[Область техники]
[0001] Настоящее изобретение относится к фармацевтической композиции для лечения злокачественного новообразования, содержащей антитело против CCR8.
[Уровень техники]
[0002] Мощные механизмы негативной регуляции, включая иммуносупрессию, опосредованную регуляторными Т–клетками (Treg–клетками) в микроокружении опухоли, являются основными препятствиями при лечении опухолей (непатентная литература 1).
Например, CD4–позитивные Treg–клетки, которые инфильтрируют опухоль, могут значительно ингибировать противоопухолевый иммунный ответ и могут стать основным препятствием для эффективного лечения злокачественного новообразования.
Опухолевая иммуносупрессия, опосредованная CD4–позитивными FoxP3–позитивными Treg–клетками, была в достаточной степени продемонстрирована на животных моделях опухолей. Сообщалось, что системное (включая внутриопухолевое) удаление Treg–клеток вызывает противоопухолевый эффект, при этом удаление приблизительно 50% инфильтрирующих опухоль Treg–клеток является не эффективным (непатентная литература 2).
[0003] Сообщалось, что внутри опухоли у пациентов с различными видами злокачественных новообразований, включая рак легкого, рак молочной железы и опухоли яичника, выявляется повышенное отношение CD4–позитивных CD25–позитивных Treg–клеток (клеточная популяция, включая Treg–клетки) ко всей популяции CD4–позитивных T–клеток у людей, и количественное соотношение отрицательно коррелирует с вероятностью выживания пациентов (непатентная литература 3–8).
[0004] Было подтверждено, что удаление CD4–позитивных CD25–позитивных Treg–клеток из опухолей с использованием анти–CD25–антитела вызывает противоопухолевый эффект. Однако это удаление не является специфичным для Treg–клеток, поскольку CD25 экспрессируется на поверхности клеток CD4–позитивных CD25–позитивных Treg–клеток, а также недавно активированных эффекторных T–клеток. Кроме того, введение мышам анти–CD25 антител вызывает ограниченный противоопухолевый эффект. На различных моделях опухолей было продемонстрировано, что введение только антитела перед опухолевой инокуляцией проявляет терапевтический эффект, при этом введение антитела после приживления опухоли у мышей редко дает терапевтический эффект. Противоопухолевый эффект ослабевал в случае начала введения анти–CD25 антитела в день 1 после трансплантации и редко наблюдался в случае начала введения анти–CD25 антитела в день 2 после трансплантации или позже (непатентная литература 9).
[0005] До настоящего времени тесты на лекарственную эффективность проводили путем введения антител мышам с целью удаления Treg–клеток. Тем не менее, есть несколько сообщений, показывающих противоопухолевый эффект. Таким образом, очень трудно подтвердить противоопухолевый терапевтический эффект, вызванный удалением Treg–клеток путем введения антител перед инокуляцией (непатентная литература 10).
[0006] CCR8, также ранее называемый CY6, CKR–L1 или TER1, представляет собой G–белок, связанный с 7 трансмембранным рецептором хемокина CC, экспрессируемый в тимусе, селезенке и тому подобное. Ген, кодирующий этот белок, находится в хромосоме человека 3р21. CCR8 человека состоит из 355 аминокислот (непатентная литература 11). CCL1 известен как эндогенный лиганд для CCR8 (непатентная литература 12). кДНК CCR8 человека состоит из нуклеотидной последовательности, представленной GenBank ACC No. M_005201.3, и кДНК CCR8 мыши состоит из нуклеотидной последовательности, представленной GenBank ACC No. NM_007720.2.
[Список литературы]
[Непатентная литература]
[0007] [Непатентная литература 1] Nat. Rev. Immunol., 2006, Vol. 6, No. 4, p. 295–307.
[Непатентная литература 2] Eur. J. Immunol., 2010, Vol. 40, p. 3325–3335.
[Непатентная литература 3] J. Clin. Oncol., 2006, Vol. 24, p. 5373–5380.
[Непатентная литература 4] Nat. Med., 2004, Vol. 10, p. 942–949.
[Non Patent Literature 5] J. Clin. Oncol., 2007, Vol. 25, p. 2586–2593.
[Непатентная литература 6] Cancer, 2006, Vol. 107, p. 2866–2872.
[Непатентная литература 7] Eur. J. Cancer, 2008, Vol. 44, p. 1875–1882.
[Непатентная литература 8] Cell. Mol. Immunol. 2011, Vol. 8, p. 59–66.
[Непатентная литература 9] Cancer Res., 1999 Jul 1; Vol. 59, No. 13, p. 3128–33.
[Непатентная литература 10] Cancer Res., 2010, Vol. 70, № 7, с. 2665–74.
[Непатентная литература 11] J. Immunol., 1996, Vol. 157, No. 7, p. 2759–63.
[Непатентная литература 12] J. Biol. Chem., 1997, Vol. 272, No. 28, p. 17251–4.
[Краткое описание изобретения]
[Техническая задача]
[0008] Целью настоящего изобретения является активация иммунитета путем ингибирования иммуносупрессии, опосредованной Treg–клетками или тому подобное, и получение фармацевтической композиции для лечения злокачественного новообразования посредством этого механизма.
[Решение задачи]
[0009] Авторы настоящего изобретения провели тщательные исследования, после было сделано настоящее изобретение на основании обнаружения, что опухоль–инфильтрирующие Treg–клетки и опухоль–инфильтрирующие макрофагальные клетки специфически экспрессируют CCR8, и введение антитела против CCR8 уменьшает количество опухоль–инфильтрирующих Treg–клеток и опухоль–инфильтрирующих клеток макрофагальные клетки и ингибирует рост опухоли.
[0010] В частности, настоящее изобретение относится к:
(1) фармацевтической композиции для лечения злокачественного новообразования, содержащей антитело против CCR8;
(2) фармацевтической композиции в соответствии с (1), где антитело против CCR8 представляет собой антитело, обладающее активностью ADCC;
(3) фармацевтической композиции в соответствии с (1) или (2), где антитело против CCR8 представляет собой CCR8–нейтрализующее антитело;
(4) фармацевтической композиции в соответствии с любым из (1)–(3), где антитело против CCR8 обладает эффектом удаления опухоль–инфильтрирующих Treg–клеток;
(5) фармацевтической композиции в соответствии с любым из (1)–(4), где антитело против CCR8 обладает эффектом удаления опухоль–инфильтрирующих макрофагальных клеток;
(6) фармацевтической композиции в соответствии с любым из (1)–(5), где злокачественное новообразование представляет собой рак молочной железы, колоректальный рак, рак почки или саркому;
(7) лекарственному средству для лечения злокачественного новообразования, содержащему комбинацию антитела против CCR8 и антитела против PD–1 или антитела против PD–L1;
(8) способу лечения злокачественного новообразования, включающему введение антитела против CCR8 в соответствии с любым из (1)–(5);
(8–1) способу лечения злокачественного новообразования, включающему введение антитела против CCR8;
(8–2) способу в соответствии с (8–1), где антитело против CCR8 представляет собой антитело, обладающее активностью ADCC;
(8–3) способу в соответствии с (8–1) или (8–2), где антитело против CCR8 представляет собой CCR8–нейтрализующее антитело;
(8–4) фармацевтической композиции в соответствии с любым из (8–1)–(8–3), где антитело против CCR8 обладает эффектом удаления опухоль–инфильтрирующих Treg–клеток;
(8–5) фармацевтической композиции в соответствии с любым из (8–1)–(8–4), где антитело против CCR8 обладает эффектом удаления опухоль–инфильтрирующих макрофагальных клеток;
(8–6) способу в соответствии с любым из (8–1)–(8–5), где рак представляет собой рак молочной железы, колоректальный рак, рак почки или саркому;
(8–7) способу в соответствии с любым из (8–1)–(8–6) с дополнительным введением антитела против PD–1 или антитела против PD–L1;
(9) антителу против CCR8 в соответствии с любым из (1)–(5) для лечения злокачественного новообразования;
(9–1) антителу против CCR8 для лечения злокачественного новообразования;
(9–2) антителу против CCR8 в соответствии с (9–1), где антитело против CCR8 представляет собой антитело, обладающее активностью ADCC;
(9–3) антителу против CCR8 в соответствии с (9–1) или (9–2), где антитело против CCR8 представляет собой CCR8–нейтрализующее антитело;
(9–4) антителу против CCR8 в соответствии с любым из (9–1)–(9–3), где антитело против CCR8 обладает эффектом удаления опухоль–инфильтрирующих Treg–клеток;
(9–5) антителу против CCR8 в соответствии с любым из (9–1)–(9–4), где антитело против CCR8 обладает эффектом удаления опухоль–инфильтрирующих макрофагальных клеток;
(9–6) антителу против CCR8 в соответствии с любым из (9–1)–(9–5), где рак представляет собой рак молочной железы, колоректальный рак, рак почки или саркому; и
(9–7) комбинации антитела против CCR8 в соответствии с любым из (9–1)–(9–6) и антитела против PD–1 или антитела против PD–L1 для применения при лечении злокачественного новообразования.
[Эффекты изобретения]
[0011] Фармацевтическая композиция, содержащая антитело по настоящему изобретению может эффективно использоваться в фармацевтике при лечении злокачественного новообразования.
[Краткое описание чертежей]
[0012] [Фигура 1] На фигуре 1 показаны результаты анализа FACS на опухоль–инфильтрирующих CD3+ CD4+ Т–клетках рака почки. Каждую молекулу CD25 и молекулу FoxP3 окрашивали антителом и оценивали по показателям их экспрессии. Было обнаружено, что CD25–экспрессирующие клетки также экспрессируют FoxP3.
[Фигура 2] На фигуре 2 показаны результаты анализа проточной цитометрии на интенсивности экспрессии CD45RA и CD25 в мононуклеарных клетках периферической крови (далее называемых РВМС) у одного и того же пациента. CD3+ CD4+ Т–клетки фракционировали на 6 фракций (от Fr1 до Fr6), как показано на чертеже, в соответствии с уровнями экспрессии CD45RA и CD25, и клетки в каждой фракции выделяли с использованием сортера. Числовые значения обозначают коэффициент содержания клеток (%) в каждой фракции. В этом случае фракциями Treg являются Fr1 и Fr2.
[Фигура 3] На фигуре 3 показаны результаты анализа проточной цитометрии на интенсивности экспрессии CD45RA и CD25 в опухоль–инфильтрирующих клетках злокачественного новообразования почки. Опухоль–инфильтрирующие CD3+ CD4+ Т–клетки разделяли на 4 фракции (от Fr2 до Fr5), как показано на чертеже, в соответствии с уровнями экспрессии CD45RA и CD25, и клетки в каждой фракции выделяли с использованием сортера. Числовые значения обозначают коэффициент содержания клеток (%) в каждой фракции.
[Фигура 4] На фигуре 4 показаны результаты проведения анализа РНК–Seq клеток в каждой из фракций, приведенных на фигурах 2 и 3, и изучения содержания какой–либо из этих фракций Treg–клеток на основе уровней экспрессии мРНК Treg–специфичных экспрессируемых генов FoxP3 и IKZF2. Ордината отображает относительный уровень экспрессии мРНК после нормализации. Для Fr2 и Fr3 наблюдалась сильная внутриопухолевая экспрессия обоих генов. Для Fr4 и Fr5 наблюдалась сильная экспрессия IL–2 или IFNγ, которые экспрессируются в эффекторных клетках.
[Фигура 5] На фигуре 5 показаны результаты анализа Treg–специфической области деметилирования (chrX, 49118000–49118500, hg19) в локусе гена FoxP3 в каждой фракции. Было обнаружено, что большинство опухоль–инфильтрирующих CD3+ CD4+ T–клеток во фракциях Fr2 и Fr3 являются Treg–клетками.
[Фигура 6] На фигуре 6 показаны результаты анализа уровня экспрессии мРНК CCR8 в каждой фракции так же, как на фигуре 4. Фракции опухоль–инфильтрирующих Treg–клеток Fr2 и Fr3 демонстрировали значительную экспрессию CCR8, причем экспрессия редко наблюдалась в Treg–клетках в мононуклеарных клетках периферической крови (РВМС).
[Фигура 7] На фигуре 7 показаны результаты анализа проточной цитометрии на клетках HEK293, экспрессирующих CCR8 мыши. Клетки HEK293 трансфицировали вектором экспрессии pcDNA3.4 со вставкой гена CCR8 мыши, и проводили селекцию по резистентности к лекарственному средству, используя G418. Что касается степени экспрессии CCR8 мыши, то экспрессию подтверждали меченым PE антимышиным CCR8 антителом. Клетки HEK293, трансфицированные вектором pcDNA3.4 и селектированные на резистентность к лекарственному средству так же, как описано выше, использовали в качестве отрицательного контроля. Было обнаружено, что почти все клетки экспрессируют CCR8 мыши.
[Фигура 8] На фигуре 8 показано, что антитело против CCR8 мыши (SA214G2) обладает способностью активировать сигнальный путь, необходимый для антителозависимой клеточной цитотоксичности (ADCC).
[Фигура 9] На фигуре 9 показано, что анти–CCR8 антитело мыши (SA214G2) обладает активностью ADCC.
[Фигура 10] На фигуре 10 показано, что анти–CCR8 антитело мыши (SA214G2) обладает активностью ингибирования внутриклеточного притока кальция, опосредованного CCR8. В качестве отрицательного контроля использовали изотипическое контрольное антитело.
[Фигура 11] На фигуре 11 показано, что антимышиное CCR8 антитело (SA214G2) не распознает клетки CT26. В качестве отрицательного контроля использовали изотипическое контрольное антитело.
[Фигура 12] На фигуре 12 показаны результаты введения контрольного антитела на 3–й день после трансплантации трем мышам BALB/c, которым были трансплантированы клетки клеточной линии CT26 колоректального злокачественного новообразования мыши, путем иссечения опухоли на 4–й или 7–й день после введения и анализируя долю Treg–клеток, присутствующих в них, с помощью проточного цитометра.
[Фигура 13] На фигуре 13 показаны результаты анализа доли CCR8+ Treg–клеток с использованием проточного цитометра в том же эксперименте, что и на фигуре 12.
[Фигура 14] На фигуре 14 показаны результаты анализа доли CCR8–позитивных клеток в клетках внутриопухолевых макрофагов CD11b+ Gr1+ CD206+ M2 с использованием проточного цитометра. В обоих случаях было обнаружено, что от 40 до 50% клеток являются CCR8–позитивными макрофагальными клетками М2.
[Фигура 15] На фигуре 15 показан ход эксперимента по введению антитела против CCR8 мыши (SA214G2) или изотипического контрольного антитела на 3–й день после трансплантации мышам BALB/c, которым были трансплантированы клетки клеточной линии CT26 колоректального злокачественного новообразования мыши, путем иссечения опухоли на 7–й или 10–й день после трансплантации и исследуя относительное содержание Т–лимфоцитов и макрофагальных клеток, присутствующих в них.
[Фигура 16] На фигуре 16 показано отношение клеток CD25+ FoxP3+ к клеткам CD45+ CD4+ на 7–й день после трансплантации (d7) или 10 (d10).
[Фигура 17] На фигуре 17 показана доля макрофагальных клеток CD11b+ F4/80+ на 7 день после трансплантации (d7).
[Фигура 18] На фигуре 18 показано относительное содержание IA/IE–положительных (IA/IE +) или IA/IE–отрицательных клеток (IA/IE–) на 7 день после трансплантации (d7).
[Фигура 19] На фигуре 19 показан ход эксперимента по введению антитела против CCR8 мыши (SA214G2) или изотипического контрольного антитела (крысиное анти–KLH) единичной дозой 400 мкг/мышь на 3–й день после трансплантации (d3) мышам BALB/c, которым трансплантировали клетки клеточной линии CT26 колоректального злокачественного новообразования, и измеряя размер опухоли каждые 3–4 дня начиная с 7–го дня после трансплантации (d7) до 21–го дня (d21).
[Фигура 20] На фигуре 20 показаны результаты определения размера солидной опухоли каждого индивида после инокуляции и расчета объема опухоли.
[Фигура 21] На фигуре 21 показан средний объем опухоли каждой группы мышей в каждый момент времени после инокуляции. Показано также стандартное отклонение. Уровень значимости *** обозначает p <0,001, а уровень значимости ** обозначает p 0,01 (t–критерий).
[Фигура 22] 2×105 клеток клеточной линии Colon26 колоректального рака трансплантировали внутрикожно в спину каждой мыши BALB/c. На 3–й день после трансплантации (d3) вводили анти–CCR8 антитело мыши (SA214G2) или контрольное изотипическое антитело в единичной дозе 400 мкг/мышь. Объем опухоли измеряли каждые 3–4 дня, начиная со дня после трансплантации 3 (d3) до 18 дня (d18). Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции.
[Фигура 23] График показывает среднюю интенсивность флуоресценции (MFI) для каждого объекта в анализе FACS. Центральные горизонтальные линии показывают среднее значение MFI для 14 случаев, а вертикальные линии показывают стандартные отклонения. Уровень значимости *** обозначает P <0,001.
[Фигура 24] На фигуре 24 показан индивид–ориентированный график зависимости отношения клеток, которые проявляли CCR8–позитивные сигналы (процент позитивности), равные или превышающие фоновый уровень, полученный в антителе контроля изотипа, к CD3+ CD4+ FoxP3+ T–клеткам или CD3+ CD4+ FoxP3– T–клеткам в раковых опухолях почек человека в 14 случаях. Центральные горизонтальные линии показывают средний процент позитивности для 14 случаев, а вертикальные линии показывают стандартные отклонения.
[Фигура 25] На фигуре 25 показана кривая Каплана–Мейера относительно вероятности выживания каждой группы, полученная путем равного деления пациентов со светлоклеточной почечно–клеточной карциномой на 2 группы с высокой экспрессией (High) и низкой экспрессией (Low) на основе уровней экспрессии мРНК CCR8 внутриопухолевых клеток с использованием базы данных Атласа Генома злокачественного новообразования (TCGA). Ордината изображает вероятность выживания, а абсцисса изображает количество месяцев. Численные значения обозначают количество людей в каждой группе. Значение P обозначает тестовое значение лог–рангового критерия.
[Фигура 26] На фигуре 26 показаны результаты анализа таким же образом, как на фигуре 25, пациентов с раком простаты.
[Фигура 27] На фигуре 27 показаны результаты анализа таким же образом, как на фигуре 25, пациентов с раком мочевого пузыря.
[Фигура 28] На фигуре 28 показано, что антитело против CCR8 мыши не распознает ни клетки MethA, ни клетки LM8, как на фигуре 11. В качестве отрицательного контроля использовали изотипическое контрольное антитело (Isotype).
[Фигура 29] 3×105 клеток клеточной линии LM8 остеосаркомы трансплантировали внутрикожно в спину каждой мыши C3H/He. На 3–й день после трансплантации (d3) вводили антитело против CCR8 мыши (SA214G2) или изотипическое контрольное антитело (контрольное антитело) в единичной дозе 400 мкг/мышь. Объем опухоли измеряли каждые 3–4 дня от 7 до 35 дней после инокуляции опухоли. Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции. Показано также стандартное отклонение. Уровень значимости *** обозначает p <0,001, уровень значимости ** обозначает p 0,01, а уровень значимости * обозначает p 0,05 (t–критерий).
[Фигура 30] 1×105 клеток MethA внутрикожно трансплантировали в спину каждой мыши Balb/c. На 3–й день после трансплантации вводили антитело против CCR8 мыши (SA214G2) или антитело контроля изотипа (контрольное антитело) в единичной дозе 400 мкг/мышь. Объем опухоли измеряли каждые 3–4 дня от 11 до 21 дней после инокуляции опухоли. Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции. Уровень значимости * обозначает p <0,05 (t–критерий).
[Фигура 31] 1×105 клеток клеточной линии EMT6 злокачественного новообразования молочной железы трансплантировали внутрикожно в спину каждой мыши Balb/c. Через 3 и 10 дней после инокуляции опухоли вводили антитело против CCR8 мыши (SA214G2) или изотипическое контрольное антитело в дозе 100 мкг/мышь. Объем опухоли измеряли каждые 3–4 дня с 4–ого по 22–ой день после инокуляции опухоли. Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции. Уровень значимости *** обозначает p <0,001, а уровень значимости ** обозначает p 0,01 (t–критерий).
[Фигура 32] 2×105 клеток клеточной линии Colon26 колоректального рака трансплантировали внутрикожно в спину каждой мыши BALB/c. Через 3 и 10 дней после инокуляции опухоли вводили анти–изотипическое контрольное антитело (изотипическое антитело), анти–CCR8 антитело мыши (SA214G2) или анти–PD–1 антитело (RMP1–14) в дозе 400 мкг/мышь. Объем опухоли измеряли каждые 3–4 дня от 3 до 24 дня после инокуляции опухоли. Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции.
[Фигура 33] 4х105 клеток, полученных из клеточной линии RAG рака почки мыши, трансплантировали внутрикожно в спину каждой мыши BALB/c. Через 6 дней после инокуляции опухоли, внутрибрюшинно вводили 100 мкг (100 мкл) изотипического контрольного антитела, анти–CCR8 антитела мыши или анти–PD–1 антитела мыши (анти–PD–1 антитело). Объем опухоли измеряли каждые 3–4 дня с 6–ого по 21–ой день после инокуляции опухоли. Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции.
[Фигура 34] 2×105 клеток клеточной линии Colon26 колоректального злокачественного новообразования трансплантировали внутрикожно в спину каждой мыши BALB/c. Через 3 и 10 дней после инокуляции опухоли вводили анти–мышиное CCR8 антитело (SA214G2) или изотипическое контрольное антитело в дозе 400 мкг/мышь. Через 24 дня после инокуляции опухоли, у мышей извлекали каждый орган и определяли его массу. Показано среднее значение для 10 случаев в каждой группе.
[Фигура 35] 1×105 клеток клеточной линии EMT6 рака молочной железы трансплантировали внутрикожно в спину каждой мыши BALB/c. Анти–CCR8 мыши вводили внутривенно через 3 и 10 дней после инокуляции опухоли, и анти–мышиное PD–1 антитело вводили внутривенно через 8 и 13 дней после инокуляции опухоли. Изотипическое контрольное антитело вводили внутривенно контрольной группе через 3 и 10 дней после инокуляции опухоли. Объем опухоли измеряли каждые 3–4 дня c 6–ого по 27–ой день после инокуляции. Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции.
[Фигура 36] На фигуре 36 показана доля особей с опухолью размером более 50 мм3 или меньше в каждый момент времени после инокуляции в каждой группе в таком же эксперименте, что и на фигуре 35.
