Область техники, к которой относится изобретение
Настоящее изобретение относится к химико-фармацевтической промышленности и медицине, а конкретно к области медицинской микробиологии и разработки антибактериальных препаратов.
Предшествующий уровень техники
Разработка новых антибактериальных препаратов, эффективных в отношении резистентных к антибиотикам бактерий и не вызывающих развития резистентности, является ключевой проблемой в области инфекционных заболеваний. В феврале 2017 года ВОЗ опубликовала список устойчивых к действию антибиотиков «приоритетных патогенов» - 12 видов бактерий, представляющих наибольшую угрозу для здоровья человека в виду отсутствия эффективных методов антибактериальной терапии. В первую категорию приоритетности в области создания новых антибиотиков «критически высокий уровень приоритетности» включена Pseudomonas aeruginosa.
Среди заболеваний, вызванных устойчивыми к антибиотикам патогенами, наибольший социально-экономический ущерб наносят внутрибольничные инфекции, инфекции мочевыводящих путей, пневмонии, хронические рецидивирующие урогенитальные инфекции, острые и хронические энтеральные инфекции.
В структуре внутрибольничных инфекций, Pseudomonas aeruginosa является второй по распространенности причиной внутрибольничных пневмоний (Am J Respir Cell Mol Biol. 2018 Apr;58(4):428-439. Pseudomonas aeruginosa Biofilms: Host Response and Clinical Implications in Lung Infections.Maurice NM, Bedi B, Sadikot RT), третьей по распространенности причиной инфекций мочевыводящих путей (Rev Chilena Infectol. 2019 Apr;36(2):180-189. [Pseudomonas aeruginosa: Pathogenicity and antimicrobial resistance in urinary tract infection].Paz-Zarza VM, Mangwani-Mordani S, Martínez-Maldonado A, Álvarez-Hernández D, Solano-Gálvez SG, Vázquez-López R; FEMS Microbiol Lett. 2017 Aug 15;364(15). The contribution of Pseudomonas aeruginosa virulence factors and host factors in the establishment of urinary tract infections. Newman JW, Floyd RV, Fothergill JL) четвертой по распространенности причиной хирургических инфекций, седьмой по распространенности инфекцией крови (Clin Microbiol Infect. 2019 Aug;25(8):964-970. Rates, predictors and mortality of community-onset bloodstream infections due to Pseudomonas aeruginosa: systematic review and meta-analysis.Rojas A, Palacios-Baena ZR, López-Cortés LE, Rodríguez-Baño J). Что наиболее важно, P.aeruginosa является лидирующей инфекцией среди антибиотикорезистентных грамотрицательных бактерий вызывающих пневмонию у госпитализированных больных (Weiner L. M. et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014 //infection control & hospital epidemiology. – 2016. – Т. 37. – №. 11. – С. 1288-1301.).
Проблемы лечения инфекций, вызванных P.aeruginosa, связаны с низкой эффективностью антибиотикотерапии в отношении резистентных штаммов; быстрым возникновением резистентности к новым препаратам во время лечения; необходимостью использования комбинации высокотоксичных антибиотиков, что приводит к возникновению опасных побочных эффектов. Появление полирезистентных псевдомонад привело к существенному ухудшению лечения внутрибольничных инфекций. Терапевтические возможности лечения этих инфекций ограничены, так как антисинегнойную активность демонстрирует очень ограниченное количество антибактериальных препаратов. Среди них: два старых препарата (колистин и фосфомицин); один более новый (тигециклин), улучшенный с точки зрения антисинегнойной ингибирующей активности; перспективный комбинированный цефалоспорин 3 генерации (цетазидим/ авибактам) и новый комбинированный антисинегнойный цефалоспорин (цефталозан/ тазобактам); меропенем-варобактам – комбинация меропенема (антибиотик из группы карбапенемов) и ингибитора β-лактамазы варобактама (Med Clin (Barc). 2020 Jan 8. pii: S0025-7753(19)30711-0. New antibiotics for the treatment of infections by multidrug-resistant microorganisms. Escolà-Vergé L, Los-Arcos I, Almirante B).
Существенное ограничение возможности антибактериальной терапии в лечении инфекций, вызванных P.aeruginosa, диктует поиск альтернативных подходов (Biotechnol Adv. 2019 Jan - Feb;37(1):177-192. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Pang Z, Raudonis R, Glick BR, Lin TJ, Cheng Z). Очевидно, что необходимо разрабатывать принципиально новые подходы и препараты для лечения бактериальных инфекций, которые могут применяться как самостоятельно, так и в комбинированной терапии, в том числе с вакцинами и антибиотиками.
Не меньшую проблему в отношении неэффективности лечения представляют бактерии рода Enterococcus. Энтерококки являются представителями нормальной микрофлоры ротовой полости, кишечника и мочеполовой системы человека. Большинство инфекций, вызываемых энтерококками, носит эндогенный характер и обусловлено инвазией микроорганизмов при избыточной колонизации данными бактериями сайтов прикрепления. Существует высокая вероятность возникновения внутрибольничных инфекций, особенно при высокой частоте применения антибактериальных препаратов широкого спектра действия. Энтерококки вызывают инфекционные осложнения после операций аортокоронарного шунтирования, реконструктивных операций на сердце и трансплантации почек. Enterococcus faecalis является патогеном, ответственным за возникновение внутрибольничных инфекций различной локализации, в первую очередь мочеполовой системы, с высоким риском развития бактериемии и сепсиса. Энтерокковые инфекции опасны тем, что возбудитель характеризуется широким спектром устойчивости ко многим антибиотикам, что делает антибиотикотерапию малоэффективной: Faron ML, Ledeboer NA, Buchan BW. Resistance Mechanisms, Epidemiology, and Approaches to Screening for Vancomycin-Resistant Enterococcus in the Health Care Setting. J Clin Microbiol. 2016; 54(10):2436-47; T.Raza,S.R. Ullah,K. Mehmood,et al. Vancomycin resistant Enterococci: A brief review. JPak Med Assoc. 2018. Vol.68, No.5; M. Ceci, G. Delpech , M. Sparo , V. Mezzina , S. S. Bruni, B. Baldaccini. Clinical and microbiological features of bacteremia caused by Enterococcus faecalis. J Infect Dev Ctries 2015; 9(11):1195-1203.
Урогенитальный хламидиоз представляет собой серьезную проблему здравоохранения, т.к. Chlamydia trachomatis является самым распространенным бактериальным возбудителем инфекций, передающихся половым путем. Хламидии вызывают широкий спектр заболеваний и, в первую очередь, связанных с тяжелыми осложнениями и переходом в хроническую форму, трахому, урогенитальный хламидиоз, бесплодие, патологию беременности, артриты. В настоящее время для лечения хламидиоза применяют этиотропную терапию, основанную на чувствительности хламидий к некоторым антибиотикам. Однако лечение хламидийных инфекций, особенно при хроническом течении, по-прежнему остается крайне актуальной и нерешенной проблемой, что обусловлено низкой чувствительностью внутриклеточно персистирующих хламидий к используемым антибиотикам. Отсутствие эффективных и безопасных препаратов для лечения разных форм хламидийных инфекций определяет необходимость разработки нового подхода для поиска лекарственных средств, основанного на выявлении принципиально новых мишеней у бактериальных патогенов: O'Connell CM, Ferone ME.Chlamydia trachomatis Genital Infections. Microb Cell. 2016;3(9):390-403; Mohammadzadeh F, Dolatian M, Jorjani M, Afrakhteh M, Majd HA, Abdi F, Pakzad R. Urogenital chlamydia trachomatis treatment failure with azithromycin: A meta-analysis. Int J Reprod Biomed. 2019;17(9):603-620; Xia Q, Wang T, Xian J, Song J, Qiao Y, Mu Z, Liu H, Sun Z. Relation of Chlamydia trachomatis infections to ectopic pregnancy: A meta-analysis and systematic review. Medicine 2020,99(1):e18489.
Сальмонеллезы человека у большинства взрослых протекают в виде острого гастроэнтерита с быстрым выздоровлением. Однако имеются группы лиц, у которых возможно развитие тяжелой системной сальмонеллезной инфекции (дети, лица пожилого возраста, а также иммунокомпроментированные пациенты). Генерализованная форма сальмонеллеза чаще других может осложняться хроническими энтеральными инфекциями, такими как гастроэнтериты и колиты, холециститы, нефриты, артриты, периоститы, перихондриты, мезенхиальные гепатиты и другие осложнения. Переход инфекции в хроническую форму обусловлен персистенцией патогена - длительным выживанием возбудителя в организме хозяина. Кроме того, устойчивые к антибиотикам сальмонеллы вызывают вспышки внутрибольничных инфекций у детей младшего возраста, характеризующиеся длительным тяжелым течением, бактериемией, нередко с генерализацией процесса и развитием септических осложнений, приводящих к высокой летальности. У взрослых инфекции, вызванные устойчивыми к антибиотикам сальмонеллами, в 28% случаев приводят к тяжелому генерализованному течению заболевания, в то время чувствительные к антибиотикам штаммы приводят к тяжелому течению инфекции в 2 раза реже.
В настоящее время для лечения хронических инфекций, вызванных сальмонеллами, а также при тифоподобных и септических формах сальмонеллезов применяют сочетанное длительное введение антибактериальных препаратов (парентерально и внутрь 10 и более дней). Однако лечение антибиотиками не всегда приводит к полному излечению тяжелых форм сальмонеллезов. Кроме того, после перенесенной сальмонеллезной инфекции может развиться бактерионосительство. Таким образом, лечение сальмонеллезных инфекций, особенно при хроническом течении, остается серьезной проблемой медицинской практики: Gal-Mor O Persistent Infection and Long-Term Carriage of Typhoidal and Nontyphoidal Salmonellae. Clin Microbiol Rev. 2018 Nov 28;32(1); Besser JM. Salmonella epidemiology: A whirlwind of change. Food Microbiol. 2018 May; 71:55-59; Zha L, Garrett S, Sun J. Salmonella Infection in Chronic Inflammation and Gastrointestinal Cancer. Diseases. 2019, 10;7(1). pii: E28.
В связи с быстрым развитием антибиотикорезистентности и отсутствием перспективных разработок новых антибиотиков на первый план выходит подход, направленный на разработку препаратов с альтернативным антибиотикам механизмом действия. Необходимо снизить селективное давление препаратов на патогены и изменить парадигму лечения инфекций – лекарство должно не убивать бактерии, а подавлять вирулентность, что принципиально снизит риск развития резистентности, при этом подавление вирулентности позволит ограничить проявления симптомокомплекса заболевания и даст возможность иммунной системе справиться с авирулентным микробом.
Среди известных мишеней для разработки таких «антивирулентных» препаратов выбираются различные факторы патогенности, такие как, токсины, секреторные системы, факторы адгезии, молекулы системы «кворум сенсинг», факторы образования биопленок. Разработка антибактериальных препаратов, подавляющих различные факторы вирулентности бактерий, бурно развивающееся направление преимущественно за рубежом, показывающее свою перспективность. При этом многие препараты находятся на стадии НИР или доклинических испытаний, лишь несколько проходят клинические исследования и единицы зарегистрированы FDA (англ. Food and Drug Administration, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов), среди которых препараты на основе иммуноглобулинов и моноклональных антител, специфически связывающихся с токсинами или мембранными факторами вирулентности.
Наиболее близким аналогом технического решения заявленного изобретения является патент РФ № 2624846, включенный в настоящее описание в качестве ссылки «Применение 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида для подавления инфекции, вызванной устойчивыми к антибиотикам штаммами Pseudomonas aeruginosa, и способ подавления этой инфекции».
В рассматриваемом патенте было экспериментально продемонстрировано, что полученное низкомолекулярное соединение, 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид, является ингибитором ССТТ Pseudomonas aeruginosa. Полученное соединение специфически ингибирует секрецию токсинов - эффекторов системы секреции III типа псевдомонад, вызывающих гибель эукариотических клеток. Блокирование in vitro системы секреции III типа Pseudomonas aeruginosa было показано для клинических штаммов с различными генотипами по генам эффекторных белков ССТТ и различным профилем антибиотикорезистентности.
Была достигнута поставленная в данном изобретении задача, а именно:
- подавление инфекции, вызванной устойчивыми к антибиотикам штаммами Pseudomonas aeruginosa с различным генотипом по генам эффекторных белков ССТТ и различным профилем антибиотикорезистентности.
- подавление развития инфекции, вызванной Pseudomonas aeruginosa у зараженного организма на модели in vivo, вне зависимости от приобретенной антибиотикорезистентности.
- отсутствие ингибирующего действия на нормальную микрофлору макроорганизма при применении 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида.
Однако можно отметить некоторые недостатки рассматриваемого изобретения:
- изучение терапевтической эффективности низкомолекулярного ингибитора было проведено на экспериментальных животных, при этом дальнейшая разработка препарата требует проведения клинических испытаний на людях, что не было проведено в данном изобретении.
- для проведения клинических испытаний с последующей регистрацией разрабатываемого препарата необходимо создание лекарственной формы для использования в виде фармацевтической композиции для дозированного приема.
- для полученного в изобретении препарата не были проведены исследования по выявлению широты действия в отношении других актуальных патогенов, возбудителей как острых, так и хронических инфекций.
- терапевтическое действие полученного в изобретении препарата было показано только на модели острой септической инфекции у мышей, вызванной псевдомонадами. Однако псевдомонады очень часто вызывают и другие нозологии, среди которых пневмония, инфекции мочевыводящих путей, протекающие как хронические рецидивирующие инфекции.
Полученные данные дают основание расширить область применения лекарственной субстанции 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид для использования в виде фармацевтической композиции.
Предпосылки создания настоящего изобретения
Разработанное нами действующее вещество по патенту РФ №2624846 «Применение 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4Н-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида для подавления инфекции, вызванной устойчивыми к антибиотикам штаммами Pseudomonas aeruginosa, и способ подавления этой инфекции» показало активность по отношению к инфекции, вызванной устойчивыми к антибиотикам штаммами Pseudomonas aeruginosa, в опытах in vitro и in vivo».
Проведение дальнейших работ показало, что можно расширить область применения действующего вещества по патенту РФ № 2624846, то есть применить его в качестве антибактериального препарата для лечения острых, рецидивирующих и хронических инфекций, таких как осложненные инфекции мочевыводящих путей, пневмонии, хронические урогенитальные инфекции, острые и хронические инфекции кишечной локализации, вызванные антибиотикорезистентными штаммами Pseudomonas aeruginosa, Enterococcus spp., Chlamydia trachomatis, Salmonella spp.
В связи с высокой актуальностью медицинской проблемы резистентности патогенов к антибиотикам, очевидна также необходимость разработки лекарственного средства в виде твердой дозированной лекарственной формы (фармацевтической композиции) для перорального применения, содержащей действующее вещество и приемлемые вспомогательные вещества, при этом она должна быть удобной для применения пациентами. Твердые пероральные фармацевтические композиции должны обеспечить поступление в организм пациента действующего вещества, в достаточном для эффективной терапии количестве, обеспечить высвобождение действующего вещества из лекарственной формы, а также обеспечить химическую стабильность действующего вещества. Для обеспечения высокого уровня удобства применения пероральная фармацевтическая композиция не должна приниматься чаще трех раз в день, а в случае если она имеет форму таблетки или капсулы, их размер не должен быть слишком большим, чтобы не вызвать затруднений при глотании. Размеры таблетки зависят от количества активного вещества, необходимого для эффективной терапии, и количества вспомогательных веществ. Тип и количество вспомогательных веществ в сочетании с технологией производства являются существенными с точки зрения свойств высвобождения, биологической доступности соединения у млекопитающих, стабильности и применимости технологии изготовления фармацевтической композиции в производственных условиях.
Раскрытие изобретения
Задачей настоящего изобретения является создание пероральной твердой фармацевтической композиции, содержащей действующее вещество в количестве, необходимом и достаточном для антимикробного действия, для лечения острых, рецидивирующих и хронических инфекций, таких как, осложненные инфекции мочевыводящих путей, пневмония, хронические урогенитальные инфекции, острые и хронические инфекции кишечной локализации, вызванные антибиотикорезистентными штаммами Pseudomonas aeruginosa и/или Enterococcus spp. и/или Chlamydia trachomatis. и/или Salmonella spp., способа ее применения для лечения рецидивирующих и хронических инфекций, вызванных этими патогенами, вне зависимости от приобретенной резистентности.
При этом фармацевтическая композиция в виде твердой лекарственной формы, должна содержать действующее вещество и обладать следующими свойствами:
- твердая лекарственная форма должна обеспечивать биодоступность и терапевтическую эффективность в модельных экспериментах не менее, чем соответствующие показатели для действующего вещества;
- проявлять токсичность, сопоставимую с токсичностью действующего вещества при пероральном введении;
- обладать приемлемыми технологическими свойствами (сыпучесть, распадаемость, растворение);
- обладать стабильностью при хранении, не менее 2 лет.
Технический результат достигается за счет того, что создана фармацевтическая композиция, включающая действующее вещество 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид в кристаллической форме с размером частиц менее 50 мкм в количестве от 150 до 600 мг в виде пероральной твердой дозированной лекарственной формы с приемлемыми вспомогательными веществами. При этом в качестве приемлемых вспомогательных веществ используют наполнители, разрыхляющие модификаторы поверхности, связывающие, скользящие при следующем соотношении компонентов, мас.%:
- Фармацевтическая композиция представляет собой пероральную твердую дозированную лекарственную форму для приема внутрь в виде таблеток, покрытых или не покрытых оболочкой, или капсул твердых, или порошка дозированного, или порошка для приготовления раствора.
