их вводят внутривенно при использовании для следовой ПЭТ визуализации для диагностики, и перорально при использовании для лечения или профилактики заболевания.
Под «фармацевтически приемлемым носителем» подразумевают нетоксичный твердый, полутвердый или жидкий наполнитель, разбавитель, инкапсулирующий материал или вспомогательное вещество любого типа.
Термин «парентеральный», используемый здесь, относится к способам введения, которые включают внутривенную, внутримышечную, внутрибрюшинную, интрастернальную, подкожную и внутрисуставную инъекцию и инфузию.
Для терапевтических целей, фармацевтическую композицию также можно соответствующим образом вводить с помощью систем с замедленным высвобождением. Подходящие примеры композиций с замедленным высвобождением включают полупроницаемые полимерные матрицы в форме формованных изделий, например, пленок или микрокапсул. Матрицы с замедленным высвобождением включают полилактиды. (патент США №3,773,919, ЕР 58 481), сополимеры L-глутаминовой кислоты и гамма-этил-L-глутамата (Sidman, U. et al., Biopolymers 22:547-556 (1983)), поли(2-гидроксиэтилметакрилат) (R. Langer et al., J. Biomed. Mater. Res. 15:167-277 (1981) и R. Langer, Chem. Tech. 12:98-105 (1982)), этилен винилацетат (R. Langer et al., Id.) или поли-D-(-)-3-гидроксимасляную кислоту (EP 133 988). Фармацевтические композиции с замедленным высвобождением могут также включать соединения, захваченные липосомами. Липосомы, содержащие фармацевтическую композицию, получают способами, известными сами по себе: DE 32 18 121; Epstein et al., Proc. Natl. Acad. Sci. (USA) 82:3688-3692 (1985); Hwang et al., Proc. Natl. Acad. Sci. (USA) 77:4030-4034 (1980); EP 52 322; EP 36 676; EP 88 046; EP 143 949; EP 142 641; Заявка на патент Японии 83-118008; патенты США №№ 4,485,045 и 4,544,545; и EP 102324. Обычно, липосомы имеют небольшой (примерно 200-800 ангстрем) однослойный тип, в котором содержание липидов превышает примерно 30% мол. холестерина, где выбранная пропорция регулируется для оптимальной терапии.
Для парентерального введения, фармацевтическую композицию, как правило, составляют путем смешивания ее до желаемой степени чистоты в стандартной дозированной форме для инъекций (растворе, суспензии или эмульсии) с фармацевтически приемлемым носителем, т.е. носителем, который не токсичен для реципиентов в применяемых дозировках и концентрациях и совместим с другими ингредиентами состава.
Как правило, составы готовят путем однородного и тесного контакта компонентов фармацевтической композиции с жидкими носителями или тонкоизмельченными твердыми носителями или с обоими. Затем, при необходимости, составу придают желаемую форму. Предпочтительно, носитель представляет собой парентеральный носитель, более предпочтительно раствор, изотонический крови реципиента. Примеры таких носителей включают воду, солевой раствор, раствор Рингера и раствор декстрозы. Не водные носители, такие как нелетучие масла и этилолеат, также применяют в настоящем документе, а также липосомы. Носитель подходящим образом содержит небольшие количества добавок, таких как вещества, повышающие изотоничность и химическую стабильность. Такие материалы нетоксичны для реципиентов в используемых дозировках и концентрациях, и включают буферы, такие как фосфатный, цитратный, сукцинатный, уксусную кислоту и другие органические кислоты или их соли; антиоксиданты, такие как аскорбиновая кислота; низкомолекулярные (менее десяти остатков) (поли)пептиды, например полиаргинин или трипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутаминовая кислота, аспарагиновая кислота или аргинин; моносахариды, дисахариды и другие углеводы, включая целлюлозу или ее производные, глюкозу, маннозу или декстрины; хелатирующие агенты, такие как ЭДТК; сахарные спирты, такие как маннит или сорбит; противоионы, такие как натрий, и/или неионные поверхностно-активные вещества, такие как полисорбаты, полоксамеры или ПЭГ.
Компоненты фармацевтической композиции, используемые для терапевтического введения, должны быть стерильными. Стерильность легко достигается путем фильтрации через стерильные фильтрационные мембраны (например, мембраны 0,2 мкм). Терапевтические компоненты фармацевтической композиции обычно помещают в контейнер, имеющий стерильный порт доступа, например, пакет для внутривенного раствора или флакон с пробкой, прокалываемой иглой для подкожных инъекций.
Компоненты фармацевтической композиции обычно хранятся в однордозовых или многодозовых контейнерах, например, в запечатанных ампулах или флаконах, в виде водного раствора или в виде лиофилизированного состава для восстановления. В качестве примера лиофилизированного состава, 10 мл флаконы заполняют 5 мл стерильно отфильтрованного 1% (масса/объем) водного раствора, и полученную смесь лиофилизируют. Раствор для инфузий готовят восстановлением лиофилизированного соединения с применением бактериостатической воды для инъекций.
Настоящее изобретение дополнительно относится к способу лечения или профилактики заболевания, связанного с агрегацией альфа-синуклеина, включающему введение терапевтически эффективного количества соединения по настоящему изобретению пациенту, нуждающемуся в этом.
Используемый здесь термин «терапевтически эффективное количество» относится к количеству, достаточному для того, чтобы вызвать желаемый биологический ответ. В настоящем изобретении, желаемый биологический ответ представляет собой ингибирование агрегации альфа-синуклеина и/или снижение количества агрегированного альфа-синуклеина, присутствующего в ткани.
Настоящее изобретение также относится к применению соединения, как определено выше, для ингибирования агрегации альфа-синуклеина in vitro или ex vivo.
Заболевание, связанное с агрегацией альфа-синуклеина, конкретно не ограничено и обычно выбирается из болезни Паркинсона, деменции с тельцами Леви и множественной системной атрофии.
Следующие примеры предназначены для иллюстрации изобретения, однако их не следует рассматривать как ограничивающие.
ПРИМЕРЫ
Пример 1: Синтез и тестирование
Методы химического синтеза
Следующие способы представлены с подробным описанием получения соединений по изобретению и иллюстративными примерами. Соединение по изобретению может быть получено из известных или коммерчески доступных исходных материалов и реагентов специалистом в области органического синтеза.
Все исходные материалы и растворители имеют коммерческую степень чистоты и используются в том виде, в каком они были получены, если не указано иное.
Тонкослойную хроматографию (ТСХ) проводят с использованием листов с предварительно нанесенным покрытием Macherey-Nagel, 0,25 мм планшетов ALUGRAM® SIL G/UV254, обнаружения с помощью УФ и/или путем карбонизации реагентом 10% масс. этанольной фосфомолибденовой кислоты с последующим нагреванием при 200°C.
Флэш-хроматографию проводят с применением силикагеля Merck 60 (0,063-0,100 мм).
Аналитическую высокоэффективную жидкостную хроматографию (ВЭЖХ) проводят с использованием системы ВЭЖХ Waters с матричным фотодиодным датчиком Waters 996. Все разделения включают подвижную фазу 0,1% трифторуксусной кислоты (ТФК) (об./об.) в воде и 0,1% ТФК в ацетонитриле. ВЭЖХ проводят с использованием колонки с обращенной фазой (ОФ) Eurospher RP 18, 100 Å, 5 мкм, 250х4,6 мм при скорости потока 1 мл⋅мин-1.
Масс-спектрометрию с ионизацией электрораспылением (ИЭР-МС) и жидкостную хроматографию/масс-спектрометрию (ЖХ/МС) проводят с использованием масс-спектрометра Waters Micromass ZQ 4000 в сочетании с аппаратом ВЭЖХ Waters, описанным выше.
Спектры ЯМР записывают на спектрометре Bruker Avance 400 МГц (Bruker AG, Rheinstetten, Germany), оснащенном z-градиентным зондом TXI HCN. Все спектры обрабатывают с помощью TOPSPIN 3.1 (Bruker AG, Karlsruhe, Germany). Химические сдвиги 1H ЯМР (δ) приведены в частях на миллион (ч./млн.) относительно CHCl3, ДМСО-d5 и TFA-d1 в качестве внутренних стандартов. Данные представлены следующим образом: химический сдвиг, мультиплетность (с=синглет, д=дублет, т=триплет, кв=квартет, кви=квинтет, дд=дублет дублетов, дт=дублет триплетов, ш=широкий, м=мультиплет), константы сочетания (J, даны в Гц), интегрирование. Химические сдвиги 13C ЯМР (δ) приведены в частях на миллион (ч./млн.) относительно CDCl3, ДМСО-d6 и ТФК-d1 в качестве внутренних стандартов. Для записи резонансов соединений используют следующие эксперименты: 1H-1D, 13C-1D ЯМР спектры и 13C-APT (тест на присоединенные протоны с одним временем J-выделения 1/145 с, спектры обработаны таким образом, что четвертичные и метиленовые группы имеют положительный знак, а метильная и метиновая группы имеют отрицательный знак). Для разрешения наложения резонансов и восстановления необнаруживаемых резонансов в спектрах 1H и APT, 2D-[13C, 1H]-HSQC (гетероядерную одноквантовую когерентность), 2D-[13C, 1H]-HMBC (гетероядерную многосвязную корреляцию связей) и 2D-NOESY записывают для некоторых соединений.