[Фигура 37] 4,5х105 клеток, полученных из клеточной линии RAG рака почки мыши, трансплантировали внутрикожно в спину каждой мыши BALB/c. Через 8 и 15 дней после инокуляции опухоли, внутривенно вводили 100 мкл физиологического раствора, анти–CCR8 антитела мыши, или анти–PD–1 антитела мыши, или анти–CCR8 антитела мыши и анти–PD–1 антитела мыши. Объем опухоли измеряли каждые 3–4 дня с 8–ого по 33–ий день после инокуляции опухоли. Показан средний объем опухоли каждой группы в каждый момент времени после инокуляции.
[Фигура 38] 2×105 клеток CT26 трансплантировали внутрикожно в спину каждой мыши дикого типа или гомозиготной мыши линии Balb/c с дефицитом гена CCR8 (N=5). После инокуляции внутривенно вводили изотипическое контрольное антитело или анти–CCR8 антитело мыши. Объем опухоли измеряли каждые 3–4 дня после инокуляции опухоли. Левая диаграмма показывает средний объем опухоли у мышей дикого типа в каждой группе в каждый момент времени после инокуляции, а правая диаграмма показывает средний объем опухоли у гомозиготной мыши с дефицитом гена CCR8 в каждой группе в каждый момент времени после инокуляции.
[Описание вариантов осуществления]
[0013] Фармацевтическая композиция по настоящему изобретению содержит антитело против CCR8.
[0014] CCR8 по настоящему изобретению включает CCR8 мышей, крыс, хомяков, морских свинок, собак, свиней и млекопитающих приматов, включая обезьян и людей. CCR8 человека является предпочтительным.
[0015] Антитело против CCR8 может быть любым антителом человека, антителом мыши, антителом крысы, антителом кролика и антителом козы, если антитело связывается с CCR8. Антитело против CCR8 может быть его поликлональным или моноклональным антителом и может быть любым из: полноразмерного антитела, фрагмента антитела (например, фрагмента F(ab')2, Fab', Fab или Fv), химерного антитела, гуманизированного антитела и полноразмерного антитела человека. Антитело, полученное у человека, гуманизированное антитело или полноразмерное антитело человека является предпочтительным.
[0016] Антитело по настоящему изобретению может быть получено в соответствии со способом получения антитела или антисыворотки, известным в данной области, с использованием полноразмерного белка или части белка CCR8 в качестве антигена. Желательно, чтобы антитело по настоящему изобретению связывалось с CCR8, экспрессируемым на поверхности клетки. Следовательно, часть белка желательно представляет собой внеклеточную область CCR8. Эти антигены могут быть получены способами экспрессии и очистки белка, известными в данной области.
[0017] Примеры антигена, отличного от описанного выше, подходящего для получения антитела против CCR8, включают клетки, индуцирующие экспрессию CCR8 под действием вектора экспрессии или тому подобного, плазмидные вектора экспрессии CCR8 и вирусные вектора экспрессии CCR8 (аденовирусные векторы и тому подобное).
[0018] Поликлональное антитело может быть получено способом, известным в данной области. Поликлональное антитело может быть получено, например, путем иммунизации соответствующего животного антигенным белком или его смесью с белком–носителем и сбора продукта, содержащего антитело против антигенного белка, от иммунизированного животного с последующим выделением и очисткой антитела. Примеры используемого животного обычно включают мышей, крыс, овец, коз, кроликов и морских свинок. Для повышения способности продуцировать антитела вместе с антигенным белком можно вводить полный адъювант Фрейнда или неполный адъювант Фрейнда. Как правило, введение осуществляют в общей сложности приблизительно от 3 до 10 раз, обычно один раз каждые приблизительно 2 недели. Поликлональное антитело может быть собрано из крови, асцитной жидкости или тому подобное у животного, иммунизированного способом, описанным выше. Титр поликлональных антител в антисыворотке можно измерить с помощью ELISA. Выделение и очистка поликлонального антитела могут быть выполнены в соответствии со способом выделения и очистки иммуноглобулина, например, способом очистки с использованием антиген–связывающей твердой фазы или активного адсорбента, такого как белок А или белок G, методом высаливания, методом осаждения спиртом, методом изоэлектрического осаждения, электрофорезом, методом адсорбции и десорбции с использованием ионообменника, методом ультрацентрифугирования или методом гель–фильтрации.
[0019] Моноклональное антитело может быть получено известным общим способом получения. В частности, млекопитающее, предпочтительно мышь, крыса, хомяк, морская свинка или кролик, иммуносенсибилизируют антигеном по настоящему изобретению, если необходимо, вместе с адъювантом Фрейнда, подкожной, внутримышечной, внутривенной, внутрь подушечки стопы или внутрибрюшиной инъекцией от одного до нескольких раз. Обычно иммунизацию осуществляют от одного до 4 раз каждые приблизительно от 1 до 21 дня после начальной иммунизации, и клетки, продуцирующие антитела, могут быть получены от сенсибилизированного иммунитетом млекопитающего приблизительно через 1–10 дней после последней иммунизации. Число иммунизаций и временной интервал могут быть соответствующим образом изменены в соответствии со свойствами и тому подобное используемого иммуногена.
[0020] Гибридомы, секретирующие моноклональное антитело, могут быть получены в соответствии со способом Kohler и Milstein (Nature, 1975, vol. 256, p. 495–497) и эквивалентным ему способом. В частности, гибридомы могут быть получены путем слияния клеток продуцирующих антитела клеток, содержащихся в селезенке, лимфатическом узле, костном мозге или миндалинах и тому подобное, предпочтительно, в селезенке млекопитающего, сенсибилизированного иммунитетом, как указано выше, причем, предпочтительно, клетки мыши, крысы, морской свинки, хомяка, кролика или млекопитающего (например, человека), более предпочтительно, клетки миеломы, полученные из мыши, крысы или человека, не обладают способностью продуцировать аутологичные антитела.
Как правило, в качестве клеток миеломы для использования при слиянии клеток может использоваться постоянная клеточная линия, полученная от мышей, например, P3–U1, NS–1, SP–2, 653, X63 или AP–1.
[0021] Скрининг гибридомного клона, продуцирующего моноклональное антитело, проводят путем культивирования гибридом, например, в микротитровальном планшете, измеряя реакционную способность супернатанта клеточной культуры в лунке, где наблюдается рост, с антигеном по настоящему изобретению, используемым при иммунной сенсибилизации мыши, упомянутой выше, с помощью метода измерения, такого как RIA, ELISA или FACS, и выбора клона, продуцирующего моноклональное антитело, которое проявляет специфическое связывание с антигеном или гаптеном. Обычно дополнительно используется способ, который включает иммобилизацию антигена на твердой фазе и обнаружение антитела в связывающем с ним супернатанте клеточной культуры с использованием вторичного антитела, меченного радиоактивным материалом, флуоресцентным материалом, ферментом или тому подобное. В случае использования антиген–экспрессирующих клеток к клеткам добавляется супернатант гибридомной культуры, и с ним затем может взаимодействовать флуоресцентно меченное вторичное антитело, после чего измеряется интенсивность флуоресценции клеток с использованием флуоресцентного устройства обнаружения, такого как проточный цитометр для обнаружения моноклонального антитела, способного связываться с антигеном по настоящему изобретению на мембранах клеток.
[0022] Моноклональное антитело может быть получено из выбранной гибридомы путем культивирования гибридомы in vitro или культивирования гибридомы в асцитной жидкости или тому подобном мыши, крысы, морской свинки, хомяка или кролика и тому подобное, предпочтительно, мыши или крысы, более предпочтительно, мыши, и выделения моноклонального антитела из полученного супернатанта клеточной культуры или асцитной жидкости млекопитающего. Для культивирования in vitro гибридому выращивают, поддерживают и сохраняют в соответствии с различными условиями, такими как характеристики типа клеток, подлежащих культивированию, цели тестирования и исследования и метода культивирования, и может культивироваться с использованием известной питательной среды, как используется для продукции моноклональных антител в супернатанте клеточной культуры, или каждой питательной среды, индуцированной и приготовленной из известной базальной среды.
[0023] Примеры базальной среды включают среду с низким содержанием кальция, такую как среда Ham 'F12, среду MCDB153 и среду с низким содержанием кальция MEM, и среду с высоким содержанием кальция, такую как среда MCDB104, среда MEM, среда D–MEM, среда RPMI1640, среда ASF104 и среда RD. Базальная среда может содержать, например, сыворотку, гормон, цитокин и/или различные неорганические или органические вещества, в зависимости от цели.
[0024] Моноклональное антитело может быть выделено и очищено, например, путем воздействия на супернатант клеточной культуры или асцитной жидкости, упомянутых выше, на насыщенный сульфат аммония, ионообменную хроматографию (DEAE или DE52 и тому подобное), или аффинной колоночной хроматографией с использованием колонки с анти–иммуноглобулином, колонки с белком А или тому подобное.
[0025] Рекомбинантное антитело, полученное путем клонирования гена антитела из антителопродуцирующих клеток, например, гибридом, интеграции гена антитела в подходящий вектор и трансфекции хозяина этим вектором с последующим получением с использованием метода рекомбинации генов может быть использовано в качестве антитела по настоящему изобретению (например, Carl et al., THERAPEUTIC MONOCLONAL ANTIBODIES, опубликовано в 1990 г.).
[0026] В частности, мРНК, кодирующая вариабельную область (V–область) антитела, выделяют из гибридом, продуцирующих интересующее антитело, или иммуноцитов, продуцирующих антитело, например клеток сенсибилизированных лимфоцитов, иммортализованных онкогеном или тому подобное. Для выделения мРНК общую РНК получают способом, известным в данной области, например, способом ультрацентрифугирования с гуанидином (Chirgwin, J.M. et al., Biochemistry (1979) 18, 5294–5299), и мРНК получают с использованием набора для очистки мРНК (производства Pharmacia Inc.) или тому подобное.
[0027] кДНК V–области антитела синтезируют из полученной мРНК с использованием обратной транскриптазы. Синтез кДНК может быть выполнен с использованием набора для синтеза первой цепи кДНК с обратной транскриптазой AMV или тому подобного. 5'–Ampli FINDER RACE Kit (производства Clontech Laboratories, Inc) и основанный на ПЦР 5'–RACE (Frohman, MA et al., Proc. Natl. Acad. Sci. USA, 1988, Vol. 85, p. 8998, etc.) могут быть использованы для синтеза и амплификации кДНК. Интересующий фрагмент ДНК очищают от полученного продукта ПЦР и лигируют с векторной ДНК. Из них дополнительно получают рекомбинантный вектор. E. coli или тому подобное трансфицируют рекомбинантным вектором и отбирают колонию для получения желаемого рекомбинантного вектора. Нуклеотидную последовательность представляющей интерес ДНК подтверждают методом, известным в данной области, например, с помощью дезокси–метода.
[0028] При условии, что ДНК, кодирующая V–область интересующего антитела, успешно получена, эту ДНК связывают с ДНК, кодирующей желаемую константную область антитела (C–область), и полученный продукт интегрируют в вектор экспрессии. Альтернативно, ДНК, кодирующая V–область антитела, может быть интегрирована в вектор экспрессии, содержащий ДНК C–области антитела. Для получения антитела по настоящему изобретению, ген антитела интегрируют в вектор экспрессии, так что ген антитела экспрессируется под контролем области контроля экспрессии, например энхансера/промотора. Затем клетки–хозяева могут быть трансформированы этим вектором экспрессии для экспрессии антитела.
[0029] Для экспрессии гена антитела, ДНК, кодирующую тяжелую цепь (Н–цепь), и ДНК, кодирующую легкую цепь (L–цепь) антитела, можно отдельно интегрировать в вектора экспрессии, которыми хозяин котрансформируется, или ДНК, кодирующую H–цепь, и ДНК, кодирующую L–цепь, можно интегрировать в общий вектор экспрессии, которым трансформируют хозяин (см. WO94/11523).
[0030] В качестве способа, отличного от описанного выше, для получения антитела по настоящему изобретению также можно использовать так называемую технологию фагового дисплея (Nature Biotechnology 23, 1105 (2005)). В частности, например, библиотека генов антител, полученная способом, известным в данной области техники, с использованием В–лимфоцитов человека или животного (например, кролика, мыши, крысы или хомяка) в качестве материала, или библиотека генов антител, полностью синтезированная путем отбора и конструирования из последовательности зародышевой линии человека или животного отображается, например, на бактериофагах, E. coil, поверхности дрожжей или клеток животных или липосомах. В этом отношении примеры формы антитела, которое должно отображаться на поверхности клетки, включают молекулы IgG, молекулы IgM, фрагменты Fab и одноцепочечные фрагменты Fv (scFv).
[0031] Полученный таким образом ген фрагмента антитела может быть рекомбинирован с соответствующей областью гена антитела IgG способом, известным в данной области техники, для получения гена антитела. Затем полученный таким образом ген может быть интегрирован в соответствующий вектор, которым трансфицирует хозяин, с последующей продукцией антитела с использованием техники рекомбинации генов (например, Carl et al., THERAPEUTIC MONOCLONAL ANTIBODIES, опубликованной в 1990 г.).
[0032] Антитело по настоящему изобретению включает антитела, искусственно созданные с целью, например, уменьшения ксеноантигенности в отношении людей, например, химерные антитела, гуманизированные антитела и полные человеческие антитела.
[0033] Антитело по настоящему изобретению может представлять собой конъюгированное антитело, в котором антитело связано с любой из различных молекул, таких как полиэтиленгликоль (ПЭГ), радиоактивные вещества, токсины и сахарные цепи. Такое конъюгированное антитело может быть получено путем химической модификации полученного антитела. Способ модификации антитела уже известен в данной области. Антитело по настоящему изобретению также охватывает эти конъюгированные антитела.
[0034] Антитело по настоящему изобретению включает антитело с областью Fc, связанной с N–гликозид–связанными цепями сахара, которые свободны от фукозы, связанной с N–ацетилглюкозамином на их восстанавливающих концах. Примеры антител с Fc–областью, связанной с N–гликозид–связанными сахарными цепями, которые свободны от фукозы, связанной с N–ацетилглюкозамином на их восстанавливающих концах, включают антитела, полученные с использованием клеток СНО с дефицитом гена α1,6–фукозилтрансферазы (международные публикации №№ WO 2005/035586 и WO 02/31140). Антитело по настоящему изобретению с областью Fc, связанной с N–гликозид–связанными сахарными цепями, которые свободны от фукозы, связанной с N–ацетилглюкозамином на их восстанавливающих концах, обладает высокой ADCC–активностью.
[0035] Антитело по настоящему изобретению может быть слито на своем N–конце или С–конце с дополнительным белком (Clinical Cancer Research, 2004, 10, 1274–1281). Белок, предназначенный для конъюгирования, может быть соответствующим образом выбран специалистами в данной области.
[0036] Фрагмент антитела представляет собой часть антитела по настоящему изобретению, указанного выше, и означает фрагмент, обладающий CCR8–специфической связывающей активностью, как в антителе. Примеры фрагментов антитела могут, в частности, включать Fab, F(ab')2, Fab', одноцепочечное антитело (scFv), дисульфид–стабилизированное антитело (dsFv), димеризованный фрагмент V–области (диатело) и CDR–содержащие пептиды (Expert Opinion on Therapeutic Patents, Vol. 6, No. 5, p. 441–456, 1996).
[0037] Альтернативно, антитело по настоящему изобретению может представлять собой биспецифическое антитело, которое имеет две разные антигенные детерминанты и связывается с разными антигенами.
[0038] Активность ADCC (антителозависимая клеточная цитотоксичность) означает активность in vivo в отношении разрушения опухолевых клеток или тому подобного путем активации эффекторных клеток посредством связывания области Fc антитела, связанного с антигеном клеточной поверхности или тому подобным на опухолевых клетках или тому подобное, чтобы представить Fc–рецептор на поверхности эффекторных клеток. Примеры эффекторных клеток включают природные клетки–киллеры и активированные макрофаги.
[0039] Антитело по настоящему изобретению, предпочтительно, представляет собой антитело, обладающее активностью ADCC против клеток, экспрессирующих CCR8, поскольку это антитело может удалять Treg–клетки или макофагальные клетки. Обладает ли антитело по настоящему изобретению такой ADCC активностью, можно определить, например, способом, описанным в примерах, указанных ниже.
[0040] Антитело против CCR8, содержащееся в фармацевтической композиции по настоящему изобретению, предпочтительно, представляет собой CCR8–нейтрализующее антитело с точки зрения супрессии внутриопухолевого накопления Treg–клеток или макрофагальных клеток. Нейтрализующее CCR8 антитело означает антитело, обладающее нейтрализующей активностью в отношении CCR8. Наличие или отсутствие нейтрализующей активности у антитела по настоящему изобретению в отношении CCR8 можно определить путем определения наличия или отсутствия подавления физиологического эффекта CCL1 на CCR8. Примеры включают, но ими не ограничиваются, определение связывания CCL1 с CCR8, миграцию CCR8–экспрессирующих клеток с помощью CCL1, повышение уровня внутриклеточного Ca++ с помощью CCL1 и изменение экспрессии гена, чувствительного к стимуляции CCL1. Это также можно определить способом, описанным в примерах, указанных ниже.
[0041] Антитело против CCR8 по настоящему изобретению предпочтительно обладает эффектом удаления опухоль–инфильтрирующих Treg–клеток. Наличие или отсутствие эффекта удаления опухоль–инфильтрирующих Treg–клеток у антитела по настоящему изобретению может быть определено, например, способом, описанным в примерах, указанных ниже.
[0042] Антитело против CCR8 по настоящему изобретению предпочтительно обладает эффектом удаления опухоль–инфильтрирующих макрофагальных клеток. Наличие или отсутствия у антитела по настоящему изобретению эффекта удаления опухоль–инфильтрирующих макрофагальных клеток, можно определить, например, способом, описанным в примерах, указанных ниже.
[0043] Антитело по настоящему изобретению может быть использовано в виде фармацевтической композиции. Так, фармацевтическая композиция, содержащая антитело по настоящему изобретению, может быть введена перорально или парентерально, системно или местно. Например, в качестве парентерального введения может быть выбрана внутривенная инъекция, такая как инфузия, внутримышечная инъекция, внутрибрюшинная инъекция, подкожная инъекция, трансназальное введение или ингаляция.
[0044] Термин «злокачественное новообразование» в случае «фармацевтической композиции для лечения злокачественного новообразования» по настоящему изобретению включает любой солидный рак и злокачественную опухоль крови. В частности, его примеры включают рак молочной железы, рак тела матки, рак шейки матки, рак яичника, рак предстательной железы, рак легкого, рак желудка (аденокарцинома желудка), немелкоклеточный рак легкого, рак селезенки, рак плоскоклеточный рак головы и шеи, рак пищевода, рак мочевого пузыря, меланому, колоректальный рак, рак почки, неходжкинскую лимфому, рак уротелия, саркому, рак клеток крови (лейкоз, лимфому и тому подобное), рак желчных протоков, рак желчного пузыря, рак щитовидной железы, рак предстательной железы, рак яичка, рак вилочковой железы и гепатокарциному. Предпочтительно, примеры включают рак молочной железы, рак матки, рак яичника, рак легкого, колоректальный рак, рак почки и саркому, и более предпочтительно, примеры включают рак молочной железы, колоректальный рак, рак почки и саркому.
«Злокачественное новообразование» в случае «фармацевтической композиции для лечения злокачественного новообразования» по настоящему изобретению, предпочтительно, представляет собой рак, экспрессирующий опухолеспецифический антиген.
[0045] Термин «злокачественное новообразование», описанный в настоящем описании, означает не только эпителиальные злокачественные опухоли, такие как рак яичника и рак желудка, но неэпителиальные злокачественные опухоли, включая гематопоэтические опухоли, такие как хронический лимфолейкоз и лимфома Ходжкина. В настоящем описании такие термины, как «злокачественное новообразование», «карцинома», «опухоль» и «новообразование», могут использоваться взаимозаменяемо друг с другом без их дифференцирования.
[0046] Антитело против CCR8 по настоящему изобретению может быть введено в качестве сопутствующего лекарственного средства в комбинации с дополнительным лекарственным средством, чтобы
(1) дополнить и/или усилить терапевтический эффект фармацевтической композиции по настоящему изобретению,
(2) улучшить фармакокинетику и абсорбцию фармацевтической композиции по настоящему изобретению и уменьшить ее дозу, и/или
(3) уменьшить побочный эффект фармацевтической композиции по настоящему изобретению.
[0047] Сопутствующее лекарственное средство антитела против CCR8 по настоящему изобретению и дополнительное лекарственное средство может быть введено в виде комбинированного лекарственного средства, содержащего оба ингредиента в одном препарате, или может быть введено в виде отдельных препаратов. Это введение в виде отдельных препаратов включает в себя одновременное введение и отсроченное во времени введение. Для отсроченного во времени введения антитело по настоящему изобретению может быть введено первым, а дополнительное лекарственное средство может быть введено позже, или дополнительное лекарственное средство может быть введено первым, а соединение по настоящему изобретению может быть введено позже. Соответствующие способы их введения могут быть одинаковыми или разными.
[0048] Примеры дополнительного лекарственного средства, которое можно использовать в комбинации с антителом против CCR8 по настоящему изобретению, включают анти–PD–1 антитела, анти–PD–L1 антитела и анти–CTLA–4 антитела. Анти–PD–1 антитела или анти–PD–L1 антитела являются предпочтительными, и анти–PD–1 антитело является более предпочтительным.
[0049] В настоящем изобретении примеры анти–PD–1 антитела включают ниволумаб и пембролизумаб.
[0050] В настоящем изобретении примеры анти–PD–L1 антитела включают атезолизумаб, авелумаб и дурвалумаб.
[0051] В настоящем изобретении примеры анти–CTLA–4 антитела включают ипилимумаб.
[0052] Предполагается, что пациент, для которого предназначена фармацевтическая композиция по настоящему изобретению, является пациентом, страдающим злокачественным новообразованием, или пациентом, у которого подозревают наличие злокачественного новообразования. Эффективную дозу выбирают из диапазона от 0,01 до 100 мг на кг массы тела на дозу. Альтернативно, доза может быть выбрана из 5–5000 мг, предпочтительно, из 10–500 мг на пациента. Однако фармацевтическая композиция, содержащая антитело по настоящему изобретению или фрагмент антитела, не ограничивается этими дозами. Кроме того, период дозирования может быть соответствующим образом выбран в соответствии с возрастом и симптомами пациента. В зависимости от пути введения фармацевтическая композиция по настоящему изобретению может дополнительно содержать фармацевтически приемлемый носитель или добавку. Примеры такого носителя и добавки включают воду, фармацевтически приемлемые органические растворители, коллаген, поливиниловый спирт, поливинилпирролидон, альгинат натрия, водорастворимый декстран, пектин, метилцеллюлозу, этилцеллюлозу, казеин, диглицерин, пропиленгликоль, полиэтиленгликоль, вазелин, человеческую сывороточный альбумин (HSA), маннит, сорбит, лактоза и поверхностно–активные вещества, приемлемые в качестве фармацевтических добавок. Используемую добавку выбирают соответствующим образом или в комбинации с добавками, которые описаны выше, в соответствии с лекарственной формой, хотя добавка не ограничивается этим.
