Настоящее изобретение относится к новой пропиленовой композиции, которая характеризуется сочетанием низкой температуры начала сваривания (ТНС), низкой общей миграции и хороших оптических свойств, таких как низкая мутность, и хорошей стерилизуемостью.
Кроме того, настоящее изобретение относится к применению полипропиленовой композиции и полученных из нее изделий.
Полипропилены подходят для множества областей применения. Например, полипропилен (ПП) применим в областях, в которых свойства свариваемости играют важную роль, например, в пищевой или медицинской упаковочной промышленности.
Термосварка является преобладающим способом получения гибких и полужестких упаковок. Важным показателем хорошего сваривания является, в частности, низкая температура начала сваривания (ТНС), которая необходима для поддержания высокой скорости упаковочных машин. Для обеспечения быстрого сваривания низкая ТНС является преимуществом. При работе при низких температурах, преимуществом является то, что свариваемое изделие не подвергают воздействию высокой температуры. Также существуют экономические преимущества, поскольку более низкие температуры, как известно, дешевле создавать и поддерживать.
Дополнительные преимущества заключаются в том, что избегают высоких температур сварки, особенно при упаковке чувствительных к температуре товаров.
Кроме того, также желательно иметь упаковочный материал с удовлетворительными оптическими свойствами, такими как низкая мутность.
В некоторых областях пищевой промышленности, где требуются стерилизуемые пакеты, или в некоторых областях медицины, необходима обработка стерилизацией. Наиболее распространенными способами стерилизации являются применение тепла (пара), излучения (бета-излучение, электроны или гамма-излучение) или химических веществ (обычно этиленоксид). Стерилизацию паром обычно осуществляют в температурном диапазоне от 120°С до 130°С. Таким образом, материал должен обладать достаточной термостойкостью, например, температура плавления должна быть значительно выше, чем обычная температура стерилизации паром приблизительно от 120°С до 130°С.
Конечно, обработка полимера в условиях стерилизации, описанных выше, может ухудшить его конечные свойства, особенно оптические свойства, такие как прозрачность, т.е. низкая мутность.
Кроме того, должны быть выполнены некоторые правила в отношении применения таких материалов в областях применения при контакте с пищевыми продуктами, поэтому соблюдение правил в отношении пищевых продуктов с точки зрения общей миграции (ОМ) является обязательным в перспективных областях применения упаковки.
Кроме того, более высокое содержание растворимых, соответственно, экстрагируемых компонентов, таких как растворимые в гексане вещества, также нежелательны в области медицинской упаковки.
Независимо от типа полимера, полимерная композиция должна в лучшем случае удовлетворять всем требуемым конечным свойствам и, кроме того, должна быть легко обрабатываемой, т.е. должна выдерживать напряжение. Однако конечные свойства и технологические свойства часто вступают в противоречие.
Часто оказывается, что улучшения одного из заданных свойств достигают за счет других свойств. Например, чтобы улучшить свойства сваривания, обычно получают низкую температуру начала сваривания (ТНС) с более высоким содержанием сомономера, однако, вызывая две проблемы:
1. высокое давление приводит к более низкой Тпл, что, в свою очередь, приводит к ухудшению стерилизации паром или даже невозможности ее осуществления, и
2. включение большего количества сомономеров, особенно для ПП на основе катализаторов типа Циглера-Натта (ЦН), вызывает проблемы, связанные с растворимыми в С6 веществами (FDA, Food and Drag Administration - Комиссия по контролю за лекарствами и питательными веществами) и общей миграции.
В ЕР 3064548 предложена полипропиленовая композиция, представляющая собой бинарную смесь, включающую фракцию П1 сополимера пропилена и гексена и фракцию П2 сополимера пропилена, гексена и этилена в определенном количестве, для достижения требуемого баланса между высокой температурой плавления и низкой температурой начала сваривания ТНС, которая к тому же обладает широким диапазоном температур сваривания, предпочтительными оптическими свойствами, низким количеством растворимых в гексане веществ и достаточной термостойкостью, обеспечивая обработку стерилизацией без отрицательного влияния на оптические свойства. Данная композиция имеет достаточно большое количество растворимой в холодном ксилоле (РХК) фракции.
В ЕР 3064514 предлагают для этой же цели композицию статистического сополимера С2 и С3, содержащую 3 полимерные фракции (А), (Б) и (В) с различным содержанием этиленового сомономера. Данная композиция снова имеет достаточно высокое количество фракции растворимой в холодном ксилоле (РХК).
Хотя в этой области была проделана большая исследовательская работа, все еще существует потребность в дальнейшем совершенствовании и, таким образом, в разработке материалов, обладающих улучшенным балансом между низкой температурой начала сваривания (ТНС), предпочтительными оптическими свойствами, низкой общей миграцией и достаточной термической стойкостью, для обеспечения возможности обработки стерилизацией без отрицательного влияния на оптические свойства.
Настоящее изобретение основано на заключении, что вышеуказанные потребности в области термосварки могут быть достигнуты с помощью особого состава полипропиленовой композиции.
Краткое описание изобретения
Таким образом, настоящее изобретение относится к полипропиленовой композиции, включающей смесь, состоящую из
(А) от 50,0 до 99,0 масс. % статистического сополимера пропилена и этилена имеющего содержание этилена от 0,1 до 12,0 масс. %, ПТР2 (230°С, 2,16 кг, ISO1133) от 0,5 до 60,0 г/10 мин и температуру плавления Тпл (ДСК) от 135°С до 155°С и
(Б) от 1,0 до 50,0 масс. % статистического сополимера пропилена и гексена, имеющего содержание гексена от 0,1 до 12,0 масс. % и ПТР2 (230°С, 2,16 кг, ISO1133) от 0,5 до 60,0 г/10 мин и температуру плавления Тпл (ДСК) от 120°С до 140°С,
где
(i) температура плавления статистического сополимера (Б) пропилена и гексена меньше, чем температура плавления статистического сополимера (А) пропилена и этилена и
(ii) ПТР2 (230°С, 2,16 кг, ISO1133) смеси составляет от 0,5 до 60,0 г/10 мин.
Неожиданно было установлено, что такие композиции имеют оптимизированный или улучшенный баланс между низкой температурой начала сваривания (ТНС), предпочтительными оптическими свойствами, низкой общей миграцией и достаточной термической стойкостью для обеспечения возможности обработки стерилизацией, благодаря которой оптические свойства поддерживают на высоком уровне до и после стерилизации.
В одном воплощении настоящего изобретения статистический сополимер (А) пропилена и этилена можно получить, предпочтительно получают в присутствии катализатора Циглера-Натта, а статистический сополимер (Б) пропилена и гексена можно получить, предпочтительно получают в присутствии металлоценового катализатора.
В еще одном воплощении изобретение относится к применению определенной выше композиции для получения изделий, и к самим изделиям.
Описание изобретения
Далее в описании отдельные компоненты определены более подробно.
Пропиленовая композиция по настоящему изобретению включает смесь из
(А) статистического сополимера пропилена и этилена и
(Б) статистического сополимера пропилена и гексена.
Компонент (А): статистический сополимер пропилена и этилена
Статистический сополимер пропилена и этилена имеет содержание этилена от 0,1 до 12,0 масс. %, предпочтительно от 0,5 до 10,0 масс. %, более предпочтительно от 1,0 до 8,0 масс. %, еще более предпочтительно от 1,5 до 7,0 масс. % и еще более предпочтительно от 2,0 до 6,0 масс. %.
ПТР2 (230°С, 2,16 кг, ISO1133) статистического сополимера пропилена и этилена составляет от 0,5 до 60,0 г/10 мин, предпочтительно от 1,0 до 55,0 г/10 мин, более предпочтительно от 5,0 до 50,0 г/10 мин, еще более предпочтительно от 10,0 до 45,0 г/10 мин и еще более предпочтительно от 20,0 до 40,0 г/10 мин.
Температура плавления Тпл статистического сополимера пропилена и этилена составляет от 135°С до 155°С, предпочтительно от 140°С до 150°С.
Для того, чтобы быть пригодным для применения в пищевой или фармацевтической упаковке, предпочтительно статистический сополимер пропилена и этилена имеет ограниченное количество растворимых и/или экстрагируемых веществ.
Таким образом, также предпочтительно содержание растворимых в холодном ксилоле веществ (РХК) в статистическом сополимере пропилена и этилена составляет от 2,0 до 13,0 масс. % более предпочтительно от 4,0 до 12,0 масс. % и еще более предпочтительно от 6,0 до 11,0 масс. %.
Статистический сополимер пропилена и этилена может быть получен на одной стадии полимеризации, включающей один ректор (Р1) полимеризации или в последовательном способе полимеризации, включающем по меньшей мере два реактора (Р1) и (Р2) полимеризации, где в первом реакторе (Р1) полимеризации получают первую фракцию пропиленового сополимера (Р-ПП1), которую затем перемещают во второй реактор (Р2) полимеризации. Во втором реакторе (Р2) полимеризации получают вторую фракцию пропиленового сополимера (Р-ПП2) в присутствии первой фракции пропиленового сополимера (Р-ПП1).
Способ полимеризации, который подходит для получения статистического сополимера пропилена и этилена, в основном включает одну или две стадии полимеризации, и каждая стадия может быть выполнена в растворе, в суспензии, в псевдоожиженном слое, в массе или в газовой фазе.
Термин «реактор полимеризации» означает, что осуществляют основную полимеризацию. Таким образом, в случае, если процесс состоит из одного или двух реакторов полимеризации, это определение не исключает возможности того, что вся система включает, например, стадию предварительной полимеризации в реакторе предварительной полимеризации. Термин «состоит из» является закрытой формулировкой только с точки зрения основных реакторов полимеризации.
Термин «последовательный способ полимеризации» означает, что статистический сополимер пропилена и этилена получают по меньшей мере в двух реакторах, соединенных последовательно. Соответственно, такая система полимеризации содержит по меньшей мере первый реактор (Р1) полимеризации и второй реактор (Р2) полимеризации, а также возможно третий реактор (Р3) полимеризации.
Первый, соответственно единственный, реактор (Р1) полимеризации предпочтительно представляет собой суспензионный реактор и может быть любым реактором непрерывного действия или реактором периодического действия с мешалкой, или петлевым реактором, работающим в массе или суспензии. «В массе» означает полимеризацию в реакционной среде, которая включает по меньшей мере 60 масс. % мономера. В соответствии с настоящим изобретением, суспензионный реактор предпочтительно является петлевым реактором (для эксплуатации в массе).
В случае, когда используют «способ последовательной полимеризации», второй реактор (Р2) полимеризации и возможно третий реактор (Р3) полимеризации представляют собой газофазные реактора (ГФР), т.е. первый газофазный ректор (ГФР1) и второй газофазный ректор (ГФР2). Газофазный ректор (ГФР) в соответствии с данным изобретением предпочтительно представляет собой реактор с псевдоожиженным слоем, реактор с быстрым псевдоожиженным слоем или реактор с неподвижным слоем, или любое их сочетание.
Предпочтительным многостадийным способом является способ «петля-газовая фаза», такой как разработанный Borealis (известный как технология BORSTAR®), описанный, например, в патентной литературе, такой как ЕР 0 887 379, WO 92/12182, WO 2004/000899, WO 2004/111095, WO 99/24478, WO 99/24479 или WO 00/68315.
Еще одним подходящим суспензионно-газофазным способом является способ Spheripol® Basell.
Статистический сополимер пропилена и этилена может быть мономодальным или мультимодальным, таким как бимодальный, с точки зрения содержания сомономера и/или ПТР2.
Если статистический сополимер пропилена и этилена является мономодальным, его предпочтительно получают на одной стадии полимеризации в единственном реакторе (Р1) полимеризации. Альтернативно, мономодальный статистический сополимер пропилена и этилена может быть получен посредством последовательного способа полимеризации с использованием одинаковых условий полимеризации во всех реакторах.
Если статистический сополимер пропилена и этилена является мультимодальным, его предпочтительно получают посредством способа полимеризации с использованием различных условий полимеризации (количество сомономера, количество водорода и т.д.) в реакторах.
Предпочтительно статистический сополимер пропилена и этилена (А) в соответствии с данным изобретением получают в присутствии катализатора Циглера-Натта.
Катализатор Циглера-Натта подают в первый, соответственно единственный, реактор (Р1) полимеризации и возможно перемещают с полимером (суспензией), полученным в первом реакторе (Р1) полимеризации, в последующие реакторы, если статистический сополимер пропилена и этилена получают посредством последовательного способа полимеризации.
Если способ также охватывает стадию предварительной полимеризации, предпочтительно весь катализатор Циглера-Натта подают в реактор предварительной полимеризации. Затем продукт предварительной полимеризации, содержащий катализатор Циглера-Натта, перемещают в первый, соответственно единственный реактор (Р1) полимеризации.
Катализатор Циглера-Натта может представлять собой любой стерео-специфический катализатор Циглера-Натта для полимеризации пропилена, который предпочтительно способен катализировать полимеризацию и сополимеризацию при давлении от 500 до 10000 кПа, в частности, от 2500 до 8000 кПа и при температуре от 40 до 110°С, в частности, от 60 до 110°С.
Предпочтительно катализатор Циглера-Натта (ЦН-К) включает тип катализатора Циглера-Натта с высоким выходом, содержащий компонент - внутренний донор, который можно использовать при высоких температурах полимеризации 80°С или более.
Такой катализатор (ЦН-К) Циглера-Натта с высоким выходом может включать сукцинат, простой диэфир, фталат и т.д., или их смеси, в качестве внутреннего донора (ВнтД), которые, например, выпускает LyondellBasell под торговой маркой Avant ZN.
Другие используемые твердые катализаторы также раскрыты в WO-A-2003/000757, WO-A-2003/000754, WO-A-2004/029112 и WO 2007/137853. Эти катализаторы представляют собой твердые катализаторы из сферических частиц с компактной структурой и низкой площадью поверхности частиц. Кроме того, эти катализаторы характеризуются равномерным распределением каталитически активных центров по частицам катализатора. Катализаторы получают способом затвердевания в эмульсии, где не требуется никаких внешних носителей. Дисперсная фаза в виде жидких капель эмульсии образуют каталитическую часть, которая превращается в твердые частицы катализатора в ходе стадии затвердевания.
Таким образом, в воплощении настоящего изобретения твердый каталитический компонент получают способом, включающим:
- приготовление раствора комплекса магния посредством взаимодействия алкоксимагниевого соединения и донора электронов или его предшественника в жидкой реакционной среде С6-С10 ароматического соединения;
- осуществление реакции указанного комплекса магния с четырехвалентным соединением титана, предпочтительно TiCl4, при температуре более 10°С и менее 50°C с получением эмульсии из плотной, дисперсной фазы, которая имеет отношение от 0,1 до 10, и непрерывной фазы, которая имеет молярное отношение Ti/Mg от 10 до 100, и
- перемешивание эмульсии, возможно в присутствии стабилизатора эмульсии и/или агента, минимизирующего турбулентность, для поддержания капель указанной дисперсной фазы в среднем диапазоне размеров от 5 до 200 мкм.
Частицы катализатора получают после отверждения указанных капель дисперсной фазы посредством нагрева, предпочтительно при температуре от 80°С до 110°С. В указанном способе добавляют соединение алкилалюминия формулы AlR3-nXn, где R является алкилом и/или алкоксигруппой, содержащей от 1 до 20, предпочтительно от 1 до 10 атомов углерода, X является галогеном и n составляет 0, 1 или 2, и оно вступает в контакт с каплями дисперсной фазы перемешиваемой эмульсии. Альтернативно, соединение алкилалюминия формулы AlR3-nXn, приводят в контакт с затвердевшими частицами на стадии промывки перед извлечением конечных твердых частиц.
Подходящими внутренними донорами электронов являются, в частности, (ди)эфиры ароматических (ди)карбоновых кислот. Указанный эфир или диэфир ароматической карбоновой кислоты может быть получен in situ посредством взаимодействия хлорида ароматической карбоновой кислоты или хлорида двухосновной кислоты с С2-С16 алканолом и/или диолом, и предпочтительным является ди-2-этилгексилфталат.
Еще одним подходящим катализатором в настоящем изобретении является твердый катализатор Циглера-Натта, который включает соединения переходного металла 4-6 группы по классификации ИЮПАК (Международный союз фундаментальной и прикладной химии), такого как титан, соединение металла 2 группы, такого как магний, и внутренний донор, представляющий собой не фталевое соединение, более предпочтительно эфир не фталевой кислоты, еще более предпочтительно, представляющий собой диэфир не фталевых дикарбоновых кислот, как описано более подробно ниже. Таким образом, катализатор абсолютно не содержит нежелательных фталевых соединений. Кроме того, твердый катализатор не содержит какого-либо материала внешнего носителя, такого как диоксид кремния или MgCl2, но катализатор является самонесущим.
Такой катализатор Циглера-Натта может быть также определен способом его получения.
Соответственно, катализатор Циглера-Натта получают способом, включающим следующие стадии:
а)
a1) предоставление раствора по меньшей мере алкоксильного соединения металла 2 группы (Ах), являющегося продуктом реакции соединения металла 2 группы и одноатомного спирта (А), включающего помимо гидроксильной группы по меньшей мере одну группу простого эфира, возможно в реакционной среде органической жидкости; или
а2) раствора по меньшей мере алкоксильного соединения металла 2 группы (Ах'), являющегося продуктом реакции соединения металла группы 2 и спиртовой смеси одноатомного спирта (А) и одноатомного спирта (В) формулы ROH, возможно в реакционной среде органической жидкости, или
а3) предоставление раствора смеси алкоксильного соединения металла 2 группы (Ах) и алкоксильного соединения металла 2 группы (Вх), являющегося продуктом реакции соединения металла 2 группы и одноатомного спирта (В), возможно в реакционной среде органической жидкости, или
а4) предоставление раствора алкоксида металла 2 группы формулы M(OR1)n(OR2)mX2-n-m или смеси алкоксидов металлов 2 группы M(OR1)n'Х2-n' и M(OR2)m'X2-m', где М-металл 2 группы, Х-галоген, R1 и R2 - различные алкильные группы, содержащие от двух до 16 атомов углерода, и 0 < n < 2, 0 < m < 2 и n+m+(2-n-m)=2, при условии, что оба n и m≠0, 0 < n' < 2 и 0 < m' < 2, и
б) добавление указанного раствора со стадии (а) по меньшей мере к одному соединению переходного металла 4-6 группы, и
в) получение частиц твердого каталитического компонента,
и добавление не фталевого внутреннего донора на любой стадии, предшествующей стадии (с).