Выбранные соединения (соединение 1, соединение 2, соединение 12, соединение 18) тритируют для анализа связывания посредством RC TRITEC AG, Teufen, Switzerland с использованием H/T-обменного мечения 99% газообразным тритием/катализатором Керра. Соединения поставляются в виде этанольных растворов в упаковке 185 МБк, в концентрации 37 МБк/мл и с удельной активностью в диапазоне от 1,1 до 2,3 ГБк/ммоль.
Способ А: синтез 1Н-пиразолов
Специалистам в данной области техники известно, что соединение 11 и соединение 11, изображенные ниже, представляют собой две таутомерные формы одного и того же соединения. Все такие таутомерные формы рассматриваются как часть настоящего изобретения. В качестве иллюстрации, все таутомерные формы пиразольной группы, как показано, например, для соединения 11 ниже, включены в настоящее изобретение. Специалистам в данной области техники будет понятно, что названия соединений, содержащиеся в настоящем документе, основаны на номенклатурной конвенции, в котором таутомерная конфигурация изображена как в отношении соединения 11 ниже. Таким образом, 1,3-бензодиоксольный заместитель находится в третьем положении. Альтернативная номенклатурная конвенция может быть основана на таутомере соединения 11 ниже, и в этой конвенции 1,3-бензодиоксольный заместитель находится в пятом положении.
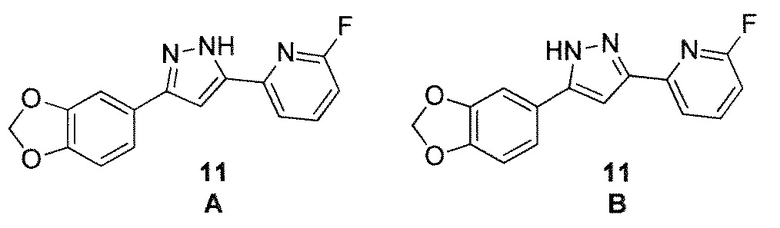
Иллюстративный пример: 2-[3-(1,3-Бензодиоксол-5-ил)-1H-пиразол-5-ил]-6-фторпиридин, соединение 11
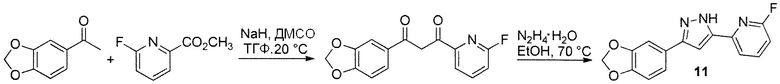
Гидрид натрия (FW 24,00, 60% в масле, 3,9 ммоль, 156 мг) добавляют к раствору 1-(1,3-бензодиоксол-5-ил)этанона (FW 164,16, 492 мг, 3,00 ммоль) и метил 6-фторпиридин-2-карбоксилат (FW 155,13, 605 мг, 3,9 ммоль) в ДМСО (7,5 мл) и ТГФ (1,9 мл) и реакционную смесь перемешивают при 20°C в течение 15 ч. Реакционную смесь выливают в 60 мл льда и воды, содержащих AcOH (450 мкл). Смесь перемешивают в течение 1 ч. Полученный осадок отфильтровывают, промывают водой (10 мл), гексаном:EtOH=5:1 (10 мл), гексаном (10 мл) и сушат на воздухе с получением неочищенного промежуточного соединения 1-(1,3-бензодиоксол-5-ил)-3-(6-фторпиридин-2-ил)пропан-1,3-диона (594 мг) в виде желтого твердого вещества. К суспензии этого неочищенного промежуточного соединения в EtOH (20 мл) добавляют гидрат гидразина (146 мкл, 150 мг, 3 ммоль). Реакционную смесь перемешивают при 70°C в течение 5 ч, охлаждают и концентрируют в вакууме. Остаток суспендируют в MeOH (10 мл), кипятят при перемешивании в течение 5 мин, охлаждают, отфильтровывают, промывают MeOH (10 мл) и сушат в высоком вакууме при 20°C в течение 15 ч с получением очищенного продукта соединения 11 (471 мг, 1,66 ммоль, 55% за две стадии) в виде белого твердого вещества.
Способ B: Синтез 1H-пиразолов
Иллюстративный пример: 4-[3-(1,3-Бензодиоксол-5-ил)-1H-пиразол-5-ил]-2-бромпиридин, соединение 9
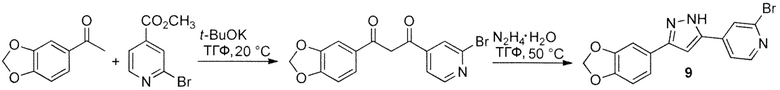
Раствор трет-бутоксида калия (FW 112,21, 281 мг, 2,5 ммоль) в сухом ТГФ (5 мл) добавляют под азотом к перемешиваемому раствору 1-(1,3-бензодиоксол-5-ил)этанона (FW 164,16, 328 мг, 2 ммоль) и метил 2-бромпиридин-4-карбоксилата (FW 216,03, 518 мг, 2,4 ммоль) в сухом ТГФ (5 мл). Реакционную смесь перемешивают в течение 15 ч при 20°C. 20 мкл аликвоту отбирают, гасят 1M фосфатным буфером pH 7, экстрагируют EtOAc и анализируют ТСХ. Наблюдают полное превращение кетона. Смесь выливают в 1M фосфатный буфер pH 7 (15 мл) и ледяную воду (15 мл) и перемешивают при 0°C в течение 30 мин. Полученное желтое твердое вещество отфильтровывают, промывают водой (5х10 мл) и сушат на воздухе с получением 656 мг (1,88 ммоль, 94%) неочищенного промежуточного соединения 1-(1,3-бензодиоксол-5-ил)-3-(2-бромпиридин-4-ил)пропан-1,3-диона, которое применяют на следующей стадии без очистки. Смесь этого промежуточного соединения и моногидрата гидразина (FW 50,06, d 1,03; 274 мкл, 282 мг, 5,64 ммоль) в ТГФ (10 мл) перемешивают при 50°C в течение 15 ч. Охлажденную смесь выливают в воду (40 мл) и перемешивают при 0°C в течение 30 мин. Полученный осадок отфильтровывают, промывают водой и сушат на воздухе. Неочищенный продукт кристаллизуют из n-BuOH (10 мл) и ДМФ (1 мл) с получением чистого продукта, соединения 9 (458 мг, 1,33 ммоль, 67% за 2 стадии) в виде белого порошка.
Способ C: Удаление защитной группы Boc
Иллюстративный пример: 4-[5-(2-Бромпиридин-4-ил)-1H-пиразол-3-ил]анилин соединение 7
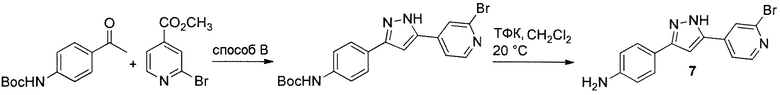
Трифторуксусную кислоту (2 мл, 2,96 г, 26 ммоль) добавляют к суспензии неочищенного трет-бутил {4-[5-(2-бромпиридин-4-ил)-1H-пиразол-3-ил]фенил}карбамата (FW 415,28, 865 мг, 2,08 ммоль), полученного из трет-бутил N-(4-ацетилфенил)карбамата и метил 2-бромпиридин-4-карбоксилата согласно способу B. Смесь перемешивают при комнатной температуре в течение 15 ч, и концентрируют в вакууме. Добавляют 1M фосфатный буфер pH 7 (20 мл), полученный осадок отфильтровывают, промывают водой (2х10 мл) и сушат на воздухе с получением 543 мг (1,72 ммоль, 69% за 3 стадии) желаемого продукта в виде желто-оранжевого твердого вещества.
Способ D: Синтез 1H-пиразолов
Иллюстративный пример: 4-[3-(1,3-Бензодиоксол-5-ил)-1H-пиразол-5-ил]-2-фторпиридин, соединение 12
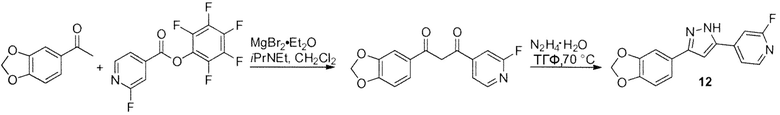
iPr2NEt (1,79 мл, 1,33 г, 10,26 ммоль) добавляют к перемешиваемой смеси 1-(1,3-бензодиоксол-5-ил)этанона (561 мг, 3,42 ммоль) и MgBr2⋅Et2O (1,56 г, 6,04 ммоль) в CH2Cl2 (35 мл). Полученную суспензию перемешивают в течение 5 мин, и затем добавляют по каплям пентафторфенил 2-фторпиридин-4-карбоксилат (1,37 г, 4,45 ммоль) в CH2Cl2 (7 мл). Реакционную смесь перемешивают в течение 48 ч. Затем добавляют 1N водную HCl (20 мл) и перемешивание продолжают в течение 5 мин. Водный слой экстрагируют CH2Cl2 (20 мл) и объединенные органические экстракты сушат (MgSO4) и концентрируют в вакууме. Остаток растирают с Et2O (10 мл) и фильтруют. Твердое вещество промывают Et2O и сушат на воздухе с получением неочищенного продукта (1,14 г, оранжевого твердого вещества). Неочищенный продукт перекристаллизовывают из EtOAc (8 мл) и гексана (4 мл) с получением очищенного промежуточного соединения 1-(1,3-бензодиоксол-5-ил)-3-(2-фторпиридин-4-ил)пропан-1,3-диона (900 мг, 3,13 ммоль, 92%) в виде оранжевого твердого вещества. Это промежуточное соединение суспендируют в ТГФ (20 мл) и добавляют моногидрат гидразина (292 мкл, 300 мг, 6 ммоль). Реакционную смесь перемешивают при 70°C в течение 5 ч, охлаждают и концентрируют в вакууме. Остаток суспендируют в Et2O (10 мл), кипятят при перемешивании в течение 5 мин, охлаждают, отфильтровывают, промывают Et2O (2х10 мл) и сушат в высоком вакууме при 20°C в течение 15 ч с получением продукта, соединения 12 (722 мг, 2,55 ммоль, 76% за 2 стадии) в виде белого твердого вещества.
