Область техники, к которой относится изобретение
Настоящее изобретение относится к новым селективным бензогидроксамовым ингибиторам фермента гистондеацетилазы 6 (HDAC6) и их фармацевтическим композициям.
Соответственно, эти соединения используются при лечении заболеваний, связанных с активностью HDAC6, таких как отторжение трансплантата, GVHD, миозит, заболевания, связанные с аномальной функцией лимфоцитов, множественная миелома, неходжкинская лимфома, периферическая нейропатия, аутоиммунные заболевания, воспалительные заболевания, рак и нейродегенеративные патологии.
Уровень техники изобретения
Генетический материал эукариотических клеток организован в сложную и динамическую структуру, состоящую из ДНК и белков, хроматина. Основными белковыми компонентами хроматина являются гистоны, основные белки, которые взаимодействуют с ДНК, образуя основную структурную единицу хроматина, нуклеосому, первый уровень хромосомной компактизации внутри ядра. Взаимодействие между основными гистоновыми остатками и кислотными остатками ДНК является решающим для определения нуклеосомной компактизации и связанной с этим доступности ДНК для молекулярных комплексов, регулирующих репликацию и транскрипцию. Это взаимодействие в основном зависит от степени ацетилирования гистонов. Деацетилирование N-концевых лизиновых остатков гистона позволяет протонировать аминогруппу, несущую положительный заряд, которая взаимодействует с отрицательными зарядами, содержащимися в ДНК. Такое взаимодействие происходит в более компактном состоянии хроматина, включая подавление экспрессии генов. И наоборот, ацетилирование этих же самых остатков предотвращает образование ионных связей, что приводит к менее компактной форме хроматина, которая обеспечивает большую доступность ДНК и взаимодействие с макромолекулярными комплексами, которые активируют транскрипцию генов.
Степень ацетилирования гистонов регулируется балансом активности двух классов ферментов: гистонацетилтрансфераз (гистонацетилтрансфераз HAT) и гистондеацетилаз (гистондеацетилаз HDAC). Нарушение этого тонкого баланса может привести к потере клеточного гомеостаза, обычно встречающегося при различных заболеваниях человека, включая рак, неврологические нарушения, воспаление и аутоиммунные заболевания.
Гистондеацетилазы были классифицированы таким образом, поскольку они обратимо катализируют деацетилирование аминогрупп N-концевых лизиновых остатков гистона. Впоследствии было обнаружено, что существует большое число субстратов этих ферментов, поскольку их активность также обусловлена негистоновым белком, который является субстратом ферментов HAT, содержащих N-ацетиллизин, таких как факторы транскрипции, ферменты репарации ДНК и другие ядерные и цитоплазматические белки.
Класс HDAC человека состоит из 18 ферментов, разделенных на две группы: цинк-зависимые HDAC и NAD-зависимые HDAC, также известные как сиртуины (класс III). Цинк-зависимые HDAC далее подразделяются на четыре класса: 1) Класс I, включающий HDAC1, 2, 3 и 8, широко распространенные изоферменты, в основном расположенные в ядре; 2) Класс IIa, включающий HDAC4, 5, 7 и 9, изоферменты, расположенные как в ядре, так и в цитоплазме; 3) Класс IIb, включающий HDAC6 и HDAC10, главным образом расположенные в цитоплазме, и 4) Класс IV, включающий только HDAC11. В отличие от класса I HDAC класс IIa и IIb имеют тканеспецифическую экспрессию.
За счет регулирования экспрессии генов и воздействия на гистоны и факторы транскрипции становится ясно, что эти ферменты вовлечены во множество клеточных функций. Кроме того, воздействуя на многочисленные другие белковые субстраты, эти ферменты, а также фосфатазы, участвуют во многих других процессах, таких как трансдукция сигнала и перегруппировка цитоскелета.
В последние десятилетия HDAC стали хорошо изученной терапевтической мишенью. Было синтезировано несколько ингибиторов HDAC, некоторые из них в настоящее время проходят поздние стадии клинических испытаний, и четыре из них одобрены для лечения различных видов рака: вориностат и ромидепсин для T-клеточной лимфомы кожи (CTLC), белиностат для периферической T-клеточной лимфомы (PTLC) и панобиностат для множественной миеломы. Эти последние ингибиторы могут в различной степени взаимодействовать с различными изоформами HDAC.
Несмотря на свою клиническую эффективность, применение пан-ингибиторов, соответственно неселективных для конкретной изоформы, ограничивается их токсичностью и побочными эффектами, наблюдаемыми как в доклинических моделях, так и, что наиболее важно, в клинических испытаниях. Следовательно, существует потребность в разработке ингибиторов HDAC с лучшим фармакологическим профилем и терапевтическим диапазоном (соотношением эффективность/токсичность).
В результате, внимание научного сообщества сосредоточено на синтезе и изучении селективных ингибиторов для отдельных изоформ HDAC с целью разработки веществ с лучшими фармакологическими возможностями.
Таким образом, использование ингибиторов HDAC может быть важным терапевтическим или диагностическим инструментом для патологий, вызванных экспрессией генов, таких как воспалительные нарушения, диабет, осложнения диабета, гомозиготная талассемия, фиброз, цирроз, острый промиелоцитарный лейкоз (APL), отторжение трансплантированного органа, аутоиммунные патологии, протозойные инфекции, раковые заболевания и т.п. Селективные ингибиторы для семейства HDAC или для конкретной изоформы, особенно HDAC6, могут быть особенно полезны для лечения патологий, связанных с пролиферативными нарушениями и накоплением белка, нарушениями иммунной системы и неврологическими и нейродегенеративными заболеваниями, такими как инсульт, болезнь Гентингтона, ALS и болезнь Альцгеймера.
В частности, для изоформы HDAC6 были идентифицированы различные субстраты, такие как α-тубулин, Hsp90 (белок теплового шока 90), кортактин, β-катенин. Модуляция ацетилирования этих белков HDAC6 коррелирует с несколькими важными процессами, такими как иммунный ответ (Wang et al., Nat. Rev. Drug Disc. (2009), 8(12), 969-981; J. Med. Chem. (2012), 55, 639-651; Mol. Cell. Biol. (2011), 31(10), 2066-2078), регуляция динамики микротрубочек, включая миграцию клеток и межклеточное взаимодействие (Aldana-Masangkay et al., J. Biomed. Biotechnol. (2011), 2011, 875824), и деградация вырожденных белков.
Кроме того, HDAC6 участвует в процессе катаболизма деградированных белков через комплекс, известный как агресома: HDAC6 способен связывать полиубиквитинированные белки и динеин, тем самым активируя своего рода доставку денатурированных белков вдоль микротрубочек к агресоме (Kawaguchi et al., Cell (2003) 115 (6), 727-738).
Изменение этой цитопротекторной активности HDAC6 коррелирует с различными нейродегенеративными патологиями, такими как болезнь Паркинсона (Outerio et al., Science (2007), 317 (5837), 516-519) и болезнь Гентингтона (Dompierre et al., J. Neurosci. (2007), 27(13), 3571-3583), при которых накопление деградированных белков является общей патологической чертой.
Кроме того, HDAC6 участвует в регуляции многих онкологических белков, особенно в гематологических опухолях, таких как различные виды лейкемии (Fiskus et al., Blood (2008), 112(7), 2896-2905; Rodriguez-Gonzales, Blood (2008), 112(11), реферат 1923) и множественная миелома (Hideshima et al., Proc. Natl. Acad. Sci. USA (2005), 102(24), 8567-8572). Регуляция ацетилирования α-тубулина с помощью HDAC6 может участвовать в развитии метастазирования, когда клеточная подвижность играет важную роль (Sakamoto et al., J. Biomed. Biotechnol. (2011), 2011, 875824).
В международной патентной заявке WO 2011/021209 описаны 1,2,3-триазольные соединения, обладающие ингибирующей активностью в отношении HDAC.
В международной патентной заявке WO 2012/178208 описаны соединения с замещенными гетероциклами, такие как бензимидазол, бензимидазолон и бензотриазол, обладающие ингибирующей активностью, селективной в отношении HDAC6.
В международной патентной заявке WO 2015/102426 описаны новые индольные производные с ингибирующей активностью в отношении HDAC.
В международной патентной заявке WO 2015/087151 описаны новые азаиндольные производные с ингибирующей активностью в отношении HDAC.
В международной патентной заявке WO 2012/106343 описаны ингибиторы HDAC и содержащие их композиции. Также описаны способы лечения заболеваний и состояний, в которых ингибирование HDAC обеспечивает преимущество, таких как рак, нейродегенеративное нарушение, периферическая нейропатия, неврологическое заболевание, травматическое повреждение мозга, инсульт, гипертония, малярия, аутоиммунное заболевание, аутизм, расстройства аутистического спектра и воспаление.
В статье «Valente et al., Journal of Medicinal Chemistry (2014), 57(14), 6259-6265» описаны гидроксаматы, содержащие 1,3,4-оксадиазол (2) и 2-аминоанилиды (3) в качестве ингибиторов гистондеацетилазы. Среди них соединения 2t, 2x и 3i описаны как наиболее мощные и селективные по отношению к HDAC1.
Определения
Если не определено иное, все термины из уровня техники, примечания и другая научная терминология, используемая в настоящем описании, имеет стандартные значения, известные специалистам в данной области техники, к которой относится настоящее изобретение. В некоторых случаях термины с общепринятыми значениями определены в настоящем описании для ясности и/или для быстрого ознакомления; таким образом, включение таких определений в данное описание не должно истолковываться как представляющее существенное отличие от обычно понимаемого в данной области техники.
Термин «галоген» относится в настоящем описании к фтору (F), хлору (Cl), брому (Br) или йоду (I).
Термин «C1-C4-алкил» относится в настоящем описании к разветвленному или линейному углеводороду, содержащему 1-4 атомов углерода. Примеры C1-C4 алкильных групп включают, без ограничения, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил.
Термин «арил» относится в настоящем описании к моно- и поликарбоциклическим ароматическим кольцевым системам (i), при этом отдельные карбоциклические кольца в поликарбоциклических кольцевых системах могут быть конденсированы или присоединены друг к другу посредством одинарной связи. Подходящие арильные группы включают, без ограничения, фенил, нафтил и бифенил.
Термин «арилокси» относится в настоящем описании к О-арильной группе, в которой «арил» является таким, как определено выше.
Термин «алкокси» относится в настоящем описании к О-алкильной группе, в которой «алкил» является таким, как определено выше.
Термин «циклоалкил» относится в настоящем описании к насыщенному или ненасыщенному углеводородному кольцу, предпочтительно имеющему 4-10 атомов углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Термин «арилалкил» относится в настоящем описании к арильному радикалу, как определено в данном документе, присоединенному к алкильному радикалу, как определено в данном документе. Примером арилалкила является бензил.
Термин «гетероцикл» относится в настоящем описании к 4-, 5-, 6-, 7- или 8-членному моноциклическому кольцу, которое является насыщенным или ненасыщенным и состоит из атомов углерода и одного или нескольких гетероатомов, выбранных из N, O и S, и при этом гетероатомы азота и серы могут быть необязательно окислены, и гетероатом азота может быть необязательно кватернизован. Гетероциклическое кольцо может быть присоединено к любому гетероатому или атому углерода, при условии, что такое присоединение приводит к образованию стабильной структуры. Термин также включает любую бициклическую систему, в которой любое из указанных выше гетероциклических колец конденсировано с арилом или другим гетероциклом. Когда гетероциклическое кольцо представляет собой ароматическое гетероциклическое кольцо, оно может быть определено как «гетероароматическое кольцо».
Термин «ненасыщенное кольцо» относится в настоящем описании к частично или полностью ненасыщенному кольцу. Например, ненасыщенное С6 моноциклическое кольцо относится к циклогексену, циклогексадиену и бензолу.
Термин «замещенный» относится в настоящем описании к моно- или полизамещению с определенным (или неопределенным) заместителем, при условии, что это одиночное или множественное замещение является химически допустимым.
Термин «физиологически приемлемый эксципиент» в данном документе относится к веществу, лишенному какого-либо собственного фармакологического эффекта, и которое не вызывает нежелательных реакций при введении млекопитающему, предпочтительно человеку. Физиологически приемлемые эксципиенты хорошо известны в области техники и описаны, например, в «Руководстве по фармацевтическим эксципиентам» (Handbook of Pharmaceutical Excipients, sixth edition 2009), включенном в настоящий документ посредством ссылки.
Термин «фармацевтически приемлемые соли или их производные» в настоящем описании относится к солям или производным, которые обладают биологической эффективностью и свойствами превращенного в соль или производного соединения, и которые не вызывают нежелательных реакций при введении млекопитающему, предпочтительно человеку. Фармацевтически приемлемые соли могут быть неорганическими или органическими солями; примеры фармацевтически приемлемых солей включают, без ограничения: карбонат, гидрохлорид, гидробромид, сульфат, гидросульфат, цитрат, малеат, фумарат, трифторацетат, 2-нафталинсульфонат и пара-толуолсульфонат. Дополнительную информацию о фармацевтически приемлемых солях можно найти в «Руководстве по фармацевтическим солям» (Handbook of pharmaceutical salts, P. Stahl, C. Wermuth, WILEY-VCH, 127-133, 2008), включенном в настоящий документ посредством ссылки. Фармацевтически приемлемые производные включают сложные эфиры, простые эфиры и N-оксиды.
Термин «содержащий», «имеющий», «включающий в себя» и «включающий» следует понимать как неограничивающие термины (означающие «включающий, без ограничения») и следует также рассматривать в качестве основы таких терминов, как «по существу состоит из», «по существу состоящий из», «состоит из» или «состоящий из».
Термин «по существу состоит из», «по существу состоящий из» следует понимать как частично ограничивающие термины, означающие, что не включен ни один другой ингредиент, влияющий на новые характеристики изобретения (соответственно, могут быть включены необязательные эксципиенты).
Термины «состоит из», «состоящий из» следует понимать как ограничивающие термины.
Термин «изомеры» относится к стереоизомерам (или пространственным изомерам), т.е. диастереоизомерам и энантиомерам.
Термин «пролекарства» относится к фармакологически неактивным производным, которые могут подвергаться метаболическому превращению in vivo с образованием активного соединения, включенного в общую формулу данного изобретения. В данной области известно много различных пролекарств (Prodrug approach: an effective solution to overcome side-effects, Patil S.J., Shirote P.J., International Journal of Medical and Pharmaceutical Sciences, 2011,1-13; Carbamate Prodrug Concept for Hydroxamate HDAC Inhibitors, Jung, Manfred et al., ChemMedChem, 2011, 1193-1198).
Термин «патология» включает одно или более из следующих аутоиммунных заболеваний или нарушений: сахарный диабет, артрит (включая ревматоидный артрит, ювенильный ревматоидный артрит, остеоартрит, псориатический артрит), рассеянный склероз, тяжелая миастения, системная красная волчанка, аутоиммунный тиреоидит, дерматит (включая атопический дерматит и экзематозный дерматит), псориаз, синдром Шегрена, включая иссушение кератоконъюнктивы, вторичное к синдрому Шегрена, очаговая алопеция, аллергические реакции, вызванные укусами членистоногих, болезнь Крона, язва желудка, ирит, конъюнктивит, кератоконъюнктивит, язвенный колит, астма, аллергическая астма, кожная красная волчанка, склеродермия, вагинит, проктит, реакция на препарат, проказа, красная волчанка, аутоиммунный увеит, аллергический энцефаломиелит, острая некротизирующая геморрагическая энцефалопатия, прогрессирующая двусторонняя идиопатическая потеря слуха, апластическая анемия, анемия, идиопатическая тромбоцитопения, полихондрит, гранулематоз Вегенера, хронический активный гепатит, синдром Стивенса-Джонсона, идиопатический спру, красный плоский лишай, офтальмопатия Грейвса, саркоидоз, первичный билиарный цирроз, задний увеит, интерстициальный легочный фиброз.
Термин «патология» относится к одному или более из следующих неврологических или нейродегенеративных заболеваний: болезнь Вильсона, спинально-церебеллярная атаксия, прионная болезнь, болезнь Паркинсона, болезнь Гентингтона, амиотрофический боковой склероз (ALS), амилоидоз, болезнь Альцгеймера, болезнь Александера, алкогольная болезнь печени, кистозный фиброз, болезнь Пика, спинальная мышечная атрофия и деменция с тельцами Леви.
Термин «патология» также включает одно или более из следующих заболеваний: ревматоидный спондилит, постишемическое реперфузионное повреждение, воспаление кишечника, хроническое воспалительное заболевание легких, экзема, астма, острый респираторный дистресс синдром, инфекционный артрит, хронический прогрессирующий артрит, деформирующий артрит, посттравматическая артропатия, подагрический артрит, синдром Рейтера, острый синовит, острый спондилит, гломерулонефрит, гемолитическая анемия, апластическая анемия, нейтропения, реакция «трансплантат против хозяина» (GVHD), отторжение трансплантата, хронический тиреоидит, болезнь Грейвса, бинарный первичный цирроз, контактный дерматит, солнечные ожоги, хроническая почечная недостаточность, синдром Гийена-Барре, увеит, отит среднего уха, периодонтит, интерстициальный легочный фиброз, бронхит, синусит, пневмокониоз, синдром легочной недостаточности, эмфизема легких, легочный фиброз, силикоз или легочные хронические воспалительные заболевания.
Термин «патология» также включает одно или более из следующих заболеваний: рак, рост опухоли, рак толстой кишки, молочной железы, кости, мозга и другие виды рака (например, остеосаркома, нейробластома, аденокарцинома толстой кишки), хронический миелоидный лейкоз (CML), острый миелоидный лейкоз (AML), острый промиелоцитарный лейкоз (APL), рак сердца (саркома, миксома, рабдомиома, фиброма, липома и тератома), рак легкого (например, бронхогенная карцинома, альвеолярная карцинома, бронхиальная аденома, саркома, лимфома, хондроматозная гамартома, мезотелиома), рак желудочно-кишечного тракта (например, рак пищевода, желудка, поджелудочной железы, тонкой кишки, толстой кишки), рак мочеполовых путей (например, рак почки, мочевого пузыря и уретры, простаты, яичка), рак печени (например, гепатоцеллюлярная карцинома, холангиокарцинома, гепатобластома, ангиосаркома, гепатоцеллюлярная аденома, гемангиома), рак кости (например, остеогенная саркома, фибросаркома, злокачественная фиброзная гистиоцитома, хондросаркома, саркома Юинга, злокачественная лимфома, множественная миелома, злокачественная гигантоклеточная опухоль, хордома, хондростеома, доброкачественная хордома, хондробластома, хондромиксофиброма, остеоид-остеома), опухоли нервной системы (например, черепа, менингит, головного мозга, спинного мозга), гинекологические опухоли (например, матки, шейки матки, яичников, вульвы и влагалища), гематологический рак (например, опухоли крови, болезнь Ходжкина, неходжкинская болезнь), рак кожи (например, злокачественная меланома, базальноклеточная карцинома, злокачественная опухоль сквамозных клеток, саркома Капоши, диспластический невус, липома, ангиома, дерматофиброма, келоид, псориаз) и опухоли надпочечников (например, нейробластома).
Описание чертежей
Фиг. 1: Ингибирование экспрессии PD-L1 в iDC (моноциты, стимулированные GMCSF-IL-4). Моноциты человека обрабатывали ингибиторами HDAC6 и стимулировали GMCSF-IL-4 в течение 5 дней. После инкубации клетки собирали и метили антителом анти-PD-L1. Затем клетки промывали и получали данные флуоресценции с помощью проточного цитометра (BD FACSVerse). Значения на графиках представляют собой среднее для 3 экспериментов, проведенных на 3 различных донорах (n=3).Экспрессия PD-L1 представлена средним геометрическим значением флуоресценции. * = P<0,05 определяется t-критерием Стьюдента.
Фиг. 2: Соединения 8 и 10 снижают рост опухоли in vivo и обладают сопоставимой эффективностью с анти-PD-1 антителом. Стрелка указывает на день начала лечения.
Фиг. 3: Ингибиторы HDAC6 снижают рост CT-26 опухоли in vivo, и их активность может быть улучшена комбинированным лечением с анти-PD-1 антителом. Статистические параметры оценивали на 30-й день с помощью t-критерия Стьюдента. *, P<0,05; **, P<0,01; ***, P<0,001. См. текст для получения дополнительной информации.
Фиг. 4: Лечение in vivo селективными ингибиторами HDAC6 вызывало специфический Т-клеточный ответ. Спленоциты животных, обработанные соединениями 8 и 10 и в сочетании с анти-PD-1 Ab, стимулировали опухолевыми пептидами, полученными из CT-26, и продукцию IFN-γ и TNF-α T-клетками CD4 определяли количественно с помощью ELISPOT.
Фиг. 5: Лечение in vivo селективными ингибиторами HDAC6 вызывало специфический Т-клеточный ответ. Спленоциты животных, обработанные соединениями 8 и 10, и комбинацией с анти-PD-1 Ab стимулировали опухолевыми пептидами, полученными из CT-26, и продукцию IFN-γ и TNF-α T-клетками CD8 определяли количественно с помощью ELISPOT.
Описание изобретения
Авторы изобретения экспериментально обнаружили, что бензогидроксамовые соединения, характеризующиеся пентагетероциклическим центральным ядром, проявляют высокую и селективную ингибирующую активность в отношении фермента HDAC6.
Эти соединения также демонстрируют низкую цитотоксичность, что позволяет осуществлять их длительное использование.
В соответствии с первым аспектом, настоящее изобретение относится к соединениям формул (I) и (II) и их фармацевтически приемлемым солям, изомерам и пролекарствам:

(I)
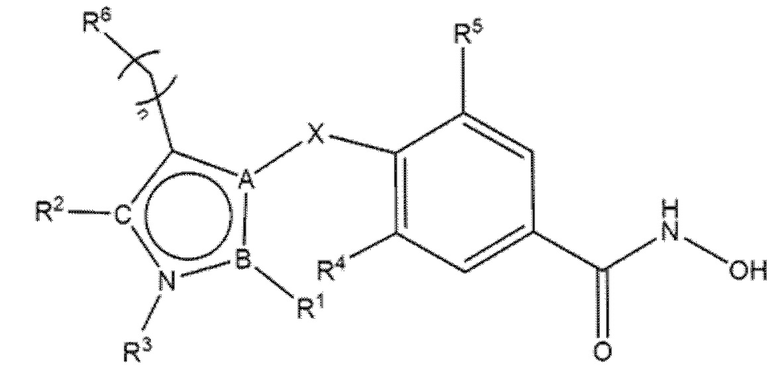
(II)
где
А=N, O, S в формуле (I), когда А=N в формуле (II);
B=C, N;
С=N, O в формуле (I), когда С=N в формуле (II);
X=CH2, S, NH, O, CD2;
n=0, 1;
когда n=1, атом углерода может быть замещен R12 и R13, независимо выбранными из группы, включающей H, D, -Me, -фенил, -F и -OH, или вместе R12 и R13 могут образовывать насыщенный циклический фрагмент, предпочтительно циклопропан, циклобутан, циклопентан или циклогексан;
когда n=1, R6 может отсутствовать;
R4=R5 =H, F;
R1 отсутствует или выбран из группы, включающей -H, -NH2, C1-C4 алкил, фенил, фенил, замещенный одним или более галогенами, арилалкил, циклоалкил, метилфуран, циклобутилметил, тетрагидрофуран-2-ил-метил, 3-(диэтиламино)пропил, 2-метоксиэтил, винил, 2-(метилсульфанил)этил, 1-циклопропилэтил, пиридин-2-ил, (пиридин-3-ил)метил, 2-(пиридин-2-ил)этил, 2- (тиофен-2-ил)этил, 3,4-диметоксифенил, 4-метоксифенил, метилфенил, 2-хлор-5-(морфолин-4-сульфонил)фенил, 4-[(дифторметил)сульфанил]фенил, 4-(морфолин-4-сульфонил)фенил, 5-(диметилсульфамоил)-2-метилфенил, 3-(трифторметил)фенил, 4-(трифторметил)фенил, 2-(морфолин-4-ил)этил, 3-(морфолин-4-ил)пропил, 1-нафтил, 2,3-дигидро-1,4-бензодиоксин-6-ил, бензгидрил, 5-инданил, тиофен и метилтиофен;
R2 отсутствует или выбран из H, алкила, циклоалкила, циклоалкилметила, гетероарила, фенила, фенила, замещенного одним или более галогенами, фенила, замещенного одной или более алкоксигруппами, фенила, замещенного одной или более нитрогруппами, бензила, алкилзамещенного бензила, (2,2-дифторциклопентил)метила, 2-бром-3-фторфенила, (2,2-диметилциклопропил)метила, 4-гидроксифенила, 2-(бензилокси)этила, 2-бром-4-метоксифенила, 2-метилхинолина, 3-метилпиридин-4-ила, 4-метансульфонил-2-метилфенила, 2-хлор-4,6-динитрофенила, 1,3-бензoдиоксол-5-илметила, или 2-бензилоксифенила;
R3 отсутствует или выбран из H, алкоксиарила, фенила, фенила, замещенного CF3, бензила, пиридила, алкила, циклоалкила, циклоалкилметила, гетероарила, фенила, замещенного одним или более галогенами, фенила, замещенного одной или более алкоксигруппами, фенила, замещенного одной или более нитрогруппами, бензила, алкилзамещенного бензила, (2,2-дифторциклопентил)метила, 2-бром-3-фторфенила, (2,2-диметилциклопропил)метила, 4-гидроксифенила, 2-(бензилокси)этила, 2-бром-4-метоксифенила, метил-2-хинолина, 3-метилпиридин-4-ила, 4-метансульфонил-2-метилфенила, 2-хлор-4,6-динитрофенила, 1,3-бензoдиоксол-5-илметила, или 2-бензилоксифенила;
R6 представляет собой замещенный или незамещенный моно- или полициклический остаток, необязательно частично или полностью ненасыщенный, содержащий атомы углерода и необязательно один или более гетероатомов, выбранных из N, S или О;
или R6 может быть выбран из:
 ;
;
при условии, что в соединениях формулы (I), когда пентагетероциклическое ядро представляет собой 1,3,4-оксадиазол, R6 не является нафтилом.
Еще один класс предпочтительных соединений включает соединения формулы (I) и (II) и их фармацевтически приемлемые соли, изомеры и фармакологически приемлемые сложные эфиры, где пентагетероциклическое ядро выбрано из группы, состоящей из тетразола, 1,2,4-триазола, 1,3,4-оксадиазола, 1,2,4-оксадиазола, 1,3,4-тиадиазола.
Другой класс предпочтительных соединений включает соединения формулы (I) и (II) и их фармацевтически приемлемые соли, изомеры и фармакологически приемлемые соли, где:
А=N, O, S в формуле (I), когда А=N в формуле (II);
B=C, N;
С=N, O в формуле (I), когда С=N в формуле (II);
X=CH2, S;
n=0, 1;
когда n=1, атом углерода может быть замещен R12 и R13, независимо выбранными из группы, включающей H, D, -Me, -фенил, -F и -OH, или вместе R12 и R13 могут образовывать насыщенный циклический фрагмент, предпочтительно циклопропан, циклобутан, циклопентан или циклогексан;
когда n=1, R6 может отсутствовать;
R4=R5 =H, F;
R1 отсутствует или выбран из группы, включающей -H, -NH2, -CH3, -CH2CH3, фенил, п-фторфенил, м-хлорфенил, п-хлорфенил, бензил, метилфуран, циклопропил, изобутил, метилфенил, трифторфенил, тиофен и 2-(морфолин-4-ил)этил;
R2 отсутствует или выбран из H, фенила или п-дихлорфенила;
R3 отсутствует или выбран из H, o-метоксифенила, п-трифторметилфенила, бензила или пиридила;
R6 выбран из группы, содержащей:

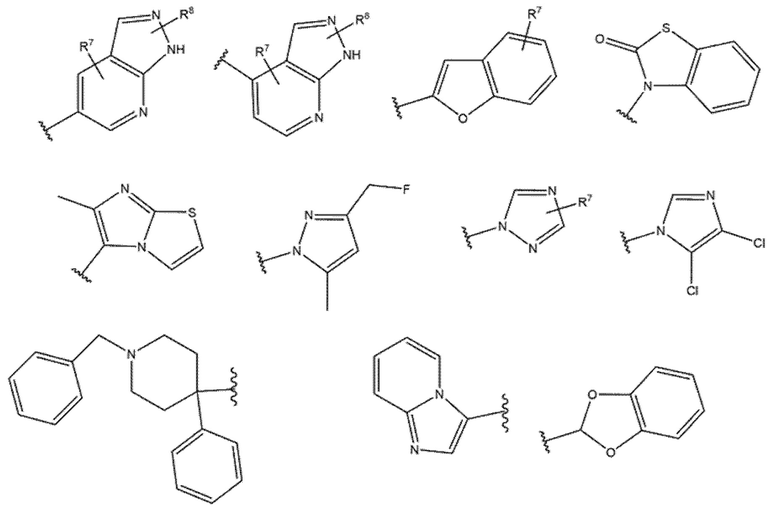
где:
R7 и R8 независимо выбраны из группы, содержащей H, D, -Cl, -F, -Br, -CF3, -Me, -Et, -OMe, -Oбензил, -SF5, -OCH2F, -CH2NH2, -NH2, -CH2NMe2, -NMe2, -N(CH2CH2OCH3)2, -COOH, -COOMe, -OH, -NHNH2, -NO2, -OEt, -OCHF2, -OiPr, -CHF2, -NEt2,