Предпочтительно внутренний донор или его предшественник добавляют в раствор на стадии (а).
В соответствии с методикой, указанной выше, катализатор Циглера-Натта может быть получен посредством способа осаждения или способа затвердевания в эмульсии (двухфазная систем жидкость/жидкость), в зависимости от физических условий, особенно от температуры, используемой на стадиях (б) и (в).
В обоих способах (осаждение или затвердевание в эмульсии) химия катализатора является одинаковой.
В способе осаждения выполняют объединение раствора стадии (а) по меньшей мере с одним соединением переходного металла на стадии (б) и всю реакционную смесь выдерживают по меньшей мере при температуре 50°С, более предпочтительно от 55°С до 110°С, более предпочтительно от 70°С до 100°С, чтобы обеспечить полное осаждение каталитического компонента в виде твердых частиц (стадия (с)).
В способе затвердевания в эмульсии на стадии (б) раствор со стадии (а) обычно добавляют по меньшей мере к одному соединению переходного металла при низкой температуре, такой как от -10°С до менее 50°С, предпочтительно от -5°С до 30°С. В ходе перемешивания температуру эмульсии обычно поддерживают на уровне от 10 до менее 40°С, предпочтительно от 5°С до 30°С. Капли дисперсной фазы эмульсии образуют композицию активного катализатора. Отверждение (стадия (с)) капель осуществляют подходящим образом посредством нагрева эмульсии до температуры от 70°С до 150°С, предпочтительно от 80°С до 110°С.
В настоящем изобретении предпочтительно используют катализатор, полученный с помощью способа затвердевания в эмульсии.
В предпочтительном воплощении на стадии (а) используют раствор (а2) или (а3), т.е. раствор (Ах') или раствор смеси (Ах) и (Вх).
Предпочтительно металлом 2 группы является магний.
Алкоксильные соединения магния (Ах), (Ах') и (Вх) можно получать in situ на первой стадии способа приготовления катализатора, стадии (а), путем взаимодействия соединения магния со спиртом (спиртами), как описано выше, или указанные алкоксильные соединения магния могут представлять собой отдельно полученные алкоксильные соединения магния, или они даже могут быть доступны в виде готовых алкоксильных соединений магния, и их используют как таковые в способе приготовления катализатора по изобретению.
Иллюстративными примерами спиртов (А) являются моноэфиры двухатомных спиртов (моноэфиры гликоля). Предпочтительными спиртами (А) являются С2-С4 гликолевые моноэфиры, в которых эфирные группы включают от 2 до 18 атомов углерода, предпочтительно от 4 до 12 атомов углерода. Предпочтительными примерами являются 2-(2-этилгексилокси)этанол, 2-бутилоксиэтанол, 2-гексилоксиэтанол и монобутиловый эфир 1,3-пропиленгликоля, 3-бутокси-2-пропанол, причем особенно предпочтительными являются 2-(2-этилгексилокси)этанол и монобутиловый эфир 1,3-пропиленгликоля, 3-бутокси-2-пропанол.
Иллюстративные одноатомные спирты (В) имеют формулу ROH, причем R представляет собой алкильный остаток C6-С10 с линейной или разветвленной цепью. Наиболее предпочтительным одноатомным спиртом является 2-этил-1-гексанол или октанол.
Предпочтительно используют смесь алкоксильных соединений Mg (Ах) и (Вх) или смесь спиртов (А) и (В), соответственно, в мольном соотношении Вх : Ах или В : А от 8:1 до 2:1, более предпочтительно от 5:1 до 3:1.
Алкоксильное соединение магния может быть продуктом реакции со спиртом (спиртвми), как определено выше, и соединением магния, выбранным из диалкилмагния, алкоксидов алкилмагния, диалкоксидов магния, галогенидов алкоксимагния и галогенидов алкилмагния. Алкильные группы могут быть одинаковыми или различными С1-С20 алкилами, предпочтительно С2-С10 алкилами. Типичными алкил-алкоксильными магниевыми соединениями, если их используют, являются бутоксид этилмагния, пентоксид бутилмагния, бутоксид октилмагния и октоксид октилмагния. Предпочтительно используют диалкилмагний. Наиболее предпочтительными соединениями диалкилмагния являются бутилоктилмагний или бутилэтилмагний.
Также возможно, что соединение магния может вступать в реакцию, помимо спирта (А) и спирта (В), также и с многоатомным спиртом (С) формулы R''(OH)m с получением указанных алкоксильных соединений магния. Предпочтительными многоатомными спиртами, если их используют, являются спирты, в которых R" представляет собой линейный, циклический или разветвленный углеводородный остаток С2-С10, a m является целым числом от 2 до 6.
Таким образом, алкоксильные соединения магния стадии (а) выбирают из группы, состоящей из диалкоксидов магния, диарилоксимагния, алкилоксигалогенидов магния, арилоксигалогенидов магния, алкоксидов алкилмагния, алкоксидов арилмагния и арилоксидов алкилмагния. Кроме того, можно использовать смесь дигалогенида магния и диалкоксида магния.
Растворители, используемые для приготовления катализатора по настоящему изобретению, могут быть выбраны из ароматических и алифатических линейных, разветвленных и циклических углеводородов, содержащих от 5 до 20 атомов углерода, более предпочтительно от 5 до 12 атомов углерода, или их смесей. Подходящие растворители включают бензол, толуол, кумол, ксилол, пентан, гексан, гептан, октан и нонан. Особенно предпочтительными являются гексаны и пентаны.
Соединение Mg обычно предоставляют в виде 10-50% раствора в растворителе, указанном выше. Обычные имеющиеся в продаже соединения Mg, особенно растворы диалкилмагния, представляют собой 20-40 масс. % растворы в толуоле или гептанах.
Реакцию для получения алкоксильного соединения магния можно осуществлять при температуре от 40°С до 70°С. Наиболее подходящую температуру выбирают в зависимости от используемого соединения Mg и спирта (спиртов).
Предпочтительно соединение переходного металла 4-6 группы представляет собой соединение титана, наиболее предпочтительно галогенид титана, такой как ИСЦ.
Не фталевый внутренний донор, который можно использовать при получении катализатора, предпочтительно выбирают из (ди)эфиров не фталевых карбоновых (ди)кислот, простых 1,3-диэфиров, их производных и их смесей. Особенно предпочтительными донорами являются диэфиры мононенасыщенных дикарбоновых кислот, в частности сложные эфиры, принадлежащие к группе, включающей малонаты, малеаты, сукцинаты, цитраконаты, глутараты, циклогексен-1,2-дикарбоксилаты и бензоаты, а также любые их производные и/или смеси. Предпочтительными примерами являются, например, замещенные малеаты и цитраконаты, наиболее предпочтительными являются цитраконаты.
В эмульсионном способе двухфазная система жидкость-жидкость может быть образована простым перемешиванием и возможно добавлением (дополнительного) растворителя (растворителей) и добавок, таких как агент минимизации турбулентности (АМТ) и/или эмульгирующие агенты и/или стабилизаторы эмульсии, такие как поверхностно-активные вещества, которые используют известным в данной области способом для облегчения образования и/или стабилизации эмульсии. Предпочтительно поверхностно-активные вещества представляют собой акриловые или метакриловые полимеры. Особенно предпочтительными являются неразветвленные С12-С20 (мет)акрилаты, такие как поли(гексадецил)метакрилат и поли(октадецил)метакрилат и их смеси. Агент минимизации турбулентности (АМТ), если его используют, выбирают из альфа-олефиновых полимеров альфа-олефиновых мономеров, содержащих от 6 до 20 атомов углерода, таких как полиоктен, полинонен, полидецен, полиундецен или полидодецен или их смеси. Наиболее предпочтительным является полидецен.
Продукт из твердых частиц, полученный осаждением или способом затвердевания в эмульсии можно промывать по меньшей мере один раз, предпочтительно, по меньшей мере дважды, наиболее предпочтительно по меньшей мере три раза ароматическими и/или алифатическими углеводородами, предпочтительно толуолом, гептаном или пентаном. Катализатор можно дополнительно высушивать, например, путем выпаривания или продувки азотом, или он может быть суспендирован с получением маслянистой жидкости без какой-либо сушки.
Полученный в итоге катализатор Циглера-Натта предпочтительно находится в форме частиц, обычно имеющих средний размер от 5 до 200 мкм, предпочтительно от 10 до 100 мкм. Частицы являются плотными и имеют низкую пористость и площадь поверхности менее 20 г/м2, более предпочтительно менее 10 г/м2. Обычно количество Ti составляет от 1 до 6 масс. %, Mg - от 10 до 20 масс. % и донора - от 10 до 40 масс. % от каталитической композиции.
Подробное описание приготовления катализатора приведено в WO 2012/007430, ЕР 2610271, ЕР 261027 и ЕР 2610272.
Катализатор Циглера-Натта возможно модифицируют с помощью так называемой BNT-technology технологии в ходе стадии предварительной полимеризации, чтобы ввести полимерный нуклеирующий агент.
Такой полимерный нуклеирующий агент предпочтительно представляет собой виниловый полимер, такой как виниловый полимер, полученный из мономеров формулы
СН2 = CH-CHR3R4,
где R3 и R4, совместно с атомом углерода, к которому они присоединены, образуют возможно замещенное, насыщенное или ненасыщенное, или ароматическое кольцо или систему конденсированных колец, где кольцо или функциональная группа с конденсированными кольцами содержит от 4 до 20 атомов углерода, предпочтительно 5-12-членное насыщенное или ненасыщенное, или ароматическое кольцо или систему конденсированных колец, или независимо представляют собой линейный или разветвленный С4-С30 алкан, С4-С20 циклоалкан или С4-С20 ароматическое кольцо. Предпочтительно R3 и R4, совместно с атомом С, к которому они присоединены, образуют пяти или шестичленное насыщенное или ненасыщенное, или ароматическое кольцо, или независимо представляют собой низшую алкильную группу, включающую от 1 до 4 атомов углерода. Предпочтительными виниловыми соединениями для получения полимерного нуклеирующего агента, используемого в соответствии с настоящим изобретением, являются, в частности, винилциклоалканы, в частности, винилциклогексан (ВЦГ), винилциклопентан и винил-2-метилциклогексан, 3 - метил-1-бутен, 3-этил-1-гексен, 3-метил-1-пентен, 4-метил-1-пентен или их смеси. ВЦГ является особенно предпочтительным мономером.
Массовое отношение винилового соединения к катализатору полимеризации на стадии модификации катализатора полимеризации предпочтительно составляет 0,3 или более, вплоть до 40,0, например, от 0,4 до 20,0 или более предпочтительно от 0,5 до 15,0, например, от 0,5 до 2,0.
Полимеризацию винилового соединения, например, ВЦГ, можно осуществлять в любой инертной текучей среде, которая на растворяет образующийся полимер (например, полиВЦГ). Важно убедиться, что вязкость конечной смеси катализатора, полимеризованного винилового соединения и инертной текучей среды достаточно высока, чтобы предотвратить осаждение частиц катализатора во время хранения и транспортировки.
Регулирование вязкости смеси можно выполнять либо до, либо после полимеризации винилового соединения. Например, можно проводить полимеризацию в масле с низкой вязкостью, а после полимеризации винилового соединения вязкость можно регулировать добавлением высоковязкого вещества. Таким высоковязким веществом может быть «воск», например, масло или смесь масла с твердым или высоковязким веществом (масло-жир). Вязкость такого вязкого вещества обычно составляет от 1000 до 15000 сП при комнатной температуре. Преимущество использования воска заключается в том, что улучшают хранение и подачу катализатора в способ. Поскольку нет необходимости в промывке, сушке, просеивании и перемещении, активность катализатора сохраняется.
Массовое соотношение между маслом и твердым или высоковязким полимером предпочтительно составляет менее 5:1.
Помимо вязких веществ, также можно использовать жидкие углеводороды, такие как изобутан, пропан, пентан и гексан, в качестве среды на стадии модификации.
Полипропилены, полученные в присутствии катализатора, модифицированного полимеризованными виниловыми соединениями, по существу не содержат свободных (непрореагировавших) виниловых соединений. Это означает, что виниловые соединения должны полностью вступать в реакцию на стадии модификации катализатора. С этой целью массовое отношение (добавленного) винилового соединения к катализатору, должно составлять от 0,05 до 10,0, предпочтительно менее 3,0, более предпочтительно приблизительно от 0,1 до 2,0 и, в частности, приблизительно от 0,1 до 1,5. Следует отметить, что при избытке использования виниловых соединений не достигают никаких преимуществ.
Кроме того, время реакции модификации катализатора посредством полимеризации виниловых соединений, должно быть достаточным для обеспечения полной реакции винилового мономера, т.е. полимеризацию продолжают до тех пор, пока количество непрореагировавших виниловых соединений в реакционной смеси (включая среду полимеризации и реагенты) не составит менее 0,5 масс. %, в частности, менее 2000 массовых частей на млн. (определенное с помощью анализа). Таким образом, когда предварительно полимеризованный катализатор содержит максимально приблизительно 0,1 масс. % винилового соединения, конечное содержание винилового соединения в полипропилене будет ниже предела определения с использованием способа ГХ-МС (газовой хроматографии-массовой спектрометрии) (<0,01 массовых частей на млн.). Обычно, при работе в промышленном масштабе, требуется время полимеризации по меньшей мере 30 мин, предпочтительно время полимеризации составляет 1 час и, в частности, по меньшей мере 5 часов. Можно даже использовать время полимеризации от 6 до 50 час. Модификацию можно осуществлять при температурах от 10°С до 60°С, предпочтительно от 15°С до 55°С.
Основные условия модификации катализатора также раскрыты в WO 00/6831, включенном в данный документ посредством ссылки, в отношении модификации катализатора полимеризации.
Предпочтительные воплощения, как описано ранее в настоящей заявке в отношении винилового соединения, применимы также в отношении катализатора полимеризации по настоящему изобретению и предпочтительной полипропиленовой композиции в соответствии с настоящим изобретением.
Подходящая среда стадии модификации включает, помимо масел, также алифатические инертные органические растворители с низкой вязкостью, такие как пентан и гептан. Кроме того, в ходе модификации можно использовать небольшое количество водорода.
Предпочтительно используют катализатор Циглера-Натта в сочетании с алкилалюминиевым сокатализатором и возможно внешними донорами.
В качестве дополнительного компонента в настоящем способе полимеризации присутствует внешний донор. Соответствующие внешние доноры включают определенные силаны, простые эфиры, сложные эфиры, амины, кетоны, гетероциклические соединения и их смеси. Особенно предпочтительно использовать силан. Наиболее предпочтительно силаны основной формулы
RapRbqSi(ORc)(4-p-q),
где Ra, Rb и Rc обозначают углеводородный радикал, в частности, алкильную или циклоалкильную группу, а р и q представляют собой числа от 0 до 3, причем их сумма р + q равна или меньше 3. Ra, Rb и Rc могут быть выбраны независимо друг от друга и могут быть одинаковыми или различными. Конкретными примерами таких силанов являются (трет-бутил)2Si(ОСН3)2, (циклогексил)(метил)Si(ОСН3)2, (фенил)2Si(ОСН3)2 и (циклопентил)2Si(ОСН3)2, или силаны основой формулы
Si(OCH2CH3)3(NR5R6),
где R5 и R6 могут быть одинаковыми или различными и представляют собой углеводородную группу, содержащую от 1 до 12 атомов углерода.
R5 и R6 независимо выбирают из группы, состоящей из линейной алифатической углеводородной группы, содержащей от 1 до 12 атомов углерода, разветвленной алифатической углеводородной группы, содержащей от 1 до 12 атомов углерода, и циклической алифатической углеводородной группы, содержащей от 1 до 12 атомов углерода. В частности, предпочтительно R5 и R6 независимо выбирают из группы, состоящей из метила, этила, н-пропила, н-бутила, октила, деканила, изопропила, изобутила, изопентила, трет-бутила, трет-амила, неопентила, циклопентила, циклогексила, метилциклопентила и циклогептила.
Более предпочтительно оба R5 и R6 являются одинаковыми, еще более предпочтительно оба R5 и R4 представляют собой этильную группу.
Особенно предпочтительными внешними донорами являются дициклопентилдиметоксисилан (D-донор) или циклогексилметилдиметоксисилан (С-донор).
Помимо катализатора Циглера-Натта и возможно внешнего донора, может быть использован сокатализатор. Сокатализатор предпочтительно представляет собой соединение 13 группы Периодической таблицы по ИЮПАК, например алюминийорганическое соединение, такое как алкилалюминий, галогенид алюминия или галогенид алкилалюминия. Соответственно, в одном конкретном варианте осуществления изобретения сокатализатором является триалкилалюминий, такой как триэтилалюминий (ТЭАЛ), хлорид диалкилалюминия или дихлорид алкилалюминия, или их смеси. В одном конкретном варианте осуществления изобретения сокатализатором является триэтилалюминий (ТЭАЛ).
Предпочтительно отношение сокатализатора (Со) к внешнему донору (ВншД) [Со/ВншД] и/или отношение сокатализатора (Со) к переходному металлу (ПМ) [Со/ПМ] следует выбирать тщательно.
