ПЕРЕКРЕСТНАЯ ССЫЛКА
[1] По настоящей заявке испрашивается приоритет предварительной заявке США с серийным номером № 62/314356, поданной 28 марта 2016 года, содержание которой включено в настоящее описание в качестве ссылки в полном объеме.
УРОВЕНЬ ТЕХНИКИ
[2] В любой конкретный момент времени многочисленные терапевтические средства, направленные против злокачественной опухоли, находятся в фазе I или фазе II клинических испытаний и оценки; однако большинство из них не проходят далее. В действительности, согласно оценке, более 90% терапевтических средств, направленных против злокачественной опухоли, оказываются неуспешными в клинических испытаниях фазы I или II. Частота неуспеха в клинических испытаниях фазы III составляет практически 50%, и стоимость разработки новых лекарственных средств от открытия до испытаний фазы III составляет от 0,8 миллиарда долларов до 1,7 миллиарда долларов, и занимает от восьми до десяти лет.
[3] Кроме того, многие пациенты не отвечают даже на стандартные лекарственные средства, для которых показано, что они являются эффективными. По причинам, которые в настоящее время не совсем ясны или которые нелегко оценить, отдельные пациенты могут не отвечать на терапию стандартным лекарственным средством. В некоторых случаях введение комбинаций лекарственных средств может быть более эффективным для лечения злокачественной опухоли, чем в случае лекарственных средств, вводимых индивидуально. Эти комбинации лекарственных средств могут действовать синергично, повышая активность лекарственных средств, направленную против злокачественной опухоли. В некоторых случаях лекарственные средства, которые являются не особенно эффективными, могут найти новые и неожиданные применения при комбинировании с дополнительными способами медикаментозной терапии.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[4] В одном аспекте изобретение относится к способу лечения злокачественной опухоли крови, включающему введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества ингибитора CDK, соответствующего формуле I:
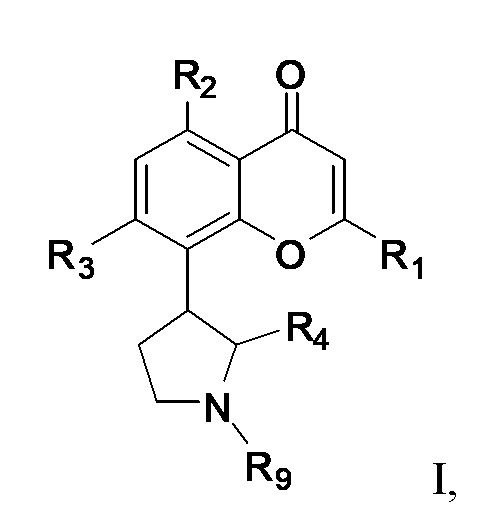
или его фармацевтически приемлемой соли, где:
R1 представляет собой необязательно замещенный фенил;
каждый из R2 и R3 независимо выбран из гидрокси и OR8, где R8 представляет собой необязательно замещенный C1-C10-алкил;
R4 представляет собой необязательно замещенный C1-C4-алкил; и
R9 представляет собой водород или необязательно замещенный C1-C4-алкил;
и терапевтически эффективного количества ингибитора BCL-2.
[5] В некоторых аспектах изобретение относится к способу лечения злокачественной опухоли, включающему введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества ингибитора CDK, соответствующего формуле I:
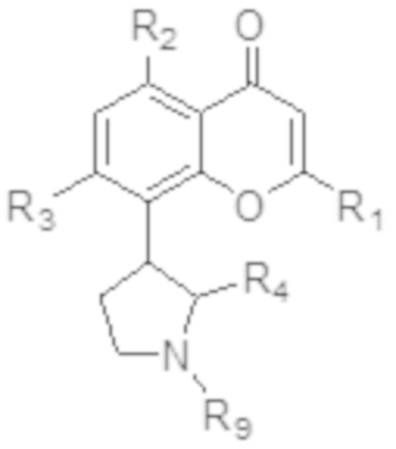 I,
I,
или его фармацевтически приемлемой соли, где:
R1 представляет собой необязательно замещенный фенил;
каждый из R2 и R3 независимо выбран из гидрокси и OR8, где R8 представляет собой необязательно замещенный C1-C10-алкил;
R4 представляет собой необязательно замещенный C1-C4-алкил; и
R9 представляет собой водород или необязательно замещенный C1-C4-алкил;
и терапевтически эффективного количества ингибитора протеасом. В определенных вариантах осуществления, злокачественная опухоль выбрана из злокачественной опухоли крови и трижды негативного рака молочной железы (TNBC).
[6] В определенных вариантах осуществления соединение формулы I представляет собой соединение формулы Ia:
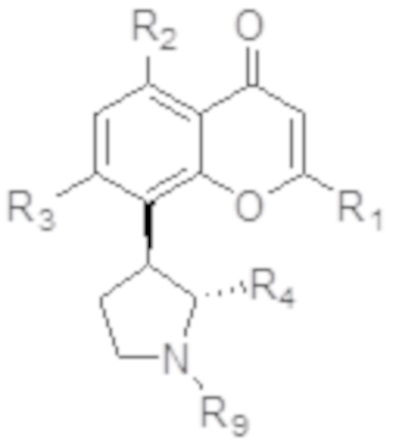 Ia или формуле его фармацевтически приемлемую соль.
Ia или формуле его фармацевтически приемлемую соль.
[7] В определенных вариантах осуществления для соединения или соли формулы I или Ia, R1 является необязательно замещенным одним или несколькими заместителями, независимо выбранными из гидрокси, циано, галогена, амино, C1-C4-алкила, C1-C4-алкокси, C1-C4-гидроксиалкила, C1-C4-галогеналкила и нитро. В определенных вариантах осуществления R1 является замещенным одним или несколькими заместителями, независимо выбранными из галогена и C1-C4-галогеналкила. В определенных вариантах осуществления R1 представляет собой 2-хлор-4-трифторметилфенил.
[8] В определенных вариантах осуществления для соединения формулы I или формулы Ia каждый из R2 и R3 независимо выбран из гидрокси и OR8, где R8 представляет собой C1-C10-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления каждый из R2 и R3 представляет собой гидрокси.
[9] В определенных вариантах осуществления для соединения формулы I или формулы Ia R4 представляет собой C1-C4-алкил, замещенный одним или несколькими заместителями, выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R4 представляет собой C1-C4-алкил, замещенный одним или несколькими заместителями, выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R4 представляет собой 2-гидроксиметил.
[10] В определенных вариантах осуществления для соединения формулы I или формулы Ia R9 представляет собой C1-C4-алкил, необязательно замещенный гидрокси, циано, галогеном, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R9 представляет собой метил. В определенных вариантах осуществления соединение формулы I представляет собой соединение формулы Ib:
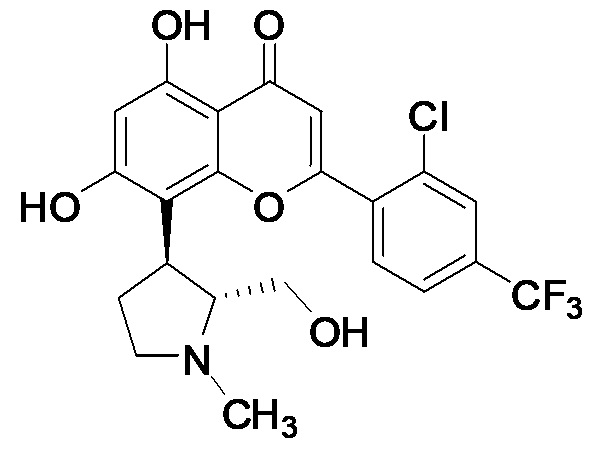 Ib
Ib
или его фармацевтически приемлемую соль.
[11] В определенных вариантах осуществления ингибитор BCL-2 согласно способам, описанным в настоящем описании, представляет собой миметик BH3. Ингибитор BCL-2 может специфически ингибировать белок Bcl-2. Ингибитор BCL-2 может быть выбран из навитоклакса, венетоклакса, A-1155463, A-1331852, ABT-737, обатоклакса, S44563, TW-37, A-1210477, AT101, HA14-1, BAM7, сабутоклакса, UMI-77, гамбогиновой кислоты, маритоклакса, MIM1, метилпреднизолона, iMAC2, ингибирующего Bax пептида V5, ингибирующего Bax пептида P5, блокатора каналов Bax и ARRY 520 трифторацетата. В определенных вариантах осуществления ингибитор BCL-2 согласно способам, описанным в настоящем описании, выбран из навитоклакса и венетоклакса или фармацевтически приемлемой соли любого из них.
[12] В определенных вариантах осуществления злокачественная опухоль крови согласно способам, описанным в настоящем описании, выбрана из острого миелоидного лейкоза (AML), хронического миелоидного лейкоза (CML), острой лимфоцитарной лимфомы (ALL) и хронического лимфоцитарного лейкоза (CLL), диффузной крупноклеточной B-клеточной лимфомы (DLBCL), первичной медиастинальной B-клеточной лимфомы, внутрисосудистой крупноклеточной B-клеточной лимфомы, фолликулярной лимфомы, мелкоклеточной лимфоцитарной лимфомы (SLL), лимфомы из клеток мантийной зоны, лимфом из B-клеток маргинальной зоны, экстранодальных лимфом из B-клеток маргинальной зоны, нодальной лимфомы из B-клеток маргинальной зоны, селезеночной лимфомы из B-клеток маргинальной зоны, лимфомы Беркитта, лимфоплазматической лимфомы и первичной лимфомы центральной нервной системы. Злокачественная опухоль крови может представлять собой диффузную крупноклеточную B-клеточную лимфому, острый миелоидный лейкоз или хронический лимфоцитарный лейкоз.
[13] Для определенных способов, описанных в настоящем описании, ингибитор CDK и ингибитор BCL-2 можно вводить одновременно. Для способов, описанных в настоящем описании, ингибитор CDK и ингибитор BCL-2 можно вводить последовательно в пределах приблизительно 12 часов друг от друга, например, в пределах приблизительно 5 часов друг от друга.
[14] Для определенных способов, описанных в настоящем описании, ингибитор CDK и ингибитор BCL-2 могут быть совместно составлены в фармацевтической композиции.
[15] Для определенных способов, описанных в настоящем описании, ингибитор CDK и ингибитор BCL-2 можно вводить каждые сутки, раз в двое суток или раз в трое суток.
[16] Для определенных способов, описанных в настоящем описании, ингибитор протеасом выбран из бортезомиба, маризомиба, иксазомиба, дисульфирама, эпигаллокатехин-3-галлата, салиноспорамида A, карфилзомиба, ONX 0912, CEP-18770, MLN9708, эпоксомицина, MG132 и фармацевтически приемлемой соли любого из них. В определенных вариантах осуществления ингибитор протеасом выбран из бортезомиба, маризомиба, иксазомиба и фармацевтически приемлемой соли любого из них.
[17] В определенных способах, описанных в настоящем описании, ингибитор CDK и ингибитор протеасом водят одновременно. Ингибитор CDK и ингибитор протеасом можно вводить последовательно в пределах приблизительно 12 часов друг от друга, например, в пределах 5 часов друг от друга.
[18] В определенных способах, описанных в настоящем описании, ингибитор CDK и ингибитор протеасом составлены совместно в фармацевтической композиции.
[19] В определенных способах, описанных в настоящем описании, ингибитор CDK и ингибитор BCL-2 вводят каждые сутки, раз в двое суток или раз в трое суток.
[20] В некоторых аспектах изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество ингибитора CDK, соответствующего формуле I:
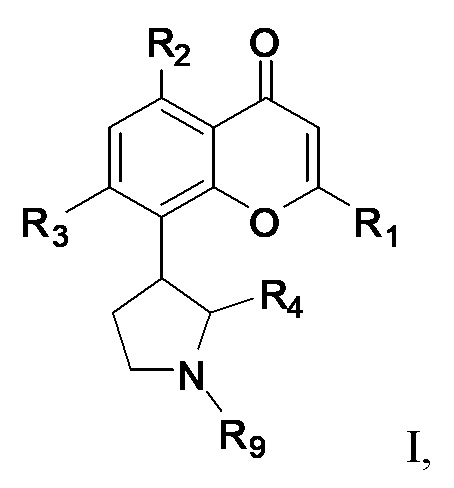
или его фармацевтически приемлемой соли, где:
R1 представляет собой необязательно замещенный фенил;
каждый из R2 и R3 независимо выбран из гидрокси и OR8, где R8 представляет собой необязательно замещенный C1-C10-алкил;
R4 представляет собой необязательно замещенный C1-C4-алкил; и
R9 представляет собой водород или необязательно замещенный C1-C4-алкил;
терапевтически эффективное количество ингибитора BCL-2 или ингибитора протеасом, и фармацевтически приемлемый эксципиент.
[21] В определенных вариантах осуществления соединение или соль формулы I соответствует формуле Ia: Ia.
[22] Для композиций, описанных в настоящем описании для соединений формулы I или формулы Ia R1 необязательно может быть замещен одним или несколькими заместителями, независимо выбранными из гидрокси, циано, галогена, амино, C1-C4-алкила, C1-C4-алкокси, C1-C4-гидроксиалкила, C1-C4-галогеналкила и нитро. В определенных вариантах осуществления R1 замещен одним или несколькими заместителями, независимо выбранными из галогена и C1-C4-галогеналкила. В определенных вариантах осуществления R1 представляет собой 2-хлор-4-трифторметилфенил.
[23] Для композиций, описанных в настоящем описании для соединений формулы I или формулы Ia, каждый из R2 и R3 может быть независимо выбран из гидрокси и OR8, где R8 представляет собой C1-C10-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления каждый из R2 и R3 представляет собой гидрокси.
[24] Для композиций, описанных в настоящем описании для соединений формулы I или формулы Ia, R4 представляет собой C1-C4-алкил, замещенный одним или несколькими заместителями, выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R4 представляет собой C1-C4-алкил, замещенный одним или несколькими заместителями, выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R4 представляет собой 2-гидроксиметил.
[25] Для композиций, описанных в настоящем описании для соединений формулы I или формулы Ia, R9 может представлять собой C1-C4-алкил, необязательно замещенный гидрокси, циано, галогеном, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R9 представляет собой метил.
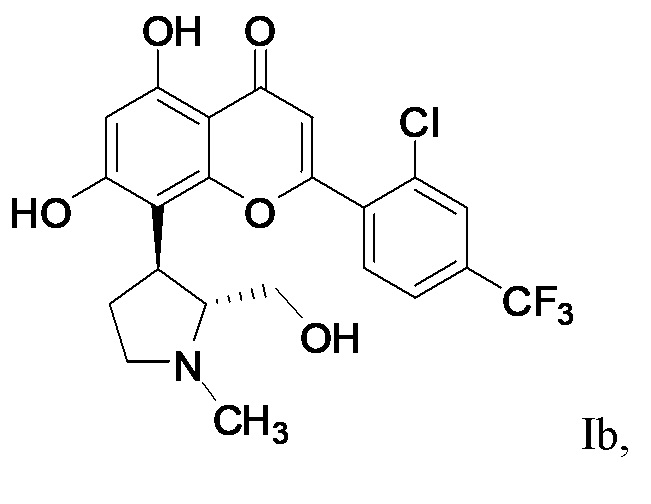
[26] Для композиций, описанных в настоящем описании, соединение формулы I может представлять собой соединение формулы Ib: 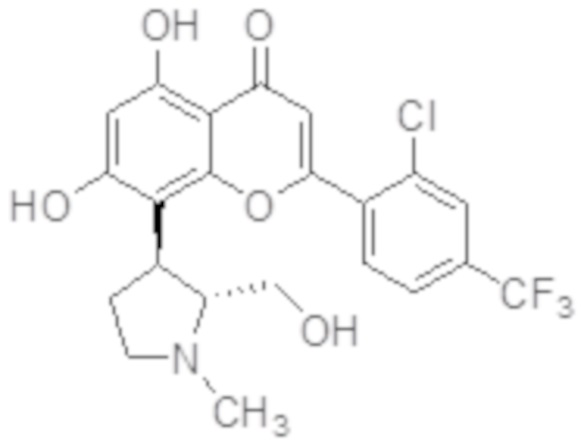 Ib, или его фармацевтически приемлемую соль.
Ib, или его фармацевтически приемлемую соль.
[27] Для композиций, описанных в настоящем описании, содержащих ингибитор BCL-2, ингибитор BCL-2 может быть выбран из навитоклакса, венетоклакса, A-1155463, A-1331852, ABT-737, обатоклакса, S44563, TW-37, A-1210477, AT101, HA14-1, BAM7, сабутоклакса, UMI-77, гамбогиновой кислоты, маритоклакса, MIM1, метилпреднизолона, iMAC2, ингибирующего Bax пептида V5, ингибирующего Bax пептида P5, блокатора каналов Bax, ARRY 520 трифторацетата и фармацевтически приемлемой соли любого из них. Ингибитор BCL-2 может быть выбран из навитоклакса и венетоклакса или фармацевтически приемлемой соли любого из них. В определенных вариантах осуществления ингибитор BCL-2 представляет собой венетоклакс.
[28] Для композиций, описанных в настоящем описании, содержащих ингибитор протеасом, ингибитор протеасом может быть выбран из бортезомиба, маризомиба, иксазомиба, дисульфирама, эпигаллокатехин-3-галлата, салиноспорамида A, карфилзомиба, ONX 0912, CEP-18770, MLN9708, эпоксомицина, MG132 и фармацевтически приемлемой соли любого из них. В определенных вариантах осуществления ингибитор протеасом выбран из бортезомиба, маризомиба, иксазомиба и фармацевтически приемлемой соли любого из них.
ВКЛЮЧЕНИЕ В КАЧЕСТВЕ ССЫЛКИ
[29] Все публикации, патенты и патентные заявки, упомянутые в настоящем описании, включены в настоящее описание в качестве ссылки в той же степени, как если бы было конкретно и индивидуально указано, что каждая индивидуальная публикация, патент или патентная заявка включены в качестве ссылок.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[30] Новые признаки изобретения указаны более конкретно в прилагаемой формуле изобретения. Лучшее понимание признаков и преимуществ настоящего изобретения может быть достигнуто с помощью следующего подробного описания, в котором указаны иллюстративные варианты осуществления, в которых используются принципы изобретения, и прилагаемых чертежей, в которых:
[31] На фиг.1 проиллюстрировано сравнение активности воруциклиба и флавопирадола против 38 киназ.
[32] На фиг.2 проиллюстрирована эффективность воруциклиба против циклин-зависимых киназ, соответствующая одноразрядному количеству нМ.
[33] На фиг.3A-3D проиллюстрирован синергический эффект воруциклиба в комбинации с венетоклаксом (ABT-199). Воруциклиб ингибирует индукцию индуцированного белка дифференцировки клеток миелоидного лейкоза (MLC-1) посредством венетоклакса в клетках диффузной крупноклеточной B-клеточной лимфомы (DLBCL) NU-DHL-1. MCL-1=красный; DAPI=синий; флуоресцентный визуализирующий маркер (FTM)=зеленый.
[34] На фиг.4A-4D проиллюстрирован повышенный апоптоз в клетках диффузной крупноклеточной B-клеточной лимфомы (DLBCL) NU-DHL-1 посредством комбинированной обработки воруциклибом и венетоклаксом. Расщепленная каспаза-3 (CC3)=красный; DAPI=синий; флуоресцентный визуализирующий маркер (FTM)=зеленый.
[35] На фиг. 5A-5D проиллюстрирован синергический эффект воруциклиба в комбинации с навитоклаксом (ABT-263). Обработка клеточной линии лимфомы Беркитта Ramos воруциклибом и навитоклаксом индуцирует апоптоз. Расщепленная каспаза-3 (CC3)=красный; DAPI=синий; флуоресцентный визуализирующий маркер (FTM)=зеленый.
[36] На фиг.6A-6E проиллюстрирован синергический эффект воруциклиба в комбинации с венетоклаксом в пяти моделях диффузной крупноклеточной B-клеточной лимфомы.
[37] На фиг.7 проиллюстрировано, что ингибирование MCL-1 через CDK9 помогает направить клетки на апоптоз.
[38] На фиг.8A-8D проиллюстрировано, что ингибирование протеасом индуцирует активацию MCL-1 при трижды негативном раке молочной железы (TNBC). На фиг.8A проиллюстрирован перечень соединений, для которых был проведен скрининг в модели с ксенотрансплантатом HCC1187 TNBC. На фиг.8B проиллюстрировано среднее % изменение индукции MCL-1 посредством обработки различными соединениями. На фиг.8C-8D проиллюстрировано окрашивание клеток в отношении CC3 (показано красным цветом) после обработки носителем и бортезомибом.
[39] На фиг.9A-9D проиллюстрирован синергический эффект воруциклиба в комбинации с маризомибом на клетки NudHL1 DLBCL. Расщепленная каспаза-3 (CC3)=красный; DAPI=синий; флуоресцентный визуализирующий маркер (FTM)=зеленый.
[40] На фиг.10A-10D проиллюстрирован синергический эффект воруциклиба в комбинации с бортезомибом на клетки NudHL1 DLBCL. Расщепленная каспаза-3 (CC3)=красный; DAPI=синий; флуоресцентный визуализирующий маркер (FTM)=зеленый.
[41] На фиг.11A-11D проиллюстрирован синергический эффект воруциклиба в комбинации с бортезомибом на клетки трижды негативного рака молочной железы. Расщепленная каспаза-3 (CC3)=красный; DAPI=синий; флуоресцентный визуализирующий маркер (FTM)=зеленый.
[42] На фиг.12A-12E проиллюстрирован синергический эффект воруциклиба в комбинации с бортезомибом на клетки трижды негативного рака молочной железы HCC1187. На фиг.12A-12D проиллюстрирован уменьшенный объем опухоли в модели на мышах HCC1187 TNBC, которым вводили воруциклиб и бортезомиб. На фиг.12E проиллюстрирован вестерн-блот, демонстрирующий сниженную экспрессию MCL-1 в клетках HCC1187 TNBC, обработанных воруциклибом и бортезомибом или воруциклибом и туникамицином.
[43] На фиг.13 проиллюстрирован эффект воруциклида и бортезомиба и комбинации воруциклиба и бортезомиба на массу тела в модели на мышах HCC1187 TNBC.
[44] На фиг.14 проиллюстрирован эффект бортезомиба в комбинации с пальбоциклибом, ингибитором CDK4/6, в модели на мышах HCC1187 TNBC.
[45] На фиг.15A-15B показано, что воруциклиб снижает индуцируемую бортезомибом экспрессию MCL-1 и убиквитин-протеинлигазы E3 XIAP. На фиг.15A проиллюстрирована предполагаемая модель ингибирования воруциклибом CDK9. На фиг.15B проиллюстрирован вестерн-блот, демонстрирующий, что воруциклиб уменьшает индуцированное бортезомибом повышение экспрессии MCL-1 и убиквитин-протеинлигазы E3 XIAP.
[46] На фиг.16A-16B проиллюстрированы клетки, резистентные к обработке бортезомибом. На фиг.16A проиллюстрированы клетки, резистентные к обработке бортезомибом, в области, в остальном очищенной от клеток посредством обработки бортезомибом. На фиг.16B проиллюстрировано, что клетки, резистентные к бортезомибу, экспрессируют GRP78 - белок, экспрессируемый в качестве части ответа на ER-стресс.
[47] На фиг.17 проиллюстрированы три различных пути ответа на ER-стресс.
[48] На фиг.18A-18B проиллюстрировано, что воруциклиб может влиять на IRE1α-зависимый каскад ответа на ER-стресс. На фиг.18A проиллюстрирован IRE1α-зависимый путь ответа на ER-стресс. На фиг.18B проиллюстрирован вестерн-блот, демонстрирующий, что индуктор ER-стресса туникамицин значительно активирует X-бокс-связывающий белок 1 (XBP1), способствующий выживанию (против смерти опухолевых клеток) белок. Этот эффект значительно снижался воруциклибом. Через 6 часов только туникамицин иллюстрирует этот эффект, однако через 24 часа этот эффект иллюстрируют как бортезомиб, так и туникамицин.
[49] На фиг.19A-19B проиллюстрировано подавление индуцируемой бортезомибом транскрипции XBP1 воруциклибом. STF083010=Ингибитор активности эндорибонуклеазы IRE1α; Tm=туникамицин.
[50] На фиг.20A-20D проиллюстрирован синергический эффект воруциклиба в комбинации с иксазомибом. Расщепленная каспаза 3 (CC3)=красный; DAPI=синий; флуоресцентный визуализирующий маркер (FTM)=зеленый.
[51] На фиг.21A-21B проиллюстрирован срез солидной опухоли, обработанный ингибиторами CDK, воруциклибом, пальбоциклибом, динациклибом и флавопирадолом в комбинации с иксазомибом. Только воруциклиб+иксазобмиб воспроизводимо обеспечивали выраженное устранение опухолевых клеток в течение 24 ч.
[52] На фиг.22А-В проиллюстрирована синергия воруциклиба и венетоклакса в модели SU-DHL-4 для диффузной крупноклеточной В-клеточной лимфомы {DLBCL).
[53] На фиг.23Ж-23С проиллюстрирована синергия воруциклиба и венетоклакса в модели SU-DHL-4, модели OCI LylO и модели U2932 диффузной крупноклеточной В-клеточной лимфомы (DLBCL).
[54] На фиг.24 проиллюстрировано, что воруциклиб подавляет
экспрессию MCL1 в моделях с ксенотрансплантатом DLBCL.
[55] На фиг.25А-25В проиллюстрирован синергический эффект
воруциклиба и венетоклакса в DLBCL ABC-типа (RIVA) у мышей.
[56] На фиг.26А-26В проиллюстрирован синергический эффект воруциклиба и венетоклакса и эффект на массу тела з модели U2 932 для DLBCL у мышей.
[57] На фиг.27А-27В проиллюстрирован синергический эффект воруциклиба и венетоклакса в модели NUDHL1 для DLBCL у мышей.
[58] На фиг.28 проиллюстрирован синергический эффект воруциклиба и венетоклакса в модели SUDHL4 для GC DLBCL,
[59] На фиг.29 проиллюстрировано, что воруциклиб восстанавливает р53, устраненный венетоклаксом.
[60] На фиг.30А-30С проиллюстрировано, что воруциклиб имеет активность в клеточных линиях AML в качестве единственного средства»
[61] На фиг.31 проиллюстрировано, что комбинация воруциклиба и венетоклакса индуцирует синергичную клеточную смерть в клеточных линиях АМН.
[62] На фиг.32 проиллюстрирован синергический эффект, состоящий в том, что комбинация воруциклиба и венетоклакса препятствует росту опухоли в ксенотрансплантатах SKM1 AML.
[63] На фиг.33 проиллюстрировано, что индуцируемый воруциклибом апоптоз коррелирует с подавлением MCL.
подробное: описание
[64] Изобретение относится к комбинированным способам терапии для лечения злокачественной опухоли. В частности, изобретение относится к комбинированным способами терапии ингибиторами CDK с другими средствами против злокачественной опухоли для лечения злокачественной опухоли. В одном аспекте изобретение относится к композициям и способам для лечения злокачественной опухоли ингибитором CDK в комбинации с ингибитором BCL-2. Такая комбинация обеспечивает синергические эффекты при лечении злокачественных опухолей и, в частности, при лечении злокачественных опухолей крови, например, лейкоза и лимфомы.
[65] В другом аспекте изобретение относится к композициям и способам для лечения злокачественной опухоли ингибитором CDK в комбинации с ингибитором протеасом. Такая комбинация обеспечивает синергический эффект при лечении злокачественной опухоли и, в частности, при лечении злокачественных опухолей крови и трижды негативного рака молочной железы.
[66] Основные термины, используемые выше и ниже, предпочтительно имеют следующие значения в контексте настоящего изобретения, если нет иных указаний. Таким образом, ниже в настоящем описании представлены определения основных терминов, как используют в контексте настоящего изобретения:
[67] Форма единственная числа включает множественное число, если контекст явно не указывает на иное.
[68] Термин "приблизительно", как используют в рамках изобретения, в общем относится к приемлемому диапазону погрешностей для конкретной величины, как определяет специалист в данной области, который может зависеть частично от того, как эту величину измеряют или определяют. Например, "приблизительно" может означать в пределах 1 или более 1 стандартного отклонения. Альтернативно "приблизительно" может означать диапазон, составляющий вплоть до 20%, вплоть до 10%, вплоть до 5%, или вплоть до 1% от данной величины. Альтернативно, в частности в отношении биологических систем или процессов, термин может означать нахождение в пределах порядка величины, в пределах 5-кратного и в пределах 2-кратного значения величины.
[69] Как используют в рамках изобретения, термин "по меньшей мере один" относится к одному или нескольким. Например, термин "по меньшей мере одно средство против злокачественной опухоли" означает, что комбинация содержит одно средство против злокачественной опухоли или больше средств против злокачественной опухоли.
[70] Термин "эффективное количество" или "терапевтически эффективное количество", как используют в рамках изобретения, в общем относится к количеству соединения, описанного в настоящем описании, которое является достаточным для обеспечения намеченного, заданного или назначенного применения, включая, но не ограничиваясь ими, лечение заболевания или состояния. Терапевтически эффективное количество может варьироваться в зависимости от применения (например, in vitro или in vivo), или индивидуума, подвергаемого лечению, и заболевания, например, массы тела и возраста индивидуума, тяжести болезненного состояния и способа введения. Также термин может относиться к дозе, которая индуцирует конкретный ответ в клетках-мишенях, например, снижение пролиферации или подавление активности белка-мишени. Конкретная доза может варьироваться в зависимости от конкретных выбранных соединений, режима дозирования, которому следуют, введения в комбинации с другими соединениями, времени введения, ткани, в которую проводят введение, и физической системы для доставки, в которой ее проводят.
[71] Как используют в рамках изобретения, термин "фармацевтически приемлемый" означает, что носитель, разбавитель, эксципиенты и/или соль должны быть совместимыми с другими ингредиентами состава и не вредоносными для их реципиента. "Фармацевтически приемлемый" также означает, что композиции или дозированные формы определяются мнением медицинского специалиста, являются пригодными для применения у животного или человека без чрезмерной токсичности, раздражения, аллергического ответа или другой проблемы или осложнения, и соответствуют приемлемому соотношению польза/риск.
[72] Как используют в рамках изобретения, термин "комбинация" или "фармацевтическая комбинация" относится к комбинированному введению средств против злокачественной опухоли. Комбинации по изобретению включают ингибитор CDK, например, соединение формулы I, Ia или Ib, и по меньшей мере одно средство против злокачественной опухоли, выбранное из ингибитора BCL-2 и ингибитора протеасом; и эти средства против злокачественной опухоли можно вводить индивидууму, нуждающемуся в этом, например, одновременно или последовательно.
[73] Термин "синергический", или "синергический эффект", или "синергизм", как используют в рамках изобретения, в общем относятся к такому эффекту, при котором один или несколько эффектов комбинации композиций превышает один или несколько эффектов каждого компонента отдельно, или он может превышать сумму одного или нескольких эффектов каждого компонента отдельно. Синергический эффект может приблизительно на 10%, 20%, 30%, 50%, 75%, 100%, 110%, 120%, 150%, 200%, 250%, 350% или 500% или более превышать эффект на индивидуума одного из компонентов отдельно, или суммарные эффекты каждого из компонентов при введении индивидуально. Эффект может представлять собой любой из поддающихся количественному определению эффектов, описанных в настоящем описании. Преимущественно такая синергия средств в комбинации может позволить применение меньших доз одного или обоих средств, может обеспечить более высокую эффективность в тех же дозах и может предотвратить или замедлить возникновение множественной резистентности к лекарственным средствам. Для определения синергического, аддитивного или антагонистического эффекта средств, используемых в комбинации, можно использовать способ показателя аддитивности (CI) Chou и Talalay. Когда величина CI меньше 1, между соединениями, используемыми в комбинации, существует синергия; когда величина CI равна 1, существует аддитивный эффект между соединениями, используемыми в комбинации, и когда величина CI превышает 1, существует антагонистический эффект. Cинергический эффект может быть достигнут путем совместного составления средств фармацевтической комбинации. Синергический эффект может быть достигнут путем введения двух или более средств в качестве отдельных составов, вводимых одновременно или последовательно.
[74] Циклин-зависимые киназы (CDK) представляют собой семейство ферментов, которые активируются в конкретных фазах клеточного цикла. CDK состоят из каталитической субъединицы (фактическая циклин-зависимая киназа или CDK) и регуляторной субъединицы (циклин). Существует по меньшей мере девять CDK (CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9 и т.д.) и по меньшей мере 15 различных типов циклинов (циклин A, B1, B2, D1, D2, D3, E, H и т.д.). Каждая стадия клеточного цикла регулируется такими комплексами CDK: переход G1/S (CDK2/циклин A, CDK4/циклин D1-D3, CDK6/циклин D3), S-фаза (CDK2/циклин A), G2-фаза 30 (CDK1/циклин A), переходная фаза G2/M (CDK1/циклин B).
[75] Как используют в рамках изобретения, термин "ингибитор CDK" относится к средству, способному ингибировать одну или несколько циклин-зависимых киназ (CDK). Аберрантная экспрессия и сверхэкспрессия этих киназ продемонстрирована при многих заболеваниях, таких как злокачественная опухоль. В контексте настоящего изобретения, ингибитор CDK фармацевтической комбинации, описанной в настоящем описании, может представлять собой соединение формулы I, Ia, или Ib или его фармацевтически приемлемую соль. Соединения по настоящему изобретению могут специфически ингибировать одно или несколько из CDK1/циклин B, CDK2/циклин E, CDK4/циклин D, CDK4/циклин D1 и CDK9/циклин T1. В определенных вариантах осуществления соединение по изобретению специфически ингибирует CDK9/циклин T1 или CDK9.
[76] В настоящем описании описаны комбинированные способы терапии для лечения злокачественной опухоли, например, лейкоза, лимфомы и рака. Способы и композиции, описанные в настоящем описании, могут включать ингибитор циклин-зависимой киназы (CDK), такой как соединение формулы I, Ia или Ib, или его фармацевтически приемлемая соль. В некоторых случаях, комбинированная терапия может включать ингибитор CDK в комбинации с ингибитором протеасом. В других случаях комбинированная терапия может включать ингибитор CDK в комбинации с ингибитором BCL-2.