или R7 и R8 вместе могут образовывать гетеропентациклический фрагмент (-OCH2O-);
R9=R10 = -H, -Me, -Et;
R11 выбран из группы, содержащей -H, -Cl, -CH3, -NO2 и -Br.
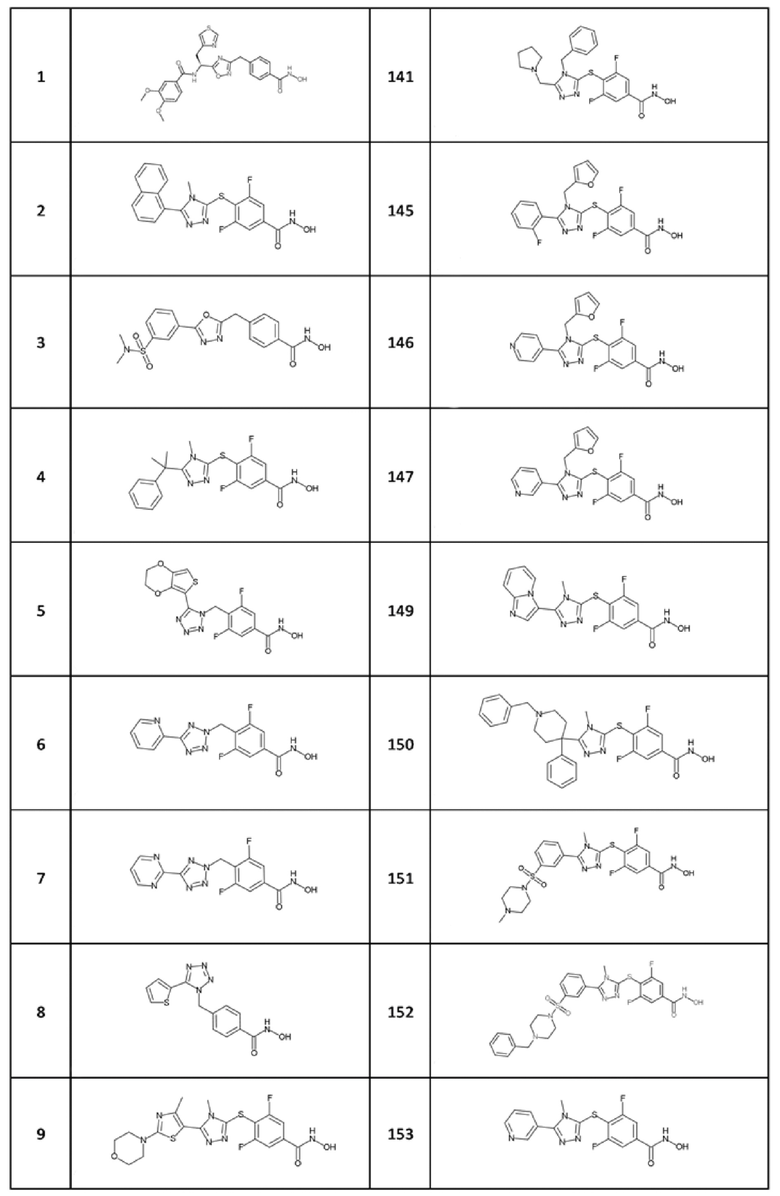
Следующие соединения формул (I) и (II) являются особенно предпочтительными:
(S)-N-(1-(3-(4-(гидроксикарбамоил)бензил)-1,2,4-оксадиазол-5-ил)-2-(тиазол-4-ил)этил)-3,4-диметоксибензамид (соед. 1);
3,5-дифтор-N-гидрокси-4-((4-метил-5-(нафталин-1-ил)-4H-1,2,4-триазол-3-ил)тио)бензамид (соед. 2);
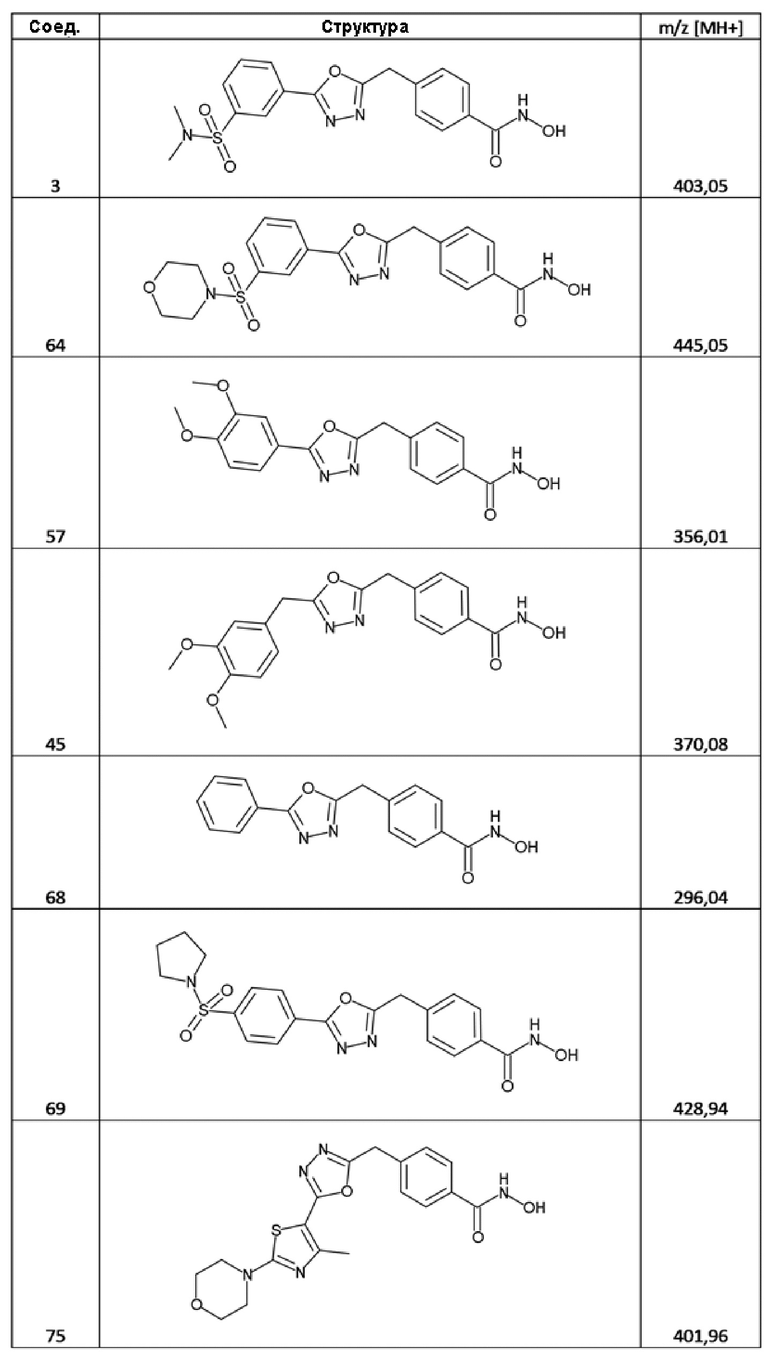
4-((5-(3-(N,N-диметилсульфамоил)фенил)-1,3,4-оксадиазол-2-ил)метил)-N-гидроксибензамид (соед. 3);
3,5-дифтор-N-гидрокси-4-((4-метил-5-(2-фенилпропан-2-ил)-4H-1,2,4-триазол-3-ил)тио)бензамид (coед. 4);
4-((5-(2,3-дигидротиено[3,4-b][1,4]диоксин-5-ил)-1H-тетразол-1-ил)метил)-3,5-дифтор-N-гидроксибензамид (соед. 5);
3,5-дифтор-N-гидрокси-4-((5-(пиридин-2-ил)-2H-тетразол-2-ил)метил)бензамид (соед. 6);
дифтор-N-гидрокси-4-((5-(пиримидин-2-ил)-2H-тетразол-2-ил)метил)бензамид (соед. 7);
N-гидрокси-4-((5-(тиофен-2-ил)-1H-тетразол-1-ил)метил)бензамид (соед. 8);
3,5-дифтор-N-гидрокси-4-((4-метил-5-(4-метил-2-морфолинoтиазол-5-ил)-4H-1,2,4-триазол-3-ил)тио)бензамид (соед. 9);
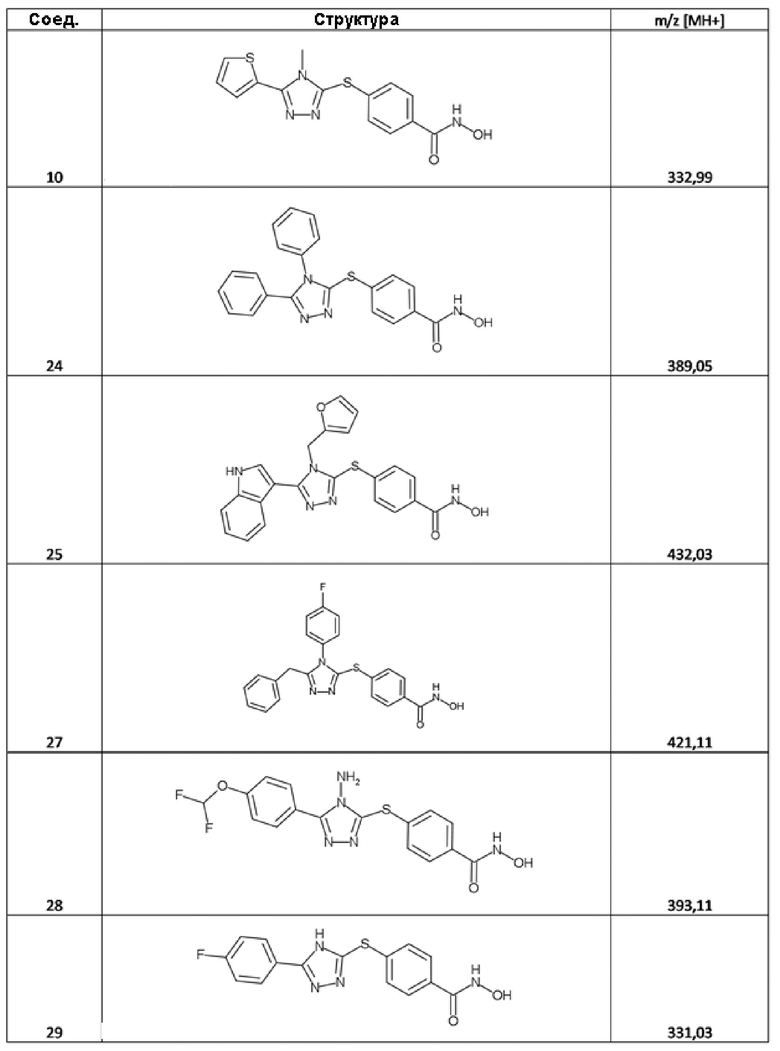
N-гидрокси-4-((4-метил-5-(тиофен-2-ил)-4H-1,2,4-триазол-3-ил)тио)бензамид (соед. 10);
4-((5-(фуран-2-ил)-2H-тетразол-2-ил)метил)-N-гидроксибензамид (соед. 12);
3,5-дифтор-N-гидрокси-4-((5-(пиридин-2-ил)-1H-тетразол-1-ил)метил)бензамид (соед. 13);
3,5-дифтор-N-гидрокси-4-((4-метил-5-(пиридин-2-ил)-4H-1,2,4-триазол-3-ил)тио)бензамид (соед. 14);
3,5-дифтор-N-гидрокси-4-((5-(тиофен-2-ил)-1H-тетразол-1-ил)метил)бензамид (соед. 15);
3,5-дифтор-N-гидрокси-4-((4-метил-5-(4-(пиперидин-1-илметил)фенил)-4H-1,2,4-триазол-3-ил)тио)бензамид (соед. 16);
3,5-дифтор-N-гидрокси-4-((4-метил-5-(тиофен-2-ил)-4H-1,2,4-триазол-3-ил)тио)бензамид (соед. 17);
3,5-дифтор-4-((5-(фуран-2-ил)-2H-тетразол-2-ил)метил)-N-гидроксибензамид (соед. 19);
N-гидрокси-4-((5-(пиридин-2-ил)-1H-тетразол-1-ил)метил)бензамид (соед. 20);
3-(3,4-диметоксифенил)-N-[(1S)-1-[3-[[4-(гидроксикарбамоил)фенил]метил]-1,2,4-оксадиазол-5-ил]-2-тиазол-4-ил-этил]пропанамид (соед. 21);
4-[[5-[4-(трифторметил)фенил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 23);
4-[(4,5-дифенил-1,2,4-триазол-3-ил)сульфанил]бензолкарбогидроксамовая кислота (соед. 24);
4-[[4-(2-фурилметил)-5-(1H-индол-3-ил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота; 2,2,2-трифторуксусная кислота (соед. 25);
4-[5-[(3,4-диметоксифенил)метил]-1,3,4-оксадиазол-2-ил]бензолкарбогидроксамовая кислота (соед. 26);
4-[[5-бензил-4-(4-фторфенил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота; 2,2,2-трифторуксусная кислота (соед. 27);
4-[[4-амино-5-[4-(дифторметокси)фенил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 28);
4-[[5-(4-фторфенил)-4H-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 29);
4-[[4-этил-5-(4-фторфенил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 30);
4-[[5-(4-хлорфенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 31);
4-[[5-(5-хлор-2-тиенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 32);
4-[[5-(2-фторфенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 33);
4-[[5-(4-фторфенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 34);
4-[[5-(4-метоксифенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 35);
4-[(5-бензилтетразол-2-ил)метил]бензолкарбогидроксамовая кислота (соед. 36);
4-[(5-бензилтетразол-1-ил)метил]бензолкарбогидроксамовая кислота (соед. 37);
4-[[5-(2,4-дихлорфенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 38);
4-[[5-(3-метил-2-тиенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 39);
4-[[5-(5-метил-2-тиенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 41);
4-[[5-(бензoтиофен-3-ил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 42);
4-[[5-(2,3-дигидротиено[3,4-b][1,4]диоксин-5-ил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 43);
4-[[5-[(3,4-диметоксифенил)метил]-2-[4-(трифторметил)фенил]-1,2,4-триазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 44);
4-[[5-[(3,4-диметоксифенил)метил]-1,3,4-оксадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 45);
4-[[5-(2-фторфенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 46);
4-[[5-[(1S)-1-амино-2-тиазол-4-ил-этил]-1,2,4-оксадиазол-3-ил]метил]бензолкарбогидроксамовая кислота; 2,2,2-трифторуксусная кислота (соед. 48);
4-[[5-(3,4-диметоксифенил)-1,2,4-оксадиазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 49);
4-[[5-(2-тиенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 50);
4-[[2-бензил-5-(4-хлорфенил)-1,2,4-триазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 51);
4-[[2-(2-пиридил)-5-(2-тиенил)-1,2,4-триазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 52);
4-[[2-(2-метоксифенил)-5-(2-тиенил)-1,2,4-триазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 53);
4-[[5-(6,6-диметил-3-метилсульфанил-4-оксо-5,7-дигидро-2-бензoтиофен-1-ил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 54);
4-[[5-(бензoтиофен-2-ил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 55);
4-[[5-(3,4-диметоксифенил)-1,3,4-оксадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 57);
4-[[5-(2,4-дифторфенил)-1,3,4-оксадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 58);
4-[[5-[3-(диметилсульфамоил)фенил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 59);
4-[(5-фенил-1,3,4-оксадиазол-2-ил)амино]бензолкарбогидроксамовая кислота (соед. 60);
4-[[4-амино-5-[3-(диэтилсульфамоил)фенил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 61);
4-[[1-(2,4-дихлорфенил)-5-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 62);
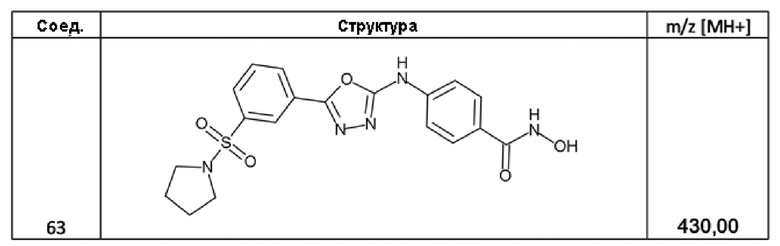
4-[[5-(3-пирролидин-1-илсульфонилфенил)-1,3,4-оксадиазол-2-ил]амино]бензолкарбогидроксамовая кислота (соед. 63);
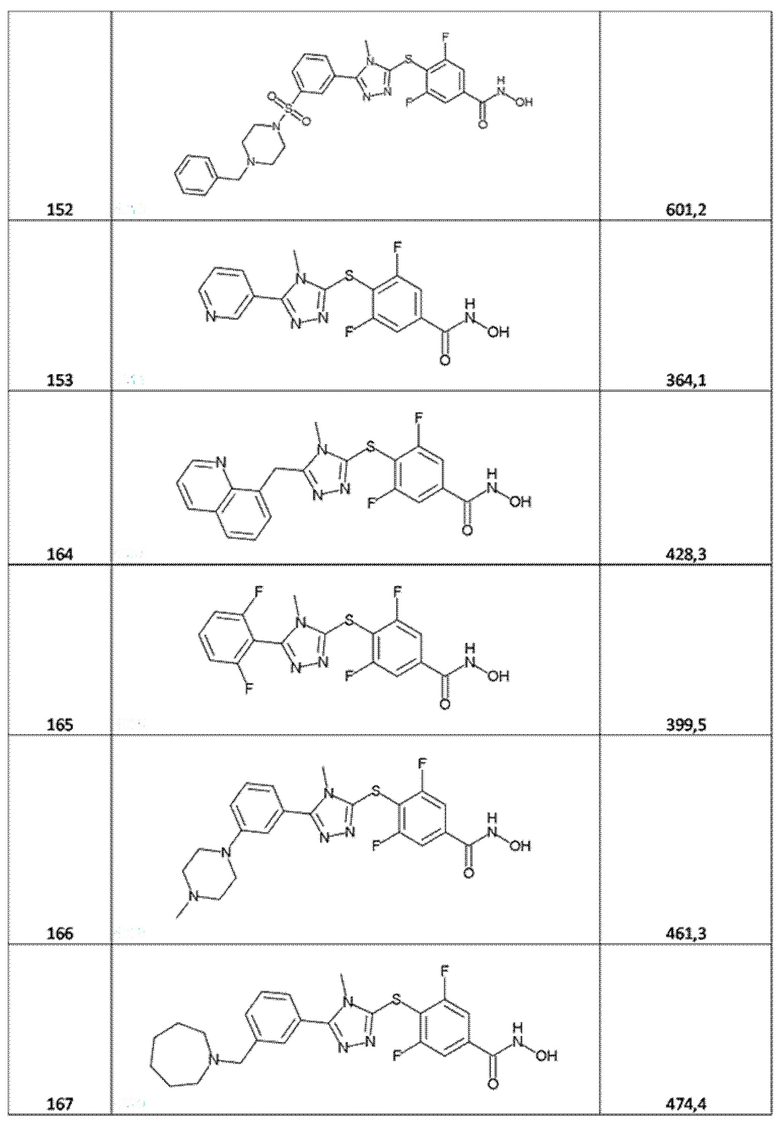
4-[[5-(3-морфолинoсульфонилфенил)-1,3,4-оксадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 64);
3,5-дифтор-4-[[5-(2-тиенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 65);
4-[[5-[3-(диэтилсульфамоил)фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 66);
4-[[4-метил-5-[2-(п-толил)-4-хинолил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 67);
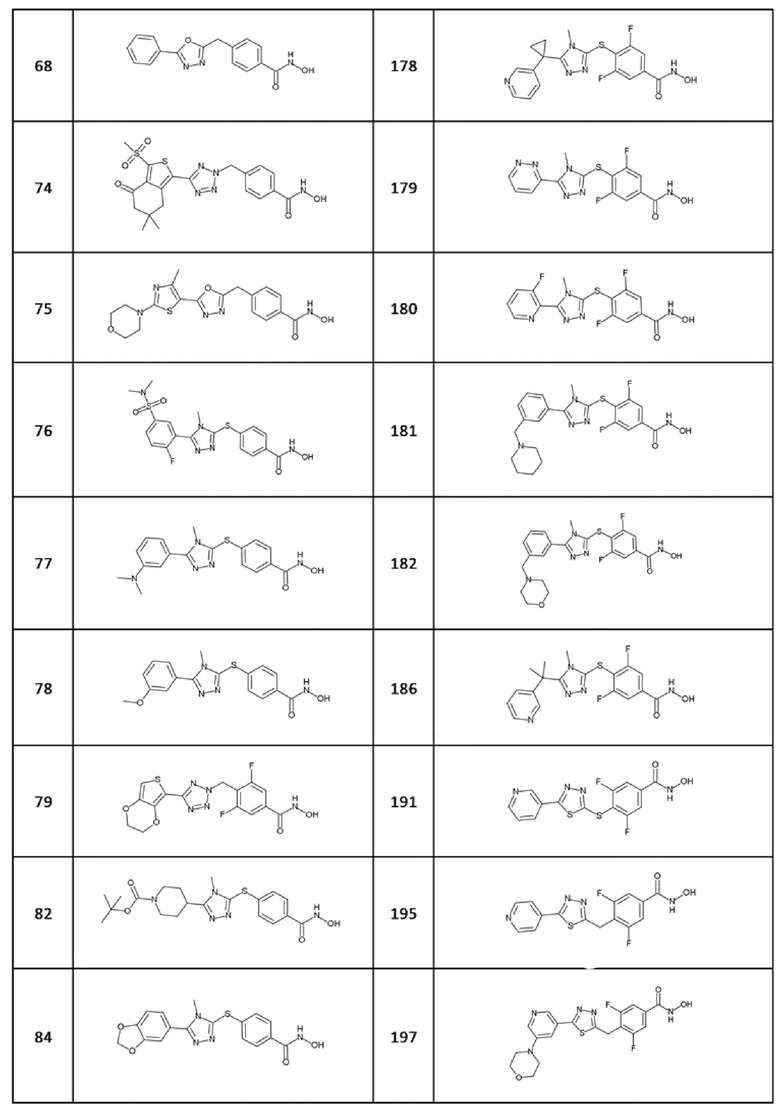
4-[(5-фенил-1,3,4-оксадиазол-2-ил)метил]бензолкарбогидроксамовая кислота (соед. 68);
4-[[5-(4-пирролидин-1-илсульфонилфенил)-1,3,4-оксадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 69);
4-[[5-(3-бензилокси-4-метокси-фенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 70);
4-[[5-(3-бензилокси-4-метокси-фенил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 71);
4-[(5-циклопропил-1-фенил-1,2,4-триазол-3-ил)сульфанил]бензолкарбогидроксамовая кислота (соед. 72);
4-[[5-[4-(диметиламино)фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 73);
4-[[5-(4-метил-2-морфолинo-тиазол-5-ил)-1,3,4-оксадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 75);
4-[[5-[3-(диметиламино)фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 77);
4-[[5-(3-метоксифенил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 78);
4-[[5-(2,3-дигидротиено[3,4-b][1,4]диоксин-5-ил)тетразол-2-ил]метил]-3,5-дифтор-бензолкарбогидроксамовая кислота (соед. 79);
4-[[5-[3-(диметиламино)фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифтор-бензолкарбогидроксамовая кислота (соед. 80);
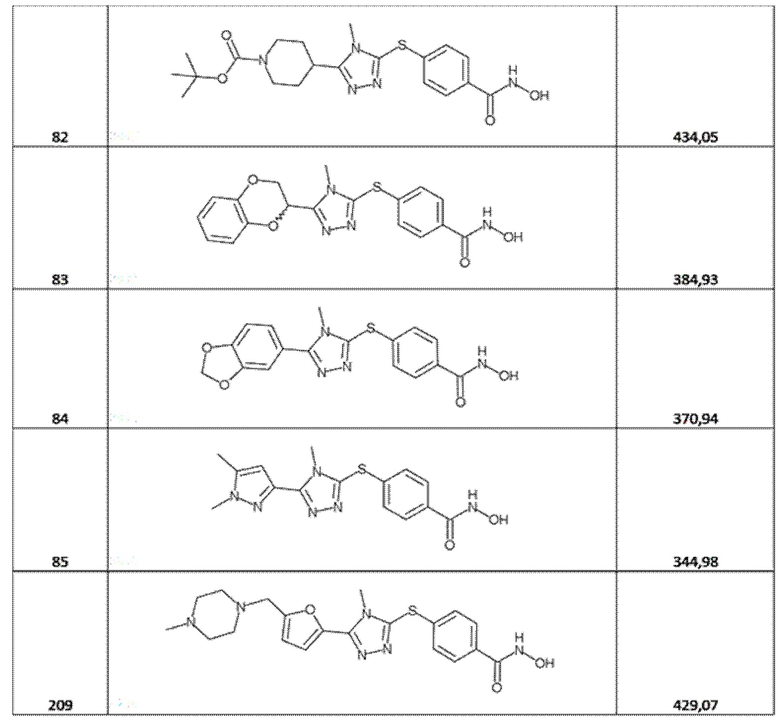
трет-бутил-4-[5-[4-(гидроксикарбамоил)фенил]сульфанил-4-метил-1,2,4-триазол-3-ил]пиперидин-1-карбоксилат (соед. 82);
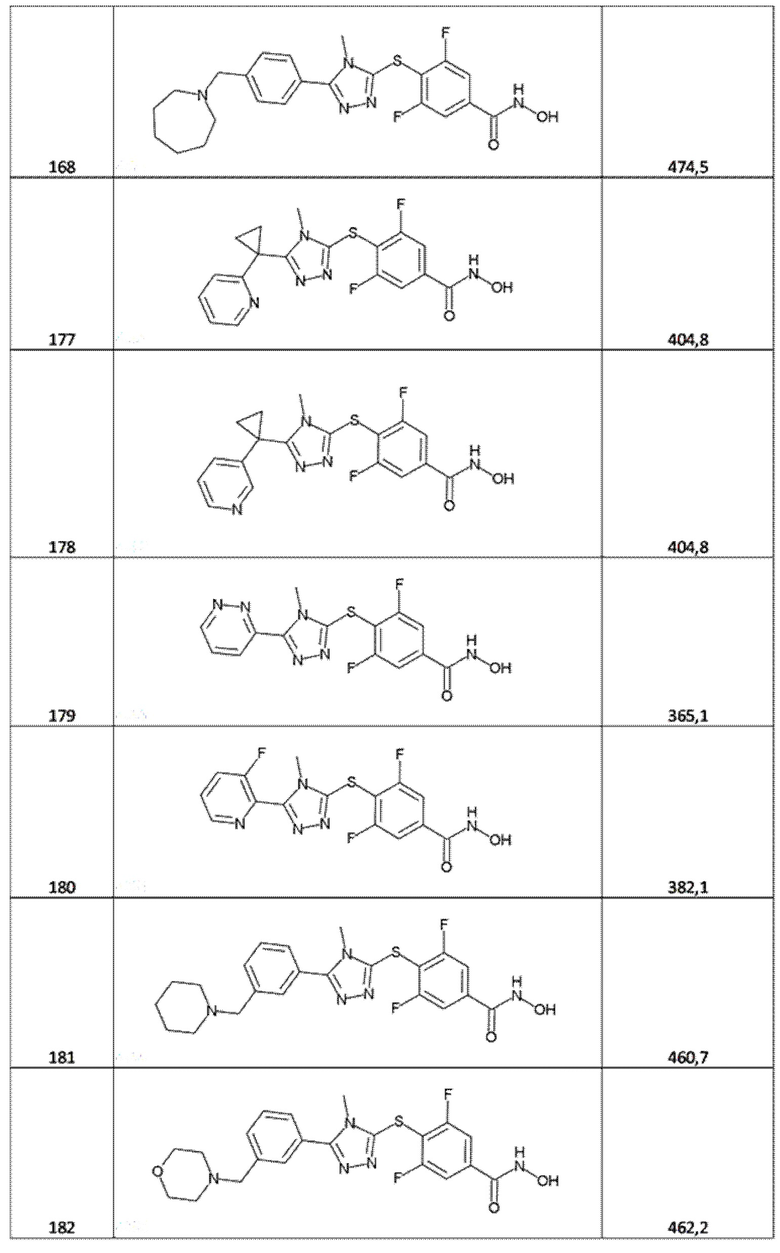
4-[[5-(2,3-дигидро-1,4-бензодиоксин-3-ил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 83);
4-[[5-(1,3-бензoдиоксол-5-ил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 84);
4-[[5-(1,5-диметилпиразол-3-ил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 85);
4-[[5-(2-фурил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 86);
4-[[5-(1-изохинолил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 87);
4-[[5-(1-изохинолил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 88);
4-[[5-(2-пиридил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 89);
4-[[5-(2-хинолил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 90);
4-[[5-(2-хинолил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 91);
3,5-дифтор-4-[[5-(2-фурил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 92);
3,5-дифтор-4-[[5-(1-изохинолил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 93);
3,5-дифтор-4-[[5-(1-изохинолил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 94);
3,5-дифтор-4-[[5-(2-хинолил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 95);
3,5-дифтор-4-[[5-(2-хинолил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 96);
3,5-дифтор-4-[[5-(2-тиенил)-4H-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 97);
4-[(5-бензгидрил-4-метил-1,2,4-триазол-3-ил)сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 98);
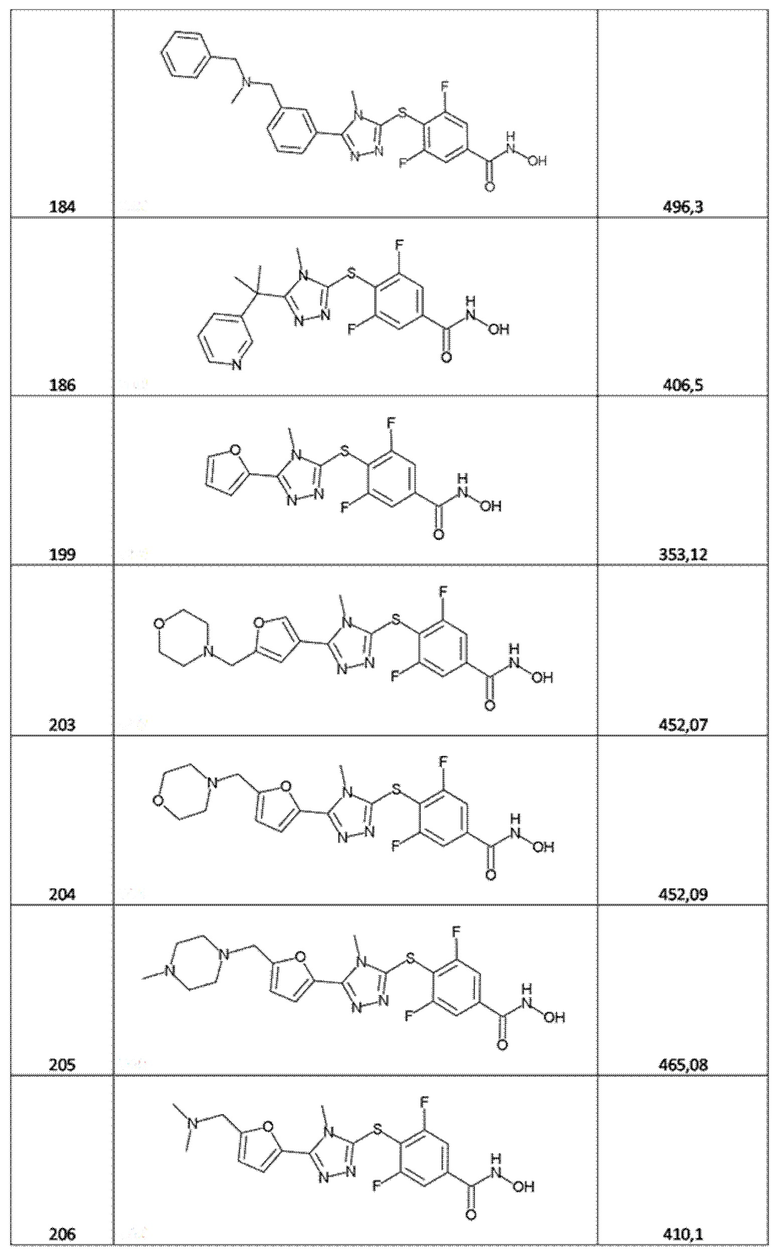
4-[[5-(3-аминотиено[2,3-b]пиридин-2-ил)-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 99);
4-[[5-(1,5-диметилпиразол-3-ил)-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 100);
3,5-дифтор-4-[[4-метил-5-(1-фенилциклобутил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 101);
3,5-дифтор-4-[[5-[1-(3-фторфенил)циклопентил]-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 102);
3,5-дифтор-4-[[5-[1-(4-метоксифенил)циклогексил]-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 103);
3,5-дифтор-4-[[5-[1-(4-метоксифенил)циклопропил]-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (coед. 104);
4-[[5-[3-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 106);
4-[[5-[3-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 107);
3,5-дифтор-4-[[5-[3-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 108);
3,5-дифтор-4-[[5-[3-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 109);
4-[[5-[4-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 110);
4-[[5-[4-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 111);
3,5-дифтор-4-[[5-[4-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 112);
3,5-дифтор-4-[[5-[4-(пентaфтор-лямбда-6-сульфанил)фенил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 113);
3,5-дифтор-4-[[4-метил-5-[3-(4-метил-4-оксидо-пиперазин-4-ий-1-ил)фенил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 114);
3,5-дифтор-4-[[4-(4-фторфенил)-5-(1-пиперидилметил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 115);
3,5-дифтор-4-[[4-(2-фурилметил)-5-пирролидин-1-ил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 116);
4-[(4-бензил-5-морфолинo-1,2,4-триазол-3-ил)сульфанил]-3,5-дифтор-бензолкарбогидроксамовая кислота (соед. 117);
4-[[5-(2,3-дигидротиено[3,4-b][1,4]диоксин-5-ил)-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифтор-бензолкарбогидроксамовая кислота (соед. 118);
3,5-дифтор-4-[[5-(1-изохинолил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 121);
3,5-дифтор-4-[[4-метил-5-(2-хинолил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 122);
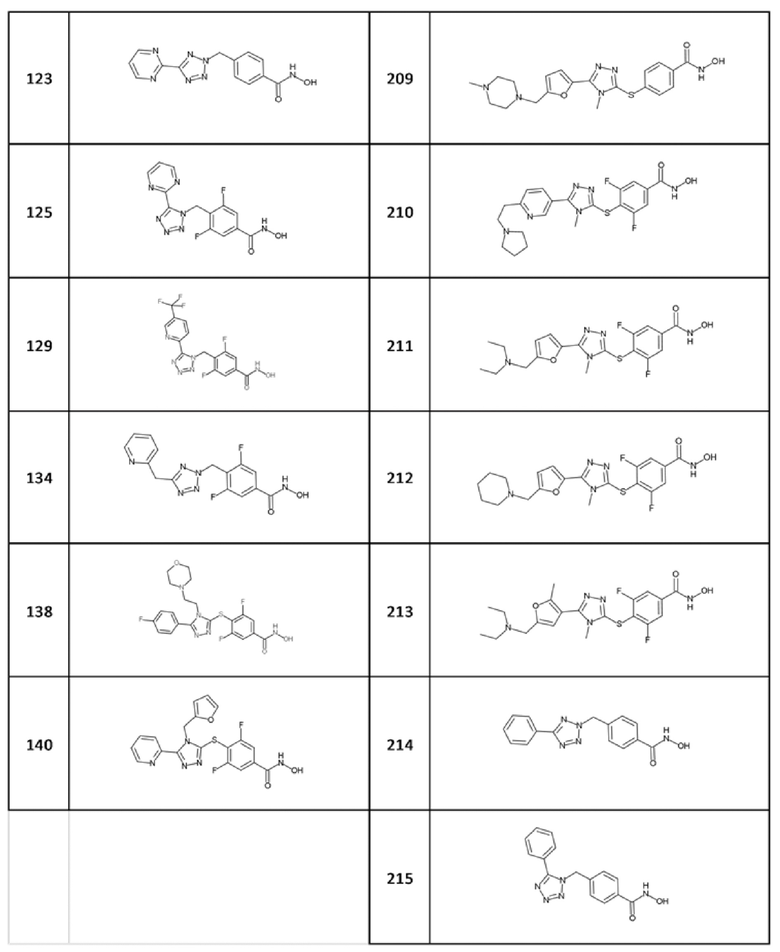
4-[(5-пиримидин-2-илтетразол-2-ил)метил]бензолкарбогидроксамовая кислота (соед. 123);
4-[(5-пиримидин-2-илтетразол-1-ил)метил]бензолкарбогидроксамовая кислота (соед. 124);
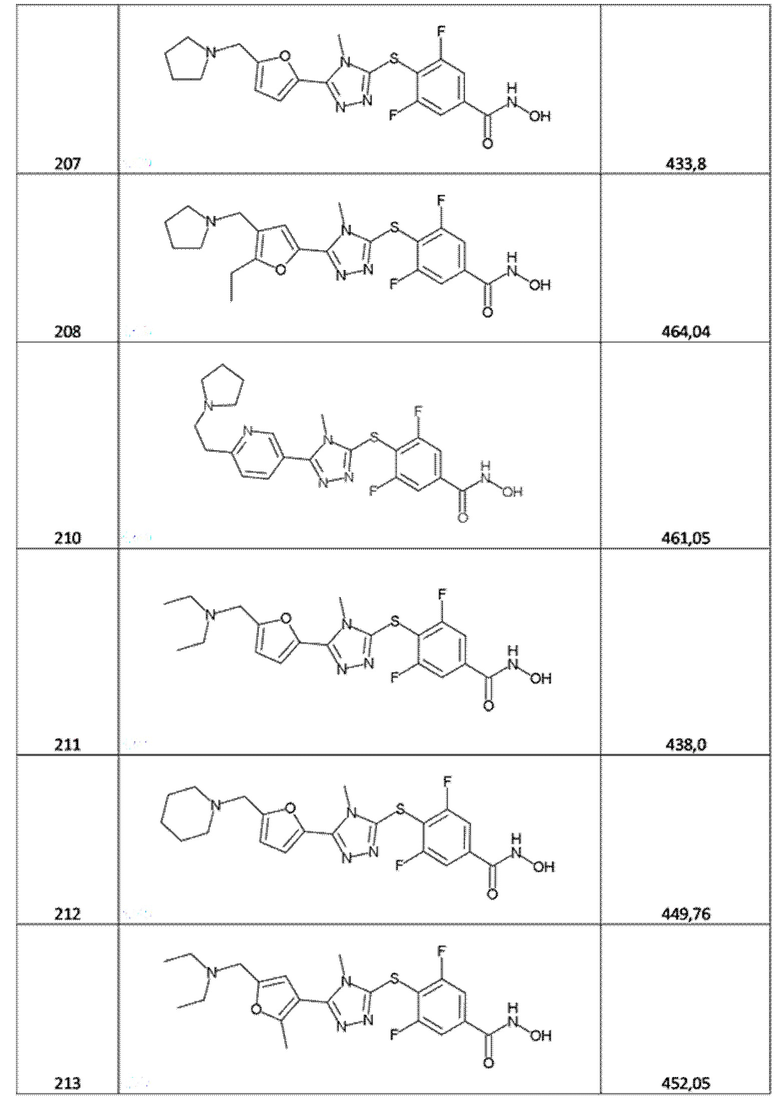
3,5-дифтор-4-[(5-пиримидин-2-илтетразол-1-ил)метил]бензолкарбогидроксамовая кислота (соед. 125);
4-[[5-[5-(трифторметил)-2-пиридил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 126);
4-[[5-[5-(трифторметил)-2-пиридил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 127);
3,5-дифтор-4-[[5-[5-(трифторметил)-2-пиридил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 128);
3,5-дифтор-4-[[5-[5-(трифторметил)-2-пиридил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 129);
4-[[5-[3-морфолинo-5-(трифторметил)-2-пиридил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 130);
4-[[5-[3-морфолинo-5-(трифторметил)-2-пиридил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 131);
4-[[5-(2-пиридилметил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота; 2,2,2-трифторуксусная кислота (соед. 132);
4-[[5-(2-пиридилметил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота; 2,2,2-трифторуксусная кислота (соед. 133);
3,5-дифтор-4-[[5-(2-пиридилметил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота; 2,2,2-трифторуксусная кислота (соед. 134);
3,5-дифтор-4-[[5-(2-пиридилметил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота; 2,2,2-трифторуксусная кислота (соед. 135);
3,5-дифтор-4-[[4-метил-5-[1-фенил-5-(2- тиенил)пиразол-3- ил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 136);
3,5-дифтор-4-[[5-(6-фтор-2-метил-3-хинолил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 137);
3,5-дифтор-4-[[5-(4-фторфенил)-4-(2-морфолинoэтил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 138);
3,5-дифтор-4-[[4-(2-фурилметил)-5-пиразин-2-ил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 139);
3,5-дифтор-4-[[4-(2-фурилметил)-5-(2-пиридил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 140);
4-[[4-бензил-5-(пирролидин-1-ил-метил)-1,2,4-триазол-3-ил]сульфанил]-3,5-дифтор-бензолкарбогидроксамовая кислота (соед. 141);
4-[[4-бензил-5-(2-фурил)-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 142);
4-[[4-бензил-5-(2-тиенил)-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 143);
3,5-дифтор-4-[[4-(2-фурилметил)-5-(2-тиенил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 144);
3,5-дифтор-4-[[5-(2-фторфенил)-4-(2-фурилметил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 145);
3,5-дифтор-4-[[4-(2-фурилметил)-5-(4-пиридил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 146);
3,5-дифтор-4-[[4-(2-фурилметил)-5-(3-пиридил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 147);
3,5-дифтор-4-[[5-(3-изохинолил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 148);
3,5-дифтор-4-[(5-имидазо[1,2-a]пиридин-3-ил-4-метил-1,2,4-триазол-3-ил)сульфанил]бензолкарбогидроксамовая кислота (соед. 149);
4-[[5-(1-бензил-4-фенил-4-пиперидил)-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 150);
3,5-дифтор-4-[[4-метил-5-[3-(4-метилпиперазин-1-ил)сульфонилфенил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 151);
4-[[5-[3-(4-бензилпиперазин-1-ил)сульфонилфенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 152);
3,5-дифтор-4-[[4-метил-5-(3-пиридил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 153);
метил-4-[[2-[[2,6-дифтор-4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]метил]бензоат (соед. 154);
метил-4-[[1-[[2,6-дифтор-4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]метил]бензоат (соед. 155);
метил-6-[2-[[4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]пиридин-3-карбоксилат (соед. 156);
метил-6-[1-[[4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]пиридин-3-карбоксилат (соед. 157);
4-[[2-[[4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]метил]бензойная кислота (соед. 158);
4-[[1-[[4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]метил]бензойная кислота (соед. 159);
4-[[2-[[2,6-дифтор-4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]метил]бензойная кислота (соед. 160);
4-[[1-[[2,6-дифтор-4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]метил]бензойная кислота (соед. 161);
6-[2-[[4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]пиридин-3-карбоновая кислота (соед. 162);
3-[2-[[4-(гидроксикарбамоил)фенил]метил]тетразол-5-ил]бензойная кислота (соед. 163);
3,5-дифтор-4-[[4-метил-5-(8-хинолилметил)-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 164);
4-[[5-(2,6-дифторфенил)-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифтор-бензолкарбогидроксамовая кислота (соед. 165);
3,5-дифтор-4-[[4-метил-5-[3-(4-метилпиперазин-1-ил)фенил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 166);
4-[[5-[3-(азепан-1-илметил)фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 167);
4-[[5-[4-(азепан-1-илметил)фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 168);
4-[[5-(4-аминофенил)тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 169);
4-[[5-(4-аминофенил)тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 170);
4-[[5-(4-аминофенил)тетразол-2-ил]метил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 171);
4-[[5-(4-аминофенил)тетразол-1-ил]метил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 172);
4-[[5-[4-(аминометил)фенил]тетразол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 173);
4-[[5-[4-(аминометил)фенил]тетразол-1-ил]метил]бензолкарбогидроксамовая кислота (соед. 174);
4-[[5-[4-(аминометил)фенил]тетразол-2-ил]метил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 175);
4-[[5-[4-(аминометил)фенил]тетразол-1-ил]метил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 176);
3,5-дифтор-4-[[4-метил-5-[1-(2-пиридил)циклопропил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 177);
3,5-дифтор-4-[[4-метил-5-[1-(3-пиридил)циклопропил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 178);
3,5-дифтор-4-[(4-метил-5-пиридазин-3-ил-1,2,4-триазол-3-ил)сульфанил]бензолкарбогидроксамовая кислота (соед. 179);
3,5-дифтор-4-[[5-(3-фтор-2-пиридил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 180);
3,5-дифтор-4-[[4-метил-5-[3-(1-пиперидилметил)фенил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 181);
3,5-дифтор-4-[[4-метил-5-[3-(морфолинoметил)фенил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 182);
4-((3-((1H-индол-3-ил)метил)-5-(тиофен-2-ил)-4H-1,2,4-триазол-4-ил)метил)-N-гидроксибензамид (соед. 183);
4-[[5-[3-[[бензил(метил)амино]метил]фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 184);
4-[[3-[(3,4-диметоксифенил)метил]-5-(2-тиенил)-1,2,4-триазол-4-ил]метил]бензолкарбогидроксамовая кислота (соед. 185);
3,5-дифтор-4-[[4-метил-5-[1-метил-1-(3-пиридил)этил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 186);
3,5-дифтор-4-[[5-[4-[метил(метилсульфонил)амино]фенил]-1,3,4-тиадиазол-2-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 187);
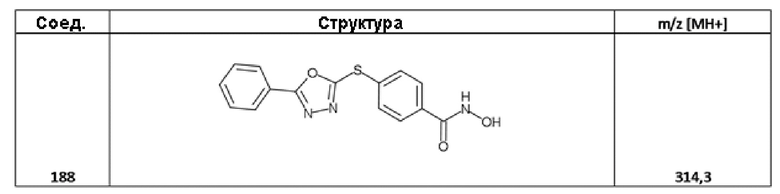
4-[(5-фенил-1,3,4-оксадиазол-2-ил)сульфанил]бензолкарбогидроксамовая кислота (соед. 188);
4-[(5-фенил-1,2,4-оксадиазол-3-ил)метил]бензолкарбогидроксамовая кислота (соед. 189);
4-[(5-фенил-1,3,4-тиадиазол-2-ил)метил]бензолкарбогидроксамовая кислота (соед. 190);
3,5-дифтор-N-гидрокси-4-((5-(пиридин-3-ил)-1,3,4-тиадиазол-2-ил)тио)бензамид (соед. 191);
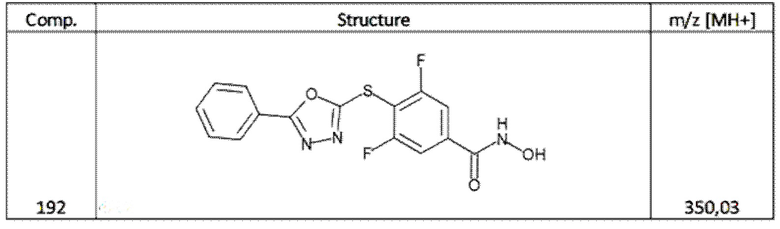
3,5-дифтор-4-[(5-фенил-1,3,4-оксадиазол-2-ил)сульфанил]бензолкарбогидроксамовая кислота (соед. 192);
4-[[5-(2-морфолинo-4-пиридил)-1,2,4-оксадиазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 193);
3,5-дифтор-N-гидрокси-4-((5-фенил-1,2,4-оксадиазол-3-ил)метил)бензамид (соед. 194);
3,5-дифтор-4-[[5-(4-пиридил)-1,3,4-тиадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 195);
4-[[5-(5-бром-3-пиридил)-1,3,4-тиадиазол-2-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 196);
3,5-дифтор-4-[[5-(5-морфолинo-3-пиридил)-1,3,4-тиадиазол-2-ил]метил]бензолкарбогидроксамовая кислота (соед. 197);
3,5-дифтор-N-гидрокси-4-((5-фенил-1,3,4-тиадиазол-2-ил)метил)бензамид (соед. 198);
3,5-дифтор-4-[[5-(2-фурил)-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 199);
4-[[5-[5-[бис(2-метоксиэтил)амино]-3-пиридил]-1,2,4-оксадиазол-3-ил]метил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 200);
3,5-дифтор-4-[[5-[5-(2-окса-6-азаспиро[3.3]гептан-6-ил)-3-пиридил]-1,2,4-оксадиазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 201);
3,5-дифтор-4-[[5-[5-(пирролидин-1-илметил)-2-фурил]-1,2,4-оксадиазол-3-ил]метил]бензолкарбогидроксамовая кислота (соед. 202);
3,5-дифтор-4-[[4-метил-5-[5-(морфолинoметил)-3-фурил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 203);
3,5-дифтор-4-[[4-метил-5-[5-(морфолинoметил)-2-фурил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 204);
3,5-дифтор-4-[[4-метил-5-[5-[(4-метилпиперазин-1-ил)метил]-2-фурил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 205);
4-[[5-[5-[(диметиламино)метил]-2-фурил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 206);
3,5-дифтор-4-[[4-метил-5-[5-(пирролидин-1-илметил)-2-фурил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 207);
4-[[5-[5-этил-4-(пирролидин-1-илметил)-2-фурил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 208);
4-[[4-метил-5-[5-[(4-метилпиперазин-1-ил)метил]-2-фурил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 209);
3,5-дифтор-4-[[4-метил-5-[6-(2-пирролидин-1-илэтил)-3-пиридил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 210);
4-[[5-[5-(диэтиламинометил)-2-фурил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 211);
3,5-дифтор-4-[[4-метил-5-[5-(1-пиперидилметил)-2-фурил]-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовая кислота (соед. 212);
4-[[5-[5-(диэтиламинометил)-2-метил-3-фурил]-4-метил-1,2,4-триазол-3-ил]сульфанил]-3,5-дифторбензолкарбогидроксамовая кислота (соед. 213);
4-[(5-фенилтетразол-2-ил)метил]бензолкарбогидроксамовая кислота (соед. 214);
4-[(5-фенилтетразол-1-ил)метил]бензолкарбогидроксамовая кислота (соед. 215);
4-[(5-фенил-4H-1,2,4-триазол-3-ил)метил]бензолкарбогидроксамовая кислота (соед. 216);
N-гидрокси-4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензамид (соед. 217).
Следующие соединения формул (I) и (II) являются особенно предпочтительными:


Соединения настоящего изобретения могут содержать один или несколько хиральных центров (асимметрических атомов углерода), поэтому они могут существовать в энантиомерной и/или диастереоизомерной формах.
Все возможные оптические изомеры, отдельно или в смеси друг с другом, входят в объем настоящего изобретения.
Соединения по изобретению могут применяться отдельно или в комбинации с другими лекарственными средствами, такими как ингибиторы протеасом, иммунохимические ингибиторы, стероиды, ингибиторы бромдомена и другие эпигенетические препараты, традиционные химиотерапевтические средства, ингибиторы киназы, такие как, например, без ограничения, семейство JAK, ингибиторы контрольных точек CTLA4, PD1 или PDL1, такие как ниволумаб, пемпролизумаб, пидилизумаб или BMS-936559 (анти-PD1), атезолизумаб или авелумаб (анти-PDL1), ипилимумаб или тремелимумаб (анти-CTLA4).
Соединения по изобретению, по отдельности или в комбинации, предпочтительно используются для лечения заболеваний, опосредованных HDAC6.
Соединения по изобретению отдельно или в комбинации предпочтительно пригодны для лечения отторжения трансплантата, GVHD, миозита, заболеваний, связанных с нарушением функции лимфоцитов, множественной миеломы, неходжкинской лимфомы, периферической нейропатии, аутоиммунных заболеваний, воспалительных заболеваний, рака и нейродегенеративных патологий, глазных болезней (например, увеита).
В связи с этим, настоящее изобретение также предлагает фармацевтические композиции, содержащие терапевтически эффективное количество соединений формулы (I) или (II) или их фармацевтически приемлемых солей, изомеров и фармакологически приемлемых пролекарств вместе с по меньшей мере одним фармацевтически приемлемым эксципиентом.
Такие композиции могут быть жидкими, подходящими для энтерального или парентерального введения, или твердыми, например, в форме капсул, таблеток, пилюль, порошков или гранул для перорального введения, или в формах, подходящих для накожного введения, таких как кремы или мази, или для ингаляционного поступления.
Фармацевтические композиции настоящего изобретения могут быть получены с использованием известных способов.
Общая схема синтеза
Соединения, описанные в настоящем изобретении, могут быть получены с использованием способов, известных специалистам в данной области.
Все исходные материалы, реагенты, кислоты, основания, растворители и катализаторы, используемые при синтезе описанных соединений, являются коммерчески доступными.
Ход реакции контролировали с помощью ВЭЖХ, СВЭЖХ или ВЭЖХ-МС анализа.
Соединения с триазолтиоловым ядром получали реакцией 1,2,4-триазолтиолов, необязательно замещенных метил-4-йодбензоатом или метил-3,4,5-трифторбензоатом, в присутствии карбоната калия в DMF при нагревании в течение ночи. Реакцию с метил-4-йодбензоатом катализировали йодидом меди и L-пролином (Схема 1) и нагревали при 120°С (Liang-Feng et al., Tetrahedron (2011), 67, 2878-2881). С другой стороны, реакция с метил-3,4,5-трифторбензоатом протекает даже в мягких условиях (55°С) и без катализа (Схема 2) (Dudutiene et al., Bioorg. Med. Chem. (2013), 21(7), 2093-2106; WO03/062225).
Такие же условия использовали для синтеза соединений с ядром 1,3,4-тиадиазол-2-тиола и 1,3,4-оксадиазол-2-тиола.
Превращения сложноэфирных производных в соответствующие гидроксамовые кислоты достигали путем обработки большим избытком водного гидроксиламина в основной среде (NaOH), в метаноле. Гидроксамовую кислоту также можно синтезировать путем гидролиза сложного метилового эфира с помощью NaOH и последующей конденсации с гидроксиламином, после активации HATU или другими связывающими реагентами.

