Область, к которой относится изобретение
Настоящее изобретение относится к новым бензо-N-гидроксиамидным соединениям, имеющим альфа-аминокислотный каркас, несущий альфа-бета-ненасыщенную карбонильную группу на аминогруппе, и к их фармацевтическим композициям.
Молекулы обладают противоопухолевой активностью и особенно активны на раковых стволовых клетках.
Предпосылки создания изобретения
Частичная или даже полная регрессия рака может достигаться у некоторых пациентов при применении существующих лечений рака. Однако за такими начальными ответами почти всегда следует рецидив, с рецидивирующим раком, резистентным к дальнейшим лечениям. Поэтому открытие терапевтических подходов, которые противодействуют рецидиву, является существенным для развития онкологии. Раковые клетки являются чрезвычайно гетерогенными даже у каждого индивидуального пациента, что касается их злокачественного потенциала, чувствительности к лекарственным средствам и их потенциала метастазировать и вызывать рецидив. Действительно, гиперзлокачественные раковые клетки, называемые раковыми стволовыми клетками или опухоль-инициирующими клетками, которые являются высокоонкогенными и метастатическими, были выделены у раковых пациентов с различными типами опухолей. Кроме того, такие раковые клетки высокой стволовости резистентны к традиционной химиотерапии и облучению.
Поэтому разработка специфической терапии, нацеленной на раковые стволовые клетки, дает надежду на улучшение выживаемости и качества жизни раковых пациентов, особенно для пациентов с метастатическим заболеванием.
US7635788 раскрывает производные гидроксамовой кислоты, содержащие альфа-аминоацильную группу и обладающие активностью ингибирования продукции провоспалительных цитокинов, в частности, TNFα. Такие соединения являются полезными для лечения воспалительных заболеваний и других расстройств, характеризующихся сверхпродукцией TNFα или других провоспалительных цитокинов. Кроме того, указанные соединения демонстрируют цитотоксическую активность в in vitro испытании на клеточной линии гепатомы человека Hep-G2.
Определения
Если не определено иначе, предполагается, что все специальные термины, обозначения и другая научная терминология, используемые в настоящей заявке, имеют значения, обычно понятные специалистам в области техники, к которой относится настоящее изобретение. В некоторых случаях термины с общепринятыми значениями определены для ясности и/или для справки; таким образом, включение таких определений в настоящее изобретение не должно истолковываться как представляющее существенную разницу по сравнению с тем, как они обычно понимаются в данной области техники.
Термин "галоген" в настоящем изобретении относится к фтору (F), хлору (Cl), брому (Br) или иоду (I).
Термин ʺC1-C4 алкилʺ в настоящем изобретении относится к разветвленному или линейному углеводороду, содержащему от 1 до 4 атомов углерода. Примеры C1-C4 алкильных групп включают, но не ограничиваются этим, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил.
Термин ʺарилʺ в настоящем изобретении относится к ароматическим моно- и поли-карбоциклическим кольцевым системам, где отдельные карбоциклические кольца в поликарбоциклических кольцевых системах могут быть конденсированы или соединены друг с другом простой связью. Подходящие арильные группы включают, но не ограничиваются этим, фенил, нафтил и бифенил.
Термин "арилокси" в настоящем изобретении относится к O-арильной группе, где "арил" определен выше.
Термин "алкокси" в настоящем изобретении относится к O-алкильной группе, где "алкил" определен выше.
Термин ʺарилалкилʺ в настоящем изобретении относится к арильному радикалу, определенному ниже, присоединенному к алкильному радикалу, определенному выше.
Термин ʺарилкарбонилʺ в настоящем изобретении относится к -C(O)-арилу, где "арил" определен выше.
Термин ʺгетероциклʺ в настоящем изобретении относится к 4-, 5-, 6-, 7- или 8-членному моноциклическому кольцу, которое является насыщенным или ненасыщенным и которое состоит из атомов углерода и одного или нескольких гетероатомов, выбранных из N, O и S, и где гетероатомы азота и серы необязательно могут быть окислены и гетероатом азота необязательно может быть кватернизирован. Гетероциклическое кольцо может быть присоединено к любому гетероатому или атому углерода, при условии, что присоединение приводит к созданию стабильной структуры. Термин также включает любую бициклическую систему, в которой любое из указанных выше гетероциклических колец является конденсированным с арилом или другим гетероциклом. Когда гетероциклическое кольцо является ароматическим гетероциклическим кольцом, оно может быть определено как ʺгетероароматическое кольцоʺ.
Термины ʺвключающийʺ, ʺимеющийʺ, ʺвключающий в себяʺ и ʺсодержащийʺ следует понимать как открытые термины (то есть означающие ʺвключающий, но не ограничивающийся этимʺ) и должны рассматриваться как обеспечивающие поддержку также для таких терминов, как ʺсостоят по существу изʺ, ʺсостоящий по существу изʺ, ʺсостоят изʺ или ʺсостоящий изʺ.
Термины ʺпо существу состоят изʺ, ʺсостоящие по существу изʺ следует понимать как полузакрытые термины, означающие, что не включены никакие другие ингредиенты и/или стадии, которые существенно влияют на основные и новые характеристики изобретения (необязательные эксципиенты, таким образом, могут быть включены).
Термины ʺсостоит изʺ, ʺсостоящий изʺ следует понимать как закрытый термин.
Термин ʺфармацевтически приемлемые солиʺ в настоящем изобретении относится к таким солям, которые обладают биологической эффективностью и свойствами солевой формы соединения и которые не вызывают побочных реакций при введении млекопитающему, предпочтительно человеку. Фармацевтически приемлемые соли могут быть неорганическими или органическими солями; примеры фармацевтически приемлемых солей включают, но не ограничиваются этим: карбонат, гидрохлорид, гидробромид, сульфат, гидросульфат, цитрат, малеат, фумарат, ацетат, трифторацетат, памоат, 2-нафталинсульфонат и пара-толуолсульфонат. Дополнительную информацию о фармацевтически приемлемых солях можно найти в, Handbook of Pharmaceutical Salts, P. Stahl, C. Wermuth, WILEY-VCH, 127-133, 2008, который включен в настоящую заявку посредством ссылки.
Термин ʺфизиологически приемлемый эксципиентʺ в настоящем изобретении относится к веществу, которое не имеет никакого собственного фармакологического эффекта и которое не вызывает побочных реакций при введении млекопитающему, предпочтительно человеку. Физиологически приемлемые эксципиенты хорошо известны в данной области техники и раскрыты, например, в Handbook of Pharmaceutical Excipients, sixth edition 2009, sixth edition 2009 г., который включен в настоящую заявку посредством ссылки.
Термин ʺизомерыʺ в настоящем изобретении относится к структурным (или конституциональным) изомерам (т.е. таутомерам) и стереоизомерам (или пространственным изомерам), то есть диастереоизомерам и энантиомерам.
Описание изобретения
Было обнаружено, что бензо-N-гидроксиамидные соединения, имеющие альфа-аминокислотный каркас, несущий альфа-бета-ненасыщенную карбонильную группу на аминогруппе, демонстрируют высокую ингибиторную активность в отношении пролиферации опухолевых клеток и стаминальных опухолевых клеток. Эти соединения особенно активны в отношении раковых стволовых клеток, что делает их полезными для лечения опухолей человека.
Настоящее изобретение предоставляет соединения формулы (I):
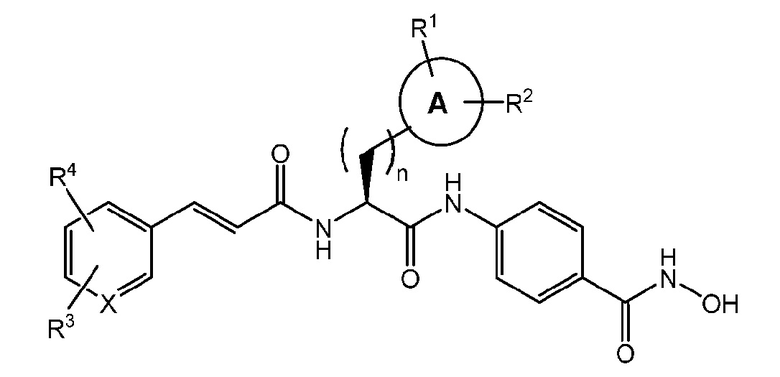
и их фармацевтически приемлемые соли, изомеры и пролекарства,
где:
n имеет значение 0, 1 или 2;
A отсутствует или представляет собой моно- или ди-карбоциклический остаток, необязательно частично или полностью ненасыщенный, включающий атомы углерода и необязательно один или более гетероатомов, выбранных из N, S или O;
R1 и R2 независимо выбраны из группы, включающей -H, -OH, -OMe, -CN, -NH2, -NO2, -Cl, -COOH, -галоген, -CF3, -N(Ra)2, линейный или разветвленный C1-C4 алкильный, арильный, арилалкильный, арилкарбонильный, алкокси, арилокси остаток, сульфониламино и -CH2N(CH2CH3)2;
Ra представляет собой линейный или разветвленный C1-C3 алкильный остаток;
X может представлять собой C или N;
R3 и R4 независимо выбраны из группы, включающей -H, -OMe, -OPh, -NO2, -NMe2, -NH2, -галоген, -CF3, -N(Ra)2, линейный или разветвленный C1-C4 алкильный, арильный, арилалкильный, арилкарбонильный, алкокси, арилокси остаток и сульфониламино, или R3 и R4 вместе могут образовывать гетеропентациклическую группу (-OCH2O-).
Соединения по настоящему изобретению могут существовать в различных изомерных формах: цис-форме и транс-форме.
Предпочтительной изомерной формой является транс-форма.
Соединения по изобретению содержат один или более хиральных центров (асимметричных атомов углерода) и, таким образом, могут существовать в энантиомерной и/или диастереоизомерной формах; все возможные оптические изомеры, отдельно или в смеси друг с другом, входят в объем настоящего изобретения.