[0053] Далее настоящее изобретение будет конкретно описано со ссылкой на примеры. Однако настоящее изобретение не ограничено приведенными ниже примерами. Способы, описанные в Molecular Cloning: A Laboratory Manual, 2nd Edition (Cold Spring Harbor Laboratory), использовали в качестве методов манипуляции генами, если не указано иное.
[Пример 1]
[0054] Экстракция и анализ опухоль–инфильтрирующих клеток рака почки, и РВМС
Следующий анализ осуществляли с использованием части первичных опухолевых тканей, полученных хирургическим путем у пациентов со светлоклеточной почечно–клеточной карциномой (ccRCC) (3 случая), которые не получали предоперационное лечение противораковым средством, облучения или тому подобное. После измерения массы опухоли, опухолевую массу с помощью ножниц нарезали на квадраты размером 2 мм и получали гомогенаты опухолевой ткани, используя набор Tumor Dissociation Kit, человека (130–095–929, Miltenyi Biotec) и диссоциатор gentleMACS (TM), (Miltenyi Biotec, 130–093–235) в соответствии с протоколом, прилагаемым к набору. Гомогенаты пропускали через клеточный фильтр размером 70 мкм и подвергали гемолизу с последующим удалением остатка и мертвых клеток в растворе 30% перколла в PBS с получением отдельных клеток опухолевой ткани.
Мононуклеарные клетки периферической крови (РВМС) одного и того же пациента выделяли из периферической крови методом центрифугирования в градиенте плотности с использованием Ficoll–Paque PLUS (GE Healthcare Japan Corp.). После определения количества клеток, выделенные внутриопухолевые клетки и PBMC обрабатывали с помощью набора Human TruStain FcX(TM)(BioLegend, Inc., 422–301) и Zombie NIR (BioLegend, Inc., 423105) в соответствии с прилагаемыми протоколами и окрашивали 30 минут на льду. Затем клетки один раз промывали 2% FCS/HEPES/HBSS и затем окрашивали следующими метящими антителами в соответствии с протоколами, прилагаемыми к метящим антителам.
[0055] Поверхность опухоль–инфильтрирующих клеток окрашивали посредством реакции в течение 30 минут на льду с использованием антитела против CD3 (BioLegend, Inc., Clone UCHT1), антитела против CD4 (BioLegend, Inc., Clone OKT4) и антитела против CD25 (BioLegend, Inc., Clone BC96). Клетки дважды промывали 2% FCS/HEPES/HBSS, а затем фиксировали и пермеабилизировали мембрану с использованием Foxp3/набора Foxp3/Transcription Factor Staining Buffer Set (eBioscience, Inc., 00–5523–00) в соответствии с протоколом, прилагаемым к набору. FoxP3 дополнительно окрашивали с использованием PE–меченного анти–FoxP3 антитела (eBioscience, Inc., Clone PCH010). Клетки промывали один раз моющим раствором, прилагаемым к набору, и затем анализировали проточной цитометрией (BD Biosciences, BD LSRFortessa). Было подтверждено, что почти все CD4+ CD25+ T–клетки в опухолях ccRCC экспрессируют FoxP3, маркер Treg–клеток (фигура 1).
[0056] Затем опухоль–инфильтрирующие клетки и PBMC, описанные выше, окрашивали анти–CD3 антителом, анти–CD4 антителом, анти–CD45RA антителом (BD Biosciences, Clone HI100) и анти–CD25 антителом. Получали двухмерную визуализацию CD3+ CD4+ T–клеток на основе уровней экспрессии CD45RA и CD25. Результаты, касающиеся PBMC, показаны на фигуре 2, и результаты, касающиеся опухоль–инфильтрирующих клеток, показаны на фигуре 3. Опухоль–инфильтрирующие клетки разделяли на 4 фракции: уверенно позитивные клетки (Fr2), слабо позитивные клетки (Fr3) и отрицательные клетки (Fr4 и Fr5), как показано на фигуре 1C, в соответствии с интенсивностью экспрессии CD3+ CD4+ CD45RA– и CD25 в качестве индекса, используя клеточный сортер (FACSAria II), и клетки, содержащиеся в каждой фракции, выделяли. Получали двухмерное изобретение PBMC, как в опухоль–инфильтрирующих клетках, и фракционировали на Fr1–Fr6, как показано на фигуре 2, в соответствии с интенсивностью экспрессии CD45RA и CD25 в качестве индекса, и клетки, содержащиеся в каждой фракции, выделяли.
[Пример 2]
[0057] Выделение РНК из фракционированных клеток и анализ последовательности кДНК
Клетки, отделенные и извлеченные из каждой фракции, лизировали в буфере RLT (Qiagen N.V.), и общую РНК экстрагировали с использованием Agencourt RNAClean XP (Beckman Coulter, Inc.). Восстановленную РНК получали в кДНК, используя набор для секвенирования SMART–Seq v4 Ultra Low Input RNA (Clontech Laboratories, Inc.), и получали библиотеку, используя набор KAPA Hyper Prep для illumina (Kapa Biosystems, Inc.). Для синтеза кДНК и получения библиотеки, постоянно проводили контроль качества, используя Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.) для подтверждения отсутствия проблем при проведении этих процедур. Готовую библиотеку кДНК титровали с использованием набора KAPA library Quantification kit Illumina Platforms (Kapa Biosystems, Inc.). Затем проводили секвенирование ДНК с помощью парных концевых считываний с использованием Hiseq 4000 (Illumina, Inc.) с получением 20000000 считываний или более данных последовательности пар из 100 оснований на образец (файл Fastq).
Исходные данные (файл Fastq) анализировали с помощью FastQC, а последовательности адаптера и повторяющиеся последовательности удаляли с использованием CutAdapt. Пары каждого спаривания и чтения сопоставляли с помощью программы cmpfastq_pe. hg38 использовали в качестве эталонной последовательности в картировании генома, и данные прочтения картировали на геном при настройке по умолчанию программы TOPHAT2 с Bowtie 2. Картированные данные прочтения сортировали по последовательности с помощью программы SAMtools и подсчитывали с помощью программы HTSEQ. Данные подсчета нормализовали с помощью программы Deseq 2. Среди полученных фракций фракция, содержащая Treg–клетки, была подтверждена следующим способом.
[0058] Известно, что Treg–клетки конститутивно экспрессируют гены FoxP3 и Ikzf2 в качестве маркерных генов и редко секретируют IFNγ или IL2, даже когда они активируются стимуляцией. Наличие или отсутствие Treg–клеток можно до некоторой степени подтвердить, изучив уровни экспрессии этих генов. В результате изучения уровней экспрессии этих генов в отношении каждой фракции опухоль–инфильтрирующих клеток и РВМС на основе данных RNA–Seq, описанных выше, было обнаружено, что Ikzf2 и FoxP3 специфически экспрессируются в Fr2 и Fr3 опухоль–инфильтрирующие клетки и Fr2 PBMCs и редко экспрессируются в других фракциях (фигура 4). Кроме того, было обнаружено, что IFNγ (IFN–гамма) и IL2 специфически экспрессируются в Fr4 и Fr5 опухоль–инфильтрирующих клеток и в Fr4 и Fr5 клеток РВМС и не экспрессируются в других фракциях (фигура 4). В заключение было обнаружено, что Treg–клетки содержатся в Fr2 и Fr3 опухоль–инфильтрирующих клеток и в Fr2 РВМС и не содержатся в других фракциях.
[Пример 3]
[0059] Измерение скорости деметилирования области FoxP3
Скорость деметилирования области FoxP3 служит показателем точного определения доли Treg–клеток. Поэтому клетки в Fr2–Fr5 опухоль–инфильтрирующих клетках злокачественного новообразования почки, полученных, как описано выше, исследовали на предмет скорости деметилирования области FoxP3. Область, деметилированная специфичным для Treg–клетки образом, находится (chrX, 49118000–49118500, hg19) в конкретной области CpG в первом интроне гена FoxP3. Клетки, содержащиеся в каждой фракции опухоль–инфильтрирующих клеток, могут быть проанализированы на предмет деметилирования этой области для проверки, состоит ли фракция, полученная на этот раз только из Treg–клеток или других клеток, которые также сосуществуют с ними.
Каждую фракцию (Fr2, Fr3, Fr4 и Fr5) опухоль–инфильтрирующих CD4+ T–клеток выделяли, и ДНК генома восстанавливали с использованием метода экстракции фенолом. ДНК генома обрабатывали бисульфитом с использованием набора MethylEasy Xceed (Human Genetic Signatures), и область интрона 1 FOXP3 (chrX, 49118000–49118500, hg19), область деметилирования, специфичную для Treg–клеток, подвергали ПЦР с ампликонами. Метилирование ДНК определяли с использованием полученного метилированного ДНК–специфичного флуоресцентного зонда FAM и специфичного к деметилированию флуоресцентного зонда VIC и цифровой системы ПЦР QuantStudio 3D (Applied Biosystems, Inc.). После ПЦР с ампликоном определяли величины светового излучения флуоресцентных зондов FAM и VIC, и скорость метилирования ДНК рассчитывали из отношения между этими величинами флуоресцентных излучений и использовали в качестве скорости метилирования каждой фракции (Fr2 до Fr5).
В результате 95% или более последовательностей CpG в области интрона 1 FOXP3 (chrX, 49118000–49118500) были деметилированы в клетках, содержащихся в Fr2 и Fr3 опухоль–инфильтрирующих клеток, тогда как скорости деметилирования Fr4 и Fr5 были 50% или менее. В заключение следует отметить, что почти все клетки, содержащиеся в Fr2 и Fr3, оказались Treg–клетками (фигура 5).
[Пример 4]
[0060] Идентификация CCR8
Для идентификации гена одной группы, специфически экспрессируемый в Treg–клетках (Fr2 опухоль–инфильтрирующих клеток), проводили иерархический кластерный анализ данных экспрессии гена на полученной из РВМС фракции CD4+ T–клеток того же пациента, что и в каждой фракции происходящих из опухолей CD4+ Т–клеток. CCR8 идентифицировали как ген, который экспрессировался в Fr2 Treg–клетках и редко экспрессировался в опухолевых Fr5 и Fr4 и Fr5 в РВМС (фигура 6).
[Пример 5]
[0061] Получение клеток, индуцированных на экспрессию CCR8 мыши
Полноразмерный ORF CCR8 мыши (в дальнейшем также называемого mCCR8) встраивали в вектор экспрессии (pcDNA3.4) для конструирования плазмиды pcDNA3.4–mCCR8. Нуклеотидную последовательность изменяли так, чтобы в ней присутствовали кодоны, чаще встречаемые у млекопитающих, без изменения аминокислот. Клетки HEK293 трансфицировали pcDNA3.4 или плазмидой экспрессии pcDNA3.4–mCCR8, используя Lipofectamine 3000, и отбирали по принципу лекарственной резистентности при концентрации генетицина (G418) 1 мг/мл в течение 2 недель.
Выжившие клетки диссоциировали трипсином и промывали средой DMEM/10% FCS. Затем добавляли меченное РЕ антитело против mCCR8 (клон SA214G2), разведенное 1/200, и проводили реакцию на льду в течение 30 минут. Затем клетки один раз промывали DMEM/10% FCS для мечения mCCR8, экспрессируемого на клеточной поверхности. Популяция клеток, экспрессирующих mCCR8, была обогащена сортировкой с использованием клеточного сортера (FACSAria II). Позитивную клеточную популяцию культивировали при 37°С в течение 2 недель в CO2–инкубаторе в присутствии DMEM/10% FCS (среда, содержащая 1 мг/мл G418). Для клеток, трансформированных pcDNA3.4, проводили только отбор лекарств, а сортировку не проводили. Для подтверждения экспрессии обе клетки окрашивали коммерчески доступным анти–PE–меченным антителом против CCR8 мыши (клон SA214G2) и анализировали с использованием проточного цитометра (FACSAria II). Результаты показаны (фигура 7). Экспрессия mCCR8 наблюдалась в 99% или более клеток, трансформированных pcDNA3.4–mCCR8, по сравнению с клетками, трансформированными pcDNA3.4.
[Пример 6]
[0062] Исследование способности антитела против CCR8 мыши (SA214G2) стимулировать FcγR
Анти–CCR8 антитело мыши (клон SA214G2, приобретенное у BioLegend, Inc.) оценивали на способность стимулировать FcgR, необходимый для его ADCC активности, с использованием набора mFcγRIV ADCC Reporter Bioassays Core (Promega Corp.). Этот набор показывает активацию FcγR на эффекторных клетках путем уровня экспрессии гена люциферазы, связанного ниже промотора NFAT в клетках. Активация сигналов FcγR может быть определена количественно путем количественной оценки этого уровня экспрессии.
Далее кратко описаны последовательности выполнения операций. 1×105 клеток/лунку клеток–мишеней HEK293, экспрессирующих mCCR8 (клетки–мишени), диссоциированных трипсином, смешивали с эффекторными клетками, экспрессирующими FcγR, прилагаемыми к набору, в соотношении 1:1,5 в 96–луночном планшете. Сразу после смешивания клеток, добавляли анти–mCCR8 антитело. Концентрация была установлена от 33 мкг/мл до 0,033 мкг/мл, как показано на фигуре 8 (N=2). В качестве отрицательного контроля были использованы только эффекторные клетки. Через 14 часов после добавления антитела клетки извлекали и измеряли активность люциферазы (фигура 8). Показано среднее значение N=2.
В результате, активность люциферазы не наблюдалась ни при одной из концентраций антител для отрицательного контроля, тогда как активность, зависящая от концентрации антител, наблюдалась в группе добавления клеток–мишеней. Ордината отображает относительную величину интенсивности люминесценции. Как видно на фигуре 8, наибольшее значение активности составляло приблизительно 6000 относительных световых единиц (R.L.U), а значение EC50 (приблизительно 3500 R.L.U) составляло приблизительно 0,1 мкг/мл (линии на чертеже). Эти результаты продемонстрировали, что антитело против CCR8 мыши (SA214G2) может активировать FcγRIV.
[Пример 7]
[0063] Измерение активности ADCC
Анти–mCCR8 антитело (SA214G2) оценивали на его цитотоксическую активность, используя клетки HEK293, стабильно экспрессирующие mCCR8, полученных в примере 5.
Выделяли селезенку мыши C57BL/6, и клетки селезенки извлекали через клеточный фильтр. Клетки промывали и затем подвергали взаимодействию с биотинилированным антителом против CD49b (клон DX5) при 4°C в течение 30 минут. После промывки, NK–клетки очищали с использованием микробусин стрептавидина (Miltenyi Biotec) и использовали в качестве эффекторных клеток. Клетки HEK293, экспрессирующие CCR8 мыши, окрашивали Cell Trace Violet (CTV) (Thermo Fisher Scientific Inc., C34557) в конечной концентрации 2,5 мкМ и использовали в качестве клеток–мишеней. Эти клетки смешивали при соотношении эффекторные клетки:клетки–мишени=5:1 (количество эффекторных клеток: 2,5×105 клеток) в 96–луночном планшете (200 мкл/лунку). Анти–мышиное CCR8 антитело или изотипическое контрольное антитело (крысиный IgG2b, клон RTK4530) добавляли в конечной концентрации 1 мкг/мл с последующим культивированием в течение ночи в CO2–инкубаторе при 37°C. Затем добавляли аннексин V с меткой PE (Annexin V–PE, Medical & Biological Laboratories Co., Ltd. (MBL), 4696–100), разбавленный 1/100, в соответствии с прилагаемым протоколом, и клетки окрашивали при 37°С в течение 30 минут и затем один раз промывали. Пропорцию аннексин V–позитивных апоптотических клеток в окрашенных CTV клетках–мишенях анализировали с использованием проточного цитометра. Тестирование проводили в трех повторах (N=3), и показаны среднее значение и его стандартное отклонение. Показан характерный пример двух сходных экспериментов (фигура 9). Добавление анти–мышиного CCR8 антитела мыши по сравнению с изотипическим контрольным антителом значительно повышало долю аннексин–V–позитивных клеток в клетках–мишенях, примерно в 6 раз. В заключение было обнаружено, что анти–мышиное CCR8 антитело (SA214G2) обладает активностью ADCC.
[Пример 8]
[0064] Измерение нейтрализующей активности против CCR8
Анти–мышиное CCR8 антитело (SA214G2) оценивали на его нейтрализующую активность против CCR8 с внутриклеточным притоком кальция, опосредованным CCL1 мыши (лигандом CCR8 мыши), в качестве индекса, используя клетки HEK293, стабильно экспрессирующие CCR8 мыши.
При измерении кальция были использованы следующие реагенты.
HEPES (Wako Pure Chemical Industries, Ltd., CAS. NO. 7365–45–9)
HBSS(+) без фенолового красного (Wako Pure Chemical Industries, Ltd.)
Fluo 3–AM (кат. F023, Dojindo Laboratories)
Пробенецид (CAS–No: 57–66–9, Nacalai Tesque, Inc.)
Pluronic F127 (P3000MP; Life Technologies Corp.)
10 мМ HEPES/HBSS/0,1% буфер BSA (HEPES (конечная концентрация: 10 мМ) и BSA (конечная концентрация: 0,1%) были добавлены к HBSS)
Fluo 3–AM и Pluronic F127 растворяли при конечных концентрациях 4 мкмоль/л и 0,04%, соответственно, в 10 мМ HEPES/HBSS–буфере. Клетки суспендировали в этом растворе и инкубировали при 37°С в течение 1 часа таким образом, чтобы клетки поглощали Fluo 3–AM. Затем клетки трижды промывали 10 мМ HEPES/HBSS /0,1% раствором BSA и суспендировали при концентрации клеток 2×105 клеток/мл в 10 мМ HEPES/HBSS/0,1% растворе BSA, содержащем 1,25 мкМ пробенецида. Затем клетки инкубировали при 37°С в течение 10 минут в CO2–инкубаторе. Антитело против mCCR8 (SA214G2) или антитело контроля изотипа (клон LTF–2, Bio X Cell) дополнительно добавляли в концентрации 5 мкг/мл. Затем клетки инкубировали при 37°С в течение 20 минут.
2 мл раствора клеток помещали в кювету из кварцевого стекла и загружали в спектрофотометр HITACHI F7000 с температурой комнаты измерения, предварительно установленной на 35°C. Условия измерения были такими, как описано ниже.
Длина волны возбуждения: 508,0 нм, длина волны флуоресценции (измерения): 527,0 нм, щель на стороне возбуждения: 5 нм, щель на стороне флуоресценции: 5 нм, напряжение фотоумножителя: 950 В, отклик: 0,5 с
Клетки инкубировали при перемешивании с использованием мешалки в течение приблизительно 30 секунд, пока длина волны флуоресценции не стабилизировалась. Когда длина волны стабилизировалась, добавляли CCL1 мыши при конечной концентрации 50 нМ (4 мкл), чтобы начать измерение. В результате измерения было обнаружено, что предварительное введение антитела против mCCR8 почти полностью подавляет внутриклеточный приток кальция, опосредованный mCCL1 (фигура 10). Такое подавление не наблюдалось при добавлении контрольного антитела. Пробелы в графиках были получены при открытии и закрытии крышки прибора для введения агониста в клетки. В заключение было обнаружено, что анти–mCCR8 антитело (SA214G2) обладает нейтрализующей активностью в отношении CCR8 мыши.
[Пример 9]
[0065] Подтверждение экспрессии mCCR8 в CT26
Клетки CT26 культивировали в 6–луночном планшете, и культуральный раствор удаляли, когда клетки стали приблизительно на 50% слитыми. Добавляли 5 мл 10 мМ EDTA/PBS, и клетки инкубировали при 37°С в течение 5 минут. В результате почти все клетки диссоциировали, суспендировали с помощью пипетки и, таким образом, они могли быть разделены на почти отдельные клетки. Клетки дважды промывали D–MEM/10% FCS, суспендировали в D–MEM/10% FCS и окрашивали на льду с помощью набора LIVE/DEAD(R) Fixable Near–IR Dead Cell Stain (Thermo Fisher Scientific Inc., Thermo Fisher Scientific Inc., L34975) и АРС–меченного антитела против mCCR8 (SA214G2) или АРС–меченного изотипического контрольного антитела. Через 1 час клетки трижды промывали D–MEM/10% FCS и анализировали на экспрессию mCCR8 с использованием проточного цитометра (FACSCanto II). Фон устанавливали с использованием изотипического контрольного антитела, и рассчитывали долю позитивных клеток (P6), равную или превышающую уровень фона и среднюю флуоресценцию APC (фигура 11). В результате не наблюдалось различий в средней интенсивности флуоресценции APC, и позитивные клетки наблюдались редко (0,2%). В заключение, клетки CT26 не распознавались антителом против mCCR8, и было подтверждено, что клетки CT26 не экспрессируют mCCR8.
[Пример 10]
[0066] Подтверждение экспрессии CCR8 в опухоль–инфильтрирующих клетках с использованием клеток линии колоректального рака линии CT26
3×105 клеток CT26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (7 недель, самка) (N=3). На 3–й день после трансплантации внутрибрюшинно вводили 400 мкг антитела против KLH крысы (гемоцианин лимфы улитки, клон LTF–2) (IgG2b). На 4 (4ый день) и 7 (7ой день) дни после введения опухоли были обнаружены у 3 индивидов (N=3). Опухолевые массы клеток CT26 резали ножницами, и опухоль–инфильтрирующие клетки получали, используя коммерчески доступные наборы (Tumor Dissociation Kit, mouse, Miltenyi Biotec and gentleMACS(TM) Dissociator, Miltenyi Biotec, cat. 130–095–929) в соответствии с протоколами, прилагаемыми к наборам.
Полученные клетки пропускали через клеточный фильтр 70 мкм и затем дважды промывали 10 мМ HEPES/HBSS/2% FBS. Затем клетки обрабатывали раствором для лизиса эритроцитов (Miltenyi Biotec) в течение 5 минут для удаления эритроцитов и дополнительно дважды промывали 2% FCS (фетальная телячья сыворотка)/10 мМ HEPES/HBSS–буфер. Опухоль–инфильтрирующие клетки разделяли на две части, одну из которых использовали для идентификации Treg–клеток, а другую использовали для идентификации миелоидных (макрофагальных) клеток. Клетки окрашивали с использованием следующего метода и антител. Используемые антитела, окрашивающие реагенты и аналитические буферы были такими, как описано ниже.
[0067] Использовали следующие антитела.
(Набор антител для подтверждения Treg–клеток)
PE анти–мышь/крыса FoxP3 (клон FJK–16s), eBioscience, Inc.