Способ E: Синтез сложных эфиров пентафторфенила
Иллюстративный пример: Пентафторфенил 2-фторпиридин-4-карбоксилат, соединение 59

ДЦК (FW 206,33, 2,27 г, 11 ммоль) добавляют к перемешиваемой суспензии 2-фторпиридин-4-карбоновой кислоты (1,41 г, 10 ммоль) и пентафторфенола (1,84 г, 10 ммоль) в 1,4-диоксане (40 мл). Перемешивание продолжают в течение 15 ч, к этому времени образуется бесцветный осадок. Смесь фильтруют через Celite® и выпаривают с получением полутвердого вещества. Хроматография над силикагелем дает сложный эфир, соединение 59 (2,21 г, 7,2 ммоль, 72%) в виде чистого бесцветного масла.
Способ F: Синтез 1H-пиразолов
Иллюстративный пример: 6-[3-(3-Бромфенил)-1H-пиразол-5-ил]-1,3-диоксоло[4,5-c]пиридин, соединение 20
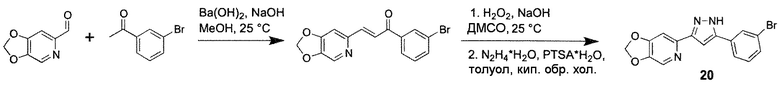
Стадия 1
К суспензии 1,3-диоксоло[4,5-c]пиридин-6-карбоксальдегида (WO/2019/208509) (35 мг, 0,23 ммоль) и 1-(3-бромфенил)этанона (46 мг, 0,23 ммоль) в метаноле (0,7 мл) добавляют Ba(OH)2⋅8H2O (5 мг) и NaOH (0,5 мг), и полученную смесь перемешивают при комнатной температуре (КТ) в течение ночи. После выпаривания метанола в вакууме, остаток растирают в воде (5 мл), твердое вещество собирают фильтрацией, промывают холодным метанолом (0,5 мл) и сушат с получением промежуточного соединения 1-(3-бромфенил)-3-([1,3]диоксоло[4,5-c]пиридин-6-ил)проп-2-ен-1-она (67 мг, 88%) в виде белого твердого вещества.
Стадия 2
К энергично перемешиваемой суспензии 1-(3-бромфенил)-3-([1,3]диоксоло[4,5-c]пиридин-6-ил)проп-2-ен-1-она (67 мг, 0,2 ммоль) в ДМСО (0,8 мл) добавляют водный раствор H2O2 (30%, 45 мг, 0,4 ммоль), затем по каплям добавляют водный NaOH (10%, 16 мкл, 0,04 ммоль). Желтую смесь перемешивают при КТ в течение 1,5 ч и выливают в холодный фосфатный буфер (20 мл, 0,1M, pH 7). Маслянистый остаток экстрагируют этилацетатом (2х15 мл), объединенные органические фракции сушат над Na2SO4, концентрируют в вакууме и остаток ресуспендируют в толуоле (0,8 мл). Суспензию в толуоле обрабатывают гидратом гидразина (35 мг, 0,7 ммоль) и гидратом PTSA (5 мг) и смесь перемешивают при кипении с обратным холодильником в течение 1,5 ч. После охлаждения, добавляют фосфатный буфер (0,3M, 20 мл) и продукт экстрагируют этилацетатом (2х20 мл). Объединенные органические фракции промывают насыщенным раствором соли (5 мл), сушат над Na2SO4 и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (15 г, силикагель 63-100, CHCl3/MeOH=100/1) с получением желтого твердого вещества, которое промывают Et2O (1 мл) с получением соединения 20 (25 мг, 36%) в виде белого твердого вещества.
Способ G: Синтез 1H-пиразолов
Иллюстративный пример: 4-[4-(1,3-Бензодиоксол-5-ил)-1H-имидазол-2-ил]-2-бромпиридин, соединение 48
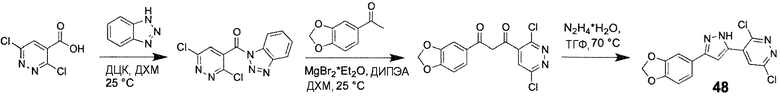
Смесь 1-(1,3-бензодиоксол-5-ил)этанона (138 мг, 0,84 ммоль), MgBr2⋅Et2O (542 мг, 2,1 ммоль) в ДХМ (5 мл) обрабатывают ДИПЭА (323 мг, 426 мкл, 2,5 ммоль) и перемешивают при КТ в течение 10 минут. Затем, неочищенную смесь 1H-бензотриазол-1-ил(3,6-дихлорпиридазин-4-ил)метанона, которую получают отдельно перемешиванием 3,6-дихлорпиридазин-4-карбоновой кислоты (203 мг, 1,05 ммоль), бензотриазола (125 мг, 1,05 ммоль) и ДЦК (216 мг, 1,05 ммоль) в сухом ДХМ (5 мл) при 25°C в течение 3 ч, добавляют по каплям в течение 5 минут. Полученную смесь перемешивают при 25°C в течение 12 ч, затем обрабатывают водной 0,5 M HCl (2 мл) и перемешивают в течение еще 10 минут при 25°C. После добавления воды (20 мл), смесь экстрагируют ДХМ (2⋅15 мл). Объединенные органические фракции промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией с получением промежуточного 1-(1,3-бензодиоксол-5-ил)-3-(3,6-дихлорпиридазин-4-ил)пропан-1,3-диона (190 мг, 67%) в виде оранжевого твердого вещества, которое применяют на следующей стадии без дальнейшей очистки. Это промежуточное соединение суспендируют в ТГФ (4 мл) и добавляют моногидрат гидразина (40 мг, 0,8 ммоль). Реакционную смесь перемешивают при 40°C в течение ночи, охлаждают и концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией (силикагель 63-100, 20 г, хлороформ/метанол=100/1) с получением соединения 48 (100 мг, 36% за две стадии) в виде светло-желтого твердого вещества.
Способ H: Синтез 1H-имидазолов
Иллюстративный пример: 4-[4-(1,3-Бензодиоксол-5-ил)-1H-имидазол-2-ил]-2-бромпиридин, соединение 33
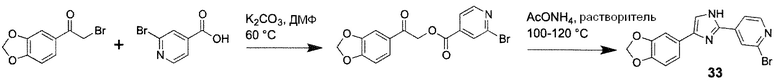
Стадия 1
Суспензию 1-(1,3-бензодиоксол-5-ил)-2-бромэтанона (729 мг, 3 ммоль), 2-бромпиридин-4-карбоновой кислоты (606 мг, 3 ммоль) и K2CO3 (414 мг, 3 ммоль) в ДМФ (6 мл) перемешивают при 55-60°C в течение 6 ч. После охлаждения, смесь выливают в воду (60 мл), перемешивают в течение 10 минут и полученный осадок собирают фильтрацией, промывают водой (20 мл) на фильтре и сушат с получением промежуточного соединения 2-(1,3-бензодиоксол-5-ил)-2-оксоэтил 2-бромпиридин-4-карбоксилата (899 мг, 87%), которое применяют на следующей стадии без дальнейшей очистки.
Стадия 2
Суспензию промежуточного соединения 2-(1,3-бензодиоксол-5-ил)-2-оксоэтил 2-бромпиридин-4-карбоксилата (364 мг, 1 ммоль) и AcONH4 (924 мг, 12 ммоль) в толуоле (7 мл) нагревают при 100°C при интенсивном перемешивании в течение 4 ч. После охлаждения, реакционную смесь обрабатывают фосфатным буфером (50 мл, 0,25 M, pH 7) и экстрагируют этилацетатом (2х50 мл). Объединенные органические фракции промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме. Остаток очищают колоночной хроматографией (силикагель 63-100, 30 г, CHCl3/MeOH=100/1 → 100/3). Очищенный продукт перекристаллизовывают из водного этанола (90%) с получением соединения 33 (140 мг, 41%) в виде бледного твердого вещества.
Способ I: Синтез 1H-1,2,4-триазолов
Иллюстративный пример 4-[3-(1,3-Бензодиоксол-5-ил)-1H-1,2,4-триазол-5-ил]-2-бромпиридин, соединение 30
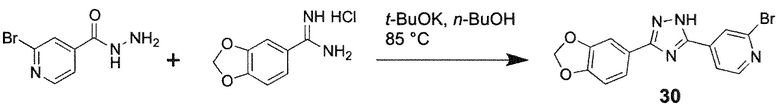
К раствору t-BuOK (84 мг, 0,75 ммоль) в n-BuOH (2 мл) добавляют бензо[1,3]диоксол-5-карбоксамидин гидрохлорид (100 мг, 0,5 ммоль) при 0°C и смесь перемешивают при КТ в течение 20 минут. После добавления гидразида 2-бромизоникотиновой кислоты (108 мг, 0,5 ммоль), желтую суспензию перемешивают при 85°C в течение 3 ч. После охлаждения до комнатной температуры, добавляют этанол (4 мл) и CO2 барботируют в течение 5 минут в суспензию. После удаления растворителей в вакууме, остаток растирают в воде (10 мл), полученный осадок собирают фильтрацией, промывают водой (5 мл) и сушат с получением соединения 30 (85 мг, 49%) в виде серого твердого вещества.
