Область техники, к которой относится изобретение
Настоящее изобретение касается класса тиазололактамных соединений и их применения при получении лекарственных средств для лечения заболеваний, связанных с ERK. В частности, настоящее изобретение касается соединений, представленных формулой (III), либо их фармацевтически приемлемых солей.
Уровень техники
Путь Ras/Raf/MEK/ERK - классический путь сигнального каскада митоген-активируемых протеинкиназ (MAPK), который участвует в передаче сигналов различных факторов роста, цитокинов, митогенов и рецепторов гормонов после активации и является одним из самых важных путей передачи сигналов для контроля роста, дифференцировки и выживания клеток.
Исследования показали, что аномальная активация пути Ras/Raf/MEK/ERK, вызванная мутацией или амплификацией, является детерминантой различных раковых заболеваний. В опухолях у человека частота мутаций RAS составляет около 22%, частота мутаций BRAF - около 7%, а частота мутаций MEK - около 1%. Поэтому белки ключевых узлов на этом пути стали важными мишенями для лечения рака (Cancer Discov. 2019, 9, 329-341). В настоящее время ряд ингибиторов BRAF и ингибиторов MEK1/2, а также их комбинированные схемы одобрены FDA США для лечения меланомы, немелкоклеточного рака легких с мутацией BRAFV600E и других раковых заболеваний. Однако применение ингибиторов BRAF и MEK для этих вышестоящих узлов может быстро вызвать проблемы лекарственной устойчивости вследствие мутации или реактивации пути, что сильно ограничивает их клиническое применение.
Регулируемые внеклеточными сигналами протеинкиназы (ERK) (особенно киназы ERK1 и ERK2) являются главными участниками и нижележащими ключевыми узлами пути Ras/Raf/MEK/ERK, и чрезмерная активация их встречается при многих раковых заболеваниях человека. У ERK, как терминальной сигнальной киназы этого пути, еще не обнаружено мутаций, вызывающих лекарственную устойчивость. Поэтому препараты, нацеленные на ERK-киназы, должны преодолеть проблему лекарственной устойчивости, вызванной применением ингибиторов вышележащих мишеней, и станут более перспективной стратегией терапии. Но пока что исследования ингибиторов ERK все еще находятся в клинической фазе, и ни один ингибитор ERK еще не был одобрен для продажи в качестве лекарственного средства.
Итак, существует настоятельная потребность в разработке безопасных и эффективных препаратов-ингибиторов ERK для удовлетворения потребности в лечении рака.
Сущность изобретения
Настоящим изобретением предусмотрены соединения, представленные формулой (III), либо их фармацевтически приемлемые соли:
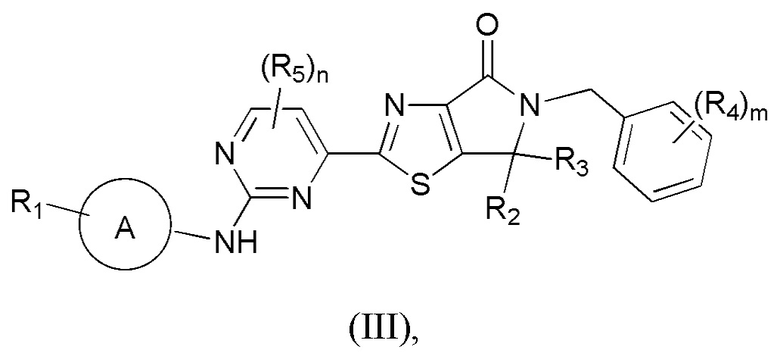
где: R1 означает H, C1-3-алкил или C3-5-циклоалкил, причем C1-3-алкил или C3-5-циклоалкил необязательно замещен 1, 2 или 3 Ra;
R2 и R3 каждый независимо означает C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Rb;
R4 означает H, F, Cl, Br, I или C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Rc;
R5 означает F, Cl, Br, I или C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Re;
m равно 0, 1 или 2;
n равно 0, 1 или 2;
кольцо A означает пиразолил или тетрагидропиранил, причем пиразолил или тетрагидропиранил необязательно замещен 1, 2 или 3 Rd;
Ra, Rb, Rc и Re каждый независимо означает D, F, Cl, Br, I, OH или OCH3;
Rd означает F, Cl, Br, I, CH3 или OCH3.
В некоторых воплощениях настоящего изобретения вышеприведенный R1 означает H, CH3 или циклопропил, причем CH3 или циклопропил необязательно замещен 1, 2 или 3 Ra, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R1 означает H, CH3, CHF2, CD3, CH2CH2OCH3 или циклопропил, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенные R2 и R3 каждый независимо означает CH3 или CH2CH3, причем CH3 или CH2CH3 необязательно замещен 1, 2 или 3 Rb, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенные R2 и R3 каждый независимо означает CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R4 означает H, F, Cl, Br, I или CH3, причем CH3 необязательно замещен 1, 2 или 3 Rc, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R4 означает H, F, Cl, Br, I или CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает F, Cl, Br, I или CH3, причем CH3 необязательно замещен 1, 2 или 3 Re, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает F, Cl, Br, I или CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает H, F, Cl, Br, I или CH3, причем CH3 необязательно замещен 1, 2 или 3 Re, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает H, F, Cl, Br, I или CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенное кольцо A означает  ,
, 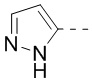 или
или 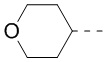 , причем , или необязательно замещено 1, 2 или 3 Rd, а другие переменные уже определены в настоящем описании.
, причем , или необязательно замещено 1, 2 или 3 Rd, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенное кольцо A означает , , 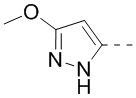 или
или 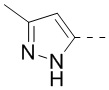 , а другие переменные уже определены в настоящем описании.
, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенная структурная группировка 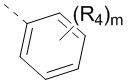 означает
означает  ,
,  или
или 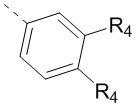 , а другие переменные уже определены в настоящем описании.
, а другие переменные уже определены в настоящем описании.
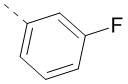
В некоторых воплощениях настоящего изобретения вышеприведенная структурная группировка означает 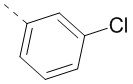 ,
,  или
или 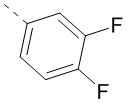 , а другие переменные уже определены в настоящем описании.
, а другие переменные уже определены в настоящем описании.
Настоящим изобретением предусмотрены соединения, представленные формулой (III), либо их фармацевтически приемлемые соли:
где: R1 означает H или C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Ra;
R2 и R3 каждый независимо означает C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Rb;
R4 означает H, F, Cl, Br, I или C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Rc;
R5 означает F, Cl, Br, I или C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Re;
m равно 0, 1 или 2;
n равно 0, 1 или 2;
кольцо A означает пиразолил или тетрагидропиранил, причем пиразолил или тетрагидропиранил необязательно замещен 1, 2 или 3 Rd;
Ra, Rb, Rc и Re каждый независимо означает F, Cl, Br, I или OH;
Rd означает F, Cl, Br, I или CH3.
В некоторых воплощениях настоящего изобретения вышеприведенный R1 означает H или CH3, причем CH3 необязательно замещен 1, 2 или 3 Ra, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R1 означает CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенные R2 и R3 каждый независимо означает CH3 или CH2CH3, причем CH3 или CH2CH3 необязательно замещен 1, 2 или 3 Rb, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенные R2 и R3 каждый независимо означает CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R4 означает H, F, Cl, Br, I или CH3, причем CH3 необязательно замещен 1, 2 или 3 Rc, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R4 означает H, F, Cl, Br, I или CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает F, Cl, Br, I или CH3, причем CH3 необязательно замещен 1, 2 или 3 Re, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает F, Cl, Br, I или CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает H, F, Cl, Br, I или CH3, причем CH3 необязательно замещен 1, 2 или 3 Re, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R5 означает H, F, Cl, Br, I или CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенное кольцо A означает , или , причем , или необязательно замещено 1, 2 или 3 Rd, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенное кольцо A означает или , а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенная структурная группировка означает , или , а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенная структурная группировка означает , или , а другие переменные уже определены в настоящем описании.
Настоящим изобретением предусмотрены соединения, представленные формулой (I), либо их фармацевтически приемлемые соли:
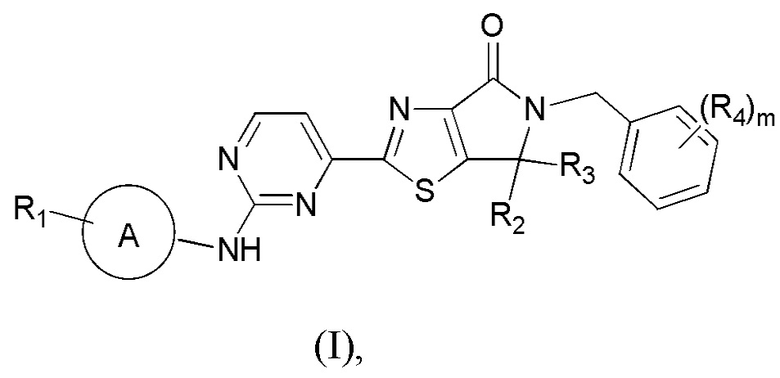
где: R1 означает H или C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Ra;
R2 и R3 каждый независимо означает C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Rb;
R4 означает H, F, Cl, Br, I или C1-3-алкил, причем C1-3-алкил необязательно замещен 1, 2 или 3 Rc;
m равно 0, 1 или 2;
кольцо A означает пиразолил или тетрагидропиранил, причем пиразолил или тетрагидропиранил необязательно замещен 1, 2 или 3 Rd;
Ra, Rb и Rc каждый независимо означает F, Cl, Br, I или OH;
Rd означает F, Cl, Br, I или CH3.
В некоторых воплощениях настоящего изобретения R1 означает H или CH3, причем CH3 необязательно замещен 1, 2 или 3 Ra, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R1 означает CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенные R2 и R3 каждый независимо означает CH3 или CH2CH3, причем CH3 или CH2CH3 необязательно замещен 1, 2 или 3 Rb, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенные R2 и R3 каждый независимо означает CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R4 означает H, F, Cl, Br, I или CH3, причем CH3 необязательно замещен 1, 2 или 3 Rc, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенный R4 означает H, F, Cl, Br, I или CH3, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенное кольцо A означает , или , причем , или необязательно замещено 1, 2 или 3 Rd, а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенное кольцо A означает или , а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенная структурная группировка означает , или , а другие переменные уже определены в настоящем описании.
В некоторых воплощениях настоящего изобретения вышеприведенная структурная группировка означает , или , а другие переменные уже определены в настоящем описании.
Настоящее изобретение также включает и некоторые воплощения, которые получаются при комбинировании каких-либо из вышеприведенных переменных.
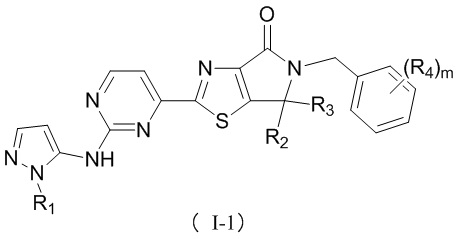
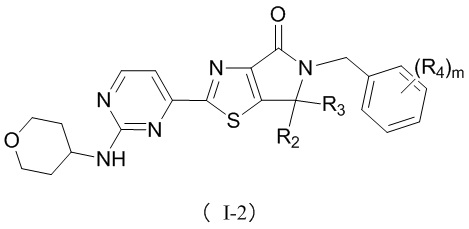
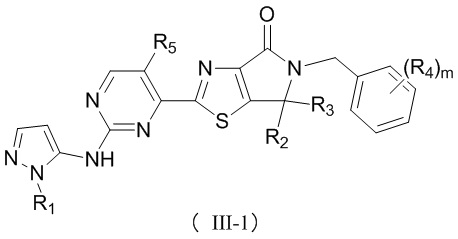
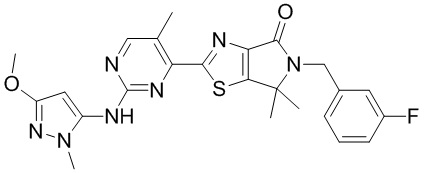
В некоторых воплощениях настоящего изобретения предусмотрены вышеприведенные соединения либо их фармацевтически приемлемые соли, которые выбраны из следующих соединений:
 ,
,  ,
,
 ,
, 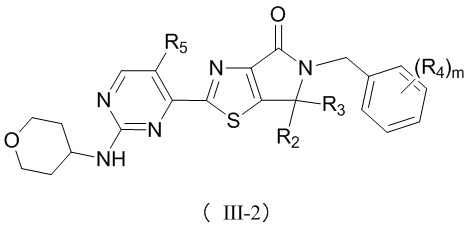 ,
,
где R1, R2, R3, R4, R5 и m уже определены в настоящем описании.
