Изобретение относится к медицине, а именно к лекарственным средствам, предназначенным для лечения сердечно-сосудистых заболеваний. Данная композиция может применяться для лечения в комплексной терапии артериальной гипертензии, начальной стадии сердечной недостаточности, гипертензии, отягощенной метаболическим синдромом с проявлением гиперхолестеролемии, гиперинсулинемии, инсулиновой резистентности, миокардиодистрофии, нарушении ритма, кардионеврозе. Согласно данным ВОЗ, сердечно-сосудистые заболевания из года в год являются причиной №1 смертности, как в развитых, так и в развивающихся странах, что указывает на принципиальную необходимость продолжения поисков новых стратегий для лечения/компенсации кардиоваскулярных патологий. Одним из наиболее распространенных (и прогрессирующих с возрастом) заболеваний является артериальная гипертензия (АГ) и ассоциированные с ее течением патологии (ишемическая болезнь сердца, гипертрофия, сердечная недостаточность). АГ имеет полиэтиологическую и многофакторную природу возникновения, что обуславливает множественность стратегий ее компенсации. Основными и наиболее часто применяемыми являются ингибиторы ангиотензин-превращающего фермента, блокаторы потенциал-чувствительных Са2+-каналов, β-адреноблокаторы, антагонисты рецепторов ангиотензина-II, тиазидные диуретики, имидазолиновые агонисты. Зачастую монотерапия препаратом из конкретной группы не является успешной, и тогда их комбинируют в составе комплексной терапии. Для ее показания должно быть соблюдено условие усиления (потенциирования) суммарного эффекта комбинации препаратов, а также минимизация возможных побочных эффектов друг друга. Основой предлагаемой композиции является совместное использование β-адреноблокаторов, агонистов имидазолиновых рецепторов 1-го типа и донора оксида азота (NO) для лечения АГ.
Артериальная гипертензия и возникающая на ее фоне гипертоническая болезнь, характеризуемые увеличенными значениями среднего систолического и диастолического давления, являются социально значимыми заболеваниями, и поражают, по разным данным, от 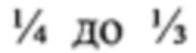 (и выше) населения Земли [1]. В силу множественности причин, приводящих к сдвигу прессорно/депрессорных механизмов регуляции сосудистого тонуса, в настоящий момент существует несколько стратегий компенсации увеличенного артериального давления, которые, зачастую, для улучшения эффектов, комбинируют между собой в составе комплексной терапии. Из патента на изобретение RU 2452363 (СПОСОБ ОЦЕНКИ ВЛИЯНИЯ АЛЬФА- И БЕТА-АДРЕНОБЛОКАТОРА НА СТРУКТУРУ РИТМА СЕРДЦА ПРИ ЛЕЧЕНИИ БОЛЬНЫХ АРТЕРИАЛЬНОЙ ГИПЕРТОНИЕЙ) известно, что при лечении АГ монотерапией β-адреноблокаторами, у значительного процента (до 40%) больных АД не снижается до целевого уровня, а в некоторых случаях регистрировалось его увеличение. Использование смешанного адреноблокатора (для β- и α1-подтипов) - карведилола улучшало некоторые показатели сердечного ритма у больных АГ, оставаясь, однако, в формате «одномерной» терапии. Кроме того, в указанном патенте, акцент делается на использование смешанного β/α1-адреноблокатора для диагностики нарушений сердечного ритма, а не их коррекции.
(и выше) населения Земли [1]. В силу множественности причин, приводящих к сдвигу прессорно/депрессорных механизмов регуляции сосудистого тонуса, в настоящий момент существует несколько стратегий компенсации увеличенного артериального давления, которые, зачастую, для улучшения эффектов, комбинируют между собой в составе комплексной терапии. Из патента на изобретение RU 2452363 (СПОСОБ ОЦЕНКИ ВЛИЯНИЯ АЛЬФА- И БЕТА-АДРЕНОБЛОКАТОРА НА СТРУКТУРУ РИТМА СЕРДЦА ПРИ ЛЕЧЕНИИ БОЛЬНЫХ АРТЕРИАЛЬНОЙ ГИПЕРТОНИЕЙ) известно, что при лечении АГ монотерапией β-адреноблокаторами, у значительного процента (до 40%) больных АД не снижается до целевого уровня, а в некоторых случаях регистрировалось его увеличение. Использование смешанного адреноблокатора (для β- и α1-подтипов) - карведилола улучшало некоторые показатели сердечного ритма у больных АГ, оставаясь, однако, в формате «одномерной» терапии. Кроме того, в указанном патенте, акцент делается на использование смешанного β/α1-адреноблокатора для диагностики нарушений сердечного ритма, а не их коррекции.
Комбинацию β-адреноблокатора (метопролола) и антагониста мускариновых рецепторов - атропина предлагается использовать для эффективной вегетативной блокады сердца в диагностических целях в патенте RU 2572339 (СПОСОБ ФАРМАКОЛОГИЧЕСКОЙ ВЕГЕТАТИВНОЙ БЛОКАДЫ СЕРДЦА). Основное преимущество комбинирования заключалось в уменьшении количества ложноположительных результатов при проведении исследований, что приводило к исключению гипердиагностики состояний, связанных с заболеванием проводящей системы сердца. Исследований по терапии / коррекции патологических состояний (АГ, ишемическая болезнь сердца, миокардиальная гипертрофия и т.д.) комбинацией β-адреноблокатор + атропин в данном случае не проводилось.
Одними из наиболее часто назначаемых препаратов при АГ и ассоциированных поражениях сердечно-сосудистой системы являются так называемые β-адреноблокаторы (бисопролол, карведилол, пиндолол, атенолол и т.д.). Их эффективность показана множественными рандомизированными исследованиями, а механизм действия сводится к ингибированию β-адренергических сайтов связывания (β-АР) в гладкомышечных клетках сосудов и сарколемме кардиомиоцитов [2]. β-адреноблокаторы обладают опосредованным гипотензивным эффектом за счет уменьшения частоты сердечных сокращений и сердечного выброса (минутного объема крови). Показано, что некоторые из них могут быть успешно применены в задачах по предупреждению и улучшению прогноза при ишемической болезни сердца [3]. β-адреноблокаторы увеличивают среднюю продолжительность жизни при сердечной недостаточности, асимптоматической дисфункции левого желудочка и у больных, перенесших инфаркт миокарда [4]. В силу того, что β-АР широко распространены в периферических тканях, где вовлекаются во множество физиологических функций, монотерапия β-адреноблокаторами может вызывать ряд нежелательных эффектов. Например, достаточно распространенным из таких эффектов является бронхоспазм, в связи с чем, большинство специалистов не рекомендуют использование β-адреноблокаторов для снижения кровяного давления при хронической обструктивной болезни легких и бронхиальной астме [5]. Основное показание к применению - комплексная терапия при сердечной недостаточности или асимптоматической дисфункции левого желудочка (карведилол и селективные β-блокаторы длительного высвобождения) и различные формы тахиаритмий, связанных с активизацией симпато-адреналовой оси при артериальной гипертензии. В клинической практике β-адреноблокаторы комбинируют с тиазидными диуретиками, что имеет весьма неплохие результаты в поддерживающей терапии при артериальной гипертензии [6]. Однако, тиазидные диуретики негативно влияют на некоторые метаболические показатели углеводного и липидного обменов, в связи с чем не рекомендуется использовать комбинацию β-адреноблокаторов и тиазидных диуретиков у лиц с метаболическим синдромом или нарушениями углеводного/липидного метаболизма [7, 8]. В обширном двойном-слепом исследовании ASCOT BPLA, регистрировалось увеличение концентрации триглицеридов и понижение липопротеинов высокой плотности в плазме при совместной терапии β-адреноблокаторами и тиазидными диуретиками. Кроме того, на 30% чаще развивался вновь возникший сахарный диабет [9]. Однако, зарегистрированный препарат "Лодоз", представляющий из себя комбинацию β-адреноблокатора бисопролола и гидрохлоротиазида до сих пор применяется в клинической практике.
