Область техники
Настоящее изобретение относится к группе конъюгатов паклитаксела и производных мурамилдипептида, и синтезу, применению указанных соединений в лечении рака. Изобретение принадлежит к области медицинской технологии.
Уровень техники
Американским Национальным Институтом Рака (NCI) было обнаружено, что Паклитаксел (также называемый таксол), выделенный из тиса коротколистного (Taxus brevifolia)[1] демонстрирует противоопухолевую активность. Первоначальные исследования механизма действия показали, что паклитаксел является митотическим ингибитором, который приостанавливает рост раковых клеток на стадии G2 и М за счет активации полимеризации и деполимеризации микротрубочек раковой клетки, таким образом предотвращая формирование веретена деления в раковой клетке121. Дальнейшие исследования механизма действия показали, что паклитаксел также может использоваться в качестве аналога бактериального липополисахарида (LPS), который проявляет свое противоопухолевое действие, оказывая влияние или изменяя функцию макрофагов в иммунной системе, например, стимулируя экспрессию фактора некроза опухоли α (TNF-α) и интерлейкина-1(IL-1) в макрофагах[3,4]. Кроме того, он демонстрирует противоопухолевый эффект, активируя МАР-2 киназу, стимулируя фосфорилирование тирозина раковых клеток [5,6].
Мурамилдипептид (N-ацетилмурамил-L-аланил-D-изоглутамин, МДП) представляет собой наименьшее структурное звено, проявляющее иммуноадъювантную активность пептидогликанов клеточной стенки микробактерий[7,8]. МДП, вводимый одновременно или перед введением антигена, усилит иммунный ответ или изменит тип иммунного ответа. Кроме того, мурамилдипептид демонстрирует и другие типы активности, такие как неспецифическая резистентность к инфекции, вызванной палочкой Фридлендера, кишечной палочкой, синегнойной палочкой, листерией мононуклеоза, грибом tritirachium album и др., неспецифическая резистентность к фибросаркоме, гепатоме и др., и иммунорегуляция [9,13]. Исследования также показали, что МДП, связанный с липополисахаридом (LPS), может существенно стимулировать экспрессию цитокинов макрофагов[14,16].
Основываясь на этом, мы предположили, что паклитаксел, связанный с мурамилдипептидом, также может демонстрировать синергичность. Мы впервые выдвигаем новую идею о том, что связывание химиотерапевтического препарата паклитаксел и иммуностимулятора мурамилдипептида приводит к образованию ряда конъюгатов, и проводятся биологические исследования для проверки предположения о расширении целей химиотерапии за счет ассоциации иммунотерапии с противоопухолевой и в тоже время противо-метастатической[17].
Мы раскрыли две группы конъюгатов в нашем предыдущем патенте[18], которые были получены за счет связывания мурамилдипептида с паклитакселом 2'-гидрокси (2'-O-МТС, Схема 1), или с 3'-амино-производным 3'-N-бензоилпаклитакселом (3'-N-МТС, Схема 1). В ходе исследований in vitro мы обнаружили, что конъюгат 2'-O-МТС не только продемонстрировал противоопухолевую активность паклитаксела, но также ассоциировался с макрофагами, что приводило к выработке αTNF- и IL-1 в существенной степени, что означает, что он потенциально предотвращает метастазирование рака. Однако, активность 3'-N-MTC конъюгата была не столь значительна, как активность 2'-O-МТС.Исходя из этого, мы предварительно определили, что оптимальное положение конъюгатов для связывания будут 2'-гидроксильная группа. К сожалению, конъюгат 2'-O-МТС не показал желаемых результатов в экспериментах in vivo, которые могли зависеть от физико-химических свойств или фармацевтических свойств молекулы. В продолжении этой дизайнерской концепции, используемой при разработке новых лекарственных средств, мы оптимизировали аналог 2'-O-МТС посредством упрощения структур молекул мурамилдипептида, и получили новую группу аналогов 2'-O-МТС, которые продемонстрировали значительную противоопухолевую и противометастатическую активность в экспериментах in vivo, что означает, что они потенциально могут разрабатываться в качестве противоопухолевых лекарств, новая группа аналога 2'-O-МТС, упомянутая выше, представляет собой группу, описанную нами в настоящем патенте

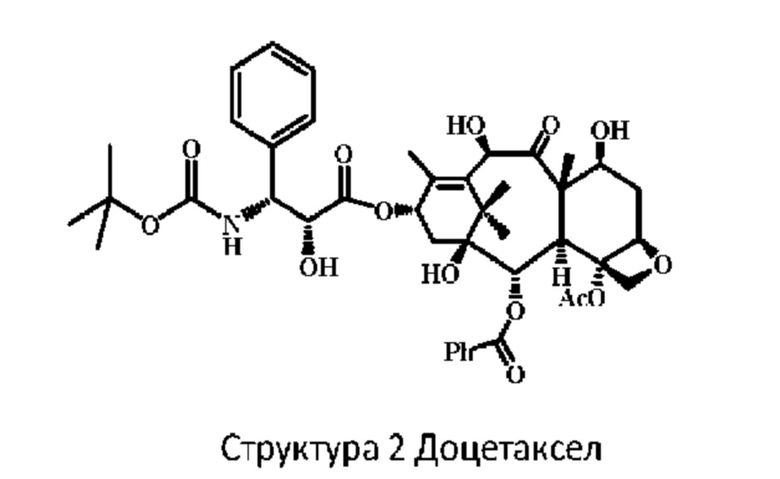
Структура 1 наша групповая патентная заявка для защиты двух типов конъюгатов Доцетаксел, полусинтетическое производное, является еще одним важным таксановым противоопухолевым препаратом, который демонстрирует ингибирующую активность на последней стадии рака молочной железы, немелкоклеточного рака легкого, рака яичников, рака поджелудочной железы, рака печени, опухолей головы и шеи. Недавние исследования показали, что доцетаксел индуцирует апоптоз раковых клеток за счет активации образования микротрубочками устойчивых полимеров и ингибирования деполимеризации[19], кроме того, он ингибирует митоз и пролиферацию раковых клеток[20]. Исследования также показали, что доцетаксел может остановить опухолевую клетку на G2/M стадии за счет активации экспрессии Вах белка и подавления экспрессии Bcl-2 белка[21].
Мурамилдипептид, стимулирующая иммунологическую реактивность часть конъюгатов паклитаксел-мурамилдипептид, демонстрирует широкий спектр биологической активности и вызывает большой интерес с момента своего открытия. Однако, мурамилдипептид проявляет несколько побочных эффектов, таких как иммуногенные индуцированные аллергические реакции, лихорадка, воспаление и сонливость, которые ограничивают его клиническое применение. Для того чтобы найти мурамил дипептид с более высокой активностью и меньшим числом побочных эффектов, ученые синтезировали сотни мурамилдипептидных соединений или аналогов и исследовали их биологическую активность. L-треонин-мурамилдипептид получают путем замены L-аланина в молекуле мурамилдипептида на L-треонин, который демонстрирует более высокую иммуноадъювантную активность, нежели чем у мурамилдипептида, но пирогенность в 100 раз ниже. При использовании в качестве адъювантной вакцины, L-треонин-мурамилдипептид не стимулирует макрофаги и противовоспалительный эффект, но стимулирует иммунный ответ введенного антигена, поэтому он является идеальной адъювантной вакциной за счет своего действия, и побочный эффект может быть эффективно отделен [22]
Мурабутид получают путем введения мурамилдипептида в длинную липотропную цепь. Мурабутид может увеличить неспецифическую антибактериальную и антивирусную инфекцию иммунной системы организма и стимулировать активность колониестимулирующего фактора. Кроме того, он хорошо переносится человеком[23-26]. По сравнению с другими экзогенными иммуномодуляторами, мурабутид не является пирогенным и способствует высвобождению цитокинами как синергетически, так и избирательно Thl цитокина, и мурабутид не вызывает воспалительной реакции[27,28] Кроме того, мурабутид в сочетании с IFN-α или IL-2 может значительно повысить противоопухолевую активность цитокинов, таким образом повышая антивирусный и противовоспалительный эффект IFN-α[29, 30]. Мурабутид может регулировать функцию макрофагов [31]. Он также может быть использован в лечении хронического гепатита С (HCV), поскольку в сочетании с IFN-α он продемонстрировал синергетический эффект в ходе экспериментов in vitro [32].
Мурамилтрипептид фосфатидилэтаноламин (МТР-РЕ) получают путем введения липофильной длинной цепи в мурамил дипептиды посредством фосфатной связи. МТР-РЕ может активировать моноциты и макрофаги, тем самым приводя к гибели опухолевых клеток. МТР-РЕ, инкапсулированный в липосомы (L-MTP-PE), вводимый внутривенно, в основном используют для активации макрофагов в легких, печени и селезенке [33]. Его активность выше от десяти до сотен раз, и пирогенность значительно снижена. Через два часа после внутривенного введения пациентам с метастатической меланомой, фактор некроза опухоли в плазме крови увеличивается в шестнадцать раз, а уровень содержания неоптерина и интерлейкина был существенно повышен [34].
MDP-Lys (L18) получают путем введения липофильной длинной цепи в мурамилдипептиды через лизин. MDP-Lys (L18) может увеличить производство цитокинов, таких как CSFs, IL-1, IL-6, -Lys (L18) может увеличить проит.д., которые играют важную роль в регулировании кроветворной системы [35, 36]. Кроме того, MDP-Lys (L18) обладает сильным антиинфекционным, противоопухолевым эффектом [37].
MDP-C получают путем введения ароматической конъюгированной системы в мурамилдипептиды посредством лизина. MDP-C способен стимулировать сильную цитотоксическую активность макрофагов против Р388 клеток лейкемии, он может также вводить стимулировать выработку мастоцитомы Р815 цитотоксическими Т-лимфоцитами (CTL). Сообщают, что MDP-C стимулирует дендритные клетки (BMDC) костного мозга мышей вырабатывать цитокины IL-2 и IL-12 (интерлейкин), и он также может быть использован как эффективный иммуностимулятор, поскольку он проявляет активность в отношении стимуляции выработки цитотоксическими Т-лимфоцитами интерферона-γ. Низкие дозы MDP-C могут значительно и синергично способствовать пролиферации лимфоцитов селезенки мыши, вызванной Конканавалином А (ConA). Кроме того, MDP-C может увеличить экспрессию молекул поверхностью дендритных клеток костного мозга, таких как CD11c, МНС I и молекулы-1 клеточной адгезии. Кроме того, MDP-C в экспериментах in vitro может быть значительно повышать за счет производства антител и специфичного поверхностного антигена вируса гепатита В (HBsAg) Т-клеток, реакция иммунной системы на HBsAg у трансгенных мышей с вирусом гепатита В была существенно усилена[38, 39].
Адамантантиламид дипептид (AdDP) получается путем соединения карбоксильного конца дипептидного фрагмента в молекуле t мурамилдипептида с амантадином. AdDP является безопасным и демонстрирует противовирусную антибактериальную активность. По сравнению с другими аналогами MDP, его биодоступность выше [40]. AdDP может улучшить гуморальный иммунитет у BALB / с мышей и кроликов в сочетании с белком иммуногена перорально или перитонеально [41]
Также были получены циклические аналоги мурамилдипептида, не содержащие сахарных остатков, путем выделения из природных веществ, такие как FK-156 и FK-565. Они продемонстрировали антибактериальную, противовирусную и противоопухолевую активность[421.
Ссылки
[1] Mansukhlal С. Wani, Harold Lawrence Taylor, Monroe E. Wall, Philip Coggon, Andrew T. McPhail; Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia; J. Am. Chem. Soc; 1971, 93(9), 2325-2327.
[2] Peter B. Schiff and Susan B. Horwitz; Taxol stabilizes microtubules in mouse fibroblast cells; Proc. Natl. Acad. Sci. USA; 1980, 77(3), 1561-1565.
[3] А. Н. Ding, F. Porteu, E. Sanchez, and C.F. Nathan; Shared actions of endotoxin and taxol on TNF receptors and TNF release; Science; 1990, 20, 370-372.
[4] Christian Bogdan and Aihao Ding; Taxol, a microtubule-stabilizing antineoplastic agent, induces expression of tumor necrosis factor a and interleukin-1 in macrophages; Journal of Leukocyte Biology; 1992, 52,119-121.
[5] B. Brugg and A. Matus; Phosphorylation determines the binding of microtubule-a- ssociated protein 2 (MAP2) to microtubules in living cells; J. Cell Biol.; 1991,114 (4), 735-743.
[6] Carboni, J., Singh, С, Tepper, M.; Cancer Institute Workshop on Taxol and Taxus, Alenandria, V.A.; NCI, 1992.
[7] Ellouz F., Adam A., Ciorbaru R., et al; Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives; Biochem. Biophys. Res. Commun.; 1974, 59,1317-1325.
[8] Adam, A., Ciorbaru, R., Ellouz, F., Petit, J.F. and Lederer, E.; Adjuvant activity of monomeric bacterial cell wall peptidoglycans; Biochem. Biophys. Res. Commun.; 1974, 56(3), 561-567.
[9] F. Audibert, L. Chedid, P. Lefrancier, J. Choay; Distinctive adjuvanticity of synthetic analogs of mycobacterial water-soluble components; Cellular Immunology; 1976, 21, 243-249.
[10] M. A. Parant, F. M. Audibert, L. A. Chedid, M. R. Level, P. L. Lefrancier, J. P. Choay, and E. Lederer; Immunostimulant activities of a lipophilic muramyl dipeptide derivative and of desmuramyl peptidolipid analogs; Infect. Immun.; 1980, 27,826-831.
[11] Adam A., Petit J. F., Chedid L; Influence of a synthetic adjuvant (MDP) on qualitative and quantitative changes of serum globulins; Immunology; 1978, 35(6), 963-970.
[12] Dietrich F.M., Hochkeppel H.K., Lukas В.; Enhancement of host resistance against virus infections by MTP-PE, a synthetic lipophilic muramyl peptide -increased survival in mice and guinea pigs after single drug administration prior to infection, and the effect of MTP-PE on interferon levels in sera and lungs; Int. J. Immunopharmacol; 1986, 8, 931-932.
[13] Adam A., Lederer E.; Muramyl peptides: immunomodulators, sleep factors, and vitamins; Med. Res. Rev., 1984, 4(2), 111-152.
[14] Anton V. Gorbachev, Nancy A. Dilulio, and Robert L.; Fairchild IL-12 augments CD81 T cell development for contact hypersensitivity responses and circumvents Anti-CD154 antibody-mediated inhibition; The Journal of Immunology, 2001,167,156-162.
[15] Alexandre A. Vetcher, Marek Napierala, Ravi R. Iyer, Paul D. Chastain, Jack D. Griffith, and Robert D.; Wells sticky DNA, a long GAA-GAA-TTC triplex that is formed intramolecularly, in the sequence of intron 1 of the frataxin gene; J. Biol. Chem.; 2002, 277, 39217-39227.
[16] C.L. Contel, N. Temime, D. J. Charron, and M.A. Parant; Modulation of lipopolysaccharide induced cytokine gene expression in mouse bone marrow-derived macrophages by muramyl dipeptide; The Journal of Immunology; 1993,150,4541-4549.
[17] Xuqin Li, Junli Yu, Song Xu, Nan Wang, Hongzhen Yang, Zheng Yan, Guifang Cheng, Gang Liu; Chemical conjugation of muramyl dipeptide and paclitaxel to explore the combination of immunotherapy and chemotherapy for cancer; Glycoconj J.; 2008, 25(5), 415-425.
[18] Патент №200510081265X.
[19] Toshiyuki Harada, Shigeaki Ogura, Koichi Yamazaki, Ichiro Kinoshita, Tomoo Itoh, Hiroshi Isobe, Katsushige Yamashiro, Hirotoshi Dosaka-Akita, Masaharu Nishimura; Predictive value of expression of P53, Bcl-2 and lung resistance-related protein for response to chemotherapy in non-small cell lung cancers; Cancer Science; 2005,94(4), 394-399.
[20] David L. Morse, Heather Gray, Claire M. Payne, and Robert J. Gillies; Docetaxel induces cell death through mitotic catastrophe in human breast cancer cells; Mol Cancer Ther; 2005, 4, 1495-1504.
[21] Yu Q, Gao J. X., He X. S., et al; Docetaxcel induces apoptosis and regulates expressions of bax an d bcl-2 protein in human breast carcinoma MCF-7 Cells; Cancer Res. Pre. Treatment, 2006, 33(6), 388-390.
[22] Deborah A. Eppstein, Noelene E. Byars, Anthony C. Allison; New adjuvants for vaccines containing purified protein antigens; Advanced Drug Delivery Reviews 1990,4, 233-253.
[23] L.A. Chedid, M.A. Parant, F.M. Audibert, G.J. Riveau, F.J. Parant, E. Lederer, J.P. Choay, and P. L. Lefrancier; Biological activity of a new synthetic muramyl peptide adjuvant devoid of pyrogenicity; Infection and Immunity; 1982, 35, 417-424.
[24] Chomel J. J., Simon-Lavoine N., Thouvenot D., Valette M., Choay J., Chedid L., Aymard M.; Prophylactic and therapeutic effects of murabutide in OF1 mice infected with influenza A and В viruses; International Journal of Immunopharmacology; 1985, 7(3), 346-347.
[25] George M. Bahr, Edith Darcissac, Dorian Bevec, Peter Dukor, Louis Chedid; Immunopharmacological activities and clinical development of muramyl peptides with particular emphasis on murabutide; International Journal of Immunopharmacology; 1995 17(2), 117-131.
[26] A. Galelli, P. Lefrancier, and L. Chedid; Colony-stimulating activity induced by synthetic muramyl peptides: variation with chemical structure and association with anti-infectious activity; Infection and Immunity; 1984,46, 495-500.
[27] George M. Bahr, Edith Darcissac, Philippe R. Pouillart, Louis A. Chedid; Synergistic effects between recombinant interleukin-2 and the synthetic immunomodulator murabutide: selective enhancement of cytokine release and potentiation of antitumor activity; Journal of Interferon and Cytokine Research; 1996,16(2), 169-178.
[28] Edith C. A. Darcissac, George M. Bahr, Philippe R. Pouillart, Gilles J. Riveau, Monique A. Parant; Selective potentiation of cytokine expression in human whole blood by murabutide, a muramyl dipeptide analogue; Cytokine, 1996,8, 658-666.
[29] George M. Bahr, Philippe R. Pouillart, Louis A. Chedid; Enhancement in vivo of the antiinflammatory and antitumor activities of type I interferon by association with the synthetic immunomodulator murabutide; Journal of Interferon and Cytokine Research; 1996, 16(4), 297-306.
[30] Philippe R. Pouillart, Francoise M. Audibert, Louis A. Chedid, Pierre L. Lefrancier, George M. Bahr; Enhancement by muramyl peptides of the protective response of interferon-α/β against encephalomyocarditis virus infection; International Journal of Immunopharmacology; 1996,18(3), 183-192.
[31] Gilles J. Riveau, Beatrice G. Brunel-Riveau, Francoise M. Audibert, Louis A. Chedid; Influence of a muramyl dipeptide on human blood leukocyte functions and their membrane antigens; Cellular Immunology; 1991,134,147-156.
[32] E. C. A. Darcissac, V. Vidal, M. Guillaume, J. J. Thebault, G. M. Bahr; Clinical tolerance and profile of cytokine induction in healthy volunteers following the simultaneous administration of IFN-a and the synthetic immunomodulator murabutide; Journal of Interferon and Cytokine Research; 2001, 21(9), 655-661.
[33] (a). Nardin A., Lefebvre M. L., Labroquere K., Faure O., Abastado J. P.; Liposomal muramyl tripeptide phosphatidylethanolamine: tTargeting and activating macrophages for adjuvant treatment of osteosarcoma; Current Cancer Drug Targets; 2006, 6,123-133.
(b). Meyers Paul A., Schwartz Cindy L., et al; A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate; J. Clin. Oncol.; 2005, 23(9), 2004-2011.
[34] Liebes L., Walsh С.M., Chachoua A., et al; Modulation of monocyte functions by muramyl triptide phosphatidylethanolamine in a phase II study in patients with metastatic melanoma; J. Natl. Cancer. Inst.; 1992,84, 694-699.
[35] Y. Osada, T. Otani, M. Sato, T. Une, K. Matsumoto, and H. Ogawa; Polymorphonuclear leukocyte activation by a synthetic muramyl dipeptide analog; Infection and Immunity; 1982, 38,848-854.
[36] Kenji Namba, Eiko Yamamura, Hironobu Nitanai, Tsuyoshi Otani, Ichiro Azuma; Romurtide, a synthetic muramyl dipeptide derivative, promotes megakaryocytopoiesis through stimulation of cytokine production in nonhuman primates with myelosuppression; Vaccine, 1997,15(4), 405-413.
[37] Ichiro Azuma, Tsukasa Seya; Development of immunoadjuvants for immunotherapy of cancer; International Immunopharmacology; 2001, 1(7), 1229-1392.
[38] Hong-Zhen Yang, Song Xu, Xue-Yan Liao, Suo-De Zhang, Zheng-Lun Liang, Bai-He Liu, Jin-Ye Bai, Chao Jiang, Jian Ding, Gui-Fang Cheng, and Gang Liu; A novel immunostimulator, N2-[α-O-Benzyl-N-(acetylmuramyl)-1-alanyl-d-isoglutaminyl]-N6-trans-(m-nitrocinnamoyl)-1-lysine, and its adjuvancy on the hepatitis В surface antigen; J. Med. Chem.; 2005, 48(16), 5112-5122.
[39] Патент CN 1609118 A.
[40] P. Walder, E. Buchar, Z. Machkova, T. Vrba, M. Flegel, I. Janku, K.  Pharmacokinetic profile of the immunomodulating compound adamantylamide dipeptide(AdDP), a muramyl dipeptide derivative in mice; Immuno- pharmacology and Immunotoxicology, 1991, 13(1 and 2), 101-119.
Pharmacokinetic profile of the immunomodulating compound adamantylamide dipeptide(AdDP), a muramyl dipeptide derivative in mice; Immuno- pharmacology and Immunotoxicology, 1991, 13(1 and 2), 101-119.
[41] Pablo D. Becker, Ricardo S. Corral, Carlos A. Guzman, Saul Grinstein; Adamantylamide dipeptide as effective immunoadjuvant in rabbits and mice; Vaccine; 2001, 19(32), 4579-4903.
[42] A. M. Kolodziejczyk, A. S. Kolodziejczyk, S. Stoev; New convenient synthesis of immunostimulating peptides containingmeso-diaminopimelic acid Syntheses of FK-565 and FK-156; International Journal of Peptide and Protein Research; 1992, 39(4), 382-387.
Подробное описание изобретения
Техническая проблема, решаемая с помощью настоящего изобретения, заключается в обеспечении ряда конъюгатов, обладающих противоопухолевой и противометастатической синергетической активностью.
Вторая техническая проблема, которая будет решена с помощью настоящего изобретения, это обеспечение способа получения таких конъюгатов.
Третьей технической проблемой, которая будет решена с помощью настоящего изобретения, является обеспечение фармацевтической композиции, содержащей такой конъюгат.
Еще одна техническая проблема, решаемая с помощью настоящего изобретения, заключается в применении такого конъюгата для получения противоопухолевых и противометастатических синергичных лекарственных средств.
Для решения технических проблем, упомянутых выше, в настоящем изобретении применены следующие решения:
По меньшей мере один активный фармацевтический ингредиент, выбранный из конъюгатов формулы I, и его фармацевтически приемлемые соли,
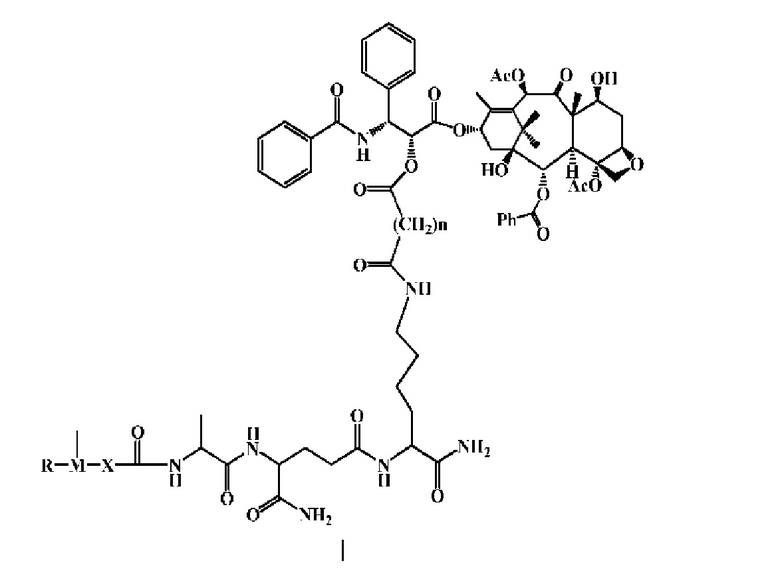
где n=2, 3,4, 5, 6, 7, 8, 9,10,11 или 12.
В предпочтительном варианте реализации n=2, 3, 4, 5, 6, 7,8, 9 или 10.
В другом предпочтительном варианте реализации n=2, 3, 4, 5, 6, 7 или 8.
В еще одном предпочтительном варианте реализации n=2, 3, 4 или 5.
Где X выбран из C1-6 алкила, C1-6 алкила и C1-6 алкила, содержащего гетероатом, причем гетероатом независимо выбран из кислорода, серы и азота; или X представляет собой одинарную связь таким образом, что М непосредственно связан с ацилом.
В предпочтительном варианте реализации X выбран из С1-4 алкила, C1-4 алкилена и C1-4 алкила, содержащего гетероатом, где гетероатом независимо выбран из кислорода и серы; или X представляет собой одинарную связь таким образом, что М непосредственно связан с ацилом.
В другом предпочтительном варианте реализации, X выбран из C1-3 алкила, C1-3 алкилена и C1-3 алкила, содержащего гетероатом, где гетероатом представляет собой является кислородом; или X представляет собой одинарную связь таким образом, что М непосредственно связан с ацилом.
В еще одном предпочтительном варианте реализации X выбран из -С=С-, -СН2-СН2-, -О-СН2- и одинарной связи.
М может представлять собой замещенный или незамещенный арил, замещенный или незамещенный гетероарил, например, М может представляет собой арил или гетероарил, термин «арил»,как показано в настоящем тексте, относится к арилу, содержащему от пяти до четырнадцати членов.
В одном варианте реализации М выбран из пятичленного арила, шестичленного арила, девятичленного конденсированного арила, десятичленного конденсированного арила, тринадцатичленного конденсированного арила и четырнадцатичленного конденсированного арила.
Термин «пятичленный арил», как показано в настоящем тексте, относится к  Термин «шестичленный арил», как показано в настоящем тексте, относится к
Термин «шестичленный арил», как показано в настоящем тексте, относится к 
Термин «девятичленный конденсированный арил», как показано в настоящем тексте, относится к 
Термин «десятичленный конденсированный арил», как показано в настоящем тексте, относится к 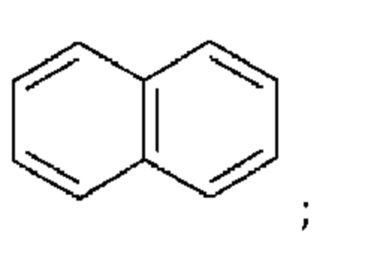
Термин «гетероарил» может, например, представлять собой гетероциклическое ароматическое кольцо, содержащее один или более, а именно один, два, три и четыре гетероатома в кольце, где гетероатом независимо выбран из азота, кислорода и серы.
В другом примере «гетероарил» может представлять собой гетероциклическое ароматическое кольцо, содержащее от пяти до четырнадцати членов и один или более, а именно один, два, три и четыре гетероатома в кольце, где гетероатом независимо выбран из азота, кислорода и серы.
В еще одном примере «гетероарил» может быть выбран из пятичленного гетероциклического ароматического кольца, шестичленного гетероциклического ароматического кольца, восьмичленного конденсированного гетероциклического ароматического кольца, девятичленного конденсированного гетероциклического ароматического кольца, десятичленного конденсированного гетероциклического ароматического кольца, все ароматические кольца, упомянутые выше, содержат один или более, а именно один, два, три и четыре гетероатома в кольце, где гетероатом независимо выбран из азота, кислорода и серы.
Термин «пятичленное гетероциклическое ароматическое кольцо», содержащее один и более, например, один, два, три или четыре гетероатома в кольце, где гетероатом независимо выбран из азота, кислорода и серы, пятичленное гетероциклическое ароматическое кольцо, описанное в настоящем тектсе, выбрано из
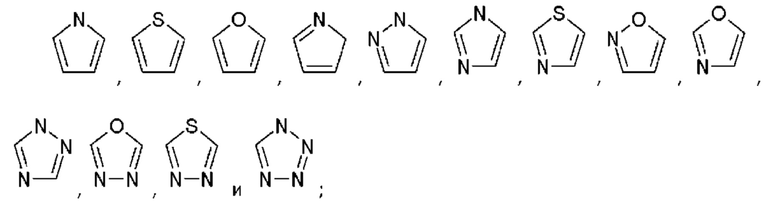
Термин «шестичленное гетероциклическое ароматическое кольцо», содержащее один и более, например, один, два, три или четыре гетероатома в ядре, где гетероатом независимо выбран из азота, кислорода и серы, шестичленное гетероциклическое ароматическое кольцо, описанное в настоящем тексте, выбрано из

Термин «восьмичленное конденсированное гетероциклическое ароматическое кольцо», содержащее один или более, например, один, два, три или четыре гетероатома в кольце, где гетероатом независимо выбран из азота, кислорода и серы, восьмичленное конденсированное гетероциклическое ароматическое кольцо, описанное в настоящем тексте, выбрано из
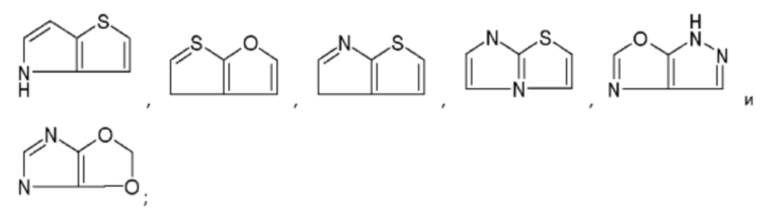
Термин «девятичленное конденсированное гетероциклическое ароматическое кольцо», содержащее один и более, например, один, два, три или четыре гетероатома в ядре, где гетероатом независимо выбран из азота, кислорода и серы, девятичленное конденсированное гетероциклическое ароматическое кольцо, описанное в настоящем тексте, выбрано из

Термин «десятичленное конденсированное гетероциклическое ароматическое кольцо», содержащее один и более, например, один, два, три или четыре гетероатома в ядре, где гетероатом независимо выбран из азота, кислорода и серы, десятичленное конденсированное гетероциклическое ароматическое кольцо, описанное в настоящем тексте, выбрано из