Настоящим изобретением также предусмотрены соединения, представленные следующими формулами, либо их фармацевтически приемлемые соли:
 ,
, 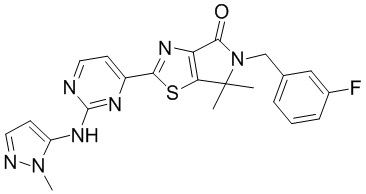 ,
,
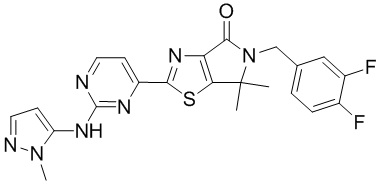 ,
, 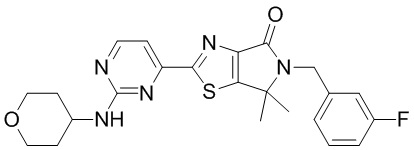 ,
,
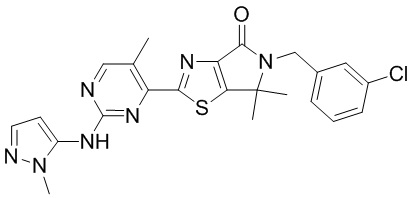 ,
, 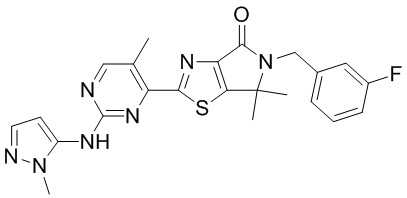 ,
,
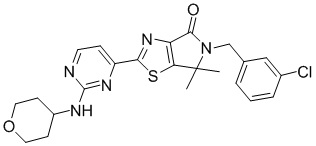 ,
, 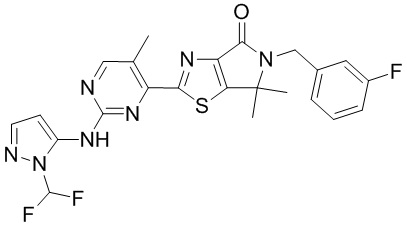 ,
,
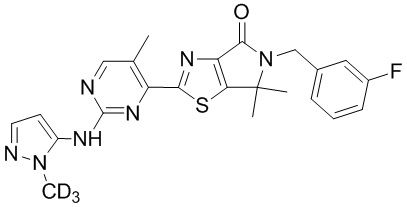 ,
, 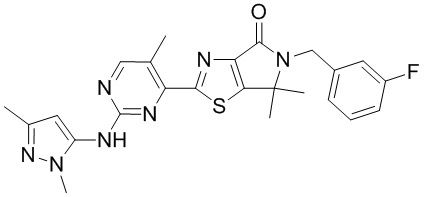 ,
,
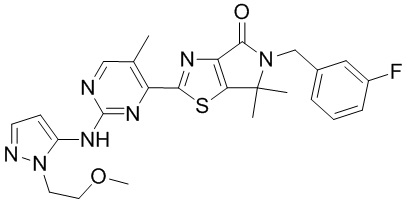 ,
, 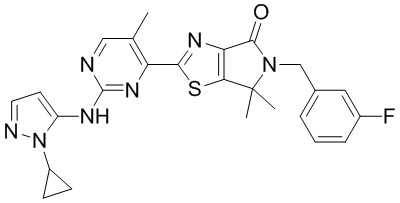 ,
,
 .
.
В некоторых воплощениях настоящего изобретения предусмотрено применение вышеприведенных соединений либо их фармацевтически приемлемых солей при изготовлении лекарственных средств для лечения заболеваний, связанных с ERK.
В некоторых воплощениях настоящего изобретения вышеприведенное применение отличается тем, что средства, относящиеся к ингибиторам ERK, представляют собой лекарственные средства для лечения солидных опухолей.
Технический эффект
Соединения по настоящему изобретению проявляют превосходную активность ингибирования киназы ERK2. Кроме того, соединения по настоящему изобретению проявляют превосходную активность ингибирования пролиферации клеток HT29. Соединения по настоящему изобретению проявляют превосходную пероральную экспозицию и биодоступность. Соединения по настоящему изобретению могут значительно ингибировать рост опухолей. При введении не наблюдается существенного снижения веса тела животных, переносимость хорошая.
Определения и термины
Если не указано иначе, следующие термины и выражения в настоящем изобретении должны иметь следующие значения. Конкретные термины или выражения не должны считаться неопределенными или неясными в отсутствие конкретного определения, а должны пониматься в общепринятом смысле. При этом торговые названия служат для обозначения соответствующих товаров либо их активных ингредиентов.
Термин “фармацевтически приемлемый” применяется здесь в отношении таких соединений, материалов, композиций и/или дозовых форм, которые подходят для применения в контакте с тканями человека и животных в рамках надежного медицинского суждения, без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений, соизмеримо с разумным соотношением польза/риск.
Термин “фармацевтически приемлемая соль” означает такие соли приведенных здесь соединений, которые получают при реакции соединений, содержащих определенные заместители, приведенные здесь, с относительно нетоксичными кислотами или основаниями. Когда приведенные здесь соединения содержат сравнительно кислые функциональные группы, можно получить соли с основаниями при обработке этих соединений достаточным количеством основания в чистом растворе или в подходящем инертном растворителе. Фармацевтически приемлемые соли с основаниями включают соли натрия, калия, кальция, аммония, органических аминов или магния либо аналогичные соли. Когда приведенные здесь соединения содержат сравнительно щелочные функциональные группы, можно получить соли с кислотами при обработке этих соединений достаточным количеством кислоты в чистом растворе или в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей с кислотами включают соли неорганических кислот, причем неорганические кислоты включают, к примеру, соляную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонаты, фосфорную кислоту, монозамещенные фосфаты, двузамещенные фосфаты, серную кислоту, бисульфаты, йодистоводородную кислоту, фосфиновую кислоту и т.п.; и соли органических кислот, причем органические кислоты включают, к примеру, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, пробковую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту, метансульфоновую кислоту и т.п.; а также соли аминокислот (типа аргинина и др.) и соли органических кислот типа глюкуроновой кислоты и др. Некоторые из приведенных здесь соединений содержат как щелочные, так и кислотные функциональные группы и могут быть преобразованы в любые соли с основаниями или кислотами.
Фармацевтически приемлемые соли, приведенные здесь, могут быть получены из исходных соединений, содержащих кислотные или щелочные группировки, стандартными химическими методами. Как правило, такие соли можно получить при реакции соединения в виде свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе либо их смеси.
Если не указано иначе, термин “изомер” охватывает геометрические изомеры, цис- или транс-изомеры, стереоизомеры, энантиомеры, оптические изомеры, диастереомеры и таутомеры.
Приведенные здесь соединения могут присутствовать в виде определенных геометрических или стереоизомеров. Настоящим изобретением предусмотрены все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереоизомеры, (D)-изомеры, (L)-изомеры, рацемические смеси и другие смеси, например, смеси, обогащенные энантиомерами или диастереоизомерами, которые все входят в рамки настоящего изобретения. Заместители типа алкилов могут содержать дополнительные асимметрические атомы углерода. Все эти изомеры и их смеси входят в рамки настоящего изобретения.
Если не указано иначе, термины “энантиомер” или “оптический изомер” означают такие стереоизомеры, которые находятся в зеркальном соотношении друг с другом.
Если не указано иначе, термины “цис-транс-изомер” или “геометрический изомер” возникают вследствие отсутствия свободного вращения у двойной связи или одинарной связи между кольцевыми атомами углерода.
Если не указано иначе, термин “диастереомер” означает такой стереоизомер, у которого в молекуле содержатся два или больше хиральных центров, которые не находятся в зеркальном соотношении между молекулами.
Если не указано иначе, “(+)” означает декстроизомер, “(-)” означает левоизомер, а “(±)” означает рацемат.
Если не указано иначе, клиновидная сплошная связь ( ) и клиновидная пунктирная связь (
) и клиновидная пунктирная связь ( ) обозначают абсолютную конфигурацию стереоцентра; прямая сплошная связь (
) обозначают абсолютную конфигурацию стереоцентра; прямая сплошная связь ( ) и прямая пунктирная связь () обозначают относительную конфигурацию стереоцентра; волнистая линия (
) и прямая пунктирная связь () обозначают относительную конфигурацию стереоцентра; волнистая линия ( ) означает клиновидную сплошную связь () или клиновидную пунктирную связь (); или же волнистая линия () означает прямую сплошную связь () либо прямую пунктирную связь ().
) означает клиновидную сплошную связь () или клиновидную пунктирную связь (); или же волнистая линия () означает прямую сплошную связь () либо прямую пунктирную связь ().
Если не указано иначе, термин “обогащенный одним изомером”, “обогащенный изомером”, “обогащенный одним энантиомером” или “обогащенный энантиомером” означает то, что содержание одного изомера или энантиомера составляет менее 100%, причем содержание изомера или энантиомера составляет 60% и более или 70% и более или 80% и более или 90% и более или 95% и более или 99% и более или 99,5% и более или 99,6% и более или 99,7% и более или 99,8% и более или 99,9% и более.
Если не указано иначе, термин “изомерный избыток” или “энантиомерный избыток” означает разницу между относительным процентным содержанием двух изомеров или двух энантиомеров. Например, если один изомер или энантиомер присутствует в количестве 90%, а другой изомер или энантиомер присутствует в количестве 10%, то изомерный избыток или энантиомерный избыток (значение ее) составляет 80%.
Оптически активные (R)- и (S)-изомеры или D- и L-изомеры можно получить с помощью хирального синтеза или хиральных реагентов или другими стандартными методами. Если нужно получить один из энантиомеров определенного соединения, приведенного здесь, то требуемый чистый энантиомер можно получить путем асимметрического синтеза или производного действия хирального вспомогательного вещества с последующим разделением полученной диастереомерной смеси и отщеплением вспомогательной группы. С другой стороны, если молекула содержит основную функциональную группу (типа аминогруппы) или кислотную функциональную группу (типа карбоксильной), то проводится реакция соединения с соответствующей оптически активной кислотой или основанием с образованием соли диастереомерного изомера, которая затем подвергается разделению диастереомеров принятым в данной области методом с получением чистого энантиомера. Кроме того, энантиомеры и диастереоизомеры обычно выделяют методами хроматографии с использованием хиральной неподвижной фазы, необязательно в сочетании с методом химической дериватизации (например, получением карбамата из амина).
Приведенные здесь соединения могут содержать неестественные доли атомных изотопов у одного или нескольких атомов, входящих в состав соединения. Например, соединение может быть помечено радиоизотопом типа трития (3H), йода-125 (125I) или C-14 (14C). В другом случае водород может быть заменен на тяжелый водород для получения дейтерированного препарата. Связь между дейтерием и углеродом прочнее, чем между простым водородом и углеродом. По сравнению с недейтерированными препаратами дейтерированные препараты обладают такими преимуществами, как снижение токсических побочных эффектов, повышение стабильности препаратов, повышение эффективности и продление биологического периода полураспада препаратов. Все изменения изотопного состава приведенных здесь соединений, независимо от радиоактивности, входят в рамки настоящего изобретения.
Термин “необязательный” или “необязательно” означает, что последующее событие или условие может произойти, но не обязательно, причем этот термин включает случаи, когда событие или условие происходит, и случаи, когда событие или условие не происходит.
Термин “замещенный” означает то, что один или несколько атомов водорода на определенном атоме замещены заместителем, включая варианты дейтерия и водорода, если только валентность данного атома остается нормальной, а замещенное соединение стабильно. Если заместителем является оксо (т.е. =O), то это значит, что замещены два атома водорода. Положения в ароматическом кольце не могут быть замещены оксогруппой. Термин “необязательно замещенный” означает то, что атом может быть замещен заместителем или нет, если не указано иначе, а вид и количество заместителей могут быть произвольными, если только они химически достижимы.
Когда какая-либо переменная (типа R) встречается в строении или структуре соединения более одного раза, то определение переменной в каждом случае является независимым. Так, например, если какая-то группа замещена 0-2 R, то она может быть необязательно замещена максимум двумя R, причем определение R в каждом случае будет независимым. Кроме того, комбинации заместителей и/или их вариантов допустимы лишь в том случае, если эти комбинации дают стабильные соединения.
Когда количество соединительных групп равно 0 типа -(CRR)0-, то это значит, что соединительная группа представляет собой простую связь.
Когда количество заместителей равно 0, то это значит, что заместитель не существует. Например, -A-(R)0 означает то, что структура фактически представляет собой -A.
Когда заместитель является вакантным, то это значит, что заместитель не существует. Например, когда X является вакантным в A-X, то структура A-X фактически представляет собой A.
Когда одна из переменных представляет собой простую связь, то это значит, что две группы, связанные простой связью, соединяются напрямую. Например, когда L в A-L-Z означает простую связь, то структура A-L-Z фактически представляет собой A-Z.
Когда связь у заместителя может быть перекрестно связана с двумя и более атомами в кольце, такой заместитель может быть связан с любым атомом в кольце. Например, в структурной группировке  или
или  представлен заместитель R, который может быть вставлен в любом положении циклогексила или циклогексадиена. Когда у пронумерованного заместителя не указано, через какой атом он связан с замещаемой группой, такой заместитель может быть связан через любой из своих атомов. Например, пиридильная группа в качестве заместителя может быть связана с замещаемой группой через любой из атомов углерода в кольце пиридина.