- Разработано применение фармацевтической композиции для лечения острых, рецидивирующих и хронических инфекций, таких как осложненные инфекции мочевыводящих путей, пневмония, хронические урогенитальные инфекции, острые и хронические инфекции кишечной локализации, вызванные антибиотикорезистентными штаммами Pseudomonas aeruginosa, Enterococcus spp., Chlamydia trachomatis, Salmonella spp.
Краткое описание чертежей
На фиг. 1
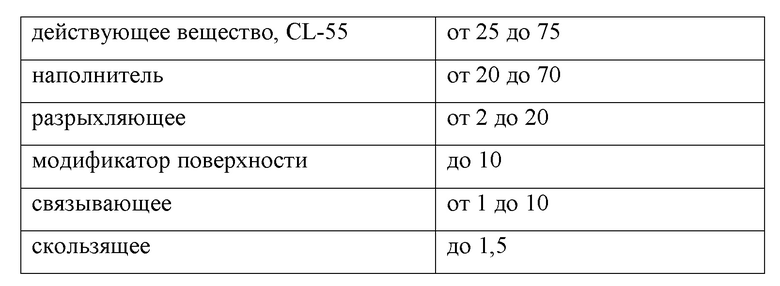
представлены данные по определению количества метаболита глюкуронида и неизмененного действующего вещества выделенного с мочой после приема 50 мг/кг фармацевтической композиции. Серые столбцы – количество выделенного с мочой неизмененного действующего вещества, заштрихованные столбцы количество выделенного с мочой метаболита глюкуронида за 12, 24 и 48 часов соответственно.
На фиг. 2
показаны сравнительные кривые зависимости концентрации в крови от времени после введения действующего вещества и заявленной фармацевтической композиции в форме таблетки. Штриховая линия отображает концентрацию в крови действующего вещества после введения таблетки, сплошная линия – после введения действующего вещества в кристаллической виде (субстанция).
На фиг.3
представлены результаты по оценке подавления активности системы секреции III типа у клинических изолятов 32461 и 41431 P. aeruginosa до и после пассирования в присутствии CL-55 и действии разных концентраций CL-55 в мкг/мл при постановке метода иммуноблотинга с сывороткой к ExoT белку. Фрагмент размером 53 кДа соответствует белку ExoT и выявляется в образцах после контакта со специфической сывороткой.
На фиг. 4
представлены результаты оценки накопления хламидий в нижних отделах урогенитального тракта мышей, инфицированных C. trachomatis serovar D, в условиях лечения в течение 7 дней фармацевтической композицией (ФК), антибиотиком ампициллином при сравнении с зараженными мышами без лечения. Данные представлены в значениях десятичного логарифма включение образующих единиц на мл (ВОЕ/мл). ВОЕ/мл определяется по высевам из вагинальных соскобов мышей на чувствительные линии клеток и детекции выросших хламидийных включений с помощью специфических моноклональных антител, меченных ФИТЦ (зеленое свечение).
На фиг.5
представлены результаты культурального метода по выделению хламидий из вагинальных соскобов мышей, зараженных C. trachomatis serovar D, получавших терапию фармацевтической композицией (ФК), азитромицином при сравнении с мышами без лечения (контроль инфекции). Лечение проводили в течение 7 дней. Забор вагинальных соскобов производили на 3, 7, 11 и 14 сутки после заражения, затем культивировали на чувствительных линиях клеток и детектировали выросшие хламидийные включения с помощью специфических моноклональных антител, меченных ФИТЦ (зеленое свечение).
На фиг. 6
представлена динамика показателей лактозоположительной кишечной палочки при микробиологическом исследовании кишечной микрофлоры мышей при введении фармацевтической композиции (ФК), гентамицина (ГЦ) и ципрофлоксацина (ЦФ). Количества высеваемых бактерий представлены в колониеобразующих единицах на 1 мл (КОЕ/мл). На оси абсцисс представлены сроки отбора проб у мышей.
На фиг. 7
представлена динамика показателей лактобактерий при микробиологическом исследовании кишечной микрофлоры мышей при введении фармацевтической композиции (ФК), гентамицина (ГЦ) и ципрофлоксацина (ЦФ). Количества высеваемых бактерий представлены в колониеобразующих единицах на 1 мл (КОЕ/мл). На оси абсцисс представлены сроки отбора проб у мышей.
На фиг. 8
представлена динамика показателей бифидобактерий при микробиологическом исследовании кишечной микрофлоры мышей при введении фармацевтической композиции (ФК), гентамицина (ГЦ) и ципрофлоксацина (ЦФ). Количества высеваемых бактерий представлены в колониеобразующих единицах на 1 мл (КОЕ/мл). На оси абсцисс представлены сроки отбора проб у мышей.
На фиг. 9
представлена динамика показателей фекального стрептококка при микробиологическом исследовании кишечной микрофлоры мышей при введении фармацевтической композиции (ФК), гентамицина (ГЦ) и ципрофлоксацина (ЦФ). Количества высеваемых бактерий представлены в колониеобразующих единицах на 1 мл (КОЕ/мл). На оси абсцисс представлены сроки отбора проб у мышей.
Реализация изобретения
Настоящее изобретение направлено на получение фармацевтической композиции твердой дозированной формы для перорального приема, включающей 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид в кристаллической форме.
Эффективное количество действующего вещества, в готовой лекарственной форме составляет 150- 600 мг. Дозировка рассчитана, исходя из экспериментов in vitro и in vivo, с использованием коэффициентов межвидового пересчета и должна обеспечивать достаточные для подавления микробного роста концентрации в биологических средах организма, а также для обеспечения возможности дозирования препарата на вес пациента, в зависимости от тяжести заболевания. Должна быть обеспечена возможность изготовления твердой лекарственной формы с различными дозировками и применение их от 1 до 5 шт. за прием, но предпочтительно не более 2 шт. за прием. Кратность приема от 1 до 6 раз в сутки, предпочтительно не чаще 3 раз. Длительность терапии определяется врачом исходя из особенностей заболевания.
Фармацевтическая композиция в контексте данного изобретения: смесь, содержащая определенное количество действующего вещества и приемлемые вспомогательные вещества, предназначенная для введения пациенту, для лечения заболеваний, вызванных резистентными микроорганизмами.
Действующее вещество: 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида (соединение CL-55) в кристаллической форме.
Фармацевтически приемлемые вспомогательные вещества - это вещества, которые пригодны для использования в лекарственных формах и безопасны при контакте с тканями человека, и которые сами по себе не оказывают значительной токсичности, раздражения, не вызывают аллергических реакций и других нежелательных явлений, связанных с осложнениями, и не вызывают значительного усиления токсичности действующего вещества, и как следствие - указанные вещества характеризуются достаточно высоким соотношением польза-риск.
Концентрация действующего вещества в фармацевтической композиции составляет определенное значение, например, равное терапевтически эффективному количеству, которое зависит от абсорбции, метаболизма и выведения из организма лекарственного средства, а также от других факторов, известных специалистам в данной области техники. Величина дозы значительно изменяется в зависимости от тяжести состояния, подлежащего лечению. Для любого конкретного реципиента следует подбирать соответствующий курс лечения в течение определенного времени с учетом индивидуальной потребности и мнения врача, который вводит фармацевтические композиции пациенту или назначает введение фармацевтических композиций. Действующее вещество в составе фармацевтической композиции можно вводить однократно или многократно в виде разделенных доз, которые вводят через различные периоды времени. Таким образом, необходимое количество, например необходимое терапевтически эффективное количество, определяет специалист в определенной области медицины.
Приемлемые вспомогательные вещества - это вещества органической или неорганической природы, которые используют в процессе производства готовых лекарственных форм для придания им необходимых свойств. Примеры вспомогательных веществ по функциональным характеристикам включают, без ограничения перечисленным, связывающие вещества, наполнители, носители, разрыхлители, скользящие, разбавители, и др. Специалисту в данной области техники понятно, что можно использовать один или более вспомогательных веществ вышеупомянутых функциональных классов, для обеспечения требуемых свойств твердой пероральной фармацевтической композиции. Вспомогательные вещества и их функциональные назначения, применяемые для получения пероральных лекарственных форм, являются общепринятыми и описаны в следующих публикациях: "The Handbook of Pharmaceutical Excipients", 4-е изд., ред. Rowe и др., American Pharmaceuticals Association (2003), и Решение Коллегии Евразийской экономической комиссии от 18.06.2019 N 103 "О справочнике функциональных назначений вспомогательных веществ, используемых при производстве лекарственных средств".
Разработанная твердая пероральная фармацевтическая композиция является лекарственной формой с обычным высвобождением (immediate release dosage form, immediate release formulation) это означает, что действующее вещество начинает высвобождаться сразу после поступления препарата в организм, у которой скорость и (или) время и (или) место высвобождения активного действующего вещества не модифицируются введением специальных вспомогательных веществ и (или) применением особой технологии производства. В связи с чем, влияние вспомогательных веществ и способа грануляции на биодоступность минимально.
Количественный и качественный состав используемых вспомогательных веществ обеспечивает возможность получения гранулята с приемлемым показателем сыпучести, позволяет изготавливать лекарственную форму с приемлемыми характеристиками (например, для таблеток – истираемость, твердость, распадаемость, растворение) и обеспечивает сохранность (химическую стабильность) действующего вещества в течение не менее 2 лет.
Приемлемые вспомогательные вещества, подходящие для использования в составе фармацевтической композиции по настоящему изобретению, включают в себя, без ограничения перечисленными (Фармацевтическая технология. Таблетки Алексеев К.В., Кедик С.А., Блынская Е.В., Алексеев В.К., Масленникова Н.В. Фармацевтическая технология. Таблетки: учебное пособие под ред. С.А. Кедика. – М.: ЗАО ИФТ. – 2015. – 672с.):
- Наполнители – вещества, используемые для придания лекарственной форме определенной массы, способствуют быстрой дезинтеграции, обеспечивают прессуемость. В качестве наполнителя могут быть использованы: лактоза, микрокристаллическая целлюлоза, крахмал, натрия гидрокарбонат, метилцеллюлоза, cилифицированная микрокристаллическая целлюлоза, натриевая соль карбоксиметил целлюлозы, кальция карбонат, кальция фосфат двузамещенный, глицин, декстрин, амилопектин, сорбит, маннит, пектин и др. или их смеси.
- Модификаторы поверхности – в качестве модификаторов поверхности используются гидрофильные поверхностно активные вещества или полимеры, понижающие поверхностное натяжение воды. Данные вспомогательные вещества используются для уменьшения потерь действующего вещества на последующих стадиях и равномерного распределения действующего вещества в грануляте.
- Связывающие вещества увеличивают взаимное сцепление отдельных частиц и обеспечивают переход порошкообразного вещества в гранулы, определяют технологические свойства гранулята (в частности однородность и сыпучесть, что коррелирует с точностью дозирования), обеспечивают прочность гранул. В качестве связующего могут быть использованы – сополимер поливиниливого спирта и полиэтилен гликоля Kollicoat IR (Kollicoat IR), поливиниливый спирт, поливинил пирролидон, полиэтиленгликоль, гипролоза, низкозамещенная гидроксипропил целюлоза, оксиэтил целлюлоза, оксипропил метилцеллюлоза и др. или их смеси.
- Разрыхляющие вещества, или дезинтегранты улучшают распадаемость твердой лекарственной формы в среде желудочно-кишечного тракта и высвобождения действующего вещества. В качестве разрыхляющих могут быть использованы – крахмал, натрия крахмал гликолят, карбоксиметил крахмал, кросповидон, аэросил, натрия лаурилсульфат, тальк, натрий-кроскармеллоза и др. или их смеси.
- Скользящие или смазывающие вещества снижают трение между частицами гранулята и фармацевтического оборудования, снижают адгезию и предотвращают прилипание гранулята к поверхностям оборудования. В качестве скользящего могут быть использованы – стеариновая кислота, кальция и магния стеарат и др.
В составе фармацевтической композиции могут использоваться вспомогательные вещества других классов, для обеспечения приемлемых технологических свойств гранулированной массы для получения твердой лекарственной формы. Так же может быть использована приемлемая оболочка для покрытия таблетки или компонентов готовой лекарственной формы.
Таблица 1. Состав фармацевтической композиции.
Готовое лекарственное средство может быть произведено в виде дозированных порошков, порошков для приготовления суспензий, таблеток, покрытых или не покрытых оболочкой, капсул. Вид лекарственной формы не играет роли с точки зрения эффективности в случае лекарственных форм немедленного высвобождения.
Гранулирование – технологический процесс, направленный на укрупнение частиц смеси, позволяющий перевести порошкообразный материал в зерна определенной величины для улучшения сыпучести массы для таблетирования, наполнения капсул, фасовки в первичную упаковку, что происходит в результате значительного уменьшения суммарной поверхности частиц при их слипании в гранулы и, следовательно, соответствующего уменьшения трения, возникающего между этими частицами при движении в емкостях технологического оборудования. Расслоение многокомпонентной порошкообразной смеси обычно происходит за счет разницы в размерах частиц и значениях удельной плотности входящих в ее состав лекарственных и вспомогательных компонентов, что вызывает в ряде случаев выделение компонента с наибольшей удельной плотностью из смеси и приводит к нарушению дозировки действующего вещества. Грануляция предотвращает эту опасность, поскольку в ее процессе происходит слипание частиц различной величины и удельной плотности. Образующийся при этом гранулят, при условии равенства размеров получаемых гранул, приобретает достаточно постоянную насыпную массу. Большую роль играет также прочность гранул: прочные гранулы меньше подвержены истиранию и обладают лучшей сыпучестью. Для получения гранул может быть использована как сухая грануляция, так и влажная грануляция или структурная грануляция.
Технология производства включает измельчение кристаллического действующего вещества, подготовку сырья (просеивание), добавление вспомогательных веществ следующих функциональных классов: наполнители, разрыхляющие/ дезинтегранты, связующие, модификаторы поверхности, скользящие и смазывающие; перемешивание, изготовление гранулята любым технологически приемлемым способом (сухая, влажная, структурная грануляция); изготовление готовой лекарственной формы (наполнение капсул, рассыпку дозированного порошка, таблетирование); проведение контроля качества готового продукта.
Перечень приведенных примеров.
1. Получение фармацевтической композиции для перорального применения.
2. Исследование фармакокинетики пероральной фармацевтической композиции.
3. Изучение токсичности фармацевтической композиции для животных.
4. Изучение безопасности фармацевтической композиции у пациентов с осложненной инфекцией мочевыводящих путей (оИМП).
5. Изучение химиотерапевтической эффективности фармацевтической композиции на модели острой пневмонии, вызванной Pseudomonas aeruginosa.
6. Сравнение химиотерапевтической эффективности фармацевтической композиции с действием антибиотиков гентамицин и ципрофлоксацин, на модели острой пневмонии, вызванной Pseudomonas aeruginosa.
7. Изучение возможности формирования резистентности к фармацевтической композиции у клинических изолятов Pseudomonas aeruginosa.
8. Применение фармацевтической композиции для лечения осложненных инфекций мочевыводящих путей (оИМП), вызванных Pseudomonas aeruginosa
9. Изучение терапевтической эффективности фармацевтической композиции на модели экспериментальной инфекции, вызванной устойчивым к антибиотикам штаммом Enterococcus faecalis.
10. Применение фармацевтической композиции для лечения осложненных инфекций мочевыводящих путей (оИМП), вызванных Enterococcus faecalis.
11. Применение фармацевтической композиции для лечения осложненных инфекций мочевыводящих путей (оИМП), при сочетанной инфекции P. aeruginosa и E. faecalis.
12. Изучение терапевтической эффективности фармацевтической композиции на модели экспериментальной острой и хронической хламидийной инфекции.
13. Изучение терапевтической и профилактической эффективности фармацевтической композиции на модели экспериментальной хронической сальмонеллезной инфекции.
14. Влияние фармацевтической композиции на количественный состав кишечной микрофлоры мышей при сравнении с действием антибиотиков гентамицин и ципрофлоксацин бактериологическим методом.
Представленные ниже примеры иллюстрируют и подтверждают возможность получения заявляемой фармацевтической композиции и решения поставленных задач.
Пример 1.
Получение фармацевтической композиции для перорального применения
Получены различные составы фармацевтической композиции разными технологическими способами. Изменение состава и соотношений вспомогательных веществ в рамках указанного в таблице 1 диапазона значений не оказывает существенного влияния на биодоступность действующего вещества в кристаллическом виде. Критическое значение для показателей качества (тест растворения) и биодоступности имеет степень измельчения действующего вещества – размер частиц должен быть менее 50 мкм.
Лекарственная форма таблетки, или капсулы, или дозированный порошок для приема внутрь изготавливается из смеси одинакового состава и характеристик, отличается только конечный этап производства – прессование массы в таблетки, или заполнение твердых желатиновых капсул, или наполнение первичной упаковки (пакетики или флаконы). Масса для получения лекарственной формы фармацевтической композиции может быть получена любой общедоступной технологией (влажная, сухая или структурная грануляция). Таблетки изготавливаются с оболочкой или без.
Получение методом грануляции в псевдоожиженом слое
Таблица 2. Состав таблеток, получаемых способом грануляции в псевдоожиженном слое (в 3 дозировках)
На весах отвешивают необходимое количество веществ для приготовления 1 загрузки таблеток, готовят раствор увлажнителя содержащий Kollicoat IR и лаурил сульфат натрия, загружают в сушилку гранулятор с псевоожиженным слоем измельченную субстанцию 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид, внутри гранулярную микрокристаллическую целлюлозу, включают поток воздуха прогревают продукт, до температуры в слое 40°С, по достижению температуры производят стадию распыления увлажнителя (водная суспензия лаурил сульфат натрия и Kollicoat IR), и сушку полученного гранулята до температуры в слое 55°С. По достижении температуры в слое, останавливают процесс сушки и выгружают гранулят.
Выгруженный сухой гранулят, калибруют на сито с размером отверстий 0,8 мм.
