Настоящее изобретение относится к ингибиторам дипептидилпептидазы IV (DP-4) на основе конденсированных циклопропилпирролидинов и к способу лечения диабета, в частности диабета типа II, а также гипергликемии, синдрома X, диабетических осложнений, гиперинсулинемии, ожирения, атеросклероза и родственных заболеваний, а также различных иммуномодуляторных заболеваний и хронического воспалительного заболевания кишечника, использующему конденсированные циклопропилпирролидины, одни или в комбинации с антидиабетическим агентом другого типа и/или терапевтическим агентом другого типа.
Дипептидилпептидаза (IV - 4) представляет собой мембраносвязанную неклассическую сериновую аминопептидазу, которая локализуется в ряде тканей (кишечник, печень, легкие, почки), а также на Т-лимфоцитах крови (где фермент известен как CD-26). Она ответственна за метаболическое расщепление эндогенных пептидов (GLP-1 (7-36), глюкагона) in vivo и проявила протеолитическую активность в отношении ряда других пептидов (GHRH, NPY, GLP-2, VIP) in vitro.
GLP-1 (7-36) представляет собой пептид из 29 аминокислот, образующийся при посттрансляционном процессировании проглюкагона в малом желудочке. GLP-1 (7-36) проявляет различную активность in vivo, включая стимуляцию секреции инсулина, ингибирование секреции глюкагона, промотирование насыщения и замедление опорожнения желудка. Исходя из его физиологического профиля, полагают, что GLP-1 (7-36) оказывает положительное воздействие по предупреждению и лечению диабета типа II и потенциально ожирения. Это положение подтверждается тем, что экзогенное введение GLP-1 (7-36) (непрерывное вливание) диабетическим больным продемонстрировало эффективность в этой группе больных. К сожалению, GLP-1 (7-36) быстро разлагается in vivo и, как было показано, имеет короткий полупериод существования in vivo (t1/2≅1,5 мин). Изучение генетически выведенных мышей DP-4 и in vivo/in vitro испытания с применением селективных ингибиторов DP-4 показали, что DP-4 является первичным ферментом GLP-1 (7-36), распадающимся in vivo. GLP-1 эффективно расщепляется с помощью DP-4 до GLP-1 (9-36), который, как предполагают, ведет себя как физиологический антагонист GLP-1 (7-36). Следовательно, ингибирование DP-4 in vivo увеличивает уровни эндогенного GLP-1 (7-36) и уменьшает образование его антагониста GLP-1 (9-36) и таким образом способствует ослаблению симптомов диабета.
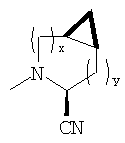
В соответствии с настоящим изобретением созданы соединения ряда конденсированных циклопропилпирролидинов, которые ингибируют DP-4 и имеют структуру I
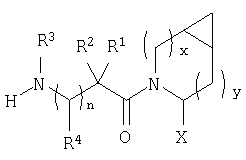
где х обозначает 0 или 1 и у обозначает 0 или 1 (при условии, что
х=1, когда у=0 и
х=0, когда у=1);
n=0 или 1;
Х обозначает Н или CN (т.е. цианогруппа);
R1, R2, R3 и R4 одинаковы или различны и независимо выбираются из Н, алкила, алкенила, алкинила, циклоалкила, бициклоалкила, трициклоалкила, алкилциклоалкила, гидроксиалкила, гидроксиалкилциклоалкила, гидроксициклоалкила, гидроксибициклоалкила, гидрокситрициклоалкила, бициклоалкилалкила, алкилтиоалкила, арилалкилтиоалкила, циклоалкенила, арила, аралкила, гетероарила, гетероарилалкила, циклогетероалкила, и циклогетероалкилалкила, все, при необходимости, имеют в качестве заместителей при соответствующем атоме углерода 1, 2, 3, 4 или 5 групп, выбираемых из водорода, галоида, алкила, полигалоидалкила, алкокси, галоидалкокси, полигалоидалкокси, алкоксикарбонила, алкенила, алкинила, циклоалкила, циклоалкилалкила, полициклоалкила, гетероариламино, ариламино, циклогетероалкила, циклогетероалкилалкила, гидрокси, гидроксиалкила, нитро, циано, амино, замещенной амино, алкиламино, диалкиламино, тиола, алкилтио, алкилкарбонила, ацила, алкоксикарбонила, аминокарбонила, алкиниламинокарбонила, алкиламинокарбонила, алкениламинокарбонила, алкилкарбонилокси, алкилкалбониламино, алкилсульфониламино, алкиламинокарбониламино, алкоксикарбониламино, алкилсульфонила, аминосульфонила, алкилсульфинила, сульфонамидо или сульфонила;
и R1 и R3 могут, при необходимости, вместе образовывать (CR5R6)m-, где m обозначает 1-6, а R5 и R6 одинаковы или различны и независимо выбираются из гидрокси, алкокси, циано, Н, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, циклоалкенила, арила, арилалкила, гетероарила, гетероарилалкила, циклогетероалкила, галоида, амино, замещенной амино, циклогетероалкилалкила, алкилкарбониламино, арилкарбониламино, алкоксикарбониламино, алкоксикарбонила или алкиламинокарбониламино, или R1 и R2, при необходимости, могут вместе образовывать (CR7R8)p, где р обозначает 2-6, а R7 и R8 одинаковы или различны и независимо выбираются из гидрокси, алкокси, циано, Н, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, циклоалкенила, арила, арилалкила, гетероарила, гетероарилалкила, циклогетероалкила, галоида, амино, замещенной амино, циклогетероалкилалкила, алкилкарбониламино, арилкарбониламино, алкоксикарбониламино, арилоксикарбониламино, алкоксикарбонила, арилоксикарбонила или алкиламинокарбониламино, или, при необходимости, R1 и R3 вместе с 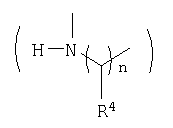 образуют 5-7-членный цикл, содержащий в целом 2-4 гетероатома, выбираемых из N, О, S, SO или SO2;
образуют 5-7-членный цикл, содержащий в целом 2-4 гетероатома, выбираемых из N, О, S, SO или SO2;
или, при необходимости, R1 и R3 вместе с образуют 4-8-членный циклогетероалкил, при этом циклогетероалкильное кольцо содержит, при необходимости, конденсированный с ним арильный цикл, или, при необходимости, конденсированный с ним 3-7-членный циклоалкил;
и включая их фармацевтически приемлемые соли и их пролекарственные сложные эфиры и все их изомеры.
Таким образом, соединения формулы I по изобретению имеют следующее строение
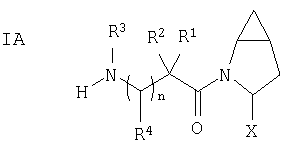
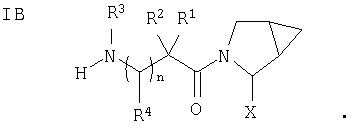
Кроме того, в соответствии с данным изобретением предлагается способ лечения диабета, в частности диабета типа III, а также нарушенный гомеостаз глюкозы, пониженную толерантность к глюкозе, бесплодие, синдром поликистоза яичников, нарушения роста, слабость, артрит, отторжение аллотрансплантата при трансплантации, аутоиммунные заболевания (такие как склеродермия и рассеянный склероз), различные иммуномодуляторные заболевания (такие как красная волчанка или псориаз), СПИД, кишечные заболевания (такие как некротический энтерит, заболевание с включениями микроворсин или заболевание брюшной полости), синдром воспаления кишечника, атрофию или поражение слизистой оболочки кишечника, вызванные химиотерапией, нервную анорексию, остеопороз, Синдром X, дисметаболический синдром, осложнения при диабете, гиперинсулинемию, ожирение, атеросклероз и родственные заболевания, а также воспалительные заболевания кишечника (такие как болезнь Крона и язвенный колит), при этом больному, нуждающемуся в лечении, назначают прием терапевтически эффективного количества соединения строения I (которое ингибирует DP 4).
Состояния, нарушения и заболевания, в целом называемые «Синдром X» или метаболический синдром, подробно описаны в Johansson J. Clin. Endocrinol. Metab., 82, 727-734 (1997).
Кроме того, согласно данному изобретению предлагается способ лечения диабета и родственных выше- и нижеуказанных заболеваний, при этом нуждающемуся в лечении больному назначают прием терапевтически эффективного количества комбинации соединения строения I и одного, двух, трех или более антидиабетических агентов других типов (которые можно применять для лечения диабета и родственных заболеваний) и/или одного, двух или трех или более терапевтических агентов других типов.
Термин «диабет и родственные заболевания» относится к диабету типа II, диабету типа I, пониженной толерантности к глюкозе, ожирению, гипергликемии, Синдрому X, дисметаболическому синдрому, осложнениям при диабете и гиперинсулинемии.
Состояния, нарушения и заболевания, в целом называемые «осложнения при диабете (осложнения, вызванные диабетом)», включают ретинопатию, невропатию и нефропатию и другие известные осложнения, вызванные диабетом.
Термин «другой(ие) тип(ы) терапевтических агентов», применяемый в данном описании, относится к одному или более диабетическому агенту (отличному от ингибиторов DP4 формулы I, одному или более агенту против ожирения, и/или одному или более липид-модулирующему агенту (включая антисклеротические агенты), и/или одному или более агенту против бесплодия, одному или более агенту для лечения синдрома поликистоза яичников, одному или более агенту для лечения нарушения роста, одному или более агенту для лечения слабости, одному или более агенту для лечения артрита, одному или более агенту для предупреждения отторжения аллотрансплантата при тансплантапии, одному или более агенту для лечения аутоиммунных заболеваний, одному или более агенту против СПИДА, одному или более агенту против остеопороза, одному или более агенту для лечения иммуномодуляторных заболеваний, одному или более агенту для лечения хронического кишечного заболевания или синдрома и/или одного или более агента для лечения нервной анорексии.
Термин «липид-модулирующий» агент, применяемый в данном описании, относится к веществам, которые понижают LDL и/или повышают HDL, и/или понижают содержание триглицеридов, и/или понижают общий холестерин, и/или другие механизмы терапевтического лечения липидных нарушений.
В вышеприведенных способах по изобретению соединения строения I применяется в весовом отношении к антидиабетическому агенту или терапевтическому агенту другого типа (в зависимости от способа его действия), примерно, 0,01:1-500:1, предпочтительно, около 0,01:1-100:1, наиболее предпочтительно, около 0,2:1-10:1.
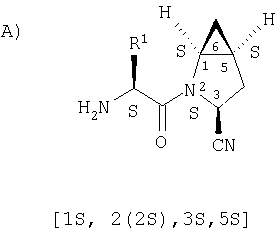
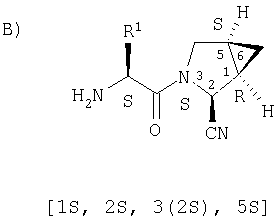
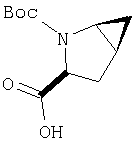
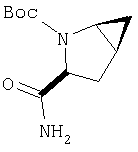
Предпочтительны соединения формулы I, в которых R3 обозначает Н или алкил, R1 обозначает Н, алкил, циклоалкил, бициклоалкил, трициклоалкил, алкилциклоалкил, гидроксиалкил, гидрокситрициклоалкил, гидроксициклоалкил, гидроксибициклоалкил или гидроксиалкилциклоалкил, R2 обозначает Н или алкил, n обозначает 0, Х обозначает CN, х обозначает 0 или 1 и у обозначает 0 или 1.
Наиболее предпочтительны соединения формулы I, описанные выше,
где Х обозначает 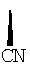 или
или 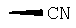 ,
,
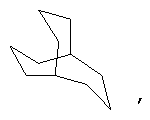
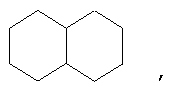
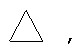
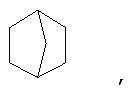
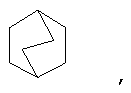
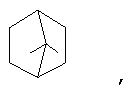
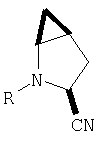
и/или где конденсированная циклопропильная группа определяется как  .
.
Следовательно, предпочтительные соединения формулы I по изобретению содержат фрагмент:
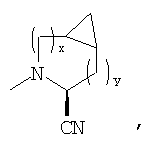
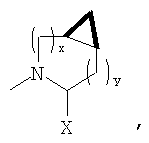
или
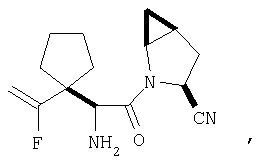
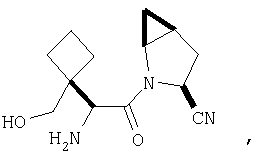
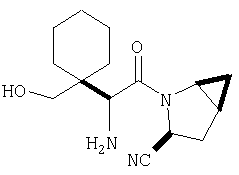
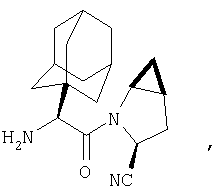
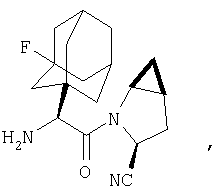
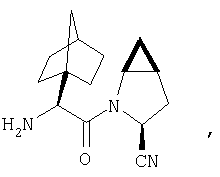
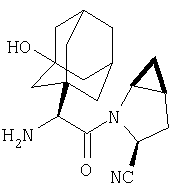
Особенно предпочтительными являются следующие соединения:
где R1 обозначает алкил, циклоалкил, бициклоалкил, трициклоалкил, гидроксиалкил, гидроксициклоалкил, гидроксиалкилциклоалкил, гидроксибициклоалкил или гидрокситрициклоалкил;
где R1 обозначает алкил, циклоалкил, бициклоалкил, трициклоалкил, гидроксибициклоалкил, гидрокситрициклоалкил, алкилциклоалкил, гидроксиалкил, гидроксициклоалкил или гидроксиалкилциклоалкил, а также следующие:
 и
и 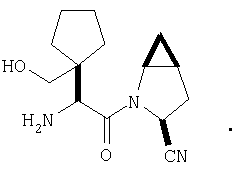
Соединения строения I можно получать способами, представленными на приведенных ниже реакционных схемах и в их описании.
Относительно Реакционной схемы 1: соединение 1, где PG1 обозначает обычную защитную группу, такую как Boc, Cbz, или FMOC, а X1 обозначает Н или CO2R9, как представлено ниже, можно получать методами, представленными в данном описании или в литературе (например, см. Sagnard et al. Tet-Lett., 1995. 36. Pp.3148-3152, Tverezovsky et al., Tetrahedron, 1997, 53, pp.14773-14792, Hanessian et al., Bioorg. Med. Chem. Lett., 1998, p. 2123-2128). Снятие защитной группы (PG1) обычными методами (например, (1) ТФК или HCl, когда PG1 обозначает Boc, или (2) Н2/Pd/С, TMSI, когда PG1 обозначает Cbz, или (3) Et2NH, когда PG1 обозначает FMOC), дает свободный амин 2. Амин 2 может присоединяться к защищенным аминокислотам, таким как 3 (где PG2 может обозначать любую из PG1 защитных групп), в стандартных условиях реакций пептидов (например, EDAC/HOAT, i-BuCOCOCl/TEA, PyBop/NMM), давая соответствующий дипептид 4. Снятие с аминогруппы защитной группы PG2 дает соединение Ia по изобретению, где Х=Н.
В случае, когда Х1=СО2R9 (где R9 обозначает алкильную или аралкильную группу, такую как метил, этил, трет-бутил или бензил), сложный эфир можно гидролизовать в разных условиях, например водной NaOH в соответствующем растворителе, таком как метанол, ТГФ или диоксан, получая кислоту 5. Превращение кислотной группы в первичный карбоксамид с образованием 6 можно активировать с помощью кислотной группы (например, применяя i-BuOCOCl/TEA или EDAC) с последующей обработкой NH3 или эквивалентом аммиака в растворителе, таком как диоксан, эфир или метанол. Амидную группу можно превратить в нитрильную рядом стандартных методов (например, POCl3/пиридин/имидазол или хлорангидрид циануровой кислоты/ДМФА или трифторуксусный ангидрид, ТГФ, пиридин) с образованием 7. Наконец, снятие PG2 защитной группы, аналогичной приведенной выше, дает соединение по изобретению Ib.
При другой последовательности (Схема 2) соединение 1, где X1 обозначает CO2R9, можно омылить до кислоты, а затем амидировать, как описано выше, получая амид 8. Снятие PG1 группы с последующим взаимодействием пептида с3 дает соединение 6, интермедиат в синтезе Ib.
Или же карбоксамидную группу в 8 можно превратить в нитрил, как описано выше, с образованием 9. Снятие PG1 дает 10, который в стандартных условиях присоединения пептидов дает 7, промежуточное соединение в синтезе Ib. Соединение 10 можно также получать окислением амина 2 (например, NCS) с последующими гидролизом и обработкой нитрила. Соединение 10 можно получать в виде смеси стереоизомеров или в виде одного изомера/стереоизомера, который можно эпимеризовать (используя соответствующие методики) с образованием смеси стереоизомеров.
Схема 1
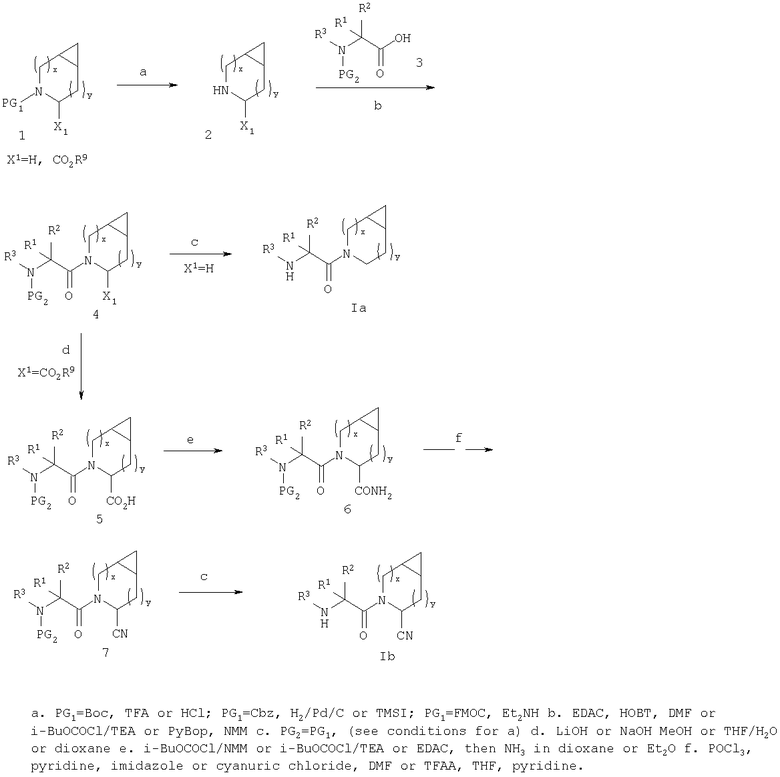
Схема 2
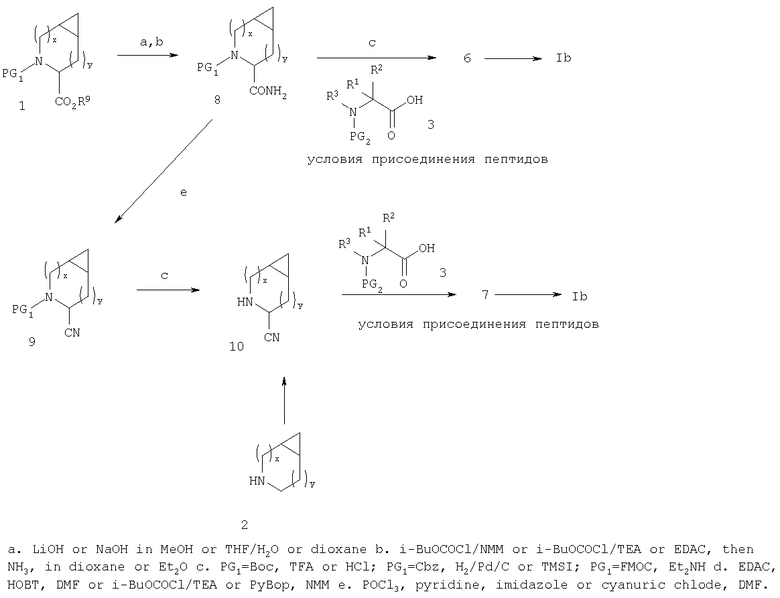
Подобным же образом β-аминокислоты, такие как
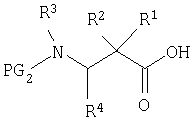
можно конденсировать с 2, свободным амином соединения 8, или 10 с образованием соответствующих амидов, которые могут быть превращены в производные аминокислот соединения Ia или Ib после аналогичной процедуры.
Если не указано иначе, термин «низший алкил», «алкил» или «alk», употребляемый в данном описании один или как часть другой группы, включает как линейные, так и разветвленные углеводородные цепи, содержащие 1-20 атомов углерода, предпочтительно, 1-10 атомов углерода, более предпочтительно, 1-8 атомов углерода в нормальной цепи, такие как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил, додецил, их различные изомеры с разветвленной цепью и т.п., а также такие группы, содержащие 1-4 заместителя, таких как галоид, например F, Br, Cl или I или CF3, алкил, алкокси, арил, арилокси, арил(арил) или диарил, арилалкил, арилалкилокси, алкенил, циклоалкил, циклоалкилалкил, циклоалкилалкилокси, амино, гидрокси, гидроксиалкил, ацил, гетероарил, гетероарилокси, гетероарилалкил, гетероарилалкокси, арилоксиалкил, алкилтио, арилалкилтио, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, нитро, циано, тиол, галоидалкил, тригалоидалкил и/или алкилтио.
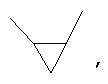
Если не указано иначе, термин «циклоалкил», применяемый в данном описании один или как часть другой группы, включает насыщенные или частично ненасыщенные (содержащие 1 или 2 двойные связи) циклические углеводородные группы, содержащие 1-3 цикла, включающие моноциклический алкил, бициклический алкил (или бициклоалкил) и трициклический алкил (трициклоалкил), имеющие всего 3-20 углеродных атомов, образующих цикл, предпочтительно, 3-10 углеродных атомов, образующих цикл, и которые могут быть конденсированы с 1 или 2 ароматическими циклами, как описано для арила, которые охватывают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил и циклододецил, циклогексенил, адамантил
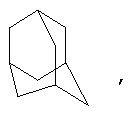
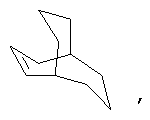
причем каждый из этой группы может, при необходимости иметь 1-4 заместителя, таких как галоген, алкил, алкокси, гидрокси, арил, арилокси. Арилалкил, циклоалкил, гидроксиалкил, алкиламидо, алканоиламино, оксо, ацил, арилкарбониламино, амино, нитро, циано, тиол и/или алкилтио и/или любой заместитель для алкила.
Термин «циклоалкенил», применяемый в данном описании один или как часть другой группы, относится к циклическим углеводородам, содержащим 3-12 атомов углерода, предпочтительно, 5-10 атомов углерода и 1 или 2 двойные связи. Примеры циклоалкенильных групп включают циклопентенил, циклогексенил, циклогептенил, циклооктенил, циклогексадиенил и циклогептадиенил, которые, при необходимости, являются замещенными, как это определено для циклоалкила.
Термин «циклоалкилен», применяемый в данном описании, относится к «циклоалкильной» группе, которая содержит свободные связи и, следовательно, является связывающей группой, такой как
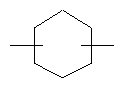 и т.п., и могут, при необходимости, быть замещенными, как определено для «циклоалкила».
и т.п., и могут, при необходимости, быть замещенными, как определено для «циклоалкила».
Термин «алканоил», применяемый в данном описании один или как часть другой группы, относится к алкилу, связанному с карбонильной группой. Если не указано иначе, термин «низший алкенил» или «алкенил», применяемый в данном описании сам по себе или как часть другой группы, относится к линейным или разветвленным радикалам, содержащим 2-20 углеродных атомов, предпочтительно, 2-12 углеродных атомов и, более предпочтительно, 1-8 атомов углерода в нормальной цепи, таким как винил, 2-пропенил, 3-бутенил, 2-бутенил, 4-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 2-гептенил, 3-гептенил, 4-гептенил, 3-октенил, 3-ноненил, 4-деценил, 3-ундеценил, 4-додеценил, 4,8,12-тетрадекатриенил и т.п., и которые, при необходимости, могут иметь 1-4 заместителя, а именно галоген, галоидалкил, алкил, алкокси, алкенил, алкинил, арил, арилалкил, циклоалкил, амино, гидрокси, гетероарил, циклогетероалкил, алканоиламино, алкиламидо, арилкарбониламино, нитро, циано, тиол, алкилтио и/или любой представленный в данном описании алкильный заместитель.
Если не указано иначе, термин «низший алкинил» или «алкинил», применяемый в данном описании самостоятельно или как часть другой группы, относится к линейным или разветвленным радикалам, содержащим 2-20 углеродных атомов, предпочтительно, 2-12 углеродных атомов и, более предпочтительно, 2-8 углеродных атомов в нормальной цепи, которые имеют одну тройную связь в нормальной цепи, таким как 2-пропинил, 3-бутинил, 2-бутинил, 4-пентинил, 3-пентинил, 2-гексинил, 3-гексинил, 2-гептинил, 3-гептинил, 4-гептинил, 3-октинил, 3-нонинил, 4-децинил, 3-ундецинил, 4-додецинил и т.п., и которые, при необходимости, могут иметь 1-4 заместитель, а именно галоген, галоидалкил, алкил, алкокси, алкенил, алкинил, арил, арилалкил, циклоалкил, амино, гетероарил, циклогетероалкил, гидрокси, алканоиламино, алкиламидо, арилкарбониламино, нитро, циано, тиол и/или алкилтио, и/или любой из представленных в данном описании алкильных заместителей.
Термины «арилалкенил» и «арилалкинил», применяемые в данном описании самостоятельно или как часть другой группы, относятся к алкенильным и алкинильным группам, описанным выше, имеющим арильный заместитель.
Если алкильные группы, описанные выше, имеют простые (ординарные) связи для связывания с другими группами у двух различных атомов углерода, они называются «алкиленовыми» группами и, при необходимости, могут иметь заместители, описанные выше для «алкила».
Если описанные выше алкенильные группы и описанные выше алкинильные группы, соответственно, имеют простые связи для присоединения у двух различных углеродных атомов, они именуются «алкениленовые группы» и «алкиниленовые группы», соответственно, и могут, при необходимости, иметь заместители, указанные выше для «алкенила» и «алкинила».
Термин «галоген» или «галоид» («гало»), применяемый в данном описании самостоятельно или как часть другой группы, относится к хлору, брому, фтору и йоду, а также к CF3, при этом предпочтительными являются хлор или фтор.
Термин «ион металла» относится к ионам щелочных металлов, таких как натрий, калий или литий, и ионам щелочно-земельных металлов, таких как магний или кальций, а также к ионам цинка и алюминия.