Пролекарства соединений формулы (I) включены в настоящее изобретение. Такие пролекарства являются биообратимыми производными соединений (I). Они являются фармакологически неактивными производными, которые могут подвергаться метаболическому преобразованию in vivo с образованием активного соединения, включенного в общую формулу настоящего изобретения. В данной области известно много различных пролекарств [Prodrug approach: an effective solution to overcome side-effects, Patil S.J., Shirote P.J., International Journal of Medical and Pharmaceutical Sciences, 2011,1-13; Carbamate Prodrug Concept for Hydroxamate HDAC Inhibitors, Jung, Manfred et al., ChemMedChem, 2011, 1193-1198].
Некоторые соединения формулы (I) содержат одну или несколько оснóвных или кислотных групп, которые могут быть преобразованы в соли обычными химическими методами, обычно обработкой соединений органической или неорганической кислотой, органическим или неорганическим основанием или при помощи ионообменной хроматографии. Примеры фармацевтически приемлемых солей включают, но не ограничиваются этим, соли, образованные из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, фосфорная, азотная и т.п., или из органических кислот, таких как уксусная, малеиновая, пропионовая, янтарная, гликолевая, стеариновая, яблочная, винная, лимонная, аскорбиновая, памовая и т.п. Другие примеры фармацевтически приемлемых солей включают, но не ограничиваются этим, соли, образованные из оснований, таких как алюминий, аммоний, кальций, медь, железо, литий, магний, калий, натрий, цинк и т.п. Фармацевтически приемлемые соли хорошо известны в данной области. Их получение подробно описано в Berg et al. ʺPharmaceutica saltsʺ, J. Pharm. Sci., 1977, 66, 1-19.
Один класс предпочтительных соединений включает соединения формулы (I) и их фармацевтически приемлемые соли, изомеры и пролекарства,
в которой:
n имеет значение 0, 1 или 2;
A отсутствует или представляет собой моно- или ди-карбоциклический остаток, необязательно частично или полностью ненасыщенный, включающий атомы углерода и необязательно один или более гетероатомов, выбранных из N, S или O;
R1 и R2 независимо выбраны из группы, включающей -H, -OH, -OMe, -CN, -NH2, -NO2, -Cl, -COOH и -CH2N(CH2CH3)2;
X может представлять собой C или N;
R3 и R4 независимо выбраны из группы, включающей -H, -OMe, -OPh, -NO2, -NMe2 и -NH2, или R3 и R4 вместе могут образовывать гетеропентациклическую группу (-OCH2O-).
Предпочтительно, A отсутствует или представляет собой моно- или ди-карбоциклический остаток, выбранный из группы, включающей фенил, нафтил, пиридил, инданил, хинолил, имидазолил, индолил, тиазолил, бензотиофенил.
Еще один класс более предпочтительных соединений включает соединения формулы (I) и их фармацевтически приемлемые соли, изомеры и пролекарства, в которой:
n имеет значение 1, X представляет собой C;
R1 и R2 независимо представляют собой -H, -Cl или -OMe;
R3 и R4 независимо представляют собой -H, -NMe2, или R3 и R4 вместе могут образовывать гетеропентациклическую группу (-OCH2O-).
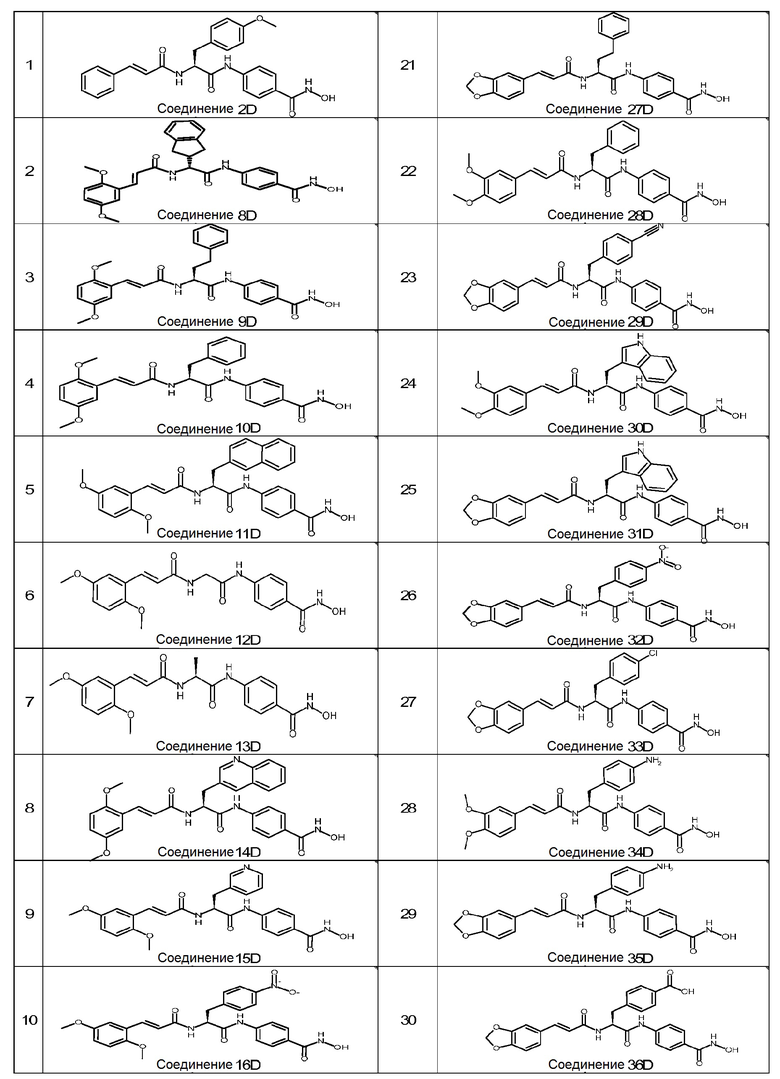
Следующие соединения формулы (I) являются особенно предпочтительными:
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)пропанамид (1D);
(2S)-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (2D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (4D);
(2S)-N-[4-(гидроксикарбамоил)фенил]-3-фенил-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (5D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-гидроксифенил)пропанамид (7D);
(E)-3-(2,5-диметоксифенил)-N-[(1R)-2-[4-(гидроксикарбамоил)анилино]-1-индан-2-ил-2-оксо-этил]проп-2-енамид (8D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-4-фенил-бутанамид (9D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (10D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(2-нафтил)пропанамид (11D);
(E)-3-(2,5-диметоксифенил)-N-[2-[4-(гидроксикарбамоил)анилино]-2-оксо-этил]проп-2-енамид (12D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (13D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-хинолил)пропанамид (14D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (15D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-нитрофенил)пропанамид (16D);
(2S)-2-[[(E)-3-(4-аминофенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (17D);
(2S)-3-(4-аминофенил)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (18D);
(2S)-3-(4-цианофенил)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (19D);
(2S)-N-[4-(гидроксикарбамоил)фенил]-2-[[(E)-3-(4-нитрофенил)проп-2-еноил]амино]-3-фенил-пропанамид (20D);
(2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(1H-имидазол-5-ил)пропанамид (21D);
(2S)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (22D);
(2S)-2-[[(E)-3-(3,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (23D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (24D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(бензотиофен-3-ил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (25D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-тиазол-4-ил-пропанамид (26D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-4-фенил-бутанамид (27D);
(2S)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (28D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(4-цианофенил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (29D);
(2S)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(1H-индол-3-ил)пропанамид (30D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(1H-индол-3-ил)пропанамид (31D);
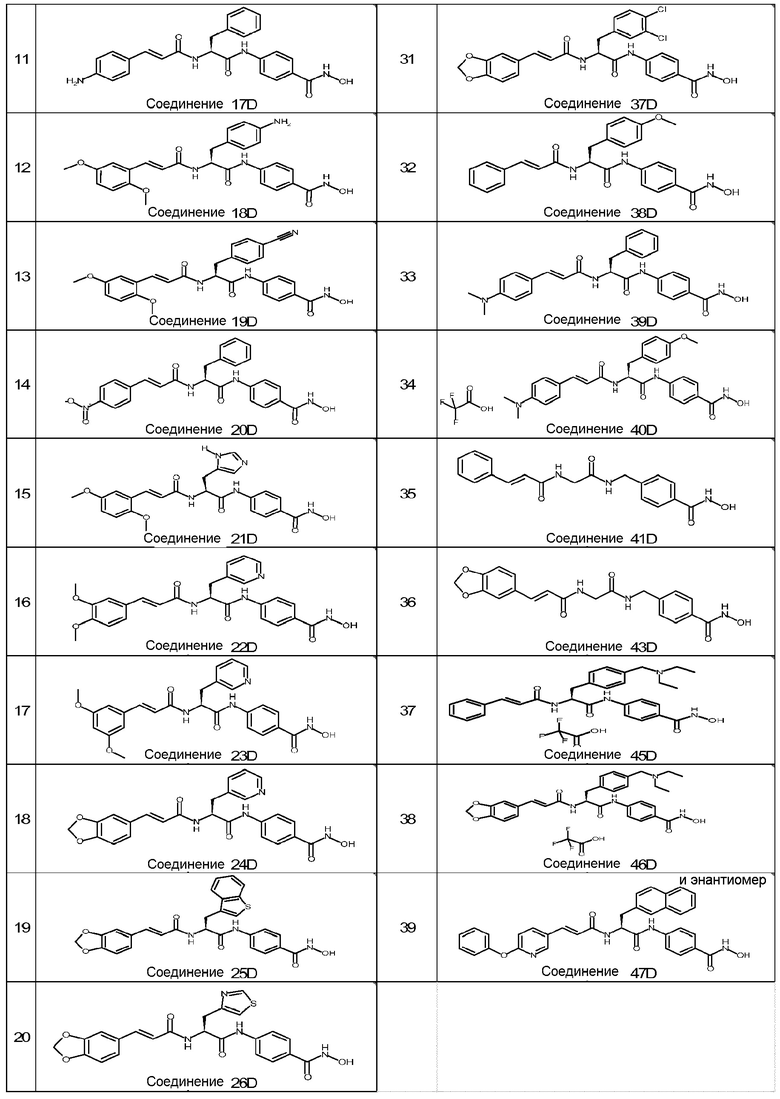
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-нитрофенил)пропанамид (32D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(4-хлорфенил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (33D);
(2S)-3-(4-аминофенил)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (34D);
(2S)-3-(4-аминофенил)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (35D);
4-[(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-[4-(гидроксикарбамоил)анилино]-3-оксо-пропил]бензойная кислота (36D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(3,4-дихлорфенил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (37D);
(2S)-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (38D);
(2S)-2-[[(E)-3-(4-диметиламинофенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (39D);
(2S)-2-[[(E)-3-(4-диметиламинофенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)пропанамид; 2,2,2-трифторуксусная кислота (40D);
(E)-N-[2-[[4-(гидроксикарбамоил)фенил]метиламино]-2-оксо-этил]-3-фенил-проп-2-енамид (41D);
(E)-3-(1,3-бензодиоксол-5-ил)-N-[2-[[4-(гидроксикарбамоил)фенил]метиламино]-2-оксо-этил]проп-2-енамид (43D);
(2S)-3-[4-(диэтиламинометил)фенил]-N-[4-(гидроксикарбамоил)фенил]-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид; 2,2,2-трифторуксусная кислота (45D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-[4-(диэтиламинометил)фенил]-N-[4-(гидроксикарбамоил)фенил]пропанамид; 2,2,2-трифторуксусная кислота (46D);
(2S)-N-[4-(гидроксикарбамоил)фенил]-3-(2-нафтил)-2-[[(E)-3-(6-фенокси-3-пиридил)проп-2-еноил]амино]пропанамид (47D).