Откалиброванный сухой гранулят переносят в смеситель, добавляют лактозы моногидрат, натрия крахмала гликолят, и микрокристаллическую целлюлозу, перемешивают заданное время, далее добавляют скользящее магния стеарат и повторно перемешивают, полученную таблеточную массу выгружают в сборник и отдают на стадию таблетирования. Полученные таблетки фасуют в банки из полимерного материала или в блистер из пленки поливинилхлоридной и фольги алюминиевой.
Получение методом влажной грануляции
Таблица 3. Состав таблеток получаемых способом влажной грануляции (в 3 дозировках)
На весах отвешивают необходимое количество веществ для приготовления 1 загрузки таблеток, сырье измельчают, готовят раствор увлажнителя, содержащий Kollicoat IR, загружают в гранулятор для влажной грануляции субстанцию 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид, микрокристаллическую целлюлозу, гранулируют. Влажный гранулят сушат на полочной сушилке с предварительно выставленной температурой 80°С и временем процесса около 4 часов. Проводят процесс до получения порошка с остаточной влажностью от 0,2 до 2%. Влагу определяют термогравиметрически. После окончания процесса сушки гранулят просеивают через сито 1,2 мм, добавляют предварительно просеянные лактозу, карбоксиметил крахмал, и перемешивают в смесителе не менее 15 минут. Добавляют скользящее магния стеарат и повторно перемешивают в течение 3 минут, полученную таблеточную массу выгружают в сборник и передают на стадию таблетирования. Полученные таблетки фасуют в банки из полимерного материала или в блистер из пленки поливинилхлоридной и фольги алюминиевой.
Таблица 4. Состав таблеток получаемых способом влажной грануляции (в 3 дозировках)
Получение методом прямого прессования
Таблица 5. Состав таблеток получаемых способом сухой грануляции (в 2 дозировках)
На весах отвешивают необходимое количество сырья для приготовления 1 загрузки таблеток, действующее вещество измельчают, добавляют ранее смешанные вспомогательные вещества (модификатор поверхности, наполнитель, разрыхляющее, связующее) тщательно перемешивают не менее 15 минут, просеивают через сито, добавляют скользящее и еще раз тщательно перемешивают в течение не менее 3 мин. Полученную массу выгружают в сборник и передают на стадию таблетирования. Полученные таблетки фасуют в банки из полимерного материала или в блистер из пленки поливинилхлоридной и фольги алюминиевой.
Изучение распадаемости таблеток
Полученные таблетки исследовали согласно ОФС.1.4.2.0013.15 «Распадаемость таблеток и капсул». Время, за которое распадались таблетки менее 20 минут.
Изучение растворения
В соответствии с рекомендациями государственной фармакопеи разработан специфический для действующего вещества тест растворения. Исследование проводили с использованием тестера растворения (аппарат II «Лопастная мешалка»): количество действующего вещества, высвободившегося в среду растворения (4% раствор натрия додецилсульфата), при температуре (37±0,5) °С, в течение 45 мин при скорости вращения лопастной мешалки 75 об/мин составило более 75% (Q) от заявленного содержания.
Стабильность
Заявленная фармацевтическая композиция стабильна при хранении в естественных условиях не менее 2 лет по результатам изучения стабильности.
Вывод. Заявленное изобретение позволяет получить фармацевтическую композицию, обладающую приемлемыми технологическими характеристиками и стабильностью при хранении, а также позволяет удобно дозировать лекарственное средство пациентам в условиях клинического применения.
Пример 2
Исследование фармакокинетики пероральной фармацевтической композиции
Для исследования фармакокинетики фармацевтической композиции были использованы таблетки соответствующего качества по показателю теста «Растворение».
Исследование фармакокинетики (всасывание, распределение и элиминация) проводили на 30 крысах самцах массой примерно 0,3 кг. Все животные при проведении эксперимента получали сбалансированный кормовой рацион и воду в неограниченном количестве. Каждой крысе вводили перорально в дозе 50 мг/кг через зонд в виде суспензии измельченную таблетку и субстанцию в кристаллическом виде. Кровь для определения концентрации аналитов (не измененное действующее вещество, и его метаболит глюкуронид) забирали из яремной вены у трех крыс на каждую точку в следующие временные точки: 1/2, 1, 2, 4, 8, 12, 24, 36, 48 часов после введения препарата. Также определяли концентрацию аналитов (не измененное действующее вещество и его метаболит) в органах крыс (печень и легкие) в следующие временные точки: 1/2, 1, 2, 4, 8, 12, 24, 36, 48 часов после введения препарата. Также определена концентрация аналитов в моче крыс в следующие временные точки: 12, 24 и 48 часов. Определение аналитов выполнено в объединенной фракции мочи собранной в течение 12, 24 и 48 часов. Фармакокинетические параметры рассчитаны с использованием модуля Pk Solver.
Далее приведены фармакокинетические параметры, полученные в результате исследования фармацевтической композиции у крыс. Изучали несвязанное / свободное действующее вещество в крови, легких, печени и моче, а так же метаболит глюкуронид действующего вещества. Данные приведены в таблицах.
Термины, использованные в тексте данного примера
Т1/2 - Период полувыведения
Cmax - Максимальная концентрация
Tmax - Время достижения максимальной концентрации
AUC0-t - Площадь под фармакокинетической кривой «концентрация-время» от 0 точки до крайней точки
AUC0-∞ - Площадь под фармакокинетической кривой «концентрация-время» от 0 точки до бесконечности
MRT0-t - Среднее время удерживания действующего вещества в крови от 0 точки до крайней точки
MRT0-∞ - Среднее время удерживания действующего вещества в крови от 0 точки до бесконечности
fT - Тканевая доступность
Таблица 6. Усредненные фармакокинетические параметры для действующего вещества и метаболита в крови крыс после приема фармацевтической композиции.
Таблица 7. Усредненные фармакокинетические параметры для действующего вещества и метаболита в легких крыс после приема фармацевтической композиции.
Таблица 8. Усредненные фармакокинетические параметры для действующего вещества и метаболита в печени крыс после приема фармацевтической композиции.
Таблица 9. Количество выведенного с мочой метаболита и действующего вещества за 12, 24 и 48 часов после приема фармацевтической композиции.
Наглядно количества выведенного с мочой свободного действующего вещества и метаболита приведены на фиг.1.
Вывод: полученная фармацевтическая композиция после перорального введения всасывается, распределяется в органах и тканях организма, метаболизирует до глюкуронида и выводится как в виде глюкуронида, так и в виде неизмененного действующего вещества.
Сравнительное исследование фармакокинетики (всасывание, распределение и элиминация) действующего вещества и фармацевтической композиции проводили на крысах самцах, 21 животное массой примерно 0,3 кг. Все животные при проведении эксперимента получали сбалансированный кормовой рацион и воду в неограниченном количестве. Каждой крысе вводили перорально в дозе 50 мг/кг через зонд в виде суспензии измельченную таблетку и субстанцию в кристаллическом виде. Кровь для определения концентрации не измененного действующего вещества забирали из яремной вены у трех крыс на каждую точку в следующие временные точки: 1/2, 1, 2, 4, 8, 12 часов после введения препарата.
Таблица 10. Усредненные фармакокинетические параметры для действующего вещества в крови крыс после приема фармацевтической композиции и действующего вещества.
Наглядно сравнительные кривые зависимости концентрации действующего вещества в крови от времени после введения действующего вещества и заявленной фармацевтической композиции в форме таблетки показаны на фиг. 2.
Вывод. Заявленное изобретение позволяет получить фармацевтическую композицию, пригодную для использования в клинической практике и обладающую более высокой биодоступностью, чем у действующего вещества, что обеспечивается измельчением действующего вещества менее 50 мкм.
Пример 3
Изучение токсичности фармацевтической композиции для животных
Изучение токсичности на животных проводилось в соответствии регуляторными требованиями: № 61-ФЗ от 12 апреля 2010 г., Руководство по проведению доклинических исследований лекарственных средств. Часть первая / Под ред. А.Н. Миронова. — М.: Гриф и К, 2013. — 944 с.
Исследование острой токсичности фармацевтической композиции
Исследование острой токсичности проводили на мышах и крысах аутбредных линий (самцах и самках) при пероральном (внутрижелудочном) способе введения. Срок наблюдения составил 14 суток. Поскольку в исследовании субстанции не была выявлена среднесмертельная доза, фармацевтическую композицию вводили животным в максимальной дозе 5000 мг/ кг (что превышает суточную терапевтическую дозу для человека в 100 раз).
Во время исследования смертности не зарегистрировано. При аутопсии животных после введения фармацевтической композиции не было обнаружено патоморфологических изменений внутренних органов и тканей. Таким образом, по результатам токсикометрии, а также по данным некропсии позволяют отнести фармацевтическую композицию на основе 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4Н-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид к практически нетоксичным лекарственным веществам.
Изучение хронической токсичности фармацевтической композиции
Исследования хронической токсичности проводили на крысах (самцы и самки) линии Вистар и кроликах (самцы и самки) породы Советская Шиншилла. Тестируемое средство вводили перорально (внутрижелудочно) крысам ежедневно 1 раз в сутки в течение 28 дней в двух дозах: терапевтическая доза – 48 мг/ кг (с учетом пересчета доз по поверхности тела с человека на крысу); десятикратная терапевтическая доза – 480 мг/ кг (для выявления возможных токсических эффектов).
Кроликам фармацевтическую композицию вводили ежедневно в течение 56 суток перорально в следующих дозах: предполагаемая однократная терапевтическая доза – 26,0 мг/ кг; предполагаемая двукратная терапевтическая доза – 53,0 мг/ кг.
При исследовании токсичности использовали общепринятые методы: интегральные и физиологические показатели, исследования клинических и биохимических показателей крови, исследования мочи, морфометрическую и гистологическую оценку внутренних органов и тканей.
Наблюдение за состоянием животных велось ежедневно. Через сутки после последнего введения часть крыс из каждой группы выводили из эксперимента с исследованием интегральных и физиологических показателей, исследованием крови и патологоанатомическим исследованием. За другой половиной крыс («отставленные» группы) наблюдали еще в течение 2 недель. После наблюдения их подвергли эвтаназии с обследованием в том же объеме, что и животных, забитых сразу после окончания введения препарата.
Было показано, что фармацевтическая композиция в дозе 480 мг/ кг на протяжении 28 дней не оказывала выраженного токсического воздействия на функцию основных физиологических систем организма, обменные процессы, показатели крови, структуру и функцию внутренних органов и тканей крыс. При использовании десятикратной дозы были выявлены функциональные изменения в печени, выражающиеся в тканевом увеличении межклеточного пространства, изменении формы клеток. Изменения носили обратимый характер. На протяжении исследования поведение и эмоциональное состояние крыс опытных групп не отличалось от контрольных животных. Многократное введение крысам фармацевтической композиции в дозе 48 мг/ кг не привело к изменению массы и структуры внутренних органов. Статистически достоверных изменений в показателях ЭКГ между контрольными и опытными группами не выявлено.
В экспериментах на кроликах после введения фармацевтической композиции в течение 56 суток перорально было установлено, что фармацевтическая композиция в условиях многократного перорального введения в дозах 26 мг кг и 53 мг/кг на протяжении 56 дней не оказала токсического воздействия на функцию основных физиологических систем организма лабораторных животных, не влияла на обменные процессы, показатели крови, структуру и функцию внутренних органов и тканей.
Исследования на двух видах животных обоего пола (крысах и кроликах) показало, что длительное внутрижелудочное введение фармацевтической композиции на протяжении 28 дней (крысы) и 56 дней (кролики) местно-раздражающего действия на слизистую желудка при макроскопическом и микроскопическом исследовании участков контакта с препаратом у животных не выявлено.
Вывод. Проведенные токсикологические исследования по изучению острой и хронической токсичности показали, что фармацевтическая композиция является малотоксичным препаратом и может быть рекомендована для проведения клинических исследований.
Пример 4
Изучение безопасности фармацевтической композиции у пациентов с осложненной инфекцией мочевыводящих путей (оИМП)
Изучение безопасности препарата фармацевтической композиции при лечении пациентов с оИМП, проводили в рамках клинического исследования (Разрешение МЗ РФ №389 от 03 августа 2018) в соответствии с Протоколом исследования (04-ФТ-2018). Исследование проводилось с целью изучить безопасность и эффективность препарата фармацевтической композиции, в комбинации с препаратом цефепим, в сравнении с плацебо в комбинации с препаратом цефепим при лечении взрослых пациентов с осложненными инфекциями мочевыводящих путей, вызванными P.aeruginosa. В исследование были включены 240 пациентов, госпитализированных по поводу обострения хронических инфекций. Все пациенты получали препарат базовой антибактериальной терапии цефепим в связи с тяжестью клинической картины и необходимостью проводить стандартную антибактериальную терапию. Препарат цефепим – цефалоспорин IV поколения эффективен в отношении грамположительных и грамотрицательных бактерий, колонизирующих мочевыводящие пути. Однако развитие резистентности к цефалоспорину существенно влияет на эффективность терапии, особенно в случае лечения хронических рецидивирующих инфекций.
Общая продолжительность исследования для каждого пациента составила 28 дней, в том числе: период скрининга (визит 0) – не более 24 часов до начала терапии ИП/ПС (0-1 день); период активной терапии (визиты 1-3) – день 7 последний день терапии ИП/ПС; период наблюдения по результатам активной терапии (амбулаторный визит): Визит ВПН – день 21 (±2 дня) с момента завершения терапии.
Пациенты были разделены на 3 группы:
- пациенты группы 1 (Г1) принимали препарат фармацевтическую композицию в дозе 300 мг/сут в сочетании с препаратом цефепим в дозе 2 г/сут, первый прием 600 мг/сут;
- пациенты группы 2 (Г2) принимали препарат фармацевтическую композицию в дозе 600 мг/сут в сочетании с препаратом цефепим в дозе 2 г/сут, первый прием 900 мг/сут;
- пациенты группы 3 (Г3) принимали плацебо в сочетании с препаратом цефепим в дозе 2 г/сут.
Всего в ходе исследования у 240 пациентов, включенных в исследование, было зарегистрировано 158 нежелательных явления (НЯ), связанных с соматическим статусом и изменением лабораторных показателей пациентов. Серьезных нежелательных явлений не выявлено. НЯ были оценены в отношении переменных: тяжесть проявления, наличие связи с исследуемым препаратом, предпринятые действия и исход явления. Все НЯ были преимущественно легкой степени тяжести, разрешились самостоятельно без медикаментозной коррекции. В 3 из 158 случаев (1,9%) причинно-следственная связь с препаратом оценена как «вероятная», в 76 случаях (48,1%) - «возможная», в 19 случаях (12,0%) - «неопределенная», в 3 случаях (1,9%) - «определенная», в 43 случаях (27,2%) - «сомнительная» и в 14 случаях (8,9%) - «условная».
Анализ данных показал, что у пациентов Г1 было зарегистрировано 47 из 158 НЯ (29,7%), Г2 - 46 из 158 (29,1%) и Г3 - 65 из 158 (41,2%).
Анализ результатов сравнительной оценки НЯ с применением Fisher exact test у пациентов показал, что общая частота нежелательных явлений в группах лечения Г1 и Г2 была статистически значимо ниже, в сравнении с группой плацебо Г3 на, соответственно 27,7% (р=0,003) и 29,2% (р=0,002). Дополнительная проверка значимости различий с применением критерия согласия Пирсона (χ2 test) подтвердила полученные оценки – р-уровни составили, соответственно, 0,002 и 0,001.
Вывод. Полученные результаты позволяют сделать заключение о благоприятном профиле безопасности фармацевтической композиции.
Пример 5
Изучение химиотерапевтической эффективности препарата фармацевтической композиции на модели острой пневмонии, вызванной Pseudomonas aeruginosa
Синегнойная палочка является значимым патогеном в возникновении нозокомиальных пневмоний, особенно в отделениях реанимации и интенсивной терапии, в том числе у пациентов с искусственной вентиляцией легких. В связи с этим, для изучения химиотерапевтической активности фармацевтической композиции использовали модель острой пневмонии на мышах чувствительной линии при заражении клиническим антибиотикорезистентным штаммом P.aeruginosa.
Была разработана модель острой пневмонии на чувствительной линии мышей A/ Sn при заражении госпитальным изолятом P.aeruginosa 1840. Была определена доза заражения для изучения динамики гибели животных при развитии пневмонии в течение 5-ти дней наблюдения, а также для определения накопления возбудителя в тканях легкого на 5-е сутки после заражения. При интраназальном введении P.aeruginosa эта доза составила 6 х 106 КОЕ (колониеобразующих единиц) / мышь, что соответствовало ЛД75.
Лечение проводили фармацевтической композицией в дозе 12,5 мг/кг; 25 мг/кг и 50 мг/кг по схеме 2-х кратного введения в течение дня. Таким образом, суточные дозы составили 25 мг/кг (150 мг); 50 мг/кг (300 мг) и 100 мг/кг (600 мг) в пересчете на человека. Кроме того, для сравнения терапевтической эффективности полученной фармацевтической композиции с действующим веществом сформировали группы животных, которым вводили перорально действующее вещество, суспендированное в 1% крахмале. Начинали лечение одновременно с заражением. Лечение проводили в течение 4-х дней, на 5-ый день выживших животных вскрывали после эвтаназии и проводили микробиологический анализ высевом из гомогенатов легких. В каждую группу закладывали по 12 мышей для оценки динамики выживаемости, а также по 6 мышей для микробиологического анализа. Эксперимент повторяли 3 раза.
Эффективность лечения препаратом проводили по динамике выживаемости, состоянию животных и освобождению организма от псевдомонад.
