ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения флутеметамола.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Инъекция флутеметамола (18F) представляет собой агент, используемый для визуализации β-амилоидных бета-бляшек в головном мозге методом позитронной эмиссионной томографии, и применяется для диагностики деменции по типу Альцгеймера.
В качестве способа получения [18F]флутеметамола, например, при использовании реактора «FASTlab» для синтеза радиофармацевтического вещества, известен способ взаимодействия АН111907 (6-этоксиметокси-2-(4'-(N-формил-N-метил)амино-3'-нитро)фенилбензотиазола) с радиоактивным фторидом для замещения нитрогруппы в АНН 1907 на 18F, с последующим превращением остатка АНН 1907 в менее жирорастворимое вещество с помощью сильного основания, снятия защитных групп с гидроксигруппы и аминогруппы продукта замещения АН111907 ионом 18F (6-этоксиметокси-2-(4'-(N-формил-N-метил)амино-3'-[18F]фторо)фенилбензотиазола), с последующим осуществлением очистки при использовании картриджа для твердофазной экстракции (WO 2011/044406).
Однако в способе, описанном в WO 2011/044406, выход [18F]флутеметамола является низким, следовательно, широкомасштабное массовое производство является трудно достижимым. Таким образом, для повышения доступности препарата [18F]флутеметамола большему количеству пациентов, потребовалось улучшить эффективность способа.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение разработано с учетом описанной выше ситуации, поэтому цель настоящего изобретения в улучшении эффективности получения [18F]флутеметамола.
Согласно одному из аспектов настоящего изобретения, предложен способ получения флутеметамола, включающий стадии:
(a) взаимодействия соединения, представленного следующей общей формулой (1), с радиоактивным фторидом с получением соединения, представленного следующей общей формулой (2);
(b) воздействия сильного основания на реакционную смесь, полученную на стадии (а), содержащую соединение, представленное следующей общей формулой (1), и соединение, представленное следующей общей формулой (2);
(c) очистки соединения, представленного общей формулой (2), после стадии (b) при использовании картриджа для обращеннофазной твердофазной экстракции и
(d) снятия защитной группы с получением [18F]флутеметамола

где R1 является защитной группой для гидроксигруппы, а C(O)R2 представляет собой защитную группу для аминогруппы
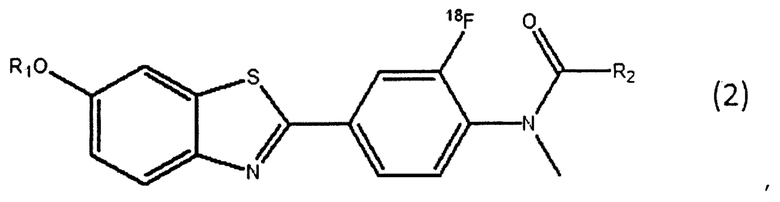
где R1 и R2 имеют те же значения, что и в соединении, представленном общей формулой (1).
Согласно настоящему изобретению, можно улучшить эффективность получения [18F]флутеметамола.
ПОДРОБНОЕ ОПИСАНИЕ
Термин «алкил», используемый в настоящей заявке самостоятельно или как часть другой группы, обозначает насыщенную или разветвленную насыщенную углеводородную группу, такую как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, н-пентил или н-гексил.
Термин «галогеналкил», используемый в настоящей заявке самостоятельно или как часть другой группы, обозначает группу, в которой один или более водородов алкильной группы замещен фтором, хлором, бромом или йодом.
Термин «алкокси», используемый в настоящей заявке самостоятельно или как часть другой группы, обозначает насыщенную или разветвленную насыщенную углеводородную группу, такую как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, н-пентокси или н-гексилокси.
Термин «арил», используемый в настоящей заявке самостоятельно или как часть другой группы, обозначает моноциклическое или сопряженное кольцо ароматического углеводорода, например, фенил или нафтил.
(а) Стадия 18F мечения
На стадии 18F мечения соединение, представленное общей формулой (1) (здесь и далее его также называют «соединение-прекурсор для мечения») взаимодействует с радиоактивным фторидом с получением соединения, представленного следующей общей формулой (2) (здесь и далее его также называют «18F меченое промежуточное соединение»).