R относится к одному или нескольким заместителям, и R может быть необязательно связан с М.
В одном из вариантов реализации R выбранирается из водорода, замещенного или незамещенного прямого неразветвленного или разветвленного C1-6 алкила, гидрокси, замещенного или незамещенного неразветвленного прямого или разветвленного C1-6 алкокси, сульфгидрила, замещенного или незамещенного неразветвленного прямого или разветвленного C1-6 алкилтио, C1-6 элкокси-C1-6 алкила, амино, замещенного или незамещенного прямого неразветвленного или разветвленного C1-6 алкиламино, включающегоий моно-алкиламино или ди-алкиламино, альдегидной группы, замещенного или незамещенного неразветвленного или разветвленного C1-6алкилацила, карбоксила, замещенного или незамещенного неразветвленного или разветвленного C1-6 алкилацилокси, карбамоила, замещенного или незамещенного неразветвленного или разветвленного C1-6алкиламида, С2-6алкена, галогена, нитро и циано;
Замещенная группа C1-6 прямой цепи или разветвленной цепи, описанной в настоящем тексте, выбрана из гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, галогена, нитро и циано;
В одном из вариантов реализации R выбран из водорода, замещенного или незамещенного неразветвленного или разветвленного С1-4 алкила, гидрокси, замещенного или незамещенного неразветвленного или разветвленного С1-4 алкокси, С1-4 алкокси-С1-4 алкила, сульфгидрила, замещенного или незамещенного неразветвленного или разветвленного С1-4 алкилтио, амино, замещенного или незамещенного неразветвленного или разветвленного С1-4 алкиламино, включающего моно-алкиламино или ди-алкиламино, альдегидной группы, замещенного или незамещенного неразветвленного или разветвленного С1-4 алкилацила, карбоксила, замещенного или незамещенного неразветвленного или разветвленного См алкилацилокси, карбамоила, замещенного или незамещенного неразветвленого или разветвленного С1-4 алкиламида, С2-4 алкена, галогена, нитро и циано;
Замещенная группа неразветвленного или разветвленного СгС4алкила, описанного в настоящем тексте, выбрана из гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, фтора, хлора, брома, нитро и циано;
В одном из вариантов реализации R выбран из водорода, неразветвленного или разветвленного С1-4 алкила, гидрокси, неразветвленного или разветвленного С1-4 алкокси, сульфгидрила, неразветвленного или разветвленного С1-4 алкилтио, амино, неразветвленного или разветвленного С1-4 алкиламино, галогена, нитро и циано;
В одном из вариантов реализации R выбран из водорода, гидроксила, сульфгидрила, амино, фтора, хлора, брома, нитро, циано, метила, этила, н-пропила, изо-пропила, метокси, этокси, н-пропокси и изо-пропокси;
В одном из вариантов реализации конъюгат формулы I, как описано в настоящем тексте, выбран из конъюгата формулы IA, как показано ниже:
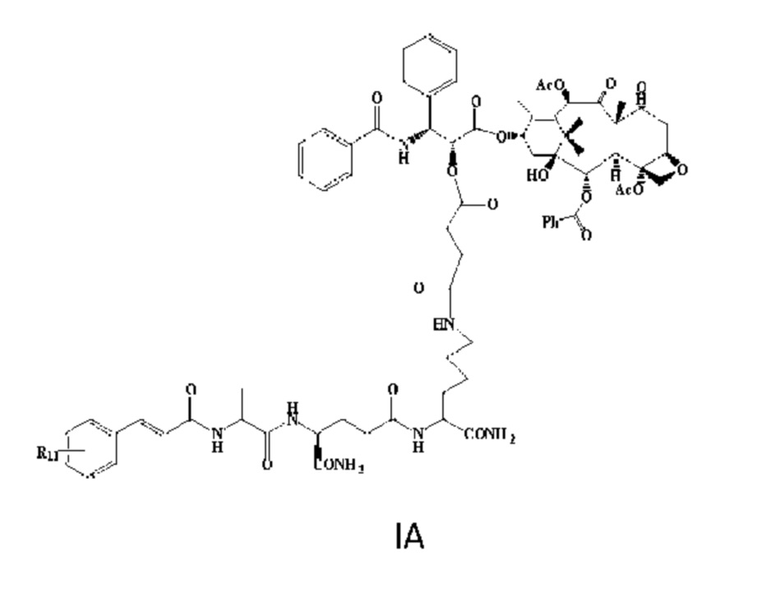
R11 относится к одному или более заместителям, и R11 может быть необязательно связан с фенилом. В одном из вариантов реализации R11 выбран из водорода, гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, галогена, нитро, циано, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино и С1-4 алкокси- С1-4 алкила.
В одном из вариантов конъюгат формулы I, как показано в настоящем тескте, выбран из конъюгата формулы IB, как представлено ниже:

R12 относится к одному или более заместителям, и R12 может быть необязательно связан с тиенилом. В одном из вариантов R12 выбран из водорода, гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, галогена, нитро, циано, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино и С1-4 алкокси- С1-4 алкила.
В одном из вариантов конъюгат формулы I, как показано в настоящем тексте, выбран из конъюгата формулы IC, как представлено ниже:

R13 относится к одному или более заместителям, и R13 может быть необязательно связан с фенилом. В одном из вариантов R13 выбран из водорода, гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, галогена, нитро, циано, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино и С1-4 алкокси- С1-4 алкила.
В одном из вариантов конъюгат формулы I, как показано в настоящем тексте, выбран из конъюгата формулы ID, как представлено ниже:

R14 относится к одному или более заместителям, и R13 может быть необязательно связан с хинолилом. В одном из вариантов R14 выбран из водорода, гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, галогена, нитро, циано, C1-4 алкила, C1-4 алкокси, C1-4 алкиламино и C1-4 алкокси-C1-4 алкила.
В одном варианте конъюгат формулы I, как показано в настоящем тексте, выбран из конъюгата формулы IE, как представлено ниже:

R15 представляет собой один или более заместителей, и R15 может быть необязательно связан с нафтилом. В одном из вариантов R15 выбран из водорода, гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, галогена, нитро, циано, C1-4 алкила, C1-4 алкокси, C1-4 алкиламино и C1-4 алкокси-C1-4 алкила.
В одном варианте неразветвленный или разветвленный C1-6 алкил, описанный в настоящем тексте, к неразветвленному или разветвленному C1-4 алкилу, или неразветвленному или разветвленному C2-5 алкилу. В другом варианте неразветвленный или разветвленный C1-6 алкил выбран из метила, этила, н-пропила, изо-пропила, н-бутила, изо-бутила, трет-бутила, пентила, нео-пентила, изо-пентила и гексила. Неразветвленный или разветвленный C1-4 алкил, описанный в настоящем тексте, предпочтительно выбран из метила, этила, н-пропила, изо-пропила, н-бутила и трет-бутила. Неразветвленный или разветвленный C2-5 алкил, описанный в настоящем тексте, предпочтительно выбран из этила, н-пропила, изо-пропила, н-бутила, трет-бутила, пентила и изо-пентила.
Замещающая группа замещенного или незамещенного неразветвленного или разветвленного C1-6 алкила, описанного в настоящем тексте, может быть выбрана из гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, галогена, нитро и циано.
Замещающая группа замещенного или незамещенного C1-4 алкила и неразветвленного или разветвленного C1-4 алкила, описанного в настоящем тексте, может быть выбрана из гидроксила, сульфгидрила, амино, альдегидной группы, карбоксила, карбамоила, фтора, хлора, брома, нитро и циано.
Термин «С2-6 алкен», как показано в настоящем тексте, относится к алкену, имеющему два, три, четыре, пять или шесть атомов углерода. Это может быть прямая цепь или разветвленная цепь. Например, С2-6 алкен может быть выбран из винила, 1-пропенила, 2-пропенила, 1-бутенила, 2-бутенила, 1-пентенила и 1-гексенила. С2-6 алкен предпочтительно выбран из C2-4 алкена.
Термин «алкокси», как показано в настоящем тексте, относится к -O-алкилу.
Термин «галоген», как показано в настоящем тексте, относится к фтору, хлору, брому или йоду. В одном из вариантов галоген предпочтительно выбран из фтора и хлора.
Группа «R-M-X-CO-» наиболее предпочтительно выбрана из п-хлорциннамоила, п-гидроксициннамоила, п-метилциннамоила, 2,4-дифторциннамоила, 3-фтор-4-хлорциннамоила, 3-хлор-4-фторциннамоила, 4-фторциннамоила, 3-фторциннамоила, 3,4-дифторциннамоила, 2-хинолинацила, 2-тиенилакрилоила, 2-нитро-4-хлорбензоила и 2-нафтилоксиацетила.
Фармацевтически приемлемая соль конъюгатов, описанных выше, представляет собой часть настоящего изобретения, основные атомы в молекулах конъюгатов в настоящем изобретении могут образовывать соли с кислотой, без особых ограничений, с любой фармацевтически приемлемой кислотой, такой как неорганические кислоты, включающие соляную кислоту, бромоводородную кислоту, серную кислоту, фосфорную кислоту, азотную кислоту и органические кислоты, включающие щавелевую кислоту, фумаровую кислоту, малеиновую кислоту, янтарную кислоту, лимонную кислоту, винную кислоту, метансульфоновую кислоту и п-толуолсульфоновую кислоту и др.
Конъюгаты аналога мурамилдипептида и паклитакселаи их соли могут быть синтезированы с помощью общих и типичных способов, как следующие:
1. синтез сложного моноэфира паклитаксел-2'-O-алкандикарбоновой кислоты посредством жидкофазного синтеза;
2. твердофазный или жидкофазный синтез аналога мурамилдепептида (MDA);
3. жидкофазный синтез конъюгатов аналога мурамилдепептида и паклитаксела.
При этом способ получения сложного моноэфира паклитаксел-2'-O-алкандикарбоновой кислоты методом жидкофазного синтеза включает следующие стадии:
(1) растворение пактитаксела, алкан-дикарбоновой кислоты диандригида и 4-N,N-диметилпиридина (DMAP) в пиридине и перемешивание в течение 4 часов при комнатной температуре (к.т.);
(2) разбавление раствора, полученного на стадии (1), этилацетатом (AcOEt), промывка слоя AcOEt последовательно насыщенным раствором CuSO4 и H2O;
(3) отделение слоя AcOEt и концентрирование под вакуумом, добавление к остатку избытка воды с выпадением белого осадка, получение сложного моноэфира паклитаксел-1'-O-алкандикарбоновой кислоты в виде белого твердого продукта после фильтрации и лиофилизации.
При этом способ получения аналога мурамилдипептида методом твердофазного синтеза и жидкофазного синтеза включает следующие стадии:
1) Твердофазный синтез:
(1) Синтез промежуточной аминокислоты Fmoc-D-iso-Gln-OH;
Схема синтеза показана на Фигуре 27, где реагенты и условия были следующими:
(а) к.т, 3 д; (b) дициклогексил карбодиимид (DCC), 0°С, 5 ч., к.т, 20 ч.; (с) NH3; -10°С, 1.5 ч.
(2) используя любую из аминосмол, такую как смола Rink AM (емкость смолы 0,88 ммоль/г], в качестве носителя твердой фазы, введение Fmoc-L-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-L-Ala-COOH и карбоновой кислоты в смолу посредством твердофазного синтеза. После завершения реакции конденсации аналог мурамилдепептида получают в результате таких стадий, как тщательная промывка и отщепление от смолы, и технический продукт очищают и т.д. Используемое здесь ацилирование является обычной реакцией конденсации с образованием амида, реакцию конденсации завершают путем добавления избыточного количества реагентов (таких как аминокислота или карбоновая кислота) и сверхактивного конденсирующего средства (такого как 2-(7-аза-1Н-бензотриазол-1-yl)-1,1,3,3-тетраметилуроний гексафторфосфат (HATU), 2-(1Н-бензотриазол-1-yl)-1,1,3,3-тетраметилуроний гексафторфосфат (HBTU), бензотриазол-1-ил-окситрис-(диметиламино)-фосфоний гексафторфосфат (ВОР), бензотриазол-1-ил-окситрипирролидинофосфоний гексафторфосфат (РуВОР). Особенностью данного способа является то, что введение карбоновой кислоты не зависит от структуры (такой как ароматические и неароматические, неразветвленная и разветвленная цепь), стерических затруднений, физико-химических свойств, электронного эффекта, кольцевой системы и линейной системы и т.д. Поэтому три вышеупомянутых аминокислоты могут быть заменены любой природной или искусственной аминокислотой, такой как Fmoc-D-Lys(Boc)-COOH, Fmoc-L-iso-Gln-COOH, Fmoc-L-Gln-COOH, Fmoc-D-Gln-COOH или Fmoc-D-Ala-COOH.
Схема синтеза показана на Фигуре 28, где реагенты и условия были следующими:
(а) 20% пиперидин/DMF; к.т., 1 ч.; (b) Fmoc-Lys(Boc)-OH, HOBt, N,N'-диизопропилкарбодиимид (DIC); к.т., 8 ч.; (С) Fmoc-D-iso-Gln-OH, HOBt, DIC; к.т, 12 ч; (d) Fmoc-Ala-OH, HOBt, DIC; к.т, 8 ч.; (е) органическая кислота©, HOBt, DIC; к.т, 8 ч.; (f) 90% трифторуксусная кислота (TFA)/H2O, к.т, 2 ч.
2) Жидкофазный синтез:
(1) Синтез промежуточной аминокислоты Boc-D-Glu(Obzl)-NH2;
Схема синтеза показана на Фигуре 29, где реагенты и условия были следующими:
(а) C6H5CH2OH, BF3⋅Et2O; к.т., 15 ч.; (b) (Вос)2O, NaHCO3; к.т., 20 ч.; (с) HOSu, DCC, NH3; -10°С, 1,5 ч.
(2) Синтез промежуточной аминокислоты Boc-Lys(Z)-NH2;
Схема синтеза показана на Фигуре 30, где реагенты и условия были следующими:
(a) HOSu, DIC, NH3;-10°C. 1,5 ч.
(3) синтез дипептидного фрагмента Boc-Ala-D-Glu(OBzl)-NH2 и трипептидного фрагмента R-Ala-D-Glu(OBzl)-NH2 посредством метода активной этерификации, и удаление защитной группы Bzl в трипептиде путем использования бромистоводной кислоты в растворе уксусной кислоты или в других подходящих кислотных/основных условиях, синтез тетрапептида R-Ala-D-iso-Gln-Lys(Z)-NH2 посредством метода активной этерификации;
(4) удаление защитной группы Z с помощью смеси этилэфирата трехфтористого бора, TFA и этантиола (V/V/V=9:9:2) с получением неочищенного продукта, и получение после очистки аналога мурамилдипептида. Схема синтеза показана на Фигуре 31, где реагенты и условия были следующими:
(а) 50% TFA/DCM; к.т. 1 ч.; (b) Boc-Ala-OH, HOSu, DIC; 0°С, 5 ч., к.т., 20 ч; (с) органическая кислота ©, HOSu, DIC; 0°С, 5 ч., к.т., 20 h; (d) HBr/HOAc; к.т., 3 ч.; (е) HOSu, DIC; 0°С, 5 ч., к.т., 20h; (f) BF3⋅Et2O,TFA, EtSH(9:9:2); к.т.2 ч.
При этом способ получения конъюгатов аналога мурамилдипептида и паклитаксела включает следующие стадии:
1) растворение сложного монозфира паклитаксел-2'-O-алкандикарбоновой кислоты, HOSu и DIC с определенным молярным соотношением (2:1-1:2) в диметилсульфоксиде (DMSO) или DMF, или N-метилпирролидоне и т.д., и осуществление реакции в растворе в течение 1-10 часов при температуре от -20°С до 50°С;
2) добавление эквимолярных количеств аналога мурамилдипептида к раствору DMSO или DMF или N-метилпирролидона и т.д., доведение рН реакционной смеси до 6-8 слабощелочным реагентом, таким как N-метилморфолин и т.д., осуществление реакции 1-10 часов с получением конъюгата после завершения реакции;
3) добавление любого растворителя, выбранного из воды, метанола, этанола, диэтилового эфира, петролейного эфира, этилбутилового эфира, в реакционный раствор, фильтрация выпавшего в осадок вещества, очистка неочищенного продукта с получением целевого продукта;
4) где способ очистки включает препаративную ВЭЖХ и перекристаллизацию.
Схема синтеза показана на Фигуре 32, где А представляет собой фенил, В представляет собой ацетокси, а реагенты и условия были следующими:
(а) ангидрид, DMAP, к.т., 4 ч.; (b) HOSu, 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDC-HCI), DMSO, к.т., 20 ч.; MDA производные, к.т., 12 ч.
При этом алкандикарбоновая кислота выбрана из C4-C14 алкандикарбоновой кислоты, диангидрид алкандикарбоновой кислоты выбран из диангидрида C4-C14 алкандикарбоновой кислоты.
Способ получения конъюгата, раскрытого в настоящем изобретении, характеризуется мягкими условиями реакции, малым временем реакции, стабильным выходом, так что он пригоден для построения библиотеки соединений посредством, например, метода комбинаторной химии, который также входит в объем притязаний согласно настоящему изобретению.
Специалисты в данной области техники могут изменить вышеуказанные стадии таким образом, чтобы повысить выход, они могут разработать схемы, исходя из базовых знаний в данной области, такие как выбор реагента, растворителя и температуры. Также они могут, используя ряд традиционных защитных групп, избежать побочной реакции и тем самым повысить выход продукта. Такие общие реакции могут быть упомянуты в книгах по химии синтеза пептидов, таких как 1) Gang LIU and Kit S. LAM, "One-bead one-compound combinatorial library method". Combinatorial Chemistry, A Practical Approach, Edited by Hicham Fenniri, OXFORD University Press, 2000, Chapter 2, pp 33-50; 2) Gang Liu, Xiaoyi Xiao, et al. Looking for combinatorial chemistry in drug research/Science Press, 2003, 6; 3) N. Leo Benoiton, Chemistry of Peptide Synthesis, published in 2005 by CRC press; 4) Miklos Bodanszky, Principles of Peptide Synthesis by Publisher of Springer Verlag (Edition: 2ND/REV). Такие модификации и изменения находятся в рамках настоящего изобретения.
В настоящем изобретении показано применение конъюгатов, раскрытых в настоящем изобретении, для изготовления лекарственных средств для профилактики и/или лечения рака. При этом указанный тип рак выбран из меланомы, рака желудка, рака легкого, рака молочной железы, рака почки, рака печени, эпидермальной карциномы полости рта, рака шейки матки, коллоидной аденофибромы, рака поджелудочной железы, рака предстательной железы и рака толстой кишки.
Поэтому настоящее изобретение также относится к композициям, содержащим терапевтическое количества конъюгата, описанного в настоящем изобретении, и один или более фармацевтически приемлемых носителей и/или наполнителей композиции. Фармацевтические носители включают по меньшей мере один в солевой раствор, буферный солевой раствор, декстрозу, воду, глицерин, этанол, далее описанные более подробно. При необходимости композиция может также содержать меньшее количество увлажняющих или эмульгирующих агентов или рН буферных агентов. Композиция может представлять собой жидкий раствор, суспензию, эмульсию, таблетки, пилюли, капсулы, препараты с замедленным высвобождением или порошки. Композиция может быть в виде суппозиторий, в которых используют традиционные связующие вещества и носители, такие как глицерид трехосновной карбоновой кислоты. В препарате для перорального приема можно использовать стандартные носители, такие как маннит, лактоза, крахмал, стеарат магния, сахарин натрия, целлюлоза и карбонат магния и др. фармацевтической степени чистоты. В соответствии с требованиями относительно изготовлений различных препаратов, стадия, предваряющая получение, может включать смешивание, гранулирование и прессование или растворение активных ингредиентов. Кроме того, композиция может быть получена в виде наночастиц.
Используемый в настоящем изобретении фармацевтический носитель может быть твердым или жидким.
Носитель или наполнитель могут представлять собой вещество замедленного растворения, известного специалистам в данной области, такое как глицерилмоностеарат или глицерилдистеарати могут включать воски, этилцеллюлозу, гидроксипропилметилцеллюлозу и метилметакрилат и др. Известный PHOSALPG-50 (фосфолипид с 1,2-пропандиолом был сконцентрирован, Д. Nattermann & Cie. GmbH) в 0,01% Твин-80, используемом для получения препарата других конъюгатов, подходящего для перорального приема, может быть также использован при получении конъюгатов, описанных в настоящем изобретении.
Конъюгаты, раскрытые в настоящем изобретении, могут быть введены в различных лекарственных формах. Если используют твердый носитель, то препарат может представлять собой таблетку, твердую капсулу с порошком или небольшими пилюлями в ней, пастилки или сахарные пастилки. Количество твердого носителя может широко варьироваться, но предпочтительно от примерно 25 мг до 1 г. Если использован жидкий носитель, то препарат может быть в виде сиропа, эмульсии, мягких желатиновых капсул, стерильных инъекционных растворов или суспензий или неводной жидкой суспензии в ампуле или флаконе.
Различные системы высвобождения известны и могут быть использованы для введения конъюгатов или различных их препаратов, эти препараты включают таблетки, капсулы, растворы для инъекций, липосомные капсулы, микрочастицы, микрокапсулы и др. Способ введения включает, но не ограничивается, кожный, внутрикожный, внутримышечный, внутрибрюшинный, внутривенный, подкожный, интраназальный, легочный, эпидуральный, офтальмологический и пероральный (предпочтительно). Конъюгаты могут быть введены любым удобным или подходящим способом, например, посредством инъекции или болюсной инъекции, поглощения через эпителиальные или слизистые оболочки (например, слизистой оболочки полости рта, прямой кишки и слизистой оболочки кишечника и т.п.), или с помощью стента, элюирующего лекарственные препараты, или может быть введен совместно с другими биологически активными веществами, или может быть использовано общее или местное введение. Для лечения или профилактики заболеваний носа, бронхиальных или легочных заболеваний, предпочтительным способом введения является пероральный, назальный или с помощью бронхиального аэрозоля или ингалятора.
Краткое описание чертежей
Фигура 1 - 50% ингибирующая рост опухоли концентрация (Gl50) и 50% летальная концентрация (LC50) МТС-220 в 60 клеточных линиях опухолей человеческого происхождения.
Фигура 2 - 50% ингибирующая рост опухоли концентрация (Gl50) и 50% летальная концентрация (LC50) МТС-302 в 60 клеточных линиях опухолей человеческого происхождения.
Фигура 3 - 50% ингибирующая рост опухоли концентрация (Gl50) и 50% летальная концентрация (LC50) МТС-213 в 60 клеточных линиях опухолей человеческого происхождения.
Фигура 4 - 50% ингибирующая рост опухоли концентрация (Gl50) и 50% летальная концентрация (LC50) МТС-219 в 60 клеточных линиях опухолей человеческого происхождения.
Фигура 5 - 50% ингибирующая рост опухоли концентрация (Gl50) и 50% летальная концентрация (LC50) МТС-233 в 60 клеточных линиях опухолей человеческого происхождения.
Фигура 6 - Противоопухолевая активность МТС-301, 302, 303 и 304 в 10 клеточных линиях опухолей in vitro.
Фигура 7 - Противоопухолевая активность МТС-305, 306, 307 и 308 в 10 клеточных линиях опухолей in vitro.
Фигура 8 - Влияние МТС-220 на массу тела мышей с MDA-MB-231 опухолью.
Фигура 9 - Торможение МТС-220 роста MDA-MB-231 опухоли у мышей.
Фигура 10 - Влияние МТС-220 на RTV мышей с MDA-MB-231 опухолью, которые подвергались лечению той же дозой при другом способе введения.
Фигура 11 - Влияние МТС-220 на массу тела мышей с MDA-MB-231 опухолью, которые подвергались лечению той же дозой при другом способе введения.
Фигура 12 - Влияние МТС-220 на массу тела мышей с H460 опухолью.
Фигура 13- Торможение МТС-220 роста H460 опухоли у мышей.
Фигура 14 - Торможение МТС-220 роста MCF-7 опухоли у мышей.
Фигура 15 - Влияние МТС-220 на массу тела мышей с MCF-7 опухолью.
Фигура 16 - Торможение МТС-220 роста А549 опухоли у мышей.
Фигура 17 - Влияние МТС-220 на массу тела мышей с А549 опухолью.
Фигура 18 - Влияние МТС-220 на массу тела мышей с H1975 опухолью.
Фигура 19 - Торможение МТС-220 роста 1-11975 опухоли у мышей.
Фигура 20 - Торможение МТС-220 роста опухоли у мышей с раком груди (1).
Фигура 21 - Влияние МТС-220 на массу тела мышей с раком груди (2).
Фигура 22 - Естественная противометастатическая активность МТС-220 у мышей с раком груди (3).
Фигура 23 - Активность МТС-220 по торможению роста опухоли у мышей с раком легких Льюиса (1).
Фигура 24 - Влияние МТС-220 на массу тела у мышей с раком легких Льюиса (2).
Фигура 25 - Естественная противометастатическая активность МТС-220 у мышей с раком легких Льюиса (3).
Фигура 26 - Искусственная противометатстатическая активность МТС-220 у мышей с раком легких Льюиса.
Фигура 27 - Схема твердофазного синтеза промежуточной аминокислоты Fmoc-D-iso-GIn-ОН.
Фигура 28 - Схема твердофазного синтеза промежуточной аминокислоты Fmoc-D-iso-GIn-ОН.
Фигура 29 - Схема жидкофазного синтеза промежуточной аминокислоты Boc-D-Glu(Obzl)-NH2.
Фигура 30 - Схема жидкофазного синтеза промежуточной аминокислоты Boc-Lys(Z)-NH2.
Фигура 31 - Схема получения очищенного аналога мурамилдипептида.
Фигура 32 - Схема синтеза конъюгатов аналога мурамилдипептида и паклитаксела.
Фигура 33 - Схема жидкофазного синтеза сложного моноэфира паклитаксел-2'-O-янтарной кислоты.
Фигура 34 - Схема синтеза Fmoc-D-iso-Gln-OH.
Фигура 35 - Схема твердофазного синтеза аналога мурамилдипептида MDA.
Фигура 36 - Схема жидкофазного синтеза аналога мурамилдипептида MDA.
Фигура 37 - Схема жидкофазного синтеза Boc-D-Glu(OBzl)-NH2.
Подробное описание примеров реализации
Настоящее изобретение далее иллюстрируется следующими примерами синтеза конъюгатов аналога мурамилдипептида и паклитаксела и биологическими исследованиями таких конъюгатов. Специалистам в данной области должно быть понятно, что эти примеры используются всего лишь в иллюстративных целях, без ограничения объема настоящего изобретения. Объем данного изобретения ограничивается только формулой изобретения. Оставаясь в рамках объема формулы изобретения, специалисты в данной области техники могут прийти к пониманию различных аспектов настоящего изобретения, различных модификаций и улучшений, при этом данные модификации и усовершенствования также будут находиться в рамках защиты настоящего изобретения.
Также, если только специально не указано, материалы и реагенты, используемые в следующих примерах, являются традиционно используемыми в данной области, и могут быть получены традиционным коммерческим путем; используемые промежуточные соединения могут быть получены традиционным коммерческим путем или изготовлены с помощью известных способов; используемые способы представляют собой традиционные способы, известные специалистам в данной области.
Химический пример
Пример 1: Жидкофазный синтез сложного моноэфира паклитаксел-2'-O-янтарной кислоты (Синтетический метод описан в CN 200510081265)
Схема синтеза показана на Фигуре 33, где реагенты и условия были следующими: янтарный ангидрид, DMAP, к.т., 4 ч.
8.53 г (1.0 eq) паклитаксела, 1.2 г (1.2 eq) янтарного ангидрида, 0.12 г (0.1 eq) 4-N,N-диметилпиридина растворили в пиридине и перемешивали при к.т.в течение 4 ч. После того как реакция завершена, раствор пиридина разбавили AcOEt. И затем слой AcOEt последовательно промыли насыщенным водным раствором CuSO4 и H2O. После этого слой AcOEt отделили. Раствор концентрировали под вакуумом, и затем избыток воды добавили к остатку, белое сухое вещество выпадает в осадок в системе. После фильтрации и лиофилизации получено 8.1 г. целевого продукта с выходом 85%, Тпл=178~180°С.
1H-NMR (600 MHz, DMSO-d6): 4.63 (1H, br.s, 1-OH), 5.40 (1H, d, J=8.4Hz, 2-H), 3.58(1H, d, J=8.4Hz, 3-H), 4.90(1H. d, J=10.8Hz, 5-H). 1.62(1H, t, J=14.4Hz, 6-Ha), 2.31(1H, m, 6-Нь),4.10(1Н, dd, J=12.0 and 8.4Hz, 7-H), 4.89(1H, d, J=10.8Hz, 7-OH). 6.29(1H, s, 10-H), 5.81(1H, t, J=10.8Hz, 13-H), 1.51(1H, m, 14-Ha), 1.81(1H, m, 14-Hb), 0.99(3H, s, 16-H), 1.02(3H, s, 17-H), 1.75(3H, s, 18-H), 1.49(3H, s, 19-H), 3.98(1H, d, J=10.2Hz, 20-Ha), 4.02(1H, d, J=10.2Hz, 20-Hb), 2.10(3H, s, 4-ОСОСН3), 2.23(3H, s, 10-ОСОСН3), 5.35(1H, d, J=10.8Hz, 2'-H), 5.54(1H, dd, J=10.8 and 10.2Hz, 3'-H), 9.21(1H, d, J=10.2Hz, 3'-NH), 7.49(2H, m, ph-o-H), 7.47(2H, m, ph-m-H), 7.54(1H, m, ph-p-Н), 7.84(2Н, d, J=10.2Hz, NBz-o-H), 7.43(2H, m, NBz-m-H), 7.19(1H, m, NBz-p-H), 7.97(2H, d, J=9.6Hz, OBz-o-H), 7.65(2H, m, OBz-m-H), 7.72(1H, m, OBz-p-H), 2.61(2H, t, J=7.2Hz, -CHb-CH2-COOH), 2.32(2H, m, -CH2-CH2-COOH), 12.23(1H, br.s, -CH2-CH2-COOH).
13C-NMR (150 MHz, DMSO-d6): 76.7(1-C), 74.5(2-C), 46.1(3-C), 80.2(4-C), 83.6(5-C), 36.5(6-C), 70.4(7-C), 57.3(8-C), 202.3(9-C), 74.7(10-C), 133.3(11-C), 139.4(12-C), 70.7(13-C), 34.4(14-C), 42.9(15-C), 26.3(16-C], 21.3(17-C], 13.8(18-C), 9.7(19-C), 75.2(20-C), 169.6(2-OCO), 169.6, 22.5(4-ОСОСН3), 168.7, 20.6(10-ОСОСН3), 169.0(1'-C), 74.7(2'-С), 53.9(3'-С), 166.4(3'-NHCO), 137.3(ph-q-C). 127.6(ph-o-C), 128.3(ph-m-C), 131.4(ph-p-C), 129.9(NBz-q-C), 127.4(NBz-o-C), 128.6(NBz-m-C), 128.2(NBz-p-C), 134.3(OBz-q-C), 129.5(OBz-o-C), 128.6(OBz-m-C), 133.4(OBz-p-C), 172.9, 28.4, 30.9,171.6(-СО-СН2-СН2-СООН).
IR: 3471.3(νOH и νNH). 3065.2(ν=C-H). 2957.5(ν-C-H), 1717.3, 1642.0(νC=O), 1602.4, 1579.8, 1525.9(νC=C), 1487.4,1370.4(ν-C-H), 1241.4(νC-O-C), 978.6, 904.7, 948.5, 776.0, 708.3(ν=CH).
ESI-MS: 954.75 [М+Н]+, 1929.13 [2M+Na]+.
HR-MS(TOF): 954.3552 [M+H]+, 976.3352 [M+Na]+, C51H55NO17.
Пример 2-3: Твердофазный синтез аналога мурамилдипептида MDA
Пример 2: Синтез Fmoc-D-iso-Gln-OH
Схема синтеза показана на Фигуре 34, где реагенты и условия были следующими:
(а) к.т., 3 д; (b) DCC, 0°С, 5 ч., к.т., 20 ч; (с) NH3; -10°С, 1.5 ч.
Стадия 1: Синтез Fmoc-D-Glu-OH
В бане с ледяной водой перемешали раствор D-глутами новой кислоты (H-D-Glu-OH, 29.4 г., 1.0 eq) в смеси с ацетоном и H2O (V/V=1:1); после того как твердое вещество полностью растворилось, NaHCO3 (23.3 г., 1.1 eq) добавили порциями, затем медленно добавили Fmoc-OSu (67.4 г., 1.0 eq), и реакционную смесь оставили перемешиваться еще на дополнительные 3 дня при к.т.. Затем смесь снова охладили в бане с ледяной водой, и довели рН до 2-3 2.0N HCl. После удаления под пониженным давлением ацетона, оставшийся раствор экстрагировали AcOEt (400 мЛ×4). Органический слой отделяли и объединили, высушили в течение ночи над MgSO4 и сконцентрировали до малого объема под пониженным давлением. Затем остаток был перекристаллизовали из смеси этилацетат-циклогексан. После фильтрации получили 59.8 г. целевого продукта в виде белого твердого вещества с выходом 81%.
Стадия 2: Синтез Fmoc-D-iso-Gln-OH
Fmoc-D-Glu-OH (59.8 г., 1.0 eq) растворили в безводном тетрагидрофуране (THF)(324 мл). Затем при перемешивании в бане с ледяной водой добавили DCC (40.1 г., 1.2 eq). Реакционную смесь оставляли подогреться до к.т.и затем продолжали перемешивание в течение еще 8 часов для получения 1,3-дициклогексилмочевины (DCU). Осадок отфильтровывали и промывали небольшим количеством THF. Затем при перемешивании в бане с NaCl льдосоляной смесью сухой газообразный аммиак пропускали через реагенты. Реакция завершается через 1.5 часа, когда больше не осаждается белое вещество. В течение еще 30 минут для растворения осадка добавили небольшое количество МеОН. Смесь охладили снова в бане с ледяной водой. Затем аккуратно и медленно добавили 2.0 N HCl для доведения рН до 2-3. Растворитель упаривали под вакуумом. Твердый остаток растворили в AcOEt и затем последовательно промыли разбавленной HCl, насыщенным водным раствором NaHCO3 и H2O. Органический слой отделили и объединили, затем в течение ночи сушили над MgSO4, отфильтровали и упаривали под вакуумом. Затем остаток был перекристализован из смеси этилацетат-циклогексан. После фильтрации получили 46.5 г. целевого продукта с выходом в 78%. Тпл=204~205°С, [α]=-4.2°(С=10 мг/мл, DMF].
1H-NMR (500 MHz, DMSO): 7.88(2H, d, J=8.0Hz), 7.72(2H, m), 7.42(2H, m), 7.40(1H, m), 7.40(1H, br.s), 7.32(2H, m, 7.02(1H, br.s),4.27(2H, m), 4.20(1H, m), 3.93(1H, dd,J=13.5 и 8.5Hz), 2.25(2H, m), 1.89(1H, m), 1.73(1H, m).
13C-NMR (125 MHz/DMSO): 173.9, 173.4, 155.9, 143.8, 140.7, 127.6, 127.0, 125.3, 120.0, 65.6,53.8,46.6,30.4,27.2.
ESI-MS: 369.03 [М+Н]+, 759.98 [2M+Na]+.
HR-MS(TOF): 369.1448 [М+Н]+, 759.2623 [2М+Na]+, C20H20N2O5.
Пример 3: Твердофазный синтез аналога мурамилдипептида MDA