Схема 1 - Синтез бензогидроксамовых производных с триазольным, тиадиазольным и оксадиазольным ядром
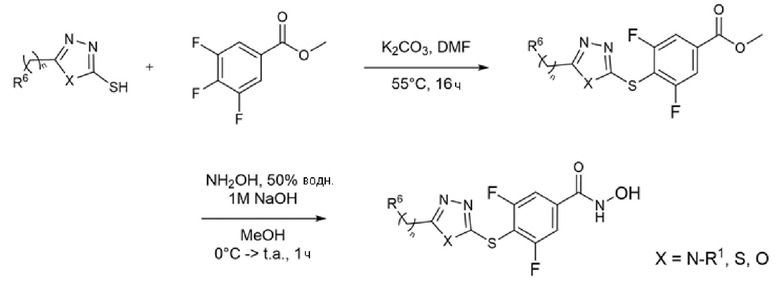
Схема 2 - Синтез 3,5-дифторбензогидроксамовых производных с триазольным, тиадиазольным и оксадиазольным ядром
Многие из исходных 1,2,4-триазолтиолов являются коммерчески доступными. В некоторых случаях их синтезировали в соответствии с двумя путями, показанными на Схеме 3. Открытое промежуточное соединение получали из карбоновой кислоты путем активации с помощью T3P и конденсации с N-замещенным гидразинкарботиоамидом в присутствии DIPEA в DMF (US 2007/0232808). Такое же промежуточное соединение получали, исходя из гидразида, который обрабатывали N-замещенным изотиоцианатом в этаноле при кипячении с обратным холодильником (Lei et al., ChemMedChem (2016), 11, 822-826; Nadjet et al., Molecules (2015), 20, 16048-16067). Циклизации открытого промежуточного соединения достигали добавлением водного NaOH в реакционную смесь.
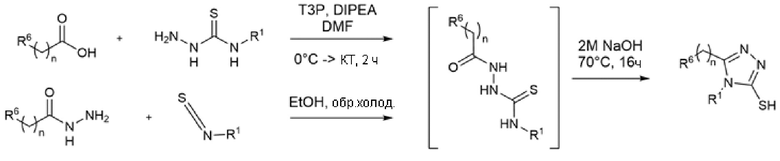
Схема 3 - Синтез 1,2,4-триазолтиолов
1,3,4-Тиадиазол-2-тиолы, которые не являются коммерчески доступными, синтезировали обработкой соответствующего гидразида КОН и CS2 при низкой температуре (0-5 °С) в течение 1 ч, и H2SO4 на второй стадии, как описано в Схеме 4.
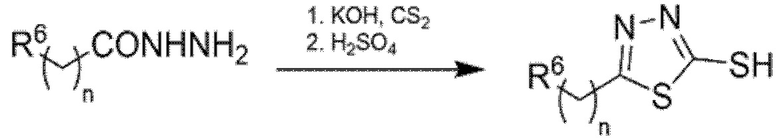
Схема 4 - Синтез 1,3,4-тиадиазолтиолов
Соединения с триазольным ядром получали, как описано в Схеме 5а, исходя из 2-(4-(метоксикарбонил)фенил) уксусной кислоты, реакцией с карбоксиимидамидом в присутствии HATU и DIPEA в DMF. После полного превращения исходных продуктов в промежуточное соединение к реакционной смеси добавляли замещенный гидразин и избыток уксусной кислоты. Образование триазольного цикла достигалось нагреванием смеси в течение ночи (Castanedo et al., J. Org. Chem. (2011), 76(4), 1177-1179).
Соединения с 1,3,4-тиадиазольным и 1,3,4-оксадиадиазольным каркасом также получали циклизацией открытого промежуточного соединения, полученного конденсацией 2-(4-(метоксикарбонил)фенил)уксусной кислоты или 2-(2,6-дифтор-4-(метоксикарбонил)фенил)уксусной кислоты с соответствующим гидразидом с помощью обычной активации HATU, DIPEA. Гидразиды были или коммерчески доступными, или могли быть легко получены из соответствующей карбоновой кислоты (Схема 5с). Реагент Лавессона использовали в качестве циклизующего агента для 1,3,4-тиадиазольных производных, в то время как то же самое промежуточное соединение циклизовали при обработке избытком реагентом Бургесса при кипячении с обратным холодильником в толуоле или THF, с получением 1,3,4-оксадиазолов (Схема 5b). Поскольку 2-(2,6-дифтор-4-(метоксикарбонил)фенил)уксусная кислота не была коммерчески доступной, ее синтезировали реакцией метил-3,4,5-трифторбензоата и ди-трет-бутилмалоната в присутствии гидрида натрия в безводном DMF. Полученный ди-трет-бутил-2-(2,6-дифтор-4-(метоксикарбонил)фенил)малонат затем декарбоксилировали обработкой с TFA при кипячении с обратным холодильником (Схема 5c).
Из-за более низкой реакционной способности 2-(2,6-дифтор-4-(метоксикарбонил)фенил)уксусной кислоты ее было необходимо активировать тионилхлоридом для достижения конденсации (Схема 5с).Превращения сложноэфирных производных в соответствующие гидроксамовые кислоты достигали путем гидроксиламинолиза, как уже описано в указанных выше случаях.
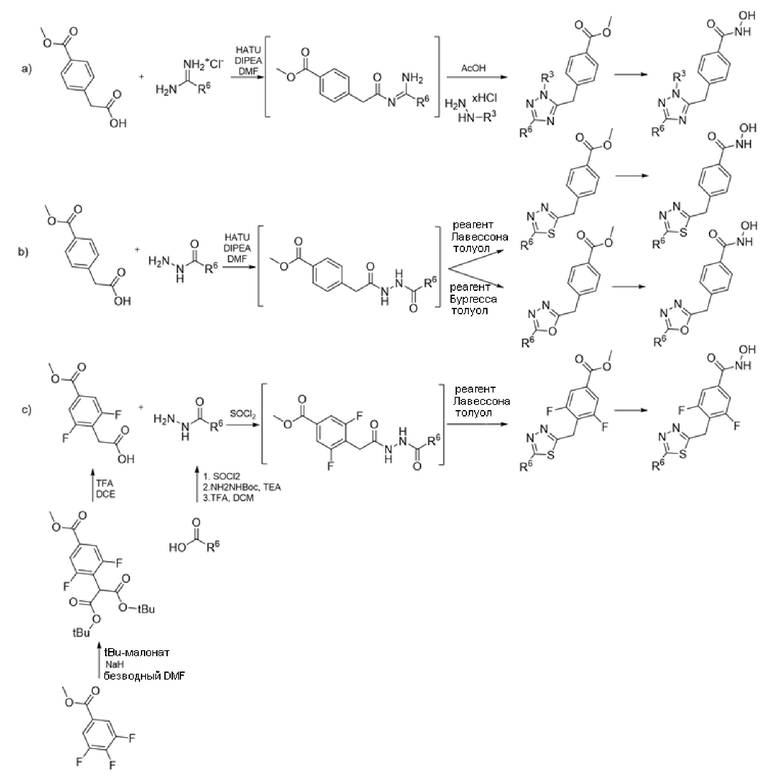
Схема 5 - Синтез бензогидроксамовых производных с триазольным, тиадиазольным и оксадиазольным ядром
Производные 1,3,4-оксадиазола использовали в качестве исходного материала для синтеза соединений, содержащих триазольное ядро. Конверсию получали нагреванием оксадиазола в THF в присутствии MeNH2, как описано в Схеме 6.
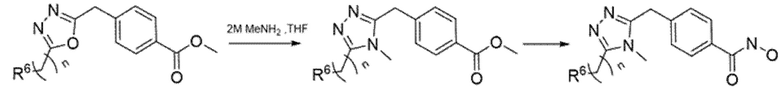
Схема 6 - Синтез 1,2,3-триазольных производных.
Соединения, содержащие 3,4,5-тризамещенный 1,2,4-триазол в качестве каркаса, получали, исходя из гидрохлорида метил-п-аминометилбензоата и соответствующего ацилхлорида в присутствии триметиламина. Полученный таким образом амид кипятили с обратным холодильником в тионилхлориде с образованием промежуточного имидоилхлорида, который давал целевой продукт при взаимодействии с соответствующим гидразидом и последующей циклизации в толуоле при кипячении с обратным холодильником (Схема 7). (WO2011106650 (A2) - 2011-09-01; Begum et al Med. Chem. Commun. 2015, 6, 80-89; Aster et al. Bioorg. Med. Chem. Lett. 2008, 18, 2799-2804.)

Схема 7- Синтез бензогидроксамовых производных с 3,4,5-тризамещенным 1,2,4-триазольным ядром
Соединения, содержащие тетразольный фрагмент, получали реакцией N-H-тетразола с метил-4-(хлорметил)бензойной кислотой или метил-4- (хлорметил)-3,5-дифторбензоатом в присутствии карбоната калия в ацетонитриле, при нагревании (Схема 8) (WO2012/106995).
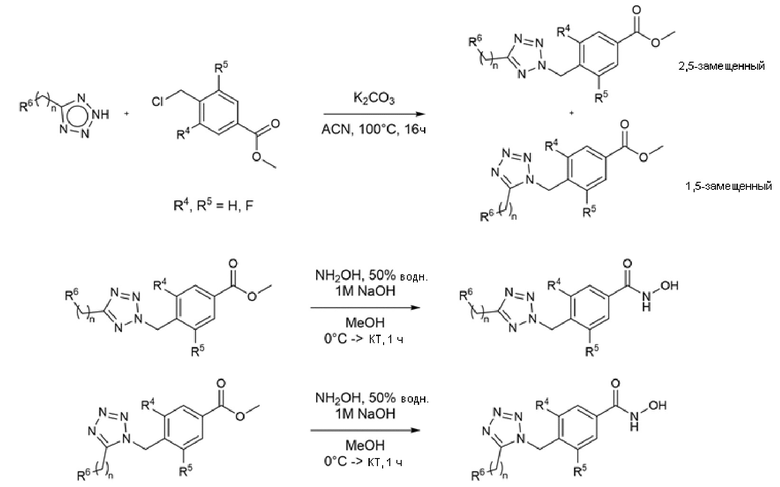
Схема 8 - Синтез бензогидроксамовых и 3,5-дифторбензогидроксамовых производных с тетразольным ядром
Региоселективность зависит от тетразольного субстрата, который обычно является 2,5-дизамещенным продуктом, в 2-10 раз более предпочтительным по сравнению с 1,5-дизамещенным продуктом. Региоизомеры, разделенные хроматографией на силикагеле, обрабатывали отдельно избытком гидроксиламина и водного гидроксида натрия с получением соответствующих гидроксамовых продуктов.
Некоторые из исходных N-Н-тетразолов являлись коммерчески доступными, тогда как другие синтезировали обработкой соответствующего нитрила азидом натрия и хлоридом аммония в DMF при нагревании (Схема 9).
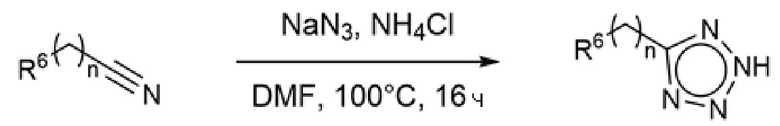
Схема 9 - Синтез N H-тетразолов
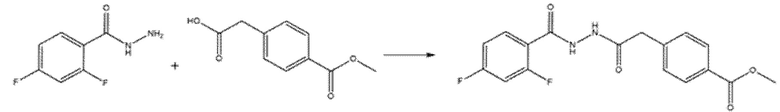
Соединения, содержащие 2-амино-1,3,4-оксадиазольный фрагмент, получали путем объединения ацилгидразида с метил-4-изоцианатобензоатом в THF при комнатной температуре (КТ) и кипячения с обратным холодильником только что образовавшегося промежуточного соединения в присутствии избытка реагента Бургесса (Схема 10) (Dolman et al., J. Org. Chem. (2006), 71(25), 9548).

Схема 10- Синтез бензогидроксамовых производных с 2-амино-1,3,4-оксадиазольным ядром
Превращения сложноэфирных соединений в гидроксамовую кислоту достигали, как уже описано в указанных выше случаях, путем гидроксиламинолиза.
Соединения с 1,2,4-оксадиазольным ядром синтезировали из 4-(цианометил)бензойной кислоты или из соответствующего сложного метилового эфира путем обработки гидрохлоридом гидроксиламина в присутствии избытка гидроксида калия или бикарбоната натрия при кипячении в этаноле с обратным холодильником (Схема 11). Полученную таким образом (Z)-4-(2-амино-2-(гидроксиимино)этил)бензойную кислоту затем подвергали реакции с подходящей карбоновой кислотой, предварительно активированной HATU и DIPEA или другими активаторами, с получением открытого промежуточного соединения, которое подвергали циклизации нагреванием при 100°С и в присутствии молекулярных сит или циклизующих агентов, таких как карбонилдиимидазол.

Схема 11- Синтез бензогидроксамовых производных с 1,2,4-оксадиазольным ядром
Превращение карбоновой кислоты в гидроксамовую кислоту может быть осуществлено любым способом, известным в данной области. Обычно ее получают активацией HATU, DCC или ацилхлоридом и реакцией активированного соединения с водным гидроксиламином. В некоторых случаях было необходимо конденсировать карбоновую кислоту с O-(тетрагидро-2H-пиран-2-ил)гидроксиламином с получением защищенной формы гидроксамовой кислоты, которая может высвобождаться при обработке TFA (Схема 12).
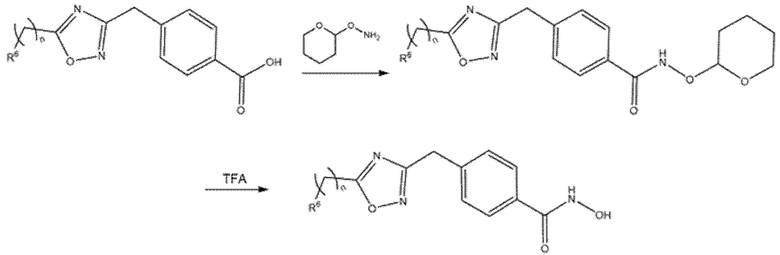
Схема 12- Превращение карбоновой кислоты в гидроксамовую кислоту через ее защищенную форму
Для синтеза соединений с 1,3,4-оксадиазольным ядром (схема 13) соответствующий гидразид получали взаимодействием соответствующей кислоты, активированной ацилхлоридом, с Boc-гидразином и последующим снятием защиты с помощью обработки TFA. Затем гидразид конденсировали с 2-(4-(метоксикарбонил)фенил)уксусной кислотой, предварительно активированной с помощью HATU и DIPEA. Циклизация открытого промежуточного соединения достигалась обработкой избытком реагента Бургесса в толуоле или ТГФ при кипячении с обратным холодильником.
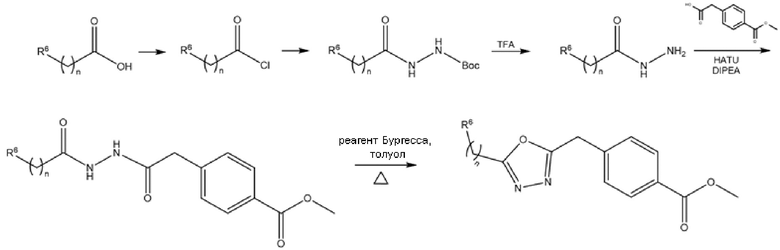
Схема 13- Синтез гидроксамовых производных с 1,3,4-оксадиазольным ядром
Как было показано ранее, конечное гидроксамовое производное можно получить гидроксиламинолизом сложного метилового эфира, путем реакции его с гидроксиламином в присутствии большого избытка гидроксида натрия.
Следующие примеры предназначены для дополнительной иллюстрации изобретения, но не для его ограничения.
ПРИМЕР 1 - Синтез (S)-N-(1-(3-(4-(гидроксикарбамоил)бензил)-1,2,4-оксадиазол-5-ил)-2-(тиазол-4ил)этил-3,4-диметоксибензамида (соед. 1)
Стадия А
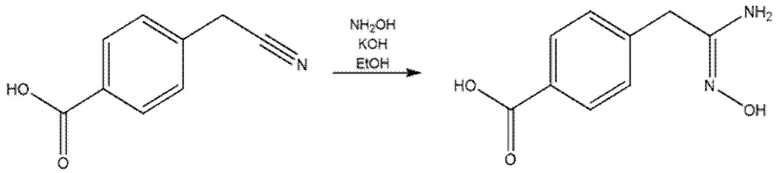
К раствору 4-(цианометил)бензойной кислоты (3,04 г, 1 экв.) в EtOH (250 мл) добавляли KOH (3,17 г, 3 экв.) и гидрохлорид гидроксилaмина (2,62 г, 2 экв.). Реакционную смесь кипятили с обратным холодильником в течение 20 ч. Затем раствор охлаждали, разбавляли водой (300 мл) и подкисляли до рН 6 с помощью конц. HCl. Осажденное белое твердое вещество отфильтровывали и высушивали под вакуумом при 50°С в течение ночи. Получали 2,6 г продукта, который использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия B