В качестве защитной группы R1 для гидроксигруппы можно использовать группы, описанные в Greene, Protective Groups in Organic Synthesis (опубликовано Wiley-Interscience, 4oe издание, 30 октября 2006). Группа, представленная как OR1, предпочтительно представляет собой алкоксиметоксигруппу, имеющую от 1 до 6 атомов углерода, примеры которой включают этоксиметоксигруппу и метоксиметоксигруппу.
R2 выбирают из водорода, алкилов, имеющих от 1 до 10 атомов углерода, галогеналкилов, имеющих от 1 до 10 атомов углерода, арилов, имеющих от 6 до 14 атомов углерода, арилалкилов, имеющих от 6 до 14 атомов углерода и -(CH2CH2O)р-CH3, где р - целое число от 1 до 10. R2 предпочтительно является водородом или алкилом, имеющим от 1 до 10 атомов углерода, более предпочтительно водородом или метилом, и наиболее предпочтительно водородом.
Соединение-прекурсор для мечения можно синтезировать, например, с помощью способа, описанного в WO 2007/020400. Предпочтительным примером соединения-прекурсора для мечения является 6-этоксиметокси-2-(4'-(N-формил-N-метил)амино-3'-нитро)фенилбензотиазол (АН111907), причем пример его способа синтеза описан в примере 1 в WO 2007/020400.
Радиоактивный фторид можно получить путем добавления катионного противоиона в водный раствор, содержащий [18F]фторид-ион, полученный из [18O]воды путем 18O(p,n)18F ядерной реакции с удалением воды. Катионный противоион предпочтительно представляет собой ион, обладающий достаточной растворимостью в безводном растворителе реакции, для сохранения растворимости [18F]фторид-иона. Примеры включают ионы тетраалкиламмония и щелочных металлов (ион натрия, ион калия, ион цезия, ион рубидия), образующих комплекс с катализатором фазового переноса (например, 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]-гексакозаном (торговое наименование: Kryptofix 2.2.2)), причем тетрабутиламмония является предпочтительным. Фторид [18F]тетрабутиламмония можно получить, например, путем пропускания [18O]воды, содержащей [18F] фторид ион, которая получена путем 18O(p,n)18F ядерной реакции, через анионообменную смолу для адсорбции [18F]фторид-иона на анионообменную смолу, элюирования его водным раствором гидрокарбоната тетрабутиламмония и образования азеотропной смеси с ацетонитрилом.
Стадию 18F мечения можно проводить в подходящем растворителе. В качестве растворителя можно использовать такие растворители, как ацетонитрил, диметилформамид, диметилсульфоксид, диметилацетамид, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, сульфолан, н-метилпирролидон, производное имидазолия, такое как гексафторфосфат 1-бутил-3-метилимидазолия, производное пиридиния, такое как гексафторборат 1-бутил-4-метилпиридиния, соединение фосфония или ионная жидкость, такая как соединение тетраалкиламмония, причем диметилсульфоксид является предпочтительным.
Стадию 18F мечения можно, например, проводить при температуре от 15 до 180°C, предпочтительно от 80 до 150°C и более предпочтительно от 120 до 140°C, причем еще более предпочтительно проводить ее при температуре примерно 130°C.
(b) Стадия разложения прекурсора
На стадии разложения прекурсора обеспечивают воздействие сильного основания на реакционную смесь на стадии 18F мечения, содержащую соединение-прекурсор для мечения и 18F меченое промежуточное соединение. Таким образом, остаток соединения-прекурсора для мечения, содержащийся в реакционной смеси на стадии 18F мечения, превращают в высокополярное соединение. Высокополярными можно считать соединения, указанные на Фиг. 1 в WO 2011/044406. На стадии разложения прекурсора 18F меченое промежуточное соединение не вступает во взаимодействие с сильным основанием.
Сильное основание включает алкоксиды щелочных металлов, гидроксиды щелочных металлов и т.п., причем предпочтительно применять метоксид натрия, этоксид натрия, гидроксид натрия, гидроксид калия, гидрид натрия или метилмеркаптан натрия. Более предпочтительным сильным основанием является метоксид натрия или этоксид натрия, и еще более предпочтительным является метоксид натрия.
Стадию разложения прекурсора предпочтительно проводят в присутствии растворителя. Растворитель включает алкиловые спирты, причем метанол является предпочтительным.