Схема синтеза показана на Фигуре 35, где реагенты и условия были следующими:
(а) 20% пиперидин/DMF; к.т., 1 ч.; (b) Fmoc-Lys(Boc)-OH, HOBt, DIC; к.т., 8 ч; (С) Fmoc-D-iso-Gln-OH, HOBt, DIC; к.т., 12 ч.; (d) Fmoc-Ala-OH, HOBt, DIC; к.т., 8 ч.; (е) 4-хлоркоричная кислота(R), HOBt, DIC; к.т., 8 ч.; (f) 90%TFA/H2O, к.т., 2 ч.
100.0 г смолы Rink-Amide AM (емкость смолы 0.88 ммоль/г, 1.0 eq) поместили в твердофазный реактор и вакуумировали при пониженном давлении в течение 1 ч. Безводный DCM (500 мл) добавили для набухания смолы на 45 минут, а затем его удалили. Fmoc группу смолы удаляют с помощью 20% (объемный процент) пиперидина/DMF в течение 1 ч. при к.т., с последующей тщательной промывкой DMF (500 мл × 6) и с DCM (500 мл × 6) последовательно. Fmoc-Lys (Boc)-COOH (61,8 г., 1,5 зкв), HOBt (17,8 г., 1,5 экв) и DIC (20,8 мл., 1,5 экв.) растворяли в DMF (500 мл), затем добавляли в реактор, первая аминокислота была связана со смолой после реакции в течение 8 ч. при к.т.Когда она была нейтрализована посредством применения нингидрина, реакция синтеза была завершена с последующей тщательной промывкой DMF (500 мл × 6) и DCM (500 мл × 6) последовательно. После Fmoc была удалена при использовании 20% (объемный процент) пиперидина/DMF. Fmoc-D-iso-Gln-OH (48.5 г., 1.5 экв), Fmoc-Ala-OH (41.0 г., 1.5 экв.), и 4-хлоркоричная кислота (24.1 г., 1.5 экв) были последовательно добавлены для ввода второй аминокислоты в твердую фазу. Реакция длилась 12 ч. и отслеживалась с использованием нингидрина. После осветления жидкой фазы 500 мл 20% (объемный процент) пиперидин / DMF был добавлен для удаления Fmoc, жидкость снова стала прозрачной спустя 1 час; смолу промывали DMF (500 мл * 6) и DCM (500 мл * 6) последовательно. Fmoc-Ala-COOH (41 г., 1.5 экв.), HOBt (17,8 г, 1,5 экв), DIC (20,8 мл, 1,5 экв) и 500 мл DMF были добавлены для ввода третей аминокислоты. Реакцию проводили 12 часов и контролировали с помощью нингидрина. После осветления жидкой фазы 500 мл 20% (объемный процент) пиперидин / DMF был добавлен для удаления Fmoc, жидкость снова отстоялась после 1 ч, смолу промывали DMF (500 мл × 6) и DCM (500 мл × 6) последовательно. Хлоркоричная кислота (24.1 г., 1.5 экв), HOBt (17.8 г., 1.5 eq), DIC (20.8 мл, 1.5 eq) и 500 мл DMF были добавлены для ввода органической кислоты. Реакция продолжалась 8 часов и контролировалась с помощью нингидрина. После осветления жидкой фазе смолу промывали DMF (500 мл * 6) и DCM (500 мл * 6) последовательно. Водный раствор TFA 90% (объемный процент) был добавлен в реактор, реакция продолжалась в течение 2 часов. После удаления жидкости водный раствор TFA 90% (объемный процент) был добавлен в реактор, реакция продолжалась в течение 2 часов, после удаления жидкости смолу промывали 200 мл DCM. Водный раствор TFA и DCM были объединены и упарены в вакууме. В бане со льдом к остатку добавили избыток диэтилового эфира, выпадает в осадок белое вещество, надосадочная жидкость удаляют. Белое твердое вещество растирали с диэтиловым эфиром несколько раз с получением неочищенного продукта (39,8) с выходом 89%. Технический продукт очищали с помощью ODS колоночной хромоторгафии с градиентным элюированием, метанол/вода с получением 35.8 г целевого продукта с 98,5% чистотой. Тпл=215 ~ 217°С, [///]=37,7° (С=11.05 мг/мл, DMF).
1H-NMR (600 MHz, DMSO-d6): 7.47(2H, d, J=8.4Hz, 2 and 6-H), 7.57(2H, d, J=8.4Hz, 3 и 5-Н), 7.39(1Н, d, J=15.9Hz, 7-H), 6.75(1H, d, J=15.9Hz, 8-H), 8.39(1H, d, J=6.6Hz. 10-H), 4.38(1H. m, 11-H), 1.26(3H, m, 12-H), 8.21(1H, d, J=8.4Hz, 14-H), 4.14(1H, m, 15-H), 6.98(1H, s, 17-Ha), 7.41(1H, s, 17-Hb), 1.71(1H, m, 18-Ha), 1.97(1H, m, 18-Hb), 2.15(2H, t, J=7.2Hz, 19-H), 7.90(1H, d, J=8.4Hz, 21-H). 4.11(1H, m, 22-H). 7.10(1H, s, 24-Ha), 7.30(1H, s, 24-Hb), 1.46(1Н, m, 25-Ha), 1.63(1H, m, 25-Hb), 1.27(2H, m, 26-H), 1.53(2H, m, 27-H), 2.73(2H, m, 28-H), 7.75(2H, br.s, 29-H).
13C-NMR (150 MHz, DMSO-d6): 134.0(1-C), 129.0(2 and 6-C), 129.2(3 and 5-C), 133.8(4-C), 137.6(7-C), 122.7(8-C), 164.7(9-C), 48.8(11-C), 18.1(12-С). 172.4(13-C), 52.2(15-C), 173.8(16-C), 27.7(18-C), 31.7(19-C), 171.6(20-C), 52.1(22-C), 173.3(23-C), 31.3(25-C), 22.4(26-C), 26.8(27-C), 38.7(28-C).
IR: 3282.3, 3202.2(νOH и νNH), 3067.3(ν=CH), 2938.0(ν-CH), 1609.5(ν-C=O), 1537.5, 1450.2(νC=C), 1199.0,1180.2, 1130.6ν-CH), 972.4, 820.4, 799.4, 720.0(ν=CH и νC-Cl).
ESI-MS: 509.60 [М+Н]+, 1017.24 [2М+Н]+.
HR-MS(TOF): 509.2292 [М+Н]+, C23H33ClN6O5.
Пример 4-10: Жидкофазный синтез аналога мурамилдипептида MDA
Схема синтеза представлена на Фигуре 36, где реагенты и условия были следующими:
(a) HOSu, DIC, NH3; -10°С, 1.5 ч.; (b) 50%TFA/DCM; к.т. 1 ч.; (с) HOSu, DIC; 0°C, 5h, к.т., 20 ч.; (d) 0°С, 5 ч., к.т, 24 ч.; (е) HBr/НОАс; к.т., 3 ч.; (f) ВР3⋅Et2O, TFA, EtSH (9:9:2); к.т. 2 ч.
Пример 4: Жидкофазный синтез Boc-D-Glu(OBzl)-NH2
Схема синтеза представлена на Фигуре 37, где реагенты и условия были следующими:
(а) C6H5CH2OH. BF3⋅Et2O; к.т., 15 ч.; (b) (Вос)2O, NaHCO3; к.т, 20 ч; (с) HOSu, DCC, NH3; -10°С, 1.5 ч.
Стадия 1: Жидкофазный синтез H-D-Glu(OBzl)-OH

К раствору 29.1 г (1.0 eq) H-D-Glu-OH в 205.6 мл (10.0 eq) бензилового спирта при перемешивании при к.т., медленно добавили 47.7 мл (2.0 eq) раствора эфирата трехфтористого бора, и 10 минут спустя весь этот субстрат растворился. Реакция была завершена через 15 часов, добавили 616.8 мл (3-хкратный избыток по отношению к объему бензилового спирта) THF, перемешали и медленно добавили 55.1 мл (2.0 eq) триэтиламина. Образовалось большое количество белого вязкого осадка. THF удаляли при пониженном давлении; остаток охладили, после добавления необходимого количества AcOEt вязкий осадок превратился в порошок. 36.6 г. целевого соединения было получено с выходом 78% после полной фильтрации. Тпл=174~176°С.
Стадия 2: Жидкофазный синтез Boc-D-Glu(OBzl)-OH

36.6 г. (1.0eq) H-D-Glu(OBzl)-OH было растворено в 500 мл диоксана/воды (о/о=1:1), 67.3 г. (2.0 eq) Boc ангидрида и 25.3 г. натрия бикарбоната (2.0eq) были последовательно добавлены; и растворения всех субстратов проводили на масляной бане. Раствор перемешивали при к.т.в течение 20 часов. После завершения реакции диоксан удаляли в вакууме, и образовалось большое количество вязкого осадка. Осадок был разбавлен 500 мл воды и перемешивали в течение еще 30 минут до полного растворения. рН раствора довели до 2 ~ 3 с помощью 2 N водного раствора HCl в бане со льдом, и смесь стала непрозрачной, и отстаивалась в течение 30 минут. Раствор экстрагировали AcOEt 5 раз, и органические фазы объединили, высушили над MgSO4 в течение ночи. После фильтрации AcOEt был удален в вакууме, и получили 48.6 г. желтого маслянистого целевого соединения с выходом 96%.
Стадия 3: Жидкофазный синтез Boc-D-Glu(OBzl)-NH2
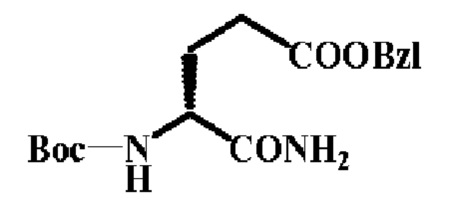
48.6 г. (1.0eq) Boc-D-Glu(OBzl)-OH растворили в тетрагидрофуране, 24.8 г. (1.5eq) HOSu и 44.5 г. (1.5 eq) DCC последовательно добавили. После перемешивания в течение 5 часов в бане со льдом, реакционную смесь нагрели до к.т.и перемешивали еще в течение 20 часов. Большое количество белого осадка (DCU) образовалось, осадок был отфильтрован и промыт небольшим количеством тетрагидрофурана. Фильтрат перемешивали в льдосоляной бане, и к раствору был добавлен безводный аммиак. Через 15 минут выпало большое количество белого осадка, смесь перемешивали еще 1.5 часа, образование белого осадка в растворе прекратилось, и реакция была завершена. Осадок был отфильтрован и промыт тетрагидрофураном, и получили желтое масло после удаления тетрагидрофурана в вакууме. Желтое масло разбавили AcOEt, и рН раствора довели до 7 2N водным раствором HCl в бане со льдом, и поставили отстаиваться в течение 30 минут. Слой AcOEt был отделен, и последовательно промыт разбавленной соляной кислотой, насыщенным бикарбонатом натрия и водой. Смесь отфильтровали, и фильтрат упарили досуха в вакууме, и остаток был перекристаллизован из смеси этилацетат-гексан с получением 34.2 г. целевого соединения с выходом 75%, Тпл=122~123°С, [α]=-1.8° (С=9.8 мг/мл, DMF)
1H-NMR (300 MHz, DMSO-d6): 1.36 (9Н, s, -С(СН3)3), 6.82 (1Н, d, J=8.4 Hz, 4-Н), 3.8 (1Н, m, 5-Н), 7.01 (1Н, s, 7-На), 7.31 (1Н, s, 7-Hb), 1.73 (1Н, m, 8-На), 1.88 (1Н, m, 8-Hb), 2.36 (2H, t, J=7.2 Hz, 9-H), 5.07 (2H, s, 11-H), 7.25-7.39 (5H, m, 12~16-H).
13C-NMR (125 MHz, DMSO-d6): 28.1 (1-С), 78.0 (2-C), 155.3 (3-C), 53.3 (5-C), 173.5 (6-C), 27.1 (8-C), 30.2 (9-C), 172.2 (10-C), 65.4 (11-C), 127.8 (12 and 16-C), 128.4 (13 and 15-C), 127.9 (14-C).
ESI-MS: 337.75 [M+H]+, 673.32 [2M+H]+.
HR-MS(TOF): 337.1754 [M+H]+, 359.1572 [M+Na]+, C17H24N2O5.
Пример 5: Жидкофазный синтез Boc-Lys(Z)-NH2
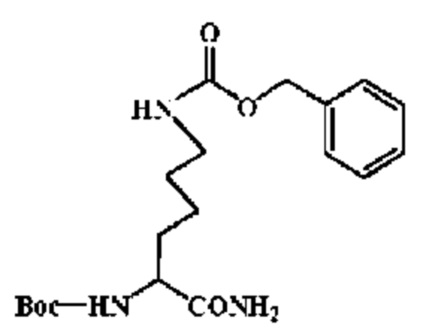
К раствору 38.0 г. (1.0 eq) Boc-Lys(Z)-OH в тетрагидрофуране было добавлено 13.8 г. (1.2 eq) HOSu и 18.9 мл (1.2 eq) DIC, смесь перемешивали на водяной бане в течение 5 часов, и затем при к.т.еще 20 часов. Большое количество белого вещества (DIU) выпало в осадок. Смесь фильтровали, и остаток промывали тетрагидрофураном. Фильтрат перемешивали в бане криогидрата хлористого натрия, и в него был добавлен безводный газообразный аммиак.. Через 15 минут большое количество белого вещества выпало в осадок, и реакцию продолжали 1.5 часа, в смеси прекратилось образование осадка, и реакция была завершена. Смесь фильтровали, остаток промыли тетрагидрофураном. Фильтрат упарили досуха в вакууме и получили белый твердый остаток. Остаток растворили в AcOEt, рН раствора довели до 7 2 N водным раствором HCl в бане со льдом, и оставили отстаиваться на 30 минут. Слой AcOEt был отделен, последовательно промыт разбавленной соляной кислотой, насыщенным водным раствором двууглекислого натрия и водой, и высушен в течение ночи с MgSO4. Смесь фильтровали, и фильтрат упаривали досуха в вакууме, остаток был перекристаллизован из AcOEt с получением 35.0 г. целевого соединения с выходом 92%, Тпл=137~138°С.
1H-NMR (300 MHz, DMSO-d6): 1.37 (9Н, br.s, 1-H), 6.71 (1Н, d, J=8.1 Hz, 4-H), 3.79 (1Н, m, 5-H), 7.23 (2H, br.s, 7-H), 1.28 (2H, m, 8-H), 1.45 (2H, m, 9-H), 1.58 (2H, m, 10-H), 2.95 (2H, m, 11-H), 6.93 (1Н, br.s, 12-H), 5.00 (2H, s, 14-H), 7.22-7.39 (5H, m, 16~20-H).
ESI-MS: 380.71 [M+H]+, 759.50 [2M+H]+.
HR-MS(TOF): 380.2201 [M+H]+, 781.4102 [2M+Na]+, C19H29N3O5.
Пример 6: Жидкофазный синтез 6ипептидного фрагмента Boc-Ala-D-Glu(OBzl)-NH2

16.9 г. (1.0 eq) Вос-Ala-ОН растворили в тетрагидрофуране, 12.3 г. (1.2 eq) HOSu и 16.9 мл (1.2 eq) DIC последовательно добавили, смесь перемешивали в течение 5 часов в бане со льдом, и затем перемешивали еще в течение 20 часов при к.т.. Образовалось большое количество белого осадка (DIU). Смесь фильтровали, и остаток промыли небольшим количеством тетрагидрофурана, и фильтрат (Boc-Ala-OSu) собрали для дальнейшего использования.
30 г (1.0 eq) Boc-D-Glu(OBzl)-NH2 растворили в 100 мл смеси трифторуксусная кислота-дихлорметан (v/v=1:1), и раствор перемешивали в течение 1 часа при к.т., чтобы удалить Вое группу. После завершения реакции, TFA удаляли в вакууме; остаток неоднократно растирали в безводном эфире, промывали и упаривали досуха, и перерастворили в тетрагидрофуране. рН раствора был установлен на 7~8 с помощью N-метилморфолина (NMM) на ледяной бане. Раствор Boc-Ala-OSu был аккуратно добавлен к раствору несколькими порциями. Смесь перемешивали в течение 5 часов на ледяной бане, и затем еще в течение 24 часов при к.т.после завершения реакции, смесь упаривали досуха. Остаток был растворен в необходимом количестве AcOEt и последовательно промыт разбавленной соляной кислотой, насыщенным водным раствором бикарбоната натрия и водой. Слой AcOEt был отделен и высушен над MgSO4 в течение ночи. Смесь была отфильтрована, и фильтрат был упарен досуха. Остаток был перекристаллизован из метанола и воды, кристаллический осадок был промыт в большом количестве эфира с получением 29.4 г. целевого соединения. Выход: 81%, Тпл=134~135°С
1H-NMR (300 MHz, DMSO-d6): 1.36 (9Н, br.s., 1-H), 7.92 (1Н, d, J=7.8 Hz, 4-H), 4.17 (1H, m, 5-H), 1.15 (3H, d, J=7.2 Hz, 6-H), 7.10 (1Н, d, J=6.6 Hz, 8-H), 3.91 (1Н, m, 9-H), 7.18 (1H, br.s, 11-Ha), 7.31 (1H, br.s, 11-Hb), 1.75 (1H, m, 12-Ha), 2.03 (1H, m, 12-Hb), 2.33 (2H, t, J=7.5 Hz, 13-H), 5.07 (2H, s, 15-H), 7.31-7.40 (5H, m, 17~21-H).
ESI-MS: 408.71 [M+H]+, 815.44 [2M+H]+.
HR-MS (TOF): 408.2137 [M+H]+, 430.1955 [M+Na]+, C20H29N3O5.
Пример 7: Жидкофазный синтез трипептидного фрагмента
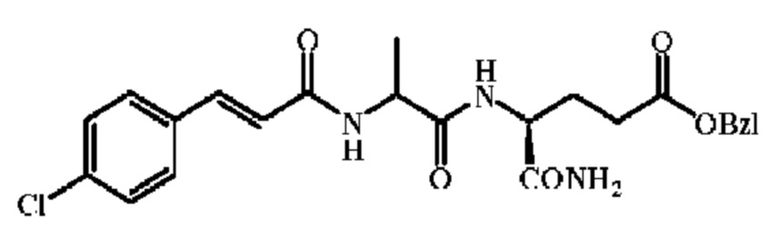
К раствору 13.2 г. (1.0 eq) 4-хлоркоричной кислоты в тетрагидрофуране было добавлено 9.9 г. (1.2 eq) HOSu и 13.6 мл (1.2 eq) DIC. Смесь перемешивали на ледяной бане в течение 5 часов, и затем при к.т. еще 20 часов. Большое количество белого вещества (DIU) выпало в осадок. Смесь фильтровали, и остаток промыли тетрагидрофураном; фильтрат (Ac-Osu) был собран для дальнейшего использования.
29.4 г. (1.0 eq) Boc-Ala-D-Glu(OBzl)-NH2 растворили в 100 мл смеси трифторуксусная кислота-дихлорметан (v/v=1:1), и раствор перемешивали в течение 1 часа, чтобы удалить Вое группу. После завершения реакции TFA удаляли в вакууме. Остаток неоднократно растирали, промывали эфиром, и упаривали досуха, и перерастворили в тетрагидрофуране. рН раствора был доведен до 7~8 с помощью N-метилморфолина (NMM) на ледяной бане. Раствор Ala-OSu был аккуратно добавлен к смеси несколькими порциями. Смесь перемешивали в течение 5 часов в на ледяной бане, и затем еще в течение 24 часов при к.т., и кипятили с обратным холодильником в течение 2 часов. После завершения реакции смесь была оставлена отстаиваться на 30 минут, и большое количество вязкого белого осадка образовалось. Смесь была отфильтрована, и осадок промыли тетрагидрофураном. Осадок растворили в AcOEt, и раствор промыли разбавленной соляной кислотой, насыщенным бикарбонатом натрия и водой. Слой AcOEt был отделен и высушен с MgSO4 в течение ночи. Смесь была отфильтрована, и фильтрат был упарен досуха. Остаток был перекристаллизиван из смеси метанол-вода, кристаллический осадок был промыт большим количестве безводного эфира с получением 26.8 г. целевого соединения. Выход: 79%, Тпл=226~228°С.
1Н-NMR (300 MHz, DMSO-d6): 7.48 (2Н, d, J=8.7 Hz, 2~6-Н), 7.59 (2Н, d, J=8.7 Hz, 3~5-Н), 7.39 (1H, d, J=15.9 Hz, 7-H), 6.76 (1Н, d, J=15.9 Hz, 8-H), 8.39 (1H, d, J=6.6 Hz, 10-H), 4.38 (1H, m, 11-H), 1.23 (3H, d, J=6.9 Hz, 12-H), 8.25 (1H, d, J=8.1 Hz, 14-H), 4.18 (1H, m, 15-H), 7.16 (1H, br.s, 17-Ha), 7.31 (1H, br.s, 17-Hb), 1.78 (1H, m, 18-Ha), 2.05 (1H, m, 18-Hb), 2.38 (2H, m, 19-H), 5.07 (2H, s, 21-H), 7.31-7.36 (5H, m, 23~27-H).
ESI-MS: 472.33 [M+H]+, 943.17 [2M+H]+.
HR-MS (TOF): 472.1635 [M+H]+, 943.3174 [2M+H]+, C24H26ClN3O5
Пример 8: Жидкофазный синтез трипептидного фрагмента

26.8 г. трипептидного фрагмента из примера 7 растворили в бромистоводородной кислоте/растворе уксусной кислоты. Раствор перемешивали в течение 2 часов для удаления защитной группы. После завершения реакции раствор выливали в ледяную воду и доводили рН смеси устанавливается до 10~11 с помощью 10% водного раствора NaOH. После экстракции AcOEt рН раствора доводили до 2~3 10% водным раствором HCl. Водную фазу экстрагировали AcOEt 3 раза, органические слои объединили, промыли насыщенным солевым раствором и высушили над NaSO4. Смесь фильтровали, и фильтрат упаривали в вакууме до небольшого объема раствора. При добавлении эфира образовалось большое количество белого твердого осссадка. Смесь фильтровали, и остаток высушли с получением 18.5 г. целевого соединения. Выход: 85%.
1H-NMR (300 MHz, DMSO-d6): 7.45 (2Н, d, J=8.1 Hz, 2~6-Н), 7.56 (2Н, d, J=8.1 Hz, 3~5-Н), 7.42 (1H, d, J=15.3 Hz, 7-H), 6.75 (1Н, d, J=15.3Hz, 8-H), 8.39 (1H, d, J=6.6 Hz, 10-H), 4.37 (1H, m, 11-H), 1.25 (3H, d, J=6.6 Hz, 12-H), 8.21 (1H, d, J=8.1 Hz, 14-H), 4.16 (1H, m, 15-H), 7.11 (1H, br.s, 17-Ha), 7.30 (1Н, br.s, 17-Hb), 1.72 (1H, m, 18-Ha), 1.98 (1H, m, 18-Hb), 2.22 (2H, m, 19-H), 12.25 (1H, br.s, 21-H).
ESI-MS: 382.17 [M+H]+, 785.04 [2M+Na]+.
HR-MS(TOF): 382.1171 [M+H]+, 785.2073 [2M+Na]+, C17H20ClN3O5.
Пример 9: Жидкофазный синтез тетрапептидного фрагмента

16.3 г. (1.0 eq) трипептидного фрагмента из примера 8 растворили в 5.9 г. (1.2 eq) HOSu и 8.1 мл (1.2 eq) DIC добавили последовательно. Смесь перемешивали в течение 5 часов на ледяной бане, а затем в течение 20 часов при к.т. Большое количество белого твердого осадка (DIU) образовалось. Смесь была профильтрована, и осадок промыт небольшим количеством тетрагидрофурана, фильтрат собрали для дальнейшего использования.
16.2 г. (1.0 eq) Boc-Lys(Z)-NH2 растворили в 100 мл смеси трифторуксусная кислота-дихлорметан (v/v=1:1), и раствор перемешивали в течение 1 часа при к.т., чтобы удалить Вое группу. После завершения реакции, TFA удаляли в вакууме, и остаток был неоднократно растирали с эфиром, промывали и упаривали досуха. Остаток перерастворили в тетрагидрофуране, и рН раствора довели до 7~8 с помощью N-метилморфолина (NMM) на ледяной бане. Фильтрат аккуратно добавили к раствору несколькими порциями и перемешивали на ледяной бане в течение 5 часов, а затем еще 24 часа при к.т. Большое количество вязкого белого осадка образовалось. Смесь была отфильтрована, и осадок промыт небольшим количеством тетрагидрофурана. Затем остаток был высушен, и было получено 14.6 г целевого соединения с выходом 74%, Тпл=195~196°С.
1H-NMR (300 MHz, DMSO-d6): 7.47 (2Н, m, 2 и 6-Н), 7.58 (2Н, m, 3 и 5-Н), 7.38 (1Н, d, J=15.3 Hz, 7-Н), 6.79 (1Н, d, J=15.3 Hz, 8-Н), 8.45 (1Н, d, J=8.1 Hz, 10-Н), 4.40 (1Н, m, 11-Н), 1.28 (3Н, m, 12-Н), 8.29 (1Н, d, J=8.1 Hz, 14-Н), 4.19 (1Н, m, 15-Н), 6.95 (1Н, s, 17а-Н), 7.41 (1Н, s, 17b-Н), 1.71 (1Н, m, 18а-Н), 1.96 (1Н, m, 18b-H), 2.14 (2Н, m, 19-Н), 7.92 (1Н, m, 21-Н), 4.12 (1Н, m, 22-Н), 7.09 (1Н, s, 24а-Н), 7.33 (1Н, m, 24b-Н), 1.49 (1Н, m, 25а-Н), 1.65 (1Н, m, 25b-Н), 1.27 (2Н, m, 26-Н), 1.53 (2Н, m, 27-Н), 2.91 (2Н, m, 28-Н), 6.91 (1Н, br.s, 29-Н), 5.00 (2Н, s, 31-Н), 7.20-7.38 (5Н, m, 33-37-Н).
13C-NMR (125 MHz, DMSO-d6): 133.9 (1-С), 129.0 (2 and 6-С), 129.2 (3 и 5-С), 133.8 (4-С), 137.6 (7-С), 122.8 (8-С), 164.7 (9-С), 48.9 (11-С), 18.1(12-С), 172.4 (13-С), 52.1 (15-С), 173.9 (16-С), 27.6 (18-С), 31.6 (19-С), 171.5 (20-С), 52.1 (22-С), 173.3 (23-С), 31.4 (25-С), 22.7 (26-С), 27.5 (27-С), 38.7 (28-С), 156.0 (30-С), 65.1 (31-С), 137.5 (32-С), 127.7 (33 и 37-С), 128.3 (34 и 36-С), 127.0 (35-С).
ESI-MS: 643.31 [M+H]+.
HR-MS (TOF): 643.2635 [M+H]+, 665.2451 [M+Na]+, C31H39ClN6O7.
Пример 10: Жидкофазный синтез аналога мурамилдипептида MDA