Следующие соединения формулы (I) являются более предпочтительными:
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)пропанамид (1D);
(2S)-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (2D);
(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (4D);
(2S)-N-[4-(гидроксикарбамоил)фенил]-3-фенил-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (5D).
Соединения по изобретению можно получить с использованием способов, известных специалистам в данной области.
Все исходные вещества, структурные элементы, реагенты, кислоты, основания, растворители и катализаторы, используемые для синтеза соединений по настоящему изобретению, являются коммерчески доступными.
Соединения формулы (I) можно получить как твердофазным синтезом (схема 1), так и способом синтеза в растворе (схема 2). В некоторых случаях возможно будет необходимо синтезировать соединение в защищенной форме и добавить одну или несколько стадий для удаления защиты.
Реакции контролируют с использованием ВЭЖХ или ЖХ-МС анализа.
Конечные продукты, как правило, очищают препаративной ВЭЖХ-МС хроматографией или методом ТФЭ (твердофазная экстракция) на обращенно-фазовом картридже.
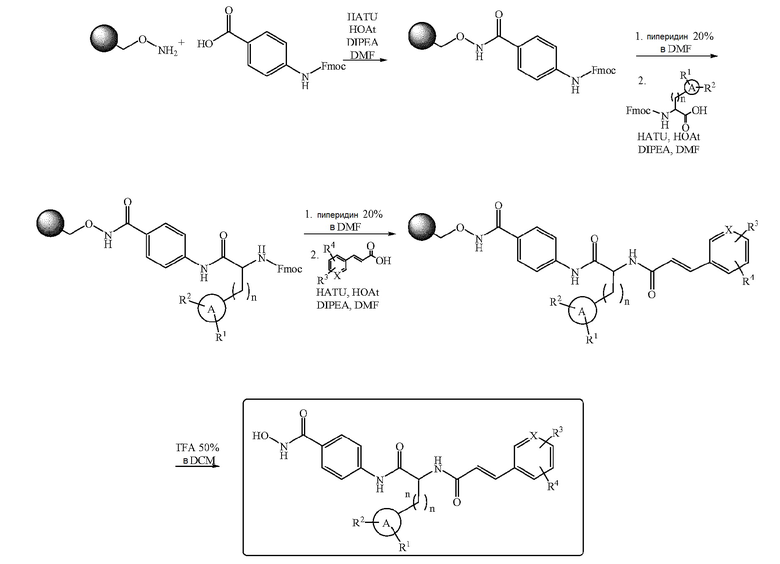
Твердофазный синтез
Соединения формулы (I) можно синтезировать с использованием твердофазного синтеза (ТФС), следуя схеме 1.
N-fmoc-4-аминобензойную кислоту загружают на связанную с гидроксиламином смолу Ванга после классической активации при помощи HATU, HOAt и DIPEA. Снятие Fmoc-защиты осуществляют путем обработки смолы 20% раствором пиперидина в DMF в течение 15 минут. Fmoc-аминокислоту затем связывают с ароматическим амином после активации при помощи HATU, HOAt и DIPEA. Осуществляют еще одну стадию удаления Fmoc-защиты и в завершение аминогруппу ацилируют путем обработки смолы активированным производным коричной кислоты. Отщепление продукта от смолы осуществляют путем обработки 50% раствором TFA в DCM.
Конечные продукты обычно очищают препаративной ЖХ-МС хроматографией или ТФЭ на заполненном C18 картридже.
Схема 1
Синтез в фазе раствора
Соединения формулы (I) также можно получить классическим способом синтеза в растворе, как показано на схеме 2. Образование амида можно осуществить с использованием одного из способов, известных специалистам в данной области. Например, после активации карбоксильной группы при помощи HATU и DIPEA Fmoc-аминокислоту подвергают взаимодействию с этил-4-аминобензоатом. Полученное соединение A подвергают процедуре удаления защиты путем обработки 20% раствором пиперидина в DMF, а затем ацилированию путем взаимодействия с активированным производным коричной кислоты, с получением промежуточного соединения B. Конечное соединение гидроксамовой кислоты можно получить, следуя другому пути синтеза. Можно осуществить непосредственный гидроксиламинолиз путем обработки сложноэфирного промежуточного соединения гидроксиламином и NaOH в метаноле.
В другом случае, можно осуществить гидролиз сложноэфирной функции при помощи NaOH и затем преобразовать полученную карбоновую кислоту в гидроксамовую кислоту путем взаимодействия с гидроксиламином после активации посредством HATU.
В некоторых случаях предпочтительно преобразовать карбоновую кислоту в защищенную форму гидроксамата для упрощения очистки соединения. В этом случае конечное соединение получают путем удаления защиты с использованием раствора TFA в метаноле.
Схема 2
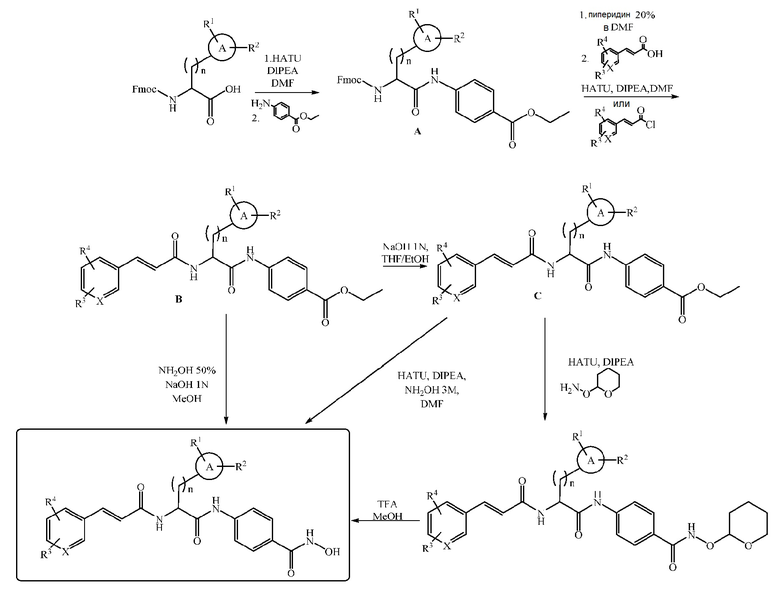
где A, R1, R2, R3, R4 и X имеют значения, указанные выше.
Препаративная ВЭЖХ-МС хроматография
Очистку осуществляли с использованием препаративной ВЭЖХ системы Waters, снабженной масс-спектрометрическим детектором (ZQ200). Для очистки использовали три разных способа в зависимости от природы R1, R2, R3 и R4 групп.
Неочищенный продукт растворяли в DMSO. Раствор фильтровали через 0,45 мкм PTFE мембрану и вводили в препаративную систему. Фракции, соответствующие пику, ассоциированному с ожидаемым молекулярным ионом ([M+H]+), собирали и концентрировали досуха.
Режим работы:
ВЭЖХ метод 1:
ВЭЖХ метод 2:
ВЭЖХ метод 3:
МС метод:
Твердофазная экстракция
Очистку осуществляли на предварительно заполненном обращенно-фазовым сорбентом ТФЭ картридже (Phenomenex Strata C18-E, 55 мм, 70A).
Неочищенный продукт растворяли в DMF и загружали на картридж. Продукт элюировали смесями ACN/вода при разных соотношениях в зависимости от природы R1, R2, R3 и R4 групп. Элюированные фракции, которые показали площадь ВЭЖХ чистоты больше 85%, собирали и концентрировали досуха.
Соединения по настоящему изобретению показали высокую ингибиторную активность в отношении пролиферации раковых стволовых клеток in vitro, c IC50 значениями в наномолярном диапазоне.
Также осуществляли исследования in vivo для отслеживания способности соединений уменьшать размер и массу опухоли, как описано, например, в Примере 7.
Эти соединения можно соответственно использовать, отдельно или вместе с другими противоопухолевыми лекарственными средствами, для профилактики и/или лечения рака.
Соединения по изобретению предпочтительно являются полезными для профилактики и/или лечения солидных опухолей, таких как колоректальный рак, рак легкого, головного мозга, предстательной железы или гинекологический рак или гематологические злокачественные заболевания.
Соединения по изобретению являются особенно активными на раковых стволовых клетках. Поэтому указанные соединения предпочтительно являются полезными для профилактики и/или лечения метастатических, рецидивирующих и лекарственно-резистентных заболеваний.
