Область техники
[0001] Настоящее изобретение относится к соединениям, которые можно применять в качестве ингибиторов протеинкиназ. Настоящее изобретение также обеспечивает фармацевтически приемлемые композиции, содержащие соединения согласно настоящему изобретению и способы применения указанных композиций в лечении различных заболеваний.
Уровень техники
[0002] Поиску новых терапевтических агентов в последние годы в значительной степени способствовало лучшее понимание структуры ферментов и других биомолекул, связанных с заболеваниями. Одним из важных классов ферментов, который интенсивно исследовался, являются протеинкиназы.
[0003] Протеинкиназы составляют обширное семейство структурно близких ферментов, ответственных за регуляцию разнообразных процессов передачи сигнала в клетке. Считается, что протеинкиназы произошли от общего гена-прародителя, поскольку они обладают общностью структуры и каталитической функции. Почти все киназы содержат каталитический домен размером 250-300 аминокислот. Киназы можно разделить на семейства в зависимости от фосфорилируемых ими субстратов (например, протеин-тирозин, протеин-серин/треонин, липидкиназы, и т.д.).
[0004] В целом, протеинкиназы опосредуют внутриклеточную передачи сигнала путем осуществления переноса фосфатной группы с нуклеозид-трифосфата на белок-акцептор, участвующий в передаче сигнала. Указанные события фосфорилирования действуют как молекулярные переключатели типа вкл./выкл., которые могут модулировать или регулировать биологическую функцию белка-мишени. В конечном счете, данные события фосфорилирования запускаются в ответ на внеклеточные и другие стимулы. Примеры таких стимулов включают сигналы из внешней среды и стрессовые сигналы (например, осмотический шок, тепловой шок, ультрафиолетовое
излучение, бактериальный цитотоксин и Н2О2, цитокины (например, интерлейкин-1 (IL-1) и фактор некроза опухолей α (TNF-α)) и факторы роста (например, гранулоцитарно-колониестимулирующий фактор гранулоцитов-макрофагов GM-CSF) и фактор роста фибробластов (FGF)). Внеклеточные стимулы могут влиять на один или более клеточных ответов, связанных с ростом, миграцией, дифференцировкой клеток, секрецией гормонов, активацией факторов транскрипции, сокращением мышц, метаболизмом глюкозы, контролем синтеза белка и регуляцией клеточного цикла.
[0005] Многие заболевания связаны с анормальными клеточными ответами, запускаемыми событиями, опосредуемыми протеинкиназами, как описано выше. Указанные заболевания включают, но не ограничиваются перечисленными: аутоиммунные заболевания, воспалительные заболевания, заболевания костей, заболевания, связанные с метаболизмом, неврологические и нейродегенеративные заболевания, рак, сердечнососудистые заболевания, аллергии и астму, болезнь Альцгеймера и заболевания, связанные с гормонами. Соответственно, сохраняется потребность в поиске ингибиторов протеинкиназ, которые можно применять в качестве терапевтических агентов.
Краткое описание изобретения
[0006] Было обнаружено, что соединения согласно настоящему изобретению и фармацевтически приемлемые композиции, содержащие указанные соединения, могут действовать как ингибиторы одной или более протеинкиназ. Такие соединения имеют общую формулу I-а или I-b:
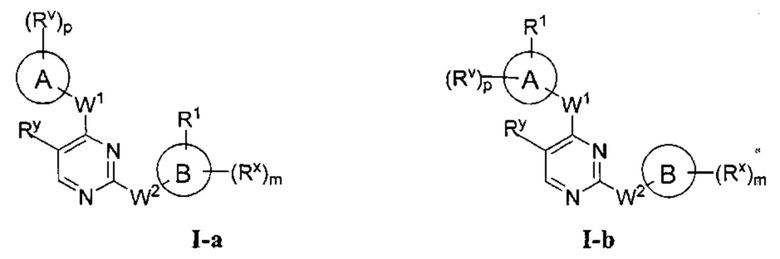
или представляют собой фармацевтически приемлемую соль соединения с такой формулой, при этом Кольцо А, Кольцо В, m, р, Rx, Ry, Rv, W1, W2 и R1 определены в настоящем тексте.
[0007] Соединения согласно настоящему изобретению и фармацевтически приемлемые композиции, содержащие указанные соединения, могут применяться для лечения различных заболеваний, нарушений или состояний, связанных с анормальными клеточными ответами, запускаемыми событиями, опосредуемыми протеинкиназами. Такие заболевания, нарушения или состояния включают заболевания, нарушения или состояния, описанные в настоящем тексте.
[0008] Соединения согласно настоящему изобретению также можно применять для исследования киназ в биологических и патологических явлениях, в исследовании путей внутриклеточной передачи сигнала, опосредуемых такими киназами, и в сравнительной оценке новых ингибиторов киназ.
Краткое описание графических материалов
На Фигуре 1 показана зависимость доза - ответ для ингибирования фосфо-фосфолипазы гамма 2 (p-plc γ 2) соединением I-2 в клетках Ramos; и результаты соединения I-2 в «отсеивающем» эксперименте.
На Фигуре 2 показана зависимость доза - ответ для ингибирования p-plc γ 2 соединением I-4 в клетках Ramos; и результаты соединения I-4в «отсеивающем» эксперименте.
На Фигуре 3 показана зависимость доза - ответ для ингибирования p-plc γ 2 соединением I-7 в клетках Ramos; и результаты соединения I-7в «отсеивающем» эксперименте.
На Фигуре 4 показана зависимость доза - ответ для ингибирования p-plc γ 2 соединением I-35 в клетках Ramos.
На Фигуре 5 показана зависимость доза - ответ для ингибирования p-plc γ 2 соединением I-38 в клетках Ramos.
На Фигуре 6 показаны результаты МС анализа, подтверждающие ковалентную модификацию киназы ТЕС в положении Cys449 под действием соединения I-2.
На Фигуре 7 показаны результаты МС анализа, подтверждающие ковалентную модификацию киназы ТЕС в положении Cys449 под действием соединения I-4.
На Фигуре 8 показаны результаты МС анализа, подтверждающие ковалентную модификацию киназы ТЕС в положении Cys449 под действием соединения I-7.
На Фигуре 9 показаны результаты соединения I-2в «отсеивающем» эксперименте в сравнении с результатами соединения 1-4 и соединения I-7 в том же «отсуивающем» эксперименте в клетках НСС827, содержащих мутантный EGFR с делецией.
На Фигуре 10 показаны результаты соединения I-7в «отсеивающем» эксперименте в сравнении с результатами для контроля с EGF в клетках А431, содержащих EGFR дикого типа.
На Фигуре 11 показаны результаты МС анализа, подтверждающие ковалентную модификацию киназы JAK-3 в положении Cys909 под действием соединения I-7.
На Фигуре 12 показана зависимость доза - ответ для ингибирования P-Stat5 соединением I-2 в клетках CTLL-2, стимулированных IL-2; и зависимость доза - ответ для ингибирования P-JAK-3 соединением I-2 в клетках CTLL-2, стимулированных IL-2.
На Фигуре 13 показана зависимость доза - ответ для ингибирования P-Stat5 соединением I-4 в клетках CTLL-2, стимулированных IL-2; и зависимость доза - ответ для ингибирования P-JAK-3 соединением I-4 в клетках CTLL-2, стимулированных IL-2.
На Фигуре 14 показана зависимость доза - ответ для ингибирования P-Stat5 соединением I-7 в клетках CTLL-2, стимулированных IL-2.
На Фигуре 15 показаны результаты МС анализа, подтверждающие ковалентную модификацию ВТК под действием соединения I-7.
На Фигуре 16 показан Вестерн-блот, демонстрирующий белок ВТК, доступный для соединения-зонда 1-215 после обработки различными количествами I-7.
На Фигуре 17 показан количественный анализ результатов Вестерн-блотинга, показанного на Фигуре 16.
На Фигуре 18 показан Вестерн-блот эксперимента с промывкой с соединением I-7 и соединением-зондом I-215.
На Фигуре 19 показан количественный анализ результатов Вестерн-блотинга, показанного на Фигуре 18.
На Фигуре 20 показана аминокислотная последовательность для ВТК (SEQ ID 1).
На Фигуре 21 показана аминокислотная последовательность для TEC (SEQ ID 2).
На Фигуре 22 показана аминокислотная последовательность для ITK (SEQ ID 3).
На Фигуре 23 показана аминокислотная последовательность для ВМХ (SEQ ID 4).
На Фигуре 24 показана аминокислотная последовательность для ТХК (SEQ ID 5).
На Фигуре 25 показана аминокислотная последовательность для JAK3 (SEQ ID 6).
Подробное описание некоторых вариантов осуществления
1. Общее описание соединений согласно настоящему изобретению
[0009] В некоторых вариантах осуществления настоящее изобретение обеспечивает соединение формулы I-а или I-b:
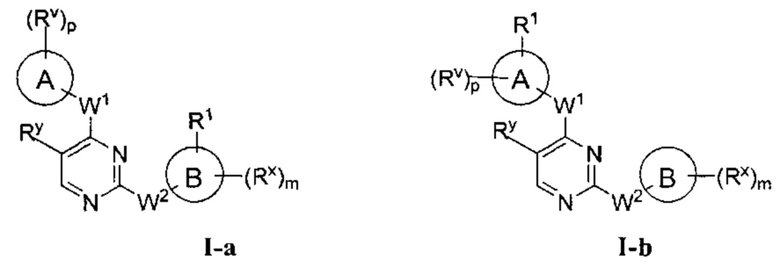
или фармацевтически приемлемую соль такого соединения, где:
Кольцо А представляет собой возможно содержащую заместители группу, выбранную из следующих: фенил, 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 8-10-членное бициклическое насыщенное, частично ненасыщенное или арильное кольцо, 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы, 4-7-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 7-10-членное бициклическое насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 8-10 членное бициклическое гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы;
Кольцо В представляет собой возможно содержащую заместители группу, выбранную из следующих: фенил, 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 8-10-членное бициклическое насыщенное, частично ненасыщенное или арильное кольцо, 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы, 4-7-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и
серы, 7-10-членное бициклическое насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 8-10 членное бициклическое гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы;
R1 представляет собой реакционноспособную группу;
Ry представляет собой водород, галоген, -CN, -CF3, С1-4 алифатическую группу, С1-4 гало-алифатическую группу, -OR, -C(O)R, или -C(O)N(R)2;
каждая группа R независимо представляет собой водород или возможно содержащую заместители группу, выбранную из следующих: С1-6 алифатическую группу, фенил, 4-7-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, независимо выбранные из азота, кислорода и серы и 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы;
W1 и W2 каждый независимо представляет собой ковалентную связь или бивдлентную С1-3 алкиленовую цепь, причем одно метиленовое звено в W1 или W2 возможно заменено на -NR2-, -N(R2)C(O)-, -C(O)N(R2)-, -N(R2)SO2-, -SO2N(R2)-, -O-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO- или -SO2-;
R2 представляет собой водород, возможно содержащую заместители С1-6 алифатическую группу, или -C(O)R, или:
R2 и заместитель на Кольце А, взятые вместе с разделяющими их атомами, образуют 4-6-членное насыщенное, частично ненасыщенное или ароматическое конденсированное кольцо, или:
R2 и Ry, взятые вместе с разделяющими их атомами, образуют 4-7-членное частично ненасыщенное или ароматическое конденсированное кольцо;
m и р независимо равны 0-4; и
Rx и Rv независимо выбраны из: -R, галогена, -OR, -O(CH2)qOR, -CN, -NO2, -SO2R, -SO2N(R)2, -SOR, -C(O)R, -CO2R, -C(O)N(R)2, -NRC(O)R, -NRC(O)NR2, -NRSO2R, или -N(R)2, причем q равно 1-4; или:
Rx и R1, если они одновременно присутствуют на Кольце В, взятые вместе с разделяющими их атомами, образуют 5-7-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо
выбранные из азота, кислорода и серы, причем указанное кольцо содержит в качестве заместителя реакционноспособную группу и 0-3 группы, независимо выбранные из оксо, галогена, -CN, или C1-6 алифатической группы; или
Rv и R1, если они одновременно присутствуют на Кольце А, взятые вместе с разделяющими их атомами, образуют 5-7-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, причем указанное кольцо содержит в качестве заместителя реакционноспособную группу и 0-3 группы, независимо выбранные из оксо, галогена, -CN, или C1-6 алифатической группы.
2. Соединения и определения
[0010] Соединения, составляющие суть этого изобретения, включают кратко описанные выше, и подробно описанные ниже с делением на классы, подклассы и виды. Данные ниже определения будут применяться во всех случаях, когда не указано иное. Для целей настоящего изобретения химические элементы идентифицировались в соответствии с Периодической таблицей элементов, CAS-номенклатурой, «Справочником по химии и физике» (75-я ред.). Кроме того, общие принципы органической химии описаны в следующих изданиях: "Organic Chemistry", Thomas SorRell, University Science Books, Sausalito: 1999; «March's Advanced Organic Chemistry», 5th Ed., Ed.: Smith, M.B. & March, J., John Wiley & Sons, New York: 2001, вследствие чего оба издания полностью включены в список источников.
[0011] Термин "алифатический" или "алифатическая группа", используемый здесь, подразумевает углеводороды с линейной (т.е. неразветвленной) или разветвленной структурой, замещенные или незамещенные углеводородные цепи, полностью насыщенные или содержащие одну/или более одной ненасыщенной связи; моноциклические или бициклические углеводороды, полностью насыщенные или содержащие одну/более одной ненасыщенной связи, но не являющиеся ароматическими (также называемыми здесь "карбоциклами" "циклоалифатическими углеводородами" или "циклоалкилами") с одной точкой крепления к остальной части молекулы. Если не указано иное, алифатические группы содержат 1-6 алифатических атомов углерода. В некоторых вариантах осуществления алифатические группы содержат 1-5 алифатических атомов углерода. В других вариантах осуществления алифатические группы содержат 1-4
алифатических атомов углерода; есть варианты осуществления с 1-3 алифатических атомов углерода; и также с 1-2 алифатическими атомами углерода. В некоторых вариантах осуществления термин "циклоалифатический" ("карбоцикл"/"циклоалкил") подразумевает моноциклический С3-С6 углеводород, полностью насыщенный или содержащий по крайней мере один ненасыщенный участок, но не ароматический, имеющий одну точку крепления к остальной части молекулы. Подходящие алифатические группы представлены (но не ограничиваются) следующими вариантами: линейные или разветвленные, замещенные или незамещенные алкилы, алкенилы, алкинилы и смешанные варианты: (циклоалкил)алкилы, (циклоалкенил)алкилы или (циклоалкил)алкенилы.
[0012] Термин "низший алкил" относится к прямой или разветвленной С1-4-алкильной группе. Примерами низших алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил и трет-бутил.
[0013] Термин "низший галоидалкил" относится к прямой или разветвленной C1-4-алкильной группе, замещенной одним или несколькими атомами галогена
[0014] Термин "гетероатом" означает одну/более одной единицы кислорода, серы, азота, фосфора, кремния (в том числе любые окисленные формы азота, серы, фосфора, кремния; или кватернизованная форма любого азотистого основания; или замещенная азотная группа гетероциклического кольца, например N (как в 3,4-дигидро-2Н-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенных пирролидинила)).
[0015] Термин "ненасыщенный", используемый здесь, означает, что соединение содержит по крайней мере одну ненасыщенную связь.
[0016] В настоящем описании термин "двухвалентная С1-8 (или С1-6) насыщенная или ненасыщенная, линейная или разветвленная углеводородная цепь" подразумевает двухвалентные алкиленовые, алкениленовые, алкиниленовые цепи, прямые или разветвленные согласно данному здесь определению.
[0017] Термин "алкилен" относится к двухвалентной алкильной группе. "Алкиленовая цепь" является полиметиленовой группой, т.е., -(СН2)n-, где N - натуральное число, предпочтительно от 1 до 6, от 1 до 4, от 1 до 3, от 1 до 2, или от 2 до 3.
Замещенная алкиленовая цепь - полиметиленовая группа, в которой один или несколько атомов водорода метиленовой группы замещены. Подходящие заместители включают описанные ниже для замещенных алифатических групп.
[0018] Термин "алкенилен" относится к двухвалентной алкениловой группе. Замещенная алкениленовая цепь - это полиметиленовая группа, содержащая по крайней мере одну двойную связь, в которой один или несколько атомов водорода замещены. Подходящие заместители включают описанные ниже для замещенных алифатических групп.
[0019] В настоящем описании термин "циклопропиленил" подразумевает двухвалентный циклопропил следующей структуры: 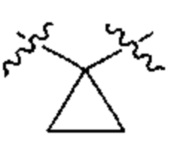
[0020] Термин "галоген"означает F, Cl, Br, или I.
[0021] Термин "арил", используемый самостоятельно или при описании фрагментов молекул, как "аралкил", "аралкокси-", или "арилоксиалкил", относится к моноциклическим и бициклическим кольцевым структурам, имеющим обычно 5-14-членные кольца, и в которых по крайней мере одно кольцо является ароматических, и каждое кольцо в системе включает 3-7 членов. Термин "арил" может использоваться наравне с термином "арильное кольцо". В некоторых вариантах изобретения понятие "арил" относится к ароматическим циклам, включающим следующие группы (но не ограниченным ими): фенил, дифенил, нафтил, антрацил и т.п., и которые могут быть однократно или многократно замещены. Также в понятие "арил", как оно понимается в настоящем документе, входит группа, в которой ароматическое кольцо сопряжено по крайней мере с одним неароматическим: инданил, флалимидил, нафтимидил, фенантридинил или тетрагидронафтидил, и т.п.
[0022] Термины "гетероарил" и "гетероар-" используемые самостоятельно или при описании фрагментов молекул, - например, "гетероаралкил", или "гетероаралкокси", - относятся к группам с 5-10 атомами в кольце (преимущественно 5, 6, 9), имеющих π-электронную систему из 6, 10, или 14 обобществленных электронов, и от одного до пяти гетероатомов, помимо атомов углерода. Термин "гетероатом" подразумевает азот, кислород и серу, а также любые окисленные формы азота или серы, и любые кватернизованные формы азотистых оснований. Гетероарильные группы включают,
помимо прочего, тиенил, фуранил, пирролил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, оксадиазолил, тиазолил, изотиазолил, тиадиазолил, пиридил, пиридазинил, пиримидинил, пиразинил, индолихинил, пуринил, нафтиридинил и птеридинил. В используемые здесь понятия "гетероарил" и "гетероар-" также входят группы, в которых гетероароматическое кольцо соединено с одним или более арильным, циклоалифатическим, или гетероциклическим кольцом, где радикал или точка крепления расположены на гетероароматическом кольце. Многочисленные примеры включают индолил, изоиндолил, бензотиенил, бензофуранил, дибензофуранил, индазолил, бензимидазолил, бензотиазолил, хинолил, изохинолил, циннолинил, фталазинил, хиназолинил, хиноксалинил, 4Н-хинолизинил, карбазолил, акридинил, феназинил, фенотиазинил, феноксазинил, тетрагидрохинолинил, тетрагидроизохинолинил и пиридо-[2,3-b]-1,4-оксазин-3(4Н)-он. Гетероарильная группа может быть моно- или бициклической. Термин "гетероарил" может использоваться наравне с терминами "гетероарильное кольцо", "гетероарильная группа", или "гетероароматический"; во всех этих случаях подразумеваются циклические соединения, которые могут быть замещенными или незамещенными. Термин "гетероаралкил" относится к алкильной группе, замещенной гетероарилом, при этом алкильные и гетероарильные фрагменты независимо и необязательно замещены.
[0023] В настоящем описании термин "гетероцикл", "гетероциклил", "гетероциклический радикал", и "гетероциклическое кольцо" взаимозаменяемы и относятся к стабильным 5-7-членным моноциклическим или 7-10-членным бициклическим гетероциклическим фрагментам, насыщенным или частично ненасыщенным, и включающим, помимо атомов углерода, по крайней мере один и преимущественно 1-4 гетероатома, в том смысле, как это определено выше. При упоминании атома, входящего в состав кольца гетероцикла, термин "азот" подразумевает в том числе замещенный азот. Например, в насыщенном или частично ненасыщенным кольце, включающем 0-3 гетероатома кислорода, серы или азота, азот может быть в форме N (как в 3,4-дигидро-2Н-пирролиле), или -NH (как в пирролидиниле), или +NR (как в N-замещенных пирролидинилах).
[0024] Гетероциклическое кольцо может присоединяться к боковой группе через любой гетероатом или атом углерода, что приводит к формированию стабильной
структуры; любой атом кольца может быть замещен. Примерами таких насыщенных или частично ненасыщенных гетероциклических радикалов могут быть, в том числе, тетрагидрофуранил, тетрагидротиофенил пирролидинил, пиперидинил, пирролинил, тетрагидрохинолинил, тетрагидроизохинолинил, деканидрохинолинил, оксазолидинил, пиперазинил, диоксанил, диоксоланил, диазепинил, оксазепинил, тиазепинил, морфолинил и хинуклидинил. Термины "гетероцикл", "гетероциклил", "гетероциклильное кольцо", "гетероциклические группы", "гетероциклический фрагмент" и "гетероциклический радикал" являются взаимозаменяемыми в настоящем документе, и включают также группы, в которых гетероциклильное кольцо соединено по крайней мере с одним арильным, гетероарильным или циклоалифатическим кольцом, например, индолинильным, 3Н-индолильным, хроманильным, фенантридинильным или тетрагидрохинолинильным, при этом радикал или точка крепления расположены на гетероциклильном кольце. Гетероциклильные группы могут быть моно- или бициклическими. Термин "гетероциклалкил" относится к гетероциклил-замещенной алкильной группе, в которой алкильная и гетероциклильная составляющие независимо друг от друга и необязательно замещены.
[0025] В настоящем описании термин "частично ненасыщенный" относится к циклическим соединениям, которые содержат по крайней мере одну двойную или тройную связь. Термин "частично ненасыщенный" охватывает циклы, имеющие несколько ненасыщенных участков, но не включает арильные или гетероарильные соединения в том смысле, в котором они определены здесь.
[0026] Согласно настоящему документу, соединения, являющиеся предметом настоящего изобретения, могут содержать «опционально замещенные» фрагменты. В целом, термин «замещенный», предварен он термином «опционально» или нет, означает, что по крайней мере один атом водорода означенного фрагмента синтезируемого соединения замещен подходящей группой;. Если не указано иное, «опционально замещенный» фрагмент может быть многократно замещен подходящими группами в каждой из возможных для замещения позиций; при этом, если замещение происходит более чем в одной позиции, и существует несколько подходящих замещающих групп, то эти замещающие группы могут быть как одинаковыми, так и разными. Настоящим изобретением предусмотрены комбинации заместителей,
предпочтительно приводящие к формированию стабильных или химически реализуемых соединений. Термин «стабильный», используемый здесь, относится к соединениям, которые не претерпевают существенных изменений в условиях, необходимых для их производства, определения и, - в некоторых вариантах - осуществления, восстановления, очистки и использования для одной или более описанных здесь целей.
[0027] Подходящие моновалентные заместители атома углерода "возможно замещенной" (возможно содержащей заместители) группы представлены независимо выбранными: галогенами, -(CH2)0-4Ro; -(CH2)0-4ORo; -O(CH2)0-4Ro, -O-(CH2)0-4C(O)ORo; -(CH2)0-4CH(ORo)2; -(CH2)0-4SRo; -(CH2)0-4Ph, который может быть замещен на Ro; -(CH2)0-4O(CH2)0-1Ph, который может быть замещен на Ro; -CH=CHPh, который может быть замещен на Ro; -(СН2)0-4О(СН2)0-1-пиридил, который может быть замещен на Ro; -NO2; -CN; -N3; -(CH2)0-4N(Ro)2; -(CH2)0-4N(Ro)C(O)Ro; -N(Ro)C(S)Ro; -(CH2)0-4N(Ro)C(O)NRo2; -N(Ro)C(S)NRo2; -(CH2)0-4N(Ro)C(O)ORo; -N(Ro)N(Ro)C(O)Ro; -N(Ro)N(Ro)C(O)NRo2; -N(Ro)N(Ro)C(O)ORo; -(CH2)0-4C(O)Ro; -C(S)Ro; -(CH2)0-4C(O)ORo; -(CH2)0-4C(O)SRo; -(CH2)0-4C(O)OSiRo3; -(CH2)0-4OC(O)Ro; -OC(O)(CH2)0-4SRo, SC(S)SRo; -(CH2)0-4SC(O)Ro; -(CH2)0-4C(O)NRo2; -C(S)NRo2; -C(S)SRo; -SC(S)SRo, -(CH2)0-4OC(O)NRo2; -C(O)N(ORo)Ro; -C(O)C(O)Ro; -C(O)CH2C(O)Ro; -C(NORo)Ro; -(CH2)0-4SSRo; -(CH2)0-4S(O)2Ro; -(CH2)0-4S(O)2ORo; -(CH2)0-4OS(O)2Ro; -S(O)2NRo2; -(CH2)0-4S(O)Ro; -N(Ro)S(O)2NRo2; -N(Ro)S(O)2Ro; -N(ORo)Ro; -C(NH)NRo2; -P(O)2Ro; -P(O)Ro2; -OP(O)Ro2; -OP(O)(ORo)2; SiRo3; -(C1-4, линейный или разветвленный алкилен)O-N(Ro)2; или -(C1-4 линейный или разветвленный алкилен)С(O)O-N(Ro)2, где каждый Ro может быть замещен, как описано ниже, и представлен независимо выбранными: водородом, C1-6 алифатическим углеводородом, -CH2Ph, -O(CH2)0-1Ph, -СН2-(5-6-членным гетероарильным кольцом), или 5-6-членным насыщенным/частично ненасыщенным/арильным кольцом с 0-4 гетероатомами, независимо скомбинированными из следующих возможных: азот, кислород, сера; или, несмотря на определение выше, два независимых появления Ro вместе с промежуточным(и) атомом(ами) образуют 3-12-членное насыщенное/частично ненасыщенное арильное моно- или бициклическое кольцо с 0-4 гетероатомами, независимо выбранными из следующих возможных: азот, кислород, сера, которые могут быть замещены, как описано ниже.
[0028] Подходящие моновалентные заместители Ro (либо кольца, образованного двумя независимыми Ro вместе с промежуточными атомами), представлены независимо выбранными группами из следующих возможных: галогены, -(CH2)0-2R•, -(галоR•), -(СН2)0-2OH, -(CH2)0-2OR•, -(CH2)0-2CH(OR•)2; -O(галоR•), -CN, -N3, -(CH2)0-2C(O)R•, -(CH2)0-2C(O)OH, -(CH2)0-2C(O)OR•, -(CH2)0-2SR•, -(CH2)0-2SH, -(CH2)0-2NH2, -(CH2)0-2NHR•, -(CH2)0-2NR•2, -NO2, -SiR•3, -OSiR•3, -C(O)SR•, -(C1-4 линейный или разветвленный алкилен)C(O)OR•, или -SSR•, где каждый R• незамещен либо, в случае уточнения "гало-", замещен одним или несколькими атомами галогена и независимо выбран из: С1-4 алифатической группы, -CH2Ph, -O(CH2)1-1Ph, или 5-6-членного насыщенного/частичного ненасыщенного/ арильного кольца с 0-4 гетероатомами, независимо выбранными из следующих: азот, кислород, сера. Подходящие бивалентные заместители насыщенного углерода: =O и =S.
[0029] Подходящие бивалентные заместители насыщенного атома углерода «возможно замещенной» группы включают: =O, =S, =NNR*2, =NNHC(O)R*, =NNHC(O)OR*, =NNHS(O)2R*, =NR*, =NOR*, -O(C(R*2))2-3O- или -S(C(R*2))2-3S- где R* независимо в каждом случае представлен: водородом, C1-6 алифатическим углеводородом, замещенным, как описано выше, или незамещенным 5-6-членным насыщенным/частично ненасыщенным/арильным кольцом с 0-4 гетероатомами, независимо выбранными из азота, кислорода и серы. Подходящие бивалентные заместители, способные связываться с соседними замещаемыми атомами углерода «возможно замещенной» группы, представлены следующими вариантами: -O(CR*2)2-3O-, где в каждом случае R* представлен следующими возможными вариантами: водород, C1-6 алифатическая цепь, незамещенная или замещенная, как описано ниже, или 5-6-членное насыщенное/частично ненасыщенное арильное кольцо с 0-4 гетероатомами, независимо выбранными из следующих: азот, кислород, сера.
[0030] Подходящие заместители алифатической группы R*: галогены, -R•, -(галоR•), -ОН, -OR•, -O(галоR•), -CN, -С(O)ОН, -C(O)OR•, -NH2, -NHR•, -NR•2, или - NO2, где каждый R• замещен либо, в случае уточнения «гало-», замещен исключительно одним или несколькими атомами галогена, и независимо выбран из групп: С1-4 алифатическая цепь, -CH2Ph, -O(CH2)0-1Ph, или 5-6-членное насыщенное/частично
ненасыщенное арильное кольцо с 0-4 гетероатомами, независимо выбранными из следующих: азот, кислород, сера.
[0031] Подходящие заместители азота в "возможно замещенной" группе включают: 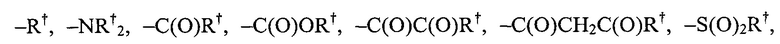
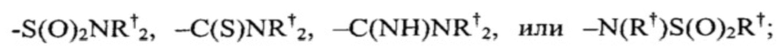 где каждый
где каждый 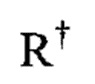 независимо представлен следующими возможными вариантами: водород, С1-6 алифатическая цепь, незамещенная/замещенная, как описано ниже; незамещенный -OPh, или незамещенное 5-6-членное насыщенное/частично ненасыщенное арильное кольцо с 0-4 гетероатомами, независимо выбранными из следующих: азот, кислород, сера; либо, несмотря на данное выше определение, два независимых
независимо представлен следующими возможными вариантами: водород, С1-6 алифатическая цепь, незамещенная/замещенная, как описано ниже; незамещенный -OPh, или незамещенное 5-6-членное насыщенное/частично ненасыщенное арильное кольцо с 0-4 гетероатомами, независимо выбранными из следующих: азот, кислород, сера; либо, несмотря на данное выше определение, два независимых 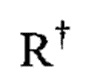 , вместе с промежуточным(и) атомом(амии) образуют незамещенное 3-12-членное насыщенное, частично ненасыщенное или арильное моно- или бициклическое кольцо 0-4 гетероатомами, независимо выбранными из азота, кислорода и серы.
, вместе с промежуточным(и) атомом(амии) образуют незамещенное 3-12-членное насыщенное, частично ненасыщенное или арильное моно- или бициклическое кольцо 0-4 гетероатомами, независимо выбранными из азота, кислорода и серы.
[0032] Подходящие заместители для алифатической группы 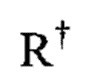 представлены независимо выбираемыми вариантами: галоген, -R•, -(галоR•), -ОН, -OR•, -O(галоR•), -CN, -С(O)ОН, -C(O)OR•, -NH2, -NHR•, -NR•2, или -NO2, где каждый R• является незамещенным или, в случае приставки "гало", замещен одним или более атомами галогена, и представлен независимыми вариантами: C1-4 алифатическая группа, -CH2Ph, -O(CH2)1-4Ph, или 5-6-членное насыщенное/частично ненасыщенное/арильное кольцо с 0-4 гетероатомами, независимо выбранными из следующих возможных: азот, кислород, сера.
представлены независимо выбираемыми вариантами: галоген, -R•, -(галоR•), -ОН, -OR•, -O(галоR•), -CN, -С(O)ОН, -C(O)OR•, -NH2, -NHR•, -NR•2, или -NO2, где каждый R• является незамещенным или, в случае приставки "гало", замещен одним или более атомами галогена, и представлен независимыми вариантами: C1-4 алифатическая группа, -CH2Ph, -O(CH2)1-4Ph, или 5-6-членное насыщенное/частично ненасыщенное/арильное кольцо с 0-4 гетероатомами, независимо выбранными из следующих возможных: азот, кислород, сера.
[0033] В настоящем описании термин "фармацевтически приемлемая соль" относится к тем солям, которые, согласно медицинскому заключению, могут быть использованы в контакте с тканями человека и животных: не являющиеся неоправданно токсичными, вызывающими раздражение, аллергические реакции и т.п., риск от использования которых соизмерим с ожидаемой пользой. Фармацевтически приемлемые соли хорошо известны. Например, такие соли описаны в работе S.М. Berge et al. (J. Pharmaceutical Sciences, 1977, 66, 1-19), включенной в список источников настоящего исследования. Фармацевтически приемлемые соли соединений, составляющих предмет данного изобретения, включают производные от соответствующих неорганических и органических кислот и оснований. Примеры фармацевтически приемлемых, нетоксичных
кислотно-аддитивных солей - аминовые соли, полученные посредством добавления неорганических кислот, таких, как соляная, бромистоводородная, фосфорная, серная и хлорная кислоты; или путем добавления органических кислот, таких как уксусная, щавелевая, малеиновая, винная, лимонная, янтарная, малоновая; или с помощью других методов, используемых в данной области, например, ионообмена. Другие фармацевтически приемлемые соли включают: адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, хемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталенсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, р-толуолсульфонат, ундеканоат, валерат и т.п.
[0034] Соли, полученные из соответствующих оснований, включают соли щелочных металлов, щелочноземельных металлов, аммонийные соли и N+(С1-4алкил)4-соли. Типичные соли щелочных и щелочноземельных металлов включают натриевые, литиевые, калийные, кальциевые, магниевые соли и т.п. Также фармацевтически приемлемыми, при необходимости, являются нетоксичные аммонийные соли, четвертичные соли аммония, соли аминовых катионов, получаемые с использованием противоионов - галогенидов, гидроксидов, карбоновой кислоты, сульфатов, фосфатов, нитратов, сульфаната низших алкилов и арил-сульфаната.
[0035] Если не указано иное, описанные здесь структуры включают также все изомерические формы данных структур (такие, как энантиомеры, дйастереомеры, геометрические (конформационные) изомеры); например, R- и S- оптические изомеры для каждого центра асимметрии, Z- и Е- дйастереомеры с разными вариантами расположения заместителей относительно плоскости двойной связи, Z- и Е-конформационные изомеры. Таким образом, изобретение включает как простые стереоизомеры, так и энантиомеры, диастереомеры и геометрические (конформационные) настоящих соединений в различных сочетаниях. Если не указано иное, все таутомеры описанных соединений также относятся к данному изобретению. Кроме того, если не указано иное, к структурам, описанным
выше, также относятся соединения, отличающиеся только наличием одного или более атома, обогащенного по изотопу. Например, соединения, имеющие описанную структуру и содержащие водород в форме дейтерия или трития; либо обогащенные 13С- или 14С-углеродом, также включены в данное изобретение. Такие соединения можно применять, например, в качестве аналитических инструментов, в качестве зондов для биологических исследований или в качестве терапевтических агентов в соответствии с настоящим изобретением. В некоторых вариантах осуществления группа R1 в формулах Ia и Ib включает один или несколько атомов дейтерия.
[00361 В настоящем описании термин "необратимый" или "необратимый ингибитор" относится к ингибиторам (т.е. соединениям), способным ковалентно связываться с протеинкиназой-мишенью по существу необратимо. В то время как обратимый ингибитор способен связываться с протеинкиназой-мишенью, но, как правило, ковалентной связью, и, следовательно, способен в дальнейшем отделиться, необратимый ингибитор после того, как произошло формирование ковалентной связи, прочно сцеплен с киназой-мишенью. Необратимые ингибиторы обычно демонстрируют временную зависимость: прямо пропорциональную зависимость эффективности ингибирования от времени контакта ингибитора с ферментом. Методы выявления необратимых ингибиторов хорошо известны специалистам. Такие методы включают, например: кинетический анализ активности целевой протеинкиназы в присутствии ингибитора, масс-спектрометрическое исследование целевого белка в присутствии ингибитора, дискретная экспозиция ("отмывание"), использование радиомеченых ингибиторов, позволяющих детектировать ковалентные модификации фермента, а также другие известные специалистам способы.
[0037] Для специалиста очевидно, что определенные функциональные группы могут выступать в качестве реакционноспособных групп ("боеголовок"). В настоящем описании термин "реакционноспособная группа" относится к функциональной группе, присутствующей в соединении согласно настоящему изобретению и способной ковалентно связываться с аминокислотными остатками (например, с остатками цистеина, лизина, гистидина или другими, способными к ковалентной модификации), присутствующими в связывающем кармане белка-мишени, и способной тем самым необратимо ингибировать белок. Следует иметь в виду, что группа -L-Y, в том смысле, в
каком она определена и описана в настоящем документе, включает «реакционноспособные группы», ковалентно и необратимо инактивирующие белок.
[0038] В настоящем описании термин "ингибитор" определяется как соединение, которое связывается с и/или ингибирует целевую протеинкиназу с достаточным сродством. В некоторых вариантах осуществления ингибитор имеет IC50 и/или константу связывания менее 50 мкМ, менее 1 мкМ, менее 500 нм, менее 100 нм, или менее 10 нм.
[0039] Термины "достаточное сродство" и "эффективное ингибирование", используемые здесь, подразумевают регистрируемые изменения активности по крайней мере для одной из следующих киназ: ErbB1, ErbB2, ErbB3, ERBB4, ТЕС, и/или JAK3 при сравнении образцов, содержащих соединение-предмет настоящего изобретения, или его компоненты плюс по крайней мере одну киназу из перечисленных: ErbB1, ErbB2, ErbB3, ERBB4, ТЕС и/или JAK3, и эквивалентный образец, содержащий по крайней мере одну киназу из перечисленных: ErbB1, ErbB2, ErbB3, ERBB4, ТЕС и/или JAK3 в отсутствие указанного соединения или его компонентов.
3. Описание примеров соединений
[0040] Согласно одному аспекту настоящее изобретение обеспечивает соединение формулы 1-а или 1-b,
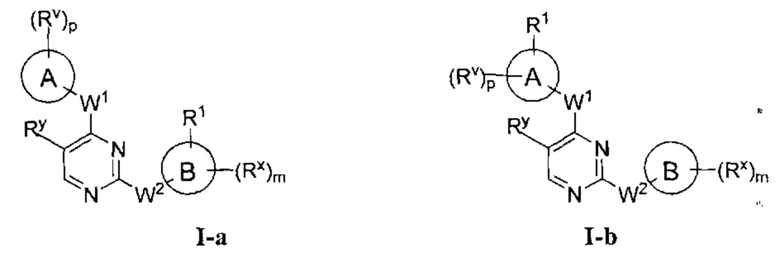
или фармацевтически приемлемую соль такого соединения, где:
Кольцо А представляет собой возможно содержащую заместители группу, выбранную из следующих: фенил, 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 8-10-членное бициклическое насыщенное, частично ненасыщенное или арильное кольцо, 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы, 4-7-членное насыщенное или частично ненасыщенное гетероциклическое
кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 7-10-членное бициклическое насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 8-10 членное бициклическое гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы;
Кольцо В представляет собой возможно содержащую заместители группу, выбранную из следующих: фенил, 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 8-10-членное бициклическое насыщенное, частично ненасыщенное или арильное кольцо, 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы, 4-7-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 7-10-членное бициклическое насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 8-10 членное бициклическое гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы;
R1 представляет собой -L-Y, где:
L представляет собой ковалентную связь или бивалентную C1-8 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь, причем одно, два или три метиленовых звена в L возможно и независимо заменены на циклопропилен, -NR-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2-, -SO2N(R)-, -O-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO-, -SO2-, -C(=S)-, -C(=NR)-, -N=N- или -C(=N2);
Y представляет собой водород, C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген или CN, или 3-10-членное моноциклическое или бициклическое насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, и при этом указанное кольцо содержит в качестве заместителей 1-4 группами, независимо выбранными из -Q-Z, оксо, NO2, галогена, CN, или С1-6 алифатическую группу, где:
Q представляет собой ковалентную связь или бивалентную С1-6 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь,
причем одно или два метиленовых звена в Q возможно и независимо заменены на -NR-, -S-, -О-, -С(О)-, -SO-, или -SO2-; и
Z представляет собой водород или С1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген или CN;
Ry представляет собой водород, галоген, -CN, -CF3, С1-4 алифатическую группу, С1-4 гало-алифатическую группу, -OR, -C(O)R или -C(O)N(R)2;
каждая группа R независимо представляет собой водород или возможно содержащую заместители группу, выбранную из следующих: C1-6 алифатическая группа, фенил, 4-7-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, независимо выбранные из азота, кислорода и серы и 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы;
W1 и W2 каждый независимо представляет собой ковалентную связь или бивалентную С1-3 алкиленовую цепь, причем одно метиленовое звено в W1 или W2 возможно заменено на -NR2-, -N(R2)C(O)-, -C(O)N(R2)-, -N(R2)SO2-, -SO2N(R2)-, -O-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO- или -SO2-;
R2 представляет собой водород, возможно содержащую заместители С1-6 алифатическую группу, или -C(O)R, или:
R2 и заместитель на Кольце А, взятые вместе с разделяющими из атомами, образуют 4-6-членное частично ненасыщенное или ароматическое конденсированное кольцо; или
R2 и Ry, взятые вместе с разделяющими их атомами, образуют; 4-6-членное насыщенное частично ненасыщенное или ароматическое конденсированное кольцо;
m и р независимо равны 0-4; и
Rx и Rv независимо выбраны из: -R, галогена, -OR, -O(CH2)qOR, -CN, -NO2, -SO2R, -SO2N(R)2, -SOR, -C(O)R, -CO2R, -C(O)N(R)2, -NRC(O)R, -NRC(O)NR2, -NRSO2R или -N(R)2, или;
Rx и R1, если они одновременно присутствуют на Кольце В, взятые вместе с разделяющими их атомами, образуют 5-7-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо
выбранных из азота, кислорода и серы, причем указанное кольцо содержит в качестве заместителя реакционноспособную группу и 0-3 группы, независимо выбранные из оксо, галогена, -CN или C1-6 алифатической группы; или
Rv и R1, если они одновременно присутствуют на Кольце А, взятые вместе с разделяющими их атомами, образуют 5-7-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, причем указанное кольцо содержит в качестве заместителя реакционноспособную группу и 0-3 группы, независимо выбранные из оксо, галогена, -CN, или C1-6 алифатическую группу.
[0041] Как определено в общем выше, Кольцо А представляет собой возможно содержащую заместители группу, выбранную из следующих: фенил, 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 8-10-членное бициклическое насыщенное, частично ненасыщенное или арильное кольцо, 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы, 4-7-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 7-10-членное бициклическое насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 8-10 членное бициклическое гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы. В некоторых вариантах осуществления Кольцо А представляет собой возможно замещенную фенильную группу. В некоторых вариантах осуществления Кольцо А представляет собой возможно замещенное нафтильное кольцо или бициклическое 8-10 членное гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы. В некоторых других вариантах осуществления Кольцо а А представляет собой возможно замещенное 3-7-членное карбоциклическое кольцо. В других вариантах осуществления Кольцо А представляет собой возможно замещенное 4-7-членное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы.
[0042] В некоторых вариантах осуществления Кольцо А замещено, как описано в настоящем тексте. В некоторых вариантах осуществления Кольцо А замещено одной,
двумя или тремя группами, выбранными из галогена, Ro, или -(CH2)0-4ORo или -O(СН2)0-4Ro, где каждый Ro определен в настоящем описании. Примеры заместителей на Кольце А включают Br, I, Cl, метил, -CF3, -С≡СН, -ОСН2фенил, -ОСН2(фторфенил), или -ОСН2пиридил.
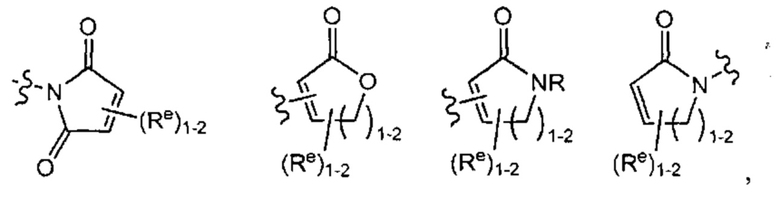
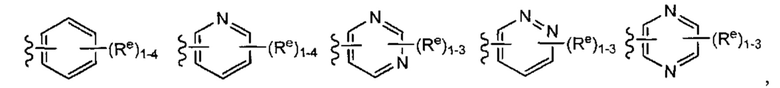
[0043] Примеры групп, представляющих Кольцо А, приведены в Таблице 1.
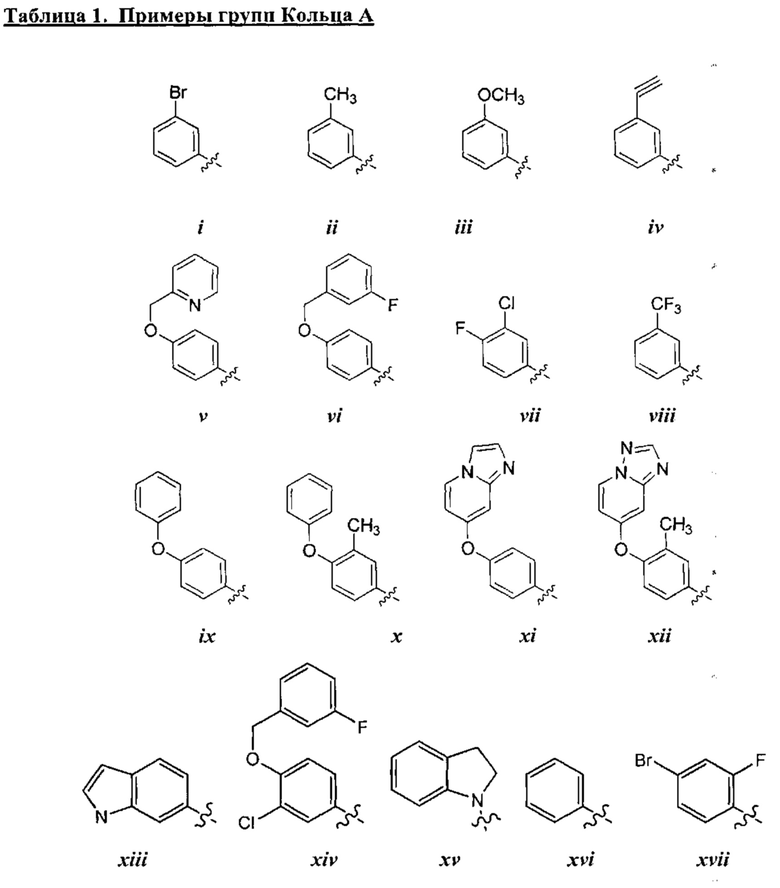
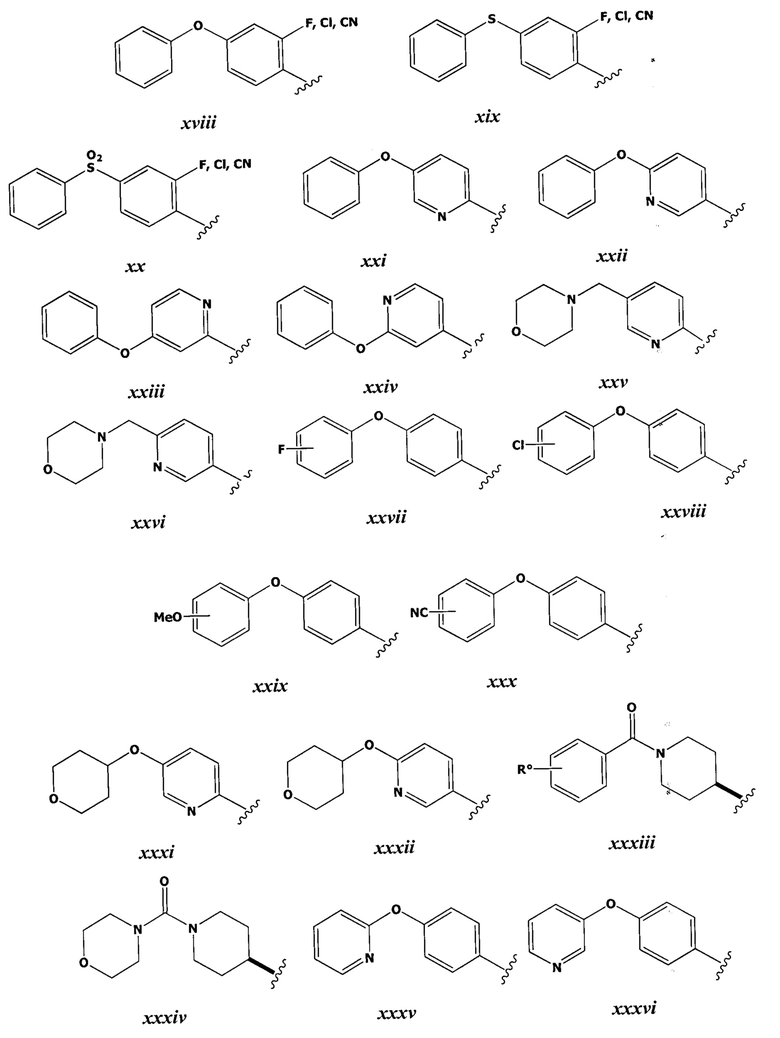
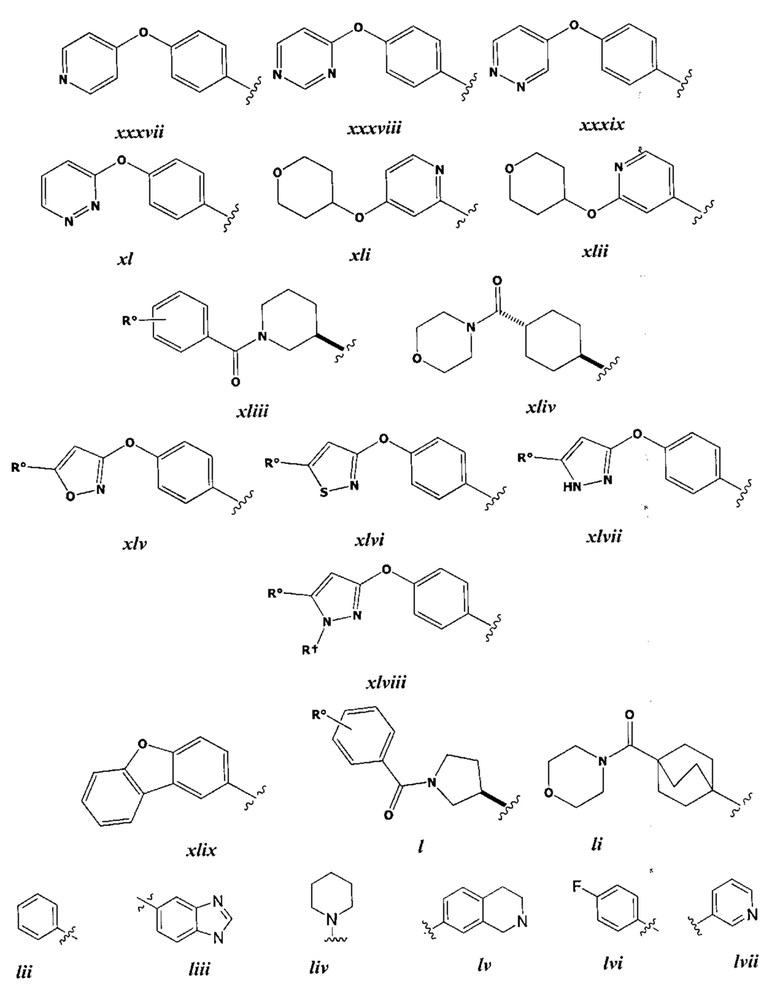
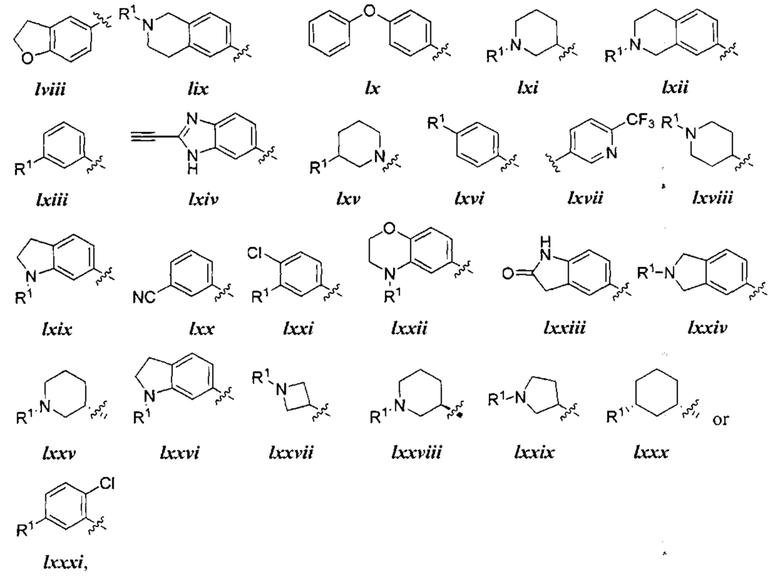
причем каждый Ro,  и R1 имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
и R1 имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
[0044] В некоторых вариантах осуществления Кольцо А выбрано из i, ii, iv, v, vi, vii, ix, xiv, xvi, 
[0045] Как определено в общем выше, Кольцо В представляет собой возможно содержащую заместители группу, выбранную из следующих: фенил, 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 8-10-членное бициклическое насыщенное, частично ненасыщенное или арильное кольцо, 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы, 4-7-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 7-10-членное бициклическое насыщенное или частично ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, 8-10 членное бициклическое
гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы. В некоторых вариантах осуществления Ring В представляет собой возможно замещенную фенильную группу. В некоторых вариантах осуществления Кольцо В представляет собой возможно замещенное нафтильное кольцо или а бициклическое 8-10-членное гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода и серы. В некоторых других вариантах осуществления Кольцо В представляет собой возможно замещенное 3-7-членное карбоциклическое кольцо. В других вариантах осуществления Кольцо В представляет собой возможно замещенное 4-7-членное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы.
[0046] В некоторых вариантах осуществления Кольцо В представляет собой фенил. В некоторых вариантах осуществления Кольцо В представляет собой 6-членное гетероарильное кольцо, содержащее 1-3 атома азота. В некоторых вариантах осуществления Кольцо В представляет собой 5~членное гетероарильное кольцо, содержащее 1 или 2 или 3 гетероатома, независимо выбранных из азота, кислорода и серы.
[0047] В некоторых вариантах осуществления Кольцо В представляет собой 5-6-членное насыщенное гетероциклическое кольцо, содержащее 1 атом азота. В некоторых вариантах осуществления Кольцо В представляет собой 9-10-членное бициклическое частично насыщенное гетероарильное кольцо, содержащее 1-3 атома азота. В некоторых вариантах осуществления Кольцо В представляет собой 9-10-членное бициклическое частично насыщенное гетероарильное кольцо, содержащее 1 атом азота. В некоторых вариантах осуществления Кольцо В 9-10-членное бициклическое частично насыщенное гетероарильное кольцо, содержащее 1 атом азота 1 атом кислорода.
[0048] В некоторых вариантах осуществления Кольцо В представляет собой возможно содержащую заместители группу, выбранную из следующих: фенил, пиридид, пиразинил, пиримидинил, имидазолил, пирролидинил, пиперидинил, индолинил, индазолил и изоиндолинил.
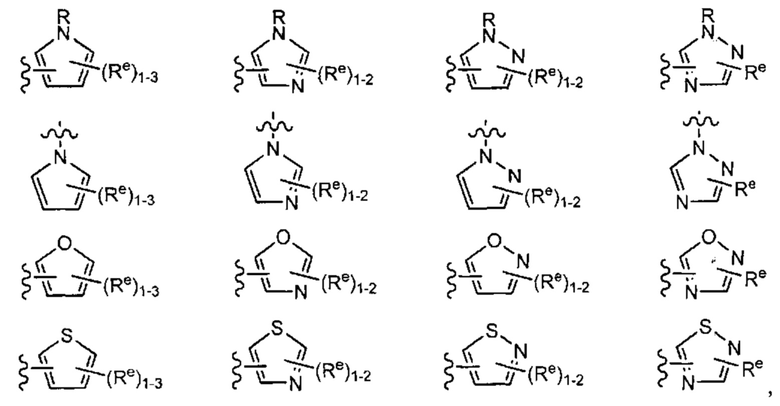
[0049] Примеры групп Кольца В приведены в Таблице 2.
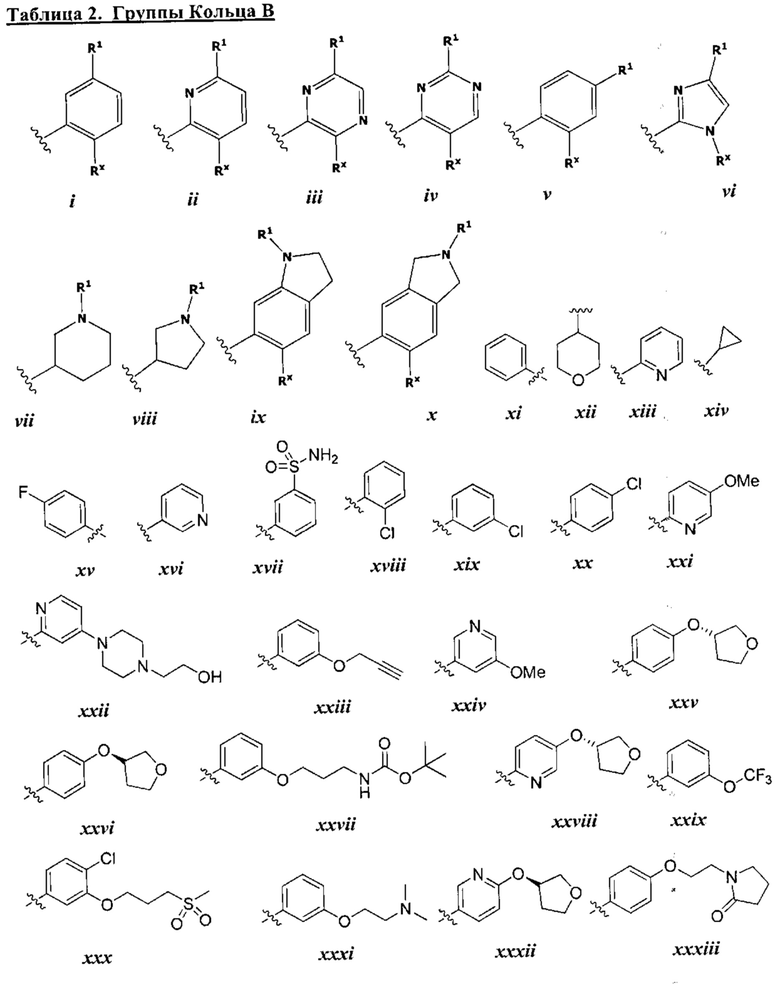
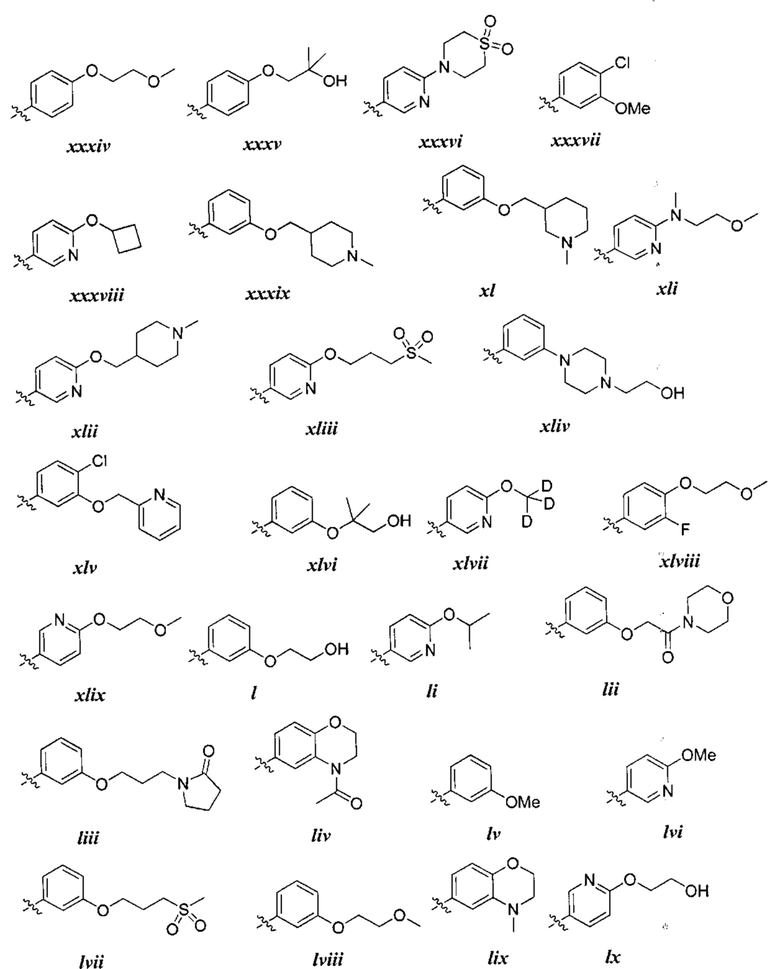
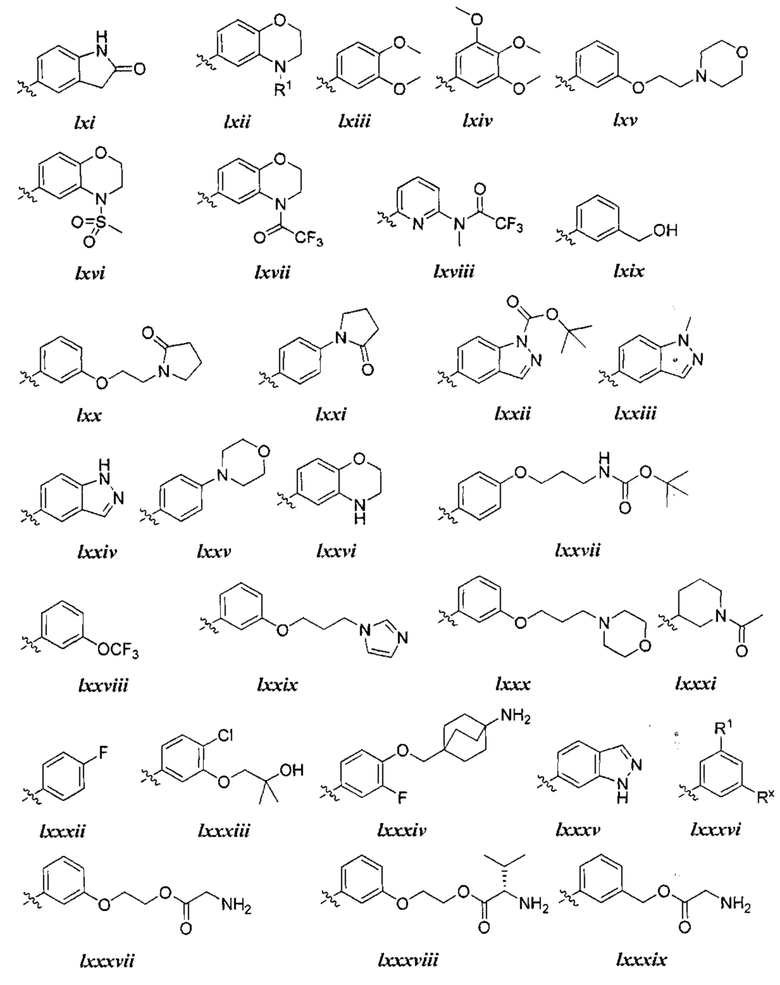
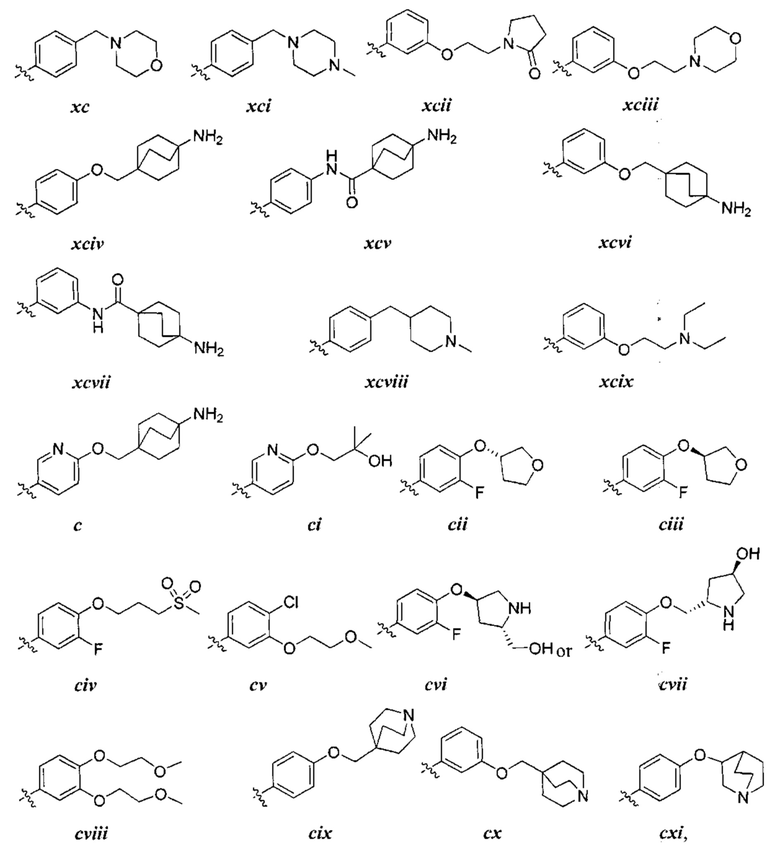
причем каждый R1 и Rx имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
[0050] В некоторых вариантах осуществления Кольцо В выбрано из i, ii, iii, iv, v, ix, х, xi, xiii, xvi, xvii, xix, xx, xxv, xxvi, xxxii, xxxiv, xxxv, xxxviii, 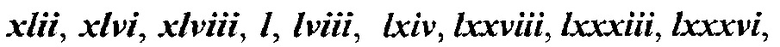 xciv, c, ci, cii, ciii, civ и cv.
xciv, c, ci, cii, ciii, civ и cv.
[0051] В некоторых вариантах осуществления m в формуле I равно 1, 2, 3 или 4. В некоторых вариантах осуществления m равно 1. В других вариантах осуществления m равно 0.
[0052] В некоторых вариантах осуществления р в формуле I равно 1, 2, 3 или 4. В некоторых вариантах осуществления р равно 1. В других вариантах осуществления р равно 0.
[0053] Как определено в общем выше каждая группа Rx в формуле I независимо выбрана -R, галогена, -OR, -O(CH2)qOR, -CN, -NO2, -SO2R, -SO2N(R)2, -SOR, -C(O)R, -CO2R, -C(O)N(R)2, -NRC(O)R, -NRC(O)NR2, -NRSO2R или -N(R)2, причем q равно 1-4, или Rx и R1, если они одновременно присутствуют на Кольце В, взятые вместе с разделяющими их атомами, образуют 5-7-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, причем указанное кольцо содержит в качестве заместителя реакционноспособную группу и 0-3 группы, независимо выбранные из оксо, галогена, CN и С1-6 алифатической группы.
[0054] В некоторых вариантах осуществления каждый пример Rx независимо выбран из следующих: -R, -OR, -O(CH2)qOR и галогена. В некоторых вариантах осуществления Rx представляет собой низший алкил, низший алкокси, низший алкоксиалкокси или галоген. Примеры групп Rx включают метил, метокси, метоксиэтокси и фтор. В некоторых вариантах осуществления Rx представляет собой водород.
[0055] Как определено в общем виде выше, каждая группа Rv в формуле I независимо выбрана из -R, галогена, -OR, -O(CH2)qOR, -CN, -NO2, -SO2R, -SO2N(R)2, -SOR, -C(O)R, -CO2R, -C(O)N(R)2, -NRC(O)R, -NRC(O)NR2, -NRSO2R и -N(R)2; причем q равно 1-4, или Rv и R1, если они одновременно присутствуют на Кольце А, взятые вместе с разделяющими их атомами, образуют 5-7-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, причем указанное кольцо содержит в качестве заместителя реакционноспособную группу и 0-3 группы, независимо выбранные из оксо, галогена, CN или C1-6 алифатической группы.
[0056] В некоторых вариантах осуществления Rv в каждом случае независимо выбран из следующих: -R, -OR, -O(CH2)qOR и галогена. В некоторых вариантах осуществления Rv представляет собой низший алкил, низший алкокси, низшей алкоксиалкокси или галоген. Примеры групп Rv включают метил, метокси, трифторметокси, метоксиэтокси и хлор. В некоторых вариантах осуществления Rv представляет собой водород.
[0057] В некоторых вариантах осуществления q равно 1, 2, 3 или 4. В некоторых вариантах осуществления q равно 1. В некоторых других вариантах осуществления q равно 2.
[0058] Как определено в общем виде выше, Ry представляет собой водород, галоген, -CN, -CF3, С1-4 алифатическую группу, С1-4 галоалифатическую группу, -OR, -C(O)R или -C(O)N(R)2, причем R имеет значение определенное выше, и описан в настоящем тексте. В некоторых вариантах осуществления Ry представляет собой водород, галоген, -CN, -CF3, низший алкил или низший галоалкил, -C≡CR и циклопропил. В других вариантах осуществления Ry представляет собой -OR, -C(O)R или -C(O)N(R)2. В некоторых вариантах осуществления Ry представляет собой -ОСН3. В некоторых других вариантах осуществления Ry представляет собой -С(O)СН3. В других вариантах осуществления Ry представляет собой -C(O)NHR. В некоторых вариантах осуществления Ry представляет собой водород. В некоторых вариантах осуществления Ry представляет собой фтор. В некоторых других вариантах осуществления Ry представляет собой метил.
[0059] Как определено в общем виде выше, W1 и W2 каждый независимо представляет собой ковалентную связь или бивалентную С1-3 алкиленовую цепь, причем одно метиленовое звено в W1 или W2 возможно заменено на -NR2-, -N(R2)C(O)-, -C(O)N(R2)-, -N(R2)SO2-, -SO2N(R2)-, -О-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO- или -SO2-. В некоторых вариантах осуществления W1 и W2 одинаковы. В некоторых вариантах осуществления W1 и W2 различны.
[0060] В некоторых вариантах осуществления W1 представляет собой ковалентную связь. В некоторых вариантах осуществления W1 представляет собой бивалентную С1-3 алкиленовую цепь, причем одно метиленовое звено в W1 возможно заменено на -NR2-, -N(R2)C(O)-, -C(O)N(R2)-, -N(R2)SO2-, -SO2N(R2)-, -О-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO- или -SO2-. В некоторых вариантах осуществления W1 представляет
собой -С(=O), -NR2-, -S-, или -O-. В некоторых вариантах осуществления W1 представляет собой -NR2-. В других вариантах осуществления W1 представляет собой -О-. В некоторых вариантах осуществления W1 представляет собой -NH-, -S- или -О-. В некоторых вариантах осуществления W1 представляет собой -СН2О-, -CH2S- или -CH2NH- В некоторых аспектах W1 представляет собой -ОСН2-, -SCH2-, -NHCH2- или -СН2СН2-.
[0061] В некоторых вариантах осуществления W2 представляет собой ковалентную связь. В некоторых вариантах осуществления W2 представляет собой бивалентную С1-3 алкиленовую цепь, причем одно метиленовое звено в W2 возможно заменено на -NR2-, -N(R2)C(O)-, -C(O)N(R2)-, -N(R2)SO2-, -SO2N(R2)-, -О-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO- или -SO2-. В некоторых вариантах осуществления W2 представляет собой -С(=O), -NR2-, -S- или -О-. В некоторых вариантах осуществления W2 представляет собой -NR2-. В других вариантах осуществления W2 представляет собой -О-. В некоторых вариантах осуществления W2 представляет собой -NH-, -S- или -О-. В некоторых вариантах осуществления W2 представляет собой -СН2О-, -CH2S- или -CH2NH-. В некоторых аспектах W представляет собой -ОСН2-, -SCH2-, -NHCH2- или -СН2СН2-.
[0062] В некоторых вариантах осуществления Кольцо В представляет собой фенил, и таким образом образует соединение формулы II-а или II-b:
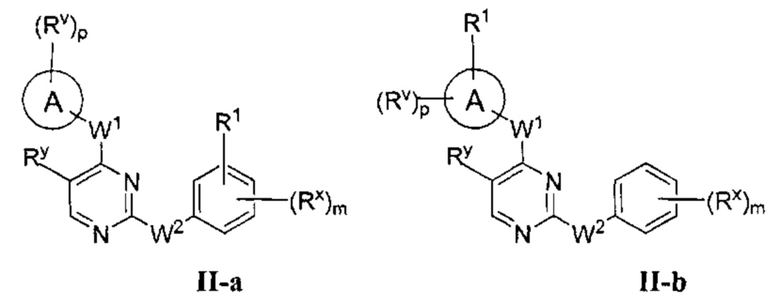
или фармацевтически приемлемая соль такого соединения, где каждое из Кольца А, m, р, Rx, Ry, Rv, W1, W2 и R1 имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
[0063] В некоторых вариантах осуществления Кольцо А представляет собой фенил, и таким образом образует соединение формулы III-а или III-b:
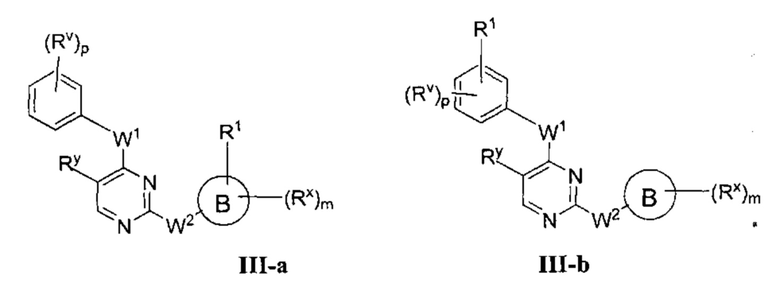
или фармацевтически приемлемая соль такого соединения, где каждый из Кольца В, m, р, Rx, Ry, Rv, W1, W2, и R1 имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
[0064] В некоторых вариантах осуществления Кольцо А представляет собой фенил и Ring В представляет собой фенил, и таким образом образует соединение формулы IV-a или IV-b:
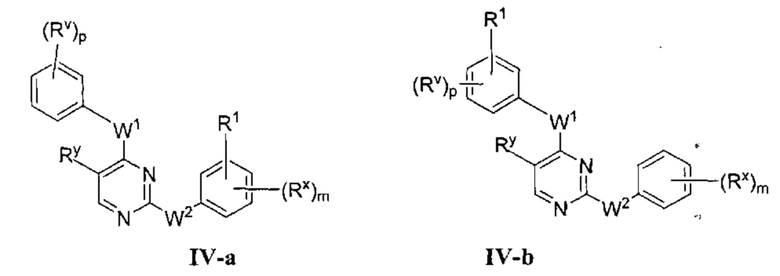
или фармацевтически приемлемая соль такого соединения, где каждый из m, р, Rx, Ry, Rv, W1, W2 и R1 имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
[0065] Как определено в общем виде выше, каждый R независимо представляет собой водород, возможно содержащую заместители C1-6 алифатическую группу или -C(O)R, или R2 и заместитель на Кольце А, взятые вместе с разделяющими из атомами, образуют 4-6-членное частично ненасыщенное или ароматическое конденсированное кольцо, или R2 и Ry, взятые вместе с разделяющими их атомами, образуют 4-6-членное насыщенное, частично ненасыщенное или ароматическое конденсированное кольцо. Согласно одному аспекту R2 представляет собой водород. Согласно другому аспекту R2 представляет собой -C(O)R, причем R2 представляет собой возможно содержащую заместители C1-6 алифатическую группу.
[0066] Согласно некоторым аспектам R2 и заместитель на Кольце А, взятые вместе с разделяющими их атомами, образуют 4-7-членное насыщенное или частично ненасыщенное кольцо, и таким образом образует соединение формулы I-а-i или I-b-i:
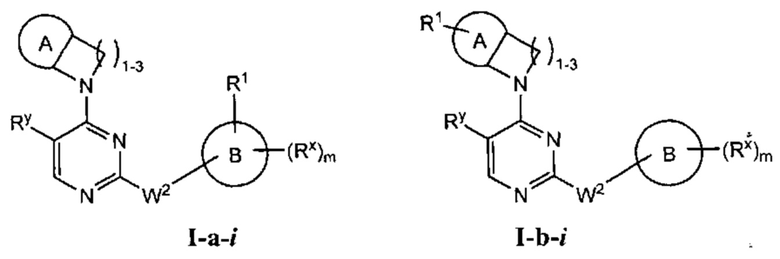
или фармацевтически приемлемая соль такого соединения, где каждый из A, R1, Rx и m имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
[0067] По аналогии с образованием соединений формул I-а-i и I-b-i выше, для специалиста в данной области очевидно, что соединения формул II-а, II-b, III-а, IIi-b, IV-а, и IV-b образуют, соответственно, соединения II-а-i, II-b-i, III-а-i, II1-b-i, IV-a-i и IV-b-i, е R2 и заместитель на Кольце А, взятые вместе с разделяющими их атомами, образуют 4-7-членное насыщенное или частично ненасыщенное кольцо.
[0068] Согласно некоторым аспектам R2 и Ry, взятые вместе с разделяющими их атомами, образуют 4-7-членное частично ненасыщенное кольцо, и таким образом образует соединение формулы I-a-ii или I-b-ii:
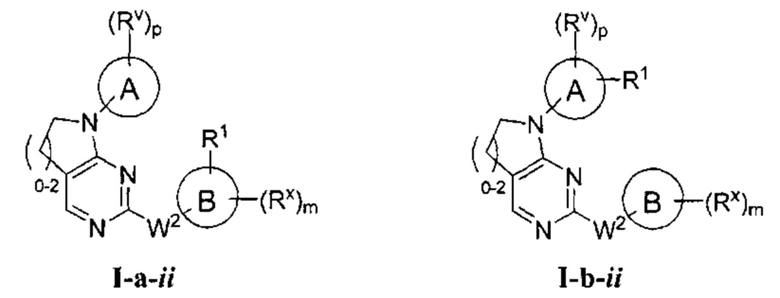
или фармацевтически приемлемая соль такого соединения, где каждый их Кольца A, R1, Rx, и m имеют значения, определенные выше, и их классы и подклассы описаны в настоящем тексте.
[0069] По аналогии с образованием соединений формул I-а-ii и I-b-ii, описанных выше, для специалиста в данной области очевидно, что соединения формул II-
a, II-b, II-а, III-b, IV-a, и IV-b образуют, соответственно, соединения формул II-а-ii, II-b-ii, III-a-ii, II1-b-ii, IV-a-ii и IV-b-ii в случаях, когда R2 и Ry, взятые вместе с разделяющими их атомами, образуют 4-7-членное частично ненасыщенное кольцо.
[0070] Как определено в общем виде выше, группа R1 в формулах I и II представляет собой -L-Y, где:
L представляет собой ковалентную связь или бивалентную C1-8 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь, причем одно, два или три метиленовых звена в L возможно и независимо заменены на циклопропилен, -NR-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2-, -SO2N(R)-, -O-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO-, -SO2-, -C(=S)-, -C(=NR)-, -N=N- или -C(=N2)-;
Y представляет собой водород, C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN, или 3-10-членное моноциклическое или бициклическое насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, и при этом указанное кольцо содержит в качестве заместителей 1-4 групп Re; и
каждый Re независимо выбран из следующих: -Q-Z, оксо, NO2, галогена, CN, подходящей уходящей группы и С1-6 алифатической группы, возможно замещенные оксо, галогеном, NO2 или CN, где:
Q представляет собой ковалентную связь или бивалентную С1-6 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь, причем одно или два метиленовых звена в Q возможно и независимо заменены на -N(R)-, -S-, -О-, -С(О)-, -ОС(О)-, -С(O)O-, -SO-, или -SO2-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2- или -SO2N(R)-; и
Z представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN.
[0071] В некоторых вариантах осуществления L представляет собой ковалентную связь.
[0072] В некоторых вариантах осуществления L представляет собой бивалентную C1-8 насыщенную или ненасыщенную, линейную или разветвленную
углеводородную цепь. В некоторых вариантах осуществления L представляет собой -СН2-.
[0073] В некоторых вариантах осуществления L представляет собой ковалентную связь, -СН2-, -NH-, -CH2NH-, -NHCH2-, -NHC(O)-, -NHC(O)CH2OC(O)-, -CH2NHC(O)-, -NHSO2-, -NHSO2CH2-, -NHC(O)CH2OC(O)- или -SO2NH-.
[0074] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и один или два дополнительных метиленовых звена L возможно и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, -C(O)O-, циклопропилен, -O-, -N(R)- или -C(O)-.
[0075] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -С(О)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-или -C(O)O-, и один или два дополнительных метиленовых звена L возможно и независимо заменены на циклопропилен, -О-, -N(R)- или -С(О)-.
[0076] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -С(О)-, и один или два дополнительных метиленовых звена L возможно и независимо заменены на циклопропилен, -О-, -N(R)- или -С(О)-.
[0077] Как описано выше, в некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь. Для среднего специалиста очевидно, что такая двойная связь может существовать в углеводородном каркасе или может быть "экзо" по отношению к каркасной цепи, что приводит к образованию алкилиденовой группы. В качестве примера, такая группа L, содержащая разветвленную алкилиденовую цепь, включает -СН2С(=СН2)СН2-. Соответственно, в некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или
разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь в составе алкилиденила. Примеры групп L включают -NHC(O)C(=CH2)CH2-.
[0078] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -С(О)-. В некоторых вариантах осуществления L представляет собой -С(O)СН=СН(СН3)-, -C(O)CH=CHCH2NH(CH3)-, -С(O)СН=СН(СН3)-, -С(O)СН=СН-, -СН2С(O)СН=СН-, -СН2С(O)СН=СН(СН3)-, -СН2СН2С(O)СН=СН-, CH2CH2C(O)CH=CHCH2-, -CH2CH2C(O)CH=CHCH2NH(CH3)-, или -СН2СН2С(O)СН=СН(СН3)- или -СН(СН3)ОС(O)СН=СН-.
[0079] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -ОС(О)-.
[0080] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, или -C(O)O-, и один или два дополнительных метиленовых звена L возможно и независимо заменены на циклопропилен, -О-, -N(R)- или -С(О)-. В некоторых вариантах осуществления L представляет собой -СН2ОС(O)СН=СНСН2- -СН2-ОС(O)СН=СН- или -СН(СН=СН2)ОС(O)СН=СН-.
[0081] В некоторых вариантах осуществления L представляет собой -NRC(O)CH=CH-, -NRC(O)CH=CHCH2N(CH3)-, -NRC(O)CH=CHCH2O-, -CH2NRC(O)CH=CH-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, -NRC(O)(C=N2)C(O)-, -NRC(O)CH=CHCH2N(CH3)-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, NRC(O)CH=CHCH2O-, -NRC(O)C(=CH2)CH2-, -CH2NRC(O)-, -CH2NRC(O)CH=CH-, -CH2CH2NRC(O)-, или -CH2NRC(O)циклопропилен-, причем каждый R независимо представляет собой водород или возможно содержащую заместители С1-6 алифатическую группу.
[0082] В некоторых вариантах осуществления L представляет собой -NHC(O)CH=CH-, -NHC(O)CH=CHCH2N(CH3)-, -NHC(O)CH=CHCH2O-, -CH2NHC(O)CH=CH-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, -NHC(O)(C=N2)C(O)-, -NHC(O)CH=CHCH2N(CH3)-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, NHC(O)CH=CHCH2O-, -NHC(O)C(=CH2)CH2-, -CH2NHC(O)-, -CH2NHC(O)CH=CH-, -CH2CH2NHC(O)- или -CH2NHC(O)циклопропилен-.
[0083] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну тройную связь. В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну тройную связь и один или два дополнительных метиленовых звена L возможно и независимо заменены на -NRC(O)-, -C(O)NR-, -S-, -S(O)-, -SO2-, -C(=S)-, -C(=NR)-, -O-, -N(R)-, или -C(O)-. В некоторых вариантах осуществления L содержит по меньшей мере одну тройную связь и по меньшей мере одно метиленовое звено в L заменено на -N(R)-, -N(R)C(O)-, -С(О)-, -С(O)O-, или -ОС(О)-, или -О-.
[0084] Примеры групп L включают -С≡С-, -С≡CCH2N(изопропил)-, -NHC(O)C≡CCH2CH2-, -СН2-C≡С-СН2-, -C≡CCH2O-, -СН2С(O)С≡С-, -С(O)C≡С-, или -СН2ОС(=O)С≡С-.
[0085] В некоторых вариантах осуществления L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем одно метиленовое звено в L заменено циклопропиленом и одно или два дополнительных метиленовых звена в L независимо заменены на -С(О)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, или -SO2N(R)-. Примеры групп L включают -NHC(O)-циклопропилен-SO2- и -NHC(O)-циклопропилен-.
[0086] Как определено в общем виде выше, Y представляет собой водород, С1-6, алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2, или CN, или 3-10-членное моноциклическое или бициклическое насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, и при этом указанное кольцо содержит в качестве заместителей 1-4 Re, каждый Re независимо выбран из следующих: -Q-Z, оксо, NO2,
галогена, CN, подходящей уходящей группы, или С1-6 алифатическую группу, причем Q представляет собой ковалентную связь или бивалентную С1-6 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь, причем одно или два метиленовых звена в Q возможно и независимо заменены на -N(R)-, -S-, -О-, -С(О)-, -ОС(О)-, -С(O)O-, -SO-, или -SO2-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2- или -SO2N(R)-; и Z представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN.
[0087] В некоторых вариантах осуществления Y представляет собой водород.
[0088] В некоторых вариантах осуществления Y представляет собой C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN. В некоторых вариантах осуществления Y представляет собой С2-6 алкенил, возможно замещенный оксо, галогеном, NO2 или CN. В других вариантах осуществления Y представляет собой С2-6 алкинил, возможно замещенный оксо, галогеном, NO2 или CN. В некоторых вариантах осуществления Y представляет собой С2-6 алкенил. В других вариантах осуществления Y представляет собой С2-4 алкинил.
[0089] В других вариантах осуществления Y представляет собой С1-6 алкил, замещенный оксо, галогеном, NO2, или CN. Такие группы Y включают -CH2F, -CH2Cl, -CH2CN и -CH2NO2.
[0090] В некоторых вариантах осуществления Y представляет собой насыщенное 3-6-членное моноциклическое кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, причем Y замещен 1-4 группами Re, где каждая Re имеет значение, определенное выше и описанное в настоящем тексте.
[0091] В некоторых вариантах осуществления Y представляет собой насыщенное 3-4-членное гетероциклическое кольцо, содержащее 1 гетероатом, выбранный из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-2 группы Re, где каждая Re имеет значение, определенное выше и описанное в настоящем тексте. Примерами таких колец являются эпоксидное и оксэтановое кольца, где каждое кольцо замещено 1-2 группами Re, причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте.
[0092] В других вариантах осуществления Y представляет собой насыщенное 5-6-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, выбранных из
кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-4 групп Re, причем каждая Re имеет значение, определенное выше и описанное в настоящем тексте. Такие кольца включают пиперидин и пирролидин, где каждое кольцо замещено 1-4 группами Re, причем каждая Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой 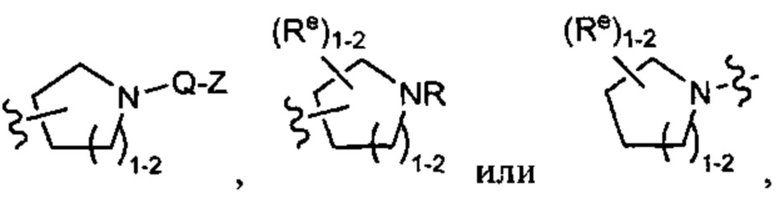 причем каждый из R, Q, Z, и Re имеет значение, определенное выше и описанное в настоящем тексте.
причем каждый из R, Q, Z, и Re имеет значение, определенное выше и описанное в настоящем тексте.
[0093] В некоторых вариантах осуществления Y представляет собой насыщенное 3-6-членное карбоциклическое кольцо, при этом указанное кольцо содержит в качестве заместителей 1-4 групп Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой циклопропил, циклобутил, циклопентил или циклогексил, где каждое кольцо содержит в качестве заместителей 1-4 группы Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой 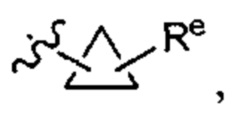 где Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой циклопропил, возможно замещенный галогеном, CN или NO2.
где Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой циклопропил, возможно замещенный галогеном, CN или NO2.
[0094] В некоторых вариантах осуществления Y представляет собой частично ненасыщенное 3-6-членное моноциклическое кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте.
[0095] В некоторых вариантах осуществления Y представляет собой частично ненасыщенное 3-6-членное карбоциклическое кольцо, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой циклопропенил, циклобутенил, циклопентенил или циклогексенил, причем каждое кольцо замещено 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y
представляет собой 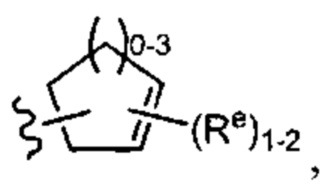 причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте.
причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте.
[0096] В некоторых вариантах осуществления Y представляет собой частично ненасыщенное 4-6-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y выбран из:
причем каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте.
[0097] В некоторых вариантах осуществления Y представляет собой 6-членное ароматическое кольцо, содержащее 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой фенил, пиридил или пиримидинил, причем каждое кольцо замещено 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте.
[0098] В некоторых вариантах осуществления Y выбран из:
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте.
[0099] В других вариантах осуществления Y представляет собой 5-членное гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-3 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y представляет собой 5-членное частично ненасыщенное или арильное кольцо, содержащее 1-3 гетероатома, независимо выбранных
из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте. Примерами таких колец являются изоксазолил, оксазолил, тиазолил, имидазолил, пиразолил, пирролил, фуранил, тиенил, тиазол, тиадиазол и оксадиазол, причемкаждое кольцо замещено 1-3 Re, где каждая группа Re имеет значение, определенное выше и описанное в настоящем тексте. В некоторых вариантах осуществления Y выбран из:
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте.
[00100] В некоторых вариантах осуществления Y представляет собой 8-10-членное бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re, где Re имеет значение, определенное выше и описанное в настоящем тексте. Согласно другому аспекту Y представляет собой 9-10-членное бициклическое, частично ненасыщенное или арильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re, причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте. Примеры таких бициклических колец включают 2,3-дигидробензо[d]изотиазол, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re, причем Re имеет значение, определенное выше и описанное в настоящем тексте.
[00101] Как определено в общем виде выше, каждая группа Re независимо выбрана из следующих: -Q-Z, оксо, NO2, галогена, CN, подходящей уходящей группы,
или C1-6 алифатической группы, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN, где Q представляет собой ковалентную связь или бивалентную C1-6 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь, причем одно или два метиленовых звена в Q возможно и независимо заменены на -N(R)-, -S-, -О-, -С(О)-, -ОС(О)-, -С(O)O-, -SO-, или -SO2-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2-, или -SO2N(R)-; и Z представляет собой водород или С1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN.
[001O2] В некоторых вариантах осуществления Re представляет собой С1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN. В других вариантах осуществления Re представляет собой оксо, NO2, галоген или CN.
[00103] В некоторых вариантах осуществления Re представляет собой -Q-Z, где Q представляет собой ковалентную связь и Z представляет собой водород (т.е., Re представляет собой водород). В других вариантах осуществления Re представляет собой -Q-Z, где Q представляет собой бивалентную С1-6 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь, причем одно или два метиленовых звена в Q возможно и независимо заменены на -NR-, -NRC(O)-, -C(O)NR-, -S-, -О-, -С(О)-, -SO- или -SO2-. В других вариантах осуществления Q представляет собой бивалентную С2-6 линейную или разветвленную углеводородную цепь, содержащую по меньшей мере одну двойную связь, причем одно или два метиленовых звена в Q возможно и независимо заменены на -NR-, -NRC(O)-, -C(O)NR-, -S-, -О-, -С(O), -SO-, или -SO2-. В некоторых вариантах осуществления группа Z группы Re представляет собой водород. В некоторых вариантах осуществления -Q-Z представляет собой -NHC(O)CH=CH2 или -С(O)СН=СН2.
[00104] В некоторых вариантах осуществления каждый Re независимо выбран из from оксо, NO2, CN, фтора, хлора, -NHC(O)CH=CH2, -С(O)СН=СН2, -СН2СН=СН2, -С≡СН, -С(O)ОСН2С1, -C(O)OCH2F, -C(O)OCH2CN, -С(O)СН2С1, -C(O)CH2F, -C(O)CH2CN или -CH2C(O)CH3.
[00105] В некоторых вариантах осуществления Re представляет собой подходящую уходящую группу, т.е. группу, способную к нуклеофильному замещению. "Подходящая уходящая" группа представляет собой химическую группу, легко замещаемую желаемой входящей химической группой, такой как тиоловая группа
представляющего интерес цистеина. Подходящие уходящие группы хорошо известны в данной области техники, см., например, "Advanced Organic Chemistry," Jerry March, 5th Ed., pp. 351-357, John Wiley и Sons, N.Y. Такие уходящие группы включают, но не ограничиваются перечисленными, галоген, алкокси, сульфонилокси, возможно замещенный алкилсульфонилокси, возможно замещенный алкенилсульфонилокси, возможно замещенный арлсульфонилокси, ацил и диазониевый фрагмент.Примеры подходящих уходящих групп включают хлор, иод, бром, фтор, ацетокси, метансульфонилокси (мезилокси), тозилокси, трифлилокси, нитро-фенилсульфонилокси (нозилокси) и бром-фенилсульфонилокси (брозилокси).
[00106] В некоторых вариантах осуществления реализуются следующие комбинации -L-Y:
(a) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и один или два дополнительных метиленовых звена L возможно и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, -C(O)O-, циклопропилен, -O-, -N(R)-, или -C(O)-; и Y представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(b) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -С(О)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, или -C(O)O-, и один или два дополнительных метиленовых звена L возможно и независимо заменены на циклопропилен, -О-, -N(R)-, или -С(О)-; и Y представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(c) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -С(О)-, и один или два дополнительных метиленовых звена L возможно и независимо заменены на циклопропилен, -О-, -N(R)-, или -С(О)-; и Y представляет собой водород или С1-6
алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(d) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -С(О)-; и Y представляет собой водород или С1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(e) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и по меньшей мере одно метиленовое звено в L заменено на -ОС(О)-; и Y представляет собой водород или С1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(f) L представляет собой -NRC(O)CH=CH-, -NRC(O)CH=CHCH2N(CH3)-, -NRC(O)CH=CHCH2O-, -CH2NRC(O)CH=CH-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, -NRC(O)(C=N2)-, -NRC(O)(C=N2)C(O)-, -NRC(O)CH=CHCH2N(CH3)-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, NRC(O)CH=CHCH2O-, -NRC(O)C(=CH2)CH2-, -CH2NRC(O)-, -CH2NRC(O)CH=CH-, -CH2CH2NRC(O)-, или -CH2NRC(O)циклопропилен-; где R представляет собой или возможно содержащую заместители С1-6 алифатическую группу; и Y представляет собой водород или С1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(g) L представляет собой -NHC(O)CH=CH-, -NHC(O)CH=CHCH2N(CH3)-, -NHC(O)CH=CHCH2O-, -CH2NHC(O)CH=CH-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, -NHC(O)(C=N2)-, -NHC(O)(C=N2)C(O)-, -NHC(O)CH=CHCH2N(CH3)-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, NHC(O)CH=CHCH2O-, -NHC(O)C(=CH2)CH2-, -CH2NHC(O)-, -CH2NHC(O)CH=CH-, -CH2CH2NHC(O)-, или -CH2NHC(O)циклопропилен-; и Y представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(h) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь в
составе алкилиденила и по меньшей мере одно метиленовое звено в L заменено на -С(О)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, или -C(O)O-, и один или два дополнительных метиленовых звена L возможно и независимо заменены на циклопропилен, -О-, -N(R)-, или -С(О)-; и Y представляет собой водород или С1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(i) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну тройную связь и один или два дополнительных метиленовых звена L возможно и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, или -C(O)O-, и Y представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(j) L представляет собой -C≡С-, -C≡CCH2N(изопропил)-, -NHC(O)C≡CCH2CH2-, -СН2-С≡С-СН2-, -С≡CCH2O-, -СН2С(O)С≡С-, -С(O)С≡С-, или -СН2ОС(=O)С≡С-; и Y представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(k) L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем одно метиленовое звено в L заменено циклопропиленом и одно или два дополнительных метиленовых звена в L независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, или -C(O)O-; и Y представляет собой водород или C1-6 алифатическую группу, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или
(l) L представляет собой ковалентную связь и Y выбран из:
(i) С1-6 алкила, замещенного оксо, галогеном, NO2 или CN;
(ii) С2-6 алкенила, возможно замещенного оксо, галогеном, NO2 или CN; или
(iii) С2-6 алкинила, возможно замещенного оксо, галогеном, NO2 или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-2 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vi) 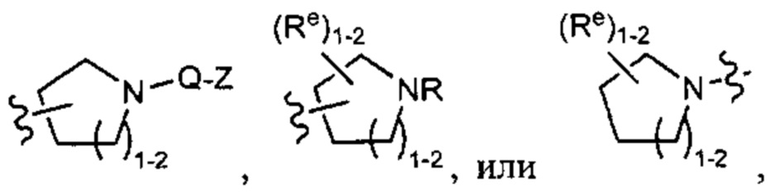 где каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(viii) частично ненасыщенного 3-6-членногомоноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(х) 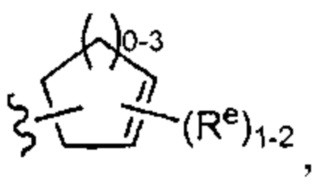 где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xi) частично ненасыщенного 4-6-членноегогетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранные из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xii) 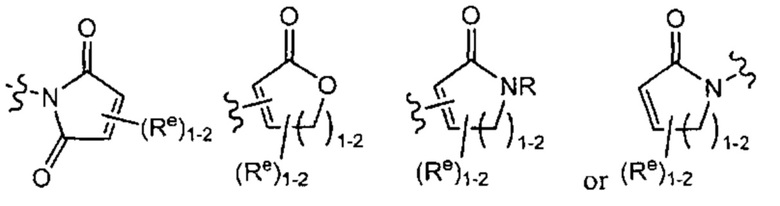
причем каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атом азота, причем указанное кольцо содержит в качестве заместителей 1-4 Re, где каждая группа Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiv) 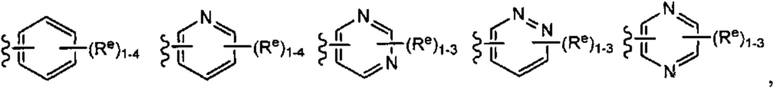
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xv) а 5-членное гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-3 Re, где каждая группа Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvi) 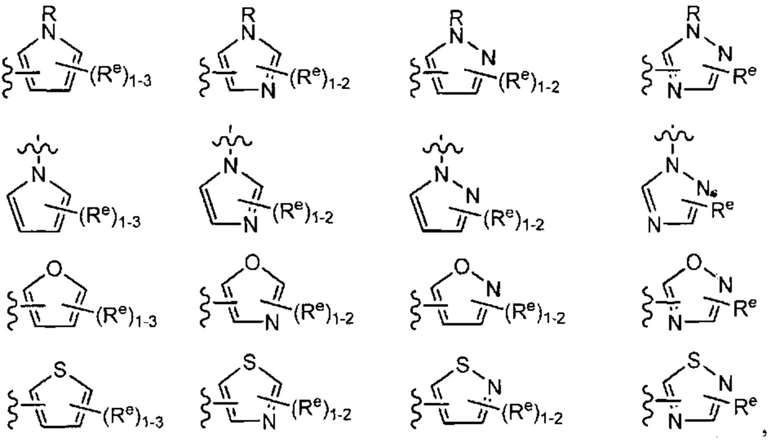
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re, причем Re имеет значение, определенное выше и описанное в настоящем тексте;
(m) L представляет собой -С(О)- и Y выбран из:
(i) C1-6 алкила, содержащего в качестве заместителей оксо, галоген, NO2, или CN; или
(ii) С2-6 алкенила, возможно содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(iii) С2-6 алкинила, возможно содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-2 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vi) 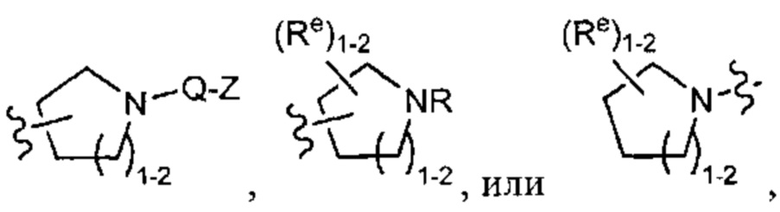 причем каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
причем каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где
каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(х) 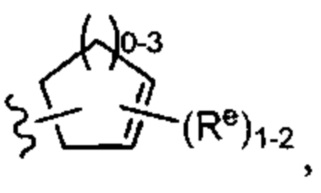 где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранные из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xii) 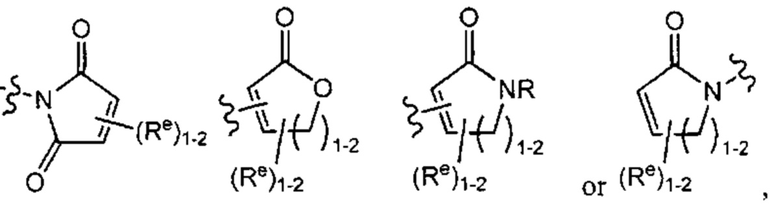
где каждый R и Re имеет значение, определенное выше и описанное в астоящем тексте; или
(xii) 6-членного ароматического кольца, содержащего 0-2 атома азота при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiv) 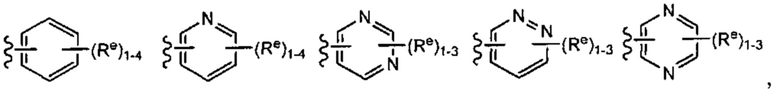
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xv) 5-членное гетероарильное кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-3 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
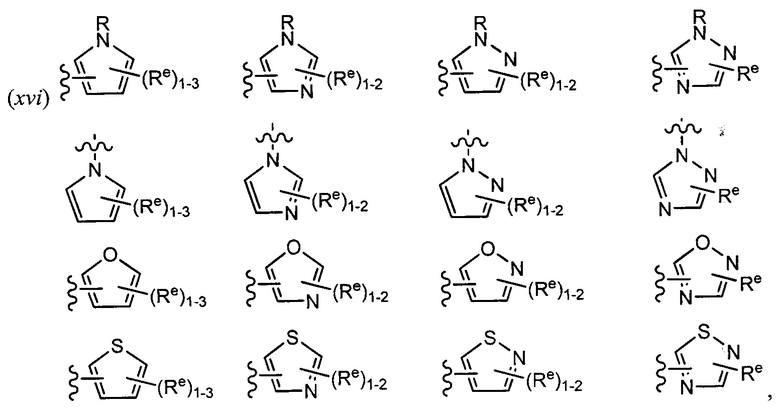
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей групп 1-4 Re, где Re имеет значение, определенное выше и описанное в настоящем тексте;
(n) L представляет собой -N(R)C(O)- и Y выбран из:
(i) C1-6 алкила, содержащего в качестве заместителя оксо, галоген, NO2, или CN; или
(ii) С2-6 алкенила, возможно содержащего в качестве заместителя оксо, галоген, NO2, или CN; или
(iii) С2-6 алкинил, возможно содержащего в качестве заместителя оксо, галоген, NO2, или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-2 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода и азота, при этом указанное кольцо
содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vi)  или
или  причем каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
причем каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(x)  где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранные из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xii) 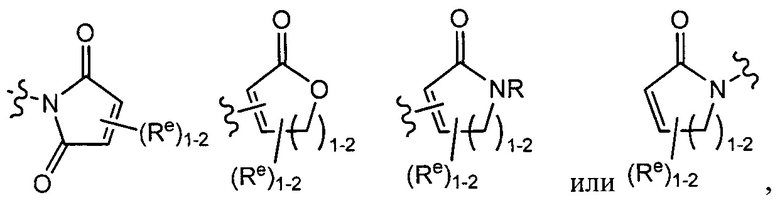
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiv) 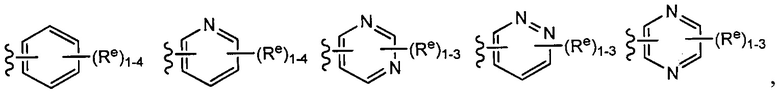
причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xv) 5--членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-3 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
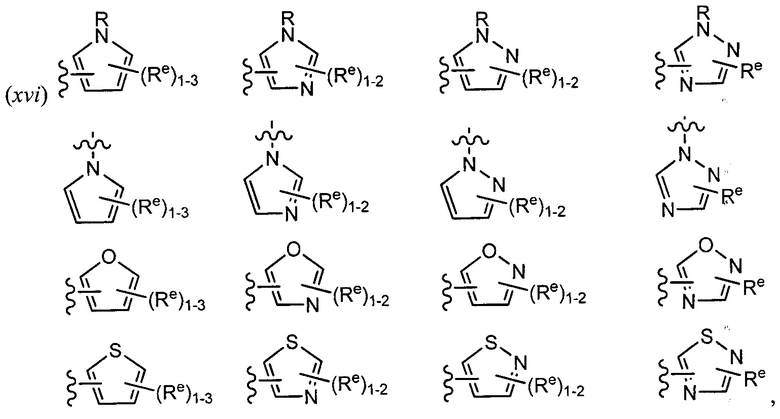
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 групп Re, причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте;
(о) L представляет собой бивалентную C1-8 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь; и Y выбран из:
(i) C1-6 алкила, содержащего в качестве заместителей оксо, галоген, NO2, или CN;
(ii) С2-6 алкенила, содержащего в качестве заместителей оксо, галоген, NO2, или CN; или
(iii) С2-6 алкинила, содержащего в качестве заместителей оксо, галоген, NO2, или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-2 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vi) 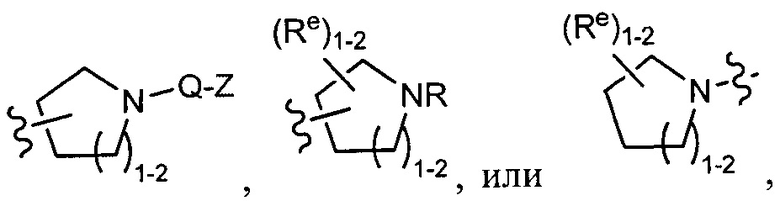 где каждый R, Q, Z, и Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый R, Q, Z, и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(x) 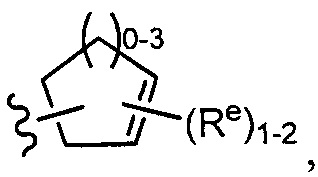 где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранные из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xii) 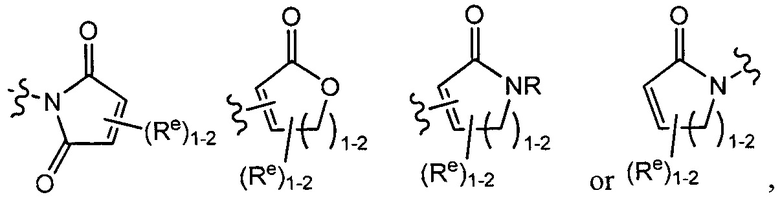
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждая группа Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiv) 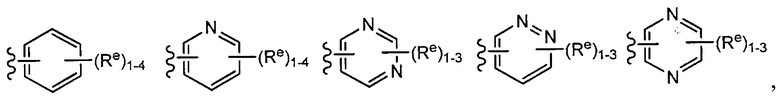
причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xv) 5--членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-3 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvi) 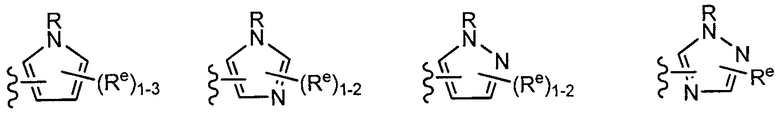
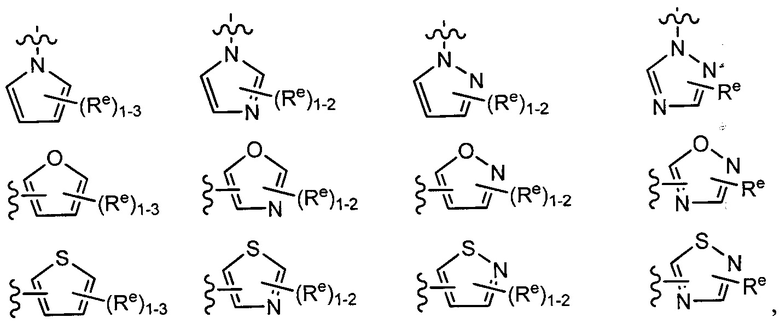
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re, где Re имеет значение, определенное выше и описанное в настоящем тексте;
(р) L представляет собой ковалентную связь, -СН2; -NH-, -С(О)-, -CH2NH-, -NHCH2-, -NHC(O)-, -NHC(O)CH2OC(O)-, -CH2NHC(O)-, -NHSO2-, -NHSO2CH2-, -NHC(O)CH2OC(O)-, или -SO2NH-; и Y выбран из:
(i) C1-6 алкила, содержащего в качестве заместителей оксо, галоген, NO2, или CN; или
(ii) С2-6 алкенила, содержащего в качестве заместителей оксо, галоген, NO2, или CN; или
(iii) С2-6 алкинила, содержащего в качестве заместителей оксо, галоген, NO2, или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-2 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода и азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vi) 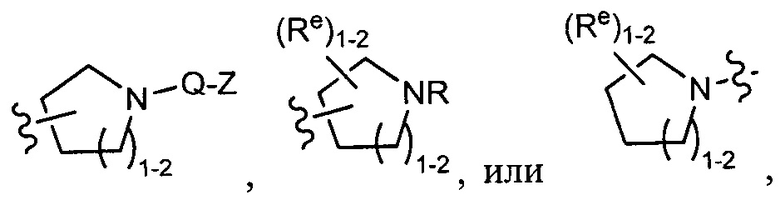 где каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый R, Q, Z и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(x) 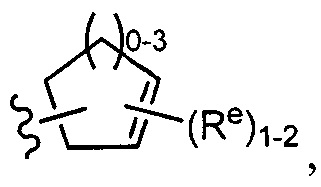 где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранные из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xii) 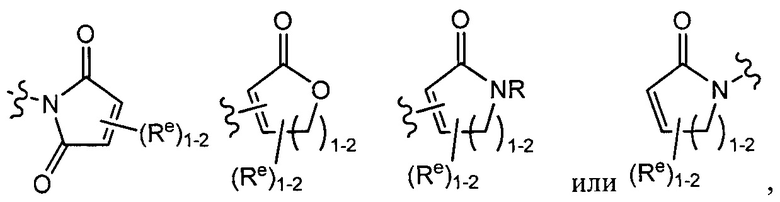
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xiv) 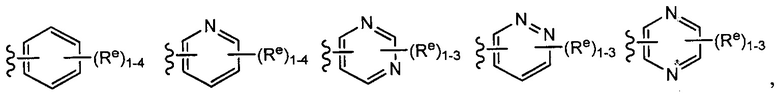
причем каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xv) 5--членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-3 Re, где каждый Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvi) 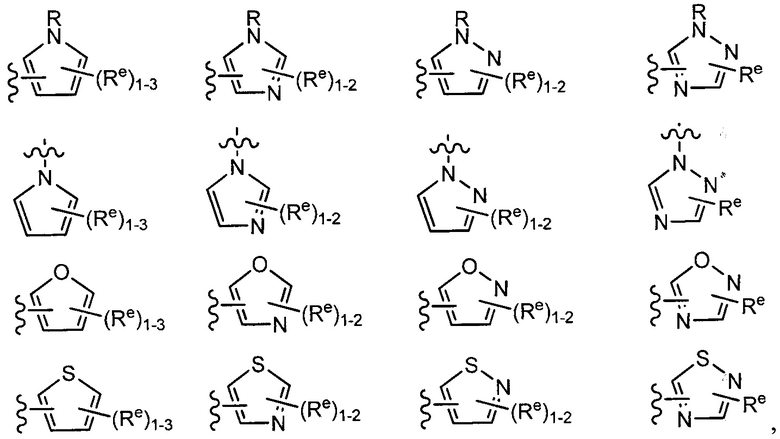
где каждый R и Re имеет значение, определенное выше и описанное в настоящем тексте; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода и серы, при этом указанное кольцо содержит в качестве заместителей 1-4 групп Re, где Re имеет значение, определенное выше и описанное в настоящем тексте.
[00107] В некоторых вариантах осуществления группа Y в формуле Ia или Ib выбрана из групп, приведенных в Таблице 3 ниже, при этом каждая волнистая линия указывает точку присоединения к остальной части молекулы.
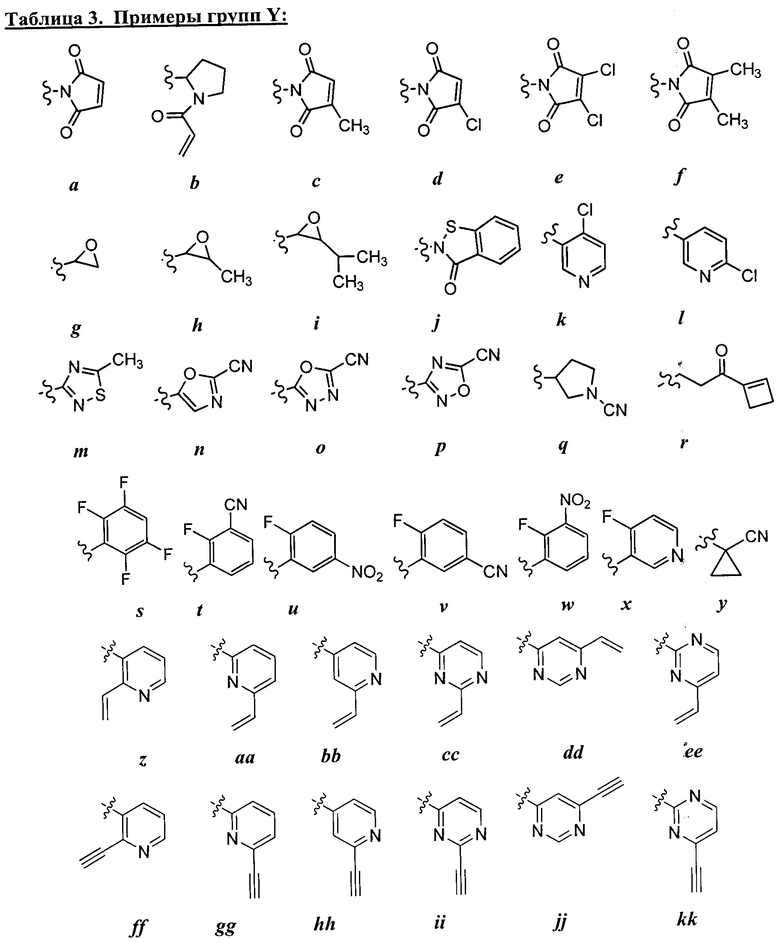
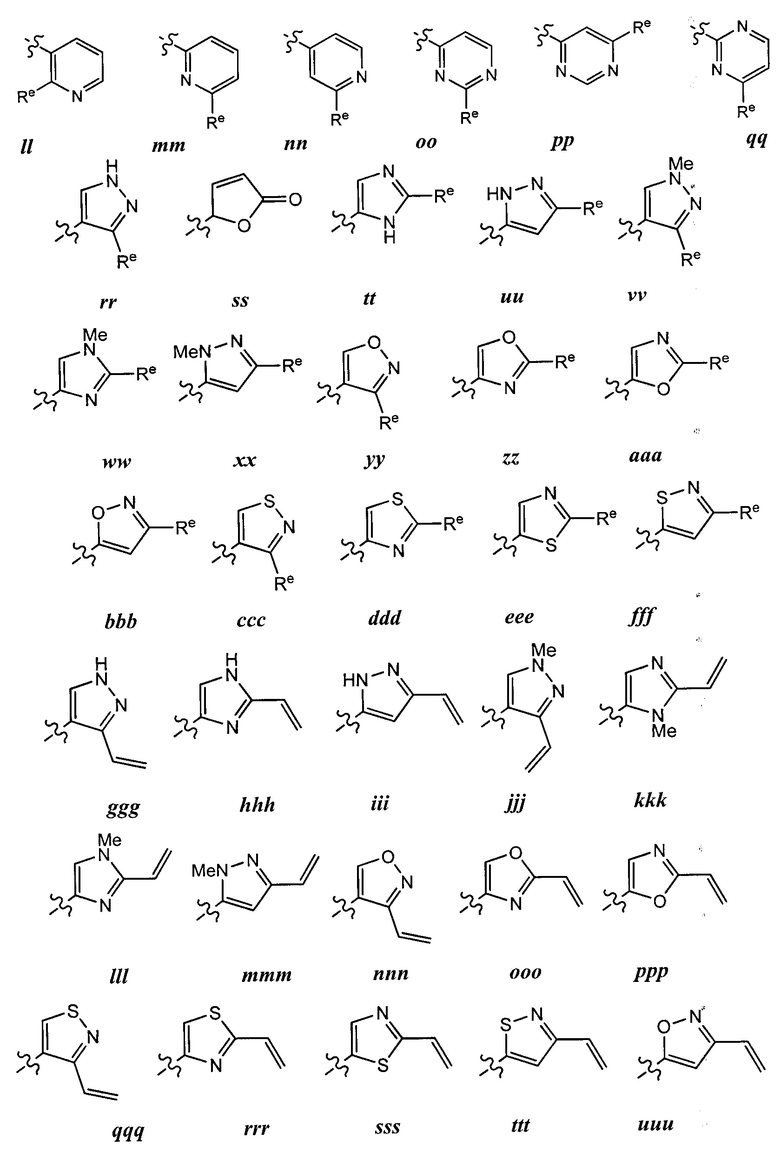
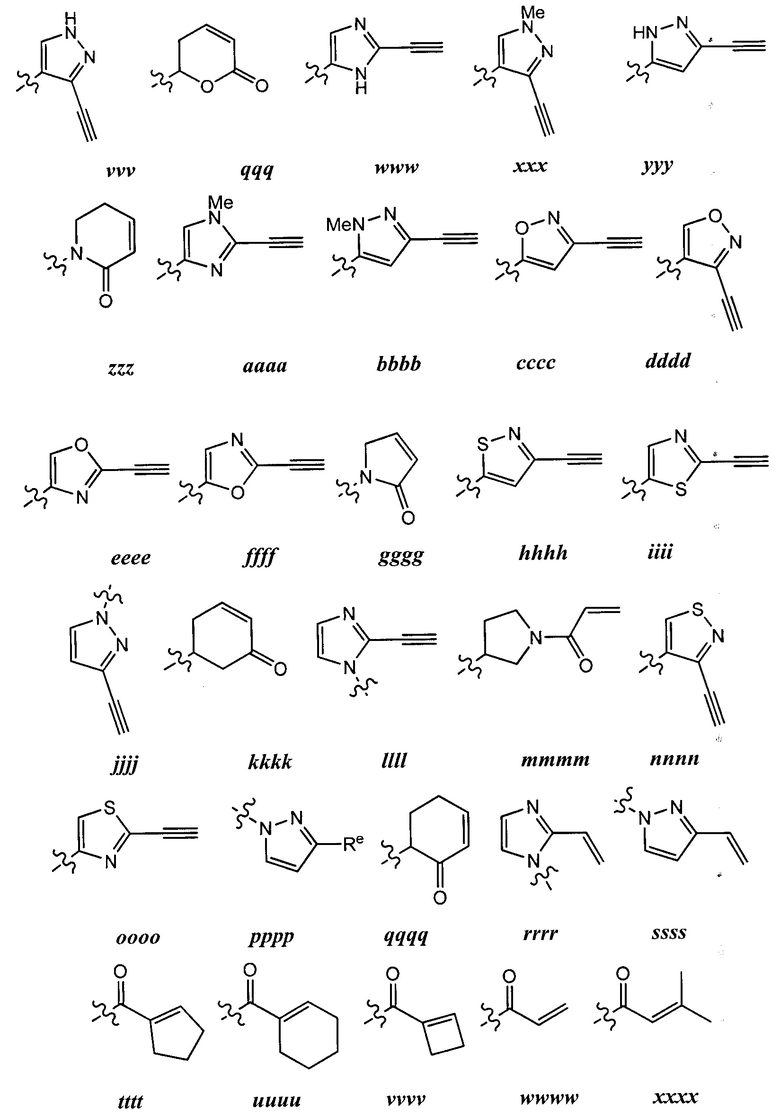
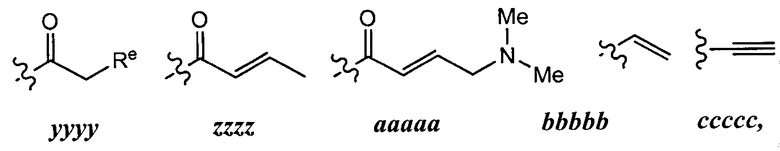
где каждый Re независимо представляет собой подходящую уходящую группу, NO2, CN или оксо.
[00108] В некоторых вариантах осуществления R1 представляет собой -С≡СН, -С≡CCH2NH(изопропил), -NHC(O)C≡CCH2CH3, -СН2-C≡С-СН3, -C≡ССН2ОН, -СН2С(O)C≡СН, -С(O)C≡СН, или -СН2ОС(=O)C≡СН. В некоторых вариантах осуществления R1 выбран из -NHC(O)CH=CH2, -NHC(O)CH=CHCH2N(CH3)2 или -CH2NHC(O)CH=CH2.
[00109] В некоторых вариантах осуществления R1 выбрана из групп, приведенных в Таблице 4 ниже, при этом каждая волнистая линия указывает точку присоединения к остальной части молекулы.
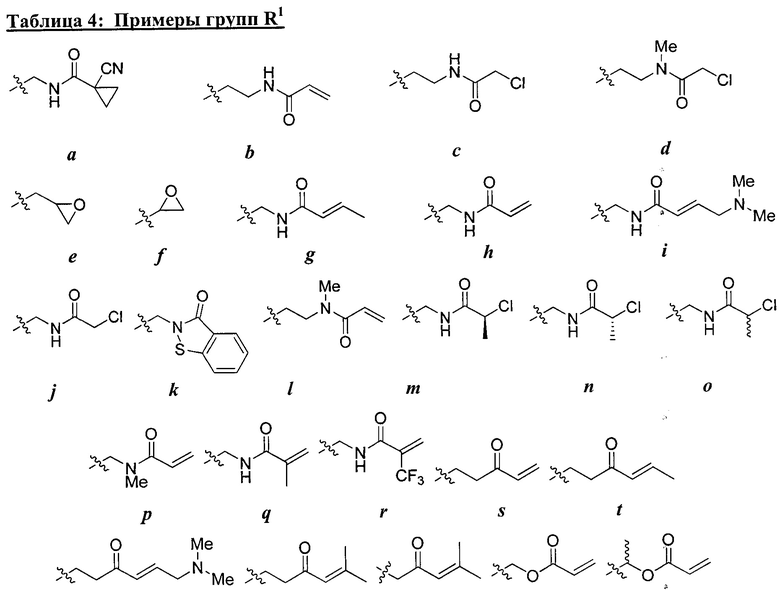
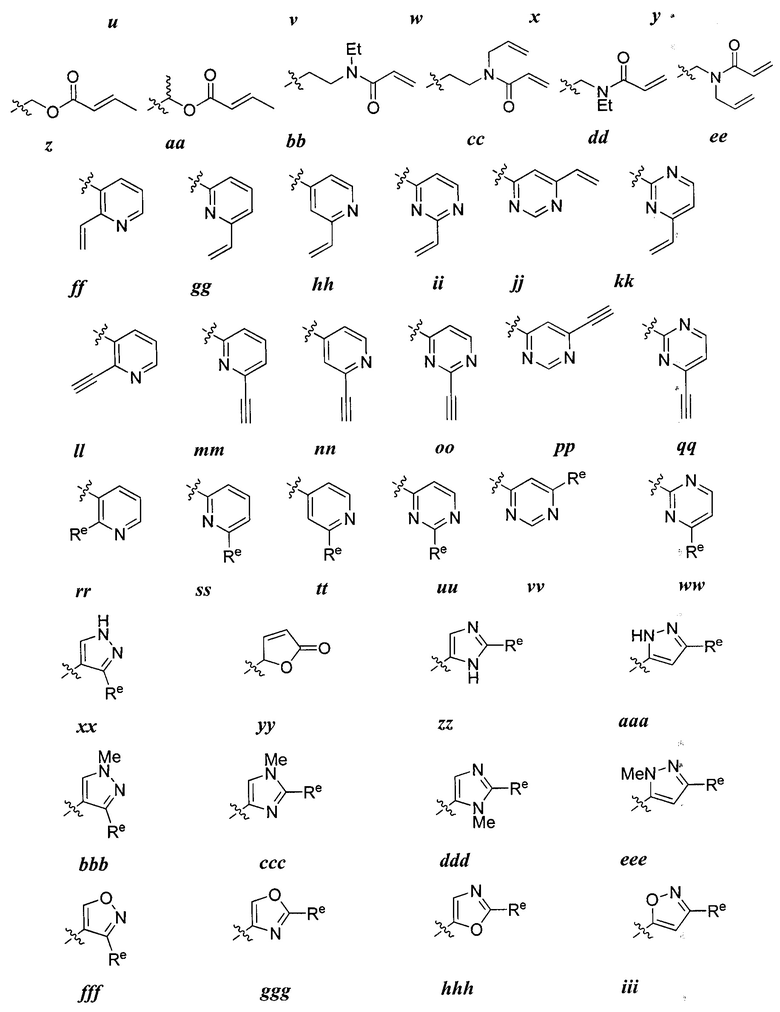
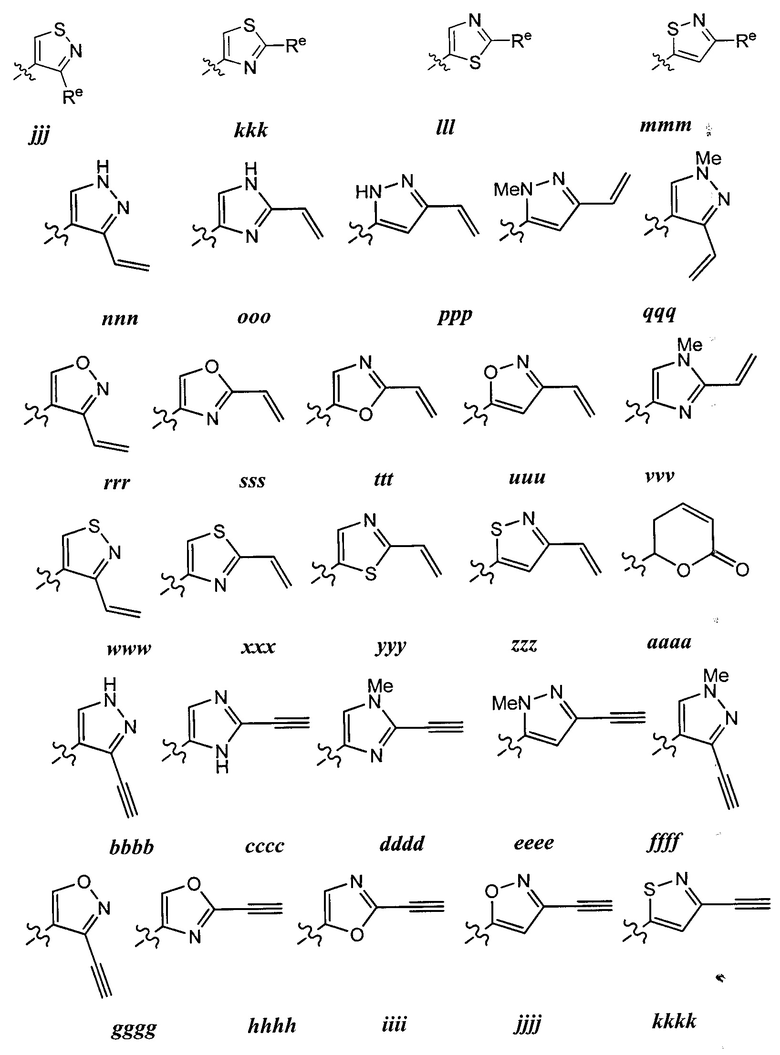
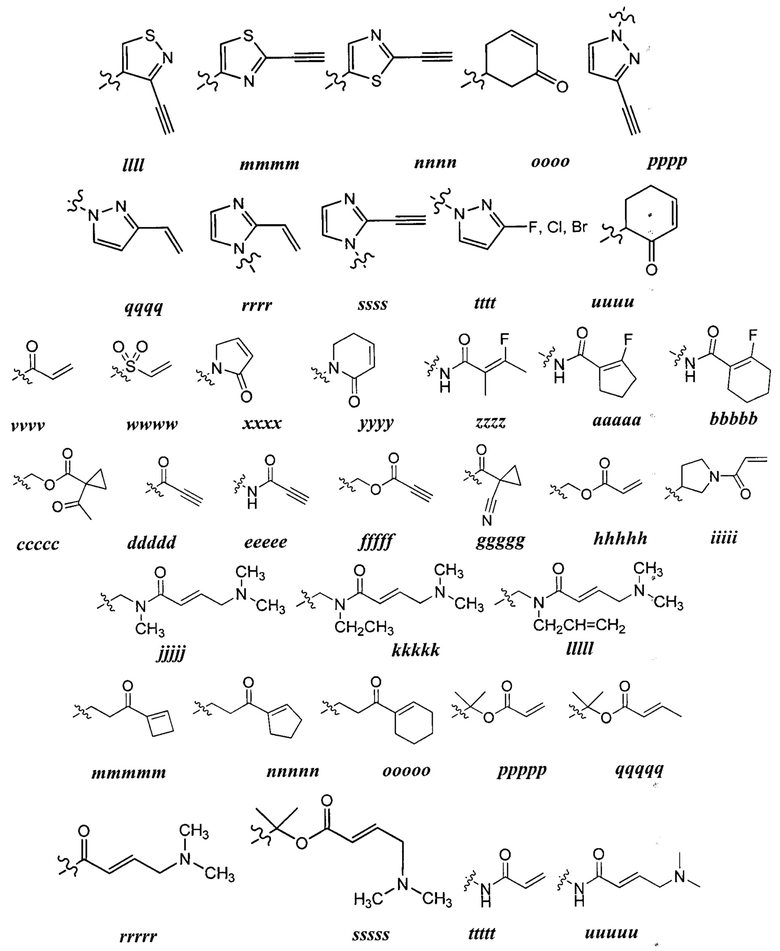
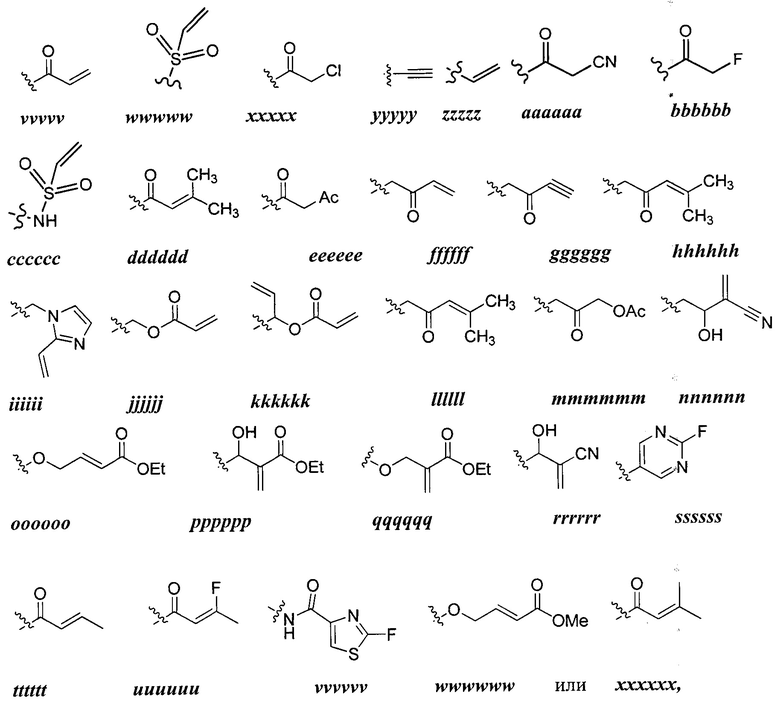
где каждый Re независимо представляет собой подходящую уходящую группу, NO2, CN или оксо.
[00110] Как определено в общем виде выше, R1 представляет собой реакционноспособную группу, или, если R1 и Rx образуют кольцо, то -Q-Z представляет собой реакционноспособную группу. Без какого-либо конкретного теоретического обоснования считается, что такие группы R1, т.е. реакционноспособные группы, особенно подходят для ковалентного связывания с ключевым остатком цистеина в связывающем домене некоторых протеинкиназы, содержащих остаток цистеина в связывающем домене, известны специалистам и включают ErbB1, ErbB2 и ErbB4 или их мутанты В некоторых вариантах осуществления соединения согласно настоящему изобретению включают
реакционно-способные группы, характеризующиеся тем, что соединения согласно настоящему изобретению направленно взаимодействуют с одним или более из следующих остатков цистеина:

[00111] Соответственно, в некоторых вариантах осуществления R1
характеризуется тем, что группа -L-Y способна ковалентно связываться с остатком цистеина, что приводит к необратимому ингибированию фермента. В некоторых вариантах осуществления остаток цистеина представляет собой Cys797 в составе ErbB1, Cys805 в составе ErbB2 и Cys803 в составе ErbB4 илимутанта указанных киназ. Нумерация остатков дана в соответствии с Uniprot (код POO533 для ErbB1; код PO4626 для ErbB2, и Q15303 для ErbB4). Очевидно, что Cys в составе ErbB1 (EGFR) может называться 773 или 797 в зависимости от того, содержит ли исходная последовательность сигнальный пептид. Таким образом, согласно настоящему изобретению соответствующие остатки цистеина ErbB1 могут быть обозначены Cys 773 или Cys 797 и употребляться взаимозаменяемо в таком контексте.
[00112] Для среднего специалиста в данной области очевидно, что разнообразные реакционноспособные группы, описанные в настоящему тексте, пригодны для такого ковалентного связывания. Такие группы R1 включают, но не ограничиваются группами, описанными в настоящему тексте и приведенными в Таблице 3 ниже.
[00113] Как показано выше в формулах I-а и I-b реакционноспособная группа R1 может находиться в орто-, мета- или пара-положении. В некоторых вариантах осуществления реакционноспособная группа R1 находится в мета-положении фенильного кольца относительно остальной молекулы.
[00114] В некоторых вариантах осуществления R1 характеризуется тем, что группа-L-Y способна ковалентно связывать остаток цистеина ТЕС, что приводит к необратимому ингибированию этого фермента. В некоторых вариантах осуществления указанный остаток цистеина представляет собой Cys 449.
[00115] В некоторых вариантах осуществления R1 характеризуется тем, что группа-L-Y способна ковалентно связывать остаток цистеина ВТК, что приводит к
необратимому ингибированию этого фермента. В некоторых вариантах осуществления указанный остаток цистеина представляет собой Cys 481.
[00116] В некоторых вариантах осуществления R1 характеризуется тем, что группа-L-Y способна ковалентно связывать остаток цистеина ITK, что приводит к необратимому ингибированию этого фермента. В некоторых вариантах осуществления указанный остаток цистеина представляет собой Cys 442.
[00117] В некоторых вариантах осуществления R1 характеризуется тем, что группа-L-Y способна ковалентно связывать остаток цистеина ВМХ, что приводит к необратимому ингибированию этого фермента. В некоторых вариантах осуществления указанный остаток цистеина представляет собой Cys 496.
[00118] В некоторых вариантах осуществления R1 характеризуется тем, что группа-L-Y способна ковалентно связывать остаток цистеина f JAK3, что приводит к необратимому ингибированию этого фермента. В некоторых вариантах осуществления указанный остаток цистеина представляет собой Cys 909.
[00119] В некоторых вариантах осуществления R1 характеризуется тем, что группа-L-Y способна ковалентно связывать остаток цистеина ТХК, что приводит к необратимому ингибированию этого фермента. В некоторых вариантах осуществления указанный остаток цистеина представляет собой Cys 350.
[00120] Для среднего специалиста в данной области очевидно, что описанные в настоящем тексте различные реакционноспособные группы пригодны для ковалентного связывания. Такие группы R1 включают группы, изображенные ниже в Таблице 3, но не ограничиваются или.
[00121] Примеры соединений согласно настоящему изобретению приведены ниже в Таблице 5.