Второй популярной стратегией является комбинация β-адреноблокаторов с антагонистами потенциал-чувствительных Са2+-каналов (так называемые "медленные" Са2+-антагонисты) [10]. Са2+-антагонисты являются метаболически нейтральными препаратами [7, 8], и в ряде случаев они снижают степень поражения органов-мишеней при артериальной гипертензии [11]. В настоящее время комплексный препарат Са2+-антагониста (амлодипина) и β-адреноблокатора бисопролола зарегистрирован под названием "Конкор AM" и применяется в клинической практике.
В патенте на изобретение RU 2304964 (СПОСОБ ЛЕЧЕНИЯ ХРОНИЧЕСКОЙ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ) предлагается в дополнение к стандартной терапии из β-адреноблокаторов / ингибиторов ангиотензин-превращающего фермента / диуретиков / ингибиторов альдостерона, назначаемых в зависимости от функционального класса сердечной недостаточности, использовать цитопротектор «Мексикор» (действующее вещество - этилметилгидроксипиридина сукцинат). Эффективность «Мексикора» обусловлена его неспецифической способностью нейтрализовывать свободные радикалы, проявляя антиоксидантные и антигипоксические свойства.
В настоящем патенте переход к «многомерной» терапии АГ и ассоциированных с ней сердечно-сосудистых патологий затрагивает (кроме β-АР) имидазолиновую рецепцию и NO-зависимую сигнальную систему. Имидазолин-связывающие сайты (рецепторы) изначально были открыты в латеральном ретикулярном ядре продолговатого мозга, как мишени для действия клонидина, отличные по структуре от адренорецепторов [12]. Их активация в центре контроля кровяного давления приводила к умеренной брадикардии и гипотензии у анестезированных животных, на основании чего было постулировано, что имидазолиновые рецепторы вовлекаются в контроль функций сердечно-сосудистой системы [12]. В настоящий момент, агонисты I1-имидазолиновых рецепторов (I1P): моксонидин ("Физиотенз") и рилменидин ("Альбарел") широко применяются в клинической практике как гипотензивные препараты второго поколения.
Не так давно, на плазматической мембране одиночных кардиомиоцитов нами был обнаружен белок Nischarin, рассматриваемый в современной литературе как основной функциональный компонент имидазолиновых рецепторов первого типа [13]. Обнаруженные I1-рецепторы функционально активны в отношении регуляции внутриклеточного уровня кальция, как основного мессенджера, отвечающего за сократимость сердечных клеток. Так, стимуляция имидазолиновых рецепторов рилменидином снижает амплитуду потенциал-зависимых Са2+-токов, однако увеличивает внутриклеточный уровень кальция в изолированных кардиомиоцитах [14]. В настоящей заявке предлагается совместное использование β-адреноблокаторов и агонистов I1-имидазолиновых рецепторов для более эффективного контроля сердечной сократимости в основе комплексного лечения артериальной гипертензии, а также ассоциированных с ней патофизиологических состояний сердечно-сосудистой системы (желудочковая гипертрофия, ишемическая болезнь сердца, сердечная недостаточность).
Также, как и монотерапия β-адреноблокаторами, применение I1-агонистов не всегда позволяет решить проблему сердечно-сосудистых патологий. Увеличение дозы действующего вещества, как самый простой способ повысить суммарную эффективность, в данном случае не работает. Так, в 2002 году потерпела неудачу III фаза клинических испытаний высоких доз моксонидина в качестве средства для борьбы с сердечной недостаточностью [15]. Испытания были вынуждены прекратить досрочно из-за почти двукратно увеличенной смертности у пациентов, принимающих моксонидин, в сравнении с плацебо, несмотря на то, что он был эффективен при лечении АГ.
В статье [16] нами был обнаружен синергистический эффект совместного влияния β-адреноблокады и I1-агониста рилменидина в отношении ингибирования потенциал-зависимых Са2+-токов в кардиомиоцитах. Это позволит существенно уменьшить эффективные дозы I1-агонистов, ослабляя их возможные побочные действия.
Входящий через потенциал-зависимые Са2+-каналы ток является определяющим для выброса депонированного Са2+из эндоплазматического ретикулума в мышечных тканях, включая сердечную и гладкомышечные клетки сосудов (так называемое Са2+-индуцированное Са2+-высвобождение). Резкое увеличение уровня кальция в цитозоле приводит к сенситизации миофибриллярных белков и инициирует акт механохимического сопряжения, приводящий к сокращению мышечных тканей при действии физиологических стимулов [17]. Хорошо известно, что регулярные и пролонгированные нарушения в регуляции работы потенциал-зависимых Са2+-каналов в сердечных клетках приводят к развитию таких патологических состояний, как гипертрофия миокарда, аритмии разного генеза, нарушения проводимости различных отделов сердца, сердечная недостаточность [17]. Одним из ключевых гуморальных регуляторов Са2+-токов в кардиомиоцитах является симпатоадреналовая ось, продуцирующая катехоламины (норадреналин и адреналин), чей синтез резко возрастает во время стресс-реакции "сражайся или спасайся" ("fight-or-flight"). Длительные увеличения плазменных концентраций катехоламинов, возникающие при психоэмоциональных перегрузках, способны менять кинетические характеристики Са2+-токов и модифицировать чувствительность Са2+-каналов. В конечном итоге, нарушения нормальной кальциевой сигнализации в кардиомиоцитах приводит к возникновению сердечно-сосудистых патологий. Так, например, было показано, что вольт-амперные кривые потенциал-зависимых Са2+-токов L-типа в кардиомиоцитах спонтанно-гипертензивных крыс (линия SHR) примерно на 10 мВ смещены в сторону гиперполяризационных потенциалов в сравнении с нормотензивными крысами [18]. Сдвиг вольт-амперных кривых влево означает, что фактический порог стимула для смещения канального воротного блока (а соответственно - и открытия ионного канала) снижен, и для генерации потенциала действия требуется меньшая амплитуда стимула. Модифицированная активность потенциал-зависимых Са2+-каналов была также показана для предсердных миоцитов и гладкомышечных клеток артерий SHR крыс, что в первом случае связывают с абнормальным Са2+-входом, который может провоцировать аритмогенные явления, а во втором - с изменением сосудистого тонуса, коррелирующим с тяжестью течения гипертензии [19, 20]. Кроме того, в сердцах SHR крыс обнаружен увеличенный уровень экспрессии Са2+-каналов L-типа [18], а недавние исследования показали изменения в регуляции статуса фосфорилирования потенциал-зависимых Са2+-каналов [21]. Отмечается, что блокада РКА-зависимого фосфорилирования или аппликация β-адреноблокаторов улучшали некоторые показатели в моделях артериальной гипертензии, в частности, уровень кровяного давления, отношение массы левого желудочка к массе сердца, общее и периферическое сопротивление сосудов [22].