Стадию разложения прекурсора можно, например, проводить при температуре от 15 до 180°C, предпочтительно от 80 до 150°C и более предпочтительно от 120 до 140°C, еще более предпочтительно проводить ее при температуре примерно 130°C.
(с) Стадия первой очистки
На стадии первой очистки, после стадии разложения прекурсора F меченое промежуточное соединение очищают при использовании картриджа для обращеннофазной твердофазной экстракции. Таким образом разделяют 18F меченое промежуточное соединение и высокополярное соединение, полученное на стадии разложения прекурсора.
В качестве картриджа для обращеннофазной твердофазной экстракции применяют картридж, в котором используют наполнитель, в котором силильная группа модифицирована алкилом, имеющим предпочтительно 8 или более атомов углерода и более предпочтительно 18 или более атомов углерода, причем наиболее предпочтительно применяют картридж для твердофазной экстракции, заполненный триаконтил-силилированным силикагелем, в котором силильная группа модифицирована группой с 30 атомами углерода. Такой картридж для обращеннофазной твердофазной экстракции коммерчески доступен, например, производства Macherey-Nagel. Картридж для обращеннофазной твердофазной экстракции предпочтительно кондиционируют перед применением ацетонитрилом и водой.
Очистка 18F меченого промежуточного соединения с применением картриджа для обращеннофазной твердофазной экстракции не является специфическим ограничением, так как очистку можно проводить с помощью технологии нормальнофазной твердофазной экстракции. Пример будет приведен ниже.
Сначала 18F меченое промежуточное соединение удерживают на картридже для обращеннофазной твердофазной экстракции [(с-1) стадия удерживания] в процессе стадии разложения прекурсора. Предпочтительно, после стадии разложения прекурсора реакционную смесь, содержащую 18F меченое промежуточное соединение и высокополярное соединение, описанное выше, разбавляют путем добавления воды и загружают в картридж для обращеннофазной твердофазной экстракции.
Затем картридж для обращеннофазной твердофазной экстракции промывают жидкой смесью из воды и одного или более органических растворителей, выбранных из группы, состоящей из ацетонитрила, тетрагидрофурана и алкиловых спиртов, имеющих от 1 до 3 атомов углерода [(с-2) стадия промывания]. Растворитель, применяемый для промывания, предпочтительно представляет собой жидкую смесь воды и ацетонитрила, причем в таком соотношении смешивания, что, например, содержание ацетонитрила может составлять от 35 до 45% (об.) и предпочтительно от 39,5 до 40,5% (об.) относительно всей жидкой смеси. Температура картриджа для обращеннофазной твердофазной экстракции предпочтительно составляет от 19 до 34°C и более предпочтительно от 20 до 30°C. Указанную стадию промывания можно повторять многократно. Таким образом, высокополярное соединение, описанное выше, можно элюировать из картриджа для обращеннофазной твердофазной экстракции, удерживая при этом 18F меченое промежуточное соединение в картридже для обращеннофазной твердофазной экстракции.
После этого 18F меченое промежуточное соединение элюируют алкиловым спиртом, имеющим от 1 до 3 атомов углерода, из картриджа для обращеннофазной твердофазной экстракции [(с-3) стадия элюирования]. Алкиловый спирт, имеющий от 1 до 3 атомов углерода, включает метанол, этанол, 1-пропанол и 2-пропанол, причем этанол является более предпочтительным с точки зрения безопасности. На этой стадии можно применять продавливание газообразным азотом со стороны впускного порта картриджа для обращеннофазной твердофазной экстракции или отсасывание со стороны выпускного порта. Полученный элюат можно использовать на следующей стадии без дальнейшей обработки или после концентрирования растворителя при нагревании или пониженном давлении.
(d) Стадия снятия защитной группы
На стадии снятия защитной группы удаляют каждую защитную группу для гидроксигруппы и аминогруппы с получением [18F] флутеметамола.
Стадию снятия защиты можно проводить в соответствии с описанием в Greene, Protective Groups in Organic Synthesis (опубликовано Wiley-Interscience, 4oe издание, 30 октября 2006), и предпочтительным является проведение кислотного гидролиза при использовании органической кислоты или неорганической кислоты. В качестве кислоты предпочтительно применять неорганическую кислоты, такую как серная кислота, соляная кислота, фосфорная кислота или бромоводородная кислота, причем более предпочтительно применять соляную кислоту.
