Уровень техники
Настоящее изобретение касается новых соединений и фармацевтических композиций, ингибирующих связывание хемокина SDF-1 (также известного как хемокин CXCL12) или I-TAC (также известного как CXCL11) с хемокиновым рецептором CXCR7. Эти соединения применимы для предотвращения пролиферации опухолевых клеток, образования опухолей, васкуляризации опухолей, метастазирования, воспалительных заболеваний, включая, без ограничения, артрит, воспалительные заболевания почек и рассеянный склероз, состояния неправильной васкуляризации, включая, без ограничения, заживление ран, лечение ВИЧ-инфекций и лечение нарушений дифференцировки и мобилизации стволовых клеток (см. также поданные параллельно заявки USSN 10/912,638, 11/407,729 и 11/050,345).
Хемокины представляют собой суперсемейство небольших, подобных цитокинам белков, индуцирующих перестройку цитоскелета, прочную адгезию к эндотелиальным клеткам и направленную миграцию, которые также могут влиять на активацию и пролиферацию клеток. Хемокины действуют скоординировано с белками клеточной поверхности, направляя специфическую наводку различных групп клеток на определенные анатомические участки.
Предшествующие научно-исследовательские работы ряда групп указывали на роль хемокинового рецептора CXCR4 в метастазировании и росте опухолей. Так, Muller et al., "Involvement of chemokine receptors in breast cancer metastasis", Nature, 410:50-56 (2001) показали, что раковые клетки молочной железы используют опосредованные хемокинами механизмы типа тех, что регулируют передвижения лейкоцитов, в процессе метастазирования. В опухолевых клетках экспрессируется особый, неслучайный профиль функционально активных рецепторов хемокинов. Сигнализация через CXCR4 опосредует полимеризацию актина и образование псевдоподий в раковых клетках молочной железы и индуцирует хемотактические и инвазивные реакции. Кроме того, органы, представляющие основные зоны метастазирования рака молочной железы (как то: лимфатические узлы, костный мозг и легкие), являются наиболее распространенными источниками лигандов для рецептора CXCR4.
Используя иммунодефицитных мышей, Muller et al. смогли уменьшить метастазирование введенных клеток рака молочной железы человека путем обработки мышей антителом, связывающим CXCR4. Их данные свидетельствуют о том, что метастазирование рака молочной железы может уменьшаться при обработке пациентов антагонистами CXCR4.
Bertolini et al., "CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin's lymphoma", Cancer Research, 62:3106-3112 (2002) показали уменьшение объема опухолей, а также продление срока жизни у иммунодефицитных мышей с введенными клетками лимфомы человека при обработке антителом против CXCR4. Они интерпретировали свои результаты таким образом, что объем опухолей может уменьшаться при обработке пациентов антагонистами CXCR4.
Более поздние исследования показали, что другой хемокиновый рецептор, CXCR7, также может быть мишенью при лечении рака. CXCR7 экспрессируется преимущественно в трансформированных клетках по сравнению с обычными клетками, при заметной экспрессии в ряде раковых опухолей у человека. Исследования in vitro показали, что пролиферация клеток, экспрессирующих CXCR7, может ингибироваться антагонистами CXCR7. Исследования in vivo на мышах свидетельствуют, что антагонисты CXCR7 могут ингибировать образование опухолей и рост опухолей.
Потенциальная значение CXCR7 иллюстрируется альтернативной интерпретацией уменьшения объема опухолей, наблюдавшегося Bertolini et al. Это уменьшение явно может быть результатом опосредованного антителами клиренса, а не результатом антител против CXCR4, как это первоначально считалось. При опосредованном антителами клиренсе любое антитело, распознающее какой-нибудь белок на поверхности клеток лимфомы, имело бы тот же эффект, который приписывается антителам против CXCR4. К сожалению, исследования Bertolini et al. не дают четкого ответа насчет того, была ли наблюдавшаяся реакция опухолей обусловлена опосредованным антителами клиренсом или взаимодействием с CXCR4.
Однако теперь уже известно, что клетки лимфомы, использовавшиеся Bertolini et al., экспрессируют и CXCR4, и CXCR7. Единственным лигандом для CXCR4 является SDF-1. Как SDF-1, так и I-TAC связываются с CXCR7. Используя антитела против SDF-1, было показано, что антагонисты CXCR7 ответственны за снижение опухолевой нагрузки и повышение выживаемости. Поскольку SDF-1 является единственным лигандом для CXCR4, то можно было бы ожидать, что нейтрализация SDF-1 антителами против SDF-1 будет эквивалентна нейтрализации CXCR4 антителами против CXCR4. Однако эксперименты с использованием антител против SDF-1 показали лишь частичное уменьшение опухолевой нагрузки и увеличение выживаемости. Поэтому вероятной мишенью является CXCR7, так как продолжительная активность, очевидно, обусловлена взаимодействием второго лиганда, I-TAC, с CXCR7.
До недавнего времени, возможное значение CXCR7 для пролиферации опухолевых клеток, роста опухолей и метастазирования не было известно. Теперь же данные указывают на способность некоторых антагонистов CXCR7 к предотвращению роста и распространения опухолей, а профили экспрессии свидетельствуют об ограниченном распределении рецептора CXCR7 в тканях, которое коррелирует с онкогенезом.
Кроме того, было обнаружено, что CXCR7 может служить сорецептором для некоторых генетически расходящихся вирусов иммунодефицита человека (ВИЧ) и вируса иммунодефицита обезьян (SIV), в частности, для Х4-тропного изолята HTV-2-ROD (Shimizu N. et al., J. Virol., (2000) 74:619-626; Balabanian K. et al., J. Biol. Chem., in press; published on August 17, 2005 as Manuscript M508234200).
К тому же было описано, что SDF-1 играет роль в мобилизации гемопоэтических клеток-предшественников и стволовых клеток, в особенности клеток, несущих рецептор CXCR4, из определенных гемопоэтических тканей, включая костный мозг (Hattori K. et al., Blood, (2000) 97:3354-3360; WO 2005/000333, содержание которых включено сюда путем ссылки). Другие недавние исследования показывают, что рецептор CXCR7 также может играть роль в процессах мобилизации стволовых клеток.
В свете вышесказанного очевидно, что соединения, способные специфически связываться с рецептором CXCR7, могут быть полезными для лечения заболеваний и других биологических состояний, которым такого взаимодействия могут пойти на пользу. В настоящем изобретении представлены такие соединения вместе с фармацевтическими композициями и связанными с ними способами лечения.
Раскрытие изобретения
Настоящим изобретением, в одном аспекте, предусмотрены соединения по формуле I:
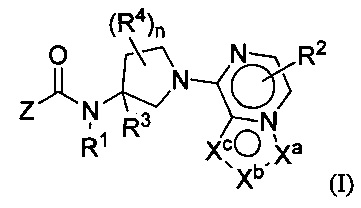
либо их фармацевтически приемлемые соли, гидраты или N-оксиды. Различные группы (например, R1, R2, R3, R4, Z, Xa, Xb, Xc и индекс n) описаны в разделе «Осуществление изобретения».
Представленные здесь соединения применимы для связывания с CXCR7 и лечения заболеваний, зависящих, по крайней мере частично, от активности CXCR7. Соответственно, настоящим изобретением в других аспектах предусмотрены композиции, содержащие одно или несколько из вышеприведенных соединений вместе с фармацевтически приемлемым наполнителем.
В следующем аспекте настоящим изобретением предусмотрены способы лечения различных заболеваний, приведенных далее, которые включают введение нуждающимся в таком лечении лицам терапевтически эффективного количества соединения по вышеприведенной формуле в течение периода времени, достаточного для лечения заболевания.
В следующем аспекте настоящим изобретением предусмотрены способы диагностики заболеваний у индивидов. В этих способах представленные здесь соединения вводятся в меченой форме субъекту, после чего следует диагностическая визуализация для определения наличия или отсутствия CXCR7. В связанном с этим аспекте способ диагностики заболеваний выполняется путем контактирования образца ткани или крови с представленным здесь меченым соединением и определения наличия, отсутствия или количества CXCR7 в образце.
В некоторых воплощениях субъекту до, после или в сочетании с соединениями по настоящему изобретению вводится некоторая доза химиотерапевтического средства или облучения. В некоторых воплощениях доза является субтерапевтической, когда химио-терапевтическое средство вводится или облучение проводится отдельно.
Осуществление изобретения
Сокращения и определения
Термин "алкил", сам по себе или как часть другого заместителя, означает, если не указано иначе, прямой или разветвленный углеводородный радикал, содержащий указанное число атомов углерода (т.е. C1-8 означает от 1 до 8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и др. Термин "алкенил" относится к ненасыщенным алкильным группам, содержащим одну или несколько двойных связей. Аналогичным образом, термин "алкинил" относится к ненасыщенным алкильным группам, содержащим одну или несколько тройных связей. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и высшие гомологи и изомеры. Термин "циклоалкил" относится к углеводородным кольцам, содержащим указанное число атомов в кольце (например, С3-6-циклоалкил), которые полностью насыщены или содержат не более одной двойной связи между вершинами кольца. "Циклоалкил" также относится к бициклическим и полициклическим углеводородным кольцам, таким, к примеру, как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и др. Термин "циклоалкенил" относится к циклоалкильным группам, содержащим по крайней мере одну двойную связь между вершинами кольца. Примерами циклоалкенилов являются циклопентенил и циклогексенил. Термин "спироциклоалкил" относится к таким циклоалкильным группам, у которых одна вершина кольца прикреплена к двум другим неводородным частям молекулы. Спироциклоалкильными являются такие заместители, у которых два атома углерода в цепи алкилена (как правило, на концах цепи алкилена) прикрепляются к одному и тому же атому углерода в остальной части молекулы. Термин "гетероциклоалкил" относится к циклоалкильным группам, содержащим от одного до пяти гетероатомов из числа N, О и S, причем атомы азота и серы необязательно окислены, а атомы азота необязательно четвертичные. Гетероциклоалкил может представлять собой моноциклическую, бициклическую или полициклическую кольцевую систему. Неограничительные примеры гетероциклоалкильных групп включают пирролидин, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, гидантоин, диоксолан, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и др. Гетероциклоалкильная группа может присоединяться к остальной части молекулы через кольцевой атом углерода или гетероатом.
Термин "алкилен" сам по себе или как часть другого заместителя означает двухвалентный радикал, происходящий из алкана, а примером служит -СН2СН2СН2СН2-. Как правило, алкильная (или алкиленовая) группа содержит от 1 до 24 атомов углерода, причем в настоящем изобретении предпочтительны группы, содержащие 10 или меньше атомов углерода. "Низший алкил" или "низший алкилен" представляет собой алкильную или алкиленовую группу с более короткой цепью и обычно содержит 4 или меньше атомов углерода. Аналогичным образом "алкенилен" и "алкинилен" означают ненасыщенные формы "алкиленов", содержащие двойные или тройные связи, соответственно.
В настоящем изобретении волнистая линия  , пересекающая простую, двойную или тройную связь в любой химической структуре, представленной здесь, обозначает точку прикрепления простой, двойной или тройной связи к остальной части молекулы.
, пересекающая простую, двойную или тройную связь в любой химической структуре, представленной здесь, обозначает точку прикрепления простой, двойной или тройной связи к остальной части молекулы.
Термины "алкокси", "алкиламино" и "алкилтио" (или тиоалкокси) применяются в их обычном смысле и обозначают такие алкильные группы, которые присоединяются к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Кроме того, в отношении диалкиламиногрупп, алкильные части могут быть одинаковыми или разными и могут объединяться с образованием 3-7-членного кольца с атомом азота, к которому они присоединены. Соответственно, группа, представленная в виде -NRaRb, может означать пиперидинил, пирролидинил, морфолинил, азетидинил и др.
Термины "гало" или "галоген", сами по себе или как часть другого заместителя, означают, если не указано иначе, атом фтора, хлора, брома или йода. Кроме того, такие термины, как "галогеналкил", также включают моногалогеналкилы и полигалогеналкилы. Например, термин "C1-4-галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.д.
Термин "арил" означает, если не указано иное, полиненасыщенную, как правило, ароматическую, углеводородную группу, которая может представлять собой одно кольцо или несколько колец (до трех колец), которые конденсированы вместе или связаны ковалентно. Термин "гетероарил" относится к таким арильным группам (или кольцам), которые содержат от 1 до 5 гетероатомов из числа N, О и S, причем атомы азота и серы необязательно окислены, а атомы азота необязательно четвертичные. Гетероарильная группа может присоединяться к остальной части молекулы через гетероатом. Неограничительные примеры арильных групп включают фенил, нафтил и бифенил, а неограничительные примеры гетероарильных групп включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолинил, фталазинил, бензотриазинил, пуринил, бензимидазолил, бензопиразолил, бензотриазолил, бензизоксазолил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридины, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Заместители для каждого из вышеприведенных арильных и гетероарильных кольцевых систем выбирают из числа приемлемых заместителей, описанных ниже.
Термин "арилалкил" охватывает такие радикалы, у которых к алкильной группе присоединена арильная группа (например, бензил, фенетил и т.п.). Аналогичным образом, термин "гетероарилалкил" охватывает такие радикалы, у которых к алкильной группе присоединена гетероарильная группа (например, пиридилметил, тиазолилэтил и т.п.).
Вышеприведенные термины (например, "алкил", "арил" и "гетероарил"), в некоторых воплощениях, охватывают как замещенные, так и незамещенные формы указанного радикала. Предпочтительные заместители для каждого типа радикалов приведены ниже.
Заместителями для алкильных радикалов (включая те группы, которые часто называют алкиленами, алкенилами, алкинилами и циклоалкилами) могут быть различные группы, выбранные из: галогена, -OR', -NR'R'', -SR', -SiR'R''R''', -OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', -NR'-C(O)NR''R''', -NR''C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NR'S(O)2R'', -CN и -NO2 в количестве от нуля до (2m'+l), где m' - общее число атомов углерода в таком радикале. R', R'' и R''' независимо друг от друга означают водород, незамещенный C1-8-алкил, незамещенный арил, арил, замещенный 1-3 галогенами, незамещенный C1-8-алкил, C1-8-алкокси или C1-8-тиоалкокси или незамещенный арил-C1-4-алкил. Когда R' и R'' присоединяются к одному и тому же атому азота, то они могут объединяться с этим атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца. Например, -NR'R'' включает в себя 1-пирролидинил и 4-морфолинил.
Точно так же заместители для арильных и гетероарильных групп могут быть различными и обычно выбираются из: галогена, -OR', -OC(O)R', -NR'R'', -SR', -R'', -CN, -NO2, -CO2R', -CONR'R'', -C(O)R', -OC(O)NR'R'', -NR''C(O)R', -NR''C(O)2R', -NR'-C(O)NR''R''', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NR'S(O)2R'', -N3, перфтор(С1-С4)алокси и перфтор(С1-С4)алкила, в количестве от нуля до общего числа открытых валентностей в ароматической кольцевой системе; причем R', R'' и R''' выбираются независимо из водорода, C1-8-алкила, C1-8-галогеналкила, С3-6-циклоалкила, С2-8-алкенила, С2-8-алкинила, незамещенного арила и гетероарила, незамещенного арил-С1-4-алкила и незамещенного арилокси-С1-4-алкила. Другими подходящими заместителями являются каждые из вышеприведенных арильных заместителей, присоединенные к атому кольца цепочкой алкилена из 1-4 атомов углерода.
Два заместителя на соседних атомах арильного или гетероарильного кольца необязательно могут быть заменены на заместители формулы -T-C(O)-(CH2)q-U-, где Т и U независимо означают -NH-, -О-, -СН2- или простую связь, a q - целое число от 0 до 2. Кроме того, два заместителя на соседних атомах арильного или гетероарильного кольца необязательно могут быть заменены на заместители формулы -А-(СН2)r-В-, где А и В независимо означают -СН2-, -О-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR'- или простую связь, а R - целое число от 1 до 3. Одна из простых связей в образовавшемся при этом новом кольце необязательно может быть заменена на двойную связь. Кроме того, два заместителя на соседних атомах арильного или гетероарильного кольца необязательно могут быть заменены на заместители формулы -(CH2)s-X-(CH2)t-, где s и t независимо означают целые числа от 0 до 3, а X означает -О-, -NR'-, -S-, -S(O)-, -S(O)2-, или -S(O)2NR'-. Заместитель R' в -NR'- и -S(O)2NR'- выбирают из водорода или незамещенного C1-6-алкила.
В настоящем изобретении термин "гетероатом" охватывает кислород (О), азот (N), серу (S) и кремний (Si).
В настоящем изобретении термины "клетки-предшественники" и "стволовые клетки" применяются взаимозаменяемым образом. "Клетки-предшественники" и "стволовые клетки" означают такие клетки, которые, в ответ на определенные раздражители, могут образовывать дифференцированные клеточные линии, в том числе, но без ограничения, гемопоэтические, мезенхимальные, эпителиальные, нейрональные, почечные или миелоидные клетки. Наличие предшественников/стволовых клеток можно определить по способности клеток в образце к образованию колониеобразующих единиц различного типа, включая, к примеру, CFU-GM (колониеобразующие единицы гранулоцитов-макрофагов); CFU-GEMM (мультипотентные колониеобразующие единицы); BFU-E (бурстобразующие единицы, эритроидные); HPP-CFC (колониеобразующие клетки с высоким пролиферативным потенциалом); или другие типы дифференцированных колоний, которые могут быть получены в культуре по известным методикам. Гемопоэтические предшественники/стволовые клетки зачастую положительны на CD34. Однако некоторые стволовые клетки не содержат этого маркера. Такие клетки CD34+ можно определить методом активируемой флуоресценцией сортировки клеток (FACS), так что их наличие в образце можно установить этим методом. С другой стороны, такие клетки можно проанализировать методом FACS на присутствие рецепторов c-kit (CD117), отсутствие линиеспецифичных маркеров (например, CD2, CD3, CD4, CD5, CD8, NK1.1, В220, TER-119 и Gr-1 у мышей и CD3, CD14, CD16, CD19, CD20 и CD56 у человека).
Термин "фармацевтически приемлемые соли" охватывает соли активных соединений, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей, находящихся на описанных здесь соединениях. Когда соединения настоящего изобретения содержат сравнительно кислые функциональные группы, то можно получить соли с основаниями путем контактирования нейтральной формы таких соединений с достаточным количеством нужного основания, в неразбавленном виде либо в подходящем инертном растворителе. Примеры солей из фармацевтически приемлемых неорганических оснований включают соли алюминия, аммония, кальция, меди, железа, лития, магния, марганца, калия, натрия, цинка и др. Соли фармацевтически приемлемых органических оснований включают соли первичных, вторичных и третичных аминов, в том числе замещенных аминов, циклических аминов, аминов природного происхождения и др., как-то аргинина, бетаина, кофеина, холина, N,N'-дибензилэтилендиамина, диэтиламина, 2-диэтиламинэтанола, 2-диметиламинэтанола, этаноламина, этилендиамина, N-этилморфолина, N-этилпиперидина, глюкамина, глюкозамина, гистидина, гидрабамина, изопропиламина, лизина, метилглюкамина, морфолина, пиперазина, пиперидина, полиаминовых смол, прокаина, пуринов, теобромина, триэтиламина, триметиламина, трипропиламина, трометамина и др. Когда соединения по настоящему изобретению содержат сравнительно основные функциональные группы, то можно получить соли с кислотами путем контактирования нейтральной формы таких соединений с достаточным количеством нужной кислоты, в неразбавленном виде либо в подходящем инертном растворителе. Примеры солей с фармацевтически приемлемыми кислотами включают соли таких неорганических кислот, как соляная, бромистоводородная, азотная, угольная, однозамещенная угольная, фосфорная, однозамещенная фосфорная, двухзамещенная фосфорная, серная, однозамещенная серная, йодистоводородная или фосфорная кислота и др., а также соли таких сравнительно нетоксичных органических кислот, как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, пробковая, фумаровая, миндальная, фталевая, бензолсульфоновая, п-толилсульфоновая, лимонная, винная, метансульфоновая и др. К ним же относятся и соли таких аминокислот, как аргинат и др., и соли таких органических кислот, как глюкуроновая или галактуроновая кислота и др. (например, см. Berge S.M. et al, "Pharmaceutical Salts", Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые конкретные соединения настоящего изобретения содержат и основные, и кислые функциональные группы, что позволяет преобразовывать эти соединения в соли как оснований, так и кислот.
Нейтральные формы соединений можно регенерировать путем контактирования соли с основанием или кислотой и выделения исходного соединения обычным способом. Исходная форма соединения отличается от различных солевых форм определенными физическими свойствами, такими как растворимость в полярных растворителях, но в остальном соли эквивалентны исходной форме соединения в целях настоящего изобретения.
Наряду с солевыми формами, настоящим изобретением предусмотрены соединения, которые находятся в пролекарственной форме. Пролекарственные формы описанных здесь соединений представляет собой такие соединения, которые легко претерпевают химические изменения в физиологических условиях, давая соединения настоящего изобретения. Кроме того, пролекарства могут быть превращены в соединения настоящего изобретения химическими или биохимическими методами в условиях ex vivo. Например, пролекарства могут медленно превращаться в соединения настоящего изобретения, если их поместить в резервуар трансдермального пластыря с подходящим ферментом или химическим реагентом.
Некоторые соединения настоящего изобретения могут существовать в несольватированных формах, наряду с сольватированными формами, включая гидратированные формы. В общем, сольватированные формы эквивалентны несольватированным формам и входят в рамки настоящего изобретения. Некоторые соединения настоящего изобретения могут существовать в нескольких кристаллических или аморфных формах. В общем, все физические формы эквивалентны для применений, предусмотренных настоящим изобретением, и входят в рамки настоящего изобретения.
Некоторые соединения настоящего изобретения содержат асимметрические атомы углерода (оптические центры) или двойные связи; в рамки настоящего изобретения входят рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры). В некоторых воплощениях, соединения по изобретению присутствуют в энантиомерно обогащенном виде, причем степень энантиомерного избытка для конкретного энантиомера рассчитывается известными методами. Получение энантиомерно обогащенных форм также хорошо известно в данной области и может осуществляться, к примеру, путем хирального разделения методом хроматографии или путем образования хиральной соли. Кроме того, настоящим изобретением предусмотрены различные конформеры, а также различные ротамеры. Конформеры представляют собой конформационные изомеры, которые могут отличаться вращением по одной или нескольким σ-связям. Ротамеры представляют собой конформеры, которые отличаются вращением только по одной σ-связи. Более того, соединения настоящего изобретения также могут содержать неестественные пропорции атомных изотопов по одному или нескольким атомам, входящим в состав таких соединений. Соответственно, в некоторых воплощениях соединения по изобретению присутствуют в изотопно обогащенном виде. Неестественные пропорции изотопа можно определить в пределах от количества, встречающегося в природе, до количества, составляющего 100% данного атома. Например, соединения могут включать радиоактивные изотопы, такие, к примеру, как тритий (3Н), йод-125 (125I) или углерод-14 (14С), или нерадиоактивные изотопы, такие как дейтерий (2Н) или углерод-13 (13С). Такие изотопные варианты могут обеспечить дополнительное применение к уже описанным в настоящей заявке. Например, изотопные варианты соединений по изобретению могут найти дополнительное применение, в том числе, но без ограничения, в качестве диагностических реагентов и/или реагентов для визуализации, либо в качестве цитотоксических/радиотоксических терапевтических средств. Кроме того, изотопные варианты соединений по изобретению могут обладать измененными фармакокинетическими и фармакодинамическими характеристиками, что может способствовать повышению безопасности, переносимости и эффективности во время лечения. Все изотопные варианты соединений настоящего изобретения, будь то радиоактивные или нет, входят в рамки настоящего изобретения.
"CXCR7", который также именуется "RDC1" или "CCXCKR2", означает предположительно связанный с G-белком рецептор (GPCR) из семи трансмембранных доменов. Первоначально был идентифицирован ортолог CXCR7 у собак в 1991 году. См. Libert et al., Science 244:569-572 (1989). Последовательность собаки описана в Libert et al., Nuc. Acids Res. 18(7):1917 (1990). Последовательность мыши описана, например, в Heesen et al., Immunogenetics 47:364-370 (1998). Последовательность человека описана, например, в Sreedharan et al., Proc. Natl. Acad. Sci. USA 88:4986-4990 (1991), где по ошибке белок описан как рецептор вазоактивного кишечного пептида. "CXCR7" включает последовательности, которые существенно близки или являются консервативно модифицированными вариантами SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 9 или SEQ ID NO: 10.
Общие сведения
Соединения настоящего изобретения могут ингибировать связывание лигандов с рецептором CXCR7 и применимы при лечении различных заболеваний, в том числе рака, особенно твердых раковых опухолей и лимфом. Совсем недавно было отмечено, что ингибирование связывания лигандов с CXCR7 уменьшает тяжесть ревматоидного артрита на модели животных.
Специалистам в данной области должно быть понятно, что средства, которые модулируют активность CCX-CKR2 (активность CXCR7), можно комбинировать в схемах лечения с другими антиангиогенными средствами и/или химиотерапевтическими средствами или облучением и/или другими средствами против артрита. В некоторых случаях доза химиотерапевтического средства или облучения была бы субтерапевтической, если бы она назначалась без комбинации с антиангиогенным средством. Специалистам в данной области должно быть известно, что "комбинации" могут включать в себя комбинации при лечении (т.е. два или несколько препаратов могут вводиться в виде смеси или по крайней мере одновременно или же вводиться субъекту в разные моменты времени, но таким образом, чтобы оба они попадали в кровоток субъекта в одно и то же время). Кроме того, композиции настоящего изобретения могут вводиться до или после второго терапевтического режима, например, до или после проведения химиотерапии или облучения.
Воплощения изобретения
А. Соединения
Настоящим изобретением, в одном аспекте, предусмотрены соединения по формуле I:
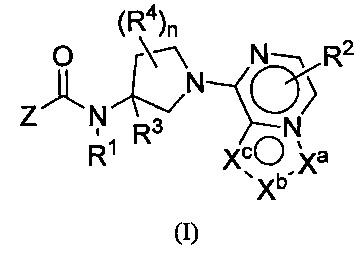
либо их фармацевтически приемлемые соли, гидраты, N-оксиды, изотопно обогащенные или энантиомерно обогащенные варианты и ротамеры. В формуле I каждая из вершин кольца Xa, Xb и Xc независимо выбрана из N, NH, N(R2), О, СН и C(R2). Кроме того, нижний индекс п означает 0, 1 или 2. Буква Z означает радикал, выбранный из:
(i) моноциклического или конденсированного бициклического арила и гетероарила, причем гетероарил содержит в кольце 1-4 гетероатома, выбранных из N, О и S; а данные арильные и гетероарильные группы необязательно замещены 1-5 заместителями R5;
(ii) моноциклического 4-, 5-, 6- или 7-членного кольца, выбранного из группы, состоящей из циклоалкана и гетероциклоалкана, причем гетероциклоалкановые кольца содержат в кольце 1-3 гетероатома, выбранных из N, О и S; а каждое из моноциклических колец Z необязательно замещено 1-3 заместителями R5;
R1 означает Н или C1-8-алкил, причем алкил необязательно замещен галогеном, -NRaRb, -ORa, -CO2Ra или -CONRaRb;
каждый R2 независимо означает Н, галоген, CN, C1-8-алкил, С1-8-галоалкил, C1-8-гидроксиалкил, -ORa, -CO2Ra, -X-CO2Ra, -NRaRb, -CONRaRb или -X-CONRaRb;
R3 означает H, C1-8-алкил, С1-8-галоалкил, C1-8-гидроксиалкил, -CO2Ra, -X-CO2Ra, -CONRaRb или -X-CONRaRb;
каждый R4, если он есть, независимо означает С1-8-алкил, С1-8-галоалкил, С1-8-гидроксиалкил, -ORa, -CO2Ra, -X-CO2Ra, -NRaRb, -CONRaRb или -X-CONRaRb;
каждый R5 независимо означает галоген, CN, -X-CN, C1-8-алкил, С3-8-циклоалкил, С3-8-циклоалкенил, С3-5-спироциклоалкил, С2-8-алкенил, С2-8-алкинил, C1-8-галоалкил, С1-8-гидроксиалкил, -ORa, -CO2Ra, -X-CO2Ra, -NRaRb, -CONRaRb, -X-CONRaRb, арил, 5- или 6-членный гетероарил или 3-, 4-, 5- или 6-членный гетероцикл, причем гетероатомы на вершинах колец гетероарила и гетероцикла выбраны из N, О и S, а арильные, гетероарильные и гетероциклические части R5 необязательно дополнительно замещены 1-3 Ra;
каждый Ra и Rb независимо означает водород, гидроксил, галоген, циано, C1-8-алкил, C1-8-алкокси, C1-8-галоалкил, С3-6-циклоалкил, С3-6-циклоалкилалкил, амино, C1-8-алкиламино, ди-C1-8-алкиламино, карбоксамид, карбоксиэфир С1-4-алкила, карбоксил или -SO2-С1-8-алкил;
каждый X означает соединительную группу С1-4-алкилена или соединительную группу по формуле -(СН2)mO(СН2)p-, где нижние индексы m и p означают целые числа от 0 до 5, a m+p составляет от 0 до 6, причем любая из метиленовых частей X необязательно замещена одной или двумя метальными группами. В одной группе воплощений каждый X независимо означает -ОСН2-, -ОСН2СН2-, -ОСН2СН2СН2-, -ОС(СН3)2-, -ОСН2С(СН3)2-, -ОСН2СН2С(СН3)2-, -СН2-, -С(СН3)2- или -СН2СН2-. В другой группе воплощений каждый X означает -О-, -СН2-, -ОСН2-, -ОСН2СН2-, -С(СН3)2- или -СН2СН2-.
В настоящем изобретении представлен целый ряд воплощений.
(A) В одной группе воплощений Z означает моноциклический или конденсированный бициклический гетероарил, содержащий в кольце 1-3 гетероатома, выбранных из N, О и S; причем данный гетероарил необязательно замещен 1-5 заместителями R5.
(B) В другой группе воплощений Z означает моноциклический или конденсированный бициклический гетероарил, выбранный из группы, состоящей из имидазола, пиразола, 1,2,3-триазола, 1,2,4-триазола, тетразола, тиазола, оксазола, оксадиазола, пиримидина, пиразина, пиридазина и хиназолина, каждый из которых необязательно замещен 1-2 заместителями R5.
(C) В еще одной группе воплощений Z означает 5-членный гетероарил, замещенный одной группой R5, выбранной из необязательно замещенного кольца арила, гетероарила, циклоалкила и гетероциклоалкила, и необязательно еще 1-2 дополнительными группами R5, выбранными из галогена, C1-4-алкила, С1-4-галоалкила и CH2CN.
(D) В других воплощениях Z выбирается из:
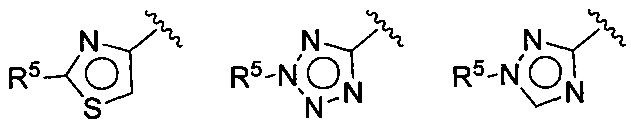

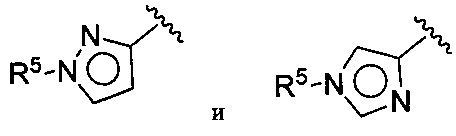
где R5 имеет значения, приведенные выше в отношении формулы I.
(Е) В следующей группе воплощений Z у соединений формулы I означает
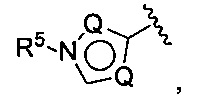
где каждый Q выбирается независимо из группы, состоящей из N, СН и C(R5), a R5 имеет значения, приведенные выше в отношении формулы I.
В рамках воплощений, представленных от (А) до (Е) или со ссылкой на формулу I, существуют и другие выбранные воплощения:
(1) в одной группе воплощений n равно нулю;
(2) в другой группе воплощений R1 означает Н.
В следующей группе воплощений соединения формулы I представлены формулой Ia:
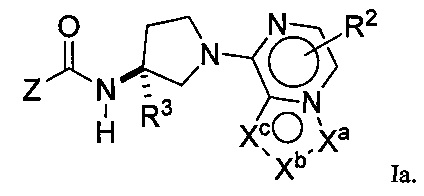
Выбранные воплощения формулы Ia включают каждое из воплощений для Z, приведенных выше от (А) до (Е).
(F) В одной конкретной группе воплощений формулы I или Ia бициклическая часть, содержащая Xa, Xb и Xc на вершинах кольца, выбирается из:
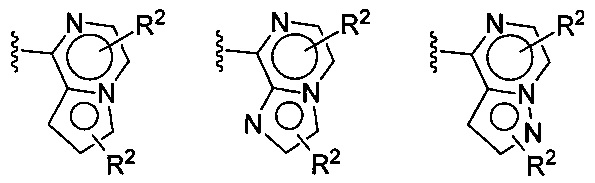
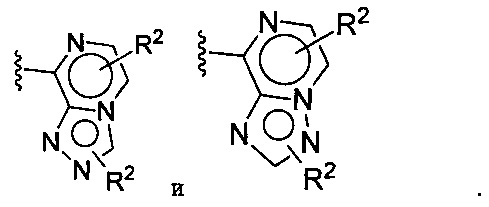
(G) В другой конкретной группе воплощений формулы Ia бициклическая часть, содержащая Xa, Xb и Xc на вершинах кольца, выбирается из:

(Н) В следующей конкретной группе воплощений формулы Ia бициклическая часть, содержащая Xa, Xb и Xc на вершинах кольца, выбирается из:
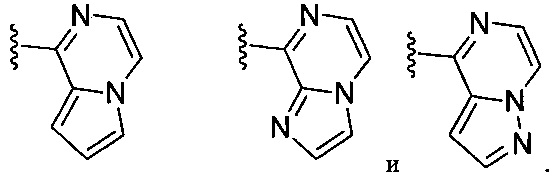
(I) В следующей конкретной группе воплощений формулы Ia бициклическая часть, содержащая Xa, Xb и Xc на вершинах кольца, выбирается из:

(J) В следующей конкретной группе воплощений формулы Ia бициклическая часть, содержащая Xa, Xb и Xc на вершинах кольца, выбирается из:
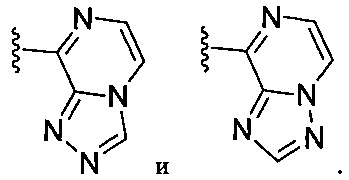
В некоторых отдельных воплощениях Z у соединений формулы Ia и воплощений, приведенных в (F), (G), (Н), (I) и (J), выбирают из воплощений, приведенных выше от (А) до (Е), особенно тех, где Z означает 5-членный гетероарил, замещенный одной группой R5, выбранной из необязательно замещенного кольца арила, гетероарила, циклоалкила или гетероциклоалкила, и необязательно еще 1-2 дополнительными группами R5, выбранными из галогена, С1-4-алкила, С1-4-галоалкла и CH2CN.
В следующей конкретной группе воплощений формулы I или Ia и воплощений, приведенных в (F), (G), (Н), (I) и (J), Z выбирают из группы, состоящей из:
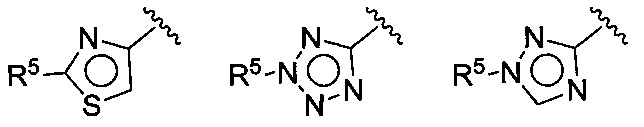
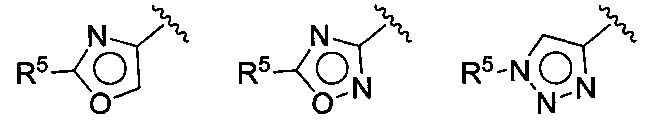
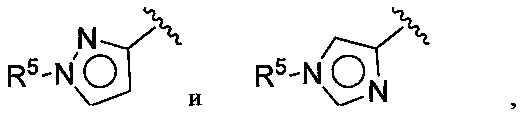
где R5 имеет значения, приведенные выше в отношении формулы I.
В следующей конкретной группе воплощений формулы I или Ia и воплощений, приведенных в (F), (G), (Н), (I) и (J), Z соответствует формуле:
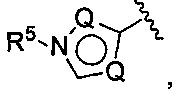
где каждый Q независимо выбран из группы, состоящей из N, СН и C(R5).
В одной конкретной группе воплощений соединения соответствуют формуле:
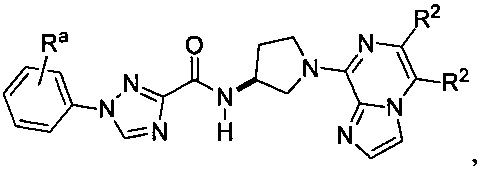
где Ra и каждый R2 имеют значения, приведенные выше в отношении формулы I.
В рамках воплощений, представленных от (А) до (J), а также воплощений, представляющих собой комбинации (к примеру, (А) и (F); (В) и (G); (А) и (Н); и т.д.), существуют и другие выбранные воплощения:
(a) в которых n=0;
(b) в которых n=0, a R1 означает Н или метил;
(c) в которых n=0, a R1 означает Н или метил, R2 означает Н или C1-8-алкил и R3 означает водород;
(d) в которых n=0, а каждый из R2 и R3 означает водород;
(e) в которых n=0, каждый R2 означает водород, a R3 выбран из группы, состоящей из метила, этила, -CONH2 и -СН2ОН;
(f) в которых каждый R2 означает водород.
Специалистам в данной области должно быть понятно, что конкретные воплощения изобретения представлены такими соединениями формулы I или Ia, у которых признаки соединений дополнительно определяются комбинациями воплощений, в том числе (А)+(F); (А)+(G); (А)+(В); (А)+(I); и (А)+(J); каждое из которых, в других выбранный воплощениях, независимо комбинируется с каждым из выбранных воплощений от (а) до (f). Точно так же, выбранные соединения формулы I или Ia представлены такими соединениями, у которых признаки соединений дополнительно определяются комбинациями воплощений, в том числе (В)+(F); (А)+(G); (А)+(Н); (А)+(I); и (В)+(J); каждое из которых, в других выбранный воплощениях, независимо комбинируется с каждым из выбранных воплощений от (а) до (f). Другие выбранные соединения формулы I или Ia представлены такими соединениями, у которых признаки соединений дополнительно определяются комбинациями воплощений, в том числе (С)+(F); (С)+(G); (С)+(Н); (С)+(I); и (С)+(J); каждое из которых, в других выбранный воплощениях, независимо комбинируется с каждым из выбранных воплощений от (а) до (f). Еще другие выбранные соединения формулы I или Ia представлены такими соединениями, у которых признаки соединений дополнительно определяются комбинациями воплощений, в том числе (D)+(F); (D)+(G); (D)+(Н); (D)+(I); и (D)+(J); каждое из которых, в других выбранный воплощениях, независимо комбинируется с каждым из выбранных воплощений от (а) до (f). А еще другие выбранные соединения формулы I или Ia представлены такими соединениями, у которых признаки соединений дополнительно определяются комбинациями воплощений, в том числе (Е)+(F); (Е)+(G); (Е)+(Н); (Е)+(I); и (Е)+(J); каждое из которых, в других выбранный воплощениях, независимо комбинируется с каждым из выбранных воплощений от (а) до (f).
В одной выбранной группе воплощений соединения выбирают из числа тех, что представлены в приведенных ниже примерах или в таблице 1.
В каждом из выбранных воплощений данные соединения могут присутствовать в виде фармацевтически приемлемой соли или в виде гидрата.
Более того, для тех соединений, которые представлены выше без стереохимии, настоящее изобретение также касается хиральных форм каждого из этих соединений, а также энантиомерно обогащенных форм данных соединений. Энантиомерно обогащенные формы могут быть получены с помощью хиральной хроматографии по хорошо известным методикам, практикуемым в данной области, или же, к примеру, путем хирального разделения с помощью хиральной формы соли. В некоторых воплощениях энантиомерный избыток энантиомерно обогащенной формы составляет по меньшей мере 10%, 20%, 30%, 40%, 50%, 60% или больше. В других воплощениях энантиомерно обогащенная форма составляет по меньшей мере 70%, 80%, 90%, 95% или больше.
Получение соединений
Некоторые соединения по изобретению могут быть получены в соответствии с методиками, описанными в разделе «Примеры» настоящего документа. Кроме того, также описан синтез некоторых промежуточных соединений, которые могут применяться при получении соединений по изобретению.
В. Композиции
Наряду с приведенными выше соединениями, композиции для модуляции активности CXCR7 у человека и животных, как правило, содержат фармацевтический носитель или разбавитель.
Термин "композиция" в настоящем изобретении охватывает препараты, содержащие определенные ингредиенты в определенных количествах, а также препараты, которые прямо или косвенно получаются при сочетании определенных ингредиентов в определенных количествах. Под "фармацевтически приемлемым" понимается то, что носитель, разбавитель или наполнитель должен быть совместимым с другими ингредиентами лекарственной формы и не быть вредным для получателя.
Фармацевтические композиции для введения соединений настоящего изобретения для удобства могут быть представлены в стандартной дозовой форме и могут быть получены любым из способов, хорошо известных в области фармацевтики и введения лекарств. Все способы включают в себя стадию приведения активного ингредиента в ассоциацию с носителем, который составляет один или несколько вспомогательных ингредиентов. В общем, фармацевтические композиции получают путем равномерного и непосредственного смешивания активного ингредиента вместе с жидким носителем или мелко измельченным твердым носителем или тем и другим, а затем, если необходимо, формования продукта в нужную лекарственную форму. В фармацевтической композиции соединение активного начала включается в количестве, достаточном для получения требуемого эффекта на процесс или состояние заболевания.
Фармацевтические композиции, содержащие активный ингредиент, могут иметь вид, подходящий для перорального применения, к примеру, в виде таблеток, пастилок, лепешек, водных или масляных суспензий, диспергирующихся порошков или гранул, эмульсий и самоэмульгирующихся препаратов, как описано в U.S. Patent Application 2002-0012680, твердых или мягких капсул, сиропов, эликсиров, растворов, буккальных пэтчей, перорального геля, жвачки, жевательных таблеток, шипучих порошков и шипучих таблеток. Композиции, предназначенные для перорального введения, могут быть получены любым способом, известным в области изготовления фармацевтических композиций, причем такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей, антиоксидантов и консервантов, с тем, чтобы получить фармацевтически элегантные и приятные на вкус препараты. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, пригодными для изготовления таблеток. Эти наполнители могут представлять собой, к примеру, инертные разбавители, такие как целлюлоза, диоксид кремния, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие средства, к примеру, кукурузный крахмал или альгиновая кислота; связующие вещества, к примеру, PVP, целлюлоза, PEG, крахмал, желатин или гуммиарабик, и смазывающие вещества, к примеру, стеарат магния, стеариновая кислота или тальк. Таблетки могут быть без оболочки или покрыты оболочкой, энтеральной или иной, известными методами, чтобы замедлить распадаемость и всасывание в желудочно-кишечном тракте и тем самым обеспечить пролонгированное действие на длительный период. Например, можно использовать материал для пролонгирующего действия типа глицерилмоностеарата или глицерилдистеарата. На них также можно наносить оболочки методами, описанными в U.S. Pat. Nos. 4,256,108; 4,166,452; и 4,265,874, с получением осмотических лечебных таблеток для контролируемого высвобождения.
Лекарственные формы для перорального применения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, к примеру, карбонатом кальция, фосфатом кальция или каолином, либо в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляной средой, к примеру, арахисовым маслом, вазелиновым маслом или оливковым маслом. Кроме того, можно получить эмульсии с несмешивающимся с водой ингредиентом типа масла и стабилизированные поверхностно-активными веществами типа моно- или диглицеридов, сложных эфиров ПЭГ и др.
Водные суспензии содержат активные вещества в смеси с наполнителями, подходящими для приготовления водных суспензий. Такими наполнителями являются суспендирующие вещества, к примеру, натриевая карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинил-пирролидон, трагакантовая камедь и гуммиарабик; диспергирующие или увлажняющие средства, которыми могут быть природные фосфатиды, например, лецитин, или продукты конденсации алкиленоксидов с жирными кислотами, к примеру, полиоксиэтиленстеарат, или же продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, к примеру, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами жирных кислот и гекситола типа полиоксиэтиленсорбитол-моноолеата, или продукты конденсации этиленоксида с частичными сложными эфирами жирных кислот и ангидридов гекситола типа полиэтиленсорбитан-моноолеата. Водные суспензии также могут содержать один или несколько консервантов, к примеру, этил- или н-пропил-пара-гидроксибензоат, один или несколько красителей, один или несколько ароматизаторов и один или несколько подсластителей типа сахарозы или сахарина.
Масляные суспензии могут быть получены путем суспендирования активного ингредиента в растительном масле, к примеру, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, либо в минеральном масле типа вазелинового масла. Масляные суспензии могут содержать загуститель, к примеру, пчелиный воск, твердый парафин или цетиловый спирт. Могут быть добавлены подсластители типа приведенных выше и ароматизаторы, чтобы получить пероральный препарат приятного вкуса. Такие композиции могут консервироваться добавлением антиоксиданта типа аскорбиновой кислоты.
Диспергируемые порошки и гранулы, подходящие для получения водных суспензий при добавлении воды, содержат активный ингредиент в смеси с диспергирующим или увлажняющим средством, суспендирующим средством и одним или несколькими консервантами. Подходящие диспергирующие или увлажняющие средства и суспендирующие средства представлены теми, что уже упоминались выше. Также могут присутствовать и другие наполнители, например, подсластители, ароматизаторы и красители.
Фармацевтические композиции настоящего изобретения также могут иметь вид эмульсий типа масло-в-воде. Масляная фаза может представлять собой растительное масло, к примеру, оливковое масло или арахисовое масло, либо минеральное масло, к примеру, вазелиновое масло, либо их смеси. Подходящими эмульгаторами могут служить природные смолы, к примеру, гуммиарабик или трагакантовая камедь, природные фосфатиды, к примеру, соевый лецитин, и сложные или неполные эфиры жирных кислот и ангидридов гекситола, к примеру, сорбитанмоноолеат, и продукты конденсации таких частичных сложных эфиров с этиленоксидом, к примеру, полиоксиэтиленсорбитол-моноолеат. Эмульсии также могут содержать подсластители и ароматизаторы.
Сиропы и эликсиры могут быть приготовлены с подсластителями, к примеру, глицерином, пропиленгликолем, сорбитом или сахарозой. Такие лекарственные формы также могут содержать средства, уменьшающие раздражение, консерванты, ароматизаторы и красители. Пероральные растворы могут быть приготовлены в комбинации, к примеру, с циклодекстрином, ПЭГ и поверхностно-активными веществами.
Фармацевтические композиции могут иметь вид стерильных водных или масляных суспензий для инъекций. Такие суспензии могут быть получены известными методами с использованием подходящих диспергирующих или увлажняющих средств и суспендирующих средств, приведенных выше. Стерильный препарат для инъекций также может представлять собой стерильный раствор для инъекций или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, к примеру, в виде раствора в 1,3-бутандиоле. В число приемлемых носителей и растворителей, которые можно использовать, входят вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно используют стерильные нелетучие масла. Для этого можно использовать любое слабое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при получении препаратов для инъекций применяются жирные кислоты типа олеиновой кислоты.
Соединения настоящего изобретения также можно вводить в виде свечей для ректального введения лекарственных средств. Такие композиции могут быть получены путем смешивания лекарственного средства с подходящим не вызывающим раздражения эксципиентом, который будет твердым при обычных температурах, но жидким при ректальной температуре и поэтому будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао и полиэтиленгликоли. Кроме того, соединения могут вводится прямо в глаза при помощи растворов или мазей. Более того, может осуществляться чрескожное введение рассматриваемых соединений при помощи ионтофоретических пластырей и т.п. Для местного применения используются кремы, мази, желе, растворы или суспензии и пр., содержащие соединения настоящего изобретения. В настоящем изобретении местное применение также включает в себя применение жидкостей для полоскания рта или горла.
Соединения настоящего изобретения также могут быть соединены с носителем типа подходящего полимера в качестве носителей для прицельной доставки лекарственных средств. Такие полимеры могут включать поливинилпирролидон, сополимеры пирана, полигидроксипропил-метакриламид-фенол, полигидроксиэтил-аспартамид-фенол или полиэтиленоксид-полилизин, замещенный пальмитоиловыми остатками. Кроме того, соединения по изобретению могут быть соединены с носителем из класса биоразлагаемых полимеров, применяемых для контролируемого высвобождения лекарственных средств, таким, к примеру, как полимолочная кислота, полигликолевая кислоты, сополимеры полимолочной и полигликолевой кислот, поли-ε-капролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатические блок-сополимеры гидрогелей. Из полимеров и полупроницаемых полимерных матриксов можно формовать профилированные изделия, такие как клапаны, стенты, трубки, протезы и др.
С. Способы применения
Не придерживаясь какой-либо определенной теории, полагаем, что соединения и композиции настоящего изобретения оказывают терапевтический эффект путем ингибирования связывания SDF-1 и/или I-TAC с рецептором CXCR7. Таким образом, соединения и композиции настоящего изобретения могут применяться при лечении или профилактике заболеваний у млекопитающих, при которых ингибирование связывания SDF-1 и/или I-TAC с рецептором CXCR7 обеспечит терапевтический эффект.
В одном воплощении предпочтительный способ ингибирования связывания хемокинов SDF-1 и/или I-TAC с рецептором CXCR7 включает контактирование одного или нескольких из вышеприведенных соединений с клетками, экспрессирующими рецептор CXCR7, в течение времени, достаточного для ингибирования связывания этих хемокинов с рецепторами CXCR7.
В некоторых воплощениях соединения и композиции по изобретению вводятся страдающим раком субъектам. В некоторых случаях вводятся модуляторы CXCR7 для лечения рака, например, карциномы, глиомы, мезотелиомы, меланомы, лимфомы, лейкемии (в том числе острой лимфобластной лейкемии), аденокарциномы, рака молочной железы, рака яичников, рака шейки матки, глиобластомы, лейкемии, лимфомы, рака простаты, лимфомы Беркитта, рака головы и шеи, рака толстой кишки, колоректального рака, немелкоклеточного рака легких, мелкоклеточного рака легких, рака пищевода, рака желудка, рака поджелудочной железы, гепатобилиарного рака, рака желчного пузыря, рака тонкой кишки, рака прямой кишки, рака почек, рака мочевого пузыря, рака предстательной железы, рака полового члена, рака мочеиспускательного канала, рака яичек, рака шейки матки, рака влагалища, рака матки, рака яичников, рака щитовидной железы, рака паращитовидной железы, рака надпочечников, рака эндокринной поджелудочной железы, карциноидов, рака костей, рака кожи, ретинобластомы, лимфомы Ходжкина, неходжкинской лимфомы (см. Cancer: Principles and Practice (DeVita V.T. et al., eds, 1997) насчет других раковых заболеваний); а также таких дисфункций мозга и нейронов, как болезнь Альцгеймера, рассеянный склероз и демиелинизирующие заболевания; таких гипертензивных заболеваний, как легочная артериальная гипертензия; дисфункции почек; ревматоидного артрита; отторжения аллотрансплантатов; атеросклероза (и повышенного уровня холестерина); астмы; гломерулонефрита; контактного дерматита; воспалительной болезни кишечника; колита; псориаза; реперфузионных повреждений; а также других описанных здесь заболеваний. В некоторых воплощениях у этих лиц нет саркомы Калоши, полицентрической болезни Кастлемана или связанной со СПИД первичной эффузионной лимфомы.
Настоящее изобретение также охватывает уменьшение ангиогенеза у нуждающихся в этом лиц путем введения соединений и композиций по изобретению. Например, снижение активности CXCR7 при контакте CXCR7 с соединениями по изобретению, а тем самым уменьшение ангиогенеза, применимо для ингибирования формирования, роста и/или метастазирования опухолей, особенно твердых опухолей. Описание воплощений, относящихся к модулированию CXCR7 и ангиогенеза, приводится, например, в U.S. Patent Application No. 11/050,345.
Другие заболевания с участием нежелательного или проблемного ангиогенеза включают ревматоидный артрит; псориаз; заболевания глазных кровеносных сосудов, например, диабетическая ретинопатия, ретинопатия при недоношенности, дегенерация желтого пятна, отторжение трансплантата роговицы, неоваскулярная глаукома, ретролентальная фиброплазия, покраснение радужной оболочки; синдром Ослера-Уэббера; ангиогенез миокарда; неоваскуляризация бляшек; телеангиэктазия; гемофилия суставов; ангиофиброма; заболевания с чрезмерной или аномальной стимуляцией эндотелиальных клеток, включая кишечные спайки, болезнь Крона, кожные заболевания, такие как псориаз, экзема и склеродермия, диабет, диабетическая ретинопатия, ретинопатия при недоношенности, возрастная дегенерация желтого пятна, атеросклероз, склеродермия, грануляция ран и гипертрофические рубцы, т.е. келоидные рубцы, а также заболевания, при которых ангиогенез является следствием патологии типа болезни кошачьих царапин и язвы (Helicobacter pylori), тоже можно лечить антителами по изобретению. Ингибиторы ангиогенеза могут применяться для предотвращения или ингибирования спайкообразования, особенно внутрибрюшинных или тазовых спаек типа возникающих после открытой или лапароскопической хирургии, и ожоговых стяжек. Другие заболевания, которые можно бы с успехом лечить с помощью ингибиторов ангиогенеза, включают предотвращение рубцевания после трансплантации, цирроз печени, фиброз легких после острого респираторного дистресс-синдрома или другого легочного фиброза у новорожденных, имплантацию временных протезов и спайки после операции между мозгом и твердой мозговой оболочкой. Эндометриоз, полипоз, гипертрофию сердца, а также ожирение тоже можно лечить путем ингибирования ангиогенеза. Такие заболевания могут включать увеличение размера или рост других типов нормальной ткани, как при фиброме матки, гипертрофии предстательной железы и амилоидозе. Соединения и композиции настоящего изобретения могут применяться профилактически или терапевтически для любого из описанных здесь заболеваний.
Снижение активности CXCR7 с помощью соединений и композиций настоящего изобретения также может применяться при профилактике неоваскуляризации для эффективного лечения многих заболеваний. Так, например, уменьшение ангиогенеза может применяться при лечении заболеваний кровеносных сосудов (например, гемангиомы и пролиферации капилляров внутри атеросклеротических бляшек), мышечных заболеваний (например, ангиогенеза миокарда, инфаркта миокарда или ангиогенеза в гладких мышцах), заболеваний суставов (например, артрита, суставной гемофилии и др.) и других заболеваний, связанных с ангиогенезом. А усиление ангиогенеза может способствовать ускорению различных физиологических процессов и лечению заболеваний, требующих усиленной васкуляризации, как-то заживления ран, переломов и ожогов, воспалительных заболеваний, ишемической болезни сердца и заболеваний периферических сосудов. Соединения настоящего изобретения также могут приносить пользу при заболеваниях, при которых ограничен нормальный кровоток, таких как легочная гипертензия.
Соединения и композиции настоящего изобретения также могут применяться для усиления заживления ран. Не намереваясь ограничивать изобретение определенным механизмом действия, возможно, что антагонизм CXCR7 позволяет эндогенным лигандам вместо этого связываться с рецепторами с меньшим сродством, тем самым способствуя усиленному заживлению ран. Например, SDF-1 связывается и с CXCR7, и с CXCR4, но с CXCR4 он связывается с меньшим сродством. Точно так же I-TAC связывается с CXCR3 с меньшим сродством, чем он связывается с CXCR7. Предотвращая связывание этих лигандов с CXCR7, антагонисты CXCR7 могут способствовать связыванию лигандов с другими рецепторами, тем самым повышая заживление ран. Таким образом, антагонизм CXCR7 для усиления заживления ран может опосредоваться другим механизмом, чем усиление заживления ран при стимуляции активности CXCR7 агонистами.
Помимо лечения заболеваний и симптомов, связанных с неоваскуляризацией, ингибирование ангиогенеза может применяться для модуляции или предотвращения возникновения нормальных физиологических состояний, связанных с неоваскуляризацией. Так, например, соединения и композиции могут применяться в качестве противозачаточного средства. В соответствии с настоящим изобретением, уменьшение активности CXCR7 в яичниках или эндометрии может ослабить неоваскуляризацию, связанную с овуляцией, имплантацией эмбриона, формированием плаценты и т.д.
Ингибиторы ангиогенеза имеют и другие терапевтические применения. Например, соединения и композиции настоящего изобретения могут применяться для следующего:
(a) удаления жировой ткани и лечения ожирения, например, см. Kolonin et al., Nature Medicine 10(6):625-632 (2004);
(b) лечения преэклампсии, например, см. Levine et al., N. Engl. J. Med. 350(7): 672-683 (2004); Maynard et al., J. Clin. Invest. 111 (5): 649-658 (2003); и
(c) лечения сердечно-сосудистых заболеваний, например, см. March et al., Am. J. Physiol. Heart Circ. Physiol. 287:H458-H463 (2004); Rehman et al., Circulation 109:1292-1298 (2004).
Способы лечения рака
В частности, настоящим изобретением также предусмотрен способ лечения рака. Предпочтительный способ лечения рака включает введение терапевтически эффективного количества одного или нескольких из вышеприведенных соединений (либо их солей) больным раком в течение времени, достаточного для лечения рака.
Для лечения композиции настоящего изобретения могут вводиться перорально, парентерально (например, внутримышечно, внутрибрюшинно, внутривенно, ICV, в виде интрацистернальной инъекции или инфузии, подкожной инъекции или имплантата), посредством ингаляции, назальным, вагинальным, ректальным, сублингвальным или местным путем введения и могут быть сформулированы, по отдельности или вместе, в подходящих составов единичных дозировок, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители, подходящие для каждого способа введения.
В некоторых воплощениях модуляторы CXCR7 по настоящему изобретению могут вводиться в комбинации с другими соответствующими терапевтическими средствами, включая, например, химиотерапевтические средства, облучение и т.д. Понятно, что такое введение может проводиться до, после или вместе со вторым терапевтическим средством с тем, чтобы терапевтические эффекты второго средства усиливались по сравнению с введением второго средства в отсутствие модулятора CXCR7. Выбор соответствующих средств для применения в комбинированной терапии может осуществляться рядовыми специалистами, в соответствии со стандартными фармацевтическими принципами. Комбинация терапевтических средств может действовать синергически при проведении лечения или профилактики различных заболеваний, таких, например, как рак, травмы, дисфункции почек, головного мозга или нейрональные дисфункции. При таком подходе можно достичь терапевтической эффективности при более низких дозах каждого из средств, тем самым снижая вероятность побочных эффектов.
Наряду с приматами, такими как человек, способом настоящего изобретению можно лечить множество других млекопитающих. Например, можно лечить млекопитающих, включая, без ограничения, коров, овец, коз, лошадей, собак, кошек, морских свинок, крыс и им подобных видов. Тем не менее, этот способ также может применяться и на других видах, таких как птицы (например, куры).
Стандартные методы анализа in vivo, показывающие, что композиции настоящего изобретения могут применяться для лечения рака, включают те, что описаны в Bertolini F. et al., Endostatin, an antiangiogenic drug, induces tumor stabilization after chemotherapy or anti-CD20 therapy in a NOD/SCID mouse model of human high-grade non-Hodgkin lymphoma. Blood, No. 1, Vol. 96, pp. 282-87 (1 July 2000); Pengnian L., Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase Tie2. Proc. Natl. Acad. Sci. USA, Vol. 95, pp. 8829-34 (July 1998); и Pulaski B. Cooperativity of Staphylococcal aureus enterotoxin В superantigen, major histocompatibility complex class II, and CD80 for immunotherapy of advanced spontaneous metastases in a clinically relevant postoperative mouse breast cancer model. Cancer Research, Vol. 60, pp. 2710-15 (May 15, 2000).
При лечении или профилактике заболеваний, требующих модуляции рецепторов хемокинов, надлежащий уровень дозировки обычно составляет от 0,001 до 100 мг на кг массы тела пациента в день, который можно вводить в виде одной или нескольких доз. Предпочтительно уровень дозировки составляет от 0,01 до 25 мг/кг в день; более предпочтительно от 0,05 до 10 мг/кг в сутки. Подходящий уровень дозировки может составлять от 0,01 до 25 мг/кг в сутки, от 0,05 до 10 мг/кг в сутки или от 0,1 до 5 мг/кг в сутки. В пределах этого диапазона дозировка может составлять от 0,005 до 0,05, от 0,05 до 0,5 или от 0,5 до 5,0 мг/кг в день. Для перорального введения композиции предпочтительно составляются в виде таблеток, содержащих от 1,0 до 1000 мг активного ингредиента, в частности 1,0, 5,0, 10,0, 15,0. 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 мг активного ингредиента для симптоматического подбора дозы для пациента, подлежащего лечению. Соединения можно вводить по схеме от 1 до 4 раз в день, предпочтительно один или два раза в день.
Однако следует иметь в виду, что конкретный уровень дозы и частота дозировки для любого конкретного пациента могут варьироваться и будут зависеть от различных факторов, в том числе от активности данного конкретного соединения, метаболической стабильности и продолжительности действия этого соединения, возраста, веса тела, наследственных признаков, общего состояния здоровья, пола и диеты субъекта, а также способа и времени введения, скорости выведения, комбинации лекарственных средств и тяжести конкретного заболевания у субъекта, проходящего лечение.
Соединения и композиции настоящего изобретения можно комбинировать с другими соединениями и композициями, обладающими аналогичным профилем, для профилактики и лечения рака и заболеваний, связанных с сигнализацией через CXCR7. Такие другие препараты могут вводиться таким же способом и в том же количестве, которое обычно применяется для этого, одновременно или последовательно с соединением или композицией настоящего изобретения. Если соединение или композиция настоящего изобретения применяется одновременно с одним или несколькими другими препаратами, то предпочтительны фармацевтические композиции, содержащие такие другие лекарственные средства в дополнение к соединению или композиции настоящего изобретения. Соответственно, фармацевтические композиции настоящего изобретения включают те, что содержат один или несколько других активных ингредиентов или терапевтических средств, наряду с соединением или композицией настоящего изобретения. Примеры таких лекарственных средств, которые можно комбинировать с соединением или композицией настоящего изобретения, при введении по отдельности или в одной и той же фармацевтической композиции, включают, без ограничения: цисплатин, паклитаксель, метотрексат, циклофосфамид, ифосфамид, хлорамбуцил, кармустин, карбоплатин, винкристин, винбластин, тиотепа, ломустин, семустин, 5-фторурацил и цитарабин. Соотношение масс соединения настоящего изобретения и второго активного ингредиента может варьироваться и будет зависеть от эффективной дозы каждого ингредиента. Как правило, используют эффективные дозы каждого из них. Так, например, если соединение настоящего изобретения применяется в сочетании со вторым противораковым средством, то соотношение масс соединения настоящего изобретения и второго средства обычно составляет от 1000:1 до 1:1000, предпочтительно от 200:1 до 1:200. Комбинации из соединения настоящего изобретения и других активных ингредиентов, как правило, тоже должны попадать в пределы вышеприведенного диапазона, но в каждом случае следует использовать эффективные дозы каждого активного ингредиента.
Способы лечения воспаления
Далее, соединения и композиции настоящего изобретения применимы для лечения воспаления и их можно комбинировать с другими соединениями и композициями, обладающими таким терапевтическим профилем, который может потребоваться для лечения до, после или одновременно с лечением рака или воспаления соединениями настоящего изобретения. Соответственно, составной частью настоящего изобретения также являются комбинированные способы и композиции для профилактики и лечения таких представляющих интерес заболеваний, как воспалительные или аутоиммунные заболевания, включая воспалительную болезнь кишечника, ревматоидный артрит, остеоартрит, псориатический артрит, полиартрит, рассеянный склероз, аллергические заболевания, псориаз, атопический дерматит и астма, а также приведенные выше патологии.
Например, при лечении или профилактике воспалительных или аутоиммунных заболеваний или, к примеру, артрита, связанного с потерей костной массы, соединения и композиции настоящего изобретения могут применяться в сочетании с такими противовоспалительными или анальгетическими средствами, как агонисты опиатов, ингибиторы липоксигеназы типа ингибиторов 5-липоксигеназы, ингибиторы циклооксигеназы типа ингибиторов циклооксигеназы-2, ингибиторы интерлейкина типа ингибиторов интерлейкина 1, антагонисты NMDA, ингибиторы оксида азота или ингибиторы синтеза оксида азота, нестероидные противовоспалительные средства или подавляющие цитокины противовоспалительные средства, например, с такими соединениями, как ацетаминофен, аспирин, кодеин, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидные анальгетики, суфентанил, сунлиндак, тенидап и др. Точно так же данные соединения и композиции можно вводить вместе с анальгетиками, перечисленными выше; потенцирующими средствами, такими как кофеин, Н2-антагонисты (например, ранитидин), симетикон, гидроксид алюминия или магния; противозастойными и противоотечными средствами, такими как фенилэфрин, фенилпропаноламин, псевдоэфедрин, оксиметазолин, адреналин, нафазолин, ксилометазолин, пропилгекседрин или лево-дезоксиэфедрин; средствами против кашля, такими как кодеин, гидрокодон, карамифен, карбетапентан или декстрометорфан; мочегонными; и седативными или не седативными антигистаминами.
Как уже отмечалось, соединения и композиции настоящего изобретения могут применяться в комбинации с другими лекарственными средствами, которые применяются для лечения, профилактики, подавления или облегчения заболеваний, при которых применимы соединения и композиции настоящего изобретения. Такие другие препараты могут вводиться таким же способом и в том же количестве, которое обычно применяется для этого, одновременно или последовательно с соединением или композицией настоящего изобретения. Если соединение или композиция настоящего изобретения применяется одновременно с одним или несколькими другими препаратами, то предпочтительны фармацевтические композиции, содержащие такие другие лекарственные средства в дополнение к соединению или композиции настоящего изобретения. Соответственно, фармацевтические композиции настоящего изобретения включают те, что содержат один или несколько других активных ингредиентов или терапевтических средств, наряду с соединением или композицией настоящего изобретения. Примеры таких лекарственных средств, которые можно комбинировать с соединением или композицией настоящего изобретения, при введении по отдельности или в одной и той же фармацевтической композиции, включают, без ограничения: (а) антагонисты VLA 4, (b) кортикостероиды, такие как беклометазон, бетаметазон, метилпреднизолон, преднизон, преднизолон, дексаметазон, флутиказон, гидрокортизон, будезонид, триамцинолон, сальметерол, сальбутамол, форметерол; (с) иммунодепрессанты, такие как циклоспорин (циклоспорин A, Sandimmune®, Neoral®), такролимус (FK-506, Prograf®), рапамицин (сиролимус, Rapamune®) и другие иммунодепрессанты типа FK-506 и рикофенолата, например, микофенолят мофетил (CellCept®); (d) антигистамины (H1-антагонисты гистамина), такие как бромфенирамин, хлорфенирамин, дексхлоифенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, пипрогептадин, антазолин, фенирамин, пириламин, астемизол, терфенадин, лоратадин, цетиризин, фексофенадин, дескарбоэтоксилоратадин и др., (е) нестероидные антиастматики (например, тербуталин, метапротеренол, фенотерол, изоэтарин, албутерол, битолтерол и пирбутерол), теофиллин, кромолин натрия, атропин, ипратропиум бромид, антагонисты лейкотриенов (например, зафлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст и SKB-106203), ингибиторы биосинтеза лейкотриенов (зилеутон, BAY-1005); (f) нестероидные противовоспалительные средства (NSAIDs), как-то производные пропионовой кислоты (например, альминопрофен, беноксапрофен, буклоксовая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (например, индометацин, ацеметацин, альклофенак, клиданак, диклофенак, фенклофенак, фенклозиновая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенамовой кислоты (например, флуфенамовая кислота, меклофенамовая кислота, мефенамовая кислота, нифлумовая кислота и толфенамовая кислота), производные бифенилкарбоновой кислоты (например, дифлунисал и флуфенисал), оксикамы (например, изоксикам, пироксикам, судоксикам и теноксикам), салицилаты (например, ацетилсалициловая кислота и сульфасалазин) и пиразолоны (например, апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон и фенилбутазон); (g) ингибиторы циклооксигеназы-2 (СОХ-2), такие как целекоксиб (Celebrex®) и рофекоксиб (Vioxx®); (h) ингибиторы фосфодиэстеразы IV типа (PDE IV); (i) соединения золота, такие как ауранофин и ауротиоглюкоза; (j) этанерцепт (Enbrel®); (k) лечебные антитела, такие как ортоклон (OKT3), даклизумаб (Zenapax®), базиликсимаб (Simulect®) и инфликсимаб (Remicade®); (l) другие антагонисты хемокиновых рецепторов, особенно CCR5, CXCR2, CXCR3, CCR2, CCR3, CCR4, CCR7, CX3CR1 и CXCR6; (m) смазывающие или смягчающие вещества, такие как вазелин и ланолин, (n) кератолитические средства (например, тазаротен), (о) производные витамина D3, например, кальципотриен или кальципотриол (Dovonex®); (p) PUVA; (q) антралин (Drithrocreme®); (r) этретинат (Tegison®) и изотретиноин; (s) лекарственные средства против рассеянного склероза, такие как интерферон β-1β (Betaseron®), интерферон (β-1α (Avonex®), азатиоприн (Imurek®, Imuran®), глатирамер ацетат (Capoxone®), глюкокортикоиды (например, преднизолон) и циклофосфамид; (t) DMARDS типа метотрексата; и (u) другие соединения, такие как 5-аминосалициловая кислота и ее пролекарственные формы; гидроксихлорохин; D-пенициламин; антиметаболиты типа азатиоприна, 6-меркаптопурина и метотрексата; ингибиторы синтеза ДНК типа гидроксимочевины, ингибиторы микротрубочек типа колхицина. Соотношение масс соединения настоящего изобретения и второго активного ингредиента может варьироваться и будет зависеть от эффективной дозы каждого ингредиента. Как правило, используют эффективные дозы каждого из них. Так, например, если соединение настоящего изобретения применяется в сочетании с NSAID, то соотношение масс соединения настоящего изобретения и NSAID обычно составляет от 1000:1 до 1:1000, предпочтительно от 200:1 до 1:200. Комбинации из соединения настоящего изобретения и других активных ингредиентов, как правило, тоже должны попадать в пределы вышеприведенного диапазона, но в каждом случае следует использовать эффективные дозы каждого активного ингредиента.
Способы индукции мобилизации предшественников/стволовых клеток
Далее, соединения и композиции настоящего изобретения могут применяться для мобилизации предшественников/стволовых клеток и тем самым для лечения или облегчения заболеваний, при которых мобилизация предшественников/стволовых клеток является эффективной или желательной, необязательно с использованием соединений настоящего изобретения в соответствии с процедурами и методиками, описанными в WO 05/000333, включенной сюда путем ссылки во всей полноте на все случаи. Заболевания, которые можно облегчить или иным образом пойти на пользу, включают, к примеру, гемопоэтические заболевания, такие как апластическая анемия, лейкемия, лекарственная анемия и гемопоэтический дефицит после химиотерапии или лучевой терапии. Кроме того, соединения и композиции по изобретению могут применяться для повышения успешности трансплантации во время и после лечения иммуносупрессорами, а также для более эффективного заживления ран и лечения бактериальных инфекций. Необязательно, после введения соединений по изобретению и после мобилизации предшественников/стволовых клеток, отбирают содержащую мобилизованные клетки кровь и необязательно очищают мобилизованные клетки и необязательно подвергают экспансии, а если нужно, то снова вводят тому же лицу или другому лицу (например, совместимому донору).
При желании можно мобилизировать самые разные типы клеток. В некоторых воплощениях после введения соединений или композиций по изобретению происходит мобилизация предшественников гемопоэтических клеток (HSCs), которые необязательно собирают и очищают от других компонентов крови. Необязательно мобилизация HSCs индуцируется путем введения по меньшей мере одного соединения по изобретению в сочетании с колониестимулирующим фактором гранулоцитов (G-CSF) и/или AMD3100 (1,1'-[1,4-фенилен-бис(метален)]-бис[1,4,8,11-тетраазациклотетрадекан]октогидробромид дигидрат) и/или их солями, рацематами или изомерами.
В некоторых воплощениях после введения соединений или композиций по изобретению происходит мобилизация предшественников эндотелиальных клеток (EPCs), которые необязательно собирают и очищают от других компонентов крови. Необязательно мобилизация EPCs индуцируется путем введения по меньшей мере одного соединения по изобретению в сочетании с фактором роста сосудистого эндотелия (VEGF) и/или агонистом VEGF (включая, без ограничения, антитела-агонисты VEGF) и/или AMD3100 и/или их солями, рацематами или изомерами.
В некоторых воплощениях после введения соединений или композиций по изобретению происходит мобилизация мезенхимальньгх стволовых клеток (MSCs) или предшественников стромальных клеток (SPCs), которые необязательно собирают и очищают от других компонентов крови. Необязательно такая мобилизация индуцируется путем введения по меньшей мере одного соединения по изобретению в сочетании с G-CSF и/или VEGF и/или агонистом VEGF (включая, без ограничения, антитела-агонисты VEGF) и/или AMD3100 и/или их солями, рацематами или изомерами.
Для мобилизации предшественников или стволовых клеток надлежащий уровень дозировки обычно составляет от 0,001 до 100 мг на кг массы тела пациента в день, которые можно вводить в виде одной или нескольких доз. Соединения можно вводить в виде однократной дозы, растянутой по времени дозы типа в/в (i.v.) или трансдермального введения либо в виде нескольких доз. Соединения по изобретению также можно использовать при проведении лечения ex vivo протоколах лечения для получения клеточных культур, которые затем используются для восполнения клеток крови у субъекта. Лечение ex vivo может проводиться на аутологичных клетках, полученных из периферической крови или костного мозга либо из аллотрансплантатов от подобранных доноров.
Настоящие соединения можно комбинировать с другими соединениями и композициями, индуцирующими активацию, пролиферацию или мобилизацию предшественников/стволовых клеток. В дополнение к тем, что описаны выше, они включают, без ограничения, лиганд родственной FMS тирозинкиназы-3 (лиганд Flt3), интерлейкин-3 (IL-3), интерлейкин-7 (IL-7), интерлейкин-20 (ИЛ-20), фактор Steel (SF) и колониестимулирующий фактор гранулоцитов-макрофагов (GM-CSF), которые могут обладать таким терапевтическим профилем, который может потребоваться или принести пользу при лечении до, после или одновременно с мобилизацией предшественников/стволовых клеток. Соответственно, составной частью настоящего изобретения также являются комбинированные способы и композиции для профилактики и лечения представляющих интерес заболеваний. Кроме того, соединения настоящего изобретения могут приносить пользу при заболеваниях, в которых может играть роль дисрегуляция мобилизации стволовых клеток, таких как болезни сердца и легочная гипертензия.
Способы диагностики заболеваний, связанных с CXCR7
Далее, соединения и композиции настоящего изобретения применимы для диагностики заболеваний, связанных с CXCR7. В частности, соединения настоящего изобретения могут быть получены в меченом виде (например, с радиоактивной меткой) и использоваться для диагностики, к примеру, рака. Меченые соединения настоящего изобретения, которые связываются с CXCR7 (например, антагонисты или агонисты), могут применяться для определения уровня CXCR7 у млекопитающих. В некоторых воплощениях страдающим раком субъектам вводятся модуляторы CXCR7. В некоторых случаях меченые соединения вводятся для выявления развития рака, например, карциномы, глиомы, мезотелиомы, меланомы, лимфомы, лейкемии, аденокарциномы, рака молочной железы, рака яичников, рака шейки матки, глиобластомы, лейкемии, лимфомы, рака простаты, лимфомы Беркитта, рака головы и шеи, рака толстой кишки, колоректального рака, немелкоклеточного рака легких, мелкоклеточного рака легких, рака пищевода, рака желудка, рака поджелудочной железы, гепатобилиарного рака, рака желчного пузыря, рака тонкой кишки, рака прямой кишки, рака почек, рака мочевого пузыря, рака предстательной железы, рака полового члена, рака мочеиспускательного канала, рака яичек, рака шейки матки, рака влагалища, рака матки, рака яичников, рака щитовидной железы, рака паращитовидной железы, рака надпочечников, рака эндокринной поджелудочной железы, карциноидов, рака костей, рака кожи, ретинобластомы, лимфомы Ходжкина, неходжкинской лимфомы (см. Cancer: Principles and Practice (DeVita V.T. et al., eds, 1997) насчет других раковых заболеваний); а также таких дисфункций мозга и нейронов, как болезнь Альцгеймера и рассеянный склероз; дисфункции почек; ревматоидного артрита; отторжения сердечных аллотрансплантатов; атеросклероза (и повышенного уровня холестерина); астмы; гломерулонефрита; контактного дерматита; воспалительной болезни кишечника; колита; псориаза; реперфузионных повреждений; а также других описанных здесь заболеваний. В некоторых воплощениях у этих субъектов нет саркомы Калоши, полицентрической болезни Кастлемана или связанной со СПИД первичной эффузионной лимфомы. Поскольку CXCR7 часто экспрессируется в раковых клетках, но не экспрессируется в нераковых клетках, то, как правило, желательно введение антагонистов CXCR7 субъектам, подверженным риску заболевания раком.
Для выявления рака могут применяться различные методы визуализации и детектирования. В некоторых воплощениях для оценки биораспределения CXCR7 в организме имеются прямые методы, такие как магнитно-резонансная томография (MRI), позитронно-эмиссионная томография (PET) и однофотонная эмиссионная компьютерная томография (SPECT). Каждым из этих методов можно детектировать распределение подходящего меченого соединения (как правило, связанного с CXCR7) в организме, если это соединение содержит атом с соответствующими свойствами ядра. MRI выявляет парамагнитные ядра; а PET и SPECT выявляют испускание частиц при распаде радионуклидов.
Для методов, связанных с PET, необходимо включение соответствующих излучающих позитроны радионуклидов. Существует сравнительно мало излучающих позитроны изотопов, пригодных для мечения терапевтических средств. Для PET применялся изотоп углерода 11С, но он имеет короткий период полураспада в 20,5 минут. Соответственно, установки для синтеза и применения, как правило, располагаются рядом с циклотроном, на котором вырабатывается содержащий 11С исходный материал-предшественник. Другой полезный изотоп, 18F, имеет период полураспада 110 минут. Это дает достаточно времени для его включения в радиоизотопный индикатор, для очистки и для введения в организм человека или животного. Другие изотопы имеют еще более короткие периоды полураспада. 13N имеет период полураспада в 10 минут, а 15О - еще более короткий период полураспада в 2 минуты. Однако излучение у них более сильное, чем у 11С, поэтому исследования методом PET проводились с этими изотопами (см. Clinical Positron Emission Tomography, Mosby Year Book, 1992, K.F. Hubner et al., Chapter 2).
При SPECT-томографии используются изотопные индикаторы, которые являются γ-излучателями. Хотя ассортимент полезных изотопов и больше, чем для PET, однако SPECT-томография дает меньшее трехмерное разрешение. Тем не менее, в некоторых случаях применяется SPECT для получения клинически значимой информации о связывании, локализации и высвобождении соединений. Одним из полезных изотопов для SPECT-томографии является 123I, γ-излучатель с периодом полураспада в 13,3 часа. Помеченные 123I соединения могут поставляться вплоть до 1000 миль от места производства или же сам изотоп можно транспортировать для синтеза на месте. 85% излучения изотопа составляют фотоны в 159 кэВ, которые легко измеряются современными приборами SPECT. Для PET- или SPECT-томографии или для обычного мечения индикаторов могут служить другие изотопы галогенов. Они включают в себя 75Br, 76Br, 77Br и 82Br, которые имеют подходящие характеристики периодов полураспада и эмиссии.
Ввиду вышеизложенного, настоящим изобретением предусмотрены способы визуализации опухолей, органов или тканей, которые включают:
(a) введение нуждающемуся в такой визуализации субъекту радиоизотопной или детектируемой формы соединения формулы I; и
(b) детектирование данного соединения для определения того, где оно концентрируется у данного субъекта.
Кроме того, настоящим изобретением предусмотрены способы выявления повышенных уровней CXCR7 в образцах, которые включают:
(a) контактирование образца с подозрением на повышенный уровень CXCR7 с радиоизотопной или детектируемой формой соединения формулы I;
(b) определение уровня соединения, связавшегося с CXCR7, присутствующим в данном образце, для определения уровня CXCR7, находящегося в данном образце; и
(c) сравнение уровня, определенного на стадии (b), с контрольным образцом, для определения того, имеется ли повышенный уровень CXCR7 в данном образце.
Как и в способах лечения, описанных здесь, введение меченых соединений может осуществляться любым из способов, обычно используемых для приведения соединений в непосредственный контакт с анализируемой тканью, которые хорошо известны специалистам. Хотя для введения определенных композиций может применяться более чем один способ введения, однако какой-то один способ зачастую может обеспечить более прямую и более эффективную диагностику, чем другой способ.
Комбинированная терапия
Ингибиторы CXCR7 могут применяться сами по себе или в сочетании с одним или несколькими другими препаратами. Возможными партнеры в комбинации могут служить, например, дополнительные антиангиогенные факторы и/или химиотерапевтические средства (например, цитотоксические средства) или облучение, противораковые вакцины, иммуномодуляторы, противососудистые средства, ингибиторы передачи сигналов, антипролиферативные средства или индукторы апоптоза.
Примеры
Следующие примеры приводятся для раскрытия, а не для ограничения заявленного изобретения.
Реагенты и растворители, используемые ниже, могут быть получены из таких коммерческих источников, как Aldrich Chemical Co. (Milwaukee, Wisconsin, США). Спектры 1Н-ЯМР регистрировали на ЯМР-спектрометре Varian Mercury 400 MHz. Значимые пики приводятся относительно TMS и представлены в следующем порядке: множественность (s, синглет; d, дублет; t, триплет, q, квартет; m, мультиплет) и количество протонов. Результаты масс-спектрометрии представлены в виде отношения массы к заряду, а после него - относительное содержание каждого иона (в скобках). В примерах приводится одно значение m/e для иона М+Н (или же М-Н), содержащего наиболее распространенные атомные изотопы. Во всех случаях изотопные профили соответствуют предполагаемым формулам. Масс-спектрометрию с ионизацией электрораспылением (ESI) проводили на электрораспылительном масс-спектрометре Hewlett-Packard MSD, используя для введения образцов HPLC HP 1100. Обычно анализируемое вещество растворяли в метаноле при 0,1 мг/мл и вводили 1 микролитр вместе с растворителем в масс-спектрометр, который сканировал от 100 до 1500 дальтон. Все соединения можно было анализировать в режиме положительного ESI, используя в качестве несущего растворителя смесь ацетонитрила и воды с 1% муравьиной кислоты. Представленные ниже соединения также можно было анализировать в режиме отрицательного ESI, используя в качестве несущей системы 2 мМ NH4OAc в смеси ацетонитрил/вода.
В примерах и по всему описанию изобретения применяются следующие сокращения: rt, комнатная температура; HPLC, жидкостная хроматография высокого давления; TFA, трифторуксусная кислота; LC-MSD, жидкостная хроматография с масс-селективным детектором; LC-MS, жидкостная хроматография/масс-спектрометрия; Pd2dba3, трис(дибензилиденацетон)дипалладий; THF, тетрагидрофуран; DMF, диметилформамид или N,N-диметилформамид; DCM, дихлорметан; DMSO, диметилсульфоксид; TLC, тонкослойная хроматография; KHMDS, гексаметилдисилазан калия; ES, электрораспыление; sat., насыщенный.
Соединения в рамках настоящего изобретения могут быть синтезированы, как описано ниже, с использованием различных реакций, известных специалистам. Специалистам также должно быть известно, что для синтеза искомых соединений настоящего изобретения можно использовать альтернативные методы, а подходы, описанные в тексте настоящего документа, не являются исчерпывающими, но действительно обеспечивают широко применимые и практические способы получения искомых соединений.
Некоторые молекулы, заявленные в настоящем патенте, могут существовать в различных энантиомерных и диастереомерных формах, причем предусмотрены все такие варианты этих соединений.
Подробное описание экспериментальных процедур, используемых для синтеза ключевых соединений в этом тексте, приводит к получению молекул, которые описаны по идентифицирующим их физическим данным, а также по связанным с ними структурным описаниям.
Специалистам в данной области также должно быть известно, что при выполнении стандартных процедур в органической химии часто используются кислоты и основания. Иногда при экспериментальных процедурах, описанных в данном патенте, получаются соли исходных соединений, если они обладают необходимой собственной кислотностью или щелочностью.
Пример 1. Синтез этил-1-(4-фторфенил)-1,2,4-триазол-3-карбоксилата
Растворяли 4-фторанилин (2,8 г, 26 ммоль) в 10% HCl и охлаждали до 0°С. В этот раствор осторожно добавляли NaNO2 (1,8 г, 26 ммоль), растворенный в 10 мл воды. В отдельной колбе в раствор этил-изоцианоацетата (2,0 г, 18 ммоль) в метаноле (80 мл) добавляли NaOAc (13 г, 96 ммоль) и воду (25 мл). Этот раствор охлаждали до 0°С и осторожно добавляли диазониевую соль 4-фторанилина на протяжении 15 минут. Продолжали перемешивание при 0°С еще 15 минут, после чего колбу вынимали из бани со льдом и продолжали перемешивание еще 2 часа. Затем реакционную смесь выливали в 500 мл воды, а образовавшийся коричневый осадок собирали фильтрованием и высушивали под вакуумом, получая 3,8 г искомого эфира (выход 90%).
Пример 2. Синтез 1-(4-фторфенил)-1,2,4-триазол-3-карбоновой кислоты
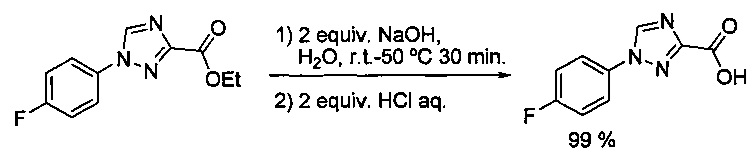
Суспендировали 1,84 г этил-1-(4-фторфенил)-1,2,4-триазол-3-карбоксилата (7,83 ммоль) в растворе 0,63 г гидроксида натрия (2 экв.) в 110 мл воды. Смесь энергично перемешивали и медленно доводили до 50°С, после чего все твердые частицы растворялись. Раствор охлаждали до комнатной температуры и разбавляли водой до общего объема 400 мл. При энергичном перемешивании смеси добавляли 1,31 мл концентрированной HCl (2 экв.). Перемешивание продолжали в течение 15 минут, при этом все твердые частицы равномерно диспергировались. Белые частицы отфильтровывали и тщательно промывали на воронке 15 мл воды, а затем высушивали в вакуумном шкафу при 50°С, получая 1,6 г искомой кислоты в виде белого порошка (выход 99%).
Пример 3. Синтез 1-(4-фторфенил)-1,2,4-триазол-3-ацетилхлорида
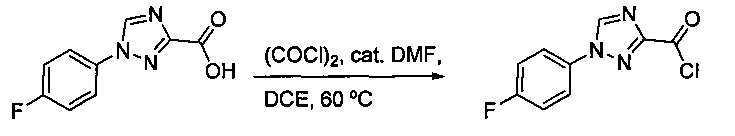
Суспендировали 1,71 г (8,24 ммоль) 1-(4-фторфенил)-1,2,4-триазол-3-карбоновой кислоты в 15 мл 1,2-дихлорэтана, а затем по каплям добавляли 1,24 мл (12,4 ммоль) оксалилхлорида при комнатной температуре, а затем еще 1,6 мкл (0,021 ммоль) DMF. Смесь перемешивали при комнатной температуре, а затем медленно доводили до 60°С. Добавляли еще 1,6 мкл DMF, что приводило к полному растворению материала через 5 минут. Раствор упаривали под вакуумом, получая 1,86 г искомого продукта в виде светло-желтого твердого вещества (выход 100%). Продукт использовали на следующих стадиях без дальнейшей очистки.
Пример 4. Синтез этил-1-(4-хлор-3-фторфенил)пиразол-3-карбоксилата
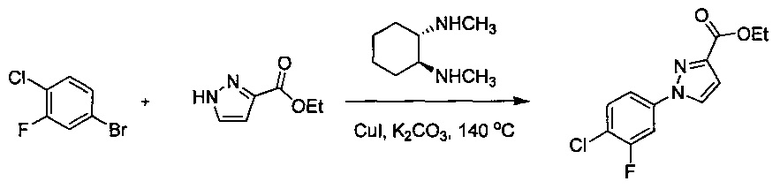
В колбу на 50 мл вносили 3,00 г 4-хлор-3-фторбензолбромида (15 ммоль), 1,40 г этил-1-Н-пиразол-3-карбоксилата (10 ммоль), 400 мг CuI (2,0 ммоль), 4,5 г K2CO3 (3,3 ммоль) и 0,9 мл транс-N,N'-диметилциклогексилдиамина (2,0 ммоль). Полученную смесь перемешивали при 140°С в течение 3 ч. После охлаждения смеси до комнатной температуры ее разбавляли 200 мл EtOAc, а затем промывали водой (2×50 мл) и насыщенным раствором NaCl (2×50 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (0-25% EtOAc в гексане), получая искомый продукт (1,2 г, 50%).
Пример 5. Синтез 1-(4-хлор-3-фторфенил)пиразол-3-карбоновой кислоты
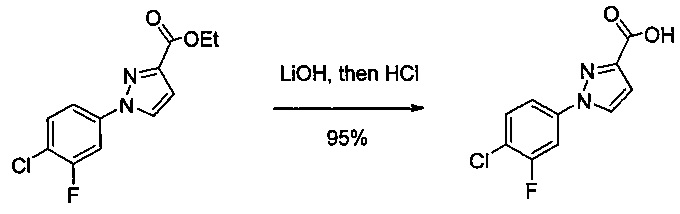
В раствор этил-1-(4-хлор-3-фторфенил)пиразол-3-карбоксилата (268 мг, 1 ммоль) в THF добавляли 3,0 мл 1,0 М LiOH (3,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч, после чего добавляли 1,0 М HCl для доведения до pH 1,0. Органические вещества экстрагировали с помощью EtOAc (2×100 мл), а затем сушили над MgSO4 и упаривали при пониженном давлении, получая белое твердое вещество (230 мг, 96%), которое использовали на следующей стадии без дальнейшей очистки.
Пример 6. Синтез этил-1-(4-хлорфенил)пиразол-3-карбоксилата
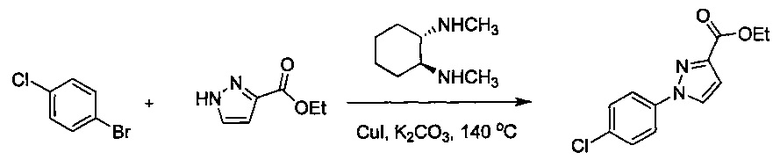
В колбу на 50 мл вносили 2,87 г 4-хлорбензолбромида (15 ммоль), 1,40 г этил-1-H-пиразол-3-карбоксилата (10 ммоль), 400 мг CuI (2,0 ммоль), 4,5 г K2CO3 (3,3 ммоль) и 0,9 мл транс-N,N'-диметилциклогексилдиамина (2,0 ммоль). Полученную смесь перемешивали при 140°С в течение 3 ч. После охлаждения смеси до комнатной температуры ее разбавляли 200 мл EtOAc, а затем промывали водой (2×50 мл) и насыщенным раствором NaCl (2×50 мл). Органический слой сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (0-25% EtOAc в гексане), получая искомый продукт (1,25 г, 50%).
Пример 7. Синтез этил-1-(4-хлорфенил)пиразол-3-карбоновой кислоты
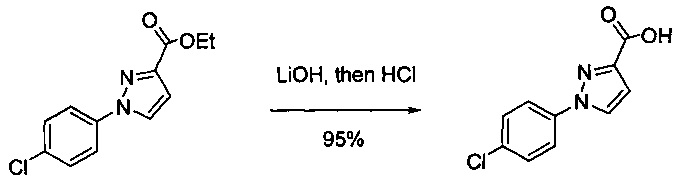
В раствор этил-1-(4-хлорфенил)пиразол-3-карбоксилата (250 мг, 1 ммоль) в THF добавляли 3,0 мл 1,0 М LiOH (3,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч, после чего добавляли 1,0 М HCl для доведения до pH 1,0. Органические вещества экстрагировали с помощью EtOAc (2×100 мл), сушили над MgSO4 и упаривали при пониженном давлении, получая белое твердое вещество (213 мг, 96%), которое использовали на следующей стадии без дальнейшей очистки.
Пример 8. Синтез этил-1-(3-фторфенил)пиразол-3-карбоксилата
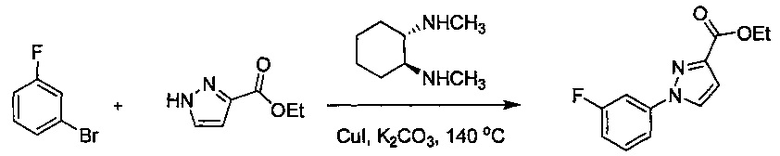
В колбу на 50 мл вносили 2,62 г 4-фторбензолбромида (15 ммоль), 1,40 г этил-1-H-пиразол-3-карбоксилата (10 ммоль), 400 мг CuI (2,0 ммоль), 4,5 г K2CO3 (3,3 ммоль) и 0,9 мл транс-N,N'-диметилциклогексилдиамина (2,0 ммоль). Полученную смесь перемешивали при 140°С в течение 3 ч. После охлаждения смеси до комнатной температуры ее разбавляли 200 мл EtOAc, а затем промывали водой (2×50 мл) и насыщенным раствором NaCl (2×50 мл). Органический слой сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (0-25% EtOAc в гексане), получая искомый продукт (1,17 г, 50%).
Пример 9. Синтез 1-(3-фторфенил)пиразол-3-карбоновой кислоты
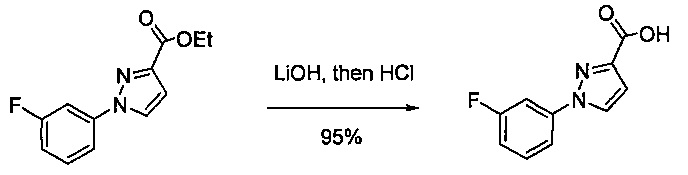
В раствор этил-1-(3-фторфенил)пиразол-3-карбоксилата (234 мг, 1 ммоль) в THF добавляли 3,0 мл 1,0 М LiOH (3,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч, а затем добавляли 1,0 М HCl для доведения до pH 1,0. Органические вещества экстрагировали с помощью EtOAc (2×100 мл), после чего сушили над MgSO4 и упаривали при пониженном давлении, получая белое твердое вещество (198 мг, 96%), которое использовали на следующей стадии без дальнейшей очистки.
Пример 10. Синтез этил-1-(4-фторфенил)-1,2,4-триазол-5-метил-3-карбоксилата
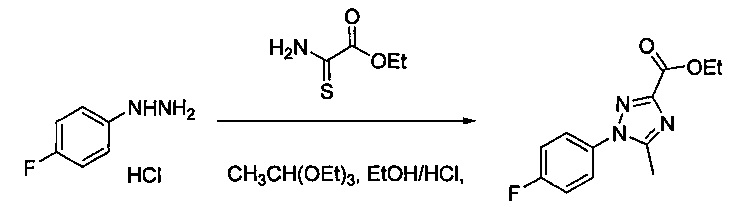
Суспендировали 4-фторгидразин гидрохлорид (12,8 г, 7,7 ммоль) и этилтиооксамат (10 г, 7,7 ммоль) в воде (80 мл), а затем охлаждали смесь до 0°С. В эту смесь по каплям добавляли триэтиламин (10,77 мл, 7,7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Твердое вещество отфильтровывали и промывали водой (2×200 мл), получая желтое вещество (13,8 г, 6,1 ммоль). 11,25 г этого желтого вещества (50 ммоль) растворяли в EtOH (50 мл) и 4М HCl (0,5 мл), после чего добавляли триэтилортоацетат (8,9 г, 55 ммоль). Полученную смесь нагревали при 80°С в течение 2 ч и собирали твердое вещество (10 г, 85%) после того, как оно охладилось до комнатной температуры.
Пример 11. Синтез 1-(4-фторфенил)-1,2,4-триазол-5-метил-3-карбоновой кислоты
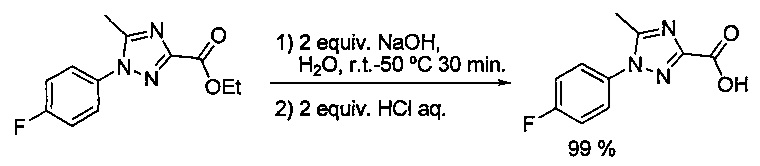
Суспендировали 1,94 г этил-1-(4-фторфенил)-1,2,4-триазол-5-метил-3-карбоксилата (7,83 ммоль) в растворе 0,63 г гидроокиси натрия (2 экв.) в 110 мл воды. Смесь энергично перемешивали и медленно доводили до 50°С, после чего все твердые вещества растворялись. Раствор охлаждали до комнатной температуры и разбавляли водой до общего объема 400 мл. Добавляли 1,31 мл концентрированной HCl (2 экв.) при энергичном перемешивании смеси. Перемешивание продолжали в течение 15 минут. Затем белое твердое вещество отфильтровывали и тщательно промывали 15 мл воды, а затем высушивали в вакуумном шкафу при 50°С, получая 1,7 г искомой кислоты в виде белого порошка (выход 99%).
Пример 12. Синтез этил-2-пирролидинтиазол-4-карбоксилата
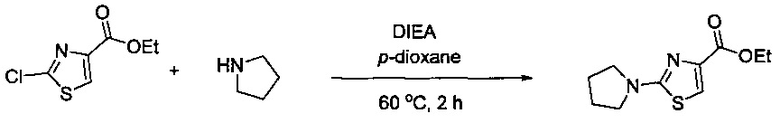
Смесь этил-2-хлортиазол-4-карбоксилата (500 мг, 2,6 ммоль), пирролидина (210 мг, 2,98 ммоль) и диэтилизопропиламина (1 мл) в n-диоксане (5 мл) нагревали до 60°С в течение 2 ч. После охлаждения до комнатной температуры раствор упаривали под вакуумом, а остаток очищали методом флэш-хроматографии (SiO2, от 2 до 5% МеОН в CH2Cl2 в качестве элюента), получая искомое соединение в виде пены (400 мг, выход 68%, использовали непосредственно на следующей стадии). MS: (ES) m/z 227,1 (М+Н+).
Пример 13. Синтез 2-пирролидинтиазол-4-карбоновой кислоты
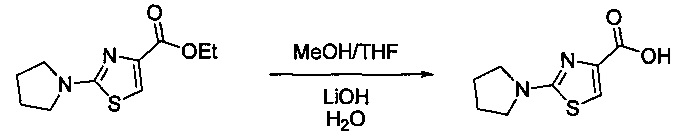
В смесь полученного выше эфира (400 мг, 1,8 ммоль), МеОН (3 мл), THF (5 мл) и дистиллированной воды (2 мл) добавляли LiOH моногидрат (210 мг, 5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли ледяной водой, доводили до рН 3 с помощью 1N HCl и экстрагировали 20% МеОН в CH2Cl2. Органический слой сушили (Na2SO4), фильтровали и упаривали под вакуумом, получая искомое соединение в виде беловатого твердого вещества (300 мг, выход 89%, использовали непосредственно на следующей стадии). MS: (ES) m/z 199,1 (М+Н+).
Пример 14. Синтез 2-(4-гидроксипиперидин-1-ил)-тиазол-4-карбоновой кислоты
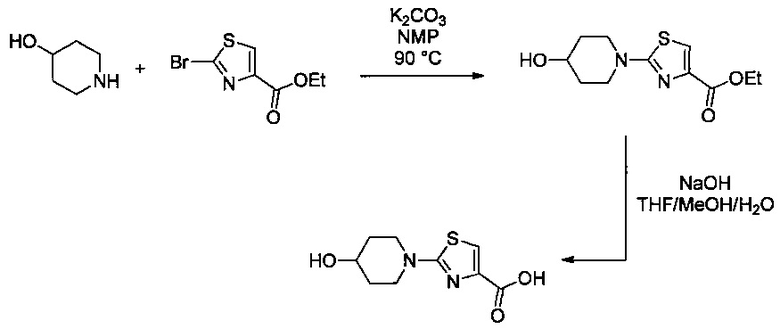
a) В колбу вносили 4-гидроксипиперидин (236 мг, 2,33 ммоль), этил-2-бромтиазол-4-карбоксилат (500 мг, 2,12 ммоль), K2CO3 (879 мг, 6,36 ммоль) и N-метилпирролидин (3,5 мл). Реакционную смесь нагревали до 90°С и перемешивали в течение ночи. Затем реакционную смесь разбавляли EtOAc (30 мл) и промывали водой (5×30 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали. Неочищенный продукт очищали на силикагеле (гексан : EtOAc от 95:5 до 20:80), получая продукт в смеси с 2 эквивалентами N-метилпирролидина (883 мг) в виде прозрачного бесцветного масла.
b) Смесь этилового эфира 2-(4-гидроксипиперидин-1-ил)-тиазол-4-карбоновой кислоты из стадии а растворяли в МеОН (10 мл). Туда же добавляли NaOH (2М, 5,00 мл). Реакционную смесь перемешивали в течение ночи, а затем разбавляли 1М NaHSO4 (30 мл). Этот раствор экстрагировали EtOAc (3×50 мл), а объединенные органические слои сушили над Na2SO4, фильтровали и упаривали, получая продукт (433 мг, 1,90 ммоль, 90%) в виде белого твердого вещества.
Пример 15. Синтез 2-(пирролидин-1-ил)тиазол-4-карбоновой кислоты
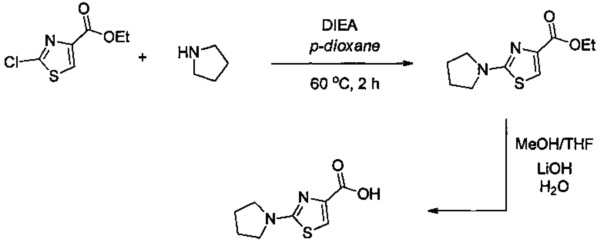
a) Смесь этил-2-хлортиазол-4-карбоксилата (500 мг, 2,6 ммоль), пирролидина (210 мг, 2,98 ммоль) и диэтилизопропиламина (1 мл) в n-диоксане (5 мл) нагревали до 60°С в течение 2 ч. После охлаждения до комнатной температуры растворитель упаривали под вакуумом, а остаток очищали методом флэш-хроматографии (SiO2, от 2 до 5% МеОН в CH2Cl2 в качестве элюента), получая искомое соединение в виде пены (400 мг, выход 68%, использовали непосредственно на следующей стадии). MS: (ES) m/z 227,1 (М+Н+).
b) В смесь полученного выше эфира (400 мг, 1,8 ммоль), МеОН (3 мл), THF (5 мл) и дистиллированной воды (2 мл) добавляли LiOH моногидрат (210 мг, 5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли ледяной водой, доводили до рН 3 с помощью 1N HCl и экстрагировали 20% МеОН в CH2Cl2. Органический слой сушили (Na2SO4), фильтровали и упаривали под вакуумом, получая искомое соединение в виде беловатого твердого вещества (300 мг, выход 89%, использовали непосредственно на следующей стадии). MS: (ES) m/z 199,1 (М+Н+).
Пример 16. Синтез метил-1-(о-фторфенил)-1,2,4-триазол-3-карбоксилата
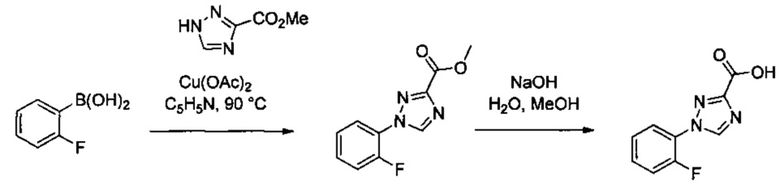
a) В раствор метил-1,2,4-триазол-3-карбоксилата (1,0 г, 7,9 ммоль) в 10 мл DMFA добавляли о-фторфенилборную кислоту (1,1 г, 7,9 ммоль), Cu(ОАс)2 (1,6 г, 8,8 ммоль) и пиридин (0,70 мл, 8,7 ммоль). Смесь ставили в масляную баню на 90°С и энергично перемешивали в течение 3,5 часов. После этого смесь разбавляли 100 мл EtOAc, фильтровали и промывали смесью конц. NH4OH-насыщенный NH4Cl=1:3 (об/об). После удаления растворителя при пониженном давлении остаток очищали методом флэш-хроматографии (SiO2, 20-80% EtOAc/гексан), получая 280 мг белого порошка (выход 16%).
b) В раствор эфира из стадии а (280 мг, 1,3 ммоль), растворенного в 4 мл МеОН, добавляли 2,5 мл 1,0 М NaOH. После перемешивания в течение 15 минут добавляли 6,0 М HCl (0,42 ммоль, 2,5 ммоль), удаляли МеОН при пониженном давлении, собирали белый осадок фильтрованием и высушивали под вакуумом, получая 150 мг искомой кислоты (выход 57%).
Пример 17. Синтез 1-(о-толил)-1,2,4-триазол-3-карбоновой кислоты