14.6 г. трипептидного фрагмента из примера 9 растворили в смеси растворителей диэтилэфирата трехфтористого брома, трифторуксусной кислоты и этанола (v:v:v=9:9:2). Раствор перемешивали в течение 2 часов при к.т.После завершения реакции растворитель упаривали досуха в вакууме. Большое количество эфира было добавлено к остатку на ледяной бане, и образовался белый твердый осадок. Смесь центрифугировали, и надосадочную жидкость отделяли. Остаток промыли большим количеством эфира, и получили 8.3 г. неочищенного продукта с выходом 72%. 8.3 г. неочищенного продукта очищали ODS колоночной хроматографией с градиентным элюированием (метанол-вода). Элюат собрали, и растворитель удаляли в вакууме, и далее высушили посредством лиофилизации, получили 6.8 г. целевого соединения с чистотой 98.5%. Тпл=215~217°С, [α]=+37.7° (С=11.05 мг/мл, DMF).
1H-NMR (600 MHz, DMSO-d6): 7.47 (2Н, d, J=8.4 Hz, 2 и 6-Н), 7.57 (2Н, d, J=8.4 Hz, 3 и 5-Н), 7.39 (1Н, d, J=15.9 Hz, 7-Н), 6.75 (1Н, d, J=15.9 Hz, 8-Н), 8.39 (1Н, d, J=6.6 Hz, 10-H), 4.38 (1Н, m, 11-H), 1.26 (3H, m, 12-H), 8.21 (1H, d, J=8.4 Hz, 14-H), 4.14 (1H, m, 15-H), 6.98 (1H, s, 17-Ha), 7.41 (1H, s, 17-Hb), 1.71 (1H, m, 18-Ha), 1.97 (1H, m, 18-Hb), 2.15 (2H, t, J=7.2 Hz, 19-H), 7.90 (1H, d, J=8.4 Hz, 21-H), 4.11 (1H, m, 22-H), 7.10 (1H, s, 24-Ha), 7.30 (1H, s, 24-Hb), 1.46 (1H, m, 25-Ha), 1.63 (1Н, m, 25-Hb), 1.27 (2H, m, 26-H), 1.53 (2H, m, 27-H), 2.73 (2H, m, 28-H), 7.75 (2H, br.s, 29-H).
13C-NMR (150 MHz, DMSO-d6): 134.0 (1-C), 129.0 (2 и 6-C), 129.2 (3 и 5-C), 133.8 (4-C), 137.6 (7-C), 122.7 (8-C), 164.7 (9-C), 48.8 (11-C), 18.1 (12-С), 172.4 (13-C), 52.2 (15-С), 173.8 (16-C), 27.7 (18-C), 31.7 (19-C), 171.6 (20-C), 52.1 (22-С), 173.3 (23-C), 31.3 (25-C), 22.4 (26-C), 26.8 (27-C), 38.7 (28-C).
IR: 3282.3, 3202.2 (νOH и νNH), 3067.3 (v=CH), 2938.0 (ν-CH), 1609.5 (ν-C=O), 1537.5, 1450.2 (νC=C), 1199.0, 1180.2, 1130.6 (δ-CH), 972.4, 820.4, 799.4, 720.0 (δ=СН and νC-Cl).
ESI-MS: 509.60 [M+H]+, 1017.24 [2M+H]+.
HR-MS(TOF): 509.2292 [M+H]+, C23H33ClN6O5.
Пример 11-22: Твердофазный синтез аналога мурамилдипептида
Пример 11: Твердофазный синтез мурамилдипептида MDA-201

Была задействована схема твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и п-гидроксикоричная кислота вводились в смолу последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель удаляли в вакууме, к остатку добавили большое количество эфира в ледяной бане, и белое твердое вещество выпало в осадок. Смесь была отфильтрована, получили неочищенный продукт, выход 85%. Технический продукт очищали посредством ODS колоночной хроматографии, и получили белое твердое вещество с чистотой 98.5% в результате липофилизации. Тпл=143~144°С.
1H-NMR (300 MHz, DMSO-d6): 9.94 (1Н, s, 1-ОН), 6.79 (2Н, d, J=8.7 Hz, 2 и 6-Н), 7.59 (2Н, d, J=8.7 Hz, 3 and 5-Н), 7.36 (1Н, d, J=15.9 Hz, 7-H), 6.51 (1H, d, J=15.9 Hz, 8-H), 8.25 (1Н, d, J=6.3 Hz, 10-H), 4.34 (1Н, m, 11-H), 1.24 (3H, m, 12-H), 8.17 (1H, d, J=8.4 Hz, 14-H), 4.12 (1H, m, 15-H), 6.98 (1Н, s, 17-Ha), 7.31 (1Н, s, 17-Hb), 1.72 (1H, m, 18-Ha), 1.98 (1H, m, 18-Hb), 2.15 (2H, m, 19-H), 7.89 (1Н, d, J=7.8 Hz, 21-H), 4.11 (1Н, m, 22-H), 7.10 (1H, s, 24-Ha), 7.3 (1Н, s, 24-Hb), 1.48 (1H, m, 25-Ha), 1.63 (1H, m, 25-Hb), 1.25 (2H, m, 26-H), 1.50 (2H, m, 27-H), 2.74 (2H, m, 28-H), 7.76 (2H, br.s, 29-H).
13C-NMR (125 MHz, DMSO-d6): 159.0 (1-C), 115.8 (2 and 6-C), 129.3 (3 и 5-C), 125.8 (4-C), 139.2 (7-C), 118.2 (8-C), 165.5 (9-C), 48.9 (11-C), 17.9 (12-C), 172.6 (13-C), 52.2 (15-C), 173.8 (16-С), 27.6 (18-C), 31.7 (19-C), 171.6 (20-C), 52.1 (22-С), 173.3 (23-C), 31.3 (25-C), 22.4 (26-C), 26.7 (27-C), 38.7 (28-C).
IR: 3273.8, 3194.6 (νOH и νNH), 3064.6 (ν=Ch), 2943.4 (ν-CH), 1663.6 (νC=O), 1605.7, 1537.3, 1515.0, 1450.4 (νC=C), 1201.6, 1180.2, 1135.7 (δ-CH), 983.8, 835.0, 800.4, 721.6 (δ=CH).
ESI-MS: 491.39 [M+H]+, 981.21 [2M+H]+.
HR-MS(TOF): 491.2597 [M+H]+, C23H34N6O6.
Пример 12: Твердофазный синтез мурамилдипептида MDA-202

Была задействована схема твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 4-метилкоричная кислота вводились в смолу последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель удаляли в вакууме, остаток поместили на ледяную баню, и к нему добавили большое количество эфира, и немедленно белое твердое вещество выпало в осадок. Смесь отфильтровали, получили неочищенный продукт, выход 86%. Неочищенный продукт очищали посредством ODS колоночной хроматографии, и белый твердый продукт с чистотой 98.5% был получен в результате липофилизации. Тпл=150~151°С.
1H-NMR (300 MHz, DMSO-d6): 2.30 (3Н, s, 1-CH3), 7.44 (2H, d, J=8.1 Hz, 2 and 6-H), 7.21 (2H, d, J=8.1 Hz, 3 and 5-H), 7.37 (1H, d, J=15.9 Hz, 7-H), 6.69 (1H, d, J=15.9 Hz, 8-H), 8.35 (1Н, d, J=6.6 Hz, 10-H), 4.37 (1Н, m, 11-H), 1.25 (3H, m, 12-H), 8.21 (1Н, d, J=8.1 Hz, 14-H), 4.12 (1Н, m, 15-H), 6.99 (1Н, s, 17-Ha), 7.32(1H, s, 17-Hb), 1.73 (1H, m, 18-Ha), 1.97 (1H, m, 18-Hb), 2.16 (2H, m, 19-H), 7.90 (1H, d, J=7.8 Hz, 21-H), 4.10 (1H, m, 22-H), 7.11 (1Н, s, 24-Ha), 7.34 (1H, s, 24-Hb), 1.49 (1H, m, 25-Ha), 1.63 (1H, m, 25-Hb), 1.28 (2H, m, 26-H), 1.51 (2H, m, 27-H), 2.74 (2H, m, 28-H), 7.80 (2H, br.s, 29-H).
13C-NMR (125 MHz, DMSO-d6): 20.9 (1-CH3), 139.0 (2 и 6-C), 129.6 (2 и 6-C), 127.5 (3 и 5-C), 132.1 (4-С), 139.3 (7-C), 120.8 (8-C), 165.2 (9-C), 48.9 (11-C), 18.0 (12-C), 172.5 (13-С), 52.2 (15-C), 173.9 (16-C), 27.6 (18-C), 31.8 (19-C), 171.7 (20-C), 52.1 (22-С), 173.4 (23-C), 31.3 (25-C), 22.4 (26-C), 26.7 (27-C), 38.7 (28-C).
IR: 3278.8, 3199.9 (νOH и νNH), 3063.3 (ν=CH), 2941.3 (ν-CH), 1656.3 (νC=O), 1540.7, 1452.5 (νC=C), 1202.2, 1184.1, 1135.3 (δ-CH), 984.0, 835.8, 813.6, 800.7, 721.6 (δ=CH).
ESI-MS: 489.48 [M+H]+, 977.29 [2M+H]+.
HR-MS (TOF): 489.2819 [M+H]+, C24H36N6O5.
Пример 13: Твердофазный синтез мурамилди пептида MDA-203
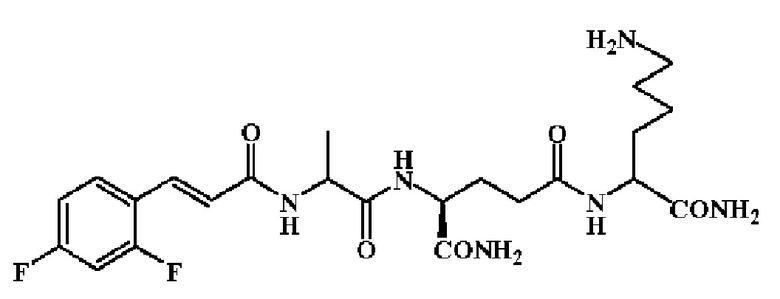
Была задействована схема твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 2.4-дифторкоричная кислота вводились в смолу последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель удаляли в вакууме, остаток помещали на ледяную баню. При добавлении к нему большого количества эфира белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, получен неочищенный продукт с выходом 80%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и белый твердый продукте чистотой 98.5% был получен в результате липофилизации. Тпл=189~190°С.
1H-NMR (300 MHz, DMSO-d6): 7.35 (1Н, m, 2-Н), 7.72 (1Н, dd, J=15.2 и 8.7 Hz, 5-Н), 7.18 (1Н, td, J=8.4 and 2.4 Hz, 6-H), 7.44 (1H, d, J=15.9 Hz, 7-H), 6.82 (1H, d, J=15.9 Hz, 8-H), 8.51 (1H, d, J=6.6 Hz, 10-H), 4.40 (1Н, m, 11-H), 1.27 (3H, d, J=7.2 Hz, 12-H), 8.24 (1H, d, J=8.1 Hz, 14-H), 4.17 (1H, m, 15-H), 7.00 (1H, s, 17-Ha), 7.33 (1H, s, 17-Hb), 1.71 (1Н, m, 18-Ha), 1.97 (1H, m, 18-Hb), 2.17 (2H, t, J=7.8 Hz, 19-H), 7.91 (1Н, d, J=8.4 Hz, 21-H), 4.13 (1H, m, 22-H), 7.07 (1H, s, 24-Ha), 7.32 (1H, s, 24-Hb), 1.49 (1H, m, 25-Ha), 1.64 (1H, m, 25-Hb), 1.29 (2H, m, 26-H), 1.50 (2H, m, 27-H), 2.75 (2H, m, 28-H).
13C-NMR (125 MHz, DMSO-d6): 163.7 (m, 1-C), 104.7 (t, J=26.0 Hz, 2-C), 159.6 (m, 3-C), 118.5 (m, 4-C), 130.6 (m, 5-C), 112.4 (d, J=18.4 Hz, 6-C), 137.4 (s, 7-C), 124.3 (s, 8-C), 164.7 (s, 9-C), 48.9 (11-C), 18.0 (12-C), 172.2 (13-C), 52.1 (15-С), 173.2 (16-C), 27.6 (18-C), 31.7 (19-C), 171.6 (20-C), 52.0 (22-C), 172.3 (23-C), 31.3 (25-C), 22.4 (26-C), 26.8 (27-C), 38.7 (28-C).
IR: 3279.8, 3198.2 (νOH и νNH), 3066.7 (ν=CH), 2939.5 (ν-CH), 1656.2 (νC=O), 1616.4, 1544.6, 1504.2, 1454.1 (νC=C), 1202.1, 1181.7, 1138.8 (νC-F and δ-СН), 967.5, 836.7, 800.7, 721.4 (νC-Cl and δ=CH).
ESI-MS: 511.28 [M+H]+, 1021.02 [2M+H]+.
HR-MS(TOF): 511.2482 [M+H]+, C24H36N6O5.
Пример 14: Твердофазный синтез мурамилдипептида MDA-204

Была задействована схема твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 4-хлор-2-фторкоричная кислота вводились в смолу последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель удаляли в вакууме, к остатку на ледяной бане добавили большое количество эфира, белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, получен неочищенный продукт с выходом 88%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и белый твердый продукт с чистотой 98.5% был получен в результате липофилизации. Тпл=149~150°С.
1H-NMR (300 MHz, DMSO-d6): 7.54 (1Н, dd, J=10.8 and 1.8 Hz, 2-H), 7.69 (1Н, t, J=8.7 Hz, 5-H), 7.36 (1Н, dd, J=10.5 и 2.1 Hz, 6-H), 7.44 (1Н, d, J=15.9 Hz, 7-H), 6.87 (1H, d, J=15.9 Hz, 8-H), 8.57 (1Н, d, J=6.6 Hz, 10-H), 4.40 (1Н, m, 11-H), 1.27 (3H, d, J=7.2 Hz, 12-H), 8.27 (1H, d, J=8.1 Hz, 14-H), 4.13 (1H, m, 15-H), 6.99 (1H, s, 17-Ha), 7.35(1H, s, 17-Hb), 1.72 (1Н, m, 18-Ha), 1.98 (1H, m, 18-Hb), 2.17 (2H, t, J=7.8 Hz, 19-H), 8.08 (1Н, d, J=8.1 Hz, 21-H), 4.10 (1H, m, 22-H), 7.12 (1H, s, 24-Ha), 7.32 (1Н, s, 24-Hb), 1.49 (1H, m, 25-Ha), 1.64 (1H, m, 25-Hb), 1.29 (2H, m, 26-H), 1.51 (2H, m, 27-H), 2.74 (2H, m, 28-H).
13C-NMR (125 MHz, DMSO-d6): 135.1 (d, J=10.9 Hz, 1-C), 117.2 (d, J=25.8 Hz, 2-C), 160.7 (d, J=252.5 Hz, 3-C), 122.1 (d, J=11.6 Hz, 4-C), 130.8 (s, 5-C), 125.9 (d, J=3.0 Hz, 6-C), 137.3 (m, 7-C), 125.8 (d, J=6.3 Hz, 8-C), 164.6 (s, 9-C), 49.4 (11-C), 18.5 (12-C), 172.8 (13-С), 52.7 (15-C), 174.3 (16-C), 28.1 (18-С), 32.2 (19-C), 172.1 (20-С), 52.6 (22-C), 173.8 (23-C), 31.8 (25-C), 22.9 (26-C), 27.5 (27-C), 38.7 (28-C).
IR: 3358.7, 3284.3, 3199.3 (νOH и νNH), 3067.3 (ν=CH), 2933.4 (ν-CH), 1654.7, 1642.5, 1642.5, 1622.9 (ν-C=O), 1540.6, 1489.9, 1453.6 (νC=C), 1202.4, 1129.9 (νC-F и δ-CH), 978.2, 815.0, 720.6, 690.2 (νC-Cl и δ=СН).
ESI-MS: 527.49 [M+H]+, 1053.17 [2M+H]+.
HR-MS(TOF): 527.2192 [М+Н]+, C23H32ClFN6O5.
Пример 15: Твердофазный синтез мурамил дипептида MDA-205
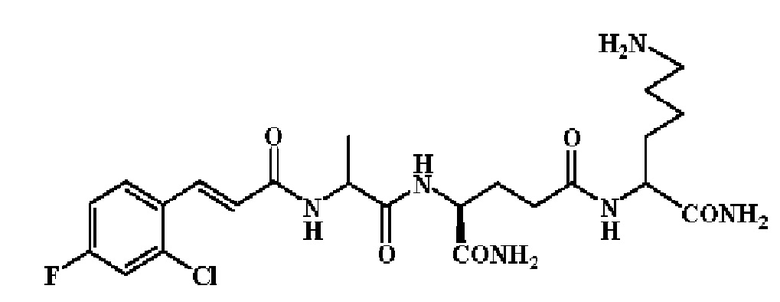
Была задействована схема твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 2-хлор-4-фторкоричная кислота вводились в смолу последовательно. После завершения конденсации смола была достаточно промыта и высушена, смола подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель был удален в вакууме, к остатку на ледяной бане было добавлено большое количество эфира, белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, получен неочищенный продукт с выходом 86%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и белый твердый продукт с чистотой 98.5% был получен в результате липофилизации. Тпл=137~138°С.
1H-NMR (300 MHz, DMSO-d6): 7.55 (1Н, dd, J=8.7 and 1.8 Hz, 2-H), 7.77 (1H, m, 5-H), 7.36 (1H, m, 6-H), 7.66 (1H, d, J=15.9 Hz, 7-H), 6.79 (1H, d, J=15.9 Hz, 8-H), 8.47 (1Н, d, J=6.6 Hz, 10-H), 4.42 (1Н, m, 11-H), 1.27 (3H, d, J=6.9 Hz, 12-H), 8.24 (1H, d, J=8.4 Hz, 14-H), 4.16 (1H, m, 15-H), 7.00 (1H, s, 17-Ha), 7.31 (1H, s, 17-Hb), 1.72 (1H, m, 18-Ha), 1.99 (1Н, m, 18-Hb), 2.17 (2H, t, J=7.8 Hz, 19-H), 7.91 (1H, d, J=8.7 Hz, 21-H), 4.13 (1H, m, 22-H), 7.12 (1H, s, 24-Ha), 7.33 (1H, s, 24-Hb), 1.49 (1Н, m, 25-Ha), 1.65 (1H, m, 25-Hb), 1.30 (2H, m, 26-H), 1.52 (2H, m, 27-H), 2.75 (2H, br.s, 28-H), 7.79(2H, br.s, 29-H).
13C-NMR (125 MHz, DMSO-d6): 162.7(d, J=250.0 Hz, 1-C), 115.9 (d, J=21.6 Hz, 2-C), 134.6 (d, J=10.0 Hz, 3-C), 129.9 (d, J=3.8 Hz, 4-C), 129.7 (d, J=10.0 Hz, 5-C), 117.7 (d, J=25.1 Hz, 3-C), 137.5 (7-C), 125.4 (8-C), 164.8 (9-C), 49.3 (11-С), 18.6 (12-C), 172.1 (13-С), 52.6 (15-C), 174.2 (16-C), 28.2 (18-C), 32.2 (19-C), 172.1 (20-C), 52.5 (22-C), 173.7 (23-C), 31.8 (25-C), 22.9 (26-C), 27.2 (27-C), 38.2 (28-C).
IR: 3279.8 (ν0H и νNH), 3066.0 (ν=CH), 2937.1 (ν-CH), 1776.1, 1656.3 (νC=O), 1537.0, 1489.0, 1452.2 (νC=C), 1238.1, 1201.1, 1181.0, 1135.6 (νC-F и δ-CH), 910.6, 835.5, 800.1, 721.3 (νC-Cl и δ=CH).
ESI-MS: 527.28 [M+H]+, 1075.00 [2M+Na]+.
HR-MS(TOF): 527.2201 [M+H]+, C23H32ClFN6O5.
Пример 16: Твердофазный синтез мурамил дипептида MDA-206
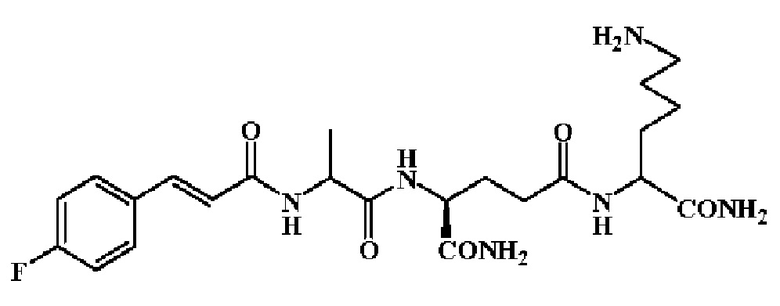
Была задействована схема твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 4-фторкоричная кислота вводились последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. К растворителю на ледяной бане было добавлено большое количество эфира, белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, получен неочищенный продукт, выход 92%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и белый твердый продукт с чистотой 98.5% был получен в результате липофилизации. Тпл=218~220°С.
1H-NMR (300 MHz, DMSO-d6): 7.26 (2Н, t, J=8.7 Hz, 2 and 6-H), 7.63 (2H, dd, J=8.4 and 5.7 Hz, 3 and 5-H), 7.42 (1H, d, J=15.9 Hz, 7-H), 6.71 (1H, d, J=15.9 Hz, 8-H), 8.37 (1H, d, J=6.6 Hz, 10-H), 4.40 (1H, m, 11-H), 1.27 (3H, d, J=7.2 Hz, 12-H), 8.21 (1H, d, J=8.1 Hz, 14-H), 4.15 (1H, m, 15-H), 7.00 (1Н, s, 17-Ha), 7.32 (1H, s, 17-Hb), 1.71 (1H, m, 18-Ha), 1.99 (1Н, m, 18-Hb), 2.17 (2H, t, J=7.8 Hz, 19-H), 7.90 (1H, d, J=8.1 Hz, 21-H), 4.14 (1H, m, 22-H), 7.12 (1H, s, 24-Ha), 7.32 (1H, s, 24-Hb), 1.49 (1H, m, 25-Ha), 1.64 (1H, m, 25-Hb), 1.29 (2H, m, 26-H), 1.52 (2H, m, 27-H), 2.76 (2H, m, 28-H), 7.71 (2H, br.s, 29-H).
13C-NMR (125 MHz,DMSO-d6): 163.2 (d, J=245.8 Hz, 1-C), 116.4 (d, J=21.6 Hz, 2 and 6-C), 130.1 (d, J=8.5 Hz, 3 and 5-C), 131.9 (4-C), 138.3 (7-C), 122.2 (8-C), 165.3 (9-C), 49.3 (11-C), 18.5 (12-C), 172.8 (13-C), 52.6 (15-C), 174.2 (16-С), 27.2 (18-C), 32.2 (19-C), 172.1 (20-С), 52.5 (22-C), 173.7 (23-C), 31.8 (25-C), 22.9 (26-C), 27.2 (27-C), 38.5 (28-C).
IR: 3278.5, 3198.1 (νOH и νNH), 3068.1 (ν=CH), 2931.9 (ν-CH), 1672.8, 1639.9 (νC=O), 1614.9, 1539.4, 1509.6, 1451.7 (νC=C), 1201.7, 1134.3 (νC-F и δ-CH), 971.4, 831.4, 800.6, 721.0 (δ=CH).
ESI-MS: 493.25 [M+H]+, 1007.02 [2M+Na]+.
HR-MS(TOF): 493.2580 [M+H]+, 515.2381 [M+Na]+, C23H33FN6O5.
Пример 17: Твердофазный синтез мурамил дипептида MDA-207

Был задействован метод твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 4-фторкоричная кислота вводились последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель удаляли в вакууме, к остатку на ледяной бане было добавлено большое количество эфира, белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, получен неочищенный продукт, выход 75%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и белый твердый продукт с чистотой 98.5% был получен в результате липофилизации. Тпл=195~196°С.
1H-NMR (300 MHz, DMSO-d6): 7.21 (1Н, s, 2-Н), 7.38 (1Н, m, 3-H), 7.41 (1H, m, 5-Н), 7.47 (1Н, m, 6-Н), 7.47 (1Н, d, J=15.9 Hz, 7-H), 6.79 (1Н, d, J=15.9 Hz, 8-H), 8.39 (1Н, d, J=6.0 Hz, 10-H), 4.38 (1H, m, 11-H), 1.26(3H, d, J=6.9Hz, 12-H), 8.22(1H, d, J=7.5Hz, 14-H), 4.13 (1H, m, 15-H), 6.97 (1H, s, 17-Ha), 7.30 (1H, s, 17-Hb), 1.65 (1H, m, 18-Ha), 1.97 (1H, m, 18-Hb), 2.15 (2H, m, 19-H), 7.90 (1H, d, J=8.4 Hz, 21-H), 4.13 (1H, m, 22-H), 7.01 (1Н, s, 24-Ha), 7.30 (1H, s, 24-Hb), 1.48 (1H, m, 25-Ha), 1.65 (1H, m, 25-Hb), 1.28 (2H, m, 26-H), 1.48 (2H, m, 27-H), 2.72 (2H, m, 28-H).
13C-NMR (125 MHz, DMSO-d6): 116.7 (d, J=21.0 Hz, 1-C), 162.9 (d, J=242.3 Hz, 2-C), 114.4 (d, J=21.4 Hz, 3-C), 137.9 (d, J=7.8 Hz, 4-C), 124.0 (d, J=22.6 Hz, 5-C), 131.4 (6-C), 138.1 (7-C), 124.0 (8-C), 165.1 (9-С), 49.3 (11-C), 18.6 (12-C), 172.8 (13-C), 52.6 (15-C), 174.3 (16-C), 28.2 (18-C), 32.2 (19-C), 172.0 (20-C), 52.5 (22-C), 173.7(23-C), 31.8 (25-C), 22.9 (26-C), 27.2 (27-C), 38.5 (28-C).
IR: 3276.4, 3201.1 (νOH и νNH), 3069.1 (ν=CH), 2938.1 (ν-CH), 1647.7 (νc=O), 1539.0, 1448.0, 1421.8 (νC=C), 1200.8, 1180.2, 1134.1 (νC-F и δ-CH), 972.1, 834.9, 798.7, 721.2 (δ=CH).
ESI-MS: 493.25 [M+H]+, 1007.09 [2M+Na]+.
HR-MS(TOF): 493.2582 [M+H]+, C23H33FN6O5.
Пример 18: Твердофазный синтез мурамилдипептида MDA-208

Был задействован метод твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 3.4-ди-фторкоричная кислота вводились в смолу последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель удаляли в вакууме, к остатку на ледяной бане было добавлено большое количество эфира, и белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, и получен неочищенный продукт с выходом 95%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и белый твердый продукт с чистотой 98.5% был получен в результате липофилизации. Тпл=139~140°С.
1H-NMR (300 MHz, DMSO-d6): 7.66 (1Н, m, 3-Н), 7.48 (1Н, m, 5-Н), 7.45 (1Н, m, 6-Н), 7.40 (1Н, d, J=15.9 Hz, 7-H), 6.75 (Н, d, J=15.9 Hz, 8-H), 8.37 (Н, d, J=6.9 Hz, 10-H), 4.40 (1Н, m, 11-H), 1.27 (3H, d, J=7.2 Hz, 12-H), 8.22 (1H, d, J=7.8 Hz, 14-H), 4.16 (1H, m, 15-H), 700 (1H, s, 17-Ha), 7.33 (1H, s, 17-Hb), 1.71 (1Н, m, 18-Ha), 1.97 (1H, m, 18-Hb), 2.17 (2H, t, J=7.8 Hz, 19-H), 7.90 (1Н, d, J=8.1 Hz, 21-H), 4.13 (1H, m, 22-H), 7.12 (1H, s, 24-Ha), 7.31 (1Н, s, 24-Hb), 1.49 (1H, m, 25-Ha), 1.65 (1H, m, 25-Hb), 1.29 (2H, m, 26-H), 1.52 (2H, m, 27-H), 2.76 (2H, m, 28-H), 7.73 (2H, br.s, 29-H).
13C-NMR (150 MHz, DMSO-d6): 149.3 (dd, J=35.6 и 12.8 Hz, 1-C), 151.2 (dd, J=38.5 и 12.9 Hz, 2-C), 118.6 (d, J=17.5 Hz, 3-C), 133.3 (m, 4-C), 125.1 (m, 5-C), 116.7 (d, J=17.4 Hz, 6-C), 137.3 (s, 7-C), 123.8 (s, 8-C), 165.0 (9-C), 49.3 (11-C), 18.6 (12-C), 172.8 (13-C), 52.6 (15-C), 174.3 (16-С), 28.2 (18-C), 31.8 (19-C), 172.1 (20-C), 52.5 (22-C), 173.7 (23-C), 31.8 (25-C), 22.9 (26-C), 27.2 (27-C), 38.2 (28-C).
IR: 3275.8, 3196.4 (νOH и νNH), 3064.8 (ν=CH), 2938.1 (ν-CH), 1673.1 (νC=O), 1612.9, 1542.1, 1516.7, 1451.5 (νC=C), 1201.6, 1135.4 (νC-F и δ-CH), 969.3, 834.3, 800.6, 721.2 (δ=СН).
ESI-MS: 511.30 [M+H]+, 1021.09 [2M+H]+.
HR-MS(TOF): 511.2479 [M+H]+, C23H32F2N6O5.
Пример 19: Твердофазный синтез мурамилдипептида MDA-113

Был задействован метод твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 2-хинолинкарбоновая кислота вводились в смолу последовательно. После завершения конденсации смола была достаточно промыта и высушена, смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель был удален в вакууме, к остатку на ледяной бане было добавлено большое количество эфира, белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, и получен неочищенный продукт, выход 80%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и MDA-113 в виде белого твердого продукта с чистотой 98.5% был получен в результате липофилизации.
Пример 20: Твердофазный синтез мурамилдипептида MDA-119

Был задействован метод твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 2-тиенилакриловая кислота вводились последовательно. После завершения конденсации смола была достаточно промыта и высушена, и смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель был удален в вакууме, к остатку на ледяной бане было добавлено большое количество эфира, белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, и получен неочищенный продукт с выходом 83%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и MDA-119 в виде белого твердого продукта с чистотой 98.5% был получен в результате липофилизации.
Пример 21: Твердофазный синтез мурамилдипептида MDA-130

Был задействован метод твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 2-тиенилакриловая кислота вводились последовательно. После завершения конденсации смола была достаточно промыта и высушена, и смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель был удален в вакууме, к остатку на ледяной бане было добавлено большое количество эфира, и белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, и получен неочищенный продукт с выходом 81%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и MDA-130 в виде белого твердого продукта с чистотой 98.5% был получен в результате липофилизации.
Пример 22: Твердофазный синтез мурамилди пептида MDA-133

Был задействован метод твердофазного синтеза. Смола Rink-Amide AM (емкость смолы 0.88 ммоль/г) была выбрана, Fmoc-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-Ala-СООН и 2-нафтоксиуксусная кислота вводились последовательно. После завершения конденсации смола была достаточно промыта и высушена, и смолу подвергали расщеплению в течение 1 часа в 90% (объемный процент) водном растворе TFA. Растворитель был удален в вакууме, к остатку на ледяной бане было добавлено большое количество эфира, и белое твердое вещество выпало в осадок немедленно. Смесь была отфильтрована, и получен неочищенный продукт с выходом 88%. Неочищенный продукт был очищен посредством ODS колоночной хроматографии, и MDA-133 в виде белого твердого продукта с чистотой 98.5% был получен в результате липофилизации.
Пример 23-35: Жидкофазный синтез МТС конъюгатов
Пример 23: Жидкофазный синтез конъюгата МТС-220
Схема синтеза показана ниже:

Реагенты и условия: (a) HOSu, EDC⋅HCl, DMSO, к.т., 20 ч.; (b) MDA, DMSO, к.т., 12 ч.
9.53 г. (1.0 экв) сложного моноэфира паклитаксел-2'-O- янтарной кислоты, 1,15 г. (1,0 экв) HOSu и 1,92 г. (1.0 экв) EDC•HCl растворили в DMSO и перемешивали при к.т.в течение 20 часов. 5,08 г. (1,0 экв) аналога мурамилдипептида MDA аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивать в течение 20 часов. После завершения реакции большое количество воды было добавлено в смесь, и белый твердый продукт выпал в осадок. Технический продукт был очищен с помощью ODS колоночной хроматографии, 11,8 г. твердого продукта было получено в результате липофилизации. Выход 82%, Тпл=180~181°С, [α]=-9.8° (С=10.1 мг/мл,0МЕ).
1H-NMR (600 MHz, DMSO-d6): 4.63 (1Н, br.s, 1-ОН), 5.42(1Н, d, J=7.2 Hz, 2-Н), 3.58 (1Н, d, J=7.2 Hz, 3-H), 4.90 (1Н, m, 5-H), 1.62 (1H, m, 6-Ha), 2.30 (1H, m, 6-Hb),4.12 (1H, m, 7-H), 4.91 (1H, m, 7-OH), 6.30 (1Н, s, 10-H), 5.82 (1H, t, J=9.0 Hz, 13-H), 1.46 (1H, m, 14-Ha), 1.79 (1Н, m, 14-Hb), 1.00 (3H, s, 16-H), 1.03 (3H, s, 17-H), 1.77 (3H, s, 18-H), 1.50 (3H, s, 19-H), 3.99 (1Н, d, J=9.0 Hz, 20-Ha), 4.02 (1H, d, J=9.0 Hz, 20-Hb), 2.24 (3H, s, 4-OCOCH3), 2.11 (3Н, s, 10-ОСОСН3), 5.34 (1Н, d, J=9.0 Hz, 2'-H), 5.54 (1H, t, J=9.0 Hz, 3'-H), 9.21 (1H, d, J=9.0 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.46 (2H, m, ph-m-H), 7.55 (1H, t, J=7.2 Hz, ph-p-H), 7.83 (2H, m, NBz-o-H), 7.44 (2H, m, NBz-m-H), 7.19 (1H, m, NBz-p-H), 7.98 (2H, d, J=7.2 Hz, OBz-o-H), 7.66 (2H, t, J=7.2 Hz, OBz-m-H), 7.74 (1H, t, J=7.2 Hz, OBz-p-H), 2.61 (2H, m, 22-H), 2.36 (2H, t, J=7.2 Hz, 23-H), 7.82 (1Н, m, 25-H), 2.90(1H, m, 26-Ha), 3.00 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.32 (2H, m, 28-H), 1.45 (1Н, m, 29-Ha), 1.63 (1H, m, 29-Hb), 4.11 (1Н, m, 30-H), 6.96 (1H, s, 32-Ha), 7.30 (1H, s, 32-Hb), 7.87 (1Н, m, 33-H), 2.16 (2H, t, J=7.2 Hz, 35-H), 1.71 (1Н, m, 36-Ha), 1.99 (1H, m, 36-Hb), 4.13 (1H, m, 37-H), 7.10 (1Н, s, 39-Ha), 7.30 (1H, s, 39-Hb), 8.21 (1H, d, J=8.4 Hz, 40-H), 4.40(1H, t, J=7.2 Hz, 42-H), 1.28 (3H, d, J=6.6 Hz, 43-Н), 8.37 (1Н, d, J=7.2 Hz, 44-H), 6.76 (1H, d, J=15.6 Hz, 46-H), 7.41 (1H, d, J=15.6 Hz, 47-H), 7.58 (2H, d, J=9.0 Hz, 49 and 53-H), 7.49 (2H, d, J=9.0Hz, 50 and 52-H).
13C-NMR (150 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-C), 133.3 (11-C), 139.2 (12-C), 70.7 (13-C), 34.7 (14-С), 42.9 (15-C), 26.3 (16-C), 21.4 (17-C), 13.9 (18-C), 9.8 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.6, 22.5 (4-OCOCH3), 168.8, 20.6 (10-OCOCH3), 169.1 (1'-C), 74.4 (2-C), 54.0 (3'-C), 166.4 (3-NHCO), 137.3 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.4 (NBz-o-C), 129.0 (NBz-m-C), 128.2 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.5 (27-C), 22.9 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.Ц37-С), 173.3 (38-C), 172.3 (41-C), 48.8 (42-C), 18.Ц43-С), 164.7 (45-C), 122.7 (46-C), 137.6 (47-C), 133.8 (48-C), 129.0 (49 and 53-C), 129.2 (50 and 52-C), 133.9 (51-C).
IR: 3316.9 (νOH и νNH), 3066.0 (ν=CH), 2935.0, 2873.1 (ν-CH), 1736.0, 1655.0 (νC=O), 1537.3, 1492.9 (νC=C), 1451.7, 1371.8 (δ=CH), 1241.5 (νC-O-C), 980.2, 906.6,822.6, 776.2, 708.9 (δ=CH).
ESI-MS: 1444.56 [M+H]+, 1466.46 [M+Na]+.
HR-MS (TOF): 1444.5645 [M+H]+, 1466.5475 [M+Na]+, C74H86ClN7O21.
Пример 24: Жидкофазный синтез конъюгата МТС-301
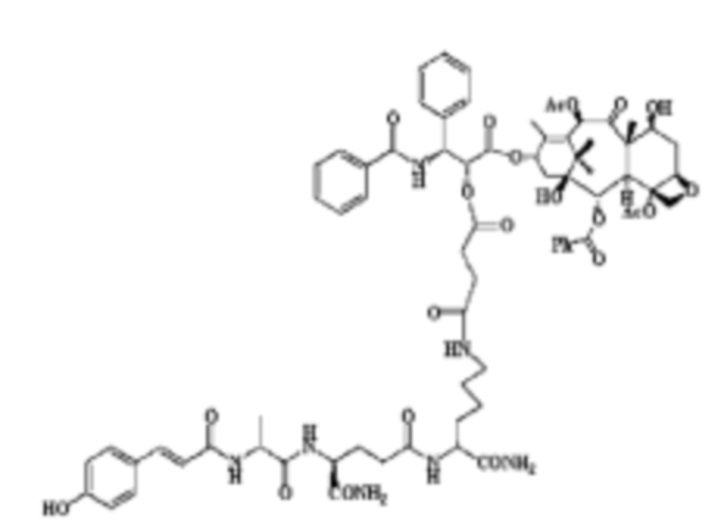
953 мг (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг (1,0 экв) HOSu и 192 мг (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 490 мг (1,0 экв) аналога мурамил дипептида MDA-201 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды было добавлено в смесь, и белый твердый продукт выпал в осадок. Технический продукт был очищен с помощью ODS колоночной хроматографии, 1,18 г твердого продукта было получено в результате липофилизации. Выход 83%, Тпл=179~180°С.
1H-NMRI (500 MHz, DMSO-d6): 4.62 (1Н, br.s, 1-OH), 5.40 (1H, d, J=7.0 Hz, 2-H), 3.56 (1H, d, J=7.0 Hz, 3-H), 4.89 (1Н, m, 5-H), 1.62 (1H, m, 6-Ha), 2.31 (1H, m, 6-Hb),4.12 (1H, m, 7-H), 4.92 (1H, m, 7-OH), 6.28 (1H, s, 10-H), 5.81 (1H, t, J=7.5 Hz, 13-H), 1.46 (1H, m, 14-Ha), 1.75 (1Н, m, 14-Hb), 1.01 (3H, s, 16-H), 1.04 (3H, s, 17-H), 1.78 (3H, s, 18-H), 1.48 (3H, s, 19-H), 3.99 (1H, d, J=8.5 Hz, 20-Ha), 4.00 (1H, d, J=8.5Hz, 20-Hb), 2.23 (3H, s, 4-ОСОСН3), 2.10 (3H, s, 10-ОСОСН3), 5.33 (1H, d, J=9.0 Hz, 2'-H), 5.52 (1H, t, J=9.0 Hz, 3'-H), 9.21 (1H, d, J=8.5 Hz, 3'-NH), 7.48 (2H, d, J=7.5 Hz, ph-o-H), 7.47 (2H, d, J=7.5Hz, ph-m-H), 7.55 (1Н, t, J=7.5 Hz, ph-p-H), 7.83 (2H, m, NBz-o-H), 7.43 (2H, m, NBz-m-H), 7.17 (1Н, m, NBz-p-H), 7.98 (2H, d, J=7.5 Hz, OBz-o-H), 7.65 (2H, t, J=8.0 Hz, OBz-m-H), 7.74 (1H, t, J=7.5 Hz, OBz-p-H), 2.72 (2H, m, 22-H), 2.35 (2H, t, J=7.0 Hz, 23-H), 7.82 (1H, m, 25-H), 2.96 (1Н, m, 26-Ha), 3.00 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.32 (2H, m, 28-H), 1.45 (1H, m, 29-Ha), 1.62 (1Н, m, 29-Hb), 4.10 (1H, m, 30-H), 6.96 (1H, s, 32-Ha), 7.30 (1H, m, 32-Hb), 7.86 (1H, m, 33-H), 2.14 (2H, t, J=8.0 Hz, 35-H), 1.75 (1H, m, 36-Ha), 1.99 (1H, m, 36-Hb), 4.11 (1Н, m, 37-H), 7.10 (1H, s, 39-Ha), 7.30 (1H, m, 39-Hb), 8.19 (1H, d, J=8.0 Hz, 40-H), 4.36 (1H, m, 42-H), 1.25 (3H, d, J=7.0 Hz, 43-H), 8.22 (1Н, d, J=6.5 Hz, 44-H), 6.51 (1H, d, J=15.5 Hz, 46-H), 7.32 (1H, d, J=15.5 Hz, 47-H), 7.46 (2H, d, J=8.5 Hz, 49 and 53-H), 6.78 (2H, d, J=8.5 Hz, 50 and 52-H), 9.85 (1H, s, 51-OH).
13C-NMR (125 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-С), 80.3 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-C), 133.3 (11-C), 139.2 (12-C), 70.4 (13-C), 34.7 (14-C), 42.9 (15-C), 26.3 (16-C), 21.4 (17-С), 13.9 (18-C), 9.8 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.6, 22.6 (4-OCOCH3), 168.8, 20.7 (10-ОСОСН3), 169.2 (1'-С), 74.4 (2'-C), 54.0 (3'-C), 166.4 (3'-NHCO), 137.4 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.5 (NBz-o-C), 129.0 (NBz-m-C), 128.2 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.5 (27-C), 22.9 (28-C), 31.6 (29-C), 52.4 (30-C), 173.9 (31-C), 171.6 (34-C), 31.8 (35-C), 27.7 (36-C), 52.1 (37-С), 173.3 (38-C), 172.3 (41-C), 48.8 (42-C), 18.0 (43-C), 164.7 (45-C), 118.2 (46-C), 137.4 (47-C), 125.8 (48-C), 127.5 (49 и 53-C), 115.8 (50 и 52-C), 158.9 (51-C).
IR: 3324.4 (νOH и νNH), 3075.1 (ν=CH), 1740.6, 1657.2 (νC=O), 1603.9, 1518.3, 1450.8 (νC=C), 1243.4 (νC-O-C), 980.6, 710.3 (δ=CH).
ESI-MS: 1426.31 [M+H]+, 1449.03 [M+Na+H]2+.
HR-MS (TOF): 1426.5974 [M+H]+, 1448.5786 [M+Na]+, C74H87N7O22.
Пример 25: Жидкофазный синтез конъюгата МТС-302

953 мг (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг (1,0 экв) HOSu и 192 мг (1.0 eq) EDC⋅HCI растворили в DMSO и перемешивали при к.т. в течение 4 часов. 488 мг (1,0 экв) аналога мурамилдипептида MDA-202 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды было добавлено в смесь, и белый твердый продукт выпал в осадок. Технический продукт был очищен с помощью ODS колоночной хроматографии, 1,09 г твердого продукта было получено в результате липофилизации. Выход 77%, Тпл=172~174°С.
1H-NMR (500 MHz, DMSO-d6): 4.63 (1Н, br.s, 1-ОН), 5.40 (1Н, d, J=7.0 Hz, 2-Н), 3.56 (1Н, d, J=7.0 Hz, 3-H), 4.89 (1Н, m, 5-H), 1.62 (1Н, m, 6-Ha), 2.31 (1H, m, 6-Hb),4.12 (1Н, m, 7-H), 4.91 (1H, m, 7-OH), 6.28 (1H, s, 10-H), 5.81 (1H, t, J=9.5 Hz, 13-H), 1.46 (1H, m, 14-Ha), 1.79 (1H, m, 14-Hb), 0.98 (3H, s, 16-H), 1.01 (3Н, s, 17-H), 1.75 (3H, s, 18-H), 1.48 (3H, s, 19-H), 3.99 (1H, d, J=8.0 Hz, 20-Ha), 4.01 (1H, d, J=8.0Hz, 20-Hb), 2.23 (3H, s, 4-OCOCH3), 2.09 (3H, s, 10-OCOCH3), 5.34 (1H, d, J=9.0 Hz, 2'-Н), 5.52 (1Н, t, J=9.0 Hz, 3'-H), 9.21 (1H, d, J=8.5 Hz, 3'-NH), 7.49 (2H, m, ph-o-H), 7.48 (2H, m, ph-m-H), 7.55 (1H, d, J=7.5 Hz, ph-p-H), 7.85 (2H, m, NBz-o-H), 7.46 (2H, m, NBz-m-H), 7.18 (1H, m, NBz-p-H), 7.97 (2H, d, J=8.0 Hz, OBz-o-H), 7.65 (2H, d, J=7.5 Hz, OBz-m-H), 7.72 (1H, d, J=7.0 Hz, OBz-p-H), 2.60 (2H, m, 22-H), 2.36 (2H, m, 23-H), 7.84 (1Н, m, 25-H), 2.91 (1H, m, 26-Ha), 2.96 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.32 (2H, m, 28-H), 1.44 (1Н, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.11 (1H, m, 30-H), 6.96 (1H, s, 32-Ha), 7.30 (1H, m, 32-Hb), 7.86 (1H, m, 33-H), 2.16 (2H, m, 35-H), 1.75 (1H, m, 36-Ha), 1.99 (1H, m, 36-Hb), 4.12 (1H, m, 37-H), 7.10 (1H, s, 39-Ha), 7.22 (1H, m, 39-Hb), 8.21 (1H, d, J=8.0 Hz, 40-H), 4.37 (1Н, m, 42-H), 1.28 (3H, d, J=7.0 Hz, 43-H), 8.31 (1H, d, J=6.5 Hz, 44-H), 6.68 (1H, d, J=15.5 Hz, 46-H), 7.43 (1H, d, J=16.0 Hz, 47-H), 7.57 (1H, m, 49 and 53-H), 7.49 (1H, m, 50 and 52-H), 2.31 (3H, m, 51-CH3).
13C-NMR (125 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-C), 133.3 (11-C), 139.4 (12-С), 70.7 (13-C), 34.7 (14-C), 42.9 (15-C), 26.3 (16-C), 21.4 (17-С), 13.9 (18-C), 9.8 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.7, 22.6 (4-ОСОСН3), 168.8, 20.7 (10-ОСОСН3), 169.1 (1'-C), 74.6 (2'-С), 54.0 (3'-С), 166.4 (3'-NHCO), 137.4 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.5 (NBz-o-C), 129.0 (NBz-m-C), 128.3 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.7 (27-C), 23.0 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.1 (37-С), 173.3 (38-C), 172.4 (41-C), 48.8 (42-C), 18.1 (43-С), 165.1 (45-С), 120.8 (46-C), 137.4 (47-C), 132.1 (48-C), 129.6 (49 and 53-C), 128.7 (50 and 52-C), 138.9 (51-C), 20.9 (51-CH3).
IR: 3324.5 (νOH и νNH), 3066.3 (ν=CH), 2938.3 (ν-CH), 1740.3, 1724.1, 1657.2 (νC=O), 1603.9, 1535.1, 1451.8 (νC=C), 1242.8 (νC-O-C), 981.3, 709.7 (δ=CH).
ESI-MS: 1424.33 [M+H]+, 1446.55 [M+Na]+.
HR-MS (TOF): 1424.6184 [M+H]+, 1446.5996 [M+Na]+, C75H89N7O21.
Пример 26: Жидкофазный синтез конъюгата МТС-303

953 мг (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг (1,0 экв) HOSu и 192 мг (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т. в течение 4 часов. 510 мг (1,0 экв) аналога мурамилдипептида MDA-203 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Технический продукт был очищен с помощью ODS колоночной хроматографии, 1.29 г. твердого продукта было получено в результате липофилизации. Выход 89%, Тпл=178~180°С.
1H-NMR (500 MHz, DMSO-d6): 4.62 (1Н, br.s, 1-ОН), 5.40 (1Н, d, J=7.0 Hz, 2-Н), 3.56 (1Н, d, J=7.0 Hz, 3-Н), 4.91 (1Н, m, 5-Н), 1.62 (1Н, m, 6-На), 2.31 (1Н, m, 6-Hb),4.13 (1Н, m, 7-Н), 4.92 (1Н, m, 7-ОН), 6.28 (1Н, s, 10-Н), 5.80 (1Н, t, J=7.5 Hz, 13-Н), 1.45 (1Н, m, 14-На), 1.77 (1Н, m, 14-Hb), 0.98 (3Н, s, 16-Н), 1.01 (3Н, s, 17-Н), 1.75 (3Н, s, 18-Н), 1.48 (3Н, s, 19-Н), 3.98 (1Н, d, J=8.0 Hz, 20-На), 4.00 (1Н, d, J=8.0 Hz, 20-Hb), 2.23 (3Н, s, 4-ОСОСН3), 2.10 (3Н, s, 10-ОСОСН3), 5.33 (1Н, d, J=9.0 Hz, 2'-H), 5.52 (1H, t, J=9.0 Hz, 3'-Н), 9.21 (1Н, d, J=8.5 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.46 (2H, m, ph-m-H), 7.55 (1H, t, J=7.5 Hz, ph-p-H), 7.82 (2H, m, NBz-o-H), 7.44 (2H, m, NBz-m-H), 7.18 (1Н, m, NBz-p-H), 7.97 (2H, d, J=7.5 Hz, OBz-o-H), 7.67 (2H, m, OBz-m-H), 7.72 (1Н, d, J=8.0 Hz, OBz-p-H), 2.60 (2H, m, 22-H), 2.36 (2H, m, 23-H), 7.82 (1H, m, 25-H), 2.90 (1H, m, 26-Ha), 2.96 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.32 (2H, m, 28-H), 1.45 (1H, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.11 (1Н, m, 30-H), 7.06 (1Н, s, 32-Ha), 7.29 (1H, m, 32-Hb), 7.87 (1H, m, 33-H), 2.14 (2H, m, 35-H), 1.75 (1H, m, 36-Ha), 2.06 (1H, m, 36-Hb), 4.13 (1H, m, 37-H), 7.11 (1H, s, 39-Ha), 7.29 (1H, m, 39-Hb), 8.23 (1H, d, J=8.5 Hz, 40-H), 4.40 (1H, m, 42-H), 1.27 (3H, m, 43-H), 8.47 (1H, d, J=6.5 Hz, 44-H), 6.89 (1H, d, J=17.0 Hz, 46-H), 7.41 (1H, d, J=16.0 Hz, 47-H), 7.34 (1H, td, J=11.5 and 2.0 Hz, 50-H), 7.17 (1H, m, 52-H),7.74 (1H, m, 53-H).
13C-NMR (125 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-0, 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-C), 133.3 (11-C), 139.6 (12-C), 70.7 (13-C), 34.7 (14-0, 42.9 (15-C), 26.3 (16-C), 21.4 (17-C), 13.9 (18-C), 9.8 (19-0, 75.3 (20-0, 165.2 (2-OCO), 169.7, 22.6 (4-OCOCH3), 168.8, 20.7 (10-OCOCH3), 169.1 (1-0, 74.6 (2-C), 54.0 (3'-O, 166.4 (3'-NHCO), 137.4 (ph-q-0, 127.7 (ph-o-C), 128.3 (ph-m-0, 131.5 (ph-p-0, 129.9 (NBz-q-0, 127.5 (NBz-o-C), 129.0 (NBz-m-O, 128.2 (NBz-p-C), 134.3 (OBz-q-0, 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-0, 172.0 (21-0, 28.8 (22-C), 29.5 (23-0, 170.0 (24-0, 38.5 (26-0, 28.7 (27-0, 23.0 (28-0, 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-0, 31.7 (35-0, 27.7 (36-0, 52.1 (37-0,173.3 (38-0, 172.3 (41-C), 48.9 (42-0, 18.1 (43-0, 164.6 (45-0, 124.4 (s, 46-0, 137.4 (s, 47-0, 118.5 (m, 48-0, 161.7 (m, 49-C), 104.6 (t, J=26.1Hz, 50-C), 163.7 (m, 51-0, 112.4 (d, J=19.9Hz, 52-0, 130.5 (m, 53-C).
IR: 3309.5 (νOH и νNH), 3067.0 (ν=CH), 2945.0 (ν-CH), 1722.0, 1653.8 (νC=O), 1531.1, 1451.5 (νC=C), 1239.9 (νC-O-C), 977.1, 708.3 (δ=CH).
ESI-MS: 1446.03 [M+H]+, 1468.26 [M+Na]+.
HR-MS (TOF): 1446.5877 [M+H]+, 1468.5646 [M+Na]+, C74H85F2N7O21
Пример 27: Жидкофазный синтез конъюгата МТС-304

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т. в течение 4 часов. 526 мг. (1,0 экв) аналога мурамилдипептида MDA-204 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.26 г. твердого продукта было получено в результате липофилизации. Выход 86%, Тпл=179~180°С.
1H-NMR (500 MHz, DMSO-d6): 4.63 (1Н, br.s, 1-ОН), 5.40 (1Н, d, J=7.5 Hz, 2-Н), 3.56 (1Н, d, J=7.0 Hz, 3-H), 4.91 (1Н, m, 5-H), 1.62 (1H, m, 6-Ha), 2.31 (1Н, m, 6-Hb),4.12 (1H, m, 7-H), 4.91 (1Н, m, 7-OH), 6.28 (1H, s, 10-H), 5.80 (1H, t, J=9.0 Hz, 13-H), 1.45 (1H, m, 14-Ha), 1.78 (1H, m, 14-Hb), 0.98 (3H, s, 16-H), 1.01 (3Н, s, 17-H), 1.77 (3H, s, 18-H), 1.48 (3H, s, 19-H), 3.98 (1H, d, J=8.0 Hz, 20-Ha), 4.01 (1H, d, J=8.0 Hz, 20-Hb), 2.23 (3H, s, 4-ОСОСН3), 2.10 (3H, s, 10-ОСОСН3), 5.33 (1H, d, J=9.0 Hz, 2'-H), 5.52 (1H, t, J=9.0 Hz, 3'-H), 9.21 (1H, d, J=8.5 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.45 (2H, m, ph-m-H), 7.55 (1Н, m, ph-p-H), 7.84 (2H, m, NBz-o-H), 7.44 (2H, m, NBz-m-H), 7.16 (1Н, m, NBz-p-H), 7.97 (2H, d, J=7.0 Hz, OBz-o-H), 7.66 (2H, m, OBz-m-H), 7.74 (1H, d, J=7.5Hz, OBz-p-H), 2.61 (2Н, m, 22-H), 2.35 (2H, m, 23-H), 7.84 (1H, m, 25-H), 2.91 (1H, m, 26-Ha), 2.96 (1H, m, 26-Hb), 1.21 (2Н, m, 27-H), 1.32 (2H, m, 28-H), 1.45 (1H, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.11 (1Н, m, 30-H), 6.96 (1Н, s, 32-Ha), 7.30 (1H, m, 32-Hb), 7.87 (1H, m, 33-H), 2.14 (2H, m, 35-H), 1.75 (1H, m, 36-Ha), 1.98 (1H, m, 36-Hb), 4.13 (1H, m, 37-H), 7.10 (1H, s, 39-Ha), 7.30 (1H, m, 39-Hb), 8.23 (1H, d, J=8.0 Hz, 40-H), 4.40 (1H, m, 42-H), 1.29 (3H, m 43-H), 8.51 (1H, d, J=6.5 Hz, 44-H), 6.85 (1H, d, J=16.0Hz, 46-H), 7.43 (1Н, d, J=16.0 Hz, 47-H), 7.54 (1H, m, 50-H), 7.35 (1H, dd, J=8.5 and 2.0 Hz, 52-H), 7.71 (1H, m, 53-H).
13C-NMR (125 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.3 (9-C), 74.7Ц0-С), 133.3 (11-C), 139.4 (12-C), 70.7 (13-C), 34.4 (14-C), 42.9Ц5-С), 26.3 (16-C), 21.4 (17-C), 13.9 (18-C), 9.7 (19-C), 75.2 (20-C), 165.2 (2-OCO), 169.6, 22.5 (4-OCOCH3), 168.7, 20.6 (10-ОСОСН3), 169.1 (1'-C), 74.7 (2'-C), 54.0 (3'-C), 166.4 (3'-NHCO), 137.3 (ph-q-C), 127.6 (ph-o-C), 128.3 (ph-m-C), 131.4 (ph-p-C), 129.9 (NBz-q-C), 127.4 (NBz-o-C), 129.0 (NBz-m-C), 128.1 (NBz-p-C), 134.2 (OBz-q-C), 129.5 (OBz-o-C), 128.6 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.7 (27-C), 22.9 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.1 (37-С), 173.2 (38-C), 172.2 (41-C), 48.9 (42-C), 18.0 (43-C), 164.4 (45-C), 125.3 (m, 46-C), 137.3 (m, 47-C), 122.1 (d, J=11.8 Hz, 48-C), 160.2 (d, J=252.6 Hz, 49-C), 116.7 (d, J=25.5 Hz, 50-C), 134.6 (d, J=10.9 Hz, 51-C), 125.4 (s, 52-C), 130.3 (s, 53-C).
IR: 3324.5 (νOH и νNH), 3066.4 (ν=CH), 2939.7 (ν-CH), 1739.5, 1724.2, 1657.7 (νC=O), 1604.5, 1534.2,1451.8 (νC=C), 1242.6 (νC-O-C), 981.6, 708.7 (δ=CH).
ESI-MS: 1462.59 [M+H]+, 1484.93 [M+Na]+.
HR-MS (TOF): 1462.5540 [M+H]+, 1484.5361 [M+Na]+, C74H86ClFN7O21.
Пример 28: Жидкофазный синтез конъюгата МТС-305

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 526 мг. (1,0 экв) аналога мурамилдипептида MDA-205 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.18 г. твердого продукта было получено в результате липофилизации. Выход 81%, Тпл=171~172°С.
1H-NMR (500 MHz, DMSO-d6): 4.63 (1Н, br.s, 1-ОН), 5.40 (1Н, d, J=7.0 Hz, 2-Н), 3.56 (1Н, d, J=7.0 Hz, 3-H), 4.91 (1Н, m, 5-H), 1.62 (1Н, m, 6-Ha), 2.31 (1H, m, 6-Hb),4.12 (1H, m, 7-H), 4.92 (1H, m, 7-OH), 6.28 (1H, s, 10-H), 5.80 (1H, t, J=9.0 Hz, 13-H), 1.46 (1H, m, 14-Ha), 1.77 (1H, m, 14-Hb), 0.98 (3H, s, 16-H), 1.01 (3H, s, 17-H), 1.75 (3H, s, 18-H), 1.48 (3H, s, 19-H), 3.99 (1H, d, J=8.0 Hz, 20-Ha), 4.02 (1H, d, J=8.0 Hz, 20-Hb), 2.23 (3H, s, 4-OCOCH3), 2.10 (3H, s, 10-ОСОСН3), 5.34 (1H, d, J=9.0 Hz, 2'-Н), 5.52 (1Н, t, J=9.0 Hz, 3'-H), 9.21 (1H, d, J=8.5Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.47 (2H, m, ph-m-H), 7.55 (1H, m, ph-p-H), 7.84 (2H, m, NBz-o-H), 7.44 (2H, m, NBz-m-H), 7.18 (1H, m, NBz-p-H), 7.97 (2H, d, J=7.5 Hz, OBz-o-H), 7.66 (2H, m OBz-m-H), 7.74 (1H, m, OBz-p-H), 2.58 (2H, m, 22-H), 2.33 (2H, t, J=7.0 Hz, 23-H), 7.82 (1H, m, 25-H), 2.91 (1H, m, 26-Ha), 2.96 (1Н, m, 26-Hb), 1.23 (2H, m, 27-H), 1.33 (2H, m, 28-H), 1.45 (1H, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.11 (1Н, m, 30-H), 6.96 (1H, s, 32-На), 7.30 (1Н, m, 32-Hb), 7.86 (1Н, m, 33-Н), 2.15 (2Н, t, J=8.0 Hz, 35-Н), 1.71 (1Н, m, 36-На), 1.99 (1Н, m, 36-Hb), 4.13 (1Н, m, 37-Н), 7.12 (1Н, s, 39-На), 7.30 (1Н, m, 39-Hb), 8.25 (1Н, d, J=8.5 Hz, 40-Н), 4.41 (1Н, m, 42-Н), 1.28 (3Н, d, J=7.0 Hz, 43-Н), 8.45 (1Н, d, J=6.5 Hz, 44-Н), 6.77 (1Н, d, J=16.0 Hz, 46-Н), 7.66 (1Н, d, J=16.0 Hz, 47-H), 7.54 (1H, m, 50-H), 7.33 (1H, td, J=8.5 and 1.5Hz, 52-H), 7.76 (1H, m, 53-H).
13C-NMR (125 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-C), 133.3 (11-C), 139.4 (12-C), 70.7 (13-C), 34.7 (14-С), 42.9 (15-С), 26.3 (16-C), 21.4 (17-C), 13.9 (18-С), 9.8 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.7, 22.6 (4-OCOCH3), 168.8, 20.7 (10-ОСОСН3), 169.1 (1'-С), 74.6 (2'-C), 54.0 (3'-C), 166.4 (3'-NHCO), 137.4 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.5 (NBz-o-C), 129.1 (NBz-m-C), 128.3 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.7 (27-C), 23.0 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.1 (37-С), 173.2 (38-C), 172.2 (41-C), 48.8 (42-C), 18.2 (43-C), 164.2 (45-C), 124.9 (46-C), 137.4 (47-C), 128.8 (48-C), 134.3 (49-C), 115.4 (d, J=21.5 Hz, 50-C), 162.2 (d, J=249.1 Hz, 51-C), 117.2 (d, J=25.1 Hz, 52-C), 129.9 (53-C).
IR: 3315.4 (νOH и νNH), 3069.3 (ν=CH), 2935.0 (ν-CH), 1722.8, 1656.5 (νC=O), 1601.8, 1534.3, 1451.5 (νC=C), 1239.3 (νC-O-C), 978.5, 709.7 (δ=CH).
ESI-MS: 1462.89 [M+H]+, 1484.21 [M+Na]+.
HR-MS (TOF): 1462.5541 [M+H]+, 1484.5350 [M+Na]+, C74HB5ClFN7O21.
Пример 29: Жидкофазный синтез конъюгата МТС-306