Стадию снятия защиты можно проводить в присутствии воды, органического растворителя, такого как алкиловый спирт, имеющий от 1 до 4 атомов углерода, или ацетонитрил или их жидкая смесь, причем предпочтительно добавлять кислоту в этанольный элюат, полученный на этапе элюирования стадии первой очистки, и затем проводить стадию снятия защиты.
Стадию снятия защиты предпочтительно проводят при температуре 100°C или более.
(е) Стадия второй очистки
На стадии второй очистки после стадии снятия защиты [18F]флутеметамол очищают при использовании картриджа для обращеннофазной твердофазной экстракции.
В качестве типа картриджа для обращеннофазной твердофазной экстракции, применяемого на стадии второй очистки, можно использовать картридж, который можно использовать на стадии первой очистки, причем предпочтительно применять картридж для твердофазной экстракции, заполненный триаконтил-силилированным силикагелем, в котором силильная группа модифицирована группой с 30 атомами углерода.
Очистка [18F]флутеметамола при использовании картриджа для обращеннофазной твердофазной экстракции не ограничивается специфически, поскольку очистку проводят также при использовании технологии нормальнофазной твердофазной экстракции. Пример будет объяснен ниже.
Сначала [18F]флутеметамол удерживается в картридже для обращеннофазной твердофазной экстракции в процессе стадии снятия защиты [(е-1) стадия удерживания]. Предпочтительно, после стадии снятия защиты неочищенный продукт [18F]флутеметамола разбавляют путем добавления воды, так что содержание органического растворителя, имеющегося с предыдущей стадии (например, этанола, имеющегося со стадии первой очистки) составляет 50% (об.) или менее, и загружают в картридж для обращеннофазной твердофазной экстракции.
Затем картридж для обращеннофазной твердофазной экстракции промывают водой или жидкой смесью воды и одного или более органических растворителей, выбранных из группы, состоящей из ацетонитрила, тетрагидрофурана и алкиловых спиртов, имеющих от 1 до 3 атомов углерода [(е-2) стадия промывания]. Растворитель, применяемый для промывания, предпочтительно представляет собой жидкую смесь воды и ацетонитрила, причем в таком соотношении смешивания, что содержание ацетонитрила может составлять, например, от 35 до 45% (об.) и предпочтительно от 39,5 до 40,5% (об.) относительно всей жидкой смеси. Температура картриджа для обращеннофазной твердофазной экстракции предпочтительно составляет от 19 до 34°C и более предпочтительно от 20 до 30°C. Стадию промывания можно повторять многократно, и при этом картридж для обращеннофазной твердофазной экстракции предпочтительно промывают водой. Таким образом, можно удалить ненужный растворитель и реагент для снятия защиты, удерживая при этом [18F]флутеметамол в картридже для обращеннофазной твердофазной экстракции.
После этого [18F]флутеметамол элюируют этанолом из картриджа для обращеннофазной твердофазной экстракции [(е-3) стадия элюирования]. После этого через картридж можно пропустить воду и объединить с элюатом. Кроме того, можно применять продавливание газообразным азотом со стороны впускного порта картриджа для обращеннофазной твердофазной экстракции или отсасывание со стороны выпускного порта.
(f) Стадия третьей очистки
На стадии третьей очистки после стадии второй очистки [18F]флутеметамол очищают с помощью картриджа для твердофазной экстракции (HILIC) с гидрофильным взаимодействием.
В качестве HILIC картриджа для твердофазной экстракции можно, например, применять картридж, заполненный силикагелем или силикагелем, в который введена высокополярная функциональная группа, такая как амино, амидо, циано, диольная, полисукцинимидное производное, цвиттер-ион или циклоцекстрин. В данной заявке предпочтительным является силикагель на основе аминной твердой фазы, причем картридж, заполненный аминопропилированным силикагелем, является более предпочтительным. Таким образом, с помощью HILIC картриджа для твердофазной экстракции можно захватывать примеси, обеспечивая при этом прохождение через него [18F]флутеметамола. Такой HILIC картридж для твердофазной экстракции коммерчески доступен, например, производства Waters, Agilent Technologies и т.п.. HILIC картридж для твердофазной экстракции предпочтительно кондиционируют путем пропускания перед применением ацетонитрила или этанола, с последующей продувкой азотом для осушения.