представлен заместитель R, который может быть вставлен в любом положении циклогексила или циклогексадиена. Когда у пронумерованного заместителя не указано, через какой атом он связан с замещаемой группой, такой заместитель может быть связан через любой из своих атомов. Например, пиридильная группа в качестве заместителя может быть связана с замещаемой группой через любой из атомов углерода в кольце пиридина.
Когда у пронумерованной соединительной группы не указано направление связи, то её направление связи является произвольным. Например, когда соединительная группа L в  представлена -M-W-, то -M-W- может соединяться с кольцом A и кольцом B в том же направлении, что и при чтении слева направо, составляя
представлена -M-W-, то -M-W- может соединяться с кольцом A и кольцом B в том же направлении, что и при чтении слева направо, составляя 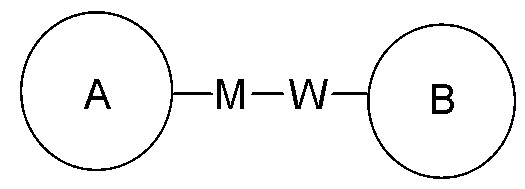 , или же может соединяться с кольцом A и кольцом B в обратном направлении, чем при чтении слева направо, составляя
, или же может соединяться с кольцом A и кольцом B в обратном направлении, чем при чтении слева направо, составляя 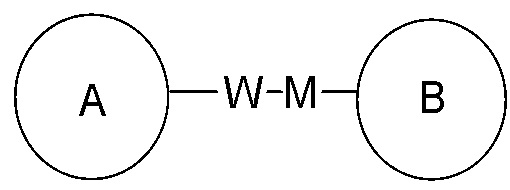 . Комбинации соединительных групп, заместителей и/или их вариантов допускаются только в том случае, если такие комбинации могут дать стабильные соединения.
. Комбинации соединительных групп, заместителей и/или их вариантов допускаются только в том случае, если такие комбинации могут дать стабильные соединения.
Если не указано иначе, когда группа содержит один или несколько соединяемых участков, то любой один или несколько участков этой группы могут соединяться с другими группами через химические связи. Если положение соединения у химической связи является переменным и в месте соединения содержатся атомы H, то при соединении соединяемых участков, содержащих атомы H, с химической связью количество атомов H в этом месте будет соответственно снижаться по мере возрастания количества связанных химических связей и группа станет группой c соответствующей валентностью. Химическая связь между этим участком и другими группами может быть представлена в виде прямой сплошной связи ( ), прямой пунктирной связи (
), прямой пунктирной связи ( ) или волнистой линии (
) или волнистой линии (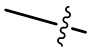 ). К примеру, прямая сплошная связь в -OCH3 означает, что эта группа соединяется с другими группами через атом кислорода в группе; прямая пунктирная связь в
). К примеру, прямая сплошная связь в -OCH3 означает, что эта группа соединяется с другими группами через атом кислорода в группе; прямая пунктирная связь в  означает, что эта группа соединяется с другими группами через два конца атома азота в группе; волнистая линия в
означает, что эта группа соединяется с другими группами через два конца атома азота в группе; волнистая линия в 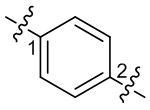 означает, что эта группа соединяется с другими группами через 1-й и 2-й атомы углерода фенильной группы;
означает, что эта группа соединяется с другими группами через 1-й и 2-й атомы углерода фенильной группы; 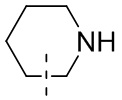 означает то, что любой соединяемый участок пиперидинильной группы может соединяться с другими группами через одну химическую связь, включая по меньшей мере четыре способа соединения:
означает то, что любой соединяемый участок пиперидинильной группы может соединяться с другими группами через одну химическую связь, включая по меньшей мере четыре способа соединения: 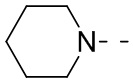 ,
, 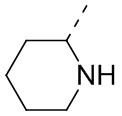 ,
, 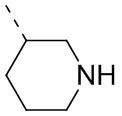 и
и  ; даже если на -N- представлен атом H, все еще включает способ соединения ; просто при соединении с одной химической связью количество Н в этом месте уменьшится на единицу и эта группа превратится в соответствующую одновалентную пиперидинильную группу.
; даже если на -N- представлен атом H, все еще включает способ соединения ; просто при соединении с одной химической связью количество Н в этом месте уменьшится на единицу и эта группа превратится в соответствующую одновалентную пиперидинильную группу.
Если не указано иначе, количество атомов в кольце обычно определяется в виде количества членов кольца, например, “5-7-членное кольцо” означает “кольцо” из 5-7 атомов, расположенных по окружности.
Если не указано иначе, термин “C1-3-алкил” применяется для обозначения линейной или разветвленной насыщенной углеводородной группы, состоящей из 1-3 атомов углерода. C1-3-алкильные группы включают C1-2- и C2-3-алкильные группы и т.п. Они могут быть одновалентными (например, метил), двухвалентными (например, метилен) или поливалентными (например, метенил). Примеры C1-3-алкильных групп включают, без ограничения, метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т.п.
Если не указано иначе, “C3-5-циклоалкил” означает насыщенную циклическую углеводородную группу, состоящую из 3-5 атомов углерода, которая представляет собой моноциклическую кольцевую систему; C3-5-циклоалкилы включают C3-4- и C4-5-циклоалкильные группы и т.п.; они могут быть одновалентными, двухвалентными или поливалентными. Примеры C3-5-циклоалкилов включают, без ограничения, циклопропил, циклобутил, циклопентил и т.п.
Термин “защитная группа” включает, без ограничения, “аминозащитные группы”, “гидроксизащитные группы” или “тиозащитные группы”. Термин “аминозащитные группы” означает защитные группы, пригодные для блокирования побочных реакций у азота аминогруппы. Репрезентативные аминозащитные группы включают, без ограничения: формил; ацилы типа алканоилов (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонилы типа трет-бутоксикарбонила (Boc); арилметоксикарбонилы типа бензилоксикарбонила (Cbz) и 9-фторенилметоксикарбонила (Fmoc); арилметилы типа бензила (Bn), тритила (Tr), 1,1-бис-(4'-метоксифенил)метила; силилы типа триметилсилила (TMS) и трет-бутилдиметилсилила (TBS) и др. Термин “гидроксизащитные группы” означает защитные группы, пригодные для блокирования побочных реакций у гидроксила. Репрезентативные гидроксизащитные группы включают, без ограничения: алкилы типа метила, этила и трет-бутила; ацилы типа алканоилов (например, ацетил); арилметилы типа бензила (Bn), п-метоксибензила (PMB), 9-фторенилметила (Fm) и дифенилметила (бензгидрила, DPM); силилы типа триметилсилила (TMS) и трет-бутилдиметилсилила (TBS) и др.
Приведенные здесь соединения могут быть получены различными методами синтеза, хорошо известными специалистам в данной области, включая следующие пронумерованные воплощения, воплощения, полученные по следующим перечисленным воплощениям в сочетании с другими методами химического синтеза, и эквивалентные замены, хорошо известные специалистам в данной области. Альтернативные воплощения включают, без ограничения, приведенные здесь воплощения.
Структура приведенных здесь соединений может быть проверена стандартными методами, хорошо известными специалистам в данной области. Если в настоящем изобретении приводится абсолютная конфигурация соединения, то абсолютная конфигурация может быть проверена стандартными методами типа рентгеновской дифракции на монокристалле (SXRD). При рентгеновской дифракции на монокристалле (SXRD) данные по интенсивности дифракции на выращенном монокристалле получают на коммерческом дифрактометре Bruker D8 с источником излучения типа CuKα в режиме сканирования ϕ/ω; после получения соответствующих данных проводится дальнейший анализ кристаллической структуры прямым методом (Shelxs97) для проверки абсолютной конфигурации.
Растворители, используемые в настоящем изобретении, коммерчески доступны.
В настоящем изобретении применяются следующие сокращения: aq означает водный; eq означает эквивалентный или эквивалентность; DCM означает дихлорметан; PE означает петролейный эфир; DMSO означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; МеОН означает метанол; Cbz означает бензилоксикарбонил, который является аминозащитной группой; BOC означает трет-бутоксикарбонил, который является аминозащитной группой; O/N означает в течение ночи; THF означает тетрагидрофуран; Boc2O означает ди-трет-бутилдикарбонат; TFA означает трифторуксусную кислоту; DIPEA означает диизопропилэтиламин; iPrOH означает 2-пропанол; к.т. означает комнатную температуру; т. пл. означает температуру плавления.
Соединения именуются согласно общим принципам обозначений в данной области или по программе ChemDraw®, а коммерчески доступные соединения именуются по их торговым названиям.
Краткое описание фигур
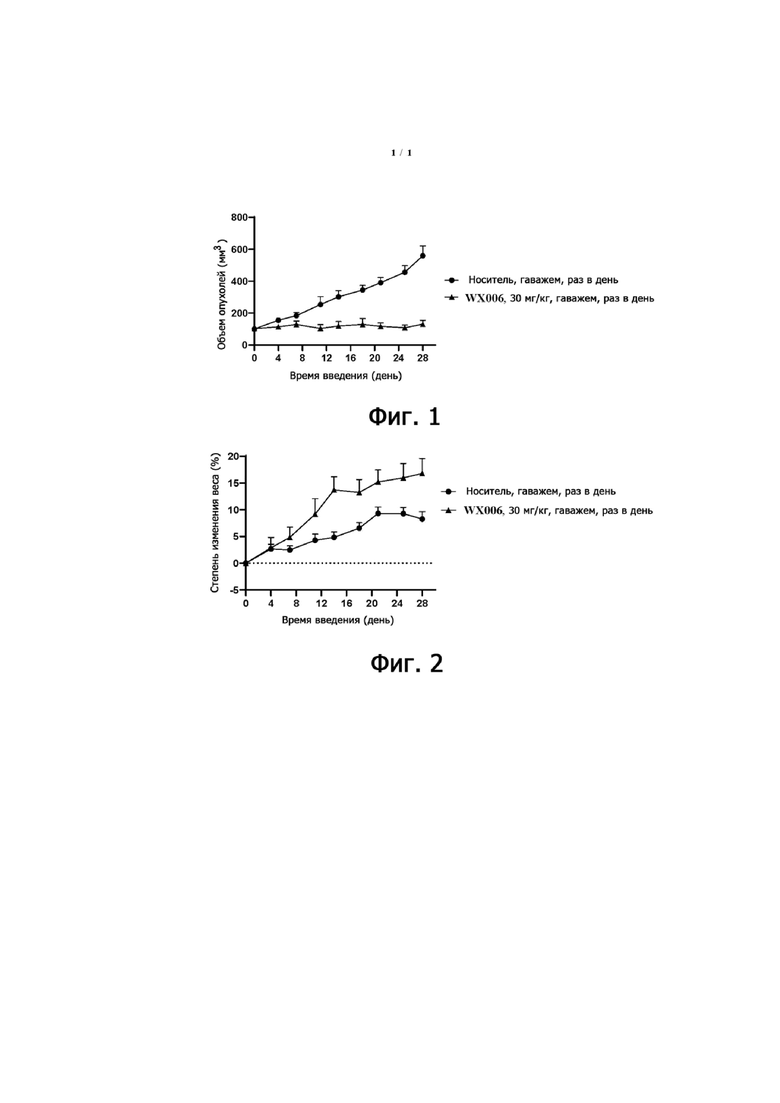
Фиг. 1. Кривая роста опухолей немелкоклеточного рака легких H358 человека на модели у животных после введения растворителя и WX006, соответственно.
Фиг. 2. Степень изменения веса (%) на модели у животных с немелкоклеточным раком легких H358 человека при введении.
Раскрытие сущности изобретения
Далее настоящее изобретение описано подробно на примерах. Однако это не означает, что данные примеры имеют какие-либо неблагоприятные ограничения для настоящего изобретения. Настоящее изобретение подробно описано здесь, и воплощения также изложены здесь. Специалистам в данной области должно быть ясно, что в приведенные здесь воплощения могут вноситься различные изменения и модификации, не выходящие за рамки изложенной здесь сущности и объема изобретения.
Контрольный пример 1. Фрагмент A-1
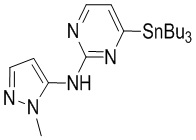
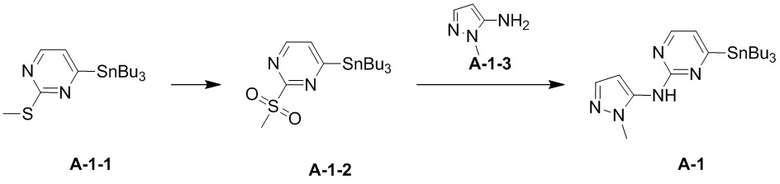
Стадия 1. Синтез соединения A-1-2
В сухую одногорлую колбу вносили раствор ацетата натрия (4,64 г, 56,60 ммоль, 5 экв.), моноперсульфата калия (13,92 г, 22,64 ммоль, 2 экв.) и воду (47 мл). Смесь охлаждали до 0°C. По каплям добавляли раствор A-1-1 (4,7 г, 11,32 ммоль, 1 экв.), растворитель тетрагидрофуран (47 мл) и метанол (47 мл) и перемешивали смесь при 0°C в течение 1 часа. Затем смесь перемешивали на масляной бане при 29°C в течение 15 часов. По завершении реакции реакционный раствор выливали в воду (200 мл) и экстрагировали водную фазу этилацетатом (3×50 мл). Органические фазы объединяли, а объединенную органическую фазу последовательно промывали насыщенным раствором NaCl (200 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат собирали и упаривали при пониженном давлении, получая остаток. Остаток очищали методом колоночной флеш-хроматографии, получая A-1-2. 1H-ЯМР (400 МГц, CDCl3) δ ppm 8,67 (d, J = 4,9 Hz, 1H), 7,64 (d, J = 4,9 Hz, 1H), 3,37 (s, 3H), 1,63-1,53 (m, 6H), 1,39-1,30 (m, 6H), 1,26-1,12 (m, 6H), 0,90 (t, J = 7,3 Hz, 9H).
