Область, к которой относится изобретение
Изобретение относится к области медицины, в частности к фармакологии, и касается средства для предупреждения развития и лечения сахарного диабета 2 типа.
Уровень техники
Сахарный диабет 2 типа (СД-2) - хроническое эндокринное заболевание, сопряженное с инсулинрезистентностью и дисфункцией β-клеток поджелудочной железы, что обусловливает стойкое и избыточное содержание глюкозы в крови (гипергликемию), которое, в свою очередь, напрямую или опосредованно запускает целый каскад нарушений всех видов метаболизма и обмена веществ. С СД-2 сопряжены нарушения эндокринной системы, повышенный липолиз в жировой ткани, увеличенная секреция глюкагона, повышенная почечная реабсорбция глюкозы и, наконец, нарушения пищевого поведения. Это заболевание сильно ухудшает качество жизни больных, а его развитие приводит к тяжелым осложнениям и последствиям - инвалидности и даже летальному исходу (Drucker, D.J., & Nauck, M.A. (2006). The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet, 368(9548), 1696-1705). Сахарный диабет представляет серьезную медико-социальную проблему во всем мире, а доля людей, страдающих этим заболеванием, непрерывно растёт. Аналитики из Международной Федерации диабета (IDF) сообщают, что к 2045 году число пациентов с СД-2 вырастет до 700 миллионов (Karuranga, S., Cho, N.H., Ohlrogge, A.W., Shaw, J.E., da Rocha Fernandes, J.D., Huang, Y., & Malanda, B. (2018). IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Research and Clinical Practice, 138, 271-281).
У людей, предрасположенных к развитию этого заболевания, и больных СД-2 в организме нарушается нормальный обмен глюкозы, повышается уровень ее концентрации, а также концентрации ее производных в крови, что влияет на обмен веществ в целом и приводит к неспособности организма осуществлять целый ряд нормальных биологических процессов. Большинство пациентов с СД-2 имеют существенно избыточный вес, что определяется нарушениями углеводного, и, как следствие, липидного метаболизма (Erion, D.M., Park, H.J., & Lee, H.Y. (2016). The role of lipids in the pathogenesis and treatment of type 2 diabetes and associated co-morbidities. BMB Reports, 49(3), 139-148).
В настоящее время на фармацевтическом рынке имеется ряд препаратов с разными механизмами действия для коррекции состояния больных. Каждый препарат из разработанных ранее (бигуаниды, тиазолидиндионы, сульфонилмочевины и др.) имеет существенные ограничения и побочные действия. Одним из новейших и востребованных подходов для лечения симптомов сахарного диабета 2-го типа является применение готовых лекарственных форм на основе ингибиторов дипептидилпептидазы-4. Самыми первыми ингибиторами ДПП-4, успешно прошедшими клинические испытания и появившимися на фармацевтическом рынке различных государств, стали ситаглиптин (https://www.lsgeotar.ru/sitagliptin.html) и вилдаглиптин (https://www.lsgeotar.ru/vildagliptin.html), и уже позднее к ним присоединились саксаглиптин (https://www.lsgeotar.ru/saxagliptin.html), алоглиптин (https://www.lsgeotar.ru/alogliptin.html), линаглиптин (https://www.lsgeotar.ru/linagliptin.html).
Клиническими исследованиями было показано, что ингибиторы ДПП-4 существенно снижают гипергликемию (уровень глюкозы в крови) постпрандиально и натощак, показатели гликированного гемоглобина в плазме крови, стимулируют синтез инсулина, а также обладают хорошей пероральной биодоступностью и относительно долгим действием и не оказывают влияния на изменение массы тела.
Известен патент РФ на изобретение № 2443687, «Новые ингибиторы дипептидилпептидазы IV, способы их получения и содержащие их фармацевтические композиции».
Известен патент РФ 2483716 на изобретение, касающееся фармацевтического состава, включающего ингибитор дипептидилпептидазы-4, предпочтительно, вилдаглиптин от 1.5 до 20% и метформин от 80 до 98.5%. При этом активные ингредиенты составляют от 60 до 98% композиции. В качестве связующего вещества используются целлюлоза или ее производные в количестве от 1 до 20%.
Известена заявка на изобретение РФ 2006121339, касающаяся фармацевтической композиции, включающей терапевтически эффективное количество (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидина или его фармацевтически приемлемой соли, предпочтительно 25-100 мг, в комбинации с одним или несколькими фармацевтически приемлемыми носителями, для лечения сердечнососудистых заболеваний или повреждений, заболеваний или повреждений почек, сердечной недостаточности или заболеваний, связанных с сердечной недостаточностью.
Известена заявка на изобретение РФ 2011146335, описывающая фармацевтическую композицию, включающую комбинацию соединения 1-тио-D-глюцитола, в которой ингибитор дипептидилпептидазы-4 представляет собой ситаглиптин или вилдаглиптин.
Известен патент РФ 2606840 на изобретение композиции, содержащей конъюгат инсулина длительного действия и конъюгат инсулинотропного пептида длительного действия, для предупреждения или лечения диабета, а также способа лечения диабета.
Известна заявка на изобретение 2015122253 «Составы таблетки с немедленным высвобождением», описывающая фармацевтический состав, содержащий дапаглифлозин и гидрохлорид метформина.
Известно изобретение РФ №2589258, описывающее фармацевтические композиции на основе средства пептидной природы, ингибирующего дипептидилпептидазу-4.
Известна заявка WO № 2012/131005 A1 «Фармацевтическая композиция ситаглиптина», выбранная авторами за прототип.
Несмотря на то, что известные лекарства на основе ингибиторов ДПП-4 (ситаглиптин или вилдаглиптин и др.) имеют неоспоримые преимущества в сравнении с другими описанными антигипергликемическими средствами (инсулин, инсулинотропные пептиды, метформин, глифозины и др.) и хорошо себя зарекомендовали с момента появления на фармацевтическом рынке, их применение ограничивается довольно короткой временнОй историей (чуть более 10 лет) в терапии заболевания, недостаточной для выявления возможных и нежелательных побочных эффектов.
В последние годы интенсивно и широко проводятся исследования физиологических профилей и клинических свойств лекарственных средств на основе ингибиторов ДПП-4, а также побочных эффектов, которые могут быть вызваны приёмом таких препаратов.
Несмотря на то, что коммерческие лекарственные препараты на основе пяти ингибиторов ДПП-4 (сита-, вилда-, сакса-, ало- и линаглиптинов) похожи в проявлении своих антигипергликемических свойств, отсутствию влияния на массу тела и риска возникновения гипогликемии и, в целом, хорошо себя зарекомендовали, в последние годы уже обнаружены и продолжают обнаруживаться некоторые специфические различия и побочные эффекты, выявленные при приеме разными пациентами лекарственных препаратов на основе структурно-различных глиптинов.
Например, дозировки ситаглиптина должны тщательно подбираться для больных со средней или тяжелой почечной недостаточностью. С особой настороженностью следует назначать это препарат больным, получающим дигоксин. Ситаглиптин противопоказан при сахарном диабете 1 типа и диабетическом кетоацидозе.
В отдельных случаях применение вилдаглиптина вызывало повышение активности аминотрансфераз в 3 раза. Клинические проявления этого факта в настоящее время тщательно изучаются. И уже известно, что при любых признаках нарушения функции печени прием этого препарата немедленно отменяется, с запретом возобновления при нормализации показателей.
При приеме саксаглиптина обнаружено учащение случаев инфекций верхних дыхательных путей, мочевых путей, развития гастроэнтерита, синусита, рвоты и головных болей. Выявленное головокружение, как побочная реакция на препарат, требует ограничений в управлении транспортными средствами или даже необходимости отказа от управления.
Кроме вышеперечисленных примеров, следует упомянуть, что еще не для всех глиптинов проводились исследования лечения детей в возрасте до 18 лет, а также беременных и кормящих, поэтому для этих групп диабетиков такие препараты противопоказаны.
Число прямых исследований, сравнивающих эффективность и побочные действия разных лекарств-ингибиторов ДПП-4, еще недостаточно. Однако, очевидно, что это направление терапии СД-2 за последние годы становится всё более популярным и востребованным, поскольку есть данные, что ещё большее число других потенциальных антидиабетических агентов подобного типа находится на различных стадиях доклинических и клинических исследований (например, тенелиглиптин (Kishimoto Syndrome, 2013), омариглиптин (Tatosian et al., 2014), эвоглиптин (Mccormack, 2015) и др.).
Таким образом, в настоящее время сохраняется необходимость в расширении арсенала известных лекарственных средств и лекарственных форм ингибиторов дипептидилпептидазы-4 с целью удовлетворения в будущем потребности в препаратах аналогичного действия, но с учетом особенностей течения заболевания, отягощения другими заболеваниями, индивидуальной переносимости у разных больных и групп больных сахарным диабетом 2-типа.
Кроме того, ни в одном из известных и представленных выше изобретений не рассматриваются композиции и лекарственные средства, обеспечивающие разнообразие высвобождения и, как одного из следствий, продолжительности действия в организме активного вещества для лечения сахарного диабета.
Развитие медицины нацелено на персонификацию в будущем. Для того чтобы лечение любого заболевания препаратами однотипного механизма действия проводилось с учетом отягощения пациентов другими заболеваниями и особенностями индивидуальной переносимости конкретных представителей, необходимо продолжение разработки новых готовых лекарственных форм, позволяющих варьировать дозировку, высвобождение и продолжительность действия в организме активного вещества, ингибитора ДПП-4.
Раскрытие изобретения
Задачей настоящего изобретения является расширение арсенала существующих фармацевтических композиций путем разработки и создания фармацевтических композиций для перорального применения на основе нового ингибитора дипептидилпептидазы-4, активного сахароснижающего и нетоксичного компонента для предупреждения развития и лечения сахарного диабета 2 типа.