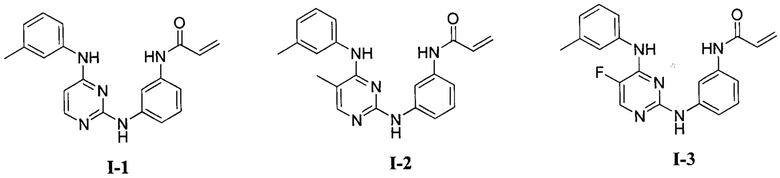
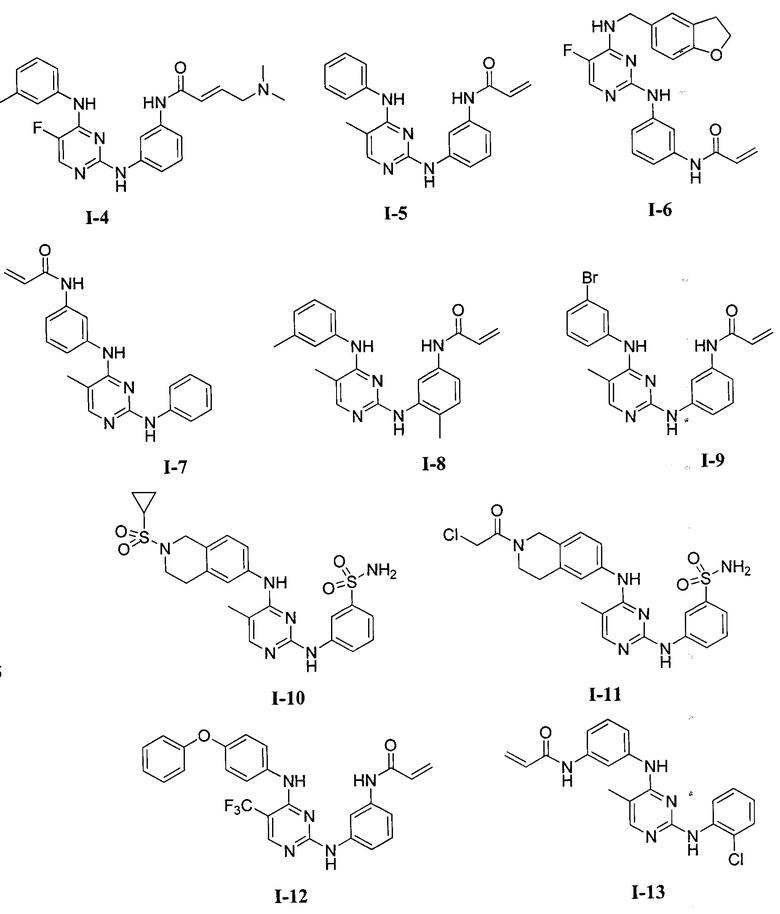
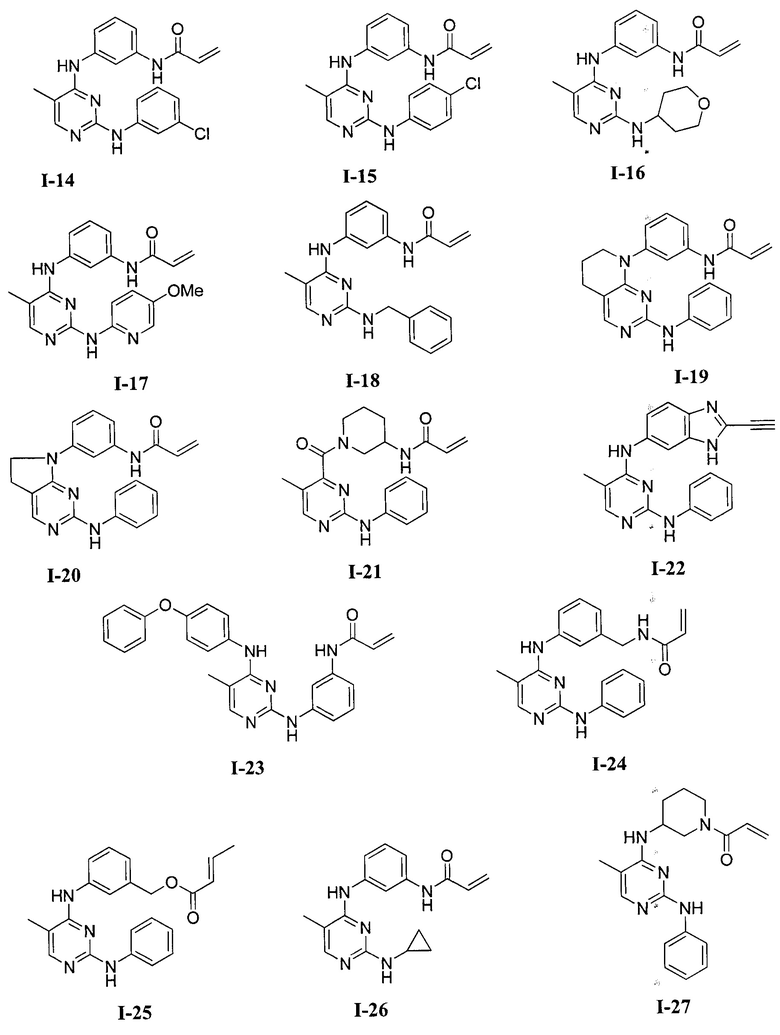
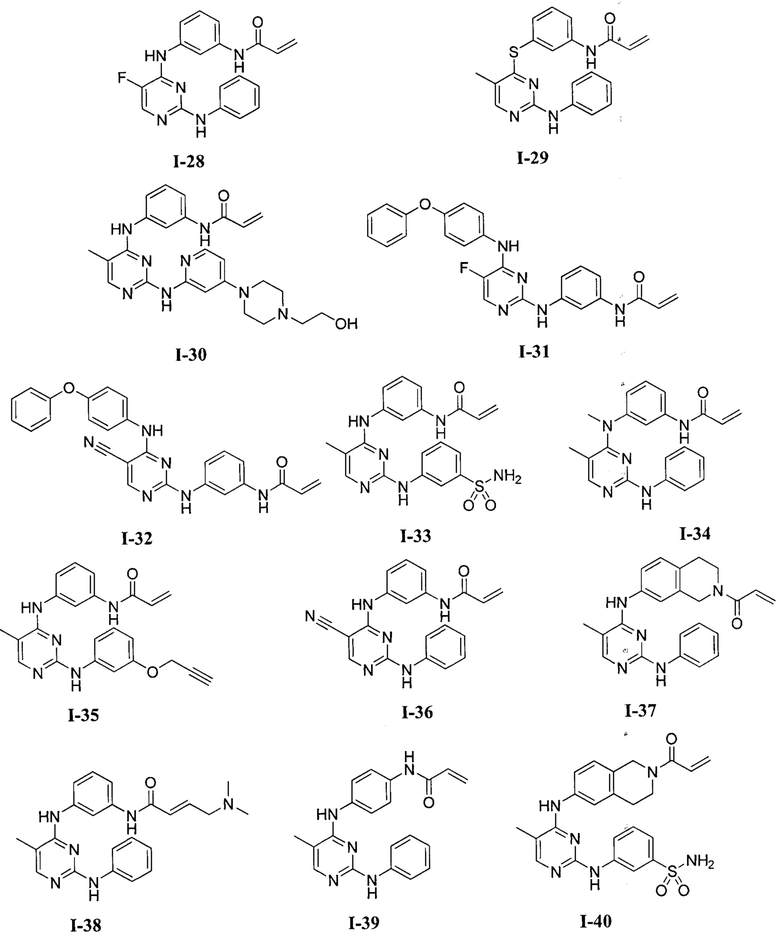
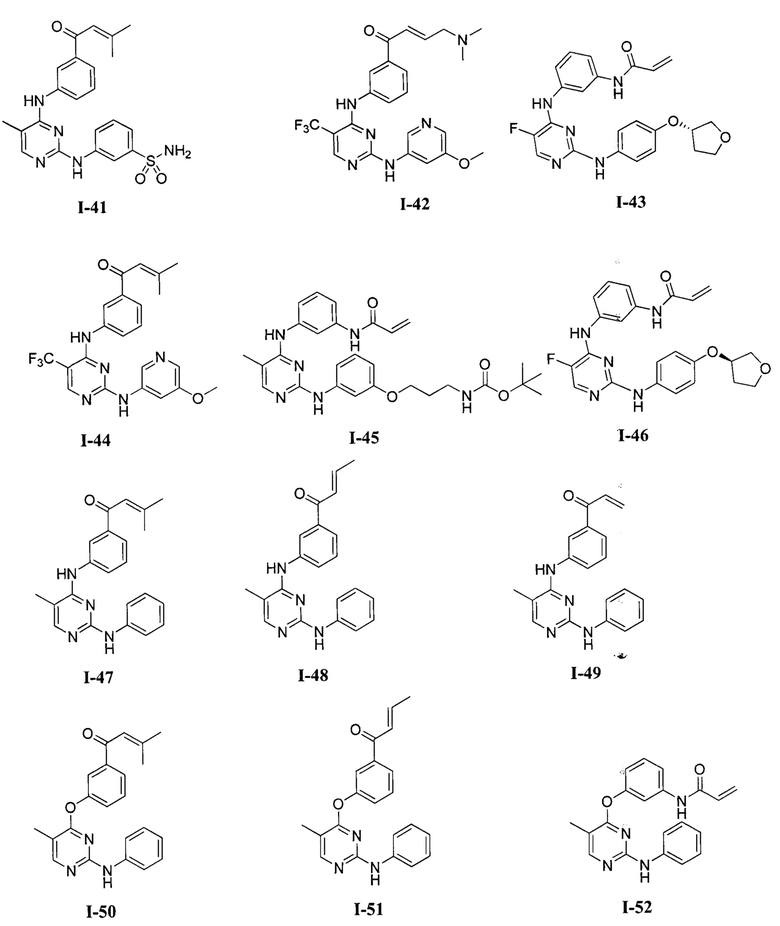
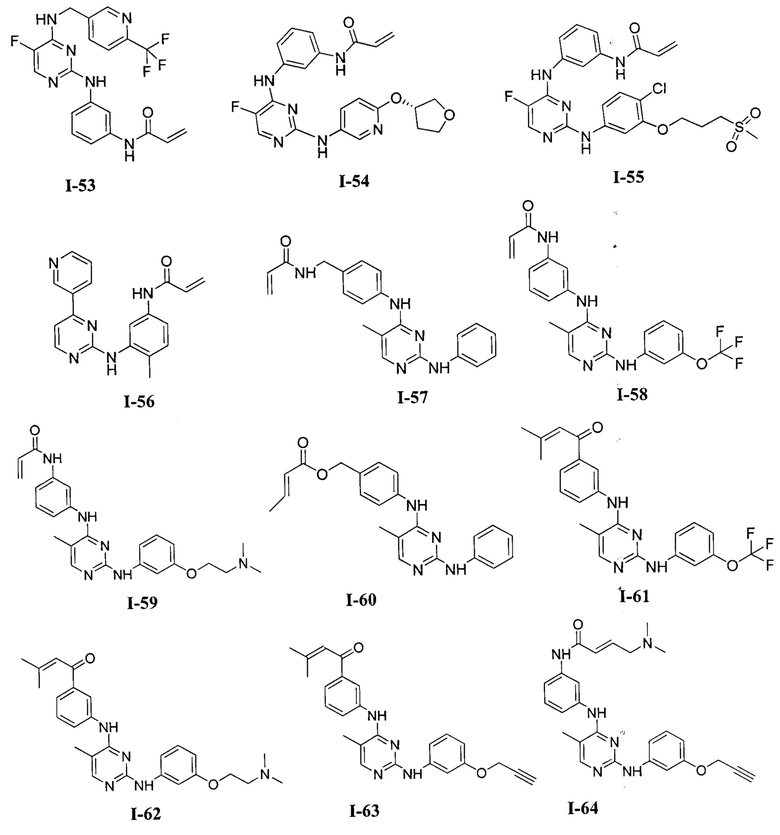
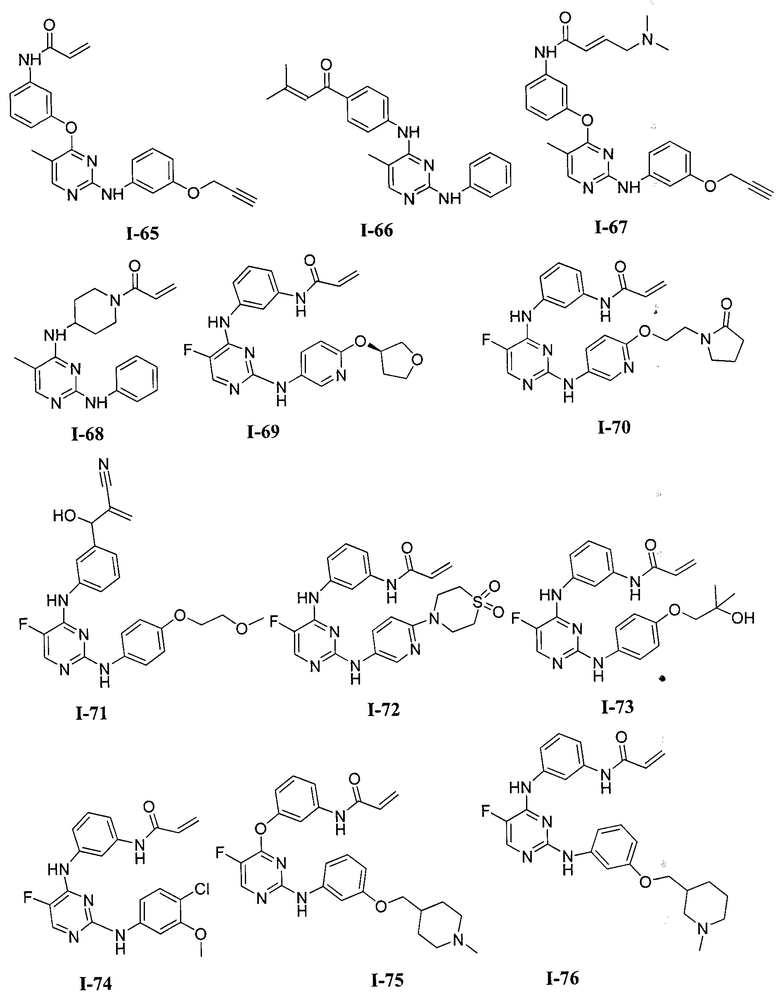
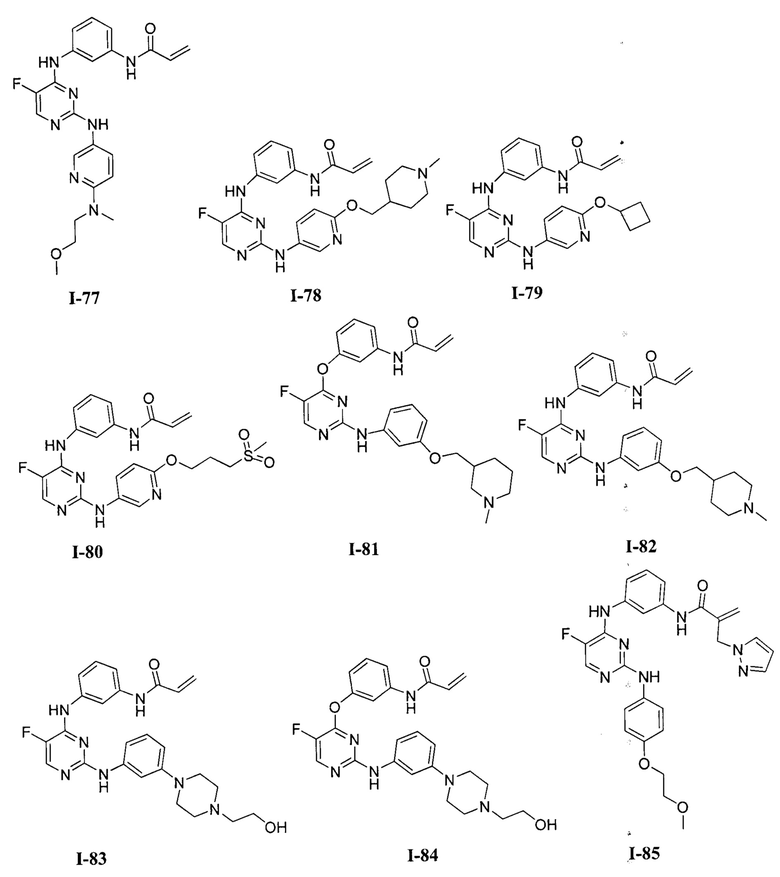
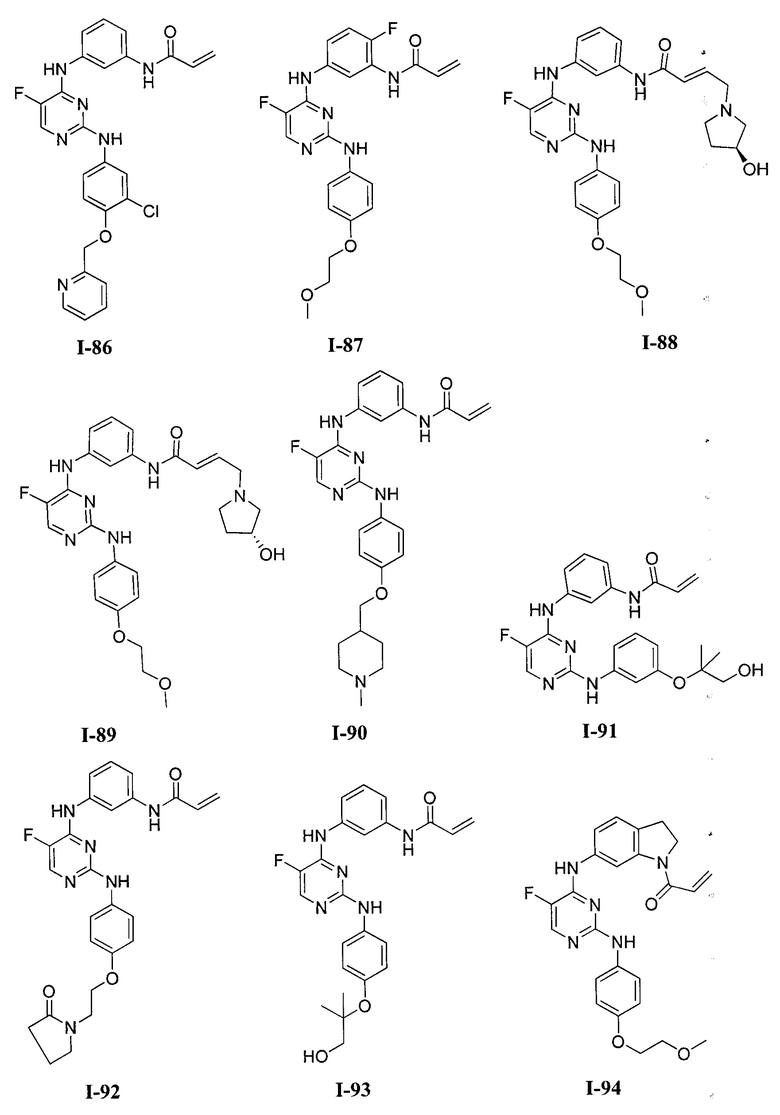
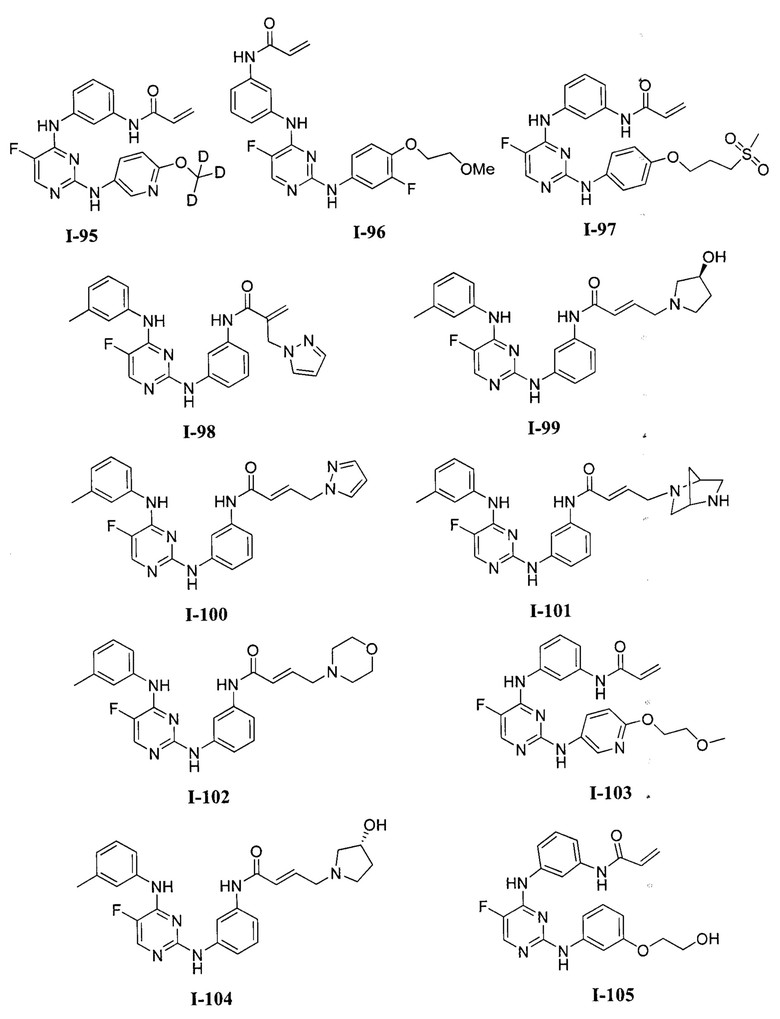
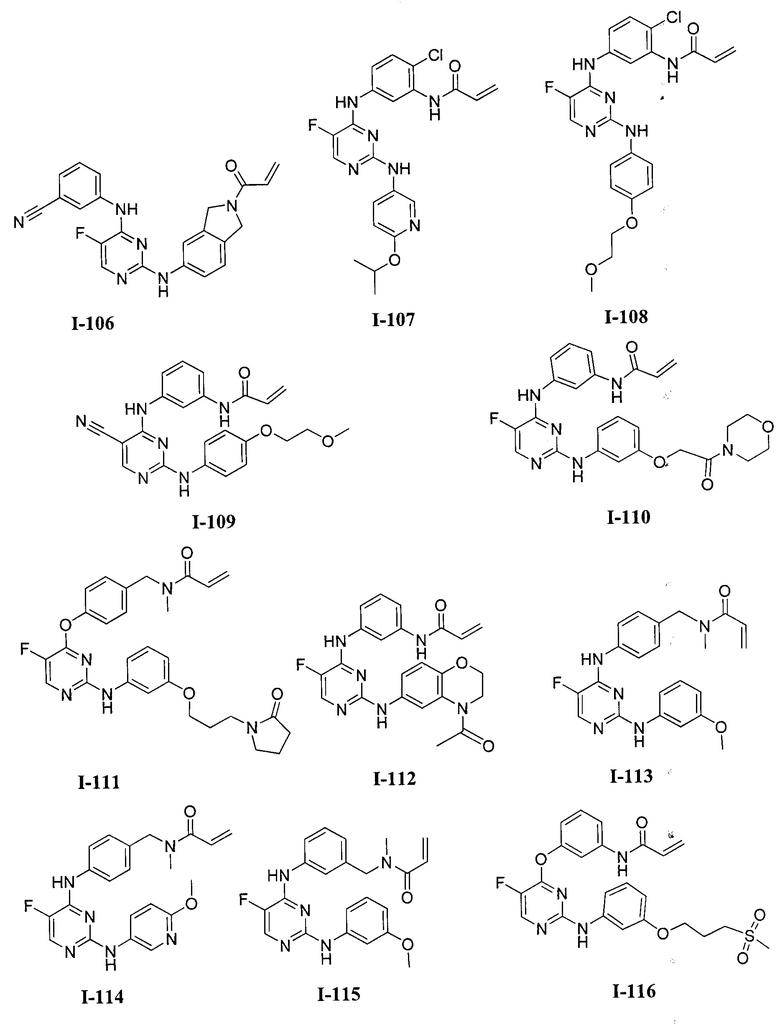
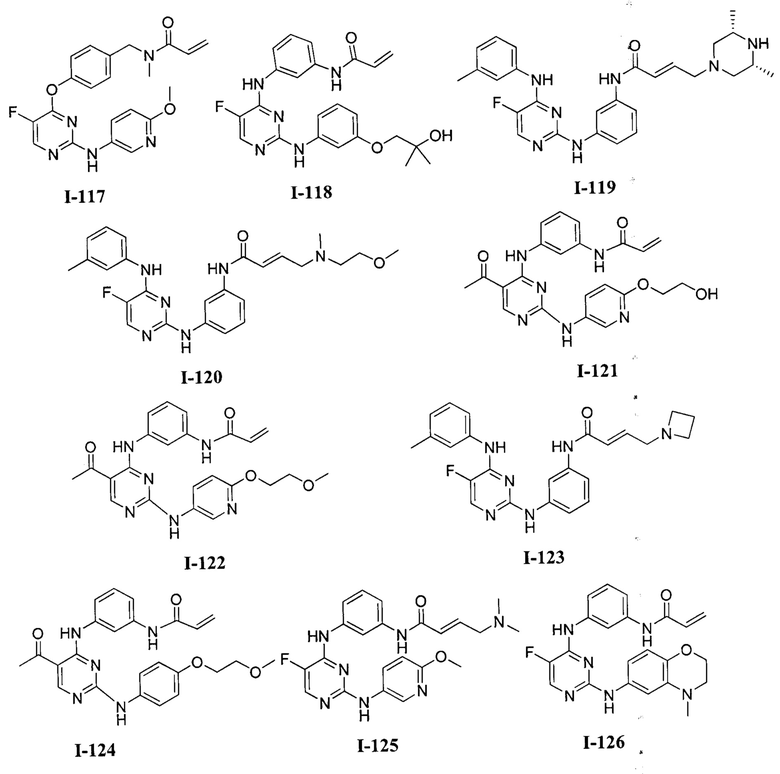
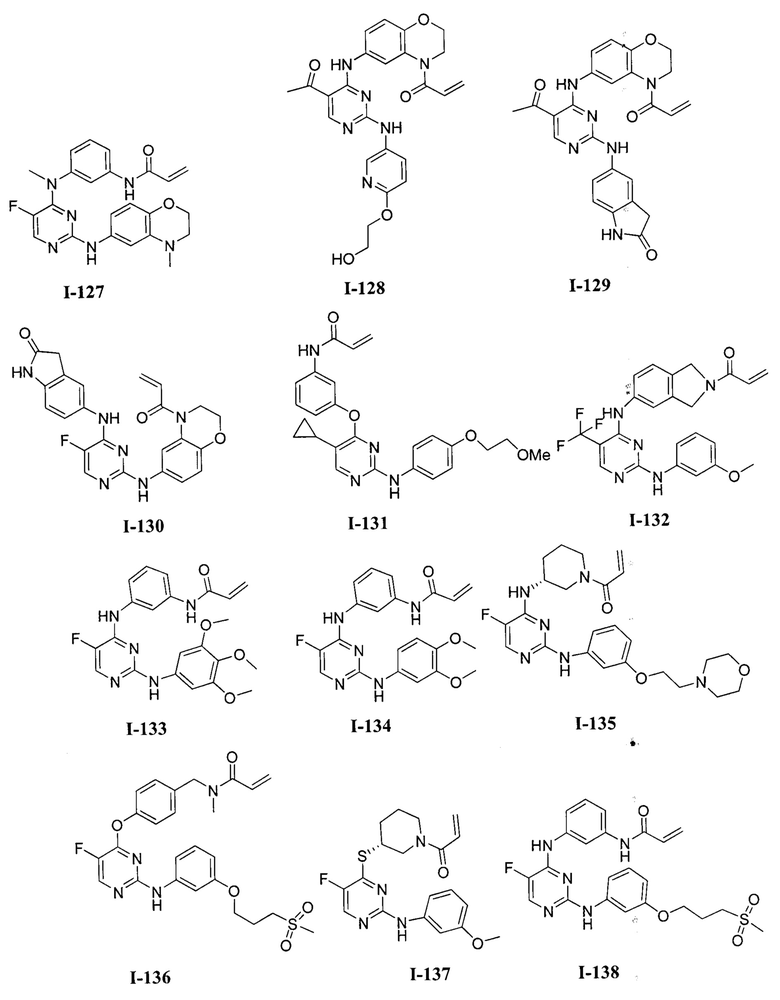
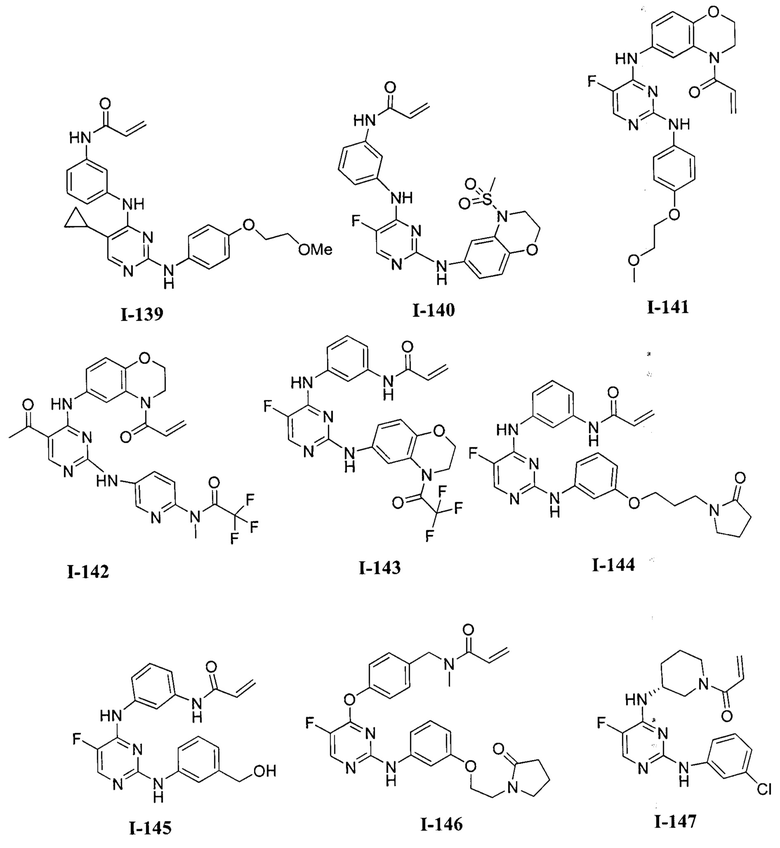
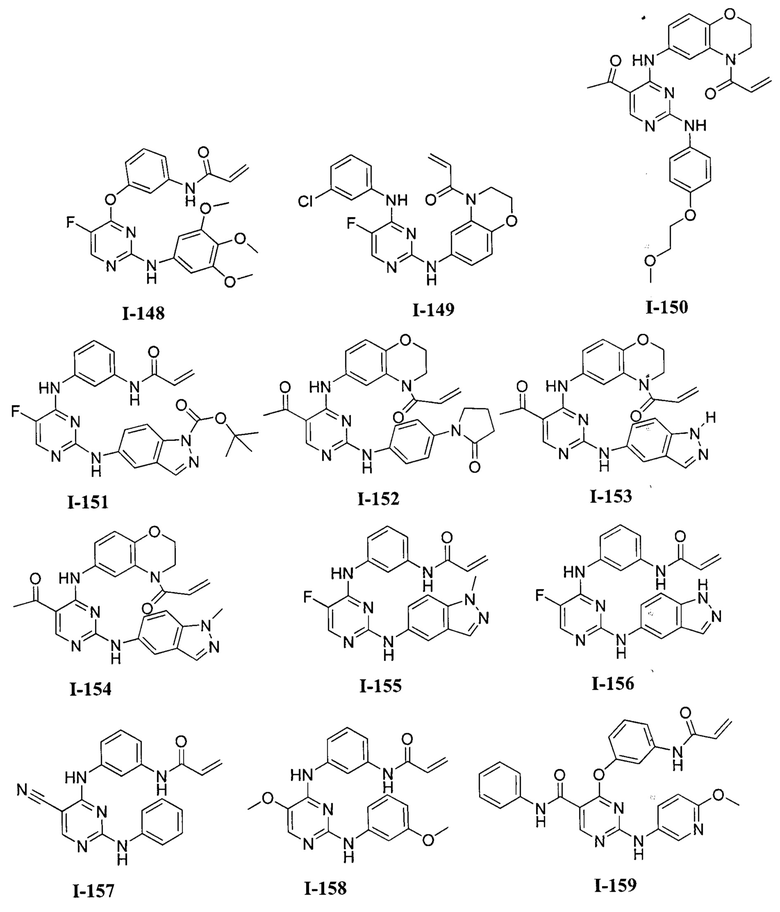
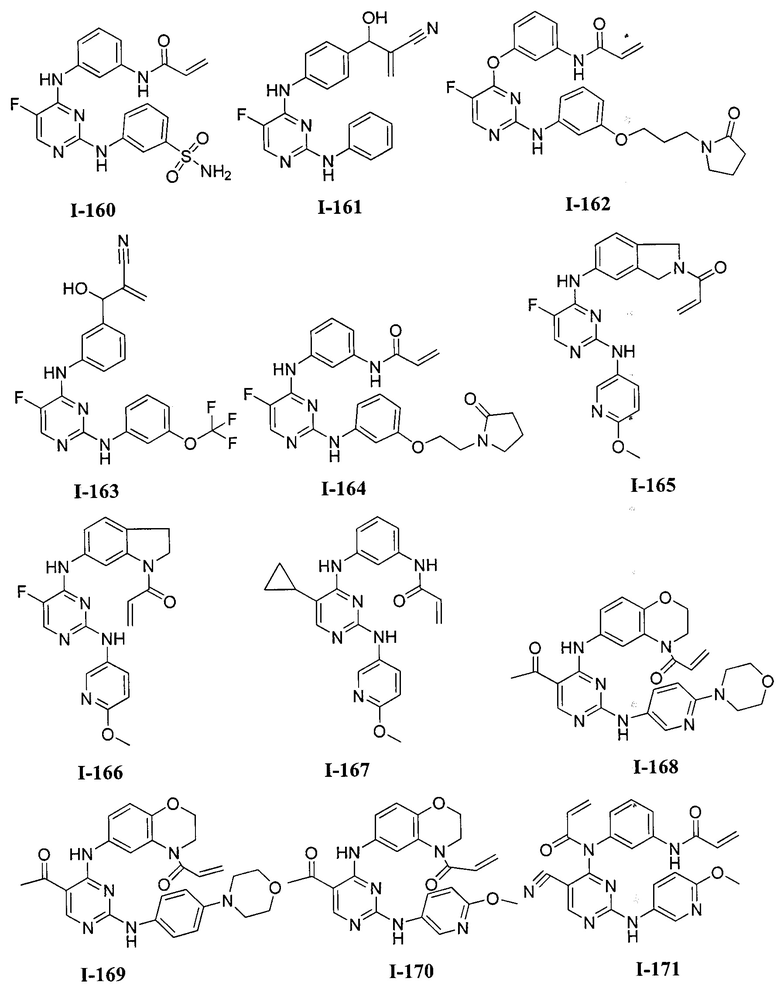
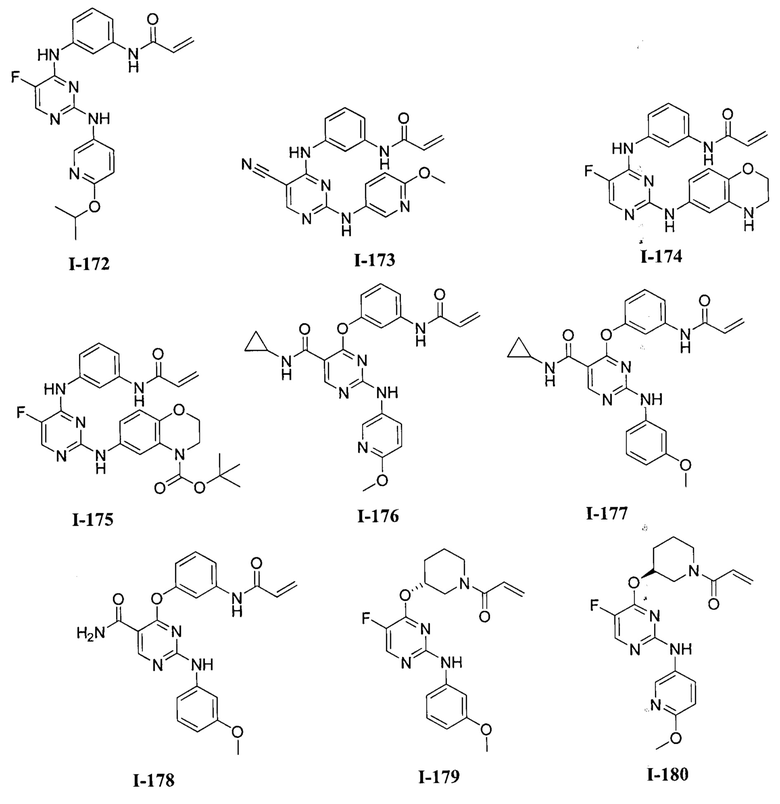
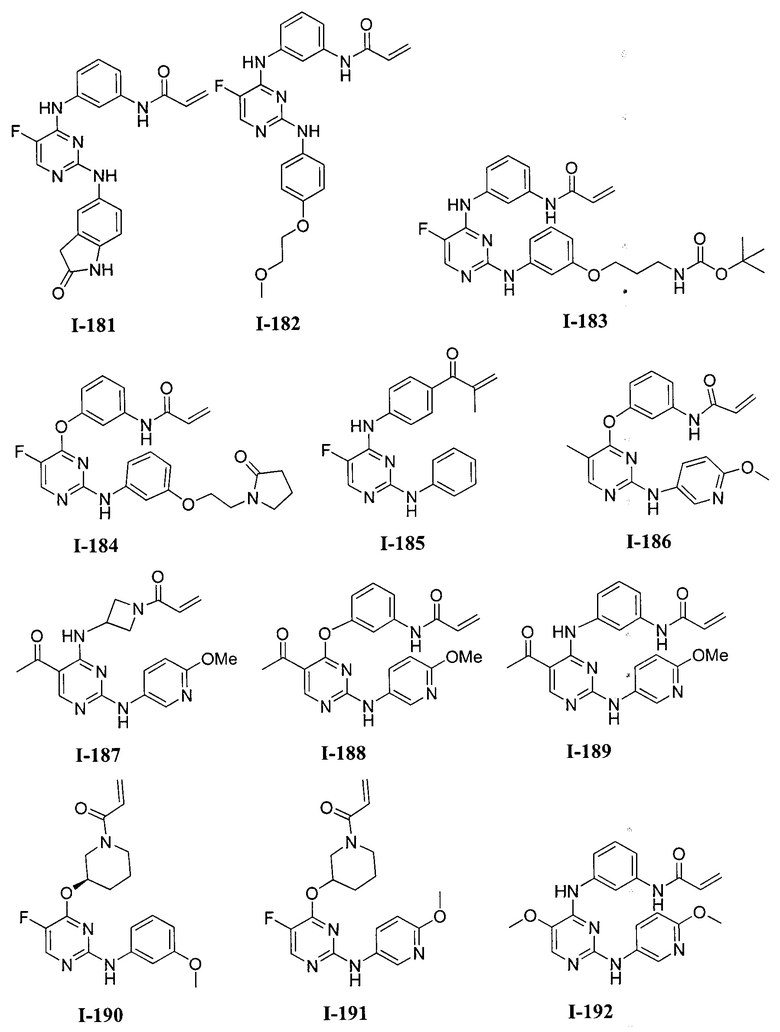
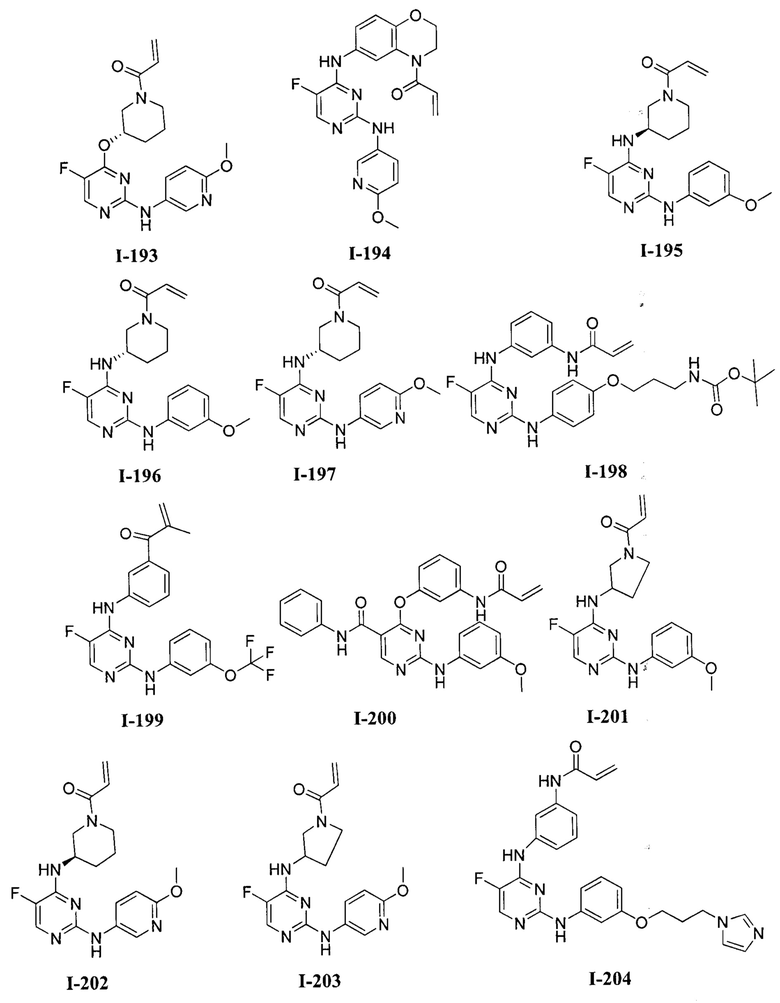
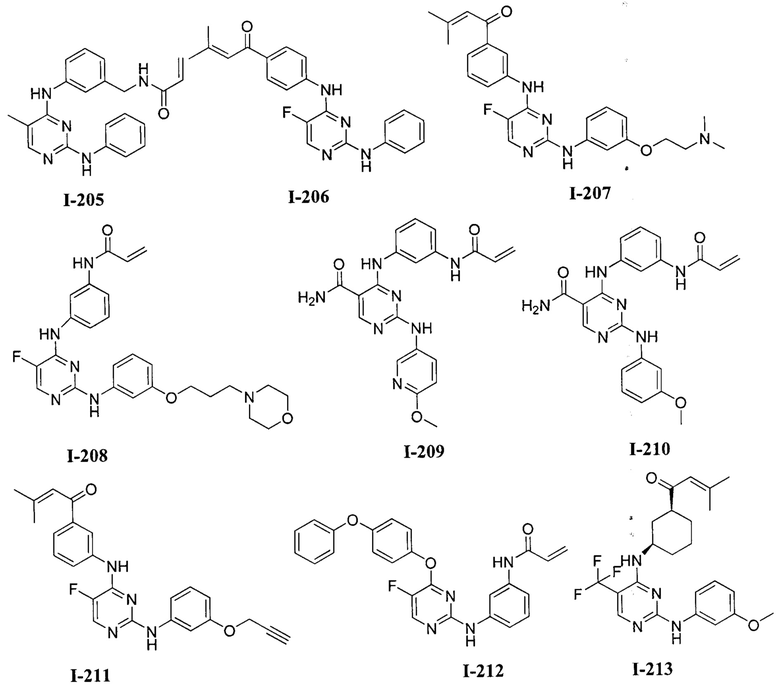
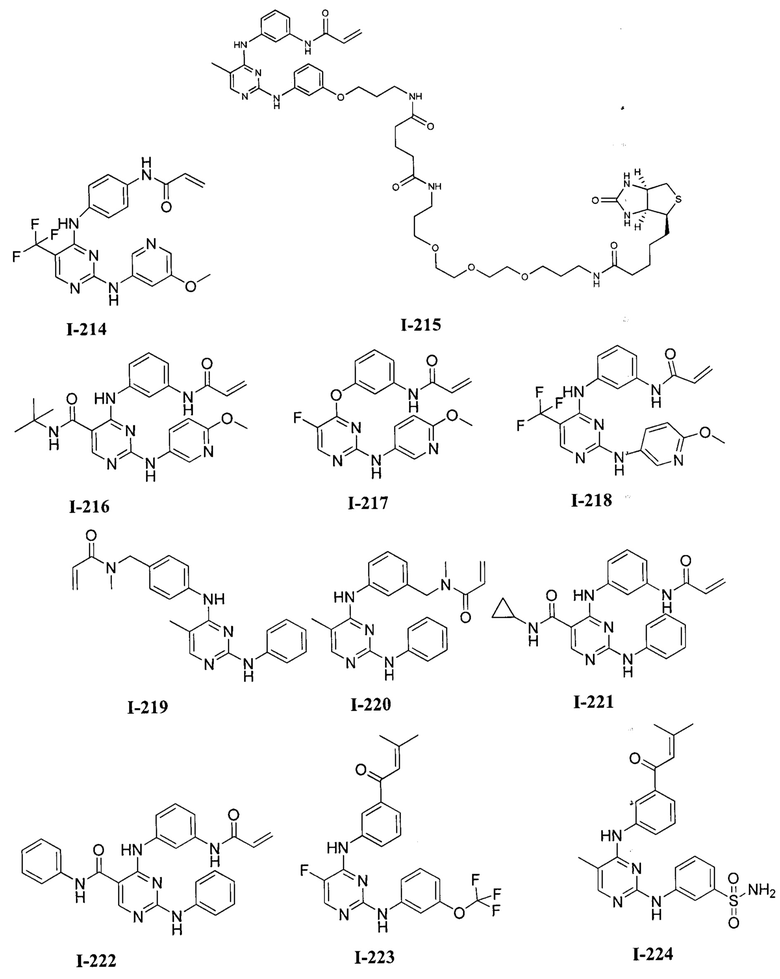
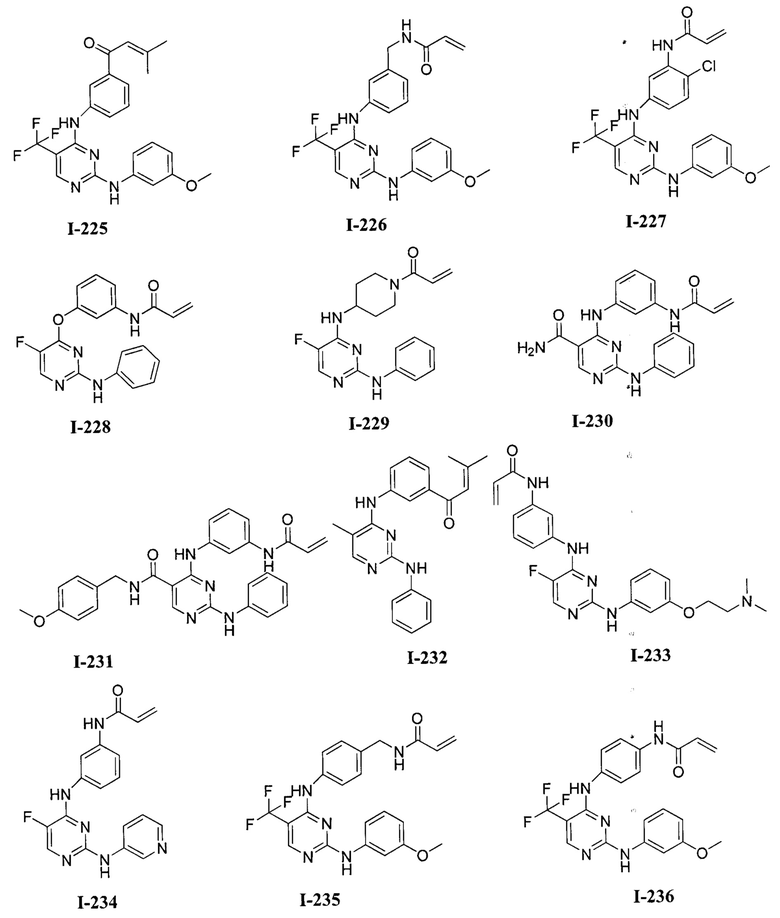
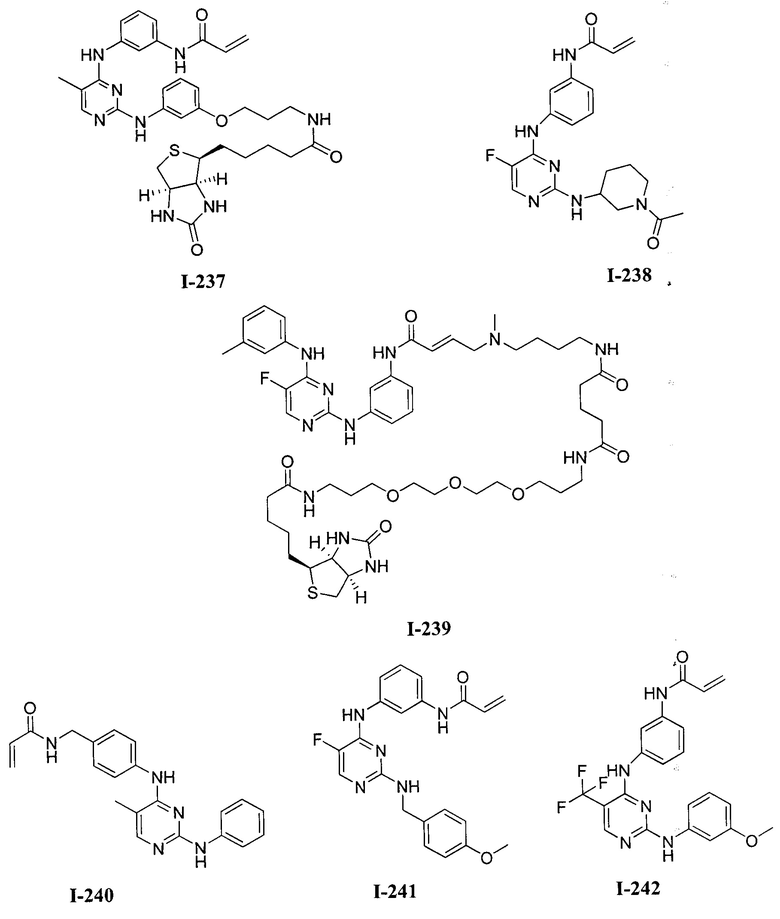
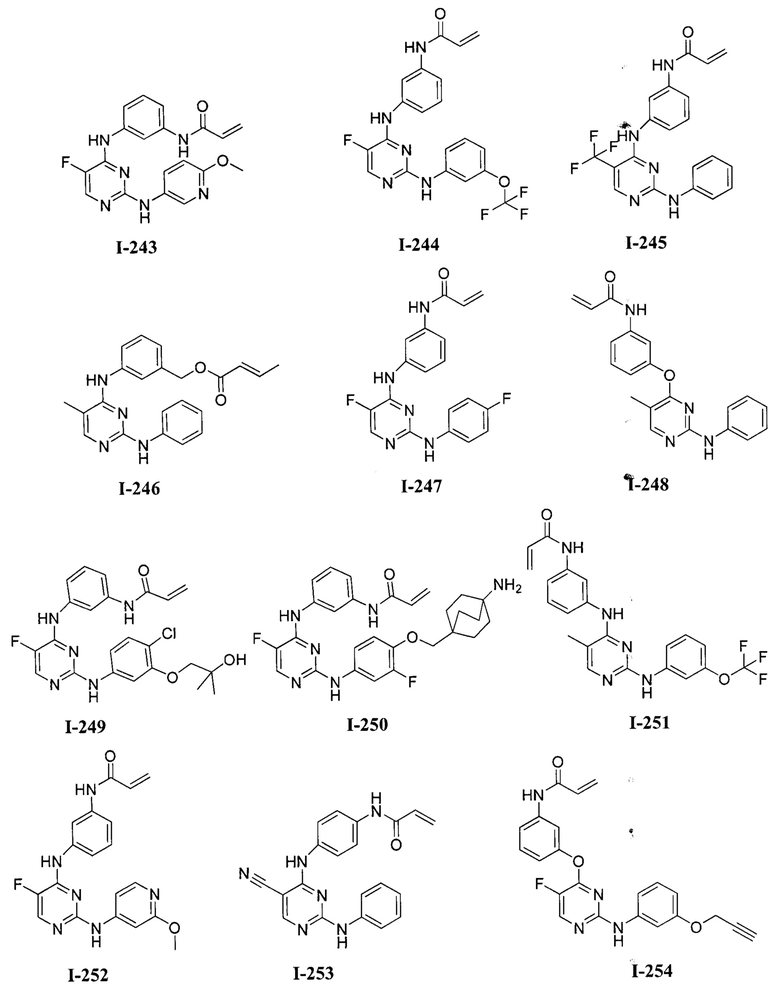
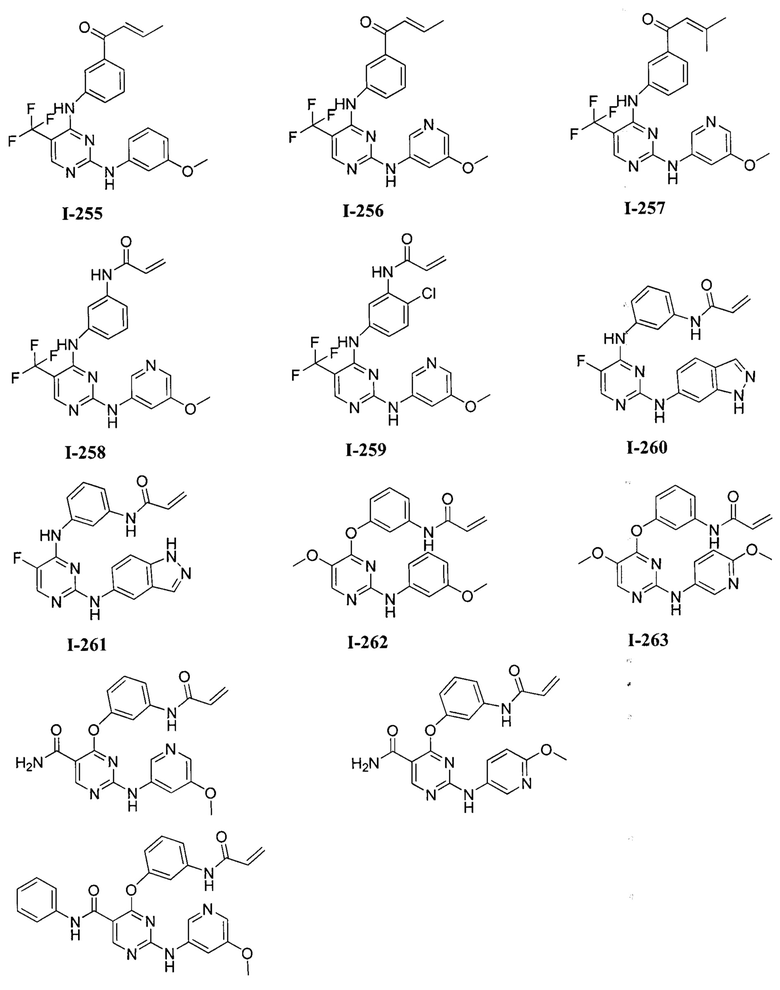
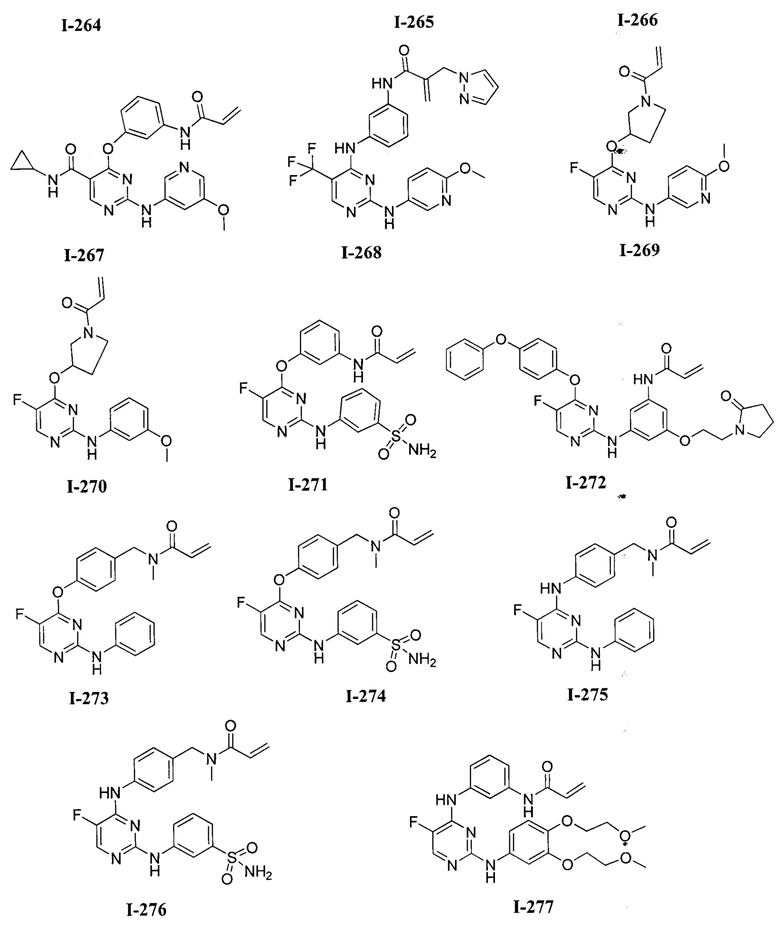
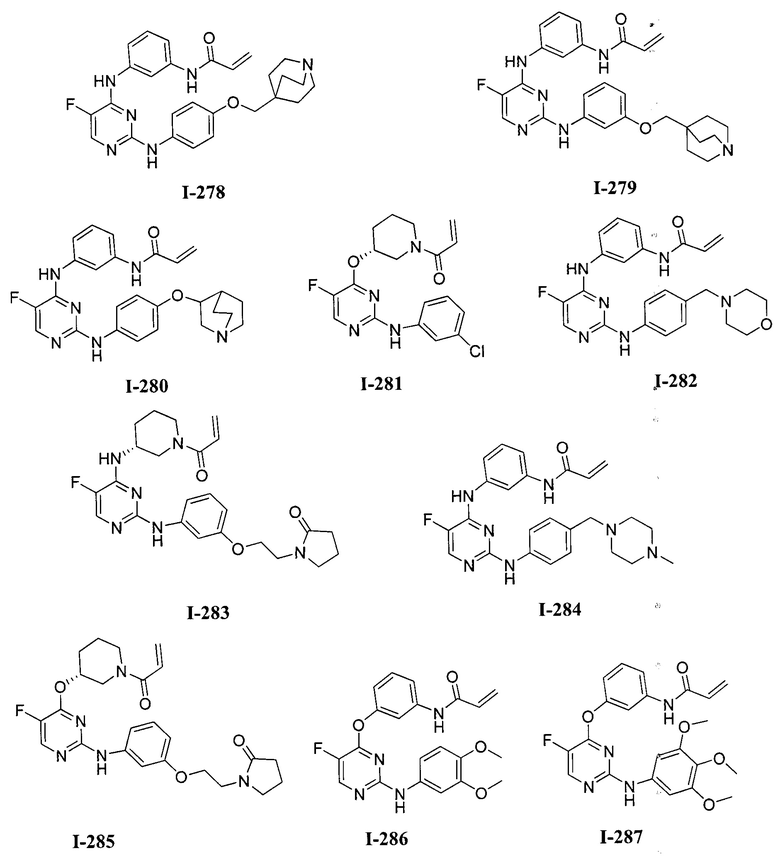
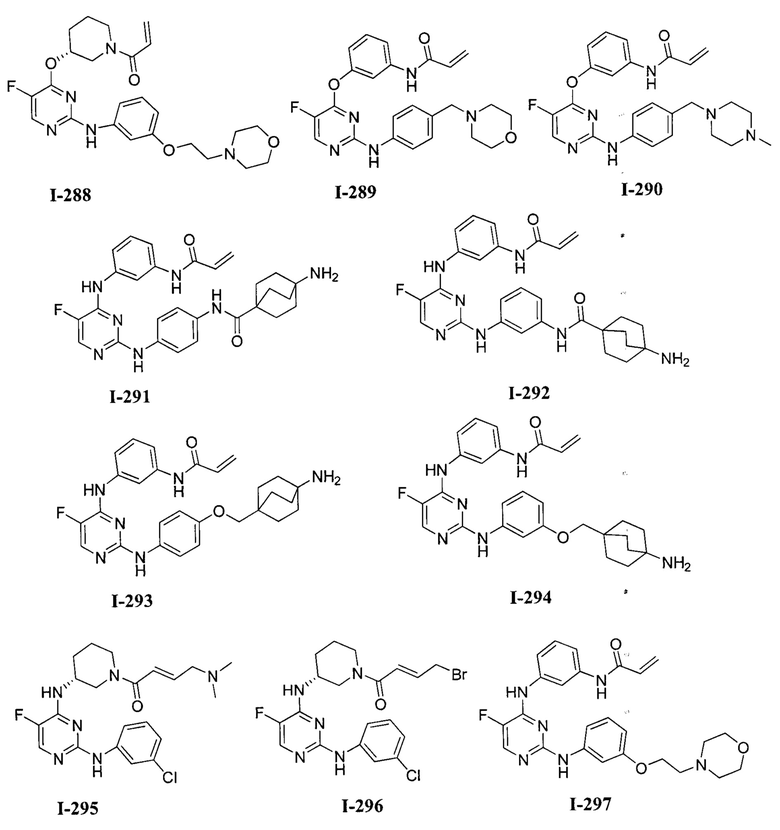
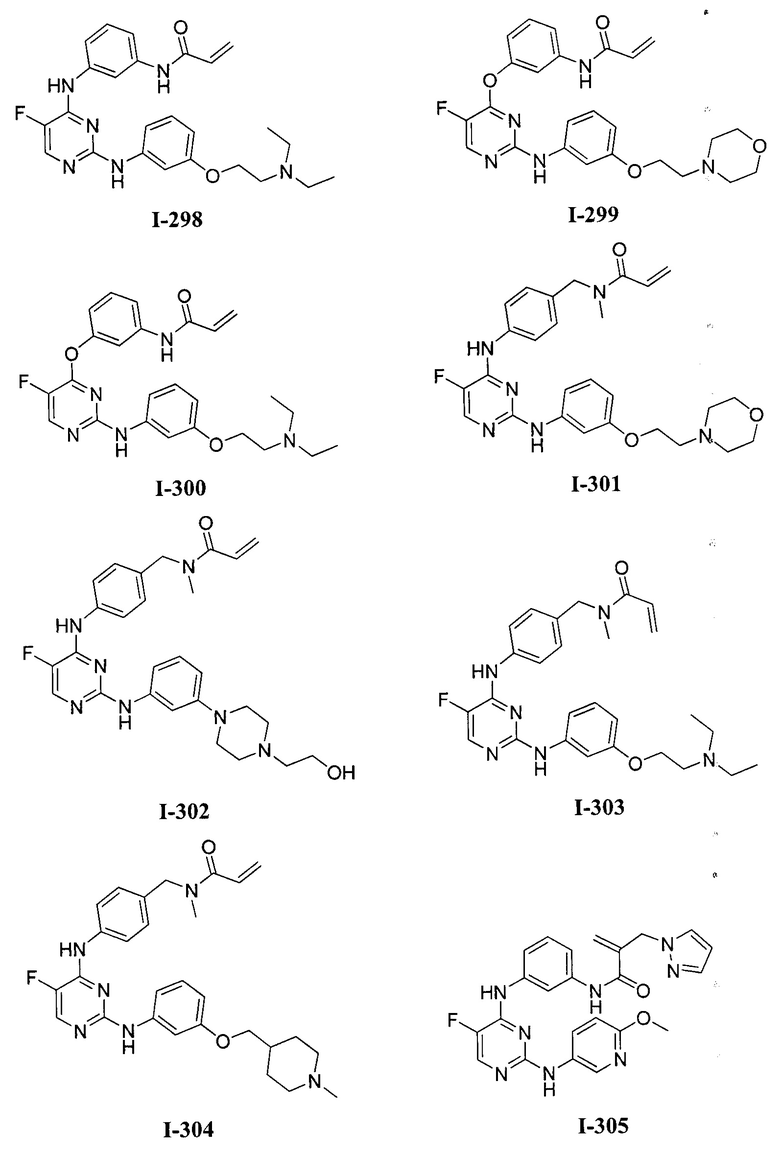
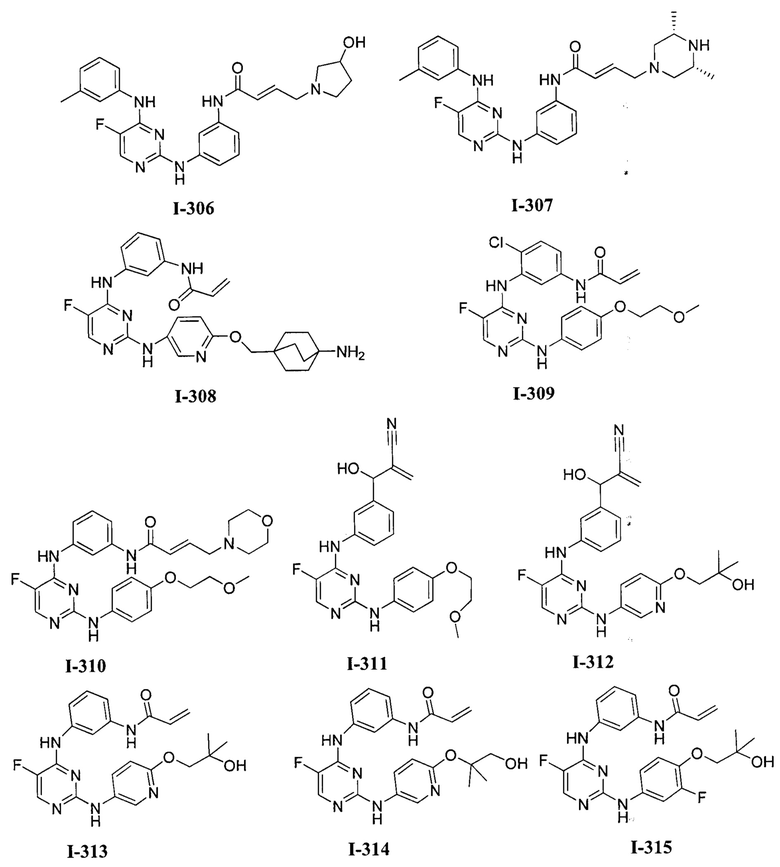
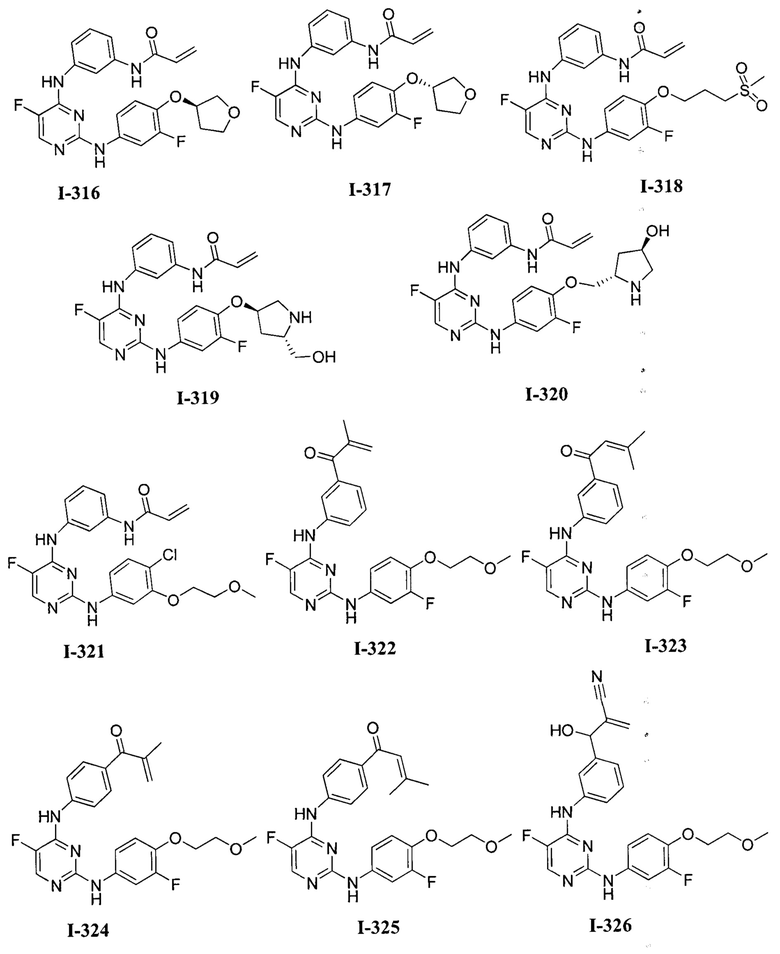
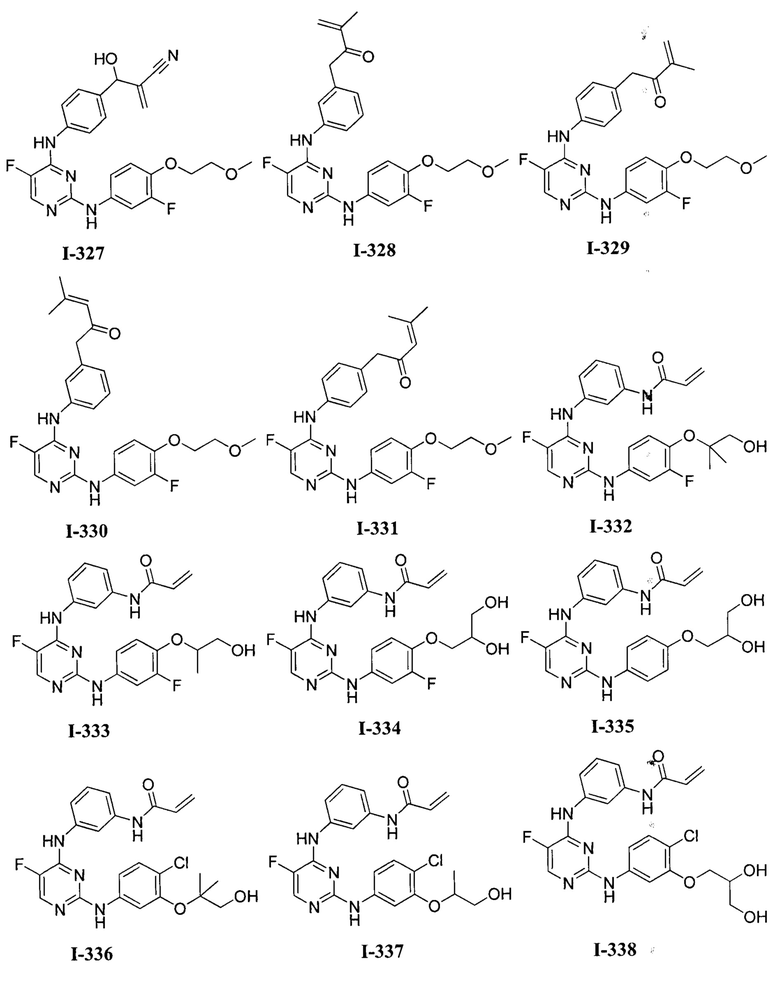
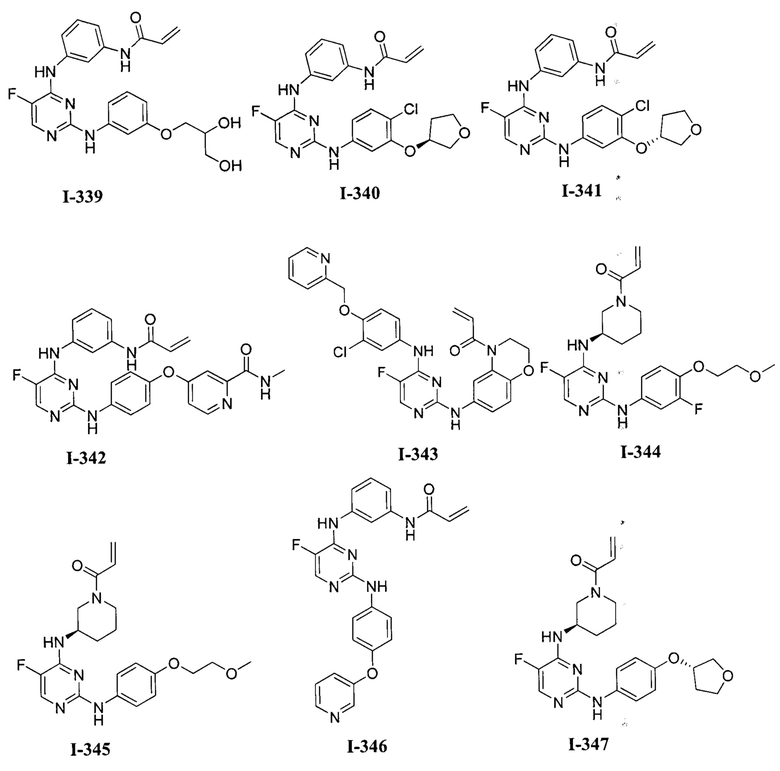
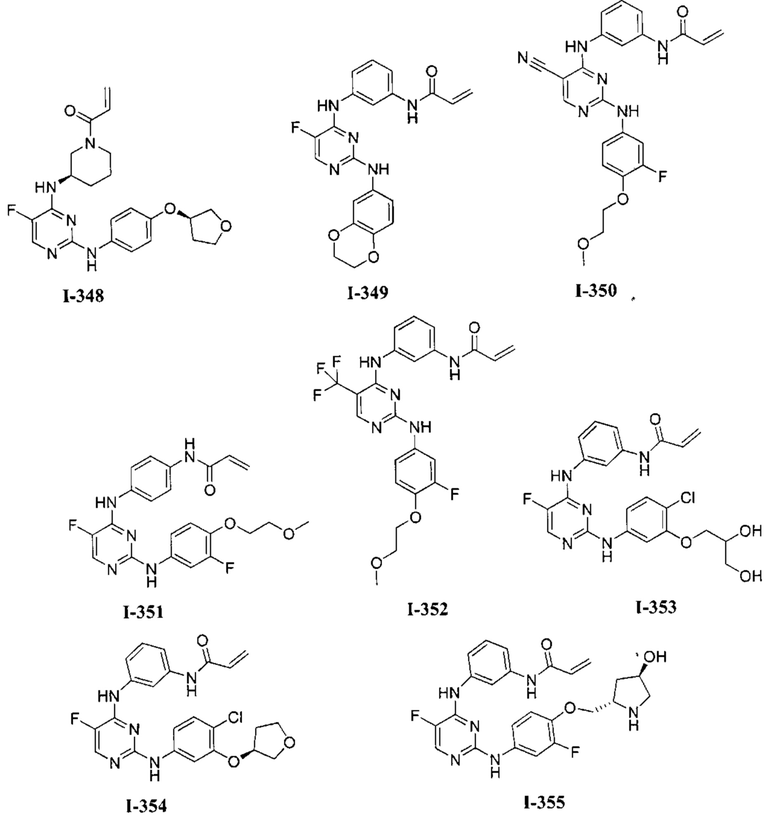
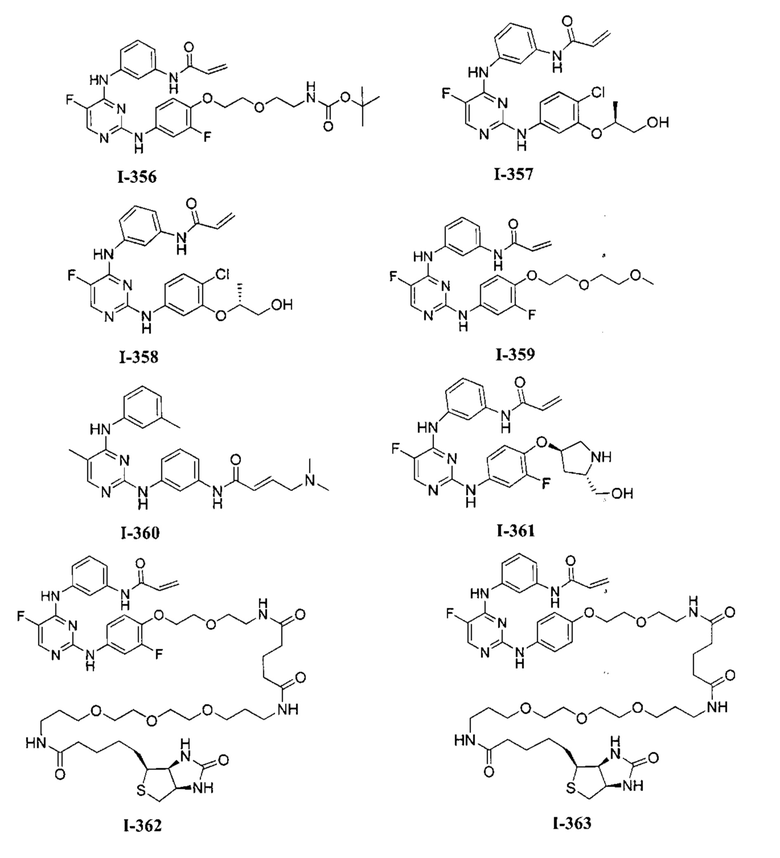
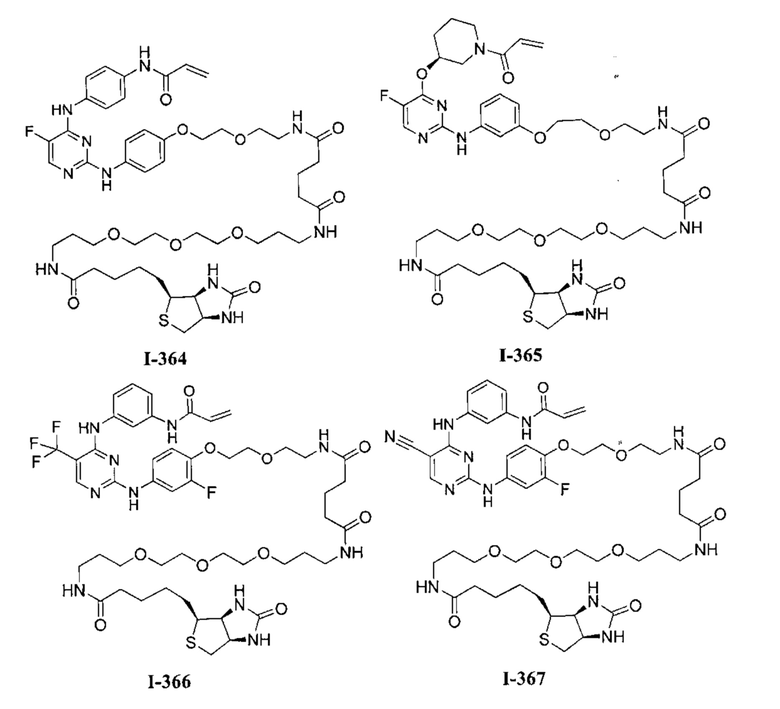
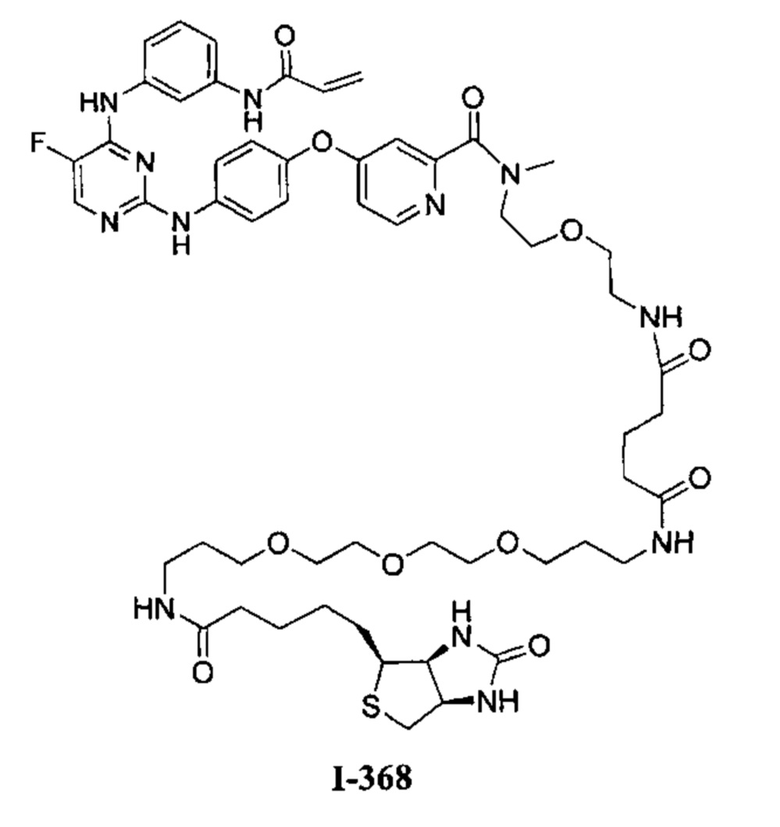
[00122] В некоторых вариантах осуществления настоящее изобретение обеспечивает любое соединение, представленное выше в Таблице 5 или фармацевтически приемлемую моль указанного соединения.
[00123] В некоторых вариантах осуществления настоящее изобретение обеспечивает соединения, выбранные из:
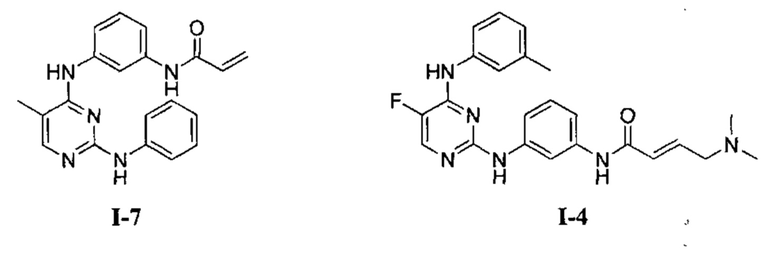
или фармацевтически приемлемая соль указанного соединения.
[00124] Как описано в настоящем тексте, соединения согласно настоящему изобретению представляют собой необратимые ингибиторы по меньшей мере одной киназы из ErbB1, ErbB2, ErbB3 и ErbB4 или мутанта указанной киназы. В некоторых вариантах осуществления соединения согласно настоящему изобретению представляют собой необратимые ингибиторы киназы ТЕС (например, BTK) и JAK3. Среднему специалисту в данной области понятно, что соединения согласно настоящему изобретению представляют собой необратимые ингибиторы. В некоторых вариантах осуществления такие соединения можно применять в качестве соединений для разделения. В других вариантах осуществления такие обратимые соединения можно применять в качестве ингибиторов ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназы и/или JAK3, или мутантов указанных киназ, и, соответственно, для лечения одного или более заболеваний, описанных в настоящей заявке. Примеры обратимых соединений согласно настоящему изобретению имеют следующую структуру.
или фармацевтически приемлемая соль указанного соединения.
4. Применения, лекарственные формы и назначения
Фармацевтически приемлемые композиции
[00125] В соответствии с другим вариантом осуществления изобретения, настоящее изобретение предоставляет композицию, включающую соединение согласно настоящему изобретению или его фармацевтически приемлемое производное, и фармацевтически приемлемый носитель, адъювант или разбавитель. Количество соединения в композиции согласно настоящему изобретению является таковым, чтобы эффективно детектируемо ингибировать протеинкиназу, в частности одну из ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназу, и/или JAK3, или их мутант, в биологическом образце или в пациенте. В некоторых вариантах осуществления изобретения количество соединения в композиции согласно настоящему изобретению является таковым, чтобы эффективно детектируемо ингибировать по крайней мере одну из ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназу, и/или JAK3, или их мутант, в биологическом образце или в пациенте. В некоторых вариантах осуществления изобретения композиция согласно настоящему изобретению изготавливается для назначения пациенту, нуждающемуся в такой композиции. В некоторых вариантах осуществления изобретения композиция согласно настоящему изобретению изготавливается для орального применения пациентом.
[00126] Термин "пациент", используемый здесь, означает животное, предпочтительно млекопитающее, и наиболее предпочтительно человека.
[00127] Термин "фармацевтически приемлемый носитель, адъювант или разбавитель" относится к нетоксичному носителю, адъюванту или разбавителю, который не разрушает
фармакологическую активность указанного соединения, вместе с которым он используется при изготовлении. Фармацевтически приемлемый носитель, адъювант или разбавитель, который может быть использован в композиции согласно настоящему изобретению, включает ионообменники, глинозем, стеарат алюминия, лецитин, белки сыворотки, такие как сывороточный альбумин человека, буферные субстанции, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, частично глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, двузамещенный фосфат натрия, двузамещенный фосфат калия, хлорид натрия, соли цинка, коллоидный кремнезем, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натриевая соль карбоксиметилцеллюлозы, полиакрилаты, воски, полиэтилен-полиоксипропилен блок-сополимеры, полиэтиленгликоль и ланолин, но не ограничивается ими.
[00128] "Фармацевтически приемлемое производное" означает любую нетоксичную соль, сложный эфир, соль сложного эфира или другое производное соединения согласно настоящему изобретению, которое, будучи назначено реципиенту, способно предоставить либо прямо, либо косвенно, соединение согласно настоящему изобретению или метаболит с ингибирующей активностью или его остаток.
[00129] В настоящем описании термин "метаболит с ингибирующей активностью или его остаток" означает, что метаболит или его остаток также является ингибитором по крайней мере одной из ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназы, и/или JAK3, или их мутанта.
[00130] Композиции согласно настоящему изобретению могут назначаться орально, парентерально, в виде спрея для ингаляции, местно, ректально, назально, букально, вагинально или с использованием имплантируемого резервуара. Термин "парентерально", используемый здесь, включает подкожную, внутривенную, внутримышечную, интраартикулярную, интрасиновиальную, внутригрудинную, внутрипеченочную и внутричерепную инъекционную или инфузионную методики, а также методику введения в пораженные ткани. Предпочтительно, указанные композиции назначаются орально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы композиции согласно настоящему изобретению могут быть водными или масляными суспензиями. Такие суспензии могут назначаться в соответствии методиками, известными в
предшествующем уровне техники, с использованием подходящих диспергирующих или смачивающих веществ и суспендирующих веществ. Стерильная инъекционная композиция также может представлять собой стерильный раствор или суспензию для инъекций в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, например раствор в 1,3-бутандиоле. Среди таких приемлемых разбавителей и растворителей, которые могут применяться, присутствуют вода, раствор Рингера и изотонический раствор хлорида натрия. Вдобавок стерильные, связанные масла традиционно применяют в качестве растворителей или среды для приготовления суспензии.
[00131] Для этой цели может применяться любое легкое связанное масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, являются подходящими для препаратов в виде инъекций, также как и природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Такие масляные растворы или суспензии также могут содержать спиртовые разбавители или диспергирующие вещества с длинной цепью, такие как карбоксиметилцеллюлоза или сходные диспергирующие вещества, которые обычно используют в лекарственных средствах в виде фармацевтически приемлемых дозированных форм, включая эмульсии и суспензии. Другие обычно используемые поверхностно-активные вещества, такие как Твин, Спан и другие эмульгирующие вещества или вещества, усиливающие биодоступность, которые обычно используют в производстве фармацевтически приемлемых твердых, жидких или других дозированных формах, также могут быть использованы для целей создания лекарственной формы.
[00132] Фармацевтически приемлемые композиции согласно настоящему изобретению могут назначаться орально в любой приемлемой для орального применения дозированной форме, включая капсулы, таблетки, водные суспензии или растворы. В случае таблеток для орального применения, обычно используемые носители включают лактозу и кукурузный крахмал, но не ограничиваются ими. Также обычно добавляют лубриканты, такие как стеарат магния. Разбавители, пригодные для орального назначения в форме капсул, включают лактозу и сухой кукурузный крахмал. В случае, когда требуются водные суспензии для орального применения, активный ингредиент комбинируют
эмульгирующими и суспендирующими агентами. При желании также могут быть добавлены некие подсластители, вкусовые вещества или красители.
[00133] С другой стороны, фармацевтически приемлемые композиции согласно настоящему изобретению могут назначаться в форме суппозиториев для ректального назначения. Они могут быть изготовлены смешением указанного агента с подходящим не раздражающим наполнителем, который находится в твердой форме при комнатной температуре, но в жидкой форме при ректальной температуре, и, следовательно, будет расплавляться в прямой кишке с высвобождением указанного лекарства. Такие материалы включают кокосовое масло, пчелиный воск и полиэтиленгликоли.
[00134] Фармацевтически приемлемые композиции согласно настоящему изобретению также могут назначаться местно, особенно когда объект для обработки включает области или органы, легко доступные для местного применения, включая заболевания глаз, кожи или нижнюю часть кишечного тракта. Соответствующие лекарственные формы легко изготавливаются для каждой из этих областей или органов.
[00135] Формы местного применения для нижней части кишечного тракта могут быть выполнены в виде ректальных суппозиториев (смотри ниже) или в виде соответствующей лекарственной формы в виде клизмы. Также могут быть использованы трансдермальные пластыри местного назначения.
[00136] Предоставляемые фармацевтически приемлемые композиции для местного применения могут быть выполнены в виде соответствующей мази, содержащей активный компонент, суспендированный или растворенный в одном или нескольких носителях. Носители для местного применения соединений согласно настоящему изобретению включают минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и воду, но не ограничиваются ими. С другой стороны, предоставляемые фармацевтически приемлемые композиции могут быть выполнены в форме соответствующего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Соответствующие носители включают минеральное масло, сорбитмоностеарат, полисорбат 60, цетиловые сложные эфиры воска, цетиариловый спирт, 2-октилдодеканол, бензиловый спирт и воду, но не ограничиваются ими.
[00137] Предоставляемые фармацевтически приемлемые композиции для офтальмологического применения могут быть выполнены в форме тонкодисперсных суспензий в изотоническом стерильном солевом растворе с доведенным рН, или, предпочтительно, в форме раствора в изотоническом стерильном солевом растворе с доведенным рН с консервантом, таким как бензалкониумхлорид, либо без него. С другой стороны, фармацевтически приемлемые композиции для офтальмологического применения могут быть выполнены в форме мази, такой как вазелин.
[00138] Фармацевтически приемлемые композиции согласно настоящему изобретению также могут назначаться в виде назального аэрозоля или ингаляции. Такие композиции изготавливают в соответствии с методами, хорошо известными из предшествующего уровня техники для фармацевтических лекарственных форм, и могут быть изготовлены в виде раствора в соляном растворе с применением бензилового спирта или других подходящих консервантов, инициаторов абсорбции для увеличения биодоступности, фторуглеродов, и/или других традиционно используемых растворяющих или диспергирующих веществ.
[00139] Наиболее предпочтительно, фармацевтически приемлемые композиции согласно настоящему изобретению изготавливают в форме для орального назначения.
[00140] Количество соединений согласно настоящему изобретению, которое может быть смешано с материалом носителя для получения композиции в форме единичной дозировки варьируется в зависимости от организма, подвергающегося лечению, от специфического способа назначения. Предпочтительно, представленные композиции могут быть изготовлены таким образом, что дозировка ингибитора в количестве от 0.01 до 100 мг на кг веса в день может быть назначена пациенту, принимающему указанные композиции.
[00141] Также должно быть ясно, что специфическая дозировка и режим лечения для любого конкретного пациента будет зависеть от множества факторов, включающих активность используемого специфического соединения, возраст, вес тела, общее состояние здоровья, пол, диету, время назначения, скорость выведения, комбинирование с другими лекарствами, решение лечащего доктора и тяжесть конкретного заболевания, которое предполагается излечить. Количество соединения согласно настоящему
изобретению в указанной композиции также будет зависеть от конкретного соединения в указанной композиции.
Использование соединений и фармацевтически приемлемых композиций
[00142] Соединения и композиции, описанные здесь, в целом пригодны для для ингибирования протеинкиназной активности одного или нескольких ферментов.
[00143] Устойчивость к лекарственным средствам является значительным вызовом для терапий, направленных на определенные мишени. Например, устойчивость к лекарственным средствам описана для Gleevec® и Iressa®, а также для нескольких других разрабатывающихся ингибиторов киназ. В дополнение, устойчивость к лекарственным средствам описана для рецепторов cKit и PDGFR. Сообщалось, что необратимые ингибиторы могут быть эффективными против форм протеинкиназ, устойчивых к лекарственных средствам (Kwak, Е.L., R. Sordella, et al. (2005). "Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib." PNAS 102(21): 7665-7670.) He стремясь быть ограниченными какой-либо конкретной теорией, предполагается, что соединения согласно настоящему изобретению могут являться эффективными ингибиторами форм протеинкиназ, устойчивых к лекарственных средствам.
[00144] В настоящем описании термин "клиническая устойчивость к лекарственным средствам" означает потерю восприимчивости мишени для лекарственного средства к обработке этим лекарственным средством вследствие мутаций в этой мишени для лекарственного средства.
[00145] В настоящем описании термин "устойчивость" относится к изменениям в последовательности природной нуклеиновой кислоты, кодирующей целевой белок, и/или последовательности целевого белка, при этом такие изменения снижают или сводят на нет ингибирующий эффект ингибитора такого целевого белка.
[00146] Примеры киназ, ингибирующихся соединениями и композициями, описанными здесь, и против которых применимы способы, описанные здесь, включают ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназу (включая BTK, ITK, ТЕС, ВМХ и RLK), и/или JAK3, или их мутанты.
[00147] Активность соединения, используемого в настоящем изобретении в качестве ингибиторов ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназы, и/или JAK3, или их мутантов, может быть проверена in vitro, in vivo или в клеточных линиях. Проверка in vitro включает
эксперименты, в которых определяется ингибирование либо фосфорилирующей активности и/или последующих функциональных следствий, или АТФ-азной активности активированных ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназы, и/или JAK3, или их мутантов. В альтернативных экспериментах in vitro производится количественный анализ способности ингибитора связываться с ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназой, и/или JAK3. Связывание ингибитора может быть измерено методом мечения ингибитора радиоактивной меткой перед связыванием, выделения комплексов ингибитор/ErbB1, ингибитор/ErbB2, ингибитор/ErbB3, ингибитор/ErbB4, ингибитор/ТЕС-киназа (например, ТЕС, BTK, ITK, RLK и ВМХ), или ингибитор/JAK3 и определения количества радиоактивных комплексов. С другой стороны, связывание ингибитора может быть проверено путем проведения эксперимента по конкурентному связыванию, когда новые ингибиторы инкубируют с ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназой, и/или JAK3, связанными с известными радиоактивно меченными лигандами. Подробные условия методики обнаружения соединения, используемого в настоящем изобретении в качестве ингибитора ErbB1, ErbB2, ErbB3, ErbB4, ТЕС-киназы, и/или JAK3, или их мутантов, изложены ниже в Примерах.
[00148] Тирозиновые протеинкиназы являются классом ферментов, катализирующих перенос фосфатной группы с АТФ или ГТФ на остаток тирозина, расположенный в белковом субстрате. Рецепторные тирозинкиназы передают сигналы снаружи внутрь клетки путем активации вторичных эффекторов, передающих сигналы, в ходе события фосфорилирования. Множество клеточных процессов активизируются этими сигналами, включая пролиферацию, утилизацию углеводов, синтез белков, ангиогенез, клеточный рост, и выживаемость клеток,
(а) Семейство ErbB
[00149] Рецепторы ErbB, основное семейство рецепторных тирозинкиназ, состоят из внеклеточного домена, связывающего лиганд, одного трансмембранного домена, и внутриклеточного домена с активностью тирозинкиназы. В семейство ErbB входят ErbB1 (общеизвестный как EGFR), ErbB2 (общеизвестный как HER2 или neu), ErbB3 (общеизвестный как HER3), и ErbB4 (общеизвестный как HER4). Идентифицировано более 10 лигандов (включая EGF, TGFα, AR, ВТС, EPR, HB-EGF, NRG-1, NRG-2, NRG-3, NRG-4) для различных членов семейства рецепторов. При связывании лиганда
конформация внутриклеточного домена изменяется, что приводит к образованию гомодимеров или гетеродимеров с другими членами семейства ErbB. Димеризация индуцирует фосфорилирование определенных остатков тирозина внутриклеточного домена, служащих местом связывания для белков-адапторов и эффекторов передающих сигнал дальше. В некоторых случаях происходит активация путей фосфатидил-инозитол-3-киназы (PI3K) и митоген-активируемой протеинкиназы, что приводит к пролиферации выживанию клеток (Lin, N.U.; Winer, Е.P., Breast Cancer Res 6: 204-210, 2004).
[00150] Взаимодействие между членами семейства влечет за собой дефицит ErbB2, лиганд которой не известен, ErbB3, которая не обладает киназной активностью. EGFR, ErbB3, и ErbB4 связываются с лигандом, что приводит к индукции гомодимеризации или гетеродимеризации рецептора ErbB, тогда как ErbB2 выступает в качестве предпочтительного партнера для димеризации. Состав парных комбинаций является важным для диверсификации сигналов, так как тождественность димеров определяет, какой из последующих путей активируется. Типичные продукты генов в последующих стадиях пути передачи сигналов ErbB включают She, Grb2, SOS1, Ras, Raf1, Mek, ERK1, ERK2, ERα, Akt, mTOR, FKHR, p27, Cyclin D1, FasL, GSK-3, Bad и STAT3.
[00151] Существуют твердые доказательства того, что EGFR и другие члены семейства ErbB связаны у человека с наличием опухолей, поскольку в более чем 60%,всех твердых опухолей наблюдается повышенная экспрессия, по крайней мере, одного из этих белков или их лигандов. Сообщалось, что конститутивно активный онкогенный EGFR vIII, мутант, содержащий укороченный внеклеточный домен, присутствует в 78% случаев карцином молочной железы, а также он был обнаружен глиобластомах. Повышенную экспрессию EGFR обычно обнаруживают в опухолях молочной железы, легких, головы и шеи, мочевого пузыря, в то время как экспрессия ErbB2 часто повышена в опухолях эпителиального происхождения человека. Активирующие мутации в домене тирозинкиназы были идентифицированы у пациентов с немелкоклеточным раком легких (Lin, N.U.; Winer, Е.P., Breast Cancer Res 6: 204-210, 2004). Амплификация ErbB1 и/или ErbB2 также происходит в эпидермоидных раках, карциномах слюнных желез, яичников и раках поджелудочной железы (Cooper, G.C. Oncogenes. 2nd ed. Sudbury: Jones and Barlett, 1995; Zhang, Y., et al., Cancer Res 66: 1025-32, 2006). Повышенная экспрессия ErbB2 обладает сильной трансформирующей активностью, вероятно ввиду его способности к
взаимодействию с другими рецепторами ErbB (Sherman, L., et al., Oncogene.18: 6692-99, 1999). В действительности, некоторые опухоли человека, в которых наблюдается повышенная экспрессия как EGFR, так и ErbB2, характеризуются худшим прогнозом, чем опухоли, в которых наблюдается повышенная экспрессия какого-либо одного рецептора.
[00152] Система передачи сигналов ErbB часто является ключевым компонентом в патогенезе рака молочной железы. Амплификация ErbB2 ассоциирована с фенотипом агрессивной опухоли, который характеризуется относительно быстрым ростом опухоли, распространением метастаз во внутренних органах и устойчивостью к лекарствам. Было показано, что ErbB2 амплифицирован в 20% случаев рака молочной железы без поражения подмышечных лимфоузлов (axillary node-negative, ANN), и такая амплификация рассматривается как независимый прогностический фактор риска рецидива в указанном раке молочной железы ANN (Andrulis, I.L., et al., J Clin Oncol 16: 1340-9, 1998).
[00153] Было показано, что целевая блокада передачи сигналов ErbB трастузумабом (trastuzumab, Herceptin), моноклональным антителом против ErbB2, улучшает выживаемость женщин ErbB2-положительным раком молочной железы в прогрессирующей стадии. К другим моноклональным антителам против рецепторов ErbB относятся цетуксимаб (cetuximab, Erbitux) и панитумумаб (panitumumab, Vectibix).
[00154] Было обнаружено, что несколько малых молекул - ингибиторов тирозинкиназы (TKIs) действуют селективно на членов семейства ErbB. К известным примерам относятся гефитиниб (gefitinib, Iressa) и эрлотиниб (erlotinib, Tarceva), оба нацеленные на EGFR. Эти малые молекулы конкурируют с АТФ за связывание с киназным доменом рецептора. По сравнению с моноклональными антителами, TKIs имеют несколько преимуществ в том, что они биодоступны орально, хорошо переносятся и производят впечатление активных против укороченных форм рецепторов ErbB2 и EGFR (например, EGFR vIII) in vitro. В дополнение, малый размер малых молекул TKIs может позволить им проникнуть в труднодоступные места, такие как центральная нервная система. Наконец, гомология между киназными доменами рецепторов ErbB позволяет разрабатывать TKIs, которые нацелены одновременно на более чем одного члена семейства ErbB, преимущества чего описаны в настоящей заявке.
[00155] Хотя некоторые злокачественные опухоли связаны с повышенной экспрессией индивидуальных рецепторов, эффективная передача сигналов основана на совместной экспрессии членов семейства рецепторов ErbB. Такая кооперация членов семейства рецепторов ErbB в передаче сигналов и трансформации злокачественных опухолей может ограничить успешность агентов, нацеленных на индивидуальные рецепторы, в лечении рака; потенциальный механизм устойчивости к агентам, нацеленным на единственный рецептор ErbB, является повышенная экспрессия других членов семейства рецепторов (Britten, С.D., Mol Cancer Ther 3: 1335-42, 2004).
[00156] Агенты, нацеленные на два или несколько рецепторов ErbB, называют общими регуляторами ErbB. ERRP является общим негативным регулятором ErbB, экспрессирующимся в большинстве доброкачественных клеток эпителия протоков и островков поджелудочной железы. Обнаружено, что в опухолях происходит прогрессирующая утрата экспрессии ERRP. То, что Эрбитукс (Erbitux) и Герцептин (Herceptin) оказываются успешными на ограниченном числе пациентов (опухоли, обладающие повышенной экспрессией EGFR или ErbB2), может быть частично из-за совместной экспрессии множества членов семейства ErbB.
[00157] В моделях как in vitro, так и in vivo стратегии, которые используют двойной подход по отношению к ErbB, кажутся обладающими большей противоопухолевой активностью, чем использование агентов, нацеленных на единичный рецептор ErbB. Таким образом, агенты, нацеленные на множество членов семейства ErbB, с большей вероятностью дадут терапевтический выигрыш на более широкой популяции пациентов (Zhang, Y., et al., Cancer Res 66: 1025-32, 2006). В определенных вариантах осуществления настоящего изобретения, предложенные соединения ингибируют один или несколько ErbB1, ErbB2, ErbB3 и ErbB4. В некоторых вариантах осуществления настоящего изобретения, предложенные соединения ингибируют два или более ErbB1, ErbB2, ErbB3 и ErbB4, или их мутантов, и поэтому являются общими ингибиторами ErbB.
[00158] Несомненно, наблюдается растущее число доказательств, подтверждающих одновременное ингибирование двух или более рецепторов ErbB (то есть, общий ErbB) в противораковой терапии. Вероятные подходы с общим ErbB с малыми молекулами включают использование комбинаций агентов, нацеленных на индивидуальные рецепторы ErbB, использование единичных агентов, нацеленных на множество рецепторов ErbB, или
использование агентов, влияющих на взаимодействия рецепторов ErbB (например, димеризацию). Дополнительные стратегии включают терапии, использующие малые молекулы в комбинации с антителами, или хемопревентивные терапии (Lin, N.U.; Winer, Е.P., Breast Cancer Res 6: 204-210, 2004).
[00159] Примером малой молекулы - общего ингибитора ErbB является CI-1033, необратимый общий ингибитор ErbB, ковалентно связывающийся с сайтом связывания АТФ внутриклеточного киназного домена. Другим необратимым общим ингибитором рецептора ErbB тирозинкиназы является HKI-272, ингибирующий рост клеток опухоли, которые экспрессируют ErbB-1 (EGFR) и ErbB-2 (HER-2) в культуре и ксенотрансплантатах, и обладающий противоопухолевой активностью в HER-2-положительном раке молочной железы (Andrulis, I.L., et al., J Clin Oncol 16: 1340-9, 1998). Необратимые ингибиторы продемонстрировали превосходную противоопухолевую активность по сравнению с обратимыми ингибиторами.
[00160] Нейрофиброматоз I типа (NF1) является по существу наследственным заболеванием, которым страдает один из 2500-3500 индивидуумов. Поражаются несколько систем органов, включая кости, кожные покровы, радужную оболочку глаз и центральную нервную систему, что проявляется в неспособности к обучению и глиомах. Признаком NF1 является развитие доброкачественных опухолей периферической нервной системы (нейрофибром), сильно различающихся по числу и размеру среди пациентов. Нейрофибромы являются гетерогенными опухолями, состоящими из Шванна, нейронов, фибробластов и других клеток, где клетки Шванна являются основным типом клеток (60-80%).
[00161] Аберрантная экспрессия EGFR ассоциирована с развитием опухоли при NF1 и в моделях NF1 на животных, что предполагает некую роль в патогенезе и представляет новую потенциальную терапевтическую мишень. Экспрессия EGFR вызывает рост опухолевых клеточных линий, полученных из пациентов с NF1, в условиях, когда EGF не является первичным фактором, вызывающим рост клеток. Эти данные наводят на мысль, что EGFR может играть важную роль в онкогенезе NF1 и трансформации клеток Шванна (DeClue, J.Е., et al., J Clin Invest 105: 1233-41, 2000).
[00162] У пациентов с NF1 развивается агрессивная неоплазма клеток Шванна, известная как злокачественная опухоль оболочек периферических нервов (MPNSTs).
Клетки Шванна являются главной популяцией вспомогательных клеток в периферической нервной системе. Неопластические клетки Шванна в этих неоплазмах с разным уровнем экспрессируют тирозинкиназы ErbB, что вызывает ответ NRG-1 (ErbB2, ErbB3, ErbB4). Белки нейрегулина-1 (NRG-1) вызывают дифференциацию, выживание, и/или пролиферацию множества типов клеток в развивающейся нервной системе, а повышенная экспрессия NRG-1 в клетках Шванна, образующих миелиновый слой, индуцирует формирование злокачественных опухолей оболочек периферических нервов (MPNSTs) (Fallon, K.В., et al., J Neuro Oncol 66: 273-84, 2004).
[00163] Нарушенная регуляция роста клеток Шванна является первичным дефектом, приводящим к развитию как доброкачественных нейрофибром, так и MPNST у пациентов с нейрофиброматозом I типа (NF1). Рост MPNSTs и транобразованных клеток Шванна мыши in vitro сильно зависит от EGF и может быть блокирован ингибиторами EGFR в условиях, когда EGF является первичным фактором роста. У некоторых MPNST линий клеток человека обнаружили конститутивное фосфорилирование ErbB. Хотя обработка ингибиторами ErbB предотвращает фосфорилирование ErbB и снижает синтез ДНК в этих линиях, эффективные хемотерапевтические режимы для MPNST остаются труднодостижимыми (Stonecypher, М.S., et al., Oncogene 24: 5589-5605, 2005).
[00164] Невриномы являются опухолями периферических нервов, почти полностью состоящими из клеток, подобных клеткам Шванна, и обычно содержащими мутации в гене супрессора опухоли нейрофиброматоза II типа (NF2). У девяноста процентов пациентов с NF2 развиваются двусторонние вестибулярные невриномы и/или спинномозговые невриномы. Увеличивающиеся невриномы могут сдавливать соседние структуры, что приводит к глухоте и другим неврологическим проблемам. Хирургическое удаление этих опухолей затруднительно и часто приводит к усилению клинических проявлений заболевания у пациентов.
[00165] Как нормальные человеческие клетки Шванна, так и клетки неврином экспрессируют рецепторы нейрегулина (то есть, рецепторы ErbB), а клетки неврином пролиферируют в ответ на нейрегулин. Возможно, что аберрантная продукция нейрегулина или ответ вносят вклад в аберрантную пролиферацию клеток невриномы (Pelton, P.D., et al., Oncogene 17: 2195-2209, 1998).
[00166] Супрессор опухоли NF2, Мерлин, является белком, ассоциированным с мембраной/цитоскелетом, вовлеченным в регуляцию активности тирозинкиназы. Генетические взаимодействия между мутациями в белке Мерлин и мутациями в пути EGFR описаны в Drosophila (LaJeunesse, D.R., et al., Genetics 158: 667-79, 2001). Другое доказательство Другое доказательство наводит на мысль, что Мерлин может ингибировать интернализацию EGFR и передачу сигнала через контакт клетка-клетка путем удерживания EGFR в мембранном компартменте, от которого не может быть передан ни сигнал, ни который не может быть интернализован (McClatchey, A.I., et al., Genes and Development 19: 2265-77, 2005; Curto, M.C., et al., J Cell Biol 177: 893-903, 2007).
[00167] Используемые здесь термины "лечение", "лечить" и "излечивание" относятся к реверсии, облегчению, замедлению проявления или ингибированию прогресса заболевания или нарушения, или одного или нескольких его симптомов, как описано в настоящем описании. В некоторых вариантах реализации настоящего изобретения, лечение может быть назначено после того, как обнаружен один или несколько симптомов. В других вариантах реализации настоящего изобретения, лечение может быть назначено в отсутствие симптомов. Например, лечение может быть назначено восприимчивому индивидууму перед наступлением симптомов (например, в свете истории симптомов и/или в свете генетических факторов или других факторов восприимчивости). Лечение может быть продолжено после того, как исчезли симптомы, например, чтобы предотвратить или замедлить ее повторение.
[00168] Предложенные соединения являются ингибиторами одного или нескольких ErbB1, ErbB2, ErbB3 и ErbB4, и поэтому пригодны для лечения одного или нескольких нарушений, ассоциированных с активностью одного или нескольких ErbB1, ErbB2, ErbB3 и ErbB4. Таким образом, в определенных вариантах реализации настоящее изобретение обеспечивает способ лечения нарушения, опосредованного ErbB1, ErbB2, ErbB3 и/или ErbB4, включающего стадию введения соединения согласно настоящему изобретению или его фармацевтически приемлемой композиции пациенту, нуждающемуся в нем/в ней.
[00169] Использованные здесь термины нарушения или состояния, «опосредованные ErbB1», «опосредованные ErbB2», «опосредованные ErbB3», и/или «опосредованные ErbB4», означают любое заболевание или другое болезнетворное состояние, для которого известно, что оно связано с одним или несколькими ErbB1, ErbB2, ErbB3 и/или ErbB4, или
их мутантами. Соответственно, другой вариант осуществления настоящего изобретения относится к лечению или снижению тяжести одного или нескольких заболеваний, для которого известно, что оно связано с одним или несколькими ErbB1, ErbB2, ErbB3 и/или ErbB4 или их мутантами. В частности, настоящее изобретение относится к способу лечения или снижению тяжести одного или нескольких заболеваний или состояний, выбранных из пролиферативного заболевания, причем указанный способ включает введение соединения согласно настоящему изобретению или его фармацевтически приемлемой композиции пациенту, нуждающемуся в нем/в ней.
[00170] В некоторых вариантах реализации настоящее изобретение обеспечивает способ лечения или снижению тяжести одного или нескольких заболеваний, представляющих собой рак. В некоторых вариантах реализации настоящего изобретения рак ассоциирован с твердой опухолью. В определенных вариантах реализации настоящего изобретения таким раком является рак молочной железы, глиобластома, рак легких, рак головы и шеи, рак толстой кишки, рак мочевого пузыря или немелкоклеточный рак легких. В некоторых вариантах реализации настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний, выбранных из карциномы сквамозных клеток, карциномы слюнных желез, карциномы яичников или рака поджелудочной железы.
[00171] В некоторых вариантах реализации настоящее изобретение обеспечивает способ лечения или снижения тяжести нейрофиброматоза I типа (NF1), нейрофиброматоза II типа (NF2), неоплазм клеток Шванна (например, MPNST's) или неврином.
(b) Семейство ТЕС
[00172] Семейство ТЕС нерецепторных тирозинкиназ, обозначенное здесь как "ТЕС-киназы", играет центральную роль в передаче сигналов через антигены-рецепторы, такие как рецепторы TCR, BCR и Fc (обзор Miller A, et al. Current Opinion in Immunology 14; 331-340 (2002). ТЕС-киназы являются необходимыми для активации Т-клеток. Три члена указанного семейства, Itk, Rlk и, активируются после контакта антигена с рецептором в Т-клетках и передают сигналы последующим эффекторам, включая PLC-γ. Совместная делеция Itk и Rlk в мышах приводит к полному ингибированию ответов TCR, включая пролиферацию, продукцию цитокинов и иммунные ответы на внутриклеточных паразитов (Toxoplasma gondii) (Schaeffer et al., Science 284; 638-641 (1999)). Внутриклеточная
передача сигналов после контакта рецептора TCR наблюдается в дефицитных по ITK/RLK Т-клетках; при этом снижаются продукция инозитолтрифосфата, мобилизация кальция и активация МАР-киназы. ТЕС-киназы также являются существенными для развития и активации В-клеток.
[00173] К ТЕС-киназам относятся пять членов семейства, которые экспрессируются главным образом в гемопоэтических клетках: ТЕС, BTK, ITK (также известный как TSK и ЕМТ), RLK (также известный как ТХК) и ВМХ (также известный как ЕТК). Другие родственные ТЕС-киназы были обнаружены в Drosophila melanogaster, в полосатом данио (Danio  ), в скате (Raja eglanteria) и морском еже (Anthocidaris crassispina).
), в скате (Raja eglanteria) и морском еже (Anthocidaris crassispina).
[00174] Предоставленные соединения являются ингибиторами одной или нескольких ТЕС-киназ и поэтому пригодны для лечения одного или нескольких заболеваний, ассоциированных с активностью одной или нескольких ТЕС-киназ. Таким образом, в определенных вариантах осуществления настоящее изобретение обеспечивает способ лечения заболевания, опосредованного ТЕС, включающий стадию введения соединения согласно настоящему изобретению или его фармацевтически приемлемой композиции пациенту, нуждающемуся в нем/в ней.
[00175] Термин «состояние, опосредованное ТЕС», использованный в настоящей заявке, означает любое заболевание или другое болезнетворное состояние, про которое известно, что оно связано с ТЕС-киназами. Такие состояния включают состояния, описанные в настоящей заявке, а также у Melcher, М et al., "The Role of TEC Family Kinases in Inflammatory Processes", Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry, Vol. 6, No. 1, pp. 61-69 (Feb. 2007). Соответственно, другой вариант осуществления настоящего изобретения относится к лечению или снижению тяжести одного или нескольких заболеваний, про которые известно, что оно связано с ТЕС-киназами. В частности, настоящее изобретение относится к способу - лечения или снижения тяжести заболевания или состояния, выбранного из аутоиммунного, воспалительного, пролиферативного и гиперпролиферативного заболеваний и иммунологически опосредованных заболеваний, включая отторжение трансплантированных органов или тканей и синдром приобретенного иммунодефицита (СПИД) (также известного как ВИЧ), при этом указанный способ включает введение
фармацевтически приемлемой композиции согласно настоящему изобретению пациенту, нуждающемуся в ней.
[00176] В некоторых вариантах реализации настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с ТЕС-киназами, включающих заболевания дыхательных путей, включая заболевания, при которых происходит обратимое закупоривание дыхательных путей, включающие астму, такую как бронхиальная, аллергическая, врожденная, неврожденная и пылевая астма, в частности хроническая или застарелая астма (например, поздняя стадия астматической гиперчувствительности дыхательных путей) и бронхит, но не ограничивающихся ими. В некоторых вариантах реализации настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с ТЕС-киназами, включая состояния, характеризующиеся воспалением носовой слизистой мембраны, включая острый ринит, аллергический, атрофический ринит и хронический ринит, включающий хронический ринит с образованием казеозных масс, гипертрофический ринит, гнойный ринит, сухой ринит и медикаментозный ринит; мембранный ринит, включая крупозный, фибринозный и превдомембранный ринит и скрофулезный рините, сезонный ринит, включая нейроринит (сенная лихорадка) и вазомоторный ринит, саркоидоз, "легкие фермера" (экзогенный аллергический альвеолит) и связанные с ним заболевания, пневмофиброз, и идиопатическая интерстициальная пневмония.
[00177] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с ТЕС-киназами, включая заболевания костей и суставов, включающих ревматоидный артрит, серонегативные спондилоартропатии (включая анкилозирующий спондилоартрит, псориатический артрит и болезнь Рейтера), болезнь Бехчета, синдром Шёгрена, системный склероз, остеопороз, рак костей и метастаз костей, но не ограничивающихся ими.
[00178] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболевании и состояний, ассоциированных с ТЕС-киназами, включая заболевания кожи, включая псориаз, системный склероз, атипичный дерматит, контактный дерматит и другие экзематозные
дерматиты, себорейный дерматит, красный плоский лишай, пемфигус, буллезный пемфигус, буллезный эпидермолиз, крапивница, ангиодермит, васкулиты, эритемы, кожную эозинофилию, увеит, аллопецию, круговой и весенний конъюнктивит, но не ограничивающихся ими.
[00179] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с ТЕС-киназами, включая заболевания и расстройства желудочно-кишечного тракта, включая глютеновую болезнь, проктит, эозинофильный гастроэнтерит, мастоцитоз, панкреатит, болезнь Крона, язвенный колит, аллергии, ассоциированные с пищей, проявляющиеся вдали от пищеварительного тракта, например мигрень, ринит и экзема, но не ограничивающихся ими.
[00180] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с ТЕС-киназами, включая заболевания и расстройства других тканей и системные заболевания, включая множественный склероз, артериосклероз, красную волчанку, системную красную волчанку, тиреоидит Хасимото, тяжелую миастению, диабет I типа, нефротический синдром, элзинофильный фасцит, гипер-IgE-синдром, лепроматозная лепра, синдром Сезари и идиопатическая розовая тромбоцитопения, последующая за рестенозом ангиопластика, опухоли (например, лейкемий, лимфомы и раки простаты) и артериосклероз, но не ограничивающихся ими.
[00181] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с ТЕС-киназами, включающих отторжение аллотрансплантата, включая острое или хроническое отторжение аллотрансплантата после, например, трансплантации почки, сердца, печени, легкого, костного мозга, кожи и роговой оболочки глаза; и хроническую реакцию "трансплантат против хозяина", но не ограничивающихся ими.
[00182] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с ТЕС-киназами, описанных выше, причем указанный способ включает введение соединения согласно настоящему изобретению или его фармацевтически приемлемой композиции пациенту, нуждающемуся в нем/в ней.
(с) Брутоновская тирозинкиназа (BTK)
[00183] Брутоновская тирозинкиназа (BTK), член семейства ТЕС-киназ, является ключевым сигнальным ферментом, экспрессирующимся во всех гемопоэтических типах клеток, за исключением Т-лимфоцитов и природных клеток-киллеров. BTK играет существенную роль в пути передачи сигналов в В-клетках, опосредуя стимуляцию рецептора на поверхности В-клетки (BCR) с последующими ответами внутри клетки.
[00184] BTK является ключевым регулятором развития, активации, передачи сигналов и выживаемости В-клеток (Kurosaki, Curr Op Imm, 2000, 276-281; Schaeffer and Schwartzberg, Curr Op Imm 2000, 282-288). В дополнение, BTK играет роль в нескольких других сигнальных путях гемопоэтических клеток, например, в продукции Toll-подобного рецептора (TLR) и опосредованной рецептором цитокина продукции TNF-α в макрофагах, в передаче сигналов рецептором IgE (Fc_epsilon_RI) в мастоцитах, ингибирование апоптотической передачи сигналов Fas/APO-1 в лимфоидных клетках В-линии, и стимулированной коллагеном агрегации тромбоцитов. Смотри, например, С.A. Jeffries, et al., (2003), Journal of Biological Chemistry 278:26258-26264; N. J. Horwood, et al., (2003), The Journal of Experimental Medicine 197: 1603- 1611; Iwaki et al. (2005), Journal of Biological Chemistry 280(48):40261-40270; Vassilev et al. (1999), Journal of Biological Chemistry 274(3): 1646-1656, и Quek et al. (1998), Current Biology 8(20): 1137-1140.
[00185] У пациентов с мутациями в BTK наблюдается полный блок в развитии В-клеток, что приводит к практически полному отсутствию зрелых В-лимфоцитов и клеток плазмы, сильному понижению уровня иммуноглобулинов и сильному ингибированию гуморального ответа на антигены тканей донора (обзор в Vihinen et al. Frontiers in Bioscience 5: d917-928). У мышей, дефицитных по BTK, также снижено количество периферических В-клеток и существенно снижены уровни IgM и IgG3 в сыворотке. Делеция BTK в мышах приводит к сильному эффекту по пролиферации В-клеток, индуцированному анти-IgM, и ингибирует иммунные ответы на тимус-независимые антигены II типа (Ellmeier et al, J Exp Med 192: 1611-1623 (2000)). BTK также играет критическую роль в активации мастоцитов через высокоаффинный рецептор IgE (Fc_epsilon_RI). У мастоцитов мыши, дефицитных по BTK, снижена дегрануляция и снижена продукция провоспалительных цитокинов, что приводит поперечным сшивкам Fc_epsilon_RI (Kawakami et al. Journal of Leukocyte Biology 65: 286-290).
[00186] Предложенные соединения являются ингибиторами BTK и поэтому являются полезными при лечении одного или нескольких заболеваний, ассоциированных с активностью BTK. Таким образом, в некоторых вариантах реализации настоящее изобретение обеспечивает способ лечения заболевания, ассоциированного с BTK, включающий введение соединения согласно настоящему изобретению или его фармацевтически приемлемой композиции пациенту, нуждающемуся в нем/в ней.
[00187] Использованный в настоящей заявке термин заболевание или состояние, «ассоциированное с BTK» означает любое заболевание или болезнетворное состояние, про которое известно, что оно связано с BTK или его мутантом. Соответственно, другой вариант осуществления настоящего изобретения относится к лечению или снижению тяжести одного или нескольких заболеваний и состояний, про которые известно, что они связано с BTK или его мутантом. В частности, настоящее изобретение относится к способу лечения или снижения тяжести заболевания или состояния, выбранного из пролиферативного заболевания или аутоиммунного нарушения, причем указанный способ включает введение соединения согласно настоящему изобретению или его композиции пациенту, нуждающемуся в нем/в ней.
[00188] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK. В некоторых вариантах осуществления настоящего изобретения таким заболеванием или состоянием является аутоиммунное заболевание, например, воспалительное заболевание кишечника, артрит, волчанка, ревматоидный артрит, псориатический артрит, остеоартрит, болезнь Стилла, подростковый артрит, диабеты, тяжелая миастения, тиреоидит Хасимото, тиреоидит Орда, базедова болезнь, синдром Шёргена, множественный склероз, синдром Гийена-Барре, острый рассеянный энцефаломиелит, болезнь Эддисона, синдром пляшущих глаз, деформирующий спондилез, синдром антифосфолипидного антитела, апластическая анемия, аутоиммунный гепатит, брюшные заболевания, синдром Гудпасчера, идиопатическая розовая тромбоцитопения, глазной неврит, склеродерма, первичный желчный цирроз, синдром Рейтера, артрит Такаясу, височный артрит, warm аутоиммунная гемолитическая анемия, гранулематоз Вегенера, псориаз, общая аллопеция, синдром Бехчета, хроническая усталость, вегетативная дистония, эндометриоз, интерстициальный цистит,
нейромиотония, склеродерма или вульводиния. В некоторых вариантах осуществления настоящего изобретения таким заболеванием или состоянием является гиперпролиферативное заболевание или иммунологически опосредованное, заболевание, включающее отторжение трансплантированных органов или тканей и синдром приобретенного иммунодефицита (СПИД, также известный как ВИЧ).
[00189] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK, причем указанное заболевание или состояние выбрано из гетероиммунных состояний или заболеваний, которые включают реакцию "трансплантат против хозяина", трансплантацию, трансфузию, анафилаксию, аллергии (например, аллергии к растительной пыльце, латексу, лекарственным препаратам, пище, ядам насекомых, шерсти животных, перхоти животных, пылевым клещам или стенкам яиц таракана), гиперчувствительность I типа, аллергический конъюнктивит, аллергический ринит и атопический дерматит, но не ограничиваются ими.
[00190] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK, причем указанное заболевание или состояние выбрано из воспалительного заболевания, например, астмы, аппендицита, блефарита, бронхиолита, бронхита, бурсита, цервицита, холангита, холецистита, колита, конъюнктивита, цистита, дакриоаденита, дерматита, дерматомиозита, энцефалита, эндокардита, эндометрита, энтерита, энтероколита, эпикондилита, эпидидимита, фасцита, фиброзита, гастрита, гастроэнтерита, гепатита, гнойного гидраденита, ларингита, мастита, менингита, миелита, миокардита, миозита, нефрита, оофорита, орхита, остеита, отита, панкреатита, паротита, перикардита, перитонита, фарингита, плеврита, флебита, пневмонита, пневмонии, проктита, простатита, пиелонефрита, ринита, сальпингита, синусита, стоматита, синовита, тендинита, тонзиллита, увеита, вагинита, васкулита или вульвита.
[00191] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK, причем указанное заболевание или состояние выбрано из рака. В одном варианте осуществления настоящего изобретения к раку относится пролиферативное заболевание В-клеток, например, диффузная лимфома больших В-
клеток, фолликулярная лимфома, хроническая лимфоцитная лимфома, хроническая лимфоцитная лейкемия, острая лимфоцитная лейкемия, пролимфоцитная лейкемия В-клеток, лимоплазмицитная лимфома / макроглобулинемия Вальденстрома, лимфома краевых зон селезенки, множественная миелома (также известная как миелома клеток плазмы), неходжскинская лимфома, ходжскинская лимфома, плазмацитома, внеузловая В-клеточная лимфома краевых зон, внутриузловая В-клеточная лимфома краевых зон, лимфома клеток коры головного мозга, медиастенальная (тимусная) лимфома больших В-клеток, внутрисосудистая лимфома больших В-клеток, первичная эффузионная лимфома, лимфома/лейкемия Буркитта или лимфоматоидный грануломатоз. В некоторых вариантах осуществления настоящего изобретения раком является рак молочной железы, рак простаты или рак мастоцитов (например, мастоцитома, лейкемия мастоцитов, саркома мастоцитов, системный мастоцитоз). В одном варианте осуществления настоящего изобретения указанным раком является рак костей. В другом варианте осуществления настоящего изобретения указанным раком является рак другого первичного происхождения с метастазами в костях.
[00192] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK, включающих заболевания костей и суставов включая ревматоидный артрит, серонегативную спондилоартропатию (включающую анкилозирующий спондилит, псориатический артрит и заболевание Рейтера), болезнь Бехчета, синдром Шёргена, системный склероз, остеопороз, рак костей и метастаз костей, но не ограничивающихся ими.
[00193] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK, где указанное заболевание или состояние выбрано из тромбоэмболических нарушений, например, инфаркт миокарда, стенокардия, реокклюзия после ангиопластики, рестеноз после ангиопластики, реокклюзия после аортокоронарного шунтирования, рестеноз после аортокоронарного шунтирования, внезапного приступа, транзиторная ишемия, окклюзивные нарушения в периферических артериях, эмболия легких или глубокий венозный тромбоз.
[00194] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK, включающих инфекционные и неинфекционные воспалительные события, а также аутоиммунные и другие воспалительные заболевания. Такие аутоиммунные и воспалительные заболевания, нарушения и синдромы включают воспалительные заболевания таза, уретрит, солнечный ожог кожи, синусит, пневмонит, энцефалит, менингит, миокардит, нефрит, остеомиелит, миозит, гепатит, гастрит, энтерит, дерматит, гингивит, аппендицит, панкреатит, хлороцистит, агаммаглобулинемию, простатит, аллергию, болезнь Крона, синдром раздраженного кишечника, язвенный колит, болезнь Шёргена, отторжение тканевого трансплантата, сверхострое отторжение трансплантированных органов, астму, аллергический ринит, хроническое закупоривающее легочное заболевание (COPD), аутоиммунное полигранулярное заболевание (также известное как аутоиммунный полигранулярный синдром), аутоиммунная аллопеция, пернициозная анемия, гломерулонефрит, дерматомозит, множественный склероз, склеродерма, васкулит, аутоиммунные гемолитические и тромбоцитопенические состояния, синдром Гудпасчера, артериосклероз, болезнь Эддисона, болезнь Паркинсона, болезнь Альцгеймера, диабет I типа, септический шок, системная красная волчанка (SLE), ревматоидный артрит, псориатический артрит, подростковый артрит, остеоартрит, хроническая идиопатическая розовая тромбоцитопения, болезнь Вальденстрема, тяжелая миастения, тиреоидит Хасимото, атопичный дерматит, дегенеративное поражение сустава, витилиго, аутоиммунный гипопитуитаризм, синдром Гийена-Барре, болезнь Бехчета, склерадерма, фунгоидный микоз, острые воспалительные ответные реакции (такие как синдром острой дыхательной недостаточности и ишемия/ реперфузионное повреждение), болезнь Грейвса.
[00195] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ лечения или снижения тяжести одного или нескольких заболеваний и состояний, ассоциированных с BTK, выбранных из ревматоидного артрита, множественного склероза, В-клеточной хронической лимфоцитной лейкемии, острой лимфоцитной лейкемии, лейкемии ворсистых клеток, не-ходжкинской лимфомы, ходжкинской лимфомы, множественной миеломы, рака костей, метастаз костей, остеопороз, синдром раздраженного кишечника, болезнь Крона, волчанка и ренальный трансплантат.
(d) ITK
[00196] Индуцируемая интерлейкином-2 Т-клеточная киназа (ITK) экспрессируется в Т-клетках, мастоцитах и природных клетках-киллерах. Она активирована в Т-клетках за счет стимулирования рецептора Т-клеток (TCR) и в мастоцитах за счет активации высокоаффинного рецептора IgE. При последующей стимуляции рецептора в Т-клетках, Lck, члена семейства Src тирозинкиназ, он фосфорилирует Y511 в петле ITK, активирующей киназный домен (S.D. Heyeck et al., 1997, J. Biol. Chem, 272, 25401-25408). Активированный ITK вместе с Zap-70 требуется для фосфорилирования и активации PLC-гамма (S.С. Bunnell et al., 2000, J. Biol. Chem., 275, 2219-2230). PLC-гамма катализирует образование инозитол-1,4,5-трифосфата и диацилглицерина, что приводит к мобилизации кальция и активации PKC, соответственно. Эти события активируют множество последующих путей и приводят в конечном итоге к дегрануляции (мастоциты) и экспрессии гена цитокина (Т-клетки) (Y. Kawakami et al., 1999, J. Leukocyte Biol., 65, 286-290).
[00197] Роль ITK в активации Т-клеток подтверждена в мышах, нокаутированных по ITK. CD4+ Т-клетки из мышей, нокаутированных по ITK, демонстрировали, ослабленный пролиферативный ответ в смешанной лимфоцитной реакции или при Con А или анти-CD3 стимуляции. (X.С. Liao and D.R. Littman, 1995, Immunity, 3, 757-769). Также, Т-клетки из мышей, нокаутированных по ITK, производили немного ИЛ-2 при стимуляции TCR, что приводило к пониженной пролиферации этих клеток. В другом исследовании CD4+ Т-клетки, дефицитные по ITK, производили пониженный уровень цитокинов, включая IL-4, IL-5 и IL-13 при стимуляции TCR, даже после примирования в индуцирующих условиях (D.J. Fowell, 1999, Immunity, 11, 399-409).
[00198] Роль ITK в PLC-гамма активации и мобилизации кальция также была подтверждена в Т-клетках этих нокаутированных мышей, у которых были значительно снижены образование IP3 и не внеклеточный транспорт кальция при стимуляции TCR (К. Liu et al., 1998, J. Exp. Med. 187, 1721-1727). Подобные исследования подтверждают ключевую роль ITK в активации Т-клеток и мастоцитов. Таким образом, ингибитор ITK будет обладать терапевтическими преимуществами в заболеваниях, опосредованных несоответствующей активацией этих клеток.
[00199] Является широко признанным тот факт, что Т-клетки играют важную роль в регулировании иммунного ответа (Powrie and Coffman, 1993, Immunology Today, 14, 270-274). Действительно, активация Т-клеток часто является инициирующим событием в иммунологических заболеваниях. Следом за активацией TCR происходит транспорт кальция, который требуется для активации Т-клеток. При активации Т-клетки производят циокины, включая IL-2, 4, 5, 9, 10 и 13, что приводит к пролиферации, дифференцировке и эффекторной функции Т-клеток. Клинические исследования с использованием ингибиторов IL-2 продемонстрировали, что взаимное влияние активации Т-клеток и пролиферации эффективно подавляет иммунный ответ in vivo (Waldmann, 1993, Immunology Today, 14, 264-270). Соответственно, агенты, которые ингибируют активацию Т-лимфоцитов и последующую продукцию цитокинов, являются терапевтически пригодными для селективного подавления иммунного ответа у пациентов, нуждающихся в такой иммуносупрессии.
[00200] Тучные клетки играют важную роль в развитии астмы и аллергических заболеваний, высвобождая медиаторы воспаления и цитокины. Антиген-опосредованная аггрегация Fc εRI, высокоаффинных рецепторов иммуноглобулина IgE, приводит к активации тучных клеток (D.B. Corry et al.., 1999, Nature, 402, В18-23), запуская таким образом сигнальную систему, ответственную за освобождение медиаторов, в том числе гистамина, протеаз, лейкотриенов и цитокинов (J. R. Gordon et al., 1990, Immunology Today, 11, 458-464). Высвобождение медиаторов приводит к повышению проницаемости сосудов, продуцированию слизи, бронхоспазму, деградации тканей и воспалению, играя, таким образом, ключевую роль в этиологии и симптоматике астмы и аллергических заболеваний.
[00201] Опубликованные данные, полученные в результате экспериментов на нокаутных по ITK- киназе мышах, позволяют предположить, что в отсутствие активной ITK происходит увеличение числа Т-клеток памяти (А.Т. Miller et al., 2002 The Journal of Immunology, 168, 2163-2172). Одним из путей увеличения эффективности вакцинации является стимуляция образования большего количества Т-клеток памяти (S.М. Kaech et al., Nature Reviews Immunology, 2, 251-262). Кроме того, делеция гена ITK у мышей приводит к снижению уровней обусловленной рецепторами Т-клеток (TCR-индуцированной) пролиферации и секреции цитокинов IL-2, ИЛ-4, IL-5, IL-10 и IFN-y
(Schaeffer et al, Science 284; 638-641 (1999)), Fowell et al, Immunity 11, 399-409 (1999), Schaeffer et al, Nature Immunology 2 (12): 1183-1188 (2001))). Иммунологические признаки аллергической астмы у ITK-делетированных (ITK(-)) мышей выражены в меньшей степени. Воспаление легких, эозинофильная инфильтрация и производство слизи в ответ на овальбумин (OVA) у ITK(-) -мышей резко снижены (Mueller et al, Journal of Immunology 170: 5056-5063 (2003)). ITK также вовлечена в формирование атопического дерматита. Было показано, что ген ITK интенсивнее экспрессируется в Т-клетках периферической крови у пациентов с умеренной и/или тяжелой формой атопического дерматита, чем в контрольной группе или в группе пациентов с мягкой формой атопического дерматита (Matsumoto et al, International Archives of Allergy and Immunology 129: 327-340 (2002)).
[00202] Спленоциты RLK-делетированных мышей секретируют в два раза меньшее количество IL-2 по сравнению с клетками животных дикого типа в ответ на предъявление TCR ((Schaeffer et al, Science 284: 638-641 (1999)); а одновременная делеция ITK и RLK у мышей приводит к выраженному ингибированию TCR-индуцированных реакций, включая пролиферацию и синтез цитокинов IL-2, IL-4, IL-5 и интерферона-гамма (Schaeffer et al, Nature Immunology 2 (12): 1183-1188 (2001), Schaeffer et al, Science 284: 638-641 (1999)). Работа внутриклеточной сигнальной системы, активируемой при предъявлении TCR, в ITK/RLK-дефицитных Т-клетках нарушена; снижены уровни производства инозитолтрифосфата, подавлены мобилизация кальция, активация МАР-киназного пути и факторов транскрипции NFAT и АР-1 (Schaeffer et al, Science 284: 638-641 (1999), Schaeffer et al, Nature Immunology 2 (12): 1183-1188 (2001)).
[00203] Данные соединения являются ингибиторами ITK и поэтому могут применяться для лечения одного или более заболеваний, связанных с активностью ITK-киназы. Таким образом, в некоторых вариантах осуществления настоящее изобретение включает способ лечения ITK-опосредованных расстройств, в том числе стадию введения пациенту, нуждающемуся в лечении указанного заболевания(ий), соединения-предмета настоящего изобретения или содержащих его фармацевтически приемлемых композиций.
[00204] В настоящем описании термин "ITK-опосредованные" заболевания или состояния означает любую болезнь или другие угрожающие здоровью состояния, возникновение которых опосредовано активностью ITK-киназы или ее
модифицированных (мутантных) версий. Таким образом, в одном из вариантов осуществления изобретение относится к лечению или уменьшения тяжести течения одного или нескольких заболеваний, возникновение которых опосредовано активностью ITK-киназы или ее модифицированных (мутантных) версий. В частности, настоящее изобретение относится к способу лечения или уменьшения тяжести заболевания или состояния, вызванных функционированием тучных клеток или базофилов, иммунными или аллергическими нарушениями, при этом указанный метод лечения подразумевает введение пациенту соединения или содержащие эти соединения композиции, являющиеся предметом настоящего изобретения.
[00205] В некоторых вариантах осуществления настоящее изобретение включает способ лечения или уменьшения тяжести одного или нескольких заболеваний или состояний, связанных с активностью ITK-киназы, а именно иммунных расстройств, в том числе воспалительных заболеваний, аутоиммунных заболеваний, отторжения трансплантированных органов и костного мозга и других нарушений, связанных с Т-клеточным или опосредованным тучными клетками иммунным ответом.
[00206] В некоторых вариантах осуществления настоящее изобретение включает способ лечения или уменьшения тяжести одного или нескольких заболеваний или состояний, связанных с активностью ITK, а именно острого или хронического воспаления, аллергий, контактного дерматита, псориаза, ревматоидного артрита, рассеянного склероза, сахарного диабета I типа, воспалительных заболеваний кишечника, синдрома Гийена-Барре, болезни Крона, язвенного колита, рака, реакции ТПХ («трансплантат против хозяина») и других форм отторжения трансплантата органа или костного мозга, красной волчанки.
[00207] В некоторых вариантах осуществления настоящее изобретение включает способ лечения или уменьшения тяжести одного или нескольких заболеваний и состояний, связанных с активностью ITK, а именно заболеваний или состояний, вызванных дисфункциями тучных клеток или базофилов, обструктивной болезни дыхательных путей, астмы, ринита, хронической обструктивной болезни легких (ХОБЛ), периферической лимфомы Т-клеток или ВИЧ [синдрома приобретенного иммунодефицита (СПИД)]. Такие состояния включают описанные у Readinger, et al., PNAS 105: 6684-6689 (2008).
(е) Семейство JAK-киназ
[00208] Янус-киназы (JAK) - семейство тирозинкиназ, состоящее из JAK1, JAK2, JAK3 и TYK2. Jak-киназы играют важную роль в цитокиновой сигнализации. Субстраты JAK-киназ включают белок-трандуктор сигнала и активатор транскрипции (STAT). JAK/STAT сигнализация задействована при различных нарушениях иммунного ответа, таких, как аллергии, астма, аутоиммунные заболевания: отторжение трансплантатов, ревматоидный артрит, боковой амиотрофический склероз и рассеянный склероз, а также злокачественные новообразования: опухоли и гемобластозы, такие, как лейкоз и лимфома Фармацевтическое вмешательства в активность JAK/ STAT сигнального пути было рассмотрено у [Frank, Mol. Med. 5: 432-456 (1999) & Seidel, et al, Oncogene 19: 2645-2656 (2000)].
[00209] JAK1, JAK2, и TYK2 экспрессируются во всех клетках, в то время как JAK3 по существу экспрессируется в кроветворных клетках. JAK3 избирательно связывается с гамма цепью цитокинового рецептора (γС) и активируется интерлейкинами IL-2, IL-4, IL-7, IL-9 и IL-15.
[00210] Было показано, что пролиферация и выживание тучных клеток мышей, индуцированных IL-4 и IL-9, зависит от JAK3 и γС-сигнализации [Suzuki et al, Blood 96: 2172-2180 (2000)].
[00211] Перекрестное связывание высокоаффинных иммуноглобулиновых IgE рецепторов в сенсибилизированных тучных клетках приводит к освобождению медиаторов воспаления, в том числе ряда вазоактивных цитокинов;⋅в результате формируется острая аллергическая реакция, или реакция гиперчувствительности немедленного типа (типа I), [Gordon et al, Nature 346: 274-276 (1990) & Galli, N. Engl. J. Med., 328: 257-265 (1993)]. Была установлена решающая роль JAK3 в опосредованном рецепторами IgE ответе тучных клеток in vitro и in vivo [Malaviya, et al, Biochem. Biophys. Res. Commun. 257: 807-813 (1999)]. Кроме того, было показано, что предотвращение реакций гиперчувствительности I типа, в том числе анафилактического шока, опосредовано активацией тучных клеток ингибированием JAK3 [Malaviya et al, J. Biol. Chem. 274: 27028-27038 (1999)]. Обработка тучных клеток ингибиторами JAK3 модулирует дегрануляцию тучных клеток in vitro и предотвращает IgE-R/антиген-опосрудованныt анафилактические реакции in vivo.
[00212] Недавние исследования успешно подтвердили роль JAK3 в подавлении иммунитета и приживлении аллотрансплантатов. Было продемонстрировано дозозависимое приживление аллотрансплантата сердца буйвола у крыс линии Вистар (Wistar Furth) при введении ингибиторов JAK3, что указывает на возможность регулирования нежелательных иммунных реакций «трансплантат против хозяина» [Kirken, Transpl. Proc. 33: 3268-3270 (2001)].
[00213] IL-4-опосредованное STAT-фосфорилирование, как установлено, вовлечено в формирование ранних и поздних стадий ревматоидного артрита (РА). Позитивная регуляция активности цитокинов воспаления синовиальной оболочки и синовиальной жидкости при РА является характеристикой данного заболевания. Показано, что IL-4-опосредованная активация IL-4/STAT пути опосредована янус-киназами (JAK 1 и 3), и что IL-4-ассоциированные JAK-киназы экспрессируются в синовиальной оболочке при РА [Muller-Ladner, et al, J. Immunol. 164: 3894-3901 (2000)]
[00214] Семейный боковой амиотрофический склероз (FALS) является фатальным нейродегенеративным расстройством, поражающим около 10% от пациентов, страдающих от бокового амиотрофического склероза. Выживаемость мышей с синдромом FALS увеличивается при введении им специфического ингибитора JAK3-киназы. Это подтверждает, что JAK3 играет определенную роль в формировании FALS [Trieu и др., Biochem. Biophys. Res. Commun. 267: 22-25 (2000)].
[00215] Белок-трансдуктор сигнала и активатор транскрипции (STAT-белки) активируются, в частности, киназами семейства JAK. Результаты недавнего исследования указывают на возможность вмешательства в JAK/STAT сигнальную систему путем модуляции активности янус-киназ специфическими ингибиторами при лечении лейкемии [Sudbeck, et al., Clin. Cancer Res. 5: 1569-1582 (1999)]. JAK3-специфические агенты препятствуют клональной пролиферации JAK3-экспрессирующих клеточных линий DAUDI, RAMOS, LCI; 19, NALM-6, MOLT-3 и HL-60. Ингибирование JAK3 и TYK 2 блокирует тирозин-фосфорилирование STAT3 и останавливает пролиферацию клеток фунгоидной гранулемы (формы кожной Т-клеточной лимфомы).
[00216] В другом варианте осуществления изобретение относится к способу лечения или уменьшения тяжести JAK3-опосредованных заболеваний или состояний,
включающий стадию введения пациенту, страдающему от указанного заболевания/состояния, композиции, являющейся предметом настоящего изобретения.
[00217] Термин "JAK3-опосредованное заболевание", используемый здесь, означает какое-либо заболевание или другое патологическое состояние, в формировании которого участвует JAK3-киназа. Соответственно, в одном из вариантов осуществления, настоящее изобретение включает способ лечения или уменьшения тяжести одного или нескольких заболеваний, в формировании которого участвует JAK3-киназа. В частности, настоящее изобретение относится к способу лечения или уменьшения тяжести заболевания или состояния из следующих перечисленных: иммунные реакции, такие, как аллергические реакции или реакции гиперчувствительности I типа; астма; аутоиммунные заболевания, такие, как отторжение трансплантатов, реакции «трансплантат против хозяина», ревматоидный артрит, боковой амиотрофический склероз, рассеянный склероз; нейродегенеративные расстройства, таких как семейный боковой амиотрофический склероз (Fals), а также злокачественные опухоли и болезни крови, такие, как лейкозы и лимфомы, когда данный способ лечения включает введение пациенту, нуждающемуся в этом, композиции, являющейся предметом настоящего изобретения.
[00218] Соединения и их композиции, в соответствии с методикой, представленной в настоящем изобретении, могут быть введены в любых дозах и любыми способами, эффективными для лечения или уменьшения тяжести рака, аутоиммунных расстройств, нейродегенеративных и неврологических расстройств, шизофрении, заболеваний костей, болезней печени и сердца. Дозировка зависит от вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного типа лекарственного вещества, формы введения и т.п. Соединения согласно настоящему изобретению предпочтительно должны быть представлены в лекарственных формах, удобных для приема и обеспечивающих однородность единиц дозирования. Термин "лекарственная форма", используемый здесь, относится к физически дискретной единице лекарственного вещества, назначаемого при терапии. Необходимо понимать, что совокупное количество соединения-предмета настоящего изобретения и его композиций, назначаемое пациенту ежедневно, должно определяться лечащим врачом по результатам тщательного медицинского обследования. Эффективная доза для каждого конкретного пациента/организма зависит от ряда факторов, включая характер заболевания и степень его тяжести, активность конкретного
используемого соединения; характеристики используемого препарата, возраст, массу тела, общее состояние здоровья, пол и рацион пациента; время введения, способ введения и скорость выведения из организма конкретного используемого соединения; длительность лечения; лекарства, используемые параллельно или в комбинации с конкретным соединением, и т.п. факторы, известные специалистам. Термин "пациент", используемый здесь, означает животных, преимущественно млекопитающих, и чаще всего людей.
[00219] Фармацевтически приемлемые композиции, составляющие предмет данного изобретения, могут быть введены людям и животным орально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей, капель), буккально (сублингвально), распылением в носовую/ротовую полость (в виде спрея) и т.п., в зависимости от тяжести инфекции. В некоторых вариантах осуществления соединения - предмет настоящего изобретения могут быть введены перорально или парентерально в дозировке 0,01-50 мг/кг, предпочтительно от 1 мг/кг до приблизительно 25 мг/кг массы тела, один или несколько раз в день, для получения желаемого терапевтического эффекта.
[00220] Жидкие лекарственные формы для перорального применения включают (но не ограничиваются ими) фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активных компонентов жидкие лекарственные формы могут содержать стандартные инертные разбавители, например, воду или другие растворители, солюбилизирующие агенты, эмульгаторы, такие, как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензил бензоат, пропиленгликоль, 1,3-бутилен гликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей пшеницы, оливковое, касторовое, кунжутное), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоль и сложные эфиры жирных кислот сорбитана, и их смеси. Кроме инертных разбавителей составы для перорального применения могут включать вспомогательные вещества, такие как увлажняющие агенты, эмульгаторы и замедлители растворения, подсластители, ароматизаторы, ароматизаторы.
[00221] Препараты для инъекций, например, стерильные инъекционные водные или масляные суспензии могут быть приготовлены в соответствии с общеизвестными методиками, с использованием подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов. Стерильный инъекционный препарат может быть также
представлен стерильным инъекционным раствором, суспензией и эмульсией в нетоксичном парентерально приемлемом разбавителе/растворителе, например, в виде раствора в 1,3-бутандиоле. Примеры приемлемых связующих/растворителей: вода, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме того, часто в качестве связующих/растворителей применяются стерильные жирные масла. С этой целью может быть использовано любое нейтральное жирное масло, в том числе синтетические моно- или диглицериды. Кроме того, для приготовления инъекций используются жирные кислоты, например, олеиновая кислота.
[00222] Составы для инъекций могут быть стерилизованы, например, путем фильтрации через бактериальный фильтр, или путем включения стерилизующих агентов в виде твердых стерильных композиций, которые перед использованием могут быть растворены или суспензированы в стерильной воде или других стерильных инъекционных средах.
[00223] Для пролонгации эффекта соединения - предмета настоящего изобретения, часто желательно замедлить всасывание действующего вещества, введенного подкожно или внутримышечно. Это может быть достигнуто путем использования жидкой суспензии кристаллического или аморфного вещества, плохо растворимого в воде. Скорость всасывания соединения в этом случае будет зависеть от скорости растворения указанного вещества, что, в свою очередь, зависит от размера кристаллов и типа кристаллизации. Кроме того, при парентеральном введении замедлить всасывание можно путем растворения или суспензирования соединения в масляном связующем. Лекарственные формы для инъекций замедленного всасывания изготавливаются путем встраивания микрокапсул с действующим веществом в биоразлагаемые полимеры, такие как полилактид-полигликоль. Скорость всасывания соединения может регулироваться, так как зависит от изменений количественного соотношения соединения и полимера, и свойств конкретного используемого полимера. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Составы для инъекций замедленного всасывания также могут быть изготовлены путем помещения действующего вещества в липосомы или путем приготовления микроэмульсий, совместимых с тканями организма.
[00224] Составы для ректального или вагинального введения, предпочтительно в форме суппозиториев, могут быть получены путем смешивания соединений, являющихся предметом данного изобретения, с подходящими нераздражающими наполнителями или
носителями, такими, как какао-масло, полиэтиленгликоль или воск; твердыми при комнатной температуре и жидкими при температуре тела, и поэтому растворяющимися в прямой кишки или вагинальномй полости, высвобождая активное вещество.
[00225] Твердые лекарственные формы для перорального применения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивают по крайней мере с одним инертным, фармацевтически приемлемым вспомогательным веществом или носителем, например, с цитратом натрия или дикальцийфосфатом и/или а.) наполнителями и добавками, такими, как крахмал, лактоза, сахароза, глюкоза, маннитол, кремниевая кислота; б) связующими веществами, такими, как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза, гуммиарабик; в.) влагоудерживаюшие вещества, такие как глицерин г.), разрыхлители, например, агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты, карбонат натрия; д.) замедлители растворения, такие как парафин; е.) ускорители абсорбции, например, соединения четвертичного аммония; ж.) увлажняющие агенты, такие, как, например, цетиловый спирт и моностеарат глицерина; з.) абсорбенты, например, каолин и бентонитовая глина; и.) любриканты, такие, как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и их смеси. В случае капсул, таблеток и пилюль лекарственная форма может также содержать буферизующие вещества.
[00226] Подобные твердые композиции могут быть также использованы при изготовлении желатиновых капсул с мягким/твердым содержимым в качестве наполнителей, в этом случае могут быть использованы лактоза (молочный сахар), полиэтиленгликоли с высокими молекулярными массами и т.п. Твердые лекарственные формы - таблетки, драже, капсулы, пилюли, гранулы - могут производиться с разнообразными покрытиями и оболочками, согласно используемым в фармацевтической промышленности рецептурам, например, кишечнорастворимыми (энтеросолюбильными) и пр. Они могут дополнительно содержать замутняющие агенты, а также могут иметь состав, обеспечивающий высвобождение активного ингредиента/ингредиентов только или преимущественно в определенной части кишечного тракта, иногда отложенное во времени. Примеры капсулирующих композиций, которые могут быть использованы при производстве лекарственных форм, включают полимерные вещества и воски. Твердые
композиции указанного типа могут быть также использованы в качестве наполнителей в желатиновых капсулах с мягким/твердым содержимым, включающим такие вспомогательные вещества, как лактоза (молочный сахар), высокомолекулярные полиэтиленгликоли и т.п..
[00227] Активные соединения могут также быть представлены микроинкапсулированными формами, в состав которых входит одно или несколько вспомогательных веществ из описанных выше. Твердые лекарственные формы - таблетки, драже, капсулы, пилюли, гранулы - могут производиться с различными покрытиями/оболочками, например, кишечнорастворимым покрытием, покрытием, контролирующим высвобождение действующего вещества и пр., согласно применяемым в фармацевтике рецептурам. В таких твердых лекарственных формах активные вещества могут быть смешаны по крайней мере с одним инертным разбавителем, таким, как сахароза, лактоза или крахмал. Обычной практикой является также добавление в такие лекарственные формы дополнительных веществ, помимо инертных разбавителей, например, смазочных веществ и других вспомогательных таблетирующих агентов, таких, как стеарат магния и микрокристаллическая целлюлоза. При производстве капсул, таблеток и пилюль лекарственные формы могут также содержать буферные агенты. Они могут дополнительно содержать замутняющие агенты, а также могут иметь состав, обеспечивающий высвобождение активного ингредиента/ингредиентов только или преимущественно в определенной части кишечного тракта, иногда отложенное во времени. Примеры капсулирующих композиций, которые могут быть использованы при производстве этих лекарственных форм, включают полимерные вещества и воски.
[00228] Лекарственные формы соединений, являющихся предметом настоящего изобретения, для местного применения или чрескожного введения включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингаляторы и пластыри. Активные компоненты смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами и буферизующими веществами. Офтальмологические препараты, ушные капли и глазные капли также входят в состав данного изобретения. Кроме того, настоящее изобретение предусматривает использование трансдермальных пластырей, которые имеют дополнительное преимущество, обеспечивая контролируемое поступление действующего вещества в организм. Такие лекарственные
формы могут быть изготовлены путем растворения или разведения действующего вещества в подходящей среде. Для увеличения эффективности проникновения через кожу могут быть использованы усилители абсорбции. Скорость проникновения можно регулировать либо при помощи мембраны, обеспечивающей определенный уровень поступления действующего вещества, либо путем диспергирования соединения в полимерной матрице или геле.
[00229] В одном из вариантов осуществления изобретение относится к способу ингибирования активности протеинкиназы в биологическом образце, включая стадию осуществления контакта биологической пробы с соединением - предметом данного изобретения, или композиции, содержащей указанное соединение.
[00230] В другом варианте осуществления изобретение относится к способу ингибирования активности ErbB1, ErbB2, ErbB3, ERBB4, ТЕС-киназы и/или JAK3, или их модифицированных (мутантных) версий в биологическом образце, включая стадию осуществления контакта биологического образца с соединением-предметом данного изобретения, или композиции, содержащей указанное соединение. В некоторых вариантах, изобретение относится к методу необратимо ингибирующие ErbB1, ErbB2, ErbB3, ERBB4, ТЭЦ-киназы, и/или JAK3, или мутант их, деятельности в биологическом образце, включающий стадию осуществления контакта биологических образцов с соединением согласно настоящему изобретению, или композиции, содержащей указанное соединение.
[00231] В настоящем описании термин "биологический образец" включает в себя неограниченные варианты культур клеток или их экстрактов, материалы биопсии, полученные из тканей млекопитающих или их экстрактов, а также кровь, слюну, мочу, кал, сперму, слезы или другие жидкости или их экстракты.
[00232] Ингибирование протеинкиназной активности, или активности протеинкиназ из следующих перечисленных: ErbB1, ErbB2, ErbB3, ERBB4, ТЕС-киназы и/или JAK3, или их модифицированных (мутантных) версий в биологических образцах может быть использовано с различными целями, очевидными для специалистов, включая следующие примеры (но не ограничиваясь ими): переливание крови, пересадка органов, хранение биологических образцов и биологических проб.
[00233] В другом варианте осуществления настоящее изобретение относится к способу ингибирования активности протеинкиназы в организме пациента, включая стадию введения указанному пациенту соединения-предмета настоящего изобретения, или композиции, содержащей указанное соединение.
[00234] В другом варианте осуществления настоящее изобретение относится к способу ингибирования активности одной или нескольких киназ из следующих перечисленных: ErbB1, ErbB2, ErbB3, ERBB4, ТЕС-киназы, и/или JAK3, или их модифицированных (мутантных) версий, в организме пациента, включая стадию введения указанному пациенту соединения - предмета настоящего изобретения, или композиции, содержащей указанное соединение. В некоторых вариантах осуществления изобретение относится к методу необратимого ингибирования активности одной или нескольких киназ из следующих перечисленных: ErbB1, ErbB2, ErbB3, ERBB4, ТЕС-киназы, и/или JAK3, или их модифицированных (мутантных) версий, в организме пациента, включая стадию введения указанному пациенту соединения-предмета настоящего изобретения, или композиции, содержащей указанное соединение. В других вариантах осуществления настоящее изобретение включает способ лечения заболеваний (расстройств), опосредованных активностью одной или нескольких киназ из следующих перечисленных: ErbB1, ErbB2, ErbB3, ERBB4, ТЕС-киназы, и/или JAK3, или их модифицированных (мутантных) версий, у пациентов, нуждающихся в этом, включая стадию введения указанному пациенту соединения-предмета настоящего изобретения или фармацевтически приемлемой композиции, включающей указанное соединение. Такие заболевания (расстройства) подробно описаны здесь.
[00235] В зависимости от конкретного целевого состояния или заболевания, в композиции-предмете данного изобретения могут присутствовать дополнительные лекарственные средства, обычно назначаемые при терапии данного состояния/заболевания. Такие дополнительные лекарственные средства, назначаемые для лечения конкретного заболевания, упоминаются здесь как "принятые для лечения данного заболевания или состояния"
[00236] Например, соединения согласно настоящему изобретению или их фармацевтически приемлемые композиции назначают в сочетании с химиотерапевтическими препаратами для лечения пролиферативных заболеваний и рака.
Примеры известных химиотерапевтических препаратов включают (не ограничиваясь перечисленными): адриамицин, дексаметазон, винкристин, циклофосфамид, фторурацил, топотекан, таксол, интерфероны, производные платины, таксан (например, паклитаксел), алкалоиды барвинка (например, винбластин), антрациклины (например, доксорубицин), эпиподофиллотоксины (например, этопозид), цисплатин, ингибиторы мTOR (например, рапамицин), метотрексат, актиномицин D, доластатин 10, колхицин, эметин, триметрексат, метоприн, циклоспорин, даунорубицин, тенипосид, амфотерицин, алкилирующие агенты (например, хлорамбуцил), 5-фторурацил, камптотецин, цисплатин, метронидазол, «Гливек»™ и др. В других вариантах осуществления соединения согласно настоящему изобретению вводят в сочетании с биологическим агентом, таким как «Авастин» или VECTIBIX.