Соответственно,
(а) молярное отношение сокатализатора (Со) к внешнему донору (ВншД) [Со/ВншД] должно составлять от 5 до 45, предпочтительно от 5 до 35, более предпочтительно от 5 до 25, и возможно
(б) молярное отношение сокатализатора (Со) к соединению титана (СТ) [Со/СТ] должно составлять от 580 до 500, предпочтительно от 100 до 350, еще более предпочтительно от 120 до 300.
Таким образом, предпочтительно статистический сополимер пропилена и этилена, используемый в соответствии с данным изобретением, получают в присутствии
(а) катализатора Циглера-Натта, включающего внутренний донор,
(б) возможно сокатализатора (Со) и
(в) возможно внешнего донора (ВншД).
Статистический сополимер пропилена и этилена, как определено в настоящем изобретении, может содержать вплоть до 5,0 масс. % добавок, таких как α-нуклеирующие агенты и антиоксиданты, а также понижающие трение добавки и агенты, препятствующие слипанию. Предпочтительно содержание добавок (без α-нуклеирующих агентов) составляет менее 3,0 масс. %, например, менее 1,0 масс. %.
Компонент (Б): статистический сополимер пропилена и гексена
Статистический сополимер пропилена и гексена имеет содержание гексена от 0,1 до 12,0 масс. %, предпочтительно от 0,5 до 11,0 масс. %, более предпочтительно от 1,0 до 10,0 масс. %, еще более предпочтительно от 1,5 до 9,0 масс. %, еще более предпочтительно от 2,0 до 8,5 масс. %, наиболее предпочтительно от 2,5 до 8,0 масс. %, например, от 3,0 до 7,0 масс. %.
ПТР2 (230°С, 2,16 кг, ISO1133) статистического сополимера пропилена и гексена составляет от 0,5 до 60,0 г/10 мин, предпочтительно от 5,0 до 55,0 г/10 мин, более предпочтительно от 10,0 до 50,0 г/10 мин, еще более предпочтительно от 20,0 до 50,0 г/10 мин и еще более предпочтительно от 25,0 до 45,0 г/10 мин.
Температура плавления Тпл статистического сополимера пропилена и гексена составляет от 120°С до 140°С, предпочтительно от 120°С до 135°С.
Предпочтительно статистический сополимер пропилена и гексена имеет молекулярно-массовое распределение (Mw/Mn) по меньшей мере 2,0, более предпочтительно от 2,0 до 4,5, еще более предпочтительно от 2,0 до 3,5.
Дополнительно или альтернативно молекулярно-массовому распределению (Mw/Mn), определенному в предыдущем абзаце, статистический сополимер пропилена и гексена предпочтительно имеет среднемассовую молекулярную массу Mw от 120 до 500 кг/моль, более предпочтительно от 130 до 400 кг/моль, например, от 135 до 300 кг/моль.
Статистический сополимер пропилена и гексена, как определено выше, может быть получен, предпочтительно получают в присутствии металлоценового катализатора.
Металлоценовый катализатор может представлять собой нанесенный катализатор, с использованием традиционных носителей, или может не содержать внешней подложки. Под тем, что катализатор не содержит внешней подложки подразумевают, что катализатор не содержит внешнего носителя, такого как неорганический носитель, например, диоксид кремния или оксид алюминия, или органический полимерный носитель.
Предпочтительно используют металлоценовые катализатора, которые не содержат внешнего носителя.
Предпочтительно металлоценовый катализатор включает: (i) комплекс формулы (I):
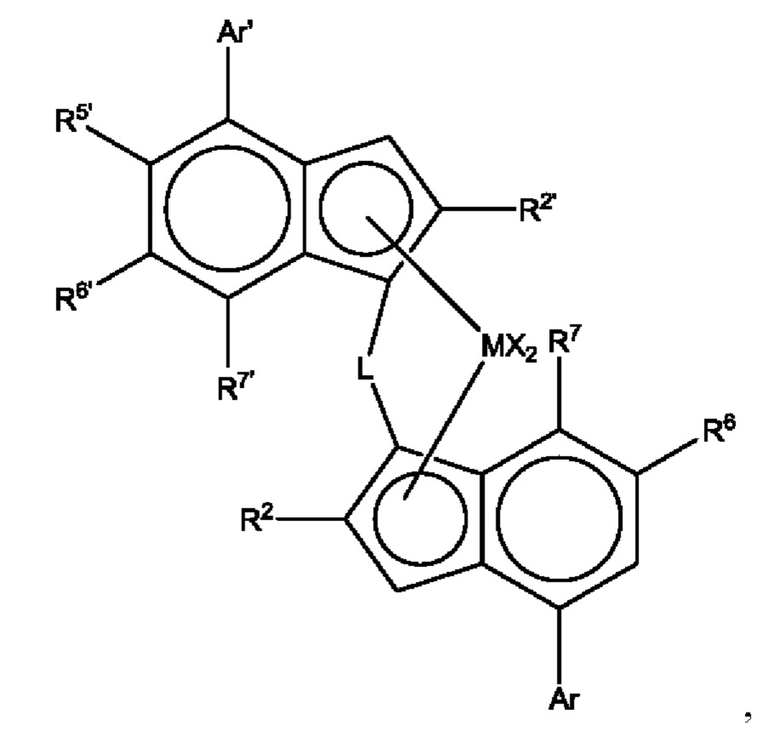
где М представляет собой цирконий или гафний;
каждый X представляет собой сигма-донорный лиганд;
L представляет собой двухвалентный мостик, выбранный из -R'2C-, -R'2C-CR'2, -R'2Si-, -R'2Si-SiR'2-, -R'2Ge-, где каждый R' независимо является атомом водорода, C1-C20-гидрокарбилом, три(C1-C20-алкил)силилом, C6-C20-арилом, C7-C20-арилалкилом или C7-C20-алкиларилом;
каждый из R2 и R2' независимо представляет собой C1-C20 гидрокарбильный радикал, возможно, содержащий один или более гетероатомов из групп 14-16;
R5' представляет собой C1-20 гидрокарбильную группу, содержащую один или более гетероатомов из групп 14-16, возможно, замещенных одним или более атомов галогена;
каждый из R6 и R6' независимо представляет собой водород или C1-20 гидрокарбильную группу, возможно содержащую один или более гетероатомов из групп 14-16;
каждый из R7 и R7' независимо представляет собой водород или C1-20 гидрокарбильную группу, возможно содержащую один или более гетероатомов из групп 14-16;
Ar независимо представляет собой арильную или гетероарильную группу, содержащую вплоть до 20 атомов углерода, возможно, замещенных одной или более группами R1;
Ar' независимо представляет собой арильную или гетероарильную группу, содержащую вплоть до 20 атомов углерода, возможно, замещенных одной или более группами R1;
каждый R1 представляет собой C1-20 гидрокарбильную группу, или две R1 группы на соседних атомах углерода вместе могут образовывать 5 или 6-членное конденсированное неароматическое кольцо с группой Ar, причем указанное кольцо возможно замещено одной или более группами R4;
каждый R4 представляет собой C1-20 гидрокарбильную группу;
и
(ii) сокатализатор, включающий соединение металла группы 13, например соединение Al или бора.
Предпочтительно катализатор, используемый для получения статистического сополимера пропилена и гексена, используемого в изобретении, находится в форме твердых частиц. Как указано выше, он может быть нанесен на традиционный носитель, известный специалисту в данной области техники. Предпочтительно используемый катализатор не содержит внешнего носителя.
В идеальном случае, катализатор получают способом, в котором
(а) образуют систему эмульсии жидкость/жидкость, причем указанная система эмульсии жидкость/жидкость включает раствор компонентов (i) и (ii) катализатора, диспергированных в растворителе с формированием дисперсных капель, и
(б) образуют твердые частицы посредством отверждения указанных дисперсных капель.
Термин «C1-20 гидрокарбильная группа» включает C1-20 алкил, C2-20 алкенил, C2-20 алкинил, C3-20 циклоалкил, C3-20 циклоалкенил, C6-20 арильные группы, C7-20 алкиларильные группы или C7-20 арилалкильные группы или, конечно, смеси этих групп, такие как циклоалкил, замещенный алкилом.
Если не указано иное, предпочтительными C1-20 гидрокарбильными группами являются C1-20 алкил, C4-20 циклоалкил, C5-20 циклоалкил-алкильные группы, C7-20 алкиларильные группы, C7-20 арилалкильные группы или C6-20 арильные группы, в особенности C1-20 алкильные группы, C6-10 арильные группы или C7-12 арилалкильные группы, например, C1-8 алкильные группы. В особенности предпочтительными гидрокарбильными группами являются метил, этил, пропил, изопропил, трет-бутил, изобутил, C5-6 циклоалкил, циклогексилметил, фенил или бензил.
Термин «гало-группа» включает группы фтора, хлора, брома и йода, в особенности группы хлора, когда речь идет об определении комплекса.
Степень окисления иона металла определяется в первую очередь природой рассматриваемого иона металла и стабильностью отдельных состояний окисления каждого иона металла.
Следует отметить, что в комплексах по изобретению ион металла М координируется с лигандами X таким образом, чтобы удовлетворять валентности иона металла и заполнить его доступные координационные центры. Природа этих σ-лигандов может сильно варьироваться.
Такие катализаторы описаны, например, в WO 2013/007650.
Предпочтительные комплексы для получения статистического сополимера пропилена и гексена имеют формулу (II') или (II)
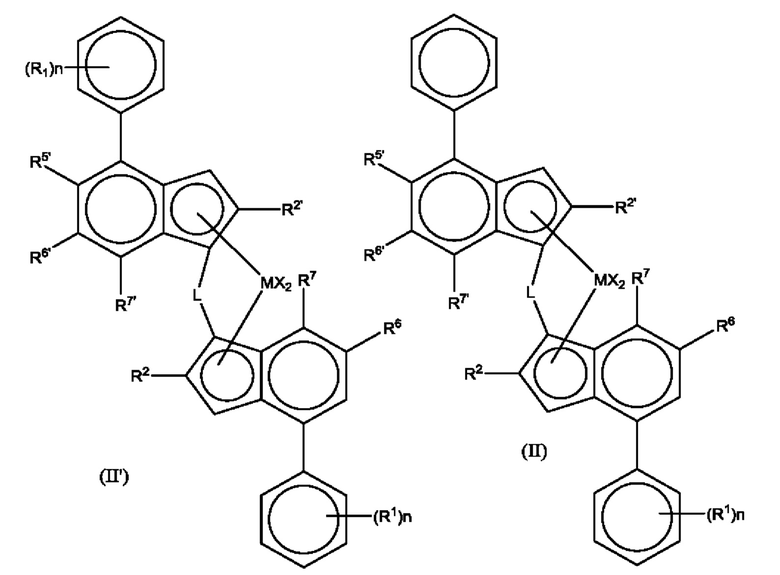
где М представляет собой цирконий или гафний;
каждый X представляет собой сигма-донорный лиганд, предпочтительно каждый X независимо является атомом водорода, атомом галогена, C1-6 алкоксильной группой, C1-6 алкильной, фенильной или бензильной группой;
L представляет собой двухвалентный мостик, выбранный из -R'2C-, -R'2C-CR'2, -R'2Si-, -R'2Si-SiR'2-, -R'2Ge-, где каждый R' независимо является атомом водорода, C1-20 алкилом, C3-10 циклоалкилом, три(C1-20 алкил)силилом, C6-20-арилом, C7-20 арилалкилом или C7-20 алкиларилом;
каждый из R2 или R2' представляет собой C1-10 алкильную группу;
R5' представляет собой C1-10 алкильную группу или Z'R3' группу;
R6 представляет собой водород или C1-10 алкильную группу;
R6' представляет собой C1-10 алкильную группу или C6-10 арильную группу;
R7 представляет собой водород, C1-6 алкильную группу или ZR3 группу;
R7' представляет собой водород или C1-10 алкильную группу;
Z и Z' независимо представляют собой О или S;
R3' представляет собой C1-10 алкильную группу или C6-10 арильную группу возможно замещенную одной или более гало-группами;
R3 представляет собой C1-10 алкильную группу;
каждый n независимо составляет от 0 до 4, например, 0, 1 или 2;
и каждый R1 независимо представляет собой C1-20 гидрокарбильную группу, например, C1-10 алкильную группу.
Другие предпочтительные комплексы для получения статистического сополимера пропилена и гексена имеют формулу (ПГ) или (III) или (ШЬ):
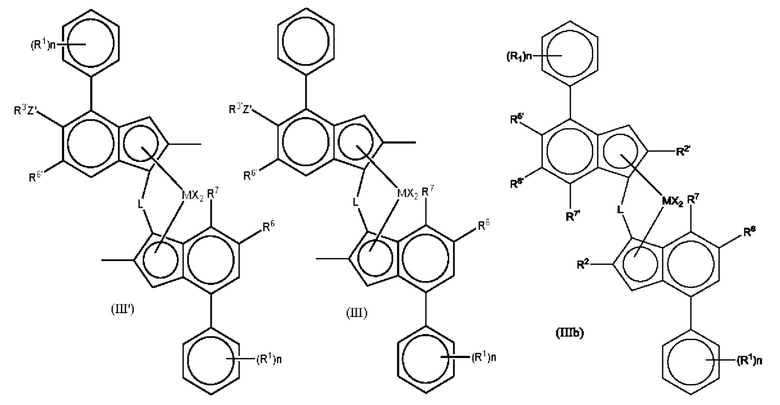
где М представляет собой цирконий или гафний;
каждый X представляет собой сигма-лиганд, предпочтительно каждый X независимо представляет собой атом водорода, атом галогена, C1-6 алкоксильную группу, C1-6 алкильную, фенильную или бензильную группу;
L представляет собой двухвалентный мостик, выбранный из -R'2C- или -R'2Si-, где каждый R' независимо представляет собой атом водорода, C1-20 алкил или C3-10 циклоалкил;
R5' представляет собой C1-6 алкильную группу;
R6 представляет собой водород или C1-10 алкильную группу;
R6' представляет собой C1-10 алкильную группу или C6-10 арильную группу;
R7 представляет собой водород, C1-6 алкил или OC1-6 алкил;
Z' представляет собой О или S;
R3' представляет собой C1-10 алкильную группу или C6-10 арильную группу, возможно замещенную одной или более гало-группами;
n независимо представляет собой от 0 до 4, например, 0, 1 или 2, и
каждый R1 независимо представляет собой C1-10 алкильную группу.
Другие предпочтительные комплексы для получения статистического сополимера пропилена и гексена имеют формулу (IV') или (IV) или (IVb):
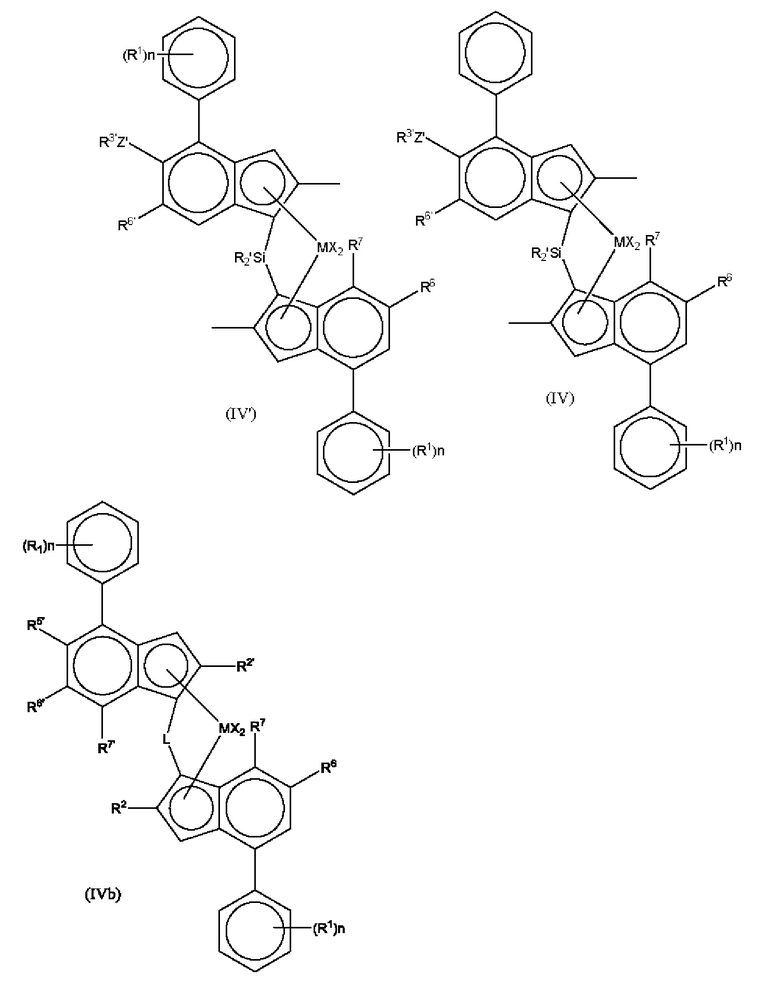
где М представляет собой цирконий или гафний;
каждый X представляет собой сигма-лиганд, предпочтительно каждый X независимо представляет собой атом водорода, атом галогена, C1-6 алкоксильную группу, C1-6 алкильную, фенильную или бензильную группу;
каждый R' независимо представляет собой атом водорода, C1-20 алкил или C3-7 циклоалкил;
R5' представляет собой C1-4 алкильную группу;
R6 представляет собой водород или C1-10 алкильную группу;
R6' представляет собой C1-10 алкильную группу или C6-10 арильную группу;
R7 представляет собой водород, C1-6 алкил или OC1-6 алкил;
Z' представляет собой О или S;
R3' представляет собой а C1-10 алкильную группу или C6-10 арильную группу, возможно замещенную одной или более гало-группами;
n независимо представляет собой 0, 1 или 2, и
каждый R1 независимо представляет собой C1-8 алкильную группу.