Анти–мышиный CD4 PerCP/Cy5.5 (клон RM4–5), eBioscience, Inc.
Анти–мышиный CD8a FITC (клон 5H10–1), BioLegend, Inc.
Bv421 анти–мышиный CD25 (клон PC61), BioLegend, Inc.
Bv510 анти–мышиный CD45 (клон 30–F11), BioLegend, Inc.
AF647 анти–мышиный CCR8 (клон SA214G2), BioLegend, Inc.
AF647 изотипический контроль (клон RTK4530), BioLegend, Inc. (CCR8–негативный контроль)
(Набор антител для подтверждения миелоидных и макрофагальных клеток)
AF647 анти–мышиный CCR8 (клон SA214G2), BioLegend, Inc.
AF647 изотипический контроль (клон RTK4530), BioLegend, Inc. (CCR8–негативный контроль)
Bv510 анти–мышиный CD45 (клон 30–F11), BioLegend, Inc.
FITC анти–мышиный Gr–1 (клон RB6–8C5), BioLegend, Inc.
Bv421 анти–мышиный F4/80 (клон BM8), BioLegend, Inc.
PECy7 анти–мышиный CD11b (клон M1/70), BioLegend, Inc.
PerCP/Cy5.5 анти–мышиный MHC класс II IA/IE (клон M5/114.15.2), BioLegend, Inc.
PE анти–мышиный CD206 (клон C068C2), BioLegend, Inc.
(Другие используемые реагенты)
Набор Zombie NIR Fixable Viability Kit (номер по каталогу 423106), BioLegend, Inc.
Буферный набор BD Pharmingen Transcription Factor buffer Set (номер по каталогу 562574)
Буфер BD Pharmingen Lysing Buffer (номер по каталогу 555899)
HBSS(–), Wako Pure Chemical Industries, Ltd., 084–08345
FCS (HyClone Laboratories Inc., номер по каталогу SH30070.03)
[0068] Метод окрашивания был следующим: инфильтрующие клетки окрашивали на льду в течение 30 минут с использованием реагента из набора Zombie NIR Fixable Viability Kit. Клетки промывали один раз 2% FCS/10 мМ HEPES/HBSS. Затем Treg– и CCR8–позитивные клетки окрашивали Bv510–меченным антителом против CD45, PerCP/Cy5.5–меченным антителом против CD4 мыши, FITC–меченным антителом против CD8 мыши, Bv421–меченным антителом против CD25 мыши и AF647– меченным антителом против CCR8 мыши (или AF647–меченным изотипическим контрольным антителом). Моноцитарные клетки окрашивали Bv510–меченным антителом против CD45, FITC антителом против Gr–1 мыши, PECy7 антителом против CD11b мыши, Bv421 антителом против F4/80 мыши, PerCP/Cy5.5–меченным антителом МНС класса 2 (IA/IE) и PE–меченным антителом против CD206 мыши.
Окрашивание проводили на льду в течение 30 минут. Клетки дважды промывали 2% FCS/HEPES/HBSS и затем фиксировали, используя коммерчески доступный набор (набор для окрашивания FoxP3, eBioscience, Inc.) в соответствии с прилагаемым протоколом, и внутриклеточный FoxP3 окрашивали с использованием PE–меченного антитела против FoxP3. Клетки промывали буфером, прилагаемым к набору, и затем анализировали с помощью проточного цитометра.
[0069] Анализировали CD45+ CD4+ Т–клетки. Область негативных клеток в CD45+ CD4+ T–клетках определяли путем окрашивания контрольным изотипическим антителом, и клетки, позитивные как к антителу против CD25 мыши, так и к антителу против FoxP3 мыши, использовали в качестве Treg–клеток для вычисления частоты присутствия через 4 дня после введения (7 дней после инокуляции) и 7 дней после введения (10 дней после инокуляции). В результате, приблизительно 23% (4d) и приблизительно 30% (7d) CD45+ CD4+ T–клеток в опухолях мыши были CD25+ FoxP3+ клетками (фигура 12).
[0070] Затем проанализировали экспрессию CCR8 в CD45+ CD4+ CD25+ FoxP3+ T–клетках. Область негативных клеток в CD45+ CD4+ T–клетках определяли путем окрашивания антителом контроля изотипа, и клетки, позитивные к антителу против CCR8 мыши, использовали в качестве CCR8+ Treg–клеток для вычисления частоты присутствия через 4 дня после введения (7 дней после инокуляции) и 7 дней после введения (10 дней после инокуляции) (фигура 13). В результате приблизительно 50% (4d) и приблизительно 67% (7d) CD45+ CD4+ CD25+ FoxP3+Т–клеток в опухолях мыши были клетками CCR8+ (фигура 13).
[0071] Что касается миелоидных клеток, миелоидную популяцию селектировали на проход клеток CD45+ и FSC/SSC, используя проточный цитометр, и анализировали на содержание клеток CCR8+ в клетках CD11b+ Gr1+ CD206+. В результате было обнаружено, что от 40 до 50% клеток как через 7 дней после инокуляции (через 4 дня после введения), так и через 10 дней после инокуляции (через 7 дней после введения) были CCR8–позитивными (фигура 14). Кроме того, скорость экспрессии CCR8 в клетках CD45+ CD11b+ F4/80+ (N=3) в отличающейся от них популяции макрофагальных клеток измеряли так же, как описано выше. В результате было подтверждено, что 45,3% (стандартное отклонение: ±8,2%) клеток экспрессируют CCR8 на 10–й день после трансплантации (7 дней). На основании этих результатов было обнаружено, что по меньшей мере CD4+ CD25+ FoxP3+ T–клетки и CD11b+ Gr1+ CD206+ макрофаги (называемые макрофагами M2) в качестве опухоль–инфильтрирующих клеток экспрессируют CCR8.
[Пример 11] [0072] Исследование влияния удаления опухоль–инфильтрирующих Treg–клеток или опухоль–инфильтрирующих макрофагальных клеток путем введения антитела против mCCR8
3×105 клеток CT26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (7 недель, самка). Через 3 дня после инокуляции 400 мкг (объем жидкости: 400 мкл) крысиного антитела против CD198 мыши (CCR8) (клон SA214G2, BioLegend, Inc.) или антитела контроля изотипа (клон LTF–2) вводили в хвостовую вену (каждая группа N=3). Через 7 дней после инокуляции опухоли (через 4 дня после введения антитела) и через 10 дней после инокуляции опухоли (через 7 дней после введения антитела) опухоли извлекали, и опухоль–инфильтрирующие клетки получали и анализировали (фигура 15).
Опухоль–инфильтрирующие Treg–клетки извлекали так же, как в примере 10. Используемые антитела были такими же, как в примере 10.
[0073] Сначала инфильтрирующие клетки окрашивали на льду в течение 30 минут с использованием набора Zombie NIR Fixable Viability. Клетки промывали один раз 2% FCS/10 мМ HEPES/HBSS и затем окрашивали Bv510–меченным антителом против CD45, PerCP/Cy5.5–меченным антителом против CD4 мыши, FITC–меченным антителом против CD8 мыши, Bv421–меченным антителом против CD25 мыши и AF647–меченным антителом против CCR8 мыши (или AF647–меченным антителом контроля изотипа). Окрашивание проводили на льду в течение 30 минут. Клетки дважды промывали 2% FCS/HEPES/HBSS и затем фиксировали, используя коммерчески доступный набор (набор для окрашивания FoxP3, eBioscience, Inc.) в соответствии с прилагаемым протоколом, и внутриклеточный FoxP3 окрашивали с использованием PE–меченного антитела против FoxP3. Клетки промывали буфером, прилагаемым к набору, и затем анализировали с помощью проточного цитометра.
[0074] Клетки CD45+ CD4+ FoxP3+ CD25+ использовали в качестве Treg–клеток мыши. Область негативных клеток в Treg–клетках определяли окрашиванием AF647–меченным антителом против изотипа, и клетки, позитивные к AF647–меченному антителу против CCR8 мыши в сравнении с контролем, использовали в качестве CCR8–позитивных клеток для расчета их частоты.
В результате, как показано на фигуре 16, процент позитивных CD45+ CD4+ CD25+ FoxP3+ T–клеток (Treg–клеток) у мышей, которым вводили антитело против CCR8 мыши (SA214G2), составлял приблизительно 80% через 7 дней после инокуляции опухоли (через 4 дня после введение антитела) и приблизительно 40% через 10 дней после инокуляции опухоли (7 дней после введения антитела 7) (фигура 16), при этом доля внутриопухолевых CD45+ CD4+ CD25+ FoxP3+ T–клеток (Treg–клеток) у мышей с данным изотипическим антителом была определена как 100% (через 10 дней после инокуляции опухоли). Уровень значимости ** составлял P <0,01 (критерий Стьюдента). Эти результаты показали, что приблизительно 60% опухоль–инфильтрирующих Treg–клеток были удалены с помощью антитела против CCR8 через 7 дней после введения антитела против CCR8.
[0075] Таким же образом, как указано выше, опухоль–инфильтрирующие клетки отделяли от опухолей на 7–й день после трансплантации (d7), и среди клеток CD45+ миелоидную популяцию подвергали селектированию на FSC/SSC (называемой FSC/SSC+) с последующим анализом CD11b+ F4/80+клеток в клетках. F4/80 (Ly719) является маркером зрелых макрофагов и моноцитов мыши. Как показано на фигуре 17, относительное содержание CD11b+ F4/80+ клеток было снижено в группе введения антител против mCCR8 (N=3) по сравнению с контролем изотипа (N=3) (t–тест; P=0,062). График показывает относительное содержание клеток F4/80+ в популяции мононуклеарных клеток CD45+ FSC/SSC+.
Относительное содержание IA/IE–позитивных или класса 2 (IA/IE)–негативных клеток в клетках F4/80+, показанных на фигуре 17, дополнительно показано для молекул MHC (антиген гистосовместимости опухоли) класса 2. Как показано на фигуре 18, в группе введения антитела против mCCR8 (N=3) в сравнении с контролем изотипа (N=3), IA/IE–негативная группа демонстрировала тенденцию к снижению, и IA/IE–позитивная группа была значительно снижена (t–тест; уровень значимости *; P <0,05). В заключение, было обнаружено, что внутриопухолевая популяция моноцитов/макрофагов CT26 мыши или часть популяции имеют пониженное количество внутриопухолевых клеток.
[Пример 12]
[0076] Оценка противоопухолевого эффекта введения антитела против mCCR8 с использованием производного колоректального злокачественного новообразования CT26
3×105 клеток колоректального злокачественного новообразования CT26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка). Через 3 дня после инокуляции опухоли внутривенно вводили 400 мкг (400 мкл) крысиного антитела против CD198 мыши (CCR8) (клон SA214G2, BioLegend, Inc.) (N=10). Для контроля вводили контрольного изотипическое антитело (N=10). Объемы опухолей измеряли каждые 3–4 дня через 8 дней после инокуляции опухоли (через 5 дней после введения антител). Объем опухоли (мм3) рассчитывали в соответствии с большая ось (мм) х малая ось (мм) х малая ось (мм)/2 (фигура 19).
В результате в группе введения анти–mCCR8 не наблюдалось значительных различий по сравнению с группой введения антитела контроля изотипа на 7–й день после трансплантации, тогда как объем опухоли в группе введения антитела против mCCR8 значительно уменьшался на 11, 14, 17 и 21 день после инокуляции опухоли (уровень значимости: ***; P <0,001 в дни 11 и 14, **; P 0,01 в дни 17 и 21). Кроме того, в группе введения антитела против CCR8 мыши объем опухоли уменьшался на 14–й день после трансплантации или позже, и на 17–й день опухоли почти полностью исчезали (данные для отдельных субъектов показаны на фигуре 20, а средние данные показаны на фигуре 21). Исходя из этих результатов был сделан вывод о том, что введение антитела против mCCR8 подавляло функции mCCR8, экспрессируемых на Treg и моноцитах/макрофагах, указанных как иммуносупрессивные клетки, или убивало (удаляло) эти экспрессирующие клетки посредством ADCC активности антитела, так что иммунитет к опухоли повышался, что приводило к регрессии и исчезновению опухолей.
[0077] Как уже сообщалось во многих литературных источниках информации и так далее, в случае введения антитела, специфичного к CD25 мыши (анти–CD25), маркера мышиных Treg–клеток, мышам и, таким образом, удаления мышиных Treg–клеток, введение перед инокуляции опухоли проявляет слабый противоопухолевый эффект, а введение на 2–й день или позже после трансплантации не оказывает противоопухолевого эффекта. Также было осуществлено введение антитела против CD25 на 3–й день после трансплантации, используя ту же систему клеток CT26, что и в этот раз, но не было обнаружено противоопухолевого эффекта. На основании этих результатов был сделан вывод о том, что анти–mCCR8 антитело обладает большей лекарственной эффективностью, чем анти–CD25 антитело.
[Пример 13]
[0078] Затем, анти–PD–1 (клон RMP1–14, Bio X Cell), антитело, специфичное к мышиному PD–1, оценивали на лекарственную эффективность, используя CT26, и сравнивали с анти–mCCR8. 2×105 клеток колоректального рака CT26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка). Анти–PD–1 антитело (200 мкг/особь, внутрибрюшинно) вводили всего три раза каждые 3–4 дня через 7 дней после трансплантации.
В результате противоопухолевый эффект наблюдался в группе, которой получавшей анти–PD–1 антитело (N=8), по сравнению с группой, получавшей контрольное изотипическое антитело (N=8). Средний объем опухоли и стандартное отклонение изотипического контроля составили 601,7±378,1 мм3, 956,3±467,7 мм3 и 1528,4±774,1 мм3 через 14, 17 и 20 дней после инокуляции опухоли, соответственно, а средний объем опухоли и стандартное отклонение в группе введения анти–PD–1 антитела составили 175,3±42,6 мм3, 174,7±55,8 мг и 209,6±99,8 мм3 через 14, 17 и 20 дней после инокуляции опухоли, соответственно. Анти–PD–1 антитело значительно подавляло увеличение объема опухоли по сравнению с контролем во все дни – 14, 17 и 20 день – после инокуляции опухоли. Однако в период наблюдения особью, у которой полностью исчезла опухоль, была 1 из 8 мышей (до 20 дня после трансплантации). С другой стороны, в тот же период при введении анти–mCCR8 антитела полное исчезновение опухолей наблюдалось во всех 10 случаях, что и выше. На основании этих результатов был сделан вывод, что при стандартном методе введения, анти–mCCR8 антитело обладает более высокой лекарственной активностью, чем анти–PD–1 антитело.
[Пример 14]
[0079] Подтверждение наличия или отсутствия индукции аутоиммунного заболевания у мыши, получавшей анти–mCCR8 антитело
На следующем этапе, здоровье мышей из примера 12 оценивали вплоть до 18ого дня после введения. Не было обнаружено существенных различий в массе тела в этот период между группой введения контрольного антитела и группой введения анти–CCR8 антитела. В обеих группах пилоэрекция не наблюдалась. Этих мышей препарировали на 18 день после введения. Не смотря на исследование наличия или отсутствия увеличения лимфатических узлов и кишечного тракта в группе, получавшей анти–CCR8 по сравнению с контролем, увеличение не отмечалось, не дела различий в группах. На основании этих результатов был сделан вывод о том, что в период, когда у мышей, получавших анти–CCR8 антитело, проявлялся противоопухолевый эффект, никаких признаков аутоиммунного заболевания не наблюдалось. В статьях сообщается, что обычно, если Treg исчезал из организма мыши до такой степени, что индуцировался противоопухолевый эффект, то через 14 дней после исчезновение возникало тяжелое аутоиммунное заболевание. Это проблема, на которую следует обратить внимание при иммунотерапии опухолей, включая терапию, основанную на супрессии Treg. Результаты, полученные в этот раз, показали, что даже на 18–й день после введения антител у мышей, у которых наблюдался сильный эффект противоопухолевого иммунитета при введении антитела против CCR8, аутоиммунное заболевание не появлялось. Одним из объяснений этого является низкая экспрессия CCR8 мыши и человека в РВМС, селезенке и лимфатическом узле по сравнению с опухолевыми тканями, согласно сообщениям. Однако ни в одном из предыдущих сообщений не указывалось, индуцируется ли или нет аутоиммунное заболевание при удалении или функциональном ингибировании Treg–клеток, экспрессирующих CCR8, в этих периферических тканях. В настоящем изобретении впервые было обнаружено, что при этих подходах не возникает аутоиммунного заболевания. Этот эффект может быть неожиданным на основании ранее известных данных.
[Пример 15]
[0080] Оценка противоопухолевого эффекта при введении анти–mCCR8 антитела, используя клетки колоректального рака Colon–26
2×105 клеток колоректального рака Colon–26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка). Через 3 дня после инокуляции опухоли внутривенно вводили 400 мкг (400 мкл) крысиного анти–мышиного CCR8 антитела (клон SA214G2, BioLegend, Inc.) (N=10). Для контроля вводили контрольного изотипическое антитело (N=10). Объемы опухолей измеряли каждые 3–4 дня через 3 дней после инокуляции опухоли (через 5 дней после введения антител). Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2. Момент времени, когда опухоль достигла объема конечной точки (800 мм3), использовали в качестве конечного показателя для каждого животного. В результате, увеличение объема опухоли подавлялось в группе введения анти–mCCR8 по сравнению с группой введения контрольного изотипического антитела через 14 и 18 дней после инокуляции опухоли. Средний объем опухоли на 14 день составил 451,3 мм3 (стандартное отклонение: ±177,5 мм3) в группе введения контрольного изотипического антитела и 322,6 мм3 (стандартное отклонение: ±146,0 мм3) в группе введения антитела против CCR8. Индивид с объемом опухоли 350 мм3 или более на 14–ый день, был в 9 из 10 случаев в контрольной изотипической группе и в 4 из 10 случаев в группе введения анти–mCCR8. Существовала значительная разница с P=0,019 в критерии хи–квадрат Пирсона в отношении этой изолированной формы. Таким образом, разница наблюдалась по количеству индивидов, у которых объем опухоли достигал 350 мм3 на 14 день. Кроме того, средний объем опухоли на 18 день после трансплантации составлял 874,7 мм3 (стандартное отклонение: ±269,2 мм3) в группе введения контрольного изотипического антитела и 585,4 мм3 (стандартное отклонение: ±401,7 мм3) в группе введения антитела против CCR8 (фигура 22). Индивид с объемом опухоли 600 мм3 или более на 18–й день, был в 9 из 10 случаев из контрольной изотипической группы и, с другой стороны, в 4 из 10 случаев из группы введения анти–mCCR8. Существовала значительная разница с P=0,019 в критерии хи–квадрат Пирсона в отношении этой изолированной формы. Таким образом, разница наблюдалась по количеству индивидов, у которых объем опухоли достигал 600 мм3 на 18 день. Кроме того, момент времени, когда объем опухоли достигал 800 мм3, был задан в качестве конечной точки. Пациент, считающийся умершим, у которого объем опухоли, превышающий 800 мм3, не наблюдался ни в одной из групп до 14 дня, и был в 7 из 10 случаев из группы с контрольным изотипом, и в 3 из 10 случаев из группы введения анти–CCR8 антитела, на 18–ый день. В результате изучения разницы в вероятности выживания в день 18 с помощью критерия хи–квадрат Пирсона, была значительная разница в вероятности выживания с P=0,025.
Противоопухолевый эффект не наблюдался в группе введения анти–PD–1 (клон RMP1–14, Bio X Cell) по сравнению с группой, которой вводили контрольное изотипическое антитело в аналогичном эксперименте с использованием той же клеточной линии, что и выше. В заключение, антитело против mCCR8 проявляло более высокий противоопухолевый эффект на клетки Colon26, устойчивые к анти–PD–1 антителу.
[Пример 16]
[0081] Анализ на экспрессию CCR8 в инфильтрирующих клетках рака почки человека
Экспрессию CCR8 анализировали в опухоль–инфильтрирующих клетках рака почки человека в 14 случаях. Информация о 14 пациентах с раком почки была следующая: пол – 11 мужчин и 3 женщины, средний возраст 68,5 лет и патологические стадии T1A для 6 пациентов, T1B для 2 пациентов, T3A для 5 пациентов и T3b для 1 пациента. В частности, первичные опухоль–инфильтрирующие клетки рака почки выделяли у 14 пациентов с раком почки (почечно–клеточный рак почки, ccRCC) таким же образом, как на фигуре 1 примера 1, окрашенных анти–CD4 (BioLegend, Inc., Clone OKT4), анти–CD3 (BioLegend, Inc., клон UCHT1), анти–CD45RA (BD Biosciences, клон HI100), анти–CD8 (BioLegend, Inc., RPA–T8), анти–CCR8 (BioLegend, Inc., клон L263G8) и анти–FoxP3 (eBioscience, Inc., клон 236A/E7) или контрольное изотипическое антитело против FoxP3 и анализируют с помощью проточной цитометрии (BD Biosciences, BD LSRFortessa). Были проанализированы CD3+ CD8+ T–клетки и CD3+ CD4+ T–клетки. CD3+ CD4+ Т–клетки затем разделяли на 2 группы в зависимости от наличия или отсутствия экспрессии FoxP3, и анализировали. Негативный по экспрессии контроль FoxP3 получали окрашиванием с контрольным изотипическим контролем. Среднее значение анализа FACS (MFI) каждого образца пациента использовали в качестве интенсивности экспрессии CCR8. В таблице 1 показано среднее значение MFI окрашивания с антителом против CCR8 или его контрольным изотипическим антителом и его стандартное отклонение.
[Таблица 1]
[0082] Было обнаружено, что CD8+ T–клетки редко экспрессируют CCR8 (таблица 1). CD4+ FoxP3– T–клетки слабо экспрессировали CCR8, тогда как CD4+ FoxP3+ T–клетки имели в 8 раз или более среднее значение MFI для CD4+ FoxP3– T–клеток, показывая, что CD4+ FoxP3+ T–клетки значительно экспрессируют CCR8 (таблица 1). На фигуре 23 показаны результаты таблицы 1 в графической форме. Каждый график показывает средний уровень экспрессии CCR8 (MFI) каждого образца пациента в проточном цитометре. Горизонтальные линии графика изображают среднее значение MFI образцов. Столбцы отображают стандартные отклонения. Уровень значимости *** обозначает P <0,001. На основании этих результатов было обнаружено, что белок CCR8 специфически экспрессируется на поверхности CD3+ CD4+ FoxP3+ T–клеток, которые проникают в опухоли при раке почки человека (ccRCC). Эти результаты также согласуются с результатами анализа экспрессии мРНК с помощью анализа RNA–Seq.