Одной из первых стратегий компенсации гипертонической болезни были предложены так называемые медленные Са2+-антагонисты, дигидропиридиновые блокаторы потенциал-зависимых (главным образом L-типа) Са2+-каналов, которые и в настоящий момент достаточно широко применяются в клинической практике. Тотальная блокада проводимости Са2+-каналов зачастую не менее критична, чем их чрезмерная активация. Потому, более предпочтительной (физиологически обоснованной) стратегией кажется не полное подавление Са2+-токов, реализуемое Са2+-антагонистами в устье канальных комплексов, а регуляция их проводящих состояний посредством процессов фосфорилирования/дефосфорилирования порообразующих и регуляторных субъединиц.
Результаты собственных исследований
Действие I1-агонистов на фоне стимуляции β-адренергической сигнализации.
Агонист β-АР - изопротеренол (1 мкМ) почти втрое увеличивает потенциал-зависимые Са2+-токи (до 286.0±15.0% относительно контрольного уровня, n=12, фиг. 1).
Резкое увеличение амплитуды и плотности входящего в клетку Са2+-тока сопровождалось также достоверным снижением времени полуинактивации канальных комплексов с 14.5±1.9 мс в контроле до 8.7±1.6 мс в присутствии изопротеренола (р≤0.05, фиг. 1е). Очевидно, что такой механизм позволяет несколько снизить входящий поток ионов кальция, хоть и недостаточен для того, чтобы справиться с состоянием кальциевой перегрузки, возникающей в цитозоле кардиомиоцитов из-за аппликации β-агониста. Активация I1Rs миллимолярными концентрациями агматина (2 мМ) частично обращает действие изопротеренола, амплитуда Са2+-токов снижается до 168.7±7.6% относительно контроля, хоть и не возвращается к базальным значениям (n=5, р≤0.05, фиг. 1б, в). Представляется интересным тот факт, что агматин (2 мМ) сам по себе снижающий Са2+-токи на 23.8±1.9% (n=9, фиг. 1в) почти в полтора раза увеличивал свою эффективность на фоне изопротеренола (1 мкМ). Так, амплитуда потенциал-зависимых Са2+-токов при действии той же концентрации агматина на кардиомиоциты, предобработанные β-агонистом, снижалась на 35.4±3.7% (n=9, р≤0.05, фиг. 1в). Агматин достоверно не влиял на величину полуинактивации Са2+-канальных комплексов (12.6±2.2 мс, р≥0.05, фиг. 1е), однако был способен увеличивать время достижения половины пиковой амплитуды при действии изопротеренола до значений, достоверно не отличимых от контроля (12.3±1.8 vs. 14.5±1.9 в контроле, р≥0.05, фиг. 1е). Действие 10 мкМ рилменидина, много более специфичного, чем агматин, агониста I1Rs в целом обнаруживает те же тенденции (фиг. 1г, д). Таким образом, активация I1-имидазолиновых рецепторов оказывается полезной при пролонгированной чрезмерной стимуляции β-адренергической системы. Несмотря на то, что при действии исследуемых агонистов, индуцированные изопротеренолом Са2+-токи сохранялись на большем уровне, чем базальные, их частичная компенсация в сторону снижения амплитуды за счет влияния на проводимость Са2+-каналов представляется важной, как с функциональной, так и фармакологической точек зрения.
Участие протеинкиназы А (PKA) в реализации эффектов I1-агонистов.
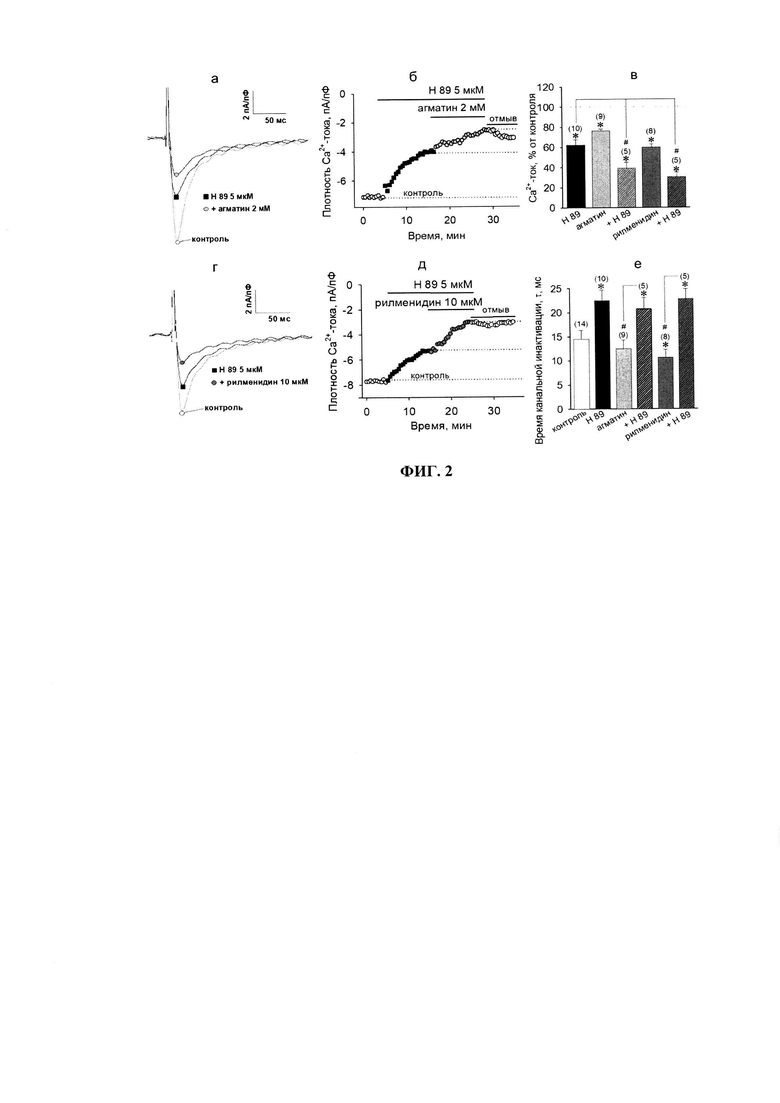
Далее был исследован возможный механизм ингибирования β-АР-сигнализации в отношении потенциал-чувствительных Са2+-токов через I1P. Известно, что основной эффект β-адреноагонистов является PKA-зависимым [2, 3]. Если сигнальные пути от имидазолиновых рецепторов приводят к ингибированию PKA-опосредованного увеличения Са2+-токов, то можно предполагать, что действие их агонистов на фоне блокады данной протеинкиназы будет отсутствовать или выражено значительно ниже. Для проверки этого предположения, нами был использован специфический PKA-блокатор - соединение Н 89. Добавление Н 89 (5 мкМ) в камеру само по себе приводило к достоверному ингибированию потенциал-зависимых Са2+-токов более, чем на треть (до 62.6±5.2% от контроля), показывая, таким образом, что базальный уровень PKA-активности в кардиоците весьма важен для его нормальной функциональности (фиг. 2).