Соответственно, настоящее изобретение также предоставляет фармацевтические композиции, содержащие терапевтически эффективное количество соединений формулы (I) или их фармацевтически приемлемых солей, изомеров и пролекарств вместе с по меньшей мере одним фармацевтически приемлемым эксципиентом.
Такие композиции могут быть жидкими, подходящими для энтерального или парентерального введения, или твердыми, например, в форме капсул, таблеток, таблеток с покрытием, порошков или гранул для перорального введения, или в формах, подходящих для кожного введения, таких как, например, кремы или мази, или для введения путем ингаляции.
Фармацевтические композиции, предоставляемые настоящим изобретением, можно получить с использованием известных способов.
Следующие примеры имеют целью дополнительную иллюстрацию изобретения, однако без его ограничения.
ПРИМЕРЫ
В представленных ниже Примерах использовали следующие аббревиатуры:
ПРИМЕР 1 (Соединение 4D)
Синтез (S,E)-4-(2-(3-(бензо[d][1,3]диоксол-5-ил)акриламидо)-3-фенилпропанамидо)-N-гидроксибензамида
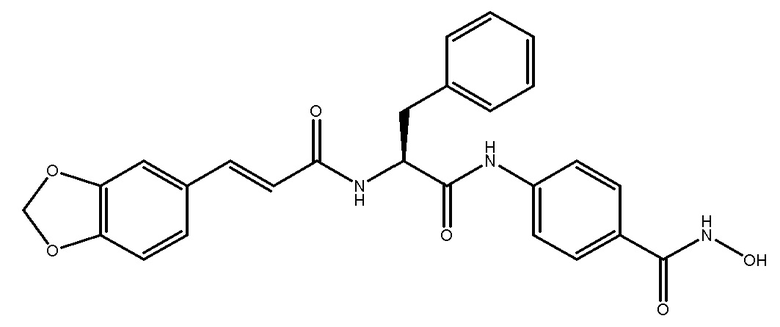
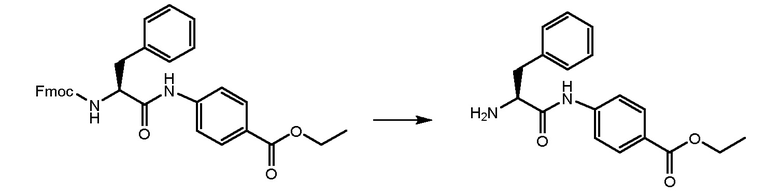
СТАДИЯ A: Этил-(S)-4-(2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-3-фенилпропанамидо)бензоат
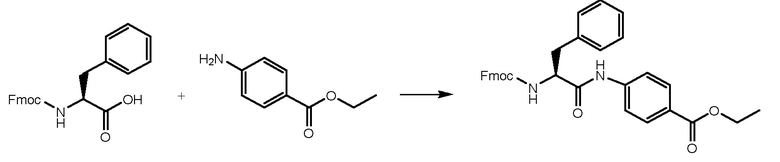
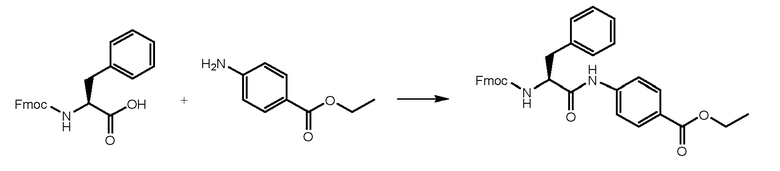
3 г Fmoc-L-фенилаланина (7,74 ммоль, 1 экв.) растворяли в 15 мл DMF и раствор охлаждали при 0°C. Добавляли HATU (3,84 г, 1,3 экв.) и DIPEA (1,75 мл, 1,3 экв.) и реакционную смесь перемешивали в течение получаса. Затем добавляли этил-4-аминобензоат (1,40 г, 1 экв.) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию контролировали при помощи ВЭЖХ анализа. Когда реакция завершалась, раствор выливали в воду (150 мл). Осажденное белое твердое вещество фильтровали и сушили. Получали 4 г чистого продукта.
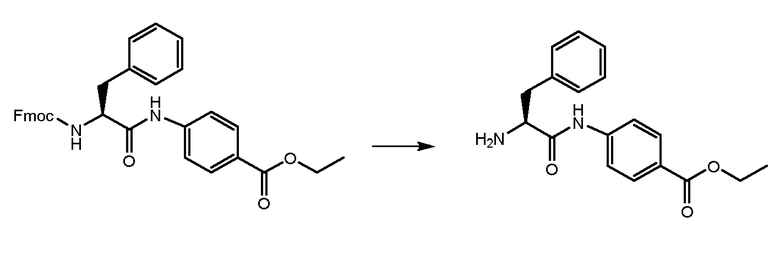
СТАДИЯ B: этил-(S)-4-(2-амино-3-фенилпропанамидо)бензоат
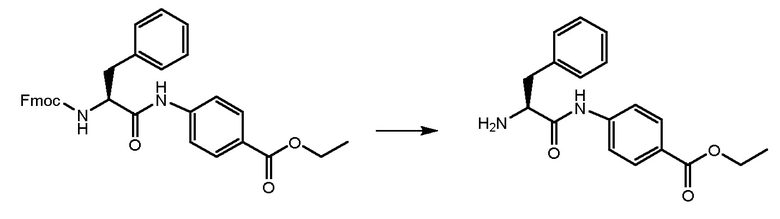
Соединение, полученное на стадии A, обрабатывали при помощи 4,6 мл DEA (6 экв.) в THF при комнатной температуре в течение ночи для удаления Fmoc-защиты. THF удаляли и остаток растворяли в н-гексане до образования твердого вещества. Растворитель удаляли и продукт промывали два раза свежим н-гексаном. Получали 2,2 г продукта.
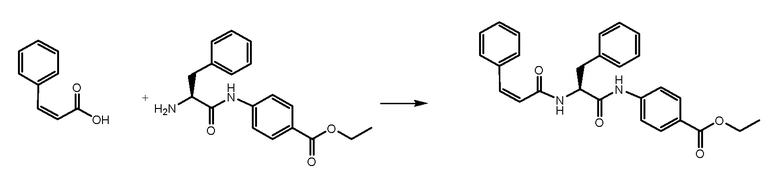
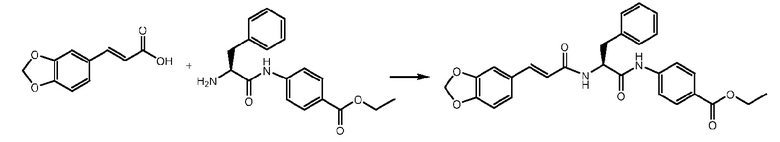
СТАДИЯ C: этил-(S,E)-4-(2-(3-(бензо[1,3]диоксол-5-ил)акриламидо)-3-фенилпропанамидо)бензоат
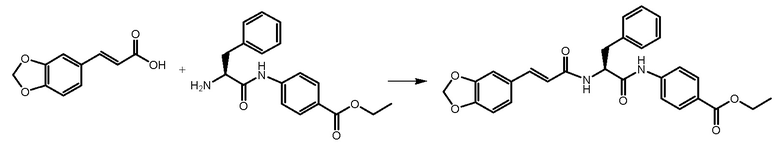
(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еновую кислоту (430 мг, 1 экв.) растворяли в 5 мл DMF, охлаждали до 0°C и добавляли HATU (1,3 экв.) и DIPEA (1,3 экв.). Через 30 минут добавляли 700 мг соединения, полученного на стадии B. Реакционную смесь доводили до комнатной температуры и перемешивали в течение 1 часа. Раствор затем выливали в воду (50 мл) и продукт экстрагировали при помощи EtOAc. После подкисления растворитель выпаривали и неочищенный продукт очищали на колонке с силикагелем.
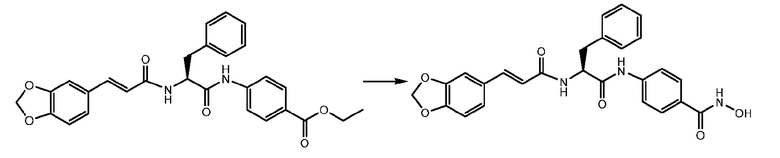
СТАДИЯ D: (S,E)-4-(2-(3-(бензо[1,3]диоксол-5-ил)акриламидо)-3-фенилпропанамидо)бензойная кислота
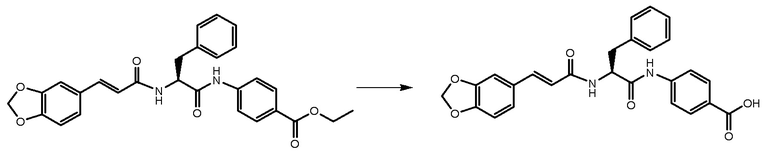
600 мг соединения, полученного на стадии C, растворяли в 30 мл EtOH/THF 1:2. К раствору добавляли NaOH 1N (3 экв.) и реакционную смесь перемешивали при кипячении с обратным холодильником в течение 4 часов.
Растворители удаляли и неочищенное вещество растворяли в воде. Раствор подкисляли при помощи HCl 6N, и наблюдалось осаждение и продукта. Отфильтрованное твердое вещество суспендировали в Et2O и фильтровали.
СТАДИЯ E: (S,E)-4-(2-(3-(бензо[d][1,3]диоксол-5-ил)акриламидо)-3-фенилпропанамидо)-N-гидроксибензамид
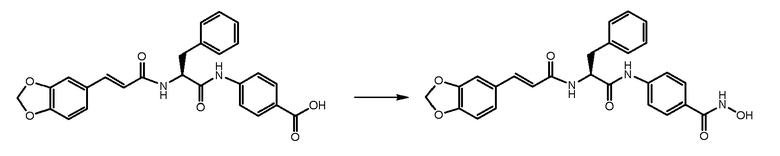
400 мг соединения, полученного на стадии D (1 экв.), растворяли в 2,5 мл DMF. После охлаждения на ледяной бане добавляли HATU (1,3 экв.) и DIPEA (1,3 экв.) и смесь перемешивали в течение 1 часа. В завершение, добавляли раствор NH2OH 3M в DMF (3 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Смесь затем выливали в воду и осажденное твердое вещество фильтровали и суспендировали в диоксане и смесь перемешивали и нагревали. Конечный продукт получали путем фильтрования.