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 492 мг. (1,0 экв) аналога мурамилдипептида MDA-206 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.24 г. твердого продукта было получено в результате липофилизации. Выход 87%, Тпл=176~178°С.
1H-NMR (500 MHz, DMSO-d6): 4.61 (1Н, br.s, 1-ОН), 5.41 (1Н, d, J=6.0 Hz, 2-H), 3.56 (1H, d, J=5.5 Hz, 3-H), 4.91 (1H, m, 5-Н), 1.62 (1Н, m, 6-Ha), 2.30 (1H, m, 6-Hb),4.11 (1Н, m, 7-H), 4.91 (1Н, m, 7-OH), 6.28 (1H, s, 10-H), 5.81 (1Н, m, 13-H), 1.49 (1H, m, 14-Ha), 1.82 (1H, m, 14-Hb), 0.99 (3H, s, 16-H), 1.01 (3H, s, 17-H), 1.76 (3H, s, 18-H), 1.49 (3H, s, 19-H), 3.99 (1H, d, J=5.5 Hz, 20-Ha), 4.00 (1Н, d, J=5.5 Hz, 20-Hb), 2.23 (3H, s, 4-ОСОСН3), 2.10 (3H, s, 10-ОСОСН3), 5.33 (1Н, d, J=8.5 Hz, 2'-H), 5.52 (1H, t, J=8.5 Hz, 3'-H), 9.20 (1H, d, J=8.0 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.46 (2H, m, ph-m-H), 7.52 (1H, m, ph-p-H), 7.84 (2H, m, NBz-o-H), 7.43 (2H, m, NBz-m-H), 7.19 (1H, m, NBz-p-H), 7.98 (2H, d, J=7.5Hz, OBz-o-H), 7.67 (2H, m, OBz-m-H), 7.72 (1H, m, OBz-p-H), 2.59 (2H, m, 22-H), 2.35 (2H, m, 23-H), 7.81 (1H, m, 25-H), 2.91 (1Н, m, 26-Ha), 2.96 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.32 (2H, m, 28-H), 1.45 (1H, m, 29-Ha), 1.62 (1Н, m, 29-Hb), 4.11 (1H, m, 30-H), 6.94 (1H, s, 32-Ha), 7.28 (1H, m, 32-Hb), 7.85 (1H, m, 33-H), 2.15 (2H, m, 35-H), 1.76 (1H, m, 36-Ha), 1.98 (1H, m, 36-Hb), 4.13 (1H, m, 37-H), 7.09 (1H, s, 39-Ha), 7.28 (1Н, m, 39-Hb), 8.20 (1H, d, J=7.5 Hz, 40-H), 4.40 (1H, m, 42-H), 1.26 (3H, m, 43-H), 8.35 (1H, d, J=4.5 Hz, 44-H), 6.79 (1H, d, J=15.5 Hz, 46-H), 7.40 (1H, d, J=15.5 Hz, 47-H), 7.81 (2H, m, 49 an 53-H), 7.39 (2H, m, 50 и 52-H).
13C-NMR (125MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.Ц3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.3 (9-C), 74.7 (10-C), 133.3 (11-C), 139.4 (12-C), 70.7 (13-C), 34.7Ц4-С), 42.9 (15-C), 26.3 (16-C), 21.4 (17-C), 13.9 (18-C), 9.7 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.6, 22.5 (4-OCOCH3), 168.8, 20.6 (10-OCOCH3), 169.1 (1'-C), 74.7 (2'-C), 54.0 (3'-C), 166.4 (3'-NHCO), 137.3 (ph-q-C), 127.6 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.4 (NBz-o-C), 129.0 (NBz-m-C), 128.3 (NBz-p-C), 134.2 (OBz-q-C), 129.5 (OBz-o-C), 128.6 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.7 (27-C), 23.0 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.1 (37-С), 173.2 (38-C), 172.3 (41-C), 48.9 (42-C), 18.1 (43-C), 164.5 (45-C), 123.5 (s, 46-C), 137.4 (s, 47-C), 133.5 (s, 48-C), 130.9 (d, J=8.3Hz, 49 и 53-C), 116.2 (d, J=21.2 Hz, 50 и 52-C), 162.4 (d, J=242.4Hz, 51-C).
IR: 3310.1 (νOH и νNH), 3063.6 (ν=CH), 2939.5 (ν-CH), 1740.5, 1724.1, 1658.2 (νC=O), 1582.5, 1536.0,1450.0 (νC=C), 1243.5 (νC-O-C), 978.0, 779.7, 709.5 (δ=CH).
ESI-MS: 1429.41 [M+2H]2+, 1451.54 [M+Na+H]2+.
HR-MS (TOF): 1428.5950 [M+H]+, 1450.5743 [M+Na]+, C74H86FN7O21.
Пример 30: Жидкофазный синтез конъюгата МТС-307

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 492 мг. (1,0 экв) аналога мурамил дипептида MDA-207 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.21 г. твердого продукта было получено в результате липофилизации. Выход 85%, Тпл=167~168°С.
1H-NMR (500 MHz, DMSO-d6): 4.63 (1Н, br.s, 1-OH), 5.40 (1H, d, J=7.0 Hz, 2-Н), 3.56 (1H, d, J=7.0 Hz, 3-H), 4.91 (1Н, m, 5-H), 1.62 (1H, m, 6-Ha), 2.30 (1Н, m, 6-Hb),4.12 (1H, m, 7-H), 4.92 (1H, m, 7-OH), 6.28 (1Н, s, 10-H), 5.81 (1H, t, J=7.5 Hz, 13-H), 1.46 (1H, m, 14-Ha), 1.78 (1Н, m, 14-Hb), 0.98 (3H, s, 16-H), 1.01 (3H, s, 17-H), 1.77 (3H, s, 18-H), 1.48 (3H, s, 19-H), 3.98 (1Н, d, J=8.5 Hz, 20-Ha), 4.01 (1Н, d, J=8.5 Hz, 20-Hb), 2.23 (3H, s, 4-OCOCH3), 2.09 (3H, s, 10-OCOCH3), 5.32 (1Н, d, J=9.0 Hz, 2'-H), 5.52 (1H, t, J=9.0 Hz, 3'-H), 9.21 (1H, d, J=8.5 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.44 (2H, m, ph-m-H), 7.55 (1H, t, J=7.5Hz, ph-p-H), 7.84 (2H, m, NBz-o-H), 7.43 (2H, m, NBz-m-H), 7.19 (1H, m, NBz-p-H), 7.97 (2H, d, J=7.0 Hz, OBz-o-H), 7.65 (2H, t, J=8.0 Hz, OBz-m-H), 7.72 (1H, t, J=7.5 Hz, OBz-p-H), 2.60 (2H, m, 22-H), 2.35 (2H, t, J=7.0 Hz, 23-H), 7.82 (1Н, m, 25-H), 2.90 (1H, m, 26-Ha), 3.00 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.33 (2H, m, 28-H), 1.46 (1Н, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.11 (1H, m, 30-H), 6.96 (1H, s, 32-Ha), 7.32 (1H, m, 32-Hb), 7.87 (1Н, m, 33-H), 2.15 (2H, t, J=8.0Hz, 35-H), 1.71 (1Н, m, 36-На), 1.99 (1Н, m, 36-Hb), 4.13 (1H, m, 37-H), 7.11 (1H, s, 39-Ha), 7.30 (1H, m, 39-Hb), 8.22 (1H, d, J=8.0 Hz, 40-H), 4.40 (1H, m, 42-H), 1.26 (3H, d, J=7.0 Hz, 43-H), 8.37 (1H, d, J=6.5 Hz, 44-H), 6.79 (1H, d, J=16.0 Hz, 46-H), 7.49 (1H, d, J=16.0 Hz, 47-H), 7.38 (1H, m, 49-H), 7.22 (1H, m, 51-H), 7.47 (1H, m, 52-H), 7.41 (1Н, m, 53-H).
13C-NMR (125 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.Ц3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-C), 133.3 (11-C), 139.4 (12-С), 70.7 (13-C), 34.7 (14-C), 42.9 (15-С), 26.3 (16-С), 21.4 (17-C), 13.9 (18-С), 9.8 (19-С), 75.3 (20-С), 165.2 (2-ОСО), 169.7, 22.6 (4-ОСОСН3), 168.8, 20.6 (10-ОСОСН3), 169.1 (1'-С), 74.4 (2'-С), 54.0 (3'-С), 166.4 (3'-NHCO), 137.5 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.5 (NBz-o-C), 129.0 (NBz-m-C), 128.2 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.5 (27-C), 23.0 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.1 (37-С), 173.3 (38-C), 172.3 (41-C),48.8 (42-C), 18.1 (43-С), 164.6 (45-C), 123.5 (46-C), 137.5 (47-C), 133.5 (48-C), 113.9 (d, J=21. 6Hz, 49-C), 162.9 (d, J=242.3Hz, 50-C), 116.7 (d, J=21.0 Hz, 51-C), 130.9 (d, J=8.5 Hz, 52-C), 123.6 (d, J=2.5 Hz,53-C)..
IR: 3320.5 (νOH и νNH), 3063.6 (ν=CH), 2939.0 (ν-CH), 1740.0, 1721.0, 1657.2 (νC=O), 1582.7, 1536.7,1450.0 (νC=C), 1243.6 (νC-O-C), 979.4, 780.5, 709.5 (δ=CH).
ESI-MS: 1429.41 [M+2H]2+, 1451.54 [M+Na+H]2+.
HR-MS (TOF): 1428.5950 [M+H]+, 1450.5736 [M+Na]+, C74H86FN7O21.
Пример 31: Жидкофазный синтез конъюгата МТС-308

953 мг. (1.0 eq) сложного моноэфира-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 510 мг. (1,0 экв) аналога мурамилдипептида MDA-208 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.14 г. твердого продукта было получено в результате липофилизации. Выход 79%, Тпл=167~168°С.
1H-NMR (500 MHz, DMSO-d6): 4.62 (1Н, br.s, 1-ОН), 5.40 (1Н, d, J=6.5 Hz, 2-Н), 3.56 (1Н, d, J=7.0 Hz, 3-H), 4.91 (1Н, m, 5-H), 1.62 (1Н, m, 6-Ha), 2.31 (1Н, m, 6-Hb),4.12 (1H, m, 7-H), 4.92 (1Н, m, 7-OH), 6.27 (1H, s, 10-H), 5.81 (1H, t, J=8.0 Hz, 13-H), 1.48 (1H, m, 14-На), 1.80 (1Н, m, 14-Hb), 0.98 (3Н, s, 16-Н), 1.01 (3Н, s, 17-Н), 1.75 (3Н, s, 18-Н), 1.48 (3Н, s, 19-Н), 3.99 (1Н, m, 20-На), 4.00 (1Н, m, 20-Hb), 2.22 (3Н, s, 4-ОСОСН3), 2.13 (3Н, s, 10-ОСОСН3), 5.32 (1Н, d, J=8.5 Hz, 2'-Н), 5.51 (1Н, t, J=8.5 Hz, 3'-Н), 9.21 (1Н, d, J=8.5 Hz, 3'-NH), 7.49 (2Н, m, ph-o-H), 7.47 (2Н, m, ph-m-H), 7.55 (1Н, m, ph-p-H), 7.84 (2Н, m, NBz-o-H), 7.43 (2Н, m, NBz-m-H), 7.17 (1Н, m, NBz-p-H), 8.06 (2Н, d, J=7.0 Hz, OBz-o-H), 7.67 (2Н, m, OBz-m-H), 7.72ЦН, d, J=8.0 Hz, OBz-p-H), 2.59 (2H, m, 22-H), 2.35 (2H, m, 23-H), 7.84 (1H, m, 25-H), 2.90 (1H, m, 26-Ha), 3.00 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.31 (2H, m, 28-H), 1.48 (1H, m, 29-Ha), 1.64 (1H, m, 29-Hb), 4.11 (1Н, m, 30-H), 6.96 (1Н, s, 32-Ha), 7.30 (1H, m, 32-Hb), 7.87 (1H, m, 33-H), 2.14 (2H, m, 35-H), 1.70 (1H, m, 36-На), 1.98 (1Н, m, 36-Hb), 4.13 (1H, m, 37-H), 7.11 (1Н, s, 39-Ha), 7.30 (1H, m, 39-Hb), 8.22 (1H, d, J=8.0 Hz, 40-H), 4.40 (1H, m, 42-H), 1.37 (3H, d, J=7.5 Hz, 43-H), 8.34 (1H, d, J=6.5 Hz, 44-H), 6.73 (1H, d, J=15.5 Hz, 46-H), 7.40 (1H, d, J=15.5 Hz, 47-H), 7.67 (1Н, m, 50-H), 7.43 (1H, m, 52-H), 7.48 (1H, m, 53-H).
13C-NMR (125 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-С), 133.3 (11-C), 139.5 (12-C), 70.7 (13-C), 34.7 (14-C), 42.9 (15-С), 26.3 (16-C), 21.4 (17-C), 13.9 (18-C), 9.8 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.7, 22.6 (4-OCOCH3), 168.8, 20.7 (10-OCOCH3), 169.2 (1'-С), 74.6 (2'-C), 54.0 (3'-C), 166.4 (3'-NHCO), 137.4 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.5 (NBz-o-C), 129.0 (NBz-m-C), 128.2 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.5 (27-C), 23.0 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.1 (37-C), 173.3 (38-C), 172.3 (41-C), 48.8 (42-C), 18.2 (43-C), 164.7 (45-C), 123.3 (s, 46-C), 137.4 (s, 47-C), 133.3 (m, 48-C), 118.6 (m, 49-C), 151.2 (m, 50-C), 149.3 (m, 51-C), 116.7 (m, 52-C), 125.1 (m, 53-C).
IR: 3306.6 (νOH и νNH), 3066.4 (ν=CH), 2932.6 (ν-CH), 1739.8, 1720.2 1658.2 (νC=O), 1535.1, 1518.5,1450.2 (νC=C), 1274.4,1243.6 (νC-O-C), 979.7, 775.8, 709.5 (δ=CH).
ESI-MS: 1446.25 [M+H]+, 1468.77 [M+Na]+.
HR-MS (TOF): 1446.5861 [M+H]+, 1468.5651 [M+Na]+, C74H85F2N7O21.
Пример 32: Жидкофазный синтез конъюгата МТС-213

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 499 мг. (1,0 экв) аналога мурамилдипептида MDA-113 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.18 г. твердого продукта было получено в результате липофилизации. Выход 82%, Тпл=167~168°С.
1H-NMR (600 MHz, DMSO-d6): 4.64 (1Н, br.s, 1-ОН), 5.40 (1Н, d, J=7.2 Hz, 2-Н), 3.56 (1Н, d, J=7.2 Hz, 3-Н), 4.91 (1Н, m, 5-H), 1.62 (1Н, m, 6-Ha), 2.31 (1Н, m, 6-Hb),4.13 (1H, m, 7-H), 4.92 (1H, m, 7-OH), 6.28 (1H, s, 10-H), 5.81 (1Н, t, J=9.0 Hz, 13-H), 1.45 (1H, m, 14-Ha), 1.79 (1H, m, 14-Hb), 0.98 (3H, s, 16-H), 0.99 (3H, s, 17-H), 1.76 (3H, s, 18-H), 1.51 (3Н, s, 19-H), 3.98 (1H, d, J=8.4 Hz, 20-Ha), 4.01 (1H, d, J=8.4Hz, 20-Hb), 2.22 (3H, s, 4-ОСОСН3), 2.09 (3H, s, 10-ОСОСН3), 5.34 (1Н, d, J=9.0 Hz, 2'-H), 5.52 (1H, t, J=9.0 Hz, 3'-H), 9.20 (1Н, d, J=9.0 Hz, 3'-NH), 7.48 (2H, d, J=7.8 Hz, ph-o-H), 7.46 (2H, m, ph-m-H), 7.55 (1H, t, J=7.8 Hz, ph-p-H), 7.82 (2H, m, NBz-o-H), 7.43 (2H, m, NBz-m-H), 7.17 (1H, m, NBz-p-H), 7.97 (2H, d, J=7.8 Hz, OBz-o-H), 7.65 (2H, t, J=7.8 Hz, OBz-m-H), 7.72 (1H, t, J=7.8 Hz, OBz-p-H), 2.61 (2H, m, 22-H), 2.35 (2H, t, J=7.2 Hz, 23-H), 7.82 (1Н, m, 25-H), 2.90 (1H, m, 26-Ha), 2.98 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.32 (2H, m, 28-H), 1.45 (1Н, m, 29-Ha), 1.64 (1H, m, 29-Hb), 4.11 (1Н, m, 30-H), 6.96 (1H, s, 32-Ha), 7.29 (1H, s, 32-Hb), 7.87 (1Н, m, 33-H), 2.11 (2Н, t, J=7.2 Hz, 35-H), 1.71 (1Н, m, 36-На), 1.99 (1Н, m, 36-Hb), 4.19 (1H, m, 37-H), 7.09 (1H, s, 39-Ha), 7.29 (1H, s, 39-Hb), 8.16 (1H, d, J=8.4 Hz, 40-H), 4.62 (1H, m, 42-H), 1.27 (3H, d, J=6.6 Hz, 43-H), 8.37 (1H, d, J=7.8 Hz, 44-H), 8.58 (1H, d, J=8.4 Hz, 47-H), 8.92 (1H, d, J=8.4 Hz, 48-H), 7.88 (1Н, m, 50-H), 7.49 (1H, m, 51-H), 7.74 (1H, m, 52-H), 8.08 (1H, d, J=8.4Hz, 53-H).
13C-NMR (150 MHz, DMSO-d3): 76.7 (1-С), 74.6 (2-C), 46.1 (3-С), 80.3 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-С), 57.4 (8-С), 202.4 (9-С), 74.7 (10-С), 133.3 (11-0), 139.5 (12-С), 70.7 (13-С), 34.4 (14-С), 42.9 (15-С), 26.3 (16-С), 21.4 (17-С), 13.9 (18-С), 9.8 (19-С), 75.3 (20-С), 165.2 (2-ОСО), 169.7, 22.6 (4-ОСОСН3), 168.8, 20.7 (10-ОСОСН3), 169.1 (1'-С), 74.5 (2'-С), 54.0 (3'-С), 166.5 (3'-NHCO), 137.4 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.5 (NBz-o-C), 128.9 (NBz-m-C), 128.2 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 171.9 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.8 (27-C), 23.0 (28-C), 31.6 (29-C), 52.3 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.8 (36-C), 52.2 (37-C), 173.2 (38-C), 172.0 (41-C), 48.6 (42-C), 19.0 (43-C), 163.3 (45-C), 149.6 (46-C), 118.5 (47-C), 138.0 (48-C), 128.1 (49-С), 128.6 (50-C), 129.2 (51-C), 130.7 (52-C), 130.3 (53-C), 146.0 (54-C).
IR: 3324.9 (νOH и νNH), 2938.5 (ν-CH), 1739.6, 1721.9, 1655.0 (νC=O), 1529.9, 1500.2, 1451.7, 1428.7 (νC=C), 1371.6, 1242.5, 1177.0, 1070.8 (δ-CH), 980.0, 776.8, 708.9 (δ=CH).
ESI-MS: 1436.75 [M+2H]2+.
HR-MS (TOF): 1435.6001 [M+H]+, 1457.5774 [M+Na]+, C75H86N8O21.
Пример 33: Жидкофазный синтез конъюгата МТС-219

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 480 мг. (1,0 экв) аналога мурамилдипептида MDA-119 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.12 г. твердого продукта было получено в результате липофилизации. Выход 79%, Тпл=169~171°С.
1H-NMR (500 MHz, DMSO-d6): 4.61 (1Н, br.s, 1-ОН), 5.41 (1Н, d, J=7.0 Hz, 2-Н), 3.56 (1H, d, J=8.5 Hz, 3-H), 4.89 (1Н, J=10 Hz, 5-H), 1.62 (1H, m, 6-Ha), 2.31 (1Н, m, 6-Hb), 4.09 (1Н, m, 7-H), 4.91 (1H, m, 7-OH), 6.28 (1H, s, 10-H), 5.81 (1Н, t, J=9.0 Hz, 13-H), 1.44 (1H, m, 14-Ha), 1.78 (1H, m, 14-Hb), 1.01 (3Н, s, 16-H), 0.99 (3H, s, 17-H), 1.76 (3H, s, 18-H), 1.49 (3H, s, 19-H), 3.98 (1Н, m, 20-Ha), 4.00 (1H, m, 20-Hb), 2.22 (3H, s, 4-ОСОСН3), 2.09 (3H, s, 10-ОСОСН3), 5.32 (1H, d, J=9.0 Hz, 2'-H), 5.52 (1Н, t, J=8.5 Hz, 3'-H), 9.19 (1H, d, J=8.5 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.43 (2H, m, ph-m-H), 7.55 (1Н, m, ph-p-H), 7.84 (2H, m, NBz-o-H), 7.49 (2H, m, NBz-m-H), 7.18 (1Н, m, NBz-p-H), 7.96 (2H, d, J=8.0Hz, OBz-o-H), 7.65 (2H, m, OBz-m-H), 7.72 (1Н, m, OBz-p-H), 2.63 (2H, m, 22-H), 2.35 (2H, m, 23-H), 7.88 (1H, m, 25-H), 2.93 (1H, m, 26-Ha), 3.21 (1Н, m, 26-Hb), 1.23 (2H, m, 27-H), 1.38 (2H, m, 28-H), 1.45 (1H, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.10 (1Н, m, 30-H), 6.95 (1H, s, 32-Ha), 7.29 (1H, s, 32-Hb), 7.87 (1H, m, 33-H), 2.26 (2H, m, 35-H), 1.76 (1H, m, 36-Ha), 1.95 (1H, m, 36-Hb), 4.12 (1H, m, 37-H), 7.03 (1H, s, 39-Ha), 7.29 (1H, s, 39-Hb), 8.24 (1H, d, J=8.0 Hz, 40-H), 4.37 (1H, m, 42-H), 1.25 (3H, m, 43-H), 8.39 (1Н, m, 44-H), 6.97 (1H, d, J=15.0 Hz, 46-H), 7.45 (1H, d, J=15.0 Hz, 47-H), 8.17 (1H, m, 50-H), 7.59 (1H, m, 51-H), 7.72 (1H, m, 52-H).
IR: 3331.9 (νOH и νNH), 2963.6, 2936.7 (ν-CH), 1739.2, 1712.5, 1649.9 (νC=O), 1538.4, 1452.3, 1438.2 (νC=C), 1370.7, 1243.8, 1172.5, 1144.1 (δ-CH), 980.0,833.2, 706.6 (δ=CH).
ESI-MS: 1417.21 [M+2H]2+.
HR-MS (TOF): 1416.5542 [M+H]+, 1438.5365 [M+Na]+, C72H85N7O21S.
Пример 34: Жидкофазный синтез конъюгата МТС-230

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т. в течение 4 часов. 553 мг. (1,0 экв) аналога мурамилдипептида MDA-130 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.28 г. твердого продукта было получено в результате липофилизации. Выход 86%, Тпл=172~173°С.
1H-NMR (600 MHz, DMSO-d6): 4.62 (1H, br.s, 1-OH), 5.40 (1H, d, J=7.2 Hz, 2-H), 3.56 (1H, d, J=7.2 Hz, 3-H), 4.90 (1H, m, 5-H), 1.62 (1H, m, 6-Ha), 2.31 (1H, m, 6-Hb),4.12 (1H, m, 7-H), 4.91 (1H, m, 7-OH), 6.28 (1H, s, 10-H), 5.81ЦН, t, J=9.0 Hz, 13-H), 1.51 (1H, m, 14-Ha), 1.79 (1H, m, 14-Hb), 0.98 (3H, s, 16-H), 0.99 (3H, s, 17-H), 1.75 (3H, s, 18-H), 1.48 (3H, s, 19-H), 3.98 (1H, d, J=7.8 Hz, 20-Ha), 4.00 (1H, d, J=7.8 Hz, 20-Hb), 2.23 (3H, s, 4-OCOCH3), 2.09 (3H, s, 10-OCOCH3), 5.33 (1H, d, J=7.8 Hz, 2'-H), 5.52 (1H, t, J=9.0 Hz, 3'-H), 9.20 (1Н, d, J=9.0 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.43 (2H, m, ph-m-H), 7.55 (1Н, t, J=7.8 Hz, ph-p-H), 7.83 (2H, m, NBz-o-H), 7.42 (2H, m, NBz-m-H), 7.18 (1Н, m, NBz-p-H), 7.98 (2H, d, J=7.2 Hz, OBz-o-H), 7.66 (2H, t, J=7.2 Hz, OBz-m-H), 7.72 (1Н, t, J=7.2 Hz, OBz-p-H), 2.60 (2H, m, 22-H), 2.35 (2H, m, 23-H), 7.82 (1H, m, 25-H), 2.91 (1H, m, 26-Ha), 2.96 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.30 (2H, m, 28-H), 1.44 (1H, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.11 (1H, m, 30-H), 6.95 (1Н, s, 32-Ha), 7.29 (1H, s, 32-Hb), 7.87 (1H, m, 33-H), 2.17 (2H, t, J=7.8 Hz, 35-H), 1.72 (1H, m, 36-Ha), 1.97 (1H, m, 36-Hb), 4.12 (1H, m, 37-H), 7.09 (1H, s, 39-Ha), 7.29 (1Н, s, 39-Hb), 8.16 (1H, d, J=7.8 Hz, 40-H), 4.46 (1H, m, 42-H), 1.30 (3H, d, J=6.6 Hz, 43-H), 8.52 (1H, d, J=6.6 Hz, 44-H), 7.70 (1H, m, 47-H), 7.84 (1H, m, 48-H), 8.97 (1H, m, 50-H).
13C-NMR (150 MHz, DMSO-d6): 76.7 (1-C), 74.5 (2-C), 46.1 (3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.4 (9-C), 74.7 (10-C), 133.3 (11-C), 139.4 (12-C), 70.7 (13-C), 34.7 (14-C), 42.9 (15-C), 26.3 (16-C), 21.5 (17-С), 13.9 (18-C), 9.8 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.6, 22.5 (4-OCOCH3), 169.6, 20.6 (10-ОСОСН3), 169.1 (1'-C), 74.4 (2'-C), 54.0 (3'-C), 166.4 (3'-NHCO), 137.4 (ph-q-C), 127.7 (ph-o-C), 128.7 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.4 (NBz-o-C), 129.0 (NBz-m-C), 128.3 (NBz-p-C), 134.3 (OBz-q-C), 129.5 (OBz-o-C), 128.6 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.7 (27-C), 22.9 (28-C), 31.5 (29-C), 52.4 (30-C), 173.9 (31-C), 171.6 (34-C), 31.7 (35-C), 27.8 (36-C), 52.1 (37-С), 173.1 (38-С), 173.2 (41-C), 48.8 (42-C), 19.7 (43-C), 164.4 (45-C), 131.5 (46-C), 130.6 (47-C), 134.5 (48-C), 147.7 (49-C), 124.0 (50-C), 149.7 (51-C).
IR: 3277.6 (νOH и νNH), 3065.0 (ν=CH), 2973.2, 2936.4 (ν-CH), 1719.3, 1646.9, 1629.8 (νC=O), 1537.1, 1452.0 (νC=C), 1350.0, 1240.9, 1151.2 (δ-CH), 978.4, 895.0, 706.3 (δ=CH).
ESI-MS: 1463.70 [M+H]+.
HR-MS (TOF): 1463.5293 [M+H]+, 1485.5120 [M+Na]+, C72H83ClN8O23.
Пример 35: Жидкофазный синтез конъюгата МТС-233