Наблюдения за животными показали, что в контрольных группах в первые 3 дня после заражения выжившие мыши теряли в весе, не питались, были малоподвижны. Их состояние становилось удовлетворительным только к 4 – 5 дням. В группах леченных фармацевтической композицией животных состояние было болезненным в первые 2 дня, но на 3-и сутки животные становились более активными, начинали питаться и постепенно прибавляли в весе. В группах лечения действующим веществом состояние мышей было удовлетворительным только при введении дозы 100 мг/кг. Результаты представлены в таблицах 11, 12, 13.
Таблица 11. Результаты изучения антибактериальной эффективности фармацевтической композиции (ФК) в суточной дозе 25 мг/кг (150 мг) при сравнении с действующим веществом (ДВ) в суточной дозе 25 мг/кг.
КОЕ/ легкое на 5-ые сутки
Лечение ФК
сравнения
Лечение ДВ
Таблица 12. Результаты изучения антибактериальной эффективности фармацевтической композиции (ФК) в суточной дозе 50 мг/кг (300 мг) при сравнении с действующим веществом (ДВ) в суточной дозе 50 мг/кг.
КОЕ/ легкое на 5-ые сутки
Лечение ФК
Лечение ДВ
Таблица 13. Результаты изучения антибактериальной эффективности фармацевтической композиции (ФК) в суточной дозе 100 мг/кг (600 мг) при сравнении с действующим веществом (ДВ) в суточной дозе 100 мг/кг.
КОЕ/ легкое на 5-ые сутки
лечение ФК
Лечение ДВ
Проведенные эксперименты по оценке антибактериальной эффективности фармацевтической композиции при пероральном введении мышам в суточных дозах 25 мг/кг (150 мг); 50 мг/кг (300 мг) и 100 мг/кг (600 мг) для лечения острой пневмонии, вызванной госпитальным штаммом P.aeruginosa, показали зависимое от дозы антибактериальное действие препарата. При интраназальном заражении изолятом 1840 в дозе 6 х 106 КОЕ/ мышь развивалась острая пневмония, вызывающая гибель 75% животных. Пероральное применение фармацевтической композиции 2 раза в день в течение 4-х дней в дозе 25 мг/ кг повысило выживаемость мышей в 2 раза. Для дозы 50 мг/ кг было показано увеличение выживаемости в 3,7 раза, а для дозы 100 мг/ кг гибели животных не наблюдали.
Проведенный анализ накопления псевдомонад в тканях легкого показал, что у контрольных животных, выживших до 5-го дня эксперимента, в легких выявлялся возбудитель в количествах до 7,5±3,2х102 КОЕ/легкое. Пероральный прием фармацевтической композиции по схеме 2 раза в день во всех испытанных дозах приводил к снижению бактериальной нагрузки в тканях легкого. Процент животных, у которых возбудитель отсутствовал в тканях легкого, для групп с лечением составил 33,3 и 50% для меньшей и большей дозы препарата соответственно.
При сравнении эффективности терапии фармацевтической композицией с терапией действующим веществом были получены лучшие показатели по выживаемости мышей и эрадикации псевдомонад из тканей легкого при лечении фармацевтической композицией.
Вывод. Полученные данные свидетельствуют об эффективности лечебного действия фармацевтической композиции на модели пневмонии, вызванной клиническим изолятом P.aeruginosa, характеризующимся множественной резистентностью к антибиотикам. Пероральное применение препарата приводило к снижению гибели животных в течение 5 дней наблюдения и к антибактериальному действию в тканях легкого после 4-х дневного курса введения.
Пример 6
Сравнение химиотерапевтической эффективности фармацевтической композиции с действием антибиотиков гентамицин и ципрофлоксацин, на модели острой пневмонии, вызванной P. aeruginosa
В эксперименты были взяты два клинических изолята синегнойной палочки, характеризующиеся различной резистентностью к антибиотикам. Изолят 1840 устойчив к гентамицину и ципрофлоксацину, а изолят 19182 обладает чувствительностью к этим антибиотикам.
Изучение эффективности действия фармацевтической композиции на модели острой пневмонии у мышей, зараженных клиническим изолятом P. aeruginosa 1840, в сравнении с действием гентамицина и ципрофлоксацина
Мышей контрольной группы заражали интраназально клиническим изолятом P. aeruginosa 1840 в дозе 5 х 105 КОЕ/мышь. Лечение 2-й опытной группы проводили фармацевтической композицией в дозе 50 мг/кг перорально по схеме двукратного введения в течение 4-х дней. 3-ю группу животных лечили гентамицином в дозе 36 мг/кг внутримышечно 2 раза в день в течение 4-х дней. 4-ю группу лечили ципрофлоксацином в дозе 100 мг/ кг перорально 2 раза в день в течение 4-х дней. На 5-й день выживших животных вскрывали после эвтаназии и проводили микробиологический анализ из тканей легкого и крови.
Состояние животных после заражения было угнетенным, у них снизился аппетит и потребление воды, появилась вялость, уменьшилась двигательная активность, шерсть была взъерошенной, дыхание поверхностным и учащённым, глаза слипшимися. Аппетит восстановился только к 5-му дню наблюдений. К этому же времени улучшилось и общее состояние мышей контрольной группы. Мыши из 2-й опытной группы, получавшие фармацевтическую композицию, на протяжении 5-и дней наблюдений сохраняли двигательную активность, уже после двух дней введения препарата начали реагировать на внешние раздражители, активнее двигаться. К 3-м суткам эксперимента животные начали активно есть и пить воду. У животных 3-й и 4-ой групп, получавших лечение гентамицином и ципрофлоксацином, общее состояние и поведение соответствовали общему состоянию и поведению мышей контрольной группы.
Оценка изменения массы зараженных животных в контрольной и опытных группах свидетельствует о том, что заражение мышей в дозе 5 х 105 КОЕ/ мышь негативно влияет на массу животных. Показано, что в 1-й контрольной группе, 3-й и 4-й группах наблюдалась тенденция к снижению массы тела, причем негативная динамика веса сохранялась на протяжении 4 суток, к 5 суткам эксперимента вес стабилизировался. Вес мышей во 2-й опытной группе, леченной фармацевтической композицией, вначале также уменьшился, но к 3-му дню наблюдений стабилизировался и к концу проведения опыта (на 5-е сутки) начал увеличиваться.
Таблица 14. Результаты сравнительного изучения выживаемости мышей при терапии фармацевтической композицией (ФК) в суточной дозе 100 мг/кг (600 мг) с гентамицином (ГЦ) и ципрофлоксацимом (ЦФ) на модели пневмонии, вызванной P.aeruginosa 1840.
Лечение: Плацебо
Лечение ФК 50 мг/ кг 2 раза в день
Лечение ГЦ 36 мг/ кг
Лечение ЦФ 100 мг/ кг 2 раза в день
Лечение гентамицином и ципрофлоксацином, начатое одновременно с заражением мышей клиническим изолятом P.aeruginosa 1840, устойчивым к гентамицину и ципрофлоксацину, в дозе 5 х105 КОЕ/ мышь, не дало положительного результата по защите животных от гибели, в отличие от мышей, леченных фармацевтической композицией, где их выживаемость на 5-е сутки составила 100% (Таблица 14).
Для проведения микробиологических исследований на 5-е сутки мышей вскрывали. При этом дополнительно закладывали большее количество мышей с тем, чтобы во всех группах на 5-е сутки число выживших мышей было достаточным для проведения статистического анализа накопления возбудителя (Таблица 15).
Таблица 15. Результаты сравнительного изучения антибактериальной эффективности фармацевтической композиции (ФК) в суточной дозе 100 мг/кг (600 мг) с гентамицином (ГЦ) и ципрофлоксацимом (ЦФ) на модели пневмонии, вызванной P.aeruginosa 1840.
Лечение: Плацебо
Лечение ФК 50 мг/ кг 2 раза в день
Лечение ГЦ 36 мг/ кг
Лечение ЦФ 100 мг/ кг 2 раза в день
Проведенный анализ накопления псевдомонад в тканях легкого и в крови после заражения клиническим изолятом P. aeruginosa 1840, устойчивым к гентамицину и ципрофлоксацину, в дозе 5 х 105 КОЕ/ мышь на фоне лечения фармацевтической композицией в сравнении с гентамицином и ципрофлоксацином показал, что фармацевтическая композиция практически полностью подавляет размножение патогена, тогда как в группе, получавшей лечение антибиотиками, накопление псевдомонад было практически одинаково по количеству КОЕ, полученном при высевах исследуемых образцов контрольной группы.
Изучение эффективности действия фармацевтической композиции на модели острой пневмонии у мышей, зараженных клиническим изолятом P.aeruginosa 19182, в сравнении с действием гентамицина и ципрофлоксацина
Мышей заражали интраназально клиническим изолятом P.aeruginosa 19182 в дозе 1,0 х 106 КОЕ/мышь (ЛД50). Лечение 2-й опытной группы проводили фармацевтической композицией в дозе 50 мг/ кг перорально по схеме двукратного введения в течение 4-х дней. 3-ю группу животных лечили гентамицином в дозе 36 мг/ кг внутримышечно 2 раза в день в течение 4-х дней. 4-ю группу лечили ципрофлоксацином в дозе 100 мг/ кг перорально 2 раза в день в течение 4-х дней. На 5-й день выживших животных вскрывали после эвтаназии и проводили микробиологический анализ из тканей легкого и крови.
Состояние всех животных в первые сутки после заражения во всех группах было угнетенным, у них снизился аппетит и потребление воды, уменьшилась двигательная активность. Дальнейшие наблюдения за мышами на протяжении 5-и дней эксперимента показали, что состояние мышей контрольной группы в первые четверо суток эксперимента изменялось мало. Аппетит восстановился только к 5-му дню наблюдений. Мыши из 2-й, 3-й и 4-й опытных групп, получавшие фармацевтическую композицию, гентамицин и ципрофлоксацин ко 2-м суткам после заражения выглядели менее больными. Мыши реагировали на внешние раздражители, дыхание восстановилось. К 3-м суткам наблюдений состояние мышей улучшилось, они начали активней двигаться, реагировать на внешние раздражители. К завершению наблюдений (на 5-сутки) мыши выглядели здоровыми.
Оценка изменения массы зараженных животных в контрольной и опытных группах свидетельствовало о том, что заражение негативно влияет на массу животных. Тенденция к снижению массы тела наблюдалась в 1-й контрольной группе, причем негативная динамика веса сохранялась на протяжении 3 – 4 суток наблюдений. Вес мышей во 2-й опытной группе, леченной фармацевтической композицией, в 3-й опытной группе, леченной гентамицином, и в 4-й, леченной ципрофлоксацином, также уменьшился на 1-е сутки, но уже к 3-м суткам стабилизировался и к концу проведения опыта (на 5-е сутки) начал увеличиваться.
Таблица 16. Результаты сравнительного изучения выживаемости мышей при терапии фармацевтической композицией (ФК) в суточной дозе 100 мг/кг (600 мг) с гентамицином (ГЦ) и ципрофлоксацином (ЦФ) на модели пневмонии, вызванной P.aeruginosa 19182.
Лечение: Плацебо
Лечение ФК 50 мг/ кг 2 раза в день
Лечение ГЦ 36 мг/ кг
Лечение ЦФ 100 мг/ кг 2 раза в день
При заражении мышей клиническим изолятом P. aeruginosa 19182 в дозе 1,0 х 106 КОЕ/ мышь в контрольной группе наблюдали гибель 50% животных. Одновременное с заражением начало лечения фармацевтической композицией дало возможность защитить животных от гибели на 100%. Лечение антибиотиками, гентамицином и ципрофлоксацином показало одинаковую эффективность в снижении гибели животных до 0 (Таблица 16).
Для проведения микробиологических исследований на 5-сутки вскрывали мышей из всех 4-х групп (Таблица 17). При этом дополнительно закладывали большее количество мышей с тем, чтобы во всех группах на 5-е сутки число выживших мышей было достаточным для проведения статистического анализа накопления возбудителя. На анализ были взяты кровь и легкие животных.
Таблица 17. Результаты сравнительного изучения антибактериальной эффективности фармацевтической композиции (ФК) в суточной дозе 100 мг/кг (600 мг) с гентамицином (ГЦ) и ципрофлоксацином (ЦФ) на модели пневмонии, вызванной P.aeruginosa 19182.
Лечение: Плацебо
Лечение ФК 50 мг/ кг 2 раза в день
Лечение ГЦ 36 мг/ кг
Лечение ЦФ 100 мг/ кг 2 раза в день
Таким образом, проведенный анализ накопления псевдомонад в тканях легкого и крови после заражения мышей клиническим изолятом P. aeruginosa 19182, чувствительным к гентамицину и ципрофлоксацину показал, что лечение фармацевтической композицией также эффективно, как и препаратами сравнения, антибиотиками гентамицином и ципрофлоксацином.
Сравнение динамики освобождения организма от псевдомонад на модели пневмонии при лечении фармацевтической композицией и ципрофлоксацином
Для сравнения эффективности антибактериального действия фармацевтической композиции с действием антибиотика ципрофлоксацин в модели острой пневмонии был выбран клинический изолят P. aeruginosa 19182, чувствительный к ципрофлоксацину.
Мышей заражали интраназально дозой 5 х 105 КОЕ/ мышь изолята 19182. Лечение начинали в день заражения. Фармацевтическая композиция в дозе 50 мг/ кг перорально 2 раза в день в течение 3-х дней; ципрофлоксацин в дозе 100 мг/ кг перорально 2 раза в день в течение 3-х дней. Вскрытие осуществляли через 24, 48 и 72 часа после заражения и проводили микробиологические высевы из легких и крови зараженных животных.
Результаты микробиологического выделения псевдомонад в высевах из легких и крови мышей, зараженных изолятом P. aeruginosa 19182 при лечении фармацевтической композицией и Ципрофлоксацином и без лечения приведены в Таблице 18.
Таблица 18. Результаты изучения динамики антибактериального действия фармацевтической композиции (ФК) в суточной дозе 100 мг/кг (600 мг) при сравнении с ципрофлоксацином (ЦФ) на модели пневмонии, вызванной P.aeruginosa 19182.
из легких, КОЕ/ орган
из крови, КОЕ/ мл
контроль заражения
50 мг/ кг 2 раза ФК
100 мг/ кг ЦФ
контроль заражения
50 мг/ кг
2 раза ФК
100 мг/ кг ЦФ
Таким образом, было показано, что лечение фармацевтической композицией и ципрофлоксацином, начатое одновременно с заражением, одинаково эффективно подавляло размножение псевдомонад в тканях легкого, а также в крови зараженных животных. Было выявлено, что динамика эрадикации возбудителя из легкого на фоне лечения фармацевтической композицией несколько отлична от действия ципрофлоксацина. Если на 1-е и 2-е сутки ципрофлоксацин более эффективно подавляет размножение псевдомонад, то на 3-ьи сутки освобождение от возбудителя при действии ципрофлоксацина и фармацевтической композицией одинаково.
Вывод. Фармацевтическая композиция обладает сопоставимым антибактериальным действием с антибиотиками при лечении инфекций, вызванных чувствительным изолятом P.aeruginosa, выражающееся в увеличении выживаемости зараженных животных и эрадикации возбудителя из тканей. Однако при лечении экспериментальной инфекции, вызванной множественно резистентным изолятом псевдомонад, эффективную терапию наблюдали только в случае лечения фармацевтической композицией. Таким образом, фармацевтическая композиция подавляет инфекционный процесс вне зависимости от приобретенной патогенами резистентности.
Пример 7
Изучение возможности формирования резистентности к препарату фармацевтической композиции
Сравнительное изучение частоты мутаций и скорости формирования резистентности при действии активной субстанции фармацевтической композиции 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4Н-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида (CL-55) и антибиотика in vitro проводили методом посева на твердые питательные среды с CL-55 и антибиотиком сравнения ципрофлоксацином (ЦФ).
Для того, чтобы выявить возникновение устойчивых к CL-55 вариантов, использовали метод накопительных культур. CL-55 в отличие от антибиотиков не вызывает гибель и не подавляет размножение патогенов, а ингибирует секрецию экзотоксинов ССТТ. Регистрировать возникновение устойчивых к CL-55 клонов прямым посевом на среды, содержащие CL-55 невозможно, так как это соединение не влияет на жизнеспособность патогенов и поэтому не создает селективный фон для отбора мутантов. Уровень секреции экзотоксинов в присутствии CL-55 можно оценить в тестах иммуноблотинга, для культур синегнойной палочки выращенных в присутствии CL-55 и без CL-55.
С целью изучить частоты образования мутаций и скорости формирования устойчивости, использовали пассажи клинических изолятов синегнойной палочки в среде, содержащей ингибирующую ССТТ концентрацию CL-55 – 50 мкг/мл и с концентрацией бактерий 1-3х109 КОЕ/мл.
Культуры 41431 и 32461 P. aeuginosa длительно пассировали в условиях индукции секреции ССТТ при добавлении CL-55 и повторяли такие пассажи 10 раз. В качестве контроля пересевали культуры в LB-бульоне без CL-55 аналогичным образом.
Далее проводили определение уровня секреции эффекторного белка ССТТ ExoT у культур 41431 и 32461, пассированных с CL-55 и без него, после 3х часового культивирования с различными концентрациями (0; 10, 30 и 50 мкг/мл). Если бы культивирование в течение 10 пассажей с CL-55 привело к отбору устойчивых к нему форм, то это могло быть обнаружено по отсутствию ингибирующего действия CL-55 на секрецию эффекторных белков или по повышению концентрации CL-55, необходимой для достижения ингибирующего действия.