Стоит отметить, что Н 89 достоверно повышал время полуинактивации Са2+-каналов с 14.5±1.9 мс в контроле до 22.5±2.2 мс (n=10, р≤0.05, фиг. 2е). Вероятно, замедление канальной инактивации было связано со снижением статуса фосфорилирования каналов и направлено на частичную компенсацию сниженного поступления ионов кальция из омывающего клетку раствора. Замедленная кинетика инактивации, индуцируемая блокатором протеинкиназы А, сохранялась и при последующей аппликации I1-агонистов: агматина (20.7±2.4 мс, n=5, р≥0.05 vs. Н 89) и рилменидина (22.8±2.1 мс, n=5, р≥0.05 vs. Н 89).
На фоне Н 89 и агматин, и рилменидин оказались способны дополнительно подавлять проводимость потенциал-зависимых Са2+-каналов в размере, сопоставимом с их действием в контроле, значения амплитуды Са2+-токов снижались до 39.5±5.6% (n=5, р≤0.05, фиг. 2в) и 30.5±4.6% (n=5, р≤0.05). Полученные результаты свидетельствуют о том, что активация I1Rs снижает входящие Са2+-токи, вероятно, не через ингибирование PKA-активности, а через какой-то альтернативный механизм. Этот механизм кумулятивен блокаде PKA-зависимого фосфорилирования, и по-видимому, не связан с G-белок-опосредуемой регуляцией активности аденилатциклазы. Известно, что флуктуации внутриклеточного уровня Са2+ тесным образом связаны с изменениями в продукции сигнальной молекулы оксида азота (NO) [13]. NO способен активировать цитоплазматическую гуанилатциклазу, ответственную за синтез циклического гуанозинмонофосфата (цГМФ), а кроме того, может напрямую нитрозилировать белки мишени (включая ионные каналы) и модулировать, таким образом, их активность [23]. Нитрозилирование потенциал-зависимых Са2+-каналов по-разному модулирует общеканальную проницаемость для кальция. Это зависит от положения нитрозилируемого остатка цистеина в структуре белка (в порообразующей или регуляторных субъединицах), конкретного подтипа каналов, функционального статуса клетки, уровня свободного кальция в цитозоле и т.д. [23].
Действие I1-агонистов на Са2+-токи не связано с модуляцией синтеза NO.
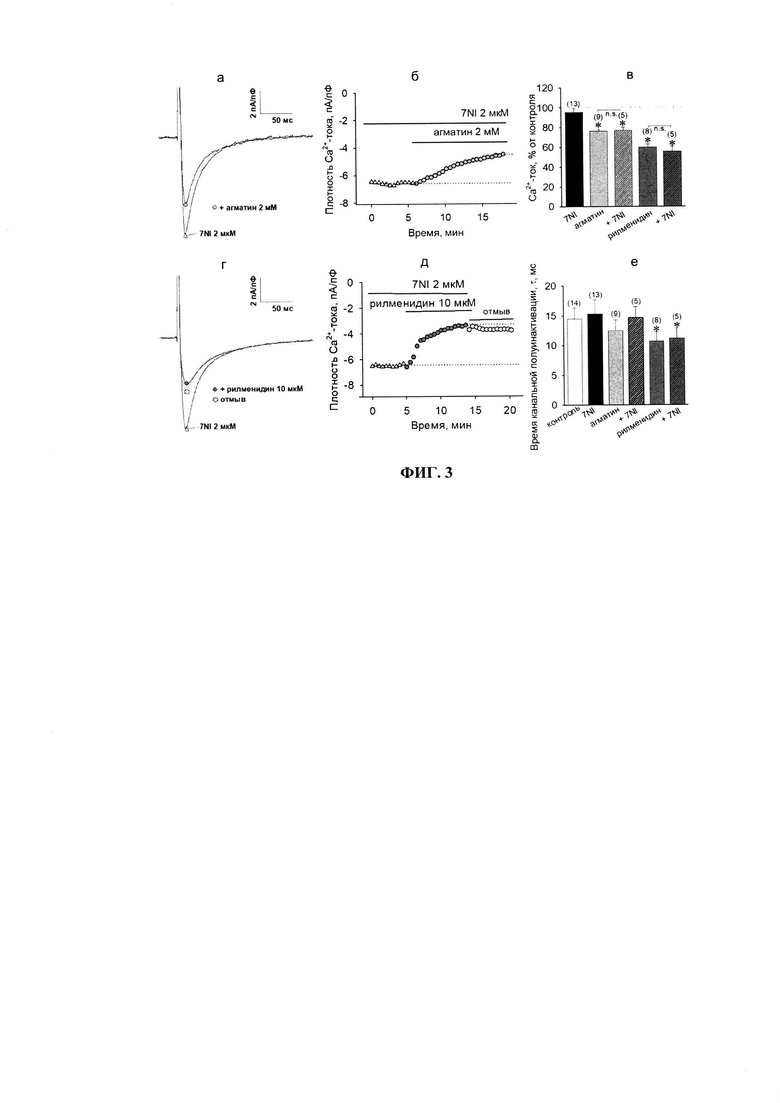
На фиг. 3 представлены результаты по действию агматина и рилменидина на фоне неспецифического блокатора изоформ NO-синтаз - 7NI (2 мкМ). В используемой концентрации, 7NI практически не влиял ни на базальный уровень Са2+-токов (95.6±3.7%, р≥0.05, фиг. 3в), ни на величину канальной полуинактивации (15.3±2.4 мс vs. 14.5±1.9 мс в контроле, р≥0.05, фиг. 3е). Подача в экспериментальную камеру 2 мМ агматина на фоне 7NI приводила к снижению амплитуды Са2+-токов, сопоставимому с действием агматина в одиночку (76.9±3.4% vs. 76.2±1.9%, р≥0.05, n=5, фиг. 3б). Достоверных различий также не было выявлено в значениях величины полуинактивации каналов (12.5±1.8 мс vs. 14.4±1.9 мс, р≥0.05, n=5, фиг. 3е).
Аналогичные результаты были получены и для рилменидина: снижение потенциал-зависимых Са2+-токов до 55.8±5.1%, n=5 (vs. 60.0±3.4%, р≥0.05, фиг. 3д); константа полуинактивации 11.2±1.7 мс (vs. 10.7±1.6 мс, р≥0.05, n=5, фиг. 3е). Таким образом, полученные результаты свидетельствуют о том, что в механизмы снижения I1-агонистами индуцированных изопротеренолом потенциал-зависимых Са2+-токов не вовлекается сигнальная система оксида азота.
Роль серин/треониновых протеинфосфатаз в реализации эффектов I1-агонистов.