[77] В определенных вариантах осуществления ингибитор CDK по изобретению соответствует соединению, описанному в патентах США № 7271193; 7915301; 8304449; 7884127; 8563596, полное содержание каждого из которых включено в настоящее описание в качестве ссылки. В определенных вариантах осуществления ингибитор CDK по изобретению представляет собой соединение формулы I:
I,
или его фармацевтически приемлемую соль, где:
R1 представляет собой необязательно замещенный фенил;
каждый из R2 и R3 независимо выбран из гидрокси и OR8, где R8 представляет собой необязательно замещенный C1-C10-алкил;
R4 представляет собой необязательно замещенный C1-C4-алкил; и
R9 представляет собой водород или необязательно замещенный C1-C4-алкил.
[78] В определенных вариантах осуществления соединение или соль формулы I соответствуют формуле Ia:
Ia.
[79] В определенных вариантах осуществления для соединения формулы I или формулы Ia, R1 необязательно замещен одним или несколькими заместителями, независимо выбранными из гидрокси, циано, галогена, амино, C1-C4-алкила, C1-C4-алкокси, C1-C4-гидроксиалкила, C1-C4-галогеналкила и нитро. В определенных вариантах осуществления R1 замещен одним или несколькими заместителями, независимо выбранными из гидрокси, циано, галогена, C1-C4-алкила и C1-C4-галогеналкила. В определенных вариантах осуществления R1 замещен одним или несколькими заместителями, независимо выбранными из галогена и C1-C4-галогеналкила. В определенных вариантах осуществления R1 представляет собой 2-хлор-4-трифторметилфенил.
[80] Термин "алкил" относится к прямому или разветвленному углеводородному радикалу, состоящему только из атомов углерода и водорода и не имеющему ненасыщенности. В определенных вариантах осуществления алкил содержит от одного до восьми атомов углерода (т.е. C1-C8 алкил). В других вариантах осуществления алкил содержит от одного до пяти атомов углерода (т.е. C1-C5 алкил). В других вариантах осуществления алкил содержит от одного до четырех атомов углерода (т.е. C1-C4 алкил). В других вариантах осуществления алкил содержит от одного до трех атомов углерода (т.е. C1-C3 алкил). В других вариантах осуществления алкил содержит от одного до двух атомов углерода (т.е. C1-C2 алкил). В других вариантах осуществления алкил содержит один атом углерода (т.е. C1 алкил). В других вариантах осуществления алкил содержит от пяти до восьми атомов углерода (т.е. C5-C8 алкил). В других вариантах осуществления алкил содержит от двух до пяти атомов углерода (т.е. C2-C5 алкил). В других вариантах осуществления алкил содержит от трех до пяти атомов углерода (т.е. C3-C5 алкил). В определенных вариантах осуществления алкильная группа выбрана из метила, этила, 1-пропила (н-пропил), 1-метилэтила (изопропил), 1-бутила (н-бутил), 1-метилпропила (втор-бутил), 2-метилпропила (изо-бутил), 1,1-диметилэтила (трет-бутил), 1-пентила (н-пентил). Алкил связан с остальной частью молекулы одинарной связью. Если в описании нет иных конкретных указаний, алкильная группа необязательно замещена одним или несколькими заместителями, такими как заместители, описанные в настоящем описании.
[81] Термин "алкокси" относится к радикалу, связанному через атом кислорода формулы -O-алкил, где алкил представляет собой алкильную цепь, как определено выше.
[82] Термин "амино" относится к группе -NR'R'', где R' и R'' независимо выбраны из водорода; и алкила, гидроксила, арила, циклоалкила, гетероциклоалкила и гетероарила, любой из которых может быть необязательно замещен одним или несколькими заместителями, такими как гидрокси, циано, галоген, амино, C1-C4-алкил, C1-C4-алкокси, C1-C4-гидроксиалкил, C1-C4-галогеналкил и нитро.
[83] Термин "Cx-y", когда его используют совместно с химической частью, такой как алкил, включает группы, которые содержат от x до y атомов углерода в цепи. Например, термин "Cx-yалкил" относится к замещенным или незамещенным насыщенным углеводородным группам, включающим прямые алкильные и разветвленные алкильные группы, которые содержат от x до y атомов углерода в цепи, включая галогеналкильные группы, такие как трифторметил и 2,2,2-трифторэтил и т.д.
[84] Термин "галогеналкил" относится к алкильной группе, которая замещена одним или несколькими галогеновыми радикалами, например, трифторметилу, дифторметилу, фторметилу, 2,2,2-трифторэтилу, 1-хлорметил-2-фторэтилу и т.п. В некоторых вариантах осуществления алкильная часть галогеналкила является дополнительно необязательно замещенной, как описано в настоящем описании.
[85] Термин "гидроксиалкил" относится к алкильной группе, которая замещена одним или несколькими гидроксирадикалами, например, гидроксиметилу, гидроксиэтилу, дигидроксиметилу и т.п. В некоторых вариантах осуществления алкильная часть гидроксиалкила является дополнительно необязательно замещенной, как описано в настоящем описании.
[86] В определенных вариантах осуществления для соединения или формулы формулы I или Ia, каждый из R2 и R3 независимо выбран из гидрокси и OR8, где R8 представляет собой C1-C10-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R8 в каждом случае выбран из необязательно замещенного C1-C6-алкила, такого как необязательно замещенный C1-C4-алкил. В определенных вариантах осуществления каждый из R2 и R3 независимо представляет собой гидрокси.
[87] В определенных вариантах осуществления для соединения или формулы формулы I или Ia, R4 представляет собой необязательно замещенный C1-C4-алкил, где R4 необязательно замещен одним или несколькими заместителями, выбранными из гидрокси, циано, галогена, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R4 представляет собой необязательно замещенный C1-C2-алкил. В определенных вариантах осуществления R4 представляет собой гидроксиалкил, например, 2-гидроксиметил.
[88] В определенных вариантах осуществления для соединения или формулы формулы I или Ia, R9 представляет собой C1-C4-алкил, необязательно замещенный гидрокси, циано, галогеном, амино, =O, =S, C1-C4-алкокси и нитро. В определенных вариантах осуществления R9 представляет собой необязательно замещенный C1-C2-алкил. В определенных вариантах осуществления R9 представляет собой метил. В определенных вариантах осуществления R9 представляет собой водород.
[89] В определенных вариантах осуществления для соединения или формулы формулы I или Ia, соединение формулы I представляет собой соединение или фармацевтически приемлемую соль, выбранные из: (+)-транс-2-(2-хлор-4-трифторметилфенил)-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-5,7-диметокси-хромен-4-она; (+)-транс-2-(2-хлор-4-трифторметилфенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метилпирролидин-3-ил)-хромен-4-она; и гидрохлорида (+)-транс-2-(2-хлор-4-трифторметилфенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-хромен-4-она.
[90] В определенных вариантах осуществления соединение формулы I или Ia представляет собой соединение формулы Ib:
или его фармацевтически приемлемую соль. В определенных вариантах осуществления соединение формулы I, Ia или Ib имеет форму кислотно-аддитивной соли, такой как соль хлористоводородной кислоты.
[91] Термин "замещенный" относится к частям, имеющим заместители, заменяющие водород на одном или нескольких атомах углерода или гетероатомах структуры. Будет понятно, что "замещение" или "замещенный посредством" включает подразумеваемое условие, что такое замещение осуществляется в соответствии с допустимой валентностью замещенного атома и заместителя, а также обеспечивает стабильное соединение, которое не легко претерпевает трансформацию, такую как перестановка, циклизация, отщепление и т.д. Как используют в рамках изобретения, термин "замещенный" включает все допустимые заместители органических соединений. В широком аспекте допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Может присутствовать один или несколько допустимых заместителей и они могут быть одинаковыми или различными для соответствующих органических соединений. Для целей настоящего изобретения гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанные в настоящем описании, которые удовлетворяют валентностям гетероатомов.
[92] Заместители могут включать любые заместители, описанные в настоящем описании, например, галоген, гидрокси, карбонил (такой как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонил (такой как сложный тиоэфир, тиоацетат или тиоформиат), алкоксил, фосфорил, фосфат, фосфонат, фосфинат, амино, амидо, амидин, имин, циано, нитро, азидо, сульфгидрил, алкилтио, сульфат, сульфонат, сульфамоил, сульфонамидо, сульфонил, гетероциклил, аралкил, карбоцикл, гетероцикл, циклоалкил, гетероциклоалкил, ароматическая и гетероароматическая часть. В некоторых вариантах осуществления заместители могут включать любые заместители, описанные в настоящем описании, например: галоген, гидрокси, оксо (=O), тиоксо (=S), циано (-CN), нитро (-NO2), имино (=N-H), оксимо (=N-OH), гидразино (=N-NH2), -Rb-ORa, -Rb-OC(O)-Ra, -Rb-OC(O)-ORa, -Rb-OC(O)-N(Ra)2, -Rb-N(Ra)2, -Rb-C(O)Ra, -Rb-C(O)ORa, -Rb-C(O)N(Ra)2, -Rb-O-Rc-C(O)N(Ra)2, -Rb-N(Ra)C(O)ORa, -Rb-N(Ra)C(O)Ra, -Rb-N(Ra)S(O)tRa (где t равен 1 или 2), -Rb-S(O)tRa (где t равен 1 или 2), -Rb-S(O)tORa (где t равен 1 или 2) и -Rb-S(O)tN(Ra)2 (где t равен 1 или 2); и алкил, алкенил, алкинил, арил, аралкил, аралкенил, аралкинил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероциклоалкилалкил, гетероарил и гетероарилалкил, любой из которых может быть необязательно замещен алкилом, алкенилом, алкинилом, галогеном, гидрокси, галогеналкилом, галогеналкенилом, галогеналкинилом, оксо (=O), тиоксо (=S), циано (-CN), нитро (-NO2), имино (=N-H), оксимо (=N-OH), гидразином (=N-NH2), -Rb-ORa, -Rb-OC(O)-Ra, -Rb-OC(O)-ORa, -Rb-OC(O)-N(Ra)2, -Rb-N(Ra)2, -Rb-C(O)Ra, -Rb-C(O)ORa, -Rb-C(O)N(Ra)2, -Rb-O-Rc-C(O)N(Ra)2, -Rb-N(Ra)C(O)ORa, -Rb-N(Ra)C(O)Ra, -Rb-N(Ra)S(O)tRa (где t равен 1 или 2), -Rb-S(O)tRa (где t равен 1 или 2), -Rb-S(O)tORa (где t равен 1 или 2) и -Rb-S(O)tN(Ra)2 (где t равен 1 или 2); где каждый Ra независимо выбран из водорода, алкила, циклоалкила, циклоалкилалкила, арила, аралкила, гетероциклоалкила, гетероциклоалкилалкила, гетероарила или гетероарилалкила, где каждый Ra, если позволяет валентность, может быть необязательно замещен алкилом, алкенилом, алкинилом, галогеном, галогеналкилом, галогеналкенилом, галогеналкинилом, оксо (=O), тиоксо (=S), циано (-CN), нитро (-NO2), имино (=N-H), оксимо (=N-OH), гидразином (=N-NH2), -Rb-ORa, -Rb-OC(O)-Ra, -Rb-OC(O)-ORa, -Rb-OC(O)-N(Ra)2, -Rb-N(Ra)2, -Rb-C(O)Ra, -Rb-C(O)ORa, -Rb-C(O)N(Ra)2, -Rb-O-Rc-C(O)N(Ra)2, -Rb-N(Ra)C(O)ORa, -Rb-N(Ra)C(O)Ra, -Rb-N(Ra)S(O)tRa (где t равен 1 или 2), -Rb-S(O)tRa (где t равен 1 или 2), -Rb-S(O)tORa (где t равен 1 или 2) и -Rb-S(O)tN(Ra)2 (где t равен 1 или 2); и где каждый Rb независимо выбран из прямой связи или прямой или разветвленной алкиленовой, алкениленовой или алкиниленовой цепи и каждый Rc представляет собой прямую или разветвленную алкиленовую, алкениленовую или алкиниленовую цепь.
[93] Способы производства соединений формулы I, Ia и Ib или их фармацевтически приемлемых солей могут быть найдены в публикации патента PCT № WO2004004632 (соответствующей патенту США 7271193) и публикации патента PCT № WO2007148158.
[94] Настоящее изобретение относится к фармацевтически приемлемым солям любого из соединений, описанных в настоящем описании, например, соединения формулы I, Ia, Ib, ингибиторов BCL-2 и ингибиторов протеасом. Фармацевтически приемлемые соли включают, например, кислотно-аддитивные соли и основно-аддитивные соли. Кислота, которую добавляют к соединению для образования кислотно-аддитивной соли, может представлять собой органическую кислоту или неорганическую кислоту. Основание, которое добавляют к соединению для образования основно-аддитивной соли. может представлять собой органическое основание или неорганическое основание. В некоторых случаях фармацевтически приемлемая соль представляет собой соль металла. В некоторых случаях фармацевтически приемлемая соль представляет собой соль аммония.
[95] Кислотно-аддитивные соли могут быть результатом добавления кислоты к соединению, описанному в настоящем описании. В некоторых случаях кислота является органической. В некоторых случаях кислота является неорганической. Неограничивающие примеры подходящих кислот включают хлористоводородную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, азотную кислоту, азотистую кислоту, серную кислоту, сернистую кислоту, фосфорную кислоту, никотиновую кислоту, изоникотиновую кислоту, молочную кислоту, салициловую кислоту, 4-аминосалициловую кислоту, виннокаменную кислоту, аскорбиновую кислоту, гентизиновую кислоту, глюконовую кислоту, глюкароновую кислоту, сахарную кислоту, муравьиную кислоту, бензойную кислоту, глутаминовую кислоту, пантотеновую кислоту, уксусную кислоту, пропионовую кислоту, масляную кислоту, фумаровую кислоту, янтарную кислоту, лимонную кислоту, щавелевую кислоту, малеиновую кислоту, гидроксималеиновую кислоту, метилмалеиновую кислоту, гликолевую кислоту, яблочную кислоту, коричную кислоту, миндальную кислоту, 2-феноксибензойную кислоту, 2-ацетоксибензойную кислоту, эмбоновую кислоту, фенилуксусную кислоту, N-циклогексилсульфаминовую кислоту, метансульфоновую кислоту, этансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, этан-1,2-дисульфоновую кислоту, 4-метилбензолсульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 2-фосфоглицериновую кислоту, 3-фосфоглицериновую кислоту, глюкоза-6-фосфорную кислоту и аминокислоту.
[96] Соли металлов могут быть образованы добавлением неорганического основания к соединению по изобретению. Неорганическое основание состоит из катиона металла в паре с основным противоионом, например, таким как гидроксид, карбонат, бикарбонат или фосфат. Металл может представлять собой щелочной металл, щелочноземельный металл и металл основной группы. В некоторых вариантах осуществления металл представляет собой литий, натрий, калий, цезий, церий, магний, марганец, железо, кальций, стронций, кобальт, титан, алюминий, медь, кадмий или цинк.
[97] В некоторых вариантах осуществления соль металла представляет собой соль лития, соль натрия, соль калия, соль цезия, соль церия, соль магния, соль марганца, соль железа, соль кальция, соль стронция, соль кобальта, соль титана, соль алюминия, соль меди, соль кадмия или соль цинка.
[98] Соли аммония могут быть образованы добавлением аммиака или органического амина к соединению, описанному в настоящем описании. Неограничивающие примеры подходящих органических аминов включают триэтиламин, диизопропиламин, этаноламин, диэтаноламин, триэтаноламин, морфолин, N-метилморфолин, пиперидин, N-метилпиперидин, N-этилпиперидин, дибензиламин, пиперазин, пиридин, пирразол, пипиразол, имидазол, пиразин, пипиразин, этилендиамин, N,N'-дибензилэтилен диамин, прокаин, хлорпрокаин, холин, дициклогексил амин и N-метилглюкамин.
[99] Неограничивающие примеры подходящих солей аммония включают соль триэтиламина, соль диизопропиламина, соль этаноламина, соль диэтаноламина, соль триэтаноламина, соль морфолина, соль N-метилморфолина, соль пиперидина, соль N-метилпиперидина, смоль N-этилпиперидина, соль дибензиламина, соль пиперазина, соль пиридина, соль пиразола, соль пипиразола, соль имидазола, соль пиразина, соль пипиразина, соль этилендиамина, соль N,N'-дибензилэтилендиамина, соль прокаина, соль хлорпрокаина, соль холина, соль дициклогексиламина и соль N-метилглюкамина.
[100] Неограничивающие примеры подходящих кислотно-аддитивных солей включают соль хлористоводородной кислоты, соль бромистоводородной кислоты, соль йодистоводородной кислоты, нитрат, нитрит, сульфат, сульфит, фосфат, гидрофосфат, дигидрофосфат, карбонат, бикарбонат, никотинат, изоникотинат, лактат, салицилат, 4-аминосалицилат, тартрат, аскорбат, гентизинат, глюконат, глюкаронат, сахарат, формиат, бензоат, глутамат, пантотенат, ацетат, пропионат, бутират, фумарат, сукцинат, цитрат, оксалат, малеат, гидроксималеат, метилмалеат, гликолят, малат, циннамат, соль миндальной кислоты, 2-феноксибензоат, 2-ацетоксибензоат, эмбонат, фенилацетат, N-циклогексилсульфамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, 2-гидроксиэтансульфонат, этан-1,2-дисульфонат, 4-метилбензолсульфонат, нафталин-2-сульфонат, нафталин-1,5-дисульфонат, 2-фосфоглицерат, 3-фосфоглицерат, глюкоза-6-фосфат и соль аминокислоты.
[101] Соединения, описанные в настоящем описании, например, соединения и соли формул I, Ia, Ib, ингибиторы BCL-2 и ингибиторы протеасом, в некоторых случаях могут существовать в качестве диастереомеров, энантиомеров или других стереоизомерных форм. Соединения, описанные в настоящем описании, включают все диастереомерные, энантиомерные и эпимерные формы, а также их соответствующие смеси. Разделение стереоизомеров можно проводить хроматографией или формированием диастереомеров и разделением перекристаллизацией или хроматографией, или любой их комбинацией. (Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc., 1981, включенная в настоящее описание в качестве ссылки). Стереоизомеры также могут быть получены стереоселективным синтезом.
[102] Соединения, описанные в настоящем описании, например, соединения и соли формул I, Ia, Ib, ингибиторы BCL-2 и ингибиторы протеасом включают использование аморфных форм, а также кристаллических форм (также известных как полиморфы). Соединения, описанные в настоящем описании, могут иметь форму фармацевтически приемлемых солей. Кроме того, в объем настоящего изобретения включены активные метаболиты этих соединений, имеющие тот же тип активности. Кроме того, соединения, описанные в настоящем описании, могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Сольватированные формы соединений, описанных в настоящем описании, также считаются раскрытыми в настоящем описании.
[103] Соединения, описанные в настоящем описании, например, соединения и соли формул I, Ia, Ib, ингибиторы BCL-2 и ингибиторы протеасом, включают соединения, которые проявляют их естественное содержание изотопов, и соединения, где один или несколько атомов искусственным образом обогащены конкретным изотопом, имеющим то же атомное число, но атомную массу или массовое число, отличные от атомной массы или атомного числа, в основном встречающихся в природе. Объем настоящего изобретения охватывает все изотопные варианты соединений по настоящему изобретению, как радиоактивные, так и не являющиеся радиоактивными. Например, водород имеет три встречающихся в природе изотопа, обозначаемых 1H (протий), 2H (дейтерий) и 3H (тритий). Протий является наиболее распространенным изотопом водорода в природе. Увеличение содержания дейтерия может обеспечить определенные терапевтические преимущества, такие как увеличенное время полужизни и/или экспозиция in vivo, или может обеспечить соединение, пригодное для исследования путей выведения и метаболизма лекарственного средства in vivo. Можно получать изотопно обогащенные соединения.
[104] Соединения, описанные в настоящем описании, например, соединения и соли формул I, Ia, Ib, ингибиторы BCL-2 и ингибиторы протеасом, где соединение имеет углерод-углеродные двойные связи или углерод-азотные двойные связи могут существовать, когда это осуществимо, в Z- или E-форме (или цис- или транс-форме). Более того, некоторые химические структуры могут существовать в различных таутомерных формах. Если нет иных указаний, химические структуры, описанные в настоящем описании, также включают все Z-, E- и таутомерные формы.
[105] В определенных случаях соединение, описанное в настоящем описании, может представлять собой пролекарство, например, где карбоновая кислота, присутствующая в исходном соединении, имеет форму сложного эфира. Термин "пролекарство" охватывает соединения, которые в физиологических условиях конвертируются в фармацевтические средства, т.е. исходное соединение, по настоящему изобретению. Одним способом получения пролекарства является включение одной или нескольких выбираемых частей, которые гидролизуются в физиологических условиях, с образованием желаемой молекулы. В определенных вариантах осуществления пролекарство конвертируется посредством ферментативной активности животного-хозяина, такой как ферментативная активность в определенных клетках-мишенях животного-хозяина. Например, предпочтительными пролекарствами по настоящему изобретению являются сложные эфиры или карбонаты (например, сложные эфиры или карбонаты спиртов или карбоновых кислот).
[106] Пролекарства часто являются полезными, поскольку в некоторых ситуациях их может быть легче вводить, чем исходное лекарственное средство. Они могут быть, например, биодоступными посредством перорального введения, в то время как исходное соединение не является таковым. Пролекарства могут обеспечивать усиленное проникновение соединений в клетки относительно исходного лекарственного средства. Например, пролекарство может иметь улучшенную клеточную проницаемость относительно исходного соединения. Пролекарство также может иметь увеличенную растворимость в фармацевтических составах относительно исходного лекарственного средства. В некоторых вариантах осуществления формат пролекарства увеличивает липофильность фармацевтического средства. В некоторых вариантах осуществления формат пролекарства повышает эффективную растворимость в воде.
[107] В определенных вариантах осуществления ингибитор циклин-зависимой киназы (CDK), например, соединение или соль формулы I, Ia или Ib, можно использовать в комбинации с ингибитором одного или нескольких белков семейства BCL-2. Ингибиторы семейства антиапоптотических белков BCL-2 изменяют по меньшей мере каскад клеточного выживания. Активация апоптоза может происходить через внешний путь, запускаемый активацией рецепторов смерти на клеточной поверхности, или внутренний путь, запускаемый обусловленными развитием сигналами и разнообразными внутриклеточными стрессами. Этот внутренний путь, также известный как стрессовый путь или митохондриальный путь, в основном регулируется семейством BCL-2, классом ключевых регуляторов активации каспазы, состоящим из антиапоптотических (способствующих выживанию) белков, имеющих домены BH1-BH4 (BCL-2, т.е. белок BCL-2, являющийся представителем семейства антиапоптотических белков BCL-2), BCL-xL, BCL-w, A1, MCL-1 и BCL-B); проапоптотических белков, имеющих домены BH1, BH2 и BH3 (BAX, BAK и BOK); и проапоптотических белков, имеющих только BH3 (BIK, BAD, BID, BIM, BMF, HRK, NOXA и PUMA) (см., например, Cory et al., Nature Reviews Cancer 2:647-56 (2002); Cory et al., Cancer Cell 8:5-6 (2005); Adams et al., Oncogene 26:1324-1337 (2007)). Активация антиапоптотических белков BCL-2 блокирует активацию проапоптотических мультидоменных белков BAX и BAK (см., например, Adams et al., Oncogene 26:1324-37 (2007)).
[108] Как используют в рамках изобретения, термин "ингибитор BCL-2" относится к средству, которое способно ингибировать один или несколько белков семейства антиапоптотических белков BCL-2, например, BCL-2, BCL-xL и BCL-w. В определенных вариантах осуществления ингибитор BCL-2 по изобретению селективно ингибирует один белок семейства BCL-2, например, ингибитор BCL-2 может селективно ингибировать BCL-2, но не BCL-xl или BCL-w.
[109] Ингибитор BCL-2, описанный в настоящем описании, может ингибировать один или несколько из BCL-2, BCL-xL и BCL-w. В определенных вариантах осуществления ингибитор антиапоптотического семейства белков BCL-2 ингибирует BCL-2. В определенных вариантах осуществления ингибитор антиапоптотического семейства белков BCL-2 ингибирует BCL-2 и не ингибирует других представителей семейства белков BCL-2, например, не ингибирует BCL-xL или BCL-w. В определенных вариантах осуществления ингибитор BCL-2 представляет собой BH3-миметик.
[110] В определенных вариантах осуществления ингибитор BCL-2 по изобретению ингибирует функцию BCL-xL. В дополнение к ингибированию BCL-xL, ингибитор также может взаимодействовать с и/или ингибировать одну или несколько функций BCL-2, как например, ингибиторы BCL-xL/BCL-2. В определенных вариантах осуществления ингибитор BCL-2 по изобретению ингибирует каждый из BCL-xL и BCL-w. В определенных вариантах осуществления ингибитор BCL-2 по изобретению ингибирует BCL-xL, BCL-2 и BCL-w.
[111] В определенных вариантах осуществления ингибитор BCL-2 препятствует взаимодействию между представителем семейства антиапоптотических белков BCL-2 и одним или несколькими лигандами или рецепторами, с которыми представитель семейства антиапоптотических белков BCL-2 связывался бы в отсутствие ингибитора. В других вариантах осуществления ингибитор одного или нескольких представителей семейства антиапоптотических белков BCL-2, где ингибитор специфически ингибирует по меньшей мере один белок BCL-2, связывается только с одним или несколькими из BCL-xL, BCL-2, BCL-w, но не с другими представителями семейства антиапоптотических белков Bcl-2, такими как Mcl-1 и BCL2A1.
[112] Аффинность связывания ингибитора BCL-2 с белками семейства BCL-2 можно количественно определять. В качестве примера, аффинность связывания ингибитора BCL-xL можно определять с использованием конкурентного флуоресцентного поляризационного анализа, в котором флуоресцентный пептид BAK с доменом BH3 инкубируют с белком BCL-xL (или другим белком семейства BCL-2) в присутствии или в отсутствие возрастающих концентраций ингибитора BCL-XL, как описано ранее (см., например, публикацию патента США 20140005190; Park et al., Cancer Res. 73:5485-96 (2013); Wang et al., Proc. Natl. Acad. Sci USA 97:7124-9 (2000); Zhang et al., Anal. Biochem. 307:70-5 (2002); Bruncko et al., J. Med. Chem. 50:641-62 (2007)). Процентное ингибирование можно определять с помощью уравнения: 1-[(величина mP для лунки - отрицательный контроль)/диапазон)] x 100%. Величину константы ингибирования (Ki) определяют по формуле: Ki=[I]50/([L]50/Kd+[P]0/Kd+1), как описано в Bruncko et al., J. Med. Chem. 50:641-62 (2007) (также см. Wang, FEBS Lett. 360:111-114 (1995)).
[113] Примеры ингибиторов BCL-2 включают ABT-263 (4-[4-[[2-(4-хлорфенил)-5,5-диметилциклогексен-1-ил]метил]пиперазин-1-ил]-N-[4-[[(2R)-4-морфолин-4-ил-1-фенилсульфанилбутан-2-ил]амино]-3-(трифторметилсульфонил)фенил]сульфонилбензамид или IUPAC, (R)-4-(4-((4'-хлор-4,4-диметил-3,4,5,6-тетрагидро-[1,1'-бифенил]-2-ил)метил)пиперазин-1-ил)-N-((4-((4-морфолино-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид) (см., например, Park et al., 2008, J. Med. Chem. 51:6902; Tse et al., Cancer Res., 2008, 68:3421; публикацию международной заявки на патент № WO 2009/155386; патенты США № 7390799, 7709467, 7906505, 8624027) и ABT-737 (4-[4-[(4'-хлор[1,1'-бифенил]-2-ил)метил]-1-пиперазинил]-N-[[4-[[(1R)-3-(диметиламино)-1-[(фенилтио)метил]пропил]амино]-3-нитрофенил]сульфонил]бензамид, бензамид, 4-[4-[(4'-хлор[1,1'-бифенил]-2-ил)метил]-1-пиперазинил]-N-[[4-[[(1R)-3-(диметиламино)-1-[(фенилтио)метил]пропил]амино]-3-нитрофенил]сульфонил]- или 4-[4-[[2-(4-хлорфенил)фенил]метил]пиперазин-1-ил]-N-[4-[[(2R)-4-(диметиламино)-1-фенилсульфанилбутан-2-ил]амино]-3-нитрофенил]сульфонилбензамид) (см., например, Oltersdorf et al., Nature, 2005, 435:677; патент США № 7973161; патент США № 7642260).
[114] В других вариантах осуществления ингибитор BCL-2 представляет собой соединение хиназолин сульфонамид (см., например, Sleebs et al., 2011, J. Med. Chem. 54:1914). В другом варианте осуществления ингибитор BCL представляет собой низкомолекулярное соединение, как описано Zhou et al., J. Med. Chem., 2012, 55:4664 (см., например, соединение 21 (R)-4-(4-хлорфенил)-3-(3-(4-(4-(4-((4-(диметиламино)-1-(фенилтио)бутан-2-ил)амино)-3-нитрофенилсульфонамидо)фенил)пиперазин-1-ил)фенил)-5-этил-1-метил-1H-пиррол-2-карбоновая кислота) и Zhou et al., J. Med. Chem., 2012, 55:6149 (см., например, соединение 14 (R)-5-(4-хлорфенил)-4-(3-(4-(4-(4-((4-(диметиламино)-1-(фенилтио)бутан-2-ил)амино)-3-нитрофенилсульфонамидо)фенил)пиперазин-1-ил)фенил)-1-этил-2-метил-1H-пиррол-3-карбоновая кислота; соединение 15 (R)-5-(4-хлорфенил)-4-(3-(4-(4-(4-((4-(диметиламино)-1-(фенилтио)бутан-2-ил)амино)-3-нитрофенилсульфонамидо)фенил)пиперазин-1-ил)фенил)-1-изопропил-2-метил-1H-пиррол-3-карбоновая кислота). В других вариантах осуществления ингибитор BCL представляет собой ингибитор BCL-2/BCL-xL, такой как BM-1074 (см., например, Aguilar et al., 2013, J. Med. Chem. 56:3048); BM-957 (см., например, Chen et al., 2012, J. Med. Chem. 55:8502); BM-1197 (см., например, Bai et al., PLoS One 2014 Jun 5;9(6):e99404. Doi: 10.1371/journal.pone. 009904); заявка на патент США № 2014/0199234; N-ацилсульфонамидные соединения (см., например, публикацию международной заявки на патент WO 2002/024636, публикацию международной заявки на патент № WO 2005/049593, публикацию международной заявки на патент № WO 2005/049594, патент США № 7767684, патент США № 7906505). В другом варианте осуществления ингибитор BCL-2 представляет собой низкомолекулярное макроциклическое соединение (см., например, публикацию международной заявки на патент № WO 2006/127364, патент США № 7777076). В другом варианте осуществления ингибитор BCL-2 представляет собой соединение изоксазолидина (см., например, публикацию международной заявки на патент № WO 2008/060569, патент США № 7851637, патент США № 7842815). В другом варианте осуществления ингибитор BCL-2 представляет собой S44563 (см., например, Loriot et. al., Cell Death and Disease, 2014, 5, e1423). В одном варианте осуществления ингибитор BCL-2 представляет собой (R)-3-((4'-хлор-[1,1'-бифенил]-2-ил)метил)-N-((4-(((R)-4-(диметиламино)-1-(фенилтио)бутан-2-ил)амино)-3-нитрофенил)сульфонил)-2,3,4,4a,5,6-гексагидро-1H-пиразинo[1,2-a]хинолин-8-карбоксамид. В другом варианте осуществления ингибитор BCL-2 представляет собой низкомолекулярное гетероциклическое соединение (см., например, патент США № 9018381).
[115] В определенных случаях ингибитор BCL-2 используют в комбинации с соединением или солью формулы I, Ia или Ib. Можно использовать любой ингибитор BCL-2, и он может демонстрировать синергический эффект при использовании в комбинации с соединением или солью формулы I, Ia или Ib. Ингибитор семейства BCL-2 может ингибировать один или несколько представителей семейства BCL-2, включая Bcl-2, Bcl-xL, Bcl-w, BAK1, BAX, BCL2, BCL2A1, BCL2L1, BCL2L2, BCL2L10, BCL2L13, BCL2L14, BOK и MCL1. В определенных вариантах осуществления соединение или соль формулы I, Ia или Ib используют в комбинации с любым из следующих: навитоклакс, венетоклакс, A-1155463, A-1331852, ABT-737, обатоклакс, TW-37, A-1210477, AT101, HA14-1, BAM7, сабутоклакс, UMI-77, гамбогиновая кислота, маритоклакс, MIM1, метилпреднизолон, iMAC2, ингибиторующий Bax пептид V5, ингибирующий Bax пептид P5, блокатор каналов Bax и ARRY 520 трифторацетат. В некоторых примерах воруциклиб используют в комбинации с навитоклаксом. В определенных вариантах осуществления воруциклиб используют в комбинации с венетоклаксом.
[116] В некоторых вариантах осуществления ингибитор BCL-2 используют в комбинации с ингибитором CDK по изобретению, например, соединением формулы I, Ia или Ib, для лечения злокачественной опухоли крови. В определенных вариантах осуществления злокачественная опухоль крови представляет собой лейкоз, такой как острый миелоидный лейкоз (AML), хронический миелоидный лейкоз (CML), острая лимфоцитарная лимфома (ALL) и хронический лимфоцитарный лейкоз (CLL). В определенных вариантах осуществления злокачественная опухоль крови представляет собой неходжкинскую лимфому, такую как B-клеточная или T-клеточная лимфома. B-клеточные лимфомы включают диффузную крупноклеточную B-клеточную лимфому (DLBCL), первичную медиастинальную B-клеточную лимфому, внутрисосудистую крупноклеточную B-клеточную лимфому, фолликулярную лимфому, мелкоклеточный лимфоцитарный лимфоцитоз (SLL), лимфому из клеток мантийной зоны, лимфомы из B-клеток маргинальной зоны, экстранодальную лимфому из B-клеток маргинальной зоны, нодальную лимфому из B-клеток маргинальной зоны, селезеночную лимфому из B-клеток маргинальной зоны, лимфому Беркитта, лимфоплазматическую лимфому и первичную лимфому центральной нервной системы. T-клеточные лимфомы включают T-лимфобластную лимфому из предшественников, периферические T-клеточные лимфомы, Т-клеточные лимфомы кожи, взрослую T-клеточную лимфому подтипов: вялотекущая хроническая, острая и лимфома, ангиоиммунобластную T-клеточную лимфому, эстранодальную лимфому из натуральных киллеров/T-клеток, назальный тип, ассоциированную с энтеропатией кишечную T-клеточную лимфому (EATL) подтипов I и II, и анапластическую крупноклеточную лимфому (ALCL). Комбинации по настоящему изобретению, например, комбинации ингибиторов CDK и ингибиторов BCL-2, описанных в настоящем описании, можно использовать для лечения злокачественной опухоли крови, описанной в настоящем описании.