В раствор этил-1-(о-толил)-1,2,4-триазол-3-карбоксилата (400 мг, 1,7 ммоль) в 3 мл THF добавляли 4 М LiOH (2 мл, 8 ммоль), а затем 3 мл метанола. Реакционную смесь перемешивали при 50°С в течение 5 минут, после чего доводили до рН 4 добавлением конц. HCl. После экстракции, сушки (MgSO4), фильтрования и высушивания при пониженном давлении получали 43 мг остатка (12%), который использовали на следующей стадии без дальнейшей очистки.
Пример 18. Синтез 1-(п-фторфенил)-1,2,4-триазол-3-карбоновой кислоты
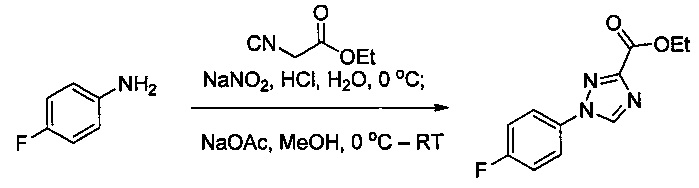
Растворяли 4-фторанилин (2,8 г, 26 ммоль) в 10% HCl и охлаждали до 0°С. В этот раствор осторожно добавляли NaNO2 (1,8 г, 26 ммоль), растворенный в 10 мл воды. В отдельной колбе в раствор этил-изоцианоацетата (2,0 г, 18 ммоль) в метаноле (80 мл) добавляли NaOAc (13 г, 96 ммоль) и воду (25 мл). Этот раствор охлаждали до 0°С и осторожно добавляли диазониевую соль 4-фторанилина на протяжении 15 минут. Продолжали перемешивание при 0°С еще 15 минут, после чего колбу вынимали из бани со льдом и продолжали перемешивание еще 2 часа. Затем реакционную смесь выливали в 500 мл воды, а образовавшийся коричневый осадок собирали фильтрованием и высушивали под вакуумом, получая 3,8 г искомого эфира (выход 90%).
Пример 19. Синтез N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-2-фенилтиазол-4-карбоксамида
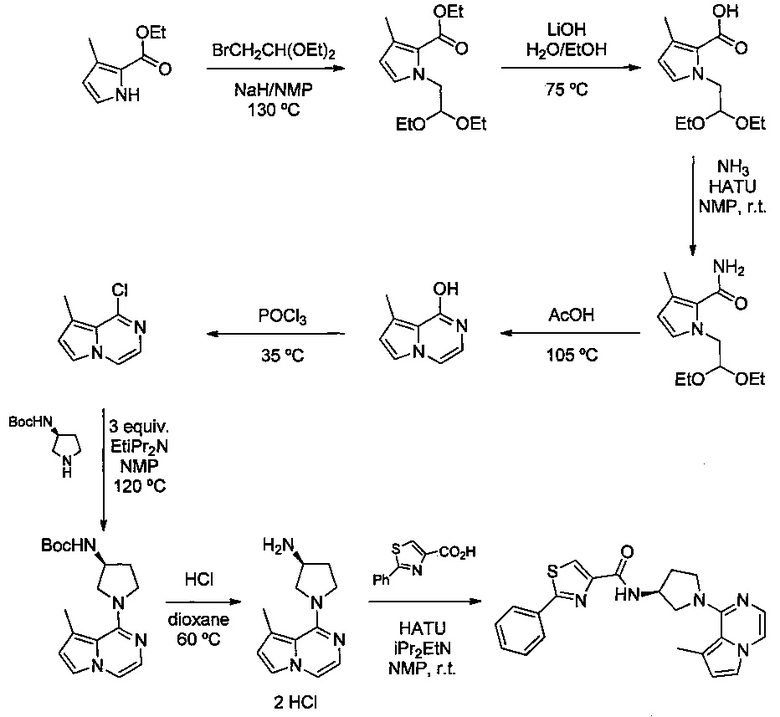
a) Растворяли этил-3-метил-1Н-ггаррол-2-карбоксилат (2,45 г, 16,0 ммоль) в смеси из 8 мл NMP и 2-бром-1,1-диэтоксиэтана (3,23 мл, 20,8 ммоль). Порциями добавляли 60% гидрид натрия (0,77 г, 19,2 ммоль) при комнатной температуре. Полученный раствор нагревали при 130°С в течение 5 часов, а затем охлаждали до комнатной температуры. После этого раствор разбавляли 100 мл воды, а полученную смесь экстрагировали одной порцией 100 мл МТВЕ. Органический слой упаривали под вакуумом и очищали методом флэш-хроматографии (SiO2, 10-40% EtOAc/гексан), получая 1,90 г искомого соединения в виде бесцветного масла (выход 44%).
b) Растворяли 1,90 г (7,06 ммоль) этил-1-(2,2-диэтоксиэил)-3-метил-1Н-пиррол-2-карбоксилата (полученного на стадии а выше) в 40 мл смеси этанол-вода (1:1). После добавления 1,48 г (35,3 ммоль) гидроксида лития моногидрата смесь нагревали при 75°С в течение 8 часов. После охлаждения большую часть этанола выпаривали под вакуумом. Полученный раствор разбавляли 30 мл воды и нейтрализовали 2,33 г уксусной кислоты (38,8 ммоль), а затем экстрагировали двумя порциями по 30 мл дихлорметана. Органическую фазу сушили над безводным сульфатом магния, фильтровали и упаривали под вакуумом, получая 1,68 г бесцветного твердого вещества (выход 99%).
c) Растворяли 1,68 г (6,97 ммоль) 1-(2,2-диэтоксиэтил)-3-метил-1Н-пиррол-2-карбоновой кислоты (полученной на стадии b выше) в смеси из 15 мл NMP и 45 мл 0,5 М раствора аммиака в диоксане. Добавляли 3,18 г HATU (8,36 ммоль) и перемешивали смесь в течение ночи при комнатной температуре. В эту смесь добавляли 10 мл NMP и упаривали диоксан под вакуумом. Добавляли другую порцию HATU (1,59 г, 4,18 ммоль) и пропускали через смесь газообразный аммиак до тех пор, пока не наблюдалось прекращение реакции. Затем смесь разбавляли 300 мл насыщенного раствора NaCl и экстрагировали одной порцией 150 мл МТВЕ. Органический слой упаривали под вакуумом и очищали методом флэш-хроматографии (SiO2, 20-100% EtOAc/гексан), получая 1,26 г искомого соединения в виде бесцветного масла (выход 75%).
d) Растворяли 1,25 г (5,21 ммоль) 1-(2,2-диэтоксиэил)-3-метил-1Н-пиррол-2-карбоксамида (полученного на стадии с выше) в 30 мл ледяной уксусной кислоты и нагревали при 105°С в течение 4 часов, а затем упаривали досуха под вакуумом. Остаток растворяли в 25 мл горячего дихлорметана, а затем добавляли 30 мл гексана и выпаривали большую часть дихлорметана. Твердые частицы отфильтровывали, промывали 5 мл гексана и высушивали, получая 730 мг чистого искомого соединения в виде желто-коричневого порошка (выход 95%).
e) Суспендировали 725 мг (4,90 ммоль) 8-метил-2Н-пирроло[1,2-а]пиразин-1-она (полученного на стадии d выше) в 7 мл фосфорилхлорида и перемешивали при комнатной температуре в течение ночи, после чего нагревали при 35°С в течение 4 часов. Затем полученный раствор упаривали досуха под вакуумом. Остаток растворяли в смеси 10 мл дихлорметана и 10 мл водного раствора бикарбоната натрия и перемешивали до тех пор, пока не прекратилось выделение газа. Органический слой отделяли, сушили над безводным сульфатом магния, фильтровали и упаривали под вакуумом. Остаток растворяли в 9 мл горячего н-гептана. Раствор декантировали от нерастворимых черных смол и упаривали под вакуумом, получая 774 мг искомого продукта в виде беловатого твердого вещества (выход 95%).
f) Смешивали 646 мг (3,87 ммоль) 1-хлор-8-метилпирроло[1,2-а]пиразина (полученного на стадии е выше), 2,16 г (11,6 ммоль) (S)-3-(Вос-амино)пирролидина, 1 мл N,N-диизопропилэтиламина и 1 мл NMP и нагревали при 120°С в течение 1 часа. Смесь охлаждали, разбавляли 50 мл воды и экстрагировали тремя порциями по 50 мл этил-ацетата. В водный слой добавляли твердый бикарбонат натрия и опять экстрагировали 50 мл этилацетата. Объединенные органические слои промывали водным раствором бикарбоната натрия, упаривали под вакуумом и очищали методом флэш-хроматографии (SiO2, 20-60% EtOAc/гексан), получая 872 мг искомого соединения в виде бесцветного масла (выход 71%).
g) Растворяли 872 мг (2,75 ммоль) трет-бутил-N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]карбамата (полученного на стадии f выше) в 3,5 мл диоксана. В этот раствор добавляли 3,5 мл раствора HCl в диоксане (4 М) и перемешивали смесь при 60°С в течение 1 ч, при этом наблюдалось выпадение бесцветного осадка. Летучие вещества удаляли под вакуумом, получая 739 мг искомого соединения без дальнейшей очистки (выход 93%).
h) Растворяли 39 мг (0,135 ммоль) (3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)-пирролидин-3-амина дигидрохлорида (полученного на стадии g выше) и 28 мг (0,135 ммоль) 2-фенилтиазол-4-карбоновой кислоты в 0,5 мл NMP. В эту смесь добавляли 77 мг (0,203 ммоль) HATU и 0,117 мл (0,675 ммоль) N,N-диизопропилэтиламина. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 3 мл DMSO и сразу же вводили в обратнофазовую полупрепаративную систему HPLC (5-60% ацетонитрил/вода, 0,1% TFA). Чистые фракции упаривали под вакуумом, получая 66 мг искомого продукта в виде соли TFA (95% выход). 1H-ЯМР (400 MHz, DMSO) δ 11.28 (s, 1H), 8.82 (d, J=7.0 Hz, 1H), 8.33 (s, 1H), 8.04-8.01 (m, 2H), 7.82 (m, 2H), 7.54-7.50 (m, 3H), 6.86 (d, J=5.5 Hz, 1H), 6.76 (d, J=2.2 Hz, 1H), 4.75-4.65 (m, 1H), 4.20-3.60 (m, 4H), 2.48 (s, 3H), 2.40-2.20 (m, 2H); MS: (ES) m/z теор. для C22H21N5OS [M+H]+ 404,2, факт. 404.
Пример 20. Синтез 1-(4-хлорфенил)-N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]пиразол-3-карбоксамида

Растворяли 40 мг (0,138 ммоль) (3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)-пирролидин-3-амина дигидрохлорида и 31 мг (0,138 ммоль) 1-(4-хлорфенил)пиразол-3-карбоновой кислоты в 0,5 мл NMP. В эту смесь добавляли 79 мг (0,207 ммоль) HATU и 0,120 мл (0,690 ммоль) N,N-диизопропилэтиламина. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 3 мл DMSO и сразу же вводили в обратно-фазовую полупрепаративную систему HPLC (5-60% ацетонитрил/вода, 0,1% TFA). Чистые фракции упаривали под вакуумом, растворяли в 1 мл метанола и пропускали через картридж с бикарбонатной смолой (PL-НСО3 MP SPE 500 мг/6 мл). В полученный раствор добавляли 15 мкл концентрированной соляной кислоты и удаляли летучие вещества под вакуумом, получая 49 мг искомого продукта в виде соли HCl (выход 78%). 1Н-ЯМР (400 MHz, DMSO) δ 11.38 (s, 1Н), 8.78 (d, J=7.0 Hz, 1H), 8.59 (d, J=3.0 Hz, 1H), 7.95 (d, J=7.0, 2H), 7.81 (m, 2H), 7.61 (d, J=8.8 Hz, 2H), 6.92 (s, 1H), 6.85 (d, J=5.5 Hz, 1H), 6.76 (d, J=2.2 Hz, 1H), 4.70-4.65 (m, 1H), 4.18-3.80 (m, 4H), 2.48 (s, 3H), 2.38-2.18 (m, 2H); MS: (ES) m/z теор. для C22H21ClN6O [M+H]+ 421,2, факт. 421.
Пример 21. Синтез 6-метил-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]хиназолин-2-карбоксамида
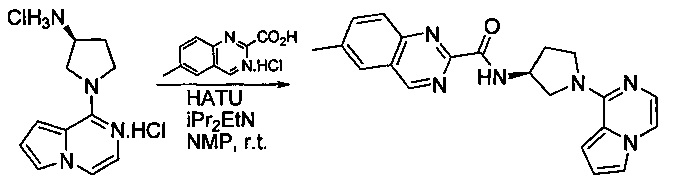
Растворяли 43 мг (0,156 ммоль) (3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амина дигидрохлорида и 33 мг (0,156 ммоль) 6-метилхиназолин-2-карбоновой кислоты гидрохлорида в 0,4 мл NMP. В эту смесь добавляли 85 мг (0,224 ммоль) HATU и 155 мкл (0,892 ммоль) N,N-диизопропилэтиламина. Смесь перемешивали при комнатной температуре в течение 30 минут, а затем разбавляли 3 мл DMSO и сразу же вводили в обратнофазовую полупрепаративную систему HPLC (5-40% ацетонитрил/вода, 0,1% TFA). Чистые фракции упаривали под вакуумом, получая 39 мг искомого продукта в виде соли TFA (51% выход). 1Н-ЯМР (400 MHz, CD3OD) δ 9.49 (s, 1Н), 8.04 (d, J=9.1 Hz, 1H), 7.93 (d, J=6.9 Hz, 2H), 7.75 (d, J=1.5 Hz, 1H), 7.70 (d, J=5.9 Hz, 1H), 7.55 (s, 1H), 6.92 (s, 1H), 6.85 (d, J=5.5 Hz, 1H), 5.00-3.70 (m, 5H), 3.30 (s, 3H), 2.60-2.30 (m, 2H); MS: (ES) m/z теор. для C21H20N6O [M+H]+ 373,2, факт. 373.
Пример 22. Синтез 1-(4-хлорфенил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]пиразол-3-карбоксамида в виде соли трифторацетата

В раствор 1-(4-хлорфенил)пиразол-3-карбоновой кислоты (49 мг, 0,22 ммоль) в 0,80 мл DMSO добавляли (3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амин дигидрохлорид (74 мг, 0,27 ммоль), а затем триэтиламин (0,12 мл, 0,86 ммоль) и HATU (92 мг, 0,24 ммоль). Через 1,5 часа реакционную смесь разбавляли 1 мл воды, фильтровали, очищали методом обратнофазовой препаративной HPLC (градиент 22-36% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомый продукт. 1Н-ЯМР (400 MHz, CD3OD) δ 8.76 (d, J=6.2 Hz, 1H), 8.31 (s, 1H), 7.86 (d, J=8.8 Hz, 2H), 7.77 (d, J=2.5 Hz, 1H), 7.72 (d, J=5.5 Hz, 1H), 7.57 (d, J=5.5 Hz, 1H), 7.50 (d, J=8.8 Hz, 2H), 6.96 (d, J=2.5 Hz, 1H), 6.93 (dd, J=2.6, 7.0 Hz, 1H), 6.84 (d, J=5.9 Hz, 1H), 4.95-4.80 (m, 1H), 4.70-3.70 (br, 4H), 2.60-2.30 (m, 2H). MS: (ES) 407,2 [M+H+].
Пример 23. Синтез 1-(о-толил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]пиразол-3-карбоксамида в виде соли трифторацетата
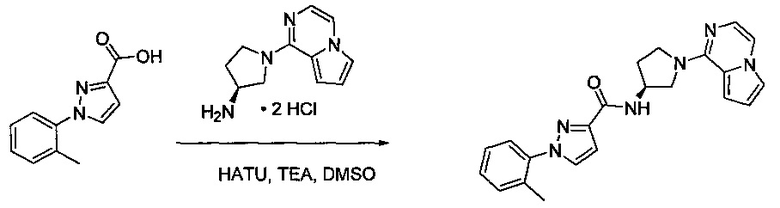
В раствор 1-(о-толил)пиразол-3-карбоновой кислоты (35 мг, 0,17 ммоль) в 0,80 мл DMSO добавляли (3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амин дигидрохлорид (46 мг, 0,17 ммоль), а затем триэтиламин (0,10 мл, 0,70 ммоль) и HATU (80 мг, 0,21 ммоль). Через 1 час реакционную смесь разбавляли дихлорметаном (70 мл), промывали водой (20 мл) и упаривали при пониженном давлении. Остаток очищали методом обратно-фазовой препаративной HPLC (градиент 20-40% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 8.62 (d, J=5.9 Hz, 1H), 7.88 (d, J=2.5 Hz, 1H), 7.75 (s, 1H), 7.70 (d, J=5.9 Hz, 1H), 7,54 (d, J=3.6 Hz, 1H), 7.40-7.25 (m, 4H), 6.94 (d, J=2.2 Hz, 1H), 6,91 (dd, J=2.6, 4.4 Hz, 1H), 6.82 (d, J=5.5 Hz, 1H), 4.95-3.65 (br, 5H), 2.58-2.30 (m, 2H), 2.20 (s, 3H). MS: (ES) 387,2 [M+H+].
Пример 24. Синтез 1-(о-толил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирро-лидин-3-ил]-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата
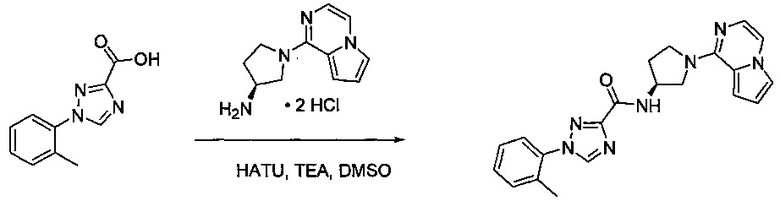
В раствор 1-(о-толил)-1,2,4-триазол-3-карбоновой кислоты (43 мг, 0,21 ммоль) в 1 мл DMSO добавляли (3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амин дигидрохлорид (60 мг, 0,22 ммоль), а затем триэтиламин (0,12 мл, 0,89 ммоль) и HATU (80 мг, 0,21 ммоль). Через 20 минут реакционную смесь гасили 10 мл воды и экстрагировали дихлорметаном. Отделяли органический слой и упаривали при пониженном давлении. Остаток очищали методом обратнофазовой препаративной HPLC (градиент 20-40% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая 20 мг искомого соединения (выход 19%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.73 (s, 1Н), 7.76 (d, J=2.6 Hz, 1H), 7.70 (d, J=5.5 Hz, 1H), 7.55 (d, J=4.0 Hz, 1H), 7.50-7.32 (m, 4H), 6.92 (dd, J=2.5, 4.4 Hz, 1H), 6.83 (d, J=5.9 Hz, 1H), 4.95-4.80 (m, 1H), 4.70-3.70 (br, 5H), 2.60-2.30 (m, 2H), 2.22 (s, 3H). MS: (ES) 388,2 [М+Н+].
Пример 25. Синтез N-[(3S)-1-(8-металпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-5-фенилпиримидин-2-карбоксамида в виде соли трифторацетата
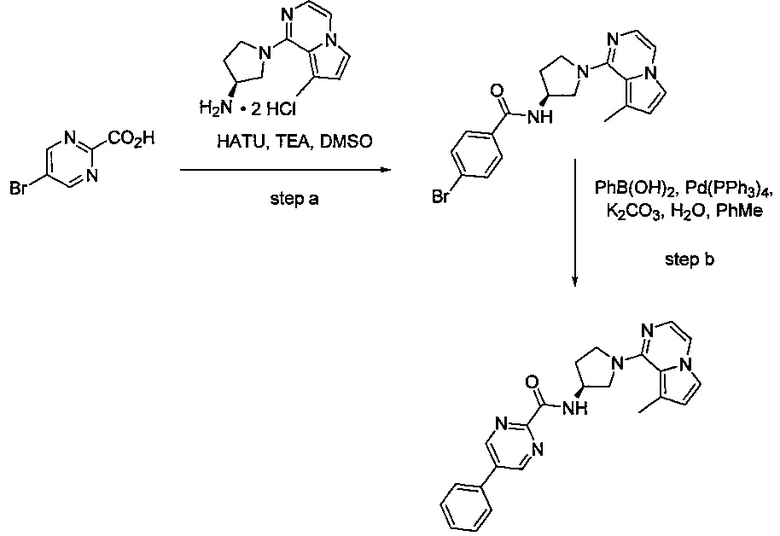
a) В раствор 5-бромпиримидин-2-карбоновой кислоты (41 мг, 0,20 ммоль) в 0,80 мл DMSO добавляли (3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (61 мг, 0,21 ммоль), а затем триэтиламин (0,12 мл, 0,86 ммоль) и HATU (81 мг, 0,21 ммоль). Через 20 минут реакционную смесь гасили 10 мл воды и экстрагировали дихлорметаном. Отделяли органический слой и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии (SiO2, 2% 7М NH3 в смеси МеОН/дихлорметан). MS: (ES) 402,2 [М+Н+].
b) Во флаконе на 4 мл смешивали продукт из стадии а с фенилборной кислотой (24 мг, 0,2 ммоль), Pd(PPh3)4 (23 мг, 0,020 ммоль), дегазированным толуолом (2 мл) и дегазированным 2М K2CO3 (0,3 мл, 0,6 ммоль). Флакон запечатывали и перемешивали реакционную смесь на предварительно нагретой плитке при 100°С в течение 1 ч. Реакционную смесь упаривали при пониженном давлении, очищали методом обратнофазовой препаративной HPLC (градиент 20-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомый продукт. 1Н-ЯМР (400 MHz, CD3OD) δ 9.18 (s, 2Н), 7.77 (d, J=7.0 Hz, 2H), 7.67 (d, J=5.9 Hz, 2H), 7.57-7.48 (m, 3H), 6.76-6.72 (m, 2H), 4.95-4.80 (m, 1H), 4.32-4.24 (m, 1H), 4.10-3.90 (m, 3H), 2,64 (s, 3H), 2,50-2,35 (m, 2H). MS: (ES) 389,2 [М+Н+].
Пример 26. Синтез N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-1-(о-толил)пиразол-3-карбоксамида в виде соли трифторацетата
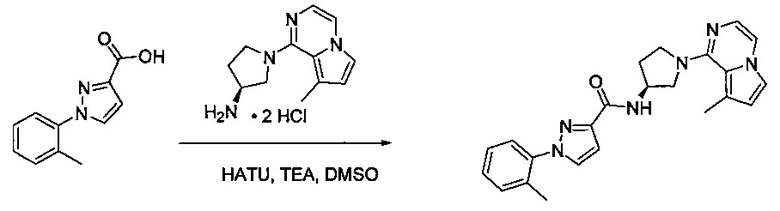
В раствор 1-(о-толил)пиразол-3-карбоновой кислоты (27 мг, 0,13 ммоль) в 0,60 мл DMSO добавляли (3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (39 мг, 0,13 ммоль), а затем триэтиламин (0,080 мл, 0,58 ммоль) и HATU (53 мг, 0,14 ммоль). Через 15 минут в реакционную смесь добавляли уксусную кислоту (0,060 мл, 1 ммоль), а затем СН3ОН (0,60 мл) и воду (0,70 мл). Смесь фильтровали и очищали методом обратнофазовой препаративной HPLC (градиент 20-40% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомый продукт. 1Н-ЯМР (400 MHz, CD3OD) δ 7.87 (s, 1H), 7.64 (m, 2H), 7.40-7.28 (m, 4H), 6.93 (s, 1H), 6.74-6.70 (m, 2H), 4.80-4.70 (m, 1H), 4.30-4.20 (m, 1H), 4.10-3.85 (m, 3H), 2.61 (s, 3H), 2.50-2.35 (m, 2H), 2.20 (s, 3H). MS: (ES) 401,2 [M+H+].
Пример 27. Синтез N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-2-фенилоксазол-4-карбоксамида в виде соли трифторацетата
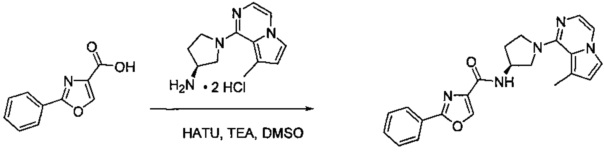
В раствор 2-фенилоксазол-4-карбоновой кислоты (24 мг, 0,13 ммоль) в 0,5 мл DMSO добавляли (3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (41 мг, 0,14 ммоль), а затем триэтиламин (0,080 мл, 0,58 ммоль) и HATU (53 мг, 0,14 ммоль). Через 15 минут в реакционную смесь добавляли уксусную кислоту (0,060 мл, 1 ммоль), а затем СН3ОН (0,60 мл) и воду (0,70 мл). Смесь фильтровали и очищали методом обратнофазовой препаративной HPLC (градиент 20-40% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомый продукт. 1Н-ЯМР (400 MHz, CD3OD) δ 8.72 (d, J=8.1 Hz, 1H), 8.45 (s, 1H), 8.06 (d, J=8.1 Hz, 2H), 7.67 (d, J=5.5 Hz, 2H), 7.52 (m, 3H), 6.76-6.73 (m, 2H), 4.85-4.75 (m, 1H), 4.30-4.22 (m, 1H), 4.10-3.90 (m, 3H), 2.64 (s, 3H), 2.55-2.35 (m, 2H). MS: (ES) 388,2 [M+H+].
Пример 28. Синтез 5-фенил-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]пиримидин-2-карбоксамида в виде соли трифторацетата
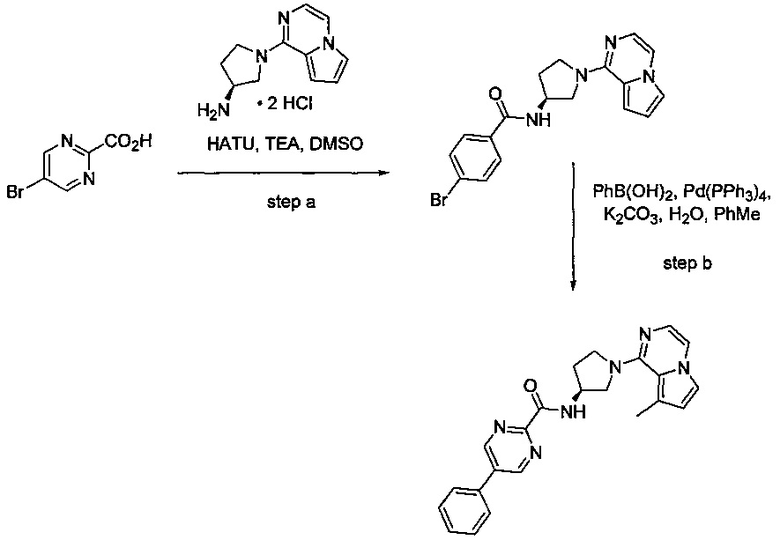
a) В раствор 5-бромпиримидин-2-карбоновой кислоты (120 мг, 0,59 ммоль) в 2,0 мл DMSO добавляли (3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амин дигидрохлорид (160 мг, 0,58 ммоль), а затем триэтиламин (0,30 мл, 2,2 ммоль) и HATU (228 мг, 0,60 ммоль). После перемешивания в течение ночи реакционную смесь гасили 10 мл воды и экстрагировали дихлорметаном. Отделяли органический слой и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии (SiO2, 2% 7М NH3 в смеси МеОН/дихлорметан). MS: (ES) 388,2 [М+Н+].
b) Во флаконе на 4 мл смешивали продукт из стадии а (80 мг, 0,21 ммоль) с фенилборной кислотой (29 мг, 0,24 ммоль), Pd(PPh3)4 (25 мг, 0,021 ммоль), дегазированным толуолом (2 мл) и дегазированным 2М K2CO3 (0,35 мл, 0,7 ммоль). Флакон запечатывали и перемешивали реакционную смесь на предварительно нагретой плитке при 100°С в течение 30 мин. Реакционную смесь упаривали при пониженном давлении, очищали методом обратнофазовой препаративной HPLC (градиент 20-40% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомый продукт. 1Н-ЯМР (400 MHz, CD3OD) δ 9.42 (d, J=7.0 Hz, 0.8H), 9.18 (s, 2H), 7.77-7.70 (m, 4H), 7.58-7.48 (m, 4H), 6.93 (dd, J=2.6, 4.4 Hz, 1H), 6.85 (d, J=5.5 Hz, 1H), 5.00-3.65 (br, 5H), 2.65-2.40 (m, 2H). MS: (ES) 385,1 [M+H+].
Пример 29. Синтез 5-фенил-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]-4H-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата

В раствор 5-фенил-4Н-1,2,4-триазол-3-карбоксилата натрия (50 мг, 0,24 ммоль) в 0,70 мл DMSO добавляли (3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амин дигидрохлорид (80 мг, 0,29 ммоль), а затем триэтиламин (0,14 мл, 1,0 ммоль) и HATU (105 мг, 0,28 ммоль). После перемешивания реакционной смеси в течение ночи ее очищали методом обратнофазовой препаративной HPLC (градиент 20-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомый продукт. 1Н-ЯМР (400 MHz, DMSO) δ 11.40 (s, 1Н), 8.05 (d, J=7.0 Hz, 2H), 7.91 (s, 1H), 7.84 (d, J=4.4 Hz, 1H), 7.60-7.40 (m, 4H), 7.00-6.90 (m, 2H), 4.85-3.50 (br, 5H), 2.45-2.20 (m, 2H). MS: (ES) 374,2 [M+H+].
Пример 30. Синтез 5-(диметиламино)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]пиримидин-2-карбоксамида в виде соли трифторацетата
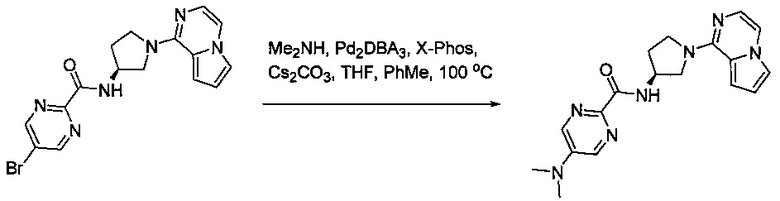
Во флаконе на 4 мл смешивали 4-бром-N-[(3S)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]бензамид (57 мг, 0,15 ммоль) с трис(дибензилиденацетон)палладием (26 мг, 0,023 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенилом ("X-Phos", 49 мг, 0,10 ммоль), Cs2CO3 (250 мг, 0,77 ммоль), дегазированным толуолом (1,0 мл) и 2М диметиламином в THF (1 мл, 2 ммоль). Флакон запечатывали и перемешивали реакционную смесь в течение 22 часов при 100°С. Смесь адсорбировали на силикагеле и очищали методом флэш-хроматографии (SiO2, 2-5% 7М NH3 в смеси МеОН/дихлорметан), а затем методом обратнофазовой препаративной HPLC (градиент 20-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомый продукт. 1Н-ЯМР (400 MHz, CD3OD) δ 8.27 (s, 2Н), 7.75 (t, J=1.1 Hz, 1H), 7.70 (d, J=5.5 Hz, 1H), 7.55 (s, 1H), 6,91 (dt, J=1.8, 2.6 Hz, 1H), 6.83 (d, J=5.5 Hz, 1H), 5.00-4.80 (m, 1H), 4.65-3.70 (br, 4H), 3.10 (s, 6H), 2.60-2.30 (m, 2H). MS: (ES) 352,2 [М+Н+].
Пример 31. Синтез 5-(1-гидроксиэтил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]пиримидин-2-карбоксамида в виде соли трифторацетата
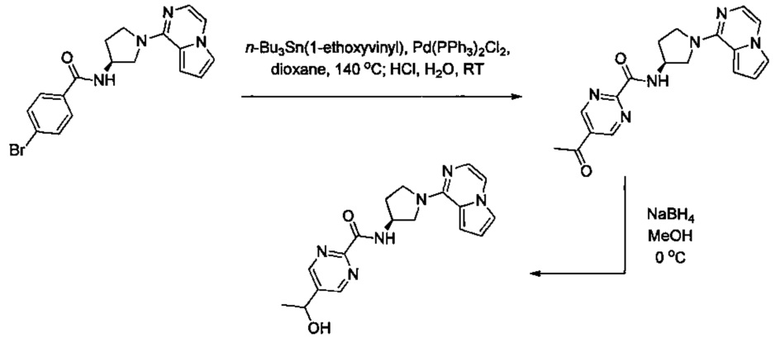
a) В толстостенный сосуд высокого давления на 15 мл вносили 4-бром-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]бензамид (289 мг, 0,747 ммоль), три-н-бутил (1-этоксивинил)олово (0,35 мл, 1,0 ммоль), дихлорбис(трифенилфосфин)палладий (30 мг, 0,042 ммоль) и дегазированный диоксан (4 мл). Сосуд запечатывали и погружали в предварительно нагретую (140°С) масляную баню. Через 25 минут сосуд вынимали из масляной бани, охлаждали до комнатной температуры, обрабатывали 6М HCl (0,40 мл, 2,4 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли 200 мл дихлорметана и 20 мл воды. Органический слой отделяли и откладывали в сторону, а водную фазу подщелачивали с помощью NaOH и экстрагировали дихлорметаном. Объединенные органические слои упаривали при пониженном давлении и оставшийся остаток очищали методом флэш-хроматографии (SiO2; 2-5% смеси СН3ОН/дихлорметан), получая 209 мг искомого соединения (выход 80%). MS: (ES) 351,2 [М+Н+].
b) Кетон, полученный на стадии а (56 мг, 0,16 ммоль), растворяли в 5 мл МеОН и охлаждали до 0°С на ледяной бане. Добавляли боргидрид натрия (7,2 мг, 0,19 ммоль) и перемешивали реакционную смесь в течение 20 минут при 0°С. Реакцию гасили 6М HCl, упаривали при пониженном давлении и сразу же очищали методом обратнофазовой препаративной HPLC (градиент 20-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 8.93 (s, 2Н), 7.79 (dd, J=1.2, 2.7 Hz, 1Н), 7.74 (d, J=5.9 Hz, 1H), 7.58 (d, J=3.5 Hz, 1H), 6.94 (dd, J=2.7, 5.7 Hz, 1H), 6.87 (d, J=5.9 Hz, 1H), 5.02-4.95 (d, J=6.6 Hz, 1H), 4.80-3.60 (br, 5H), 2.62-2.38 (m, 2H), 1.53 (d, J=6.6 Hz, 3H). MS: (ES) 353,2 [M+H+].
Пример 32. Синтез N-[(3R)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]-1-фенил-1,2,4-триазол-3-карбоксамида
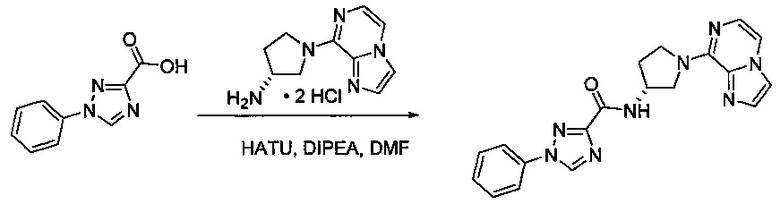
Смешивали 1-фенил-1,2,4-триазол-3-карбоновую кислоту (1,2 г, 6,3 ммоль), (3R)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-амин дигидрохлорид (2,0 г, 7,4 ммоль) и диизопропилэтиламин (13 мл, 75 ммоль) в 50 мл DMF. В эту смесь добавляли HATU (2,5 г, 6,6 ммоль). После перемешивания в течение 3 мин анализ пробы методом LCMS показал, что реакция завершилась. DIPEA и большую часть DMFA удаляли при пониженном давлении. Остаток разбавляли 400 мл EtOAc и промывали 50 мл насыщенного NaH2PO4. Было установлено, что большое количество искомого продукта находится в органической фазе, поэтому ее экстрагировали 400 мл 10% об. PrOH/CHCl3. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали. Некочищенньш продукт очищали методом флэш-хроматографии (SiO2, 2-5% смеси МеОН/дихлорметан), получая 936 мг искомого продукта (выход 39%). 1Н-ЯМР (400 MHz, CDCl3) δ 8.54 (s, 1Н), 7.74 (td, J=1.1, 7.5 Hz, 2H), 7.56-7.48 (m, 4H), 7.47-7.40 (m, 2H), 7.37 (d, J=8.2 Hz, 1H), 7.33 (d, J=4.3 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-4.35 (br, 1H), 4.30-4.15 (br, 3H), 3.45-3.30 (m, 1H), 2.90-2.75 (m, 1H), 2.50-2.38 (m, 1H), 2.25-2.15 (m, 1H). MS: (ES) 375,2 [М+Н+].
Пример 33. Синтез N-[(3S)-1-(3-этилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-1-фенил-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата
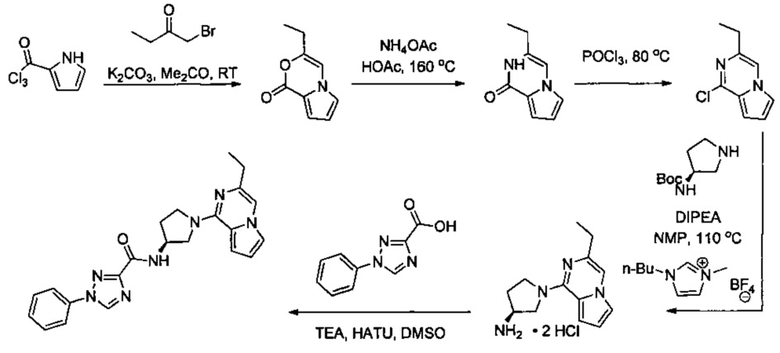
a) В кашицу из 2-(трихлорацетил)пиррола (380 мг, 1,79 ммоль) и K2CO3 (780 мг, 5,7 ммоль) в 5 мл ацетона добавляли раствор 1-бром-2-бутанона (400 мг, 2,7 ммоль) в 2 мл ацетона. Смесь перемешивали в течение 19 часов при комнатной температуре, после чего фильтровали и упаривали при пониженном давлении, получая 3-этил-1Н-пирроло [2,1-с][1,4]оксазин-1-он (298 мг, количественно), который использовали на следующей стадии без очистки.
b) В толстостенный сосуд высокого давления на 150 мл вносили лактон из стадии а, 3-этил-1Н-пирроло[2,1-с][1,4]оксазин-1-он (2,6 г, 16 ммоль), NH4OAC (6,0 г, 78 ммоль) и уксусную кислоту (40 мл). Сосуд запечатывали, погружали в предварительно нагретую масляную баню на 160°С и перемешивали в течение 3 ч. После этого удаляли уксусную кислоту при пониженном давлении, а остаток очищали методом флэш-хроматографии (SiO2, 2% МеОН/дихлорметан), получая 3-этилпирроло[1,2-а]пиразин-1(2Н)-он (760 мг, выход 29%). MS: (ES) 163,1 [М+Н+].
c) Смешивали 3-этилпирроло[1,2-а]пиразин-1(2Н)-он из стадии b (760 мг, 4,7 ммоль) с POCl3 (20 мл) и перемешивали при 80°С в течение 20 мин. Удаляли POCl3 при пониженном давлении. Остаток растворяли в DCM и дважды промывали насыщенным раствором бикарбоната натрия. После этого органическую фазу сушили над сульфатом магния, фильтровали и упаривали при пониженном давлении. Остаток использовали на следующей стадии без очистки.
d) Во флаконе на 20 мл смешили трет-бутил-N-[(3S)-пирролидин-3-ил]карбамат (610 мг, 3,3 ммоль), 1-хлор-3-этилпирроло[1,2-а]пиразин из стадии с (470 мг, 2,6 ммоль), DIPEA (2,0 ммоль, 11 ммоль) и 1-бутил-3-метилимидазолий тетрафторборат (100 мг, 0,4 ммоль) и нагревали смесь в течение 45 мин при 110°С. Добавляли 1-метилпирролидинон (1 мл), а затем еще трет-бутил-N-[(3S)-пирролидин-3-ил]карбамат (200 мг, 1,1 ммоль) и перемешивали реакцию еще 1 час при 110°С. Затем еще добавляли 400 мг (2,2 ммоль) аминопирролидона и перемешивали смесь при 110°С еще 20 мин. Реакционную смесь высушивали под вакуумом и очищали методом флэш-хроматографии (SiO2, 2% 7М NH3 в смеси МеОН/дихлорметан). После высушивания получали требуемое Вос-защищенное промежуточное соединение. Остаток растворяли в минимальном количестве метанола и дихлорметана и обрабатывали 4М HCl в диоксане (3 мл, 12 ммоль). Смесь нагревали при 50°С и перемешивали в течение 30 мин, после чего колбу вынимали из нагревателя, давали остыть до комнатной температуры и добавляли эфир. Образовавшийся коричневый осадок (3S)-1-(3-этилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амина дигидрохлорида собирали фильтрованием и высушивали под вакуумом; 638 мг, выход 86%.
е) Смешивали 1-фенил-1,2,4-триазол-3-карбоновую кислоту (34 мг, 0,18 ммоль), (3S)-1-(3-этилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (63 мг, 0,21 ммоль) и триэтиламин (0,13 мл, 0,93 ммоль) в 1 мл DMSO. Добавляли HATU (74 мг, 0,19 ммоль) и перемешивали смесь в течение 40 мин. Удаляли DMSO при пониженном давлении, а остаток очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 9.14 (s, 1Н), 9.05 (d, J=6.7 Hz, 1H), 7.88 (d, J=7.4 Hz, 2H), 7.71 (s, 1H), 7.60-7.46 (m, 5H), 6.89 (dd, J=2.8, 4.7 Hz, 1H), 4.95-4.80 (m, 1H), 4.78-3.80 (br, 4H), 2.68 (d, J=7.4 Hz, 2H), 2.60-2.40 (m, 2H), 1,33 (t, J=7.4 Hz, 3H). MS: (ES) 402,2 [М+Н+].
Пример 34. Синтез N-[(3S)-1-(3-этилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-1-(4-фторфенил)-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата

Смешивали 1-(4-фторфенил)-1,2,4-триазол-3-карбоновую кислоту (35 мг, 0,17 ммоль), (3S)-1-(3-этилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (60 мг, 0,20 ммоль) и триэтиламин (0,12 мл, 0,86 ммоль) в 1 мл DMSO. Добавляли HATU (72 мг, 0,19 ммоль) и перемешивали смесь в течение 50 мин. Удаляли DMSO при пониженном давлении, а остаток очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 9.10 (s, 1Н), 7.92-7.88 (m, 2Н), 7.71 (s, 1Н), 7.53 (s, 2H), 7.35 (t, J=8.8 Hz, 2H), 6.89 (dd, J=2.4, 4.7 Hz, 1H), 4.95-4.80 (m, 1H), 4.78-3.80 (br, 4H), 2.68 (d, J=7.4 Hz, 2H), 2.60-2.35 (m, 2H), 1,33 (t, J=7.4 Hz, 3H). MS: (ES) 420,2 [M+H+].
Пример 35. Синтез N-[(3S)-1-(3-этилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-2-(4-гидрокси-1-пиперидил)тиазол-4-карбоксамида в виде соли трифторацетата
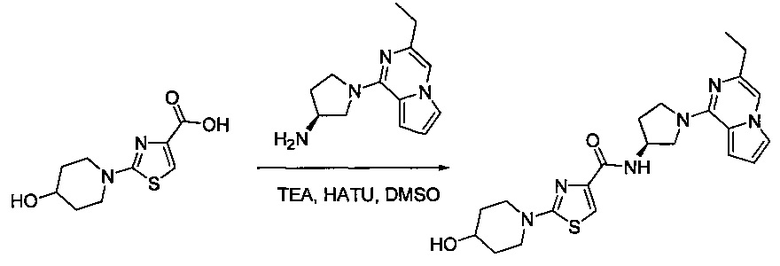
Смешивали 2-(4-гидрокси-1-пиперидил)тиазол-4-карбоновую кислоту (40 мг, 0,18 ммоль), (3S)-1-(3-этилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (60 мг, 0,20 ммоль) и триэтиламин (0,13 мл, 0,93 ммоль) в 1 мл DMSO. Добавляли HATU (77 мг, 0,20 ммоль) и перемешивали смесь в течение 40 мин. Удаляли DMSO при пониженном давлении, а остаток очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 7.70 (d, J=2.4 Hz, 1H), 7.52-7.50 (m, 2H), 7.41 (s, 1H), 6.89 (dd, J=2.4,4.3 Hz, 1H), 4.95-4.78 (m, 2H), 4.60-3.70 (br, 4H), 3.95-3.80 (m, 2H), 3.36-3.20 (m, 2H), 2.68 (d, J=7.4 Hz, 2H), 2.60-2.30 (m, 2H), 1.98-1.90 (m, 2H), 1.62-1.55 (m, 2H), 1,33 (t, J=7.4 Hz, 3H). MS: (ES) 441,2 [М+Н+].
Пример 36. Синтез 1-(4-фторфенил)-N-[(3R)-1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил]-1,2,4-триазол-3-карбоксамида

Смешивали 1-(4-фторфенил)-1,2,4-триазол-3-карбоновую кислоту (300 мг, 1,4 ммоль), (3R)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амин дигидрохлорид (400 мг, 1,4 ммоль) и триэтиламин (1,5 мл, 11 ммоль) в 8 мл DMF. Смесь перемешивали в течение 30 мин, после чего добавляли HATU (576 мг, 1,5 ммоль) и перемешивали смесь при 60°С в течение 1 ч. Смесь подвергали хроматографии (SiO2, 5-20% MeOH/EtOAc). Часть остатка, полученного при флэш-хроматографии, очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и упаривали при пониженном давлении, получая 70 мг соли TFA искомого соединения. Соль TFA превращали в свободное основание путем суспендирования ее в насыщенном растворе NaCl (1 мл), добавления 1М NaOH (0,6 мл, 0,6 ммоль) и экстрагирования дихлорметаном. Удаление дихлорметана давало стеклообразный остаток. Образец растворяли в CH3CN/H2O и высушивали (лиофилизировали), получая белый порошок (28 мг, выход 5%). 1Н-ЯМР (400 MHz, DMSO) δ 9.31 (s, 1Н), 8.92 (d, J=5.9 Hz, 1H), 7.92-7.85 (m, 3H), 7.76 (s, 1H), 7.53 (s, 1H), 7.41 (t, J=9.0 Hz, 2H), 7.21 (d, J=4.7 Hz, 1H), 4.65-4.50 (m, 1H), 4.48-3.65 (br, 4H), 2.30-2.05 (m, 2H). MS: (ES) 393,2 [М+Н+].
Пример 37. Синтез N-[(3S)-1-(3-изопропилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-1-фенил-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата

a) В кашицу из 2-(трихлорацетил)пиррола (5,3 г, 25 ммоль) и K2CO3 (11 г, 76 ммоль) в 100 мл ацетона добавляли раствор 1-бром-3-метилбутан-2-она (4,5 г, 27 ммоль) в 30 мл ацетона. Смесь перемешивали в течение 1 ч при 50°С, а затем фильтровали и упаривали при пониженном давлении, получая 3-изопропил-1Н-пирроло[2,1-с][1,4]оксазин-1-он (4,1 г, 93%), который использовали на следующей стадии без очистки.
b) В толстостенный стеклянный сосуд высокого давления на 150 мл вносили лактон из стадии а, 3-изопропил-1Н-пирроло[2,1-с][1,4]оксазин-1-он (4,1 г, 23 ммоль), NH4OAc (9,4 г, 122 ммоль) и уксусную кислоту (25 мл). Сосуд запечатывали тефлоновой резьбовой пробкой, погружали в масляную баню, предварительно нагретую до 160°С, и перемешивали в течение 9 часов при этой температуре. Сосуд вынимали из бани, добавляли еще 10 г (130 ммоль) NH4OAc и перемешивали реакцию при 160°С еще в течение 3 часов. После этого уксусную кислоту удаляли при пониженном давлении, а остаток суспендировали в 150 мл DCM и 30 мл насыщенного NaHCO3. После фильтрования через целит водную фазу отделяли и отбрасывали, а органическую фазу упаривали и очищали методом флэш-хроматографии (SiO2, 1% МеОН/дихлорметан), получая 2,0 г 3-изопропилпирроло[1,2-а]пиразин-1(2Н)-она (выход 49%). MS: (ES) 177,2 [М+Н+].
c) Смешивали 3-изопропилпирроло[1,2-а]пиразин-1(2Н)-он из стадии b (1,6 г, 4,7 ммоль) с POCl3 (20 мл) и перемешивали при 70°С в течение 20 мин. POCl3 удаляли при пониженном давлении. Остаток растворяли в DCM и дважды промывали насыщенным раствором бикарбоната натрия. После этого органическую фазу сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии (SiO2, 5-10% МТВЕ/гексан), получая хлор-3-изопропилпирроло[1,2-а]пиразин (1,09 г, выход 62%).
d) Во флаконе на 20 мл смешивали трет-бутил-N-[(3S)-пирролидин-3-ил]карбамат (2,06 г, 11,1 ммоль), 1-хлор-3-изопропилпирроло[1,2-а]пиразин из стадии с (1,09 г, 5,6 ммоль), DIPEA (4,0 ммоль, 23 ммоль) и 1-метилпирролидинон (1 мл) и нагревали смесь с перемешиванием во флаконе при 110°С в течение 1 ч. Температуру понижали до 90°С и перемешивали реакционную смесь еще в течение 16 часов. После этого органическую фазу реакционной смеси разбавляли DCM (100 мл), промывали водой (20 мл), сушили (Na2SO4), фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографи (SiO2, 2% 7М NH3 в смеси МеОН/дихлорметан). После высушивания получали искомое Вос-защищенное промежуточное соединение. Его суспендировали в минимальном количестве метанола и дихлорметана и обрабатывали 4М HCl в диоксане (4 мл, 16 ммоль). Смесь нагревали при 50°С и перемешивали в течение 20 мин, после чего колбу вынимали из нагревателя, давали остыть до комнатной температуры и добавляли диоксан. Образовавшийся белый осадок (3S)-1-(3-изопропилпирроло[1,2-а]пиразин-1-ил)-пирролидин-3-амина дигидрохлорида собирали фильтрованием и высушивали под вакуумом; 925 мг, выход 52%.
e) Смешивали 1-фенил-1,2,4-триазол-3-карбоновую кислоту (54 мг, 0,29 ммоль), (3S)-1-(3-изопропилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (98 мг, 0,31 ммоль) и триэтиламин (0,16 мл, 1,2 ммоль) в 0,6 мл DMSO. Добавляли HATU (110 мг, 0,29 ммоль) и перемешивали смесь в течение 1 ч. DMSO удаляли при пониженном давлении, а остаток очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1H-ЯМР (400 MHz, CD3OD) δ 9.14 (s, 1Н), 7.89 (d, J=7.4 Hz, 2H), 7.73 (s, 1H), 7.60-7.46 (m, 5H), 6.90 (dd, J=2.7, 4.3 Hz, 1H), 5.00-4.80 (m, 1H), 4.60-3.85 (br, 4H), 3.05-2.95 (m, 1H), 2.60-2.38 (m, 2H), 1.35 (d, J=6.6 Hz, 6H). MS: (ES) 416,2 [M+H+].
Пример 38. Синтез 1-(4-фторфенил)-N-[(3S)-1-(3-изопропилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата
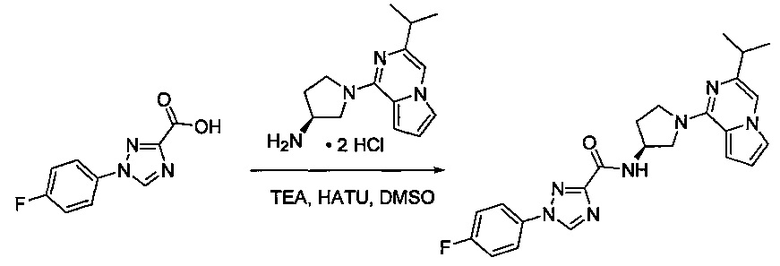
Смешивали 1-(4-фторфенил)-1,2,4-триазол-3-карбоновую кислоту (53 мг, 0,26 ммоль), (3S)-1-(3-изопропилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (91 мг, 0,29 ммоль) и триэтиламин (0,16 мл, 1,15 ммоль) в 0,6 мл DMSO. Добавляли HATU (104 мг, 0,27 ммоль) и перемешивали смесь в течение 1 ч. DMSO удаляли при пониженном давлении, а остаток очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1H-ЯМР (400 MHz, CD3OD) δ 9.10 (s, 1H), 7.92 (dd, J=4.7, 7.0 Hz, 2H), 7.73 (s, 1H), 7.57 (s, 1H), 7.54 (d, J=4.3 Hz, 1H), 7.35 (t, J=8.6 Hz, 2H), 6.91 (dd, J=2.7, 4.3 Hz, 1H), 5.00-4.85 (m, 1H), 4.60-3.65 (br, 4H), 3.05-2.95 (m, 1H), 2.60-2.38 (m, 2H), 1.35 (d, J=7.0 Hz, 6H). MS: (ES) 434,2 [M+H+].
Пример 39. Синтез 2-(4-гидрокси-1-пиперидил)-N-[(3S)-1-(3-изопропилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]тиазол-4-карбоксамида в виде соли трифторацетата
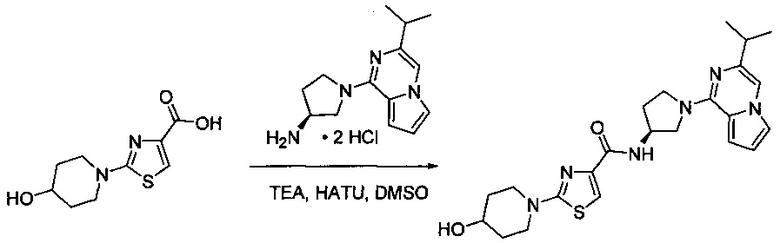
Смешивали 2-(4-гидрокси-1-пиперидил)тиазол-4-карбоновую кислоту (54 мг, 0,24 ммоль), (3S)-1-(3-изопропилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин дигидрохлорид (81 мг, 0,26 ммоль) и триэтиламин (0,14 мл, 1,0 ммоль) в 0,6 мл DMSO. Добавляли HATU (104 мг, 0,27 ммоль) и перемешивали смесь в течение 25 мин. DMSO удаляли при пониженном давлении, а остаток очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 7.72 (d, J=2.4 Hz, 1H), 7.56 (s, 1H), 7.52 (d, J=4.3 Hz, 1H), 7.41 (s, 1H), 6.90-6.87 (m, 1H), 4.95-4.70 (m, 2H), 4.60-3.80 (br, 4H), 3.95-3.80 (m, 2H), 3.36-3.20 (m, 2H), 3.05-2.95 (m, 1H), 2.60-2.30 (m, 2H), 1.98-1.90 (m, 2H), 1.62-1.55 (m, 2H), 1.35 (d, J=6.6 Hz, 6H). MS: (ES) 455,2 [M+H+].
Пример 40. Синтез 1-(2-фторфенил)-N-[(3S)-1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил]-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата
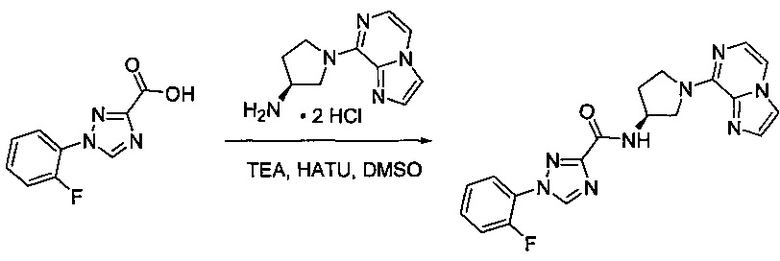
Смешивали 1-(2-фторфенил)-1,2,4-триазол-3-карбоновую кислоту (41 мг, 0,20 ммоль), (3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-амин дигидрохлорид (69 мг, 0,25 ммоль) и триэтиламин (0,11 мл, 0,80 ммоль) в 0,5 мл DMSO. Добавляли HATU (80 мг, 0,21 ммоль) и перемешивали смесь в течение 10 мин. После этого добавляли уксусную кислоту (0,1 мл), МеОН (0,4 мл) и воду (1 мл), а затем фильтровали и очищали остаток методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая 40 мг искомого соединения (выход 40%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.96 (d, J=2.7 Hz, 1H), 8.05 (s, 1H), 7.95-7.82 (m, 3H), 7.56-7.38 (m, 3H), 7.17 (d, J=5.5 Hz, 1H), 5.00-3.65 (br, 5H), 2.60-2.38 (m, 2H). MS: (ES) 393,2 [M+H+].
Пример 41. Синтез N-[(3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]-1-(4-метилсульфонилфенил)-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата
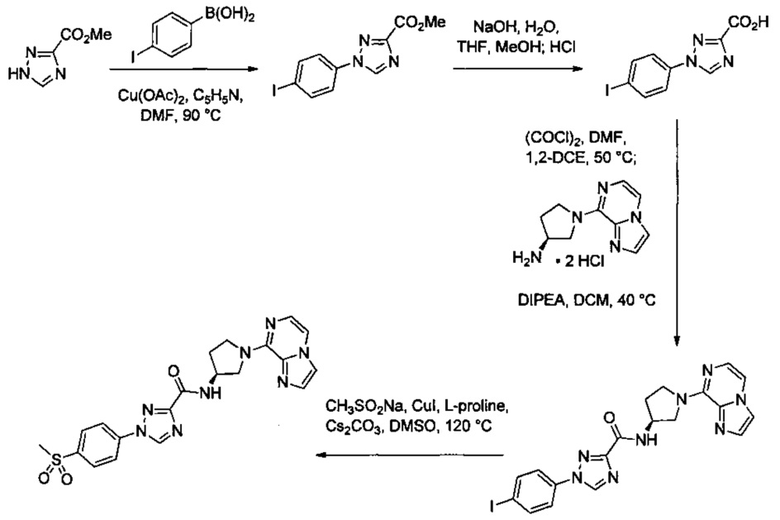
a) В колбу на 100 мл вносили метил-1Н-1,2,4-триазол-3-карбоксилат (2,8 г, 16 ммоль), 4-йодфенилборную кислоту (4,0 г, 16 ммоль), Cu(ОАс)2 (3,3 г, 18 ммоль), пиридин (1,5 мл, 19 ммоль) и DMF (30 мл). Смесь перемешивали на предварительно нагретой до 90°С масляной бане в течение 30 мин, при этом раствор изменял окраску с темно-синей на светло-зеленую. Смесь разбавляли EtOAc (200 мл), фильтровали и промывали смесью насыщенный NH4Cl-30% NH4OH 3:1 об/об. Органическую фазу упаривали под вакуумом, а полученный остаток очищали методом флэш-хроматографии (SiO2, 20%-60% EtOAc/гексан), получая белый порошок метил-1-(4-йодфенил)-1,2,4-триазол-3-карбоксилата (830 мг, выход 16%).
b) Растворяли метил-1-(4-йодфенил)-1,2,4-триазол-3-карбоксилат (830 мг, 2,5 ммоль) в МеОН (20 мл) и THF (10 мл). Добавляли 1М NaOH (2,5 мл, 2,5 ммоль) и перемешивали смесь при комнатной температуре в течение 2 ч. Добавляли еще 1М NaOH (2,5 мл, 2,5 ммоль) и нагревали реакционную смесь при 50°С в течение 30 минут. После охлаждения до комнатной температуры доводили до рН 4 добавлением 6М HCl, удаляли МеОН и THF при пониженном давлении, а образовавшийся белый осадок собирали фильтрованием и сушили под вакуумом, получая 680 мг (выход 86%) 1-(4-йодфенил)-1,2,4-триазол-3-карбоновой кислоты.
c) В суспензию 1-(4-йодфенил)-1,2,4-триазол-3-карбоновой кислоты (680 мг, 2,15 ммоль) в 1,2-дихлорэтане (8 мл) добавляли оксалилхлорид (0,30 мл, 3,4 ммоль), а затем DMF (0,020 мл, 0,26 ммоль). Смесь перемешивали на масляной бане при 50°С в течение 5 мин, а затем упаривали при пониженном давлении и высушивали под вакуумом. Полученный остаток суспендировали в DCM (10 мл) и добавляли к нему (3S)-1-имидазо-[1,2-а]пиразин-8-илпирролидин-3-амин дигидрохлорид (650 мг, 2,36 ммоль), а затем еще DIPEA (1,5 мл, 8,6 ммоль). Смесь перемешивали, быстро доводили до кипения, снимали с нагревателя и перемешивали еще 20 минут. После упаривания при пониженном давлении остаток очищали методом флэш-хроматографии (SiO2; 2% МеОН/дихлорметан), получая 886 мг белой пены (выход 82%).
d) Смешивали N-[(3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]-1-(4-йодфенил)-1,2,4-триазол-3-карбоксамид из стадии с (73 мг, 0,15 ммоль), метансульфинат натрия (31 мг, 0,30 ммоль), CuI (5,4 мг, 0,028 ммоль), пролин (6,8 мг, 0,059 ммоль), Cs2CO3 (23 мг, 0,70 ммоль) и DMSO (0,40 мл) во флаконе на 4 мл и перемешивали при 120°С в течение 3 ч. Реакционную смесь очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 9.31 (s, 1Н), 9.15 (d, J=7.0 Hz, 0.5H), 8.20-8.10 (m, 4H), 8.05 (s, 1H), 7.95 (d, J=5.4 Hz, 1H), 7.82 (s, 1H), 7.18 (d, J=5.8 Hz, 1H), 5.00-3.65 (br, 5H), 3.18 (s, 3H), 2.60-2.38 (m, 2H). MS: (ES) 453,2 [M+H+].
Пример 42. Синтез N-[(3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]-1-(п-толил)-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата
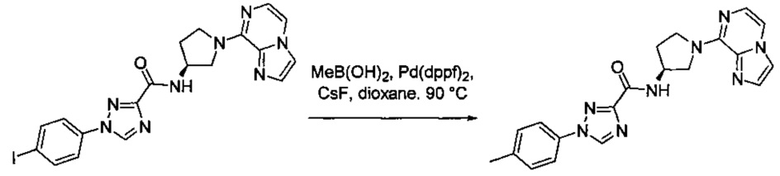
Во флаконе на 4 мл смешивали N-[(3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]-1-(4-йодфенил)-1,2,4-триазол-3-карбоксамид (64 мг, 0,13 ммоль), метилборную кислоту (32 мг, 0,53 ммоль), (1,1'-бис(дифенилфосфино)ферроцен)палладий (II) дихлорид (9,5 мг, 0,013 ммоль), CSF (67 мг, 0,44 ммоль) и дегазированный диоксан (0,80 мл). Флакон запечатывали и перемешивали при 90°С в течение 2 ч. Диоксан удаляли при пониженном давлении, а остаток очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1H-ЯМР (400 MHz, CD3OD) δ 9.07 (s, 1Н), 8.05 (s, 1H), 7.94 (d, J=5.9 Hz, 1H), 7.82 (s, 1H), 7.74 (d, J=8.6 Hz, 2H), 7.38 (d, J=8.6 Hz, 2H), 7.17 (d, J=5.4 Hz, 1H), 5.00-3.65 (br, 5H), 2.60-2.38 (m, 2H), 2.41 (s, 1H). MS: (ES) 389,2 [M+H+].
Пример 43. Синтез 1-(4-фторфенил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]-1,2,4-триазол-3-карбоксамида в виде соли трифторацетата
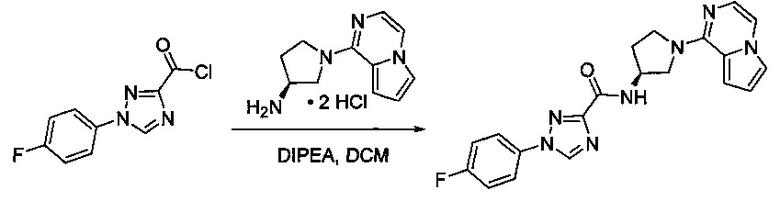
В суспензию 1-(4-фторфенил)-1,2,4-триазол-3-карбонилхлорида (70 мг, 0,31 ммоль) в DCM (10 мл) добавляли (3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-амин дигидрохлорид (104 мг, 0,38 ммоль), а затем DIPEA (0,70 мл, 4,0 ммоль). Реакционную смесь перемешивали в течение 10 мин, упаривали при пониженном давлении, очищали методом обратнофазовой препаративной HPLC (градиент 10-30% CH3CN/H2O с добавлением 0,1% TFA) и высушивали (лиофилизировали), получая искомое соединение. 1Н-ЯМР (400 MHz, CD3OD) δ 9.10 (s, 1Н), 7.91-7.88 (m, 2H), 7.79 (dd, J=1.2, 2.8 Hz, 1H), 7.74 (d, J=5.9 Hz, 1H), 7.59 (d, J=4.3 Hz, 1H), 7.34 (t, J=8.6 Hz, 2H), 6.95 (dd, J=2.8, 4.7 Hz, 1H), 6.86 (d, J=5.5 Hz, 1H), 5.00-4.85 (m, 1H), 4.80-3.65 (br, 4H), 2.60-2.38 (m, 2H). MS: (ES) 392,2 [M+H+].
Пример 44. Синтез 1-(4-цианофенил)-N-[(3S)-1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил]-1,2,4-триазол-3-карбоксамида
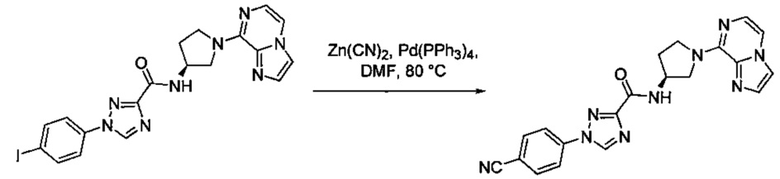
Во флакон на 4 мл вносили N-[(3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]-1-(4-йодфенил)-1,2,4-триазол-3-карбоксамид (82 мг, 0,16 ммоль), Zn(CN)2 (24 мг, 0,20 ммоль), тетракис(трифенилфосфин)палладий(0) (23 мг, 0,020 ммоль) и дегазированный DMF. Флакон запечатывали и перемешивали смесь при 80°С в течение 1 ч. DMF удаляли при пониженном давлении, а остаток очищали методом флэш-хроматографии (SiO2, 7М NH3 в МеОН/дихлорметане), получая 48 мг белого порошка искомого продукта (выход 74%). 1Н-ЯМР (400 MHz, CDCl3) δ 8.64 (s, 1Н), 7.94 (d, J=6.6 Hz, 2H), 7.85 (d, J=6.6 Hz, 2H), 7.55 (s, 1H), 7.50 (s, 1H), 7.44 (d, J=4.3 Hz, 1H), 7.40-7.30 (m, 2H), 5.00-4.85 (m, 1H), 4.50-4.35 (br, 1H), 4.30-4.15 (br, 3H), 2.50-2.38 (m, 1H), 2.25-2.15 (m, 1H). MS: (ES) 400,1 [М+Н+].
Пример 45. Синтез 1-(4-хлорфенил)-N-[(3S)-3-(гидроксиметил)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]пиразол-3-карбоксамида
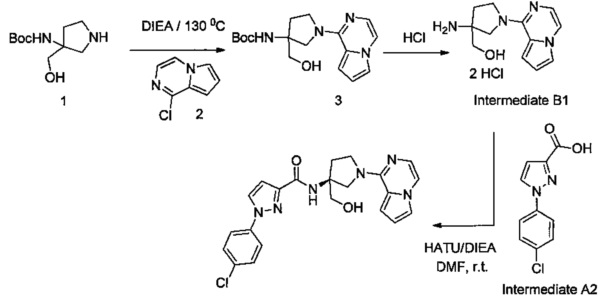
a) В смесь из соединения 1 (1,0 г, 4,6 ммоль) и 580 мг соединения 2 (580 мг, 3,8 ммоль) добавляли 1,7 мл основания Хюнига (9,5 ммоль). Полученную смесь перемешивали при 130°С в течение 3 ч. После охлаждения смеси до комнатной температуры добавляли 200 мл изопропанол/хлороформа (1:2), а органические слои промывали насыщенным водным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×50 мл). Органический слой сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (0-10% МеОН в EtOAc), получая искомый продукт 3 в виде коричневого порошка (700 мг, 46%).
b) Смесь из соединения 3 (332 мг, 1,0 ммоль) и 3 мл 4,0 М HCl в диоксане (12 ммоль) перемешивали при 50°С в течение 1 ч. Затем смесь упаривали при пониженном давлении, получая коричневый порошок (300 мг, 98%), который использовали на следующей стадии без дальнейшей очистки.
c) Во флакон на 10 мл вносили промежуточное соединение А2 (92 мг, 0,417 ммоль), В1 (122 мг, 0,40 ммоль), HATU (166 мг, 0,437 ммоль), основание Хюнига (146 мг, 1,12 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (105 мг, 60%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.28 (d, J=2.5 Hz, 1H), 7.85 (dd, J=2.2, 6.9 Hz, 2H), 7.48 (dd, J=2.2, 6.9 Hz, 2H), 7.42 (d, J=5.2 Hz, 1H), 7.39 (dd, J=1.1, 2.6 Hz, 1H), 6.98 (d, J=4.4 Hz, 1H), 6.93 (d, J=2.6 Hz, 1H), 6.86 (d, J=5.1 Hz, 1H), 6.65 (dd, J=2.5, 4.1 Hz, 1H), 4.34 (d, J=11.4 Hz, 1H), 4.15 (d, J=11.4 Hz, 1H), 4.02-3.88 (m, 4H), 2.65-2.55 (m, 1H), 2.45-2.36 (m, 1H). MS: (ES) m/z теор. для C22H21ClN6O2 [M+H]+ 437,2, факт. 437.
Пример 46. Синтез 1-(4-фторфенил)-N-[(3S)-3-(гидроксиметил)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил] пиразол-3-карбоксамида
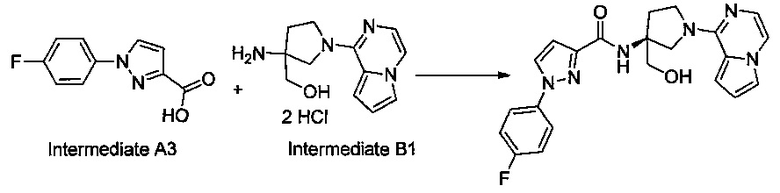
Во флакон на 10 мл вносили промежуточное соединение A3 (83 мг, 0,417 ммоль), В1 (122 мг, 0,40 ммоль), HATU (166 мг, 0,437 ммоль), основание Хюнига (146 мг, 1,12 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом HPLC, получая искомый продукт (120 мг, 71%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.22 (d, J=2.6 Hz, 1H), 7.86-7.82 (m, 2H), 7.42 (d, J=4.7 Hz, 1H), 7.38 (t, J=1.5 Hz, 1H), 7.21 (dt, J=2.2, 8.4 Hz, 2H), 6.98 (d, J=4.4 Hz, 1H), 6.91 (d, J=2.5 Hz, 1H), 6.86 (d, J=4.7 Hz, 1H), 6.64 (dd, J=2.5, 4.0 Hz, 1H), 4.34 (d, J=11.4 Hz, 1H), 4.15 (d, J=11.4 Hz, 1H), 4.02-3.88 (m, 4H), 2.65-2.55 (m, 1H), 2.45-2.36 (m, 1H). MS: (ES) m/z теор. для C22H21FN6O2 [M+H]+ 421,2, факт. 421.
Пример 47. Синтез N-[(3S)-3-(гидроксиметил)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]-2-(4-гидрокси-1-пиперидил)тиазол-4-карбоксамида
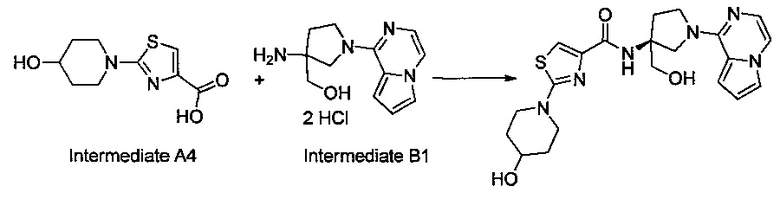
Во флакон на 10 мл вносили промежуточное соединение А4 (90 мг, 0,395 ммоль), В1 (122 мг, 0,40 ммоль), HATU (166 мг, 0,437 ммоль), основание Хюнига (146 мг, 1,12 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом HPLC, получая искомый продукт (85 мг, 49%). 1H-ЯМР (400 MHz, CD3OD) δ 8.07 (s, 1Н), 7.80-7.76 (m, 1H), 7.72 (d, J=5.6 Hz, 1H), 7.56 (d, J=4.4 Hz, 1H), 7.37 (s, 1H), 6.94 (dd, J=2.4, 4.4 Hz, 1H), 6.85 (d, J=6.0 Hz, 1H), 4.80-4.60 (br, 1H), 4.50-4.20 (br, 2H), 4.02-3.80 (m, 4H), 3.35-3.25 (m, 4H), 2.78-2.66 (m, 1H), 2.56-2.45 (m, 1H), 1.98-1.88 (m, 2H), 1.64-1.52 (m, 2H). MS: (ES) m/z теор. для C21H26N6O3S [M+H]+ 443,2, факт. 443.
Пример 48. Синтез N-[(3S)-3-(гидроксиметил)-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил]-1-фенил-1,2,4-триазол-3-карбоксамида
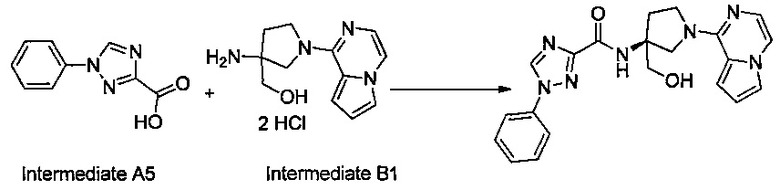
Во флакон на 10 мл вносили промежуточное соединение А5 (76 мг, 0,40 ммоль), В1 (122 мг, 0,40 ммоль), HATU (166 мг, 0,437 ммоль), основание Хюнига (146 мг, 1,12 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (110 мг, 62%). 1H-ЯМР (400 MHz, CD3OD) δ 9.12 (s, 1Н), 7.85 (td, J=1.5, 7.4 Hz, 2H), 7.76 (d, J=2.2 Hz, 1H), 7.70 (d, J=5.5 Hz, 2H), 7.55 (m, 2H), 7.46 (m, 1H), 6.92 (s, 1H), 6.83 (d, J=5.5 Hz, 1H), 4.60-3.80 (br, 4H), 4.02 (s, 2H), 2.82-2.70 (m, 1H), 2.58-2.48 (m, 1H). MS: (ES) m/z теор. для C21H21N7O2 [M+H]+ 404,2, факт. 404.
Пример 49. Синтез 1-(4-фторфенил)-N-[(3S)-3-(гидроксиметил)-1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил]пиразол-3-карбоксамида
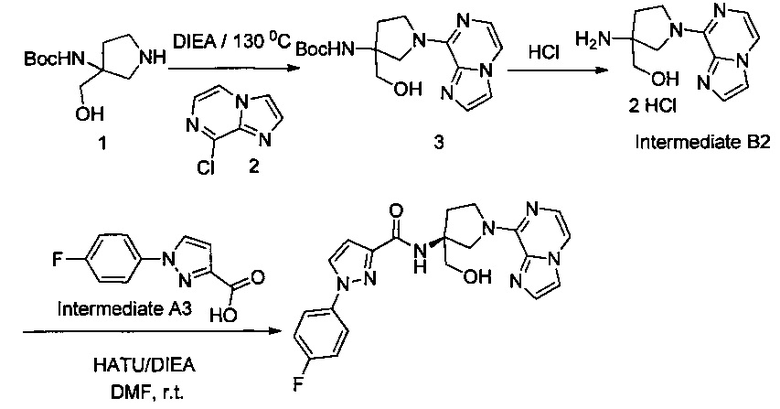
a) В смесь из соединения 1 (1,0 г, 4,6 ммоль) и 580 мг соединения 2 (580 мг, 3,8 ммоль) добавляли 1,7 мл основания Хюнига (9,5 ммоль). Полученную смесь перемешивали при 130°С в течение 3 ч. После охлаждения смеси до комнатной температуры добавляли 200 мл смеси изопропанол/хлороформ (1:2), а органические слои промывали насыщенным водным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×50 мл). Органический слой сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (0-10% МеОН в EtOAc), получая искомый продукт 3 в виде коричневого порошка (900 мг, 58%).
b) Смесь из соединения 3 (333 мг, 1 ммоль) и 3 мл 4,0 М HCl в диоксане (12 ммоль) перемешивали при 50°С в течение 1 ч. Затем смесь упаривали при пониженном давлении, получая коричневый порошок (300 мг, 98%), который использовали на следующей стадии без дальнейшей очистки.
c) Во флакон на 10 мл вносили промежуточное соединение A3 (86 мг, 0,417 ммоль), В2 (120 мг, 0,40 ммоль), HATU (166 мг, 0,437 ммоль), основание Хюнига (146 мг, 1,12 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом HPLC, получая искомый продукт (118 мг, 71%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.23 (d, J=2.5 Hz, 1H), 8.01 (d, J=1.1 Hz, 1H), 7.89 (d, J=5.7 Hz, 1H), 7.86-7.82 (m, 2H), 7.78 (s, 1H), 7.22 (t, J=8.8 Hz, 2H), 7.12 (d, J=5.5 Hz, 1H), 6.91 (d, J=2.6 Hz, 1H), 4.60-3.80 (br, 4H), 4.00 (s, 2H), 2.80-2.70 (m, 1H), 2.56-2.46 (m, 1H). MS: (ES) m/z теор. для C21H20FN7O2 [M+H]+ 422,2, факт. 422.
Пример 50. Синтез N-[(3S)-3-(гидроксиметил)-1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил]-2-(4-гидрокси-1-пиперидил)тиазол-4-карбоксамида
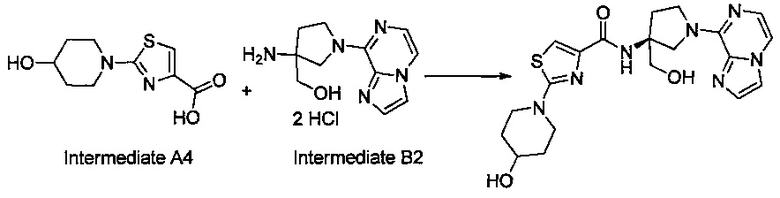
Во флакон на 10 мл вносили промежуточное соединение А4 (90 мг, 0,395 ммоль), В2 (120 мг, 0,40 ммоль), HATU (166 мг, 0,437 ммоль), основание Хюнига (146 мг, 1,12 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (81 мг, 49%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.02 (s, 1Н), 7.90 (d, J=5.5 Hz, 1H), 7.79 (s, 1H), 7.35 (s, 2H), 7.14 (d, J=5.8 Hz, 1H), 4.60-3.80 (br, 4H), 3.94 (s, 2H), 3.90-3.80 (m, 3H), 3.32-3.20 (m, 2H), 2.75-2.65 (m, 1H), 2.52-2.45 (m, 1H), 1.97-1.87 (m, 2H), 1.64-1.50 (m, 2H). MS: (ES) m/z теор. для C20H25N7O3S [M+H]+ 444,2, факт. 444.
Пример 51. Синтез N-[(3S)-3-(гидроксиметил)-1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил]-1-фенил-1,2,4-триазол-3-карбоксамида
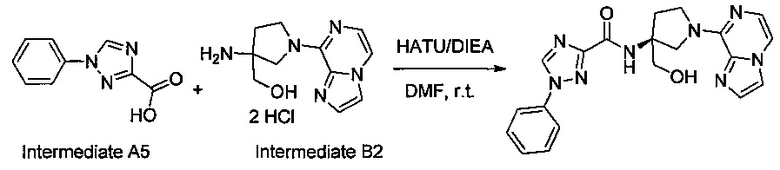
Во флакон на 10 мл вносили промежуточное соединение А5 (76 мг, 0,40 ммоль), В2 (122 мг, 0,40 ммоль), HATU (166 мг, 0,437 ммоль), основание Хюнига (146 мг, 1,12 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (110 мг, 62%). 1H-ЯМР (400 MHz, CD3OD) δ 9.12 (s, 1Н), 8.02 (d, J=1.1 Hz, 1H), 7.90 (d, J=5.9 Hz, 1H), 7.84 (dd, J=1.5, 8.8 Hz, 2H), 7.79 (s, 1H), 7.55 (t, J=7.3 Hz, 2H), 7.46 (t, J=7.3 Hz, 1H), 7.13 (d, J=5.5 Hz, 1H), 4.02 (s, 2H), 4.60-3.80 (br, 4H), 2.80-2.70 (m, 1H), 2.58-2.48 (m, 1H). MS: (ES) m/z теор. для C20H20N8O2 [M+H]+ 405,2, факт. 405.
Пример 52. Синтез 1-(4-фторфенил)-N-[(3R,4S)-4-гидрокси-1-пирроло[1,2-а]-пиразин-1-ил-пирролидин-3-ил]пиразол-3-карбоксамида
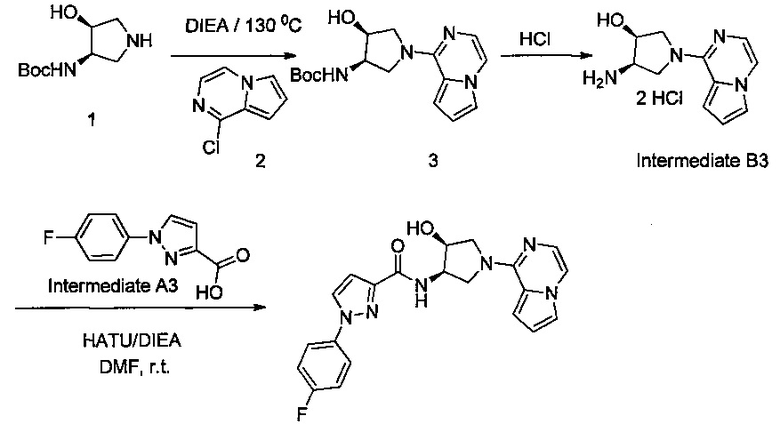
a) В смесь из соединения 1 (120 мг, 0,59 ммоль) и 2 (90 мг, 0,59 ммоль) добавляли 1,0 мл основания Хюнига (5,5 ммоль). Полученную смесь перемешивали при 130°С в течение 3 ч. После охлаждения смеси до комнатной температуры добавляли 200 мл смеси изопропанол/хлороформ (1:2), а органические слои промывали насыщенным водным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×50 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (0-10% МеОН в EtOAc), получая искомый продукт 3 в виде коричневого порошка (150 мг, 79%).
b) Смесь из соединения 3 (150 мг, 0,47 ммоль) и 2 мл 4,0 М HCl в диоксане (8,0 ммоль) перемешивали при 50°С в течение 1 ч. Затем смесь упаривали при пониженном давлении, получая коричневый порошок (130 мг, 95%), который использовали на следующей стадии без дальнейшей очистки.
c) Во флакон на 10 мл вносили промежуточное соединение A3 (43 мг, 0,209 ммоль), В2 (60 мг, 0,20 ммоль), HATU (83 мг, 0,218 ммоль), основание Хюнига (73 мг, 0,56 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (54 мг, 50%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.29 (d, J=2.6 Hz, 1H), 7.89-7.84 (m, 2H), 7.77 (d, J=1.5 Hz, 1H), 7.72 (d, J=5.9 Hz, 1H), 7.57 (d, J=4.4 Hz, 1H), 7.26 (t, J=8.8 Hz, 2H), 6.98 (d, J=2.2 Hz, 1H), 6.92 (t, J=3.0 Hz, 1H), 6.84 (d, J=5.8 Hz, 1H), 4.85 (m, 1H), 4.64 (m, 1H), 4.70-3.60 (br, 4H). MS: (ES) теор. для C21H19FN6O2 [M+H]+ 407,2, факт. 407.
Пример 53. Синтез промежуточного соединения A1
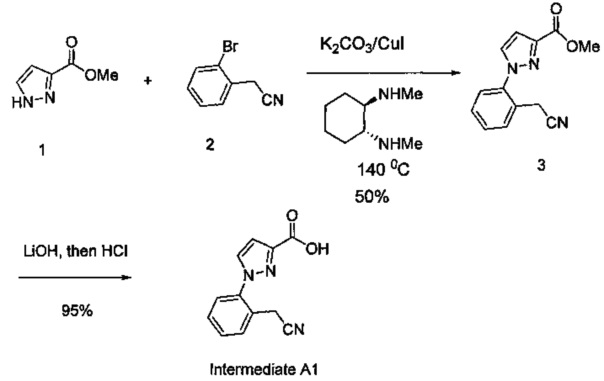
a) В колбу на 50 мл вносили 1,89 г исходного вещества 1 (15 ммоль), 1,96 г исходного вещества 2 (10 ммоль), 400 мг CuI (2,0 ммоль), 4,5 г K2CO3 (3,3 ммоль) и 0,9 мл транс-N,N'-диметилциклогексилдиамина (2,0 ммоль). Полученную смесь перемешивали при 140°С в течение 3 ч. После охлаждения смеси до комнатной температуры добавляли 200 мл EtOAc, а органические слои промывали водой (2×50 мл) и насыщенным раствором NaCl (2×50 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле (0-25% EtOAc в гексане), получая искомый продукт 3 (1,2 г, 50%).
b) В раствор сложного эфира 3 (240 мг, 1 ммоль) в THF добавляли 3,0 мл 1,0 М LiOH (3,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли 1,0 М HCl для доведения до рН 1,0, а органические слои экстрагировали EtOAc (2×100 мл), после чего сушили над MgSO4. После упаривания при пониженном давлении получали продукт виде белого твердого вещества (217 мг, 96%), которое использовали на следующей стадии без дальнейшей очистки.
Пример 54. Синтез 1-[2-(цианометил)фенил]-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]пиразол-3-карбоксамида
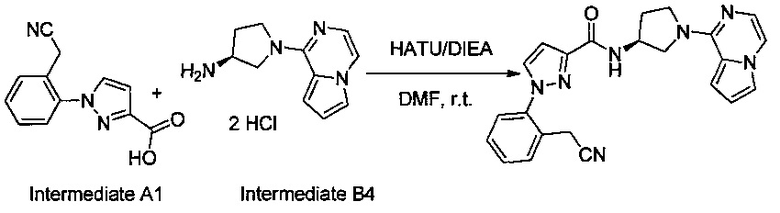
Во флакон на 10 мл вносили промежуточное соединение А1 (45 мг, 0,20 ммоль), В4 (55 мг, 0,20 ммоль), HATU (83 мг, 0,22 ммоль), основание Хюнига (73 мг, 0,6 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (56 мг, 68%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.03 (d, J=2.2 Hz, 1H), 7.75 (s, 1H), 7.70 (d, J=5.5 Hz, 1H), 7.60-7.48 (m, 5H), 6.98 (d, J=2.5 Hz, 1H), 6.91 (dd, J=2.6, 4.4 Hz, 1H), 6.82 (d, J=5.8 Hz, 1H), 4.95-3.65 (br, 5H), 3.93 (s, 2H), 2.55-2.30 (m, 2H). MS: (ES) m/z теор. для C23H21N7O [M+H]+ 412,2, факт. 412.
Пример 55. Синтез 1-[2-(цианометил)фенил]-N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]пиразол-3-карбоксамида
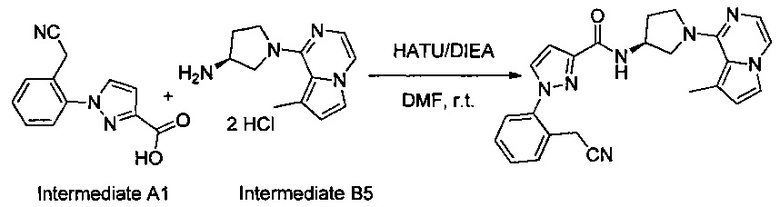
Во флакон на 10 мл вносили промежуточное соединение А1 (45 мг, 0,20 ммоль), В5 (58 мг, 0,20 ммоль), HATU (83 мг, 0,22 ммоль), основание Хюнига (78 мг, 0,6 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (60 мг, 56%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.02 (d, J=2.2 Hz, 1H), 7.65-7.57 (m, 3H), 7.54-7.46 (m, 3H), 6.97 (d, J=2.2 Hz, 1H), 6.75-6.72 (m, 2H), 4.82-4.75 (m, 1H), 4.28-4.24 (m, 1H), 4.08-4.00 (m, 1H), 3.98-3.90 (m, 4H), 2.61 (s, 3H), 2.52-2.30 (m, 2H). MS: (ES) m/z теор. для C24H23N7O [M+H]+ 426,2, факт. 426.
Пример 56. Синтез 1-(4-хлор-3-фторфенил)-N-[(3S)-1-(8-метилпирроло[1,2-а]-пиразин-1-ил)пирролидин-3-ил]пиразол-3-карбоксамида и 1-(3-фторфенил)-N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]пиразол-3-карбоксамида
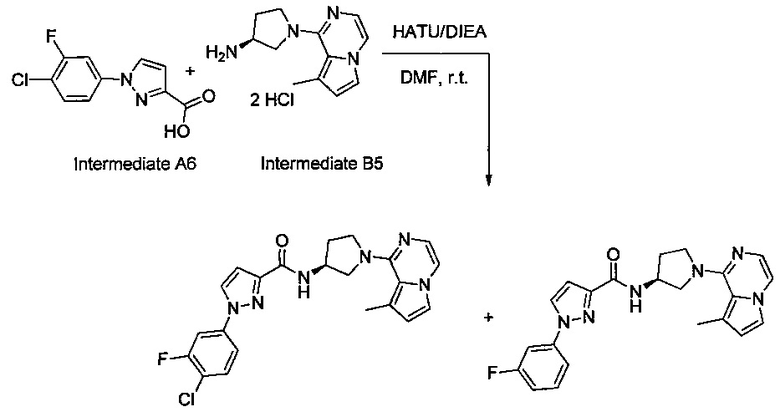
Во флакон на 10 мл вносили промежуточное соединение А6 (48 мг, 0,20 ммоль), В5 (58 мг, 0,20 ммоль), HATU (83 мг, 0,22 ммоль), основание Хюнига (78 мг, 0,6 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая хлорфтор-замещенный (40 мг, 46%) и фтор-замещенный (5 мг, 6%) продукты. 1-(4-Хлор-3-фторфенил)-N[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]пиразол-3-карбоксамид: 1Н-ЯМР (400 MHz, CD3OD) δ 8.36 (d, J=2.6 Hz, 1H), 7.90 (dd, J=2.6, 10.3 Hz, 1H), 7.72-7.64 (m, 3H), 7.61 (t, J=8.0 Hz, 1H), 6.95 (d, J=2.6 Hz, 1H), 6.77-6.73 (m, 2H), 4.82-4.75 (m, 1H), 4.30-4.24 (m, 1H), 4.08-3.92 (m, 3H), 2.63 (s, 3H), 2.52-2.30 (m, 2H); MS: (ES) m/z теор. для C22H20ClFN6O [M+H]+ 439,2, факт. 439. 1-(3-Фторфенил)-N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]пиразол-3-карбоксамид: 1Н-ЯМР (400 MHz, CD3OD) δ 8.35 (d, J=2.6 Hz, 1H), 7.76-7.64 (m, 3H), 7.53 (dt, 6.2, 8.4 Hz, 2H), 7.13 (dt, J=2.5, 8.4 Hz, 1H), 6.95 (d, J=2.6 Hz, 1H), 6.77-6.73 (m, 2H), 4.82-4.75 (m, 1H), 4.30-4.24 (m, 1H), 4.08-3.92 (m, 3H), 2.63 (s, 3H), 2.52-2.30 (m, 2H); MS: (ES) m/z теор. для C22H21FN6O [M+H]+ 405,2, факт. 405.
Пример 57. Синтез 2-(4-гидрокси-1-пиперидил)-N-[(3S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]тиазол-4-карбоксамида

Во флакон на 10 мл вносили промежуточное соединение А4 (45 мг, 0,20 ммоль), В5 (58 мг, 0,20 ммоль), HATU (83 мг, 0,22 ммоль), основание Хюнига (78 мг, 0,6 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (35 мг, 41%). 1H-ЯМР (400 MHz, CD3OD) δ 7.68-7.65 (m, 2Н), 7.38 (s, 1Н), 6.76-6.74 (m, 2H), 4.75-4.65 (m, 1H), 4.25-4.15 (m, 1H), 4.06-3.80 (m, 5H), 3.35-3.20 (m, 3H), 2.62 (s, 1H), 2.50-2.30 (m, 2H), 1.98-1.90 (m, 2H), 1.65-1.52 (m, 2H). MS: (ES) m/z теор. для C21H26N6O2S [M+H]+ 427,2, факт. 427.
Пример 58. Синтез 1-(4-хлор-3-фторфенил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]пиразол-3-карбоксамида и 1-(3-фторфенил)-N-[(3S)-1-пирроло-[1,2-а]пиразин-1-илпирролидин-3-ил]пиразол-3-карбоксамида
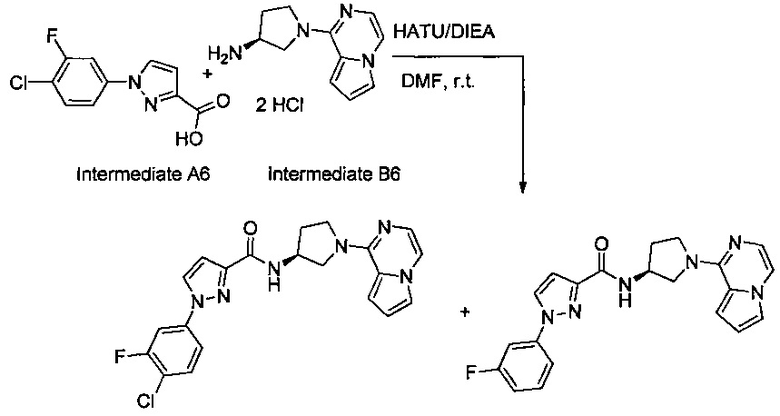
Во флакон на 10 мл вносили промежуточное соединение А6 (48 мг, 0,20 ммоль), В6 (58 мг, 0,20 ммоль), HATU (83 мг, 0,22 ммоль), основание Хюнига (78 мг, 0,6 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая 4-хлор-3-фтор-замещенный продукт (38 мг, 45%) и 3-фтор-замещенный продукт (8 мг, 10%). 1-(4-Хлор-3-фторфенил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]пиразол-3-карбоксамид: 1H-ЯМР (400 MHz, CD3OD) δ 8.37 (d, J=2.9 Hz, 1H), 7.91 (dd, J=2.6, 10.3 Hz, 1H), 7.77 (s, 1H), 7.71-7.68 (m, 2H), 7.61-7.55 (m, 2H), 6.97 (d, J=2.6 Hz, 1H), 6.92 (dd, J=2.6, 4.4 Hz, 1H), 6.84 (d, J=5.9 Hz, 1H), 4.95-3.70 (br, 5H), 2.60-2.36 (m, 2H); MS: (ES) m/z теор. для C21H18ClFN6O [M+H]+ 425,2, факт. 425. 1-(3-Фторфенил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]пиразол-3-карбоксамид: 1Н-ЯМР (400 MHz, CD3OD) δ 8.80 (d, J=5.9 Hz, 1H), 8.38 (d, J=2.3 Hz, 1H), 7.79-7.68 (m, 4H), 7.59-7.48 (m, 2H), 7.14 (dt, 7=2.8, 8.6 Hz, 1H), 6.98 (d, J=2.4 Hz, 1H), 6.95 (dd, J=2.6, 4.4 Hz, 1H), 6.86 (d, J=5.5 Hz, 1H), 4.95-3.70 (br, 5H), 2.60-2.36 (m, 2H); MS: (ES) m/z теор. для C21H19FN6O [M+H]+ 391,2, факт. 391.
Пример 59. Синтез 2-(4-гидрокси-1-пиперидил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]тиазол-4-карбоксамида
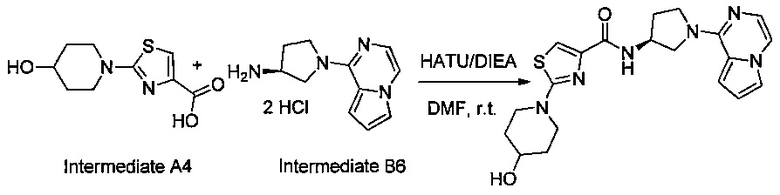
Во флакон на 10 мл вносили промежуточное соединение А4 (45 мг, 0,20 ммоль), В6 (55 мг, 0,20 ммоль), HATU (83 мг, 0,22 ммоль), основание Хюнига (78 мг, 0,6 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (65 мг, 79%). 1Н-ЯМР (400 MHz, CD3OD) δ 7.76 (s, 1Н), 7.70 (d, J=5.8 Hz, 1H), 7.54 (d, J=3.7 Hz, 1H), 7.39 (s, 1H), 7.53 (dd, J=2.5, 4.0 Hz, 1H), 6.84 (d, J=5.5 Hz, 1H), 4.82-4.75 (m, 1H), 4.70-3.60 (br, 4H), 3.90-3.80 (m, 2H), 3.40-3.20 (m, 3H), 2.52-2.30 (m, 2H), 1.96-1.90 (m, 2H), 1.65-1.52 (m, 2H); MS: (ES) m/z теор. для C20H24FN6O2S [M+H]+ 413,2, факт. 413.
Пример 60. Синтез 1-(5-хлор-2-пиридил)-N-[(3S)-1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил]пиразол-3-карбоксамида
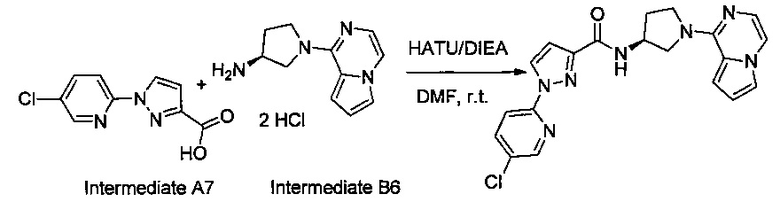
Во флакон на 10 мл вносили промежуточное соединение А7 (45 мг, 0,20 ммоль), В6 (55 мг, 0,20 ммоль), HATU (83 мг, 0,22 ммоль), основание Хюнига (78 мг, 0,6 ммоль) и 2 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (45 мг, 56%). 1H-ЯМР (400 MHz, CD3OD) δ 8.61 (s, 1H), 8.44 (s, 1H), 8.07 (d, J=8.8 Hz, 1H), 7.99 (d, J=8.8 Hz, 1H), 7.77 (s, 1H), 7.72 (d, J=5.9 Hz, 1H), 7.57 (d, J=4.4 Hz, 1H), 6.96 (s, 1H), 6.94-6.91 (m, 1H), 6.85 (d, J=5.9 Hz, 1H), 5.00-3.65 (br, 5H), 2.60-2.30 (m, 2H); MS: (ES) m/z теор. для C20H18ClN7O [M+H]+ 408,2, факт. 408.
Пример 61. Синтез 2-(4-гидрокси-1-пиперидил)-N-[(3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]тиазол-4-карбоксамида
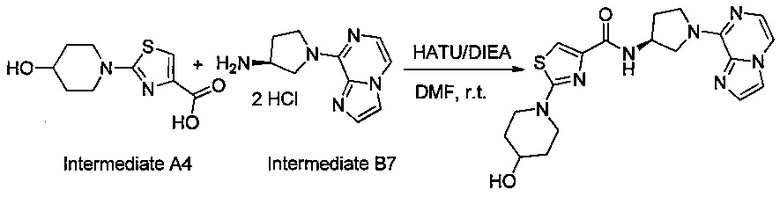
Во флакон на 10 мл вносили промежуточное соединение А4 (90 мг, 0,40 ммоль), В7 (100 мг, 0,40 ммоль), HATU (166 мг, 0,40 ммоль), основание Хюнига (156 мг, 1,2 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (80 мг, 48%). 1Н-ЯМР (400 MHz, CD3OD) δ 7.74 (s, 1Н), 7.66 (d, J=4.7 Hz, 1H), 7.50 (s, 1H), 7.36 (s, 1H), 7.22 (d, J=4.9 Hz, 1H), 4.76-4.65 (m, 1H), 4.40-4.30 (m, 1H), 4.20-3.75 (m, 4H), 3.40-3.20 (m, 4H), 2.42-2.10 (m, 2H), 1.95-1.85 (m, 2H), 1.65-1.50 (m, 2H); MS: (ES) m/z теор. для C19H23N7O2S [M+H]+ 414,2, факт. 414.
Пример 62. Синтез 1-(4-фторфенил)-N-[(3S)-1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил]-5-метил-1,2,4-триазол-3-карбоксамида
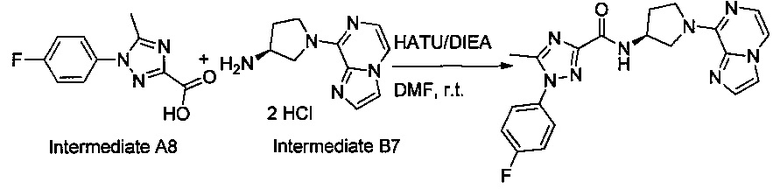
Во флакон на 10 мл вносили промежуточное соединение А8 (82 мг, 0,30 ммоль), В7 (82 мг, 0,30 ммоль), HATU (125 мг, 0,33 ммоль), основание Хюнига (160 мг, 1,23 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (65 мг, 53%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.95 (d, J=6.3 Hz, 0.2H), 8.05 (s, 1H), 7.94 (d, J=5.5 Hz, 1H), 7.81 (s, 1H), 7.64-7.59 (m, 2H), 7.36-7.30 (m, 2H), 7.17 (d, J=5.4 Hz, 1H), 5.00-4.85 (m, 1H), 4.80-3.65 (br, 4H), 2.51 (s, 3H), 2.60-2.38 (m, 2H); MS: (ES) m/z теор. для C20H19FN8O [M+H]+ 407,2, факт. 407.
Пример 63. Синтез 2-(4-гидрокси-1-пиперидил)-N-[(3S)-1-имидазо[1,2-а]пиразин-8-илпирролидин-3-ил]тиазол-4-карбоксамида
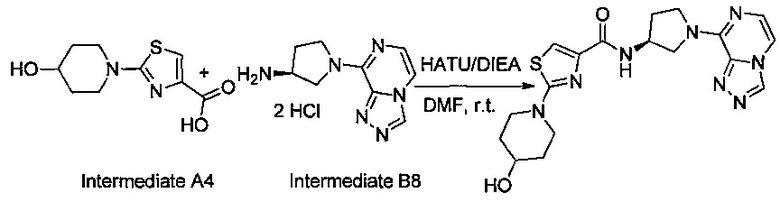
Во флакон на 10 мл вносили промежуточное соединение А4 (90 мг, 0,40 ммоль), В 8 (100 мг, 0,40 ммоль), HATU (166 мг, 0,40 ммоль), основание Хюнига (156 мг, 1,2 ммоль) и 4 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси изопропанол/хлороформ (1:2), после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной HPLC, получая искомый продукт (75 мг, 45%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.06 (s, 1Н), 7.64 (d, J=4.8 Hz, 1H), 7.36 (s, 1H), 7.26 (d, J=4.8 Hz, 1H), 4.73-4.68 (m, 1H), 4.60-3.60 (br, 4H), 3.96-3.80 (m, 3H), 3.35-3.20 (m, 2H), 2.45-2.35 (m, 1H), 2.30-2.18 (m, 1H), 1.95-1.88 (m, 2H), 1.62-1.50 (m, 2H); MS: (ES) m/z теор. для C18H22N8O2S [M+H]+ 415,2, факт. 415.
Пример 64. Синтез 3-[[1-(4-фторфенил)пиразол-3-карбонил]амино]-1-пирроло-[1,2-а]пиразин-1-илпирролидин-3-карбоновой кислоты и N-3-карбамоил-1-пирроло-[1,2-а]пиразин-1-ил-пирролидин-3-ил-1-(4-фторфенил)пиразол-3-карбоксамида
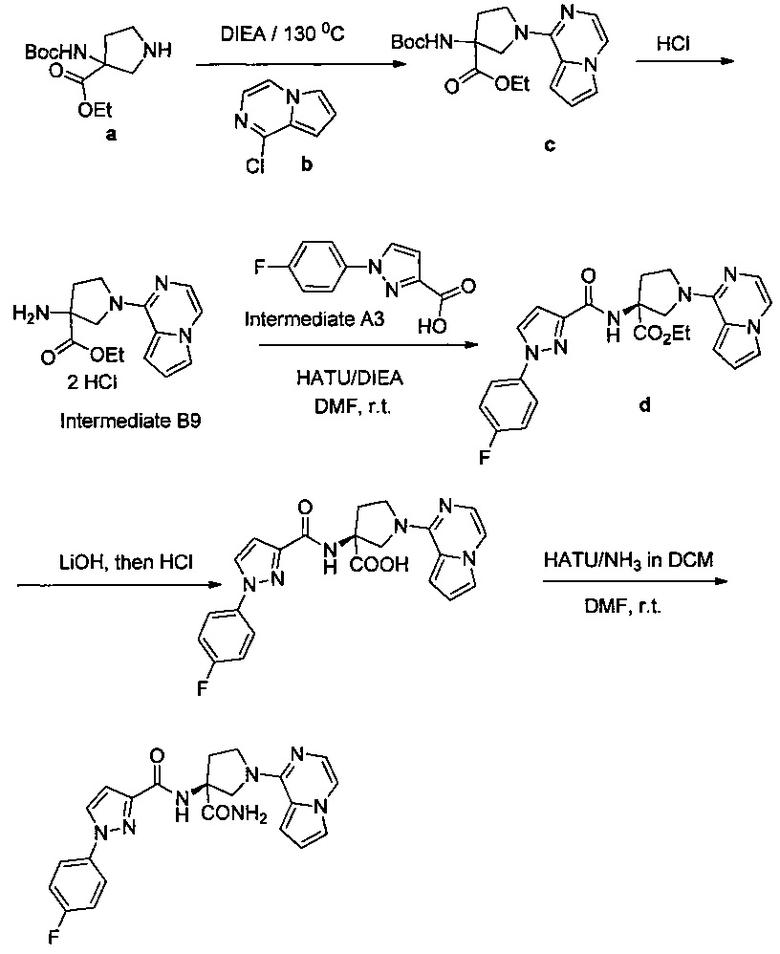
a) В смесь из соединения а (248 мг, 1,0 ммоль) и b (152 мг, 1,0 ммоль) добавляли 2,0 мл основания Хюнига (9,5 ммоль). Полученную смесь перемешивали при 130°С в течение 3 ч. После охлаждения смеси до комнатной температуры добавляли 200 мл смеси изопропанол/хлороформ (1:2), а затем промывали смесь насыщенным водным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×50 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (0-10% МеОН в EtOAc), получая искомый продукт с в виде коричневого порошка (250 мг, 67%).
b) Смесь из соединения с (250 мг, 0,67 ммоль) и 2 мл 4,0 М HCl в диоксане (8,0 ммоль) перемешивали при 50°С в течение 1 ч. Затем смесь упаривали при пониженном давлении, получая коричневый порошок (228 мг, 98%), который использовали на следующей стадии без дальнейшей очистки.
c) Во флакон на 10 мл вносили промежуточное соединение A3 (136 мг, 0,66 ммоль), В9 (228 мг, 0,66 ммоль), HATU (274 мг, 0,72 ммоль), основание Хюнига (257 мг, 1,98 ммоль) и 5 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси EtOAc, после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле (50-100% EtOAc в гексане), получая искомый продукт d (220 мг, 72%).
d) Во флакон на 10 мл вносили соединение d (230 мг, 0,50 ммоль), 1N LiOH (3 мл) и 3 мл МеОН. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем доводили до рН 3,0. После этого смесь экстрагировали 100 мл EtOAc, а затем промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле (100% EtOAc в гексане), получая искомый продукт (205 мг, 95%). 3-[[1-(4-фторфенил)пиразол-3-карбонил]амино]-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-карбоновая кислота: 1Н-ЯМР (400 MHz, CD3OD) δ 8.29 (d, J=2.7 Hz, 1H), 7.89 (m, 2H), 7.78 (dd, J=1.2, 2.7 Hz, 1H), 7.73 (d, J=5.4 Hz, 1H), 7.55 (d, J=3.5 Hz, 1H), 7.30-7.22 (m, 2H), 6.97 (d, J=2.7 Hz, 1H), 6.94 (dd, J=2.8, 4.3 Hz, 1H), 6.84 (d, J=5.5 Hz, 1H), 4.60-3.80 (br, 4H), 2.90-2.82 (m, 1H), 2.80-2.70 (m, 1H); MS: (ES) m/z теор. для C22H19FN6O3 [M+H]+ 435,2, факт. 435.
e) Во флакон на 10 мл вносили 3-[[1-(4-фторфенил)пиразол-3-карбонил]амино]-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-карбоновую кислоту (100 мг, 0,25 ммоль), HATU (114 мг, 0,3 ммоль), насыщенный раствор NH3 в DCM (2 мл) и 5 мл DMF. Смесь перемешивали при комнатной температуре в течение 1 ч, а затем разбавляли 100 мл смеси EtOAc, после чего промывали водой (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле (50-100% EtOAc в гексане), получая искомый продукт (75 мг, 69%). N-3-карбамоил-1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил-1-(4-фторфенил)пиразол-3-карбоксамид: 1H-ЯМР (400 MHz, CD3OD) δ 8.30 (d, J=2.4 Hz, 1H), 7.88 (m, 2H), 7.78 (q, J=1.2 Hz, 1H), 7.72 (d, J=5.9 Hz, 1H), 7.55 (d, J=3.9 Hz, 1H), 7.30-7.22 (m, 2H), 6.99 (d, J=2.7 Hz, 1H), 6.93 (dd, J=2.7,4.3 Hz, 1H), 6.83 (d, J=5.5 Hz, 1H), 4.60-3.80 (br, 4H), 2.90-2.80 (m, 1H), 2.78-2.68 (m, 1H); MS: (ES) m/z теор. для C22H20FN7O2 [M+H]+ 434,2, факт. 434.
Пример 65. Синтез этил-2-фенил-2Н-тетразол-5-карбоксилата
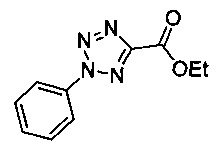
В раствор анилина (0,58 г, 6,2 ммоль) в 9,2 мл EtOH/Н2О (1:1) при 0°С добавляли 2,3 мл конц. HCl, а затем NaNO2 (0,47 г, 6,8 ммоль, 1,1 экв.). Смесь перемешивали при 0°С в течение 30 мин. В отдельный раствор этилглиоксалата (1,74 г, 17 ммоль, 3,1 экв) в 34 мл EtOH добавляли п-толуолсульфонилгидразид (1,0 г, 5,4 ммоль, 1 экв.). Смесь перемешивали при комнатной температуре в течение 30 мин, а затем упаривали. Остаток растворяли в 34 мл пиридина и охлаждали до 0°С. В охлажденный раствор добавляли образовавшуюся соль диазония. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, а затем гасили Н2О. Содержимое экстрагировали этилацетатом, а органический слой сушили над Na2SO4 и упаривали под вакуумом. После очистки методом флэш-хроматографии на силикагеле (Нех : EtOAc = 9:1) получали этил-2-фенил-2Н-тетразол-5-карбоксилат (0,84 г, 3,9 ммоль, 71%).
Пример 66. Синтез 2-фенил-2Н-тетразол-5-карбоновой кислоты
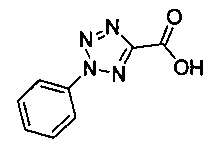
В раствор этил-2-фенил-2Н-тетразол-5-карбоксилата (0,84 г, 3,9 ммоль, 1 экв.) в 7,6 мл EtOH/Н2О (1,5:1) добавляли раствор NaOH (0,31 г, 7,8 ммоль, 2 экв.). Реакционную смесь нагревали при 60°С в течение 30 мин, а затем гасили 0,65 мл конц. HCl. Содержимое фильтровали и промывали МеОН, а фильтрат сушили над Na2SO4 и упаривали под вакуумом. После очистки методом флэш-хроматографии на силикагеле (100% EtOAc) получали 2-фенил-2Н-тетразол-5-карбоновую кислоту (0,41 г, 2,2 ммоль, 55%).
Пример 67. Синтез (S)-трет-бутил(1-(имидазо[1,2-а]пиразин-8-ил)пирроли-дин-3-ил)карбамата
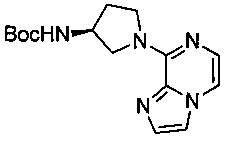
В раствор (S5)-трет-бутилпирролидин-3-илкарбамата (80,2 г, 0,43 моль, 1,1 экв.) в 75 мл диизопропиламина и 34 мл NMP порциями добавляли 8-хлоримидазо[1,2-а]пиразин (60 г, 0,39 моль, 1 экв.). Реакционную смесь нагревали при 100°С в течение 4 ч, а затем разбавляли 1,2 л этилацетата. Органический слой промывали Н2О и насыщенным раствором NaCl, сушили над Na2SO4 и упаривали, получая (S)-трет-бутил-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)карбамат (111,7 г, 0,37 моль, 94%).
Пример 68. Синтез (S)-1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-амина
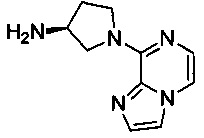
В раствор ацетилхлорида (114 мл, 1,6 моль) в 290 мл МеОН при 0°С добавляли раствор (S)-трет-бутил-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)карбамата (111,7 г, 0,37 моль) в 450 мл МеОН. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем нагревали при 60°С в течение 30 мин. Продукт отфильтровывали, промывали 200 мл МеОН и высушивали в вакуумной печи, получая (S)-1-(имидазо[1,2-а] пиразин-8-ил)пирролидин-3-амин (101,2 г, 1,6 моль, 100%).
Пример 69. Синтез (S)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-2-фенил-2Н-тетразол-5-карбоксамида
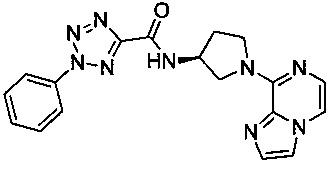
В раствор 2-фенил-2Н-тетразол-5-карбоновой кислоты (0,10 г, 0,53 ммоль, 1 экв.) и (S)-1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-амина (0,15 г, 0,53 ммоль, 1 экв.) в 1 мл DMSO добавляли триэтиламин (0,22 мл, 0,58 ммоль, 1,1 экв.) и HATU (0,22 г, 0,58 ммоль, 3 экв.). Смесь перемешивали при комнатной температуре в течение 1 ч, а затем упаривали и очищали методом HPLC, получая (S)-N-(1-(имидазо[1,2-a]пиразин-8-ил)пирролидин-3-ил)-2-фенил-2Н-тетразол-5-карбоксамид (0,10 г, 0,26 ммоль, 49%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.19 (d, J=7.0 Hz, 2H), 8.06 (s, 1H), 7.95 (d, J=5.5 Hz, 1H), 7.82 (s, 1H), 7.70-7.58 (m, 3H), 7.19 (d, J=5.8 Hz, 1H), 5.00-4.90 (m, 1H), 4.80-3.80 (br, 4H), 2.62-2.38 (m, 2H); MS: (ES) m/z теор. для C18H17N9O [M+H]+ 376,2, факт. 376.
Пример 70. Синтез метил-6-(4-гидроксипиперидин-1-ил)пиридазин-3-карбоксилата