Если не указано иначе, термин «арил», применяемый в данном описании самостоятельно или как часть другой группы, относится к моноциклическим и бициклическим ароматическим группам, содержащим 6-10 атомов углерода в циклическом фрагменте (таком как фенил или нафтил, включая 1-нафтил и 2-нафтил), и, при необходимости, могущим иметь один-три дополнительных цикла, конденсированных с карбоциклическим фрагментом или гетероциклическим фрагментом (такие как арил, циклоалкил, гетероарил или циклогетероалкил, например

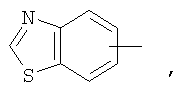
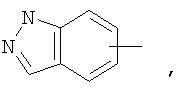
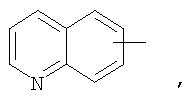
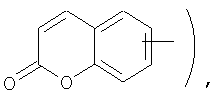
и, при необходимости, могущие иметь, при соответствующих атомах углерода, иметь в качестве заместителей 1, 2 или 3 группы, выбираемые из водорода, галоида, галоидалкила, алкила, алкокси, галоидалкокси, алкенила, алкенила, трифторметила, трифторметокси, алкинила, циклоалкилалкила, циклогетероалкила, циклогетероалкилалкила, арила, гетероарила, арилалкила, арилокси, арилоксиалкила, арилалкокси, арилтио, арилазо, гетероарилалкенила, гетероарилгетероарила, гетероарилокси, гидрокси, нитро, циано, амино, замещенная аминогруппа, причем амино включает 1 или 2 заместителя (которыми являются алкил, арил или любой другой из указанных в определениях арильных соединений), тиол, алкилтио, арилтио, гетероарилтио, арилтиоалкил, алкоксиарилтио, алкилкарбонил, алкиламинокарбонил, ариламинокарбонил, алкоксикарбонил, аминокарбонил, алкилкарбонилокси, арилкарбонилокси, алкилкарбониламино, арилкарбониламино, арилсульфинил, арилсульфинилалкил, арилсульфониламино или арилсульфон - аминокарбонила и/или любого из алкильных заместителей, представленного в данном описании.
Если не указано иначе, термин «низший алкокси», «алкокси», «арилокси» или «аралкокси», применяемый в данном описании самостоятельно или как часть другой группы, включает любую из вышеприведенных алкильных, аралкильных или арильных групп, связанную с атомом кислорода.
Если не указано иначе, термин «замещенный(ая) амино», применяемый в данном описании самостоятельно или как часть другой группы, относится к аминогруппе, имеющей один или два заместителя, которые могут быть одинаковыми или различными, такими как алкил, арил, аралкил, гетероарил, гетероарилалкил, циклогетероалкил, циклогетероалкилалкил, циклоалкил, циклоалкилалкил, галоидалкил, гидроксиалкил, алкоксиалкил или тиоалкил. Эти заместители могут быть дополнительно замещены любой из R1-групп или заместителей для R1, указанных выше. Кроме того, заместители при аминогруппе вместе с атомом азота, с которым они связаны, образовывать 1-пирролидинил, 1-пиперидинил, 1-азепинил, 4-морфолинил, 4-тиаморфолинил, 1-пиперазинил, 4-алкил-1-пиперазинил, 4-арилалкил-1-пиперазинил, 4-диарилалкил-1-пиперазинил, 1-пирролидинил, 1-пиперидинил или 1-азепинил, при необходимости, имеющий заместители алкил, алкокси, алкилтио, галоид, трифторметил или гидрокси.
Если не указано иначе, термин «низший алкилтио», «алкилтио», «арилтио» или «аралкилтио», применяемый в данном описании самостоятельно или как часть другой группы, включает любую из вышеприведенных алкильных, аралкильных или арильных групп, связанную с атомом серы.
Если не указано иначе, термин «низший алкиламино», «алкиламино», «ариламино» или «арилалкиламино», применяемый в данном описании самостоятельно или как часть другой группы, включает любую из вышеприведенных алкильных, арильных или арилалкильных групп, связанную с атомом азота.
Если не указано иначе, термин «ацил», применяемый в данном описании самостоятельно или как часть другой группы по определению в данном описании, относится к органическому радикалу, связанному с карбонильной 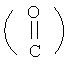 группой; примеры ацильных групп включают любую R1-группу, связанную с карбонилом, такую как алканоил, алкеноил, ароил, аралканоил, гетероароил, циклоалканоил, циклогетероалканоил и т.п.
группой; примеры ацильных групп включают любую R1-группу, связанную с карбонилом, такую как алканоил, алкеноил, ароил, аралканоил, гетероароил, циклоалканоил, циклогетероалканоил и т.п.
Если не указано иначе, термин «циклогетероалкил», применяемый в данном описании самостоятельно или как часть другой группы, относится к 5-, 6- или 7-членному насыщенному или частично ненасыщенному циклу, который содержит 1-2 гетероатома, таких как азот, кислород и/или сера, связанных через атом углерода или гетероатом, где это возможно, при необходимости, через линкер (СН2)r (где r обозначает 1, 2 или 3), например:
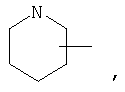
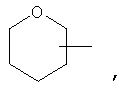
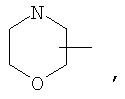
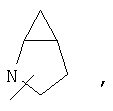
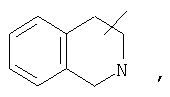
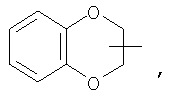
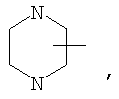
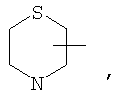
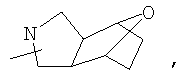
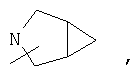
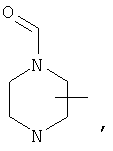
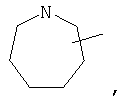
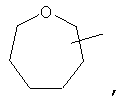
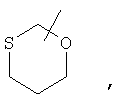
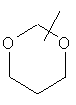
и т.п. Вышеприведенные группы могут включать 1-4 заместителя, таких как алкил, галоид, оксо и/или любой из алкильных заместителей, представленных в данном описании. Кроме того, любой из циклогетероалкильных циклов может быть конденсирован с циклоакильным, арильным, гетероарильным или циклогетероалкильным кольцом.
Если не указано иначе, термин «гетероарил», применяемый в данном описании самостоятельно или как часть другой группы, относится к 5- или 6-членному ароматическому циклу, который содержит 1, 2, 3 или 4 гетероатома, таких как азот, кислород или сера, и такие циклы конденсированы с арильным, циклоалкильным, гетероарильным или циклогетероалкильным кольцом (например, безотиофенил, индолил) и могут включать N-оксиды. Гетероарильная группа может, при необходимости, включать 1-4 заместителя, такого как любой из заместителей, указанных выше для алкила. Примеры гетероарильных групп включают следующие:
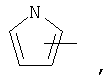
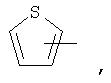
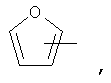
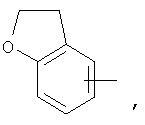
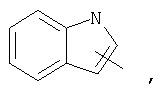
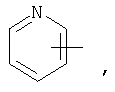
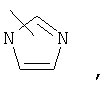
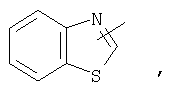
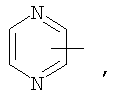
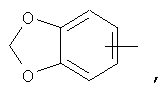
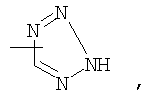
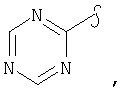
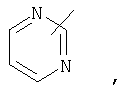
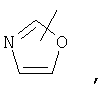
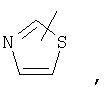
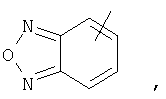
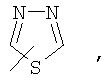
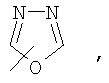
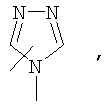
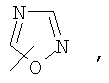
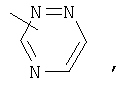
Термин «циклогетероалкилалкил», применяемый в данном описании самостоятельно или как часть другой группы, относится к циклогетероалкильным группам по определению выше, связанным через атом С или гетероатом с цепью (СН2)r.
Термин «гетероарилалкил» или «гетероарилалкенил», применяемый в данном описании самостоятельно или как часть другой группы, относится к гетероарильной группе по определению (см. выше), связанной через атом С или гетероатом с -(CH2)r-цепью, алкиленом или алкениленом по определению (см. выше).
Термин «полигалоидалкил», применяемый в данном описании, относится к «алкильной» группе по определению выше, которая содержит 2-9, предпочтительно, 2-5, галоидных заместителей, таких как F или Cl, предпочтительно, F, например, CF3СН2, CF3 или CF3CF3СН2.
Термин «полигалоидалкокси», применяемый в данном описании, относится к «алкокси» или «алкилокси»-группе по определению выше, которая содержит 2-9, предпочтительно, 2-5 галоидных заместителей, таких как F или Cl, предпочтительно, F, такой как CF3СН2О, CF3O или CF3CF3СН2О.
Охватываются все стереоизомеры соединений по данному изобретению, либо в смеси, либо чистом или практически чистом виде. Соединения по данному изобретению могут иметь асимметрические центры при любом атоме углерода, включая любой из заместителей R. Следовательно, соединения формулы I могут существовать в энантиомерной или диастереомерной формах или в виде их смесей. В процессе получения можно использовать рацематы, энантиомеры или диастереомеры в качестве исходных веществ. Когда получают диастереомерные или энантиомерные продукты, их можно разделять обычными методами, например, хроматографией или фракционной кристаллизацией.
Если требуется, соединения строения I можно применять в комбинации с одним или более антидиабетических агентов других типов (применяемых для лечения диабета и родственных заболеваний) и/или одним или более терапевтических агентов других типов, которые можно вводить перорально в той же лекарственной форме, в виде отдельной пероральной лекарственной формы или в виде инъекции.
Антидиабетический агент другого типа, который можно, при необходимости, применять в комбинации с ингибитором DP4 формулы I, может представлять собой 1, 2, 3 или более антидиабетических агентов или антигипергликемических агентов, включая средства, усиливающие секрецию инсулина, или сенсибилизаторы инсулина, или другие антидиабетические агенты, предпочтительно, имеющие механизм действия, отличный от ингибирования DP4, и может включать бигуаниды, сульфонилмочевины, ингибиторы глюкозидазы, PPAR γ агонисты. Такие как тиазолидиндионы, ингибиторы SGLT2, PPAR α/γ двойные агонисты, ингибиторы аР2, ингибиторы гликогенфосфорилазы, ингибиторы конечных продуктов «прогрессивного» гликозилирования (AGE), и/или меглитиниды, а также инсулин и/или глюкагоноподобный пептид-1 (GLP-1) или их миметики.
Полагают, что применение соединений строения I в сочетании с 1, 2, 3 или более других антидиабетических агентов дает лучшие антигипергликемические результаты, чем можно ожидать в случае самостоятельного применения каждого из этих медикаментов, и лучшие, чем аддитивный гипергликемический эффект этих медикаментов.
Другой антидиабетический агент может представлять собой пероральный антигипергликемический агент, предпочтительно, бигуанид, такой как метформин или фенформин или их соли, предпочтительно, метформин HCl.
Если другой антидиабетический агент представляет собой бигуанид, соединения структуры I применяют в весовом отношении к бигуаниду, примерно, 0,01:1-100:1, предпочтительно. Около 0,1:1-5:1.
Другой антидиабетический агент может также предпочтительно представлять собой сульфонилмочевину, такую как глибурид (также известный как глибенкламид), глимепирид (описанный в Патенте США 4379785), глипизид, гликлазид или хлорпропамид, другие известные сульфонилмочевины или другие антигипергликемические агенты, которые действуют на АТФ-зависимый канал β-клеток, при этом предпочтительными являются глибурид и глипизид, которые можно вводить в виде общей или раздельных пероральных лекарственных форм.
Соединения структуры I применяют с сулфонилмочевинами в весовом соотношении, примерно, 0,01:1-100:1, предпочтительно, около 0,05:1-5:1.
Пероральный диабетический агент может также представлять собой ингибитор глюкозидазы, такой как акарбоза (описанная в Патенте США 4904769) или миглитол (описанный в Патенте США 4639436), которые можно вводить в виде общей или раздельных пероральных лекарственных доз.
Соединения структуры I применяют в весовом отношении к ингибитору глюкозидазы, примерно, 0,01:1-100:1, предпочтительно, около 0,2:1-50:1.
Соединения структуры I можно применять в комбинации с PPAR γ агонистом, таким как пероральный антидиабетический агент ряда тиазолидиндиона, или другими сенсибилизаторами инсулина (которые повышают чувствительность к инсулину у больных NIDDM), такими как троглитазон (Rezulin® фирмы Warner - Lambert, раскрываемый в Патенте США 4572912), розиглитазон (SKB), пиоглитазон (Takeda), MCC-555 фирмы Mitsubishi (раскрываемый в Патенте США 5594016), GL-262570 фирмы Glaxo - Wellcome, эглитазон (СР-68722, Pfizer) или дарглитазон (СР-86325, Pfizer), изаглитазон (MIT/J and J), JTT-501 (JPNT/P and U), L-895645 (Merck), R-119702 (Sankyo/WL), NN-2344 (Dr. Reddy/NN) или YM (Yamanouchi), предпочтительно, розиглитазон и пиоглитазон.
Соединения структуры I применяются в весовом отношении к тиазолидиндиону, примерно, 0,01:1-100:1, предпочтительно, около 0,1:1-10:1.
Сульфонилмочевина и тиазолидиндион в количествах менее, примерно, 150 мг перорального антидиабетического агента могут входить в состав единой таблетки с соединениями строения I.
Соединения строения I могут также применяться в комбинации с антигипергликемическим агентом, таким как инсулин, или с глюкагоноподобным пептидом-1 (GLP-1), таким как амид GLP-1 (1-36), амид GLP-1 (7-36), амид GLP-1 (7-37) (раскрываемый в Патенте США 5614492, выданном Habener, который вводится в данное описание в качестве ссылки) или имитатор (миметик) GLP-1, такой как АС2993 или Exendin - 4 (Amylin) и LY-315902 или LY-307167 ((Lilly) и NN2211 (Novo - Nordisk), которые можно вводить с помощью инъекции, интраназально или с помощью трансдермальных или внутриротовых устройств.
Если присутствуют метформин, сульфонилмочевины, такие как глибурид, глимепирид, глипирид, глипизид, хлорпропамид и гликлазид, и ингибиторы глюкозидазы акарбоза или миглитол, или инсулин (инъецируемая, легочная, внутриротовая или пероральная форма), то они применяются в препаратах, как описано выше, в количествах и дозировках, указанных в Настольном врачебном справочнике (PDR).
Если присутствуют метформин или его соль, то они применяются в количествах около 500-2000 мг в день, которые можно вводить в виде разовой дозы или в виде разделенных доз один-четыре раза в день.
Антидиабетический агент ряда тиазолидиндиона, при его наличии, может применяться в количествах около 0,01-2000 мг/день, которые можно вводить в виде однократной (разовой) или разделенных доз один-четыре раза в день.
Инсулин, при его наличии, можно применять в рецептурах, количествах и дозировках, указанных в Настольном врачебном справочнике.
Пептиды GLP-1, при их наличии, можно вводить в виде трансбуккальных (внутриротовых) рецептур, назально (например, спрей для ингаляции) или парентерально, как описано в Патентах США 5346701 (TheraTech), 5614492 и 5631224, которые вводятся в данное описание в качестве ссылки.
Другой антидиабетический агент может также представлять собой PPAR α/γ двойной агонист, такой как AR-НO39242 (Astra/Zeneca), GW-409544 (Glaxo - Wellcome), KRP297 (Kyorin Merck), а также антидиабетические агенты, описанные Murakami et al., "A Novel Insulin Sensitizer Acts As a Coligand for Peroxisome Proliferation - Activated Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Activation on Abnormal Lipid Metabolism in Liver of Zucker Fatty Rats", Diabetes 47,1841-1847 (1998), и в заявке на Патент США 09/664598, поданной 18 сентября 2000 года (№ дела у поверенного LA29NP), которые вводятся в данное описание во всей полноте в качестве ссылки, применяемые в указанных там дозах, при этом соединения, признанные в этих ссылках предпочтительными, являются предпочтительными для применения по данному описанию.
Другим антидиабетическим агентом может быть ингибитор SGTL2, такой как раскрываемый в заявке на Патент США 09/679027, поданной 4 октября 2000 года (№ дела у поверенного LA49NP), которая вводится в данное описание в качестве ссылки, применяющийся в указанных в данном описании дозах. Предпочтительными являются соединения, признанные предпочтительными в вышеуказанной заявке.
Другим антидиабетическим агентом, который можно, при необходимости, применять в комбинации с ингибитором DP4 формулы I, может быть ингибитор аР2, такой как раскрываемый в заявке на Патент США 09/391053, поданной 7 сентября 1999 года, и в заявке на Патент США 09/519079, поданной 6 марта 2000 года (дело у патентного поверенного ДФ27NP), которые вводятся в данное описание в качестве ссылки, в дозировках по данному описанию.
Другим диабетическим агентом, который, при необходимости, можно применять в комбинации с ингибитором DP4 формулы I, может быть ингибитор гликогенфосфорилазы, такой как раскрываемый в Международных заявках WO 96/39384, WO 96/39385, ЕР 978279, WO 2000/47206, WO 99/43663 и заявках на Патент США 5952322 и 5998463, Международной заявке WO 99/26659 и Европейской заявке ЕР 1041068.
Меглитинид, который может, при необходимости, применяться в комбинации с соединением формулы I по изобретению, может представлять собой репаглинид, натеглинид (Novartis) или KAD (PF/Kissei), при этом репаглинид является предпочтительным.
Ингибитор DP4 формулы I применяют в примерном весовом соотношении с меглитинидом, PPAR α/γ двойным агонистом, ингибитором SGLT2, ингибитором аР2 или ингибитором гликогенфосфорилазы 0,01:1-100:1, предпочтительно, около 0,1:1-10:1.
Гиполипидемический агент или липид-модулирующий агент, который, при необходимости, можно применять в комбинации с соединениями формулы I по изобретению, может включать 1, 2, 3 или более ингибиторов МТР, ингибиторы HMG СоА редуктазы, ингибиторы сквален-синтетазы, производные фибриновой кислоты, ингибиторы АСАТ, ингибиторы липоксигеназы, ингибиторы всасывания (абсорбции) холестерина, ингибиторы котранспортера Na+ / желчные кислоты в подвздошной кишке, положительные регуляторы LDL-рецепторной активности, ингибиторы цитрат-лиазы АТФ, белковые ингибиторы переноса холестериновых сложных эфиров, вещества, усиливающие секрецию желчных кислот, и/или никотиновую кислоту и ее производные.
Ингибиторы МТР, применяемые по данному описанию, охватывают ингибиторы МТР, раскрываемые в Патентах США 5595872, 5739135, 5712279, 5760246, 5827875, 5885983 и заявке на Патент США 09/175180, поданной 20 октября 1998 года, в настоящее время Патент США 5962440. Предпочтительными является каждый предпочтительный ингибитор МТР, раскрываемый в каждой из вышеуказанных патентов и заявок.
Все вышеуказанные Патенты и заявки на Патенты США вводятся в данное описание в качестве ссылки.
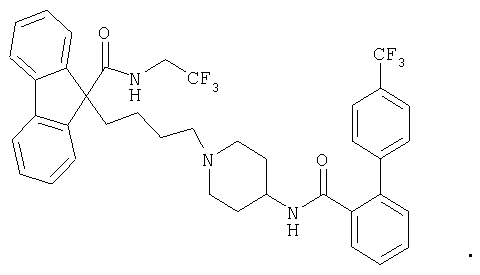
Наиболее предпочтительные ингибиторы для применения в соответствии с настоящим изобретением включают предпочтительные ингибиторы МТР, подробно описанные в Патентах США 5739135, 5712279 и 5760246, а также имплитапид (Bayer). Наиболее предпочтительным ингибитором МТР является 9-[4-[4-[[2-(2,2,2-трифторэтокси)бензоил]амино]-1-пиперидинил]бутил]-N-(2,2,2-трифторэтил)-9Н-флуорен-9-карбоксамид.
Гиполипидемический агент может являться ингибитором HMG СоА редуктазы, который включает, без ограничения, мевастатин и родственные соединения, раскрываемые в Патенте США 3983140, ловастатин (мевинолин) и родственные соединения, раскрываемые в Патенте США 4231938, правастатин и родственные соединения, например, такие, которые раскрываются в Патенте США 4346227, симвастатин и родственные соединения. Раскрываемые в Патентах США 4448784 и 4450171. Другие ингибиторы HMG СоА редуктазы, которые можно применять по данному описанию, включают, но без ограничения, флувастатин, раскрываемый в Патенте США 5354772, церивастатин, описанный в Патентах США 5006530 и 5177080, аторвастатин, описанный в Патентах США 4681893, 5273995, 5385929 и 5686104, атавастатин (нисвастатин фирмы Nissan/Sankyo (NK-104)), описанный в Патенте США 5011930, визастатин (visastatin) Shionogi - Astra/Zeneca (ZD-4522), раскрываемый в Патенте США 5260440.
Ингибиторы сквален-синтетазы, пригодные для применения по данному описанию, включают, но без ограничения, α-фосфонсульфонаты, раскрываемые в Патенте США 5712396, α-фосфонсульфонаты, описанные Biller et al., J. Med. Chem., 1988, Vol.31, No.10, pp 1869-1871, включая изопреноидные (фосфинилметил)фосфонаты, а также другие известные ингибиторы сквален-синтетазы, например, описываемые в Патентах США 4871721 и 4924024 и в Biller, S.A., Neuenschwander, K., Ponpipom, M.M., and Poulter, C.D., Current Pharmaceutical Design, 2, 1-40 (1996).
Далее, другие ингибиторы сквален-синтетазы, пригодные для применения по данному описанию, охватывают терпеноидные пирофосфаты, описанные Р.Ortiz de Montellano et al., J. Med. Chem. 1977, 20, 243-249, аналог А фарнезилдифосфата и аналоги прескваленпирофосфата (PSQ-РР), описанные Corey and Volante, J. Am. Chem. Soc., 1976, 98, 1291-249, фосфинилфосфонаты, о которых сообщает McClard, R.W. et al., J.А.С.S., 1987, 109, 5544, и циклопропаны, описанные Capson, T.L., PhD dissertation, June, 1987, Dept. Med. Chem. U of Utah, Abstract, Table of Contents, pp. 16, 17, 40-43, 48-51, Summary.
Другие гиполипидемические агенты, пригодные для применения по данному описанию, включают, но без ограничения, производные фибриновой кислоты, такие как фенофибрат, гемфиброзил, клофибрат, безафибрат, пипрофибрат, клинофибрат и т.п., пробукол и родственные соединения, раскрываемые в Патенте США 3674836, при этом предпочтительными являются пробукол и гемфиброзил, вещества, усиливающие секрецию желчных кислот, такие как холестирамин, колестипол и DEAE - Sephadex (Secholex®, Policexide® ), а также липостабил (Rhone - Poulenc), Eisai E-5050 ( производное N-замещенного этаноламина), иманиксил (НОЕ-402), тетрагидролипстатин (THL), истигмастанилфосфорилхолин (SPC, Roche), аминоциклодекстрин (Tanabe Seioku), Ajimoto AJ-814 (производные азулена), мелинамид (Sumimoto), Sandoz 58-035, CL-277082 и CL-283546 фирмы American Cyanamid (производные дизамещенной мочевины), никотиновую кислоту, аципимокс, ацифран, неомицин, п-аминосалициловую кислоту, аспирин, производные поли(диаллилметиламина), такие как описанные в Патенте США 4759923, соль четвертичного аммониевого основания поли(диаллилдиметиламмонийхлорид) и иононы, такие как раскрываемые в Патенте США 4027009, и другие известные агенты, понижающие содержание холестерина в сыворотке.
Другой гиполипидемический агент может представлять собой ингибитор АСАТ, такой как описанный в Drugs in Future 24, 9-15 (1999), (Avasimibe); " The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters", Nicolosi et al., Atherosclerosis (Shannonb, Irel). (1998), 137(1), 77-85; The pharmalogical prophile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of hepatic secretion of АроВ100 - containing lipoprotein", Ghiselli, giancarlo, Cardiovasc. Drug Rev. (1998), 16(1), 16-30; "RP 73163: a bioavailable alkylsulfinyl - diphenylimidazole ACAT inhibitor", Smith, C., et al., Bioorg. Med. Chem. Lett. (1996), 6(1), 47-50; "ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals", Krause et al., Editor(s): Ruffolo, Robert., Jr; Hollinger, Mannfred A., Inflammation: Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton? Fla.; "ACAT inhibitors: potential anti-atherosclerotic agents", Sliskovic et al., Curr. Med. Chem. (1994), 1(3), 204-25; "Inhibitors of acyi - CoA: cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. Inhibitors of acyi-CoA: cholesterol acyltransferase (ACAT). 7. Development of a series substituted N-phenyl N'-[(1-phenylcyclopentyl)methyl] ureas with enhanced hypocholesterolemic activity". Stout et al., Chemtracts: Org. Chem. (1995), 8(6), 359-62, или TS-962 (Taisho Pharmaceutical Co. Ltd.).
Гиполипидемический агент может быть позитивным регулятором LD2 рецепторной активности, таким как MD-700 (Taisho Pharmaceutical Co Ltd.) и LY295427 (Eli Lilly).
Гиполипидемический агент может быть ингибитором всасывания холестерина, предпочтительно, SCH484461 фирмы Schering - Plough, а также ингибиторы всасывания холестерина, раскрываемые в Atherosclerosis 115,45-63 (1995) и J. Med. Chem. 41, 973 (1998).
Гиполипидемический агент может представлять собой ингибитор котранспортера Na+/желчные кислоты, таким как описанные в Drugs in Future, 24, 425 430 (1999).
Липид-модулирующий агент может представлять собой ингибитор белка - носителя эфиров холестерина (СЕТР), такой как СР 529414 фирмы Pfizer (Международная заявка WO/0038722 и Европейская заявка ЕР 818448) и SC-744 и SC-795 фирмы Pharmacia.
Ингибитор АТФ - цитрат-лиазы, который можно применять в комбинации по изобретению, может включать, например, ингибитор, раскрываемый в Патенте США 5447954.
Предпочтительными гиполипидемическими агентами являются правастатин, ловастатин, симвастатин, аторвастатин, флувастатин, церивастатин и ZD-4522.
Вышеуказанные патенты США вводятся в данное описание в качестве ссылок. Применяются количества и дозы, указанные в Настольном врачебном справочнике и/или в приведенных выше патентах.
Соединения формулы I по изобретению применяются в весовом отношении к гиполипидемическому агенту (если он присутствует), примерно, 500:1-1:500, предпочтительно, около 100:1-1:100.
Применяемую дозу следует тщательно корректировать в соответствии с возрастом, весом и состоянием больного, а также со способом применения, лекарственной формой и схемой приема и ожидаемым результатом.
Дозы и рецептуры гиполипидемического агента такие, как описываемые в различных патентах и заявках, обсуждаемых выше.