ПРИМЕР 2 (Соединение 5D)
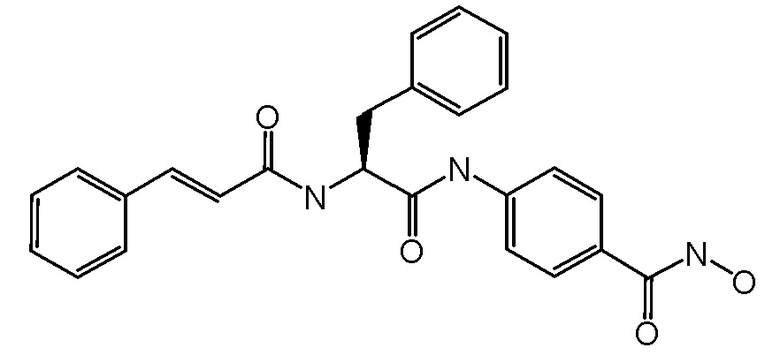
Синтез (S)-4-(2-циннамамидо-3-фенилпропанамидо)-N-гидроксибензамида
СТАДИЯ A: Этил-(S)-4-(2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-3-фенилпропанамидо)бензоат
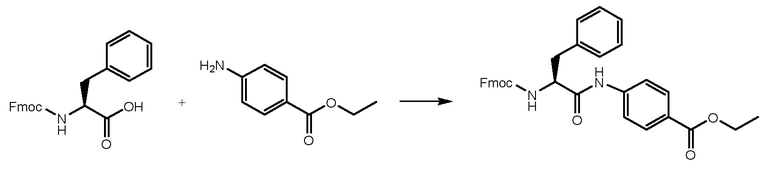
3 г Fmoc-L-фенилаланина (7,74 ммоль, 1 экв.) растворяли в 15 мл DMF и раствор охлаждали при 0°C. Добавляли HATU (3,84 г, 1,3 экв.) и DIPEA (1,75 мл, 1,3 экв.) и реакционную смесь перемешивали в течение получаса. Затем добавляли этил-4-аминобензоат (1,40 г, 1 экв.) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию контролировали при помощи ВЭЖХ анализа. Когда реакция завершалась, раствор выливали в воду (150 мл). Осажденное белое твердое вещество фильтровали и сушили. Получали 4 г чистого продукта.
СТАДИЯ B: этил-(S)-4-(2-амино-3-фенилпропанамидо)бензоат
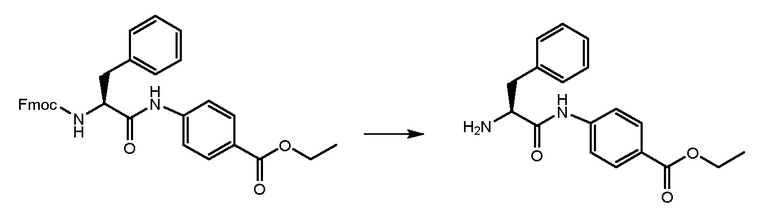
Соединение, полученное на стадии A, обрабатывали при помощи 4,6 мл DEA (6 экв.) в THF при комнатной температуре в течение ночи для удаления Fmoc-защиты. THF удаляли и остаток растворяли в н-гексане до образования твердого вещества. Растворитель удаляли и продукт промывали два раза свежим н-гексаном. Получали 2,2 г продукта.
СТАДИЯ C: этил-(S)-4-(2-циннамамидо-3-фенилпропанамидо)бензоат.
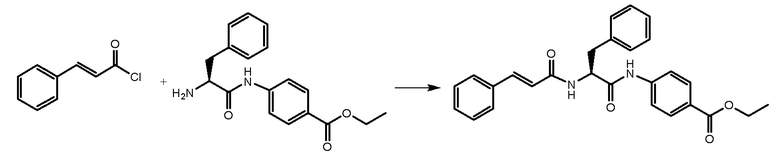
900 мг продукта, полученного на стадии B (1 экв.), растворяли в DCM (25 мл) и раствор охлаждали при 0°C. Добавляли TEA (1 экв.). В завершение добавляли раствор циннамоилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции раствор промывали водой, HCl 1N и NaHCO3 5% в воде. Органическую фазу сушили при помощи CaCl2, фильтровали и упаривали. Неочищенное вещество очищали на колонке с силикагелем с использованием смеси толуол/EtOH в качестве подвижной фазы.
СТАДИЯ D: (S)-4-(2-циннамамидо-3-фенилпропанамидо)бензойная кислота
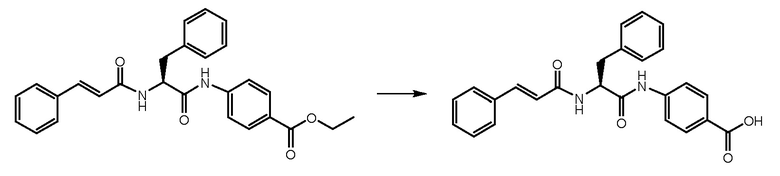
Этил-(S)-4-(2-циннамамидо-3-фенилпропанамидо)бензоат, полученный на стадии C (400 мг, 1 экв.), растворяли в смеси THF/EtOH и обрабатывали при помощи NaOH 1N (3 экв.) в течение 1 часа. Растворитель удаляли и неочищенное вещество растворяли в воде. Раствор подкисляли концентрированной HCl. Получали твердое вещество, которое фильтровали и использовали на следующей стадии.
СТАДИЯ E: 4-((S)-2-циннамамидо-3-фенилпропанамидо)-N-((тетрагидро-2H-пиран-2-ил)окси)бензамид
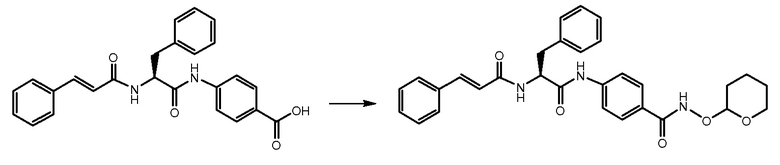
400 мг соединения, полученного на стадии D (1 экв.), растворяли в 2,5 мл DMF. После охлаждения на ледяной бане добавляли HATU (1,3 экв.) и DIPEA (1,3 экв.) и смесь перемешивали в течение 1 часа. В завершение добавляли O-(тетрагидро-2H-пиран-2-ил)гидроксиламин (3 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Смесь затем выливали в воду и осажденное твердое вещество фильтровали и очищали на колонке с силикагелем с использованием смеси DCM/MeOH/NH3 25:1:0,1 в качестве подвижной фазы.
СТАДИЯ F: (S)-4-(2-циннамамидо-3-фенилпропанамидо)-N-гидроксибензамид
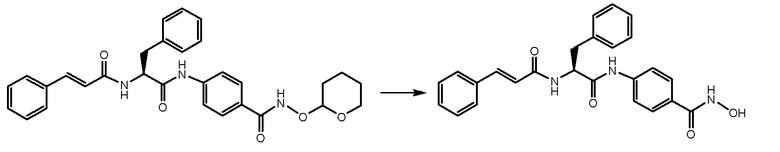
Соединение, полученное на стадии E (200 мг, 1 экв.), растворяли в MeOH (50 мл) и обрабатывали при помощи TFA (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель и избыток TFA удаляли путем выпаривания и неочищенное вещество суспендировали в Et2O и фильтровали. Получали 120 мг чистого продукта.
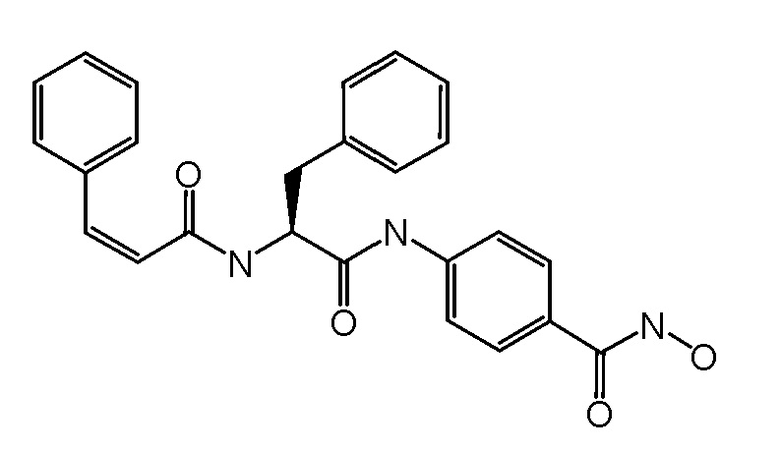
ПРИМЕР 3 (Соединение 5DZ)
Синтез (S,Z)-N-гидрокси-4-(3-фенил-2-(3-фенилакриламидо)пропанамидо)бензамида
СТАДИЯ A: Этил-(S)-4-(2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-3-фенилпропанамидо)бензоат
3 г Fmoc-L-фенилаланина (7,74 ммоль, 1 экв.) растворяли в 15 мл DMF и раствор охлаждали при 0°C. Добавляли HATU (3,84 г, 1,3 экв.) и DIPEA (1,75 мл, 1,3 экв.) и реакционную смесь перемешивали в течение получаса. Затем добавляли этил-4-аминобензоат (1,40 г, 1 экв.) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию контролировали при помощи ВЭЖХ анализа. Когда реакция завершалась, раствор выливали в воду (150 мл). Осажденное белое твердое вещество фильтровали и сушили. Получали 4 г чистого продукта.