Затем обеспечивают пропускание через HILIC картридж для твердофазной экстракции элюата, полученного на второй стадии очистки. После этого через него можно пропустить воду и можно объединить элюаты. Кроме того, можно применять продавливание газообразным азотом со стороны впускного порта картриджа для обращеннофазной твердофазной экстракции или отсасывание со стороны выпускного порта.
Полученный элюат может содержать фармацевтически приемлемый носитель, разбавитель, эмульсию, эксципиент, наполнитель, диспергент, буфер, консервант, солюбилизатор, антисептик, краситель, стабилизатор и т.п., чтобы иметь форму, подходящую для введения [18F]флутеметамола в живой организм, предпочтительно форму инъекции. Полученный содержащий [18F]флутеметамол раствор желательно отфильтровать с помощью мембранного фильтра.
Пример препарата [18F]флутеметамола раскрыт, например, в W0 2009/027452.
Согласно способу по настоящему изобретению, описанному выше, стадию первой очистки, которую обычно проводят после стадии снятия защиты, проводят после стадии 18F мечения и перед стадией снятия защиты. Когда стадию первой очистки поводят после стадии снятия защиты, присутствует много примесей, содержащих высокополярное соединение, полученное из соединения-прекурсора для мечения, так что при удалении примесей возрастают потери [18F]флутеметамола. С другой стороны, когда стадию первой очистки проводят перед стадией снятия защиты, высокополярное соединение, полученное из соединения-прекурсора для мечения, можно удалить перед стадией снятия защиты. Таким образом, можно очистить 18F меченое промежуточное соединение, предотвращая при этом потери 18F меченого промежуточного соединения, которое является промежуточным соединением [18F]флутеметамола, то есть можно получить [18F]флутеметамол с более высоким выходом, чем обычно, и с качеством, равным обычному. Таким образом, согласно настоящему изобретению, можно улучшить эффективность получения [18F] флутеметамола.
ПРИМЕРЫ
В данной заявке настоящее изобретение будет подробно описано ниже со ссылкой на примеры, но настоящее изобретение не ограничивается содержанием указанных примеров. В данной заявке в качестве реагента и элемента колонки, используемого в примерах, использовали компонент реактора FASTlab для синтеза радиофармацевтического вещества (для синтеза флутеметамола) производства GE Healthcare или эквивалентный ему компонент.
Примеры 1-3
(a) Стадия 18F мечения
Содержащую [18F] фторид-ион [18O]воду, полученную путем протонного облучения [18O]воды при использовании циклотрона, пропускали через анионообменную колонку и адсорбировали и собирали [18F]фторид-ион. После этого колонку промывали водой (3 мл), затем элюировали водным раствором гидрокарбоната тетрабутиламмония с концентрацией 0,15 моль/л (0,35 мл) и ацетонитрила (1 мл) и упаривали полученный элюат. К нему добавляли раствор 6-этоксиметокси-2-(4'-(N-формил-N-метил)амино-3'-нитро)фенилбензотиазола (АН111907) (75 мкмоль) в диметилсульфоксиде (1 мл) и нагревали смесь при температуре 130°C в течение 15 минут, затем охлаждали.
(b) стадия разложения прекурсора
Раствор метоксида натрия (11% (масс.), 1 мл) в метаноле добавляли в жидкую реакционную среду после охлаждения на стадии (а) и нагревали смесь при температуре 130°C в течение 5 минут, затем охлаждали.
(c) стадия первой очистки
Воду (2 мл) добавляли в жидкую реакционную среду после охлаждения на стадии (b) и пропускали смесь через (C30) колонку с триаконтил-силилированным силикагелем для удерживания 18F меченого промежуточного соединения. Затем проводили промывание путем пропускания 40%-ного (об.) водного раствора ацетонитрила (6 мл) через реакционный сосуд и через C30 колонку, с последующим промыванием путем непосредственного пропускания 40%-ного (об.) водного раствора ацетонитрила (6 мл) через C30 колонку. Для сбора элюата через указанную C30 колонку пропускали этанол (2 мл).