Стадия 2. Синтез соединения A-1
В реакционную колбу вносили A-1-2 (3,9 г, 8,72 ммоль, 1 экв.), A-1-3 (1,02 г, 10,46 ммоль, 1,2 экв.) и тетрагидрофуран (117 мл). Заменяли атмосферу на газообразный азот, а затем по каплям добавляли гексаметилдисилазид лития (1 М, 18,31 мл, 2,1 экв.) при -35°C. Раствор этой смеси инкубировали при -35°C в течение 10 минут. По завершении реакции реакционный раствор гасили насыщенным водным раствором хлористого аммония (100 мл) и экстрагировали этилацетатом (2×100 мл) и дихлорметаном (100 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали досуха на роторном испарителе, получая неочищенный продукт. Неочищенный продукт очищали методом колоночной хроматографии, получая A-1. 1H-ЯМР (400 МГц, CDCl3) δ ppm 8.17 (d, J=4.85 Hz, 1H), 7.46 (d, J=1.76 Hz, 1H), 6.91 (d, J=4.63 Hz, 1H), 6.60 (s, 1H), 6.32 (d, J=1.98 Hz, 1H), 3.79 (s, 3H), 1.52-1.61 (m, 6H), 1.28-1.40 (m, 6H), 1.03-1.20 (m, 6H), 0.89 (t, J=7.28 Hz, 9H).
Пример 1
Схема синтеза
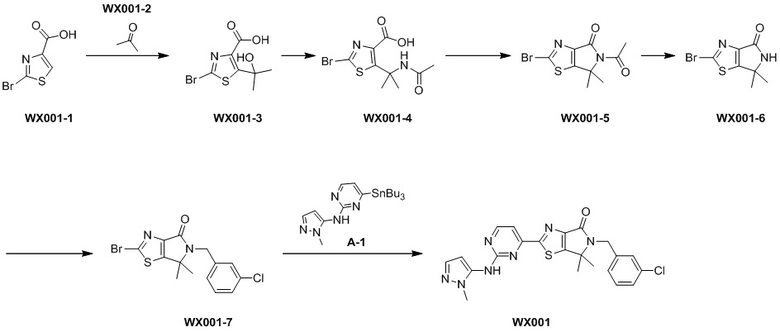
Стадия 1. Синтез WX001-3
В реакционную колбу при -78°C в атмосфере азота вносили раствор диизопропиламида лития (2М, 2,88 мл, 2,4 экв.) в тетрагидрофуране (5 мл), а затем медленно добавляли раствор WX001-1 (500 мг, 2,40 ммоль, 1 экв.) и тетраметилэтилендиамина (418,94 мг, 3,61 ммоль, 544,08 мкл, 1,5 экв.) в тетрагидрофуране (1 мл). Смесь инкубировали при -78°C в течение 0,5 часа, а затем добавляли WX001-2 (279,18 мг, 4,81 ммоль, 353,40 мкл, 2 экв.). Смесь инкубировали при -78°C еще в течение 2 часов. По завершении реакции реакционный раствор медленно выливали в 30 мл насыщенного водного раствора хлористого аммония при 0°C, доводили до рН 3-4 соляной кислотой (2 моль/л) и экстрагировали этилацетатом (3×20 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая WX001-3.
Стадия 2. Синтез WX001-4
В реакционную колбу вносили WX001-3 (250 мг, 939,45 мкмоль, 1 экв.) и ацетонитрил (8 мл). Заменяли атмосферу газообразным азотом, а затем добавляли концентрированную серную кислоту (110,57 мг, 1,13 ммоль, 60,09 мкл, 1,2 экв.) при 0°C. Раствор этой смеси инкубировали при 25°C в течение 16 часов. По завершении реакции реакционный раствор разбавляли 10 мл воды и экстрагировали этилацетатом (3×30 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (3×30мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая WX001-4.
Стадия 3. Синтез WX001-5
В сухую реакционную колбу вносили WX001-4 (200 мг, 651,12 мкмоль, 1 экв.) и уксусный ангидрид (2 мл). Заменяли атмосферу газообразным азотом, а затем инкубировали смесь при 90°C в течение 16 часов. По завершении реакции реакционный раствор упаривали при пониженном давлении с помощью масляного насоса при 45°C, получая WX001-5.
Стадия 4. Синтез WX001-6
В сухую реакционную колбу вносили WX001-5 (60 мг, 207,51 мкмоль, 1 экв.), соляную кислоту (2 М, 2 мл, 19,28 экв.) и этанол (2 мл). Смесь инкубировали при 50°C в течение 16 часов, а затем инкубировали при 70°C еще в течение 4 часов. По завершении реакции реакционный раствор упаривали при пониженном давлении с помощью водяного насоса при 45°C и экстрагировали этилацетатом (3×30 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (3×30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX001-6. 1H-ЯМР (400 МГц, DMSO-d6) δ ppm 8.85 (br s, 1H), 1.48 (s, 6H).
Стадия 5. Синтез WX001-7
В сухую реакционную колбу вносили WX001-6 (60 мг, 242,80 мкмоль, 1 экв.) и N,N-диметилформамид (1 мл). Заменяли атмосферу газообразным азотом, а затем добавляли гидрид натрия (14,57 мг, 364,21 мкмоль, чистота 60%, 1,5 экв.) при 0°C. Смесь инкубировали при 0°C в течение 0,5 часа, а затем добавляли 3-хлорбензилбромид (49,89 мг, 242,80 мкмоль, 31,78 мкл, 1 экв.). Реакционный раствор медленно нагревали до 25°C и инкубировали еще 2 часа. По завершении реакции в реакционный раствор добавляли 20 мл воды и экстрагировали смесь этилацетатом (3×10 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX001-7. 1H-ЯМР (400 МГц, CDCl3) δ ppm 7.26 (s, 1H), 7.17 (s, 3H), 4.62 (s, 2H), 1.37 (s, 6H).
Стадия 6. Синтез WX001
В реакционную колбу вносили WX001-7 (35 мг, 94,17 мкмоль, 1 экв.), A-1 (56,28 мг, 103,58 мкмоль, 1,1 экв.) и толуол (2 мл) и заменяли атмосферу газообразным азотом. Смесь нагревали до 125°C, а затем медленно добавляли тетракис(трифенилфосфин)палладий (21,76 мг, 18,83 мкмоль, 0,2 экв.). Смесь инкубировали при 125°C в течение 48 часов. По завершении реакции реакционный раствор упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX001.
Пример 2
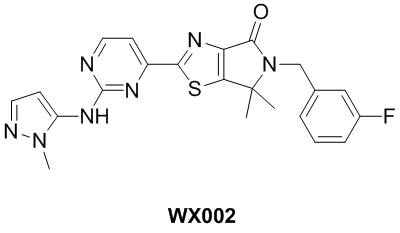
Схема синтеза
Стадия 1. Синтез WX002-2
В сухую реакционную колбу вносили WX001-6 (50 мг, 202,34 мкмоль, 1 экв.) и диметилформамид (1 мл). Заменяли атмосферу газообразным азотом, а затем добавляли гидрид натрия (12,14 мг, 303,51 мкмоль, чистота 60%, 1,5 экв.) при 0°C. Смесь инкубировали при 0°C в течение 0,5 часа, затем добавляли WX002-1 (38,25 мг, 202,34 мкмоль, 24,84 мкл, 1 экв.). Реакционный раствор медленно нагревали до 20°C и инкубировали еще 2 часа. По завершении реакции в реакционный раствор добавляли 20 мл воды и экстрагировали смесь этилацетатом (3×10 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX002-2. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 7.32-7.40 (m, 1H), 7.15-7.23 (m, 2H), 7.03-7.11 (m, 1H), 4.67 (s, 2H), 1.48 (d, J = 3.8 Hz, 6H).
Стадия 2. Синтез WX002
В реакционную колбу вносили WX002-2 (60 мг, 168,91 мкмоль, 1 экв.), A-1 (100,94 мг, 185,80 мкмоль, 1,1 экв.) и толуол (1 мл) и заменяли атмосферу газообразным азотом. Смесь нагревали до 125°C, а затем медленно добавляли тетракис(трифенилфосфин)палладий (39,04 мг, 33,78 мкмоль, 0,2 экв.). Смесь инкубировали при 125°C в течение 48 часов. По завершении реакции реакционный раствор упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX002.
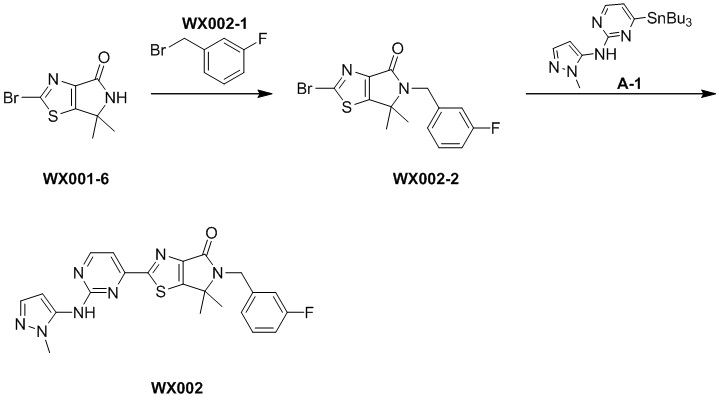
Пример 3
Схема синтеза
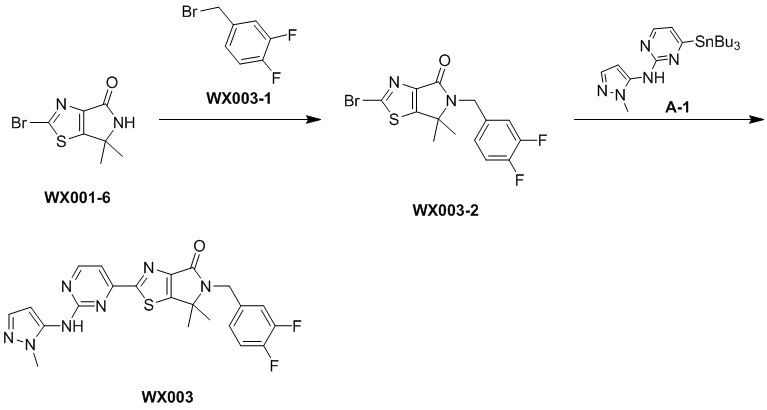
Стадия 1. Синтез WX003-2
В сухую реакционную колбу вносили WX001-6 (80 мг, 325,04 мкмоль, 1 экв.) и диметилформамид (4 мл). Заменяли атмосферу газообразным азотом, а затем добавляли гидрид натрия (19,50 мг, 487,56 мкмоль, чистота 60%, 1,5 экв.) при 0°C. Смесь инкубировали при 0°C в течение 0,5 часа, затем добавляли WX003-1 (67,29 мг, 325,04 мкмоль, 41,54 мкл, 1 экв.). Реакционный раствор медленно нагревали до 20°C и инкубировали еще 2 часа. По завершении реакции в реакционный раствор добавляли 20 мл воды и экстрагировали смесь этилацетатом (3×20 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом колоночной хроматографии, получая WX003-2. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 7.33-7.47 (m, 2H), 7.23 (m, 1H), 4.65 (s, 2H), 1.41-1.52 (m, 6H).
Стадия 2. Синтез WX003
В реакционную колбу вносили WX003-2 (70 мг, 187,56 мкмоль, 1 экв.), A-1 (112,09 мг, 206,32 мкмоль, 1,1 экв.) и толуол (2 мл) и заменяли атмосферу газообразным азотом. Смесь нагревали до 125°C, а затем медленно добавляли тетракис(трифенилфосфин)палладий (43,35 мг, 37,51 мкмоль, 0,2 экв.). Смесь инкубировали при 125°C в течение 48 часов. По завершении реакции реакционный раствор упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX003.
Пример 4
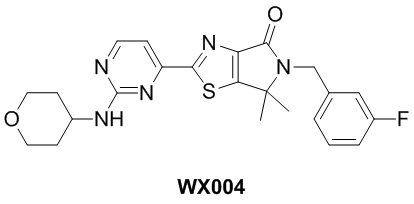
Схема синтеза
Стадия 1. Синтез WX004-1
В реакционную колбу вносили WX002-2 (70 мг, 197,06 мкмоль, 1 экв.), A-1-2 (113,45 мг, 216,76 мкмоль, 1,1 экв.) и толуол (2 мл) и заменяли атмосферу газообразным азотом. Смесь нагревали до 125°C, а затем медленно добавляли тетракис(трифенилфосфин)палладий (45,54 мг, 39,41 мкмоль, 0,2 экв.). Смесь инкубировали при 125°C в течение 48 часов. По завершении реакции реакционный раствор упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX004-1. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 9.26 (d, J = 5.1 Hz, 1H), 8.43 (d, J = 5.3 Hz, 1H), 7.33-7.42 (m, 1H), 7.16-7.28 (m, 2H), 7.08 (td, J = 8.6, 2.0 Hz, 1H), 4.73 (s, 2H), 3.50 (s, 3H), 1.55 (s, 6H).