Способ J: Синтез 1H-1,2,4-триазолов
Иллюстративный пример 4-[3-(2-Бромпиридин-4-ил)-1H-1,2,4-триазол-5-ил]-N, N-диметиланилин, соединение 44
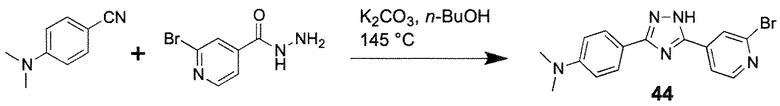
Смесь гидразида 2-бромизоникотиновой кислоты (151 мг, 0,7 ммоль), 4-(диметиламино)бензонитрила (307 мг, 2,1 ммоль) и K2CO3 (48 мг, 0,5 ммоль) в n-BuOH (2 мл) перемешивают при 145°C в течение 8 ч. Смесь концентрируют в вакууме, и неочищенный продукт очищают двумя колоночными хроматографиями (силикагель 63-100, 20 г, CHCl3/MeOH=100/1) и (силикагель 63-100, 20 г, ацетон/гексан=1/5) с получением соединения 44 (20 мг, 8%) в виде бежевого твердого вещества.
Способ K: Синтез 4-хлор-1H-пиразолов
4-[3-(1,3-Бензодиоксол-5-ил)-4-хлор-1H-пиразол-5-ил]-2-бромпиридин, соединение 28
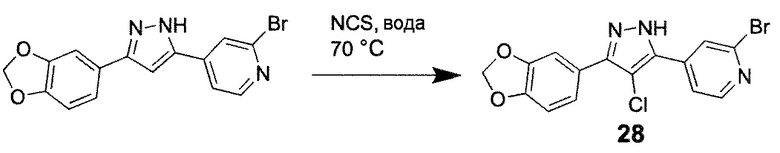
Суспензию соединения 9 (100 мг, 0,29 ммоль) и NCS (80 мг, 0,6 ммоль) в воде (3 мл) перемешивают при 70°C в течение 16 ч, ТСХ показала поглощение исходного материала. После охлаждения до КТ, осадок собирают фильтрацией, промывают водой (2х5 мл) и перекристаллизовывают из этанола (8 мл) с получением соединения 28 (52 мг, 47%) в виде серого твердого вещества.
Способ L: Синтез 1-[4-(4-R-пиперазин-1-ил)фенил]этанонов
Иллюстративный пример 1-{4-[4-(2-Метоксиэтил)пиперазин-1-ил]фенил}этенон
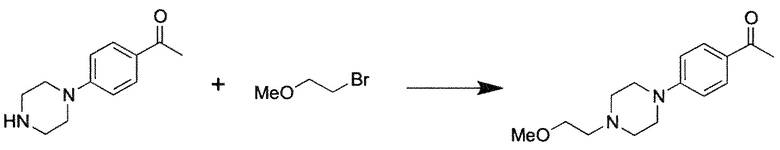
Смесь 1-[4-(пиперазин-1-ил)фенил]этанона (408 мг, 2 ммоль), 1-бром-2-метоксиэтана (417 мг, 3 ммоль) и Cs2CO3 (1304 мг, 4 ммоль) в ДМФ (5 мл) перемешивают при 25°C в течение ночи. После выпаривания ДМФ в вакууме, остаток разделяют между водой (25 мл) и этилацетатом (35 мл), водную фазу экстрагируют этилацетатом (25 мл), и объединенные органические фракции промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией (30 г силикагель 63-100, хлороформ → хлороформ/MeOH=100/2) с получением 1-{4-[4-(2-метоксиэтил)пиперазин-1-ил]фенил}этенона (525 мг, 99%) в виде светло-желтого твердого вещества.
2-Метоксиэтил 2,6-дибромпиридин-4-карбоксилат и 2-метоксиэтил 2,6-дихлорпиридин-4-карбоксилат получают в соответствии с опубликованным протоколом (Journal of Medicinal Chemistry (2012), 55, 10564-10571)
трет-Бутил (4-ацетилфенил)метилкарбамат, трет-бутил (4-ацетилфенил)-N-(2H3)метилкарбамат, трет-бутил (4-ацетилфенил)этилкарбамат и трет-бутил (4-ацетилфенил)(2-фторэтил)карбамат получают в соответствии с опубликованным протоколом (Organic Letters (2020), 22, 5522-5527).
1-[4-(4-Фторпиперидин-1-ил)фенил]этанон и 1-[4-(3-фторазетидин-1-ил)фенил]этанон получают в соответствии с опубликованным протоколом (WO 2011071570 A1).
Пентафторфенил 2-фторпиридин-4-карбоксилат и пентафторфенил 2-хлорпиридин-4-карбоксилат получают согласно способу E. 1-{4-[4-(2-Метоксиэтил)пиперазин-1-ил]фенил}этенон и 1-{4-[4-(2-фторэтил)пиперазин-1-ил]фенил}этенон получают согласно способу L.
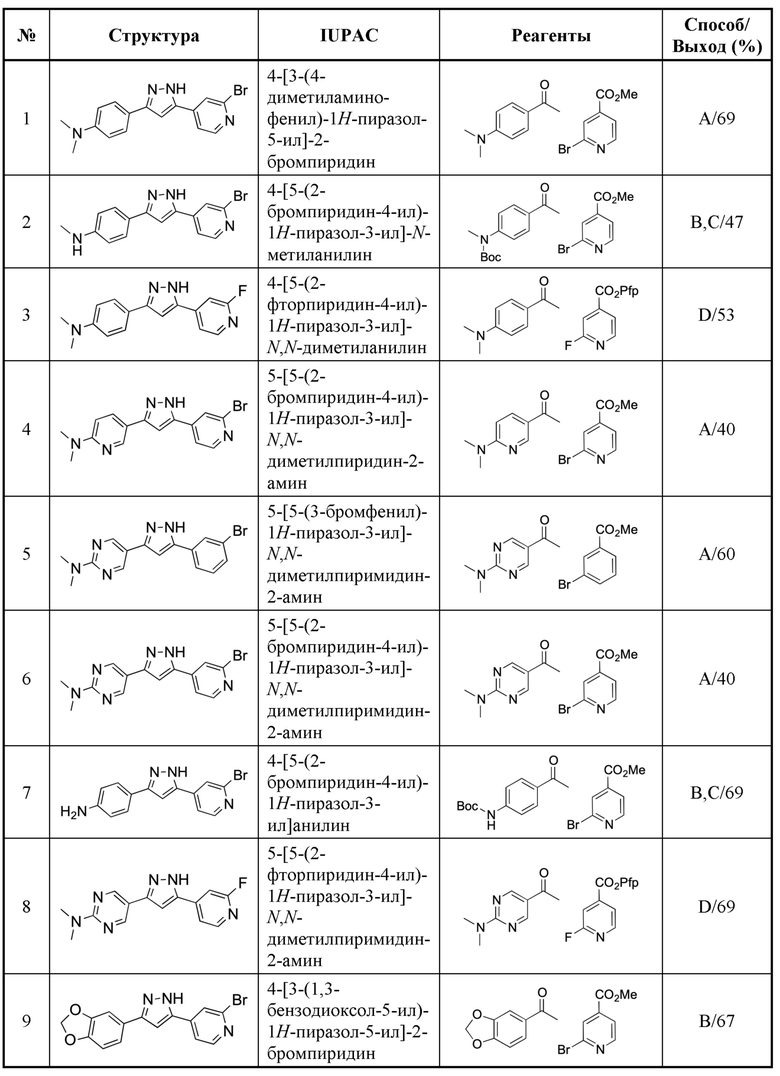
Типовые соединения по настоящему изобретению показаны в таблицах 1 и 2. В таблице 1 показаны название, структура, название IUPAC, исходные материалы для получения, способ синтеза и химический выход для конкретного примера соединения. В таблице 2 показано время удержания высокоэффективной жидкостной хроматографии (ВЭЖХ), молекулярная масса, определенная с помощью масс-спектрометрии низкого разрешения в сочетании с ВЭЖХ, и протонный ядерно-магнитный резонанс (1H-ЯМР) для конкретного примера соединения.