[117] Термины "лечить," "лечение" или "лечащий", как используют в рамках изобретения, могут включать облегчение, ослабление или смягчение симптомов заболевания или состояния, предупреждение дополнительных симптомов, смягчение или предупреждение основных причин симптомов, ингибирование заболевания или состояния, например, остановку развития заболевания или состояния, смягчение заболевания или состояния, обеспечение регрессии заболевания или состояния, смягчение состояния, вызванного заболеванием или состоянием, или остановку симптомов заболевания или состояния либо профилактически и/либо терапевтически.
[118] Изобретение относится к способам предупреждения или снижения вероятности рецидива злокачественной опухоли у индивидуума, нуждающегося в этом. В определенных вариантах осуществления термин "предупреждать" или "предупреждение", связанный с заболеванием или нарушением, может относиться к соединению или комбинации, которые в статистической выборке снижают частоту возникновения нарушения или состояния в подвергаемой лечению выборке относительно контрольной выборки без лечения, или замедляют возникновение или снижают тяжесть одного или нескольких симптомов нарушения или состояния относительно контрольной выборки без лечения. Способ включает проведение комбинированной терапии, описанной в настоящем описании, для лечения минимальной резидуальной болезни, и/или в качестве поддерживающей терапии, например, в качестве пролонгированной или расширенной терапии после прекращения другой терапии злокачественной опухоли. Например, комбинированную терапию можно проводить после прекращения другой терапии злокачественной опухоли, такой как химиотерапия, лучевая терапия и/или хирургическая операция.
[119] В некоторых аспектах ингибитор протеасом можно комбинировать или использовать в комбинации с ингибитором CDK по изобретению, например, соединением или солью любой из формул I, Ia или Ib. В эукариотических клетках каскад убиквитин (Ub)-протеасома (UPS) вовлекает модификацию Ub и последующую деградацию белковых субстратов. UPS контролирует уровни многих клеточных регуляторных белков, в том числе факторов транскрипции, регулирующих клеточный цикл белков и факторов, участвующих в различных клеточных процессах. Общим признаком пути UPS является то, что высококонсервативный Ub ковалентно связывается с белками-мишенями через серии ферментов, а именно Ub-активирующий фермент E1, Ub-конъюгирующий фермент E2 и Ub-лигазу E3. E1 сначала активирует Ub и переносит ее на E2. С фермента E2 Ub переносится прямо на белок-мишень или непрямо через Ub-лигазу E3. Полиубиквитинилированный белок распознается и деградируется протеасомой 26S, большим комплексом с множеством видов протеолитической активности.
[120] Как используют в рамках изобретения, термин "ингибитор протеасом" относится к средству, которое блокирует действие протеасомы. Ингибирование протеасомы может препятствовать деградации проапоптотических факторов, таких как белок p53, позволяя активацию запрограммированной клеточной смерти в неопластических клетках, зависимых от подавления проапоптотических каскадов.
[121] Можно использовать любой ингибитор протеасом, и он может демонстрировать синергический эффект при использовании в комбинации с ингибитором CDK, например, соединением или солью формулы I, Ia или Ib. Неограничивающие примеры ингибиторов протеасом могут включать: бортезомиб, маризомиб, иксазомиб, дисульфирам, эпигаллокатехин-3-галлат, салиноспорамид A, карфилзомиб, ONX 0912, CEP-18770, MLN9708, эпоксомицин и MG132.
[122] В некоторых вариантах осуществления ингибитор протеасом используют в комбинации с ингибитором CDK по изобретению, например, соединением формулы I, Ia или Ib, для лечения злокачественной опухоли крови, такой как крупноклеточная B-клеточная лимфома или трижды негативный рак молочной железы.
[123] В некоторых аспектах комбинации, описанные в настоящем описании, например, комбинации ингибиторов CDK с ингибиторами BCL-2 или ингибиторами протеасом, можно использовать для лечения злокачественной опухоли. Комбинированная терапия, описанная в настоящем описании, может снижать вероятность метастазов у индивидуума, нуждающегося в этом. В некоторых вариантах осуществления метастаз представляет собой солидную опухоль. В некоторых вариантах осуществления метастаз представляет собой жидкостную опухоль. Злокачественные опухоли, которые представляют собой жидкостные опухоли, могут представляют собой опухоли, которые возникают, например, в крови, костном мозге и лимфатических узлах, и могут включать, например, лейкоз, миелоидный лейкоз, лимфоцитарный лейкоз, лимфому, лимфому Ходжкина, меланому и множественную миелому. Лейкозы включают, например, острый лимфобластный лейкоз (ALL), острый миелоидный лейкоз (AML), хронический лимфоцитарный лейкоз (CLL), хронический миелогенный лейкоз (CML) и волосатоклеточный лейкоз. Злокачественные опухоли, которые представляют собой солидные опухоли, включают, например, рак предстательной железы, рак яичка, рак молочной железы, рак головного мозга, рак поджелудочной железы, рак толстого кишечника, рак щитовидной железы, рак желудка, рак легкого, рак яичника, саркому Капоши, рак кожи, плоскоклеточный рак кожи, рак почки, рак головы и шеи, рак горла, плоскоклеточные карциномы, которые образуют влажные слизистые выстилки носа, рта, горла, рак мочевого пузыря, остеосаркому, рак шейки матки, рак эндометрия, рак пищевода, рак печени и рак почки. В некоторых вариантах осуществления состояние, подвергаемое лечению способами, описанными в настоящем описании, представляет собой метастаз клеток меланомы, клеток рака предстательной железы, клеток рака яичка, клеток рака молочной железы, клеток рака головного мозга, клеток рака поджелудочной железы, клеток рака толстого кишечника, клеток рака щитовидной железы, клеток рака желудка, клеток рака легкого, клеток рака яичника, клеток саркомы Капоши, клеток рака кожи, клеток рака почки, клеток рака головы и шеи, клеток рака горла, клеток плоскоклеточной карциномы, клеток рака мочевого пузыря, клеток остеосаркомы, клеток рака шейки матки, клеток рака эндометрия, клеток рака пищевода, клеток рака печени или клеток рака почки.
[124] Способы, описанные в настоящем описании, также можно использовать для ингибирования прогрессирования метастазирующих злокачественных опухолей. Неограничивающие примеры злокачественных опухолей включают карциному надпочечников, детскую карциному надпочечников, связанные со СПИД злокачественные опухоли, рак анального канала, рак аппендикса, базально-клеточную карциному, детскую базально-клеточную карциному, рак мочевого пузыря, детский рак мочевого пузыря, рак кости, опухоль головного мозга, детские астроцитомы, детскую глиому ствола головного мозга, детскую атипичную тератоидную/рабдоидную опухоль центральной нервной системы, детские эмбриональные опухоли центральной нервной системы, детские герминогенные опухоли центральной нервной системы, детскую краниофарингиому головного мозга, детскую эпендимому головного мозга, рак молочной железы, детские бронхиальные опухоли, карциноидную опухоль, детскую карциноидную опухоль, карциноидную опухоль желудочно-кишечного тракта, карциному неизвестного происхождения, детскую карциному неизвестного происхождения, детские опухоли сердца, рак шейки матки, детский рак шейки матки, детскую хордому, хронические миелопролиферативные нарушения, рак толстого кишечника, рак ободочной и прямой кишки, детский рак ободочной и прямой кишки, внепеченочный рак желчных протоков, карциному протоков in situ (DCIS), рак эндометрия, рак пищевода, детский рак пищевода, детскую эстезионейробластому, злокачественную опухоль глаза, злокачественную фиброзную гистиоцитому кости, рак желчного пузыря, гастральный рак (желудка), детский рак желудка, желудочно-кишечные стромальные опухоли (GIST), детские желудочно-кишечные стромальные опухоли (GIST), детскую экстракраниальную герминогенную опухоль, внегонадную герминогенную опухоль, гестационную трофобластическую опухоль, глиому, рак головы и шеи, детский рак головы и шеи, печеночноклеточный рак, гипофарингеальный рак, рак почки, ренальный рак, опухоль Вильмса, детские опухоли почек, гистиоцитоз из клеток Лангерганса, рак гортани, детский рак гортани, лейкоз, острый лимфобластный лейкоз (ALL), острый миелоидный лейкоз (AML), хронический лимфоцитарный лейкоз (CLL), хронический миелогенный лейкоз (cml), волосатоклеточный лейкоз, рак губы, рак печени (первичный), детский рак печени (первичный), долевую карциному in situ (LCIS), рак легкого, немелкоклеточный рак легкого, мелкоклеточный рак легкого, лимфому, связанную со СПИД лимфому, лимфому Беркитта, Т-клеточную лимфому кожи, лимфому Ходжкина, неходжкинскую лимфому, первичную лимфому центральной нервной системы (ЦНС), меланому, детскую меланому, внутриглазную меланому, карциному из клеток Меркеля, злокачественную мезотелиому, детскую злокачественную мезотелиому, метастазирующий плоскоклеточный рак шеи со скрытой первичной карциномой средней линии, вовлекающей ген NUT, рак полости рта, детские синдромы множественной эндокринной неоплазии, фунгоидный микоз, миелодиспластические синдромы, миелодиспластические новообразования, миелопролиферативные новообразования, множественную миелому, рак носовой полости, рак носоглотки, детский рак носоглотки, нейробластому, рак полости рта, детский рак полости рта, рак ротоглотки, рак яичника, детский рак яичника, эпителиальный рак яичника, низкозлокачественный рак яичника, рак поджелудочной железы, детский рак поджелудочной железы, нейроэндокринные опухоли поджелудочной железы (опухоли островковых клеток), детский папилломатоз, параганглиому, рак параназального синуса, рак паращитовидной железы, рак полового члена, рак глотки, феохромацитому, опухоль гипофиза, плазмаклеточное новообразование, детскую плевролегочную бластому, рак предстательной железы, рак прямой кишки, рак переходных клеток почечной лоханки, ретинобластому, рак слюной железы, детский рак слюной железы, семейство опухолей саркомы Юинга, саркому Капоши, остеосаркому, рабдомиосаркому, детскую рабдомиосаркому, саркому мягких тканей, саркому матки, синдром Сезари, детский рак кожи, немеланомный рак кожи, рак тонкого кишечника, плоскоклеточную карциному, детскую плоскоклеточную карциному, рак яичка, детский рак яичка, рак горла, тимому и тимическую карциному, детскую тимому и тимическую карциному, рак щитовидной железы, детский рак щитовидной железы, рак переходных клеток мочеточника, рак уретры, эндометриальный рак тела матки, рак влагалища, рак вульвы и макроглобулинемию Вальденстрема.
[125] Комбинированные способы терапии, описанные в настоящем описании, можно использовать вместе с другими способами терапии, такими как лучевая терапия. Режимы химиотерапии и лучевой терапии могут включать конечное количество циклов терапии с лекарственным средством, за которой следует терапия без лекарственного средства, или могут включать конечные временные рамки, в которые проводят химиотерапию или лучевую терапию. Протоколы могут определяться клиническими испытаниями, ярлыками лекарственных средств и клиническим персоналом совместно с индивидуумом, подвергаемым лечению. Количество циклов химиотерапии или лучевой терапии, или общая длительность режима химиотерапии или лучевой терапии могут варьироваться в зависимости от ответа индивидуума на терапию злокачественной опухоли. Фармацевтическое средство, описанное в настоящем описании, можно вводить после завершения режима химиотерапии химиотерапия или лучевой терапии.
[126] В некоторых аспектах комбинации, описанные в настоящем описании, можно использовать для лечения индивидуума, нуждающегося в этом. В некоторых случаях индивидуум, подвергаемый лечению способами и композициями, описанными в настоящем описании, может представлять собой человека. Индивидуум, подвергаемый лечению способами и композициями, описанными в настоящем описании, может представлять собой не являющееся человеком животное. Неограничивающие примеры не являющихся человеком животных могут включать не являющегося человеком примата, домашний скот, домашнее животное и лабораторное животное.
[127] В определенных вариантах осуществления комбинированную терапию, описанную в настоящем описании, можно вводить в качестве отдельных средств или можно комбинировать в единую фармацевтическую композицию. Например, комбинацию ингибитора CDK, например, соединения или соли формулы I, Ia или Ib, и ингибитора BCL-2, например, венетоклакса или навитоклакса, можно составлять в качестве двух отдельных фармацевтических композиций или два средства можно совместно составлять в качестве единой фармацевтической композиции.
[128] В определенных вариантах осуществления ингибитор CDK, например, соединение или соль формулы I, Ia или Ib, составляют совместно с ингибитором BCL-2 или ингибитором протеасом. В некоторых случаях, соединение формулы I, Ia или Ib составляют совместно с любым из навитоклакса, венетоклакса, бортезомиба, маризомиба или иксазомиба, или их комбинации.
[129] В определенных вариантах осуществления изобретение относится к фармацевтической композиции, например, для перорального или парентерального введения, содержащей соединение или соль формулы I, Ia или Ib. В некоторых аспектах фармацевтическая композиция содержит соединение или соль формулы I, Ia или Ib в количестве, составляющем по меньшей мере от приблизительно 1 мг до приблизительно 1000 мг, от приблизительно 100 мг до приблизительно 400 мг, от приблизительно 100 мг до приблизительно 200 мг, от приблизительно 200 мг до приблизительно 400 мг, или от приблизительно 250 мг до приблизительно 350 мг. Например, фармацевтическая композиция по изобретению может содержать приблизительно 100 мг, приблизительно 120 мг, приблизительно 140 мг, приблизительно 160 мг, приблизительно 180 мг, приблизительно 200 мг, приблизительно 220 мг, приблизительно 240 мг, приблизительно 260 мг, приблизительно 280 мг, приблизительно 300 мг, приблизительно 320 мг, приблизительно 340 мг, приблизительно 360 мг, приблизительно 380 мг, приблизительно 400 мг, приблизительно 420 мг, приблизительно 440 мг, приблизительно 460 мг, приблизительно 480 мг или приблизительно 500 мг соединения формулы I, Ia или Ib. Для соединения, описанного в настоящем описании, например, соединения формулы Ib, составленного в фармацевтическую композицию в форме соли, количество соединения может отражать массу свободного основания, а не массу солевой формы. В определенных вариантах осуществления фармацевтическая композиция соединения или соли формулы I, Ia или Ib не включает дополнительное средство против злокачественной опухоли, например, ингибитор BCL-2 или ингибитор протеасом. В определенных вариантах осуществления фармацевтическая композиция включает дополнительное средство против злокачественной опухоли, например, ингибитор BCL-2 или ингибитор протеасом.
[130] Терапевтически эффективное количество соединения по изобретению, например, соединения или соли формулы I, Ia или Ib, можно выражать в мг соединения на кг массы тела индивидуума. В некоторых случаях доза терапевтически эффективного количества может составлять по меньшей мере от приблизительно 0,1 мг/кг до приблизительно 20 мг/кг, например, приблизительно 0,1 мг/кг, 0,2 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг, 0,6 мг/кг, 0,7 мг/кг, 0,8 мг/кг, 0,9 мг/кг, 1 мг/кг, приблизительно 2 мг/кг, приблизительно 3 мг/кг, приблизительно 4 мг/кг, приблизительно 5 мг/кг, приблизительно 6 мг/кг, приблизительно 7 мг/кг, приблизительно 8 мг/кг, приблизительно 9 мг/кг, приблизительно 10 мг/кг или приблизительно 20 мг/кг. Для соединения, описанного в настоящем описании, например, соединения формулы Ib, составленного в фармацевтическую композицию в форме соли, терапевтически эффективное количество соединения может отражать массу свободного основания, а не массу формы соли.
[131] В определенных вариантах осуществления изобретение относится к фармацевтической композиции, например, для перорального или парентерального введения, содержащей ингибитор BCL-2, например, венетоклакс или навитоклакс. Фармацевтическая композиция может содержать ингибитор BCL-2 в количестве по меньшей мере от приблизительно 1 мг до приблизительно 1000 мг, от приблизительно 100 мг до приблизительно 1000 мг, от приблизительно 100 мг до приблизительно 800 мг, от приблизительно 200 мг до приблизительно 800 мг, или от приблизительно 300 мг до приблизительно 8000 мг. Например, фармацевтическая композиция по изобретению может содержать приблизительно 100 мг, приблизительно 120 мг, приблизительно 140 мг, приблизительно 160 мг, приблизительно 180 мг, приблизительно 200 мг, приблизительно 220 мг, приблизительно 240 мг, приблизительно 260 мг, приблизительно 280 мг, приблизительно 300 мг, приблизительно 320 мг, приблизительно 340 мг, приблизительно 360 мг, приблизительно 380 мг, приблизительно 400 мг, приблизительно 420 мг, приблизительно 440 мг, приблизительно 460 мг, приблизительно 480 мг, приблизительно 500 мг, приблизительно 520 мг, приблизительно 540 мг, приблизительно 560 мг, приблизительно 580 мг, приблизительно 600 мг, приблизительно 620 мг, приблизительно 640 мг, приблизительно 660 мг, приблизительно 680 мг, приблизительно 700 мг, приблизительно 720 мг, приблизительно 740 мг, приблизительно 760 мг, приблизительно 780 мг, приблизительно 800 мг, приблизительно 820 мг, приблизительно 840 мг, приблизительно 860 мг, приблизительно 880 мг, приблизительно 900 мг, приблизительно 920 мг, приблизительно 940 мг, приблизительно 960 мг, приблизительно 980 мг, или приблизительно 1000 мг ингибитора BCL-2, например, венетоклакса или навитоклакса.
[132] В определенных вариантах осуществления изобретение относится к фармацевтической композиции, например, для перорального или парентерального введения, содержащей ингибитор протеасом, например, бортезомиб, маризомиб или иксазомиб. Фармацевтическая композиция может содержать ингибитор протеасом в количестве по меньшей мере от приблизительно 0,5 мг до приблизительно 50 мг, от приблизительно 1 мг до приблизительно 30 мг, от приблизительно 1 мг до приблизительно 20 мг, от приблизительно 1 мг до приблизительно 10 мг, или от приблизительно 1 мг до приблизительно 5 мг. Например, фармацевтическая композиция по изобретению может содержать приблизительно 0,5 мг, приблизительно 1 мг, приблизительно 2 мг, приблизительно 3 мг, приблизительно 4 мг, приблизительно 5 мг, приблизительно 6 мг, приблизительно 7 мг, приблизительно 8 мг, приблизительно 9 мг, приблизительно 10 мг, приблизительно 11 мг, приблизительно 12 мг, приблизительно 13 мг, приблизительно 14 мг, приблизительно 15 мг, приблизительно 16 мг, приблизительно 17 мг, приблизительно 18 мг, приблизительно 19 мг или приблизительно 20 мг ингибитора протеасом, например, бортезомиба, маризомиба или иксазомиба.
[133] В определенных вариантах осуществления составы по изобретению содержат соединение или соль формулы I, Ia или Ib, ингибитор BCL-2 или ингибитор протеасом, где соединение или соль на от приблизительно 70% до приблизительно 99,99%, от приблизительно 80% до приблизительно 99,9%, от приблизительно 85% до приблизительно 99%, от приблизительно 90% до приблизительно 99%, от приблизительно 95% до приблизительно 99%, от приблизительно 97% до приблизительно 99%, от приблизительно 98% до приблизительно 99%, от приблизительно 98% до приблизительно 99,9%, от приблизительно 99% до приблизительно 99,99%, от приблизительно 99,5% до приблизительно 99,99%, от приблизительно 99,6% до приблизительно 99,99%, от приблизительно 99,8 до приблизительно 99,99%, или от приблизительно 99,9% до приблизительно 99,99% свободна от примесей.
[134] В определенных вариантах осуществления фармацевтическая композиция по изобретению содержит как соединение или соль формулы I, Ia или Ib, так и ингибитор BCL-2 в таких количествах, которые описаны в настоящем описании, как например, фармацевтическая композиция с 100-400 мг соединения или соли формулы Ib и от 200 до 800 мг ингибитора BCL-2, например, венетоклакса.
[135] В определенных вариантах осуществления фармацевтическая композиция по изобретению содержит как соединение или соль формулы I, Ia или Ib, так и ингибитор протеасом таких количествах, которые описаны в настоящем описании, как например, фармацевтическая композиция с 100-400 мг соединения или соли формулы Ib и 1-10 мг ингибитора протеасом, например, иксазомиба.
[136] Фармацевтические композиции, описанные в настоящем описании, могут иметь форму жидкого состава, твердого состава или их комбинации. Неограничивающие примеры составов могут включать таблетку, капсулу, пилюлю, гель, пасту, жидкий раствор и крем. В некоторых случаях терапевтическое средство, например, соединение или соль формулы I, Ia или Ib, ингибитор BCL-2 или ингибитор протеасом, могут быть в кристаллизованной форме. В фармацевтических композициях, содержащих два или более терапевтических средств, каждое средство может быть кристаллизовано отдельно, а затем они могут быть объединены, или они могут быть кристаллизованы вместе. Композиции могут содержать два или более терапевтических средств в одном или нескольких физических состояниях. Например, композиция может представлять собой таблетку, содержащую одно терапевтическое средство в твердом составе и другое терапевтическое средство или лекарственное средство в составе геля. В определенных вариантах осуществления композиция представляет собой единую фармацевтическую композицию, содержащую соединение или соль формулы I, Ia или Ib в первом физическом состоянии и ингибитор семейства BCL-2 или ингибитор протеасом во втором физическом состоянии.
[137] Композиции по настоящему изобретению, кроме того, могут содержать эксципиент или добавку. Эксципиенты могут включать любые и все растворители, покрытия, хелатирующие агенты, вкусовые добавки, красители, смазывающие вещества, разрыхлители, консерванты, подсластители, пеногасители, буферные средства, полимеры, антиоксиданты, связующие вещества, разбавители и наполнители (или носители). Как правило, эксципиент совместим с терапевтическими композициями по настоящему изобретению.
[138] Жидкие препараты для перорального введения могут иметь форму, например, растворов, сиропов или суспензий, или они могут быть предоставлены в качестве сухого продукта для восстановления водой или другими подходящими носителями перед применением. Такие жидкие препараты можно получать с использованием общепринятых подходов с фармацевтически приемлемыми добавками, такими как суспендирующие вещества (например, сорбит сироп, метил целлюлоза или гидрогенизированные пищевые жиры); эмульгаторы (например, лецитин или гуммиарабик); неводные носители (например, миндальное масло, маслообразные сложные эфиры или этиловый спирт); консерванты (например, метил или пропил п-гидроксибензоаты или сорбиновая кислота); и искусственные или натуральные красители и/или подсластители.
[139] Кроме того, настоящее изобретение охватывает безводные композиции и дозированные формы, содержащие активный ингредиент, поскольку вода может способствовать деградации некоторых соединений. Безводные композиции и дозированные формы по настоящему изобретению можно получать с использованием безводных или имеющих низкое содержание влаги ингредиентов и условий с низким содержанием влаги или с низкой сыростью. Композиции и дозированные формы по настоящему изобретению, которые содержат лактозу, могут быть изготовлены безводными, если ожидается существенный контакт с влагой и/или сыростью в ходе производства, упаковывания и/или хранения. Безводную композицию можно получать и хранить так, чтобы сохранялась ее безводная природа. Таким образом, безводные композиции можно упаковывать с использованием материалов, которые препятствуют воздействию воды, так чтобы они могли быть включены в подходящие наборы. Примеры подходящей упаковки включают, но не ограничиваются ими, герметично запаянную фольгу, контейнеры для единичных доз, блистерные упаковки и контурные упаковки.
[140] Ингредиент, описанный в настоящем описании, можно комбинировать в однородной смеси с фармацевтическим носителем согласно общепринятым способам составления фармацевтических препаратов. Носитель может иметь различные формы в зависимости от желаемой формы препарата для введения. При получении композиций для пероральной дозированной формы в качестве носителя можно использовать любую из обычных фармацевтических сред, например, такую как вода, гликоли, масла, спирты, вкусовые добавки, консерванты, красители, в случае пероральных жидких препаратов (таких как суспензии, растворы и эликсиры) или аэрозолей; или такие носители, как крахмалы, сахара, микрокристаллическая, разбавители, гранулирующие вещества, смазывающие вещества, связующие вещества и дезинтегрирующие вещества, можно использовать в случае пероральных твердых препаратов, в некоторых вариантах осуществления без использования лактозы. Например, в случае твердых пероральных препаратов подходящие носители включают порошки, капсулы и таблетки. Если желательно, таблетки можно покрывать стандартными водными или неводными способами.
[141] Некоторые примеры материалов, которые служат в качестве фармацевтически приемлемых носителей, включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлоза и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошковый трагакант; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и воски суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные средства, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) свободную от пирогенов воду; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатно-солевые буферы; и (21) другие нетоксичные совместимые вещества, используемые в области составления фармацевтических составов.
[142] Связующие вещества, пригодные для применения в дозированных формах, включают, но не ограничиваются ими, кукурузный крахмал, картофельный крахмал или другие крахмалы, желатин, природные и синтетические камеди, такие как гуммиарабик, альгинат натрия, альгиновая кислота, другие альгинаты, порошковый трагакант, гуаровая камедь, целлюлозу и ее производные (например, этилцеллюлоза, ацетат целлюлозы, кальций карбоксиметилцеллюлоза, натрий карбоксиметилцеллюлоза), поливинилпирролидон, метилцеллюлозу, прежелатинизированный крахмал, гидроксипропилметилцеллюлозу, микрокристаллическую целлюлозу и их смеси.
[143] Примеры подходящих наполнителей для применения в композициях и дозированных формах, описанных в настоящем описании, включают, но не ограничиваются ими, тальк, карбонат кальция (например, гранулы или порошок), микрокристаллическую целлюлозу, порошковую целлюлозу, декстраты, каолин, маннит, кремниевую кислоту, сорбит, крахмал, прежелатинизированный крахмал и их смеси.
[144] Когда для введения являются желательными водные суспензии и/или эликсиры, активный ингредиент в них можно комбинировать с различными подсластителями или вкусовыми добавками, красителями или пигментами и, если желательно, эмульгирующими и/или суспендирующими веществами, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и их различные комбинации.
[145] В одном варианте осуществления композиция может включать солюбилизатор для обеспечения высокой солюбилизации и/или растворения соединения по настоящему изобретению и для минимизации выпадения соединения по настоящему изобретению в осадок. Это может быть особенно важным для композиций не для перорального применения, например, для композиций для инъекции. Cолюбилизатор также можно добавлять для повышения растворимости гидрофильного лекарственное средство и/или других компонентов, таких как поверхностно-активные вещества, или для поддержания композиции в виде стабильного или однородного раствора или дисперсии.
[146] Фармацевтические композиции, описанные в настоящем описании, могут быть пригодными для введения индивидууму, нуждающемуся в этом. В некоторых случаях можно получать составы с замедленным высвобождением для перорального введения с целью достижения контролируемого высвобождения активного вещества в контакте жидкостями организма в желудочно-кишечном тракте и для обеспечения по существу постоянного и эффективного уровня активного вещества в плазме крови. Для этой цели кристаллическая форма может быть заключена в полимерную матрицу из биологически деградируемого полимера, растворимого в воде полимера или их смеси, и необязательно подходящих поверхностно-активных веществ. В этом контексте заключение может означать включение микрочастиц в матрицу из полимеров. Составы с контролируемым высвобождением также получают путем инкапсулирования диспергированных микрочастиц или эмульгированных микрокапель известными способами диспергирования или эмульгирования.
[147] В некоторых вариантах осуществления композиции можно составлять в виде пищевой композиции. Например, композиции могут представлять собой напиток или другие жидкости, твердые продукты питания, полутвердые продукты питания, с пищевым носителем или без него. Например, композиции могут включать черный чай с добавлением любой из композиций, описанных в настоящем описании. Композиция может представлять собой молочный продукт с добавлением любой из композиций, описанных в настоящем описании. В некоторых вариантах осуществления композиции могут быть составлены в виде пищевой композиции. Например, композиции могут содержать напиток, твердый продукт питания, полутвердый продукт питания или пищевой носитель.
[148] В определенных вариантах осуществления фармацевтические составы могут иметь форму, пригодную для парентеральной инъекции, такую как стерильная суспензия, раствор или эмульсия в масляных или водных носителях, и может содержать средства для составления, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Фармацевтические составы для парентерального введения включают, например, водные растворы активных соединений в растворимой в воде форме. Суспензии активных соединений можно получать, например, в качестве масляных суспензий для инъекций. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат, изопропилпальмитат, или триглицериды средней цепи, или липосомы. В предпочтительных вариантах осуществления состав для парентерального введения представляет собой водную суспензию.
[149] Соединение, описанное в настоящем описании, может присутствовать в композиции в диапазоне концентраций, причем диапазон определяется верхней и нижней величиной, выбранной из описанных выше концентраций. Например, соединение или соль по изобретению могут присутствовать в составе в концентрации от приблизительно 1 нМ до приблизительно 100 мМ, от приблизительно 10 нМ до приблизительно 10 мМ, от приблизительно 100 нМ до приблизительно 1 мМ, от приблизительно500 нМ до приблизительно 1 мМ, от приблизительно 1 мМ до приблизительно 50 мМ, от приблизительно 10 мМ до приблизительно 40 мМ, от приблизительно 20 мМ до приблизительно 35 мМ, или от приблизительно 20 мМ до приблизительно 30 мМ.
[150] Способы получения композиций, содержащих соединения, описанные в настоящем описании, могут включать составление соединений с одним или несколькими инертными фармацевтически приемлемыми эксципиентами. Жидкие композиции включают, например, растворы, в которых растворяют соединение, эмульсии, содержащие соединение, или раствор, содержащий липосомы, мицеллы или наночастицы, содержащие соединение, как описано в настоящем описании. Эти композиции также могут содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие вещества или эмульгаторы, pH-буферные средства и другие фармацевтически приемлемые добавки.
[151] Составы для инъекций могут быть предоставлены в единичной дозированной форме, например, в ампулах или контейнерах для многократных доз с добавлением консерванта. Композиции могут иметь такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать средства для составления, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Композиции могут быть предоставлены в контейнерах для однократной дозы или для многократных доз, например, в запаянных ампулах или флаконах, и их можно хранить в порошковой форме или в высушенном сублимационной сушкой (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, солевого раствора или стерильной свободной от пирогенов воды, непосредственно перед применением. Из стерильных порошков, гранул и таблеток описанного выше типа можно получать инъекционные растворы и суспензии для немедленного применения.
[152] Фармацевтические составы для парентерального введения включают водные и неводные (масляные) стерильные растворы для инъекции активных соединений, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, которые обеспечивают изотоничность состава с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие вещества и загустители. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы.
[153] Композицию, описанную в настоящем описании, например, фармацевтическую композицию соединения или соли формулы I, Ia или Ib, или ингибитора BCL-2 или ингибитора протеасом, или совместный состав соединения формулы I, Ia или Ib с ингибитором BCL-2 или ингибитором протеасом можно вводить один раз или более одного раза в сутки. Композицию можно вводить периодически (например, каждые сутки без перерыва на протяжении режима лечения). В некоторых случаях режим лечения может составлять менее недели, неделю, две недели, три недели, месяц или более месяца. В некоторых случаях композицию по изобретению вводят в течение по меньшей мере 12 недель. В других случаях композицию вводят в течение суток, по меньшей мере двух последовательных суток, по меньшей мере трех последовательных суток, по меньшей мере четырех последовательных суток, по меньшей мере пяти последовательных суток, по меньшей мере шести последовательных суток, по меньшей мере семи последовательных суток, по меньшей мере восьми последовательных суток, по меньшей мере девяти последовательных суток, по меньшей мере десяти последовательных суток или по меньшей мере более чем десяти последовательных суток. В некоторых случаях терапевтически эффективное количество можно вводить один раз в неделю, два раза в неделю, три раза в неделю, четыре раза в неделю, пять раз в неделю, шесть раз в неделю, семь раз в неделю, восемь раз в неделю, девять раз в неделю, 10 раз в неделю, 11 раз в неделю, 12 раз в неделю, 13 раз в неделю, 14 раз в неделю, 15 раз в неделю, 16 раз в неделю, 17 раз в неделю, 18 раз в неделю, 19 раз в неделю, 20 раз в неделю, 25 раз в неделю, 30 раз в неделю, 35 раз в неделю, 40 раз в неделю или более 40 раз в неделю. В некоторых случаях терапевтически эффективное количество можно вводить один раз в сутки, два раза в сутки, три раза в сутки, четыре раза в сутки, пять раз в сутки, шесть раз в сутки, семь раз в сутки, восемь раз в сутки, девять раз в сутки, 10 раз в сутки или более 10 раз в сутки. В некоторых случаях композицию вводят по меньшей мере два раза в сутки. В других случаях композицию вводят по меньшей мере каждый час, по меньшей мере каждые два часа, по меньшей мере каждые три часа, по меньшей мере каждые четыре часа, по меньшей мере каждые пять часов, по меньшей мере каждые шесть часов, по меньшей мере каждые семь часов, по меньшей мере каждые восемь часов, по меньшей мере каждые девять часов, по меньшей мере каждые 10 часов, по меньшей мере каждые 11 часов, по меньшей мере каждые 12 часов, по меньшей мере каждые 13 часов, по меньшей мере каждые 14 часов, по меньшей мере каждые 15 часов, по меньшей мере каждые 16 часов, по меньшей мере каждые 17 часов, по меньшей мере каждые 18 часов, по меньшей мере каждые 19 часов, по меньшей мере каждые 20 часов, по меньшей мере каждые 21 час, по меньшей мере каждые 22 часа, по меньшей мере каждые 23 часа или по меньшей мере каждые сутки.