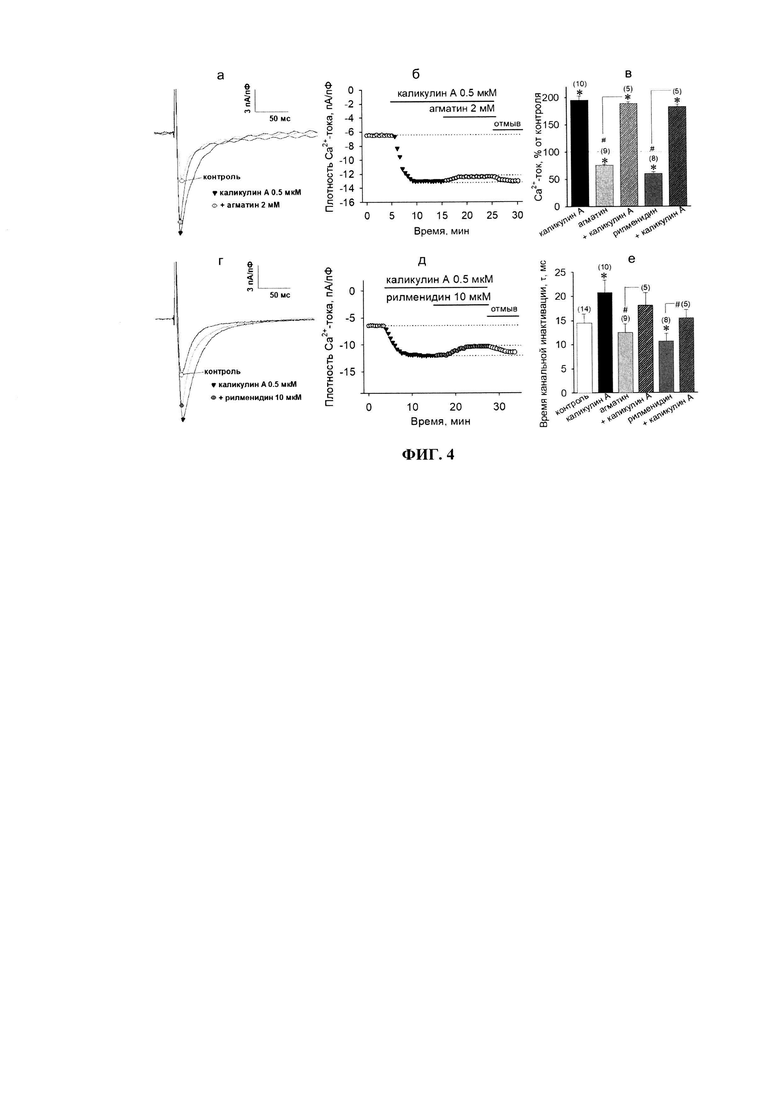
При активации I1P происходит стимуляции активности протеинкиназы С (PKC) [24]. PKC является одним из ключевых Са2+-сенсоров внутри клетки, поскольку ее активность может существенно модулироваться при низкоамплитудных (физиологических) концентрациях Са2+ в цитозоле [24]. Известно, что PKC, среди прочих сигналов, прямо или опосредованно может приводить к активации протеинфосфатаз. Так, например, PKC фосфорилирует 17-кДа ингибиторный белок, ассоциированный с протеинфосфатазой-1 (РР1), вследствие чего этот белок за счет остатка фосфорной кислоты приобретает дополнительный отрицательный заряд и отсоединяется от РР1-комплекса, значительно увеличивая его фосфатазную активность [25]. Дефосфорилирование потенциал-зависимых Са2+-каналов фосфатазами (преимущественно РР1 и РР2А) по остаткам серина и/или треонина - не менее существенный механизм регуляции Са2+-тока в кардиомиоцитах, чем протеинкиназные влияния [26]. Поэтому, мы предположили, что в действие агонистов I1P могут вовлекаться фосфатазы, меняя, таким образом, киназно-фосфатазный баланс. Для проверки участия РР1 и/или РР2А в трансдукции сигналов от I1P был использован общий ингибитор этих фосфатаз - каликулин А (0.5 мкМ). Его апплицирование на клетки приводило к почти двукратному увеличению амплитуды потенциал-зависимых Са2+-токов (до 195.6±7.8%, n=10), что, вероятно, было связано со сдвигом киназно-фосфатазного баланса в сторону фосфорилирования (фиг. 4).
Несмотря на увеличенные значения амплитуды Са2+-тока, входящего в клетку при действии каликулина А, величина канальной полуинактивации достоверно увеличивалась до 20.8±2.6 мс (vs. 14.5±1.9 мс в контроле, n=5, р≤0.05, фиг. 4е), что говорит о замедлении перехода каналов в неактивное состояние, индуцируемое блокадой статуса дефосфорилирования. Агматин (2 мМ) на фоне каликулина А снижает амплитуду Са2+-тока до 188.8±3.6% (n=5, фиг. 4в), что достоверно не отличается от действия ингибитора протеинфосфатаз в одиночку, и свидетельствует о вовлечении активности фосфатаз в I1-опосредуемую сигнализацию. Кроме того, агматин предотвращал вызванное каликулином А замедление перехода Са2+-каналов в инактивированное состояние, возвращая его к значениям не отличающимся от контроля (18.1±2.6 мс, р≥0.05, n=5, фиг. 4е). Аналогичные результаты были получены с использованием рилменидина (фиг. 4д, е).
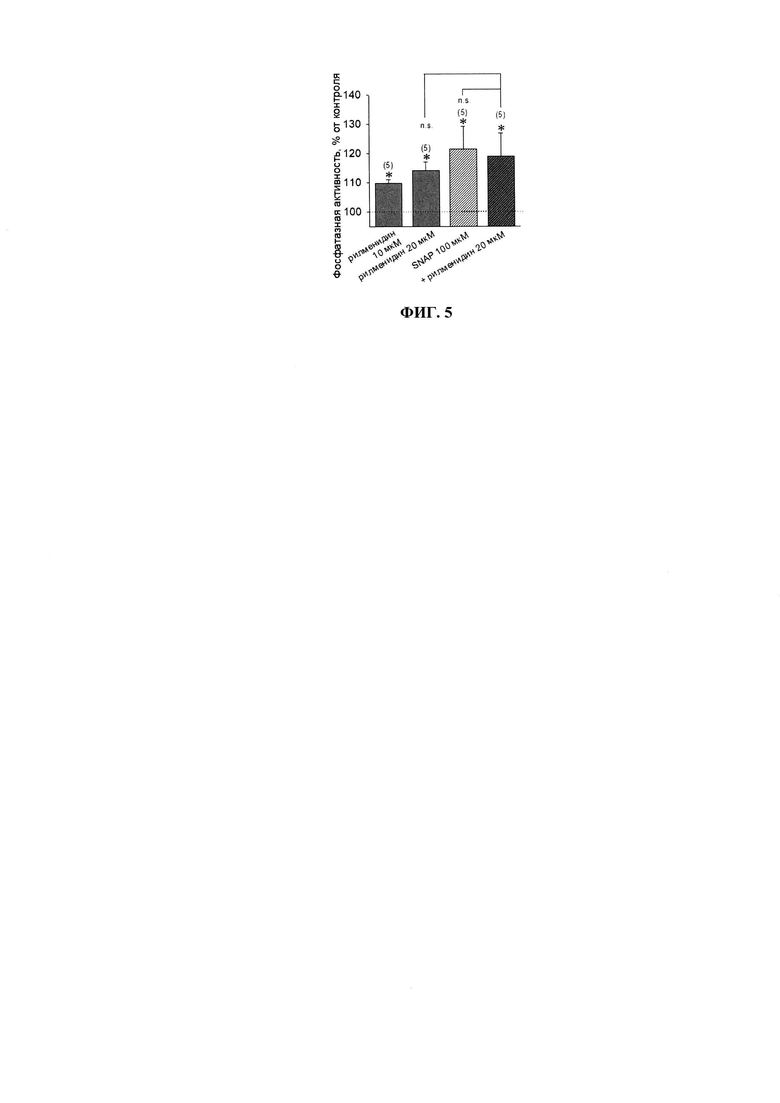
Далее были проведены прямые измерения активности серин/треониновых фосфатаз в миокардиальной ткани левого желудочка. Рилменидин дозо-зависимым образом увеличивает в миокарде величину фосфатазной реакции (фиг. 5).