[00237] В некоторых вариантах осуществления соединения, являющиеся предметом настоящего изобретения или их фармацевтически приемлемые композиции вводятся в сочетании с одним или несколькими антипролиферативными или химиотерапевтическими веществами, выбранными из следующих: абареликс, альдеслейкин, алемтузумаб, алитретиноин, аллопуринол, альтретамин, амифостин, анастрозол, триоксид мышьяка, аспарагиназа, азацитидин, живая БЦЖ, бевацизумаб, фторурацил, бексаротен, блеомицин, бортезомиб, бусульфан, калустерон, капецитабин, камптотецин, карбоплатин, кармустин, целекоксиб, цетуксимаб, хлорамбуцил, кладрибин, клофарабин, циклофосфамид, цитарабин, дактиномицин, дарбепоэтин альфа, даунорубицин, денилейкин, дексразоксан, доцетаксел, доксорубицин (нейтральный), доксорубицина гидрохлорид, дромостанолон пропионат, эпирубицин, эпоэтин альфа, эрлотиниб, эстрамустин, этопозида фосфат, этопозид, экземестан, филграстим, флоксуридин, флударабин, фулвестрант, гефитиниб, гемцитабин, гемтузумаб, госерелина ацетат, гистрелина ацетат, гидроксимочевина, ибритумомаб, идарубицин, ифосфамид, иматиниба мезилат, интерферон альфа-2а, интерферон альфа-2b, иринотекан, леналидомид, летрозол, лейковорин, лейпролида ацетат, левамизол, ломустин, мегестрол ацетат, мелфалан, меркаптопурин, 6-меркаптопурин, месна, метотрексат, метоксален, митомицин С, митотан, митоксантрон, нандролон, неларабин, нофетумомаб, опрелвекин, оксалиплатин, паклитаксел, палифермин, памидронат, пегадемаза, пегаспаргаза, пэгфилграстим, пеметрексед динатрия, пентостатин, пипоброман, пликамицин, порфимер натрия, прокарбазин,
акрихин, расбуриказа, ритуксимаб, сарграмостим, сорафениб, стрептозоцин, сунитиниб малеат, тальк, тамоксифен, темозоломид, тенипозид, VM-26, тестолактон, тиогуанин, 6-тиогуанин, тиотепа, топотекана, торемифен, тоситумомаб, трастузумаб, третиноин, ATRA, урамустин, вальрубицин, винбластин, винкристин, винорельбин, золедронат или золедроновая кислота.
[00238] Примеры других агентов, с которыми могут быть скомбинированы ингибиторы, являющиеся предметом данного изобретения, включают (без ограничения): препараты для лечения болезни Альцгеймера, такие, как донепезила гидрохлорид (Арисепт®) и ривастигмин (Экселон®); препараты для лечения болезни Паркинсона, такие как леводопа/карбидопа, энтакапон, ропинирол, прамипексол, бромокриптин, перголид, тригексифенидил и амантадин; средства для лечения рассеянного склероза, такие как бета-интерферон (например, Авонекс® и Ребиф®), глатирамера ацетат (Копаксон®) и митоксантрон; препараты для лечения астмы, такие, как альбутерол и монтелукаст (Сингуляр®); препараты для лечения шизофрении, такие, как зипрекса, риспердал, сероквель и галоперидол; противовоспалительные средства, такие как кортикостероиды, блокаторы ФНО (фактора некроза опухолей), ИЛ-1 РА (блокаторы интелейкина-1), азатиоприн, циклофосфамид и сульфасалазин; иммуномодулирующие и иммуносупрессивные препараты, такие, как циклоспорин, такролимус, рапамицин, микофенолата мофетил, интерфероны, кортикостероиды, циклофосфамид, азатиоприн, сульфасалазин; и нейротрофические факторы, такие, как ингибиторы ацетилхолинэстеразы, ингибиторы моноаминоксидазы, интерфероны, противосудорожные препараты, блокаторы ионных каналов, рилузол; препараты для лечения болезни Паркинсона; препараты для лечения сердечно-сосудистых заболеваний - бета-блокаторы, ингибиторы АПФ (ангиотензинпревращающего фермента), диуретики, нитраты, блокаторы кальциевых каналов и статины; препараты для лечения заболеваний печени - кортикостероиды, холестирамин, интерфероны; антивирусные препараты; препараты для лечения заболеваний крови - кортикостероиды, антилейкемические препараты, факторы роста, а также препараты для лечения иммунодефицитных состояний, такие, как гамма-глобулин.
[00239] В некоторых вариантах осуществления соединения согласно (настоящему изобретению или их фармацевтически приемлемые композиции вводят в сочетании с моноклональными антителами или препаратами миРНК.
[00240] Эти дополнительные средства могут вводиться отдельно от композиций, содержащих соединения - предмет данного изобретения, в составе комплексной схемы лечения. Также эти средства могут быть частью одной лекарственной формы, составляя единую композицию вместе с соединением-предметом этого изобретения. При назначении в составе комплексной схемы, два активных вещества могут приниматься одновременно, последовательно или с временным интервалом, обычно равным пяти часам
[00241] Используемые здесь термины "сочетание", «комбинация», "комбинированный" и связанные с ними термины относятся к одновременному или последовательному назначению лекарственных средств в соответствии с настоящим изобретением. Например, соединения, являющиеся предметом настоящего изобретения, могут быть введены одновременно или последовательно с другим лекарственным средством, в виде отдельных лекарственных форм или в составе одной лекарственной формы. Таким образом, настоящее изобретение касается дозированных единичных лекарственных форм, включающих указанные соединения, дополнительные лекарственные средства и фармацевтически приемлемые вещества-носители, вспомогательные или связующие вещества.
[00242] Количество обоих веществ: соединения-предмета изобретения и дополнительного лекарственного средства (в тех композициях, которые включают дополнительное лекарственное средство, как описано выше), которое может быть соединено с веществами-носителями для производства одной лекарственной формы, будет меняться в зависимости от целевого объекта и способа введения. Преимущественно, рецептура композиций согласно настоящему изобретению должна обеспечивать возможность введения указанной композиции в дозировке 0,01-100 мг/кг массы тела в день.
[00243] В композициях, включающих дополнительное лекарственное средство, данное дополнительное лекарственное средство и соединение-предмет данного изобретения могут действовать синергически (взаимно усиливая эффект от применения). Таким образом, количество дополнительного лекарственного средства в таких композициях
будет меньше, чем требуется при мототерапии с использованием одного указанного лекарственного средства. При терапии с использованием указанных композиций может назначаться дополнительное лекарственное средство в дозировках 0,01-1,000 иг/кг массы тела в день.
[00244] Количество дополнительного лекарственного средства в составах, являющихся предметом данного изобретения, не будет превышать количество, в норме присутствующее в композициях, где лекарственное средство является единственным активным веществом. Преимущественно, количество дополнительного лекарственного средства в данных композициях будет варьировать от 50% до 100% от количества, в норме присутствующего в композициях, где лекарственное средство является единственным активным веществом.
[00245] Соединения, являющиеся предметом настоящего изобретения, или фармацевтические композиции, в состав которых входят указанные соединения, также могут быть включены в составы для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, сосудистые импланты, стенты и катетеры. Сосудистые стенты, например, используются для профилактики рестеноза (повторного сужения просвета сосуда). Однако при использовании стентов или других имплантируемых устройств существует риск образования тромбов или активации тромбоцитов. Эти нежелательные эффекты могут быть предотвращены или смягчены путем предварительного покрытия имплантируемого устройства фармацевтически приемлемым составом, содержащим ингибитор киназной активности. Имплантируемые устройства, покрытые соединением-предметом данного изобретения, также являются вариантом осуществления настоящего изобретения.
5. Соединения-зонды
[00246] В некоторых аспектах соединение согласно настоящему изобретению может быть связано с детектируемой группой с образованием соединения-зонда. В одном аспекте соединение-зонд согласно настоящему изобретению включает необратимый ингибитор протеинкиназы формулы I-а или I-b, как описано выше, детектируемую группу и связывающую группу, которая присоединяет ингибитор к детектируемой группе.
[00247] В некоторых вариантах осуществления такие соединения-зонды включают предложенное соединение формулы I-а или I-b, связанное с детектируемой группой, Rt,
бивалентной связывающей группой, Связывающая группа может быть присоединены к соединению формулы I-а или I-b через Кольцо А, Кольцо В или, что если связывающая группа присоединена к R1, R1 представляет собой бивалентную реакционную группу, обозначенную R1'. В некоторых вариантах осуществления предложенное соединение-зонд выбрано из соединений любой из формул V-a, V-b, VI-a, VI-b, VII-a или VII-b:
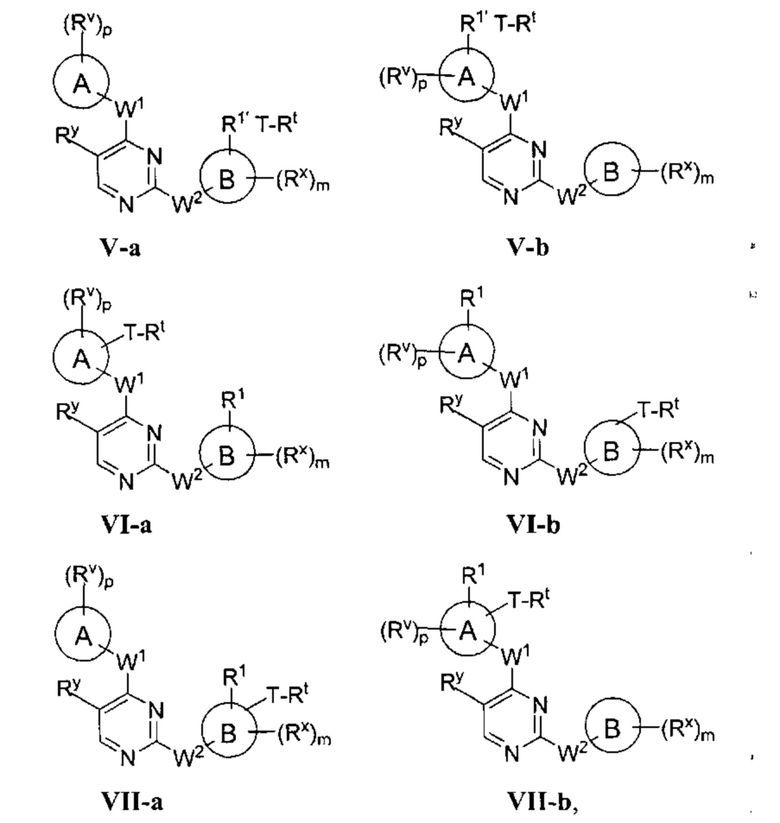
где каждый из Кольца А, Кольца В, R1, m, р, Rx, Ry, Rv, W1 и W2 имеет значение, приведенные выше для формул I-а и I-b и описанные в классах и подклассах в настоящему тексте R1' представляет собой бивалентную реакционноспособную группу, Т представляет собой бивалентную соединительную группу; и Rt представляет собой бивалентную детектируемую группу.
[00248] В некоторых вариантах осуществления Rt представляет собой бивалентную детектируемую группу, выбранную из первичной метки и вторичной метки. В некоторых вариантах осуществления Rt представляет собой бивалентную детектируемую группу,
выбранную из флюоресцентной метки (например, флюоресцентного красителя или флюорофора), масс-метки, хемилюминесцентной группы, хромофора, электронно-плотной группы или вещества, переносящего энергию.
[00249] В настоящем тексте термин "детектируемая метка" используется взаимозаменяемо с терминами "метка" и "репортер" и относится к любой группе, которую можно детектировать, например, к первичным метками и вторичным меткам. Присутствие детектируемой группы моет быть измерено с использованием методов количественного анализа (в абсолютных, приблизительных или относительных единицах) детектируемой группы в исследуемой системе. В некоторых вариантах осуществления такие методы хорошо известны средним специалистам в данной области и включают любые методы количественного анализа репортерной группы (например, метки, красителя, агента, образующего поперечные сшивки под действием света, цитотоксического соединения, лекарственного средства, аффинной метки, реактивного соединения, антитела или фрагмента антитела, биоматериала, наночастицы, спиновой метки, флюорофора, металлсодержащего соединения, радиоактивной группы, квантовой точки (точек), новой функциональной группы, группы, которая ковалентно или нековалентно взаимодействует с другими молекулами, фотосвязываемой группы, группы, возбуждаемой актиничным излучением, лиганда, фотоизомеризуемого соединения, биотина, аналога биотина (например, биотин сульфоксида), фрагмента, содержащего тяжелый атом, химически расщепляемой группы, фоторасщепляемой группы, редокс-активного агента, группы, меченной изотопом, биофизического зонда, фосфоресцентной группы, хемилюминесцентной группы, электронноплотной группы, магнитной группы, интеркалирующей группы, хромофора, агента, переносящего энергию, биологически активного вещества, детектируемой метки и любой комбинации вышеперечисленного).
[00250] Первичные метки, такие как радиоизотопы (например, тритий, 32Р, 35Р, 35S, 14С, 123I, 124I, 125I или 131I), масс-метки, включая, без ограничения, стабильные изотопы (например, 13С, 2Н, 17О, 18О, 15N, 19F и 127I), позитрон-испускающие изотопы (например, 11С, 18F, 13N, 124I и 15O) и флюоресцентные метки, представляют собой репортерные группы, генерирующие сигнал, которые моно детектировать без дополнительных модификаций. Детектируемые группы можно анализировать способами, включающими, но не ограничивающимися перечисленными: флюоресценция, позитронно-эмиссионная
томография, медицинская визуализация SPECT, хемилюминесценция, электрон-спиновый резонанс, спектроскопия поглощения в УФ- и видимой области, масс-спектрометрия, ядерный магнитный резонанс, магнитный резонанс, проточная цитометрия, авторадиография, регистрация сцинтилляции и методы электрохимии.
[00251] Термин «вторичная метка» в настоящем тексте относится к группам, таким как биотин и антигены различных белков, которые генерируют детектируемый сигнал только в присутствии второго промежуточного соединения. Для биотина вторичным промежуточным соединением могут быть конъюгаты стрептавидин-фермент. Для меток, являющихся антигенами, вторичные промежуточные соединения могут включать конъюгаты антитело-фермент. Некоторые флюоресцентные группы действуют как вторичные метки, поскольку они переносят энергию на другую группу в процессе нерадиоактивного резонансного переноса энергии (FRET), и указанная вторая группа генерирует сигнал.
[00252] Термины "флюоресцентная метка", "флюоресцентный краситель" и "флюорофор" в настоящем тексте относятся к группам, которые поглощают энергию света на определенной длине волны возбуждения и испускают световую энергию на другой длине волны. Примеры флюоресцентных меток включают, но не ограничиваются перечисленными: (Alexa Fluor 350, Alexa Fluor 488, Alexa Fluor 532, Alexa Fluor 546, Alexa Fluor 568, Alexa Fluor 594, Alexa Fluor 633, Alexa Fluor 660 и Alexa Fluor 680), AMCA, AMCA-S, BODIPY dyes (BODIPY FL, BODIPY R6G, BODIPY TMR, BODIPY TR, BODIPY 493/503, BODIPY 530/550, BODIPY 558/568, BODIPY 564/570, BODIPY 576/589, BODIPY 581/591, BODIPY 630/650, BODIPY 650/665), Карбоксиродамин 6G, карбокси-Х-родамин (ROX), Cascade Blue, Cascade Yellow, Coumarin 343, цианиновые красители (Су3, Cy5, Су3.5, Су5.5), Dansyl, Dapoxyl, диалкиламинокумарин, 4',5'-дихлор-2',7'-диметокси-флюоресцин, DM-NERF, Eosin, Erythrosin, Fluorescein, FAM, Гидроксикумерин, IRDyes (IRD40, IRD 700, IRD 800), JOE, Lissaraine rhodamine B, Marina Blue, Methoxycoumarin, Naphthofluorescein, Oregon Green 488, Oregon Green 500, Oregon Green 514; Pacific Blue, PyMPO, Pyrene, Rhodamine B, Rhodamine 6G, Rhodamine Green, Rhodamine Red, Rhodol Green, 2',4',5',7'-тетра-бромсульфон-флюоресцеин, тетраметил-родамин (TMR), карбокситетраметилродамин (TAMRA), Texas Red, Texas Red-X, 5(6)-карбоксифлюоресцеин, 2,7-Dichlorofluorescein, N,N-Bis(2,4,6-trimethylphenyl)-3,4:9,10-
perylenebis(dicarboximide, HPTS, Ethyl Eosin, DY-490XL MegaStokes, DY-485XL MegaStokes, Adirondack Green 520, ATTO 465, ATTO 488, ATTO 495, YOYO-1,5-FAM, BCECF, дихлорфлюоресцеин, родамин 110, родамин 123, YO-PRO-1, SYTOX Green, Sodium Green, SYBR Green I, Alexa Fluor 500, FITC, Fluo-3, Fluo-4, fluoro-emerald, YoYo-1 ssDNA, YoYo-1 dsDNA, YoYo-1, SYTO RNASelect, Diversa Green-FP, Dragon Green, EvaGreen, Surf Green EX, Spectrum Green, NeuroTrace 500525, NBD-X, MitoTracker Green FM, LysoTracker Green DND-26, CBQCA, PA-GFP (пост-активационный), WEGFP (пост-активационный), FlASH-CCXXCC, Azami Green мономерный, Azami Green, зеленый флюоресцирующий белок (GFP), EGFP (Campbell Tsien 2003), EGFP (Patterson 2001), Kaede Green, 7-бензиламино-4-нитробенз-2-окса-1,3-диазол, Bexl, Doxorubicin, Lumio Green и SuperGlo GFP.
[00253] Термин "масс-метка" в настоящем тексте относится к любой группе, которую возмоно однозначно детектировать по ее массе посредством методик детектирования на основе масс-спектрометрии (MS). Примеры масс-меток включают метки, высвобождающие электрофор, такие как N-[3-[4'-[(p-метокситетрафторбензил)окси]венил]-3-метилглицеронил]изонипекотиновая кислота, 4'-[2,3,5,6-тетрафтор-4-(пентафторфеноксил)]метил ацетофенон и их производные. Синтез и использование этих масс-меток описаны в патентах США 4,650,750, 4,709,016, 5,360,8191, 5,516,931, 5,602,273, 5,604,104, 5,610,020 и 5,650,270. Другие примеры масс-меток включают, но не ограничиваются перечисленными: нуклеотиды, дидеоксинуклеотиды, олигонуклеотиды различной длины и состава оснований, олигопептиды, олигосахариды и другие синтетические полимер, имеющие различную длину и состав мономеров. Разнообразные органические молекулы, как нейтральные так и заряженные (биомолекулы и синтетические соединения), масса которых лежит в подходящем диапазоне (100-2000 Дальтон), также моно использовать в качестве масс-меток. Таке в качестве масс-меток моно использовать стабильные изотопы (например, 13C, 2H, 17О, 18О, и 15N).
[00254] Термин "хемилюминесцентная группа" в настоящем тексте относится к группе, которая испускает свет в результате химической реакции без добавления тепла. В качестве пример, люмино(5-амино-2,3-дигидро-1,4-фталазиндион) реагирует с окисляющими веществами, такими как перекись водорода (H2O2) в присутствии основания и
металлического катализатора с образованием продукта в возбужденном состоянии (3-аминофталат, 3-АРА).
[00255] Термин «хромофор" в настоящем тексте относится к молекуле, которая поглощает свет видимых длин волн, УФ длин волн или ИК длин волн.
[00256] Термин «краситель" в настоящем тексте относится к растворимому красящему веществу, которое включает хромофор.
[00257] Термин «электронно-плотная группа" в настоящем тексте относится к группе, которая при облучении пучком электронов рассеивает электроны. Такие группы включают, но не ограничиваются перечисленными: молибдат аммония, субнитрат висмута, иодид кадмия, карбогидразид, гексагидрат хлорида железа, гексаметилен тетрамин, безводный трихлорид индия, нитрат лантана, тригидрат цитрата свинца, нитрат свинца, периодная кислота, фосфомолибденовая кислота, фосфовольфрамовая кислота, феррицианид калия, рутениевый красный, нитрат серебра, протеинат серебра (Тест на Ag: 8.0-8.5%) "Strong", тетрафенилпорфин серебра (S-TPPS), хлораурат натрия, вольфрамат натрия, нитрат таллия, тиосемикарбазид (TSC), уранилацетат, уранил сульфат и ванадил сульфат.
[00258] Термин «вещество, переносящее энергию" в настоящем тексте относится к молекуле, которая либо отдает, либо принимает энергию от другой молекулы. Исключительно в качестве примера: флюоресцентный резонансный перенос энергии (FRET) представляет собой процесс диполь-дипольного взаимодействия, в результате которого энергия возбужденного состояния флюоресцентной молекулы-донора нерадиоактивным путем переносится на невозбужденную молекулу-акцептор, которая затем испускает полученную энергию на более длинных волнах (флюоресцирует).
[00259] Термин «группа, включающая тяжелый атом" в настоящем тексте относится к группе, которая включает ион или атом, который обычно тяжелее углерода. В некоторых вариантах осуществления такие ионы или атомы включают, но не ограничены перечисленными: кремний, вольфрам, золото, свинец и уран.
[00260] Термин «фотоаффинная метка" в настоящем тексте относится к метке с группой, которая под воздействием света образует связь с молекулой, к которой эта метка обладает аффинностью.
[00261] Термин «фотосвязываемая группа" в настоящем тексте относится к группе, которая после освещения на определенной длине волны ковалентно или нековалентно связывается с другими ионами или молекулами.
[00262] Термин «фотоизомеризуемая группа" в настоящем тексте относится к группе, которая после освещения светом переходит из одной изоформы в другую.
[00263] Термин «радиоактивная группа" в настоящем тексте относится к группе, ядра которой спонтанно выдают ядерное излучение, такое как альфа-, бета-, или гамма-частицы; где альфа-частицы - это ядра гелия, бета-частицы - это электроны, а гамма-частицы - это фотоны с высокой энергией.
[00264] Термин «спиновая метка" в настоящем тексте относится к молекулам, которые содержат атом или группу атомов, демонстрирующих неспаренный электронный спин (т.е. стабильная парамагнитная группа), которую в некоторых вариантах осуществления детектируют путем электрон-спиновой резонансной спектроскопии, а в других вариантах реализации присоединяют к другой молекуле. Такие спин-метящие молекулы включают, но не ограничиваются перечисленными: нитрильные радикалы и нитроксиды и, в некоторых вариантах осуществления представляют собой отдельные спиновые метки или двойные спиновые метки.
[00265] Термин «квантовые точки" в настоящем тексте относится к коллоидным полупроводниковым нанокристаллам, которые в некоторых вариантах реализации детектируют в ближнем инфракрасном свете и которые обладают исключительно высокими квантовыми выходами (т.е., очень ярко светятся после умеренного освещения).
[00266] Для специалиста в данной области очевидно, что детектируемая группа может быть присоединена к предложенному соединению через подходящий заместитель. В настоящем тексте термин «подходящий заместитель" относится к группе, которая способна к ковалентному присоединению детектируемой группы. Такие группы хорошо известны специалистам и включают группы, включающие например, карбоксилатнную группу, аминогруппу, тиольную группу, гидроксильную группу или одну из многих других групп. Очевидно, что такие группа могут быть присоединены к предложенному соединению непосредственно или через связывающую группу, такую как бивалентная насыщенная или ненасыщенная углеводородная цепь.
[00267] В некоторых вариантах осуществления детектируемые группы присоединены к предложенному соединению посредством приемов «клик»-химии. В некоторых вариантах осуществления такие группы присоединены через 1,3-циклоприсоединение азида с алкином, возможно в присутствии медного катализатора. Методы использования клик-химии известны и включают методы, описанные в ст.Rostovtsev et ah, Angew. Chem. Int. Ed. 2002, 41, 2596-99 и Sun et ah, Bioconjugate Chem., 2006, 17, 52-57. некоторых вариантах осуществления обеспечивают клик-доступную ингибирующую группу и осуществляют ее реакцию с клик-доступной группой-T-Rt. В настоящем тексте "клик-доступный" относится к группе, содержащей азид или алкин для использования в реакции клик-химиию В некоторых вариантах осуществления клик-доступная ингибиторная группа включает азид. В некоторых вариантах осуществления клик-доступная группа-Т-Rt включает напряженный циклооктин для использования в реакции клик-химии без использования меди (например, с использованием методов, описанных в Baskin et ah, Proc. Natl. Acad. Sci. USA 2007, 104, 16793-16797).
[00268] В некоторых вариантах осуществления клик-доступная группа ингибитора имеет одну из следующих формул:
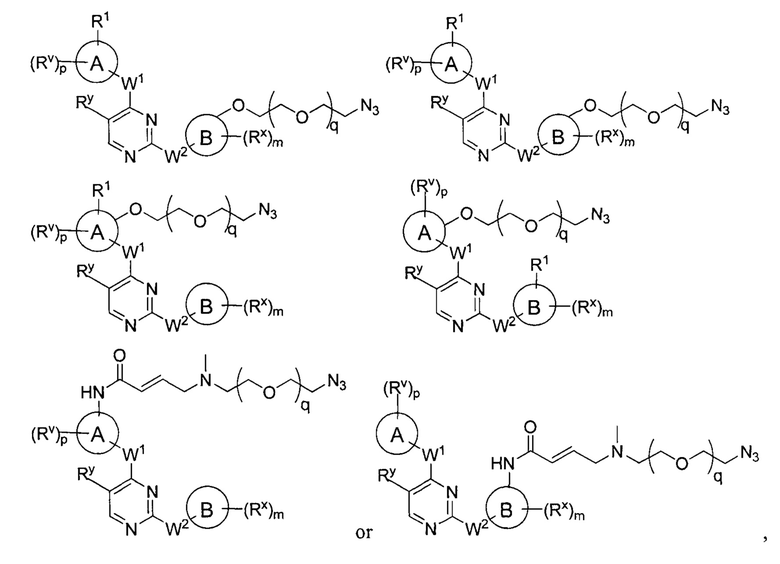
где кольцо А, кольцо В, W1, W2, Ry, Rv, р, Rx и m имеют значения, определенные выше для Формулы I и описанные в настоящем тексте, a q равно 1, 2 или 3.
[00269] Примеры клик-доступных ингибиторов включают:
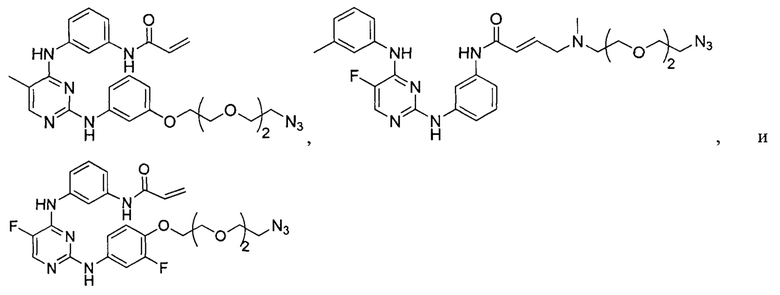
[00270] В некоторых вариантах осуществления клик-доступная группа -T-Rt имеет формулу:
[00271] Примером реакций, в которых клик-доступная группа ингибитора и клик-доступная группа -T-Rt присоединяются друг к другу путем [2+3]-циклоприсоединения, является следующая реакция:
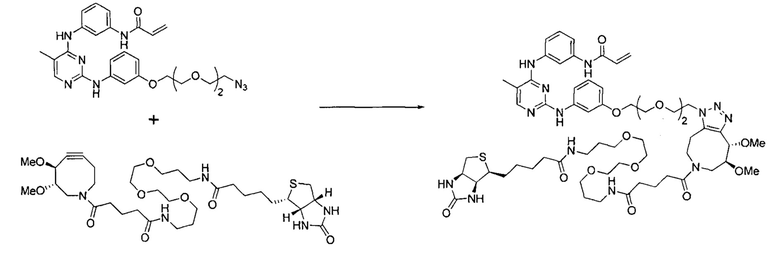
[00272] В некоторых вариантах осуществления детектируемая группа, Rt, выбрана из метки, красителя, агента, образующего поперечные сшивки под действием света,
цитотоксического соединения, лекарственного средства, аффинной метки, реактивного соединения, антитела или фрагмента антитела, биоматериала, наночастицы, спиновой метки, флюорофора, металл-содержащего соединения, радиоактивной группы, квантовой точки (точек), новой функциональной группы, группы, которая ковалентно или нековалентно взаимодействует с другими молекулами, фотосвязываемой группы, группы, возбуждаемой актиничным излучением, лиганда, фотоизомеризуемого соединения, биотина, аналога биотина (например, биотин сульфоксида), фрагмента, содержащего тяжелый атом, химически расщепляемой группы, фоторасщепляемой группы, редокс-активного агента, группы, меченной изотопом, биофизического зонда, фосфоресцентной группы, хемилюминесцентной группы, электронноплотной группы, магнитной группы, интеркалирующей группы, хромофора, агента, переносящего энергию, биологически активного вещества, детектируемой метки и любой комбинации вышеперечисленного.
[00273] В некоторых вариантах осуществления Rt представляет собой биотин или его аналог. В некоторых вариантах осуществления Rt представляет собой биотин. В некоторых других вариантах осуществления Rt представляет собой биотин сульфоксид. [00274] В другом варианте осуществления Rt представляет собой флюорофор. В дальнейшем варианте осуществления флюорофор из красителей Alexa Fluor (Alexa Fluor 350, Alexa Fluor 488, Alexa Fluor 532, Alexa Fluor 546, Alexa Fluor 568, Alexa Fluor 594, Alexa Fluor 633, Alexa Fluor 660 и Alexa Fluor 680), AMCA, AMCA-S, красителей BODIPY (BODIPY FL, BODIPY R6G, BODIPY TMR, BODIPY TR, BODIPY 493/503, BODIPY 530/550, BODIPY 558/568, BODIPY 564/570, BODIPY 576/589, BODIPY 581/591, BODIPY 630/650, BODIPY 650/665), Карбоксиродамина 6G, крабокси-Х-родамина (ROX), Cascade Blue, Cascade Yellow, Кумарина 343, цианиновых красителей Cyanine (Су3, Cy5, Су3.5, Су5.5), Дазила, Дапоксила, Диалкиламинокумарина, 4',5'-дихлор-2',7'-диметокси-флюоресцеина, DM-NERF, Эозина, Эритрозина, Флюоресцеина, FAM, гидроксикумарина IRDyes (IRD40, IRD 700, IRD 800), JOE, Lissamine rhodamine В, Marina Blue, метоксикумарин, нафтофлюоресцеин, Oregon Green 488, Oregon Green 500, Oregon Green 514, Pacific Blue, PyMPO, Pyrene, Родамин В, Родамин 6G, Rhodamine Green, Rhodamine Red, Rhodol Green, 2',4',5',7'-тетра-бромсульфон-флюоресцеин, тетраметил-родамин (TMR), карбокситетраметилродамин С (TAMRA), Texas Red, Texas Red-X, 5(6)-Карбоксифлуоресцеин, 2,7-дихлорфлюоресцеин, N,N-бис(2,4,6-триметилфенил)-3,4:9,10-
периленбис(дикарбоксимид, HPTS, Ethyl Eosin, DY-490XL MegaStokes, DY-485XL MegaStokes, Adirondack Green 520, ATTO 465, ATTO 488, ATTO 495, YOYO-l,5-FAM, BCECF, дихлорфлюоресцеин, Родамин 110, Родамин 123, YO-PRO-1, SYTOX Green, Sodium Green, SYBR Green I, Alexa Fluor 500, FITC, Fluo-3, Fluo-4, fluoro-emerald, YoYo-1 ssDNA, YoYo-1 dsDNA, YoYo-1, SYTO RNASelect, Diversa Green-FP, Dragon Green, EvaGreen, Surf Green EX, Spectrum Green, NeuroTrace 500525, NBD-X, MitoTracker Green FM, LysoTracker Green DND-26, CBQCA, PA-GFP (пост-активационный), WEGFP (пост-активационный), F1ASH-CCXXCC, Azami Green мономерный, Azami Green, зеленый флюоресцентный белок (GFP), EGFP (Campbell Tsien 2003), EGFP (Patterson 2001), Kaede Green, 7-бензиламино-4-нитробелнз-2-окса-1,3-диазол, Bexl, Doxorubicin, Lumio Green, или SuperGlo GFP.
[00275] Как в общих чертах описано выше, предложенные соединения-зонды включают связывающую группу, -Т-, которая присоединяет необратимый ингибитор к детектируемой группе. В настоящем тексте термин «нить" или "связывающая группа" относится к любому бивалентному химическому спейсеру, включая, но не ограничиваясь перечисленными: ковалентная связь, полимер, водорастворимый полимер, возможно содержащий заместители алкил, возможно содержащий заместители гетероалкил, возможно содержащий заместители гетероциклоалкил, возможно содержащий заместители циклоалкил, возможно содержащий заместители гетероциклил, возможно содержащий заместители гетероциклоалкилалкил, возможно содержащий заместители гетероциклоалкилалкенил, возможно содержащий заместители арил, возможно содержащий заместители гетероарил, возможно содержащий заместители гетероциклоалкилалкенилалкил, возможно содержащая заместители амидная группа, эфирная группа, кетонная группа, сложноэфирная группа, возможно содержащая заместители карбаматная группа, возможно содержащая заместители гидразонная группа, возможно содержащая заместители гидразинная группа, возможно содержащая заместители оксимная группа, дисульфидная группа, возможно содержащая заместители иминная группа, возможно содержащая заместители сульфонамидная группа, сульфонная группа, сульфоксидная группа, тиоэфирная группа или любая комбинация перечисленного.
[00276] В некоторых вариантах осуществления связывающая группа, -Т-, выбрана из ковалентной связи, полимера, водорастворимого полимера, возможно 'содержащего заместители алкила, возможно содержащего заместители гетероалкила, возможно содержащего заместители гетероциклоалкила, возможно содержащего заместители циклоалкила, возможно содержащего заместители гетероциклоалкилалкила, возможно содержащего заместители гетероциклоалкилалкенила, возможно содержащего заместители арила, возможно содержащего заместители гетероарила, и возможно содержащего заместители гетероциклоалкилалкенилалкила. В некоторых вариантах осуществления связывающая группа представляет собой возможно замещенный гетероцикл. В других вариантах осуществления гетероцикл выбран из азиридина, оксирана, эписульфида, азетидина, оксетана, пирролина, тетрагидрофурана, тетрагидротиофена, пирролидина, пиразола, пиррола, имидазола, триазола, тетразола, оксазола, изоксазола, оксирена, тиазола, изотиазола, дитиолана, фурана, тиофена, пиперидина, тетрагидропирана, тиана, пиридина, пирана, тиапирана, пиразина, пиримидина, пиперазина, оксазина, тиазина, дитиана и диоксана. В дальнейших вариантах осуществления связывающая группа возможно содержит в качестве заместителей галоген, -CN, -ОН, -NO2, алкил, S(O) и S(O)2. В других вариантах осуществления водорастворимый полимер представляет собой ПЭГ группу.
[00277] В других вариантах осуществления связывающая группа обеспечивает достаточной пространственное разделение детектируемой группы и группы ингибитора протеинкиназы. В других вариантах осуществления связывающая группа стабильна. В еще одном варианте осуществления связывающая группа не оказывает существленного влияния на ответ детектируемой группы. В других вариантах осуществления связывающая группа обеспечивает химическую стабильность соединения-зонда. В других вариантах осуществления связывающая группа обеспечивает достаточную растворимость соединения-зонда.
[00278] В некоторых вариантах осуществления связывающая группа, -Т-, такая как водорастворимый полимер одним концом присоединена к необратимому ингибитору согласно настоящему изобретению, а другим концом к детектируемой группе, Rt. В других вариантах осуществления водорастворимый полимер присоединен через функциональную группу или заместитель необратимого ингибитора согласно
настоящему изобретению. В других вариантах осуществления водорастворимый полимер присоединен через функциональную группу или заместитель репортерной группы.
[00279] В некоторых вариантах осуществления примеры гидрофильных полимеров для использования в связывающей группе -Т- включают, но не ограничиваются перечисленными: полиалкиловые эфиры и их кэпированные по карбокси-концу аналоги (например, полиоксиэтиленгликоль, полиоксиэтилен/пропиленгликоль, а также их метокси- и этокси-кэпированные аналоги, полиоксиэтиленгликоль также известен как полиэтиленгликоль или ПЭГ); поливинлипирролидоны; поливинилалкиловые эфиры; полиоксазолины, полиалкилоксазолины и полигидроксиалкил оксазолины, полиакриламиды, полиалкилакриламиды и полигидроксиалкил акриламиды (например, полигидроксипропилметакриламид и их производные); полигидроксиалкилакрилаты, полисиаловые кислоты и их аналоги, гидрофильные пептиные последовательности, полисахариды и их производные, включая производные декстана, например карбоксиметилдекстран, декстрансульфаты, аминодекстран; целлюлозу и ее производные, например, карбоксиметилцеллюлозу, гидроксиалкилцеллюлозы; хитин и его производные, хитозан, сукцинил хитозан, карбоксиметилхитин, карбоксиметилхитозан; гиалуроновую кислоту и ее производные; крахмалы; альгинаты; хондроитин сульфат; альбумин; пуллулан и карбоксиметилпуллулан; полиаминокислоты и их производные, например, полиглутаминовые кислоты, полилизины, полиаспарагиновые кислоты, полиаспартамида; сополимеры малеинового ангидрида, такие как: сополимер малеинового ангидрида и стирола, сополимер дивинилэтилового эвира и малеинового ангидрида; поливиниовые спирты; их сополимеры, их терполимеры, их смеси и полизводные перечисленных выше соединений. В других вариантах осуществления водорастворимый полимер имеет структурную форму, включающую, но не ограничивающуюся перечисленными: линейную, раздваивающуюся и разветвленную. В других вариантах осуществления многофункциональные производные полимера включают, но не ограничиваются перечисленными: линейные полимеры, имеющие два конца, каждый из которых связан с функциональной группой, причем группы могут быть одинаковыми или различными.
[00280] В некоторых вариантах осуществления водный полимер включает фрагмент поли(этиленгликоль). В других вариантах осуществления молекулярная масса полимера лежит в широком диапазоне, включая, без ограничения, от приблизительно 100 Да до
приблизительно 100,000 Да или больше. В других вариантах осуществления молекулярная масса полимера составляет в диапазоне от приблизительно 100 Да до приблизительно 100,000 Да, включая, без ограничения, приблизительно 100,000 Да, приблизительно 95,000 Да, приблизительно 90,000 Да, приблизительно 85,000 Да, приблизительно 80,000 Да, приблизительно 75,000 Да, приблизительно 70,000 Да, приблизительно 65,000 Да, приблизительно 60,000 Да, приблизительно 55,000 Да, приблизительно 50,000 Да, приблизительно 45,000 Да, приблизительно 40,000 Да, приблизительно 35,000 Да, 30,000 Да, приблизительно 25,000 Да, приблизительно 20,000 Да, приблизительно 15,000 Да, приблизительно 10,000 Да, приблизительно 9,000 Да, приблизительно 8,000 Да, приблизительно 7,000 Да, приблизительно 6,000 Да, приблизительно 5,000 Да, приблизительно 4,000 Да, приблизительно 3,000 Да, приблизительно 2,000 Да, приблизительно 1,000 Да, приблизительно 900 Да, приблизительно 800 Да, приблизительно 700 Да, приблизительно 600 Да, приблизительно 500 Да, приблизительно 400 Да, приблизительно 300 Да, приблизительно 200 Да и приблизительно 100 Да. В некоторых вариантах осуществления молекулярная масса полимера составляет от приблизительно 100 Да до 50,000 Да. В некоторых вариантах осуществления молекулярная масса полимера составляет от приблизительно 100 Да до приблизительно 40,000 Да. В некоторых вариантах осуществления молекулярная масса полимера составляет приблизительно от 1,000 Да до 40,000 Да. В некоторых вариантах осуществления молекулярная масса полимера составляет приблизительно от 5,000 Да до 40,000 Да. В некоторых вариантах осуществления молекулярная масса полимера составляет приблизительно от 10,000 Да до 40,000 Да. В некоторых вариантах осуществления молекула полиэтиленгликоля представляет собой разветвленный полимер. В других вариантах осуществления молекулярная масса разветвленной цепи ПЭГ составляет от приблизительно 1,000 Да до приблизительно 100,000 Да, включая, без ограничения, приблизительно 100,000 Да, приблизительно 95,000 Да, приблизительно 90,000 Да, приблизительно 85,000 Да, приблизительно 80,000 Да, приблизительно 75,000 Да, приблизительно 70,000 Да, приблизительно 65,000 Да, приблизительно 60,000 Да, приблизительно 55,000 Да, приблизительно 50,000 Да, приблизительно 45,000 Да, приблизительно 40,000 Да, приблизительно 35,000 Да, приблизительно 30,000 Да, приблизительно 25,000 Да, приблизительно 20,000 Да, приблизительно 15,000 Да,
приблизительно 10,000 Да, приблизительно 9,000 Да, приблизительно 8,000 Да, приблизительно 7,000 Да, приблизительно 6,000 Да, приблизительно 5,000 Да, приблизительно 4,000 Да, приблизительно 3,000 Да, приблизительно 2,000 Да, и приблизительно 1,000 Да. В некоторых вариантах осуществления молекулярная масса разветвленной цепи ПЭГ составляет от приблизительно 1,000 Да до приблизительно 50,000 Да. В некоторых вариантах осуществления молекулярная масса разветвленной цепи ПЭГ составляет от приблизительно 1,000 Да до приблизительно 40,000 Да. В некоторых вариантах осуществления молекулярная масса разветвленной цепи ПЭГ составляет от приблизительно 5,000 Да до приблизительно 40,000 Да. В некоторых вариантах осуществления молекулярная масса разветвленной цепи ПЭГ составляет от приблизительно 5,000 Да до приблизительно 20,000 Да. Ниже приведен список существенных водорастворимых каркасов, который не является исчерпывающим и представляет собой лишь иллюстрацию, в некоторых вариантах осуществления для использования в способах и композициях согласно настоящему изобретению подходят материалы, обладающие с описанными выше свойствами.
[00281] Для среднего специалиста в данной области очевидно, что если -T-Rt присоединена к соединению формулы I-а или I-b через реакционноспособную группу R1, получаемая в результате связывающая группа содержит указанную реакционноспособную группу R1. В настоящем описании фраза "содержит реакционноспособную группу" означает, что связывающая группа, образованная -R1'-Т- формулы V-a или V-b либо содерит реакционноспособную группу в качестве заместителя, либо реакционноспособная группа входит в состав связывающей группы. Например, связывающая группа, образованная -R1'-Т-, может содержать в качестве заместителя реакционноспособную группу -L-Y, причем каждая группа определена в настоящем описании. В альтернативном варианте связывающая группа, образованная -R1'-Т- обладает необходимыми свойствами реакционноспособной группы, включенной в эту связывающую группу. Например, связывающая группа, образованная -R1'-Т-, может включать один или ненасыщенных фрагментов и необязательных заместителей и/или гетероатомов, которые, в комбинации, образуют группу, способную к ковалентной модификации протеинкиназы согласно настоящему изобретению. Такая связывающая группа -R1'-T- представлена ниже.
[00282] В некоторых вариантах осуществления метиленовое звено связывающей группы -R1'-Т- заменено на бивалентную группу -L-Y'- с образованием соединения формулы V-a-iii или V-b-iii:
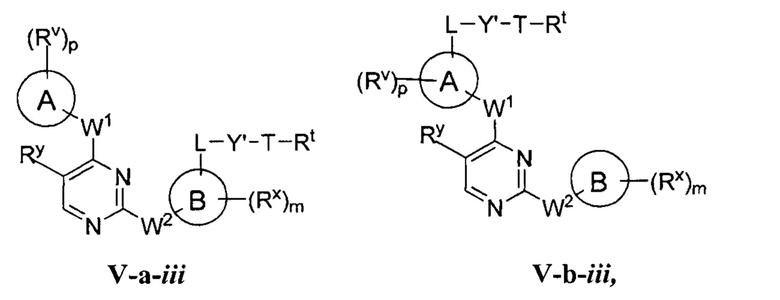
где каждый из Кольца А, Кольца В, m, р, Rx, Ry, Rv, W1, W2, Т, L, Y' и Rt определен выше и описан в настоящем текста в классах и подклассах, и Y' представляет собой бивалентный вариант группы Y, определенной в классах и подклассах в настоящем тексте.
[00283] В некоторых вариантах осуществления метиленовое звено связывающей группы - R1'-Т- заменено на группу -L(Y)- с получением соединения формулы V-a-iv или V-b-iv:
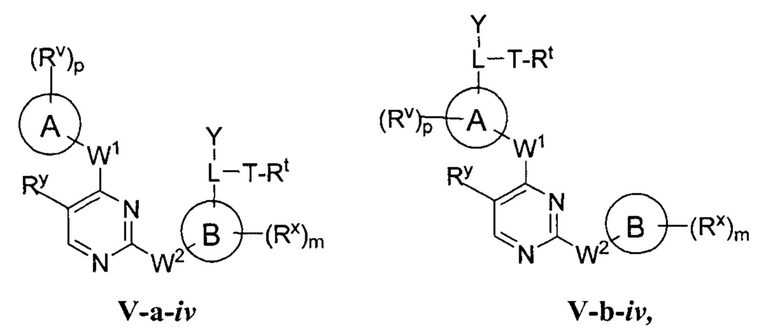
где каждый из Кольца А, Кольца В, m, р, Rx, Ry, Rv, W1, W2, T, L, Y' и Rt определен выше и описан в настоящем тексте в классах и подклассах.
[00284] В некоторых вариантах осуществления связывающая группа содержит в качестве заместителя группу L-Y, в результате чего образуется соединение формулы V-a-v или V-b-v:
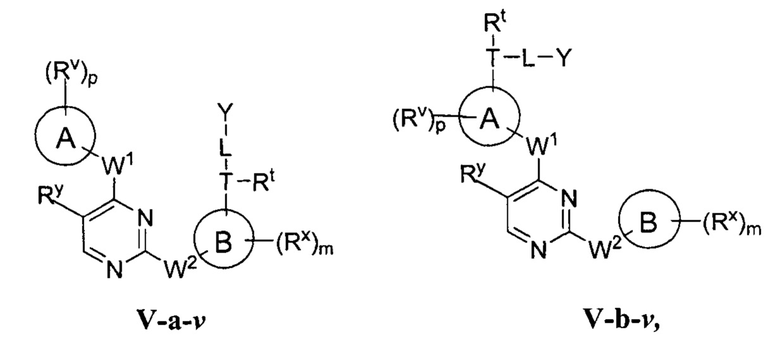
где каждый из Кольца А, Кольца В, m, р, Rx, Ry, Rv, W1, W2, T, L, Y и Rt определен выше и описан в настоящем тексте в классах и подклассах.
[00285] В некоторых вариантах осуществления связывающая группа, -Т-, имеет одну из следующих структур:
[00286] В некоторых вариантах осуществления связывающая группа, -Т-, имеет следующую структуру:
[00287] В других вариантах осуществления связывающая группа, -Т-, имеет следующую структуру:
[00288] В некоторых других вариантах осуществления связывающая группа, -Т-, имеет следующую структуру е:
[00289] В других вариантах осуществления связывающая группа, -Т-, имеет следующую структуру:
[00290] В некоторых вариантах осуществления связывающая группа, -Т-, имеет следующую структуру:
[00291] В некоторых вариантах осуществления -T-Rt имеет следующую структуру:
[00292] В других вариантах осуществления -T-Rt имеет следующую структуру:
[00293] В некоторых вариантах осуществления -T-Rt имеет следующую структуру:
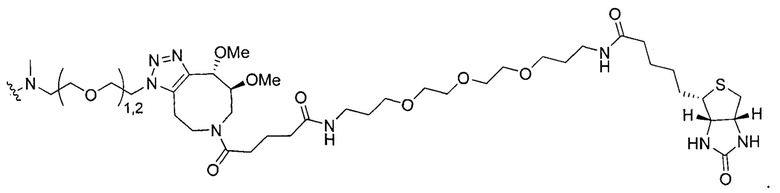
[00294] В некоторых вариантах осуществления соединение-зонд формулы V-a, V-b, VI-а, VI-b, VII-а или VII-b является производной одного из соединений в Таблице 5.
[00295] В некоторых вариантах осуществления соединение-зонд имеет одну из следующих структур:
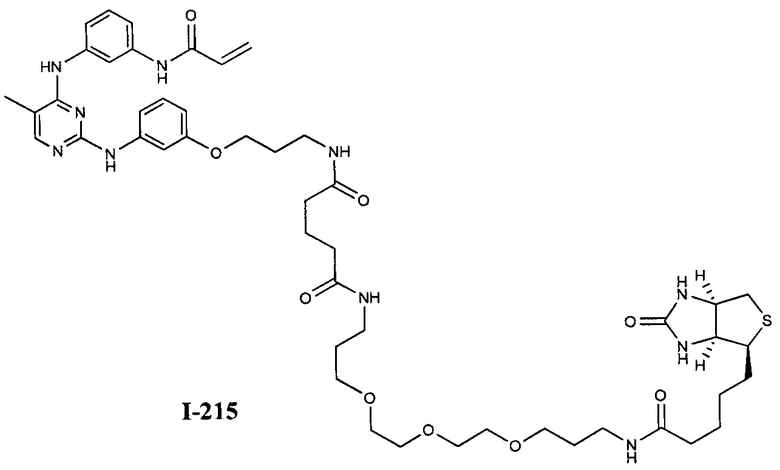
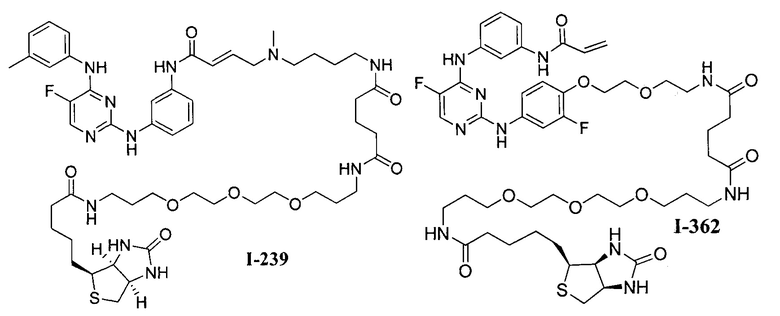
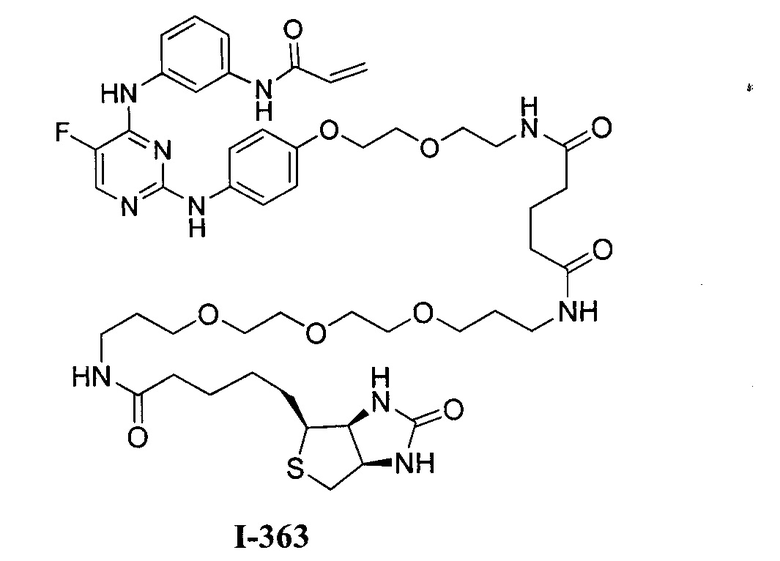
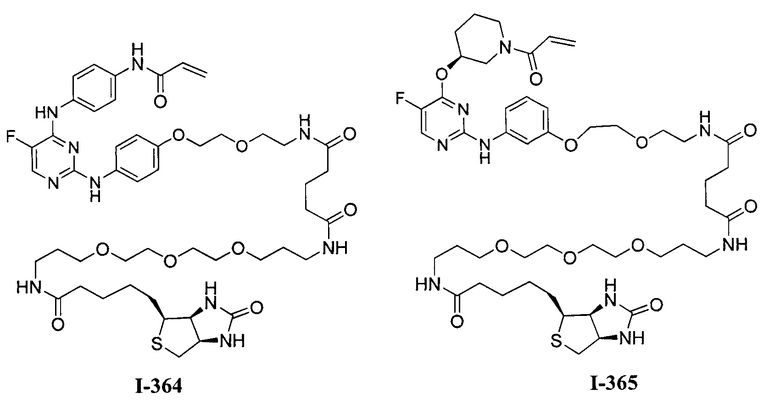
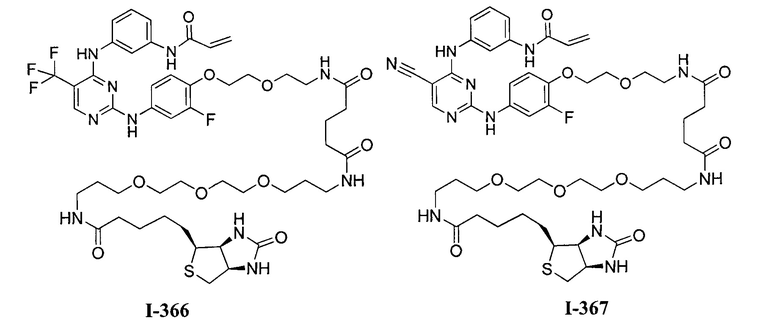
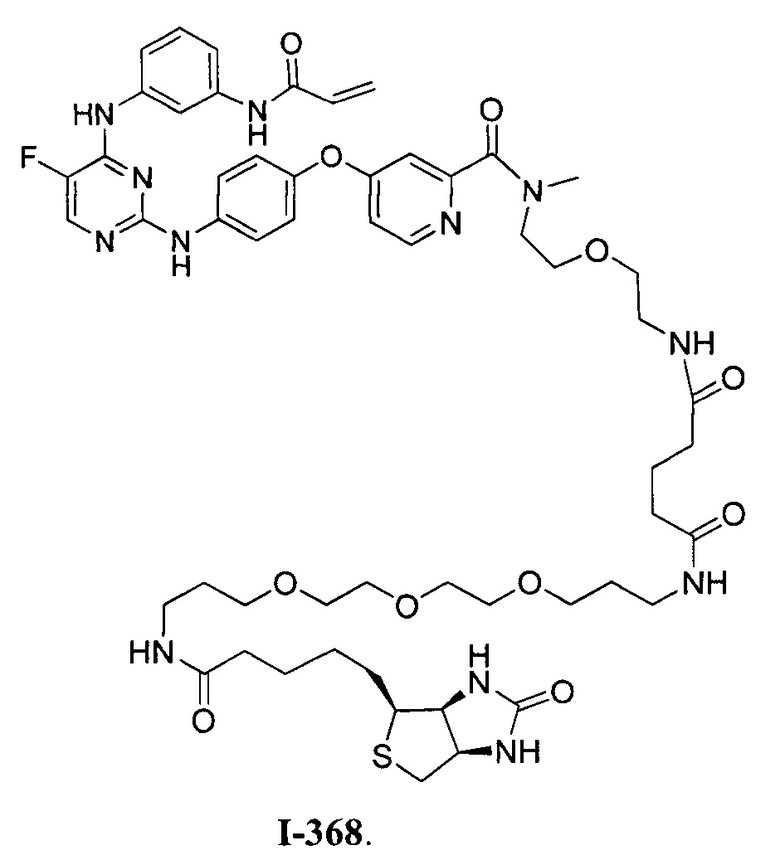
[00296] Очевидно, что многие реагенты-T-Rt доступны для приобретения. Например, множество биотинилирующих реагентов с различной длиной цепи можно приобрести, например, в Thermo Scientific. Такие реагенты включают NHS-PEG4-Biotin и NHS-PEG12-Biotin.
[00297] В некоторых вариантах осуществления структуры зондов, аналогичные приведенным в качестве примеров выше, получают с использованием клик-доступных групп-T-Rt, как описано в настоящем тексте.
[00298] В некоторых вариантах осуществления предложенные соединения-зонды ковалентно модифицируют фосфорилированную конформацию протеинкиназы. В одном аспекте фосфорилированная конформация протеинкиназы представляет собой либо активную, либо неактивную форму протеинкиназы. В некоторых вариантах осуществления фосфорилированная конформация протеинкиназы представляет собой активную форму указанной киназы. В некоторых вариантах осуществления зонд может проникать в клетки.
[00299] В некоторых вариантах осуществления настоящее изобретение обеспечивает занятости протеинкиназы необратимым ингибитором (т.е., Соединением-формулы I-а или I-b) в организме пациента, включающий обеспечение одной или больше тканей, типов клеток или их лизатов, полученных у пациента, которому ввели по меньшей мере одну
дозу необратимого ингибитора, осуществление контакта указанной ткани, типа клеток или их лизата с соединением указанного необратимого ингибитора (т.е., Соединением формулы V-a, V-b, VI-a, VI-b, VII-a или VII-b), что приводит к ковалентной модификации по меньшей мере одной протеинкиназы в указанном лизате, и измерение количества указанной протеинкиназы ковалентно модифицированной указанным соединением-зондом, что позволяет определить занятость указанной протеинкиназы указанным соединением формулы I-а или I-b в сравнении с занятостью указанной протеинкиназы указанным соединением-зондом. В некоторых вариантах осуществления способ дополнительно содержит этап подбора дозы соединения формулы I-а или I-b для повышения занятости протеинкиназы. В некоторых других вариантах осуществления указанный способ дополнительно включает этап подбора дозы соединения' формулы I-а или I-b для снижения занятости протеинкиназы
[00300] В настоящем описании термины "занятость" или "занимать" относятся к степени, до которой протеинкиназа модифицируется предложенными ковалентным ингибитором. Для среднего специалиста очевидно, что желательно вводить самую низкую дозу, обеспечивающую желаемый эффект протеинкиназы.
[00301] В некоторых вариантах осуществления модифицируемая протеинкиназа представляет собой ВТК. В других вариантах осуществления модифицируемая протеинкиназа представляет собой EGFR. В некоторых вариантах осуществления модифицируемая протеинкиназа представляет собой JAK. В некоторых других вариантах осуществления модифицируемая протеинкиназа представляет собой одну или более из ErbB1, ErbB2 или ErbB4. В других вариантах осуществления протеинкиназа представляет собой TEC, ITK или ВМХ.
[00302] В некоторых вариантах осуществления соединение-зонд представляет собой необратимый ингибитор, для которого определяют занятость.
[00303] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ оценки эффективности необратимого ингибитора согласно настоящему изобретению у млекопитающих, включающий введение необратимого ингибитора согласно настоящему изобретению млекопитающему, введение соединения-зонда согласно настоящему изобретению в такни или клетки, выделенные из указанного
млекопитающего, или их лизат, измерение активности детектируемой группы соединения-зонда и сравнение активности детектируемой группы со стандартом.
[00304] В других вариантах осуществления настоящее изобретение обеспечивает способ исследования фармакодинамики необратимого ингибитора согласно настоящему изобретению в организме млекопитающего, включающий введение необратимого ингибитора согласно настоящему изобретению млекопитающему, введение соединения-зонда, описанного здесь, в один или более типов клеток, тканей или из лизатов, выделенных из организма млекопитающего, и измерение активности соединения-зонда в разные моменты времени после введения ингибитора.
[00305] В других вариантах осуществления настоящее изобретение обеспечивает способ in vitro мечения протеинкиназы, включающий осуществление контакта указанной протеинкиназы с описанным здесь соединением-зондом. В одном варианте осуществления этап приведения в контакт включает инкубирование протеинкиназы с соединением-зондом, описанным здесь.
[00306] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ настоящее изобретение обеспечивает способ in vitro мечения протеинкиназы, включающий осуществление одной или больше клеток или тканей, экспрессирующих протеинкиназу, или их лизатов, с описанным здесь соединением-зондом.
[00307] В некоторых других вариантах осуществления настоящее изобретение обеспечивает способ, детектирования меченой протеинкиназы, включающий разделение белков, включающих протеинкиназу меченную зондом, описанным здесь, путем электрофореза и детектирование соединения-зонда по флюоресценции.
[00308] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ исследования фармакодинамики необратимого ингибитора согласно настоящему изобретению in vitro, включающий инкубацию необратимого ингибитора согласно настоящему изобретению с целевой протеинкиназой, добавление описанного здесь соединения-зонда к указанной протеинкиназе и определение количества мишени, модифицированной соединением-зондом.
[00309] В некоторых вариантах осуществления соединение-зонд детектируют путем связывания с авидином, стрептавидином или каптавидином.
[00310] В некоторых вариантах осуществления зонд детектируют методом Вестерн-блот. В других вариантах осуществления зонд детектируют методом твердофазного ИФА (ELISA). В некоторых вариантах осуществления зонд детектируют путем проточной цитометрии.
[00311] В других вариантах осуществления настоящее изобретение обеспечивает способ зондирования кинома (полного комплемента киназ) необратимым ингибитором, включающий инкубирование одного или больше типов клеток или их лизатов биотинилированным соединением-зондом с получением белков, модифицированных фрагментом биотина, расщепление белков, связывание авидином или его аналогом и осуществление многомерного анализа ЖХ-МС-МС для идентификации протеинкиназ, модифицированных соединением-зондом и сайтов присоединения указанных киназ.
[00312] В некоторых вариантах осуществления настоящее изобретение обеспечивает способ измерения синтеза белка в клетке, включающий инкубирование клетки с необратимым ингибитором белка-мишени, получение лизатов указанных клеток в определенные моменты времени и инкубацию указанных лизатов клеток с соединениями-зондами согласно настоящему изобретению, что позволяет измерить появление свободного белка на протяжении продолжительного периода времени.
[00313] В других вариантах осуществления настоящее изобретение обеспечивает способ определения режима дозирования у млекопитающего для максимизации занятости целевой протеинкиназы включающий анализ одного или более типов клеток или лизатов клеток, выделенных из организма млекопитающего (полученные, например, из спленоцитов, периферических В-лимфоцитов, цельной крови, лимфатических узлов, интестициальной ткани и других тканей), полученных от нормального млекопитающего, которому введи необратимый ингибитор формулы I-а или I-b, причем указанный этап анализа включает осуществление контакта указанной одной или более тканей, типов клеток или их лизатов с соединением-зондом согласно настоящему изобретению и измерение количестве протеинкиназы, ковалентно модифицированной соединением-зондом.
ПРИМЕРЫ
[00314] Как описано ниже в разделе Примеры, в некоторых вариантах осуществления соединение получают в соответствии с приведенными ниже общими процедурами. Очевидно, что несмотря на то, что общие способы описывают синтез отдельных соединений, следуя описанным способами и другим известным методам специалист сможет реализовать все соединения, подклассы и виды каждого из соединений, описанных в настоящем тексте.
[00315] Номера соединений, используемые ниже в разделе Примеры, соответствуют номерам, приведенным в Таблице 5, выше.
ПРИМЕР 1
[00316] Получение N-(3-(5-метил-2-(фениламино)пиримидин-4-иламино)фенил)акриламида I-7
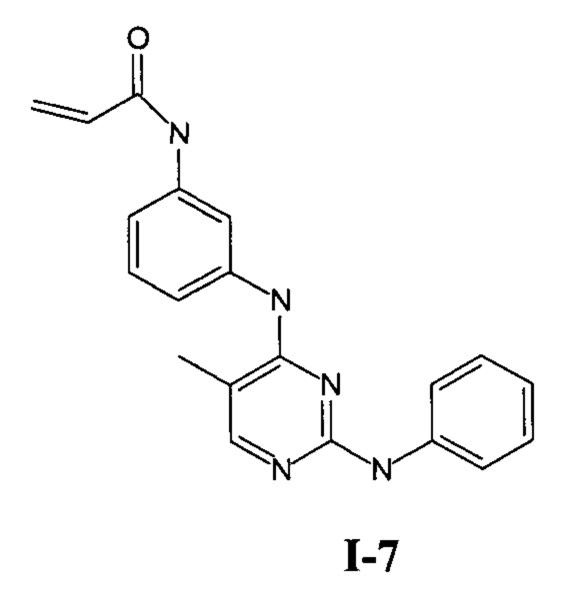
[00317] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
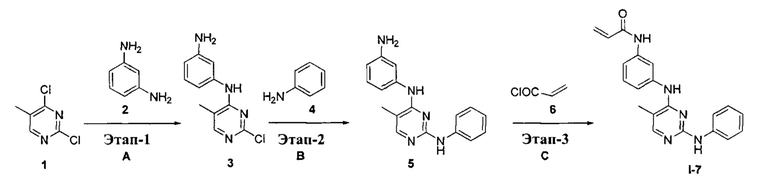
A) DIPEA, n-BuOH, 120°С, 30 минут, MB; В) NMP, 200°С, 10 минут, MB; С) NMP, 0°С-30 минут, комн. т.-30 минут.
[00318] Этап-1
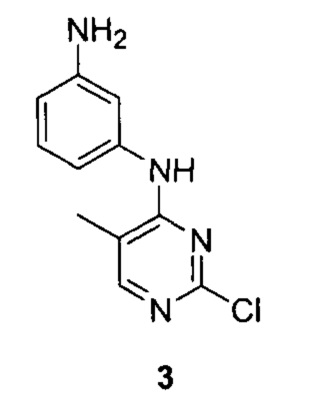
[00319] Раствор 1 (2.0 г, 0.012 моль), 1,3-фениленедиамина (2.0 г, 0.018 ммоль), DIPEA (2.33 г, 0.018 моль) в n-BuOH (20 мл) облучали в микроволновой печи при 120°С в течение 30 минут. Реакционную смесь гасили водой (100 мл), экстрагировали EtOAc (3×100 мл). Объединенные экстракты EtOAc промывали водой (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении.
Полученный остаток далее очищали методом колоночной хроматографии (SiO2, меш 60-120, EtOAc/CHCl3: 15/85) с получением 3 (1.3 г, 45%) в форме темно-коричневого твердого вещества.
[00320] Этап-2
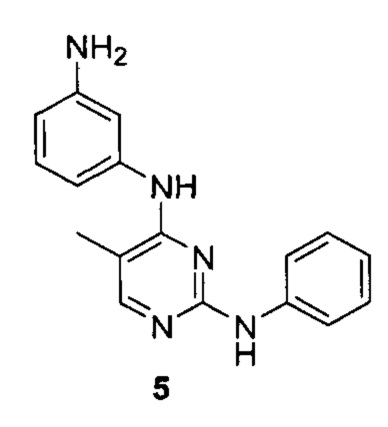
[00321] Раствор 3 (1.0 г, 4.27 ммоль), 4 (1.5 г, 16.12 ммоль) в NMP (10.0 мл) облучали в микроволновой печи (200°С, 10 минут). Реакционную смесь охлаждали, разбавляли водой (100 мл) и экстрагировали EtOAc (3×100 мл). Объединенный экстракт этил ацетата промывали водой (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением остатка. Сырой остаток затем очищали методом колоночной хроматографии (SiO2, CHCl3/МеОН: 98/2) с получением 5 (0,5 г, 40,3%) в форме светло-коричневого твердого вещества.
[00322] Этап-3
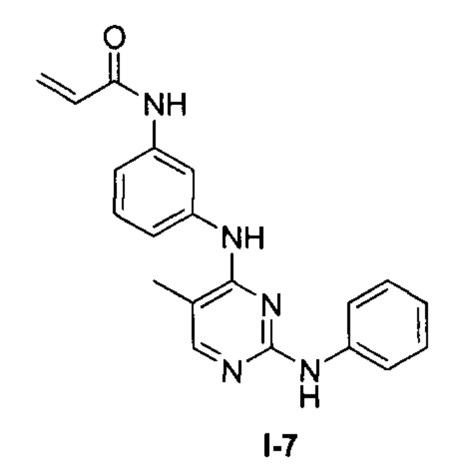
[00323] К раствору 5 (200 мг, 0.68 ммоль) в NMP (2.0 мл) при температуре 0°С при помешивании добавляли акрилоил хлорид (248 мг, 0.2.74 ммоль), и помешивали реакционную смесь при температуре 0°С в течение 60 минут. Реакционную смесь затем перемешивали с гексаном в течение 1/2 часа, и затем гексан удаляли из реакционной смеси путем декантирования, а остаток гасили водой (10 мл). Водный раствор приводили к форме основания при помощи насыщенного раствора NaHCO3 и затем экстрагировали EtOAc (3×10 мл). Объединенные экстракты EtOAc промывали водой (10 мл), солевым раствором (10 мл), сушили Na2SO4 и концентрировали при пониженном
давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, 230-400, МеОН/ CHCl3: 10/90) с выходом I-7 (110 мг, 46.4%) в форме твердого коричневого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.10 (s, 3Н), 5.73 (dd, 1.88 & 10.42 Гц, 1Н), 6.24 (dd, J=1.88 & 17 Гц, 1Н), 6.44 (dd, J=10.08 & 16.92 Гц, 1H), 6.78 (t, J=7.36 Гц, 1Н), 7.06-7.11 (m, 2Н), 7.26 (t, J=8.08 Гц, 1H), 7.38-7.40 (bm, 2Н), 7.65 (d,J=8.52 Гц, 2Н), 7.88 (s, 1Н), 7.92 (s, 1Н), 8.37 (s, 1Н), 8.91 (s, 1H), 10.09 (s, 1H); ЖХМС: m/е 346.8 (М+1).
ПРИМЕР 2
[00324] Приготовление N-(3-(4-(m-толиламино)пиримидин-2-иламино)фенил) акриламида I-1
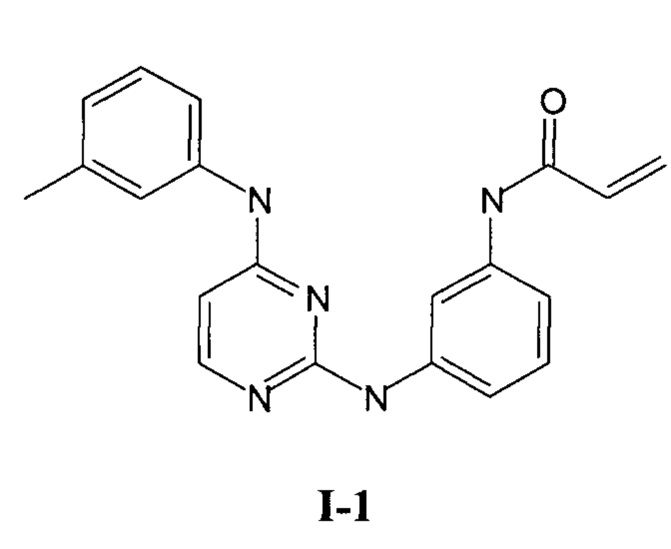
[00325] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
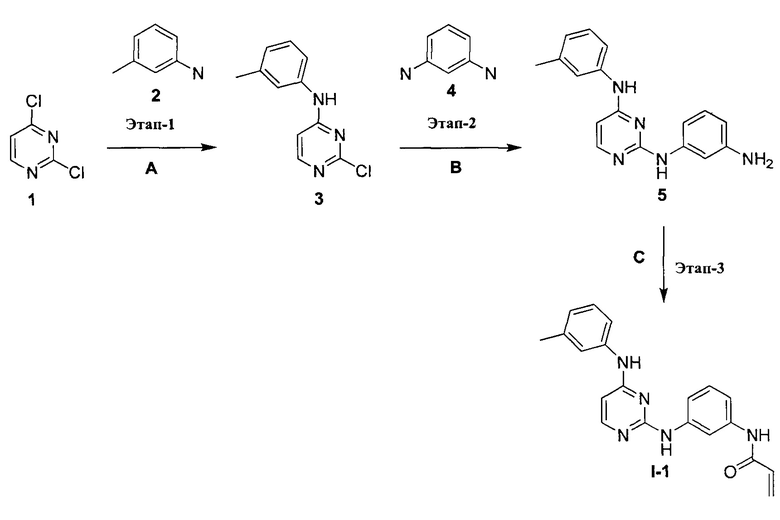
A) DIEA, n-BuOH, 110°C, 30 минут, микроволн.; В) NMP, 200°С, 10 минут, микроволн.; С) акрилоил хлорид, NMP, 0°С-30 минут, комн. т. - 30 минут.
[00326] Этап-1
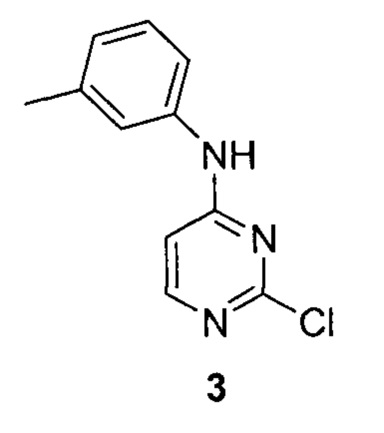
[00327] Раствор 1 (0.5 г, 3.35 ммоль), m-толуидина (0.36 г, 3.35 ммоль), DIEA (0.65 г, 5.0 ммоль) в n-BuOH (2.0 мл) облучали в микроволновой печи при 110°С в течение 30 мин. Реакционную смесь затем концентрировали при пониженном давлении, гасили водой (5 мл) и экстрагировали EtOAc (3×20 мл). Объединенные экстракты EtOAc промывали водой (5 мл), солевым раствором (5 мл), сушили Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, меш 60-120, CHCl3/МеОН: 99/1) с получением 3 (0,4 г, 54,2%) в форме желтого твердого вещества.
[00328] Этап-1
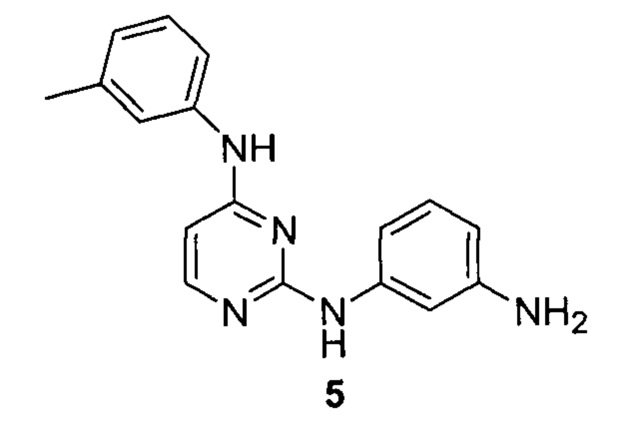
[00329] Раствор 3 (0,2 г, 0,91 ммоль), 4 (0,2 г, 1,8 ммоль) в NMP (2,0 мл) облучали в микроволновой печи (200°С, 10 минут). Реакционную смесь охлаждали, разбавляли водой (10 мл) и экстрагировали CH2Cl2 (3×15 мл). Объединенный экстракт CH2Cl2 промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением остатка. Сырой остаток затем очищали методом колоночной хроматографии (SiO2, CHCl3/МеОН: 98/2) с получением 5 (0,14 г, 53%) в форме светло-желтого твердого вещества.
[00330] Этап-3
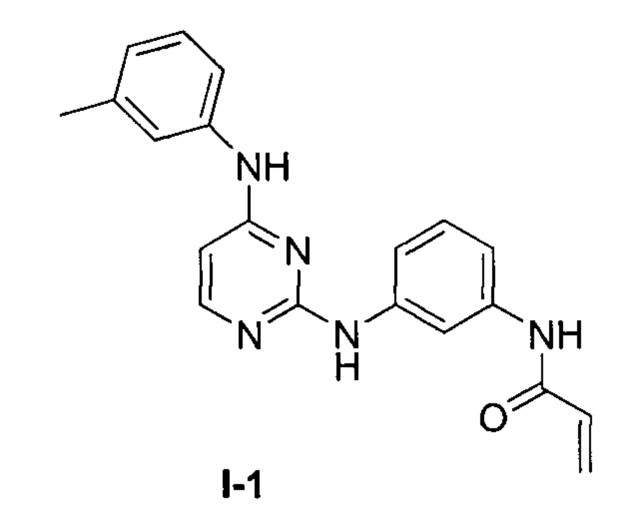
[00331] К раствору 5 (0.075 г, 0.25 ммоль) в NMP (1.0 мл) при помешивании при температуре 0°С добавляли акрилоил хлорид (0.19 г, 2.0 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 30 минут после чего помешивали при комнатной температуре в течение 30 минут. Беспримесную реакционную смесь затем очищали методом колоночной хроматографии (нейтральный Al2O3, CHCl3/МеОН: 98/2) с получением -1 (0,04 г, 45%) в форме белого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.56 (s, 3Н), 5.71 (dd, J=2.0 & 10.08 Гц, 1H), 6.20-6.25 (m, 2Н), 6.45 (dd, J=10.12 & 17.00 Гц, 1Н), 6.78 (d,J=7.52 Гц, 1H), 7.12-7.19 (m, 2Н), 7.31 (d, J=8.44 Гц, 1Н), 7.46-7.53 (m, 3Н), 7.87 (s, 1H), 7.99 (d, J=5.76 Гц, 1H), 9.15 (s, 1H), 9.24 (s, 1H), 10.03 (s, 1H); ЖХМС: m/е 346.4 (М+1).
ПРИМЕР 3
[00332] Приготовление N-(3-(5-(m-толиламино)пиримидин-2-иламино)фенил)акриламида I-2
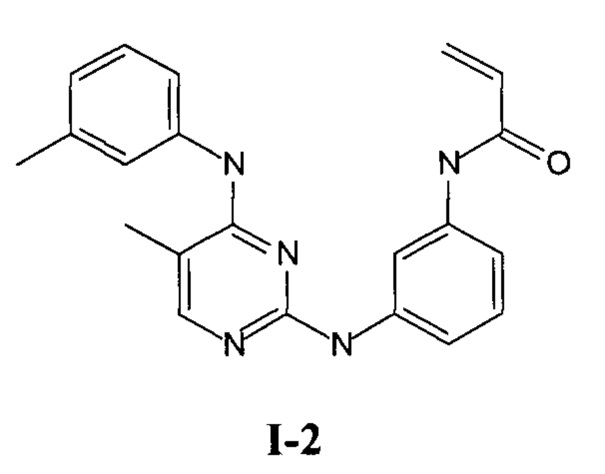
[00333] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
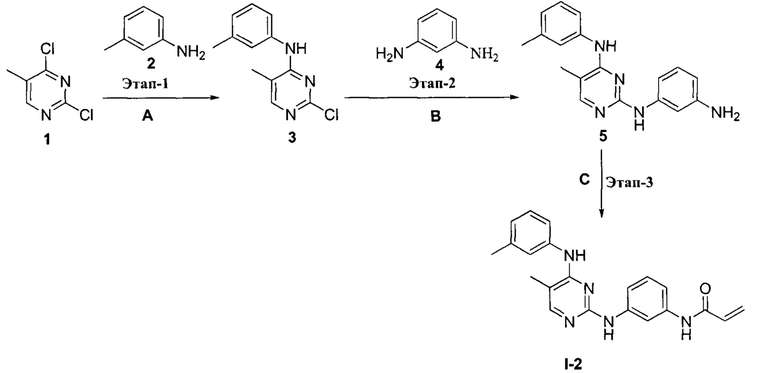
A) DIPEA, n-BuOH, 110°С, 30 минут, MB; В) NMP, 200°С, 15 минут, MB; С) акрилоил хлорид, NMP, 0°С-30 минут, комн. т.-30 минут.
[00334] Этап-1
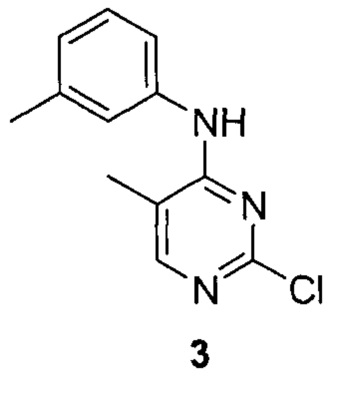
[00335] Раствор 1 (0.1 г, 0.613 ммоль), 2 (0.066 г, 0.613 ммоль), DIPEA (0.118 г, 0.919 ммоль) в n-BuOH (2.0 мл) облучали в микроволновой печи при 110°С в течение 90 минут. Реакционную смесь охлаждали, концентрировали при пониженном давлении, и полученный остаток затем очищали методом колоночной хроматографии (SiO2, смеси Метанол/хлороформа) с получением 3 (0.05 г, 34%) в форме беловатого твердого вещества.
[00336] Этап-2
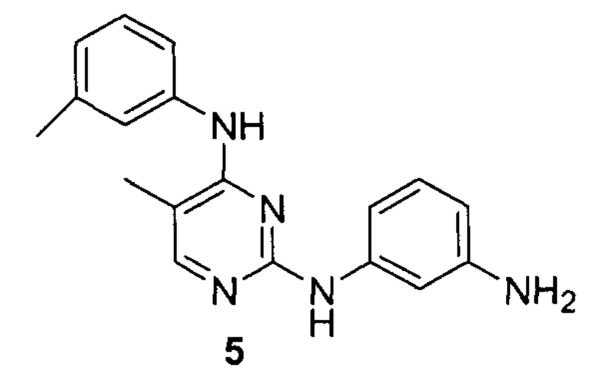
[00337] Раствор 3 (0,05 г, 0,213 ммоль), 4 (0,046 г, 0,427 ммоль) в NMP (2,0 мл) облучали в микроволновой печи (200°С, 15 минут). Реакционную смесь охлаждали, разбавляли водой (15 мл) и экстрагировали EtOAc (3×15 мл). Объединенные экстракты EtOAc промывали водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с выпадением осадка. Сырой остаток затем очищали методом колоночной хроматографии (SiO2, CHCl3/МеОН: 98/2) с получением 5 (0,03 г, 46%) в форме серого твердого вещества.
[00338] Этап-3
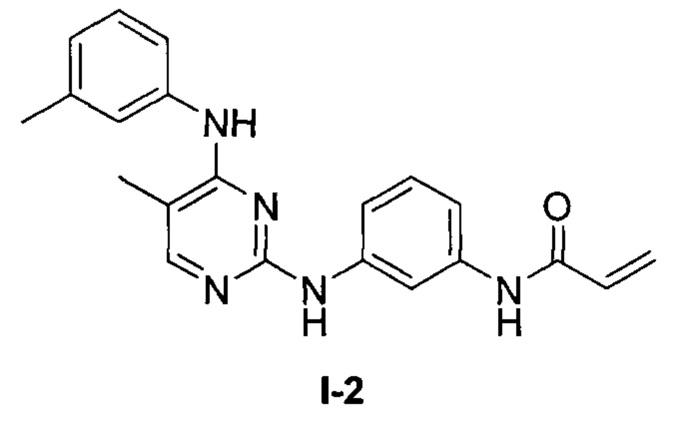
[00339] К раствору 5 (0. 025 г, 0,082 ммоль) в NMP (0,5 мл) при помешивании при температуре 0°С добавляли акрилоил хлорид (0. 073 г, 0,821 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 30 минут после чего помешивали при комнатной температуре в течение 30 минут. Сырую реакционную смесь затем очищали методом колоночной хроматографии с оксидом алюминия (нейтральный Al2O3, смеси хлороформ/метанол) с получением I-2 (0.012 г, 41%) в форме бледно-коричневого
твердого вещества: 1Н-ЯМР (ДМСО-d6) δ ppm: 2.10 (s, 3Н), 2.27 (s, 3Н), 5.72 (dd, J=2 & 10.04 Гц, 1H), 6.22 (dd, J=1.96 & 16.92 Гц, 1H), 6.45 (dd, J=10.08 & 16.92 Гц, 1Н), 6.83 (d, J=7.36 Гц, 1Н), 7.09 (t, J=8.06 Гц, 1Н), 7.17 (t, J=7.78 Гц, 1H), 7.26 (d, J=7.80 Гц, 1Н), 7.47 (d, J=1.08 Гц, 1Н), 7.53 (s, 1Н), 7.58 (d, J=8.60 Гц, 1Н), 7.78 (s, 1Н), 7.88 (s, 1Н), 8.15 (s, 1Н), 9.01 (s, 1Н), 9.99 (s, 1Н); ЖХМС: m/е 360.1 (М+1).
ПРИМЕР 4
[00340] Приготовление N-(3-(5-фтор-4-(m-толиламино)пиримидин-2-иламино)фенил) акриламида I-2
[00341] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
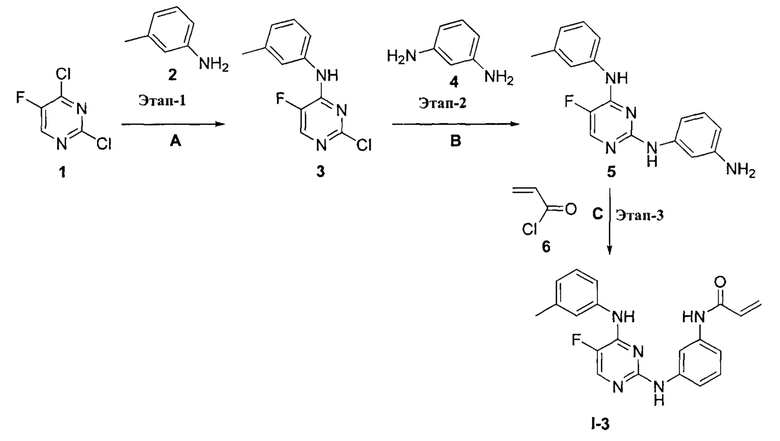
А) n-бутанол, DIPEA, 110°С, 45 минут, MB; В) NMP, 200°С, 10 минут, MB; С) NMP, DMAP, 0°С, 30 минут.
[00342] Этап-1
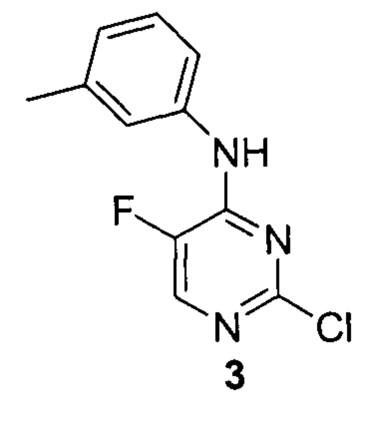
[00343] К раствору 1 (0.5 г, 3 ммоль) в п-бутаноле (5.0 мл) добавляли 2 (0.64 г, 0.6 ммоль), DIPEA (0.116 г, 0.8 ммоль) и облучали реакционную смесь в микроволновой печи при температуре 110°С в течение 45 минут. Его охлаждали, тушили водой (50 мл) и экстрагировали EtOAc (2×25 мл). Комбинированный экстракт EtOAc промывали водой (25 мл), солевым раствором (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3 (0.45 г, 63%), который использовали на следующем этапе без дополнительной очистки.
[00344] Этап-2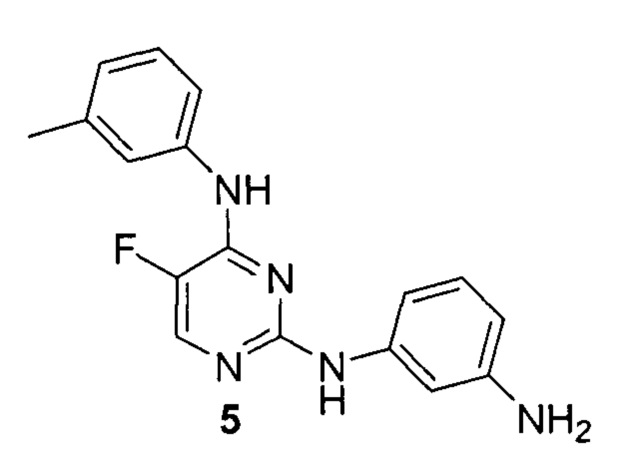
[00345] Раствор 3 (0.45 г, 1.8 ммоль) и 4 (0.41 г, 3.7 ммоль) в NMP (4.5 мл) облучали в микроволновой печи при 200°С в течение 10 минут. Затем его охлаждали, разбавляли водой (25 мл) и экстрагировали EtOAc (3×25 мл). Объединенные экстракты EtOAc промывали водой (2×25 мл), солевым раствором (25 мл), сушили Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, 60-120, Хлороформ/этил ацетат: 90/10) с получением 5 (0,23 г, 41%) в форме светло-желтого твердого вещества.
[00346] Этап-1
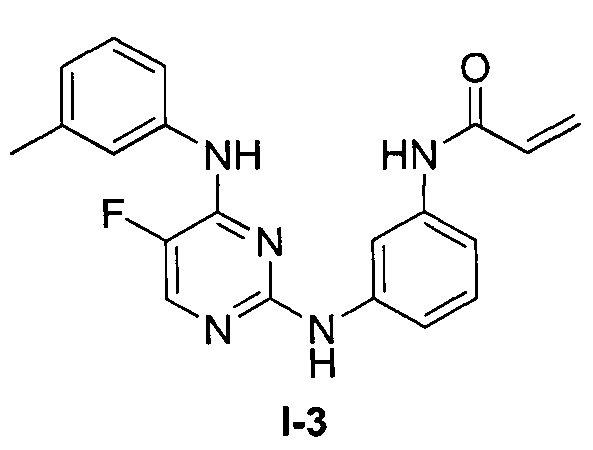
[00347] К раствору 5 (0.075 г, 0.24 ммоль) в NMP(1.5 мл) при помешивании при 0°С в атмосфере N2 добавляли DMAP (0.059 г, 0.48 ммоль) и акрилоил хлорид (0.064 г, 0.725 ммоль) и реакционную смесь держали при этой температуре в течение 30 минут. Затем ее гасили водой (7.5 мл) и экстрагировали EtOAc (3×25 мл). Объединенные экстракты EtOAc промывали 5% лимонной кислотой (10 мл), водой (2×10 мл), солевым раствором (10 мл), сушили Na2SO4 и концентрировали при пониженном давлении. Сырой остаток далее очищали методом колоночной хроматографии (Al2O3, Хлороформ / метанол): 98/2) с получением I-3 (0,01 г, 11,3%) в форме беловатого твердого вещества. 1H-ЯМР (ДМСО-d6) δ ppm: 2.27 (s, 3Н), 5.72 (d, J=9.84 Гц, 1H), 6.22 (d, J=16.92 Гц, 1Н), 6.44 (dd, J=10.2 & 17.02 Гц, 1H), 6.85 (d, J=7.12 Гц, 1H), 7.12-7.19 (m, 2H), 7.29 (d, J=7.68 Гц, 1H), 7.43 (d, J=7.92 Гц, 1H), 7.61-7.63 (m, 2H), 7.82 (s, 1H), 8.08 (s, 1H), 9.23 (bs, 2H), 10.03 (s, 1H); ЖХМС: m/e 364.2 (M+1).
ПРИМЕР 5
[00348] Приготовление (Е)-4-(диметиламино)-N-(3-(5-фтор-4-(m-толиламино)пиримидин-2-иламино)фенил)бут-2-енамида I-4
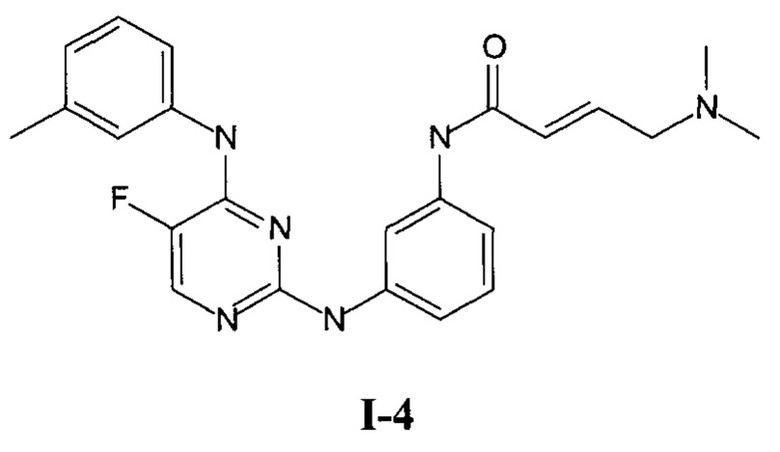
[00349] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
А) n-бутанол, DIPEA, 110°С, 45 минут., MB; В) NMP, 200°С, 10 минут., MB; С) оксалил хлорид, CH3CN, 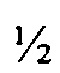 at 0°C, 2 часа at 25°C, 5 минут при температуре 45°С; D) NMP, 0°С to 10°C, 30 минут.
at 0°C, 2 часа at 25°C, 5 минут при температуре 45°С; D) NMP, 0°С to 10°C, 30 минут.
[00350] Этап-1
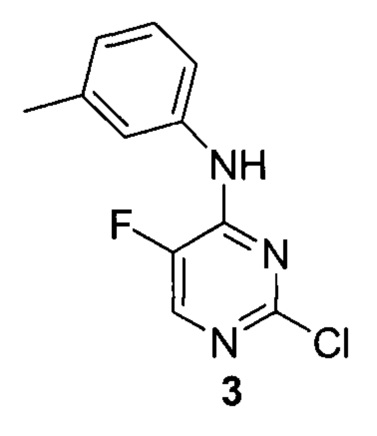
[00351] Раствор 1 (0,5 г, 3,0 ммоль), 2 (0,32 г, 3,0 ммоль) в п-бутаноле (5,0 мл) облучали в микроволновой печи (110°С, 45 минут). Охлаждали, тушили водой (50 мл) и экстрагировали EtOAc (2×25 мл). Объединенный экстракт EtOAc промывали водой (25 мл), солевым раствором (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3 (0.45 г, 63%), который использовали на следующем этапе без дополнительной очистки.
[00352] Этап-1
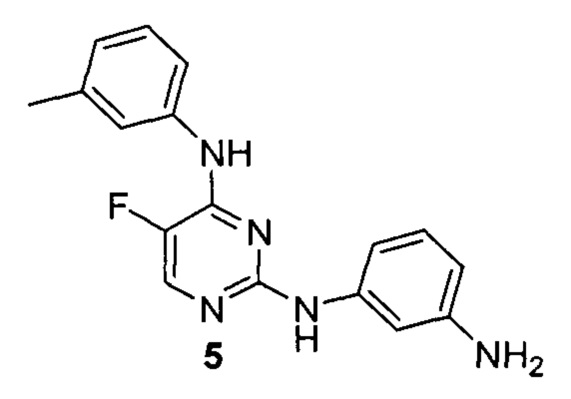
[00353] Раствор 3 (0,45 г, 1,8 ммоль), 4 (0,41 г, 3,7 ммоль) в NMP (4,5 мл) облучали в микроволновой печи (200°С, 10 минут). Охлаждали, разбавляли водой (25 мл) и экстрагировали EtOAc (3×25 мл). Объединенные экстракты этил ацетата промывали водой (2×25 мл), солевым раствором (25 мл), сушили Na2SO4 и концентрировали при пониженном давлении. Остаток далее очищали методом колоночной хроматографии (SiO2, Хлороформ/этил ацетат: 90/10) с получением 5 (0,23 г, 41%) в форме светло-желтого твердого вещества.
[00354] Этап-3а
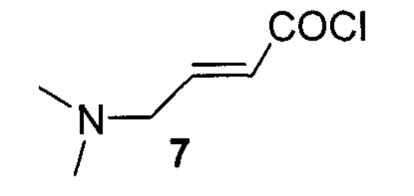
[00355] К раствору 6 (0.13 г, 0.80 ммоль) в CH3CN (1.0 мл) при помешивании добавляли оксалил хлорид (0.122 г, 0.96 ммоль) при температуре 0°С. Реакционную смесь оставляли с перемешиванием при температуре 0°С в течение получаса, а затем при комнатной температуре в течение 2 часа. Окончательно реакционную смесь грели при 45°С в течение 5 минут, охлаждали и использовали на следующем этапе без дальнейшей очистки.
[00356] Этап-3
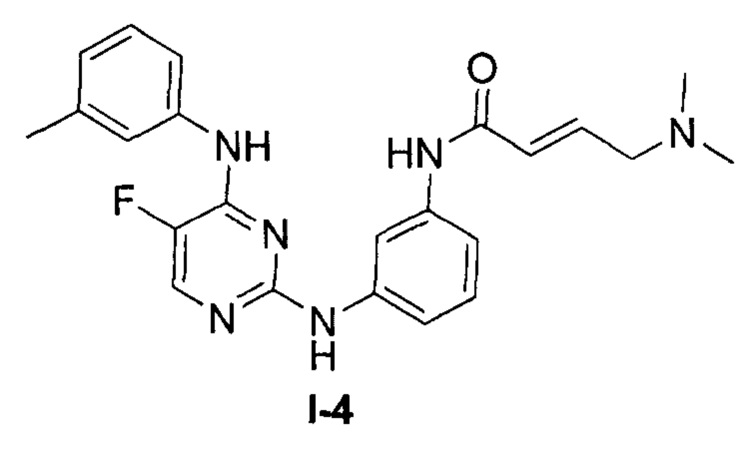
[00357] К раствору 5 (0,05 г, 0,16 ммоль) в NMP (1.0 мл) при помешивании добавляли 7 при температуре 0°С. Реакционную смесь помешивали при температуре 0°С в течение 30 минут и при температуре 10°С в течение 30 минут. Гасили насыщенным раствором
натрия бикарбоната (5 мл) и экстрагировали CH2Cl2 (3×5 мл). Объединенный органический экстракт промывали водой (1 мл), солевым раствором (1 мл) и сушили над Na2SO4. Концентрировали при пониженном давлении, после чего очищали методом колоночной хроматографии (SiO2, 230-400, CHCl3/МеОН, 95/5) с получением -4 (0.02 г, 29.4%) в форме белого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.21 (s, 6Н), 2.28 (s, 3Н), 3.08 (bd, J=5.6 Гц, 2Н), 6.29 (d, J=15.60 Гц, 1H), 6.67-6.74 (m, 1Н), 6.86 (d, J=7.20 Гц, 1Н), 7.12-7.20 (m, 2Н), 7.27 (d, J=8.00 Гц, 1Н), 7.43 (d, J=8.00 Гц, 1Н), 7.62-7.64 (m, 2Н), 7.82 (s, 1Н), 8.08 (d, J=3.6 Гц, 1Н), 9.23 (s, 1Н), 9.24 (s, 1H), 9.96 (s, 1Н); ЖХМС: m/е 421.2 (М+1).
ПРИМЕР 6
[00358] Приготовление N-(3-(5-метил-4-(фениламино)пиримидин-2-иламино)фенил) акриламида I-5
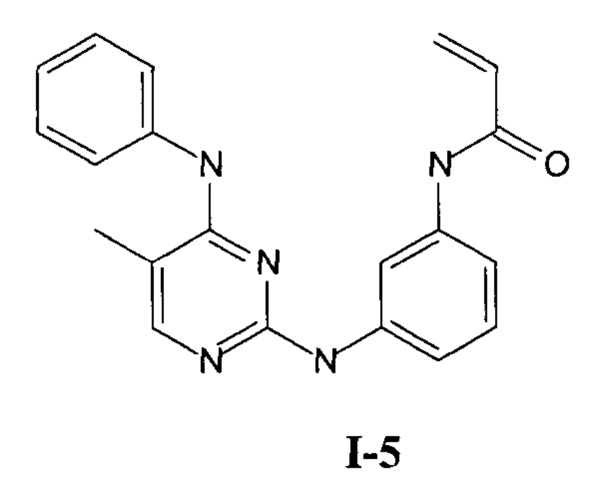
[00359] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
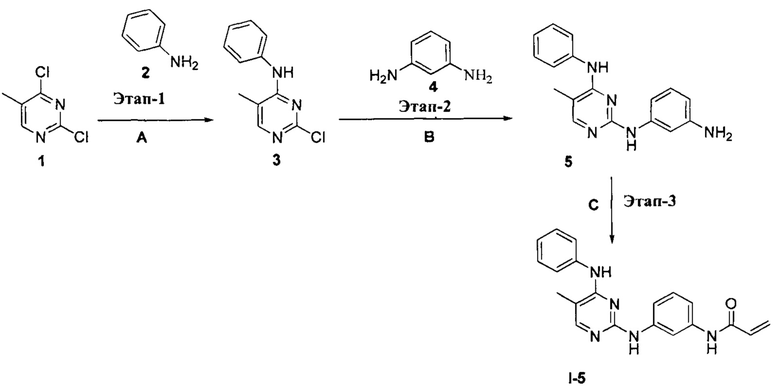
A) DIPEA, n-BuOH, 110°C, 30 минут, MB; В) NMP, 200°C, 15 минут, MB; С) акрилоил хлорид, NMP, 0°C-30 минут, комн. т.-30 минут.
[00360] Этап-1
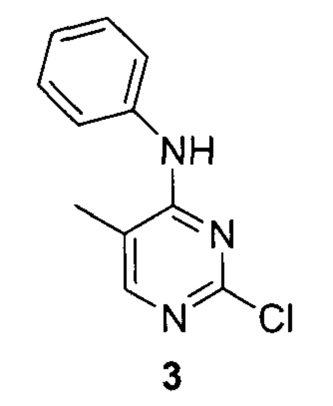
[00361] Раствор 1 (0.1 г, 0.613 ммоль), 2 (0. 114 г, 1.226 ммоль), DIPEA (0.118 г, 0.919 ммоль) в n-BuOH (2.0 мл) облучали в микроволновой печи при 110°С в течение 90 минут. Реакционную смесь охлаждали, концентрировали при пониженном давлении и полученный остаток затем очищали методом колоночной хроматографии (SiO2, Метанол/хлороформ: 1/9) с получением 3 (0,08 г, 59%) в форме белого твердого вещества
[00362] Этап-2
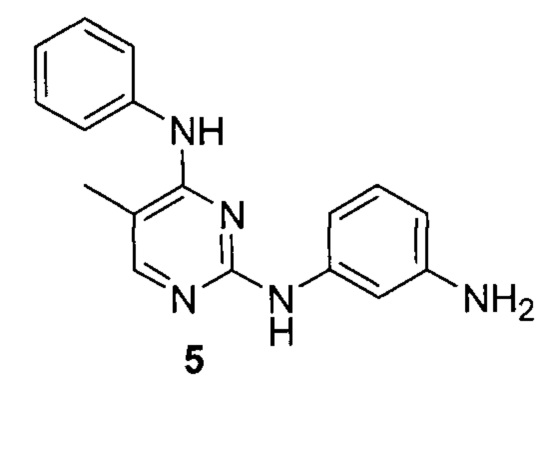
[00363] Раствор 3 (0,08 г, 0,364 ммоль), 4 (0,059 г, 0,546 ммоль) в NMP (2.0 мл) облучали в микроволновой печи (200°С, 15 минут). Реакционную смесь охлаждали, разбавляли водой (15 мл) и экстрагировали EtOAc (3×15 мл). Объединенные экстракты этил ацетата промывали водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4H концентрировали при пониженном давлении с выпадением осадка. Сырой остаток затем очищали методом колоночной хроматографии (SiO2, 60-120, CHCl3/МеОН: 98/2) с получением 5 (0,06 г, 60%) в форме светло-серого твердого вещества. 1 Н-ЯМР (ДМСО-с1б) δ ppm: 2.09 (s, 3Н), 4.74 (s, 2Н), 6.09-6.11 (m, 1H), 6.77-6.85 (m, 2Н), 6.91 (t, J=1.72 Гц, 1H), 7.02 (t, J=7.36 Гц, 1Н), 7.31 (t, J=7.52 Гц, 2Н), 7.75 (d, J=7.68 Гц, 2Н), 7.84 (s, 1H), 8.18 (s, 1H), 8.65 (s, 1Н); ЖХМС: m/е 293.2 (М+1).
[00364] Этап-3
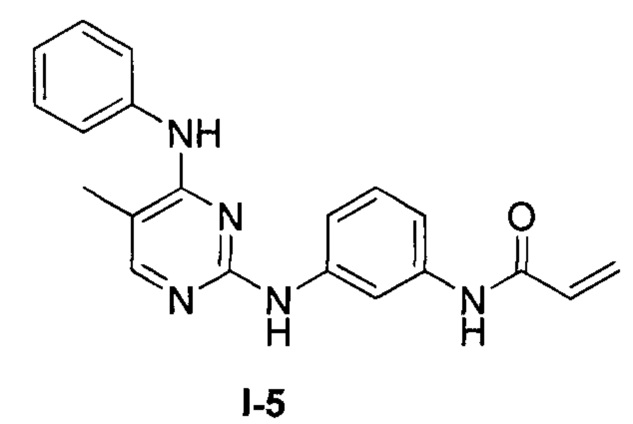
[00365] К раствору 5 (60 мг, 0,205 ммоль) в NMP (2.0 мл) при температуре 0°С при помешивании добавляли акрилоил хлорид (0,148 г, 1.64 ммоль), и помешивали реакционную смесь при температуре 0°С в течение 30 минут. Беспримесную реакционную смесь пропускали через колонку с оксидом алюминия (нейтральный Al2O3, хлороформ/метанол, 99/1) с получением I-5 (0.013 г, 18.5%) в форме беловатого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.11 (s, 3Н), 5.72 (dd, J=1.92 & 10.04 Гц, 1H), 6.22 (dd, J=1.92 & 16.92 Гц, 1Н), 6.45 (dd, J=9.32 & 16.92 Гц, 1Н), 7.00 (t, J=7.28 Гц, 1H), 7.09 (t, J=8.04 Гц, 1Н), 7.23-7.30 (m, 3Н), 7.43 (d, J=8.04 Гц, 1H), 7.75-7.77 (m, 2Н), 7.83 (s, 1Н), 7.88 (s, 1Н), 8.22 (s, 1Н), 9.00 (s, 1Н), 9.99 (s, 1Н); ЖХМС: m/е 346 (М+1).
ПРИМЕР 7
[00366] Получение N-метил-3-(5-метил-4-(m-толиламино)пиримидин-2-иламино) фенил)акриламида I-8
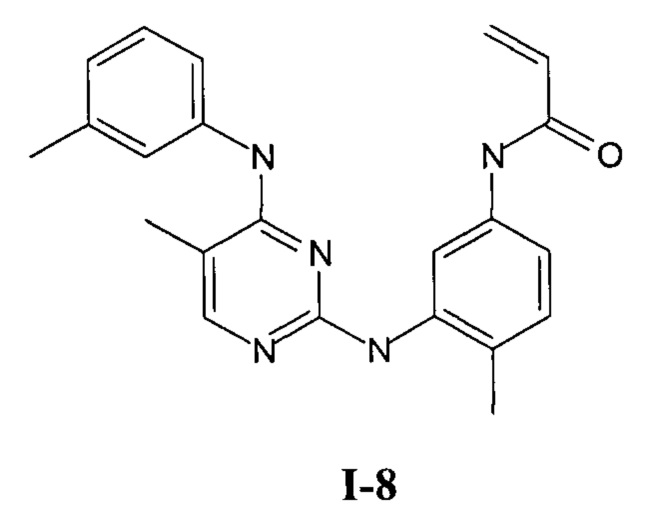
[00367] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
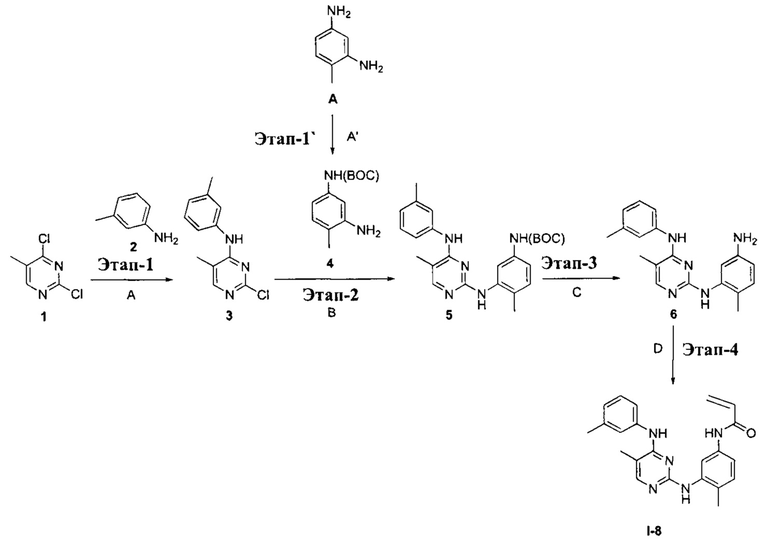
A) DIPEA, n-BuOH, 120°С, 60 минут., MB; А') (ВОС)20, МеОН, -10°С, 4 часа; В) Pd(OAc)2, BIN АР, Cs2CO3, толуол, 110°С, 12 часов; С) ТФА, CH2Cl2, 0°С-30 минут, комн. т.-2 часа; D) акрилоил хлорид, NMP, 0°С-30 минут, комн. т.-30 минут.
[00368] Этап-1'
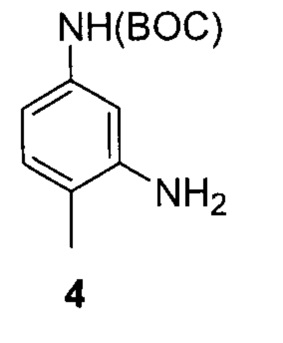
[00369] К раствору А (5 г, 0.04 ммоль) в МеОН (75 мл) при помешивании медленно добавляли (ВОС)2O (11.59 г, 0.050 ммоль) при температуре -10°С. Реакционную смесь помешивали при этой температуре в течение 4 часа, после чего концентрировали при пониженном давлении. Полученный осадок добавляли к EtOAc (300 мл). Его промывали водой (25 мл), солевым раствором (25 мл) и сушили над Na2SO4. Фильтровали, после чего концентрировали при пониженном давлении с получением 4 (2.5 г, 27%) в форме беловатого твердого вещества.
[00370] Этап-1
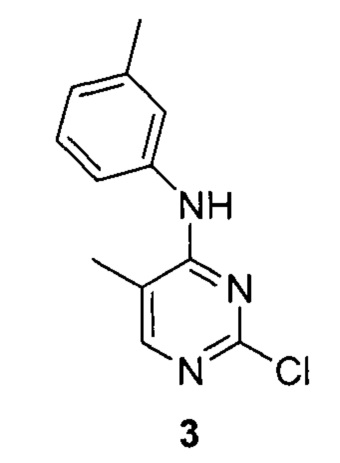
[00371] Раствор 1 (0,5 г, 3,06 ммоль), 2 (0,39 г, 3,06 ммоль), DIPEA (0.59 г, 4.5 ммоль) в n-BuOH (5 мл) облучали в микроволновой печи (120°С, 30 минут). Реакционную смесь охлаждали, удаляли растворители при пониженном давлении, и тушили полученный осадок водой (5 мл). Его экстрагировали EtOAc (3×20 мл) и объединенный слой EtOAc промывали водой (5 мл), солевым раствором (5 мл) и сушили над Na2SO4. Фильтровали, после чего концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, CHCl3/МеОН: 9/1) с получением 3 (0,35 г, 49%) в форме беловатого твердого вещества
[00372] Этап-2
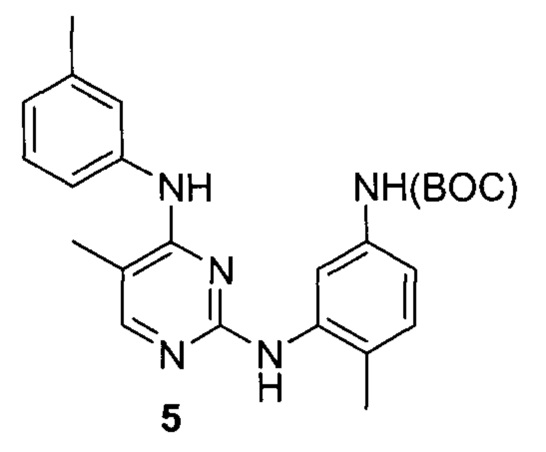
[00373] Раствор 3 (0.1 г, 0.43 ммоль), 4 (0.14 г, 0.64 ммоль), Pd(OAc)2 (10 мг, 0.043 ммоль), BINAP (0.013 г, 0.021 ммоль) и Cs2CO3 (0.2 г, 1.06 ммоль) в дегазированном толуоле (толуол продували N2 в течение 15 минут) нагревали с обратным холодильником
в течение 12 часов в атмосфере N2. Реакционную смесь охлаждали и пропускали через короткую пластинку целлита®. Фильтрат разбавляли EtOAc (25 мл) промывали водой (5 мл), солевым раствором (5 мл) и сушили над Na2SO4. Фильтровали, после чего концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, CHCl3/МеОН: 9/1) с получением 5 (40 мг, 22%) в форме беловатого твердого вещества.
[00374] Этап-3
[00375] К раствору 5 (0.04 г, 0.095 ммоль) в сухом CH2Cl2 (2 мл) при температуре 0°С при помешивании добавляли CF3COOH (0.2 мл, 5 объемов), затем реакционную смесь держали при этой температуре в течение 30 минут. Доводили до комнатной температуре и помешивали при комнатной температуре в течение 2 часа. Затем реакционную смесь гасили охлажденной льдом водой (2 мл), приводили к форме основания при помощи раствора натрия бикарбоната и экстрагировали EtOAc (2×10 мл). Объединенный экстракт EtOAc промывали водой (2 мл), солевым раствором (2 мл) и сушили над Na2SO4. Фильтровали, после чего концентрировали при пониженном давлении с получением 6 (22 мг, 73%) в форме светло-коричневого твердого вещества.
[00376] Этап-4
[00377] К раствору 6 (0,2 г, 0,63 ммоль) в NMP (4 мл) при помешивании добавляли акрилоил хлорид (0.12 г, 1.25 ммоль) при температуре 0°С. Реакционную смесь держали при этой температуре в течение 30 минут, а затем при комнатной температуре в течение
30 минут. Затем ее гасили охлажденной льдом водой (2 мл) и экстрагировали (2×10 мл). Объединенный экстракт EtOAc промывали водой (2 мл), солевым раствором (2 мл) и сушили над Na2SO4. Фильтровали, после чего концентрировали при - пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 230-400, CHCl3/МеОН: 9/1) с получением I-8 (10 мг,.4%) в форме белого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.07 (s, 3Н), 2.13 (s, 6Н), 5.70 (dd, J=1.92 & 10.08 Гц, 1Н), 6.20 (dd, J=1.96 & 16.88 Гц, 1Н), 6.41 (dd, J=10.16 & 16.96 Гц, 1Н), 6.69 (d, J=7.36 Гц, 1Н), 6.98 (t, J=7.76 Гц, 1H), 7.11 (d, J=8.24 Гц, 1Н), 7.41 (q, J=9.92 Гц, 1Н), 7.49-7.51 (m, 2Н), 7.73 (s, 1Н), 7.80 (s, 1H), 7.97 (s, 1Н), 8.16 (s, 1Н), 10.00 (s, 1Н); ЖХМС: m/е 374 (М+1).
ПРИМЕР 8
[00378] Получение N-(3-(4-(3-бромофениламино)-5-метилпиримидин-2-иламино)фенил) акриламида I-9
[00379] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
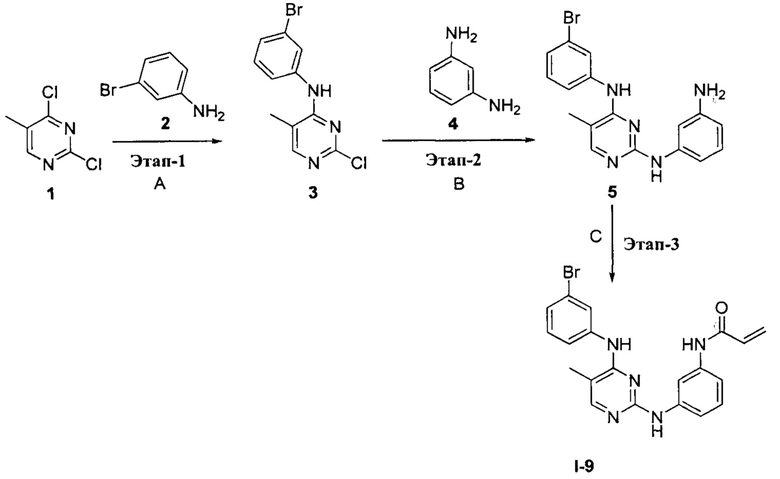
A) DIPEA, n-бутанол, 110°С, 1 час, MB; В) 1.5 N HCl, этанол, 90°С, 30 минут., MB; С) акрилоил хлорид, NMP, 0°С, 30 минут.
[00380] Этап-1
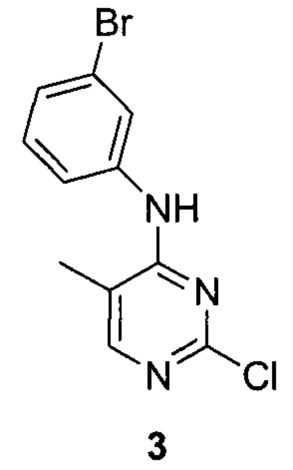
[00381] Раствор 1 (0,5 г, 3,06 ммоль), 2 (0,53 г, 3,06 ммоль), и DIPEA (0.80 мл, 4.06 ммоль) в п-бутаноле (5 мл) облучали в микроволновой печи (110°С, 1 час). Реакционную смесь охлаждали и концентрировали при пониженном давлении с получением осадка. Остаток добавляли к EtOAc (5 мл) и промывали раствором NaHCO3 (2 мл), водой (2 мл) и солевым раствором (2 мл). Сушили над Na2SO4, после чего концентрировали при пониженном давлении с получением сырого 3, которое затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 3 (0.125 г, 13%) в форме коричневого твердого вещества.
[00382] Этап-1
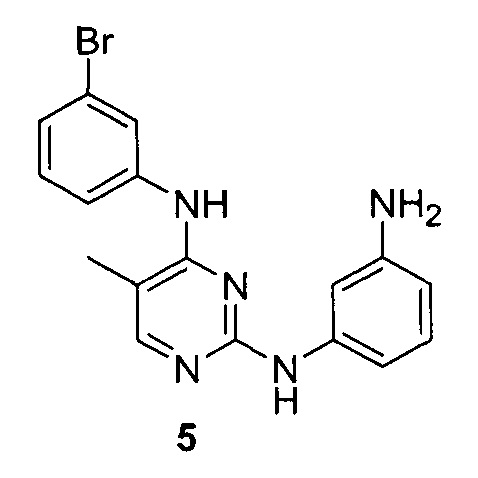
[00383] К раствору 3 (0.15 г, 0.5 ммоль) в EtOH (3 мл) добавляли 4 (0.081 г, 0.75 ммоль) а затем 1.5 N HCl (0.055 г, 1.5 ммоль). Реакционную смесь облучали в микроволновой печи (90°С, 30 минут), охлаждали и концентрировали при пониженном давлении. Полученный остаток добавляли к EtOAc (5 мл) и промывали раствором NaHCO3 (2 мл), водой (2 мл) и солевым раствором (2 мл). Сушили над Na2SO4, фильтровали, после чего концентрировали при пониженном давлении с получением сырого 5, которое затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 5 (0.06 г, 32%) в форме светло-коричневого твердого вещества.
[00384] Этап-3
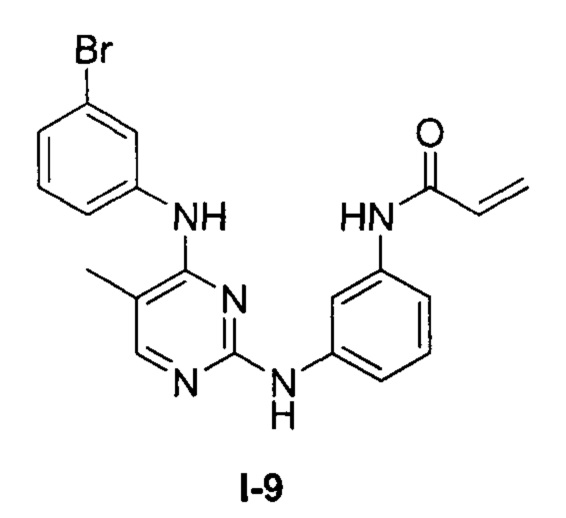
[00385] К раствору 5 (0,06 г, 0,16 ммоль) в NMP (1 мл) при помешивании добавляли акрилоил хлорид (0.117 г, 1,29 ммоль) при температуре 0°С. Реакционную смесь оставляли при перемешивании при этой температуре в течение 30 минут, после чего ее добавляли к дихлорметану (2 мл). Промывали раствором NaHCO3 (1 мл), водой (1 мл) и солевым раствором (1 мл). Затем его сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением I-9 (0.016 г, 23%) в форме бледно-коричневого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.16 (s, 3Н), 5.75 (dd, J=1.72
& 10 Гц, 1Н), 6.23 (dd, J=1.76 & 16.88, Гц, 1H), 6.45 (dd, J=10.08 & 16.92 Гц, 1H), 7.22-7.34 (m, 4H), 7.38 (d, J=8.00 Гц, 1H), 7.63 (d, J=8.08 Гц, 1H), 7.77 (s, 1H), 7.82 (s, 1H), 7.93 (s, 1H), 9.68 (s, 1H), 10.26 (s, 1H), 10.34 (s, 1H); ЖХМС: m/e 426 (M+1).
ПРИМЕР 9
[00386] Получение 3-(4-(2-(циклопропил сульфонил)-1,2,3,4-тетрагидроизохинолин-6-иламино)-5-метилпиримидин-2-иламино)бензолсульфонамида I-10
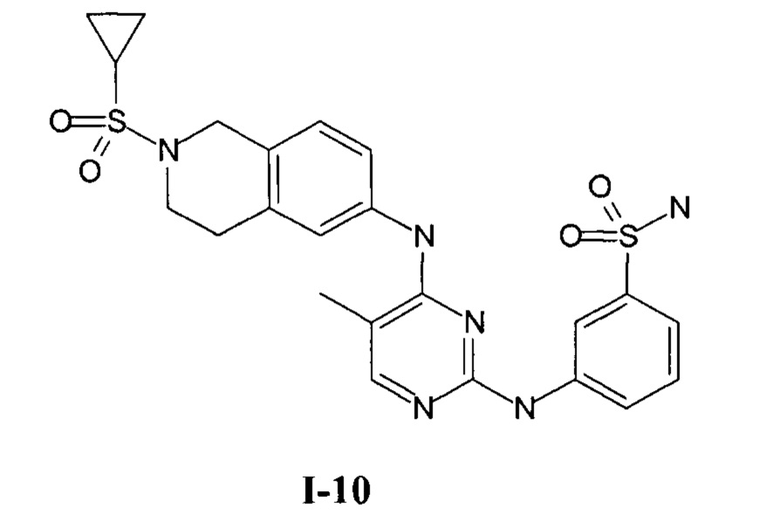
[00387] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
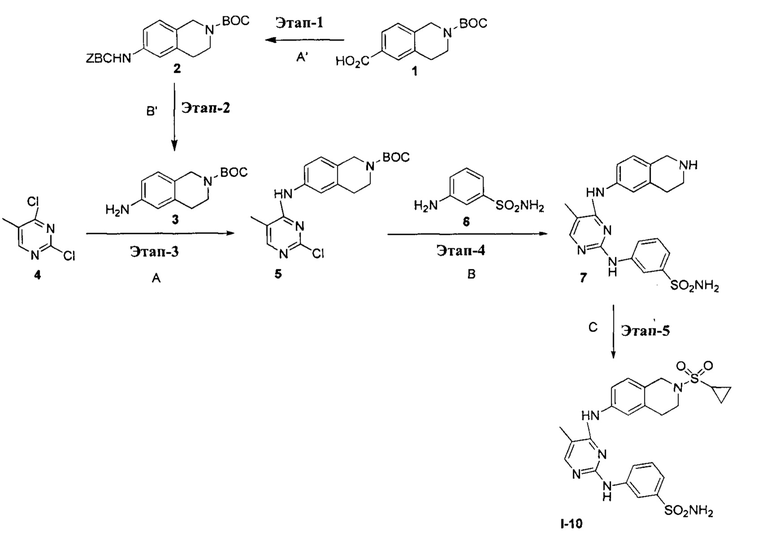
A') DPPA, Бензиловый спирт, Et3N, толуол, 110°С, 12 часов.; В') Pd(OH)2, Аммония формат, EtOH, с обратным холодильником, 6 часов; A) DIPEA, n-BuOH, 120°С, 1 час, MB; В) 1.5 N HCl, EtOH, с обратным холодильником 12 часов.; С) Циклопропилсульфонил хлорид, DIPEA, ТГФ, комн. т., 12 часов.
[00388] Этапы I-4 Процедура синтеза остова 7 описана в эксперименте для Соединения I-11 в данном описании.
[00389] Этап-5
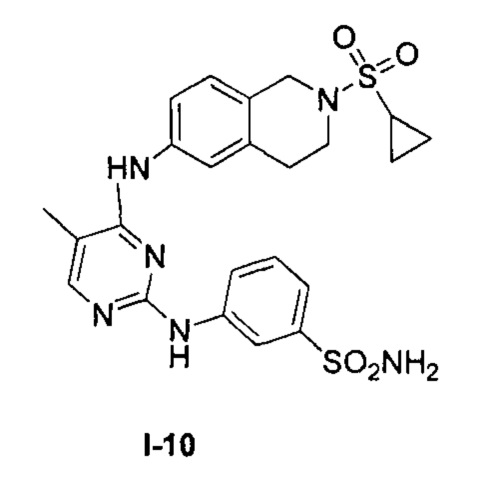
[00390] К раствору 7 (0.05 г, 0.0121 ммоль) в ТГФ (4 мл) при температуре 0°С при помешивании добавляли DIPEA (0.023 г, 0.182 ммоль), а затем циклопропилсульфонил хлорид (0.031 г, 0.182 ммоль) в атмосфере N2. Реакционную смесь оставляли до тех пор,
пока она доходила до комнатной температуры, и выдерживали при этой температуре в течение 12 часов. Затем ее добавляли к EtOAc (10 мл), промывали водой (5 мл), солевым раствором (5 мл) и сушили над Na2SO4. Фильтровали, и затем концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 6/4) с получением I-10 (0.035 г, 56%) в форме желтого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 0.97-0.1.00 (m, 4Н), 2.12 (s, 3Н), 2.60-2.66 (m, 1Н), 2.90 (t, J=5.2 Гц, 2Н), 3.52 (t, J=6 Гц, 2Н), 4.42 (s, 2Н), 7.16 (d, J=8.4 Гц, 1Н), 7.27 (s, 2Н), 7.31-7.35 (m, 2Н), 7.53 (s, 1H), 7.59 (d, J=8.4 Гц, 1H), 7.92 (s, 1Н), 8.03-8.04 (m, 2Н), 8.45 (s, 1Н), 9.40 (s, 1Н); ЖХМС: m/е 515 (М+1).
ПРИМЕР 10
[00391] Получение 3-(4-(2-(2-хлороацетил)-1,2,3,4-тетрагидроизохинолин-6-иламино)-5-метилпиримидин-2-иламино)бензолсульфонамида I-11
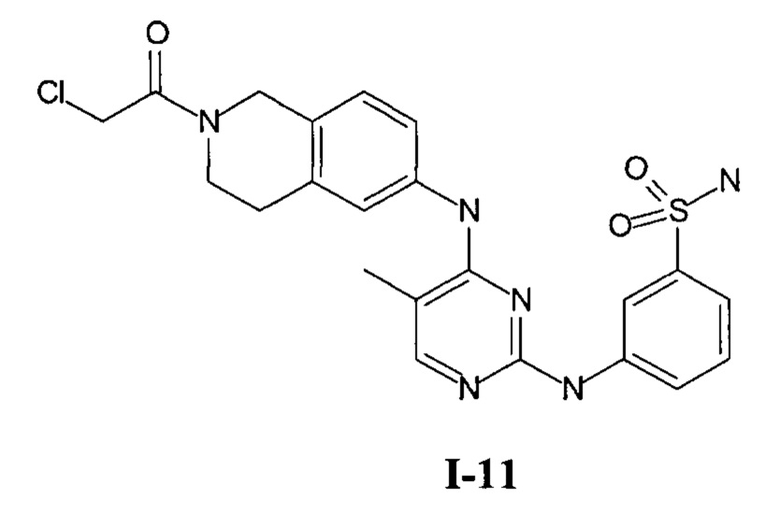
[00392] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
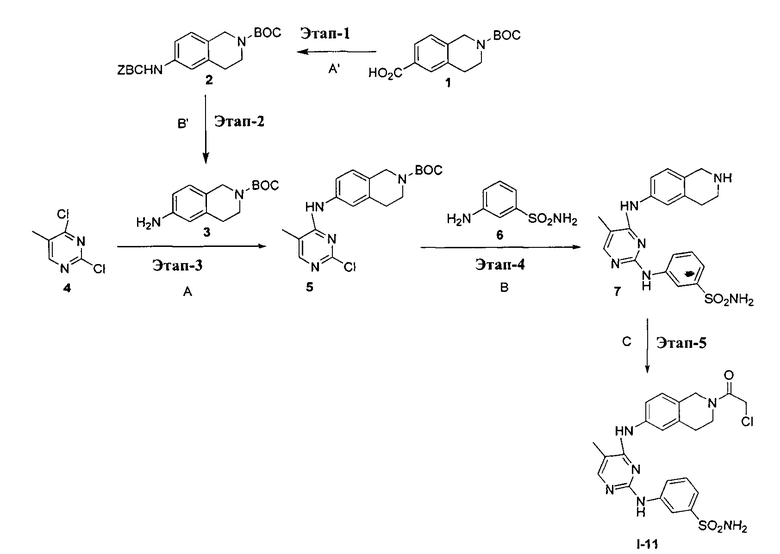
A') DPP А, Бензиловый спирт, Et3N, толуол, 110°С, 12 часов.; В') Pd(OH)2, Аммония формат, EtOH, с обратным холодильником, 6 часов; A) DIPEA, n-BuOH, 120°С, 1 час, MB; В) 1.5 N НС1, EtOH, с обратным холодильником 12 часов.; С) Cl-СН2-COCl, Et3N, ТГФ, комн. т., 12 часов.
[00393] Этап-1
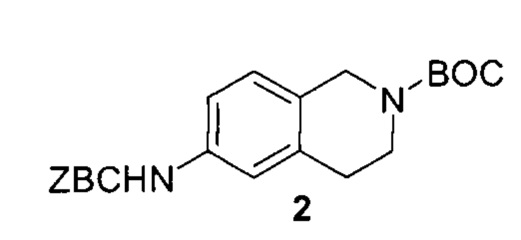
[00394] К раствору 1 (1.5 г, 5.4 ммоль) в толуоле (15 мл) добавляли при помешивании DPPA (2.17 г, 8.11 ммоль), Et3N (1.05 мл, 8.11 ммоль) и бензиловый спирт (0.876 г, 8.11 ммоль) а атмосфере N2. Реакционную смесь оставляли нагреваться с обратным холодильником в течение 12 часов, охлаждали и разбавляли этил ацетатом (100 мл). Его промывали водой (5 мл), солевым раствором (5 мл) и сушили над Na2SO4. Затем фильтровали и концентрировали при пониженном давлении, и осадок очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 2 (2.0 г, 97%) в форме белого твердого вещества.
[00395] Этап-1
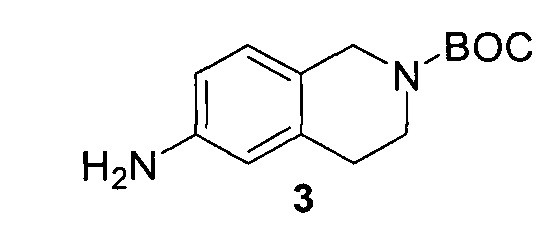
[00396] К раствору 2 (2.2 г, 5.75 ммоль) в EtOH (25 мл) при помешивании добавляли формиат аммония (3.68 г, 57.5 ммоль) и нагревали реакционную смесь с обратным холодильником в течение 6 часов. Затем охлаждали, фильтровали через целитную пластину ® и концентрировали фильтрат при пониженном давлении с получением 3 (1.3 г, 91%) в форме темно-коричневого масла, которое использовалось без дальнейшей очистки.
[00397] Этап-3
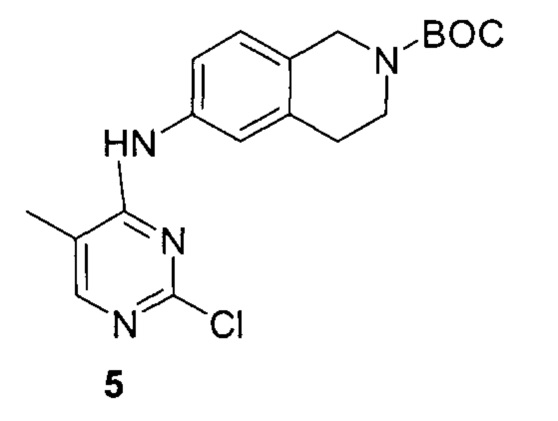
[00398] Раствор 3 (1.4 г, 5.56 ммоль), 4 (0.912 г, 5.56 ммоль) и DIPEA (1.077 г, 8.3 ммоль) в n-BuOH (15 мл) облучали в микроволновой печи при 120°С в течение 45 минут. Реакционную смесь охладлали и концентрировали при пониженном давлении. Остаток добавляли к этил ацетату (20 мл) и промывали водой (5 мл) и солевым раствором (5 мл). Сушили над Na2SO4, после чего концентрировали при пониженном давлении с получением остатка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 5 (1.1 г, 52%) в форме твердого вещества кремового цвета.
[00399] Этап-4
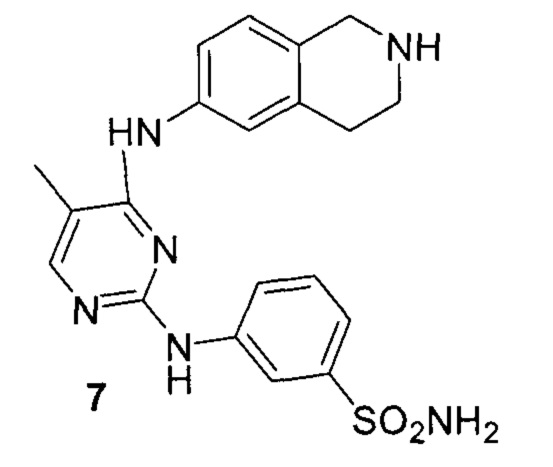
[00400] К раствору 5 (0.25 г, 0.66 ммоль) в этаноле (5 мл) при помешивании добавляли 6 (0.126 г, 0.73 ммоль) и каталитическое количество водного HCl, после чего реакционную смесь нагревали с обратным холодильником в течение 12 часов при 100°С. Ее охлаждали, полученное твердое вещество фильтровали и промывали диэтиловым эфиром и сушили под высоким вакуумом с получением 7 (0.24 г, 82%) в форме светор-желтого вешества.
[00401] Этап-5
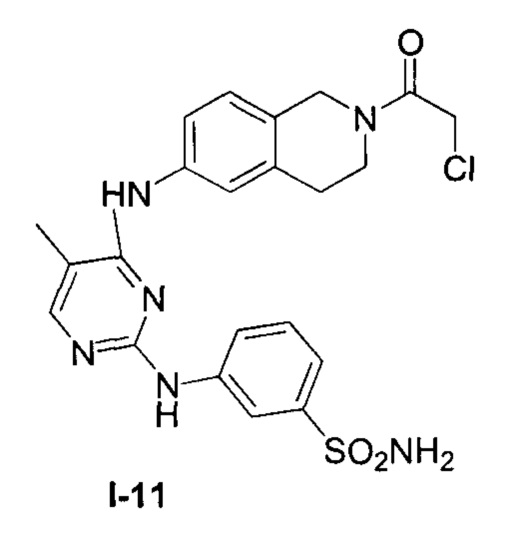
[00402] К раствору 7 (0,2 г, 0,487 ммоль) в NMP (5 мл) при помешивании добавляли Et3N (0.094 г, 0.731 ммоль). Раствор охлаждали до 0°С и добавляли хлороацетилхлорид (0.082 г, 0.731 ммоль). Реакционную смесь оставляли дойти до комнатной температуры и помешивать при комнатной температуре в течение 12 часов. Затем ее тушили охлажденной льдом водой (2 мл) и экстрагировали этил ацетатом (3×5 мл). Объединенный экстракт этил ацетата промывали солевым раствором (2 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением I-11 (0.038 г, 16%) в форме светло-желтого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.11 (s, 3Н), 2.77-2.89 (m, 2Н), 3.70-3.72 (m, 2Н), 4.49 (d, J=2.92 Гц, 2Н), 4.63 (d, J=23.56 Гц, 2Н), 7.15-7.17 (m, 1H), 7.24 (s, 2Н), 7.30-7.32 (m, 2Н), 7.50-7.65 (m, 2Н), 7.91 (s, 1H), 8.04-8.05 (m, 2Н), 8.27 (s, 1Н), 9.31 (s, 1H), ЖХМС: m/е 486.8 (МН+).
ПРИМЕР 11
[00403] Получение N-(3-(5-метил-4-(4-феноксифениламино)пиримидин-2-иламино) фенил)акриламида I-23
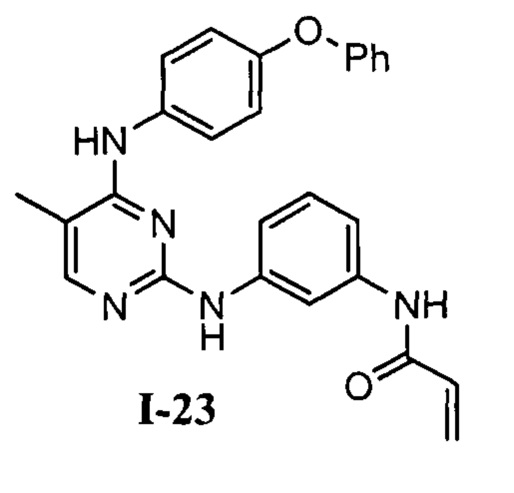
[00404] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
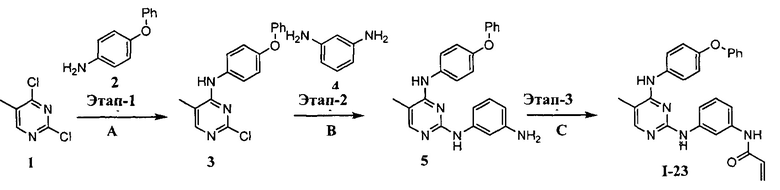
A) DIPEA, п-бутанол, 100°С, 1 час, MB; В) конц. HCl, n-BuOH, 160°С, 20 минут., MB; С) Акрилоил хлорид 0°С, комн. т., 1 час.
[00405] Этап-1
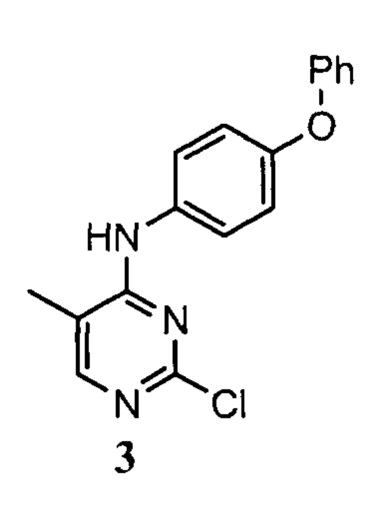
[00406] Раствор 1 (0,2 г, 1,2 ммоль), 2 (0,12 г, 0,95 ммоль) и DIPEA (0,23 г, 1,78 ммоль) в n-BuOH (2 мл) облучали в микроволновой печи (100°С в течение i час). Затем реакционную смесь охлаждали, концентрировали при пониженном давлении и остаток добавляли в EtOAc (5 мл). Промывали раствором NaHCO3 (2 мл), водой (2 мл) и солевым раствором (2 мл), после чего сушили над безводным Na2SO4. Концентрировали при повышенном давлении и очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 3 (0.11 г, 28,9%) в форме светло-коричневого твердого вещества.
[00407] Этап-1
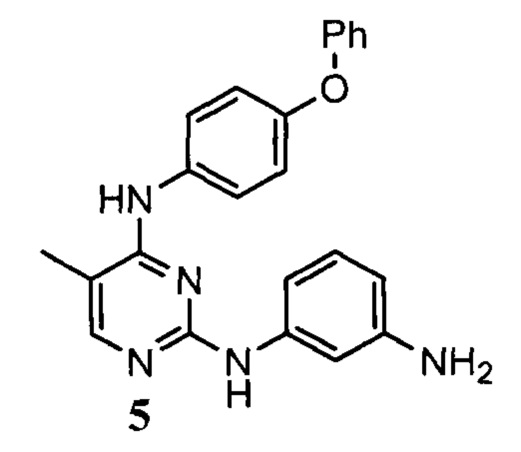
[00408] К раствору 3 (0.11 г, 0.3 ммоль), 4 (0.114 г, 1.05 ммоль) в п-бутанол (1 мл) добавляли концентрированную HCl (1 каплю) и подвергали смесь облучению в микроволномой печи (165°С а течение 10 минут). Реакционную смесь охлаждали, концентрировали при пониженном давлении и остаток добавляли в EtOAc (5 мл). Промывали раствором NaHCO3 (2 мл), водой (2 мл) и солевым раствором (2 мл). Сушили над Na2SO4, после чего концентрировали при пониженном давлении с получением остатка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 5 (0,08 г, 65%) в форме коричневого твердого вещества.
[00409] Этап-3
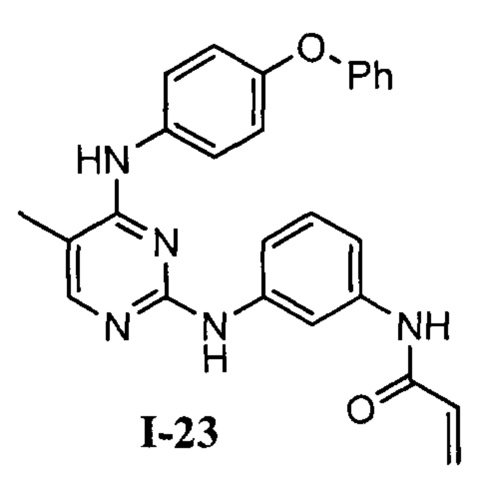
[00410] К раствору 5 (0,015 г, 0,03 ммоль) в NMP (1 мл) при помешивании добавляли акрилоил хлорид (0.005 г, 0,05 ммоль) при температуре 0°С. Реакционную смесь оставляли дойти до комнатной температуры и держать при этой температуре в течение 1 часа. Затем ее разбавляли дихлорметаном (2 мл) и промывали раствором NaHCO3 (1 мл), водой (1 мл) и солевым раствором (1 мл). Сушили над Na2SO4, после чего концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением I-23 (0.004 г, 23%) в форме коричневого твердого вещества. 400 МГц, MeOD: 5 2.14 (s, 3Н), 5.71 (d, J=11.20 Гц, 1H), 6.30-6.44 (m, 2Н), 6.94-6.99 (m, 4Н), 7.07-7.15 (т,
2Н), 7.22 (d, J=7.2 Гц, 1H), 7.34-7.36 (m, 3H), 7.63 (d, J=8.8 Гц, 2H), 7.79 (s, 2H); ЖХМС: m/e 437 (M+l).
ПРИМЕР 12
[00411] Получение N-(3-(5-метил-2-(3-сульфамоилфениламино)пиримидин-4-иламино)фенил)акриламида I-33
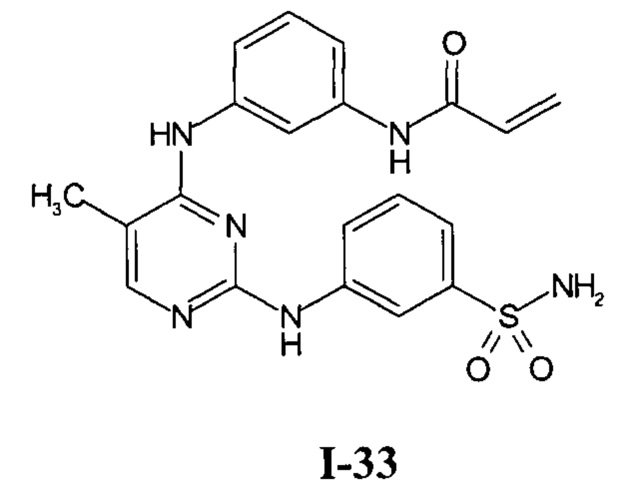
[00412] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
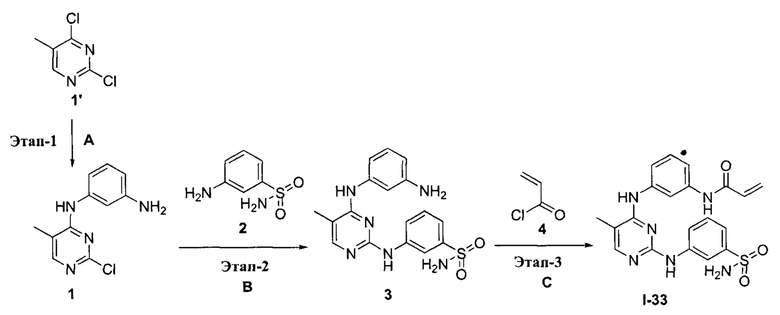
A) DIPEA, n-BuOH, 120°С, 30 минут., MB; В) 1.5 N HCl, Этанол, 100°С, 12 часов; С) NMP, 0°С до комн. т., 1 час.
[00413] Этап-1
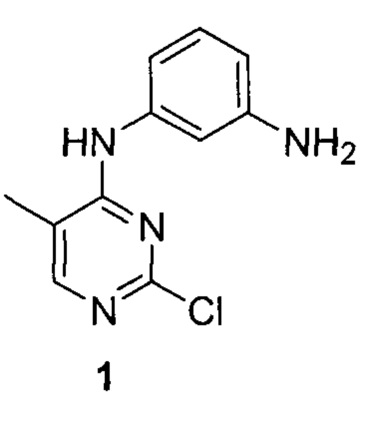
[00414] Раствор 1 (0,5 г, 3,06 ммоль), 1 (0,49 г, 4,59 ммоль), DIPEA (0.59 г, 4,59 ммоль) в п-бутаноле (8 мл) облучали в микроволновой печи (120°С, 30 минут). Охлаждали, тушили водой (5 мл) и экстрагировали этил ацетатом (3×20 мл). Объединенный слой этил ацетата промывали солевым раствором (5 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/этил ацетат, 9/1) с получением 1 (0.25 г, 34.77%) в форме светло-коричневого твердого вещества.
[00415] Этап-2
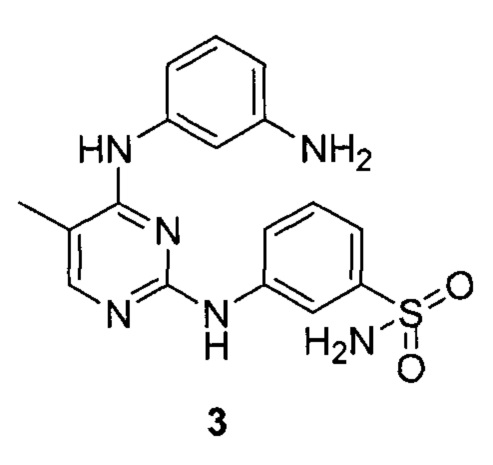
[00416] К раствору 3 (0.1 г, 0.48 ммоль), в этаноле (2 мл) при помешивании добавляли 4 (0.070 г, 0.42 ммоль) и каталитическое количество 1.5 N HCl (3 капли), затем нагревали до 100°С, в течение 12 часов. Затем реакционную смесь охлаждали, твердую фазу отделяли, фильтровали и промывали эфиром 5 (0.1 g сырого), после чего использовали на следующем этапе без дополнительной очистки.
[00417] Этап-3
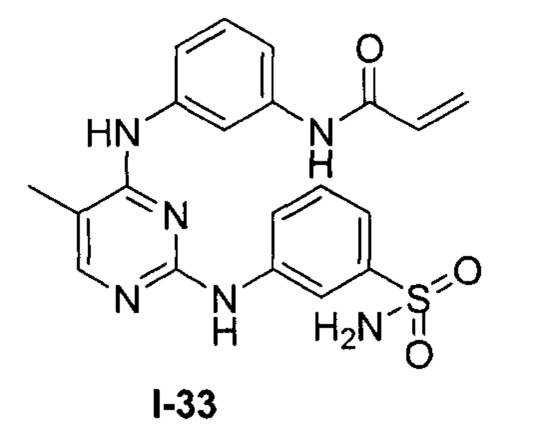
[00418] К раствору 5 (0.1 г, 0.27 ммоль) в NMP (2 мл) при помешивании добавляли акрилоил хлорид (0.037 г, 0.425 ммоль) при температуре 0°С, после чего помешивали при комнатной температуре в течение 1 часа, затем реакционную смесь тушили вдодй (4 мл) и приводили к форме основания NaHCO3, после чего экстрагировали этил ацетатом (5 мл), объединенный органический слой промывали солевым раствором (1 мл), сушили над
безводным Na2SO4, фильтровали и концентрировали, затем очищали сырой продукт методом препаративной ЖХВД с получением I-33 (0.07 г, 6%) в форме беловатого твердого вещества. 1Н-ЯМР (MeOD) δ ppm: 2.17 (s, 3Н), 5.78 (dd, J=2.36 & 9.52 Гц, 1H), 6.34-6.48 (m, 2H), 7.26-7.43 (m, 5H), 7.87 (s, 1H), 7.96-8.03 (m, 3Н); ЖХМС: m/e 425 (M+1).
ПРИМЕР 13
[00419] Получение N-(3-(метил(5-метил-2-(фениламино)пиримидин-4-ил)амино)фенил)акриламида I-34
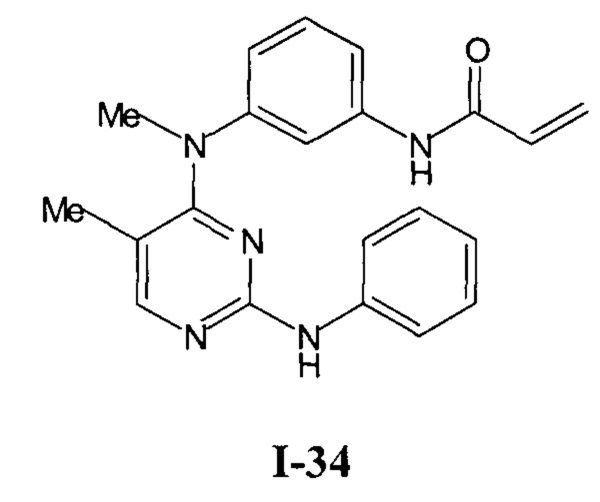
[00420] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
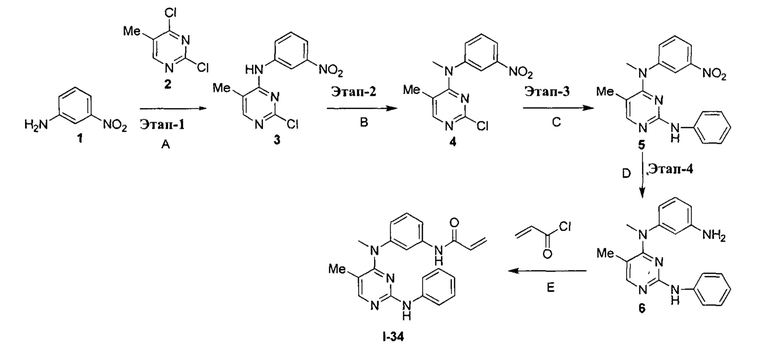
A) Pd(OAC)2 ,BINAP, Cs2CO3, Toluene, 100°С, 16 часов; В) NaH, CH3I, ТГФ, 0°С-30 минут, комн. т.-16 часов; С) Анилин, конц. HCl, Этанол, 90°С, 60 мин; D) Н2, Pd/C, Этанол, 16 часов; Е) акрилоил хлорид, NMP, 0°С, 1 час.
[00421] Этап-1
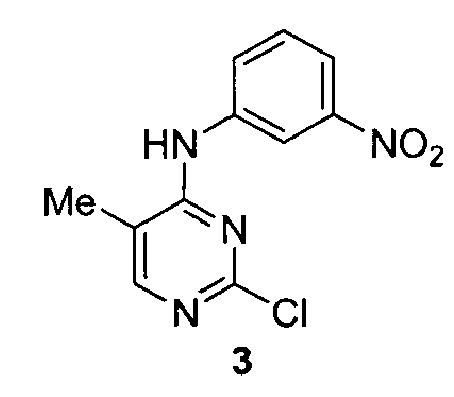
[00422] К раствору 2 (1.0 г, 6.0 ммоль), в толуоле (30.0 мл) при помешивании добавляли 1 (0.84 г, 6.0 ммоль), BINAP (0.186 г, 0.3 ммоль), Cs2CO3 (4.87 г, 15.0 ммоль). Реакционную смесь дегазировали, продувая N2 в течение 15 минут. Затем к реакционной смеси добавляли Pd(ОАс)2 (0.134 г, 0.6 ммоль), и нагревали при 100°C в течение 16 часов в атмосфере N2. Затем ее охлаждали, разбавляли этил ацетатом (30 мл) и фильтровали через целит®. Фильтрат промывали водой (2×25 мл), солевым раствором (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, меш 60-120, этилацетат/гексан: 10/90) с получением твердого вещество, которое промывали эфиром с получением 3 (0.6 г, 37%) в форме светло-желтого твердого вещества.
[00423] Этап-2
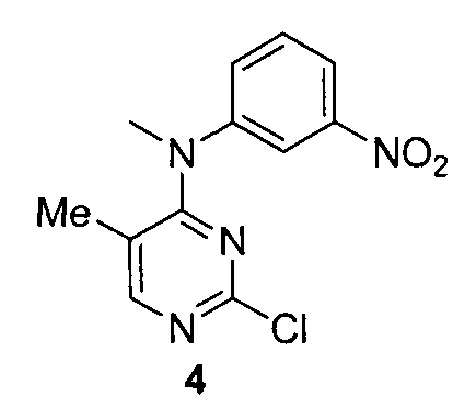
[00424] К смеси NaH (0.1 г, 2.5 ммоль, 60% дисперсия в парафиновом масле) в сухом ТГФ (10.0 мл) при помешивании добавляли 3 (0.5 г, 1.89 ммоль) при температуре 0°C, и помешивали реакционную смесь при этой температуре в течение 30 минут. Добавляли CH3I (0.305 г, 2.15 ммоль), после чего реакционную смесь разрешалось довести до комнатной температуры и помешивать при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли водой (25 мл) и экстрагировали EtOAc (3×25 мл). Объединенные экстракты EtOAc промывали водой (25 мл), солевым раствором (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с выпадением осадка. Сырой остаток затем очищали методом колоночной хроматографии (SiO2, CHCl3/МеОН: 99/1) с получением 4 (0,12 г, 22,7%) в форме светло-желтого твердого вещества.
[00425] Этап-2
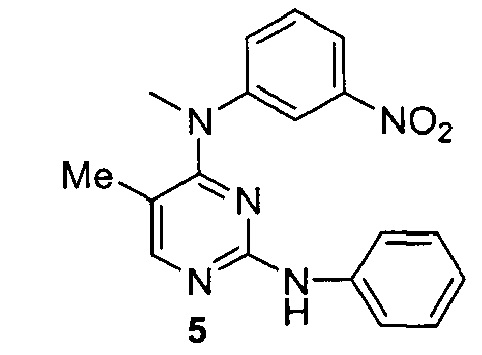
[00426] К раствору 4 (120 мг, 0.431 ммоль) в EtOH (2 мл) добавляли концентрированную HCl (0.044 г, 1.2 ммоль) и анилин (0.16 г, 1.72 ммоль) и грели реакционную смесь в запечатанной колбе при 90°C в течение 1 час. Реакционную смесь охлаждали, растворители удаляли методом концентрирования при пониженном давлении и полученный осадок разбавляли 10% NaHCO3 (10.0 мл). Его экстрагировали EtOAc (3×15 мл) и объединенный экстракт EtOAc промывали водой (15 мл), солевым раствором (15 мл) и сушили над Na2SO4. Концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, CHCl3/МеОН: 99/1) с получением 5 (0,11 г, 76%) в форме светло-желтого твердого вещества.
[00427] Этап-4
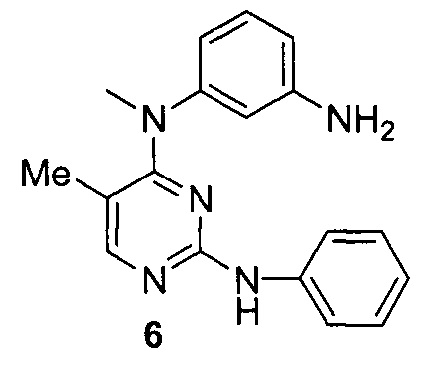
[00428] К раствору 5 (0.110 г, 0.328 ммоль), в этаноле (50 мл) добавляли 10% палладий на угле (0.022 г) и помешивали реакционную смесь в атмосфере Н2 (1.5 Кг) при комнатной температуре в течение 16 часов. Фильтровали через целит® и концентрировали при пониженном давлении с получением остатка. Остаток очищали методом колоночной хроматографии (SiO2, 60-120, метанол/хлороформ: 1/99) с получением 6 (0,07 г, 69,9%) в форме бесцветной вязкой жидкости.
[00429] Этап-5
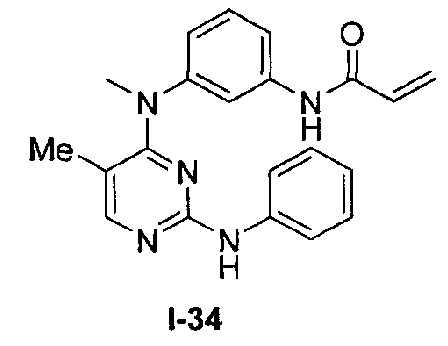
[00430] К раствору 6 (0.070 г, 0.23 ммоль) в NMP (1.5 мл) при помешивании при 0°C добавляли акрилоил хлорид (0.083 г, 0.916 ммоль) и реакционную смесь помешивали при 0°C в течение 1 часа. Затем ее гасили 10% раствором натрия бикарбоната (15 мл) после чего отфильтровывали твердую фазу, промывали холодной водой (5 мл), гексаном (5 мл). Твердое вещество сушили в течение 2 часа при пониженном давлении с получением 1-34 (0.033 г, 40%) в форме бледно-желтого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: δ 1.47 (s, 3Н), 3.45 (s, 3Н), 5.74 (dd, J=Гц, 1Н), 6.22 (dd, J=2.0 & 16.98 Гц, 1Н), 6.38 (dd, J=10 & 16.94 Гц, 1H), 6.85-6.91 (m, 2Н), 7.21-7.25 (m, 2Н), 7.32 (t, J=8.02 Гц, 1Н), 7.43-7.47 (m, 2Н), 7.77-7.79 (m, 2Н), 7.90 (s, 1H), 9.22 (s, 1H), 10.18 (s, 1Н); ЖХМС: m/е 360.8 (М+1).