Сравнение данных иммуноблотинга для культур 41431 и 32461, предварительно инкубированных в присутствии CL-55 в течение 21 пассажа, или без CL-55, показало, что как в первой, так и во второй группе происходило одинаковое подавление секреции эффекторного белка ExoТ под действием CL-55 в концентрациях 10, 30 или 50 мкг/мл (фиг.3). Полученные данные могут свидетельствовать о том, что длительное пассирование культур в присутствии CL-55 не приводило к накоплению устойчивых к CL-55 форм.
Для сравнения исследовали процесс образования мутантов устойчивых к ципрофлоксацину у культур штаммов 41431 и 32461, трижды пассированных в LB-бульоне, содержащим 0.5 МИК ципрофлоксацина (0,4 мкг/мл), и без ципрокфлоксацина. МИК ципрофлоксацина для культур 41431 и 32461 предварительно была определена экспериментально путем посева культур в пробирки с LB-бульоном, содержащим ципрофлоксацин в конечных концентрациях 8,0; 4,0; 2,0; 1,0; 0,8; 0,4 и 0,2 мкг/мл.
Для определения скорости и частоты образования мутантов устойчивых к ЦФ высевали культуры 41431 и 32461с концентрацией 2.0х109 KOE/мл по 0.5 мл на чашки Петри с LB–агаром, содержащим МИК и двукратно возрастающие концентрации ЦФ (0,8; 1,6; 3,2 и 6,4 мкг/мл). Титр культур был предварительно определен посевом из разведений 10-7 на LB–агар без ЦФ.
Таблица 19. Количество колоний при посевах культур 41431 и 32461с концентрацией 2.0х109 KOE/мл по 0.5 мл на чашки Петри с LB–агаром, содержащим МИК и двукратно возрастающие концентрации ЦФ.
с ЦФ (мкг/мл)
Таким образом, мутации устойчивости к ципрофлоксацину возникали у культур 41431 и 32461(таблица 16) как после 3-х пассажей с 0,5 МИК (0,8 мкг/мл) в бульоне, с частотой около 106, соответственно. Скорость формирования мутаций и уровень устойчивости к ЦФ были выше у культур 41431 и 32461 после 3-х пассажей с 0,5 МИК ЦФ. Известно, что формирование устойчивости к фторхинолонам, в том числе к ЦФ, имеет ступенчатый характер вследствие наличия у патогенов трех мишеней, мутации в которых приводят к возникновению соответствующей антибиотикорезистентности. Поэтому предварительное пассирование культур синегнойной палочки с субингибиторной концентрацией ЦФ ускоряет процесс формирования антибиотикорезистентных вариантов.
Вывод. В проведенных экспериментах не было выявлено изменений в ингибирующей активности CL-55 в отношении культур синегнойной палочки после длительного (10 пассажей) пассирования в присутствии CL-55. Сравнение данных иммуноблотинга для культур 41431 и 32461, предварительно инкубированных в присутствии CL-55 в течение 10 пассажей, или без CL-55, показало, что как в первой, так и во второй группе происходило одинаковое подавление секреции эффекторного белка ExoТ под действием CL-55 в концентрациях 10, 30 или 50 мкг/мл, что свидетельствует об отсутствии накопления устойчивых к фармацевтической композиции форм. В то время как для антибиотика ципрофлоксацина было показано, что даже короткое (3 пассажа) пассирование в среде, содержащей субингибиторную концентрацию ципрофлоксацина, приводило к повышению скорости формирования и накоплению мутантов, устойчивых к антибиотику.
Пример 8
Применение фармацевтической композиции для лечения осложненных инфекций мочевыводящих путей (оИМП), вызванных Pseudomonas aeruginosa
Изучение терапевтической эффективности препарата фармацевтической композиции при лечении пациентов с оИМП, вызванных Р.aeruginosa, проводили в рамках клинического исследования (Разрешение МЗ РФ №389 от 03 августа 2018) в соответствии с Протоколом исследования (04-ФТ-2018). Исследование проводилось с целью изучить безопасность и эффективность препарата фармацевтической композиции, в комбинации с препаратом цефепим, в сравнении с плацебо в комбинации с препаратом цефепим при лечении взрослых пациентов с осложненными инфекциями мочевыводящих путей, вызванными P.aeruginosa. В исследование были включены 160 пациентов, госпитализированных по поводу обострения хронических инфекций. Все пациенты получали препарат базовой антибактериальной терапии цефепим в связи с тяжестью клинической картины и необходимостью проводить стандартную антибактериальную терапию. Препарат цефепим – цефалоспорин IV поколения эффективен в отношении грамположительных и грамотрицательных бактерий, колонизирующих мочевыводящие пути. Однако развитие резистентности к цефалоспорину существенно влияет на эффективность терапии, особенно в случае лечения хронических рецидивирующих инфекций.
Общая продолжительность исследования для каждого пациента составила 28 дней, в том числе: период скрининга (визит 0) – не более 24 часов до начала терапии ИП/ПС (0-1 день); период активной терапии (визиты 1-3) – день 7 последний день терапии ИП/ПС; период наблюдения по результатам активной терапии (амбулаторный визит): визит ВПН – день 21 (±2 дня) с момента завершения терапии.
Пациенты были разделены на 2 группы:
- пациенты первой группы принимали фармацевтическую композицию (ФК) в дозе 600 мг/сут , первый прием 900 мг/сут, в сочетании с препаратом цефепим в дозе 2 г/сут;
-пациенты второй группы принимали плацебо в сочетании с препаратом цефепим в дозе 2 г/сут.
В исследование эффективности вошло 14 пациентов (ФК - 600 мг/сут) и 13 пациентов (плацебо), у которых на исходном уровне (визит 0) была выделена Р.aeruginosa из мочи. Первичную конечную точку эффективности оценивали по общему ответу, включающему клиническую эффективность и микробиологическую эрадикацию, на визите последующего наблюдения (ВПН), через 21 день после завершения терапии.
Таблица 20. Показатели общего клинического ответа у пациентов, имевших Р.aeruginosa в моче на исходном уровне, в исследуемых группах на Визите через 21 день после завершения терапии.
Анализ данных по клиническому излечению и микробиологической эрадикации в популяции пациентов, имевших выделенную Р.aeruginosa на исходном уровне, на визите ВПН показал, что терапия с применением фармацевтической композиции в комбинации с препаратом цефепим в дозе 600 мг/сут показывает выраженную тенденцию к увеличению эффективности терапии при сравнении с группой, получавшей плацебо в комбинации с препаратом цефепим.
Анализ доли пациентов с ответом в виде микробиологической эрадикации Р.aeruginosa в популяциях пациентов группы, принимавших ФК -600 мг/сут и группы (плацебо), на визите ВПН представлены в таблице 21.
Таблица 21. Показатели пациентов с ответом в виде микробиологической эрадикации Р.aeruginosa в исследуемых группах на Визите через 21 день после завершения терапии.
Проведенный сравнительный анализ доли пациентов с ответом в виде микробиологической эрадикации на визите ВПН показал выраженную тенденцию к более эффективной антибактериальной терапии в отношении Р.aeruginosa в группе пациентов, получавших фармацевтическую композицию в комбинации с препаратом цефепим, при сравнении с группой пациентов, получавших плацебо в комбинации с препаратом цефепим.
Анализ доли пациентов с ответом в виде клинического излечения в популяциях пациентов группы, принимавших ФК в дозе 600 мг/сут и группы плацебо, на визите ВПН представлены в таблице 22.
Таблица 22. Показатели пациентов с ответом в виде клинического излечения в исследуемых в группах на Визите через 21 день после завершения терапии.
Вывод. Изучение терапевтической эффективности фармацевтической композиции при лечении рецидивирующих инфекций мочевыводящих путей, вызванных Р.aeruginosa, показывает выраженную тенденцию к увеличению эффективности терапии при сравнении с группой, получавшей плацебо в комбинации с препаратом цефепим. Через 21 день после окончания терапии в группе с лечением фармацевтической композицией 600 мг/сут наблюдали рецидивы в 1 случае из 14. В тоже время, в группе пациентов, принимавших с плацебо в комбинации с препаратом цефепим, на эти же сроки выявили микробиологический рецидив псевдомонадной этиологии в 3 случаях из 13. Кроме того, в группе пациентов, принимавших плацебо в сочетании с препаратом цефепим на визите ВПН у двух пациентов произошла колонизация псевдомонадами, что привело к рецидиву клинических симптомов. В группах лечения фармацевтической композицией колонизация псевдомонадами не наблюдалась. Тем самым, наблюдается выраженная тенденция антибактериального действия фармацевтической композиции в отношении P.aeruginosa.
Пример 9
Изучение терапевтической эффективности фармацевтической композиции на модели экспериментальной инфекции, вызванной устойчивым к антибиотикам штаммом Enterococcus faecalis
Enterococcus faecalis является патогеном, ответственным за возникновение нозокомиальных инфекций различной локализации с высоким риском развития бактериемии и сепсиса. Энтерокковые инфекции опасны тем, что возбудитель характеризуется широким спектром устойчивости ко многим антибиотикам, что делает антибиотикотерапию малоэффективной. Химиотерапевтическое действие фармацевтической композиции проводили на модели генерализованной инфекции у мышей при заражении клиническим изолятом Enterococcus faecalis.
Исследования были выполнены на мышах инбредной линии DBA/2 с массой тела 16 – 18 грамм. Количество животных, используемых в исследовании – по 8 голов в группе, что является достаточным для полной регистрации изучаемых эффектов. Дополнительно в каждый опыт закладывали большее количество мышей с тем, чтобы во всех группах число выживших мышей было достаточным для проведения статистического анализа накопления возбудителя в органах.
Бактериальную культуру штамма выращивали на жидкой среде Luria-Bertani (LB - бульон), при 37°С 18 часов, центрифугировали (6000 об/ мин, 10 мин.), осадок ресуспендировали в физиологическом растворе и использовали для заражения. Для определения количества КОЕ бактерий отбирали аликвоту полученной суспензии (100 мкл), готовили серию последовательных разведений и 500 мкл разведенной культуры наносили на чашки Петри с LB-агаром. Чашки Петри культивировали при 37°С. Через 24 часа культивирования производили подсчет колоний. Концентрация бактерий выражалась числом КОЕ/ мл.
Для выявления E. faecalis в легких, образцы органов гомогенизировали в 1 мл физиологического раствора, полученные гомогенаты центрифугировали 10 мин при 800 об/ мин (0,8 rcf) для удаления грубых остатков тканей. Затем готовили серии 10-кратных разведений исходной суспензии гомогенатов легких в физиологическом растворе, и 500 мкл каждого разведения помещали на чашку Петри, покрытую LB-агаром. Чашки Петри культивировали при 37 ºС, через 24 часа культивирования производили подсчет колоний. Концентрация псевдомонад в органах выражалась числом колоний образующих единиц на орган. Чашки Петри инкубировали при 37ºС и через 24 часа культивирования производили подсчет колоний. Концентрацию энтерококков выражали числом КОЕ в 1 мл гомогената.
Животных заражали внутрибрюшинно культурой E. faecalis 366 объёмом 500 мкл в дозе 4 х 109 КОЕ/ мышь.
При пероральном введении препарата применяли атравматичный металлический зонд (так называемую гаважную иглу), который вводили в пищевод до желудка. Таблетку с содержанием 300 мг фармацевтической композиции растирали в ступке. Затем постепенно при интенсивном перемешивании добавляли небольшие порции 2% крахмального клейстера и растирали до однородной массы. Контрольные животные получали эквивалентные объемы крахмального клейстера. Мышам вводили по 200 мкл суспензии препарата.
Целью данного этапа работы явилось определение эффективности лечебного действия фармацевтической композиции на модели генерализованной инфекции по динамике выживаемости, состоянию животных и освобождению организма от E. faecalis. Лечение начинали одновременно с заражением, фармацевтическую композицию вводили животным в дозе 100 мг/ кг один раз в день в течение 8-ми дней. Для учета количества энтероккоков в легких животных вскрывали на 6-й день и 8-й день и проводили микробиологический анализ высевом из гомогенатов легких. Эксперимент повторяли 3 раза.
Наблюдения за животными показали, что в контрольных группах в первые 3 дня после заражения выжившие мыши теряли в весе, не питались, были малоподвижны. Их состояние становилось удовлетворительным только к 4 – 5 дням. В группах леченных препаратом животных состояние было болезненным в первые 2 дня, но на 3-и сутки животные становились более активными, начинали питаться и постепенно прибавляли в весе (Таблица 23).
Таблица 23. Результаты наблюдения за динамикой гибели и освобождению организма мышей от энтерококков, при заражении клиническим изолятом E. faecalis 366 и лечении фармацевтической композицией (ФК).
Лечение ФК 100 мг/кг 1 раз в день
Примечание: данные представлены как среднее арифметическое ± стандартная ошибка среднего арифметического.
При заражении мышей клиническим изолятом E. faecalis 366 в дозе 4 х 109 КОЕ/ мышь в контрольной группе наблюдали гибель 37,5% животных. В группе лечения гибель животных отсутствовала. Проведенный анализ накопления энтероккоков в тканях легкого показал, что у контрольных животных, выживших до 6-го дня эксперимента, в легких выявлялся возбудитель в количествах до 4,1 х 104 КОЕ/ легкое. На 8-й день эксперимента при анализе количества энтерококков в тканях легкого было показано, что среднее количество бактерий составило 1,2 х 103 КОЕ/ легкое.
Пероральный прием препарата фармацевтической композиции приводил к снижению бактериальной нагрузки в тканях легкого. В группе лечения на 6-й день после заражения количество энтероккоков в легких составило 3,8х1,2 х 102 КОЕ/ легкое, а на 8-й день после заражения 0,15 х 101 КОЕ/ легкое.
Статистический анализ количества псевдомонад в высевах из легких мышей выявляли с помощью непараметрического критерия Манна-Уитни. Разница между контрольной и опытной группами у мышей, зараженных и леченых фармацевтической композицией, является статистически достоверной (p < 0,05).
Полученные данные свидетельствуют об эффективности терапевтического действия препарата фармацевтической композиции на модели генерализованной инфекции, вызванной клиническим изолятом E. faecalis. Было показано, что пероральное применение препарата защищало 100% животных в группе лечения. При анализе накопления бактерий в тканях легкого отмечали, что 50% мышей из группы лечения были свободны от энтероккоков. В тоже время в группе контроля инфекции все животные имели бактериальную нагрузку. Для остальных 50% в группе лечения было показано значительное снижение количества бактерий в легких при сравнении с контрольной группой.
Вывод. Лечение генерализованной энтерококковой инфекции препаратом фармацевтической композиции показало эффективное терапевтическое действие, что выражалось в достоверном снижении количества энтероккоков в легких животных, увеличении средней продолжительности жизни и 100% защите животных от гибели.
Пример 10
Применение фармацевтической композиции для лечения осложненных инфекций мочевыводящих путей (оИМП), вызванных Enterococcus faecalis
Изучение терапевтической эффективности фармацевтической композиции при лечении пациентов с оИМП, вызванных E.faecalis, проводили в рамках клинического исследования (Разрешение МЗ РФ №389 от 03 августа 2018) в соответствии с Протоколом исследования (04-ФТ-2018). Исследование проводилось с целью изучить безопасность и эффективность препарата фармацевтической композиции, в комбинации с препаратом цефепим, в сравнении с плацебо в комбинации с препаратом цефепим при лечении взрослых пациентов с осложненными инфекциями мочевыводящих путей, вызванными P.aeruginosa. В исследование были включены 160 пациентов, госпитализированных по поводу обострения хронических инфекций. Все пациенты получали препарат базовой антибактериальной терапии цефепим в связи с тяжестью клинической картины и необходимостью проводить стандартную антибактериальную терапию. Препарат цефепим – цефалоспорин IV поколения эффективен в отношении грамположительных и грамотрицательных бактерий, колонизирующих мочевыводящие пути. Однако развитие резистентности к Цефалоспорину существенно влияет на эффективность терапии, особенно в случае лечения хронических рецидивирующих инфекций.
Общая продолжительность исследования для каждого пациента составила 28 дней, в том числе: период скрининга (визит 0) – не более 24 часов до начала терапии ИП/ПС (0-1 день); цериод активной терапии (визиты 1-3) – день 7 последний день терапии ИП/ПС; период наблюдения по результатам активной терапии (амбулаторный визит): визит ВПН – день 21 (-±2 дня) с момента завершения терапии.
Пациенты были разделены на 2 группы:
- пациенты первой группы принимали препарат фармацевтической композиции (ФК) в дозе 600 мг/сут, первый прием 900 мг/сут, в сочетании с препаратом цефепим в дозе 2 г/сут;
- пациенты второй группы принимали плацебо в сочетании с препаратом цефепим в дозе 2 г/сут.
В исследование вошли 21 пациент (ФК- 600 мг/сут) и 24 пациента (плацебо), у которых на исходном уровне (визит 0) была выделена E.faecalis из мочи. Первичную конечную точку эффективности оценивали по общему ответу, включающему клиническую эффективность и микробиологическую эрадикацию, на визите последующего наблюдения (ВПН), через 21 день после завершения терапии.
Таблица 24. Показатели общего ответа у пациентов, имевших в моче E.faecalis на исходном уровне, в исследуемых группах на Визите через 21 день после завершения терапии.
Антибактериальное действие фармацевтической композиции в отношении E.faecalis оценивали на визите 3, после окончания терапии, и на визите последующего наблюдения (ВПН), через 21 день после завершения терапии.
Таблица 25. Показатели пациентов с ответом в виде микробиологической эрадикации E. faecalis на визите 3 и на визите ВПН.
При анализе доли пациентов, у которых наблюдали микробиологическую эрадикацию энтерококков, в группе плацебо показатели в день 7 и на визите ВПН были достоверно ниже при сравнении с группой пациентов, принимавших препарат фармацевтической композиции. Эрадикация энтерококков в группе, получавших плацебо в комбинации с препаратом цефепим, на 7 день и на визите ВПН составила 58,3% и 66,7%, соответственно. В группе пациентов, получавших фармацевтическую композицию 600 мг/сут в комбинации с препаратом цефепим, эрадикация энтерококков на 7 день и на визите ВПН составила 81% и 76,2%, соответственно.