В концентрациях 10 и 20 мкМ он повышает общую активность серин/треониновых фосфатаз на 9.9±1.2% (n=5, р≤0.05) и 14.3±2.9% (n=5, р≤0.05), соответственно. Для дополнительного валидирования протокола определения фосфатазной активности, был использован прямой донор NO - SNAP (S-нитрозо-N-ацетил-пеницилламин), который, как известно, увеличивает активность РР2А, РР2В и некоторых других. SNAP (100 мкМ) повышал величину фосфатазной реакции на 21.5±7.8% (n=5, р≤0.05, фиг. 5), и его совместная аппликация с 20 мкМ рилменидина достоверно не влияла на этот эффект (19.2±8.0%), n=5, р≤0.05). Таким образом, активация I1-имидазолиновой системы системы в условиях избыточной активации β-адренорецепторов в кардиомиоцитах, PKA-независимо снижает потенциал-чувствительные Са2+-токи, и одним из ключевых механизмов наблюдаемого подавления работы Са2+-каналов является I1P-опосредуемая активация серин/треониновых фосфатаз.
Внутриклеточный уровень Са2+, имидазолиновые агонисты и оксид азота (NO).
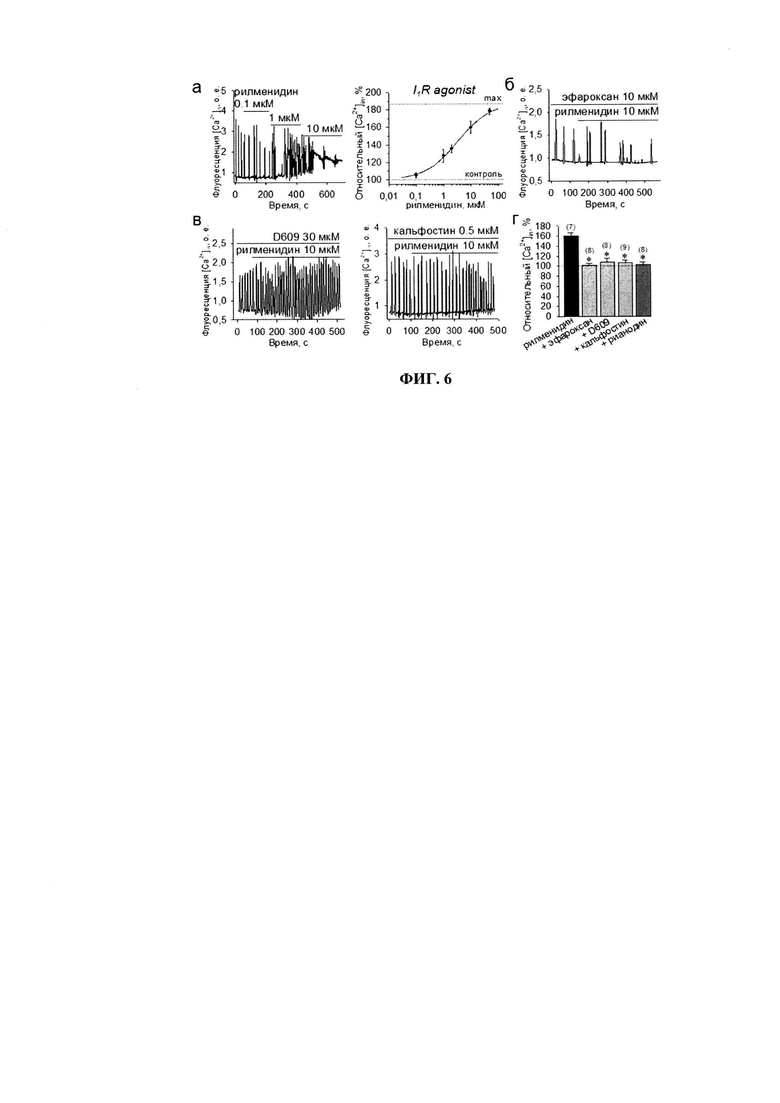
Имидазолиновые агонисты сами по себе, несмотря на снижение потенциал-зависимых Са2+-токов, увеличивают свободный уровень кальция в цитозоле. Так, рилменидин в концентрациях 0.1, 1, 5, 10 и 50 мкМ дозо-зависимо повышал уровень [Ca2+]in на 5.2±3.1% (n=6), 27.4±7.4% (n=9), 33.8±4.1% (n=6), 59.7±6.9% (n=7) и 78.5±3.1% (n=8) соответственно, и мог увеличивать частоту спонтанных волн (фиг. 6а). Дозо-зависимое действие рилменидина хорошо аппроксимировалось уравнением Хилла с коэффициентом 0.7±0.1, величиной полуэффективности ЕС50=3.3±0.4 мкМ и максимальной активацией до 87.3±2.7%.
Эффект 10 мкМ рилменидина на спонтанные Са2+-волны в кардиомиоцитах полностью предотвращался антагонистом I1P - эфароксаном (с 59.7±6.9% до 1.2±4.8%), n=8, р<0.05, фиг. 6б), блокатором PC-PLC - D609 (до 8.4±7.8%, n=8, р<0.05, фиг. 6в), ингибитором PKC изоформ - кальфостином С (6.5±5.7%, n=9, р<0.05, фиг. 6в) и ингибирующими концентрациями рианодина (3.5±4.8%), р<0.05, фиг. 6г).