[154] Фармацевтические композиции по изобретению можно вводить либо кратковременно, либо длительно. Фармацевтические композиции по изобретению можно вводить в качестве однократного лечения или в качестве курса лечения. Лечение можно проводить один раз в сутки, два раза в сутки, три раза в сутки, утром, вечером, до сна или непрерывно на протяжении суток. Лечение можно проводить каждые сутки, раз в двое суток, раз в трое суток, два раза в неделю, один раз в неделю, раз в две недели, раз в месяц, раз в шесть недель, раз в два месяца, раз в три месяца, раз в шесть месяцев, раз в год, раз в два года, раз в 5 лет или при необходимости.
[155] В определенных вариантах осуществления вводимую дозу лекарственного средства можно временно снижать или временно отменять на определенный период времени. В определенных вариантах осуществления пациент имеет выходные от лекарственного средства, когда пациент не получает лекарственное средство или получает уменьшенное количество лекарственного средства в течение некоторого периода времени. Выходные от лекарственного средства могут составлять, например, от 2 суток до 1 года, включая, только в качестве примера, 2 суток, 3 суток, 4 суток, 5 суток, 6 суток, 7 суток, 10 суток, 12 суток, 15 суток, 20 суток, 28 суток или более 28 суток. Выходные от лекарственного средства могут составлять приблизительно 1 месяц, приблизительно 2 месяца, приблизительно 3 месяца, приблизительно 4 месяца, приблизительно 5 месяцев, приблизительно 6 месяцев, приблизительно 7 месяцев, приблизительно 8 месяцев, приблизительно 9 месяцев, приблизительно 10 месяцев, приблизительно 11 месяцев или приблизительно 12 месяцев. Уменьшение дозы в ходе выходных от лекарственного средства может составлять, например 10%-100% от первоначальной вводимой дозы, включая, только в качестве примера, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% и 100%. Для дальнейших примеров снижение дозы может составлять от 10% до 100%, от 20% до 80%, от 30% до 70%, от 50% до 90%, от 80% до 100% или от 90% до 100%.
[156] После улучшения состояния пациента при необходимости можно вводить поддерживающую дозу. Затем дозировку или частоту введения, или обе из них, можно снижать в зависимости от симптомов, до уровня, на котором улучшение заболевания, нарушения или состояния может сохраняться.
[157] Дополнительные способы введения составов, описанных в настоящем описании, включают, например, доставку энтеральным путем, включая пероральный, желудочную или дуоденальную питающую трубку, ректальный суппозиторий, ректальную клизму, парентеральным путем, инъекцией, инфузией, внутриартериальным, внутрисердечным, внутрикожным, интрадуоденальным, интрамедуллярным, внутримышечным, внутрикостным, внутрибрюшинным, интратекальным, внутрисосудистым, внутривенным, интравитреальным, внутрикамерным, эпидуральным, подкожным, ингаляционным, трансдермальным, трансмукозальным, сублингвальным, буккальным, местным, накожным, дермальным, посредством ушных капель, интраназальным и вагинальным введением. Соединения, описанные в настоящем описании, можно вводить локально в область, нуждающуюся в лечении, например, посредством локальной инфузии в ходе хирургической операции, местного нанесения, такого как посредством кремов или мазей, инъекции, катетера или имплантата. Введение также можно осуществлять посредством прямой инъекции в область пораженной заболеванием ткани или органа.
[158] Длительность введения и/или вводимые количества могут определяться врачом или клиническим специалистом любого другого типа. Врач или клинический специалист может наблюдать ответ индивидуума на вводимые композиции и корректировать дозирование на основе показателей индивидуума. Например, дозировку для индивидуумов, которые демонстрируют сниженные эффекты в отношении регуляции энергии, можно повышать для достижения желаемых результатов.
[159] В некоторых вариантах осуществления комбинированную терапию, описанную в настоящем описании, можно проводить совместно в одно и то же время одним и тем же путем, или ее можно проводить по отдельности. В некоторых вариантах осуществления компоненты в композициях можно вводить с использованием одного и того же или различных путей введения.
[160] В некоторых вариантах осуществления изобретение также относится к способам производства композиций, описанных в настоящем описании. В некоторых вариантах осуществления производство композиции, описанной в настоящем описании, включает смешение или комбинирование двух или более компонентов.
[161] В некоторых вариантах осуществления композиции можно комбинировать или смешивать с фармацевтически активным или терапевтическим средством, носителем и/или эксципиентом. Примеры таких компонентов описаны в настоящем описании. Комбинированным композициям можно придавать форму единичных дозировок, таких как таблетки, капсулы, желатиновые капсулы, таблетки с замедленным высвобождением и т.п.
[162] В некоторых вариантах осуществления композицию получают так, что получают твердую композицию, содержащую по существу однородную смесь одного или нескольких компонентов, так что один или несколько компонентов равномерно диспергированы в композиции, так что композицию можно без труда подразделять на равноэффективные единичные дозированные формы, такие как таблетки, пилюли и капсулы.
[163] Единичную дозу можно упаковывать в контейнер для транспортировки к пользователю. Единичную дозу можно упаковывать в пробирку, банку, коробку, флакон, мешок, лоток, бидон, бутылку, шприц или консервную банку.
[164] Другой аспект изобретения относится к достижению желаемых эффектов у одного или нескольких индивидуумов после введения комбинированной композиции, описанной в настоящем описании, в течение определенного периода времени. Например, полезные эффекты композиций, описанных в настоящем описании, можно наблюдать после введении композиций индивидууму в течение 1, 2, 3, 4, 6, 8, 10, 12, 24 или 52 недель.
[165] В определенных вариантах осуществления комбинированные способы терапии, описанные в настоящем описании, можно проводить посредством комбинированного режима лечения. Комбинированный режим лечения может охватывать режимы лечения, в которых введение соединения, описанного в настоящем описании, или его фармацевтически приемлемой соли начинают до, в процессе или после введения второго средства, описанного в настоящем описании, и его продолжают до любого момента лечения вторым средством или после завершения лечения вторым средством. Также изобретение относится к способам лечения, в которых соединение, описанное в настоящем описании, или его фармацевтически приемлемую соль и второе средство, используемое в комбинации, вводят одновременно или в различные моменты времени и/или с уменьшающимися или возрастающими интервалами в ходе периода лечения. Комбинированное лечение, кроме того, включает периодическое лечение, которое начинает и останавливается в различные моменты времени, способствуя клиническому контролю пациента.
[166] В определенных вариантах осуществления комбинированная терапия может обеспечить терапевтическое преимущество ввиду дифференциальной токсичности, ассоциированной с двумя способами лечения. Например, лечение ингибиторами CDK, такими как ингибиторы CDK, описанные в настоящем описании, может приводить к конкретной токсичности, которая не наблюдается в случае средства против злокачественной опухоли, например, ингибитора BCL-2 или ингибитора протеасом, и наоборот. По существу, эта дифференциальная токсичность может позволить проведение каждого лечения в дозе, в которой указанная токсичность не существует или минимально, так что комбинированная терапия обеспечивает терапевтическую дозу, избегая токсичности каждого из компонентов комбинированных средств. Более того, когда терапевтические эффекты, достигаемые в результате комбинированного лечения, являются синергичными, дозы каждого из средств можно снижать еще больше, таким образом, снижая ассоциированную с ними токсичность в еще большей степени.
[167] Соединения, описанные в настоящем описании, или их фармацевтически приемлемые соли, а также комбинированные способы терапии можно проводить до, в процессе или после возникновения заболевания или состояния и время введения композиций, содержащих соединение, варьируется. Соединения, описанные в настоящем описании, можно использовать в качестве профилактического средства и можно вводить непрерывно индивидуумам, предрасположенным к развитию состояний или заболеваний для предупреждения возникновения заболевания или состояния. Соединения, описанные в настоящем описании, и их композиции можно вводить индивидууму при наличии или насколько возможно ранее после возникновения симптомов. Соединение, описанное в настоящем описании, можно вводить насколько рано, насколько это применимо на практике после обнаружения или предположения возникновения заболевания или состояния, и в течение периода времени, необходимого для лечения заболевания.
ПРИМЕРЫ
[168] Приведенные ниже примеры предоставлены для иллюстрации различных вариантов осуществления изобретения и не ограничивают настоящее изобретение никоим образом. Приведенные пример, а также способы, описанные в настоящем описании, являются отражают предпочтительные варианты осуществления, являются иллюстративными и не ограничивают объем изобретения. Их изменения и другие применения, которые охватываются сущностью изобретения, как определено объемом формулы изобретения, станут понятными специалистам в данной области.
[169] Для всех исследований механизма действия лекарственных средств in vitro использовали материалы и способы, описанные ниже.
[170] Клеточные линии, представленные в таблице 1, прикрепленные или в виде суспензии, обрабатывали в 6-ячеечных чашках или флаконах T10, соответственно, с плотностью посева в количестве один миллион на мл перед обработкой в общих объемах 3-10 мл. Все средства, использованные для обработки клеток in vitro, составляли в воде/DMSO для достижения требуемых концентраций (как указано на соответствующих подписях/легендах к фигурам), причем концентрации DMSO не превышали 0,01%. Источники лекарственных средств указаны в таблице в столбце "исследования микроинъекции CIVO". Клетки собирали в экспериментальные моменты времени: через 2, 6 или 24 часов после обработки, как указано на соответствующих фигурах, сначала путем промывания клеток два раза ледяным PBS в 15-мл пробирках falcon, центрифугирования при 1000 об/мин в течение 5 мин, за которым следовал лизис соответствующих клеточных осадков, как описано в разделе ниже. Примечание: резистентные к лекарственным средствам клеточные линии NUDHL1 Dox 1000 нМ и Toledo Dox 1000 нМ были получены в Presage путем культивирования исходных линий в среде с пошаговым увеличением концентрации доксорубицина на протяжении нескольких пассажей. Эти клетки использовали для получения ксенотрансплантата, когда достигали резистентности к 1 мкМ Dox.
Таблица 1
[171] Для всех исследований с анализом с использованием вестерн-блоттинга использовали материалы и способы, описанные ниже.
[172] Белковые лизаты получали добавлением ледяного 1X буфера RIPA (из 10X, Millipore) с 1X-3X коктейлем ингибиторов протеаз и фосфатаз Halt (100X, Pierce) к клеточным осадкам с последующей обработкой импульсным ультразвуковым излучением (~3-5 с) три раза. Лизаты очищали центрифугированием при 14000 об/мин в течение 15-20 мин и супернатант (лизат) собирали для дальнейшего анализа. Для получения лизатов из криоконсервированной опухолевой ткани трансплантата ткань сначала гомогенизировали с использованием ручного гомгогенизатора тканей (Cole Palmer) в описанном выше лизирующем буфере, а затем проводили импульсную обработку ультразвуковым излучением и центрифугирование, как описано выше. Описанные выше стадии проводили в условиях охлаждения льдом. Образцы белка количественно определяли с использованием анализа Брэдфорда (Biorad) в разведениях 1:10 или выше с использованием соответствующих пустых контролей с 1x RIPA. 20-25 микрограмм белка из каждого образца подвергали SDS-PAGE с использованием 4-12% Bis-Tris гелей BOLT (Thermofisher) и переносили на мембраны с размером пор либо 0,45, либо 0,2 микрометра (в зависимости от молекулярного размера блека), и либо нитроцеллюлозы (NC), либо pvdf (Thermofisher) с использованием протокола изготовителя, блокировали в течение одного часа в 5% молоке при комнатной температуре, добавляли первичные антитела (таблица 2) при 4°C в течение ночи и соответствующие конъюгированные с HRP вторичные антитела (Jackson Immunoresearch, разведения 1:10000-1:20000) на 1 час при комнатной температуре. После каждой инкубации с антителом проводили три промывания PBS/0,01% Tween-20 (5-10 мин каждое). Для обнаружения белковых сигналов использовали хемилюминесцентный субстрат ECL (Pierce или GE) и авторадиографическую пленку (Thermofisher).
Таблица 2
[173] Для всех исследований in vivo использовали материалы и способы, приведенные ниже.
[174] Вся работа на мышах была одобрена IACUC Board of Presage Biosciences, Seattle, WA. Все связанные с ними процедуры проводили под анестезии и были приложены все усилия для минимизации боли и страдания. Ни одна из мышей, участвовавших в испытании, не заболела и не погибла до конца эксперимента и все мыши, которым проводили лечение лекарственными средствами, как описано ниже, подвергались плановому мониторингу состояния здоровья, и были гуманным образом умерщвлены в конце эксперимента. Подкожные ксенотрансплантаты в боку получали с использованием клеточных линий/линий мышей, приведенных в таблице ниже (таблица 3). 200 мкл суспензии клеток в соотношении 1:1 с матригелем (Corning) инъецировали в правый бок с использованием 1-мл шприцов (BD 309659) и игл 27G (BD 305109).
Таблица 3
[175] Для всех исследований микроинъекции CIVO использовали материалы и способы, описанные ниже.
[176] Мышей включали в исследования лекарственных средств CIVO, когда объем опухоли достигал приблизительно 1000 мм3. Исследования микроинъекции проводили с использованием устройства CIVO, как описано ранее (Klinghoffer et al, Science Translational Medicine 2015). Устройство было оборудовано 6 иглами для инъекций и настроено для длины инъекции 6 мм и общего объема доставки 3 мкл. В каждый резервуар для лекарственного средства в носитель для доставки добавляли флуоресцентный визуализирующий маркер (FTM) вместе с каждым лекарственным средством или комбинацией лекарственных средств. Все микродозы были эквивалентными или меньшими, чем дозы, которые были бы допустимы согласно руководству FDA для испытаний Исследуемых IND (Исследуемое новое лекарственное средство) и вследствие растворимости лекарственного средства в носителе. Общие инъецированные количества средств приведены в таблице 4.
Таблица 4
[177] Для всех исследований системной эффективности лекарственных средств использовали материалы и способы, приведенные ниже.
[178] Мышей включали в исследование, когда опухоли достигали среднего объема 150-200 мм3. Объем опухоли вычисляли как V=длина × ширина × высота, все три размера измеряли с использованием цифровых толщиномеров одновременно с массой тела. Исследование животного прекращали, когда любой из трех измеренных размеров превышал 2 см, объем превышал 2500 мм3, наблюдалось изъязвление или снижение массы тела более чем на 20%.
Ингибирование роста опухоли % (TGI) определяют как:
 ,
,
где размеры усреднены среди опухолей в соответствующих группах. Критерий суммы рангов Уилкоксона или критерий Манна-Уитни использовали в качестве статистического критерия для определения различий между группами.
[179] Дозы лекарственных средств, пути введения, схемы введения и протоколы составления описаны ниже. Для кормления через пероральный зонд использовали трубки для кормления Instech, Ref: FTP-20-38, а для в/в использовали инсулиновые шприцы BD.
Исследования DLBCL
- RIVA: Воруциклиб 200 мг/кг (п/о) 6 введение/ 1 отдых и ABT 199 (п/о) 1 мг/кг 2x/неделя в течение 4 недель
- SUDHL4: Воруциклиб 200 мг/кг (п/о) 6 введение/ 1 отдых и ABT 199 (п/о) 25 мг/кг 6 введение/ 1 отдых в течение 4 недель
- U2932: Воруциклиб 200 мг/кг (п/о) 6 введение/ 1 отдых и ABT 199 (п/о) 10 мг/кг 2x/неделя в течение 4 недель
Исследования AML
- SKM1: Воруциклиб 200 мг/кг (п/о) 6 введение/ 1 отдых+ABT 199 (п/о) 10 мг/кг 6 введение/ 1 отдых в течение 3 недель
Исследования TNBC
- HCC1187: Воруциклиб 200 мг/кг (п/о) 5 введение/ 2 отдых+иксазомиб/MLN2238 (п/о) 3 мг/кг сутки 3, 6, 10, 13 в течение 2 недель
- HCC1187: пальбоциклиб 120 мг/кг (п/о) QD × 14+бортезомиб 0,42 мг/кг (в/в), сутки 1, 4, 8, 11 в течение 2 недель
- HCC1187: Воруциклиб 200 мг/кг (п/о)+бортезомиб 0,42 мг/кг (в/в), сутки 1, 4, 8, 11 в течение 2 недель
Пример 1. Воруциклиб является мощным ингибитором циклин-зависимой киназы-9 (CDK9)
[180] Полагают, что воруциклиб является мощным ингибитором CDK4/6. Активность воруциклиба в отношении 38 различных киназ тестировали по сравнению с флавопирадолом, известным ингибитором CDK9. На фиг.1 представлены структуры воруциклиба и флавопиридола, а также сравнение профилей ингибирования воруциклиба и флавопирадола. На фиг.1 продемонстрировано, что аналогично флавопирадолу, воруциклиб является мощным ингибитором CDK9 (Ki < 10 нМ). Однако воруциклиб является более специфичным к CDK, чем флавопирадол. На фиг.1 показано, что флавопирадол является мощным ингибитором серин/треониновых протеинкиназ MAK и ICK, в то время как воруциклиб не демонстрирует мощного ингибирования ни MAK, ни ICK. MAK и ICK ассоциированы с эпителиальными клетками кишечника и могут быть причиной тяжелой диареи, ассоциированной с лечением флавопирадолом. Воруциклиб, с другой стороны, может демонстрировать меньше желудочно-кишечных побочных эффектов. Скрининг DiscoveRx и Thermofisher: процентное ингибирование киназной активности для 468 анализов киназ в панели киназ DiscoveRx ScanMax и 414 анализов киназ в панели киназ ThermoFisher SelectScreen при 10 мкМ и 50 нМ. Исследуемое соединение предварительно отвешивают в Presage в силанизированные емкости из темного стекла с навинчивающейся крышкой и транспортируют в DiscoveRx и Reaction Biology. Для ThermoFisher соединение взвешивают и растворяют в концентрации 10 мМ в DMSO перед транспортировкой в условиях окружающей среды. При получении флаконы хранят при -20°C до применения. Порошок растворяют в DMSO в концентрации 10 мМ.
[181] На фиг.2 представлена проиллюстрирована эффективность воруциклиба против CDK 1, 4, 6, и 9, соответствующая одноразрядному количеству нМ, причем наиболее сильным является ингибирование CDK9. Определение профиля Reaction Biology: задачей исследования было определение рангового порядка чувствительности 48 киназ к воруциклибу гидрохлориду. Тестирование проводили в Reaction Biology Corp. Киназную активность количественно определяли с использованием анализа связывания с фильтром с радиоактивным γ-33P-ATP в качестве донора фосфатов. Концентрация ATP была близкой к Km для соответствующей киназы. Для каждой киназы величину IC50 вычисляли из кривой концентрации из 10 точек для исследуемого соединения и конвертировали в значения Ki. 48 киназ, исследованных в данном примере, были идентифицированы в предшествующих скрининговых экспериментах в качестве наиболее перспективных кандидатов.
Пример 2. Воруциклиб демонстрирует синергический эффект в комбинации с ингибитором семейства Bcl-2 венетоклаксом (ABT-199)
[182] Клетки диффузной крупноклеточной B-клеточной лимфомы (DLBCL) NU-DHL-1 обрабатывали носителем, воруциклибом отдельно, венетоклаксом (ABT-199) отдельно или комбинацией воруциклиба и венетоклакса. На фиг.3A-3D представлены клетки NU-DHL-1, окрашенные в отношении антиапоптотического белка, индуцированного белка дифференцировки клеток миелоидного лейкоза Mcl-1 (MCL-1) (показано красным цветом). На фиг.3A показаны клетки, которые обрабатывали только носителем. Фиг.3B: воруциклиб отдельно снижал экспрессию MCL-1, как показано увеличением более темных областей на фрагменте 301. Венетоклакс отдельно повышал экспрессию MCL-1, как показано светлыми областями на фрагменте 302 фиг.3C. Комбинация воруциклиба и венетоклакса приводила к выраженному снижению экспрессии MCL-1, как показано на фиг.3D, показанное темными областями на фрагменте 303.
[183] На фиг.4A-4D продемонстрирована корреляция между подавлением MCL-1 и синергической индукцией апоптоза. Клетки диффузной крупноклеточной B-клеточной лимфомы (DLBCL) NU-DHL-1 обрабатывали носителем, воруциклибом отдельно, венетоклаксом (ABT-199) отдельно или комбинацией воруциклиба и венетоклакса. Клетки окрашивали на расщепленную каспазу-3 (CC3) (показано красным цветом) в качестве маркера апоптоза. На фиг.4A показаны клетки, которые обрабатывали только носителем. Воруциклиб отдельно, как показано на фиг.4B, продемонстрировал несколько увеличенную экспрессию CC3, как показано светлыми областями на фиг.401. Аналогично венетоклакс отдельно, как показано на фиг.4C, продемонстрировал небольшое увеличение экспрессии CC3, как показано светлыми областями на фрагменте 402. Комбинация воруциклиба и венетоклакса на фиг.4D продемонстрировало синергичное увеличение экспрессии CC3, на что указывает большая светлая область на фрагменте 403, что позволяет предположить об увеличенной индукции апоптоза в обработанных клетках.
[184] На фиг.6A-6E продемонстрировано, что синергический эффект воруциклиба и венетоклакса на апоптоз является воспроизводимым в множестве моделей DLBCL. В кратком изложении, клетки RIVA, Toledo, Toledo/Dox1000, NUDHL и NUDHL/Dox1000 обрабатывали воруциклибом отдельно, венетоклаксом отдельно или воруциклибом и венетоклаксом в комбинации, как показано на фиг.6A, фиг.6B, фиг.6C, фиг.6D и фиг.6E, соответственно. Воруциклиб отдельно и венетоклакс отдельно имели небольшой эффект на экспрессию CC3, однако комбинация воруциклиба и венетоклакса продемонстрировала синергичную индукцию экспрессии CC3 во всех пяти моделях DLBCL.
[185] На фиг.7 представлена предложенная модель для этого синергического эффекта. Воруциклиб, посредством ингибирования MCL-1 через CDK9, может направлять клетки на апоптоз при комбинировании с ингибированием Bcl-2.
Пример 3. Воруциклиб демонстрирует синергический эффект в комбинации с ингибитором семейства Bcl-2 навитоклаксом
[186] Клетки лимфомы Беркитта Ramos обрабатывали носителем, воруциклибом отдельно, навитоклаксом (ABT-263) отдельно или комбинацией воруциклиба и навитоклакса. На фиг.5A-5D показаны клетки лимфомы Беркитта Ramos, окрашенные на расщепленную каспазу-3 (CC3) (показано красным) в качестве маркера апоптоза. На фиг.5A показаны клетки, обработанные только носителем. Воруциклиб отдельно, как показано на фиг.5B, продемонстрировал небольшое увеличение экспрессии CC3, как показано светлыми областями на фрагменте 501. Аналогично, венетоклакс отдельно, как показано на фиг.5C, продемонстировал небольшое увеличение экспрессии CC3, как показано светлыми областями на фрагменте 502. Комбинация воруциклиба и навитоклакса продемонстрировала выраженное увеличение экспрессии CC3, как показано большим количеством светлых областей на фрагменте 503 фиг.5D, что указывает на повышенную индукцию апоптоза в обработанных клетках.
Пример 4. Воруциклиб демонстрирует синергический эффект в комбинации с ингибитором протеасом
[187] Скрининг лекарственных средств проводили для исследования множества лекарственных средств одновременно. На фиг.8A представлен перечень различных соединений, которые инъецировали в модели с ксенотрансплантатом трижды негативного рака молочной железы (TNBC) HCC1187. На фиг.8B представлено среднее % изменение индукции MCL-1 в ксенотрансплантатах. Ингибитор протеасом бортезомиб продемонстрировал увеличение экспрессии MCL-1. На фиг.8C показаны ксенотрансплантаты, которые обрабатывали носителем. На фиг.8D представлены ксенотрансплантаты, которые обрабатывали бортезомибом и окрашивали на экспрессию MCL-1 (показано красным цвеом). Бортезомиб значительно повышал экспрессию MCL-1 в этих клетках, как показано увеличением светлых областей на фрагменте 801.
Пример 5. Воруциклиб демонстрирует синергический эффект в комбинации с ингибитором протеасом маризомибом
[188] Ксенотрансплантаты DLBCL NudHL1 обрабатывали в течение 6 часов носителем, воруциклибом отдельно, маризомибом отдельно или воруциклибом и маризомибом в комбинации. На фиг.9A-9D представлены клетки, которые были обрабатывали, как описано выше, и окрашивали на расщепленную каспазу-3 (CC3) в качестве маркера апоптоза (показано красным цветом). На фиг.9A показаны клетки, которые обрабатывали только носителем. Клетки, обработанные воруциклибом отдельно, как показано на фиг.9B, имели либо небольшое повышение экспрессии CC3, либо не имели его, на что указывает небольшое количество светлых областей на фрагменте 901. Аналогично, клетки, обработанные только маризомибом, как показано на фиг.9C, имели либо небольшое повышение экспрессии CC3, либо не имели его, на что указывает небольшое количество светлых областей на фрагменте 902. Клетки, обработанные комбинацией воруциклиба и маризомиба, продемонстрировали выраженное повышение экспрессии CC3, на что указывает большое количество светлых областей на фрагменте 903 фиг.9D, позволяя сделать предположение о синергическом эффекте на индукцию апоптоза в этих клетках.
Пример 6. Воруциклиб демонстрирует синергический эффект в комбинации с ингибитором протеасом бортезомибом
[189] в кратком изложении, ксенотрансплантаты DLBCL NudHL1 обрабатывали в течение 6 часов носителем, воруциклибом отдельно, бортезомибом отдельно или воруциклибом и бортезомибом в комбинации. На фиг.10A-10D представлены клетки, которые обрабатывали, как описано выше, и окрашивали на расщепленную каспазу-3 (CC3) в качестве маркера апоптоза (показано красным цветом).
[190] На фиг.10A показаны клетки, которые обрабатывали только носителем. Клетки, обработанные воруциклибом отдельно, как показано на фиг.10B, имели либо небольшое повышение экспрессии CC3, либо не имели его. Аналогично, клетки, обработанные только бортезомибом, как показано на фиг.10C, имели либо небольшое повышение экспрессии CC3, либо не имели его, на что указывает небольшое количество светлых областей на фрагменте 1001. Клетки, обработанные комбинацией воруциклиба и бортезомиба, продемонстрировали выраженное повышение экспрессии CC3, на что указывает большое количество светлых областей на фрагменте 1002 фиг.10D, позволяя сделать предположение о синергическом эффекте на индукцию апоптоза в этих клетках.
[191] На фиг.11 представлен сходный эксперимент, проведенный на модели с ксенотранспланатом трижды негативного рака молочной железы (TNBC) HCC1187. В кратком изложении, клетки обрабатывали носителем, воруциклибом отдельно, бортезомибом отдельно, или воруциклибом и бортезомибом в комбинации. Затем клетки окрашивали на экспрессию CC3 (показано красным цветом) в качестве маркера апоптоза. На фиг.11A показаны клетки, которые обрабатывали только носителем. Клетки, обработанные воруциклибом отдельно, как показано на фиг.11B, имели либо небольшое повышение экспрессии CC3, либо не имели его. Аналогично, клетки, обработанные бортезомибом отдельно, как показано на фиг.11C, имели либо небольшое повышение экспрессии CC3, либо не имели его. Клетки, обработанные комбинацией воруциклиба и бортезомиба, продемонстрировали выраженное повышение экспрессии CC3, на что указывает большое количество светлых областей на фрагменте 1101 фиг.11D, позволяя сделать предположение о синергическом эффекте на индукцию апоптоза в этих клетках.
[192] Модельным мышам с ксенотрансплантатами трижды негативного рака молочной железы (TNBC) HCC1187 вводили носитель, воруциклиб (200 мг/кг), бортезомиб (0,42 мг/кг) или воруциклиб (200 мг/кг) и бортезомиб (0,42 мг/кг) в комбинации. Проводили измерение объема опухоли у каждой мыши с течением времени и результаты представлены на фиг.12A-12D. Мыши, которым вводили комбинацию воруциклиба и бортезомиба (фиг.12D), продемонстрировали выраженное снижение нормализованного объема опухоли с течением времени по сравнению с мышами, которым вводили воруциклиб отдельно (фиг.12B) или бортезомиб отдельно (фиг.12C). На фиг.12E представлен вестерн-блот для экспрессии белка MLC-1. В клетках, в случае которых вводили воруциклиб, была снижена экспрессия белка MCL-1 по сравнению с образцом без введения, в то время как клетки, в случае которых вводили бортезомиб, имели увеличенную экспрессию MCL-1 по сравнению с образцом без введения. Кроме того, воруциклиб снижал индуцируемое бортезомибом увеличение экспрессии MCL-1.
[193] На фиг.13 продемонстрировано, что масса тела у мышей не изменялась в результате введения воруциклиба отдельно, бортезомиба отдельно или воруциклиба и бортезомиба в комбинации. Эти данные могут указывать на то, что комбинированная терапия воруциклибом и бортезомибом может демонстрировать либо малую токсичность, либо ее отсутствие.
[194] Кроме того, воруциклиб препятствует индуцируемой бортезомибом активации способствующих выживанию белков, MCL-1 и убиквитин-протеинлигазы E3 XIAP. На фиг.15A представлена предполагаемая модель ингибирования CDK9 воруциклибом. На фиг.15B показаны уровни белка MCL-1 и XIAP в клетках, в случае которых вводили воруциклиб отдельно, бортезомиб отдельно или воруциклиб и бортезомиб в комбинации. Вестерн-блот демонстрирует, что воруциклиб снижает индуцируемую бортезомибом активацию как белка MCL-1, так и белка XIAP в этих клетках.
Пример 7. Пальбоциклиб, ингибитор CDK 4/6, не демонстрирует синергического эффекта в комбинации с бортезомибом
[195] Модельным мышам с ксенотрансплантатами трижды негативного рака молочной железы (TNBC) HCC1187 вводили носитель, пальбоциклиб (120 мг/кг), бортезомиб (0,42 мг/кг) или пальбоциклиб (120 мг/кг) и бортезомиб (0,42 мг/кг) в комбинации. Проводили измерение объема опухоли у каждой мыши с течением времени и результаты представлены на фиг.14. В отличие от воруциклиба, пальбоциклиб не демонстрировал синергического эффекта на объем опухоли в комбинации с бортезомибом. Пальбоциклиб является специфическим ингибитором CDK4/6, в то время как воруциклиб нацелен на CDK9. Таким образом, это различие может быть специфическим для CDK9.
Пример 8. Каскад стрессового ответа эндоплазматической сети (ER) может играть роль в синергических эффектах воруциклиба в комбинации с бортезомибом
[196] На фиг.16A представлен срез ткани, который обрабатывали бортезомибом. Бортезомиб индуцирует апоптоз в некоторых клетках, в то время как некоторые клетки оказались резистентными к обработке бортезомибом. Клетки окрашивали на белок-шаперон ER GRP178/BiP. На фиг.16B показано, что клетки, резистентные к апоптозу под действием бортезомиба, экспрессируют GRP178/BiP.
[197] Далее клетки трижды негативного рака молочной железы HCC1187 обрабатывали воруциклибом отдельно, бортезомибом отдельно, туникамицином отдельно (индуктор ER-стресса) или в комбинации. Туникамицин отдельно сильно индуцировал X-бокс связывающий белок 1 (XBP1), белок ER-стресса из IRE1α-звена каскада ER-стресса, через 6 часов. Кроме того, туникамицин и бортезомиб сильно индуцировали экспрессию XBP1 через 24 часа (фиг.18B). Комбинация воруциклиба либо с бортезомибом, либо с туникамицином, снижала активацию XBP1. Эти результаты указывают на то, что воруциклиб может ингибировать IRE1α-звено пути ER-стресса (фиг.18B). Воруциклиб также снижает индуцируемую бортезомибом индукцию транскрипции XBP1 (фиг.19A-19B). Клетки HCC1187 обрабатывали воруциклибом (1,5 мкМ), бортезомибом (10 нМ), ингибитором эндорибонуклеазной активности IRE1a STF083010 (Millipore) (60 мкМ) или туникамицином (Sigma) (100 нМ) в течение 4 часов. мРНК собирали с использованием набора Qiagen RNAeasy Kit, количественно определяли с использованием спектрофотометра Nanodrop. 1 мкг мРНК каждых условий использовали для получения кДНК с использованием набора для High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) и протокола изготовителя. 100 нг кДНК использовали для проведения анализа ПЦР для амплификации несплайсированных или сплайсированных форм XBP1 (Thermofisher PCR Supermix) - 94°C в течение 2 мин, (94°C в течение 15 с, 55°C в течение 30 с, 72°C в течение 1 мин)×30 циклов, 72°C в течение 7 минут, удержание при 4°C. Выявляли сплайсированные и несплайсированные формы XBP1.
Пример 9. Воруциклиб демонстрирует синергический эффект в комбинации с ингибитором протеасом иксазомибом
[198] На фиг.20A-20D представлена модель с ксенотрансплантатом трижды негативного рака молочной железы (TNBC) HCC1187. В кратком изложении, клетки обрабатывали носителем, воруциклибом отдельно, иксазомибом отдельно или воруциклибом и иксазомибом в комбинации. Затем клетки окрашивали на экспрессию CC3 (показано красным цветом) в качестве маркера апоптоза.
[199] На фиг.20A показаны клетки, которые обрабатывали только носителем. Клетки, обработанные воруциклибом отдельно, как показано на фиг.20B, имели либо небольшое повышение экспрессии CC3, либо не имели его, на что указывает небольшое количество светлых областей на фрагменте 2001. Аналогично, клетки, обработанные иксазомибом отдельно, как показано на фиг.20C, имели либо небольшое повышение экспрессии CC3, либо не имели его, на что указывает небольшое количество светлых областей на фрагменте 2002. Клетки, обработанные комбинацией воруциклиба и иксазомиба, продемонстрировали выраженное повышение экспрессии CC3, на что указывает большое количество светлых областей на фрагменте 2003 фиг.20D, позволяя сделать предположение о синергическом эффекте на индукцию апоптоза в этих клетках.