Дозы и препараты другого применяемого гиполипидемического агента, в случае его применения, берут из последнего издания Настольного врачебного справочника.
В случае перорального применения удовлетворительных результатов можно достичь, применяя ингибитор МТР в количествах около 0,01 мг/кг - 500 мг, предпочтительно, около 0,1 мг - 100 мг один - четыре раза в день.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор МТР в количестве около 1-500 мг, предпочтительно, около 2-400 мг и, наиболее предпочтительно, около 5-250 мг один - четыре раза в день.
При пероральном приеме удовлетворительные результаты достигаются при применении ингибитора HMG СоА редуктазы, например, правастатина, ловастатина, симвастатина, аторвастатина, флувастатина или церивастатина в дозах, предписываемых Настольным врачебным справочником, например, в количествах около 1-2000 мг, и предпочтительно, около 4-200 мг.
Ингибитор сквален-синтетазы можно применять в дозах, примерно, 10-2000 мг, и предпочтительно, около 25-200 мг.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор в количестве около 0,1-100 мг, предпочтительно, около 5-80 мг, и более предпочтительно, около 10-40 мг.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор сквален-синтетазы в количестве около 10-500 мг, предпочтительно, около 25-200 мг.
Другой гиполипидемический агент может также быть ингибитором липоксигеназы, включая ингибитор 15-липоксигеназы (15-LO), такой как производные бензимидазола, описываемые в Международной заявке WO 97/12615, ингибиторы 15-LO, описываемые WO 97/12613, изотиазолоны, подобные раскрываемым в WO 96/38144, и ингибиторы 15-LO, описываемые Sendobry et al., "Attenuation of diet - induced atherosclerosis in rabbits with a highly selective 15 - Lipoxygenase inhibitor lacking significant antioxidant properties", Brit. J. Pharmacology (1997) 120б 1199 - 1206б, и Comicelli et al., "15 - Lipoxygenase and its Inhibition: A Novel Therapeutic Target for Vascular Disease", Current Pharmaceutical Design, 1999, 5, 11-20.
Соединения формулы I и гиполипидемический агент могут применяться вместе в виде одной и той же лекарственной формы или в виде раздельных лекарственных форм, принимаемых одновременно.
Вышеописанные композиции можно принимать в представленных выше лекарственных формах в виде однократной или разделенных доз один - четыре раза в день. Желательно больному начинать с комбинации низких доз и постепенно доходить до комбинации высоких доз.
Предпочтительным гиполипидемическим агентом является правастатин, симвастатин, ловастатин, аторвастатин, флувастатин или церивастатин.
Другим типом терапевтического агента, который, при необходимости, можно применять с ингибитором DP4 формулы I, может являться 1, 2, 3 или более агентов против ожирения, включая бета 3 адренергический агонист, ингибитор липазы, ингибитор повторного поглощения серотонина (и дофамина), препарат тироидного рецептора бета, средство, снижающее аппетит, и/или позитивный регулятор окисления жирных кислот.
Бета 3 адренергический агонист, который можно, при необходимости, применять в комбинации с соединением формулы I, может представлять собой AJ9677 (Takeda/Dainippon), L750355 (Merck) или СР331648 (Pfizer) или другой известный бета 3 агонист, описанный в Патентах США 5541204, 5770615, 5491134, 5776983 и 5488064, при этом предпочтительными являются AJ9677, 1750355 и СР331648.
Ингибитор липазы, который можно, при необходимости, применять в комбинации с соединением формулы I, может представлять собой орлистат или ATL-962 (Alizyme), при этом предпочтительным является орлистат.
Ингибитор повторного поглощения серотонина (и дофамина), который, при необходимости, может применяться в комбинации с соединением формулы I, может представлять собой сибутрамин, топирамат (Johnson and Johnson) или аксокин (Regeneron), при этом предпочтительными являются сибутрамин и топирамат.
Тироидный рецептор бета, который, при необходимости, можно применять в комбинации с соединением формулы I, может представлять собой лиганд тироидного рецептора, описанный в WO 97/21993 (U. Cal SF), WO 99/00353 (KaroBio) и в английском патенте GB 98/284425 (KaroBio), при этом предпочтительными являются соединения заявителей KaroBio.
Веществом, снижающим аппетит, который, при необходимости, можно применять в комбинации с соединением формулы I, может быть дексамфетамин, фентермин, фенилпропаноламин или мазиндол, причем предпочтительными является дексамфетамин.
Позитивный регулятор (активатор) окисления жирных кислот, который, при необходимости, применяют в комбинации с соединением формулы I, может представлять собой фамоксин (Genset).
Различные вышеописанные вещества против ожирения могут применяться в одной лекарственной форме с соединением формулы I или в различных лекарственных формах, дозы и схемы приема обычно известны из уровня техники или из PDR.
Агент против бесплодия, который, при необходимости, может применяться в комбинации с ингибитором DP4 по изобретению, может представлять собой 1, 2 агента или более из кломифен цитрата (Clomid®, Aventis), бромкриптина мезилат (Parlodel®, Novartis), аналоги LHRH, Lupron (TAP Pharm.), даназол, Danocrine (Sanofi), прогестогены или глюкокортикоиды, которые могут применяться в количествах, предписываемых PDR.
Агент против синдрома поликистоза яичников, который, при необходимости, можно применять в комбинации с ингибитором DP4 по изобретению, может представлять собой 1,2 или более агентов, выбираемых из гонадотропин-релизинг-гормона (GnRH), лейпролида (Lupron®), Clomid®, Parlodel®, пероральных контрацептивов или сенсибилизаторов инсулина, таких как агонисты PPAR, или других обычно применяемых для этой цели агентов, которые назначают в количествах, предписываемых PDR.
Агент для лечения нарушений роста и/или слабости, который можно, при необходимости, применять в сочетании с ингибитором DP4 по изобретению, может представлять собой 1, 2 или более агентов, выбираемых из гормона роста или вещества, стимулирующего секрецию гормона роста, такого как МК-677 (Merck), CP-424391 (Pfizer) и соединения, описанные в заявке на Патент США 09/506749, поданной 18 февраля 2000 года (№ дела у патентного поверенного LA26), а также из селективных модуляторов рецепторов андрогена (SARM), которые вводятся в данное описание в качестве ссылки, и их можно применять в количествах, рекомендуемых в PDR.
Агент для лечения артрита, который можно, при необходимости, применять в комбинации с ингибитором DP4 по изобретению, может представлять собой 1, 2 или более веществ, выбираемых из аспирина, индометацина, ибупрофена, диклофенак-натрия, напроксена, набуметона (Relafen®, SmithKline), толметин-натрия (Tolectin®, Ortho - McNeil), пироксикама (Feldene®, Phizer), кеторолак-трометамина (Toradol®, Roche), целекоксиба (Celebrex®, Searle), рофекоксиба (Vioxx®, Merck) и т.п., которые можно назначать в количествах, предписываемых PDR.
Обычные агенты для предупреждения отторжения аллотрансплантата при трансплантации, такие как циклоспорин, Sandimmune (Novartis), азатиоприн, Immuran (Faro) или метотрексат, можно, при необходимости, применять с ингибиторами по изобретению в количествах, рекомендуемых в PDR.
Общепринятые агенты для лечения аутоиммунных заболеваний, таких как рассеянный склероз, и иммуномодуляторных заболеваний, таких как красная волчанка, псориаз, например азатиоприн, иммуран, циклофосфамид, НПВС (NSAIDS), такие как ибупрофен, ингибиторы сох 2, такие как Vioxx и Celebrex, глюкокортикоиды и гидроксихлорокин, можно, при необходимости, применять в комбинации с ингибитором DP4 по изобретению в количествах, рекомендуемых в PDR.
Агенты против СПИД'а, которые можно, при необходимости, применять с ингибитором DP4 по изобретению, могут представлять собой ненуклеозидный ингибитор обратной транскриптазы, нуклеозидный ингибитор обратной транскриптазы, ингибитор протеаз и/или противоинфекционные средства при СПИД'е, могут представлять собой 1, 2 или более агентов, выбираемых из дронабинола (Marinol®, Roxane Labs), диданозина (Videx®, Bristol - Myers Squibb), магестрола ацетата (Megace®, Bristol - Myers Squibb), ставудина (Zerit®, Bristol - Myers Squibb), делавердинмезилата (Rescriptor®, Pharmacia), ламивудина/зидовудина (Combivir ™, Glaxo), ламивудина (Epivir™, Glaxo), зальцитабина (Hivid®, Roche), зидувидина (Retrovir®, Glaxo), индинавира сульфата (Crixivan®, Merck), саквинавира (Fortovase™, Roche), саквиновирмезилата (hivirase®, Roche), ритиновира (Norvir®, Abbot), нельфинавира (Viracept®, Agouron).
Вышеперечисленные агенты против СПИД'а можно применять в количествах, рекомендуемых в PDR.
Агент для лечения воспалительного кишечного заболевания или синдрома, который, при необходимости, можно применять в комбинации с ингибитором DP4 по изобретению, может представлять собой 1, 2 или более агентов из сульфасалазина, салицилатов, мезаламина (Asacol®, P and G) или Zeimac® (Bristol - Myers Squibb), которые можно назначать в количествах, рекомендуемых в PDR или каким-либо другим способом известных из уровня техники.
Агент для лечения остеопороза, который, при необходимости, применять в комбинации с ингибитором DP4 по изобретению, может представлять собой 1, 2 или более агентов из группы: алендронат-натрий (Fosamax®, Merck), тилудронат (Skelid®, Sanofi), этидронат-динатрий (Didronel®, P and G), ралоксифен HCl (Evista®, Lilly), которые можно применять в количествах, определенных в PDR.
При осуществлении способа по изобретению применяется фармацевтическая композиция, содержащая соединения строения I, с другим антидиабетическим агентом и/или терапевтическим агентом другого типа или без этих агентов, в сочетании с фармацевтическим носителем или разбавителем. Можно приготовить фармацевтическую композицию, использующую обычные твердые или жидкие носители или разбавители и фармацевтические добавки, соответствующие заданному способу применения. Соединения можно вводить различным видам млекопитающих, включая человека, обезьян, собак и т.д., перорально, например, в форме таблеток, капсул, гранул или порошков, или их можно вводить парентерально в виде инъецируемых препаратов. Доза для взрослых, предпочтительно, составляет около 10-1000 мг в день, это количество можно вводить в виде разовой дозы или в виде раздельных доз 1-4 раза в день.
Типичная капсула для перорального применения содержит соединения строения I (250 мг), лактозу (75 мг) и стеарат магния (15 мг). Смесь пропускают через сита 60 меш и помещают в желатиновую капсулу №1.
Типичный инъецируемый препарат готовят помещая в асептических условиях 250 мг соединений строения I в ампулу, лиофилизируют и запаивают в асептических условиях. В момент использования содержимое ампулы смешивают с 2 мл физиологического раствора, получая инъецируемый раствор.
DP4-ингибиторную активность соединений по изобретению можно определять, применяя in vitro аналитическую систему, которая измеряет потенциирование ингибирования DP4. Ингибиторные константы (Ki) для ингибиторов DP4 по изобретению можно определять описанным ниже способом.
Очистка дипептидилпептидазы IV свиньи.
Фермент свиньи очищают как описано ранее (1) с некоторыми изменениями. Получают почки 15-20 животных, отсекают корковое вещество и замораживают при -80°С. Замороженные ткани (2000-2500 г) гомогенизируют в 12 л 0,25 М сахарозы в гомогенизаторе Уоринга. Затем гомогенат оставляют при 38°С на 18 часов для более легкого отщепления DP-4 от клеточных мембран. После стадии расщепления гомогенат осветляют при 7000Х g в течение 20 мин при 4°С, а супернатант собирают. Добавляют твердый сульфат аммония до 60% насыщения и осадок собирают центрифугированием при 10000Х g и отбрасывают. Снова насыщают супернатант сульфатом аммония до 80% и собирают 80% дебриса и растворяют в 20 мМ Na2HPO4, pH 7,4.
После диализа против 20 мМ Na2HPO4, pH 7,4, препарат осветляют центрифугированием при 100000Х g. Затем осветленный препарат добавляют к 300 мл ConA-сефарозы, уравновешенной в том же буфере. После отмывания в том же буфере до постоянного А280 колонку элюируют 5% (вес/объем) раствором α-D-маннопиранозида. Активные фракции объединяют, концентрируют и подвергают диализу против 5 мМ ацетата натрия, pH 5,0. Затем продукт диализа пропускают через 100 мл колонку Pharmacia Resource S, уравновешенную в том же буфере. Элюат собирают, и он содержит основную часть ферментной активности. Активный материал снова концентрируют и подвергают диализу в 20 мМ Na2HPO4, pH 7,4. Наконец, сконцентрированный фермент хроматографируют на колонке для гель-фильтрации Pharmacia S-200 для удаления низкомолекулярных примесей. Чистоту фракций с колонки анализируют с помощью восстановительного SDS-PAGE и самые чистые фракции собирают и концентрируют. Очищенный фермент хранят в 20% глицерине при -80°С.
Анализ дипептидилпептидазы IV свиньи.
Фермент анализируют в стационарных условиях, как описано ранее (2) с гли-про-п-нитроанилидом в качестве субстрата, со следующими изменениями. Реакционная смесь в конечном объеме 100 мкл содержит 100 мМ Aces, 52 мМ TRIS, 52 мМ этаноламина, 500 мкМ гли-про-п-нитроанилида, 2% ДМСО и 4,5 нМ фермента при 25°С, рН 7,4. Для отдельного анализа при 10 мкМ испытуемого соединения буфер, соединение и фермент помещают в лунки 96-луночного титрационного микропланшета и инкубируют при комнатной температуре 5 минут. Реакция начинается при добавлении субстрата. Непрерывное образование п-нитроанилина определяют при 405 нМ в течение 15 минут с помощью планшет-ридера Molecular Device Tmax, считывая каждые 9 секунд. Линейную скорость образования п-нитроанилина наблюдают на линейном участке кривой. Стандартную кривую оптической плотности п-нитроанилина строят в начале каждого эксперимента, и по стандартной кривой количественно определяют катализируемое ферментом образование п-нитроанилина. Для последующего анализа отбирают соединения, ингибирующие более чем на 50%.
Для анализа позитивных соединений определяют стационарные кинетические ингибиторные константы как функцию концентрации как субстрата, так и ингибитора. Кривые насыщения для субстрата получают при концентрациях гли-про-п-нитроанилида 60-3600 мкМ. Также снимают дополнительные кривые насыщения в присутствии ингибитора. Полный эксперимент по ингибированию включает 11 концентраций субстратов и 7 концентраций ингибиторов, при трехкратном повторении определения по всем планшетам. Для более тесного связывания ингибиторов при значении Кi менее 20 нМ концентрацию фермента уменьшают до 0,5 нМ, а время реакции увеличивают до 120 минут. Полученный набор данных для трех планшетов вводят в соответствующее уравнение конкурентного и неконкурентного ингибирования.
(1) Rahfeld, J. Schutkowski, M., J. Faust, J., Neubert., Barth., and Heins, J. (1991) Biol.Chem. Hoppe - Seyler, 372, 313-318.
(2) Nagatsu, Т., Hino, M., Ffuyamada, H., Hayakawa, Т., Sakakibara, S., Nakagawa, Y., and Takemoto, T. (1976) Anal. Biochem., 74, 466-476.
В Примерах и по всему данному описанию применяются следующие сокращения:
Ph = фенил
Bn = бензил
i-Bu = изо-бутил
Me = метил
Et = этил
Pr = пропил
Bu = бутил
ТМС, TMS = триметилсилил
FMOC = флуоренилметоксикарбонил
Boc или ВОС = трет-бутоксикарбонил
Cbz = карбобензилокси или карбобензокси или бензилоксикарбонил
НОАс или АсОН = уксусная кислота
ДМФА = N,N-диметилформамид
EtOAc = этилацетат
ТГФ = тетрагидрофуран
ТФК, TFA = трифторуксусная кислота
Et2NH = диэтиламин
NMM = N-метилморфолин
Н-BuLi = н-бутиллитий
Pd/C = палладий на угле
PtO2 = двуокись платины, оксид платины
TEA, ТЭА = триэтиламин
EDAC = 3-этил-3'-(диметиламино)пропил-карбодиимид гидрохлорид (или 1-[3-(диметил)амино)пропил])-3-этилкарбодиимид гидрохлорид)
НОВТ или НОВТ•Н2О = 1-гидроксибензотриазол гидрат
НОАТ = 1-гидрокси-7-азабензотриазол
РуВОР реагент = бензотриазол-1-илокситрипирролидинофосфония гексафторфосфат
мин = минута(ы)
ч, час = час(ы)
л = литр
мл = миллилитр
мкл = микролитр
г = грамм
мг = миллиграмм
мол = моль (моли)
ммол = миллимоль (миллимоли)
мэкв = миллиэквивалент
rt = комнатная температура
нас (sat или sat'd) = насыщенный
aq. = водный
ТСХ = тонкослойная хроматография
ВЭЖХ = высокоэффективная жидкостная хроматография
LC/MS (ЖХ/Масс-спектрометрия) = высокоэффективная жидкостная хроматография/масс-спектрометрия
MS или масс-спектр = масс-спектрометрия
ЯМР = ядерный магнитный резонанс
mp, т.пл. = температура плавления
В нижеследующих Примерах представлены предпочтительные варианты изобретения.
Пример 1.
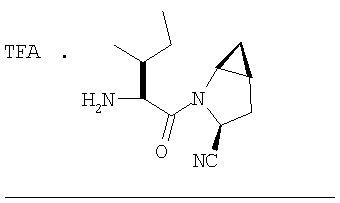
Стадия 1
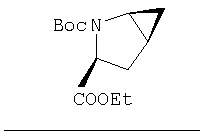
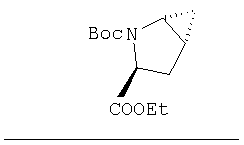
Соединение стадии 1 синтезируют по литературной методике [Stephen Hanessian, Ulrich Reinold, Michel Saulnier, and Stephen Claridge; Bioorganic and Medicinal Chemistry 8 (1998) 2123-2128] или с нижеследующими изменениями. Этиловый эфир L-пироглутаминовой кислоты защищают по атому азота, превращая в трет-бутилкарбамат (Вос2О, DMAP или NaH), а затем дегидратируют до этилового эфира 4,5-дегидропролина в одной колбе восстановлением карбонила (триэтилборгидрид, толуол, -78°С) с последующей дегидратацией (TFAA, лутидин). Заглавное соединение получают циклопропанированием этилового эфира 4,5-дегидропролина (Et2Zn, CICH2I, 1,2-дихлорэтан, -15°С). Ниже дано более подробное описание.
Синтез этилового эфира 4,5-дегидро-L-пролина: Этиловый эфир L-пироглутаминовой кислоты (200 г, 1,27 мол) растворяют в 1,2 литра хлористого метилена и обрабатывают последовательно дитрет-бутилдикарбонатом (297 г, 1,36 мол) и катализатором DMAP (1,55 г, 0,013 мол) при комнатной температуре. Через 6 ч к смеси добавляют (гасят) рассол и органическую фазу сушат (Na2SO4) и фильтруют через короткую колонку с силикагелем, получая 323 г (100%) этилового эфира N-Boc-L-пироглутаминовой кислоты. Этиловый эфир N-Boc-L-пироглутаминовой кислоты (160 г, 0,62 мол) растворяют в 1 литре толуола, охлаждают до -78°С и обрабатывают триэтилборгидридом (666 мл 1 М раствора в ТГФ), добавляя по каплям в течение 90 минут. Через 3 часа по каплям добавляют 2,6-лутидин (423 мл, 3,73 мол), а затем DMAP (0,2 г, 0,0016 мол). К этой смеси прибавляют TFAA (157 г, 0,74 мол) и реакционную смесь оставляют на 2 ч при комнатной температуре. Смесь разбавляют EtOH, водой и органический слой промывают 3N HCl, водой, водным бикарбонатом и рассолом и сушат (Na2SO4) фильтруют через слой силикагеля, получая 165 г сырого этилового эфира 4,5-дегидропролина, который очищают флеш-хроматографией на силикагеле, элюент этилацетат: смесь гексанов 1:5, получают 120 г, 75% олефина.
Циклопропанирование этилового эфира 4,5-дегидро-L-пролина: Этиловый эфир 4,5-дегидро-L-пролина (35,0 г, 0,145 мол) добавляют к раствору чистого Et2Zn (35,8 г, 0,209 мол) в 1 литре дихлорэтана при -15°С. К этой смеси по каплям добавляют ClCH2I (102 г, 0,58 мол) за 1 час и смесь перемешивают при -15°С 18 ч. К реакционной смеси прибавляют насыщенный водный раствор бикарбоната и растворитель упаривают, добавляют EtOAc, промывают рассолом и очищают хроматографией на силикагеле, применяя постадийный градиент от 20% EtOAc/смесь гексанов до 50% EtOAc/смесь гексанов, получают 17,5 г (50%) диастереомерно чистого соединения стадии 1.
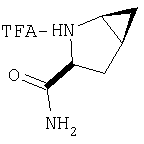
Стадия 2.
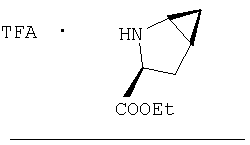
К перемешиваемому раствору соединения Стадии 1 (411 мг, 1,61 ммол) в CH2Cl2 (1,5 мл) при комнатной температуре добавляют ТФК (1,5 мл). Реакционную смесь перемешивают при комнатной температуре 2 часа и упаривают. Остаток разбавляют СН2Cl2, а затем упаривают и повторяют упаривание три раза, получают заглавное соединение в виде бесцветного масла, 433 мг, выход 100%.
Стадия 3.
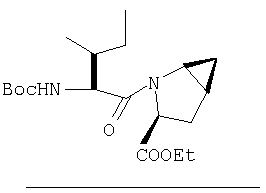
К перемешиваемому раствору (S)-N-трет-бутоксикарбонилизолейцина (372,6 мг, 1,61 ммол) и гексафтопирфосфата бензотриазол-1-илоксипирролидинфосфония (1,25 г, 2,42 ммол) в CH2Cl2 (6 мл) под азотом при комнатной температуре добавляют 4-метилморфолин (NMM) (0,36 мл, 3,2 ммол). Через 5 мин добавляют раствор соединения Стадии 2 (433 мг, 1,61 ммол) и NMM (0,27 мл, 2,4 ммол) в CH2Cl2 (1 мл). После прибавления реакционную смесь перемешивают под азотом при комнатной температуре в течение ночи. Реакционную смесь разбавляют CH2Cl2 (40 мл) и промывают 4% KHSO4 (10 мл), водным раствором NaHCO3 (10 мл) и рассолом (10 мл), сушат (Na2SO4) и упаривают. Очистка с помощью флеш-хроматографии (1:4 EtOAc/гексан) дает заглавное соединение в виде бесцветного масла, 530 мг, выход 89%).
Стадия 4
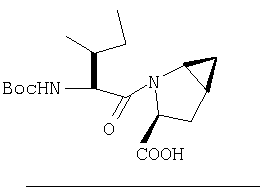
К перемешиваемому раствору соединения Стадии 3 (530 мг, 1,44 ммол) в МеОН (4 мл) и Н2О (4 мл) при комнатной температуре добавляют LiOH-Н2О (91 мг, 2,16 ммол). Реакционную смесь перемешивают при комнатной температуре в течение ночи и упаривают. К остатку добавляют воду (10 мл) и экстрагируют Et2O (2×10 мл). Водный слой подкисляют до рН ˜4 добавлением по каплям 4% KHSO4. Молочный «раствор» экстрагируют EtOAc (15 мл × 3). Объединенные вытяжки EtOAc промывают рассолом, сушат Na2SO4 и упаривают, получают заглавное соединение в виде белого твердого вещества, 440 мг, выход 90%.
Стадия 5
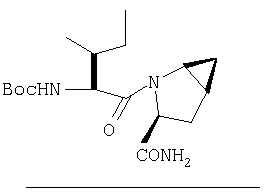
К перемешиваемому раствору соединения Стадии 4 (300 мг, 0,88 ммол) в ТГФ (6 мл) при -15°С под азотом прибавляют 4-метилморфолин (0,12 мл, 1,06 ммол), а затем изобутилхлорформиат (0,13 мл, 0,97 ммол) за 2 мин. Образуется белый осадок. Реакционную смесь перемешивают при -15°С под азотом в течение 25 мин и добавляют раствор NH3, в диоксане (8,8 мл, 4,4 ммол). Реакционную смесь перемешивают при -15°С 30 мин, доводят до комнатной температуры и перемешивают в течение ночи при rt. Реакцию прекращают (гасят), добавляя 4% KHSO4 до рН ˜4 и экстрагируют EtOAc (20 мл × 3). Объединенные вытяжки промывают рассолом (10 мл), сушат (Na2SO4) и упаривают. Очистка с помощью флеш-хроматографии на колонке (1:1 EtOAc/гексан) дает заглавное соединение в виде белой пены, 268 мг, выход 90%.
Стадия 6
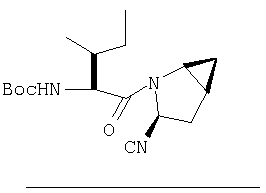
К перемешиваемому раствору соединения Стадии 5 (248 мг, 1,38 ммол) и имидазола (94 мг, 1,38 ммол) в сухом пиридине (12 мл) при 35°С под азотом по каплям добавляют POCl3 (0,26 мл, 2,76 ммол). Реакционную смесь перемешивают при - 35÷-20°С 1 час и упаривают. Добавляют CH2Cl2 (10 мл), образуется белый осадок. Фильтруют, фильтрат упаривают и очищают флеш-хроматографией (2:5 EtOAc/гексан), получают заглавное соединение в виде бесцветного масла, 196 мг, выход 88%.
Стадия 7
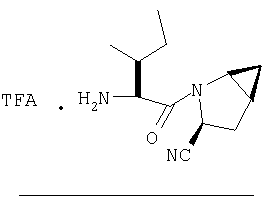
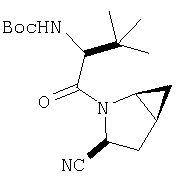
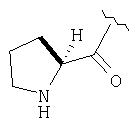
К перемешиваемому раствору соединения Стадии 6 (130 мг, 0,4 ммол) в CH2Cl2 (2 мл) при комнатной температуре добавляют ТФК (2 мл). Реакционную смесь перемешивают при этой температуре 2 ч и медленно прибавляют к предварительно охлажденной эмульсии NaHCO3 (3,8 г) в Н2О (3 мл). Смесь экстрагируют СН2Cl2 (6 мл × 5), объединенные CH2Cl2 вытяжки упаривают и очищают препаративной ВЭЖХ, получая заглавное соединение в виде белого порошка, 77 мг, выход 57%, т.пл 141-143°С. LC/MS дает корректный молекулярный ион [(М+Н)+=222] заданного соединения.
Пример 2.
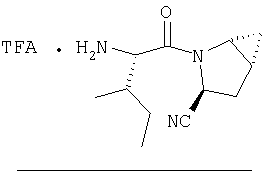
Стадия 1
Заглавный продукт Стадии 1 синтезируют по известной методике [Stephen Hanessian, Ulrich Reinhold, Michel Saulnier and Stephen Claridge; Bioorganic and Medicinal Chemistry Letters 8 (1998) 2123-2128.