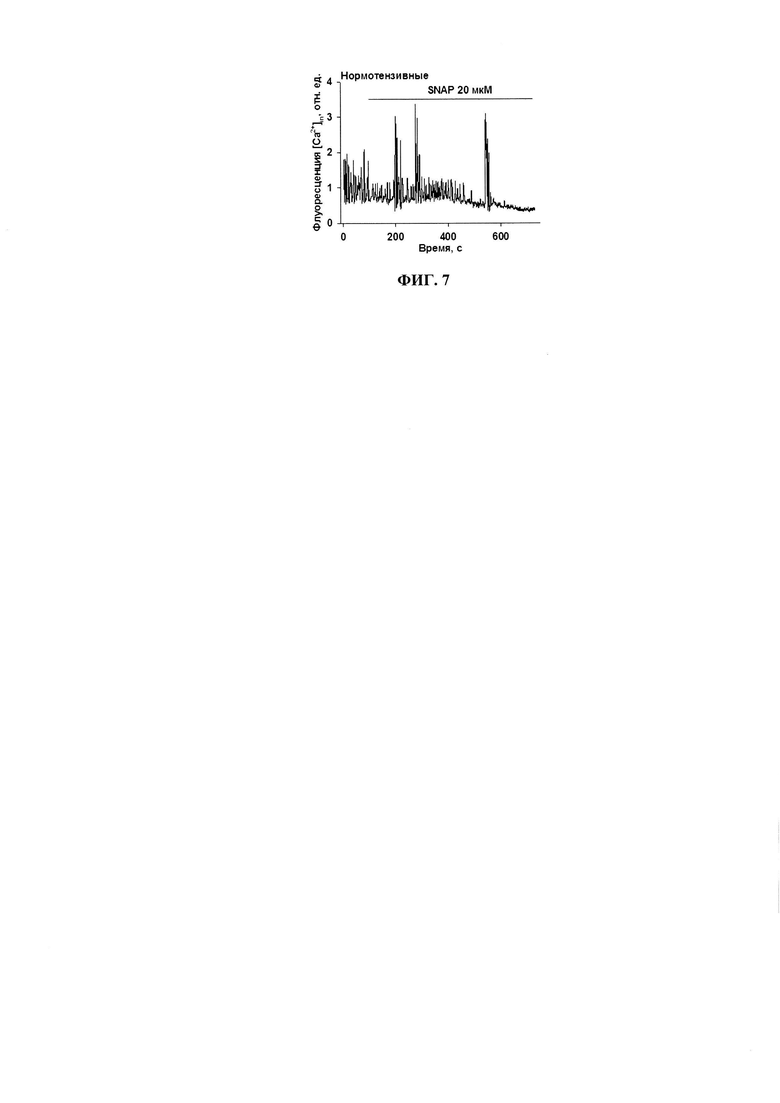
Известно, что NO является вазодилататором, расслабляющим стенки кровеносных сосудов через релаксацию гладкомышечных клеток. Некоторые доноры NO (нитроглицерин) применяются в клинической практике для снятия приступов стенокардии и улучшению расслабляемости сердечной мышцы. Кроме того, мы показали, что NO приводит к утилизации свободного кальция из цитозоля за счет стимуляции Са2+-АТФазы саркоплазматического ретикулума (SERCA) (фиг. 7).
Таким образом, доноры NO могут предотвратить нежелательные эффекты аппликации имидазолиновых агонистов, связанные с накоплением повышенных концентраций кальция в цитозоле.
Преимущества фиксированных комбинаций препаратов в одной таблетке ранее были исследованы и показана их эффективность. Так, Egan и соавт. обнаружили, что использование фиксированной комбинации, в отличие от свободной комбинации тех же препаратов, приводит к значительному улучшению контроля уровня артериального давления [10], а по данным метаанализа Gupta и соавт. к повышению приверженности больных терапии [27].
В итоге, при использовании нашей композиции при лечении сердечно-сосудистых заболеваний мы получаем следующие преимущества по сравнению с имеющимися аналогами:
1) Синергизм (усиление) совместного действия β-адреноблокаторов и имидазолиновых агонистов позволит снизить эффективные терапевтические дозы каждого из соединений, что неизбежно приведет к минимизации возможных побочных эффектов, связанных с приемом данных соединений.
2) Третий компонент в виде донора NO минимизирует негативное действие имидазолиновых агонистов на внутриклеточный уровень кальция, что приводит к более эффективному контролю над сократимостью миокарда и снижает риски возникновения кальциевой перегрузки рабочих кардиомиоцитов, предотвращая развитие патофизиологических событий в сердце.
Литература.
[1] World Health Statistics 2016: Monitoring health for the SDGs. Geneva: World Health Organization; 2016. http://www.who.int/gho/publications/world_health_statistics/2016/en/
[2] C. McCune, P. McKavanagh, I.B. Menown, A review of the key clinical trials of 2015: results and implications, Cardiol. Ther. 5 (2016) 109-132.
[3] A. Cannavo, G. Rengo, D. Liccardo, A. Pun, E. Gao, A.J. George, G. Gambino, A. Rapacciuolo, D. Leosco, B. Ibanez, N. Ferrara, N. Paolocci, W.J. Koch, β1-Blockade Prevents Post-Ischemic Myocardial Decompensation Via β3AR-Dependent Protective Sphingosine-1 Phosphate Signaling, J. Am. Coll. Cardiol. 70 (2017) 182-192.
[4] J. Hong, A.R. Barry, Long-term beta-blocker therapy after myocardial infarction in the reperfusion era: a systematic review, Pharmacotherapy 38 (2018) 546-554.
[5] J.G. Baker, R.G. Wilcox, β-blockers, heart disease and COPD: current controversies and uncertainties, Thorax 72 (2017) 271-276.
[6] L. Hansson, L.H. Lindholm, T. Ekbom,  J. Lanke,
J. Lanke,  P.O. Wester, T. Hedner, U. de Faire, Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension-2 study, Lancet 354 (1999) 1751-1756.
P.O. Wester, T. Hedner, U. de Faire, Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension-2 study, Lancet 354 (1999) 1751-1756.
[7] Brook R.D., Mechanism of Differential Effects of Antihypertensive Agents on Serum Lipids, Curr Hypertens Rep.2 (2000) 370-377.
[8] J.H. Karnes, R.M. Cooper-DeHoff, Antihypertensive medications: benefits of blood pressure lowering and hazards of metabolic effects, Expert Rev Cardiovasc Ther. 7 (2009) 689-702.
[9]  P.S. Sever, N.R. Poulter et al., Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial, Lancet. 366 (2005) 895-906.
P.S. Sever, N.R. Poulter et al., Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial, Lancet. 366 (2005) 895-906.
[10] B.M. Egan, D. Bandyopadhyay, S.R. Shaftman et al., Initial monotherapy and combination therapy and hypertension control the first year, Hypertension. 59 (2012) 1124-1131.
[11] A.U. Klingbeil, M. Schneider, P. Martus et al., A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension, Am J Med. 115 (2003) 41-46.
[12] P. Bousquet, J. Feldman, D. Atlas, An endogenous, non-catecholamine clonidine antagonist, increases mean arterial blood pressure, Eur. J. Pharmacol. 124 (1986) 167-170.
[13] A.V. Maltsev, Y.M. Kokoz, E.V. Evdokimovskii, et al., Alpha-2 adrenoceptors and imidazoline receptors in cardiomyocytes mediate counterbalancing effect of agmatine on NO synthesis and intracellular calcium handling, J. Mol. Cell. Cardiol. 68 (2014) 66-74.
[14] A.V. Maltsev, M.N. Nenov, O.Y. Pimenov, et al., Modulation of L-type Ca2+ currents and intracellular calcium by agmatine in rat cardiomyocytes, Biochem. (Moscow) Suppl. Series A: Membr. Cell. Biol. 7 (2013) 100-112.
[15] J.N. Cohn, M.A. Pfeffer, J. Rouleau, N. Sharpe, K. Swedberg, M. Straub, et al., Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON), Eur. J. of Heart Fail. 5 (2003) 659-667.
[16] A.V. Maltsev, E.V. Evdokimovskii, Y.M. Kokoz, Synergism of myocardial β-adrenoceptor blockade and I1-imidazoline receptor-driven signaling: Kinase-phosphatase switching, Biochem. Biophys. Res. Commun. 511 (2019) 363-368.
[17] D.M. Bers, Calcium cycling and signaling in cardiac myocytes, Annu. Rev. Physiol. 70 (2008) 23-49.
[18] Z.Z. Tang, P. Liao, G. Li, et al., Differential splicing patterns of L-type calcium channel Cav 1.2. subunit in hearts of Spontaneously Hypertensive Rats and Wistar Kyoto Rats, Biochim. Biophys. Acta 1783 (2008) 118-130.