Наиболее предпочтительно, комплекс для получения статистического сополимера пропилена и гексена имеет формулы (V') или (V) или (Vb):
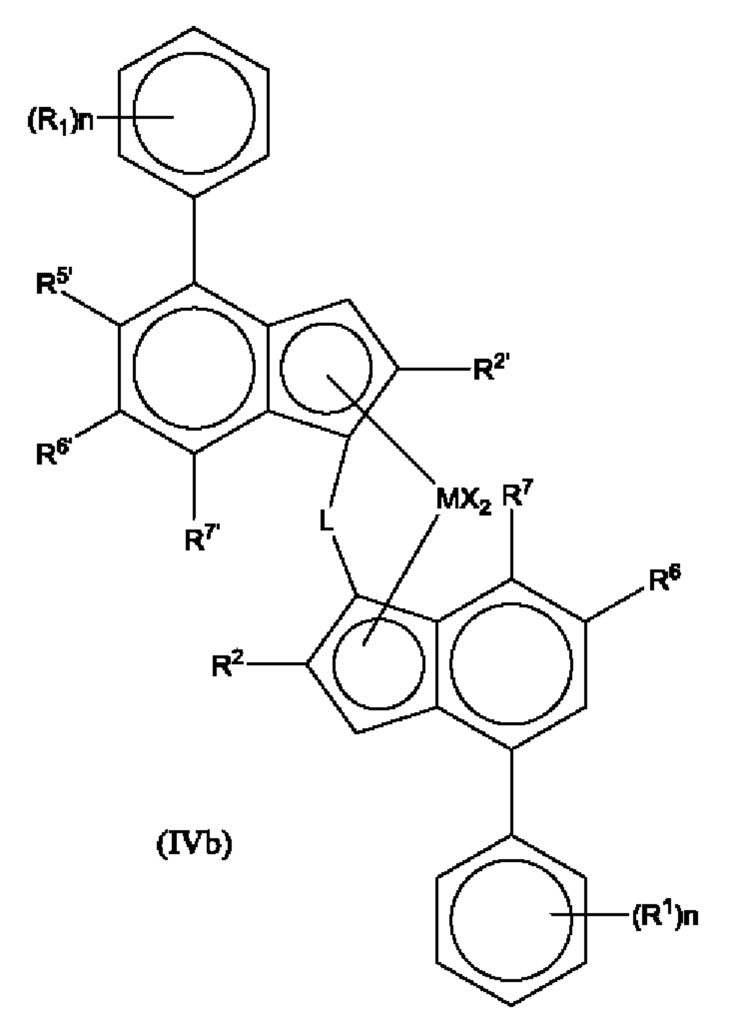
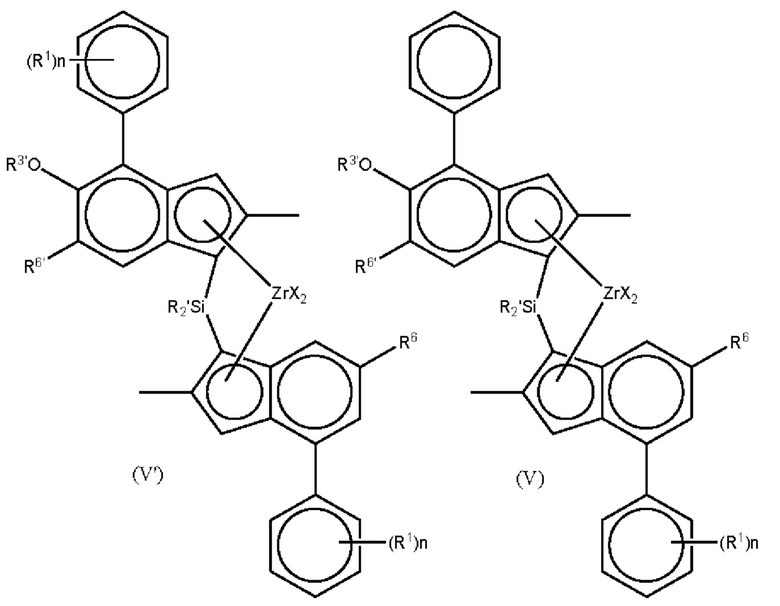
где каждый X представляет собой сигма-лиганд, предпочтительно каждый X независимо представляет собой атом водорода, атом галогена, C1-6 алкоксильную группу, C1-6 алкильную, фенильную или бензильную группу;
R' независимо представляет собой C1-6 алкил или C3-10 циклоалкил;
R1 независимо представляет собой C1-8 алкил;
R5' представляет собой алкильную группу
R6 представляет собой водород или C1-8 алкильную группу;
R6' представляет собой C1-8 алкильную группу или C6-10 арильную группу;
R3' представляет собой C1-6 алкильную группу или C6-10 арильную группу, возможно замещенную одной или более гало-группами, и
n независимо представляет собой 0, 1 или 2.
Синтез данных материалов описан, например, в WO2013/007650.
Лиганды, необходимые для образования комплексов и, следовательно, катализаторов по изобретению, могут быть синтезированы любым способом, и квалифицированный химик-органик сможет разработать различные синтетические протоколы для изготовления необходимых материалов для лигандов. Например, в WO 2007/116034 раскрыта необходимая химия. Протоколы синтеза также могут быть в общем найдены в WO 2002/02576, WO 2011/135004, WO 2012/084961, WO 2012/001052, WO 2011/076780 и WO 2015/158790. Раздел примеров обеспечивает специалисту в данной области техники достаточные инструкции.
Например, может быть использована следующая основная схема синтеза:
Подходящие реагенты для этого превращения приведены в разделе примеров.
Хотя эта схема относится к конкретным соединениям, общие принципы, представленные здесь, применимы ко всем металлоценам, описанным выше. Если лиганды асимметричны, обычная реакция с SiMe2Cl2 не может быть осуществлена для соединения мостиком двух лигандов, поскольку это приводит к получению симметричных продуктов. Вместо этого, лиганды должны быть присоединены к мостику поэтапно, с контролем стехиометрии реакции.
Сокатализатор
Для получения соединений, обладающих каталитической активностью, обычно необходимо использовать сокатализатор, как это хорошо известно в технике. Со катализаторы, включающие одно или более соединений металлов 13 группы, таких как алюминийорганические соединения или бораты, используемые для активации металлоценовых катализаторов, подходят для применения в данной изобретении. Таким образом, сокатализатор предпочтительно является алюмоксаном, таким как МАО. Также могут быть использованы сокатализаторы на основе боратов. Применение В(C6F5)3, C6H5N(CH3)2H:B(C6F5)4, (С6Н5)3С:В(C6F5)4 или Ni(CN)4[B(C6F5)3]42- является особенно предпочтительным. Походящие сокатализаторы описаны в WO 2013/007650.
Соответствующее количество сокатализатора должно быть хорошо известно специалисту в данной области техники.
Получение
Катализатор, используемый для получения гетерофазных сополимеров по изобретению, в идеальном случае обеспечивают в форме твердых частиц, но не нанесенных, т.е. внешний носитель не используют. Для того, чтобы обеспечить катализатор по изобретению в твердой форме, но без использования внешнего носителя, предпочтительно использовать систему эмульсии жидкость-жидкость. Способ включает формирование дисперсии компонентов катализатора (i) и (ii) в растворителе, и отверждение указанных дисперсных капель с формированием твердых частиц.
В частности, способ включает приготовление раствора одного или более компонентов катализатора; диспергирование указанного раствора в растворителе с получением эмульсии, в которой указанный один или более компонентов катализатора присутствуют в виде капель дисперсной фазы; иммобилизацию компонентов катализатора в дисперсных каплях, в отсутствие внешнего пористого носителя в виде частиц, с формированием твердых частиц, включающих указанный катализатор, и необязательно извлечение указанных частиц.
Данный способ позволяет получить активные частицы катализатора с улучшенной морфологией, например, с заданной сферической формой и размером частиц, и без использования какого-либо добавляемого внешнего пористого материала носителя, такого как неорганический оксид, например, диоксид кремния. Также могут быть получены предпочтительные свойства поверхности. Исчерпывающие подробности данного способа можно также найти в WO 2013/007650.
Предварительная полимеризация катализатора Применение гетерогенных, ненанесенных катализаторов (т.е. «самонесущих» катализаторов) может иметь недостаток, заключающийся в тенденции к растворению до некоторой степени в среде полимеризации, т.е. некоторые активные компоненты катализатора могут выщелачиваться из частиц катализатора в ходе суспензионной полимеризации, в результате чего может быть утрачена исходная хорошая морфология катализатора. Такие выщелоченные компоненты катализатора очень активны, и могут вызывать проблемы во время полимеризации. Поэтому количество выщелачиваемых компонентов должно быть сведено к минимуму, т.е. все компоненты катализатора необходимо поддерживать в гетерогенной форме.
Кроме того, самонесущие катализаторы создают, благодаря большому количеству каталитически активных веществ в каталитической системе, высокие температуры в начале полимеризации, которые могут вызвать плавление получаемого материала. Оба эффекта, т.е. частичное растворение каталитической системы и выделение тепла, могут привести к загрязнению, расслоению и ухудшению морфологии полимерного материала.
Чтобы минимизировать возможные проблемы, связанные с высокой активностью или выщелачиванием, предпочтительно «предварительно полимеризировать» катализатор перед использованием его в процессе полимеризации. Следует отметить, что предварительная полимеризация в этом отношении является частью способа приготовления катализатора, причем стадию осуществляют после получения твердого катализатора. Эта стадия предварительной полимеризации катализатора не является частью компоновки процесса фактической полимеризации, которая также может включать в себя стадию традиционного способа предварительной полимеризации. После стадии предварительной полимеризации катализатора получают твердый катализатор и используют его при полимеризации.
«Предварительную полимеризацию» катализатора осуществляют после стадии отверждения описанного выше способа с использованием эмульсии жидкость-жидкость. Предварительную полимеризацию можно осуществлять известными способами, описанными в технике, такими как описаны в WO 2010/052263, WO 2010/052260 или WO 2010/052264. В данном документе описаны предпочтительные воплощения данного аспекта изобретения.
В качестве мономеров на стадии предварительной полимеризации катализатора предпочтительно используют альфа-олефины. Предпочтительно используют C2-C10олефины, такие как этилен, пропилен, 1-бутен, 1-пентен, 1-гексен, 4-метил-1-пентен, 1-гептен, 1-октен, 1-нонен, 1-децен, стирол и винилциклогексен. Наиболее предпочтительными альфа-олефинами являются этилен и пропилен.
Предварительную полимеризацию катализатора осуществляют в газовой фазе или в инертном разбавителе, обычно в масле или фторированном углеводороде, предпочтительно во фторированных углеводородах или смеси фторированных углеводородов. Предпочтительно используют перфторированные углеводороды. Температура плавления таких (пер)фторированных углеводородов обычно составляет от 0°С до 140°С, предпочтительно от 30°С до 120°С, например, от 50°С до 110°С.
Если предварительную полимеризацию катализатора осуществляют во фторированных углеводородах, то температура на стадии предварительной полимеризации составляет менее 70°С, например от -30°С до 70°С, предпочтительно от 0°С до 65°С и более предпочтительно от 20°С до 55°С.
Давление внутри емкости для предварительной полимеризации предпочтительно выше, чем атмосферное давление, чтобы свести к минимуму возможное попадание воздуха и/или влаги в сосуд для катализатора. Предпочтительно давление составляет от 0,1 до 1,5 МПа (от 1 до 15 бар), предпочтительно от 0,2 до 1 МПа (от 2 до 10 бар). Емкость для предварительной полимеризации выдерживают в инертной атмосфере, такой как азот, аргон или аналогичной атмосфере.
Предварительную полимеризацию продолжают до тех пор, пока не будет достигнута степень предварительной полимеризации, определяемой как масса полимерной матрицы/массы твердого катализатора перед стадией предварительной полимеризации. Степень составляет менее 25, предпочтительно от 0,5 до 10,0, более предпочтительно от 1,0 до 8,0, наиболее предпочтительно от 2,0 до 6,0.
Применение стадии предварительной полимеризации катализатора дает преимущество, заключающееся в минимизации выщелачивания компонентов катализатора и следовательно, локального перегрева.
После предварительной полимеризации, катализатор может быть извлечен и отправлен на хранение.
Статистический сополимер пропилена и гексена может быть получен на одной стадии полимеризации, включающей один реактор (Р1) полимеризации, или посредством последовательного способа полимеризации, включающего по меньшей мере два реактора (Р1) и (Р2) полимеризации, причем в первом реакторе (Р1) полимеризации получают первую фракцию пропиленового сополимера (Р-ПП1), которую затем переносят во второй реактор (Р2) полимеризации. Во втором реакторе (Р2) полимеризации получают вторую фракцию пропиленового сополимера (Р-ПП2) в присутствии первой фракции пропиленового сополимера (R-ПП1).
Способы полимеризации, которые подходят для получения статистического сополимера пропилена и гексена, обычно включают одну или две стадии полимеризации, и каждая стадия может быть выполнена в растворе, в суспензии, в псевдоожиженном слое, в массе или в газовой фазе.
Термин «реактор полимеризации» определен, как указано выше для компонента (А).
Термин «способ последовательной полимеризации» определен, как указано выше, для компонента (А).
Первый, соответственно единственный, реактор (Р1) полимеризации предпочтительно представляет собой суспензионный реактор и может быть любым реактором непрерывного действия или реактором периодического действия с мешалкой, или петлевым реактором, работающим в массе или суспензии. «В массе» означает полимеризацию в реакционной среде, которая включает по меньшей мере 60 масс. % мономера. В соответствии с настоящим изобретением, суспензионный реактор предпочтительно является петлевым реактором (для эксплуатации в массе).
В случае, когда используют «способ последовательной полимеризации», второй реактор (Р2) полимеризации и возможно третий реактор (Р3) полимеризации представляют собой газофазные реактора (ГФР), т.е. первый газофазный ректор (ГФР1) и второй газофазный ректор (ГФР2). Газофазный ректор (ГФР) в соответствии с данным изобретением предпочтительно представляет собой реактор с псевдоожиженным слоем, реактор с быстрым псевдоожиженным слоем или реактор с неподвижным слоем, или любое их сочетание.
Предпочтительным многостадийным способом является способ «петля-газовая фаза», такой как разработанный Borealis (известный как технология BORSTAR®), описанный, например, в патентной литературе, такой как ЕР 0 887 379, WO 92/12182, WO 2004/000899, WO 2004/111095, WO 99/24478, WO 99/24479 или WO 00/68315.
Еще одним подходящим суспензионно-газофазным способом является способ Spheripol® Basell.
Статистический сополимер пропилена и гексена может быть мономодальным и мультимодальным, например, бимодальным, с точки зрения содержания сомономера и/или ПТР2.
Если статистический сополимер пропилена и гексена является мономодальным, предпочтительно его получают на одной стадии полимеризации в одном реакторе (Р1) полимеризации. В качестве альтернативы, мономодальный статистический сополимер пропилена и гексена может быть получен в ходе последовательного способа полимеризации с использованием одинаковых условий полимеризации во всех реакторах.
Если статистический сополимер пропилена и гексена является мультимодальным, его предпочтительно получают в ходе последовательного способа полимеризации с использованием различных условий полимеризации (количество сомономера, количество водорода и т.д.) в реакторах.
Предпочтительно статистический сополимер пропилена и гексена, используемый в соответствии с настоящим изобретением, является мономодальным.
Статистический сополимер пропилена и гексена, как определено в настоящем изобретении, может содержать вплоть до 5,0 масс. % добавок, таких как α- нуклеирующие агенты и антиоксиданты, а также понижающие трение добавки и агенты, препятствующие слипанию. Предпочтительно содержание добавок (без α-нуклеирующих агентов) составляет менее 3,0 масс. %, например, менее 1,0 масс. %.
Полипропиленовая композиция Полипропиленовая композиция в соответствии с настоящим изобретением может быть получена посредством смешивания (в расплаве) отдельных фракций, т.е. статистического сополимера (А) пропилена и этилена и статистического сополимера (Б) пропилена и гексена. В течение смешивания в расплаве могут быть дополнительно введены соответствующие добавки. Для смешивания может быть использовано традиционное компаундирующее или смесительное устройство, например, смеситель Бенбери, двухвальцовая мельница для резинового производства, месильная машина Бусс с двумя шнеками, вращающимися в одном направлении, одношнековый экструдер со специальными смесительными секциями или двухшнековый экструдер. Полимерная композиция, извлеченная из экструдера, обычно находится в форме гранул.
В смеси для полипропиленовой композиции в соответствии с настоящим изобретением компонент (А) присутствует в количестве от 50,0 до 99,0 масс. %, а компонент (Б) присутствует в количестве от 1,0 до 50,0 масс. %.
Предпочтительно компонент (А) присутствует в количестве от 52,0 до 95,0 масс. % и более предпочтительно в количестве от 54,0 до 90,0 масс. %.
Таким образом компонент (Б) предпочтительно присутствует в количестве от 5,0 до 48,0 масс. % и более предпочтительно в количестве от 10,0 до 46,0 масс. %.
Для приготовления композиции в является существенным то, что используют компонент (Б), который имеет температуру Тпл плавления меньше, чем температура Тпл плавления компонента (А).
Общий показатель текучести расплава, т.е. показатель текучести расплава ПТР2 (230°С), измеренный в соответствии со стандартом ISO 1133, пропиленовой композиции составляет от 0,5 до 60,0 г/10 мин, предпочтительно от 0,5 г/10 мин до 50,0 г/10 мин, более предпочтительно от 1,0 г/10 мин до 40,0 г/10 мин и еще более предпочтительно от 1,0 до 30,0 г/10 мин.
Следует отметить, что полипропиленовая композиция по изобретению имеет температуру плавления от 128°С до 150°С, предпочтительно 130°С до 148°С, и более предпочтительно от 132°С до 145°С.
Дополнительно следует отметить, что полипропиленовая композиция по изобретению имеет температуру кристаллизации от 85°С до 115°С, предпочтительно от 90°С до 110°С и более предпочтительно от 95°С до 105°С.