ПРИМЕР 14
[00431] Получение N-(3-(5-метил-2-(3-(проп-2-инилокси)фениламино)пиримидин-4-иламино)фенил) акриламида 1-35
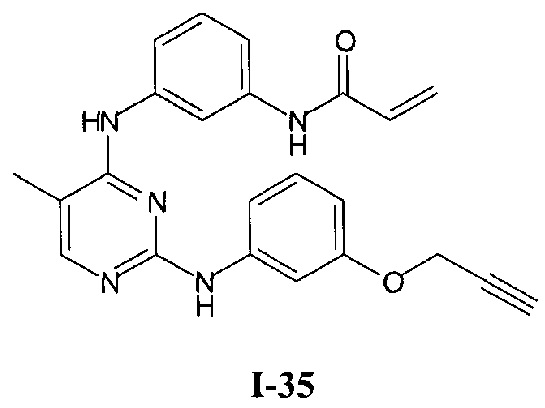
[00432] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
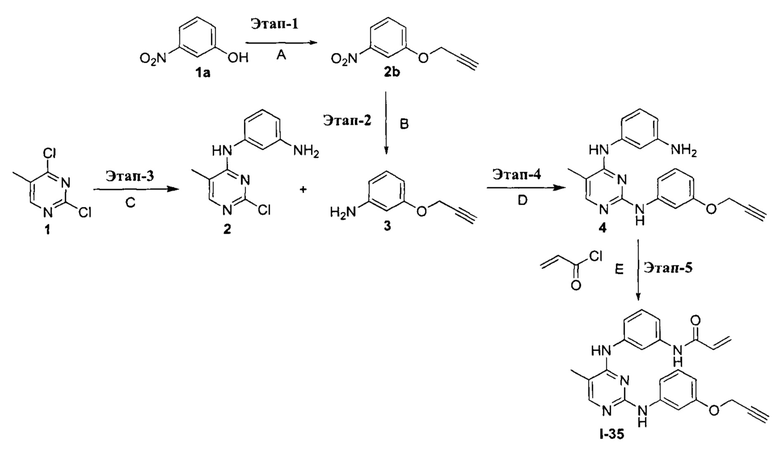
A) K2CO3, CH3CN, 65°C, 8 часов; В) порошок Fe, NH4Cl, МеОН, H2O, 80°C, 4 часа; С) 1,3-фенеилендиамин, DIPEA, n-BuOH, 120°C, 30 минут, MB; D) Конц. HCl, абсолютный этанол, 110°C, 2 часа; Е) NMP, 0°C, 1 час.
[00433] Этап-1
[00434] К раствору 1а (4 г, 0.0287 моль) и K2CO3 (5.6 г, 0.0574 моль) в CH3CN (15 мл) при помешивании добавляли пропаргил бромид (4.1 г, 0.0345 моль) и полученную смесь разрешалось нагревать с обратным холодильником в течение 8 часов. Реакционную смесь затем охлаждали, тушили водой и экстрагировали EtOAc (3×50 мл). Объединенный экстракт EtOAc промывали водой (20 мл), солевым раствором (20 мл) и сушили над Na2SO4. Фильтровали и концентрировали при пониженном давлении с получением 2b в форме коричневатого твердого вещество, которое использовали без дальнейшей очистки.
[00435] Этап-2
[00436] К раствору 2b в смеси метанола (30 мл) и воды (30 мл) при помешивании последовательно добавляли, NH4Cl (10.3 г, 0.194 моль) и железный порошок (6.8 г, 0.121
моль). Полученную смесь нагревали с обратным холодильником при 80°C в течение 4 часа. Реакционную смесь охлаждали, разбавляли метанолом и фильтровали через пластину целлита®. Фильтрат концентрировали при пониженном давлении и остаток добавляли в EtOAc. Затем промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении с получением осадка. Осадок затем очищали методом колоночной хроматографии (SiO2, 60-120, колоночной хроматографии с гравитационным элюированием, и ожидаемый продукт извлекали из адсорбента CHCl3/МеОН 96/4) с получением 3 (3,2 г, 91%) в форме коричневатого вещества.
[00437] Этап-3
[00438] Раствор 2, 4-дихлоро-5-метил пиримидина 1 (0.3 г, 0.0018 моль), 1,3-фенилен диамина (0.24 г, 0022 моль), DIPEA (0.35 г, 0.0027 моль) в n-BuOH (3 мл) подвергали облучению в микроволновой печи (120°C, 30 минут). Реакционную смесь охлаждали, разбавляли водой (15 мл) и экстрагировали EtOAc (3×15 мл). Объединенные экстракты EtOAc промывали водой (20 мл), солевым раствором (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный осадок далее очищали методом колоночной хроматографии (SiO2, 60-120) с получением 2 (0.15 г, 35%) в форме коричневатого твердого вещества.
[00439] Этап-4
2 (0.15 г, 0.006 моль) и 3 (0.37 г, 0.0025 моль) помещали в нагнетательную трубку и в нее добавляли абс. EtOH (3 мл) а затем концентрированная HCl (0.04 г, 0.0012 моль). Трубку плотно закручивали и грели при 120°C в течение 2 часа. Затем реакционную смесь
охлаждали, при пониженном давлении удаляли растворители, и полученный остаток добавляли к (10 мл). Промывали водой (4 мл), раствором NaHCO3 (4 мл), и солевым раствором (5 мл). Сушили над Na2SO4, после чего концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, колоночной хроматографии с гравитационным элюированием, элюировали ожидаемое вещество CHCl3/МеОН: 94/6) с получением 4 (125 мг, 56%) в форме светло-коричневого твердого вещества.
[00440] Этап-5
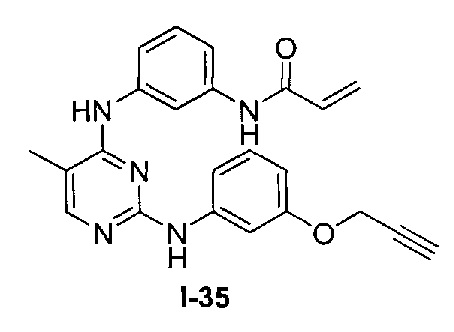
[00441] К раствору 4 (0,1 г, 0,002 ммоль) в NMP (8 мл) при помешивании добавляли акрилоил хлорид (0.1 г, 0.001 моль) по каплям при температуре 0°C. Реакционную смесь держали при этой температуре в течение 10 минут, после чего разрешалось помешивать при этой температуре в течение 1,5 часа. Затем ее гасили 10% раствором натрия бикарбоната и экстрагировали EtOAc (2×15 мл). Объединенные экстракты EtOAc промывали водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный осадок затем очищали методом колоночной хроматографии (SiO2, 60-120, колоночной хроматографии с гравитационным элюированием, и ожидаемое соединение элюировали CHCl3/МеОН 90/10) с получением I-35 (20 мг, 18%) в форме беловатого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.11 (s, 3Н), 3.51 (s, 1Н), 4.61 (s, 2Н), 5.74 (d, J=9.08 Гц, 1Н), 6.25 (d, J=15.84 Гц, 1Н), 6.45 (s, 2Н), 7.02 (s, 1Н), 7.27-7.45 (m, 5Н), 7.91 (d, J=8.84 Гц, 2Н), 8.36 (s, 1H), 8.93 (s, 1H), 10.09 (s, 1Н), ЖХМС: m/е 400 (М+1).
ПРИМЕР 15
[00442] Получение (Е)-4-(диметиламино)-N-(3-(5-метил-2-(фениламино)пиримидин-4-иламино) фенил)бут-2-енамида I-38
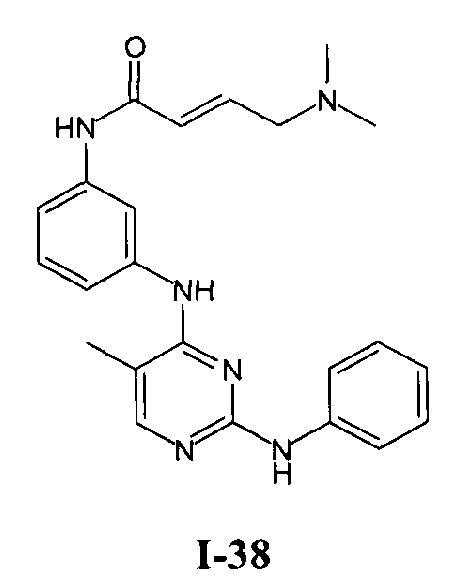
[00443] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
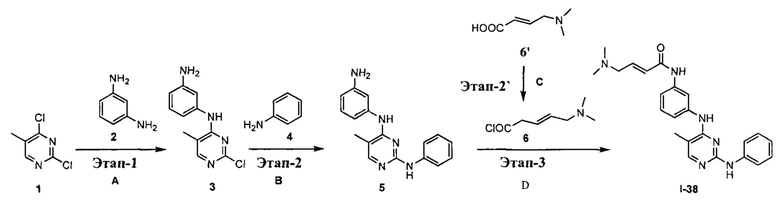
A) DIEA, n-BuOH, 120°C, 30 минут, MB; В) NMP, 200°C, 10 минут, MB;, С)) оксалил хлорид, CH3CN, 30 минут при температуре 0°C, 2 часа при температуре 25°C, 5 минут при температуре 45°C, D) NMP, 0°C, 1 час.
[00444] Этап-1
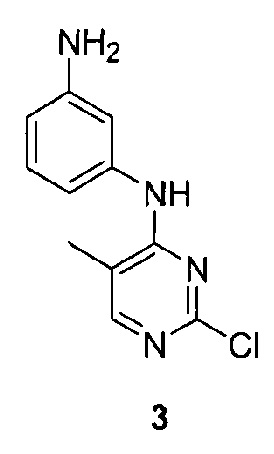
[00445] Раствор 1 (2,0 г, 12 ммоль), 2 (2,0 г, 18 ммоль), DIEA (2,33 г, 18 ммоль) в n-ВиОН (20,0 мл) облучали в микроволновой печи при 120°C в течение 30 мин. Реакционную смесь затем гасили водой (100 мл) и экстрагировали EtOAc (3×20 мл). Объединенные экстракты EtOAc промывали водой (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток
затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/этил ацетат, 15/85) с получением 3 (1,3 г, 45%) в форме темно-коричневого твердого вещества.
[00446] Этап-2
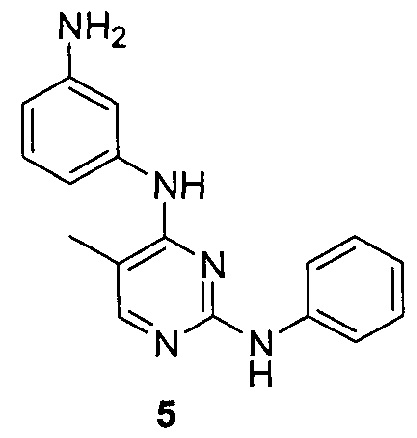
[00447] Раствор 3 (1.0 г, 4.27 ммоль), 4 (1.5 г, 16.12 ммоль) в NMP (10 мл) облучали в микроволновой печи (200°C, 10 минут). Реакционную смесь охлаждали, разбавляли водой (100 мл) и экстрагировали EtOAc (3×100 мл). Объединенные экстракты EtOAc промывали водой (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с выпадением осадка. Сырой остаток затем очищали методом колоночной хроматографии (SiO2, 60-120, CHCl3/МеОН: 98/2) с получением 5 (0,5 г, 40,3%) в форме светло-коричневого твердого вещества.
[00448] Этап-2'
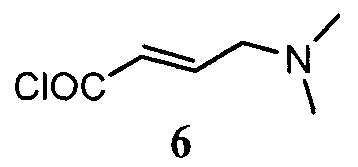
[00449] К раствору 6' (70 мг, 0,42 ммоль) в CH3CN (1.0 мл) при помешивании добавляли оксалил хлорид (80 мг, 0.62 ммоль) при температуре 0°C. Реакционную смесь оставляли с перемешиванием при температуре 0°C в течение получаса, а затем при комнатной температуре в течение 2 часа. Окончательно реакционную смесь грели при 45°C в течение 5 минут, охлаждали и использовали на следующем этапе без дальнейшей очистки.
[00450] Этап-3
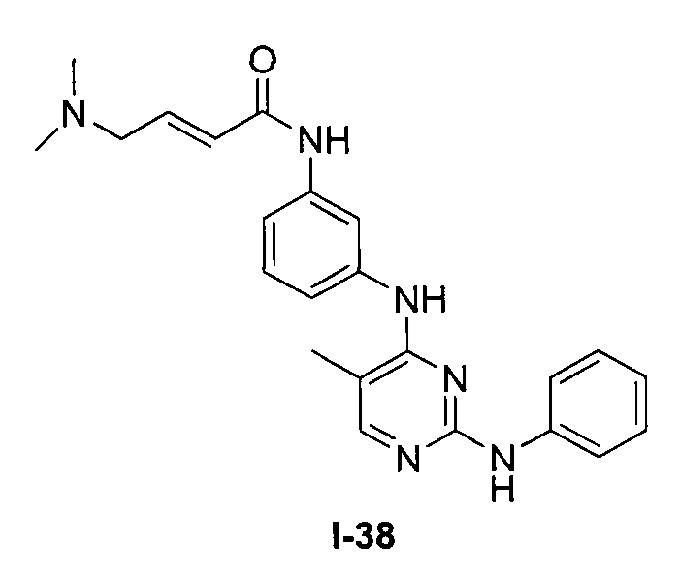
[00451] К раствору 5 (75 мг, 0,12 ммоль) в NMP (1 мл) при помешивании добавляли 6 при температуре 0°C. Реакционную смесь помешивали при 0°C в течение 1 часа, гасили холодной водой (5 мД), приводили к форме основания Et3N и экстрагировали CH2Cl2 (3×10 мл). Объединенный органический экстракт промывали водой (5 мл), солевым раствором (5 мл) и сушили над Na2SO4. Концентрировали при пониженном давлении, затем очищали над силикагелем (60-120) при помощи 5% раствора метанола в хлороформе с получением сырого соединения (20 мг) в форме коричневого смолистого твердого вещества, которое затем снова добавляли в дихлорметан и перемешивали с 10% раствором бикарбоната в течение 30 минут, отделяли слой дихлорметана, сушили над Na2SO4 и концентрировали с получением I-38 (8 мг, 17%) в форме коричневого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.15 (s, 3Н), 2.32 (s, 6Н), 3.21 (d, J=5.76 Гц, 2Н), 6.27 (d, J=15.36 Гц, 1Н), 6.84-6.93 (m, 2Н), 7.14 (t, J=7.52 Гц, 2Н), 7.27-7.33 (m, 2Н), 7.44 (dd, J=2.04 Гц & 5.08 Гц, 1Н), 7.53 (d, J=7.72 Гц, 2Н), 7.80 (s, 1Н), 8.00 (s, 1Н); ЖХМС: m/е 402.8 (М+1).
ПРИМЕР 16
[00452] Получение N-(4-(5-метил-2-(фениламино)пиримидин-4-иламино)фенил) акриламида I-39
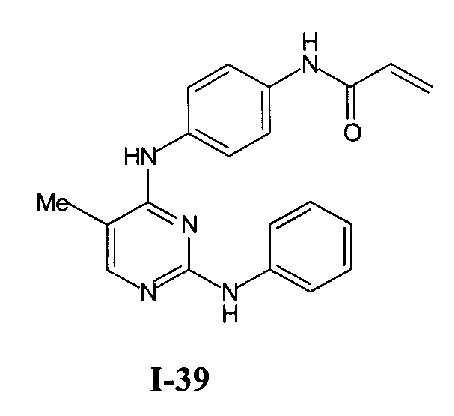
[00453] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
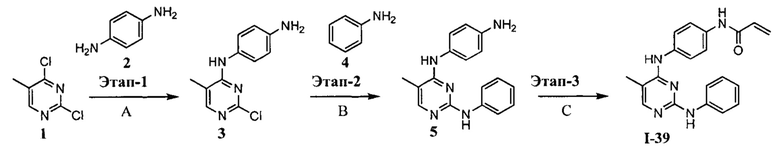
A) DIPEA, n-BuOH, 110°C, 45 минут, MB; В) Конц. HCl, n-BuOH, 150°C, 10 минут, MB; С) Акрилоил хлорид, NMP, 0°C-30 минут, комн. т.-2 часа.
[00454] Этап-1
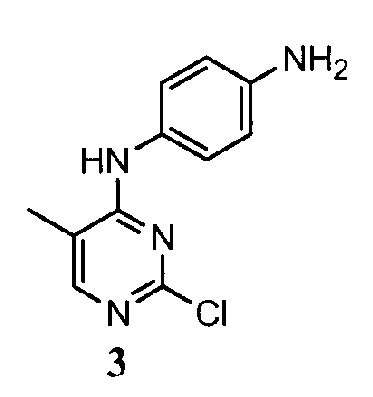
[00455] Раствор 1 (0,4 г, 2,4 ммоль), 2 (0,3 г, 2,6 ммоль), DIPEA (0,46 г, 3,6 ммоль) в n-ВиОН (10 мл) облучали в микроволновой печи (110°C, 45 минут). Реакционную смесь охлаждали, разбавляли водой (20 мл) и экстрагировали EtOAc (3×15 мл). Объединенные экстракты EtOAc промывали водой (20 мл), солевым раствором (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии (SiO2, 60-120, CHCl3/МеОН: 99/1) с получением 3 (350 мг, 62%) в форме беловатого твердого вещества.
[00456] Этап-2
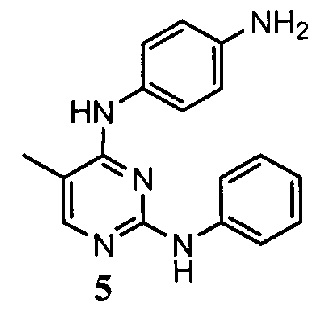
[00457] Раствор 3 (0,2 г, 0,8 ммоль), 4 (0,63 г, 6,8 ммоль), и концентрированной HCl (0,03 г, 0,8 ммоль) в n-BuOH (10 мл) облучали в микроволновой печи (150°C, 10 минут). Реакционную смесь охлаждали, разбавляли водой (10 мл), приводили к форме основания раствором натрия бикарбоната и экстрагировали EtOAc (3×15 мл). Объединенные экстракты EtOAc промывали водой (15 мл), солевым раствором (15 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Сырой остаток очищали методом колоночной хроматографии (SiO2, 60-120, CHCl3/МеОН: 97/3) с получением 5 (110 мг, 47%) в форме коричневого смолистого твердого вещества.
[00458] Этап-3
[00459] К раствору 5 (0,06 г, 0,2 ммоль) в NMP (2 мл) при помешивании добавляли акрилоил хлорид (0.03 г, 0.3 ммоль) при температуре 0°C. Оставляли для перемешивания при той же температуре в течение 20 минут, а затем при комнатной температуре в течение 2 часа. Реакционную смесь тушили водой, приводили к форме основания 10% раствором натрия бикарбоната и экстрагировали EtOAc (3×10 мл). Объединенный слой EtOAc промывали водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии (SiO2, 60-120) и окончательно методом препаративной ЖХВД с выходой I-39 (10 мг, 16%) в форме беловатого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.10 (s, 3Н), 5.71-5.76 (m, 1H), 6.25 (dd, J 2.04 & 16.96 Гц, 1H), 6.45 (dd, J=10.08 & 16.92 Гц, 1H),
6.84 (t, J=7.30 Гц, 1H), 7.14-7.18 (m, 2H), 7.62-7.68 (m, 6H), 7.86 (s, 1H), 8.26 (s, 1H), 8.94 (s, 1H), 10.11 (s, 1H), ЖХМС: m/e 346 (M+1).
ПРИМЕР 17
[00460] Получение N-(3-(5-метил-2-(фениламино)пиримидин-4-иламино)фенил)пропионамида IR-7
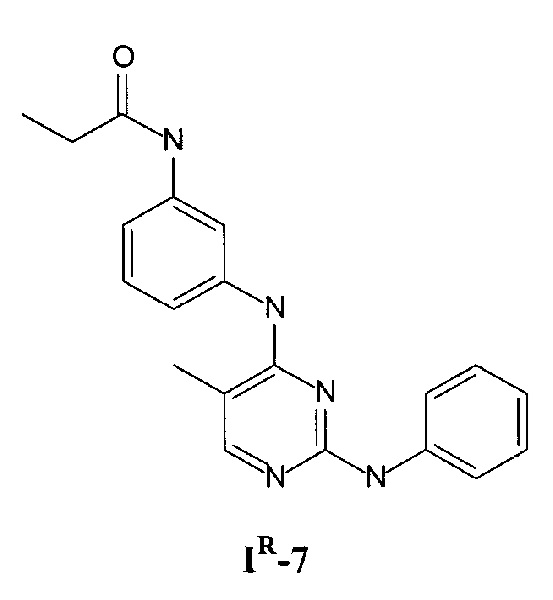
[00461] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
A) DIPEA, n-BuOH, 120°C, 30 минут, MB; В) NMP, 200°C, 10 минут, MB; С) 6, NMP, 0°C, 60 минут.
[00462] Этап-1
[00463] Раствор 1 (2,0 г, 12 ммоль), 2 (2,0 г, 18 ммоль), DIEA (2,33 г, 18 ммоль) в n-ВиОН (20,0 мл) облучали в микроволновой печи при 120°C в течение 30 мин. Реакционную смесь затем гасили водой (100 мл) и экстрагировали EtOAc (3×20 мл). Объединенные экстракты EtOAc промывали водой (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, меш 60-120, EtOAc/CHCl3: 15/85) с получением 3 (1.3 г, 45%) в форме темно-коричневого твердого вещества.
[00464] Этап-2
[00465] Раствор 3 (1.0 г, 4.27 ммоль), 4 (1.5 г, 16.12 ммоль) в NMP (10 мл) облучали в микроволновой печи (200°C, 10 минут). Реакционную смесь охлаждали, разбавляли водой (100 мл) и экстрагировали EtOAc (3×100 мл). Объединенные экстракты EtOAc промывали водой (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с выпадением осадка. Сырой остаток затем очищали методом колоночной хроматографии (SiO2, CHCl3/МеОН: 98/2) с получением 5 (0,5 г, 40,3%) в форме светло-коричневого твердого вещества.
[00466] Этап-3
[00467] К раствору 5 (75 мг, 0.25 ммоль) в NMP (1.0 мл) при помешивании при температуре 0°C добавляли пропаноил хлорид (6) (72 мг, 0.75 ммоль) и помешивали реакционную смесь при температуре 0°C в течение 60 минут. Затем реакционную смесь гасили водой (5 мл), приводили к форме основания Et3N и экстрагировали EtOAc (3×10 мл). Объединенные экстракты EtOAc промывали водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, 230-400, метанол/хлороформ: 2/98) с получением IR-7 (0,025 г, 28,73%) в форме беловатого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.08 (t, J=7.6 Гц, 3Н), 2.11 (s, 3Н), 2.31 (q, 7=7.6 Гц, 2Н), 6.81 (t, J=7.2 Гц, 1Н), 7.11 (t, J=8 Гц, 2Н), 7.21-7.25 (m, 1H), 7.31 (d, J=8.40 Гц, 1Н), 7.36 (d, J=8.00 Гц, 1Н), 7.66 (d, J=8.40 Гц, 2Н), 7.86 (s, 1Н), 7.89 (s, 1Н), 8.35 (s, 1H), 8.93 (s, 1Н), 9.81 (s, 1Н); ЖХМС: m/е 348.3 (М+1).
ПРИМЕР 18
[00468] Получение N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)акриламида I-56
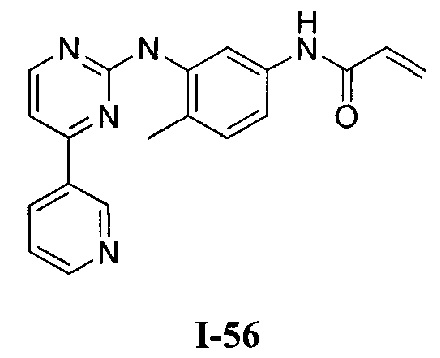
[00469] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
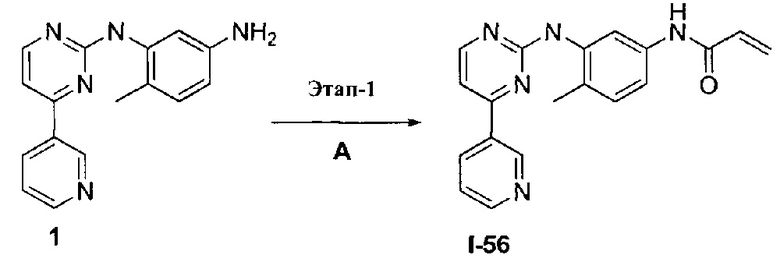
А) акрилоил хлорид, Et3N, ДМФ, комн. т., 12 часов
[00470] Этап-1
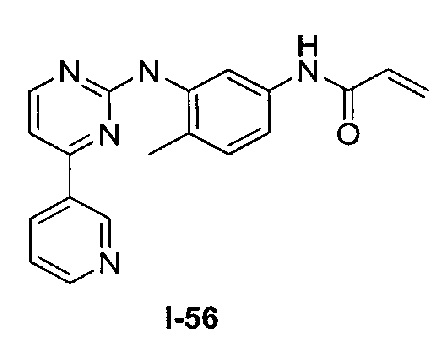
[00471] К раствору 1 (0.15 г, 0.54 ммоль) и Et3N (0.11 г, 1.08 ммоль) в ДМФ (1 мл) при помешивании при температуре 0°C добавляли акрилоил хлорид (0.09 г, 1.08 ммоль), по каплям в атмосфере N2. Реакционную смесь оставляли дойти до комнатной температуры и помешивать последующие 12 часов. Затем ее тушили охлажденной льдом водой (2 мл) и экстрагировали EtOAc (2×15 мл). Объединенный экстракт EtOAc промывали солевым раствором (2 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с выпадением сырого осадка. Осадок далее очищали методом препаративной ЖХВД с выходом I-56 (0.060 г, 33%) в форме бледно-желтого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.19 (s, 3Н), 5.72 (dd, J=2 & 10.08 Гц, 1H), 6.22 (dd, J=2 & 16.92 Гц, 1H), 6.45 (dd, J=10 & 17 Гц, 1H), 7.16 (d, J=8.36 Гц, 1H), 7.32 (dd, J=1.92 & 8.16 Гц, 1H), 7.42 (d, J=5.12 Гц, 1H), 7.50-7.53 (m, 1H), 7.95 (d, J=1.68 Гц, 1H), 8.45 (dd, J=6.16 & 8.16 Гц, 1H), 8.49 (d, J=5.16 Гц, 1H), 8.68 (dd, J=1.56 & 4.76 Гц, 1H), 8.95 (s, 1H), 9.25 (d, J=1.56 Гц, 1H), 10.08 (s, 1H); ЖХМС: m/e 332.4 (M+1).
ПРИМЕР 19
[00472] Общая схема получения соединений, имеющих енон-содержащую реакционноспособную группу, например, 3-метил-1-(3-(5-метил-2-(фениламино)пиримидин-4-иламино)фенил)бут-2-ен-1-она I-47
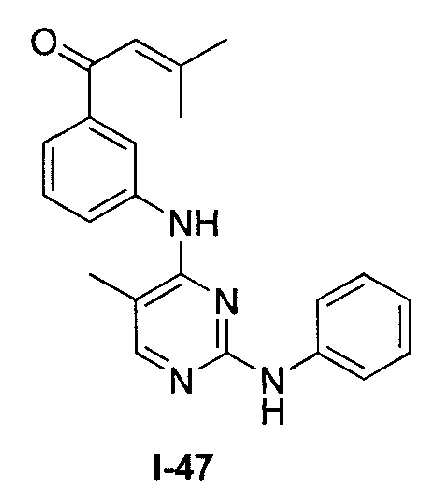
[00473] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже. Специалисты в данной области техники понимают, что I-47 является примером соединения, имеющего енон-содержащие реакционноспособные группы, и что другие соединения, имеющие енон-содержащие реакционноспособные группы, могут быть синтезированы по существу тем же образом, с использованием схем, этапов и промежуточных соединений, описанных ниже.
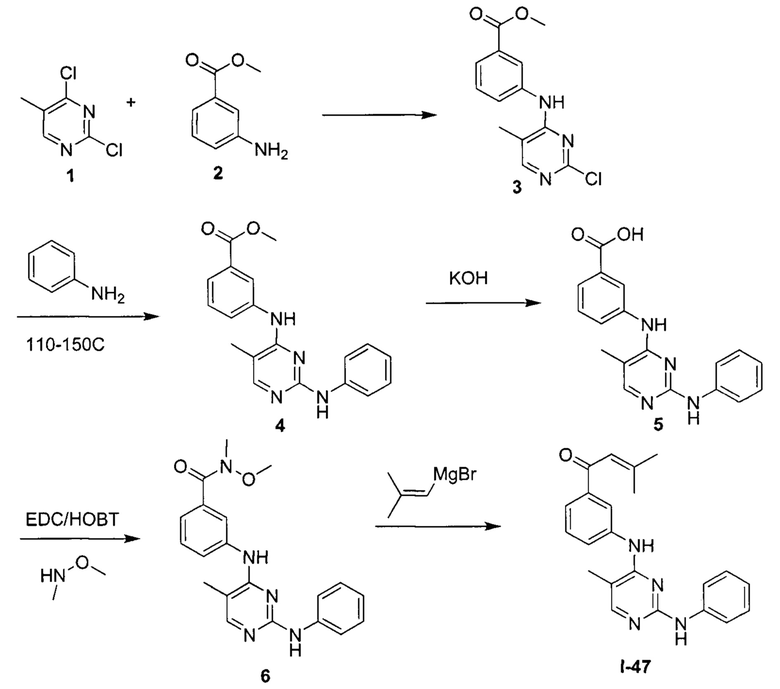
[00474] Соединения 1 и 2 сочетают в присутствии триэтиламина с получением 3. Соединение 3 обрабатывают аналином при повышенной температуре, с получением соединения 4. Омыление соединения 4 при помощи гидроксида калия дает кислотное соединение 5, которое сочетают с N-O-диметилгидроксиламином при помощи EDC с получением соединения 6. Обработка соединения 6 при низкой температуре дает образцовое соединение I-47.
ПРИМЕР 20
[00475] Получение N-(3-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-182
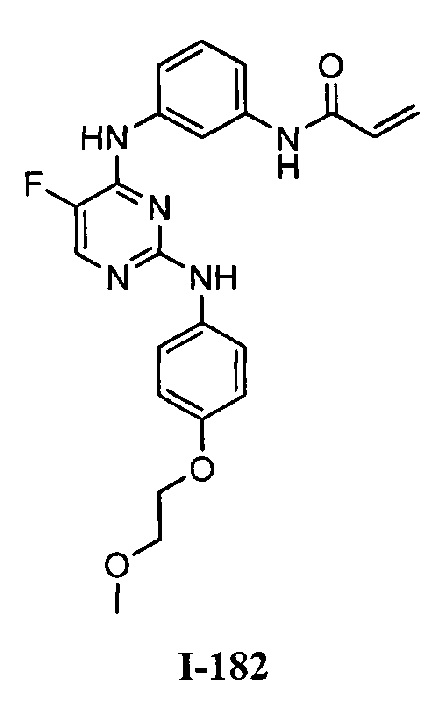
[00476] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
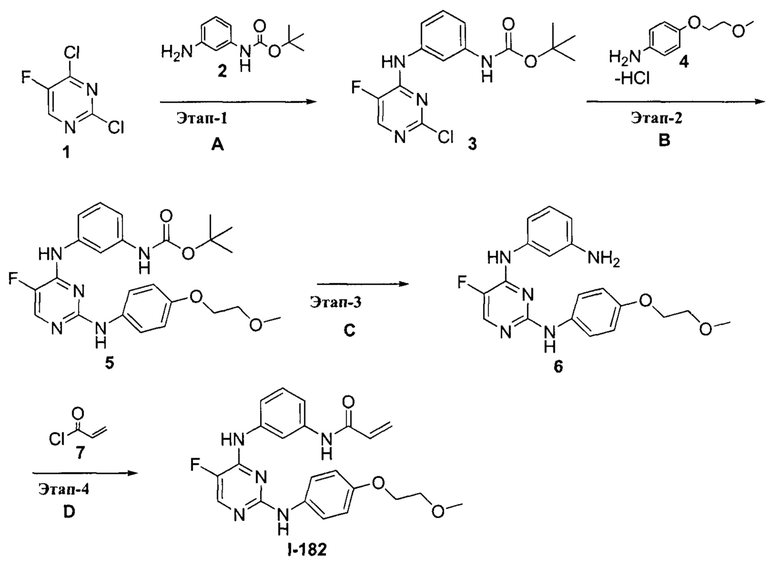
А) 2, DIPEA, ТГФ, с обратным холодильником; В) 4, t-амиловый спирт, НОАс, с обратным холодильником; С) ТФА, ДХМ; D) 7, DIPEA, ТГФ, -10°C.
[00477] Этап-1
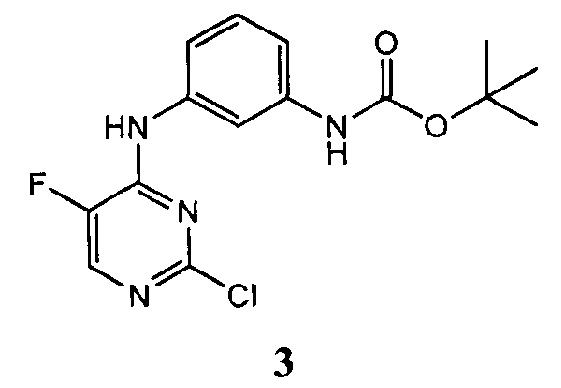
[00478] 1 (800 мг, 4.8 ммоль), 2 (996 мг, 4.8 ммоль) и основание Хунига (948 мкл, 5.75 ммоль) растворяли в ТГФ (20 мл). Реакционную смесь нагревали с обратным холодильником в течение ночи. После охлаждения, разделяли между водой/солевым раствором (10 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения. Титрировали EtOAc и гептаном с получением после фильтрации белого твердого вещества, 1 г. ЖХ/МС (RT=2.03/(M+1)) 339.1.
[00479] Этап-2
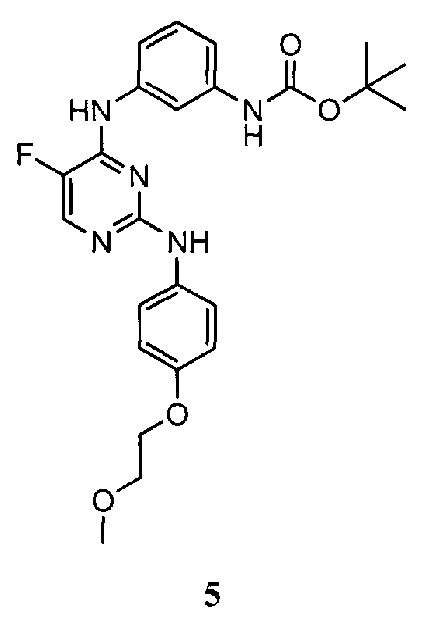
[00480] 3 (800 мг, 2.37 ммоль) и 4 (576 мг, 2.84 ммоль) суспендировали в трет-амиловом спирте (14 мл) и уксусной кислоте (5 капли). Нагревали с обратным холодильником в течение 4х часов. Охлаждали, удаляли растворители роторным испарением. Темное масло разделяли между водой, солевым раствором и ТГФ (по 10 мл каждого), встряхивали и разделяли слои, после чего органическую фазу сушили над сульфатом натрия. Растворитель удаляли методом роторного испарения с получением фиолетового твердого
вещества, 0.55 г. ЖХ/МС (RT=2.997/(М+1)) 470.2. Дополнительные 150 мг продукта без защитной группы (ВОС) кристаллизовали из водного слоя.
[00481] Этап-3
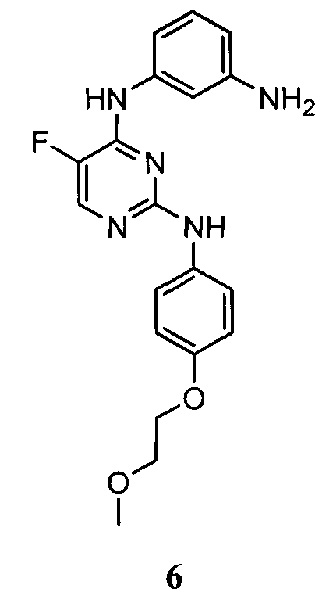
[00482] К раствор 6 (550 мг, 1.17 ммоль) в ДХМ (20 мл) добавляли ТФА (2 мл). Помешивали в течение 30 минут, при комнатной температуре в течение 4 часа, удаляли растворитель методом роторного испарения и разделяли масли с холодным (0°C) насыщенным натрия бикарбонатом (10 мл) и EtOAc (10 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения с получением темного масла. Очищали методом флэш-хроматографии с 20%-100% градиента Гептан/EtOAc при помощи системы комбифлэш с получением 309 мг светло-розового твердого вещества. ЖХ/МС (RT=2.78/(М+1)) 370.2.
[00483] Этап-4
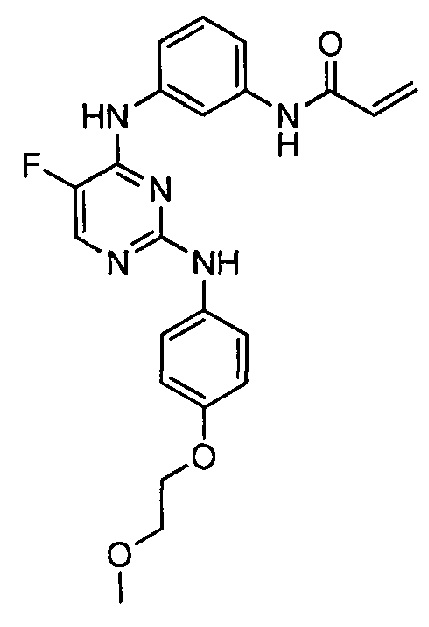
[00484] Раствор 6 (309 мг, 0.84 ммоль) в ТГФ (10 мл) охлаждали в ванне вода/лед-МеОН (-10°C). К нему добавляли 7 (71 мкл, 0.88 ммоль), помешивали в течение 10 минут, затем добавляли основание Хунига (145 мкл, 0.88 ммоль), и помешивали в течение 10 минут. Разделяли между водой/солевым раствором (10 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия. Удаляли растворители методом роторного испарения и толкли с диэтиловым эфиром с получением после фильтрации 285 мг (80%) в форме беловатого твердого вещества. ЖХ/МС (RT=2.79/(М+Н)) 424.2.
ПРИМЕР 21
[00485] Получение N-(3-(2-(3-хлоро-4-(пиридин-2-илметокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-86
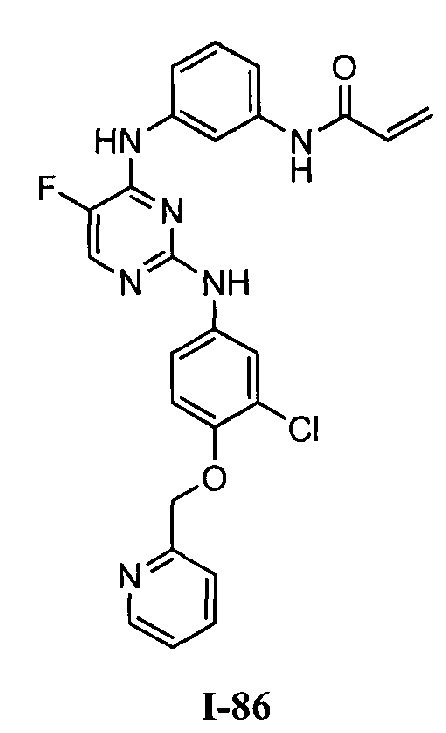
[00486] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20 с использованием 3-хлоро-4-(пиридин-2-илметокси)анилина вместо 4 на этапе 2. ЖХ/МС (RT=2.87/(М+Н)) 491.1.
ПРИМЕР 22
[00487] Получение N-(3-(5-фтор-2-(4-(2-(2-оксипирролидин-1-ил)этокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-92
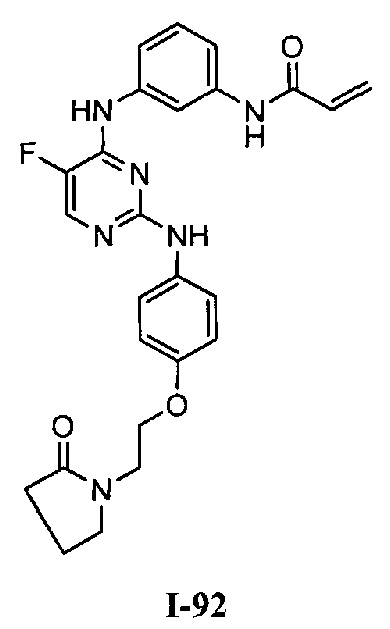
[00488] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 1-(2-(4-аминофенокси)этил)пирролидин-2-она вместо 4 на этапе 2. ЖХ/МС (RT=2.718/(М+Н)) 477.1.
ПРИМЕР 23
[00489] Получение N-(5-фтор-2-(4-(1-гидрокси-2-метилпропан-2-илокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-93
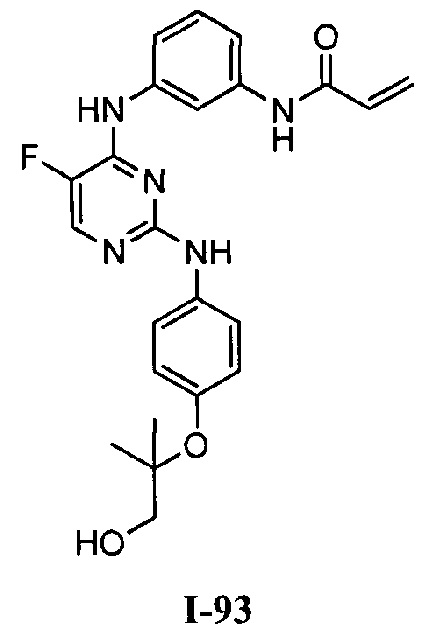
[00490] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 2-(4-аминофенокси)-2-метилпропан-1-ола вместо 4 на этапе 2. ЖХ/МС (RT=2.724/(М+Н)) 438.1.
ПРИМЕР 24
[00491] Получение N-(3-(5-фтор-2-(6-изопропоксипиридин-3-иламино}пиримидин-4-иламино)фенил)акриламида I-172
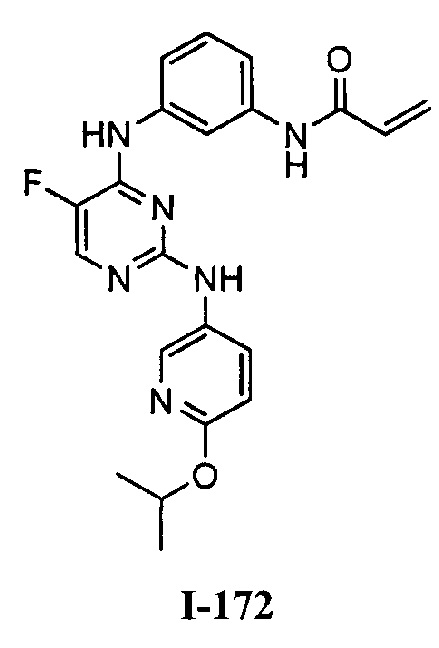
[00492] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 6-изопропоксипиридин-3-амина вместо 4 на этапе 2. ЖХ/МС (RT=2.878/(М+Н)) 409.2.
ПРИМЕР 25
[00493] Получение N-(3-(5-фтор-2-(2-оксоиндолин-5-иламино)пиримидин-4-иламино)фенил)акриламида I-181
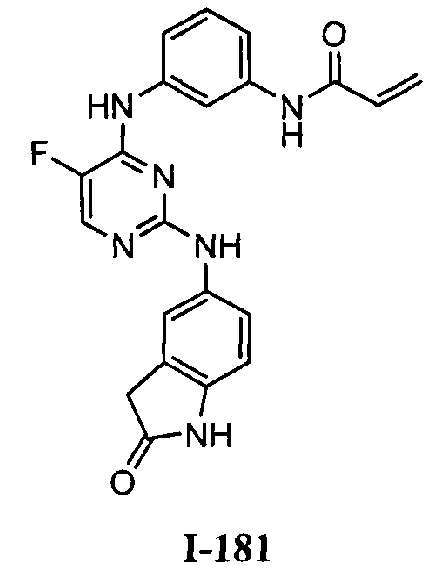
[00494] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 5-аминоиндолин -2-она вместо 4 на этапе 2. ЖХ/МС (RT=2.617/(М+Н)) 405.1.
ПРИМЕР 26
[00495] Получение N-(2-хлоро-5-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-108
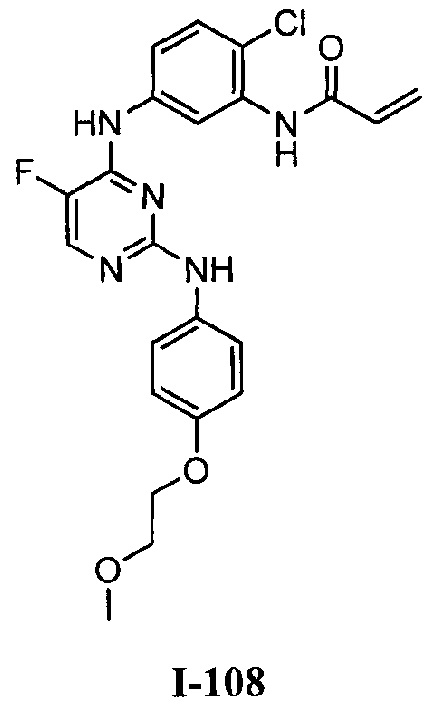
[00496] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием трет-бутил 5-амино-2-хлорофенилкарбамата вместо 4 на этапе 1. ЖХ/МС (RT=2.852/(М+Н)) 458.1.
ПРИМЕР 27
[00497] Получение N-(2-хлоро-5-(5-фтор-2-(6-изопропоксипиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-107
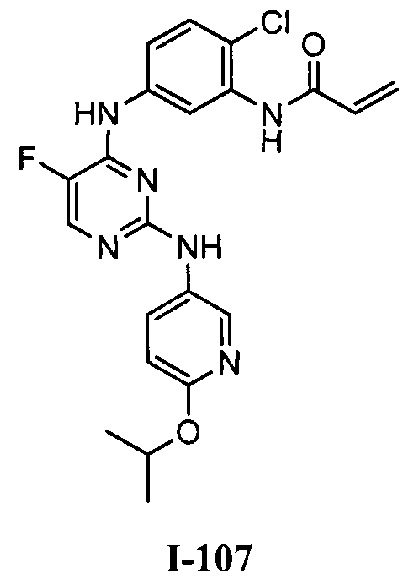
[00498] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием трет-бутил 5-амино-2-фторфенилкарбамата вместо 2 на этапе 1 и 6-изопропоксипиридин-3-амина вместо 4 на этапе 2. ЖХ/МС (RT=2.938/(М+Н)) 443.1.
ПРИМЕР 28
[00499] Получение N-фтор-5-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-87
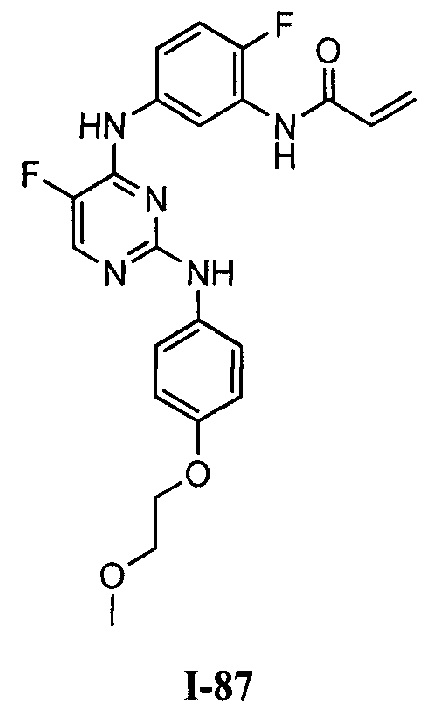
[00500] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием трет-бутил 5-амино-2-фторфенилкарбамата вместо 2 на этапе 1. ЖХ/МС (RT=2.797/(М+Н)) 442.0.
ПРИМЕР 29
[00501] Получение N-(3-(5-фтор-2-(4-((1-метилпиперидин-4-ил)метокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-90
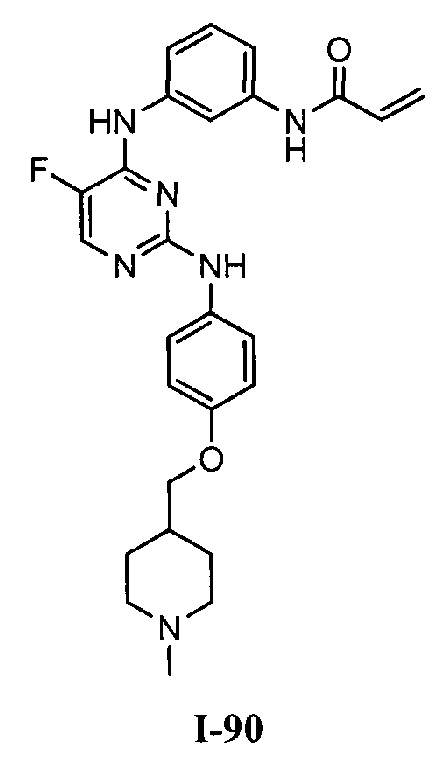
[00502] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
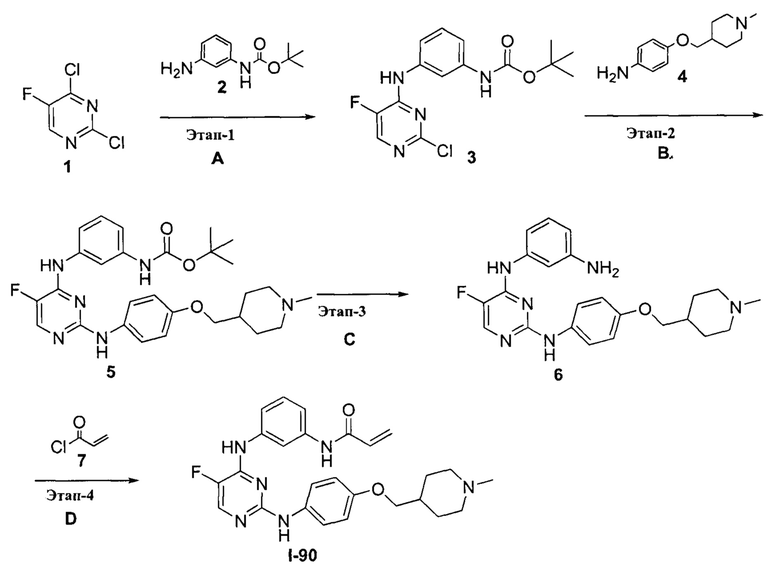
А) 2, DIPEA, ТГФ, с обратным холодильником; В) 4, Pd(OAc)2, X-Phos, CsCO3, диоксан, с обратным холодильником, 12 часов; С) ТФА, ДХМ; D) 7, DIPEA, ТГФ, -10°C.
[00503] Этап-1
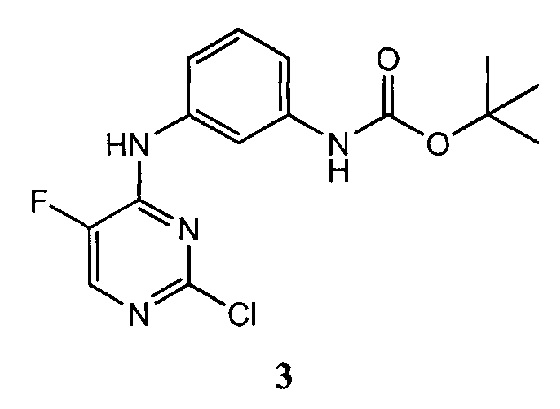
[00504] 1 (800 мг, 4.8 ммоль), 2 (996 мг, 4.8 ммоль) и основание Хунига (948 мкл, 5.75 ммоль) растворяли в ТГФ (20 мл). Реакционную смесь нагревали с обратным холодильником в течение ночи. После охлаждения, разделяли между водой/солевым раствором (10 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения. Титрировали
EtOAc и гептаном с получением после фильтрации белого твердого вещества, 1 г. ЖХ/МС (RT=2.03/(M+1)) 339.1.
[00505] Этап-2
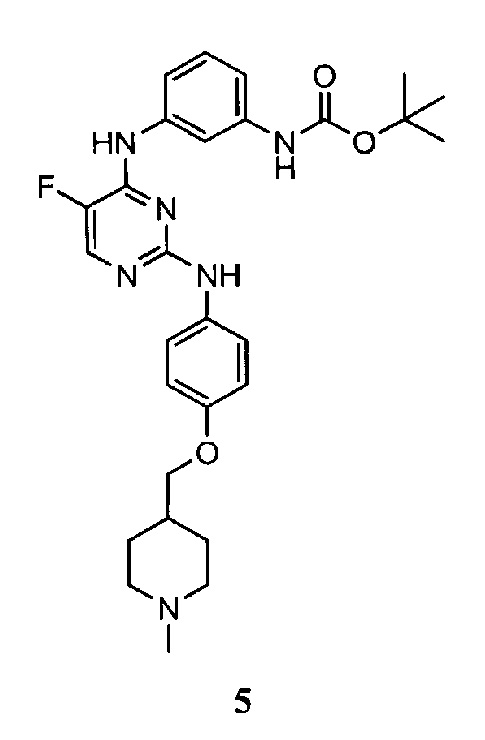
[00506] 3 (205 мг, 0.61 ммоль) и 4 (150 мг, 0.73 ммоль) растворяли в диоксане (4 мл). Дегазировали раствор в течение 1 минуты. Добавляли палладия ацетат (20 мг, 5 моль %), X-Phos лиганд (35 мг, 10 моль %) и CsCO3 (325 мг, 1.2 ммоль) в указанном порядке. Дегазировали суспензию в течение 1 минуты и нагревали с обратным холодильником в атмосфере аргона в течение 12 часов. После охлаждения удаляли растворитель методом роторного испарения. Темное масло разделяли между водой/солевым раствором и EtOAc (по 5 мл каждого), встряхивали, отфильтровывали осадок и разделяли слои фильтрата. Органическую фазу сушили над сульфатом натрия. Растворитель удаляли методом роторного испарения с получением темного масла. Очищали методом флэш-хроматографии с использованием 0-30% градиента Гептан/ EtOAc и получали светло-желтое масло. ЖХ/МС (RT=3,043/(М+1)) 523,2.
[00507] Этап-3
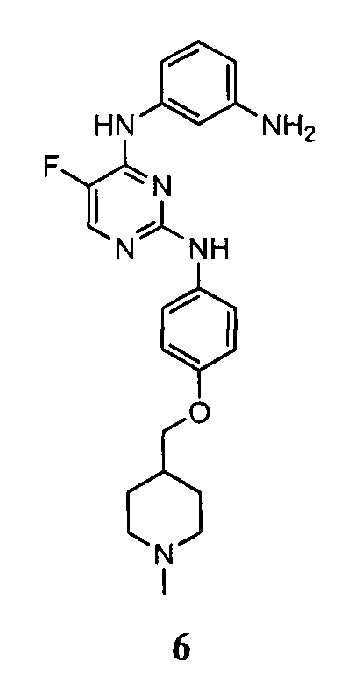
[00508] К раствору 5 (144 мг, 0,27 ммоль) в ДХМ (10 мл) добавляли ТФА (1 мл). Помешивали в течение 30 минут, при комнатной температуре в течение 12 часов, удаляли растворитель методом роторного испарения и разделяли масли с холодным (0°C) насыщенным натрия бикарбонатом (5 мл) и EtOAc (5 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения с получением светло-желтой пены. ЖХ/МС (RT=2,723/(М+1)) 423,1.
[00509] Этап-4
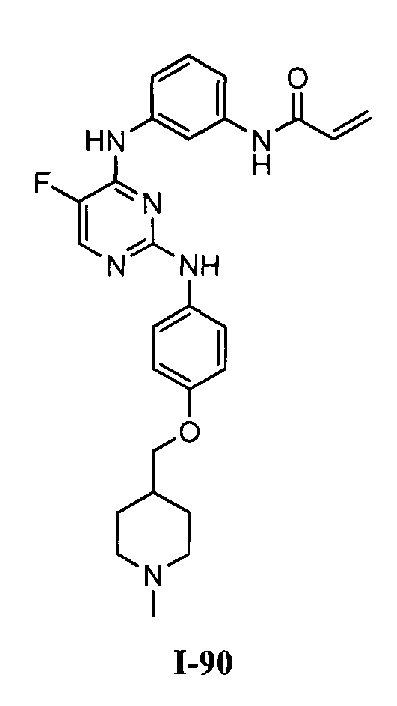
[00510] Раствор 6 (105 мг, 0,25 ммоль) в ТГФ (3 мл) охлаждали в ванне вода/лед-МеОН (-10°C). К нему добавляли 7 (21 мкл, 0,26 ммоль), помешивали в течение 10 минут, затем
добавляли основание Хунига ((51 мкл, 0.26 ммоль), и помешивали в течение 5 минут. Разделяли между водой/солевым раствором (10 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия. Растворитель удаляли методом роторного испарения с положением светло-желтой пены. ЖХ/МС (RT=2,726/(М+Н)) 477.1.
ПРИМЕР 30
[00511] Получение N-(3-(5-фтор-2-(6-((2-метоксиэтил)(метил)амино)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-77
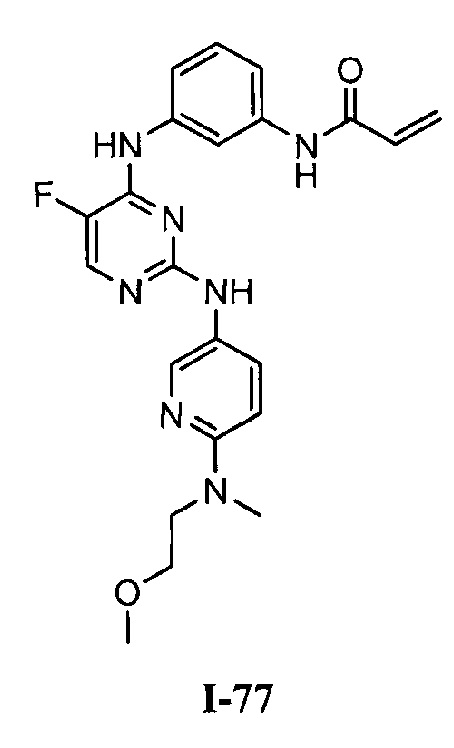
[00512] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 29, с использованием N2-(2-метоксиэтил)-N2-метилпиридин-2,5-диамина вместо 4 на этапе 2. ЖХ/МС (RT=2.739/(М+Н)) 438.1.
ПРИМЕР 31
[00513] Получение 1-(6-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)-2Н-бензо[b][1,4]оксазин-4(3Н)-ил)проп-2-эн-1-она I-194
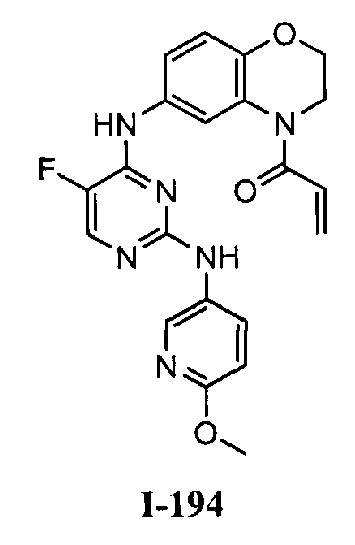
[00514] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
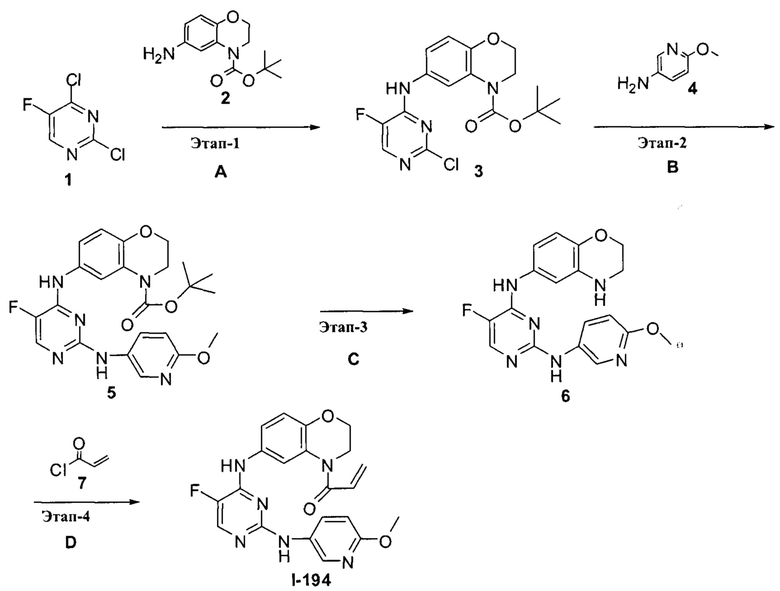
А) 2, DIPEA, ТГФ, с обратным холодильником; В) 4, НОАс, трет-амиловый спирт, с обратным холодильником, 12 часов; С) ТФА, ДХМ; D) 7, DIPEA, ДХМ, NMP, -10°C.
[00515] Этап-1
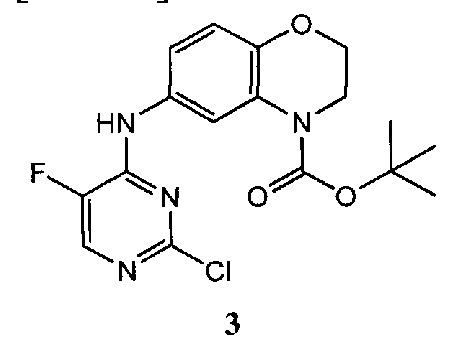
[00516] 1 (186 мг, 1.1 ммоль), 2 (280 мг, 1.1 ммоль) и основание Хунига 220 мкл, 1.3 ммоль) растворяли в ТГФ (6 мл). Реакционную смесь нагревали с обратным холодильником в течение ночи. После охлаждения, разделяли между водой/солевым раствором (6 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения с получением палевого твердого вещества. ЖХ/МС (RT=3,008/(М+1)) 381,1.
[00517] Этап-2
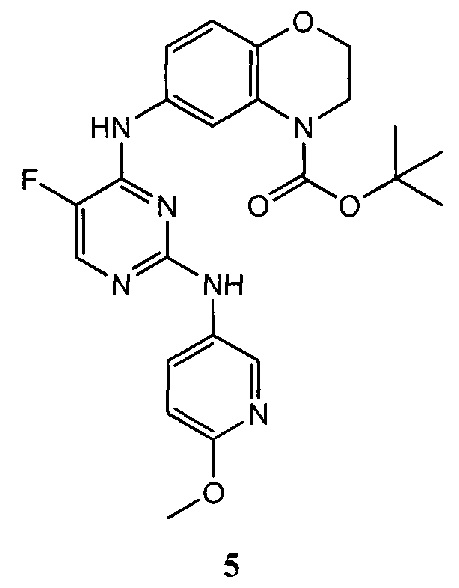
[00518] 3 (215 мг, 0,56 ммоль) и 4 (83 мг, 0,66 ммоль) суспендировали в трет-амиловом спирте (6 мл) и уксусной кислоте (3 капли). Нагревали с обратным холодильником в течение 12 часов. Охлаждали и удаляли растворитель методом роторного испарения. Темное масло разделяли между водой, солевым раствором и EtOAc (по 5 мл каждого), встряхивали и разделяли слои, после чего органическую фазу сушили над сульфатом натрия. Растворитель удаляли методом роторного испарения с получением масла. Очищали методом флэш-хроматографии и использованием 30-70% градиента гептан/этил
ацетат при помощи системы комбифлэш с получением палевого твердого вещества. ЖХ/МС (RT=2,011/(М+1)) 469,2.
[00519] Этап-3
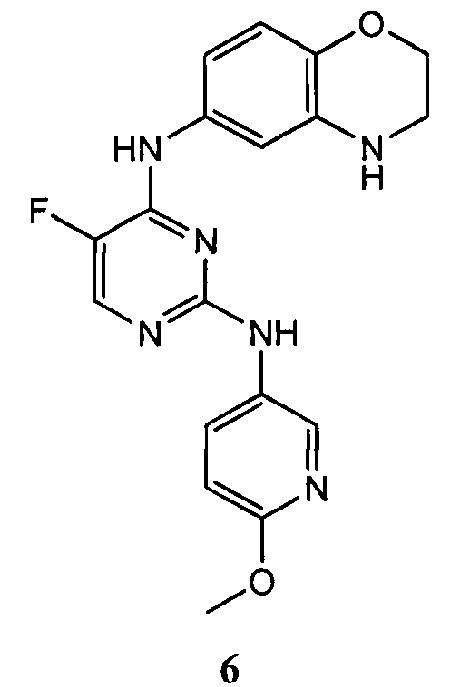
[00520] К раствору 5 (200 мг, 0,43 ммоль) в ДХМ (10 мл) добавляли ТФА (1 мл). Помешивали в течение 30 минут, при комнатной температуре в течение 12 часов, удаляли растворитель методом роторного испарения и разделяли масли с холодным (0°C) насыщенным натрия бикарбонатом (5 мл) и EtOAc (5 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения с получением розового твердого вещества. ЖХ/МС (RT=2,782/(М+1)) 369,1.
[00521] Этап-4
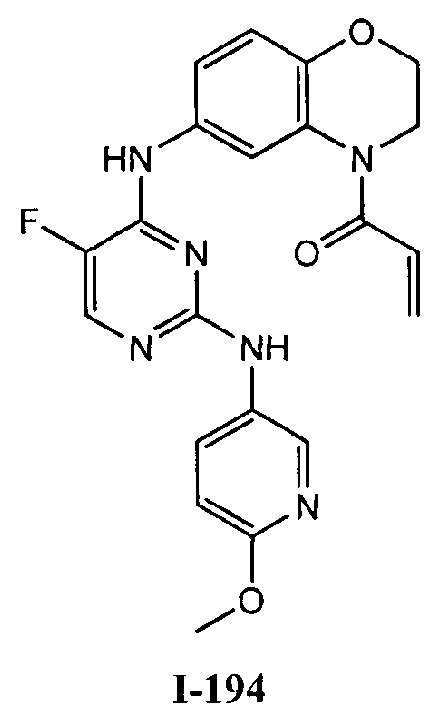
[00522] Раствор 6 (150 мг, 0,41 ммоль) в ДХМ (2 мл) и NMP (0.5 мл) охлаждали в ванне вода/лед-МеОН (-10°C). К нему добавляли 7 (34 мкл, 0,43 ммоль), помешивали в течение 10 минут, затем добавляли основание Хунига (70 мкл, 0,43 ммоль), и помешивали в течение 10 минут. Разделяли между водой/солевым раствором (5 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия. Непосредственно очищали методом флэш-хроматографии с использованием 20-80% градиента гептан/этил ацетат с получением розового твердого вещества. ЖХ/МС (RT=2,8/(М+Н)) 423,1.
ПРИМЕР 32
[00523] Получение 1-(6-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)-2Н-бензо[b][1,4]оксазин-4(3Н)-ил)проп-2-эн-1-она I-141
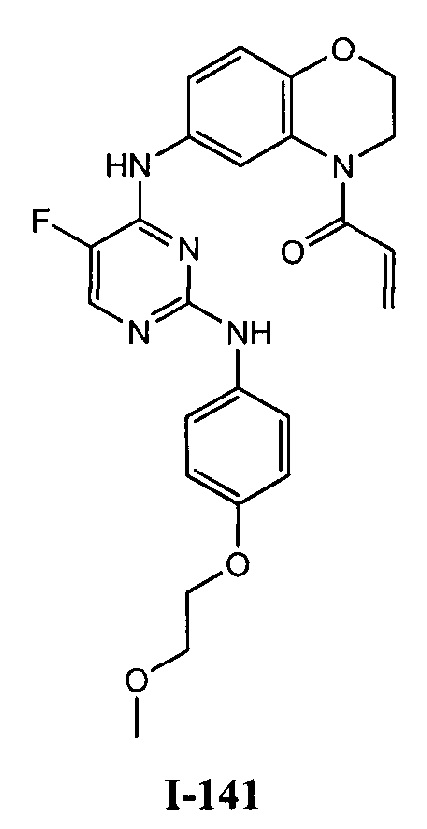
[00524] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 31, с использованием 4-метоксиэтокси)анилина вместо 4 на этапе 2. ЖХ/МС (RT=2.845/(М+Н)) 466,2.
ПРИМЕР 33
[00525] Получение 1-(6-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)индолин-1-ил)проп-2-эн-1-она I-166
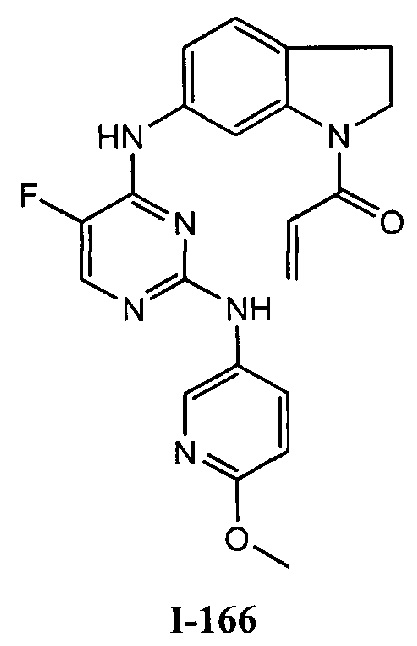
[00526] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 31, с использованием трет-бутил 6-аминоиндолин-1-карбоксилата вместо 2 на этапе 1. ЖХ/МС (RT=2.825/(М+Н)) 407,1.
ПРИМЕР 34
[00527] Получение 1-(5-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)изоиндолин-2-ил)проп-2-эн-1-она I-165
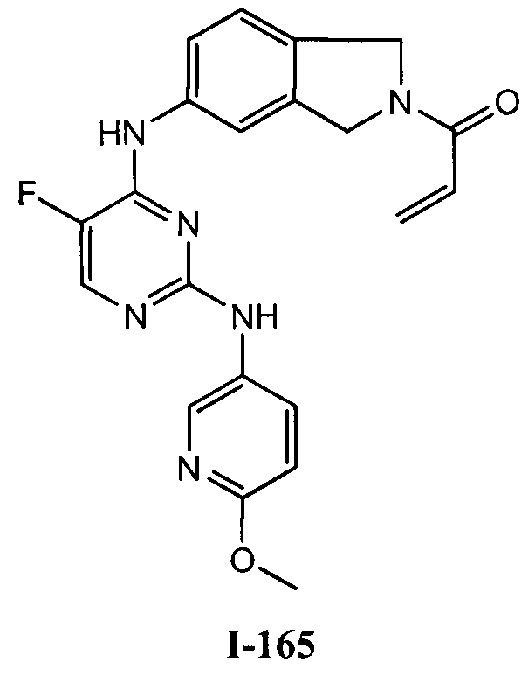
[00528] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 31, с использованием трет-бутил 5-аминоизоиндолин-1-карбоксилата вместо 2 на этапе 1. ЖХ/МС (RT=2.751/(М+Н)) 407,1.
ПРИМЕР 35
[00529] Получение 1-(6-(4-(3-хлорфениламино)-5-фторпиримидин-2-иламино)-2Н-бензо[b][1,4]оксазин-4(3Н)-ил)проп-2-эн-1-она I-149
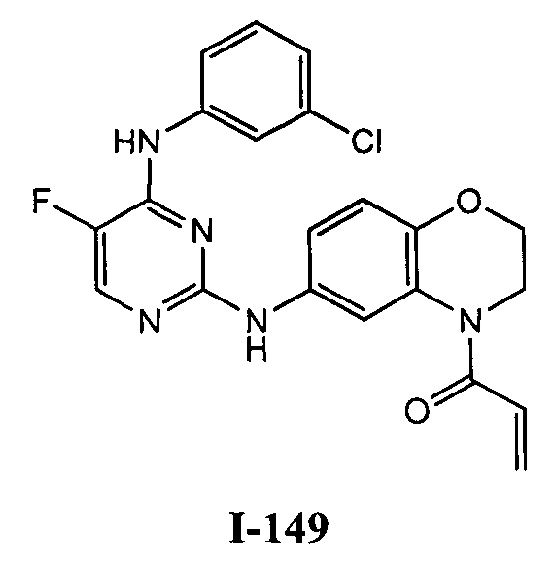
[00530] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
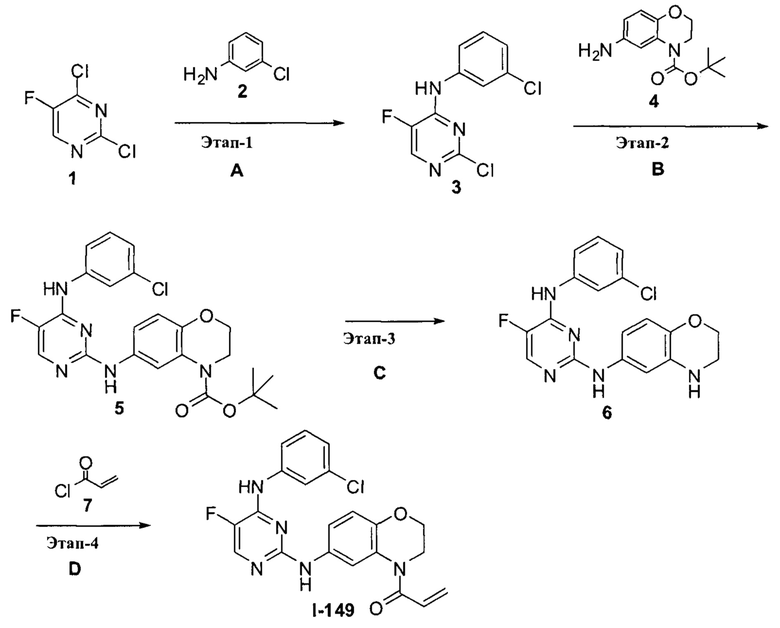
А) 2, DIPEA, ТГФ, с обратным холодильником; В) 4, НОАс, трет-амиловый спирт, с обратным холодильником, 12 часов; С) ТФА, ДХМ; D) 7, DIPEA, ТГФ, -10°C.
[00531] Этап-4
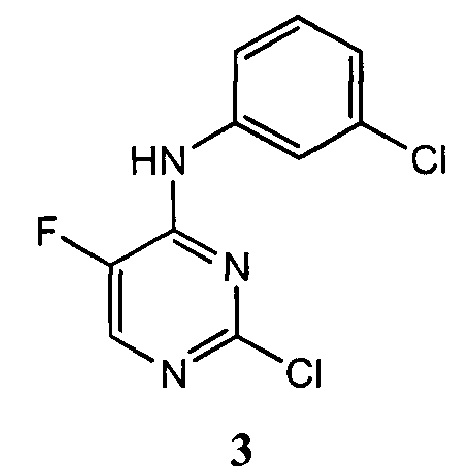
[00532] 1 (484 мг, 2,9 ммоль), 2 (305 мг, 2.9 ммоль) и основание Хунига (526 мкл, 3,5 ммоль) растворяли в ТГФ (10 мл). Реакционную смесь нагревали с обратным холодильником в течение ночи. После охлаждения, разделяли меду водой/солевым раствором (10 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения. Очищали методом флэш-хроматографии и использованием 0-30% градиента гептан/этил ацетат при помощи системы комбифлэш с получением белого твердого вещества. ЖХ/МС (RT=2.03/(М+1)) 339.1.
[00533] Этап-2
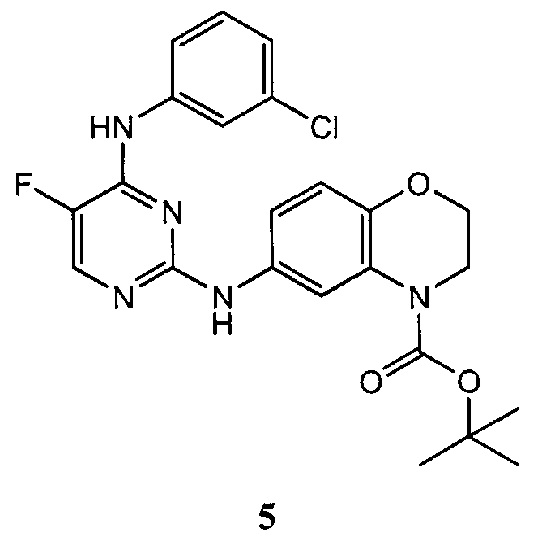
[00534] 3 (150 мг, 0,58 ммоль) и 4 (175 мг, 0,7 ммоль) суспендировали в трет-амиловом спирте (8 мл) и уксусной кислоте (3 капли). Нагревали с обратным холодильником в течение 12 часов. Охлаждали, удаляли растворители роторным испарением. Темное масло разделяли между водой, солевым раствором и EtOAc (по 5 мл каждого), встряхивали и разделяли слои, после чего органическую фазу сушили над сульфатом натрия. Растворитель удаляли методом роторного испарения с получением темного масла. Очищали методом флэш-хроматографии и использованием 0-25% градиента
гептан/этил ацетат при помощи системы комбифлэш с получением белого твердого вещества. ЖХ/МС (RT=2,997/(М+1)) 470,2.
[00535] Этап-3
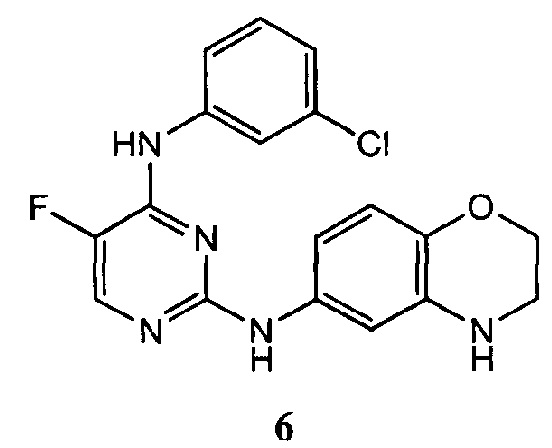
[00536] К раствору 5 (180 мг, 0,38 ммоль) в ДХМ (10 мл) добавляли ТФА (1 мл). Помешивали в течение 30 минут, при комнатной температуре в течение 4 часа, удаляли растворитель методом роторного испарения и разделяли масло между холодным (0°C) насыщенным натрия бикарбонатом (5 мл) и EtOAc (5 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия и удаляли растворитель методом роторного испарения с получением светло-желтого твердого вещества. ЖХ/МС (RT=2,723/(М+1)) 423,1.
[00537] Этап-4
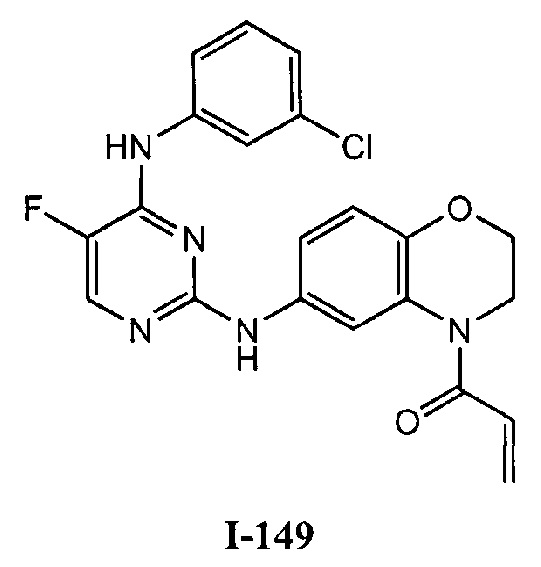
[00538] Раствор 6 (150 мг, 0,4 ммоль) в ТГФ (3 мл) охлаждали в ванне вода/лед-МеОН (-10°C). К нему добавляли 7 (34 мкл, 0.42 ммоль), помешивали в течение 10 минут, затем добавляли основание Хунига (70 мкл, 0.42 ммоль), и помешивали в течение 10 минут. Разделяли между водой/полевым раствором (5 мл), встряхивали и разделяли слои. Органическую фазу сушили над сульфатом натрия. Растворитель удаляли методом роторного испарения с получением светло-желтого твердого вещества. Очищали методом
флэш-хроматографии и использованием 10-50% градиента гептан/этил ацетат при помощи системы комбифлэш с получением белого твердого вещества. ЖХ/МС (RT=2,945/(М+Н)) 426.
ПРИМЕР 36
[00539] Получение 5-(2-(4-акрилоил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-иламино)-5-фторпиримидин-4-иламино)индолин-2-она I-130
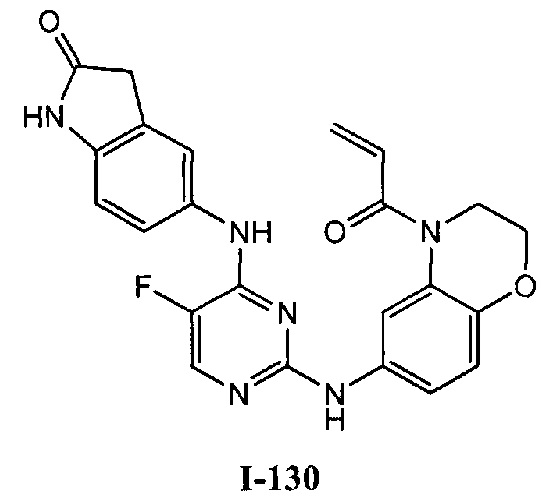
[00540] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 35, с использованием 5-аминоиндолин -2-она вместо 2 на этапе 1. ЖХ/МС (RT=2.673/(М+Н)) 447,1.
ПРИМЕР 37
[00541] Получение 4-(3-акриламидофениламино)-2-(фениламино)пиримидин-5-карбоксиламида I-230
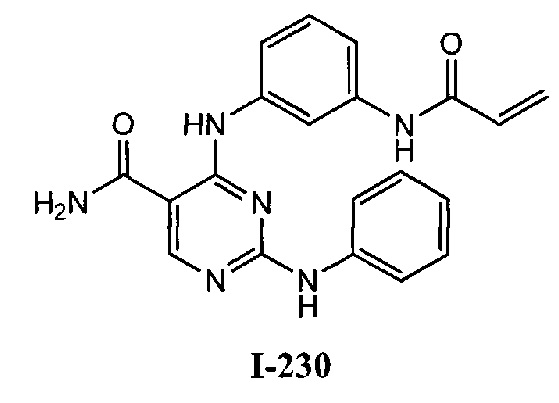
[00542] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
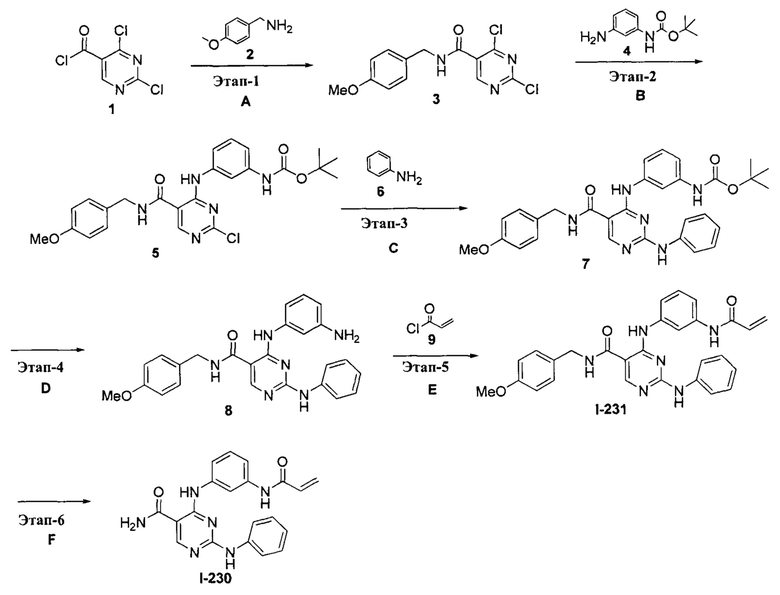
А) 2, NEt3, ДХМ, 0°C до комн. т.; В) 4, DIPEA, ТГФ, комн. т., 12 часов; С) 6, DIPEA, t-амиловый спирт, с обратным холодильником, 4 часа; D) ТФА, ДХМ, комн. т.; Е) 7, NEt3, ТГФ, 0°C; F) ТФА, TFOH, ДХМ, комн. т.
[00543] Этап-1
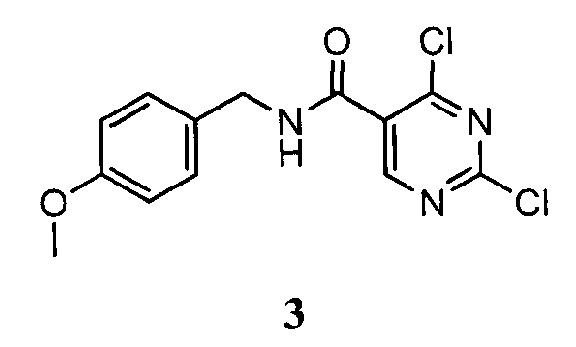
[00544] 1 (500 мг, 2.4 ммоль, полученный из 2,4-дигидрохупиримидин-5-карбоновой кислоты согласно J. Med. Chem. 50: 591 (2007) и US 2007/0072851) растворяли в ДХМ (10 мл) и охлаждали а водяной/ледяной ванне (0°C). добавляли 2 (309 мкл, 2.4 ммоль) и помешивали смесь в течение 10 минут. Добавляли триэтиламин (365 мкл, 2.6 ммоль), после чего смесь оставляли для нагревания при комнатной температуре в течение 30
минут. Методом роторного испарения уменьшали объем растворителя и непосредственно очищали методом флэш-хроматографии с использованием градиента -30% гептан/этил ацетат при помощи системы комбифлэш с получением белого твердого вещества. ЖХ/МС (RT=2,789/(M+1))312.
[00545] Этап-2
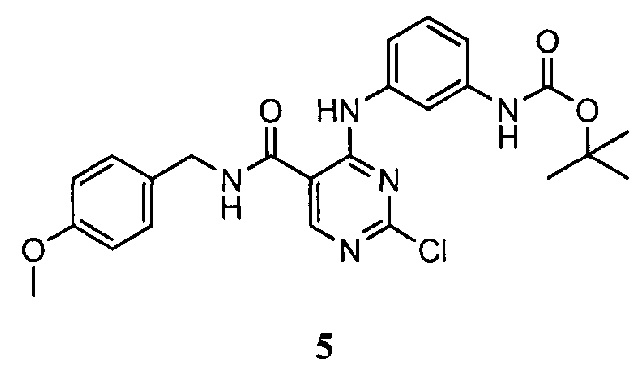
[00546] 3 (170 мг, 0,55 ммоль), 4 (113 мг, 0,55 ммоль) и основание Хунига (108 мкл, 0,65 ммоль) растворяли в ТГФ (6 мл). Помешивали в течение 12 часов. Разделяли между водой/солевым раствором, встряхивали и разделяли слои, после чего органическую фазу сушили над сульфатом натрия. Удаляли растворитель методом роторного испарения с получением после титрования с EtOAc белого твердого вещества. ЖХ/МС (RT=3,123/(М+1))484.
[00547] Этап-3
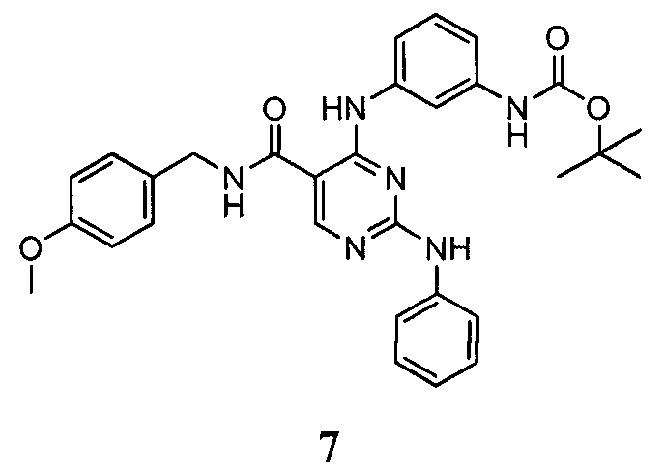
[00548] 5 (230 мг, 0,48 ммоль), 6 (280 мг, 1.1 ммоль) и основание Хунига 94 мкл, 0,57 ммоль) растворяли в t-амиловом спирте (6 мл). Нагревали с обратным холодильником, охлаждали и добавляли к твердой массе воду. Встряхивали, фильтровали и сушили с получением белого твердого вещества. ЖХ/МС (RT=3,182/(М+1)) 541,2.
[00549] Этап-4
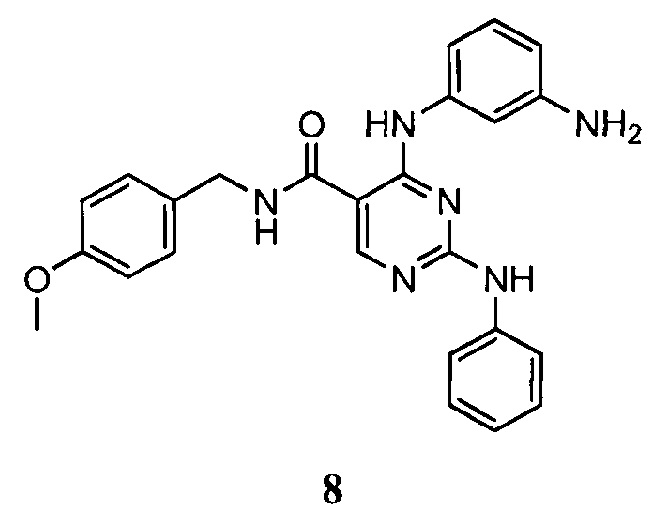
[00550] 7 (180 мг, 0,33 ммоль) суспендировали в ДХМ (10 мл) и обрабатывали ТФА (1 мл). Помешивали в течение ночи при комнатной температуре. Разбавляли ДХМ (40 мл) и промывали NaOH (1N, 25 мл). Встряхивали до образования осадка, фильтровали и сушили с получением белого твердого вещества. ЖХ/МС (RT=2,934/(М+1)) 441,1.
[00551] Этап-5
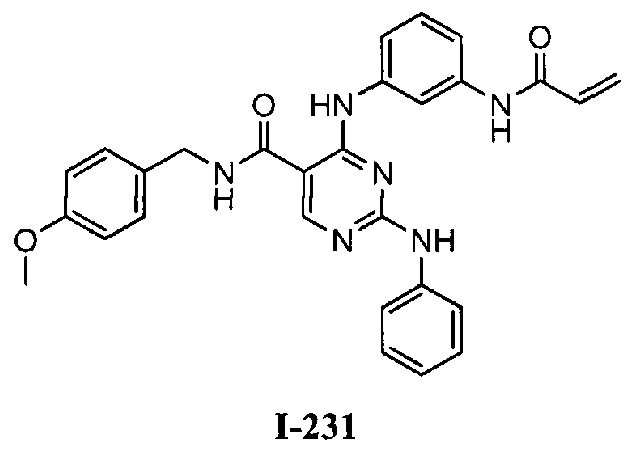
[00552] Суспензию 8 (130 мг, 0,29 ммоль) в ТГФ (6 мл) охлаждали в ванне с водой/льдом-МеОН (0°C). К нему добавляли 9 (25 мкл (плюс дополнительные 5 мкл), 0.38 ммоль (всего)), затем добавляли триэтиламин (43 мкл (плюс дополнительные 11 мкл), 0.38 ммоль (всего)), и помешивали, суммарное время помешивания составляло 1 час. Добавляли воду, встряхивали, отфильтровывали и выбрасывали оставшийся осадок. Фильтрат сушили над сульфатом натрия. Растворитель удаляли методом роторного испарения с получением желтого твердого вещества. Очищали методом флэш-хроматографии и использованием 0-25% градиента гептан/этил ацетат при помощи системы комбифлэш с получением белого твердого вещества. ЖХ/МС (RT=2,964/(М+Н)) 495,1.
[00553] Этап-6
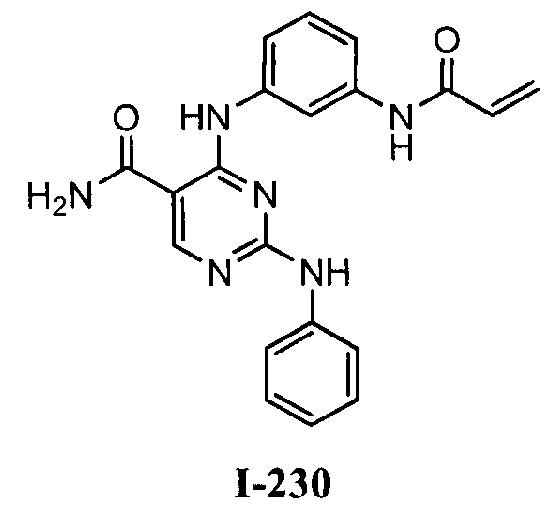
[00554] К суспензии I-231 (30 мг, 0.061 ммоль) в ДХМ (4 мл) добавляли ТФА (200 мкл) и трифлатную кислоту (68 мкл, 0.61 ммоль). Помешивали при комнатной температуре в течение 1 часа. Удаляли растворитель методом роторного испарения при пониженном давлении и разделяли между холодным (0°C) насыщенным натрия бикарбонатом (10 мл) и EtOAc (10 мл), встряхивали и разделяли слои. Сушили органический слой над сульфатом натрия и удаляли растворитель методом роторного испарения, титрировали с диэтиловым эфиром с получением белого твердого вещества. ЖХ/МС (RT=2,715/(М+Н)) 375,1.
ПРИМЕР 38
[00555] Получение 4-(3-акриламидофениламино)-N-фенил-2-(фениламино)пиримидин-5-карбоксиламида I-222
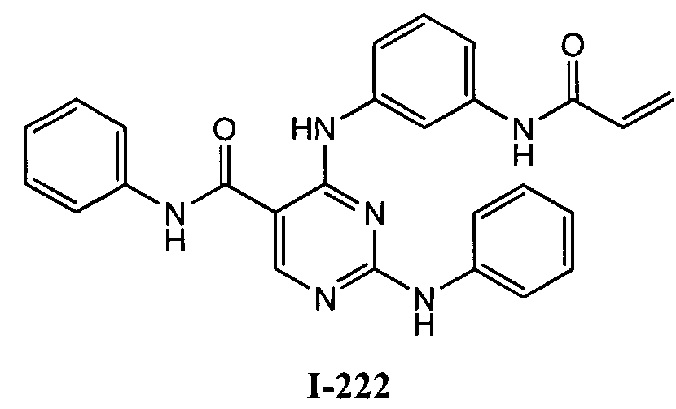
[00556] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 37, с использованием анилина вместо 2 на этапе 1, и пропуская этап 6. ЖХ/МС (RT=2.991/(М+Н)) 451.2.
ПРИМЕР 39
[00557] Получение 4-(3-акриламидофениламино)-N-циклопропил-2-(фениламино)пиримидин-5-карбоксиламида I-221
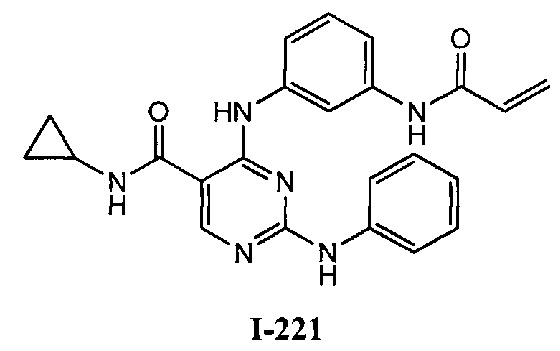
[00558] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 37, с использованием циклопропиламина вместо 2 на этапе 1, и пропуская этап 6. ЖХ/МС (RT=2.838/(М+Н)) 415.2.
ПРИМЕР 40
[00559] Получение 4-(3-акриламидофениламино)-2-(3-метоксифениламино)пиримидин-5-карбоксиламида I-210
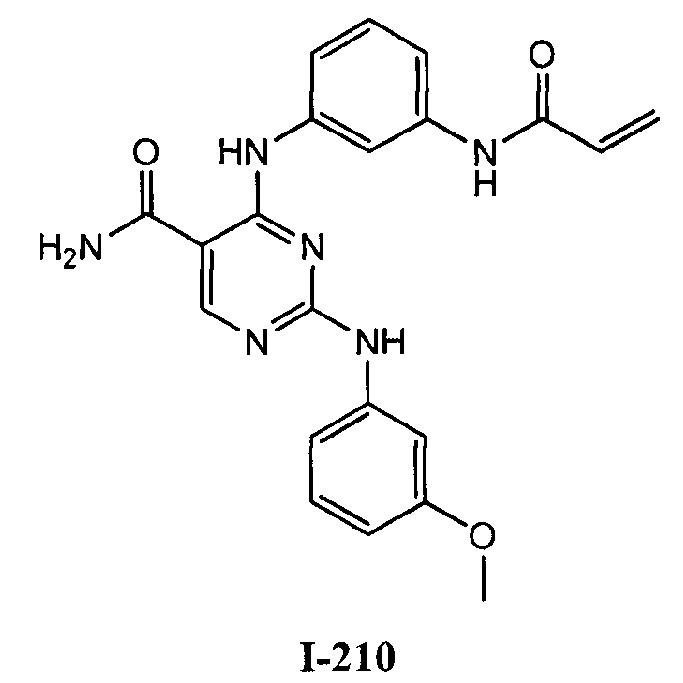
[00560] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 37, с использованием 3-метоксиамина вместо 6 на этапе 3. ЖХ/МС (RT=2.743/(М+Н)) 405.1.
ПРИМЕР 41
[00561] Получение 4-(3-акриламидофениламино)-2-(6-метоксипиридин-3-иламино)пиримидин-5-карбоксиламида I-209
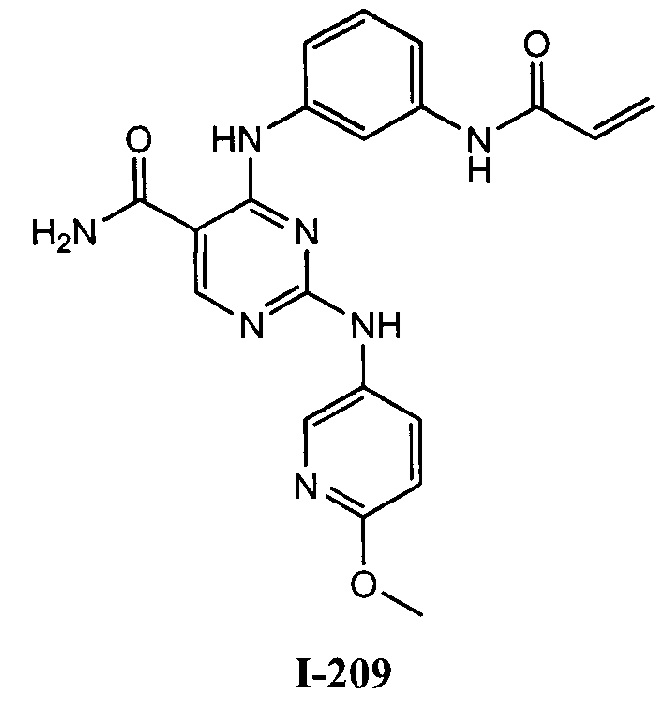
[00562] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 37, с использованием 6-метоксипиридин-3-амина вместо 6 на этапе 3. ЖХ/МС (RT=2.657/(М+Н)) 406.2.
ПРИМЕР 42
[00563] Получение 1-{6-[5-Ацетил-2-(6-метокси-пиридин-3-иламино)-пиримидин-4-иламино]-2,3-дигидро-бензо[1,4]оксазин-4-ил}-пропенона I-170
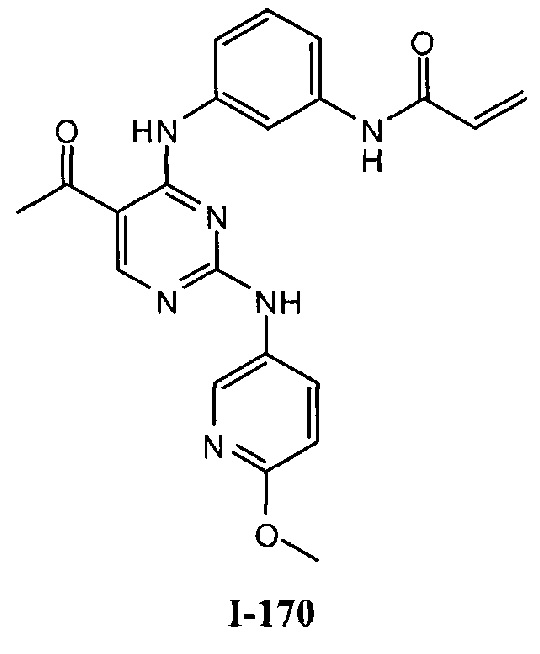
[00564] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
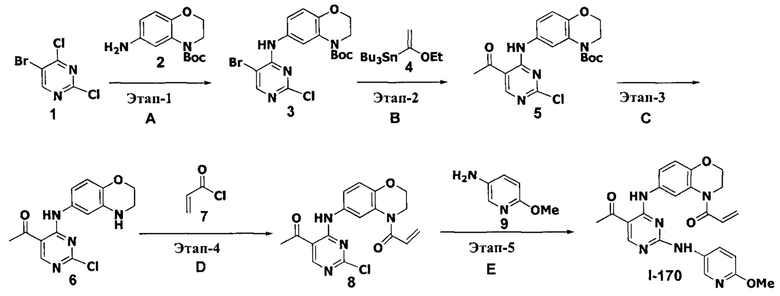
А) 2, DIPEA, ТГФ, 70°C, 16 часов; В) (а) 4, PdCl2(PPh3)2, ДМФ, 70°C; (b) 1N HCl, ацетон, 60°C, 15 минут; С) HCl/диоксан, ДХМ; D) 7, DIPEA, NMP, ДХМ, -20°C до комн. т.; Е) 9, pTsOH, диоксан, 100°C, 15 минут.
[00565] Этап-1
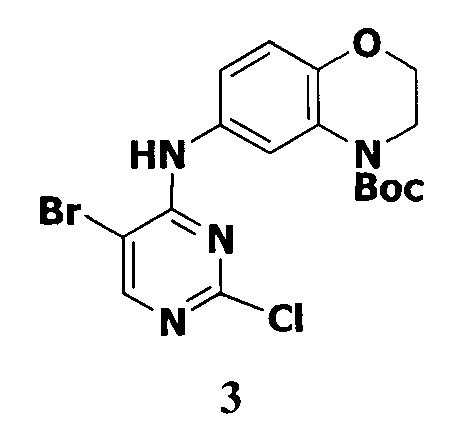
[00566] Смесь 499 мг 1 (2.19 ммоль), 547 мг 2 (2.19 ммоль), и 500 мкл N,N-диизопропилэтиламина в 20 мл безводного тетрагидрофурана грели при температуре 70°C в течение ночи. После охлаждения, реакционную смесь концентрировали и обрабатывали водным EtOAc 50 мД, раствором натрия бикарбоната 20 мл, солевым раствором, и сушили над безводным сульфатом натрия. После фильтрации и концентрации, остаток пропускали через короткий картридж с оксидом кремния, в качестве элюента брали гептан/EtOAc (объем/объем 3/1), с получением 815 мг слегка желтоватого твердого вещества (84%). ЖХ-МС: m/z 441.0 (ES+), 439.0 (ES-).
[00567] Этап-2
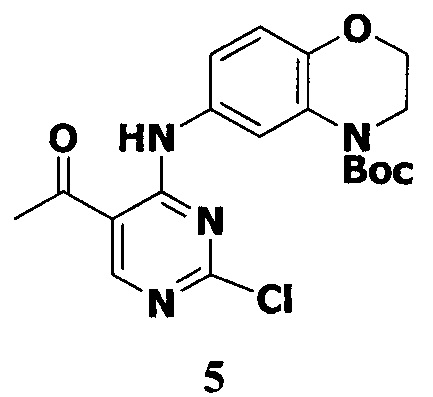
[00568] Раствор промежуточного соединения 3 (815 мг, 1.85 ммоль), 4 (740 мг, 1.1 экв.), 27 мг дихлоробис(трифенилфосфин)палладия (II) (2% моль) в 6 мл безводного ДМФ продували азотом в течение 30 минут. Реакционную смесь грели при температуре 70°C в течение ночи. Анализ методом ЖХ-МС показал 70% превращение. После охлаждения, добавляли 30 мл этил ацетата и 760 мл калия фторида в 5 мл водя, и помешивали реакционную смесь при комнатной температуре по крайней мере в течение 2 часа. Белый осадок отфильтровывали, и разделяли органический слой, промывали водой, солевым раствором, и сушили над безводным сульфатом натрия.
[00569] Фильтровали и концентрировали, затем остаток растворяли в 20 мл ацетона, после чего добавляли 3 мл 1.0N водного раствора HCl. Реакционную смесь грели при температуре 60°C в течение 15 минут, и концентрировали при пониженном давлении. Обрабатывали 50 мл EtOAc, 10 мл насыщенным раствором натрия бикарбоната, солевым раствором, безводным сульфатом натрия. Концентрировали, очищали остаток методом колоночной флэш-хроматографии на силикагеле, с получением 405 мг желтого твердого вещества (70% состоящего из исходного результата), а также промежуточного соединения 3 183 мг. ЖХ-МС: m/z 405.1 (ES+), 403.1 (ES-).
[00570] Этап-3
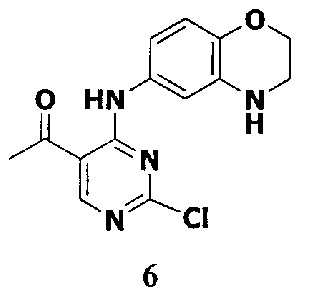
[00571] К смеси 1.28 г промежуточного соединения I-2 в 10 мл Дихлорметана, добавляли 10 мл 4.0 N HCl в диоксане. Перемешивали при комнатной температуре в
течение ночи, удаляли растворитель, и сушили остаток под вакуумом. ЖХ-МС: m/z 305.1 (ES+), 303.1 (ES-).
[00572] Этап-4
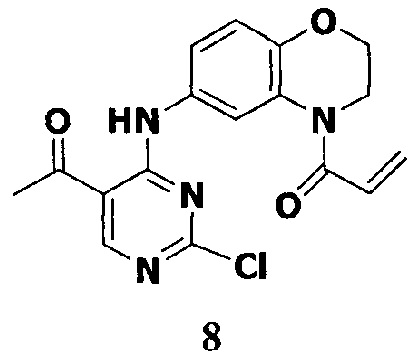
[00573] В атмосфере азота к смеси промежуточного соединения 6, полученного выше, 1 мл DIPEA в 10 мл NMP и 10 мл дихлорметана при температуре -20°C, добавляли 275 мкл 7 (1.1 эквив.). Реакция продолжалась 5 минут, после чего ее гасили 1 мл изопропилового спирта. Реакционную смесь подогревали до комнатной температуры и экстрагировали 100 мл EtOAc, промывали водой, 10 мл × 2, солевым раствором, сушили над сульфатом натрия. Фильтровали и концентрировали, после чего остаток очищали метдом колоночной флэш-хроматографии с гептанами/EtOAc (объем/объем 2/3) в качестве подвижной фазы, с получением желтого твердого вещества 1-3 450 мг (40%). ЖХ-МС: m/z 359.1 (ES+), 357.1 (ES-).
[00574] Этап-5
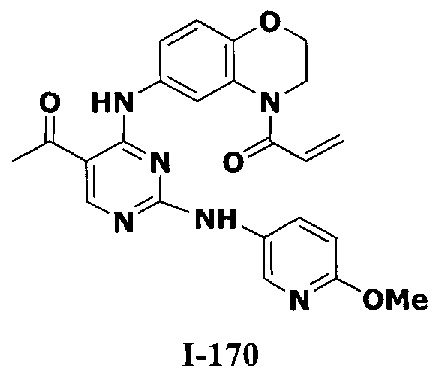
[00575] Смесь 30 мг промежуточного продукта 8 (84 мкМоль) и 13 мг 9 (1.2 эквив) в 1 мл раствора 0.08 М p-TsOH диоксана грели при температуре 100°C в течение 15 минут. Охлаждали, после чего реакционную смесь обрабатывали 50 мл EtOAc, водным натрия бикарбонатом, солевым раствором, и сушили над безводным сульфатом натрия. Концентрировали и остаток очищали методом колоночной хроматографии на силикагеле с
элюентом гептан/EtOAc (объем/объем 1/4) с получением 22.8 бледного белого твердого вещества (61%). ЖХ-МС: m/z=447.1 (ES+), 445.2 (ES-).
ПРИМЕР 43
[00576] Получение 1-{6-[5-Ацетил-2-(4-морфолин-4-ил-фениламино)-пиримидин-4-иламино]-2,3-дигидро-бензо[1,4]оксазин-4-ил}-пропенона I-169
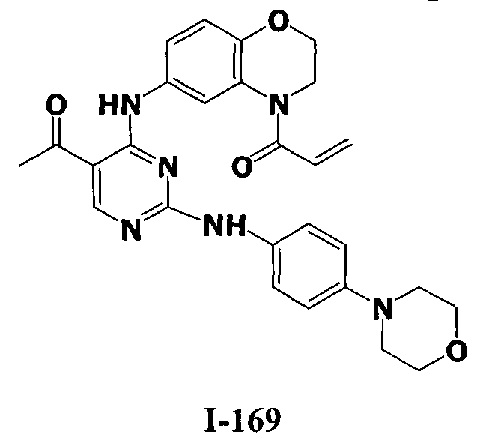
[00577] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 4-морфолин-4-ил-фениламина вместо 9 на этапе 5. ЖХ-МС: m/z 501.1 (ES+), 499.2 (ES-).
ПРИМЕР 44
[00578] Получение 1-{6-[5-Ацетил-2-(6-морфолин-4-ил-пиридин-3-иламино)-пиримидин-4-иламино]-2,3-дигидро-бензо[1,4]оксазин-4-ил}-пропенона I-168
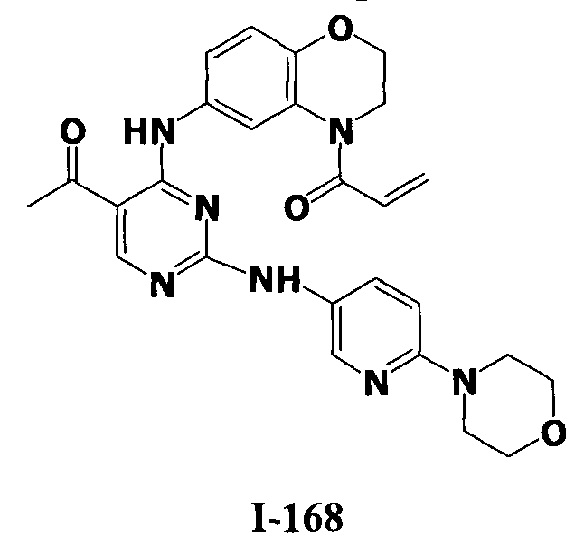
[00579] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 3-амино-[6-морфолин-4-ил]-пиридина вместо 9 на этапе 5. ЖХ-МС: m/z 502.2 (ES+), 500.3 (ES-).
ПРИМЕР 45
[00580] Получение 1-{6-[5-Ацетил-2-(1-метил-1Н-индазол-6-иламино)-лиримидин-4-иламино]-2,3-дигидро-бензо[1,4]оксазин-4-ил}-пропенона I-154
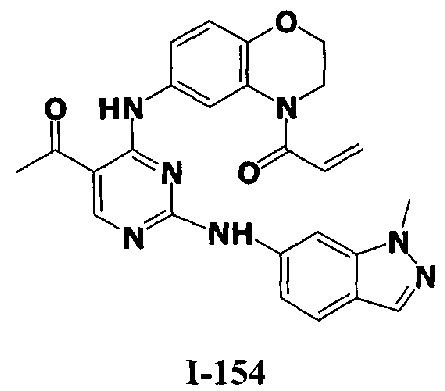
[00581] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 1-метил-1Н-индазол-6-иламина вместо 9 на этапе 5. ЖХ-МС: m/z 470.1 (ES+), 468.1 (ES-).
ПРИМЕР 46
[00582] Получение 1-{6-[5-Ацетил-2-(1Н-индазол-6-иламино)-пиримидин-4-иламино]-2,3-дигидро-бензо[1,4]оксазин-4-ил}-пропенона I-153
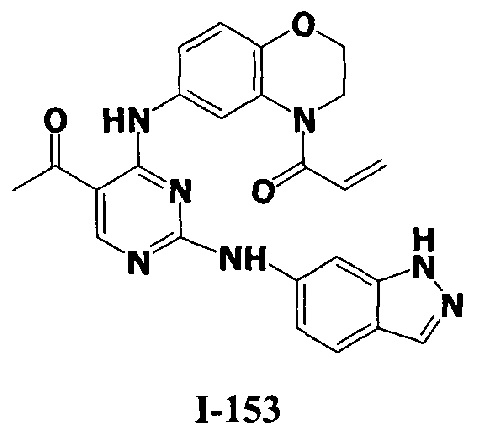
[00583] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 1H-индазол-6-иламина вместо 9 на этапе 5. ЖХ-МС: m/z 456.1 (ES+), 454.2 (ES-).
ПРИМЕР 47
[00584] Получение 1-{4-[5-Ацетил-4-(4-акрилоил-3,4-дигидро-2Н-бензо[1,4]оксазин-6-иламино)-пиримидин-2-иламино]-фенил}-пирролидин-2-она I-152
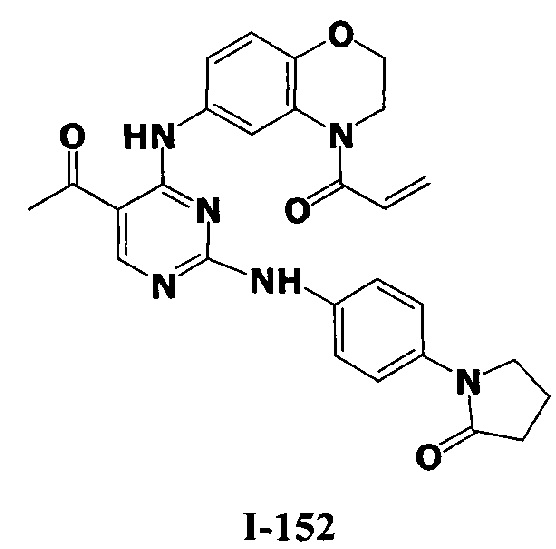
[00585] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 1-(4-амино-фенил)-пирролидин-2-она вместо 9 на этапе 5. ЖХ-МС: m/z 456.1 (ES+), 454.2 (ES-).
ПРИМЕР 48
[00586] Получение 1-(6-{5-Ацетил-2-[4-(2-метокси-этокси)-фениламино]-пиримидин-4-иламино}-2,3-дигидро-бензо[1,4]оксазин-4-ил)-пропенона I-150
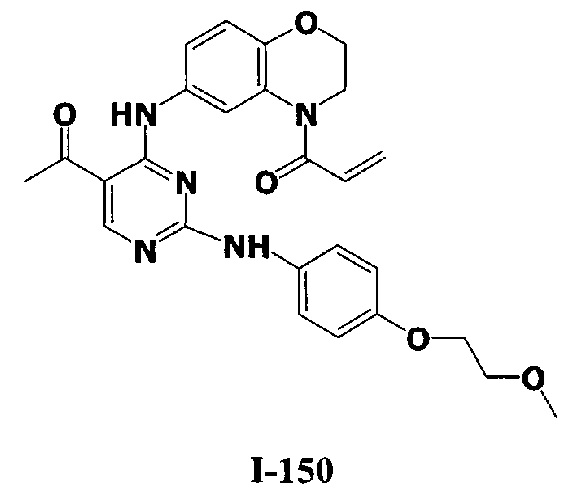
[00587] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 4-(2-метокси-этокси)-фениламина вместо 9 на этапе 5. ЖХ-МС: m/z 490.2 (ES+), 488.3 (ES-).
ПРИМЕР 49
[00588] Получение 5-[5-Ацетил-4-(4-акрилоил-3,4-дигидро-2Н-бензо[1,4]оксазин-6-иламино)-пиримидин-2-иламино]-1,3-дигидро-индол-2-она I-129
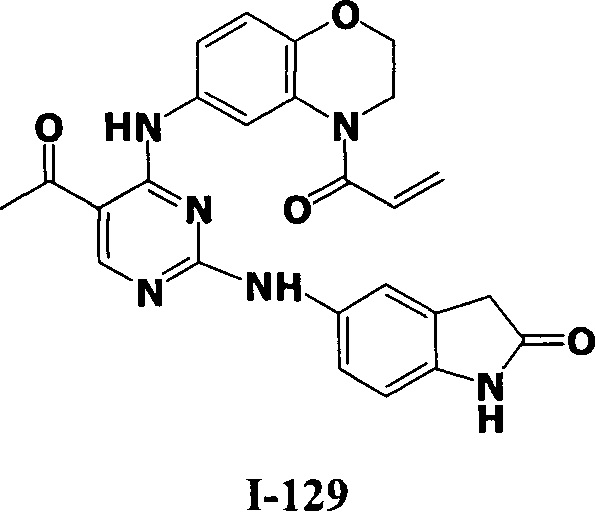
[00589] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 5-амино-1,3-дигидро-индол-2-она вместо 9 на этапе 5. ЖХ-МС: m/z 471.1 (ES+), 469.2 (ES-).
ПРИМЕР 50
[00590] Получение 1-(6-{5-Ацетил-2-[6-(2-гидрокси-этокси)-пиридин-3-иламино]-пиримидин-4-иламино}-2,3-дигидро-бензо[1,4]оксазин-4-ил)-пропенона I-128
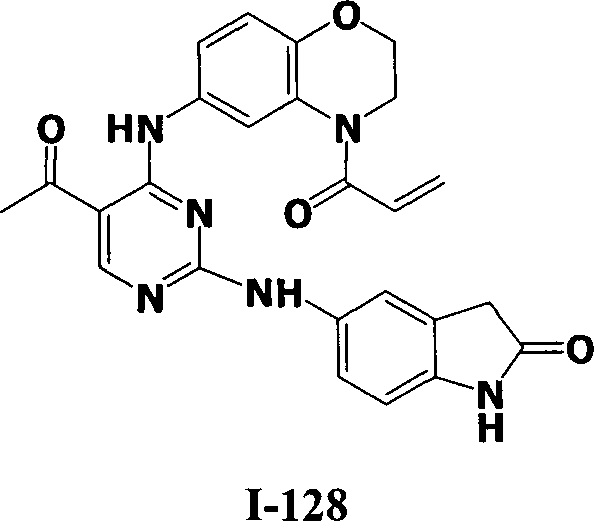
[00591] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 2-(5-амино-пиридин-2-илокси)-этанола вместо 9 на этапе 5. ЖХ-МС: m/z 477.1 (ES+), 475.2 (ES-).
ПРИМЕР 51
[00592] Получение N-{3-[5-Ацетил-2-(6-метокси-пиридин-3-иламино)-пиримидин-4-иламино]-фенил}-акриламида I-189
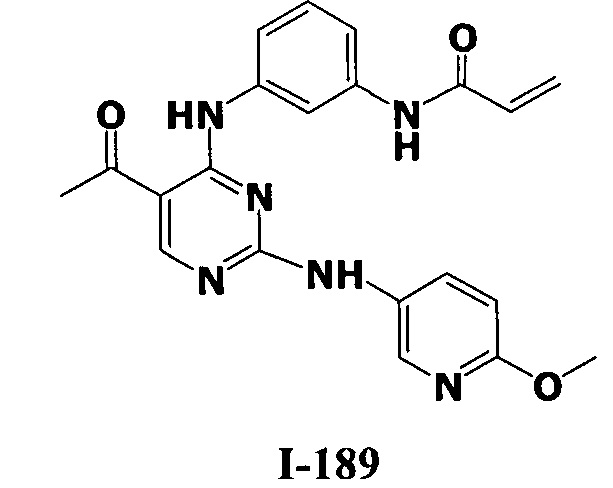
[00593] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием трет-бутил 3-аминофенилкарбамата вместо 9 на этапе 5. ЖХ/МС: m/z 405.1 (ES+), 403.2 (ES-).
ПРИМЕР 52
[00594] Получение N-{3-[5-Ацетил-2-(6-метокси-пиридин-3-иламино)-пиримидин-4-илокси]-фенил}-акриламида I-188
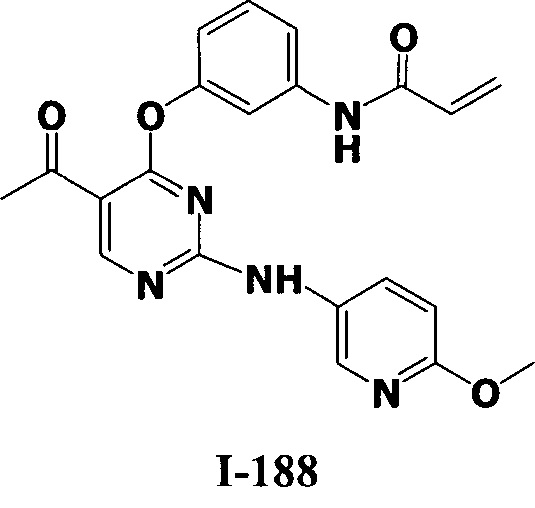
[00595] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием трет-бутил 3-гидроксифенилкарбамата вместо 2 на этапе 1 и 5-амино-2-метоксипиридина вместо 9 на этапе 5. ЖХ/МС: m/z 406.2 (ES+), 404.1 (ES-).
ПРИМЕР 53
[00596] Получение 1-{3-[5-Ацетил-2-(6-метокси-пиридин-3-иламино)-пиримидин-4-иламино]-азетидин-1-ил}-пропенона I-187
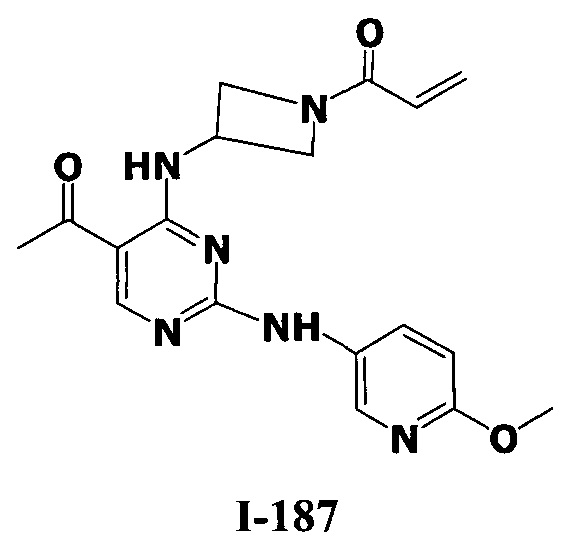
[00597] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 3-амино-N-Boc-азетидина вместо 9 на этапе 5. ЖХ/МС: m/z 369.1 (ES+), 367.2 (ES-).
ПРИМЕР 54
[00598] Получение N-(3-{5-Ацетил-2-[4-(2-метокси-этокси)-фениламино]-пиримидин-4-иламино}-фенил)-акриламида I-124
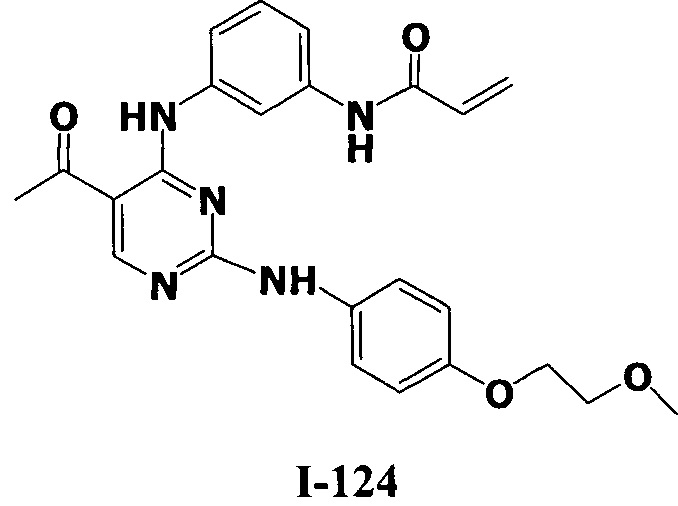
[00599] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием трет-бутил 3-аминофенилкарбамата вместо 2 на этапе 1 и 4-(2-метокси-этокси)-фениламина вместо 9 на этапе 5. ЖХ/МС: m/z 448.2 (ES+), 446.3 (ES-).
ПРИМЕР 55
[00600] Получение N-(3-{5-Ацетил-2-[6-(2-метокси-этокси)-пиридин-3-иламино]-пиримидин-4-иламино}-фенил)-акриламида I-122
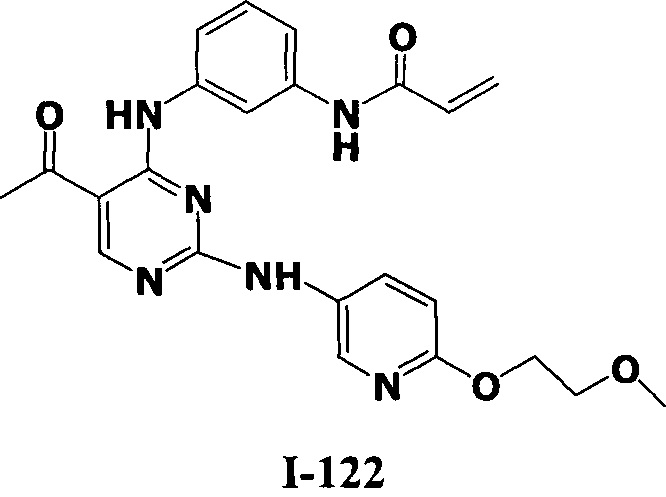
[00601] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием трет-бутил 3-аминофенилкарбамата вместо 2 на этапе 1 и 6-(2-метокси-этокси)-пиридин-3-иламину вместо 9 на этапе 5. ЖХ/МС: m/z 449.2 (ES+), 447.1 (ES-).
ПРИМЕР 56
[00602] Получение N-(3-{5-Ацетил-2-[6-(2-гидрокси-этокси)-пиридин-3-иламино]-пиримидин-4-иламино}-фенил)-акриламид I-121
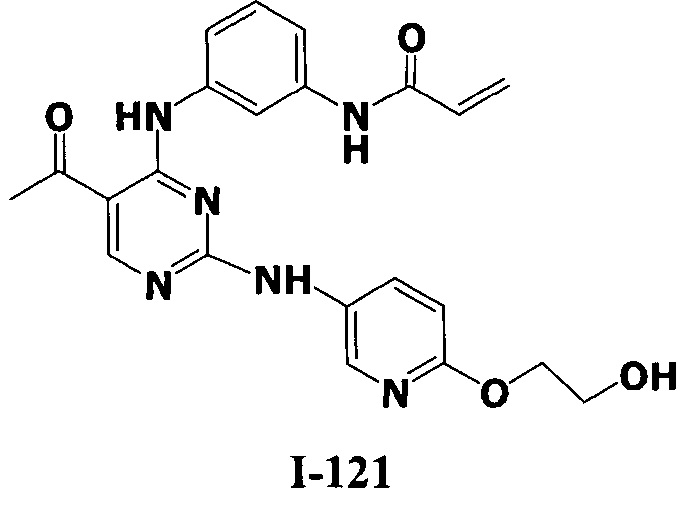
[00603] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием трет-бутил 3-аминофенилкарбамата вместо 2 на этапе 1 и 2-(5-амино-пиридин-2-илокси)-этанола вместо 9 на этапе 5. ЖХ/МС: m/z 435.1 (ES+), 433.2 (ES-).
ПРИМЕР 57
[00604] Получение 4-(3 -акриламидофенокси)-2-(3 -метоксифениламино)-пиримидин-5-карбоновой кислоты фениламида I-200
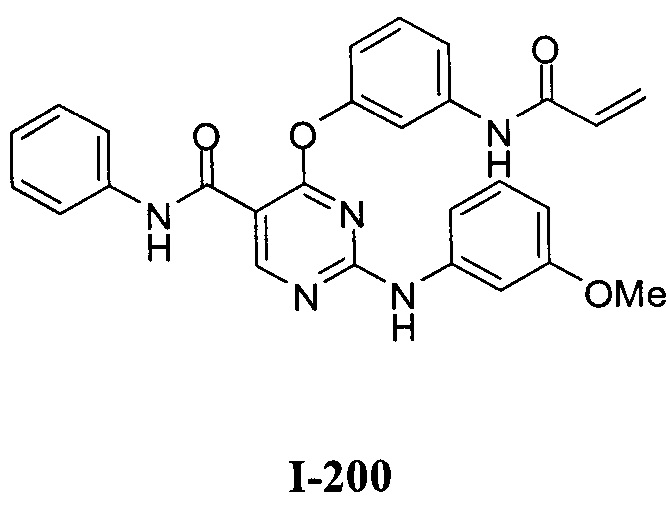
[00605] Указанное в названии соединение получали с использованием схем, этапов и 5 промежуточных соединений, описанных ниже.
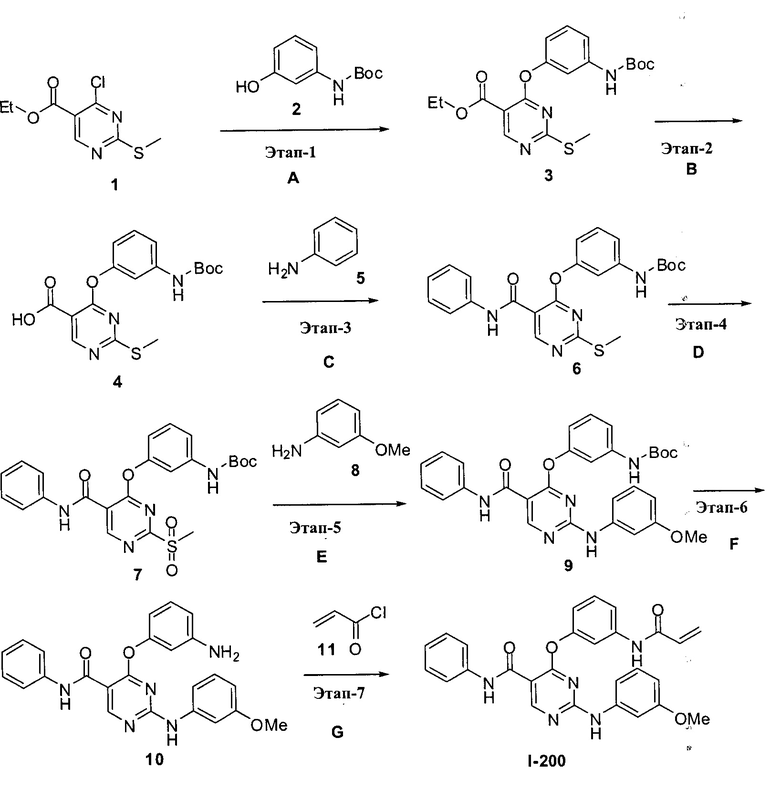
А) 2, NaH, ТГФ, 0°С; В) NaOH, ТГФ, МеОН; С) 5, TBTU, DIPEA, CH3CN, 0°С; D) МСРВА, CH2Cl2, 0°С; Е) 8, 50°С, 3 часа; F) ТФА, CH2Cl2; г) 11, DIPEA, CH2Cl2
[00606] Этап-1
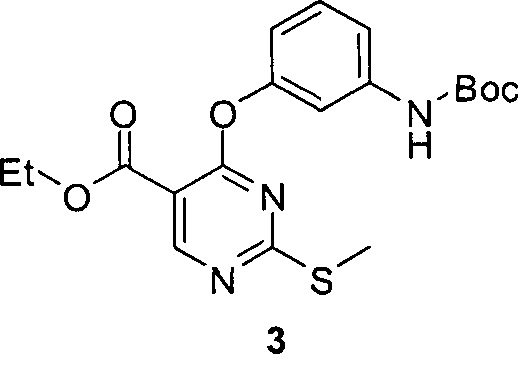
[00607] К раствору (3-гидроксифенил)карбаминовой кислоты трет-бутил эфира 2 (1.79 г, 8.59 ммоль) при температуре 0°С при помешивании добавляли суспензию натрия гидрида (60% дисперсия в минеральном масле) (0.34 г, 8.9 ммоль) в безводном ТГФ (30 мл). Смесь помешивали при температуре 0°С в течение 20 минут. Затем к раствору 4-хлоро-(2-метилсульфанил)пиримидин-5-карбоновой кислоты этил эфира 1 (2 г, 8.59 ммоль) в ТГФ (20 мл) по каплям добавляли раствор феноксида при температуре 0°С. Смесь помешивали при температуре 0°С в течение 2 часа. Реакционную смесь разбавляли этил ацетатом (150 мл) и промывали водой (50 мл) и солевым раствором (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом. Сырой продукт промывали CH2Cl2 : гексан (1:9) с получением указанного в названии соединения 3 в форме белого твердого вещества (2.43 г, 70%).
[00608] Этап-2
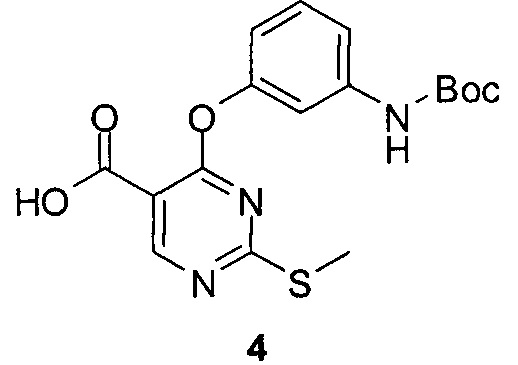
[00609] К раствору 4-(3-трет-бутоксикарбониламинофенокси)-2-(метилсульфанил-пиримидин)-5-карбоновой кислоты этил эфира 3 (2 г, 4.93 ммоль) в ТГФ (60 мл), при помешивании добавляли метанол (60 мл) при температуре -10°С, а затем водным натрия гидроксидом (0.3 г, 30 мл воды, 7.5 ммоль). Реакционную смесь оставляли дойти до комнатной температуры, после чего ее помешивали в течение 1 часа. Реакционную смесь разбавляли водой (50 мл), подкисляли лимонной кислотой и получившееся твердое
вещество собирали методом фильтрации и промывали ледяной водой (50 мл) с получением 4 в форме белого твердого вещества. (1.52 г, 82%).
[00610] Этап-3
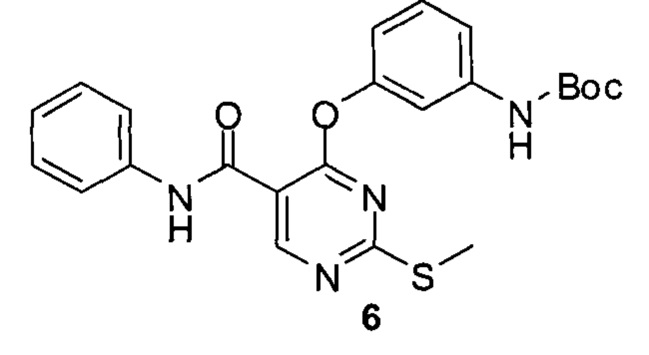
[00611] К раствору 4-(3-трет-бутоксикарбониламинофенокси)-2-(метилсульфанил)пиримидин-5-карбоновой кислоты 4 (2.0 г, 5.29 ммоль) и TBTU (2.55 г, 7.94 ммоль) в ацетонитриле (30 мл) при температуре 0°С при помешивании добавляли DIPEA (1.36g, 10.6 ммоль) а затем анилин 5 (0.60 г, 6.35 ммоль). Смесь помешивали при комнатной температуре в течение 2 часа. После завершения реакции, реакционную смесь выливали в ледяную воду (100 мл) и собирали получение белое твердое вещество методом фильтрации, промывали ледяной водой (20 мл), сушили под вакуумом и получали указанное в названии соединение 6 (1.79 г, 75%).
[00612] Этап-4
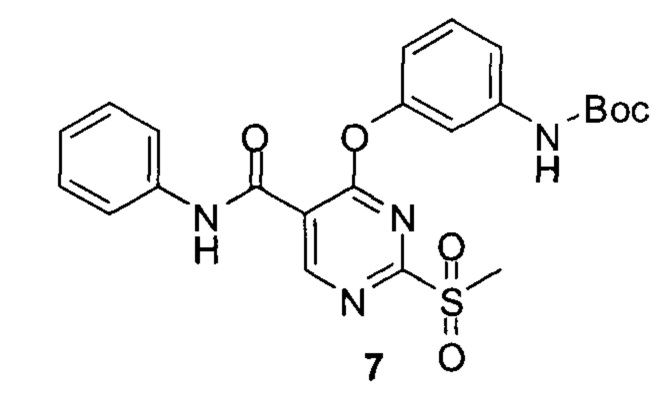
[00613] К раствору [3-(2-метилсульфанил-5-фенилкарбамоилпиримидин-4-илокси)-фенил]-карбаминовой кислоты трет-бутил эфира 6 (1.5 г, 3.31 ммоль) в CH2Cl2 при температуре 0°С при помешивании добавляли раствор m-СРВА (70%, 1.62 г, 2 эк) в CH2Cl2 (10 мл). Реакционную смесь оставляли нагреваться до комнатной температуры, затем ее помешивали в течение 12 часов. Реакционную смесь тушили насыщенным водным NaHCO3, и все вместе экстрагировали EtOAc. Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Сырой продукт промывали CH2Cl2 : гексан (1:9) с получением указанного в названии соединения 7 в форме белого твердого вещества (1,16 г, 73%).
[00614] Этап-5
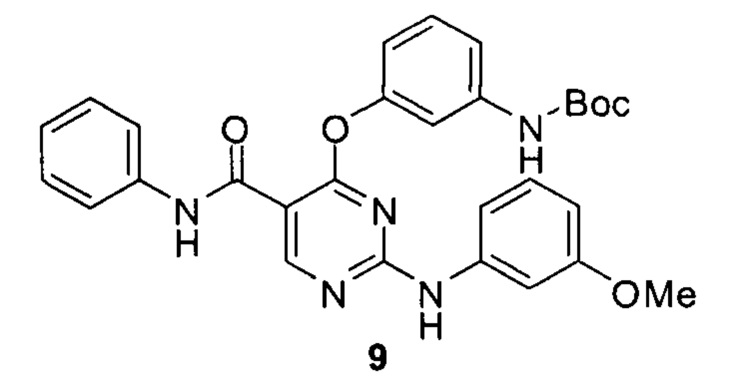
[00615] Избыток 3-метоксианилина (8) (2 мл) добавляли к твердому [3-(2-метансульфонил-5-фенилкарбамоил-пиримидин-4-илокси)-фенил]-карбаминовой кислоты трет-бутил эфиру 7 (0.5 г, 1.03 ммоль) и полученную смесь грели при температуре 50°С в атмосфере аргона в течение 3 часа. Реакционную смесь остужали до комнатной температуры и разбавляли этил ацетат/гексаном (1:1, 20 мл) после чего полученный осадок фильтровали и промывали этил ацетат/гексаном (1:1, 10 мл) с получением желаемого продукта 9 в форме белого твердого вещества (0.40 г, 75% выход).
[00616] Этап-6
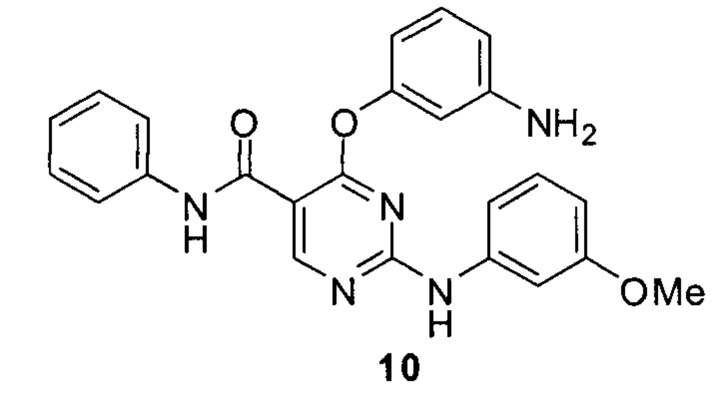
[00617] К раствору {3-[2-(3-метокси-фениламино)-5-фенилкарбамоил-пиримидин-4-илокси]-фенил}-карбаминовой кислоты трет-бутил эфира 9 (0.3 г, 0.56 ммоль) в CH2Cl2 (10 мл) добавляли трифторуксусную кислоту (2 мл) и помешивали смесь при комнатной температуре в течение 1 часа. Удаляли растворители при пониженном давлении, и растворяли остаток в CH2Cl2, промывали 10% водным раствором NaHCO3, сушили (Na2SO4), фильтровали, и упаривали при пониженном давлении с получением свободного амина 10 в форме белого твердого вещества.
[00618] Этап-7
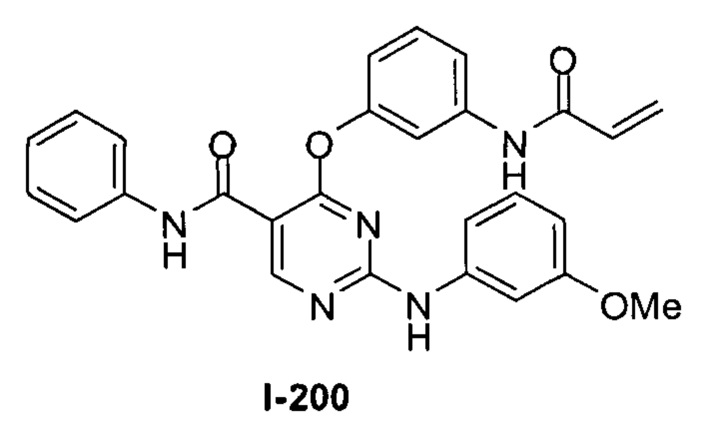
[00619] К раствору амина 10 (0.24 г, 0.56 ммоль) в дихлорметане (20 мл) в атмосфере аргона, охлажденному до -70°С при помешивании добавляли DIPEA (0.072 г, 0.56 ммоль) после чего по каплям добавляли акрилоил хлорид (0.050 г, 0.56 ммоль). Полученную смесь помешивали при температуре -70°С в течение 5 минут, и разбавляли реакционную смесь CH2Cl2 (50 мл) после чего промывали насыщенным водным раствором. NaCl (10 мл). Органический слой сушили (Na2SO4), фильтровали и упаривали при пониженном давлении. Осадок очищали методом флэш-хроматографии на силикагеле с использованием (МеОН-CHCl3 5:95) в качестве элюента, с получением требуемого соединения 11 (0.094 г, 35%) в форме белого твердого вещества: 1Н-ЯМР (200 МГц, ДМФ-d7) δ 8.9 (s, 1H), 8.10-7.70 (m, 6H), 7.60-7.10 (m, 6H), 6.60 (m, 2H) 6.40 (dd, 1H, J=8.0, 2.0 Гц), 5.80 (m, 2H), 3.70 (s, 3H).
ПРИМЕР 58
[00620] Получение 4-(3-акриламидофенокси)-2-(6-метоксипиридин-3-иламино)-пиримидин-5-карбоновой кислоты фениламида I-159
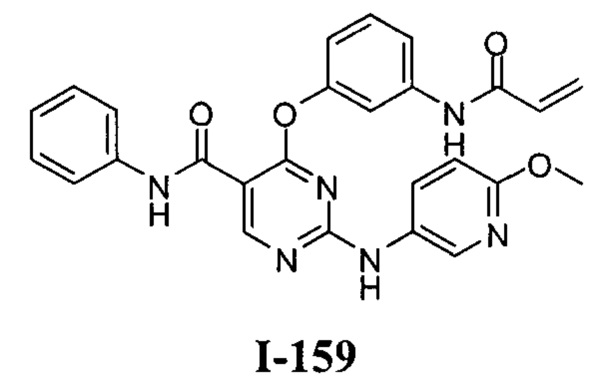
[00621] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 57 с использованием 6-метокси-3-аминопиридина вместо 8 на этапе 5. 1Н-ЯМР (200 МГц, ДМСО-d6 δ 8.90 (s, 1H), 8.20 (brs, 1Н), 7.90-7.60 (m, 4Н), 7.45 (m, 4Н), 7.10 (m, 2Н), 6.50 (m, 1Н), 6.20 (m, 2Н), 5.90 (dd, J=8.0,2.0 Гц, 1H), 3.90 (s, 3Н).
ПРИМЕР 59
[00622] Получение 4-(3-акриламидофенокси)-2-(3-метоксифениламино)-пиримидин-5-карбоновой кислоты циклопропиламида I-177
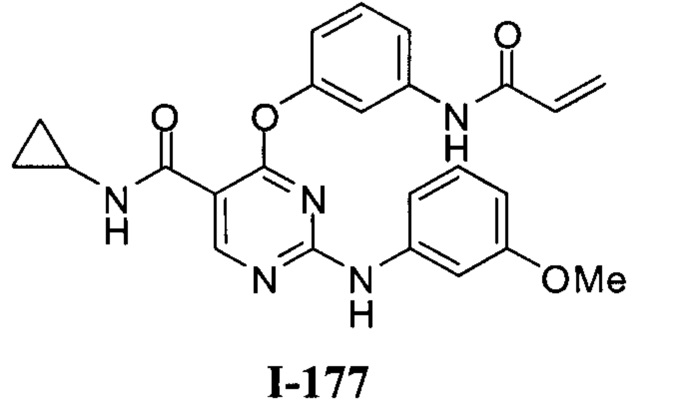
[00623] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 57 с использованием циклопропиламина вместо 5 на этапе 3. 1Н-ЯМР (200 МГц, CD3OD) δ 9.0 (s, 1H), 7.90 (brs, 1H), 7.50 (m, 3H), 7.0 (m, 4H), 6.50 (m, 1H), 6.40 (d, J=8.0 Гц, 2H), 5.80 (dd, J=8.2, 3.0 Гц, 1H), 3.60 (s, 3H), 0.90 (m, 2H), 0.62 (m, 2H).
ПРИМЕР 60
[00624] Получение 4-(3-акриламидофенокси)-2-(6-метоксипиридин-3-иламино)-пиримидин-5-карбоновой кислоты циклопропиламида I-176
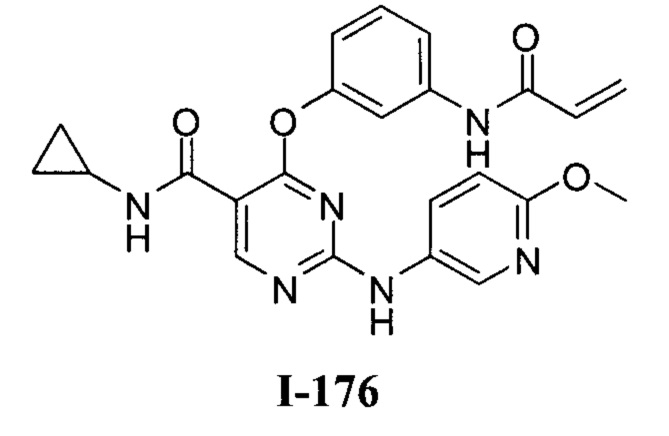
[00625] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 57 с использованием циклопропиламина вместо 5 на этапе 3 и 6-метокси-3-аминопиридине вместо 8 на этапе 5. 1Н-ЯМР (200 МГц, CD3OD) δ 8.90 (s, 1Н), 7.95 (brs, 1H), 7.90-7.82 (m, 3Н), 7.40 (m, 3Н), 6.98 (d, J=6.0 Гц, 1H), 6.42 (m, 2Н), 5.90 (dd, J=8.0, 2.0 Гц, 1H), 3.90 (s, 3Н), 0.95 (m, 2Н), 0.83 (m, 2Н).
ПРИМЕР 61
[00626] Получение 4-(3-акриламидофенокси)-2-(3-метоксифениламино)пиримидин-5-карбоновой кислоты амида I-178
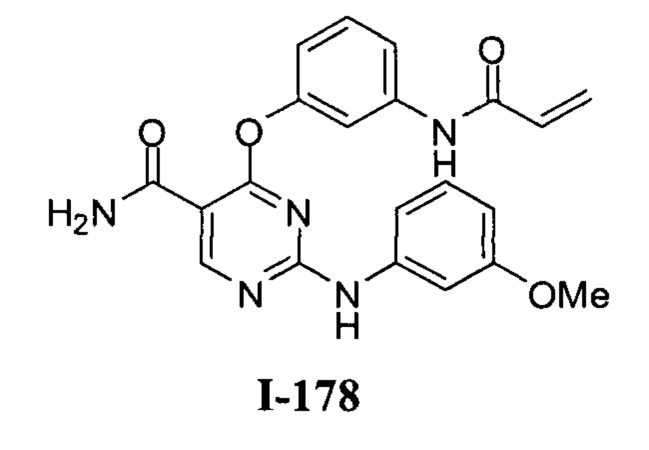
[00627] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
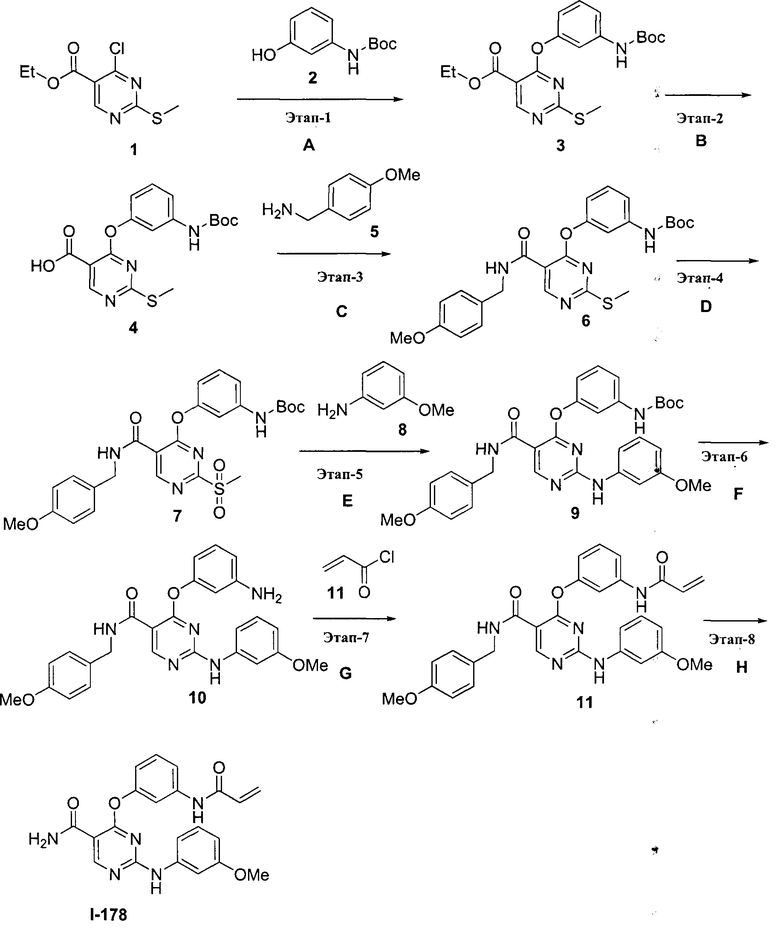
А) 2, NaH, ТГФ, 0°С; В) LiOH, ТГФ, H2O; С) 5, TBTU, DIPEA, CH3CN; D) МСРВА, CHCl3, 0°С; Е) 8, DMA, 90°С, 24 часа; F) 4N HCl, диоксан; г) 11, CH2Cl2; Н) трифлатная кислота, ТФА, CH2Cl2
[00628] Этап-1
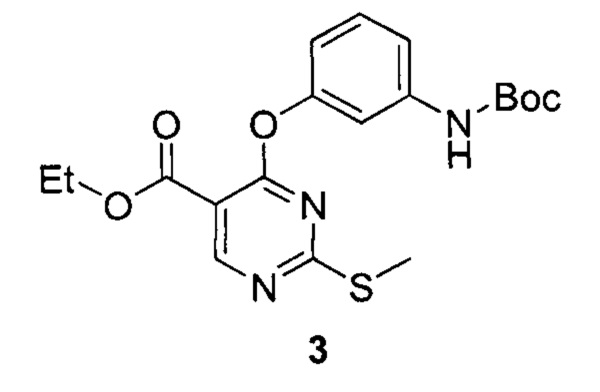
[00629] Этап 1 осуществляли также, как Этап 1 Примера 57.
[00630] Этап-2
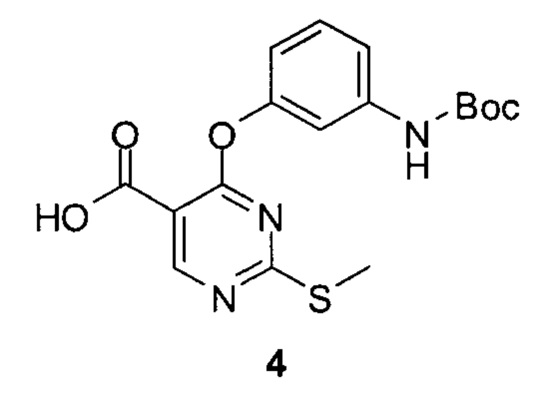
[00631] Омыляли соединение 3 (4.58 г, 11.3 ммоль) LiOH (500 мг, 20 ммоль) в 80 мл ТГФ/H2O (1:1) и после стандартной обработки 1 N HCl получали свободную кислоту 4.
[00632] Этап-3
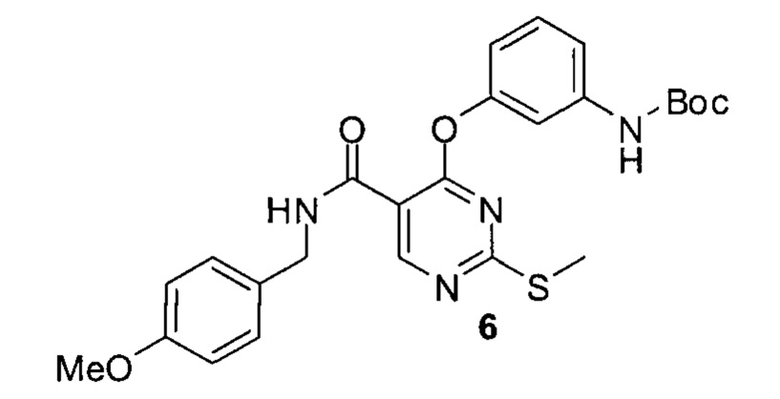
[00633] Кислоту 4 непосредственно смешивали с 4-метоксибензиламином (1.55 г, 11.3 ммоль), TBTU (5.4 г, 16.8 ммоль) и DIEA (2.4 мл, 13.4 ммоль) в 100 мл MeCN при комнатной температур. Реакционную смесь перегоняли в течение ночи и очищали методом флэш-хроматографии (EtOAc-гексан) с получением 6 в форме белого твердого вещества (4.2 г, 8.5 ммоль).
[00634] Этап-4
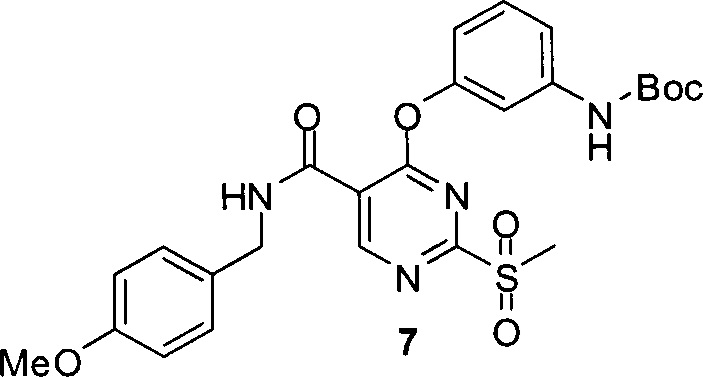
[00635] Этап 4 осуществляли также, как и этап 4 в примере 57 но применяя CHCl3 вместо CH2Cl2 в качестве растворителя.
[00636] Этап-5
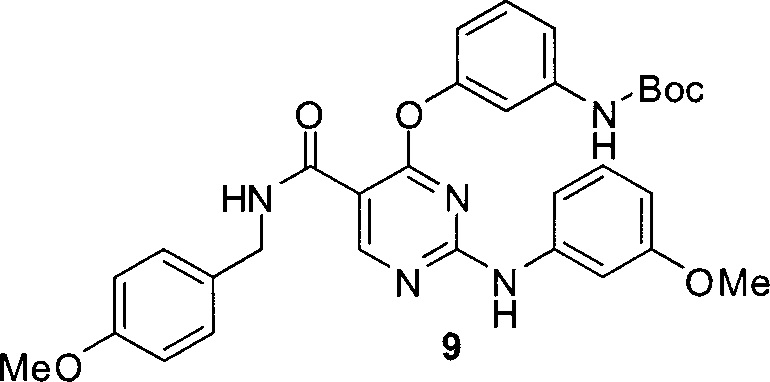
[00637] 2-метилсульфон соединения 7 (1.0 г, 1.9 ммоль) смешивали с 3-метоксианилином (420 мг, 3.4 ммоль) в DMA и смесь нагревали при температуре 90°С а течение 24 часа. Обработку проводили так же, как и на этапе 5 примера 57, с получением 9 (300 мг, 0.52 ммоль).
[00638] Этапы-6, 7 и 8
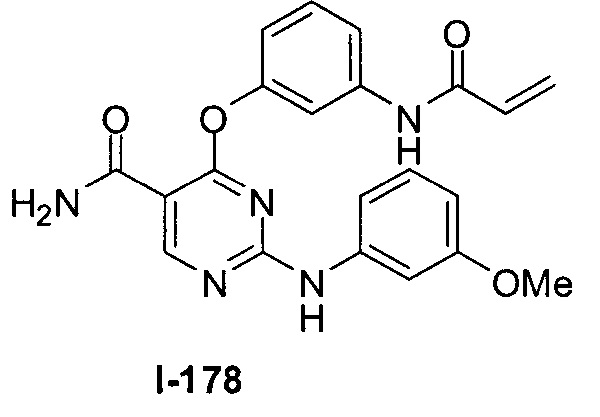
[00639] Группу ВОС удаляли из соединения 9 обработкой 4 N HCl в диоксане. Продукт (300 мг, 0.52 ммоль) немедленно обрабатывали акрилоил хлоридом (43 мкл, 0.52 ммоль) в 15 мл ДХМ при температуре -40°С. Это промежуточное соединение очищали методом флэш-хроматографии и подвергали реакции с трифлатной кислотой ТФА в ДХМ с получением сырого бензиламина 11 (120 мг, 0.228 ммоль). Это промежуточное соединение (120 мг, 0.228 ммоль) превращали в I-178 используя трифлатную кислоту (305
μL, 3.44 ммоль) в ТФА/ДХМ (5 мл, 1:1) при комнатной температуре, очищали методом колоночной хроматографии и получали ~35 мг результирующего соединения I-178 в форме серого порошка (16% выход после 3х этапов). MS: m/z=405.
ПРИМЕР 62
[00640] Получение трет-бутил 3-(3-(4-(3-акриламидофениламино)-5-метйлпиримидин-2-иламино)фенокси)пропилкарбамата I-45
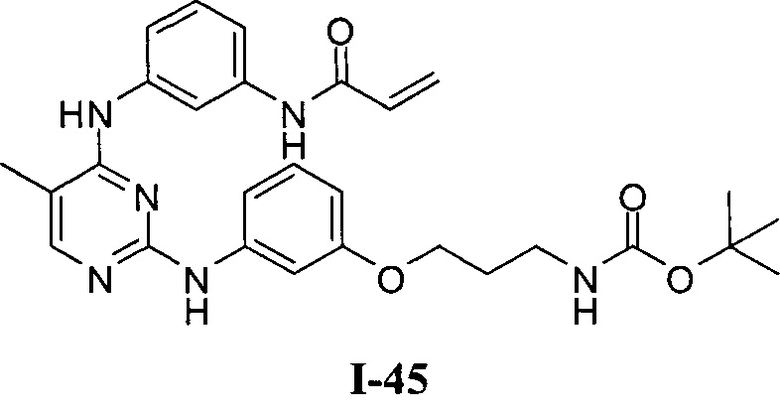
[00641] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
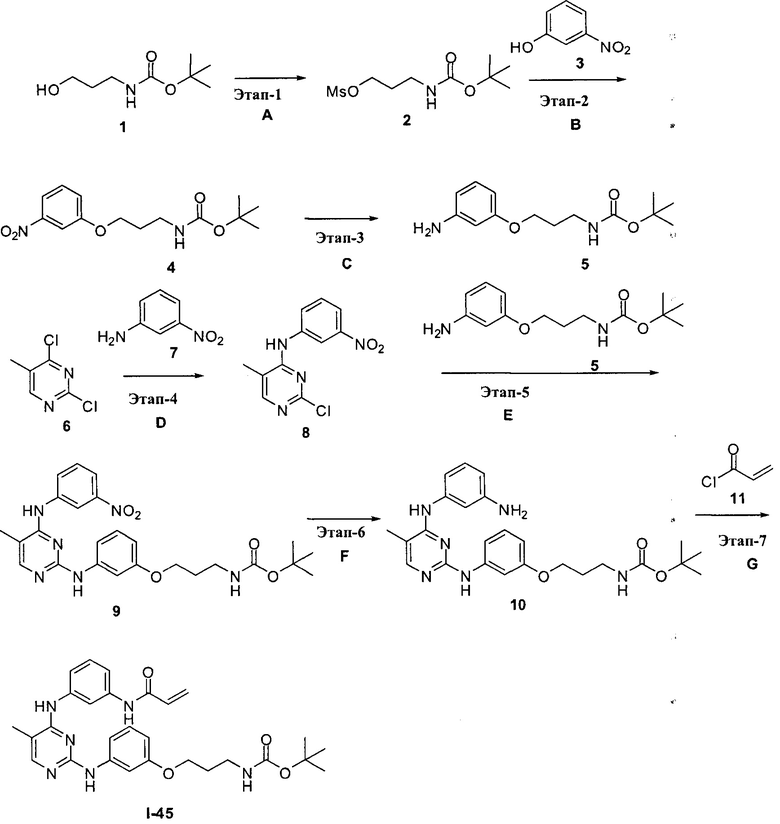
А) 1, метансульфонил хлорид, CH2Cl2, Et3N, комн. т., 1 час; В) 3, K2CO3, ДМФ, 60°С; С) Н2, Pd/C, EtOH, комн. т., 16 часов; D) 6, 7, Pd(OAc)2, ВINАР, Cs2CO3, толуол, 100°С, 16 часов; Е) 5, АсОН, EtOH, 90°С, 16 часов; F) Н2, Pd/C, EtOH, комн. т., 16 часов; г) 11, NMP, 0°С, 15 минут
[00642] Этап-1
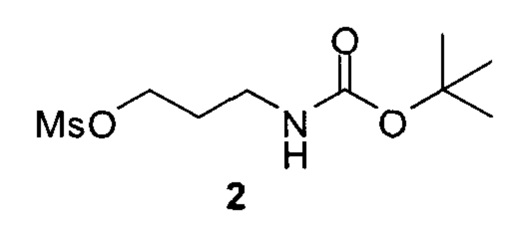
[00643] К раствору 1 (1,0 г, 5,7 ммоль) в дихлорметане (20,0 мл) при помешивании добавляли Et3N (1.15 г, 11.41 ммоль) и метансульфонил хлорид (0.98 г, 8.56 ммоль). Реакционную смесь помешивали в атмосфере азота при комнатной температуре в течение 60 минут. Затем гасили водой (20 мл) и экстрагировали EtOAc (2×50 мл). Объединенный экстракт EtOAc промывали 10% раствором NaHCO3. (25 мл), водой (25 мл), солевым раствором (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 2 (1.36 г, 94%) в форме бесцветной вязкой жидкости. Ее использовали на следующем этапе без дополнительной очистки.
[00644] Этап-2
[00645] К раствору 2 (0.749 г, 5.39 ммоль) и K2CO3 (0.99 г, 7.19 ммоль) в сухом ДМФ (20 мл) при помешивании добавляли 3 (1.36 г, 5.39 ммоль) и реакционную смесь нагревали при температуре 60°С в течение 16 часов в атмосфере азота. Реакционную смесь охлаждали, концентрировали при пониженном давлении и остаток добавляли в этил ацетат (25 мл). Раствор этил ацетата промывали водой (2×10 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 4 (1.2 г, 75%) в форме желтоватой вязкой жидкости. Ее использовали на следующем этапе без дополнительной очистки.
[00646] Этап-3
[00647] К раствору 4 (1.20 г, 4.05 ммоль) в этаноле (25 мл)) добавляли Pd/C (0.12 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (1.5 Кг давление водорода) при комнатной температуре в течение 16 часов. Реакционную
смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 5 (0.95 г, 88%) в форме коричневатого вязкого масла. Его использовали на следующем этапе без дополнительной очистки.
[00648] Этап-4
[00649] К раствору 6 (1,69 г, 12,26 ммоль), в толуоле (50,0 мл) добавляли 7 (2,0 г, 12,26 ммоль), BINAP (0.3 г, 0.49 ммоль) карбонат цезия (7,9 г, 24,5 ммоль). Раствор дегазировали (продувая N2 в течение 15 минут) и добавляли к нему Pd(ОАс)2 (0.054 г, 0.25 ммоль). Реакционную смесь помешивали при температуре 100°С в течение 16 часов в атмосфере азота. Затем ее охлаждали, разбавляли этил ацетатом (100 мл) и фильтровали через целит®. Фильтрат промывали водой (2×25 мл), солевым раствором (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, меш 60-120, этилацетат/гексан: 15/85). Твердое вещество, полученное после упаривания необходимых фракций, промывали диэтиловым эфиром и сушили под высоким вакуумом с получением 8 (1.2 г, 37%) в форме желтого твердого вещества.
[00650] Этап-5
[00651] К раствору 8 (0.5 г, 1.89 ммоль) и 5 (0.805 г, 3.0 ммоль) в этаноле (10.0 мл) добавляли ледяную уксусную кислоту (0.056 г, 0.95 ммоль), и помешивали реакционную смесь в запечатанной трубке в течение 16 часов при температуре 90°С. Реакционную смесь остужали, концентрировали при пониженном давлении. Осадок тушили 10% раствором натрия бикарбоната. (10.0 мл) и экстрагировали этил ацетатом (3×15 мл).
Объединенный экстракт этил ацетата промывали водой (15 мл), солевым раствором (15 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением осадка. Сырой осадок затем очищали методом колоночной хроматографии (SiO2, EtOAc/Гексан: 50/50) с получением 9 (0,57 г, 61%) в форме светло-желтого твердого вещества.
[00652] Этап-6
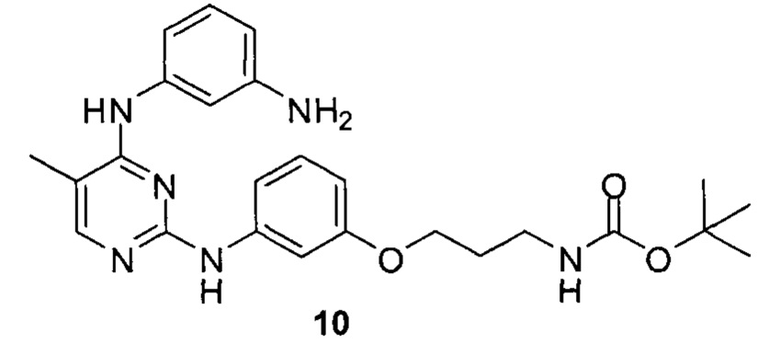
[00653] К раствору 9 (0,56 г, 1,13 ммоль) в этаноле (25 мл)) добавляли 10% Pd/C (0,068 г), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (1.5 Кг давление водорода) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 10 (0,45 г, 85%) в форме коричневатого твердого вещества. Его использовали на следующем этапе без дополнительной очистки.
[00654] Этап-7
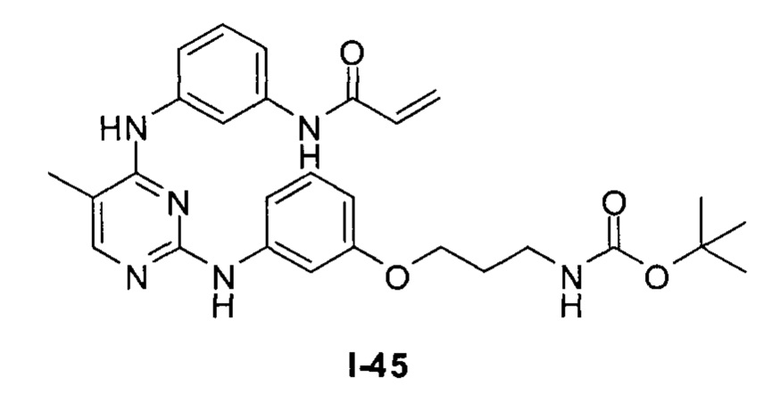
[00655] К раствору 10 (0.25 г, 0.5382 ммоль) в NMP (2.5 мл) при температуре 0°С при помешивании добавляли акрилоил хлорид (0.073 г, 0.807 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 15 минут. Реакционную смесь добавляли по каплям в холодный 10% раствор NaHCO3 при помешивании. После полного добавления раствор помешивали в течение еще 30 минут при температуре 0°С, после чего фильтровали через воронку Бухнера, чтобы отделить твердый осадок. Твердую фазу промывали холодной водой и гексаном. Ее растворяли в смеси метанол: дихлорметан (50:50, 10 мл) и концентрировали при пониженном давлении. Полученный осадок
суспендировали в холодной воде (50 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×100 мл). Объединенный экстракт этил ацетата промывали водой (50 мл), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением I-45 (0.100 г, 35.8%) в форме беловатого осадка. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.37 (s, 9Н), 1.70-1.80 (m, 2Н), 2.10 (s, 3Н), 3.00-3.06 (m, 2Н), 3.79 (t, J=6.24 Гц, 2H), 5.74 (d, J=11.92 Гц, 1H), 6.24 (dd, J=1.84 & 15.16 Гц, 1H), 6.35-6.47 (m, 2H), 6.80-6.90 (bs, 1H), 6.97 (t, J=8.28 Гц, 1H), 7.23-7.27 (m, 2H), 7.31 (s, 1H), 7.37 (d, J=8.2 Гц, 1H), 7.46 (d, J=7.48 Гц, 1H), 7.90-7.90-7.91 (m, 2H), 8.36 (s, 1H), 8.87 (s, 1H), 10.07 (s, 1H); ЖХМС: m/е 519 (М+1).
ПРИМЕР 63
[00656] Получение трет-бутил 3-(3-(4-(3-акриламидофениламино)-5-фторопиримидин-2-иламино)фенокси)пропилкарбамата I-183
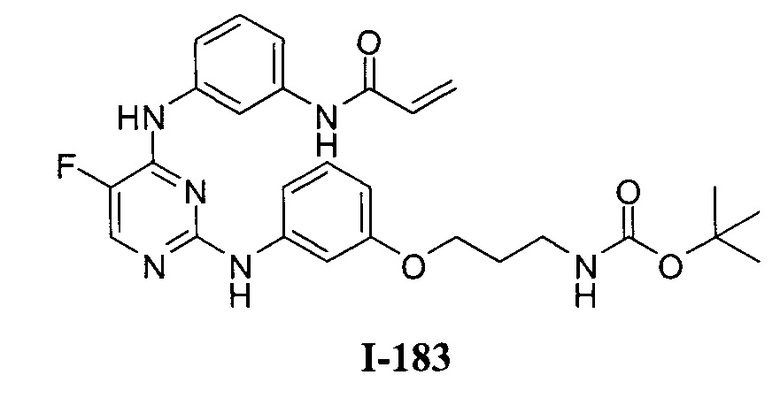
[00657] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
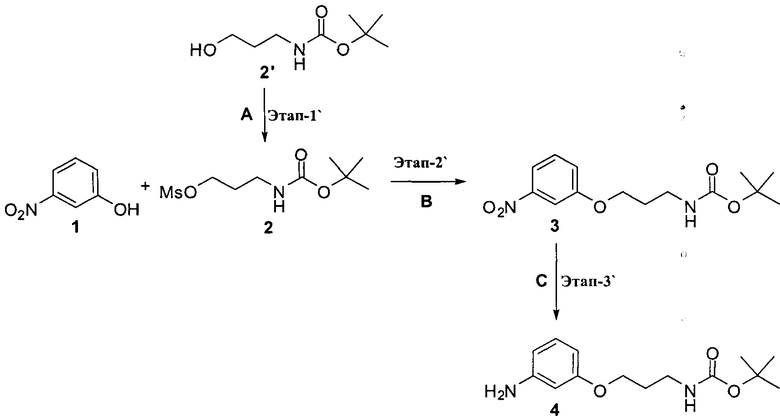
А) метансульфонил хлорид, CH2Cl2, Et3N, комн. т., 1 час; В) K2CO3, ДМФ, 60°С, 16 часов; С) Pd-C, Н2, этанол, комн. т., 16 часов.
[00658] Этап 1'
[00659] К раствору 2' (4.0 г, 22.8 ммоль) в дихлорметане (80.0 мл) при помешивании добавляли Et3N (4.6 г, 45.5 ммоль) и метансульфонил хлорид (3.92 г, 34.2 ммоль), и помешивали реакционную смесь в атмосфере азота при комнатной температуре в течение 60 минут. Реакционную смесь гасили водой (50 мл) и экстрагировали EtOAc (2×100 мл). Объединенные экстракты промывали 10% раствором NaHCO3 (50 мл), водой (50 мл), и солевым раствором (50 мл), сушили над Na2SO4, и концентрировали при пониженном давлении с получением 2 (5.5 г, 95.2%) в форме светло-желтой вязкой жидкости. Соединение 2 использовали на следующем этапе без дополнительной очистки.
[00660] Этап 2'
[00661] К раствору 1 (2.3 г, 16.5 ммоль) и K2CO3 (4,6 г, 33,3 ммоль) в сухом ДМФ (100 мл) при помешивании добавляли 2 (5,5 г, 21.7 ммоль) и реакционную смесь нагревали при
температуре 60 С в течение 16 часов в атмосфере азота. Реакционную смесь охлаждали, разбавляли водой (250 мл) и экстрагировали EtOAc (2×100 мл). Объединенные экстракты промывали 10% раствором NaHCO3 (100 мл), водой (3×100 мл), и солевым раствором (50 мл), сушили над Na2SO4, и концентрировали при пониженном давлении с получением 3 (4,0 г, 81,6%) в форме светло-желтой вязкой жидкости. Соединение 3 использовали на следующем этапе без дополнительной очистки.
[00662] Этап 3'
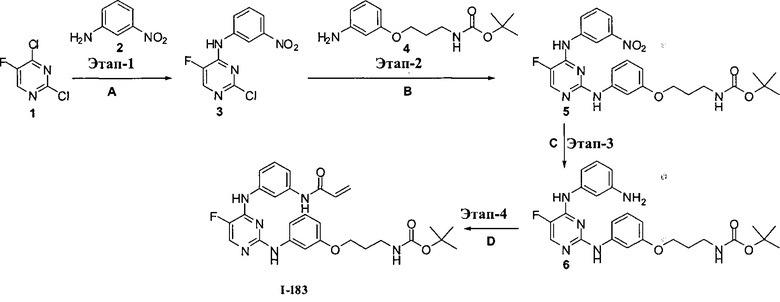
[00663] К раствору 3 (4,0 г, 13.4 ммоль) в этаноле (50 мл) добавляли Pd/C (0,8 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (1.5 Кг давление водорода) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 4 (3,3 г, 91,9%) в форме коричневатого вязкого масла. Соединение 4 использовали на следующем этапе без дополнительной очистки.
[00664] Этап 1
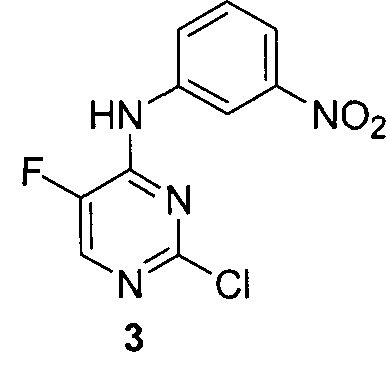
[00665] В нагнетательную трубку помещали соединения 2 (10.0 г, 0.072 mol), 1 (24.1 г, 0.145 mol), n-BuOH (100 мл) и DIPEA (13.9 г, 0.108 mol), и помешивали содержимое при температуре 120°С в течение 2 часа. Реакционную смесь охлаждали, и отделяли твердый осадок методом фильтрации через воронку Бухнера, промывали холодным гексаном и сушили с получением 3 (12.5 г, 64%) в форме желтого твердого вещества. Соединение 3 использовали на следующем этапе без дополнительной очистки.
[00666] Этап 2
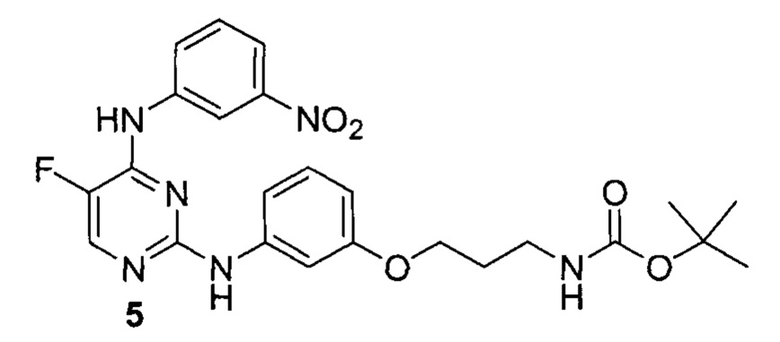
[00667] К раствору 3 (1.5 г, 5.58 ммоль) и 4 (1.48 г, 5.58 ммоль) в этаноле (30.0 мл) добавляли ледяную уксусную кислоту (0.167 г, 2.79 ммоль), и перемешивали реакционную смесь в нагнетательной трубке при температуре 90°С в течение 48 часов. Реакционную смесь охлаждали и концентрировали при пониженном давлении; осадок гасили 10% раствором натрия бикарбоната (20.0 мл) и экстрагировали этил ацетат (2×50 мл). Объединенные экстракты промывали водой (25 мл) и солевым раствором (25 мл), сушили над Na2SO4, и концентрировали при пониженном давлении с получением 5. Сырой остаток очищали методом колоночной хроматографии (нейтральный Al2O3; МеОН/Хлороформ: 0.5/99.5) с получением 5 (1.4 г, 50.3%) в форме коричневого твердого вещества.
[00668] Этап 3
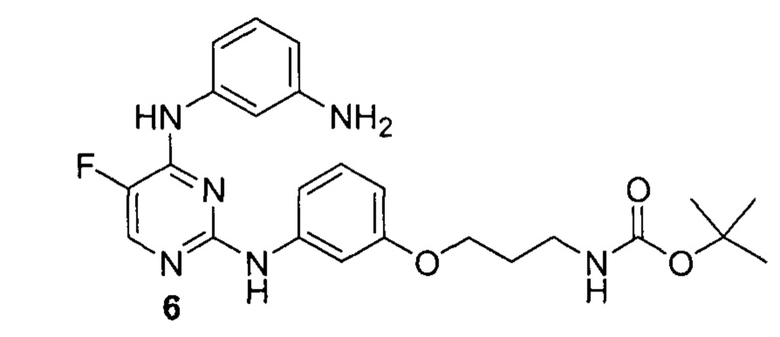
[00669] К раствору 5 (1,4 г, 2,8 ммоль) в этаноле (50 мл) добавляли 10% Pd/C (0,28 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (1.5 Кг давление водорода) при комнатной температуре в течение 16 часов.
Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением осадка. Сырой остаток далее очищали методом колоночной хроматографии (Al2O3, МеОН/Хлороформ): 0.5/99.5) с получением твердого вещества, которое промывали смесью дихлорметан/гексан с получением 6 (0.7 г, 53.4%) в форме бледно-коричневого твердого вещества.
[00670] Этап 4
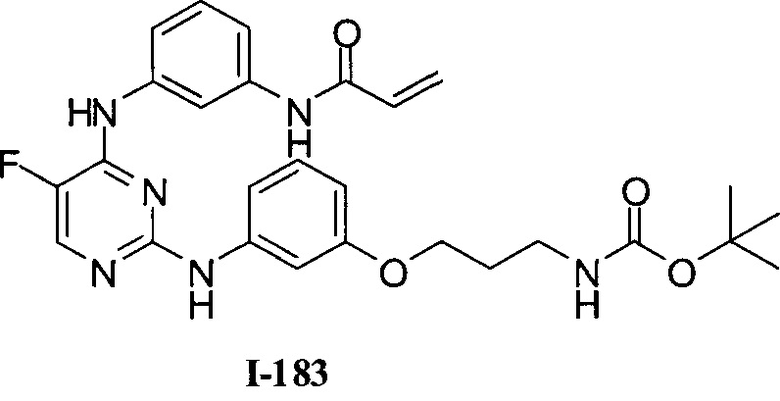
[00671] трет-бутил 3-(3-(4-(3-акриламидофениламино)-5-фторопиримидин-2-иламино)фенокси)пропилкарбамата. К раствору 6 (0,25 г, 0,533 ммоль) и калия карбоната (0.138 г, 1.02 ммоль) в NMP (2,5 мл) при температуре 0°С при помешивании добавляли акрилоил хлорид (0,060 г, 0,665 ммоль), и помешивали реакционную смесь при температуре 0°С в течение 30 минут. Реакционную смесь добавляли по каплям при помешивании к 10% раствору NaHCO3 и помешивали при той же температуре (0°С) в течение 30 минут. Белый твердый осадок отделяли при помощи воронки Бухнера. Затем твердый осадок промывали холодной водой и гексаном растворяли в смеси метанол/дихлорметан (50:50, 10 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (25 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×50 мл). Объединенные экстракты этил ацетата промывали водой (50 мл), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением I-183 (0.255 г, 91,4%) в форме светло-желтого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.36 (s, 9Н), 1.78 (quin, J=6.4 Гц, 2Н), 3.01-3.06 (m, 2Н), 3.83 (t, J=6.12 Гц, 2Н), 5.74 (dd, J=1.4 & 10.04 Гц, 1Н), 6.24 (d, J=16.84 Гц, 1Н), 6.41-6.48 (m, 2Н), 6.88 (s, 1Н), 7.03 (t, J=8.24 Гц, 1H); 7.23-7.31 (m, 3Н), 7.41 (d, J=8.28 Гц, 1Н), 7.56 (d, J=7.96 Гц, 1H), 7.90 (s, 1Н), 8.11 (d, J=3.56 Гц, 1H), 9.11 (s, 1Н), 9.43 (s, 1H), 10.10 (s, 1Н); ЖХМС: m/е 523.1 (М+1).
ПРИМЕР 64
[00672] Получение трет-бутил 3-(4-(4-(3-акриламидофениламино)-5-фторпиримидин-2-иламино)фенокси)пропилкарбамата I-198
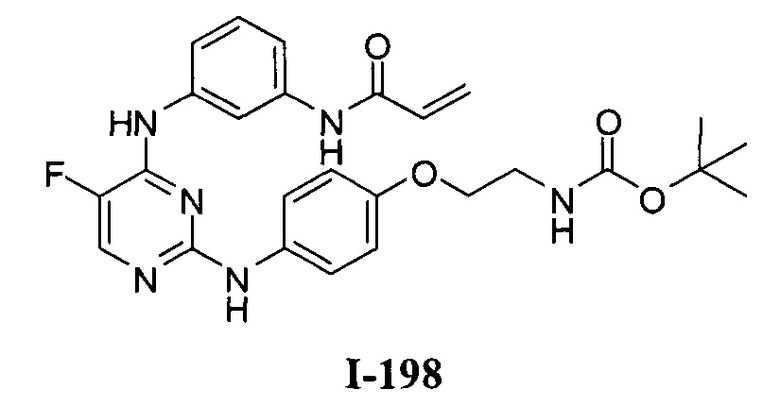
[00673] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 63, с использованием трет-бутил 3-(4-аминофенокси)пропилкарбамата вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.37 (s, 9Н), 1.78 (quin, J=6.36 Гц, 2Н), 3.05 (q, J=6.24 Гц, 2Н), 3.86 (t, J=6.2 Гц, 2Н), 5.75 (dd, J=1.92 & 10.04 Гц, 1Н), 6.24 (dd, J=1.92 & 16.92 Гц, 1H), 6.45 (dd, J=10.08 & 16.92 Гц, 1Н), 6.72 (d, J=9 Гц, 2Н), 6.89 (t, J=5.4 Гц, 1Н), 7.26 (t, J=8.08 Гц, 1H), 7.40 (d, J=8.12 Гц, 1H), 7.48-7.52 (m, 3Н), 7.92 (s, 1H), 8.05 (d, J=3.72 Гц, 1H), 8.95 (s, 1Н), 9.36 (s, 1H), 10.12 (s, 1H); ЖХМС: m/e 523.2 (M+1).
[00674] Промежуточное соединение трет-бутил 3-(4аминофенокси)пропилкарбамат получали с использованием схемы описанной ниже.
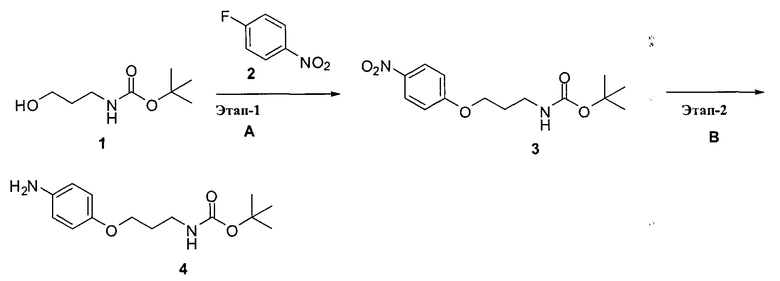
A) NaH, ТГФ, комн. т., 16 часов; В) Н2, Pd/C, EtOH, комн. т., 16 часов
[00675] Этап-1
[00676] К раствору 1 (1.7 г, 9.7 ммоль) в сухом ТГФ (40 мл) при помешивании добавляли NaH (0.72 г, 18.0 ммоль, 60% дисперсия в парафиновом масле) при температуре
0°С и помешивали реакционную смесь при комнатной температуре в течение 15 минут в атмосфере азота. К нему добавляли 2 (2.0 г, 13.87 ммоль) и помешивали реакционную смесь при комнатной температуре в течение 16 часов. Тушили холодной водой (20 мл) и экстрагировали этил ацетатом (25 мл). Экстракт этил ацетата промывали водой (2×10 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением маслянистой жидкости, которую толкли с гексаном с получением 3 (2.0g, 69.5%) в форме желтого кристаллического твердого вещества.
[00677] Этап-2
[00678] К раствору 3 (2,0 г, 6,749 ммоль) в этаноле (30 мл) добавляли 10% Pd/C (0,4 г, 20% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (1.5 Кг давление водорода) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 4 (1.6 г, 89.3%) в форме розоватого вязкого масла. Его использовали на следующем этапе без дополнительной очистки.
ПРИМЕР 65
[00679] Получение 4-(3акриламидофениламино)-5-фтор-2-(3,4-диметоксифениламино)-пиримидина I-134
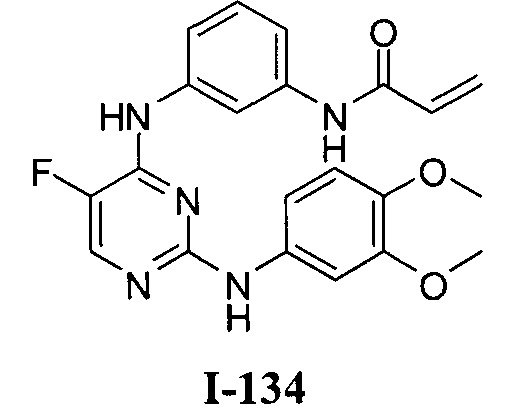
[00680] Указанное в названии соединение получали с использованием схем, этапов и промежуточных продуктов, описанных в Примере 20 с использованием 3,4-диметоксианилина вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CD3OD) δ 8.50 (s, 1H), 7.80 (d, J=6.5 Гц, 1Н), 7.70-7.66 (m, 2H), 7.20 (m, 1H), 7.0 (m, 2H), 6.41 (m, 2H), 5.92 (dd, J=8.0, 2.0 Гц, 1H), 3.89 (s, 6H).
ПРИМЕР 66
[00681] Получение 4-(3-акриламидофениламино)-5-фтор-2-(3,4,5-триметоксифениламино)-пиримидина I-133
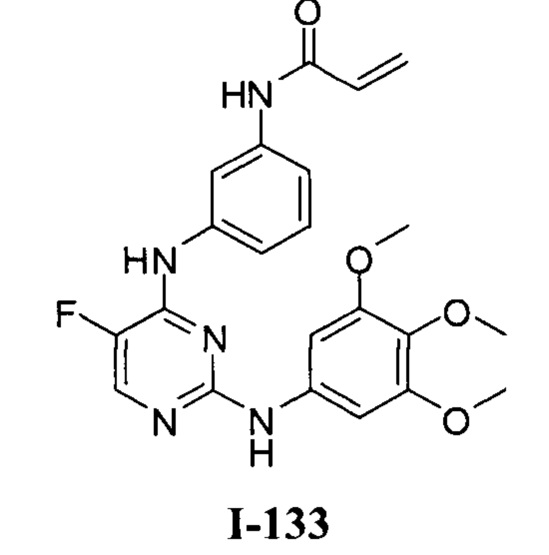
[00682] Указанное в названии соединение получали с использованием схем, этапов и промежуточных продуктов, описанных в Примере 20 с использованием 3,4,5-триметоксианилина вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CD3OD) δ 8.10 (s, 1H), 8.0 (d, J=6.0 Гц, 1H), 7.50 (m, 2H), 7.30 (m, 1H), 7.0 (m, 2H), 6.45 (m, 2H), 5.90 (dd, J=8.0, 2.0 Гц, 1H), 3.90 (s, 3H), 3.89 (s, 9H).
ПРИМЕР 67
[00683] Получение 4-(3-акриламидофениламино)-5-фтор-2-(3-(гидроксиметил)фениламино)-пиримидина I-145
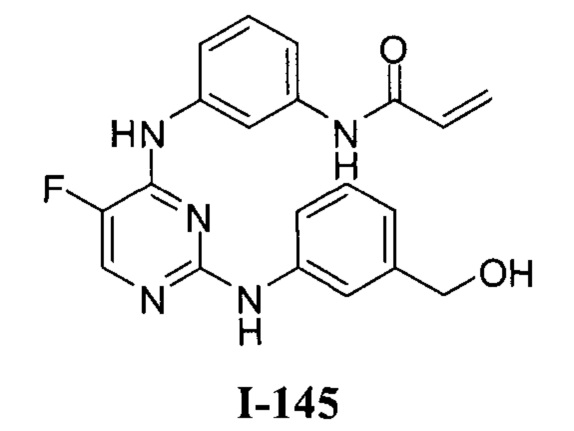
[00684] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-гидроксиметиланилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 4.38 (d, J=5.6 Гц, 2Н), 5.07 (t, J=5.68 Гц, 1H), 5.75 (d, J=10.84 Гц, 1Н), 6.24 (dd, J=16.96 Гц, 1H), 6.44 (dt, J=10.04 & 17.0 Гц, 1Н), 6.83 (d,7=7.4 Гц, 1H), 7.10 (t, J=7.72 Гц, 1Н), 7.28 (г, J=8.16 Гц,
1Н), 7.40 (d, 7=8.08 Гц, 1H), 7.55-7.59 (m, 3Н), 7.92 (s, 1H), 8.09 (d, 7=3.6 Гц, 1H), 9.11 (s, 1H), 9.40 (s, 1H), 10.1 (s, 1H); ЖХМС: m/e 378.0 (M+1).
ПРИМЕР 68
[00685] Получение 4-(3-акриламидофениламино)-5-фтор-2-(3-(3-(2-оксопирролидин-1-ил)пропокси)фениламино)-пиримидина I-144
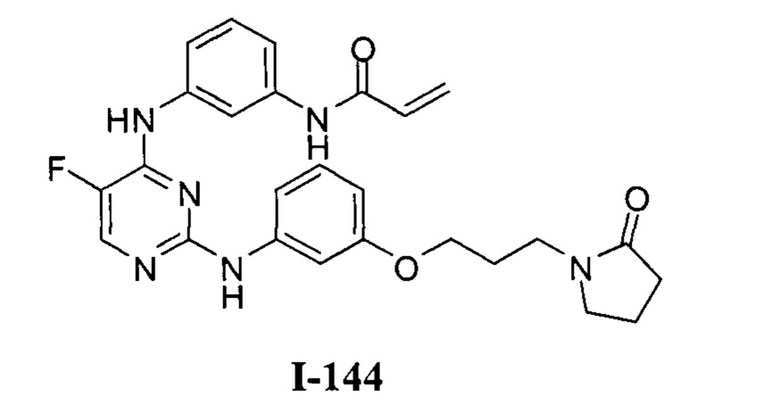
[00686] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(2-оксопирролидин-1-ил)пропоксианилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.8-1.94 (m, 4Н), 2.18 (q, J=8.08 Гц, 2Н), 3.26-3.40 (m, 4Н), 3.80 (t, J=6 Гц, 2Н), 5.74 (d, J=10.72 Гц, 1Н), 6.24 (d, J=15.64 Гц, 1Н), 6.41-6.80 (m, 2Н), 7.04 (t, J=8.16 Гц, 1Н), 7.22-7.29 (m, 2Н), 7.33 (s, 1H), 7.42 (d, J=8.08 Гц, 1Н), 7.55 (d, J=7.52 Гц, 1Н), 7.91 (s, 1Н), 8.11 (d, J=3.48 Гц, 1H), 9.13 (s, 1H), 9.43 (s, 1H), 10.11 (s, 1H); ЖХМС: m/е 491 (М+1).
ПРИМЕР 69
[00687] Получение 4-(3-акриламидофениламино)-5-фтор-2-(3-(3-(метилсульфонил)пропокси)фениламино)-пиримидина I-138
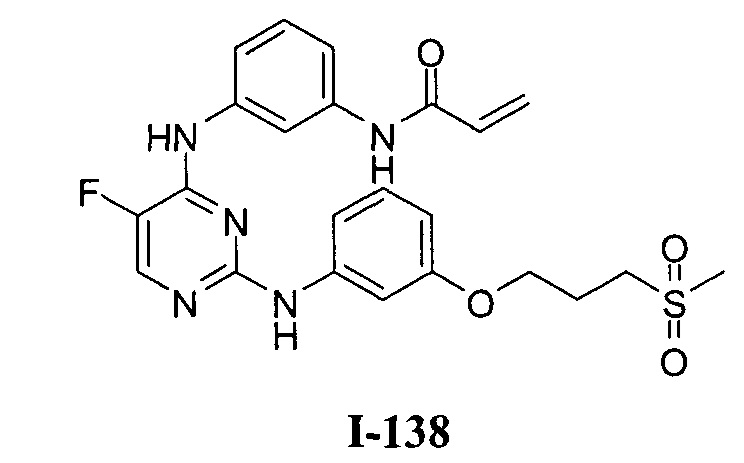
[00688] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(3-
(метилсульфонил)пропоксианилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.05-2.15 (m, 2Н), 3.0 (s, 3Н), 3.22 (t, J=7.76 Гц, 2Н), 3.93 (t, J=6.08 Гц, 2Н), 5.74 (dd, J=1.88 & 10 Гц, 1H), 6.25 (dd, J=1.8 & 16.88 Гц, 1Н), 6.44 (dd, J=10.16 & 16.84 Гц, 2Н), 7.05 (t, J=8.16 Гц, 1Н), 7.24-7.30 (m, 2Н), 7.35 (s, 1Н), 7.42 (d, J=8.2 Гц, 1H), 7.55 (d, J=8 Гц, 1H), 7.90 (s, 1Н), 8.11 (d, J=3.6 Гц, 1H), 9.14 (s, 1Н), 9.43 (s, 1Н), 10.10 (s, 1Н); ЖХМС: m/е 484 (М+1).
[00689] Промежуточное соединение 3-(3-(метилсульфонил)пропоксианилин получали с использованием схемы описанной ниже.
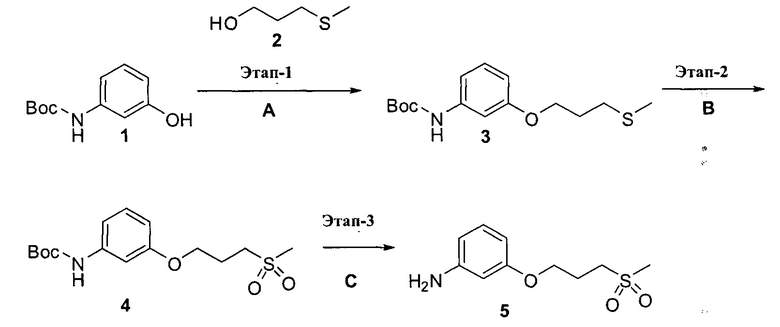
A) DEAD, Ph3P, Et3N, ТГФ, комн. т., 1 час; В) МСРВА, CH2Cl2, комн. т., 30 минут; С) ТФА, CH2Cl2, комн. т., 1 час
[00690] Этап-1
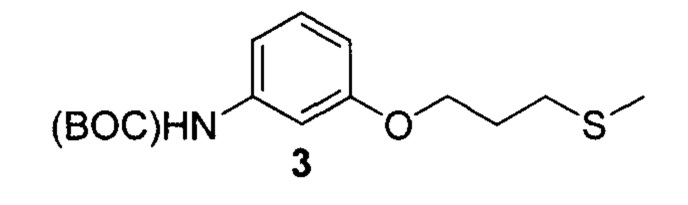
[00691] К раствору 2 (1.1 г, 10.3 ммоль) в ТГФ (20 мл) добавляли при помешивании 1 (2,18 г, 10,3 ммоль), PPh3 (2.98 г, 11.3 ммоль) и Et3N (1,68 г, 15 ммоль) а атмосфере N2. Реакционную смесь охлаждали до 0°С и добавляли к ней DEAD (1.98 г, 11.3 ммоль). Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали 1 час. Затем тушили водой, экстрагировали этил ацетатом (3×25 мл) и промывали объединенные экстракты EtOAc водой и солевым раствором (по 5 мл каждого). Полученный осадок концентрировали при пониженном давлении и очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 8/2) с получением 3 (2 g 60.6%) в форме белого твердого вещества.
[00692] Этап-2
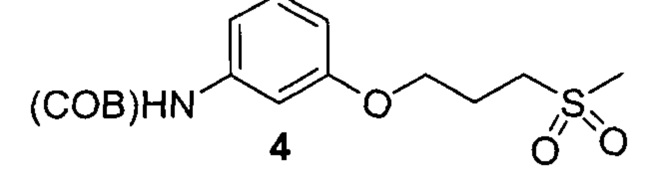
[00693] К раствору 3 (2 г, 6,7 ммоль) в CH2Cl2 (25 мл) при помешивании добавляли m-СРВА (4.13 г, 26.7 ммоль) при температуре -10°С. Реакционную смесь оставляли дойти до комнатной температуры, после чего ее помешивали в течение 30 минут. Затем тушили раствором Na2CO3 (10 мл), экстрагировали CH2Cl2 (10 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 4 (0.016 г, 68,8%) в форме желтого масла
[00694] Этап-3
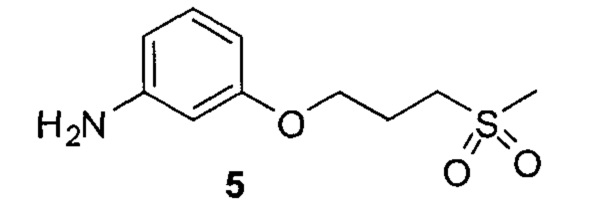
[00695] К раствору 4 (0,75 г, 2,2 ммоль) в CH2Cl2 (7,5 мл) при помешивании добавляли ТФА (3 vol.) при температуре 0°С. Реакционную смесь оставляли дойти до комнатной температуре, после чего ее помешивали 1 час. Затем концентрировали при пониженном давлении, приводили к форме основания раствором NaHCO3 (5 мл) и экстрагировали CH2Cl2 (3×10 мл). Объединенный органический экстракт промывали водой (2 мл) и солевым раствором (2 мл). Сушили над Na2SO4 затем фильтровали и концентрировали при пониженном давлении с получением 5 (500 мг, 96%) в форме коричневого твердого вещества.
ПРИМЕР 70
[00696] Получение N-(3-(5-фтор-2-(3-(2-гидроксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-105
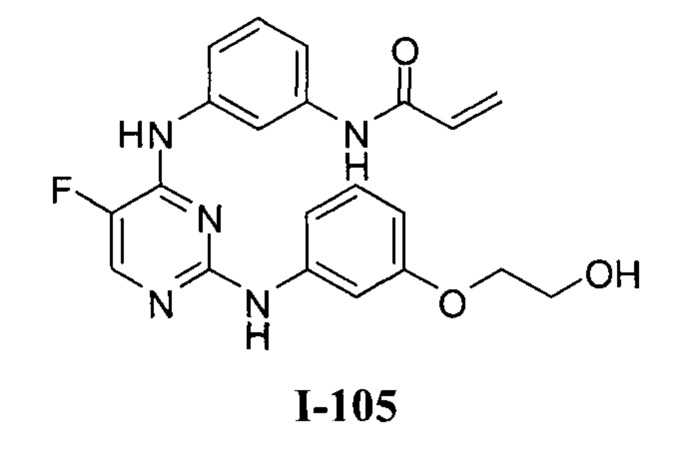
[00697] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(2-гидрокси)этоксианилин вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.67 (dd, J=4.5 & 10 Гц, 2Н), 3.85-3.87 (m, 2Н), 4.83 (t, J=5.6 Гц, 1Н), 5.75 (bd, J=10 Гц, 1H), 6.25 (d, J=15.6 Гц, 1Н), 6.42-6.46 (m, 2Н), 7.05 (t, J=8.4 Гц, 1H), 7.26 (d, J=8 Гц, 1Н), 7.30 (d, J=8 Гц, 1Н), 7.33 (s, 1H), 7.41 (d, J=8 Гц, 1Н), 7.57 (d, J=8 Гц, 1Н), 7.92 (s, 1H), 8.11 (d, J=3.6 Гц, 1Н), 9.11 (s, 1Н), 9.42 (s, 1H), 10.11 (s, 1Н); ЖХМС: m/е 409.9 (М+1).
[00698] Промежуточное соединение 3-(2-гидрокси)этоксианилин получали с использованием схемы описанной ниже.
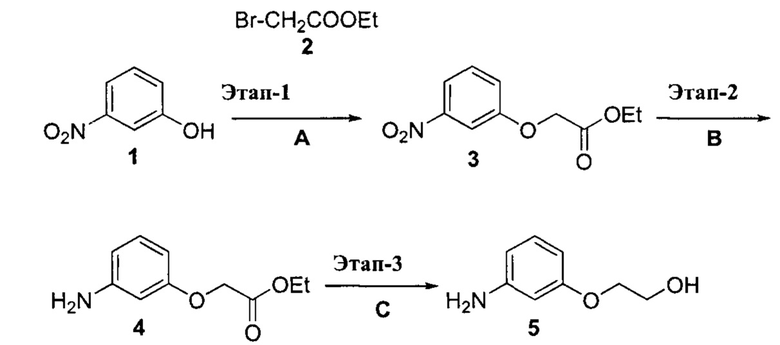
А) K2CO3, ДМФ, 70°С, 12 часов; В) Pd-C, H2, этанол, комн. т., 10 часов; С) 1М раствора LAH, ТГФ, -15°С, 45 минут.
[00699] Этап-1
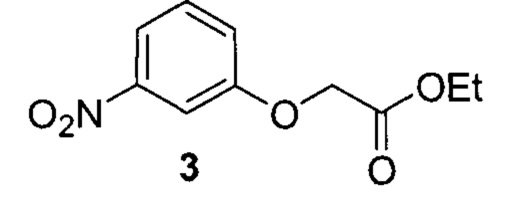
[00700] К раствору 1 (2,0 г, 14,37 ммоль) и K2CO3 (3,95 г, 28,6 ммоль) в сухом ДМФ (15 мл) при помешивании добавляли 2 (2,88 г, 17,25 ммоль) и реакционную смесь нагревали при температуре 70°С в течение 12 часов в атмосфере азота. Реакционную смесь охлаждали, концентрировали при пониженном давлении и осадок разбавляли этил ацетатом (50 мл). Ее промывали водой (2×10 мл), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением I3 (0.255 г, 78%) в форме светло-коричневой жидкости. Его использовали на следующем этапе без дополнительной очистки.
[00701] Этап-2
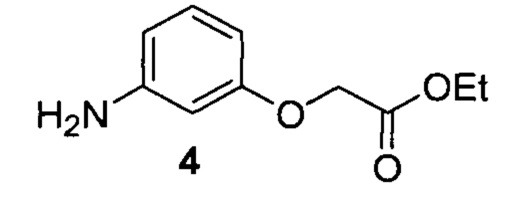
[00702] К раствору 3 (2,0 г, 8,88 ммоль) в этаноле (20 мл) добавляли Pd/C (0,2 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (1,0 Кг давление водорода) при комнатной температуре в течение 10 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 4 (1,6 г, 94%) в форме светло-коричневой жидкости. Его использовали на следующем этапе без дополнительной очистки.
[00703] Этап-3
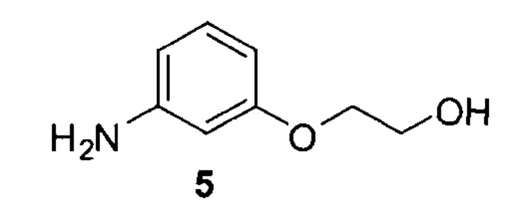
[00704] К раствору 4 (1.2 г, 6.14 ммоль) в сухом ТГФ (12 мл) при помешивании жлюавляли литий алюминий гидрид (9.2 мл, 9.20 ммоль, 1.0 М раствор в ТГФ) при температуре -15°С, в атмосфере N2. Реакционную смесь оставляли дойти до комнатной температуры, после чего ее помешивали в течение 45 минут. Реакционную смесь тушили насыщенным раствором аммония хлорида и фильтровали через целитную® пластину и экстрагировали EtOAc (2×20 мл).Объединенный органический слой промывали солевым раствором (10 мл) и концентрировали при пониженном давлении с получением А (0.9 г, 95%) в форме темно-коричневой жидкости. Его использовали на следующем этапе без дополнительной очистки.
ПРИМЕР 71
[00705] Получение N-(3-(5-фтор-2-(3-(2-гидрокси-2-метилпропокси)фениламино) пиримидин-4-иламино)фенил)акриламида I-118
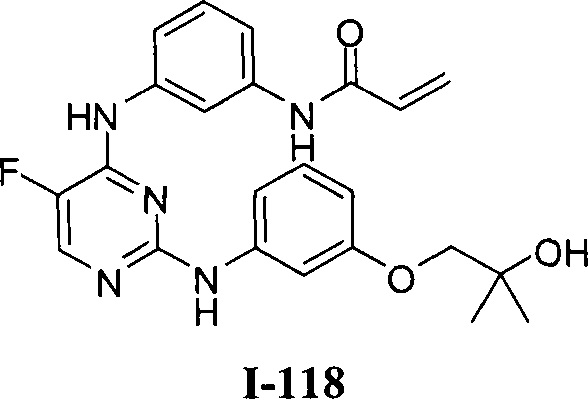
[00706] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
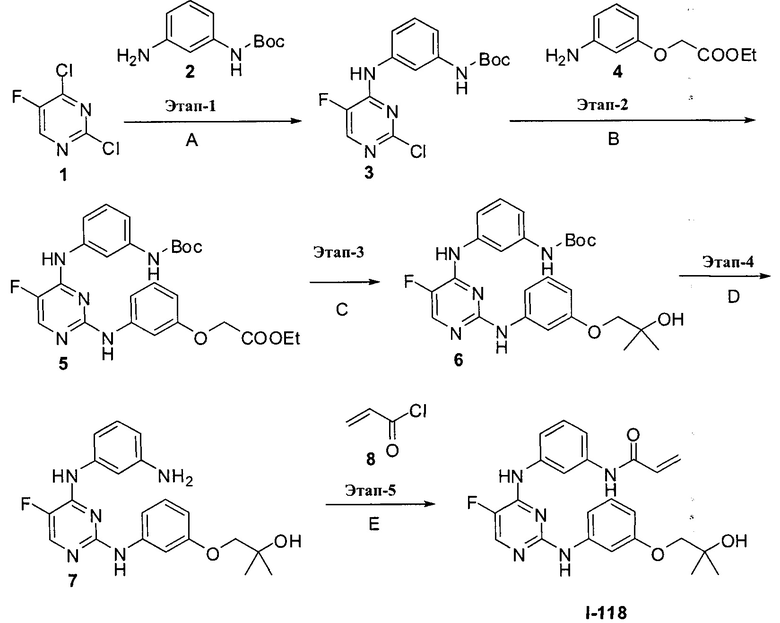
A) DIPEA, n-BuOH, 110°С, 16 часов; В) Pd(OAc)2, BINАР, Cs2CO3, толуол, 100°С, 16 часов; С) MeMgBr (3М раствор в эфире), ТГФ, -78°С, 3 часа; D) ТФА, CH2Cl2, комн. т., 3 часа.
Е) K2CO3, NMP, комн. т., 45 минут.
[00707] Этап-1
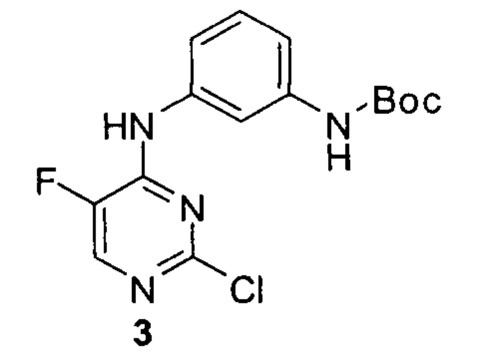
[00708] Соединение 3 получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20.
[00709] Этап-2
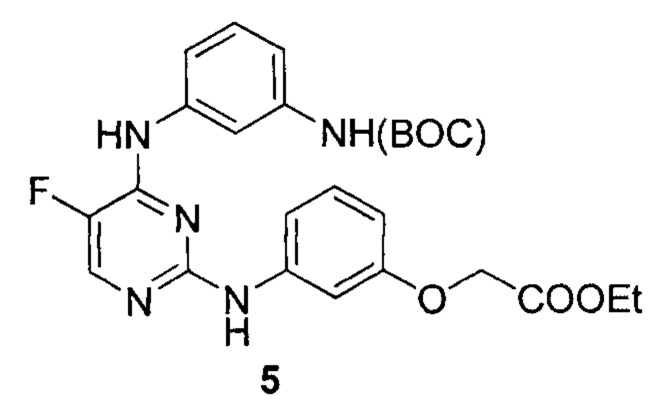
[00710] Раствор 3 (0,7 г, 3,5 ммоль), 4 (1,45g, 4,3 ммоль), Pd(OAc)2 (0,03 г, 0.14 ммоль), BINAP (0.013 г, 0.021 ммоль) и Cs2CO3 (2,8 г, 8,7 ммоль) в дегазированном толуоле (толуол продували N2 в течение 30 минут) нагревали с обратным холодильником в течение 16 чаосов при температуре 100°С в атмосфере N2. Реакционную смеет охлаждали, разбавляли EtOAc (15 мл) промывали водой (10 мл), солевым раствором (10 мл) и сушили над Na2SO4. Фильтровали, и затем концентрировали при, пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 6/4) с получением 5 (700 мг, 40%) в форме белого твердого вещества.
[00711] Этап-3
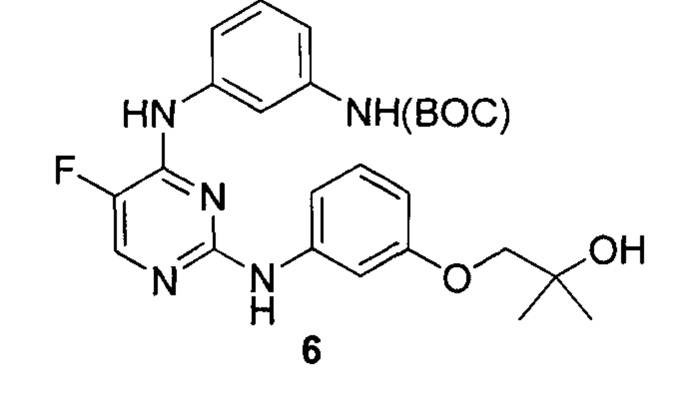
[00712] К раствору 5 (0.4 г, 0.8 ммоль) в ТГФ (10 мл) при помешивании добавляли метил магний бромид (3 М раствор в эфире, 1.6 мл, 4.8 ммоль) при температуре -78°С. Реакционную смесь оставляли нагреваться до -30°С на 3 часа, после чего снова охлаждали до -78°С насыщенным раствором аммония хлорида (5 мл). Реакционную смесь фильтровали через Целит® и концентрировали фильтрат при пониженном давлении с получением 6 в форме бледно-желтого твердого вещества (300 мг, 78%) которое использовали на следующем этапе без дополнительной очистки.
[00713] Этап-4
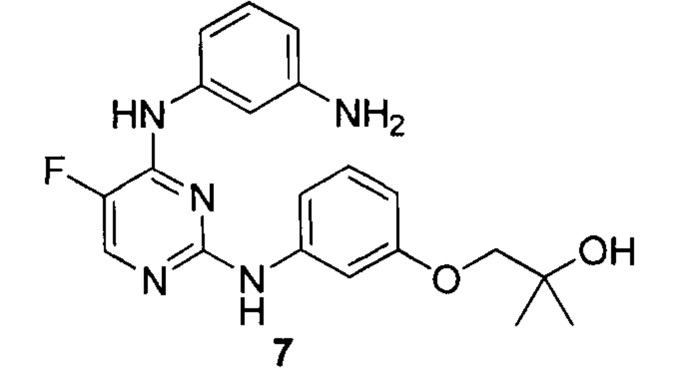
[00714] К раствору 6 (0,2 г, 0,4 ммоль) в CH2Cl2 (7,5 мл) при помешивании добавляли ТФА (3 vol.) при температуре 0°С. Реакционную смесь оставляли дойти до комнатной температуре, после чего ее помешивали 3 час. Затем концентрировали при пониженном давлении, приводили к форме основания раствором NaHCO3 (5 мл) и экстрагировали CH2Cl2 (3×10 мл). Объединенный органический экстракт промывали водой (2 мл) и солевым раствором (2 мл). Сушили над Na2SO4, фильтровали, и затем концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 6/4) с получением 7 (130 мг, 86%) в форме белого твердого вещества.
[00715] Этап-5
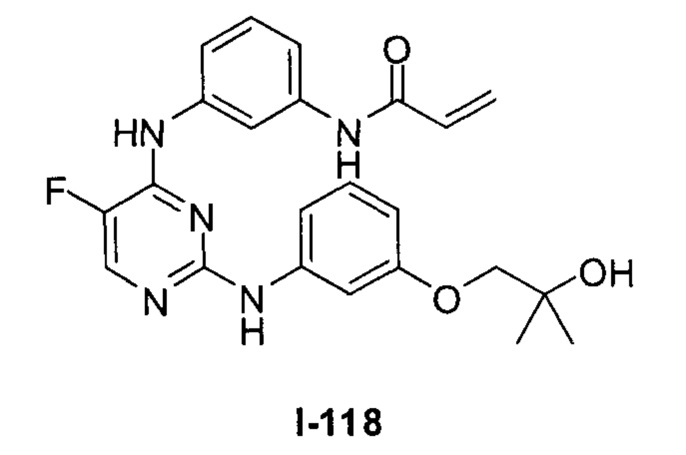
[00716] К раствору 7 (0,08 г, 0,2 ммоль) и калия карбоната (0.11 г, 0,8 ммоль) в NMP (1 мл) при температуре 0°С при помешивании добавляли 8 (0,023 г, 0.22 ммоль), и
помешивали реакционную смесь при температуре 0°С в течение 45 минут. Реакционную смесь по каплям при помешивании добавляли в холодный 10% раствор NaHCO3 и помешивали при той же температуре (0°С) в течение 30 минут. Твердой осадок отделяли методом фильтрации через воронку Бухнера. Затем твердый осадок промывали холодной водой и гексаном растворяли в смеси метанол/дихлорметан (50:50, 5 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (10 мл), добавляли к нему Et3N и затем экстрагировали этил "ацетатом (2×5 мл). Объединенный экстракт этил ацетата промывали водой (2 мл), солевым раствором (2 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный осадок очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 5/5) с получением -118 (35 мг 38%) в форме белого твердого вещества. 1Н-ЯМР (CD3OD) δ ppm: 1.27 (s, 6Н), 3.67 (s, 2Н), 5.76 (dd, J=2.4 & 9.6 Гц, 1Н), 6.34 (dd, J=2 & 16.8 Гц, 1H), 6.42 (dd, J=9.6 & 16.8 Гц, 1Н), 6.54 (td, J=2 & 7.2 Гц, 1Н), 7.07-7.12 (m, 2Н), 7.27-7.31 (m, 2Н), 7.40 (d, J=8 Гц, 1Н), 7.45 (d, J=8 Гц, 1H), 7.92 (d, J=4 Гц, 1Н), 8.07 (d, J=2 Гц, 1Н); ЖХМС: m/е 436.2 (М-1).
ПРИМЕР 72
[00717] Получение 1 N-(3-(5-фтор-2-(3-(2-морфолино-2-охоэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-110
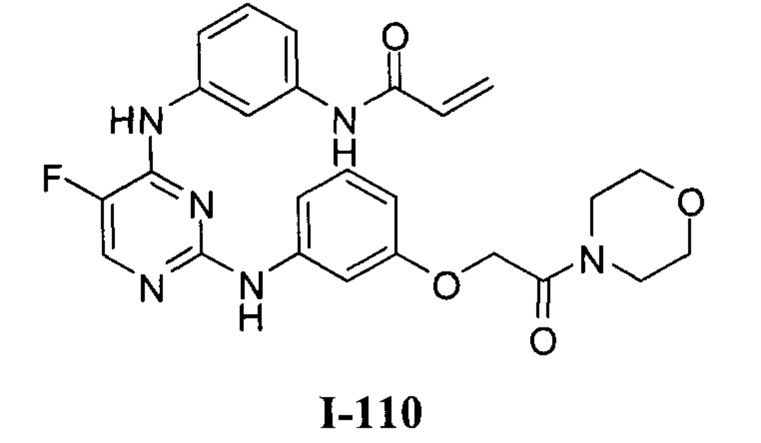
[00718] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-[(3-аминофенокси)Ацетил]-морфолина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.4-3.5 (bm, 4Н), 3.5-3.6 (bm, 4Н), 4.69 (s, 2Н), 5.75 (dd, J=2 & 10 Гц, 1H), 6.25 (dd, J=2 & 17.2 Гц, 1Н), 6.42-6.49 (m, 2Н), 7.05 (t, J=8 Гц, 1Н), 7.29 (t, J=8 Гц, 3Н), 7.41 (d, J=8 Гц, 1H),
7.57 (d, J=8.8 Гц, 1H), 7.91 (s, 1H), 8.12 (d, J=3.6 Гц, 1H), 9.15 (s, 1H), 9.45 (s, 1H), 10.12 (s, 1H); ЖХМС: m/e 491.0 (M-2).
[00719] Промежуточное соединение 4-[(3-аминофенокси)Ацетил]-морфолин получали с использованием схемы описанной ниже.
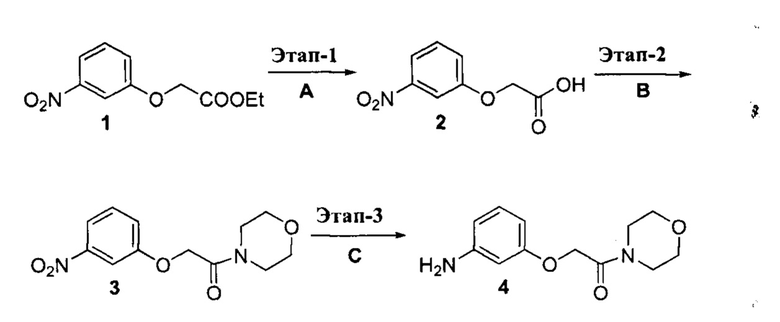
A) LiOH, ТГФ, МеОН, H2O, комн. т., 4 часа; В) SOCl2, 85°С, морфолин, 0°С, 30 минут; С) Pd-C, Н2, этил ацетат, комн. т., 2 часа.
[00720] Этап-1
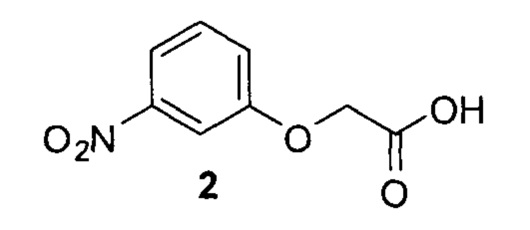
[00721] К раствору 1 (1.0 г, 4.44 ммоль) в смеси метанол/ТГФ/вода: 5 мл/»5 мл/5 мл при помешивании добавляли LiOH моногидрат (0.75 г, 17.76 ммоль) и помешивали реакционную смесь при комнатной температуре в течение 4 часа. Концентрировали при пониженном давлении, осадок разбавляли водой (10 мл), подкисляли 1.0 N HCl (РН -5-6) и экстрагировали эфиром (2×20 мл). Объединенный экстракт эфира промывали водой (20 мл), солевым раствором (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 2 (0.8 г, 91.43%) в форме беловатого твердого вещества.
[00722] Этап-2
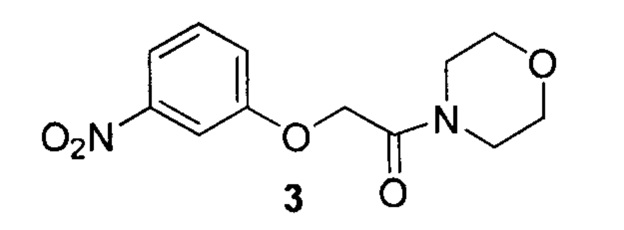
[00723] Тионил хлорид (2.0 ml, 27.56 ммоль) добавляли к 2 (0.2 г, 1.014 ммоль) в атмосфере азота. К смеси добавляли каплю N,N Диметилформамида и помешивали содержимое при температуре 85°С в течение 2 часа. После охлаждения до комнатной
температуры тионил хлорид удаляли методом концентрации при пониженном давлении. Осадок охлаждали до 0°С, добавляли маленькими порциями морфолин (0.5 г, 5.74 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 30 минут. Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали в течение 30 минут, охлаждали и тушили водой (10 мл). Содержимое экстрагировали эфиром (2×10 мл) и промывали объединенный экстракт эфира водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3 (0.180 г, 66.67%) в форме желтого твердого вещества.
[00724] Этап-3
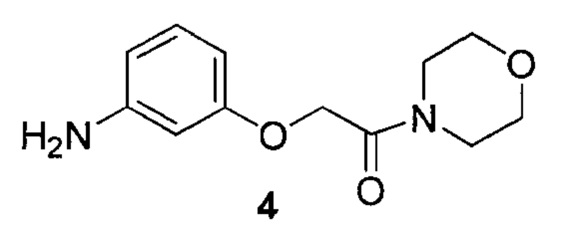
[00725] К раствору 3 (0.180 г, 0.676 ммоль) в этил ацетате (10 мл) добавляли Pd/C (0.036 г, 20% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (давление водорода 1.0 Кг) при комнатной температуре в течение 2 часа. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением А (0.14 г, 87.67%)) в форме беловатого твердого вещества. Его использовали на следующем этапе без дополнительной очистки.
ПРИМЕР 73
[00726] Получение N-(3-(5-фтор-2-(3-(1-гидрокси-2-метилпропан-2-илокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-91
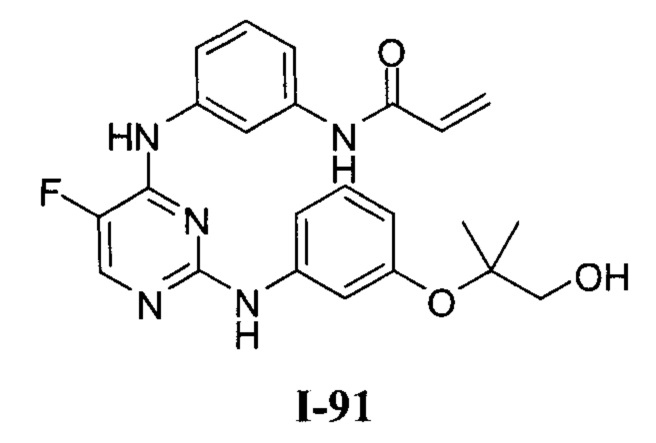
[00727] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(1-гидрокси-2-метилпропан-2-илокси)анилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.16 (s, 6Н), 3.32-3.35 (m, 2Н), 4.81 (t, J=5.74 Гц, 1H), 5.74 (dd, J=1.84& 10.04 Гц, 1H), 6.24 (dd,
J=1.88 & 16.96 Гц, 1H), 6.44 (dd, J=10.12 & 16.96 Гц, 1H), 6.50 (dd, J=2.12 & 7.96 Гц, 1H), 7.02 (t, J=8.12 Гц, 1H), 7.26-7.30 (m, 2H), 7.41 (d, J=8.16 Гц, 1H), 7.48 (d, J=8.24 Гц, 1H), 7.57 (d, J=8.12 Гц, 1H), 7.92 (s, 1H), 8.09 (d, J=3.6 Гц, 1H), 9.07 (s, 1H), 9.41 (s, 1H), 10.09 (s, 1H); ЖХМС: m/e 438.0 (M+1).
[00728] Промежуточное соединение 3-(1-гидрокси-2-метилпропан-2-илокси)анилин получали с использованием схемы описанной ниже.
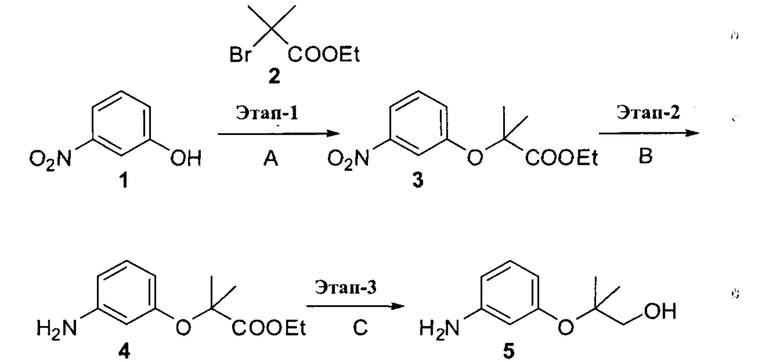
А) K2CO3, ДМФ, 16 часов, комн. т.; В) Pd/C, этанол, 5 часов, комн. т.; С) LAH (1М в ТГФ раствор), 0°С до комн. т., 2 часа.
[00729] Этап-1
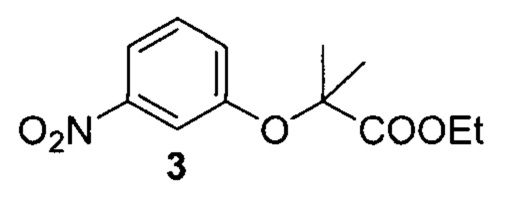
[00730] К раствору 1 (0.5 г, 3.59 ммоль) и 2 (0.84 г, 4.316 ммоль) в ДМФ добавляли K2CO3 (0.99 г, 7.194 ммоль). Помешивали при комнатной температуре в течение 16 часов, затем реакционную смесь концентрировали при пониженном давлении. Осадок промывали этил ацетатом (10 мл) и промывали 10% раствором NaOH (5 мл), водой (5 мл) и солевым раствором (5 мл). Сушили над Na2SO4, после чего концентрировали при пониженном давлении с получением 3 в форме красно-коричневой жидкости (0.5 г, 52%).
[00731] Этап-2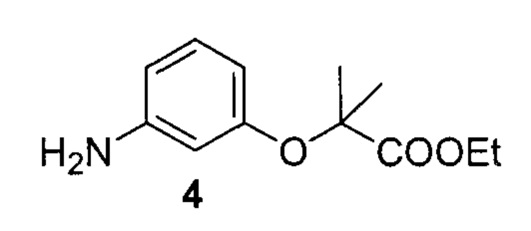
[00732] К раствору 3 (0.45 г, 1.77 ммоль) в этаноле (5 мл) при помешивании добавляли Pd/C (45 мг) после чего реакционную смесь гидрогенезировали (давление в пневмокамере,
~1.5 Кг) в течение 5 часов. Реакционную смесь пропускали через целитную® пластину и концентрировали под вакуумом с получением 4 (0.35 г, 88%) в форме бесцветной жидкости.
[00733] Этап-3
[00734] К раствору 4 (0.25 г, 1.15 ммоль) в ТГФ (5 мл)в атмосфере N2 при помешивании добавляли LAH (3.45 мл, 3.35 ммоль, 1М раствор в ТГФ,) при температуре 0°С. Реакционную смесь оставляли дойти до комнатной температуры, после чего ее помешивали в течение 2 часа. Затем аккуратно гасили насыщенным раствором Na2SO4 (2 мл), фильтровали и концентрировали. Полученный осадок очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 6/4) с получением 5 (0,15 g 71%) в форме светло-коричневого твердого вещества.
ПРИМЕР 74
[00735] Получение N-(3-(5-фтор-2-(3-(2-(2-оксопирролидин-1-ил)этокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-164
[00736] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(2-(2-оксопирролидин-1-ил)этокси)анилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.89 (quin, J=7.6 Гц, 2Н), 2.21 (t, J=8 Гц, 2Н), 3.40 (t, J=6.8 Гц, 2Н), 3.50 (t, J=5.6 Гц, 2Н), 3.93 (t, J=5.2 Гц, 2Н), 5.75 (dd, J=2 & 10 Гц, 1Н), 6.25 (dd, J=2& 16.84 Гц, 1Н), 6.42-6.49 (m, 2Н), 7.05 (t, J=8.4 Гц, 1H), 7.28 (t, J=8 Гц, 2Н), 7.33 (s, 1Н), 7.43 (d, J=8 Гц, 1H), 7.57 (d, J=8 Гц, 1H), 7.92 (s, 1H), 8.12 (d, J=3.6 Гц, 1H), 9.15 (s, 1H), 9.45 (s, 1H), 10.13 (s, 1H); ЖХМС: m/e 475 (M-2).
ПРИМЕР 75
[00737] Получение N-(3-(5-фтор-2-(6-(3-(метилсульфонил)пропокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-80
[00738] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-амино-6-(3-(метилсульфонил)пропокси)пиридина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.05-2.20 (m, 2Н), 3.00 (s, 3Н), 3.24 (t, J=7.46 Гц, 2Н), 4.27 (t, J=6.32 Гц, 2Н), 5.75 (dd, J=1.76 & 10 Гц, 1H), 6.25 (dd, J=1.8 & 16.96 Гц, 1Н), 6.45 (dd, J = 10.04 & 16.92 Гц, 1Н), 6.65 (d, J=8.88 Гц, 1Н), 7.27 (t, J=8.08 Гц, 1Н), 7.39 (d, J=8.08 Гц, 1Н), 7.49 (d, J=8 Гц, 1Н), 7.92 (s, 1H), 7.99 (dd, J=2.6 & 8.76 Гц, 1H), 8.07 (d, J=3.64 Гц, 1H), 8.31 (d, J=2.28 Гц, 1H), 9.10(s, 1Н), 10.11 (s, 1Н); ЖХМС: m/e 486.9 (M+1).
ПРИМЕР 76
[00739] Получение N-(3-(2-(6-циклобутоксипиридин-3-иламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-79
[00740] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-амино-6-циклобутоксипиридина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.57-1.66 (m, 1H), 1.71-1.78 (m, 1Н), 1.94-2.04 (m, 2Н), 2.32-2.38 (m, 2Н), 4.95-5.05 (m, 1Н), 5.73-5.76 (m, 1H),
6.25 (dd, J=1.92 & 16.92 Гц, 1H), 6.45 (dd, J=10.08 & 16.92 Гц, 1H), 6.58 (d, J=8.84 Гц, 1H), 7.25 (t, J=8.04 Гц, 1H), 7.39 (d, J=7.84 Гц, 1H), 7.45-7.55 (m, 1H), 7.90 (s, 1H), 7.94 (dd, J=2.72 & 8.88 Гц, 1H), 8.06 (d, J=3.68 Гц, 1H), 8.27 (d, J=2.6 Гц, 1H), 9.04 (s, 1H), 9.41 (s, 1H), 10.1 (s, 1H); ЖХМС: m/e 421.2 (M+1).
ПРИМЕР 77
[00741] Получение N-(3-(5-фтор-2-(6-((1-метилпиперидин-4-ил)метокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-78
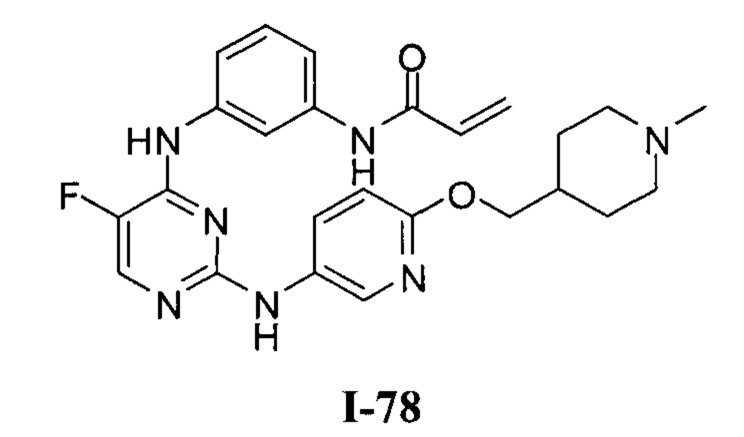
[00742] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-амино-6-(1-метилпиперидин-4-ил)метоксипиридина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.23-1.27 (m, 3Н), 1.65-1.69 (m, 2Н), 1.83 (t, J=11.72 Гц, 2Н), 2.14 (s, 3Н), 2.75 (d, J=11.24 Гц, 2Н), 4.0 (d, J=6.2 Гц, 2Н), 5.74 (dd, J=2 & 10.04 Гц, 1Н), 6.24 (dd, J=1.96 & 16.92 Гц, 1Н), 6.45 (dd, J=10.08 & 16.92 Гц, 1Н), 6.62 (d, J=8.88 Гц, 1Н), 7.27 (t, J=8.08 Гц, 1H), 7.40 (d, J=8.88 Гц, 1H), 7.47 (d, J=7.6 Гц, 1Н), 7.92 (s, 1H), 7.97 (dd, J=2.76 & 8.92 Гц, 1Н), 8.07 (d, J=3.72 Гц, 1H), 8.28 (d, J=2.64 Гц, 1H), 9.07 (s, 1Н), 9.41 (s, 1Н), 10.11 (s, 1H); ЖХМС: m/е 478.0 (М+1).
ПРИМЕР 77
[00743] Получение N-(3-(5-фтор-2-(4-хлоро-3-метоксифениламино)пиримидин-4-иламино)фенил)акриламида I-74
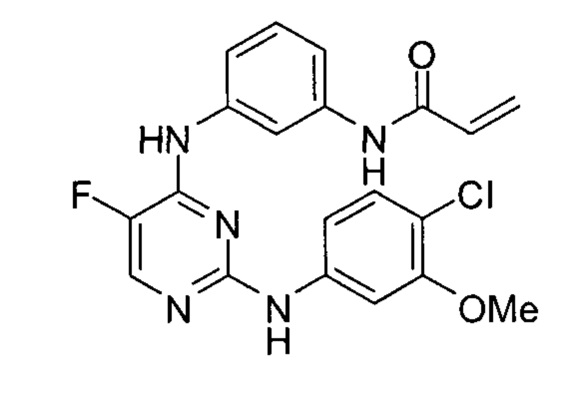
[00744] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-хлоро-3-метоксианилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.64 (s, 3Н), 5.74 (dd, J=2.12 & 9.96 Гц, 1Н),6.24 (dd, J=1.84 & 17 Гц, 1Н), 6.44 (dd, J=10 & 16.84 Гц, 1H),7.13 (s, 1Н), 7.28 (t, J=8.08 Гц, 1Н), 7.36 (dd, J=2.12 & 8.68 Гц, 1Н), 7.40 (d, J=7.64 Гц, 1Н), 7.45 (d, J=2.04 Гц, 1Н), 7.50 (d, J=8.12 Гц, 1Н), 7.91 (s, 1Н), 8.13 (d, J=3.52 Гц, 1H), 9.28 (s, 1Н), 9.48 (s, 1Н), 10.12 (s, 1Н); ЖХМС: m/е 414.0 (М+1).
ПРИМЕР 78
[00745] Получение N-(3-(5-фтор-2-(4-(2-гидрокси-2-метилпропокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-73
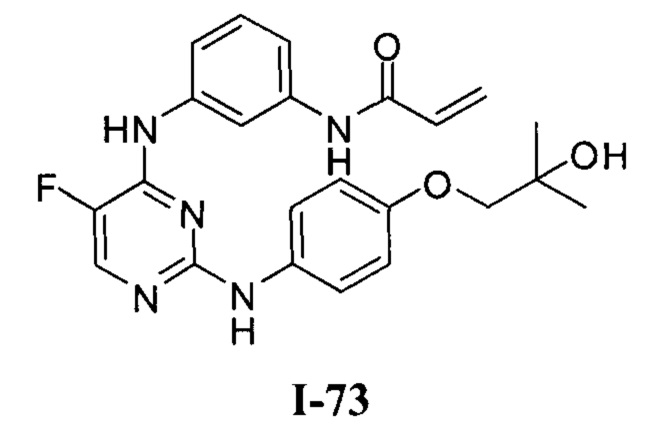
[00746] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-(2-гидрокси-2-метилпропокси)анилин вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.33 (s, 6Н), 3.75 (s, 2Н), 5.80 (dd, J=3.28 & 10.64 Гц, 1H), 6.39 (dd, J=2.24 & 16.96 Гц, 1Н), 6.47 (dd, J=9.6 & 16.96 Гц, 1Н), 6.84 (td, J=3.48 & 9.0 Гц, 2Н), 7.30 (t, J=7.72 Гц, 1Н), 7.41-7.50 (m, 4Н), 7.89 (d, J=3.88 Гц, 1Н), 8.09 (s, 1Н); ЖХМС: m/е 438 (М+1).
ПРИМЕР 79
[00747] Получение N-(3-(5-фтор-2-(6-(1,1-диоксидотиоморфолин-4-ил)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-72
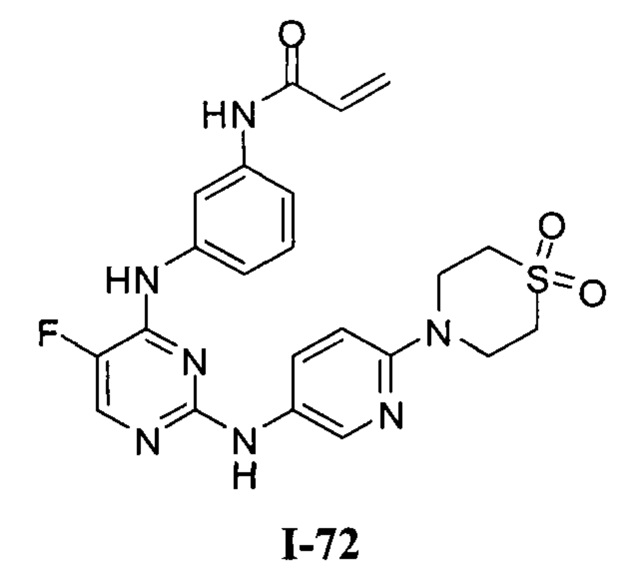
[00748] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-амино-6-(1,1-диоксидотиоморфолин-4-ил)пиридина вместо 4 на этапе-2. 1H-ЯМР (ДМСО-d6) δ ppm: 3.00-3.15 (bm, 4Н), 3.90-4.10 (bm, 4Н), 5.76 (dd, J=1.64 & 10.04 Гц, 1Н), 6.26 (dd, J=1.72 & 16.92 Гц, 1Н), 6.46 (dd, J=10.04 & 16.88 Гц, 1Н), 6.87 (d, J=9.04 Гц, 1Н), 7.20 (t, J=8.04 Гц, 1H), 7.39 (d, J=8.24 Гц, 1H), 7.50 (d, J=7.68 Гц, 1H), 7.90-7.93 (m, 2Н), 8.06 (d, J=3.6 Гц, 1H), 8.35 (d, J=2.4 Гц, 1Н), 9.0 (s, 1H), 9.40 (s, 1Н), 10.12 (s, 1H); ЖХМС: m/е 484 (М+1).
ПРИМЕР 80
[00749] Получение N-(3-(5-фтор-2-(6-(2-(2-оксопирролидин-1-ил)этокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-70
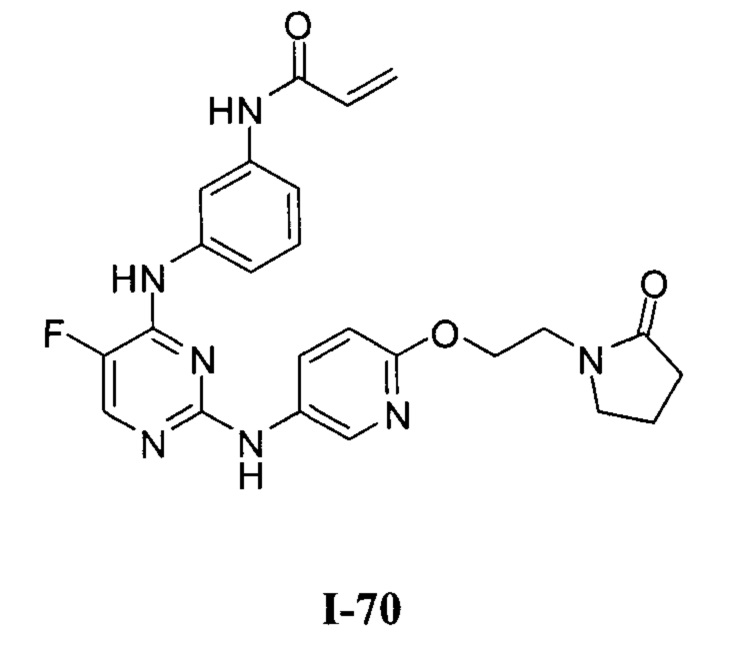
[00750] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-амино-6-(2-(2-оксопирролидин-1-ил)этокси)пиридина вместо 4 на этапе-2. 1H-ЯМР (ДМСО-d6) 8 ppm:
1.90 (quintet, J=7.6 Гц, 2H), 2.19 (t, J=8.04 Гц, 2H), 3.41 (t, J=6.88 Гц, 2H), 3.50 (t, J=5.36 Гц, 2H), 4.27 (t, J=5.48 Гц, 2H), 5.75 (d, J=10.92 Гц, 1H), 6.25 (d, J=17.04 Гц, 1H), 6.45 (dd, J=10.12 & 16.84 Гц, 1H), 6.63 (d, J=8.96 Гц, 1H), 7.27 (t, J=8.04 Гц, 1H), 7.39 (d, J=7.56 Гц, 1H), 7.47 (d, J=7.32 Гц, 1H), 7.92 (s, 1H), 7.98 (dd, J=2.36 & 8.84 Гц, 1H), 8.08 (d, J=3.3 Гц, 1H), 8.31 (d, J=2.24 Гц, 1H), 9.10 (s, 1H), 9.44 (s, 1H), 10.11 (s, 1H); ЖХМС: m/e 478.0 (M+1).
ПРИМЕР 81
[00751] Получение (R)-N-(3-(5-фтор-2-(6-(тетрагидрофуран-3-илокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-69
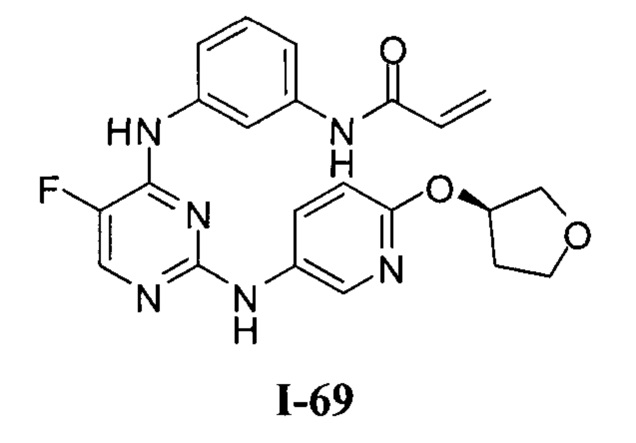
[00752] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (к)-3-амино-6-(тетрагидрофуран-3-илокси)пиридина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.91-1.99 (m, 1Н), 2.14-2.23 (m, 1Н), 3.70-3.77 (m, 2Н), 3.81 (dd, J=7.90 & 15.48 Гц, 1Н), 3.88 (dd, J=4.76 & 10.16 Гц, 1H), 5.38 (t, J=4.68 Гц, 1Н), 5.75 (dd, J=1.72 & 10.08 Гц, 1Н), 6.24 (d, J=16.92 Гц, 1Н), 6.45 (dd, J=10.16 & 16.88 Гц, 1Н), 6.63 (d, J=8.84 Гц, 1Н), 7.26 (d, J=7.64 Гц, 1Н), 7.39 (d, J=7.92 Гц, 1H), 7.46 (d, J=7.64 Гц, 1H), 7.92 (s, 1H), 7.97 (dd, J=2.6 & 8.83 Гц, 1Н), 8.07 (d, J=3.6 Гц, 1H), 8.32 (d, J=2.48 Гц, 1H), 9.08 (s, 1Н), 9.42 (s, 1H), 10.10 (s, 1Н); ЖХМС: m/е 437.2 (М+1).
ПРИМЕР 82
[00753] Получение N-(3-(2-(4-хлоро-3-(3-(метилсульфонил)пропокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-55
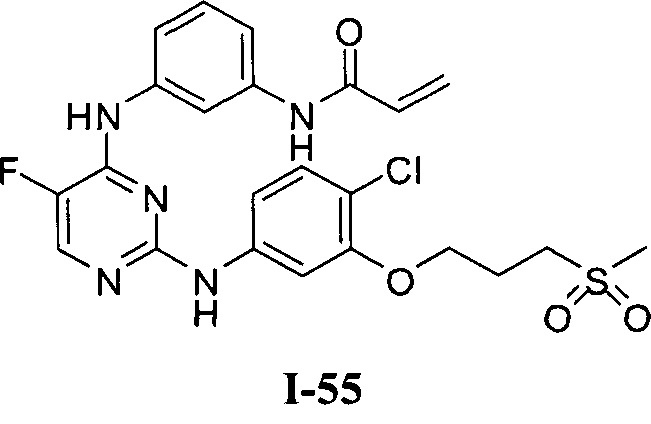
[00754] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-хлоро-3-(3-(метилсульфонил)пропокси)анилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.07-2.14 (m, 2Н), 3.0 (s, 3Н), 3.22 (t, J=1.12 Гц, 2Н), 3.90 (t, J=6.08 Гц, 2Н), 5.75 (dd, J=1.88 & 10.08 Гц, 1Н), 6.24 (dd,J=1.84 & 16.92 Гц, 1H), 6.44 (dd, J=10.12 & 16.96 Гц, 1H), 7.15 (d, J=8.72 Гц, 1H), 7.30 (t, J=8.08 Гц, 1H), 7.35 (dd, J=2.2 & 8.8 Гц, 1Н), 7.43 (d, J=8 Гц, 1Н), 7.45-7.55 (m, 2Н), 7.91 (s, 1Н), 8.14 (d, J=3.56 Гц, 1Н), 9.31 (s, 1H), 9.49 (s, 1Н), 10.14 (s, 1H); ЖХМС: m/е 520.0 (М+1).
ПРИМЕР 83
[00755] Получение N-(3-(2-(3-фтор-4-(2-метоксиэтокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-96
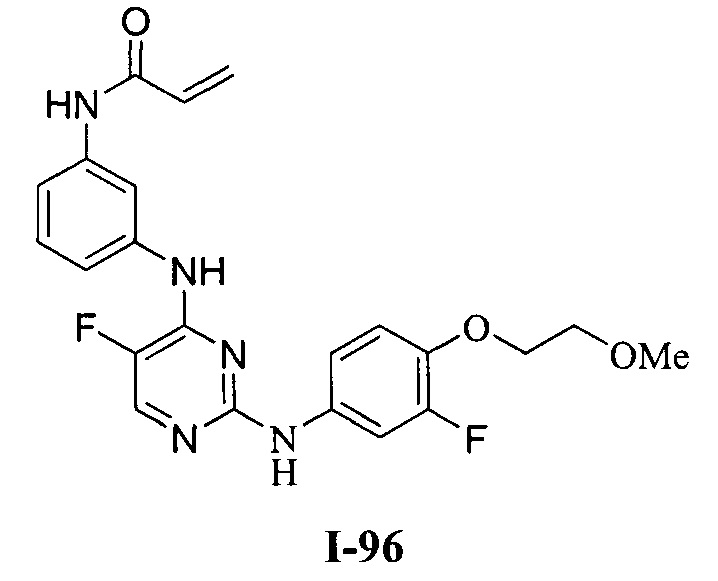
[00756] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе-2. 1Н-ЯМР (ДМСО, 400 МГц) δ 10.13 (s, 1H), 9.43 (s, 1H), 9.18 (s, 1Н), 8.09 (d, 1H, J=3.68 Гц), 7.92 (s, 1H), 7.65 (dd, 1H, J=2.3, 14.2 Гц), 7.47 (d, 1H, J=8.24 Гц), 7.41 (d, 1H, J=8.28 Гц), 7.27 (t, 2Н, J=8.0 Гц), 6.94 (t, 1H, J=9.4
Гц), 6.44 (dd, 1H, J=16.96, 10.1 Гц), 6.23 (dd, 1Н, J=1.84, 16.96 Гц), 5.73 (dd, 1H, J=1.4, 10.1 Гц), 4.04 (m, 2H), 3.61 (m, 2H), 3.29 (s, 3H). MS m/z: 442.0 (M+H+).
ПРИМЕР 84
[00757] Получение N-(3-(2-(4-трет-бутоксикарбонил-2,3-дигидробензо[1,4]оксазин-6-ил)амино-5-фторпиримидин-4-иламино)фенил)акриламида I-175
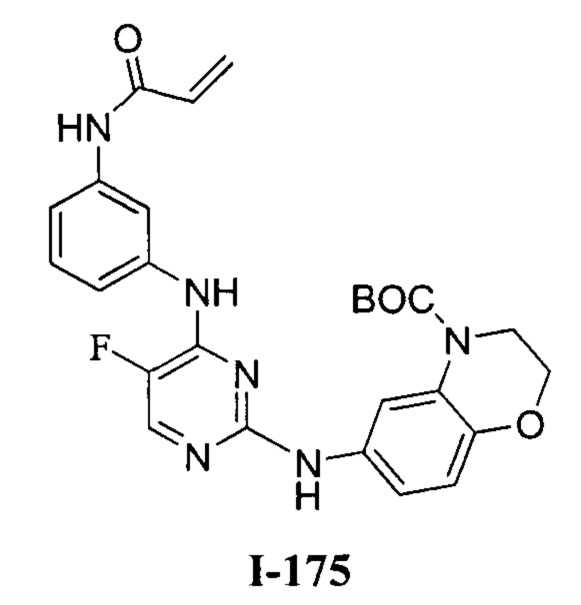
[00758] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 6-амино-4-трет-бутоксикарбонил-2,3-дигидробензо[1,4]оксаксина вместо 4 на этапе-2. 507.1 (М+Н+).
ПРИМЕР 85
[00759] Получение N-(3-(2-(4-трет-бутоксикарбонил-2,3-дигидробензо[1,4]оксазин-6-ил)амино-5-фторпиримидин-4-иламино)фенил)акриламида I-174
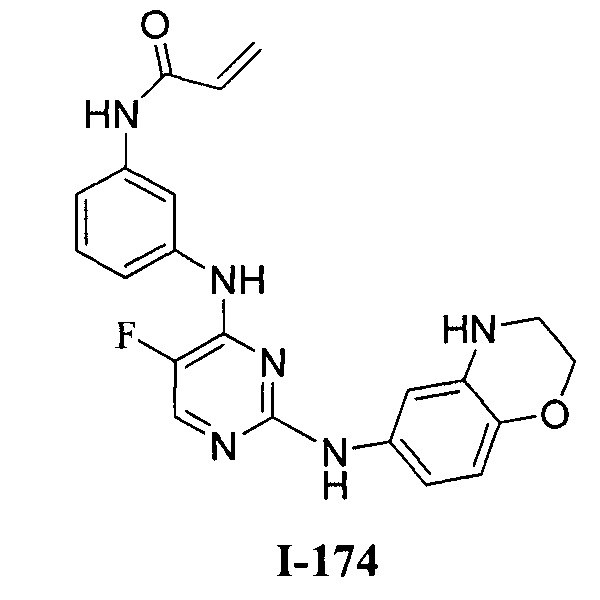
[00760] Указанное в названии соединение получали обработкой продукта из примера 83 4N HCl в диоксане при комнатной температуре в течение 1 часа, после чего удаляли растворители под вакуумом. MS m/z: 407.1 (М+Н+).
ПРИМЕР 86
[00761] Получение N-(3-(2-(4-трифторАцетил-2,3-дигидробензо[1,4]оксазин-6-ил)амино-5-фторпиримидин-4-иламино)фенил)акриламида I-143
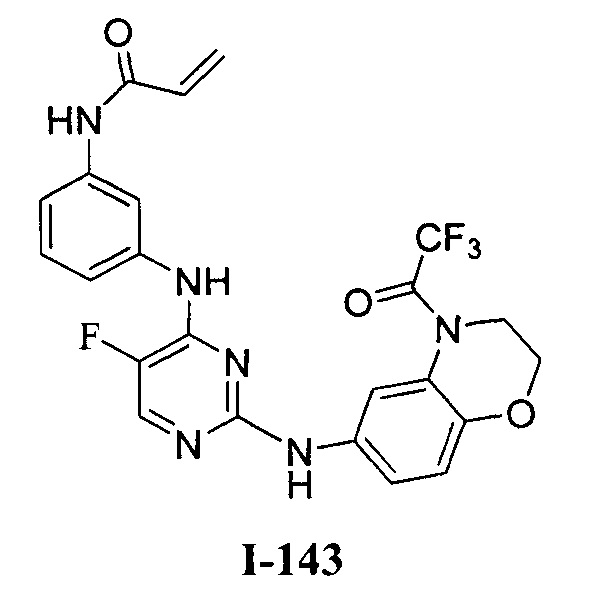
[00762] Указанное в названии соединение получали обработкой продукта из примера 85 трифторуксусным ангидридом в диоксане при комнатной температуре в течение 1 часа, после чего удаляли растворители под вакуумом. MS m/z: 503.1 (М+Н+).
ПРИМЕР 87
[00763] Получение N-(3-(2-(4-метилсульфонил-2,3-дигидробензо[1,4]оксазин-6-ил)амино-5-фторпиримидин-4-иламино)фенил)акриламида I-140
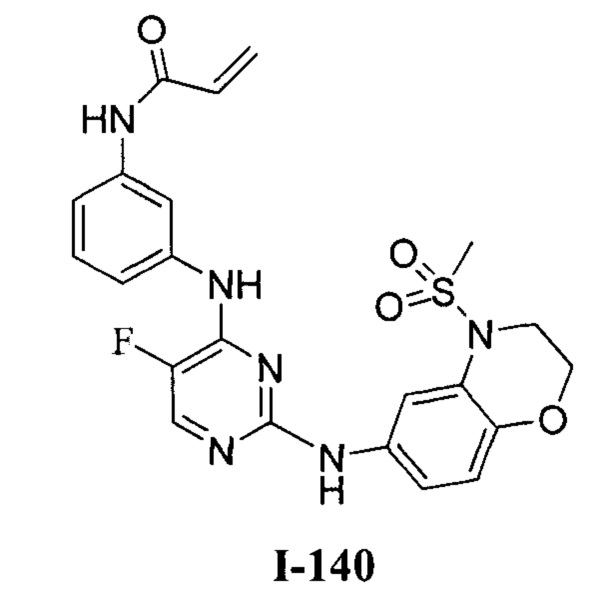
[00764] Указанное в названии соединение получали обработкой продукта из Примера 85 метилсульфонил хлорид Et3N в CH2Cl2 при температуре 0°С в течение 30 минут, после чего промывали водным NaHCO3, сушили Na2SO4 и удаляли растворители под вакуумом. MS m/z: 485.1 (М+Н+).
ПРИМЕР 88
[00765] Получение N-(3-(2-(4-метил-2,3-дигидробензо[1,4]оксазин-6-ил)амино-5-фторпиримидин-4-иламино)фенил)акриламида I-126
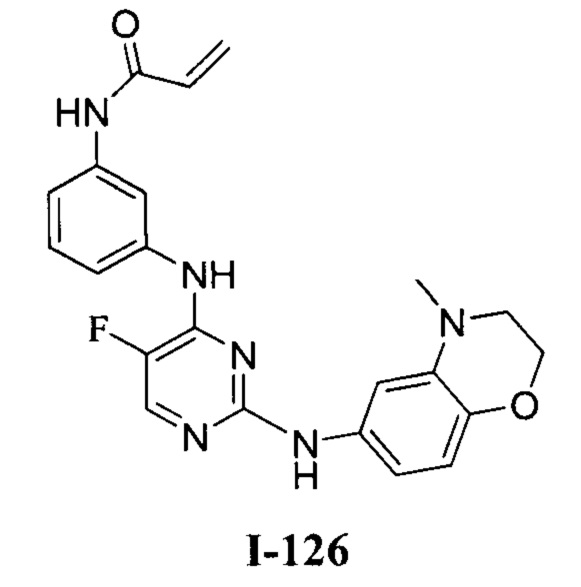
[00766] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 6-амино-4-метил-2,3-дигидробензо[1,4]оксазина вместо 4 на этапе-2 MS m/z: 421.1 (М+Н+).
ПРИМЕР 89
[00767] Получение N-(3-(2-(4-Ацетил-2,3-дигидробензо[1,4]оксазин-6-ил)амино-5-фторпиримидин-4-иламино)фенил)акриламида I-112
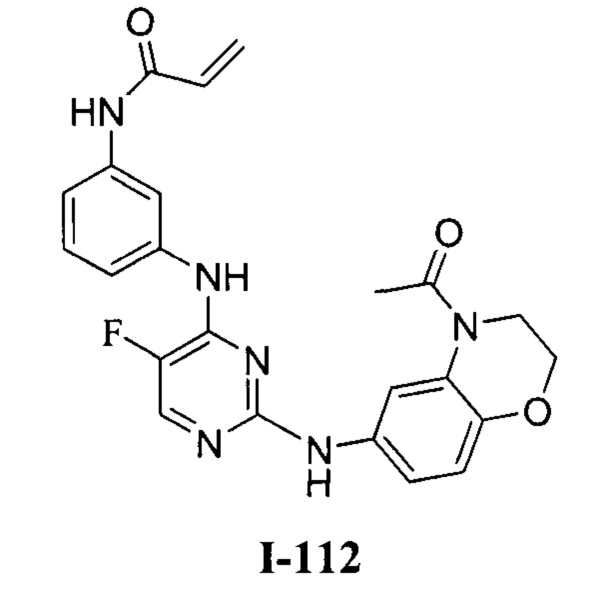
[00768] Указанное в названии соединение получали обработкой продукта из Примера 85 уксусным ангидридом и пиридином в CH2Cl2 при комнатной температуре в течение 1 часа, после чего промывали водным NaHCOs, сушили Na2SO4 и удаляли растворители под вакуумом. MS m/z: 449.1 (М+Н+).
ПРИМЕР 90
[00769] Получение N-(3-(2-(1-трет-бутоксикарбонил-1Н-индазол-5-ил)амино)-5-фторпиримидин-4-иламино)фенил)акриламида I-151
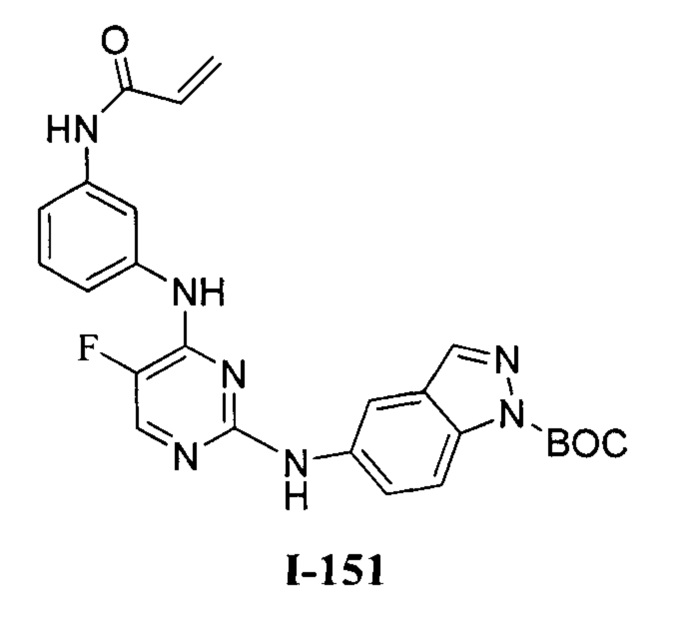
[00770] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 5-амино-N-(трет-бутоксикарбонил)-1H-индазол вместо 4 на этапе-2 MS m/z: 490.2 (М+Н+).
ПРИМЕР 91
[00771] Получение N-(3-(2-(1Н-индазол-5-ил)амино)-5-фторпиримидин-4-иламино)фенил)акриламида I-156
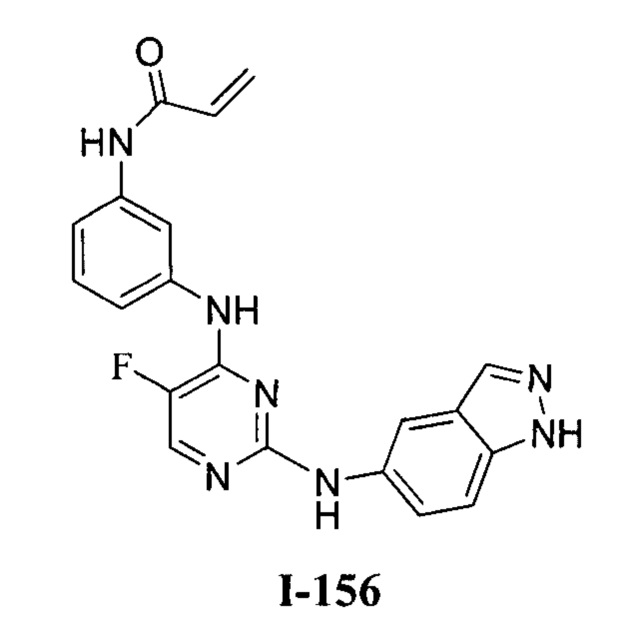
[00772] Указанное в названии соединение получали обработкой продукта из Примера 90 4N HCl в диоксане при комнатной температуре в течение 1 часа, после чего удаляли растворители под вакуумом. MS m/z: 390.1 (М+Н+).
ПРИМЕР 92
[00773] Получение N-(3-(2-(1-метил-1Н-индазол-5-ил)амино)-5-фторпиримидин-4-иламино)фенил)акриламида I-155
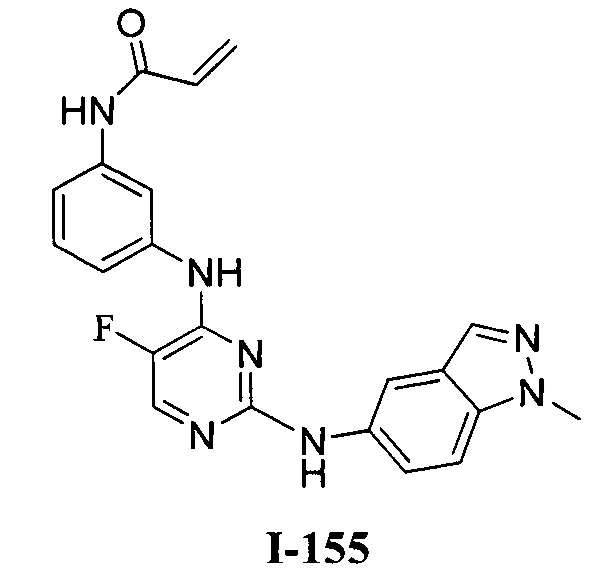
[00774] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 5-амино-1-метил-1H-индазола вместо 4 на этапе-2 MS m/z: 404.2 (М+Н+).
ПРИМЕР 93
[00775] Получение N-(3-(5-фтор-2-(3-сульфамоилфениламино)пиримидин-4-иламино)фенил)акриламида I-160
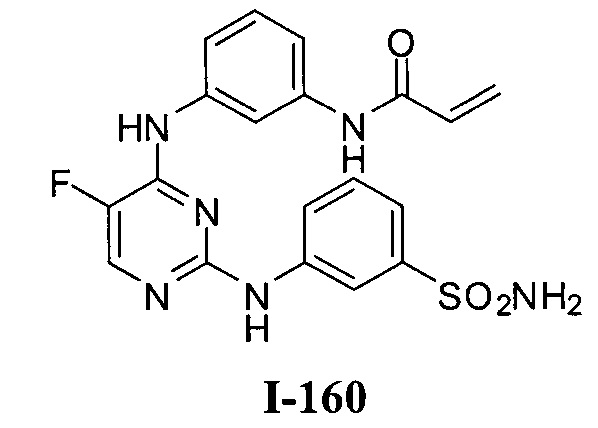
[00776] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
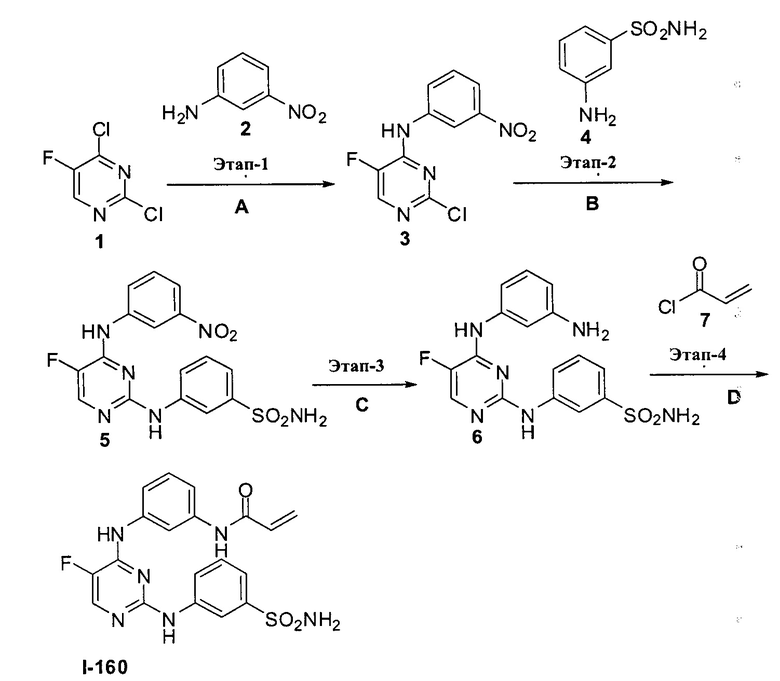
A) DIPEA, n-бутанол, 120°С, 2 часа, нагнетательная трубка; В) АсОН, этанол, 90°С, 16 часов; С) Pd-C, Н2, этанол, комн. т., 3 часа; D) акрилоил хлорид, K2CO3, NMP, 0°С, 60 минут.
[00777] Этап-1
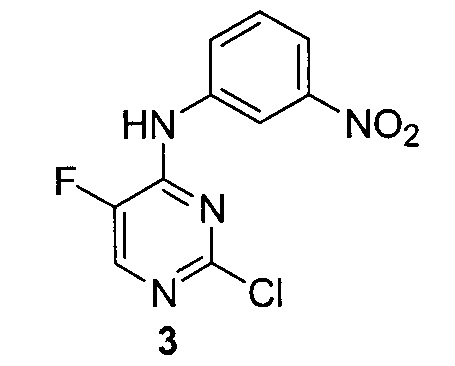
[00778] В нагнетательную трубку помещали соединения 2 (10.0 г, 0.072 mol), 1 (24.1 г, 0.145 mol), n-BuOH (100 мл) и DIPEA (13.9 г, 0.108 mol), и помешивали содержимое при температуре 120°С в течение 2 часа. Реакционную смесь охлаждали, и отделяли твердый осадок методом фильтрации через воронку Бухнера, промывали холодным гексаном и
сушили с получением 3 (12.5 г, 64%) в форме желтого твердого вещества. Его использовали на следующем этапе без дополнительной очистки.
[00779] Этап-2
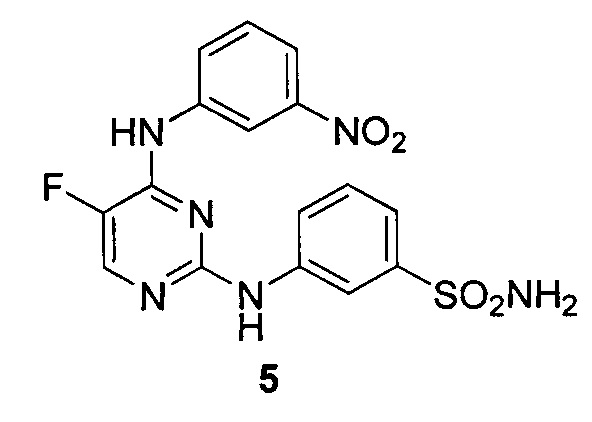
[00780] К раствору 3 (0,25 г, 0,93 ммоль) и 4 (0.805 г, 0,93 ммоль) в этаноле (2,5 мл) добавляли ледяную уксусную кислоту (0.083 г, 1,39 ммоль), и помешивали реакционную смесь в нагнетательной трубке в течение 16 часов при температуре 90°С. Его остужали, образовавшийся твердой осадок отделяли методом фильтрации через воронку Бухнера, промывали холодным эфиром и сушили с получением 5 (0.245 г, 65%) в форме коричневого твердого вещества. Его использовали на следующем этапе без дополнительной очистки.
[00781] Этап-3
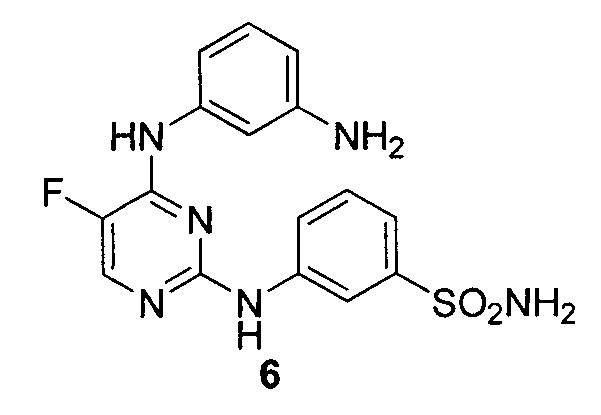
[00782] К раствору 5 (0,1 г, 0,24 ммоль) в метаноле (4 мл) добавляли 10% Pd/C (0,2 г, 20% масс./масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (1,5 Кг давление водорода) при комнатной температуре в течение 3 часа. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 6 (0,076 г, 82%) в форме коричневой жидкости. Его использовали на следующем этапе без дополнительной очистки.
[00783] Этап-1
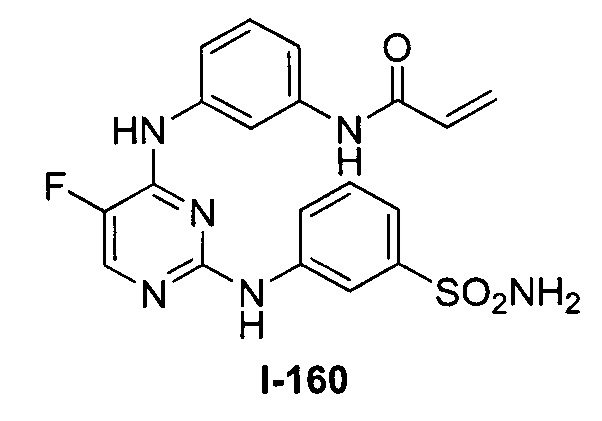
[00784] К раствору 6 (0.07 г, 0.18 ммоль) и калия карбоната (0.051 г, 0.37 ммоль) в NMP (0.7 мл) при температуре 0°С при помешивании добавляли акрилоил хлорид (0.021 г, 0.23 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 60 минут. Реакционную смесь по каплям добавляли при помешивании к холодному 10% раствору NaHCO3 и держали при этой же температуре (0°С) в течение 30 минут. Твердой осадок отделяли методом фильтрации через воронку Бухнера. Затем твердый осадок промывали холодной водой и гексаном растворяли в смеси метанол/дихлорметан (50:50, 5 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (10 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×10 мл). Объединенный экстракт этил ацетата промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением осадка. Сырой остаток далее очищали методом колоночной хроматографии (Al2O3, МеОН/Хлороформ): 3/97) с получением -160 (0,028 г, 35%) в форме светло-коричневого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 5.75 (dd, J=1.68 & 10.24 Гц, 1Н), 6.25 (dd, J=1.8 & 17 Гц, 1Н), 6.43 (dd, J=10 & 16.92 Гц, 1H), 7.27-7.35 (m, 5H), 7.40 (d, J=8 Гц, 1H), 7.60 (d, J=8.16 Гц, 1H), 7.92 (s, 1H), 7.95-8.05 (m, 1H), 8.07 (s, 1H), 8.14 (d, J=3.52 Гц, 1H), 9.50 (s, 2H), 10.12 (s, 1H); ЖХМС: m/e 428.9 (M+1).
ПРИМЕР 94
[00785] Получение N-(3-(5-циано-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-109
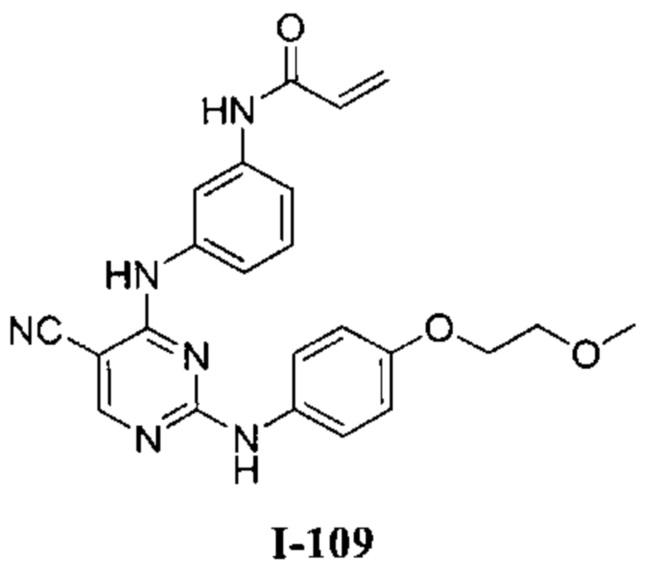
[00786] Указанное в названии соединение получали с использованием этапов и промежуточных соединений, описанных ниже.
A) DMA, K2CO3, комн. т., 10 h, нагнетательная трубка; В) PTSA, диоксан, 100°С, 2 часа; С) Zn(CN)2, Ph3P, ДМФ, 120°С, 12 часов; D) 4N HCl, диоксан, комн. т., 1 час; затем акрилоил хлорид, Et3N, ДХМ, -10°С, 10 минут.
00787] Этап-1
[00788] К раствору 5-бромо-2,4-дихлоропиримидина (0.45 г, 2.0 ммоль) и трет-бутил 3-аминофенилкарбамата (0.44 г, 2.1 ммоль) в DMA (3 мл) добавляли K2CO3 (0.55 г, 4.0 ммоль). Суспензию помешивали в течение 10 часов. Добавляли воду (10 мл) и собирали осадок методом фильтрации. Осадок промывали эфиром и сушили с получением 0.8 г соединения 3. MS: m/e=399.1, 401.2 (M+1).
[00789] Этап-2
[00790] К раствору соединения 3 (400 мг, 1.0 ммоль) и 4-(2-метоксиэтокси)анилина (0.2 г, 1.2 ммоль) в 8 мл диоксана добавляли 4-метилбензолсульфоновой кислоты моногидрат (0.15g, 0.8 ммоль). Смесь помешивали при температуре 100°С в течение двух часов. Растворитель выпаривали. Остаток растворяли в 30 мл этил ацетата и промывали водным раствором NaHCO3, водой и солевым раствором.: Органические слои разделяли и сушили над Na2SO4. После удаления растворителя, сырой продукт очищали методом хроматографии на силикагеле (гексан : EtOAc = 1:1). С получением 0.40 г указанного в названии соединения: MS m/z: 530.1, 532.1(М+Н+).
[00791] Этап-3
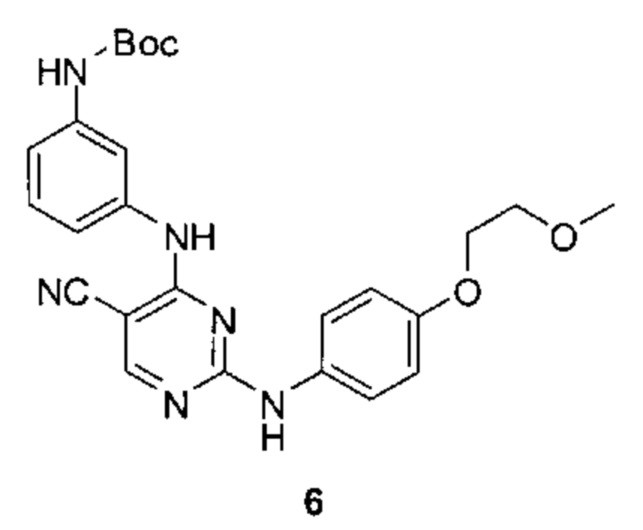
[00792] К суспензии Zn(CN)2 (0.24g, 2.0 ммоль), Pd(PPh3)4 (60 мг, 0.05 ммоль) в 3 мл ДМФ добавляли к 5 (0.25 г, 0.5 ммоль). Реакционную смесь дегазировали и запечатывали в атмосфере аргона, после чего нагревали до 120°С в течение 12 часов. Добавляли воду
(10 мл) и собирали осадок методом фильтрации. Осадок промывали эфиром и сушили с получением 0.2 г соединения 6. MS: m/e=477.1 (M+1).
[00793] Этап-4
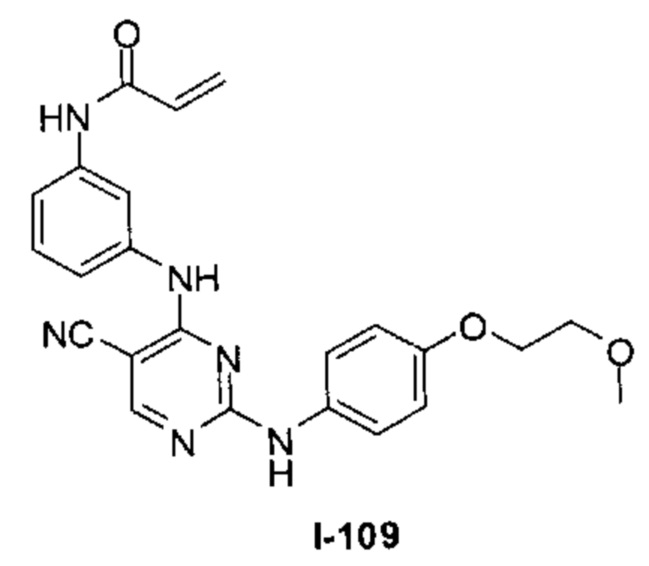
[00794] Соединение 6 (0.10 г, 0.21 ммоль) растворяли в 4 N HCl (2 мл) в диоксане. Смесь помешивали при комнатной температуре в течение 1 часа. После удаления растворителей, вливали 5-мл ДХМ, после чего упаривали до сухого состояния. Процедуру добавления ДХМ и упаривания повторяли три раза с получением а твердого осадка, который непосредственно использовали на следующем этапе: MS m/z: 377.0 (М+Н+).
[00795] К раствору полученного ранее промежуточного соединения, триэтиламин (0.1 ml, 0.8 ммоль) в 2 мл дихлорметана, добавляли акрилоил хлорид (19 мг, 0.21 ммоль) при температуре -10°С. Реакционную смесь помешивали в течение 10 минут при температуре -10°С и тушили водным раствором NaHCO3. Добавляли этил ацетата (10 мл) и промывали водным раствором NaHCO3, водой и солевым раствором.: Органические слои разделяли и сушили над Na2SO4. После удаления растворителя, сырой продукт очищали методом хроматографии на силикагеле (гексан:ЕЮАс = 1:2) с получением 30 мг указанного в названии соединения. MS m/z: 431.1 (М+Н+).
ПРИМЕР 95
[00796] Получение N-(3-(5-циано-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)фенил)акрил амида I-173
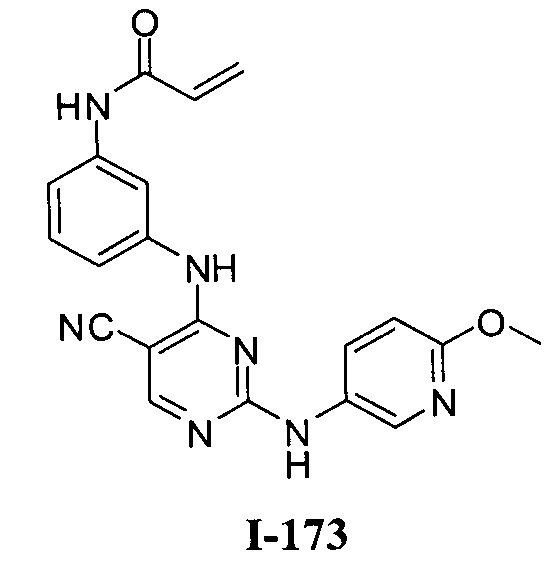
[00797] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 94, с использованием 3-амино-6-метоксипиридина вместо 4 на этапе-2 MS m/z: 388.2 (М+Н+).
ПРИМЕР 96
[00798] Получение N-(3-(5-циклопропил-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-139
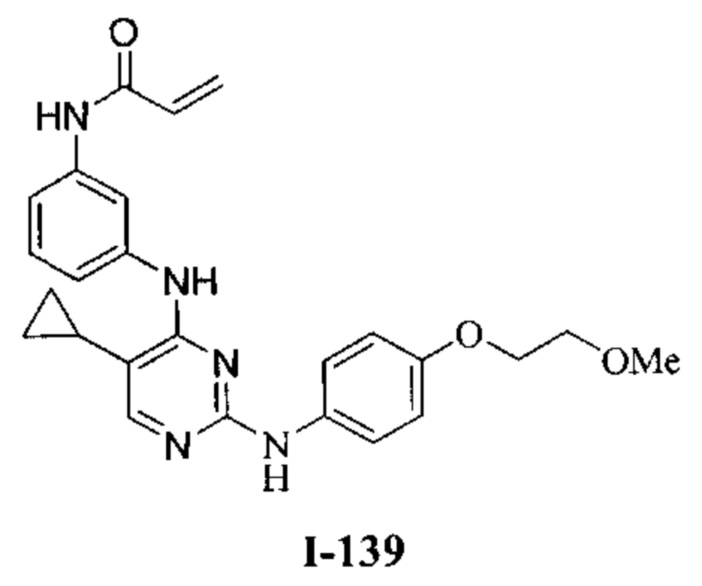
[00799] Указанное в названии соединение получали с использованием этапов и промежуточных соединений, описанных ниже.
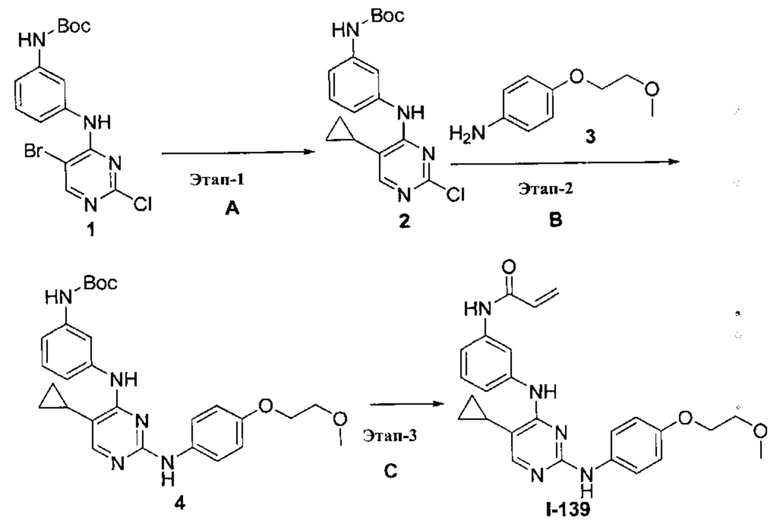
А) Калия циклопропилтрифторборат, Pd(OAc)2, Xanphos, Cs2(CO3), толуол, 100°С, 12 часов; В) PTSA, диоксан, 100°С, 2 часа; С) Zn(CN)2, Ph3P, ДМФ, 120°С, 12 часов; D) 4N HCl, диоксан, комн. т., 1 час; затем акрилоил хлорид, Et3N, ДХМ, -10°С, 10 минут.
[00800] Этап-1
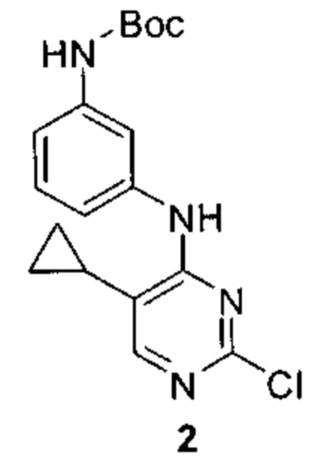
[00801] Калия циклопропилтрифторборат (0.4g, 3.0 ммоль), Соединение 1 (1.0g, 2.5 ммоль), палладия ацетат (34 мг, 0.15 ммоль), Xanphos (0.17 г, 0.3 ммоль) и Cs2CO3 (2.4g, 7.5 ммоль) суспендировали в 25 мл толуола и 5 мл воды. Реакционную смесь дегазировали и запечатывали в атмосфере аргона, после чего нагревали до 100°С в течение 12 часов. Добавляли 50 мл этил ацетата и промывали водным раствором NaHCO3, водой и солевым раствором. Органический слой разделяли и сушили над Na2SO4. После удаления растворителя, сырой продукт очищали методом хроматографии на силикагеле
(гексан : EtOAc = 3:2). с получением 0.54 г указанного в названии соединения 2: MS m/z: 361.2 (М+Н+).
[00802] Этап-2
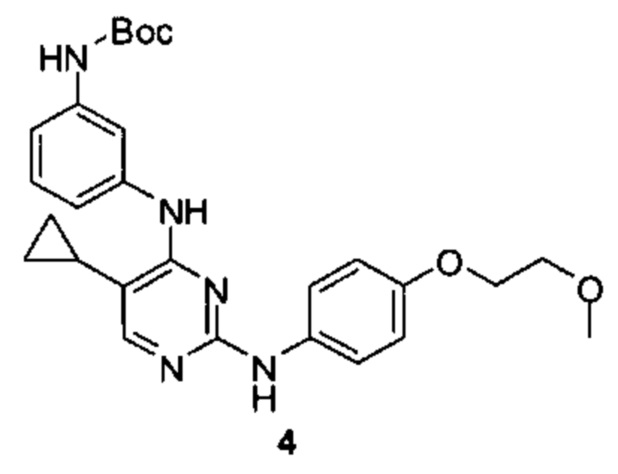
[00803] Соединение 4 получали из соединений 2 и 3 следуя процедуре, описанной в этапе 2 Примера 94. MS m/z: 492.2 (М+Н+).
[00804] Этап-3
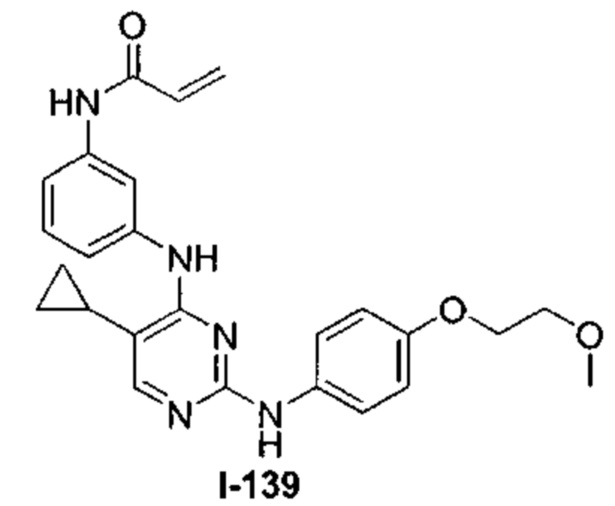
[00805] Указанное в названии соединение I-139 получали из соединения 4 следуя процедуре, описанной в этапе 4 Примера 94. MS m/z: 446.1 (М+Н+).
ПРИМЕР 97
[00806] Получение N-(3-(5-циклопропил-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-167
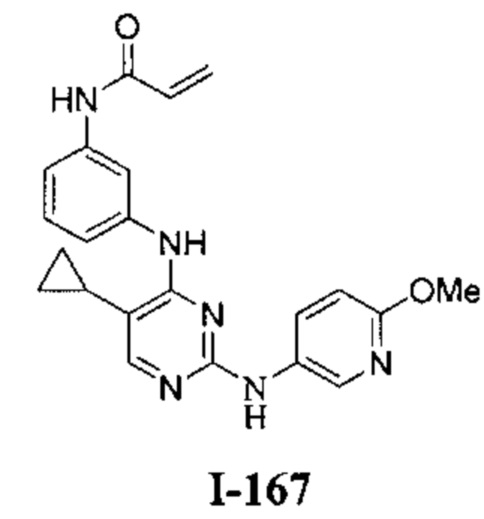
[00807] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 96, с использованием 3-амино-6-метоксипиридина вместо 3 на этапе-2 MS m/z: 403.2 (М+Н+).
ПРИМЕР 98
[00808] Получение N-(3-(5-фтор-2-(3-(3-(2-оксопирролидин-1-ил)пропокси)фениламино)пиримидин-4-илокси)фенил)акриламида I-162
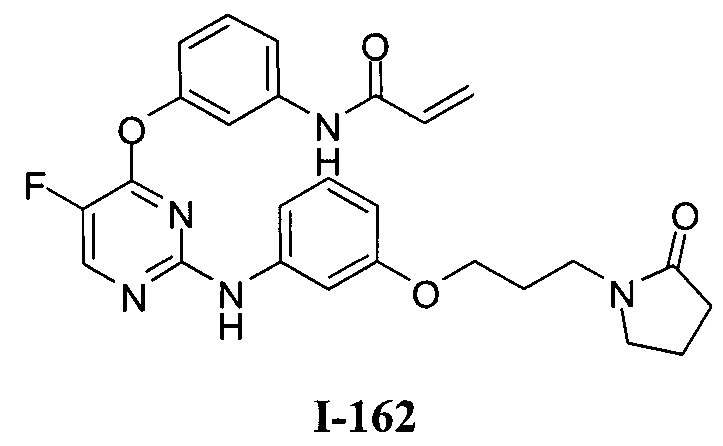
[00809] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
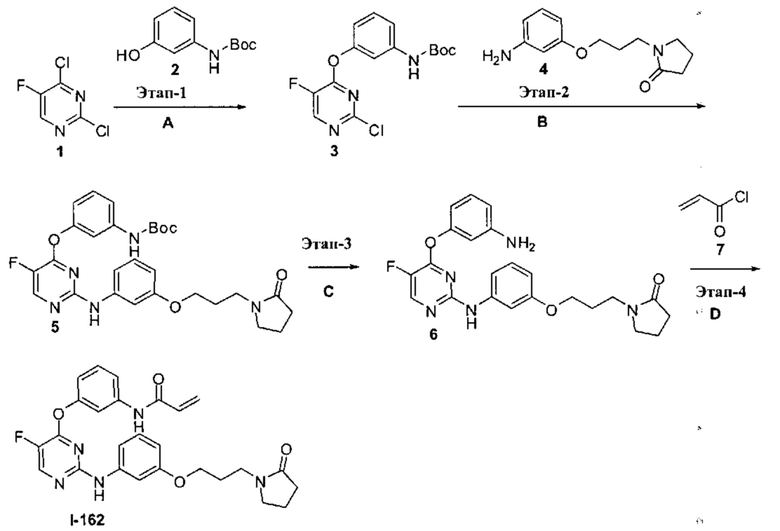
A) DIPEA, n-BuOH, 110°С, 16 часов; В) Pd(OAc)2, BINAP, Cs2CO3, толуол, 100°С, 16 часов; С) ТФА, CH2Cl2, комн. т. 2 часа; D) K2CO3, NMP, комн. т., 45 минут.
[00810] Этап-1
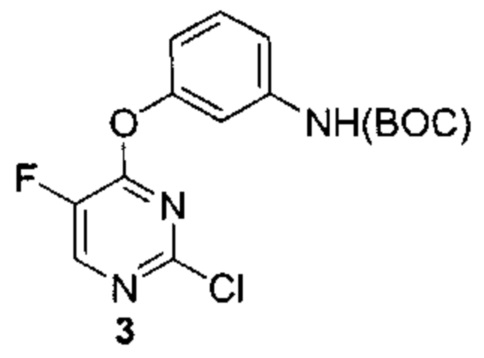
[00811] В нагнетательную трубку помещали 2 (2.0 г, 9.61 ммоль), 1 (3.21 г, 19.23 ммоль), n-BuOH (30 мл) и DIPEA (1.86 г, 14.42 ммоль), после чего содержимое помешивали при температуре 110°С в течение 16 часом. Реакционную смесь охлаждали, концентрировали при пониженном давлении, тушили водой (30 мл) экстрагировали этил ацетатом (2×30 мл). Объединенный экстракт этил ацетата промывали водой (20 мл), солевым раствором (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением остатка. Затем толкли с гексаном с получением 3 (2.5 г, 96%) в форме желтого твердого вещества.
[00812] Этап-2
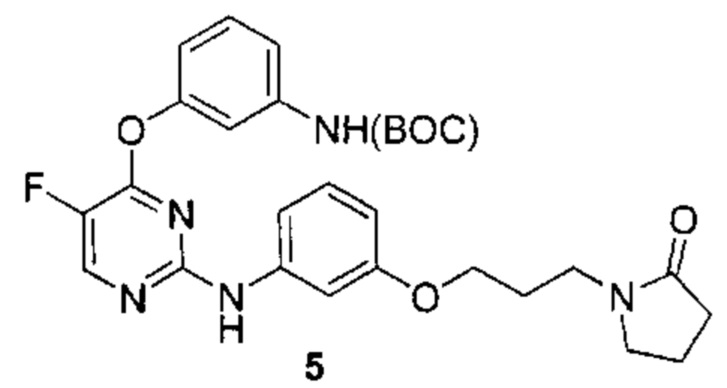
[00813] К раствору 3 (0.36 г, 1.1 ммоль) в толуоле (15 мл) добавляли 3-(3-(2-оксопирролидин-1-ил)пропоксианилин 4 (0.25 г, 1.1 ммоль) а затем BINAP (0.031 г, 0.05 ммоль), палладия ацетат (0.0022 г, 0.01 ммоль), и Cs2CO3 (0.82 г, 2.5 ммоль). Реакционную смесь перемешивали и пропускали через нее N2 в течение 15 минут. Нагревали при температуре 100°С в течение 8 часов в атмосфере N2. Реакционную смесь охлаждали до комнатной температуры, разбавляли этил ацетатом (30 мл), промывали водой (15 мл), солевым раствором (15 мл), и сушили над Na2SO4. Концентрировали при пониженном давлении с получением осадка, который очищали методом колоночной хроматографии (SiO2, 60-120, используя в качестве элюента 3% метанол/хлороформ: 3/97) с получением 5 (0.3 г, 60%) в форме желтого твердого вещества.
[00814] Этап-1
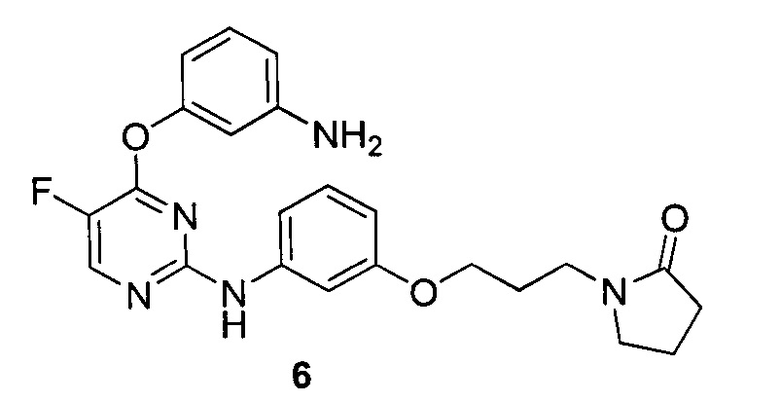
[00815] К раствору 5 (0,25 г, 0,46 ммоль) в CH2Cl2 (10 мл) при помешивании добавляли ТФА (1,0 мл) при температуре 0°С в атмосфере азота. Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали при той же температуре в течение 2 часа. Сырую реакционную смесь выливали в ледяную воду (10 мл), приводили к форме основания раствором натрия бикарбоната и экстрагировали этил ацетатом (3×15 мл). Объединенный экстракт этил ацетата промывали водой (15 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 6 (0.130 г, 65%) в форме желтого твердого вещества. Его использовали на следующем этапе без дополнительной очистки.
[00816] Этап-4
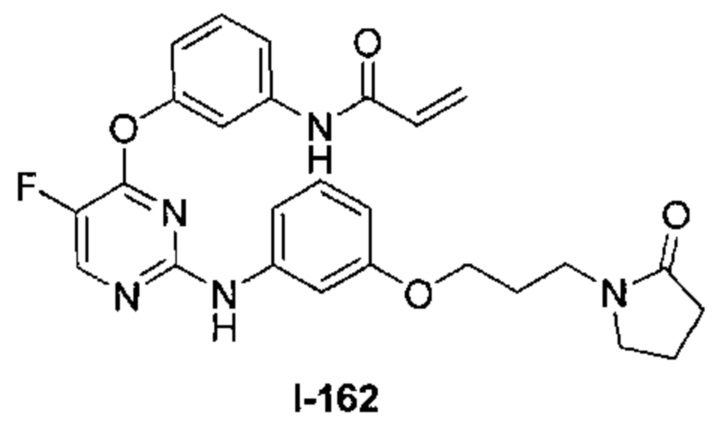
[00817] К раствору 6 (0,08 г, 0.18 ммоль) и калия карбоната (0.124 г, 0,9 ммоль) в NMP (1,2 мл) при температуре 0°С при помешивании добавляли акрилоил хлорид (0.020 г, 0,22 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 45 минут. Реакционную смесь по каплям добавляли при помешивании к холодному 10% раствору NaHCO3 и держали при этой же температуре (0°С) в течение 30 минут. Твердый осадок отделяли методом фильтрации через воронку Бухнера. Затем твердый осадок промывали холодной водой и гексаном растворяли в смеси метанол/дихлорметан (50:50, 5 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (10 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×
10 мл). Объединенный экстракт этил ацетата промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением I-162 (0.050 мг, 56%). 1Н-ЯМР (ДМСО-d6) δ ppm: 1.81-1.91 (m, 4Н), 2.19 (t, J=7.84 Гц, 2Н), 3.26-3.35 (m, 4Н), 3.73 (t, J=6.04 Гц, 2Н), 5.76 (dd, J=1.92 & 10.04 Гц, 1Н), 6.25 (dd, J=1.88 & 16.9 Гц, 1Н), 6.38-6.45 (m, 2Н), 6.93 (t, J=8.12 Гц, 1Н), 7.02-7.04 (m, 2Н), 7.11 (s, 1Н), 7.43 (t, J=8.16 Гц, 1Н), 7.55 (d, J=8.24 Гц, 1Н), 7.68 (d, J=1.8 Гц, 1Н), 8.56 (d, J=2.88 Гц, 1Н), 9.56 (s, 1H), 10.34 (s, 1Н); ЖХМС: m/е 490.0 (М-2).
[00818] Промежуточное соединение 3-(3-(2-оксопирролидин-1-ил)пропоксианилин получали с использованием схемы описанной ниже.
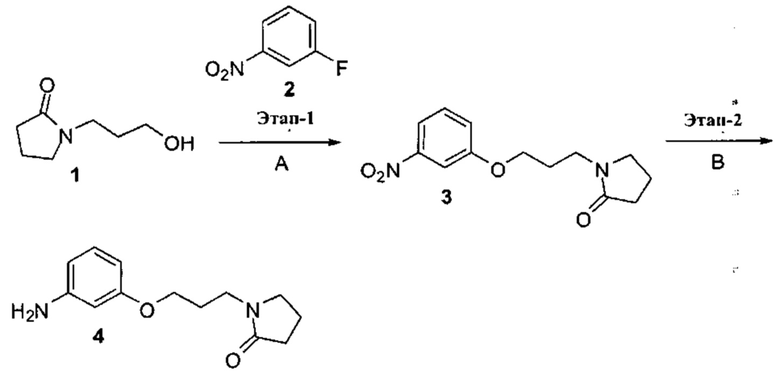
A) NaH, ДМФ, комн. т., 16 часов; В) SnCl2, Конц. HCl, 50°С, 2 часа.
[00819] Этап-1
[00820] К раствору NaH (1.0 г, 20.94 ммоль) в ДМФ (10 мл) при помешивании добавляли 1 (2.0 г, 13.96 ммоль) при температуре 0°С. Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали при той же температуре в течение 30 минут. К реакционной смеси медленно добавляли 2 (1.96 г, 13.96 ммоль), после чего реакционную смесь оставляли для перемешивания при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении и разбавляли этил ацетатом (20 мл). Его промывали водой (2×5 мл), солевым раствором (5 мл) и сушили над Na2SO4. Фильтровали, концентрировали при пониженном давлении с
получением сырого 3 (2 г, 55.5%) который использовали на следующем этапе без дополнительной очистки.
[00821] Этап-2
[00822] К раствору 3 (2 г, 7,57 ммоль) в концентрированной HCl (20 мл) при помешивании маленькими порциями добавляли SnCl2 (7.5 г, 34.06 ммоль). Реакционную смесь помешивали при температуре 50°С в течение 2 часа, охлаждали и приводили к форме основания NaHCO3. Экстрагировали этил ацетатом (3×25 мл), промывали водой (5 мл), солевым раствором (5 мл) и сушили над безводным Na2SO4. Фильтровали, концентрировали при пониженном давлении с получением 4 (1.65 г, 93%) в форме темно-коричневого твердого вещества, которое использовали на следующем этапе без дополнительной обработки.
ПРИМЕР 99
[00823] Получение N-(4-(5-фтор-2-(3-(2-(2-оксопирролидин-1-ил)этокси)фениламино)пиримидин-4-илокси)бензил)-N-метилакриламида I-146
[00824] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98 с использованием (4-гидроксибензил)(метил)карбаминовой кислоты трет-бутил эфир вместо 2 на этапе-1 и 3-(2-(2-оксопирролидин-1-ил)этоксианилина вместо 4 на Этапе 2. 1Н-ЯМР (CDCl3) δ ppm: 2.03 (quin, J=7.4 Гц, 2Н), 2.39 (t, J=8 Гц, 2Н), 3.07 & 3.06 (s, together 3Н), 3.57 (t, J=6.96 Гц, 2Н), 3.66 (t, J=5.08 Гц, 2Н), 4.03-4.04 (bd, J=4.96 Гц, 2Н), 4.67 & 4.72 (s, together 2Н),
5.70-5.85 (m, 1H), 6.43 (d, J=16.72 Гц, 1H), 6.50 (d, J=5.72 Гц, 1H), 6.60-6.75 (m, 1H), 6.89-6.96 (m, 2H), 7.06-7.08 (m, 2H), 7.18-7.30 (m, 2H), 7.37 (d, J=8.44 Гц, 1H), 8.21 (d, J=2.36 Гц, 1H); ЖХМС: m/e 506.2 (M+1).
ПРИМЕР 100
[00825] Получение N-(4-(5-фтор-2-(3-(3-(метилсульфонил)пропокси)фениламино)пиримидин-4-илокси)бензил)-N-метилакриламида I-136
[00826] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98 с использованием (4-гидроксибензил)(метил)карбаминовой кислоты трет-бутил эфир вместо 2 на этапе-1 и 3-(3-(3-метилсульфонил)пропоксианилина вместо 4 на Этапе 2. 1Н-ЯМР (CDCl3) δ ppm: 2.25-2.40 (m, 2Н), 2.97 (s, 3Н), 3.07 (s, 3Н), 3.20-3.30 (m, 2Н), 3.98-4.05 (m, 2Н), 4.67 (s, 1Н), 4.72 (s, 1H), 5.7-5.82 (m, 1H), 6.43 (dd, J=1.96 & 16.96 Гц, 1Н), 6.49-6.53 (m, 1Н), 6.6-6.75 (m, 1Н), 6.85-7.00 (m, 2Н), 7.05-7.15 (m, 2Н), 7.18-7.25 (m, 2Н), 7.36 (d, J=8.36 Гц, 1Н), 8.21 (bd, J=2.52 Гц, 1Н); ЖХМС: m/е 515.0 (М+1).
ПРИМЕР 101
[00827] Получение N-(4-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-илокси)бензил)-N-метилакриламида I-117
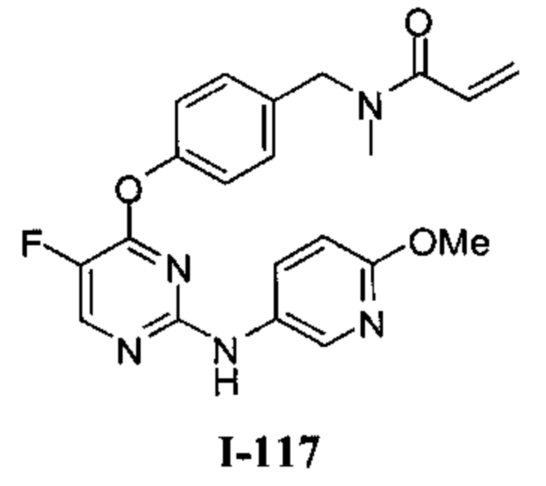
[00828] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98 с использованием (4-гидроксибензил)(метил)карбаминовой кислоты трет-бутил эфира вместо 2 на этапе-1 и 6-метокси-3-аминопиридина вместо 4 на Этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.92 & 3.07 (s, together 3Н), 3.76 (s, 3Н), 4.62 & 4.74 (s, together 2Н), 5.69 & 5.75 (dd, J=1.6 & 10.4 Гц, together 1H), 6.20 (dd, J=1.2 & 16.4 Гц, 1H), 6.56 (d, J=8.8 Гц, 1H), 6.82-6.90 (m, 1H), 7.28-7.35 (m, 4H), 7.71 (bd, J=7.6 Гц, 1H), 8.14 (s, 1H), 8.44 (bd, J=2.8 Гц, 1H), 9.48 (s, 1H); ЖХМС: m/e 410 (M+1).
ПРИМЕР 102
[00829] Получение N-(4-(5-фтор-2-(3-(3-(2-оксопирролидин-1-ил)пропокси)фениламино)пиримидин-4-илокси)бензил)-N-метилакриламида I-111
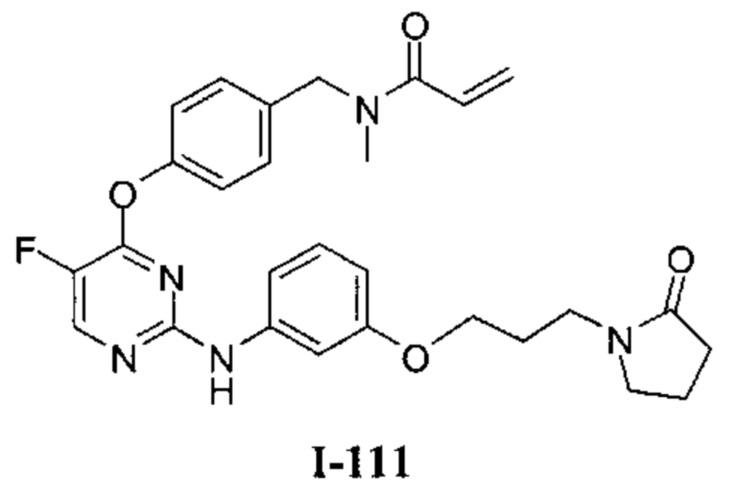
[00830] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98 с использованием (4-гидроксибензил)(метил)карбаминовой кислоты трет-бутил эфира вместо 2 на этапе-1 и 3-(3-(2-оксопирролидин-1-ил)пропокси)анилина вместо 4 на Этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.80-2.6 (m, 4Н), 2.20 (t, J=7.6 Гц, 2Н), 2.92 & 3.06 (s, together 3Н), 3.20-3.40 (m, 4Н), 3.75-3.90 (m, 2Н), 4.62 & 4.73 (s, together 2Н), 5.65-5.77 (m, 1H), 6.20 (dd, J=2.4 & 16.8 Гц,
1H), 6.24 (bd, J=8 Гц, 1H), 6.86 (dd, J=10.4 & 16.8 Гц, 1H), 6.93 (t, J=8 Гц, 1H), 7.08 (t, J=8 Гц, 2H), 7.28-7.36 (m, 4H), 8.48 (d, J=2.8 Гц, 1H), 9.51 (s, 1H); ЖХМС: m/e 520.2 (M+1).
ПРИМЕР 103
[00831] Получение N-(3-(5-фтор-2-(3-(2-(2-оксопирролидин-1-ил)этокси)фениламино)пиримидин-4-илокси)фенил)акриламида I-184
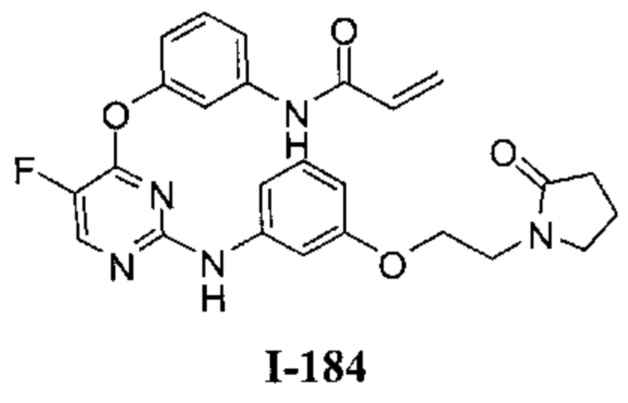
[00832] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
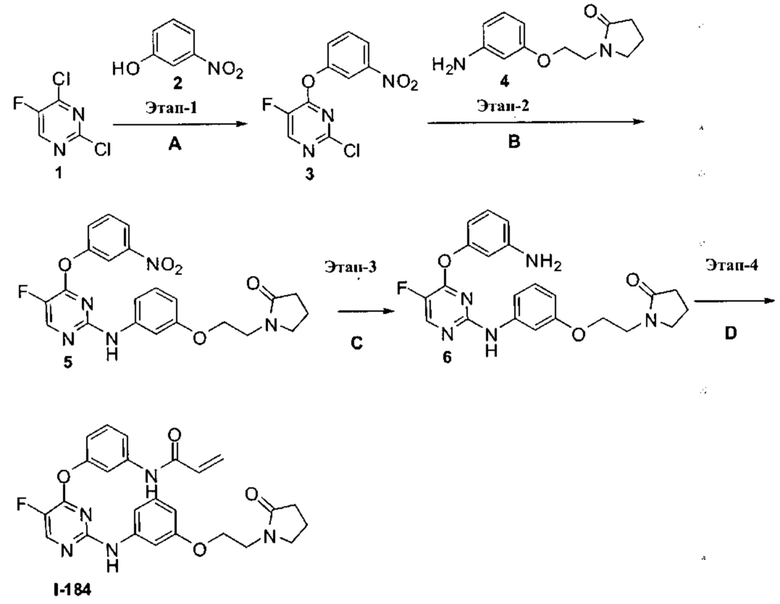
А)) K2CO3, ДМФ, комн. т., 16 часов; В) Pd(OAc)2, BINAP, Cs2CO3, толуол, 100°С, 8 часов; С) Pd-C, Н2, метанол, комн. т., 16 часов. D)) акрилоил хлорид, K2CO3, NMP, 0°С, 30 минут.
[00833] Этап-1
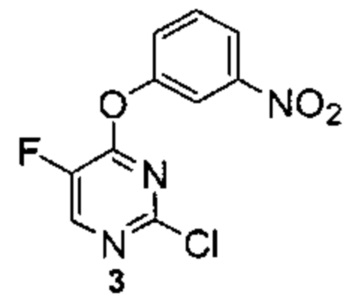
[00834] К раствору 2 (24 г, 143.7 ммоль) и K2CO3 (20 г, 143,6 ммоль) в сухом ДМФ (300 мл) при помешивании добавляли 2 (10 г, 71.8 ммоль) и реакционную смесь нагревали при комнатной температуре в течение 16 часов в атмосфере азота. Охлаждали и гасили водой (600 мл). Белый твердый осадок отделяли методом фильтрации через воронку Бухнера и сушили в вакууме с получением 3 (13 г, 68%) в форме белого твердого вещества.
[00835] Этап-2
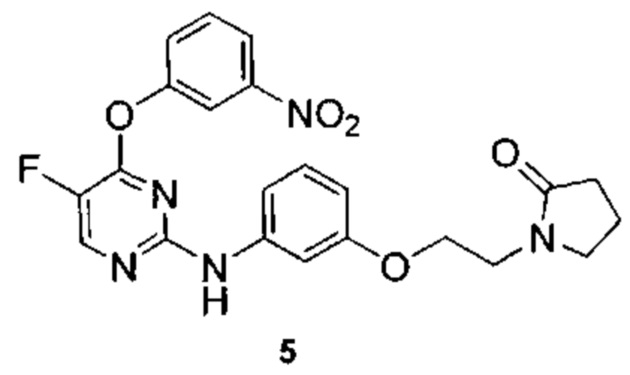
[00836] К раствору 3 (0,9 г, 3,3 ммоль) в толуоле (30 мл) добавляли 4 (950 мг, 4.3 ммоль) а затем BINAP (0.12 г, 0,19 ммоль), палладия ацетат (0,02 г, 0,09 ммоль), и Cs2CO3 (2,7 г, 8,2 ммоль). Реакционную смесь перемешивали и пропускали через нее N2 в течение 15 минут. Нагревали при температуре 100°С в течение 8 часов в атмосфере N2. Реакционную смесь охлаждали до комнатной температуры, разбавляли этил ацетатом (60 мл), промывали водой (35 мл), солевым раствором (35 мл), и сушили над Na2SO4. Концентрировали при пониженном давлении с получением осадка, который очищали методом колоночной хроматографии (SiO2, 60-120, используя в качестве элюента 3% метанол/хлороформ: 8/92) с получением 5 (0,50 г, 33%) в форме белого твердого вещества.
[00837] Этап-3
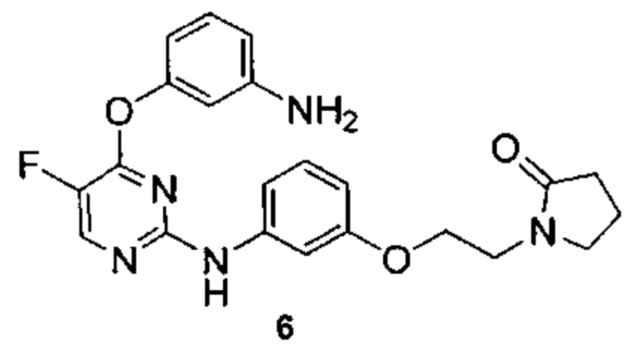
[00838] К раствору 5 (0,5 г, 1.1 ммоль) в метаноле (50 мл) добавляли 10% Pd/C (0,05 г, 10% масс./масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (давление водорода 1.5 Кг) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 6 (0,3 г, 65%) в форме бесцветной вязкой жидкости.
[00839] Этап-4
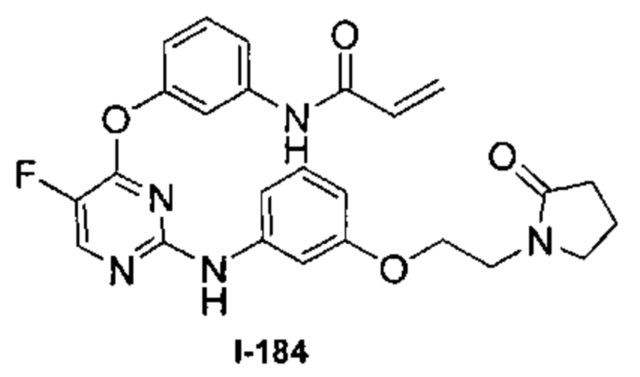
[00840] К раствору 6 (0,21 г, 0,5 ммоль) и калия карбоната (0.27 г, 2,0 ммоль) в NMP (2,5 мл) при температуре 0°С при помешивании добавляли акрилоил хлорид (0.053 г, 0,6 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 30 минут. Реакционную смесь по каплям добавляли при помешивании к холодному 10% раствору NaHCO3 и держали при этой же температуре (0°С) в течение 30 минут. Твердый белый осадок отделяли методом фильтрации через воронку Бухнера. Затем твердый осадок промывали холодной водой и гексаном растворяли в смеси метанол/дихлорметан (50:50, 10 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (25 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×50 мл). Объединенный экстракт этил ацетата промывали водой (50 мл), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением I-184 (0.050 мг, 65%) в форме белого твердого вещества. 1H-ЯМР (ДМСО-d6) δ ppm: 1.89 (quin, J=7.2 Гц, 2Н), 2.21 (t, J=7.6 Гц, 2Н), 3.39 (t, J=7.2 Гц, 2Н),
3.49 (t, J=5.2 Гц, 2H), 3.87 (t, 5.6 Гц, 2H), 5.77 (dd, J=1.6 & 10.4 Гц, 1H), 6.26 (dd, J=1.6 & 17.2 Гц, 1H), 6.39-6.46 (m, 2H), 6.95 (t, J=8.4 Гц, 1H), 7.03-7.12 (m, 3H), 7.44 (t, J=8.4 Гц, 1H), 7.56 (d, J=8.4 Гц, 1H), 7.70 (s, 1H), 8.51 (d, J=2.8 Гц, 1H), 9.56 (s, 1H), 10.35 (s, 1H); ЖХМС: m/e 478 (M+1).
[00841] Промежуточное соединение 3-(2-(2-оксопирролидин-1-ил)этоксианилин 4 получали с использованием схемы описанной ниже.
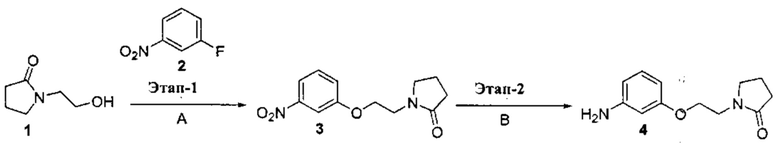
A) NaH, ТГФ, комн. т., 16 часов; В) Pd-C, Н2, метанол, комн. т., 16 часов.
[00842] Этап-1
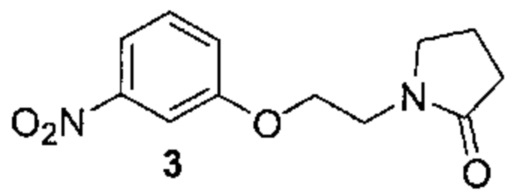
[00843] К раствору NaH (3.4 г, 141.6 ммоль, 60% дисперсия в парафиновом масле) в сухом ТГФ (50 мл) при помешивании добавляли 1 (6 г, 46.0 ммоль) при температуре 0°С после чего реакционную смесь помешивали в течение 15 минут в атмосфере азота. К нему добавляли раствор 2 (5.0 г, 35.4 ммоль) в ТГФ (10 мл) после чего реакционную смесь помешивали при комнатной температуре в течение 16 часов. Затем тушили холодной водой (40 мл), экстрагировали этил ацетатом (35 мл). Экстракт этил ацетата промывали водой (2×25 мл), солевым раствором (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением осадка который очищали методом колоночной хроматографии (SiO2, 60-120, в качестве элюента использовали метанол/хлороформ: 10/90) с получением 3 (2,5 г, 30%) в форме коричневатой жидкости.
[00844] Этап-2
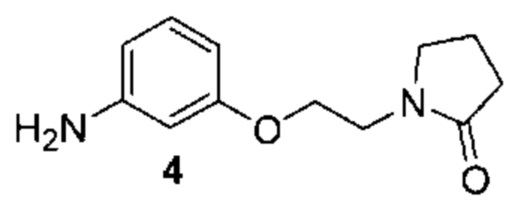
[00845] К раствору 3 (2,2 г, 8,8 ммоль) в метаноле (50 мл) добавляли 10% Pd/C (22 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (давление водорода 1.5 Кг) при комнатной температуре в течение 16 часов.
Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 4 (1,7 г, 89%) в форме желтоватой жидкости. Его использовали на следующем этапе без дальнейшей очистки.
ПРИМЕР 104
[00846] Получение N-(3-(2-(6-метоксипиридин-3-иламино)-5-метилпиримидин-4-илокси)фенил)акриламида I-186
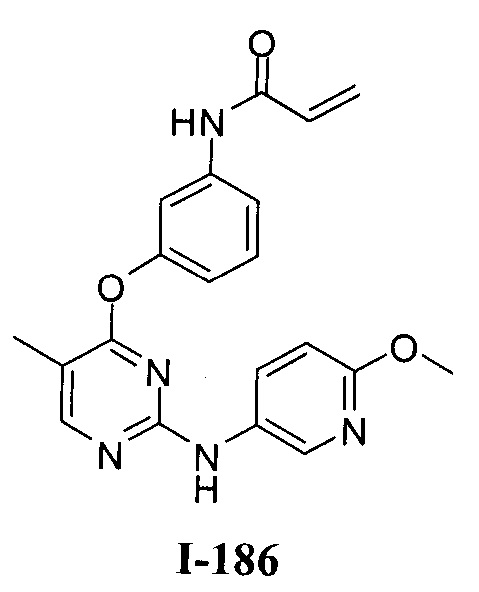
[00847] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 103 с использованием 2,4-дихлоро-5-метилпиримидина вместо 1 на этапе-1 и 6-метокси-3-аминопиридина вместо 4 на Этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.16 (s, 3Н), 3.73 (s, 3Н), 5.76 (dd, J=1.92 & 10.04 Гц, 1Н), 6.24 (dd, J=1.92 & 16.92 Гц, 1Н), 6.39-6.49 (m, 2Н), 6.92 (dd, J=1.48 & 8 Гц, 1H), 7.40 (t, J=8.08 Гц, 1Н), 7.49 (d, J=8.16 Гц, 1Н), 7.64 (d, J=1.84 Гц, 1Н), 7.77-7.79 (m, 1Н), 8.17 (bs, 1Н), 8.20 (s, 1H), 9.27 (s, 1H), 10.29 (s, 1Н); ЖХМС: m/е 378 (М+1).
ПРИМЕР 105
[00848] Получение N-(3-(5-метил-2-(фениламино)пиримидин-4-илокси)фенил)акриламида I-248
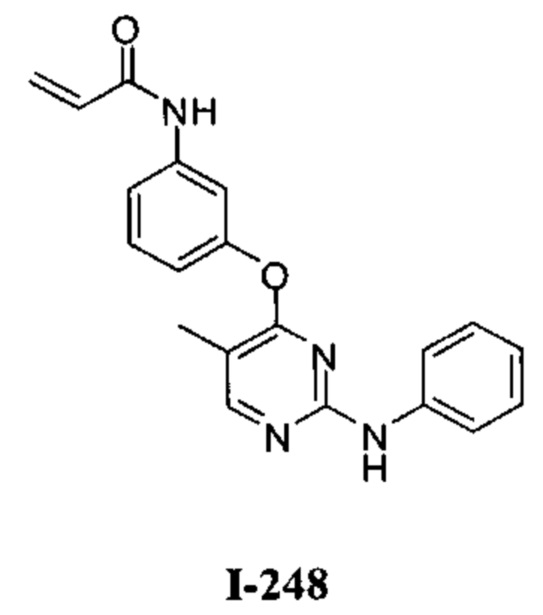
[00849] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
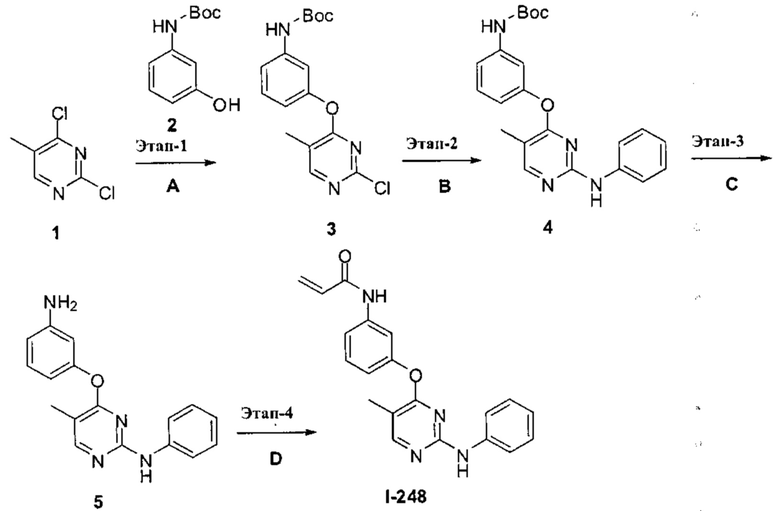
А) K2CO3, ДМФ, комн. т., 24 часа; А') (Вос)2O, ТГФ, 60°С, 2 часа; В) анилин, конц. HCl, EtOH, 80°С, 1 час; С) ТФА, СН2С12, 0°С до комн. т.,  часа; D) акрилоил хлорид, NMP, 0°С, 10 минут.
часа; D) акрилоил хлорид, NMP, 0°С, 10 минут.
[00850] Этап-1
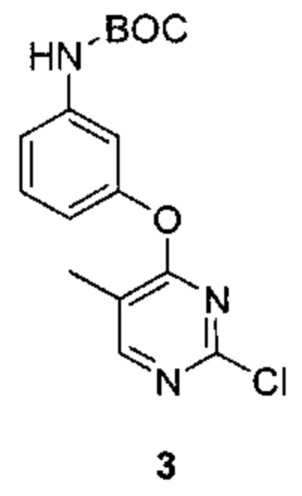
[00851] К раствору 2 (100 мг, 0,48 ммоль) и K2CO3 (99,2 мг, 0,717 ммоль) в сухом ДМФ (5 мл) при помешивании добавляли 1 (78 мг, 0,478 ммоль) и реакционную смесь нагревали при комнатной температуре в течение 24 часа в атмосфере азота. Реакционную смесь концентрировали при пониженном давлении и остаток разбавляли этил ацетатом (10 мл). Промывали водой (2×5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3 (120 мг, 75%) в форме белого твердого вещества. Его использовали на следующем этапе без дальнейшей очистки.
[00852] Этап-2
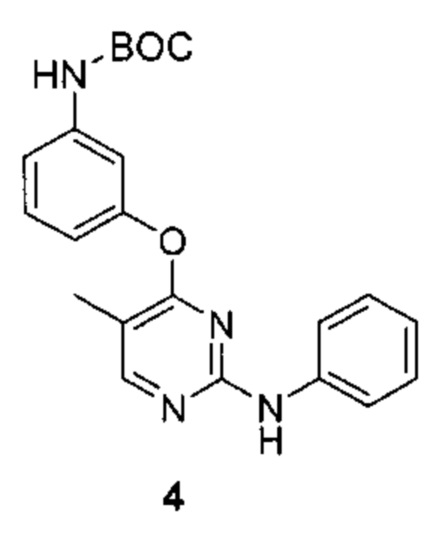
[00853] В нагнетательную колбу помещали 3 (75 мг, 0.224 ммоль), концентрированную. HCl (40 мг, 0.4 ммоль), анилин (83 мг, 0.89 ммоль) и этанол (2.0 мл) Закручивали крышку трубки и помешивали содержимое при 80°С в течение 60 минут. Реакционную смесь охлаждали, концентрировали при пониженном давлении и тушили осадок водой (50 мл). Приводили к форме основания 10% раствором NaHCO3 и экстрагировали этил ацетатом (3×10 мл). Объединенный слой EtOAc промывали водой (2×5 мл), солевым, раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, меш 60-120,
этилацетат/гексан: 50/50) с получением 4 (0,04 г, 45,9%) в форме беловатого твердого вещества.
[00854] Этап-3
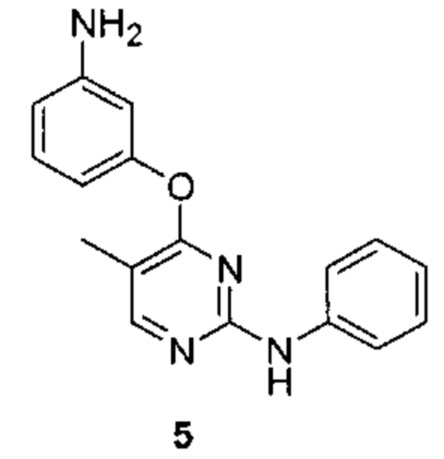
[00855] К раствору 4 (160 мг, 0.40 ммоль) в дихлорометане (4.0 мл) при помешивании добавляли при температуре 0°С, трифторуксусную Кислоту (0.8 мл). Продолжали помешивать при той же температуре в течение 30 минут, затее реакционную смесь концентрировали при пониженном давлении и растворяли осадок в воде (5.0 мл), приводили к форме основания 10% раствором NaHCO3 и экстрагировали дихлорметаном (2×5 мл). Экстракт дихлорметана промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 5 (110 мг, 93.2%) в форме беловатого твердого вещества. Его использовали на следующем этапе без дальнейшей очистки.
[00856] Этап-4
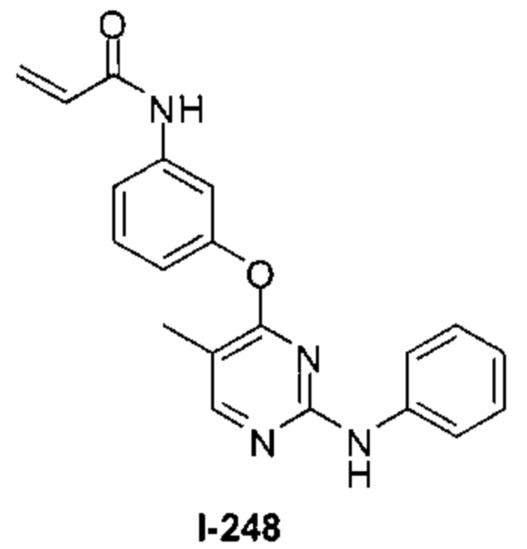
[00857] К раствору 5 (75 мг, 0,256 ммоль) в NMP (0,8 мл) при помешивании при температуре 0°С добавляли акрилоил хлорид (34,8 мг, 0.38 ммоль) и помешивали реакционную смесь при температуре 0°С в течение 10 минут. Затем реакционную смесь гасили водой (4,0 мл), приводили к форме основания 10% раствором NaHCO3 и экстрагировали дихлорметаном (2×5 мл). Экстракт дихлорметан промывали водой (5 мл),
солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток далее очищали методом колоночной хроматографии (SiO2, меш 60-120, CHCl3/МеОН: 99/1) с получением I-248 (0.035 г, 39.6%) в форме белого окрашенного твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.15 (s, 3Н), 5.75 (dd, J=1.92 & 10.04 Гц, 1H), 6.24 (dd, J=1.96 & 16.96 Гц, 1H), 6.41 (dd, J=10.6 & 17 Гц, 1Н), 6.78-6.8 (m, 1H), 6.94 (dd, J=1.44 & 8.04 Гц, 1Н), 7.00 (t, J=7.52 Гц, 2Н), 7.40-7.44 (m, 3Н), 7.53 (d, J=8.24 Гц, 1H), 7.6 (t, J=2 Гц, 1Н), 8.23 (d, J=1.04 Гц, 1H), 9.36 (s, 1Н), 10.30 (s, 1Н); ЖХМС: m/е 346.8 (М+1).
ПРИМЕР 106
[00858] Получение 1-(4-(5-фтор-2-(фениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-229
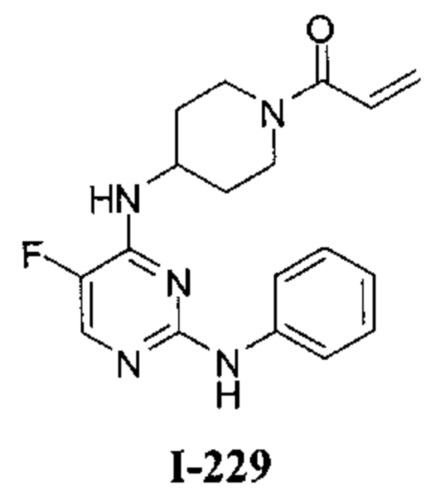
[00859] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20 с использованием 1-трет-бутилоксикарбонил-4-аминопиперидина вместо 2 на Этапе 1 и анилин вместо 4 на Этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.35-1.50 (m, 2Н), 1.90-2.05 (m, 2Н), 2.7-2.85 (m, 1H), 3.10-3.20 (m, 1H), 4.11-4.15 (m, 2Н), 4.46 (bd, J=13.72 Гц, 1H), 5.67 (dd, J=2.44 & 10.4 Гц, 1H), 6.10 (dd, J=2.44 & 16.6 Гц, 1H), 6.82-6.88 (m, 2H), 7.22 (t, J=7.44 Гц, 2H), 7.35 (d, J=7.56 Гц, 1H), 7.70 (d, J=7.72 Гц, 2H), 7.87 (d, i=3.76 Гц, 1H), 9.07 (s, 1H); ЖХМС: m/e 341.383 (M+1).
ПРИМЕР 107
[00860] Получение 2-((3-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)(гидрокси)метил)акрилонитрил I-71
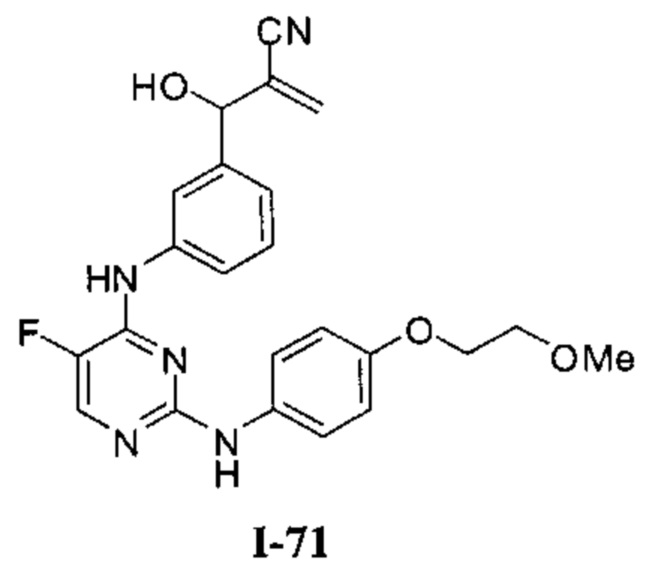
[00861] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
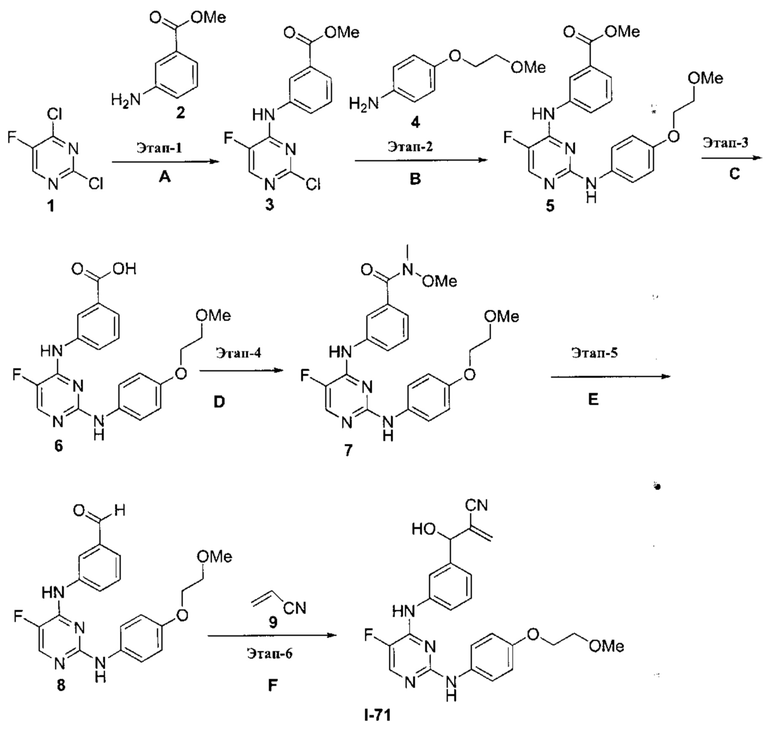
А) 2, DIPEA, n-BuOH, 120°C, 12 часов; В) конц. HCl, этанол, 100°С, 5 часов; С) LiOH, МеОН/ТГФ/H2O, комн. т., 6 часов; D) Me-NH-OMe.HCl, EDCI.HCl, НОВТ, DIPEA, ДМФ, комн. т., 8 часов; Е) LAH (1.0 М раствор в ТГФ), -78°С, 30 минут; F) DABCO, 1,4-диоксан/вода, комн. т., 48 часов.
[00862] Этап-1
[00863] Раствор 1 (0.50 г, 2.99 ммоль), 2 (0.45 г, 2.99 ммоль) и DIPEA (0.57 г, 4.48 ммоль) в n-бутаноле (5.0 мл) нагревали в в нагнетательной трубке (120°С, 16 часов). Охлаждали, тушили водой (5 мл) и экстрагировали EtOAc (2×25 мл). Объединенный экстракт EtOAc промывали водой (2 мл), солевым раствором (2 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3 (0,70 г, 83,3%) в форме беловатого твердого вещества.
[00864] Этап-2
[00865] Раствор 3 (0.5 г, 1.77 ммоль) и 4 (0.29 г, 1.77 ммоль) в этаноле (2.5 мл) помещали в нагнетательную трубку и добавляли к нему уксусную кислоту (0.1 мл). Плотно закрывали трубку и помешивали содержимое при 100°С в течение 5 минут. Реакционную смесь охлаждали, концентрировали при пониженном давлении и добавляли осадок в этил ацетат (50 мл). Промывали раствором NaHCO3 (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Твердый осадок отделяли методом фильтрации. Его сушили под вакуумом с получением 5 (0.6 г, 80%).
[00866] Этап-3
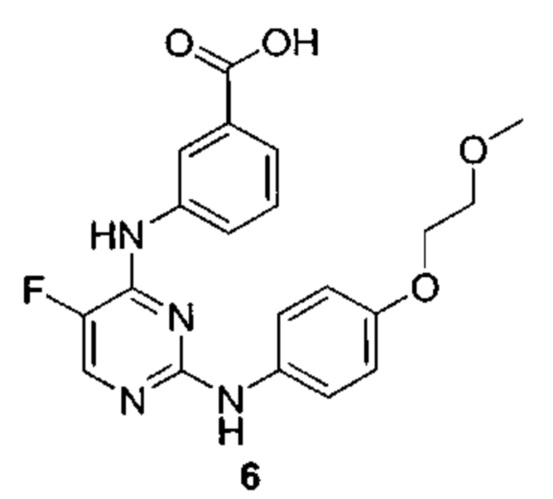
[00867] К раствору 5 (0,6 г, 1,4 ммоль) в смеси метанол/ТГФ/вода : 6 мл/6 мл/3 мл добавляли LiOH (0.298 г, 7 ммоль) после чего реакционную смесь помешивали при комнатной температуре в течение 2 часа. Ее концентрировали при пониженном давлении; осадок разбавляли водой (2 мл) и экстрагировали диэтиловым эфиром (5 мл). Водный слой разделяли и подкисляли 1.5 N HCl (рН ~4-5), концентрировали и сушили под вакуумом с получением 6 (0.4 г, 70%) в форме белого твердого вещества, которое использовалось на следующем этапе без дополнительной очистки.
[00868] Этап-4
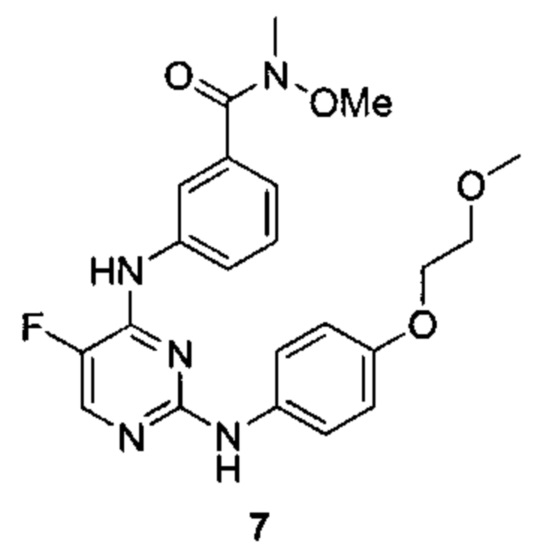
[00869] К старвору 6 (0.4 г, 1 ммоль) в ДМФ (3 мл) при помешивании добавляли MeNH-OMe.HCl (0.102 г, 0.1 ммоль), EDCI.HCl (0.003g, 1.5 ммоль), НОВТ (71 мг, 0.5 ммоль) и DIPEA (0.204 г, 1.5 ммоль). Реакционную смесь помешивали при комнатной температуре в течение 8 часов, тушили водой и экстрагировали EtOAc (2×5 мл). Объединенный органический слой промывали солевым раствором, и сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 7 (0.4 г, 90.9%) в форме белого твердого вещества.
[00870] Этап-5
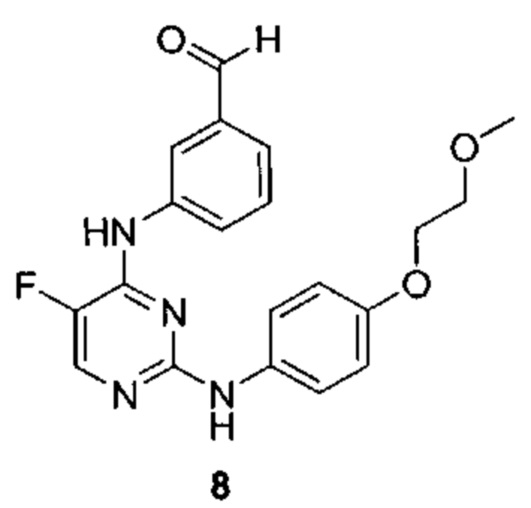
[00871] К раствору 7 (0,4 г, 0.9 ммоль) в ТГФ (10 мл) при помешивании добавляли LAH (1.8 мл, 1,8 ммоль) при температуре -78°С. Реакционную смесь оставляли для перемешивания при той же температуре 30 минут, после чего ее тушили раствором Na2SO4 (2 мл) и экстрагировали этил ацетатом (10 мл). Слой этил ацетата отделяли и промывали водой (2 мл), солевым раствором (2 мл) и сушили над безводным Na2SO4. Фильтровали, и затем концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 7/3) с получением 8 (200 мг, 58%) в форме желтого твердого вещества.
[00872] Этап-6
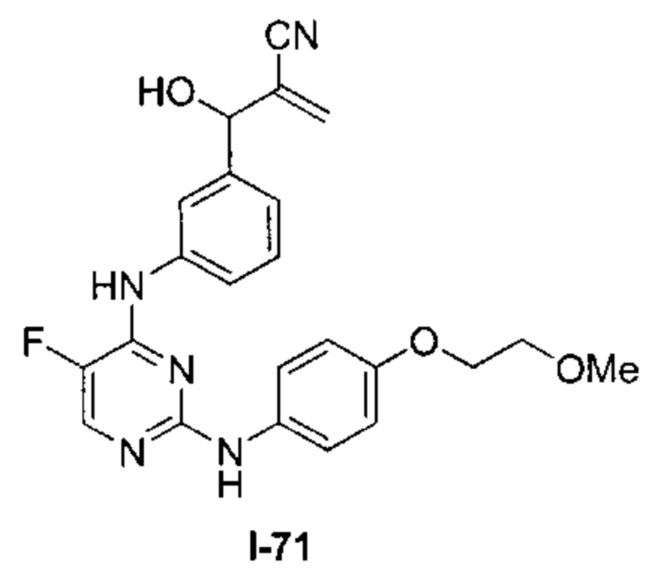
[00873] К раствору 8 (200 мг, 0.523 ммоль) и 9 (69 мг, 1.3 ммоль) в 1,4-диоксана/Н2О (1.4 мл/0.6 мл) при помешивании добавляли DABCO (50 мг, 0.2523 ммоль) при комнатной температуре. Продолжали помешивать при комнатной температуре в течение 48 часов, после чего реакционную смесь концентрировали при пониженном давлении. Полученный осадок далее очищали методом колоночной хроматографии (SiO2, петролейный эфир/ этил ацетат, 6/4) с получением I-71 в форме зеленоватого смолистого материала (0.05 г, 22.7%). 1Н-ЯМР (ДМСО-d6) δ ppm: 3.48 (s, 3Н), 3.77 (t, J=4.4 Гц, 2Н), 4.11-4.16 (m, 2Н),
5.11 (s, 1H), 5.99 (s, 1H), 6.06 (s, 1H), 6.85 (s, 1H), 6.91 (d, 7=8.84 Гц, 2H), 7.15 (d, 7=7.44 Гц, 1H), 7.30-7.40 (m,; ЖХМС: m/e (M+1).
ПРИМЕР 108
[00874] Получение 2-((4-(5-фтор-2-(фениламино)пиримидин-4-иламино)фенил)(гидрокси)метил)акрилонитрила I-161
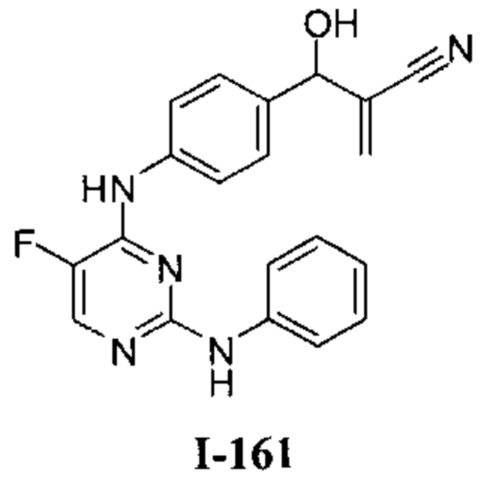
[00875] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 107, с использованием анилин вместо 4 на этапе 2. 1Н-ЯМР (CDCl3) δ ppm: 5.32 (s, 1H), 6.07 (d, J=0.8 Гц, 1H), 6.15 (d, J=1.6 Гц, 1H), 6.84 (d, J=2.8 Гц, 1H), 7.03-7.06 (m, 2H), 7.29 (t, J=1.6 Гц, 2H), 7.38 (d, J=8.44 Гц, 2H), 7.52 (d, J=8.8 Гц, 2H), 7.67 (dd, J=1.6 & 6.4 Гц, 2H), 7.96 (d, J=3.2 Гц, 1H); ЖХМС: m/e 361.8 (M+1).
ПРИМЕР 109
[00876] Получение 2-((4-(5-фтор-2-(3-трифторметоксифениламино)пиримидин-4-иламино)фенил)(гидрокси)метил)акрилонитрила I-163
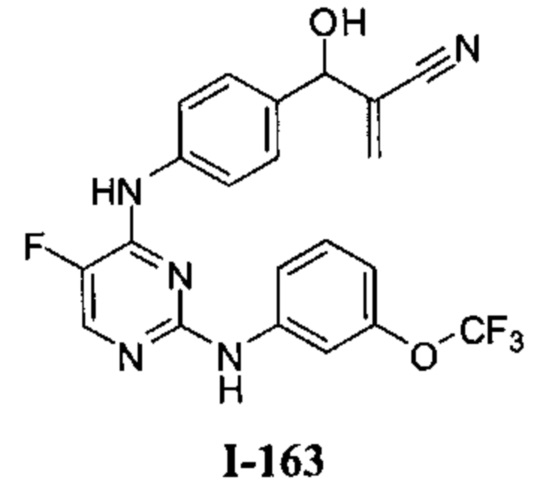
[00877] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 107, с использованием 3-
трифторметоксианилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 5.29 (d, J=3.8 Гц, 1Н), 6.13 (s, 1H), 6.19 (s, 1Н), 6.31 (dd, J=3.8 Гц, 1Н), 6.83 (d, J=7.76 Гц, 1Н), 7.11 (d, J=7.8 Гц, 1Н), 7.30-7.37 (m, 2Н), 7.61-7.63 (m, 2Н), 7.81 (s, 1H), 7.90 (d, J=7.4 Гц, 1H), 8.16 (dd, J=1.44 & 3.56 Гц, 1Н), 9.45 (s, 1H), 9.53 (s, 1Н); ЖХМС: m/е 446 (М+1).
ПРИМЕР 110
[00878] Получение N-(3-(5-фтор-2-(3-(3-(метилсульфонил)пропокси)фениламино)пиримидин-4-илокси)фенил)акриламида I-116
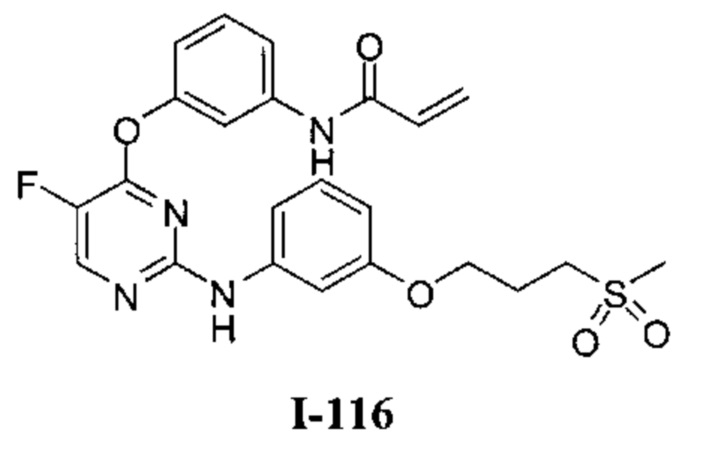
[00879] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием 3-(3-(метилсульфонил)пропоксианилина вместо 4 на этапе 4. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.02-2.15 (m, 2Н), 3.01 (s, 3Н), 3.22 (t, J=7.56 Гц, 2Н), 3.88 (t, J=6.12 Гц, 2Н), 5.77 (dd, J=1.84 & 10.12 Гц, 1Н), 6.25 (dd, J=1.72 & 16.88 Гц, 1H), 6.43 (d, J=9.96 & 16.76 Гц, 2Н), 6.95 (t, J=8.12 Гц, 1Н), 7.06 (t, J=7.48 Гц, 2Н), 7.13 (s, 1Н), 7.44 (t, J=8.12 Гц, 1H), 7.56 (d, J=8.44 Гц, 1H), 7.68 (s, 1Н), 8.50 (d, J=2.84 Гц, 1Н), 9.57 (s, 1Н), 10.34 (s, 1H); ЖХМС: m/е 487.0 (М+2).
ПРИМЕР 111
[00880] Получение N-(3-(5-циклопропил-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-илокси)фенил)акриламида I-131
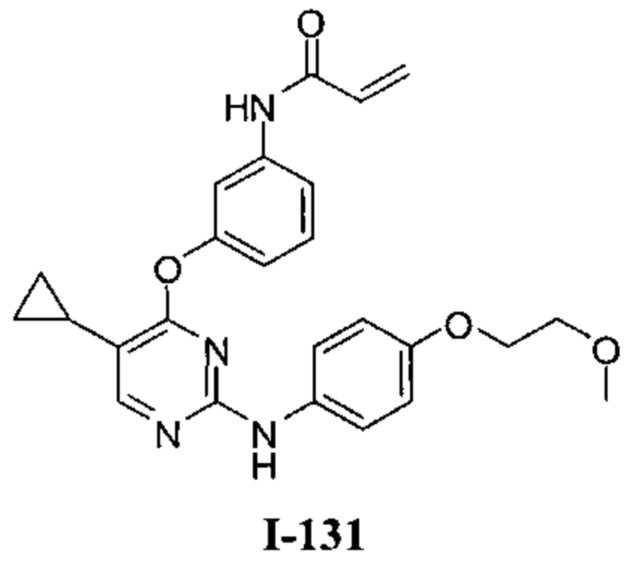
[00881] Указанное в названии соединение получали с использованием этапов и промежуточных соединений, описанных ниже.
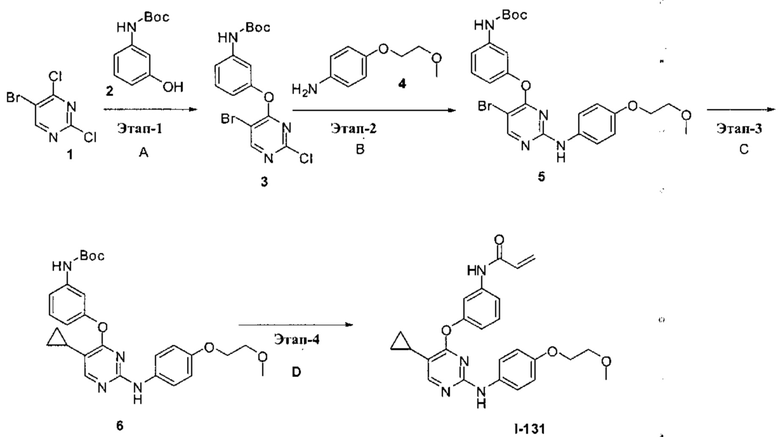
А) K2CO3, DMA, комн. т., 5 часов; В) PTSA, диоксан, 100°С, 2 часа; С) калия циклопропилтрифторборат, Pd(OAc)2, Xanphos, Cs2CO3, толуол, 100°С, 12 часов; D) 4N HCL, диоксан, комн. т., 1 час; затем акрилоил хлорид, Et3N, ДХМ, -10°С, 10 минут
[00882] Этап-1
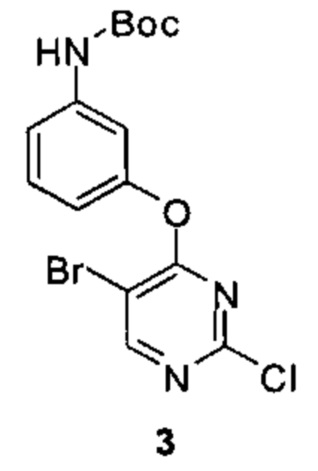
[00883] К раствору 5-бромо-2,4-дихлоропиримидина (0,68g, 3,0 ммоль) и трет-бутил 3-гидроксифенилкарбамата (0,65 г, 3,1 ммоль) в DMA (4 мл) добавляли K2CO3 (0,83g, 6,0 ммоль). Суспензию помешивали в течение 5 часов. Добавляли воду (15 мл) и собирали осадок методом фильтрации. Осадок промывали эфиром и сушили с получением 1.2 г соединения 3. MS: m/e=400.2,402.2 (M+1).
[00884] Этап-2
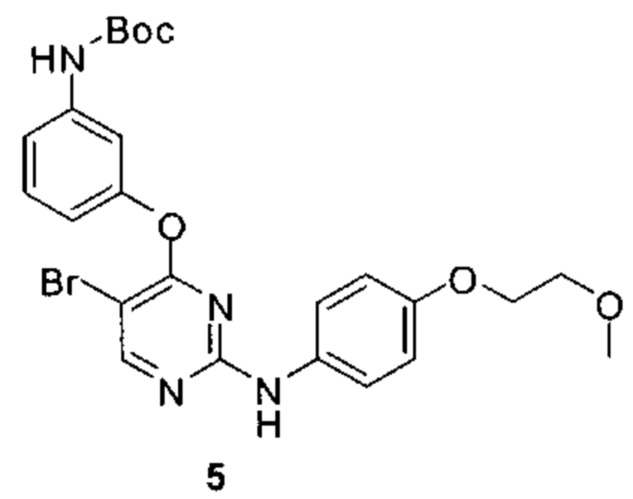
[00885] К раствору соединения 3 (200 мг, 0,5 ммоль) и 4-(2-метоксиэтокси)анилина (0,1 г, 0,6 ммоль) в 8 мл диоксана добавляли 4-метилбензолсульфоновой кислоты моногидрат (0,08 г, 0,4 ммоль). Смесь помешивали при температуре 100°С в течение двух часов. Растворитель выпаривали. Остаток растворяли в 20 мл этил ацетата и промывали водным раствором NaHCO3, водой и солевым раствором. Органические слои разделяли и сушили над Na2SO4. После удаления растворителя, сырой продукт очищали методом хроматографии на силикагеле (гексан : EtOAc = 1:1). получали 0.10 g соединения 5: MS m/z: 531.1, 531.0 (М+Н+).
[008861 Этап-1
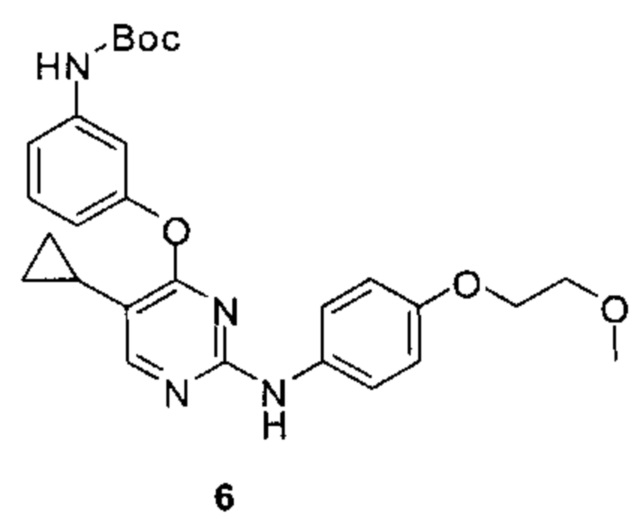
[00887] Калия циклопропилтрифторборат (36 мг, 0,25 ммоль), Соединение 5 (0,10 г, 0,19 ммоль), палладия ацетат (3,4 мг, 0,015 ммоль), Xantphos (17,5 мг, 0,03 ммоль) и Cs2CO3 (186 мг, 0,57 ммоль) суспендировали в 5 мл толуола и 1 мл воды. Реакционную смесь дегазировали и запечатывали в атмосфере аргона, после чего нагревали до 100°С в течение 12 часов. Добавляли этил ацетата (20 мл) и промывали водным раствором NaHCO3, водой и солевым раствором.: Органические слои разделяли и сушили над Na2SO4. После удаления растворителя, сырой продукт очищали методом хроматографии на силикагеле (гексан : EtOAc = 1:1). получали 50 мг соединения 6: MS m/z: 493.2 (М+Н+).
[00888] Этап-4
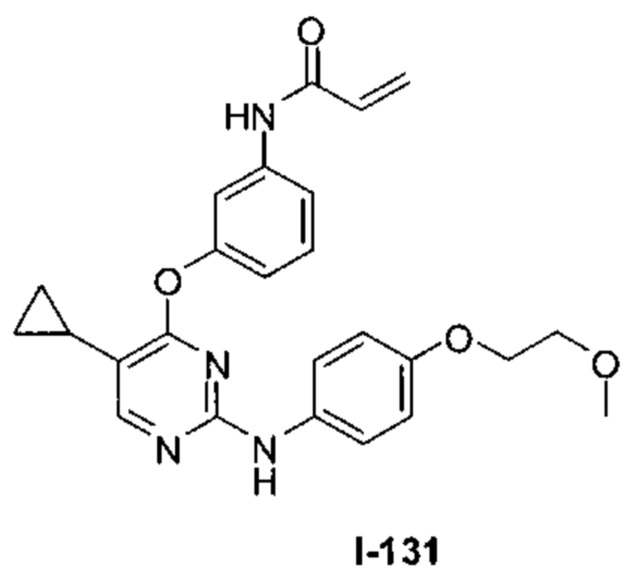
[00889] Указанное в названии соединение получали из соединения 6 следуя процедуре, описанной в Примера 96. MS m/z: 447.1 (М+Н+).
ПРИМЕР 112
[00890] Получение 1-(4-(5-фтор-2-(3-(2-диметиламиноэтокси)фениламино)пиримидин-4-иламино)фенил)-2-метилprop-2-ен-1-она I-207
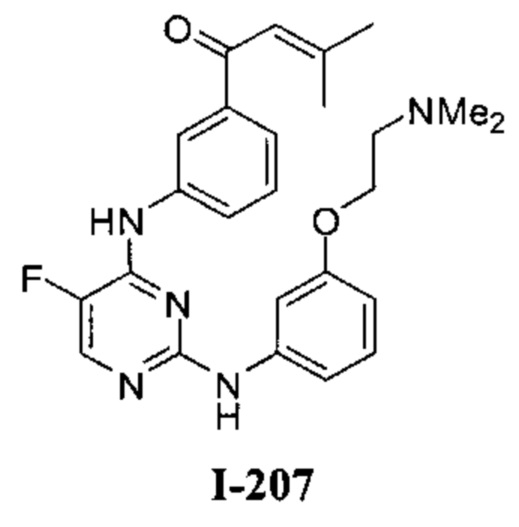
[00891] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
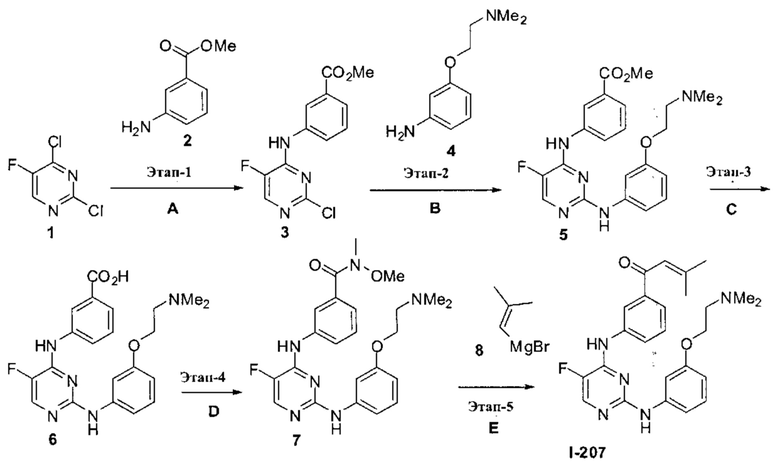
А) 2, DIPEA, n-BuOH, 90°С, 12 часов; В) 4, Pd(OAc)2, BINAP, Cs2CO3, толуол, 110°С, 16 часов; С) LiOH, МеОН/ТГФ/H2O, комн. т., 6 часов; D) MeNHOMe.HCl, EDCI.HCl, НОВТ, DIPEA, ДМФ, комн. т., 3 часа; Е) 8, ТГФ, 0°С до комн. т., 2 часа.
[00892] Этап-1
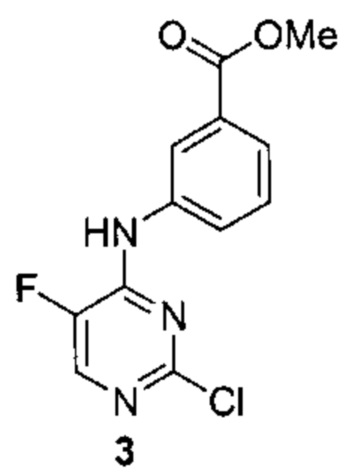
[00893] Раствор 1 (4 г, 23,9 ммоль), 2 (3,6 г, 23,7 ммоль) и DIPEA (4,6 г, 35,58 ммоль) в n-бутаноле (40 мл) нагревали в в нагнетательной трубке (90°С, 12 часов). Охлаждали, тушили водой (5 мл) и экстрагировали EtOAc (2×5 мл). Объединенный экстракт EtOAc промывали водой (60 мл), солевым раствором (40 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3 (5,5 г, 82%) в форме беловатого твердого вещества.
[00894] Этап-2
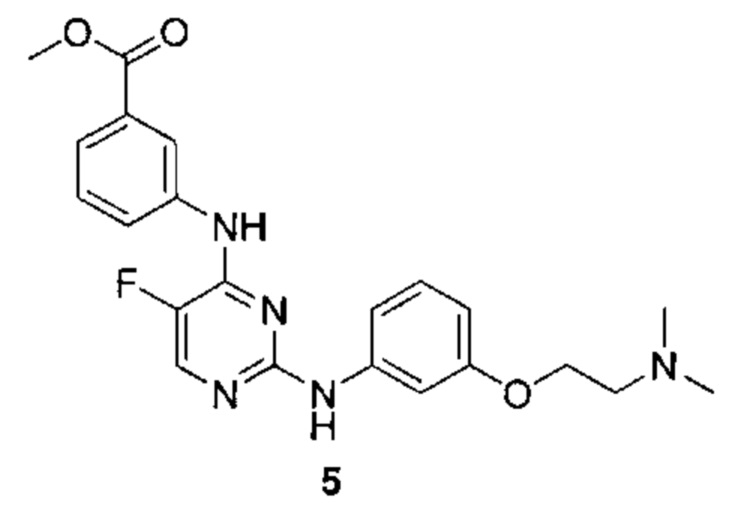
[00895] Раствор 3 (0,319 г, 1,76 ммоль), 4 (0,5 г, 1,76 ммоль), Pd(OAc)2 (0,039 г, 0,17 ммоль), BINAP (0.055 г, 0.08 ммоль) и Cs2CO3 (1,44 г, 4,42 ммоль) в дегазированном толуоле (толуол продували N2 в течение 30 минут) нагревали в течение, 16 часов при температуре 100°С в атмосфере N2. Реакционную смесь охлаждали, разбавляли EtOAc (25 мл) промывали водой (5 мл), солевым раствором (2 мл) и сушили над Na2SO4. Фильтровали, и затем концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, хлороформ/метанол, 9/1) с получением 4 (0.63 г, 84%) в форме желтого твердого вещества.
[00896] Этап-1
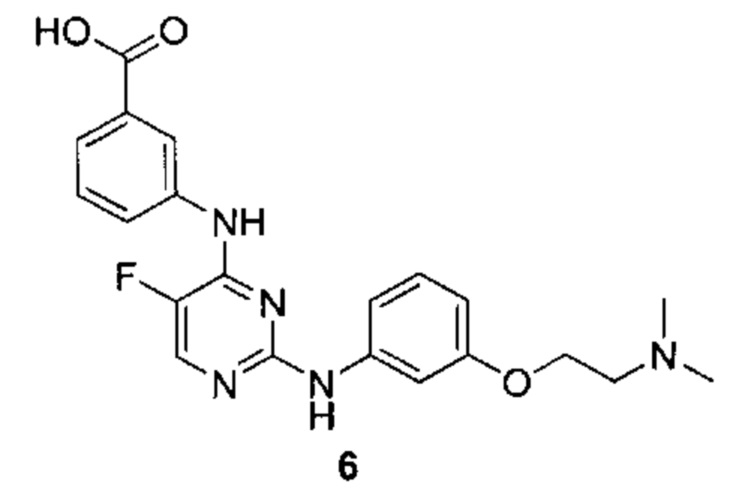
[00897] К раствору 5 (0,3 г, 0,70 ммоль) в смеси метанол/ТГФ/вода : 1 мл/1 мл/0,5 мл добавляли LiOH (0.147 г, 3,52 ммоль) после чего реакционную смесь помешивали при комнатной температуре в течение 6 часов. Ее концентрировали при пониженном давлении; осадок разбавляли водой (2 мл) и экстрагировали диэтиловым эфиром (5 мл). Водный слой разделяли и подкисляли 1.5 N HCl (рН ~4-5), концентрировали и сушили под вакуумом с получением 6 (0,31 г, сырого) в форме желтого смолистого твердого вещества, которое использовалось на следующем этапе без дополнительной очистки.
[00898] Этап-4
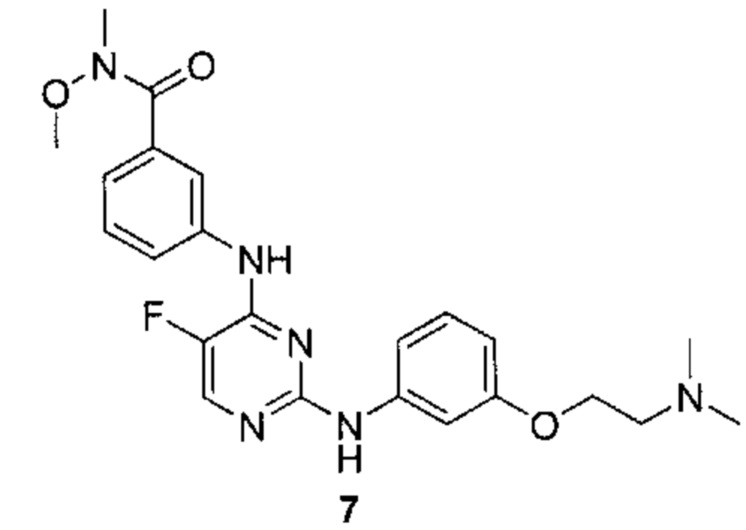
[00899] К раствору 6 (0,29 г, 0,70 ммоль) в ДМФ (3 мл) при помешивании добавляли MeNH-OMe.HCl (0. 068 г, 0,70 ммоль), EDCI.HCl (0.202g, 1,05 ммоль), НОВТ (0,047 г, 0,35 ммоль) и DIPEA (0.136 г, 1,05 ммоль). Реакционную смесь помешивали при комнатной температуре в течение 3 часа, тушили водой и экстрагировали EtOAc (2×5 мл). Объединенный органический слой промывали солевым раствором, и сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии (SiO2, 60-120, метанол/хлороформ: 20/80) с получением 7 (0,061 г, 19%) в форме желтого смолистого твердого вещества.
[00900] Этап-5
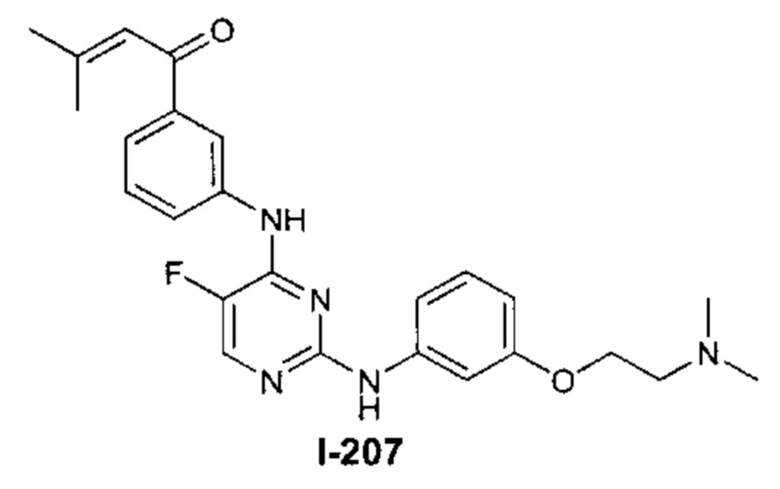
[00901] К раствору 7 (100 мг, 0,22 ммоль) в ТГФ (1 мл) при помешивании добавляли 8 (17.6 г, 8.80 ммоль) при температуре 0°С. Реакционную смесь оставляли для перемешивания при комнатной температуре в течение 2 часа. Затем ее тушили насыщенным раствором NH4Cl (0.5 мл) и экстрагировали EtOAc (2×3 мл). Объединенный органический слой промывали солевым раствором, и сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением белого твердого вещества. Затем его очищали методом колоночной хроматографии (SiO2, 60-120, использовали в качестве элюента 20% метанол/хлороформ) с получением I-207 (9 mg. 9%) в форме желтого смолистого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.90 (s, 3Н), 2.01 (s, 3Н), 2.19 (s, 6Н), 2.57 (t, J=5.64 Гц, 2Н), 3.87 (t, J=5.84 Гц, 2Н), 6.45 (dd, J=1.64 & 8.08 Гц, 1Н), 6.82 (s, 1Н), 7.04 (t, J=8.16 Гц, 1Н), 7.18 (d, J=8.16 Гц, 1Н), 7.33 (s, 1Н), 7.47 (t, J=7.88 Гц, 1Н), 7.62 (d, J=7.72 Гц, 1H), 8.13-8.16 (m, 3Н), 9.24 (s, 1H), 9.56 (s, 1Н); ЖХМС: m/е 450.1 (М+1).
ПРИМЕР 113
[00902] Получение 1-(4-(5-фтор-2-(фениламино)пиримидин-4-иламино)фенил)-3-метилбут-2-ен-1-она I-206
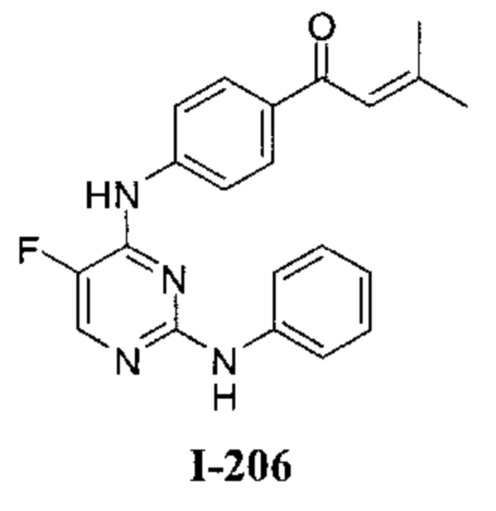
[00903] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием метил 4-аминобензоата вместо 4 на этапе 1 и анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.98 (s, 3Н), 2.12 (s, 3Н), 6.90-7.00 (m, 2Н), 7.20-7.30 (m, 2Н), 7.65 (d, J=8.16 Гц, 2Н), 7.90 (d, J=8.56 Гц, 2Н), 7.98 (d, J=8.68 Гц, 2Н), 8.18 (bs, 1H), 9.31 (s, 1Н), 9.68 (s, 1H); ЖХМС: m/е 363.0 (М+1).
ПРИМЕР 114
[00904] Получение 1-(3-(5-фтор-2-(3-(проп-2-инилокси)фениламино)пиримидин-4-иламино)фенил)-3-метилбут-2-ен-1-она I-211
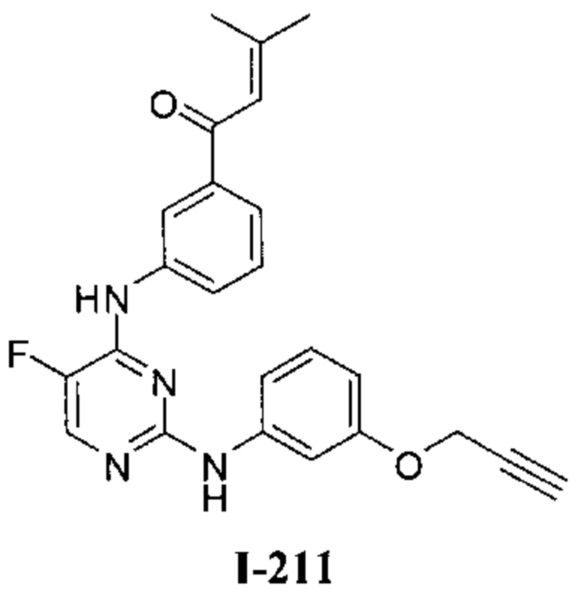
[00905] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием 3-prop-2-инилоксианилина вместо 4 на этапе 4. 1Н-ЯМР (CD3OD) δ ppm: 2.0 (d, J=1 Гц, 3Н), 2.21 (d, J=1.04 Гц, 3Н), 2.94-2.96 (d, J=2.44 Гц, 1H), 4.59 (d, J=2.36 Гц, 2Н), 6.79-6.81 (m, 2Н), 7.03 (dd, J=3.12 & 8.04 Гц, 1Н), 7.14 (t, J=2.2 Гц, 1Н), 7.23 (t, J=8.12 Гц, 1H), 7.54 (t, J=7.92 Гц, 1Н), 7.83 (d, J=7.96 Гц, 1H), 7.86 (dd, J=2.08 & 8.08 Гц, 1Н), 8.03 (d, J=4.96 Гц, 1H), 8.21 (t, J=1.88 Гц, 1Н); ЖХМС: m/е 417.0 (М+1).
ПРИМЕР 115
[00906] Получение 1-(3-(5-фтор-2-(3-(трифторметокси)фениламино)пиримидин-4-иламино)фенил)-3-метилбут-2-ен-1-она I-223
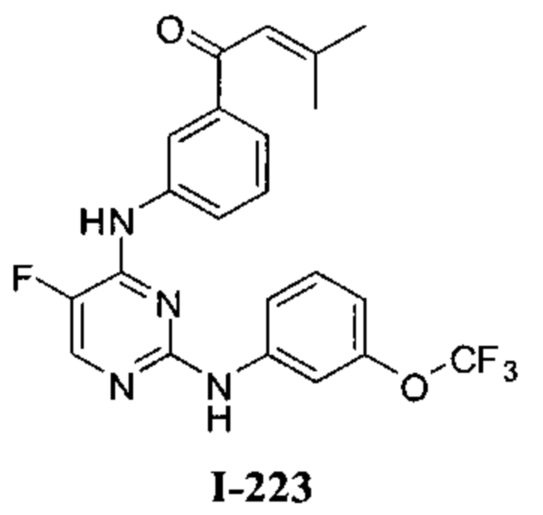
[00907] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием 3-трифторметоксианилина вместо 4 на этапе 2. 1Н-ЯМР (CDCl3) δ ppm: 2.0 (d, J=1.08 Гц, 3Н), 2.24 (d, J=1.04 Гц, 3Н), 6.74 (t, J=1.24 Гц, 1Н), 6.85 (dd, J=1.08 & 7.0 Гц, 1Н), 6.90 (s, 1H), 7.08 (s, 1H), 7.24-7.28 (m, 1H), 7.35 (td, J=1.2 & 7.44 Гц, 1H), 7.49 (t, J=7.88 Гц, 1H), 7.63 (s, 1Н), 7.72 (td, J=1.04 & 7.76 Гц, 1H), 7.90-7.92 (m, 1H), 8.00-8.05 (m, 2H); ЖХМС: m/e 447 (M+1).
ПРИМЕР 116
[00908] Получение 1-(3-(5-фтор-2-(3-(трифторметокси)фениламино)пиримидин-4-иламино)фенил)-2-метилпроп-2-ен-1-она I-199
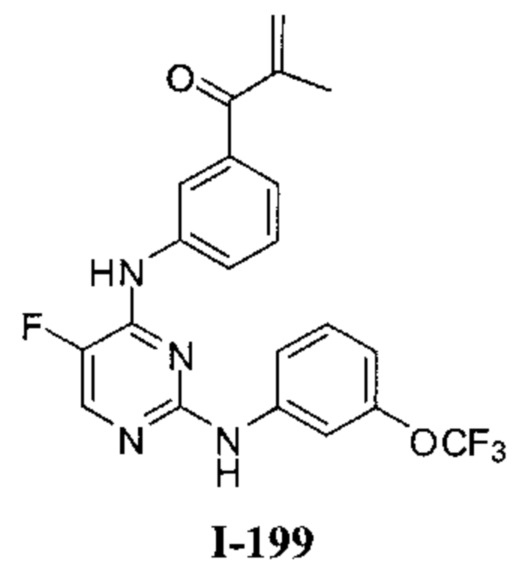
[00909] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием 3-трифторметоксианилина вместо 4 на этапе 2 и изопропенилмагния бромид вместо 8 на Этапе 5. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.97 (s, 3Н), 5.6 (s, 1Н), 6.0 (d, J=0.96 Гц, 1H), 6.82 (d, J=8.08 Гц, 1H), 7.27 (t, J=8.2 Гц, 1H), 7.41 (dd, J=1.12 & 7.56 Гц, 1H), 7.48 (t, J=7.76
Гц, 1Н), 7.60 (dd, J=1.28 & 7.88 Гц, 1H), 7.78 (s, 1H), 7.90 (d, J=1.64 Гц, 1H), 8.15 (d, J=8 Гц, 1H), 8.19 (d, J=3.64 Гц, 1H), 9.55 (s, 1H), 9.65 (s, 1H); ЖХМС: m/e 433 (M+1).
ПРИМЕР 117
[00910] Получение 1-(4-(5-фтор-2-(фениламино)пиримидин-4-иламино)фенил)-2-метилprop-2-ен-1-она I-185
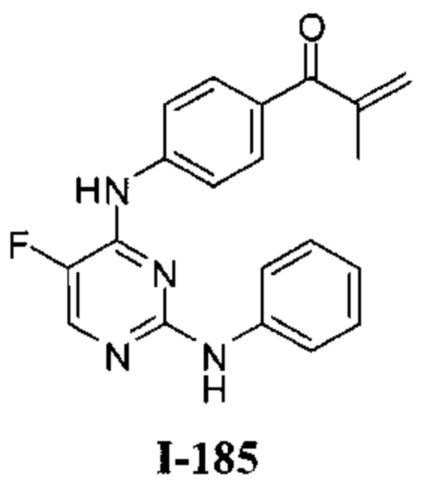
[00911] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием метил 4-аминобензоата вместо 2 на этапе 1 и анилина вместо 4 на этапе 2 и изопропенилмагния бромида вместо 8 на Этапе 5. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.99 (s, 3Н), 5.54 (s, 1H), 1.01 (s, 1H), 6.93 (t, J=7.36 Гц, 1H), 7.24 (t, J=7.52 Гц, 2Н), 7.66-7.72 (m, 4Н), 8.01 (d, J=8.72 Гц, 2Н), 8.19 (d, J=3.6 Гц, 1H), 9.32 (s, 1Н), 9.72 (s, 1H); ЖХМС: m/е 348.8 (М+1).
ПРИМЕР 118
[00912] Получение N-(3-(2-(3-(2-(диметиламино)этокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-233
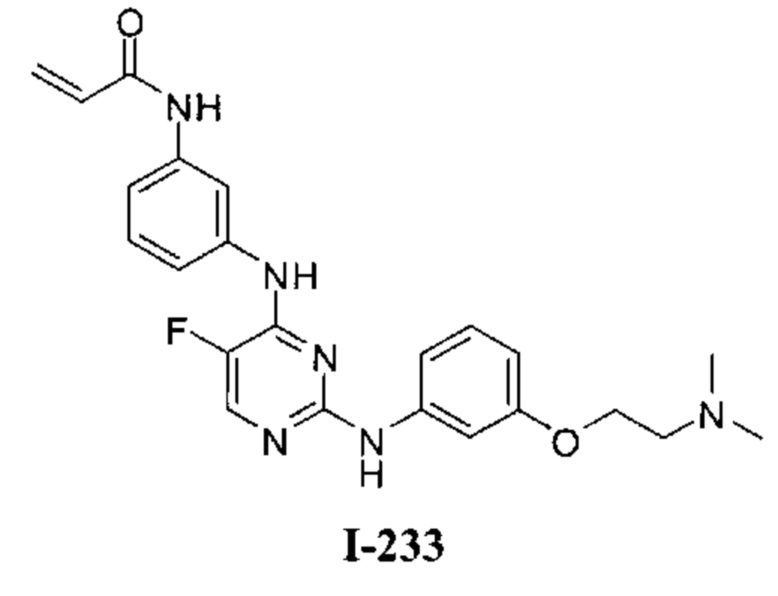
[00913] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(2-диметиламиноэтокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (CD3OD) δ ppm: 2.31 (s, 6Н), 2.76 (t, J=5.6 Гц, 2Н), 3.97 (t, J=5.2 Гц, 2Н), 5.78 (dd, J=2 & 9.2 Гц, 1Н), 6.38-6.42 (m, 2Н), 6.52-6.55 (m, 1Н), 7.1-7.11 (m 2Н), 7.30 (t, J=8.0 Гц, 1H), 7.36 (s, 1Н), 7.42-7.48 (m, 2Н), 7.94 (d, 7=3.6 Гц, 1H), 8.05 (s, 1Н); ЖХМС: m/е 437 (М+1).
ПРИМЕР 119
[00914] Получение N-(3-(5-фтор-4-(4-феноксифенокси)пиримидин-2-иламино)фенил)акриламида I-130
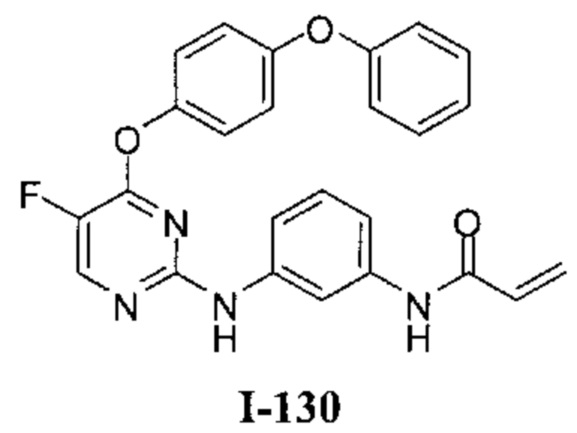
[00915] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 11, с использованием 5-фтор-2,4-дихлоропиримидина вместо 1 на этапе 1. 1Н-ЯМР (ДМСО-d6) δ ppm: 5.71 (dd, J=1.6 & 10 Гц, 1Н), 6.22 (dd, J=1.6 & 16.8 Гц, 1H), 6.44 (dd, J=10.4 & 17.2 Гц, 1Н), 6.98-7.05 (m, 3Н), 7.1-7.12 (m, 2Н), 7.17 (t, J=7.2 Гц, 1Н), 7.24 (t, J=7.6 Гц, 2Н), 7.35-7.37 (m, 2Н), 7.42 (t, J=8.4 Гц, 2Н), 7.71 (s, 1Н), 8.5 (s, 1Н), 9.6 (s, 1H), 10.05 (s, 1Н); ЖХМС: m/е 443,0 (М+1).
ПРИМЕР 120
[00916] Получение (S)-N-(3-(5-фтор-2-(4- (тетрагидрофуран-3-илокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-43
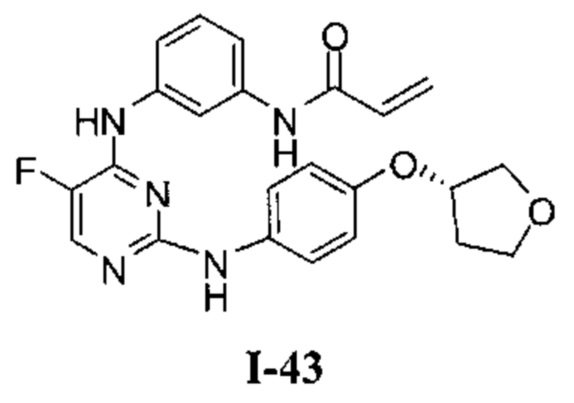
[00917] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-4-(тетрагидрофуран-3-илоксианилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: δ 10.10 (s, 1Н), 9.35 (s, 1H), 8.95 (s, 1H), 8.05 (d, J=4.0 Гц, 1H), 7.92 (s, 1H), 7.52 (d, J=9.0 Гц, 2H), 7.47 (d, J=7.5 Гц, 1H), 7.41 (d, J=8.5 Гц, 1H), 7.27 (t, J=8.0 Гц, 1H), 6.72 (d, J=9.0 Гц, 2H), 6.45 (dd, J=1.5, 17.0 Гц, 1H), 6.25 (dd, J=1.1, 16.5 Гц, 1H), 5.75 (dd, J=1.1, 10.0 Гц, 1H), 4.93-4.84 (m, 1H), 3.88-3.72 (m, 4H), 2.20-2.10 (m, 1H), 1.97-1.90 (m, 1H). MS m/e = 436 [M++1]
ПРИМЕР 121
[00918] Получение (R)-N-(3-(5-фтор-2-(4-(тетрагидрофуран-3-илокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-46
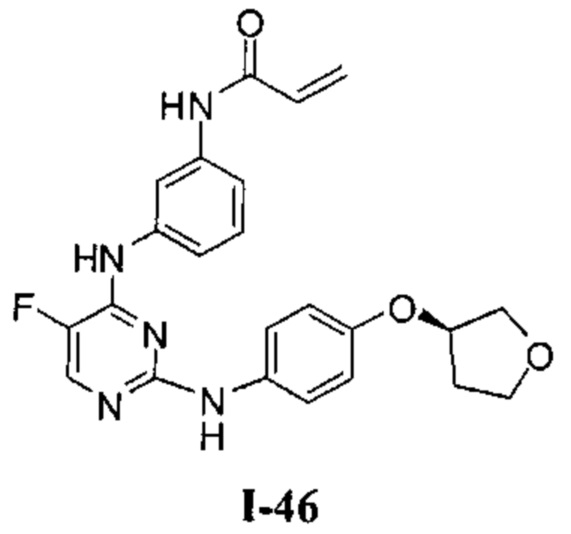
[00919] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (R)-4-(тетрагидрофуран-3-илоксианилина вместо 4 на этапе 2. 1H-ЯМР (ДМСО-d6) δ ppm: δ 10.10 (s, 1H), 9.35 (s, 1H), 8.95 (s, 1H), 8.05 (d, J=4.0 Гц, 1H), 7.92 (s, 1H), 7.52 (d, J=9.0 Гц, 2E), 7.47 (d, J=7.5 Гц, 1H), 7.41 (d, J=8.5 Гц, 1H), 7.27 (t, J=8.0 Гц, 1H), 6.72 (d, J=9.0 Гц, 2H), 6.45 (dd, J=1.5, 17.0 Гц, 1H), 6.25 (dd, J=1.1, 16.5 Гц, 1H), 5.75 (dd, J=1.1, 10.0 Гц, 1H), 4.93-4.84 (m, 1H), 3.88-3.72 (m, 4E), 2.22-2.14 (m, 1H), 1.97-1.90 (m, 1H). MS: m/e=436 [M++1]
ПРИМЕР 122
[00920] Получение N-(3-(5-фтор-2-(3-((1-метилпиперидин-3-ил)метокси)фениламино)пиримидин-4-иламино)фенил)-акриламида I-76)
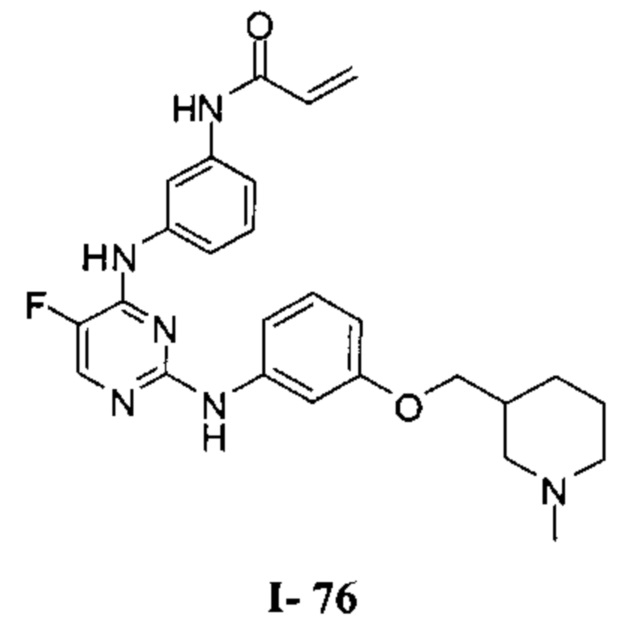
[00921] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(1-метилпиперидин-3-ил)метоксианилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: δ 10.08 (s, 1Н), 9.41 (s, 1Н), 9.09 (s, 1H), 8.11 (d, J=3.5 Гц, 1Н), 7.90 (s, 1Н), 7.57 (d, J=8.0 Гц, 1H), 7.41 (d, J=8.0 Гц, 1H), 7.32 (s, 1Н), 7.30-7.20 (m, 2Н), 7.03 (t, J=8.0 Гц, 1Н), 6.50-6.40 (m, 2Н), 6.24 (dd, J=1.5, 17.0 Гц, 1H), 5.74 (dd, J=2.0, 10.5 Гц, 1Н), 3.75-3.65 (m, 2Н), 2.73 (d, J=10 Гц, 1Н), 2.60 (d, J=10.5 Гц, 1Н), 2.13 (s, 3Н), 1.98-1.83 (m, 2Н), 1.75-1.58 (m, 3Н), 1.55-1.45 (m, 1H), 1.05-0.95 (m, 1H). MS: m/е=477 (М++1).
ПРИМЕР 123
[00922] Получение N-(3-(5-фтор-2-(3-((1-метилпиперидин-4-ил)метокси)фениламино)пиримидин-4-иламино)фенил)-акриламида I-82
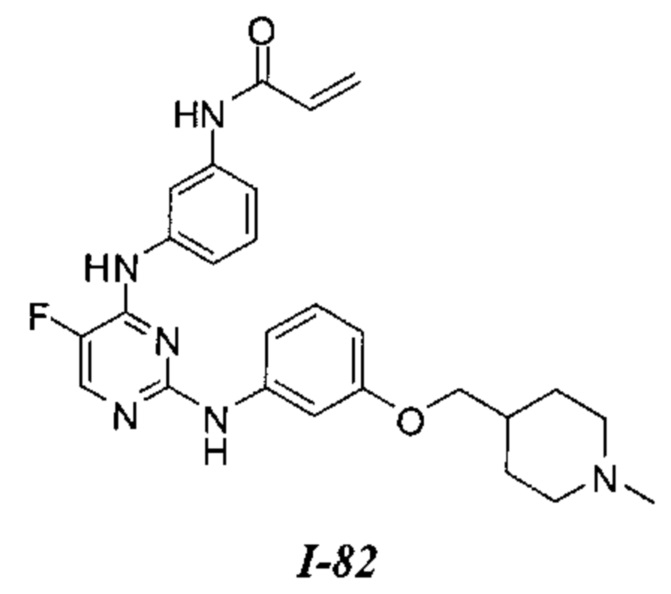
[00923] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(1-метилпиперидин-4-ил)метоксианилина вместо 4 на этапе 2. 1Н-ЯМР (CDCl3+ДМСО-D6,
500 МГц): δ 9.04 (bs, 1Н), 8.30 (s, 1H), 7.96 (s, 1H), 7.75 (s, 1H), 7.63 (s, 1H), 7.59 (d, J=7.0 Гц, 1H), 7.38-7.33 (m, 2H), 7.27 (t, J=8.5 Гц, 1H), 7.16 (t, J=8 Гц, 1H), 7.09 (d, J=8.5 Гц, 1H), 6.52 (d, J=7.0 Гц, 1H), 6.51-6.38 (m, 2H), 5.73 (dd, J=2.0, 9.0 Гц, 1H), 3.77 (d, J=6.0 Гц, 2H), 2.90-2.84 (m, 2H), 2.28 (s, 3H), 1.95 (t, J=10.0 Гц, 1H), 1.85-1.74 (m, 2H), 1.45-1.32 (m, 2H), 1.28-1.25 (m, 2H). MS: m/e=477 (M++1).
ПРИМЕР 124
[00924] Получение N-(3-(2-(3-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-83
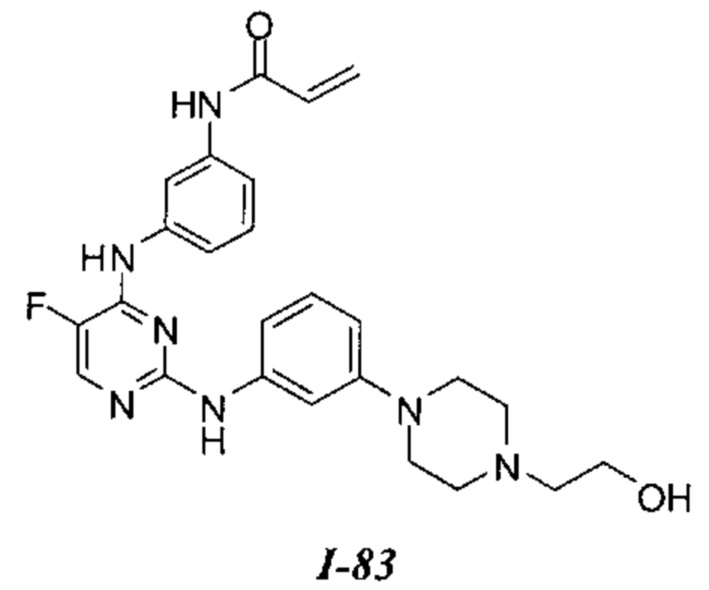
[00925] Указанное в названии соединение получали используя этапы, схемы и промежуточные соединения, описанные в Примере 20, но использовали 3-(4-(2-т-бутилдиметилсилилоксиэтил)пиперазин-1-иланилина вместо 4 на Этапе 2 и удаляли защиту TBS эфира с помощью ТФА в ДХМ в качестве этапа 5. 1Н-ЯМР (ДМСО-d6, 500 МГц): δ 8.99 (s, 1Н), 8.85 (s, 1Н), 8.04 (d, J=4.0 Гц, 1H), 7.28-7.19 (m, 2Н), 7.08-7.00 (m, 2Н), 6.94 (t, J=3.5 Гц, 2Н), 6.48 (dd, J=2.0, 8.0 Гц, 1Н), 6.35-6.31 (m, 1H), 4.94 (s, 2Н), 3.71 (t, J=6.0 Гц, 2Н), 3.02 (t, J=4.5 Гц, 4Н), 2.57-2.50 (m, 4Н), 2.46 (t, J=6.0 Гц, 2Н), 0.87 (s, 9Н), 0.05 (s, 6Н). MS: m/е=538 (М++1).
ПРИМЕР 125
[00926] Получение N-(4-(5-фтор-2-(3-метоксифениламино)пиримидин-4-иламино)бензил)-N-метилакриламида I-113
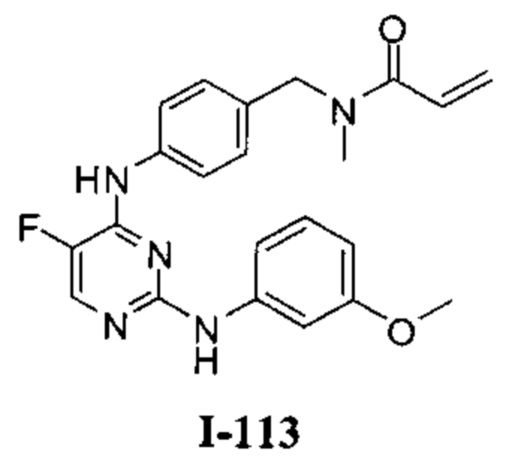
[00927] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием метил 4-(N-метил-N-трет-бутилоксикарбониламино)метиланилина вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6, 200 МГц): δ 9.36 (bs, 1Н), 9.16 (bs, 1Н), 8.10 (d, J=3.4 Гц, 1Н), 7.83-7.70 (m, 2Н), 7.34 (bs, 1Н), 7.26-7.01 (m, 4Н), 6.86-6.73 (m, 1H), 6.48 (d, J=8.0 Гц, 1Н), 6.21 (dd, J=16.4, 2.2 Гц, 1Н), 5.76-5.64 (m, 1Н), 4.64-4.53 (два s, 2Н), 3.65 (s, 3Н), 3.00-2.88 (два s, 3Н). MS: m/е=408.2 [М++1].
ПРИМЕР 126
[00928] Получение N-(4-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)бензил)-N-метилакриламида I-114
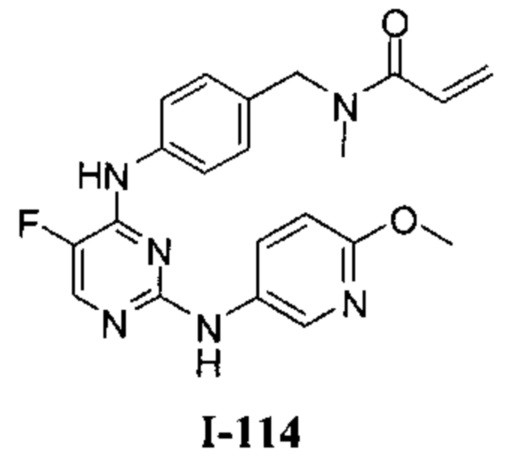
[00929] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием метил 4-(N-метил-N-трет-бутилоксикарбониламино)метиланилина вместо 2 на этапе 1 и 6-метокси-3-аминопиридина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6, 200 МГц): δ 9.36 (bs, 1H), 9.09 (bs, 1H), 8.32 (bs, 1H), 8.06 (dd, J=3.8 Гц, 1H), 7.97-7.92 (m, 1H), 7.76-7.68 (m, 2Н), 7.20-7.08 (m, 2Н), 6.87-6.75 (m, 1H), 6.72 (d, J=8.4 Гц, 1H), 6.22 (dd, J=19.4, 2.6 Гц, 1H), 5.74-5.65 (m, 1H), 4.64-4.53 (два s, 2Н), 3.78 (s, 3Н), 3.00-2.88 (два s, 3Н). MS: m/е=409 (М++1).
ПРИМЕР 127
[00930] Получение N-(3-(5-фтор-2-(3-метиоксифениламино)пиримидин-4-иламино)бензил)-N-метилакриламида I-115
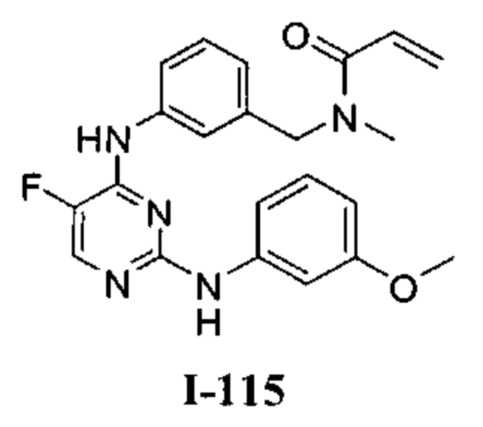
[00931] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием метил 3-(N-метил-N-трет-бутилоксикарбониламино)метиланилина вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6, 200 МГц): δ 9.50-9.30 (m, 1Н), 9.15-8.96 (m, 1Н), 8.15 (bs, 1H), 7.82-7.59 (m, 2Н), 7.45-7.00 (m, 4Н), 6.97-6.65 (m, 2Н), 6.55-6.45 (m, 1Н), 6.26-6.12 (m, 1H), 5.78-5.60 (m, 1Н), 4.68 (s, 1Н), 4.55 & 3.75 (два s, 3Н), 2.90 & 3.00 (два s, 3Н). MS: m/е=408.2 [М++1].
ПРИМЕР 128
[00932] Получение N-(3-(5-фтор-2-(3-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)пиримидин-4-илокси)фенил)акриламида I-84
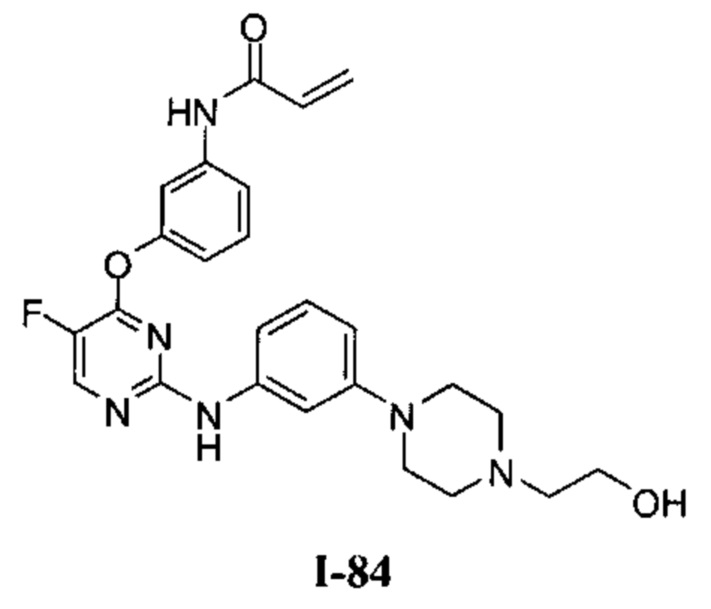
[00933] Указанное в названии соединение получали используя этапы, схемы и промежуточные соединения, описанные в Примере 98, но использовали 3-(4-(2-т-бутилдиметилсилилоксиэтил)пиперазин-1-иланилина вместо 4 на Этапе 2 и удаляли
защиту TBS эфира с помощью ТФА в ДХМ в качестве этапа 5. 1Н-ЯМР (ДМСО-d6, 500 МГц): δ 8.19 (s, 1Н), 7.75-7.70 (m, 2Н), 7.42-7.35 (m, 2Н), 7.12-7.05 (m, 2Н), 6.97 (dd, J=2.0, 10.5 Гц, 1Н), 6.93 (s, 1H), 6.72 (d, J=6.5 Гц, 1H), 6.57-6.54 (m, 1Н), 6.44 (d, J=17.0 Гц, 1Н), 6.29-6.20 (m, 1Н), 5.78 (d, J=10.0 Гц, 1Н), 3.68 (t, J=5.5 Гц, 2Н), 3.00-2.94 (m, 4Н), 2.63-2.56 (m, 6Н). MS: m/е=479 (М++1).
ПРИМЕР 129
[00934] Получение N-(3-(5-фтор-2-(3-((1-метилпиперидин-3-ил)метокси)фениламино)пиримидин-4-илокси)фенил)акриламида I-81
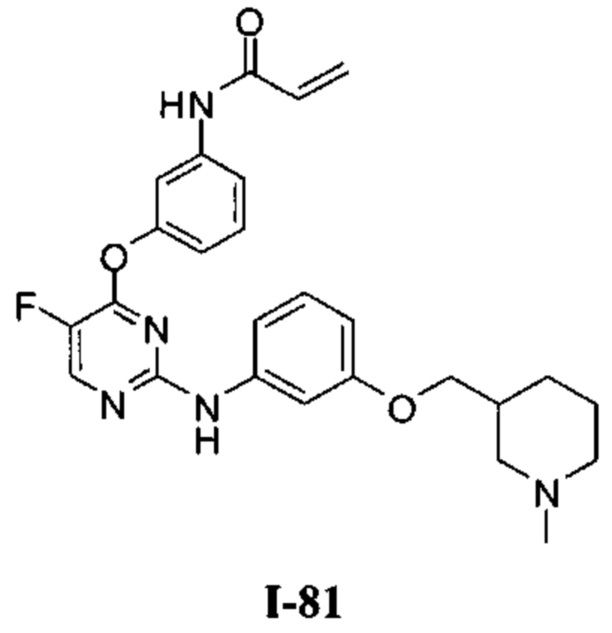
[00935] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием 3-(1-метилпиперидин-3-ил)метоксианилина вместо 4 на этапе 2. 1Н-ЯМР (CDCl3, 500 МГц): δ 8.19 (d, J=2.5 Гц, 1H), 7.82-7.75 (m, 2Н), 7.42-7.36 (m, 2Н), 7.08-7.02 (m, 3Н), 6.99 (d, J=7.0 Гц, 1Н), 6.91 (s, 1Н), 6.79 (d, J=7.5 Гц, 1Н), 6.48-6.44 (m, 1Н), 6.42 (s, 1H), 6.29-6.23 (m, 1Н), 5.77 (d, J=10 Гц, 1Н), 3.67-3.62 (m, 2Н), 2.95-2.91 (m, 1Н), 2.82-2.76 (m, 1H), 2.28 (s, 3Н), 2.11-2.05 (m, 1H), 1.98-1.92 (m, 1H), 1.79-1.70 (m, 3Н), 1.11-1.04 (m, 1H). MS: m/e=478 (M++1).
ПРИМЕР 130
[00936] Получение N-(3-(5-фтор-2-(3-((1-метилпиперидин-4-ил)метокси)фениламино)пиримидин-4-илокси)фенил)-акриламида I-75
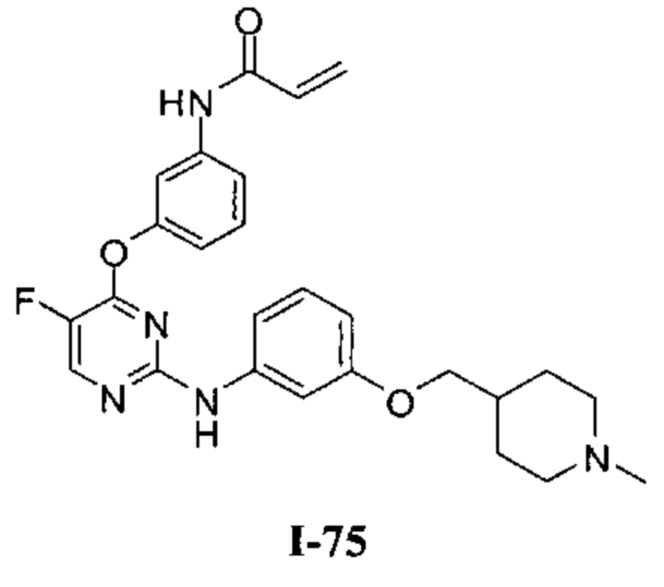
[00937] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием 3-(1-метилпиперидин-4-ил)метоксианилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-D6, 500 МГц): δ 10.31 (s, 1H), 9.50 (s, 1Н), 8.50 (s, 1Н), 7.67 (s, 1H), 7.55 (d, J=8.0 Гц, 1H), 7.42 (t, J=8.0 Гц, 1Н), 7.10 (s, 1Н), 7.04 (d, J=7.0 Гц, 2Н), 6.94 (t, J=8.0 Гц, 1Н), 6.45-6.39 (m, 2Н), 6.27 (d, J=15.0 Гц, 1Н), 5.78 (dd, J=2.0, 10.5 Гц, 1Н), 3.64 (d, J=6.0 Гц, 2Н), 2.75 (d, J=6.5 Гц, 2Н), 2.14 (s, 3Н), 1.83 (t, J=10.5 Гц, 2Н), 1.66-1.64 (m, 3Н), 1.25-1.23 (m, 2Н). MS: m/e=478 (М++1).
ПРИМЕР 131
[00938] Получение N-(3-(5-циано-2-(фениламино)пиримидин-4-иламино)фенил)акриламида I-157
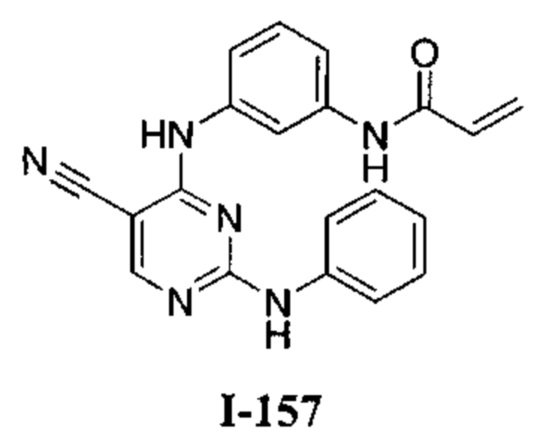
[00939] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 94, с использованием 2,4-дихлоро-5-суапопиримидина вместо 4 на этапе 2. MS 379.1 (M+Na).
ПРИМЕР 132
[00940] Получение N-(3-(5-фтор-2-(3-(трифторметокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-244
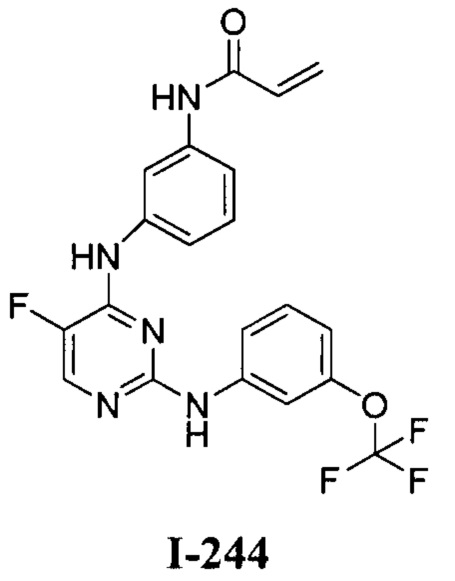
[00941] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(трифторметокси)анилин вместо 4 на этапе MS 434.1 (М+1).
ПРИМЕР 133
[00942] Получение N-(3-(5-фтор-2-(пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-234
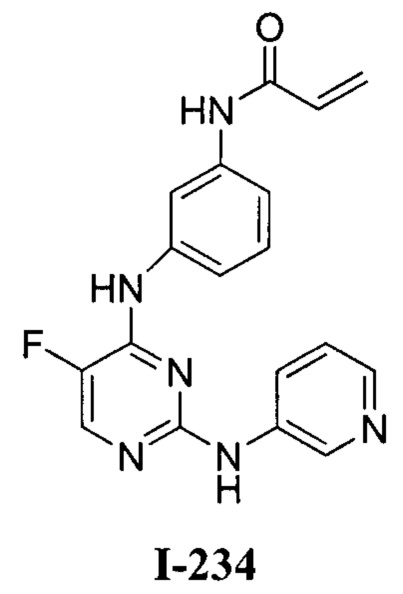
[00943] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-аминопиридине вместо 4 на этапе 2. MS 351.1 (М+1).
ПРИМЕР 134
[00944] Получение N-(3-(5-фтор-2-(4-фторфениламино)пиримидин-4-иламино)фенил)акриламида I-247
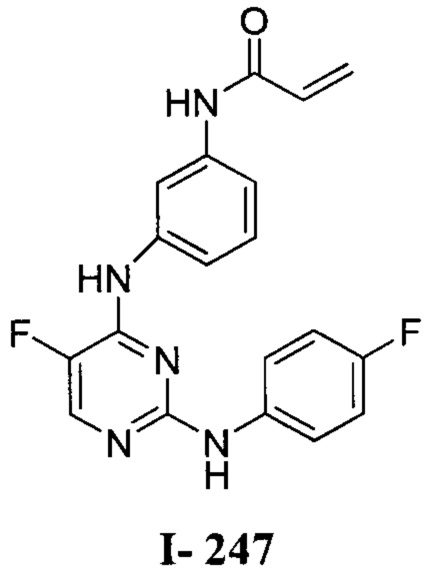
[00945] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-фторанилина вместо 4 на этапе 2. MS 368,1 (М+1).
ПРИМЕР 135
[00946] Получение N-(3-(5-фтор-2-(3-(3-морфолинопропокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-208
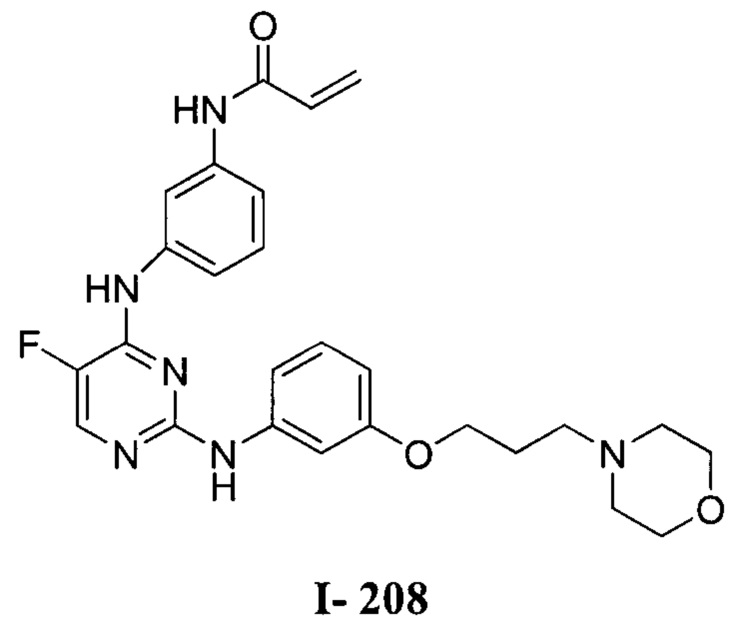
[00947] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(3-морфолинопропокси)анилина вместо 4 на этапе MS 515,3 (M+Na).
ПРИМЕР 136
[00948] Получение N-(3-(2-(3-(3-(1Н-имидазол-1-ил)пропокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-204
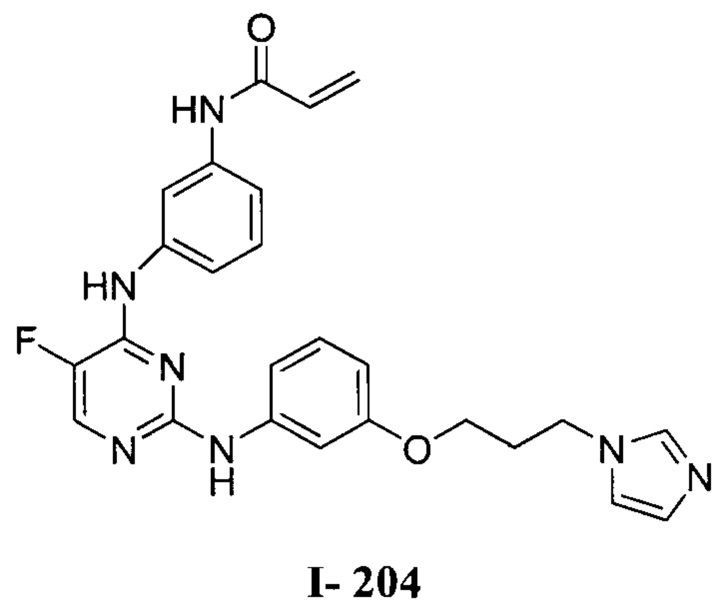
[00949] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(3-(1Н-имидазол-1-ил)пропокси)анилин вместо 4 на этапе 2. MS 474,3 (M+Na).
ПРИМЕР 137
[00950] Получение N-(3-(2-(1-ацетилпиперидин-3-иламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-238
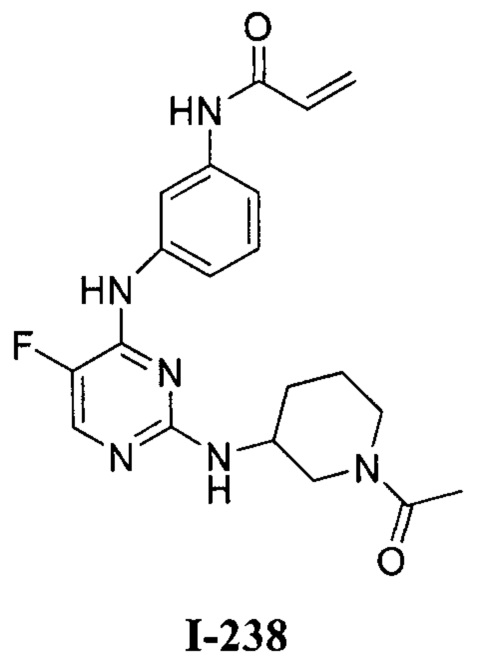
[00951] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 1-(3-аминопиперидин-1-ил)этанона вместо 4 на этапе 2. MS 421,1 (M+Na).
ПРИМЕР 138
[00952] Получение N-(3-(5-фтор-2-(фениламино)пиримидин-4-илокси)фенил)акриламида I-228
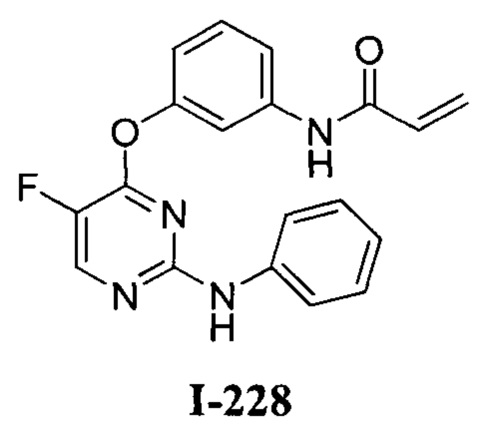
[00953] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием анилина вместо 4 на этапе 2. MS 351,3 (М+1).
ПРИМЕР 139
[00954] Получение N-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-243
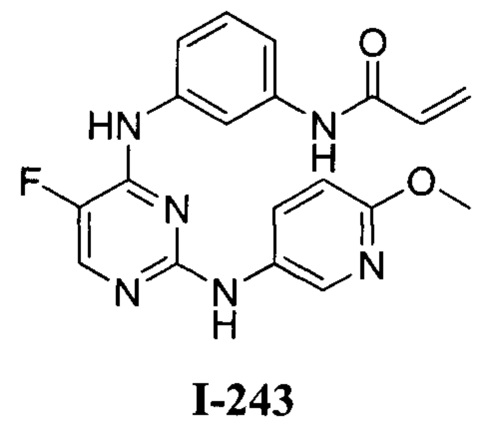
[00955] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 6-метоксипиридин-3-амина вместо 4 на этапе 2. MS 381,1 (М+1).
ПРИМЕР 140
[00956] Получение N-(3-(5-метокси-2-(3-метоксифениламино)пиримидин-4-иламино)фенил)акриламида I-158
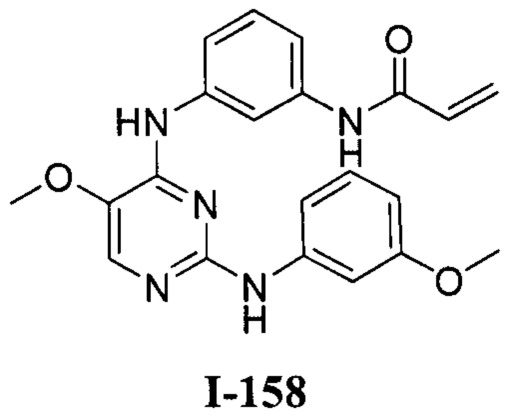
[00957] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 1, с использованием 5-метокси-2,4-дихлоропиримидина вместо 1 на этапе 1 и 3-метоксианилин вместо 4 на этапе 2. MS 392,3 (М+1).
ПРИМЕР 141
[00958] Получение N-(3-(5-метокси-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-192
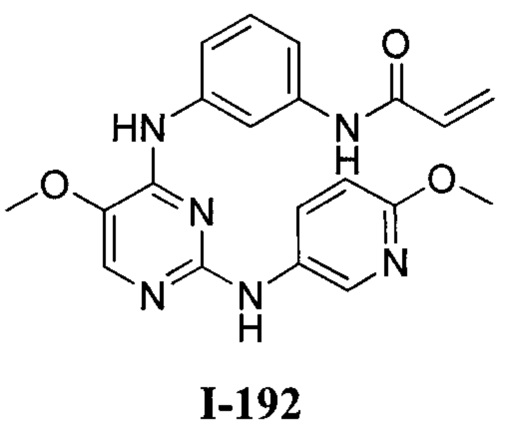
[00959] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 1, с использованием 5-метокси-2,4-дихлоропиримидина вместо 1 на этапе 1 и 5-амино-2-метоксипиридина вместо 4 на этапе 2. MS 393,3 (М+1).
ПРИМЕР 142
[00960] Получение N-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-илокси)фенил)акриламида I-222
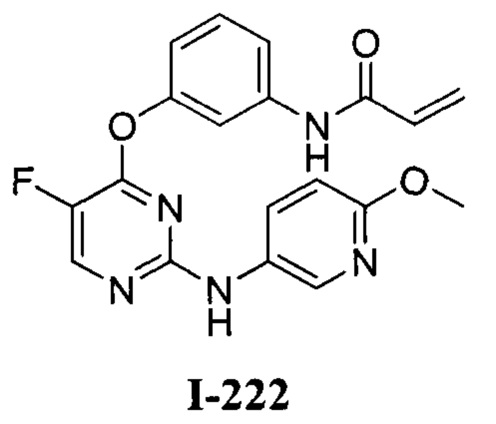
[00961] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием 3-амино-6-метоксипиридина вместо 4 на этапе 2. MS 382,3 (М+1).
ПРИМЕР 143
[00962] Получение 4-(3-акриламидофениламино)-N-трет-бутил-2-(6-метоксипиридин-3-иламино)пиримидин-5-карбоксамида I-216
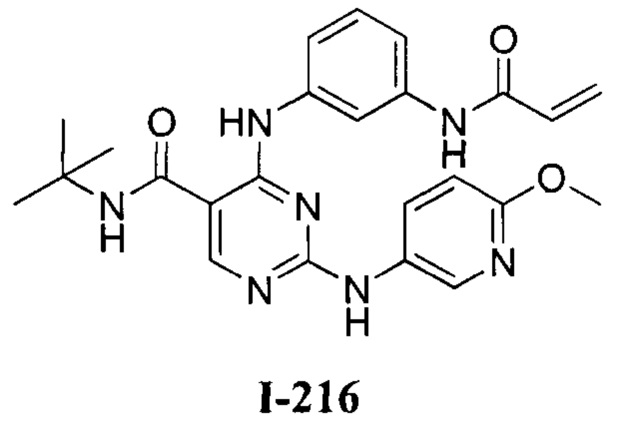
[00963] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 37, с использованием трет-бутиламина вместо 2 на этапе 1, и пропуская этап 6. MS 484,3 (M+Na).
ПРИМЕР 144
[00964] Получение (R)-1-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-202
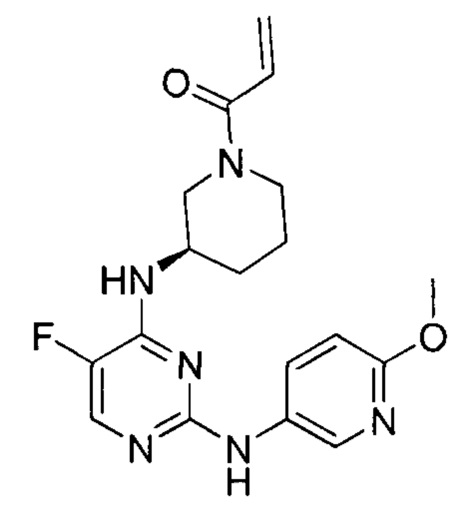
I-202
[00965] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (R)-трет-бутил 3-аминопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-амино-6-метоксипиридин вместо 4 на этапе 2. MS 395.3 (M+Na).
ПРИМЕР 145
[00966] Получение (R)-1-(3-(5-фтор-2-(3-метоксифениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-195
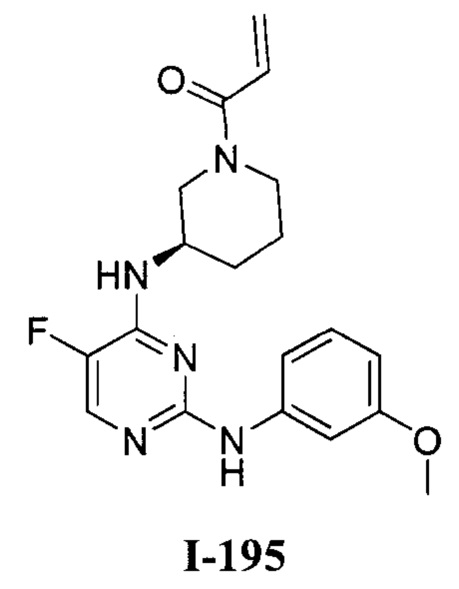
[00967] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (R)-трет-бутил 3-аминопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. MS 394,3 (M+Na).
ПРИМЕР 146
[00968] Получение (S)-1-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-197
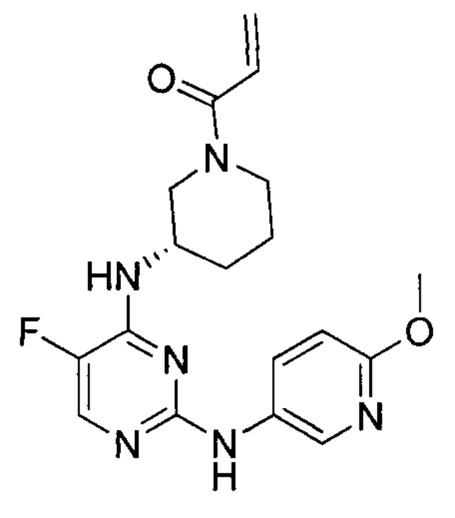
I-197
[00969] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-трет-бутил 3-аминопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-амино-6-метоксипиридин вместо 4 на этапе 2. MS 373,3 (М+1).
ПРИМЕР 147
[00970] Получение (S)-1-(3-(5-фтор-2-(3-метоксифениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-196
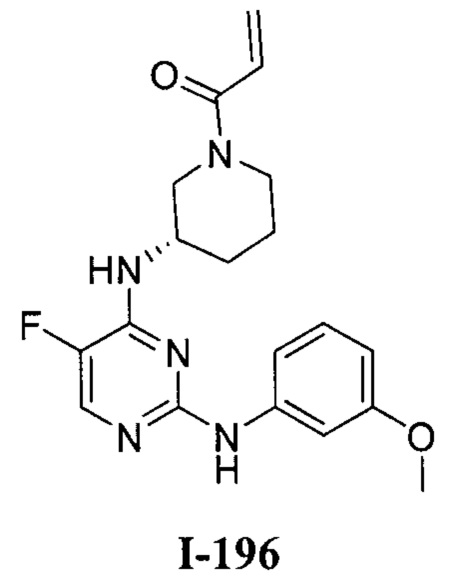
[00971] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-трет-бутил 3-аминопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. MS 372,3 (М+1).
ПРИМЕР 148
[00972] Получение (R)-1-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-илокси)пиперидин-1-ил)проп-2-ен-1-она I-180
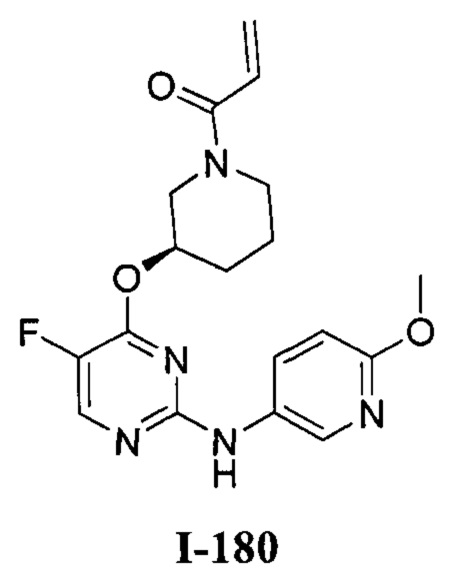
[00973] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием (R)-трет-бутил 3-гидроксипиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-амино-6-метоксипиридин вместо 4 на этапе 2. MS 374,3 (М+1).
ПРИМЕР 149
[00974] Получение (R)-1-(3-(5-фтор-2-(3-метоксифениламино)пиримидин-4-илокси)пиперидин-1-ил)проп-2-ен-1-она I-190
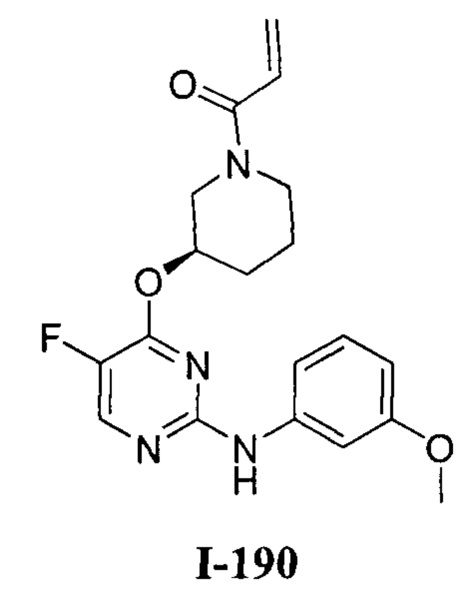
[00975] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием (R)-трет-бутил 3-гидроксипиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. MS 395,3 (M+Na).
ПРИМЕР 150
[00976] Получение (S)-1-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-илокси)пиперидин-1-ил)проп-2-ен-1-она I-193
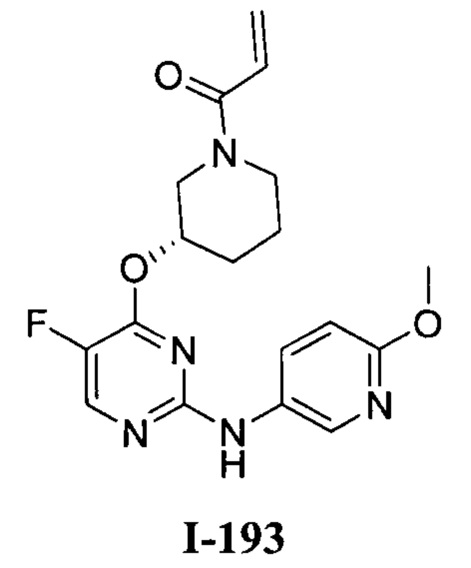
[00977] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием (S)-трет-бутил 3-гидроксипиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-амино-6-метоксипиридина вместо 4 на этапе 2. MS 396,3 (M+Na).
ПРИМЕР 151
[00978] Получение (S)-1-(3-(5-фтор-2-(3-метоксифениламино)пиримидин-4-илокси)пиперидин-1-ил)проп-2-ен-1-она I-179
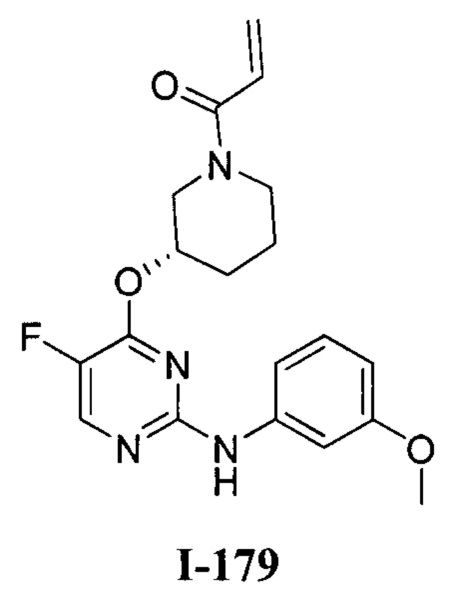
[00979] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием (S)-трет-бутил 3-гидроксипиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. MS 395,3 (M+Na).
ПРИМЕР 152
[00980] Получение 1-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)пирролидин-1-ил)проп-2-ен-1-она I-203
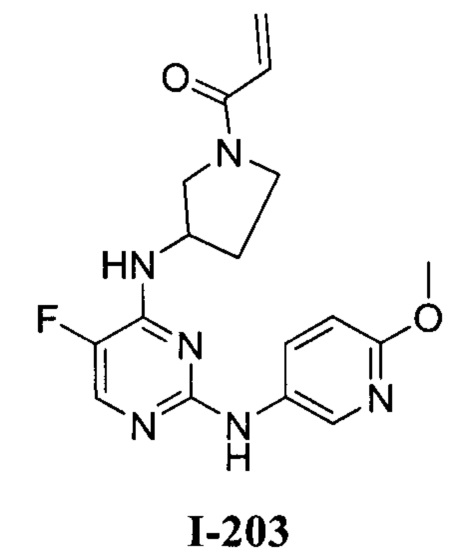
[00981] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-трет-бутил 3-аминопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-амино-6-метоксипиридина вместо 4 на этапе 2. MS 381,3 (M+Na).
ПРИМЕР 153
[00982] Получение 1-(3-(5-фтор-2-(3-метоксифениламино)пиримидин-4-иламино)пирролидин-1-ил)проп-2-ен-1-она I-201
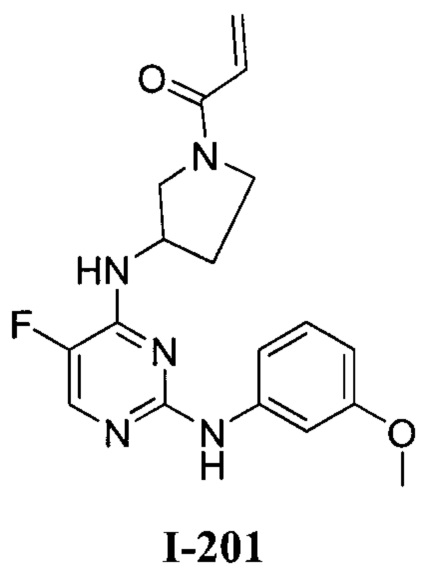
[00983] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-трет-бутил 3-аминопирролидин-1-карбоксилата вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. MS 358,3 (М+1).
ПРИМЕР 154
[00984] Получение (R)-1-(3-(5-фтор-2-(3-метоксифениламино)пиримидин-4-илтио)пиперидин-1-ил)проп-2-ен-1-он I-137
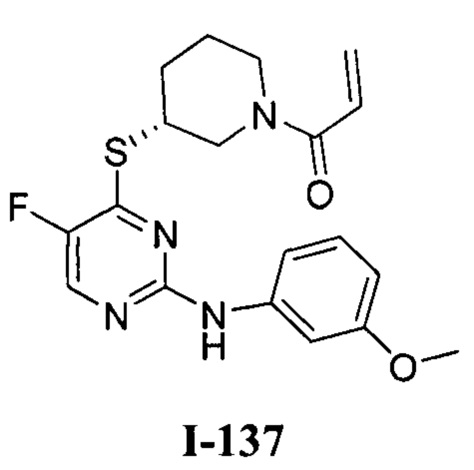
[00985] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-трет-бутил 3-меркаптопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-метоксианилина вместо 4 на этапе 2. MS 411,1 (M+Na).
ПРИМЕР 155
[00986] Получение (R)-1-(3-(2-(3-хлорфениламино)-5-фторпиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-147
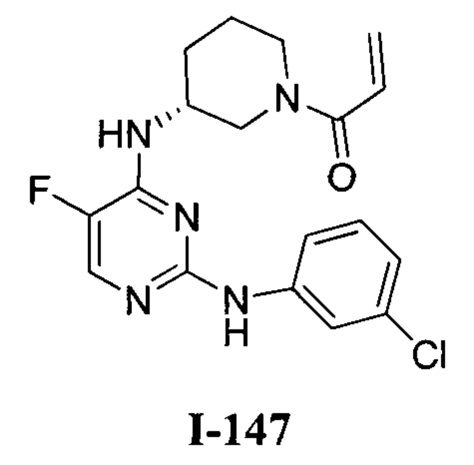
[00987] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-трет-бутил 3-аминопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-хлороанилина вместо 4 на этапе 2. MS 376,1 (М+1).
ПРИМЕР 156
[00988] Получение (R)-1-(3-(5-фтор-2-(3-(2-морфолиноэтокси)фениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-135
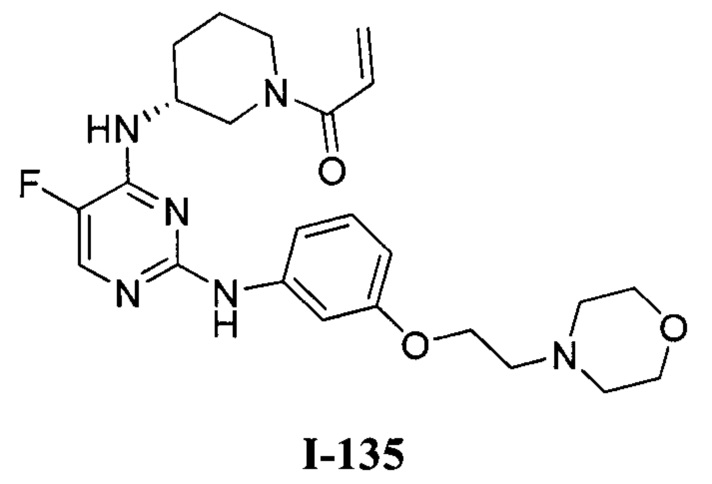
[00989] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-трет-бутил 3-аминопиперидин-1-карбоксилата вместо 2 на этапе 1 и 3-(2-морфолиноэтокси)анилина вместо 4 на этапе 2. MS 471,3 (М+1).
ПРИМЕР 157
[00990] Получение (Е)-4-(диметиламино)-N-(3-(5-фтор-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)фенил)бут-2-енамида I-125
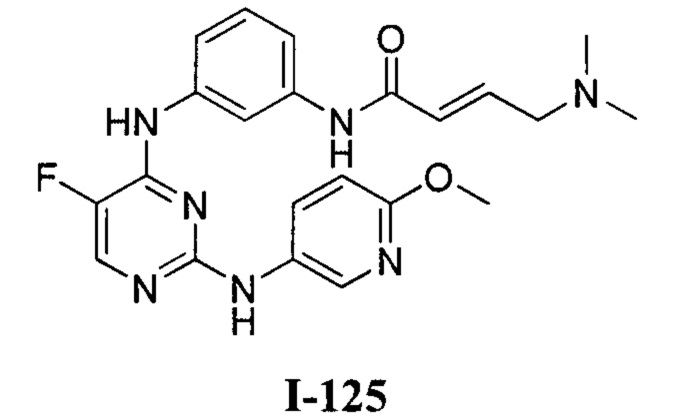
[00991] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 139, с использованием (Е)-4-(диметиламино)бут-2-еноил хлорида вместо 7 на этапе 4. MS 460,1 (M+Na).
ПРИМЕР 158
[00992] Получение 2-((1Н-пиразол-1-ил)метил)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)акриламида I-98
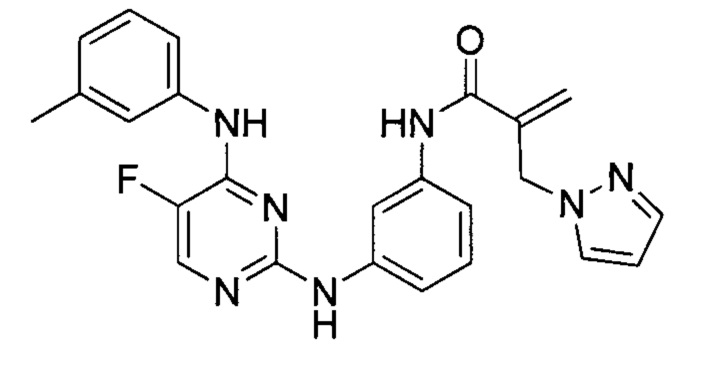
I-98
[00993] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием 2-((1Н-пиразол-1-ил)метил)акрилоил хлорида вместо 6 на этапе 3. MS 466,1 (M+Na).
ПРИМЕР 159
[00994] Получение (Е)-4-(азетидин-1-ил)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)бут-2-енамида I-123
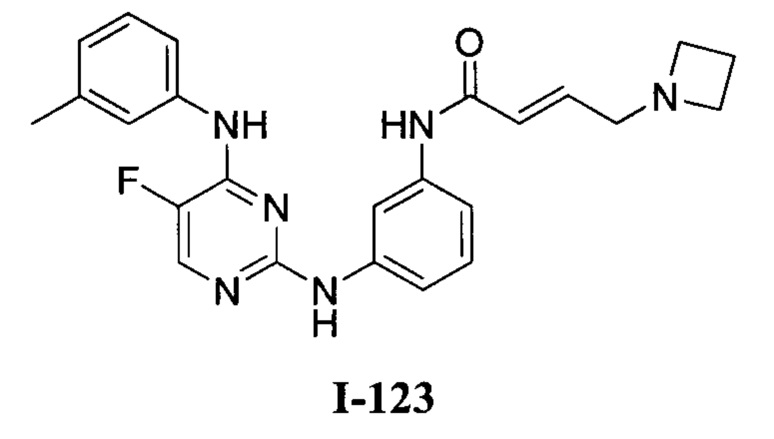
[00995] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (Е)-4-(азетидин-1-ил)бут-2-еноил хлорида вместо 6 на этапе 3. MS 455,1 (M+Na).
ПРИМЕР 160
[00996] Получение (Е)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)-4-морфолинобут-2-енамида I-102
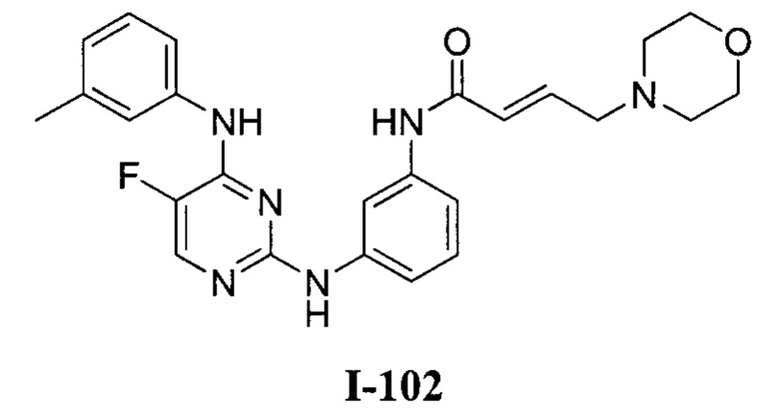
[00997] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (Е)-4-(морфолин-4-ил)бут-2-еноил хлорида вместо 6 на этапе 3. MS 485,3 (M+Na).
ПРИМЕР 161
[00998] Получение (Е)-4-((1S,4S)-2,5-диазабицикло[2.2.1]гептан-2-ил)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)бут-2-енамида I-101
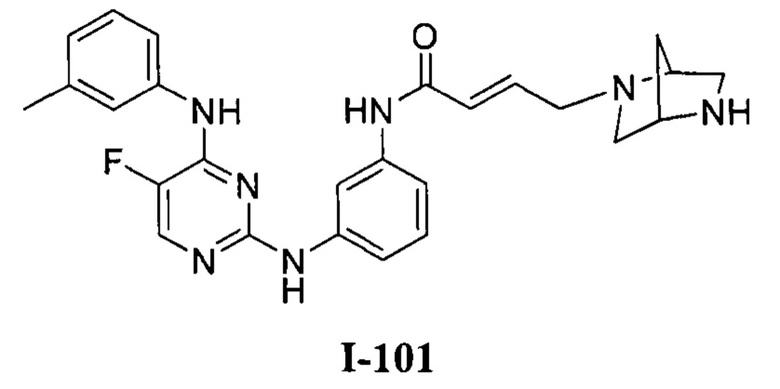
[00999] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (E)-4-((1S,4S)-2,5-диазабицикло[2.2.1]гептан-2-ил)бут-2-еноил хлорида вместо 6 на этапе 3. MS 496,1 (M+Na).
ПРИМЕР 162
[001000] Получение (Е)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)-4-((2-метоксиэтил)(метил)амино)бут-2-енамида I-120
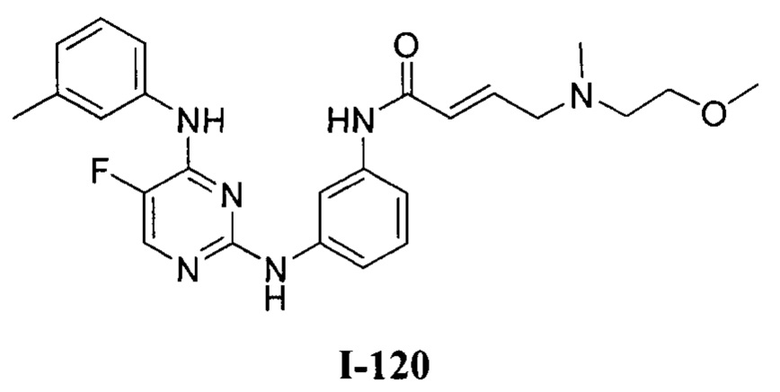
[001001] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (Е)-4-((2-метоксиэтил)(метил)амино)бут-2-еноил хлорида вместо 6 на этапе 3. MS 487,3 (M+Na).
ПРИМЕР 163
[001002] Получение (S,Е)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)-4-(3-гидроксипирролидин-1-ил)бут-2-енамида I-99
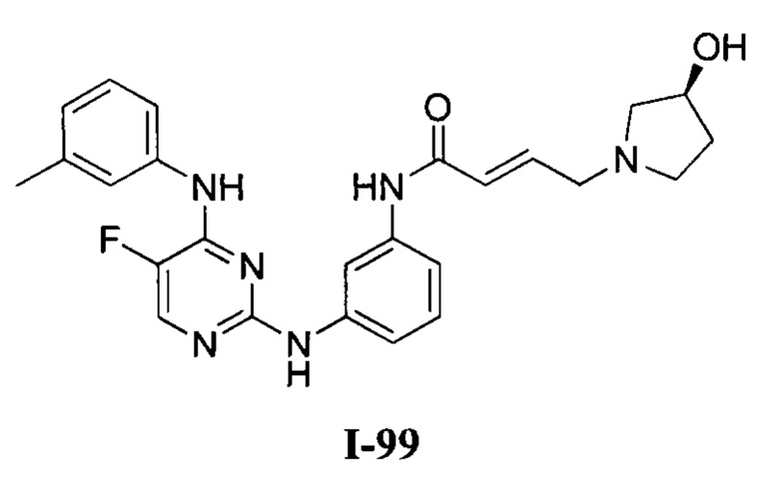
[001003] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (S,E)-4-(3-гидроксипирролидин-1-ил)бут-2-еноил хлорида вместо 6 на этапе 3. MS 485,3 (M+Na).
ПРИМЕР 164
[001004] Получение (R,E)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)-4-(3-гидроксипирролидин-1-ил)бут-2-енамида I-104
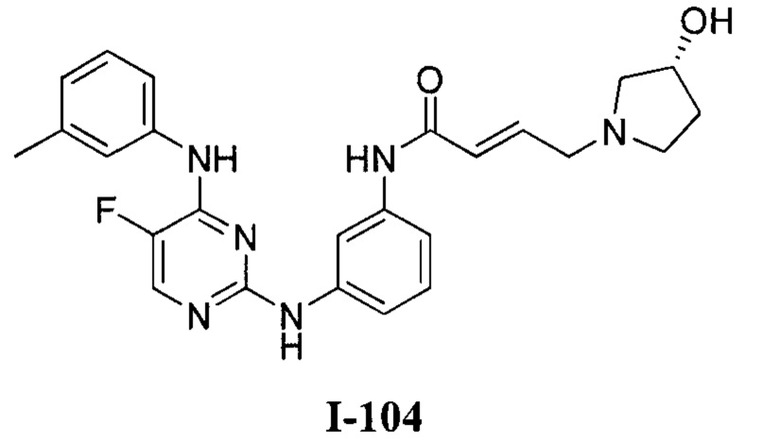
[001005] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (R,E)-4-(3-гидроксипирролидин-1-ил)бут-2-еноила вместо 6 на этапе 3. MS 485,3 (M+Na).
ПРИМЕР 165
[001006] Получение (Е)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)-4-(1Н-пиразол-1-ил)бут-2-енамида I-100
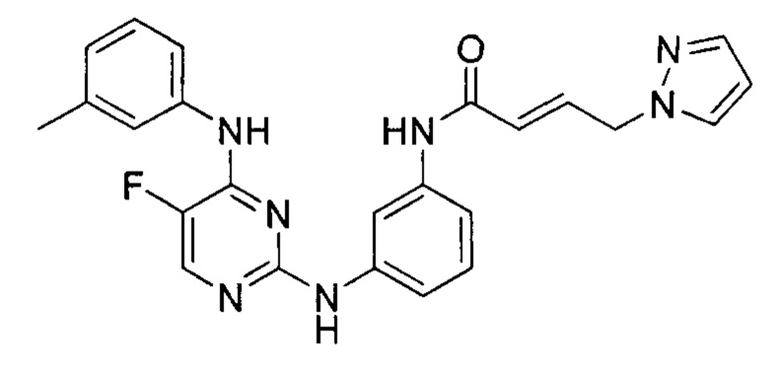
I-100
[001007] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (Е)-4-(1Н-имидазол -1-ил)бут-2-еноил хлорида вместо 6 на этапе 3. MS 466,1 (M+Na).
ПРИМЕР 166
[001008] Получение (R,E)-N-(3-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-4-(3-гидроксипирролидин-1-ил)бут-2-енамида I-89
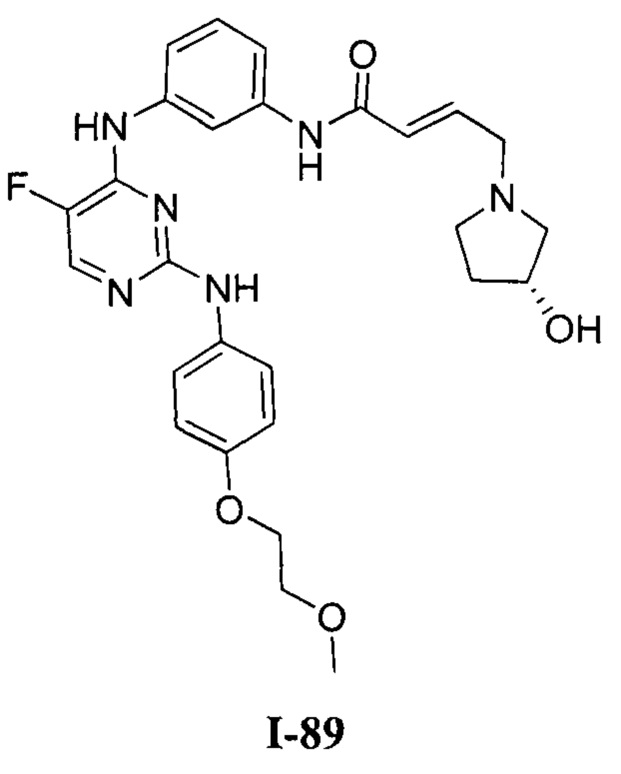
[001009] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (R,E)-4-(3-гидроксипирролидин-1-ил)бут-2-еноил хлорида вместо 7 на этапе 4. MS 545,3 (M+Na).
ПРИМЕР 167
[001010] Получение (S,Е)-N-(3-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-4-(3-гидроксипирролидин-1-ил)бут-2-енамида I-88
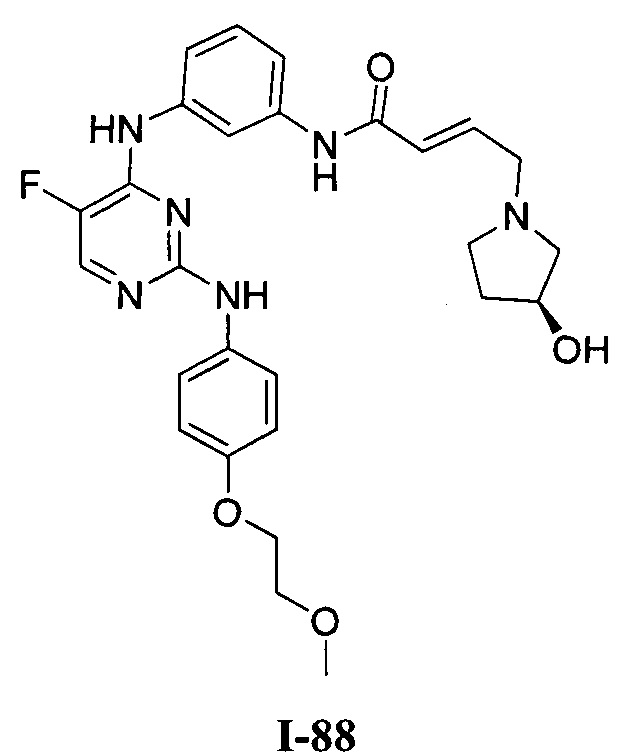
[001011] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S,E)-4-(3-гидроксипирролидин-1-ил)бут-2-еноил хлорида вместо 7 на этапе 4. MS 545,3 (M+Na).
ПРИМЕР 168
[001012] Получение 2-((1Н-пиразол-1-ил)метил)-N-(3-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-85
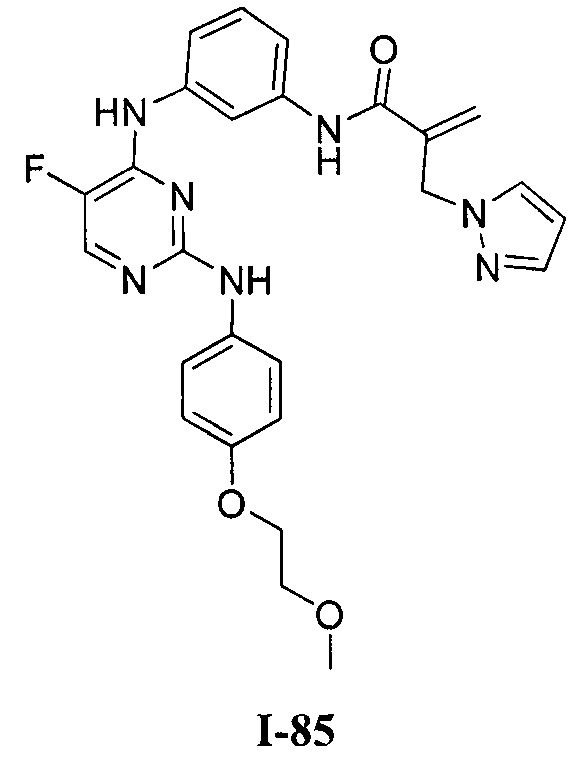
[001013] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 2-((1Н-пиразол-1-ил)метил)акрилоил хлорида вместо 7 на этапе 4. MS 526,1 (M+Na).
ПРИМЕР 169
[001014] Получение N-(3-(5-фтор-2-(фениламино)пиримидин-4-иламино)фенил)акриламида I-28
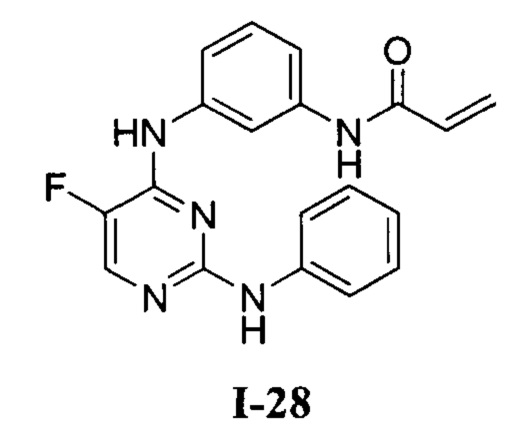
[001015] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием анилина вместо 4 на этапе 2. MS 372,1 (M+Na).
ПРИМЕР 170
[001016] Получение (Е)-4-((3R,5S)-3,5-диметилпиперазин-1-ил)-N-(3-(5-фтор-4-(м-толиламино)пиримидин-2-иламино)фенил)бут-2-енамида I-119
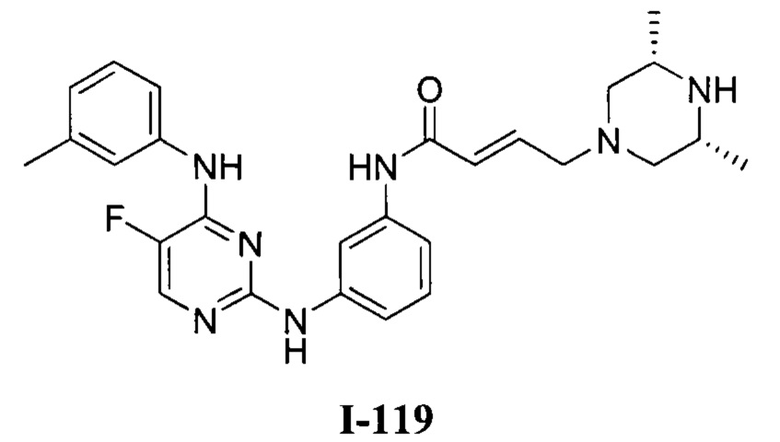
[001017] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 4, с использованием (E)-4-((3R,5S)-3,5-диметилпиперазин-1-ил)бут-2-еноил хлорида вместо 6 на этапе 3. MS 512,3 (M+Na).
ПРИМЕР 171
[001018] Получение 1-(3-(5-метил-2-(3-аминосульфонилфениламино)пиримидин-4-иламино)фенил)-3-метилбут-2-ен-1-она I-224
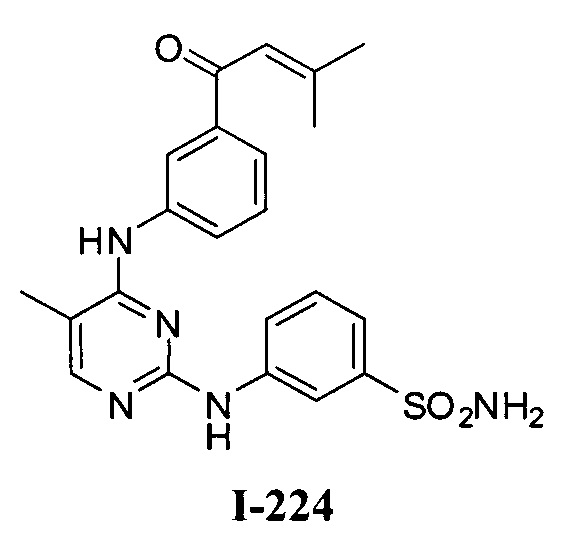
[001019] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 112 с использованием 2,4-дихлоро-5-метилпиримин вместо 1 на этапе 1 и 3-аминобензолсульфонамид вместо 4 на этапе-2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.97 (s, 3Н), 2.14 (s, 6Н), 6.88 (s, 1H), 7.25-7.30 (m, 4Н), 7.47 (t, J=7.92 Гц, 1Н), 7.62 (d, J=7.72 Гц. 1H), 7.96 (s, 1H), 8.0-8.07 (m, 3Н), 8.20 (t, J=7.36 Гц, 1Н), 8.55 (s, 1Н), 9.37 (s, 1Н); ЖХМС: m/е 438 (М+1).
ПРИМЕР 172
[001020] Получение N-(3-акриламидофенил)-N-(5-циано-2-(6-метоксипиридин-3-иламино)пиримидин-4-ил)акриламида I-171
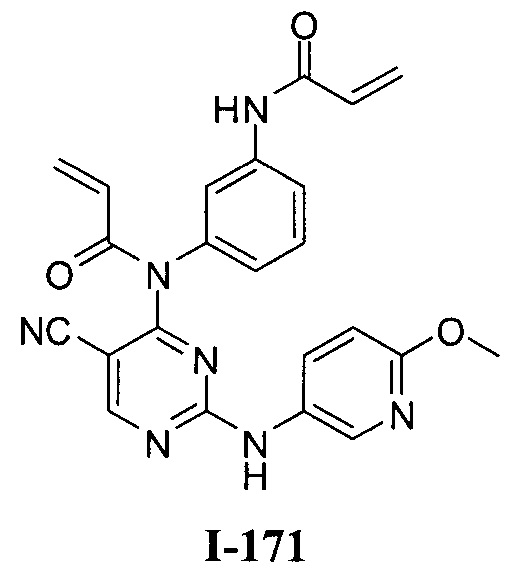
[001021] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 95, с использованием избытка акрилоил хлорида вместо -4 на этапе 2. MS m/z. 442.1 (М+Н+).
ПРИМЕР 173
[001022] Получение N-3-(N-метил-N-(5-фтор-2-(4-метил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-иламино)пиримидин-4-ил)аминофенилакриламида I-127
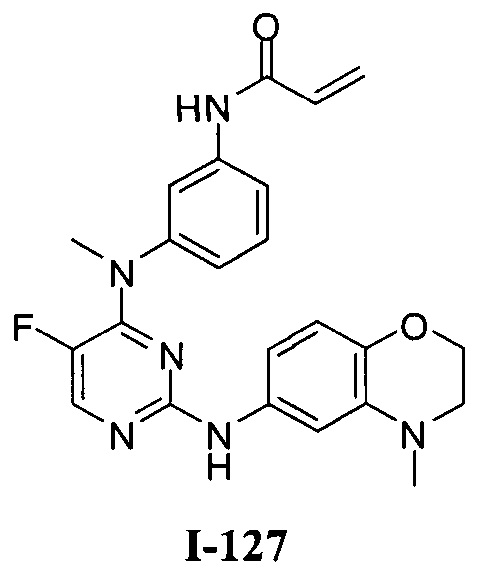
[001023] Указанное в названии соединение получали обработкой продукта Примера 88 избытком формальдегида и NaBH3CN (2 эквив.) в ацетонитриле и уксусной кислоте (4:1). MS m/z: 435.1 (М+Н+).
ПРИМЕР 174
[001024] Получение N-(3-(5-метил-2-(фениламино)пиримидин-4-иламино)бензил)акриламид I-205
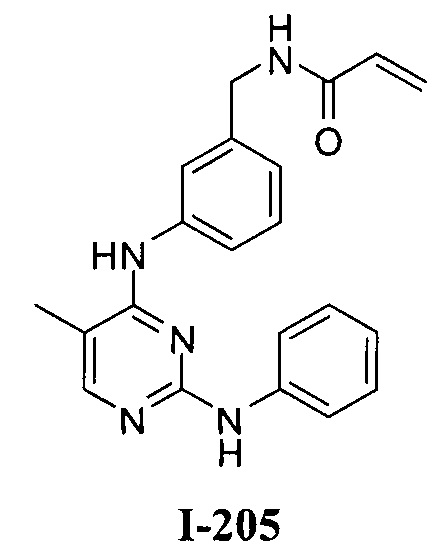
[001025] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений описанных в Примере 20 с использованием 2,4-дихлоро-5-метилпиримидина вместо 1 и 3-(трет-бутоксикарбониламино)метиланилина вместо 2 на этапе-1, и анилина вместо 4 на этапе 2. 1H-NMR (CDCl3, 500 МГц): δ 7.91 (s, 1H), 7.77 (s, 1Н), 7.57 (d, J=9.0 Гц, 2Н), 7.41 (d, J=8.0 Гц, 1Н), 7.36-7.26 (m, 2Н), 7.13-6.96 (m, 4Н), 6.36-6.25 (m, 2Н), 5.97 (dd, J=10.5, 17.0 Гц, 1H), 5.78 (bs, 1H), 5.63 (d, J=10.5 Гц, 1Н), 4.51 (d, J=6.0 Гц, 2Н), 2.12 (s, 3Н). MS: m/е=360 (М++1).
ПРИМЕР 175
[001026] Получение (Е)-3-(5-метил-2-(фениламино)пиримидин-4-иламино) бензил бут-2-еноата I-246
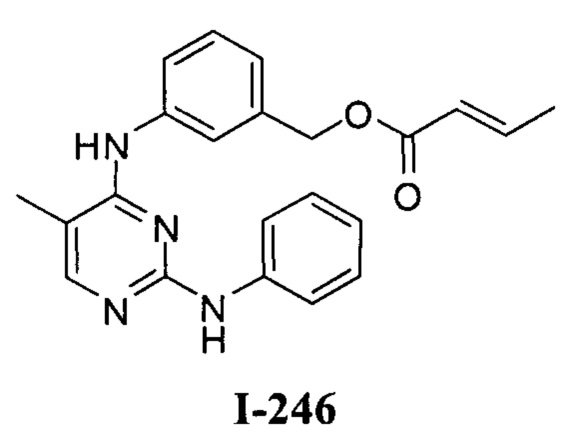
[001027] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
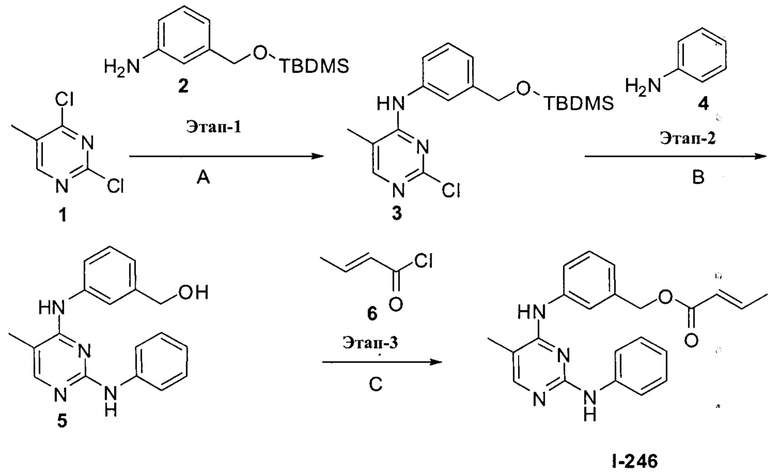
А) 2, Xanthophos, Pd2(dba)3, Cs2CO3, CH3CN, 90°C, 12 часов; В) 4, t-BuOH, 90°C, 4 часов; С) 6, ТЭА, ДХМ, -30°С, 5 минут
[001028] Этап-1
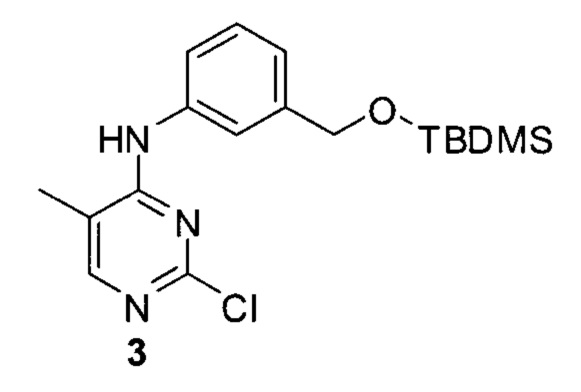
[001029] К раствору 1 (0.34 г, 2.08 ммоль) в ацетонитриле (5 мл) добавляли при помешивании Cs2CO3 (1,09 г, 3,35 ммоль), Xanthophos (0,024 г, 0,041 ммоль) и Pd2(dba)3 (38 мг, 0.04 ммоль) и 2 (0.5 г, 2.1 ммоль) при комнатной температуре а атмосфере N2. Аргон продували в реакционную смесь в течение 1 часа, и помешивали при температуре 90°С в течение 12 часов. Степень завершенности реакции определяли методом ТСХ. Реакционную смесь фильтровали через целитную пластину, после чего фильтрат концентрировали при пониженном давлении. Сырое соединение очищали методом колоночной хроматографии на силикагеле с получением 3 (0.56 г, 29.16%) в форме светло-желтого твердого вещества. 1H-NMR (CDCl3, 500 МГц): δ 8.0 (s, 1Н), 7.55 (s, 1H), 7.51 (d, J=8.0 Гц, 1H), (t, J=7.5 Гц, 1H), (d, J=7.5 Гц, 1H), 6.48 (s, 1H), 4.76 (s, 2H), 2.18 (s, 3H), 0.95 (s, 9H), 0.10 (s, 6H). MS: m/e=364 [M++1].
[001030] Этап-2
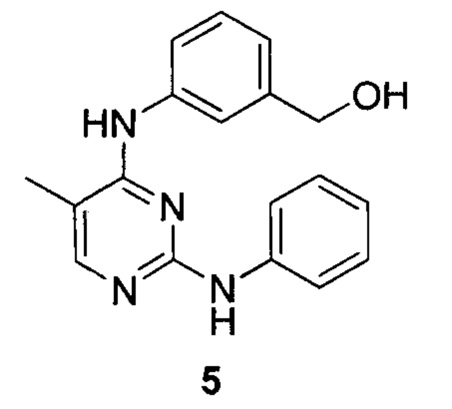
[001031] К раствору 3 (0.07 г, 0.19 ммоль) в t-BuOH (1.5 мл) при помешивании добавляли 4 (0,018 г, 0,19 ммоль) при комнатной температуре. Реакционную смесь нагревали до 90°С и помешивали в течение 4 часа при той же температуре. Степень завершения реакции определяли методом ТСХ. После готовности начальных материалов, летучие фазы удаляли при пониженном давлении с получением 5 (0.033 г, 55.9%) в форме светло-желтого твердого вещества. 1H-NMR (ДМСО-d6, 500 МГц): δ 10.45 (s, 1Н), 9.82 (s, 1Н), 7.91 (s, 1H), 7.58-7.01 (m, 9Н), 4.50 (s, 2Н), 2.16 (s, 3Н). MS: m/е=307 [М++1].
[001032] Этап-3
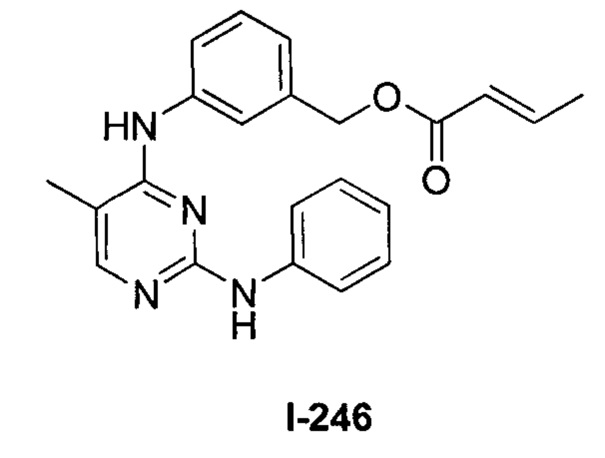
[001033] К раствору 5 (0.5 г, 1.63 ммоль) в ДХМ (5 мл) при помешивании добавляли 6 (0.18 г, 1.72 ммоль) и затем ТЭА (0.66 мл, 4.78 ммоль) при температуре -30°С в атмосфере N2. Реакционную смесь помешивали в течение 5 минут при температуре -30°С а степень завершения реакции определяли методом ТСХ. После завершения реакции, тушили водой и экстрагировали ДХМ (2×50 мл). Органические слои разделяли, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Сырой материал очищали методом колоночной хроматографии на силикагеле с получением 50 мг изомерного раствора указанного в названии соединения. Смесь в ДХМ (2 мл) обрабатывали DBU (0.02 г, 0.127 ммоль) при комнатной температуре. Реакционную смесь помешивали при комнатной температуре в течение 2 часа, тушили водой и экстрагировали ДХМ (2×10 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-246 (0.05 г, 10%) в форме светло-желтого вещества. 1H-NMR (CDCl3, 500 МГц): δ 7.87 (s, 1H), 7.65 (s, 1H), 7.58-7.51 (m, 3Н), 7.34 (t, J=7.5 Гц, 1Н), 7.30-7.23 (m, 3Н), 7.13 (d, J=7.5 Гц, 1Н), 7.08-6.96 (m, 2Н), 6.40 (s, 1Н), 5.87 (dd, J=1.5, 15.5 Гц, 1H), 5.16 (s, 2Н), 2.13 (s, 3Н), 1.87 (dd, J=2.0, 7.0 Гц, 3Н). 13C-NMR (CDCl3, 125 МГц): δ 166.3, 159.1, 158.4, 155.4, 145.3, 139.9, 138.9, 137.0, 128.9, 128.7, 123.2, 122.4, 121.9, 121.2, 121.0, 119.3, 105.2, 65.7, 17.9, 13.2. MS: m/е=375 [М++1].
ПРИМЕР 176
[001034] Получение (Е)-4-(5-метил-2-(фениламино) пиримидин-4-иламино) бензил бут-2-еноата I-60
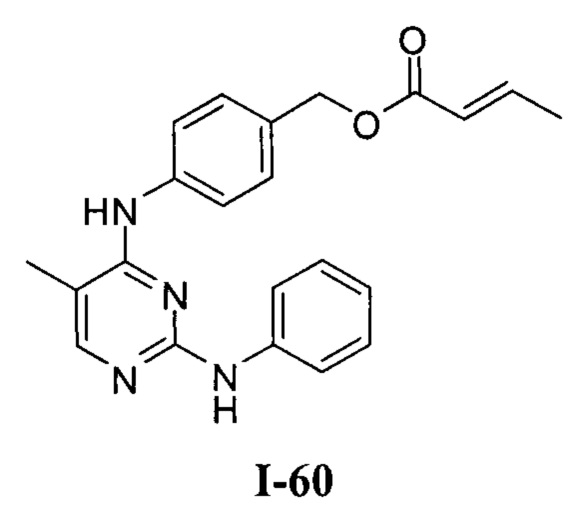
[001035] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 175, с использованием 4-((трет-бутилдиметилсилилокси) метил) анилина вместо 2 на этапе 1. 1Н-ЯМР (CDCl3, 500 МГц): δ
8.19 (bs, 1H), 7.81 (s, 1H), 7.56 (d, J=8.5 Гц, 2H), 7.53 (d, J=7.5 Гц, 2H), 7.42-7.36 (m, 3H), 7.28-7.22 (m, 1H), 7.08-6.98 (m, 2H), 6.54 (s, 1H), 5.89 (dd, J=12.5, 14.0 Гц, 1H), 5.17 (s, 2H), 2.15 (s, 3H), 1.89 (dd, 7=1.5, 7.0 Гц, 3H). MS: m/e=375 [M++1].
ПРИМЕР 177
[001036] Получение N-метил-N-(3-(5-метил-2-(фениламино)пиримидин-4-иламино)бензил)акриламида I-220