СТАДИЯ B: этил-(S)-4-(2-амино-3-фенилпропанамидо)бензоат
Соединение, полученное на стадии A, обрабатывали при помощи 4,6 мл DEA (6 экв.) в THF при комнатной температуре в течение ночи для удаления Fmoc-защиты. THF удаляли и остаток растворяли в н-гексане до образования твердого вещества. Растворитель удаляли и продукт промывали два раза свежим н-гексаном. Получали 2,2 г продукта.
СТАДИЯ C: этил-(S,Z)-4-(3-фенил-2-(3-фенилакриламидо)пропанамидо)бензоат
(Z)-3-фенилакриловую кислоту (1 экв.) растворяли в 5 мл DMF и раствор охлаждали при 0°C. Добавляли HATU (1,3 экв.) и DIPEA (1,3 экв.) и реакционную смесь перемешивали в течение одного часа. Затем добавляли этил-(S)-4-(2-амино-3-фенилпропанамидо)бензоат (1 экв.), полученный на стадии B, и смесь перемешивали при комнатной температуре в течение ночи. Реакцию контролировали при помощи ВЭЖХ анализа. Когда реакция завершалась, раствор выливали в воду. Неочищенный продукт экстрагировали при помощи EtOAc и очищали на колонке с силикагелем, с использованием смеси толуол/EtOAC 6:4 в качестве подвижной фазы.
СТАДИЯ D: (S,Z)-4-(3-фенил-2-(3-фенилакриламидо)пропанамидо)бензойная кислота
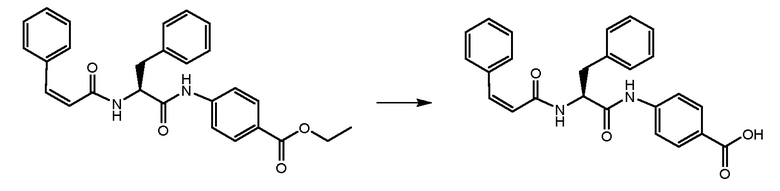
Этил-(S,Z)-4-(3-фенил-2-(3-фенилакриламидо)пропанамидо)бензоат, полученный на стадии C (250 мг, 1 экв.), растворяли в смеси THF/EtOH и обрабатывали при помощи NaOH 1N (3 экв.) в течение 24 часов. Растворитель удаляли и неочищенное вещество растворяли в воде. Неочищенный продукт очищали на колонке с силикагелем, элюируя смесью толуол/EtOAc.
СТАДИЯ E: 4-((S)-3-фенил-2-((Z)-3-фенилакриламидо)пропанамидо)-N-((тетрагидро-2H-пиран-2-ил)окси)бензамид
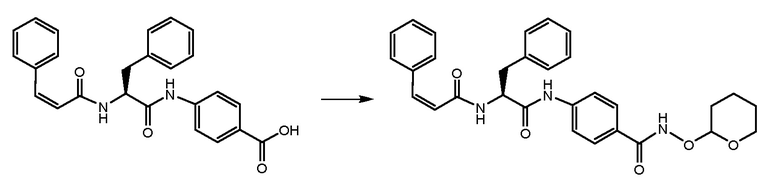
200 мг соединения, полученного на стадии D (1 экв.), растворяли в 1 мл DMF. После охлаждения на ледяной бане добавляли HATU (1,3 экв.) и DIPEA (1,3 экв.) и смесь перемешивали в течение 1 часа. В завершение добавляли O-(тетрагидро-2H-пиран-2-ил)гидроксиламин (1 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Смесь затем выливали в воду и осажденное твердое вещество фильтровали и очищали на колонке с силикагелем с использованием смеси EtOAc/толуол 7:3 в качестве подвижной фазы.
СТАДИЯ F: (S,Z)-N-гидрокси-4-(3-фенил-2-(3-фенилакриламидо)пропанамидо)бензамид
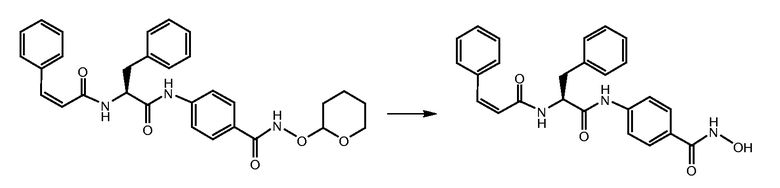
Соединение, полученное на стадии E (110 мг, 1 экв.), растворяли в MeOH (25 мл) и обрабатывали при помощи TFA (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель и избыток TFA удаляли путем выпаривания и неочищенное вещество суспендировали в Et2O и фильтровали. Получали 80 мг чистого продукта.
ПРИМЕР 4 (Соединение 1D)
Синтез (S,E)-4-(2-(3-(бензо[1,3]диоксол-5-ил)акриламидо)-3-(4-метоксифенил)пропанамидо)-N-гидроксибензамида
СТАДИЯ A: Этил-(S)-4-(2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-3-фенилпропанамидо)бензоат

3 г Fmoc-L-фенилаланина (7,74 ммоль, 1 экв.) растворяли в 15 мл DMF и раствор охлаждали при 0°C. Добавляли HATU (3,84 г, 1,3 экв.) и DIPEA (1,75 мл, 1,3 экв.) и реакционную смесь перемешивали в течение получаса. Затем добавляли этил-4-аминобензоат (1,40 г, 1 экв.) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию контролировали при помощи ВЭЖХ анализа. Когда реакция завершалась, раствор выливали в воду (150 мл). Осажденное белое твердое вещество фильтровали и сушили. Получали 4 г чистого продукта.
СТАДИЯ B: этил-(S)-4-(2-амино-3-фенилпропанамидо)бензоат
Соединение, полученное на стадии A, обрабатывали при помощи 4,6 мл DEA (6 экв.) в THF при комнатной температуре в течение ночи для удаления Fmoc-защиты. THF удаляли и остаток растворяли в н-гексане до образования твердого вещества. Растворитель удаляли и продукт промывали два раза свежим н-гексаном. Получали 2,2 г продукта.
СТАДИЯ C: этил-(S,E)-4-(2-(3-(бензо[1,3]диоксол-5-ил)акриламидо)-3-фенилпропанамидо)бензоат
(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еновую кислоту (430 мг, 1 экв.) растворяли в 5 мл DMF, охлаждали до 0°C и подвергали взаимодействию с HATU (1,3 экв.) и DIPEA (1,3 экв.). Через 30 минут добавляли 700 мг соединения, полученного на стадии B. Реакционную смесь доводили до комнатной температуры и перемешивали в течение 1 часа. Раствор затем выливали в воду (50 мл) и продукт экстрагировали при помощи EtOAc. После подкисления растворитель выпаривали и неочищенный продукт очищали на колонке с силикагелем.
СТАДИЯ D: (S,E)-4-(2-(3-(бензо[1,3]диоксол-5-ил)акриламидо)-3-(4-метоксифенил)пропанамидо)-N-гидроксибензамид
Раствор соединения, полученного на стадии D, в MeOH охлаждали на ледяной бане. Добавляли раствор гидроксиламина 50% в воде (15 экв.) и NaOH 1N (10 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Раствор затем нейтрализовали при помощи HCl 1N. Происходило образование осадка. Чистый продукт получали просто с использованием фильтрования.
ПРИМЕР 5 (Соединение 7D)
Синтез (S,E)-4-(2-(3-(2,5-диметоксифенил)акриламидо)-3-(4-гидроксифенил)пропанамидо)-N-гидроксибензамида

СТАДИЯ A: загрузка (9H-флуорен-9-ил)метил (4-(гидроксикарбамоил)фенил)карбамата на связанную с гидроксиламином смолу Ванга
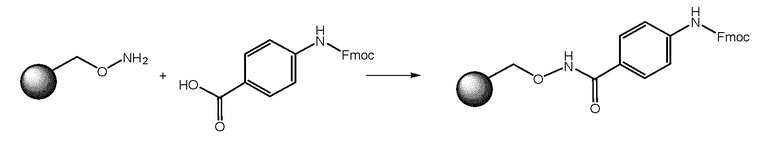
Реакцию осуществляли в очищенной пластиковой фильтровальной пробирке для ТФЭ с использованием синтезатора Activotec PLS 4×6 Organic Synthesizer.
После набухания смолы при использовании DCM и DMF добавляли раствор 4-((((9H-флуорен-9-ил)метокси)карбонил)амино)бензойной кислоты (4 экв.), HATU (4 экв.), HOAt (4 экв.) и DIPEA (8 экв.) в DMF. Реакционную смесь встряхивали при комнатной температуре в течение ночи. Смолу затем фильтровали и промывали при помощи DMF и DCM.
СТАДИЯ B: первое связывание

Смолу со стадии A (200 мг, 1 экв.) обрабатывали в DMF для набухания, затем фильтровали. Удаление Fmoc-защиты осуществляли путем обработки смолы два раза раствором пиперидина 20% в DMF в течение 30 минут. Раствор отфильтровывали и смолу промывали при помощи DMF. К смоле добавляли раствор (S)-2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-3-(4-((трет-бутоксикарбонил)окси)фенил)пропановой кислоты (4 экв.), HATU (4 экв.), HOAt (4 экв.) и DIPEA (8 экв.) в DMF. Реакционную смесь встряхивали при комнатной температуре в течение ночи. Смолу фильтровали и промывали при помощи DMF и DCM.
СТАДИЯ C: второе связывание
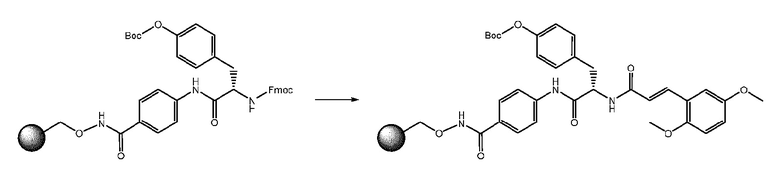
Смолу со стадии B (1 экв.) обрабатывали в DMF для набухания, затем фильтровали. Удаление Fmoc-защиты осуществляли путем обработки смолы два раза раствором пиперидина 20% в DMF в течение 30 минут. Раствор отфильтровывали и смолу промывали при помощи DMF. К смоле добавляли раствор (E)-3-(2,5-диметоксифенил)акриловой кислоты (4 экв.), HATU (4 экв.), HOAt (4 экв.) и DIPEA (8 экв.) в DMF. Реакционную смесь встряхивали при комнатной температуре в течение ночи. Смолу фильтровали и промывали при помощи DMF и DCM.