Более того, полипропиленовая композиция изобретения отличается низким уровнем общей миграции. Так, полипропиленовая композиция изобретения имеет уровень общей миграции, определяемый в соответствии со стандартом EN ISO 1186-14:2002 на полученных инжекционным формованием пластинах размером 60×60×1 мм3 менее 20,0 мг/дм2, предпочтительно менее 17,0 мг/дм2, более предпочтительно менее 14,0 мг/дм2 и еще более предпочтительно менее 12,0 мг/дм2.
Растворимая в холодном ксилоле (РХК) фракция настоящей полимерной композиции составляет от 2,0 до 20,0 масс. % (определено при температуре 25°С в соответствии со стандартом ISO 16152; первая редакция; 2005), предпочтительно от 3,0 до 20,0 масс. % и более предпочтительно от 4,0 до 10,0 масс. %.
Полипропиленовая композиция, как определено в настоящем изобретении, может содержать вплоть до 5,0 масс. % добавок, таких как α-нуклеирующие агенты и антиоксиданты, а также понижающие трение добавки и агенты, препятствующие слипанию. Предпочтительно содержание добавок (без α-нуклеирующих агентов) составляет менее 3,0 масс. %, например, менее 1,0 масс. %.
Кроме того, настоящее изобретение относится к способу получения полипропиленовой композиции, определенной выше, включающему следующие стадии:
(i) приготовление компонента (А) посредством полимеризации пропилена и этилена в присутствии катализатора Циглера-Натта;
(ii) приготовление компонента (Б) посредством полимеризации пропилена и гексена в присутствии металлоценового катализатора;
(iii) смешивание указанного статистического сополимера (А) пропилена и этилена со статистическим сополимером (Б) пропилена и гексена с получением смешанной массы (А) и (Б), и
(iv) экструдирование указанной смешанной массы с получением смеси (А) и (Б).
Области применения
Настоящее изобретение относится не только к рассматриваемой полипропиленовой композиции, а также к применению полипропиленовой композиции для получения изделий и к изделиям, включающим полипропиленовую композицию.
В общем, подходящими изделиями являются пленки для гибкой упаковки, такой как мешки или пакеты для упаковки пищевых продуктов и фармацевтической продукции или медицинских изделий.
В одном воплощении настоящее изобретение также относится к неориентированным пленками и слоям пленок многослойных пленочных конструкций, включающим полипропиленовую композицию по изобретению. Соответственно, настоящее изобретение также относится к неориентированным пленкам, таким как литые пленки или получаемые экструзией с раздувом пленки, например, пленки, получаемые экструзией с раздувом при воздушном охлаждении, включающим по меньшей мере 90 масс. %, предпочтительно включающим по меньшей мере 95 масс. %, еще более предпочтительно включающим по меньшей мере 99 масс. % полипропиленовой композиции по настоящему изобретению.
Было обнаружено, что такая полипропиленовая композиция в соответствии с настоящим изобретением позволяет обеспечить изготовленный из нее пленочный материал, который характеризуется сочетанием низкой температуры начала сваривания (ТНС), преимущественных оптических свойств, низкого количества растворимых в гексане веществ и термостойкости, достаточной для обеспечения возможности обработки стерилизацией, при которой оптические свойства сохраняются на высоком уровне до и после стерилизации.
Таким образом, пленки, соответственно изделия, в соответствии с настоящим изобретением, включающие вышеуказанную полипропиленовую композицию, подходят для проведения их стерилизации без отрицательного влияния на оптические свойства.
Следовательно, настоящее изобретение также относится к стерилизуемому или стерилизованному изделию, предпочтительно к стерилизуемой или стерилизованной пленке, например, к стерилизуемой или стерилизованной пленке. Более предпочтительно изобретение относится к контейнерам, т.е. пакетам, особенно к стерилизуемым паром или стерилизованным паром контейнерам, т.е. пакетам, предпочтительно включающим пленку, более предпочтительно состоящим из пленки, как она определена в настоящем документе. В частности контейнер представляет собой пакет. Кроме того, указанный контейнер, т.е. пакет, подвергают стерилизации паром в диапазоне температур от 120°С до 130°С.
Кроме того, изобретение также относится к многослойной пленочной конструкции, включающей неориентированную пленку, определенную выше, в качестве внешнего слоя.
Чтобы служить в качестве герметизирующего слоя в многослойной пленочной конструкции, такая неориентированная пленка, включающая полипропиленовую композицию по изобретению, предпочтительно должна иметь температуру начала сваривания (ТНС) от 90°С до 125°С, более предпочтительно от 95°С до менее 122°С, например, от 100°С до менее 121°С.
Кроме того, такая неориентированная пленка, включающая полипропиленовую композицию по изобретению, предпочтительно должна иметь мутность, определяемую на литой пленке толщиной 50 мкм, менее 80,0%, предпочтительно менее 70,0% и более предпочтительно менее 66,0%.
Прозрачность, определяемая на литой пленке толщиной 50 мкм, включающей полипропиленовую композицию по изобретению, предпочтительно должна составлять более 90,0%.
Пленки в соответствии с изобретением более предпочтительно имеет
величину мутности (определяемую на литой пленке толщиной 50 мкм) после стерилизации при температуре 121°С в течение 30 мин менее 80,0%, предпочтительно менее 70,0% и более предпочтительно менее 66,0%
и
прозрачность (определяемую на литой пленке толщиной 50 мкм) после стерилизации при температуре 121°С в течение 30 мин по меньшей мере 85,0%.
Многослойную пленочную конструкцию, включающую по меньшей мере один слой, включающий полипропиленовую композицию по изобретению, предпочтительно получают многослойной совместной экструзией с последующим наливом пленки или экструзией пленки с раздувом. В данном случае, по меньшей мере один из внешних слоев указанной многослойной пленочной конструкции, служащий в качестве припойного слоя (слоев), должен включать полипропиленовую композицию по изобретению, как указано выше. Многослойная пленочная конструкция по изобретению предпочтительно должна иметь толщину от 30 до 500 мкм, более предпочтительно от 50 до 400 мкм, например, от 60 до 300 мкм. Припойный слой (слои), включающий полипропиленовую композицию по изобретению, предпочтительно должен иметь толщину от 3 до 50 мкм, более предпочтительно от 5 до 30 мкм, например, от 8 до 25 мкм.
Пленки и/или многослойные пленочные конструкции в соответствии с настоящим изобретением предпочтительно следует использовать в системах гибкой упаковки, таких как мешки или пакеты, в основном, для упаковки пищевых продуктов и фармацевтической продукции или медицинских изделий.
В случае, когда пленку получают посредством технологии налива пенки, расплавленную полипропиленовую композицию экструдируют через щелевую экструзионную головку на охлаждающий валок для охлаждения полипропиленовой композиции с получением твердой пленки. Обычно полипропиленовую композицию сначала сжимают и приводят в жидкое состояние в экструдере, причем любые добавки могут быть уже добавлены к полипропиленовой композиции, или их можно вводить на этой стадии в составе маточной смеси. Затем расплав проталкивают через плоскую головку для пленок (щелевую головку), и экструдированную пленку снимают с одного или более приемных валков, при этом она охлаждается и затвердевает. Было показано, что особенно благоприятно поддерживать приемный валок или валки, с помощью которых экструдированную пленку охлаждают и отверждают, при температуре от 10°С до 50°С, предпочтительно от 15°С до 40°С.
В способе экструзии пленки с раздувом расплав полипропиленовой композиции экструдируют через кольцевую головку и выдувают в виде трубчатой пленки, посредством формирования пузыря, который лопается между прижимными валками после затвердевания. Предпочтительно экструзию с раздувом можно выполнять при температуре от 160°С до 240°С и осуществлять охлаждение водой или предпочтительно газом продувки (обычно воздухом) при температуре от 10°С до 50°С, чтобы обеспечивать высоту мутной линии пленки, составляющую от 0,5 до 8 диаметров головки. Обычно коэффициент раздува должен составлять от 1,5 до 4, например, от 2 до 4, предпочтительно от 2,5 до 3,5.
Методы
Растворимая в ксилоле фракция при комнатной температуре (РХК, масс. %): Количество полимера, растворимого в ксилоле, определяют в соответствии со стандартом ISO 16152; 2005, 5-е издание;
ПТР2 (230°С) измеряют в соответствии со стандартом ISO 1133 (230°С, нагрузка 2,16 кг). Показатель текучести расплава измеряют как ПТР2 в соответствии со стандартом ISO 1133 15 (при температуре 230°С, нагрузка 2,16 кг) для полипропилена. ПТР представляет собой показатель текучести и, следовательно, обрабатываемости полимера. Чем выше показатель текучести расплава, тем ниже вязкость полимера.
Определение сомономера с помощью ЯМР спектроскопии (определение С2)
Количественная ядерная магнитная резонансная (ЯМР) спектроскопия также была использована для количественного определения содержания сомономеров и распределения сомономеров по порядку расположения в полимерах. Регистрировали количественные 13С{1Н} ЯМР спектры в состоянии раствора с помощью спектрометра Bruker Advance III 400 ЯМР, работающего на частотах 400,15 и 100,62 МГц для 1Н и 13С соответственно. Все спектры записывали с использованием оптимизированной системы измерения линейных величин 13С расширенного температурного зонда размером 10 мм, при температуре 125°C с использованием газообразного азота для всех пневматических устройств. Приблизительно 200 мг материала растворяли в 3 мл 1,2-тетрахлорэтана-d2 (ТХЭ-d2) вместе с ацетилацетонатом хрома (III) (Cr (ацац)3), с получением 65 мМ раствора релаксационного агента в растворителе (Singh, G., Kothari, A., Gupta, V., Polymer Testing 28 5 (2009), 475). Для обеспечения однородности раствора после первоначальной подготовки образца в термостате, трубку ЯМР дополнительно нагревали во вращающейся печи в течение по меньшей мере 1 часа. При введении в магнит трубка вращалась с частотой 10 Гц. Указанная установка была выбрана главным образом по причине высокого разрешения и необходимости точной количественной оценки содержания этилена. Стандартное одноимпульсное возбуждение использовали без ЯЭО (ядерного эффекта Оверхаузера), с использованием оптимизированного угла наконечника, задержкой между сканами 1 с и двухуровневой схемой развязки WALTZ 16 (Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, В., J. Mag. Reson. 187 (2007) 225; Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol. Rapid Commun. 2007, 28, 1128). Всего было получено 6144 (6k) переходных процесса на спектр. Количественные спектры ЯМР 13С{1Н} были обработаны, интегрированы и соответствующие количественные свойства определены из интегралов с использованием собственных компьютерных программ. Все химические сдвиги были косвенно связаны с центральной метиленовой группой этиленового блока (ЕЕЕ) при 30,00 ppm при использовании химического сдвига растворителя. Такой подход позволил провести сравнение с эталоном даже в отсутствие этой структурной единицы. Характеристические сигналы, соответствующие встраиванию этилена, наблюдали в Cheng, Н. N., Macromolecules 17 (1984), 1950).
При характеристических сигналах, соответствующих наблюдаемым 2,1-эритрорегиодефектам (как описано в L. Resconi, L. Cavallo, A. Fait, F. Piemontesi, Chem. Rev. 2000, 100 (4), 1253, in Cheng, H. N., Macromolecules 1984, 17, 1950 и в W-J. Wang and S. Zhu, Macromolecules 2000, 33 1157) была необходима коррекция влияния региодефектов на определяемые свойства. Характеристические сигналы, соответствующие другим типам региодефектов, не наблюдали.
Количество фракции сомономера определяли способом Wang и др. (Wang, W-J., Zhu, S., Macromolecules 33 (2000), 1157) путем интегрирования множества сигналов по всей спектральной области 13С{1Н} спектров. Этот способ был выбран из-за его надежности и способности при необходимости учитывать присутствие региодефектов. Области интегрирования слегка регулировали для повышения применимости способа ко всему диапазону полученного содержания сомономеров.
Для исследования систем, в которых наблюдали только изолированный этилен в последовательностях РРЕРР, способ Wang и др. был модифицирован для снижения влияния ненулевых интегралов на центрах, которые, как известно, отсутствуют. Такой подход снижает переоцененное содержание этилена в таких системах и включает уменьшение количества центров, используемых для определения абсолютного содержания этилена, до следующего выражения:
Е = 0,5(Sββ + Sβγ + Sβδ + 0,5(Sαβ + Sαγ))
Благодаря использованию этого набора центров соответствующее интегральное уравнение приобретает следующий вид:
Е = 0,5(IH + IG + 0,5(IC + ID))
при использовании таких же условных обозначений, как в статье Wang и др. (Wang, W-J., Zhu, S., Macromolecules 33 (2000), 1157). Уравнения, используемые для определения абсолютного содержания пропилена, не изменяли.
Молярное процентное содержание включаемого сомономера вычисляли, исходя из мольной доли:
Е [mol%] = 100 * fE
Массовое процентное содержание включаемого сомономера вычисляли, исходя из мольной доли:
Е [масс. %] = 100 * (fE * 28.06) / ((fE * 28.06) + ((1-fE) * 42.08))
Распределение последовательностей сомономеров на уровне триады определяли аналитическим способом, предложенным Kakugo и др. (Kakugo, М., Naito, Y., Mizunuma, K., Miyatake, Т. Macromolecules 15 (1982) 1150). Этот способ был выбран из-за его надежности, и области интегрирования слегка регулировали для повышения применимости способа к более широкому диапазону содержания сомономеров.
Определение сомономера: содержание гексена - 13С ЯМР спектроскопия Количественные 13С{1Н} ЯМР спектры записывали в расплавленном состоянии с использованием спектрометра Bruker Avance III 500 ЯМР, работающего при частотах 500,13 и 125,76 МГц для 1Н и 13С, соответственно. Все спектры записывали, применяя оптимизированную 7-мм измерительную головку для 13C с вращением образца под магическим углом (МУ) при 180°С, подавая газообразный азот во все пневматические устройства. Приблизительно 200 мг материала помещали с уплотнением в ротор для вращения под магическим углом, полученный из оксида циркония, имеющий внешний диаметр 7 мм, и вращали при 4 кГц. Такой режим был выбран в основном для достижения высокой чувствительности, необходимой для быстрой идентификации и точного количественного определения (Klimke, K., Parkinson, М., Piel, С., Kaminsky, W., Spiess, H.W., Wilhelm, М., Macromol. Chem. Phys. 2006; 207:382., Parkinson, M., Klimke, K., Spiess, H.W., Wilhelm, M., Macromol. Chem. Phys. 2007; 208:2128., Castignolles, P., Graf, R., Parkinson, M., Wilhelm, M., Gaborieau, M., Polymer 50 (2009) 2373). Использовали стандартное одно импульсное возбуждение с применением ЯЭО (ядерного эффекта Оверхаузера) с малым временем ожидания восстановления, составляющим 3 сек (Klimke, K., Parkinson, М., Piel, С., Kaminsky, W., Spiess, H.W., Wilhelm, М., Macromol. Chem. Phys. 2006; 207:382., Pollard, M., Klimke, K., Graf, R., Spiess, H.W., Wilhelm, M., Sperber, O., Piel, C., Kaminsky, W., Macromolecules 2004; 37:813.) и схему расщепления RS-HEPT (много импульсный способ Хана с синхронизаций ротора) (Filip, X., Tripon, С., Filip, С, J. Mag. Resn. 2005, 176, 239., Griffin, J.M., Tripon, C., Samoson, A., Filip, C., and Brown, S.P., Mag. Res. in Chem. 2007 45, SI, S198). В общей сложности, было получено 16384 (16к) перехода на спектр.
Количественные спектры ЯМР 13С{1Н} были обработаны, интегрированы, и из интегралов были определены соответствующие количественные характеристики. Все химические сдвиги соотносили с внутренним эталоном, которым являлся сигнал метальной изотактической пентады (mmmm) при 21,85 ppm.
Наблюдали характеристические сигналы, соответствующие встраиванию 1-гексена, и количественно определяли содержание сомономера следующим образом.
Количество 1-гексена, включенного в локализованные последовательности РНР, количественно определяли с использованием интеграла участка αВ4 при 44,2 ppm с учетом количества выявленных участков на сомономер:
Н = IαВ4 / 2
Количество 1-гексена, включенного в двойные непрерывные последовательности РННР, определяли с использованием интеграла участка ααВ4 при 41,7 ppm, в соответствии с количеством выявленных участков на сомономер:
НН = 2 * IααВ4
Когда наблюдали двойные непрерывные последовательности, необходимо было компенсировать количество 1-гексена, включенного в локализованные последовательности РНР, вследствие наложения сигналов αВ4 и αВ4В4 при 44.4 ppm:
Н = (IαВ4 - 2 * IααВ4) / 2
Общее содержание 1-гесена рассчитывали на основе суммы локализованного и непрерывно встроенного 1-гесена:
Нобщ = Н + НН
Когда не наблюдали участков, указывающих на непрерывное встраивание, общее содержание 1-гексенового сомономера рассчитывали исключительно на это количество:
Нобщ = Н
Наблюдали характеристические сигналы, указывающие на 2,1-эритрорегиодефекты (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253).
На наличие 2,1-эритрорегиодефектов указывали метальные участки Pαβ (21е8) и Pαγ (21е6) при 17,7 и 17,2 ppm, и это подтверждалось другими характеристическими сигналами.