Разработка включает фармацевтические композиции, содержащие вспомогательные вещества и носители, обеспечивающие различные механизмы высвобождения активного компонента, а именно, немедленное, пролонгированное и модифицированное высвобождение активного действующего вещества. Другой задачей настоящего изобретения является разработка получения фармацевтически приемлемых солевых форм активного вещества - основы для композиций и ГЛФ.
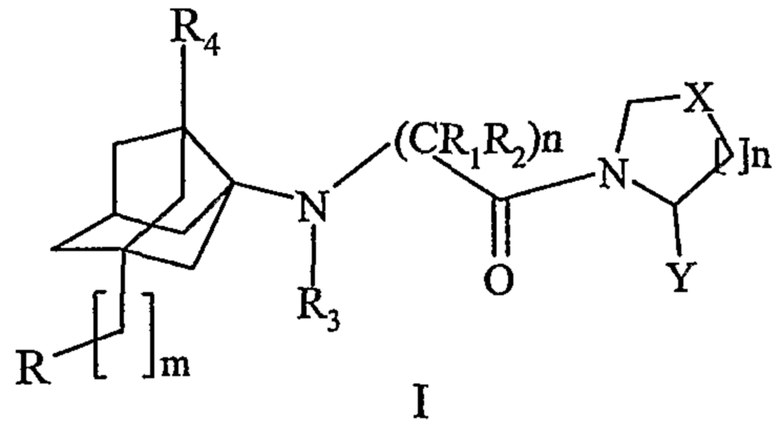
Задача настоящего изобретения решена за счет создания фармацевтических композиций для перорального применения, содержащих активный компонент, высокоэффективный ингибитор ДПП-4 экзо-(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил - вещество I в виде фармацевтически приемлемых солевых форм, вспомогательные вещества и носители, обеспечивающие немедленное, пролонгированное и модифицированное высвобождение активного компонента для лечения больных сахарным диабетом 2 типа.
Мол. масса 337
Структура действующего активного компонента - вещества I.
Краткое описание фигур
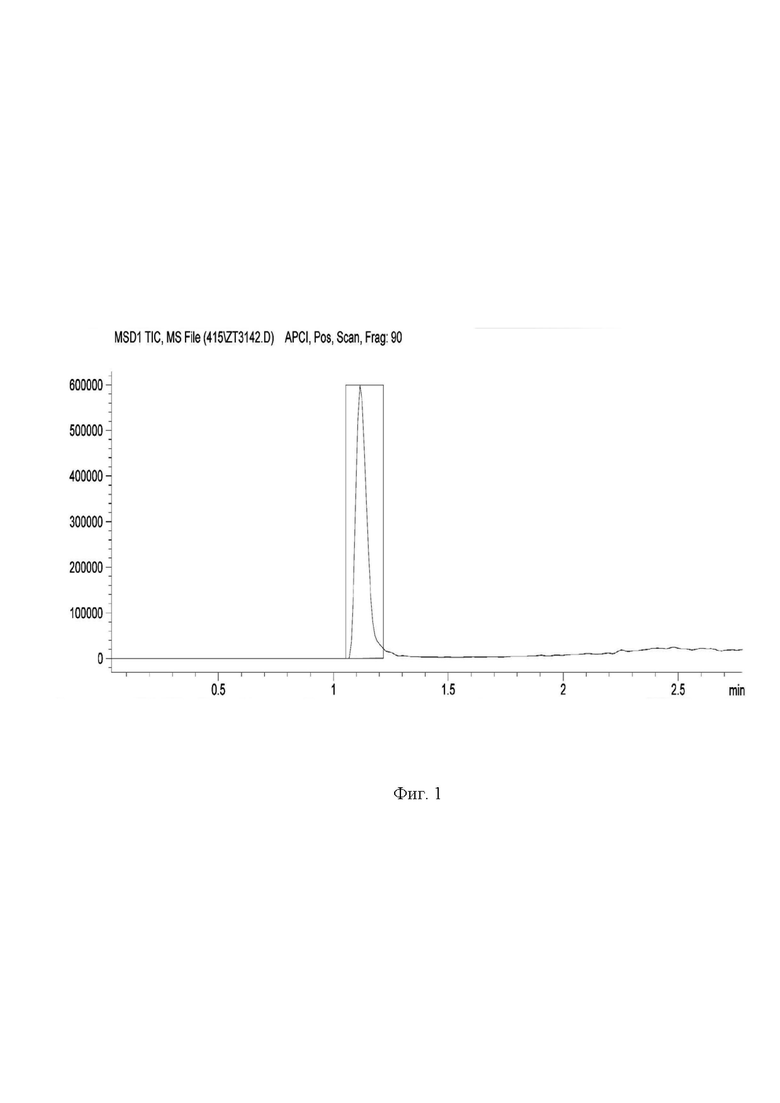
На фиг. 1
представлена типичная хроматограмма вещества I, полученная методом LC/MS c масс-детектором. В хроматограммах, полученных методом LC/MS с использованием масс-детектора, во всех случаях (далее примеры 2-4) наблюдалось по одному пику вещества I с чистотой 100%. Молекулярная масса (М) определена масс-детектором и в точности соответствовала рассчитанной для вещества I.
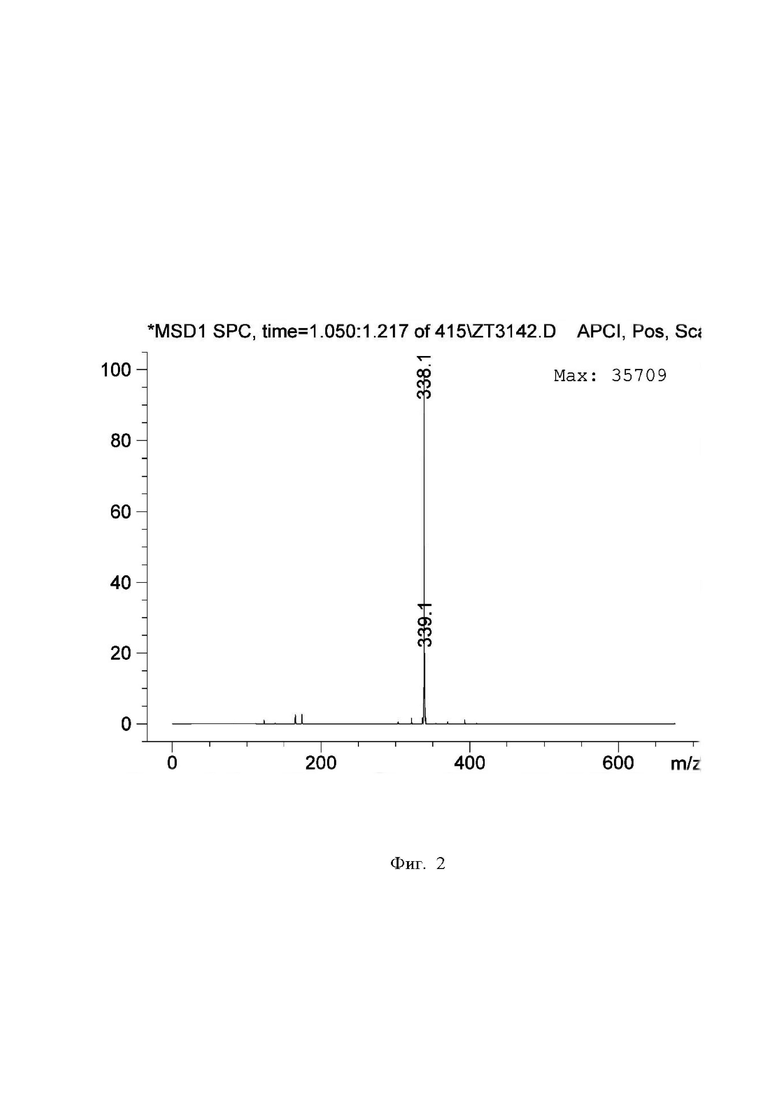
На фиг. 2
представлен масс-спектр вещества I, полученный методом LC/MS с APCI-ионизацией. Приведен масс-спектр пика из хроматограммы, изображенной на Фиг.1. Значение m/z [M+H]+ = 338 получено с применением APCI-ионизации и в точности соответствует молекулярной массе вещества I (М=337).