[200] Различные ингибиторы CDK тестировали в комбинации с иксазомибом, как показано на фиг.21A и 21B. Только воруциклиб продемонстрировал синергический эффект на апоптоз опухолевых клеток в комбинации с иксазомибом, как показано светлыми областями на фрагментах 2101 и 2102. Ни один из носителя отдельно, иксазомиба отдельно, пальбоциклиба и иксазомиба, динациклиба и иксазомиба, и флавопирадола и иксазомиба не демонстрировал синергический эффект на апоптоз опухолевых клеток, который демонстрировала комбинация воруциклиба и иксазомиба.
Пример 10. Воруциклиб демонстрирует синергический эффект в комбинации с венетоклаксом
[201] На фиг.22A-B проиллюстрирована синергия воруциклиба и венетоклакса в модели DLBCL SU-DHL-4. Расщепленный PARP (cPARP) можно использовать в качестве биомаркера апоптоза. Для оценки эффективности комбинации двух средств воруциклиба и венетоклакса, низкие дозы каждого из них (150 нМ для воруциклиба и 20 нМ для венетоклакса) исследовали в качестве единственных средств или в качестве комбинации в модели DLBCL SUDHL4. Клетки подвергали воздействию лекарственного средства в течение 24 ч перед лизисом клеток и вестерн-блоттингом с использованием антител, специфичных к расщепленной форме PARP, MCL-1, C-MYC и бета-актину (в качестве белкового нагрузочного контроля) на фиг.22A и BCL2 в качестве дополнительного белкового нагрузочного контроля на фиг.22B. На фиг.22A проиллюстрировано, что воруциклиб подавляет MCL1 в концентрации 1,5 мкМ в модели DLBCL.
[202] На фиг.22B представлено использование концентрации воруциклиба 0,15 мкМ, которая составляла 1/10 от концентрации, для которой показано, что она эффективно индуцирует апоптоз в качестве единственного средства. Этот уровень воруциклиба сохраняет некоторую эффективность в отношении подавления экспрессии MCL-1 (см. дорожки 1 и 2 фиг.22B), в то время как воздействие на клетки венетоклакса индуцирует экспрессию MCL-1 (см. дорожки 1 и 3 фиг.22B). Низкая экспозиция воруциклиба также подавляет индуцируемый венетоклаксом MCL-1 (см. дорожки 3 и 4 фиг.22B).
[203] На фиг.23A-C проиллюстрирована синергия воруциклиба и венетоклакса в модели SU-DHL-4, модели OCI Ly10 и модели U2932 DLBCL. Расщепленный PARP (cPARP) можно использовать в качестве биомаркера апоптоза. Для оценки эффективности комбинации двух средств воруциклиба и венетоклакса, низкие дозы каждого из них исследовали в качестве единственных средств или в качестве комбинации в трех моделях DLBCL. Клетки подвергали воздействию лекарственного средства в течение 24 ч перед лизисом клеток и вестерн-блоттингом с использованием антител, специфичных к расщепленной форме PARP и бета-актину (в качестве белкового нагрузочного контроля).
[204] На фиг.23A проиллюстрировано, что комбинация 0,15 мкМ воруциклиба и 20 нМ венетоклакса демонстрирует синергию в отношении индукции апоптоза в модели DLBCL SUDHL4, которую не индуцирует ни одно из этих соединений в качестве единственного средства. На фиг.23B проиллюстрировано, что комбинация 5 мкМ воруциклиба и 50 нМ венетоклакса демонстрирует синергию в отношении индукции апоптоза в модели DLBCL OCI Ly10, где ни одно из соединений в качестве единственного средства не индуцирует апоптоз в значительной степени. На фиг.23C проиллюстрировано, что комбинация 5 мкМ воруциклиба и 50 нМ венетоклакса демонстрирует синергию в отношении индукции апоптоза в модели DLBCL U2932, где комбинация двух соединений значительно превосходит добавление каждого соединения в качестве единственного средства.
Пример 11. Воруциклиб подавляет экспрессию MCL1 в модели с ксенотрансплантатом DLBCL у мышей
[205] На фиг.24 проиллюстрировано, что воруциклиб подавляет экспрессию MCL1 в ксенотрансплантатах опухолей DLBCL у мышей. Мышам, имеющим ксенотрансплантаты опухолей DLBCL (OCILY10), вводили воруциклиб или контроль в виде носителя через желудочный зонд каждые сутки в течение 5 суток и опухоли извлекали для анализа экспрессии MCL-1 через 4 ч после последней дозы. Образцы опухолей разрезали пополам, причем одну половину солюбилизировали в лизирующем буфере для анализа с использованием вестерн-блоттинга, а другую половину фиксировали в формалине и подготавливали для иммуногистохимии. Вестерн-блоттинг лизатов опухолей проводили для 3 индивидуальных мышей на группу введения с использованием антител, специфичных к MCL-1 или бета-актину (белковый нагрузочный контроль). На графике проиллюстрировано, что воруциклиб в качестве единственного средства подавлял экспрессию MCL1 в опухолях с ксенотрансплантатом DLBCL в количестве 100 мг/кг.
Пример 12. Воруциклиб и венетоклакс препятствуют росту ксенотрансплантатов опухолей DLBCL у мышей
[206] На фиг.25A-B проиллюстрирован эффект комбинации воруциклиба и венетоклакса на препятствование росту ксенотрансплантатов опухолей DLBCL. Иммунодефицитным мышам NOD SCID имплантировали RIVA (активированные B-клетки или ABC), являющиеся моделью DLBCL. Ксенотрансплантатам опухолей позволяли расти в хозяине NOD SCID до достижения ими 200 мм3, после чего животных с ксенотрансплантатами случайным образом распределяли на 4 исследуемых группы: 1. Контроль в виде носителя; 2. Воруциклиб в дозе 200 мг/кг; 3. Венетоклакс в дозе 1 мг/кг; и 4. Комбинация воруциклиба в дозе 200 мг/кг и венетоклакса в дозе 1 мг/кг. Воруциклиб дозировали через желудочный зонд каждые сутки в течение 6 дней недели с одними сутками отдыха между курсами. Венетоклакс дозировали через желудочный зонд на 1 и 4 сутки каждого недельного курса. Эффекты лекарственных средств оценивал технический специалист, не имевший информации о лечении в каждой исследуемой группе, посредством измерения объема опухоли два раза в неделю. N=5-6 животных на группу введения.
[207] На фиг.25A представлены графики среднего объема опухоли (мм3) для каждой из 4 исследуемых групп: носитель, единственное средство воруциклиб, единственное средство венетоклакс и комбинация воруциклиба и венетоклакса. Введение средств по отдельности продемонстрировало более медленное снижение объема опухоли в дни исследования. Комбинированное введение продемонстрировало значительно большее снижение объема опухоли, более чем 2-кратное снижение среднего объема опухоли через 3 недели исследования.
[208] На фиг.25B проиллюстрированы изображения опухолей у животных из каждой из 4 исследуемых групп: носитель, воруциклиб в качестве единственного средства, венетоклакс в качестве единственного средства и комбинация воруциклиба и венетоклакса. Размер опухоли в случае носителя был наибольшим, как представлено на фрагменте 2501. Опухоль в случае воруциклиба в качестве единственного средства является меньшей, как представлено на фрагменте 2502. Опухоль в случае венетоклакса в качестве единственного средства также является меньшей, чем в случае носителя, как представлено на фрагменте 2503. Опухоль в случае комбинированной терапии представлена на фрагменте 2504, и является значительно меньшей, чем в случае носителя или любой из терапий единственными средством, что указывает на синергический эффект.