Общее количество вторичного (2,1-эритро) встроенного пропена количественно определяли, исходя из метиленового участка αα21e9 при 42,4 ppm:
Р21 = Iαα21e9
Общее количество первичного (1,2) встроенного пропена количественно определяли, исходя из главных метиленовых участков Sαα при 46,7 ppm и компенсации относительного количества неучтенного 2,1-эритро, αВ4 и ααВ4В4 метиленового звена пропена (следует отметить, что расчетное количество Н и НН мономеров гексена на последовательность не является количеством последовательностей):
Р12 = ISαα = 2*Р21 + Н + НН / 2
Общее количество пропена определяли как сумму первичного (1,2) и вторичного (2,1-эритро) встроенного пропена:
Робщ = Р12 + Р21 = ISαα + 3* Iαα21e9 + (IαB4 - 2 * IααB4) / 2 + IααB4
Это упрощается до:
Робщ = ISαα + 3* Iαα21e9 + 0.5*IαB4
Затем рассчитывали общую мольную долю 1-гесена в полимере:
fH = Нобщ / (Нобщ + Робщ)
Полное интегральное уравнение для мольной доли 1-гексена в полимере имеет вид:
fH = (((IαB4 - 2 * IααB4) / 2) + (2 * IααВ4) /((ISαα + 3* Iαα21е9 + 0.5*IαВ4) + ((IαВ4 - 2 * IααВ4) / 2) + (2 * IααВ4))
Это упрощается до:
fH = (IαВ4/2 + IααВ4) / (ISαα + 3* Iαα21е9 + IαВ4 + IααВ4)
Общее содержание сомономера 1-гексена в мольных процентах рассчитывали исходя из молярной доли обычным способом:
Н [моль%] = 100 * fH
Общее содержание сомономера 1-гексена в массовых процентах рассчитывали исходя из мольной доли стандартным способом:
Н [масс. %] = 100 * (fH * 84.16) / ((fH * 84.16) + ((1 - fH) * 42.08))
Анализ ДСК, температура плавления (Тпл) и температура кристаллизации (Ткр)
Эти параметры определяли с помощью прибора ТА Instrument Q2000 дифференциальной сканирующей калориметрии (ДСК) на образцах массой от 5 до 7 мг. ДСК проводили в соответствии со стандартом ISO 11357 / часть 3 /метод С2 в цикле нагрев/охлаждение/нагрев со скоростью сканирования 10°С/мин в диапазоне температур от -30°С до +225°С.
Температуру кристаллизации и теплоту кристаллизации (Нкр) определяют на стадии охлаждения, при этом температуру плавления и теплоту плавления (Нпл) определяют на второй стадии нагрева.
Общая миграция
Общую миграцию определяли в соответствии со стандартом EN ISO 1186-14:2002 на пластинах, полученных инжекционным формованием, размером 60×60×1 мм3.
Среднечисленную молекулярную массу (Mn), среднемассовую молекулярную массу (Mw) и полидисперсность (Mw/Mn) определяли с помощью гель-проникающей хроматографии (ГПХ) по следующей методике:
Среднемассовую молекулярную массу (Mw) и полидисперсность (Mw/Mn), где Mn представляет собой среднечисленную молекулярную массу, a Mw представляет собой среднемассовую молекулярную массу, измеряют методом, основанным на стандартах ISO 16014-1:2003 и ISO 16014-4:2003.
Использовали прибор Waters Alliance GPCV 2000, оборудованный детектором показателя преломления и онлайн-вискозиметром, с 3 колонками TSK-gel (GMHXL-HT), от TosoHaas и 1,2,4-трихлорбензолом (ТХБ, стабилизированным 200 мг/л 2,6-ди-трет-бутил-4-метилфенолом) в качестве растворителя, при е 145°С и постоянном расходе 1 мл/мин. На один анализ вводили 1 раствор образца объемом 216,5 мкл. Комплект колонок калибровали с использованием относительной калибровки, с помощью 19 стандартов полистирола (ПС) с узким ММР в диапазоне от 0,5 кг/моль до 11500 кг/моль и комплекта хорошо изученных широких стандартов полипропилена.
Все образцы получали посредством растворения 5-10 мг полимера в стабилизированном ТХБ (таком же, как подвижная фаза) и выдерживания в течение 3 часов при непрерывном встряхивании перед отбором проб в приборе ГПХ.
Мутность и прозрачность определяли в соответствии со стандартом ASTM D1003-00 на пластинах размером 60×60×1 мм3, полученных инжекционным формованием, в соответствии со стандартом EN ISO 1873-2, при использовании температуры расплава 200°С, и на литых пленках толщиной 50 мкм, полученных из многослойной литой пленки при температуре плавления 220°С и температуре охлаждающего валка 20°С.
Температура начала сваривания (ТНС), температура окончания сваривания (ТОС); диапазон температур сваривания
Эксперименты по дифференциальной сканирующей калориметрии (ДСК) проводили на приборе ТА Instruments Q2000, калиброванном с помощью индия, цинка и олова, в соответствии со стандартом ISO 11357/1. Измерения проводили в атмосфере азота (50 мл/мин) на образцах массой 5±0,5 мг в цикле нагрев/охлаждение/нагрев со скоростью сканирования 10°С/мин при температуре от -30°С до 225°С, в соответствии со стандартом ISO 11357/3. За температуру плавления (Тпл) и кристаллизации (Ткр) принимали пики эндотерм и экзотерм в ходе цикла охлаждения и второго цикла нагрева, соответственно. Температуру начала сваривания (ТНС) прогнозировали путем анализа кривой второго нагрева в соответствии со следующей методикой: первый предел интегрирования устанавливали при температуре 16°С, второй предел при температуре Тпл+20°С и регистрировали полную энтальпию плавления. Температуру Т1 определяют как температуру, при которой было получено 19% энтальпии плавления с указанными выше пределами интегрирования. Параметр ТНС окончательно вычисляется как:
ТНС = 1,0596 × T1 + 3,8501
Стерилизацию паром осуществляли в машине серии Systec D (Systec Inc., США). Образцы нагревали при скорости нагрева 5°С/мин, начиная с температуры 23°С. После выдерживания в течение 30 мин при температуре 121°С, их немедленно извлекали из парового стерилизатора и выдерживали при комнатной температуре до последующей обработки.
2. Примеры
Компонент (А)
В качестве компонента (А) получали статистический сополимер пропилена и этилена в экспериментальной установке Borstar® с реактором предварительной полимеризации и одним суспензионным петлевым реактором.
В качестве катализатора использовали ZN180M от Basell. Сокатализатор представлял собой ТЭАЛ (триэтилалюминий), а внешний донор был донором D.

Компонент Б
Катализатор: Синтез металлоцена МС-1
2-хлор-4-метилбензальдегид
165 мл (413 ммоль) 2,5 М раствора nBuLi в гексан добавляли по капле в течение 1 ч к 82,2 г (400 ммоль) раствора 3-хлор-4-бром-толуола в 400 мл тетрагидрофурана (ТГФ), охлажденного до температуры -88°С. Полученную смесь перемешивали в течение 30 мин при этой температуре и затем по капле добавляли 44,0 г (602 ммоль) диметилфталата (ДМФ) в течение 10 мин при интенсивном перемешивании. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем охлаждали до температуры 0°С в ледяной бане и затем добавляли 100 мл воды и 400 мл 3Н HCl. Органический слой отделяли и водный слой экстрагировали 2×125 мл дихлорметана. Объединенный органический экстракт сушили высушивали над K2CO3 и затем пропускали через тонкий слой силикагеля 60 (40-63 мкм). Слой силикагеля дополнительно промывали 50 мл дихлорметана.
Объединенный органический элюат выпаривали досуха с получением слабооранжевой жидкости, которую затем перегоняли в вакууме с получением 58,0 г целевого продукта (94%) (температура кипения 99-102°С/11 мм рт.ст.) в виде бесцветной жидкости, которая кристаллизовалась в течение ночи при комнатной температуре.
Аналитический расчет для C8H7ClO: С: 62,15; Н: 4,56. Установлено: С: 62,24; Н: 4,45.
1H ЯМР (CDCl3): δ 10,4 (s, 1H, СНО), 7,80 (d, J=7,8 Гц, 1H, 6-Н), 7,25 (s, 1Н, 3-Н), 7,17 (d, J=7,8 Гц, 1H, 5-Н), 2,40 (s, 3Н, 4-Ме).
(2-хлор-4-метилфенил)метанол

375 мл метанола добавляли по капле при интенсивном перемешивании в течение 5 ч к смеси 116 г (0,75 моль) 2-хлор-4-метилбензальдегида и 43,0 г (1,14 моль) NaBH4, в 750 мл ТГФ при температуре 0-5°С. Эту смесь перемешивали в течение ночи при комнатной температуре и затем выпаривали досуха. Полученную маслянистую массу подкисляли посредством 1200 мл 2 М HCl до рН~1, и образованный продукт впоследствии экстрагировали 3×400 мл дихлорметана. Объединенный органический экстракт сушили над Na2SO4 и выпаривали досуха. Этот продукт использовали без дополнительной очистки.
1Н ЯМР (CDCl3): δ 7,29 (d, J=7,8 Гц, 1Н, 5-Н), 7,15 (s, 1H, 3-Н), 7,04 (d, J=7,8 Гц, 1H, 6-Н), 4,67 (s, 2Н, CH2OH), 2,59 (br.s, 1H, CH2OH), 2,30 (s, 3Н, 4-Ме). 13С{1Н} ЯМР (CDCl3): δ 138,9; 135,0; 132,4; 129,7; 128,6; 127,6; 62,5; 20,7.
2-Хлор-1-(хлорметил)-4-метилбензен
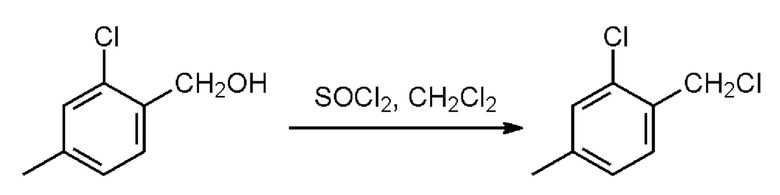
Полученный выше 2-хлор-4-метилбензиловый спирт, растворенный в 750 мл дихлорметана, добавляли по капле к 55 мл (754 ммоль) тионилхлорида при температуре +5°С. Полученный раствор перемешивали в течение ночи при комнатной температуре и затем выпаривали досуха. Остаток растворяли в 500 мл дихлорметана и полученный раствор промывали 250 мл воды. Органический слой отделяли, водный слой экстрагировали 2×150 мл дихлорметана. Объединенный органический экстракт сушили над Na2SO4, пропускали через тонкий слой силикагеля 60 (40-63 мкм) и затем выпаривали досуха. Неочищенный продукт перегоняли в вакууме с получением 114 г (87%) целевого продукта в виде бесцветной жидкости, температура кипения 92-95°С/5 мм рт.ст.
Аналитический расчет для C8H8Cl2: С: 54,89; Н: 4,61. Установлено: С: 54,80; Н: 4,65.
1Н ЯМР (CDCl3): δ 7,30 (d, J=7,8 Гц, 1Н, 5-Н), 7,19 (s, 1H, 3-Н), 7,04 (d, J=7,8 Гц, 1H, 6-Н), 4,64 (s, 2Н, CH2Cl), 2,30 (s, 3Н, Me). 13С{1Н} ЯМР (CDCl3): δ 140,3; 133,7; 131,9; 130,6; 130,2; 127,9; 43,5; 20,8.
3-(2-Хлор-4-метилфенил)-2-метилпропаноил хлорид
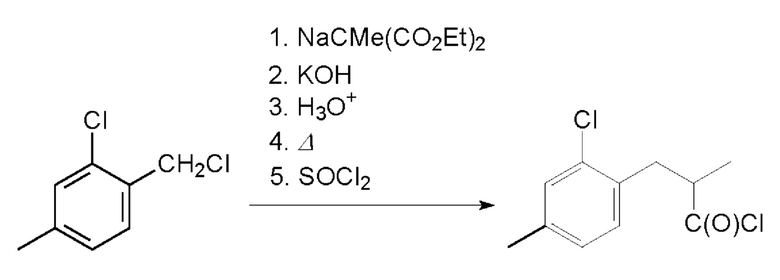
119 г (0,68 моль) диэтилметилмалоната добавляли к раствору этоксида натрия, полученному из 17,0 г (0,74 моль)металлического натрия и 600 мл сухого этанола. Полученную смесь перемешивали в течение 15 мин и затем добавляли 114 г (0,651 моль) 2-хлор-1-(хлорметил)-4-метилбензена при интенсивном перемешивании с такой скоростью, чтобы поддерживать легкое орошение. Полученную смесь кипятили с обратным холодильником в течение 2 ч и затем охлаждали до комнатной температуры. Добавляли раствор 135 г КОН в 550 мл воды. Данную смесь кипятили с обратным холодильником в течение 4 ч для омыления полученного эфира. Этанол и воду отгоняли до тех пор, пока температура не достигала 95°С, затем к остатку добавляли 3000 мл воды и затем 12 М HCl (до рН~1). Осажденную замещенную метилмалоновую кислоту отфильтровывали и промывали водой. Эту дикислоту декарбоксилировали при 160-180°C с получением слабооранжевого масла, которое кристаллизовалось при комнатной температуре. Смесь полученной кислоты и 166 мл тионилхлорида перемешивали в течение 24 ч при комнатной температуре. После выпаривания избытка тионилхлорида остаток перегоняли в вакууме с получением 123 г (82%) целевого продукта, температура кипения 105-117°С/5 мм рт.ст.
Аналитический расчет для C11H12Cl2O: С: 57,16; Н: 5,23. Установлено: С: 57,36; Н:, 5,38.
1Н ЯМР (CDCl3): δ 7,19 (s, 1H, 3-Н), 7,10 (d, J=7,7 Гц, 1H, 5-Н), 7,00 (d, J=7,7 Гц, 1Н, 6-Н), 3,20-3,32 (m, 2Н, CHH' и CHMe), 2,82 (dd, J=12.8 Гц, J=6.4 Гц, 1H, CHH'), 2,30 (s, 3Н, 4-Ме), 1,30 (d, J=6,8 Гц, 3Н, СНМе). 13C{1H} ЯМР (CDCl3): δ 177,1; 138,6; 133,8; 132,1; 131,2; 130,2; 127,7; 51,4; 36,5; 20,7; 16,7.
4-Хлор-2,6-диметилиндан-1-он
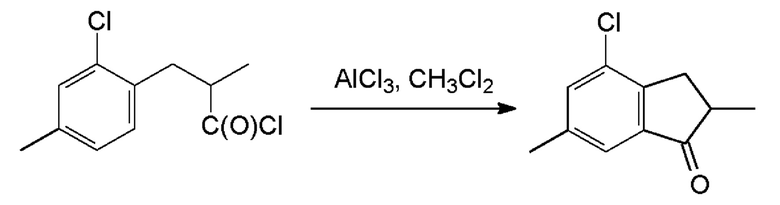
123 г (531 ммоль) раствора 3-(2-хлор-4-метилфенил)-2-метилпропаноил хлорида в 100 мл дихлорметана добавляли по капле к перемешиваемой суспензии из 85,0 г (638 ммоль) AlCl3 в 500 мл дихлорметана при 5°С. Эту смесь перемешивали в течение ночи при комнатной температуре и затем выливали на 500 г измельченного льда. Органический слой отделяли и водный слой экстрагировали 3×100 мл дихлорметана. Объединенный органический экстракт промывали водным K2CO3, сушили над K2CO3, пропускали через тонкий слой силикагеля 60 (40-63 мкм), и затем выпаривали досуха. Неочищенный продукт перегоняли в вакууме с получением 98,4 г (95%) бесцветной жидкости, температура кипения 131-132°С/8 мм рт.ст.
Аналитический расчет для С11Н11С10: С: 67,87; Н: 5,70. Установлено: С: 68,01; Н:, 5,69.
1Н ЯМР (CDCl3): δ 7,42 (s, 1H, 7-Н), 7,38 (s, 1H, 5-Н), 3,32 (dd, J=17,3 Гц, J=7,8 Гц, 1Н, 3-CHH'), 2,68-2,76 (m, 1Н, 2-Н), 2,62 (dd, 1H, J=17,3 Гц, J=3,6 Гц, 3-CHH'), 2,38 (s, 3Н, 6-Ме), 1,31 (d, J=7,5 Гц, 3Н, 2-Ме). 13С{1Н} ЯМР (CDCl3): δ 208,2; 148,0; 139,3; 138,1; 135,0; 132,1; 122,2; 42,0; 33,3; 20,7; 16,1.
4-Хлор-1-метокси-2,6-диметилиндан
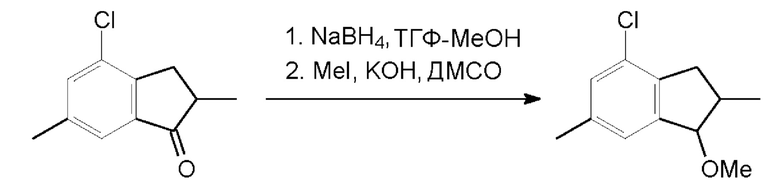
205 мл метанола добавляли по капле при интенсивном перемешивании в течение 5 ч к смеси из 98,4 г (0,505 моль) 4-хлор-2,6-диметилиндан-1-она и 29,0 г (0,767 моль) NaBH4 в 510 мл ТГФ при е 0-5°С. Эту смесь перемешивали в течение ночи при комнатной температуре и затем выпаривали досуха. Остаток подкисляли 2 М HCl до рН 5-6, и полученный 4-хлор-2,6-диметилиндан-1-ол экстрагировали 3×300 мл дихлорметана. Объединенный органический экстракт сушили над Na2SO4 и выпаривали досуха с получением белого твердого вещества. Добавляли 132 г (2,35 моль) КОН и 163 г (1,15 моль) Mel к раствору таким образом полученного белого вещества в 800 мл диметилсульфоксида (ДМСО). Эту смесь перемешивали в течение 5 ч при температуре окружающей среды. Раствор декантировали от избытка КОН, и после этого дополнительно промывали 3×350 мл дихлорметана. Объединенный органический экстракт промывали 3000 мл воды. Органический слой отделяли, а водный слой экстрагировали 3×300 мл дихлорметана. Объединенный органический экстракт промывали 7×1500 мл воды, сушили над Na2SO4, а затем выпаривали досуха. Остаток перегоняли в вакууме с получением 99,9 г (94%) целевого продукта, состоящего из двух пар энантиомеров, температура кипения 104-105°С/8 мм рт.ст.
Аналитический расчет для C12H15ClO: С: 68,40; Н: 7,18. Установлено: С: 68,58; Н: 7,25.
Син-изомеры. 1H ЯМР (CDCl3): δ 7,05 (s, 2Н, 5-Н и 7-Н), 4,51 (d, J=5,7 Гц, 1Н, 1-Н), 3,41 (s, 3Н, ОМе), 2,92 (dd, J=15,3 Гц, J=6,4 Гц, 1H, 3-CHH'), 2,68-2,59 (m, 2Н, 3-CHH' и 2-Н), 2,32 (s, 3Н, 6-Ме), 1,07 (d, J=6,8 Гц, 3Н, 2-Ме). 13С{1Н} ЯМР (CDCl3): δ 144,6; 138,3; 137,8; 130,7; 128,7; 124,1, 86,4; 57,0; 38,2; 36,9; 21,0; 13,5.
Анти-изомеры. 1Н ЯМР (CDCl3): δ 7,05 (s, 1Н, 7-Н), 7,07 (s, 1H, 5-Н), 4,37 (d, J=3,9 Гц, 1H, 1-Н), 3,45 (s, 3Н, ОМе), 3,19 (dd, J=16.2 Гц, J=7,6 Гц, 1Н, 3-CHH'), 2,50 (m, 1H, 2-Н), 2,42 (dd, J=16,2 Гц, J=5,0 Гц, 1H, 3-CHH'), 2,32 (s, 3Н, 6-Ме), 1,16 (d, J=6,8 Гц, 3Н, 2-Ме). 13С{1Н} ЯМР (CDCl3): δ 144,2, 138,1 (два резонанса), 130,7; 128,9; 124,2; 91,8; 56,6; 39,4; 37,2; 21,0; 19,3.
4-(3,5-Диметилфенил)-1-метокси-2,6-диметилидан
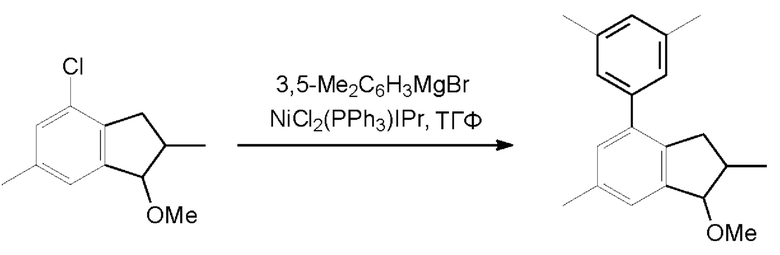
200 мл (200 ммоль) 1,0 М раствора 3,5-ди-метилфенилмагния бромида в ТГФ добавляли при комнатной температуре к смеси 2, 10 г (2,69 ммоль, 2,0 моль%) NiCl2(PPh3)IPr и 28,4 г (134,7 ммоль) 4-хлор-1-метокси-2,6-диметилиндана. Полученную смесь кипятили с обратным холодильником в течение 1,5 ч, затем охлаждали до комнатной температуры и добавляют 100 мл воды. Большую часть ТГФ отгоняли во вращающемся испарителе. К остатку добавляли 500 мл дихлорметана и 1000 мл 1 М HCl. Органический слой отделяли, затем водный слой экстрагировали 2×100 мл дихлорметана. Объединенный органический экстракт выпаривали досуха с получением желтого масла. Продукт выделяли посредством флэш-хроматографии на силикагеле 60 (40-63 мкм; элюент : гексаны-дихлорметан = 2:1 об., затем 1:1 об.). В результате получали 33,8 г (90%) 4-(3,5-диметилфенил)-1-метокси-2,6-диметилиндана в виде бесцветного густого масла, включающего два диастереомера.
Аналитический расчет для С20Н24О: С: 85,67; Н: 8,63. Установлено: С: 86,03; Н: 8,80.
1Н ЯМР (CDCl3), смесь изомеров: δ 7,20-6,93 (набор сигналов, сумма 5Н); 4,51 (d, J -=5,7 Гц) и 4,39 (d, J -=3,9 Гц) {сумма 1H}; 3,49 (s) и 3,45 (s) {сумма 3Н}; 3,29-3,17 (m), 2,94-2,84 (m), 2,80-2,70 (m) и 2,60-2.37 (m) {сумма 3Н}; 2,38 (s) и 2,35 (s) {сумма 9Н}; 1,12 (d, J=6,9 Гц) и 1,06 (d, J=7,1 Гц) {сумма 3Н}. 13С{1Н} ЯМР (CDCl3), смесь изомеров: δ 143,50; 143,00; 140,91; 138,68; 138,58; 138,09; 137,64; 136,40; 136,03; 129,51; 129,17; 128,48; 126,35; 124,66; 91,42; 86,23; 56,82; 56,62; 40,12; 39,06; 38,00; 37,85; 21,36; 21,25; 19,17; 13,53.
7-(3,5-Диметилфенил)-2,5-диметил-1H-инден
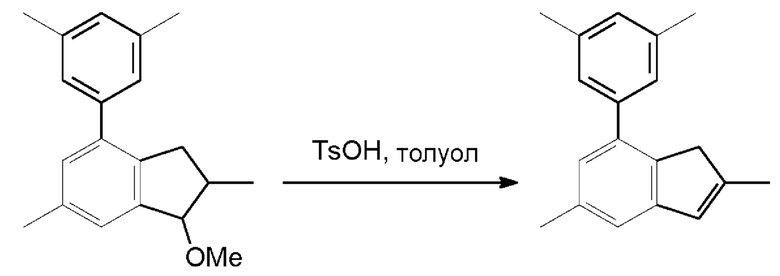
300 мг TsOH добавляли к раствору 33,8 г (120,6 ммоль) 4-(3,5-диметилфенил)-1-метокси-2,6-диметилиндана в 300 мл толуола, и полученную смесь кипятили с обратным холодильником, используя насадку Дина-Старка, в течение 10 мин. Затем добавляли еще одну порцию TsOH массой 150 мг и вновь полученный раствор кипятили с обратным холодильником в течение 10 мин. После охлаждения до комнатной температуры реакционную смесь промывали 200 мл 10% K2CO3. Органический слой отделяли и затем водный слой экстрагировали 2×100 мл дихлорметана. Объединенный органический экстракт сушили над безводным K2CO3 и выпаривали. Полученное желтое масло растворяли в гексане. Полученный раствор пропускали через тонкий слой силикагеля 60 (40-63 мкм), и элюат выпаривали досуха с получением 29,1 г (97%) 7-(3,5-диметилфенил)-2,5-диметил-1H-индена в виде слегка желтоватого масла.
Аналитический расчет для С19Н20: С: 91,88; Н: 8,12. Установлено: С: 92,11; Н: 8,34.
1Н ЯМР (CDCl3): δ 7,13 (s, 2Н), 7,05 (s, 1H), 6,98 (s, 1H), 6,93 (s, 1H), 6,47 (m, 1H), 3,33 (s, 2Н), 2,40 (s, 3Н), 2,37 (s, 6Н), 2,12 (s, 3Н), 13С{1Н} ЯМР (CDCl3): δ 146,65; 146,45; 141,43; 137,84; 137,75; 137,21; 136,41; 128,55; 127,04; 126,23; 125,04; 119,58; 42,41; 21,40; 16,741.
1 - два сигнала в алифатической области были объединены
[6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метил-1Н-инден-1-ил](хлор)-диметилсилан

14,6 мл (35,5 ммоль) 2,43 М раствора nBuLi в гексанах добавляли одной порцией к раствору 11,3 г (35,3 ммоль) 5-трет-бутил-7-(3,5-диметилфенил)-6-метокси-2-метил-1Н-индена в 200 мл простого эфира, охлажденного до 50°С. Полученный оранжевый раствор перемешивали в течение ночи при комнатной температуре, затем полученный оранжевый раствор, содержащий желтоватый осадок, охлаждали до -78°С (осадок почти полностью исчезал), и добавляли 22,8 г (177 ммоль, 5 экв.) дихлордиметилсилана одной порцией. Полученный раствор подогревали до комнатной температуры и перемешивали в течение ночи при комнатной температуре. Полученную смесь фильтровали через стеклянную крошку (G4). Осадок дополнительно промывали 2×10 мл простого эфира. Объединенный фильтрат выпаривали досуха с получением целевого материала в виде слегка оранжевого масла, которое использовали без дополнительной очистки.
1Н ЯМР (CDCl3): δ 7,38 (s, 1H), 7,08 (s, 2Н), 6,98 (s, 1H), 6,43 (s, 1H), 3,53 (s, 1H), 3,25 (s, 3Н), 2,37 (s, 6Н), 2,19 (s, 3Н), 1,43 (s, 9Н), 0,43 (s, 3Н), 0,17 (s, 3Н), 13С{1Н} ЯМР (CDCl3): δ 155,79; 145,87; 143,74; 137,99; 137,55; 137,49; 136,75; 128,32; 127,87; 127,55; 126,65; 120,86; 60,46; 49,99; 35,15; 31,17; 21,42; 17,56; 1,11; -0,58.
[2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-1Н-инден-1-ил][2,6-диметил-4-(3,5-диметилфенил)-1Н-инден-1-ил]диметилсилан
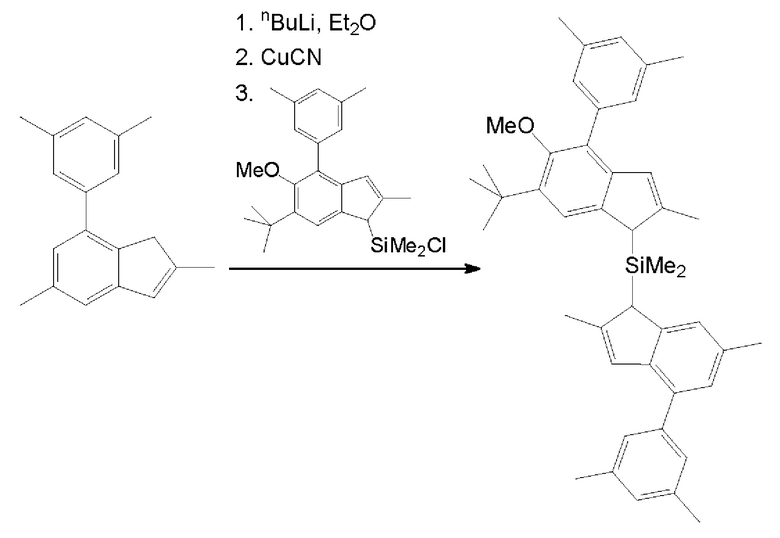
14,6 мл (35,5 ммоль) 2,43 М nBuLi раствора в гексанах добавляли одной порцией в раствор 8,78 г (35,35 ммоль) 7-(3,5-диметилфенил)-2,5-диметил-1Н-индена в 200 мл простого эфира при 50°С. Эту смесь перемешивали в течение ночи при комнатной температуре, затем полученный желтоватый раствор, содержащий большое количество желтого осадка, охлаждали до -50°С, затем последовательно добавляли 40 мл ТГФ и 200 мг CuCN. Полученную смесь перемешивали в течение 0,5 ч при -25°С, затем добавляли одной порцией раствор [6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метил-1Н-инден-1-ил](хлор)-диметилсилана (35,32 ммоль) в 200 мл простого эфира. Эту смесь перемешивали в течение ночи при температуре окружающей среды, затем фильтровали через стеклянную крошку (G4), и полученный желтый раствор выпаривали досуха. Целевой продукт выделяли посредством флэш-хроматографии на силикагеле 60 (40-63 мкм; элюэнт : гексаны-дихлорметан = 10:1 об., затем 3:1 об). В результате получали 17,5 г (79%) [2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-1Н-инден-1-ил][2,6-диметил-4-(3,5-диметилфенил)-1H-инден-1-ил]диметилсилана.
Аналитический расчет для C44H52OSi: С: 84,56; Н: 8,39. Установлено: С: 84,85; Н: 8,78.
1H ЯМР (CDCl3): δ 7,51-7,02 (группа сигналов, сумма 7Н), 6,99 (s, 2Н), 6,79 (s, 1Н), 6,45 (s, 1Н), 3,68 и 3,66 (2m, сумма 2Н), 3,28 и 3,26 (2s, сумма 3Н), 2,44-2,32 (группа сигналов, 15Н), 2,18 и 2,15 (2s, сумма 6Н), 1,43 и 1,42 (2s, сумма 9Н), -0,16, -0,18, -0,19 и -0,25 (4s, сумма 6Н). 13С{1H} ЯМР (CDCl3): δ 155,36; 147,36; 147,28; 146,50; 146,25; 146,00; 143,75; 143,70; 141,41; 140,42; 139,21; 138,24; 137,76; 137,53; 137,16; 137,09; 133,94; 132,44; 132,32; 128,34; 128,24; 127,94; 127,53; 127,15; 126,74; 126,41; 126,10; 126,05; 125,84; 125,75; 123,04; 122,84; 120,56; 120,50; 60,51; 47,37; 47,30; 47,23; 47,15; 35,16; 31,27; 31,23; 21,68; 21,59; 21,43; 17,95; 17,85; -5,27; -5,28; -5,37; -5,85
рац-анти-Me2Si(2,6-Me2-4-(3,5-Me2Ph)Ind)(2-Me-4-(3,5-Me2Ph)-5-OMe-6-tBu-Ind)ZrCl2

23,1 мл (56,1 ммоль) 2,43 М раствора nBuLi в гексанах добавляли одной порцией к раствору 17,53 г (28,05 ммоль) [2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-1Н-инден-1 -ил] [2,6-диметил-4-(3,5-диметилфенил)-1Н-инден-1-ил]диметилсилана в 200 мл простого эфира, охлажденного до-50°С. Эту смесь перемешивали в течение ночи при комнатной температуре. Полученный красноватый раствор, содержащий желтоватый осадок, охлаждали до -50°С и добавляли 6,54 г (28,06 ммоль) ZrCl4. Реакционную смесь перемешивали в течение 24 ч при комнатной температуре, с получением светло-красной суспензии с оранжевым осадком. Эту смесь выпаривали досуха, а остаток обрабатывали 200 мл горячего толуола. Смесь фильтровали в горячем состоянии через стеклянную крошку (G4), и фильтрат выпаривали до приблизительно 60 мл. Оранжевый кристаллический порошок, осажденный из этого раствора в течение 3 ч при комнатной температуре, собирали и сушили в вакууме. В результате получали 3,60 г чистого анти-диметилсиландиил[2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-инден-1-ил][2,6-диметил-4-(3,5-диметилфенил)-инден-1-ил]дихлорида циркония. Маточный раствор выпаривали почти досуха, а остаток растирали с 50 мл простого эфира. Нерастворимый оранжевый осадок отфильтровывали (G3) с получением 5,01 г смеси анти-/син-комплексов в отношении приблизительно 93:7. Красноватый кристаллический порошок, осажденный в течение ночи при 30°С из фильтрата, собирали и сушили в вакууме. В результате получали 1,98 г чистого син-диметилсиландиил[2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-инден-1-ил] [2,6-диметил-4-(3,5-диметилфенил)-инден-1-ил]дихлорида циркония. После выдержки в течение нескольких дней при комнатной температуре из маточного раствора осаждали еще одну порцию красного кристаллического порошка. Этот осадок отфильтровывали с получением 4,91 г смеси син/анти-комплексов при отношении приблизительно 96:4. Таким образом, общий выход син- и анти-комплексов, выделенных в этом синтезе, составил 15,5 г (70%).
Анти-диметилсиландиил[2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-инден-1-ил] [2,6-диметил-4-(3,5-диметилфенил)-инден-1-ил]дихлорид циркония
Аналитический расчет для C44H50Cl2OSiZr: С: 67,31; Н: 6,42. Установлено: С: 67,58; Н: 6,59.
1Н ЯМР (CDCl3, 400 МГц, 27°С): δ 7,49 (s, 1H), 7,36 (s, 1Н), 7,28 (s, 2Н), 7,22 (s, 1Н), 7,32-7,12 (два очень широких сигнала, 2Н), 6,98 (s, 1H), 6,95 (2s, сумма 2Н) 6,57 (s, 1Н), 3,43 (s, 3Н), 2,35 (2s, сумма 9Н), 2,32 (s, 6Н), 2,23 (s, 3Н), 2,17 (s, 3Н), 1,39 (s, 9Н), 1,30 (s, 3Н), 1,29 (s, 3Н). 13С{1H} ЯМР (CDCl3,): δ 159,81; 144,25; 139,44; 138,46; 138,02; 137,90; 136,75; 135,59; 135,44; 134,26; 133,57; 130,51; 129,36; 129,03; 128,86; 128,73; 128,22; 127,77; 127,39; 127,08; 126,41; 123,16; 122,59; 122,03; 121,72; 120,81; 81,98; 81,95; 62,61; 35,77; 30,40; 22,11; 21,45; 21,35; 18,40; 18,25; 2,68; 2,52.
Син-диметилсиландиил[2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-инден-1-ил][2,6-диметил-4-(3,5-диметилфенил)-инден-1-ил]дихлорид циркония
Аналитический расчет для C44H50Cl2OSiZr: С: 67,31; Н: 6,42. Установлено: С: 67,56; Н: 6,60.
1Н ЯМР (CDCl3, 400 МГц, 27°С): δ 7,50 (s, 1H), 7,35 (s, 1H), 7,25 (s, 2Н), 7,31-7,08 (два очень широких сигнала, 2Н), 7,01 (s, 1H), 6,96 (s, 1Н), 6,95 (s, 1H) 6,84 (s, 1H), 6,48 (s, 1Н), 3,26 (s, 3Н), 2,42 (s, 3Н), 2,36, 2,35 и 2,34 (3s, сумма 15Н), 2,30 (s, 3Н), 1,43 (s, 3Н), 1,35 (s, 9H), 1,20 (s, 3Н). 13С{1H} ЯМР (CDCl3,): δ 158,95; 143,13; 139,34; 137,91; 137,78; 137,59; 136,81; 136,15; 135,78; 135,11; 134,48; 132,32; 129,25; 129,21; 128,80; 128,35; 127,33; 126,32; 124,00; 122,89; 121,45; 121,24; 121,00; 83,74; 83,67; 62,36; 35,55; 30,31; 22,72; 21,44; 18,53; 18,45; 2,92; 2,65,
Катализатор: синтез катализатора
Внутри перчаточного бокса 72,0 мг сухого и дегазированного раствора поверхностно-активного вещества (1Н,1Н-перфтор(2-метил-3-оксагексан-1-ол, (номер CAS 26537-88-2), (Apollo Scientific), дегазированный посредством барботажа аргона перед использованием) смешивали с 2 мл 30 масс. % раствора МАО от Chemtura в сосуде с перегородкой и оставили реагировать на ночь. На следующий день 39,8 мг (0,051 ммоль, 1 экв.) МС-1 растворяли в 4 мл 30 масс. % раствора МАО от Chemtura в другом сосуде с перегородкой и оставляли перемешиваться внутри перчаточного бокса.
По прошествии 60 мин, 1 мл раствора поверхностно-активного вещества и 4 мл раствора МАО-металлоцена последовательно добавляли в стеклянный реактор, содержащий 40 мл PFC при -10°С и оборудованный верхней мешалкой (скорость перемешивания = 600 об/мин). Общее количество МАО составляет 5 мл (300 экв.). Сразу же получали красную эмульсию и перемешивали в течение 15 мин при температуре/скорости -10°С/ 600 об/мин. Затем эмульсию переносили через тефлоновую трубку 2/4 на 100 мл горячего PFC при 90°С и перемешивали при скорости 600 об/мин до завершения переноса, а затем скорость снижали до 300 об/мин. Через 15 минут перемешивания масляную ванну удаляли и мешалку выключали. Катализатор оставляли оседать поверх PFC и через 35 минут растворитель откачивали. Оставшийся катализатор сушили в течение 2 часов при 50°С в потоке аргона. Получали 0,72 г красного сыпучего порошка
Al/Zr = 450, Al/поверхностно-активное вещество = 250
Методика автономной предварительной полимеризации
Катализатор МС-1 предварительно полимеризировали в соответствии со следующей методикой. Эксперимент по автономной предварительной полимеризации осуществляли в реакторе под давлением объемом 125 мл, оборудованном линиями подачи газа и верхней мешалкой. Сухой и дегазированный перфтор-1,3-диметилциклогексан (15 см) и требуемое количество катализатора, подлежащего предварительной полимеризации, загружали в реактор внутри перчаточного бокса и герметизировали реактор. Затем реактор вынимали из перчаточного бокса и помещали в ванну с водяным охлаждением, выдерживаемую при 20°С. Верхнюю мешалку и питающие линии соединяли и устанавливали скорость перемешивания 450 об/мин. Эксперимент начинали посредством открытия подачи пропилена в реактор. Общее давление в реакторе повышали примерно до 0,5 МПа (5 бар) и поддерживали постоянным путем подачи пропилена через регулятор массового расхода до достижения целевой степени полимеризации (10 мин). Реакцию останавливали путем мгновенного испарения летучих компонентов. Реактор открывали внутри перчаточного бокса и содержимое выливали в стеклянный сосуд. Перфтор-1,3-диметилциклогексан выпаривали до достижения постоянной массы с получением предварительно полимеризованного катализатора.

Примеры полимеризации компонента Б
В автоклав с перемешиванием (оборудованный спирально-лопастной мешалкой) общим объемом 21,2 дм3, содержащий 0,2 бар изб. пропилена (качество 2,3; очищенный через колонки, заполненные PolyMax301 Т-4427 В (60°С; Cu/CuO), MS13X-APG 1/16 и Selexsorb COS), дополнительно вводили 4,45 кг пропилена и выбранное количество 1-гексена. Количество триэтилалюминия составляло 0,4 мл ТЭА. После добавления триэтилалюминия (0,62 М раствор в н-гептане), используя поток 250 г пропилена, раствор перемешивали при 20°С и 250 об/мин в течение по меньшей мере 20 мин. Затем реактор доводили до заданной температуры предварительной полимеризации (30°С) и впрыскивали катализатор.
Твердый, предварительно полимеризованный катализатор, полученный, как указано выше, загружали в сосуд из нержавеющей стали объемом 5 мл внутри перчаточного бокса. Сосуд присоединяли к автоклаву, затем сверху устанавливали второй сосуд объемом 5 мл, содержащий 4 мл н-гептана и находящийся под давлением 0,1 МПа (10 бар) N2. Выбранное количество Н2 дозировали в реактор через регулятор потока. Выбранное количество гексена и Н2 зависит от заданных конечных свойств компонента (Б), и специалист в данной области техники может легко его подобрать.
Вентиль между двумя сосудами открывали, и твердый катализатор вступал в контакт с гептаном под давлением N2 в течение 2 с, а затем все смывали в реактор с помощью 250 г пропилена. Поддерживали скорость перемешивания 250 об/мин и осуществляли предварительную полимеризацию в течение заданного времени. Теперь температуру полимеризации повышали до 75°С. Температуру реактора поддерживали постоянной на протяжении всей полимеризации. Время полимеризации измеряли, начиная с момента, когда температура была на 2°С ниже заданной температуры полимеризации. По истечении времени полимеризации 60 мин реакцию останавливали путем введения 5 мл этанола, охлаждали реактор и мгновенно испаряли летучие компоненты. После продувки реактора 3 раза N2 и одного цикла вакуум/N2, продукт извлекали и сушили в течение ночи в вытяжном шкафу.
Были получены два сополимера Б1 и Б2 пропилена и гексена, имеющие следующие основные свойства:
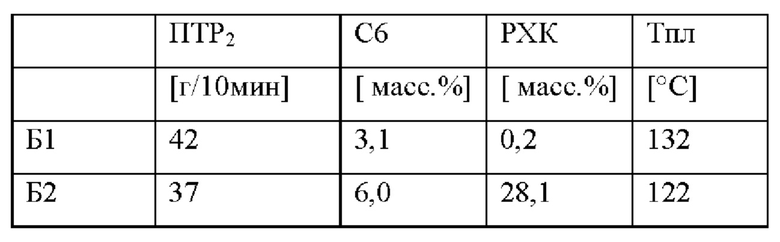
Компонент (А) и компонент (Б) и их смеси перемешивали в двухшнековом экструдере со шнеками, вращающимися в одном направлении, Coperion TSE 16, при 220°C с 0,15 масс. % антиоксиданта (Irganox B215FF otBASF AG, Германия; который представляет собой смесь 1:2 пентаэритрит-тетракис(3-(3',5'-ди-трет-бутил-4-гидроксифенил)-пропионата, номер CAS6683-19-8, и трис(2,4-ди-трет-бутилфенил)фосфита, номер CAS 31570-04-4); 0,05 масс. % стеарата Са (номер CAS1592-23-0, от Faci, Италия).
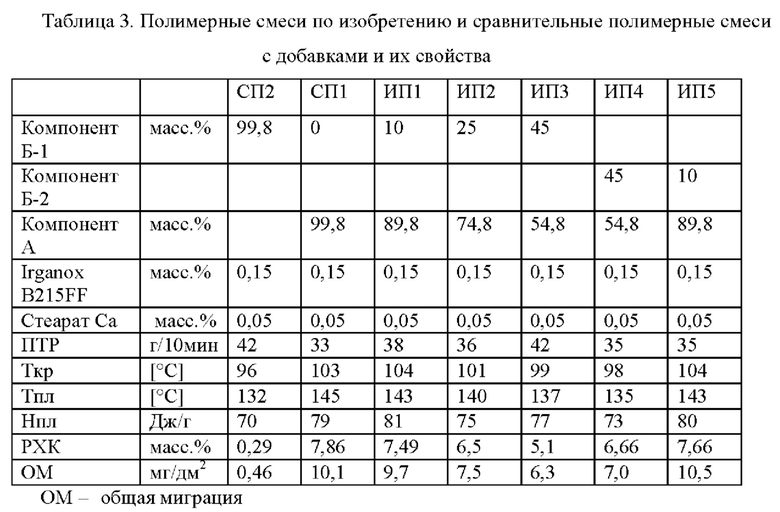
Полимерные смеси по изобретению и сравнительные полимерные смеси перерабатывали в многослойные литые пленки толщиной 50 мкм на линии литья РМ30 (лабораторный экструдер, выпускаемый компанией Plastik Maschinenbau GmbH., Германия). Оборудование состоит из экструдера, охлаждающего валка с воздушным ракелем и намоточной машиной.
ПП шнек с тремя зонами диаметром 30 мм, длиной 25D, экструзионной головкой 200 мм, зазором головки 0,5 мм используют в сочетании с выносной щелевидной головкой с угловым подводящим каналом в сборе.
Использовали следующие параметры экструзии:
Профиль температуры экструдера: 220°С/240°С/250°С/260°С/260°С (температура плавления 250°С; давление плавления 0,61 МПа (61 бар))
Скорость экструдера: 50 об/мин
Температура охлаждающего валка: 20°С
Скорость отрыва: 10,2 м/мин
В таблице 4 приведены оптические параметры (до стерилизации, д.с.), а также характеристики сваривания

Пленки дополнительно стерилизовали паром
Стерилизацию паром осуществляли в машине серии Systec D (Systec Inc., США). Образцы нагревали со скоростью нагрева 5°С/мин, начиная от 23°С. После выдержки в течение 30 мин при температуре 121°С, их сразу извлекали из парового стерилизатора и хранили при комнатной температуре до начала последующей обработки.
Оптические параметры после стерилизации (п.с.) приведены в таблице 5, причем относительное изменение мутности рассчитывали следующим образом:
Δ мутности = (мутность п.с. - мутность д.с.) / мутность д.с. * 100%
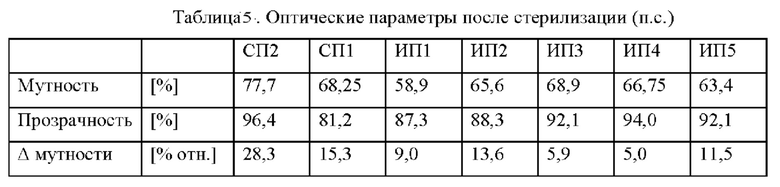
Из приведенных выше таблиц ясно видно, что полимерные смеси по изобретению отличаются преимущественным сочетанием характеристик сваривания, содержания экстрагируемых веществ, оптических свойств и их изменения при стерилизации паром.
| название | год | авторы | номер документа |
|---|---|---|---|
| Полипропиленовая композиция | 2018 |
|
RU2744581C1 |
| Пропиленовая композиция для вспенивания с улучшенными механическими свойствами | 2020 |
|
RU2806522C1 |
| Композиция пропиленового сополимера с превосходными оптическими и механическими свойствами | 2019 |
|
RU2775266C1 |
| НОВОЕ МЕТАЛЛОЦЕНОВОЕ СОЕДИНЕНИЕ, СОДЕРЖАЩАЯ ЕГО КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРОВ НА ОСНОВЕ ОЛЕФИНОВ С ЕЕ ПРИМЕНЕНИЕМ | 2012 |
|
RU2529020C2 |
| Армированная волокном полипропиленовая композиция | 2022 |
|
RU2824348C1 |
| ПОЛИМЕРНАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ПРОПИЛЕНА | 2018 |
|
RU2735731C1 |
| МЕТАЛЛОЦЕНЫ, СПОСОБ СОПОЛИМЕРИЗАЦИИ ПРОПИЛЕНА И ЭТИЛЕНА, СТАТИСТИЧЕСКИЕ СОПОЛИМЕРЫ ПРОПИЛЕНА С ЭТИЛЕНОМ | 2000 |
|
RU2276671C2 |
| Усовершенствованная система катализаторов для получения сополимеров полиэтилена в способе высокотемпературной растворной полимеризации | 2015 |
|
RU2693453C2 |
| Полипропиленовая композиция с низкой усадкой в широком интервале температур применения | 2018 |
|
RU2739399C1 |
| ГЕТЕРОФАЗНЫЙ ПОЛИПРОПИЛЕН СО СТАТИСТИЧЕСКИМ СОПОЛИМЕРОМ ПРОПИЛЕНА И ГЕКСЕНА В КАЧЕСТВЕ МАТРИЦЫ | 2019 |
|
RU2759852C1 |
Изобретение относится к пропиленовым композициям для получения упаковочных материалов и пленок. Предложена полипропиленовая композиция, включающая смесь (А) 50-90 мас.% статистического сополимера пропилена и этилена, имеющего содержание этилена 2-6 мас.%, и (Б) 10-50 мас.% статистического сополимера пропилена и гексена, имеющего содержание гексена 3-7 мас.%, где температура плавления сополимера (Б) меньше температуры плавления сополимера (А). Предложены также способ получения указанной композиции, применение предложенной композиции для получения изделий, выбранных из упаковочных материалов и пленок, и соответствующие изделия. Технический результат - получение материала, обладающего улучшенным балансом между низкой температурой начала сваривания, низкой общей миграцией, хорошими оптическими характеристиками и достаточной термической стойкостью. 4 н. и 10 з.п. ф-лы, 5 табл.
1. Полипропиленовая композиция для получения упаковочных материалов и пленок, включающая смесь
(А) от 50,0 до 90,0 мас.% статистического сополимера пропилена и этилена, имеющего содержание этилена от 2,0 до 6,0 мас.%, ПТР2, измеренное при 230°С и нагрузке 2,16 кг согласно ISO 1133, от 20,0 до 40,0 г/10 мин и температуру плавления Тпл, измеренную посредством ДСК, от 135°С до 155°С, и
(Б) от 10,0 до 50,0 мас.% статистического сополимера пропилена и гексена, имеющего содержание гексена от 3,0 до 7,0 мас.%, ПТР2, измеренное при 230°С и нагрузке 2,16 кг согласно ISO 1133, от 20,0 до 50,0 г/10 мин и температуру плавления Тпл, измеренную посредством ДСК, от 120°С до 140°С, где
(i) температура плавления статистического сополимера (Б) пропилена и гексена меньше, чем температура плавления статистического сополимера (А) пропилена и этилена, и
(ii) ПТР2, измеренное при 230°С и нагрузке 2,16 кг согласно ISO 1133, смеси составляет от 30,0 до 50,0 г/10 мин.
2. Полипропиленовая композиция по п. 1, в которой количество растворимых в холодном ксилоле (РХК) веществ в статистическом сополимере пропилена и этилена составляет от 2,0 до 13,0 мас.%, измеренное в соответствии с ISO 16152, первая редакция, 2005, при 25°С.
3. Полипропиленовая композиция по любому из пп. 1, 2, которая имеет температуру плавления от 128°С до 150°С.
4. Полипропиленовая композиция по любому из пп. 1-3, которая имеет температуру кристаллизации от 85°С до 115°С.
5. Полипропиленовая композиция по любому из предшествующих пунктов, которая имеет величину общей миграции менее 20 мг/дм2, измеренную в соответствии с EN-ISO 116-14:2002 на полученных инжекционным формованием пластинах размером 60×60×1 мм3.
6. Полипропиленовая композиция по любому из предшествующих пунктов, в которой количество растворимой в холодном ксилоле (РХК) фракции составляет от 2,0 до 20,0 мас.%, измеренное в соответствии с ISO 16152, первая редакция, 2005, при 25°С.
7. Способ получения полипропиленовой композиции по любому из предшествующих пунктов, включающий стадии:
(i) получение статистического сополимера (А) пропилена и этилена посредством полимеризации пропилена и этилена в присутствии катализатора Циглера-Натта;
(ii) получение статистического сополимера (Б) пропилена и гексена посредством полимеризации пропилена и гексена в присутствии катализатора с единым центром полимеризации на металле;
(iii) смешивание указанного статистического сополимера (А) пропилена и этилена со статистическим сополимером (Б) пропилена и гексена с получением смешанной массы (А) и (Б) и
(iv) экструдирование указанной смешанной массы с получением смеси (А) и (Б).
8. Применение полипропиленовой композиции по пп. 1-6 для получения изделий, выбранных из упаковочных материалов и пленок.
9. Применение полипропиленовой композиции по п. 8 для получения стерилизуемых изделий, выбранных из упаковочных материалов и пленок.
10. Изделие, включающее полипропиленовую композиции по любому из пп. 1-6, где изделие выбрано из упаковочных материалов и пленок.
11. Изделие по п. 10, включающее неориентированную пленку, содержащую более 90% смеси по любому из предшествующих пп. 1-6, где пленка представляет собой литую пленку или полученную экструзией с раздувом пленку.
12. Изделие по п. 11, в котором пленка имеет температуру начала сваривания, рассчитанную из ДСК, от 90°С до 125°С.
13. Изделие по п. 11 или 12, где пленка была подвергнута обработке стерилизацией.
14. Изделие по любому из пп. 11-13, в котором пленка а) перед обработкой стерилизацией имеет мутность, определяемую на литой пленке толщиной 50 мкм, менее 80,0% и прозрачность, определяемую на литой пленке толщиной 50 мкм, более 90,0% и б) после обработки стерилизацией при температуре 121°С в течение 30 мин имеет величину мутности, определяемую на литой пленке толщиной 50 мкм, менее 80,0% и прозрачность, определяемую на литой пленке толщиной 50 мкм, по меньшей мере 85,0%.
| WO 2017097579 A1, 15.06.2017 | |||
| ТРУБЫ И ПОЛИОЛЕФИНОВАЯ КОМПОЗИЦИЯ ДЛЯ ИХ ИЗГОТОВЛЕНИЯ | 2011 |
|
RU2567750C2 |
| ПОЛИМЕРНАЯ ПЛЕНКА, СОДЕРЖАЩАЯ СТАТИСТИЧЕСКИЙ СОПОЛИМЕР ПРОПИЛЕНА | 2002 |
|
RU2334766C2 |