Enterococcus faecalis - крайне актуальный возбудитель госпитальных инфекций различной локализации, в том числе эндокардитов и инфекций мочевыводящих путей, с высоким риском развития бактериемии и сепсиса. Как показали результаты наших исследований энтерококк встречается примерно у 30% пациентов с осложненными инфекциями мочевыводящих путей. Энтерокковые инфекции опасны тем, что возбудитель характеризуется широким спектром устойчивости ко многим антибиотикам, что делает антибиотикотерапию малоэффективной.
Вывод. Для фармацевтической композиции выявлена выраженная тенденция к демонстрации антибактериального действия в отношении крайне актуального возбудителя госпитальных инфекций – E.faecalis. Оценка эффективности препарата фармацевтической композиции по общему ответу у пациентов, имевших выделенный патоген E.faecalis на исходном уровне в моче, продемонстировала тенденцию к улучшению данного показателя по доле пациентов, имеющих устойчивое клиническое излечение и устойчивую микробиологическую эрадикацию в группе с лечением препаратом фармацевтической композиции в дозе 600 мг в сутки в комбинации с препаратом цефепим (2 г в сутки парентерально) по сравнению с группой плацебо в комбинации с препаратом цефепим (2 г в сутки парентерально): 75% по сравнению с 56,5%, соответственно.
Пример 11
Применение фармацевтической композиции для лечения осложненных инфекций мочевыводящих путей (оИМП), при сочетанной инфекции P. aeruginosa и E. Faecalis
Изучение терапевтической эффективности фармацевтической композиции при лечении пациентов с оИМП, вызванных E.faecalis, проводили в рамках клинического исследования (Разрешение МЗ РФ №389 от 03 августа 2018) в соответствии с Протоколом исследования (04-ФТ-2018). Исследование проводилось с целью изучить безопасность и эффективность препарата фармацевтической композиции, в комбинации с препаратом цефепим, в сравнении с плацебо в комбинации с препаратом цефепим при лечении взрослых пациентов с осложненными инфекциями мочевыводящих путей, вызванными P.aeruginosa. В исследование были включены 160 пациентов, госпитализированных по поводу обострения хронических инфекций. Все пациенты получали препарат базовой антибактериальной терапии цефепим в связи с тяжестью клинической картины и необходимостью проводить стандартную антибактериальную терапию. Препарат цефепим – цефалоспорин IV поколения эффективен в отношении грамположительных и грамотрицательных бактерий, колонизирующих мочевыводящие пути. Однако развитие резистентности к цефалоспорину существенно влияет на эффективность терапии, особенно в случае лечения хронических рецидивирующих инфекций.
Общая продолжительность исследования для каждого пациента составила 28 дней, в том числе: период скрининга (визит 0) – не более 24 часов до начала терапии ИП/ПС (0-1 день); период активной терапии (визиты 1-3) – день 7 последний день терапии ИП/ПС; период наблюдения по результатам активной терапии (амбулаторный визит): визит ВПН – день 21 (±2 дня) с момента завершения терапии.
Пациенты были разделены на 2 группы:
- пациенты первой группы принимали препарат фармацевтической композиции (ФК) в дозе 600 мг/сут, первый прием 900 мг/сут, в сочетании с препаратом цефепим в дозе 2 г/сут;
- пациенты второй группы принимали плацебо в сочетании с препаратом цефепим в дозе 2 г/сут.
В исследование вошли 8 пациентов, принимавших фармацевтическую композицию 600 мг/сут, и 6 пациентов, принимавших плацебо, у которых на исходном уровне (визит 0) были выделены одновременно P.aeruginosa и E.faecalis из мочи. Первичную конечную точку эффективности оценивали по общему ответу, включающему клиническую эффективность и микробиологическую эрадикацию, на визите последующего наблюдения (ВПН), через 21 день после завершения терапии.
Таблица 26. Показатели общего ответа у пациентов, имевших в моче P.aeruginosa и E.faecalis на исходном уровне, в исследуемых группах на Визите через 21 день после завершения терапии.
Результаты показывают повышение эффективности терапии фармацевтической композицией при лечении сочетанных инфекций по сравнению с плацебо.
Анализ доли пациентов с ответом в виде микробиологической эрадикации Р.aeruginosa и E.faecalis в популяциях пациентов группы, принимавших ФК -600 мг/сут и группы (плацебо), на визите ВПН представлены в таблице 27.
Таблица 27. Показатели пациентов с ответом в виде микробиологической эрадикации Р.aeruginosa и E.faecalis в исследуемых группах на Визите через 21 день после завершения терапии.
Проведенный сравнительный анализ доли пациентов с ответом в виде микробиологической эрадикации на визите ВПН показал выраженную тенденцию к более эффективной антибактериальной терапии в отношении Р.aeruginosa и E.faecalis в группе пациентов, получавших фармацевтическую композицию в комбинации с препаратом цефепим, при сравнении с группой пациентов, получавших плацебо в комбинации с препаратом цефепим.
Вывод. Проведенный анализ по оценке терапевтической эффективности фармацевтической композиции в комбинации с препаратом цефепим при лечении сочетанных инфекций, вызванных Р.aeruginosa и E.faecalis, показал тенденцию к лучшим показателям по общему ответу и по микробиологической эрадикации при сравнении с группой пациентов, получавших плацебо в комбинации с препаратом цефепим.
Пример 12
Изучение терапевтической эффективности фармацевтической композиции на модели экспериментальной острой и хронической хламидийной инфекции
Для изучения хронической хламидийной инфекции была разработана модель восходящей хламидийной инфекции, вызванной C. trachomatis серовара D на мышах линии DBA/2. C. trachomatis серовар D является возбудителем хламидиоза у человека и вызывает уретриты, воспаление верхних отделов урогенитального тракта, неонатальную пневмонию и конъюнктивиты.
Оценка терапевтического действия фармацевтической композиции на ранних сроках развития острой урогенитальной инфекции в нижних отделах урогенитального тракта (УГТ).
Для изучения действия фармацевтической композиции мышей заражали C. trachomatis серовара D в дозе 106 ВОЕ/мышь. Лечение проводили в течение 7 дней с момента заражения препаратом фармацевтической композиции в дозе 100 мг/кг перорально и антибиотиком азитромицином в дозе - 10 мг/кг перорально, как препаратом сравнения. В качестве контрольной группы использовали группу зараженных мышей.
Развитие острой урогенитальной хламидийной инфекции в нижних отделах урогенитального тракта оценивали на третьи, седьмые, одиннадцатые и четырнадцатые сутки после заражения. Для этого у мышей отбирали вагинальные смывы и определяли наличие в них хламидийной инфекции с помощью культурального метода исследования (выделение хламидий на клеточной линии McCoy), и иммунофлуоресцентным методом при окраске специфическими моноклональными антителами (ПИФ).
Было показано, что фармацевтическая композиция оказывала антибактериальное действие и значительное снижение количества жизнеспособных хламидий в нижних отделах УГТ при сравнении с контрольной группой инфицированных животных. Разница была наиболее заметна на 7 сутки после заражения (фиг.4). Течение инфекционного процесса у контрольной (нелеченой) группы животных, в отличие от экспериментальных групп, характеризовалось воспалительными процессами в нижних отделах урогенитального тракта, стекловидными или слизисто-гнойными выделениями из влагалища. Начиная с 3 суток по 14 сутки лечения, как фармацевтической композицией, так и антибиотиком высеваемость хламидий из урогенитального тракта была в значительной степени снижена.
Полученные выше данные были подтверждены при микроскопическом анализе культуральных высевов смывов из влагалища, полученных от исследуемых групп мышей (фиг.5).
Для оценки течения инфекционного процесса в экспериментальной и контрольных группах был определен процент инфицированных животных на начальных этапах лечения в нижних отделах УГТ.
Таблица 28. Количество инфицированных животных в группе при лечении фармацевтической композицией и антибиотиком в сравнении с контрольной группой.
Как видно из таблицы 28 уже на 3 сутки в группе лечения фармацевтической композицией количество мышей, у которых развитие хламидийной инфекции было подавлено, уменьшилось в 2 раза по сравнению с контрольной группой. В группе лечения азитромицином инфекция отсутствовала на всех этапах времени до 14 суток.
Таким образом, с использованием разработанной модели урогенитальной хламидийной инфекции при заражении мышей линии DBA/2 клинически значимым штаммом C. trachomatis серовар D было показано, что фармацевтическая композиция эффективно подавляет развитие острой урогенитальной хламидийной инфекции, что выражалось в подавлении накопления возбудителя и отсутствием воспалительных процессов в нижних отделах урогенитального тракта мышей.
Изучение терапевтической эффективности фармацевтической композиции на модели хронической урогенитальной инфекции, вызванной C. trachomatis серовар D
Для изучения терапевтической эффективности фармацевтической композиции при лечении хронической инфекции были проведены опыты по оценке ее протективного действия на модели хронической урогенитальной инфекции, вызванной C. trachomatis серовар D. Мышей линии DBA/2 самок заражали интравагинально C. trachomatis серовар D в дозе 106 ВОЕ/мышь. Лечение проводили, начиная с 19 дня с момента заражения в течение 10 дней. Фармацевтическую композицию вводили перорально в дозе 50 мг/кг и 100 мг/кг в объеме 0,2 мл. Накопление хламидийной инфекции в верхних отделах УГТ проводили на 30 сутки с момента заражения методом ПЦР-РВ, для чего из матки, яичника и эндоцервикса выделяли ДНК.
Таблица 29. Результаты определения количества геном эквивалентов (ГЭ) C.trachomatis в верхних отделах УГТ методом количественного анализа ПЦР-РВ после терапии фармацевтической композицией (ФК).
Результаты ПЦР показали заметное снижение накопления хламидий в группе с лечением фармацевтической композицией в дозе 100 мг/кг на 30 день после заражения, что подтверждает терапевтический эффект данной дозы препарата на модели хронической урогенитальной инфекции. Использованный метод определения ДНК является единственным информативным в отношении выявления хламидий в верхних отделах УГТ при развитии хронической инфекции, т.к. на этой стадии инфекционного процесса хламидии переходят в некультивируемое состояние, что не позволяет использовать метод культурального выявления.
Гистологическое изучение образцов тканей матки, маточных труб и яичников показало, что урогенитальный хламидиоз вызывает у мышей острый патологический процесс, характеризующийся воспалением всех изучаемых отделов. У зараженных животных, по сравнению с нативным контролем выявлено нарушение гистоархитектоники органов, воспалительный инфильтрат представленный эозинофиллами, лимфоцитами и нейтрофиллами, сосуды органов кровенаполненны. Слизистые оболочки отёчны и часть эпителиальных клеток в состоянии апоптоза. У животных, получавших лечение фармацевтической композицией, наблюдается положительная динамика. Элиминация возбудителя из тканей пораженных органов привела к снижению патологических процессов. Отечность органов и количество инфильтрата значительно снижены по сравнению с контрольной инфицированной группой.
Вывод. Показан антибактериальный эффект фармацевтической композиции на модели острой и хронической хламидийной инфекции в нижних и верхних отделах урогенитального тракта. Терапия фармацевтической композицией восходящей хламидийной инфекции при введении препарата в дозе 100 мк/кг в течение 10 дней подавляла размножение хламидий у 5 мышей из 6. Снижение количества геном эквивалентов хламидий у одной леченной мыши по сравнению с контролем составило 500 раз. Гистологические исследования показали, что лечение хронической инфекции фармацевтической композицией приводило к значительному уменьшению признаков воспалительной патологии в гистоархитектонике тканей матки в сравнении с контрольной группой.
Пример 13
Изучение терапевтической и профилактической эффективности фармацевтической композиции на модели экспериментальной хронической сальмонеллезной инфекции
Важной частью проводимых исследований является изучение эффективности терапии фармацевтической композицией хронических инфекций, плохо поддающихся лечению антибиотиками. Было проведено исследование терапевтической активности фармацевтической композиции в модели хронической сальмонеллезной инфекции.
Заражение мышей перорально (per os), то есть таким образом, как это происходит естественным путем, приводит к развитию системной хронической сальмонеллезной инфекции у животных. Было показано, что заражение штаммом IE147 S. enterica серовар Typhimurium приводит к персистенции сальмонелл в течение как минимум 4 недель в организме мышей линии BALB/cJCit(BALB/c. Количество сальмонелл, высеваемых из органов мышей, было максимальным (2х103 - 5х105 КОЕ/мл) на ранних стадиях инфекции (7-12 сутки после заражения), что коррелировало с выраженной спленомегалией. На 2 и 3 неделе количество сальмонелл, высеваемых из органов, постепенно снижалось. На 4 неделе после заражения сальмонеллы высевались преимущественно из мезентериальных лимфатических узлов (МЛУ).
Сравнительная терапевтическая эффективность фармацевтической композиции и антибиотика ампициллина на модели хронической сальмонеллёзной инфекции
Исследования были выполнены на мышах инбредной линии BALB/cJCit(BALB/c). Терапию начинали через 7 дней после заражения, поскольку при моделировании хронической инфекции на этих сроках были показаны наиболее репрезентативные манифестации в развитии патологического процесса для высокочувствительных к сальмонелльной инфекции мышей BALB/с. Введение фармацевтической композиции осуществляли в дозе 50 мг/кг перорально в объеме 0,5 мл. Введение водного раствора ампициллина (для инъекций) осуществляли в дозе 50 мг/кг перорально в объеме 0,5 мл. Оценку терапевтической активности проводили по количеству сальмонелл, высеваемых из селезенок зараженных животных, получавших и не получавших лечение.
Мышей распределяли на 3 группы, каждая по 6 животных. 1-й группе вводили перорально раствор фармацевтической композиции; 2-й группе вводили перорально водный раствор антибиотика ампициллина; 3-я группа была контрольной (без лечения). Лечение проводили в течение 7 дней, затем после эвтаназии животных вскрывали, гомогенизировали селезенки и определяли количество сальмонелл в группах, получавших лечение, а также контрольной группы. Высевы сальмонелл из гомогенизированных в 1 мл физраствора селезенок производили из разведений гомогенатов с десятикратным шагом на селективную среду SS-агар (Pronadisa, Испания). Исследования проводили в 3-х повторах.
Посев гомогенатов селезенок на селективную среду показал сравнимый по эффективности терапевтический эффект фармацевтической композиции (подавление инфекции на 91%) и ампициллина (подавление инфекции на 90%). Результаты исследования представлены в таблице 30.
Таблица 30. Количество сальмонелл, высеваемых из селезенок мышей, получавших перорально терапию фармацевтической композицией в дозе 50 мг/кг или ампициллином в дозе 50 мг/кг по 5-ти дневной схеме лечения, и мышей контрольной группы в модели хронического сальмонеллеза.
КОЕ/селезенка
Таким образом, на модели хронической сальмонеллезной инфекции на высокочувствительных к сальмонеллезу мышах линии BALB/c пероральное введение фармацевтической композиции оказывало сравнимое по эффективности с антибиотиком антибактериальное действие, выражающееся в подавлении накопления возбудителя в селезенке на 90%.
Профилактические режимы терапии фармацевтической композицией и антибиотиком ампициллином в экспериментальных моделях острого и хронического сальмонеллеза
Введение ингибитора фармацевтической композиции осуществляли в дозе 50 мг/кг перорально за 1, 2 и 5 суток до заражения. Профилактику антибиотиком ампициллином проводили параллельно в той же дозе 50 мг/кг и по той же схеме введения. Профилактическую эффективность исследуемых препаратов оценивали по количеству сальмонелл, высеваемых из селезенок, зараженных животных, получавших и не получавших лекарственное средство.
Изучение профилактической активности фармацевтической композиции проводили на модели острого и хронического сальмонеллеза, вызванного при заражении штаммом IE147 S.enterica серовар Typhimurium мышей линии BALB/cJCit(BALB/c). Профилактику фармацевтической композицией проводили в течение 5 дней до заражения. Каждая группа состояла из 6 животных. После профилактического введения исследуемого лекарственного средства мышей заражали для индукции хронической сальмонеллезной инфекции, как описано выше. Для индукции острой инфекции суспензию сальмонелл вводили внутрибрюшинно в дозе 103 КОЕ/мл. Количество сальмонелл в селезенках определяли через 3 дня после заражения у мышей каждой группы путем высева на питательные среды. Исследования были выполнены в 3-х повторах.
Результаты исследования показали высокую профилактическую эффективность фармацевтической композиции в отношении эрадикации возбудителя из организма зараженных животных в модели хронического сальмонеллеза. Профилактика в течение 5 дней показала полное подавление накопления сальмонелл в селезенке животных Подавление инфекции в модели острого сальмонеллеза было менее эффективным и составило около 77%.
Таблица 31. Количество сальмонелл, высеваемых из селезенок мышей, получавших профилактику фармацевтической композицией (ФК) в течение 5 суток до заражения в дозе 50 мг/кг, и мышей контрольной группы.
КОЕ/селезенка
ФК
ФК
Профилактическую эффективность фармацевтической композиции оценивали на модели хронической сальмонеллезной инфекции при сравнении с профилактикой антибиотиком ампициллином. Мышей заражали штаммом IE147 S.enterica серовар Typhimurium мышей линии BALB/cJCit(BALB/c). Профилактику фармацевтической композицией проводили в течение 1 или 2 дней до заражения. Каждая группа состояла из 6 животных. После профилактического введения исследуемых лекарственных средств мышей заражали для индукции хронической сальмонеллезной инфекции, как описано выше. Количество сальмонелл в селезенках определяли через 3 дня после заражения у мышей каждой группы путем высева на питательные среды. Исследования были выполнены в 3-х повторах.