Опухоль–инфильтрирующие CD4+ T–клетки в 14 образцах ccRCC, описанных выше, подвергали анализу проточной цитометрией с использованием FoxP3 и CCR8. Отношение CCR8–позитивных клеток к FoxP3–позитивным клеткам и отношение CCR8–позитивных клеток к FoxP3–негативным клеткам были построены на выборочной основе (фигура 24). Окрашивание с антителом контроля изотипа использовали в качестве отрицательных стандартов как для FoxP3, так и для CCR8, а клетки, имеющие значение, равное или превышающее этот порог, использовали в качестве положительных клеток. В результате уровень экспрессии CCR8 внутриопухолевых CD3+ CD4+ FoxP3+T–клеток составил приблизительно 75%, и уровень экспрессии CCR8 CD3+ CD4+ FoxP3–T–клеток составил приблизительно 10%.
На основании этих результатов было обнаружено, что CCR8 экспрессируется в большинстве экспрессирующих FoxP3 клеток Treg среди клеток, инфильтрирующих опухоль злокачественного новообразования почки человека, и экспрессируется примерно в 10% CD4–позитивных Т–клеток, отличных от Treg–клеток. Исходя из этих результатов, скорость экспрессии CCR8 человеческих внутриопухолевых FoxP3–позитивных Treg–клеток была аналогична таковой у мышиных внутриопухолевых Treg–клеток, что указывает на возможность того, что специфическое антитело против CCR8 человека может удалить большую часть проникающих в опухоль FoxP3–позитивных Treg–клеток, как у мышей.
[Пример 17]
[0083] Корреляция скорости экспрессии CCR8 в опухоль–инфильтрирующих клетках при различных видах злокачественных новообразований с вероятностью выживания
Ген FoxP3 был идентифицирован как ген, который специфически экспрессируется в Treg–клетках и не экспрессируется в опухолевых клетках или большинстве нормальных клеток человека. Например, ген FoxP3 в качестве маркерного гена Treg–клеток, ген CD3G в качестве маркерного гена T–клеток и NK–клеток и ген CD8A в качестве маркерного гена CD8–позитивных Т–клеток известны в качестве так называемые маркерных генов, которые экспрессируется только в определенных специфических клетках, как указано выше.
В отношении гена FoxP3, маркерного гена клеток Treg, также сообщалось, что уровень экспрессии мРНК гена FoxP3 в каждой опухоли может быть измерен и, таким образом, использован в качестве показателя для отношения численности Treg–клеток в опухоли (Cell, 2015, Vol. 160, p. 48–61).
Как также сообщается в этой статье, количественное соотношение внутриопухолевых Treg–клеток к вероятности выживания может быть проанализировано путем построения кривой выживаемости Каплана–Мейера в отношении скорости внутриопухолевой экспрессии (отношение численности Treg) маркерного гена и вероятности выживания пациентов за счет использования базы данных RNA–Seq, такой как TCGA. Данные RNA–Seq об опухолевых массах представляют собой смешанные данные о мРНК, экспрессированной как в опухолевых клетках, так и в инфильтрирующих клетках, присутствующих в них (лимфоциты, сосудистые клетки и тому подобное). Однако ген, который не экспрессируется в опухолевых клетках, можно рассматривать как ген, экспрессируемый в опухоль–инфильтрирующих клетках. Посредством их использования опухоль–инфильтрирующие клетки могут быть идентифицированы с помощью анализа, как описано выше, то есть анализа экспрессии маркерного гена с использованием данных RNA–Seq по опухолевым массам. Кроме того, уровень экспрессии маркерного гена в опухолевой массе можно рассматривать как произведение количества экспрессирующих клеток определенных клеток, соответствующего маркерному гену, инфильтрирующего опухолевую массу, и уровня экспрессии маркерного гена в каждой экспрессирующей клетке.
В этом контексте, если уровень экспрессии маркерного гена в каждой клетке у индивидов практически постоянен, то уровень экспрессии прямо пропорционален количеству инфильтрирующих клеток. Таким образом, количество внутриопухолевых экспрессирующих клеток может быть рассчитано индивидуально с использованием этого уровня экспрессии и может быть сравнено между индивидами.
[0084] (Анализ экспрессии CCR8 на клеточном уровне)
Данные об экспрессии РНК из 1037 различных типов человеческих клеточных линий зарегистрированы в общедоступной базе данных CCLE (Cancer Cell Line Encyclopedia). Наличие или отсутствие экспрессии гена CCR8 или CD3G в злокачественных клетках, отличных от Т–клеток или нормальных клеток, анализировали с использованием базы данных.
Экспрессию мРНК CD3G и CCR8 анализировали в отношении линий клеток, вызванных раком почки, раком предстательной железы и мочевого пузыря, с использованием базы данных CCLE.
Исследованные клеточные линии представляли собой 40 клеточных линий, полученных из рака почки:
VMRCRCW, SKRC20, SNU34, SKRC31, UOK10, SLR20, OSRC2, TUHR14TKB, SLR24, HK2, A498, RCC4, KMRC1, RCC10RGB, ACHN, SLR25, SNU1272, UMRC6, SLR23, 769P, SLR21, HEKTE, CAKI1, TUHR4TKB, KMRC2, VMRCRCZ, KMRC3, KMRC20, CAKI2, BFTC909, 786O, A704, TUHR10TKB, SLR26, UMRC2, CAL54, FURPNT1, FURPNT2, HEK293 и G402;
8 клеточных линий рака простаты:
VCAP, LNCAPCLONEFGC, DU145, PC3, 22RV1, PRECLH, MDAPCA2B и NCIH660; и
2 клеточные линии рака мочевого пузыря:
TCBC14TK и TCBC2TKB. Во всех этих исследованных клеточных линиях солидного рака, экспрессия CCR8 и CD3G была на том же уровне, что и фоновый уровень, и экспрессия мРНК не наблюдалась (даже самое большое значение, указывающее на экспрессию, составляло 1/500 или менее от уровня экспрессии G3PDH, и все остальные значения составляли 1/1000 или менее от уровня экспрессии G3PDH). Вкратце, было подтверждено, что CCR8 и CD3G редко экспрессируются на солидных раковых клетках. Первичные нормальные клетки, полученные из каждой ткани человека, также анализировали так же, как описано выше. Обнаружено, что CCR8 и CD3G экспрессируются только в некоторых кроветворных клетках и редко экспрессируются в первичных нормальных клетках других тканей.
Эти результаты показали, что клетки этих трех видов злокачественных новообразований не экспрессируют ни CCR8, ни CD3G. Таким образом, был сделан вывод, что данные об экспрессии РНК TCGA, используемые для опухолевых масс рака почки, рака предстательной железы и рака мочевого пузыря, отражают экспрессию мРНК CCR8 и CD3G в инфильтрирующих нормальных клетках, отличных от раковых клеток, присутствующих в опухолевых массах.
[0085] (Анализ с использованием общедоступной базы данных TCGA)
Затем, отношение гена CCR8 к гену CD3G (CCR8/CD3G), экспрессируемого в опухоли рака почки, рака простаты или рака мочевого пузыря, и вероятности выживания пациентов анализировали, используя общедоступную базу данных TCGA. Обнаружено, что ген, который наиболее сильно коррелирует (корреляция Пирсона) с точки зрения экспрессии с генами CCR8 и CD3G в этих 3 опухолях, представляет собой различные гены, специфически экспрессируемые в Т–клетках (FoxP3, CD5, IL7R и т.д., с коэффициентом корреляции r 0,7 или больше). Эти результаты указывают на то, что CCR8 или CD3G не экспрессируются на собственно опухолевых клетках и специфически экспрессируются на опухоль–инфильтрирующих клетках (в частности, Т–клетках). Однако, в данном случае использовали клеточную популяцию, экспрессирующую CCR8, поскольку это не противоречит тому, что CCR8 экспрессируется на инфильтрирующих клетках, отличных от Т–клеток. CD3G, как уже сообщалось в статьях и тому подобное, специфически экспрессируется на Т–клетках и NK–клетках. Кроме того, Т–клетки являются основными опухоль–инфильтрирующими клетками. Следовательно, количество инфильтрирующих Т–клеток можно предположить на основе уровня экспрессии CD3G. Таким образом, значение CCR8/CD3G можно определить как число клеток, экспрессирующих CCR8, в расчете на количество Т–клеток, присутствующих в опухоли.
Отношение CCR8/CD3G и вероятности выживания пациентов были проанализированы в отношении этих 3 карцином с использованием кривой Каплана–Мейера. При раке почки использовали данные почечно–клеточной карциномы почек (TCGA, Provisional) в данных TCGA, и 523 случая имели полные данные о экспрессии РНК и данные о вероятности выживания пациентов. Аналогично, для рака простаты использовали данные аденокарциномы простаты (TCGA, Provisional) в данных TCGA, и 490 случаев имели полные данные экспрессии РНК и данные вероятности выживания пациентов.
Кроме того, для рака мочевого пузыря использовали данные по уротелиальной карциноме мочевого пузыря (TCGA, Provisional) в данных TCGA, и 392 случая имели полные данные об экспрессии РНК и данные о вероятности выживания пациентов.
Пациенты с каждым видом рака поровну разделяли на 2 группы (пациентов с раком почки было нечетное количество, и, следовательно, они были разделены на 261:262) с высокими значениями экспрессии CCR8/CD3G и с низкими значениями экспрессии CCR8/CD3G, после чего следовал анализ с кривой выживаемости Каплана–Мейера с использованием аналитического программного обеспечения R (R–Studio). Логарифмический тест был проведен как тест на значимое различие. Результаты по поводу рака почки показаны на фигуре 25, результаты по поводу рака предстательной железы показаны на фигуре 26, а результаты по поводу рака мочевого пузыря показаны на фигуре 27. Вертикальные линии на графиках показывают, что пациенты выжили, но были классифицированы как исключенные из исследования (в соответствии с так называемым цензором) в данный момент времени, поскольку период оценки был закончен в этот момент времени. Значения на абсциссе показывают количество месяцев на всех графиках.
[0086] В результате в случаях всех 3 карцином группы с высокими значениями CCR8/CD3G имели значительно более низкие вероятности выживания пациентов. Обнаружено, что группы с высоким отношением опухоль–инфильтрующих клеток, экспрессирующих CCR8, и Т–клеток имеют пониженную вероятность выживания. Это говорит о том, что и у людей CCR8–экспрессирующие клетки оказывают подавляющее влияние на иммунитет против опухоли. Это наводит на мысль о том, что, как и при противоопухолевом действии антитела против mCCR8, вводимого мышам, внутриопухолевые клетки, экспрессирующие CCR8, у людей специфически удаляются или уничтожаются каким–то способом, тем самым повышая иммунитет против опухоли и повышая вероятность выживания.
[Пример 18]
[0087] Подтверждение экспрессии CCR8 мыши в клетках LM8 и клетках MethA
Клетки LM8, полученные из остеосаркомы, или клетки MethA, полученные из фибросаркомы кожи, культивировали в 6–луночном планшете, и культуральный раствор удаляли, когда клетки становились приблизительно на 50% слитыми. Добавляли 5 мл 10 мМ EDTA/PBS, и клетки инкубировали при 37°С в течение 5 минут. В результате почти все клетки диссоциировали, суспендировали с помощью пипетки и, таким образом, они могли быть разделены на почти отдельные клетки. Клетки дважды промывали D–MEM/10% FCS, суспендировали в D–MEM/10% FCS и окрашивали на льду с помощью набора LIVE/DEAD(R) Fixable Near–IR Dead Cell Stain (Thermo Fisher Scientific Inc., Thermo Fisher Scientific Inc., L34975) и анти–CCR8 мыши (SA214G2) или контрольного изотипического антитела. Через 1 час клетки трижды промывали D–MEM/10% FCS и анализировали на экспрессию CCR8, используя проточный цитометр (FACSCanto II). Фон устанавливали с помощью контрольного изотипического антитела, и рассчитывали долю позитивных клеток, равную или превышающую уровень фона и среднюю флуоресценцию (фигура 28). В результате никакой разницы в средней интенсивности флуоресценции PE не наблюдалось в обеих клетках, и позитивные клетки не наблюдались. В заключение, эти клетки не распознавались антителом против CCR8 мыши, и было подтверждено, что они не экспрессируют CCR8 мыши и не сохраняют эпитоп, реагирующий с антителом.
[Пример 19]
[0088] Оценка противоопухолевого эффекта при введении анти–мышиного CCR8 антитела с использованием LM8, полученного из остеосаркомы
3×105 клеток LM8 мыши, полученных из остеосаркомы (50 мкл), внутрикожно трансплантировали в спину каждой мыши C3H/He (возраст 7 недель, самец). Через 3 дня после инокуляции опухоли, внутривенно вводили 400 мкг (400 мкл) крысиного антитела против CCR8 мыши (клон SA214G2, BioLegend, Inc.) (N=11). Для контроля вводили контрольного изотипическое антитело (N=10). Объемы опухолей измеряли каждые 3–4 дня через 7 дней после инокуляции опухоли (через 4 дня после введения антител). Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2 (фигура 29). В результате средний объем опухоли в группе введения анти–mCCR8 по сравнению с группой введения контрольного антитела изотипа был значительно снижен во всех временных точках измерения на 18 день после трансплантации или позже (уровень значимости: *; P < 0,05 в день 18, **; P <0,01 в дни 21, 24, 27 и 31, ***; P 0,001 в день 35). Кроме того, в группе введения анти–антитела против CCR8 мыши опухоли исчезли у 6 из 11 мышей и в группе введения изотипичного антитела у 1 из 10 мышей на 31 день после введения антитела. Существовало значимое различие с P=0,031 в критерии хи–квадрат Пирсона в отношении этой изолированной формы.
[Пример 20]
[0089] Оценка противоопухолевого эффекта введения антитела против CCR8 мыши с использованием MethA, полученного из фибросаркомы кожи
1×105 клеток MethA, полученных из фибросаркомы кожи (50 мкл), внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка). Через 3 дня после инокуляции опухоли внутривенно вводили 400 мкг (400 мкл) крысиного антитела против CCR8 мыши (клон SA214G2, BioLegend, Inc.) (N=5). Для контроля вводили контрольное изотипическое антитело (N=5). Объемы опухолей измеряли каждые 3–4 дня через 11 дней после инокуляции опухоли (через 8 дня после введения антител). Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2 (фигура 30).
В результате средний объем опухоли в группе введения анти–CCR8 мыши по сравнению с группой введения контрольного антитела изотипа был значительно уменьшен во всех точках времени измерения на 11 день после трансплантации или позже (уровень значимости: *; P <0,05 во все моменты времени). Кроме того, в группе введения анти–антитела против CCR8 мыши опухоли исчезли у 5 из 5 мышей и в группе введения изотипичного антитела у 0 из 5 мышей на 21 день после введения антитела. Существовала значимое различие с P=0,0016 в критерии хи–квадрат Пирсона в отношении этой изолированной формы.
[Пример 21]
[0090] Оценка противоопухолевого эффекта введения антитела против CCR8 мыши с использованием EMT6, полученного из рака молочной железы
1×105 клеток злокачественного новообразования молочной железы EMT6 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка). Через 3 и 10 дней после инокуляции опухоли внутрибрюшинно вводили 100 мкг (100 мкл) крысиного анти–мышиного CCR8 антитела (клон SA214G2, BioLegend, Inc.) (N=20). Для контроля вводили контрольное изотипическое антитело (N=20). Объемы опухолей измеряли каждые 3–4 дня через 4 дней после инокуляции опухоли (через 1 день после введения антитела). Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2 (фигура 31).
В результате средний объем опухоли в группе введения анти–CCR8 мыши по сравнению с группой введения контрольного изотипического антитела был значительно уменьшен во всех точках времени измерения на 10–й день после трансплантации или позже (уровень значимости: **; P <0,01 в день 10, ***; P 0,001 в дни 14, 17 и 21). Кроме того, в группе введения анти–антитела против CCR8 мыши опухоли исчезли у 19 из 20 мышей и в группе введения изотипного антитела у 2 из 20 мышей на 21 день после введения антитела. Существовала значимое различие (P <0,0001) в критерии хи–квадрат Пирсона в отношении этой изолированной формы.
[Пример 22]
[0091] Подтверждение преимущества анти–CCR8 антитела мыши над анти–PD–1 антителом
2×105 клеток колоректального рака Colon26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка). Через 3 и 10 дней после инокуляции опухоли 400 мкг (400 мкл), контрольное изотипическое антитело, крысиное анти–мышиное CCR8 антитело (клон SA214G2, BioLegend, Inc.) или анти–мышиное PD–1 антитело (RMP1–14, Bio X Cell) вводили внутривенно (N=10). Объемы опухолей измеряли каждые 3–4 дня через 3 дня после инокуляции опухоли. Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2 (фигура 32). В результате объем опухоли в группе введения анти–мышиного CCR8 антитела по сравнению с группой введения изотипического антитела был значительно уменьшен на 17, 20 и 24 дни (непараметрический тест Стила: уровень значимости P <0,05). Никакого значимого различия не наблюдалось в группе введения анти–PD–1 антитела по сравнению с группой введения изотипического антитела в любой момент времени.
Особей мышей с опухолью объемом 1000 мм3 или более на 24 день после введения антитела было 7 из 10 мышей в группе введения изотипического антитела, 2 из 10 мышей в группе введения антитела против CCR8 мыши и 7 из 10 мышей в группе введения анти–PD–1. Группа введения анти–CCR8 имела существенное отличие как от группы введения изотипического антитела, так и от группы введения антитела против PD–1 в тесте Хи–квадрат Пирсона в отношении изолированной формы (P=0,025 для обоих). В заключение было подтверждено, что введение анти–мышиного CCR8 оказывает противоопухолевый терапевтический эффект на клеточную линию колоректального рака Colon26.
Кроме того, объем опухоли в группе введения анти–мышиного CCR8 по сравнению с группой введения анти–PD–1 антитела мыши был значительно уменьшен через 20 и 24 дня после инокуляции опухоли (непараметрический тест Steel–Dwass; уровень значимости P <0,05). В заключение следует отметить, что более сильный противоопухолевый терапевтический эффект на линию колоректальных клеток мыши наблюдался в группе введения анти–мышиных CCR8 антител по сравнению с группой введения анти–PD–1 антител.
[Пример 23]
[0092] Оценка противоопухолевого эффекта при введении анти–мышиного CCR8 антитела с использованием линии клеток, полученных из рака почки RAG
Аналогичное исследование было проведено с использованием линии клеток, полученных из рака почки мыши RAG. 4×105 клеток, полученных из рака почки RAG (50 мкл), внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 8 недель, самка). Через 6 дней после инокуляции опухоли внутрибрюшинно вводили 100 мкг (100 мкл) контрольного изотипического антитела (N=10, за исключением N=9 на 21–й день), крысиного анти–мышиного CCR8 антитела (N=10) (клон SA214G2, BioLegend, Inc.) или анти–мышиного PD–1 антитела (N=10) (RMP1–14, Bio X Cell). Объемы опухолей измеряли каждые 3–4 дня через 6 дней после инокуляции опухоли. Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2 (фигура 33). В результате объем опухоли в группе введения анти–мышиного CCR8 по сравнению с группой введения изотипического антитела был значительно уменьшен на 14, 17 и 21 день (непараметрический тест Стила: уровень значимости P <0,05). Никакого значимого различия не наблюдалось в группе введения анти–PD–1 антитела по сравнению с группой введения изотипического антитела. В заключение, было подтверждено, что введение анти–мышиного CCR8 антитела оказывает противоопухолевый терапевтический эффект на клеточную линию рака почки. Кроме того, объем опухоли в группе введения анти–мышиного CCR8 по сравнению с группой введения анти–PD–1 антитела мыши был значительно уменьшен на 14 день после трансплантации (непараметрический тест Steel–Dwass; уровень значимости P <0,05). В заключение следует отметить, что более сильный противоопухолевый терапевтический эффект на клеточную линию рака почки мыши наблюдался в группе введения антитела против CCR8 мыши по сравнению с группой введения антитела против PD–1 мыши.
[Пример 24]
[0093] Анализ на наличие или отсутствие воспалительного ответа у мыши, получавшей анти–CCR8 мыши
2×105 клеток колоректального рака Colon26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка). Через 3 и 10 дней после инокуляции опухоли внутривенно вводили 400 мкг (400 мкл) крысиного анти–мышиного CD198 антитела (CCR8) (клон SA214G2, BioLegend, Inc.) или контрольного изотипического антитела (LTF–2, Bio X Cell) (N=10). Массу тела и массу каждого органа мыши (легкого, печени, селезенки, тонкой кишки и пахового узла) измеряли на 24 день после трансплантации (фигура 34). В результате, как показано на фигуре 34, не наблюдалось значимого различия в массе тела и массе каждого органа между контрольной группой введения (N=10) и группой введения анти–мышиное CCR8 (N=10). На основании этих результатов был сделан вывод, что введение анти–мышиного CCR8 антитела не вызывало ни воспалительного ответа, ни аутоиммунного заболевания.
[Пример 25]
[0094] Анализ на экспрессию CCR8 в различных клинических опухоль–инфильтрирующих клетках
Экспрессию CCR8 анализировали в опухоль–инфильтрирующих клетках рака почки человека, рака яичника, рака матки, колоректального рака и рака легких. Число пациентов с различными клиническими опухолями, использованных в анализе экспрессии, составляло 12 пациентов с раком почки, 14 пациентов с раком яичника, 21 пациент с раком матки, 10 пациентов с колоректальным раком и 4 пациента с раком легкого. Различные клинические опухоль–инфильтрирующие клетки выделяли таким же образом, как на фигуре 1 примера 1, и окрашивали с анти–CD45 (BioLegend, Inc., Clone H130) и анти–CCR8 (BioLegend, Inc., Clone L263G8) антителами, а затем измеряли методом проточной цитометрии (BD Biosciences, BD LSRFortessa). Анализировали количество CCR8–позитивных клеток на массу опухоли и соотношение CCR8–позитивных клеток к CD45–позитивным лейкоцитам.
В таблице 2 показано среднее количество CCR8–позитивных клеток на массу опухоли и их стандартное отклонение. В таблице 3 показано среднее соотношение CCR8–позитивных клеток к CD45–позитивным лейкоцитам и их стандартное отклонение.
[Таблица 2]
[Таблица 3]
[0095] В различных клинических опухолях с раком почки в качестве эталона, число CCR8–позитивных клеток на массу опухоли, в случае рака яичника и колоректального рака было ниже среднего значения, чем в случае рака почки, и в случае рака тела матки и рака легкого было выше среднего значения, чем в случае рака почки. Что касается соотношения CCR8–позитивных клеток к CD45–позитивным лейкоцитам, в случае рака яичника среднее значение было эквивалентно раку почки, а в случае рака легкого оно было ниже значения для рака почки. Кроме того, в случае рака тела матки и колоректального рака оно было выше значения при раке почки. Экспрессия CCR8 была подтверждена в опухоль–инфильтрирующих клетках рака яичника, рака матки, колоректального рака и рака легкого, в дополнение к опухолевым клеткам рака почки человека. Эти результаты указывают на возможность того, что при раке почки, а также раке яичника, раке матки, раке ободочной и прямой кишки и раке легкого CCR8–позитивные опухоль–инфильтрирующие клетки могут быть удалены с использованием специфического антитела против CCR8 человека.