СТАДИЯ D: отщепление
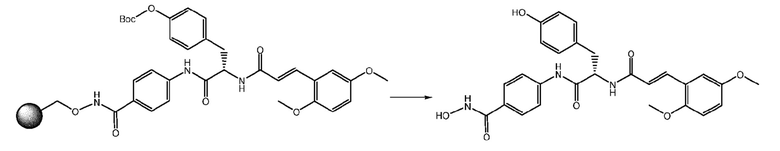
Отщепление продукта от смолы осуществляли путем обработки при помощи TFA 50% в DCM при комнатной температуре в течение 30 минут. На этой стадии также удаляли Boc-защиту.
Неочищенное вещество очищали на ТФЭ картридже.
ПРИМЕР 6
Следующие соединения были получены с использованием процедуры, описанной в примере 5:
ПРИМЕР 7
Цитотоксическая активность in vitro
Соединения по настоящему изобретению представляют собой малые молекулы формулы (I), характеризующиеся присутствием металл-хелатирующего бензогидроксаматного фрагмента, α-аминокислотного N-ацилированного ядра и α,β-ненасыщенности по ацильной группе. Именно эта структурная особенность по-видимому является ответственной за высокую ингибиторную активность на линиях раковых стволовых клеток и на HCT116 клетках (ATCC CCL-247), клеточной линии колоректальной карциномы человека, широко используемой в онкобиологии как in vitro, так и in vivo (Botchkina Cancer Genom Proteom 6, 19-30, 2009; Yeung PNAS 107, 3722-3727, 2010).
Цитотоксическую активность оценивали следующим образом:
- Пред-B лейкозную клеточную линию 697 высевали при 2×105 клеток/лунка;
- Клеточные линии карциномы толстой кишки HCT116, HT29 и COLO2015 высевали при 4×103, 4×103 и 10×103 клеток/лунка, соответственно;
- Первичные клетки почки человека высевали при 1,5×103 и 6,5×103 в двух разных экспериментах;
- Человеческие PBMC высевали при 5×105 клеток/лунка;
- Стволовые клетки рака толстой кишки (CSC) высевали при 3×103;
Испытываемые соединения добавляли через 24 часа и инкубировали в течение 72 часов (48 часов для 697 клеточной линии). Концентрации молекул составляли от 10000 нМ до 1,5 нМ (10000-1 нМ для 697 клеточной линии). Цитотоксическую активность соединений оценивали с использованием анализа CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega), который измеряет функциональность митохондрий, в соответствии с инструкциями изготовителя. Для стволовых клеток рака толстой кишки жизнеспособность определяли с использованием люминесцентного анализа клеточной жизнеспособности CellTiter-Glo (Promega).
В первичном скрининговом анализе цитотоксичности, осуществляемом на 697 клеточной линии, активность соединений D была такой же или выше, чем активность насыщенных соединений.
Ингибирующая пролиферацию активность ненасыщенных соединений по настоящему изобретению на HCT116 клетках в 30-80 раз выше, чем активность, демонстрируемая насыщенными аналогами предшествующего уровня техники US7635788, при этом она является по меньшей мере аналогичной или в 3,5-40 раз выше на стволовых клетках (см. таблицу 1). В частности, все соединения по изобретению оказались более активными, чем насыщенные аналоги предшествующего уровня техники, на HCT116 клетках.
Цитотоксичность соединений была подтверждена дополнительными анализами на двух других клеточных линиях рака толстой кишки HT29 (ATCC HTB38) и Colo205 (ATCC CCL222) и на стволовых клетках рака толстой кишки (CSCs) (см. таблица 2 и 3). Следует отметить, что цитотоксическая активность в отношении первичных клеток почки человека и мононуклеарных клеток периферической крови (PBMC), выделенных у здоровых доноров, была ниже по сравнению с цитотоксичностью в отношении опухолевых клеток (см. таблицу 4).
Цис-форма, полученная в соответствии с Примером 3, оказалась менее эффективной на HCT116 клетках, чем транс-форма.
Кроме того, цис-форма химически менее стабильна, чем транс-форма. В испытании форсированного разложения все соединения в транс-форме показали высокую стабильность даже при высоких температурах (15 дней при 80°C) и при низком pH (15 дней при pH 2), тогда как цис-аналог был несколько менее стабилен при 80°C и значительно менее стабилен при pH 2.
Таблица 1: Сравнительные результаты на клеточных линиях рака толстой кишки
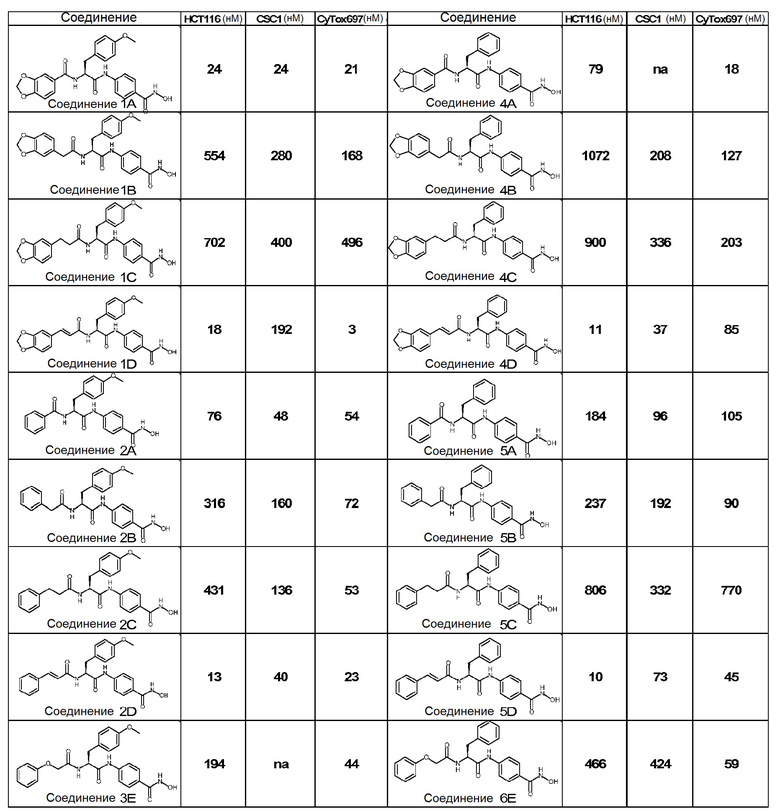
Цитотоксическая активность соединений на клеточных линиях рака толстой кишки
na=нет данных
Активность соединений при любой дозе рассчитывают как процент ингибирования относительно обработанного носителем контроля (= 0). Значения IC50 экстраполировали из кривой ингибирования доза/ответ с использованием программы GraphPad Prism.
Цитотоксическая активность соединений на стволовых клетках рака толстой кишки
Цитотоксическая активность соединений на первичных клетках почки и PBMC
Противоопухолевая активность in vivo
Соединения по настоящему изобретению также показали активность in vivo в модели ксенотрансплантата рака толстой, кишки, где клеточную линию HTC116 колоректальной карциномы человека вводили подкожно (п/к) самкам бестимусных мышей CD1 (см. таблицу 5).
Самок бестимусных мышей CD-1 возраста 5 недель с массой тела 20-22 г помещали в виварий Italfarmaco Research Centre. Мышей содержали в микро-изоляторных клетках и обеспечивали стерильной пищей и водой в стандартных условиях.
Ксенотрансплантацию осуществляли путем п/к инъекции 7×106 HCT116 клеток в правый бок животных.
Размеры опухолей определяли при помощи циркуля и рассчитывали объемы опухолей в соответствии со следующей формулой:
Объем опухоли (мм3)=(w2 × l)/2
где ʺwʺ означает ширину, а ʺlʺ означает длину карциномы в мм.
Рост опухоли отслеживали путем измерения три раза в неделю.
Масса опухоли достигала измеряемого размера через три недели после трансплантации, в это время начинали лечение.
В эксперименте также использовали два эталонных соединения. Производное гидроксамовой кислоты панобиностат и 5-FU. Соединения вводили при MTD в соответствии с предыдущими экспериментами или как описано в литературе. Как показано в таблице 5, соединения по настоящему изобретению способны уменьшать размер и объем опухоли, при этом их активность сопоставима с активностью двух эталонных соединений.
Следует отметить, что лечение эталонным соединением 5-FU приводило к значительной лейкопении (70,4%) в конце лечения, в то время как соединение 4D показало сопоставимую эффективность (уменьшение опухоли на 54% по сравнению с 61%, полученным с 5-FU), но гораздо менее выраженную лейкопению (23,7%).
Кроме того, тромбоцитопению и потерю массы тела не наблюдали в конце лечения, и это указывает на то, что при эффективных дозах соединения, являющиеся объектом настоящего изобретения, демонстрировали благоприятное терапевтическое окно в этой животной модели.
Соединения по настоящему изобретению демонстрируют хорошую метаболическую стабильность in vitro как при инкубации с человеческой S9-фракцией, так и в плазме человека. Они также показывают хороший фармакокинетический профиль в предварительных исследованиях в предклинических состояниях.