Раствор 6-хлорпиридазин-3-карбоновой кислоты (0,50 г, 3,2 ммоль) в 11 мл SOCl2 нагревали при 75°С в течение 2 ч, а затем упаривали и растворяли в 5 мл метанола. В этот раствор добавляли 25%-й раствор метоксида натрия (0,75 мл, 3,5 ммоль, 1,1 экв.) в МеОН. Реакционную смесь перемешивали при комнатной температуре в течение 20 ч, а затем гасили Н2О и экстрагировали дихлорметаном. Остаток подвергали флэш-хроматографии на силикагеле (EtOAc/MeOH=90:10), получая смесь из 6-хлорпиридазин-3-карбоновой кислоты и 6-метоксипиридазин-3-карбоновой кислоты 2:1 (0,160 г). В раствор этой смеси эфира и хлорида в 1,9 мл n-диоксана добавляли 4-гидроксипиперидин (0,094 г, 93 ммоль), а затем диизопропилэтиламин (0,49 мл, 2,8 ммоль). Смесь нагревали при 100°С в течение 20 ч, а затем упаривали. После флэш-хроматографии на силикагеле получали метил-6-(4-гидроксипиперидин-1-ил)пиридазин-3-карбоксилат (0,15 г, 63 ммоль).
Пример 71. Синтез 6-(4-гидроксипиперидин-1-ил)пиридазин-3-карбоновой кислоты
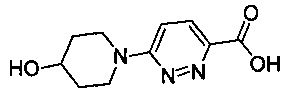
В раствор сложного эфира (0,15 г, 0,63 ммоль, 1 экв.) в 2,5 мл EtOH/Н2О (1,5:1) добавляли раствор NaOH (0,08 г, 1,9 ммоль, 3 экв.). Реакционную смесь нагревали при 50°С в течение 1 ч, а затем гасили 0,16 мл конц. HCl. Содержимое фильтровали и промывали МеОН, а фильтрат сушили над Na2SO4 и упаривали под вакуумом, получая 6-(4-гидроксипиперидин-1-ил)пиридазин-3-карбоновую кислоту.
Пример 72. Синтез (S)-6-(4-гидроксипиперидин-1-ил)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)пиридазин-3-карбоксамида
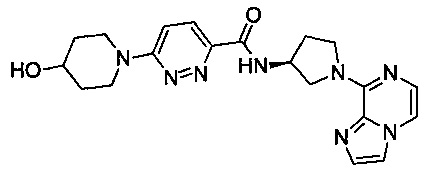
В раствор 6-(4-гидроксипиперидин-1-ил)пиридазин-3-карбоновой кислоты (0,10 г, 0,45 ммоль, 1 экв.) и (S)-1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-амина (0,12 г, 0,44 ммоль, 1 экв.) в 1 мл DMSO добавляли триэтиламин (0,19 мл, 0,49 ммоль, 1,1 экв.) и HATU (0,19 г, 0,49 ммоль, 3 экв.). Смесь перемешивали при комнатной температуре в течение 30 мин, а затем упаривали и очищали методом HPLC, получая (S)-6-(4-гидроксипиперидин-1-ил)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)пиридазин-3-карбоксамид (0,12 г, 0,29 ммоль, 66%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.05 (s, 1Н), 8.02 (d, J=9.8 Hz, 1H), 7.94 (d, J=5.5 Hz, 1H), 7.81 (s, 1H), 7.5 (d, J=9.8 Hz, 1H), 7.18 (d, J=5.8 Hz, 1H), 4.98-4.80 (m, 1H), 4.80-3.80 (br, 4H), 4.20-4.10 (m, 2H), 4.00-3.90 (m, 1H), 3.55-3.45 (m, 2H), 2.60-2.35 (m, 2H), 2.05-1.95 (m, 2H), 1.68-1,55 (m, 2H); MC: (ES) m/z теор. для C20H24N8O2 [М+Н]+ 409,2, факт. 409.
Пример 73. Синтез этил-1-(3,4-дифторфенил)-1Н-1,2,4-триазол-3-карбоксилата
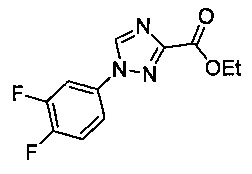
В раствор 3,4-дифторанилина (1 мл, 10 ммоль) в 5,4 мл Н2О при 0°С добавляли 2,8 мл конц. HCl, а затем NaNO2 (1,0 г, 15 ммоль, 1,5 экв.). Смесь перемешивали при 0°С в течение 30 мин. Добавляли еще NaNO2 (0,35 г, 5 ммоль, 0,5 экв.) и перемешивали смесь в течение 1 ч при 0°С. В отдельный раствор NaOAc (8,9 г, 108 ммоль, 11 экв.) в 13 мл EtOH/Н2О (12:1) добавляли этил-2-изоцианоацетат (1,1 мл, 10 ммоль, 1 экв.). Смесь охлаждали до 0°С и по каплям добавляли смесь диазониевых солей. После перемешивания в течение 1 ч реакцию гасили Н2О и экстрагировали этил ацетатом. Органический слой сушили над Na2SO4 и упаривали под вакуумом. После очистки методом флэш-хроматографии на силикагеле (гексан : EtOAc = 1:1) получали этил-1-(3,4-дифторфенил)-1Н-1,2,4-триазол-3-карбоксилат (0,30 г, 1,3 ммоль, 13%).
Пример 74. Синтез 1-(3,4-дифторфенил)-1Н-1,2,4-триазол-3-карбоновой кислоты
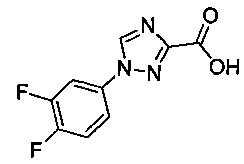
В раствор этил-1-(3,4-дифторфенил)-1Н-1,2,4-триазол-3-карбоксилата (0,30 г, 1,2 ммоль, 1 экв.) в 5 мл EtOH/Н2О (1,5:1) добавляли раствор NaOH (0,095 г, 2,4 ммоль, 2 экв.). Реакционную смесь нагревали при 50°С в течение 1 ч, а затем гасили 0,20 мл конц. HCl. Содержимое фильтровали и промывали EtOAc, а затем МеОН, и фильтрат после МеОН сушили над Na2SO4 и упаривали под вакуумом, получая 1-(3,4-дифторфенил)-1Н-1,2,4-триазол-3-карбоновую кислоту (0,22 г, 0,98 ммоль, 84%).
Пример 75. Синтез (S)-1-(3,4-дифторфенил)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-1Н-1,2,4-триазол-3-карбоксамида
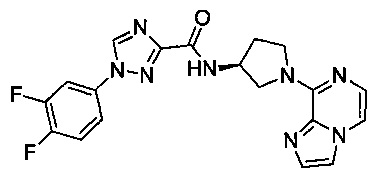
В раствор 1-(3,4-дифторфенил)-1Н-1,2,4-триазол-3-карбоновой кислоты (0,10 г, 0,44 ммоль, 1 экв.) и (S)-1-(имидазо[1,2-]ггиразин-8-ил)гшрролидин-3-амина (0,12 г, 0,44 ммоль, 1 экв.) в 1 мл DMSO добавляли триэтиламин (0,19 мл, 0,49 ммоль, 1,1 экв.) и HATU (0,19 г, 1,3 ммоль, 3 экв.). После перемешивания в течение 1 ч при комнатной температуре смесь упаривали и очищали методом HPLC, получая (S)-1-(3,4-дифторфенил)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-1Н-1,2,4-триазол-3-карбоксамид (0, 046 г, 0,11 ммоль, 25%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.14 (s, 1H), 8.05 (s, 1H), 7.96-7.88 (m, 2H), 7.81 (s, 1H), 7.75-7.71 (m, 1H), 7.52 (q, J=8.6 Hz, 1H), 7.19 (d, J=5.5 Hz, 1H), 4.98-4.80 (m, 1H), 4.80-3.80 (br, 4H), 2.62-2.40 (m, 2H); MS: (ES) m/z теор. для C19H16F2N8O [M+H]+ 411,2, факт. 411.
Пример 76. Синтез (S)-трет-бутил(1-(пиразоло[1,5-а]пиразин-4-ил)пирролидин-3-ил)карбамата

В раствор (S)-трет-бутил-пирролидин-3-илкарбамата (0,50 г, 2,7 ммоль, 1 экв.) и диизопропиламина (0,66 мл, 3,8 ммоль, 1,4 экв.) в 0,2 мл NMP добавляли 4-хлорпиразоло [1,5-а]пиразин (0,47 мл, 3,1 ммоль, 1,1 экв.). После перемешивания в течение 1 ч при комнатной температуре смесь упаривали и очищали методом колоночной хроматографии на силикагеле (100% EtOAc), получая (S)-трет-бутил-(1-(пиразоло[1,5-а]пиразин-4-ил)-пирролидин-3-ил)карбамат (0740 г, 2,4 ммоль, 91%).
Пример 77. Синтез (S)-1-(пиразоло[1,5-а]пиразин-4-ил)пирролидин-3-амина
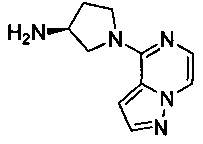
В раствор (S)-трет-бутил(1-(пиразоло[1,5-а]пиразин-4-ил)пирролидин-3-ил)карбамата (0,74 г, 2,4 ммоль, 1 экв.) в 3 мл диоксана добавляли раствор 4,0 М HCl в диоксане (3 мл, 20 ммоль, 4 экв.). После нагревания в течение 3 ч при 60°С смесь упаривали, получая (S)-1-(пиразоло[1,5-а]пиразин-4-ил)пирролидин-3-амин (0,670 г, 2,4 ммоль, 100%).
Пример 78. Синтез (S)-1-фенил-N-(1-(пиразоло[1,5-а]пиразин-4-ил)пирроли-дин-3-ил)-1Н-1,2,4-триазол-3-карбоксамида
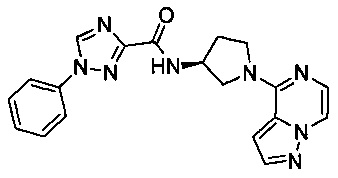
В раствор 1-фенил-1Н-1,2,4-триазол-3-карбоновой кислоты (0,093 г, 0,49 ммоль, 1,4 экв.) и (S-1-(пиразоло[1,5-а]пиразин-4-ил)пирролидин-3-амина (0,10 г, 0,36 ммоль, 1 экв.) в 1 мл DMSO добавляли триэтиламин (0,20 мл, 1,4 ммоль, 2,6 экв.) и НАТО (0,21 г, 0,55 ммоль, 1,5 экв.). После перемешивания в течение 1 ч при комнатной температуре смесь упаривали и очищали методом HPLC, получая (S)-1-фенил-N-(1-(пиразоло[1,5-а]пиразин-4-ил)пирролидин-3-ил)-1Н-1,2,4-триазол-3-карбоксамид (0,095 г, 0,25 ммоль, 52%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.13 (s, 1Н), 8.16 (d, J=2.4 Hz, 1H), 8.09 (d, J=5.8 Hz, 1H), 7.86 (d, J=7.8 Hz, 2H), 7.60-7.46 (m, 4H), 7.19 (d, 7=5.5 Hz, 1H), 4.95-4.80 (m, 1H), 4.78-3.80 (br, 4H), 2.62-2.40 (m, 2H); MS: (ES) m/z теор. для C19H18N8O [M+H]+ 375,2, факт. 375.
Пример 79. Синтез (S)-трет-бутил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)карбамата
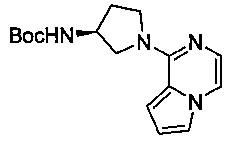
В раствор (S)-трет-бутил-пирролидин-3-илкарбамата (0,91 г, 4,9 ммоль, 1 экв.) в 2,6 мл DIPEA добавляли 1-хлорпирроло[1,2-а]пиразин (0,75 г, 4,9 ммоль, 1 экв.) и 1 каплю 1-бутил-3-метилимидазолия тетрафторбората. После нагревания в течение 3 ч при 110°С смесь упаривали и очищали методом флэш-хроматографии на силикагеле (100% EtOAc), получая (S)-трет-бутил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)карбамат (0,88 г, 2,9 ммоль, 59%).
Пример 80. Синтез (S)-1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-амина
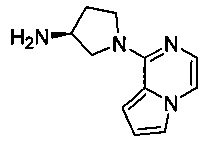
В раствор (S)-трет-бутил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-карбамата (0,88 г, 2,9 ммоль, 1 экв.) в 3,6 мл диоксана добавляли 4,0 М раствор HCl в диоксане (3,6 мл, 14,5 ммоль, 5 экв.). После нагревания в течение 1 ч при 60°С смесь упаривали, получая (S)-1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин (0,79 г, 2,9 ммоль, 100%).
Пример 81. Синтез (S)-5-бром-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида
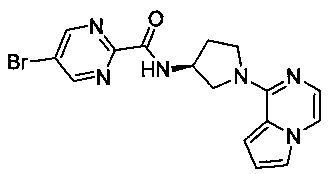
В раствор 5-бромпиримидин-2-карбоновой кислоты (2,0 г, 9,9 ммоль, 1 экв.) и (S)-1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-амина (2,7 г, 9,9 ммоль, 1 экв.) в 20 мл DMSO добавляли триэтиламин (5,4 мл, 39 ммоль, 3,9 экв.) и HATU (4,1 г, 11 ммоль, 1,1 экв.). После перемешивания в течение 1 ч при комнатной температуре смесь упаривали и очищали методом флэш-хроматографии на силикагеле (EtOAc:МеОН = 2:3), получая (S)-5-бром-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамид (5,0 г, 1,3 ммоль, 13%).
Пример 82. Синтез (S)-5-(циклопент-1-ен-1-ил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида
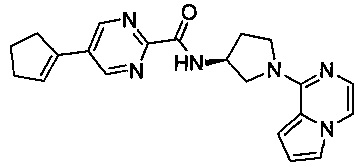
В раствор (S)-5-бром-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида (0,10 г, 0,27 ммоль, 1 экв.) в 1,8 мл DMF добавляли 2-(циклопент-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,15 г, 0,77 ммоль, 2,8 экв.), а затем K2CO3 (0,18 г, 1,3 ммоль, 4,8 экв.) в 0,2 мл H2O и Pd(dppf)Cl2 (0,02 г, 0,03 ммоль, 0,09 экв.). После нагревания в течение 1 ч при 120°С смесь упаривали и очищали методом флэш-хроматографии на силикагеле (EtOAc:МеОН = 1:1), а затем HPLC, получая (S)-5-(циклопент-1-ен-1-ил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамид (0,070 г, 0,19 ммоль, 69%).
Пример 83. Синтез (S)-5-циклопентy-l-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида
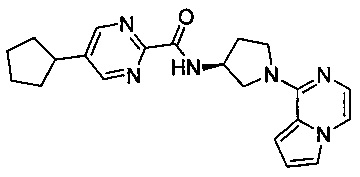
В раствор (S)-5-(циклопент-1-ен-1-ил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида (0,07 г, 0,19 ммоль, 1 экв.) в 1 мл МеОН добавляли 10% Pd/C (0,02 г, 0,02 ммоль, 0,1 экв.). Реакционную смесь подсоединяли к баллону с Н2 и перемешивали в течение 3 ч при комнатной температуре. Содержимое фильтровали и очищали методом HPLC, получая (S)-5-циклопентал-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамид (0,005 г, 0,013 ммоль, 7%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.83 (s, 2Н), 7.78 (q, J=1.1 Hz, 1H), 7.73 (d, J=5.8 Hz, 1H), 7.57 (d, J=3.9 Hz, 1H), 6.94 (dd, J=2.7, 4.7 Hz, 1H), 6.86 (d, J=5.9 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-3.50 (br, 4H), 3.20-3.10 (m, 1H), 2.60-2.48 (m, 1H), 2.48-2.38 (br, 1H), 2.22-2.12 (m, 2H), 1.95-1.60 (m, 6 H); MS: (ES) m/z теор. для C21H24N6O [M+H]+ 377,2, факт. 377.
Пример 84. Синтез (S)-N-(1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-ил)-6-метилхиназолин-2-карбоксамида
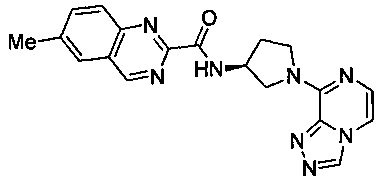
В раствор 6-метилхиназолин-2-карбоновой кислоты (0,040 г, 0,21 ммоль, 1,2 экв.) и (S)-1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-амина (0,49 г, 0,18 ммоль, 1 экв.) в 1 мл DMSO добавляли триэтиламин (0,10 мл, 0,7 ммоль, 4 экв.) и HATU (0,075 г, 0,20 ммоль, 1,1 экв.). После перемешивания в течение 1 ч при комнатной температуре смесь упаривали и очищали методом HPLC, получая (S)-N-(1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-ил)-6-метилхиназолин-2-карбоксамид (0,028 г, 0,07 ммоль, 42%). 1H-ЯМР (400 MHz, DMSO) δ 9.62 (s, 1Н), 9.31 (t, J=3.9 Hz, 1H), 8.08-8.00 (m, 2H), 8.00-7.92 (m, 1H), 7.85 (d, J=5.1 Hz, 1H), 7.29 (d, J=5.0 Hz, 1H), 4.82-4.70 (m, 1H), 4.70-3.60 (br, 4H), 2.56 (s, 3H), 2.40-2.20 (br, 2H); MS: (ES) m/z теор. для C19H18N8O [M+H]+ 375,2, факт. 375.
Пример 85. Синтез (S)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-6-метилхиназолин-2-карбоксамида
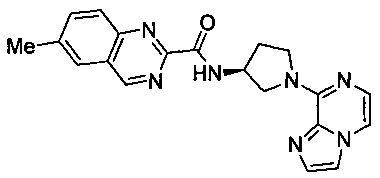
В раствор 6-метилхиназолин-2-карбоновой кислоты (0,040 г, 0,21 ммоль, 1,2 экв.) и (S)-1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-амина (0,49 г 0,18 ммоль, 1 экв.) в 1 мл DMSO добавляли триэтиламин (0,10 мл, 0,7 ммоль, 4 экв.) и HATU (0,075 г, 0,20 ммоль, 1,1 экв.). После перемешивания в течение 1 ч при комнатной температуре смесь упаривали и очищали методом HPLC, получая (S)-N-(1-(имидазо[1,2-а]пиразин-8-ил)-пирролидин-3-ил)-6-метилхиназолин-2-карбоксамид (0,057 г, 0,15 ммоль, 86%). 1H-ЯМР (400 MHz, CD3OD) δ 9.48 (s, 1H), 8.02 (d, J=1.6 Hz, 2H), 7.98-7.88 (m, 3H), 7.78 (s, 1H), 7.17 (d, J=5.8 Hz, 1H), 5.00-3.80 (br, 4H), 2.60 (s, 3H), 2.65-2.40 (br, 2H); MS: (ES) m/z теор. для C20H19N7O [M+H]+ 374,2, факт. 374.
Пример 86. Синтез этил-2-(4-хлор-2-формил-анилино)-2-оксоацетата
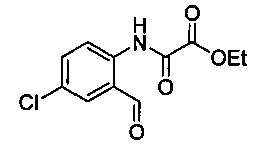
В раствор 2-амино-5-хлор-бензальдегида (0,10 г, 0,65 ммоль, 1 экв.) в 1,6 мл DCM при 0°С добавляли этилхлорацетат (0,09 мл, 0,85 ммоль, 1,3 экв.) и пиридин (0,16 мл, 2,0 ммоль, 3 экв.). После перемешивания при комнатной температуре в течение 1 ч реакцию гасили НгО и экстрагировали этилацетатом. Органический слой промывали 10% лимонной кислотой, затем насыщенным NaHCO3, после чего сушили над Na2SO4 и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (гексан:EtOAc, 3:2), получая этил-2-(4-хлор-2-формил-анилино)-2-оксоацетат (0,14 г, 0,55 ммоль, 84%).
Пример 87. Синтез этил-6-хлорхиназолин-2-карбоксилата
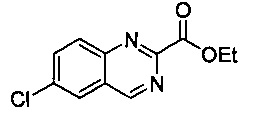
В раствор этил-2-(4-хлор-2-формил-анилино)-2-оксоацетата (0,14 г, 0,55 ммоль, 1 экв.) в 5,5 мл уксусной кислоты добавляли ацетат аммония (0,42 г, 5,4 ммоль, 10 экв.). После нагревания при 115°С в течение 1 ч смесь упаривали, а затем разбавляли Н2О и экстрагировали этилацетатом. Органический слой упаривали, а остаток очищали методом колоночной хроматографии на силикагеле (гексан : EtOAc, 3:2), получая этил-6-хлорхиназолин-2-карбоксилат (0,11 г, 0,48 ммоль, 88%).
Пример 88. Синтез (S)-трет-бутил-3-(6-хлорхиназолин-2-карбоксамидо)-пирролидин-1-карбоксилата
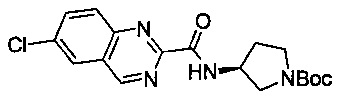
В раствор этил-6-хлорхиназолин-2-карбоксилата (0,200 g, 0,85 ммоль, 1 экв.) в 0,42 мл NMP добавляли (S)-трет-бутил-3-аминопирролидин-1-карбоксилат (0,16 г, 0,86 ммоль, 1 экв.) и диизопропилэтиламин (0,6 мл, 3,4 ммоль, 4 экв.). После перемешивания при 150°С в течение 20 ч смесь упаривали и очищали методом флэш-хроматографии на силикагеле, получая (S)-трет-бутил-3-(6-хлорхиназолин-2-карбоксамидо)пирролидин-1-карбоксилат (0,08 г, 0,21 ммоль, 25%).
Пример 89. Синтез (S)-6-хлор-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)хиназолин-2-карбоксамида
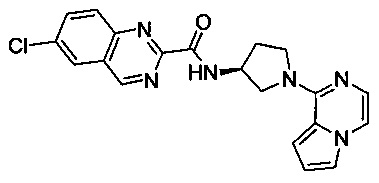
В раствор (S)-трет-бутил-3-(6-хлорхиназолин-2-карбоксамидо)пирролидин-1-карбоксилата (0086 г, 0,23 ммоль, 1 экв.) в 1 мл n-диоксана добавляли 4,0 М раствор HCl в диоксане (0,29 мл, 1,2 ммоль, 5 экв.). После перемешивания при 60°С в течение 1 ч смесь упари, получая (S)-6-хлор-N-(пирролидин-3-ил)хиназолин-2-карбоксамид, который переносили на следующую стадию без дальнейшей очистки.
В раствор неочищенного амина в 0,16 мл диизопропилэтиламина добавляли 1-хлорпирроло[1,2-а]пиразин (0,047 г, 0,31 ммоль), а затем еще 1 каплю 1-бутил-3-метил-имидазолия тетрафторбората. Реакционную смесь нагревали при 90°С в течение 20 ч. Остаток очищали методом HPLC, получая (S)-6-хлор-N-(1-(пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил)хиназолин-2-карбоксамид (0,05 г, 0,013 ммоль, 6%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.60 (s, 1Н), 8.26 (s, 1H), 8.17 (d, J=9.1 Hz, 1H), 8.07 (dd, J=1.8, 9.1 Hz, 1H), 7.76 (d, J=1.5 Hz, 1H), 7.72 (d, J=5.8 Hz, 1H), 7.57 (s, 1H), 6.93 (m, 1H), 6.85 (d, J=5.8 Hz, 1H), 5.06-4.84 (m, 1H), 4.82-3.60 (br, 4H), 2.65-2.40 (br, 2H); MS: (ES) m/z теор. для C20H17ClN6O [M+H]+ 393,2, факт. 393.
Пример 90. Синтез (S)-5-метил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида
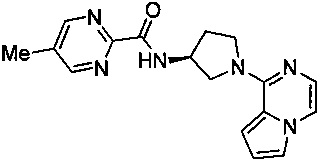
В раствор 5-метилпиримидин-2-карбоновой кислоты (0,03 г, 0,22 ммоль, 1 экв.) и (S)-1-(пирроло [1,2-а]пиразин-1-ил)пирролидин-3-амина (0,060 г, 0,22 ммоль, 1 экв.) в 1 мл DMSO добавляли триэтиламин (0,12 мл, 0,86 ммоль, 4 экв.) и HATU (0,09 г, 0,24 ммоль, 1,1 экв.). После перемешивания в течение 1 ч при комнатной температуре смесь упаривали и очищали методом HPLC, получая (S)-5-метил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамид (0,029 г, 0,09 ммоль, 41%). 1H-ЯМР (400 MHz, CD3OD) δ 8.75 (s, 2Н), 7.76 (s, 1Н), 7.71 (d, J=5.5 Hz, 1H), 7.54 (s, 1H), 6.91 (s, 1H), 6.85 (d, J=5.8 Hz, 1H), 5.00-3.60 (br, 5H), 2.65-2.30 (br, 2H), 2.40 (s, 3H); MS: (ES) m/z теор. для C17H18N6O [М+Н]+ 323,2, факт. 323.
Пример 91. Синтез (S)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-5-винилпиримидин-2-карбоксамида
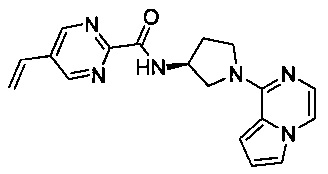
В раствор (S)-5-бром-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида (0,10 г, 0,26 ммоль, 1 экв.) в 1 мл DMF добавляли 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолан (0,13 г, 0,84 ммоль, 3,2 экв.), а затем K2CO3 (0,18 г, 1,3 ммоль, 5 экв.) в 0,2 мл Н2О и Pd(dppf)Cl2 (0,02 г, 0,02 ммоль, 0,1 экв.). После нагревания в течение 1 ч при 120°С смесь упаривали и очищали методом колоночной хроматографии на силикагеле (EtOAc : МеОН = 1:1), а затем еще HPLC, получая (S)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-5-винилпиримидин-2-карбоксамид (0,040 г, 0,12 ммоль, 46%).
Пример 92. Синтез (S)-5-этил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида
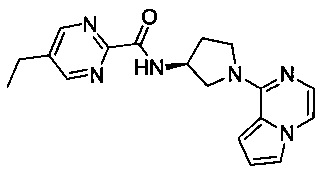
В раствор (S)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-5-винилпиримидин-2-карбоксамида (0,04 г, 0,12 ммоль, 1 экв.) в 1 мл МеОН добавляли 10% Pd/C (0,013 г, 0,01 ммоль, 0,1 экв.). Реакционную смесь подсоединяли к баллону с Н2 и перемешивали в течение 1 ч при комнатной температуре. Содержимое фильтровали и очищали методом HPLC, получая (S)-5-этил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамид (0,009 г, 0,25 ммоль, 22%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.80 (s, 2Н), 7.76 (m, 1Н), 7.71 (d, J=5.8 Hz, 1H), 7.54 (s, 1H), 6.92 (dd, J=2.6, 4.4 Hz, 1H), 6.84 (d, J=5.5 Hz, 1H), 5.00-3.60 (br, 5H), 2.81 (q, J=7.4 Hz, 2H), 2.65-2.30 (br, 2H), 1.33 (t, J=7.4 Hz, 3H); MS: (ES) m/z теор. для C17H18N6O [M+H]+ 337,2, факт. 337.
Пример 93. Синтез (S)-5-циклопропил-N-(1-(пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил)пиримидин-2-карбоксамида
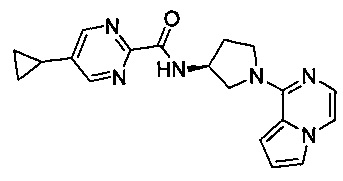
В раствор (S)-5-бром-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида (0,25 г, 0,65 ммоль, 1 экв.) в 2 мл толуола добавляли 2-циклопропил-4,4,5,5-тетршетил-1,3,2-диоксаборолан (0,16 г, 1,86 ммоль, 2,8 экв.), а затем PCy3 (0,033 г, 0,11 ммоль, 0,2 экв.), K3PO4 (0,48 г, 2,3 ммоль, 3,5 экв.) в 0,1 мл H2O и Pd(OAc)2 (0,013 г, 0,06 ммоль, 0,1 экв.). После нагревания в течение 1 ч при 120°С смесь фильтровали и очищали методом HPLC, получая (S)-5-циклопропил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамид (0,009 г, 0,026 ммоль, 4%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.64 (s, 2Н), 7.76 (m, 1Н), 7.71 (d, J=5.4 Hz, 1H), 7.54 (s, 1H), 6.92 (dd, J=2.6, 4.4 Hz, 1H), 6.84 (d, J=5.5 Hz, 1H), 5.00-3.60 (br, 5H), 2.65-2.30 (br, 2H), 2.07-2.02 (m, 1H), 1.23-1.18 (m, 2H), 0.96-0.91 (m, 2H); MS: (ES) m/z теор. для C19H20N6O [M+H]+ 349,2, факт. 349.
Пример 94. Синтез (S)-5-(4-фторфенил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил)пиримидин-2-карбоксамида
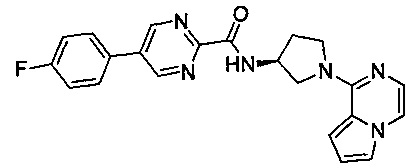
В раствор (S)-5-бром-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида (0,036 г, 0,093 ммоль, 1 экв.) в 1,8 мл DMF добавляли (4-фторфенил)борную кислоту (0,10 г, 0,71 ммоль, 7,7 экв.), а затем K2CO3 (0,18 г, 1,3 ммоль, 14 экв.) в 0,2 мл H2O и Pd(dppf)Cl2 (0,02 г, 0,03 ммоль, 0,3 экв.). После нагревания в течение 1 ч при 120°С смесь фильтровали и очищали методом HPLC, получая (S)-5-(4-фторфенил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамид (0,014 г, 0,035 ммоль, 37%). 1H-ЯМР (400 MHz, CD3OD) δ 9.17 (s, 2Н), 7.83 (m, 3Н), 7.71 (d, J=5.4 Hz, 1H), 7.56 (s, 1H), 7.31 (t, J=8.5 Hz, 2H), 6.92 (s, 1H), 6.85 (d, J=5.9 Hz, 1H), 5.00-3.60 (br, 5H), 2.65-2.30 (br, 2H); MS: (ES) m/z теор. для C22H19FN6O [M+H]+ 403,2, факт. 403.
Пример 95. Синтез (S)-5-(проп-1-ен-2-ил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил)пиримидин-2-карбоксамида
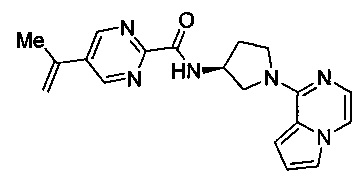
В раствор (S)-5-бром-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида (0,075 г, 0,19 ммоль, 1 экв.) в 1,8 мл DMF добавляли 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолан (0,10 г, 0,6 ммоль, 3 экв.), а затем K2CO3 (0,14 г, 0,99 ммоль, 5 экв.) в 0,2 мл H2O и Pd(dppf)Cl2 (0,02 г, 0,02 ммоль, 0,1 экв.). После нагревания в течение 2 ч при 120°С смесь фильтровали, упаривали и переносили на следующую стадию.
Пример 96. Синтез (S)-5-изопропил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида
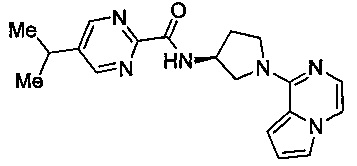
В раствор (S)-5-(проп-1-ен-2-ил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)пиримидин-2-карбоксамида (0,090 г, 0,26 ммоль, 1 экв.) в 1 мл МеОН добавляли 10% Pd/C (0,027 г, 0,03 ммоль, 0,1 экв.). Реакционную смесь подсоединяли к баллону с Н2 и перемешивали в течение 1 ч при комнатной температуре. Содержимое фильтровали и очищали методом HPLC, получая (S)-5-изопропил-N-(1-(пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил)пиримидин-2-карбоксамид (0,048 г, 0,14 ммоль, 53%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.81 (s, 2Н), 7.76 (s, 1Н), 7.71 (d, J=5.8 Hz, 1H), 7.55 (s, 1H), 6.92 (s, 1H), 6.84 (d, J=5.9 Hz, 1H), 5.00-3.60 (br, 5H), 3.18-3.00 (m, 1H), 2.65-2.30 (br, 2H), 1.36 (d, J=7.0 Hz, 6H); MS: (ES) m/z теор. для C19H22N6O [M+H]+ 351,2, факт. 351.
Пример 97. Синтез этил-1-(4-фторфенил)пиразол-3-карбоксилата
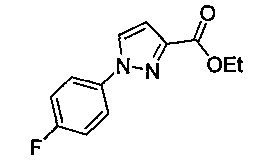
В раствор 1-фтор-4-йод-бензола (1,47 г, 6,6 ммоль, 1,3 экв.) и этил-1Н-пиразол-3-карбоксилата (0,71 г, 5,1 ммоль, 1 экв.) в 15 мл толуола добавляли CuI (0,19 г, 1,0 ммоль, 0,2 экв.), транс-N,N-диметилциклогексан-1,2-диамин (0,4 мл, 2,5 ммоль, 0,2 экв.) и карбонат калия (1,4 г, 10 ммоль, 2 экв.). Реакционную смесь нагревали при 110°С в течение 2 дней, а затем фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (гексан : EtOAc = 4:1), получая этил-1-(4-фторфенил)пиразол-3-карбоксилат (0,92 г, 3,9 ммоль, 77%).
Пример 98. Синтез 1-(4-фторфенил)пиразол-3-карбоновой кислоты
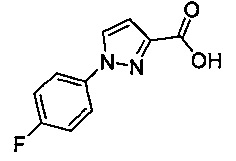
В раствор этил-1-(4-фторфенил)пиразол-3-карбоксилата (0,92 г, 3,9 ммоль, 1 экв.) в 18 мл EtOH/H2O (1,5:1) добавляли NaOH (0,47 г, 11,8 ммоль, 3 экв.). Реакционную смесь нагревали при 50°С в течение 1 ч, а затем гасили H2O и экстрагировали этилацетатом. Объединенные органические слои упаривали и очищали методом колоночной хроматографии на силикагеле (EtOAc : МеОН = 3:1), получая 1-(4-фторфенил)пиразол-3-карбоновую кислоту (0,42 г, 2,0 ммоль, 52%).
Пример 99. Синтез (S)-1-(4-фторфенил)-N-(1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-1Н-пиразол-3-карбоксамида

В раствор 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты (0,04 г, 0,19 ммоль, 1,5 экв.) в 1 мл DCM добавляли (S)-1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-амин (0,036 г, 0,13 ммоль, 1 экв.), пропилфосфоновый ангидрид (0,3 мл, 0,47 ммоль, 2,5 экв.) и N-метилморфолин (0,15 мл, 1,36 ммоль, 10,5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем гасили Н2O и экстрагировали этилацетатом. Объединенные органические слои упаривали и очищали методом HPLC, получая (S)-1-(4-фторфенил)-N-(1-(8-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-1Н-пиразол-3-карбоксамид (0,030 г, 0,074 ммоль, 57%). 1Н-ЯМР (400 MHz, CDCCl3) δ 7.91 (d, J=7.3 Hz, 1Н), 7.82 (s, 1H), 7.68 (dd, J=4.4, 8.8 Hz, 2H), 7.33 (d, J=4.8 Hz, 1H), 7.19 (t, J=8.8 Hz, 3H), 7.00 (m, 2H), 6.50 (s, 1H), 4.75 (s, 1H), 3.90-3.56 (m, 4H), 2.57 (s, 3H), 2.42-2.38 (m, 1H), 2.02-1.80 (m, 1H); MS: (ES) m/z теор. для C22H21FN6O [M+H]+ 405,2, факт. 405.
Пример 100. Синтез (S)-N-(1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-ил)-1-фенил-1Н-1,2,4-триазол-3-карбоксамида
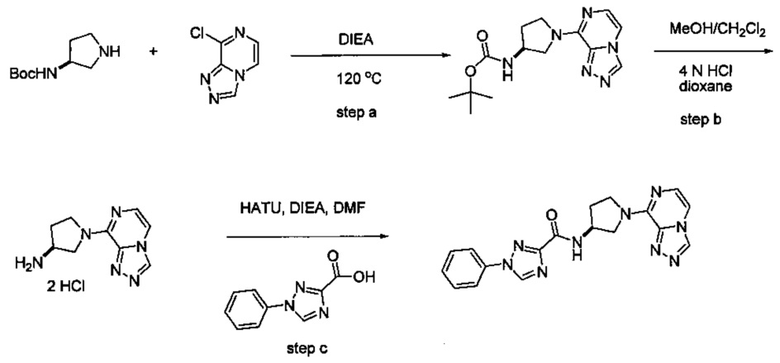
a) Смесь 8-хлор-[1,2,4]триазоло[4,3-а]пиразина (620 мг, 4,0 ммоль) и (S)-трет-бутил-пирролидин-3-илкарбамата (1,2 г, 6,5 ммоль) в диэтилизопропиламине (3 мл) нагревали при 120°С в течение 2 ч. После охлаждения до комнатной температуры смесь разбавляли 10% МеОН в EtOAc и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом флэш-хроматографии (SiO2, от 2 до 5% МеОН в CH2Cl2 в качестве элюента), получая искомое соединение в виде пены (1,15 г, выход 95%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 305,2 [M+H+].
b) Полученный выше Вос-амин растворяли в CH2Cl2 (4 мл) и МеОН (3 мл) при комнатной температуре, после чего добавляли 4N HCl в диоксане (9 мл, 36 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч. Полученную суспензию разбавляли EtOAc. После фильтрования и высушивания на воздухе получали искомое соединение в виде беловатого твердого вещества (1,05 г, количественно), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 205,1 [М+Н+].
c) В суспензию полученного выше (S)-1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)-пирролидин-3-амина гидрохлорида (90 мг, 0,32 ммоль) и 1-фенил-1Н-1,2,4-триазол-3-карбоновой кислоты (58 мг, 0,30 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 23 мг искомого соединения (выход 20%) виде белого твердого вещества. 1Н-ЯМР (400 MHz, DMSO-d6) δ 2.10-2.38 (два набора m, 2Н), 3.50-4.60 (br, 4Н), 4.65 (m, 1Н), 7.29 (d, J=4.4 Hz, 1H), 7.46 (t, J=7.3 Hz, 1H), 7.59 (t, J=7.3 Hz, 2H), 7.75 (d, J=4.7 Hz, 1H), 7.90 (d, J=8.0 Hz, 2H), 9.00 (d, J=7.0 H, 1H), 9.20 (s, 1H), 9.40 (s, 1H). MS: (ES) теор. для C18H17N9O [M+H]+ 376,2, факт. 376,2.
Пример 101. Синтез (S)-N-(1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-ил)-1-(4-фторфенил)-1Н-1,2,4-триазол-3-карбоксамида
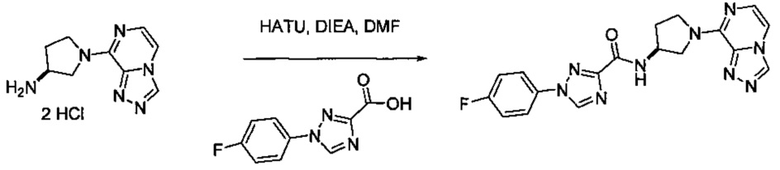
В суспензию (S)-1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-амина гидрохлорида (63 мг, 0,23 ммоль) и 1-(4-фторфенил)-1Н-1,2,4-триазол-3-карбоновой кислоты (42 мг, 0,2 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (76 мг, 0,2 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 32 мг искомого соединения (выход 40%) в виде белого твердого вещества. MS: (ES) m/z факт. 394,2.
Пример 102. Синтез (S)-N-(1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-ил)-1-(4-фторфенил)-1Н-пиразол-3-карбоксамида
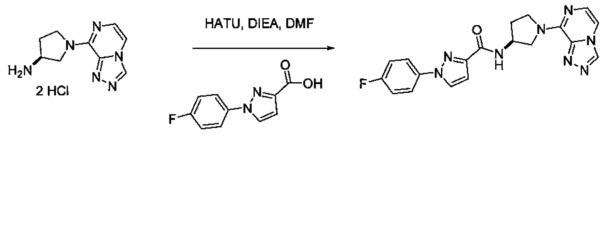
В суспензию (S)-1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-амина гидрохлорида (94 мг, 0,34 ммоль) и 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты (63 мг, 0,3 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 50 мг искомого соединения (выход 42%) в виде белого твердого вещества. MS: (ES) m/z факт. 393,2.
Пример 103. Синтез (S)-N-(1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-ил)-1-фенил-1Н-имидазол-4-карбоксамида
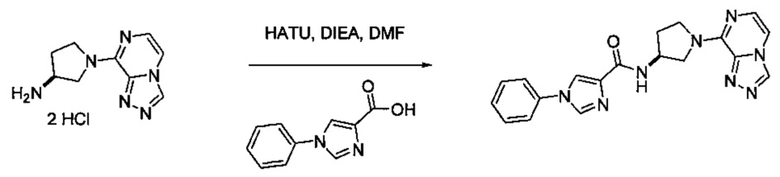
В суспензию (S)-1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-амина гидрохлорида (109 мг, 0,4 ммоль) и 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты (76 мг, 0,4 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (160 мг, 0,41 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 100 мг искомого соединения (выход 67%) виде белого твердого вещества. MS: (ES) m/z факт. 375,1.
Пример 104. Синтез (S)-N-(1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-ил)-5-изопропилпиримидин-2-карбоксамида
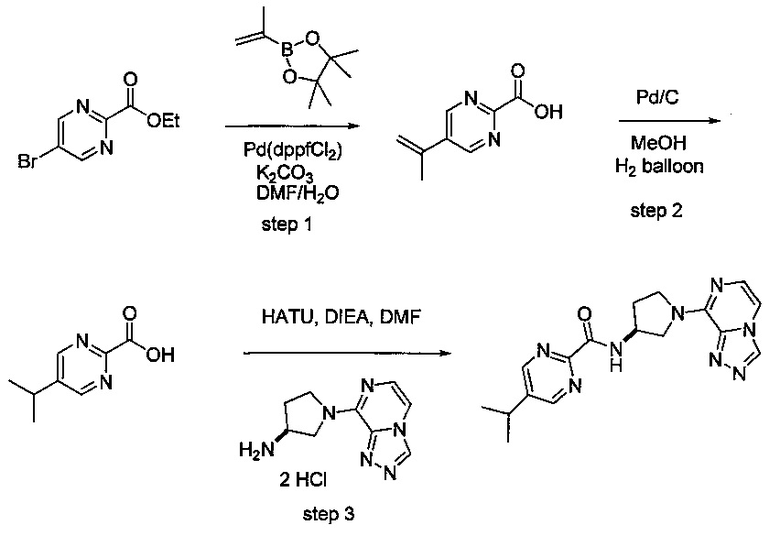
a) В суспензию этил-5-бромпиримидин-2-карбоксилата (700 мг, 3 ммоль), 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолана (1,5 г, 9 ммоль) и K2CO3 (2,1 г, 15 ммоль) в DMF (5 мл) и воде (1 мл) добавляли Pd(dppfCl2) (250 мг, 0,3 ммоль). Полученную смесь дегазировали (N2) в течение 2 мин, а затем нагревали при 120°С в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли МеОН и фильтровали через целит. Фильтрат использовали непосредственно на следующей стадии. MS: (ES) m/z 165,2 [M+H+].
b) В полученный выше фильтрат добавляли 10% Pd/C (влажный, 300 мг) и перемешивали смесь с подсоединенным Н2-баллоном в течение ночи. Твердое вещество отфильтровывали. Фильтрат разбавляли CH2Cl2 и промывали 1N HCl и насыщенным раствором NaCl. Органический слой упаривали под вакуумом, получая 300 мг искомого соединения (выход 60%) в виде светло-коричневого твердого вещества. MS: (ES) m/z 167,2 [M+H+].
c) В суспензию (S)-1-([1,2,4]триазоло[4,3-а]пиразин-8-ил)пирролидин-3-амина гидрохлорида (84 мг, 0,3 ммоль) и 5-изопропилпиримидин-2-карбоновой кислоты (51 мг, 0,3 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 6 мг искомого соединения (выход 5%) виде белого твердого вещества. MS: (ES) m/z факт. 353,1.
Пример 105. Синтез (S)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-5-изопропилпиримидин-2-карбоксамида
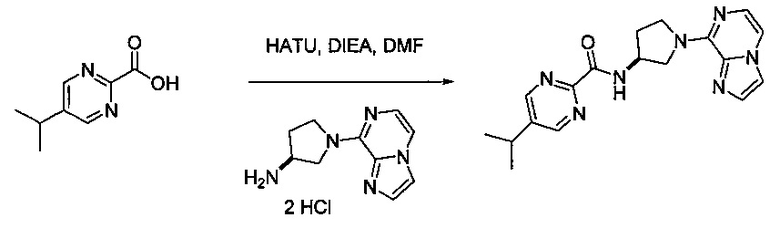
В суспензию (S)-1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-амина гидрохлорида (84 мг, 0,3 ммоль) и 5-изопропилпиримидин-2-карбоновой кислоты (85 мг, 0,5 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (190 мг, 0,5 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 35 мг искомого соединения (выход 32%) виде белого твердого вещества. MS: (ES) m/z факт. 352,1.
Пример 106. Синтез (±)-N-(3-(гидроксиметил)-1-(пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил)-5-изопропилпиримидин-2-карбоксамида
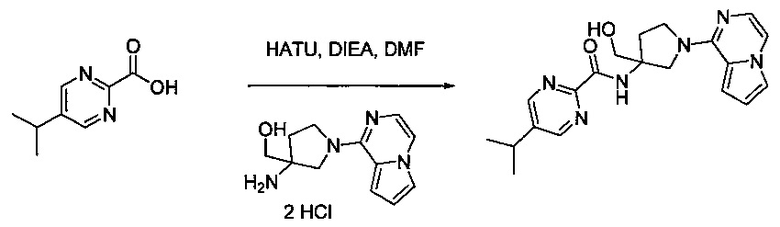
В суспензию (±)-(3-амино-1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)метанола гидрохлорида (62 мг, 0,2 ммоль) и 5-изопропилпиримидин-2-карбоновой кислоты (40 мг, 0,22 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (76 мг, 0,2 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 25 мг искомого соединения (выход 32%) виде белого твердого вещества. MS: (ES) m/z факт. 381,1.
Пример 107. Синтез (S)-N-(1-([1,2,4]триазоло[1,5-а]пиразин-8-ил)пирролидин-3-ил)-1-(4-фторфенил)-1Н-пиразол-3-карбоксамида
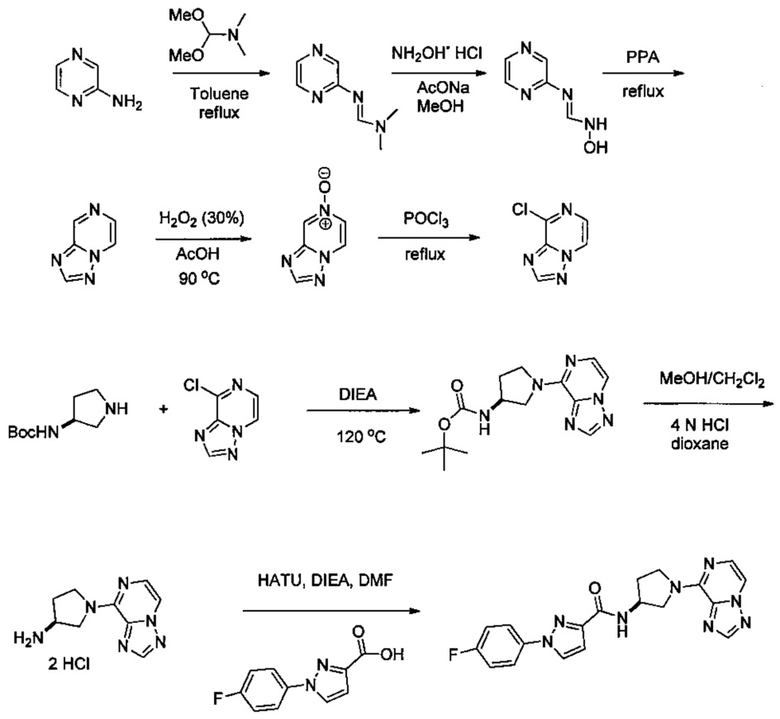
a) Смесь 2-аминопиразина (1,9 г, 20 ммоль) и диметилацеталя N,N-диметилформамида (2,62 г, 22 ммоль) в толуоле нагревали до кипения в течение 6 ч. Растворитель выпаривали, а остаток использовали непосредственно на следующей стадии.
b) В раствор полученного выше соединения в МеОН (30 мл) на ледяной бане добавляли NaOAc тригидрат (3,65 г, 25 ммоль), после чего медленно добавляли NH2OH⋅HCl (1,74 г, 25 ммоль). Смесь оставляли медленно нагреваться до комнатной температуры в течение 6 ч, а затем разбавляли DCM и 20% 7N раствором аммиака в МеОН. Твердое вещество отфильтровывали, фильтрат упаривали под вакуумом, а остаток растирали с EtOH. После фильтрования и высушивания на воздухе получали искомое соединение в виде беловатого твердого вещества (1,9 г, выход 69%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 139,1 [M+H+].
c) Смесь полученного выше вещества (1,9 г, 13,8 ммоль) и РРА (15 мл) нагревали при 90°С в течение 6 ч, а затем разбавляли ледяной водой и доводили до рН 8 водным раствором аммиака и NaHCO3. Затем смесь экстрагировали 10% МеОН в CH2Cl2. Органический слой сушили (Na2SO4), фильтровали и упаривали под вакуумом, получая искомое соединение в виде беловатого твердого вещества (1,2 г, выход 75%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 121,1 [M+H+].
d) В смесь из полученного выше вещества (600 мг, 5 ммоль) в АсОН медленно добавляли 30%-й водный раствор Н2О2 (1,8 мл, 19 ммоль) при комнатной температуре. Полученную смесь нагревали при 90°С в течение 6 ч, а затем выливали в ледяную воду и доводили до рН 8 с помощью 1N NaOH. Смесь экстрагировали 10% МеОН в CH2Cl2. Органический слой сушили (Na2SO4), фильтровали и упаривали под вакуумом, получая искомое соединение в виде пены (120 мг, выход 17%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 137,2 [M+H+].
e) Смесь полученной выше пены (120 мг, 0,9 ммоль) и POCl3 (5 мл) нагревали при 120°С в течение 6 ч, а затем упаривали под вакуумом. Остаток разбавляли EtOAc и промывали насыщенным водным NaHCO3. Органический слой сушили (Na2SO4), фильтровали и упаривали под вакуумом, получая искомое соединение в виде светло-коричневого твердого вещества (50 мг, выход 36%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 155,0 [M+H+].
f) Смесь полученного выше 8-хлор-[1,2,4]триазоло[1,5-а]пиразина (50 мг, 0,32 ммоль) и (S)-трет-бутил-пирролидин-3-илкарбамата (100 мг, 0,53 ммоль) в диэтилизо-пропиламине (2 мл) нагревали при 120°С в течение 2 ч. После охлаждения до комнатной температуры смесь разбавляли 10% МеОН в EtOAc и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом флэш-хроматографии (SiO2, от 2 до 5% МеОН в CH2Cl2 в качестве элюента), получая искомое соединение в виде пены (80 мг, выход 82%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 305,2 [M+H+].
g) Полученный выше Вос-амин растворяли в CH2Cl2 (3 мл) и МеОН (1 мл) при комнатной температуре, после чего добавляли 4N HCl в диоксане (4 мл, 16 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч, а затем упаривали под вакуумом, получая искомое соединение в виде светло-желтого твердого вещества (72 мг, количественно), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 205,1 [M+H+].
h) В суспензию полученного выше амина гидрохлорида (60 мг, 0,22 ммоль) и 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты (46 мг, 0,22 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (86 мг, 0,22 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом флэш-хроматографии на силикагеле (от 2 до 5% МеОН в CH2Cl2 в качестве элюента), а затем методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 28 мг искомого соединения (выход 32%) виде белого твердого вещества. 1Н-ЯМР (400 MHz, DMSO-d6) δ 2.05-2.38 (два набора m, 2Н), 3.60-4.58 (br, 4Н), 4.63 (m, 1Н), 6.89 (d, J=2.2 Hz, 1Н), 7.36 (t, J=6.6 Hz, 2H), 7.29 (d, J=4.4 Hz, 1H), 7.46 (t, J=7.3 Hz, 1H), 7.57 (d, J=4.4 Hz, 1H), 7.97-7.92 (m, 2H) (проверка F-взаимодействия), 8.13 (d, J=4.4 Hz, 1H), 8.46 (s, 1H), 8.52 (d, J=2.6 Hz, 1H), 8.55 (d, J=7.0 Hz, 1H). MS: (ES) m/z теор. для C19H17FN8O [M+H]+ 393,2, факт. 393.
Пример 108. Синтез (S)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-1-фенил-1Н-1,2,4-триазол-3-карбоксамида

a) Смесь 8-хлоримидазо[1,2-а]пиразина (500 мг, 3,25 ммоль) и (S)-трет-бутил-пирролидин-3-илкарбамата (1,0 г, 5,38 ммоль) в диэтилизопропиламине (3 мл) нагревали при 120°С в течение 3 ч. После охлаждения до комнатной температуры смесь разбавляли 10% МеОН в EtOAc и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом флэш-хроматографии (SiO2, от 2 до 5% МеОН в CH2Cl2 в качестве элюента), получая искомое соединение в виде пены (985 мг, количественно), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 304,1 [M+H+].
b) Полученный выше Вос-амин растворяли в CH2Cl2 (5 мл) и МеОН (3 мл) при комнатной температуре, после чего добавляли 4N HCl в диоксане (9 мл, 16 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч. Полученную суспензию разбавляли EtOAc. После фильтрования и высушивания на воздухе получали искомое соединение в виде беловатого твердого вещества (895 мг, количественно), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 204,1 [M+H+].
с) В суспензию полученного выше (S)-1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-амина гидрохлорида (84 мг, 0,3 ммоль) и 1-фенил-1Н-1,2,4-триазол-3-карбоновой кислоты (57 мг, 0,3 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом флэш-хроматографии на силикагеле (от 2 до 5% МеОН в CH2Cl2 в качестве элюента), а затем методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 60 мг искомого соединения (выход 54%) виде белого твердого вещества. MS: (ES) m/z теор. для C19H18N8O [М+Н]+ 375,2, факт. 375,2.
Пример 109. Синтез (S)-1-(4-фторфенил)-N-(1-(имидазо[1,2-а]пиразин-8-ил)-пирролидин-3-ил)-1Н-1,2,4-триазол-3-карбоксамида
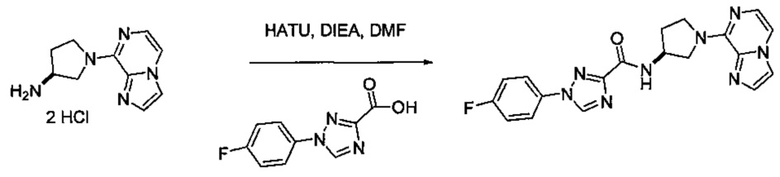
В суспензию (S)-1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-амина гидрохлорида (55 мг, 0,2 ммоль) и 1-(4-фторфенил)-1Н-1,2,4-триазол-3-карбоновой кислоты (42 мг, 0,2 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (76 мг, 0,2 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 45 мг искомого соединения (выход 57%) виде белого твердого вещества. MS: (ES) m/z факт. 393,1.
Пример 110. Синтез (S)-1-(3-фторфенил)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-1Н-1,2,4-триазол-3-карбоксамида
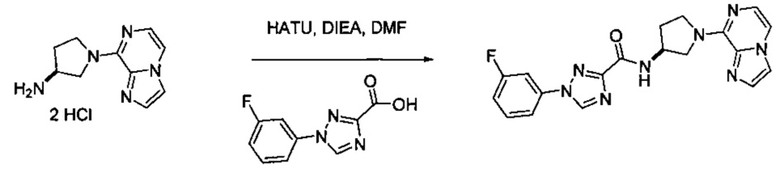
В суспензию (S)-1-(имидазо[1,2-a]пиразин-8-ил)пирролидин-3-амина гидрохлорида (84 мг, 0,3 ммоль) и 1-(3-фторфенил)-1Н-1,2,4-триазол-3-карбоновой кислоты (63 мг, 0,3 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 88 мг искомого соединения (выход 75%) виде белого твердого вещества. MS: (ES) m/z факт. 393,1.
Пример 111. Синтез (S)-N-(1-(имидазо[1,2-а]пиразин-8-ил)пирролидин-3-ил)-5-фенил-1,2,4-оксадиазол-3-карбоксамида