Стадия 2. Синтез WX004
В сухую реакционную колбу вносили WX004-1 (50 мг, 115,61 мкмоль, 1 экв.) и WX004-2 (11,69 мг, 115,61 мкмоль, 1 экв.), а затем растворяли смесь в диметилсульфоксиде (1 мл). Смесь инкубировали с перемешиванием при 100°C в течение 14 часов. По завершении реакции реакционный раствор упаривали, а остаток очищали методом тонкослойной препаративной хроматографии, получая WX004.
Пример 5
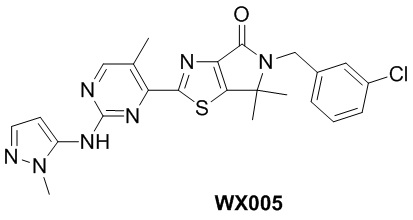
Схема синтеза
Стадия 1. Синтез WX005-2
В реакционную колбу вносили WX001-7 (530 мг, 1,43 ммоль, 1 экв.), хлорид цинка (0,7 М, 1,83 мл, 0,9 экв.) и тетрагидрофуран (3,5 мл). Заменяли атмосферу газообразным азотом, а затем добавляли н-бутиллитий (2,5 М, 855,58 мкл, 1,5 экв.) при -25°C. Раствор этой смеси инкубировали при 20°C в течение 1 часа. По каплям при -25°C добавляли раствор WX005-1 (379,45 мг, 1,43 ммоль, 1 экв.), трис(дибензилиденацетон)дипалладия (65,29 мг, 71,30 мкмоль, 0,05 экв.) и три(2-фурил)фосфина (33,11 мг, 142,60 мкмоль, 0,1 экв.) в тетрагидрофуране (0,5 мл) и инкубировали смесь при 20°C в течение 10 часов. Добавляли еще тетракис(трифенилфосфин)палладий (82,5 мг, 71,5 мкмоль, 0,05 экв.) и инкубировали смесь при 65°C в течение 20 часов. По завершении реакции реакционный раствор гасили метанолом и упаривали, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии, получая WX005-2. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 8.75 (s, 1H), 7.46 (s, 1H), 7.26-7.41 (m, 3H), 4.72 (s, 2H), 2.65 (s, 3H), 2.59 (s, 3H), 1.53 (s, 6H).
Стадия 2. Синтез WX005-3
В реакционную колбу вносили WX005-2 (140 мг, 324,85 мкмоль, 1 экв.) и дихлорметан (6 мл). Заменяли атмосферу газообразным азотом, а затем добавляли м-хлорпероксибензойную кислоту (210,22 мг, 974,54 мкмоль, чистота 80%, 3 экв.) при 0°C. Раствор этой смеси медленно нагревали до 20°C и инкубировали в течение 10 часов. По завершении реакции реакционный раствор гасили насыщенным раствором сульфита натрия (15 мл) и насыщенным водным раствором бикарбоната натрия (15 мл) до щелочного значения pH, пока не цвет индикаторной бумаги с KI больше не менялся, и экстрагировали смесь дихлорметаном (3×3 мл). Органическую фазу упаривали досуха при пониженном давлении с помощью водяного насоса при 45°C, получая коричневое твердое вещество. Затем коричневое вещество растирали с этилацетатом (3 мл), получая WX005-3. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 9.18 (s, 1H), 7.47 (s, 1H), 7.30-7.39 (m, 3H), 4.73 (s, 2H), 3.48 (s, 3H), 2.83 (s, 3H), 1.55 (s, 6H).
Стадия 3. Синтез WX005
В реакционную колбу вносили A-1-3 (15,73 мг, 162,00 мкмоль, 1,5 экв.) и N,N-диметилформамид (1 мл). Заменяли атмосферу газообразным азотом, а затем добавляли гидрид натрия (6,48 мг, 162,00 мкмоль, чистота 60%, 1,5 экв.) при 0°C. Через 0,5 часа добавляли раствор WX005-3 (50 мг, 108,00 мкмоль, 1 экв.) в N,N-диметилформамиде (1 мл) и инкубировали эту смесь при 20°C в течение 10 часов. По завершении реакции реакционный раствор гасили насыщенным водным раствором хлористого аммония (5 мл) и экстрагировали дихлорметаном (3×1 мл). Органическую фазу упаривали досуха при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной хроматографии, получая WX005.
Пример 6
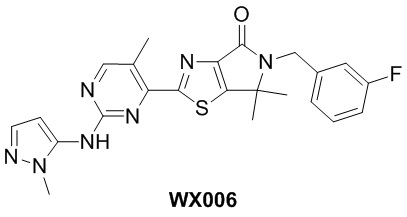
Схема синтеза
Стадия 1. Синтез WX006-1
В сухую реакционную колбу вносили WX002-2 (0,456 г, 1,28 ммоль, 1 экв.), хлорид цинка (0,7 М, 1,65 мл, 0,9 экв.) и тетрагидрофуран (10,5 мл). Заменяли атмосферу газообразным азотом, а затем реакционную систему охлаждали до -25°C и добавляли н-бутиллитий (2,5 М, 770,22 мкл, 1,5 экв.). Раствор этой смеси инкубировали с перемешиванием при 20°C в течение 1 часа. Добавляли раствор WX005-1 (341,59 мг, 1,28 ммоль, 1 экв.) и тетракис(трифенилфосфин)палладия (74,17 мг, 64,18 мкмоль, 0,05 экв.) в тетрагидрофуране (1,5 мл) при -25°C. Смесь инкубировали с перемешиванием при 60°C в течение 12 часов. По завершении реакции реакционный раствор гасили 3 мл метанола и упаривали досуха на роторном испарителе, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной хроматографии на покрытой силикагелем пластинке, получая WX006-1.
Стадия 2. Синтез WX006-2
В сухую реакционную колбу вносили WX006-1 (210 мг, 506,61 мкмоль, 1 экв.) и дихлорметан (5 мл). Реакционную систему охлаждали до 0°C и добавляли м-хлорпероксибензойную кислоту (327,85 мг, 1,52 ммоль, чистота 80%, 3 экв.). Смесь медленно нагревали до комнатной температуры (20°C) и инкубировали с перемешиванием в течение 12 часов. По завершении реакции реакционный раствор доводили до рН около 8 водным раствором бикарбоната натрия (5 мл). Добавляли насыщенный раствор сульфита натрия до тех пор, пока индикаторная бумажка с крахмалом и KI не посинеет. Раствор этой смеси экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (2×10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной хроматографии на покрытой силикагелем пластинке, получая WX006-2.
Стадия 3. Синтез WX006
В сухую реакционную колбу вносили A-1-3 (26,10 мг, 268,75 мкмоль, 1,2 экв.) и N,N-диметилформамид (3 мл). Реакционную систему охлаждали до 0°C. Затем добавляли гидрид натрия (13,44 мг, 335,93 мкмоль, чистота 60%, 1,5 экв.). Смесь инкубировали с перемешиванием при 0°C в течение 0,5 часа. Добавляли WX006-2 (100 мг, 223,96 мкмоль, 1 экв.) и инкубировали смесь с перемешиванием при 0°C в течение 0,5 часа. По завершении реакции реакционный раствор гасили водой (5 мл) и экстрагировали этилацетатом (3×5 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (4×5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной хроматографии на покрытой силикагелем пластинке, получая WX006.
Пример 7
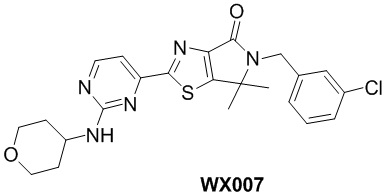
Схема синтеза
Стадия 1. Синтез WX007-1
В реакционную колбу вносили WX001-7 (50 мг, 134,52 мкмоль, 1 экв.), A-1-2 (77,45 мг, 147,98 мкмоль, 1,1 экв.) и толуол (2 мл) и заменяли атмосферу газообразным азотом. Смесь нагревали до 125°C, а затем медленно добавляли тетракис(трифенилфосфин)палладий (31,09 мг, 26,90 мкмоль, 0,2 экв.). Смесь инкубировали при 125°C в течение 48 часов. По завершении реакции реакционный раствор упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной хроматографии на покрытой силикагелем пластинке, получая WX007-1. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 9.26 (d, J = 5.1 Hz, 1H), 8.43 (d, J = 5.1 Hz, 1H), 7.47 (s, 1H), 7.28-7.39 (m, 3H), 4.73 (s, 2H), 3.50 (s, 3H), 1.55 (s, 6H).
Стадия 2. Синтез WX007
В сухую реакционную колбу вносили WX007-1 (30 мг, 66,82 мкмоль, 1 экв.) и WX004-2 (6,76 мг, 66,82 мкмоль, 1 экв.), а затем растворяли смесь в диметилсульфоксиде (1 мл). Смесь инкубировали с перемешиванием при 100°C в течение 16 часов. По завершении реакции реакционный раствор упаривали, а остаток очищали методом тонкослойной препаративной хроматографии, получая WX007.
Пример 8
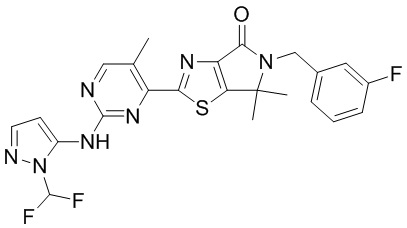
Схема синтеза
Стадия 1. Синтез WX008-2
В реакционную колбу вносили WX008-1 (9 г, 79,59 ммоль, 1 экв.), карбонат калия (13,20 г, 95,51 ммоль, 1,2 экв.) и N,N-диметилформамид (100 мл) и заменяли атмосферу газообразным азотом. Смесь нагревали до 120°C и перемешивали в течение 10 минут. Затем по частям добавляли 2-хлор-2,2-дифторацетат натрия (24,27 г, 159,19 ммоль, 2 экв.). Раствор этой смеси инкубировали при 120°C в течение 20 минут. По завершении реакции реакционный раствор разбавляли водой (800 мл) и экстрагировали этилацетатом (3×200 мл). Органическую фазу промывали насыщенным раствором NaCl (300 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса, а остаток очищали методом тонкослойной хроматографии на покрытой силикагелем пластинке, получая WX008-2. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 7.96-8.28 (m, 2H), 7.50 (d, J = 2.0 Hz, 1H).
Стадия 2. Синтез WX008-3
В реакционную колбу вносили палладий на угле (50 мг, 367,91 мкмоль, чистота 10%, 1 экв.) и метанол (1 мл). Заменяли атмосферу газообразным азотом, а затем добавляли WX008-2 (60 мг, 367,91 мкмоль, 1 экв.). Затем заменяли атмосферу газообразным водородом и инкубировали раствор этой смеси под давлением 15 psi в атмосфере водорода (741,67 мкг, 367,91 мкмоль, 1 экв.) при 25°C в течение 1 часа. По завершении реакции реакционный раствор фильтровали через целит, а фильтрат упаривали досуха при пониженном давлении с помощью водяного насоса, получая WX008-3. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 7.36-7.69 (m, 1H), 7.33 (s, 1H), 5.84 (br s, 2H), 5.31 (d, J = 1.3 Hz, 1H).
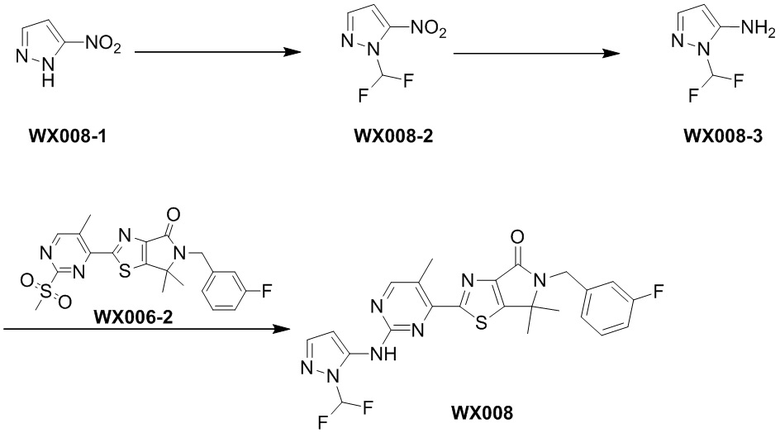
Стадия 3. Синтез WX008
В реакционную колбу вносили WX008-3 (17,88 мг, 134,37 мкмоль, 1,5 экв.), WX006-2 (40 мг, 89,58 мкмоль, 1 экв.), дихлорметан (0 ,5 мл) и тетрагидрофуран (0,5 мл). Заменяли атмосферу газообразным азотом и охлаждали смесь до 0°C. Затем медленно по каплям добавляли гексаметилдисилазид лития (1 М, 188,12 мкл, 2,1 экв.) и инкубировали раствор этой смеси при 0°C в течение 0,5 часа. По завершении реакции реакционный раствор упаривали, а остаток очищали методом тонкослойной препаративной хроматографии, получая WX008.