Стадия 2
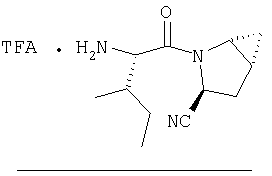
Заглавное соединение получают из соединения Стадии 1, используя методику, описанную в Примере 1, Стадии 2-6. LC/MS дает корректный молекулярный ион [(М+Н)+=222] заданного соединения.
Пример 3.
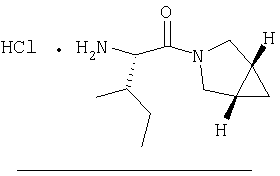
Стадия 1
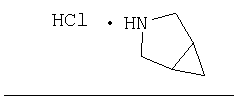
Заглавное соединение Стадии 1 получают по известной методике. [Willy D. Kollmeyer, Патент США 4183857].
Стадия 2
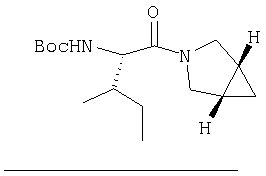
К перемешиваемому раствору (S)-трет-бутоксикарбонилизолейцина (231 мг, 1 ммол) и бензотриазол-1-илокситрипирролидинфосфония гексафторфосфата (780 мг, 1,5 ммол) в CH2Cl2 (6 мл) под азотом при комнатной температуре добавляют 4-метилморфолин (0,33 мл, 3 ммол). Через 5 мин добавляют сразу все количество соединения Стадии 1 (120 мг, 1 ммол). Реакционную смесь перемешивают под азотом при комнатной температуре в течение ночи, а затем разбавляют CH2Cl2 (30 мл), промывают 4,1% раствором KHSO4 (10 мл), водным раствором NaHCO3 (10 мл), рассолом (10 мл), сушат (Na2SO4) и упаривают. Очистка флеш-хроматографией на силикагеле (колонка 2,4×20 см, EtOAc/гексан 1:3) дает заглавное соединение в виде бесцветное масло, 290 мг, выход 90%. LC/MS дает корректный молекулярный ион [(+H)+=297] заданного соединения.
Стадия 3
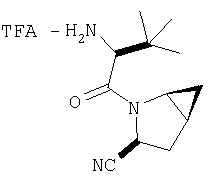
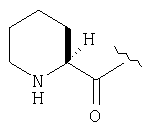
Реакционную смесь соединения Стадии 2 (220 мг, 0.74 ммол) и 4 М HCl в диоксане (1,5 мл, 6 ммол) перемешивают при комнатной температуре 2 часа и упаривают в вакууме. К остатку добавляют Et2O, образуется осадок. Осадок сушат в вакууме. Осадок сушат в вакууме, получают заглавное соединение в виде белого порошка, 130 мг (выход 76%), т.пл 205-206°С. LC/MS дает корректный молекулярный ион [(М+H)+=197] заданного соединения.
Примеры 4-4А
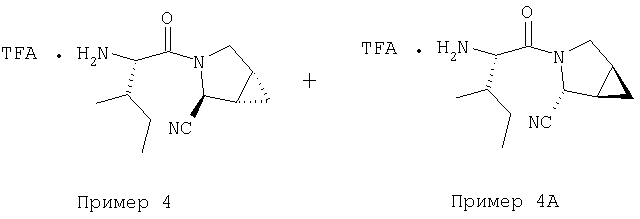
Стадия 1
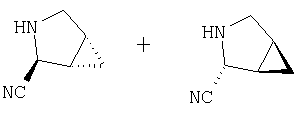
Соединение Стадии 1 получают в виде смеси энантиомеров 1:1 получают по известной методике. [Willy D. Kollmeyer, Патент США 4183857].
Стадия 2
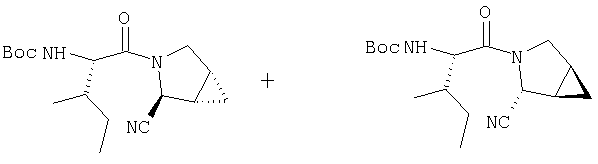
Суспензию (S)-N-трет-бутоксикарбонилизолейцина (92,5 мг, 0,4 ммол), 1-[(3-(диметил)амино)пропил]-3-этилкарбодиимида (77 мг, 0,4 ммол) и НОАТ (54,4 мг, 0,4 ммол) в ClCH2CH2Cl (0,3 мл) перемешивают под азотом при комнатной температуре 1 ч, добавляют соединение Стадии 1 (22 мг, 0,2 ммол), а затем Et3N (0,015 мл, 0,1 ммол). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем разбавляют CH2Cl2 (3 мл), промывают Н2О (1 мл), водным раствором NaHCO3 (1 мл) и рассолом (1 мл), сушат (Na2SO4) и упаривают. Очисткой флеш-хроматографией на силикагеле (колонка 2,4×12 см, EtOAc/гексан 2:7) получают заглавное соединение в виде бесцветного масла, 33 мг, выход 51%. LC/MS дает корректный молекулярный ион [(М+H)+=322] заданного соединения.
Стадия 3
К перемешиваемому раствору соединения Стадии 2 (30 мг, 0,4 ммол) в CH2Cl2 (0,5 мл) при комнатной температуре добавляют ТФК (0,5 мл). Реакционную смесь перемешивают при комнатной температуре 2 ч. Реакционную смесь медленно добавляют к предварительно охлажденной суспензии NaHCO3 (0,8 г) в Н2О (1 мл). Смесь экстрагируют CH2Cl2 (2 мл × 5), объединенные CH2Cl2 - вытяжки упаривают и очищают препаративной ВЭЖХ, получают заглавные соединения с соотношением стереоизомеров 1:1, 22 мг, выход 73%. LC/MS дает корректный молекулярный ион [(М+Н)+=222] заданных соединений.
Примеры 5-5A
Стадия 1
К раствору соединения Стадии 1 из Примера 4 (150 мг, 1,39 ммол) в 2-пропаноле (0,8 мл) добавляют NaCN (40 мг, 1,0 ммол). Реакционную смесь кипятят 3 часа. По охлаждении до комнатной температуры реакционную смесь упаривают, а затем суспендируют в Et2O (5 мл), фильтруют, фильтрат упаривают, получают смесь соединения Стадии 1 Примера 4 и Стадии 1 Примера 5 (140 мг, 98%) при соотношении диастереоизомеров 2:1, причем каждый является рацемической смесью.
Стадия 2
Суспензию (S)-N-трет-бутоксикарбонилизолейцина (595 мг, 2,57 ммол), 1-[(3-(диметил)амино)пропил]-3-этилкарбодиимида (493 мг, 2,57 ммол) и 1-гидрокси-7-азабензотриазола (350 мг, 2,57 ммол) в ClCH2CH2Cl (2 мл) перемешивают под азотом при комнатной температуре 1 час, затем прибавляют смесь соединений Стадии 1 (139 мг, 1,28 ммол). Реакционную смесь перемешивают под азотом при комнатной температуре в течение ночи, затем разбавляют CH2Cl2 (30 мл), промывают Н2О (10 мл), насыщенным водным раствором NaHCO3 (10 мл) и рассолом (10 мл), сушат (Na2SO4) и упаривают.
Очисткой с помощью флеш-хроматографии на силикагеле (колонка 2,4×20 см, EtOAc/гексан) получают соединение Стадии 2 из Примера 4 (260 мг) и заглавные соединения (105 мг) с соотношением диастереомеров 1:1. LC/MS дает корректный молекулярный ион [(М+Н)+=322] для заданных соединений.
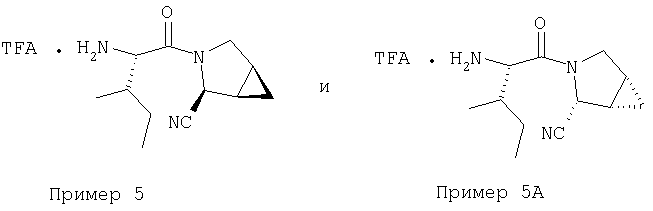
Стадия 3
К перемешиваемому раствору соединений Стадии 2 (104 мг, 0,32 ммол) в СН2Cl2 (1 мл) при комнатной температуре прибавляют ТФК (1 мл). Реакционную смесь перемешивают при этой температуре 2 часа и медленно прибавляют к предварительно охлажденной суспензии NaHCO3 (2 г) в Н2О (2 мл). Смесь экстрагируют CH2Cl2 (4 мл × 4), объединенные CH2Cl2-вытяжки упаривают и очищают препаративной ВЭЖХ, получают заглавное соединение Примера 5 (36 мг) и Примера 5А (36 мг). LC/MS дает корректный молекулярный ион [(М+H)+=222] для заданных соединений.
Пример 6
Общий метод А: Синтетические методы получения ингибиторов из промышленных аминокислот аналогичны. Как показано на Схеме 3, сложный эфир 11, описанный в Примере 1 Стадия 1, омыляют до кислоты, действуя LiOH в ТГФ/Н2О, и превращают в амид 12 обработкой изобутилхлорформиатом/NMM, а затем аммиаком в диоксане. Защитную группу, Boc, снимают в кислой среде, применяя ТФК в хлористом метилене, получают 13. Соль ТФК реагирует с Boc-трет.-бутилглицином либо под действием EDAC/НОВТ/ДМФА, либо в присутствии EDAC/DMAP/CH2Cl2, давая 14. Амид дегидратируют до нитрила 15, применяя POCl3/ имидазол в пиридине при -20°С, и, наконец, снимают защиту с помощью ТФК в СН2Cl2 при комнатной температуре, получая целевое соединение 16.
Схема 3, Общий метод А (Примеры 6-27)
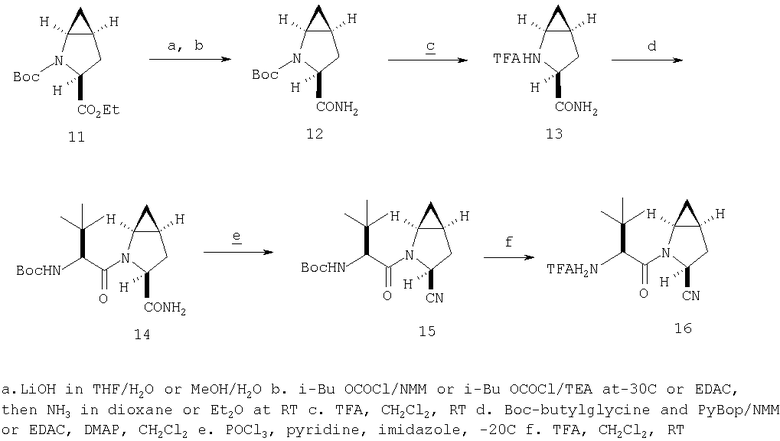
Стадия 1
К перемешиваемому раствору соединения Стадии 1 Примера 1 (1,40 г, 5,49 ммол) в 40 мл водного метанола (1:1) при комнатной температуре добавляют гидроксид лития (0,20 г, 8,30 ммол). Реакционную смесь перемешивают при этой температуре 18 ч, а затем 2 ч греют при 50°С. Смесь разбавляют равными объемами эфира и воды (50 мл), а затем подкисляют KHSO4 до рН 3. «Молочный раствор» экстрагируют эфиром (3×20 мл). Объединенные эфирные вытяжки сушат Na2SO4 и упаривают. Остаток летучих отгоняют с толуолом (2×10 мл), сушат в вакууме, получают заглавное соединение в виде вязкого сиропа, 1,20 г, 96%.
Стадия 2
К перемешиваемому раствору соединения Стадии 1 (1,20 г, 5,28 ммол) в ТГФ (20 мл) при -15°С под азотом прибавляют 4-метилморфолин (0,71 мл, 6,50 ммол), а затем за 5 мин прибавляют изобутилхлорформиат (0,78 мл, 6,00 ммол). Реакционную смесь перемешивают при -15° 30 мин, охлаждают до -30° и обрабатывают раствором NH3 в диоксане (50 мл, 25 ммол). Перемешивают при -30°С 30 мин, доводят до комнатной температуры и перемешивают в течение ночи. Добавляют раствор лимонной кислоты (рН 4) и экстрагируют эфиром (3×50 мл). Объединенные органические вытяжки промывают рассолом, сушат Na2SO4 и концентрируют. После очистки флеш-хроматографией на колонке с силикагелем с элюентом - EtOAc получают соединение стадии 2, 1,00 г, 84%.
Стадия 3
К перемешиваемому раствору соединения Стадии 2 (0,90 г, 4,00 ммол) в CH2Cl2 (3 мл) при 0°С добавляют ТФК (3 мл). Реакционную смесь перемешивают при 0°С 18 ч, упаривают в вакууме, получая заглавное соединение в виде вязкого масла, 0,98 г, 100%. Масло постепенно кристаллизуется при продолжительном стоянии.
Стадия 4
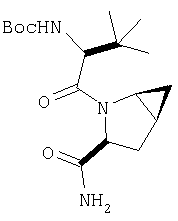
В высушенную в сушильном шкафу пробирку на 15 мл помещают соединение Стадии 3 (56 мг, 0,22 ммол), N-трет-бутоксикарбонил-(L)-трет-лейцин (53 мг, 23 ммол), диметиламинопиридин (0,11 г, 0,88 ммол) и CH2Cl2 (4 мл). Пробирку герметично закрывают под азотом, прибавляют 1-[(3-(диметил)амино)пропил]-3-этилкарбодиимид (84 мг, 0,44 ммол). Смесь помещают в вибратор (качалку) и встряхивают в течение ночи. Продукт очищают экстракцией из твердой фазы, применяя колонку United Technology SCX (2 г сорбента в колонке 6 мл), материал наносят на SCX - ионообменную колонку и последовательно промывают СН2Cl2 (5 мл), 30%-ным метанолом в CH2Cl2 (5 мл), 50%-ным метанолом в CH2Cl2 (5 мл) и метанолом (10 мл). Фракции, содержащие продукт, упаривают в вакууме, получая заданный амид. Дополнительная очистка обращенно-фазовой препаративной хроматографией на колонке YMC S5 ODS 20 Х 250 мм дает заглавное соединение, 50 мг (выход 68%). Условия очистки: градиентная элюция от 30% метанол /вода /0,1 ТФК до 91% метанол/вода/0,1 ТФК за 15 мин. 5 мин выдерживают с элюентом 90% метанол/вода/0,1 ТФК. Скорость потока 20 мл/мин. Детектируют при длине волны 220. Время удерживания 14 мин.
Стадия 5
В высушенную в сушильном шкафу пробирку на 15 мл помещают соединение Стадии 4 (50 мг, 0,15 ммол), имидазол (31 мг, 0,46 ммол) и пиридин (1 мл). Пробирку герметично закрывают под азотом и охлаждают до - 30°С.Медленное прибавление POCl3 (141 мг, 88 мкл, 0,92 ммол) дает при перемешивании густую суспензию. Содержимое пробирки перемешивают при - 30°С 3 час и летучие упаривают. Продукт очищают твердофазной экстракцией на экстракционной колонке United Technology с силикагелем (2 г сорбента в колонке на 6 мл), материал наносят на колонку с силикагелем и последовательно промывают СН2Cl2 (5 мл), 7% метанолом в СН2Cl2 (5 мл) (10 мл). Фракции, содержащие продукт, объединяют и упаривают в вакууме, получая заглавное соединение, 46 мг, 96%.
Стадия 6
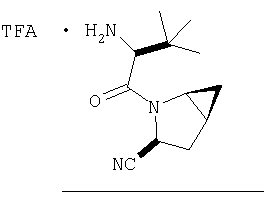
В высушенную в сушильном шкафу пробирку на 15 мл помещают соединение Стадии 5 (0,45 мг, 0,14 ммол), CH2Cl2 (1 мл) и ТФК (1 мл). Реакционную смесь встряхивают 40 мин при комнатной температуре, добавляют толуол (4 мл) и упаривают в вакууме до густого масла. Продукт очищают обращенно-фазовой препаративной хроматографией на колонке YMC S5 ODS 20 Х 250 мм, получая конечное соединение Примера 6, 14 мг, 35%. Условия очистки: градиентная элюция от 10% метанол/вода/0,1 ТФК за 15 мин; 5-минутная выдержка при 90% метанол/вода/0,1 ТФК. Скорость тока: 20 мл/мин. Определение при длине волны 220. Время удерживания: 10 мин.
Соединения в Примерах 7-27 получают из выпускаемых промышленностью аминокислот по методике из Примера 6.
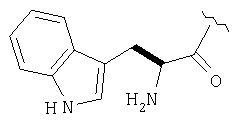
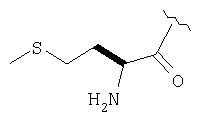
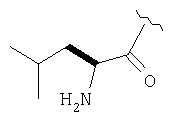
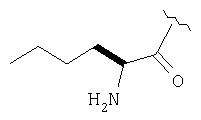
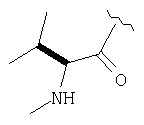
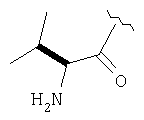
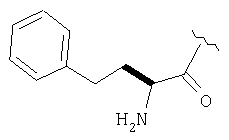
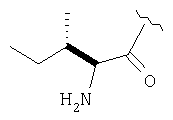
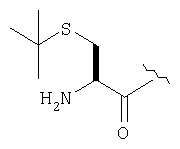
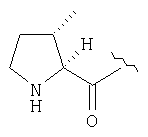
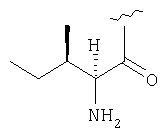
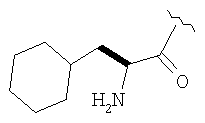
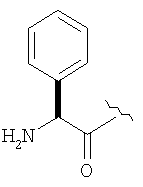
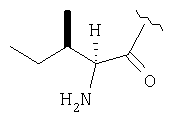
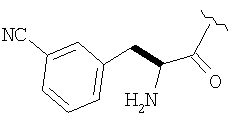
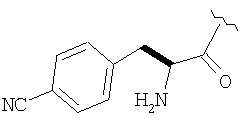
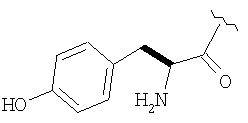
Пример 27
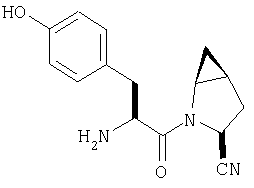
Стадия 1
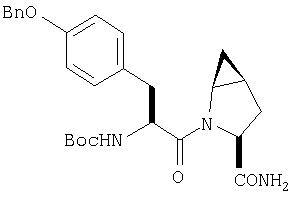
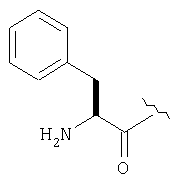
ТФК-соль карбоксиламида (2S, 4S, 5S)-4,5-метан-L-пролина (53 мг, 0,22 ммол) реагирует с N-Boc-L-тирозинбензиловым эфиром (82 мг, 0,22 ммол) в присутствии PyBop (172 мг), 33 ммол) N-метилморфолина (67 мг, 66 ммол) в 4 мл CH2Cl2. Реакционную смесь перемешивают 16 ч, добавляют EtOAc, промывают H2O, 1 N водной HCl, рассолом, затем упаривают и очищают флеш-хроматографией на силикагеле, получают продукт конденсации (MS, ББА: МН+ 480).
Стадия 2
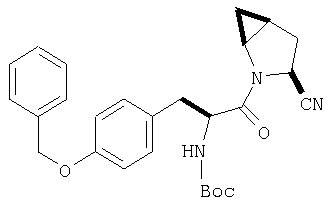
Амид, полученный на Стадии 1, дегидратируют до нитрила согласно общему методу С (описан после Примера 29) (ББА МН+ 462).
Стадия 3
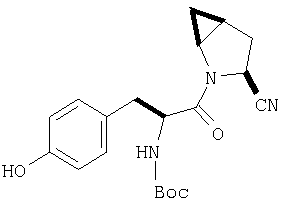
Бензиловый эфир, полученный на Стадий 2, расщепляют каталитическим гидрогенолизом при давлении водорода 1 атмосфера в метаноле в присутствии палладия на угле при комнатной температуре в течение 1,5 ч. Реакционную смесь фильтруют через целит и упаривают, получают масло, которое используют без дополнительной очистки (ББА MH+ 372).
Стадия 4
N-[N-Boc-L-Тирозин-]-(2S,4S,5S)-2-циано-4,5-метан-L-пролиламид (продукт Стадии 3) растворяют в CH2Cl2 и при комнатной температуре добавляют ТФК. Реакционную смесь перемешивают 1 час, упаривают и очищают препаративной ВЭЖХ, как описано в общем методе В (описан после Примера 29), получают заглавное соединение (ББА MH+ 272).
Пример 28.
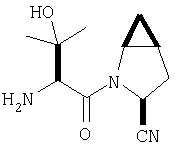
Заглавное соединение получают реакцией ТФК соли карбоксиламида(2S,4S,5S)-метан-L-пролина, описанной в Примере 6, Стадия 3, с N-(трет-бутилоксикарбонилгидроксивалином. После защиты гидроксильной группы с помощью триэтилсилилхлорида, дегидратации амида в присутствии POCl3 / имидазол в пиридине и снятия защиты (N-концевой азот и гидроксил валина) под действием ТФК по общему методу С (ББА MH+ 224) получают заглавное соединение.
Пример 29
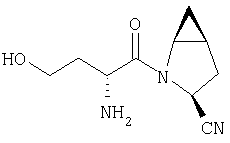
Стадия 1
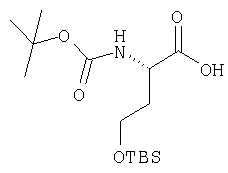
Густую суспензию N-Boc-L-гомосерина (1,20 г, 5,47 ммол), трет-бутилдиметилсилилхлорида (1,67 г, 11,04 ммол) и имидазола (938 мг, 13,8 ммол) в ТГФ (17 мл) перемешивают под азотом 48 ч. Растворитель упаривают, сырой продукт растворяют в МеОН (10 мл). Полученный раствор перемешивают 2 ч при комнатной температуре. Растворитель упаривают и сырой продукт растворяют в CH2Cl2 (50 мл) и обрабатывают 0,1 N HCl (2×10 мл). Слой CH2Cl2 промывают рассолом и сушат MgSO4. Удаление летучих дает заглавное соединение в виде масла (1,8 г), которое используют без дополнительной очистки (LC/Macc-спектр., + ион): 334 (М+Н).
Стадия 2
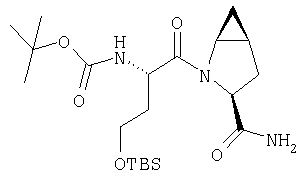
К раствору соединения Стадии 2 (333 мг, 1,0 ммол) в 6 мл CH2Cl2 при перемешивании прибавляют 1-[3-(диметиламино)-пропил]-3-этилкарбодиимида гидрохлорида (256 мг, 1,32 ммол). Раствор перемешивают при комнатной температуре 30 мин, а затем добавляют ТФК-соль амина из Примера 6, Стадия 3 (160 мг, 0,66 ммол) и 4-(диметиламино)пиридин (244 мг, 2,0 ммол). Раствор перемешивают при комнатной температуре в течение ночи. К смеси добавляют СН2Cl2 (5 мл) и промывают последовательно Н2О, 10% лимонной кислотой, рассолом, сушат Na2SO4 и упаривают, получают заглавное соединение (350 мг), которое используют без дополнительной очистки (LC/Macc-спектр., + ион): 442 (М+Н).
Стадия 3
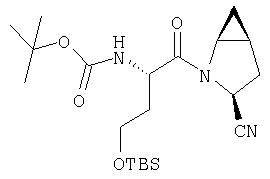
В высушенную сушильном шкафу круглодонную колбу на 10 мл помещают соединение Стадии 2 (350 мг, 0,79 ммол), имидазол (108 мг, 1,58 ммол), пиридин (3 мл). Колбу под аргоном охлаждают до -30°С. При медленном прибавлении POCl3 при перемешивании густая эмульсия. Эмульсию перемешивают 3 часа при -30°С и летучие упаривают. Затем добавляют хлористый метилен (5 мл) и нерастворимые твердые вещества отфильтровывают. Органический слой промывают H2O, 10% лимонной кислотой, рассолом, сушат Na2SO4. Удаление растворителя дает сырой заданный нитрил (330 мг) (LC/Macc-спектр. + ион): 424 (М+Н).
Стадия 4
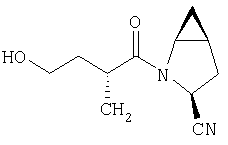
К раствору соединения Стадии 3 (330 мг, 0,58 ммол) в 3,3 мл CH2Cl2 при перемешивании прибавляют трифторуксусную кислоту (3,3 мл). Раствор перемешивают при комнатной температуре 30 мин, добавляют несколько капель воды и смесь перемешивают 0,5 ч, разбавляют CH2Cl2 (5 мл) и упаривают в вакууме до густого масла. Продукт очищают обращенно-фазовой препаративной хроматографией на колонке YMC S5 ODS 20×100, получают заглавное соединение, 59 мг.17%. Условия очистки: градиентная элюция от 10% метанол/вода/0,1 ТФК до 90% метанол/вода/0,1 ТФК за 15 мин; выдерживают 5 мин при 90% метанол/вода/0,1 ТФК. Скорость тока: 20 мл/мин. Длина волны при детектировании: 220. Время удерживания 10 мин. (LC/Macc-спектр, + ион): 210 (М+Н).
Общий метод В: Перегруппировка Кляйзена дает Вос-защищенные аминокислоты.
Общий метод В дает четвертичные Вос-защищенные аминокислоты. В Примерах 30-47 даны аминокислоты с винильной группой в боковой цепи, представителем является соединение 20 на Схеме 4. Циклопентанон «олефинируют» по реакции Хорнера - Эванса, получая соединение 17, которое восстанавливают до аллилового спирта 18 с помощью DEBAL-Н в толуоле от -78°С до комнатной температуры. Аллиловый спирт 18 этерифицируют N-Вос-глицином в присутствии DCC/DMAP в СН2Cl2, получают соединение 19. Глициновый эфир 19 в результате перегруппировки Кляйзена под действием кислоты Льюиса, а именно путем образования комплекса с безводным хлористым цинком и депротонирования при -78°С диизопропиламидом лития с последующим нагреванием до комнатной температуры получают 20.
Схема 4, Общий метод В, Примеры 30-47
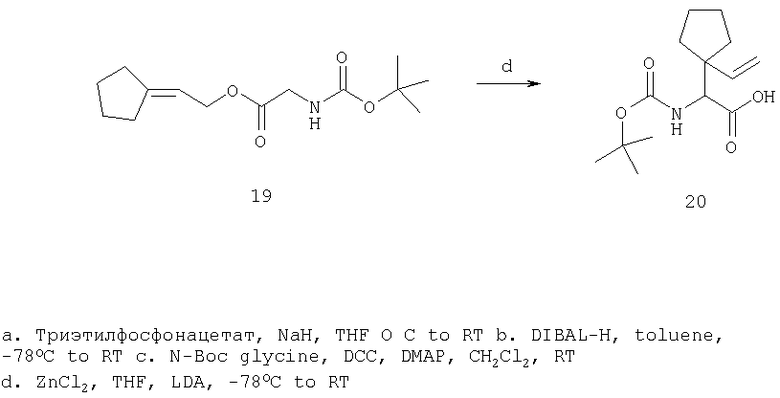
Стадия 1
Этиловый эфир диклопентилиденуксусной кислоты.