Реализация изобретения
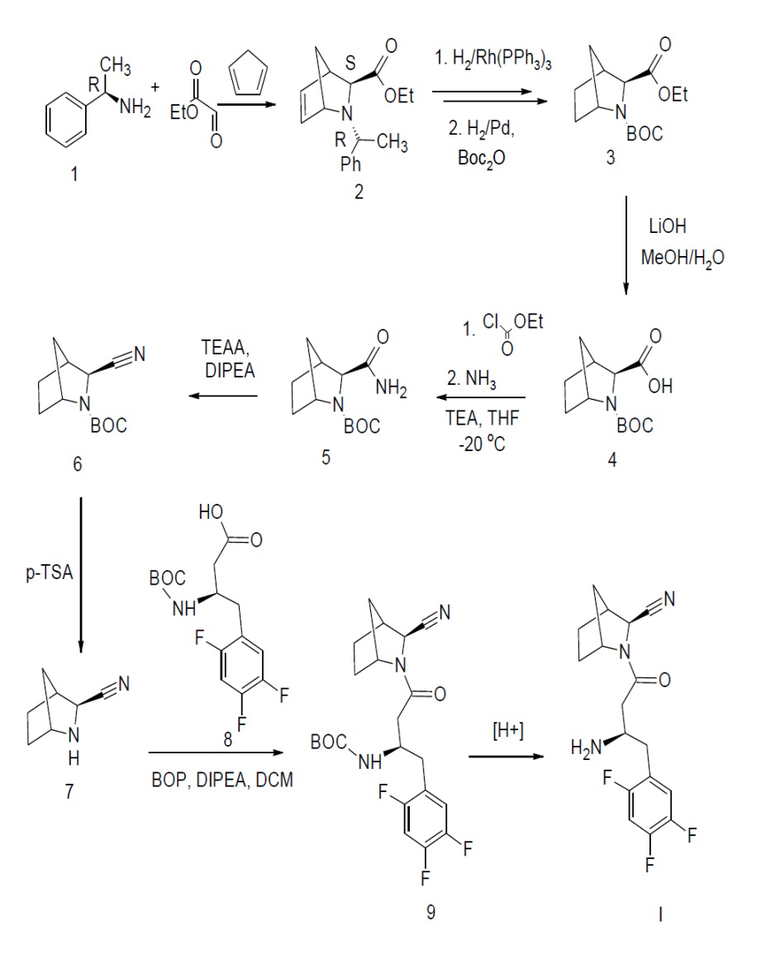
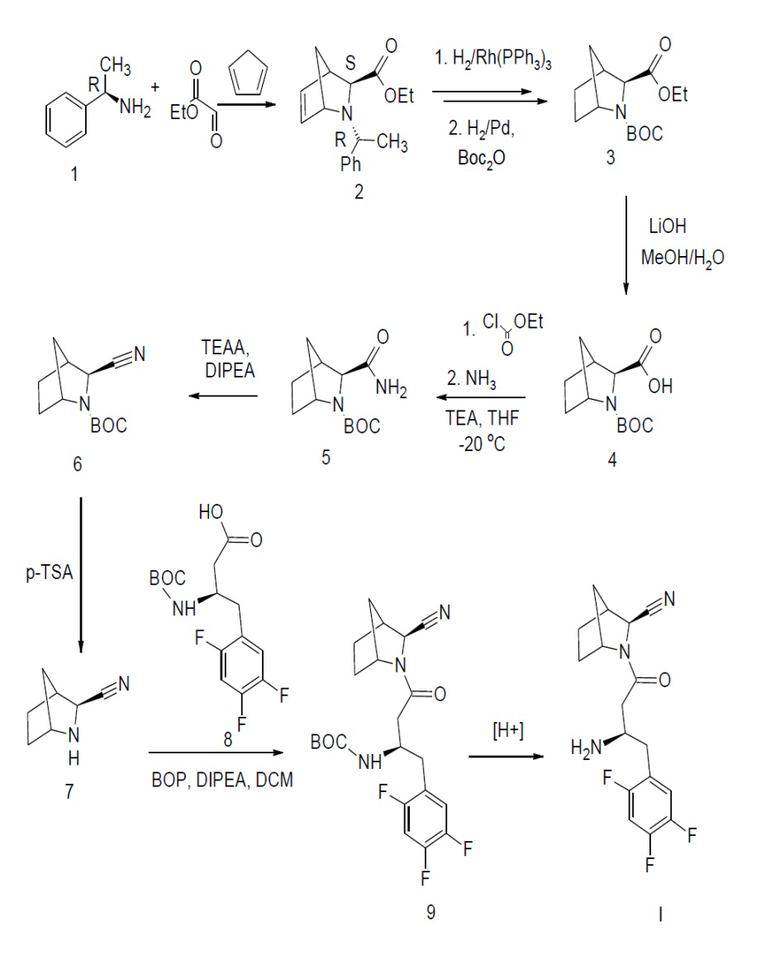
Синтез вещества I осуществляли согласно схеме:
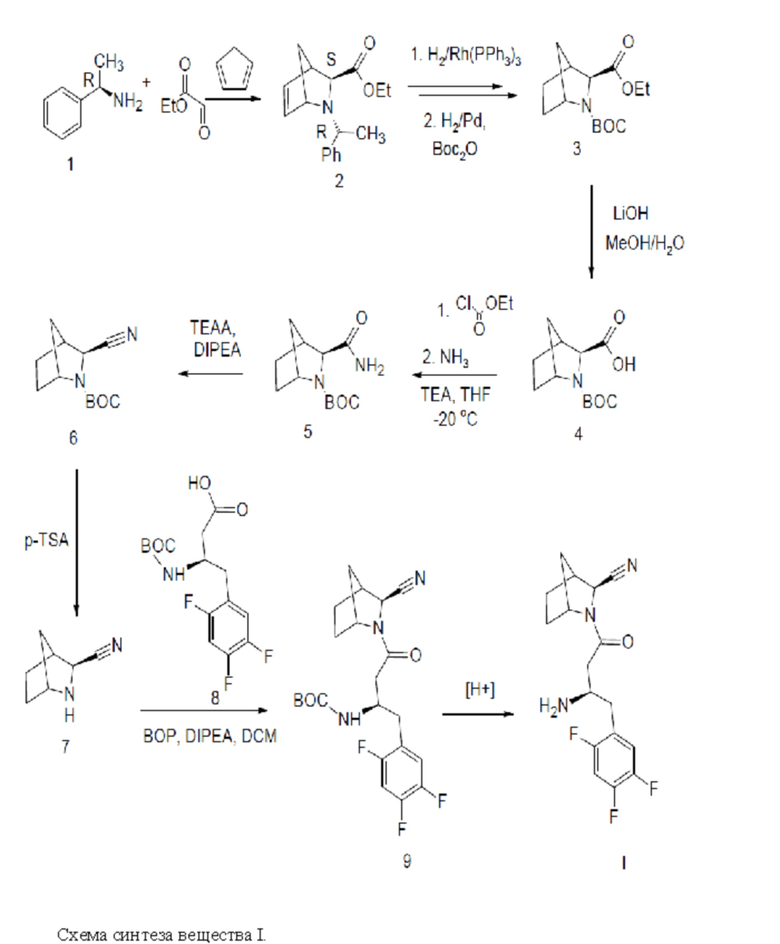
Способ синтеза вещества I.
Пример 1.
Синтез вещества I в виде соли трифторацетата.
Этил-(2S,3S)-3-(1-фенилэтил)-3-азабицикло[2.2.1]гепт-5-ен-2-карбоксилат (2).
В литровую колбу поместили 50%-ный раствор этилглиоксилата в толуоле (19.6 мл, 0.096 моль), толуол (300 мл) и бензиламин 1 (10.3 мл, 0.08 моль), перемешивали в течении 2 часов до выделения воды, затем воду удалили при помощи сульфата натрия и растворитель упарили. Полученный остаток растворили в диметилформамиде (100 мл), прибавили при охлаждении в ледяной бане раствор трифторуксусной кислоты (6.12 мл, 0.08 моль) в диметилформамиде. Перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Затем при охлаждении в ледяной бане одной порцией прибавили свежеперегнанный циклопентадиен (10.6 г, 0.16 моль). Выдержали реакционную смесь 24 часа при комнатной температуре. Через 24 часа реакционную смесь разбавили 400 мл 10%-ного раствора поташа при охлаждении в ледяной бане, экстрагировали этилацетатом 3 раза по 100 мл, органический слой высушили сульфатом натрия и упарили при температуре бани не выше 30°C. Колоночной хроматографией на силикагеле в системе этилацетат : петролейный фир 1:10 выделили 8.1 г (0.0299 моль) чистой фракции соединения 2 . Выход 37.3%.
(2S)-3-трет-бутоксикарбонил-3-азабицикло[2.2.1]гептен-2-карбоновой кислоты этиловый эфир (3).
В 200 мл бензола растворили соединение 2 (8.1 г, 0.0299 моль) и прибавили трис(трифенилфосфин)родийхлорид (0.405 г, 5 % масс). Гидрировали на аппарате Парра при 20psi до прекращения поглощения водорода (около 4 часов). По данным LCMS контролировали полноту протекания гидрирования. После окончания реакции раствор пропустили через слой силикагеля, смывая продукт системой этилацетат : петролейный эфир 1:4. Полученный раствор упарили, добавили 200 мл этанола, 5%-ный Pd на угле (0.8 г) и Вос2О (6.9 г, 0.0316 моль). Далее гидрировали при 20psi на аппарате Парра. Контроль полноты реакции осуществляли методом LCMS. Полученную реакционную смесь использовали в следующей стадии.
(2S)-3-трет-бутоксикарбонил-3-азабицикло[2.2.1]гептен-2-карбоновая кислота (4)
Этанольный раствор соединения 3 с предыдущей стадии после завершения гидрирования отфильтровали от катализатора и прибавили к нему 50 мл (0.075 моль) водного раствора гидроксида натрия. Полученный раствор нагревали 1 час при 60°C. После полной конверсии эфира (контроль ТСХ) упарили этанол, растворили остаток в воде, подкислили раствор 2М соляной кислотой до рН 3, экстрагировали продукт этилацетатом, промыли органические вытяжки водой, высушили сульфатом натрия и упарили. Продукт перекристаллизовывали из гексана. Получили 5.42 г (0.022 моль) кислоты 4. Выход составил 90%. Далее использовали этот продукт без дополнительной очистки.
(2S)-Экзо-трет-бутил-3-карбамоил-2-азабицикло[2.2.1]гептан-2-карбоксилат (5)
К раствору кислоты 4 (5.42 г) в сухом ТГФ при охлаждении до -20°С в атмосфере аргона добавляли триэтиламин 3.45 мл (2.5 г) затем по каплям добавляли этилхлорформиат (2.68 г, 0.0247 моль) в течение 10 мин. Выдерживали реакционную смесь при охлаждении в течение 40 мин. Затем пропускали аммиак из баллона 1 ч. Упарили растворитель, обработали остаток раствором лимонной кислоты до pH 4, экстрагировали этилацетатом, этилацетатные экстракты промывали раствором соды, сушили сульфатом натрия и концентрировали. Получили 5.29 г бесцветного кристаллического амида 5. Выход количественный.
(2S)-Экзо-третбутил-3-циано-2-азабицикло[2.2.1]гептан-2-карбоксилат (6)
К суспензии исходного амида 5 (5.29 г) в сухом ТГФ, при температуре не выше 4°С добавляли 2 эквивалента триэтиламина (6.13 мл, 4.45 г), ангидрид трифторуксусной кислоты (6.93 г, 0.033 моль) в течение 10 минут, по ТСХ контролировали степень протекания реакции. Реакционную смесь выдерживали при охлаждении в течение 3 часов. Реакционную смесь упаривали, наносили на силикагель и разделяли на хроматографической колонке. Элюент - смесь петролейный эфир: этилацетат 4:1. Получили 4.29 г (87%) целевого нитрила 6 в виде бледно-желтого густого маслообразного вещества.