Пример 9
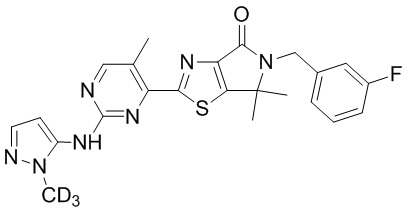
Схема синтеза
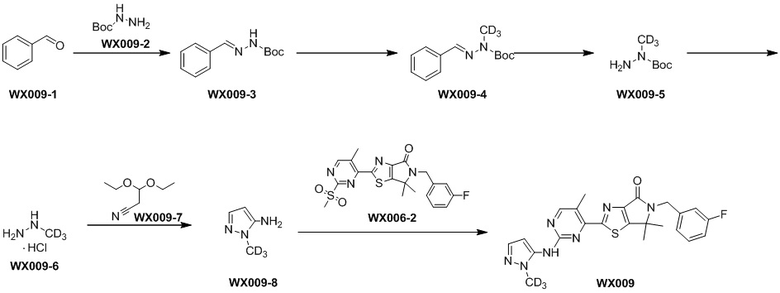
Стадия 1. Синтез WX009-3
В реакционную колбу вносили WX009-1 (8,03 г, 75,67 ммоль, 7,65 мл, 1 экв.), WX009-2 (10 г, 75,67 ммоль, 1 экв.) и тетрагидрофуран (100 мл). Заменяли атмосферу газообразным азотом и инкубировали раствор этой смеси при 25°C в течение 4 часов. По завершении реакции реакционный раствор упаривали досуха на роторном испарителе, получая WX009-3. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 10.86 (br s, 1H), 8.00 (s, 1H), 7.59 (br d, J = 6.6 Hz, 2H), 7.31-7.45 (m, 3H), 1.47 (s, 9H).
Стадия 2. Синтез WX009-4
В реакционную колбу вносили WX009-3 (10 г, 45,40 ммоль, 1 экв.), трет-бутоксид калия (6,11 г, 54,48 ммоль, 1,2 экв.) и тетрагидрофуран (160 мл). Заменяли атмосферу газообразным азотом, а затем медленно по каплям добавляли дейтерированный йодометан (7,90 г, 54,48 ммоль, 3,39 мл, 1,2 экв.). Раствор этой смеси инкубировали при 25°C в течение 16 часов. По завершении реакции реакционный раствор разбавляли водой (50 мл) и экстрагировали этилацетатом (3×100 мл). Органическую фазу промывали насыщенным раствором NaCl (100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса, получая WX009-4. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 7.81 (s, 1H), 7.64-7.72 (m, 2H), 7.32-7.46 (m, 3H), 1.50 (s, 9H).
Стадия 3. Синтез WX009-5
В реакционную колбу вносили влажный палладий на угле (2 г, чистота 10%) и метанол (100 мл). Заменяли атмосферу газообразным водородом, а затем добавляли WX009-4 (10,5 г, 44,25 ммоль, 1 экв.). Раствор этой смеси инкубировали в атмосфере водорода (89,19 мг, 44,25 ммоль, 1 экв.) под давлением 50 psi при 50°C в течение 48 часов. По завершении реакции реакционный раствор фильтровали через целит и промывали метанолом (2×20 мл). Фильтрат упаривали досуха при пониженном давлении с помощью водяного насоса, получая WX009-5. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 4.48 (s, 2H), 1.40 (s, 9H).
Стадия 4. Синтез WX009-6
В реакционную колбу вносили WX009-5 (6,6 г, 44,23 ммоль, 1 экв.) и раствор соляной кислоты в этилацетате (4 М, 33,18 мл, 3 экв.) и инкубировали раствор этой смеси при 25°C в течение 16 часов. Реакционный раствор упаривали досуха на роторном испарителе, получая WX009-6. 1H-ЯМР (400 МГц, DMSO-d6): δ ppm 4.95 (br s, 3H).
Стадия 5. Синтез WX009-8
В реакционную колбу вносили WX009-6 (1,5 г, 17,53 ммоль, 1 экв., HCl), WX009-7 (2,51 г, 17,53 ммоль, 2,63 мл, 1 экв.), уксусную кислоту (2,11 г, 35,07 ммоль, 2,01 мл, 2 экв.), сульфат магния (5,02 г, 41,73 ммоль, 2,38 экв.) и этанол (75 мл). Заменяли атмосферу газообразным азотом, а затем инкубировали раствор этой смеси при 90°C в течение 2 часов. По завершении реакции реакционный раствор разбавляли насыщенным водным раствором бикарбоната натрия (50 мл) и экстрагировали дихлорметаном (3×50 мл). Органическую фазу промывали насыщенным раствором NaCl (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая WX009-8. 1H-ЯМР (400 МГц, DMSO-d6): δ (ppm) 6.98 (d, J = 1.6 Hz, 1H), 5.24 (d, J = 1.8 Hz, 1H), 5.09 (br s, 2H).
Стадия 6. Синтез WX009
В реакционную колбу вносили WX009-8 (13,46 мг, 134,37 мкмоль, 1,5 экв.), WX006-2 (40 мг, 89,58 мкмоль, 1 экв.), дихлорметан (0,5 мл) и тетрагидрофуран (0,5 мл). Заменяли атмосферу газообразным азотом и охлаждали смесь до 0°C. Затем медленно по каплям добавляли гексаметилдисилазид лития (1 М, 188,12 мкл, 2,1 экв.) и инкубировали раствор этой смеси при 0°C в течение 0,5 часа. По завершении реакции реакционный раствор разбавляли насыщенным водным раствором хлористого аммония (5 мл) и экстрагировали дихлорметаном (3×5 мл). Органическую фазу промывали насыщенным раствором NaCl (5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении с помощью водяного насоса при 45°C, а остаток очищали методом тонкослойной препаративной хроматографии, получая WX009.
Пример 10
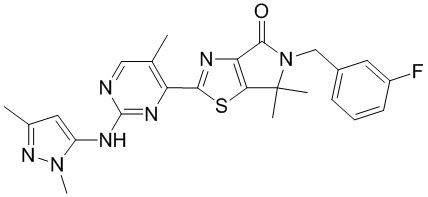
Схема синтеза
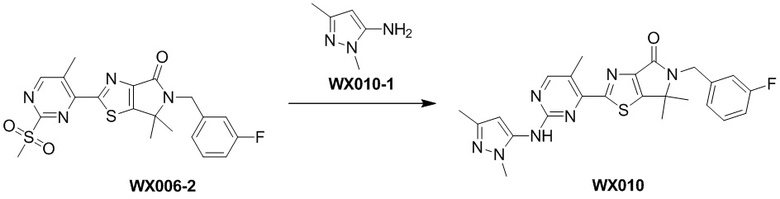
Стадия 1. Синтез WX010
В сухую реакционную колбу вносили WX006-2 (30 мг, 67,19 мкмоль, 1 экв.) и WX010-1 (15,68 мг, 141,09 мкмоль, 2,1 экв.) в смесь тетрагидрофурана (1,5 мл) и дихлорметана (1,5 мл). Заменяли атмосферу газообразным азотом и охлаждали смесь до 0°C. Добавляли гексаметилдисилазид лития (1 М, 134,37 мкл, 2 экв.), перемешивали смесь при 0°C в течение 0,5 часа, нагревали до 25°C и перемешивали еще 1,5 часа. По завершении реакции реакционный раствор разбавляли водой (10 мл) и экстрагировали дихлорметаном (3×10 мл). Проводили разделение слоев. Затем отбирали органическую фазу, которую последовательно промывали насыщенным раствором NaCl (3×10 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом тонкослойной препаративной хроматографии, получая WX010.
Пример 11
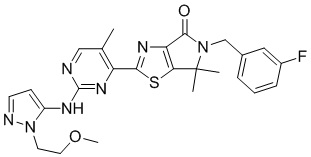
Схема синтеза
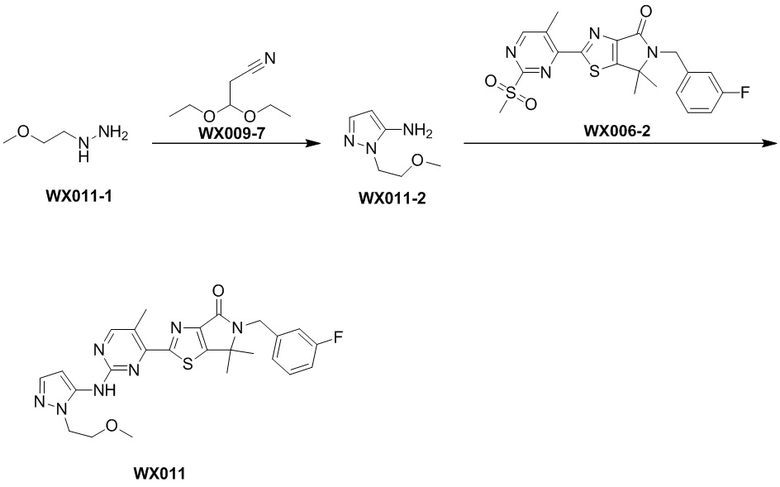
Стадия 1. Синтез WX011-2
В сухую реакционную колбу вносили WX009-7 (226,22 мг, 1,58 ммоль, 237,13 мкл, 1 экв.), сульфат магния (452,63 мг, 3,76 ммоль, 2,38 экв.), уксусную кислоту (189,75 мг, 3,16 ммоль, 180,72 мкл, 2 экв.) и этанол (2,5 мл). Заменяли атмосферу газообразным азотом и инкубировали смесь при 25°C в течение 1 часа. Затем добавляли WX011-1 (200 мг, 1,58 ммоль, 1 экв., HCl) и инкубировали смесь при 80°C в течение 2 часов и при 90°C еще 12 часов. По завершении реакции реакционный раствор выливали в 3 мл насыщенного водного раствора бикарбоната натрия. Смесь экстрагировали дихлорметаном (2× 3 мл). Органические фазы объединяли, промывали 3 мл насыщенного раствора NaCl, сушили над безводным сульфатом натрия и упаривали при пониженном давлении, получая неочищенный продукт. Неочищенный продукт очищали методом тонкослойной хроматографии на покрытой силикагелем пластинке, получая WX011-2. 1H-ЯМР (400 МГц, CH3Cl-d): δ (ppm) 7.25 (d, J = 1.8 Hz, 1H), 5.50 (d, J = 2.0 Hz, 1H), 4.15-4.22 (m, 2H), 3.83-4.11 (m, 2H), 3.68-3.73 (m, 2H), 3.34 (s, 3H).
Стадия 2. Синтез WX011
В сухую реакционную колбу вносили WX006-2 (40 мг, 89,58 мкмоль, 1 экв.) и WX011-2 (26,56 мг, 188,12 мкмоль, 2,1 экв.) в смесь тетрагидрофурана (2 мл) и дихлорметана (2 мл). Заменяли атмосферу газообразным азотом и охлаждали смесь до 0°C. Добавляли гексаметилдисилазид лития (1 М, 179,16 мкл, 2 экв.), перемешивали смесь при 0°C в течение 0,5 часа, нагревали до 25°C и перемешивали еще 1 час. По завершении реакции реакционный раствор разбавляли водой (5 мл) и экстрагировали дихлорметаном (3×5 мл). Проводили разделение слоев. Затем отбирали органическую фазу, которую последовательно промывали насыщенным раствором NaCl (3×5 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом тонкослойной препаративной хроматографии, получая WX011.
Пример 12
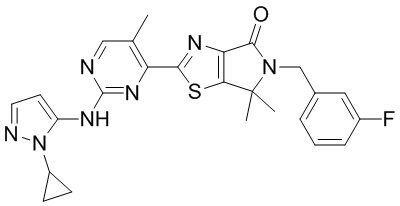
Схема синтеза
Стадия 1. Синтез WX012
В реакционную колбу вносили WX006-2 (40 мг, 89,58 мкмоль, 1 экв.), WX012-1 (16,55 мг, 134,37 мкмоль, 1,5 экв.), дихлорметан (1 мл) и тетрагидрофуран (1 мл). Заменяли атмосферу газообразным азотом и охлаждали смесь до 0°C. Медленно по каплям добавляли гексаметилдисилазид лития (1 М, 188,12 мкл, 2,1 экв.) и инкубировали раствор этой смеси при 0°C в течение 0,5 часа. По завершении реакции реакционный раствор упаривали, а остаток очищали методом тонкослойной препаративной хроматографии, получая WX012.
Пример 13
Схема синтеза

Стадия 1. Синтез WX013-2
Растворяли WX013-1 (800 мг, 4,70 ммоль, 1 экв.) и гидроксид лития моногидрат (986,42 мг, 23,51 ммоль, 5 экв.) в смеси воды (8 мл) и тетрагидрофурана (8 мл). Смесь перемешивали при 25°C в течение 2 часов. По завершении реакции реакционный раствор непосредственно упаривали досуха на роторном испарителе, а затем экстрагировали водой (10 мл) и дихлорметаном (3×10 мл). Отбирали органическую фазу, промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали досуха на роторном испарителе, получая WX013-2.