[209] На фиг.26A-B иллюстрируется эффект комбинации воруциклиба и венетоклакса на рост ксенотрансплантатов опухолей DLBCL. Иммунодефицитным мышам NOD SCID имплантировали U2932, являющиеся моделью DLBCL. Ксенотрансплантатам опухолей позволяли расти в хозяине NOD SCID до достижения ими 200 мм3, после чего животных с ксенотрансплантатами случайным образом распределяли на 4 исследуемых группы: 1. Контроль в виде носителя; 2. Воруциклиб в дозе 200 мг/кг; 3. Венетоклакс в дозе 10 мг/кг; и 4. Комбинация воруциклиба в дозе 200 мг/кг и венетоклакса в дозе 10 мг/кг. Воруциклиб дозировали через желудочный зонд каждые сутки в течение 6 дней недели с одними сутками отдыха между курсами. Венетоклакс дозировали через желудочный зонд на 1 и 4 сутки каждого недельного курса. Эффекты лекарственных средств оценивал технический специалист, не имевший информации о лечении в каждой исследуемой группе, посредством измерения объема опухоли два раза в неделю. N=5-6 животных на группу введения.
[210] На фиг.26A представлены графики среднего объема опухоли (мм3) для каждой из 4 исследуемых групп. Введение средств по отдельности продемонстрировало более медленное снижение объема опухоли в дни исследования. Комбинированное введение продемонстрировало практически отсутствие роста опухоли, причем средний объем опухоли оставался постоянным в ходе периода исследования на протяжении 29 суток.
[211] На фиг.26B представлены графики средней массы тела в граммах (г) для индивидуумов в каждой из 4 исследуемых групп. Все 4 исследуемых группы продемонстрировали стабильную и постоянную массу тела, что является основным признаком здоровья и безопасности.
[212] На фиг.27A-B иллюстрируется эффект комбинации воруциклиба и венетоклакса на рост ксенотрансплантатов опухолей DLBCL. Иммунодефицитным мышам NOD SCID имплантировали NUDHL1, являющиеся моделью DLBCL. Ксенотрансплантатам опухолей позволяли расти в хозяине NOD SCID до достижения ими 200 мм3, после чего животных с ксенотрансплантатами случайным образом распределяли на 4 исследуемых группы: 1. Контроль в виде носителя; 2. Воруциклиб в дозе 200 мг/кг; 3. Венетоклакс в дозе 50 мг/кг; и 4. Комбинация воруциклиба в дозе 200 мг/кг и венетоклакса в дозе 50 мг/кг. Воруциклиб дозировали через желудочный зонд каждые сутки в течение 6 дней недели с одними сутками отдыха между курсами. Венетоклакс дозировали через желудочный зонд на 1 и 4 сутки каждого недельного курса. Эффекты лекарственных средств оценивал технический специалист, не имевший информации о лечении в каждой исследуемой группе, посредством измерения объема опухоли два раза в неделю. N=5-6 животных на группу введения.
[213] На фиг.27A представлены графики среднего объема опухоли (мм3) для каждой из 4 исследуемых групп. Введение средств по отдельности продемонстрировало более медленное снижение объема опухоли в дни исследования. Комбинированное введение продемонстрировало значительно большее снижение объема опухоли, более чем 2-кратное снижение среднего объема опухоли через 3 недели исследования.
[214] На фиг.27B проиллюстрированы изображения опухолей животных из каждой из 4 исследуемых групп: носитель, воруциклиб в качестве единственного средства, венетоклакс в качестве единственного средства и комбинация воруциклиба и венетоклакса. Размер опухоли в случае носителя был наибольшим, как представлено на фрагменте 2701. Опухоль в случае единственного средства воруциклиба является меньшей, как представлено на фрагменте 2702. Опухоль в случае венетоклакса в качестве единственного средства также является меньшей, чем в случае носителя, как представлено на фрагменте 2703. Опухоль в случае комбинированной терапии представлена на фрагменте 2704, и является значительно меньшей, чем в случае носителя или любой из терапий единственными средствами, что указывает на синергический эффект.
[215] На фиг.28 иллюстрируется эффект комбинации воруциклиба и венетоклакса на рост ксенотрансплантатов опухолей DLBCL. Иммунодефицитным мышам NOD SCID имплантировали SUDHL4, являющиеся моделью DLBCL. Ксенотрансплантатам опухолей позволяли расти в хозяине NOD SCID до достижения ими 200 мм3, после чего животных с ксенотрансплантатами случайным образом распределяли на 4 исследуемых группы: 1. Контроль в виде носителя; 2. Воруциклиб в дозе 200 мг/кг; 3. Венетоклакс в дозе 25 мг/кг; и 4. Комбинация воруциклиба в дозе 200 мг/кг и венетоклакса в дозе 25 мг/кг. Воруциклиб дозировали через желудочный зонд каждые сутки в течение 6 дней недели с одними сутками отдыха между курсами. Венетоклакс дозировали через желудочный зонд каждые сутки в течение 6 дней недели с одними сутками отдыха между курсами. Эффекты лекарственных средств оценивал технический специалист, не имевший информации о лечении в каждой исследуемой группе, посредством измерения объема опухоли два раза в неделю. N=5-6 животных на группу введения. На фиг.28 представлены графики нормализованного объема опухоли (мм3) для каждой из 4 исследуемых групп. Проведение терапии единственными средствами
продемонстрировало увеличение объема опухоли в дни исследования. Комбинированное введение продемонстрировало меньший рост опухоли, чем введение единственных средств и введение носителя.
[216] Каждая дорожка на фиг.29 соответствует индивидуальному ксенотраксплантату SUDHL4. В совокупности, вестери-блоттинг иллюстрирует, что воруцикдиб восстанавливает р53, устраненный венетоклактом. Опухоли извлекали на 26 сутки через 4 часа после введения. Каждую опухоль фрагментировали на 2 половины. Одну половину использовали для анализа IHC па основе FFPE, а другую половину использовали для анализа с использованием вестерн-блоттинга.
Пример 13. Воруциклиб подавляет экспрессию MCL1 в клеточных
[217] На фиг.30А-30С проиллюстрировано, что воруциклиб обладает активностью в качестве единственного средства в клеточных линиях AML. Клеточные линии AML (SKM-1, MV-4-11 и OCI-AML-3) либо оставляли необработанными, либо подвергали воздействию клинически достижимых уровней воруциклиба (1,5 и 5,0 микромоль/л) в течение 2, 6 или 24 ч. В данные моменты времени клетки лизировали и подвергали анализу с использованием вестерн-блоттинга с антителами, которые специфически распознают фосфорилированную РНК-полимеразу II (pSer2), MCL-1, расщепленный PARP и бета-актин. Во всех трех клеточных линиях AML (SKM-1, MV-4-11 и OCI-AML-3), воруциклиб подавляет MCL-1 и индуцирует апоптоз, как показано на фрагментах 3001, 3003 и 3005. Расщепленный PARP (cPARP) можно использовать в качестве биомаркера апоптоза. Во всех трех клеточных линиях AML (SKM-1, MV-4-11 и OCI-AML-3) воруциклиб индуцирует апоптоз, как показано на фрагментах 3002, 3004 и 3006.
Пример 14. Комбинация воруциклиба и венетоклакса индуцирует клеточную смерть в клеточных линиях AML
[218] На фиг.31 проиллюстрировано, что комбинация воруциклиба и венетоклакса индуцирует синергичную клеточную смерть в клеточных линиях AML. Клеточные линии AML (SKM-1, MV-4-11 и OCI-AML-3) либо оставляли необработанными, либо подвергали воздействию уровней воруциклиба (1,5 микромоль/л), венетоклакса (100 нМ), для которых ранее было показано, что они ниже уровней, требуемых для индукции апоптоза единственным средством, или двух лекарственных средств в комбинации в этих субэффективных концентрациях в течение 24 ч. В данные моменты времени клетки лизировали и подвергали анализу с использованием вестерн-блоттинга с антителами, которые специфически распознают фосфорилированную РНК-полимеразу II (pSer2), MCL-1, расщепленный PARP и бета-актин. Как показано на фрагментах 3101 и 3102, комбинация воруциклиба и венетоклакса индуцирует апоптоз (PARP), когда ни одно из средств отдельно не индуцирует апоптоз. Обнаружено, что венетоклакс индуцирует активацию MCL-1 во всех трех клеточных линиях. Воруциклиб устраняет обеспечиваемый MCL-1 механизм резистентности во всех трех клеточных линиях.