В высушенную на пламени (горелки) круглодонную колбу на 500 мл, в которую помещен NaH (5,10 г 60% суспензии в минеральном масле, 128 ммол 1,10 эквив) в 120 мл сухого ТГФ, при 0°С под аргоном из капельной воронки добавляют триэтилфосфонацетат (25,6 мл, 128 ммол, 1,10 эквив). Смесь доводят до комнатной температуры, перемешивая еще 1 ч. По каплям в течение 20 мин добавляют циклопентанон (10,3 мл, 116 ммол) в 10 мл безводного ТГФ и смесь перемешивают при комнатной температуре 2,5 ч. Затем добавляют эфир (200 мл) и воду (100 мл), слои разделяют. Органическую фазу промывают последовательно водой (100 мл) и рассолом (100 мл), сушат (Na2SO4) и упаривают в вакууме, получая 17,5 г (98%) заданного эфира в виде бесцветного масла.
Стадия 2
2-Циклопентинилиденэтанол
В высушенную на пламени круглодонную колбу на 500 мл с помещенным в нее этиловым эфиром циклопентилиденуксусной кислоты (17,5 г, 113 ммол) в 100 мл безводного толуола при -78°С под аргоном по каплям из капельной воронки за 30 мин добавляют DIBAL-Н (189 мл 1,5 М раствора в толуоле, 284 ммол, 2,50 эквив), доводят смесь до комнатной температуры, перемешивают 18 ч. Затем реакционную смесь снова охлаждают до -78°С и разбавляют, осторожно добавляя 30 мл безводного МеОН. Доводят до комнатной температуры, добавляют 1 N соль Rochelle (100 мл) и перемешивают смесь 90 мин. Двухфазную реакционную смесь разбавляют Et2O (200 мл) в делительной воронке и слои разделяют. Органический слой промывают рассолом (100 мл), сушат (Na2SO4) и упаривают в вакууме. Очистка флеш-хроматографией (силикагель, CH2Cl2/EtOAc, 10:1) дает 11,6 г (92%) заданного аллилового спирта в виде бесцветного масла.
Стадия 3
(2-Циклопентилиденэтил)N-(трет-бутилоксикарбонил)глицинат
В высушенную на пламени круглодонную колбу на 500 мл, содержащую N-(трет-бутилоксикарбонил)глицин (13,45 г, 76,75 ммол) в 100 мл CH2Cl2 при комнатной температуре добавляют соединение Стадии 2 (8,61 г, 76,75 ммол, 1 эквив) в 20 мл СН2Cl2, а затем дициклогексилкарбодиимид (16,63 мг, ммол, 1,05 эквив) в 80 мл СН2Cl2. К этой реакционной смеси добавляют диметиламинопиридин (0,94 мг, ммол, 0,10 эквив), смесь перемешивают в течение ночи, фильтруют через воронку со стеклянным фильтром, промывают 100 мл СН2С2 и упаривают в вакууме. Сырой продукт очищают флеш-хроматографией (силикагель, смесь гексанов/EtOAc, градиент от 20:1 до 1:1), получают 19,43 г (94%) заданного глицинилового эфира в виде бесцветного масла.
Стадия 4
N-(трет-бутилоксикарбонил)(1'-винилциклопентил)-глицин
В высушенную на пламени круглодонную колбу на 500 мл под аргоном помещают ZnCl2 (11,8 г, ммол, 1,20 эквив) и 20 мл толуола. Смесь нагревают в вакууме при энергичном перемешивании, отгоняя следы влаги азеотропной перегонкой с толуолом и повторяя этот процесс (2 х). Колбу охлаждают под аргоном до комнатной температуры, добавляют (2-циклопентилиденэтил)-N-(трет-бутилксикарбонил)глицинат (19,36 г, 71,88 ммол) в виде раствора в 180 мл ТГФ через иглу, смесь охлаждают до -78°С. В отдельной высушенной на пламени круглодонной колбе на 200 мл, содержащей диизопропиламин (26,3 мл, ммол, 2,60 эквив) в 90 мл ТГФ при -78°С добавляют н-бутиллитий (71,89 мл 2,5 М раствора в смеси гексанов, ммол, 2,5 эквив) и температуру смеси доводят до 0°С за 30 мин, прежде чем охладить ее до -78°С. Полученный таким образом литийдиизопропиламин добавляют через иглу по каплям к ZnCl2 - эфирной смеси с постоянной скоростью за 40 мин, образующуюся реакционную смесь медленно доводят до комнатной температуры и перемешивают в течение ночи. Желтую реакционную смесь выливают в делительную воронку, разбавляют 300 мл Et2O и образующийся органический раствор промывают последовательно 300 мл 1 N HCl и 300 мл рассола, сушат (Na2SO4) и упаривают в вакууме. Очистка флеш-хроматографией (силикагель, 3% МеОН в CH2Cl2 с 0,5% НОАс) дает 17,8 г (92%) заданного аминокислотного продукта в виде белого твердого вещества. (ББА МН+ 270).
Пример 30
Общий метод С: Конденсация пептида с 4,5-метанпролинамидом, дегидратация амида и конечное снятие защиты.
ТФК-соль амида 13 конденсируют с рядом рацемических защищенных аминокислот с четвертичным атомом азота в присутствии HOBT/EDC в ДМФА при комнатной температуре, получают смесь D/L диастереоизомеров по N-концевой аминокислоте. Заданный L-диастереоизомер выделяют хроматографически либо как амид 21, либо как нитрил 22. Нитрил 22 получают обработкой амида POCl3/имидазол в пиридине при -20°С. Конечный целевой 23 получают в кислой среде, применяя ТФК в CH2Cl2.
Схема 5, Общий Метод С
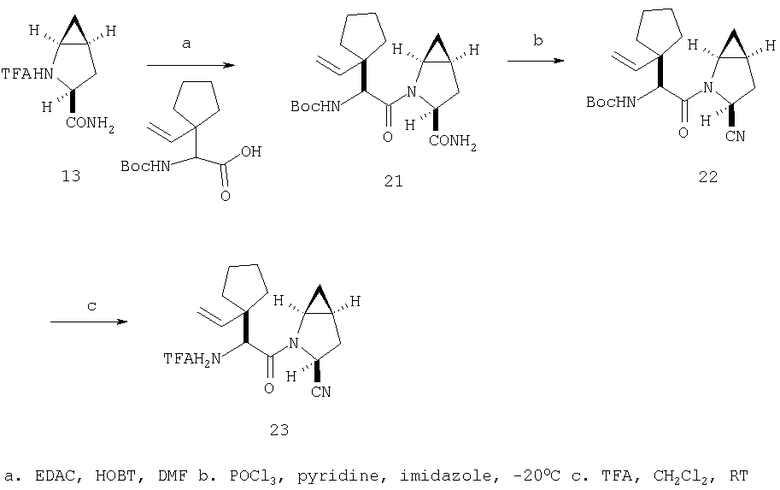
Стадия 1
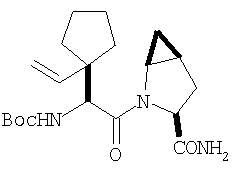
Соединение Стадии 3 Примера 6 (877 мг, 3,65 ммол) и N-Boc-циклопентилвиниламинокислоту, описанную на Стадии 4 Общего метода В (1,13 г, 4,20 ммол) растворяют в 20 мл безводного ДМФА, охлаждают до 0°С и к этой смеси добавляют EDAC (1,62 г, 8,4 ммол), HOBT гидрат (2,54 г, 12,6 ммол) и TEA (1,27 г, 12,6 ммол), реакционную смесь доводят до комнатной температуры и перемешивают 24 часа. Добавляют EtOAc (100 мл), промывают Н2О (3×20 мл), сушат (Na2SO4) и очищают флеш-хроматографией на колонке (100% EtOAc), получают 1,38 г (86%) соединение Стадии 1 (МН+, 378).
Стадия 2
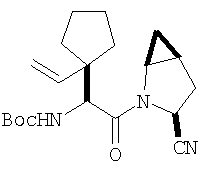
Соединение Стадии 1 (1,38 г, 3,65 ммол) и имидазол (497 мг, 7,30 ммол) сушат азеотропной отгонкой воды с толуолом (5 мл х 2), растворяют в 10 мл сухого пиридина, охлаждают до -30°С под азотом и шприцом добавляют POCl3 (2,23 г, 14,60 ммол). Реакция заканчивается через 1 час, реакционную смесь упаривают досуха, остаток очищают повторяемой дважды флеш-хроматографией на колонке с силикагелем. Первую колонку (100% EtOAc) применяют для очистки смеси стереоизомеров (1,15 г, 88%) от побочных продуктов реакции. На второй колонке (градиент от 25% EtOAc/смесь гексанов до 50% EtOAc/смесь гексанов) разделяют смесь диастереомеров и получают 504 мг заданного нитрила Стадии 2 (МН+ 360).
Стадия 3
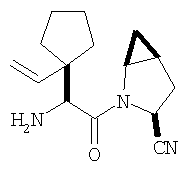
Соединение Стадии 2 (32 мг, 0,09 ммол) растворяют в 1 мл СН2Cl2 и добавляют 1 мл ТФК, реакционную смесь перемешивают 30 мин при комнатной температуре и упаривают досуха. Продукт очищают обращенно-фазовой препаративной хроматографией на колонке YMC S5 ODS 20 Х 250 мм, получают 12 мг ТФК-соли (лиофилизированной из воды или выделенной после упаривания элюента и затирания с эфиром) заглавного соединения. Условия очистки: градиентная элюция из 10% метанол/вода/0,1 ТФК до 90% метанол/вода/0,1 ТФК за 18 мин; 5 мин выдерживают при 90% метанол/вода/0,1 трифторуксусной кислоты. Скорость потока: 20 мл/мин. Определяемая длина волны: 220.
Соединения примеров 30-39 получают методами, описанными в Общем методе В и в Общем методе С, исходными являются циклопентанон, циклобутанон, циклогексанон, циклогептанон, циклооктанон, цис-3,4,-диметилциклопентанон и 4-пиранон, циклопропанэтилполуацеталь, ацетон и 3-пентанон соответственно.
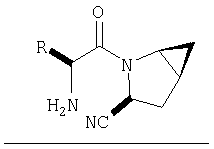
Пример 40.
Стадия 1
Соединение Стадии 1 получают, применяя общий метод В, исходя из циклопентанона и 2-фторфосфонацетата вместо триэтилфосфонацетата
Стадия 2
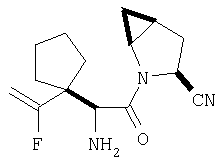
Заглавное соединение получают пептидным синтезом из кислоты Стадии 1 с последующей дегидратацией и конечным снятием защиты по общему методу С [MS (М+Н) 278].
Пример 41.
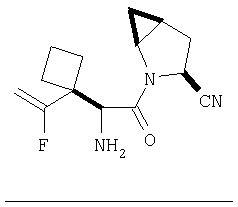
Стадия 1
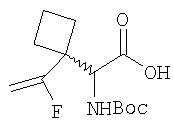
Соединение Стадии 1 получают по общему методу В из исходных циклобутанона и 2-фтортриэтилфосфонацетата вместо триэтилфосфонацетата.
Стадия 2
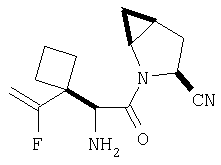
Заглавное соединение получают пептидным синтезом из кислоты Стадии 1 с последующей дегидратацией и финальным снятием защиты по общему методу С. MS (М+Н) 264.
Пример 42
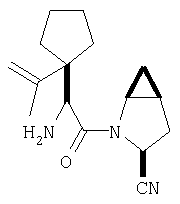
Стадия 1
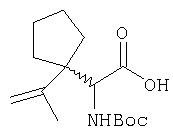
Соединение Стадии 1 получают по общему методу В из исходных циклопентанона и триэтилфосфонопропионата вместо триэтилфосфонацетата.
Стадия 2
Заглавное соединение получают пептидным синтезом из кислоты Стадии 1 с последующей дегидратацией и финальным снятием защиты по общему методу С. MS (М+Н) 274.
Пример 43.
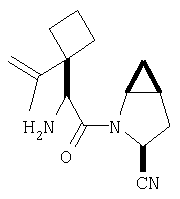
Стадия 1
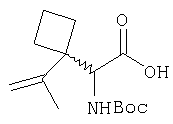
Соединение Стадии 1 получают по общему методу В из исходных циклобутанона и триэтилфосфонпропионата вместо триэтилфосфонацетата.
Стадия 2
Заглавное соединение получают пептидным синтезом из кислоты Стадии 1 с последующей дегидратацией и финальным снятием защиты по общему методу С. MS (М+Н) 260.
Пример 44.
Общий Метод D: Окислительное расщепление винильного заместителя с помощью озонолиза. Защищенный циклопентилвинильный нитрил 22 обрабатывают озоном 6-8 минут и подвергают восстановительному разложению с непосредственным образованием гидроксиметильного аналога 24. Это соединение депротекционируют в кислой среде ТФК в CH2Cl2 при 0°С, получая заглавное соединение 25.
Схема 6. Общий Метод D, Примеры 44, 46, 48
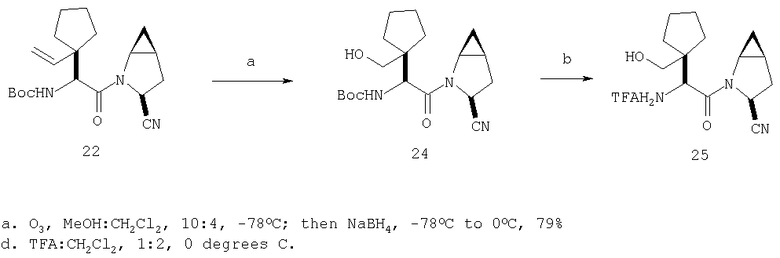
Стадия 1
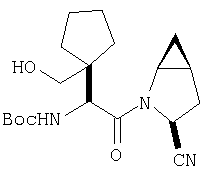
Циклопентилвинильное соединение, полученное на Стадии 2 общего метода С (1,28 г, 3,60 ммол), растворяют в 56 мл смеси CH2Cl2: метанол 2:5, охлаждают до -78°С и пропускают ток озона до появления голубой окраски, в это время добавляют NaBH4 (566 мг, 15,0 ммол, 4,2 эквив) и доводят температуру реакционной смеси до 0°С. Через 30 мин добавляют 2 мл насыщенного водного раствора NaHCO3 и доводят до комнатной температуры. Реакционную смесь упаривают досуха и добавляют EtOAc. Добавляют небольшое количество воды для растворения неорганических примесей и разделяют слои. EtOAc слой сушат (Na2SO4), фильтруют и упаривают, получая масло, которое очищают колоночной флеш-хроматографией на силикагеле с EtOAc, получают 922 мг (71%) соединения Стадии 1. MS (M+H) 364.
Стадия 2
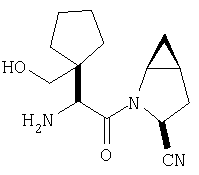
Соединение стадии 1 (900 мг, 2,48 ммол) растворяют в 60 мл СН2Cl2, охлаждают до 0°С и обрабатывают 20 мл свежеперегнанной ТФК. Реакция заканчивается через 80 мин, и смесь упаривают досуха и очищают препаративной ВЭЖХ (YMC S5 ODS 30×100 мм, 18-минутный градиент от 80% Раств. А: Раств. В до 100% Раств. В. Растворитель А=10% МеОН - 90% Н2О - 0,1% ТФК, Растворитель В=90% МеОН - 10% Н2О - 0,1% ТФК), собирают продукт от 5,1-6,5 мин, после лиофилизации из воды получают 660 мг (71%) ТФК соли заглавного соединения в виде белого лиофилизата. (МН+ 264).
Пример 45
Общий метод Е: Окислительное расщепление винильного заместителя тетроксидом осмия - периодатом натрия с последующим восстановлением натрийборгидридом до спирта. Циклобутилолефин 26 обрабатывают тетроксидом осмия и периодатом натрия в смеси ТГФ: вода, 1: 1, и промежуточный альдегид выделяют в сыром виде и сразу же восстанавливают натрийборгидридом, получая 27 с выходом 56%. Стандартные условия депротекционирования с применением ТФК дают целевое соединение 28.
Схема 7, общий метод Е, Примеры 45, 47
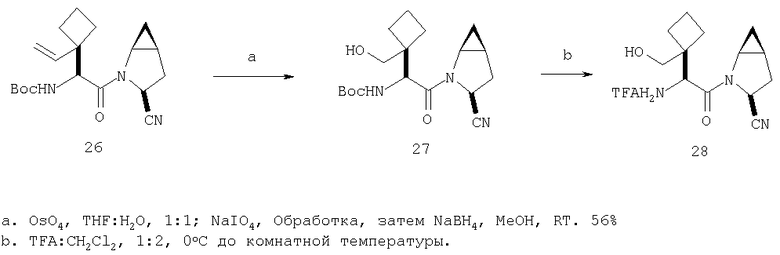
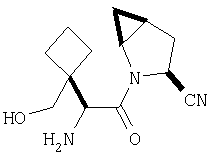
Стадия 1
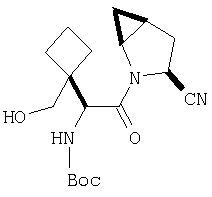
Циклобутилвинильное соединение с N-Boc-защитной группой (Пример 31, получено общим методом С) (0,16 г, 0,46 ммол) растворяют в 10 мл смеси ТГФ: вода (1:1) и обрабатывают OsO4 (12 мг, катализатор) и NaIO4 (0,59 г, 2,76 ммол, 6 эквив). Через 2 ч реакционную смесь разбавляют 50 мл эфира и 10 мл воды. Слои уравновешивают и органическую фракцию промывают один раз раствором NaHCO3, сушат MgSO4 и упаривают, получая темное масло. Масло разбавляют 10 мл метанола и обрабатывают NaBH4 (0,08 г, 2,0 ммол). Смесь становится очень темной, и через 30 мин ее разбавляют эфиром и добавляют к ней водный раствор NaHCO3. Смесь уравновешивают и слои разделяют. Органическую фракцию промывают раствором NaHCO3 и 0,1 М HCl. Органические вытяжки сушат MgSO4 и упаривают, получают 90 мг (56%) соединения Стадии 1 в виде темного масла.
Стадия 2
Соединение Стадии 1 (90 мг, 0,26 ммол) растворяют в 3 мл CH2Cl2, охлаждают до 0°С и обрабатывают 3 мл свежеперегнанной ТФК. Реакция заканчивается через 30 мин, реакционную смесь упаривают досуха и очищают препаративной ВЭЖХ (YMC S5 ODS 30×100 мм, 10-минутный градиент от 100% А до 100% В, Растворитель А=10% МеОН - 90% Н2О - 0,1% ТФК, Растворитель В=90% МеОН - 10% Н2O - 0,1% ТФК), получают, после удаления растворителя, 50 мг (60%) заглавного соединения. (МН+ 250).
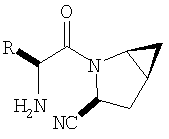
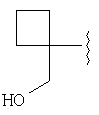
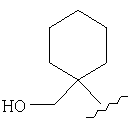
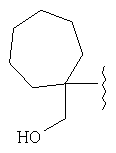
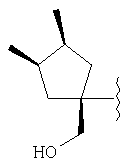
Пример 49
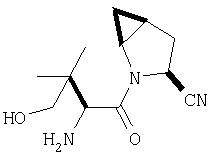
Стадия 1
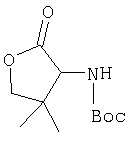
Часть А. В колбу на 50 мл помещают дигидро-4,4-диметил-2,3-фурандион (5,0 г, 39 ммол). уксусную кислоту (10 мл), ацетатнатрия (3,82 г, 39 ммол) и гидроксиламин гидрохлорид (2,71 г, 39,0 ммол). Реакционную смесь перемешивают 2 ч при комнатной температуре и упаривают в вакууме для удаления основной части уксусной кислоты. Остаток выливают в воду (100 мл) и водную фазу экстрагируют EtOAc (3×40 мл). Органический слой сушат Na2SO4 и упаривают, получая бесцветное масло, затвердевающее при стоянии.
Часть В. В круглодонную колбу на 200 мл помещают твердое вещество Части А (˜39 ммол) и добавляют 80 мл этанола и 39 мл 2 N HCl (78 ммол). Смесь обрабатывают 1,0 г 5% Pd/C и дегазируют. Колбу помещают в атмосферу Н2 на 8 ч, фильтруют через целит и фильтрат упаривают, получая грязно-белое твердое вещество.
Часть С. В круглодонную колбу на 250 мл помещают твердое вещество из Части В и добавляют ТГФ (50 мл) и воду (15 мл). Смесь обрабатывают дитрет-бутилкарбонатом (12,7 г, 117 ммол) и бикарбонатом натрия (10,0 г, 117 ммол). После 4-часового перемешивания к смеси добавляют 50 мл эфира и 50 мл воды. Разделяют слои и органический слой сушат MgSO4 и упаривают. Остаток очищают флеш-хроматографией на колонке с силикагелем с 30% EtOAc в смеси гексанов, получают 2,00 г (22% общий выход) соединения Стадии 1 в виде белого твердого вещества.
Стадия 2
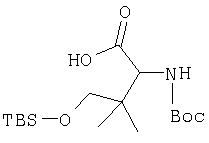
К раствору соединения Стадии 1 (1,00 г, 3,80 ммол) в ТГФ (20 мл) при комнатной температуре под азотом при перемешивании добавляют LiOH гидрат (0,16 г, 3,80 ммол), а затем воду (5 мл). Реакционную смесь перемешивают при 40°С 0,5 ч и охлаждают до комнатной температуры. Смесь упаривают досуха и остаток упаривают из ТГФ (2Х), толуола (2Х) и ТГФ (1X). Остаток - стекловидное вещество - разбавляют 5 мл ТГФ и добавляют имидазол (0,63 г, 9,19 ммол), а затем трет-бутилдиметилсилилхлорид (1,26 г, 8,36 ммол). Реакционную смесь перемешивают в течение ночи и добавляют 10 мл метанола. Перемешивают 1 час и упаривают. Добавляют еще одну порцию метанола и смесь упаривают. Масло разбавляют эфиром и 0,1 N HCl (рН 2). Слои уравновешивают и водный слой отбрасывают. Органическую фракцию сушат MgSO4 и упаривают, получают 1,25 г (83%) соединения Стадии 2 в виде бесцветного стекловидного вещества.
Стадия 3
Заглавное соединение получают пептидным синтезом из карбоновой кислоты Стадии 2 и амина из Примера 6 Стадия 3 с последующей дегидратацией и депротекционированием по Общему методу С. MS (M+H) 238.
Общий Метод F: Каталитическое гидрирование винильного заместителя. Как показано на Схеме 8, защищенная аминокислота 20 превращается в соответствующий насыщенный аналог 29 каталитическим гидрированием на 10% Pd/C при атмосферном давлении водорода.
Схема 8, Общий Метод F, Примеры 50-56
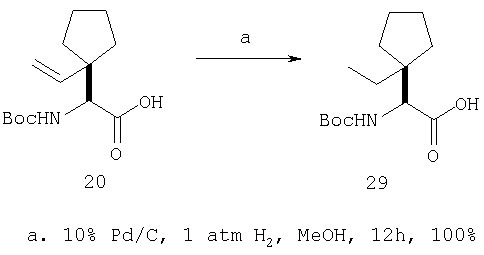
Стадия 1.
N-(трет-Бутилоксикарбонил)(1'-винилциклопентил)глицин (2,23 г, 8,30 ммол) растворяют в 50 мл МеОН и помещают в сосуд для гидрирования, продутый аргоном. К этой смеси добавляют 10% Pd/С (224 мг, 10 вес %) и перемешивают при 1 атм Н; при комнатной температуре 12 час. Реакционную смесь фильтруют через целит и упаривают, очищают флеш-хроматографией на колонке с силикагелем, элюент 1:9 метанол: СН2Cl2, получают стекловидное соединение Стадии 1. (ББА МН+ 272).
Соединения из Примеров 50-56 получают пептидным синтезом из аминокислот (в которых винильный заместитель гидрируют по Общему методу F) с последующей дегидратацией и снятием защиты по Общему методу С.
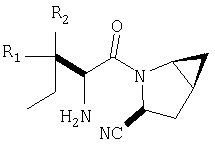
Пример 57.
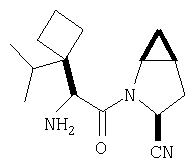
Заглавное соединение в Примере 57 получают пептидным синтезом из изопропилциклобутановой аминокислоты, в которой олефиновый заместитель гидрирован по Общему методу F) с последующей дегидратацией и депротекционированием по общему методу С.
Пример 58.
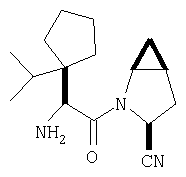
Заглавное соединение в Примере 58 получают пептидным синтезом из изопропилциклопентановой аминокислоты (в которой олефиновый заместитель гидрирован по Общему методу F) с последующей дегидратацией и депротекционированием по Общему методу С. MS (M+H) 276.
Общий Метод G: L-Аминокислоты, полученные асимметрическим синтезом Штреккера. Получаемые промышленно адамантилкарбоновые кислоты этерифицируют либо в метаноле с HCl при кипячении, либо в присутствии триметилсилилдиазометана в Et2О/метаноле, получая 30. Простой эфир восстанавливают в спирт 31 LAH в ТГФ, а затем окисляют по Сверну, получая альдегид 32. Альдегид 32 в условиях асимметрического синтеза Штреккера в присутствии KCN, NaHSO3 и R-(-)-2-фенилглицинола превращается в 33. Нитрил 33 в присутствии сильной кислоты - 12 М HCl в НОАс - дает 34. Хиральное соединение каталитическим восстановлением в присутствии катализатора Перлмана в метаноле, содержащем кислоту, при давлении водорода 50 psi (344, 738 кПа), превращают в 35, а образующуюся аминогруппу защищают в виде трет-бутилкарбамата, соединение 36.