а) В смесь этил-2-оксиминоксамата (1,32 г, 10 ммоль) и диэтилизопропиламина (4 мл, 23 ммоль) в CH2Cl2 (30 мл) при -15°С медленно добавляли бензоилхлорид (1,41 г, 10 ммоль) на протяжении 2 ч. Смесь оставляли медленно нагреваться до комнатной температуры в течение 5 ч, а затем выливали в ледяную воду и экстрагировали CH2Cl2. Органический слой сушили (Na2SO4), фильтровали и упаривали, получая искомое соединение в виде белого твердого вещества (2,36 г, количественно), которое использовали непосредственно на следующей стадии.
b) Смесь полученного выше вещества (1,2 г, 5 ммоль) и пиридина (6 мл) нагревали при 120°С в течение 10 ч. После испарения растворителя остаток очищали методом флэш-хроматографии на силикагеле (от 10 до 25% EtOAc в гексане в качестве элюента), получая искомое соединение в виде беловатого твердого вещества (900 мг, выход 83%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 219,1 [M+H+].
c) В смесь полученного выше эфира (900 мг, 4,1 ммоль), МеОН (5 мл), THF (5 мл) и дистиллированной воды (5 мл) добавляли LiOH моногидрат (420 мг, 10 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. Затем разбавляли смесь ледяной водой, доводили до рН 3 с помощью 1N HCl, после чего экстрагировали 10% МеОН в CH2Cl2. Органический слой сушили (Na2SO4), фильтровали и упаривали под вакуумом, получая искомое соединение в виде беловатого твердого вещества (900 мг, выход 83%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 219,1 [M+H+].
d) В суспензию полученной выше 5-фенил-1,2,4-оксадиазол-3-карбоновой кислоты (58 мг, 0,3 ммоль) и (S)-1-([1,2,4]триазоло[1,5-а]пиразин-5-ил)пирролидин-3-амина гидрохлорида (90 мг, 0,32 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 26 мг искомого соединения (выход 23%) в виде белого твердого вещества. MS: (ES) m/z факт. 376,2 [M+H+].
Пример 112. Синтез (S)-1-(4-хлорфенил)-N-(1-(пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил)-1Н-1,2,4-триазол-3-карбоксамида
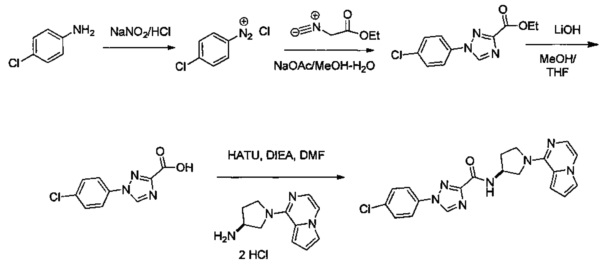
a) В смесь 4-хлоранилина (2,56 г, 20 ммоль) и конц. HCl (15 мл) в Н2О (30 мл) при -5°С по каплям добавляли раствор NaNO2 (1,38 г, 20 ммоль) в Н2О (5 мл), а затем перемешивали при 0°С в течение 10 мин, получая соль диазония.
b) Полученную выше смесь медленно добавляли в раствор этил-изоцианоацетата (2,26 г, 20 ммоль) в МеОН (100 мл) и H2O (10 мл) на ледяной бане. Смесь оставляли медленно нагреваться до комнатной температуры в течение 5 ч, а затем выпаривали растворитель. Остаток разбавляли 10% МеОН в CH2Cl2 и промывали насыщенным водным раствором NaHCO3, а затем насыщенным раствором NaCl. Органический слой сушили (Na2SO4), фильтровали и упаривали при пониженном давлении, получая искомое соединение в виде желтого твердого вещества (1,5 г, выход 29%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 252,2 [M+H+].
c) В смесь полученного выше эфира (1,5 г, 6 ммоль), МеОН (30 мл), THF (50 мл) и дистиллированной воды (15 мл) добавляли LiOH моногидрат (2,5 г, 60 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь разбавляли ледяной водой, доводили до рН 3 с помощью 1N HCl, а затем экстрагировали 15% iPrOH в CH2Cl2. Органический слой сушили (Na2SO4), фильтровали и упаривали под вакуумом, получая искомое соединение в виде беловатого твердого вещества (900 мг, выход 83%), которое использовали непосредственно на следующей стадии. MS: (ES) m/z 219,1 [M+H+].
d) В суспензию полученной выше 5-фенил-1,2,4-оксадиазол-3-карбоновой кислоты (58 мг, 0,3 ммоль) и (S)-1-([1,2,4]триазоло[1,5-а]пиразин-5-ил)пирролидин-3-амина гидрохлорида (90 мг, 0,32 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 26 мг искомого соединения (выход 23%) в виде белого твердого вещества. MS: (ES) m/z факт. 376.2 [M+H+].
Пример 113. Синтез (S)-5-фенил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-1,2,4-оксадиазол-3-карбоксамида
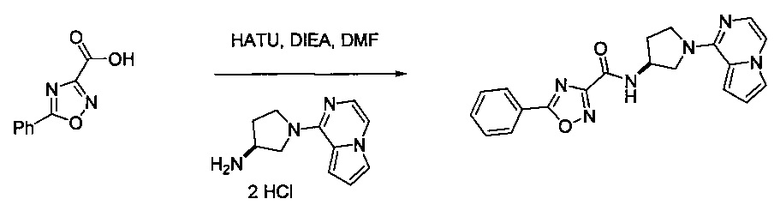
В суспензию (S)-1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-амина гидрохлорида (90 мг, 0,32 ммоль) и 5-фенил-1,2,4-оксадиазол-3-карбоновой кислоты (58 мг, 0,3 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (120 мг, 0,31 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом флэш-хроматографии на силикагеле (от 2 до 5% МеОН в CH2Cl2 в качестве элюента), а затем методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 15 мг искомого соединения (выход 18%) виде белого твердого вещества. MS: (ES) m/z теор. для C19H18N8O [М+Н]+ 375,2, факт. 374,1.
Пример 114. Синтез (S)-1-фенил-N-(1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил)-1Н-1,2,4-триазол-3-карбоксамида
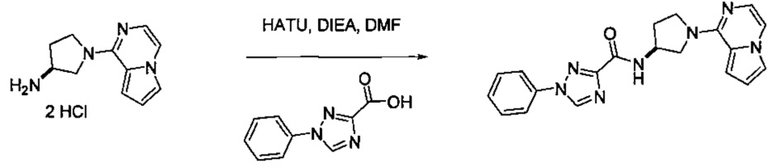
В суспензию (S)-1-(пирроло[1,2-а]пиразин-1-ил)пирролидин-3-амина гидрохлорида (60 мг, 0,21 ммоль) и 1-фенил-1Н-1,2,4-триазол-3-карбоновой кислоты (40 мг, 0,21 ммоль) в DMF (5 мл) добавляли диэтилизопропиламин (650 мг, 5 ммоль), а затем HATU (80 мг, 0,21 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенными водными растворами KH2PO4 и NaHCO3, а затем насыщенным раствором NaCl. Органический слой упаривали под вакуумом, а остаток очищали методом обратнофазовой HPLC (ацетонитрил-Н2О с 0,1% TFA в качестве элюента), получая 15 мг искомого соединения (выход 18%) виде белого твердого вещества. MS: (ES) m/z факт. 374,1.
Пример 115. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты
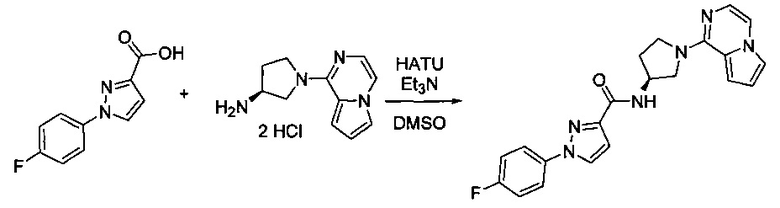
В смесь 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты (41 мг, 0,20 ммоль) и 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламина дигидрохлорида (55 мг, 0,20 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,112 мл, 0,80 ммоль), а затем HATU (84 мг, 0,22 ммоль). Смесь перемешивали в течение 30 мин, разбавляли дихлорметаном (1,0 мл), а затем промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (9,5 мг, 0,02 ммоль, выход 9%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.26 (s, 1Н), 7.87 (dd, J=4.8, 9.2 Hz, 2H), 7.77 (d, J=2.6 Hz, 1H), 7.71 (d, J=5.5 Hz, 1H), 7.56 (d, J=4.4 Hz, 1H), 7.25 (t, J=9.2 Hz, 2H), 6.95 (d, J=2.5 Hz, 1H), 6.92 (dd, J=2.6, 4.4 Hz, 1H), 6.84 (d, J=5.5 Hz, 1H), 4.95-3.65 (br, 5H), 2.58-2.30 (m, 2H); MS: (ES) m/z теор. для C21H19FN6O [M+H]+ 391,2, факт. 391.
Пример 116. Синтез 2-фенил-оксазол-4-карбоновой кислоты

Растворяли этиловый эфир 2-фенил-оксазол-4-карбоновой кислоты (500 мг, 2,30 ммоль) в смеси THF (2,3 мл) и МеОН (2,3 мл). Туда же добавляли NaOH (10% в воде, 2,3 мл). Реакционную смесь перемешивали в течение 2 ч, а затем разбавляли EtOAc и промывали IN HCl. Водный слой экстрагировали EtOAc, а объединенные органические слои сушили над Na2SO4, фильтровали и упаривали, получая продукт в виде белого твердого вещества (366 мг, 84%).
Пример 117. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 2-фенил-оксазол-4-карбоновой кислоты
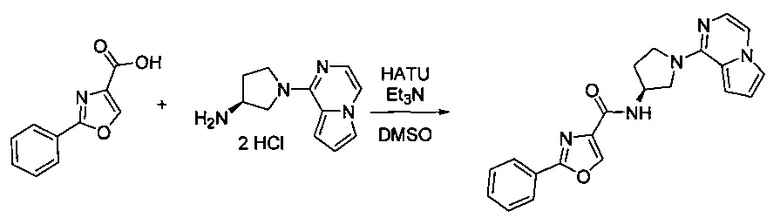
В смесь 2-фенил-оксазол-4-карбоновой кислоты (38 мг, 0,20 ммоль) и 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламина дигидрохлорида (55 мг, 0,20 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,112 мл, 0,80 ммоль), а затем HATU (84 мг, 0,22 ммоль). Смесь перемешивали в течение 30 мин, разбавляли дихлорметаном (1,0 мл), а затем промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали, получая неочищенный материал. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (27 мг, 0,06 ммоль, выход 28%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.76 (d, J=6.6 Hz, 1H), 8.49 (s, 1H), 8.10-8.06 (m, 2H), 7.79 (dd, 7=1.2, 2.4 Hz, 1H), 7.74 (d, J=5.5 Hz, 1H), 7.59-7.50 (m, 4H), 6.95 (dd, J=2.7, 4.2 Hz, 1H), 6.87 (d, J=5.4 Hz, 1H), 4.95-3.65 (br, 5H), 2.60-2.30 (m, 2H); MS: (ES) m/z теор. для C21H19N5O2 [M+H]+ 374,2, факт. 374.
Пример 118. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 2-фенил-тиазол-4-карбоновой кислоты
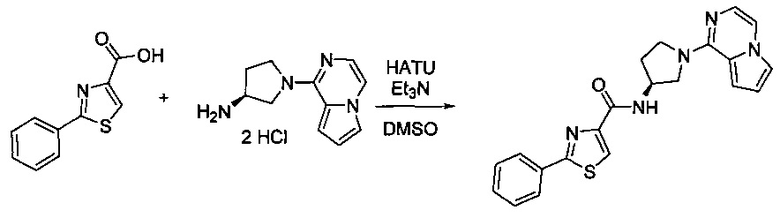
В смесь 2-фенил-тиазол-4-карбоновой кислоты (41 мг, 0,20 ммоль) и 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламина дигидрохлорида (55 мг, 0,20 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,112 мл, 0,80 ммоль), а затем HATU (84 мг, 0,22 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл) и промывали водой (3×1,0 мл). Органические слои сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая неочищенный продукт. Неочищенный продукт очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (34 мг, 0,07 ммоль, выход 34%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.87 (d, J=6.7 Hz, 1H), 8.26 (s, 1H), 8.07-8.01 (m, 2H), 7.79 (dd, J=1.2, 2.7 Hz, 1H), 7.74 (d, J=5.5 Hz, 1H), 7.59 (d, J=4.3 Hz, 1H), 7.51-7.46 (m, 3H), 6.95 (dd, J=2.3, 4.4 Hz, 1H), 6.87 (d, J=5.5 Hz, 1H), 4.95-3.65 (br, 5H), 2.60-2.38 (m, 2H); MS: (ES) m/z теор. для C21H19N5OS [M+H]+ 390,2, факт. 390.
Пример 119. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 1-о-толил-1H-пиразол-3-карбоновой кислоты
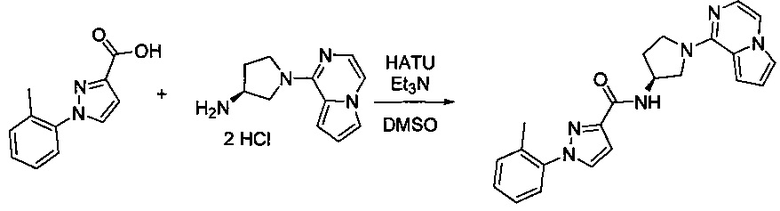
В смесь 1-о-толил-1H-пиразол-3-карбоновой кислоты (15 мг, 0,07 ммоль) и 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламина дигидрохлорида (19 мг, 0,07 ммоль) добавляли DMSO (0,4 мл). Туда же добавляли триэтиламин (0,039 мл, 0,28 ммоль), а затем HATU (30 мг, 0,08 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл) и промывали водой (3×1,0 мл). Органические слои сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая неочищенный продукт. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (12 мг, 0,02 ммоль, выход 34%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.62 (d, J=5.9 Hz, 1H), 7.88 (d, J=2.5 Hz, 1H), 7.75 (s, 1H), 7.70 (d, J=5.9 Hz, 1H), 7.54 (d, J=3.6 Hz, 1H), 7.40-7.25 (m, 4H), 6.94 (d, J=2.2 Hz, 1H), 6.91 (dd, J=2.6, 4.4 Hz, 1H), 6.82 (d, J=5.5 Hz, 1H), 4.95-3.65 (br, 5H), 2.58-2.30 (m, 2H), 2.20 (s, 3H); MS: (ES) m/z теор. для C22H22N6O [M+H]+ 387,2, факт. 387.
Пример 120. Синтез [1-(8-метил-пирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил]амида 2-(4-фторфенил)-тиазол-4-карбоновой кислоты
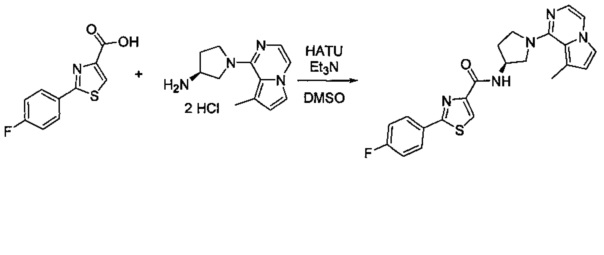
В смесь 2-(4-фторфенил)-тиазол-4-карбоновой кислоты (31 мг, 0,14 ммоль) и 1-(8-метилпирроло[1,2-а]пиразин-1-ил)-пирролидин-3-иламина дигидрохлорида (40 мг, 0,14 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,078 мл, 0,56 ммоль), а затем HATU (57 мг, 0,15 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл) и промывали водой (3×1,0 мл). Органические слои сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая неочищенный продукт. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Объединенные фракции упаривали, разводили в CH2Cl2, промывали NaHCO3, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученное масло затем очищали методом флэш-хроматографии на силикагеле (градиент CH2Cl2:МеОН от 99:1 до 90:10). Содержащие продукт фракции объединяли и упаривали, а затем лиофилизировали из смеси MeCN и 1М HCl, получая HCl-соль продукта в виде белого твердого вещества (32 мг, 0,06 ммоль, выход 43%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.22 (s, 1Н), 8.09 (dd, J=5.1, 8.8 Hz, 2H), 7.67 (d, J=5.5 Hz, 2H), 7.24 (t, J=8.8 Hz, 2H), 6.75 (d, J=5.5 Hz, 2H), 4.85-4.75 (m, 1H), 4.30-4.22 (m, 1H), 4.10-3.90 (m, 3H), 2.64 (s, 3H), 2.55-2.35 (m, 2H); MS: (ES) m/z теор. для C22H20FN5OS [M+H]+ 422,2, факт. 422.
Пример 121. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 2-(4-фторфенил)-тиазол-4-карбоновой кислоты
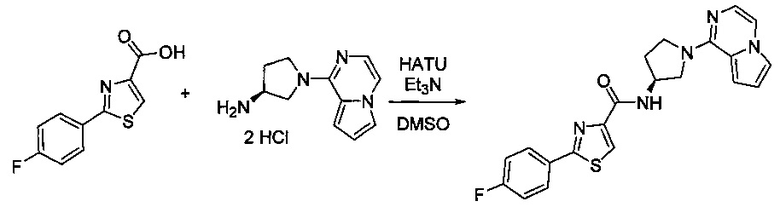
В смесь 2-(4-фторфенил)-тиазол-4-карбоновой кислоты (56 мг, 0,25 ммоль) и 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламина дигидрохлорида (70 мг, 0,25 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,139 мл, 1,0 ммоль), а затем HATU (106 мг, 0,28 ммоль). Смесь перемешивали в течение 30 мин, разбавляли дихлорметаном (1,0 мл), а затем промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая неочищенный продукт. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде желтого твердого вещества (67 мг, 0,13 ммоль, выход 51%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.85 (d, J=6.6 Hz, 1H), 8.23 (s, 1H), 8.09-8.05 (m, 2H), 7.76 (s, 1H), 7.72 (d, J=5.9 Hz, 1H), 7.56 (s, 1H), 7.23 (t, J=8.8 Hz, 1H), 6.93 (dd, J=2.5,4.4 Hz, 1H), 6.85 (d, J=5.9 Hz, 1H), 5.00-3.60 (m, 5H), 2.60-2.35 (m, 2H); MS: (ES) m/z теор. для C21H18FN5OS [M+H]+ 408,2, факт. 408.
Пример 122. Синтез 1-(тетрагидропиран-4-ил)-1Н-пиразол-3-карбоновой кислоты

a) В колбу вносили 4-гидрокситетрагидропиран (3,0 г, 29,4 ммоль), метан-сульфонилхлорид (2,39 мл, 30,9 ммоль), триэтиламин (8,20 мл, 58,8 ммоль) и CH2Cl2. Полученную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь промывали насыщенным водным NaHCO3 (100 мл) и водой (100 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный при этом неочищенный продукт использовали без дальнейшей очистки.
b) В сосуд вносили этил-1Н-пиразол-3-карбоксилат (3,74 г, 26,7 ммоль), неочищенный тетрагидропиран-4-иловый эфир метансульфоновой кислоты (29,4 ммоль), Cs2CO3 (17,4 г, 53,4 ммоль) и DMF (100 мл). Реакционную смесь нагревали до 80°С и перемешивали в течение ночи. Затем реакционную смесь распределяли между CH2Cl2 (250 мл) и водой (250 мл), а органический слой промывали водой (5×250 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Неочищенный продукт, содержащий 2 региоизомера, очищали методом флэш-хроматографии на силикагеле (от 95:5 до 50:50 гексан : EtOAc) и выделяли выходящий последним изомер. Его очищали на силикагеле еще раз (от 85:15 до 60:40 гексан : EtOAc), получая чистый искомый продукт (1,14 г, 19%).
c) Растворяли этиловый эфир 1-(тетрагидропиран-4-ил)-1Н-пиразол-3-карбоновой кислоты (1,14 g, 5,09 ммоль) в смеси THF (5,0 мл) и МеОН (5,0 мл). Туда же добавляли NaOH (20% в воде, 5,0 мл). Реакционную смесь перемешивали в течение ночи, а затем разбавляли EtOAc и промывали 1М NaHSO4. Водный слой экстрагировали EtOAc, а объединенные органические слои сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая продукт в виде белого твердого вещества (802 мг, 80%).
Пример 123. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 1-(тетрагидропиран-4-ил)-1Н-пиразол-3-карбоновой кислоты
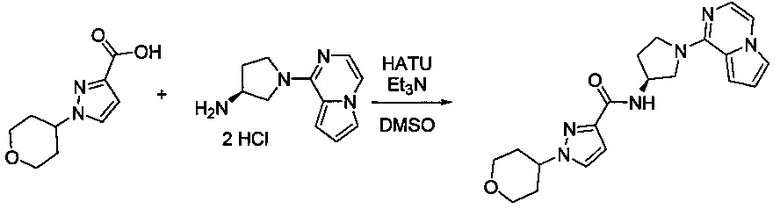
В смесь 1-(тетрагидропиран-4-ил)-1Н-пиразол-3-карбоновой кислоты (31 мг, 0,16 ммоль) и 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламина дигидрохлорида (45 мг, 0,16 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,089 мл, 0,64 ммоль), а затем HATU (68 мг, 0,18 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде бледно-желтого твердого вещества (29 мг, 0,06 ммоль, выход 37%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.48 (d, J=6.2 Hz, 1H), 7.78-7.70 (m, 3H), 7.55 (s, 1H), 6.91 (dd, J=2.5, 4.4 Hz, 1H), 6.83 (d, J=5.5 Hz, 1H), 6.73 (d, J=2.2 Hz, 1H), 5.00-3.60 (m, 8H), 3.55 (dt, J=2.2, 11.7 Hz, 2H), 2.60-2.39 (m, 2H), 2.20-1.95 (m, 4H); MS: (ES) m/z теор. для C20H24N6O2 [M+H]+ 381,2, факт. 381.
Пример 124. Синтез (1-[1,2,4]триазоло[4,3-а]пиразин-8-ил-пирролидин-3-ил)амида 1-(тетрагидропиран-4-ил)-1H-пиразол-3-карбоновой кислоты
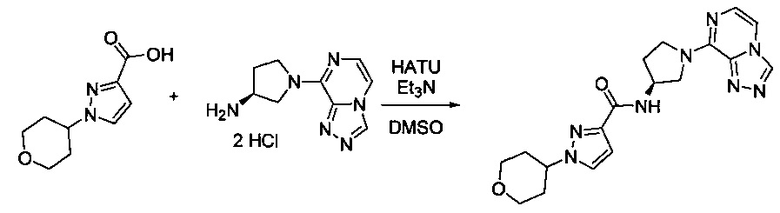
В смесь 1-(тетрагидропиран-4-ил)-1Н-пиразол-3-карбоновой кислоты (35 мг, 0,18 ммоль) и 1-[1,2,4]триазоло[4,3-а]пиразин-8-ил-пирролидин-3-иламина (50 мг, 0,18 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,100 мл, 0,72 ммоль), а затем HATU (76 мг, 0,20 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (16 мг, 0,03 ммоль, выход 18%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.25 (s, 1Н), 7.85 (d, J=5.1 Hz, 1H), 7.73 (d, J=2.6 Hz, 1H), 7.20 (d, J=5.5 Hz, 1H), 6.73 (d, J=2.5 Hz, 1H), 5.00-3.60 (m, 8H), 3.55 (dt, J=2.2, 11.7 Hz, 2H), 2.60-2.39 (m, 2H), 2.20-1.95 (m, 4H); MS: (ES) m/z теор. для C18H22N8O2 [M+H]+ 383,2, факт. 383.
Пример 125. Синтез (1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил)амида 1-(тетрагидропиран-4-ил)-1Н-пиразол-3-карбоновой кислоты
В смесь 1-(тетрагидропиран-4-ил)-1Н-пиразол-3-карбоновой кислоты (35 мг, 0,18 ммоль) и 1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-иламина дигидрохлорида (50 мг, 0,18 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,100 мл, 0,72 ммоль), а затем HATU (76 мг, 0,20 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде бледно-желтого масла (35 мг, 0,07 ммоль, выход 39%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.02 (s, 1Н), 7.90 (d, J=5.9 Hz, 1H), 7.79 (s, 1H), 7.73 (d, J=2.5 Hz, 1H), 7.14 (d, J=5.5 Hz, 1H), 6.72 (d, J=2.6 Hz, 1H), 5.00-3.70 (m, 8H), 3.55 (dt, J=2.2, 11.7 Hz, 2H), 2.60-2.30 (m, 2H), 2.20-1.95 (m, 4H); MS: (ES) m/z теор. для C19H23N7O2 [M+H]+ 382,2, факт. 382.
Пример 126. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 2-о-толилоксазол-4-карбоновой кислоты
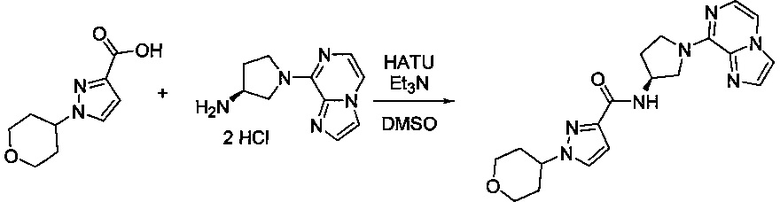
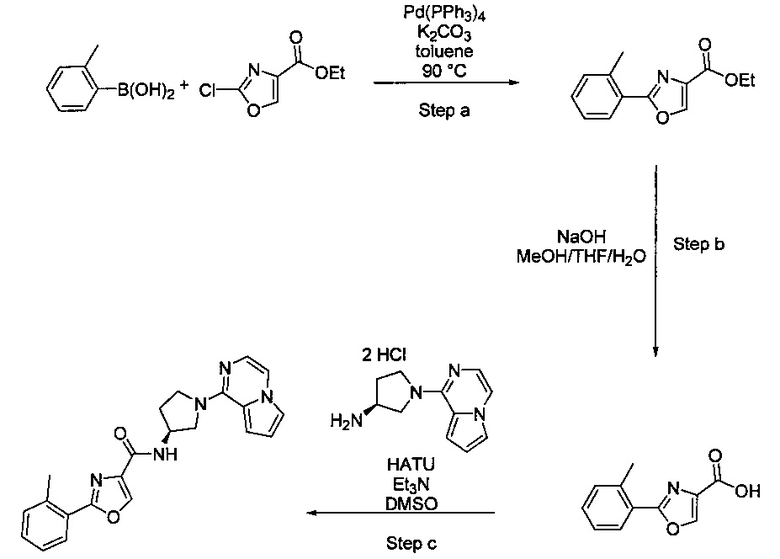
a) В колбу вносили этил-2-хлороксазол-4-карбоксилат (500 мг, 2,85 ммоль), 2-метилбензолбороновую кислоту (387 мг, 2,85 ммоль), Pd(PPh3)4 (132 мг, 0,114 ммоль), K2CO3 (787 мг, 5,7 ммоль) и толуол (29 мл). Реакционную смесь дегазировали струей N2, а затем нагревали при 90°С в течение 1 ч. Затем реакцию разбавляли EtOAc и промывали 1М водным NaOH. Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом флэш-хроматографии на силикагеле (гексан:EtOAc), получая смесь 2:1 продукта с этил-2-хлороксазол-4-карбо-ксилатом (360 мг), которую переносили на следующую стадию без дальнейшей очистки.
b) Смесь 2:1 этилового эфира 2-о-толил-оксазол-4-карбоновой кислоты с этил-2-хлороксазол-4-карбоксилатом (360 мг) растворяли в смеси THF (1,0 мл) и МеОН (1,0 мл). Туда же добавляли NaOH (20% в воде, 1,0 мл). Реакционную смесь перемешивали в течение ночи, а затем разбавляли EtOAc (50 мл) и промывали 1М NaHSO4 (50 мл). Водный слой экстрагировали EtOAc (2×50 мл), а объединенные органические слои сушили над Na2SO4, фильтровали и упаривали, получая смесь 2:1 продукта и 2-хлор-оксазол-4-карбоновой кислоты (360 мг).
c) Смесь 2:1 кислот из стадии b (37 мг) растворяли в DMSO (0,5 мл) и добавляли 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламин дигидрохлорид (50 мг, 0,18 ммоль). Туда же добавляли триэтиламин (0,100 мл, 0,72 ммоль), а затем HATU (76 мг, 0,20 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:Н2О + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде желтого твердого вещества (26 мг, 0,05 ммоль, выход 29%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.61 (d, J=6.2 Hz, 1H), 8.49 (s, 1H), 7.94 (d, J=7.7 Hz, 1H), 7.76 (d, О=1.1 Hz, 1H), 7.71 (d, J=5.4 Hz, 1H), 7.56 (d, J=2.9 Hz, 1H), 7.40-7.26 (m, 3H), 6.92 (dt, J=1.8,2.6 Hz, 1H), 6.83 (d, J=5.5 Hz, 1H), 5.00-3.60 (m, 5H), 2.66 (s, 3H), 2.60-2.38 (m, 2H); MS: (ES) m/z теор. для C22H21N5O2 [M+H]+ 388,2, факт. 388.
Пример 127. Синтез (1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил)амида 2-фенилоксазол-4-карбоновой кислоты

В смесь 2-фенилоксазол-4-карбоновой кислоты (42 мг, 0,22 ммоль) и 1-имид-азо[1,2-а]пиразин-8-илпирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:Н2О + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (62 мг, 0,13 ммоль, выход 58%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.71 (d, J=6.3 Hz, 1H), 8.46 (s, 1H), 8.06-8.02 (m, 3H), 7.91 (d, J=5.5 Hz, 1H), 7.80 (s, 1H), 7.49 (m, 3H), 7.15 (d, J=5.9 Hz, 1H), 5.00-3.80 (m, 5H), 2.60-2.38 (m, 2H); MS: (ES) m/z теор. для C20H18N6O2 [M+H]+ 375,2, факт. 375.
Пример 128. Синтез (1-[1,2,4]триазоло[4,3-а]пиразин-8-ил-пирролидин-3-ил)-амида 2-фенилоксазол-4-карбоновой кислоты
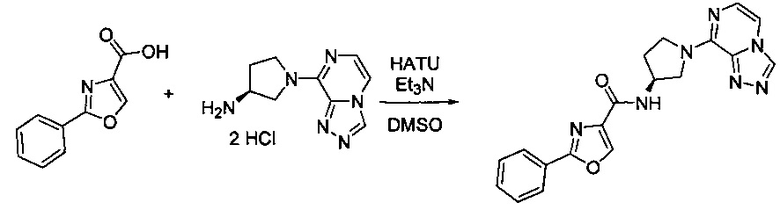
В смесь 2-фенилоксазол-4-карбоновой кислоты (42 мг, 0,22 ммоль) и 1-[1,2,4]триазоло[4,3-а]пиразин-8-ил-пирролидин-3-иламина (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (58 мг, 0,12 ммоль, выход 54%). 1H-ЯМР (400 MHz, CD3OD) δ 9.26 (s, 1Н), 8.70 (d, J=5.9 Hz, 0.5H), 8.45 (s, 1H), 8.04 (d, J=5.5 Hz, 2H), 7.86 (d, J=5.5 Hz, 1H), 7.49 (m, 3H), 7.21 (d, J=5.5 Hz, 1H), 5.00-3.80 (m, 5H), 2.60-2.38 (m, 2H); MS: (ES) m/z теор. для C19H17N7O2 [M+H]+ 376,2, факт. 376.
Пример 129. Синтез (1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил)амида 2-фенилтиазол-4-карбоновой кислоты
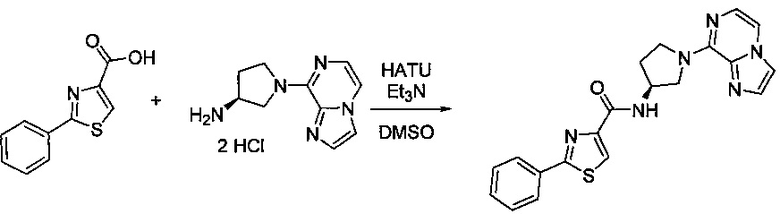
В смесь 2-фенилтиазол-4-карбоновой кислоты (45 мг, 0,22 ммоль) и 1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой хроматографии (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде бледно-желтого твердого вещества (74 мг, 0,15 ммоль, выход 67%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.82 (d, J=6.3 Hz, 1H), 8.23 (s, 1H), 8.03-8.01 (m, 3H), 7.92-7.90 (m, 1H), 7.88 (s, 1H), 7.48-7.45 (m, 3H), 7.15 (d, J=5.8 Hz, 1H), 4.98-3.80 (m, 5H), 2.60-2.40 (m, 2H); MS: (ES) m/z теор. для C20H18N6OS [M+H]+ 391,2, факт. 391.
Пример 130. Синтез (1-[1,2,4]триазоло[4,3-а]пиразин-8-ил-пирролидин-3-ил)-амида 2-фенилтиазол-4-карбоновой кислоты
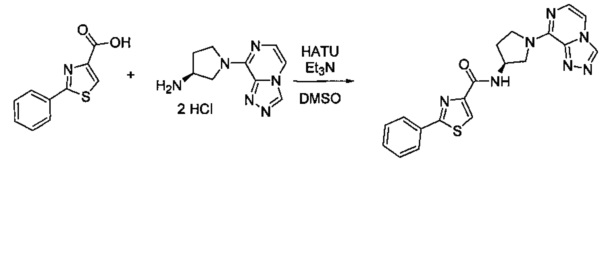
В смесь 2-фенилтиазол-4-карбоновой кислоты (42 мг, 0,22 ммоль) и 1-[1,2,4]триазоло[4,3-а]пиразин-8-ил-пирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном, промывали водой (3×), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал растирали с MeCN и фильтровали, а твердое вещество промывали MeCN. Твердое вещество разводили в MeCN и 1М HCl, а затем лиофилизировали, получая HCl-соль продукта в виде белого твердого вещества (17 мг, 0,04 ммоль, выход 18%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.31 (s, 1Н), 8.85 (d, J=3.6 Hz, 1H), 8.24 (s, 1H), 8.02 (d, J=3.6 Hz, 2H), 7.92 (d, J=5.4 Hz, 1H), 7.47 (s, 3H), 7.19 (d, J=5.4 Hz, 1H), 5.00-3.80 (m, 5H), 2.60-2.40 (m, 2H); MS: (ES) m/z теор. для C19H17N7OS [M+H]+ 392,2, факт. 392.
Пример 131. Синтез 1-о-толил-1H-пиразол-3-карбоновой кислоты
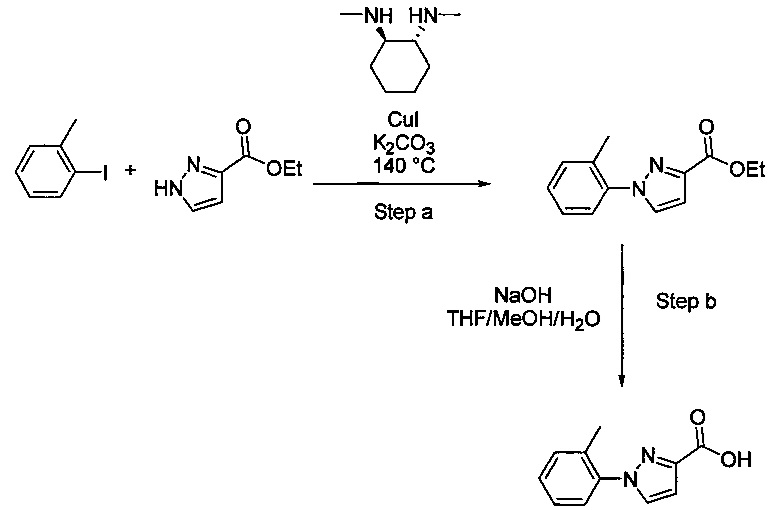
а) В колбу вносили 2-йодтолуол (0,964 мл, 7,57 ммоль), этил-1H-пиразол-3-карбоксилат (1,00 г, 7,14 ммоль), CuI (272 мг, 1,43 ммоль), транс-N,N'-диметилциклогексан-1,2-диамин (0,451 мл, 2,86 ммоль) и K2CO3 (3,15 г, 22,8 ммоль). Реакционную смесь нагревали при 140°С в течение 3 ч. Затем реакционную смесь распределяли между CH2Cl2 и насыщенным водным NH4Cl и разделяли фазы. Органический слой промывали водой, сушили над Na2SO4, фильтровали и упаривали. Неочищенный продукт очищали методом флэш-хроматографии на силикагеле (от 90:10 до 70:30 гексан : метил-трет-бутиловый эфир), получая продукт (238 мг, 1,03 ммоль, 14%) в виде бесцветного масла.
b) Растворяли этиловый эфир 1-о-толил-1Н-пиразол-3-карбоновой кислоты (238 мг, 1,03 ммоль) в смеси THF (2,0 мл) и МеОН (2,0 мл). Туда же добавляли NaOH (20% в воде, 1,0 мл). Реакционную смесь перемешивали в течение 2 ч, а затем разбавляли EtOAc и промывали 1М HCl. Водный слой экстрагировали EtOAc, а объединенные органические слои сушили над Na2SO4, фильтровали и упаривали, получая продукт в виде белого твердого вещества (164 мг, 0,81 ммоль, 79%).
Пример 132. Синтез (1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил)амида 1-о-толил-1H-пиразол-3-карбоновой кислоты
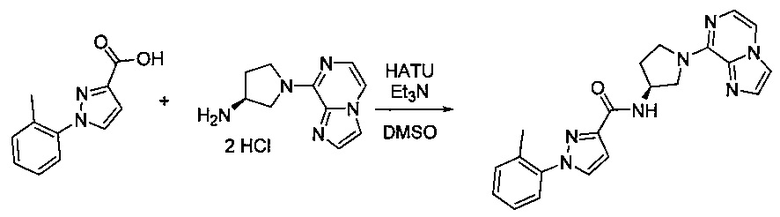
В смесь 1-о-толил-1Н-пиразол-3-карбоновой кислоты (44 мг, 0,22 ммоль) и 1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный продукт очищали методом обратнофазовой хроматографии (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде желтого твердого вещества (29 мг, 0,06 ммоль, выход 26%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.58 (d, J=7.3 Hz, 1H), 8.01 (d, J=0.7 Hz, 1H), 7.92-7.87 (m, 2H), 7.78 (d, J=1.1 Hz, 1H), 7.40-7.28 (m, 4H), 7.12 (d, J=5.8 Hz, 1H), 6.93 (d, J=2.2 Hz, 1H), 4.94-3.80 (m, 5H), 2.56-2.32 (m, 2H), 2.20 (s, 3H); MS: (ES) m/z теор. для C21H21N7O [M+H]+ 388,2, факт. 388.
Пример 133. Синтез (1-[1,2,4]триазоло[4,3-д]пиразин-8-ил-пирролидин-3-ил)-амида 1-о-толил-1H-пиразол-3-карбоновой кислоты

В смесь 1-о-толил-1Н-пиразол-3-карбоновой кислоты (44 мг, 0,22 ммоль) и 1-[1,2,4]триазоло[4,3-а]пиразин-8-ил-пирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном, промывали водой (3×), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал растирали с MeCN и фильтровали, а твердое вещество промывали MeCN. Твердое вещество разводили в MeCN и 1М HCl, а затем лиофилизировали, получая HCl-соль продукта в виде желтого твердого вещества (13 мг, 0,03 ммоль, выход 12%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.30 (s, 1Н), 7.91 (d, J=5.8 Hz, 1H), 7.88 (s, 1H), 7.40-7.28 (m, 4H), 7.17 (m, 1H), 6.95 (s, 1H), 4.94-4.70 (m, 4H), 4.24-3.84 (m, 3H), 2.62-2.38 (m, 2H), 2.20 (s, 3H); MS: (ES) m/z теор. для C20H20N8O [M+H]+ 389,2, факт. 389.
Пример 134. Синтез 2-о-толилтиазол-4-карбоновой кислоты
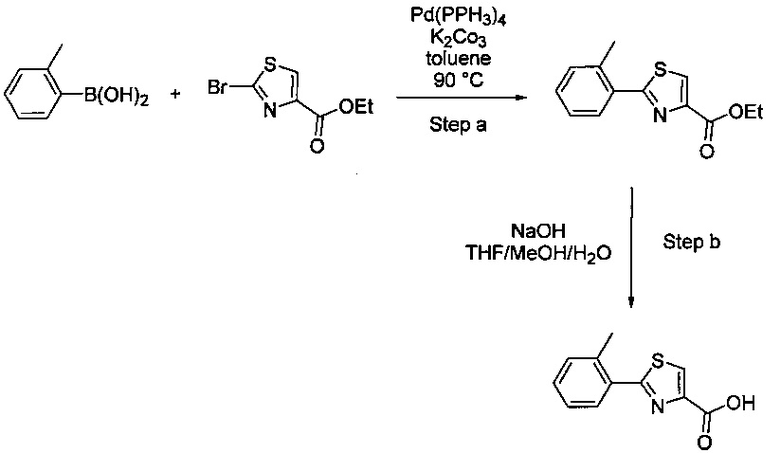
а) В колбу вносили 2-метилбензолбороновую кислоту (500 мг, 3,68 ммоль), этил-2-бромтиазол-4-карбоксилат (869 мг, 3,68 ммоль), Pd(PPh3)4 (173 мг, 0,150 ммоль), K2CO3 (2М в воде, 3,68 мл, 7,36 ммоль) и толуол (37 мл). Реакционную смесь дегазировали струей N2, а затем нагревали до 90°С и перемешивали в течение ночи. Затем реакцию разбавляли EtOAc и промывали 1М водным NaOH. Водный слой экстрагировали EtOAc (2×100 мл), а объединенные органические слои сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом флэш-хроматографии на силикагеле (от 95:5 до 70:30 гексан : EtOAc), получая продукт (550 мг, 2,22 ммоль, 60%) в виде бледно-желтого масла.
b) Растворяли этиловый эфир 2-о-толилтиазол-3-карбоновой кислоты (550 мг, 2,22 ммоль) в смеси THF (1,0 мл) и МеОН (1,0 мл). Туда же добавляли NaOH (20% в воде, 0,444 мл). Реакционную смесь перемешивали в течение ночи, а затем разбавляли CH2Cl2 (30 мл) и промывали 1М NaHSO4 (30 мл). Водный слой экстрагировали CH2Cl2 (2×30 мл), а объединенные органические слои промывали водой (1×30 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая продукт (434 мг, 1,98 ммоль, 89%) в виде белого твердого вещества.
Пример 135. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 2-о-толилтиазол-4-карбоновой кислоты

В смесь 2-о-толилтиазол-4-карбоновой кислоты (48 мг, 0,22 ммоль) и 1-пирроло-[1,2-а]пиразин-1-илпирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, разбавляли дихлорметаном (1,0 мл), а затем промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:Н2О + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде желтого твердого вещества (10 мг, 0,02 ммоль, выход 9%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.74 (d, J=6.7 Hz, 1H), 8.34 (s, 1H), 7.78 (dd, J=1.2, 2.7 Hz, 1H), 7.76-7.70 (m, 2H), 7.57 (d, J=4.3 Hz, 1H), 7.42-7.26 (m, 3H), 6.93 (dd, J=2.8, 4.7 Hz, 1H), 6.84 (d, J=5.9 Hz, 1H), 5.00-4.80 (m, 1H), 4.60-3.60 (br, 4H), 2.60-2.36 (m, 2H), 2.55 (s, 3H); MS: (ES) m/z теор. для C22H21N5OS [M+H]+ 404,2, факт. 404.
Пример 136. Синтез (1-имидазо[1,2-а]пиразин-8-ил-пирролидин-3-ил)амида 2-о-толилтиазол-4-карбоновой кислоты
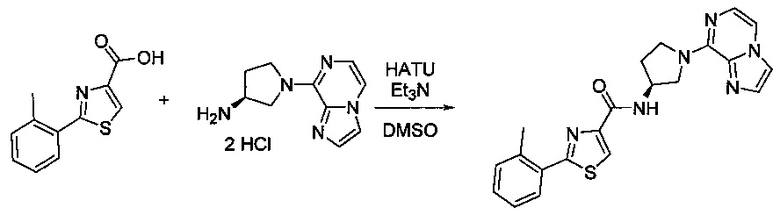
В смесь 2-о-толилтиазол-4-карбоновой кислоты (48 мг, 0,22 ммоль) и 1-имидазо-[1,2-а]пиразин-8-ил-пирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Содержащий неочищенный продукт материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде желтого твердого вещества (58 мг, 0,11 ммоль, выход 51%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.33 (s, 1Н), 8.04 (d, J=1.2 Hz, 1H), 7.92 (d, J=5.5 Hz, 1H), 7.81 (d, J=1.2 Hz, 1H), 7.73 (dd, J=1.5, 7.8 Hz, 1H), 7.42-7.26 (m, 3H), 7.16 (d, J=5.5 Hz, 1H), 5.00-4.80 (m, 1H), 4.60-3.60 (br, 4H), 2.60-2.38 (m, 2H), 2.55 (s, 3H); MS: (ES) m/z теор. для C21H20N6OS [M+H]+ 405,2, факт .405.
Пример 137. Синтез 1-(3-метилпирроло[1,2-а]пиразин-1-ил)-пирролидин-3-иламина дигидрохлорида
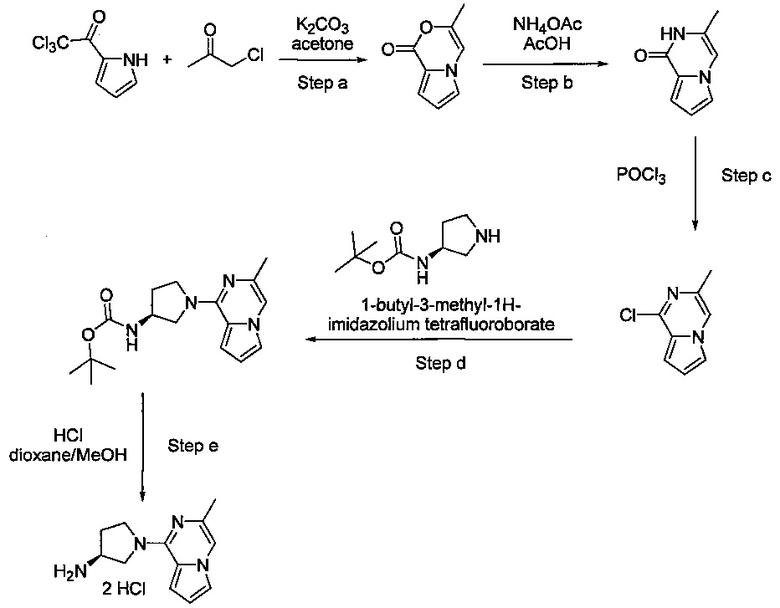
a) В кашицу из 2-(трихлорацетил)пиррола (5,00 g, 23,5 ммоль), ацетона (70 мл) и K2CO3 (9,74 g, 70,5 ммоль) по каплям добавляли раствор хлорацетона (2,81 мл, 35,3 ммоль) в ацетоне (50 мл). Смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь фильтровали, а твердое вещество промывали ацетоном. Фильтрат упаривали, а остаток распределяли между водой (100 мл) и этилацетатом (100 мл). Разделяли фазы, и водный слой экстрагировали этилацетатом (100 мл). Объединенные органические слои промывали водой (3×100 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом флэш-хроматографии на силикагеле с помощью смеси гексан : этилацетат, получая продукт (2,35 г, 15,8 ммоль, 67%).
b) Смесь 3-метил-пирроло[2,1-с][1,4]оксазин-1-она (2,35 г, 15,8 ммоль), NH4OAc (5,32 г, 69,0 ммоль) и АсОН (13,8 мл) нагревали при 160°С в запечатанном флаконе в течение 48 ч. Растворитель удаляли под вакуумом, а неочищенный продукт разводили в CH2Cl2. Его промывали насыщенным раствором NaHCO3, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. После очистки методом флэш-хроматографии (на силикагеле с помощью смеси гексан:EtOAc) получали продукт (710 мг, 4,79 ммоль, 35%).
c) В 3-метил-2Н-пирроло[1,2-а] пиразин-1-он (710 мг, 4,79 ммоль) добавляли POCl3 (2 мл) и нагревали раствор при 105°С в течение 3 ч. Реакционную смесь упаривали под вакуумом. Образовавшееся масло разводили в CH2Cl2 и промывали насыщенным водным NaHCO3, а затем водой. Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. После очистки методом флэш-хроматографии на силикагеле (от 95:5 до 70:30 гексан:EtOAc) получали продукт (798 мг, 4,79 ммоль, количественный выход) в виде желтого твердого вещества.
d) Во флакон вносили 1-хлор-3-метилпирроло[1,2-а]пиразин (798 мг, 4,79 ммоль), (5)-3-(Вос-амино)пирролидин (901 мг, 4,84 ммоль), iPr2NEt (2,52 мл, 14,52 ммоль) и 1-бутил-3-метил-1Н-имидазолий тетрафторборат (каталитическое количество) и нагревали при 110°С в течение 15 ч. Реакционную смесь упаривали под вакуумом и очищали методом флэш-хроматографии на силикагеле (от 99:1 до 90:10 CH2Cl2:МеОН), получая продукт (965 мг, 3,05 ммоль, 64%).
e) В раствор трет-бутилового эфира [1-(3-метилпирроло[1,2-а]пиразин-1-ил)-пирролидин-3-ил]карбаминовой кислоты (965 мг, 3,04 ммоль) в МеОН (10 мл) добавляли HCl (4М в диоксане, 3,80 мл, 15,2 ммоль). Все это перемешивали в течение ночи, а затем упаривали, получая продукт (860 мг, 2,97 ммоль, 98%) в виде белого твердого вещества.
Пример 138. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты
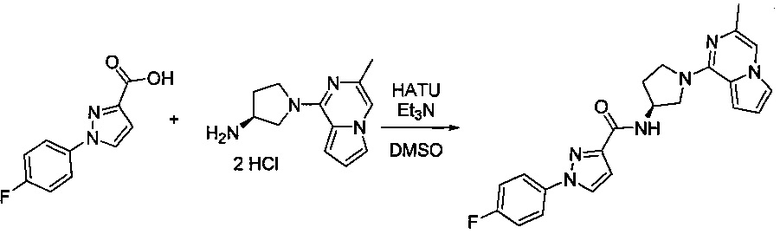
В смесь 1-(4-фторфенил)-1Н-пиразол-3-карбоновой кислоты (50 мг, 0,24 ммоль) и 1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (69 мг, 0,29 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,134 мл, 0,96 ммоль), а затем HATU (99 мг, 0,26 ммоль). Смесь перемешивали в течение 30 мин, а затем разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (31 мг, 0,06 ммоль, выход 25%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.75 (d, J=7.0 Hz, 1H), 8.28 (d, J=2.7 Hz, 1H), 7.88 (m, 2H), 7.68 (q, J=1.2 Hz, 1H), 7.52 (d, J=4.3 Hz, 1H), 7.49 (s, 1H), 7.27-7.22 (m, 2H), 6.96 (d, J=2.4 Hz, 1H), 6.87 (dd, J=2.7, 4.3 Hz, 1H), 5.00-4.80 (m, 1H), 4.60-3.60 (br, 4H), 2.60-2.38 (m, 2H), 2.30 (d, J=1.1 Hz, 3H); MS: (ES) m/z теор. для C22H21FN6O [M+H]+ 405,2, факт. 405.
Пример 139. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 1-фенил-1Н-[1,2,4]триазол-3-карбоновой кислоты