(d) стадия снятия защиты
В элюат, собранный на стадии (с), добавляли соляную кислоту (2,0 мл) с концентрацией 4 моль/л и нагревали смесь при температуре 125°C в течение 5 минут с получением неочищенного раствора [18F] флутеметамола.
(e) вторая стадия очистки
Неочищенный раствор [18F]флутеметамола, полученный на стадии (d), охлаждали, затем к нему добавляли воду (10 мл) и для удерживания [18F]флутеметамола на C30 колонке пропускали смесь через не использованную ранее C30 колонку, отличную от колонки, использованной на стадии (b). Промывание проводили путем пропускания 40%-ного (об.) водного раствора ацетонитрила (6 мл) через C30 колонку, затем путем пропускания воды (6 мл). [18F]флутеметамол с C30 колонки элюировали этанолом (3,5 мл).
(f) третья стадия очистки
Элюат на стадии (е) пропускали через колонку (NH2 колонка), заполненную аминопропилированным силикагелем. Промывание проводили путем пропускания воды (9,3 мл) сначала через C30 колонку, использованную на стадии (е), и затем NH2 колонку в указанном порядке, причем каждый элюат собирали в сосуд, в который добавляли фосфорнокислый буфер (37,2 мл) с концентрацией 18,8 ммоль/л, содержащий 0,7% (масс./об.) полисорбата 80 и 1,2% (масс./об.) хлорида натрия.
Сравнительные примеры 1 и 2
Проводили стадии (а) и (b) из Примеров 1-3 и стадии, указанные далее.
(с') стадия снятия защиты
В жидкую реакционную среду, полученную на стадии (b), добавляли соляную кислоту (0,6 мл) с концентрацией 4 моль/л и нагревали смесь при температуре 125°C в течение 5 минут с получением неочищенного раствора [18F]флутеметамола.
(d') первая стадия очистки
Неочищенный раствор [18F]флутеметамола, полученный на стадии (с'), охлаждали, затем к нему добавляли воду (2 мл) и пропускали смесь через C30 колонку для удерживания [18F]флутеметамола. Затем проводили промывание путем пропускания 40%-ного (об.) водного раствора ацетонитрила (12 мл) через реакционный сосуд и через C30 колонку, с последующим пропусканием воды (5 мл) через C30 колонку. Для сбора элюата через указанную C30 колонку пропускали ацетонитрил (2 мл).
(е') вторая стадия очистки
Элюат, полученный на стадии (d'), очищали путем пропускания через NH2 колонку, с последующим дополнительным пропусканием через NH2 колонку раствора ацетонитрила (1 мл), причем указанные растворы смешивали.
(f') третья стадия очистки
В раствор, полученный на стадии (е'), добавляли воду (5 мл) и пропускали через неиспользованную C30 колонку, отличную от колонки, использованной на стадии (d'), для удерживания [18F]флутеметамола на C30 колонке, затем проводили промывание путем пропускания воды (4 мл) через C30 колонку три раза. Через C30 колонку пропускали этанол (3,5 мл) и через C30 колонку пропускали воду (9,3 мл). Каждый элюат собирали в сосуд, в который добавляли фосфорнокислый буфер (37,2 мл) с концентрацией 18,8 ммоль/л, содержащий 0,7% (масс./об.) полисорбата 80 и 1,2% (масс./об.) хлорида натрия.
Результаты, полученные в Примерах 1-3 и сравнительных Примерах 1 и 2, приведены в Таблице 1. В Таблице 1, «величина радиоактивности (МБк)» обозначает величину радиоактивности [18F]фторид-иона в начале синтеза, характерную для каждого примера и сравнительного примера; «время синтеза (в минутах)» представляет собой время, необходимое для осуществления каждого примера и сравнительного примера; «радиохимический выход (%)» означает радиохимический выход [18F]флутеметамола на основе величины радиоактивности [18F]фторид-иона после коррекции затухания в начале синтеза; «радиохимическая чистота (%)» обозначает радиохимическую чистоту [18F]флутеметамола, а «общее количество нерадиоактивных примесей (мкг/мл)» представляет концентрацию нерадиоактивных примесей в полученном растворе [18F] флутеметамола.
Радиохимическую чистоту [18F]флутеметамола и концентрацию нерадиоактивных примесей определяли с помощью аналитических методов, приведенных ниже.