[Пример 26]
[0096] Оценка противоопухолевого эффекта комбинированного введения анти–мышиного CCR8 антитела и анти–PD–1 антитела, используя EMT6, полученную из рака молочной железы
1×105 клеток злокачественного новообразования молочной железы EMT6 (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 7 недель, самка).
В группе введения одного анти–мышиного CCR8 антитела внутривенно вводили 15 мкг крысиного анти–мышиного CCR8 антитела (клон SA214G2, BioLegend, Inc.) (100 мкл) через 3 и 10 дней после инокуляции опухоли и 200 мкг (100 мкл) контрольного изотипического антитела вводили через 8 и 13 дней после инокуляции опухоли (N=10). В группе введения только анти–PD–1 антитела внутривенно вводили 15 мкг (100 мкл) контрольного изотипического антитела через 3 и 10 дней после инокуляции опухоли, и внутривенно вводили 200 мкг (100 мкл) анти–мышиного PD–1 антитела (RMP1–14, Bio X Cell) через 8 и 13 дней после инокуляции опухоли (N=10). В группе комбинированного введения анти–PD–1 антитела и анти–мышиного CCR8 антитела внутривенно вводили 15 мкг (100 мкл) крысиного анти–мышиного CCR8 антитела через 3 и 10 дней после инокуляции опухоли и 200 мкг (100 мкл) анти–PD–1 антитела вводили внутривенно через 8 и 13 дней после инокуляции опухоли (N=10). В контрольной группе, внутривенно вводили 15 мкг (100 мкл) контрольного изотипического антитела через 3 и 10 дней после инокуляции опухоли, и внутривенно вводили 100 мкл PBS через 8 и 13 дней после инокуляции опухоли (N=10). Объемы опухолей измеряли каждые 3–4 дня через 3 дней после инокуляции опухоли (через 1 день после введения антитела). Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2 (фигура 35).
При сравнении среднего объема опухоли между группами введения, средний объем опухоли был значительно меньше в группе введения анти–мышиного CCR8 антитела по сравнению с группой введения анти–PD–1 антитела в дни 10, 14, 17, 20, 23 и 27 (метод Даннета, уровень значимости: P <0,05). Кроме того, опухоли были небольшими в группе комбинированного введения по сравнению с каждой группой отдельного введения.
Также сравнивали полную ремиссию опухолей через 17 и 27 дней после инокуляции. На 17–й день после трансплантации полная ремиссия опухолей была обнаружена у 0 из 10 мышей в контрольной группе и в группе введения антител против PD–1 и у 1 из 10 мышей в группе введения анти–мышиного CCR8, тогда как опухоли полностью исчезали у 6 из 10 мышей в группе комбинированного введения анти–PD–1 антитела и анти–мышиного CCR8. На 27–й день после трансплантации полная ремиссия опухолей была показана у 2 и 3 из 10 мышей в контрольной группе и группе введения анти–PD–1 антитела, соответственно, и у 7 из 10 мышей в группе введения анти–мышиного CCR8 антитела, тогда как опухоли полностью исчезали у 9 из 10 мышей в группе комбинированного введения анти–PD–1 антитела и анти–мышиного CCR8 антитела.
Затем была вычислена доля индивидов с опухолью более 50 мм3 или меньше (фигура 36). Опухоли более 50 мм3 или меньше у всех индивидов в группе комбинированного введения анти–PD–1 антитела и анти–мышиного CCR8 (100%) на 17–й день после трансплантации и затем были 50 мм3 или меньше до 27–го дня, тогда как в группе введения анти–PD–1 антитела на 17 и 27 день, соответственно, эта доля составляла 10% и 30%, а в группе введения анти–мышиного CCR8 антитела составляла 17% как через 17, так и через 27 дней после инокуляции опухоли.
Эти результаты продемонстрировали, что в группе с комбинированным введением для регрессии опухоли требовалось меньше времени и у нее был сильный регрессирующий эффект по сравнению с другими группами введения.
[Пример 27]
[0097] Оценка противоопухолевого эффекта комбинированного введения анти–мышиного CCR8 антитела и анти–PD–1 антитела, используя клеточную линию рака мыши RAG
4,5×105 клеток рака почки RAG (50 мкл) внутрикожно трансплантировали в спину каждой мыши Balb/c (возраст 6 недель, самка). Используемые RAG–клетки представляли собой RAG–клетки (акклиматизированная клеточная линия) с эффективностью подкожного приживления у мыши, повышенной путем трансплантации, опять мыши, опухоли, которая была успешно привита заранее путем подкожной инокуляции мыши Balb/c и повторения этой операции дважды.
Группе отдельного введения анти–PD–1 антитела внутривенно вводили 50 мкг (100 мкл) анти–PD–1 антитела (RMP1–14, Bio X Cell) через 8 и 15 дней после инокуляции опухоли (N=10). Группе отдельного введения анти–мышиного CCR8 антитела, внутривенно вводили 25 мкг (100 мкл) крысиного анти–мышиного CCR8 антитела (клон SA214G2, BioLegend, Inc.) через 8 и 15 дней после инокуляции опухоли (N=10). Группе комбинированного введения анти–PD–1 антитела и анти–мышиного CCR8 антитела смешивали (100 мкл) и внутривенно вводили 50 мкг анти–PD–1 антитела (RMP1–14, Bio X Cell) и 25 мкг крысиного анти–мышиного CCR8 антитела (клон SA214G2, BioLegend, Inc.) через 8 и 15 дней после инокуляции опухоли (N=10). Контрольной группе внутривенно вводили 100 мкл физиологического раствора через 8 и 15 дней после инокуляции опухоли (N=10).