(2S)-Экзо-2-азабицикло[2.2.1]гептан-3-карбонитрилтозилат (7).
К исходному ВОС-нитрилу 6 (4.29 г, 0.019 моль) в 30 мл ацетонитрила добавили двукратный избыток п-толуолсульфоновой кислоты (п-ТСК) (6.54 г, 0.038 моль) и оставили перемешиваться на ночь. Отгоняли ацетонитрил, остаток растирали с диэтиловым эфиром (3-4 обработки с декантацией). Упаривали эфир. Получили 4.58 г кристаллического целевого вещества 7 виде соли тозилата.
трет-бутил-N-[(1R)-3-[(2S)-2-циано-3-азабицикло[2.2.1]гептан-3-ил]-3-оксо-1-[(2,4,5-трифторфенил)метил]пропил]карбамат ( 9).
К раствору кислоты 8 (5.19 г, 0.0156 моль) в 60 мл дихлорметана прибавили BOP (8.27 г, 0.0187 моль) и триэтиламин (4.74 мл, 0.0468 моль, 3 экв.). Через 15 минут при комнатной температуре прибавили тозилат амина 7 (4.57 г, 0.0156 моль). Через 2 часа LCMS показала полную конверсию. Дихлорметан упарили, остаток растворили в этилацетате и промыли 10%-ным раствором поташа, затем водой, высушили сульфатом натрия и упарили. Добавили к остатку небольшое количество петролейного эфира и выкристаллизовавшийся продукт отфильтровали. Получили 5.08 г соединения 9. Выход 75%.
Удаление N-защитной группы в BOC-производном 9 проводят в любых стандартных условиях с использованием подходящих кислот НХ, хорошо известных в органической химии. Получают вещество I в виде соли соответствующей кислоты.
экзо-(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (I) в виде соли трифторацетата.
Соединение 9 (0.21 г, 0.65 ммоль) растворяли в DCM (20 мл) и добавляли TFA (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель упаривали на роторном испарителе досуха. К остатку добавляли Et2O (20 мл). Выпавший осадок отфильтровывали и сушили в вакууме. Выход вещества I в виде соли трифторацетата - 0.18 г (90%). LC/MS анализ: m/z [M+H] + = 338.
Пример 2.
Синтез вещества I в виде соли тозилата.
Суспендировали соединение 9 (4.37 г, 0.01 моль) в 100 мл ацетонитрила и прибавили двукратный избыток гидрата толуолсульфокислоты (3.8 г, 0.02 моль). Перемешивали ночь при комнатной температуре. Отфильтровали осадок и промыли ацетонитрилом. Выход вещества I в виде соли тозилата - 4.42 г (87%). LC/MS анализ: m/z [M+H] + = 338.
Пример 3.
Синтез вещества I в виде соли гидрохлорида.
К соединению 9 (6 г, 0.013 моль) в 25 мл ацетонитрила добавили 4М раствор НСl в диоксане (10 мл) и оставляли перемешиваться на 2 часа. Отгоняли растворитель до стеклообразного остатка. Остаток растворяли в ацетонитриле (20 мл) при 50оС и раствор оставляли охлаждаться до комнатной температуры. Выпавший осадок отфильтровывали и сушили в вакууме. Выход вещества I в виде соли гидрохлорида - 3.54 г (73%). LC/MS анализ: m/z [M+H] + = 338.
Условия проведения LC/MS анализов: колонка - Onix C18 50x4.6 мм; элюент 1 - 0.1% TFA в воде; элюент 2 - 0.1% TFA в ацетонитриле, градиент - элюент 1 - 2.9 мин, элюент 2 - 0.2 мин, элюент 1 - промывка, скорость потока - 3.75 мл/мин, детекция - УФ (254 нм) и масс-спектрометрия.
Типичная хроматограмма вещества I (Примеры 2-4), полученная методом LC/MS анализа с масс-детекцией.
Разработаны и получены фармацевтически приемлемые для ГЛФ бессолевая (А) и солевая (Б) формы вещества I.
Фармакологически приемлемые солевые формы вещества I включают соли с неорганическими кислотами - хлористоводородной кислотой (гидрохлорид), бромистоводородной кислотой, азотной кислотой, серной кислотой и фосфорной кислотой; и органическими кислотами и их производными из числа сульфо-, алкилокси-, гидрокси-, оксо-замещенных кислот: муравьиной кислотой, уксусной кислотой, фумаровой кислотой, щавелевой кислотой, винной кислотой, малеиновой кислотой, лимонной кислотой, янтарной кислотой, яблочной кислотой, метансульфоновой кислотой, трифторметансульфоновой кислотой, бензолсульфоновой кислотой и пара-толуолсульфоновой кислотой (тозилат), нафтилсульфоновой кислотой и сульфосалициловой кислотой, молочной кислотой, виноградной кислотой, адипиновой кислотой, глюконовой кислотой, глюкогептоновой кислотой, глюкуроновой кислотой, терефталевой кислотой, гиппуровой кислотой, 1,2-этандисульфоновой кислотой, изэтионовой кислотой, лактобионовой кислотой, олеиновой кислотой, памовой кислотой, полигалактуроновой кислотой, стеариновой кислотой, дубильной кислотой, или аминокислотами из числа всех известных природных аминокислот - глицин, аланин, валин, лейцин, изолейцин, пролин, цистеин, метионин, фенилаланин, триптофан, серин, треонин, глютамин, тирозин, аспарагин, гистидин, лизин, аргинин, аспарагиновая кислота, аспарагиновая кислота.
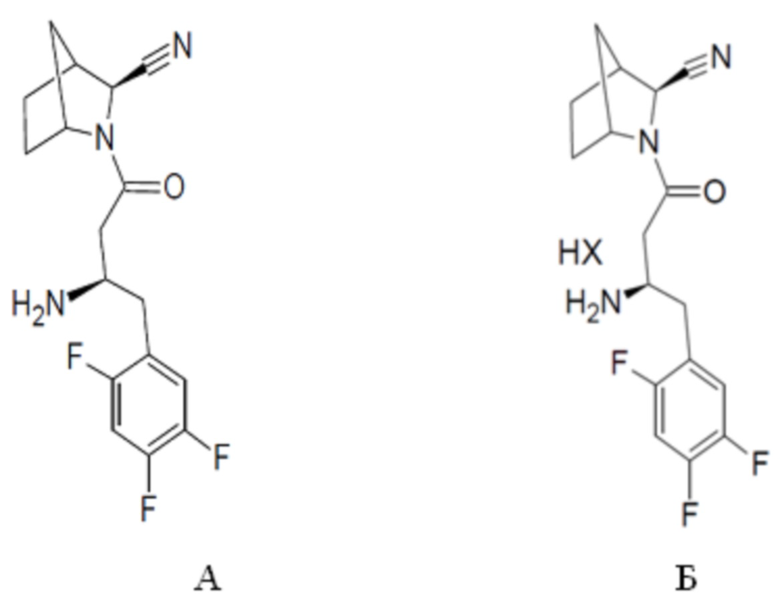
Бессолевая А (свободное основание) и солевая форма Б вещества I в общем виде, где НХ - любая из описанных выше кислот.
Бессолевая форма А вещества I получается выделением свободного основания после обработки солевых форм основными реагентами любыми стандартными, химическими и физико-химическими методами, применяемыми на основе базовых знаний по органической химии.
Пример 4.
Получение вещества I в виде свободного основания.
К раствору вещества I в виде трифторацетата (0.45 г, 1 ммоль) в воде (10 мл) добавляли поташ (0.3 г, 2 ммоль) и хлористый метилен (10 мл), перемешивали 1 час. Отделяли органический слой, промывали 10%-ным раствором поташа, затем водой, высушивали сульфатом натрия и упаривали. Остаток сушили в вакууме. Получили вещество I в виде свободного основания. Выход 0.29 г (86%). LC/MS анализ: m/z [M+H] + = 338.
Любые из фармакологически приемлемых солевых форм Б вещества I получаются стандартной процедурой либо на завершающей стадии синтеза (удаления NH2-защитной группы), как это показано на Примерах 1-3, либо любым легко осуществимым и стандартным способом (Пример 5), смешением эквимолярных количеств любой соответствующей кислоты, ее сольвата или гидрата и аминосодержащего вещества (в данном случае, вещества I) или солевым обменом с использованием хорошо известных физико-химических методов, например, ионообменная хроматография.
Пример 5.
Получение вещества I в виде соли аминокислоты - глицината.
К раствору вещества I в виде свободного основания (0.34 г, 1 ммоль) в этиловом спирте (5 мл) добавляли раствор глицина (0.076 г) в воде (1 мл). Смесь перемешивали 1 час, растворитель упаривали. Остаток сушили в вакууме. Получили вещество I в виде соли глицината - 0.42 г. LC/MS анализ: m/z [M+H] + = 338.
Высокая ДПП-4 ингибирующая активность вещества I в солевых и бессолевой формах экспериментально подтверждена в экспериментах in vitro в сравнении с сертифицированными (Sigma-Aldrich) образцами коммерческих ингибиторов- фармацевтических композиций - вилда-, сита-, ало- и линаглиптинов. Результаты получены с помощью спектрофотометрического метода, а также программ Excel и GraphPad Prism 8, в сравнении с диапазонами допустимых значений IC50, известных для коммерческих ингибиторов из литературных данных. В экспериментах использовали рекомбинантный фермент ДПП-4 D4943 (Sigma), субстрат Gly-Pro-pNA и буферную систему, содержащую 50 мМ Tris-HCl, 50 мМ NaCl, 0.01% Triton, pH=7.6. Каждое вещество проанализировано в диапазоне концентраций от 10-4 до 10-11 М. Все полученные значения показателей IC50 вычислены по показателям оптической плотности через 30 минут после начала инкубации при 37оС. Результаты оценки IC50 приведены в Таблице 1.