1. Анализ радиохимической чистоты [18F]флутеметамола
Анализ проводили методом ТСХ (тонкослойной хроматографии). Условия были следующие.
ТСХ пластина: Silica Gel 60 F254 (производства Merck)
Подвижная фаза: Этилацетат/диэтиламин = 100/1
Измерительное устройство: Rita Star (производства raytest)
2. Анализ концентрации нерадиоактивных примесей в растворе [18F] флутеметамола
Анализ проводили методом ВЭЖХ (высокоэффективной жидкостной хроматографии) с УФ-детектором. Условия были следующие.
Колонка: Luna С18(2) (производства Phenomenex, размером: 4,6×150 мм, 3 мкм)
Подвижная фаза: 20 ммоль аммонийноацетатный буфер (рН 6,0)/ацетонитрил = 62/38 → 40/10 (0-9 минут), 40/10 → 10/90 (9 → 10 минут), 10/90 (10 → 20 минут), 10/90 → 62/38 (20 → 20,5 минут), 62/38 (20,5 → 30 минут)
Скорость потока: 1,0 мл/мин
Детектор: фотометр поглощения для УФ-видимой области спектра (длина волны для детектирования: 330 нм)
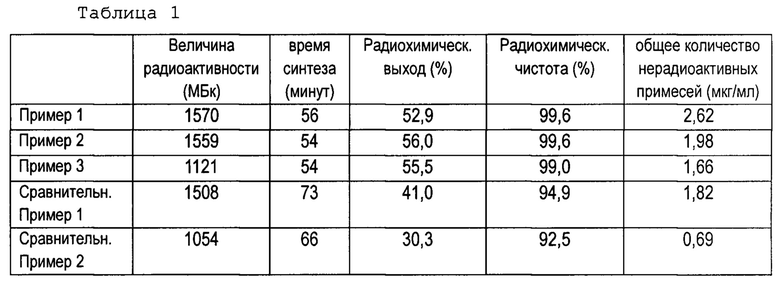
Как показано в Таблице 1, радиохимический выход [18F] флутеметамола улучшился в способах из примеров 1-3, при этом время синтеза можно было сократить. Кроме того, была улучшена радиохимическая чистота, при этом не было обнаружено заметного возрастания общего количества примесей. Таким образом, было показано, что в соответствии с настоящим изобретением, можно получить [18F]флутеметамол такого же качества, улучшив при этом больше, чем обычно, эффективность его получения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ РАДИОМЕЧЕНИЯ | 2014 |
|
RU2690848C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРЗАМЕЩЕННОГО ОРГАНИЧЕСКОГО АЛИФАТИЧЕСКОГО СОЕДИНЕНИЯ И СПОСОБ ОЧИСТКИ ФТОРЗАМЕЩЕННОГО ОРГАНИЧЕСКОГО АЛИФАТИЧЕСКОГО СОЕДИНЕНИЯ | 2015 |
|
RU2710558C2 |
| Раствор элюента | 2011 |
|
RU2608932C2 |
| Способ синтеза F-меченых биомолекул | 2012 |
|
RU2620598C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОАКТИВНОГО ФТОР-МЕЧЕННОГО ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ | 2007 |
|
RU2434846C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОАКТИВНОГО, МЕЧЕННОГО ФТОРОМ ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ | 2008 |
|
RU2476423C2 |
| НОВЫЕ ПРЕДШЕСТВЕННИКИ ПРОИЗВОДНЫХ ГЛУТАМАТА | 2012 |
|
RU2600981C2 |
| Получение 18F-флуцикловина | 2013 |
|
RU2640805C2 |
| НУКЛЕОФИЛЬНОЕ ФТОРИРОВАНИЕ В ТВЕРДОЙ ФАЗЕ | 2002 |
|
RU2315769C9 |
| РЕАГЕНТЫ И СПОСОБЫ ВВЕДЕНИЯ РАДИОАКТИВНОЙ МЕТКИ | 2010 |
|
RU2524284C2 |
Изобретение относится к способу получения [18F]флутеметамола, включающему стадии (a) взаимодействия соединения, представленного следующей общей формулой (1), с радиоактивным фторидом с получением реакционной смеси, содержащей соединение, представленное следующей общей формулой (2), и соединение, представленное следующей общей формулой (1); (b) воздействия сильного основания на реакционную смесь, полученную на стадии (а); (c) очистки соединения, представленного общей формулой (2), после стадии (b) при использовании картриджа для обращеннофазной твердофазной экстракции и (d) снятия обеих защитных групп с получением [18F]флутеметамола, где R1 является защитной группой для гидроксигруппы, а C(O)R2 представляет собой защитную группу для аминогруппы, где R1 и R2 имеют те же значения, что и в соединении, представленном общей формулой (1). Технический результат – получение [18F]флутеметамола с высоким выходом. 7 з.п. ф-лы, 1 табл., 5 пр.