Стадия 2. Синтез WX013-3
Растворяли WX013-2 (500 мг, 3,20 ммоль, 1 экв.), дифенилфосфорилазид (889,00 мг, 3,23 ммоль, 700 мкл, 1,01 экв.) и триэтиламин (1,45 г, 14,37 ммоль, 2 мл, 4,49 экв.) в трет-бутаноле (10 мл). Трижды заменяли атмосферу газообразным азотом, а затем перемешивали смесь при 85°C в течение 16 часов. По завершении реакции реакционный раствор упаривали досуха на роторном испарителе, получая неочищенный продукт. Неочищенный продукт очищали методом колоночной хроматографии, получая WX013-3. 1H-ЯМР (400 МГц, CD3Cl): δ ppm 1.50 (s, 9H), 3.58 (s, 3H), 3.84 (s, 3H), 5.61 (br s, 1H).
Стадия 3. Синтез WX013-4
В реакционную колбу вносили WX013-3 (520 мг, 2,29 ммоль, 1 экв.) и раствор соляной кислоты в этилацетате (4 М, 5 мл, 8,74 экв.) и перемешивали смесь при 25°C в течение 2 часов. По завершении реакции реакционный раствор экстрагировали водой (10 мл). Отбирали водную фазу, а затем упаривали досуха на роторном испарителе, получая неочищенный продукт. Неочищенный продукт очищали методом колоночной хроматографии, получая WX013-4. 1H-ЯМР (400 МГц, CD3Cl): δ ppm 3.53 (s, 3H), 3.82 (s, 3H), 5.01 (s, 1H).
Стадия 4. Синтез WX013
В сухую реакционную колбу вносили WX006-2 (50 мг, 111,98 мкмоль, 1 экв.), WX013-4 (29,90 мг, 235,15 мкмоль, 2,1 экв.), дихлорметан (1 мл) и тетрагидрофуран (1 мл). Заменяли атмосферу газообразным азотом и охлаждали смесь до 0°C. По каплям добавляли гексаметилдисилазид лития (1 М, 223,96 мкл, 2 экв.). Смесь инкубировали при 0°C в течение 0,5 часа и при 25°C еще 1 час. По завершении реакции реакционный раствор гасили 10 мл воды и экстрагировали 20 мл дихлорметана. Проводили разделение слоев. Отбирали органическую фазу, а водную фазу экстрагировали дихлорметаном (3×20 мл). Органические фазы объединяли и последовательно промывали насыщенным раствором NaCl (3×20 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. По завершении упаривания остаток очищали методом тонкослойной препаративной хроматографии, получая WX013.
В таблице 1 представлены данные спектров 1H-ЯМР и масс-спектров из каждого примера.
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+1]+
[M+1]+
[M+1]+
[M+1]+
[M+1]+
[M+1]+
[M+1]+
[M+1]+
[M+1]+
Тест-пример 1. Анализ активности киназы in vitro
1. Цель исследования
Измерение способности соединений ингибировать активность киназы ERK2.
2. Буфер для анализа
20 мМ Hepes (рН 7,5), 10 мМ MgCl2, 1 мМ этиленбис(оксиэтиленнитрило)тетрауксусная кислота (EGTA), 0,02% Brij35, 0,02 мг/мл бычьего сывороточного альбумина (BSA), 0,1 мМ Na3VO4, 2 мМ дитиотреитол (DTT), 1% DMSO.
3. Обработка соединений
Исследуемые соединения растворяли в 100% DMSO, получая маточные растворы в определенной концентрации. Делали серийные разведения соединений в растворе DMSO с помощью смарт-пипетки Integra Viaflo Assist.
4. Методика исследования
1) Готовили субстрат MBP в свежеприготовленном буфере для реакции.
2) В данный раствор MBP добавляли киназу ERK2 и осторожно перемешивали.
3) В систему для киназной реакции вносили соединения, растворенные в 100% DMSO, по ультразвуковой технологии (Echo550; диапазон: нанолитры), и инкубировали смеси при комнатной температуре в течение 20 минут.
4) В реакционную систему добавляли 33P-АТФ (удельная концентрация: 10 мкКи/мкл) и сразу же запускали реакцию.
5) Инкубировали смеси при комнатной температуре в течение 2 часов.
6) Определяли уровень радиоактивности методом связывания на фильтре.
7) Рассчитывали активность киназы ERK2 как отношение остающейся в исследуемом образце активности киназы к активности киназы в контрольной группе (DMSO). Строили кривые с помощью Prism (программное обеспечение GraphPad) и рассчитывали значения IC50.
5. Результаты исследования представлены в таблице 2.
Вывод. Соединения по настоящему изобретению проявляют превосходную активность ингибирования киназы ERK2.
Тест-пример 2. Анализ ингибирования пролиферации клеток in vitro
1. Цель исследования
Измерение способности соединений ингибировать пролиферацию раковых клеток НТ29.
2. Обработка соединений
Исследуемые соединения растворяли в 100% DMSO, получая 10 мМ маточные растворы.
3. Методика и стадии исследования
1) Включали УФ-освещение в кабине биологической безопасности и начинали отсчет 30 минут.
2) На водяной бане подогревали среду RPMI 1640 и трипсин при 37°C.
3) По завершении УФ-облучения открывали кабину биологической безопасности. Подогретую среду, трипсин и фосфатно-солевой буфер (PBS) и т.д. протирали спиртом и помещали в кабину биологической безопасности.
4) Извлекали клетки НТ29 из инкубатора и удаляли старую среду в кабине биологической безопасности. Добавляли 10 мл PBS. Смесь осторожно встряхивали, а затем удаляли PBS.
5) Добавляли 1,5 мл подогретого 0,25% трипсина. Встряхивали горизонтально сосуд для культивирования с тем, чтобы трипсин равномерно покрывал клетки на дне, и помещали в инкубатор на 2 минуты.
6) Останавливали расщепление клеток добавлением полной среды и доводили суспензию клеток до гомогенности пипеткой и проводили подсчет клеток.
7) По результатам подсчета клеток доводили плотность клеточной суспензии до 1500 клеток на лунку и высеивали суспензию клеток по 50 мкл на лунку.
8) Делали серийные разведения маточных растворов соединений в растворе DMSO и вносили соединения на планшет с помощью Tecan.
9) Уравновешивали планшет с клетками и добавленными соединениями и CellTiterGlo при комнатной температуре, а затем в каждую лунку добавляли 25 мкл CellTiterGlo. Планшет с клетками встряхивали 1-2 минуты, а затем оставляли на 10 минут. Затем определяли значения сигналов. Данные анализировали с помощью XL-Fit и рассчитывали IC50 для каждого соединения.
4. Результаты исследования представлены в таблице 3.
Вывод. Соединения по настоящему изобретению проявляют превосходную активность ингибирования пролиферации клеток HT29.
Тест-пример 3. Исследование DMPK in vivo
Исследование DMPK in vivo на мышах.
1. Цель исследования
Определение концентрации соединений в крови и оценка фармакокинетического поведения после однократного введения, используя самок мышей BALB/c в качестве исследуемых животных.
2. Процедура исследования
Отбирали 8 здоровых взрослых самок мышей BALB/c, причем 4 мыши были в группе внутривенного введения и 4 мыши - в группе перорального введения. Носителем в группе внутривенного введения был 5% DMSO + 95% (20% HP-β-CD). Исследуемые соединения смешивали с соответствующим количеством носителя для внутривенной инъекции, обрабатывали на вибромешалке и ультразвуком, получая прозрачный раствор в 0,5 мг/мл. Прозрачный раствор фильтровали через микропористую мембрану и готовили к использованию. Носителем в группе перорального введения был 5% DMSO + 95% (20% HP-β-CD). Исследуемые соединения смешивали с носителем, обрабатывали на вибромешалке и ультразвуком, получая раствор в 0,3 мг/мл. Мышам вводили по 1 мг/кг внутривенно или 3 мг/кг перорально, а затем в течение определенного времени собирали цельную кровь. Выделяли плазму. Концентрацию препаратов анализировали методом LC-MS/MS, а фармакокинетические параметры рассчитывали с помощью программы Phoenix WinNonlin (Pharsight, США).
Примечания. DMSO: диметилсульфоксид; HP-β-CD: гидроксипропил-β-циклодекстрин.
3. Результаты исследования представлены в таблице 4.
(нМ)
(нМ·ч/mpk)
(л/кг)
(мл/мин/кг)
(ч)
Примечания. Cmax - максимальная концентрация; F% - пероральная биодоступность; DNAUC = AUCPO/доза, где AUCPO - AUC при пероральном введении, а доза - доза препарата; Vdss - объем распределения; Cl - скорость клиренса; T½ - период полувыведения; а NA означает, что исследование не проводилось.
Вывод. Соединения по настоящему изобретению проявляют превосходную пероральную экспозицию и биодоступность.
Тест-пример 4. Анализ эффективности in vivo на модели H358 у мышей
1. Цель исследования
Оценка противоопухолевого действия WX006 на модели подкожных ксенотрансплантатов клеток H358 немелкоклеточного рака легких человека у мышей nude.
2. Животные для исследования
Вид: мышь; линия: мыши BALB/c nude; возраст: 6-7 недель; пол: самки; вес: 20 грамм; поставщик: Shanghai Sippe-Bk Lab Animal Co., Ltd.; сертификат на животных № 20180006017149.
3. Условия содержания
Животных содержали в клетках IVC (независимая система подачи воздуха, постоянная температура и влажность) (по 4 животных в клетке) в помещении вивария для животных класса SPF при температуре 20-26°C и влажности 40-70%. Клетки были изготовлены из поликарбоната и имели объем 300 мм × 180 мм × 150 мм. Материал подстилки из сердцевины кукурузных початков, его меняли раз в неделю. Исследуемые животные имели свободный доступ к пище (стерилизованный облучением, сухой корм в гранулах) на всем протяжении исследования. Животные имели свободный доступ к питьевой стерилизованной воде. Идентификация клеток: в карточке информации о животных для каждой клетки должно быть указано количество, пол, линия, дата поступления животных в клетку, номер исследования, схема введения, группа и дата начала исследования. Исследуемых животных идентифицировали по ушным биркам.
4. Процедура исследования
1) Клетки и их культивирование. Клетки H358 немелкоклеточного рака легких человека культивировали в монослое in vitro. Условия культивирования: среда 1640 плюс 10% фетальной телячьей сыворотки и инкубатор на 37°C с 5% CO2. Обычно для пересева проводилась обработка клеток трипсином с ЭДТА три раза в неделю. Когда конфлюэнтность клеток достигала 80%-90% и их количество соответствовало потребности, клетки собирали, подсчитывали и высеивали.
2) Инокуляция опухолевой ткани и разбивка на группы. Инокулировали подкожно по 0,1 мл (5×105) клеток H358 в правую подмышечную ямку каждой мыши. Когда средний объем опухолей достигал 100 мм3, животных случайным образом разбивали на 2 группы и начинали введение. Схема распределения по группам и введения при исследовании представлена в таблице 5.
3) Ежедневное наблюдение исследуемых животных. Разработка данной методики исследования и её модификаций проводилась с одобрения Институтского комитета по содержанию и использованию животных (IACUC). Использование и благосостояние исследуемых животных контролировалось в соответствии с правилами Ассоциации по оценке и аккредитации лабораторий по содержанию животных (AAALAC). Животных ежедневно обследовали на предмет здоровья и смертности. Плановые обследования включали отслеживание роста опухолей и влияния приема препаратов на повседневное поведение животных типа поведенческой активности, приема пищи и воды (только визуальный осмотр), изменения веса (измеряли вес два раза в неделю), внешних признаков или других аномалий. Отмечали гибель животных и побочные эффекты в каждой группе, исходя из количества животных в каждой группе.
4) Рецептуры исследуемых соединений. Группа носителя: 5% DMSO + 95% (20% HP-β-CD). Группа исследуемого соединения: делали навеску исследуемого соединения в рецептурном флаконе. Добавляли соответствующий объем DMSO, а затем обрабатывали смесь на вибромешалке до получения прозрачного раствора. Добавляли соответствующий объем 20% HP-β-CD, а затем обрабатывали смесь на вибромешалке до получения однородной суспензии. Соединения готовили через каждые три дня.
5) Измерение опухолей и показателей при исследовании. Измеряли диаметр опухолей два раза в неделю с помощью штангенциркуля. Рассчитывали объем опухолей по формуле: TV=1/2×a×b2, где a и b -длинный и короткий диаметр опухоли, соответственно. Определяли эффективность ингибирования опухолей соединениями по TGI (%).TGI (%) отражает степень ингибирования роста опухоли. TGI (%) рассчитывали следующим образом: TGI (%) = {[1 - (средний объем опухолей под конец введения в опытной группе - средний объем опухолей в начале введения в той же опытной группе)]/(средний объем опухолей под конец обработки в контрольной группе растворителя - средний объем опухолей в начале обработки в контрольной группе растворителя)}×100%.