Объемы опухолей измеряли каждые 3–4 дня через 8 дней после инокуляции опухоли. Объем опухоли (мм3) рассчитывали по большой оси (мм) х малой оси (мм) х малой оси (мм)/2 (фигура 37).
В результате было обнаружено, что опухоли уменьшаются в размере в группе комбинированного введения анти–PD–1 антитела и анти–мышиного CCR8 антитела по сравнению с группой отдельного введения анти–PD–1 антитела или анти–мышиного CCR8 антитела.
[Пример 28]
[0098] Анализ на специфичность анти–мышиного CCR8 антитела с использованием гомозиготной мыши с дефицитом гена CCR8
3×105 клеток колоректального рака Colon26 (50 мкл) внутрикожно трансплантировали в спину каждой мыши дикого типа (N 10) или гомозиготно дефицитной по гену CCR8 мыши (N=5) линии Balb/c. Мышам дикого типа внутривенно вводили 100 мкг (100 мкл) крысиного анти–мышиного CCR8 антитела (клон SA214G2, BioLegend, Inc.) или контрольного изотипического антитела (LTF–2, Bio X Cell) через 3 и 10 дней после инокуляции опухоли (N=5). Гомозиготной мыши с дефицитом гена CCR8 также внутривенно вводили 100 мкг (100 мкл) крысиного анти–мышиного CCR8 антитела (клон SA214G2, BioLegend, Inc.) или контрольного изотипического антитела (LTF–2, Bio X Cell) через 3 и 10 дней после инокуляции опухоли (N=5). Размеры опухолей измеряли с 7–ого дня после введения.
В результате значительная регрессия опухоли и окончательная полная регрессия опухоли наблюдались у всех мышей дикого типа при введении анти–антитела против CCR8 мыши по сравнению с введением контрольного изотипичного антитела. С другой стороны, ни изменений в объеме опухоли, ни регрессии опухоли не наблюдалось у гомозиготных мышей с дефицитом гена CCR8 в группе введения анти–мышиного CCR8 антитела по сравнению с группой введения изотипического антитела (фигура 38).
Противоопухолевый эффект анти–мышиного CCR8 антитела полностью исчезал у гомозиготных мышей с дефицитом гена CCR8, демонстрируя, что используемое анти–мышиное CCR8 антитело (SA214G2) оказывает противоопухолевый эффект через CCR8.
[Промышленная применимость]
[0099] Антитело против CCR8 по настоящему изобретению обладает эффектом активации иммунитета путем уменьшения количества опухоль–инфильтрующих Treg–клеток или тому подобного и, таким образом, может использоваться в фармацевтике для лечения злокачественного новообразования.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИСПЕЦИФИЧЕСКИЕ АНТИТЕЛА К CD25 И Fc ГАММА-РЕЦЕПТОРУ ДЛЯ ЭЛИМИНАЦИИ ОПУХОЛЕСПЕЦИФИЧЕСКИХ КЛЕТОК | 2017 |
|
RU2759970C2 |
| НОВЫЕ МОЛЕКУЛЫ АНТАГОНИСТИЧЕСКИХ АНТИ-TNFR2-АНТИТЕЛ | 2019 |
|
RU2829587C2 |
| АНТИ-ICOS АНТИТЕЛА | 2017 |
|
RU2764548C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА С ИСПОЛЬЗОВАНИЕМ АНТАГОНИСТОВ, СВЯЗЫВАЮЩИХ С ОСЬЮ PD-1, И ИНГИБИТОРОВ TIGIT | 2014 |
|
RU2702108C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА С ИСПОЛЬЗОВАНИЕМ АНТАГОНИСТОВ, СВЯЗЫВАЮЩИХ С ОСЬЮ PD-1, И ИНГИБИТОРОВ TIGIT | 2014 |
|
RU2825390C2 |
| ГУМАНИЗИРОВАННЫЕ АНТИТЕЛА К OX40 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2709742C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ TUSC2-ИММУНОТЕРАПИИ | 2017 |
|
RU2755903C2 |
| АНТИТЕЛО ПРОТИВ TNFR2 И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2793165C1 |
| FC-ОПТИМИЗИРОВАННЫЕ АНТИТЕЛА К CD25 ДЛЯ ИСТОЩЕНИЯ ОПУХОЛЕСПЕЦИФИЧЕСКИХ КЛЕТОК | 2018 |
|
RU2839380C1 |
| КОМБИНИРОВАННОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ РАКА С ИСПОЛЬЗОВАНИЕМ ПОКСВИРУСА, ЭКСПРЕССИРУЮЩЕГО ОПУХОЛЕВЫЙ АНТИГЕН, И АНТАГОНИСТА И/ИЛИ АГОНИСТА ИНГИБИТОРА ИМУННОЙ КОНТРОЛЬНОЙ ТОЧКИ | 2014 |
|
RU2714142C2 |
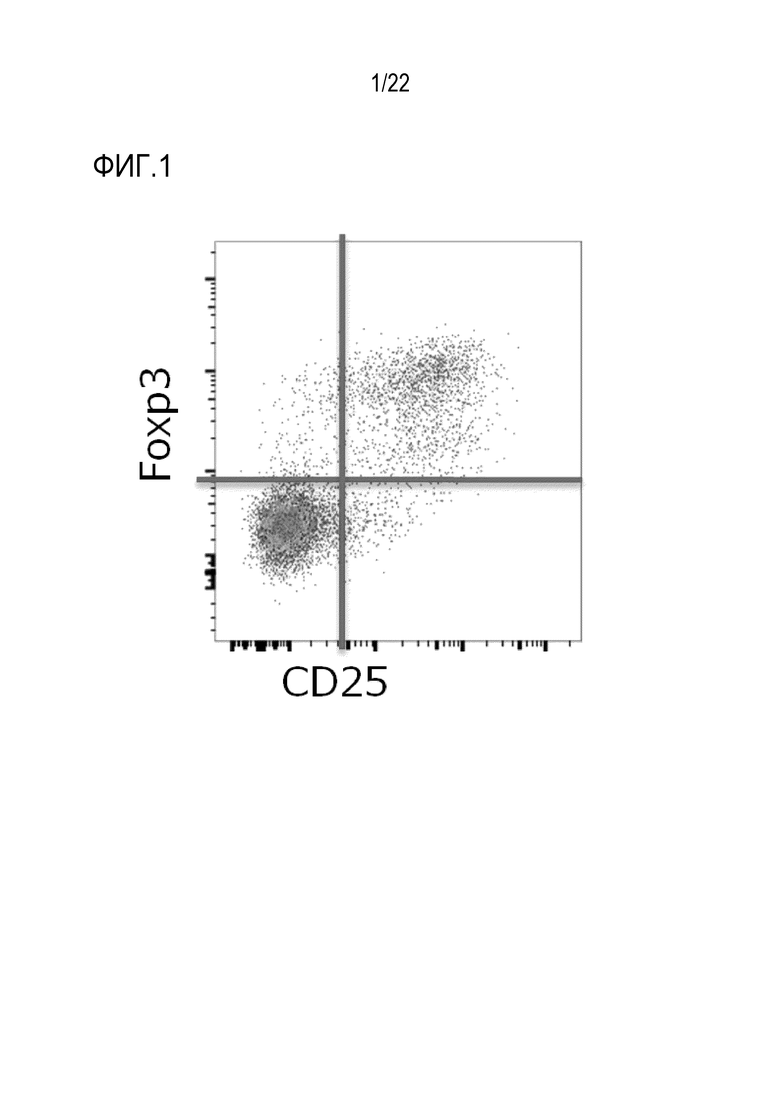
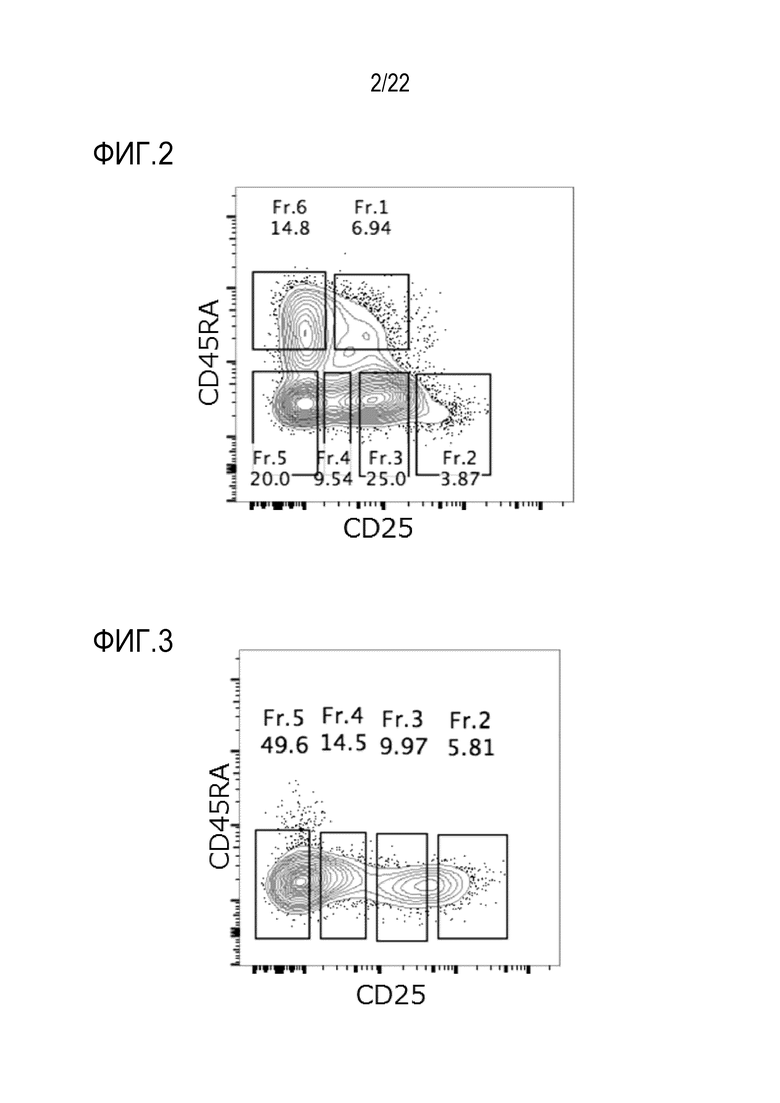
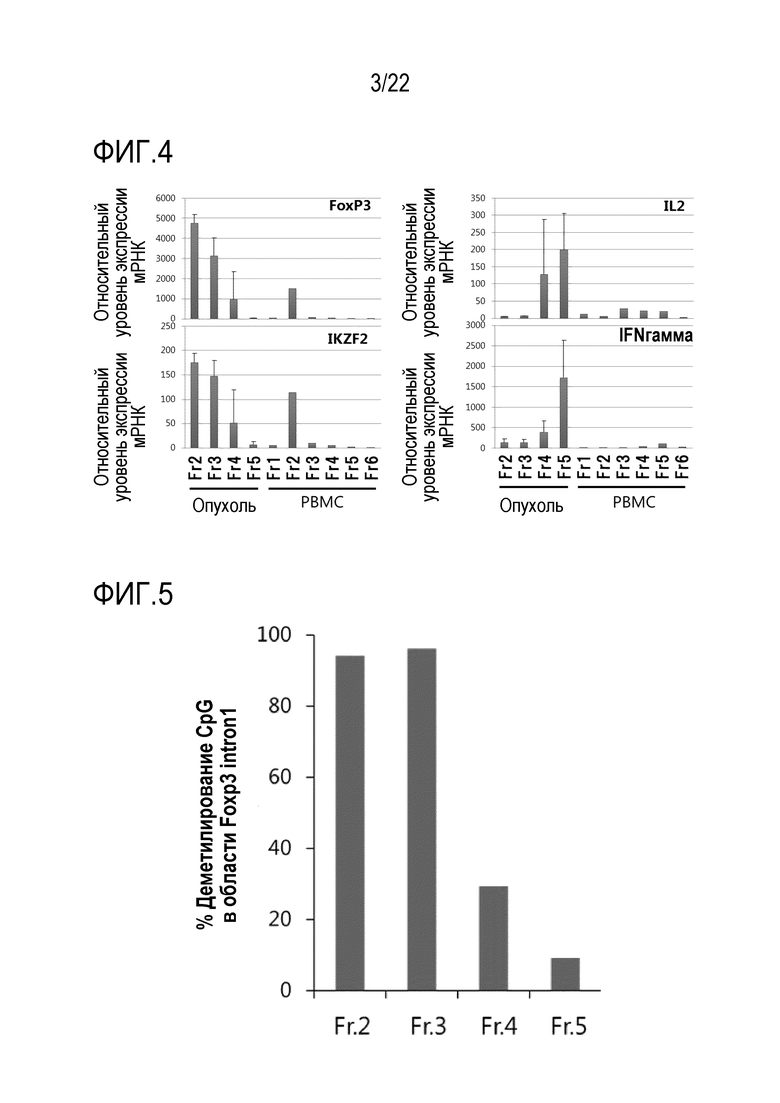
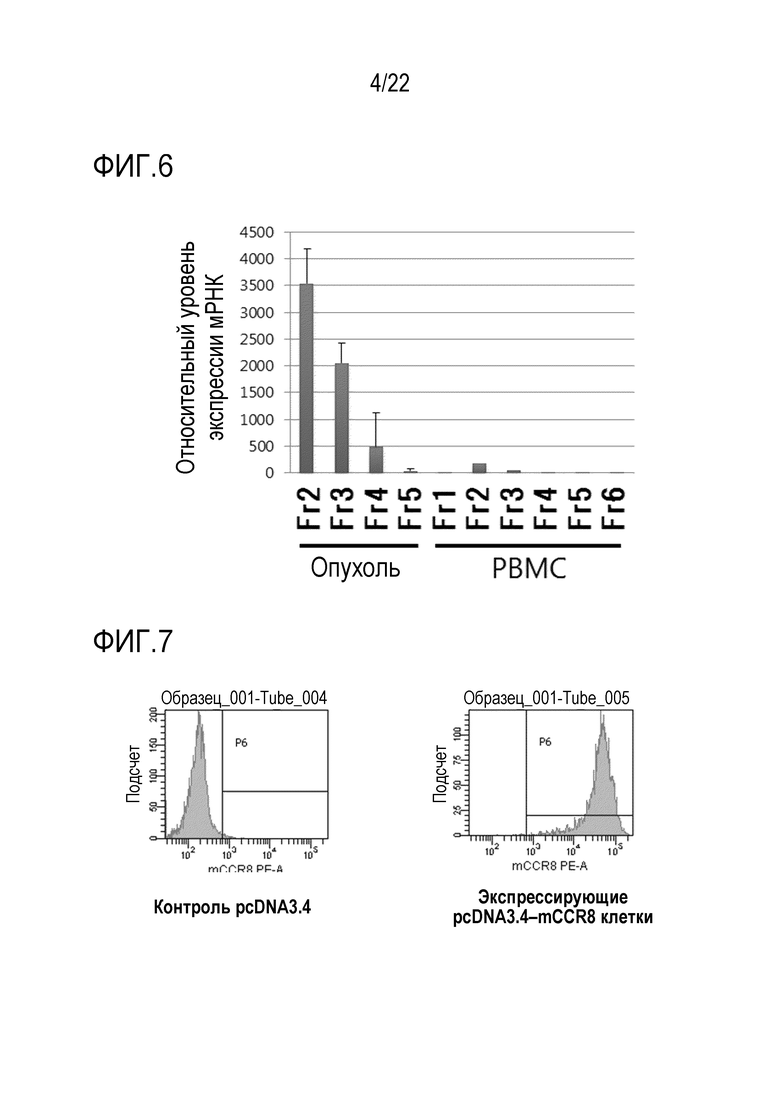
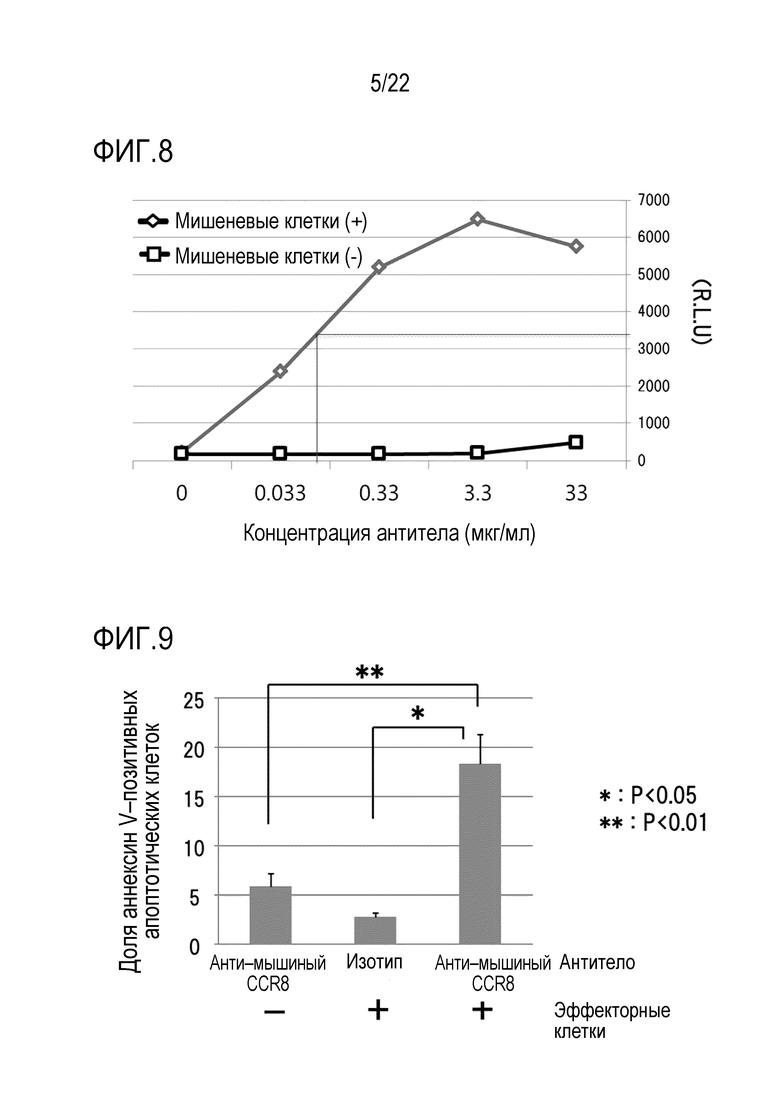
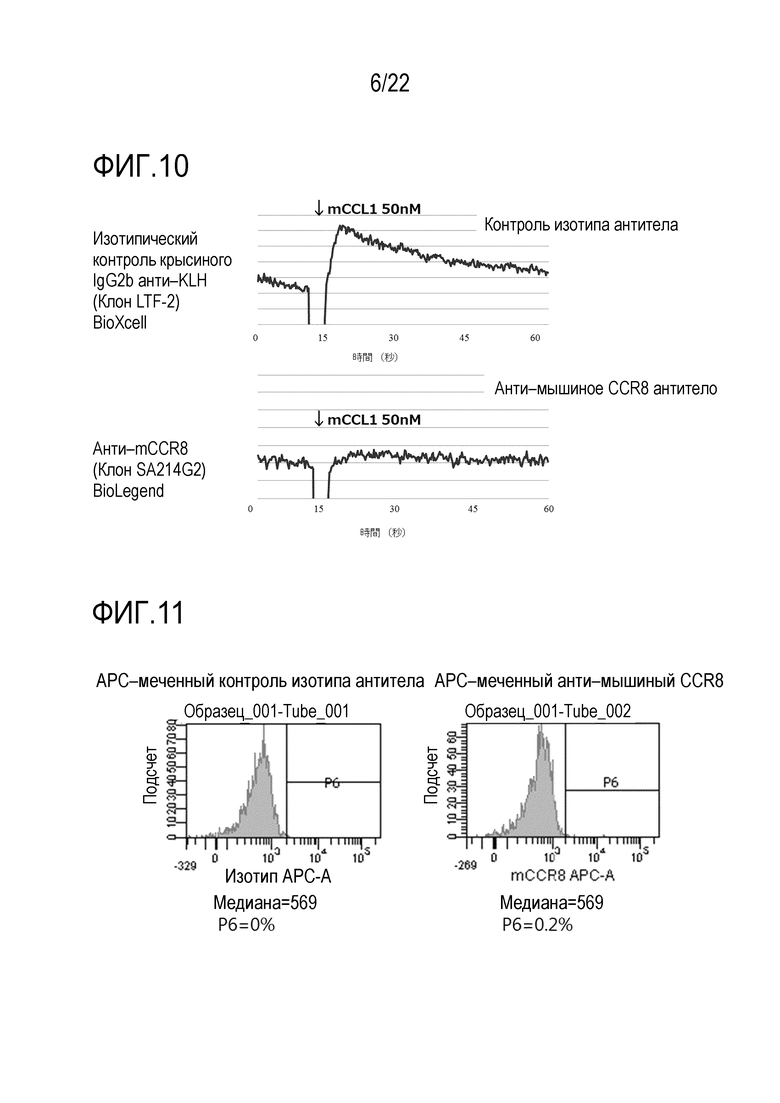
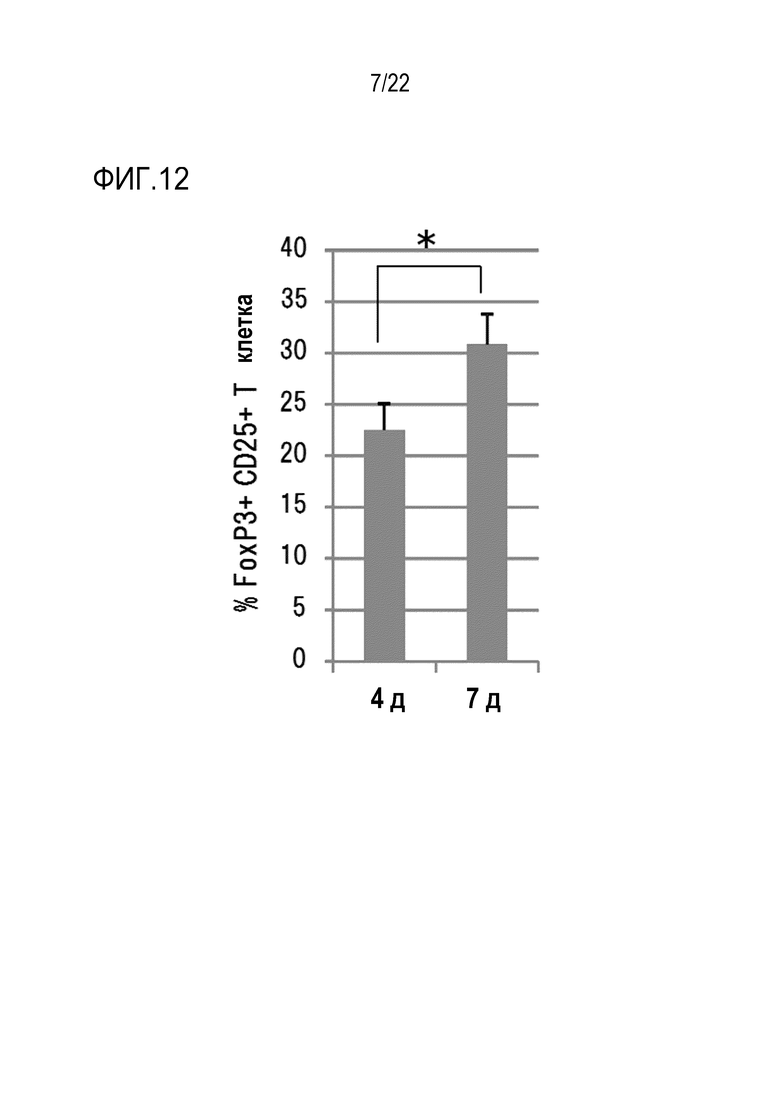
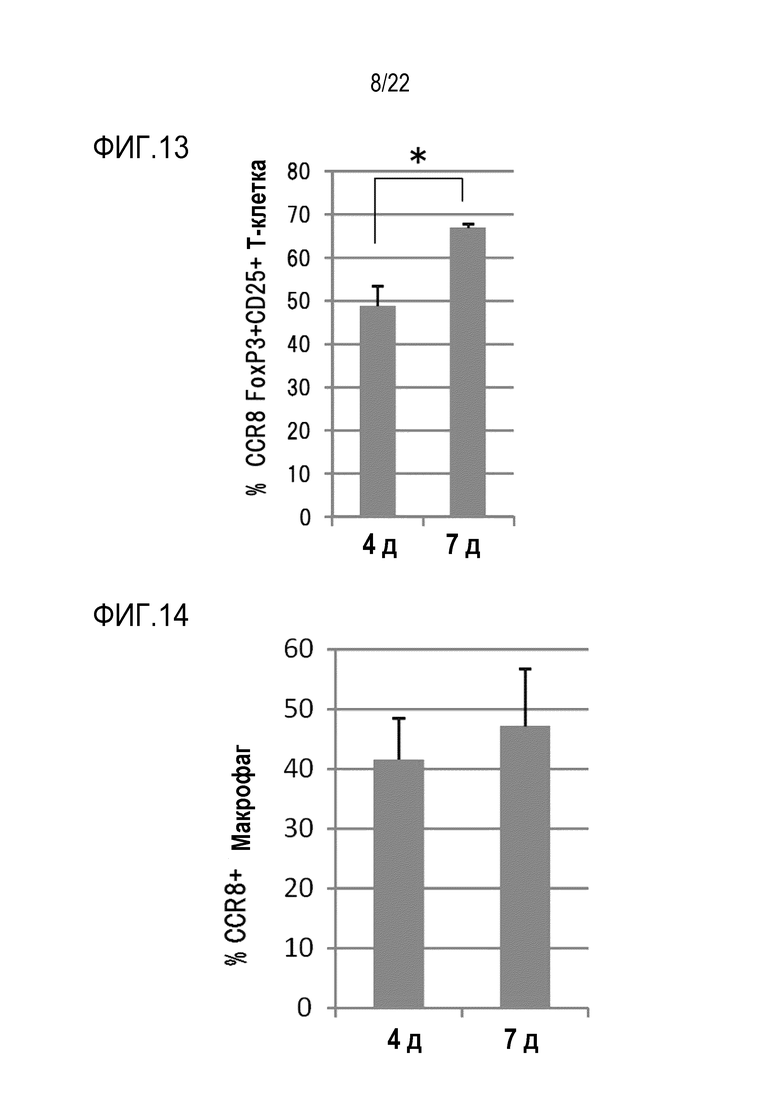
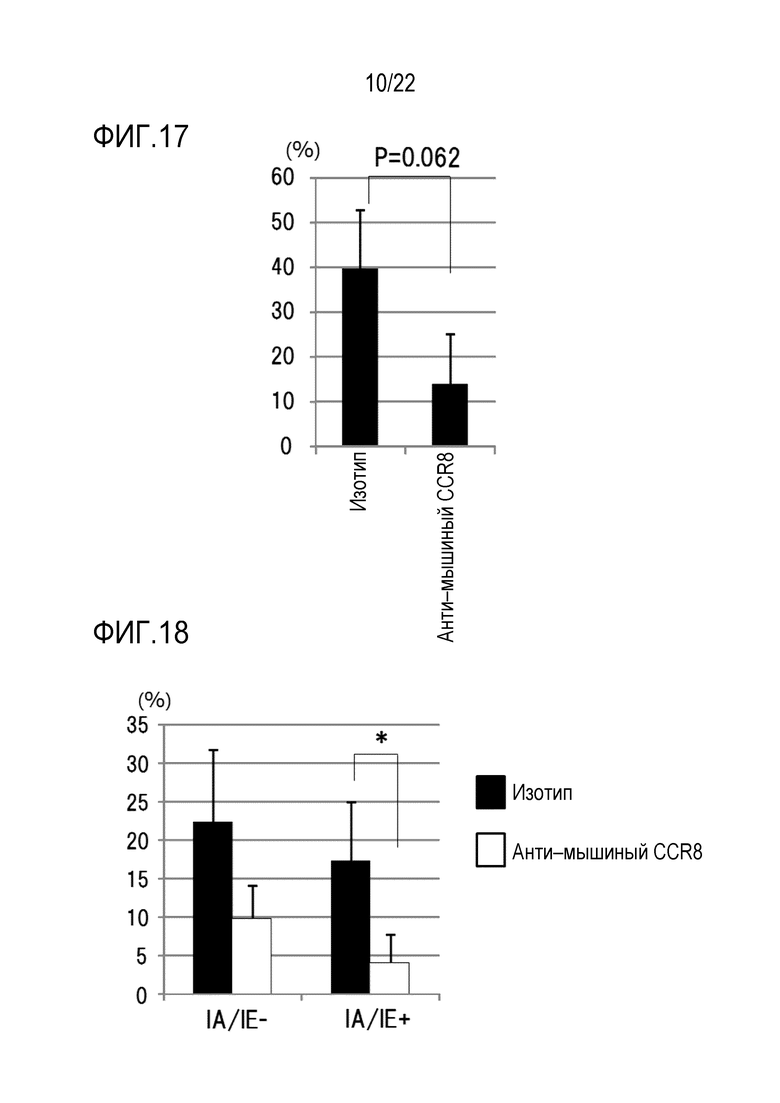
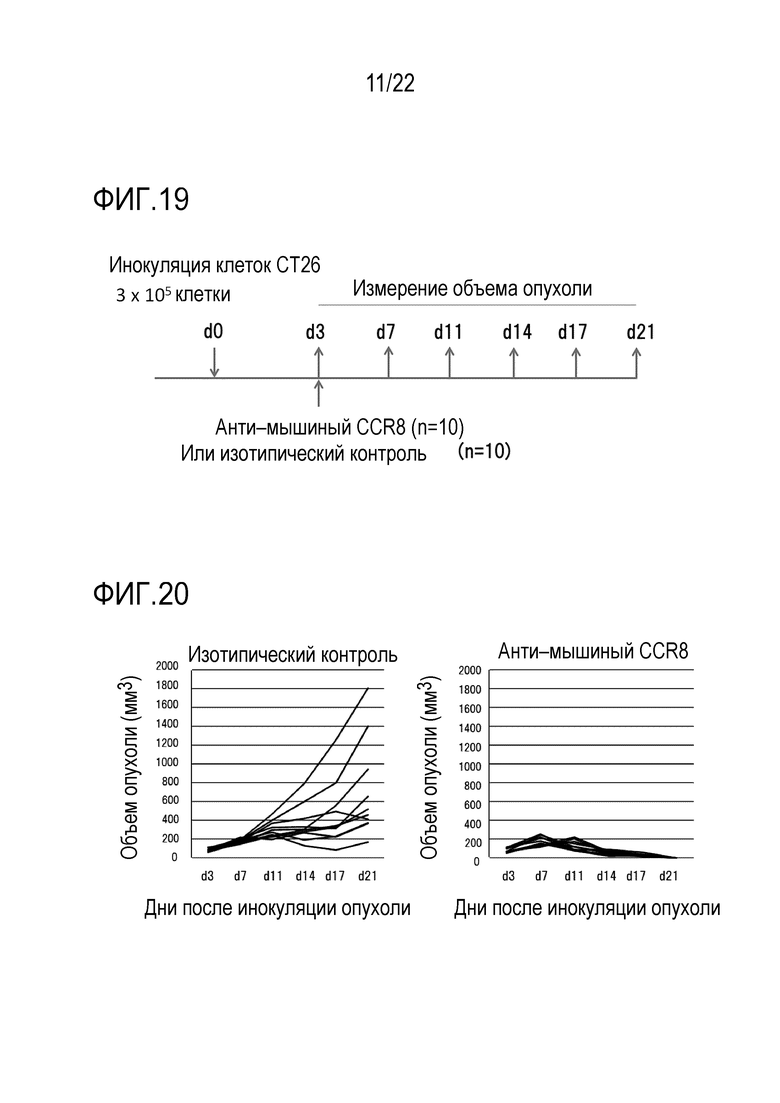
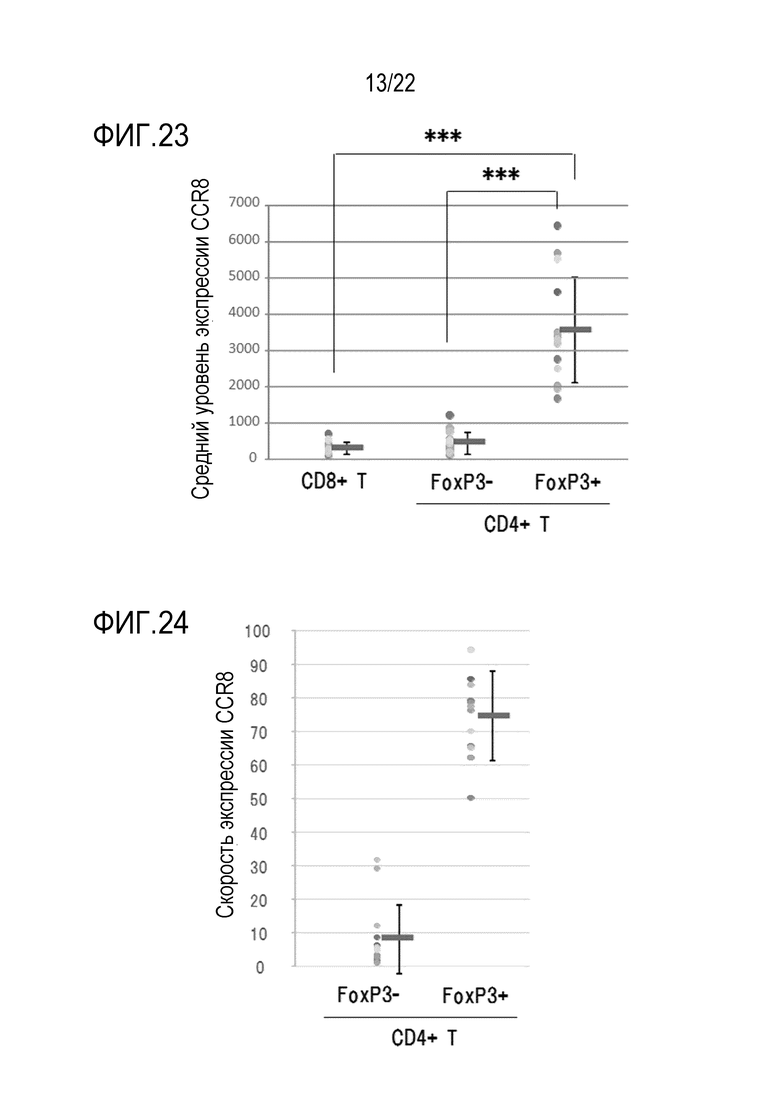
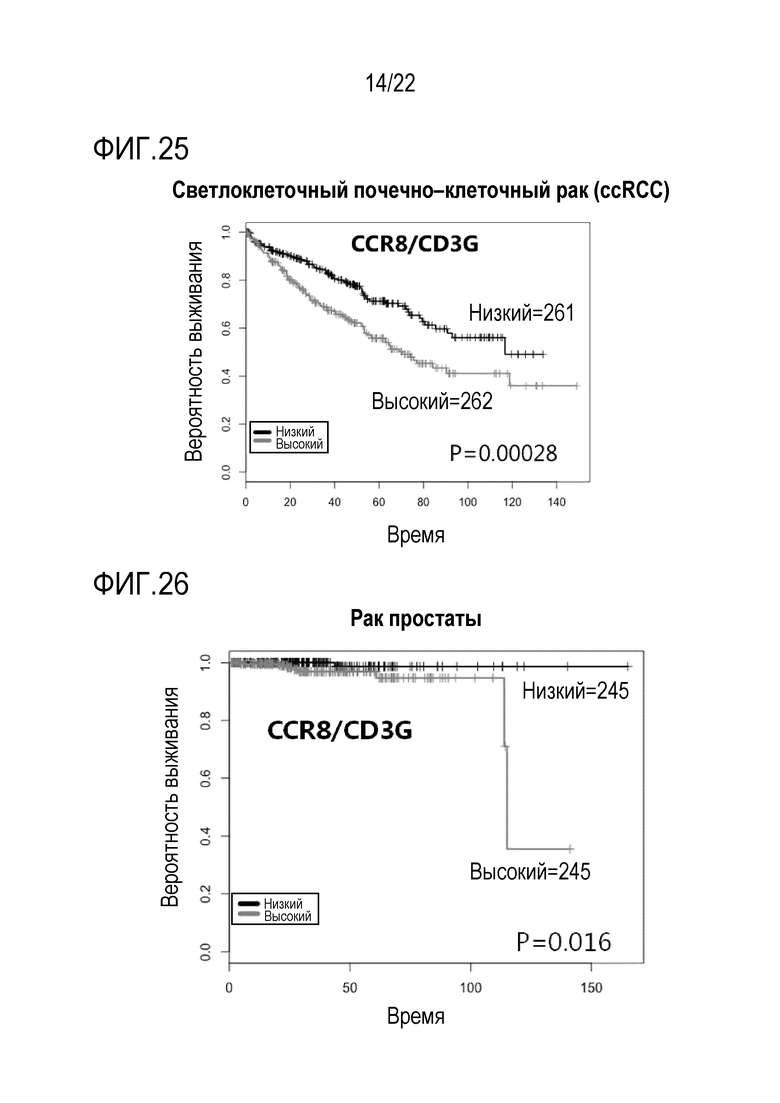
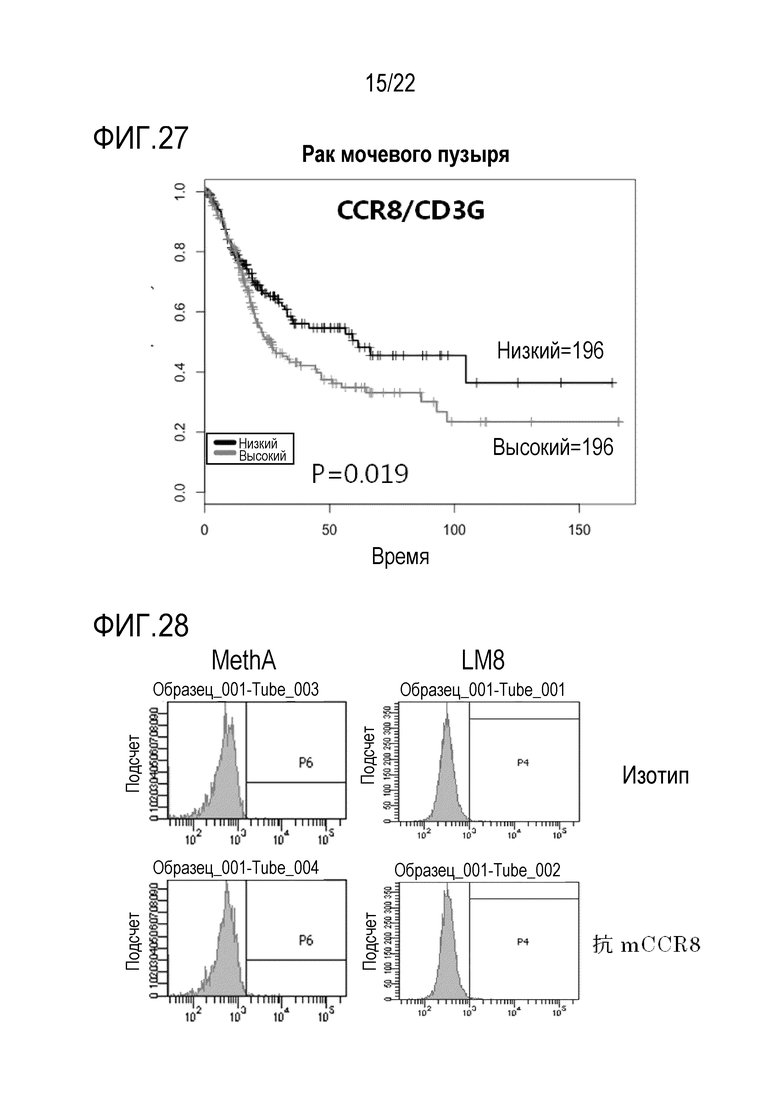
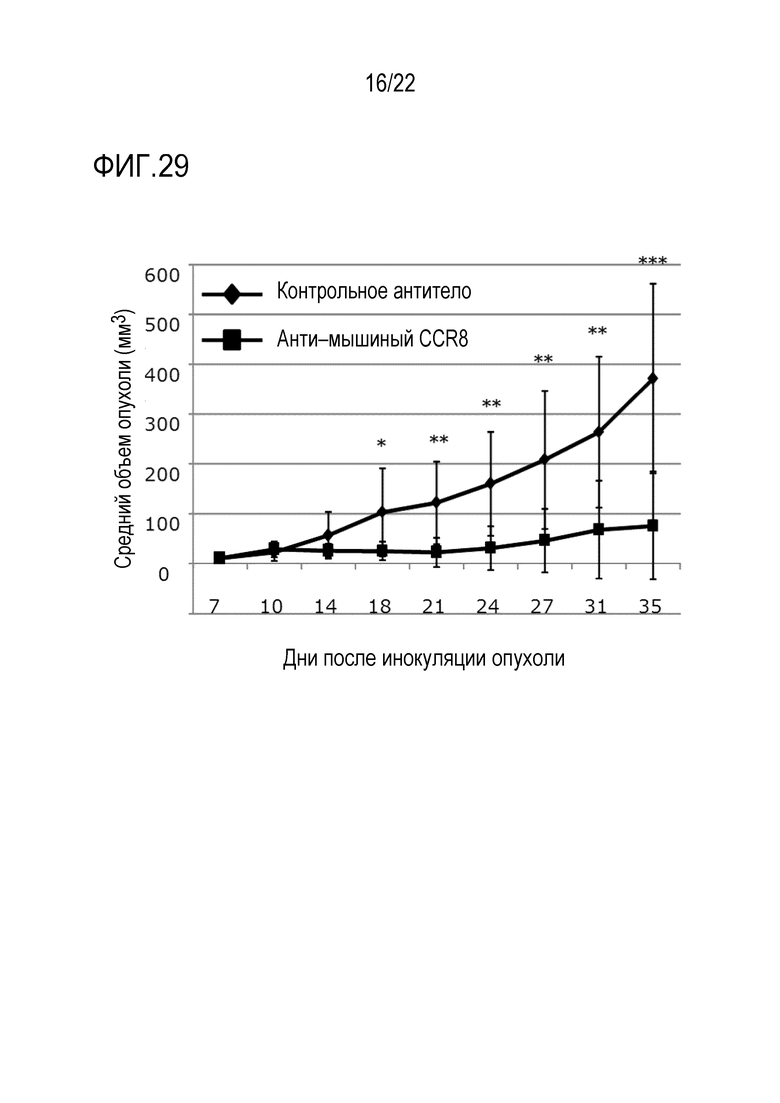
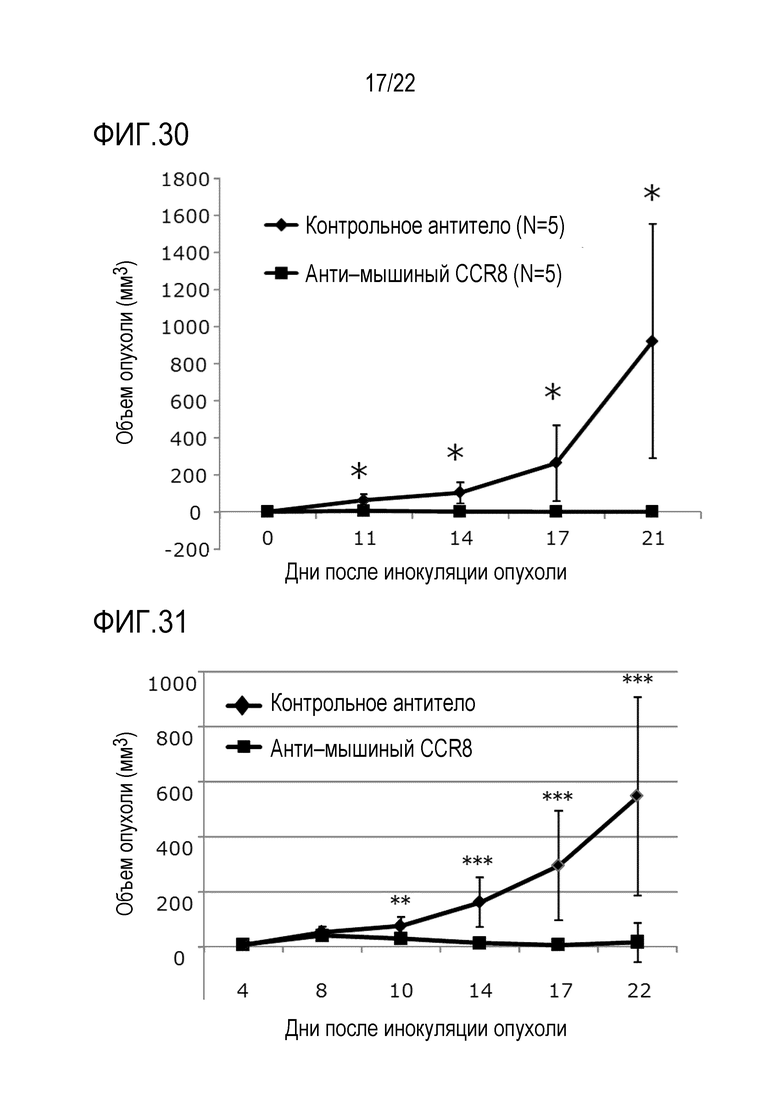
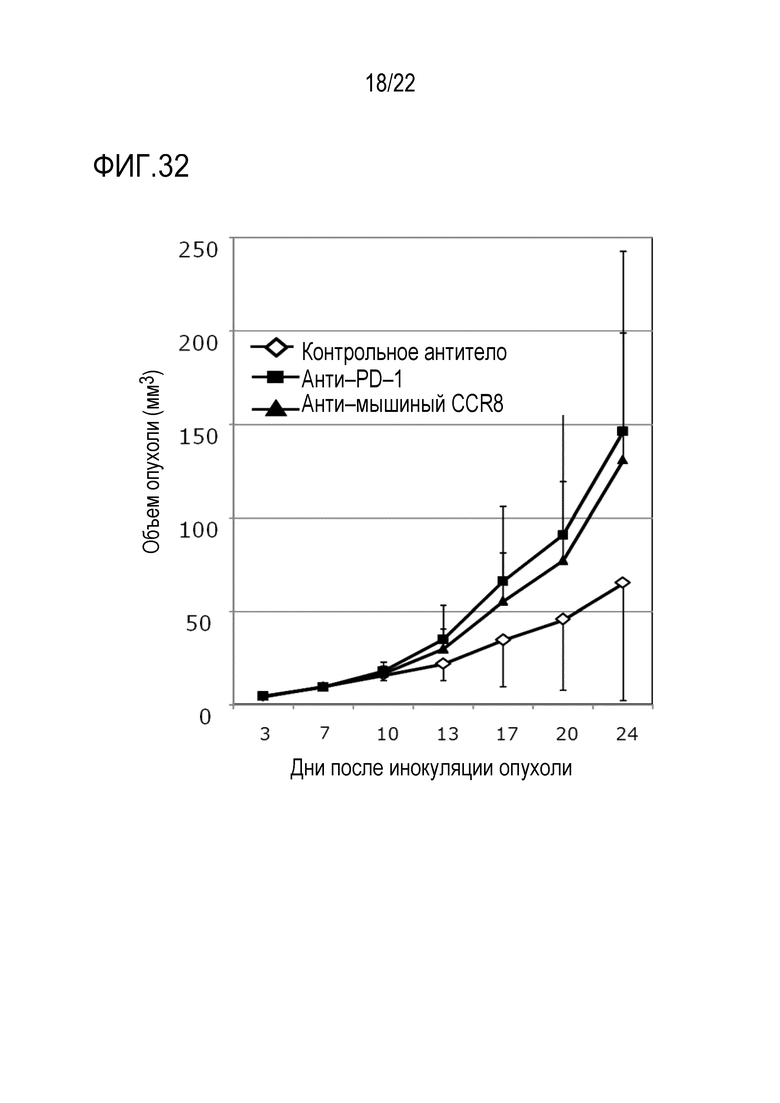
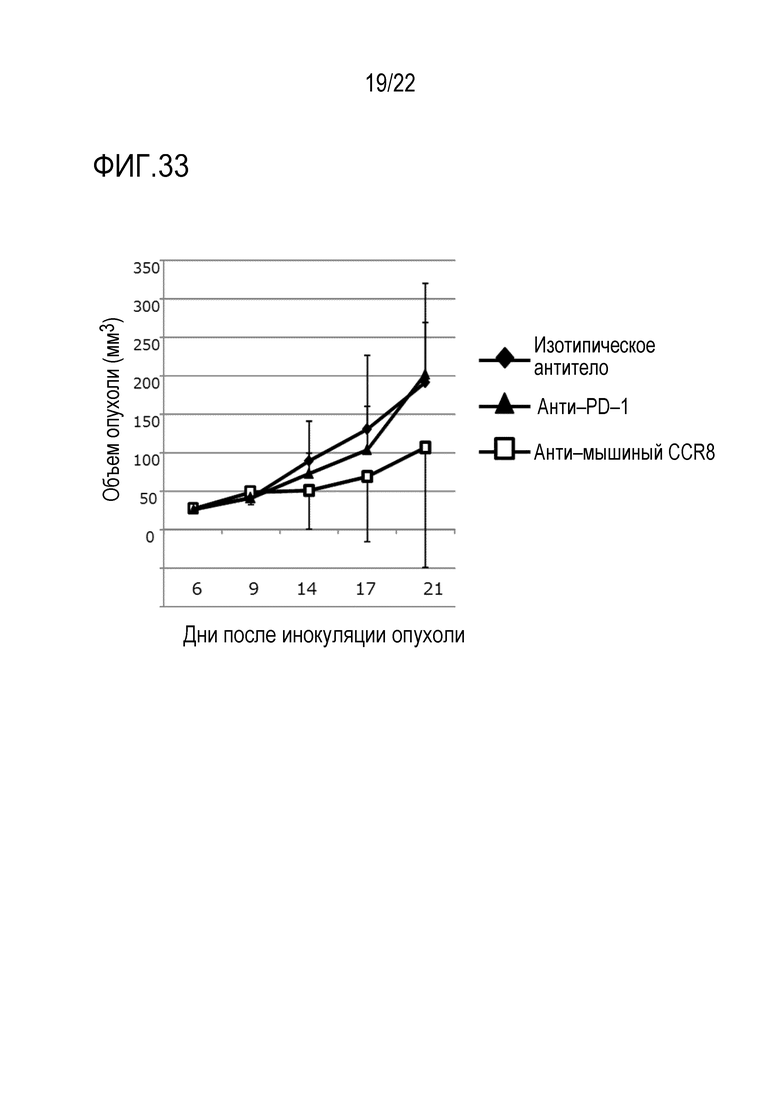

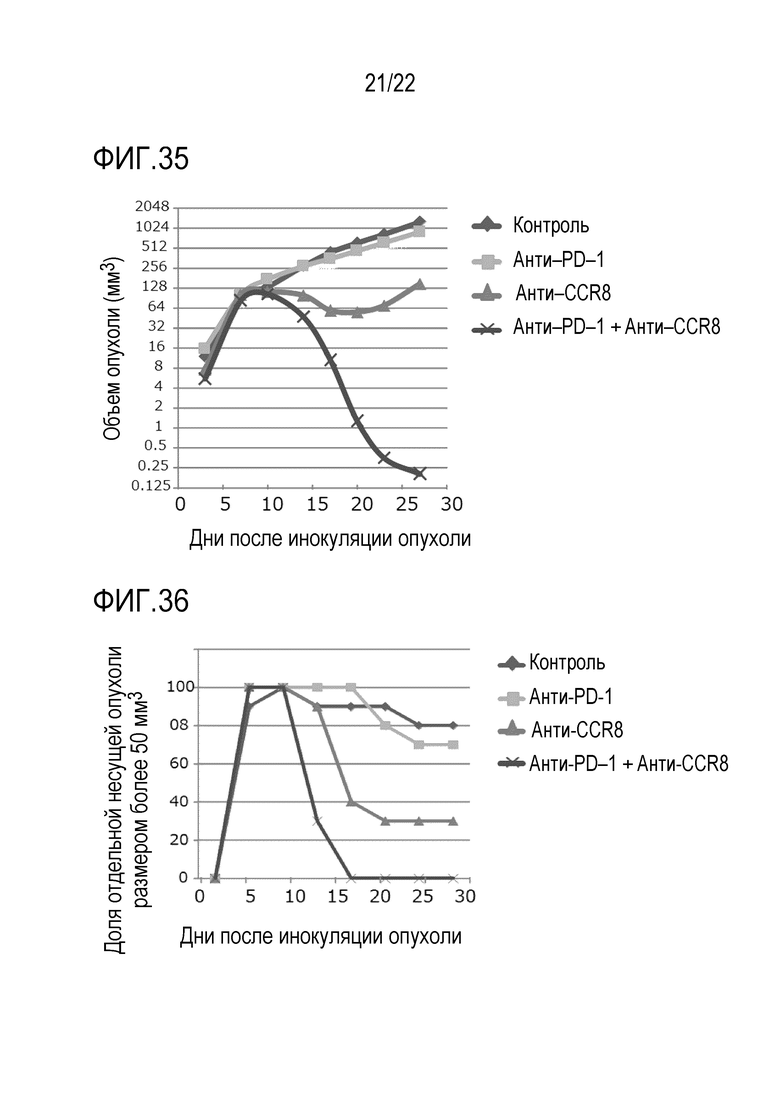
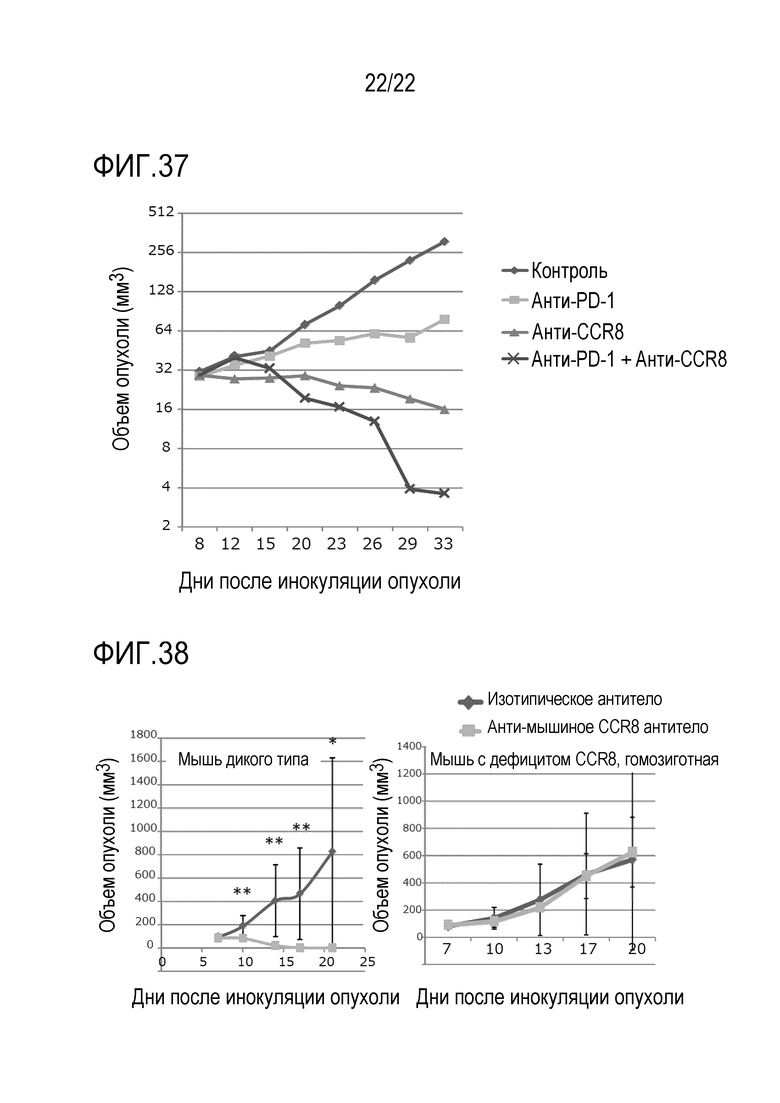
Группа изобретений относится к медицине и касается лекарственного средства для лечения злокачественного новообразования, содержащего комбинацию антитела против CCR8 и анти-PD-1 антитела или анти-PD-L1 антитела, где антитело против CCR8 представляет собой антитело с активностью ADCC и представляет собой CCR8-нейтрализующее антитело. Группа изобретений также касается применения лекарственного средства, содержащего антитело против CCR8 в комбинации с анти-PD-1 антителом или анти-PD-L1 антителом, для лечения злокачественного новообразования; фармацевтической композиции для лечения злокачественного новообразования, содержащей антитело против CCR8. Группа изобретений обеспечивает регрессию опухоли. 4 н. и 22 з.п. ф-лы, 28 пр., 3 табл., 38 ил.