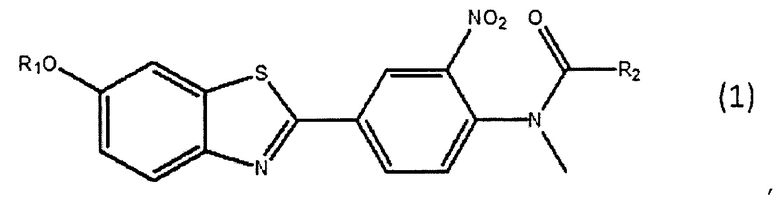

1. Способ получения [18F]флутеметамола, включающий стадии:
(a) взаимодействия соединения, представленного следующей общей формулой (1), с радиоактивным фторидом с получением реакционной смеси, содержащей соединение, представленное следующей общей формулой (2), и соединение, представленное следующей общей формулой (1);
(b) воздействия сильного основания на реакционную смесь, полученную на стадии (а);
(c) очистки соединения, представленного общей формулой (2), после стадии (b) при использовании картриджа для обращеннофазной твердофазной экстракции и
(d) снятия обеих защитных групп с получением [18F]флутеметамола

где R1 является защитной группой для гидроксигруппы, а C(O)R2 представляет собой защитную группу для аминогруппы

где R1 и R2 имеют те же значения, что и в соединении, представленном общей формулой (1).
2. Способ получения [18F]флутеметамола по п. 1, в котором стадия (с) включает стадии:
(с-1) удерживания соединения, представленного общей формулой (2), в картридже для обращеннофазной твердофазной экстракции;
(с-2) промывания картриджа для обращеннофазной твердофазной экстракции жидкой смесью из одного или более органических растворителей, выбранных из воды и группы, состоящей из ацетонитрила, тетрагидрофурана и алкиловых спиртов, имеющих от 1 до 3 атомов углерода; и
(с-3) элюирования соединения, представленного общей формулой (2), алкиловым спиртом, имеющим от 1 до 3 атомов углерода, с картриджа для обращеннофазной твердофазной экстракции.
3. Способ получения [18F]флутеметамола по п. 2, в котором на стадии (с-2) картридж для обращеннофазной твердофазной экстракции промывают жидкой смесью воды и ацетонитрила.
4. Способ получения [18F]флутеметамола по п. 2 или 3, в котором на стадии (с-3) алкиловый спирт, имеющий от 1 до 3 атомов углерода, представляет собой этанол.
5. Способ получения [18F]флутеметамола по любому из пп. 1-4, в котором картридж для обращеннофазной твердофазной экстракции на стадии (с) представляет собой картридж, заполненный триаконтил-силилированным силикагелем.
6. Способ получения [18F]флутеметамола по любому из пп. 1-5, дополнительно включающий стадии:
(e) очистки [18F]флутеметамола после стадии (d) при использовании картриджа для обращеннофазной твердофазной экстракции и
(f) очистки [18F]флутеметамола после стадии (е) при использовании картриджа для твердофазной экстракции с гидрофильным взаимодействием.
7. Способ получения [18F]флутеметамола по п. 6, в котором картридж для обращеннофазной твердофазной экстракции на стадии (е) представляет собой картридж, заполненный триаконтил-силилированным силикагелем.
8. Способ получения [18F]флутеметамола по п. 6 или 7, в котором картридж для твердофазной экстракции с гидрофильным взаимодействием на стадии (f) представляет собой картридж, заполненный аминопропилированным силикагелем.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| РАДИОФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2008 |
|
RU2475267C2 |