953 мг. (1.0 eq) сложного моноэфира паклитаксел-2'-O-янтарной кислоты, 115 мг. (1,0 экв) HOSu и 192 мг. (1.0 eq) EDC⋅HCl растворили в DMSO и перемешивали при к.т.в течение 4 часов. 528 мг. (1,0 экв) аналога мурамилдипептида MDA-133 аккуратно добавили к смеси несколькими порциями. рН смеси довели до 7~8 N-метилморфолином, и продолжили перемешивание в течение 4 часов. После завершения реакции большое количество воды добавили в смесь, и белый твердый продукт выпал в осадок. Смесь фильтровали, и получили неочищенный продукт. Этот неочищенный продукт был очищен с помощью ODS колоночной хроматографии, 1.17 г. твердого продукта было получено в результате липофилизации. Выход 80%, Тпл=155~156°С.
1H-NMR (600 MHz, DMSO-d6): 4.61 (1Н, br.s, 1-ОН), 5.40 (1Н, d, J=7.2 Hz, 2-Н), 3.56 (1Н, d, J=7.2 Hz, 3-H), 4.90 (1Н, m, 5-H), 1.62 (1H, m, 6-Ha), 2.31 (1Н, m, 6-Hb),4.12 (1H, m, 7-H), 4.91 (1Н, m, 7-OH), 6.28 (1H, s, 10-H), 5.81 (1H, t, J=9.0 Hz, 13-H), 1.48 (1Н, m, 14-Ha), 1.79 (1H, m, 14-Hb), 0.98 (3H, s, 16-H), 0.99 (3H, s, 17-H), 1.75 (3H, s, 18-H), 1.49 (3H, s, 19-H), 3.98 (1H, d, J=8.4 Hz, 20-Ha), 4.00 (1H, d, J=8.4 Hz, 20-Hb), 2.22 (3H, s, 4-OCOCH3), 2.09 (3H, s, 10-OCOCH3), 5.33 (1H, d, J=9.0 Hz, 2'-H), 5.52 (1H, t, J=9.0 Hz, 3'-H), 9.19 (1H, d, J=9.0 Hz, 3'-NH), 7.48 (2H, m, ph-o-H), 7.43 (2H, m, ph-m-H), 7.56 (1Н, m, ph-p-H), 7.83 (2H, m, NBz-o-H), 7.42 (2H, m, NBz-m-H), 7.18 (1Н, m, NBz-p-H), 7.97 (2H, d, J=7.2 Hz, OBz-o-H), 7.66 (2H, m, OBz-m-H), 7.72 (1Н, m, OBz-p-H), 2.60 (2H, m, 22-H), 2.35 (2H, t, J=7.2 Hz, 23-H), 7.82 (1H, m, 25-H), 2.90 (1H, m, 26-Ha), 2.96 (1H, m, 26-Hb), 1.22 (2H, m, 27-H), 1.33 (2H, m, 28-H), 1.44 (1H, m, 29-Ha), 1.62 (1H, m, 29-Hb), 4.11 (1Н, m, 30-H), 6.94 (1H, s, 32-Ha), 7.37 (1H, s, 32-Hb), 7.87 (1H, m, 33-H), 2.15 (2H, t, J=7.8 Hz, 35-H), 1.70 (1H, m, 36-Ha), 1.97 (1H, m, 36-Hb), 4.12 (1H, m, 37-H), 7.09 (1H, s, 39-Ha), 7.32 (1Н, s, 39-Hb), 8.21 (1H, d, J=8.4 Hz, 40-H), 4.43 (1H, m, 42-H), 1.29 (3H, d, J=6.6 Hz, 43-H), 8.28 (1H, d, J=7.8 Hz, 44-H), 4.73 (1H, s, 46-H), 6.20 (1H, d, J=7.8 Hz, 48-H), 7.32 (1Н, m, 49-H), 7.38 (1H, m, 50-H), 7.97 (1H, m, 52-H), 7.49 (1H, m, 53-H), 7.54 (1H, m, 54-H), 8.30 (1Н, m, 55-H).
13C-NMR (150 MHz, DMSO-d6): 76.7 (1-С), 74.5 (2-C), 46.1 (3-С), 80.2 (4-C), 83.6 (5-C), 36.5 (6-C), 70.4 (7-C), 57.4 (8-C), 202.3 (9-C), 74.7 (10-C), 133.3 (11-С), 139.4 (12-C), 70.7 (13-C), 34.4 (14-C), 42.9 (15-C), 26.3 (16-С), 21.4 (17-C), 13.9 (18-C), 9.8 (19-C), 75.3 (20-C), 165.2 (2-OCO), 169.6, 22.5 (4-ОСОСН3), 168.8, 20.6 (10-ОСОСН3), 169.1 (1'-C), 74.4 (2'-С), 54.0 (3'-С), 166.4 (3'-NHCO), 137.4 (ph-q-C), 127.7 (ph-o-C), 128.3 (ph-m-C), 131.5 (ph-p-C), 129.9 (NBz-q-C), 127.4 (NBz-o-C), 129.0 (NBz-m-C), 128.2 (NBz-p-C), 134.3 (OBz-q-C), 129.6 (OBz-o-C), 128.7 (OBz-m-C), 133.5 (OBz-p-C), 172.0 (21-C), 28.8 (22-C), 29.5 (23-C), 170.0 (24-C), 38.5 (26-C), 28.7 (27-C), 22.9 (28-C), 31.6 (29-C), 52.4 (30-C), 173.9 (31-C), 171.5 (34-C), 31.7 (35-C), 27.7 (36-C), 52.2 (37-C), 173.2 (38-C), 173.3 (41-C),48.2 (42-C), 18.4 (43-C), 167.2 (45-C), 67.2 (46-C), 153.1 (47-C), 105.7 (48-C), 126.1 (49-C), 120.7 (50-C), 134.0 (51-C), 127.6 (52-C), 126.1 (53-C), 125.4 (54-C), 121.7 (55-C), 127.4 (56-C).
IR: 3289.3 (νOH и νNH), 3065.7 (ν=CH), 2937.8 (ν-CH), 1739.5, 1720.9, 1647.6 (νC=O), 1577.5, 1537.2, 1450.4 (νC=C), 1265.1, 1239.5, 1154.1 (δ-CH), 905.9,853.3, 792.9, 771.3, 707.4 (δ=CH).
ESI-MS: 1465.32 [M+2H]2+.
HR-MS (TOF): 1464.6128 [M+H]+, 1486.5942 [M+Na]+, C77H89N7O22.
Биологический пример
Определение активности in vitro
Пример 36:
Пять соединений согласно настоящему изобретению, МТС-220, МТС-302, МТС-213, МТС-219 и МТС-233 были отправлены в Национальный институт рака США (NCl) для скрининга их противоопухолевой активности in vitro. Результаты экспериментов показывают, что 50% ингибирующая рост опухоли концентрация (Gl50) для конъюгатов этого типа в 60 клеточных линиях опухолей человека находилась в том же диапазоне величины, как у паклитаксела, и 50% летальная концентрация (LC50) была более 10 μM. Результаты экспериментов отражены на Фигурах 1-5.
Соединения согласно настоящему изобретению, МТС-301, МТС-302, МТС-303, МТС-304, МТС-305, МТС-306 и МТС-307 исследовали с целью определения их противоопухолевой активности в 10 клеточных линиях опухолей человека. 50% ингибирующая рост опухоли концентрация (Gl50) для конъюгированных соединений этого типа находилась в том же диапазоне величины, как и у паклитаксела или доцетаксела. Результаты экспериментов показаны на Фигурах 6-7.
Биологическое действие in vivo.
Пример 37: ингибирование роста опухоли с МТС-220 в ксенографтных моделях у бестимусных мышей с использованием линии MDA-MB-231 рака молочной железы человека
Материалы и подопытные животные для проведения эксперимента:
1. МТС-220, бесцветная и прозрачная жидкость с концентрацией 1.0 мг/мл, 1.5 мг/мл, 2.0 мг/мл, была перепакована в стерильных условиях и может быть непосредственно использована, хранится при температуре 4°С. Были установлены следующие дозы вводимого лекарственного средства: 10 мг/кг, 15 мг/кг и 20 мк/кг, объем вводимого лекарственного средства был 0.2 мл/20 г.
2. Паклитаксела для инъекций, продукция фармацевтической фабрики Beijing Union Pharmaceutical Factory, Регистрационный номер: Н10980069, номер партии продукта: 080704, спецификация 5 мл: 30 мг.
3. Taxol+MDA [Пептид MDA(P) 0.54 мг/мл (0.001М)+Таксол(Т) 0.9 мг/мл (0.001М)], был получен на заказ, может быть непосредственно использован после переупаковки в стерильных условиях, хранится при температуре 4°С. В соответствии с планом эксперимента объем вводимого лекарственного препарата составил 0,2 мл/20 кг.
4. MDA [Пептид (Р) 0.54 мг/мл (0.001М), Пример 10], бесцветная и прозрачная жидкость, была получена на заказ, может непосредственно использоваться после переупаковки в стерильных условиях, хранится при температуре 4°С.
Опухолевые линии: Обширно метастазированные линии MDA-MB-231 рака молочной железы человека были имплантированы бестимусным мышам, и мыши, имеющие онкологические опухоли, были получены из Crown Bioscience Co. Ltd. (Пекин), выращивалась и хранились в нашей лаборатории.
Животные: «голые» мыши BALB/c,  , 4~5 недельного возраста, были получены из института, занимающегося лабораторными животными, Китайской Академии Медицинских Наук. Сертификат NO. SCXK (Пекин) 2005-0013.
, 4~5 недельного возраста, были получены из института, занимающегося лабораторными животными, Китайской Академии Медицинских Наук. Сертификат NO. SCXK (Пекин) 2005-0013.
Оборудование для кормления: центр экспериментальных животных, Китайская Академия Медицинских Наук, Зоо Лаборатория SPF уровень, Сертификат NO. SYSK (Пекин) 2004-0001.
Экспериментальные методы:
Мыши с онкологией с хорошим ростом опухоли и хорошим общим физическим состоянием были отобраны и умерщвлены путем смещения шейных позвонков. Опухоль была изолирована в стерильных условиях и разрезана на фрагменты (диаметром около 2-3 мм) хирургическим ножом. Фрагменты затем были гиподермально инокулированы в задне подмышечную область бестимусных мышей. Опухоль росла нормально. Мыши были разделены на группы и через 11 дней им вводили лекарственное средство. Длину и ширину опухолей измеряли с помощью штангенциркуля, и делили на группы по объему опухоли.
Мыши были разделены на восемь групп, в каждой группе было по 6-8 мышей. Мышам с относительно большими объемами опухоли по сравнению с обычнами (со средним объемом в 340 мм3) вводили МТС-220 в дозе 30 мг/кг (группа МТС-220 30 мг/кг). Другие мыши были разделены на 7 групп по размерам опухоли, в каждой группе было по 6-8 мышей. Группы включали отрицательный контроль; Паклитаксел-группа, в которой вводили паклитаксел в дозе 24 мг/кг с перерывами; три МТС группы, в которых вводили МТС в дозе 10 мг/кг, 15 мг/кг и 20 мг/кг, соответственно; MDA группа; Таксол + MDA группа. Размеры опухолей вышеуказанных 7 групп мышей были относительно равны со средним объемом около 140 мм3. Всем мышам вводили лекарственный препарат после распределения по группам внутрибрюшинной инъекцией один раз в день в зависимости от веса их тела.
Первый день группирования и введения лекарственного препарат был обозначен D1, размеры опухоли (длина и ширина) и вес тела мыши измеряли каждые три дня. В контрольной группе паклитаксела введение проводили с перерывами 4 раза, тогда как в группе МТС-220 30 мг/кг лекарственный препарат прекратили вводить после последовательного введения 12 раз. В других группах введение проводили последовательно 24 раза. Эксперимент был завершен через 24 часа после последнего введения.
Мыши были умерщвлены путем смещения шейных позвонков, и опухоли были выделены, и измерили их вес, и была рассчитана скорость ингибирования роста опухоли лекарственными препаратами. Статистическая значимость веса опухоли, объем опухоли и уровня RTV были оценены с помощью t-теста. Расчетные методы и формулы опущены.
Противоопухолевая активность была оценена показателем относительной скорости распространения опухоли Т/С (%)
Критерий оценки терапевтического эффекта: Т/С (%)>40 был признан недействительным;
Т/С(%)≤40 и согласно Статистической оценке Р<0.05 был признан действительным. Результаты эксперимента:
Во время наблюдения за ходом эксперимента масса тела мышей в группе отрицательного контроля постепенно возрастала. Средняя масса тела выросла на 3.5 г. по сравнению с началом деления на группы. В контрольной группе, получающей паклитаксел, вводили препарат с перерывами, масса тела сохранялась в допустимых пределах для токсического и побочного эффектов. В группе МТС-220 30 мг/кг вводили препарат 12 раз последовательно в течение 12 дней, и масса тела мышей оставалась по существу на том же уровне, когда их делили по группам. Масса тела стала постепенно возрастала после отмены лекарственного средства, и в конце эксперимента вес тела увеличился на 2.6 г по сравнению с началом, когда делили на группы. Рост массы тела в группе МТС-220 30 мг/кг был таким же, как и в группе МТС-220 с дозой 15 мг/кг, которая получала препарат 24 раза (в последней группе была 2.7 г.), в обеих группах общее доза введенного лекарственного препарата было схожей. В то время как группа МТС-220 с дозой препарата 20 мг/кг получала лекарственный препарат последовательно 24 дня, вес тела мышей в данной группе вырос на 1.9 г., что меньше, чем масса тела в группе отрицательного контроля. Масса тела группы «Т(0.9 мг/мл)+Р (0.54 мг/мл)» при последовательном приеме препарата была близка на начальной стадии массе тела в группе, принимающей паклитаксел, но токсический и побочный эффекты у мышей проявлялись постепенно в течение непрерывного введения препарата, они включают вздутие живота, понижение подвижности, потерю веса и др. На двадцатый день 2/3 мышей в этой группе умерли.
Кривая роста опухоли у мышей показала, что скорость роста опухоли в группе, принимающей раствор MDA [Р (0.54 мг/мл)] была ниже, чем скорость роста опухоли в группе отрицательного контроля, и относительная скорость распространения опухоли (Т/С) была 83.5%. Рост опухоли в существенной степени связан с вводимой дозой препарата МТС-220 10 мг/кг, 15 мг/кг и 20 мг/кг. В конце эксперимента скорость ингибирования роста опухоли в трех группах были 37.3%, 57.4% и 72.2% по отдельности. Относительные скорости распространения опухоли были 70.0%, 39.5% и 29.4% по отдельности, при этом группа МТС 15 мг/кг и группа МТС 20 мг/кг были оценены как действительные.
В группе МТС-220 30 мг/кг, которая получала препарат последовательно 12 раз, общая доза была такой же, как в группе МТС 15 мг/кг, которая получала препарат последовательно 24 раза. Хотя объем опухоли в группе МТС-220 30 мг/кг на начальном этапе эксперимента был несколько больше, он постепенно снизился по мере применения препарата. Скорость роста также была достаточно медленной после отмены препарата. В конце эксперимента разница ингибирования роста опухоли группы МТС-220 30 мг/кг и группы МТС-220 15 мг/кг значительно возросла (у группы МТС-220 15 мг/кг было 57.4%, у группы МТС-220 30 мг/кг было >87%), и относительная скорость распространения опухоли (Т/С) существенно снизилась (у группы МТС-220 15 мг/кг было 37.5%, у группы МТС-220 15 мг/кг было 6.16%). Для сравнения: препарат в группе МТС-220 с дозой 30 мг/кг последовательно вводился 12 раз, а в группе МТС-220 с дозой 20 мг/кг препарат вводился 24 раза, и доза препарата в группе МТС-220 30 мг/кг была меньше, но скорость ингибирования у группы МТС-220 30 мг/кг была выше, относительная скорость распространения опухоли (Т/С) также была значительно снижена, и физическое состояние мышей было лучше. Все вышесказанное свидетельствует о том, что мышей с раковой опухолью вводили подходящую дозу, которая не только контролировала рост опухоли, но также представляла собой необходимо меньшую дозу, и характеризовалась менее длительным лечением и снижеными токсическим и побочными эффектами.
Итоги эксперимента: ингибирование рака молочной железы человека MDA-MB-231 у бестимусных мышей с опухолями было значительным после того, как мышам внутрибрюшинно вводили МТС-220 10 мг/кг, 15 мг/кг и 20 мг/кг последовательно. Рост MDA-MB-231 опухолевых линий ингибировался значительно, и ингибирующие эффекты были связаны с вводимой дозой. Эффект от введения 15 мг/кг и 20 мг/кг были оценены как действительные в данной серии экспериментов.
Группа МТС-220 30 мг/кг получала препарат последовательно 12 раз, ингибирование роста опухоли MDA-MB-231 было значительным. Опухоль росла медленно после отмены препарата, и показатели физического состояния хорошо восстанавливались. Период лечения был более коротким, и эффект ингибирования опухоли был более значительным по сравнению с тем, что в группе МТС-220 15 мг/кг. Результаты эксперимента показаны на Фигурах 8-11 и в Таблицах 1-2.
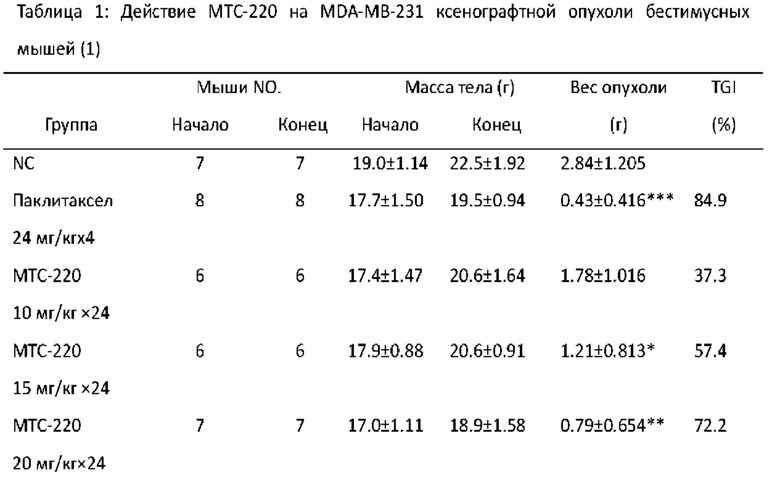

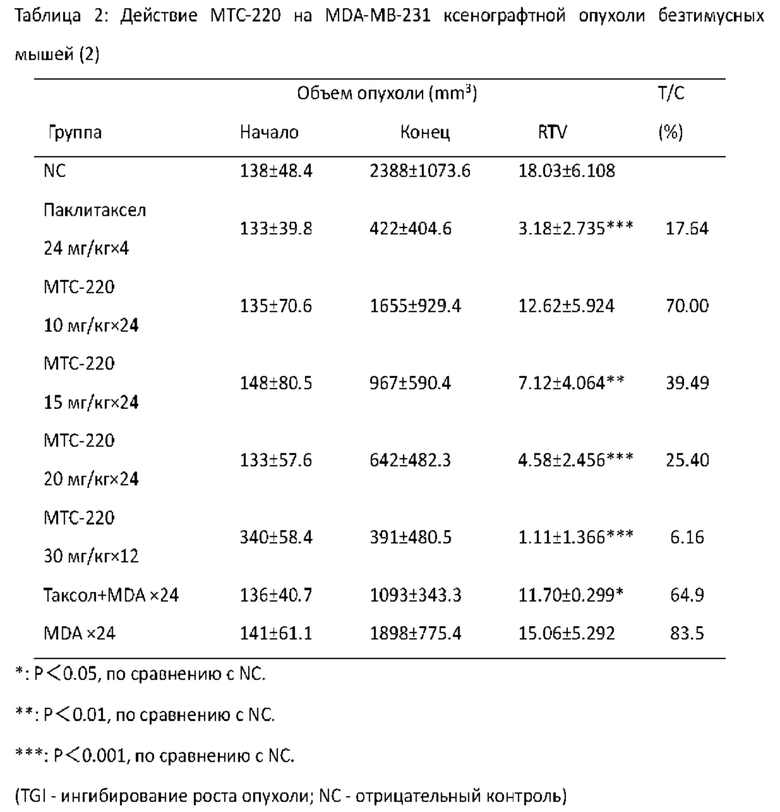
Пример 38: ингибирование роста опухоли с МТС-220 в ксенографтных моделях у бестимусных мышей с использованием линии рака легких человека Н460
Экспериментальные материалы и подопытные животные:
МТС-220: Получен на заказ, в трех концентрациях 1.0 мг/мл, 1.5 мг/мл и 2.0 мг/мл, бесцветная и прозрачная жидкость, была переупакована в стерильных условиях и может быть непосредственно использована, хранится при температуре 4°С.
Паклитаксел для инъекций: продукция фармацевтической фабрики Beijing Union Pharmaceutical Factory, Регистрационный номер: Н10980069, номер партии продукта: 080704, спецификация 5 мл: 30 мг. Раствор наполнителя: (смесь физиологического солевого раствора, содержащая 50% DMSO и 5% продукта конденсации полиоксиэтиленового спирта и касторового масла (Cremphor EL)), был расфасован в стерильных условиях и может быть непосредственно использован, температура хранения 4°С.
Опухолевые линии: Клеточные линии рака легких человека Н460 были получены из АТСС и культивировались и хранились в лаборатории. Выращенная культура клеток in vitro опухоль была инокулирована бестимусным мышам, опухоль росла и была передана для проведения экспериментов.
Животные: «голые» мыши BALB/c,  ,4~5 недельного возраста, были получены из экспериментальной лаборатории, Китайская Академия Медицинских Наук, Сертификат NO. SCXK (Пекин) 2005-0013.
,4~5 недельного возраста, были получены из экспериментальной лаборатории, Китайская Академия Медицинских Наук, Сертификат NO. SCXK (Пекин) 2005-0013.
Оборудование для кормления: Центр Экспериментальных Животных, SPF уровень Зоо Лаборатории, Китайская Академия Медицинских Наук, Сертификат NO. SYSK (Пекин) 2004-0001.
Экспериментальный метод:
Мыши с опухолью с хорошим ростом опухоли и в хорошем общем физическом состоянии были отобраны и умерщвлены путем смещения шейных позвонков. Опухоль была изолирована в стерильных условиях и разрезана на фрагменты (диаметром около 2-3 мм) хирургическим ножом. Фрагменты затем были подкожно инокулированы в задней подмышечной области бестимусных мышей с помощью троакара.
После того как опухоли нормально росли в течение восьми дней, им была дана возможность вырасти до среднего объема 130 мм3. Длина и ширина опухоли была измерена с использованием штангенциркуля, и мыши были разделены на несколько групп по объему опухоли.
Мыши были разделены для наблюдения на пять групп, в каждой из которой было по восемь мышей. Группа отрицательного контроля получала раствор наполнителя, и три другие группы получали МТС-220 5 мг/кг, 10 мг/кг, 20 мг/кг по отдельности. Группа положительного контроля получала инъекции паклитаксела в дозе 24 мг/кг один раз в три дня. Каждая группа получала препарате момента формирования группы.
День формирования групп был определен как D1, введение препарат в контрольной группе, получающей паклитаксел, проводилось с промежутками 4 раза, тогда как в группах МТС-220 введение проводили 25 раз последовательно. Эксперимент был остановлен спустя 24 часа после последнего введения.
Во время эксперимента размеры опухоли (длина и ширина) и массу тела мышей измеряли каждые три дня. Объем опухоли (TV) и относительный объем опухоли (RTV) были подсчитаны по методу сравнения, и была составлен график направления роста объема опухоли.
В конце эксперимента мыши были умерщвлены путем смещения шейных позвонков. Опухоли были удалены и взвешены, и была рассчитана скорость ингибирования роста опухоли с помощью лекарственных препаратов. Статистическое значение массы опухоли, объема опухоли и уровня RTV были оценены посредством t-теста.
Расчетная формула:

(С - средний вес опухоли контрольной группы; Т - средний вес опухоли группы, получавшей препарат)
Объем опухоли (TV)=длина×ширина2/2
Формула расчета относительного объема опухоли (RTV): Vt/Vo
(Vo является объемом TV в начале деления на группы, и Vt является объемом TV, измеряемым каждый раз)
Противоопухолевая активность была оценена по относительной скорости распространения опухоли Т/С (%)

(administrated group - группа, получавшая препарат, negative control group - группа отрицательного контроля)
Критерий оценки терапевтического эффекта: Т/С (%)>40, был признан недействительным;
Т/С(%)≤40, и согласно статистической оценке Р<0.05, был признан действительным.
Результаты эксперимента:
Результаты наблюдений показали, что в течение 25 дней масса тела в группе отрицательного контроля постепенно повышалась, и общий статус не изменился. Опухоль Н460 росла быстрее в сравнении с объемом опухоли в начале деления по группам, средний объем опухоли в группе отрицательного контроля был 33.3 в конце эксперимента.
Группа положительного контроля, в которой вводили паклитаксел в дозе 24 мг/кг дважды в день, показала, что он подавляет рост опухоли Н460. Ингибирование роста опухоли постепенно увеличивалось с увеличением частоты приема препарата. По сравнению с группой отрицательного контроля, ингибирование роста опухоли достигло 65% после четвертого введения. Терапевтический эффект сохранялся в течение недели, и постепенно снижался после отмены препарата. В конце эксперимента статистический результат указывал на то, что скорость ингибирования веса опухоли составил 61%, и относительная скорость распространения опухоли (Т/С) была 35,6%. Терапевтический эффект в группе положительного контроля был лучше, чем у группы отрицательного контроля. Также в ходе эксперимента наблюдали, что после введения паклитаксела в дозе 24 мг/кг с перерывами два раза за период, мыши похудели, и вес постепенно снижался, что поддерживалось на уровне 2 г ниже, чем средний вес в начале деления по группам. Масса тела восстановилась через неделю после отмены препарата.
В начале 20 дней вес мышей был по существу одинаков между группой отрицательного контроля и двумя группами, в которых вводили МТС-200 10 мг/кг и 5 мг/кг по отдельности. Масса тела в двух группах, в которых применяли препараты, несколько уменьшилась по сравнению с группой отрицательного контроля в течение непрерывного введения. Через 25 дней последовательного введения МТС-220 в дозе 5 мг/кг скорость увеличения объема опухоли существенно не отличалась по сравнению с группой отрицательного контроля. Через 2 недели после последовательного введения дозы 10 мг/кг, измеренный объем опухоли Н460 отличался от группы отрицательного контроля. В конце эксперимента ингибирование объема опухоли в группе с применением дозы 10 мг/кг составила 18,8%, а скорость ингибирования увеличения веса опухоли составляла 17,3%.
После 10 дней лечения препаратом МТС-220 в дозе 20 мг/кг результат измеренного объема опухоли отличался от группы отрицательного контроля. Опухоль росла медленней при непрерывном введении, и ингибирование роста опухоли постепенно увеличивалось. До конца эксперимента ингибирование веса опухоли было 52.9%, и относительная скорость распространения опухоли (Т/С) была 50.1%, это было статистически значимо в сравнении с группой отрицательного контроля. Результаты эксперимента показаны на Фигурах 12-13 и таблицах 3-4.


Результаты эксперимента: Мыши с Н460 опухолью легкого человека получали внутрибрюшинно инъекции МТС-220 последовательно в дозе 5 мг/кг, 10 мг/кг, 20 мг/кг в течение 25 дней. Образец МТС ингибировал рост опухоли Н460, и эффект ингибирования опухоли зависел от дозы препарата. В конце эксперимента ингибирование веса опухоли в группы, получающей дозу препарата 20 мг/кг, составила 52,9%, относительная скорость распространения опухоли составила 50,1%, что статистически значительно отличается по сравнению с такими показателями группы отрицательного контроля.
Пример 39: Результаты скрининга действия МТС-220 на ксенографтные опухоли у бестимусных мышей с использованием линий клеток чувствительных опухолей
Цель эксперимента: Проверить влияние МТС-200 на ксенотрансплантированную опухоль бестимусной мыши с помощью клеточных линий рака молочной железы, рака легких и рака яичников in vivo. Исследовали опухолевые линии, которые показали существенную восприимчивость к МТС-220, и наблюдали ответную реакцию бестимусной мыши мыши при последовательном введении препарата.
Подопытные животные: Бестимусные белые мыши BALB/c были получены из Института лабораторных животных Академии медицинских наук КНР. №сертификата: SCXK(BeiJing)2005-0013. Клеточные линии: Опухолевые клеточные линии были пересеяны в нашей лаборатории и хранились в ней, некоторые из них были получены от АТСС.
Опухолевые клеточные линии включали: Рак молочной железы человека МХ-1 и MCF-7.
Рак яичников человека А2780 и чистый рак яичников человека ES-2,
Рак легких человека Н1975 и А549.
Проведение эксперимента:
1. Мыши были разделены на группу отрицательного контроля и группу, принимавшую МТС-220.
2. Методика была в целом аналогичной, применявшейся в Примерах 52 и 53, которые не описаны в текущем эксперименте.
3. Доза приема и ход терапии был получен в ходе эксперимента профазы, который показал определенный эффект и прогресс при кратчайшей терапии при дозе 30 мг/кг/день, а период приема для каждого эксперимента в серии не превышало 12 дней.
Результаты эксперимента (1): После введения МТС-220 опухоль MCF-7 у мышей уменьшилась в размере. При введении десятой дозы объем опухоли был крайне небольшим, после чего препарат был отменен с последующим наблюдением динамики опухоли MCF-7 у мышей. Через неделю опухоль в группе исчезла постепенноно. Через три недели непрерывного наблюдения опухоль в организме мышей не была выявлена. Только опухоль молочной железы MCF-7 показала медленный рост после прививки в течение 50 дней. Объем опухоли в группе отрицательного контроля не превысил 600 мм3. Мы прекратили наблюдение, поскольку результат эксперимента был очевидным.
Изменение массы тела отражено на Фигурах, а препарат показал очевидный эффект на массу тела, которая сократилась во время приема препарата. После прекращения приема препарата масса тела возросла, и такое изменение по существу соответствовало изменениям в группе отрицательного контроля. Результаты эксперимента отражены на Фигурах 14-15 и в Таблицах 5-6.
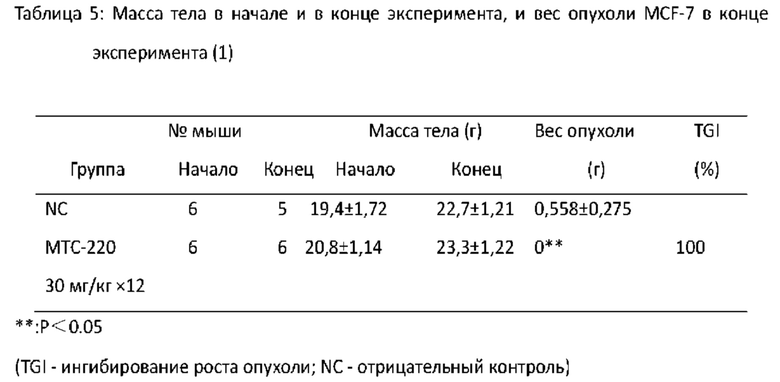
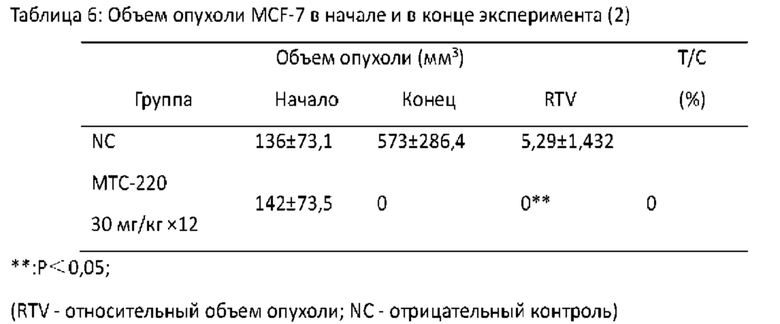
Результаты эксперимента (2): Во время приема МТС-220 опухоль А549 уменьшалась в размерах, но не исчезла. После отмены приема препарата на одну неделю опухоль исчезла у одной мыши. После отмены приема препарата на две недели средний объем в группе, принимавшей препарат, сохранился как в начале эксперимента без увеличения.
Изменение массы тела отражено на Фигурах, и препарат показал очевидный эффект на массу тела, которая уменьшилась значительно во время приема препарата. Масса тела также уменьшилась после отмены препарата, одна из мышей умерла через неделю, а масса тела других мышей постепенно восстанавливалась. Результаты эксперимента отражены на Фигурах 16-17 и в Таблицах 7-8.
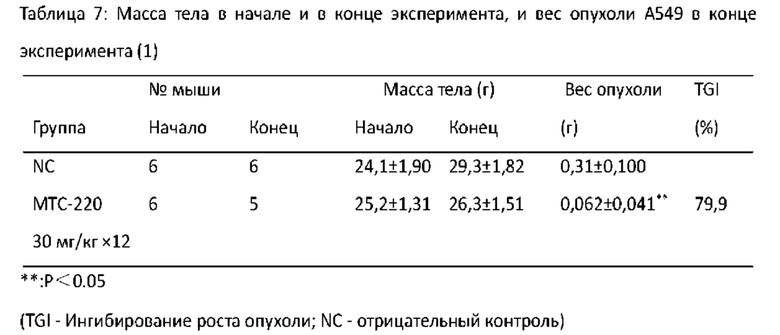

Результаты эксперимента (3): Прием МТС-220 существенно подавил рост опухоли Н1975. Во время приема препарата объем опухоли у группы, получавшей терапию, постепенно сокращался, и затем у некоторых мышей опухоль исчезла. Результаты эксперимента отражены на Фигурах 18-19 и в Таблицах 9-10.
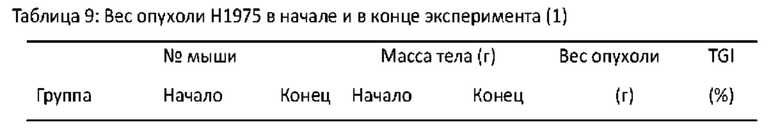


Вывод результатов скрининга действия МТС-220 на опухоль:
МТС-220 применяли в отношении опухоли рака молочной железы человека, рака легких, рака яичников у ксенографтных бестимусных мышей, основываясь на результатах скрининга предварительного эксперимента; мыши получали интраперитональную инъекцию МТС-220 в дозе 30 мг/кг 10~12 раз, и образцы МТС имели разный ингибирующий эффект на рост выбранной опухоли в скрининговвых исследованиях.
Согласно наблюдениям в эксперименте, ингибирующее действие МТС-200 в отношении роста рака молочной железы МХ-1 было слабым, ингибирование МТС-220 рака яичников А2780 и ES-2 наблюдалось, но не достигло допустимых критериев. МТС-200 показал существенный ингибирующий эффект в отношении опухолей рака молочной железы MCF-7, рака легких А549 и опухоли Н1975. Наблюдаемые результаты показали, что при использовании опухолевых линий, восприимчивых к МТС-220, объем опухоли у мыши-носителя уменьшился, после отмены приема препарата опухоль продолжала уменьшаться, а у некоторых мышей опухоли исчезли. При этом, в конце эксперимента в модели рака легких А549 и Н1975 ингибирование роста опухоли было выше 80%, относительная скорость распространения опухоли составила менее 30%, что статистически значительно отличалось по сравнению с группой отрицательного контроля. МТС-220 существенно подавлял рост опухоли MCF-7, а опухоль у мышей экспериментальной группы исчезла через 10 последовательных приемов препарата.
Вывод: МТС-220 существенно подавлял рак молочной железы и рак легких. В частности, опухолевые линии MDA-MB-231, MCF-7, Н460, Н1975 и А549 были наиболее чувствительны.
Пример 40: Противоопухолевой естественный метастатический эффект МТС-220 у мышей с раком молочной железы
Клеточная линия рака молочной железы у мышей (4Т1, АТСС CRL2539) стала щедрым подарком профессора Вей Ли Яня из Института биофизики Академии наук КНР. Клетка была выращена в среде 1640 (Gibco), содержащей 10% эмбриональной бычьей сыворотки (ЭБС), 1% глутамина и 1% пенициллина.
Клетки 4Т1 были собраны в логарифмической фазе, и концентрация зафиксирована на уровне 2×106 /мл. Опухоли 4Т1 были введены самкам мыши BALB/c посредством подкожной инъекции в четвертое скопление жировой ткани правой брюшной молочной железы в дозе 2×105 /0,1 мл. Через пять дней после имплантации клеток опухоли 4Т1 мыши были разделены на пять групп в случайном порядке, по восемь мышей в каждой, и мыши отдельно получали интраперитональные дозы паклитаксела (3 мг/кг), МТС-220 (2,5 мг/кг, 5 мг/кг, 10 мг/кг) или наполнителя в качестве контроля один раз в день. Начиная с 9-го дня после имплантации, рост первичной опухоли измеряли через каждые 2 дня с помощью штангенциркуля для определения наибольшего и наименьшего диаметра опухоли. Объем опухоли рассчитывался по формуле (1/2)×наименьший диаметр2×наибольший диаметр. После прекращения приема препарата через 28 дней после имплантации все мыши были умерщвлены посредством перелома шейных позвонков, и измеряли массу тела. Опухоли, селезенка и легкое были удалены и взвешены. Легкие были зафиксированы в фиксирующей смеси Буэна на 24 ч. Количество узлов легочных метастаз было подсчитано, а статистическая оценка проведена с помощью U-критерия Манна-Уитни.
Результаты показали, что МТС-220 значительно понижает количество узлов легочных метастаз 4Т1 мышей со статистическим уровнем значимости (р<0.01) по сравнению с контрольной группой, получающей наполнитель, и результат зависит от вводимой дозы. Не было существенного изменения узлов легочных метастаз в группе, получающей Таксол. И МТС-220, и Таксол в значительной степени ингибировали рост опухоли по сравнению с контрольной группой, получающей наполнитель. Во время наблюдений в ходе эксперимента, у МТС-200 не было ни токсического, ни побочных эффектов. Результаты эксперимента отражены на Фигурах 20-22 и в Таблице 11.