Таблица 1. Сравнение полученных и литературных данных для коммерческих фармацевтических композиций - ингибиторов ДПП-4 с результатами, полученными для вещества (I) в бессолевой и солевых формах.
Для лечения сахарного диабета 2 типа нет ограничений для видов фармацевтических композиций. Согласно данному изобретению выбран наиболее предпочтительный способ приема для лечения сахарного диабета - пероральный.
Настоящее изобретение включает в себя описание фармацевтических композиций в виде следующих лекарственных форм - таблетки, гранулы, гранулы или пеллеты в виде твердых желатиновых капсул.
Поскольку варианты осуществления изобретения описаны в тексте, настоящее изобретение не ограничивается ими, поскольку, как представляют себе специалисты, могут быть сделаны многие модификации или вариации. Такие модификации или вариации, не рассмотренные здесь в деталях, считаются очевидными эквивалентами настоящего изобретения.
Фармацевтическая композиция для лечения сахарного диабета согласно данному изобретению включает в себя активный компонент вещество I в виде свободного основания и/или в виде фармакологически приемлемой солевой формы из числа перечисленных выше, фармакологически допустимые носители и добавочные компоненты, вспомогательные вещества, связующие вещества, смазывающие вещества, покрывающие вещества, водные растворители, солюбилизирующие вещества, которые обеспечивают получение приемлемой фармацевтической композиции, отвечающей требованиям Государственной фармакопеи.
Способ приема фармацевтической композиции для лечения сахарного диабета согласно данному изобретению, представлен лекарственными формами для внутреннего применения (пероральным), но не ограничивается этим. Предпочтительный прием - пероральный.
Дозировка состава для лечения сахарного диабета согласно настоящему
изобретению соответствующим образом определена на основе состояния больного, тяжести симптомов, возраста, способа приема и результатов диагностики врачами. Предпочтительными в случае перорального применения являются дозировки от 1 мг до 50 мг вещества I в день, наиболее предпочтительно 5 мг. Вышеуказанная доза согласно данному изобретению принимается пациентом единоразово или разделяется на несколько доз на период от 2-4 до 8-12 часов, в зависимости от тяжести симптомов и диагноза врача.
Препарат изготавливают в виде следующих лекарственных форм - таблеток, гранул, гранул или пеллет в виде твердых желатиновых капсул, обеспечивающих немедленное (Примеры 6-8), модифицированное (примеры 9-14) и пролонгированное (Примеры 15-18) действие.
Пример 6.
Получение фармацевтической композиции в виде таблетки с немедленным высвобождением активного компонента (вещества I) на основе вещества I в бессолевой форме (свободного основания; получение - Пример 4).
Пероральная фармацевтическая композиция, представляющая таблетку, покрытую пленочной оболочкой, содержащую вещество I в бессолевой форме (свободное основание) в дозировке 5 мг в соотношении от 5 до 94% от общей массы таблетки при следующем соотношении ингредиентов на 100 мг, масс %, указанных в таблце 2.
Таблица 2
Способ получения лекарственного средства, изготовленного на основе фармацевтической композиции, содержащей вещество I в бессолевой форме (свободное основание) в качестве единственного действующего вещества в дозировке от 1 и 50 мг (в пересчете на основание) в соотношении от 5 до 94% от общей массы таблетки, при осуществлении которого просеивают магния стеарат, вещество I в бессолевой форме (свободное основание), целлюлозу микрокристаллическую (тип 21 или аналогичная), целлюлозу 2-гидроксипропиловый эфир (низкозамещённый, тип LH-21 или аналогичная) и кремния диоксид коллоидный (тип А 200 или аналогичный), готовят смесь из вещества I в бессолевой форме (свободное основание), целлюлозы микрокристаллической, целлюлозы 2-гидроксипропиловый эфир (низкозамещённую) (25-60%) и кремния диоксида коллоидного, магния стеарата (35-65%), перемешивают компоненты до однородного состояния (контроль визуальный) 3-24 минут при скорости перемешивания 7-25 об/мин, переносят в устройство для компактирования и полученную смесь подвергают сухой грануляции (компактированию) и полученный гранулят просеивают через сито 0,71 мм и 0,25 мм, при этом, фракцию выше 0,71 мм и ниже 0,25 мм смешивают и компактируют повторно до тех пор, пока количество мелкой фракции составит не более 10-15%, причем параметры перемешивания зависят от объема лабораторной серии, скорость роликов составляет не более 2-6 об/мин, давление - не более 37,0 бар, скорость мельницы - не менее 56 и не более 70,0 об/мин, диаметр ячеек сита не более 1,0 мм, далее в смеситель загружают полученный гранулят, оставшуюся часть целлюлозы 2-гидроксипропиловый эфир (низкозамещённую тип LH-21 или аналогичная), оставшуюся часть кремния диоксида коллоидного (тип А 200 или аналогичный) и проводят перемешивание компонентов до однородного состояния (контроль визуальный) 3-24 минут при скорости перемешивания 7-25 об/мин, помещают в устройство для компактирования, после чего в смеситель загружают оставшуюся часть магния стеарата и перемешивают компоненты до однородного состояния (контроль визуальный), параметры перемешивания те же, далее, смесь для таблетирования выгружают в емкость, отбирают репрезентативную пробу для проведения контроля, процесс таблетирования осуществляют на таблеточном прессе, для чего используют пресс-инструмент с геометрией 8,00-13,00 мм, круглой, овальной формы (с риской или без), после настройки средней массы таблеток осуществляют настройку таблеточного пресса по таким показателям, как твердость - 80-100 Н; истираемость - не более 0,5%; распадаемость - не более 1 минуты; отклонения от средней массы не должны превышать 5,0 %, затем осуществляют процесс нанесения оболочки в псевдоожиженном слое, где в процессе нанесения пленочной оболочки используют 18-22% пленочной суспензии для нанесения покрытия, следующего состава: гипромеллозы, титана диоксида, полидекстрозы, талька, полиэтиленгликоля, перед осуществлением процесса нанесения оболочки нагревают таблетки до 35-41°С при периодическом перемешивании в барабане коатера, по достижении температуры таблеток начинают распыление пленочной суспензии, в течение процесса контролируют внешний вид таблеток и привес оболочки, процесс нанесения оболочки осуществляют до получения средней заданной массы таблеток (± 5,0%), при достижении заданной массы таблеток процесс останавливают и охлаждают таблетки до температуры не выше 33°С, после окончания процесса емкости закрывают.
Полученная фармацевтическая композиция, как лекарственная форма обеспечивает профиль немедленного высвобождения, указанный в таблице 3.
Таблица 3
Пример 7.
Получение таблетки с немедленным высвобождением активного компонента (вещества I) на основе вещества I в виде соли гидрохлорида (получение - Пример 3 или 5).
Пероральная фармацевтические композиции, представляющая таблетку, покрытую пленочной оболочкой, содержащую вещество I в виде соли гидрохлорида была приготовлена аналогично процедуре, описанной в Примере 6, с пересчетом загрузки солевой формы гидрохлорид на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 2.
Пример 8.
Получение таблетки с немедленным высвобождением активного компонента (вещества I) на основе вещества I в виде соли тозилата (получение - Пример 2 или 5).
Пероральная фармацевтические композиции, представляющая таблетку, покрытую пленочной оболочкой, содержащую вещество I в виде соли тозилата была приготовлена аналогично процедуре, описанной в Примере 6, с пересчетом загрузки солевой формы тозилат на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 2.
Гранулы с модифицированным высвобождением готовятся по следующим методикам (Примеры 9-11 и/или 12-14).
Пример 9.
Получение гранул с модифицированным высвобождением активного компонента (вещества I) на основе вещества I в бессолевой форме (свободного основания; получение - Пример 4).
Пероральная фармацевтическая композиция, представляющая капсулу, содержащую гранулы, содержащие вещество I в бессолевой форме (свободное основание) в дозировке 5 мг на 1 капсулу (в пересчете на основание) в соотношении от 5 до 94% от общей массы смеси при следующем соотношении ингредиентов на 100 мг, масс %, указанных в таблице 4.
Таблица 4.
Способ получения фармацевтической композиции, содержащей вещество I в бессолевой форме (свободное основание) в качестве единственного действующего вещества в дозировке 5 мг на 1 капсулу (в пересчете на основание) в соотношении от 5 до 94% от общей массы смеси, при осуществлении которого просеивают вещество I в бессолевой форме (свободное основание), лактозы моногидрат 200, гипромеллозу (например тип 2208), повидон-К30 на сите с размером отверстий 0,25-0,5 мм. При этом в капсуле находятся гранулы, покрытые кишечнорастворимой оболочкой и оболочкой, обеспечивающей замедленное высвобождение. Соотношение пеллет колеблется от 5:90 до 90:5.
Далее готовят раствор увлажнителя на основе повидона-К30. Перемешивание проводят до полного растворения (контроль визуальный). В полученный раствор добавляют расчетное количество субстанции.
В смеситель загружают вещество I в бессолевой форме (свободное основание), гипромеллозу (например тип 2208), лактозы моногидрат и крахмал кукурузный. Проводят перемешивание смеси порошков.
Далее смесь порошков переносят в установку для получения гранул путем псевдоожижения. Гранулы получают путем распыления увлажняющей смеси, в которой предварительно растворили вещество I в бессолевой форме (свободное основание) в псевдоожиженном слое.
Полученный гранулят калибруют через сито 0,25-0,5 мм. Фракции гранул выше 0,5 мм и ниже 0,25 мм считаются некондиционной продукцией.
На следующем этапе проводят покрытие полученных гранул кишечнорастворимой оболочкой. В воду очищенную вносят гипромеллозу (например, тип 2208) перемешивают, после этого добавляют полисорбат-80. Все действия проводят при температуре 40-55 градусов. Далее добавляют расчетное количество талька и опять тщательно перемешивают в течение 5-15 минут. После этого добавляют пленочное покрытие нейтрального сополимера на основе этилакрилата и метилметакрилата (например EUDRAGIT NM 30 D / NE 30 D), перемешивают 5 минут и подвергают фильтрации через сита с размером пор 0,5 мм.