(S)-2-(N-Fmoc-амино)-3-(тиазол-4-ил)пропановую кислоту (2 г, 1 экв.) активировали обработкой HATU (2,5 г, 1,3 экв.) и DIPEA (1,4 мл) в DMA при комнатной температуре в течение 1 ч. Затем к реакционной смеси добавляли дополнительный DIPEA (1,4 мл) и (Z)-4-(2-амино-2-(гидроксиимино)этил)бензойную кислоту (985 мг, 1 экв.). После полного растворения исходных продуктов добавляли молекулярные сита, чтобы удалить образующуюся воду и способствовать циклизации открытого промежуточного соединения. Через два часа молекулярные сита удаляли фильтрованием и растворитель выпаривали при пониженном давлении. Остаток растворяли в метаноле. Отделяющееся белое твердое вещество удаляли фильтрованием. Растворитель частично выпаривали. Наблюдалось дополнительное осаждение белого твердого вещества, которое отфильтровывали. Раствор выпаривали досуха и остаток очищали флэш-хроматографией с обращенной фазой (С18) в градиенте H2O/ACN/TFA.
Стадия C
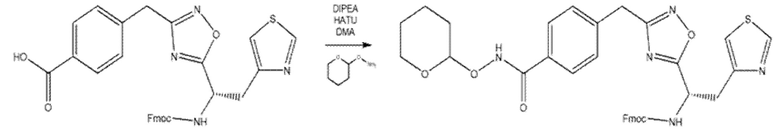
Кислоту, полученную на стадии B (82 мг, 1 экв.), активировали обработкой HATU (73 мг, 1,3 экв.) и DIPEA (41 µ 1,3 экв.) в DMF при комнатной температуре. Затем к реакционной смеси добавляли O-(тетрагидро-2H-пиран-2-ил)гидроксилaмин (17 мг, 1 экв.). После 2 ч перемешивания при комнатной температуре растворитель выпаривали в вакуумной центрифуге. Остаток использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия D
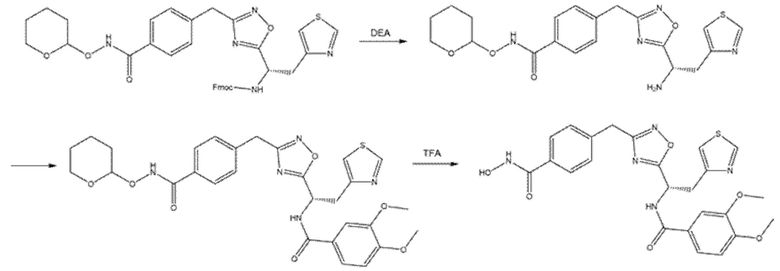
Продукт, полученный на стадии С, разбавляли в 1 мл THF и обрабатывали DEA (70 мкл, 4,5 экв.). После 4 ч перемешивания при 40°C растворитель и избыток DEA удаляли выпариванием при пониженном давлении. Остаток растворяли в 1 мл DMF и 3,4-диметоксибензойную кислоту (27 мг, 1 экв.), предварительно активированную HATU (74 мг, 1,3 экв.) и DIPEA (41 µ 1,3 экв.) в DMF (1 мл), добавляли в раствор. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Наконец, 0,4 мл TFA добавляли для снятия защиты с гидроксамовых функциональных групп. Через 4 ч растворитель и избыток TFA удаляли выпариванием и остаток очищали с помощью полупрепаративной ЖХМС (m/z 509,84 [MH+]).
Следующее соединение синтезировали с использованием той же самой методики:
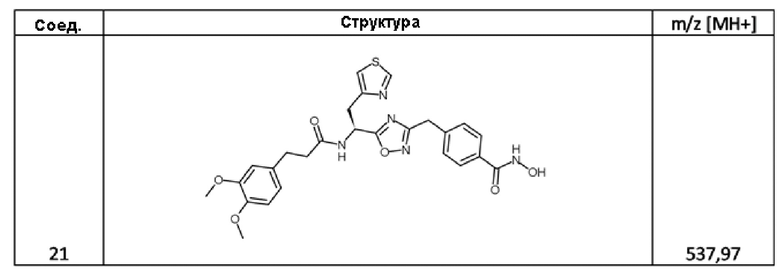
ПРИМЕР 2 - Синтез (S)-4-((5-(1-амино-2-(тиазол-4-ил)этил)-1,2,4-оксадиазол-3-ил)метил)-N-гидроксибензамида 2,2,2-трифторацетата (соед. 48)
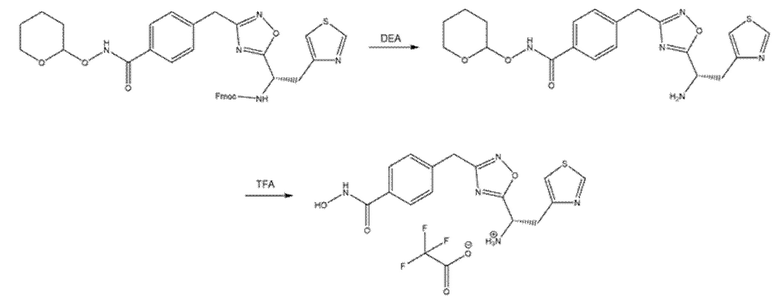
(9H-Флуорен-9-ил)метил((1S)-1-(3-(4-(((тетрагидро-2H-пиран-2-ил)окси)карбамоил)бензил)-1,2,4-оксадиазол-5-ил)-2-(тиазол-4-ил)этил)карбамат (полученный на стадии C синтеза соединения 1) (222 мг, 1 экв.) обрабатывали DEA (159 мкл, 4,5 экв.) в DMF (1 мл) в течение ночи при КТ. Затем к реакционной смеси добавляли 0,520 мл TFA (20 экв.). Растворитель удаляли выпариванием и остаток очищали в полупрепаративной ЖХМС (m/z 346,04 [MH+]).
Пример 3 - Синтез 4-[[5-(3,4-диметоксифенил)-1,2,4-оксадиазол-3-ил]метил]бензолкарбогидроксамовой кислоты (соед. 49)
Стадия А
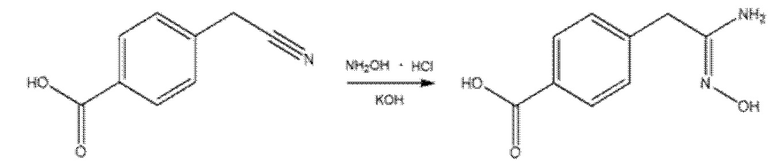
Смесь 4-(цианометил)бензойной кислоты (3 г, 1 экв.), гидрохлорида гидроксилaмина (2,6 г, 2 экв.) и гидроксида калия (3,2 г, 3 экв.) в этаноле (250 мл) нагревали с обратным холодильником в течение ночи. После охлаждения до КТ к реакционной смеси добавляли 300 мл воды и 15 мл 1 н. HCl (pH ≈ 5). Целевой продукт, полученный в виде осадка, отфильтровывали на перегородке из спеченного материала и сушили под вакуумом в течение ночи. Извлекали 320 мг чистого продукта.
Стадия B

(Z)-4-(2-амино-2-(гидроксиимино)этил)бензойную кислоту (319 мг, 1,1 экв.), полученную на стадии A, растворяли в толуоле (6 мл) и добавляли пиридин (3 мл). 3,4-Диметоксибензоилхлорид (300 мг, 1 экв.), полученный ранее путем реакции 3,4-диметоксибензойной кислоты с избытком тионилхлорида, добавляли к реакционной смеси. Реакционную смесь кипятили с обратным холодильником в течение 4 ч. Растворитель выпаривали при пониженном давлении и продукт очищали полупрепаративной ЖХМС.
Стадия C

4-((5-(3,4-диметоксифенил)-1,2,4-оксадиазол-3-ил)метил)бензойную кислоту (71 мг, 1 экв.), полученную на стадии B, активировали нагреванием с HATU (103 мг, 1,3 экв.) и DIPEA (47 мкл, 1,3 экв.) в DMF (1 мл) 30 мин при комнатной температуре. Затем к реакционной смеси добавляли гидрохлорид гидроксиламина (14 мг, 1 экв.) и дополнительное количество DIPEA (47 мкл, 1,3 экв.). После перемешивания при комнатной температуре в течение ночи растворитель удаляли выпариванием при пониженном давлении и остаток очищали полупрепаративной ЖХМС. Извлекали 33 мг чистого продукта (m/z 356,08 [MH+]).
Пример 4. Синтез 4-((5-(2,4-дифторфенил)-1,3,4-оксадиазол-2-ил)метил)-N-гидроксибензамида (соед. 58)
Стадия А

Раствор Boc-гидразина (150 мг, 1 экв.) в ACN (2 мл) и 95 мг NaHCO3 (1 экв.) добавляли к раствору 2,4-дифторбензоилхлорида (200 мг, 1 экв.) в ACN (3 мл). После 3 ч при КТ растворитель выпаривали в потоке воздуха. Остаток обрабатывали TFA в течение 3 ч. Кислоту удаляли в потоке воздуха, и остаток растворяли в EtOAc и промывали 2,5% раствором NaHCO3. Объединенные органические фазы сушили над Na2SO4, фильтровали и выпаривали досуха. Получали 159 мг продукта, который использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия B
HATU (439 мг, 1,3 экв.) и DIPEA (0,4 мл, 2,6 экв.) добавляли к раствору 2-(4-(метоксикарбонил)фенил)уксусной кислоты (224 мг, 1,3 экв.) в 5 мл THF). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч до полного растворения реагентов. Затем к смеси добавляли раствор 2,4-дифторбензогидразида (153 мг, 1 экв.) в THF (2 мл). Через 4 ч при КТ наблюдали полное превращение исходных реагентов в целевой продукт. Растворитель удаляли выпариванием в потоке воздуха. Остаток растворяли в H2O и образовавшийся осадок фильтровали через перегородку из спеченного материала. Продукт (149 мг) использовали на последующей стадии без какой-либо дополнительной очистки.
Стадия C

175 мг реагента Бургесса (1,72 экв.) добавляли к суспензии соединения, полученного на стадии B (149 мг, 1 экв.) в 5 мл сухого толуола, нагретого при кипячении с обратным холодильником. Через 1 ч наблюдали полное превращение исходного соединения в циклический продукт. Растворитель удаляли выпариванием под вакуумом. Остаток растворяли в DCM и промывали 1 н. HCl и H2O. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха. Извлекали 132,3 мг продукта, который использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия D

0,707 мл водного гидроксилaмина (60 экв.) добавляли к раствору соединения, полученного на стадии C (132 мг, 1 экв.) в 4 мл MeOH/THF. Медленно по каплям добавляли 1,998 мл 1 н. NaOH (5 экв). Примерно через 1 ч систему нейтрализовали добавлением 1 н. HCl (2 мл). Растворитель выпаривали под вакуумом и остаток разбавляли 2,5% раствором NaHCO3, фильтровали и промывали H2O. Твердое вещество суспендировали в Et2O и фильтровали. Получали 53 мг чистого продукта (m/z 332,01 [MH+]).
Следующие соединения синтезировали с использованием той же самой методики:
Пример 5. Синтез 4-[(5-фенил-1,3,4-оксадиазол-2-ил)амино]бензолкарбогидроксамовой кислоты (соед. 60)
Стадия А
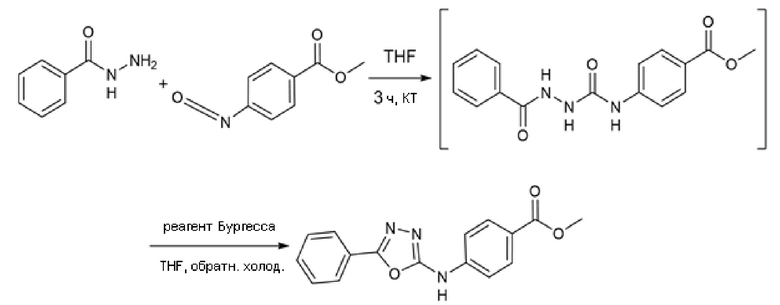
68 мг бензогидразида (1 экв.) и метил-4-изоцианoбензоат (88,5 мг, 1 экв.) смешивали в THF (5 мл) при комнатной температуре. Полученный раствор перемешивали в течение 3 ч. Промежуточное соединение верифицировали с помощью ВЭЖХ и ЖХМС. Растворитель удаляли выпариванием при пониженном давлении. Остаток растворяли в толуоле. Смесь кипятили с обратным холодильником и небольшими порциями добавляли реагент Бургесса (298 мг, 2,5 экв.) до полного превращения промежуточного соединения в циклический продукт. После охлаждения до комнатной температуры проводили промывку водой. Органическую фазу высушивали, фильтровали и выпаривали досуха. Продукт очищали кристаллизацией из DCM. Получали 172 мг чистого продукта (Dolman et al., J. Org. Chem. (2006), 71(25), 9548).
Стадия B
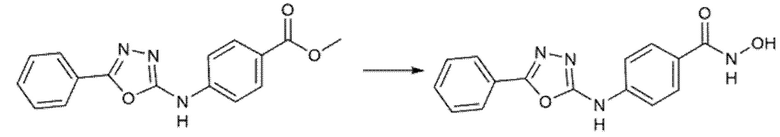
Полученный на стадии А сложный эфир (172 мг, 1 экв.) суспендировали в 4 мл метанола и реакционную смесь охлаждали на ледяной бане при 0°С и перемешивали магнитной мешалкой. После добавления гидроксиламина (50%, водный раствор, 1,365 мл, 40 экв.) медленно по каплям добавляли 1 М водный раствор гидроксида натрия (6 мл, 10 экв.). Ледяную баню убирали, позволяя раствору достичь комнатной температуры. Превращение исходного продукта в гидроксамовую кислоту подтверждали ВЭЖХ через 1 час. Метанольную часть удаляли выпариванием при пониженном давлении и затем реакцию гасили, добавляя 6 мл 1 М водного раствора HCl и 6 мл этилацетата. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (2×), насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Получали 26 мг чистого продукта (m/z 297,09 [MH+]).
Следующее соединение синтезировали с использованием той же самой методики:
Пример 6. Синтез 3,5-дифтор-N-гидрокси-4-((4-метил-5-(пиридин-2-ил)-4H-1,2,4-триазол-3-ил)тио)бензамида (соед. 14)
Стадия А
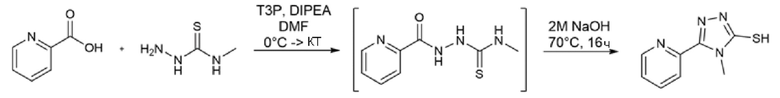
2-Пиридилкарбоновую кислоту (123 мг, 1 экв.) и 4-метил-3-тиосемикарбазид (116 мг, 1,1 экв.) суспендировали в 2 мл DMF и смесь охлаждали до 0°С с помощью ледяной бани. T3P (50% раствор DMF, 893 мкл, 1,5 экв.) и диизопропилэтиламин (310 мкл, 1,78 экв.) медленно добавляли к реакционной смеси при перемешивании. Ледяную баню удаляли, и смесь реагировала при комнатной температуре в течение 16 ч. Полное превращение исходного материала подтверждали с помощью ВЭЖХ. К реакционной смеси добавляли 2 мл этилацетата, 2 мл воды и 2 мл 4 М водного раствора NaOH. Фазы разделяли и органический слой повторно экстрагировали 4 М водным раствором NaOH. Объединенные водные фазы перемешивали 16 ч при 70°С. Превращение открытого промежуточного соединения в целевой продукт подтверждали с помощью ЖХМС. Величину pH реакционной смеси доводили до 5 добавлением по каплям конц. соляной кислоты при перемешивании. Осадок собирали фильтрованием.
Получали 157 мг продукта, который использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия B
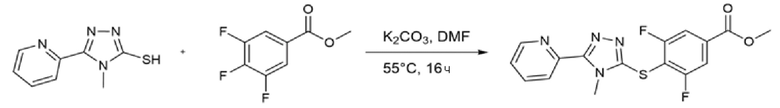
4-Метил-5-(пиридин-2-ил)-4H-1,2,4-триазол-3-тиол (157 мг, 1 экв.), метил-3,4,5-трифторбензоат (156 мг, экв.) и карбонат калия (261 мг, 2,3 экв.) суспендировали в 2 мл DMF в атмосфере аргона. Полученную смесь нагревали до 40°С и перемешивали в течение ночи.
Реакционную смесь разбавляли 10 мл этилацетата и 10 мл воды. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали.
Неочищенную реакционную смесь очищали флэш-хроматографией (Grace Reveleris X2, гексан:этилацетат). Получали 149 мг чистого продукта (Dudutiene et al., Bioorg. Med. Chem. (2013), 21(7), 2093-2106; международная патентная заявка WO03/062225).
Стадия C

Полученный на стадии В сложный эфир (149 мг, 1 экв.) суспендировали в 5 мл метанола и реакционную смесь охлаждали на ледяной бане при 0°С и перемешивали магнитной мешалкой. После добавления гидроксиламина (50%, водный раствор, 0,97 мл, 40 экв.) по каплям добавляли 1 М водный раствор гидроксида натрия (4,1 мл, 10 экв.). Ледяную баню убирали, позволяя раствору достичь комнатной температуры. Превращение исходного продукта в гидроксамовую кислоту подтверждали ВЭЖХ через 1 час. Метанольную часть удаляли выпариванием при пониженном давлении и затем реакцию гасили, добавляя 4,1 мл 1 М водного раствора соляной кислоты и 6 мл этилацетата. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (2×), насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Получали 113 мг чистого продукта (m/z 363,94 [MH+]).
Следующие соединения синтезировали с использованием данной методики:



Следующее соединение синтезировали с использованием данной методики, исходя из 2-меркапто-1,3,4-оксадиазола вместо 2-меркапто-1,3,4-триазола:
Пример 7. Синтез 4-[[5-[3-(диэтилсульфамоил)фенил]-4-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовой кислоты (соед. 66)
Стадия А

К раствору йодида меди (10 мг, 0,05 экв.), L-пролина (11 мг, 0,1 экв.) и карбоната калия (152 мг, 1,1 экв.) в 1 мл DMF в атмосфере аргона последовательно добавляли метил-4-йодбензоат (288 мг, 1,1 экв.) и N,N-диэтил-3-(5-меркапто-4-метил-4H-1,3,4-триазол-3-ил)бензолсульфонамид (326 мг, 1 экв.). Реакционную смесь нагревали при 120°C и перемешивали в течение ночи. Расход гетероароматического тиола наблюдали с помощью ВЭЖХ.
Реакционную смесь разбавляли 10 мл этилацетата и 10 мл воды. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали.
Неочищенный продукт очищали флэш-хроматографией (Grace Reveleris X2, гексан:этилацетат). Получали 236 мг продукта.
Стадия B
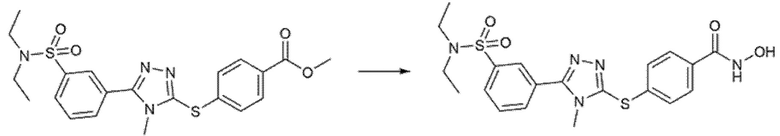
Полученный на стадии А сложный эфир (236 мг, 1 экв.) суспендировали в 15 мл метанола и реакционную смесь охлаждали на ледяной бане при 0°С и перемешивали магнитной мешалкой. После добавления гидроксиламина (50%, водный раствор, 1,2 мл, 40 экв.) по каплям добавляли 1 М водный раствор гидроксида натрия (4,1 мл, 10 экв.). Ледяную баню убирали, позволяя раствору достичь комнатной температуры. Превращение исходного продукта в гидроксамовую кислоту подтверждали ВЭЖХ через 1 час. Метанольную часть удаляли выпариванием при пониженном давлении и затем реакцию гасили, добавляя 4,1 мл 1 М водного раствора соляной кислоты и 6 мл этилацетата. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (2×), насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Получали 207 мг чистого продукта (m/z 432,00 [MH+]).
Следующие соединения синтезировали с использованием данной методики:

Следующее соединение синтезировали с использованием данной методики, исходя из 2-меркапто-1,3,4-оксадиазола вместо 2-меркапто-1,3,4-триазола:
Пример 8. Синтез 4-[[1-(2,4-дихлорфенил)-5-метил-1,2,4-триазол-3-ил]сульфанил]бензолкарбогидроксамовой кислоты (соед. 62)
Стадия А
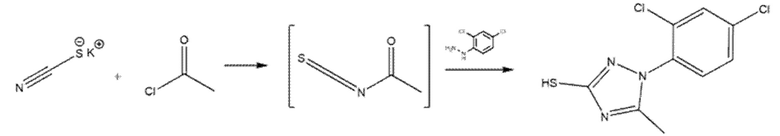
К раствору тиоцианата калия (194 мг, 1 экв.) в сухом ацетонитриле (6 мл) медленно добавляли ацетилхлорид (143 мкл, 1 экв.). Смесь кипятили с обратным холодильником в течение 1 ч, затем образовавшийся хлорид калия удаляли фильтрованием. К раствору добавляли (2,4-дихлорфенил)гидразин (427 мг, 1 экв.) и реакционную смесь нагревали с обратным холодильником. Через 1,5 ч анализ ЖХМС показал полное расходование гидразина. Реакционную смесь обильно разбавляли холодной водой (50 мл) и осажденное твердое вещество извлекали фильтрованием. Продукт очищали кристаллизацией из н-Hex/EtOAc 75:25. Извлекали 60 мг продукта.
Стадия B
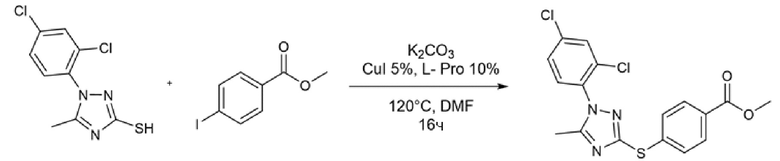
К раствору йодида меди (2 мг, 0,05 экв.), L-пролина (3 мг, 0,1 экв.) и карбоната калия (35 мг, 1,1 экв.) в 2 мл DMF в атмосфере аргона добавляли метил-4-йодбензоат (66,5 мг, 1,1 экв.) и 1-(2,4-дихлорфенил)-5-метил-1H-1,2,4-триазол-3-тиол (60 мг, экв.). Реакционную смесь нагревали при 120°C и перемешивали в течение ночи. Расход гетероароматического тиола наблюдали с помощью ВЭЖХ.
Реакционную смесь разбавляли 6 мл этилацетата и 6 мл воды. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия C

Полученный на стадии В сложный эфир (40 мг, 1 экв.) суспендировали в 6 мл метанола и реакционную смесь охлаждали на ледяной бане при 0°С и перемешивали магнитной мешалкой. После добавления гидроксиламина (50%, водный раствор, 236 мкл, 40 экв.) по каплям добавляли 1 М водный раствор гидроксида натрия (1 мл, 10 экв.). Ледяную баню убирали, позволяя раствору достичь комнатной температуры. Превращение исходного продукта в гидроксамовую кислоту подтверждали ВЭЖХ через 1 час. Метанольную часть удаляли выпариванием при пониженном давлении и затем реакцию гасили, добавляя 1 мл 1 М водного раствора соляной кислоты и 1 мл этилацетата. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (2×), насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Получали 30 мг чистого продукта (m/z 396,89 [MH+]).
Пример 9. Синтез 4-[[5-[(3,4-диметоксифенил)метил]-2-[4-(трифторметил)фенил]-1,2,4-триазол-3-ил]метил]бензолкарбогидроксамовой кислоты (соед. 44)
Стадия А
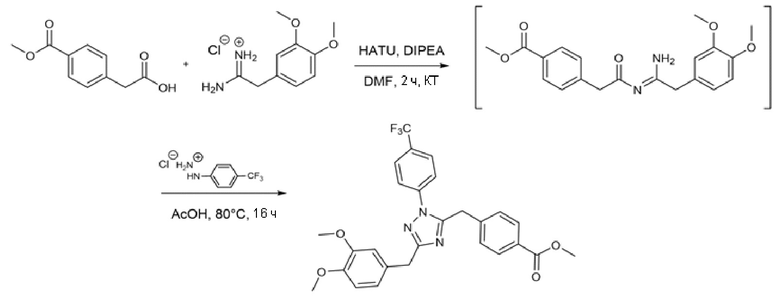
Во флакон с завинчивающейся крышкой загружали 2-(4-(метоксикарбонил)фенил)уксусную кислоту (97 мг, 0,5 ммоль), 1-амино-2-(3,4-диметоксифенил)этан-1-иминогидрохлорид (200 мг, 1,73 экв) и HATU (209 мг, 1,1 экв). 2 мл DMF и DIPEA (248 мкл, 3 экв.) добавляли последовательно в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре и проверяли ВЭЖХ расход карбоновой кислоты и образование промежуточного ациламидина. Полное превращение в промежуточное соединение наблюдали в пределах 2-3 ч.
Затем к реакционной смеси добавляли гидрохлорид (4-(трифторметил)фенил)гидразина (187 мг, 1,76 экв.) и уксусную кислоту (286 мкл, 10 экв.). Флакон закрывали и смесь нагревали до 80°С и перемешивали в течение ночи.
Потребление промежуточного ациламидина наблюдали с помощью ВЭЖХ.
Смеси давали возможность нагреться до комнатной температуры, затем разбавляли этилацетатом и последовательно промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором. Органический слой высушивали над сульфатом натрия, фильтровали и концентрировали досуха.
Продукт очищали флэш-хроматографией (гексан:этилацетат) (Castanedo et al., J. Org. Chem. (2011), 76(4), 1177-1179).
Стадия B
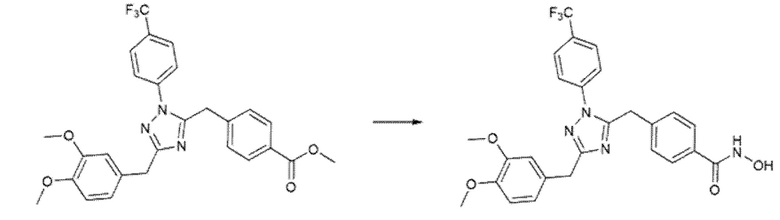
Полученный на стадии А сложный эфир (82 мг, 1 экв.) суспендировали в 5 мл метанола и полученную реакционную смесь охлаждали на ледяной бане при 0°С и перемешивали магнитной мешалкой. После добавления гидроксиламина (50%, водный раствор, 189 мкл, 20 экв.) медленно по каплям добавляли 1 М водный раствор гидроксида натрия (1,6 мл, 10 экв.). Ледяную баню убирали, позволяя раствору достичь комнатной температуры. Превращение исходного продукта в гидроксамовую кислоту подтверждали ВЭЖХ через 1 час. Метанольную часть удаляли выпариванием при пониженном давлении и затем реакцию гасили, добавляя 1,6 мл 1 М водного раствора соляной кислоты и 3 мл этилацетата. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (2×), насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Извлекали 27 мг чистого продукта (m/z 513,18 [MH+]).
Следующие соединения синтезировали с использованием данной методики:

Пример 10. Синтез 4-((5-(фуран-2-ил)-2H-тетразол-2-ил)метил)-N-гидроксибензамида (соед. 12)
Стадия А

Фуран-2-карбонитрил (500 мг, 1 экв.) растворяли в 10 мл DMF. Азид натрия (770 мг, 2,2 экв.) и хлорид аммония (631 мг, 2,2 экв.) добавляли в реакционную смесь при комнатной температуре при перемешивании магнитной мешалкой. Суспензию нагревали при 120°C и перемешивали в течение ночи. Полное превращение исходного материала наблюдали с помощью ВЭЖХ.
Смесь охлаждали до 0°С на ледяной бане, разбавляли 10 мл воды и подкисляли 1 М водным раствором соляной кислоты. Образовавшийся осадок собирали фильтрованием и дважды промывали водой перед сушкой под вакуумом. Получали 720 мг продукта (международная патентная заявка WO2006/003096).
Стадия B
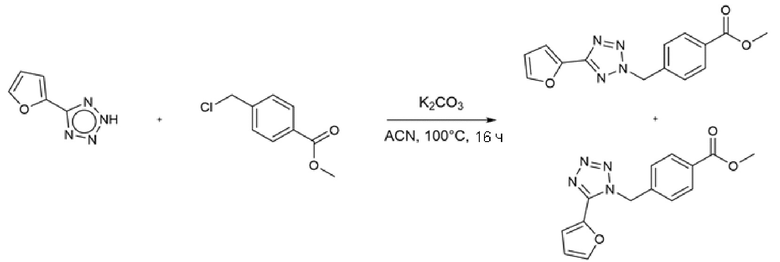
В реакционный сосуд загружали карбонат калия (742 мг, 1 экв.) и 5 мл ацетонитрила. Тетразол, полученный на стадии А (364 мг, 1 экв.), добавляли в виде твердого вещества при перемешивании магнитной мешалкой при комнатной температуре, в то время как метил-4-хлорметилбензоат (1,1 экв.) добавляли в виде раствора в 5 мл ацетонитрила. Смесь нагревали при 100°C и перемешивали в течение ночи. Полное превращение исходного материала в два региоизомерных продукта проверяли ЖХМС. Нерастворимый материал удаляли фильтрованием и фильтрат выпаривали при пониженном давлении. Два региоизомера выделяли колоночной хроматографией на силикагеле (толуол:этилацетат). Получали 384 мг 2,5-дизамещенного изомера и 234 мг 1,5-дизамещенного изомера (международная патентная заявка WO 2012/106995).
Стадия C
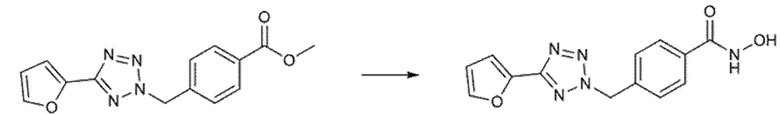
Полученный на стадии В сложный эфир (100 мг, 1 экв.) суспендировали в 10 мл метанола и полученную реакционную смесь охлаждали на ледяной бане при 0°С и перемешивали магнитной мешалкой. После добавления гидроксиламина (50%, водный раствор, 700 мкл, 30 экв.) по каплям добавляли 1 М водный раствор гидроксида натрия (3,52 мл, 10 экв.). Ледяную баню убирали, позволяя раствору достичь комнатной температуры. Превращение исходного продукта в гидроксамовую кислоту подтверждали ВЭЖХ через 1 час. Метанольную часть удаляли выпариванием при пониженном давлении и затем реакцию гасили, добавляя 3,52 мл 1 М водного раствора соляной кислоты и 6 мл этилацетата. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (2×), насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Извлекали 93,5 мг чистого продукта (m/z 286,02 [MH+]).
Следующие соединения синтезировали с использованием данной методики:

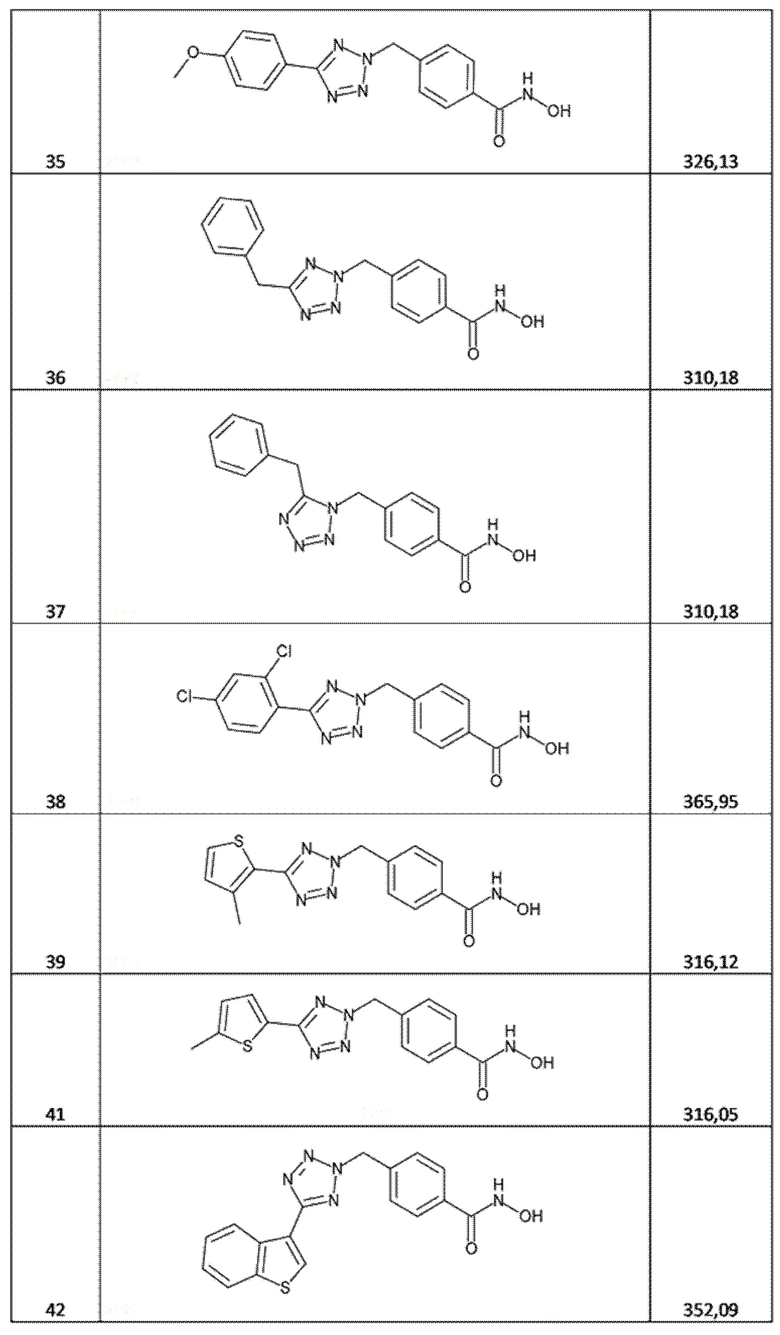
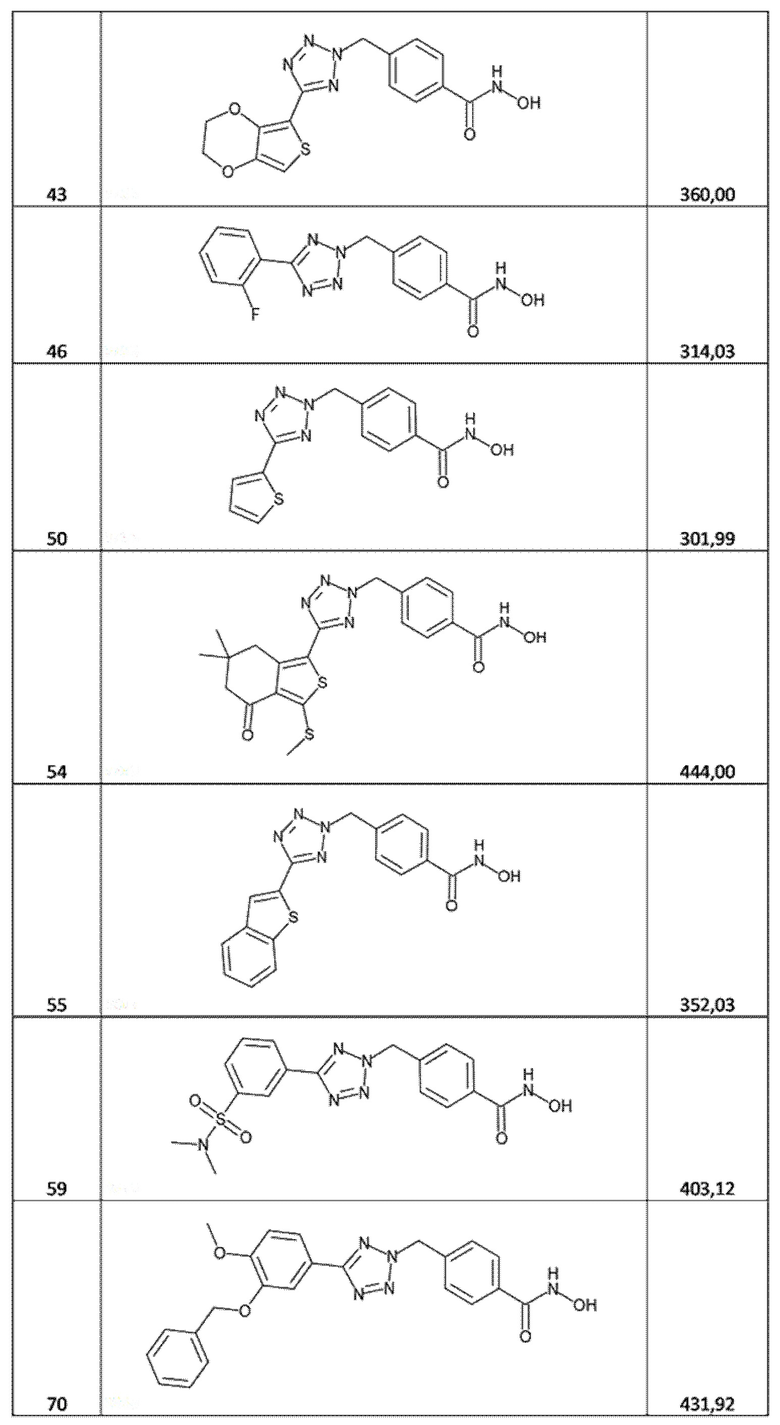
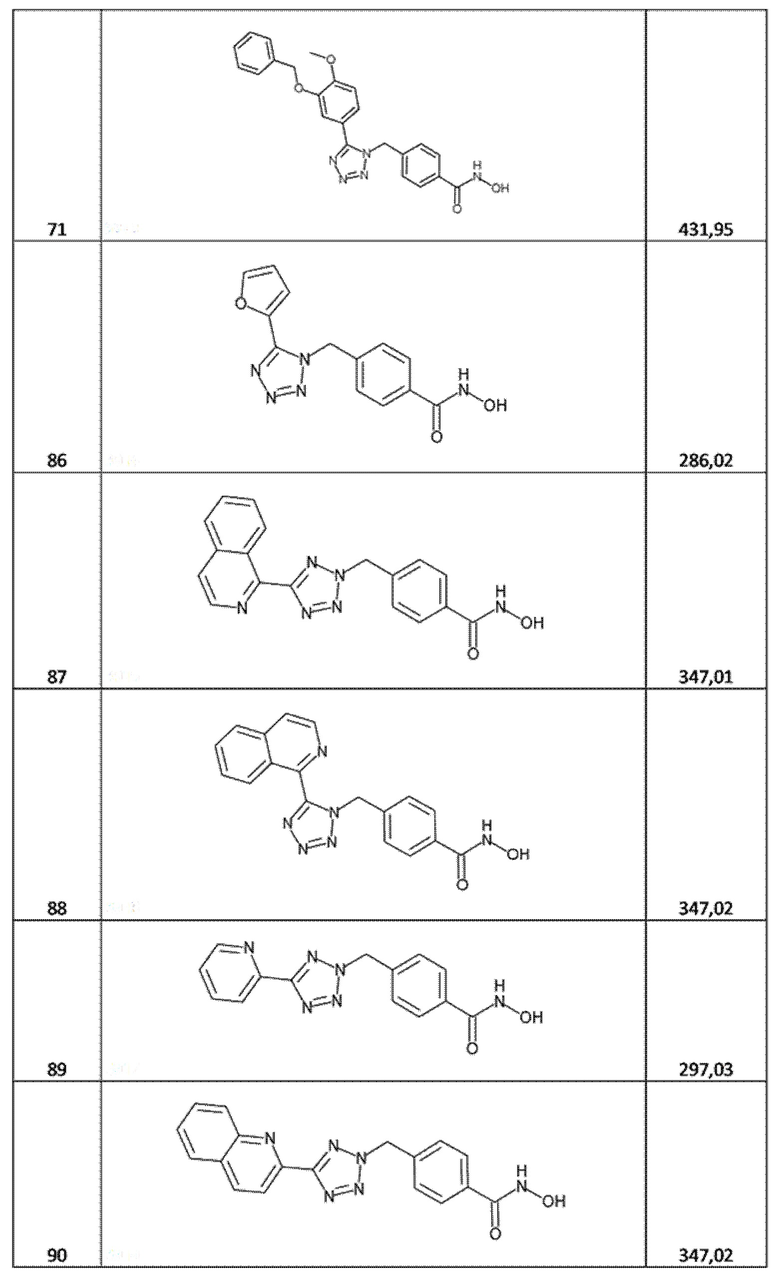
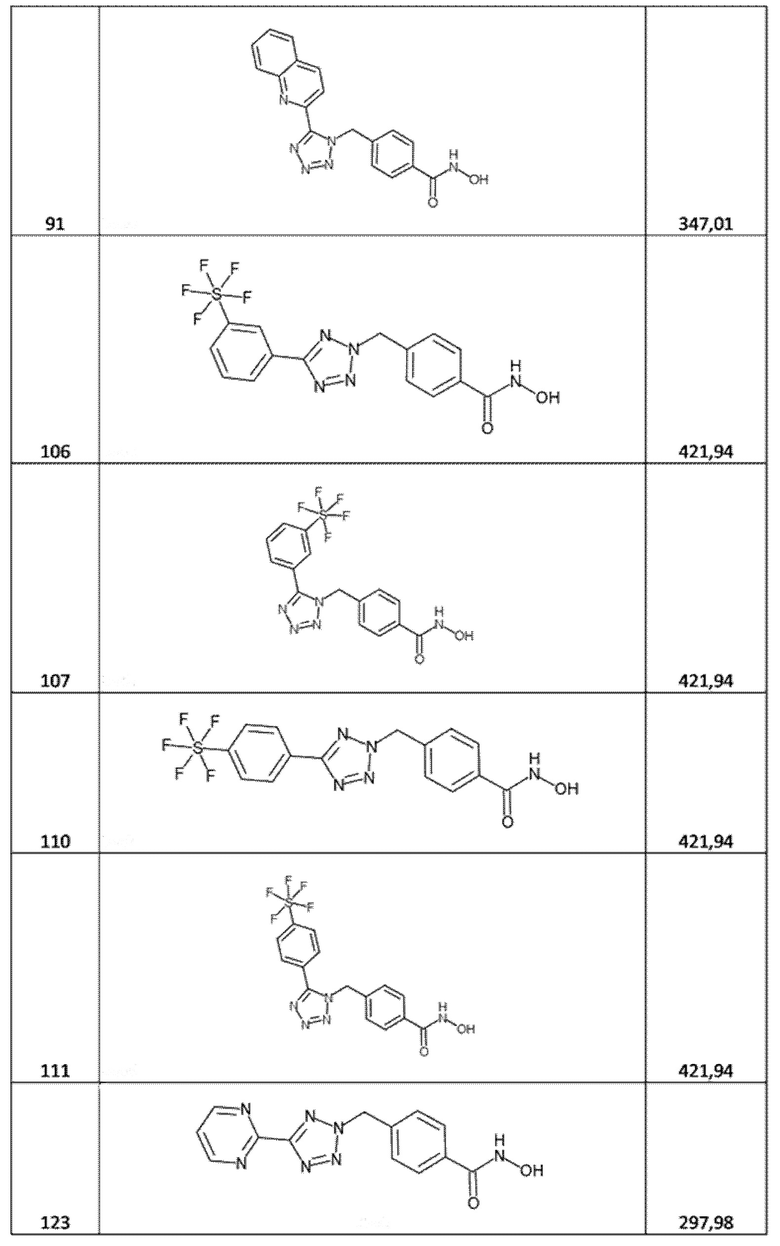
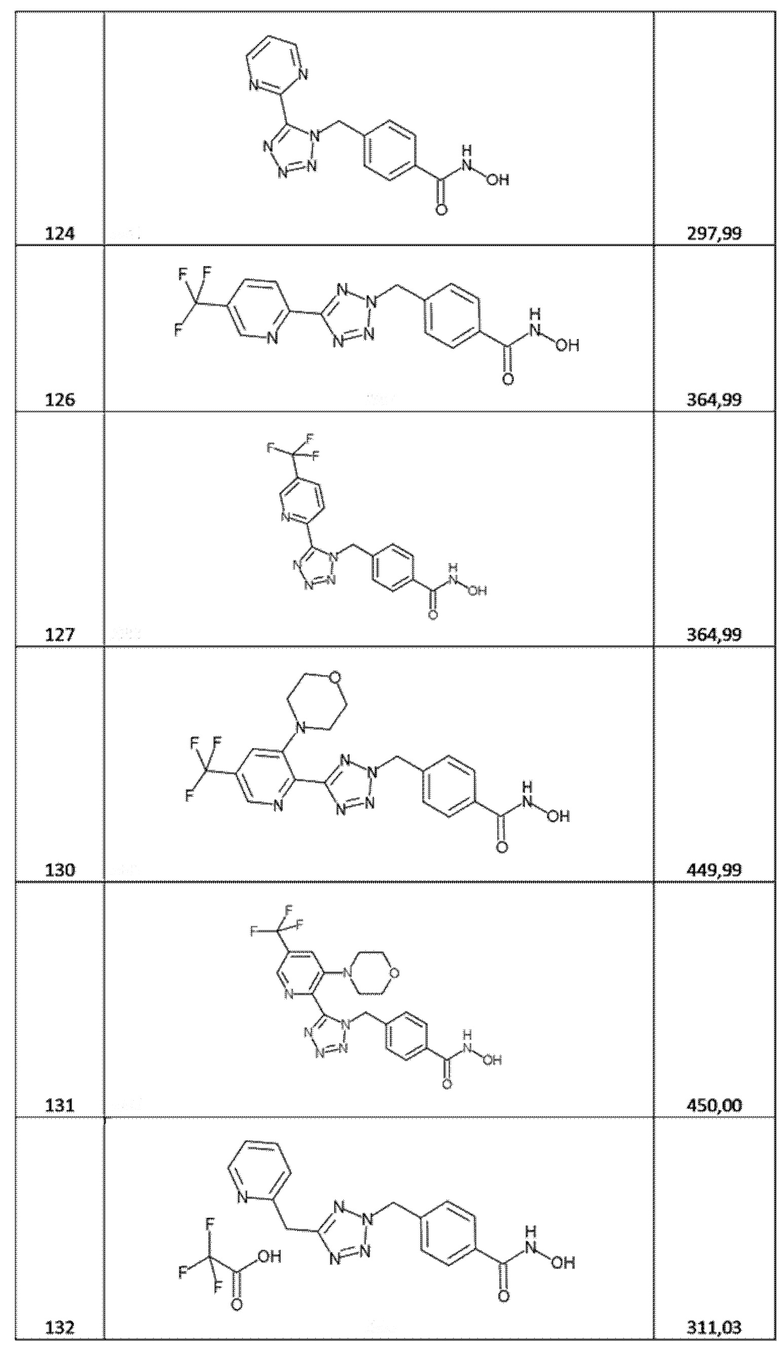