1. Лекарственное средство для лечения злокачественного новообразования, содержащее комбинацию антитела против CCR8 и анти-PD-1 антитела или анти-PD-L1 антитела,
где антитело против CCR8 представляет собой антитело с активностью ADCC и представляет собой CCR8-нейтрализующее антитело.
2. Применение лекарственного средства, содержащего антитело против CCR8 в комбинации с анти-PD-1 антителом или анти-PD-L1 антителом, для лечения злокачественного новообразования,
где антитело против CCR8 представляет собой антитело с активностью ADCC и представляет собой CCR8-нейтрализующее антитело.
3. Применение лекарственного средства, содержащего анти-PD-1 антитело или анти-PD-L1 антитело в комбинации с антителом против CCR8, для лечения злокачественного новообразования,
где антитело против CCDR8 представляет собой антитело с активностью ADCC и представляет собой CCR8-нейтрализующее антитело.
4. Лекарственное средство по любому из пп.1-3, где антитело против CCR8 обладает эффектом удаления Treg-клеток, инфильтрирующих опухоль.
5. Лекарственное средство по любому из пп.1-4, где антитело против CCR8 обладает эффектом удаления макрофагальных клеток, инфильтрирующих опухоль.
6. Лекарственное средство по любому из пп.1-5, где злокачественное новообразование представляет собой рак молочной железы, рак тела матки, рак шейки матки, рак яичников, рак предстательной железы, рак легкого, рак желудка, немелкоклеточный рак легкого, рак селезенки, плоскоклеточный рак головы и шеи, рак пищевода, рак мочевого пузыря, меланому, колоректальный рак, рак почки, неходжкинскую лимфому, уротелиальный рак, саркому, злокачественные новообразования клеток крови, такие как лейкоз или лимфома, рак желчных протоков, карциному желчного пузыря, рак щитовидной железы, рак яичек, рак тимуса или гепатокарцинома.
7. Лекарственное средство по п.6, где злокачественным новообразованием является рак молочной железы, рак тела матки, рак яичников, рак легкого, колоректальный рак, рак почки или саркома.
8. Лекарственное средство по п.7, где злокачественным новообразованием является рак молочной железы.
9. Лекарственное средство по п.7, где злокачественным новообразованием является рак тела матки.
10. Лекарственное средство по п.7, где злокачественным новообразованием является рак яичников.
11. Лекарственное средство по п.7, где злокачественным новообразованием является рак легких.
12. Лекарственное средство по п.7, где злокачественным новообразованием является колоректальный рак.
13. Лекарственное средство по п.7, где злокачественным новообразованием является рак почки.
14. Лекарственное средство по п.7, где злокачественным новообразованием является саркома.
15. Фармацевтическая композиция для лечения злокачественного новообразования, содержащая антитело против CCR8, где антитело против CCR8 представляет собой антитело с активностью ADCC и представляет собой CCR8-нейтрализующее антитело.
16. Фармацевтическая композиция по п.15, где антитело против CCR8 обладает эффектом удаления Treg-клеток, инфильтрирующих опухоль.
17. Фармацевтическая композиция по любому из пп.1-16, где антитело против CCR8 обладает эффектом удаления макрофагальных клеток, инфильтрирующих опухоль.
18. Фармацевтическая композиция по любому из пп.1-17, где злокачественное новообразование представляет собой рак молочной железы, рак тела матки, рак шейки матки, рак яичников, рак предстательной железы, рак легкого, рак желудка, немелкоклеточный рак легкого, рак селезенки, плоскоклеточный рак головы и шеи, рак пищевода, рак мочевого пузыря, меланому, колоректальный рак, рак почки, неходжкинскую лимфому, уротелиальный рак, саркому, злокачественные новообразования клеток крови, такие как лейкоз или лимфома, рак желчных протоков, карциному желчного пузыря, рак щитовидной железы, рак яичек, рак тимуса или гепатокарцинома.
19. Фармацевтическая композиция по п.18, где злокачественным новообразованием является рак молочной железы, рак тела матки, рак яичников, рак легкого, колоректальный рак, рак почки или саркома.
20. Фармацевтическая композиция по п.19, где злокачественным новообразованием является рак молочной железы.
21. Фармацевтическая композиция по п.19, где злокачественным новообразованием является рак тела матки.
22. Фармацевтическая композиция по п.19, где злокачественным новообразованием является рак яичников.
23. Фармацевтическая композиция по п.19, где злокачественным новообразованием является рак легкого.
24. Фармацевтическая композиция по п.19, где злокачественным новообразованием является колоректальный рак.
25. Фармацевтическая композиция по п.19, где злокачественным новообразованием является рак почки.
26. Фармацевтическая композиция по п.19, где злокачественным новообразованием является саркома.
| WO2007044756 A2, 19.04.2007 | |||
| WO2016069727 A1, 06.05.2016 | |||
| PLITAS G., et al., Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer.Immunity | |||
| Токарный резец | 1924 |
|
SU2016A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| ERUSLANOV E., et al., Expansion of CCR8(+) inflammatory myeloid cells in cancer patients with urothelial and renal | |||