Пример 41: Противоопухолевой естественный метастатический эффект МТС-220 у мышей с раком легких
Мыши C57BI/6 с раком легких Льюиса были получены и пассированы, после чего убиты путем перелома шейных позвонков с последующим удалением опухоли. Суспензию клеток опухоли (5×106 клеток/мл) была подготовлена в стерильных условиях. Суспензия (0,2 мл/мышь, 1×106 клеток опухоли) была привита подкожно в подмышечную область 24 мышам C57BI/6 Через три дня после имплантации мыши были разделены на три группы в случайном порядке, каждая группа состояла из восьми мышей, и мыши отдельно получали интраперитональные дозы паклитаксела (6 мг/кг), МТС-220 (10 мг/кг) или наполнителя в качестве контроля один раз в день. Начиная с 7-го дня после имплантации, через каждые 2 дня измеряли наибольший и наименьший диаметры опухоли. Объем опухоли рассчитывался по формуле (1/2)×наименьший диаметр2×наибольший диаметр. После прекращения приема препарата через 28 дней после имплантации все мыши были убиты посредством перелома шейных позвонков с последующим измерением массы тела. Опухоли, селезенка и легкое были удалены и взвешены. Легкие были зафиксированы в фиксирующей смеси Буэна на 24 ч. Количество узлов легочных метастаз было подсчитано, а статистическая оценка проведена с помощью U-критерия Манна-Уитни.
Результаты показали, что МТС-220 значительно снижает количество узлов легочных метастаз у мышей LLC со статистическим уровнем значимости (р<0,05) по сравнению с контрольной группой. Существенных улучшений в отношении узлов легочных метастаз в группе, принимавшей Таксол, отмечено не было. И МТС-220, и Таксол в значительной степени ингибировали рост опухоли по сравнению с контрольной группой, получавшей наполнитель. Во время наблюдений в ходе эксперимента у МТС-200 не было обнаружено ни токсического, ни побочного эффектов, а масса тела мышей постепенно возрастала. Результаты эксперимента отражены на Фигурах 23-25 и в Таблице 12.

Пример 42: Противоопухолевая искусственная метастатическая активность МТС-220 в отношении рака легких Льюиса у мышей
Мыши C57BI/6) с раком легких Льюиса были получены и пассированы, после чего убиты путем перелома шейных позвонков с последующим удалением опухоли. Суспензию клеток опухоли (5×106 клеток/мл) была подготовлена в стерильных условиях. Суспензия (0,2 мл/мышь, 3×105 клеток опухоли) была привита под кожу в подмышечную область 50 мышам C57BI/6. Через два дня после имплантации мыши были разделены на пять групп в случайном порядке, каждая группа состояла из десяти мышей, и мыши отдельно получали интраперитональные дозы паклитаксела (3 мг/кг), МТС-220 (2,5 мг/кг, 5 мг/кг или 10 мг/кг) или наполнителя в качестве контроля. После прекращения приема препарата через 28 дней последовательного приема все мыши были убиты посредством перелома шейных позвонков с последующим измерением массы тела. Опухоли, селезенка и легкое были удалены и взвешены. Легкие были зафиксированы в фиксирующей смеси Буэна на 24 ч. Количество узлов легочных метастаз было подсчитано, а статистическая оценка проведена с помощью U-критерия Манна-Уитни.
Результаты показали, что МТС-220 значительно снижает количество узлов легочных метастаз у мышей LLC со статистическим уровнем значимости по сравнению с контрольной группой, получавшей наполнитель, и результаты зависели от вводимой дозы. Существенных улучшений в отношении узлов метастаз легкого в группе, принимавшей Таксол, отмечено не было. Результаты эксперимента отражены на Фигуре 26 и в Таблице 13.

Пример 43: Тест на токсичность МТС-220 по однократной дозе
Методика проведения эксперимента:
Смотрите публикацию Государственного управления по контролю за качеством медикаментов и продуктов питания "Технические указания по проведению неклинических испытаний цитоксичных противораковых препаратов", а также "Технические указания по проведению испытаний острой токсичности химических препаратов". Максимальная вводимая доза МТС-200 у мышей ICR определялась посредством наблюдения за токсичностью после внутривенного введения однократной дозы.
Результаты эксперимента:
После внутривенной инъекции МТС-220 в дозе 112,5 мг⋅кг-1 самостоятельные передвижения в группе, которая получала препарат, были уменьшены, у некоторых мышей проявлялись симптомы прыжка, и мыши восстановливались через 10 минут. Необычных явлений в группе, получавшей наполнитель (эпоксидированное касторовое масло : диметилсульфоксид : физиологический раствор = 5:5:90, объемное соотношение) и контрольной группе на наблюдалось. После непрерывного наблюдения в течение 14 дней поведение животных, самостоятельные передвижения и физические признаки каждой группы были в норме, смертей зафиксировано не было.
Масса тела в группе, получавшей препарат, и группе, получавшей наполнитель, существенно не отличались от контрольной. Анатомический осмотр включал в себя: сердце, печень, селезенку, легкие, почки, органы желудочно-кишечного тракта и прочие органы животного, все они не имели патологических изменений.
Результаты эксперимента:
После внутривенного введения в хвост мыши ICR МТС-220 в однократной дозе 112,5 мг⋅кг-1 существенные симптомы токсичности или смерть не наблюдались. Был сделан вывод, что для испытуемых мышей ICR максимально переносимая доза (МПД) при внутривенном введении образца была выше максимальной введенной дозы (112,5 мг⋅кг-1).
Вышеуказанные результаты фармакологических экспериментов, а также результаты теста по исследованию токсичности в тесте с однократной дозой показали, что направление разработки конъюгата противоопухолевого средства таксана и аналога мурамилдипептида было выбрано правильно. Он представляет собой серию безопасных и новых соединений, которые можно разрабатывать для новых препаратов двойного действия - противоопухолевым и противометастаическим эффектами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИМИЧЕСКИЙ СИНТЕЗ И ПРОТИВООПУХОЛЕВЫЙ И ПРОТИВОМЕТАСТАТИЧЕСКИЙ ЭФФЕКТЫ КОНЪЮГАТА ДВОЙНОГО ДЕЙСТВИЯ | 2011 |
|
RU2604718C2 |
| Средство для ингибирования фермента тирозил-ДНК-фосфодиэстеразы 1 человека на основе производных пентафуранозилнуклеозидов | 2019 |
|
RU2748103C1 |
| СОЕДИНЕНИЕ ПЕПТИДНОЙ ПРИРОДЫ, ОБЛАДАЮЩЕЕ СПОСОБНОСТЬЮ СВЯЗЫВАТЬСЯ С ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2823164C2 |
| Соединение-антагонист PD-L1 | 2020 |
|
RU2823231C1 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2609018C2 |
| АНТАГОНИСТЫ CXCR7 | 2013 |
|
RU2649004C2 |
| Тиазололактамные соединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2805569C1 |
| КОНДЕНСИРОВАННЫЕ ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПИРИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПРОИЗВОДСТВА И ПРИМЕНЕНИЕ | 2014 |
|
RU2668074C2 |
| Спиросоединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2800042C1 |
| БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2565079C2 |












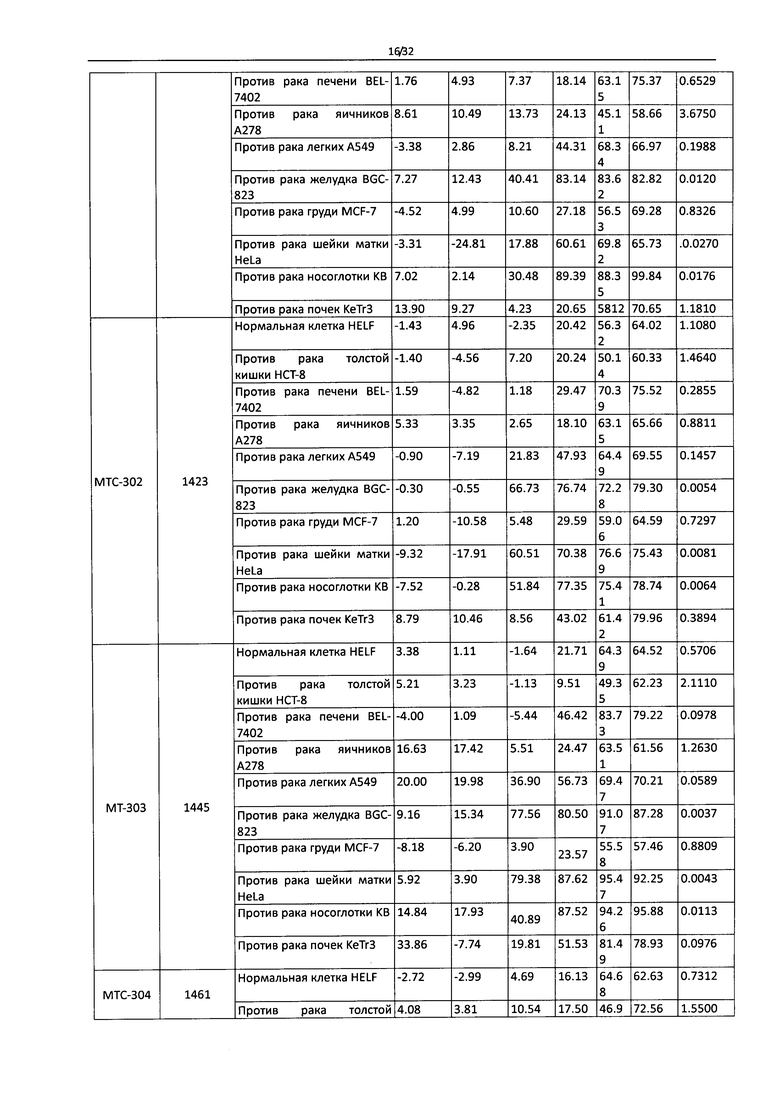


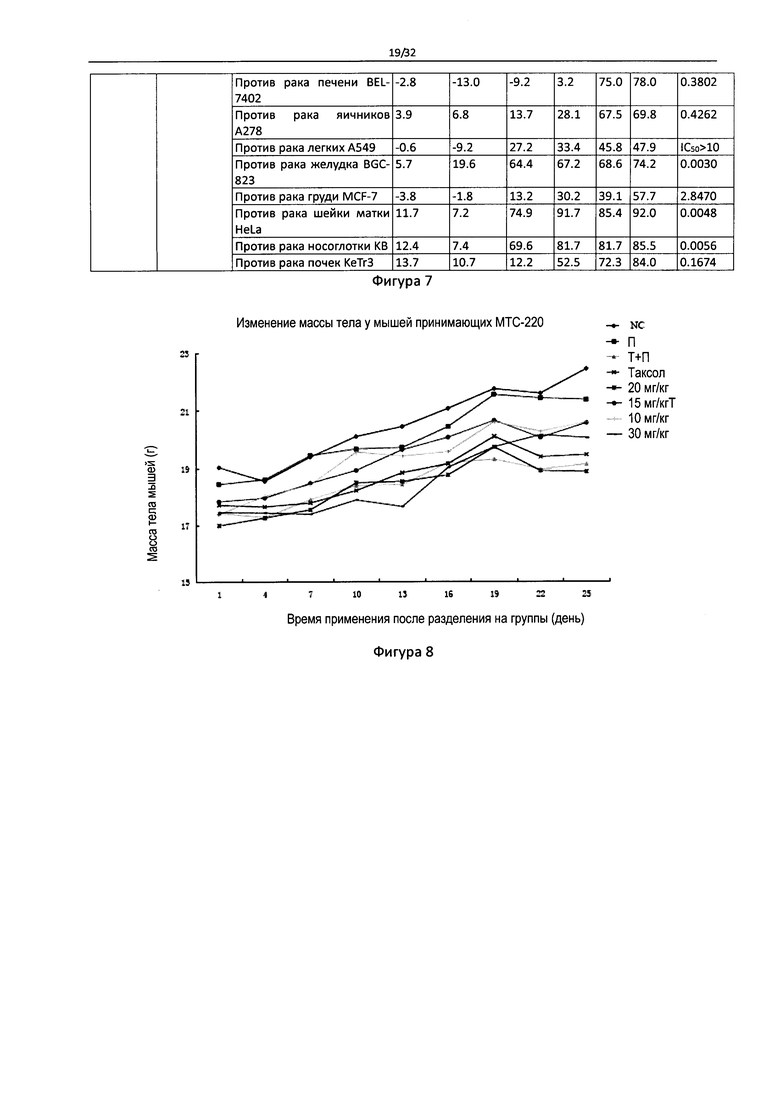




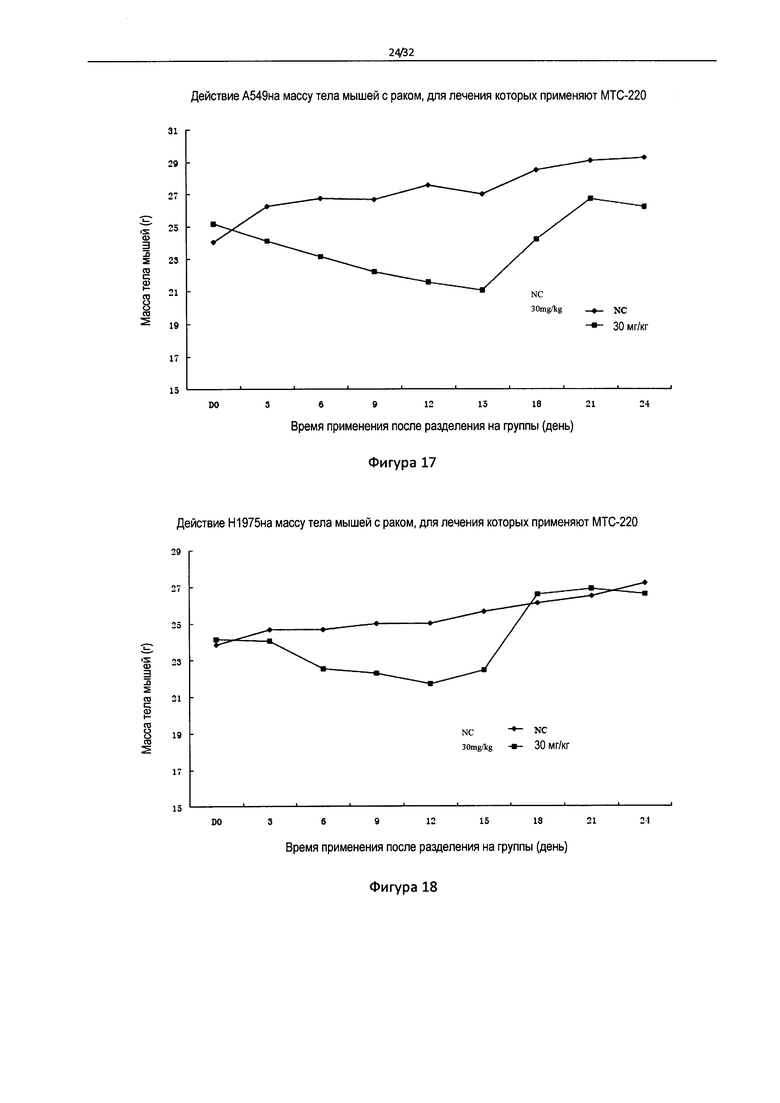



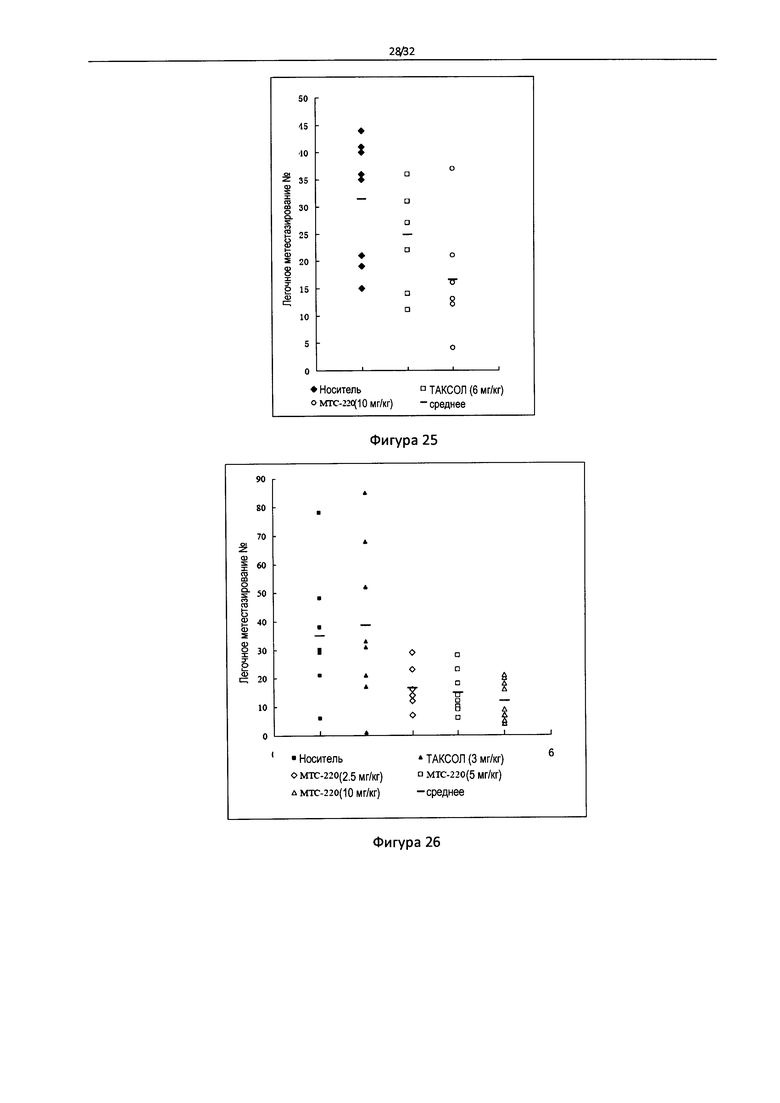


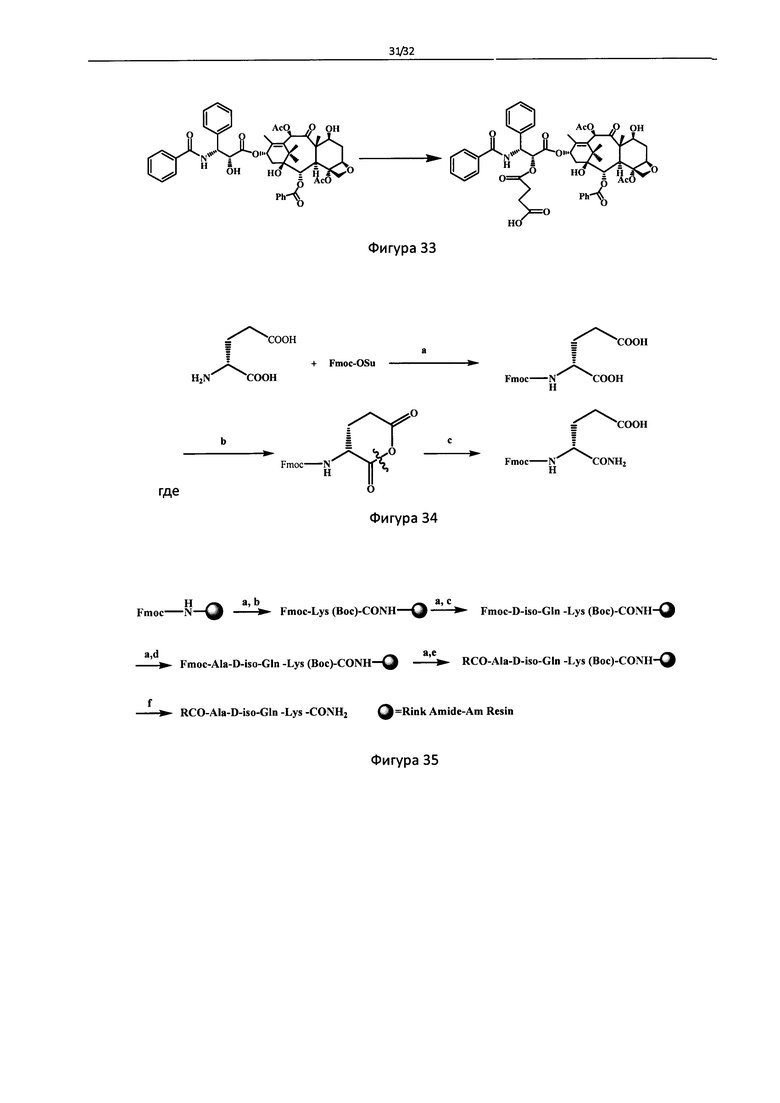

В настоящем изобретении предложены химический синтез, противоопухолевый и противометастатический эффекты конъюгата двойного действия, представленного формулой I. Синтез конъюгата формулы I осуществляют путем комбинации твердофазного и жидкофазного синтеза, и указанный конъюгат можно применять для получения противоопухолевых лекарственных средств, что подтверждено достоверными результатами биологических исследований. 6 н. и 16 з.п. ф-лы, 37 ил., 13 табл., 43 пр.

1. Соединение формулы I или его фармацевтически приемлемая соль,

формула I
где n = 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12;
X выбран из C1-6 алкила, группы C=C- и C1-6 алкила, содержащего гетероатом, где указанный гетероатом представляет собой кислород; или X представляет собой одинарную связь таким образом, что M непосредственно связан с ацилом;
M выбран из фенила, нафтила и гетероарила, выбранного из пятичленного гетероциклического ароматического кольца, содержащего атом серы, или десятичленного конденсированного гетероциклического ароматического кольца, содержащего атом азота;
R выбран из водорода, незамещенного неразветвленного или разветвленного C1-6 алкила, гидрокси, галогена и нитро.
2. Соединение или его фармацевтически приемлемая соль по п. 1, в котором n = 2, 3, 4, 5, 6, 7, 8, 9 или 10.
3. Соединение или его фармацевтически приемлемая соль по п. 2, в котором n = 2, 3, 4, 5, 6, 7 или 8.
4. Соединение или его фармацевтически приемлемая соль по п. 3, в котором n = 2, 3, 4 или 5.
5. Соединение или его фармацевтически приемлемая соль по п. 1, в котором десятичленное конденсированное гетероциклическое ароматическое кольцо выбрано из
 ,
, , и
, и .
.
6. Соединение или его фармацевтически приемлемая соль по п. 1, в котором указанное соединение выбрано из соединений формулы IA
 ,
,
формула IA
где R11 выбран из водорода, незамещённого неразветвлённого или разветвлённого С1-6 алкила, гидрокси, галогена и нитро.
7. Соединение или его фармацевтически приемлемая соль по п. 1, в котором указанное соединение выбрано из соединений формулы IВ
 ,
,
формула IB
где R12 выбран из водорода, незамещённого неразветвлённого или разветвлённого С1-6 алкила, гидрокси, галогена и нитро.
8. Соединение или его фармацевтически приемлемая соль по п. 1, в котором указанное соединение выбрано из соединений формулы IС
 ,
,
формула IC
где R13 выбран из водорода, незамещённого неразветвлённого или разветвлённого С1-6 алкила, гидрокси, галогена и нитро.
9. Соединение или его фармацевтически приемлемая соль по п. 1, в котором указанное соединение выбрано из соединений формулы ID
 ,
,
формула ID
где R14 выбран из водорода, незамещённого неразветвлённого или разветвлённого С1-6 алкила, гидрокси, галогена и нитро.
10. Соединение или его фармацевтически приемлемая соль по п. 1, в котором указанное соединение выбрано из соединений формулы IЕ
 ,
,
формула IE
где R15 выбран из водорода, незамещённого неразветвлённого или разветвлённого С1-6 алкила, гидрокси, галогена и нитро.
11. Соединение или его фармацевтически приемлемая соль по п. 1, в котором указанное соединение выбрано из:
 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, , и
, и .
.
12. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-11, в котором фармацевтически приемлемые соли выбраны из гидрохлорида, гидробромида, сульфата, фосфата, нитрата, оксалата, фумарата, малеата, сукцината, цитрата, тартата, мезилата и p-толуолсульфоната.
13. Фармацевтическая композиция для получения лекарственного средства для регуляции иммунитета, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-12 и по меньшей мере один фармацевтически приемлемый носитель.
14. Фармацевтическая композиция для получения лекарственного средства для профилактики и/или лечения рака, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-12 и по меньшей мере один фармацевтически приемлемый носитель.
15. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-12 для получения лекарственного средства для регуляции иммунитета.
16. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-12 для получения лекарственного средства для профилактики и/или лечения рака.
17. Применение по п. 16, в котором рак выбран из меланомы, рака желудка, рака легких, рака молочной железы, рака почки, рака печени, эпидермальной карциномы ротовой полости, рака шейки матки, коллоидной аденофибромы, рака поджелудочной железы, рака простаты и рака толстой кишки.
18. Способ получения соединения или его фармацевтически приемлемой соли по любому из пп. 1-12, который включает в себя следующие стадии:
1) синтез сложного моноэфира паклитаксел-2′-O-алкандикарбоновой кислоты с помощью жидкофазного синтеза;
2) твердофазный или жидкофазный синтез аналога мурамил дипептида;
3) жидкофазный синтез конъюгатов аналога мурамил дипептида и паклитаксела.
19. Способ по п. 18, отличающийся тем, что стадия 1) способа получения сложного моноэфира паклитаксел-2’-O-алкандикарбоновой кислоты включает:
(1) растворение паклитаксела, алкандикарбоновой кислоты диангидрида и 4-N,N-диметилпиридина в пиридине и перемешивание в течение 4 ч при комнатной температуре;
(2) разбавление раствора пиридина этилацетатом с последующей промывкой этилацетатного слоя последовательно насыщенным водным раствором CuSO4 и H2O;
(3) отделение этилацетатного слоя и концентрирование в вакууме, добавление к остатку избытка воды с выпадением белого осадка и последующая фильтрация и лиофилизация белого осадка с получением сложного моноэфира паклитаксел-2’-O-алкандикарбоновой кислоты.
20. Способ по п. 18, отличающийся тем, что стадия 2) способа получения аналога мурамил дипептида включает следующие стадии:
1) твердофазный синтез:
(1) синтез промежуточного соединения Fmoc-D-iso-Gln-OH с помощью жидкофазного синтеза;
(2) введение Fmoc-L-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-L-Ala-COOH и карбоновую кислоту в твердофазный носитель аминосмолу Rink-Amide AM с помощью твердофазного синтеза,
где реакция конденсации представляет собой традиционную реакцию конденсации с образованием амида; где реакцию конденсации проводят до конца путем добавления избыточного количества вышеуказанных трех аминокислот или любой карбоновой кислоты и любого конденсирующего агента HATU или HBTU, BOP или PyBOP; и
получение аналога мурамил дипептида путем выполнения стадий, включающих тщательную промывку смолы и отщепление продукта от смолы, и очистку сырого продукта;
2) жидкофазный синтез:
(1) синтез промежуточных соединений Boc-D-Glu(OBzl)-NH2 и Boc-Lys(Z)-NH2;
(2) синтез дипептидного фрагмента Boc-Ala-D-Glu(OBzl)-NH2 и трипептидного фрагмента R-Ala-D-Glu(OBzl)-NH2 с помощью способа активной этерификации, и удаление защитной группы Bzl действием уксуснокислого раствора бромоводородной кислоты или другой кислоты или основания, синтез тетрапептида R-Ala-D-iso-Gln-Lys(Z)-NH2 способом активной этерификации;
(3) удаление защитной группы Z с помощью смешанного раствора этилэфирата трифторида бора, трифторуксусной кислоты и этантиола (об./об./об.=9:9:2) с получением сырого продукта, и очистка сырого продукта с получением аналога мурамил дипептида.
21. Способ по п. 20, в котором аминокислота Fmoc-L-Lys(Boc)-COOH, Fmoc-D-iso-Gln-COOH, Fmoc-L-Ala-COOH в твердофазном синтезе заменена любой природной или искусственной аминокислотой.
22. Способ по п. 18, в котором способ получения конъюгатов аналога мурамил дипептида и паклитаксела включает следующие этапы:
1) растворение сложного моноэфира паклитаксел-2’-O-алкандикарбоновой кислоты, HOSu и DIC с молярным соотношением (2:1-1:2) в растворе диметилсульфоксида или N,N-диметилформамида или N-метилпирролидона, и осуществление реакции указанного раствора в течение 1-10 часов при температуре от -20°C до +50°C;
2) добавление эквимолярных количеств аналога мурамил дипептида в раствор диметилсульфоксида или N,N-диметилформамида или N-метилпирролидона, доведение pH реакционной системы до 6-8 с помощью слабощелочного реагента N-метилморфолина, осуществление реакции в течение 1-10 часов с получением конъюгата после завершения реакции;
3) добавление в реакционный раствор любого из веществ, выбранных из воды, метанола, этанола, диэтилового эфира, петролейного эфира и этилбутилового эфира, фильтрация выпавшего в осадок твердого вещества, и очистка сырого продукта препаративной ВЭЖХ или перекристаллизацией с получением целевого продукта.
| RU 2008120699 A, 20.01.2010 | |||
| Биореактор для микробиологического разложения растительного сырья | 1989 |
|
SU1712399A1 |
| XUQIN LI et al, "Chemical conjugation of muramyl dipeptide and paclitaxel to explore the combination of immunotherapy and chemotherapy for cancer", GLYCOCONJ J., 2008, vol | |||
| Видоизменение пишущей машины для тюркско-арабского шрифта | 1923 |
|
SU25A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |