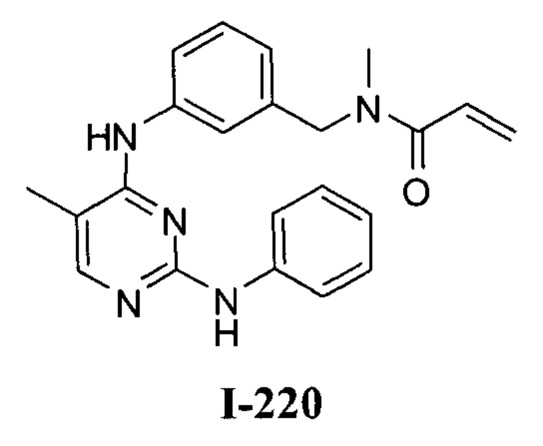
[001037] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
А) 2, Xanthophos, Pd2(dba)3, Cs2CO3, CH3CN, 100°C, 12 часов; В) 4, t-BuOH, 90°C, 4 часов; С) 10 N HCl, ДХМ, комн. т., 30 минут: D) 7, ТЭА, ДХМ, -10°С, 10 минут.
[001038] Этап-1
[001039] К раствору 1 (2,6 г, 15,7 ммоль) в ацетонитриле (26,7 мл) добавляли при помешивании 2 (2.67 г, 11.3 ммоль), Pd2 (dba)3 (0.31 г, 0.33 ммоль), Xanthophos (0.52 г, 0.89 ммоль) и Cs2CO3 (6.6 г, 20.0 ммоль). Реакционную смесь дегазировали, продувая через нее аргон в течение 1 часа, затем нагревали до 100°С в течение 12 часов. После завершения реакции (согласно ТСХ), реакционную смесь фильтровали через целитную пластину и концентрировали фильтрат при пониженном давлении. Полученный сырой материал очищали методом колоночной хроматографии (меш 60-120 силикагель; 20% этил ацетат/Гексан) с получением 3 (2.32 г, 56.71%) в форме светло-коричневого твердого вещества. 1H-NMR (CDCl3, 500 МГц): δ 8.01 (s, 1Н), 7.56 (d, J=7.5 Гц, 1H), 7.43 (s, 1H), 7.35 (t, J=7.0 Гц, 1H), 7.05 (s, 1H), 6.80 (bs, 1H), 4.45 (s, 2H), 2.87 (s, 3H), 2.30 (s, 3H), 1.48 (s, 9H).
[001040] Этап-2
[001041] К раствору 3 (2.32 г, 6.0 ммоль) в t-BuOH (11.6 мл) при помешивании добавляли 4 (0.65 г, 6.9 ммоль) при комнатной температуре, после чего реакционную смесь нагревали с обратным холодильником в течение 48 часов. Прогресс реакции отслеживали методом ТСХ. После завершения реакции, t-BuOH концентрировали при пониженном давлении с получением 5 (2.3 г, 85.82%) в форме светло-желтого твердого вещества. 1H-NMR (ДМСО-d6, 500 МГц): δ 10.32 (s, 1H), 9.78 (s, 1H), 7.91 (s, 1Н), 7.50 (d,
J=8.0 Гц, 1H), 7.45-7.30 (m, 4H), 7.24 (t, J=7.0 Гц, 2H), 7.15-7.06 (m, 2H), 4.36 (s, 2H), 2.72 (s, 3H), 2.17 (s, 3H), 1.41, 1.34 (два s, 9H). MS: m/e=420 (M++1).
[001042] Этап-3
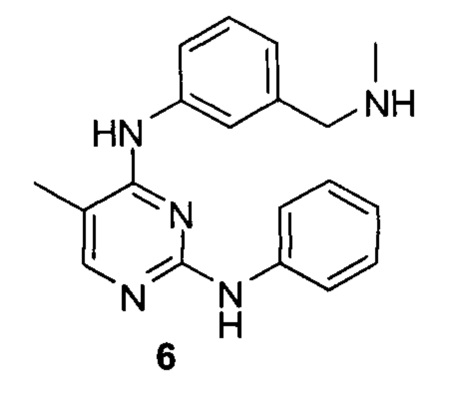
[001043] К раствору 5 (0.05 мг, 0.11 ммоль) в ДХМ (5 мл) при помешивании добавляли 37% HCl (1.0 мл) и помешивали при комнатной температуре в течение 30 минут. После завершения реакции (согласно ТСХ), летучие фазы; удаляли при пониженном давлении. Водный слой охлаждали до 0°С, приводили к форме основания до уровня рН ~ 8-9 10% раствором NaOH и экстрагировали ДХМ (50 мл). Органический слой отделяли, промывали водой, солевым раствором, сушили над безводным Na2SO4 и выпаривали при пониженном давлении с получением 6 (0,015 г, 48,36%) в форме светло-желтого вещества. 1H-NMR (CDCl3, 500 МГц): δ 7.95-7.85 (m, 2Н), 7.68-7.60 (m, 3Н), 7.42 (d, J=8.0 Гц, 1Н), 7.32-7.20 (m, 4Н), 7.05 (d, J=7.5 Гц, 1Н), 7.00-6.95 (m, 1H), 6.40 (s, 1Н), 3.84 (s, 2Н), 2.48 (s, 3Н), 2.06 (s, 3Н). MS: m/е=320 (М++1).
[001044] Этап-4
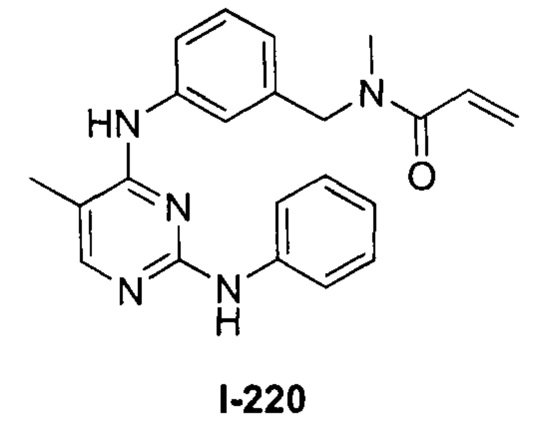
[001045] К раствору 6 (0.3 г, 0.94 ммоль) в ДХМ (12 мл) при помешивании добавляли ТЭА (0.10 г, 0.99 ммоль) и 7 (0.08 г, 0.88 ммоль) по каплям в течение 5 минут при температуре -10°С в атмосфере инертного газа. Реакционную смесь помешивали при температуре -10°С в течение 5-10 минут. После завершения реакции (согласно ТСХ), реакционную смесь тушили холодной водой (5 мл) и экстрагировали ДХМ (2×50 мл). Слой ДХМ промывали водой, солевым раствором, сушили над безводным Na2SO4 и
концентрировали при пониженном давлении. Полученный сырой материал очищали методом колоночной хроматографии (меш 60-120 силикагель; 30% этил ацетат/Гексан) с получением I -220 (0,15 г, 42,85%) в форме беловатого твердого вещества. 1H-NMR (ДМСО-d6, 500 МГц, at 80°С): δ 8.48 (bs, 1H), 8.07 (s, 1Н), 7.88 (s, 1Н), 7.69-7.58 (m, 4Н), 7.28 (t, J=8.0 Гц, 1Н), 7.16 (t, J=8.0 Гц, 2Н), 6.91-6.85 (m, 2Н), 6.75 (dd, J=2.5, 15.3 Гц, 1Н), 6.13 (d, J=15.0 Гц, 1H), 5.66 (d, J=7.5 Гц, 1Н), 4.60 (s, 2Н), 2.95 (s, 3Н), 2.12 (s, 3Н). MS: m/е=374 (М++1).
ПРИМЕР 178
[001046] Получение N-метил-N-(4-(5-метил-2-(фениламино)пиримидин-4-иламино)бензил)акриламида I-219
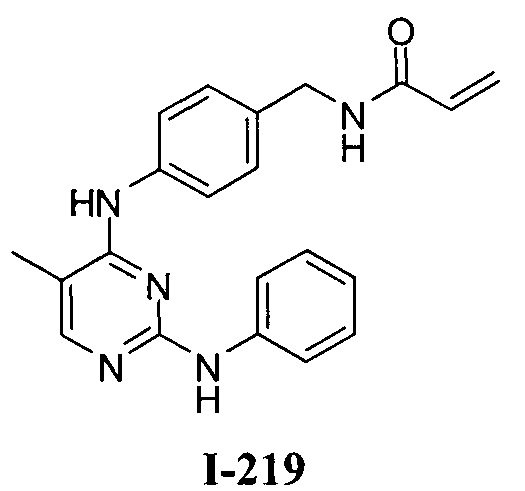
[001047] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 177, с использованием трет-бутил-4-аминобензил(метил)-карбамата вместо 2 на этапе 1. 1Н-ЯМР (ДМСО-d6, 500 МГц at 80°С): δ 8.55 (s, 1H), 8.03 (s, 1Н), 7.86 (s, 1H), 7.65 (d, J=8.0 Гц, 2Н), 7.62 (d, J=8.0 Гц, 2Н), 7.20-7.13 (m, 4Н), 6.86-6.75 (m, 2Н), 6.14 (dd, J=2.5, 17.0 Гц, 1H), 5.67 (d, J=15.0 Гц, 1H), 4.58 (s, 2Н), 2.96 (s, 3Н), 2.10 (s, 3Н). MS: m/е=374 (М++1).
ПРИМЕР 179
[001048] Получение N-(5-(5-ацетил-4-(4-акрилоил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-иламино)пиримидин-2-иламино)пиридин-2-ил)-2,2,2-трифтор-N-метилацетамида I-142
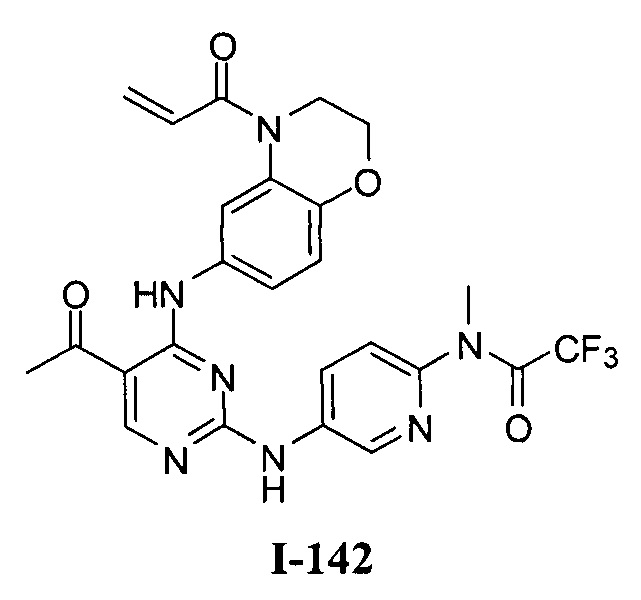
[001049] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 42, с использованием 5-амино-2(2,2,2-трифторацетамидо)пиридина вместо 9 на этапе -5. ЖХ-МС: m/z 542.2 (ES+), 540.2.2 (ES-).
ПРИМЕР 180
[001050] Получение 1-(6-(5-фтор-2-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)индолин-1-ил)проп-2-ен-1-она I-94
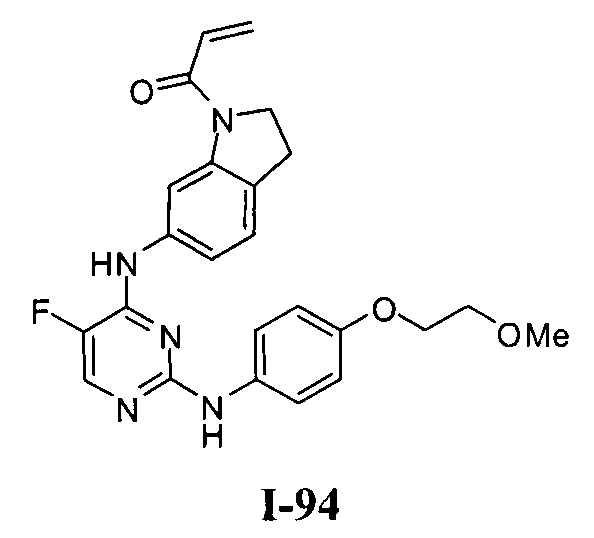
[001051] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием N-Boc-6-аминоиндолина вместо 2 на этапе -1. ЖХ-МС: m/z 450.1 (ES+), 448.1 (ES-).
ПРИМЕР 181
[001052] Получение N-(3-(5-фтор-2-(6-(2-метоксиэтокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-103
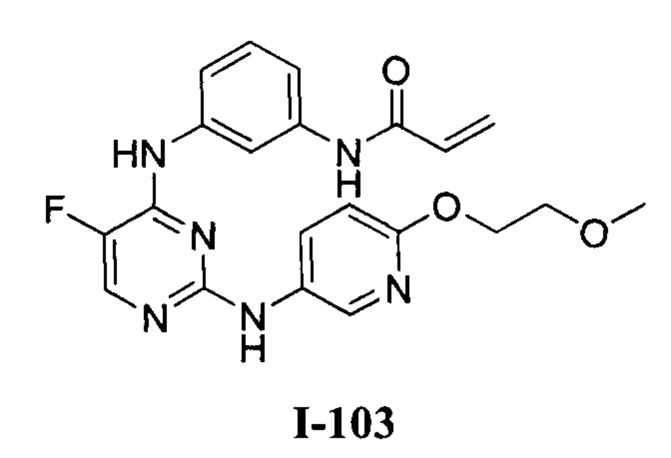
[001053] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-амино-6-(2-метоксиэтокси)пиридина вместо 4 на этапе-2. 1Н-ЯМР (CDCl3 + следовое количество ДМСО-d6) δ ppm: 3.44 (s, 3Н), 3.75 (t, J=4.4 Гц, 2Н), 4.43 (t, J=4.4 Гц, 2Н), 5.81 (dd, J=1.8 & 9.6 Гц, 1Н), 6.45 (m, 1Н), 6.80 (m, 3Н), 7.17 (m, 1Н), 7.29 (m, 1Н), 7.43 (m, 1Н), 7.49 (m, 1Н), 7.60 (m, 1Н), 7.80 (dd, J=2.8 & 9.2 Гц, 1Н), 7.94 (d, J=3.1 Гц, 1Н), 8.14 (s, 1Н), 8.32 (d, J=2.9 Гц, 1Н); ЖХМС: m/е 425.1 (М+1).
ПРИМЕР 182
[001054] Получение N-(3-(5-фтор-2-(4-(3-метилсульфонилпропокси)фенил)аминопиримидин-4-иламино)фенил)акриламида I-97
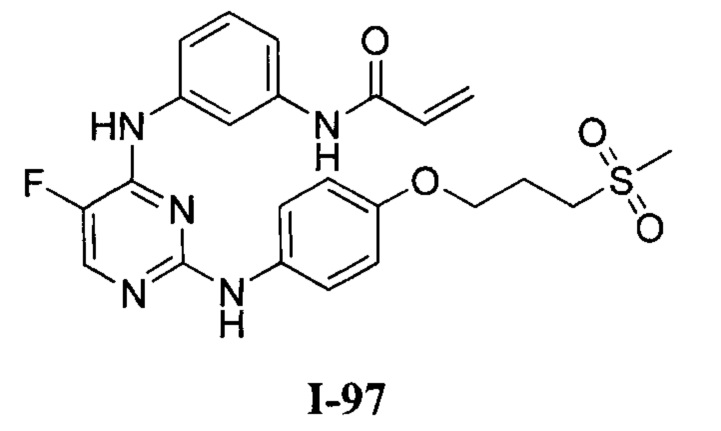
[001055] Указанное в названии соединение получали как соль ТФА с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-(3-метилсульфонилпропокси)анилина вместо 4 на этапе-2. 1Н-ЯМР (CDCl3 + следовое количество ДМСО-d6) δ ppm: 1.95 (m, 2Н), 2.67 (s, 3Н), 2.98 (m, 5Н), 3.74 (t, J=6.0 Гц, 2Н), 5.45 (dd, J=4.1 & 7.3 Гц, 1Н), 6.07 (m, 2Н), 6.48 (d, J=8.2 Гц, 1Н), 6.77 (m, 4Н), 7.09 (d,J=7.4Tn, 1H), 7.51 (d, J=4.1 Гц, 1Н), 7.70 (br, 1Н); ЖХМС: m/е 486.1 (М+1).
ПРИМЕР 183
[001056] Получение N-(3-(5-фтор-2-(6-(тридейтерометокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-95
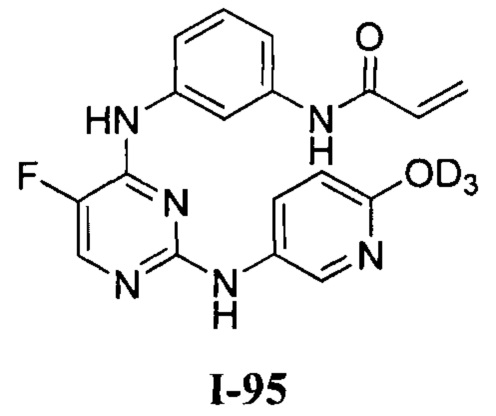
[001057] Указанное в названии соединение получали как соль ТФА с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 6-(тридейтерометокси)пиридин-3-амина вместо 4 на этапе-2. 1Н-ЯМР (CDCl3 + следовое количество ДМСО-d6) δ ppm: 5.78 (dd, J=3.7 & 7.8 Гц, 1H), 6.40 (m, 2Н), 6.71 (d, J=8.7 Гц, 1H), 7.3 (m, 3Н), 7.75 (dd, J=2.7 & 8.7 Гц, 1Н), 7.82 (d, J=4.6 Гц, 1H), 7.95 (s, 1Н), 8.33 (d, J=2.3 Гц, 1H); ЖХМС: m/е 384.1 (М+1).
[001058] Промежуточное соединение 6-(тридейтратметокси)пиридин-3-амина получали с использованием схемы описанной ниже.
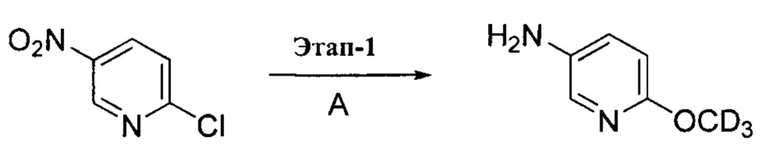
A)NaH, CD3OD, комн. т.; BH3⋅NMe3, Pd(OH)2
[001059] Этап-1
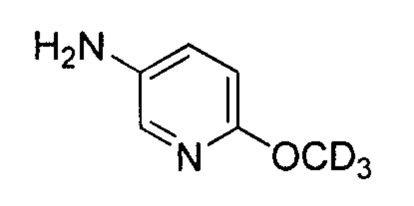
[001060] К NaH (60%, 0.30 г) в 5 мл CD3OD при температуре 0°С добавляли 2-хлоро-5-нитропиридин (1.0 г). Смесь помешивали при комнатной температуре в течение ночи. К этой смеси добавляли BH3⋅NMe3 (550 мг) и Pd(OH)2 (100 мг). Полученную смесь нагревали с обратным холодильником в течение 2 часа. Охлаждали, после чего концентрировали методом хроматографии на силикагеле с получением желаемого 6-(тредейтратметокси)пиридин-3-амина (130 мг). 1Н-ЯМР (CDCl3) δ ppm: 3.30 (br, 2Н), 6.60 (d, J=8.7 Гц, 1H), 7.03 (dd, J=3.2 & 8.7 Гц, 1H), 7.66 (d, J=3.2 Гц, 1H).
ПРИМЕР 184
[001061] Получение N-(3-(5-фтор-2(3,4,5-триметоксифениламино)пиримидин-4-илокси)фенил)акриламида I-148,
[001062] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 98, с использованием 3,4,5-триметоксианилин вместо 4 на этапе -2. MS: m/e 441 [М+1].
ПРИМЕР 185
[001063] Получение 3-метил-1-(3-(5-метил-2-(фениламино)пиримидин-4-иламино)фенил)бут-2-ен-1-она I-232
[001064] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112 с использованием 2,4-дихлоро-5-метилпиримидина вместо 1 на этапе-1 и анилина вместо 4 на Этапе -2. 1Н-ЯМР (CDCl3) δ ppm: 1.97 (s, 3H),2.15(s, 3Н), 2.22 (s, 3Н), 6.47 (s, 1Н),6.71 (s, 1H), 6.97 (t, J=9.8 Гц, 1H), 7.17 (s, 1H), 7.24 (t, J=10.36 Гц, 1H), 7.27 (s, 1H), 7.44 (t, J=10.64 Гц, 1H), 7.53 (d, J=10.48 Гц, 2H), 7.68 (d, J=10.28 Гц, 1H), 7.94 (d, J=10 Гц, 1H), 7.98 (s, 1H); ЖХМС: m/e 359 (M+1).
ПРИМЕР 186
[001065] Получение 1-(3-(5-метил-2-фениламино)пиримидин-4-иламино(пиперидин-1-ил)проп-2-ен-она I-27
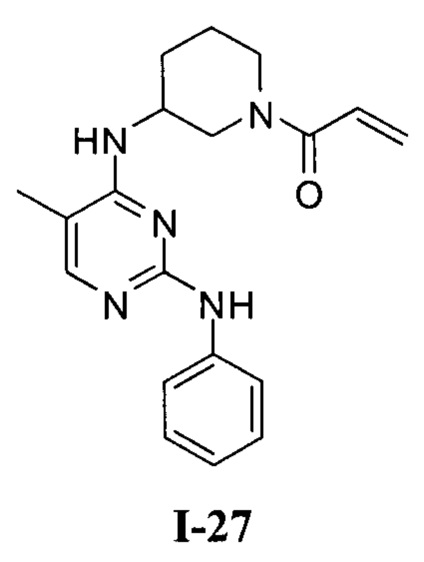
[001066] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 1, с использованием 1-фтор-3-дихлоропиримидина вместо 1 на этапе -1. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.30-1.50 (m, 1Н), 1.55-1.75 (m, 1H), 1.75-1.90 (m, 1Н), 1.92 (s, 3Н), 1.95-2.05 (m, 1Н), 2.75-3.31 (m, 2Н), 3.99-4.09 (m, 2Н), 4.10-4.15 & 4.40-4.47 (m, 1Н), 5.49 & 5.70 (d, J=10.8 Гц & d, J=9.2 Гц соответственно, вместе 1Н), 6.02 & 6.13 (d, J=17.6 Гц & d, J=16.8 Гц соответственно, вместе 1Н), 6.25-6.40 (m, 1Н), 6.63 & 6.80-6.90 (dd, J=10.8, 16.8 Гц & m соответственно, вместе 1H), 6.75-6.85 (m, 1Н), 7.15 (t, J=8 Гц, 2Н), 7.69 (bs, 3Н), 8.81 (s, 1H); ЖХМС: m/е 337.8 (М+1).
ПРИМЕР 187
[001067] Получение 3-(4-(2-акрилоил-1,2,3,4-тетрагидроизохинолин-6-иламино)-5-метилпиримидин-2-иламино)бензолсульфонамида I-40
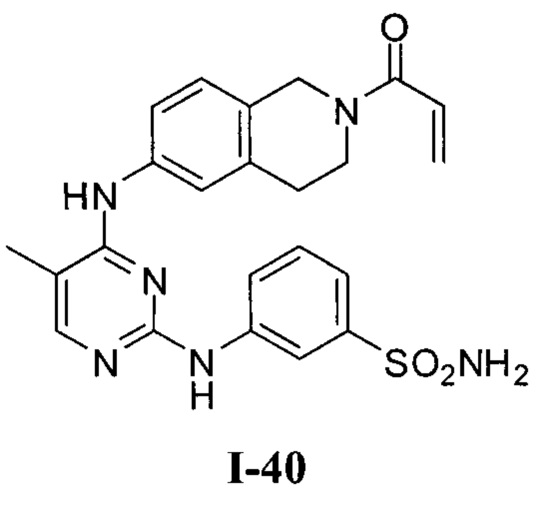
[001068] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 1, с использованием 6-амино-2-трет-бутоксикарбонил-1,2,3,4-тетрагидроизохинолина вместо 1 на этапе -1. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.10 (s, 3Н), 2.80-2.83 (m, 2Н), 3.75-3.90 (m, 2Н), 4.66 (s, 1Н), 4.76 (s, 1H), 5.71-5.74 (m, 1H), 6.16 (dd, J=2.32 & 16.76 Гц, 1Н), 6.87-6.91 (m, 1Н), 7.13-7.18 (m, 1H), 7.25-7.31 (m, 4Н), 7.53-7.57 (m, 2Н), 7.90 (s, 1H), 8.05 (s, 2Н), 8.28 (s, 1H), 9.31 (s, 1H); ЖХМС: m/е 464.8 (М+1).
ПРИМЕР 188
[001069] Получение (S)-N-(3-(5-фтор-2-(тетрагидроfuran-3-илокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-54
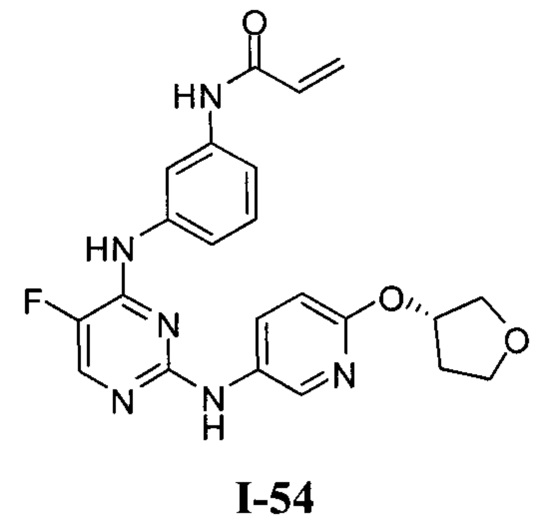
[001070] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-3-амино-6-(тетрагидрофуран-3-илокси)пиридина вместо 4 на этапе -2. MS: m/e=437 [М+1].
ПРИМЕР 189
[001071] Получение N-(3-(5-трифторметил-2-(фениламино)пиримидин-4-иламино)фенил)акриламида I-245
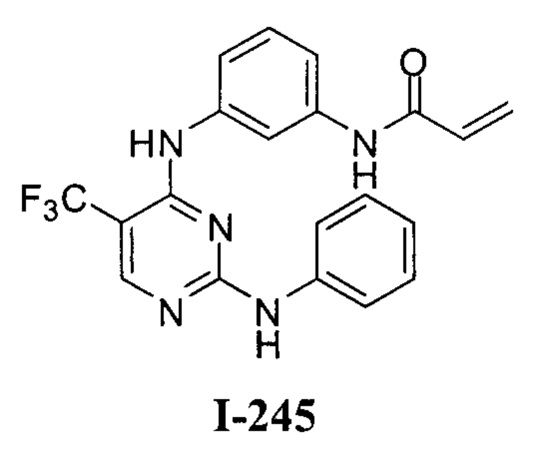
[001072] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
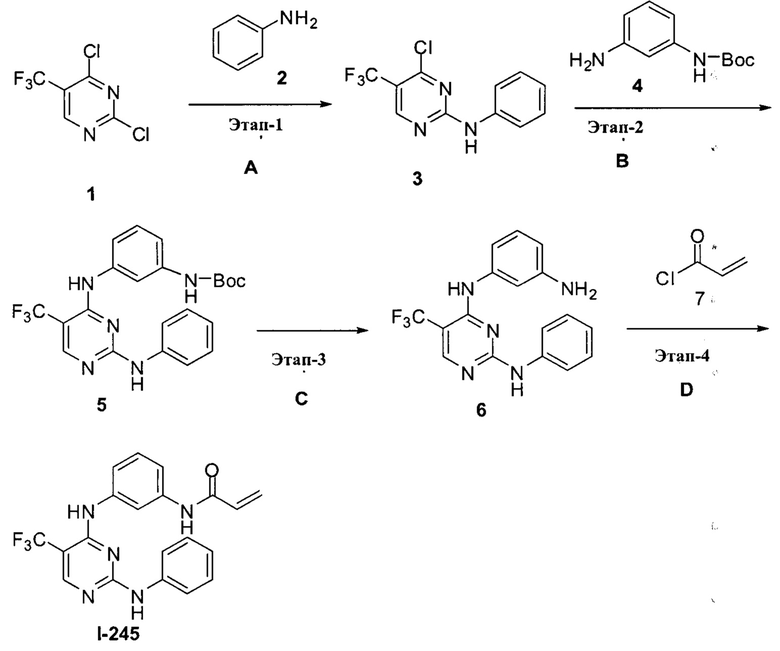
А) 2, ZnCl2, ДХЭ, t-BuOH (1:1), 0°С, 30 минут; В) 4, ДМФ, DIEPA, 70°С, 16 часов; С) ТФА, ДХМ, комн. т., 1 час; D) 7, ТЭА, ДХМ.
[001073] Этап-1
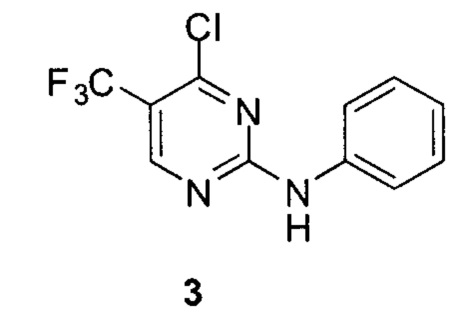
[001074] К холодному (0°С) раствору 1 (2 г, 9.2 ммоль) в 80 мл 1:1 смеси tBuOH/ДХЭ добавляли цинка хлорид (11 мл 1 М раствора в эфире, 1.2 экв.). Через час добавляли 2 (0.858 g, 9.2 ммоль), а затем по каплям добавляли триэтиламин (1.03 г; 1.1 эк) в 10 мл ДХЭ/t-BuOH. Помешивали в течение 30 минут, удаляли растворители при пониженном
давлении, и растворяли остаток в этил ацетате (50 мл), после чего промывали водой и солевым раствором (10 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом. Желаемый продукт 3 получали в форме белого твердого вещества, после рекристаллизации из EtOAc/гексан (1:9), (2g, 80%).
[001075] Этап-2
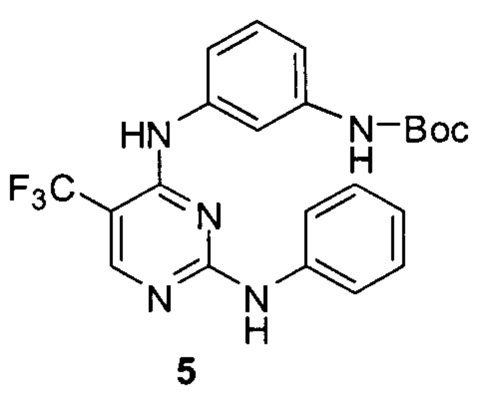
[001076] К раствору 3 (0.5 г, 1.82 ммоль) и 4 (0.38 г, 1.83 ммоль) в ДМФ (10 мл) добавляли DIPEA (0.283 г, 2.192 ммоль) и смесь нагревали до 60°С в атмосфере аргона в течение 16 часов. Растворитель удаляли методом дистилляции, после чего осадок растворяли в этил ацетате (50 мл) и промывали солевым раствором (10 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом. Сырую смесь очищали методом колоночной хроматографии (элюент: EtOAc/гексан 1:1) с получением 5 в форме белого твердого вещества (0.48 г, 60%).
[001077] Этап-3
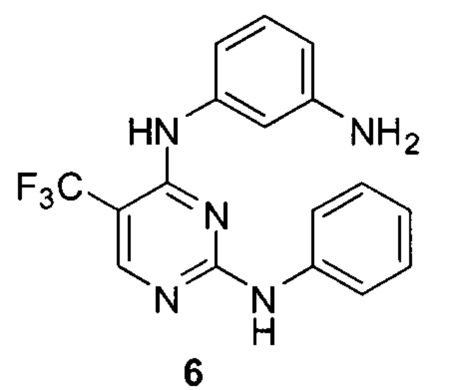
[001078] К раствору 6 (0.25 г, 0.63 ммоль) в CH2Cl2 (10 мл) добавляли трифторуксусную кислоту (2 мл) после чего смесь помешивали при комнатной температуре в течение 1 часа. Удаляли растворители при пониженном давлении, и растворяли остаток в CH2Cl2, промывали 10% водным раствором NaHCO3, сушили (Na2SO4), фильтровали, и упаривали при пониженном давлении с получением свободного амина 10 в форме белого твердого вещества.
[001079] Этап-4
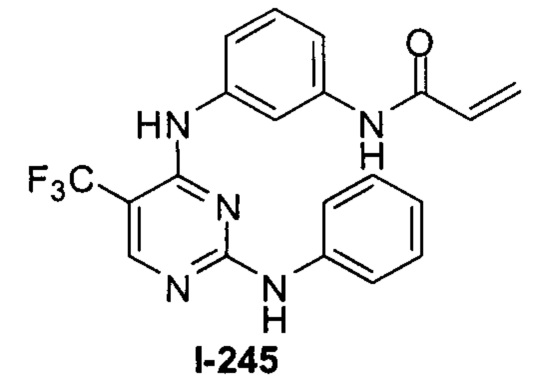
[001080] К раствору 6 (0.2 г) в ДХМ (20 мл) в атмосфере аргона охлажденной до -40°С при помешивании добавляли триэтиламин, после чего по каплям добавляли 7 (0.069 г, 0.686 ммоль). Полученную смесь помешивали при -40°С в течение 10 минут. Реакционную смесь разбавляли ДХМ (50 мл) и промывали солевым раствором (10 мл). Органический слой сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле с (МеОН-EtOAc 5:95) в качестве элюента с получением желаемого соединения I-245. 1Н-ЯМР (200 МГц, CD3OD) δ 8.25 (s, 1Н), 7.80 (s, 1H), 7.60-7.05 (m, 7Н), 6.90 (m, 1H), 6.35 (m, 2Н), 5.75 (dd, J=8.0, 2.0 Гц, 1Н).
ПРИМЕР 190
[001081] Получение N-(3-(5-трифторметил-2-(3-метоксифениламино)пиримидин-4-иламино)фенил)акриламида I-242
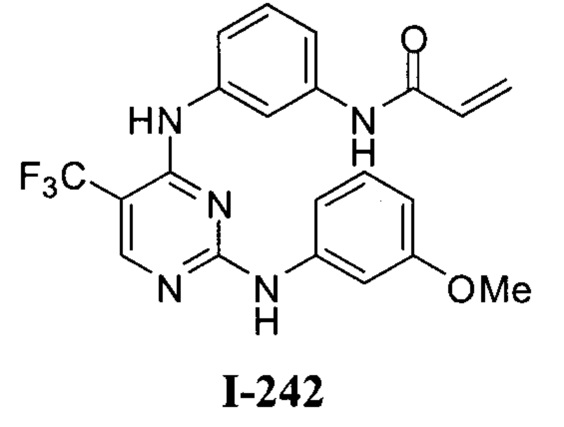
[001082] Указанное в названии соединение получали с использованием схем, этапов и промежуточных продуктов, описанных в Примере 189 с использованием 3-диметоксианилина вместо 2 на этапе 1. 1Н-ЯМР (200 МГц, CD3OD) δ 8.31 (s, 1Н), 7.84 (s, 1Н), 7.59 (m, 1H), 7.37-7.09 (m, 5Н) 6.53 (m, 1H), 6.41 (m, 2Н), 5.79 (dd, J=8.0, 2.0, Гц, 1Н), 3.66 (s, 3Н).
ПРИМЕР 191
[001083] Получение N-(4-(5-трифторметил-2-(3-метоксифениламино)пиримидин-4-иламино)фенил)акриламида I-236
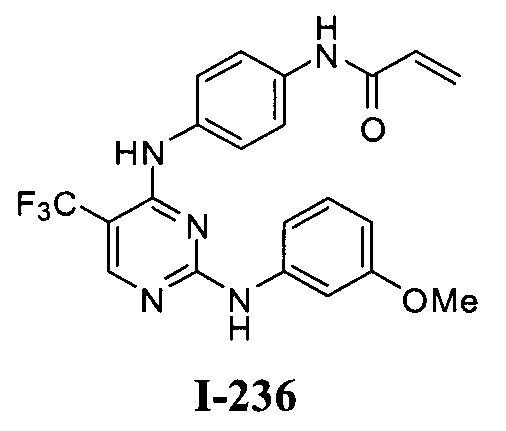
[001084] Указанное в названии соединение получали с использованием схем, этапов и промежуточных продуктов, описанных в Примере 189 с использованием 3-метоксианилин вместо 2 на этапе 1 и 4-амино-N-трет-бутоксикарбониланилина вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CD3OD) δ 8.27 (s, 1Н), 7.70 (d, J=6.0Hz) 1H), 7.46 (d, J=6.0Hz, 1H), 7.09 (brs, 1H), 7.07 (m, 2H) 6.51 (m, 1H), 6.44 (m, 2H), 5.80 (dd, J=8.0, 2.0 Гц, 1H), 3.56 (s, 3H).
ПРИМЕР 192
[001085] Получение N-(4-(5-трифторметил-2-(3-метоксифениламино)пиримидин-4-иламино)фенил)метилакриламида I-235
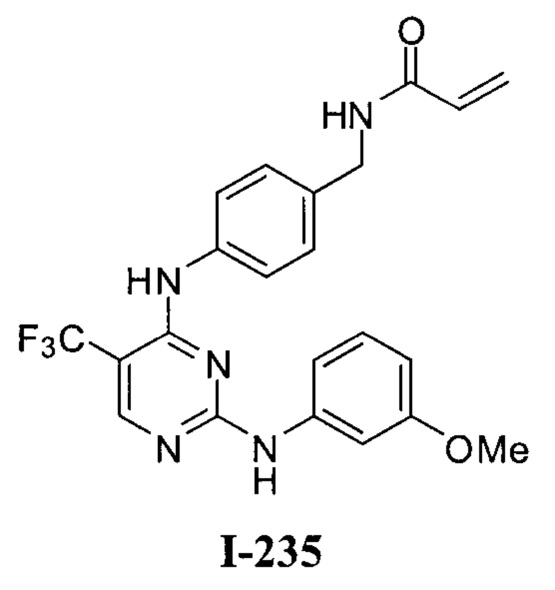
[001086] Указанное в названии соединение получали с использованием схем, этапов и промежуточных продуктов, описанных в Примере 189 с использованием 3-метоксианилин вместо 2 на этапе 1 и 4-аминофенилметил-N-трет-бутоксикарбониламин вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CD3OD) δ 8.30 (s, 1H), 7.49 (d, J=8.0 Гц, 1H), 7.37 (d, J=8.0 Гц, 1H), 7.10 (m, 3H), 6.60 (m, 1H) 6.34 (m, 2H), 5.75 (dd, J=8.0, 2.0 Гц, 1H), 4.51(s,2H), 3.68 (s, 3H).
ПРИМЕР 193
[001087] Получение N-(4-хлоро-3-(5-трифторметил-2-(3-метоксифениламино)пиримидин-4-иламино)фенил)акриламида I-227
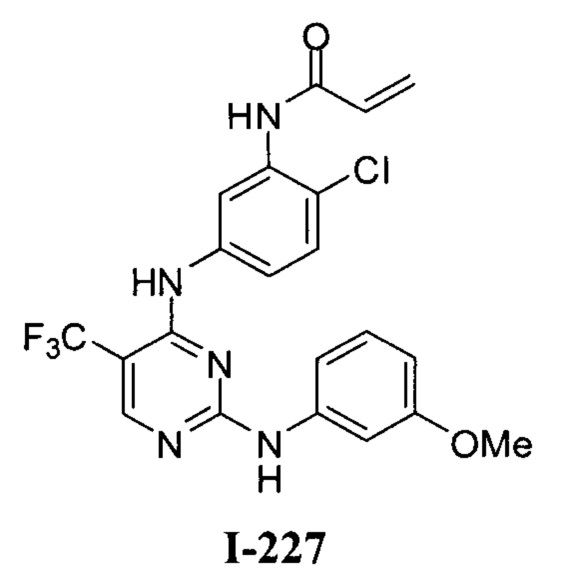
[001088] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 189 с использованием 3-метоксианилина вместо 2 на этапе-1 и N-трет-бутоксикарбонил-3-амино-6-хлороанилина вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CD3OD) δ 8.33 (s, 1H), δ 8.08 (s, 1Н), 7.45 (m, 2Н), 7.21-7.07 (m, 3Н), 6.60-6.36 (m, 3Н), 5.84 (dd, J=8.0, 2.0 Гц, 1Н), 3.71 (s, 3Н).
ПРИМЕР 194
[001089] Получение N-(3-(5-трифторметил-2-(3-метоксифениламино)пиримидин-4-иламино)фенил)метилакриламида I-226
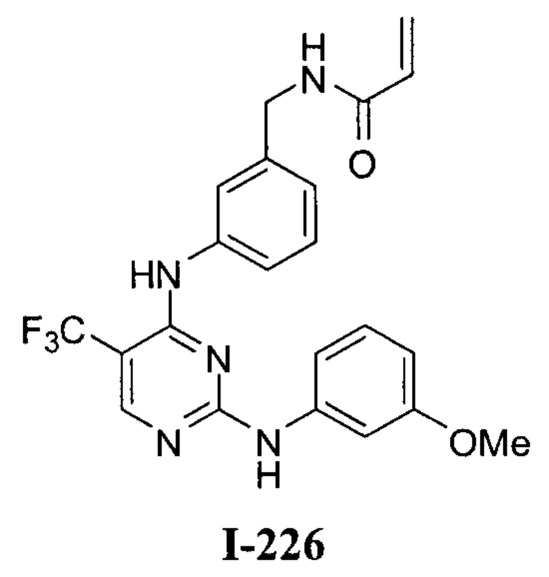
[001090] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 189 с использованием 3-метоксианилина вместо 2 на этапе-1 и 3-аминофенилметил-N-трет-бутоксикарбониламин вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CD3OD) δ 8.31 (s, 1H), 7.71-7.33 (m, 3Н), 7.21-7.08
(m, 4H), 6.57 (m, 1H), 6.26 (d, J=4Hz, 2H), 5.69 (dd, J=8.0, 2.0 Гц, 1H), 4.47 (s, 2H), 3.67 (s, 3H).
ПРИМЕР 195
[001091] Получение N-(4-(5-трифторметил-2-(6-метоксипиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-218
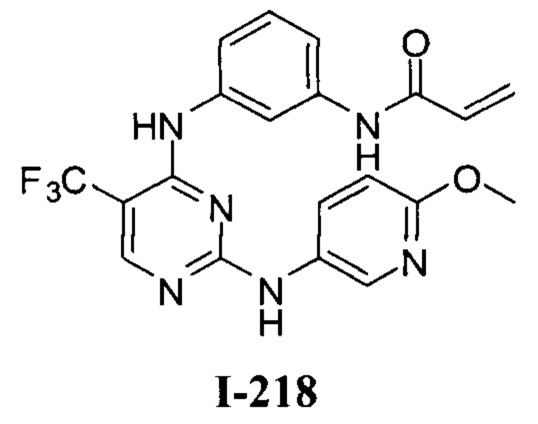
[001092] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 189, с использованием 3-амино-6-метоксипиридина вместо 2 на этапе -1. 1Н-ЯМР (200 МГц, CD3OD) δ 8.21 (s, 1H), 7.90-7.78 (m, 3Н), 7.48 (m, 2Н), 7.30 (m, 2Н), 6.40 (m, 2Н), 5.75 (dd, J=8.0, 2.0 Гц, 1H), 3.81 (s, 3Н).
ПРИМЕР 196
[001093] Получение N-(4-(5-трифторметил-2-(5-метоксипиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-214
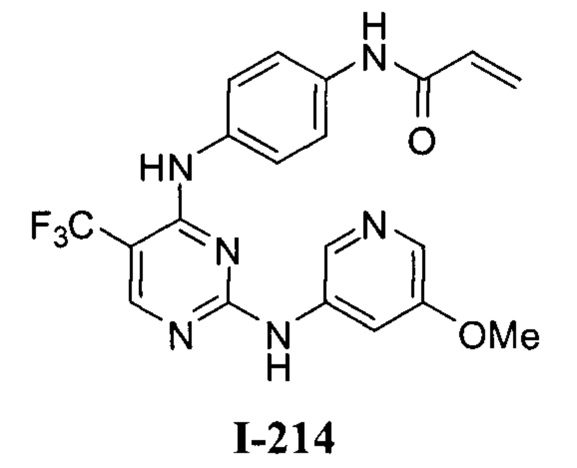
[001094] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 189 с использованием 3-амино-5-метоксипиридина вместо 2 на этапе-1 и 4-амино-N-трет-бутоксикарбониланилина вместо 4 на этапе-2. MS: m/e=431 [М+1].
ПРИМЕР 197
[001095] Получение 1-(3-(5-трифторметил-2-(3-метоксифениламино)пиримидин-4-иламино)фенил)-3-метил-бут-2-ен-1-она I-225
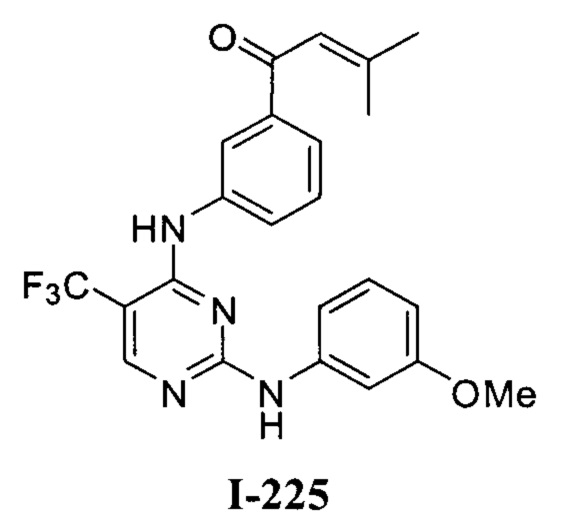 в
в
[001096] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 189 с использованием 3-метоксианилина вместо 2 на этапе-1 и 1-(3-аминофенил)-3-метилбут-2-ен-1-она вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CDCl3) δ 8.33 (s, 1Н), δ 8.38 (s, 1Н), 7.99-7.77 (m, 4H), 7.50 (m, 2H), 7.20-7.01 (m, 4H), 6.68-6.60(m, 2H), 3.68 (s, 3H), 2.25 (s, 3H), 1.99 (s, 3H).
[001097] 1-(3-Аминофенил)-3-метилбут-2-ен-1-он получали с использованием схем, этапов, промежуточных соединений описанных ниже.
A) Boc2O, NEt3, DMAP, ДХМ; В) NHMe(OMe)-HCl, TBTU, ДХМ, 0°С до комн. т.; С) 4, ТГФ, 0°С до комн. т.; D) ТФА, ДХМ.
[001098] Этап-1
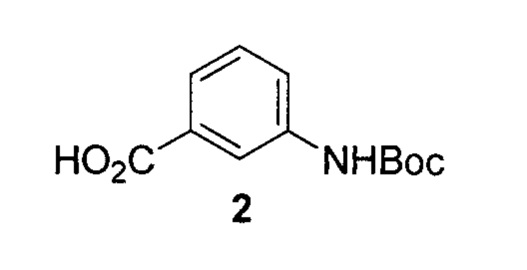
[001099] Ди-трега-бутилдикарбонат (6.54 г, 30 ммоль, 1.5 экв.) добавляли к раствору 1 (2.70 г, 20 ммоль) в CH2Cl2 (100 мл) содержащему Et3N (3.4 мл, 24 ммоль, 1.2 экв.) и DMAP (122 мг, 1.0 ммоль, 5 mol%). Раствор помешивали в течение ночи в сушильной трубке с CaCl2. Растворители выпаривали и разделяли осадок между эфиром (50 мл) и водой (50 мл). Водную фазу экстрагировали эфиром и подкисляли до уровня рН 3 1N HCl после чего экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой и солевым раствором и сушили над NaSO4. Концентрировали с получением сырого продукта, который рекристаллизовали из EtOAc/гексаны с получением 2 (2.92 г, 62%).
[001100] Этап-2
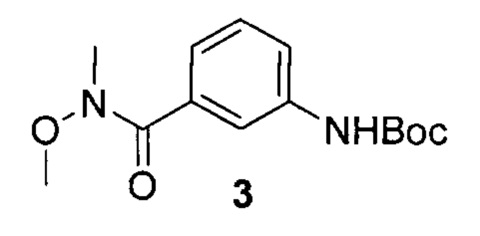
[001101] К смеси 2 (1.5 г, 6.33 ммоль), N-метокси-N-метиламин гидрохлорида (614 мг, 6.33 ммоль), и TBTU (2.05 г, 6.33 ммоль) в ДХМ (30 мл) при температуре 0°С добавляли NEt3 (2.7 мл, 19 ммоль, 3 экв.). Смесь помешивали в течение 30 минут при температуре 0°С, а затем при комнатной температуре в течение 2 часа, когда анализ методом HPLC показывали завершение реакции. Реакционную смесь наливали в 150 мл холодной воды, после чего выпавший белый осадок собирали и промывали водой. Продукт сушили в вакуумной печи в течение ночи с получением 3 (1.34 г, 75%) в форме белого твердого вещества.
[001102] Этап-3
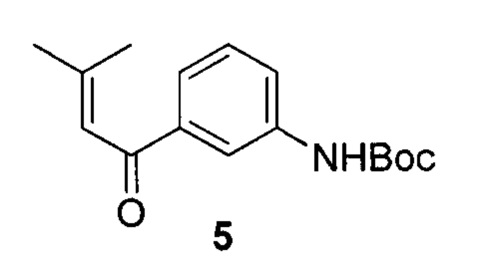
[001103] К раствору 3 (546 мг, 1.95 ммоль) в ТГФ (3 мл) при температуре 0°С в атмосфере Ar по каплям добавляли 4 (0.5 М в ТГФ, 9.75 мл, 4.9 ммоль, 2.5 экв.). Реакционную смесь помешивали при температуре 0°С в течение 30 -минут, затем охлаждающую ванну удаляли и помешивали реакционную смесь при комнатной температуре в течение 2 часа. Реакционную смесь охлаждали до температуры 0°С и гасили 5% раствором лимонной кислоты. Разбавляли водой (10 мл) и экстрагировали
водную фазу эфиром (2×15 мл) затем объединенные органические слои промывали водой и солевым раствором и сушили над NaSO4. Концентрировали с получением 5 (82%) в форме желтого твердого вещества, которое было достаточно чистым, чтобы непосредственно использовать на следующем этапе.
[001104] Этап-4
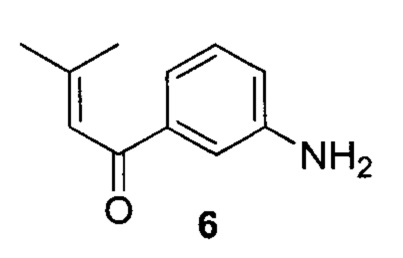
[001105] Образец соединения 5 весом 400 мг обрабатывали 5 мл 1:3 CH2Cl2: Трифторуксусная кислота, и полученный раствор помешивали при комнатной температуре в течение 15 минут. Растворители удаляли под вакуумом, и растворяли осадок в CH2Cl2, после чего выпаривали еще три раза. Осадок снова помещали в CH2Cl2, и промывали смесь насыщенным раствором бикарбоната натрия. Слой CH2Cl2 сушили над сульфатом натрия, фильтровали и выпаривали с получением 6 в форме белого твердого вещества, которое непосредственно использовали в следующее реакции.
ПРИМЕР 198
[001106] Получение 1-(3-(2-(3-Метокси-фениламино)-5-трифторметил-пиримидин-4-иламино)-циклогексил)-3-метил-бут-2-ен-1-она I-213
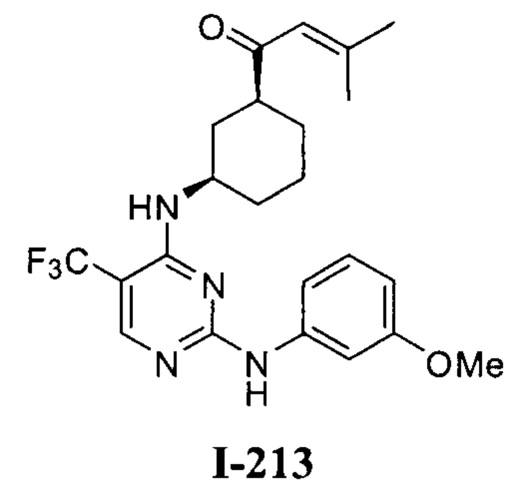
[001107] Указанное в названии соединения получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 189 с использованием 3-метоксианилина вместо 2 на этапе-1 и (d,I)-цис-1-(3-Амино-циклогексил)-3-метил-бут-2-ен-1-она вместо 4 на этапе-2. 1Н-ЯМР (200 МГц, CDCl3) δ 8.07 (s, 1Н), 7.33 (s, 1Н), 7.19-7.0 (m, 3Н), 6.54 (d, J=2.7 Гц, 1H), 6.02 (s, 1Н), 4.04 (m, 1H), 3.75 (s, 3Н), 2.50 (m, 1Н), 2.10 (m, 1Н), 1.89-1.15 (m, 14Н).
[001108] (D,L)-цис-1-(3-Амино-циклогексил)-3-метил-бут-2-ен-1-он получали с использованием схем, этапов, промежуточных соединений описанных ниже.
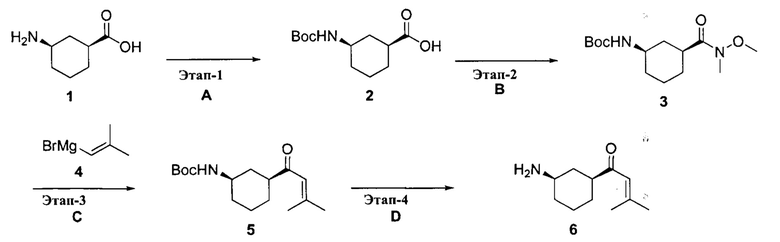
А) Boc2O, Na2CO3, ацетон, H2O; В) NHMe(OMe)-HCl, TBTU, ДХМ, 0°С до "комн. т.; С) 4, ТГФ, 0°С до комн. т.; D) ТФА, ДХМ.
[001109] Этап-1
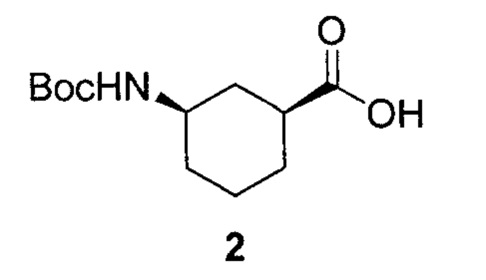
[001110] К раствору (±)-1 (4.05 г, 28.2 ммоль) в воде (150 мл) содержащем Na2CO3 (3.0 г, 28.2 ммоль) и ацетон (100 мл) при помешивании добавляли BOC2O (7.4 г, 33.8 ммоль, 1.2 эк) и помешивали реакционную смесь при температуре 25°С в течение ночи. Ацетон удаляли и водные слои экстрагировали эфиром (2Х). Водный слой подкисляли до уровня рН 3, собирали осадок и промывали его водой. Продукт сушили в вакуумной печи в течение ночи с получением 2 (5,90 г, 85%) в форме белого твердого вещества.
[001111] Этап-2
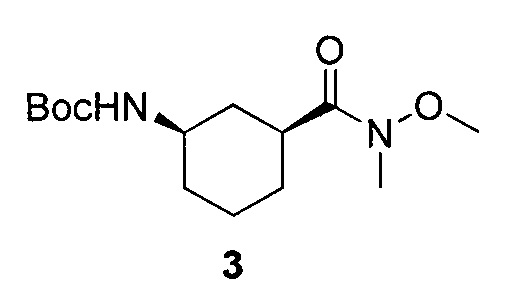
[001112] К раствору 2 (2.45 г, 10.1 ммоль), TBTU (3.44 г, 10.6 ммоль, 1.05 эк) и N-метил-N-метоксиамин гидрохлорида (1.03 г, 10.6 ммоль, 1.05 эк) в CH2Cl2 (40 мл) при температуре 0°С при помешивании добавляли триэтиламин (4.25 мл, 30.3 ммоль, 3 эк). Смесь помешивали при температуре 0°С в течение 20 минут, и удаляли ванну, после чего продолжали помешивать в течение 3 часа при температуре 25°С. Затем тушили водой, CH2Cl2 удаляли, и остаток разделяли между эфиром и водой. Водную фазу экстрагировали эфиром и промывали объединенные органические слои водой и солевым раствором, после чего сушили над MgSO4. Выпаривали растворители с получением 3 (2.37 г, 82%) в форме белого твердого вещества.
[001113] Этап-3
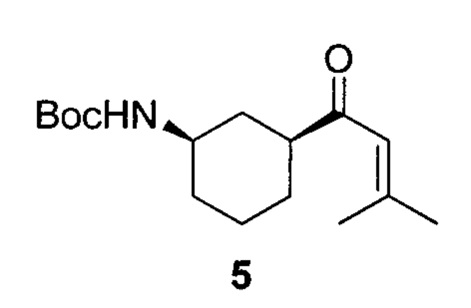
[001114] К раствору 3 (1,23 г, 4,32 ммоль) в ТГФ (20 мл) при температуре 0°С в атмосфере аргона по каплям добавляли 4 (27 мл, 0.5 М в ТГФ, 10.8 ммоль, 2.5 экв.). После добавления смесь помешивали при температуре 0°С в течение 30 минут, а затем при комнатной температуре в течение 1 часа. Реакционную смесь охлаждали до 0°С затем тушили 5% раствором лимонной кислоты (5 мл). Разбавляли водой и экстрагировали смесь эфиром (2 X) затем объединенные органические слои промывали водой и солевым раствором и сушили над NaSO4. После упаривания оставался оранжевый осадок, который очищали методом хроматографии на силикагеле, с 20% раствором EtOAc в гексанах в качестве элюента с получением 5 (600 мг, 56%) в форме светло-желтого твердого вещества.
[001115] Этап-4
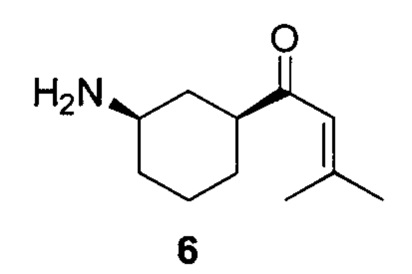
[001116] Образец соединения 5 весом 500 мг обрабатывали 6 мл 1:3 CH2Cl2: Трифторуксусная кислота, и полученный раствор помешивали при комнатной температуре в течение 15 минут. Растворители удаляли под вакуумом, и растворяли осадок в CH2Cl2, после чего выпаривали еще три раза. Осадок снова помещали в CH2Cl2, и промывали смесь насыщенным раствором бикарбоната натрия. Слой CH2Cl2 сушили над сульфатом натрия, фильтровали и выпаривали с получением 6 в форме белого твердого вещества, которое непосредственно использовали без очистки.
ПРИМЕР 199
[001117] Получение 1-(5-(5-трифторметил-2-(3-метоксифениламино)пиримидин-4-ил)амино-1,3-дигидроизоиндол-2-ил)2-пропен-1-она I-132
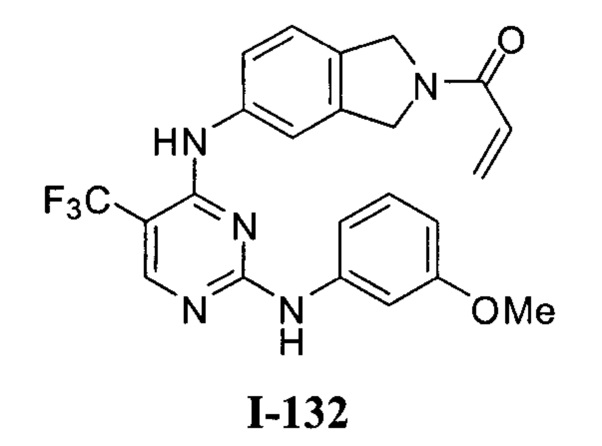
[001118] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 189 с использованием 3-метоксианилина вместо 2 на этапе-1 и 4-амино-N-трет-бутоксикарбониланилина вместо 2-(N-трет-бутоксикарбонил)-5-аминоизоиндолина вместо 4 на этапе-2. MS m/e=456 [М+1].
ПРИМЕР 200
[001119] Получение 3-(2-(2-акрилоилизоиндолин-5-иламино)-5-фторпиримидин-4-иламино)бензонитрила I-106
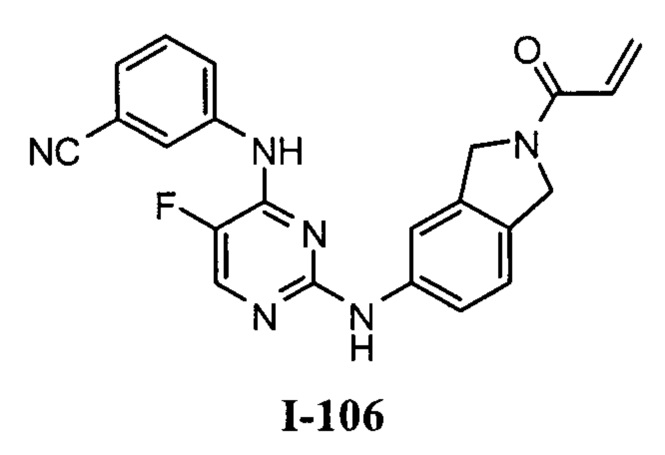
[001120] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 2 с использованием 3-аминобензонитрила вместо 2 на этапе-1 и 4-амино-N-трет-бутоксикарбониланилина вместо 2-(трет-бутоксикарбонил-5-аминоизоиндолина вместо 4 на этапе-2. ЖХ/МС (RT=2.82/(М+Н)) 401.1
ПРИМЕР 201
[001121] Получение N-(3-(5-фтор-4-((6-(трифторметил)пиридин-3-ил)метиламино)пиримидин-2-иламино)фенил)акриламида I-53
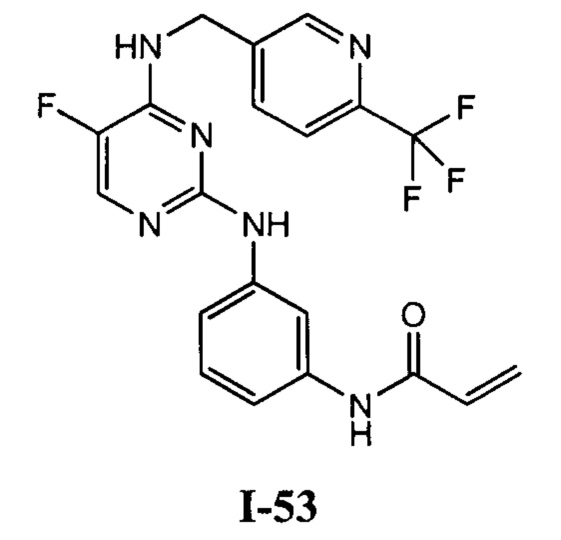
[001122] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 2 с использованием 5-фтор-2,4-дихлоропиримидина вместо 1 и 3-аминометил-6-трифторметилпиридина вместо 2 на этапе-1. ЖХ/МС (RT=2.805/(М+Н)) 433.0
ПРИМЕР 202
[001123] Получение N-(3-(4-((2,3-дигидробензофуран-5-ил)метиламино)-5-фторпиримидин-2-иламино)фенил)акриламида I-6
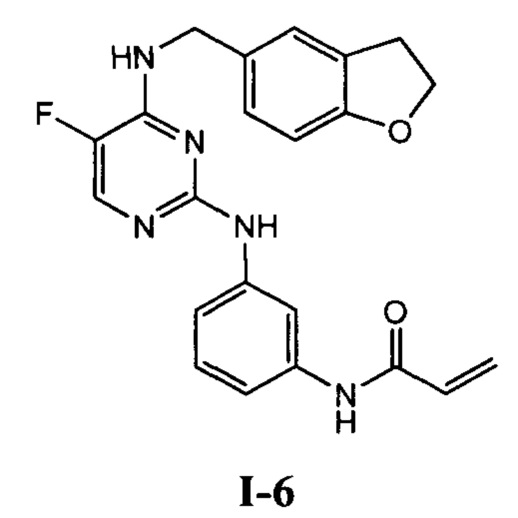
[001124] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в примере 2 с использованием 5-фтор-2,4-дихлоропиримидина вместо 1 и 3-аминометил-2,3-дигидробензофурана вместо 2 на этапе-1. ЖХ/МС (RT=2.815/(М+Н)) 406.2
ПРИМЕР 203
[001125] Получение N-(3-(5-фтор-2-(4-метоксибензиламино)пиримидин-4-иламино)фенил)акриламида I-241
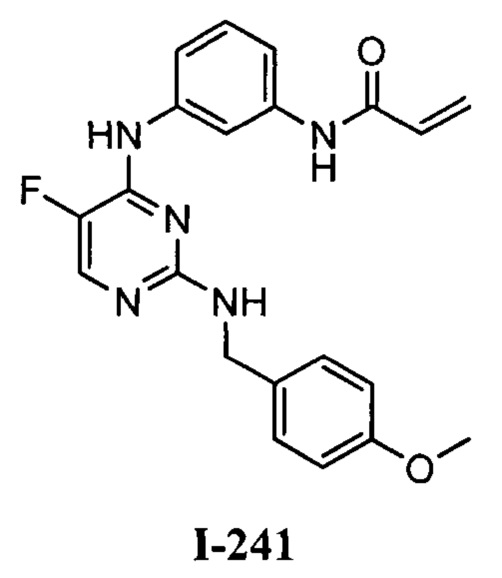
[001126] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-метоксибензиламина вместо 4 на этапе-2. ЖХ/МС (RT=2.801/(М+Н)) 394.2
ПРИМЕР 204
[001127] Получение N1-(3-(3-(4-(3-акриламидофениламино)-5-метилпиримидин-2-иламино)феноски)пропил)-N5-(15-оксо-19-((3aR,4R,6aS)-2-оксогексагидро-1H-тиено[3,4-d]имидазол-4-ил)-4,7,10-триокса-14-азанонадецил)глутарамида I-215
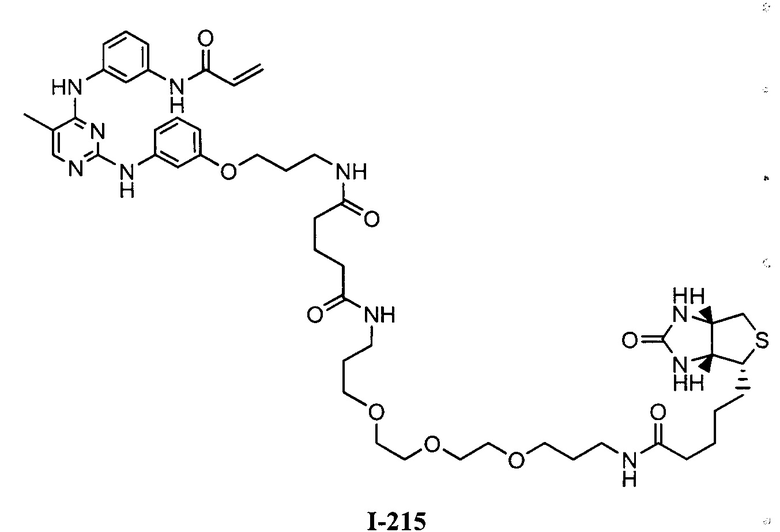
[001128] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
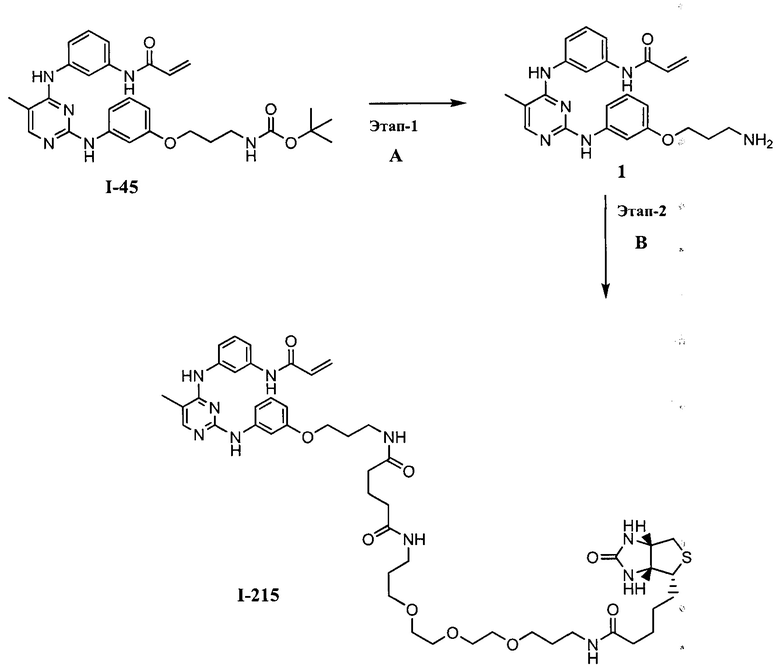
А) ТФА, ДХМ; В) N-Биотинил-NH-(PEG)2-COOH-DIPEA, HOBt, EDC, NMM, ДМФ.
[001129] Этап-1
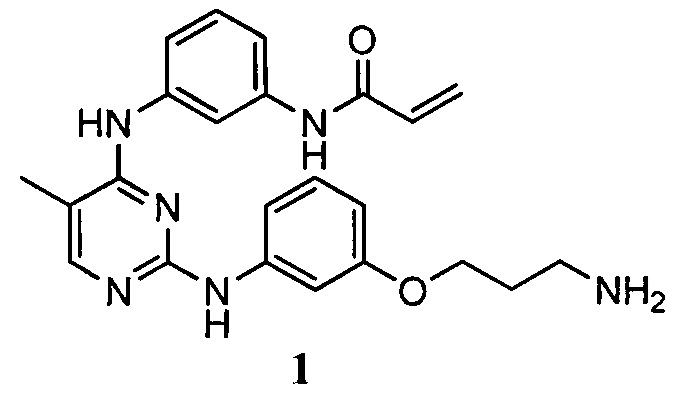
[001130] I-45 (97 мг, 0.19 ммоль; синтез I-45 приведен в примере 62)' растворяли в ДХМ (10 мл). Добавляли трифторуксусную кислоту (200 мкл), после чего смесь оставляли для перемешивания при комнатной температуре в течение 24 часов. Растворитель удаляли методом роторного испарения с получением палево-коричневой пены (130 mg), которую использовали в следующей реакции без дальнейшей очистки. ЖХ/МС (RT=2.63/(МН+) 419.2)
[001131] Этап-2
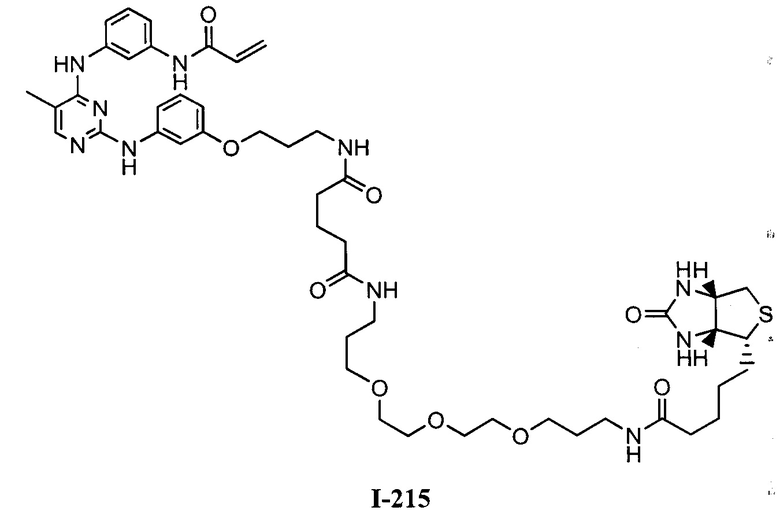
1 (80 мг, 0.15 ммоль) растворяли в ДМФ (2 мл). К смеси добавляли N-Биотинил-NH-(PEG)2-COOH-DIPEA (114 мг, 0.16 ммоль) и HOBt (25 мг, 0.16 ммоль (89%)), после чего реакционную смесь охлаждали в бане с ледяной водой. Добавляли EDC (32 мг, 0.16 ммоль), и затем N-метилморфолин (50 мкл, 0.45 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуре и продолжать помешивать в течение 30 минут. Непосредственно очищали методом флэш-хроматографии с использованием 10% раствором МеОН в ДХМ с получением 40 мг I-215 в форме желтой пленки. ЖХ/МС (RT=2.654/(МН+) 961.3).
ПРИМЕР 205
[001132] Получение N-(3-(3-(4-(3-акриламидофениламино)-5-метилпиримидин-2-иламино)феноски)пропил)-5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)пентанамида I-237
[001133] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 204, с использованием Б-(+)-биотин вместо N-Биотинил-NH-(PEG)2-COOH-DIPEA на этапе-2. ЖХ/МС (RT=2.686/(М+Н)) 645.2
ПРИМЕР 206
[001134] Получение (R)-N-(3-(5-фтор-2-(3-фтор-4-(тетрагидрофуран-3-илокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-316
[001135] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (R)-3-фтор-4-(тетрагидрофуран-3-илоксианилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.85-2.00 (m, 1H), 2.20 (m, 1Н), 3.70-3.90 (m, 4Н), 4.90 (s, 1H), 5.73 (dd, J=1.56 & 10.04 Гц, 1H), 6.23 (dd, J=1.76 & 17.00 Гц, 1H), 6.44 (dd, J=10.08 & 16.88 Гц, 1H), 7.28 (t, J=8.04 Гц, 1Н), 7.40-7.47 (m, 2Н), 7.67-7.71 (m, 2Н), 7.68 (dd, J=1.96 & 14.08 Гц, 1H), 7.92 (s, 1Н), 8.1 (d, J=3.64 Гц, 1Н), 9.21 (s, 1Н), 9.44 (s, 1Н), 10.12 (s, 1Н); ЖХМС: т/е 452.0 (м-l).
ПРИМЕР 207
[001136] Получение 1-(4-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-3-метилбут-2-ен-1-она I-325
[001137] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием метил 4-аминобензоата вместо 2 на этапе 1 и 3-фтор-4-(2-метоксиэтокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.01 (s, 3Н), 2.15 (d, J=0.72 Гц, 3Н), 3.31 (s, 3Н), 3.66 (dd, J=3.64 & 4.56 Гц, 2Н), 4.11 (dd, J=44,4 & 6.12 Гц, 2Н), 6.93 (s, 1Н), 7.08 (t, J=9.44 Гц, 1H), 7.27 (d, J=8.88 Гц, 1H), 7.74 (dd, J=2.44 & 14.24 Гц, 1H), 7.93 (d, J=8.96 Гц, 2H), 7.98 (d, J=8.92 Гц, 2H), 8.20 (d, J=3.64 Гц, 1H), 9.35 (s, 1H), 9.73 (s, 1H); ЖХМС: m/e 455 (M+l).
ПРИМЕР 208
[001138] Получение 1-(3-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-3-метилбут-2-ен-1-она I-323
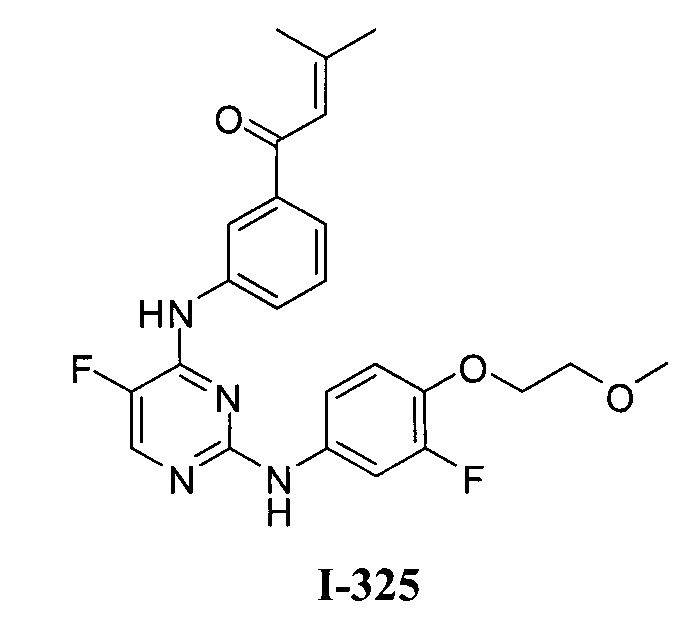
[001139] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием 2-(3-фтор-4-(2-метоксиэтокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.95 (s, 3Н), 2.14 (s, 3Н), 3.31 (s, 3Н), 3.63 (t, J=4.64 Гц, 2Н), 4.06 (t, J=4.36 Гц, 2Н), 6.85 (bs, 1Н), 6.97 (t, J=9.52 Гц, 1Н), 7.26 (bd, J=8.32 Гц, 1Н), 7.48 (t, J=7.92 Гц, 1H), 7.62-7.67 (m, 2Н), 8.08 (bd, J=7.04 Гц, 1H), 8.15-8.16 (m, 2Н), 9.29 (s, 1Н), 9.58 (s, 1H); ЖХМС: т/е455 (М+l).
ПРИМЕР 209
[001140] Получение 1-(4-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-2-метилпроп-2-ен-1-она I-324
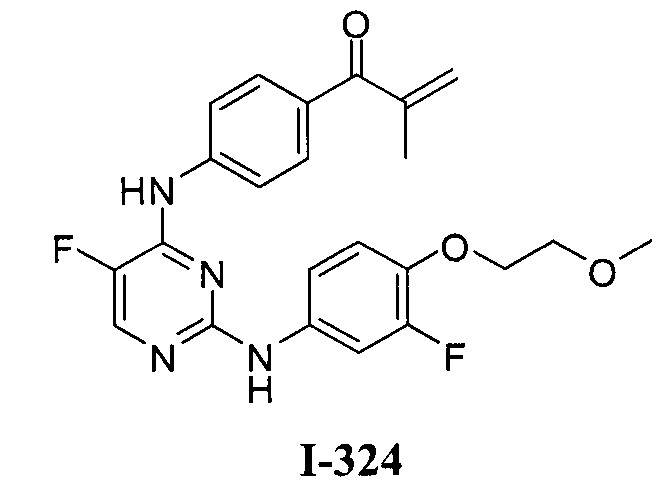
[001141] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием метил 4-аминобензоата вместо 2 на этапе 1 и 3-фтор-4-(2-метоксиэтокси)анилин вместо 4 на этапе 2 и и изопропенилмагния бромида вместо 8 на этапе 5. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.0 (s, 3Н), 3.32 (s, 3Н), 3.66 (t, J=4.36 Гц, 2Н), 4.11 (t, J=4.44 Гц, 2Н), 5.55 (s, 1Н), 5.94 (s, 1H), 7.07 (t, J=9.32 Гц, 1H), 7.26 (t, J=9.4 Гц, 1H), 7.72-7.76 (m, 3H), 7.99 (d, J=8.44 Гц, 2H), 8.21 (d, J=3.56 Гц, 1H), 9.38 (s, 1H), 9.75 (s, 1H); ЖХМС: m/e 441.2 (M+l).
ПРИМЕР 210
[001142] Получение 1-(4-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-3-метилбут-3-ен-2-она I-329
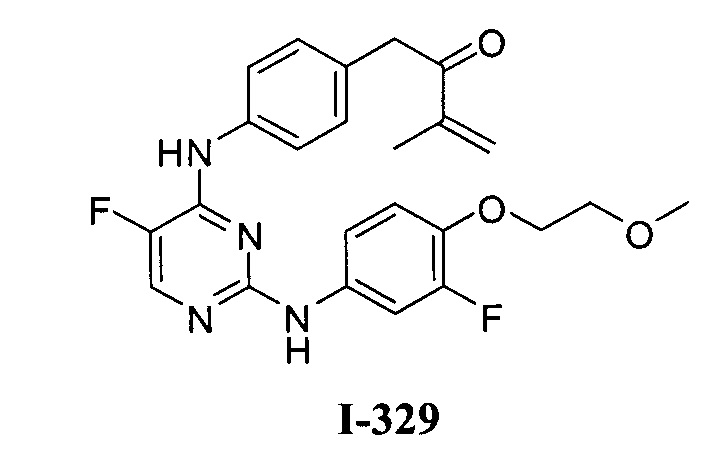
[001143] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием метил 4-аминофенилацетата вместо 2 на этапе 1, 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе 2 и изопропенилмагния бромида вместо 8 на этапе 5. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.79 (s, 3Н), 3.30 (s, 3Н), 3.62-3.65 (m, 2Н), 4.06 (s, 2Н), 4.08-4.10 (m, 2Н), 5.95 (d,J=1 Гц, 1Н), 6.27 (s, 1H), 7.01 (t, J=9.44 Гц, 1Н), 7.15 (d, J=8.52 Гц, 2Н), 7.28 (d, J=8.88 Гц, 1Н), 7.64-7.70 (m, 3Н), 8.08 (d, J=3.72 Гц, 1Н), 9.19 (s, 1Н), 9.33 (s, 1Н); ЖХМС: т/е 455.3 (М+l).
ПРИМЕР 211
[001144] Получение 1-(4-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-4-метилпент-3-эн-2-она I-331
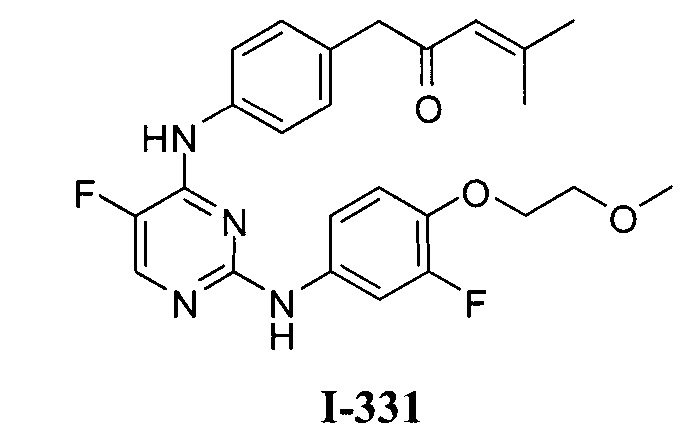
[001145] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием этил 4-аминофенилацетата вместо 2 на этапе1 и 3-фтор-4-(2-метоксиэтокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.84 (d,J=1 Гц, 3Н), 2.05 (d, J=0.92 Гц, 3Н), 3.30 (s, 3Н), 3.62-3.64 (m, 2Н), 3.68 (s, 2Н), 4.07-4.09 (m, 2Н), 6.21 (t, J=1.2 Гц, 1Н), 7.01 (t, J=8.68 Гц, 1Н), 7.16 (d, J=8.48 Гц, 2Н), 7.26 (d, J=8.96 Гц, 1Н), 7.65-7.72 (m, 3Н), 8.08 (d, J=3.72 Гц, 1H), 9.20 (s, 1H), 9.34 (s, 1Н); ЖХМС: т/е 469.3 (М+l).
ПРИМЕР 212
[001146] Получение 1-(3-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-2-метилпроп-2-ен-1-она I-322
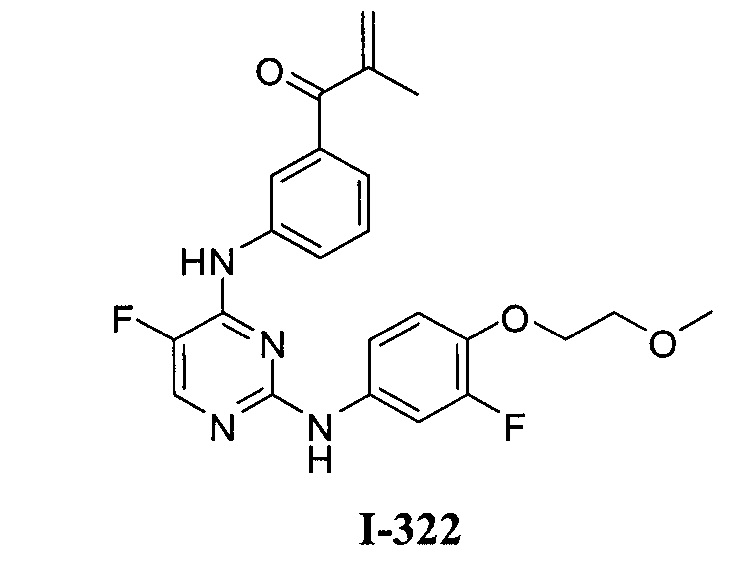
[001147] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием 1,3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе 2 и изопропенилмагния бромида вместо 8 на этапе 5. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.96 (s, 3Н), 3.30 (s, 3Н), 3.63 (t, J=4.6 Гц, 2Н), 4.07 (t, J=4.36 Гц, 2Н), 5.62 (s, 1Н), 6.00 (s, 1Н), 6.99 (t, J=9.24 Гц, 1Н), 7.26 (d, J=8.92 Гц, 1Н), 7.39 (d, J=7.56 Гц, 1Н), 7.46 (t, J=7.72 Гц, 1H), 7.62 (bd, J=14.4 Гц, 1H), 7.93 (s, 1H), 8.12-8.14 (m, 2Н), 9.25 (s, 1Н), 9.58 (s, 1Н); ЖХМС: т/е 441.2 (М+l).
ПРИМЕР 213
[001148] Получение 1-(3-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-3-метилбут-3-эн-2-она I-328
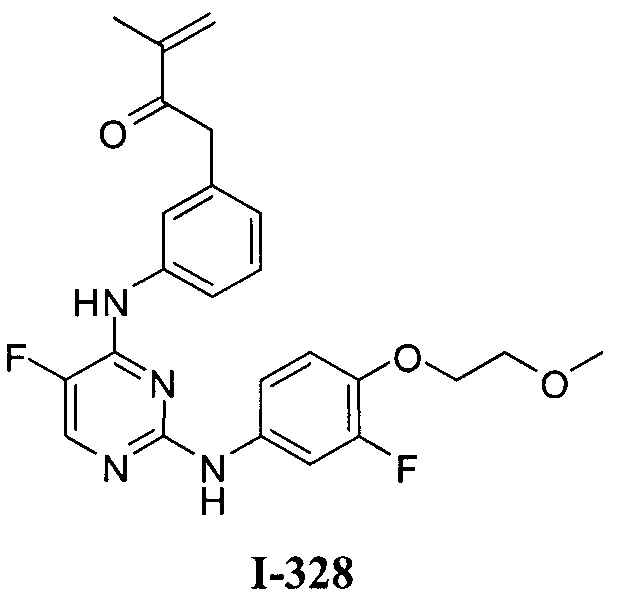
[001149] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием этил 3-
аминофенилацетата вместо 2 на этапе1, 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе 2 и изопропенилмагния бромида вместо 8 на этапе 5. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.8 (s, 3Н), 3.31 (s, 3Н), 3.64 (t, J=4.56 Гц, 2Н), 4.06 (s, 2Н), 4.09 (t, J=4.37 Гц, 2Н), 5.95 (s, 1Н), 6.23 (s, 1H), 6.92 (d, J=7.52 Гц, 1H), 7.02 (t, J=9.4 Гц, 1H), 7.27 (t, J=7.8 Гц, 2H), 7.50 (s, 1H), 7.66-7.72 (m, 2H), 8.10 (d, J=3.56 Гц, 1H), 9.21 (s, 1H), 9.36 (s, 1H); ЖХМС: m/e 455.1 (M+l).
ПРИМЕР 214
[001150] Получение 2-((3-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)(гидроки)метил)акрилонитрила I-326
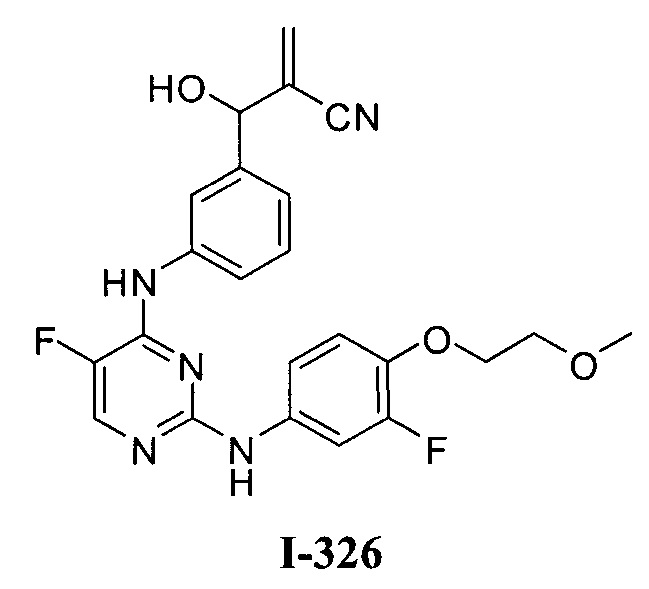
[001151] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 107, с использованием 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.30 (s, 3Н), 3.62-3.65 (m, 2Н), 4.09 (t, J=4.6 Гц, 2Н), 5.29 (d, J=3.84 Гц, 1H), 6.13 (s, 1H), 6.19 (s, 1H), 6.31 (d, J=4.04 Гц, 1H), 7.03 (t, J=9.24 Гц, 1H), 7.10 (d, J=7.44 Гц, 1H), 7.28 (d, J=8.72 Гц, 1H), 7.36 (t, J=7.8 Гц, 1H), 7.62 (s, 1H), 7.66 (dd, J=2.32 & 14.44 Гц, 1H), 7.89 (d, J=8.2 Гц, 1H), 8.10 (d, J=3.68 Гц, 1H), 9.14 (s, 1H), 9.45 (s, 1H); ЖХМС: m/e 454 (M+l).
ПРИМЕР 215
[001152] Получение 2-((4-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)(гидроки)метил)акрилонитрила I-327
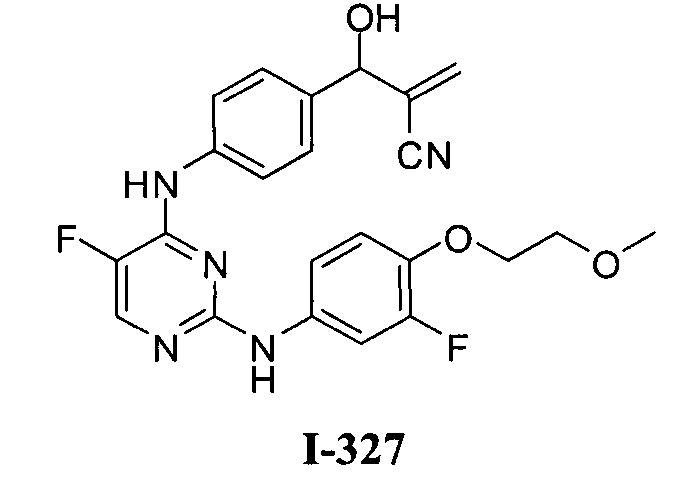
[001153] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 107, с использованием метил 4-аминобензоата вместо 2 на этапе 1 и 3-фтор-4-(2-метоксиэтокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.30 (s, 3Н), 3.64 (dd, J=2.96 & 4.56 Гц, 2Н), 4.08 (t, J=4.48 Гц, 2Н), 5.29 (d, J=3.8 Гц, 1H), 6.11 (s, 1H), 6.21 (s, 1H), 6.24 (d, J=4.12 Гц, 1H), 7.01 (t, J=9.4 Гц, 1H), 7.29-7.34 (m, 3H), 7.68 (dd, J=2.2 & 14.16 Гц, 1H), 7.77 (d, J=8.52 Гц, 2H), 8.11 (d, J=3.68 Гц, 1H), 9.23 (s, 1H), 9.42 (s, 1H); ЖХМС: m/e 454.0 (M+l).
ПРИМЕР 216
[001154] Получение N-(3-(2-(4-хлоро-3-(2-гидроки-2-метилпропокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-249
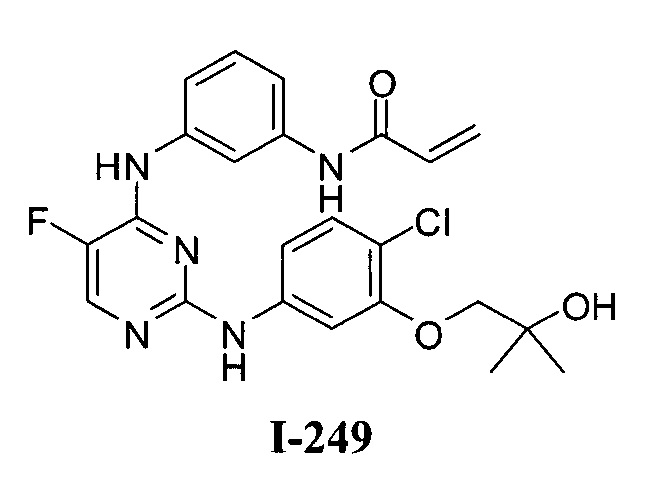
[001155] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-хлоро-3-(2-гидроки-2-метилпропокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.20 (s, 6Н), 3.61 (s, 2Н),4.61 (s, 1Н), 5.75 (d,J=11.4 Гц, 1H), 6.24 (d,J=18.36 Гц, 1H), 6.44 (dd,J=10.32 & 17.08 Гц, 1H), 7.13 (d, J=8.64 Гц, 1H), 7.28 (t, J=8 Гц, 1H), 7.37-7.44 (m, 3H), 7.55 (d, J=7.08 Гц, 1H), 7.93 (s, 1H), 8.12 (d, J=3.44 Гц, 1H), 9.23 (s, 1H), 9.47 (s, 1H), 10.11 (s, 1H); ЖХМС: m/e 472.0 (M+l).
ПРИМЕР 217
[001156] Получение N-(3-(5-фтор-2-(3-фтор-4-(2-гидроки-2-метилпропокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-315
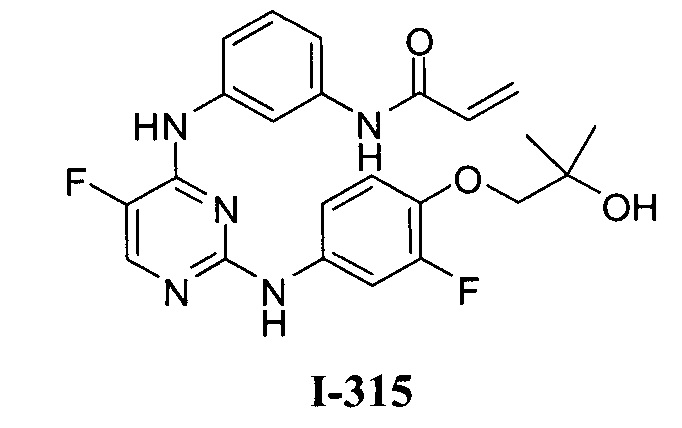
[001157] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-фтор-4-(2-гидроки-2-метилпропокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.19 (s, 6Н), 3.67 (s, 2Н), 4.62 (s, 1H), 5.75 (d, J=10.4 Гц, 1Н), 6.25 (d, J=17.2 Гц, 1H), 6.45 (dd, J=10 & 16.8 Гц, 1Н), 6.94 (t, J=9.2 Гц, 1H), 7.29 (t, J=8 Гц, 2Н), 7.43 (d, J=8.4 Гц, 1Н), 7.49 (d, J=7.6 Гц, 1H), 7.67 (d, J=13.6 Гц, 1Н), 7.94 (s, 1Н), 8.11 (d, J=3.6 Гц, 1H), 9.19 (s, 1H), 9.45 (s, 1H), 10.14 (s, 1H); ЖХМС: т/е 456 (М-l).
ПРИМЕР 218
[001158] Получение N-(3-(5-фтор-2-(3-фтор-4-(1-гидрокипропан-2-илокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-333
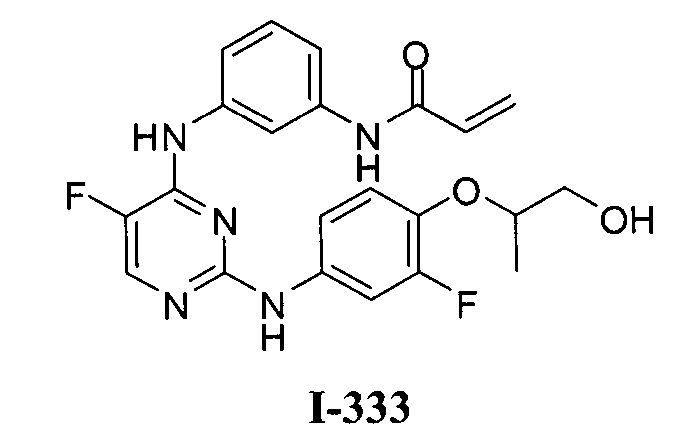
[001159] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-фтор-4-(2-гидроки-1-метилэтокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.16 (d, J=6.12 Гц, 3Н), 3.40-3.46 (m, 1H), 3.50-3.56 (m, 1H), 4.22 (sextet, J=5.6 Гц, 1H), 4.84 (t, J=5.68 Гц, 1H), 5.75 (dd, J=1.96 & 10.08 Гц, 1Н), 6.25 (dd, J=1.92 & 16.92 Гц, 1Н), 6.46 (dd,
J=10.08 & 16.92 Гц, 1H), 6.98 (t, J=9.32 Гц, 1H), 7.26-7.31 (m, 2H), 7.43 (d, J=8.76 Гц, 1H), 7.49 (d,J=S Гц, 1H), 7.68 (dd, J=2.44 & 14.28 Гц, 1H), 7.95 (s, 1H), 8.11 (d, J=3.68 Гц, 1H), 9.23 (s, 1H), 9.46 (s, 1H), 10.17 (s, 1H); ЖХМС: m/e 442.2 (M+l).
ПРИМЕР 219
[001160] Получение N-(3-(2-(4-(2,3-дигидрокипропокси)-3-фторфениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-334
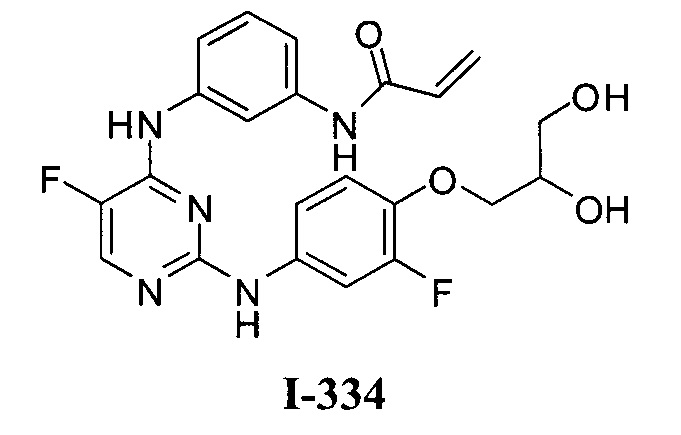
[001161] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-(2,3-дигидрокипропокси)-3-фторанилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.42 (t, J=5.6 Гц, 2Н), 3.7-3.8 (m, 1Н), 3.8-3.9 (m, 1H), 3.94 (dd, J=4.36 & 9.92 Гц, 1H), 4.65 (t, J=5.64 Гц, 1H), 4.93(d, J=5.08 Гц, 1Н), 5.7-5.8 (m, 1Н), 6.24 (dd,7=1.64 & 16.84 Гц, 1Н), 6.44 (dd, J=10 & 16.96 Гц, 1H), 6.94 (t, J=9.32 Гц, 1H), 7.28 (t, J=7.96 Гц, 2Н), 7.40 (d, J=8.28 Гц, 1H), 7.49 (d, J=7.44 Гц, 1Н), 7.66 (d, J=14.24 Гц, 1Н), 7.92 (s, 1Н), 8.09 (d, J=3.6 Гц, 1Н), 9.17 (s, 1Н), 9.45 (s, 1Н), 10.15 (s, 1Н); ЖХМС: т/е 456 (М-l).
ПРИМЕР 220
[001162] Получение N-(3-(2-(4-хлоро-3-(1-гидроки-2-метилпропан-2-илокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-336
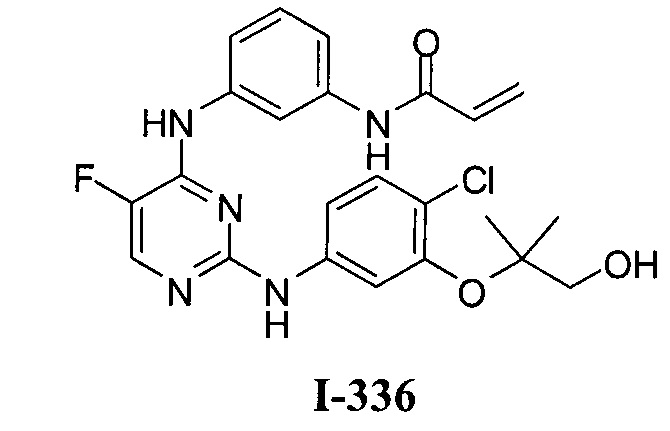
[001163] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-хлоро-3-(1-гидроки-2- метилпропан-2-илокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.22 (s, 6Н), 3.47 (d, J=5.88 Гц, 2Н), 4.88 (t, J=5.84 Гц, 1Н), 5.75 (dd, J=3.24 & 10 Гц, 1Н), 6.25 (dd, J=2 & 16.92 Гц, 1Н), 6.46 (dd, J=10.12 & 17.08 Гц, 1Н), 7.15 (d, J=8.8 Гц, 1Н), 7.31 (t, J=8.2 Гц, 1Н), 7.40-7.45 (m, 1H), 7.51-7.60 (m, 3Н), 7.93 (s, 1H), 8.13 (d, J=3.56 Гц, 1H), 9.24 (s, 1H), 9.47 (s, 1H), 10.12 (s, 1Н); ЖХМС: т/е 472.2 (М+l).
ПРИМЕР 221
[001164] Получение N-(3-(2-(4-хлоро-3-(1-гидрокипропан-2-илокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-337
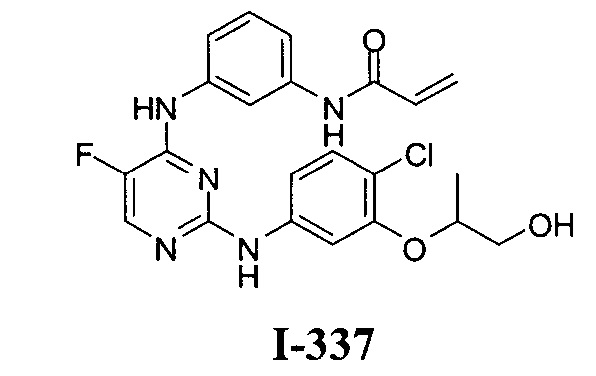
[001165] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-хлоро-3-(1-гидрокипропан-2-илокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.18 (d, J=6.12 Гц, 3Н), 3.40-3.47 (m, 1Н), 3.50-3.56 (m, 1H), 4.20-4.30 (m, 1Н), 4.82 (t, J=5.6 Гц, 1H), 5.75 (dd, J=1.88 & 10.08 Гц, 1H), 6.25 (dd, J=1.92 & 16.92 Гц, 1Н), 6.45 (dd, J=10.08 & 16.92 Гц, 1Н), 7.12 (d, J=8.76 Гц, 1Н), 7.29 (t, J=8.08 Гц, 1H), 7.40-7.44 (m, 3Н), 7.52 (d, J=8.44 Гц, 1Н), 7.91 (s, 1Н), 8.12 (d, J=3.64 Гц, 1Н), 9.21 (s, 1H), 9.45 (s, 1Н), 10.12 (s, 1H); ЖХМС: т/е 458.0 (М+l).
ПРИМЕР 222
[001166] Получение N-(3-(2-(4-(2,3-дигидрокипропокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-335
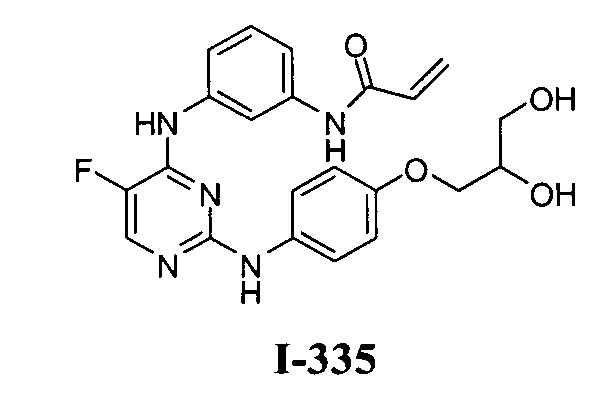
[001167] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-(1-гидрокипропан-2-илокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.43 (dd, J=0.64 & 6.84 Гц, 2Н), 3.70-3.80 (m, 2Н), 3.85-3.95 (m, 1H), 4.62 (t, J=5.6 Гц, 1H), 4.88 (d, J=4.76 Гц, 1Н), 5.75 (dd, J=1.76 & 10.08 Гц, 1H), 6.25 (dd, J=1.72 & 16.92 Гц, 1H), 6.45 (dd, J=10.08 & 16.88 Гц, 1Н), 6.74 (d, J=9 Гц, 2Н), 7.27 (t, J=8.08 Гц, 1H), 7.39 (d, J=8.04 Гц, 1Н), 7.38-7.53 (m, 3Н), 7.93 (s, 1Н), 8.05 (d, J=3.68 Гц, 1Н), 8.93 (s, 1H), 9.35 (s, 1H), 10.11 (s, 1Н); ЖХМС: т/е 440.3 (М+l).
ПРИМЕР 223
[001168] Получение (R)-N-(3-(2-(4-хлоро-3-(тетрагидрофуран-3-илокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-341
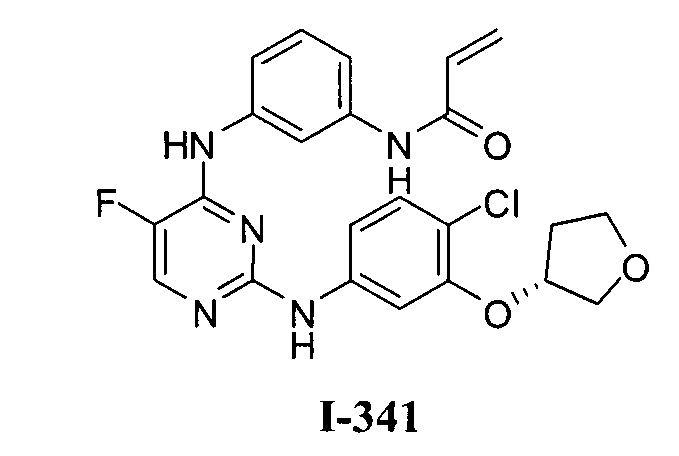
[001169] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (R)- 4-хлоро-3-(тетрагидрофуран-3-илокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.85-1.95 (m, 1Н), 2.0-2.15 (m, 1Н), 3.60-3.70 (m, 1Н), 3.73-3.83 (m, 3Н), 4.68 (s, 1H), 5.74 (dt, J=1.92 & 10.0 Гц, 1H), 6.23 (dd, J=1.88 & 16.92 Гц, 1H), 6.43 (dd, J=10.12 & 16.96 Гц, 1Н), 7.14 (d, J=8.72 Гц, 1Н), 7.29 (t, J=8.08 Гц, 1H), 7.33 (dd, J=4.16 & 8.76 Гц, 1Н), 7.41-7.47 (m, 3Н), 7.90 (s, 1Н), 8.13 (d, J=3.56 Гц, 1Н), 9.28 (s, 1Н), 9.47 (s, 1Н), 10.13 (s, 1H); ЖХМС: т/е 469.8 (М+l).
ПРИМЕР 224
[001170] Получение N-(3-(5-фтор-2-(3-фтор-4-(1-гидроки-2-метилпропан-2-илокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-332
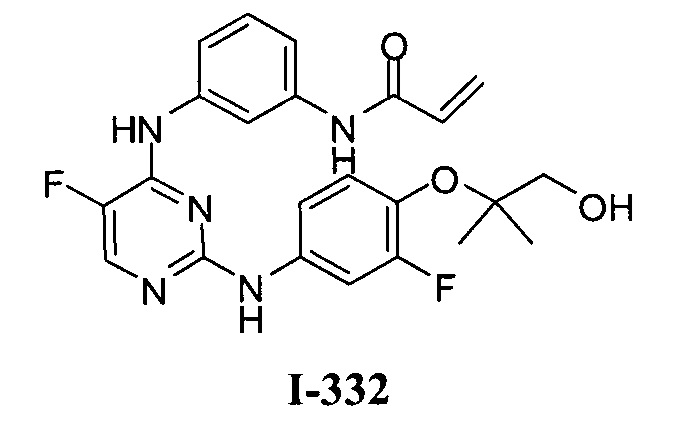
[001171] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-фтор-4-(1-гидроки-2- метилпропан-2-илокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.13 (s, 6Н), 3.36 (d, J=5.84 Гц, 2Н), 4.86 (t, J=5.84 Гц, 1H), 5.73 (dd, J=1.96 & 10.04 Гц, 1H), 6.24 (dd, J=1.96 & 16.96 Гц, 1H), 6.44 (dd, J=10.08 & 16.92 Гц, 1Н), 6.95 (t, J=9.16 Гц, 1Н), 7.23 (dd, J=1.64 & 8.96 Гц, 1Н), 7.28 (t, J=8.12 Гц, 1Н), 7.43 (d, J=8.8 Гц, 1Н), 7.48 (d, J=7.92 Гц, 1Н), 7.70 (dd, J=2.48 & 13.84 Гц, 1Н), 7.92 (s, 1Н), 8.11 (d, J=3.68 Гц, 1Н), 9.25 (s, 1H), 9.45 (s, 1Н), 10.10 (s, 1H); ЖХМС: т/е 456.2 (М+l).
ПРИМЕР 225
[001172] Получение N-(3-(2-(3-(2,3-дигидрокипропокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-339
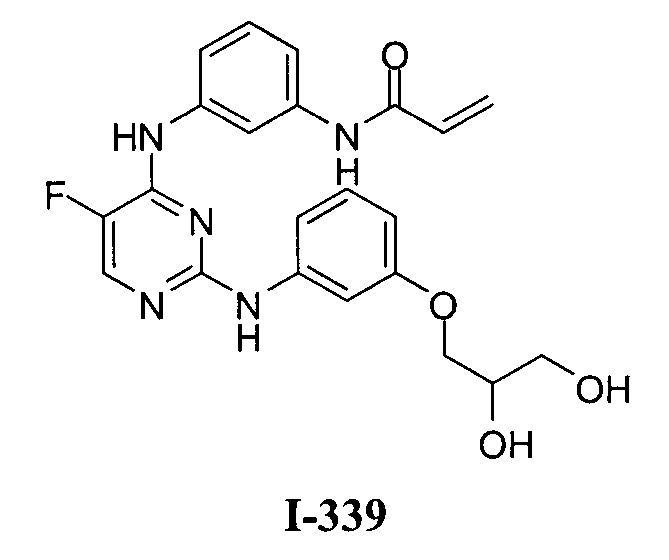
[001173] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-(2,3-дигидрокипропокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (MeOD) δ ppm: 3.59-3.69 (m, 2Н), 3.88-3.98 (m, 3Н), 5.78 (dd, J=2.16 & 9.6 Гц, 1Н), 6.36 (dd, J=2.24 & 17.04 Гц, 1H),
6.44 (dd, J=9.56 & 16.96 Гц, 1H), 6.54-6.57 (m, 1H), 7.08-7.12 (m, 2H), 7.32 (t, J=7.92 Гц, 2H), 7.44 (dd, J=7.88 & 13.4 Гц, 2H), 7.94 (d, J=3.8 Гц, 1H), 8.09 (s, 1H); ЖХМС: m/e 440.1 (M+l).
ПРИМЕР 226
[001174] Получение N-(4-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-351
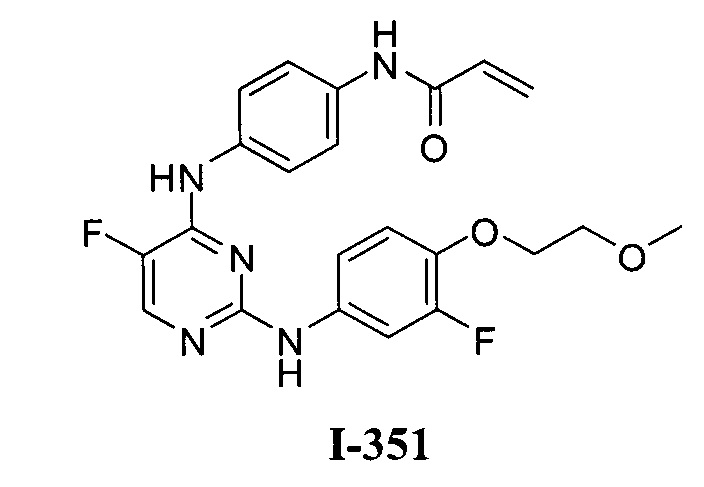
[001175] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием метил tert-бутоксикарбониламино-4-аминоанилина вместо 2 на этапе 1 и 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.30 (s, 3Н), 3.63 (t, J=4.6 Гц, 2Н), 4.08 (t, J=4.48 Гц, 2Н), 5.74 (dd, J=2 & 10.08 Гц, 1H), 6.25 (dd, J=1.96 & 16.92 Гц, 1H), 6.44 (dd, J=10.04 & 16.96 Гц, 1Н), 7.02 (t, J=9.48 Гц, 1Н), 7.23 (bd, J=7.44 Гц, 1H), 7.64 (d, J=9 Гц, 2Н), 7.70-7.74 (m, 3Н), 8.07 (d, J=3.72 Гц, 1H), 9.19 (s, 1H), 9.34 (s, 1Н), 10.13 (s, 1H); ЖХМС: т/е 442.0 (М+l).
ПРИМЕР 227
[001176] Получение 2-((3-(5-фтор-2-(6-(2-гидроки-2-метилпропокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)(гидроки)метил)акрилонитрила I-312.
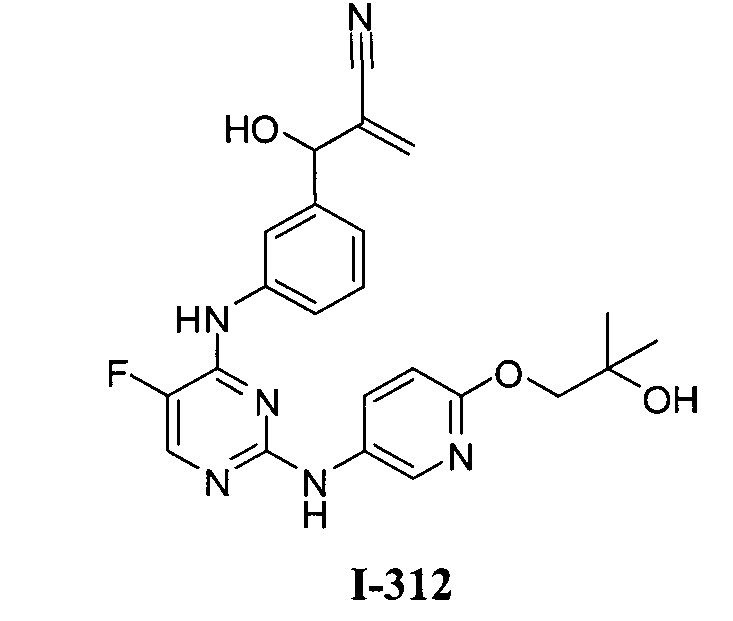
[001177] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 107, с использованием 3- амино -6-(2-гидроки-2-метилпропокси)пиридина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.18 (s, 6Н), 3.98 (s, 2Н), 4.61 (s, 1Н), 5.31 (d, J=3.88 Гц, 1H), 6.13 (s, 1Н), 6.19 (s, 1H), 6.32 (d, J=4 Гц, 1Н), 6.75 (d, J=8.88 Гц, 1Н), 7.10 (d, J=7.72 Гц, 1H), 7.33 (t, J=7.84 Гц, 1Н), 7.68 (s, 1H), 7.84 (d, J=7.52 Гц, 1H), 7.96 (dd, J=2.72 & 8.88 Гц, 1H), 8.08 (d, J=3.64 Гц, 1H), 8.33 (d,J=1.76 Гц, 1Н), 9.06 (s, 1H), 9.44 (s, 1Н); ЖХМС: т/е 451 (М+l).
ПРИМЕР 228
[001178] Получение 4-(4-(4-(3-акриламидофениламино)-5-фторпиримидин-2-иламино)фенокси)-N-метилпиколинамида I-342
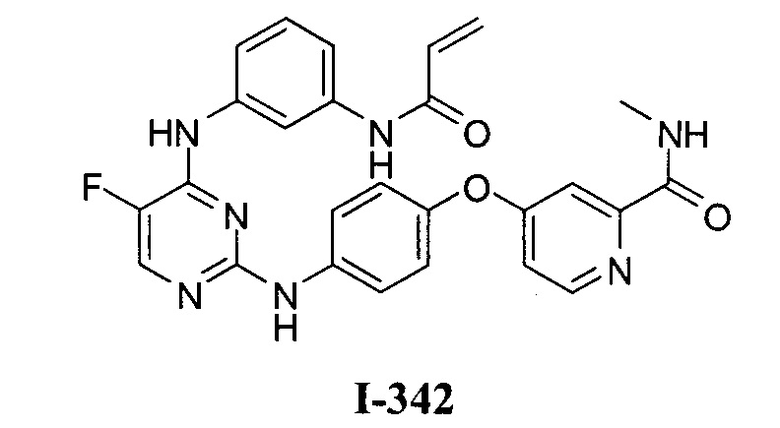
[001179] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-(4-аминофенокси)-N-метилпиколинамид вместо 4 на этапе 2. ЖХ/МС (М+Н) 500.2
ПРИМЕР 229
[001180] Получение (R)-1-(3-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-344
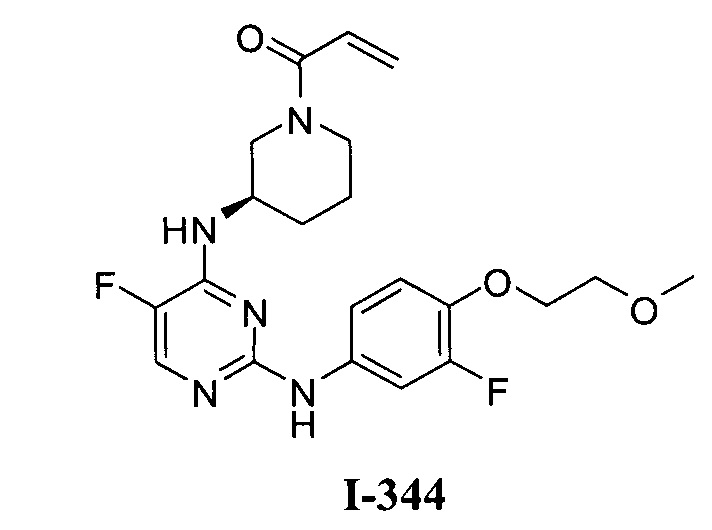
[001181] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20 с использованием (R)-1-трет-бутоксикарбонил-4-аминопиперидина вместо 2 на Этапе 1 и 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на Этапе 2. ЖХ/МС (М+Н) 434.1.
ПРИМЕР 230
[001182] Получение (R)-1-(3-(4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-345
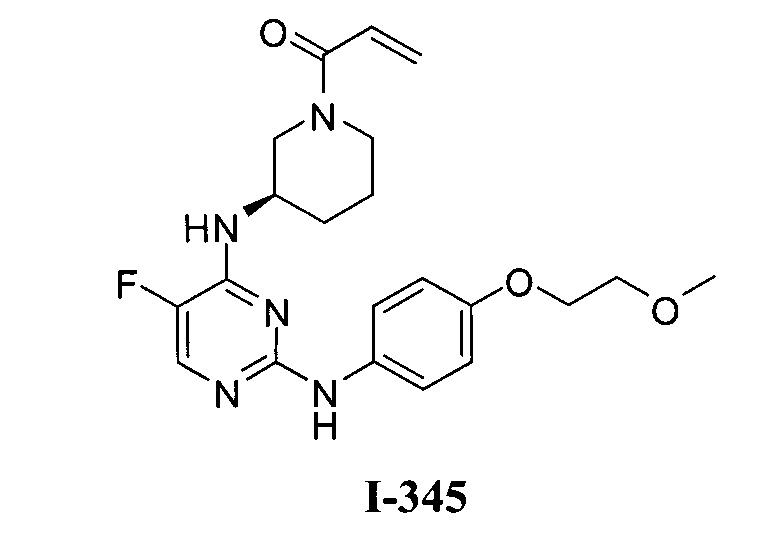
[001183] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20 с использованием (R)-1-трет-бутоксикарбонил-4-аминопиперидина вместо 2 на Этапе 1 и 4-(2-метоксиэтокси)анилина вместо 4 на Этапе 2. ЖХ/МС (М+Н) 416,2.
ПРИМЕР 231
[001184] Получение 4-(4-(4-(3-акриламидофениламино)-5-фторпиримидин-2-иламино)фенокси) пиридина I-346
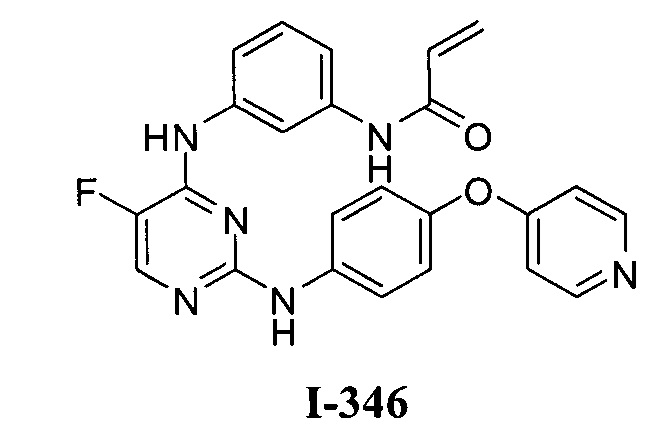
[001185] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-(4-аминофенокси)пиридина вместо 4 на этапе 2. ЖХ/МС (RT=2.802/(М+Н)) 500.2
ПРИМЕР 232
[001186] Получение 1-((R)-3-(5-фтор-2-(4-((S)-тетрагидрофуран-3-илокси)фениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-он I-347
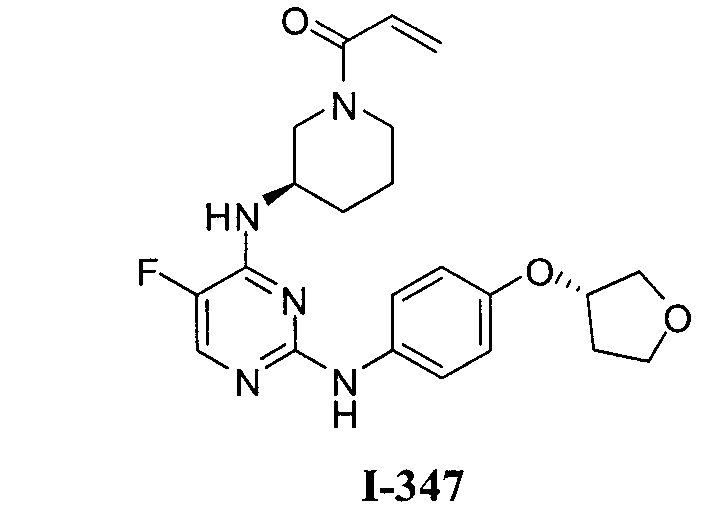
[001187] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20 с использованием (R)-1-трет-бутоксикарбонил-4-аминопиперидина вместо 2 на Этапе 1 и 4-(S)-(тетрагидрофуран-3-илокси)анилина вместо 4 на Этапе -3. ЖХ/МС (М+Н) 428.3.
ПРИМЕР 233
[001188] Получение 1-((R)-3-(5-фтор-2-(4-((R)-тетрагидрофуран-3-илокси)фениламино)пиримидин-4-иламино)пиперидин-1-ил)проп-2-ен-1-она I-348
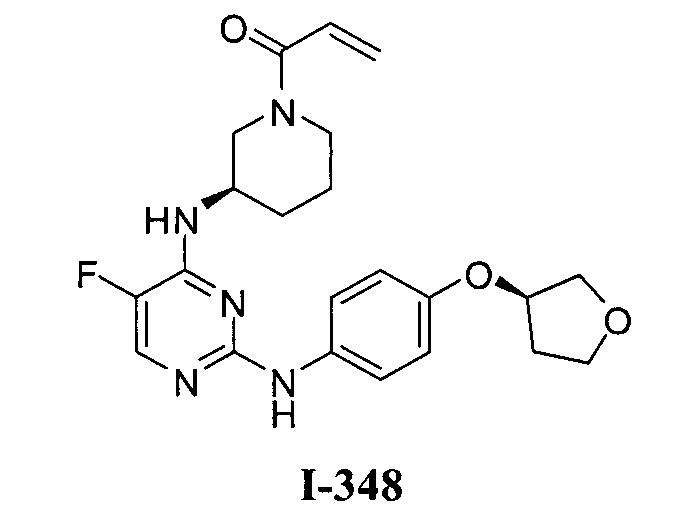
[001189] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20 с использованием (R)-1-трет-бутоксикарбонил-4-аминопиперидина вместо 2 на Этапе 1 и 4-(R)-(тетрагидрофуран-3-илокси)анилина вместо 4 на Этапе 2. ЖХ/МС (М+Н) 428.3.
ПРИМЕР 234
[001190] Получение N-(3-(2-(2,3-дигидробензо[b]1,4]диоксин-6-иламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-349
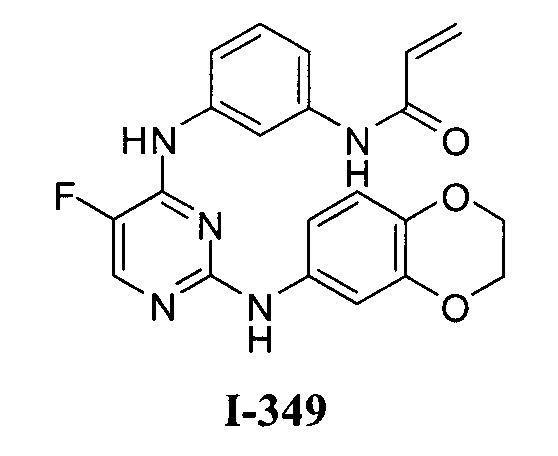
[001191] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 6- амино -2,3-дигидробензо[b]1,4]диоксана вместо 4 на этапе 2. ЖХ/МС (М+Н) 408
ПРИМЕР 235
[001192] Получение 1-(6-(4-(3-хлоро-4-(пиридин-2-илметокси)фениламино)-5-фторпиримидин-2-иламино)-2H-бензо[b][1,4]оксазин-4(3H)-ил)проп-2-ен-1-она I-343
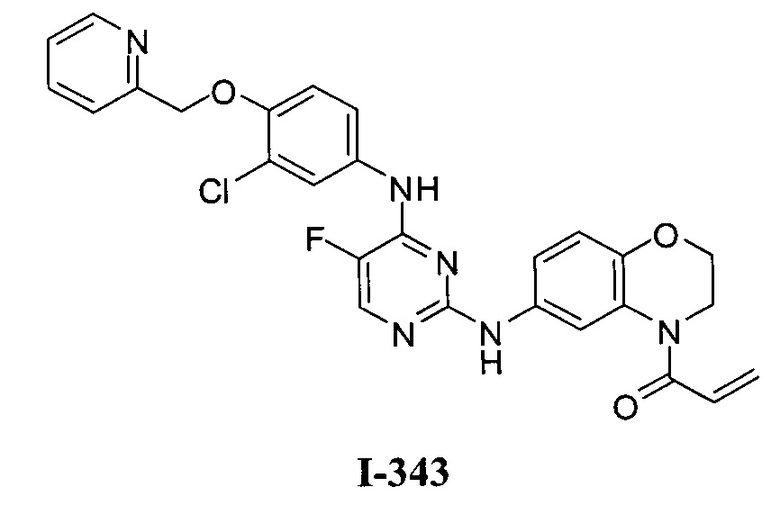
[001193] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 35, с использованием 3-хлоро-4-(пиридин-2-илметокси)анилина вместо 2 на этапе 1. ЖХ/МС (М+Н) 533,1
ПРИМЕР 236
[001194] Получение N-(3-(5-циано-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-350
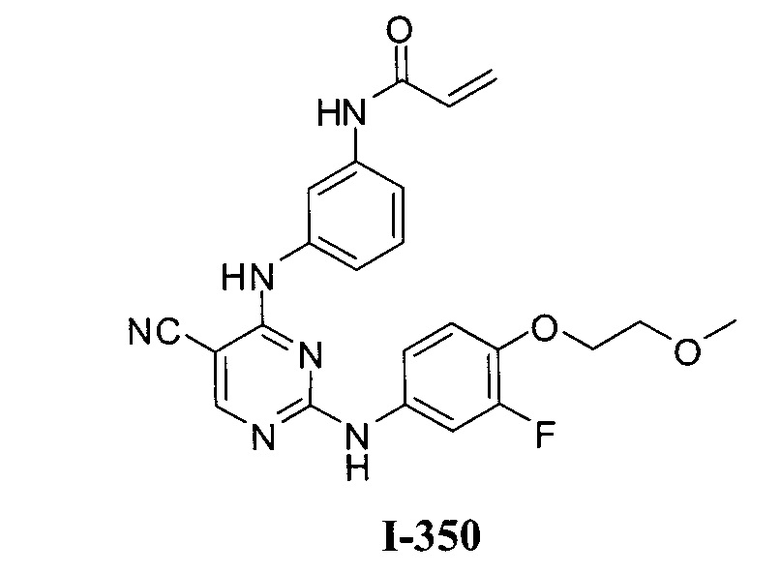
[001195] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 94, с использованием 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе 2. ЖХ/МС (М+Н) 449,1
ПРИМЕР 237
[001196] Получение N-(3-(5-трифторметил-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-352
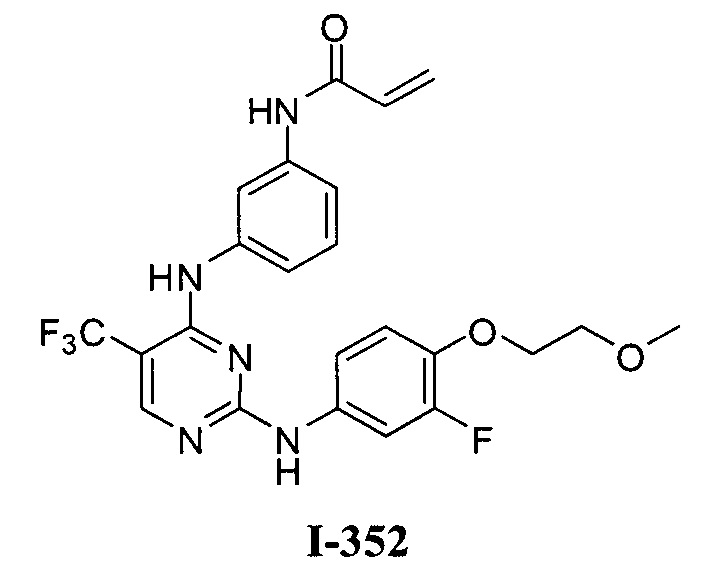
[001197] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 189, с использованием 3-фтор-4-(2-метоксиэтокси)анилина вместо 2 на этапе 1. ЖХ/МС (М+Н) 492,1
ПРИМЕР 238
[001198] Получение N-(3-(2-(4-хлоро-3-(2-метоксиэтокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-321
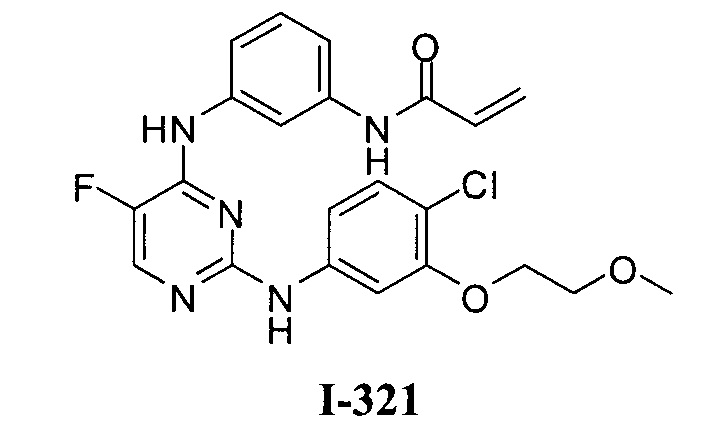
[001199] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 4-хлоро-3-(2-метоксиэтокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.30 (s, 3Н), 3.60 (t, J=4.56 Гц, 2Н), 3.88 (t, J=3.48 Гц, 2Н), 5.74 (dd, J=4.36 & 10.0 Гц, 1Н), 6.24 (dd, J=1.8 & 16.88 Гц, 1H), 6.44 (dd, J=4.36 & 10.0 Гц, 1H), 7.13 (d, J=8.72 Гц, 1H), 7.28 (t, J=8.04 Гц, 1H), 7.33 (dd, J=2.16 & 8.8 Гц, 1H), 7.41 (d, J=7.96 Гц, 1H), 7.47-7.49 (m, 2H), 7.84 (s, 1H), 8.13 (d, J=3.6 Гц, 1H), 9.27 (s, 1H), 9.47 (s, 1H), 10.12 (s, 1H); ЖХМС: m/e 458.0 (M+l).
ПРИМЕР 239
[001200] Получение N-(3-(5-фтор-2-(6-(2-гидроки-2-метилпропокси)пиридин-3-иламино)пиримидин-4-иламино)фенил)акриламида I-313
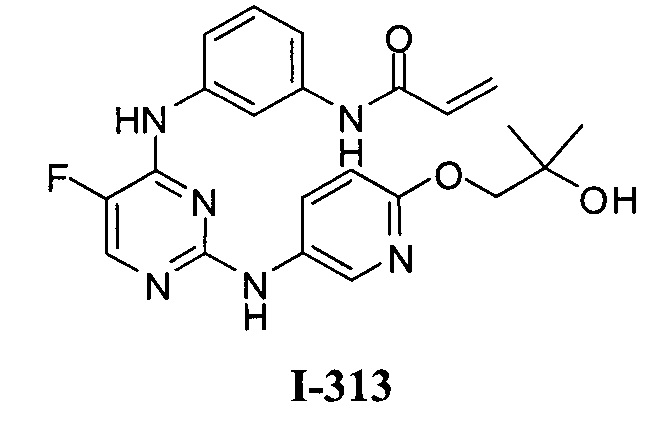
[001201] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3- амино -6-(2-гидроки-2-метилпропокси)пиридина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.16 (s, 6Н), 3.93 (s, 2Н), 4.57 (s, 1Н), 5.74 (dd, J=1.68 & 10.04 Гц, 1Н), 6.24 (dd, J=1.84 & 16.92 Гц, 1H), 6.45 (dd, J=10.04 & 16.88 Гц, 1H), 6.65 (d, J=8.88 Гц, 1H), 7.26 (t, J=8.04 Гц, 1H), 7.39 (d, J=8 Гц, 1H), 7.48 (d, J=7.8 Гц, 1H), 7.91 (s, 1H), 7.99 (dd, J=2.72 & 8.92 Гц, 1H), 8.07 (d, J=3.68 Гц, 1H), 8.27 (d, J=2.52 Гц, 1H), 9.06 (s, 1H), 9.41 (s, 1H), 10.1 (s, 1H); ЖХМС: m/e 439.0 (M+l).
ПРИМЕР 240
[001202] Получение N-(3-(5-фтор-2-(3-фтор-4-(3-(метилсульфонил)пропокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-318
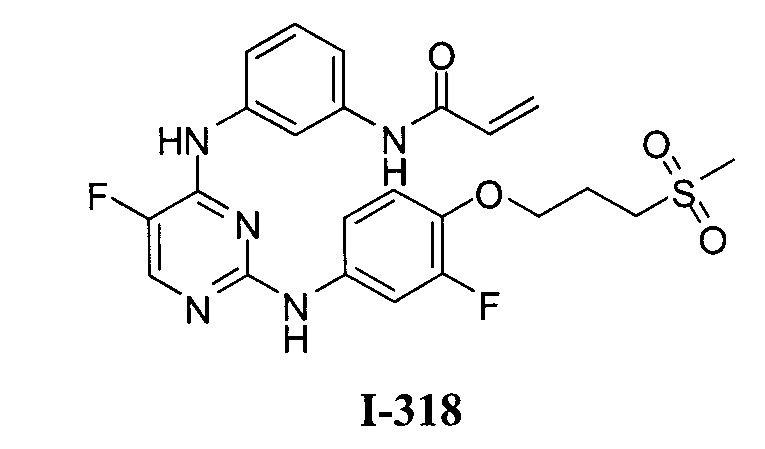
[001203] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием 3-фтор-4-(3-(метилсульфонил)пропокси)анилин вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 2.05-2.15 (m, 2Н), 3.01 (s, 3Н), 3.24 (t, J=7.56 Гц, 2Н), 4.05 (t, J=6.12 Гц, 2Н), 5.74 (dd, J=1.84 & 9.72 Гц, 1Н), 6.24 (dd, J=1.72 & 16.96 Гц, 1H), 6.44 (dd, J=10 & 16.84 Гц, 1Н), 6.96 (t, J=
=9.36 Гц, 1Н), 7.28 (t, J=8.04 Гц, 2H), 7.40 (d, J=7.8 Гц, 1H), 7.48 (d, J=8.32 Гц, 1H), 7.69 (dd, J=2.2 & 14.4 Гц, 1H), 7.91 (s, 1H), 8.10 (d, J=3.64 Гц, 1H), 9.20 (s, 1H), 9.44 (s, 1H), 10.12 (s, 1H); ЖХМС: m/e 504.2 (M+l).
ПРИМЕР 241
[001204] Получение 1-(3-(5-фтор-2-(3-фтор-4-(2-метоксиэтокси)фениламино)пиримидин-4-иламино)фенил)-4-метилпент-3-эн-2-она I-330
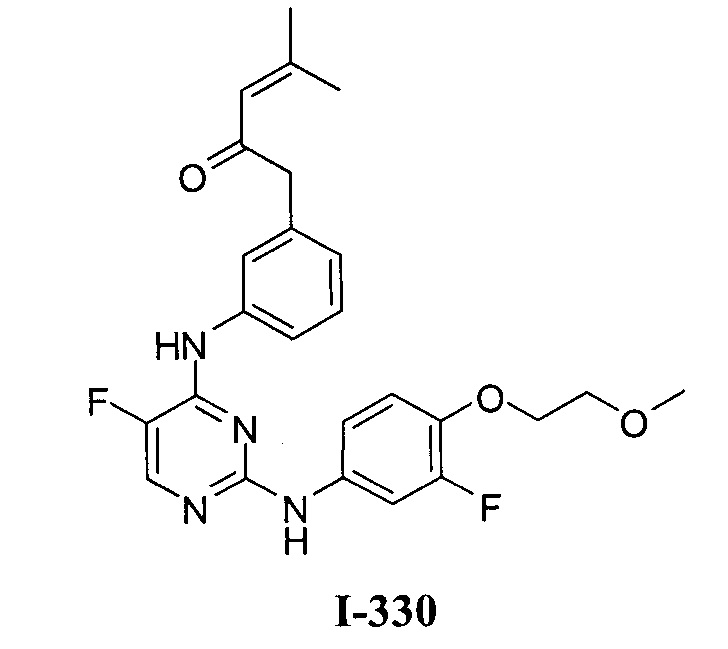
[001205] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 112, с использованием этил 4-аминометилбензоата вместо 2 на этапе 1 и 3-фтор-4-(2-метоксиэтокси)анилина вместо 4 на этапе 2. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.83 (s, 3Н), 2.05 (s, 3Н), 3.31 (s, 3Н), 3.64 (t, J=4.56 Гц, 2Н), 3.69 (s, 2Н), 4.08 (t, J=4.4 Гц, 2Н), 6.18 (s, 1Н), 6.92 (d, J=7.44 Гц, 1Н), 7.01 (t, J=9.36 Гц, 1H), 7.27 (t, J=7.84 Гц, 2H), 7.51 (s, 1H), 7.64-7.71 (m, 2H), 8.09 (d, J=3.64 Гц, 1H), 9.19 (s, 1H), 9.34 (s, 1H); ЖХМС: m/e 469.1 (M+l).
ПРИМЕР 242
[001206] Получение N-(3-(2-(4-хлоро-3-(2,3-дигидрокипропокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-353
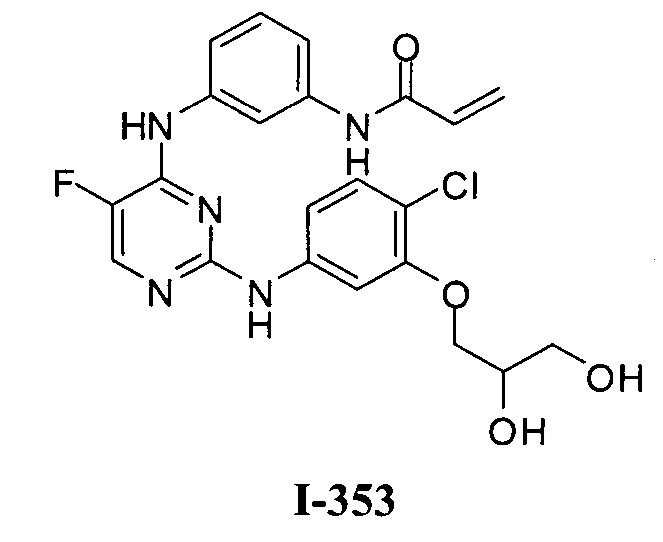
[001207] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
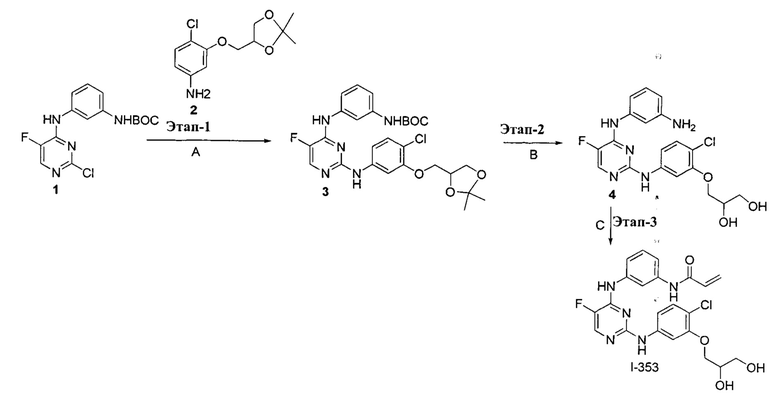
A) Pd(OAc)2, BINAP, Cs2CO3, толуол, 110°C 16 часов; В) ТФА, CH2Cl2, комн. т., 2 часа; С) акрилоил хлорид, K2CO3, NMP, комн. т., 45 минут.
[001208] Этап-1
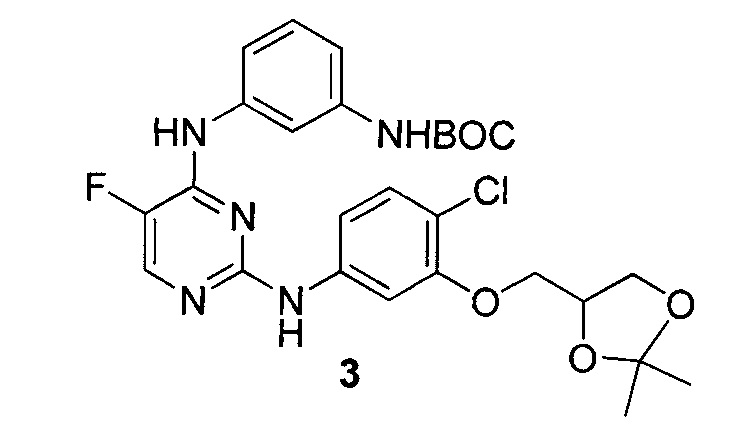
[001209] Раствор 2 (200 мг, 0.77 ммоль), 1 (262 мг, 0.77 ммоль), Pd(OAc)2 (17.3 мг, 0.07 ммоль), BINAP (24 мг, 0.038 ммоль) и Cs2CO3 (630 мг, 1.9 ммоль) в дегазированном
толуоле (через толуол пропускали N2 в течение 30 минут) нагревали в течение 16 часов при 100°С в атмосфере N2. Реакционную смесь охлаждали, разбавляли EtOAc (15 мл) и фильтровали через целит®. Фильтрат промывали водой (5 мл), солевым раствором (3 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3 (0.3 г, 69%) в форме желтого твердого вещества.
[001210] Этап-2
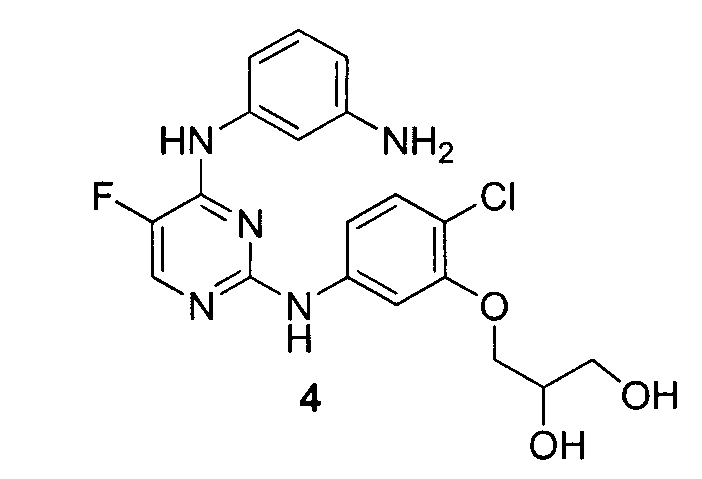
[001211] К раствору 3 (300 мг, 0.5 ммоль) в сухом CH2Cl2 (6 мл) при температуре 0°С добавляли CF3COOH (3 мл), и держали реакционную смесь при этой температуре в течение 30 минут. Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали при той же температуре в течение 3 часа. Реакционную смесь концентрировали при пониженном давлении, и тушили остаток водой (5 мл), приводили к форме основания раствором NaCO3, и экстрагировали этил ацетатом (2×10 мл). Объединенные экстракты промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 4 (200 мг, 88%) в форме желтого твердого вещества.
[001212] Этап-3
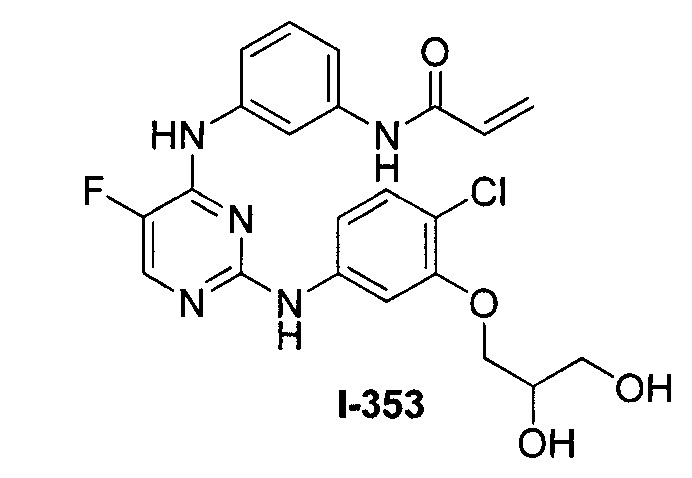
[001213] К раствору 4 (240 мг, 0,5 ммоль) и NMP (1,5 мл) при температуре 0°С при помешивании добавляли калия карбонат (780 мг, 5,7 ммоль) и акрилоил хлорид (57 мг, 0.5
ммоль), и помешивали реакционную смесь при температуре 0°С в течение 3 часа. Реакционную смесь помешивали при комнатной температуре в течение еще 30 минут, и тушили, добавляли по капле в холодный 10% раствор NaHCO3 при помешивании, после чего помешивали при температуре 0°С еще 30 минут. Твердой осадок отделяли методом фильтрации через воронку Бухнера. Твердый осадок промывали холодной водой, растворяли в EtOAc (20 мл) и приводили к форме основания используя триэтиламин, после чего промывали водой (2 мл), солевым раствором (1 мл), сушили над Na2SO4 и концентрировали при пониженном давлении.: Осадок очищали методом препаративной HPLC с получением указанного в названии соединения (45 мг, 15.5%) в форме белого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.43-3.50 (m, 2Н), 3.78-3.85 (m, 2Н), 3.89-3.92 (m, 1Н), 4.65 (t, J=5.6 Гц, 1H), 4.93 (d, J=4.8 Гц, 1H), 5.76 (dd, J=1.92 & 10.04 Гц, 1H), 6.26 (dd, J=1.92 & 16.92 Гц, 1H), 6.46 (dd, J=10.08 & 16.92 Гц, 1H), 7.14 (d, J=8.72 Гц, 1H), 7.30 (t, J=8.08 Гц, 1H), 7.39-7.43 (m, 2H), 7.46 (dd, J=2.2 & 8.72 Гц, 1H), 7.56 (d, J=8.04 Гц, 1H), 7.94 (s, 1H), 8.14 (d, J=3.6 Гц, 1H), 9.24 (s, 1H), 9.48 (s, 1H), 10.13 (s, 1H); ЖХМС: m/e 473.8 (M+).:
[001214] Получение промежуточного соединения 2
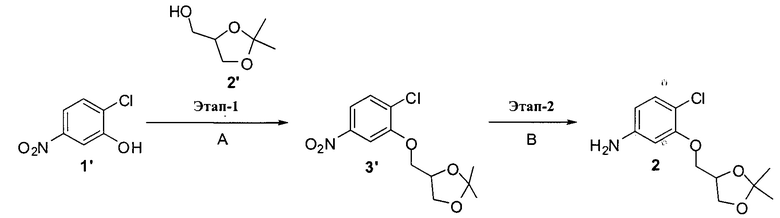
A) DIAD, PPh3, Et3N, сухой ТГФ, комн. т., 1 час; В) Н2, Ra Ni, метанол, 2 часа.
[001215] Этап-1
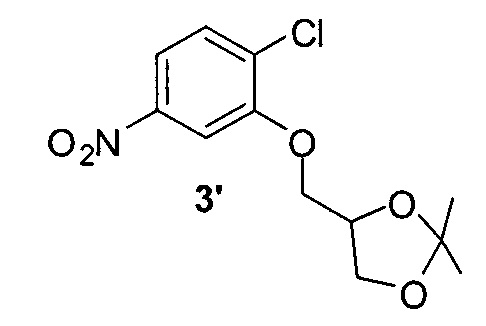
[001216] К раствору 2' (0,640 г, 3,7 ммоль) в ТГФ (20 мл) добавляли при помешивании 1' (0,5 г, 3,7 ммоль), PPh3 (1,09 г, 4,1 ммоль) и Et3N (1,68 г, 5,6 ммоль) а
атмосфере N2. Реакционную смесь охлаждали до 0°С и добавляли DIAD (0.84 г, 4.1 ммоль). Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали 1 час. Затем тушили водой, экстрагировали этил ацетатом (3×10 мл) и промывали объединенные экстракты водой и солевым раствором (по 5 мл каждого). Полученный осадок концентрировали при пониженном давлении и очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 9/1) с получением 3' (0,6 г 60%) в форме белого твердого вещества.
[001217] Этап-2
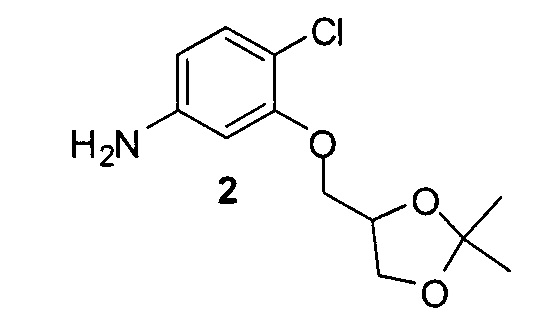
[001218] К раствору 3' (0.3 г, 1.04 ммоль) в метаноле добавляли Никель Ренея (60 мг, 20% масс/масс.) в атмосфере N2, и держали реакционную смесь в атмосфере Н2 (давление в пневмокамере) в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину, и концентрировали фильтрат при пониженном давлении. Осадок разбавляли 1.5 N HCl (2 мл) и промывали этил ацетатом (5 мл) чтобы удалить органические примеси. Водный слой приводили к форме основания раствором NaHCO3 (5 мл), экстрагировали этил ацетатом, промывали водой (2 мл) и солевым раствором (2 мл), и сушили над безводным Na2SO4. Фильтровали, после чего концентрировали при пониженном давлении с получением 2 (0,2 г, 76,9%) в форме коричневой жидкости.
ПРИМЕР 243
[001219] Получение (S)-N-(3-(2-(4-хлоро-3-(тетрагидрофуран-3-илокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-354
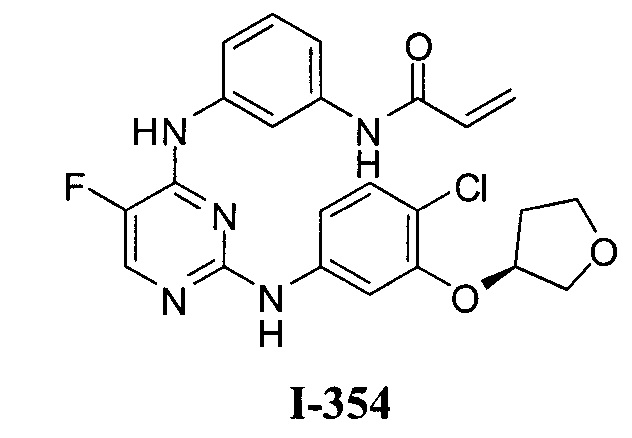
[001220] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 20, с использованием (S)-4-хлоро-3-(тетрагидрофуран-3-илокси)анилина вместо 4 на этапе 2. ЖХМС: т/е 469.8 (М+l).
ПРИМЕР 244
[001221] Получение (N-(3-(5-фтор-2-(3-фтор-4-(((2S,4R)-4-гидрокипирролидин-2-ил)метокси)фениламино)пиримидин-4-иламино)фенил)акриламида I-355
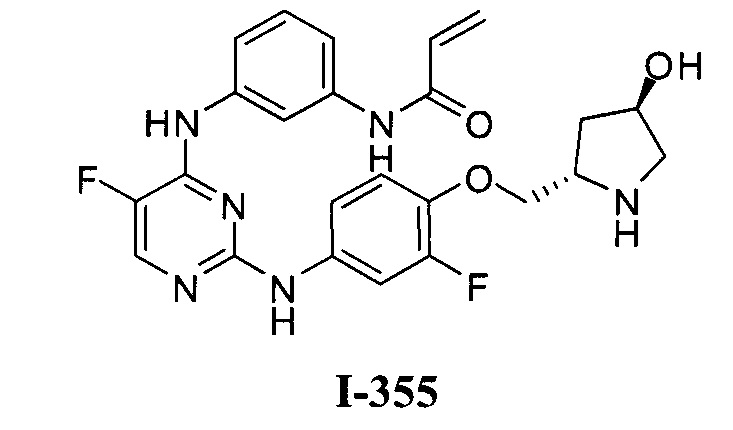
[001222] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
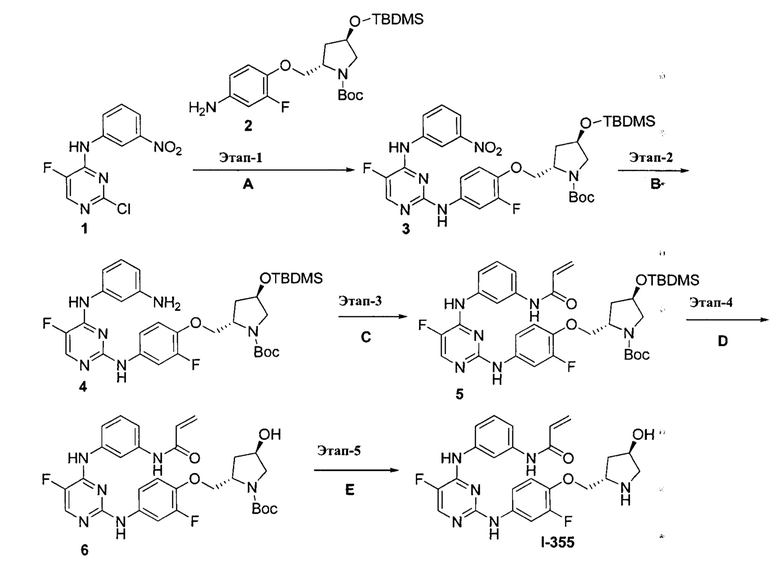
A) Pd(OAc)2, BINAP, Cs2CO3, толуол, 110°C, 6 часов; В) ТФА, CH2Cl2, комн. т., 1 час; С) (Вос)2O, 30 минут затем акрилоил хлорид, K2CO3, NMP, 0°С, 90 минут; D) HF (49% aq. Solution), CH3CN, комн. т., 2 часа; Е) ТФА, ДХМ, комн. т., 2 часа.
[001223] Этап-1
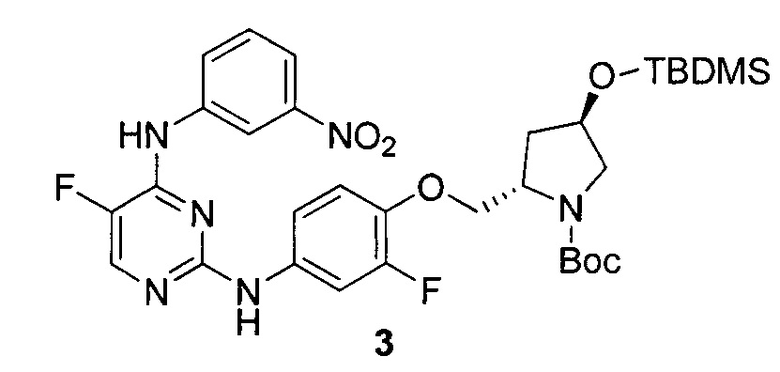
[001224] Раствор 2 (0,50 г, 1,13 ммоль), 1 (0,30g, 1,13 ммоль), Pd(OAc)2 (0,0025 г, 0.1 ммоль), BINAP (0.0035 г, 0,05 ммоль) и Cs2CO3 (0,92 г, 2,8 ммоль) в дегазированном толуоле (толуол продували N2 в течение 30 минут) нагревали в течение 16 часов при температуре 110°С в атмосфере N2. Реакционную смесь охлаждали, разбавляли EtOAc (20 мл) промывали водой (10 мл), солевым раствором (10 мл) и сушили над Na2SO4.
Фильтровали и концентрировали при пониженном давлении с получением осадка, которые промывали гексаном с получением 3 (0.3 г, 42.8%) в форме желтого твердого вещества.
[001225] Этап-2
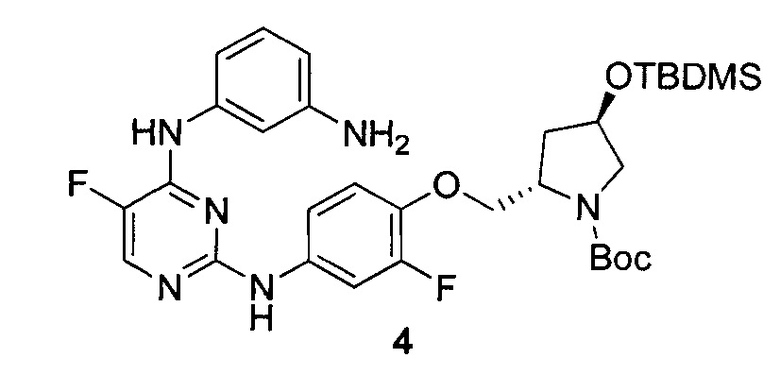
[001226] К раствору 3 (0,3 г, 0.44 ммоль) в этаноле (5 мл)) добавляли Pd/C (0,030 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере H2 (balloon) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 4 (0,19 г, 67,6%) в форме желтого твердого вещества.
[001227] Этап-3
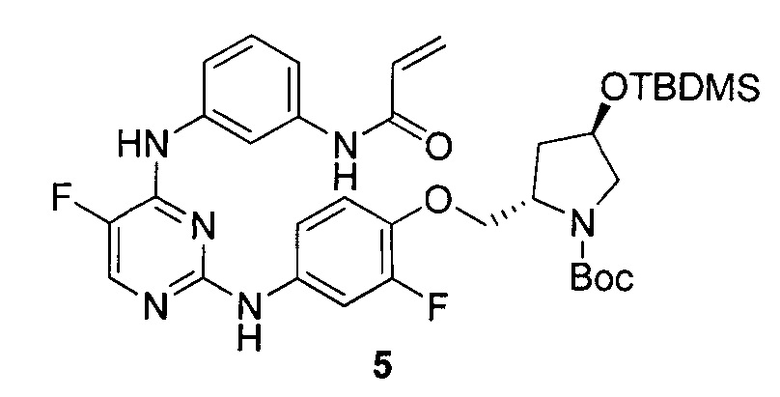
[001228] К раствору 4 (0,1 г, 0,15 ммоль) в NMP (1,0 мл) при комнатной температуре при помешивании Boc ангидрид (0,046 г, 0.212 ммоль), и помешивали реакционную смесь при комнатной температуре в течение 60 минут. Затем его охлаждали до 0°С и добавляли K2CO3 (0.107 г, 0.77 ммоль), акрилоил хлорид (0.016 г, 0.18 ммоль), после чего реакционную смесь помешивали при температуре 0°С в течение 90 минут. Реакционная смесь по каплям добавляли при помешивании в холодный 10% раствор NaHCO3. После добавления, раствор помешивали в течение еще 30 минут при температуре 0°С, и отделяли твердый осадок с помощью воронки Бухнера. Полученное твердое вещество промывали холодной водой, гексаном и растворяли в смеси метанол: дихлорметан (50:50, 10 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (5 мл), добавляли к нему Et3N и затем экстрагировали
этил ацетатом (2×10 мл). Объединенный экстракт этил ацетата дихлорметан промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток далее очищали методом колоночной хроматографии (SiO2, метанол/хлороформ: 4/96) с получением 5 (0,075 г, 71,4%) в форме желтого твердого вещества.
[001229] Этап-4
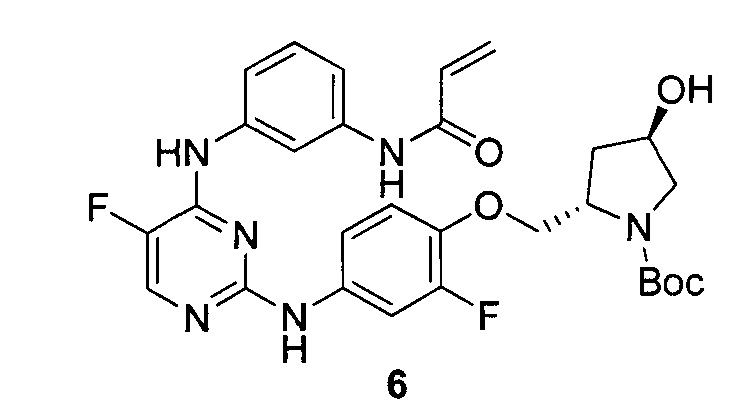
[001230] К раствору 5 (15 мг, 0.02 ммоль) в ацетонитриле добавляли, HF (49% вод. раствор. 0.0048 мл, 0.024 ммоль) при температуре 0°С. Реакционную смесь помешивали при комнатной температуре в течение 2 часов, экстрагировали этил ацетатом (2 мл), промывали водой (1 мл), и сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Осадок показал 60% чистого вещества согласно данным анализа ЖХМС и использовался на следующем этапе без дальнейшей очистки.
[001231] Этап-5
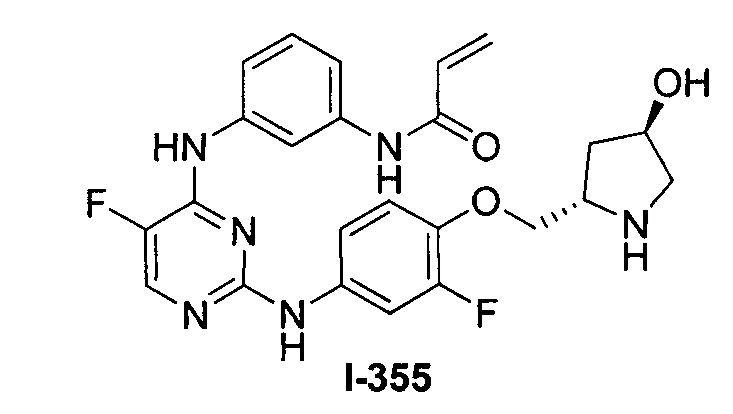
[001232] К раствору 6 (0,008 г, 0,013 ммоль) в CH2Cl2 (0,024 мл) при помешивании добавляли ТФА (0,016 мл) при температуре 0°С. Реакционную смесь оставляли дойти до комнатной температуры, после чего ее помешивали в течение дополнительных 2 часа. Затем концентрировали и помешивали с холодным 10% NaHCO3 (1.0 мл). Экстрагировали EtOAc (2×2 мл) и объединенный экстракт EtOAc промывали солевым раствором (1 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Сырой остаток далее очищали методом колоночной хроматографии (SiO2, метанол/хлороформ: 2/98) и далее
очищали методом препаративной ТСХ с получением указанного в названии соединения (2 мг, 81% чистота согласно HPLC, и 79% чистота согласно ЖХМС) в форме белого твердого вещества. ЖХМС: т/е 483 (М+).
[001233] Соединение 2 получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
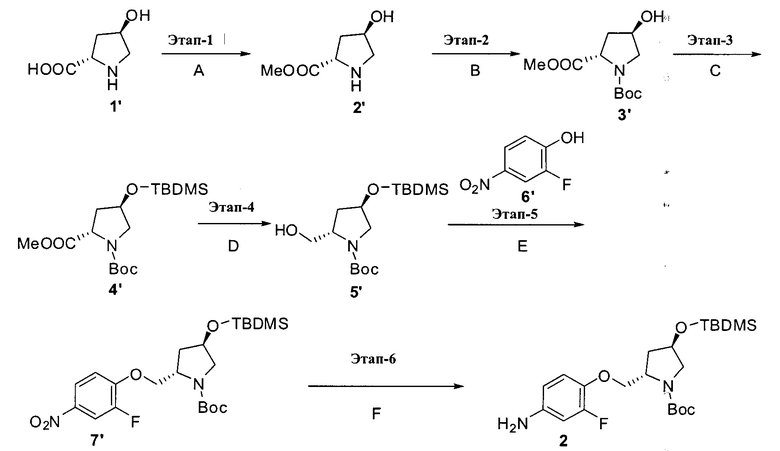
A) MeOH, SOCl2, с обратным холодильником, 5 часов; В) (Boc)2О, Et3N, CH2Cl2, комн. т.,
5 часов; С) TBDMS-Cl, имидазол, ДМФ, комн. т., 16 часов; D) LAH раствор (1М в ТГФ), - 20°С, 20 минут; Е) DIAD, PPh3, Et3N, ТГФ, 16 часов; F) Н2, Pd/C, метанол, комн. т., 16 часов.
[001234] Этап-1
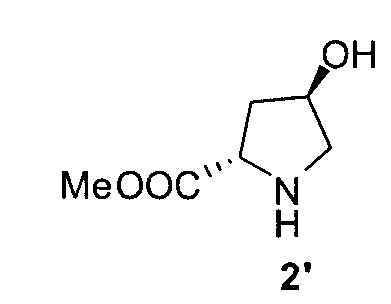
[001235] К раствору 1' (2 г, 15.26 ммоль) при помешивании добавляли раствор приготовленный добавлением тионил хлорида (2 мл) в метанол (20 мл). Реакционную смесь нагревали с обратным холодильником в течение 5 часов. После завершение
реакции, при пониженном давлении удаляли метанол с получением 2' в форме бесцветной соли (3.0 г), которую без дополнительной очистки использовали на следующем этапе.
[001236] Этап-2
[001237] К раствору 2' (3,0 г, 12,24 ммоль) в ДХМ (30 мл) при помешивании добавляли Et3N (1,85 г, 18,31 ммоль) и Вое ангидрид (2,92 г, 13,46 ммоль). Помешивание при комнатной температуре продолжали в течение 5 часов, после чего реакционную смесь тушили водой. Органический слой отделяли, сушили и концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии (SiO2, 60-120, 100% этил ацетат) с получением 3' (3.2 г, 64%) в форме белого твердого вещества.
[001238] Этап-3
[001239] К раствору 3' (3 г, 12.24 ммоль) в ДМФ (30 мл) при помешивании добавляли имидазол (1.2 г, 18.36 ммоль) а затем TBDMS хлорид (1.84 г, 12.24 г). Помешивание продолжали в течение 16 часов. Реакционную смесь разбавляли этил ацетатом (50 мл) после чего отделяли слой этил ацетата. Его промывали водой (5 мл), солевым раствором (5 мл) и сушили над Na2SO4. Фильтровали, после чего концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир /этил ацетат: 6/4) с получением 4' (3,2 г, 80%) в форме бесцветной жидкости.
[001240] Этап-4
[001241] К раствору 4' (0,5 г, 1,39 ммоль) в ТГФ (5 мл) при помешивании добавляли LAH (1,39 мл, 1М раствор, 1,39 ммоль) при температуре -20°С. Реакция продолжалась при той же температуре в течение 15 минут, после чего ее тушили раствором Na2SO4. Реакционную массу фильтровали через целит ® и концентрировали фильтрат при пониженном давлении. Осадок разбавляли этил ацетатом (10 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 5' (0.3 г, 65%) в форме бесцветной жидкости.
[001242] Этап-5
[001243] К раствору 5' (0,1 г, 0,3 ммоль) в ТГФ (6 мл) добавляли при помешивании 6' (0,047 г, 0,3 ммоль), PPh3 (0,16 г, 0,64 ммоль) и Et3N (0,048 г, 0,48 ммоль) а атмосфере N2. Реакционную смесь охлаждали до 0°С и добавляли к ней DIAD (0,094 г, 0,48 ммоль). Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали 1 час. Затем тушили водой, экстрагировали этил ацетатом (3×5 мл) и промывали объединенный экстракт этил ацетата водой и солевым раствором (по 5 мл каждого). Полученный осадок концентрировали при пониженном давлении и очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 9/1) с получением 7' (0,120 г 85%) в форме желтого твердого вещества.
[001244] Этап-6
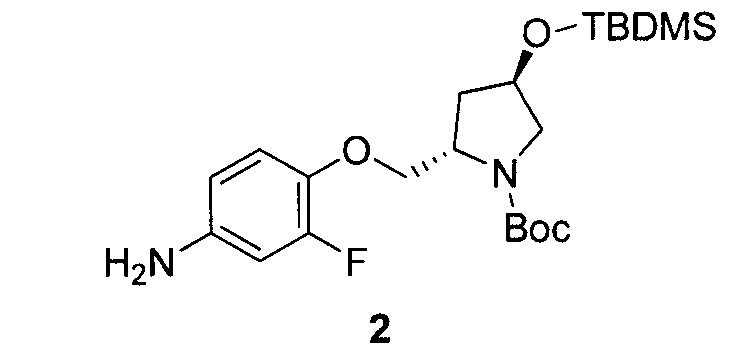
[001245] К раствору 7' (0,1 г, 0,21 ммоль) в этаноле (5 мл)) добавляли Pd/C (0,010 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (пневмокамера) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при
пониженном давлении с получением 2 (0,085 г, 91%) в форме коричневатого вязкого масла. Его использовали на следующем этапе без дальнейшей очистки.
ПРИМЕР 245
[001246] Получение трет-бутил 2-(2-(4-(4-(3-акриламидофениламино)-5-фторпиримидин-2-иламино)-2-фторфенокси)этокси)этилкарбамата I-356
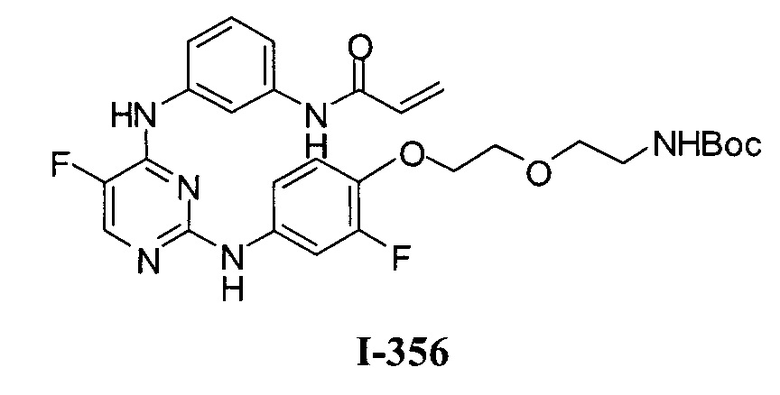
[001247] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
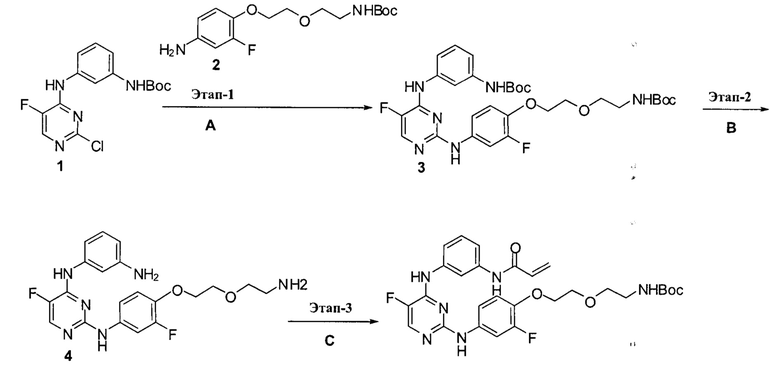
A) Pd(OAc)2, BINAP, Cs2CO3, толуол, 110°C, 6 часов; В) ТФА, CH2Cl2, Комн. Т. 1 час; С) (Вос)2O, 30 минут затем акрилоил хлорид, K2CO3, NMP, 0°С, 90 минут.
[001248] Этап-1
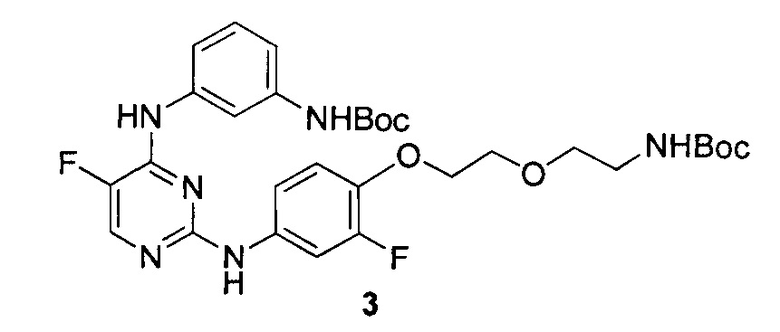
[001249] Раствор 2 (0,050 г, 0,159 ммоль), 1 (0,053 г, 0,159 ммоль), Pd(OAc)2 (0,0035 г, 0,01590 ммоль), BINAP (0.0049 г, 0,0079 ммоль) и Cs2CO3 (0,129 г, 0.3975 ммоль) в дегазированном толуоле (толуол продували N2 в течение 30 минут) нагревали в течение 16 часов при температуре 110°С в атмосфере N2. Реакционную смесь охлаждали, разбавляли EtOAc (20 мл) промывали водой (10 мл), солевым раствором (10 мл) и сушили над Na2SO4. Фильтровали и концентрировали при пониженном давлении с получением осадка, которые промывали гексаном с получением 3 (0,049 г, 50%) в форме коричневого твердого вещества.
[001250] Этап-2
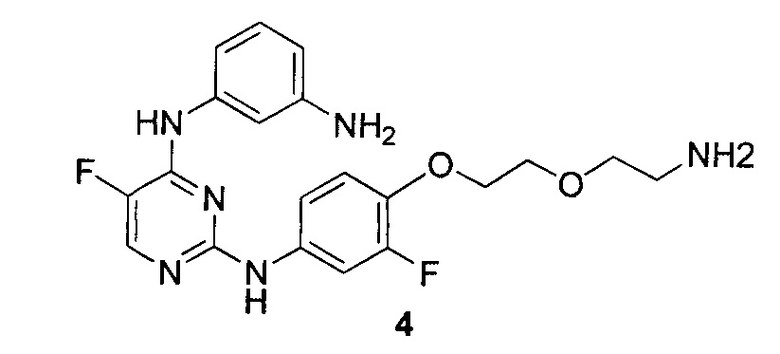
[001251] К раствору 3 (0.047 г, 0.0762 ммоль) в сухом CH2Cl2 (3 мл) при температуре 0°С при помешивании добавляли CF3COOH (1.0 мл) и реакционную смесь помешивали при температуре 0°С в течение 30 минут. Реакционную смесь оставляли дойти до до комнатной температуры, после чего ее помешивали в течение 1 часа. Затем концентрировали при пониженном давлении и тушили остаток раствором NaHCO3 (3 мл). Содержимое экстрагировали этил ацетатом (3×10 мл) и объединенный экстракт EtOAc промывали водой (10 мл), а затем 10% раствором лимонной кислоты (3×10 мл). Объединенный экстракт лимонной кислоты приводили к форме основания 10% раствором NaOH и экстрагировали EtOAc (3×25 мл). Экстракт EtOAc промывали водой (20 мл), солевым раствором (10 мл) и сушили над Na2SO4 с получением 4 (0.028 г, 88%) в форме светло-желтого твердого вещества.
[001252] Этап-3
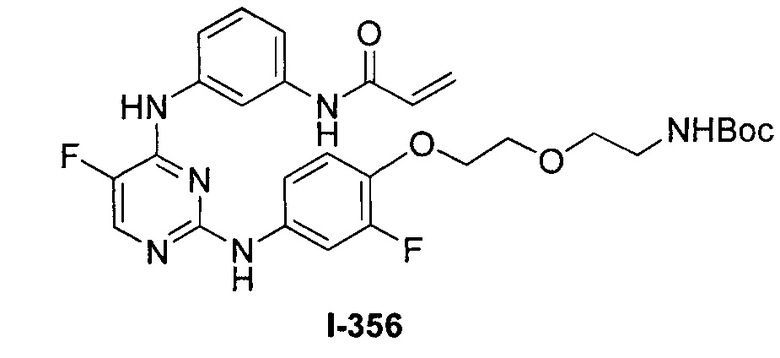
[001253] К раствору 4 (0,028 г, 0,06731 ммоль) в NMP (1,0 мл) при комнатной температуре при помешивании добавляли (Вос)2О (0,016 г, 0. 07404 ммоль), и помешивали реакционную смесь при комнатной температуре в течение 30 минут. Охлаждали до температуры 0°С и к ней добавляли K2CO3 (0.051 г, 0.372 ммоль) и акрилоил хлорид (0.0067 г, 0.07404 ммоль) и реакционную смесь помешивали при температуре 0°С в течение 30 минут. Реакционная смесь по каплям добавляли при помешивании в холодный 10% раствор NaHCO3. После добавления раствор помешивали в течение еще 30 минут при температуре 0°С, и отделяли твердый осадок с помощью воронки Бухнера. Полученное твердое вещество промывали холодной водой, гексаном и растворяли в смеси метанол: дихлорметан (50:50, 5 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной- воде (3 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×5 мл). Объединенный экстракт этил ацетата промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением указанного в названии соединения (0,016 г, 42%) в форме серого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.37 (s, 9Н), 3.09 (d, J=5.5 Гц, 2H), 3.43 (d, J=5.84 Гц, 2H), 3.69 (s, 2Н), 4.05 (s, 2Н), 5.75 (d, J=11.12 Гц, 1Н), 6.25 (d, J=16.76 Гц, 1H), 6.46 (dd, J=10.12 & 16.84 Гц, 1H), 6.81 (s, 1H), 6.96 (t, J=9.12 Гц, 1H), 7.24-7.31 (m, 2H), 7.43 (d, J=7.96 Гц, 1H), 7.49 (d, J=7.56 Гц, 1H), 7.68 (d, J=14 Гц, 1H), 7.94 (s, 1H), 8.11 (d, J=3.24 Гц, 1H), 9.21 (s, 1H), 9.46 (s, 1H), 10.15 (s, 1H); ЖХМС: m/e 571.
(М+l).
[001254] Промежуточное соединение 2 получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
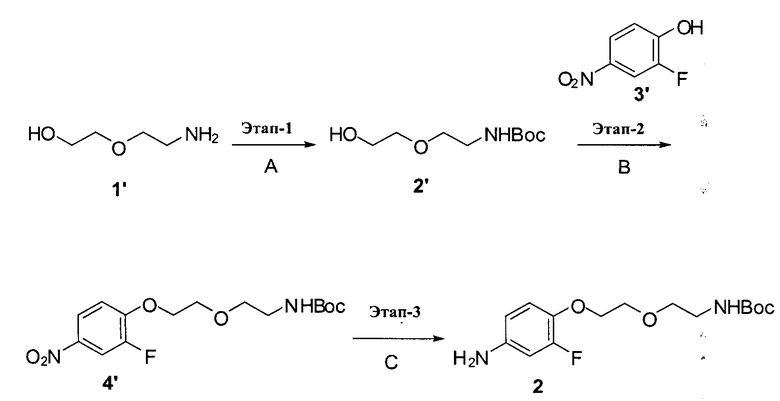
А) (Boc)2О, aq. NaOH, комн. т., 16 часов; В) DIAD, PPh3, Et3N, сухой ТГФ, комн. т., 1 час; С) Н2, Pd/C, этанол, комн. т., 16 часов.
[001255] Этап-1
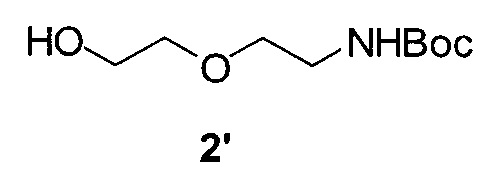
[001256] К раствору NaOH (0.76 г, 0.019 ммоль) в воде (9.6 мл) при комнатной температуре добавляли 1' (2.0 г, 19.022 ммоль) и реакционную смесь помешивали в течение 30 минут. К нему добавляли раствор Вос-ангидрида (4.561 г, 20.92 ммоль) в ТГФ (12.0 мл) по каплям добавляли в течение 5 минут. Реакционную смесь помешивали при комнатной температуре в течение 16 часов. Концентрировали при пониженном давлении, разбавляли водой (20 мл) и экстрагировали EtOAc (4×50 мл).: Объединенный экстракт EtOAc промывали водой (50 мл), солевым раствором (50 мл) и сушили над Na2SO4 с получением 2' (3,2 г, 82%) в форме вязкого масла.
[001257] Этап-2
[001258] К раствору 2' (0,38 г, 1,851 ммоль) в ТГФ (6 мл) добавляли при помешивании 3' (0,29 г, 1,851 ммоль), PPh3 (0,534 г, 2,0361 ммоль) и Et3N (0,280 г, 2,776 ммоль) а атмосфере N2. Реакционную смесь охлаждали до 0°С и добавляли к ней DIAD
(0,411 г, 2,0361 ммоль). Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали 1 час. Затем тушили водой, экстрагировали этил ацетатом (3×5 мл) и промывали объединенный экстракт этил ацетата водой и солевым раствором (по 5 мл каждого). Полученный осадок концентрировали при пониженном давлении и очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 9/1) с получением 4' (0,360 г сырого) в форме желтого твердого вещества.
[001259] Этап-3
[001260] К раствору 4' (0.360 г, 1.0456 ммоль) в этаноле (10 мл) добавляли Pd/C (0.072 г, 20% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (давление водорода 1,5 Кг) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 2 (0,28 г, 85%) в форме коричневатого вязкого масла. Его использовали на следующем этапе без дальнейшей очистки.
ПРИМЕР 246
[001261] Получение N1-(2-(2-(4-(4-(3-акриламидофениламино)-5-фторпиримидин-2-иламино)-2-фторфенокси)этокси)этил)-N5-(15-оксо-18-((3aR,4R,6aS)-2-оксогексагидро-1H-тиено[3,4-d]имидазол-4-ил)-4,7,10-триокса-14-азаостадецил)глутарамид I-362
[001262] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений описанных в Примере 204, с использованием трет-бутил 2-(2-(4-(4-(3-акриламидофениламино)-5-фторпиримидин-2-иламино)-2-фторфенокси)этокси)этилкарбамата (I-356, описано в примере 245) вместо I-45 на этапе 1.
1Н-ЯМР (ДМСО-d6) δ ppm: 10.1 (s, 1H), 9.87 (s, 1H), 9.52 (s, 1H), 8.09 (d, J=4.1 Гц, 1H), 7.86 (s, 1H), 7.78 (t, J=5.5 Гц, 1H), 7.66 (m, 3H), 7.48 (dd, J=2.3 & 13.8 Гц, 1H), 7.33 (m, 2H), 7.21 (t, J=7.8 Гц, 1H), 7.11 (d, J=9.2 Гц, 1H), 6.90 (t, J=9.2 Гц, 1H), 6.34 (m, 2H), 6.14 (dd, J=2.3 & 17.0 Гц, 1H), 5.66 (dd, J=2.3 & 17.0 Гц, 1H), 4.20 (dd, J=5.0 & 7.3 Гц, 1H), 3.99 (m, 3H), 3.61 (m, 2H), 3.12 (q, J=6.0 Гц, 2H), 2.97 (m, 9H), 2.72 (m, 2H), 2.46 (m, 2H), 1.95 (m, 9H), 1.1-1.6 (m, 18H); ЖХМС: m/e 1013. (M+l).
ПРИМЕР 247
[001263] Получение N-(3-(5-фтор-2-(3-фтор-4-(2-(2-метоксиэтокси)этокси)-фениламино)пиримидин-4-иламино)фенил)акриламида I-359
[001264] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
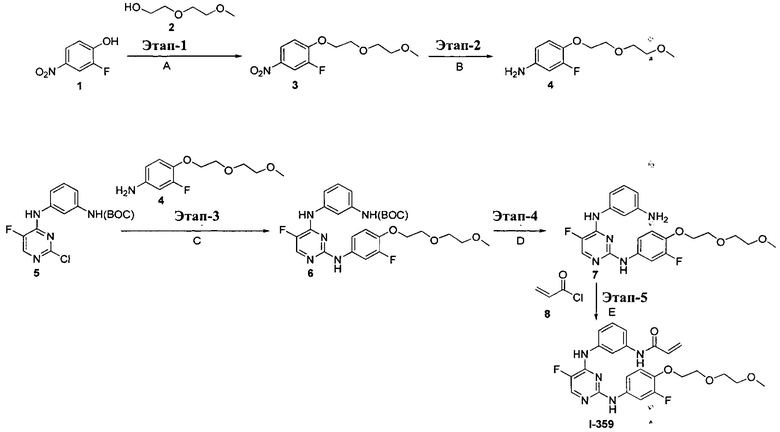
A) DIAD, PPh3, Et3N, сухой ТГФ, комн. т., 1 час; В) Н2, Pd/C, метанол, комн. т., 16 часов; С) Pd(OAc)2, BINAP, Cs2CO3 толуол, 110°С, 6 часов; D) ТФА, CH2Cl2, комн. т., 1 час; Е) (ВОС)2O, 30 минут, затем K2CO3, NMP, 0°С, 15 минут.
[001265] Этап-1
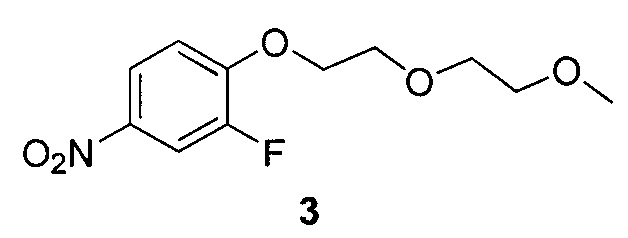
[001266] К раствору 1 (0,5 г, 3,18 ммоль) в ТГФ (10 мл) добавляли при помешивании 2 (0,38 г, 3,18 ммоль), PPh3 (0,91 г, 3,498 ммоль) и Et3N (0,48 г, 4,776 ммоль) а атмосфере N2. Реакционную смесь охлаждали до 0°С и добавляли к ней DIAD (0,707 г, 3,5 ммоль). Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали 1 час. Затем тушили водой, экстрагировали этил ацетатом (3×5 мл) и промывали объединенный экстракт этил ацетата водой и солевым раствором (по 5 мл каждого). Полученный осадок концентрировали при пониженном давлении и очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 7/3) с получением 3 (0,61 г, 65%) в форме белого твердого вещества.
[001267] Этап-2
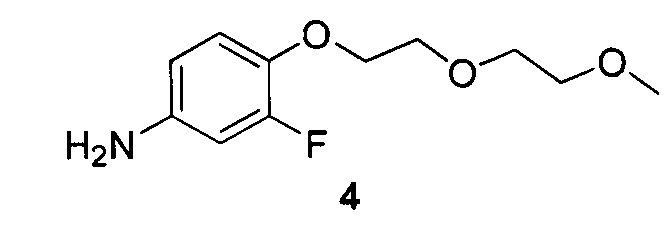
[001268] К раствору 3 (0.6 г, 2,31 ммоль) в этаноле (20 мл) добавляли Pd/C (0.060 г, 10% масс/масс.), после чего реакционную смесь оставляли при перемешивании в атмосфере Н2 (давление в пневмокамере) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении с получением 4 (0,375 г, 70,7%) в форме коричневатого вязкого масла.
[001269] Этап-3
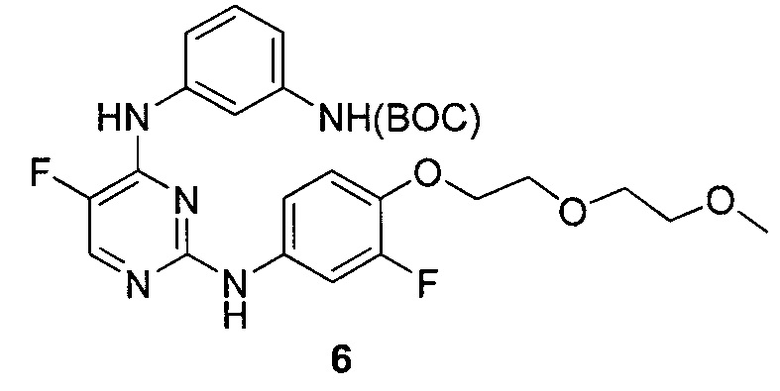
[001270] Раствор 4 (0,275 г, 1,19 ммоль), 5 (0,403 г, 1,19 ммоль), приготовленный как указано в Примере 20, Pd(OAc)2 (0,0026 г, 0,11 ммоль), BINAP (0.0037 г, 0,059 ммоль) и Cs2CO3 (0,969 г, 2,95 ммоль) в дегазированном толуоле (толуол продували N2 в течение 30 минут) нагревали в течение 16 часов при температуре 100°С в атмосфере N2. Реакционную смесь охлаждали, разбавляли EtOAc (20 мл) промывали водой (10 мл), солевым раствором (10 мл) и сушили над Na2SO4. Фильтровали, и затем концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 5/5) с получением 6 (0,350 г, 55%) в форме желтого твердого вещества.
[001271] Этап-4
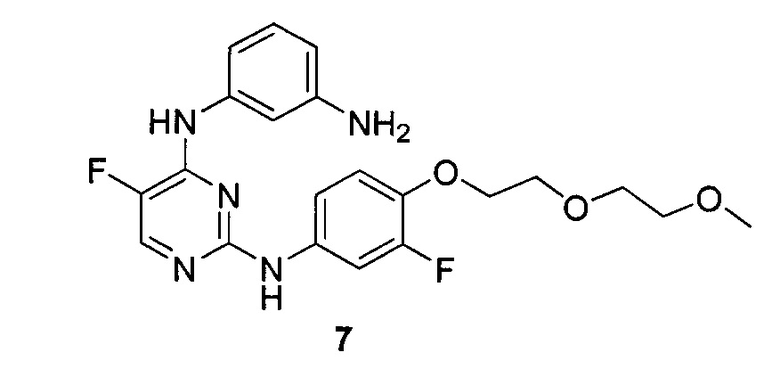
[001272] К раствору 6 (0.3 г, 0.56 ммоль) в сухом CH2Cl2 (3 мл) при температуре 0°С при помешивании добавляли CF3COOH (1.0 мл) и реакционную смесь помешивали при температуре 0°С в течение 30 минут. Реакционную смесь оставляли дойти до комнатной температуры, после чего ее помешивали 1 час. Затем концентрировали при пониженном давлении, гасили раствором NaHCO3 (3 мл) и экстрагировали CH2Cl2 (3×25 мл). Экстракт EtOAc промывали водой (20 мл), солевым раствором (10 мл) и сушили над Na2SO4 с получением 7 (0.15 г, 62,5%) в форме коричневатой вязкой жидкости.
[001273] Этап-5
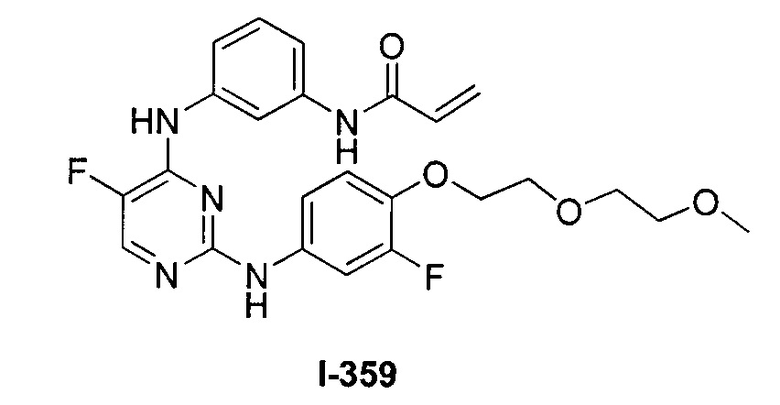
[001274] К охлажденному раствору 7 (0.1 г, 0.23 ммоль) в NMP (1.0 мл) при температуре примерно 0°С добавляли K2CO3 (0.15 г, 1.1 ммоль), акрилоил хлорид (0.0022 г, 0.25 ммоль) и реакционную смесь помешивали при температуре 0°С в течение 30 минут. Реакционная смесь по каплям добавляли при помешивании в холодный 10% раствор NaHCO3. После добавления, раствор помешивали в течение еще 30 минут при температуре 0°С, и отделяли твердый осадок с помощью воронки Бухнера. Полученное твердое вещество промывали холодной водой, гексаном и растворяли в смеси метанол: дихлорметан (50:50, 25 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (3 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×5 мл). Объединенный экстракт этил ацетата промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при
пониженном давлении с получением указанного в названии соединения (0,055 г, 50%) в форме желтого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 3.24 (s, 3Н), 3.44 (t, J=4.88 Гц, 2Н), 3.57 (t, J=4.04 Гц, 2Н), 3.69 (t, J=4.24 Гц, 2Н), 4.04 (t, J=4.04 Гц, 2Н), 5.73 (d, J=10.12 Гц, 1Н), 6.23 (d, J=16.8 Гц, 1H), 6.45 (dd, J=10.12 & 16.92 Гц, 1H), 6.95 (t, J=9.4 Гц, 1H), 7.28-7.30 (m, 2H), 7.42 (d, J=8.04 Гц, 1H), 7.48 (d, J=7.48 Гц, 1H), 7.67 (d, J=14.36 Гц, 1H), 7.93 (s, 1H), 8.10 (d, J=3.44 Гц, 1H), 9.20 (s, 1H), 9.44 (s, 1H),. 10.14 (s, 1H); ЖХМС: m/e 486.1 (M+l).
ПРИМЕР 248
[001275] Получение (S)-N-(3-(2-(4-хлоро-3-(1-гидрокипропан-2-илокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-357
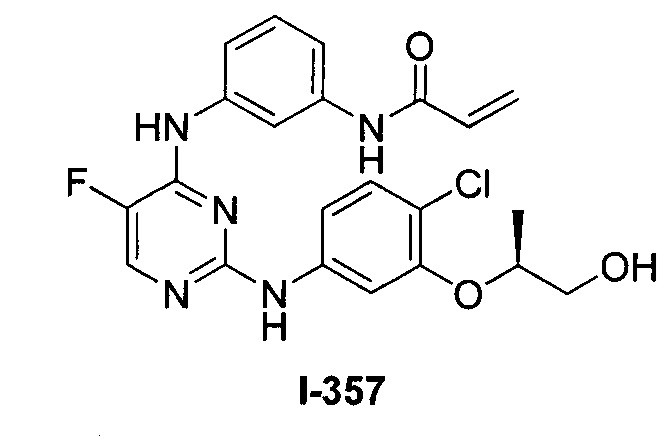
[001276] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных ниже.
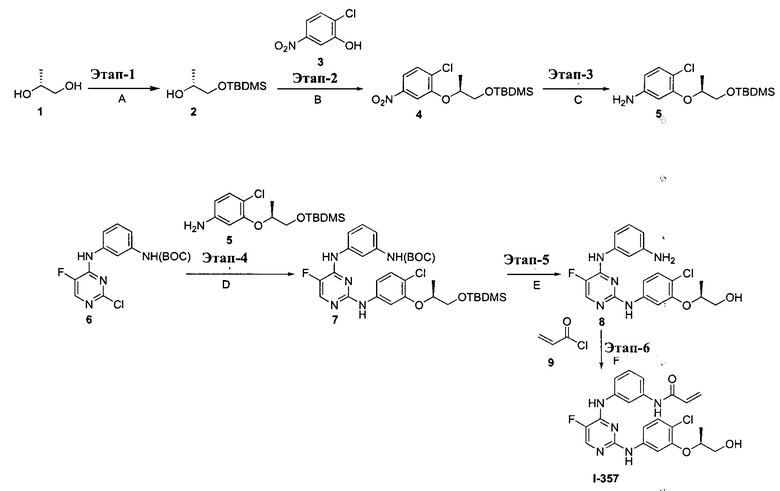
A) TBDMSCl, имидазоле, CH2Cl2, 0°C, 2 часа; В) DIAD, PPh3, Et3N, сухой ТГФ, комн. т., 1 час; С) Н2, Никель Ренея, МеОН, 2 часа; D) Pd(OAc)2, BINAP, Cs2CO3, толуол, 110°С, 6 часов; Е) ТФА, CH2Cl2, комн. т., 1 час; Г) (ВОС)2O, 30 минут, затем K2CO3, NMP, 0°С, 15 минут.
[001277] Этап-1
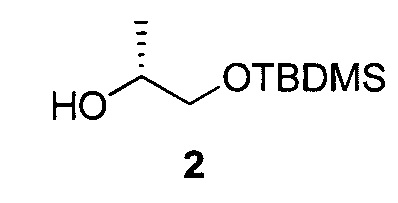
[001278] К раствору 1 (1 г, 13.1 ммоль) в ДХМ при помешивании добавляли при температуре 0°С имидазол (0.875 г, 13.1 ммоль) и трет-бутилдиметилсилил хлорид (1.98 г, 13.1 ммоль). Ту же температуру поддерживали в течение 2 часа, после чего реакционную смесь охлаждали и концентрировали. Остаток очищали методом колоночной хроматографии (нейтральный оксид алюминия, петролейный эфир/этил ацетат 7/3) с получением 2 (1.4 г, 56%) в форме бесцветной жидкости.
[001279] Этап-2
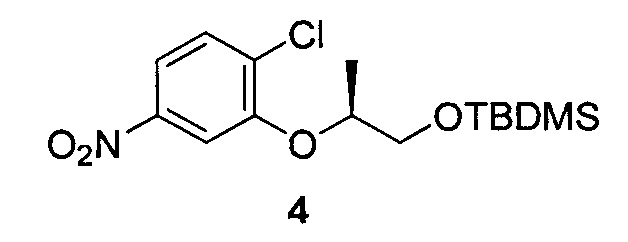
[001280] К раствору 7,89 (1,5 г, 3 ммоль) в ТГФ (15 мл) добавляли при помешивании 2 (1,36 г, 3,18 ммоль), PPh3 (2,27 г, 8,6 ммоль) и Et3N (1,19 г, 11,1 ммоль) а атмосфере N2. Реакционную смесь охлаждали до 0°С и добавляли к ней DIAD (1,75 г, 8,6 ммоль). Реакционную смесь оставляли дойти до комнатной температуры, после чего помешивали 1 час. Затем тушили водой, экстрагировали этил ацетатом (3×5 мл) и промывали объединенный экстракт этил ацетата водой и солевым раствором (по 5 мл каждого). Полученный осадок концентрировали при пониженном давлении и очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 7/3) с получением 4 (2,1 g 76,9%) в форме желтого масла.
[001281] Этап-3
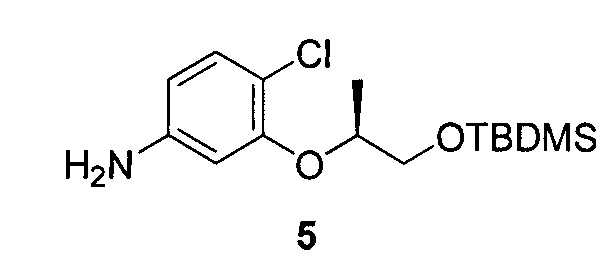
[001282] К раствору 4 (2 г, 5.7 ммоль) в метаноле (20 мл)) добавляли Никель Ренея (3 г). Реакционную смесь оставляли для перемешивания в атмосфере H2 (давление в пневмокамере) при комнатной температуре в течение 2 часа. Реакционную смесь фильтровали через целитную® пластину и концентрировали при пониженном давлении, после чего остаток очищали методом колоночной хроматографии (нейтральный оксид алюминия, петролейный эфир/ этил ацетат, 8/2) с получением 5 (1.4 г, 77%) в форме коричневатого вязкого масла.
[001283] Этап-4
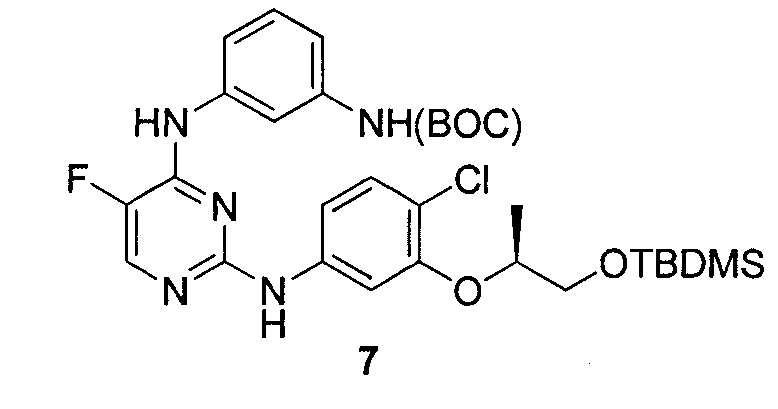
[001284] Раствор 6 (0,2 г, 0,63 ммоль), 5 (0,403 г, 1,19 ммоль), приготовленный как указано в Примере 20, Pd(OAc)2 (0,014 г, 0,063 ммоль), BINAP (0,0019 г, 0,031 ммоль) и Cs2CO3 (0,511 г, 1,5 ммоль) в дегазированном толуоле (толуол продували N2 в течение 30 минут) нагревали в течение 16 часов при температуре 100°С в атмосфере N2. Реакционную смесь охлаждали, разбавляли EtOAc (20 мл) промывали водой (10 мл), солевым раствором (10 мл) и сушили над Na2SO4. Фильтровали, и затем концентрировали при пониженном давлении с получением осадка, который затем очищали методом колоночной хроматографии (SiO2, 60-120, петролейный эфир/этил ацетат, 7/3) с получением 7 (0,15 г, 38,4%) в форме желтого твердого вещества.
[001285] Этап-5
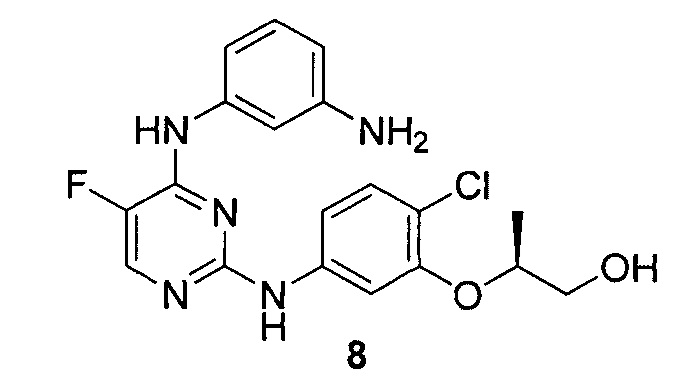
[001286] К раствору 7 (0,15 г, 0,24 ммоль) в сухом CH2Cl2 (5 мл) при температуре 0°С при помешивании добавляли CF3COOH (1,5 мл) и реакционную смесь помешивали при температуре 0°С в течение 30 минут. Реакционную смесь оставляли дойти до комнатной температуры, после чего ее помешивали 1 час. Затем концентрировали при пониженном давлении, гасили раствором NaHCO3 (3 мл) и экстрагировали CH2Cl2 (3×25 мл). Объединенный экстракт EtOAc промывали водой (20 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 8 (0,085 г, 86,7%) в форме белого твердого вещества.
[001287] Этап-6
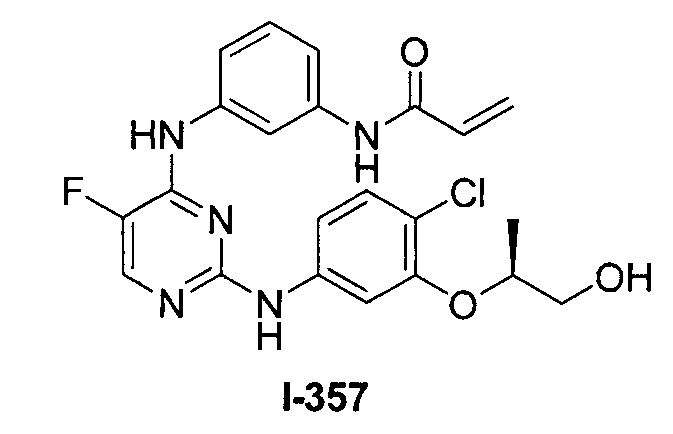
[001288] Раствор 8 (0.085 г, 0.21 ммоль) в NMP (2.0 мл) охлаждали до температуры 0°С и добавляли к нему K2CO3 (0.29 г, 2.1 ммоль) и акрилоил хлорид (1 М раствор в ТГФ,
0.21 мл, 0.21 ммоль) и реакционную смесь помешивали при температуре 0°С в течение 30 минут. Реакционная смесь по каплям добавляли при помешивании в холодный 10% раствор NaHCO3. После добавления, раствор помешивали в течение еще 30 минут при температуре 0°С, и отделяли твердый осадок с помощью воронки Бухнера. Полученное твердое вещество промывали холодной водой, гексаном и растворяли в смеси метанол: дихлорметан (50:50, 25 мл) и концентрировали при пониженном давлении. Полученный осадок суспендировали в холодной воде (3 мл), добавляли к нему Et3N и затем экстрагировали этил ацетатом (2×5 мл). Объединенный экстракт этил ацетата промывали водой (5 мл), солевым раствором (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением указанного в названии соединения (65 мг, 67%) в форме желтого твердого вещества. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.18 (d, J=6.12 Гц, 3Н), 3.40-3.47 (m, 1Н), 3.50-3.56 (m, 1H), 4.20-4.30 (m, 1Н), 4.82 (t, J=5.6 Гц, 1Н), 5.75 (dd, J=1.88 & 10.08 Гц, 1H), 6.25 (dd, J=1.92 & 16.92 Гц, 1Н), 6.45 (dd, J=10.08 & 16.92 Гц, 1Н), 7.12 (d, J=8.76 Гц, 1Н), 7.29 (t, J=8.08 Гц, 1H), 7.40-7.44 (m, 3Н), 7.52 (d, J=8.44 Гц, 1Н), 7.91 (s, 1Н), 8.12 (d, J=3.64 Гц, 1Н), 9.21 (s, 1Н), 9.45 (s, 1Н), 10.12 (s, 1H); ЖХМС: т/е 458.0 (М+l).
ПРИМЕР 249
[001289] Получение (R)-N-(3-(2-(4-хлоро-3-(1-гидрокипропан-2-илокси)фениламино)-5-фторпиримидин-4-иламино)фенил)акриламида I-35
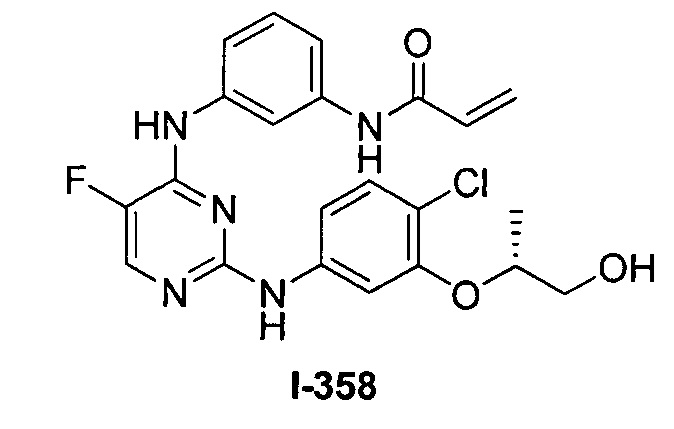
[001290] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 248, с использованием (R)-пропан-1,2-диола вместо 1 на этапе-1. 1Н-ЯМР (ДМСО-d6) δ ppm: 1.18 (d, J=6.12 Гц, 3Н), 3.40-3.47 (m, 1H), 3.50-3.56 (m, 1H), 4.20-4.30 (m, 1H), 4.82 (t, J=5.6 Гц, 1H), 5.75 (dd, J=1.88 & 10.08 Гц, 1Н), 6.25 (dd, J=1.92 & 16.92 Гц, 1Н), 6.45 (dd, J=10.08 & 16.92 Гц, 1H), 7.12 (d, J=8.76 Гц, 1H), 7.29 (t, J=8.08 Гц, 1Н), 7.40-7.44 (m, 3Н), 7.52 (d, J=8.44 Гц, 1H),
7.91 (s, 1H), 8.12 (d, J=3.64 Гц, 1H), 9.21 (s, 1H), 9.45 (s, 1H), 10.12 (s, 1H); ЖХМС: m/e 458.0 (M+l).
ПРИМЕР 250
[001291] Получение (Е)-4-(диметиламино)-N-(5-метил-4-(m-толиламино)пиримидин-2-иламино)фенил)бут-2-енамида I-360
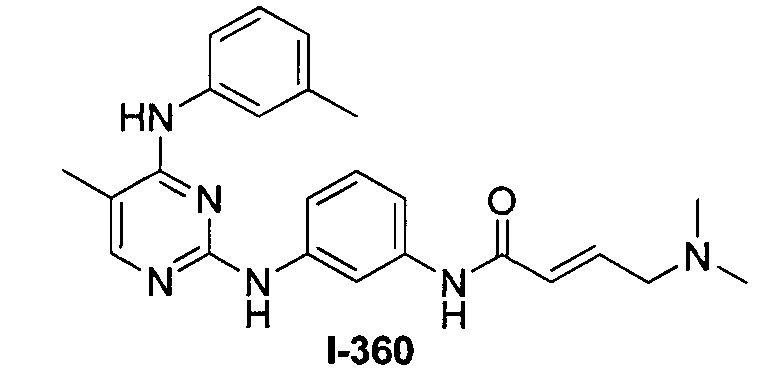
[001292] Указанное в названии соединение получали с использованием схем, этапов и промежуточных соединений, описанных в Примере 3, с использованием (Е)-4-(диметиламино)бут-2-еноил хлорида вместо акрилоил хлорида на этапе-3. 1Н-ЯМР (ДМСО-d6) δ ppm: 7.91 (s, 1Н), 7.85 (s, 1Н), 7.52 (d, J=6.4 Гц, 1Н), 7.45 (d, J=7.8 Гц, 1H), 7.34 (s, 1Н), 7.31-7.26 (m, 1Н), 7.21 (dd, J=8.2, 8.0 Гц, 1Н), 7.01-6.92 (m, 4Н); 6.27 (s, 1Н), 6.06 (d, J=15.1 Гц, 1H), 3.14 (d, J=5.5 Гц, 2Н), 2.37 (s, 3Н), 2.31 (s, 6Н), 2.13 (s, 3Н); ЖХМС m/z 417 (М+l).
Биологические примеры
[001293] Ниже описаны тесты, использованные для измерения биологической активности предложенных соединений в качестве ингибиторов f BTK, TEC, ITK, ВМХ, ErbB1 (EGFR), ErbB2, ErbB4 и JAK3.
ПРИМЕР 251
Общий протокол анализа эффективности против BTK
[001294] Ниже описаны протокол с использованием EGFR-WT и EGFR-T790M/L858R и затем в условиях с оптимизацией реагентов для BTK.
[001295] Принцип действия платформу теста лучше всего описан на веб-сатйе поставщика (Invitrogen, Карлсбад, Калифорния, США): URL: www.invitrogen.com/content.cfm?pageid=11338 или www.invitrogen.com/site/us/en/home/Products-and-Services/Applications/Drug-
Discovery/Target-and-Lead-Identification-and-Validation/KinaseBiology/KB-Misc/Biochemical-Assays/Omnia-Kinase-Assays.html.
[001296] Вкратце, готовили 10X маточные растворы EGFR-WT (PV3872), полученный из Invitrogen, и EGFR-T790M/L858R (40350), полученный из BPS Bioscience, Сан-Диего, Калифорния, 1.13Х ATP (AS001A), и соответствующих пептидных субстратов, конъюгированных с Tyr-Sox (KCZ1001) в 1X буфере для реакций с киназами, состоящем из 20 мМ Tris, рН 7.5, 5 мМ MgCl2, 1 мМ EGTA, 5 мМ β-глицерофосфата, 5% глицерина (10Х маточный раствор, KB002A) и 0.2 мМ DTT (DS001A). 5, мкл каждого субстрата предынкубировали в 384-луночом белом микротитрационном планшете с несвязывающей поверхностью Corning (#3574) (Corning, NY) в течение 30 мин. при 27°С с 0.5 мкл 50% ДМСО и серийными разбавлениями соединений, приготовленными в 50% ДМСО. Киназные реакции инициировали путем добавления 45 мкл смеси АТФ/Tyr-Sox-пептидный субстрат и мониторировали каждые 30-90 секунд в течение 60 минут при λex360/λem485 в считывающем устройстве для планшетов Synergy4 производства BioTek (Винуски, Вермонт, США). В заключение каждого теста кривые процесса в каждой лунке анализировали согласно линейной кинетике реакции и с использованием статистической аппроксимации (R2, 95% доверительный интервал, модуль суммы квадратов). Начальную скорость (0 минут до ~30 минут) для каждой реакционной смеси определяли по углу наклона графика относительных единиц флюоресценции от времени (минуты) и затем строили график зависимости начальной скорости от концентрации ингибитора, по которому оценивали IC50 от log [Ингибитора] vs ответ, модель Variable Slope (переменный угол наклона) в программе GraphPad Prism от GraphPad Software (Сан-Диего, Калифорния, США).
[001297] Условия с оптимизацией реагентов для BTK приведены ниже:
[BTK] = 5 нМ, [АТФ] = 40 мМ, [Y5-Sox] = 10 мМ (АТФ KMapp ~ 36 мМ).
ПРИМЕР 252
[001298] В Таблице 6 показана активность отобранных соединений согласно настоящему изобретению в тесте на ингибирование ВТК. Номера соединений соответствуют номерам соединений в Таблице 5. Соединения, активность которых обозначена "А", продемонстрировали IC50 ≤10 нМ; соединения, активность которых
обозначена "В", продемонстрировали IC50 10-100 нМ; соединения, активность которых обозначена "С", продемонстрировали IC50 100-1000 нМ; соединения, активность которых обозначена "D", продемонстрировали IC50 1000-10,000 нМ; и соединения, активность которых обозначена "Е", продемонстрировали IC50 ≥10,000 нМ. Таблица 6. Данные по ингибированию BTK
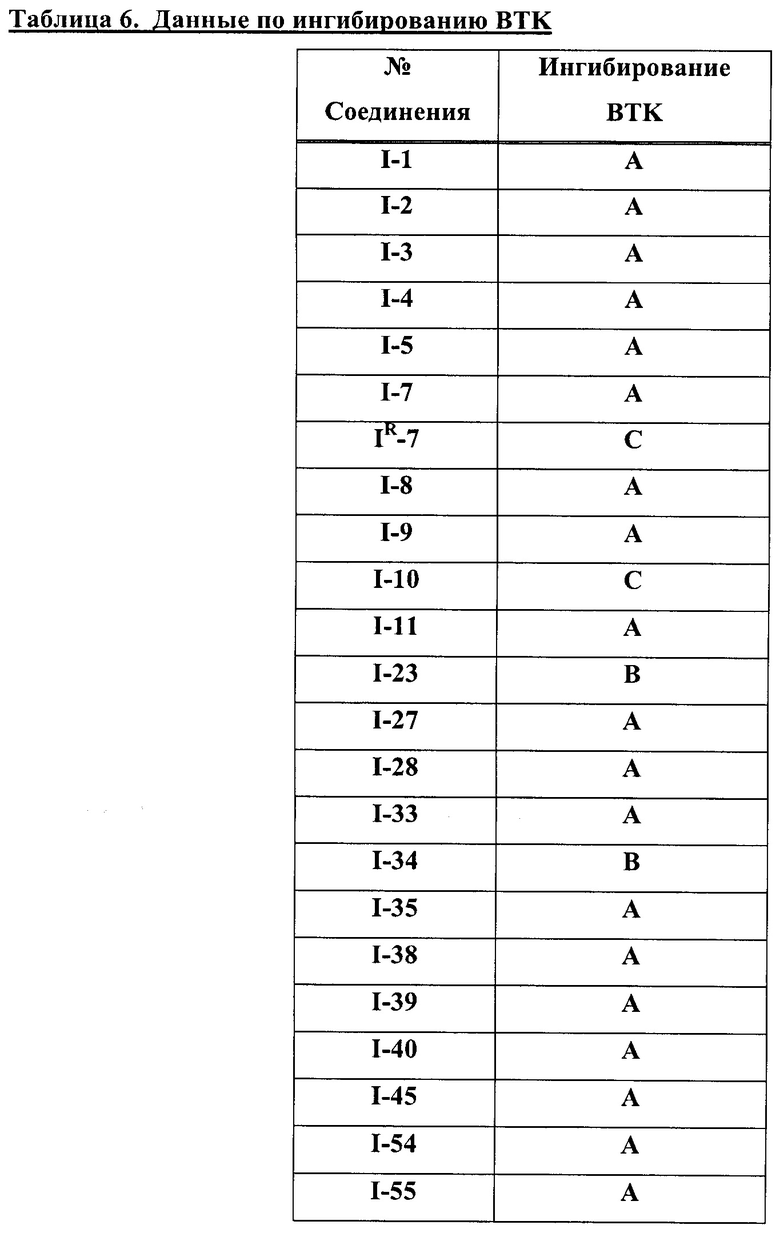