Таблица 1:
(растворитель, T)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% DCl, 313K)
(ДМСО-d6+1% DCl, 313K)
(ТФК-d1, 313K)
(ТФК-d1, 298K)
(ТФК-d1, 298K)
(ДМСО-d6, 298K)
(ТФК-d1, 298K, две формы в соотношении 10/1, основная форма)
(ДМСО-d6+1% DCl, 313K)
(ДМСО-d6+1% DCl, 313K)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% DCl)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% DCl, 313K)
(ДМСО-d6+1% DCl, 298K)
(ТФК-d1, 298K)
(ДМСО-d6+1% DCl, 313K)
(ДМСО-d6, 298K)
(CDCl3, 298K)
346,2 (100%)
(ДМСО-d6+1% DCl, 298K)
331,1 (100%)
(ТФК-d1, 298K)
287,2 (34%)
(ДМСО-d6+1% DCl, 298K)
345,2 (100%)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% DCl, 298K)
363,2 (100%)
(ДМСО-d6+1% DCl, 298K)
400,4 (100%)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% DCl, 298K)
380,1 (100%)
(ДМСО-d6+1% DCl, 298K)
346,1 (98%)
(ДМСО-d6+1% DCl, 298K)
347,2 (93%)
(ДМСО-d6+1% DCl, 298K)
345,1 (98%)
(ДМСО-d6+1% ТФК-d1, 298K)
347,1 (100%)
(ДМСО-d6+1% DCl, 298K)
346,2 (96%)
(ДМСО-d6+1% ТФК-d1, 298K)
346,1 (100%)
(ДМСО-d6+1% DCl, 298K)
327,4 (35%)
(ДМСО-d6+1% ТФК-d1, 298K)
288,2 (33%)
(ДМСО-d6+1% DCl, 298K)
409,1 (100%)
411,1 (50%)
(ДМСО-d6+1% DCl, 298K)
345,1 (98%)
(ДМСО-d6+1% DCl, 298K)
288,2 (34%)
(ДМСО-d6+1% DCl, 298K)
288,2 (30%)
(ДМСО-d6+1% DCl, 298K)
287,2 (34%)
(ДМСО-d6+1% DCl, 298K)
334,1 (61%)
335,1 (61%)
(ДМСО-d6+1% DCl, 298K)
302,2 (33%)
(ДМСО-d6+2% уксусная кислота-d4, 298K)
346,3 (100%)
(ДМСО-d6+1% DCl, 298K)
345,2 (98%)
(ДМСО-d6+1% ТФК-d1, 298K)
424,1 (100%)
426,1 (49%)
(ДМСО-d6+1% DCl, 298K)
360,1 (98%)
(ДМСО-d6+1% DCl, 298K)
337,2 (66%)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% ТФК-d1, 298K)
388,3 (37%)
(ДМСО-d6+1% DCl, 298K)
444,2 (100%)
(ДМСО-d6+1% DCl, 298K)
403,2 (98%)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% DCl, 298K)
(ДМСО-d6+1% DCl, 298K)
331,1 (33%)
(ДМСО-d6, 298K)
331,1 (98%)
(ДМСО-d6+1% DCl, 298K)
331,1 (98%)
(ДМСО-d6, 298K)
331,1 (98%)
(ДМСО-d6+1% DCl, 298K)
a градиент 50% CH3CN/50% H2O → 100% CH3CN за 30 мин
b градиент 5% CH3CN /100% H2O → 100% CH3CN за 30 мин
Пример 2: Исследования связывания с агрегированным альфа-синуклеином, тау и А бета
Для анализа аффинности связывания и селективности соединений к мишени используют два типа анализов связывания фибрилл in vitro: анализ насыщения и конкурентный анализ.
1) Подготовка фибрилл
Альфа-синуклеин и тау46 очищают из E. coli в соответствии с установленными протоколами (Nuscher B, et al., J. Biol. Chem., 2004; 279(21):21966-75), Aβ1-42 получают от из rPeptide, Watkinsville, GA, USA. Фибриллы получают путем агрегации рекомбинантного белка при постоянном перемешивании. В частности, 70 мкМ альфа-синуклеина, смешанного со 100 мМ NaCl в 50 мМ Трис, рН 7,0+0,02% NaN3 инкубируют при 1400 об/мин, 37°C в течение 96 ч. 10 мкМ тау46 инкубируют с 0,03 мг/мл гепарина в 50 мМ Трис, pH=7,0 при 1000 об/мин, 37°C в течение 72 ч. Aβ1-42 растворяют в 20 мМ NaPi, pH=8,0+0,2 мМ ЭДТК+0,02% NaN3 (Deeg AA, et al., Biochim. Biophys. Acta, 2015; 1850(9): 1884-90; Goedert M, et al. Nature, 1996; 383(6600): 550-3).
2a) Анализ насыщения связывания
Обработанные ультразвуком фибриллы αSYN (0,04 мкМ) или тау46 (0,4 мкМ) или Aβ1-42 (6 мкМ), разведенные в PBS, инкубируют с уменьшающимися концентрациями [3H]-меченого соединения (48 нМ или 24 нМ - 23 пМ) в 50 мМ Tris-основании, 10% этаноле, 0,05% Tween20, pH 7,4. Для определения неспецифического связывания, соответствующее не меченое соединение (400 нМ) добавляют к дублирующему набору реакций связывания.
Планшеты для анализа инкубируют при перемешивании при 37°C в течение 2 ч, покрывают пластиковой пленкой (Resealable Tape, PerkinElmer, Waltham, MA, USA). инкубируют с 5 мг/мл полиэтиленимина (PEI, раствор поли(этиленимина), Sigma Aldrich Chemie GmbH, Taufkirchen, Germany) при 4°C в течение 30 мин. После инкубации, связанные и свободные лиганды разделяют вакуумной фильтрацией с использованием сборщика клеток (Filtermate harvester, PerkinElmer, Waltham, MA, USA). Фильтр трижды промывают буфером, охлажденным до 4°C, и затем сушат в микроволновой печи в течение 2 минут при средней мощности. Пластины сплавленного сцинтиллятора (MeltiLex™ B/HS, PerkinElmer, Waltham, MA, USA) вплавляют в фильтр с помощью нагревательной пластины, установленной на 120°C. После затвердевания при комнатной температуре, фильтр запечатывают в пластиковый пакет (пакет для образцов для MicroBeta®, PerkinElmer, Waltham, MA, USA). Накопление трития сразу же подсчитывают в жидкостном сцинтилляционном счетчике (Wallac MicroBeta® TriLux, 1450 LSC & Luminescence Counter, PerkinElmer, Waltham, MA, USA). Радиоактивность наносят на график к повышающимся концентрациях меченого тритием соединения или холодного соединения. Данные подгоняют с применением анализа нелинейной регрессии в GraphPad Prism (GraphPad Software, Inc., версия 7.03, La Jolla, CA, USA).
2b) Модифицированный анализ насыщения связывания
Для анализа насыщения связывания, фиксированную концентрацию обработанных ультразвуком рекомбинантных αSYN (15 нМ/лунку) или тау46 (250 нМ/лунку) или Aβ1-42 (1 мкМ/лунку) фибрилл человека, разведенных в фосфатно-солевом буфере (PBS), инкубируют в планшетах с низким связыванием (96-луночный микропланшет для испытаний, Ratiolab GmbH, Dreieich, Germany) с [3H]-соединением 1 или [3H]-соединением 2 в возрастающих концентрациях (от 0,05 нМ до 12 нМ/24 нМ) в 30 мМ Tris HCl, 10% этанола, 0,05% Tween20, pH 7,4 (далее названном инкубационным буфером) в общем объеме 200 мкл/лунку. Неспецифическое связывание меченого атома определяют путем совместной инкубации с 400 нМ не меченого соединения 1 или соединения 2. Оптимальные концентрации фибрилл определяют с использованием анализа определения концентрации.
Планшеты, покрытые съемной герметизирующей лентой (PerkinElmer), инкубируют на шейкере (MaxQ™ 6000, диаметр орбиты 1,9 см, Thermo Fisher Scientific Inc., Marietta, OH, USA) при 45 об/мин в течение двух часов при 37°C. После инкубации, связанные и свободные радиолиганды разделяют вакуумной фильтрацией через стекловолоконный плоский фильтр В (PerkinElmer) с использованием сборщика клеток для плоских фильтров (PerkinElmer). Для сбора клеток из планшетов, содержащих фибриллы αSYN и Aβ1-42, плоский фильтр дополнительно инкубируют с 5 мг/мл полиэтиленимина в течение 30 минут при 4C перед сбором клеток. Фильтр трижды промывают 100 мл (примерно 1 мл/лунку) ледяного буфера для инкубации и затем сушат в микроволновой печи в течение 2,5 минут при средней мощности. Расплав на сцинтилляционных листах (MeltiLex™ B/HS, PerkinElmer) вплавляют в фильтр с помощью нагревательной пластины, установленной на 120°C. После затвердевания при комнатной температуре, фильтр запечатывали в пластиковый пакет для образцов (PerkinElmer). Накопление трития сразу же подсчитывают на жидкостном сцинтилляционном счетчике Wallac MicroBeta® TriLux (PerkinElmer). Радиоактивность наносят на график к концентрации 3H-меченного соединения. Данные подгоняют с использованием нелинейного регрессионного анализа в GraphPad Prism (GraphPad Software, Inc., версия 7.03, La Jolla (CA), USA).
3a) Анализ конкурентного связывания
Обработанные ультразвуком рекомбинантные αSYN (200 нМ/лунку) или тау46 (208 нМ/лунку) фибриллы человека в фиксированной концентрации, разведенные в фосфатно-солевом буфере (PBS), инкубируют в планшетах с низким связыванием (96-луночный микропланшет для испытаний, Ratiolab GmbH, Dreieich, Germany) вместе с 1 нМ [3H]-соединения 1 и серийными разведениями 1:4 представляющего интерес холодного соединения, начиная с 1 мкМ, разведенного в 50 мМ Tris-основании, 10% EtOH, 0,05% Tween20, pH 7,4. Для расчета значений Ki, значение KD соединения 1 по отношению к обработанным ультразвуком рекомбинантным фибриллам αSYN человека устанавливают равным 3 нМ.