5. Результаты исследования
1) Как видно из таблицы 6 и фиг. 1, на модели подкожных ксенотрансплантатов клеток H358 немелкоклеточного рака легких человека у мышей nude, при пероральном введении до 28-го дня, WX006 в дозе 30 мг/кг оказывал значительное ингибирующее действие на рост опухолей со значением TGI = 94%.
2) В качестве показателя для косвенного определения токсичности препарата использовали вес тела исследуемых животных. Как видно из фиг. 2, при введении до 28-го дня, у всех животных в контрольной группе растворителя и в группе WX006 не было существенного снижения веса тела и не отмечалось болезненности или смертности.
6. Заключение. WX006 может значительно ингибировать рост опухолей при вводимой дозе. При введении не наблюдается существенного снижения веса тела животных, переносимость хорошая.
| название | год | авторы | номер документа |
|---|---|---|---|
| Спиросоединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2800042C1 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ДЕЙСТВУЮЩИЕ НА БЕЛКИ CRBN | 2019 |
|
RU2801068C2 |
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| ГЛУТАРИМИДНОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ КОЛЬЦОМ, КОНДЕНСИРОВАННЫМ С ФУРАНОМ | 2022 |
|
RU2836921C1 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
| СОЕДИНЕНИЯ АЛЬФА-АМИНОАМИДНЫХ ПРОИЗВОДНЫХ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2015 |
|
RU2673622C2 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
Изобретение относится к тиазололактамным соединениям формулы (III), где значения R1-R5, n, m определены в формуле изобретения, и их применению при получении лекарственных средств для лечения заболеваний, связанных с ERK. 3 н. и 13 з.п. ф-лы, 2 ил., 6 табл., 13 пр.

1. Соединение, представленное формулой (III), или его фармацевтически приемлемая соль:
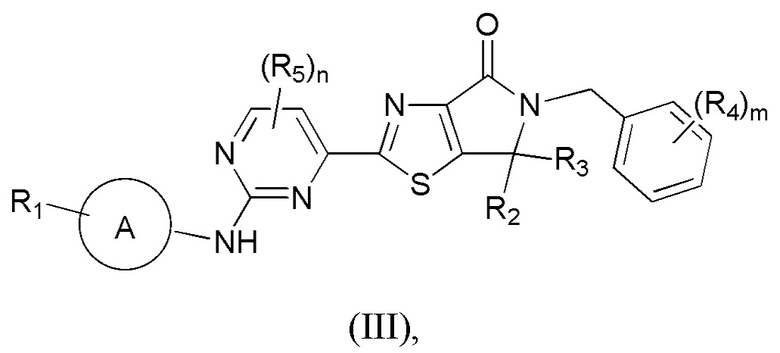
где R1 означает H, C1-3-алкил или C3-5-циклоалкил, причем C1-3-алкил или C3-5-циклоалкил необязательно замещен 1, 2 или 3 Ra;
R2 и R3 каждый независимо означает C1-3-алкил;
R4 выбран из F и Cl;
R5 выбран из C1-3-алкила;
m равно 0, 1 или 2;
n равно 0, 1 или 2;
кольцо A означает пиразолил или тетрагидропиранил, причем пиразолил или тетрагидропиранил необязательно замещен 1, 2 или 3 Rd;
Ra каждый независимо означает D, F или OCH3;
Rd выбран из CH3 или OCH3.
2. Соединение или его фармацевтически приемлемая соль по п. 1, при этом R1 означает H, CH3 или циклопропил, причем CH3 или циклопропил необязательно замещен 1, 2 или 3 Ra.
3. Соединение или его фармацевтически приемлемая соль по п. 2, при этом R1 означает H, CH3, CHF2, CD3, CH2CH2OCH3 или циклопропил.
4. Соединение или его фармацевтически приемлемая соль по п. 1, при этом R2 и R3 каждый независимо выбран из CH3 или CH2CH3.
5. Соединение или его фармацевтически приемлемая соль по п. 4, при этом R2 и R3 каждый независимо означает CH3.
6. Соединение или его фармацевтически приемлемая соль по п. 1, при этом R4 означает F.
7. Соединение или его фармацевтически приемлемая соль по п. 1, при этом R4 означает Cl.
8. Соединение или его фармацевтически приемлемая соль по п. 1, при этом R5 означает CH3.
9. Соединение или его фармацевтически приемлемая соль по п. 1, при этом кольцо A означает 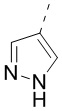 ,
, 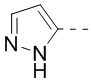 или
или 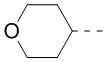 , причем , или необязательно замещено 1, 2 или 3 Rd.
, причем , или необязательно замещено 1, 2 или 3 Rd.
10. Соединение или его фармацевтически приемлемая соль по п. 9, при этом кольцо A означает , , 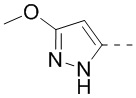 или
или 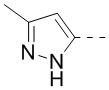 .
.
11. Соединение или его фармацевтически приемлемая соль по п. 1, при этом структурная группировка  означает
означает 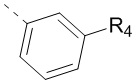 ,
, 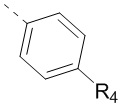 или
или 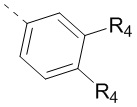 .
.
12. Соединение или его фармацевтически приемлемая соль по п. 11, при этом структурная группировка означает 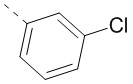 ,
, 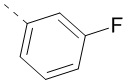 или
или 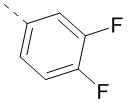 .
.
13. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-8, при этом соединение выбрано из:
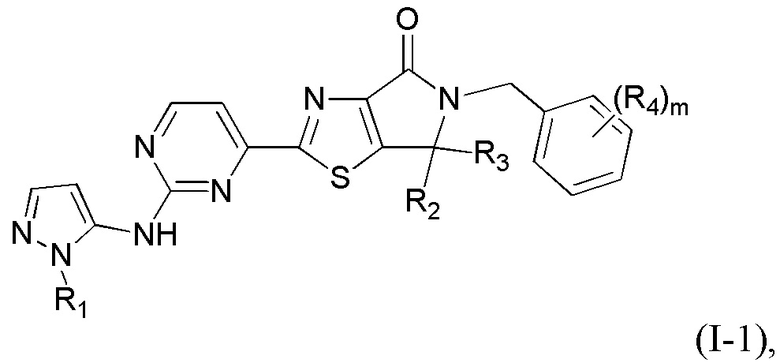
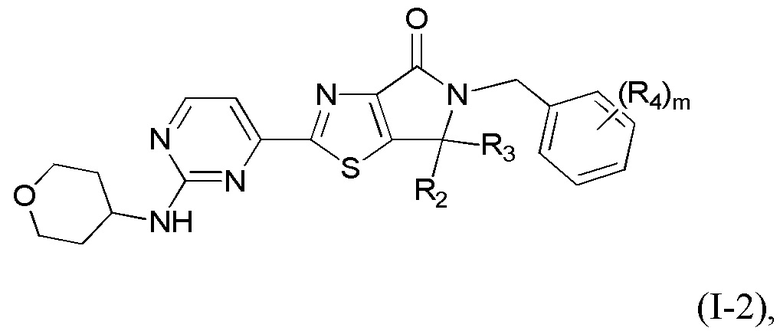
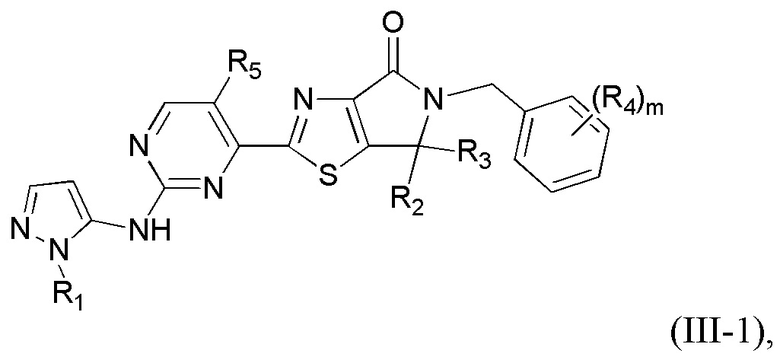
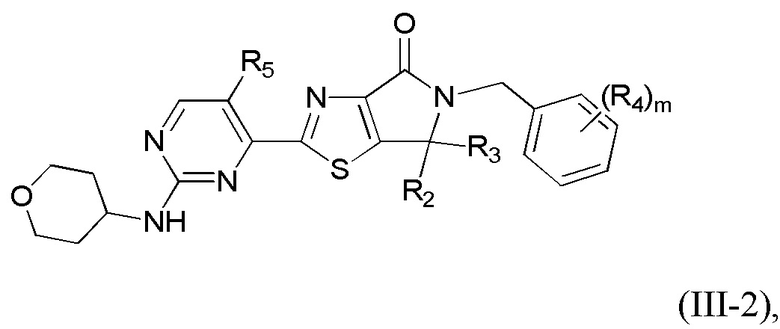
где R1 уже определен в любом из пп. 1-3; R2 и R3 определены в любом из пп. 1, 4, 5; R4 определен в любом из пп. 1, 6, 7; R5 определен в п. 1 или 8; а m - в п. 1.
14. Соединение, представленное следующей формулой, или его фармацевтически приемлемая соль:
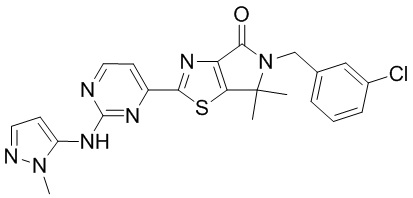 ,
,  ,
, 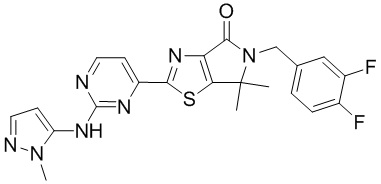 ,
,  ,
, 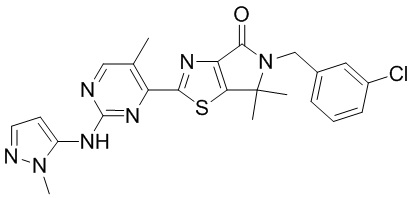 ,
, 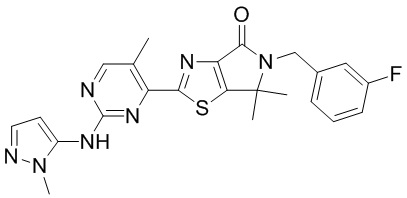 ,
,
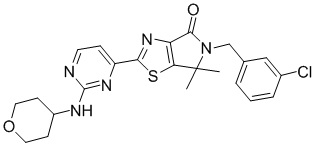 ,
, 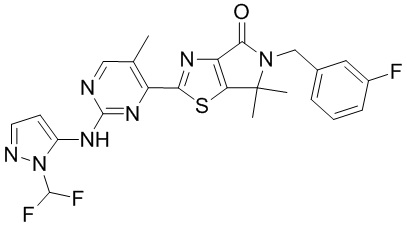 ,
, 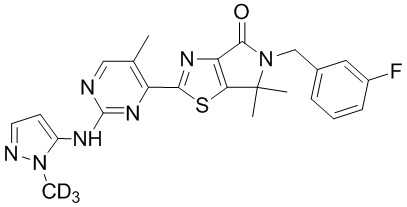 ,
, 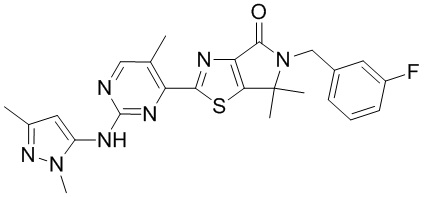 ,
, 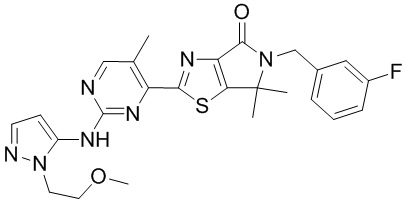 ,
, 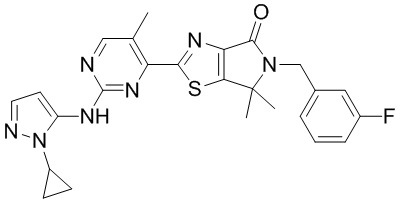 ,
, 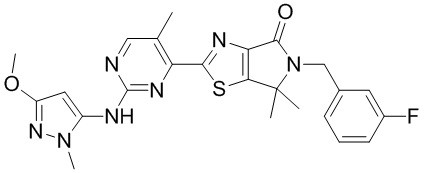 .
.
15. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-14 при изготовлении лекарственного средства для лечения заболеваний, связанных с ERK.
16. Применение по п. 15, отличающееся тем, что лекарственное средство для лечения заболеваний, связанных с ERK, является средством для лечения солидных опухолей.
| WO 2019223632 A1, 28.11.2019 | |||
| EA 201891063 A1, 28.12.2018 | |||
| CN 109608444 A, 12.04.2019 | |||
| Аппарат для измерения количества протекающей по трубе жидкости | 1932 |
|
SU32754A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2015 |
|
RU2684102C2 |
| Устройство для учета количества тепла, отдаваемого теплоносителем | 1932 |
|
SU31659A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2332415C2 |