Схема 9, Общий Метод G, Примеры 59-64
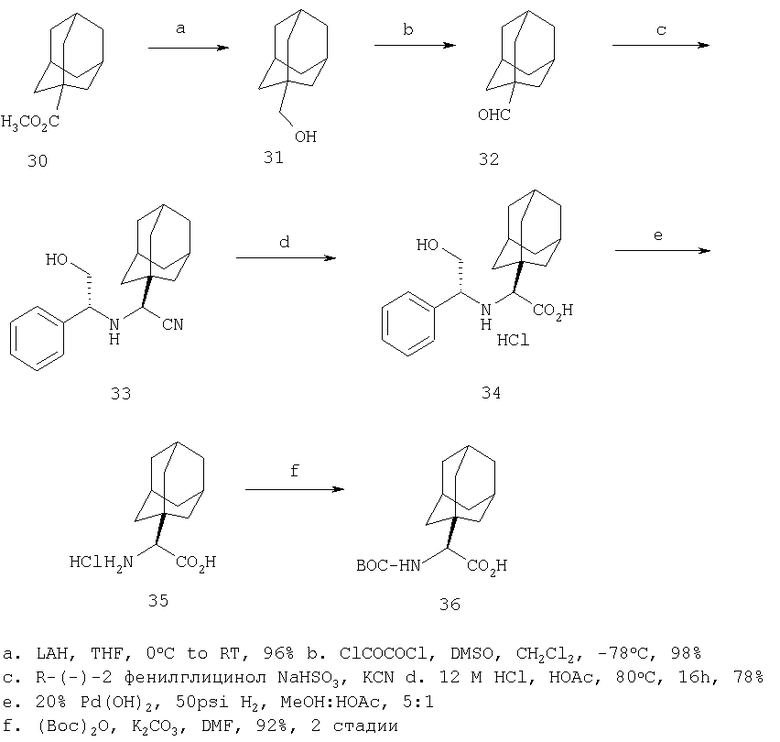
Стадия 1
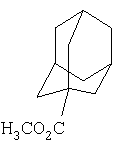
Адамантан-1-карбоновую кислоту (10,0 г, 55 ммол, 1 эквив) растворяют в смеси Et2O (160 мл) и МеОН (40 мл), прибавляют триметилсилилдиазометан (2,0 М в гексане, 30 мл, 60 ммол, 1,1 эквив) и перемешивают при комнатной температуре 3 ч. Летучие отгоняют на роторном испарителе и продукт очищают флеш-хроматографией на колонке с силикагелем (5×15 см), элюент 40% СН2Cl2/смесь гексанов, получают продукт в виде белого кристаллического вещества (10,7 г, 100%).
Стадия 2
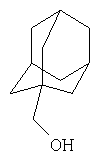
Соединение Стадии 1 (10,7 г, 0,055 ммол, 1 эквив) растворяют в безводном ТГФ (150 мл) под аргоном и обрабатывают раствором LiAlH4 (1 M в ТГФ, 69 мл, 69 ммол, 1,25 эквив). Перемешивают при комнатной температуре 1,5 ч, охлаждают до 0°С и последовательно промывают Н2О (5,1 мл), 15% водн. NaOH (5,1 мл) и Н2О (10,2 мл). Перемешивают 15 мин при комнатной температуре, суспензию фильтруют под вакуумом, твердый осадок промывают EtOAc (2×100 мл). Фильтрат упаривают на роторном испарителе, а твердый остаток очищают флеш-хроматографией на колонке с силикагелем (5×15 см), элюент 10% EtOAc/CH2Cl2. При этом получают продукт Стадии 2 в виде белого твердого вещества (8,74 г, 96%).
Стадия 3
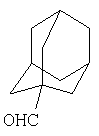
В высушенную в сушильном шкафу трехгорлую колбу с капельной воронкой на 125 мл под аргоном помещают сухой CH2Cl2 (150 мл) и сухой ДМСО (10,3 мл, 0,145 мол, 2,5 эквив) и охлаждают до -78°С. По каплям медленно прибавляют оксалилхлорид (6,7 мл, 0,0768 мол, 1,32 эквив), перемешивают 15 мин, получая активированный аддукт с ДМСО. Прибавляют раствор соединения Стадии 2 (9,67 г, 58,2 ммол, 1 эквив) в сухом CH2Cl2 (75 мл) и перемешивают реакционную смесь 1 ч. К образовавшейся смеси белого цвета по каплям прибавляют триэтиламин (40,5 мл, 0,291 мол, 5 эквив). Через 30 мин отставляют охлаждающую баню, и реакционную смесь промывают последовательно холодным 20% водным раствором КН2РО4 (25 мл) и холодной водой (150 мл). Перемешивают при комнатной температуре 15 мин, добавляют Et2О (400 мл) и слои разделяют. Органический слой промывают холодным 10%-ным водным раствором КН2PO4 (3×150 мл) и насыщенным водным раствором NaCl (100 мл), сушат Na2SO4, фильтруют и упаривают. Остаток очищают флеш-хроматографией на колонке с силикагелем, элюент СН2Cl2, получают соединение Стадии 3 в виде белого твердого вещества (9,40 г, 98%).
Стадия 4
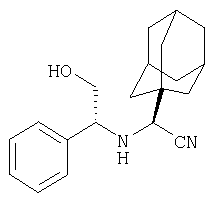
Соединение Стадии 3 (9,40 г, 57 ммол, 1 эквив) суспендируют в Н2О (145 мл) и охлаждают до 0°С. Смесь обрабатывают NaHSO3 (5,95 г, 57 ммол, 1 эквив), KCN (4,0 г, 59 ммол, 1,04 эквив) и раствором (R)-(-)-фенилглицинола (8,01 г, 57 ммол, 1 эквив) в МеОН (55 мл). Образующуюся смесь 2 часа перемешивают при комнатной температуре, затем кипятят 16 ч. Смесь охлаждают до комнатной температуры и добавляют 200 мл EtOAc. Перемешивают 15 мин и слои разделяют. Водную фракцию экстрагируют EtOAc. Объединенные EtOAc вытяжки промывают рассолом (50 мл) сушат безводным Na2SO4, фильтруют и фильтрат упаривают. Продукт очищают флеш-хроматографией на колонке с силикагелем (6,4×20 см), элюент 20% EtOAc/смесь гексанов, получают заданный (R,S)-продукт в виде белого твердого вещества (11,6 г, 37,4 ммол, 65%): MS m/e 311 (M+H)+.
Стадия 5
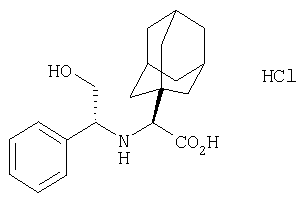
Нитрил Стадии 4 (5,65 г, 18 ммол) нагревают в концентрированной HCl (120 мл) и НОАс (30 мл) при 80°С 18 ч. Реакционную смесь охлаждают в бане со льдом. Фильтрование под вакуумом дает заданный продукт в виде белого твердого вещества (5,21 г, 14 ммол, 78%). MS m/e 330 (M+H)+.
Стадия 6
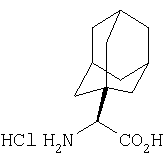
Соединение стадии 5 (5,21 г, 14 ммол) растворяют в МеОН (50 мл) и НОАс (10 мл) и гидрируют Н2 (50 psi) в присутствии катализатора Перлмана (20% Pd(OH)2, 1,04 г, 20 вес.%) 18 ч. Реакционную смесь фильтруют через мембранный фильтр PTFE и катализатор промывают МеОН (3×25 мл). Фильтрат упаривают на роторном испарителе, получают белое твердое вещество. Продукт используют на Стадии 7 без дополнительной очистки.
Стадия 7
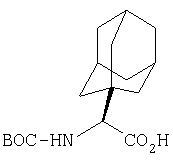
Сырое соединение Стадии 6 (˜14 ммол) растворяют в безводном ДМФА (50 мл) под аргоном и обрабатывают К2СО3 (5,90 г, 42 ммол, 3 эквив) и дитрет-бутилкарбонатом (3,14 г, 14 ммол, 1 эквив) под аргоном при комнатной температуре. Через 19 ч ДМФА отгоняют на роторном испарителе (насос) и остаток сушат дополнительно в вакууме, смешивают с водой (100 мл) и Et2О (100 мл), слои разделяют и щелочной водный раствор промывают Et2O для удаления побочных продуктов гидрогенолиза. Водный слой охлаждают до 0°С, добавляют EtOAc (200 мл) и при энергичном перемешивании подкисляют 1 N водн HCl до рН 3. Слои разделяют и водный слой экстрагируют EtOAc (100 мл). Объединенные EtOAc-вытяжки промывают рассолом (50 мл), сушат (Na2SO4), фильтруют и фильтрат упаривают на роторном испарителе. Остаток очищают флеш-хроматографией на колонке с SiO2 (5×12 см), элюент 5% МеОН/СН2Cl2+0,5% НОАс. Продукт затирают с гексаном, получают белую пену (4,07 г, 13 ммол, 92%). MS m/e 310 (М+Н)+.
Пример 59.
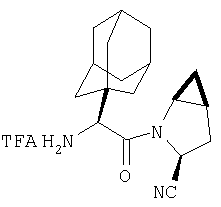
Заглавное соединение в Примере 59 получают пептидным синтезом из соединения Стадии 7 по Общему методу G с последующей дегидратацией и депротекционированием по Общему методу С. MS m/e 300 (m+H)+.
Пример 60.
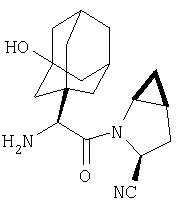
Стадия 1
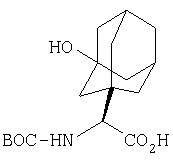
Раствор KMnO4 (337 мг, 2,13 ммол, 1,1 эквив) в 2% водн. КОН (6 мл) нагревают до 60°С и порциями добавляют соединение Стадии 7, полученное по Общему методу G (600 мг, 1,94 ммол, 1 эквив). Нагревают до 90°С. Через 1,5 ч реакционную смесь охлаждают до 0°С, добавляют EtOAc (50 мл) и смесь осторожно подкисляют 1 N HCl до рН 3. Слои разделяют и водный слой экстрагируют EtOAc (50 мл). Объединенные органические вытяжки промывают рассолом, сушат Na2SO4, фильтруют и упаривают. Остаток очищают флеш-хроматографией на колонке с силикагелем (3,8×15 см), элюент 2% (200 мл), 3% (200 мл), 4% (200 мл) и 5% (500 мл) МеОН/СН2Cl2+0,5% НОАс. После выделения продукт затирают с гексаном, получают белое твердое вещество (324 мг, 51%): MS m/e 326 (m+H)+.
Стадия 2
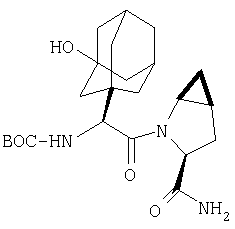
Соединение Стадии 1 (404 мг, 1,24 ммол, 1 эквив) растворяют в безводном ДМФА (10 мл) под аргоном и охлаждают до 0°С. Следующие агенты добавляют в таком порядке: соль из Примера 6 Стадия 3 (328 мг, 1,37 ммол, 1,1 эквив), НОВТ (520 мг, 3,85 ммол, 3,1 эквив), EDAC (510 мг, 2,61 ммол, 2,1 эквив) и TEA (0,54 мл, 3,85 ммол, 3,1 эквив). Реакционную смесь в течение ночи доводят до комнатной температуры и отгоняют ДМФА на роторном испарителе (насос). Остаток сушат дополнительно в вакууме, растворяют в EtOAc (100 мл), промывают нас. водн. NaCl (25 мл), сушат безводным Na2SO4, фильтруют и упаривают на роторном испарителе. Продукт очищают флеш-хроматографией на колонке с силикагелем (3,8×15 см) с градиентом 6% (200 мл), 7% (200 мл) и *% (500 мл) MeOH/CH2Cl2, получают продукт в виде белого твердого вещества (460 мг, 1,06 ммол, 85%): MS m/e 434 (m+H)+.
Стадия 3
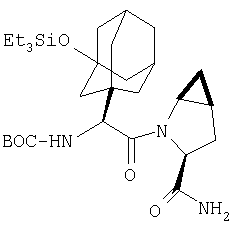
Соединение Стадии 2 (95 мг, 0,22 ммол, 1 эквив) растворяют в безводном CH2Cl2 (2,5 мл) под аргоном и охлаждают до -78°С. Смесь обрабатывают диизопропилэтиламином (65 мкл, 37 ммол, 1,7 эквив) и триэтилсилилтрифлатом (75 мкл, 0,33 ммол, 1,5 эквив) и перемешивают при 0°С 1,5 ч. Смешивают с МеОН (0,5 мл), силикагелем (200 мг) и Н2О (2 капли) и перемешивают при комнатной температуре 18 ч. Растворитель отгоняют на роторном испарителе и остаток очищают флеш-хроматографией на колонке с силикагелем (2,5×10 см), элюент MeOH/CH2Cl2, получают продукт (92 мг, 0,17 ммол, 77%): MS m/e 548 (m+H)+.
Стадия 4
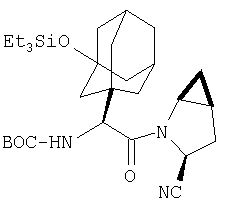
Соединение Стадии 3 (90 мг, 0,16 ммол, 1 эквив) под аргоном растворяют в безводном пиридине (2 мл) и охлаждают до -30°С. Обработка имидазолом (24 мг, 0,35 ммол, 2,1 эквив) хлорокисью фосфора (ббмкл, 0,67 ммол, 4,1 эквив) и последующее нагревание при -30°С в течение 45 мин дает густую суспензию. Летучие удаляют отгонкой на роторном испарителе, а твердый остаток сушат в вакууме. Продукт очищают флеш-хроматографией на колонке с силикагелем (2,5×10 см), элюент 7% EtOAc/СН2Cl2. Получают белую пену (76 мг, 87%): MS m/e 530 (m+H)+.
Стадия 5
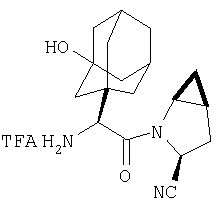
Соединение Стадии 4 (76 мг, 0,14 ммол) растворяют в безводном CH2Cl2 (1 мл) и охлаждают до 0°С, добавляют ТФК (1 мл) и Н2О (2 капли) и перемешивают 1,5 ч при 0°С. Растворители удаляют отгонкой на роторном испарителе, к остатку добавляют толуол (5 мл) и сушат в вакууме. Затирают с Et2O и получают заглавное соединение в виде белого твердого вещества (54 мг, 88%): MS m/e 316 (m+H)+.
Пример 61.
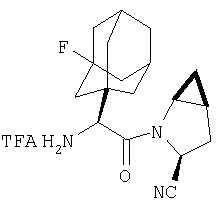
Стадия 1
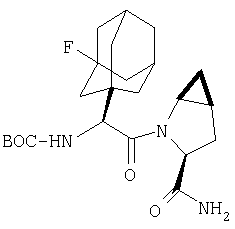
В высушенную в сушильном шкафу, продутую аргоном колбу помещают безводный СН2Cl2 (3 мл) и охлаждают до -78°С. Прибавляют диэтиламиносульфуртрифторид (DAST, 60 мкл, 45 ммол, 1,5 эквив), а затем раствор соединения Стадии 2 Пример 60 (131 мг, 0,30 ммол, 1 эквив) в сухом CH2Cl2 (3 мл). Через 15 мин реакционную смесь выливают в делительную воронку, содержащую нас. водн. NaHCO3 (25 мл) и слои разделяют. Водный слой экстрагируют СН2Cl2 (25 мл), объединенные органические вытяжки промывают рассолом (10 мл), сушат (Na2SO4), отфильтровывают и упаривают. Продукт очищают флеш-хроматографией на колонке с силикагелем (2,5×10 см), элюент 5% MeOH/CH2Cl2, получают соединение Стадии 1 (124 мг, 0,29 ммол, 94%): MS m/e 436 (m+H)+.
Стадия 2
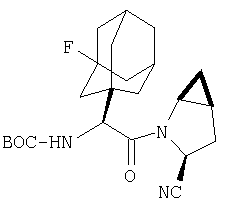
Фторсодержащий амид Стадии 1 (161 мг, 0,37 ммол, 1 эквив) растворяют в безводном пиридине (4 мл) под аргоном и охлаждают до -30°С. К смеси добавляют имидазол (54 мг, 0,77 ммол, 2,1 эквив) и хлорокись фосфора (143 мкл, 1,52 ммол, 4,1 эквив) и перемешивают при -30°С 40 мин. Растворитель отгоняют на роторном испарителе дополнительно сушат в вакууме. Продукт очищают флеш-хроматографией на колонке с силикагелем (2,5×10 см), элюент 5% EtOAc/CH2Cl2. Получают соединение Стадии 2 в виде белой пены (126 мг, 82%): MS m/e 418 (m+H)+.
Стадия 3
Соединение Стадии 2 (125 мг, 0,30 ммол) растворяют в ТФК/СН2Cl2 (1:1 об/об, 2 мл) и перемешивают при комнатной температуре. Через 30 мин растворители отгоняют на роторном испарителе, дважды добавляют и отгоняют толуол (2×5 мл), твердый остаток сушат в вакууме. Затирают с Et2О, получают заглавное соединение в виде белого твердого вещества (93 мг, 0,21 ммол, 72%): MS m/e 318 (m+H)+.
Пример 62.
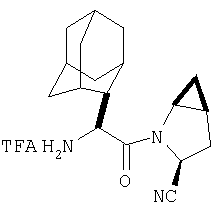
Стадия 1
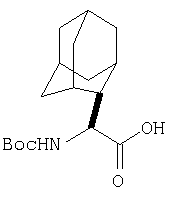
Соединение Стадии 1, гомохиральную Boc-аминокислоту, получают из 2-адамантаналя асимметрическим синтезом Штреккера по Общему методу G.
Стадия 2
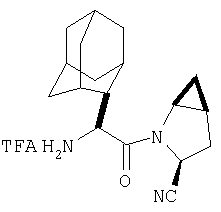
Заглавное соединение в Примере 62 получают пептидным синтезом из 2-адамантиламинокислоты, описанной на Стадии 1, с последующей дегидратацией и депротекционированием по Общему методу С. MS (M+H) 300.
Пример 63.
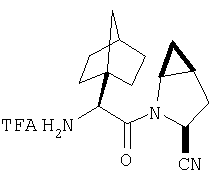
Стадия 1
В высушенную в сушильном шкафу колбу с холодильником и осушительной трубкой помещают норборнан-2-карбоновую кислоту (4,92 г, 35 ммол, 1 эквив) и добавляют бром (2,1 мл, 41 ммол, 1,15 эквив) и треххлористый фосфор 90.153 мл, 1,8 ммол, 0,05 эквив). Смесь греют при 85°С 7 ч в темноте. Добавляют бром (0,4 мл, 7,8 ммол, 0,22 эквив) и греют еще 1 час. Смесь охлаждают до комнатной температуры и добавляют Et2О (100 мл). Смесь промывают 10 NaHSO3 (50 мл), Н2О (2×50 мл) и рассолом (25 мл). Эфирный слой сушат (Na2SO4), фильтруют и упаривают на роторном испарителе. Продукт очищают флеш-хроматографией на колонке с силикагелем (5×15 см), элюент 2-4% MeOH/СН2Cl2+0,5% НОАс. Выделенный продукт содержит два неразделяющихся компонента (4,7 г) и его используют в следующей стадии без дополнительной очистки.
Сырой продукт, полученный выше, экзо-2-бромнорборнан-1 карбоновую кислоту (4,7 г, неочищенная) в Et2О (80 мл) и МеОН (20 мл) смешивают с триметилсилилдиазометаном (2,0 М в гексане, 11,8 мл, 23,6 мол) и перемешивают при комнатной температуре 1 час. Растворитель отгоняют на роторном испарителе, масло очищают флеш-хроматографией на колонке с силикагелем (5×18 см) в градиенте СН2Cl2/смеси гексанов (600 мл каждого, 20% и 30%), затем СН2Cl2, получают продукт в виде белого твердого вещества (3,97 г, 0,017 мол, 79% на 2 стадии): MS m/e 233/235 (m+Н)+.
Метиловый эфир экзо-2-бромборнан-1-карбоновой кислоты (2,0 г, 8,58 ммол, 1 эквив) растворяют в безводном ТГФ (50 мл) в колбе, высушенной в сушильном шкафу, снабженной холодильником и продутой аргоном. К смеси добавляют AIBN (288 мг, 1,71 ммол, 0,2 эквив), а затем греют при кипении 2 ч. Колбу охлаждают до комнатной температуры, ТГФ отгоняют на роторном испарителе и получают сырой продукт. Очищают флеш-хроматографией на колонке с силикагелем (5×10 см), элюент 5% EtOAc/смесь гексанов. Образующийся продукт используют на следующей стадии без дополнительной очистки.
Стадия 2
Соединение Стадии 2, гомохиральную Boc-аминокислоту получают исходя из 1-норборнилметилкарбоксилата асимметрическим синтезом Штреккера по Общему методу G.
Стадия 3
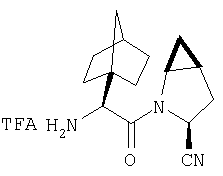
Заглавное соединение в Примере 63 получают пептидным синтезом из описанной на Стадии 1 1-норборниламинокислоты с последующей дегидратацией и снятием защиты по Общему методу С. MS (M+H) 260.
Пример 64.
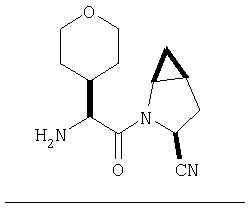
Стадия 1
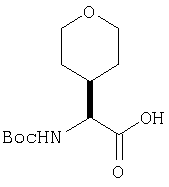
Соединение Стадии 1, гомохиральную Boc-аминокислоту, получают исходя из 4-формилпирана, с применением асимметрического синтеза Штреккера по Общему методу G.
Стадия 2
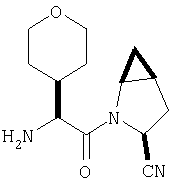
Заглавное соединение в Примере 64 получают пептидным синтезом описанной на стадии 2 4-пираниламинокислоты с последующей дегидратацией и депротекционированием по Общему методу С. MS (M+H) 250.
Общий Метод Н: Синтез рацемических аминокислот по Штреккеру.
Схема 10, Общий Метод Н, Примеры 65-66
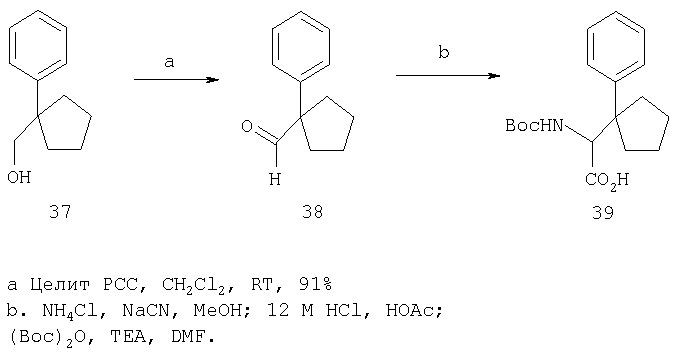
Стадия 1
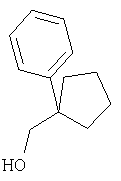
К раствору 1-фенилцикло-1-пентанкарбоновой кислоты (5,00 г, 26,3 ммол) в 25 мл ТГФ при 0°С при перемешивании добавляют LAH (52 мл, 52 ммол, 1 М) в ТГФ. Реакционную смесь медленно доводят до комнатной температуры, а затем кипятят 18 ч. Реакцию прекращают по методу Физера: осторожно добавляют 2 мл Н2О; 6 мл 15% NaOH в воде; и 2 мл воды. Двухфазную смесь разбавляют 100 мл эфира и отфильтровывают белые гранулы. Эфирную фракцию сушат над Na2SO4 и упаривают, получают 4,30 г (93%) соединения Стадии 1.
Стадия 2
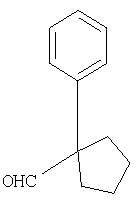
К раствору соединения Стадии 1 (0,80 г, 4,50 ммол) в 15 мл СН2Cl2 при перемешивании при комнатной температуре добавляют целит (5 г), а затем РСС (1,95 г, 5,00 ммол). Перемешивают при этой температуре 3 ч, добавляют 40 мл СН2Cl2 и фильтруют через целит. Фильтрат еще раз фильтруют через силикагель, получают бесцветный фильтрат. СН2Cl2 - раствор упаривают, получают 0,72 г (91%) альдегида в виде бесцветного масла.
Стадия 3
К соединению Стадии 2 (0,72 г, 4,20 ммол) в круглодонной колбе на 50 мл в 8 мл воды при комнатной температуре добавляют NaCN (0,20 г, 4,20 ммол), а затем NH4Cl (0,20 г, 5,00 ммол). К этой реакционной смеси добавляют метанол (8 мл) и перемешивают ее в течение ночи. Экстрагируют эфиром (2×15 мл), сушат (MgSO4) и упаривают в вакууме, получают сырой продукт реакции Штреккера.
В круглодонную колбу на 100 мл, содержащую продукт Штреккера, помещают 10 мл НОАс и 10 мл конц. HCl. Смесь кипятят в течение ночи, упаривают в вакууме, получают желтый твердый остаток. Его затирают с 5 мл смеси эфир: смесь гексанов 1:1. К белому твердому веществу добавляют триэтиламин (1,4 мл, 9,99 ммол) и дитрет-бутилкарбонат (1,00 г, 4,60 ммол) в 50 мл ДМФА. Через 4 ч рН смеси доводят до 9 насыщенным раствором Na2CO3. Перемешивают 3 ч и экстрагируют смесью эфир: смесь гексанов 1:1, водный слой подкисляют 5% раствором KHSO3 до рН 2. Водную фазу промывают эфиром (2×40 мл), органические вытяжки сушат (MgSO4) и упаривают, получают масло, которое очищают флеш-хроматографией на колонке с силикагелем, элюент метанол: СН2Cl2 8:92, получают 0,3 г (23%) Boc-защищенной аминокислоты в виде легкого масла (М-Н, 318).
Пример 65.
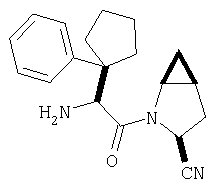
Стадия 1
Синтез соединения Стадии 1 описан при описании общего метода Н для синтеза рацемических аминокислот по Штреккеру.
Стадия 2
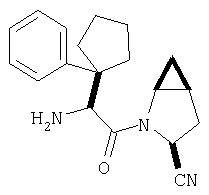
Заглавное соединение в Примере 65 получают пептидным синтезом из циклопентилфениламинокислоты, описанной на Стадии 1, по общему методу Н с последующими дегидратацией и снятием защиты по Общему методу С. MS (M+H) 310.
Пример 66.
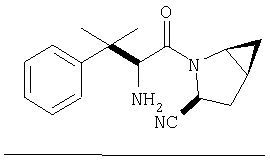
Стадия 1
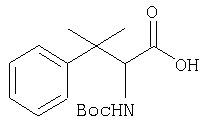
Соединение Стадии 1 получают по методу Штреккера для синтеза рацемических соединений по Общему методу Н из 2,2-диметилфенилуксусной кислоты.
Стадия 2
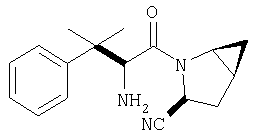
Заглавное соединение в Примере 66 получают пептидным синтезом из диметилфениламинокислоты, описанной на Стадии 1, с последующими дегидратацией и снятием защиты по Общему методу С. MS (M+H) 284.