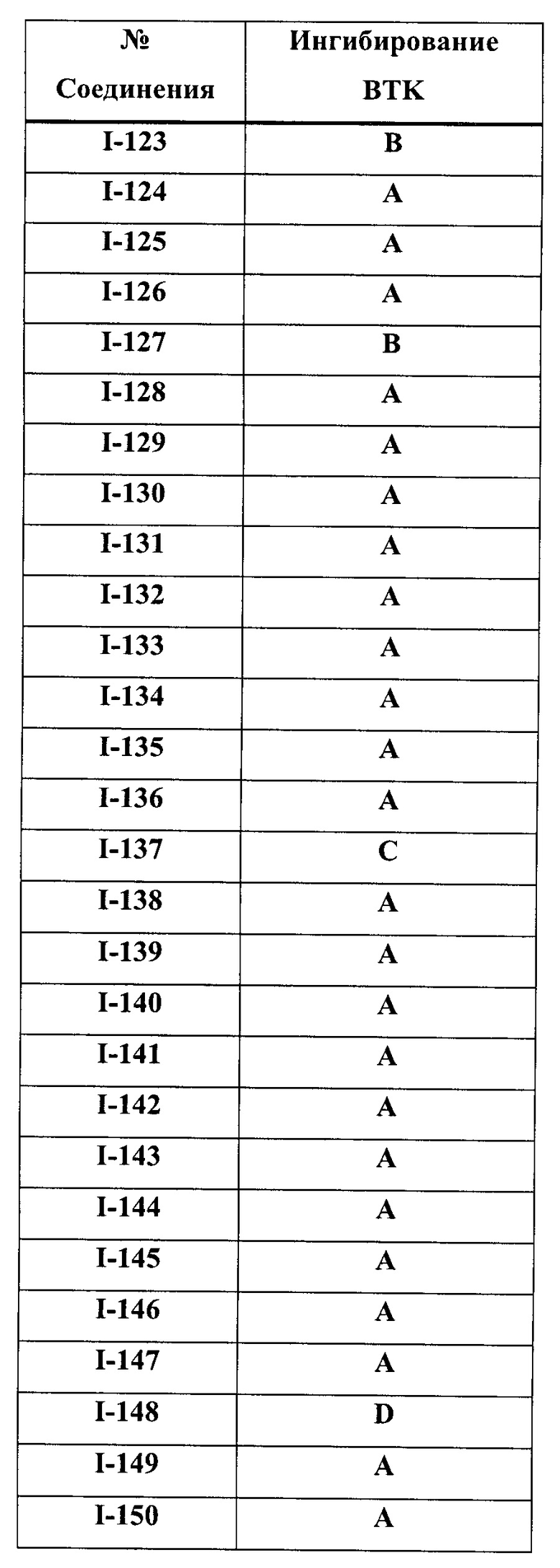

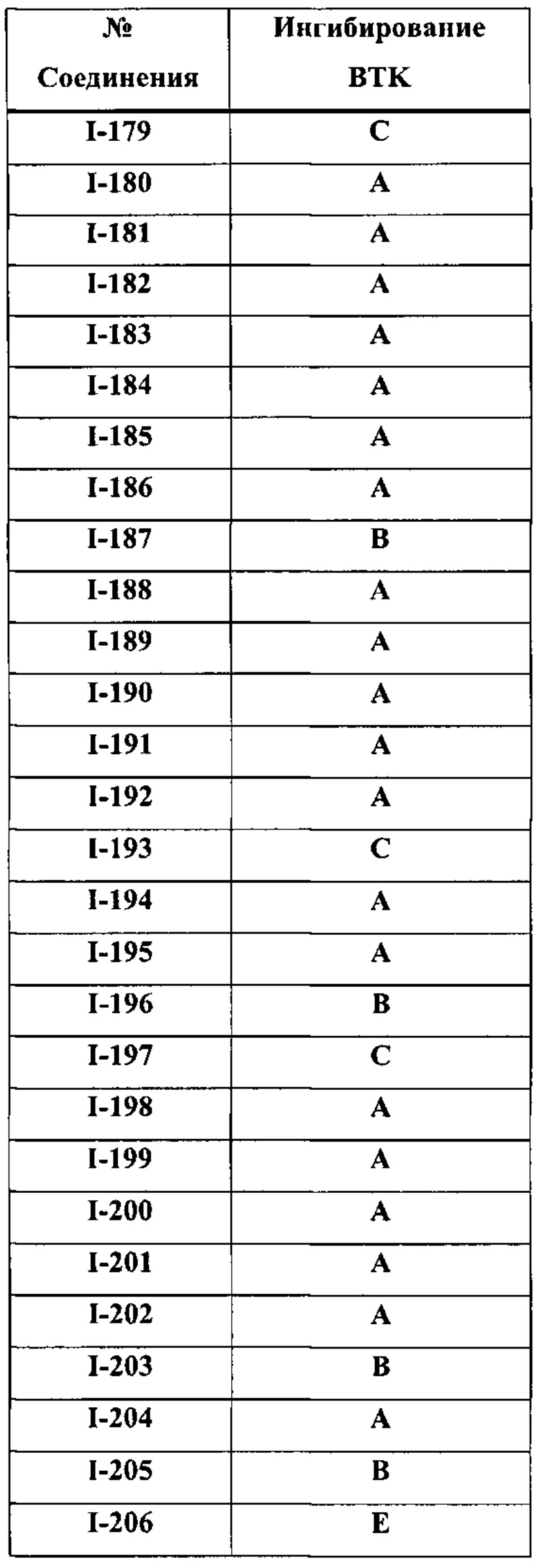


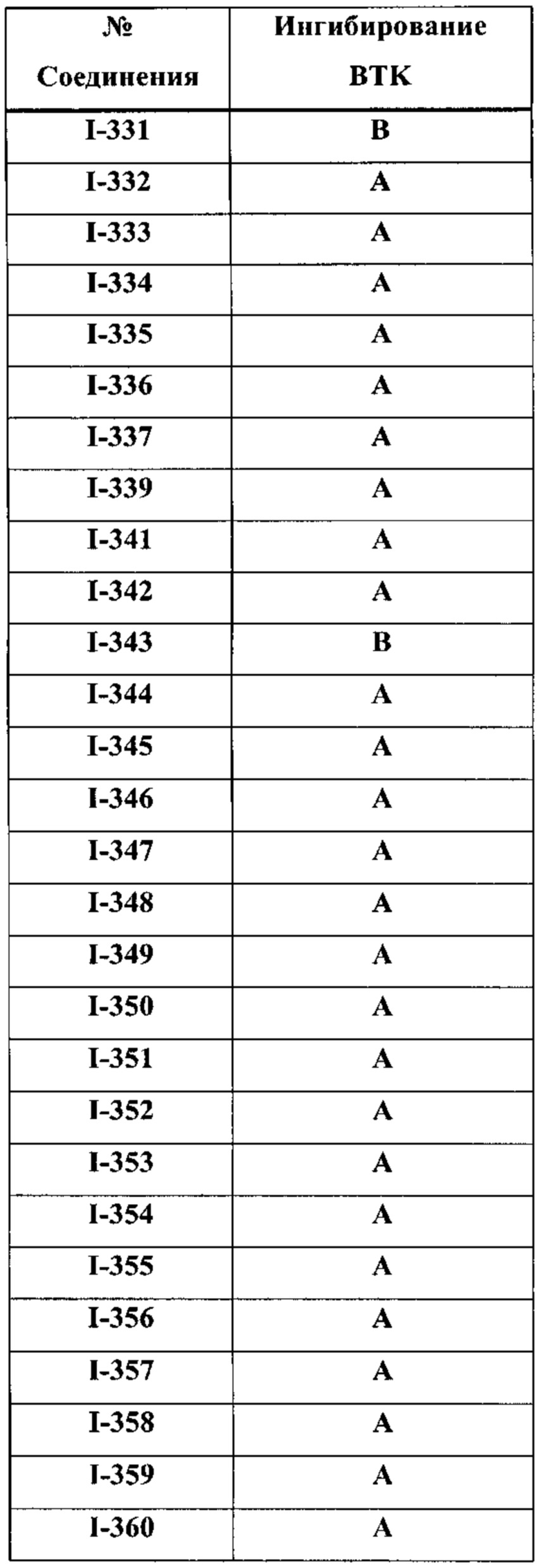
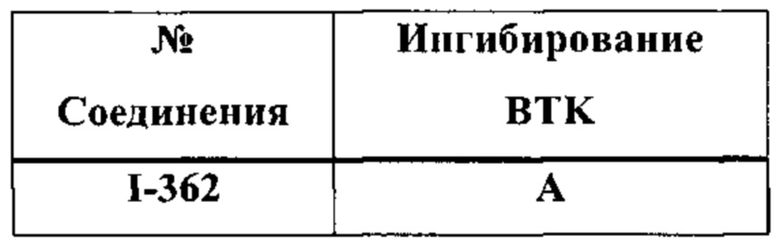
ПРИМЕР 253
Тест ВТК с клетками Ramos
[001299] Соединения I-2, I-4 и I-7 тестировали на человеческих клетках лимфомы Беркитта Ramos. Клетки Ramos выращивали в суспензии во флаконах Т225, центрифугировали, ресуспендировали в 50 мл бессывороточной среды и инкубировали в течение 1 часа. Соединения добавляли к клеткам Ramos в бессывороточной среде до конечной концентрации 1, 0.1, 0.01, или 0.001 мкМ. Клетки Ramos инкубировали с соединением в течение 1 часа, промывали еще раз и ресуспендировали в 100 мкл бессывороточной среды. Затем клетки стимулировали 1 мкг F(ab')2 козы к IgM человека и инкубировали на льду в течение 10 минут, чтобы активировать рецепторные сигнальные пути В-лимфоцитов. Через 10 минут, клетки однократно промывали ФБР и затем лизировали на льду буфером для экстракции клеток производства Invitrogen Cell Extraction. 16 мкг общего белкам из лизатов загружали на гель и блоты тестировали на фосфорилирование субстрата ВТК - PLCγ2. Дозозависимое ингибирование передачи сигнала ВТК в клетках Ramos представлено на Фигурах 1, 2, 3, 4 и 5.
[001300] В Таблице 7 показана активность примеров соединений согласно настоящему изобретению в тесте на ингибирование ВТК в клетках Ramos. Номера соединений соответствуют номерам соединений в Таблице 5. Соединения, активность которых обозначена "А", продемонстрировали IC50≤10 нМ; соединения, активность которых обозначена "В", продемонстрировали IC50 10-100 нМ; соединения, активность которых обозначена "С", продемонстрировали IC50 100-1000 нМ; соединения, активность которых обозначена "D", продемонстрировали IC50 1000-10,000 нМ; и соединения, активность которых обозначена "Е", продемонстрировали IC50≥10,000 нМ.
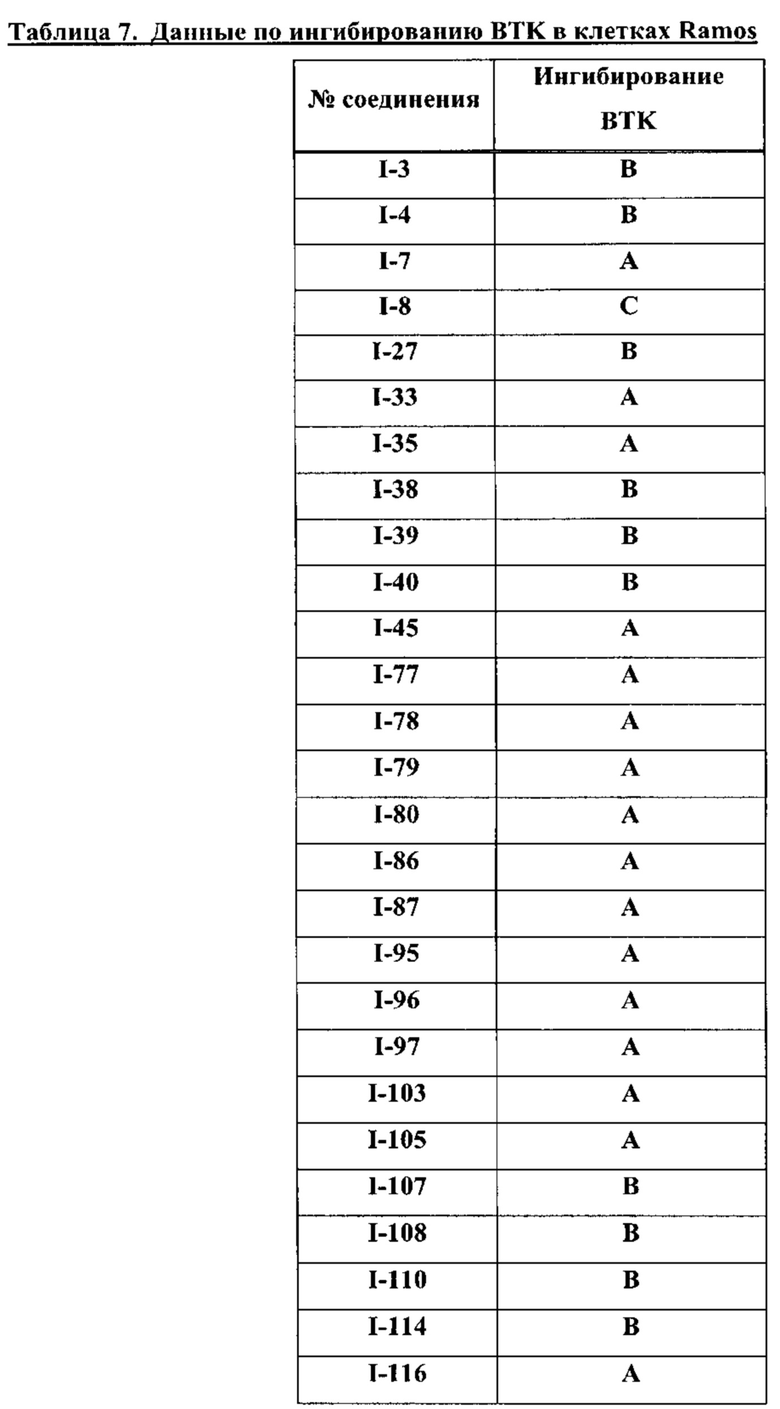
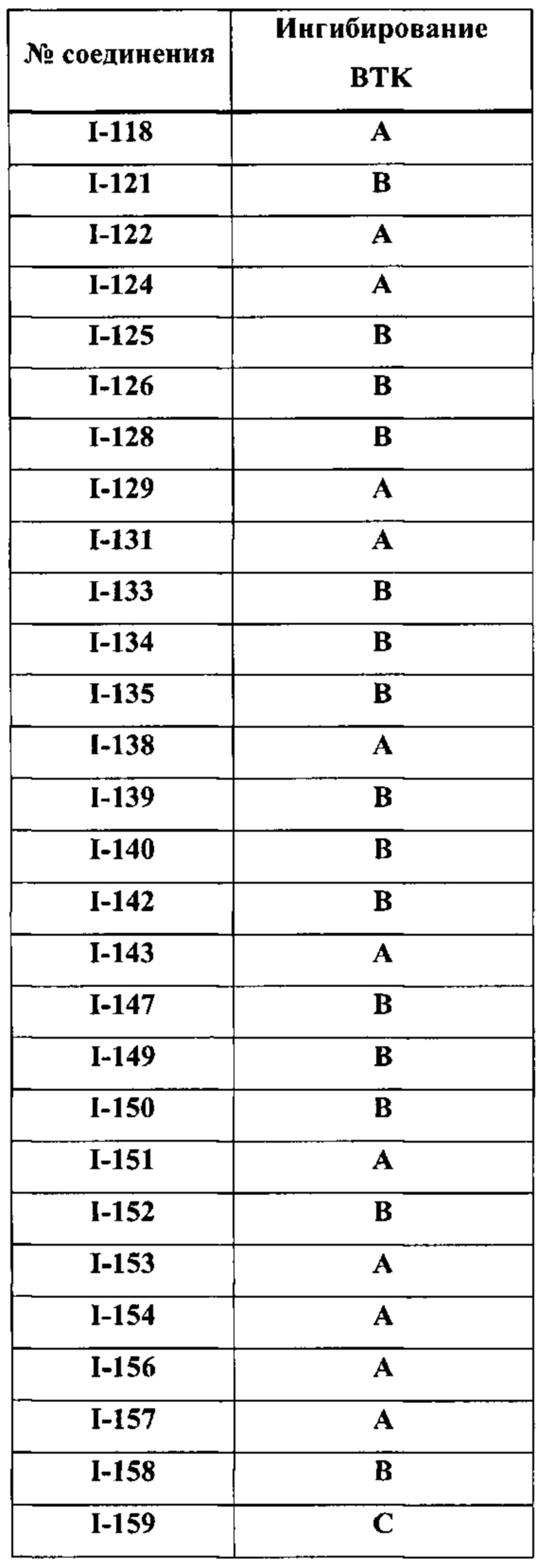
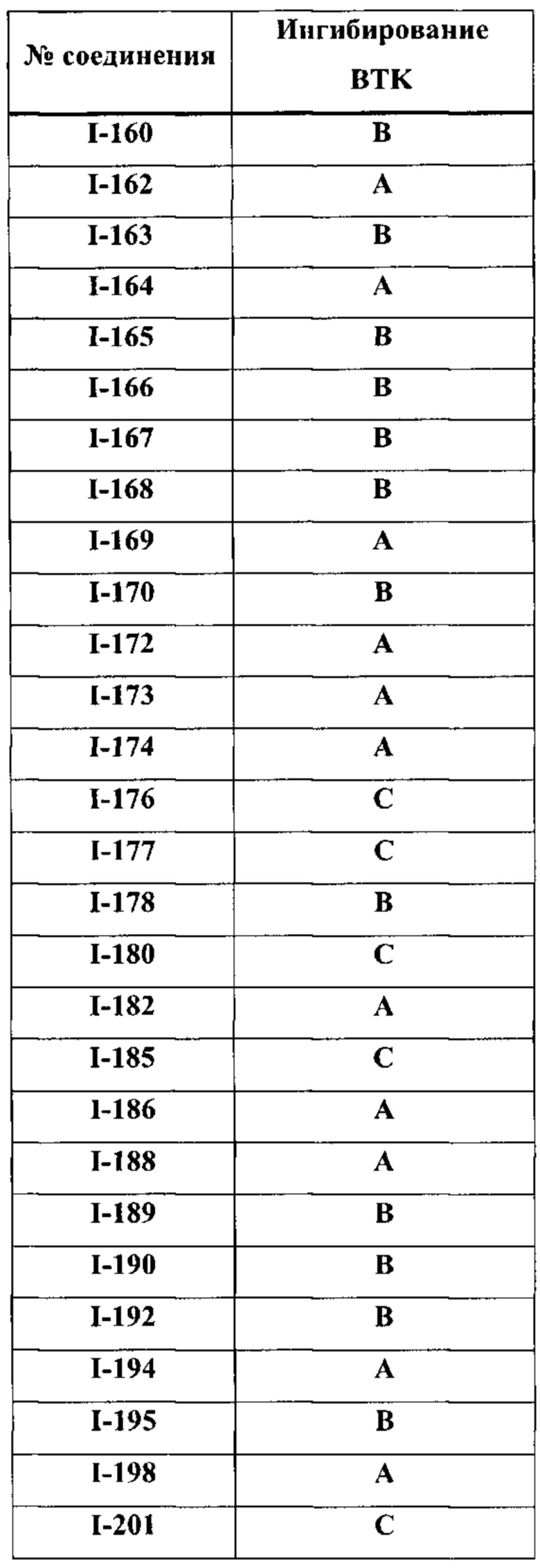

ПРИМЕР 254 Эксперимент с отмывкой на клетках Ramos
[001301] Клетки Ramos выдерживали без сыворотки в течение одного часа в среде RPMI+1% глютамин при 37°С. После этого клетки Ramos обрабатывали 100 нМ соединения, разбавленного в бессывороточной среде RPMI в течение 1 часа. После обработки соединением среду удаляли и клетки промывали средой без соединения. Затем клетки Ramos промывали каждые 2 часа и ресуспендировали в свежей среде без соединения. Клетки собирали в указанные моменты времени с использованием 1 мкг IgM к антителам человека (Southern Biotech кат. №2022-01) в течение 10 минут на льду, чтобы стимулировать передачу сигнала с участием BCR, а затем промывали в ФБР. Затем клетки Ramos лизировали в буфере Cell Extraction Buffer (Invitrogen FNN0011), добавляли таблетки полного ингибитора протеаз Roche (Roche 11697498001) и ингибиторы фосфатаз (Roche 04 906 837 001), и загружали по 18 мкг полного белкового лизата на каждую дорожку. Ингибирование активности киназы ВТК определяли путем измерения фосфорилирования ее субстрата (PLCγ2) методом вестерн-блот с использованием фосфо-специфичных антител производства Cell Signaling Technologies (кат. №3871). Результаты этого эксперимента с соединениями 1-2,1-4 и 1-7 представлены на Фигурах 1, 2 и 3.
[001302] В Таблице 8 приведены результаты теста с отмывкой на клетках Ramos для отобранных соединений.
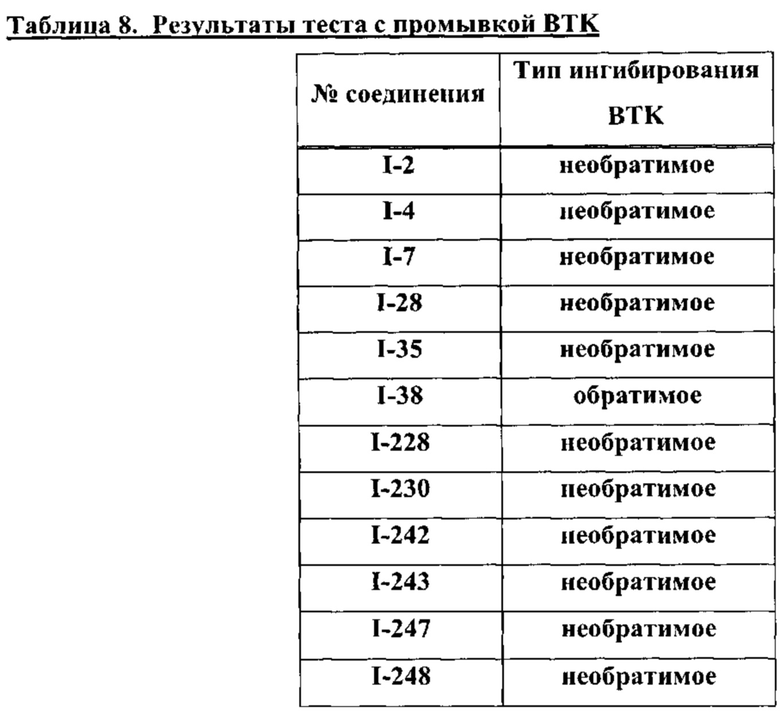
ПРИМЕР 255
Масс-спектрометрия на BTK
[001303] Интактную BTK инкубировали в течение 1 часа при 10-кратном избытке I-7 по отношению к белку. Аликвоты (2 мкл) образцов разбавляли 10 мкл 0.1%-ТФА, а затем наносили при помощи наконечников С4 ZipTip непосредственно на мишень MALDIc использованием синапиновой кислоты в качестве десорбирующего (10 мг/мл в 0.1% смеси ТФА : ацетонитрил 20:80). См. Фиг. 15. На верхнем рисунке показан масс-спектр интактного белка ВТК (m/z 81,032 Да). На нижней панели показан масс-спектр в случае, когда BTK инкубировали с I-7 (ММ=345.4). Центр масс (m/z=81,403 Да) демонстрирует положительный сдвиг около 371.1 Да, что указывает на полную модификацию BTK под действием I-7. Другие соединения, которые полностью модифицируют BTK, включают I-96, I-71, I-149, I-161, I-163, I-182, I-195, I-207, I-219, и I-244.
ПРИМЕР 256
Тест на пролиферацию первичных В-лимфоцитов человека
[001304] Не подвергавшиеся воздействию (наивные) В-лимфоциты человека выделяли из 100 мл цельной крови с использованием набора для очистки MACS, предназначенного для выделения CD19+, IgD+ клеток путем отрицательного отбора. Очищенные наивные В-лимфоциты ресуспендировали в полной RPMI и стимулировали 5 мкг/мл α-IgM в течени 72 часов. 3Н-Тимидин включали в среду для культивирования на последние 16 часов, клетки собирали и измеряли включение 3Н. Ингибирование пролиферации В-клеток коррелирует с ингибированием фосфорилирования субстрата BTK после стимуляции α-IgM. Важно, что молекула с таким же каркасом, что и I-7, но не обладающая биохимической активностью в отношении BTK, IR-7, не активна в тесте на пролиферацию наивных В-лимфоцитов.

ПРИМЕР 257 Тест на пролиферацию В-клеточной лимфомы
[001305] Предложенные соединения ингибируют пролиферацию различных линий клеток В-клеточной лимфомы, как показано в Таблице 10. Номера соединений соответствуют номерам соединений в Таблице 5. Соединения, активность которых обозначена "А", продемонстрировали ЕС50<0.1 мкМ; соединения, активность которых обозначена "В", продемонстрировали ЕС50 0.1-1 мкМ; соединения, активность которых обозначена "С", продемонстрировали ЕС50 1-10 мкМ; и соединения, активность которых обозначена "D", продемонстрировали ЕС50>10 мкМ.
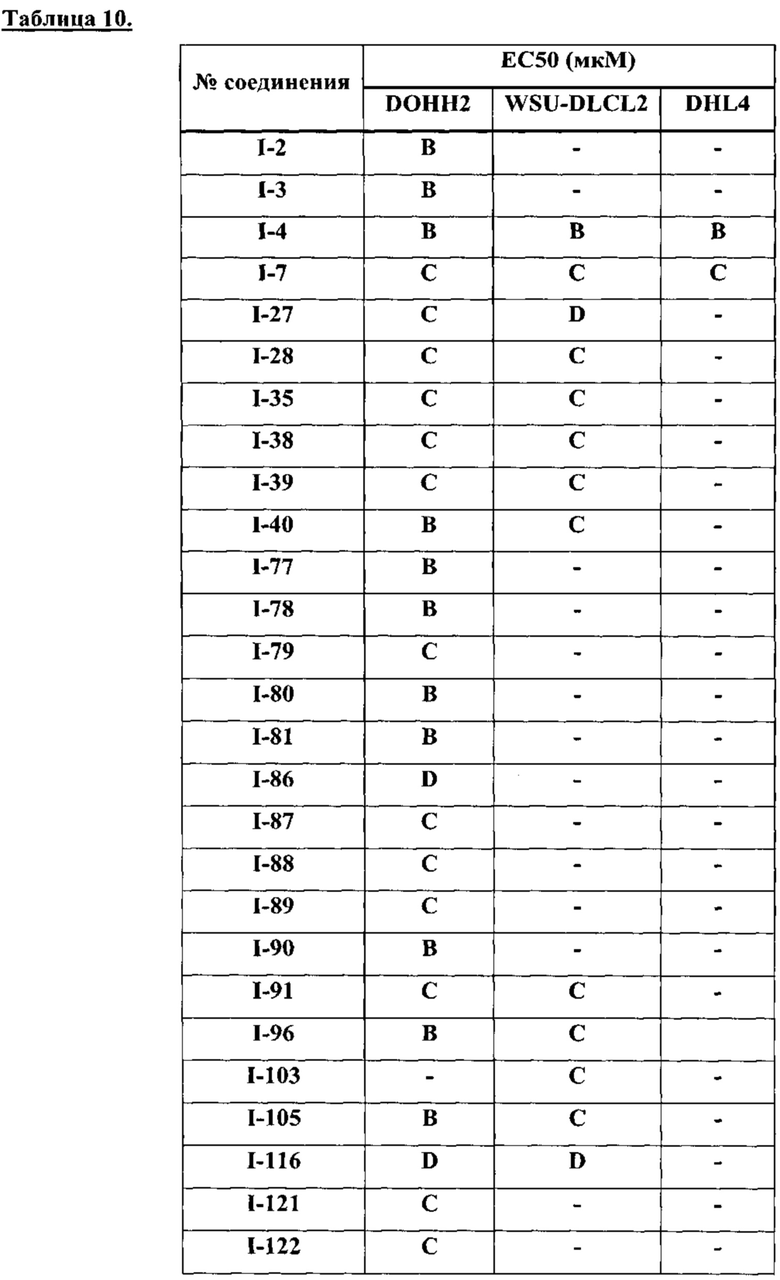
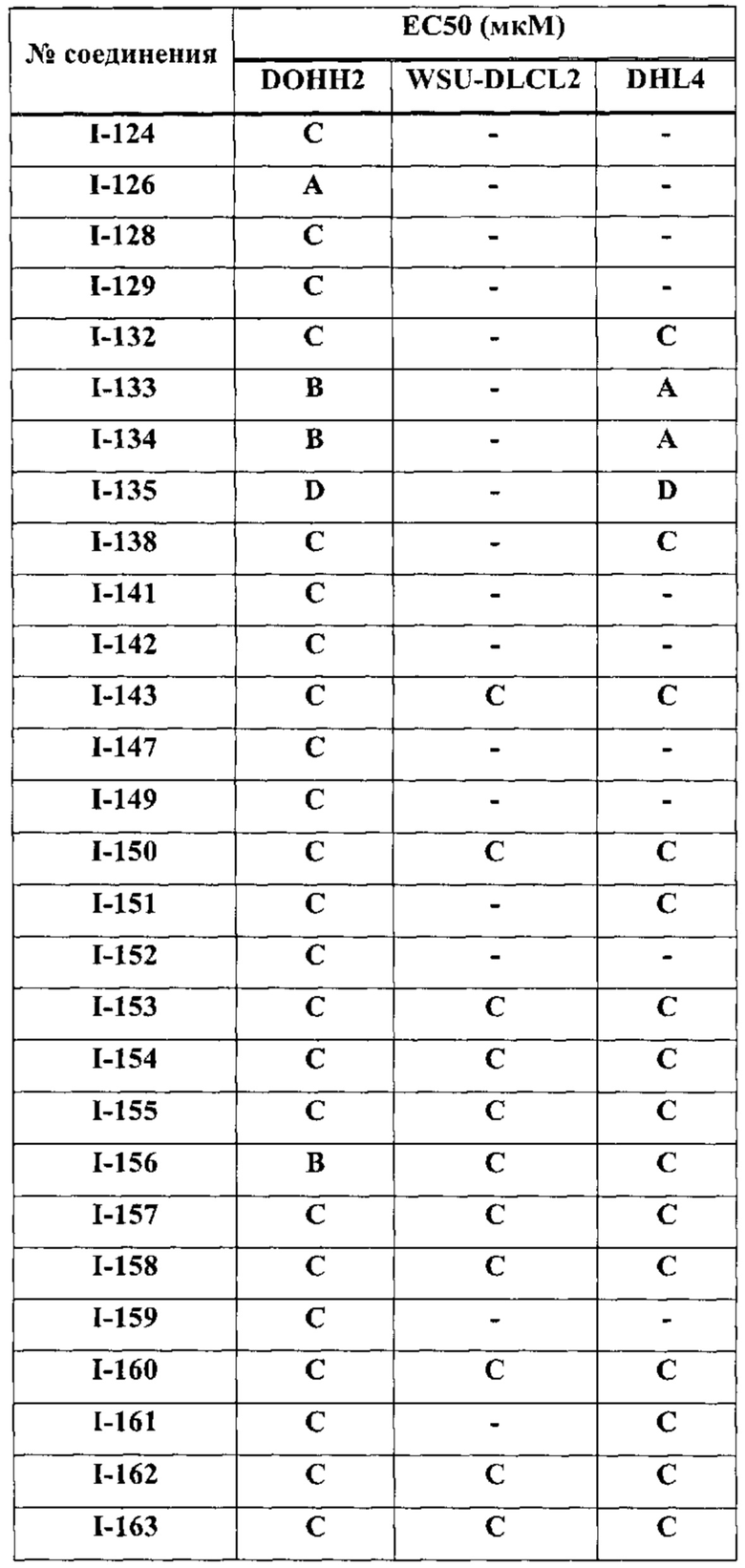
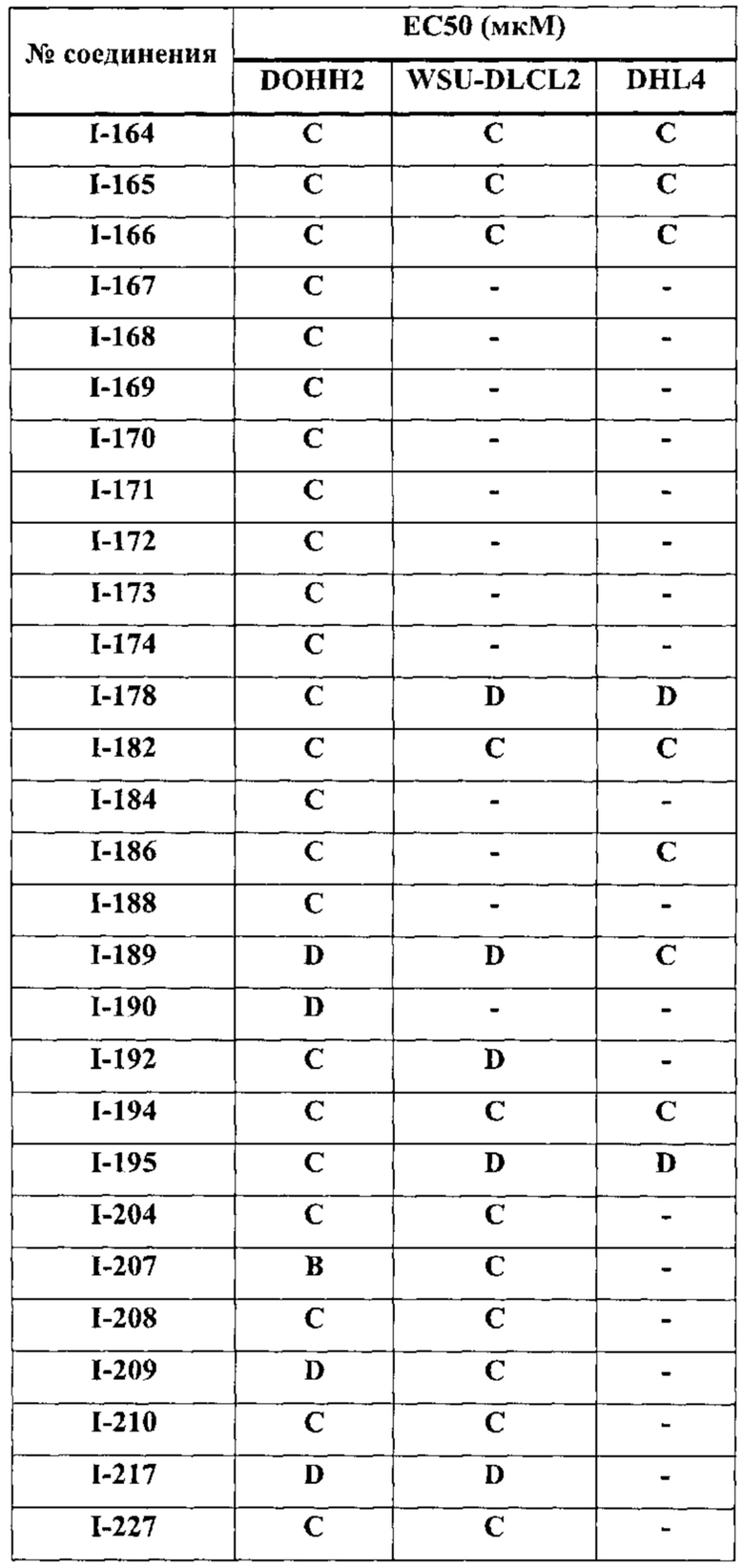
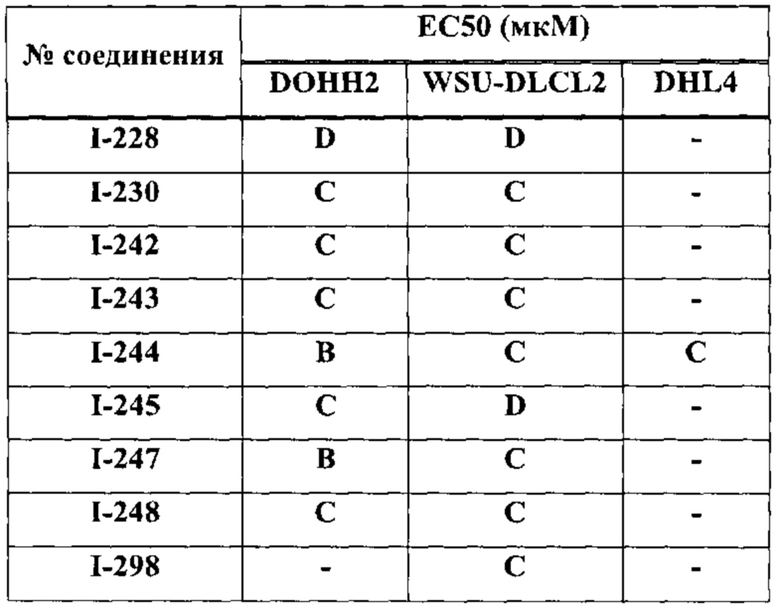
ПРИМЕР 258
Тимус-независимая активация В-лимфоцитов In vivo (TI-2)
[001306] Мышам С57/В6 ежедневно вводили 100 мг/кг соответствующего соединения в дни с 0 по 5. Мышей иммунизировали 25 мкг TNP-Ficoll в день 1, сыворотку собирали в день 6 и исследовали на продукцию циркулирующих антител α-TNP IgM (разбавление сыворотки 1:1600) и IgG3 (разбавление сыворотки 1:200) методом твердофазного иммуноферментного анализа. Результаты представлены в форме среднего по 10 мышам, составляющим группу обработки, и приведены в таблице 11 в форме % ингибирования TI-2-независимой активации В-лимфоцитов.

ПРИМЕР 259
Модель артрита, индуцированного антителом к коллагену
[001307] В день 0 проводили фоновые измерения подушечек лап и делили животных на экспериментальные группы таким образом, чтобы получить группы, которые не имели существенных отличий друг от друга. Затем каждому животному внутривенно вводили 2 мг коктейля моноклональных антител Arthritomab. В этот время начинали обработку исследуемыми агентами. В день 6 каждому животному путем внутривенной инъекции вводили 50 мкг ЛПС в 200 мкл стерильного ФБР. Измерения подушечек и клиническую оценку осуществляли в Дни 6, 7, 8, 9, 10, 11, 12, 14, 18, и 21. Результаты показаны в Таблице 12.

ПРИМЕР 260
Модель артритаPG-PS
[001308] В день 0, самкам крыс Lewis делали внутрибрюшинные болюсные инъекции (IP) пептидгликан - полисахарида (PG-PS) в количестве 15 мкг/г массы тела крысы. Крысам, представляющим контроль фона, делали внутрибрюшинную болюсную инъекцию ФБР. Группе, получавшей среду, и группе лечения вводили соединения путем орального гаважа непосредственно перед введением PG-PS. Ежедневное введение среды и соединения продолжали до дня 22. На протяжении исследования осуществляли измерение максимальной толщины лодыжки на обеих задних конечностях. В день 23 исследование завершали, рассчитывали конечное изменение опухания лодыжки, и сравнивали с контролем (мыши, получавшие среду). В Таблице 13 показаны результаты для двух соединений (n = число экспериментов).