Далее проводят процесс нанесения оболочки на установке псевдоожиженного слоя. Перед проведением процесса нанесения оболочки гранулы прогревают до 35-37°С при периодическом перемешивании в кипящем слое.
По достижении заданной температуры гранул начинают распыление пленочной суспензии. В процессе контролируют внешний вид гранул.
Параметры процесса: температура продукта 27-37°С.
После полного расхода суспензии процесс останавливают и охлаждают гранулы до температуры не выше 33°С. После окончания процесса емкости закрывают. Проводят репрезентативный отбор гранул для проведения промежуточного контроля, согласно спецификации.
Далее проводят технологические операции по нанесению второго типа покрытия на гранулы.
Используют водный раствор метакриловой кислоты и этилакрилата, талька, диоксида титана, оксида железа (желтый), диоксида кремния, натрий двууглекислого и лаурилсульфат натрия (например Acryl-EZE). Суспензию перемешивают до однородного состояния (контроль визуальный).
Далее проводят процесс нанесения оболочки на установке псевдоожиженного слоя. Перед проведением процесса нанесения оболочки гранулы нагревают до 35-37°С при периодическом перемешивании в кипящем слое.
По достижении заданной температуры гранул начинают распыление пленочной суспензии. В процессе контролируют внешний вид пеллет.
Параметры процесса: температура продукта 27-37°С
После полного расхода суспензии процесс останавливают и охлаждают гранулы до температуры не выше 33°С. После окончания процесса, емкости закрывают. Проводят репрезентативный отбор гранул для проведения промежуточного контроля, согласно спецификации.
Далее проводят капсулирование полученных гранул, исходя из расчета, что соотношение пеллет колеблется от 5:90 до 90:5. При этом дозировка действующего вещества I в 1 капсуле должна составлять 5 мг.
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, указанный в таблице 5.
Таблица 5.
Пример 10.
Получение гранул на основе вещества I в виде соли гидрохлорида (получение - Пример 3) с модифицированным высвобождением активного компонента (вещества I).
Пероральная фармацевтическая композиция, представляющая собой гранулы для внутреннего применения, содержащие вещество I в виде соли гидрохлорида была приготовлена аналогично процедуре, описанной в Примере 9, с пересчетом загрузки солевой формы гидрохлорид на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 3.
Пример 11.
Получение гранул на основе вещества I в виде соли тозилата (получение - Пример 2) с модифицированным высвобождением активного компонента (вещества I).
Пероральная фармацевтическая композиция, представляющая собой гранулы для внутреннего применения, содержащие вещество I в виде соли тозилата была приготовлена аналогично процедуре, описанной в Примере 9, с пересчетом загрузки солевой формы гидрохлорид на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 5.
Гранулы в виде твердых желатиновых капсул с модифицированным высвобождением готовятся по следующей методике.
Пример 12.
Получение гранул в виде твердых желатиновых капсул с модифицированным высвобождением активного компонента (вещества I) на основе вещества I в бессолевой форме (свободного основания; получение - Пример 4).
Пероральная фармацевтическая композиция, представляющая собой гранулы в виде твердых желатиновых капсул для внутреннего применения, содержащие вещество I в бессолевой форме в дозировке 5 мг на 1 дозу в соотношении от 5 до 94% от общей массы смеси при следующем соотношении ингредиентов на 100 мг, масс %, указанным в таблице 6.
Таблица 6.
Способ получения фармацевтической композиции, содержащей вещество I в бессолевой форме (свободное основание) в качестве единственного действующего вещества в дозировке 5 мг на 1 дозу (в пересчете на основание) в соотношении от 5 до 94% от общей массы смеси, при осуществлении которого просеивают вещество I в бессолевой форме (свободное основание), лактозы моногидрат 200, гипромеллозу (например тип 2208), на сите с размером отверстий 0,25-0,5 мм. Соотношение гранул в 1 дозе на прием колеблется от 5:90 до 90:5.
Далее готовят раствор увлажнителя на основе гипромеллозы. В части воды (1/3) проводят набухание полимера, после чего добавляют оставшуюся часть(2/3) и с помощью механической мешалки проводят гомогенизацию. В полученный раствор добавляют расчетное количество субстанции. Качество растворения и набухания полимера оценивают после приготовления раствора (контроль визуальный). В смеситель загружают лактозу моногидрат и крахмал кукурузный. Проводят перемешивание смеси порошков.
Далее смесь порошков переносят в установку для получения гранул путем псевдоожижения. Гранулы получают путем распыления увлажняющей смеси, в которой предварительно растворили вещество I в бессолевой форме (свободное основание) в псевдоожиженном слое.
Полученный влажный гранулят калибруют через сито 0,25-0,5 мм. Фракции гранул выше 0,5 мм и ниже 0,25 мм считаются некондиционной продукцией.
На следующем этапе проводят покрытие полученных гранул пленочным покрытием из привитого сополимера макрогола и поливинилового спирта (например, Kollicoat IR). В воду очищенную вносят расчетное количество (1,5-9,8%) Kollicoat IR и проводят перемешивание 5 минут и подвергают фильтрации через сита с размером пор 0,5 мм.
Далее проводят процесс нанесения оболочки на установке псевдоожиженного слоя. Перед проведением процесса нанесения оболочки гранулы прогревают до 35-37°С при периодическом перемешивании в кипящем слое.
По достижении заданной температуры гранул начинают распыление пленочной суспензии. В процессе контролируют внешний вид гранул.
Параметры процесса: температура продукта 27-37°С.
После полного расхода суспензии процесс останавливают и охлаждают гранулы до температуры не выше 33°С. После окончания процесса емкости закрывают. Проводят репрезентативный отбор гранул для проведения промежуточного контроля, согласно спецификации.
Далее проводят технологические операции по нанесению второго типа покрытия на гранулы.
Используют водный раствор сополимера метакриловой кислоты и этилакрилат (1: 1) (например Vivacoat E). Для приготовления суспензии полимера используют верхнеприводную мешалку. Отмеренное количество воды помещают в стакан и включают мешалку таким образом, чтобы образовался конус. Затем в образовавшийся конус постепенно высыпают покрытие. После чего продолжают перемешивание еще 45 минут. После этого готовый раствор фильтруют через сито с размером отверстий 0,5 мм.
Далее проводят процесс нанесения оболочки на установке псевдоожиженного слоя. Перед проведением процесса нанесения оболочки гранулы нагревают до 35-37°С при периодическом перемешивании в кипящем слое.
По достижении заданной температуры гранул начинают распыление пленочной суспензии. В процессе контролируют внешний вид пеллет.
Параметры процесса: температура продукта 27-37°С
После полного расхода суспензии процесс останавливают и охлаждают гранулы до температуры не выше 33°С. После окончания процесса, емкости закрывают. Проводят репрезентативный отбор гранул для проведения промежуточного контроля, согласно спецификации.
Далее проводят дозирование полученных гранул, исходя из расчета, что соотношение гранул колеблется от 5:90 до 90:5. При этом дозировка действующего веществ в 1 прием должна составлять 5 мг.
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, указанный в таблице 7.
Таблица 7.
Пример 13.
Получение гранул в виде твердых желатиновых капсул на основе вещества I в виде соли гидрохлорида (получение - Пример 3) с модифицированным высвобождением активного компонента (вещества I).
Пероральная фармацевтическая композиция, представляющая собой гранулы для внутреннего применения, содержащие вещество I в виде соли гидрохлорида была приготовлена аналогично процедуре, описанной в Примере 12, с пересчетом загрузки солевой формы гидрохлорид на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 4.
Пример 14.
Получение гранул на основе вещества I в виде соли тозилата (получение - Пример 2) с модифицированным высвобождением активного компонента (вещества I).
Пероральная фармацевтическая композиция, представляющая собой гранулы для внутреннего применения, содержащие вещество I в виде соли тозилата была приготовлена аналогично процедуре, описанной в Примере 12, с пересчетом загрузки солевой формы гидрохлорид на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 4.
Получение пеллет пролонгированного высвобождения проводят по следующей методике.
Пример 15. Получение пеллет пролонгированного высвобождения активного компонента (вещества I) на основе вещества I в бессолевой форме (свободного основания; получение - Пример 4).
Пероральная фармацевтическая композиция, представляющая капсулу, содержащую пеллеты, содержащие вещество I в бессолевой форме в дозировке 5 мг на 1 капсулу в соотношении от 5 до 94% от общей массы смеси. Соотношения указаны в таблице 8.
Таблица 8
Способ получения фармацевтической композиции, изготовленной на основе фармацевтической композиции, содержащей вещество I в бессолевой форме (свободное основание) в качестве единственного действующего вещества в дозировке 5 мг на 1 капсулу в соотношении от 5 до 94% от общей массы смеси, при осуществлении которого просеивают вещество I в бессолевой форме (свободное основание), гипромеллозу (например тип 2208) на сите с размером отверстий 0,25-0,5 мм. При этом в капсуле находятся пеллеты, покрытые рН-независимой оболочкой.
Далее готовят раствор увлажнителя на основе гипромеллозы. В части воды (1/3) проводят набухание полимера, после чего добавляют оставшуюся часть (2/3) и с помощью механической мешалки проводят гомогенизацию. В полученный раствор добавляют расчетное количество субстанции. Качество растворения и набухания полимера оценивают после приготовления раствора (контроль визуальный).
Далее в установку для получения пеллет путем псевдоожижения помещают расчетное количество пеллет с размером фракции 0,25-0,5 мм (20-90%) пеллеты прогревают до температуры 40-45 градусов. Далее проводят распыление смеси гипромеллозы с веществом I в бессолевой форме (свободное основание). Скорость распыления составляет 12-20 г/мин. Температура входного воздушного потока составляет 60-65 градусов. Процесс распыления продолжают до полного израсходования раствора пленкообразователя. После этого продолжают сушку пеллет 10-15 минут, в токе теплого воздуха. После чего проводят выгрузку полученных пеллет.