Пример 67.
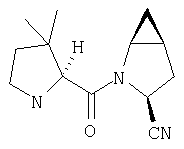
Стадия 1
N-(Бензилоксикарбонил) сукцинимид (5,6 г, 22,4 ммол) растворяют в CH2Cl2 (25 мл) и прибавляют этот раствор к перемешиваемому охлажденному (0°С) раствору диэтиламиномалоната гидрохлорида (5,0 г, 23,6 ммол) и триэтиламина (13,4 мл, 95 ммол) в CH2Cl2 (125 мл). Образовавшийся раствор перемешивают при 0°С 10 мин, а затем при комнатной температуре 1 час. Раствор промывают 10% лимонной кислотой (2×50 мл), 10% бикарбонатом натрия (2×50 мл) и водой (50 мл), сушат (Na2SO4) и упаривают, получают диэтиловый эфир N-бензилоксикарбониламиномалоновой кислоты в виде бесцветного масла, которое кристаллизуется при стоянии при 0°С (6,3 г) (LC/Macc, + ион): 310 (M+H).
Стадия 2
Соединение стадии 1 (6,18 г, 20 ммол) растворяют в сухом этаноле (30 мл) и прибавляют к раствору этилата (этоксида) натрия (2,85 г, 8,8 ммол; 21 вес.% раствор в этаноле (6 мл). Добавляют раствор 3-метил-2-бутеналя (1,68 г, 20 ммол) в этаноле (12 ммол) и перемешивают 24 ч при 25°С. Добавляют уксусную кислоту и гидрируют при 50 psi 24 ч с 10% Pd/C (2,0 г) в качестве катализатора. Раствор фильтруют, упаривают и остаток хроматографируют на силикагеле, элюент CH2Cl2/EtOAc (9:1), получают 2,2-дикарбэтокси-3,3-диметилпирролидин (1,6 г) (LC/Macc, + ион): 244 (M+H).
Этот диэфир (850 мг) 8 ч кипятят в 5 М соляной кислоте (10 мл)/ТФК (1 мл), получают, после упаривания, белый порошок. Перекристаллизацией из смеси метанол/эфир получают 3,3-диметил-d1-пролин (190 мг) в виде белых кристаллов, т.пл 110-112°С.
Стадия 3
Соединение Стадии 2 (173 мг, 0,97 ммол) растворяют в смеси ДМФА (3 мл)/вода (3 мл). К этому прозрачному раствору добавляют триэтиламин (0,46 мл, 3,18 ммол) и дитрет-бутилдикарбонат (0,23 г, 1,06 ммол) и перемешивают при комнатной температуре 5 ч. Раствор упаривают и остаток хроматографируют на колонке с силикагелем, элюент CH2Cl2/метанол (9:1), получают трет-бутилоксикарбонил-3,3-диметил-d1-пролин (200 мг) в виде масла (LC/Macc, + ион): 244 (М+Н).
Стадия 4
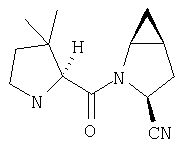
Заглавное соединение в Примере 67 получают пептидным синтезом аминокислоты, трет-бутилоксикарбонил-3,3-диметил-d1-пролина, описанной на Стадии 3, с последующими дегидратацией и депротекционированием по Общему методу С. MS (М+Н) 220.
Пример 68.
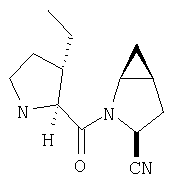
Стадия 1
К раствору диэтилацетамидмалоната (4,31 г, 19,8 ммол) в EtOH (23 мл) при перемешивании при комнатной температуре под аргоном прибавляют этилат натрия (940 мг 21 вес.% раствора в этаноле, 2,9 ммол). Реакционную смесь охлаждают до 0°С и по каплям прибавляют транс-2-пентеналь (1,51 г, 18,0 ммол), поддерживая температуру <5°С. Доводят до комнатной температуры, перемешивают 4 ч, добавляют уксусную кислоту (460 мкл). Раствор упаривают в вакууме, остаток растворяют в EtOAc (25 мл), промывают 10% раствором NaHCO3 (2×5 мл), рассолом и сушат (MgSO4). Раствор фильтруют и упаривают до объема 10 мл, затем нагревают до кипения и разбавляют гексаном (20 мл). По охлаждении до комнатной температуры заглавное соединение выпадает. Осадок собирают, получают 3,0 г (50%) соединения Стадии 1 (т.пл 106-109°С LC/Macc: + ионы, 324 M+Na).
Стадия 2
К раствору соединения Стадии 1 (2,87 г, 9,5 ммол) и триэтилсилана (2,28 мл, 14,3 ммол) в CH2Cl2 (30 мл) под аргоном при перемешивании прибавляют по каплям ТФК (7,35 мл, 95,3 ммол), поддерживая внутри колбы температуру 25°С с помощью бани со льдом. После 4 часов перемешивания при комнатной температуре раствор упаривают. Остаток разбавляют CH2Cl2 (100 мл), при энергичном перемешивании добавляют воду (50 мл) и твердую Na2СО3, фильтруют, упаривают, получают соединение Стадии 2 в виде желтого масла, которое используют без дополнительной очистки (LC/Macc: + ионы, 308 M+Na).
Стадия 3
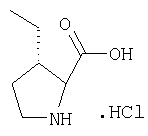
Соединение Стадии 2 (3,73 г, 9,5 ммол) суспендируют в 6 N HCl (20 мл) и НОАс (5 мл) и кипятят 20 ч. Реакционную смесь охлаждают, промывают EtOAc (20 мл), упаривают, получают масло, которое кристаллизуется при затирании с эфиром, давая заглавное соединение (1,2 г, 70,6%) (LC/Macc, + ион): 144 (М+Н).
Стадия 4
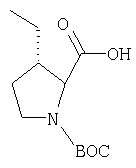
Соединение Стадии 3 (695 мг, 3,76 ммол) растворяют в смеси ацетон (12 мл)/вода (12 мл). К этому прозрачному раствору прибавляют триэтиламин (1,9 мл, 12,8 ммол) и дитрет-бутилдикарбонат (928 мг, 4,24 ммол). Перемешивают 18 ч при комнатной температуре. Растворители упаривают, а остаток хроматографируют на силикагеле смесью метанол: CH2Cl2 1:9, получают соединение Стадии 4 в виде масла (LC/Macc: + ионы, 266 M+Na).
Стадия 5
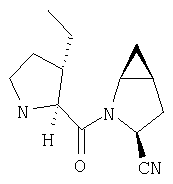
Соединение Примера 68 получают пептидным синтезом из аминокислоты Стадии 4 с последующими дегидратацией и депротекционированием по Общему методу С (MS (М+Н) 234).
Пример 69.
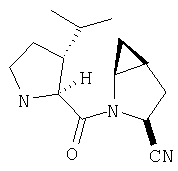
Стадия 1
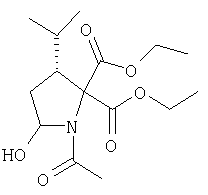
К раствору диэтилацетамидмалоната (4,31 г, 19,8 ммол) в EtOH (23 мл) при перемешивании при комнатной температуре под аргоном прибавляют этилат натрия (940 мг, 2,9 ммол; 21 вес.% раствор в этаноле) в этаноле (2 мл). Реакционную смесь охлаждают до 0°С; прибавляют по каплям 4-метил-2-пентеналь (1,77 г, 18,0 ммол), поддерживая температуру в ней <5°С. Доводят до комнатной температуры, перемешивают 4 ч, прибавляют уксусную кислоту (460 мкл). Раствор упаривают, остаток растворяют в EtOAc (25 мл). Органический слой промывают 10% раствором NaHCO3 (2×5 мл), рассолом и сушат (MgSO4). Раствор фильтруют и упаривают до объема 10 мл, нагревают до кипения и обрабатывают гексаном (20 мл). По охлаждении соединение Стадии 1 выпадает и его собирают (3,3 г) (LC/Macc, + ион) 338 (M+Na).
Стадия 2
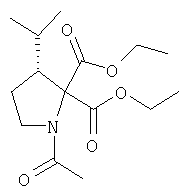
К раствору соединения Стадии 1 (3,0 г, 9,5 ммол) и триэтилсилана (2,28 мл, 14,3 ммол) в CH2Cl2 (30 мл) при перемешивании под аргоном по каплям прибавляют ТФК (7,35 мл, 95,3 ммол), поддерживая температуру в колбе 25°С с помощью бани со льдом. После четырехчасового перемешивания при комнатной температуре раствор упаривают, к остатку добавляют CH2Cl2 (100 мл), обрабатывают водой (50 мл) и твердым Na2CO3 при энергичном перемешивании до щелочной реакции среды (смеси). Органический слой отделяют, сушат (Na2SO4), упаривают, получают заглавное соединение в виде масла, которое используют без дополнительной очистки (LC/Macc: + ионы, 300 М+Н).
Стадия 3
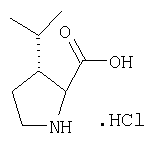
Соединение Стадии 2 (3,8 г, 9,5 ммол) суспендируют в 6 N HCl (20 мл) и НОАс (5 мл) и кипятят 20 ч. Реакционную смесь охлаждают, промывают EtOAc (20 мл), упаривают, получают масло, которое кристаллизуется при затирании с эфиром. Получают 1,4 г (76,0%) соединения Стадии 3. LC/Macc: + ионы, 158 (М+Н).
Стадия 4
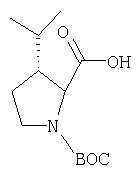
Соединение Стадии 3 (728 мг, 3,76 ммол) растворяют в смеси ацетон/вода 1:1 (24 мл). К этому прозрачному раствору добавляют триэтиламин (19 мл, 128 ммол) и дитрет-бутилдикарбонат (928 мг, 4,24 ммол). Реакционную смесь перемешивают при комнатной температуре 18 ч. Растворители упаривают, остаток хроматографируют на колонке с силикагелем, элюент CH2Cl2/метанол (9:1), получают заглавное соединение в виде масла (LC/Macc, + ион): 258 (М+Н).
Стадия 5
Соединение из Примера 69 получают пептидным синтезом аминокислоты Стадии 4 с последующими дегидратацией и снятием защиты по Общему методу С (MS (M+H) 248).
Пример 70.
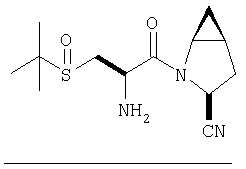
Стадия 1
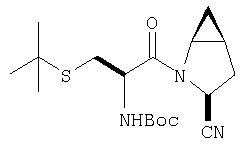
Соединение 1 получают по методике, описанной в Общем Методе С, с N-Вос-S-трет-бутилцистеином в качестве исходного.
Стадия 2
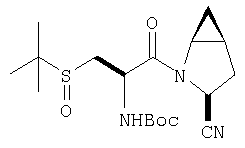
В круглодонную колбу на 25 мл, снабженную магнитной мешалкой и вводом для азота, помещают соединение Стадии 1 (78 мг, 0,21 ммол) и хлороформ (3 мл). Смесь охлаждают до 0°С и обрабатывают м-хлорбензойной кислотой (85 мг, 0,44 ммол) в CHCl3 (2 мл). Через 3 час к раствору прибавляют CHCl3 (7 мл), промывают 5% NaHCO3 (2×5 мл), Н2О и сушат Na2SO4. Отгонка растворителя дает сырой сульфоксид, который используют без дополнительной очистки (LC/Macc, + ионы): 384 (M+H).
Стадия 3
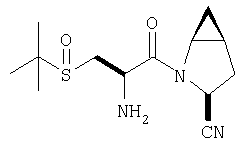
К охлажденному до 0°С раствору соединения Стадии 2 (100 мг, 0,26 ммол) в 5 мл CH2Cl2 прибавляют трифторуксусную кислоту (1,5 мл). Раствор перемешивают при 0°С 1,5 ч и упаривают в вакууме, получают вязкое масло. Продукт очищают обращенно-фазовой препаративной хроматографией на колонке YMC S5 ODS 20×100 мм, получают заглавное соединение Примера 70,17 мг, 16%. Условия очистки: градиентная элюция от 10% метанол/вода/0,1 ТФК до 90% метанол/вода/0,1 ТФК за 15 мин, 5 мин при 90% метанол/вода/0,1 ТФК. Скорость тока: 20 мл/мин. Длина волны при детектировании: 220. Время удерживания 10 мин (LC/Macc, + ион): 284 (М+Н).
Пример 71.
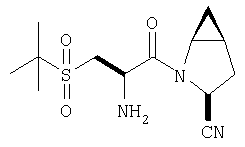
Стадия 1
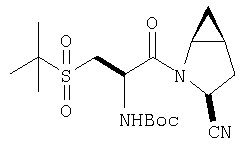
В круглодонную колбу на 25 мл, снабженную магнитной мешалкой и вводом для азота, помещают соединение Стадии 1, Пример 70 (78 мг, 0,21 ммол) в хлороформе (3 мл). Смесь охлаждают до 0°С и добавляют м-хлорбензойную кислоту (144 мг, 0,84 ммол) в CHCl3 (2 мл). Через 30 мин при комнатной температуре добавляют CHCl3 (7 мл), промывают 5% NaHCO3 (2×10 мл), Н2O и сушат Na2SO4. После удаления растворителя получают сырой сульфон (110 мг), который используют далее без дополнительной очистки (LC/Macc, + ион): 344 (М+Н - Bu).
Стадия 2
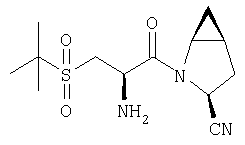
К охлажденному (0°С) перемешиваемому раствору соединения Стадии 1 (100 мг, 0,26 ммол) в 5 мл CH2Cl2 прибавляют трифторуксусную кислоту (1,5 мл). Раствор перемешивают при 0°С 30 мин, добавляют (5 мл) и упаривают в вакууме, получают вязкое масло. Продукт очищают обращенно-фазовой препаративной хроматографией на колонке YMC S5 ODS 20×100 мл, получают заглавное соединение, 14 мг, 17%. Условия очистки: градиентная элюция от 10% метанол/вода/0,1 ТФК до 90% метанол/вода/0,1 ТФК. Скорость тока: 20 мл/мин. Длина волны при детектировании: 220. Время удерживания 10 мин. (LC/Macc, + ион): 300 (М+Н).
Пример 72.
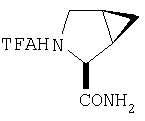
Заглавное соединение получают по известной методике (Sasaki et al. Tetrahedron Lett. 1995, 36, 3149, Sasaki et al. Tetrahedron 1994, 50, 7093), применяемой для синтеза (2S,3R,4S)-N-Вос-3,4-метан-L-пролинкарбоксилата. Соответствующий амид получают по Общему методу А и депротекционируют, действуя солью ТФК, также по Общему методу А.
Пример 73.
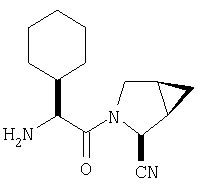
Заглавное соединение получают конденсацией (2S,3R,4S)-3,4-метан-L-пролинкарбоксамид-N-трифторацетата, описанного в Примере 72, с L-циклогексилглицином, а затем дегидратируют до амида POCl3/имидазолом и депротекционируют N-концевой азот) с помощью ТФК по Общему методу С (ББА МН+248).
Пример 74.
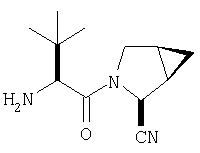
Заглавное соединение получают конденсацией (2S,3R,4S)-3,4-метан-L-пролинкарбоксамид-N-трифторацетата, описанного в Примере 72, с L-трет-бутилглицином, а затем дегидратируют в амид, действуя POCl3/имидазолом, и депротекционируют (N-концевой азот) ТФК по Общему методу С (ББА МН+222).
Пример 75.
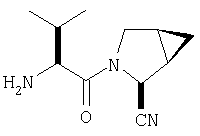
Заглавное соединение получают конденсацией (2S,3R,4S)-3,4-метан-L-пролинкабоксамид-N-трифторацетата, описанного в Примере 72, с L-валином, а затем депротекционируют (N-концевой азот) ИФК по Общему методу С (ББА МН+207).
Пример 76.
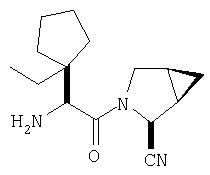
Заглавное соединение получают конденсацией (2S,3R,4S)-3,4-метан-L-пролинкарбоксамид-N-трифторацетата, описанного в Примере 72, с N-(трет-бутилоксикарбонил)-(1'-этилциклопентил) глицином, описанным в Общем методе В, а затем дегидратируют до амида с помощью POCl3/имидазола и депротекционируют (N-концевой азот) ТФК по Общему методу С (ББА МН+262).
Пример 77.
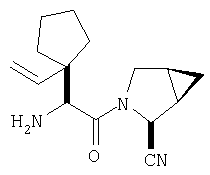
Заглавное соединение получают конденсацией (2S,3R,4S)-3,4-метан-L-пролинкарбоксамид-N-трифторацетата, описанного в Примере 72, с N-(трет-бутилоксикарбонил)-(1'-винилциклопентил)глицином, описанным в Общем методе В, а затем дегидратируют в амид POCl3/имидазолом и депротекционируют (N-концевой азот) ТФК по Общему методу С (ББА МН+260).
Пример 78.
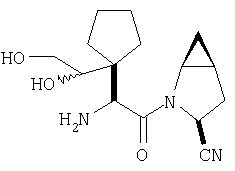
N-[((S)-циклопентилвинил)-N-трет-бутоксикарбонилглицинил]-(2S,4S,5S)-2-циан-4,5-метан-L-пролиламид (70 мг, 0,19 ммол), описанный в Общем методе С, Стадия 2, растворяют в смеси 2 мл трет-BuOH/3 мл ТГФ и добавляют N-метилморфолин-N-оксид (33 мг, 0,28 ммол), а затем тетраоксид осмия (0,1 ммол, 50 мол.%). К реакционной смеси добавляют 1 мл 10% водного раствора Na2SO4, экстрагируют EtOAc и промывают Н2О (5 мл), сушат (Na2SO4), фильтруют, упаривают и очищают флеш-хроматографией на колонке с силикагелем (5% МеОН/CH2Cl2), получают 41 мг (55%) защищенного диола в виде масла. Заглавное соединение получают депротекционированием аминогруппы ТФК по Общему методу С (ББА МН+294).
Пример 79.
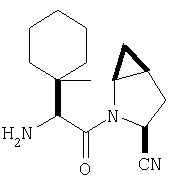
Общая методика I: Синтез четвертичных аминокислот присоединением к малонатам по реакции Михаэля с последующими селективным гидролизом и перегруппировкой Курциуса. Примеры 79-84.
Циклогексанон конденсируют с диэтилмалонатом по Кневенагелю в присутствии четыреххлористого титана в ТГФ и CCl4 с образованием 40. Реакцией Гриньяра в присутствии меди (I) из метилмагнийбромида получают 41, который селективно омыляют до 42. Перегруппировкой Курциуса с «захватом» бензилового спирта получают 43, который превращают в 44 стандартным методом депротекционирования-протекционирования. Эфир 44 омыляют, получая четвертичную аминокислоту 45.
Схема 11, Общий Метод I
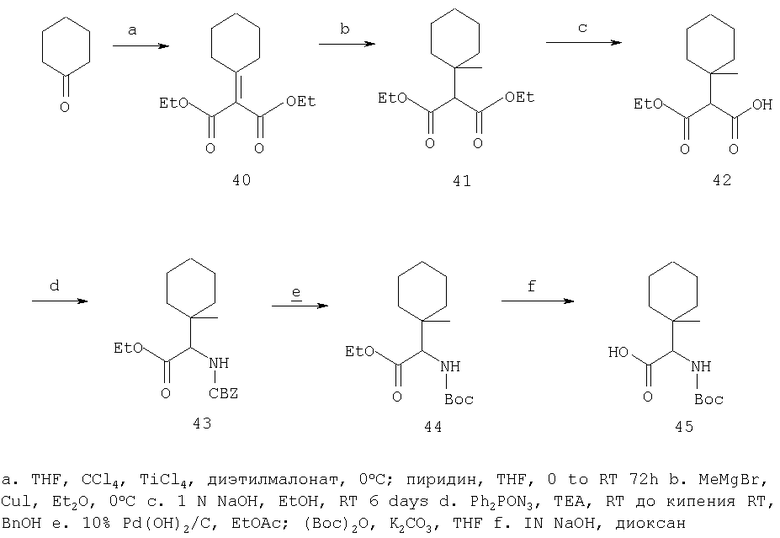
Стадия 1
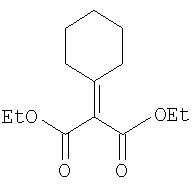
По литературной методике (Tetrahedron 1973, 29, 435) смесь сухого тетрагидрофурана (400 мл) и сухого четыреххлористого углерода (50 мл) охлаждают до 0°С (снег + соль) и добавляют четыреххлористый титан (22,0 мл, 0,2 мол). Образующуюся желтую суспензию перемешивают при 0°С 5 мин, прибавляют последовательно циклогексанон (10,3 мл, 0,1 мол) и перегнанный диэтилмалонат (15,2 мл, 0,1 мол), перемешивают при 0°С 30 мин. Реакционную смесь обрабатывают раствором сухого пиридина (32 мл, 0,40 мол) в сухом ТГФ (60 мл), перемешивают при 0°С 1,0 час, затем при комнатной температуре 72 ч. К реакционной смеси добавляют воду (100 мл), перемешивают 5 мин, затем экстрагируют эфиром (2×200 мл). Объединенные эфирные вытяжки промывают насыщенным раствором хлористого натрия (100 мл), насыщенным раствором бикарбоната натрия (100 мл) и рассолом (100 мл), фильтруют и упаривают. Флеш-хроматографией с 5% EtOAc в гексане в качестве элюента получают соединение Стадии 1 в виде легкого желтого масла. Выход: 5,25 г (22%). MS (M+Na) 263.
Стадия 2
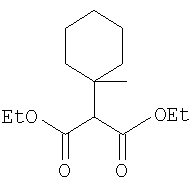
В соответствии с литературной методикой (Org.Syn. VI, 442, 1988; LiebigsAnn. Chem. 1981, 748) смесь 3,0 M метилмагнийиодида (3,1 мл, 0,36 ммол) и хлорида меди (9 мг) перемешивают при 0°С (баня соль - снег), прибавляют за 5 мин раствор соединения Стадии 1 (1,5 г, 6,24 ммол) в сухом эфире (1,8 мл) и перемешивают при 0°С 1 час, затем при комнатной температуре 40 мин. Смесь медленно выливают в смесь льда с водой, прибавляют по каплям 10% HCl (3,7 мл), экстрагируют EtOAc (3×25 мл). Объединенные органические вытяжки промывают 1% тиосульфатом натрия (2,0 мл) и насыщенным раствором хлористого натрия (2,0 мл), сушат безводным сульфатом натрия, фильтруют и упаривают. Флеш-хроматографией на колонке с силикагелем (элюент 5% эфир в гексане) получают продукт Стадии 2 в виде прозрачного сиропа. Выход: 1,09 г (68%). MS (М+Н) 257.
Стадия 3
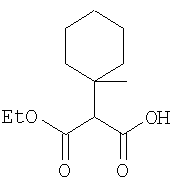
К раствору соединения Стадии 2 (1,09 г, 4,03 ммол) в смеси метанола (5,4 мл) и воды (2,7 мл) прибавляют 1 N раствор гидроксида натрия (4,84 мл, 4,84 ммол или 1,2 эквив) и перемешивают при комнатной температуре 6 дней. В реакционной смеси все еще имеется исходное, поэтому добавляют ТГФ (4,0 мл) и смесь перемешивают еще 2 дня. Раствор упаривают досуха и образовавшийся сироп распределяют между водой (8,0 мл) и эфиром (15 мл). Водную фазу подкисляют 1 N соляной кислотой (4,8 мл) до рН 2-3 и экстрагируют EtOAc (3×25). Объединенные органические вытяжки промывают рассолом (10,0 мл), сушат безводным сульфатом магния, фильтруют и упаривают, получают соединение Стадии 3 в виде вязкого сиропа. Выход: 875 мг (95,1%). MS (M+H) 229.
Или же (альтернатива): растворы диэфира в смеси этанола, ТГФ, диоксана и воды или их смеси можно гидролизовать гидроксидом натрия.
Стадия 4
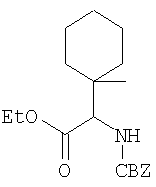
По литературной методике (J. Org. Chem 1994, 59, 8215) к раствору соединения Стадии 3 (0,875 г, 3,83 мл) в сухом бензоле (4.0 мл) прибавляют триэтиламин (0,52 мл, 3,83 ммол) и дифенилфосфорилазид (0,85 мл, 3,83 ммол), кипятят под азотом 1 час и охлаждают до комнатной температуры. Прибавляют бензиловый спирт (0,60 мл, 5,75 ммол или 1,5 эквив), кипятят 17 час, охлаждают и добавляют эфир (40 мл). Раствор промывают 10% водной лимонной кислотой (2×3 мл), снова промывают эфиром (40 мл) слой лимонной кислоты. Объединенные органические вытяжки промывают 5% раствором бикарбоната натрия (2×3 мл), сушат (MgSO4), фильтруют и упаривают. Флеш-хроматографией сырого продукта на силикагеле (10% EtOAc в гексане, 1,0 л) получают соединение Стадии 4 в виде прозрачного сиропа. Выход: 1,15 г (90%). Ms (M+H) 334.
Стадия 5
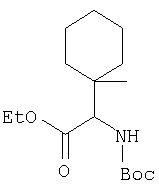
К раствору соединения Стадии 4 (1,15 г, 3,46 ммол) в EtOAc (60 мл) прибавляют гидроксид палладия на угле (298 мг) и гидрируют при комнатной температуре 20 ч. Смесь фильтруют через целит, целит промывают EtOAc (3×25 мл), фильтрат упаривают, получают свободный амин. Раствор амина в тетрагидрофуране (12 мл) обрабатывают дитрет-бутилдикарбонатом (1,0 г, 4,58 ммол или 1,48 эквив) и карбонатом калия (854 мг, 6,18 ммол или 2,0 эквив), перемешивают при комнатной температуре 20 ч. Реакционную смесь распределяют между водой (8 мл) и диэтиловым эфиром (3×40 мл) и объединенные органические вытяжки промывают рассолом (8 мл), сушат (MgSO4), фильтруют и упаривают. Флеш-хроматографией сырого продукта 10% EtOAc в гексане (1 л) получают соединение Стадии 5 в виде прозрачного вязкого сиропа. Выход: 1,18 г (100%). MS: (М+Н) 300.
Можно также применять другие методы, например: метод, приведенный в Tetrahedron Lett. 1988, 29, 2983, где раствор бензилкарбамата в этаноле можно обрабатывать триэтилсиланом (2 эквив), дитрет-бутилдикарбонатом (1,1 эквив), катализатором ацетатом палладия и триэтиламином (0,3 эквив), при этом получают ВОС-защищенный амин «в одной колбе».