Планшеты для анализа инкубируют при перемешивании при 37°C в течение 2 ч, накрыв пластиковой пленкой (Resealable Tape, PerkinElmer, Waltham, MA, USA). До сбора клеток, фильтр (принтованный плоский фильтр В, PerkinElmer, Waltham, MA, USA) инкубируют с 5 мг/мл полиэтиленимина (PEI, раствор поли(этиленимина), Sigma Aldrich Chemie GmbH, Taufkirchen, Germany) при 4°C в течение 30 мин. После инкубации, связанные и свободные лиганды разделяют вакуумной фильтрацией с использованием сборщика клеток (сборщик клеток с плоского фильтра, PerkinElmer, Waltham, MA, USA). Фильтр трижды промывают буфером, охлажденным до 4°C, и затем сушат в микроволновой печи в течение 2 мин при средней мощности. Фильтр запечатывали в пластиковый пакет (пакет для образцов для MicroBeta®, PerkinElmer, Waltham, MA, USA) вместе с добавленным сцинтиллятором (BETAPLATE SCINT, PerkinElmer, Waltham, MA, USA). Накопление трития сразу же подсчитывают в жидкостном сцинтилляционном счетчике (Wallac MicroBeta® TriLux, 1450 LSC & Luminescence Counter, PerkinElmer, Waltham, MA, USA). Радиоактивность наносят на график к увеличению концентрации меченного тритием соединения или холодного соединения. Точки данных подгоняют с использованием нелинейного регрессионного анализа в GraphPad Prism (GraphPad Software, Inc., версия 7.03, La Jolla, CA, USA).
3b) Модифицированный анализ конкурентного связывания
Для экспериментов по конкурентному связыванию, фиксированную концентрацию рекомбинантных фибрилл αSYN (обработанных ультразвуком, 15 нМ/лунку) инкубируют с 1 нМ [3H]-соединения 1 или [3H]-соединения 2 и уменьшающимися концентрациями серийного разведения не меченого конкурента (серийное разведение 1:4 или 1:3,5 или 1:3 с указанием диапазонов концентраций не меченого конкурента 1 мкМ-1 пМ или 1 мкМ-4 нМ или 1 мкМ-17 пМ, соответственно) в 30 мМ Tris-HCl, 10% этаноле, 0,05% Tween20, pH 7,4, в общем объеме 200 мкл/лунку. Планшеты инкубируют в течение 4,5 часов при комнатной температуре, накрыв съемной герметизирующей лентой (PerkinElmer, Waltham, MA, USA).
Фильтрацию и считывание проводят, как описано для анализов насыщения связывания. Связывание [3H]-меченого лиганда [в CMP] наносят на график к возрастающим концентрациям конкурента, и данные подгоняют с использованием нелинейного регрессионного анализа для расчета значений IC50 и Ki (GraphPad Software, Inc., версия 7.03, La Jolla (CA), USA). В анализе конкурентного связывания с обработанными ультразвуком рекомбинантными фибриллами αSYN, расчет значений Ki проводят на основе значений KD, равных 0,6 нМ и 0,2 нМ для соединения 1 и соединения 2, соответственно.
Анализ насыщения связывания фибрилл дает значения KD. Этот анализ проводят с непосредственно радиоактивно меченными соединениями (например, мечение 3H). Значения KD показаны в таблицах 3a и 3b. Анализы связывания насыщения проводят с использованием в качестве структур-мишеней агрегатов фибрилл, полученных из рекомбинантного альфа-синуклеина человека, Aβ1-42 и тау46, соответственно.
Результаты показывают высокую аффинность к фибриллам альфа-синуклеина. Соединение 1, соединение 2 и соединение 12 показали очень высокую аффинность при значениях KD ниже 10 нМ. Соединение 1 и соединение 2 также тестируют в отношении связывания с фибриллами A бета и тау46, и они показывают селективность от хорошей до превосходной, что доказывает пригодность этих соединений для диагностического обнаружения агрегированного альфа-синуклеина.
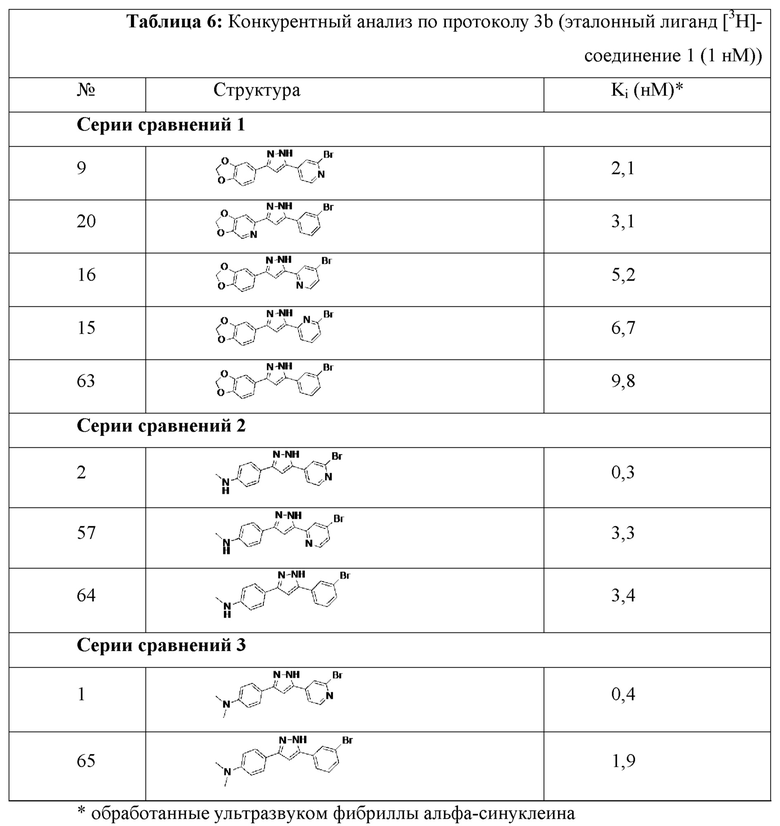
Конкурентный анализ фибрилл дает значения Ki. Ki представляет собой количественную меру, которая представляет собой концентрацию не радиоактивного тестируемого соединения (лиганд-конкурент), необходимую для замены 50% эталонного соединения, в данном случае [3H]-соединения 1 (1 нМ) или [3H]-соединения 2 (1 нМ), которое связано с структурами-мишенями (1 нМ), в нашем случае, рекомбинантным альфа-синуклеином и фибриллами тау46. Этот анализ подходит для скрининга не радиоактивных лигандов. Значения Ki, полученные для тестируемых соединений, приведены в таблицах 4а, 4b и 5.
* повторение
Таблица 5: Конкурентный анализ по протоколу 3b (эталонный лиганд [3H]-соединение 2 (1 нМ))
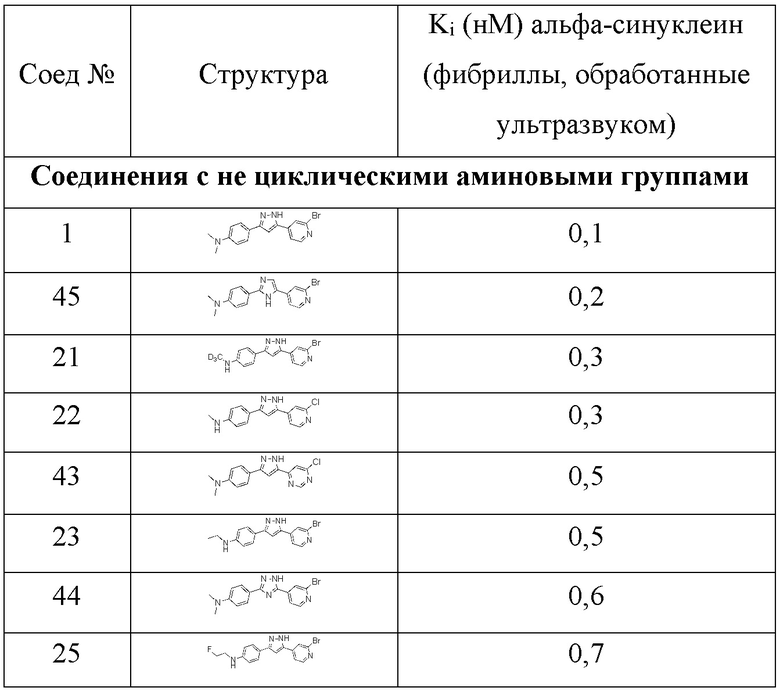

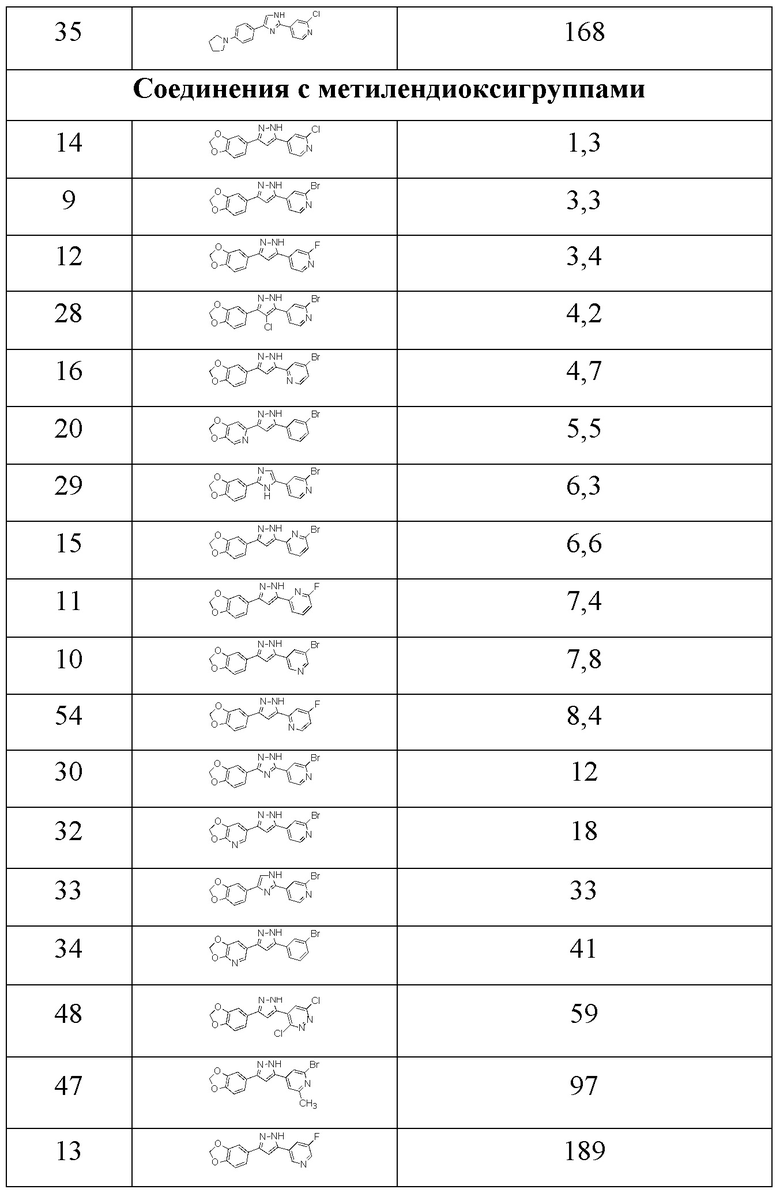
Результаты различных конкурентных анализов указывают на высокую аффинность связывания с фибриллами альфа-синуклеина для тестируемых соединений (более низкие значения Ki указывают на более высокую аффинность связывания). В случае конкурентного анализа относительно соединения 1 (таблица 4а), аналогичные значения Ki для фибрилл альфа-синуклеина и тау46 указывают на аналогичную селективность, как у соединения 1, для соединений с более высокими значениями Ki для фибрилл тау46, а не для фибрилл альфа-синуклеина, где данные предполагают дальнейшее улучшение селективности.