Противоопухолевая активность in vivo
после 44 дней лечения
после 44 дней лечения
(vs до начала лечения)
после 44 дней лечения
(vs до начала лечения)
после 65 дней лечения
(-7,2)
(-20,3)
(-15,7)
(-22,9)
(-29,4)
(-27,3)
(-26,9)
(-22,7)
(-89,4)
(-31,9)
(-42,1)
(-25)
(-67)
(-12,2)
(-25,5)
(-13,2)
(49,7)
(-29,8)
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ HDAC6 | 2018 |
|
RU2764718C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
| ЛИГАНДЫ РЕЦЕПТОРА НЕЙРОТЕНЗИНА | 2013 |
|
RU2671970C2 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2629957C2 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| СПОСОБ ЛЕЧЕНИЯ РЕТРОВИРУСНЫХ ИНФЕКЦИЙ У МЛЕКОПИТАЮЩИХ (ВАРИАНТЫ) | 1995 |
|
RU2166317C2 |
| ПАРА-АМИНОБЕНЗИЛЬНЫЕ ЛИНКЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КОНЪЮГАТАХ | 2021 |
|
RU2839675C1 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ НУКЛЕИНОВЫХ КИСЛОТ | 2011 |
|
RU2582235C2 |
Настоящее изобретение относится к бензо-N-гидроксиамидным соединениям формулы (I), их фармацевтически приемлемым солям или изомерам, обладающим ингибирующей активностью в отношении пролиферации опухолевых клеток, а также к фармацевтической композиции на их основе. Соединения могут найти применение для лечения опулевых заболеваний, в частности колоректального рака или гематологических злокачественных заболеваний. В общей формуле (I) n имеет значение 0, 1 или 2; A отсутствует или представляет собой 5-6-членный моно- или 9-10-членный ди-карбоциклический остаток, необязательно частично или полностью ненасыщенный, содержащий атомы углерода и необязательно один или два гетероатома, выбранных из N и S; R1 и R2 независимо выбраны из группы, содержащей -H, -OH, -OMe, -CN, -NH2, -NO2, -Cl, -COOH и -CH2N(CH2CH3)2; X может представлять собой C или N; R3 и R4 независимо выбраны из группы, содержащей -H, -OMe, -OPh, -NO2, -NMe2 и -NH2, или R3 и R4 вместе могут образовывать гетеропентациклическую группу (-OCH2O-). 3 н. и 7 з.п. ф-лы, 5 табл., 6 пр.

(I)
1. Соединение формулы (I)
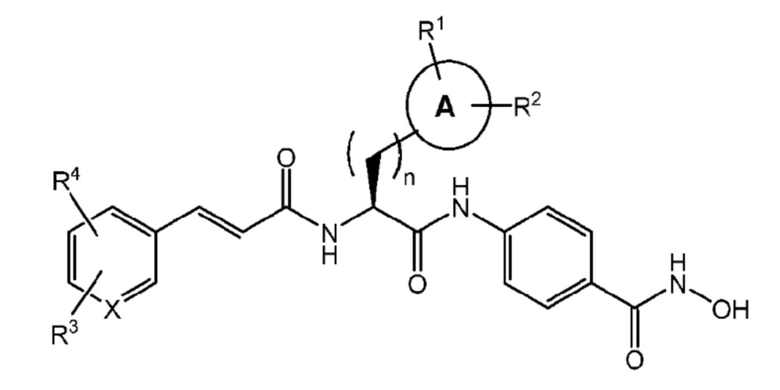
(I)
или его фармацевтически приемлемые соли или изомеры,
где:
n имеет значение 0, 1 или 2;
A отсутствует или представляет собой 5-6-членный моно- или 9-10-членный ди-карбоциклический остаток, необязательно частично или полностью ненасыщенный, содержащий атомы углерода и необязательно один или два гетероатома, выбранных из N и S;
R1 и R2 независимо выбраны из группы, содержащей -H, -OH, -OMe, -CN, -NH2, -NO2, -Cl, -COOH и -CH2N(CH2CH3)2;
X может представлять собой C или N;
R3 и R4 независимо выбраны из группы, содержащей -H, -OMe, -OPh, -NO2, -NMe2 и -NH2, или R3 и R4 вместе могут образовывать гетеропентациклическую группу (-OCH2O-).
2. Соединение по п. 1, в котором:
n имеет значение 0, 1 или 2;
A представляет собой 5-6-членный моно- или 9-10-членный ди-карбоциклический остаток, необязательно частично или полностью ненасыщенный, включающий атомы углерода и необязательно один или два гетероатома, выбранных из N и S;
R1 и R2 независимо выбраны из группы, содержащей -H, -OH, -OMe, -CN, -NH2, -NO2, -Cl, -COOH и -CH2N(CH2CH3)2;
X может представлять собой C или N;
R3 и R4 независимо выбраны из группы, содержащей -H, -OMe, -OPh, -NO2, -NMe2 и -NH2, или R3 и R4 вместе могут образовывать гетеропентациклическую группу (-OCH2O-).
3. Соединение по п. 1 или 2, в котором
n имеет значение 1, X представляет собой C;
R1 и R2 независимо представляют собой -H, -Cl или -OMe;
R3 и R4 независимо представляют собой -H, -NMe2, или R3 и R4 вместе могут образовывать гетеропентациклическую группу (-OCH2O-).
4. Соединение, выбранное из следующих:
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)пропанамид (1D);
- (2S)-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (2D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (4D);
- (2S)-N-[4-(гидроксикарбамоил)фенил]-3-фенил-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (5D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-гидроксифенил)пропанамид (7D);
- (E)-3-(2,5-диметоксифенил)-N-[(1R)-2-[4-(гидроксикарбамоил)анилино]-1-индан-2-ил-2-оксо-этил]проп-2-енамид (8D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-4-фенил-бутанамид (9D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (10D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(2-нафтил)пропанамид (11D);
- (E)-3-(2,5-диметоксифенил)-N-[2-[4-(гидроксикарбамоил)анилино]-2-оксо-этил]проп-2-енамид (12D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (13D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-хинолил)пропанамид (14D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (15D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-нитрофенил)пропанамид (16D);
- (2S)-2-[[(E)-3-(4-аминофенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (17D);
- (2S)-3-(4-аминофенил)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (18D);
- (2S)-3-(4-цианофенил)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (19D);
- (2S)-N-[4-(гидроксикарбамоил)фенил]-2-[[(E)-3-(4-нитрофенил)проп-2-еноил]амино]-3-фенил-пропанамид (20D);
- (2S)-2-[[(E)-3-(2,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(1H-имидазол-5-ил)пропанамид (21D);
- (2S)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (22D);
- (2S)-2-[[(E)-3-(3,5-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (23D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(3-пиридил)пропанамид (24D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(бензотиофен-3-ил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (25D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-тиазол-4-ил-пропанамид (26D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-4-фенил-бутанамид (27D);
- (2S)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (28D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(4-цианофенил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (29D);
- (2S)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(1H-индол-3-ил)пропанамид (30D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(1H-индол-3-ил)пропанамид (31D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-нитрофенил)пропанамид (32D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(4-хлорфенил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (33D);
- (2S)-3-(4-аминофенил)-2-[[(E)-3-(3,4-диметоксифенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (34D);
- (2S)-3-(4-аминофенил)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]пропанамид (35D);
- 4-[(2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-[4-(гидроксикарбамоил)анилино]-3-оксо-пропил]бензойная кислота (36D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-(3,4-дихлорфенил)-N-[4-(гидроксикарбамоил)фенил]пропанамид (37D);
- (2S)-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид (38D);
- (2S)-2-[[(E)-3-(4-диметиламинофенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-фенил-пропанамид (39D);
- (2S)-2-[[(E)-3-(4-диметиламинофенил)проп-2-еноил]амино]-N-[4-(гидроксикарбамоил)фенил]-3-(4-метоксифенил)пропанамид; 2,2,2-трифторуксусная кислота (40D);
- (E)-N-[2-[[4-(гидроксикарбамоил)фенил]метиламино]-2-оксо-этил]-3-фенил-проп-2-енамид (41D);
- (E)-3-(1,3-бензодиоксол-5-ил)-N-[2-[[4-(гидроксикарбамоил)фенил]метиламино]-2-оксо-этил]проп-2-енамид (43D);
- (2S)-3-[4-(диэтиламинометил)фенил]-N-[4-(гидроксикарбамоил)фенил]-2-[[(E)-3-фенилпроп-2-еноил]амино]пропанамид; 2,2,2-трифторуксусная кислота (45D);
- (2S)-2-[[(E)-3-(1,3-бензодиоксол-5-ил)проп-2-еноил]амино]-3-[4-(диэтиламинометил)фенил]-N-[4-(гидроксикарбамоил)фенил]пропанамид; 2,2,2-трифторуксусная кислота (46D);
- (2S)-N-[4-(гидроксикарбамоил)фенил]-3-(2-нафтил)-2-[[(E)-3-(6-фенокси-3-пиридил)проп-2-еноил]амино]пропанамид (47D).
5. Соединение по любому из пп. 1-4, в котором изомерная форма представляет собой транс-форму.
6. Соединение по любому из пп. 1-5 для применения в качестве лекарственного средства для профилактики и/или лечения колоректального рака и гематологических злокачественных заболеваний.
7. Соединение по любому из пп. 1-5 для применения в профилактике и/или лечении колоректального рака или гематологических злокачественных заболеваний.
8. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая терапевтически эффективное количество по меньшей мере одного из соединений формулы (I) или их фармацевтически приемлемых солей или изомеров по любому из пп. 1-5 вместе с по меньшей мере одним фармацевтически приемлемым эксципиентом.
9. Фармацевтическая композиция по п. 8, подходящая для введения энтеральным путем, парентеральным путем, пероральным путем, местным путем или путем ингаляции.
10. Фармацевтическая композиция по п. 8 или 9 в форме жидкости или твердого вещества, предпочтительно в форме капсул, таблеток, таблеток с покрытием, порошков, гранул, кремов или мазей.
| WO 2006003068 A2, 12.01.2006 | |||
| LI XIAOYANG ET AL, "Development ofN-hydroxybenzamide derivatives with indole-containing cap group as histone deacetylases inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY,Vol | |||
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Видоизменение устройства для движения судна реакцией выходящей из трубчатых насадок кожуха смеси пара с топочными газами парового котла, воздухом и водой | 1927 |
|
SU19951A1 |