Результаты исследования приведены в таблице 32 и демонстрируют высокую профилактическую эффективность фармацевтической композиции в модели хронического сальмонеллеза по сравнению с антибиотиком ампициллином.
Таблица 32. Количество сальмонелл, высеваемых из селезенок мышей, получавших профилактику фармацевтической композицией (ФК) в течение 1 и 2 суток до заражения в дозе 50 мг/кг, и профилактику ампициллином в дозе 50 мг/кг в течение 1 и 2 суток до заражения, и мышей контрольной группы через 3 дня после заражения.
КОЕ/селезенка
Из таблицы видно, что даже 1 профилактическое введение фармацевтической композиции оказывает выраженное протективное действие и приводит к снижению количества сальмонелл в селезенках на 95% по сравнению с группой без профилактики. Профилактическое действие фармацевтической композиции приводит к большему на один порядок снижению количества сальмонелл в селезенках по сравнению с профилактическим действием ампициллина.
Вывод. На модели хронической сальмонеллезной инфекции у мышей было показано, что фармацевтическая композиция характеризуется антибактериальным действием, приводя к подавлению сальмонелл в селезенке. Наиболее эффективной оказалась схема лечения для хронической сальмонеллезной инфекции, включающая однократное введение препарата в течение 7 дней в дозе 50 мг/ кг, приводящая к 100% эрадикации возбудителя. Кроме этого, была показана эффективность профилактического действия фармацевтической композиции, что приводило к подавлению накопления возбудителя сальмонеллезной инфекции в селезенке на 90%, при хроническом течении. При этом показано преимущество фармацевтической композиции в качестве как терапевтического, так и профилактического средства по сравнению с антибиотиком ампициллином на моделях сальмонеллезной инфекции.
Пример 14
Влияние фармацевтической композиции на количественный состав кишечной микрофлоры в сравнении с действием антибиотиков гентамицин и ципрофлоксацин бактериологическим методом
Мишенью препарата фармацевтической композиции является система секреции III типа, которая присутствует только у патогенных бактерий. Кроме того, ранее было показано, что препарат не обладает прямым антибактериальным действием. Эти обстоятельства являются существенным преимуществом фармацевтической композиции перед антибиотиками в отношении влияния на нормальную микрофлору.
Было сформировано 4 группы мышей линии BALB/c по 6 голов. 1-ая группа – контрольная. Мышам 2-й группы перорально вводили фармацевтическую композицию в дозе 50 мг/кг 2 раза в день 5 дней. Мышам из 3-й группы был проведен курс внутримышечных инъекций гентамицином (ГЦ) в дозе 5 мг/кг 2 раза в день 5 дней. Мыши 4-й группы получали антибиотик ципрофлоксацин (ЦФ) по 100 мг/ кг 2 раза в день в течение 5-ти дней per os. Сбор экскрементов и микробиологическое исследование кала производилось 3 раза: до начала введения препаратов, на 15-е и 28-е сутки после начала введения препаратов.
В обеспечении колонизационной резистентности участвуют облигатные представители нормальной микрофлоры, поэтому интерес представляли изменения в популяциях у таких эубионтов, как эшерихии, лактобактерии, бифидобактерии и фекальный энтерококк.
Проведенная сравнительная характеристика действия фармацевтической композиции (ФК) и антибиотиков сравнения ГЦ и ЦФ на количественный состав кишечной микрофлоры позволила выявить достоверные различия во влиянии изучаемого препарата и антибиотиков на разные сроки эксперимента.
В динамике эксперимента обнаруживались значительные изменения количества лактозоположительных эшерихий. Было показано, что при применении фармацевтической композиции их количество уменьшилось в 11 раз (c 4,28 х 108 до 3,83 х 107 КОЕ/г) на 15-е сутки после начала введения препарата, а к окончанию эксперимента восстановилось практически до начальных значений. В группах мышей, получавших ГЦ и ЦФ, на 15-е сутки эксперимента наблюдалось сокращение общего количества эшерихий на 2 порядка по сравнению с показателем до начала лечения. К окончанию эксперимента численность лактозоположительной кишечной палочки в этих группах не увеличилась (Фиг.6).
При изучении динамики лактобактерий в кишечном биоценозе мышей было выявлено, что при использовании фармацевтической композиции на 15-е сутки наблюдений количество лактобактерий снизилось незначительно (3,9 х 107 КОЕ/г против 7,5 х 107 КОЕ/ г до начала введения). На 28-е сутки их количество практически восстановилось до уровня начала наблюдений (фиг.7). В группах мышей, получавших антибиотикотерапию, снижение количества лактобактерий было более выраженным к 15-м суткам наблюдений (5,58 х 106 против 6,4 х 107 КОЕ/ г в начале эксперимента в группе с ГЦ и 6,0 х 106 против 5,8 х 107 КОЕ/ г в начале эксперимента в группе с ЦФ). К окончанию опыта показатели содержания лактобактерий в группе с ГЦ практически не изменились. В группе с применением ЦФ количество лактобактерий начало восстанавливаться и достигло 3,1 х 107 КОЕ/ г.
Проведенные исследования количественного состава бифидобактерий показали, что их численность подвергалась незначительным изменениям во всех опытных группах на протяжении всего срока наблюдений. Колебания их количества оставались в пределах одного порядка (фиг.8).
Наблюдения за изменением показателей фекального стрептококка обнаружили незначительное увеличение его количества в группе с применением ФТ на протяжении всего срока эксперимента (фиг.9). Выраженные изменения количества фекального стрептококка произошли в результате антибиотикотерапии в обеих опытных группах мышей, получавших ГЦ и ЦФ. По сравнению с начальным уровнем, на 15-е сутки наблюдений содержание фекального стрептококка в группе с ГЦ снизилось приблизительно в 11 раз, в группе с ЦФ – в 11,6 раз. На 28-е сутки наблюдений количество фекального стрептококка в группе с ГЦ продолжало уменьшаться, а в группе с ЦФ возросло.
Таким образом, проведенные эксперименты по сравнительной оценке количественного и качественного состава кишечной микрофлоры при действии ФТ и антибиотиков сравнения ГЦ и ЦФ свидетельствовали о том, что применение ФТ в дозе 50 мг/ кг два раза в день перорально на протяжении 5-и дней не оказывало негативного бактерицидного действия на нормальную микрофлору кишечника животных на протяжении всего срока исследований. При изучении динамики изменения состава кишечной микрофлоры, обусловленном применением антибиотиков широкого спектра действия ГЦ и ЦФ, были обнаружены значительные изменения. Они характеризовались уменьшением количества постоянных (резидентных) представителей микрофлоры: лактобактерий, эшерихий, фекального стрептококка. В частности, были выявлены существенные нарушения микробиоценоза кишечника, вызванные применением ГЦ, которые проявились в значительном подавлении жизнедеятельности лактозоположительной кишечной палочки, снижении количеств лактобактерий и фекального стрептококка на 15-е сутки эксперимента. К концу срока наблюдения численность этих представителей микробиоты не восстановилась.