Кроме того, соединения 63, 64 и 65 были протестированы в прямом сравнении с соответствующими соединениями, содержащими атом азота в одном из фенильных колец. В то время как в таблице 6 для соединения 1 получено значение Ki, равное 0,4 нМ, значение Ki для соединения 65 (у которого отсутствует атом азота в фенильном кольце) было намного выше (1,9 нМ), что указывает на значительно улучшенную аффинность связывания для соединения 1. Анализ свойств связывания на основе значений Ki показал значительно улучшенную аффинность связывания для N-содержащих соединений. Значения Ki, полученные для тестируемых соединений в анализе конкурентного связывания с эталонным лигандом [3H]-соединением 1 (1 нМ), показаны в таблице 6.
Пример 3: Терапевтический эффект в клеточной культуре
Для идентификации потенциальных терапевтически полезных эффектов, соединения тестируют на двух клеточных моделях агрегированной альфа-синуклеин-зависимой токсичности в клетках H4. Эти клеточные модели допускают индуцируемую сверхэкспрессию слитых конструкций альфа-синуклеина полу-Venus (V1S+SV2) или полноразмерного альфа-синуклеина (модель подробно описана в: Bartels M, Weckbecker D, Kuhn PH, Ryazanov S, Leonov A, Griesinger C, Lichtenthaler SF, Bötzel K, Giese A: Iron-mediated aggregation and toxicity in a novel neuronal cell culture model with inducible alpha-synuclein expression, Sci. Rep. 2019 Jun 24; 9(1):9100). Кроме того, добавление ДМСО и FeCl3 способствует агрегации альфа-синуклеина, что связано с увеличением цитотоксичности. Анализ клеточной культуры использует эти эффекты через определение изменения числа клеток и изменения доли конденсированных ядер, что свидетельствует об апоптозе, чтобы получить информацию о том, какие соединения демонстрируют наиболее многообещающие эффекты и, таким образом, могут быть терапевтически полезными.
Данные обобщены ниже в Таблице 7. В этих экспериментах клетки инкубируют с 100 мкМ FeCl3 и 0,75% ДМСО в присутствии 10 мкМ anle 138c в качестве положительного контроля, соединений 1-19 (10 мкМ) или ДМСО в качестве отрицательного контроля. Через 48 ч клетки визуализируют высокопроизводительной системой визуализации OPERA и анализируют с помощью программного обеспечения Acapella (Perkin Elmer). В таблице показано изменение числа клеток и изменение доли клеток с конденсированными ядрами. (т.е. апоптотических клеток) по сравнению с контролем ДМСО. Уменьшение доли конденсированных ядер (при отсутствии сильного уменьшения количества клеток) свидетельствует о благоприятном терапевтическом эффекте.
Пример 4: In vivo биораспределение [11C]-меченого соединения 1 и соединения 2
[11C]-меченое соединение 1 и соединение 2, соответственно, вводят внутривенно в хвостовую вену мышей. Затем мышей визуализируют с помощью ПЭТ прибора для мелких животных. Для обоих соединений наблюдают хорошее проникновение через гематоэнцефалический барьер с значениями SUV >1,5. Кроме того, было обнаружено быстрое вымывание из головного мозга. Период полувыведения для [11C]-меченого соединения 1 и соединения составляет 12 минут и 9 минут, соответственно.
Пример 5: Ауторадиография с использованием ткани головного мозга человека
Ауторадиографию проводят на гистологических срезах замороженной ткани головного мозга с использованием тритированного соединения 1 и ткани головного мозга, полученной из поясной извилины больного деменцией с тельцами Леви. При использовании [3H]-соединения 1 в концентрации 3 нМ для инкубации ткани с последующими стадиями промывания, можно наблюдать предпочтительное связывание с серым веществом, которое является моделью, идентичной известному распределению агрегированного альфа-синуклеина (фигура 1, левая панель). Когда специфическое связывание блокируется избытком не тритированного (т.е. «холодного») соединения 1, можно увидеть отсутствие или очень слабое неспецифическое связывание соединения 1 с тканью головного мозга (фигура 1, правая панель).
Эти данные показывают, что соединение 1 специфически и с высокой аффинностью связывается с патологически агрегированным альфа-синуклеином, присутствующим у пациента-человека с синуклеинопатией, и позволяет диагностическое обнаружение агрегированного альфа-синуклеина.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ФУРО[3,2-В]ПИРАНА, ПРИМЕНИМЫЕ В СИНТЕЗЕ АНАЛОГОВ | 2011 |
|
RU2579511C2 |
| N-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ АНИЛИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ НА СИНТАЗУ ОКИСЛА АЗОТА, ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ, ПРОИЗВОДНЫЕ НИТРОФЕНИЛКАРБОНОВОЙ КИСЛОТЫ | 1995 |
|
RU2167858C2 |
| ПРОТИВОГЕЛЬМИНТНЫЕ АЗА-БЕНЗОТИОФЕНОВЫЕ И АЗА-БЕНЗОФУРАНОВЫЕ СОЕДИНЕНИЯ | 2020 |
|
RU2833610C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2014 |
|
RU2687276C2 |
| РАДИОАКТИВНО МЕЧЕННЫЕ ПРОИЗВОДНЫЕ 2-АМИНО-6-ФТОР-N-[5-ФТОР-ПИРИДИН-3-ИЛ]-ПИРАЗОЛО[1, 5-А]ПИРИМИДИН-3-КАРБОКСАМИДА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРА ATR КИНАЗЫ, ПРЕПАРАТЫ НА ОСНОВЕ ЭТОГО СОЕДИНЕНИЯ И ЕГО РАЗЛИЧНЫЕ ТВЕРДЫЕ ФОРМЫ | 2015 |
|
RU2719583C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ATR КИНАЗЫ (ВАРИАНТЫ) | 2014 |
|
RU2720408C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2020 |
|
RU2750148C1 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛСУЛЬФОНАМИДОВ В КАЧЕСТВЕ ОБРАТНЫХ АГОНИСТОВ СВЯЗАННОГО С РЕТИНОИДАМИ ОРФАННОГО РЕЦЕПТОРА ГАММА ROR GAMMA (Т) | 2015 |
|
RU2738843C2 |
| ИНСЕКТИЦИДНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2493148C2 |
| ЛЕЧЕНИЕ ИЛИ ПРОФИЛАКТИКА ПРОЛИФЕРАТИВНЫХ СОСТОЯНИЙ | 2010 |
|
RU2705548C2 |
Изобретение относится к соединениям, представленным общими формулами Ia, Ib, IIa или IIb:
 , фармацевтической композиции на их основе, а также примнению и способу визуализации альфа-синуклеина и для диагностики заболеваний, которые связаны с агрегацией альфа-синуклеина. Технический результат: получены новые соединения, которые подходят для лечения и профилактики заболеваний, связанных с агрегацией альфа-синуклеина. 8 н. и 12 з.п. ф-лы, 5 пр., 7 табл., 1 ил.
, фармацевтической композиции на их основе, а также примнению и способу визуализации альфа-синуклеина и для диагностики заболеваний, которые связаны с агрегацией альфа-синуклеина. Технический результат: получены новые соединения, которые подходят для лечения и профилактики заболеваний, связанных с агрегацией альфа-синуклеина. 8 н. и 12 з.п. ф-лы, 5 пр., 7 табл., 1 ил.