ПРИМЕР 261
Масс-спектрометрия для киназы ТЕС (Соединение 1-2)
[001309] Киназу ТЕС (45 пмоль; Invitrogen) инкубировали с (1-2) (450 пмоль) в течение 3 часов при 10Х избытке, после чего осуществляли расщепление трипсином. После инкубации с соединением использовали иодацетамид в качестве алкилирующего агента. Также готовили контрольный образец (45 пмоль), в который не добавляли (I-2). Аликвоты фрагментов, полученных в результате расщепления трипсином, объемом 5 мкл (7.5 пмоль) разбавляли 15 мкл 0.1% ТФА, а затем наносили в микроколичествах с использованием наконечника С18 Zip Tip непосредственно на мишень MALDI с использованием альфа-циано-4-гидрокси-коричной кислоты в качеств матрицы (5 мкг/мл в 0.1% смеси ТФА : Ацетонитрил 50:50).
[001310] Как показано на Фигуре 6, пептид, который, как ожидалось, будет модифицирован (GCLLNFLR), обнаруживали сразу после реакции с I-2 (масса MI 359.17 Да) при МН+ 1294.72. Также было четко установлено, что этот пептид был модифицирован в контрольном образце на МН+ 992.56. Интересно, что модифицированный иодоацетамидом пептид не обнаруживался в расщепленном белке после реакции с соединением I-2, что указывает на то, что реакция завершалась. Следов присутствия других модифицированных пептидов обнаружено не было.
[001311] Присутствие соединения I-2 наблюдали при МН+ 360.17 в диапазоне низких масс спектра. Спектр фрагментации пика 360.17 не выявил диагностических фрагментов, которые были видны в спектре PSD модифицированного пептида на 1294.72 (См. Фиг. 6).
[001312] Чтобы убедиться в присутствии модифицированных пептидов, пептиды меченные иодацетамидом (992.56) и меченные I-2 (1294.72) подвергали анализу методом
PSD (MC/MC). Поиск в базе данных NCBI Homo sapien с использованием программы для поиска ионов MC/MC Ion Search показал, что в обоих случаях ожидаемыми были пептиды с высоким совпадением.
Аппаратура:
[001313] Для продуктов расщепления трипсином аппарат устанавливали в режим Reflection с настройкой пульсовой экстракции 2200. Калибровку проводили с использованием стандартов Laser Biolabs Pep Mix (1046.54, 1296.69, 1672.92, 2093.09, 2465.20). Для анализа CID/PSD пептид отбирали, используя курсор для того, чтобы задавать интервалы пропускания ионов и фрагментацию, которые имеют место при энергии лазера выше 20%, в качестве газа столкновения для CID использовали гелий. Калибровку для фрагментов осуществляли с использованием калибровки P14R для режима Curved field Reflectron.
ПРИМЕР 262
Масс-спектрометрия для киназы ТЕС (Соединение I-4)
[001314] Киназу ТЕС (45 пмоль; Invitrogen) инкубировали с (I-4) (450 пмоль) в течение 3 часов при 10Х избытке, после чего осуществляли расщепление трипсином. После инкубации с соединением использовали иодацетамид в качестве алкилирующего агента. Также готовили контрольный образец (45 пмоль), в который не добавляли (I-4). Аликвоты фрагментов, полученных в результате расщепления трипсином, объемом 5 мкл (7.5 пмоль) разбавляли 15 мкл 0.1% ТФА, а затем наносили в макроколичествах с использованием наконечника С18 Zip Tip непосредственно на мишень MALDI с использованием альфа-циано-4-гидрокси-коричной кислоты в качестве матрицы (5 мкг/мл в 0.1% смеси ТФА : Ацетонитрил 50:50).
[001315] Как показано на Фигуре 7, пептид, который, как ожидалось, будет модифицирован (GCLLNFLR), обнаруживали при МН+ 1355.72. Эта масса соответствует тому, что соединение I-4 с прибавленной массой 420.21 добавляют к пептиду массой 935.51. Также этот пептид, модифицированный иодоацетамидом, был выявлен в контрольном образце на МН+ 992.56. Интересно, что модифицированный иодоацетамидом пептид не обнаруживался в расщепленном белке после реакции с
соединением I-4, что указывает на то, что реакция завершалась. Следов присутствия других модифицированных пептидов обнаружено не было.
[001316] Присутствие соединения I-4 наблюдали при МН+ 421.35 в диапазоне низких масс спектра. Спектр фрагментации прика 360.17 не выявил диагностических фрагментов, которые были видны в спектре PSD модифицированного пептида на 1294.72 (См. Фиг. 6). Спектр фрагментации пика 421.35 выявил два значительных пика, которые были видны в PSD-спектре модифицированного пептида на 1355.72 (См. Фиг. 7).
[001317] Чтобы убедиться в присутствии модифицированного пептида, I-4, пептиды на МН+ 1355.72 подвергали PSD-анализу (МС/МС). Из-за низкой интенсивности фрагментов, корреляция с базой данных невозможна. Однако, диагностические фрагменты самой молекулы I-4 обеспечили достоверную идентификацию. Диагностические фрагменты на МН+ 376.38 и 421.83 - фрагменты I-4.
Аппаратура:
[001318] Для продуктов расщепления трипсином аппарат устанавливали в режим Reflectron с настройкой пульсовой экстракции 2200. Калибровку проводили с использованием стандартов Laser Biolabs Pep Mix (1046.54, 1296.69, 1672.92, 2093.09, 2465.20). Для анализа CID/PSD пептид отбирали, используя курсор для того, чтобы задавать интервалы пропускания ионов и фрагментацию, которые имеют место при энергии лазера выше 20%, в качестве газа столкновения для CID использовали гелий. Калибровку для фрагментов осуществляли с использованием калибровки P14R для режима Curved field Reflectron.
ПРИМЕР 263
Масс-спектрометрия для киназы ТЕС (Соединение I-7)
[001319] ТЕС-киназу (45 пмоль; Invitrogen) инкубировали с (I-7) (450 пмоль) в течение 3 часов при 10Х избытке, после чего осуществляли расщепление трипсином. После инкубации с соединением использовали иодацетамид в качестве алкилирующего агента. Также готовили контрольный образец (45 пмоль), в который не добавляли (1-7). Аликвоты фрагментов, полученных в результате расщепления трипсином, объемом 5 мкл (7.5 пмоль) разбавляли 15 мкл 0.1% ТФА, а затем наносили в макроколичествах с использованием наконечника С18 Zip Tip непосредственно на мишень MALDI с
использованием альфа-циано-4-гидрокси-коричной кислоты в качестве матрицы (5 мкг/мл в 0.1% смеси ТФА : Ацетонитрил 50:50).
[001320] Как показано на Фигуре 8, пептид, который, как ожидалось, будет модифицирован (GCLLNFLR), обнаруживали при МН+ 1355.72. Эта масса соответствует тому, что соединение I-7 с прибавленной массой 345.16, добавляют к пептиду массой 935.51. Также этот пептид, модифицированный иодацетамидом, был выявлен в контрольном образце на МН+ 992.56. Интересно, что модифицированный иодацетамидом пептид не обнаруживался в расщепленном белке после реакции с соединением I-7, что указывает на то, что реакция завершалась. Следов присутствия других модифицированных пептидов на МН+ 1985.93 обнаружено не было (TIDELVECEETFGR).
[001321] Присутствие соединения I-7 наблюдали при МН+ 346.32 в диапазоне низких масс спектра. Спектр фрагментации пика 346.32 не выявил диагностических фрагментов, которые были видны в спектре PSD двух модифицированных пептидов (См. Фиг. 8).
[001322] Чтобы дополнительно убедиться в присутствии пептидов, модифицированных соединением I-7, пептиды на МН+ 1280.73 и 1985.93 подвергали PSD-анализу (МС/МС). Из-за низкой интенсивности фрагментов, корреляция с базой данных невозможна. Анализ корреляции с базой данных по белкам человека выявил, правильные пептиды, модифицированные I-7.
Аппаратура:
[001323] Для продуктов расщепления трипсином аппарат устанавливали в режим Reflectron с настройкой пульсовой экстракции 2200. Калибровку проводили с использованием стандартов Laser Biolabs Pep Mix (1046.54, 1296.69, 1672.92, 2093.09, 2465.20). Для анализа CID/PSD пептид отбирали, используя курсор для того, чтобы задавать интервалы пропускания ионов и фрагментацию, которые имеют место при энергии лазера выше 20%, в качестве газа столкновения для CID использовали гелий. Калибровку для фрагментов осуществляли с использованием калибровки P14R для режима Curved field Reflectron.
ПРИМЕР 264
Резюме протокола теста для оценки эффективности в отношении активных форм киназы ITK
[001324] В этом примере описаны тесты с непрерывным считыванием для измерения эффективности соединений в отношении активных форм ферментов ITK, осуществляемых как описано выше в примере 251, за исключением того, что использовались условия с оптимизацией реагентов по ITK:
[ITK]=10 нМ, [АТФ]=25 мкМ, [Y6-Sox]=10 мкМ (АТФ KМарр=33 мкМ).
ПРИМЕР 265
[001325] В Таблице 14 показана активность примеров соединений согласно настоящему изобретению в тесте на ингибирование ITK. Номера соединений соответствуют номерам соединений в Таблице 5. Соединения, активность которых обозначена "А", продемонстрировали IC50≤10 нМ; соединения, активность которых обозначена "В", продемонстрировали IC50 10-100 нМ; соединения, активность которых обозначена "С", продемонстрировали IC50 100-1000 нМ; соединения, активность которых обозначена "D", продемонстрировали IC50 1000-10,000 нМ; и соединения, активность которых обозначена "Е", продемонстрировали IC50≥10,000 нМ.
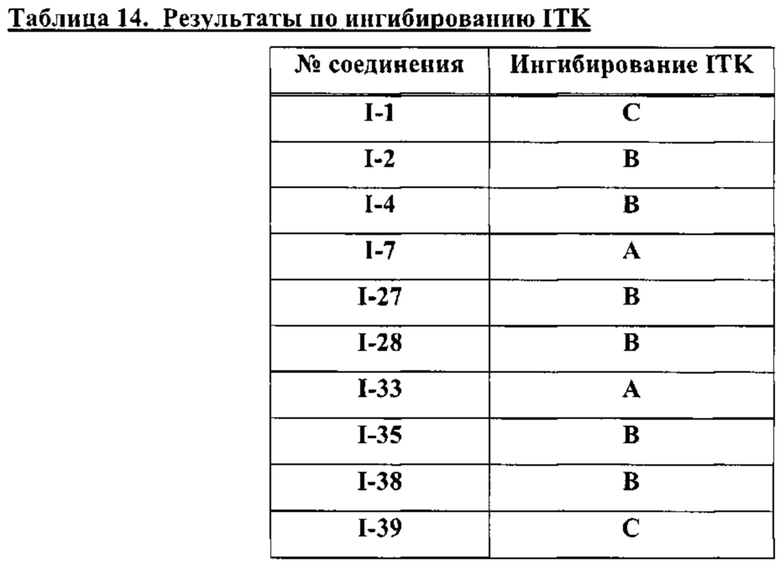
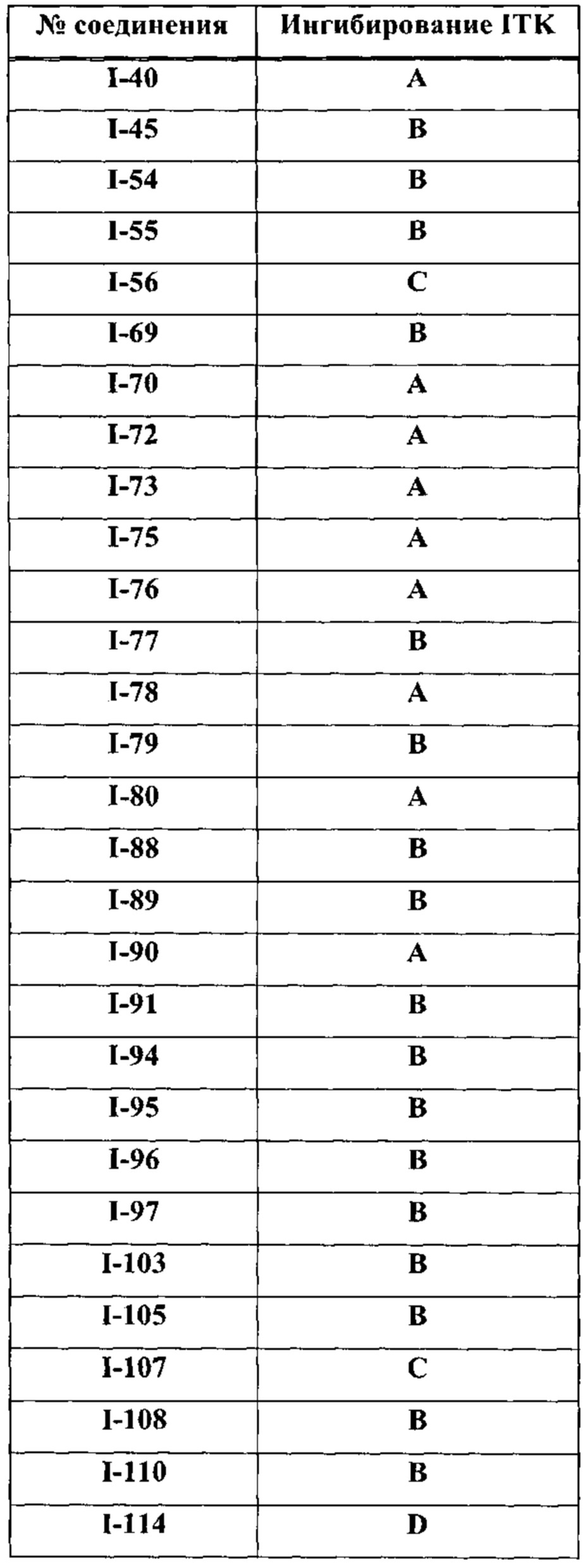
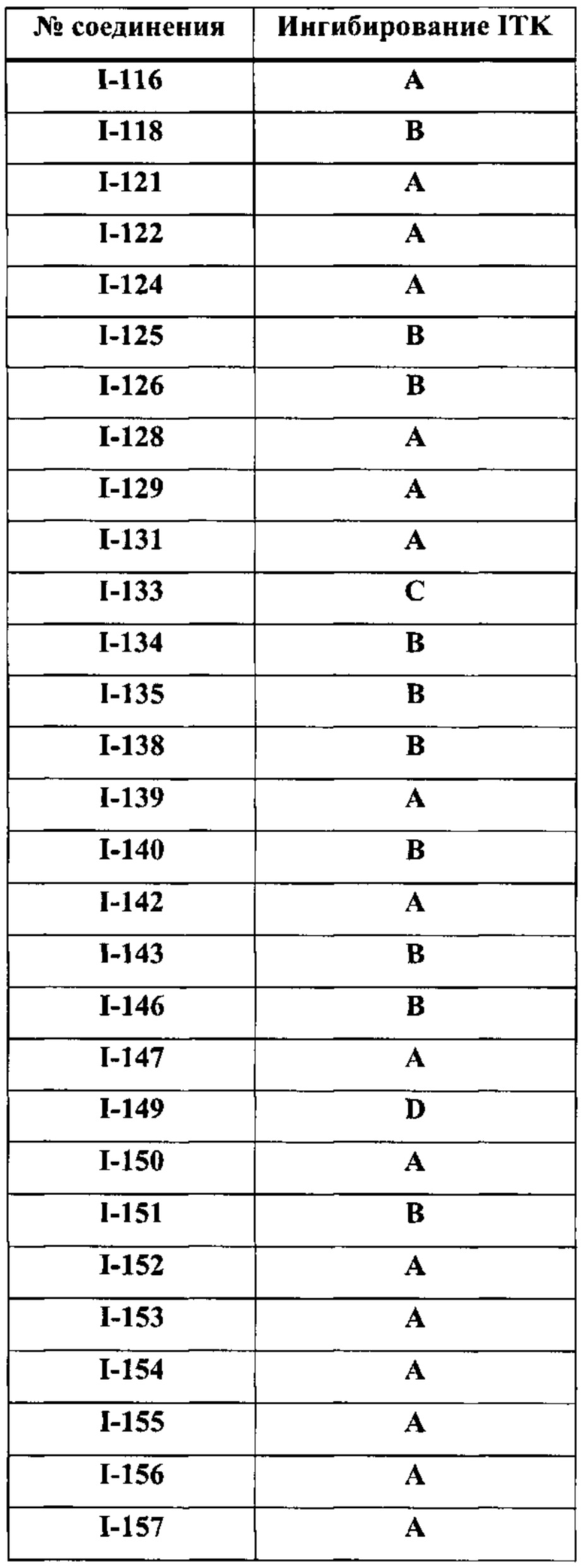

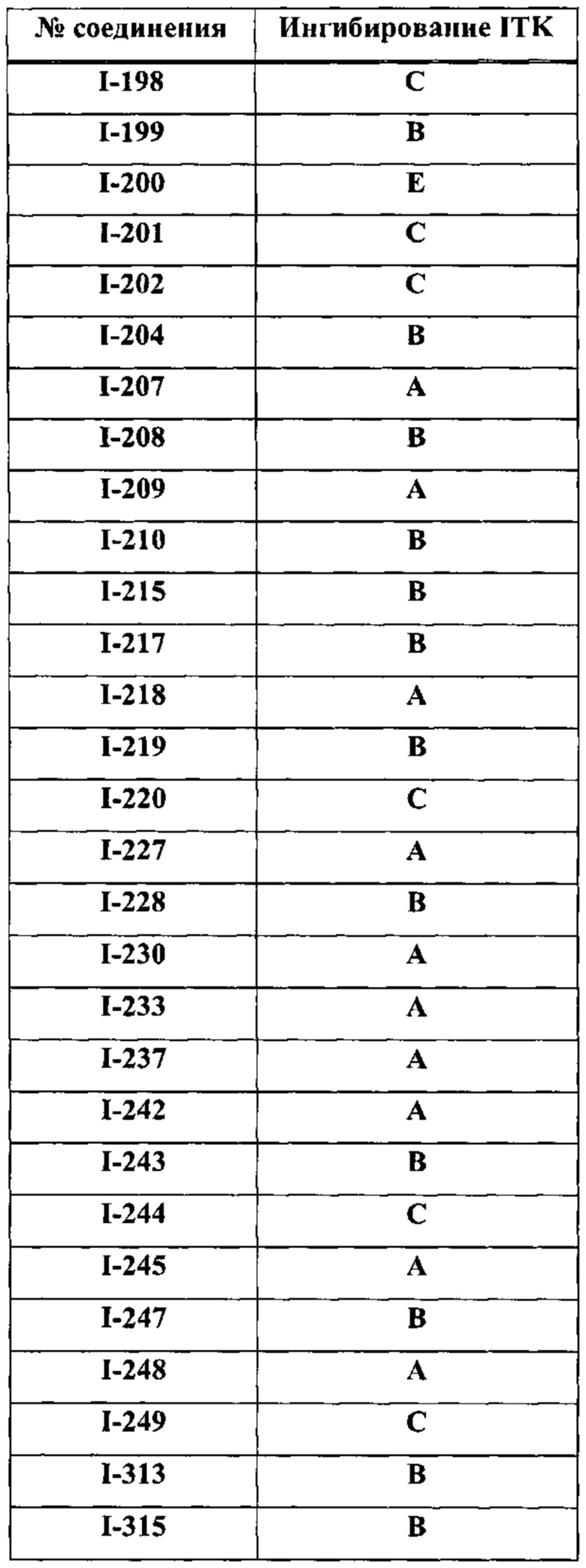

ПРИМЕР 266
Резюме протокола теста для оценки эффективности в отношении акттивных форм киназы ВМХ
[001326] В этом примере описаны тесты с непрерывным считыванием для измерения эффективности соединений в отношении активных форм ферментов ВМХ, осуществляемых как описано выше в примере 251, за исключением того, что использовались условия с оптимизацией реагентов по ВМХ:
[ВМХ]=2.5 нМ, [АТФ]=100 мкМ, [Y5-Sox]=7.5 мкМ (АТФ KМарр=107 мкМ).
ПРИМЕР 267
[001327] В Таблице 15 показана активность примеров соединений согласно настоящему изобретению в тесте на ингибирование ВМХ. Номера соединений
соответствуют номерам соединений в Таблице 5. Соединения, активность которых обозначена "А", продемонстрировали IC50≤10 нМ; соединения, активность которых обозначена "В", продемонстрировали IC50 10-100 нМ; соединения, активность которых обозначена "С", продемонстрировали IC50 100-1000 нМ; соединения, активность которых обозначена "D", продемонстрировали IC50 1000-10,000 нМ; и соединения, активность которых обозначена "Е", продемонстрировали IC50≥10,000 нМ.
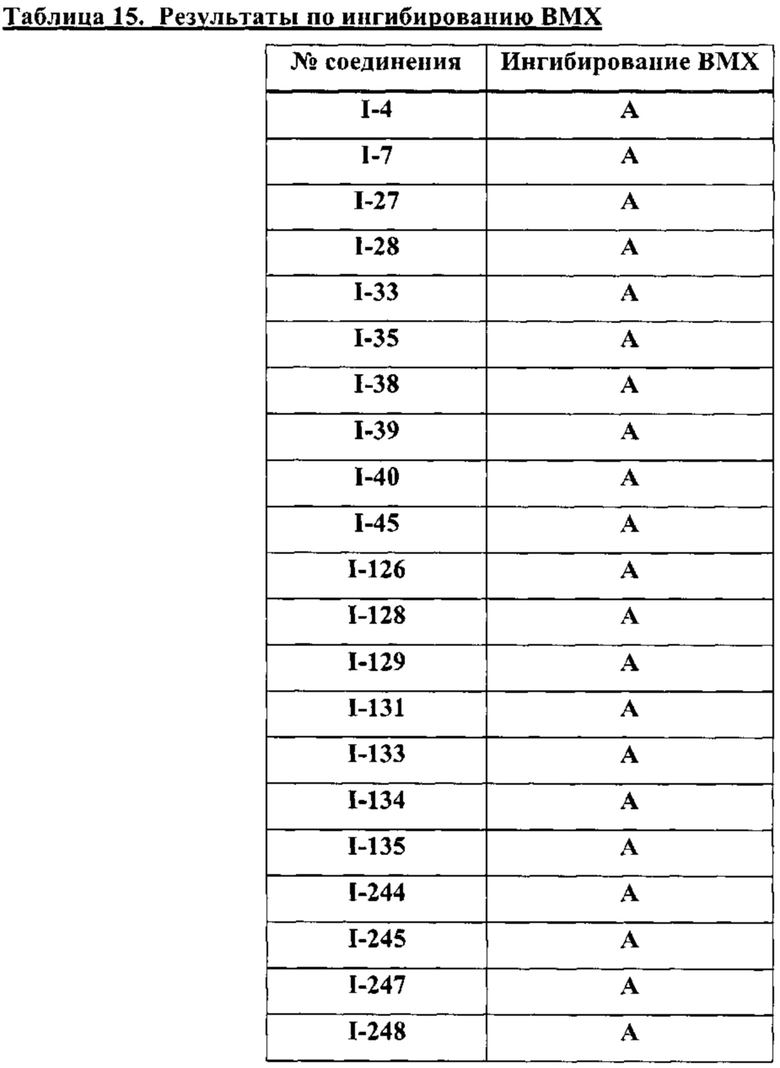
ПРИМЕР 268
Клонирование, экспрессия и очистка мутантных EGFR-WT и EGFR C797S с использованием бакуловируса и клеток насекомого
(i) Субклонирование EGFR-WT и мутантных киназных доменов
[001328] Аминокислоты с 696 по 1022 киназного домена EGFR-WT (NM_005228, NP_005219.2) субклонировали в сайты NcoI и HindIII вектора pFastHTa (Invitrogen, Карлсбад, Калифорния, США). Для получения мутантного белка EGFR цистин в положении 797 заменяли на серии, используя набор Stratagene QuikChange kit (Stratagene, Cedar Creek, TX) в соответствии с инструкциями производителя.
(ii) Экспрессия
[001329] Маточный раствор бакуловируса Р1 нарабатывали в клетках SF9 в соответствии с протоколом трансфекции в суспензии Blue Sky Biotech's (Worcester, Массачусетс, США). Анализ экспрессии осуществляли в 125 мл культуры клеток насекомого SF21 ((выращенный в SF900I SFM (Invitrogen, кат.№10902-088), дополненной 10 мг/л гентамицина (Invitrogen, Карлсбад, Калифорния, кат.№15710-064)), при вирусной нагрузке 0.1 мл вируса на 100 мл суспензии клеток. Экспрессию оптимизировали с использованием системы мониторинга кинетики инфекции Blue Sky Biotech's Infection Kinetics Monitoring (Worcester, Массачусетс, США).
(iii) Очистка
[001330] Инфицированные клетки насекомого осаждали. Клеточные осадки ресуспендировали в буфере для лизирования пр-ва Blue Sky Biotech (Worcester, Массачусетс, США, 1X WX; солюбилизирующий буфер, содержащий коктейль ингибиторов протеаз из лейпептина, пепстатина, PMSF (фенилметансульфонилфторид), апротинин и ЭДТА) в отношении 10 мл на грамм влажной суспензии клеток. Клетки лизировали путем обработки ультразвуком и лизат осветляли путем центрифугирования при 9,000 ОБ./МИН. в течение 30 минут в роторе GSA. К слою смолы NiNTA (Qiagen, Valencia, СА) объемом 500 мкл добавляли супернатанты и партию, связывавшуюся в течение двух часов при постоянном встряхивании. Материал переносили под действием силы тяжести на пустую колонку объемом 2 мл. Колонку промывали 2 мл промывного буфера (Blue Sky Biotech, Worcester, Массачусетс, США, 1X WX, 25 мМ имидазола). Булок элюировали 1X WX + имидазол при варьирующихся концентрациях: Элюат 1: 75 мМ
имидазола (2 фракции, 1 объем колонки); Элюат 2 : 150 мМ имидазола (2 фракции, 1 объем колонки); Элюат 3 : 300 мМ имидазола (2 фракции, 1 объем колонки). Все элюированные фракции исследовали методом электрофореза на полиакриламидном геле с додецилсульфатом натрия с последующим окрашиванием Кумасси и Вестерн-блоттингом с использованием антитела к пента-гистидину (Qiagen, Валенсия, Калифорния, США). Из некоторых очищенных белков удаляли карбокси-концевую метку из шести гистидинов с использованием набора AcTEV Protease (Invitrogen, Карлсбад, Калифорния, Кат. №12575-015) в соответствии с инструкциями производителя. Все образцы (до и после расщепления Tev) исследовали методом электрофореза на полиакриламидном геле с додецилсульфатом натрия с последующим окрашиванием Кумасси и Вестерн-блоттингом, как описано выше.
ПРИМЕР 269
Масс-спектрометрия EGFR
[001331] EGFR дикого типа и EGFR (мутант C797S) инкубировали с 10-кратньш избытком исследуемого соединения в течение 1 часа и 3 часов. Аликвоты образцов объемом 1 мкл (общий объем 5-8 мкл) разбавляли 10 мкл 0.1% ТФА перед нанесением с использованием наконечника С4 ZipTip непосредственно на мишень MALDI с синаприновой кислотой в качестве матрицы десорбции (10 мкг/мл в 0.1% смеси ТФА : ацетонитрил 50:50). Измерение массы интактного соединения показало, что номинальная масса дикого типа составляет около 37557, а у мутанта - несколько ниже, на 37500. Реактивность наблюдали только для EGFR дикого типа, при этом появлялся новый пик массы, соответствующей ковалентной модификации в единственном сайте род действием тестируемого соединения, имеющего массу 410 Да.
ПРИМЕР 270
Общий протокол теста для оценки эффективности в отношении активных ферментов EGFR (WT) и EGFR (T790M/L858R)
[001332] Общий протокол теста для оценки эффективности в отношении EGFR осуществляют как описано выше в Примере 251 за тем исключением, что использовали условия с оптимизацией реагентов для EGFR-WT и EGFR T790M/L858R-модиф.).:
[001333] [EGFR-WT]=5 нМ, [АТФ]=15 мМ, [Y12-Sox]=5 мМ (АТФ KМарр ~ 12 мМ); и [EGFR-T790M/L858R]=3 нМ, [АТФ]=50 мМ, [Y12-Sox]=5 мМ (АТФ KМарр ~ 45 мМ).
ПРИМЕР 271
[001334] В Таблицах 16 и 17 показана активность отобранных соединений согласно настоящему изобретению в тесте на ингибирование EGFR. В Таблице 16 показаны данные для EGFR дикого типа; в Таблице 17 показаны данные для двух мутантов EGFR. Номера соединений соответствуют номерам соединений в Таблице 5. Соединения, активность которых обозначена "А", продемонстрировали IC50≤10 нМ; соединения, активность которых обозначена "В", продемонстрировали IC50 10-100 нМ; соединения, активность которых обозначена "С", продемонстрировали IC50 100-1000 нМ; соединения, активность которых обозначена "D", продемонстрировали IC50 1000-10,000 нМ; и соединения, активность которых обозначена "Е", продемонстрировали IC50≥10,000 нМ.
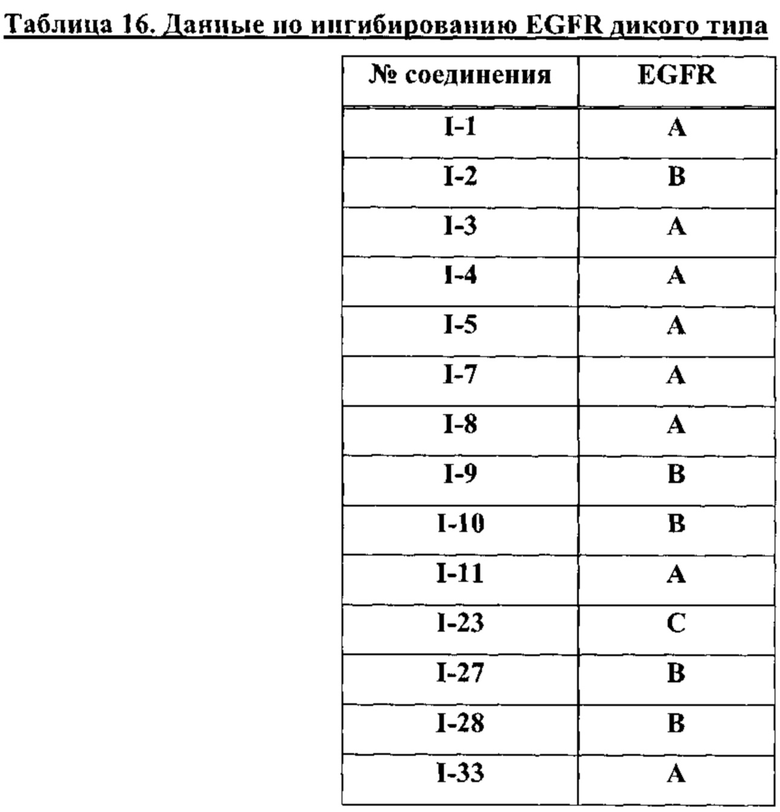
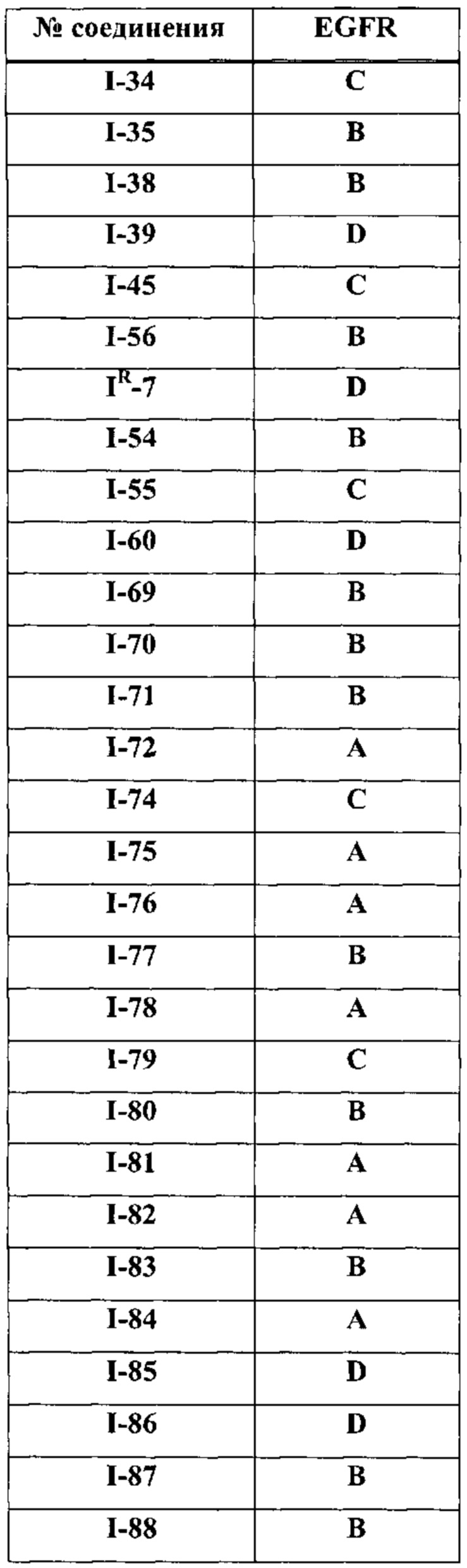

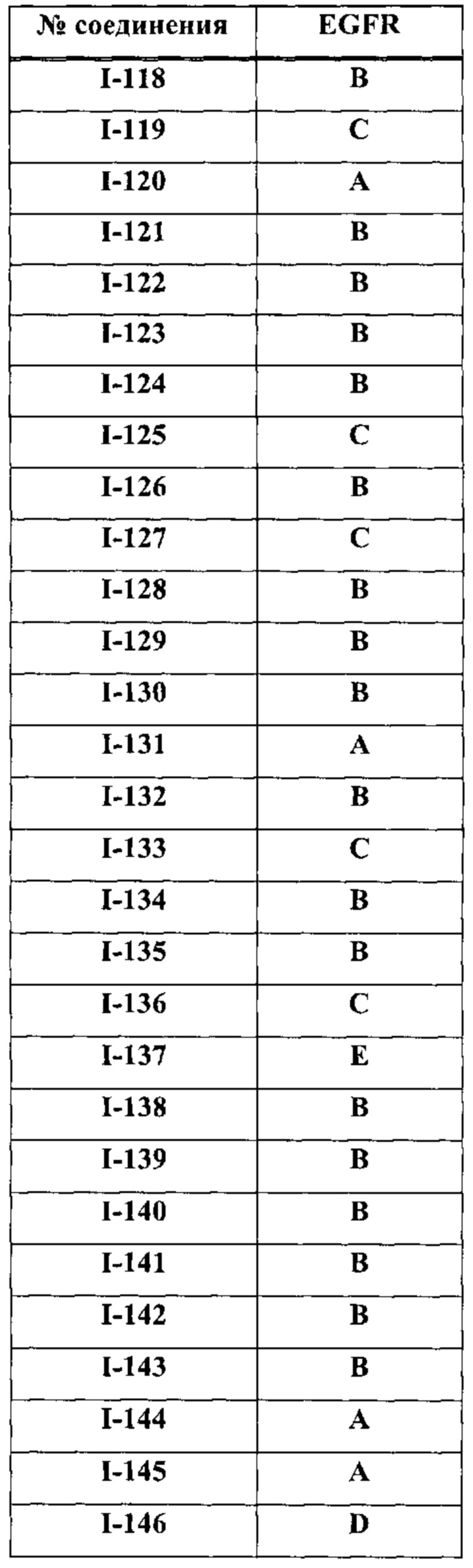

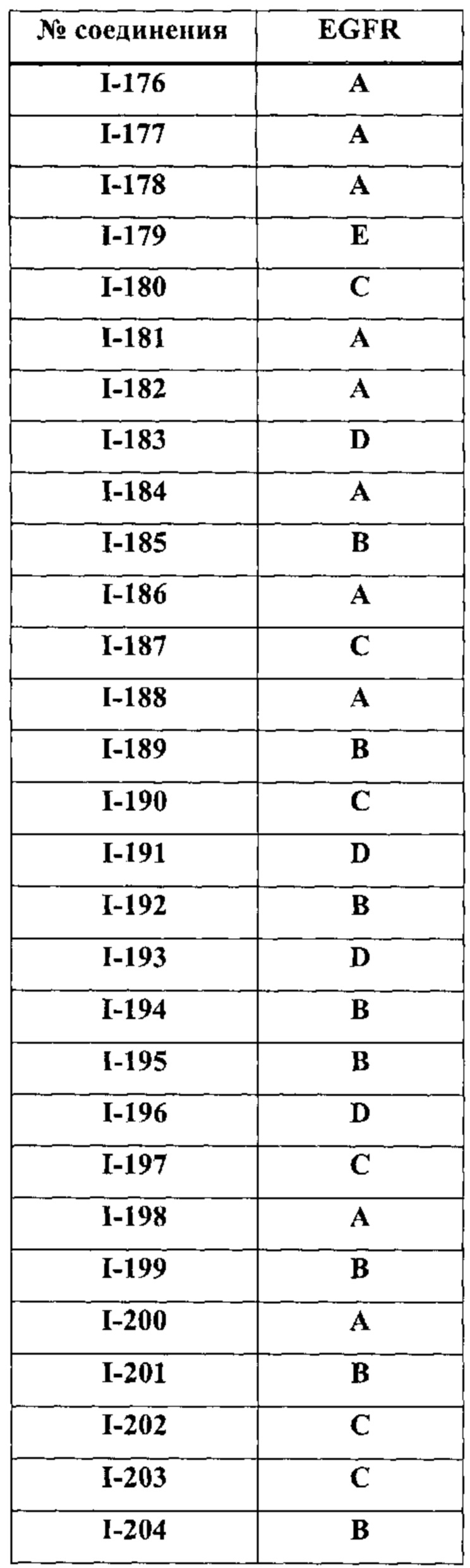
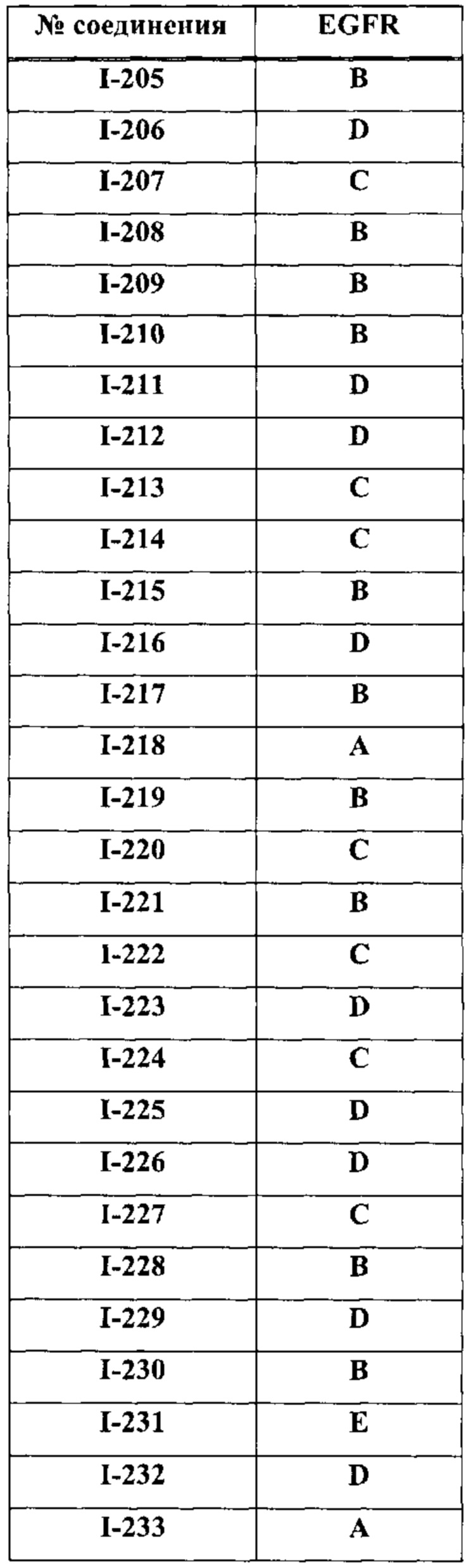
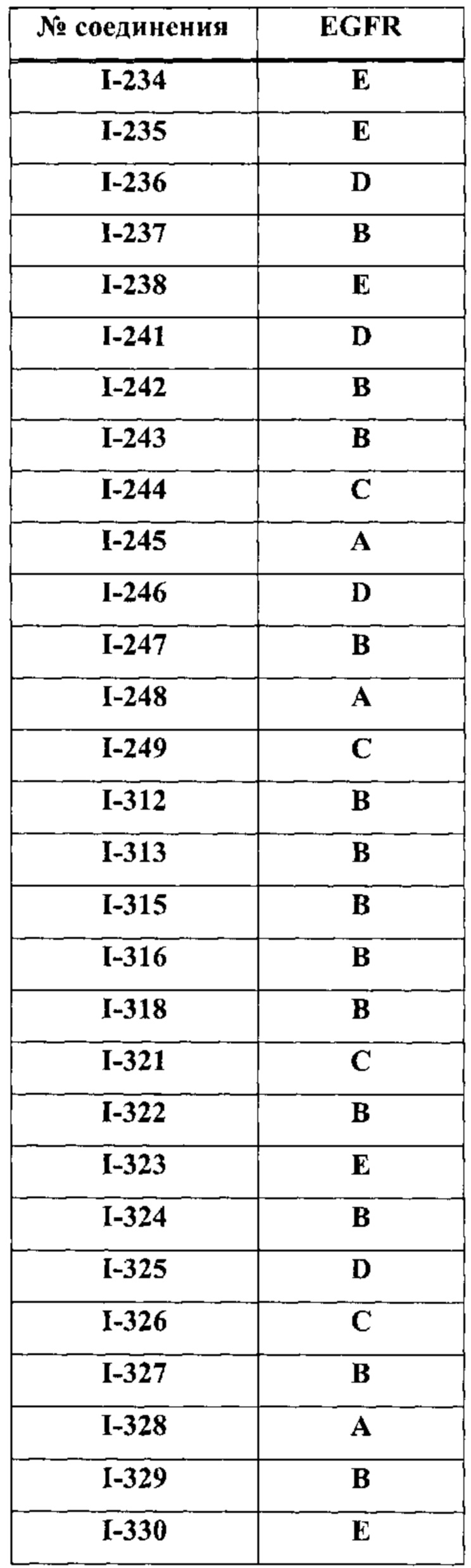
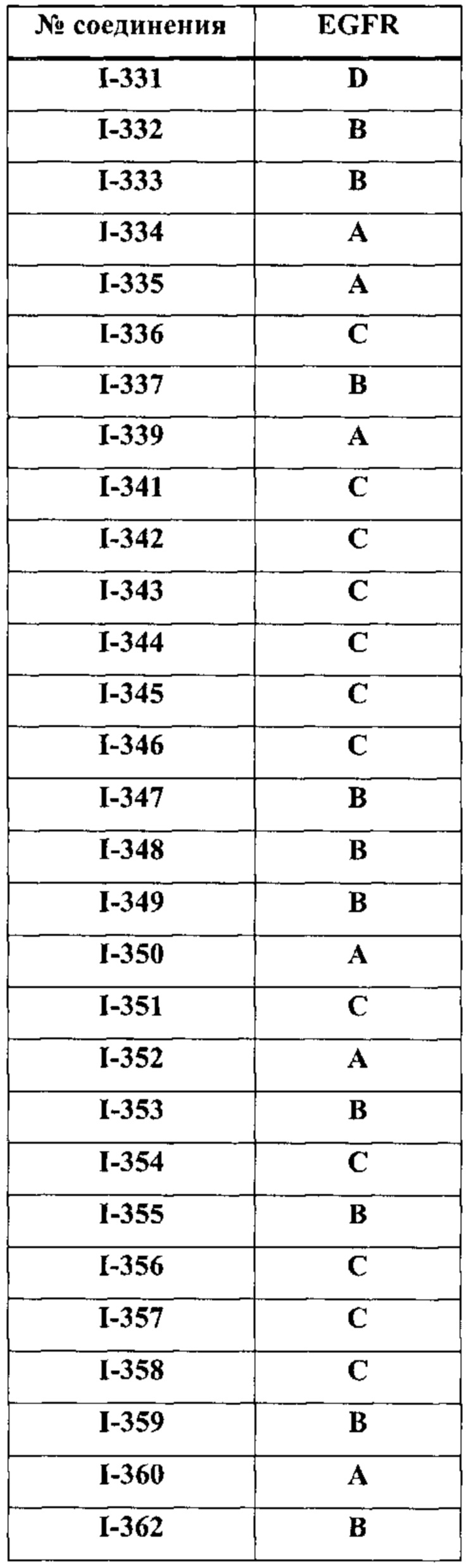
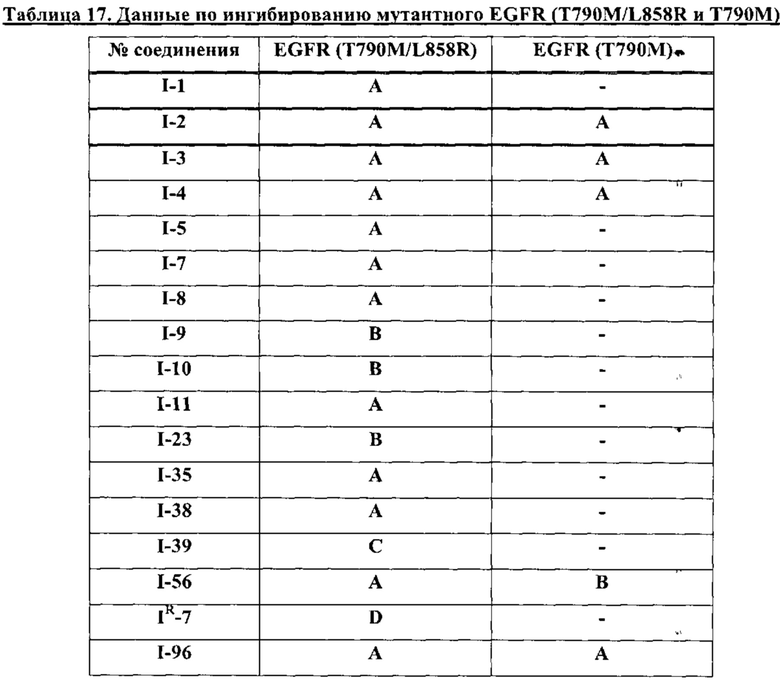
ПРИМЕР 272
Тесты на активность EGFR с использованием клеток
[001335] Соединения исследовали на клетках А431 эпидермоидной карциномы человека с использованием метода, по существу аналогичного методу, описанного Fry, et al., Proc. Natl. Acad. Sci. USA Vol 95, pp 12022-12027, 1998. Конкретно, клетки эпидермоидной карциномы человека А431 выращивали в 6-луночных планшетах до непрерывности слоя 90% и затем инкубировали в бессывороточной среде в течение 18 часов. Наборы клеток в дупликатах обрабатывали 1 мкМ обозначенного соединения в течение 2, 5, 10, 30 или 60 мин. Клетки отмывали от соединения подогретой бессывороточной средой, инкубировали в течение 2 ч, промывали еще раз, инкубировали
еще 2 ч, промывали еще раз и затем инкубировали еще 2 ч промывали еще раз и инкубировали в течение дополнительных 2 ч, после чего стимулировали путем обработки 100 нг/мл EGF в течение 5 мин. Экстракты готовили, как описано у Fry et al.
[001336] Соединения тестировали на клетках эпидермоидной карциномы человека А431 с использованием метода, по существу аналогичного методу, описанного Fry, et al. Конкретно, клетки эпидермоидной карциномы человека А431 выращивали в 6-луночных планшетах до непрерывности слоя 90% и затем инкубировали в бессывороточной среде в течение 18 часов. Затем клетки обрабатывали 10, 1, 0.1, 0.01 или 0.001 мкМ тестируемого соединения в течение 1 ч. Затем клетки стимулировали 100 нг/мл EGF в течение 5 мин и готовили экстракты, как описано у Fry et al. 20 мкг общего белка из лизатов загружали на гель и зондировали либо на фосфорилирование EGFR, либо на фосфорилирование р42/р44 Erk.
ПРИМЕР 273
Эксперименты с отмывкой на активность EGFR
[001337] Клетки эпидермоидной карциномы человека А431 выращивали в 6-луночных планшетах до непрерывности слоя 90% и затем инкубировали в бессывороточной среде в течение 18 часов Наборы клеток в дупликатах обрабатывали 1 мкМ обозначенного соединения в течение 1 часа. Затем один набор клеток стимулировали 100 нг/мл EGF в течение 5 минут и готовили экстракты, как описано. Другой набор клеток отмывали от соединения I-7 подогретой бессывороточной средой, инкубировали в течение 2 часов, промывали еще раз, инкубировали еще 2 ч, промывали еще раз, и затем инкубировали еще 2 часа, после чего промывали еще раз и инкубировали в течение дополнительных 2 часов и затем стимулировали EGF. Результаты этого эксперимента с соединением I-7 изображены на Фигуре 10.
ПРИМЕР 274
Эксперименты с промывкой на клетках НСС827, содержащих мутант EGFR НСС827 с делецией
[001338] Клетки (АТСС, Манассас, Виржиния, США) высевали в планшет в среде Growth Media (RPM 1640), дополненной 10% ФБР, 10 мкМ HEPES, 2 мМ 1-глютамином,
1 мМ Na-пируватом и pen/strep (Invitrogen, Карлсбад, СА) при плотности 2.5 × 105 клеток на лунку в 6-луночных культуральных планшетах. Спустя двадцать четыре часа дважды промывали ФБР, а затем выдергивали без сыворотки в течение ночи в основной среде Basal Media (среда для выращивания без ФБР).
[001339] На следующее утро среду удаляли и лунки (в дупликатах) добавляли 2 мл свежей основной среды, содержащей 1 мкМ соединения в 0.1% ДМСО. Через 1 час одну лунку с клетками обрабатывали 100 нг/мл EGF в течение 5 минут, промывали ФБР, после чего осуществляли лизирование путем разрушения в 75 мкл в буфере Cell Extraction Buffer (Invitrogen, Карлсбад, Калифорния, США) плюс ингибитор фосфатаз PhosSTOP и полный ингибитор протеаз Complete Protease Inhibitor (Roche, Indianapolis, Индианаполис, США) в момент времени 0 ч. Соединение удаляли из второго набора лунок и промывали эти лунки дважды Основной Средой. Клетки промывали Основной средой каждый 2 часа до 8 часов, когда их обрабатывали EGF и лизировали как в момент времени 0 ч.
[001340] Концентрации белка в лизатах определяли с использованием теста ВСА Assay (Pierce, Rockford, Иллинойс, США), 10 мкг каждого лизата отделяли с использованием 4-12% градиента в SDS-ПААГ (Invitrogen), переносили на мембрану Immobilon-FL (Millipore) и зондировали антителами кролика к Фосфо-EGFR (Туг 1068) (Zymed, Invitrogen) и антителами мыши к EGFR (Cell Signaling Technologies, Дэнверс, Массачусетс, США). Сигналы Фосфо-белка измеряли с использованием системы инфракрасной визуализации Odyssey (Li-Cor Biosciences, Линкольн, Небраска, США). Результаты этих экспериментов представлены на Фигуре 9, на которой показано сравнение результатов для соединения I-2 с результатами для соединения I-4 и соединения I-7 в одном и том же эксперименте с отмывкой.
ПРИМЕР 275
Масс-спектрометрия ERBB4
[001341] Киназный домен Erbb4 (Upstate) инкубировали с соединением в течение 60 минут при 10-клратном избытке соединений I-4 и I-11 относительно белка. Аликвоты образцов объемом 1 мкл (общий объем 4.24 мкл) разбавляли 10 мкл 0.1% ТФА, а затем наносили при помощи наконечника С4 ZipTip непосредственно на мишень MALDI с синапиновой кислотой в качестве матрицы (10 мкг/мл в 0.1% ТФА : ацетонитрил 50:50).
Для измерения массы интактного белка прибор устанавливали в линейный режим (Linear) и использованием настройки импульсной экстракции 16,952 для стандарта миоглобина для калибровки прибора (Shimadzu Axima TOF2).
[001342] Интактный белок ErbB4 наблюдают при МН+ 35850, соответствующие продукты присоединения синопиновой кислоты (матрицы) имеют массу примерно на 200 Да выше. Стехиометрическое включение исследуемого соединения (I-4 и I-11) (ММ 410 Да) дало новый пика массы примерно на 410 Да выше (МН+ 36260). Это соответствует ковалентной модификации ErbB4 соединениями I-4 и I-11.
ПРИМЕР 276
Ингибирование киназ ErbB1, ErbB2 и/или ErbB4
[001343] Соединения согласно настоящему изобретению исследовали в качестве ингибиторов одной или более киназ ErbB1, ErbB2 и/или ErbB4 по существу аналогично методу, описанного Invitrogen Corp (Invitrogen Corporation, 1600 Faraday Avenue, Карлсбад, Калифорния; http://www.invitrogen.com/downloads/Z-LYTE_Brochure_1205.pdf) с использованием процедуры биохимического анализа Z'-LYTE™ или аналогичного биохимического анализа. Биохимическом анализ Z'-LYTE™ использует флюоресцентный формат со связанным ферментом и основан на различной чувствительности фосфорилированного и нефосфорилированного пептидов к протеолитическому расщеплению. При помощи этого анализа было обнаружено, что соединение I-56 ингибирует ERBB1 с IC50, равной 2,233 нМ. При помощи этого анализа было обнаружено, что соединение I-56 ингибирует ERBB4 (HER4) с IC50, равной 2,165 нМ.
ПРИМЕР 277
Масс-спетрометрия для киназы Janus-3 (JAK3)
[001344] Киназу JAK3 (33 пмоль; Invitrogen) инкубировали с (I-7) (327 пмоль) в течение 3 часов при 10Х избытке, после чего осуществляли расщепление трипсином. После инкубации с соединением использовали иодацетамид в качестве алкилирующего агента. Аликвоты продуктов расщепления трипсином объемом 5 мкл (5.5 пмоль) разбавляли 15 мкл 0.1% ТФА, а затем наносили с использованием микро-наконечника
C18 Zip Tip на мишень MALDI с альфа-циано-4-гидрокси-коричной кислотой в качестве матрицы (5 мкг/мл в 0.1% смеси ТФА : ацетонитрил 50:50).
[001345] Как показано на Фигуре 11, ожидаемый модифицированный пептид (LVMEYLPSGCLR) немедленно обнаруживали в форме самого крупного пика на МН+ 1725.88. Эта масса соответствует ожидаемой для случая, когда соединение I-7, с присоединяемой массой 345.16, добавляют к массе пептида 1380.70. Интересно, что пептида, модифицированный иодоацетамидом не обнаруживали при МН+ 1437.73 в продуктах расщепления после реакции с соединением I-7, что указывает, на неполное завершение реакции. Также обнаружили признаки присутствия других модифицированных, однако сигналы были низкими.
[001346] Признаки присутствия соединения I-7 наблюдали при МН+ 346.12 в диапазоне низких масс спектра. Спектр фрагментации пика 346.12 не выявил диагностических фрагментов, которые были видны в PSD-спектре модифицированных пептидов (См. Фиг. 11).
[001347] Чтобы дополнительно удостовериться в присутствии модифицированных пептидов при использовании соединения I-7, пептиды с МН+ 1725.88 и 1118.55 подвергали PSD-анализу (МС/МС). Анализ корреляции с базой данных для человека позволил идентифицировать соответствующие пептиды как модифицированные соединением I-7. Соединение I-11 также тестировали с использованием той же процедуры и обнаружили измеряемую модификацию.
Аппаратура:
[001348] Для продуктов расщепления трипсином аппарат устанавливали в режим Reflectron с настройкой пульсовой экстракции 2200. Калибровку проводили с использованием стандартов Laser Biolabs Pep Mix (1046.54, 1296.69, 1672.92, 2093.09, 2465.20). Для анализа CID/PSD пептид отбирали, используя курсор для того, чтобы задавать интервалы пропускания ионов и фрагментацию, которые имеют место при энергии лазера выше 20%, в качестве газа столкновения для CID использовали гелий. Калибровку для фрагментов осуществляли с использованием калибровки P14R для режима Curved field Reflectron.
ПРИМЕР 278,
Протокола общего теста для оценки эффективности в отношении активных форм JAK3:
[001349] Общий протокол тестирования для оценки эффективности в отношении JAK3 осуществляли по существу аналогично протоколу, описанному выше в Примере 251, за тем исключением, что использовали следующие условия реакции, оптимизированные для JAK3:
[JAK3]=5 нМ, [АТФ]=5 мкМ, [Y12-Sox]=5 мкМ (АТФ KМарр ~5 мкМ).
ПРИМЕР 279
[001350] В Таблице 18 показана активность отобранных соединений согласно настоящему изобретению в тесте на ингибирование JAK3. Номера соединений соответствуют номерам соединений в Таблице 5. Соединения, активность которых обозначена "А", продемонстрировали IC50≤10 нМ; соединения, активность которых обозначена "В", продемонстрировали IC50 10-100 нМ; соединения, активность которых обозначена "С", продемонстрировали IC50 100-1000 нМ; соединения, активность которых обозначена "D", продемонстрировали IC50 1000-10,000 нМ; и соединения, активность которых обозначена "Е", продемонстрировали IC50≥10,000 нМ.
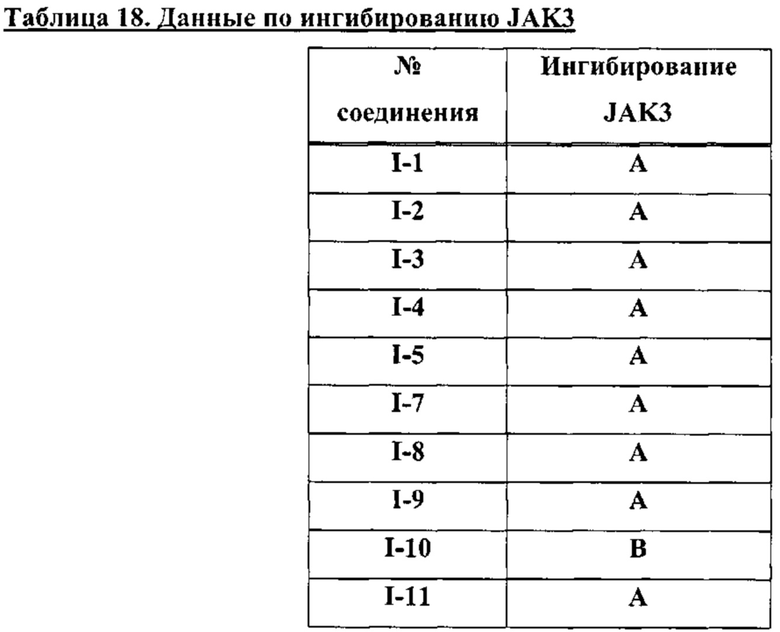

ПРИМЕР 280
Протокол анализа JAK3 с использованием клеток CTLL2
[001351] Соединения I-2, I-4 и I-7 исследовались в соответствии со следующим протоколом.
CTLL2: линия клеток лимфомы мыши АТСС: TIB-214. 5×106 клеток/образец выдерживали без IL-2 в среде RPMI-1640 в течение 2 часов. Расщепленные образцы затем обрабатывали соединением в течение 90 минут. Затем образцы, за исключением контроля с ДМСО стимулировали 100 нМ IL-2 в течение 10 минут. Образцы лизировали и подвергали анализу методом вестерн-блот. Результаты представлены на Фигуре 12, Фигуре 13 и Фигуре 14.
ПРИМЕР 281
Занятие BTK клетках Ramos соединениями I-7 и I-215 с использованием гранул со стрептавидином
[001352] Клетки Ramos инкубировали с 0.1, 0.05, 0.01 или 0.001 мкМ 1-7 в бессывороточной среде в течение 1 часа при 37°С. Клетки осаждали путем центрифугирования и лизировали в буфере для экстракции клеток (Invitrogen) в течение 10 минут на льду, центрифугировали (10 минут при 14,000 об./мин.) и собирали супернатанты. Лизаты клеток инкубировали с 1 мкМ 1-215 в течение 1 часа при комнатной температуре, а затем инкубировали с агарозными гранулами, связанными со стрептавидином (ThermoFisher) в течение ночи при 4°С. Гранулы трижды промывали лизирующим буфером и выпаривали связавшийся белок с гранул при 95°С в течение 5 минут в 4Х буфере для образцов LDS Sample Buffer. Количество ВТК, связанной с зондом I-215, оценивали путем вестерн-блоттинга на ВТК. Все значения нормировали по образцу, обработанному ДМСО, которому присваивали значение 100%. На Фигуре 16 показан вестерн-блот; на Фигуре 17 показан результат количественного анализа данных, представленных на Фигуре 16, демонстрирующие, что незанятый белок ВТК доступен для зонда I-215 при обработке клеток низкими концентрациями (10 нМ, 1 нМ) I-7, но при более высоких концентрациях I-7 белок ВТК полностью занят и не может взаимодействовать I-215.
ПРИМЕР 282
Эксперименты с отмывкой с 1-7 и соединением-зондом I-215
[001353] Клетки Ramos инкубировали 0.1 мкМ I-7 или контрольного соединения - обратимого ингибитора BTK в бессывороточной среде в течение 1 часа при 37°С. Затем клетки промывали в бессывороточной среде и лизировали через 0, 4, 6 или 8 часов после удаления соединения. Лизаты клеток инкубировали с 1 мкМ 1-215 в течение 1 часа при комнатной температуре, а затем в течение ночи при 4°С с агарозными гранулами, связанными со стрептавидином. Белки выпаривали с гранул и оценивали связь с BTK методом вестерн-блот. На Фигуре 18 показан Вестерн-блот; на Фигуре 19 показан результат количественного анализа данных, представленных на Фигуре 18, эта фигура демонстрирует, что весь белок BTK остается занятым соединением I-7 на протяжении более 8 часов. Это указывает на то, что временные рамки повторного синтеза детектируемого белка BTK в клетках Ramos превышают 8 часов. В контроле с обратимым ингибитором, напротив, 45% белка ВТК не связано и доступно для зонда в момент времени 0, а к 4 часам 100% белка BTK не связано и доступно для связывания с зондом. Все образцы нормировали по клеткам, обработанным ДМСО, которые собирали в 0 часов.
ПРИМЕР 283
Измерение занятости BTK в in vitro образцах методом твердофазного ИФА (ELISA)
[001354] Для того, чтобы определить количество свободной BTK в лизатах клеток или тканей, применяли протокол ELISA с использованием биотинилированного соединения-зонда, который связывается только со свободным, незанятым BTK. Конъюгированный биотин иммобилизуют на покрытом стрептавидином планшете для ELISA и детектируют антителом мыши к BTK (Becton Dickinson, Franklin Lakes, Нью-Джерси, США) и вторичным антителом козы к антителу мыши с HRP antibody (Zymed, South San Francisco, Калифорния, США).
[001355] Все образцы готовили с одинаковыми концентрациями буфера для лизиса Biorad (Неrсмклеs, Калифорния, США), 0.5% бычьего сывороточного альбумина в ФБР с 0.05% Tween-20, с конечной концентрацией 1 мкМ I-215. Образцы инкубировали в планшете для смешивания в течение 1 часа при комнатной температуре при
перемешивании, чтобы обеспечить связывание соединения I-215 со свободной BTK. После инкубации с I-215 образцы добавляли в промытый планшет для ELISA, покрытый стрептавидином (Pierce, Rockford, Иллинойс, США) и инкубировали в течение 1 часа при комнатной температуре при встряхивании. Затем планшеты промывали ФБР, содержащим 0.05% Tween-20, с использованием автоматического промывателя для планшетов. Антитело к BTK готовили при разбавлении 1:1000 в 0.5% БСА в ФБР (0.05% Tween-20) и добавляли в планшет для ELISA. Планшет инкубировали в течение 1 часа при комнатной температуре со встряхиванием. Планшет промывали, как описано выше, и готовили вторичное антитело HRP с разбавлением 1:5000 в 0.5% БСА в ФБР (0.05% Tween-20). Планшет инкубировали и промывали, как описано выше. В планшет добавляли ТМВ и мониторировали ОП650 до достижения 1 единицы ОП. Затем реакцию останавливали путем добавления H2SO4. Планшет анализировали с использованием программы Gen 5 software, и 4-параметрической логистической кривой для количественного анализа образцов. Для построения стандартной кривой использовали рекомбинантную ВТК (Invitrogen, Карлсбад, Калифорния, США).
[001356] В Таблице 19 показаны результаты, полученные с использованием клеток Ramos, представленные в форме концентрации, при которой занято >50% или >90% BTK. Концентрация, обозначенная "А" больше 1 нМ; концентрация, обозначенная "В" - больше 10 нМ; и концентрация, обозначенная "С" - больше 50 нМ.
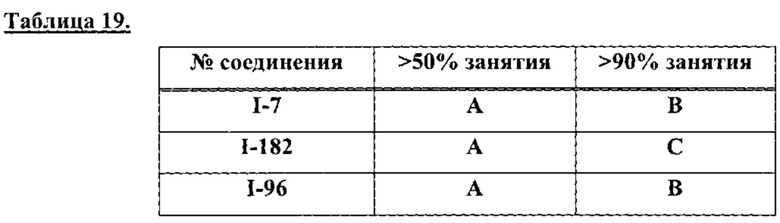
ПРИМЕР 284
Ковалентная занятость зонда в первичных В-клетках человека in vitro
[001357] Первичные В-клетки человека выделяли, как описано в Примере 256, а затем ресуспендировали в среде RPMI (10% сыворотки). К среде добавляли анализируемое соединение в разбавлении 1:1000. Клетки инкубировали с соединением в инкубаторе для культур клеток в течение 1 ч при 37°С. После инкубации клетки
осаждали, промывали 1X ФБР и лизировали на льду в течение 45 мин, время от времени встряхивая. Образцы центрифугировали в охлаждаемой микроцентрифуге в течение 30 мин при 14,000 об./мин. и выделяли супернатант. Супернатант исследовали, как описано в примере 283 с использование I-215. I-96 и I-182 занимали по меньшей мере 50% BTK при концентрациях более 10 нМ.
ПРИМЕР 285
Ковалентная занятость зонда в первичных В-клетках собаки in vitro
[001358] Цельную кровь собаки (30 мл) разбавляли до общего объема 50 мл 1X ФБР и наслаивали на Histopaque-1077 (Sigma Aldrich). Цельную кровь на Histopaque центрифугировали при 400 х g в течение 30 мин в центрифуге Beckman без перерыва Мононуклеары периферической крови (РВМС) собирали и осаждали при 400 х g в течение 15 мин. Эритроциты лизировали с использованием 2.5 мл буфера для лизирования эритроцитов (Boston Bioproducts) и оставшиеся РВМС трижды промывали IX ФБР при 250 х g. РВМС обрабатывали соединением при разбавлении 1:1000 в течение одного часа при 37°С, промывали ФБР и лизировали на льду в течение 45 минут. Лизат центрифугировали в течение 30 минут при14,000 х g и собирали супернатант. Супернатант анализировали, как описано в Примере 283 с использованием I-215. I-96 занимал по меньшей мере 50% BTK при концентрации больше 10 нМ.
ПРИМЕР 286
Измерение занятости BTK из образцов in vivo методом ELISA
[001359] Крысам перорально вводили 30 мг/кг соединения и собирали селезенки через 2 или 24 часа после введения соединения. Селезенки крыс разрушали между двумя стеклами, покрытыми матовым стеклом, чтобы получить суспензии отдельных клеток. Эритроциты лизировали путем инкубации с буфером для лизиса эритроцитов (Boston Bioproducts) в течение 2 минут при комнатной температуре, затем клетки ресуспендировали в полной среде RPMI и осаждали путем центрифугирования. В-клетки крысы выделяли путем положительного отбора с использованием конъюгатов В220+ антитело-магнитные гранулы, очищали с использованием колонки - MACS и лизировали в буфере для лизиса Bio-Rad при концентрации 10 миллионов клеток/100 мкл. Лизаты
анализировали с использованием биотинилированного соединения - зонда I-215 в протоколе ELISA, подробно описанном в Примере 278. В Таблице 20 показаны результаты.
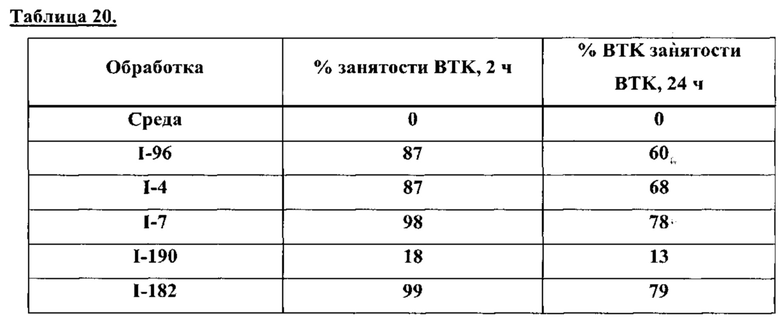
ПРИМЕР 286
Анализ методами протеомики
[001360] Белки, которые ковалентно связываются с I-215 в лизате клеток идентифицируют методом масс-спектрометрии. Лизаты клеток инкубирубт с 1 мкМ I-215 в течение 1 часа при комнатной температуре с последующим добавлением агарозных гранул, связанных со стрептавидином. Белки, отличные от BTK, идентифицируют путем масс-спектрометрии. Они соответствуют потенциальным «нецелевым» взаимодействиям.
[001361] Хотя в настоящем тексте описан рад вариантов осуществления настоящего изобретения, очевидно, что приведенные базовые примеры могут быть изменены, в результате чего будут реализованы другие варианты осуществления с использованием способов и соединений согласно настоящему изобретению. Соответственно, очевидно, что объем настоящего изобретения определяется сопутствующей формулой изобретение, а не конкретными вариантами осуществления, которые были приведены в качестве примеров.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2536584C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2636584C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ПУТИ JAK | 2011 |
|
RU2672100C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА | 2013 |
|
RU2637925C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОАРИЛЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2552114C2 |
| ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2701206C2 |
| Новые модулирующие регуляцию дыхания соединения и способы их получения и применения | 2016 |
|
RU2734506C2 |
| ИНГИБИТОРЫ PI3-КИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2595718C2 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
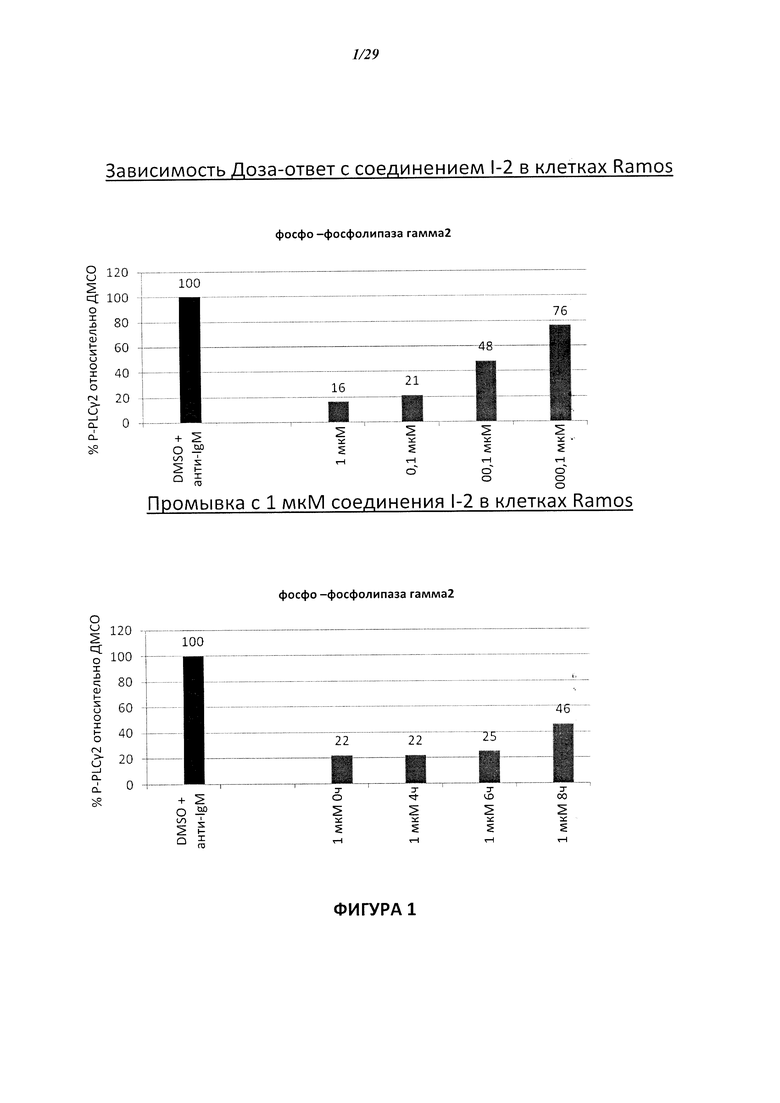
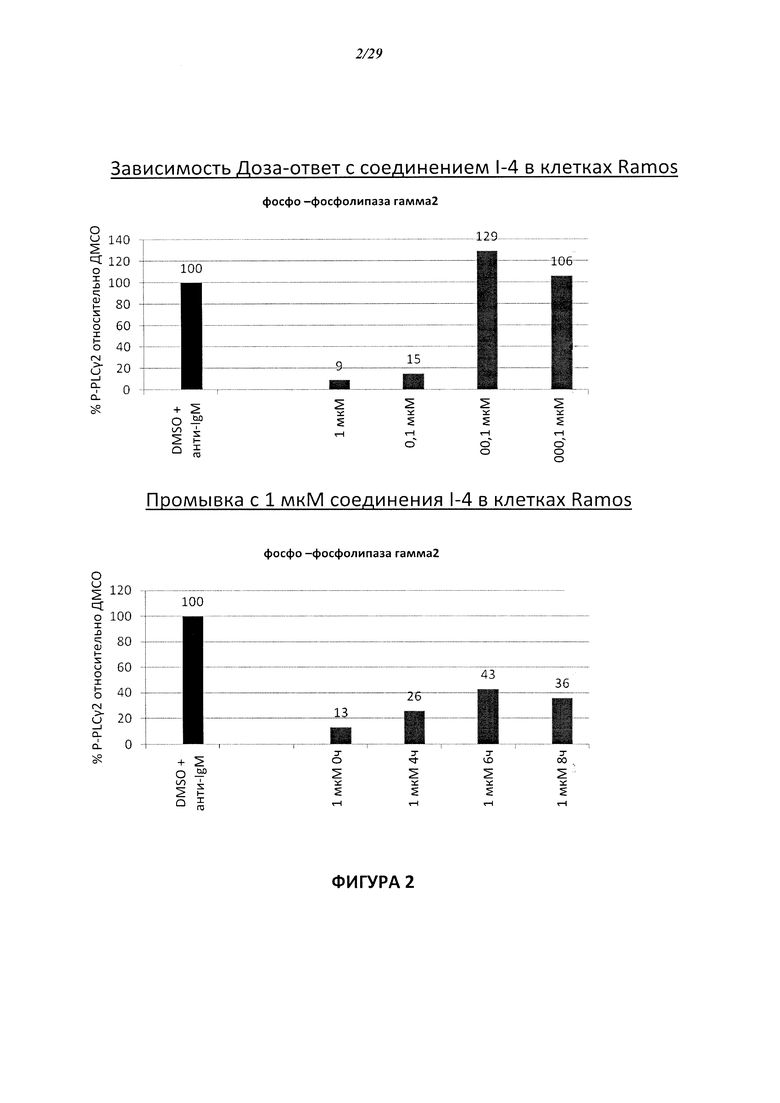
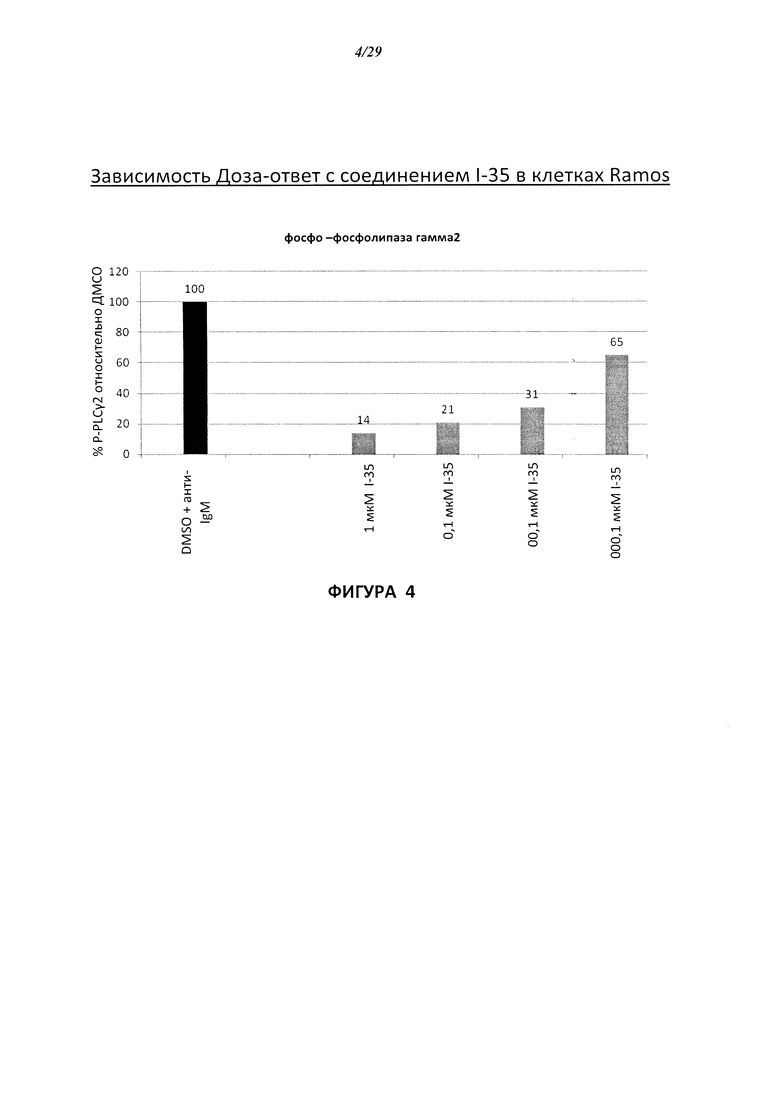
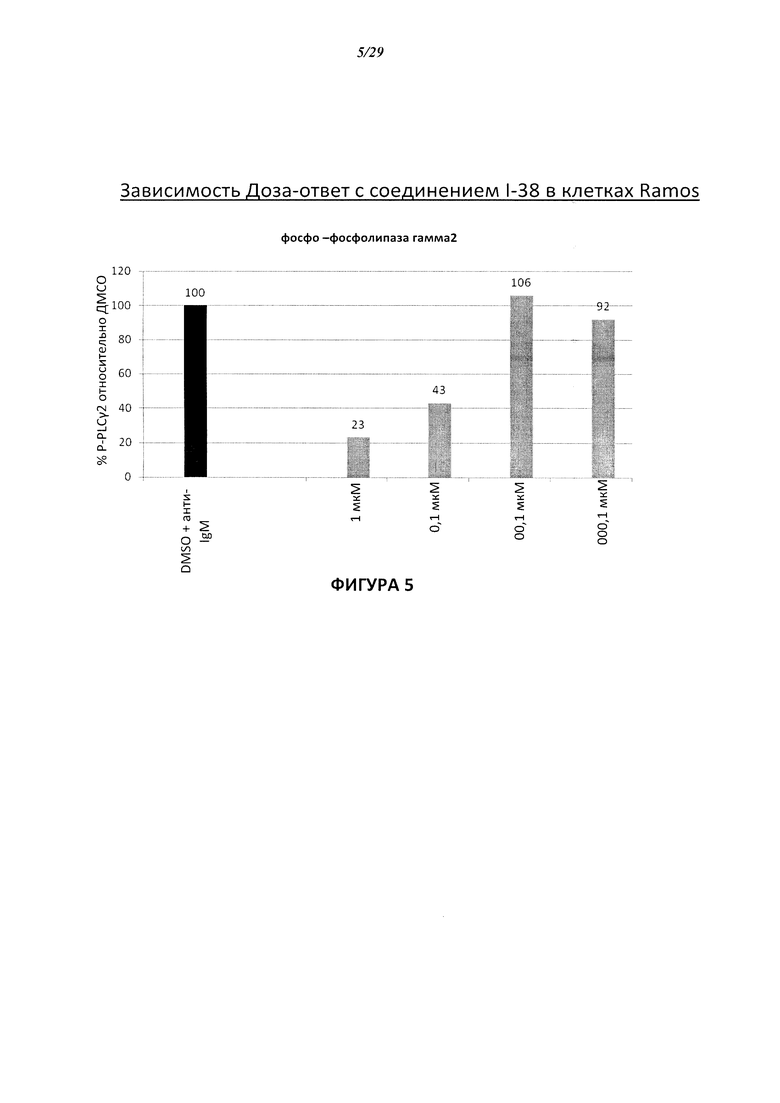

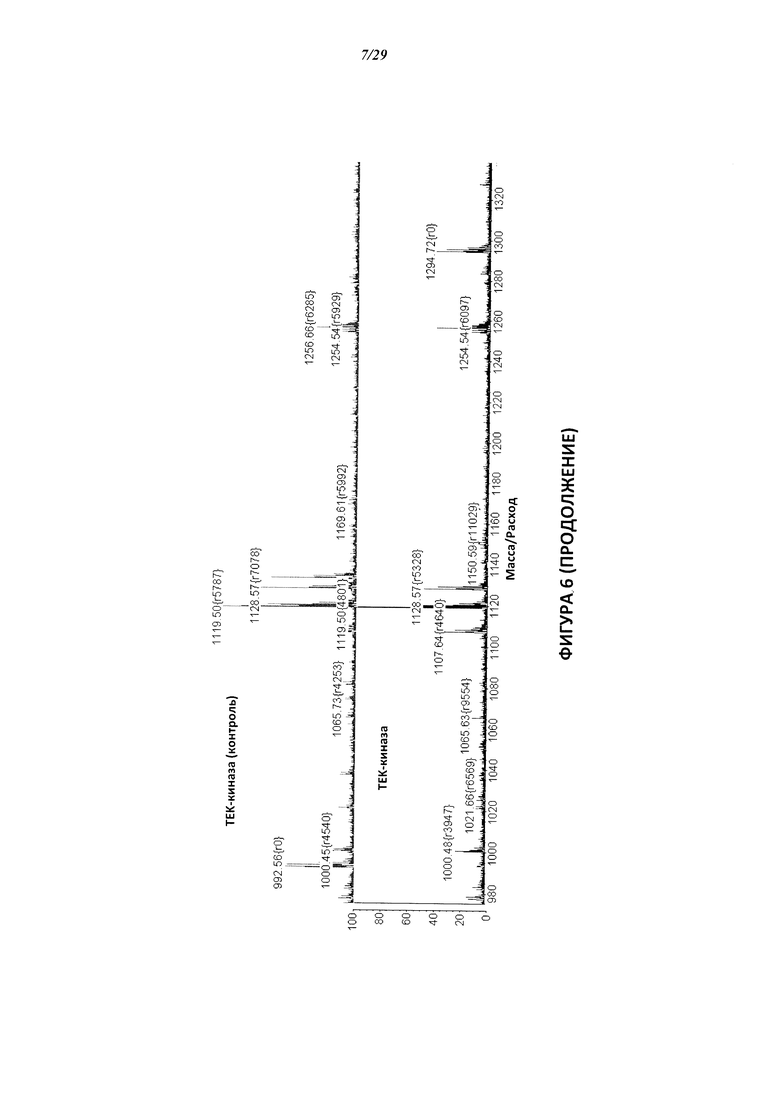

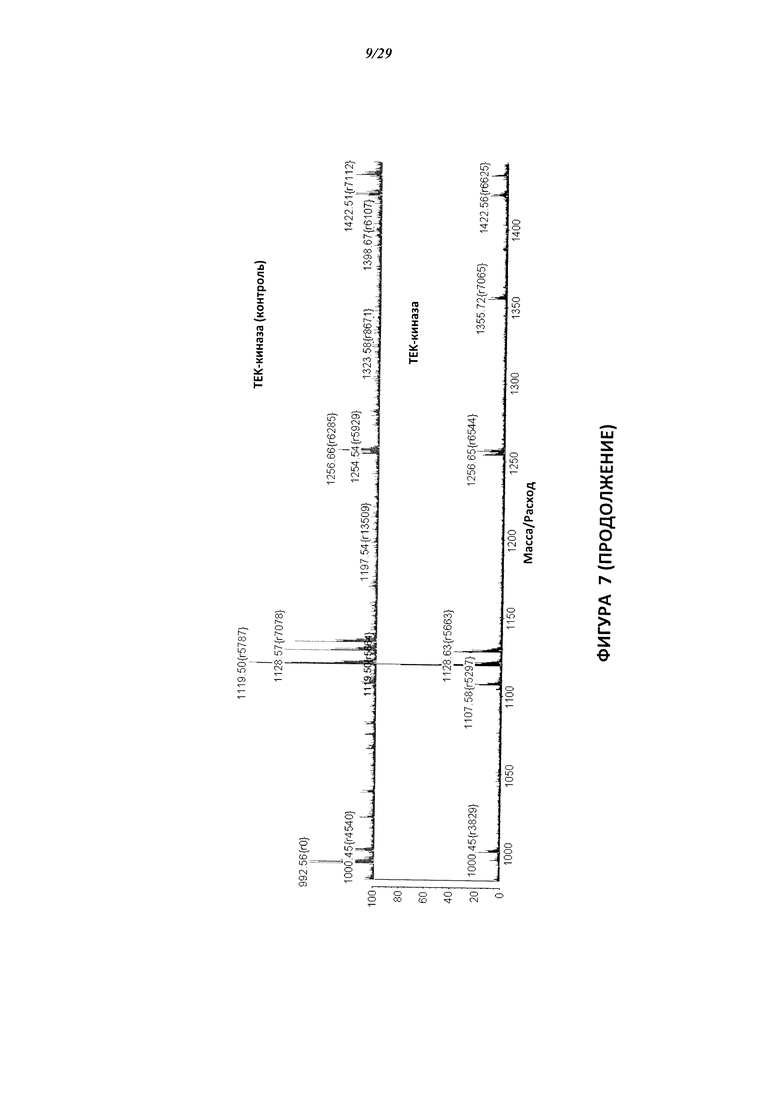

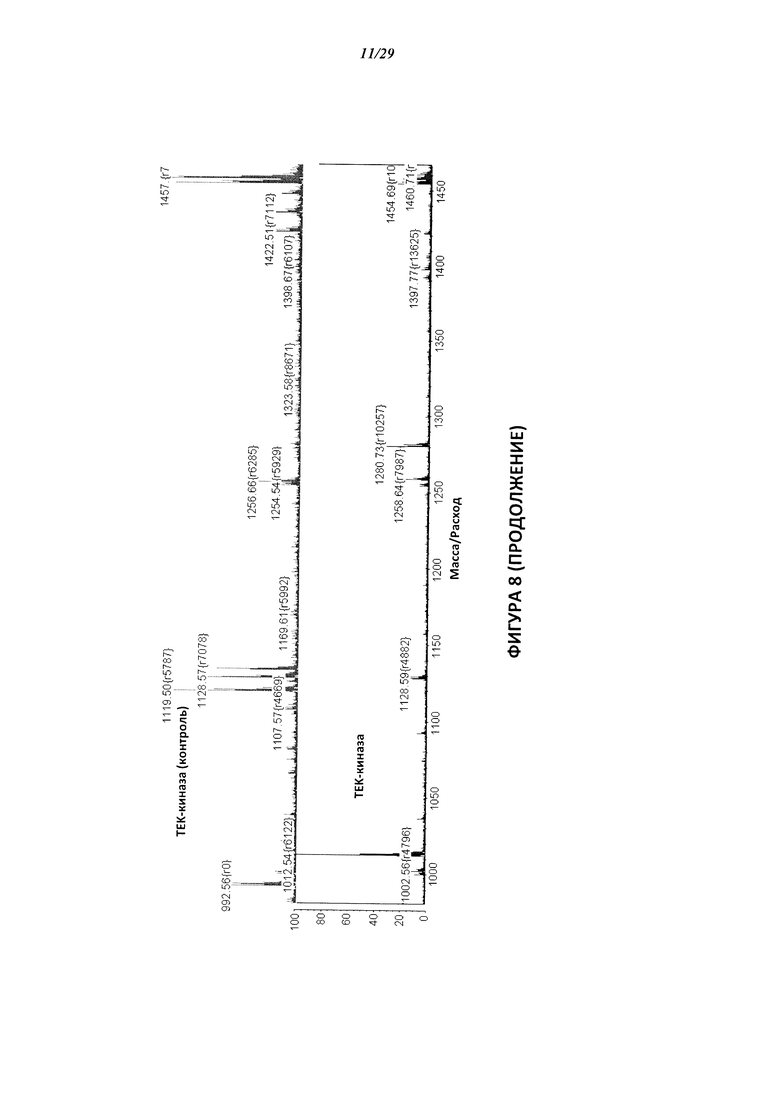

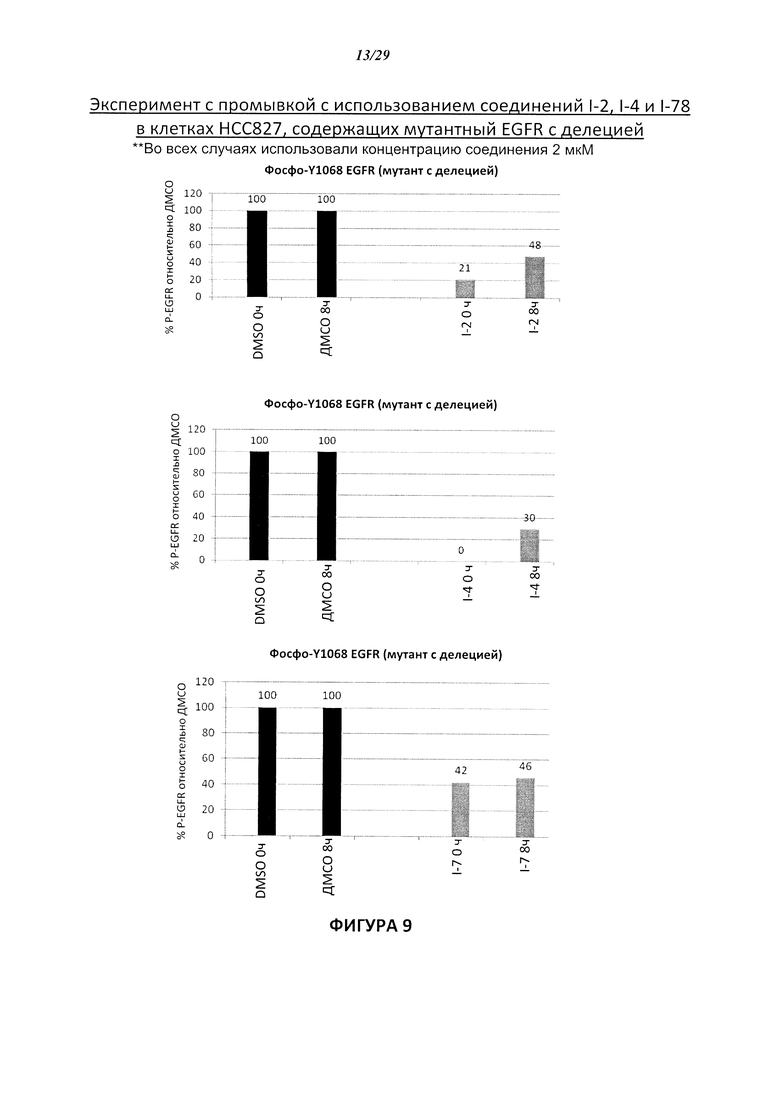
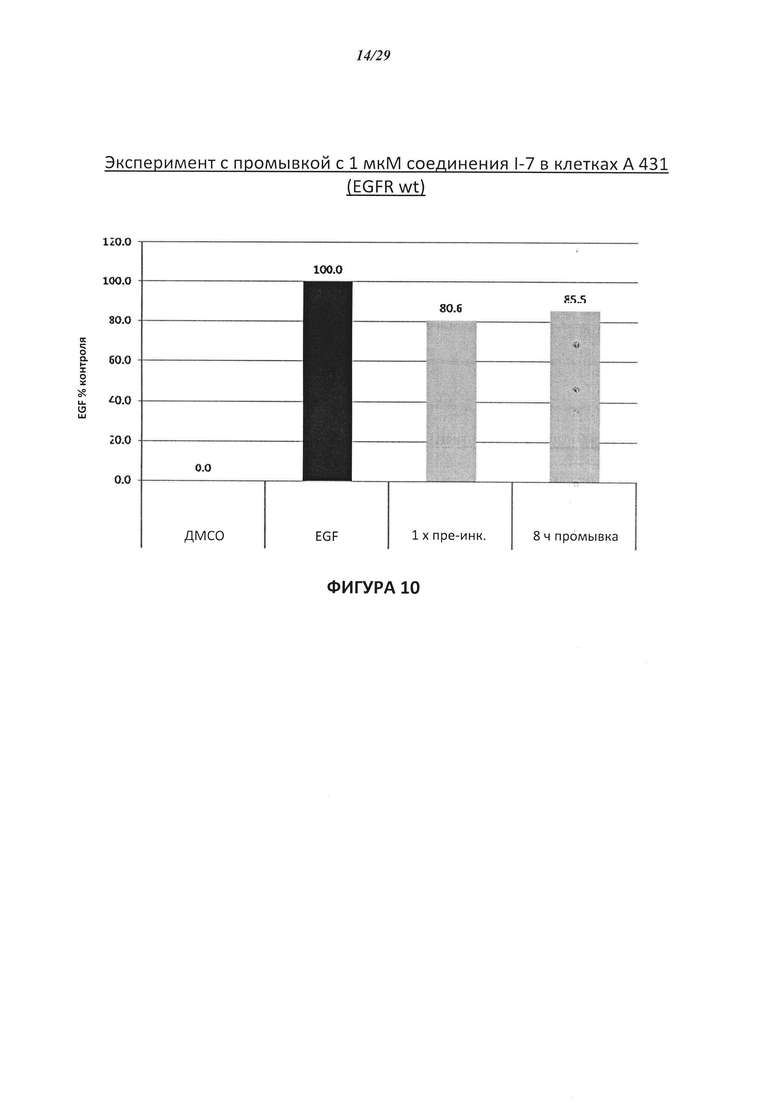

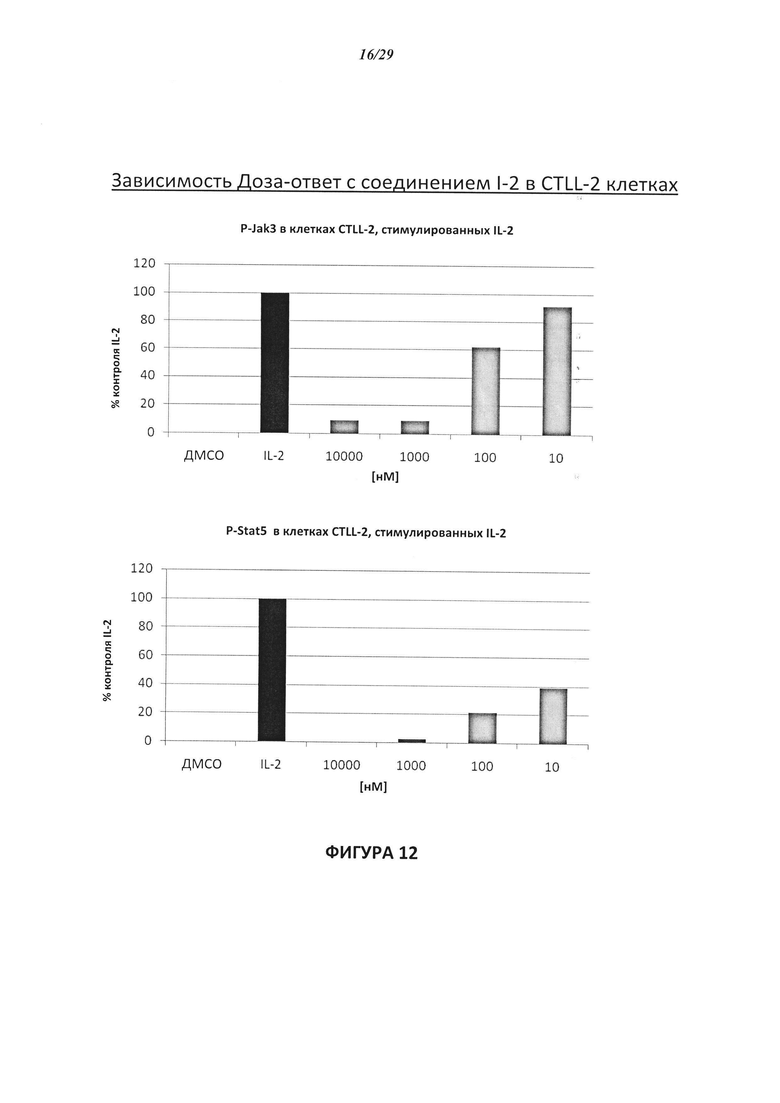
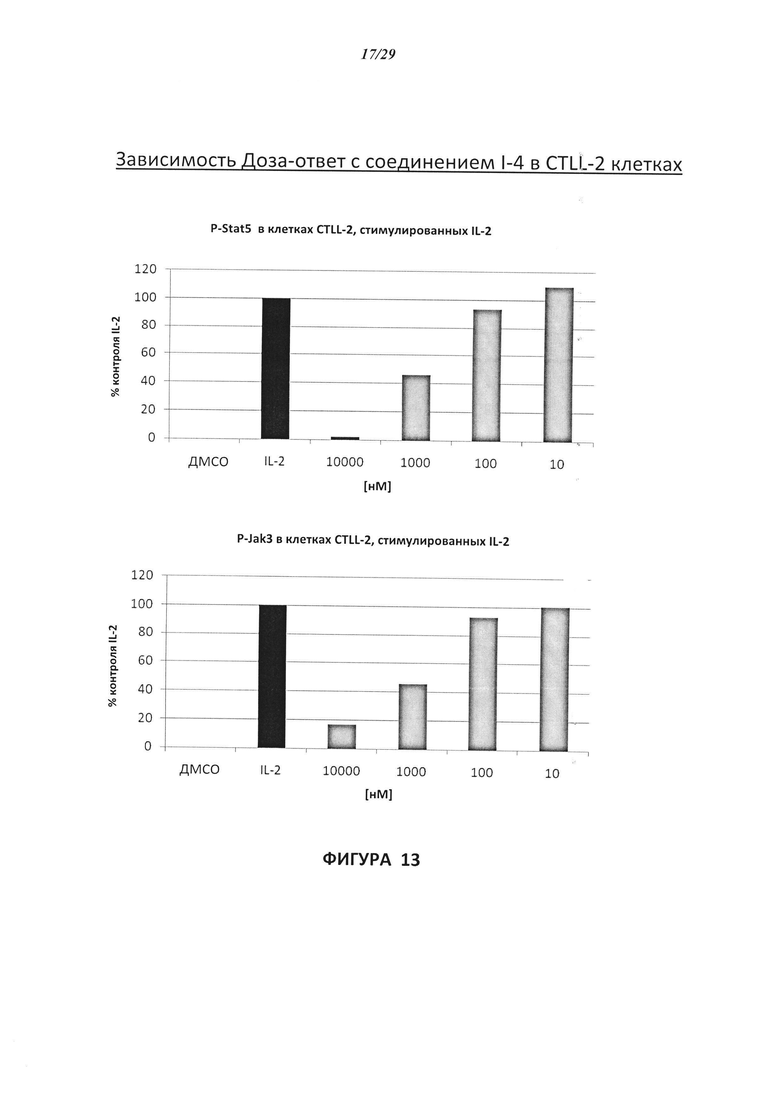
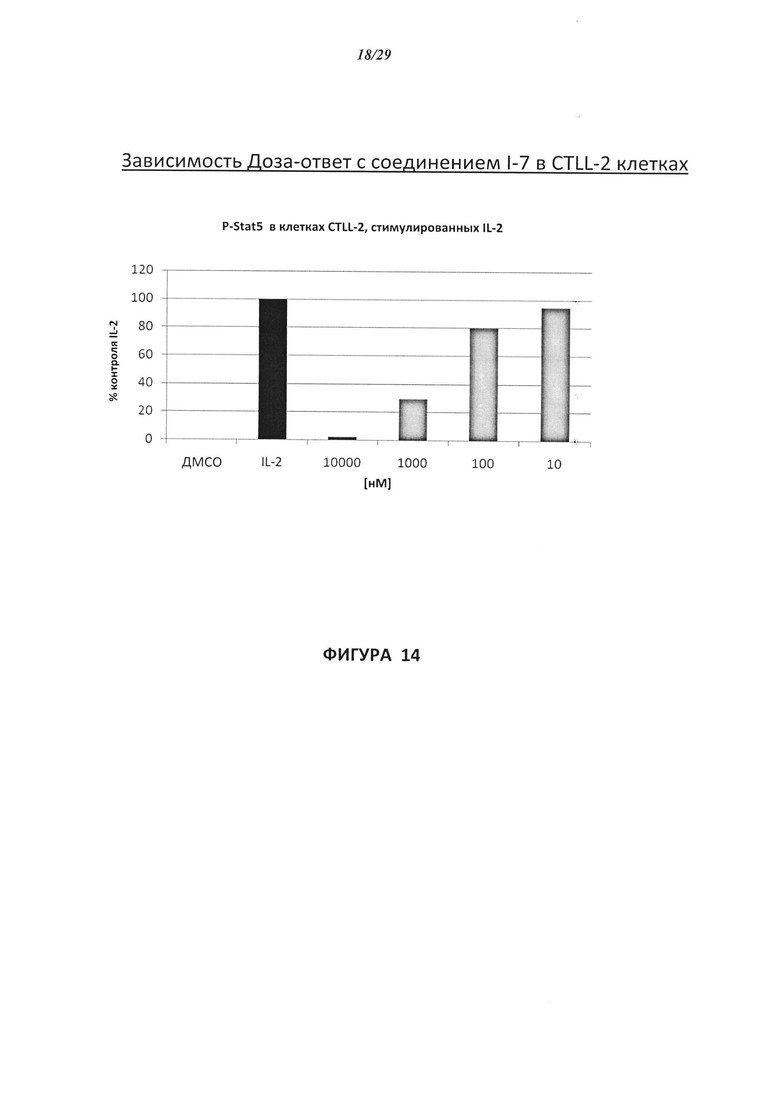
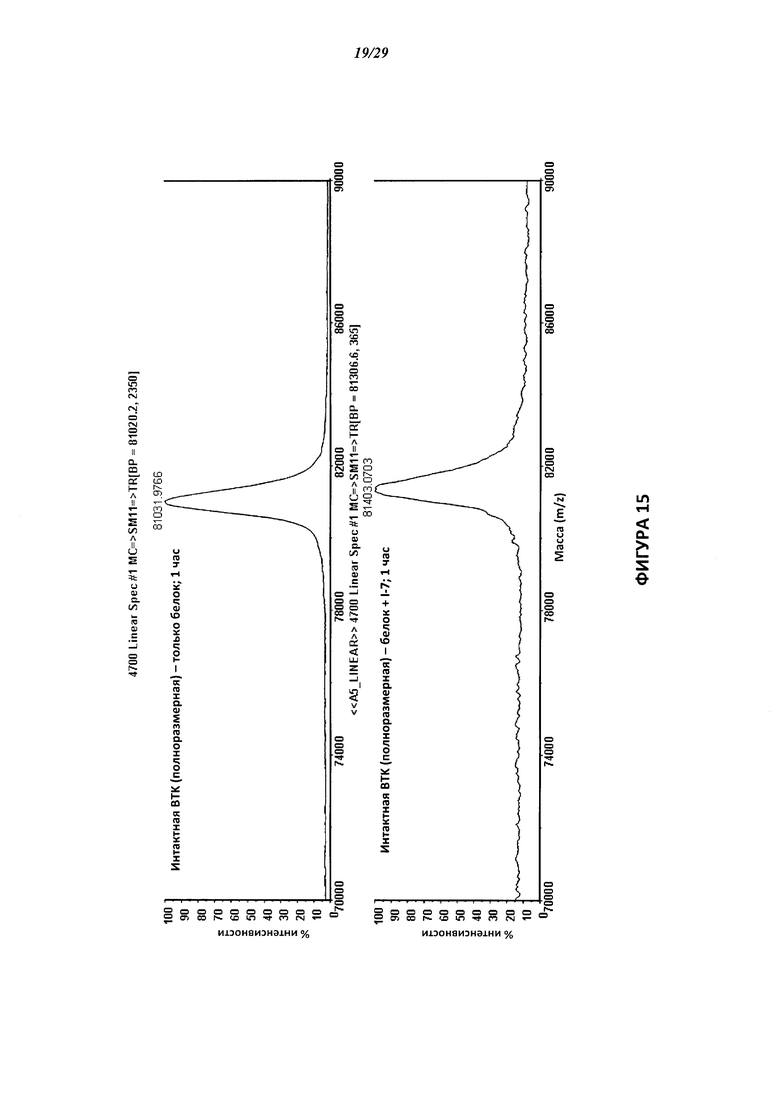
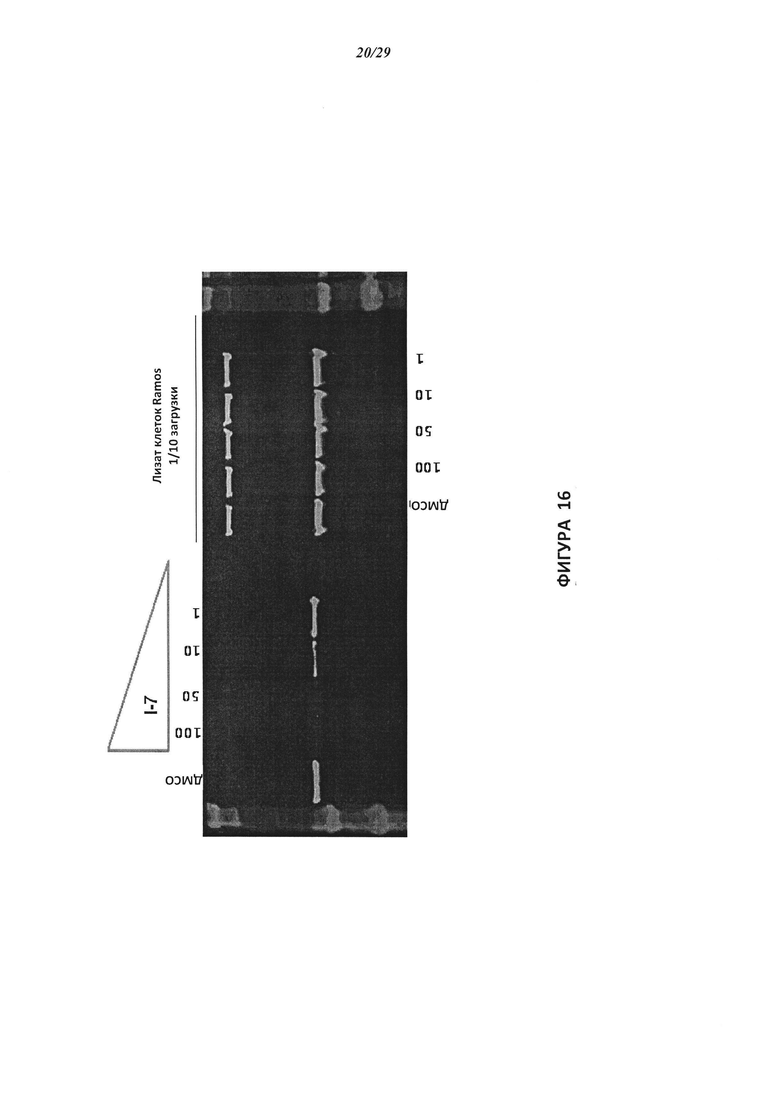
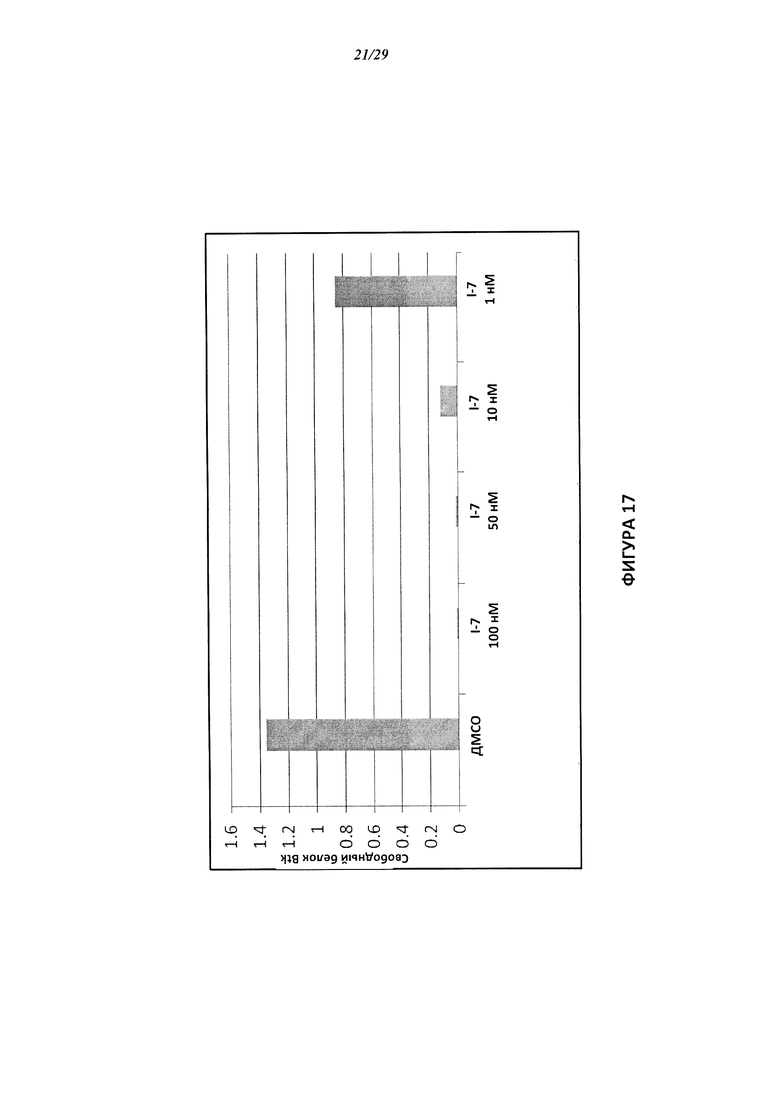

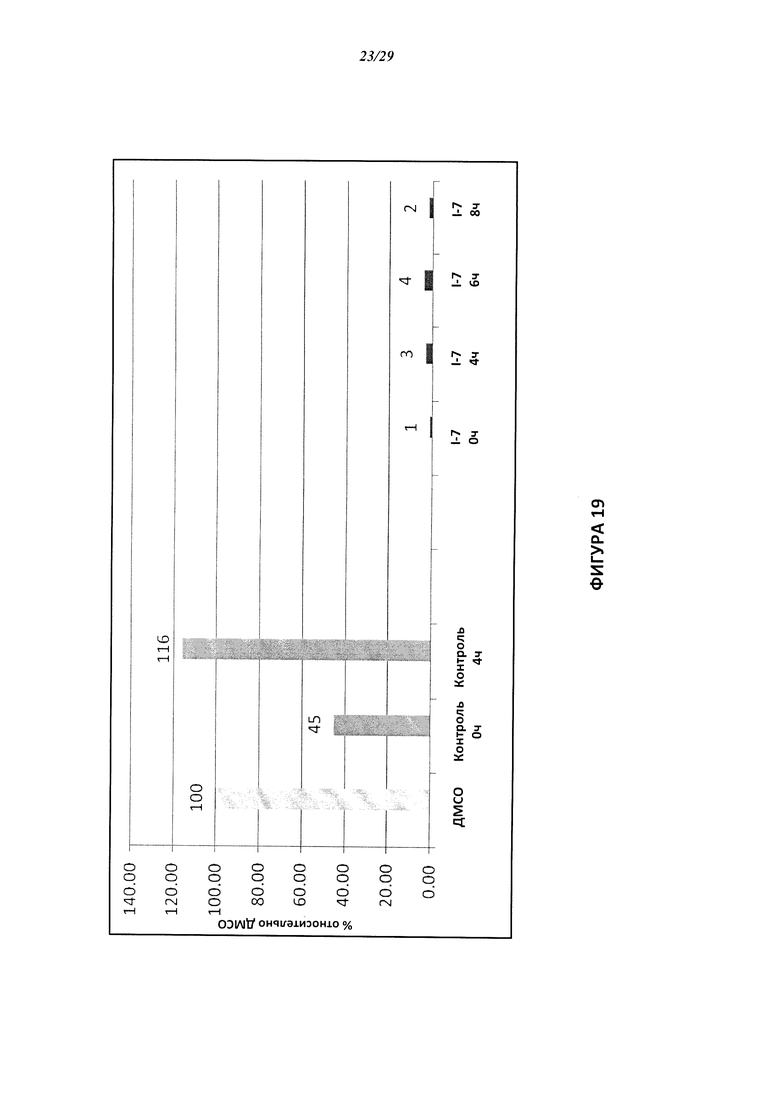






Изобретение относится к соединениям и их фармацевтически приемлемым солям, где в формуле I-b кольцо А и В представляют собой фенил; R1 представляет собой реакционноспособную группу -L-Y, где R1 присоединен к кольцу А в положении, отличном от атома, смежного к атому, присоединенному к W1, L и Y являются такими, как указано в формуле изобретения, W1 и W2 каждый представляет собой -NR2-; R2 представляет собой водород, С1-6 алифатическую группу или -C(O)R; m и р независимо равны 0-4; Ry представляет собой галоген; Rx независимо выбран из -R, -OR, -O(CH2)qOR или галогена, где q равно 1, 2, 3 или 4; и Rv независимо выбран из -R, галогена, -OR, -O(CH2)qOR, -CN, -NO2, -SO2R, -SO2N(R)2, -SOR, -C(O)R, -CO2R, -C(O)N(R)2, -NRC(O)R, -NRC(O)N(R)2, -NRSO2R или -N(R)2, где q равно 1-4. Также изобретение относится к композициям для ингибирования по меньшей мере одной киназы, выбранной из BTK, TEC, ВМХ, ITK, ErbB1, ErbB2, ErbB3, ErbB4 и JAK3 или их мутантов, способам ингибирования указанных киназ и их мутантов, а также к способам лечения нарушений, опосредуемых этими киназами. Технический результат – соединения формулы I-b в качестве ингибиторов активности по меньшей мере одной киназы, выбранной из BTK, TEC, ВМХ, ITK, ErbB1, ErbB2, ErbB3, ErbB4 и JAK3 или их мутантов. 11 н. и 36 з.п. ф-лы, 25 ил., 20 табл., 286 пр.
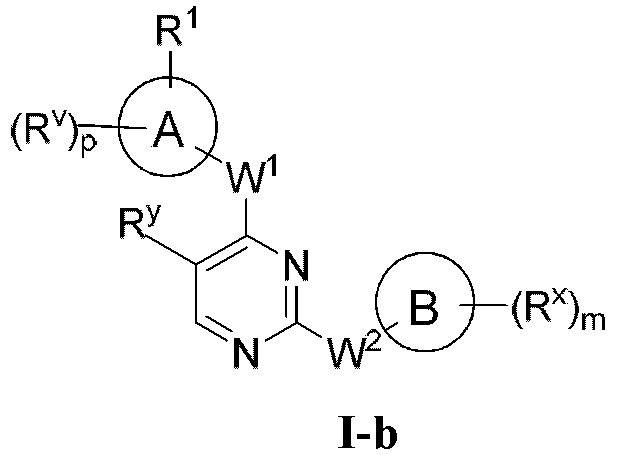
1. Соединение формулы I-b
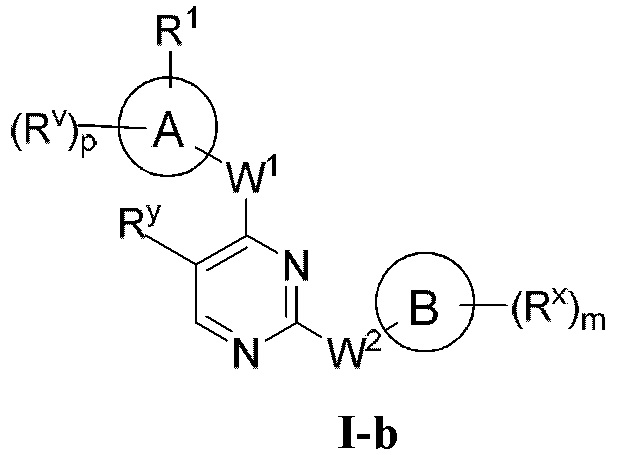
или фармацевтически приемлемая соль такого соединения,
где Кольцо А представляет собой фенил;
Кольцо В представляет собой фенил;
R1 представляет собой реакционноспособную группу -L-Y, где R1 присоединен к кольцу А в положении, отличном от атома, смежного к атому, присоединенному к W1;
-L-Y выбран из 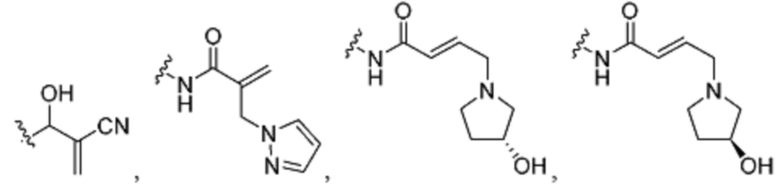 или
или 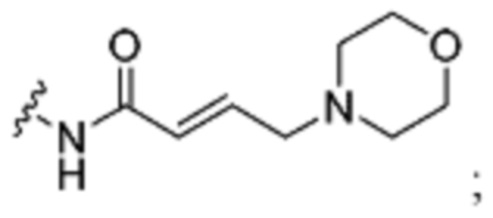 или
или
L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь, и один или два дополнительных метиленовых звена в L возможно и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, -C(O)O-, циклопропилен, -O-, -N(R)- или -C(O)-; и Y представляет собой C1-6 алифатическую группу, возможно замещенную оксо, галогеном, NO2 или CN; или 3-10-членное моноциклическое или бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, и где указанное кольцо замещено 1-4 группами Re; или
L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь и одно метиленовое звено в L заменено на -С(О)-, -NRC(O)-, -C(O)NR-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -C(O)O-; или
L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь, и одно метиленовое звено в L заменено на -С(О)-, и одно дополнительное метиленовое звено в L возможно заменено на циклопропилен, -О-, -N(R)- или -С(О)-; или
L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь в составе алкилиденила, и по меньшей мере одно метиленовое звено в L заменено на -С(О)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -C(O)O-, и один или два дополнительных метиленовых звена в L возможно и независимо заменены на циклопропилен, -О-, -N(R)- или -С(О)-; или
L представляет собой -NRC(O)CH=CHCH2N(CH3)-, -NRC(O)CH=CHCH2O- или -NRSO2CH=CHCH2-; и
Y представляет собой водород, C1-6 алифатическую группу, возможно замещенную оксо, галогеном, NO2 или CN, или 3-10-членное моноциклическое или бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, и где указанное кольцо замещено 1-4 группами Re; или
L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем одно метиленовое звено в L заменено циклопропиленом, и одно или два дополнительных метиленовых звена в L независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -C(O)O-; и
Y представляет собой C1-6 алифатическую группу, возможно замещенную оксо, галогеном, NO2 или CN; или
L представляет собой ковалентную связь; и Y выбран из:
(ii) С2-6 алкенила, замещенного оксо, галогеном, NO2 или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-2 группы Re; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, причем указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xvi)
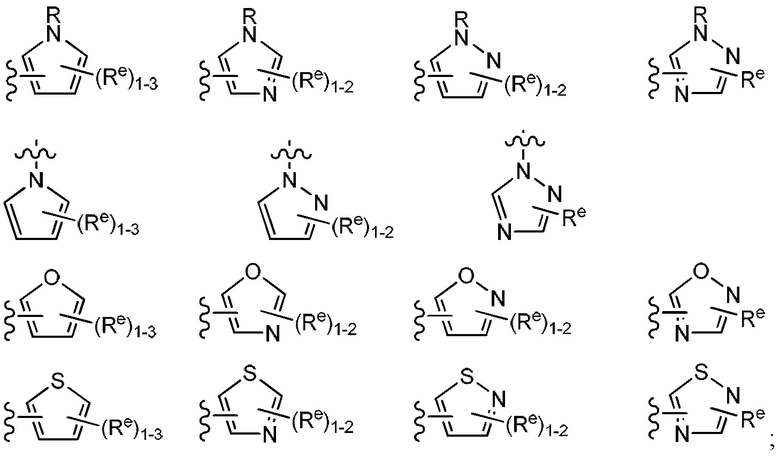 или
или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
L представляет собой -С(О)-; и Y выбран из:
(i) С1-6 алкила, содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(ii) С2-6 алкенила, возможно содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(iii) С2-6 алкинила, возможно содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-2 группы Re; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xv) 5-членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-3 группы Re; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1 -4 группы Re; или
L представляет собой -N(R)C(O)-; и Y выбран из:
(i) C1-6 алкила, содержащего в качестве заместителя оксо, галоген, NO2 или CN; или
(ii) С2-6 алкенила, возможно содержащего в качестве заместителя оксо, галоген, NO2 или CN; или
(iii) С2-6 алкинила, возможно содержащего в качестве заместителя оксо, галоген, NO2 или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-2 группы Re; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xv) 5-членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-3 группы Re; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
L представляет собой бивалентную C1-8 насыщенную или ненасыщенную, линейную или разветвленную углеводородную цепь; и Y выбран из:
(i) С1-6 алкила, содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(ii) С2-6 алкенила, возможно содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-2 группы Re; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xv) 5-членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-3 группы Re; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
L представляет собой -CH2-, -NH-, -С(О)-, -CH2NH-, -NHCH2-, -NHC(O)-, -NHC(O)CH2OC(O)-, -CH2NHC(O)-, -NHSO2CH2- или -SO2NH-; и Y выбран из:
(ii) С2-6 алкенила, содержащего в качестве заместителей оксо, галоген, NO2 или CN; или
(iv) насыщенного 3-4-членного гетероциклического кольца, содержащего 1 гетероатом, выбранный из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-2 группы Re; или
(v) насыщенного 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из кислорода или азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(vii) насыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(viii) частично ненасыщенного 3-6-членного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(ix) частично ненасыщенного 3-6-членного карбоциклического кольца, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xi) частично ненасыщенного 4-6-членного гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xiii) 6-членного ароматического кольца, содержащего 0-2 атома азота, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re; или
(xv) 5-членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-3 группы Re; или
(xvii) 8-10-членного бициклического насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re;
каждый Re независимо выбран из следующих: -Q-Z, оксо, NO2, галогена, CN, C1-6 насыщенной алифатической группы, содержащей в качестве заместителей оксо, галоген, NO2 или CN; C2-6 ненасыщенной алифатической группы, возможно содержащей в качестве заместителей оксо, галоген, NO2 или CN; или подходящей уходящей группы, выбранной из алкокси, сульфонилокси, алкилсульфонилокси, алкенилсульфонилокси, арилсульфонилокси, ацила или диазония,
где:
Q представляет собой бивалентную С2-6 ненасыщенную, линейную или разветвленную углеводородную цепь, причем одно или два метиленовых звена в Q возможно и независимо заменены на -N(R)-, -S-, -О-, -С(О)-, -ОС(О)-, -С(O)O-, -SO- или -SO2-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2- или -SO2N(R)-; и
каждый Z независимо представляет собой водород или C1-6 алифатическую группу, содержащую в качестве заместителей оксо, галоген, NO2 или CN;
Ry представляет собой галоген;
каждая группа R независимо представляет собой водород или возможно содержащую заместители группу, выбранную из следующих: C1-6 алифатическая группа, фенил, 4-7-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, или 5-6-членное моноциклическое гетероарильное кольцо, содержащее 1-4 гетероатома, независимо выбранных из азота, кислорода или серы;
заместитель возможно замещенного атома углерода возможно замещенной группы R независимо выбран из галогена, -(CH2)0-4R°, -(СН2)0-4N(R°)C(O)OR°, -(СН2)0-4S(O)2R°, -(CH2)0-4N(R°)2, -(CH2)0-4OR°, -(CH2)0-4C(O)R° и -(CH2)0-4N(R°)C(O)R°; где каждый R° независимо выбран из водорода, C1-6 алифатической группы или 5-6-членного насыщенного или арильного кольца, содержащего 0-4 гетероатома, независимо выбранных из азота, кислорода или серы; или два независимых R° вместе с промежуточным(и) атомом(ами) образуют 3-12-членное насыщенное моно- или бициклическое кольцо с 0-4 гетероатомами, независимо выбранными из азота, кислорода или серы; где каждый R° возможно замещен моновалентным заместителем, независимо выбранным из -(СН2)0-2R• и -(СН2)0-2ОН; где каждый R• представляет собой C1-4 алифатическую группу; или каждый R° возможно замещен при насыщенном атоме углерода группой =O,
заместитель возможно замещенного атома азота возможно замещенной группы R независимо представляет собой  или
или  где каждый
где каждый  независимо выбран из C1-6 алифатической группы, необязательно замещенной -ОН;
независимо выбран из C1-6 алифатической группы, необязательно замещенной -ОН;
W1 и W2 каждый представляет собой -NR2-;
R2 представляет собой водород, С1-6 алифатическую группу или -C(O)R;
m и р независимо равны 0-4; и
в каждом случае Rx независимо выбран из -R, -OR, -O(CH2)qOR или галогена, где q равно 1, 2, 3 или 4; и
в каждом случае Rv независимо выбран из -R, галогена, -OR, -O(CH2)qOR, -CN, -NO2, -SO2R, -SO2N(R)2, -SOR, -C(O)R, -CO2R, -C(O)N(R)2, -NRC(O)R, -NRC(O)N(R)2, -NRSO2R или -N(R)2, где q равно 1-4.
2. Соединение по п. 1, отличающееся тем, что Кольцо А выбрано из:
3. Соединение по п. 2, отличающееся тем, что Кольцо А выбрано из lxiii, lxxi и lxxxi.
4. Соединение по п. 1, отличающееся тем, что Кольцо В выбрано из:
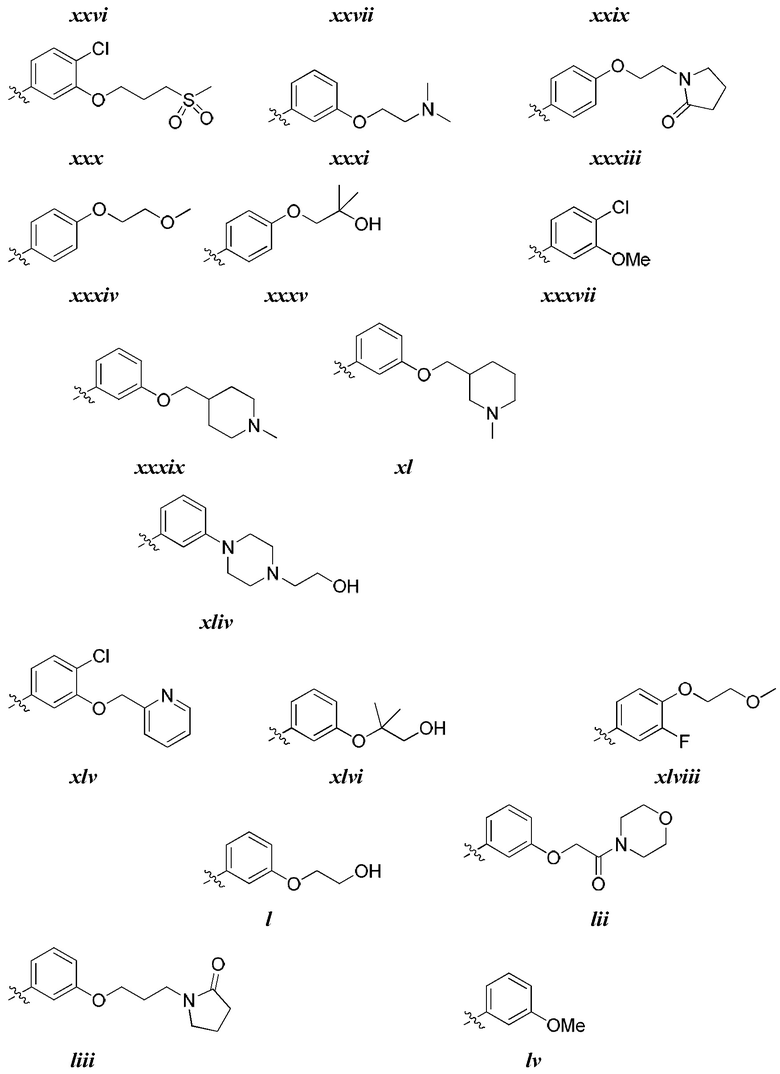
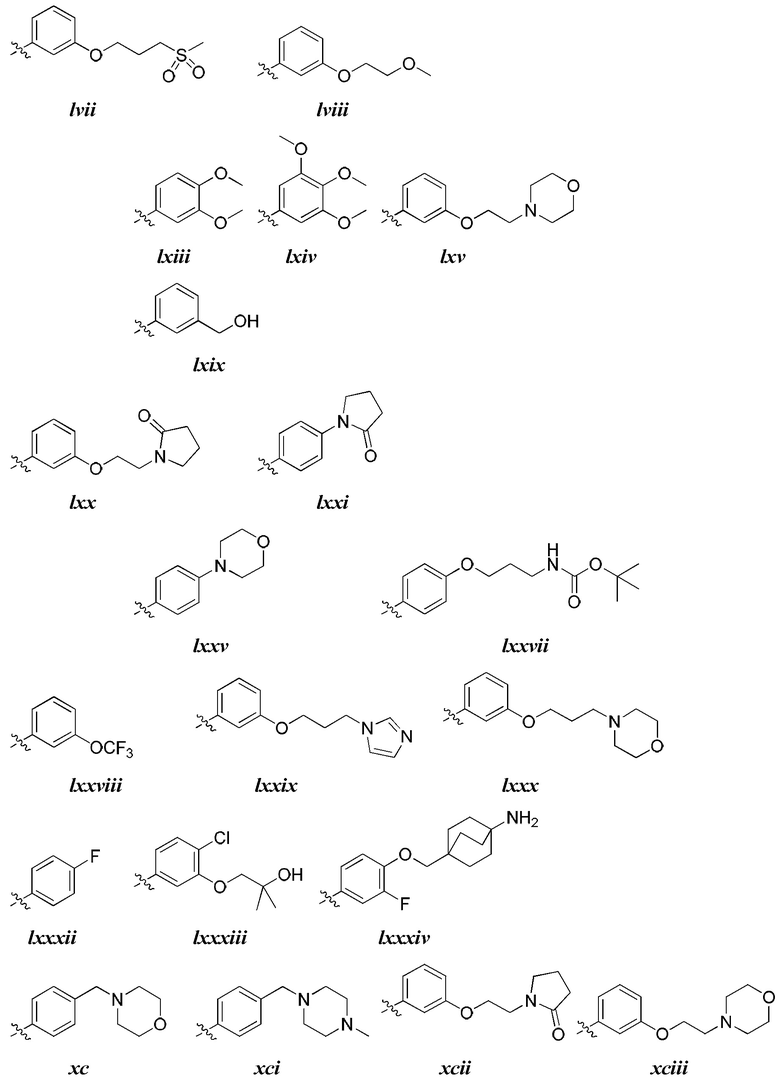
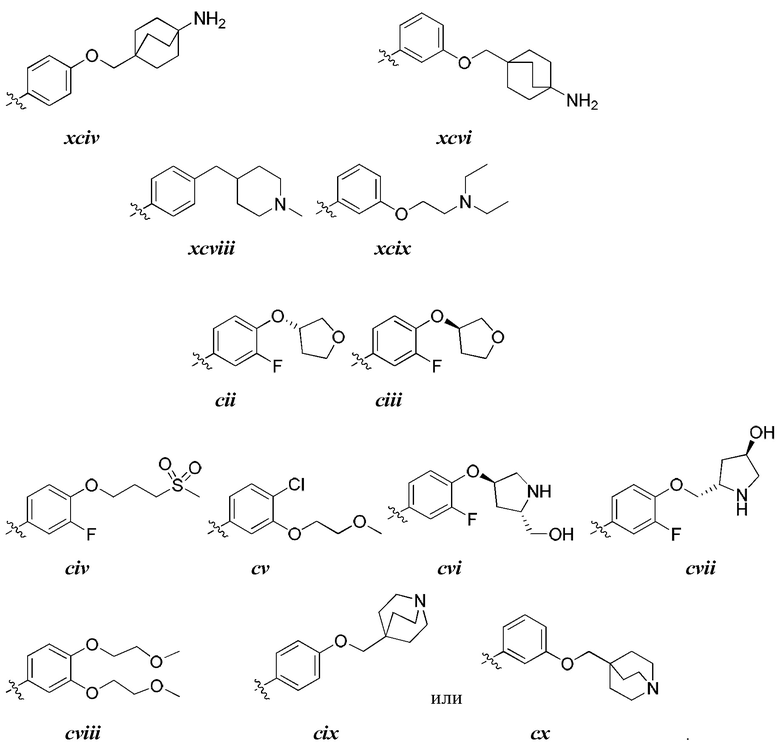
5. Соединение по п. 4, отличающееся тем, что Кольцо В выбрано из xi, xix, хх, xxv, xxvi, xxxiv, xxxv, xlvi, xlviii, l, lviii, lxiv, lxxviii, lxxxiii, xciv, cii, ciii, civ и cv.
6. Соединение по п. 1, отличающееся тем, что:
L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь, и один или два дополнительных метиленовых звена в L возможно и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, -C(O)O-, циклопропилен, -O-, -N(R)- или -C(O)-; и
Y представляет собой C1-6 алифатическую группу, возможно содержащую в качестве заместителя оксо, галоген, NO2 или CN, или 3-10-членное моноциклическое или бициклическое насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, и при этом указанное кольцо содержит в качестве заместителей 1-4 группы Re.
7. Соединение по п. 1, отличающееся тем, что:
L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь, и одно метиленовое звено в L заменено на -С(О)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -C(O)O-; и
Y представляет собой водород или C1-6 алифатическую группу, возможно содержащую в качестве заместителей оксо, галоген, NO2 или CN.
8. Соединение по п. 1, отличающееся тем, что L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь, и одно метиленовое звено в L заменено на -С(О)-, и одно дополнительное метиленовое звено в L возможно заменено на циклопропилен, -О-, -N(R)- или -С(О)-.
9. Соединение по п. 7, отличающееся тем, что L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь, и одно метиленовое звено в L заменено на -ОС(О)-.
10. Соединение по п. 1, отличающееся тем, что L представляет собой -NRC(O)CH=CH-, -NRC(O)CH=CHCH2N(CH3)-, -NRC(O)CH=CHCH2O-, -CH2NRC(O)CH=CH-, -NRSO2CH=CH-, -NRSO2CH=CHCH2- или -NRC(O)C(=CH2)CH2-; и при этом R в L представляет собой Н или C1-6 алифатическую группу; и Y представляет собой водород или C1-6 алифатическую группу, возможно содержащую в качестве заместителей оксо, галоген, NO2 или CN.
11. Соединение по п. 10, отличающееся тем, что L представляет собой -NHC(O)CH=CH-, -NHC(O)CH=CHCH2N(CH3)-, -NHC(O)CH=CHCH2O-, -CH2NHC(O)CH=CH-, -NHSO2CH=CH-, -NHSO2CH=CHCH2- или -NHC(O)C(=CH2)CH2-.
12. Соединение по п. 1, отличающееся тем, что L представляет собой бивалентную С2-8 линейную или разветвленную углеводородную цепь, причем L содержит по меньшей мере одну двойную связь в составе алкилиденила, и по меньшей мере одно метиленовое звено в L заменено на -С(О)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -C(O)O-, и один или два дополнительных метиленовых звена в L возможно и независимо заменены на циклопропилен, -О-, -N(R)- или -С(О)-.
13. Соединение по п. 1, отличающееся тем, что -L-Y представляет собой -NHC(O)CH=CH2.
14. Соединение по п. 13, отличающееся тем, что каждый R2 представляет собой водород.
15. Соединение по п. 14, отличающееся тем, что m равно 1.
16. Соединение по п. 15, отличающееся тем, что Rx представляет собой C1-6 алкил, -OC1-6 алкил, -O(CH2)qOC1-6 алкил или галоген.
17. Соединение по п. 16, отличающееся тем, что Rx представляет собой метил, метокси, метоксиэтокси или фтор.
18. Соединение по п. 17, отличающееся тем, что Rx представляет собой метоксиэтокси.
19. Соединение по п. 18, отличающееся тем, что Ry представляет собой фтор.
20. Соединение по п. 1, где соединение выбрано из группы, состоящей из:
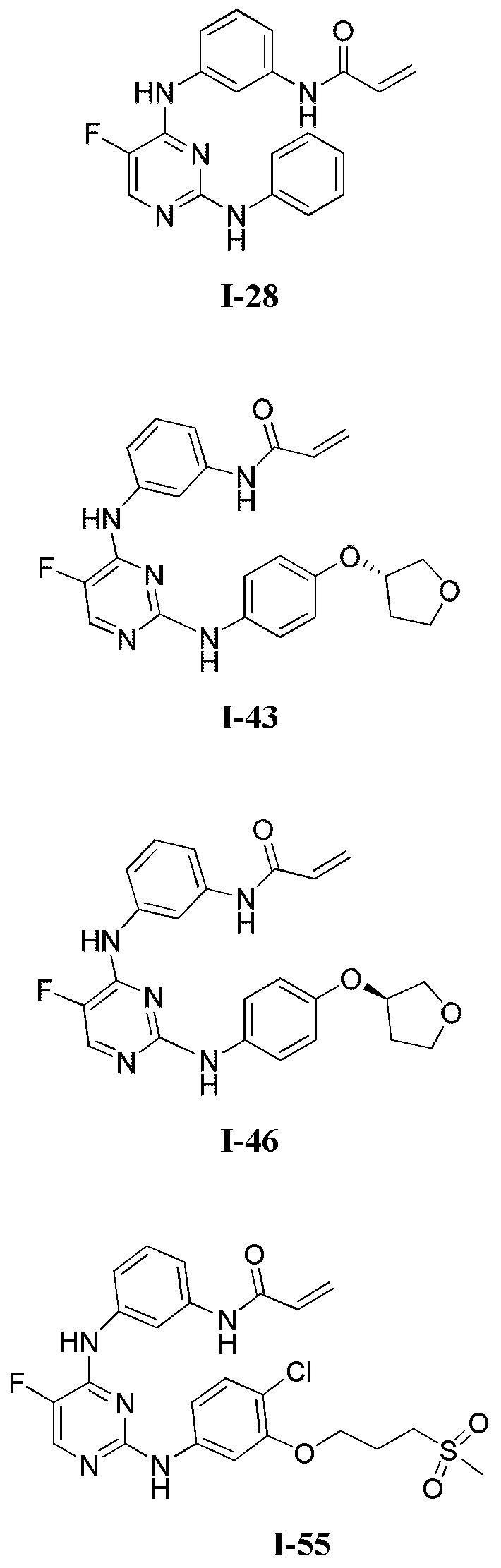
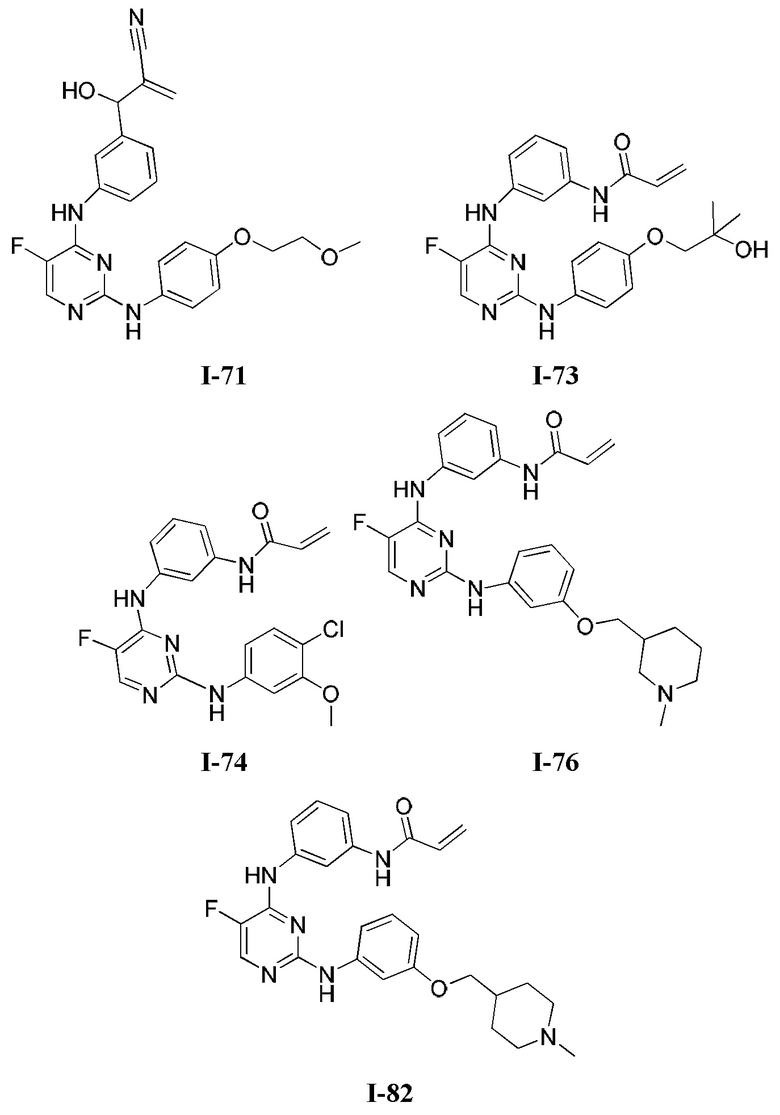
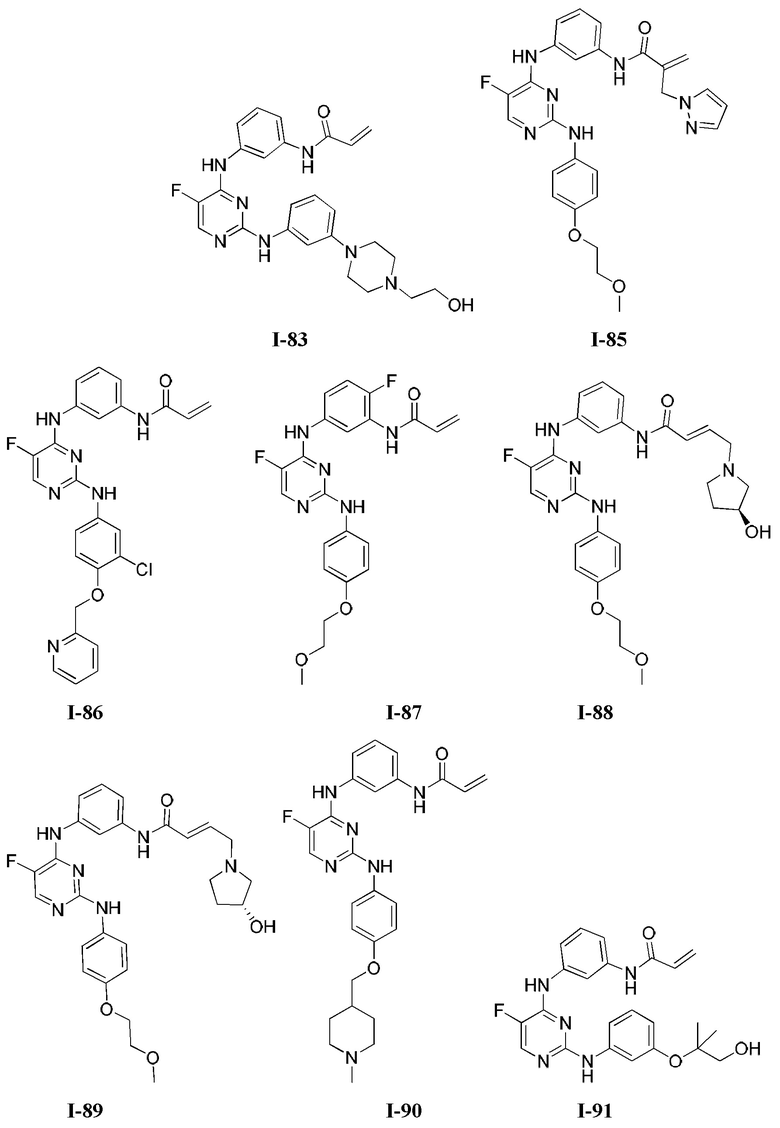
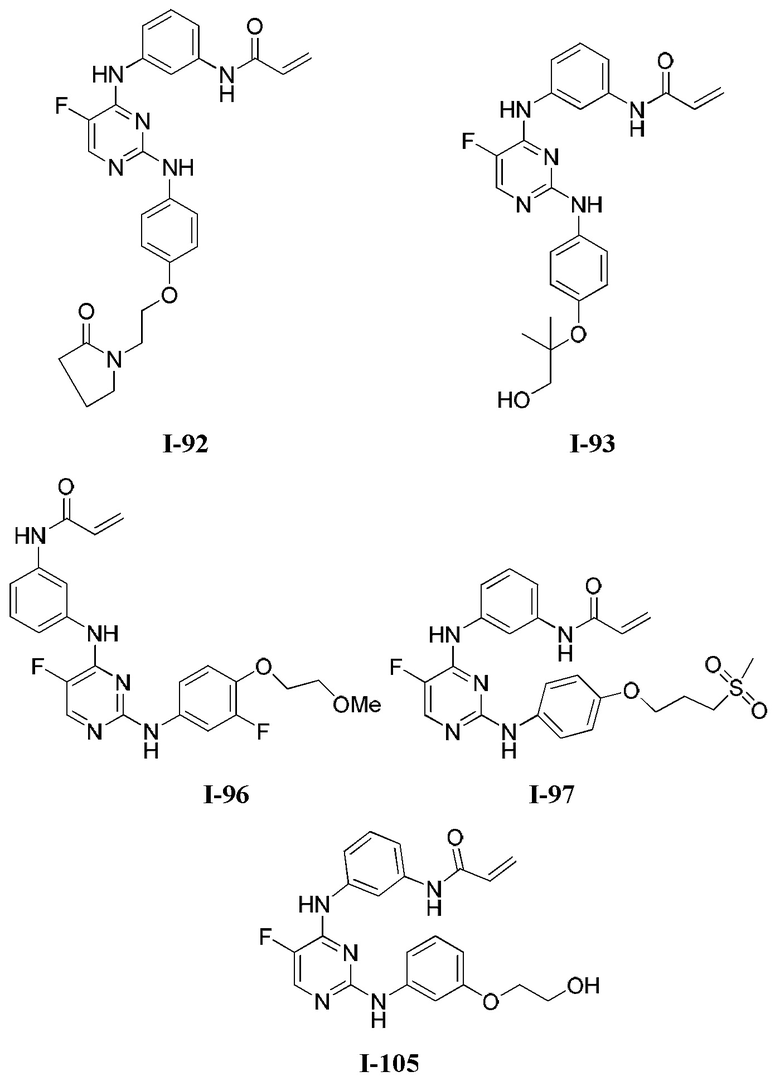
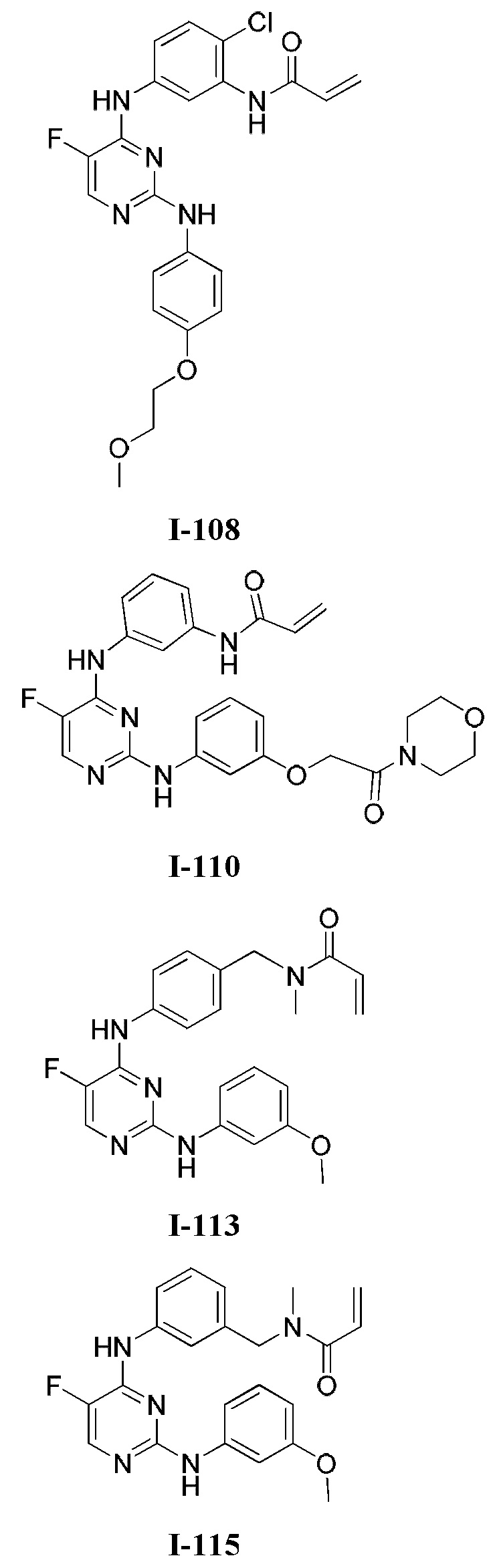
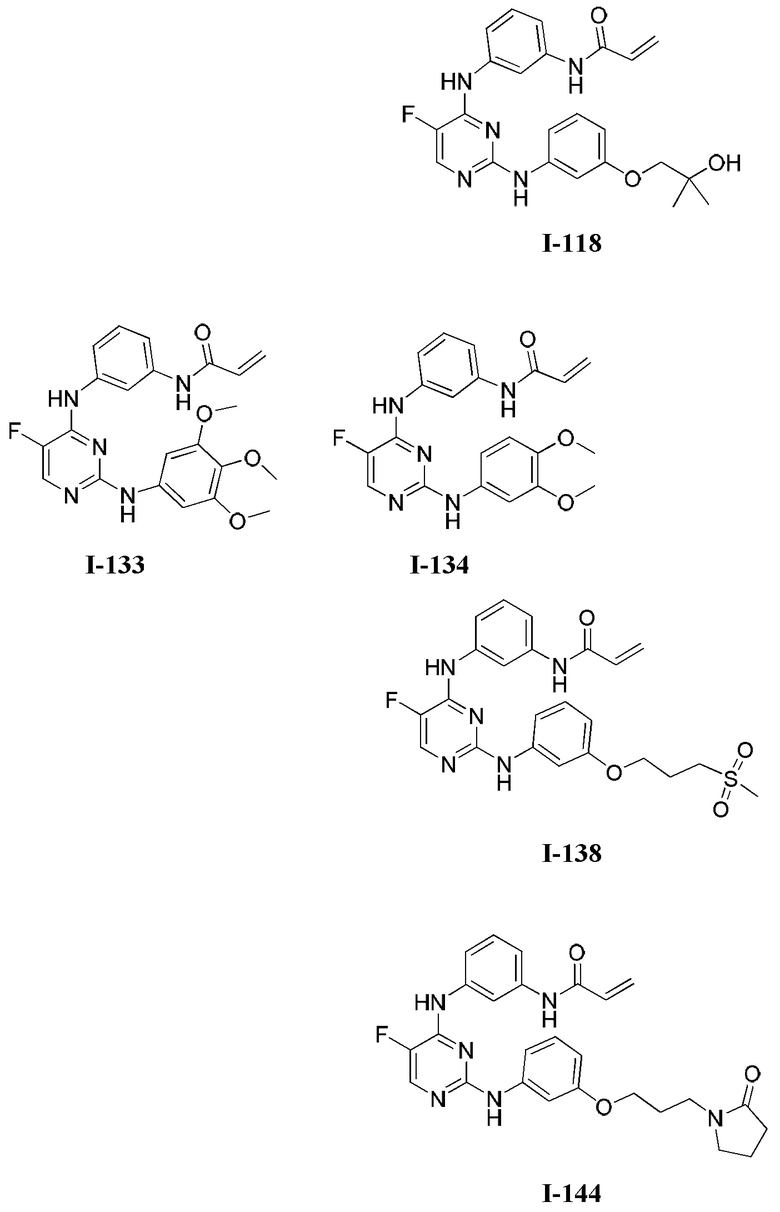
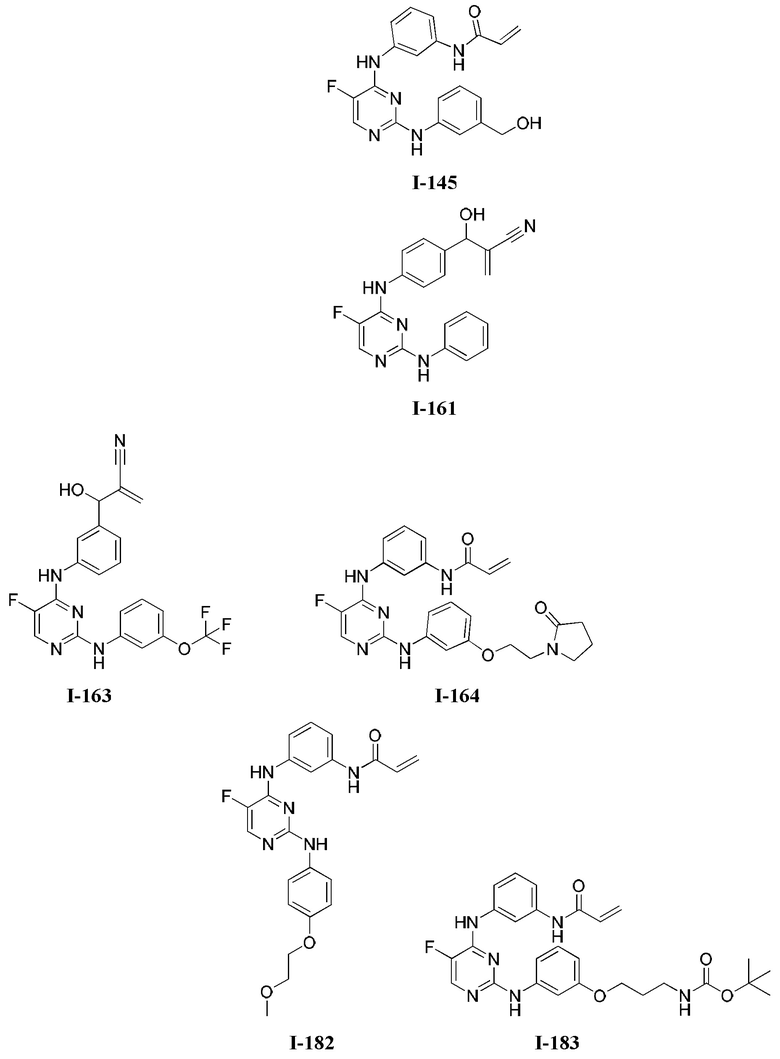
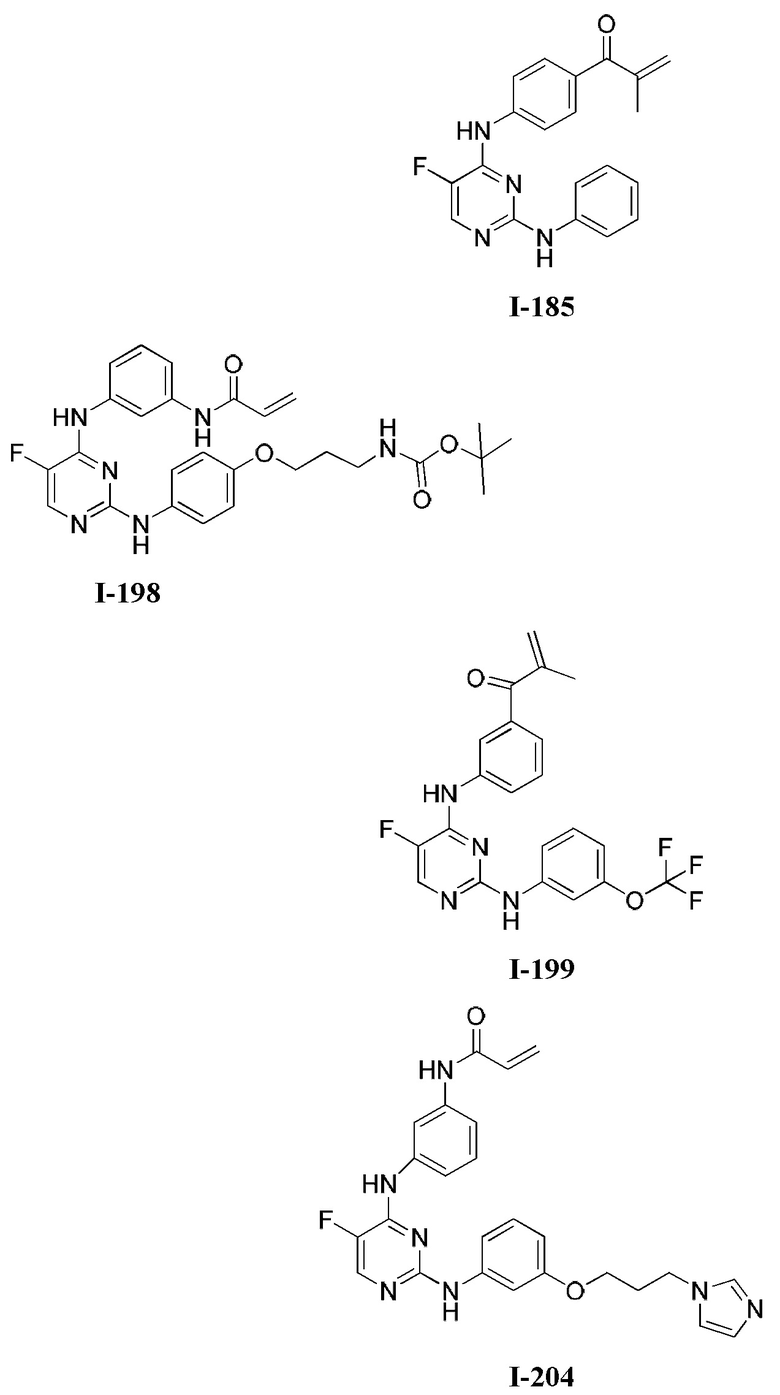
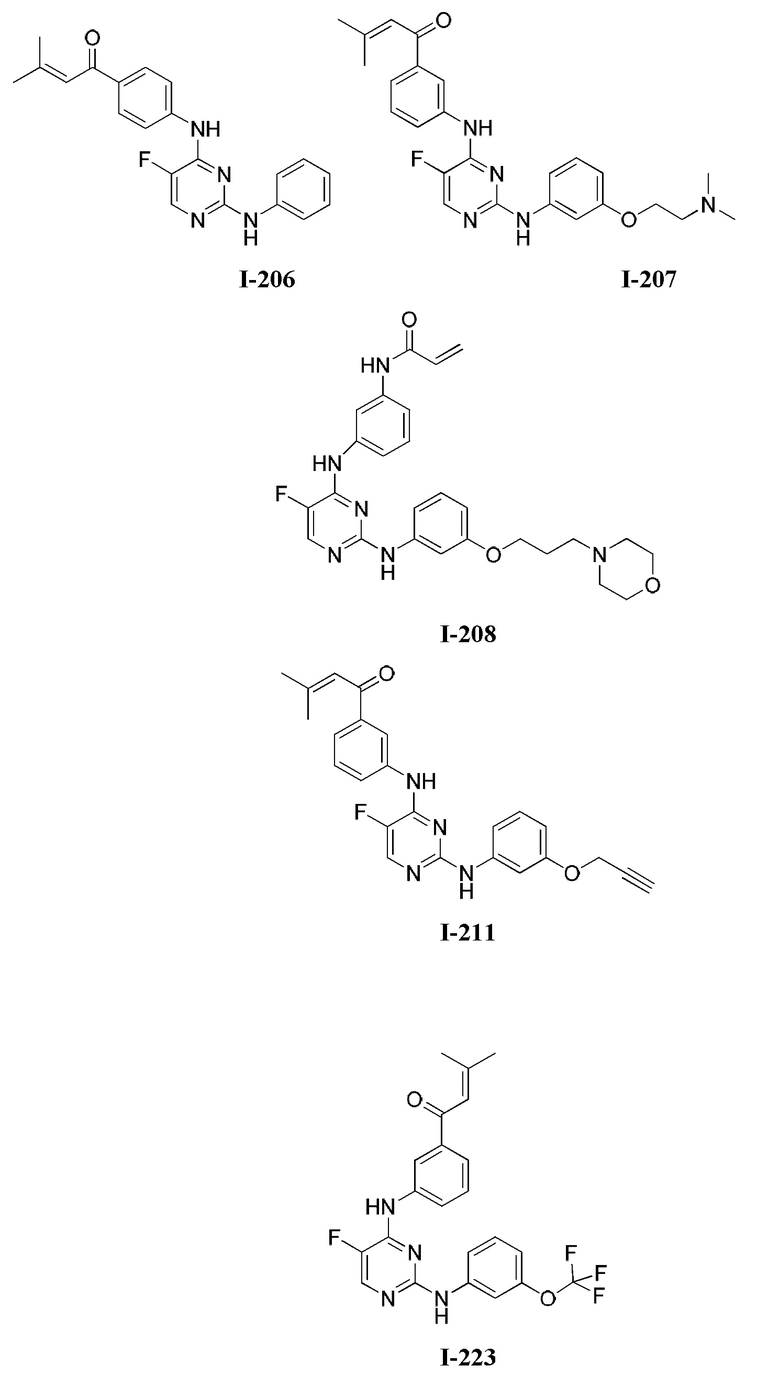
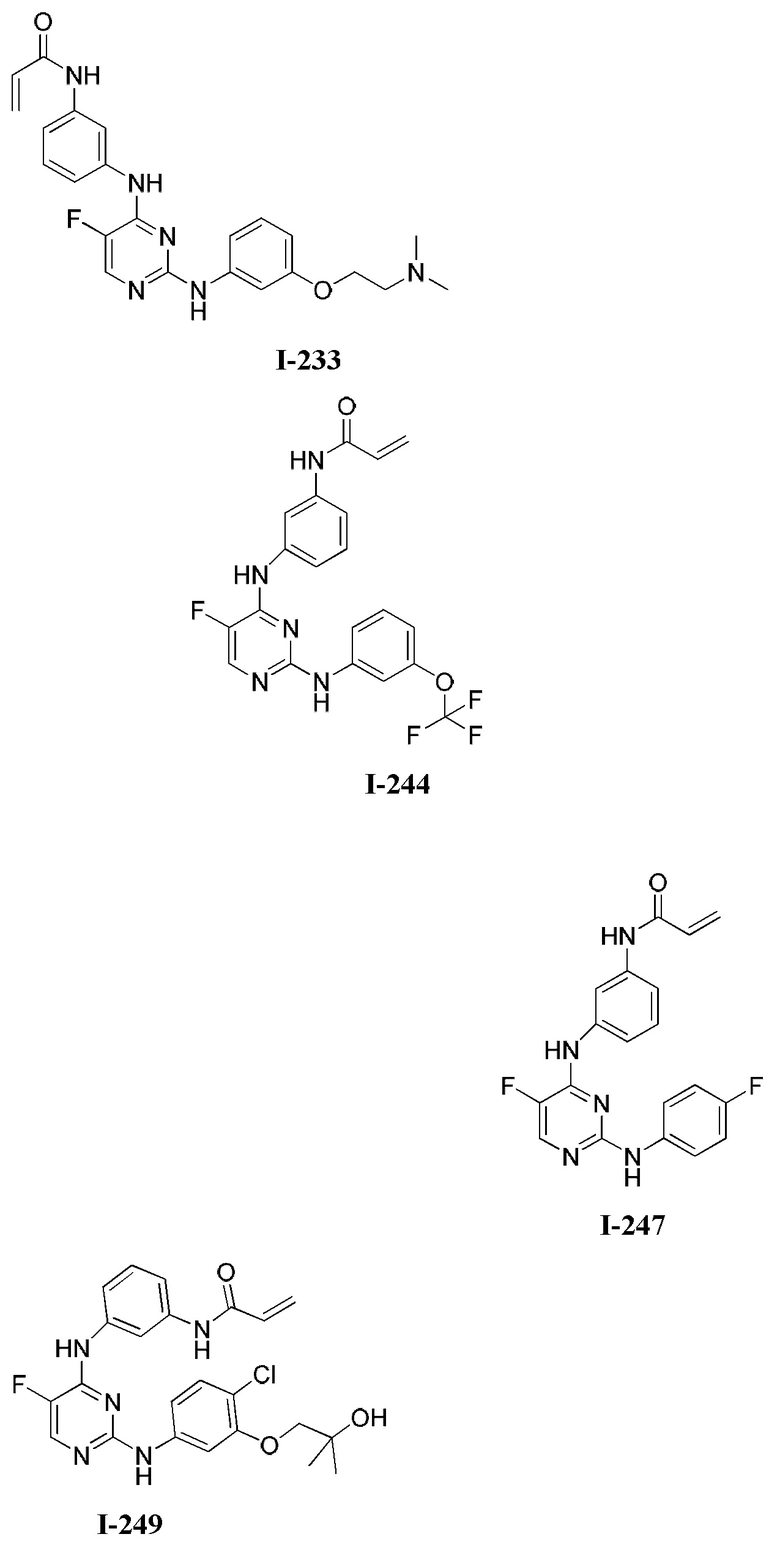
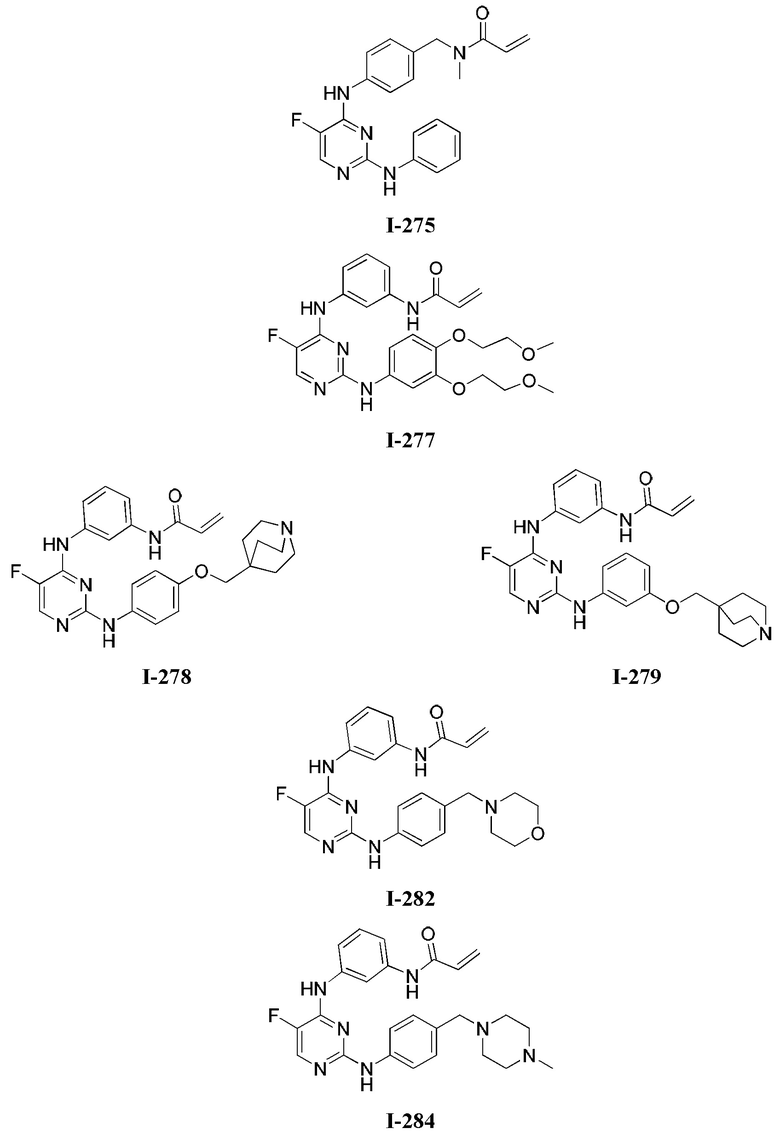
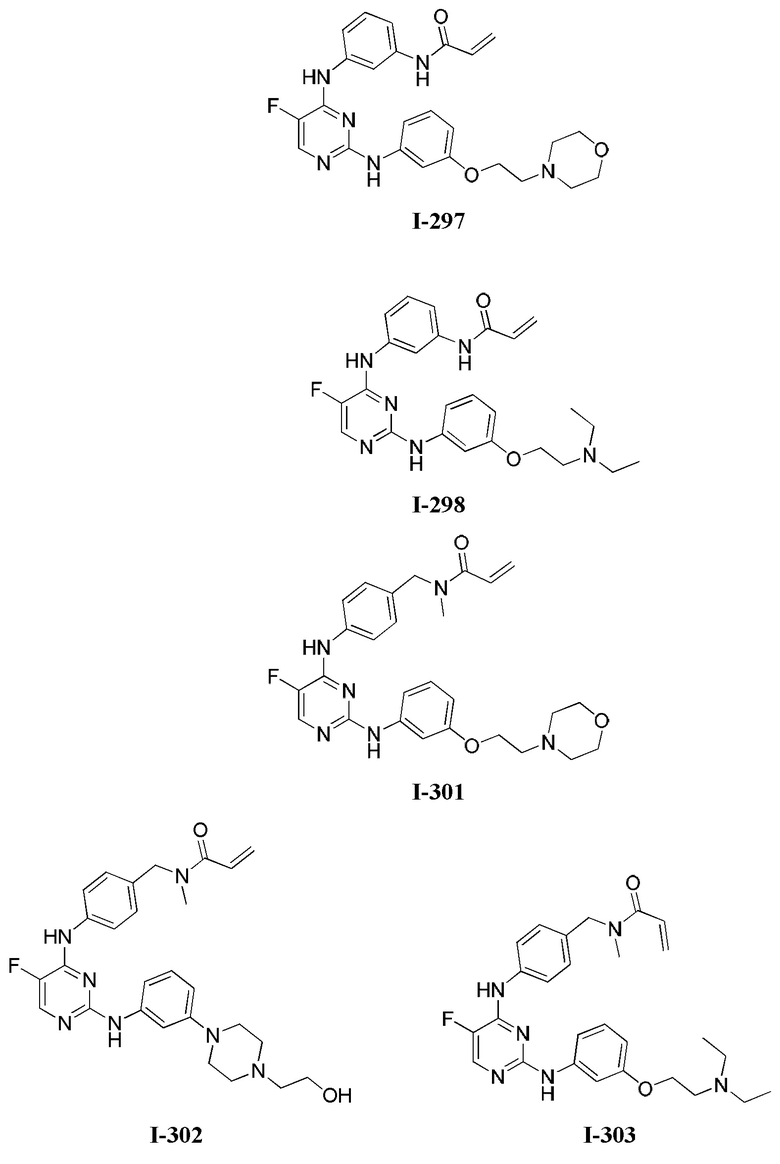
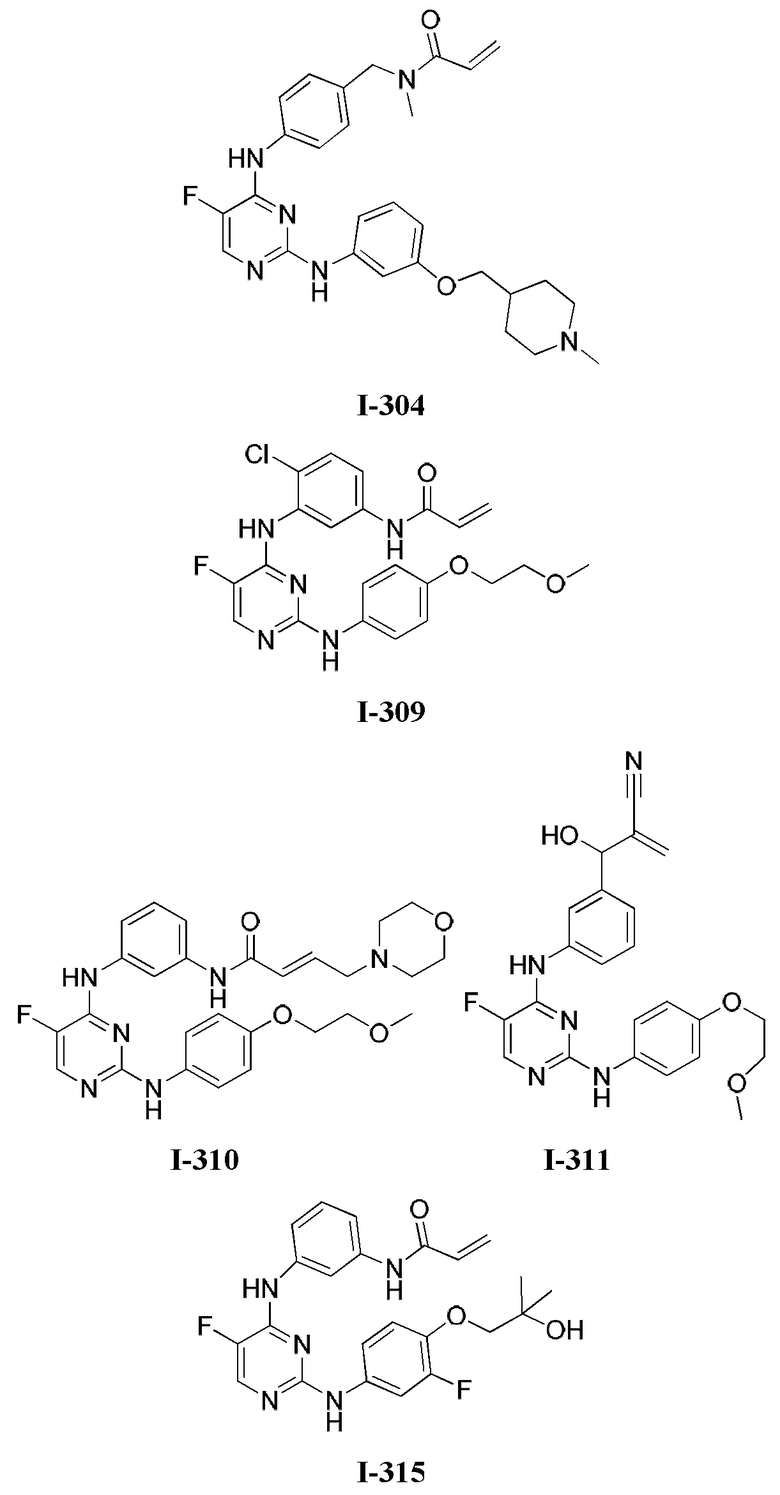
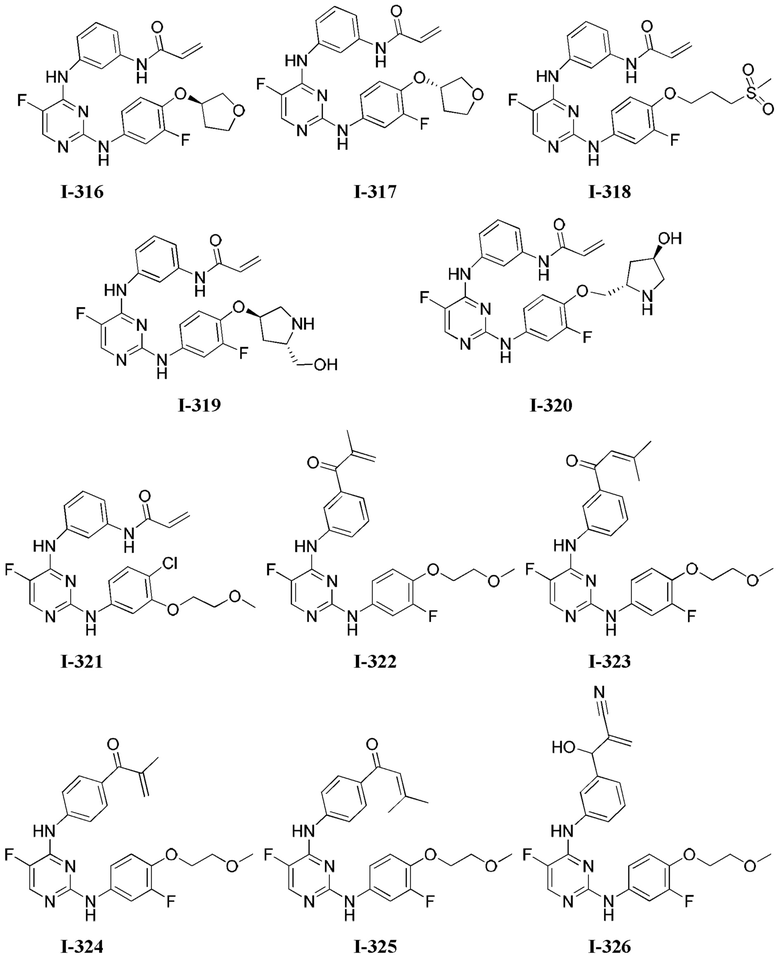
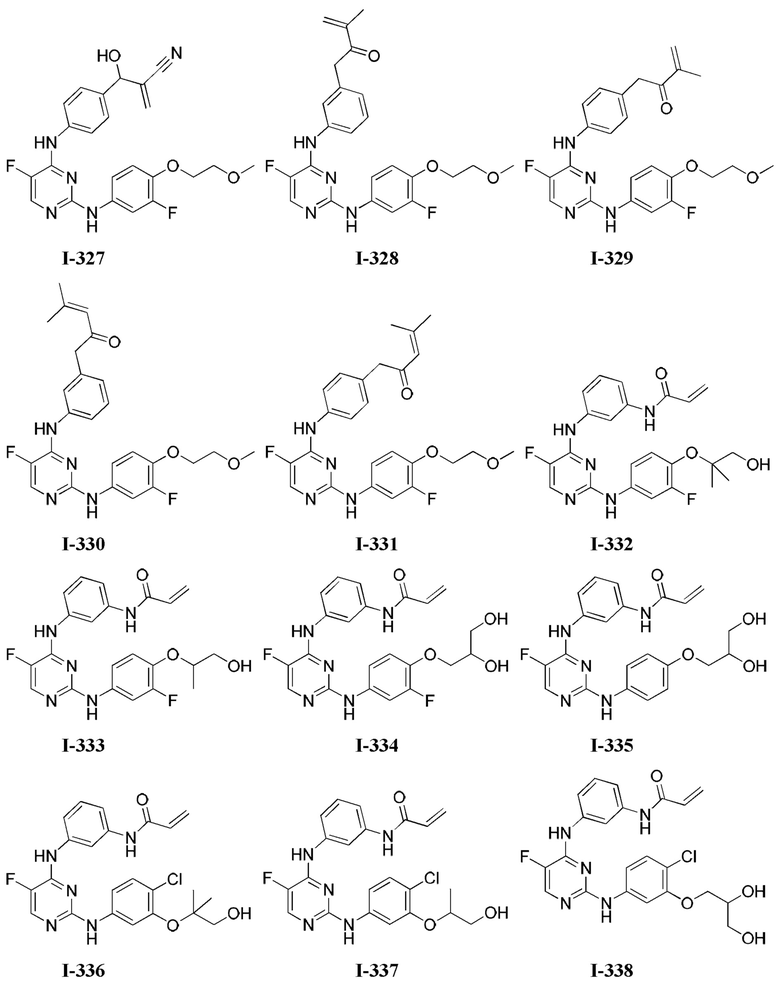
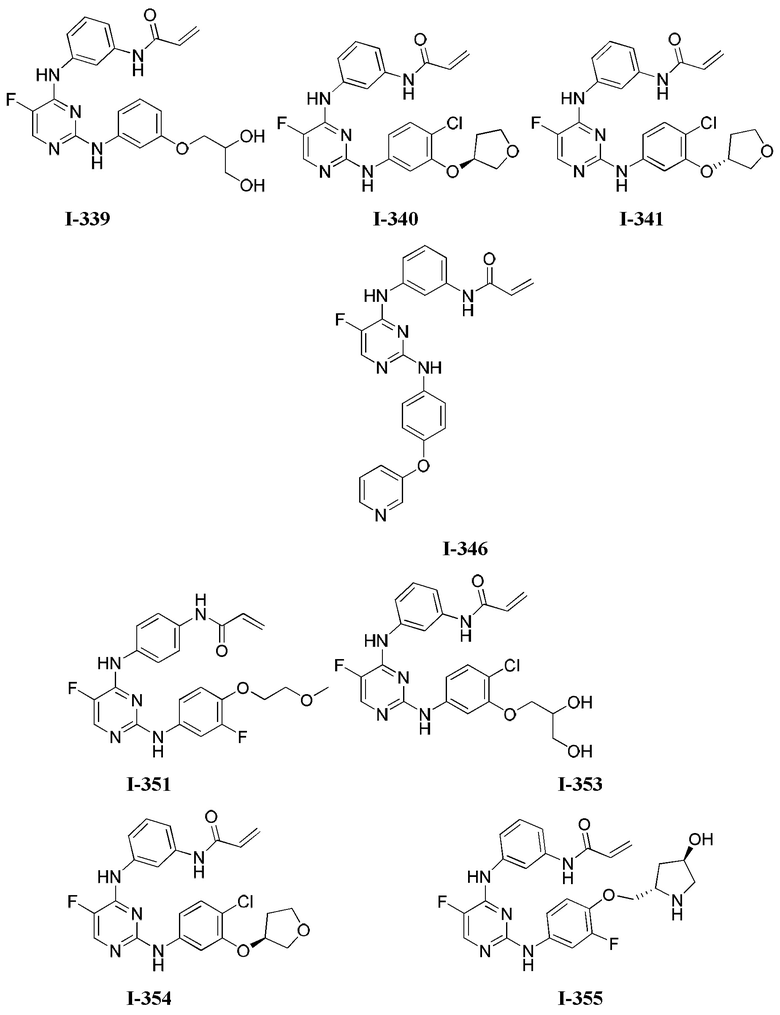
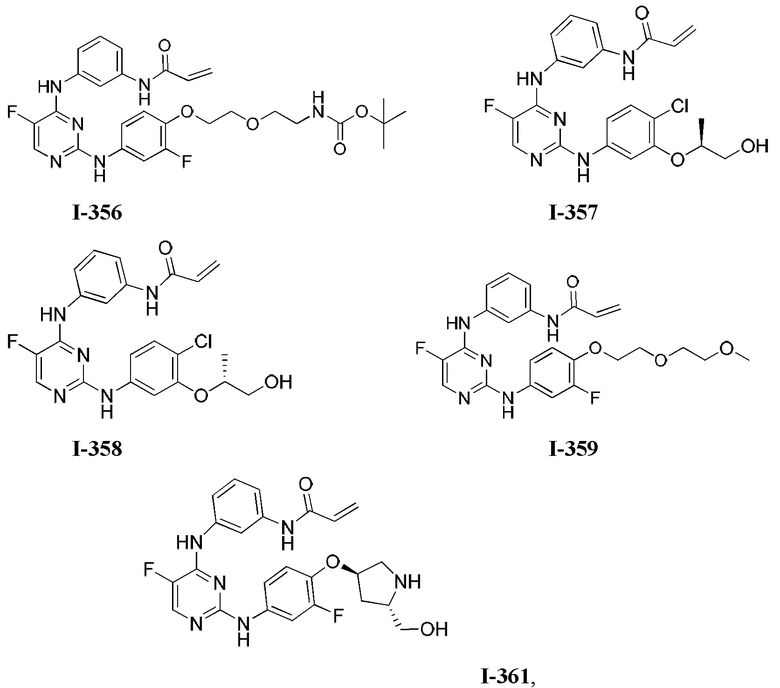
или фармацевтическая приемлемая соль указанного соединения.
21. Соединение по п. 20, имеющее структуру:
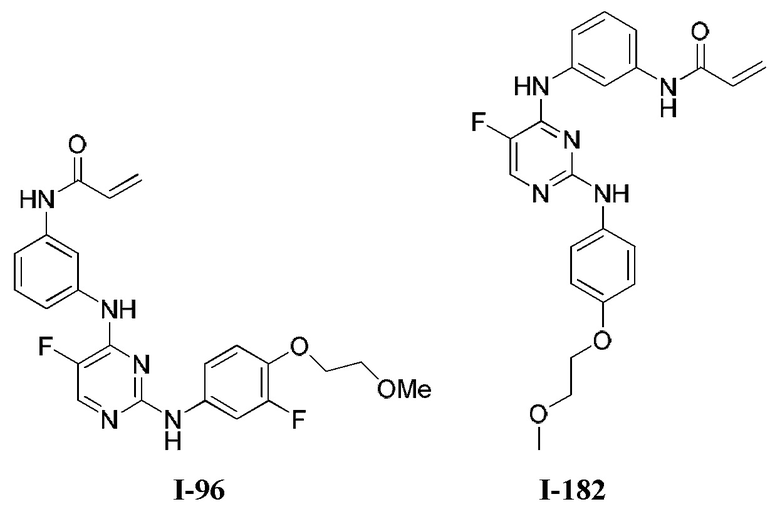
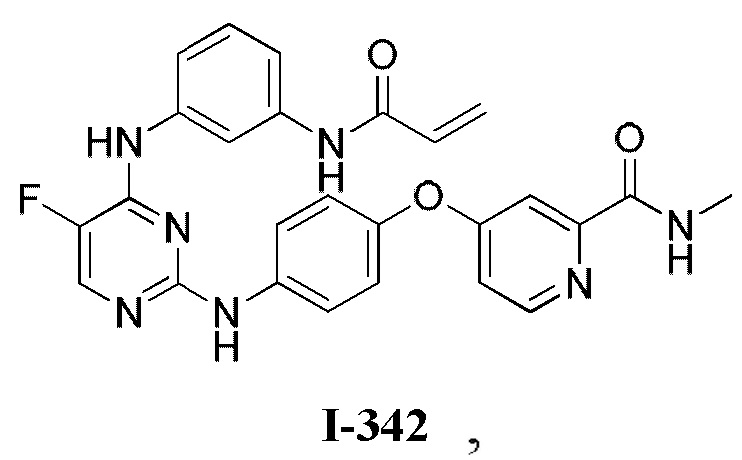
или фармацевтическая приемлемая соль указанного соединения.
22. Соединение по п. 1, имеющее структуру:
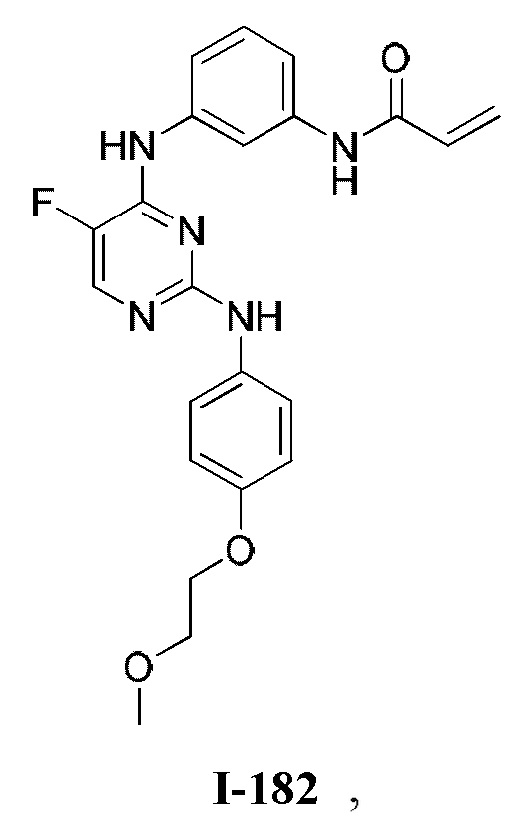
или фармацевтическая приемлемая соль указанного соединения.
23. Композиция для ингибирования по меньшей мере одной киназы, выбранной из BTK, TEC, ВМХ, ITK, ErbB1, ErbB2, ErbB3, ErbB4 и JAK3, содержащая эффективное количество соединения по п. 1 и фармацевтически приемлемый адъювант, носитель или несущую среду.
24. Композиция для ингибирования по меньшей мере одной киназы, выбранной из BTK, ТЕС, ВМХ, ITK, ErbB1, ErbB2, ErbB3, ErbB4 и JAK3, содержащая эффективное количество соединения по п. 22 и фармацевтически приемлемый адъювант, носитель или несущую среду.
25. Способ ингибирования активности киназы BTK, или ее мутанта, в организме пациента или в биологическом образце, включающий этап введения указанному пациенту соединения по п. 1 или приведение в контакт указанного биологического образца с соединением по п. 1.
26. Способ по п. 25, отличающийся тем, что активность киназы BTK или мутанта киназы BTK ингибируют необратимо.
27. Способ по п. 26, отличающийся тем, что активность киназы BTK или мутанта киназы ВТК ингибируют необратимо путем ковалентной модификации Cys 481 BTK.
28. Способ лечения опосредуемого киназой BTK нарушения у нуждающегося в этом пациента, включающий этап введения указанному пациенту соединения по п. 1.
29. Способ по п. 28, отличающийся тем, что нарушение представляет собой аутоиммунное заболевание, гетероиммунное заболевание, воспалительное заболевание, рак, заболевание костей и суставов или тромбоэмболическое нарушение.
30. Способ по п. 29, отличающийся тем, что нарушение выбрано из ревматоидного артрита, рассеянного склероза, хронического В-клеточного лимфоцитарного лейкоза, острого лимфоцитарного лейкоза, волосатоклеточного лейкоза, неходжкинской лимфомы, лимфомы Ходжкина, множественной миеломы, рака костей, метастазов в кости, остеопороза, синдрома раздраженного кишечника, болезни Крона, волчанки или нарушений, связанных с трансплантатом почки.
31. Способ ингибирования активности одной или более киназ ТЕС или мутантов киназ ТЕС в организме пациента или в биологическом образце, включающий этап введения указанному пациенту соединения по п. 1 или приведение в контакт биологического образца с соединением по п. 1.
32. Способ по п. 31, отличающийся тем, что активность киназы ТЕС или мутанта киназы ТЕС ингибируют необратимо.
33. Способ по п. 32, отличающийся тем, что киназа ТЕС выбрана из одной или более из TEC, ITK или ВМХ.
34. Способ по п. 33, отличающийся тем, что активность одной или более из киназ TEC, ITK или ВМХ ингибируют необратимо путем ковалентной модификации Cys 449 киназы ТЕС, Cys 442 киназы ITK или Cys 496 киназы ВМХ.
35. Способ лечения опосредуемого киназой ТЕС нарушения у нуждающегося в этом пациента, включающий этап введения указанному пациенту соединения по п. 1.
36. Способ по п. 35, отличающийся тем, что нарушение представляет собой аутоиммунное нарушение, воспалительное нарушение, пролиферативное нарушение, гиперпролиферативное нарушение, иммунологически опосредуемое заболевание, заболевание дыхательных путей, заболевание костей и суставов, заболевание кожи, заболевание желудочно-кишечного тракта, системное заболевание или отторжение аллотрансплантата.
37. Способ ингибирования активности одной или более из киназ ErbB1, ErbB2, ErbB3 и ErbB4, или их мутантов, в организме пациента или в биологическом образце, включающий этап введения указанному пациенту соединения по п. 1 или приведения указанного биологического образца в контакт с соединением по п. 1.
38. Способ по п. 37, отличающийся тем, что активность одной или более из киназ ErbB1, ErbB2 или ErbB4 или их мутантов ингибируют необратимо.
39. Способ по п. 38, отличающийся тем, что активность одной или более из киназ ErbB1, ErbB2 и ErbB4 или их мутантов ингибируют необратимо путем ковалентной модификации Cys797 киназы ErbB1, Cys805 киназы ErbB2 или Cys803 киназы ErbB4.
40. Способ лечения нарушения, опосредуемого киназой ErbB1, ErbB2, ErbB3 или ErbB4, у пациента, нуждающегося в этом, включающий этап введения указанному пациенту соединения по п. 1.
41. Способ по п. 40, отличающийся тем, что нарушение представляет собой карциному, выбранную из рака груди, глиобластомы, рака легких, рака головы и шеи, рака прямой и толстой кишки, рака мочевого пузыря, немелкоклеточного рака легких, плоскоклеточной карциномы, карциномы слюнной железы, карциномы яичников или рака поджелудочной железы.
42. Способ по п. 41, отличающийся тем, что нарушение выбрано из нейрофиброматоза типа I (NF1), нейрофиброматоза типа II (NF2), неоплазий шванновских клеток (например, злокачественные опухоли оболочки периферических нервов (MPNST)) или шванномы.
43. Способ ингибирования активности киназы JAK3 или мутанта киназы JAK3 в организме пациента или в биологическом образце, включающий этап введения указанному пациенту соединения по п. 1 или приведения указанного биологического образца в контакт с соединением по п. 1.
44. Способ по п. 43, отличающийся тем, что активность киназы JAK3 или мутанта киназы JAK3 ингибируют необратимо.
45. Способ по п. 44, отличающийся тем, что активность киназы JAK3 или мутанта киназы JAK3 ингибируют необратимо путем ковалентной модификации Cys 909 в JAK3.
46. Способ лечения нарушения, опосредуемого киназой JAK3, у пациента, нуждающегося в этом, включающий этап введения указанному пациенту соединения по п. 1.
47. Способ по п. 46, отличающийся тем, что нарушение выбрано из аутоиммунного нарушения, воспалительного нарушения, нейродегенеративного нарушения, солидного злокачественного заболевания и злокачественного заболевания крови.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Angiolelli, Mary E.: "Palladium-catalyzed cross-coupling of benzylzinc reagents with methylthio N-heterocycles: a new coupling reaction with unusual selectivity" Synlett, 2000, vol.6, pp.905-907 | |||
| Database CASREACT [online], Portnyagina, V | |||
| A | |||
| et al.: "Pyrimidine derivatives as possible anticandidiasis agents" | |||