Пример 15. Комбинация воруциклиба и венетоклакса препятствует росту опухоли в ксенотрансплантатах AML
[219] На фиг.32 проиллюстрировано, что комбинация воруциклиба и венетоклакса препятствует росту опухоли в ксенотрансплантатах AML SKM1. Терапия венетоклаксом в качестве единственного средства имеет нормализованный объем опухоли, сходный с объемом опухоли в случае носителя. Введение воруциклиба в качестве единственного средства демонстрирует несколько более низкий объем опухоли, в то время как комбинация воруциклиба и венетоклакса в значительной степени препятствует росту опухоли, причем конечный объем опухоли приблизительно в два раза меньше, чем в случае носителя, на 22 сутки исследования.
Пример 16. Воруциклиб индуцирует апоптоз в качестве единственного средства в моделях DLBCL
[220] На фиг.33 проиллюстрировано, что индуцированный воруциклибом апоптоз коррелирует с подавлением MCL-1. Использовали три модели DLBCL: SUDHL4, RIvA и U2932. Расщепленный PARP (cPARP) можно использовать в качестве биомаркера апоптоза. Вестерн-блоттинг демонстрирует, что апоптоз возрастает при увеличении концентрации воруциклиба. Аналогично, воруциклиб подавляет MCL-1 и индуцирует апоптоз при увеличении концентрации воруциклиба.
[221] Хотя предпочтительные варианты осуществления настоящего изобретения показаны и описаны в настоящем описании, специалистам в данной области будет очевидно, что такие варианты осуществления предоставлены только в качестве примера. Специалистам в данной области теперь будут понятны многочисленные варианты, изменения или замены без отклонения от изобретения. Следует понимать, что при применении настоящего изобретения на практике можно использовать различные альтернативы вариантам осуществления изобретения, описанным в настоящем описании. Следует понимать, что приведенная ниже формула изобретения определяет объем изобретения и что оно охватывает способы и структуры, входящие в объем этих пунктов формулы изобретения, и их эквиваленты.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ ЛИМФОМЫ | 2019 |
|
RU2784243C2 |
| КОМБИНАЦИЯ, ВКЛЮЧАЮЩАЯ ПО МЕНЬШЕЙ МЕРЕ ОДИН МОДУЛЯТОР СПЛАЙСОСОМЫ И ПО МЕНЬШЕЙ МЕРЕ ОДИН ИНГИБИТОР, ВЫБРАННЫЙ ИЗ ИНГИБИТОРОВ BCL2, ИНГИБИТОРОВ BCL2/BCLXL И ИНГИБИТОРОВ BCLXL, А ТАКЖЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2018 |
|
RU2783239C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА С ИСПОЛЬЗОВАНИЕМ АПИЛИМОДА | 2016 |
|
RU2738934C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ | 2007 |
|
RU2482877C2 |
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ С ИСПОЛЬЗОВАНИЕМ МОДУЛЯТОРОВ ИЗОФОРМ PI3-КИНАЗЫ | 2013 |
|
RU2702908C2 |
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ С ИСПОЛЬЗОВАНИЕМ МОДУЛЯТОРОВ ИЗОФОРМ PIЗ-КИНАЗЫ | 2014 |
|
RU2705204C2 |
| ТВЕРДЫЕ ДИСПЕРСИИ, СОДЕРЖАЩИЕ СПОСОБСТВУЮЩЕЕ АПОПТОЗУ | 2010 |
|
RU2550134C2 |
| КОМБИНАЦИЯ BCL-2 ИНГИБИТОРА И MCL-1 ИНГИБИТОРА, ИХ ПРИМЕНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2017 |
|
RU2746705C2 |
| КОМБИНАЦИЯ ИНГИБИТОРА MCL-1 И МИДОСТАУРИНА, ЕЕ ПРИМЕНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2019 |
|
RU2818453C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ АНТИТЕЛАМИ АНТИ-CD20 ТИПА II В СОЧЕТАНИИ С ИНГИБИТОРОМ ПРОТЕАСОМ | 2008 |
|
RU2520757C2 |
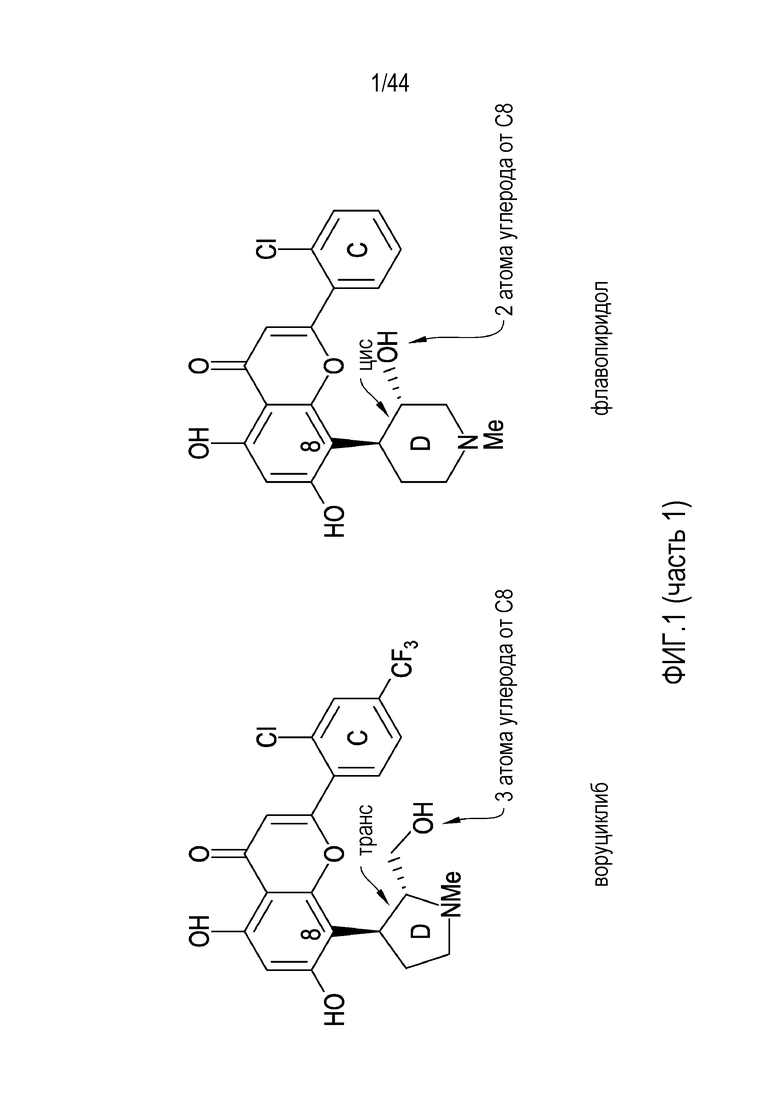
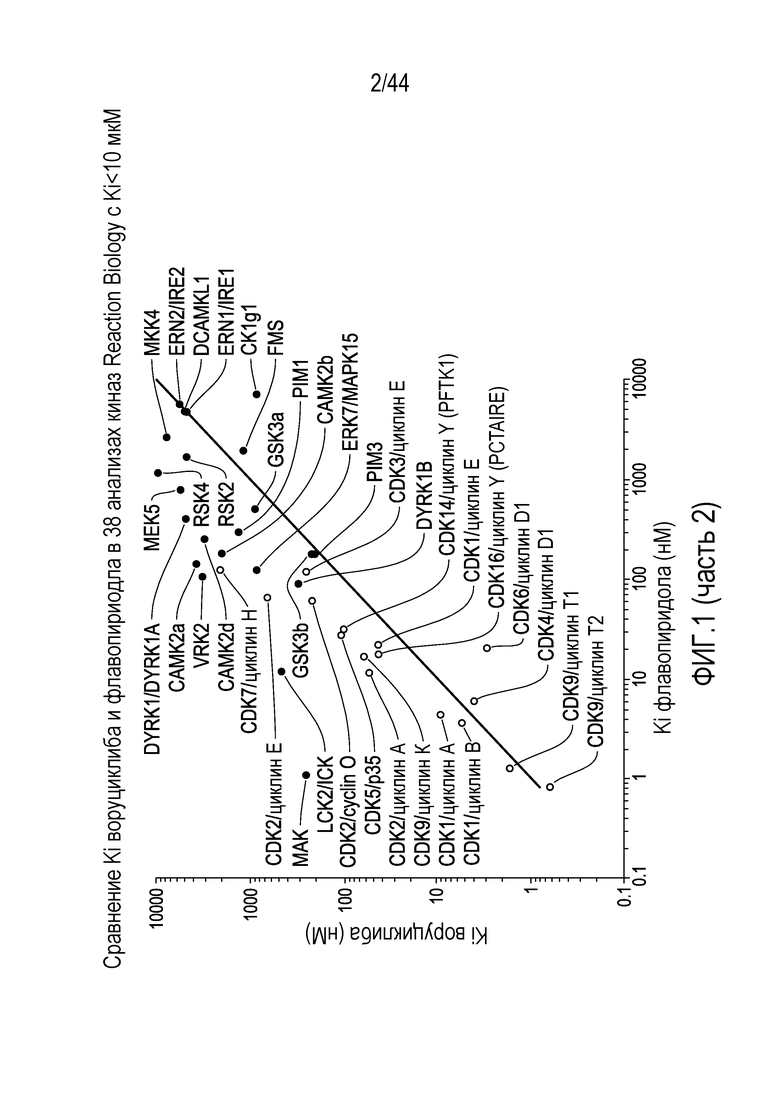
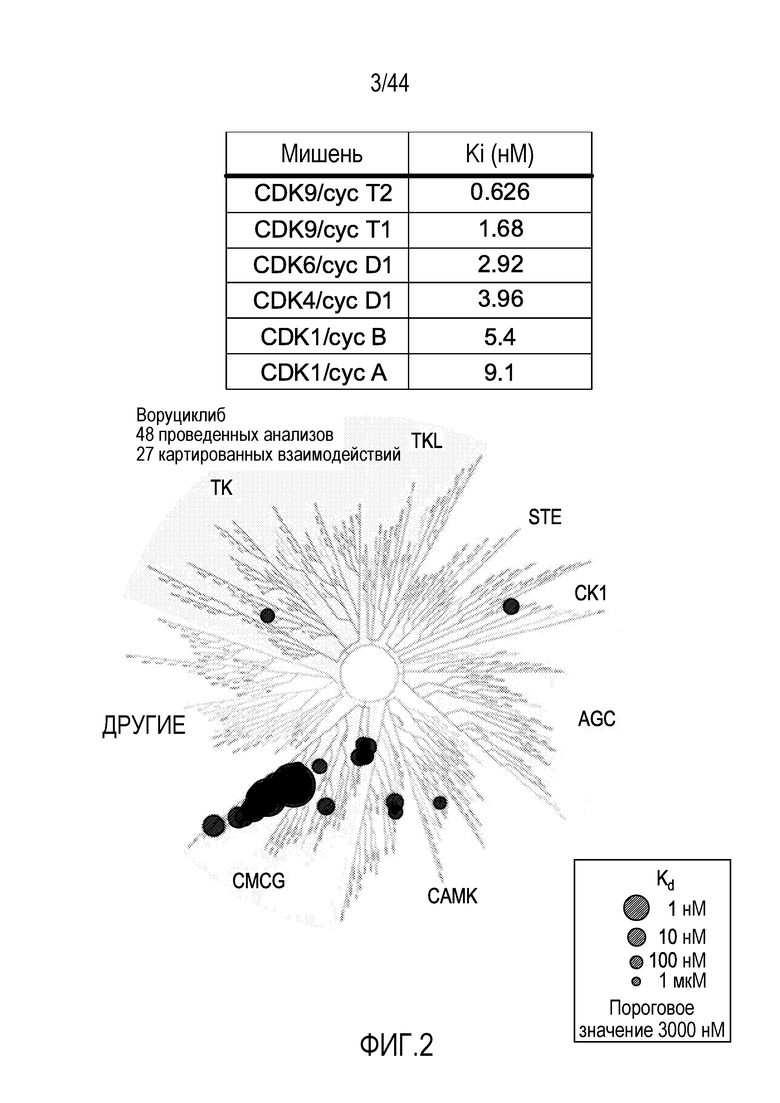

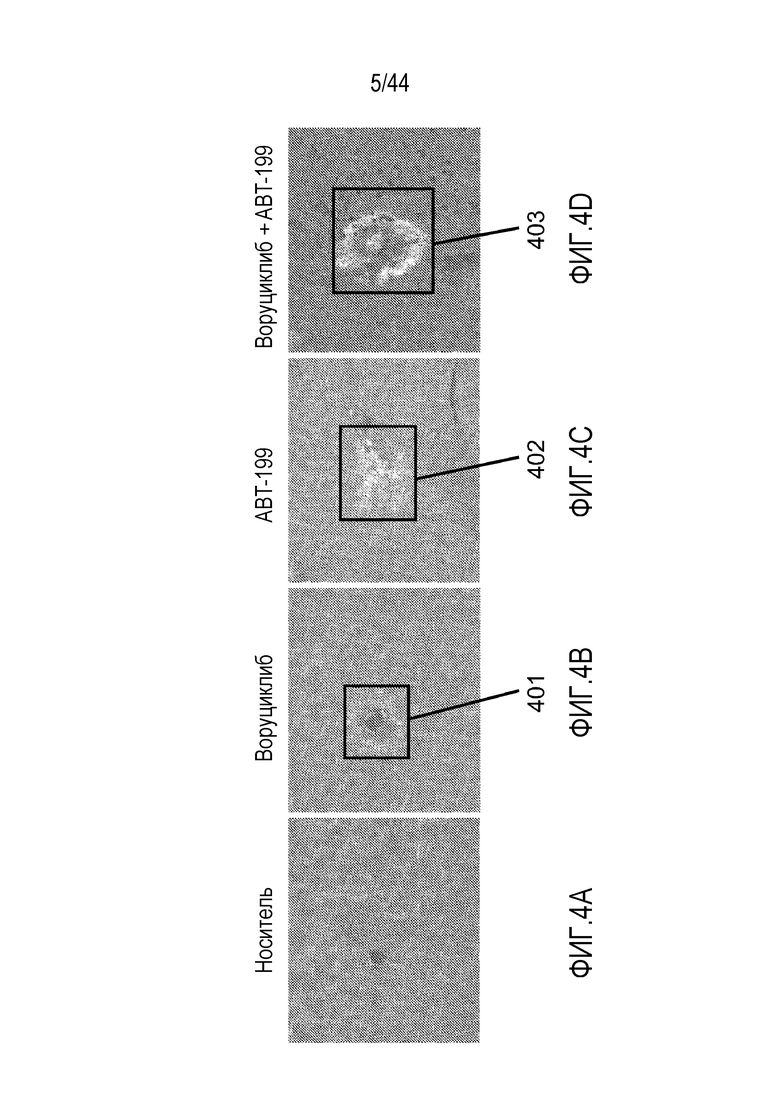
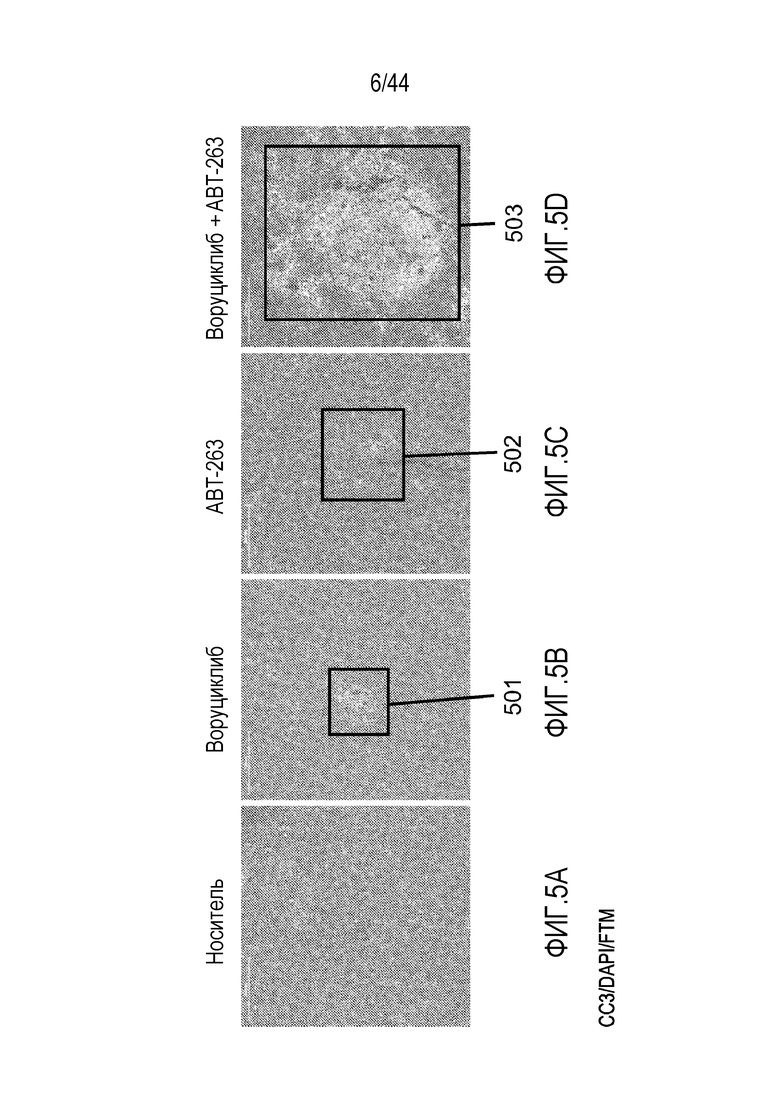
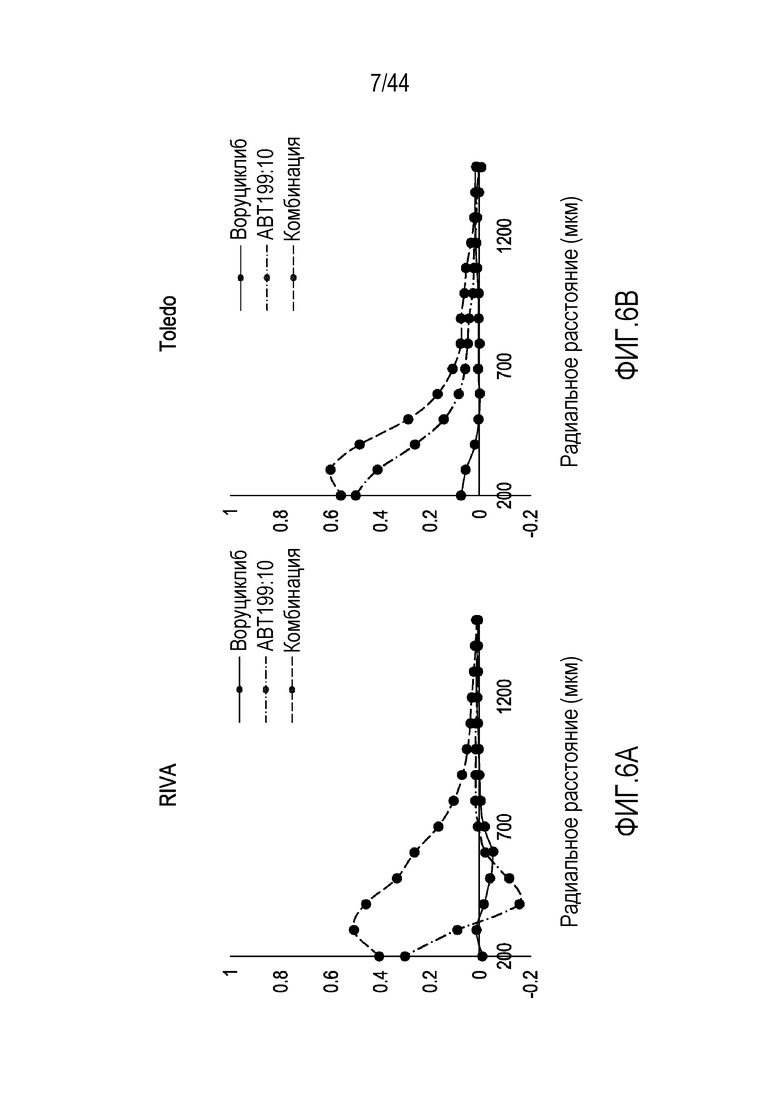
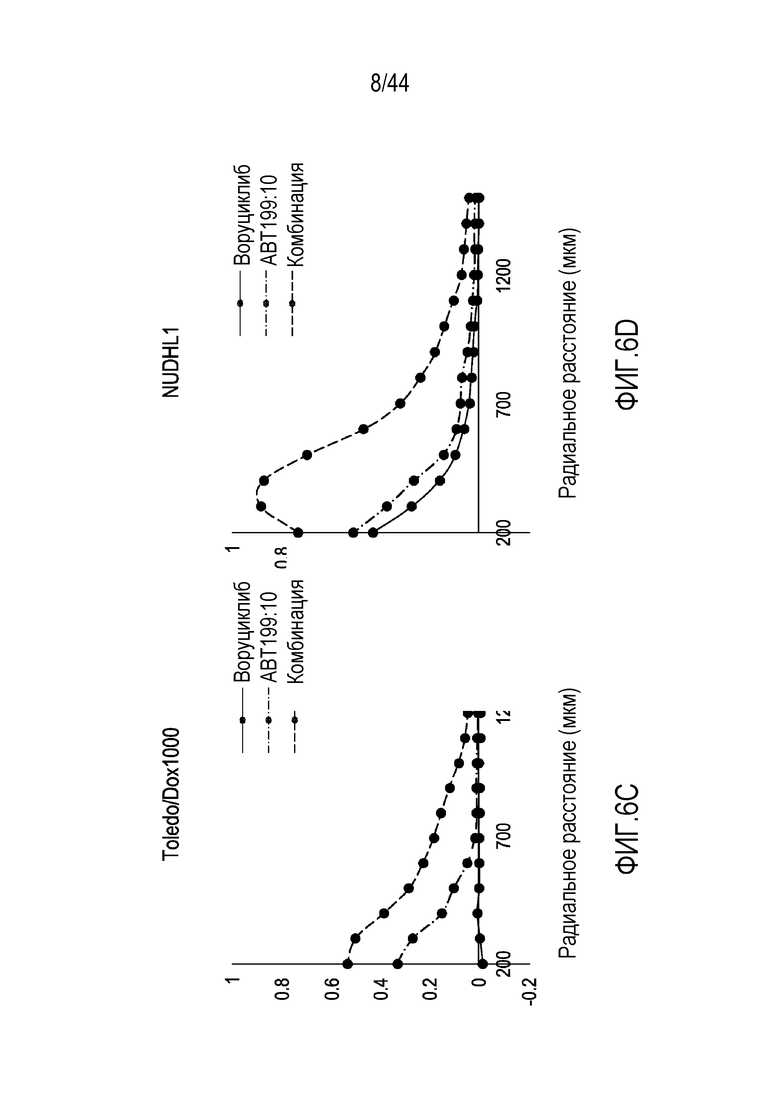
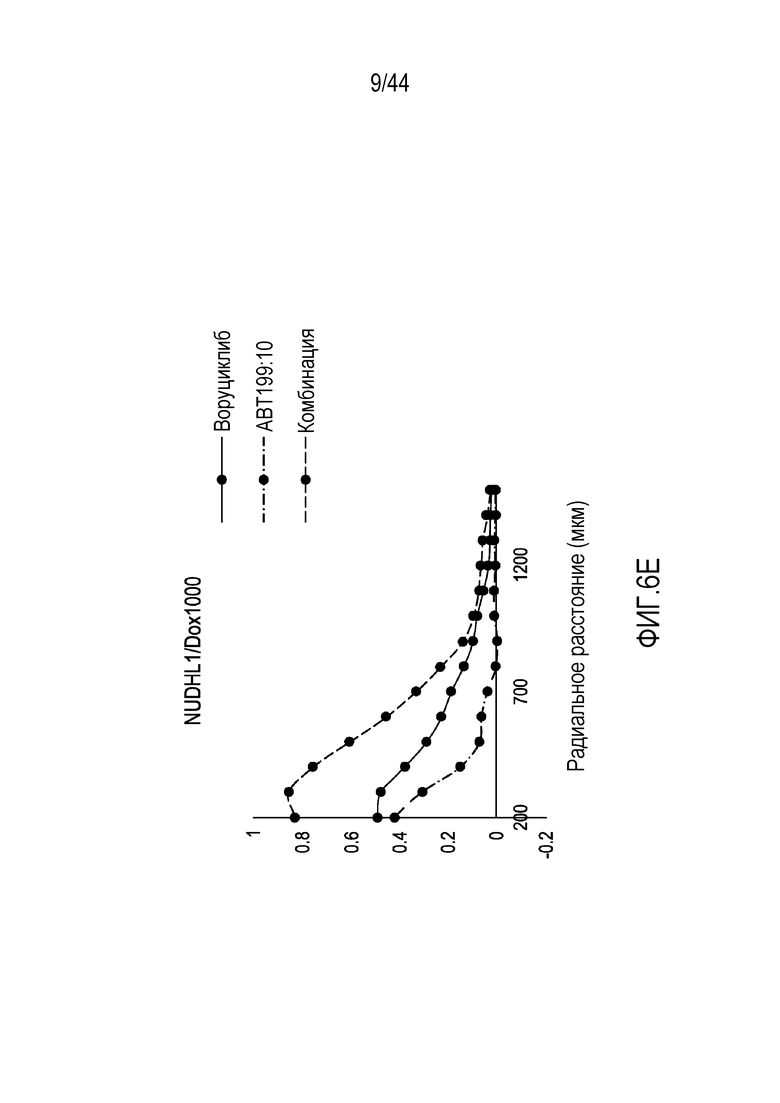
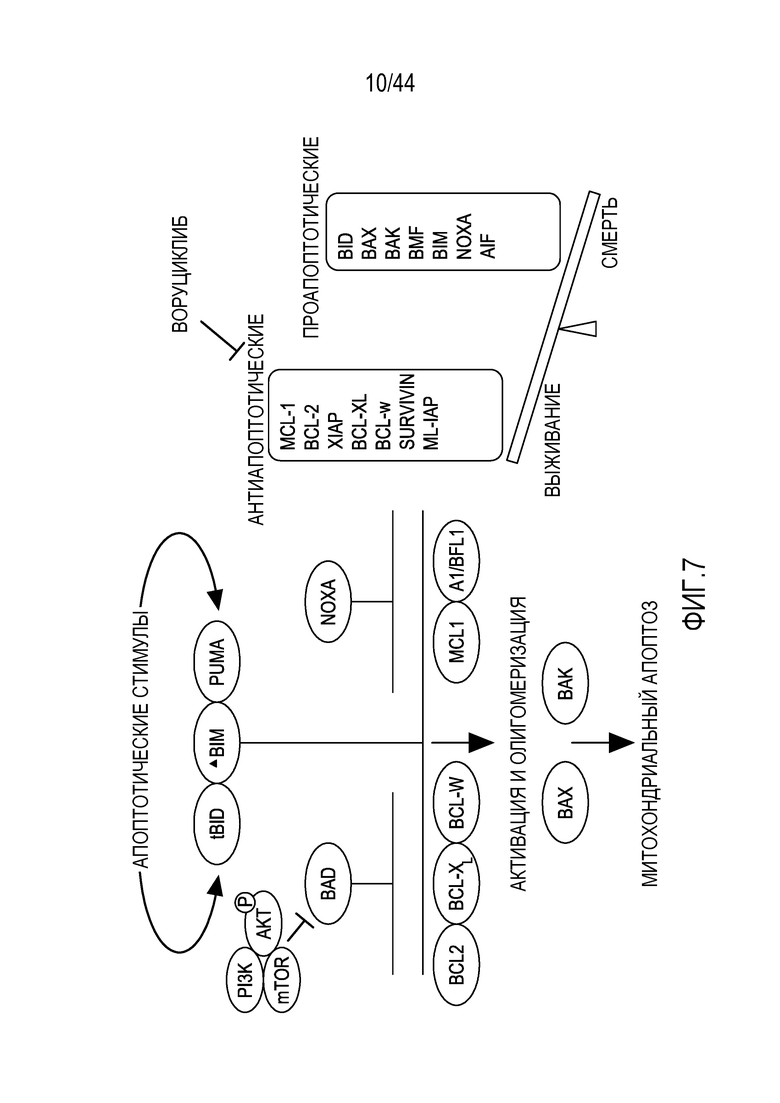

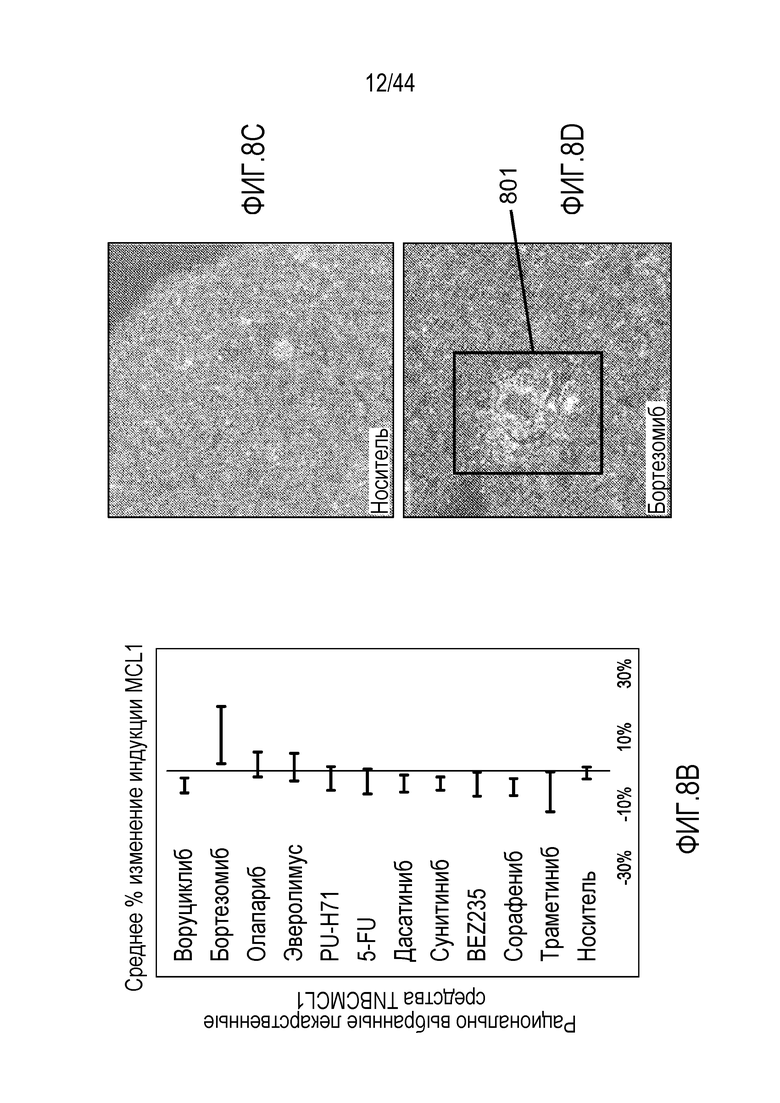
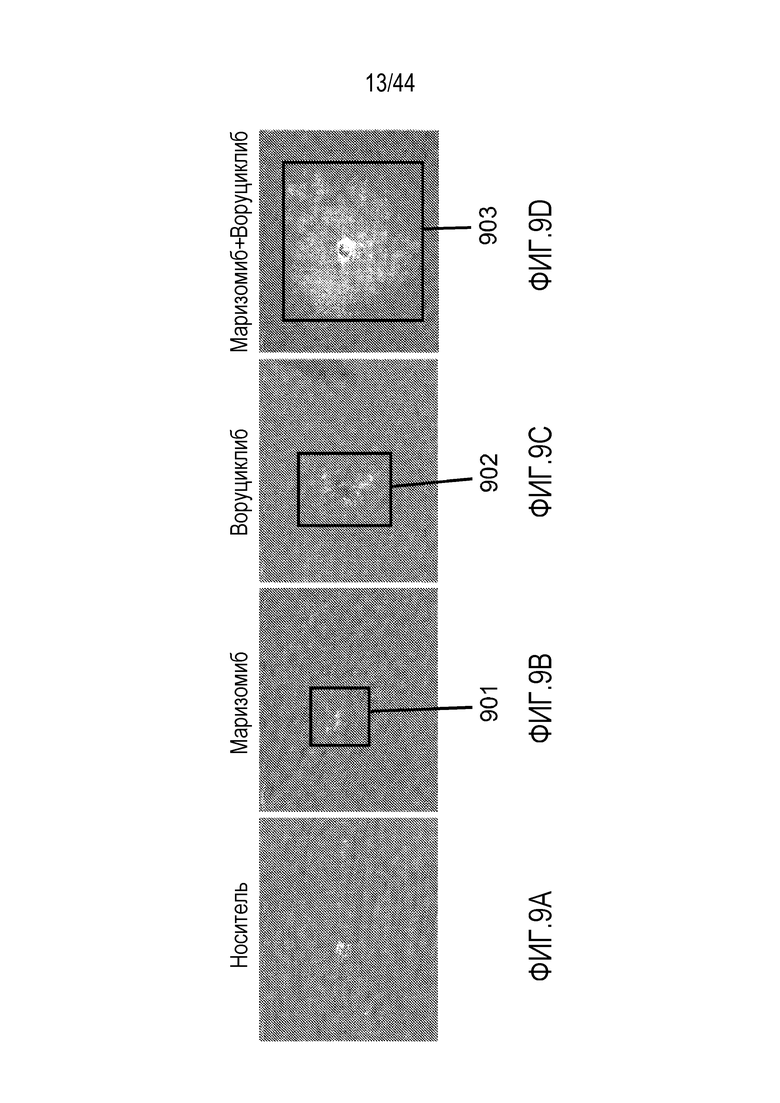
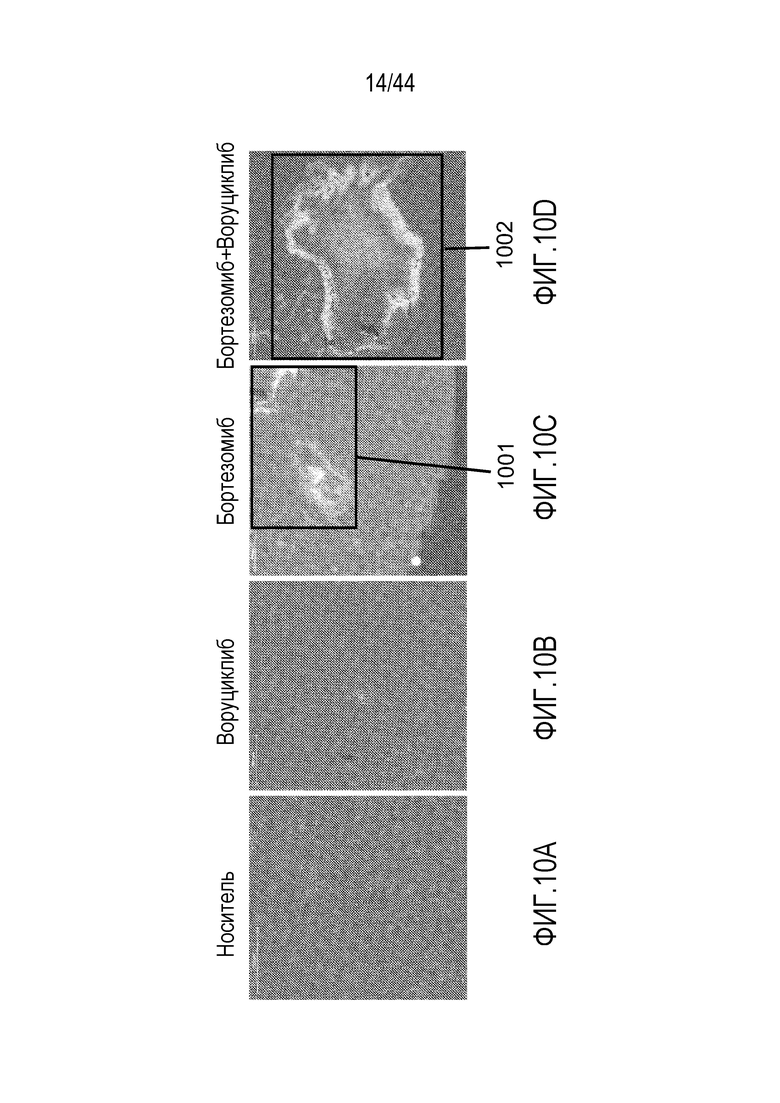
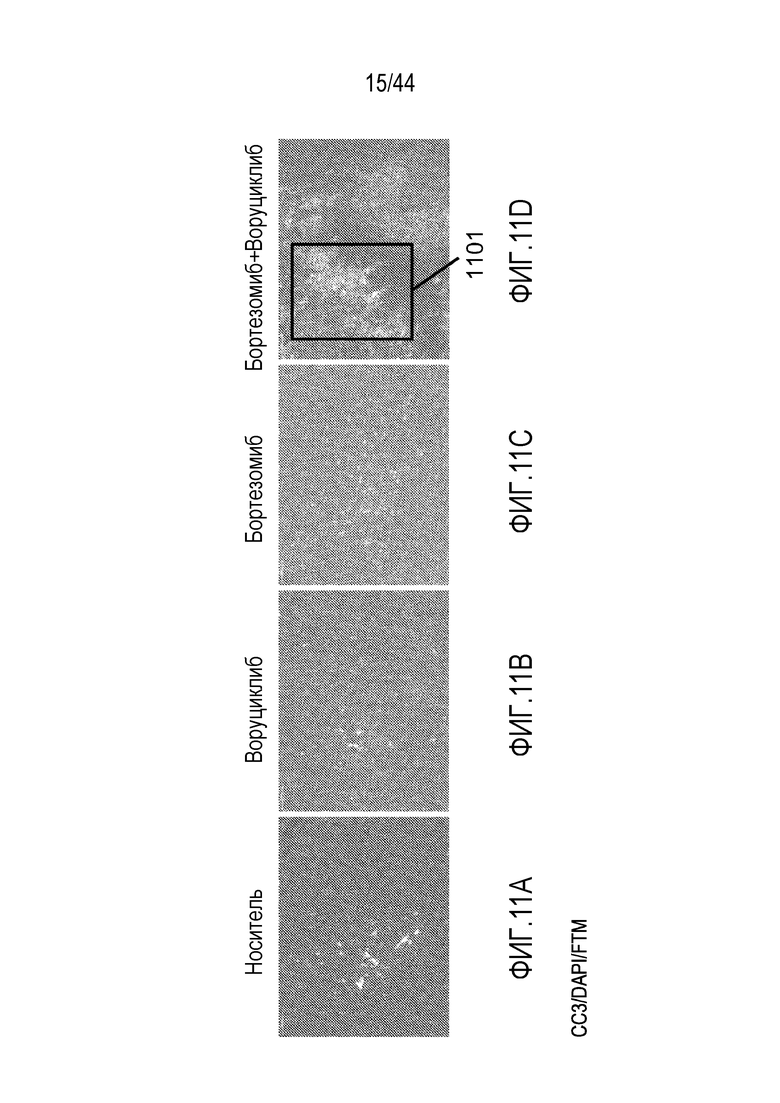
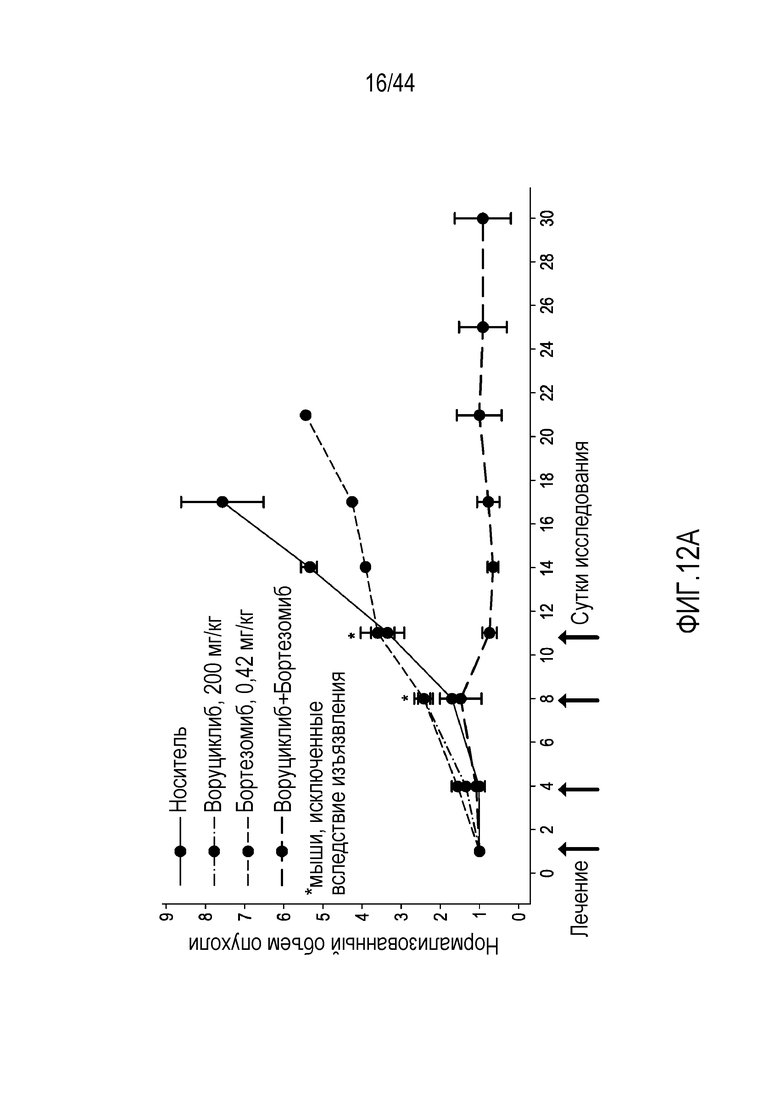
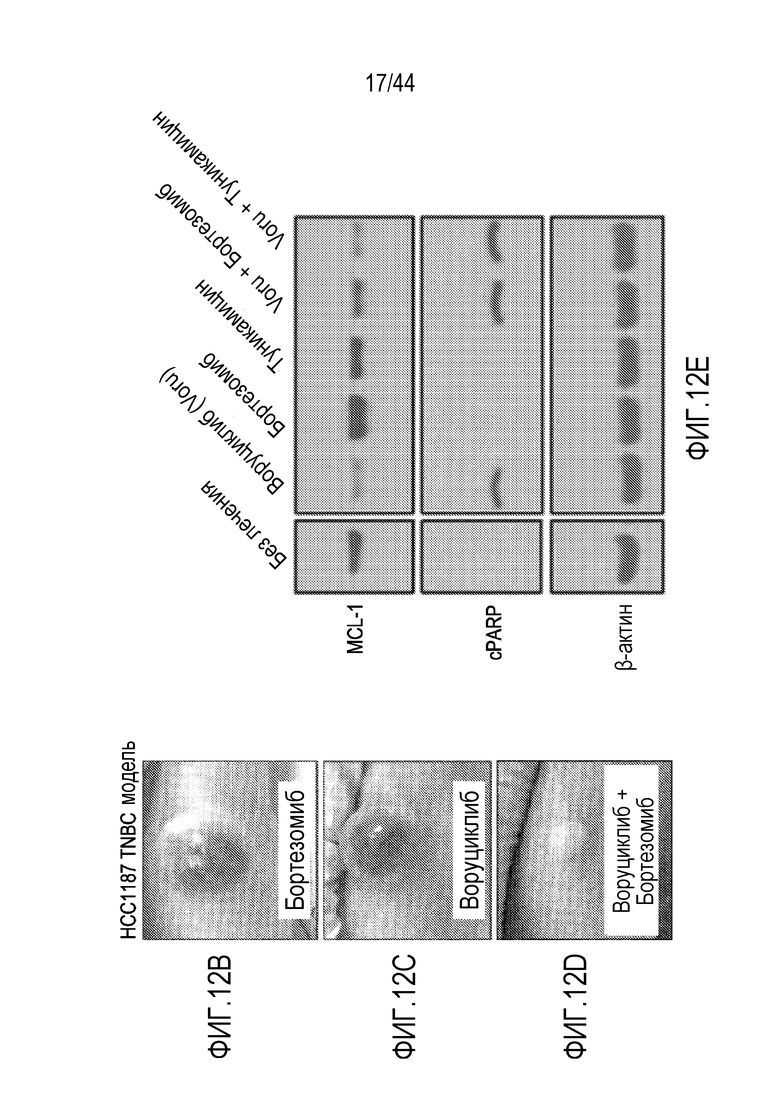
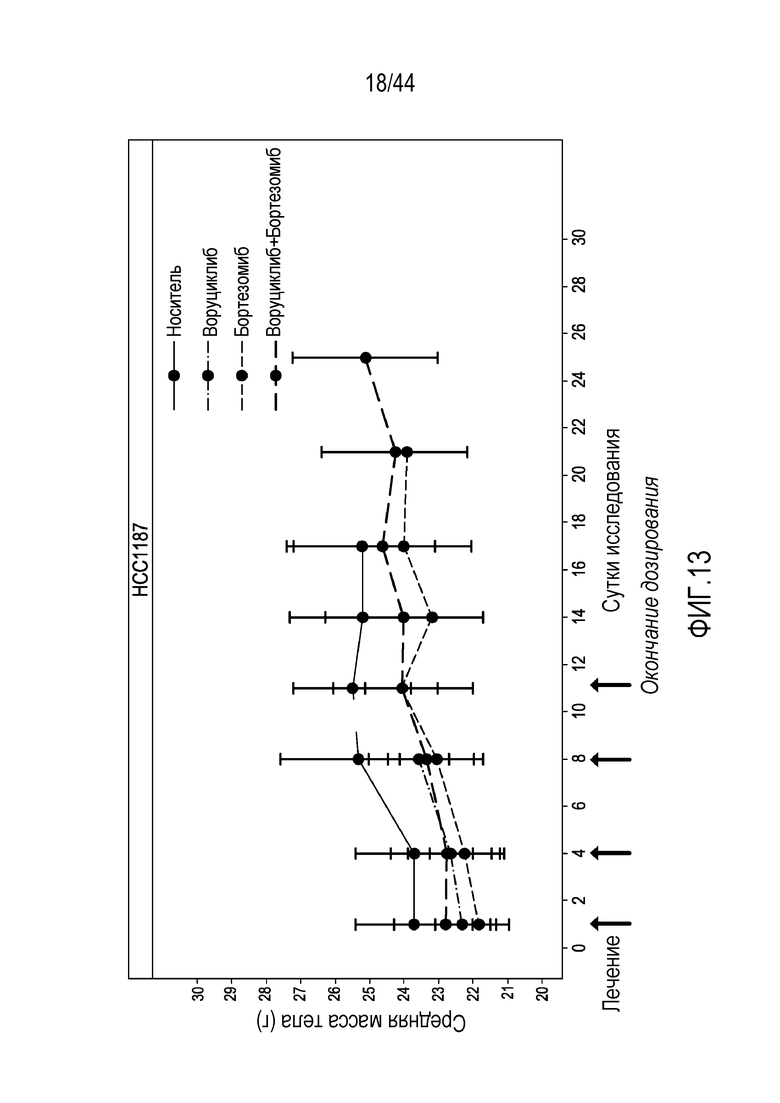
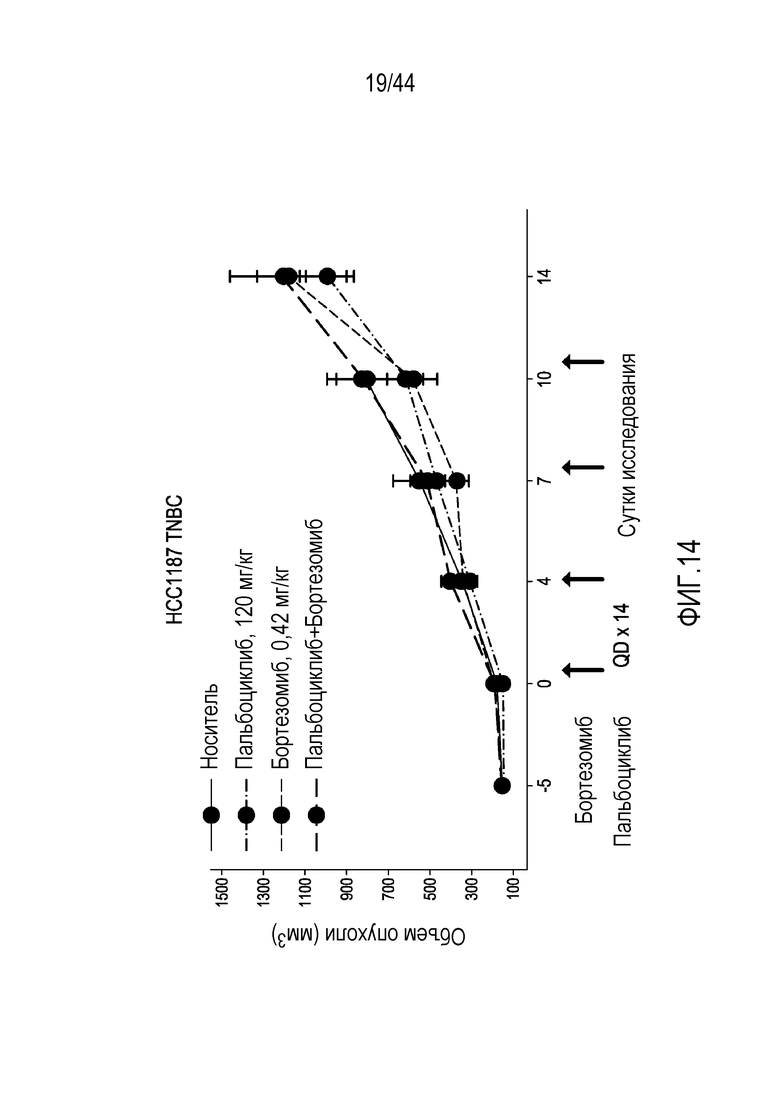
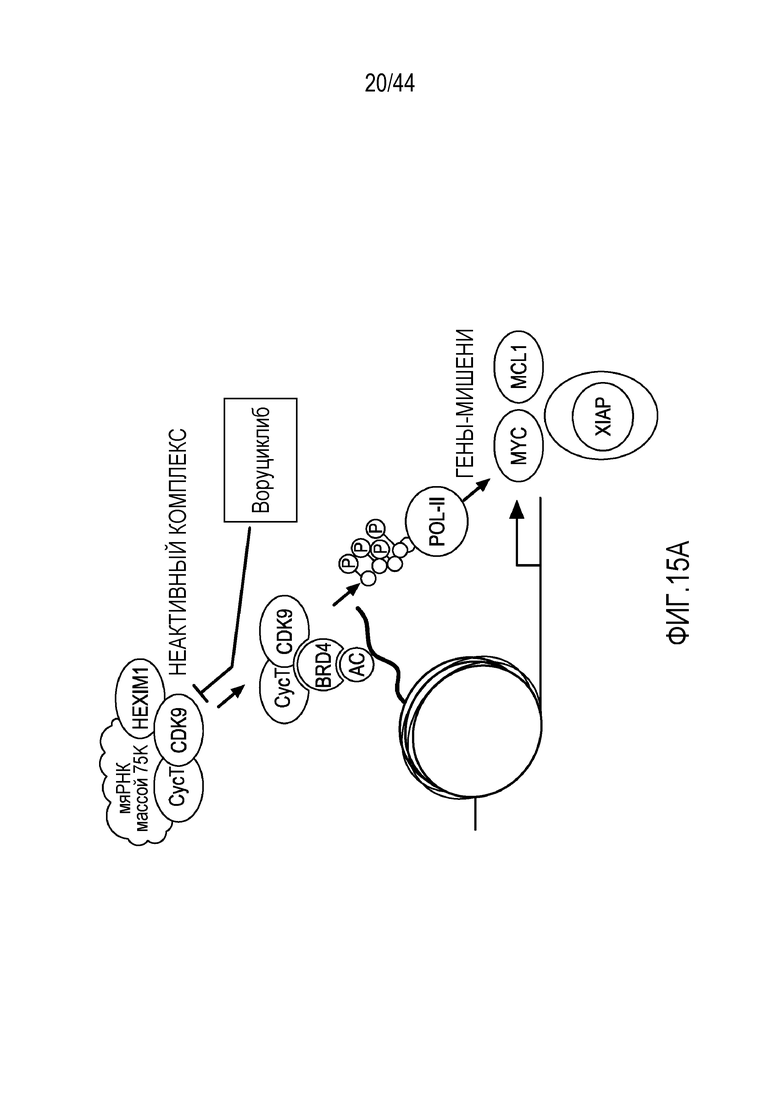

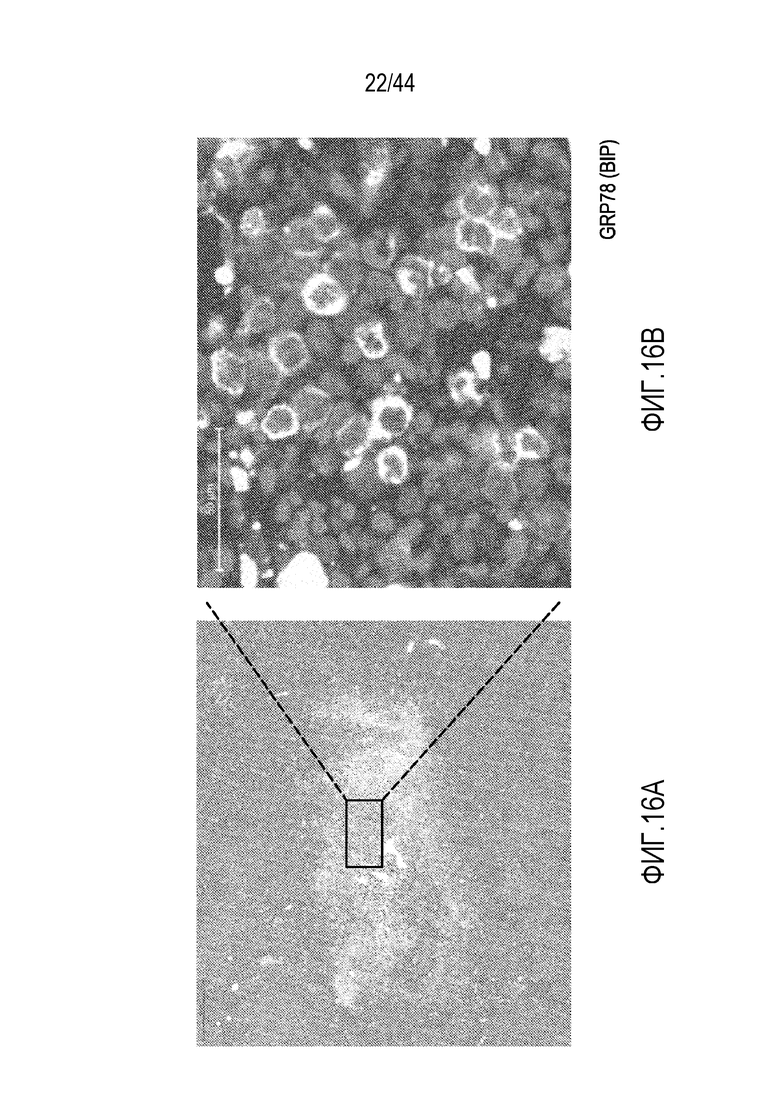
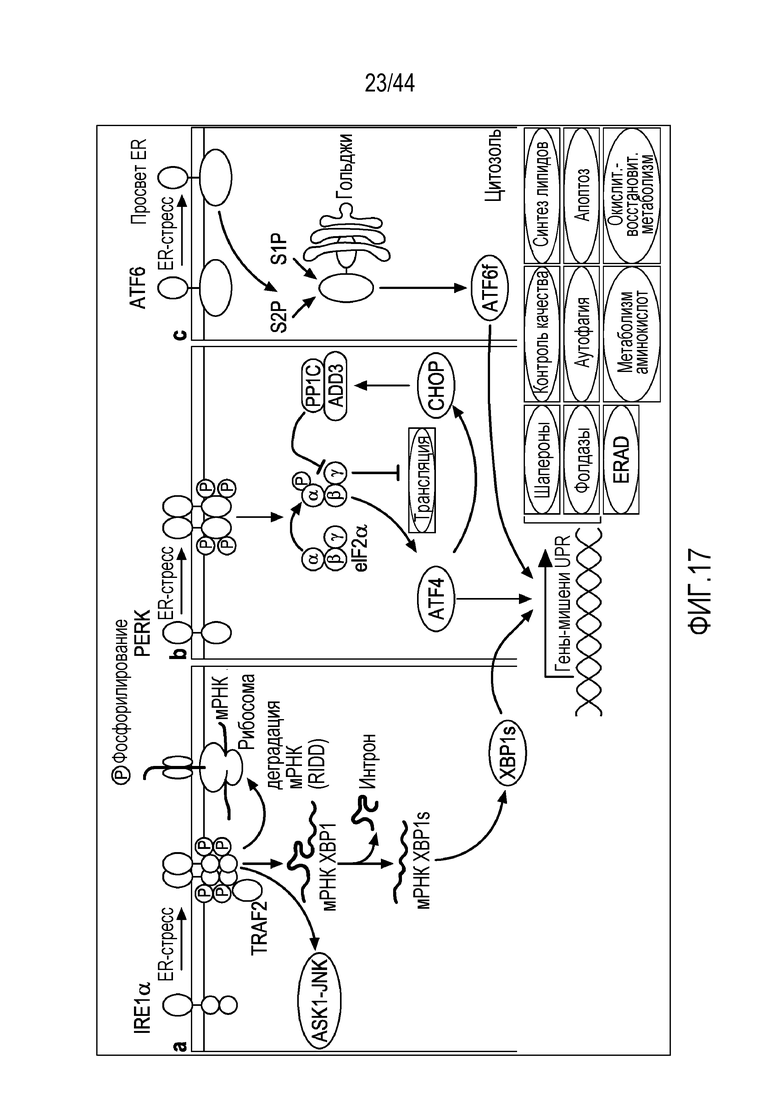

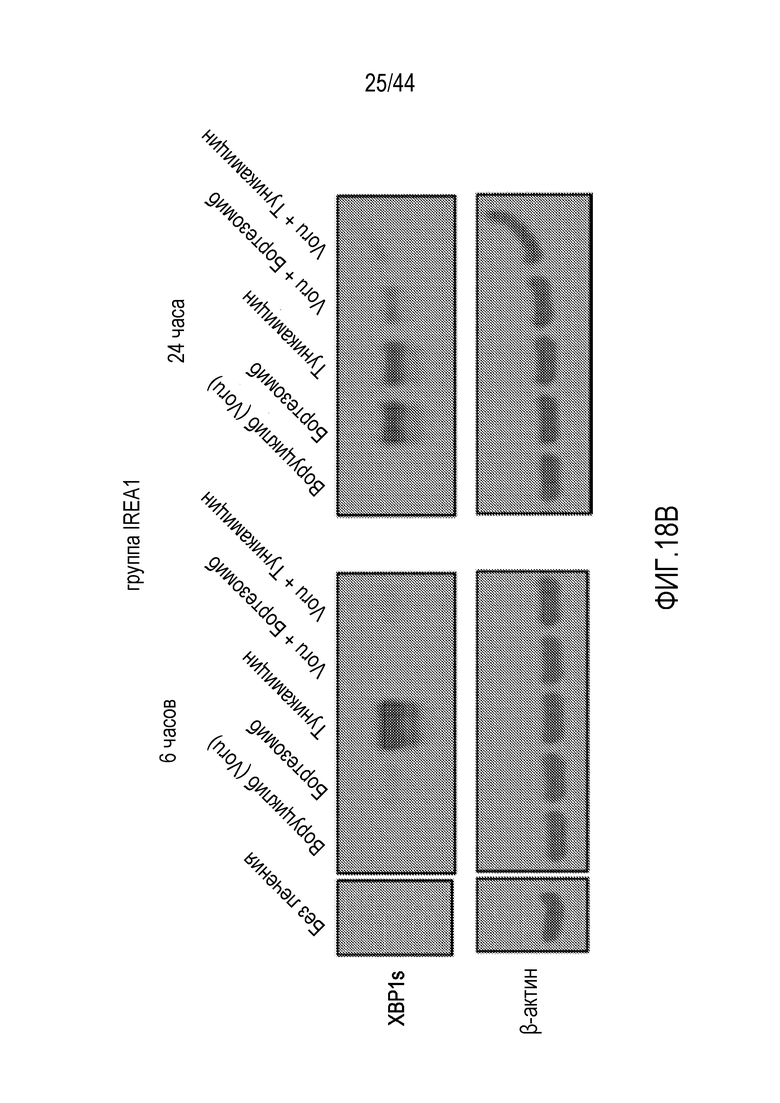
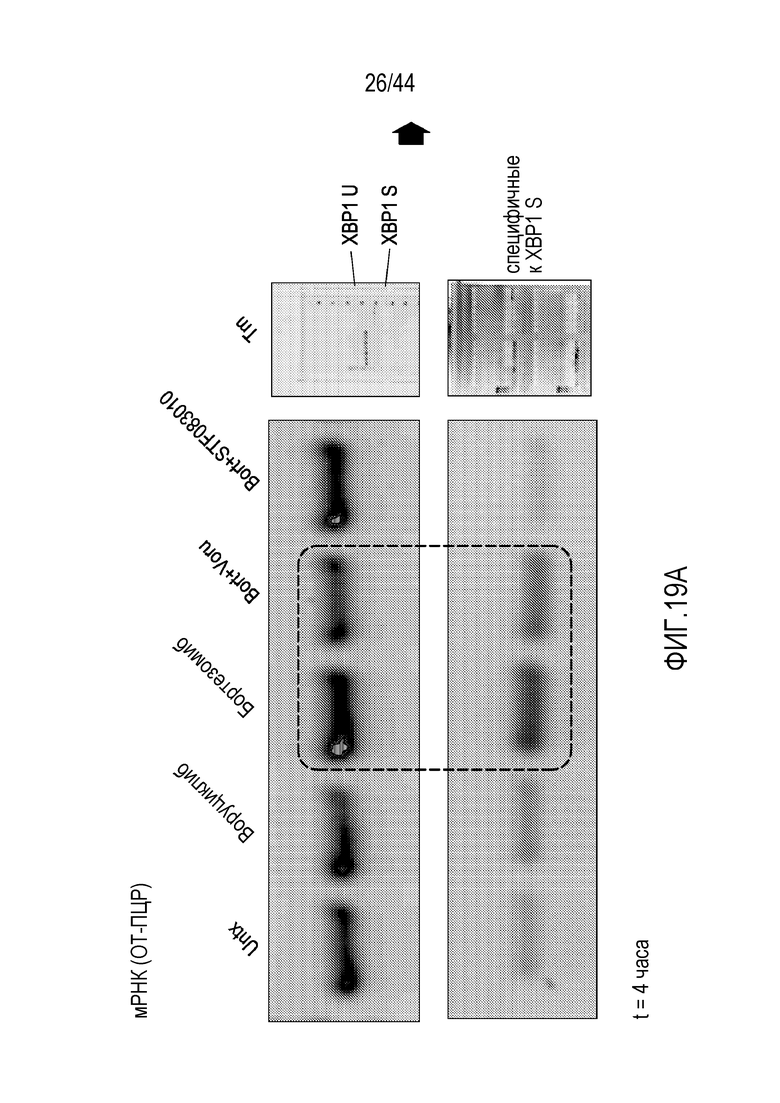
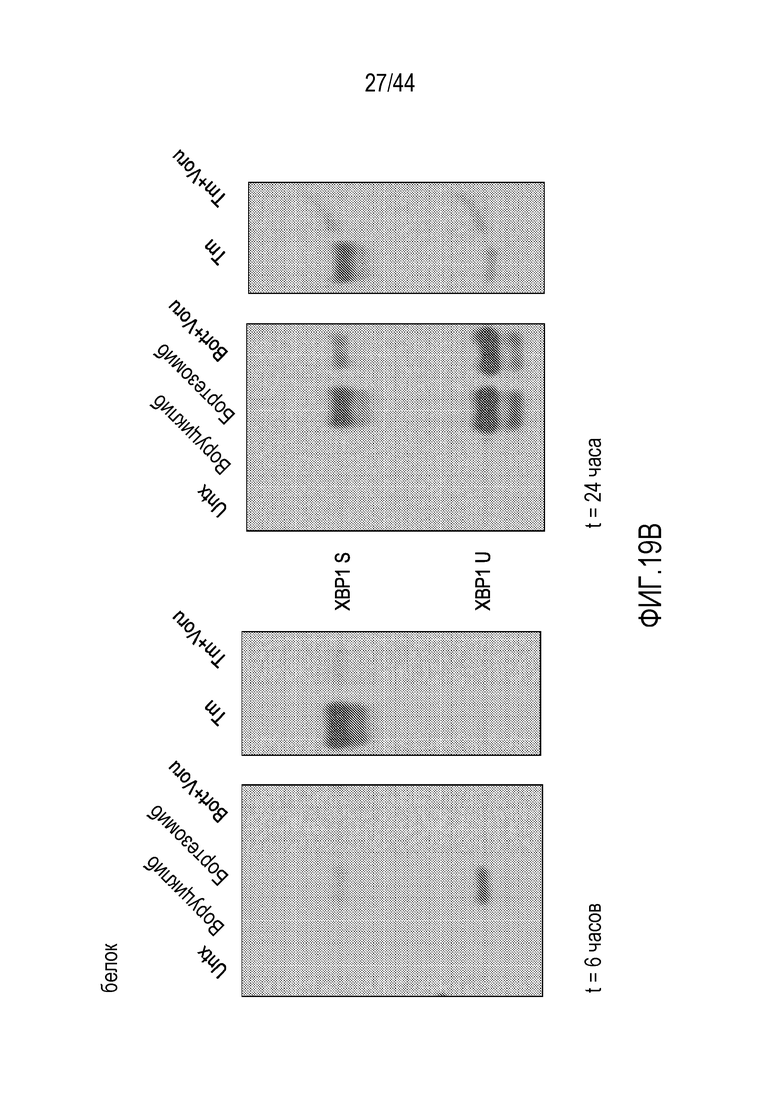
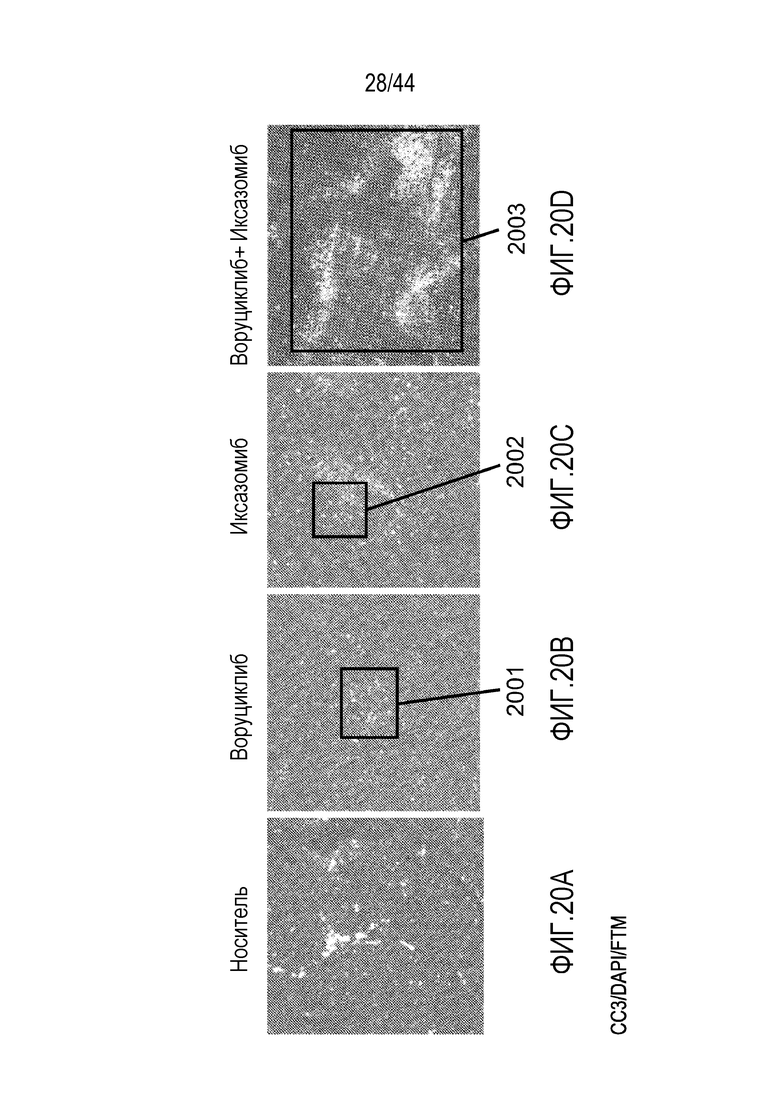

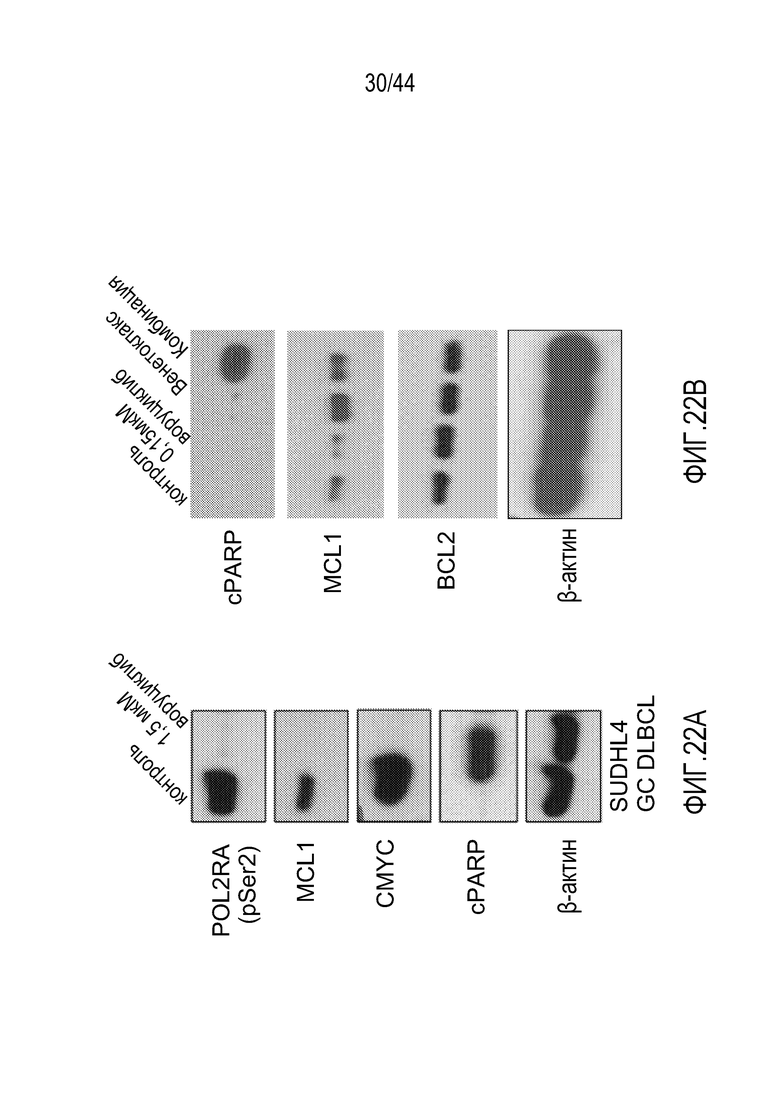
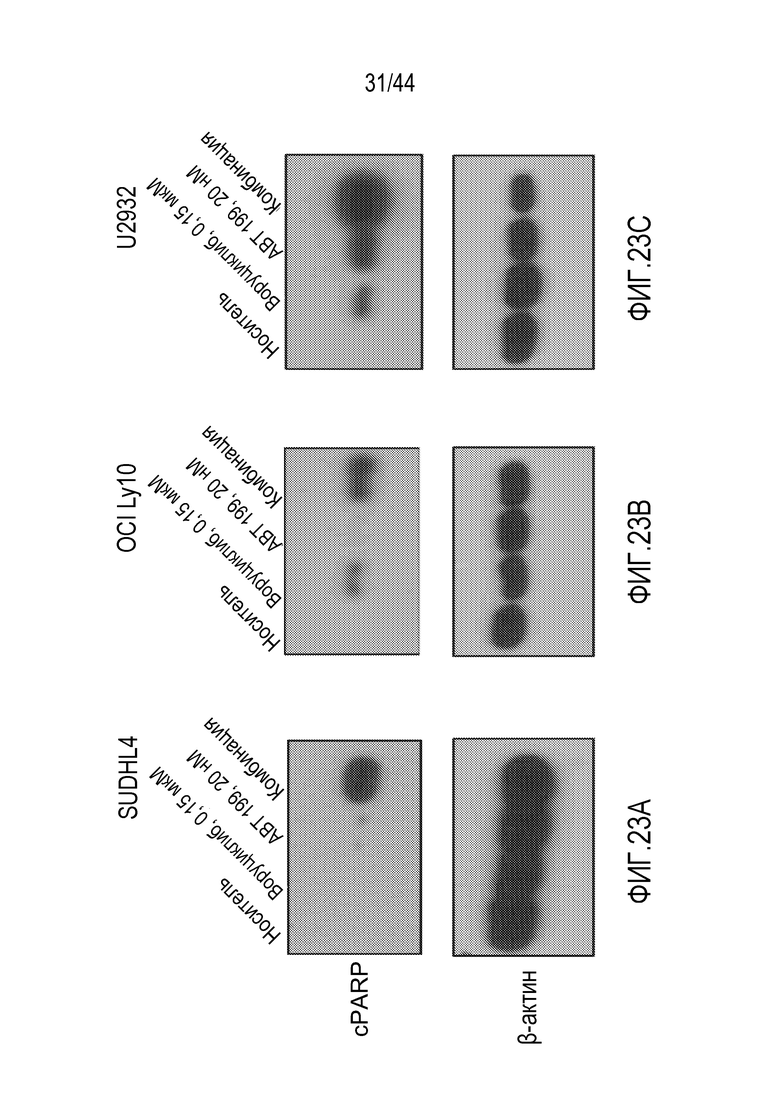
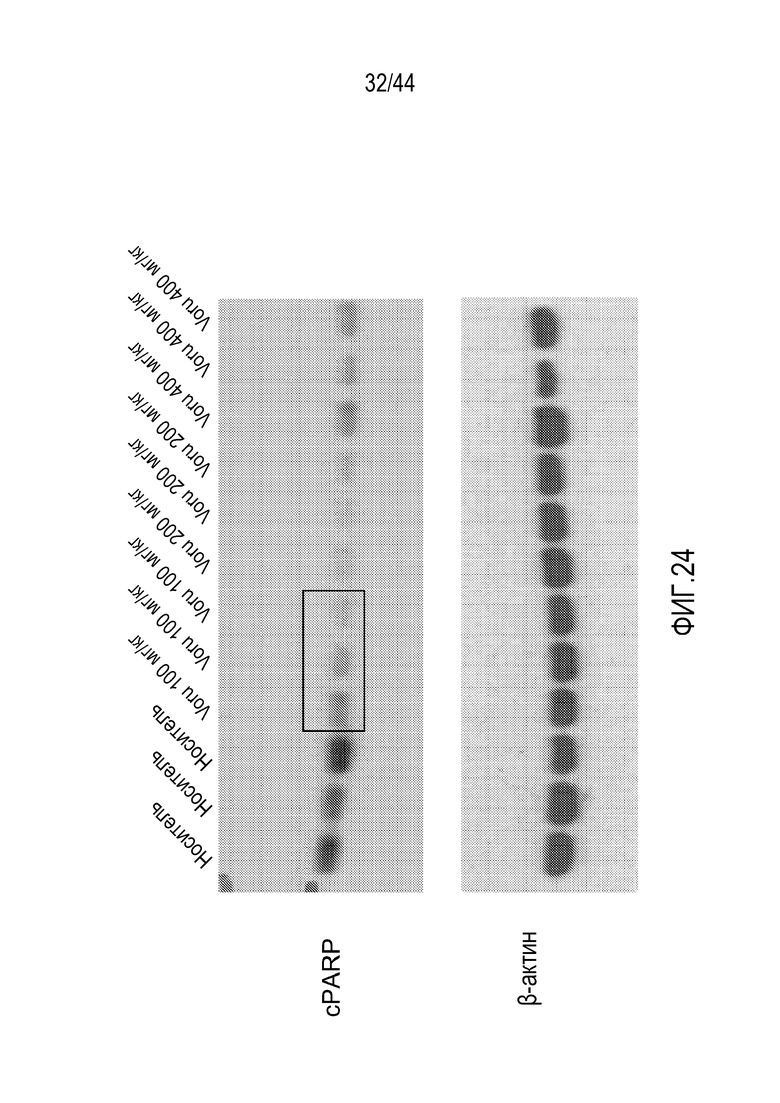
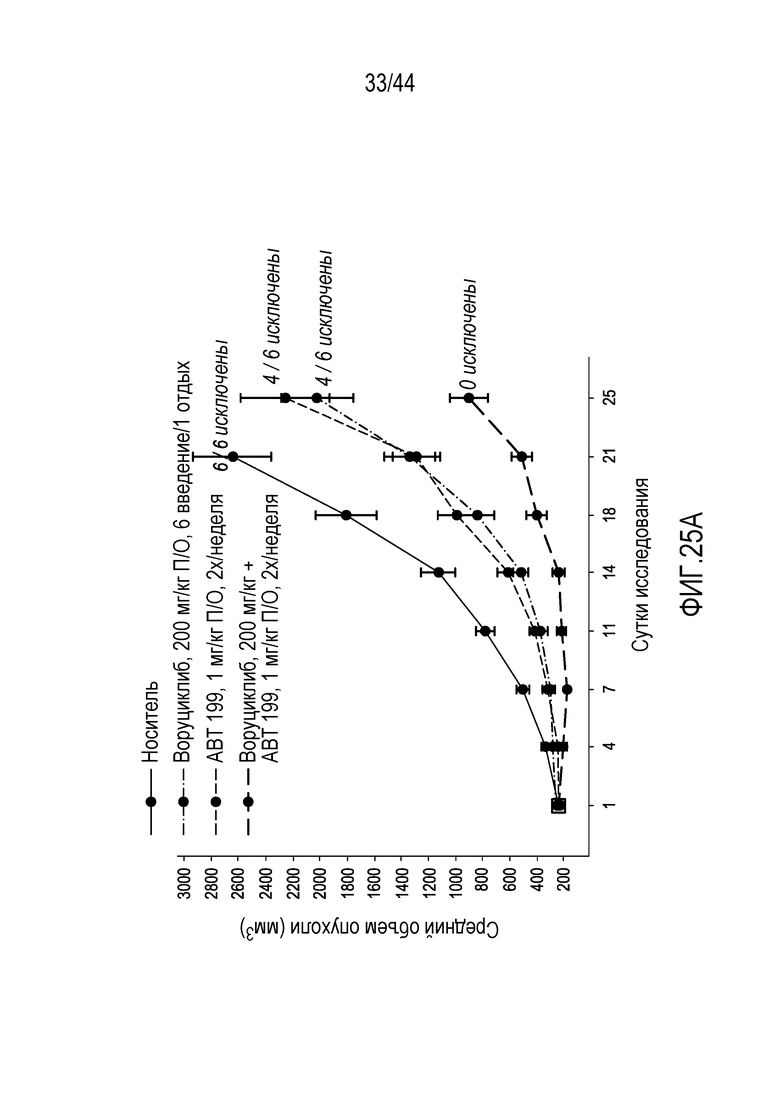

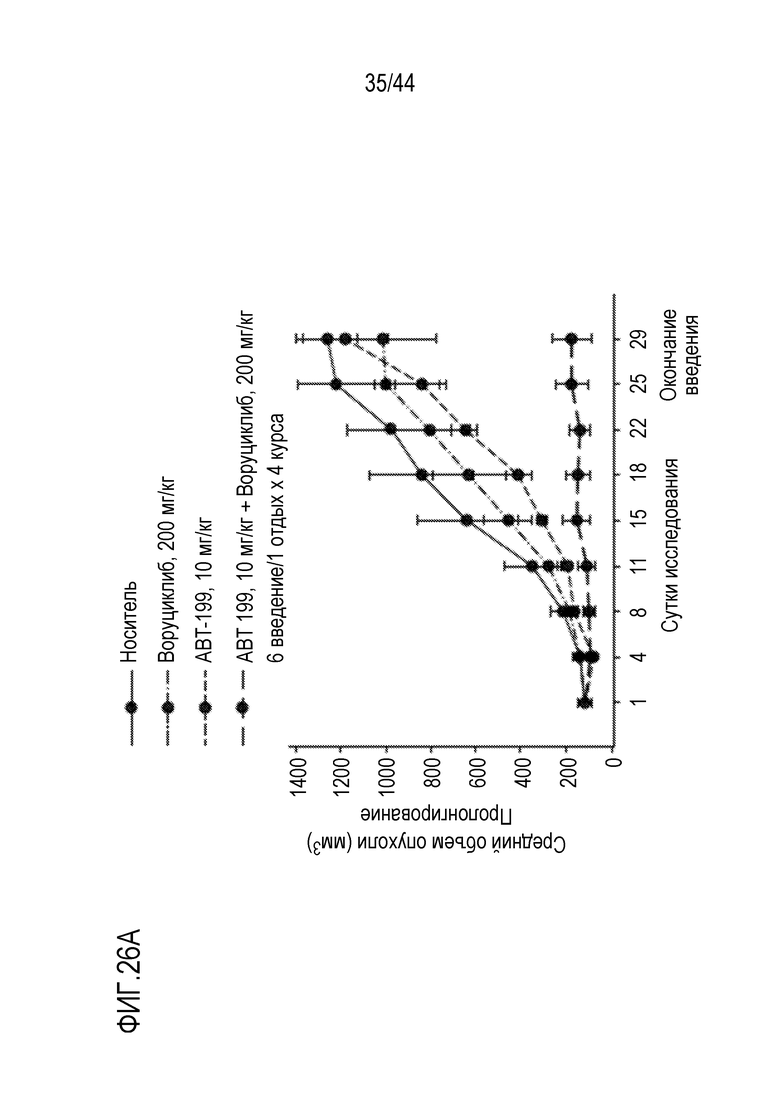
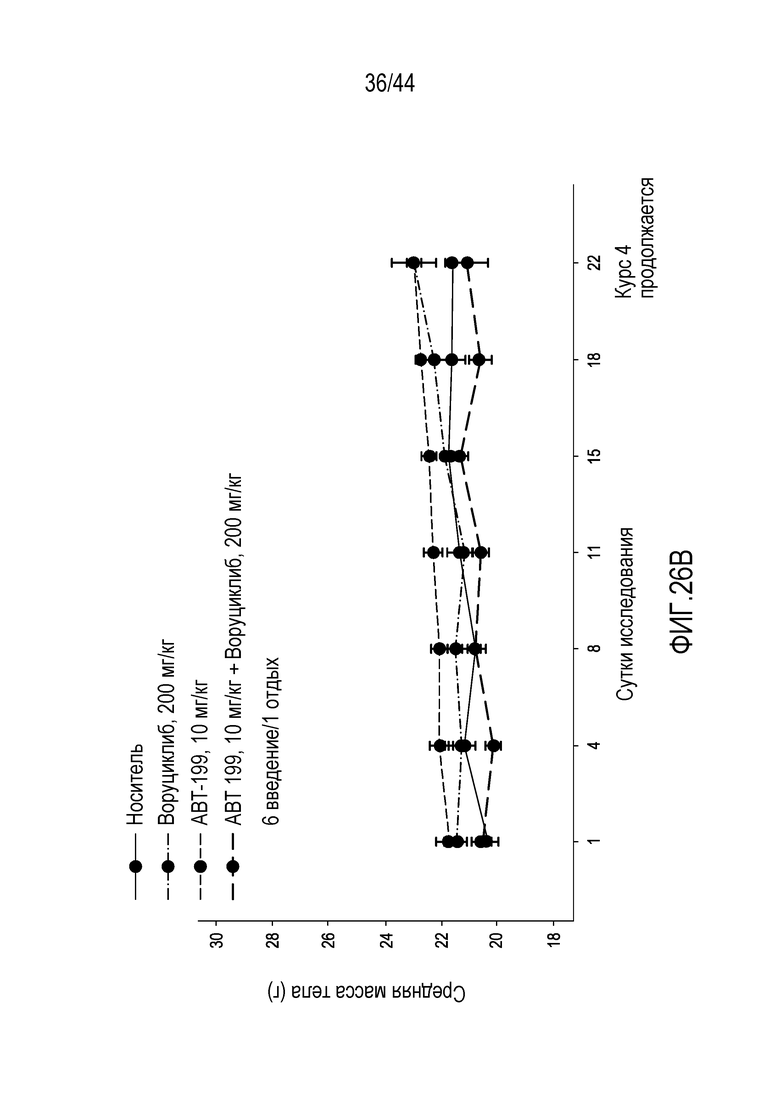
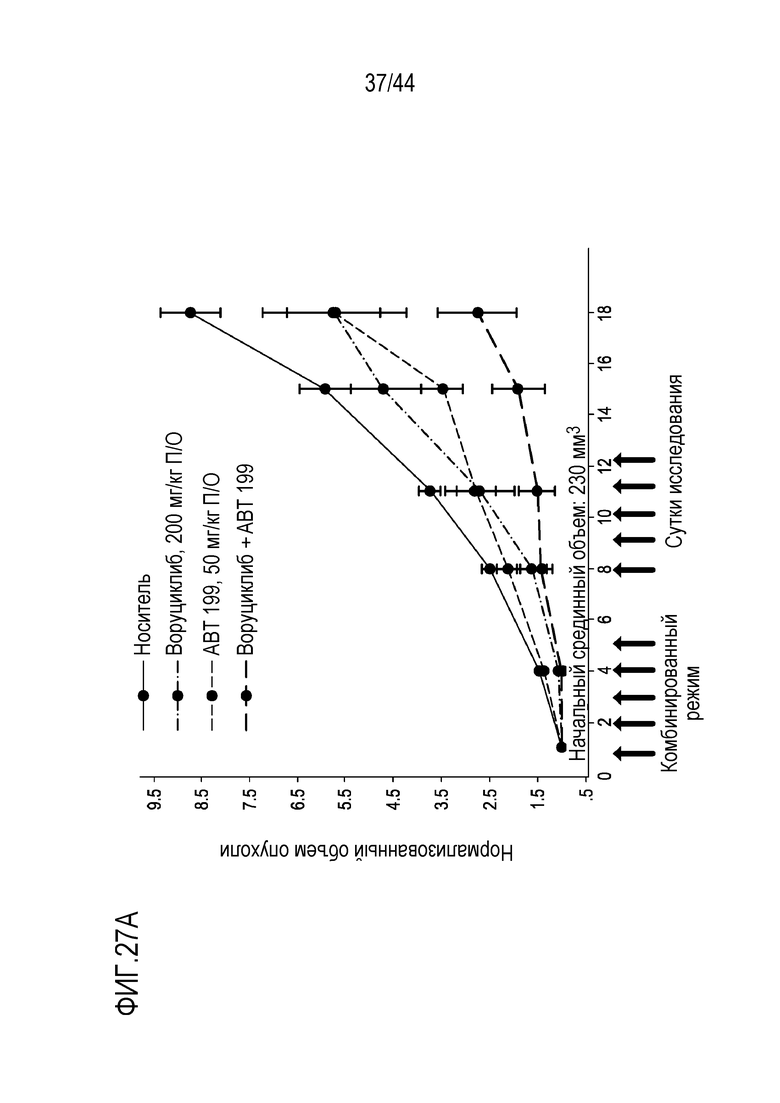
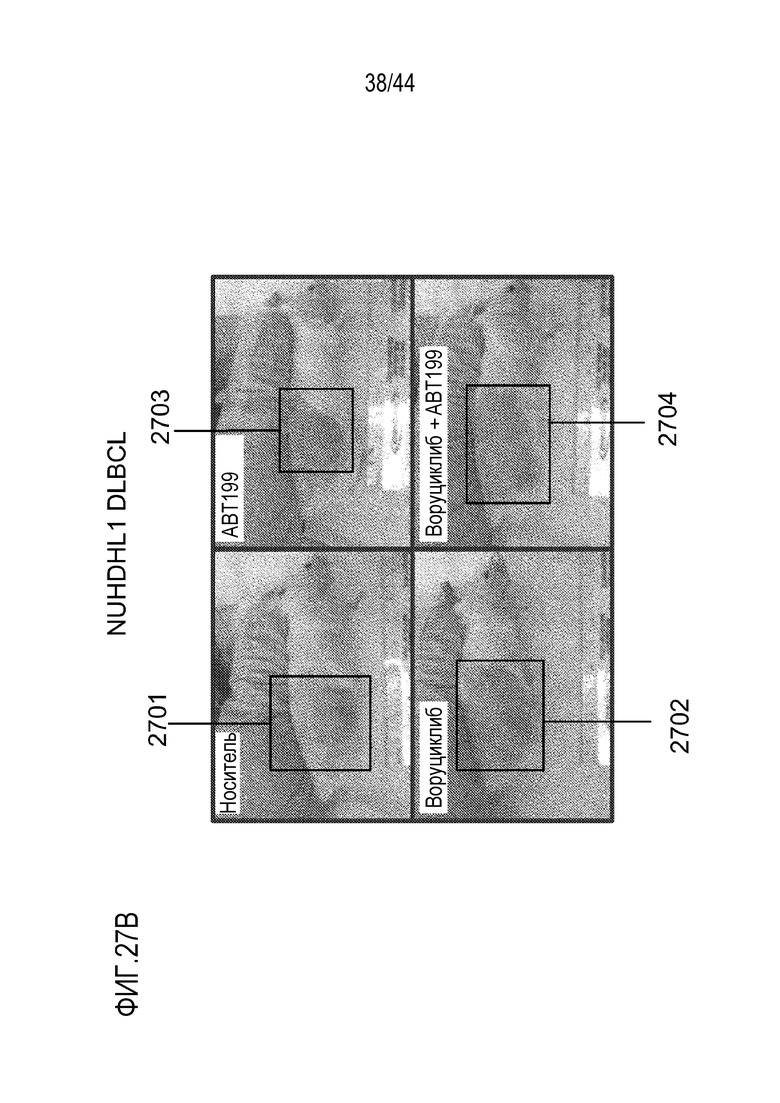
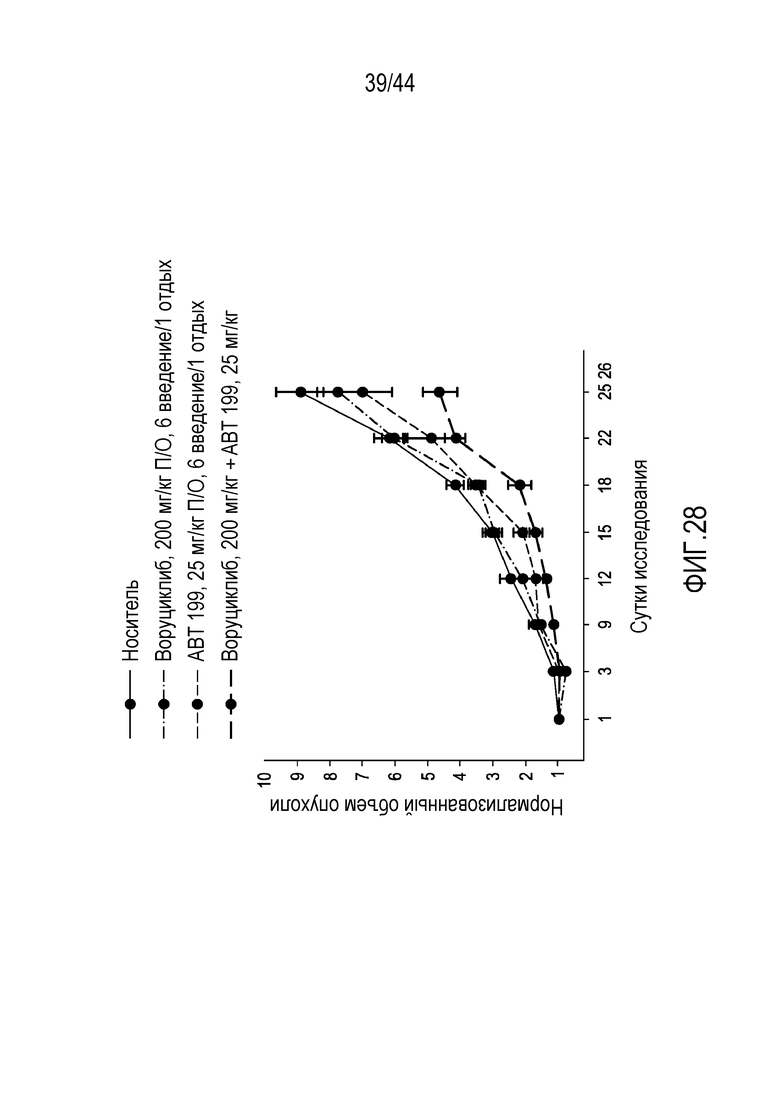
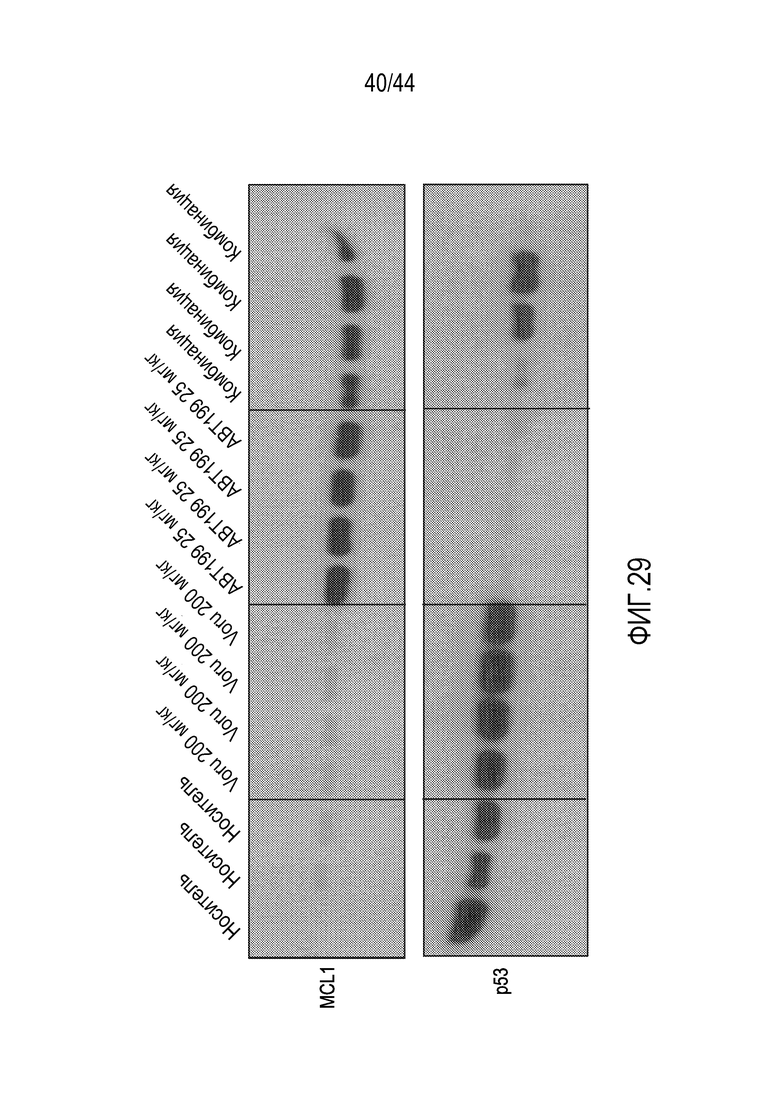
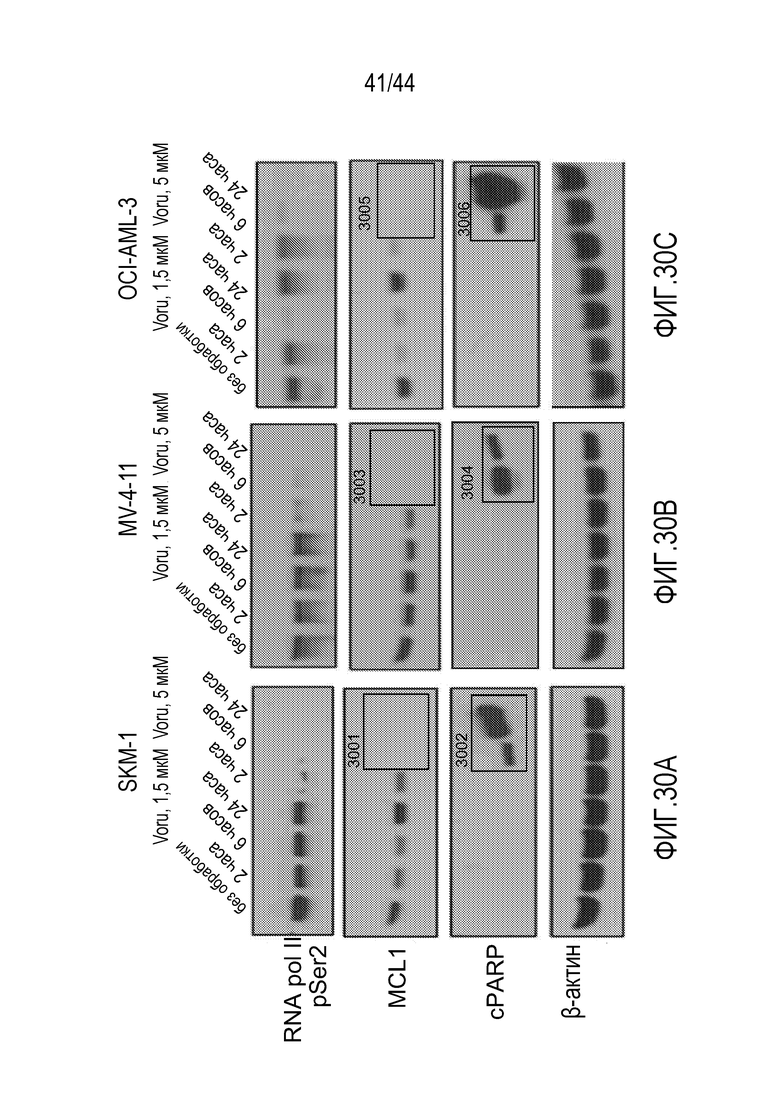
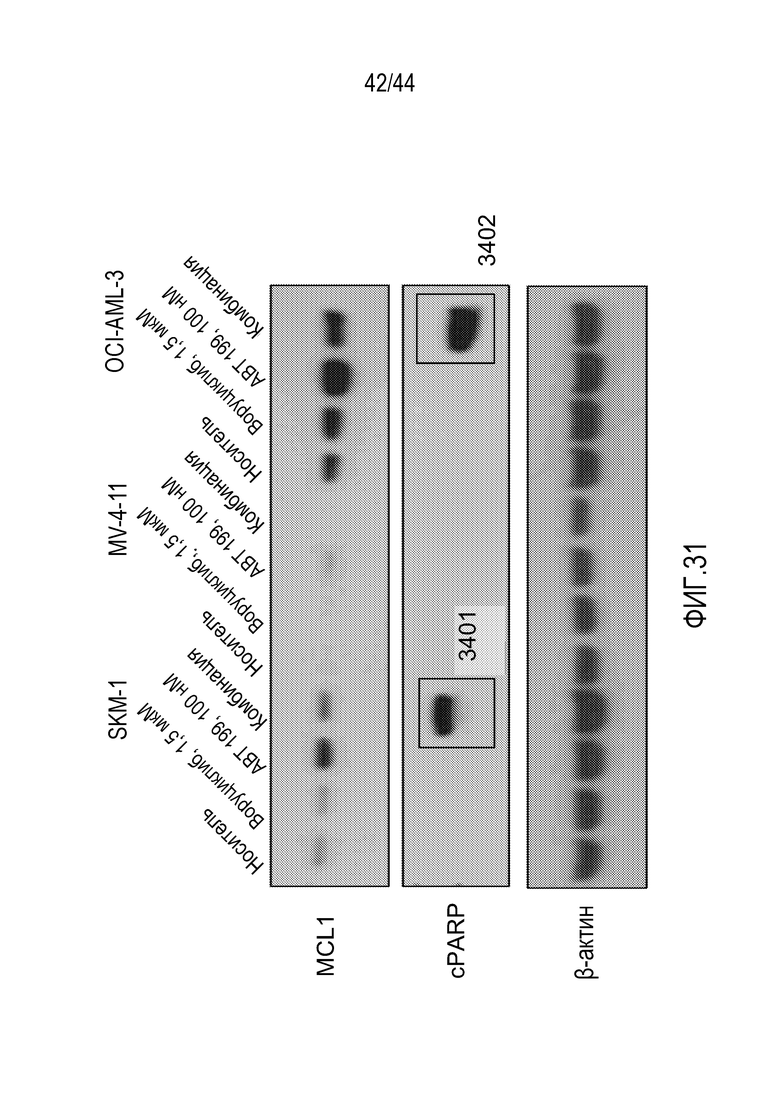
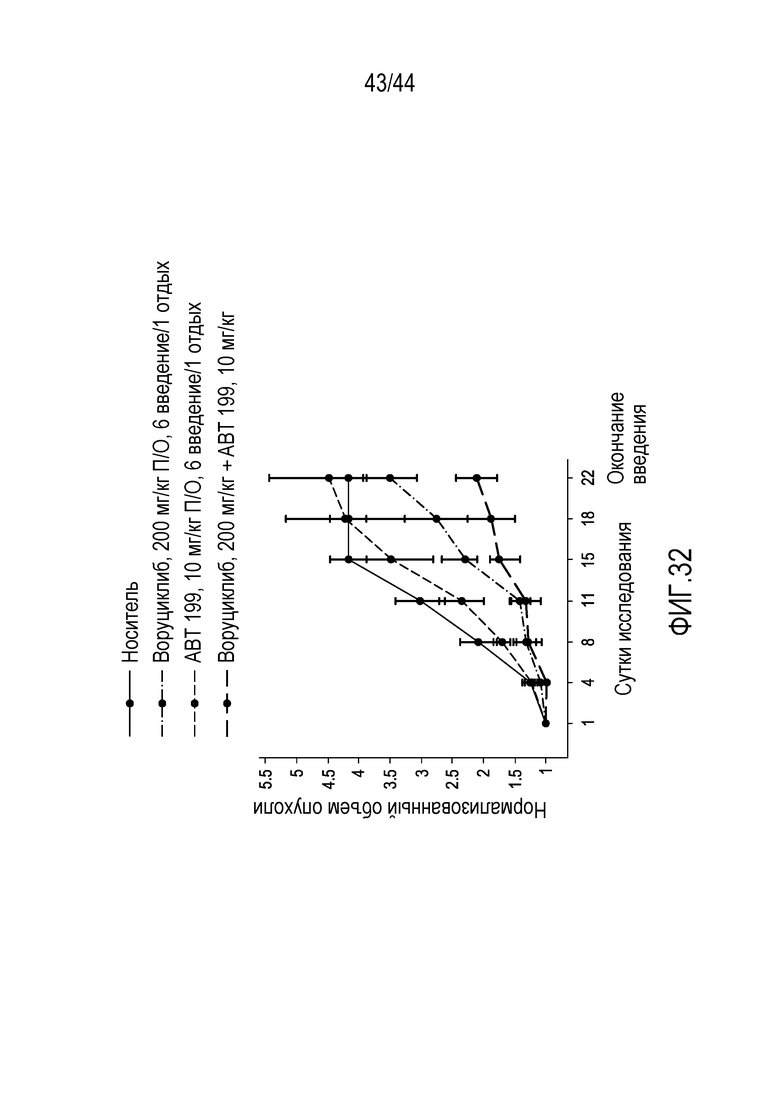
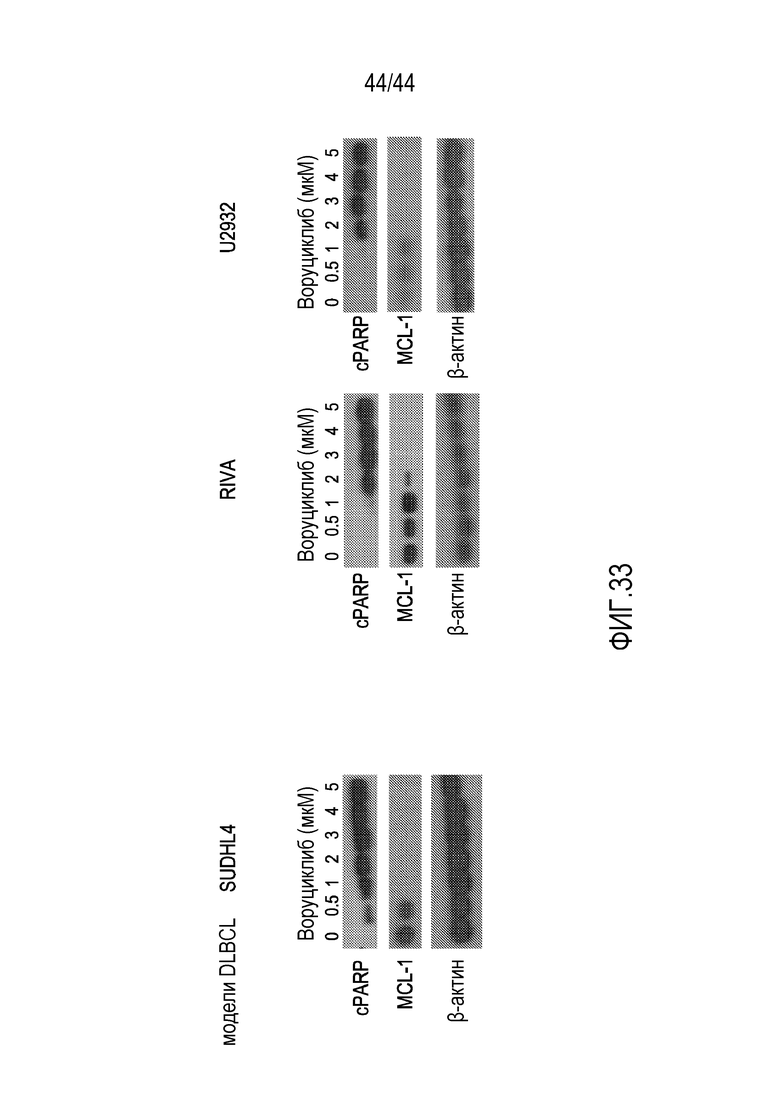
Группа изобретений относится к области медицины и фармацевтики. Первое изобретение представляет собой способ лечения злокачественной опухоли крови, включающий введение индивидууму ингибитора CDK, соответствующего формуле:
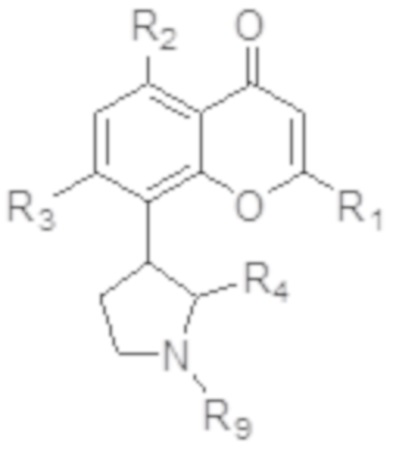 ,
,
или его фармацевтически приемлемой соли, где: R1 представляет собой фенил, замещенный одним или двумя заместителями, независимо выбранными из галогена и галогенметила; каждый из R2 и R3 независимо выбран из гидрокси и -OR8, где R8 представляет собой C1-C10-алкил; R4 представляет собой -CH2OH; и R9 представляет собой водород или метил; и венетоклакса или его фармацевтически приемлемой соли. Второе изобретение представляет собой способ лечения злокачественной опухоли, включающий введение индивидууму указанного ингибитора CDK и ингибитора протеасом, выбранного из бортезомиба, маризомиба, иксазомиба и их фармацевтически приемлемых солей. Третье изобертение представляет собой фармацевтическую композицию, включающую указанный ингибитор CDK и венетоклакс или ингибитор протеосом, выбранный из бортезомиба, маризомиба, иксазомиба или их фармацевтически приемлемых солей. 3 н. и 38 з.п. ф-лы, 33 ил., 4 табл., 16 пр.
1. Способ лечения злокачественной опухоли крови, включающий введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества ингибитора CDK, соответствующего формуле I:
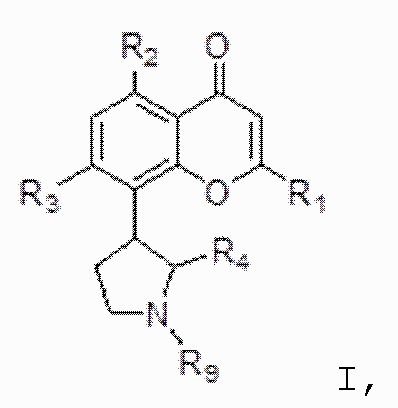
или его фармацевтически приемлемой соли, где:
R1 представляет собой фенил, замещенный одним или двумя заместителями, независимо выбранными из галогена и галогенметила;
каждый из R2 и R3 независимо выбран из гидрокси и -OR8, где R8 представляет собой C1-C10-алкил;
R4 представляет собой -CH2OH; и
R9 представляет собой водород или метил;
и терапевтически эффективного количества ингибитора BCL-2, где ингибитор BCL-2 представляет собой венетоклакс или его фармацевтически приемлемую соль.
2. Способ по п.1, где соединение или соль формулы I соответствует формуле Ia:
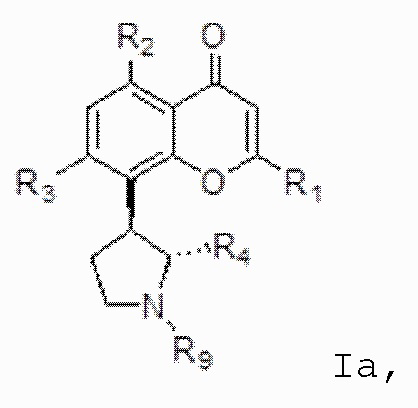
где R1, R2, R3, R4 и R9 определены в п.1.
3. Способ по п.1 или 2, где R1 замещен одним или двумя заместителями, независимо выбранными из галогена и галогенметила.
4. Способ по п.3, где R1 представляет собой 2-хлор-4-трифторметилфенил.
5. Способ по п.4, где каждый из R2 и R3 представляет собой гидрокси.
6. Способ по п.4, где R4 представляет собой -CH2OH.
7. Способ по п.6, где R9 представляет собой метил.
8. Способ по п.1, где соединение формулы I представляет собой соединение формулы Ib:
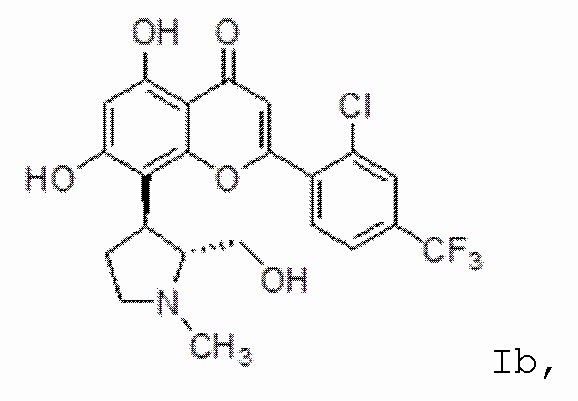
или его фармацевтически приемлемую соль.