1. Соединение, представленное общей формулой Ia, Ib, IIa или IIb:

или его соль,
где
 выбран из
выбран из  и
и 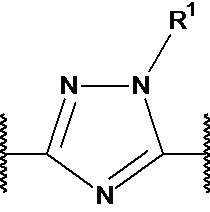 и
и
 выбран из
выбран из 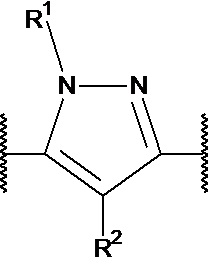 ,
,  и
и 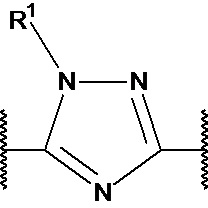 ;
;
Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 независимо выбраны из CR3 и N при условии, что по меньшей мере один из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N, и при условии, что в формулах Ia или Ib по меньшей мере один из Y1, Y3, Y4 и Y6 представляет собой N;
R1 независимо выбирают из водорода, C1-4 алкила и -(CH2)-O-P(=O)(OR)(OR), где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами;
R2 независимо выбирают из водорода, галогена и C1-4 алкила, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами;
R представляет собой водород или катион;
R3 представляет собой водород;
R4 и R5 независимо выбраны из H и C1-4 алкила, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами или где R4 и R5 вместе с атомом азота, с которым они связаны, образуют 4-6-членное насыщенное гетероциклическое кольцо, выбранное из азетидинила, пиперидинила, пиперазинила и морфолинила, где 4-6-членное насыщенное гетероциклическое кольцо может быть необязательно замещено одним или несколькими R6;
R6 независимо выбран из галогена и C1-4 алкила, где C1-4 алкил может быть необязательно замещён одним или несколькими галогенами; и
Hal представляет собой галоген,
или соединение представляет собой 
при условии, что соединение не представляет собой
 .
.
2. Соединение по п. 1, отличающееся тем, что
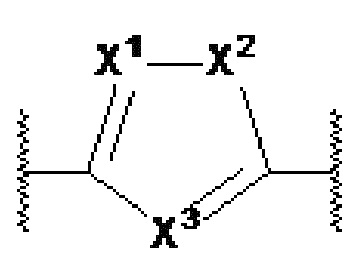 представляет собой
представляет собой 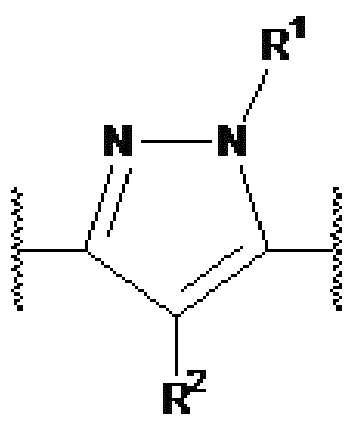 или
или
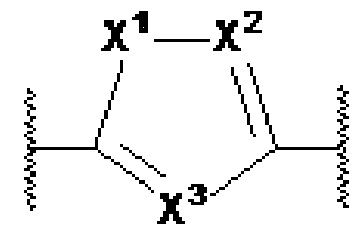 представляет собой
представляет собой 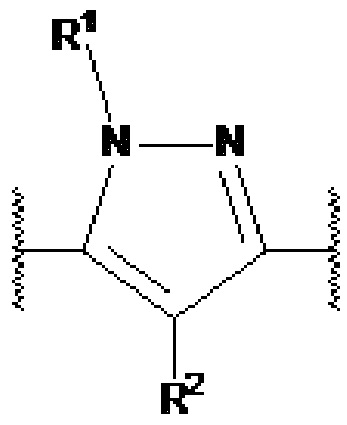 или
или  ,
,
отличающееся тем, что R1 и R2 такие, как определены в п. 1.
3. Соединение по п. 2, отличающееся тем, что
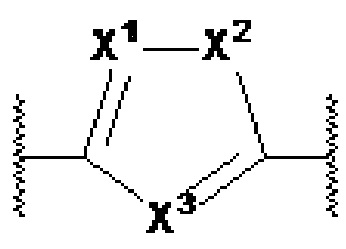 представляет собой
представляет собой 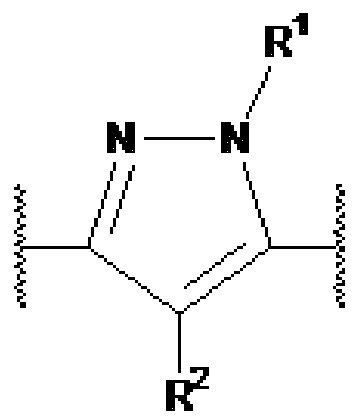 или
или 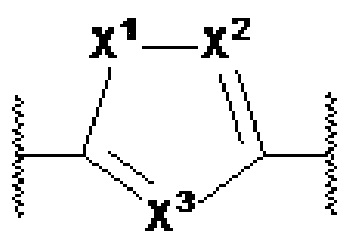 представляет собой
представляет собой  ,
,
отличающееся тем, что R1 и R2 такие, как определены в п. 1.
4. Соединение по любому из пп. 1-3, отличающееся тем, что один, или два, или три из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой N.
5. Соединение по любому из пп. 1-4, отличающееся тем, что
в формулах Ia или Ib Y1 представляет собой N и по меньшей мере один из Y3, Y4 и Y6 представляет собой N; или
в формулах IIa или IIb Y1 представляет собой N и по меньшей мере один из Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N.
6. Соединение по любому из пп. 1-5, отличающееся тем, что Y1 представляет собой N и Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой CR3, предпочтительно Y1 представляет собой N и Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляют собой СН.
7. Соединение по любому из пп. 1-6, отличающееся тем, что R2 представляет собой H.
8. Соединение по любому из пп. 1-7, отличающееся тем, что по меньшей мере один из R4 и R5 представляет собой C1-4 алкил, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами.
9. Соединение, представленное общей формулой Ia, Ib, IIa или IIb:
или его соль,
где
выбран из и и
выбран из , и ;
Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 независимо выбраны из CR3 и N при условии, что по меньшей мере один из Y1, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 представляет собой N, и при условии, что в формулах Ia или Ib по меньшей мере один из Y1, Y3, Y4 и Y6 представляет собой N;
R1 независимо выбирают из водорода, C1-4 алкила и -(CH2)-O-P(=O)(OR)(OR), где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами;
R2 независимо выбирают из водорода, галогена и C1-4 алкила, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами;
R представляет собой водород или катион;
R3 представляет собой водород;
R4 и R5 независимо выбраны из H и C1-4 алкила, где C1-4 алкил может быть необязательно замещен одним или несколькими галогенами или где R4 и R5 вместе с атомом азота, с которым они связаны, образуют 4-6-членное насыщенное гетероциклическое кольцо, выбранное из азетидинила, пиперидинила и морфолинила; и
Hal представляет собой галоген,
отличающееся тем, что соединение является обнаруживаемо меченным 18F, 11C, 125I, 123I, 131I, 77Br и 76Br.
10. Соединение по п. 9, отличающееся тем, что соединение является обнаруживаемо меченным 18F или 11C.
11. Диагностическая композиция для диагностики заболевания, связанного с агрегацией альфа-синуклеина, содержащая диагностически эффективное количество соединения, определенного в любом из пп. 1-9, или его соли и, необязательно, фармацевтически приемлемый носитель.
12. Фармацевтическая композиция для лечения заболевания, связанного с агрегацией альфа-синуклеина, содержащая терапевтически эффективное количество соединения, определенного в любом из пп. 1-8, или его соли и, необязательно, фармацевтически приемлемый носитель.
13. Соединение по любому из пп. 1-9 или его соль для применения в диагностике заболевания, связанного с агрегацией альфа-синуклеина.
14. Соединение по любому из пп. 1-8 или его соль для применения в лечении заболевания, связанного с агрегацией альфа-синуклеина.
15. Соединение для применения по п. 13 или 14, отличающееся тем, что заболевание, связанное с агрегацией альфа-синуклеина, выбрано из болезни Паркинсона, деменции с тельцами Леви и множественной системной атрофии.
16. Применение соединения по любому из пп. 1-9 или его соли для ингибирования агрегации альфа-синуклеина in vitro.
17. Соединение по любому из пп. 1-9 или его соль для применения для визуализации агрегатов альфа-синуклеина.
18. Способ визуализации отложений агрегированного альфа-синуклеина, где способ включает следующие стадии:
(i) введение субъекту обнаруживаемого количества обнаруживаемо меченного соединения по п. 9 субъекту;
(ii) предоставление достаточного количества времени для связывания соединения с агрегированным альфа-синуклеином; и
(iii) обнаружение соединения, связанного с агрегированным альфа-синуклеином.
19. Способ получения обнаруживаемо меченного соединения по п. 9 или его соли, где способ включает взаимодействие двух предшественников соединений, где первое соединение-предшественник представляет собой соединение по любому из пп. 1-8 или его соль, а второе соединение-предшественник представляет собой 2-[18F]фторэтилтозилат, TBA[18F], [11C]MeI, [11C]CH2O или 99% газообразный тритий/катализатор Керра.
20. Способ получения обнаруживаемо меченного соединения по п. 9 или его соли, где способ включает взаимодействие двух предшественников соединений, где первое соединение-предшественник представляет собой соединение 24
или его соль, а второе соединение-предшественник представляет собой 18FK.
| US2005075375A1, 07.04.2005 | |||
| НОВЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ИНГИБИРОВАНИЯ АГРЕГАЦИИ БЕЛКОВ, ВОВЛЕЧЕННЫХ В ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С АГРЕГАЦИЕЙ БЕЛКОВ, И НЕЙРОДЕГЕНЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2531915C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ЛЕЧЕНИЯ БЕТА-АМИЛОИДНЫХ ЗАБОЛЕВАНИЙ И СИНУКЛЕИНОПАТИЙ | 2009 |
|
RU2501792C2 |