[19] L. Shi, H. Zhang, Y. Chen, et al., Chronic exercise normalizes changes in Cav 1.2 and KCa 1.1 channels in mesenteric arteries from spontaneously hypertensive rats, Br. J. Pharmacol. 172 (2015) 1846-1858.
[20] F. Pluteanu,  J. Plackic, et al., Early subcellular Ca2+ remodelling and increased propensity for Ca2+ alternans in left atrial myocytes from hypertensive rats, Cardiovasc. Res. 106 (2015) 87-97.
J. Plackic, et al., Early subcellular Ca2+ remodelling and increased propensity for Ca2+ alternans in left atrial myocytes from hypertensive rats, Cardiovasc. Res. 106 (2015) 87-97.
[21] M. Poomvanicha, J. Matthes, K. Domes, et al., Beta-adrenergic regulation of the heart expressing the Ser1700A/Thr1704A mutated Cav 1.2 channel, J. Mol. Cell. Cardiol. 111 (2017) 10-16.
[22] S.M. MacDonnell, H. Kubo, D.L. Crabbe, et al., Improved myocardial beta-adrenergic responsiveness and signaling with exercise training in hypertension, Circulation 111 (2005)3420-3428.
[23] R. Fischmeister, L. Castro, A. Abi-Gerges, et al., Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels, Comp. Biochem. Physiol. A Mol. Integr. Physiol. 142 (2005) 136-143.
[24] A.C. Newton, C.E. Antal, S.F. Steinberg, Protein kinase С mechanisms that contribute to cardiac remodeling, Clin. Sci. (Lond.) 130 (2016) 1499-1510.
[25] J.C. Braz, K. Gregory, A. Pathak, et al., PKC-alpha regulates cardiac contractility and propensity toward heart failure, Nat. Med. 10 (2004) 248-254.
[26] W.H. duBell, T.B. Rogers, Protein phosphatase 1 and an opposing protein kinase regulate steady-state L-type Ca2+ current in mouse cardiac myocytes, J. Physiol. 556 (2004) 79-93.
[27] A.K. Gupta, S. Arshad, N.R. Poulter, Compliance, safety, and effectiveness of fixed-dose combinations of antihypertensive agents: a meta-analysis, Hypertension. 55 (2010) 399-407.
| название | год | авторы | номер документа |
|---|---|---|---|
| Фармакологическая композиция для компенсации артериальной гипертензии и ассоциированных с ней патологий сердечно-сосудистой системы | 2021 |
|
RU2792884C1 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ЭФФЕКТИВНОСТИ ЛЕЧЕНИЯ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ СЕЛЕКТИВНЫМИ АГОНИСТАМИ ЦЕНТРАЛЬНЫХ I-ИМИДАЗОЛИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2363378C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ СЕЛЕКТИВНЫЙ АГОНИСТ ИМИДАЗОЛИНОВОГО РЕЦЕПТОРА I1 И БЛОКАТОР РЕЦЕПТОРА АНГИОТЕНЗИНА II | 2004 |
|
RU2362561C2 |
| СПОСОБ ДИФФЕРЕНЦИРОВАННОГО НАЗНАЧЕНИЯ МОКСОНИДИНА У ПАЦИЕНТОВ С АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИЕЙ НА ФОНЕ МЕТАБОЛИЧЕСКОГО СИНДРОМА | 2010 |
|
RU2426543C1 |
| СПОСОБ ВЫБОРА ТАКТИКИ ПРОВЕДЕНИЯ ЛЕЧЕБНЫХ МЕРОПРИЯТИЙ У БОЛЬНЫХ С МЕТАБОЛИЧЕСКИМ СИНДРОМОМ НА ФОНЕ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ | 2007 |
|
RU2337612C1 |
| СПОСОБ ОЦЕНКИ ВЛИЯНИЯ АЛЬФА- И БЕТА-АДРЕНОБЛОКАТОРА НА СТРУКТУРУ РИТМА СЕРДЦА ПРИ ЛЕЧЕНИИ БОЛЬНЫХ АРТЕРИАЛЬНОЙ ГИПЕРТОНИЕЙ | 2011 |
|
RU2452363C1 |
| СПОСОБ ИНДИВИДУАЛЬНОГО ВЫБОРА ГИПОТЕНЗИВНОГО ПРЕПАРАТА ДЛЯ ЛЕЧЕНИЯ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ | 1999 |
|
RU2232543C2 |
| ПРИМЕНЕНИЕ ЦИКЛИЧЕСКИХ БИОИЗОСТЕРОВ ПРОИЗВОДНЫХ ПУРИНОВОЙ СИСТЕМЫ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, ВЫЗВАННЫХ НАРУШЕНИЯМИ НИТРЕРГИЧЕСКОЙ И ДОФАМИНЕРГИЧЕСКОЙ СИСТЕМ | 2004 |
|
RU2326870C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ РИСКА РАЗВИТИЯ ЭССЕНЦИАЛЬНОЙ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ | 2019 |
|
RU2703559C1 |
| Способ дифференцированного выбора фармакотерапии артериальной гипертензии на фоне дорсопатии у лиц операторских профессий | 2024 |
|
RU2826094C1 |

Настоящее изобретение относится к медицине, а именно к композиции для лечения артериальной гипертензии и ассоциированных с ней патологий сердечно-сосудистой системы, состоящей из взятых в терапевтически эффективных количествах β-адреноблокатора, агониста I1-имидазолиновых рецепторов, донора оксида азота, где β-адреноблокатор выбирают из бисопролола, карведилола, пиндолола, атенолола, где агонист I1-имидазолиновых рецепторов выбирают из моксонидина и рилменидина, где донор оксида азота выбирают из S-нитрозо-N-ацетилпеницилламина и натрия нитропруссида. Настоящее изобретение обеспечивает синергизм совместного действия β–адреноблокаторов и имидазолиновых агонистов, который позволит снизить эффективные терапевтические дозы каждого из соединений, что неизбежно приведет к минимизации возможных побочных эффектов, связанных с приемом данных соединений, а третий компонент в виде донора NO минимизирует негативное действие имидазолиновых агонистов на внутриклеточный уровень кальция, что приводит к более эффективному контролю над сократимостью миокарда и снижает риски возникновения кальциевой перегрузки рабочих кардиомиоцинтов, предотвращая развитие патофизиологических событий в сердце. 7 ил.
Композиция для лечения артериальной гипертензии и ассоциированных с ней патологий сердечно-сосудистой системы, состоящая из взятых в терапевтически эффективных количествах β-адреноблокатора, агониста I1-имидазолиновых рецепторов, донора оксида азота, где β-адреноблокатор выбирают из бисопролола, карведилола, пиндолола, атенолола, где агонист I1-имидазолиновых рецепторов выбирают из моксонидина и рилменидина, где донор оксида азота выбирают из S-нитрозо-N-ацетилпеницилламина и натрия нитропруссида.
| WO 2013010241 A1, 24.01.2013 | |||
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ СЕЛЕКТИВНЫЙ АГОНИСТ ИМИДАЗОЛИНОВОГО РЕЦЕПТОРА I1 И БЛОКАТОР РЕЦЕПТОРА АНГИОТЕНЗИНА II | 2004 |
|
RU2362561C2 |