9. Способ по любому из пп.1-8, где злокачественная опухоль крови выбрана из острого миелоидного лейкоза (AML), хронического миелоидного лейкоза (CML), острой лимфоцитарной лимфомы (ALL) и хронического лимфоцитарного лейкоза (CLL), диффузной крупноклеточной B-клеточной лимфомы (DLBCL), первичной медиастинальной B-клеточной лимфомы, внутрисосудистой крупноклеточной B-клеточной лимфомы, фолликулярной лимфомы, мелкоклеточной лимфоцитарной лимфомы (SLL), лимфомы из клеток мантийной зоны, лимфом из B-клеток маргинальной зоны, экстранодальных лимфом из B-клеток маргинальной зоны, нодальной лимфомы из B-клеток маргинальной зоны, селезеночной лимфомы из B-клеток маргинальной зоны, лимфомы Беркитта, лимфоплазматической лимфомы и первичной лимфомы центральной нервной системы.
10. Способ по п.9, где злокачественная опухоль крови представляет собой диффузную крупноклеточную B-клеточную лимфому, острый миелоидный лейкоз или хронический лимфоцитарный лейкоз.
11. Способ по любому из пп.1-10, где ингибитор CDK и ингибитор BCL-2 вводят одновременно.
12. Способ по любому из пп.1-10, где ингибитор CDK и ингибитор BCL-2 вводят последовательно в пределах приблизительно 12 часов друг от друга.
13. Способ по п.12, где ингибитор CDK и ингибитор BCL-2 вводят последовательно в пределах приблизительно 5 часов друг от друга.
14. Способ по любому из пп.1-10, где ингибитор CDK и ингибитор BCL-2 составлены совместно в фармацевтической композиции.
15. Способ по любому из пп.1-14, где ингибитор CDK и ингибитор BCL-2 вводят каждые сутки, раз в двое суток или раз в трое суток.
16. Способ лечения злокачественной опухоли, включающий введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества ингибитора CDK, соответствующего формуле I:
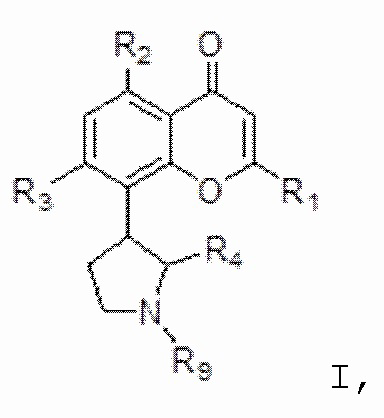
или его фармацевтически приемлемой соли, где:
R1 представляет собой фенил, замещенный одним или двумя заместителями, независимо выбранными из галогена и галогенметила;
каждый из R2 и R3 независимо выбран из гидрокси и -OR8, где R8 представляет собой C1-C10-алкил;
R4 представляет собой -CH2OH; и
R9 представляет собой водород или метил;
и терапевтически эффективного количества ингибитора протеасом, где ингибитор протеасом выбран из бортезомиба, маризомиба, иксазомиба и фармацевтически приемлемой соли любого из них.
17. Способ по п.16, где соединение или соль формулы I соответствует формуле Ia:
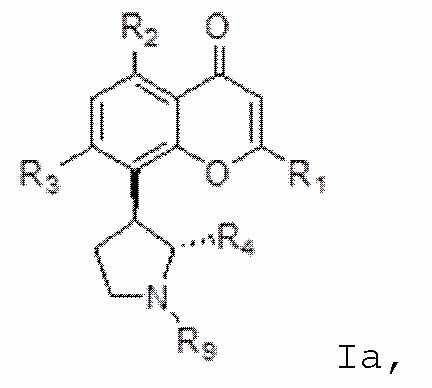
где R1, R2, R3, R4 и R9 определены в п.16.
18. Способ по п.16 или 17, где R1 замещен одним или двумя заместителями, независимо выбранными из галогена и галогенметила.
19. Способ по п.18, где R1 представляет собой 2-хлор-4-трифторметилфенил.
20. Способ по п.19, где каждый из R2 и R3 представляет собой гидрокси.
21. Способ по п.20, где R4 представляет собой -CH2OH.
22. Способ по п.21, где R9 представляет собой метил.
23. Способ по п.16, где соединение формулы I представляет собой соединение формулы Ib:
или его фармацевтически приемлемую соль.
24. Способ по любому из пп.16-23, где злокачественная опухоль представляет собой злокачественную опухоль крови.
25. Способ по п.24, где злокачественная опухоль крови выбрана из острого миелоидного лейкоза (AML), хронического миелоидного лейкоза (CML), острой лимфоцитарной лимфомы (ALL) и хронического лимфоцитарного лейкоза (CLL), диффузной крупноклеточной B-клеточной лимфомы (DLBCL), первичной медиастинальной B-клеточной лимфомы, внутрисосудистой крупноклеточной B-клеточной лимфомы, фолликулярной лимфомы, мелкоклеточной лимфоцитарной лимфомы (SLL), лимфомы из клеток мантийной зоны, лимфом из B-клеток маргинальной зоны, экстранодальных лимфом из B-клеток маргинальной зоны, нодальной лимфомы из B-клеток маргинальной зоны, селезеночной лимфомы из B-клеток маргинальной зоны, лимфомы Беркитта, лимфоплазматической лимфомы и первичной лимфомы центральной нервной системы.
26. Способ по п.25, где злокачественная опухоль крови представляет собой диффузную крупноклеточную B-клеточную лимфому, острый миелоидный лейкоз или хронический лимфоцитарный лейкоз.
27. Способ по любому из пп.16-23, где злокачественная опухоль представляет собой трижды негативный рак молочной железы (TNBC).
28. Способ по любому из пп.16-27, где ингибитор CDK и ингибитор протеасом вводят одновременно.
29. Способ по любому из пп.16-27, где ингибитор CDK и ингибитор протеасом вводят последовательно в пределах приблизительно 12 часов друг от друга.
30. Способ по п.29, где ингибитор CDK и ингибитор протеасом вводят последовательно в пределах приблизительно 5 часов друг от друга.
31. Способ по любому из пп.16-27, где ингибитор CDK и ингибитор протеасом составляют совместно в фармацевтической композиции.
32. Способ по любому из пп.16-31, где ингибитор CDK и ингибитор протеасом вводят каждые сутки, раз в двое суток или раз в трое суток.
33. Фармацевтическая композиция для лечения злокачественной опухоли, содержащая терапевтически эффективное количество ингибитора CDK, соответствующего формуле I:
или его фармацевтически приемлемой соли, где:
R1 представляет собой фенил, замещенный одним или двумя заместителями, независимо выбранными из галогена и галогенметила;
каждый из R2 и R3 независимо выбран из гидрокси и -OR8, где R8 представляет собой C1-C10-алкил;
R4 представляет собой -CH2OH; и
R9 представляет собой водород или метил;
терапевтически эффективное количество ингибитора BCL-2 или ингибитора протеасом и фармацевтически приемлемый эксципиент,
где ингибитор BCL-2 представляет собой венетоклакс или его фармацевтически приемлемую соль и где ингибитор протеасом выбран из бортезомиба, маризомиба, иксазомиба и фармацевтически приемлемой соли любого из них.
34. Фармацевтическая композиция по п.33, где соединение или соль формулы I соответствует формуле Ia:
где R1, R2, R3, R4 и R9 определен в п.33.
35. Фармацевтическая композиция по п.33 или 34, где R1 замещен одним или двумя заместителями, независимо выбранными из галогена и галогенметила.
36. Фармацевтическая композиция по п.35, где R1 представляет собой 2-хлор-4-трифторметилфенил.
37. Фармацевтическая композиция по п.36, где каждый из R2 и R3 представляет собой гидрокси.
38. Фармацевтическая композиция по п.37, где R4 представляет собой -CH2OH.
39. Фармацевтическая композиция по п.38, где R9 представляет собой метил.
40. Фармацевтическая композиция по п.33, где соединение формулы I представляет собой соединение формулы Ib:
или его фармацевтически приемлемую соль.
41. Фармацевтическая композиция по любому из пп.33-40, где композиция содержит ингибитор протеасом и ингибитор протеасом представляет собой бортезомиба или его фармацевтически приемлемую соль.
| ХАРКЕВИЧ Д.А | |||
| Фармакология | |||
| М.: "Медицина" | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
| С | |||
| Способ запрессовки не выдержавших гидравлической пробы отливок | 1923 |
|
SU51A1 |
| US 2014080838 A1, 20.03.2014 | |||
| US 2009142337 A1, 04.06.2009 | |||
| BOGENBERGER J | |||
| et al | |||
| Combined venetoclax and alvocidib in acute myeloid leukemia // Oncotarget | |||
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Vol | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| P | |||
| Устройство для управления возбуждением генераторов главного привода реверсивного прокатного стана типа блуминг и т.п. | 1956 |
|
SU107206A1 |
| СПОСОБ ОБРАБОТКИ НЕКРОТИЧЕСКОЙ ТКАНИ | 2001 |
|
RU2219867C2 |
| WO 2016024230 A1, 18.02.2016 | |||
| WO 2009061345 A2, 14.05.2009 | |||
| US 2013237582 | |||