Вывод. При изучении количественного состава кишечной микрофлоры мышей при действии фармацевтической композиции и антибиотиков сравнения ГЦ и ЦФ было показано, что применение фармацевтической композиции в дозе 50 мг/ кг два раза в день перорально на протяжении 5-ти дней не оказывало негативного бактерицидного действия на нормальную микрофлору кишечника животных на протяжении всего срока исследований. При применении фармацевтической композиции условно-патогенная флора не выявлялась, что в сочетании с отсутствием негативного влияния на микробиоценоз кишечника является его принципиальным преимуществом перед используемыми антибиотиками.
Приведенные примеры подтверждают что коллективом изобретателей выполнена поставленная задача настоящего изобретения по созданию пероральной твердой фармацевтической композиции, содержащей действующее вещество в количестве, необходимом и достаточном для антимикробного действия, для лечения острых, рецидивирующих и хронических инфекций, таких как осложненные инфекции мочевыводящих путей, пневмонии, хронические урогенитальные инфекции, острые и хронические инфекции кишечной локализации, вызванные антибиотикорезистентными штаммами Pseudomonas aeruginosa и/или Enterococcus spp. и/или Chlamydia trachomatis. и/или Salmonella spp., способа ее применения для лечения рецидивирующих и хронических инфекций, вызванных этими патогенами, вне зависимости от приобретенной резистентности.
Промышленная применимость
Все приведенные примеры подтверждают промышленную применимость данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Твердая дисперсия 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида для изготовления лекарственной формы и способ лечения хронических инфекционных заболеваний | 2020 |
|
RU2743927C1 |
| Применение 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4Н-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида для подавления инфекции, вызванной устойчивыми к антибиотикам штаммами Pseudomonas aeruginosa, и способ подавления этой инфекции | 2016 |
|
RU2624846C1 |
| Лекарственное средство для лечения бактериальных инфекций | 2019 |
|
RU2778521C2 |
| СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЧЕЛОВЕКА, ВЫЗВАННЫХ ГРАМПОЛОЖИТЕЛЬНЫМИ БАКТЕРИЯМИ | 2017 |
|
RU2661613C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И НАБОР ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2011 |
|
RU2496475C2 |
| СПОСОБ СНИЖЕНИЯ ТЯЖЕСТИ ТОКСОПЛАЗМОЗА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ РИФАМИЦИНА ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ ДЛЯ ЛЕЧЕНИЯ ТОКСОПЛАЗМОЗА | 1994 |
|
RU2121838C1 |
| СПОСОБ И СРЕДСТВО АКТИВАЦИИ IRF-3 ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ, ВЫЗЫВАЕМЫХ (+) PHK-СОДЕРЖАЩИМИ ВИРУСАМИ | 2012 |
|
RU2518314C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ РИФАБУТИНА С ПОВЫШЕННОЙ БИОДОСТУПНОСТЬЮ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ МИКОБАКТЕРИОЗОВ | 2013 |
|
RU2520603C1 |
| ПАЛЛАДИЕВО-МЕДНЫЕ КАТАЛИЗАТОРЫ ГОМОГЕННОГО СЕЛЕКТИВНОГО ОКИСЛЕНИЯ ТИОЛЬНЫХ ГРУПП, КОМБИНАЦИЯ И КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ТЕРАПЕВТИЧЕСКОГО ВОЗДЕЙСТВИЯ | 2011 |
|
RU2451010C1 |
| Способ получения полиморфной формы 4-(3-этокси-4-гидроксибензил)-N-(2,4-дифторфенил)-5-оксо-5,6-дигидро-4H-1,3,4-тиадиазин-2-карбоксамида | 2022 |
|
RU2785195C1 |
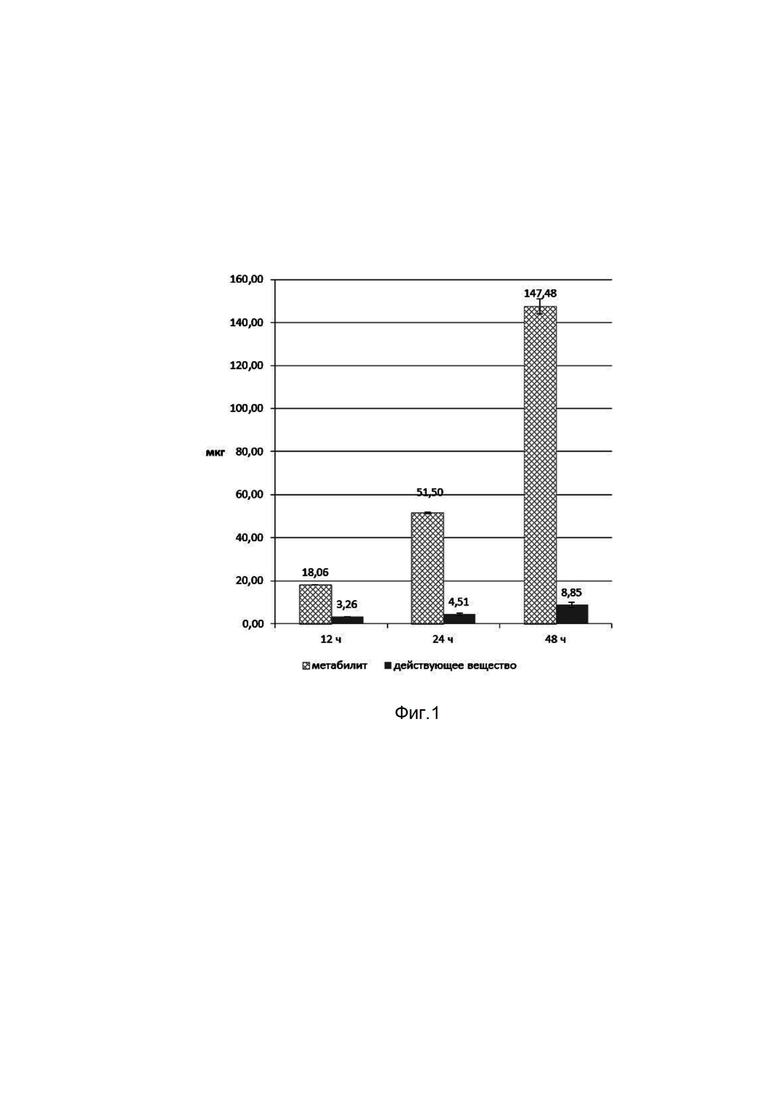
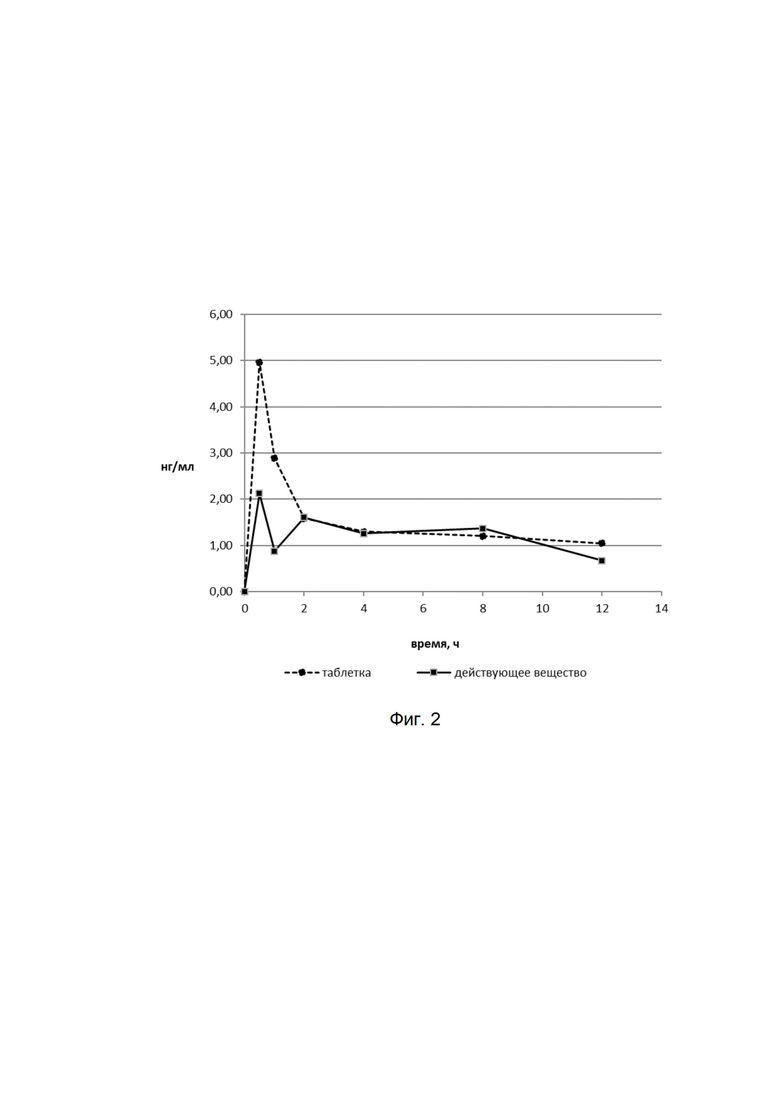

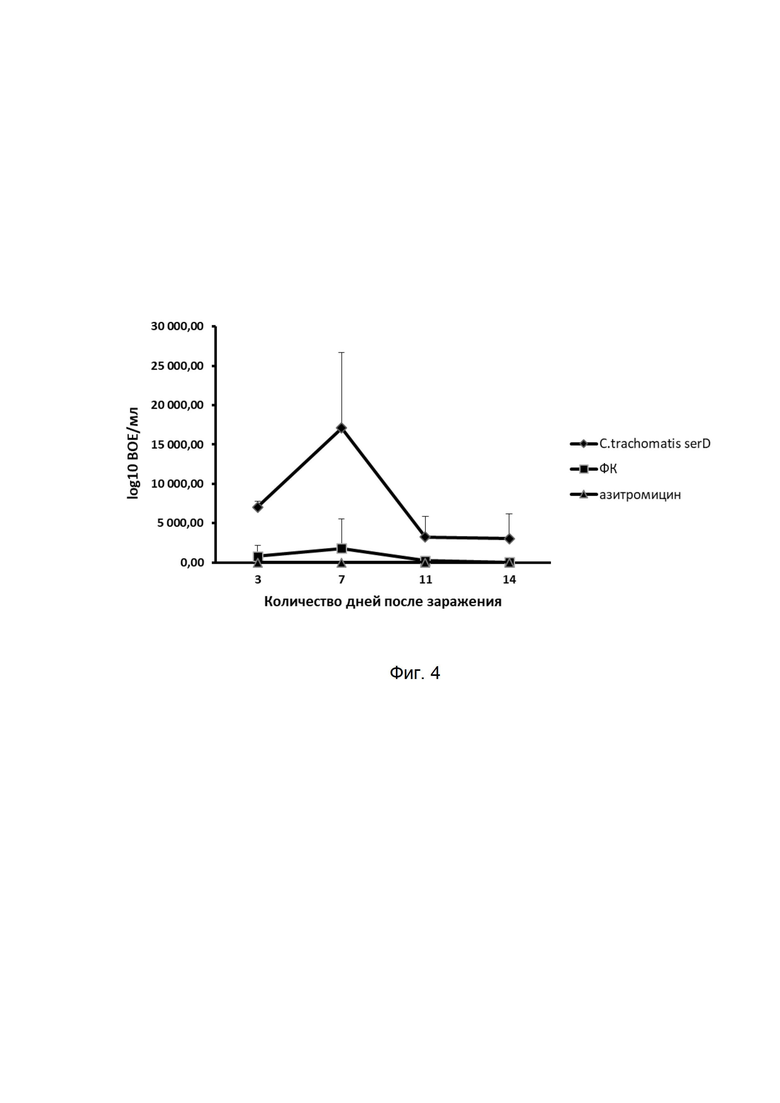
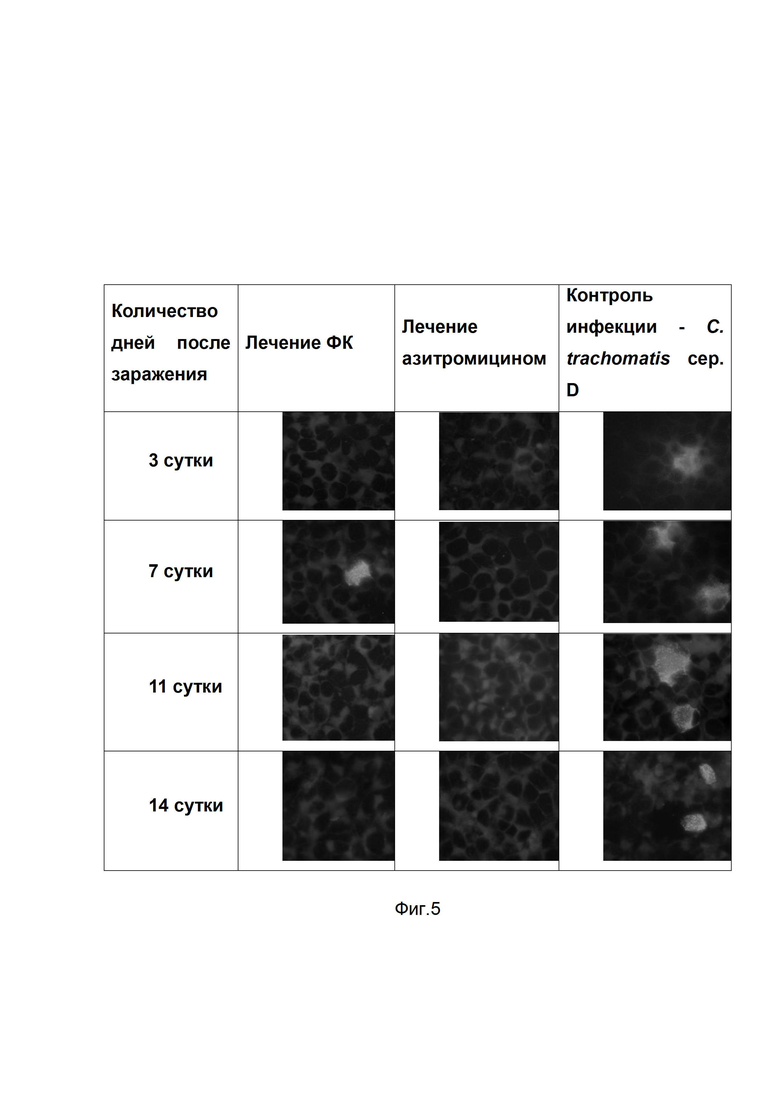
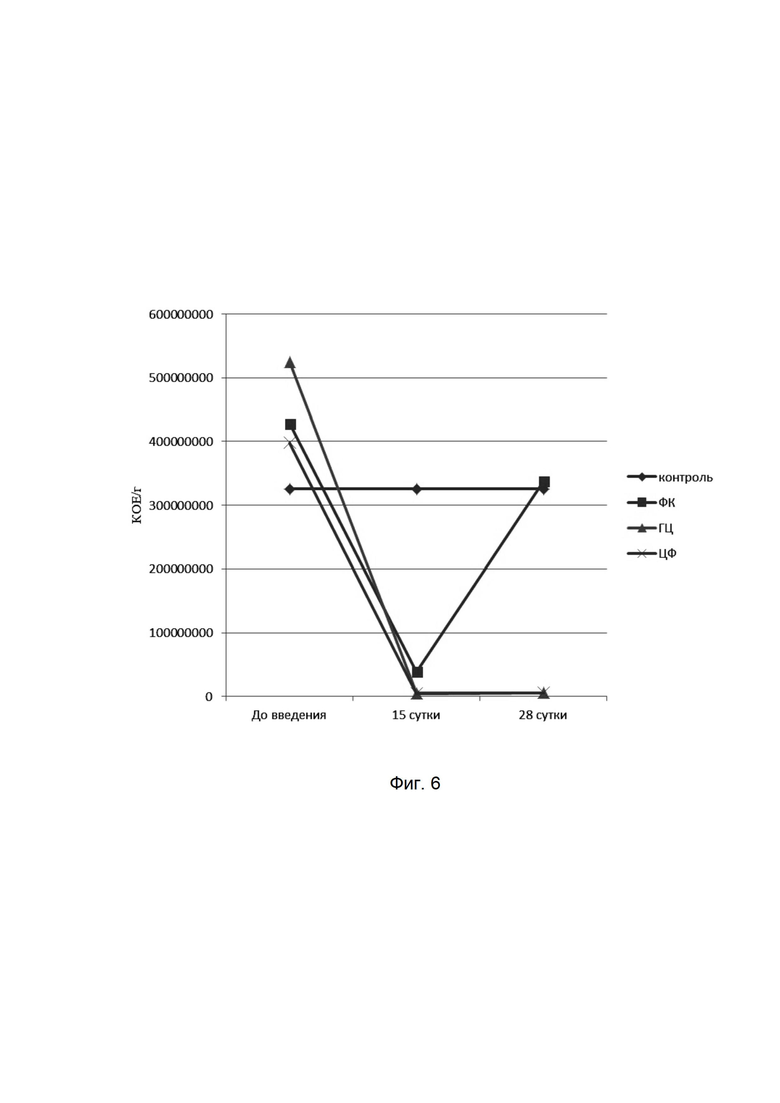
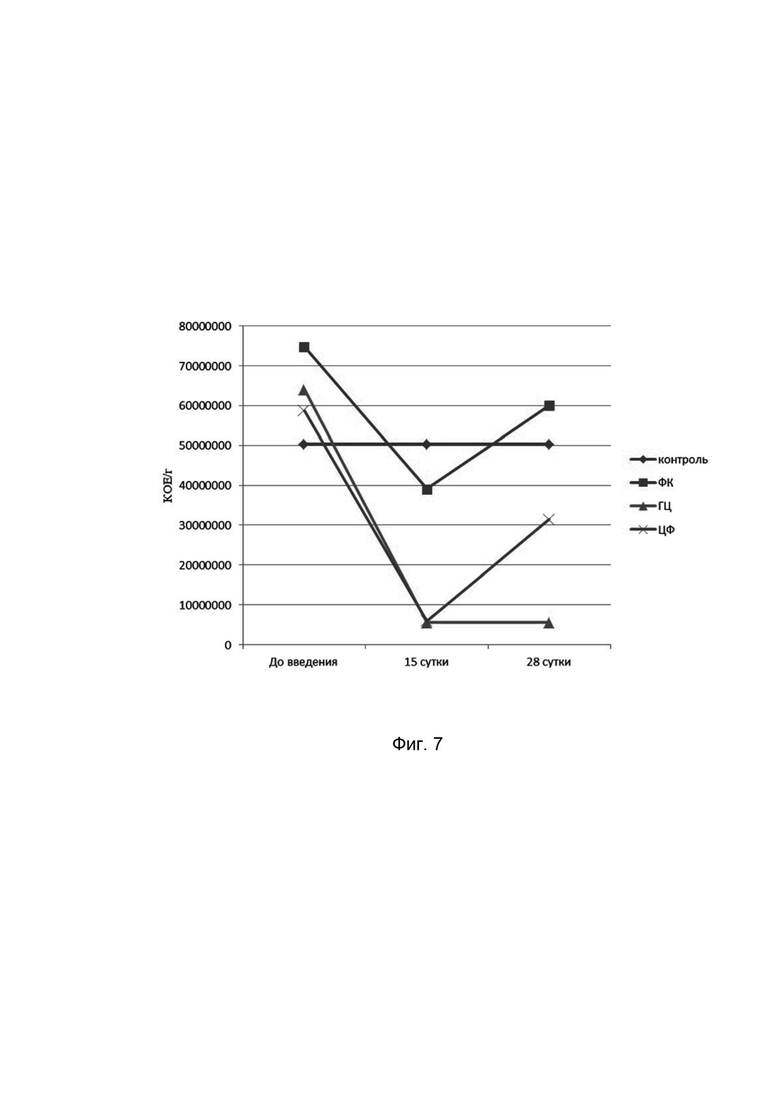
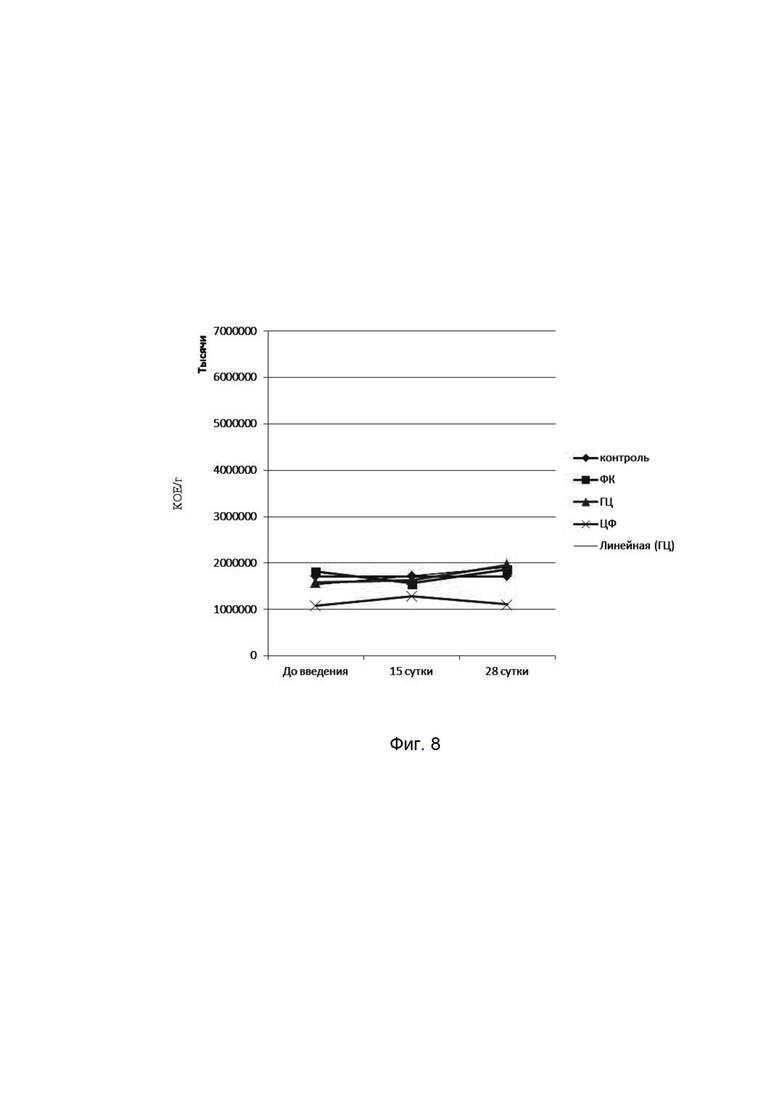
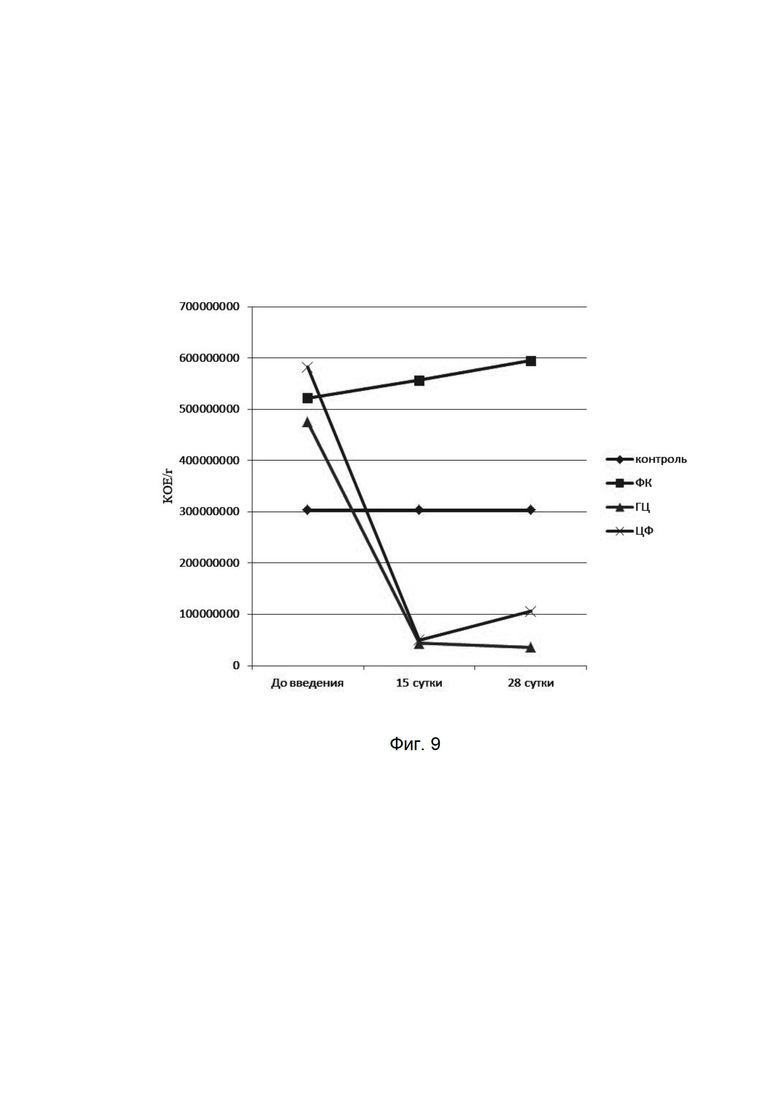
Изобретение относится к фармацевтической композиции антибактериального действия, включающей действующее вещество 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид в кристаллической форме с размером частиц менее 50 мкм в количестве от 150 до 600 мг в виде пероральной твердой дозированной лекарственной формы с приемлемыми вспомогательными веществами. Изобретение также относится к применению указанной фармацевтической композиции для лечения острых, рецидивирующих и хронических инфекций, таких как осложненные инфекции мочевыводящих путей, пневмония, хронические урогенитальные инфекции, острые и хронические инфекции кишечной локализации, вызванные антибиотикорезистентными штаммами Pseudomonas aeruginosa, Enterococcus spp., Chlamydia trachomatis, Salmonella spp. Технический результат – получена новая фармацевтическая композиция, которая может найти свое применение в медицине. 2 н. и 2 з.п. ф-лы, 32 табл., 14 пр., 9 ил.
1. Фармацевтическая композиция антибактериального действия, включающая действующее вещество 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4H-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамид в кристаллической форме с размером частиц менее 50 мкм в количестве от 150 до 600 мг в виде пероральной твердой дозированной лекарственной формы с приемлемыми вспомогательными веществами.
2. Фармацевтическая композиция по п. 1, в которой в качестве приемлемых вспомогательных веществ используют наполнители, разрыхляющие, модификаторы поверхности, связывающие, скользящие, при следующем соотношении компонентов, мас.%:
3. Фармацевтическая композиция по п. 1, представляющая собой пероральную твердую дозированную лекарственную форму для приема внутрь в виде таблеток, покрытых или не покрытых оболочкой, или капсул твердых, или порошка дозированного, или порошка для приготовления раствора.
4. Применение фармацевтической композиции по п. 1 для лечения острых, рецидивирующих и хронических инфекций, таких как осложненные инфекции мочевыводящих путей, пневмония, хронические урогенитальные инфекции, острые и хронические инфекции кишечной локализации, вызванные антибиотикорезистентными штаммами Pseudomonas aeruginosa, Enterococcus spp., Chlamydia trachomatis, Salmonella spp.
| Применение 4-(3-этокси-4-гидроксибензил)-5-оксо-5,6-дигидро-4Н-[1,3,4]-тиадиазин-2-(2,4-дифторфенил)-карбоксамида для подавления инфекции, вызванной устойчивыми к антибиотикам штаммами Pseudomonas aeruginosa, и способ подавления этой инфекции | 2016 |
|
RU2624846C1 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ВЕЩЕСТВА, ПОДАВЛЯЮЩИЕ ПАТОГЕННЫЕ БАКТЕРИИ, И СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ III ТИПА У ПАТОГЕННЫХ БАКТЕРИЙ | 2010 |
|
RU2447066C2 |
| KOROLEVA E.A | |||
| et al | |||
| BioMed Research International, Article ID 484853, 2015, 7 pages | |||
| NESTERENKO L.N | |||
| et al | |||
| Journal of Antibiotics, 69(6), 2016, pp | |||
| Стрелочный контрольный замок | 1924 |
|
SU422A1 |
| US 4626536 A, 02.12.1986. | |||