Полученные пеллеты калибруют через сито 0,25-0,5 мм. Фракции гранул выше 0,5 мм и ниже 0,25 мм считаются некондиционной продукцией.
Проводят репрезентативный отбор пеллет для проведения промежуточного контроля, согласно спецификации.
Далее проводят капсулирование полученных пеллет, исходя из расчета, что действующего веществ в 1 капсуле должна быть 5 мг.
Полученная фармацевтическая композиция обеспечивает пролонгированный профиль высвобождения, указанный в таблице 9.
Таблица 9
Пример 16.
Получение гранул пролонгированного высвобождения активного компонента (вещества I) на основе вещества I в солевой форме гидрохлорид (получение - Пример 3).
Пероральная фармацевтическая композиция, представляющая собой гранулы для внутреннего применения, содержащие вещество I в виде соли гидрохлорида была приготовлена аналогично процедуре, описанной в Примере 15, с пересчетом загрузки солевой формы гидрохлорид на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 5.
Пример 17.
Получение пеллет пролонгированного высвобождения активного компонента (вещества I) на основе вещества I в солевой форме тозилат (получение - Пример 2).
Пероральная фармацевтическая композиция, представляющая собой пеллеты для внутреннего применения, содержащие вещество I в виде соли тозилат была приготовлена аналогично процедуре, описанной в Примере 15, с пересчетом загрузки солевой формы гидрохлорид на свободное основание так, чтобы обеспечивалась та же дозировка вещества I (5 мг).
Полученная фармацевтическая композиция обеспечивает профиль высвобождения, аналогичный указанному в таблице 9.
Другая цель изобретения, состоящая в применении разработанных композиций и ГЛФ в качестве сахароснижающего средства для предупреждения развития и лечения сахарного диабета 2 типа, была достигнута изучением эффектов и показателей в экспериментах in vivo на крысах. Соблюдались этические правила гуманного обращения с животными, изложенные в директивах Совета Европейского сообщества 86/609/ЕЕС.
Тесты по выявлению эффектов вещества I в составе ГЛФ проводили в экспериментах in vivo с определением основных показателей:
уровень гликозилированного гемоглобина (HbA1c)
уровень глюкозы
динамика массы тела
Показатели уровней гликозилированного гемоглобина (HbA1c) и глюкозы, а также динамику массы тела животных оценивали в экспериментах с применением композиции, представленной в Примере 7.
Показатель острая токсичность оценивали в экспериментах с применением вещества I в виде солевой формы гидрохлорид.
Техническим результатом изобретения является сахароснижающий эффект и положительная динамика массы тела у животных на выбранных моделях крыс - стрептозотоцин-индуцированного (STZ) сахарного диабета и модели сахарной нагрузки. Другим результатом является низкая токсичность при пероральном введении (4-й класс токсичности), выявленная в экспериментах по острой токсичности на крысах.
Полученные результаты иллюстрируются следующими примерами.
Пример 18.
Эффект вещества I в составе композиции, описанной в Примере 7, на модели STZ-индуцированного сахарного диабета у крыс.
В эксперименте на модели STZ-индуцированного сахарного диабета использовали один вид животных: самцы белых аутбредных крыс CD.
При составлении плана и дизайна экспериментов учитывали, что вероятность индуцирования диабета у крыс с помощью STZ не всегда является 100 %-ной. По этой причине, из включенных в эксперимент 60-ти самцов крыс, 50-ти животным был введен STZ, а 10 животных составляли контрольную группу (здоровый контроль).
Диабет индуцировали однократным внутрибрюшинным введением 60 мг/кг стрептозотоцина (растворенного в 0,1М натрий-цитратном буфере). Контрольной группе (Здоровый контроль) вводили равное количество натрий-цитратного буфера.
После введения стрептозотоцина за животными наблюдали 96 часов. Индуцированный диабет подтверждали путем измерения уровня глюкозы в крови с помощью глюкометра Акку-Чек Актив. Крыс с уровнем глюкозы в крови более 15 мМ/л включали в исследование.
Животных с подтвержденным диабетом распределяли по трем группам случайным образом, используя в качестве критерия массу тела.
Исследуемый препарат, композиция, описанная в Примере 7, для перорального введения готовили ежедневно в начале исследования на 1% крахмале, в концентрациях, обеспечивающих дозировку вещества I (в пересчете на свободное основание) в дозах 5 и 20 мг/кг и вводили в первой половине дня ежедневно после индукции STZ перорально, так как данный путь введения соответствует клиническому. Животным группы диабетического и здорового контроля в качестве плацебо вводили 1% крахмал.
Оценку специфической фармакологической активности вещества I в составе композиции проводили по уровню гликозилированного гемоглобина (HbA1c). На шестидесятые сутки проводили плановый забой животных с забором крови на анализ HbA1c. Уровень гликозилированного гемоглобина в группах при введении Композиции, описанной в Примере 7, в дозах 5 мг/кг и 20 мг/кг статистически значимо уменьшался по сравнению с группой контрольных животных (группа диабета) (Таблица 10).
Таблица 10. Уровень гликозилированного гемоглобина в модели STZ- диабета (M±SD).
Еженедельно измеряли уровень сахара в крови животных. Начиная с сорок девятых суток, в экспериментальных группах уровень сахара в крови статистически отличается от группы диабета (p<0,05). (Таблица 11)
Таблица 11. Динамика уровня сахара в крови у крыс в модели STZ- диабета (M±SD).
Одновременно проводили наблюдение за изменением массы животных. Начиная с седьмого дня после индукции STZ диабета, наблюдали уменьшение массы животных в опытных группах и группе диабетического контроля по сравнению с группой здорового контроля. На седьмые сутки индукции STZ диабета животные во всех группах отставали в динамике набора веса. На тридцать пятые сутки животные экспериментальных групп набирали вес быстрее по сравнению с группой диабетического контроля (p<0,05). Положительная динамика набора веса продолжилась на протяжении всего исследования. (Таблица 12).
Таблица 12. Динамика веса крыс в модели STZ- диабета (M±SD).
±16,3
Пример 19.
Эффект препарата - композиции, описанной в Примере 7, на модели сахарной нагрузки.
В эксперименте на модели сахарной нагрузки использовали один вид животных: самцы белых аутбредных крыс CD.
Сахарную нагрузку вызывали пероральным введением 5 г глюкозы на крысу. Глюкозу готовили на 10% крахмале. Контрольной группе (Здоровый контроль) вводили равное количество 10% крахмала.
После введения глюкозы за животными наблюдали 4 часа. Глюкозу измеряли глюкометром Акку-Чек Актив через временные интервалы 0,5, 1, 1,5 2, 3 и 4 часа после сахарной нагрузки.
Исследуемая композиция, описанная в Примере 7, и препарат сравнения вилдаглиптин для перорального введения готовили в начале исследования на 10% крахмале, в концентрациях, обеспечивающих дозировку исследуемого препарата в дозе 20 мг/кг. Препараты вводили за полчаса до сахарной нагрузки в объеме 1 мл/жив. Животным группы диабетического и здорового контроля в качестве плацебо вводили физраствор.
Оценку специфической фармакологической активности проводили по уровню глюкозы в крови. Глюкозу определяли глюкометром Акку-Чек Актив.
Начиная с 30 минут исследования препарат, композиция, описанная в Примере 7, в дозе активного вещества I 20 мг/кг статистически значимо снижает уровень сахара в крови по сравнению с контролем. (Таблица 13).
Таблица 13. Уровень сахара в крови при сахарной нагрузке (M±SD).
В модели STZ-диабета доза 20 мг/кг эффективнее дозы 5 мг/кг по показателям сахара в крови, массе тела и уровня гликозилированного гемоглобина. Уровень сахара в группах при введении Композиции, описанной в Примере 7 в дозах 5 мг/кг и 20 мг/кг статистически значимо уменьшался по сравнению с группой контрольных животных (группа диабета).
На модели сахарной нагрузки препарат - композиция, описанная в Примере 7, показал активность выше, чем вилдаглиптин.
В экспериментах, приведенных в Примерах 18 и 19, с использованием композиции, описанной в Примере 7, в дозах активного вещества I 5 и 20 мг/кг, токсического действия выявлено не было - гибели животных не зафиксировано.
Токсическое действие дополнительно исследовали в экспериментах по острой токсичности при однократном введении крысам больших доз вещества I в виде соли гидрохлорид.
Пример 20.
Исследование токсичности (острой) при однократном введении препарата, вещества I в виде соли гидрохлорид.
Исследования токсичности при однократном введении проводятся с целью качественной и количественной оценки токсических реакций, возникающих после однократного введения (обычно перорально или внутривенно) большой дозы вещества. Исследования обычно проводятся на грызунах. Крыса является предпочтительным для исследований видом.
Исследования токсичности при однократном введении препарата, вещества I в виде соли гидрохлорид проводили при пероральном введении на аутбредных крысах обоих полов. Препарат вводился однократно перорально аутбредным крысам CD в дозах 500, 1000 и 2000 мг/кг. В первые часы после введения наблюдались явные признаки интоксикации: снижение мышечного тонуса, снижение спонтанной двигательной активности, развитие общей депрессии. Гибели не зафиксировано (Таблица 10), поэтому расчет полулетальных доз оказался невозможен, LD50>2000 мг/кг. Согласно классификации токсичности по Hodge и Sterner (1943) разработанная фармацевтическая композиция, содержащая вещество I, при пероральном введении на крысах относится к 4 классу токсичности (малотоксичные).
Таблица 14. Результаты эксперимента по острой токсичности на крысах (самцы и самки) CD после однократного перорального введения исследуемой фармацевтической композиции как лекарственного средства, вещества I в виде соли гидрохлорид.
Гиб/жив
Измеренные биохимические показатели и показатели гематологии не отличались достоверно между опытными и контрольной группами и колебались в пределах физиологической нормы (Таблицы 15 и 16).
Таблица 15. Усредненные гематологические показатели крыс CD.