Или же: Растворы бензилкарбамата в метаноле можно подвергнуть гидрогенолизу в присутствии дитрет-бутилдикарбоната с образованием ВОС-защищенного амина «в одной колбе».
Стадия 6
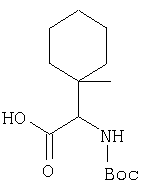
К раствору соединения Стадии 5 (1,18 г, 3,09 ммол) в диоксане (8 мл) прибавляют IN раствор гидроксида натрия (9,1 мл, 9,1 ммол или 3,0 эквив) и перемешивают при 60°С (масляная баня) 28 ч. Реакционную смесь упаривают до сиропа, который растворяют вводе (15 мл) и экстрагируют эфиром (25 мл). Водный слой подкисляют до рН 2-3 1 N соляной кислотой (9,2 мл), экстрагируют EtOAc (3×50 мл). Объединенные органические экстракты промывают насыщенным раствором хлористого натрия (10 мл), сушат (MgSO4), фильтруют и упаривают, получают соединение Стадии 6 желтовато-белого цвета. Выход: 808 мг (96%). MS (M+H) 272.
Стадия 7
Заглавное соединение получают из соединения Стадии 6 по методике Общего метода С, в которой конденсируют аминокислоту, амиддегидратируют и защитную группу удаляют, получая заглавное соединение. MS (M+H) 262.
Соединения 90-100 получают по Общему методу I и по Общему методу С, в качестве исходных берут циклогексанон, циклопентанон и циклобутанон, в качестве реагентов Гриньяра используют метил -, этил-, аллил- и пропилмагнийгалогениды
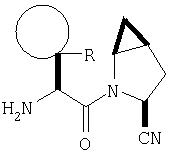
Пример 85.
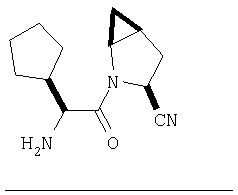
Стадия 1
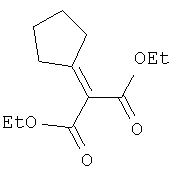
По методике из Примера 79: Сухой четыреххлористый углерод (50 мл) (смесь ?) охлаждают до 0°С (баня лед + соль) и добавляют четыреххлористый титан (11,0 мл, 0,1 мол). Образовавшуюся желтую суспензию перемешивают при 0°С 5 мин, прибавляют последовательно циклопентанон (4,42 мл, 0,05 мол) и перегнанный диэтилмалонат (7,6 мл, 0,05 мол), перемешивают при 0°С 30 мин. К реакционной смеси прибавляют сухой пиридин (16 мл, 0,20 мол) в сухом ТГФ (30 мл), перемешивают при 0°С 1 час, затем при комнатной температуре 20 ч. Реакционную смесь разбавляют водой (50 мл), перемешивают 5 мин и экстрагируют эфиром (2×100 мл). Объединенные органические вытяжки промывают насыщенным раствором хлористого натрия (50 мл), насыщенным раствором бикарбоната натрия (50 мл) и рассолом (50 мл), сушат (MgSO4), фильтруют и упаривают. Флеш-хроматографией с 5% EtOAc в гексане получают соединение Стадии 1 в виде легкого желтого масла. Выход: 7,67 г (68%). MS (M+H) 226.
Стадия 2
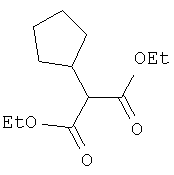
К раствору соединения Стадии 1 (1,00 г, 4,42 ммол) в метаноле (50 мл) добавляют 10% Pd/C (0,20 г, 10 мол.%) и гидрируют (баллон) при комнатной температуре 20 ч. Добавляют метанол и фильтруют через слой целита. Фильтрат упаривают и очищают флеш-хроматографией на силикагеле, элюент 7% EtOAc в смеси гексанов, получают 0,84 г (91%) соединения Стадии 2. MS (M+H) 229.
Стадия 3
Соединение Стадии 3 получают по методу, описанному в Общем Методе Н, где сложный эфир претерпевает гидролиз, далее следует перегруппировка Курциуса, снятие защитной группы и снова гидролиз сложного эфира.
Стадия 4
Заглавное соединение получают из соединения Стадии 3 по методике из Общего Метода С, по которой конденсируют аминокислоту, дегидратируют амид и снимают защитную группу заглавного соединения. MS (M+H) 234.
Соединения из Примеров 86 и 87 получают по методикам из Примера 85, применяя в качестве исходных циклогексанон и циклобутанон, соответственно.
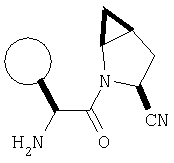
Пример 89.
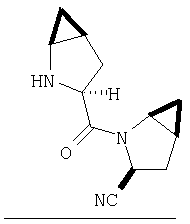
Стадия 1
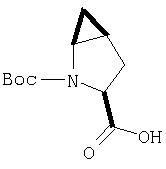
Соединение Стадии 1 получают по методике из Примера 6 Стадия 1.
Стадия 2
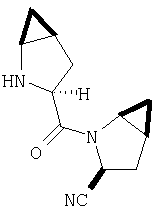
Заглавное соединение получают из соединения Стадии 1 по методике из Общего Метода С, в которой карбоновая кислота вступает в реакцию пептидного синтеза, далее идет дегидратация амида и снятие защитной группы. MS (M+H) 218.
Примеры 90-99.
Соединения в Примерах, где Х=Н, включают следующие, которые можно получить по представленным ранее в данном описании методикам.
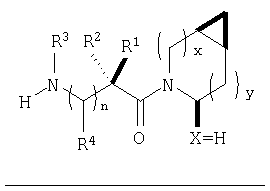
Примеры 100-109.
Соединения в примерах, где n=1, включают следующие, которые можно получать по методикам, представленным в данном описании ранее.
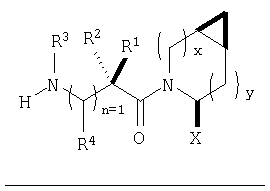
Пример 110
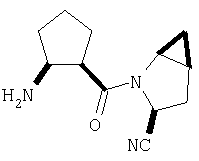
A.
трет-Бутил-2-((1S,3S,5S)-3-карбамоил-2-азабицикло[3.1.0]гексан-2-карбонил)циклометилкарбамат (смесь двух диастереомеров)
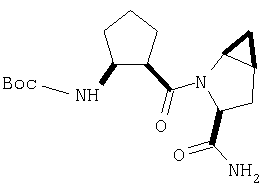
Смесь рацемической 2-(трет-бутоксикарбониламино)циклопентанкарбоновой кислоты (47 мг, 0.21 ммоля), (1S,3S,5S)-2-азабицикло[3.1.0]гексан-2-карбоксамида (HCl соль, гомохиральная, 34 мг, 0.21 ммоля), EDC (59 мг, 0.31 ммоля), DIPEA (100 мкл) и DMAP (38 мг, 0.31 ммоля) в DCM (3 мл) перемешивают при rt. в течение ночи. Реакционную смесь разбавляют EtOAc, промывают 1 N раствором HCl, насыщенным раствором бикарбоната натрия, сушат MgSO4, фильтруют и упаривают в вакууме, получают титульное соединение в виде смеси двух диастереомеров (44 мг). (М+Н)=360.
В.
трет-Бутил-2-((1S,3S,5S)-3-циано-2-азабицикло[3.1.0]гексан-2-карбонил)циклометилкарбамат (смесь двух диастереомеров)
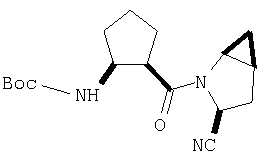
К раствору соединения из раздела А (смесь двух изомеров, 44 мг, 0.13 ммоля) в ТГФ (300 мкл) прибавляют пиридин (100 мкл). Смесь охлаждают до -10°С и одной порцией прибавляют TFAA (71 мкл). Смесь перемешивают 1 минуту при -10°С и отставляют охлаждающую баню. Реакционную смесь перемешивают 25 минут. LC-MS показывает, что заданный продукт реагирует с ТФК (TFA), давая TFA амид заданного продукта. Для гидролиза этих амидов прибавляют насыщенный раствор К2СО3 (300 мкл) и КОН (47 мг) и перемешивают при rt. в течение 24 ч. Смесь экстрагируют EtOAc. Органический слой промывают рассолом, сушат MgSO4, фильтруют и упаривают в вакууме, получают титульное соединение (37 мг) в виде смеси двух диастереомеров, изомер А, ТСХ (50% EtOAc/гексан) Rt=0.53 и изомер В, ТСХ (50% EtOAc/гексан) Rf=0.27. Смесь используют в неочищенном виде.
С.
(1S,3S,5S)-2-((1R,2S)-2-Аминоциклопентанкарбонил)-2-азабицикло[3.1.0]гексан-3-карбонитрил (один диастереомер)
Соединение из раздела В (смесь двух диастереомеров, 37 мг) обрабатывают 20% TFA/DCM (1.5 мл) при rt. в течение 2 минут. Реакционную смесь упаривают и остаток очищают обращенно-фазовой препаративной ВЭЖХ (Phenomenex Luna 5 мк С 18 4.6×50 мм, 0-100% растворителя В в 10 мин градиенте, скорость потока 4 мл/мин, длина волны 220 нм, растворитель А=10% МеОН + 90% воды + 0.1% TFA (ТФК), растворитель В=90% МеОН + 10% воды + 0.1% TFA), получают два диастереомера. Изомер А, титульное соединение (10.4 мг), LCMS (M+H)+=220.10, rt=1.08 мин (YMC ODS S5 С18 4.6×50 мм, 4 мин градиент, 0-100% В, скорость потока 4 мл/мин, А=10% МеОН/Вода + 0.1% TFA, В=90% МеОН/Вода + 0.1% TFA). Изомер В (8.1 мг), LCMS (M+H)=220.1, rt=0.75 мин.
Пример 111
Дихлорэтан (100 мкл) прибавляют в реакционную пробирку, содержащую N-α,β-дотрет-бутоксикарбонил-L-α,β-диаминопропионовую кислоту (13.09 мг, 0.043 ммоля). Прибавляют раствор 1,3- диизопропилкарбодиимида (7.07 мкл, 0.045 ммоля) и 1-гидрокси-7-азабензотриазола (6.15 мг, 0.045 ммоля) в дихлорэтане: прибавляют раствор N,N-диметилформамид (1:1) (100 мкл). Прибавляют раствор триэтиламина (5.77 мкл, 0.041 ммоля) в дихлорэтане (50 мкл). Прибавляют раствор вторичного амина 52939-103-31 (4.50 мг, 0.038 ммоля) в N,N-диметилформамиде (100 мкл).
Реакционную пробирку плотно закрывают и смесь встряхивают в течение 14 часов. Встряхивание прекращают. Прибавляют аминометильную смолу (15 мг), а затем дихлорэтан (200 мкл). Раствор встряхивают 3 часа и сливают в микропробирку. Смолу промывают дихлорэтаном (200 мкл), который сливают в микропробирку. Раствор упаривают на концентраторе speed-vac. В микропробирку, содержащую интермедиат с защитной Boc-группой, прибавляют раствор трифторуксусной кислоты и дихлорэтана (1:2, 600 мкл). Пробирку закрывают и реакционную смесь встряхивают в течение 4 ч. Затем раствор концентрируют на speed-vac. Остаток растворяют в МеОН (1.0 мл) и остаток помещают сверху на слой SCX (сильная катионообменная смола) (300 мг). Смолу промывают МеОН (2×1.5 мл) и 0.1 М раствором аммиака в МеОН (1.5 мл). Затем продукт элюируют 2 М раствором аммиака в МеОН (1.5 мл). Раствор, содержащий продукт, упаривают с помощью speed-vac. Сырой продукт очищают препаративной ВЭЖХ (10-100% градиент растворителя В за 10 мин при скорости потока 30 мл/мин, колонка = YMC S5 ODS 30×100 мм; растворитель А=90% Н2О, 10% МеОН, 0.1% TFA; растворитель В=10% Н2O 90% МеОН, 0.1% TFA). Получают 5.5 мг (выход 36%) BMS-644105. Масс-спектрометрия: (М+Н)+ вычислено = 170, найдено = 170.
Пример 112
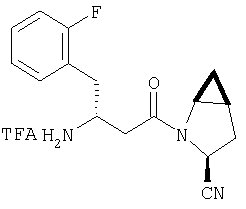
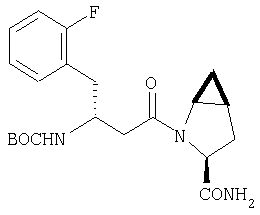
A.
К раствору (R)-3-(трет-бутоксикарбониламино)-4-(2-фторфенил)бутановой кислоты (200 мг, 0.673 ммоля) и (1S,3S,5S)-2-азабицикло[3.1.0]гексан-2-карбоксамида (132 мг, 0.81 ммоля) в сухом хлористом метилене (дихлорметане, DCM, 7 мл) прибавляют РуВОР (562 мг, 1.08 ммоля) и 4-метилморфолин (0.22 мл, 2.0 ммоля), затем перемешивают при комнатной температуре в течение 17.0 ч. Смесь распределяют между 5% бисульфатом калия (10 мл) и хлористым метиленом (3×50 мл), снова промывают объединенные органические вытяжки 5% бисульфатом калия (10 мл). Органическую фазу перемешивают с 2.0 М раствором карбоната натрия (25 мл) в течение 20 минут, промывают рассолом (10 мл), сушат (безводным сульфатом натрия), фильтруют и упаривают досуха. Очистка колоночной хроматографией (40 г; градиент СН3ОН:СН2Cl2) дает титульное соединение в виде белой твердой пены (175.4 мг, 64.3%). LC/MS дает корректный молекулярный ион [(М+Н)+=406] для заданного соединения.
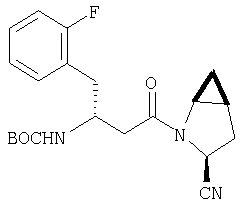
B.
К охлажденному (-30°С) раствору соединения из раздела А (75 мг, 0.185 ммоля) и имидазола (26 мг, 0.38 ммоля) в сухом пиридине (2.6 мл) прибавляют оксихлорид фосфора (66.5 мкл, 0.71 ммоля) и перемешивают при температуре -20°С÷-30°С в течение 1.0 часа. Реакционную смесь упаривают, получают сироп, который промывают хлористым метиленом (25 мл) и эфиром (25 мл). Сироп растирают с хлористым метиленом (2×10 мл), получают твердый осадок, который отфильтровывают. Фильтрат упаривают, получают сироп, очистка которого хроматографией (12 г, градиент EtOAc: Гексан) дает титульное соединение в виде сиропа (52.9 мг, 73.8%). LC/MS дает корректный молекулярный ион [(М+Н)+=388] для заданного соединения.
С.
Раствор соединения из раздела В (53 мг, 0.137 ммоля) в сухом хлористом метилене (0.90 мл) и трифторуксусной кислоте (0.22 мл) перемешивают при комнатной температуре в течение 1.0 часа. К реакционной смеси прибавляют хлористый метилен (1.0 мл), упаривают досуха и полученный сироп промывают (chase) толуолом (3.0 мл) и эфиром (2×10 мл). Сырой продукт растирают с эфиром (0.8 мл), получают желтую пену (48 мг, 87%). Препаративной хроматографией 24 мг получают титульное соединение в виде белого лиофилизата (11.0 мг). LC/MS дает корректный молекулярный ион [(M+H+)=288] для заданного соединения.
Пример 113
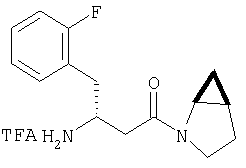
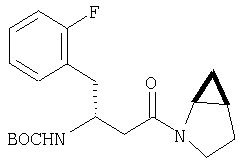
А.
К раствору (R)-3-(трет-бутоксикарбониламино)-4-(2-фторфенил)бутановой кислоты (40 мг, 0.13 ммоля) и гидрохлорида (1S,5R)-2-азабицикло[3.1.0]гексана (18.3 мг, 0.15 ммоля) в сухом хлористом метилене (дихлорметане, DCM, 1.2 мл) прибавляют РуВОР (108 мг, 0.2 ммоля) и 4-метилморфолин (42 мкл, 0.31 ммоля), затем перемешивают при комнатной температуре в течение 24 ч. Смесь распределяют между 5% бисульфатом калия (1.3 мл) и хлористым метиленом (3×10 мл) и органическую фазу перемешивают с 2.0 М раствором карбоната натрия (2.5 мл) в течение 5 минут, промывают рассолом (2 мл), сушат (безводным сульфатом натрия), фильтруют и упаривают досуха. Очистка колоночной хроматографией (12 г; градиент CH3OH:CH2Cl2, затем 4.0 г; EtOAc: Гексан) дает титульное соединение в виде белой твердой пены (43.8 мг, 93%). LC/MS дает корректный молекулярный ион [(М+Н)+=363] для заданного соединения.
В.
Раствор соединения из раздела А (43.8 мг, 0.12 ммоля) в сухом хлористом метилене (0.80 мл) и трифторуксусной кислоте (0.20 мл) перемешивают при комнатной температуре в течение 1.0 часа. К реакционной смеси прибавляют хлористый метилен (1.0 мл), упаривают досуха и полученный сироп промывают (chase) толуолом (3.0 мл) и эфиром (2×10 мл). Сырой продукт растирают с эфиром (0.8 мл), затем лиофилизируют, получают белый лиофилизат (37.0 мг, 82%). LC/MS дает корректный молекулярный ион [(М+Н)+=263] для заданного соединения.
В нижеследующей таблице 1* представлены сведения об ингибирующей (DP4) активности отдельных представителей заявленных соединений.
In Vitro ингибиторные константы свиной DPP-IV (Дипептидилпептидазы IV)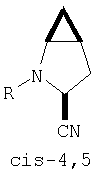
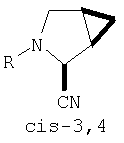
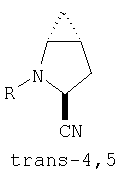
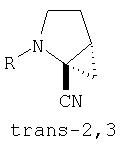
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АЛЬФА-(N-СУЛЬФОНАМИДО)АЦЕТАМИДА КАК ИНГИБИТОРЫ БЕТА-АМИЛОИДА | 2002 |
|
RU2300518C2 |
| ОКСА- И ТИАЗОЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТИДИАБЕТИЧЕСКИХ АГЕНТОВ И АГЕНТОВ ПРОТИВ ОЖИРЕНИЯ | 2000 |
|
RU2279427C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНКИНАЗ | 2000 |
|
RU2260592C9 |
| ЛИГАНДЫ ТИРОИДНОГО РЕЦЕПТОРА - ПРОИЗВОДНЫЕ АНИЛИНА | 2001 |
|
RU2260586C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ДЛЯ ИНГИБИРОВАНИЯ ВЗАИМОДЕЙСТВИЯ BCL БЕЛКОВ С КОМПОНЕНТАМИ ПО СВЯЗЫВАНИЮ | 2005 |
|
RU2416606C2 |
| ИНГИБИТОРЫ MEK И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2812929C1 |
| ТРИПЕПТИД ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ СОЛЬВАТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АМИДАЗЫ ТРОМБИНА | 1994 |
|
RU2105010C1 |
| МОДУЛЯТОРЫ CCR2 | 2016 |
|
RU2726206C2 |
| Производные 2H-[1,4]оксазино- и 2H-[1,4]тиазино[3,2-c]хинолин-3(4H)-онов в качестве ингибиторов ATM и DNA-PK киназ для лечения и/или профилактики онкологических заболеваний | 2024 |
|
RU2835676C1 |
| ОКСА- И ТИАЗОЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТИДИАБЕТИЧЕСКИХ АГЕНТОВ И АГЕНТОВ ПРОТИВ ОЖИРЕНИЯ | 2000 |
|
RU2327692C2 |
Изобретение относится к новым конденсированным циклопропилпирролидинам формулы:
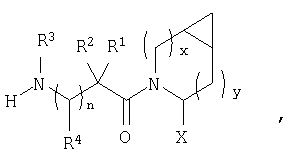
где х обозначает 0 или 1 и у обозначает 0 или 1, при условии, что х обозначает 1, когда у обозначает 0 и х обозначает 0, когда у обозначает 1; и где n обозначает 0 или 1; Х обозначает Н или CN; R1, R2, R3 и R4 одинаковы или различны и независимо выбираются из Н, C1-С10 алкила, C2-C12 алкенила, насыщенного C3-С10 циклоалкила, насыщенного C3-С10 циклоалкил C1-С10 алкила, насыщенного C3-С10 бициклоалкила, насыщенного C3-С10 трициклоалкила, гидроксил C1-С10 алкил насыщенного С3-С10 циклоалкила, C1-С10 алкилтио C1-С10 алкила, C6-С10 арил C1-С10 алкилтио C1-С10 алкила, C6-С10 арил C1-С10 алкила, 5- или 6- членного гетероарила, содержащего один атом N или один атом О, 5- или 6-членного гетероарила, содержащего один атом N конденсированный с C6-С10 арильным кольцом, 5- или 6-членного гетероарила, содержащего один атом N или один атом О C1-С10 алкила, циклогетероалкила, который является C5-С8 насыщенным кольцом, содержащим один гетероатом, такой как N или О; все, при необходимости, имеют в качестве заместителей при соответствующем атоме углерода 1, 2, 3, 4 или 5 групп, выбираемых из галоида, C1-С10 алкила, C2-C12 алкенила, гидрокси, гидрокси C1-С10 алкила или циано, и R1 и R4 могут, при необходимости, вместе образовывать - (CR5R6)m-, где m обозначает 2-6, а R5 и R6 одинаковы или различны и независимо выбираются из гидрокси, Н или C1-С10 алкила, включая все их стереоизомеры; и их фармацевтически приемлемые соли, или пролекарственные сложные эфиры и все их стереоизомеры. Соединения ингибируют дипептидазу IV, что позволяет использовать их в фармацевтических композициях при лечении диабета типа 2. 3 н. и 17 з.п. ф-лы, 6 табл.
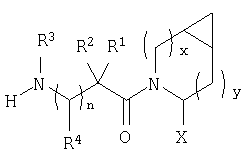
где х обозначает 0 или 1 и у обозначает 0 или 1, при условии, что
х обозначает 1, когда у обозначает 0 и
х обозначает 0, когда у обозначает 1; и где
n обозначает 0 или 1;
Х обозначает Н или CN;
R1, R2, R3 и R4 одинаковы или различны и независимо выбираются из водорода, C1-С10 алкила, C2-С12 алкенила, насыщенного C3-С10 циклоалкила, насыщенного C3-С10 циклоалкил C1-С10 алкила, насыщенного C3-С10 бициклоалкила, насыщенного C3-С10 трициклоалкила, гидроксил C1-С10 алкил насыщенного C3-С10 циклоалкила, C1-С10 алкилтио C1-С10 алкила, C6-С10 арил C1-С10 алкилтио C1-С10 алкила, C6-С10 арил C1-С10 алкила, 5 или 6-членного гетероарила, содержащего один атом N или один атом О, 5 или 6-членного гетероарила, содержащего один атом N конденсированный с C6-С10 арильным кольцом, 5 или 6-членного гетероарила, содержащего один атом N или один атом О C1-С10 алкила, циклогетероалкила, который является C5-С8 насыщенным кольцом, содержащим один гетероатом, такой как N или О; все при необходимости, имеют в качестве заместителей при соответствующем атоме углерода 1, 2, 3, 4 или 5 групп, выбираемых из галоида, C1-С10 алкила, C2-С12 алкенила, гидрокси, гидрокси C1-С10 алкила или циано, и R1 и R4 могут, при необходимости, вместе образовывать -(CR5R6)m-, где m обозначает 2-6, а R5 и R6 одинаковы или различны и независимо выбираются из гидрокси, Н или C1-С10 алкила,
включая все их стереоизомеры;
и их фармацевтически приемлемые соли, или пролекарственные сложные эфиры и все их стереоизомеры.
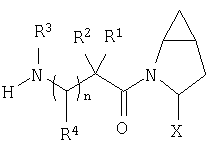
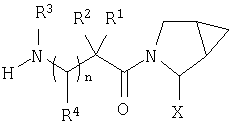
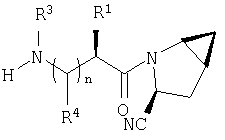
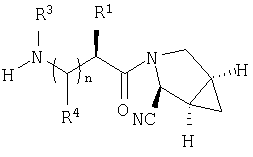
R3 обозначает Н, R1 обозначает Н, C1-С10 алкил, насыщенный C3-С10 циклоалкил, насыщенный C3-С10 бициклоалкил, насыщенный C3-С10 трициклоалкил, гидрокси C1-С10 алкил насыщенный C3-С10 циклоалкил, который может быть замещен гидрокси или гидрокси C1-С10 алкилом;
R2 обозначает Н или C1-С10 алкил, n обозначает О,
Х обозначает CN.
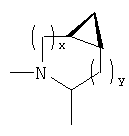
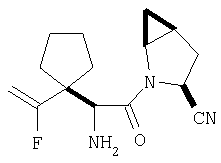
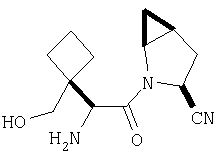
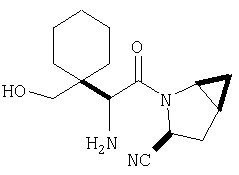
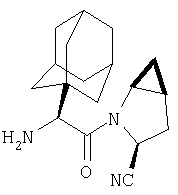
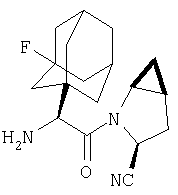
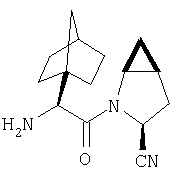
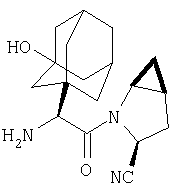
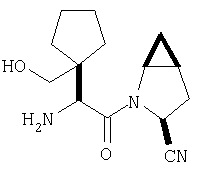
или его фармацевтически приемлемая соль.
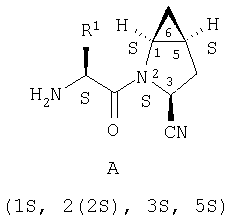
или
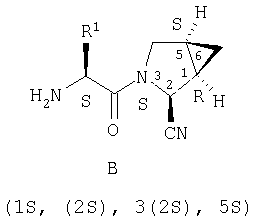
где R1 обозначает C1-С10 алкил, насыщенный C3-С10 циклоалкил, насыщенный C3-С10 бициклоалкил, насыщенный C3-С10 трициклоалкил, гидрокси C1-С10 алкил насыщенный C3-С10 циклоалкил.
| US 6011155, 04.01.2000 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| НЕКОТОРЫЕ КОНДЕНСИРОВАННЫЕ ПИРРОЛКАРБОКСАНИЛИДЫ, НОВЫЙ КЛАСС ЛИГАНДОВ МОЗГОВЫХ РЕЦЕПТОРОВ ГАМК | 1994 |
|
RU2125989C1 |