В смесь 1-фенил-1Н-1,2,4-триазол-3-карбоновой кислоты (50 мг, 0,26 ммоль) и 1-(3-метилпирроло[1,2-а]пиразин-1-ил)гшрролидин-3-иламина дигидрохлорида (74 мг, 0,26 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,145 мл, 1,04 ммоль), а затем HATU (ПО мг, 0,29 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде коричневого твердого вещества (66 мг, 0,13 ммоль, выход 51%). 1H-ЯМР (400 MHz, CD3OD) δ 9.14 (s, 1Н), 7.88 (td, J=1.2, 6.2 Hz, 2H), 7.68 (q, J=1.2 Hz, 1H), 7.62-7.44 (m, 6H), 6.88 (dd, J=2.3,4.3 Hz, 1H), 5.00-4.80 (m, 1H), 4.60-3.60 (br, 4H), 2.60-2.38 (m, 2H), 2.30 (d, J=1.1 Hz, 3H); MS: (ES) m/z теор. для C21H21N7O [M+H]+ 388,2, факт. 388.
Пример 140. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 1-фенил-1H-имидазол-4-карбоновой кислоты
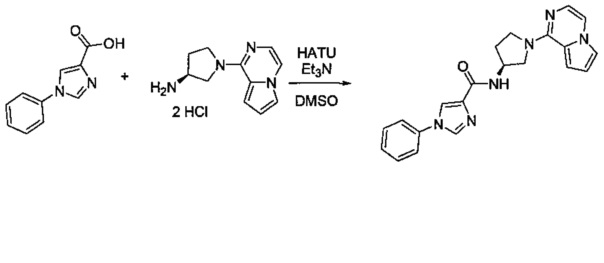
В смесь 1-фенил-1Н-имидазол-4-карбоновой кислоты (34 мг, 0,18 ммоль) и 1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-иламина дигидрохлорида (50 мг, 0,18 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,100 мл, 0,72 ммоль), а затем HATU (76 мг, 0,20 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:Н2О + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде бледно-желтого твердого вещества (49 мг, 0,10 ммоль, выход 56%). 1H-ЯМР (400 MHz, CD3OD) δ 8.23 (d, J=1.1 Hz, 1H), 8.15 (d, J=1.6 Hz, 1H), 7.79 (dd, J=1.2, 2.8 Hz, 1H), 7.73 (d, J=5.5 Hz, 1H), 7.64-7.52 (m, 5H), 7.50-7.42 (m, 1H), 6.94 (dd, J=2.3,4.3 Hz, 1H), 6.86 (d, J=5.5 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-3.50 (br, 4H), 2.58-2.48 (m, 1H), 2.48-2.35 (br, 1H); MS: (ES) m/z теор. для C21H20N6O [M+H]+ 373,2, факт. 373.
Пример 141. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 2-(4-гидроксипиперидин-1-ил)-тиазол-4-карбоновой кислоты
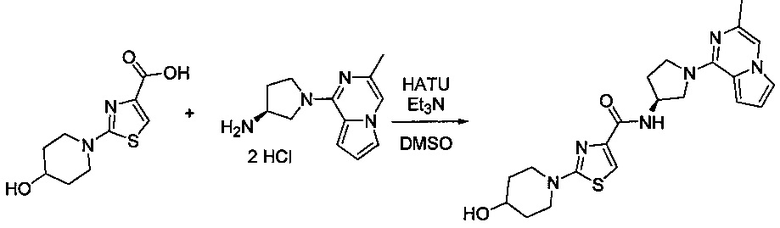
В смесь 2-(4-гидроксипиперидин-1-ил)тиазол-4-карбоновой кислоты (48 мг, 0,21 ммоль) и 1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (60 мг, 0,21 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,117 мл, 0,84 ммоль), а затем HATU (87 мг, 0,23 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Объединенные фракции упаривали, разводили в CH2Cl2, промывали NaHCO3, сушили над Na2SO4, фильтровали и упаривали. Образовавшееся масло затем очищали методом флэш-хроматографии на силикагеле (градиент CH2Cl2:МеОН от 99:1 до 90:10). Содержащие продукт фракции объединяли и упаривали, а затем лиофилизировали из смеси MeCN и 1 М HCl, получая HCl-соль продукта в виде желтого твердого вещества (22 мг, 0,04 ммоль, выход 19%). 1Н-ЯМР (400 MHz, CD3OD) δ 7.37 (s, 1Н), 7.28 (q, J=1.2 Hz, 1H), 7.25 (s, 1H), 6.86 (d, J=4.3 Hz, 1H), 6.57 (dd, J=2.7, 4.3 Hz, 1H), 5.48 (s, 2H), 4.70-4.60 (m, 1H), 4.22-3.78 (m, 8H), 3.28-3.10 (m, 1H), 2.40-2.28 (m, 1H), 2.22-2.12 (m, 4H), 1.96-1.88 (m, 2H); 1.62-1.52 (m, 2H); MS: (ES) m/z теор. для C21H26N6O2S [M+H]+ 427,2, факт. 427.
Пример 142. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 1-фенил-1Н-имидазол-4-карбоновой кислоты
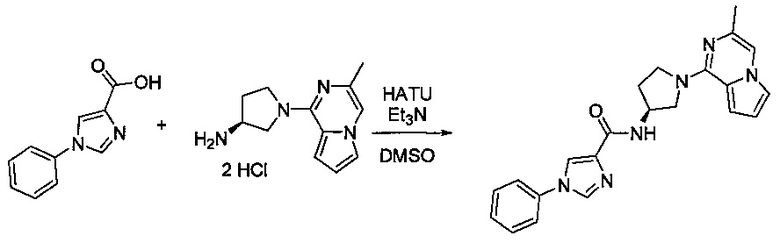
В смесь 1-фенил-1H-имидазол-4-карбоновой кислоты (32 мг, 0,17 ммоль) и 1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (50 мг, 0,17 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,095 мл, 0,68 ммоль), а затем HATU (72 мг, 0,19 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде светло-коричневого твердого вещества (33 мг, 0,07 ммоль, выход 39%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.23 (d, J=1.6 Hz, 1H), 8.15 (d, J=1.6 Hz, 1H), 7.67 (q, J=1.2 Hz, 1H), 7.62-7.45 (m, 6H), 6.88 (q, J=2.3 Hz, 1H), 5.00-4.80 (m, 1H), 4.60-3.60 (br, 4H), 2.58-2.48 (m, 1H), 2.48-2.38 (br, 1H), 2.30 (d, J=1.1 Hz, 3H); MS: (ES) m/z теор. для C22H22N6O [M+H]+ 387,2, факт. 387.
Пример 143. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 1-(4-хлорфенил)-1Н-пиразол-3-карбоновой кислоты
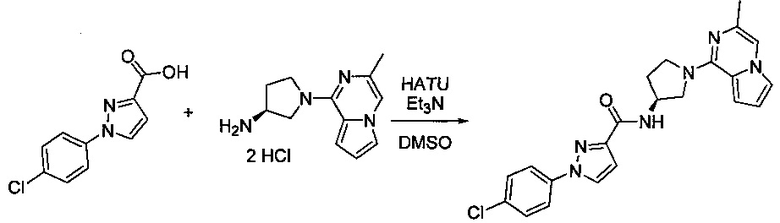
В смесь 1-(4-хлорфенил)-1Н-пиразол-3-карбоновой кислоты (38 мг, 0,17 ммоль) и 1-(3-метилпирроло[1,2-a]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (50 мг, 0,17 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,095 мл, 0,68 ммоль), а затем HATU (72 мг, 0,19 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде белого твердого вещества (50 мг, 0,09 ммоль, выход 55%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.78 (d, J=7.0 Hz, 0.5Н), 8.33 (d, J=2.7 Hz, 1H), 7.87 (td, J=2.4, 9.0 Hz, 2H), 7.67 (q, J=1.2 Hz, 1H), 7.51 (td, J=3.1, 9.0 Hz, 4H), 6.98 (d, J=2.8 Hz, 1H), 6.87 (q, J=2.8, 1.9 Hz, 1H), 4.50-3.50 (br, 5H), 2.58-2.48 (m, 1H), 2.48-2.38 (br, 1H), 2.30 (d, J=0.7 Hz, 3H); MS: (ES) /и/z теор. для C22H21ClN6O [M+H]+ 421,2, факт. 421.
Пример 144. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 2-пирролидин-1-илтиазол-4-карбоновой кислоты
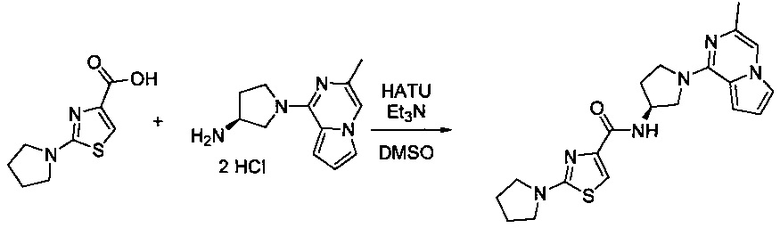
В смесь 2-пирролидин-1-илтиазол-4-карбоновой кислоты (34 мг, 0,17 ммоль) и 1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (50 мг, 0,17 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,095 мл, 0,68 ммоль), а затем HATU (72 мг, 0,19 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде красноватого твердого вещества (20 мг, 0,04 ммоль, выход 23%). 1Н-ЯМР (400 MHz, CD3OD) δ 7.67 (dd, J=1.2, 2.3 Hz, 1H), 7.52 (d, J=4.6 Hz, 1H), 7.49 (s, 1H), 7.36 (s, 1H), 6.87 (dd, J=2.7, 4.7 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-3.50 (br, 4H), 3.50-3.40 (m, 3H), 2.58-2.48 (m, 1H), 2.48-2.38 (br, 1H), 2.30 (d, J=0.7 Hz, 3H), 2.10-2.00 (m, 4H); MS: (ES) m/z теор. для C20H24N6OS [M+H]+ 397,2, факт. 397.
Пример 145. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 2-фенилоксазол-4-карбоновой кислоты
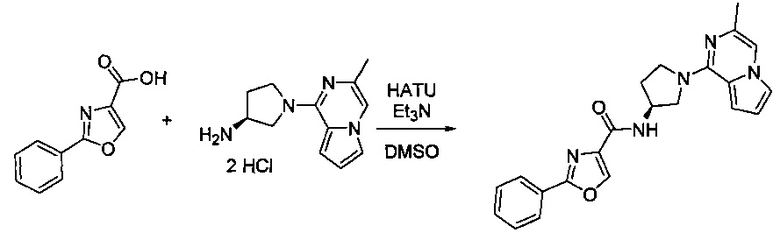
В смесь 2-фенилоксазол-4-карбоновой кислоты (32 мг, 0,17 ммоль) и 1-(3-метил-пирроло[1,2-а]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (50 мг, 0,17 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,095 мл, 0,68 ммоль), а затем HATU (72 мг, 0,19 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде светло-коричневого твердого вещества (42 мг, 0,08 ммоль, выход 49%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.74 (d, J=6.1 Hz, 1H), 8.49 (s, 1H), 8.10-8.06 (m, 2H), 7.68 (dd, J=1.2, 2.7 Hz, 1H), 7.57-7.50 (m, 4H), 6.88 (dd, J=2.7, 4.7 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-3.50 (br, 4H), 2.58-2.48 (m, 1H), 2.48-2.38 (br, 1H), 2.31 (d, J=1.1 Hz, 3H); MS: (ES) m/z теор. для C22H21N5O2 [M+H]+ 388,2, факт. 388.
Пример 146. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 2-фенилтиазол-4-карбоновой кислоты
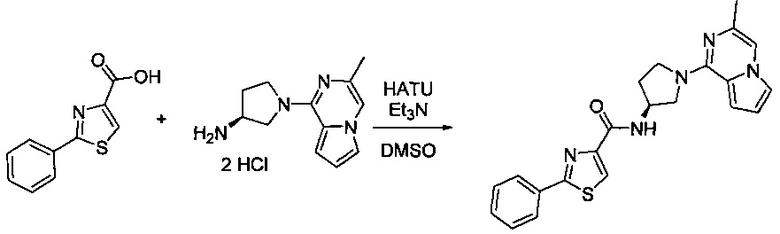
В смесь 2-фенилтиазол-4-карбоновой кислоты (43 мг, 0,21 ммоль) и 1-(3-метил-пирроло[1,2-а]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (60 мг, 0,21 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,117 мл, 0,84 ммоль), а затем HATU (87 мг, 0,23 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде серого твердого вещества (30 мг, 0,06 ммоль, выход 28%). 1Н-ЯМР (400 MHz, CD3OD) δ 8.87 (d, J=6.6 Hz, 1H), 8.26 (s, 1H), 8.06-8.03 (m, 2H), 7.68 (dd, J=1.2, 2.7 Hz, 1H), 7.54-7.45 (m, 5H), 6.88 (dd, J=2.4, 6.7 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-3.50 (br, 4H), 2.60-2.40 (m, 2H), 2.31 (d, J=1.1 Hz, 3H); MS: (ES) m/z теор. для C22H21N5OS [M+H]+ 404,2, факт. 404.
Пример 147. Синтез [1-(3-метилпирроло[1,2-а]пиразин-1-ил)пирролидин-3-ил]-амида 1-(4-фторфенил)-1Н-[1,2,4]триазол-3-карбоновой кислоты
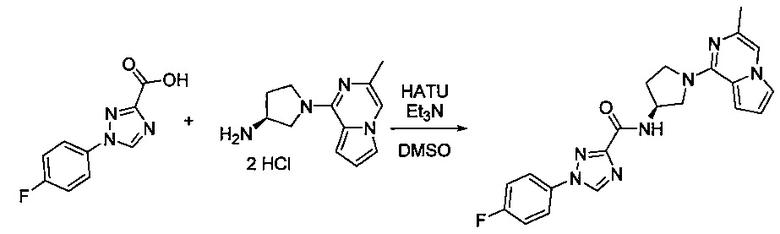
В смесь 1-(4-фторфенил)-1Н-[1,2,4]триазол-3-карбоновой кислоты и 1-(3-метил-пирроло[1,2-а]пиразин-1-ил)пирролидин-3-иламина дигидрохлорида (60 мг, 0,21 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,150 мл, 1,05 ммоль), а затем HATU (87 мг, 0,23 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде желтого твердого вещества (11 мг, 0,02 ммоль, выход 10%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.10 (s, 1Н), 9.07 (d, J=6.2 Hz, 1H), 7.92-7.88 (m, 2H), 7.68 (dd, J=1.2, 2.6 Hz, 1H), 7.53 (d, J=4.4 Hz, 1H), 7.50 (s, 1H), 7.34 (t, J=8.6 Hz, 2H), 6.89 (dd, J=2.4, 4.4 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-3.50 (br, 4H), 2.60-2.40 (m, 2H), 2.31 (d, J=1.1 Hz, 3H); MS: (ES) m/z теор. для C21H20FN7O [M+H]+ 406,2, факт. 406.
Пример 148. Синтез (1-пирроло[1,2-а]пиразин-1-ил-пирролидин-3-ил)амида 6-метилхиназолин-2-карбоновой кислоты
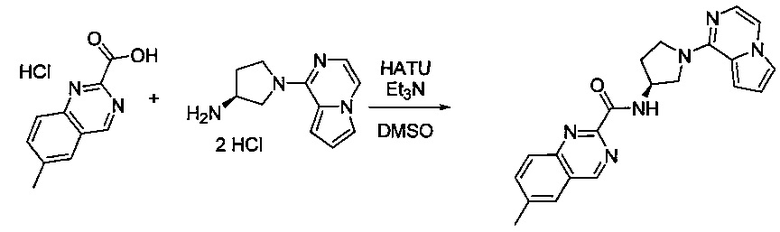
В смесь 6-метилхиназолин-2-карбоновой кислоты⋅HCl (27 мг, 0,12 ммоль) и 1-пирроло[1,2-а]пиразин-1-илпирролидин-3-иламина дигидрохлорида (33 мг, 0,12 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,084 мл, 0,60 ммоль), а затем HATU (49 мг, 0,13 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде красноватого твердого вещества (13 мг, 0,03 ммоль, выход 22%). 1H-ЯМР (400 MHz, CD3OD) δ 9.55 (s, 1Н), 8.11 (d, J=9.4 Hz, 1H), 7.98 (d, J=9.4 Hz, 2H), 7.53 (dd, J=1.2, 2.4 Hz, 1H), 7.55 (d, J=4.8 Hz, 1H), 7.50 (s, 1H), 6.89 (dd, J=2.3, 4.8 Hz, 1H), 5.00-4.80 (m, 1H), 4.50-3.50 (br, 4H), 2.63 (s, 3H), 2.60-2.40 (m, 2H), 2.31 (d, J=1.1 Hz, 3H); MS: (ES) m/z теор. для C22H22N6O [M+H]+ 387,2, факт. 387.
Пример 149. Синтез (1-пирроло[1,2-а]пиразин-1-илпирролидин-3-ил)амида 1-фенил-1H-[1,2,3]триазол-4-карбоновой кислоты

В смесь 1-фенил-1Н-[1,2,3]триазол-4-карбоновой кислоты (42 мг, 0,22 ммоль) и 1-пирроло[1,2-a]пиразин-1-илпирролидин-3-иламина дигидрохлорида (60 мг, 0,22 ммоль) добавляли DMSO (0,5 мл). Туда же добавляли триэтиламин (0,123 мл, 0,88 ммоль), а затем HATU (91 мг, 0,24 ммоль). Смесь перемешивали в течение 30 мин, после чего разбавляли дихлорметаном (1,0 мл), промывали водой (3×1,0 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали методом обратнофазовой HPLC (от 10:90 до 95:5 MeCN:H2O + 0,1% TFA). Содержащие продукт фракции объединяли и лиофилизировали, получая TFA-соль продукта в виде бледно-желтого твердого вещества (68 мг, 0,14 ммоль, выход 63%). 1Н-ЯМР (400 MHz, CD3OD) δ 9.03 (d, J=6.7 Hz, 1H), 8.97 (s, 1H), 7.89 (d, J=8.2 Hz, 2H), 7.79 (dd, J=1.2, 2.7 Hz, 1H), 7.74 (d, J=5.9 Hz, 1H), 7.65-7.50 (m, 4H), 6.95 (dd, J=2.8, 4.7 Hz, 1H), 6.87 (d, J=5.9 Hz, 1H), 5.00-4.80 (m, 1H), 4.70-3.70 (br, 4H), 2.60-2.38 (m, 2H); MS: (ES) m/z теор. для C20H19N7O [M+H]+ 374,2, факт. 374.
Пример 150
Соединения в приведенной ниже таблице получали, как описано выше. Для каждого из них представлены характеризующие их данные (ЯМР).
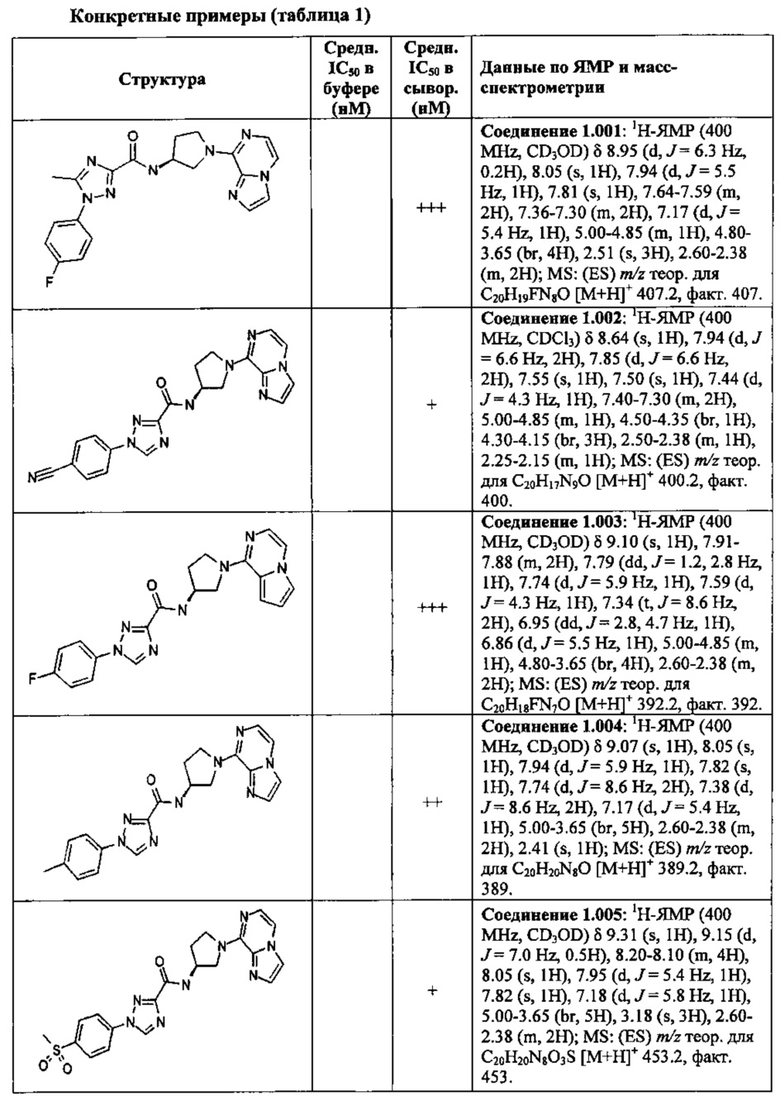
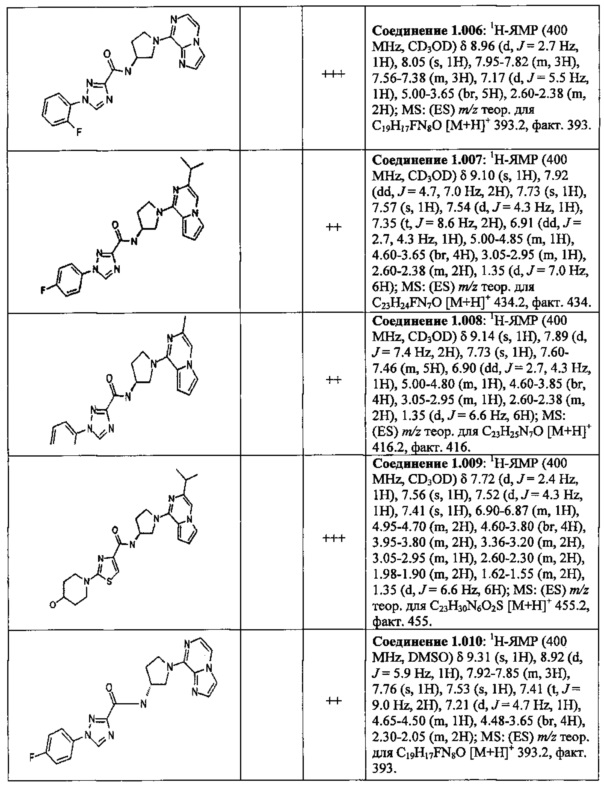
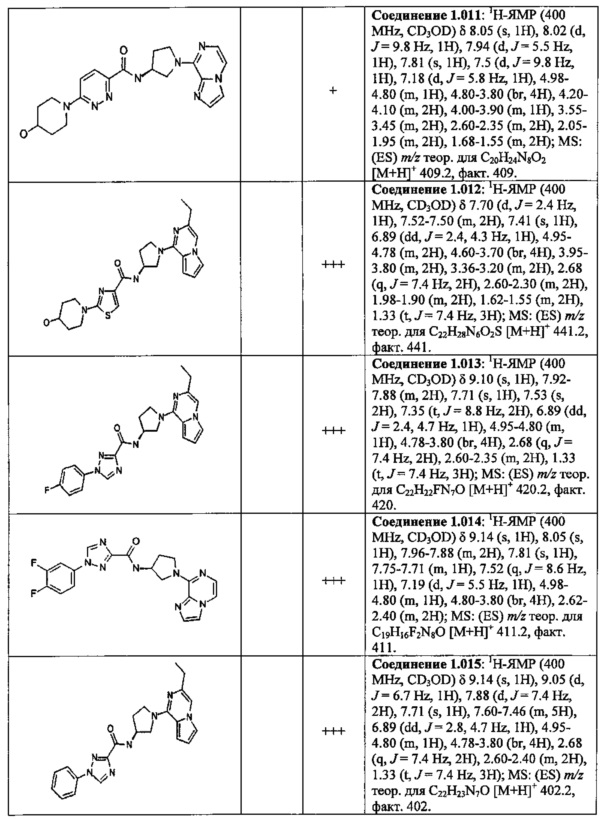
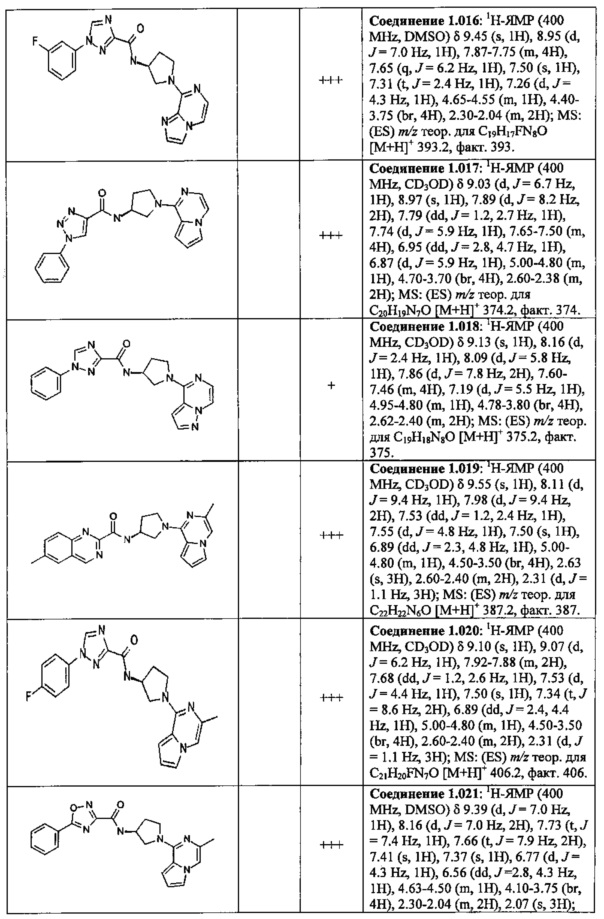
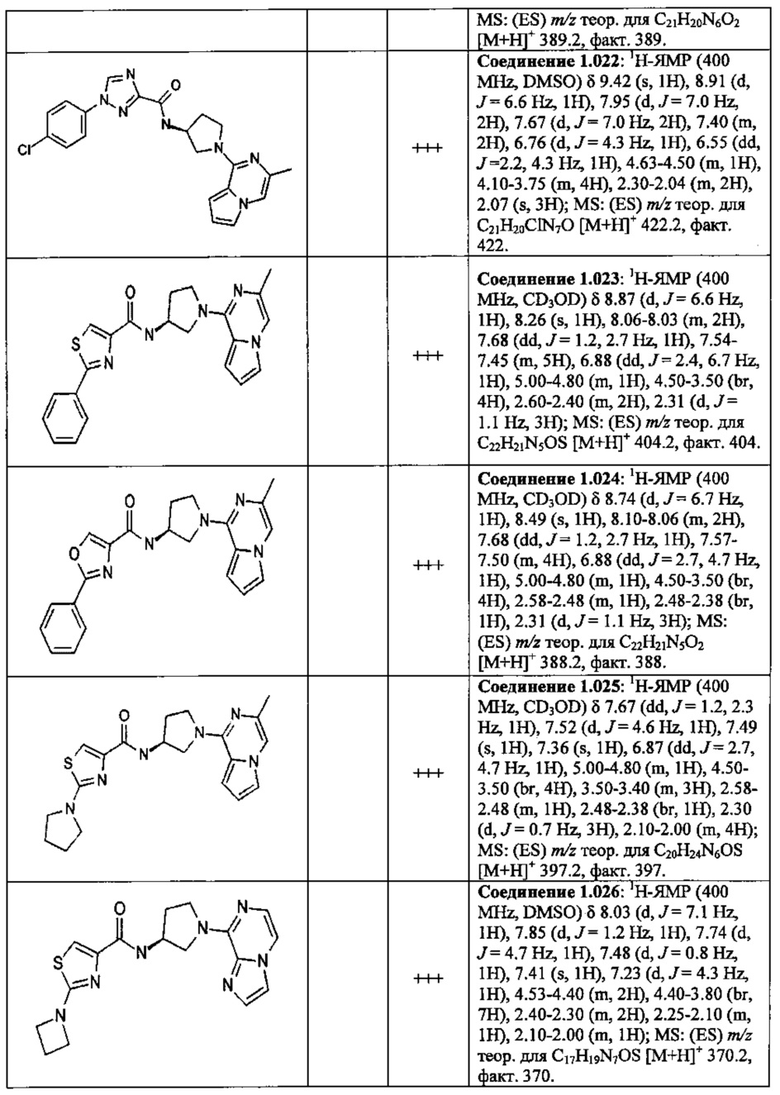
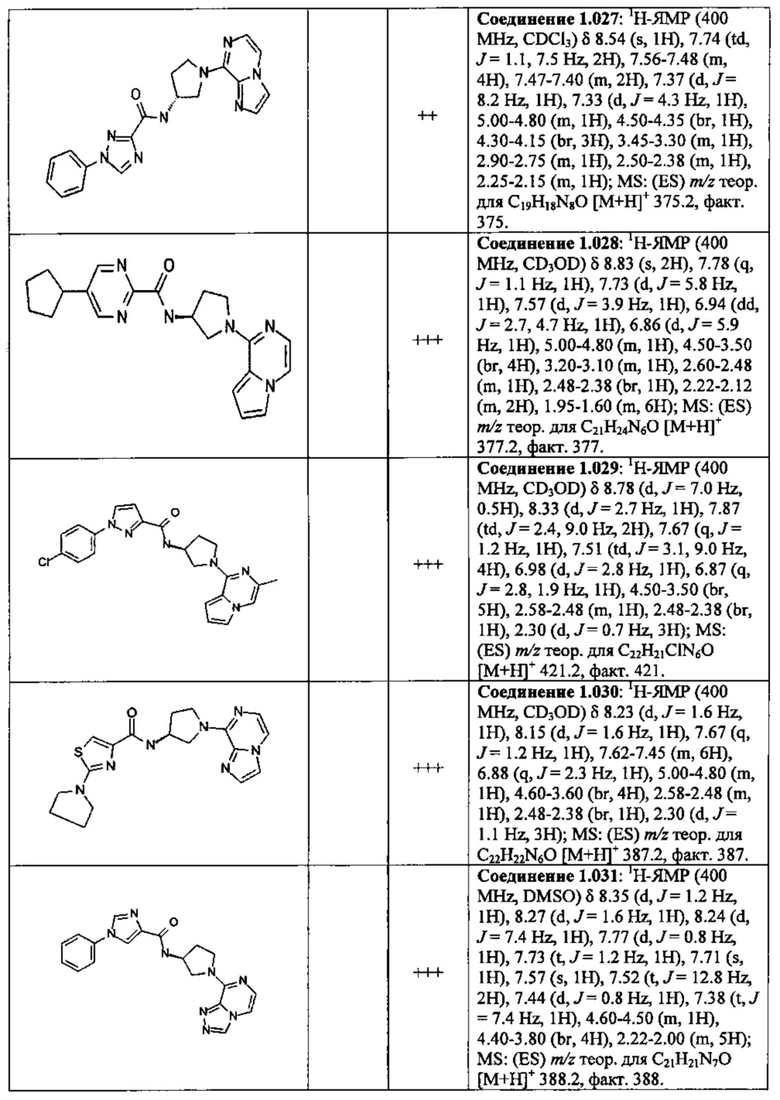
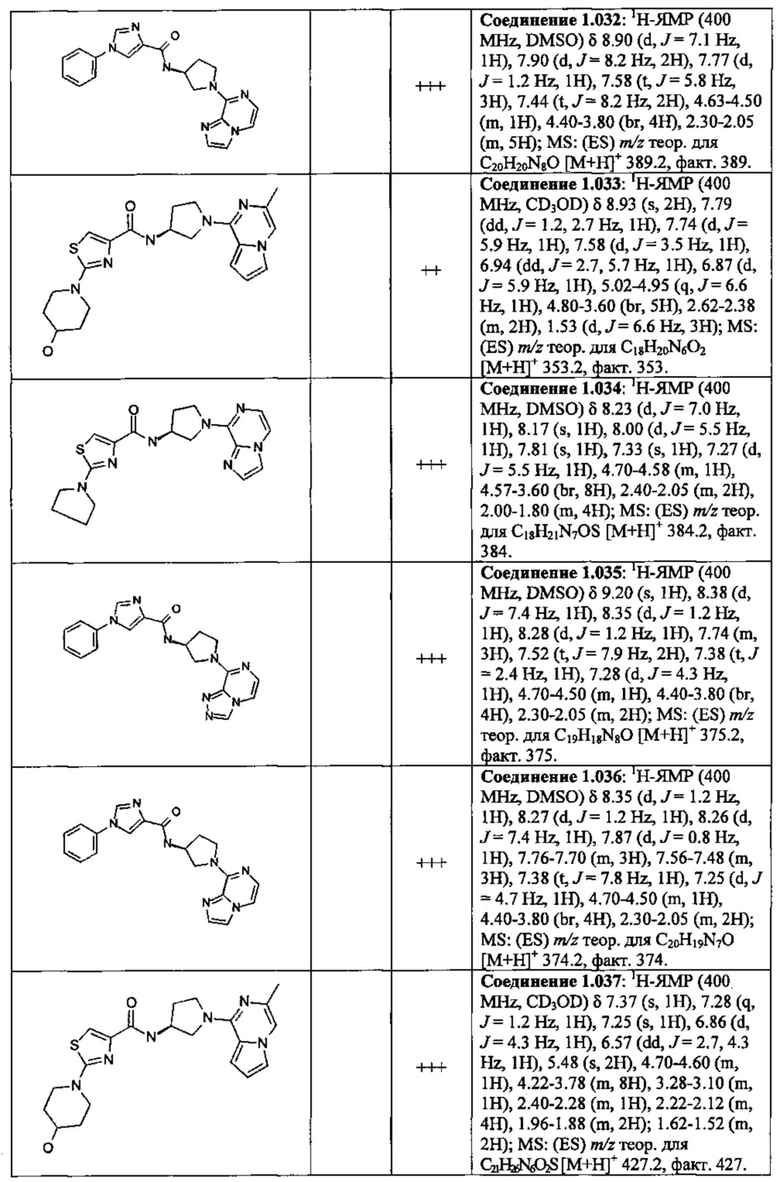
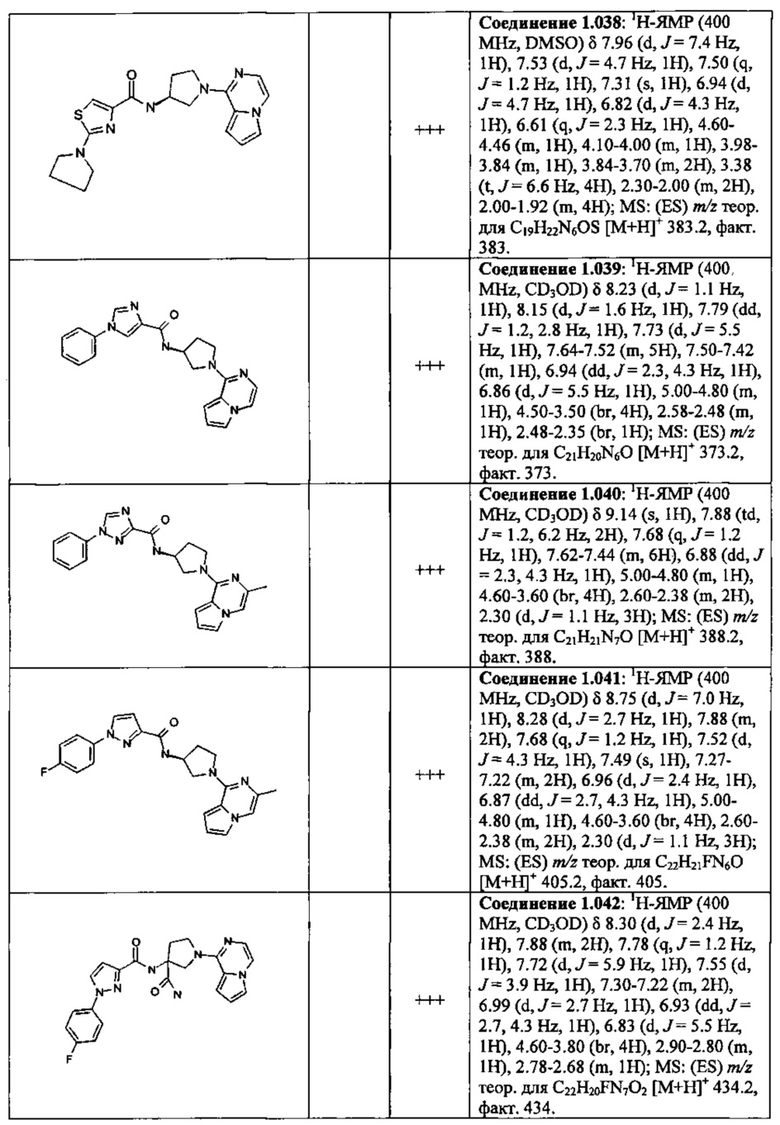
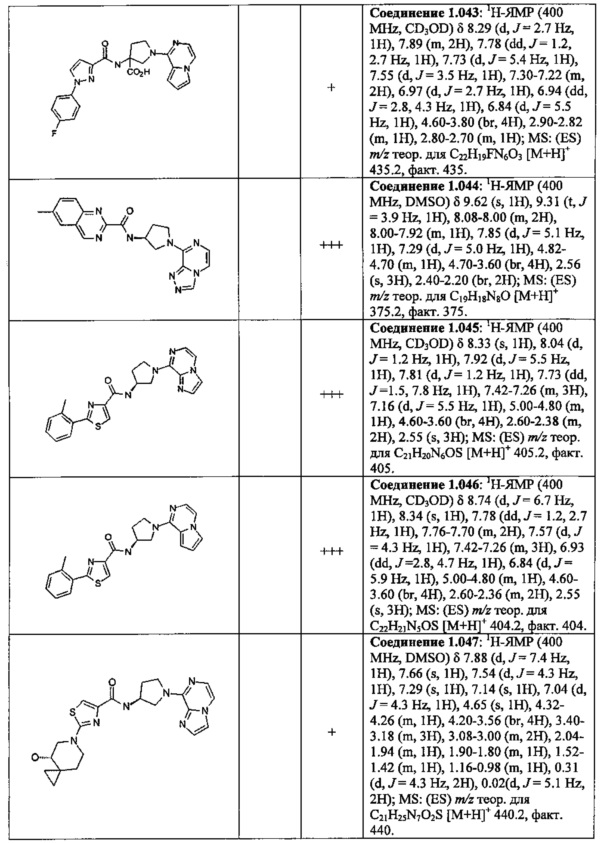
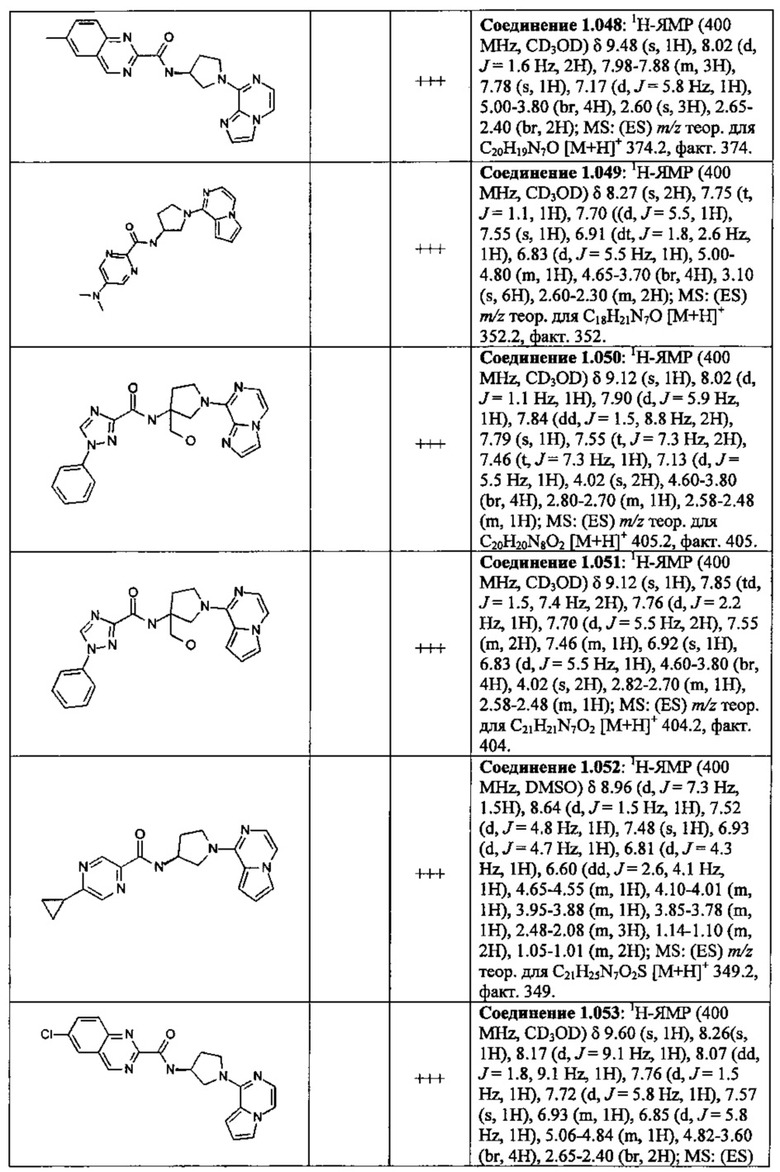
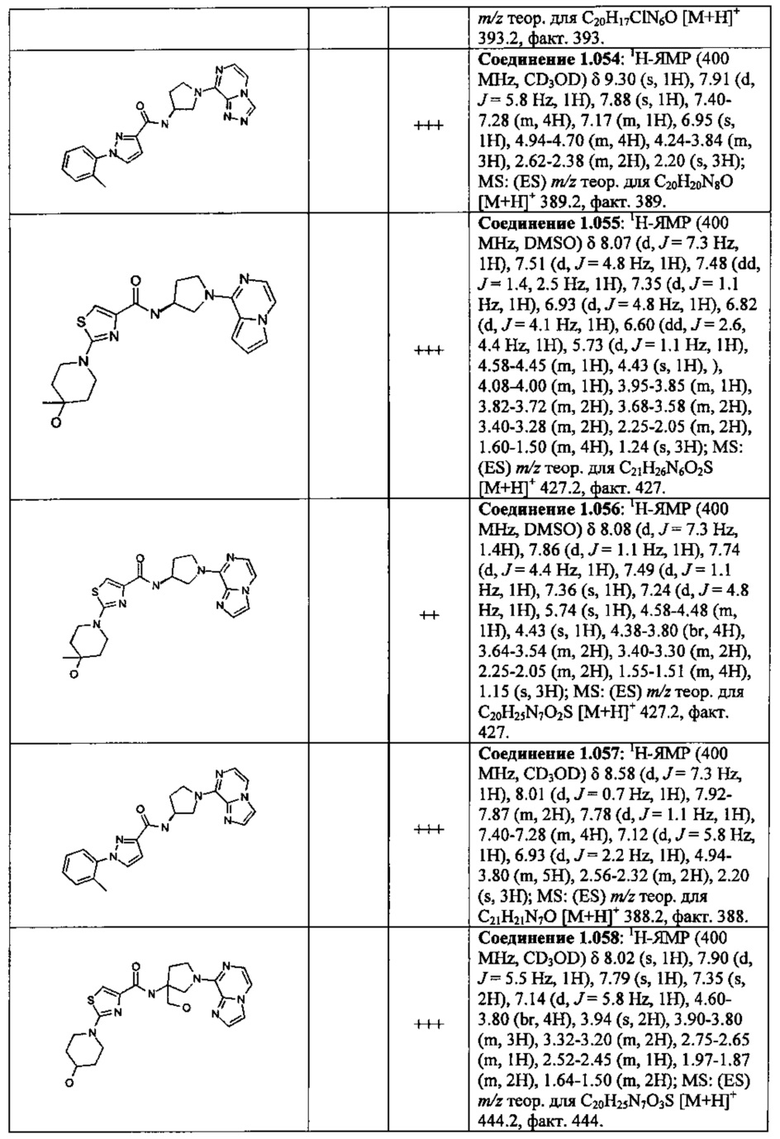
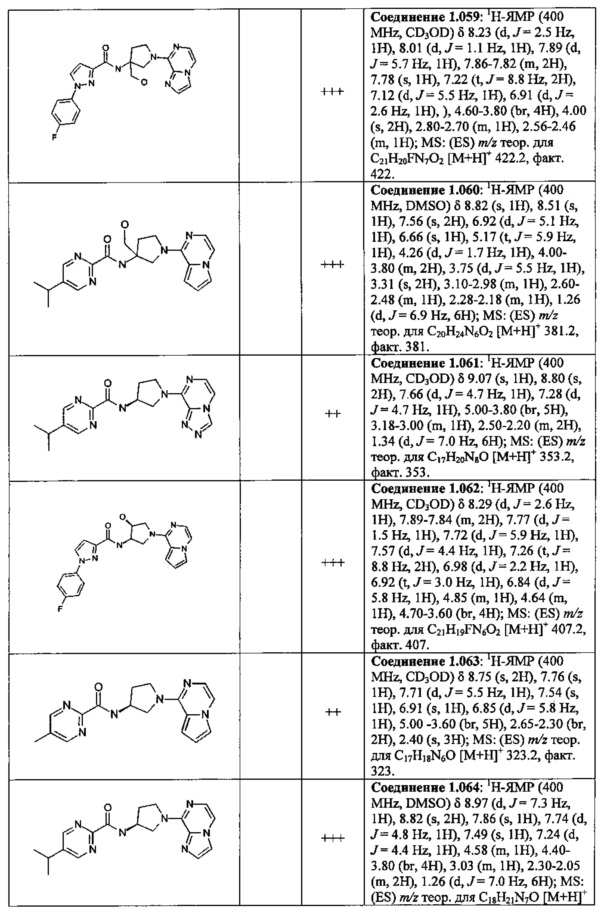
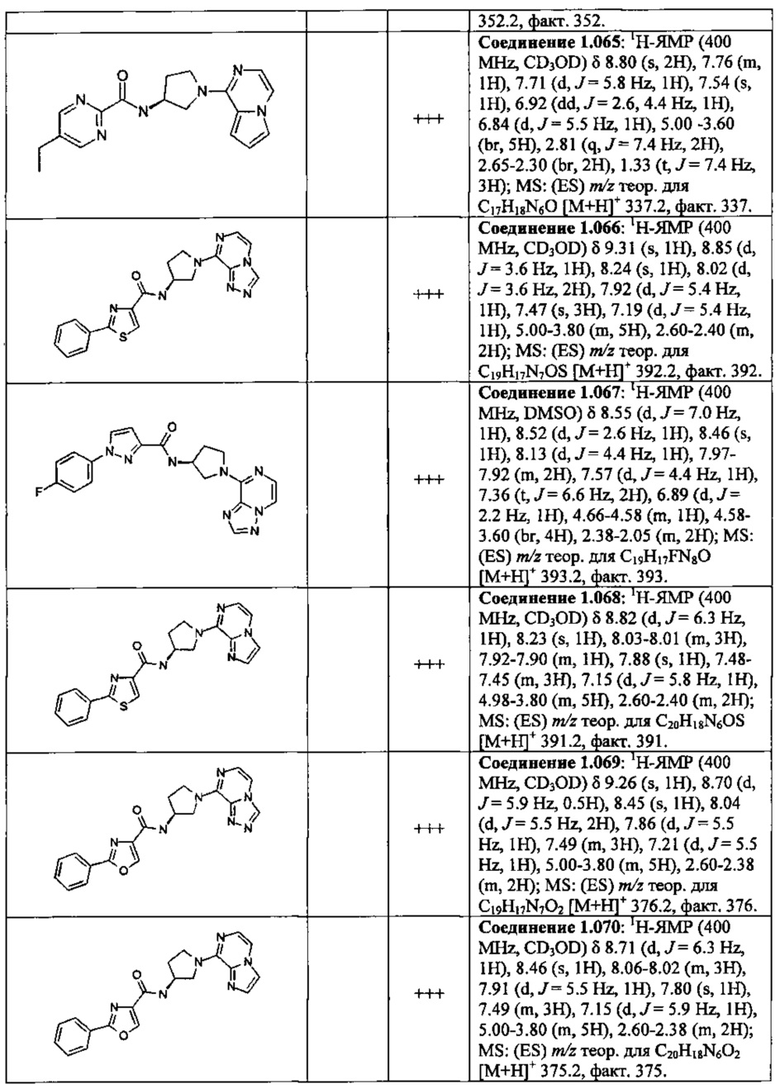

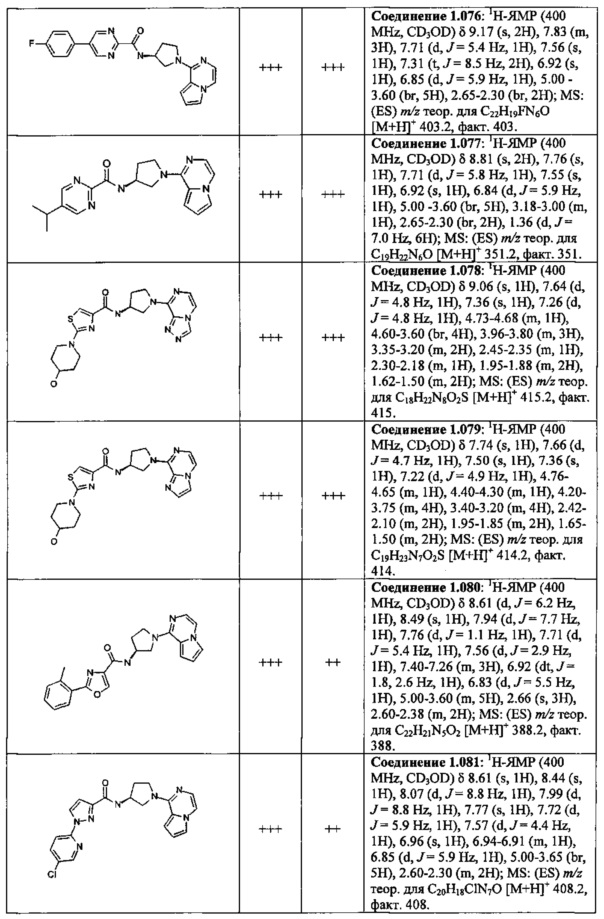
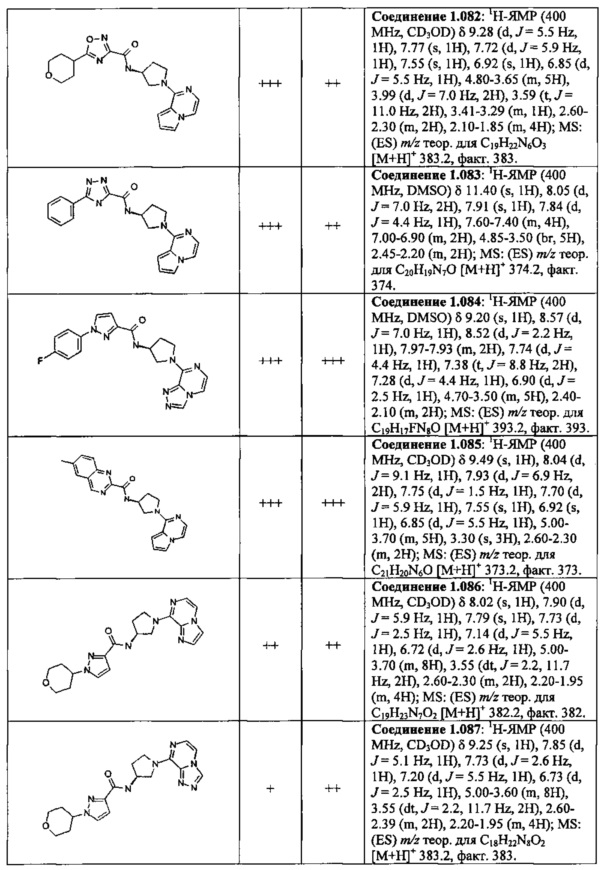
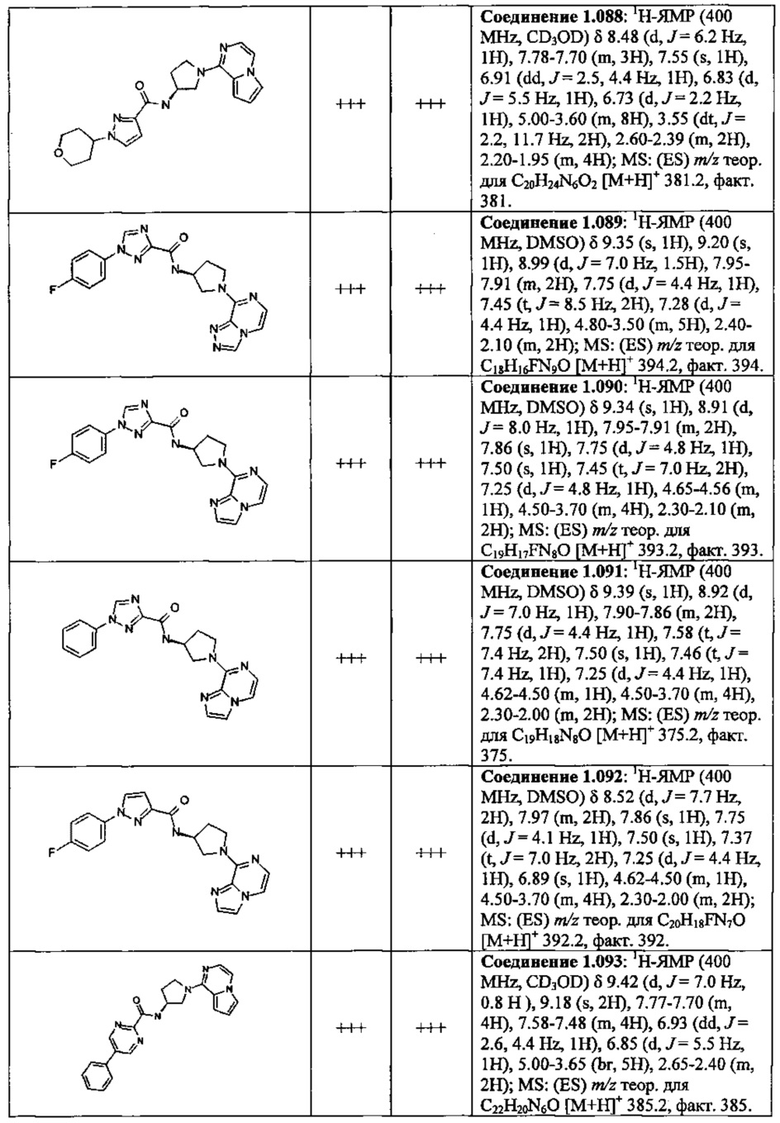