Таблица 16. Усредненные биохимические показатели крыс CD.
Таким образом, задачи настоящего изобретения решены за счет разработки фармацевтических композиций и готовых лекарственных форм для перорального применения, содержащих неактивные вспомогательные вещества и носители, обеспечивающие немедленное и/или пролонгированное, и/или модифицированное высвобождение активного компонента вещества I - высокоэффективного ингибитора дипептидилпептидазы-4, малотоксичного (4 класс токсичности) и проявляющего активное сахароснижающее действие, для предупреждения развития и лечения сахарного диабета 2 типа. Подтверждено примерами.
Промышленная применимость
Все приведенные примеры подтверждают промышленную применимость данного изобретения.
Список сокращений.
СД - сахарный диабет
ДПП-4 - дипептидилпептидаза-4
DCM - дихлорметан (хлористый метилен)
МеОН - метанол
EtOH - этанол
CHCl3 - хлороформ
п-ТСК - пара-толуолсульфокислота
HCl - хлористый водород
Et2O - диэтиловый эфир
Вос2О - ди-трет-бутилдикарбонат
TFA - трифторуксусная кислота
NaHCO3 - натрия гидрокарбонат
TEA - триэтиламин
DIPEA - диизопропилэтиламин
Na2SO4 - натрия сульфат
ВОР - бензотриазол-1-илокси-трис(диметиламино)фосфоний гексафторфосфат
LC/MS - высокоэффективная жидкостная хроматография с масс-детекцией
DMSO - диметилсульфоксид
TFAA - ангидрид трифторуксусной кислоты
THF - тетрагидрофуран
HbA1c - гликозилированный гемоглобин.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ ПРОТИВОДИАБЕТИЧЕСКАЯ КОМПОЗИЦИЯ ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ | 2011 |
|
RU2465896C2 |
| НОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ТАМСУЛОЗИНА И ДУТАСТЕРИДА | 2019 |
|
RU2795928C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ АКОТИАМИДА И ИНГИБИТОРА ПРОТОННОЙ ПОМПЫ | 2019 |
|
RU2820820C2 |
| ЛЕКАРСТВЕННАЯ ФОРМА ЗАМЕДЛЕННОГО ВЫСВОБОЖДЕНИЯ ГЛЮКОЗАМИНА | 2011 |
|
RU2521231C2 |
| СИСТЕМЫ ДОСТАВКИ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ, ВКЛЮЧАЮЩИЕ В СЕБЯ СЛАБООСНОВНЫЕ ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА И ОРГАНИЧЕСКИЕ КИСЛОТЫ | 2009 |
|
RU2504362C2 |
| СПОСОБ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ОСЛОЖНЕНИЙ САХАРНОГО ДИАБЕТА, СВЯЗАННЫХ С РАЗВИТИЕМ ДЕГЕНЕРАТИВНЫХ ПРОЦЕССОВ В НЕРВНОЙ ТКАНИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ НЕЙРОПРОТЕКТИВНОЙ ТЕРАПИИ ТАКИХ ОСЛОЖНЕНИЙ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2012 |
|
RU2484844C1 |
| КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ СЛАБООСНОВНЫЕ ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА, И ЛЕКАРСТВЕННЫЕ ФОРМЫ С КОНТРОЛИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ | 2009 |
|
RU2548748C2 |
| ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ-4 ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА | 2015 |
|
RU2628573C2 |
| Способ лечения гипергликемии | 2017 |
|
RU2739255C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2010 |
|
RU2532330C2 |
Изобретение относится к медицине и фармацевтической промышленности, а именно к композициям, предназначенным для перорального приема, в целях предупреждения развития и лечения сахарного диабета 2 типа. Сущность изобретения заключается в том, что созданная фармацевтическая композиция содержат активный компонент антигипергликемического действия - высокоэффективный ингибитор дипептидилпептидазы-4 (ДПП-4), экзо-(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил - вещество I в виде фармацевтически приемлемых солевых форм, вспомогательные вещества и носители, обеспечивающие немедленное, пролонгированное и модифицированное высвобождение активного компонента для лечения больных сахарным диабетом 2 типа.
Мол. масса 337
Композиции включают также неактивные компоненты, с одной стороны, сохраняющие эффективность действующего вещества, а с другой - позволяющие варьировать дозировку, высвобождение и продолжительность его действия в организме. 12 з.п. ф-лы, 20 примеров, 16 таблиц, 2 фиг.
1. Фармацевтическая композиция для лечения сахарного диабета 2 типа и сопутствующих состояний, в патогенез которых вовлечен фермент ДПП-4, профилактики, предупреждения или лечения различных нарушений, в том числе, связанных с нарушениями массы тела при сахарном диабете, а также заживления, рубцевания ран, диабетической стопы на основе активного компонента высокоэффективного ингибитора дипептидилпептидазы-4 вещества I, экзо-(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрила, содержащие фармацевтически приемлемые носители, при этом активный компонент имеет следующую структуру:

2. Фармацевтическая композиция по п. 1, отличающаяся тем, что действующее вещество I находится в виде свободного основания или фармацевтически приемлемой солевой формы.
3. Фармацевтическая композиция по пп. 1 и 2, отличающаяся тем, что солевые формы действующего вещества I представляют соли с неорганическими кислотами - хлористоводородной кислотой - гидрохлорид, или бромистоводородной кислотой, или азотной кислотой, или серной кислотой, или фосфорной кислотой; или органическими кислотами и их производными из числа сульфо-, или алкилокси-, или гидрокси-, или оксо-замещенных кислот - муравьиной кислотой, или уксусной кислотой, или фумаровой кислотой, или щавелевой кислотой, или винной кислотой, или малеиновой кислотой, или лимонной кислотой, или янтарной кислотой, или яблочной кислотой, или метансульфоновой кислотой, или трифторметансульфоновой кислотой, или бензолсульфоновой кислотой, или пара-толуолсульфоновой кислотой (тозилат), или нафтилсульфоновой кислотой, или сульфосалициловой кислотой, или молочной кислотой, или виноградной кислотой, или адипиновой кислотой, или глюконовой кислотой, или глюкогептоновой кислотой, или глюкуроновой кислотой, или терефталевой кислотой, или гиппуровой кислотой, или 1,2-этандисульфоновой кислотой, или изэтионовой кислотой, или лактобионовой кислотой, или олеиновой кислотой, или памовой кислотой, или полигалактуроновой кислотой, или стеариновой кислотой, или дубильной кислотой; или аминокислотами из числа всех известных природных аминокислот - глицин, или аланин, или валин, или лейцин, или изолейцин, или пролин, или цистеин, или метионин, или фенилаланин, или триптофан, или серин, или треонин, или глютамин, или тирозин, или аспарагин, или гистидин, или лизин, или аргинин, или аспарагиновая кислота, или аспарагиновая кислота.
4. Фармацевтическая композиция по п. 1, отличающаяся тем, что количество действующего вещества в единичной дозе средства составляет: от 1 мг до 50 мг, более предпочтительно 5 мг.
5. Фармацевтическая композиция по п. 1, отличающаяся тем, что для перорального применения она представляет собой одну из следующих лекарственных форм: таблетки, капсулы, содержащие гранулы и, или пеллеты.
6. Фармацевтическая композиция по п. 1, отличающаяся тем, что она дополнительно содержит фармацевтически приемлемые вспомогательные вещества, обеспечивающие немедленное и, или пролонгированное, и, или модифицированное высвобождение активного вещества.
7. Фармацевтическая композиция по пп. 1 и 6, отличающаяся тем, что в качестве вспомогательных веществ используют фармакологически допустимые носители, связующие вещества, смазывающие вещества, покрывающие вещества, водный растворитель, солюбилизирующие вещества.
8. Фармацевтическая композиция по пп. 1, 6 и 7, отличающаяся тем, что в качестве носителей используют целлюлозу микрокристаллическую, целлюлозу 2- гидроксипропиловый эфир низкозамещенный, тип LH-21, лактозу, моногидрат 200, крахмал кукурузный, гипромеллоза, пеллеты с размером фракции 0,25-0,5 мм из микрокристаллической целлюлозы, сахара.
9. Фармацевтическая композиция по пп. 1, 6 и 7, отличающаяся тем, что в качестве связующих веществ используют целлюлозу микрокристаллическую, целлюлозу 2-гидроксипропиловый эфир низкозамещенный, тип LH-21, повидон-К30.
10. Фармацевтическая композиция по пп. 1, 6 и 7, отличающаяся тем, что в качестве смазывающего вещества используют магния стеарат, тальк.
11. Фармацевтическая композиция по пп. 1, 6 и 7, отличающаяся тем, что в качестве покрывающего вещества используют смесь компонентов для нанесения покрытия, следующего состава: гипромеллозы, титана диоксида, полидекстрозы, талька, полиэтиленгликоля; водный раствор метакриловой кислоты и этилакрилата, талька, диоксида титана, оксида железа (желтый), диоксида кремния, натрий двууглекислого и лаурилсульфат натрия, пленочное покрытие нейтрального сополимера на основе этилакрилата и метилметакрилата, водный раствор сополимера метакриловой кислоты и этилакрилат (1: 1), пленочное покрытие из привитого сополимера макрогола и поливинилового спирта, поливинилацетат.
12. Фармацевтическая композиция по пп. 1, 6 и 7, отличающаяся тем, что в качестве растворителя используют воду очищенную.
13. Фармацевтическая композиция по пп. 1, 6 и 7, отличающаяся тем, что в качестве солюбилизирующего вещества используют твин-80.
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV НА ОСНОВЕ КОНДЕНСИРОВАННЫХ ЦИКЛОПРОПИЛПИРРОЛИДИНОВ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2286986C2 |
| НОВЫЙ СОСТАВ | 2006 |
|
RU2483716C2 |
| WO 2017186934 A2, 02.11.2017 | |||
| WO 2015104602 A2, 2015.07.16 | |||
| ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ-4 ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА, СОЕДИНЕНИЯ (ВАРИАНТЫ) | 2018 |
|
RU2712097C1 |