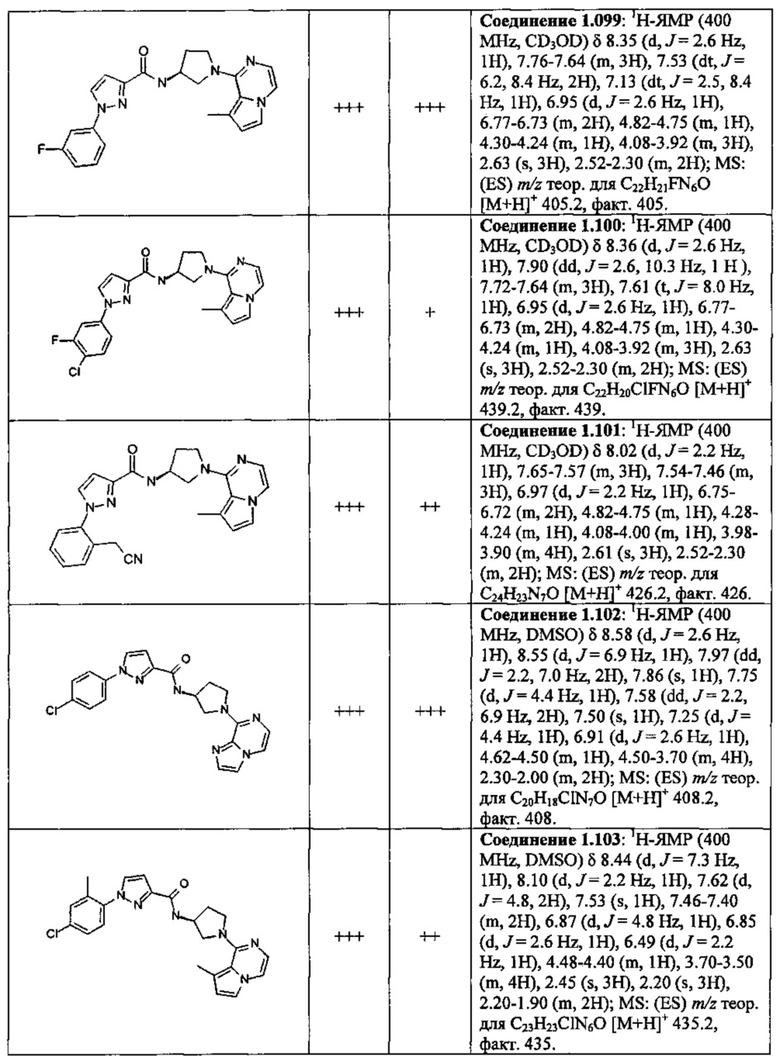
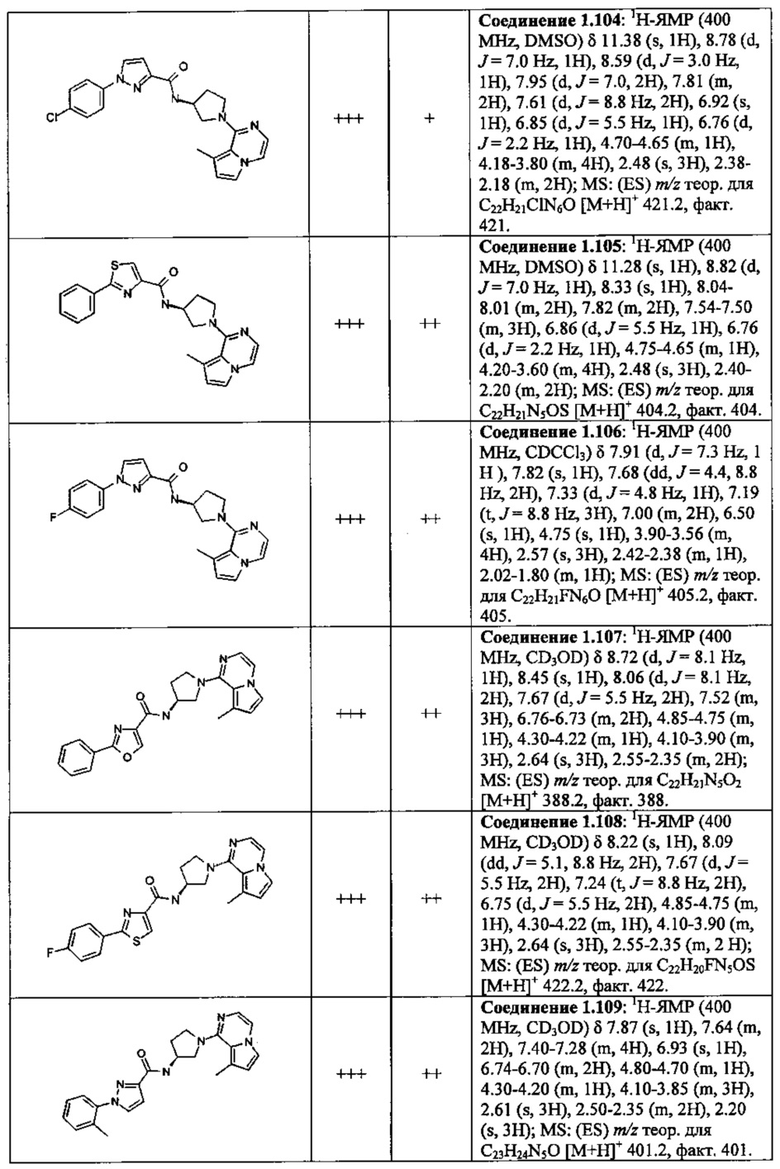
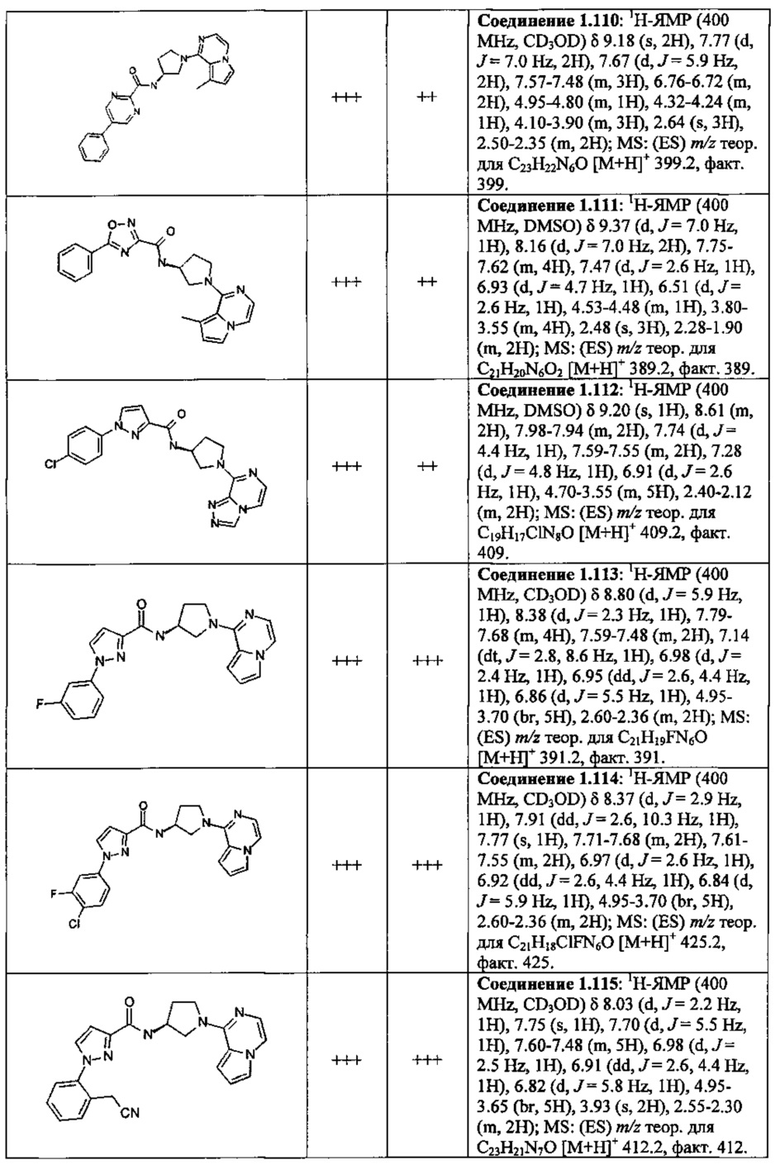
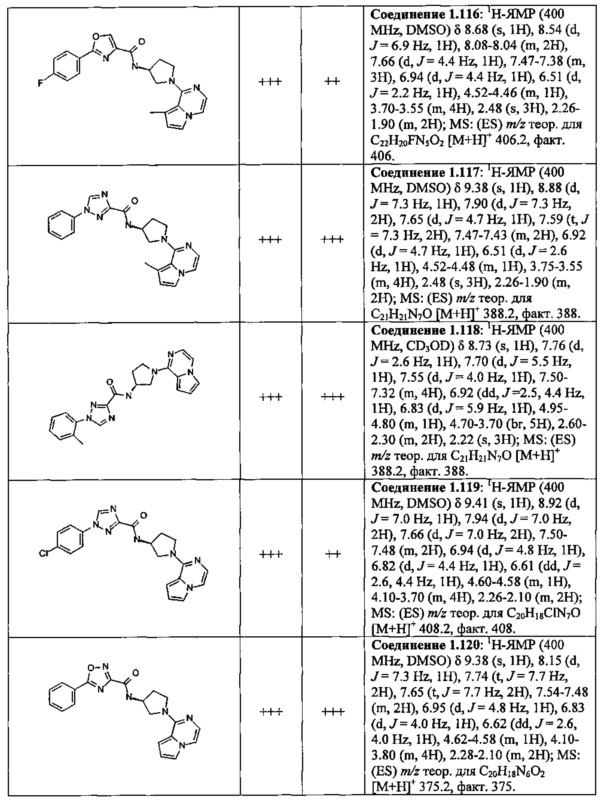
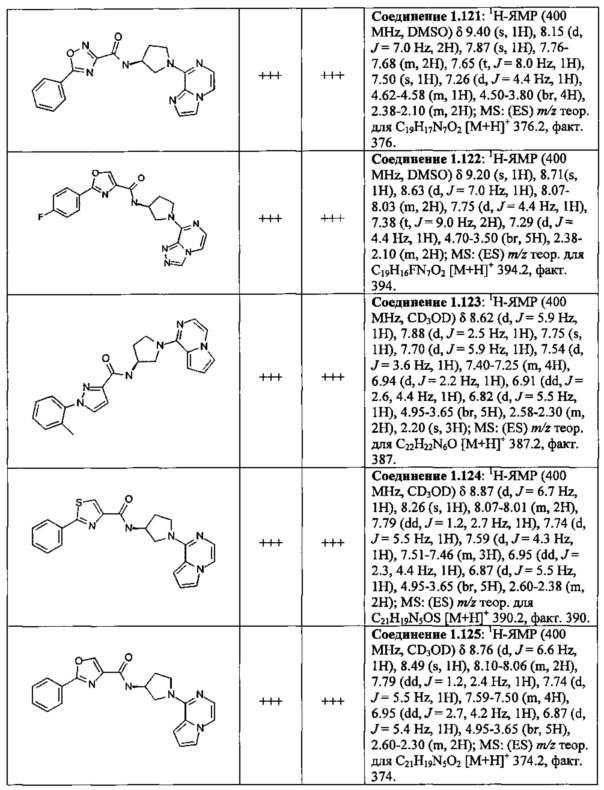
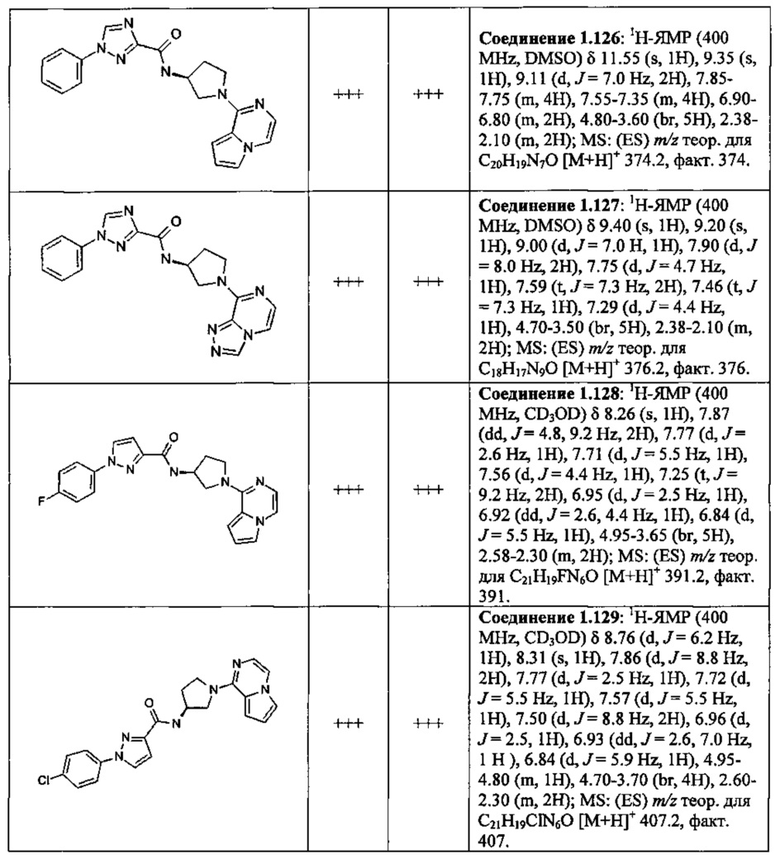
Биологический пример 1
Для того чтобы продемонстрировать, что описанные выше соединения являются полезными модуляторами связывания хемокинов с CXCR7, соединения подвергали скринингу in vitro для определения их способности вытеснять SDF-1 из рецептора CXCR7 при нескольких концентрациях. Соединения смешивали с клетками, экспрессирующими рецептор CXCR7 (например, клетками MCF или трансфецированными клетками для экспрессии CXCR7), в присутствии 125I-меченного хемокина SDF-1, как описано в разделе «Определение значений IC50. Реагенты и клетки» (см. ниже). Затем определяли способность соединения вытеснять меченый хемокин из рецепторных сайтов CXCR7 при нескольких концентрациях в процессе скрининга.
Эффективными модуляторами считались такие соединения, которые способны вытеснить по меньшей мере 50% SDF-1 из рецептора CXCR7 при концентрации на уровне или меньше 15 мкМ, но >2500 нМ (+); еще более предпочтительно при концентрации от >500 нМ до ≤2500 (++). В настоящее время наиболее предпочтительные соединения могут вытеснить по меньшей мере 50% SDF-1 из рецептора CXCR7 при концентрациях, равных или меньше 500 нм (+++). Типичные соединения, удовлетворяющие этим критериям, представлены в таблице 1 и приведенных выше Примерах. В таблице 1 "Средн. IC50 в буфере" относится к «Анализу связывания в буфере», описанному ниже, а "Средн. IC50 в сывор." относится к «Анализу связывания в сыворотке», описанному ниже. Все соединения получали, как описано в приведенных выше Примерах, либо аналогичными способами с использованием легко доступных исходных веществ.
Определение значений IC50
Реагенты и клетки. 125I-меченный SDF-1 приобретали у Perkin-Elmer Life Sciences, Inc. (Boston, MA). Клетки линии MCF-7 (аденокарцинома; молочная железа) получали из Американской коллекции типовых культур (АТСС, Manassas, VA) и культивировали в среде DMEM (Mediatech, Herndon, VA) с добавлением 10% эмбриональной телячьей сыворотки (FBS) (HyClone Logan, UT) и бычьего инсулина (0,01 мг/мл) (Sigma, St. Louis, МО) при 37°С в инкубаторе с увлажненным воздухом с 5% СО2. Трансфецированные CXCR7 клетки MDA-MB-435S получали, как описано ниже. Клетки MDA-MB-435S раковой линии молочной железы человека получали из АТСС и культивировали в среде DMEM с 10% FBS. Полную кодирующую последовательность гена, кодирующего CXCR7 (иначе CCXCKR2, hRDC1), выделяли из клеток MCF-7 с помощью набора μMACs для выделения мРНК (Miltenyi Biotec, Auburn, СА). Примесь ДНК удаляли путем расщепления ДНКазой на колонках RNeasy (Qiagen, Inc., Valencia, СА), а кДНК получали с помощью набора GeneAmp RNA PCR Core Kit (Applied Biosystems, Foster City, СА). Образцы кДНК подвергали ГЩР с помощью набора Taq PCR Master Mix kit (Qiagen, Inc.) и праймеров hRDC1, содержащих 5'- и 3'-сайты NotI (hRDC1F: 5'-GAATGCGGCCGCTATGGATCTGCATCTCTTCGACT-3', hRDC1R: 5'-GAATGCGGCCGCTCATTTGGTGCTCTGCTCCAAG-3'). Расщепленные NotI продукты ГЩР лигировали в расщепленную NotI плазмиду pcDNA3.1(+) (Invitrogen, Carlsbad, СА) и подвергали скринингу для подтверждения ориентации и последовательности. Затем из ночных бактериальных культур выделяли плазмидную ДНК с помощью набора Maxiprep (Qiagen, Inc.). Плазмидную ДНК (10 мкг) добавляли к клеткам MDA-MB-435S и подвергали клетки электропорации (0,22 кВ, 960 мкФ) на приборе Gene Pulser (BioRad Laboratories, Hercules, СА). Через 48 ч после электропорации клетки переносили в селективную среду (600 мкг/мл G418).
Анализ связывания в буфере. Искомые соединения тестировали на их способность связываться с сайтами CXCR7 на трансфецированных CXCR7 клетках MCF-7 и/или MDA-MB-435S. Проводили связывание радиолиганда с максимальной эффективностью по методике с фильтрованием, как описано в Dairaghi DJ et al., HHV8-encoded vMIP-I selectively engages chemokine receptor CCR5. Agonist and antagonist profiles of viral chemokines. J. Biol. Chem. 1999 Jul 30, 274(31): 21569-74; и Gosling J et al. Cutting edge: identification of a novel chemokine receptor that binds dendritic cell- and T cell-active chemokines including ELC, SLC, and TECK. J. Immunol. 2000 Mar 15, 164(6): 2851-6.
При этом проводили инкубацию клеток MCF-7 и/или MDA-MB-435S с исследуемыми соединениями и определяли способность этих соединений вытеснять 125I-меченный SDF-1 по методике, описанной в Dairaghi и Gosling. Искомые соединения вносили на планшет при указанной концентрации, а затем инкубировали с клетками, после чего добавляли хемокин с радиоактивной меткой (125I-SDF-1) на 3 часа при 4°С в следующей среде связывания (25 мМ HEPES, 140 мМ NaCl, 1 мМ CaCl2, 5 мМ MgCl2 и 0,2% бычьего сывороточного альбумина, доведенная до рН 7,1). Затем все пробы инкубировали 3 часа при 4°С с легким встряхиванием. После инкубации реакционные смеси во всех пробах переносили на обработанные PEI стеклянные фильтры GF/B (Packard) с помощью коллектора клеток (Packard) и дважды промывали (25 мМ HEPES, 500 мМ NaCl, 1 мМ CaCl2, 5 мМ MgCl2, доведенный до рН 7,1). В лунки добавляли сцинтиллятор (MicroScint 10, Packard) и определяли радиоактивность на фильтрах на сцинтилляционном счетчике Packard Topcount. Данные анализировали и строили графики с помощью GraphPad Prism (GraphPad Software).
Анализ связывания в сыворотке. Для того, чтобы более точно оценить эффективность связывания у соединений, проводили анализ связывания радиолиганда в присутствии 100% сыворотки человека, что более точно отражает условия in vivo. Хотя обычно при этом наблюдаются меньшие значения IC50, чем при анализе связывания в буфере, но повышается значимость анализа для ранжирования соединений по эффективности. Искомые соединения тестировали на их способность связываться с сайтами CXCR7 на трансфецированных CXCR7 клетках MCF-7 и/или MDA-MB-435S. Проводили связывание радиолиганда с максимальной эффективностью по методике с фильтрованием, как описано в Dairaghi DJ et al., HHV8-encoded vMIP-I selectively engages chemokine receptor CCR5. Agonist and antagonist profiles of viral chemokines. J. Biol. Chem. 1999 Jul 30, 274(31): 21569-74; и Gosling J et al. Cutting edge: identification of a novel chemokine receptor that binds dendritic cell- and T cell-active chemokines including ELC, SLC, and TECK. J. Immunol. 2000 Mar 15, 164(6): 2851-6.
При этом проводили инкубацию клеток MCF-7 и/или MDA-MB-435S с иссле-дуемыми соединениями и определяли способность этих соединений вытеснять 125I-меченный SDF-1 по методике, описанной в Dairaghi и Gosling. Искомые соединения вносили на планшет при указанной концентрации, после чего добавляли клетки и хемокин с радиоактивной меткой (125I-SDF-1), оба в следующей среде (сыворотка АВ человека с добавлением 10 мМ HEPES для стабилизации при рН 7,4). Затем все пробы инкубировали 3 часа при 4°С с легким встряхиванием. После инкубации реакционные смеси во всех пробах переносили на обработанные PEI стеклянные фильтры GF/B (Packard) с помощью коллектора клеток (Packard) и дважды промывали (25 мМ HEPES, 500 мМ NaCl, 1 мМ CaCl2, 5 мМ MgCl2, доведенный до рН 7,1). В лунки добавляли сцинтиллятор (MicroScint 10, Packard) и определяли радиоактивность на фильтрах на сцинтилляционном счетчике Packard Topcount. Данные анализировали и строили графики с помощью GraphPad Prism (GraphPad Software).
Анализ трансэндотелиальной миграции. Соединения по изобретению также можно оценить на их способность подавлять миграцию клеток при анализе трансэндотелиальной миграции. При этом проводится анализ способности клеток мигрировать через слой эндотелиальньгх клеток к источнику хемокинов. В одном примере такого анализа 100000 эндотелиальных клеток из пупочной вены человека (HUVECs, доступны от Lonza) засеивали в верхнюю камеру транслуночного планшета с размером пор у фильтра 5 мкм (Corning Costar). Добавляли среду и ставили планшеты на всю ночь в инкубатор с 5% СО2 при 37°С. После того, как HUVECs прикрепились к фильтру за ночь и образовали монослой, в нижнюю камеру добавляли среду, содержащую хемокин (например, SDF-1, конечная концентрация 10 нМ). Затем в верхнюю камеру добавляли 500000 клеток NC-37 (из АТСС) в присутствии или в отсутствие исследуемого соединения и планшеты возвращали в инкубатор на время от 3 ч до всей ночи. Можно добавлять различные концентрации соединения в различные лунки, чтобы получить зависимость от дозы. По окончании инкубации верхнюю камеру снимали и определяли количество клеток в нижней камере. Клетки можно оценивать количественно, к примеру, путем маркировки их флуоресцентным красителем типа CyQuant® (Invitrogen, СА), а затем количественного определения флуоресценции на соответствующем считывающем устройстве. Данные можно анализировать и наносить на график с помощью GraphPad Prism (GraphPad Software). Эффективность соединения определяется как его способность ингибировать миграцию данных клеток в нижнюю камеру.
Эффективность in vivo
Модель деструктивного воспаления суставов на кроликах
Исследование с LPS на кроликах проводится в основном так, как описано в Podolin et al., ibid. Самкам кроликов New Zealand (примерно 2 кг) вводили LPS (10 нг) внутри-суставно в оба колена. Исследуемые соединения (например, в виде состава в 1% метоцеле) или носитель (1% метоцель) вводили перорально в объеме дозы 5 мл/кг за два раза (за 2 часа до внутрисуставного введения LPS и через 4 часа после внутрисуставного введения LPS). Через 16 часов после введения LPS проводили лаваж коленей и определяли количество клеток. Положительные эффекты обработки определяли по уменьшению количества воспалительных клеток, привлеченных в воспаленную синовиальную жидкость коленных суставов. Обработка исследуемым соединением приводит к значительному снижению рекрутмента воспалительных клеток.
Оценка соединений на модели индуцированного коллагеном артрита у крыс
Для оценки эффекта исследуемых соединений на вызванное артритом клиническое опухание лодыжек можно провести 17-дневное исследование вызванного коллагеном II типа артрита. Коллагеновый артрит у крыс является экспериментальной моделью полиартрита, которая широко применялась для доклинических испытаний многочисленных противоартритных средств (см. Trentham et al., J. Exp. Med. 146(3): 857-868 (1977); Bendele et al., Toxicologic Pathol. 27:134-142 (1999); Bendele et al., Arthritis Rheum. 42:498-506 (1999)). Отличительными чертами этой модели являются надежное возникновение и прогрессирование устойчивого, легко измеримого полисуставного воспаления, заметное разрушение хрящей в связи с образованием паннуса и легкая или умеренная резорбция кости и периостальная пролиферация костной ткани.
Самок крыс Lewis (примерно 0,2 кг) анестезировали изофлураном и вводили неполный адъювант Фрейнда, содержащий 2 мг/мл бычьего коллагена II типа, у основания хвоста и в две точки на спине в 0-й и 6-й день этого 17-дневного исследования. Исследуемые соединения вводили подкожно раз в день с 0-го по 17-й день в эффективной дозе. Проводили измерения диаметра голеностопного сустава штангенциркулем, а в качестве меры эффективности принимали снижение опухания суставов.
Оценка соединений на модели заживления ран у мышей
В исследованиях по заживлению ран использовали полученных из ICR самцов мышей (24±2 г). На период тестирования животных размещали по одному в отдельных клетках. Под гексобарбиталовым (90 мг/кг, в/б) наркозом у каждого животного сбривали плечи и часть спины. Для удаления кожи, включая panniculus carnosus и сращенные ткани, использовали острый перфоратор (внутр. диаметр 12 мм). Тестируемое соединение или носитель наносили на это место сразу же после повреждения кожи раз в день в течение 10 дней подряд. Положительный контроль, к примеру, агонист аденозиновых А2-рецепторов (CGS-21680; 10 мкг/мышь), также можно вводить топически раз в день на протяжении всего эксперимента. Площадь раны, выведенной на прозрачные пластиковые пластины, измеряли с помощью анализатора изображений (Vista, Life Science Resources, version 3.0) в 1-й, 3, 5, 7, 9 и 11-й день. Рассчитывали процент (%) закрытия раны, определяли полупериод закрытия раны (СТ50) и анализировали методом линейной регрессии с помощью GraphPad Prism (GraphPad Software). Для сравнения между группой обработки и группой, получавшей носитель, по каждой временной точке измерения применяли непарный t-критерий Стьюдента. Различия считались статистически значимыми при р<0,05.
Оценка соединений на модели карциномы легких у мышей
Известны многие модели опухолей у животных, которые можно использовать для оценки исследуемых соединений. Например, на модели ксенотрансплантатов карциномы легких в подкожное пространство у мышей nude имплантируют фрагменты опухолей А549 (30-40 мг). Опухолям дают расти размером примерно до 150 мг (от 100 до 200 мг), в этот момент мышей включают в исследование и начинается обработка. Мышам вводится исследуемое соединение или контрольный носитель. В качестве положительного контроля можно включать мелфалан (9 мг/кг, внутрибрюшинно, Q4Dx3). Опухоли измеряют по два раза в неделю штангенциркулем в двух измерениях и вычисляют массу опухолей по формуле для вытянутого эллипсоида (а×b2/2), где а означает больший размер, а b - меньший размер, принимая плотность равной единице (1 мм3=1 мг). Также можно измерять вес тела по два раза в неделю, чтобы оценить любые неблагоприятные эффекты введения соединений. Противоопухолевая активность оценивается по замедлению роста опухолей в группе обработки по сравнению с контрольной группой, получавшей носитель.
Модель привитого экспериментального аутоиммунного энцефаломиелита на грызунах
ЕАЕ на грызунах - это экспериментальная модель рассеянного склероза (MS), которая широко применялась для доклинических испытаний многочисленных средств для лечения рецидивирующего, ремиттирующего и прогрессирующего MS. Отличительными чертами этой модели являются надежное возникновение и прогрессирование устойчивого, легко измеримого паралича хвоста и конечностей, воспаление нейронов, заметная демие-линизация в ответ на нейронные антигены.
Мышам вводится соответствующий нейронный антиген (например, основной белок миелина, гликопротеин миелина олигодендроцитов, протеолипидный белок) в полном адъюванте Фрейнда в день 0. После введения CFA/антигена выделяют иммунные клетки и стимулируют их ex vivo цитокинами и нейронным антигеном, получая линию Т-клеток со специфичностью к нейронному антигену. Затем эти клетки переносят мышам-реципиентам. Исследуемые соединения вводятся подкожно, внутрибрюшинно или перорально раз в день от 0-го дня до окончания исследования в эффективной дозе. В качестве меры эффективности принимаются ежедневные наблюдения степени паралича.
Оценка соединений на модели глиобластомы у мышей
Известны многие модели опухолей у животных, которые можно использовать для оценки исследуемых соединений. Например, на модели глиобластомы у мышей в головной мозг мышей nude посредством стереотаксической инъекции имплантируют 1×106 клеток U251MG. Через 20 дней опухоли облучают дозой излучения в 1-15 Gy. После облучения мышей обрабатывают (например, подкожно, внутрибрюшинно, перорально, парентерально или иным способом) соединением или контрольным носителем и дают развиваться опухолям. За все оставшееся время исследования отслеживают рост опухолей и/или смертность. Опухоли измеряют по два раза в неделю штангенциркулем в двух измерениях и вычисляют массу опухолей по формуле для вытянутого эллипсоида (a×b2/2), где а означает больший размер, а b - меньший размер, принимая плотность равной единице (1 мм3=1 мг). Также можно измерять вес тела по два раза в неделю, чтобы оценить любые неблагоприятные эффекты введения соединений. Противоопухолевая активность оценивается по замедлению роста опухолей в группе обработки по сравнению с контрольной группой, получавшей носитель.
Оценка соединений на модели глиобластомы у крыс
Известны многие модели опухолей у животных, которые можно использовать для оценки исследуемых соединений. Например, на модели глиобластомы С6 у крыс в головной мозг крыс Sprague-Dawley посредством стереотаксической инъекции имплантировали 1×106 клеток С6. Через 7 дней опухоли облучали дозой излучения в 5-20 Gy. После облучения крыс обрабатывали (например, подкожно, внутрибрюшинно, перорально, парентерально или иным способом) соединением или контрольным носителем и давали развиваться опухолям. Животных отслеживали до наступления смерти или потери >20% веса либо отбирали для тестирования на вызванные опухолями нейродефипиты, например, судороги и неподвижность, по соответствующим правилам и стандартам. Активность соединений определяли методом анализа выживаемости согласно Kaplan-Meier, причем соединение 1.090 в таблице 1 обладало сильной противоопухолевой активностью при оценке по замедлению роста опухолей в группе обработки по сравнению с получавшей носитель контрольной группой.
Оценка соединений на модели гипертензии у мышей
Известны многие модели легочной дисфункции и гипертензии у животных, которые можно использовать для оценки исследуемых соединений. Например, на модели легочной гипертензии, вызванной хронической гипоксией, новорожденных мышей (FVB/NJ) в случайном порядке подвергают нормобарической нормоксии (обычный воздух (RA)) или гипоксии (НА) (FiO2=0,12) в течение 2 недель. После 1-недельной RA или НА мышей обрабатывают исследуемым соединением, например, в виде ежедневных подкожных инъекций носителя или соединения с 7-го по 14-й день после рождения. Степень легочной гипертензии можно определить путем измерения систолического давления в правом желудочке (RVSP). Вкратце, проводится торакотомия и в правый желудочек вставляется игла 25 калибра, подсоединенная к датчику давления (Gould Instruments, ОН), и регистрируется RSVP. Сразу же после измерения RVSP мышей забивают, извлекают и вскрывают сердце. Оценивают гипертрофию правого желудочка (RVH) по соотношению массы правого желудочка и массы левого желудочка + перегородка (RV/LV+S). Улучшение показателей RSVP и RVH означает, что соединение-кандидат обладает терапевтическим потенциалом.
Проверка адекватности
Соединения, которые первоначально идентифицированы как представляющие интерес по любому из вышеприведенных способов скрининга, можно дополнительно протестировать для проверки отмеченной активности in vivo. Предпочтительно такие опыты проводятся на подходящих животных моделях. Базовый формат таких методов включает введение перспективного соединения, идентифицированного при первичном скрининге, тем животным, которые служат моделью для заболевания у человека, а затем определение того, будет ли заболевание (например, рак, инфаркт миокарда, заживление ран, воспалительные заболевания или другие заболевания, связанные с CXCR7) действительно модулироваться и/или улучшаться. Животные на моделях, используемых при проверке адекватности, как правило, представлены какими-либо млекопитающими. Конкретные примеры подходящих животных включают, без ограничения, приматов, мышей, крыс и рыбок данио.





Рядовым специалистам в данной области должно быть понятно из приведенного описания, чертежей и примеров, что в любых воплощениях изобретения могут производиться модификации и изменения, не выходящие за рамки объема изобретения, установленного нижеследующей формулой изобретения и ее эквивалентами.
Все указанные здесь патенты, патентные заявки, публикации и презентации включены сюда путем ссылки во всей полноте. Любые противоречия между приведенными здесь ссылками и положениями настоящего описания должны разрешаться в пользу последнего. Точно так же любые противоречия между принятыми в данной области определениями терминов или выражений и их определениями, приведенными в настоящем описании, должны разрешаться в пользу последнего.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ GRP119 | 2014 |
|
RU2642429C2 |
| ПИРРОЛО[2,3-В]ПИРАЗИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ SYK | 2013 |
|
RU2656853C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811408C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения | 2019 |
|
RU2717101C1 |
| 6-СУЛЬФАМОИЛХИНОЛИН-4-КАРБОНОВЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И КОМБИНАТОРНАЯ БИБЛИОТЕКА | 2003 |
|
RU2229475C1 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2589878C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
Изобретение относится к соединениям формулы I:
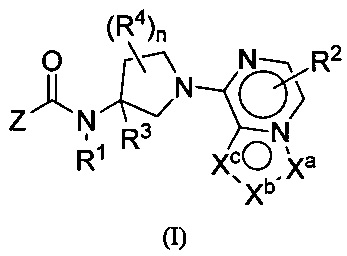 ,
,
а также к их фармацевтически приемлемым солям. Технический результат: получены новые соединения, а также фармацевтические композиции, которые применимы для связывания с CXCR7 и лечения медиируемых заболеваний, таких как рак, воспалительное заболевание. 5 н. и 23 з.п. ф-лы, 1 табл., 151 пр.
1. Соединение по формуле I:
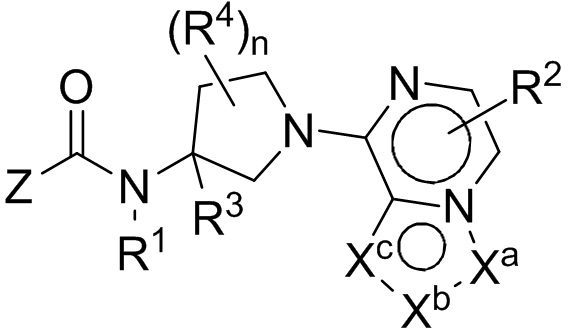
(I)
либо его фармацевтически приемлемая соль, где:
каждая из вершин кольца Ха, Xb и Хс независимо выбрана из группы, состоящей из N, CH и C(R2);
нижний индекс n означает 0;
Z выбран из группы, состоящей из:
моноциклического или конденсированного бициклического и 5-10-членного гетероарила, причем гетероарил содержит в кольце 1-3 гетероатома, выбранных из N, О и S; и эти арильные и гетероарильные группы необязательно замещены 1-2 заместителями R5;
R1 - H;
каждый R2 независимо выбран из группы, состоящей из H и C1-2-алкила;
R3 выбран из группы, состоящей из H, C1-2-гидроксиалкила и -CONRaRb;
каждый R5 независимо выбран из группы, состоящей из -X-CN, C1-3-алкила, C3-циклоалкила, С6-арила и 5- или 6-членного гетероциклоарила, причем гетероатомы на вершинах колец гетероциклоарила выбраны из N и О, а арильные и гетероциклические части R5 необязательно дополнительно замещены 1-3 Ra;
каждый Ra и Rb независимо выбран из группы, состоящей из водорода, гидроксила, галогена, циано, C1-2-алкила и -SO2-C1-2-алкила;
каждый Х означает соединительную группу C1-2-алкилена.
2. Соединение по п. 1, где Z означает моноциклический или конденсированный бициклический 5-10-членный гетероарил, содержащий в кольце 1-3 гетероатома, выбранных из N, О и S; причем данный гетероарил необязательно замещен 1-2 заместителями R5.
3. Соединение по п. 1, где Z означает моноциклический или конденсированный бициклический гетероарил, выбранный из группы, состоящей из имидазола, пиразола, 1,2,3-триазола, 1,2,4-триазола, тиазола, оксазола, оксадиазола, пиримидина, пиразина, пиридазина и хиназолина, каждый из которых необязательно замещен 1-2 заместителями R5.
4. Соединение по п. 1, где R3 выбран из группы, состоящей из H, CH2OH и C(O)NH2.
5. Соединение по п. 1, имеющее структуру:
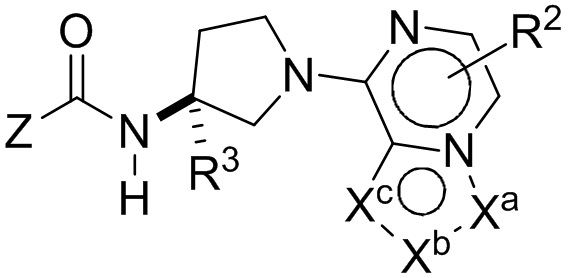 .
.
6. Соединение по п. 5, где бициклическая часть, содержащая Xa, Xb и Xc на вершинах кольца, выбрана из группы, состоящей из:
 .
.
и
7. Соединение по п. 5, где Z означает 5-членный гетероарил, замещенный одной группой R5, выбранной из необязательно замещенного кольца арила, 5-10-членного гетероарила, С3-циклоалкила и 5-10-членного гетероциклоалкила, и необязательно еще 1-2 дополнительными группами R5, выбранными из C1-3-алкила и CH2CN.
8. Соединение по п. 7, где Z выбран из группы, состоящей из:
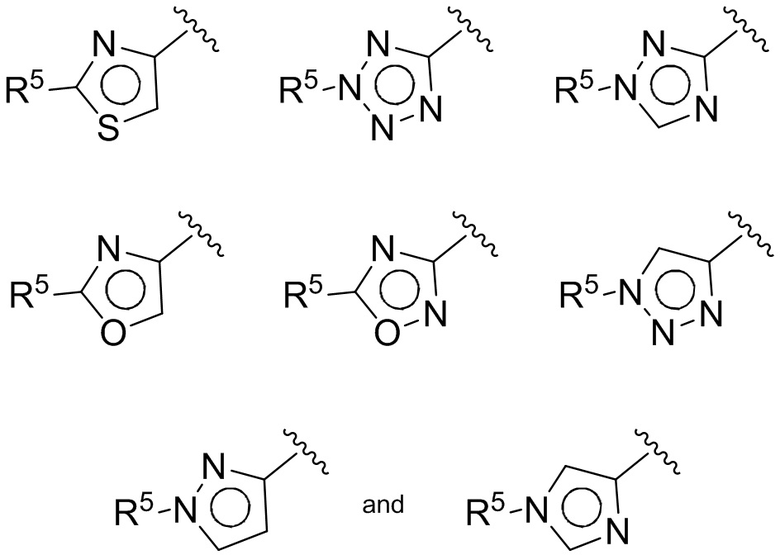
и
9. Соединение по п. 8, где Z соответствует формуле:
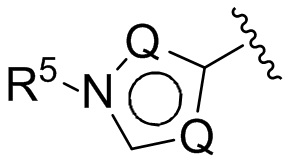
причем каждый Q независимо выбран из группы, состоящей из N, CH и C(R5), где каждый R5 независимо выбран из группы, состоящей из -ХСN, C1-3-алкила, С3-циклоалкила, С6-арила и 6-членного гетероциклоалкильного кольца.
10. Соединение по п. 1, соответствующее формуле:
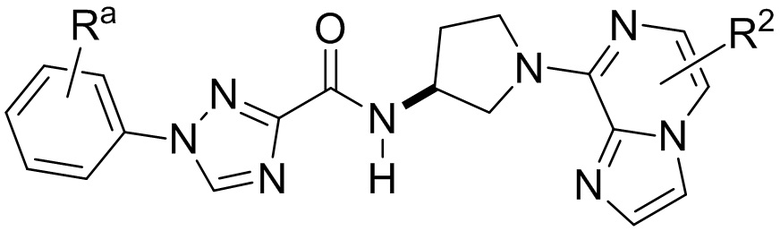 ,
,
либо его фармацевтически приемлемая соль.
11. Соединение по п. 10, где Ra выбран из группы, состоящей из водорода, галогена, циано, C1-2-алкила и -SO2-C1-2-алкила.
12. Соединение по п. 10, где R2 - H.
13. Соединение по п. 10, где Ra выбран из группы, состоящей из водорода, галогена, циано, C1-2-алкила и -SO2-C1-2-алкила; а R2 - H.
14. Соединение по п. 1, выбранное из группы, состоящей из:
или его фармацевтически приемлемой соли.
 ,
,
 ,
,
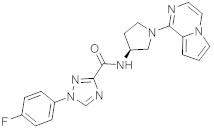 ,
,
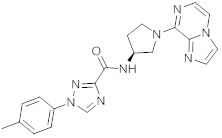 ,
,
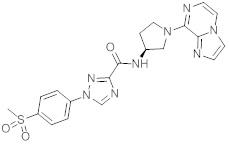 ,
,
 ,
,
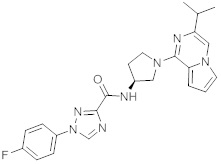 ,
,
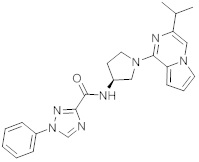 ,
,
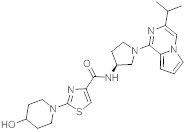 ,
,
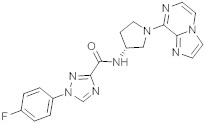 ,
,
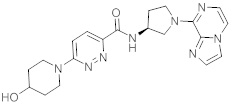 ,
,
 ,
,
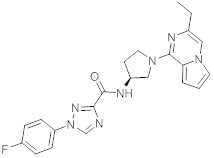 ,
,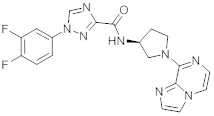 ,
,
 ,
,
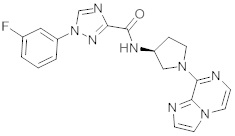 ,
,
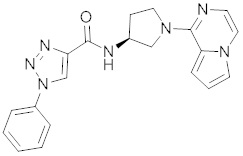 ,
,
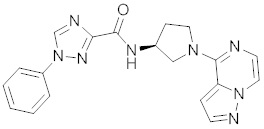 ,
,
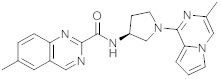 ,
,
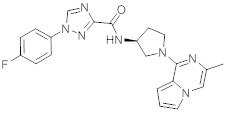 ,
,
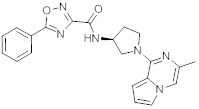 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
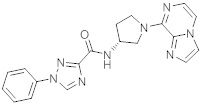 ,
,
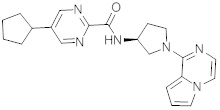 ,
,
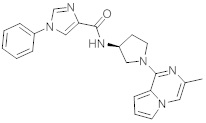 ,
,
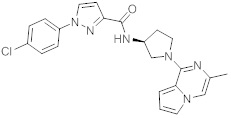 ,
,
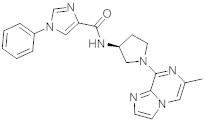 ,
,
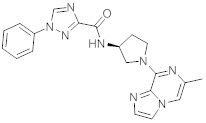 ,
,
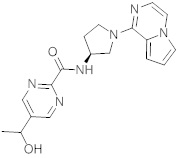 ,
,
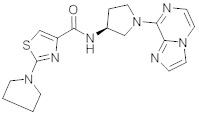 ,
,
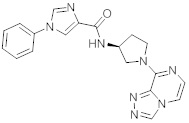 ,
,
 ,
,
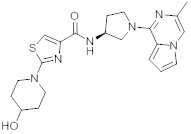 ,
,
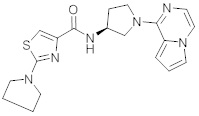 ,
,
 ,
,
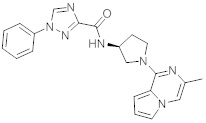 ,
,
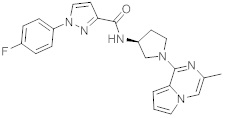 ,
,
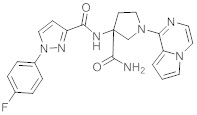 ,
,
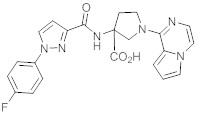 ,
,
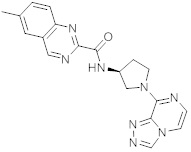 ,
,
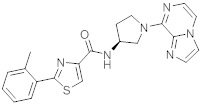 ,
,
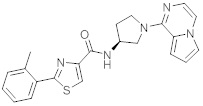 ,
,
 ,
,
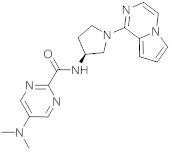 ,
,
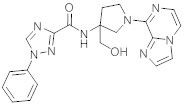 ,
,
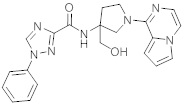 ,
,
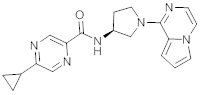 ,
,
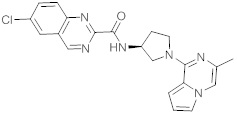 ,
,
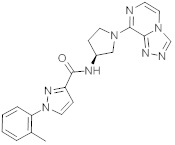 ,
,
 ,
,
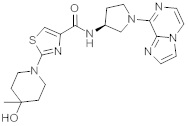 ,
,
 ,
,
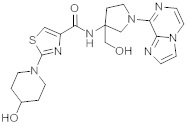 ,
,
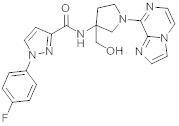 ,
,
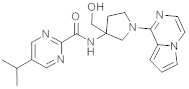 ,
,
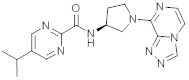 ,
,
 ,
,
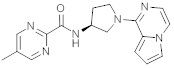 ,
,
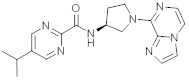 ,
,
 ,
,
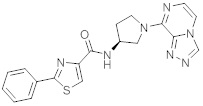 ,
,
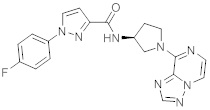 ,
,
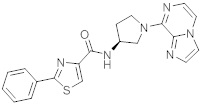 ,
,
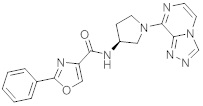 ,
,
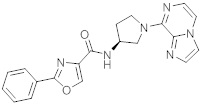 ,
,
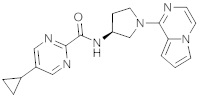 ,
,
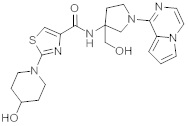 ,
,
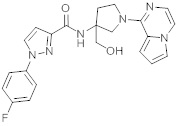 ,
,
 ,
,
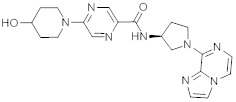 ,
,
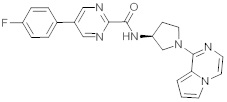 ,
,
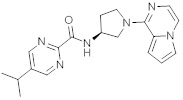 ,
,
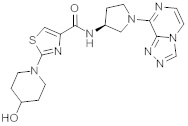 ,
,
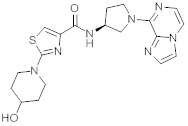 ,
,
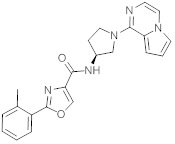 ,
,
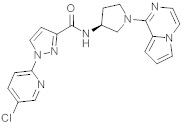 ,
,
 ,
,
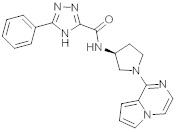 ,
,
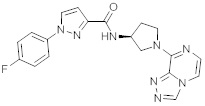 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
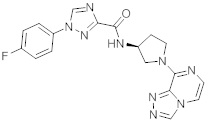 ,
,
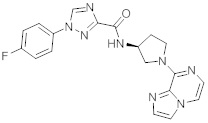 ,
,
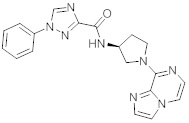 ,
,
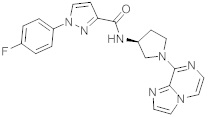 ,
,
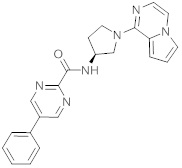 ,
,
 ,
,
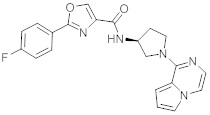 ,
,
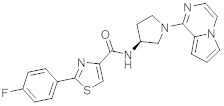 ,
,
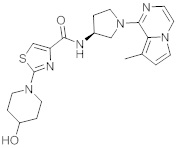 ,
,
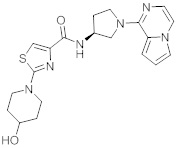 ,
,
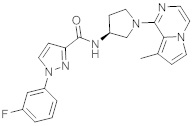 ,
,
 ,
,
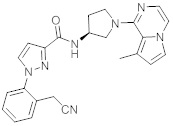 ,
,
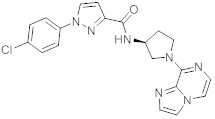 ,
,
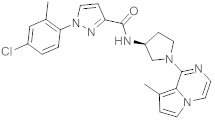 ,
,
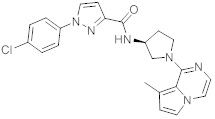 ,
,
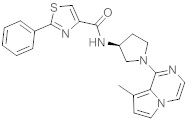 ,
,
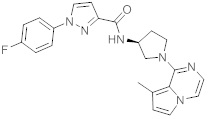 ,
,
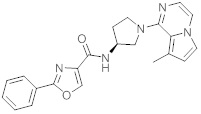 ,
,
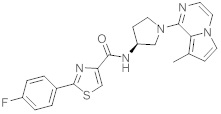 ,
,
 ,
,
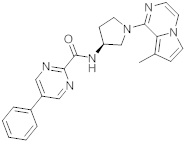 ,
,
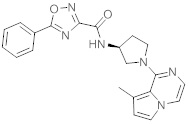 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
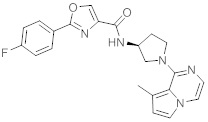 ,
,
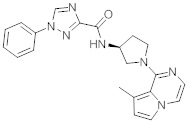 ,
,
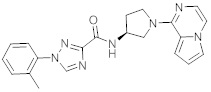 ,
,
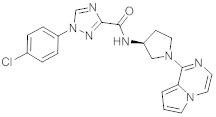 ,
,
 ,
,
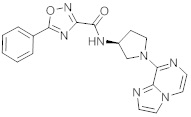 ,
,
 ,
,
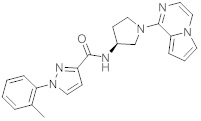 ,
,
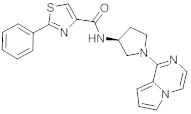 ,
,
 ,
,
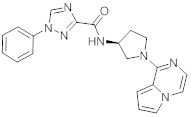 ,
,
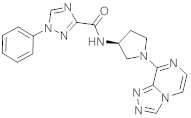 ,
,
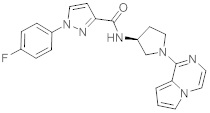 , и
, и
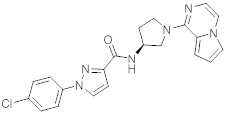 .
.
или его фармацевтически приемлевой соли.
15. Соединение по п. 1, имеющее структуру
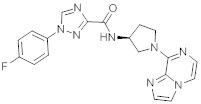
16. Соединение по п. 1, имеющее структуру
17. Фармацевтическая композиция для лечения заболеваний или нарушений, медиируемых/опосредуемых CXR7, включающая эффективное количество соединения по любому из пп. 1-16 и фармацевтически приемлемый наполнитель-эксципиент, где медиируемые CXR7 заболевания или нарушения выбраны из группы, состоящей из рака, воспалительного заболевания, демиенилирующего заболевания и гипертензивного заболевания.
18. Способ лечения медиируемых CXR7 заболеваний у млекопитающих, который включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-16 или фармацевтической композиции по п. 17 в течение периода времени, достаточного для лечения этого заболевания.
19. Способ по п. 18, в котором соединение представлено соединением по любому из пп. 14, 15 или 16.
20. Способ по п. 18, где указанный рак выбран из группы, состоящей из рака простаты, рака молочной железы, рака желудка, рака прямой кишки, лейкемии, лимфомы, меланомы, рака мочевого пузыря, рака прямой кишки, эзоофагального рака, рака щитовидной железы, рака легкого, рака горла/шеи, глиобластомы.
21. Способ по п. 18, в котором рак представляет собой рак горла/шеи.
22. Способ по п. 18, в котором рак представляет собой глиобластому.
23. Способ по п. 18, в котором воспалительным заболеванием является ревматоидный артрит.
24. Способ по п. 18, в котором димиелимирующим заболеванием является рассеянный склероз.
25. Способ ингибирования связывания хемокина I-TAC или SDF-1 с рецептором CXCR7, включающий контактирование соединения по любому из пп. 1-16 с клетками, экспрессирующими рецептор CXCR7, в течение времени, достаточного для ингибирования связывания этих хемокинов с рецепторами CXCR7.
26. Способ по п. 25, в котором соединение представлено соединением по любому из пп. 14, 15 или 16.
27. Способ выявления повышенного уровня CXCR7 в образце, который включает:
(a) контактирование образца с подозрением на повышенный уровень CXCR7 с радиоизотопной или детектируемой формой соединения по любому из пп. 1-16;
(b) определение уровня соединения, связавшегося с CXCR7, присутствующим в данном образце, для определения уровня CXCR7, находящегося в образце; и
(c) сравнение уровня, определенного на стадии (b), с контрольным образцом для определения того, имеется ли повышенный уровень CXCR7 в образце.
28. Способ по п. 27, в котором соединение помечено радиоактивной меткой.
| US 2010150831 A1, 17.06.2010 | |||
| US 2007275965 A1, 29.11.2007 | |||
| US 2007167443 A1, 19.07.2007 | |||
| WO 2012022265 A1, 23.02.2012 | |||
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА И ПИПЕРИДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2178414C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА КАК ИНГИБИТОРЫ ПРЕНИЛТРАНСФЕРАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 1999 |
|
RU2241712C9 |