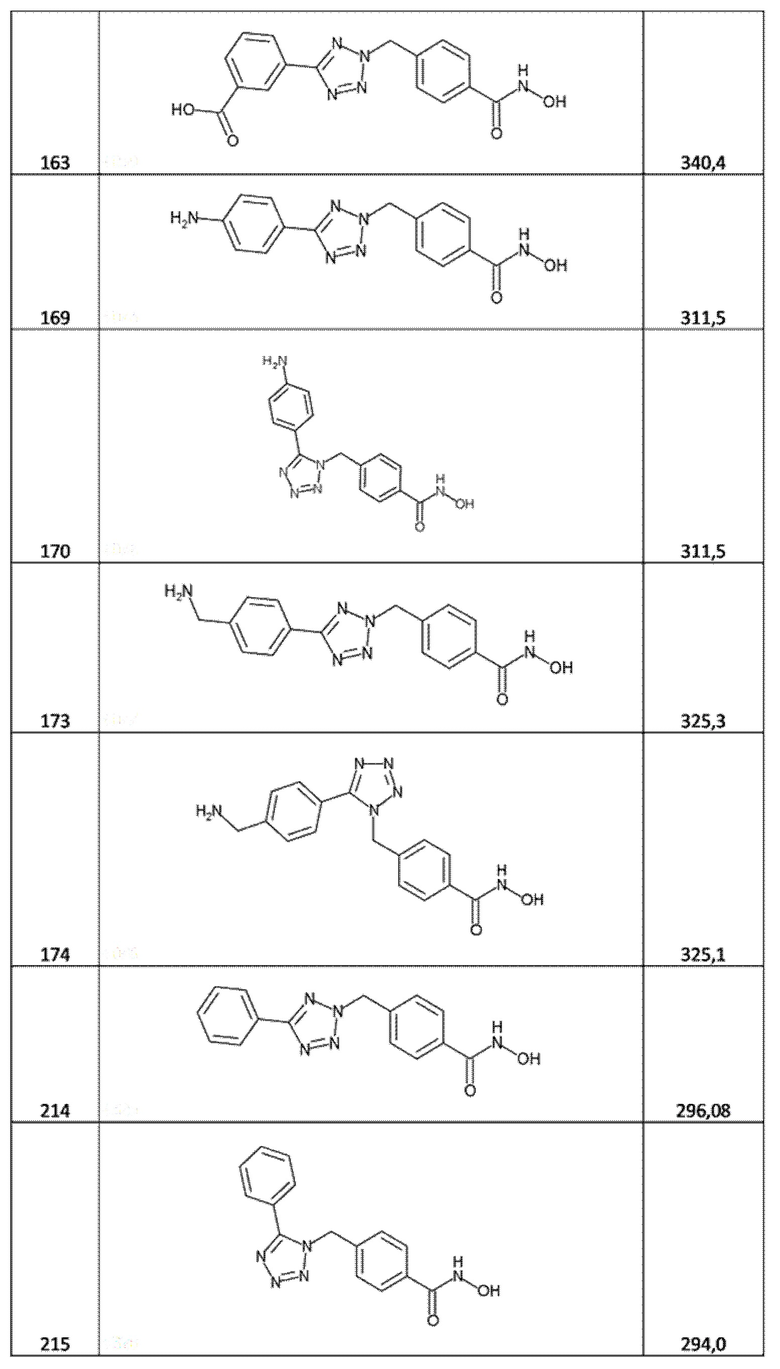
Пример 11. Синтез 4-((5-(2,3-дигидротиено[3,4-b][1,4]диоксин-5-ил)-1H-тетразол-1-ил)метил)-3,5-дифтор-N-гидроксибензамида (соед. 5)
Стадия А

В реакционный сосуд загружали карбонат калия (85 мг, 1 экв.) и 2 мл ацетонитрила. Тетразол (105 мг, 1 экв.), добавляли в виде твердого вещества при перемешивании магнитной мешалкой при комнатной температуре, тогда как метил-3,5-дифтор-4-хлорметилбензоат (122,3 мг, 1,1 экв) добавляли в виде раствора в 2 мл ацетонитрила. Смесь нагревали при 100°C и перемешивали в течение ночи. Полное превращение исходного материала в два региоизомерных продукта проверяли ЖХМС. Нерастворимый материал удаляли фильтрованием и фильтрат выпаривали при пониженном давлении. Два региоизомера выделяли колоночной хроматографией на силикагеле (толуол:этилацетат). Получали 23 мг 2,5-дизамещенного изомера и 52 мг 1,5-дизамещенного изомера (международная патентная заявка WO 2012/106995).
Стадия B

Полученный на стадии А сложный эфир (52 мг, 1 экв.) суспендировали в 2 мл метанола и полученную реакционную смесь охлаждали на ледяной бане при 0°С и перемешивали магнитной мешалкой. После добавления гидроксиламина (50%, водный раствор, 311 мкл, 40 экв.) по каплям добавляли 1 М водный раствор гидроксида натрия (1,3 мл, 10 экв.). Ледяную баню убирали, позволяя раствору достичь комнатной температуры. Превращение исходного продукта в гидроксамовую кислоту подтверждали ВЭЖХ через 1 час. Метанольную часть удаляли выпариванием при пониженном давлении и затем реакцию гасили, добавляя 1,3 мл 1 М водного раствора соляной кислоты и 2 мл этилацетата. Фазы разделяли и водный слой повторно экстрагировали дополнительным количеством этилацетата (3×). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (2×), насыщенным солевым раствором (2×), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Извлекали 32 мг чистого продукта (m/z 395,91 [MH+]).
Следующие соединения синтезировали с использованием данной методики:
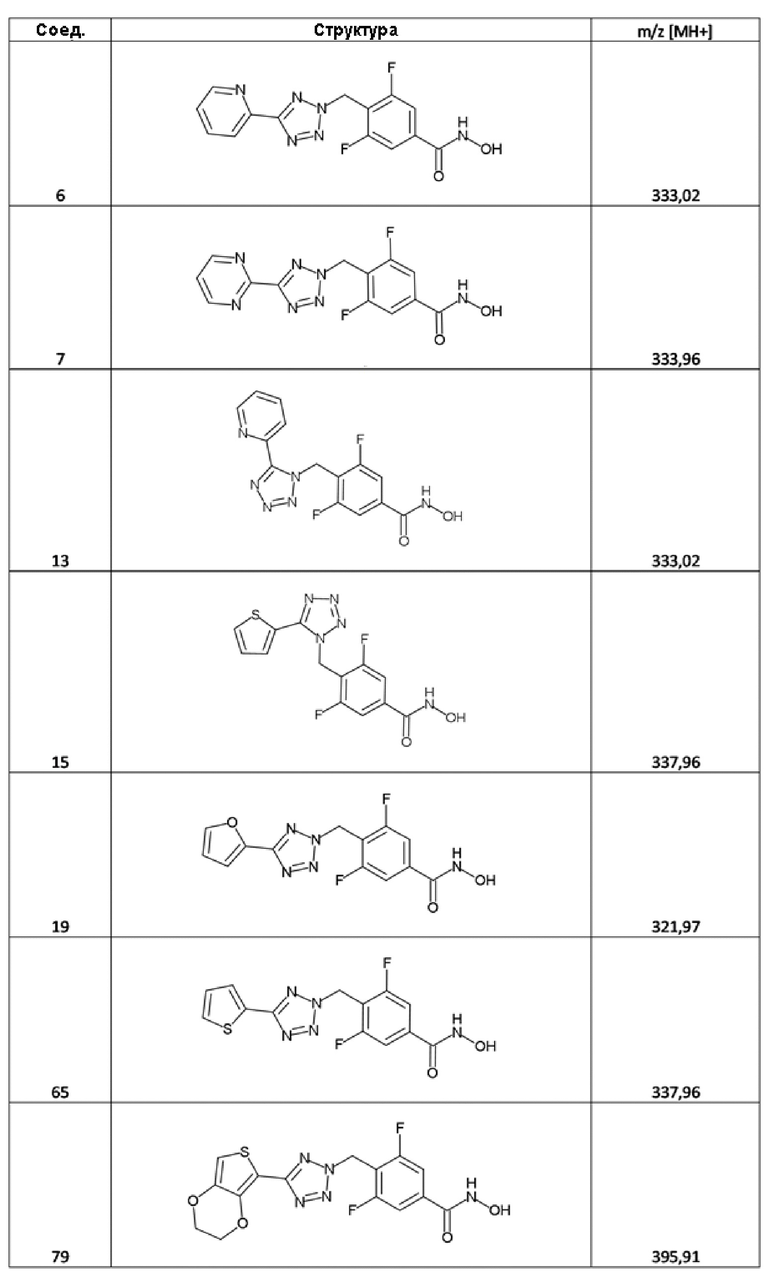



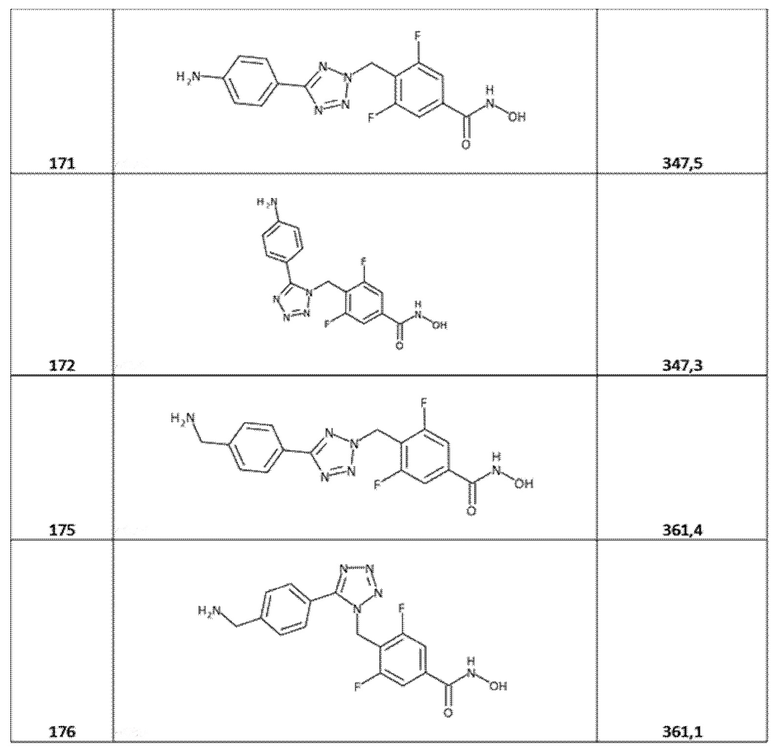
Пример 12 - Синтез 3,5-дифтор-N-гидрокси-4-((5-(пиридин-3-ил)-1,3,4-тиадиазол-2-ил)тио)бензамида (соед. 191)
Стадия А
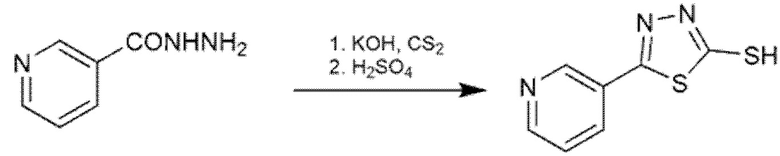
КОН (1,48 г, 26,47 ммоль, 1,1 экв.) растворяли в 45 мл безводного этанола. Добавляли гидразид (3,30 г, 24,06 ммоль, 1 экв.) и реакционную смесь охлаждали до 0-5 °C. По каплям добавляли CS2 (1,66 мл, 27,67 ммоль, 1,15 экв.) и реакционную смесь перемешивали при 0-5 °C в течение 1 ч. Полученный осадок собирали, промывали холодным ацетоном и сушили, получая 5,50 г желтого твердого вещества. Полученное промежуточное соединение небольшими порциями добавляли к 25 мл серной кислоты, охлажденной до 0-5 °С. Через 1 ч при 0-5 °С реакционную смесь выливали в ледяную воду и полученный осадок собирали, промывали водой и сушили.
Стадия B
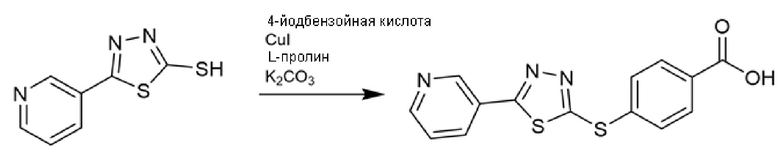
Смесь 5-(пиридин-3-ил)-1,3,4-тиадиазол-2-тиола, полученного на стадии A (0,8 г, 4,1 ммоль, 1 экв.), 4-йодбензойной кислоты (1,22 г, 4,92 ммоль, 1,2 экв.), L-пролина (0,047 г, 0,4 ммоль, 0,1 экв.) и K2CO3 (2,26 г, 16,4 ммоль, 4 экв.) в 20 мл безводного DMF дегазировали и добавляли CuI (0,039 г, 0,2 ммоль, 0,05 экв.). Реакционный сосуд герметично закрывали и реакционную смесь перемешивали при 120°С в течение 48 ч. Полное превращение исходного тиола контролировали с помощью ЖХМС. Реакционную смесь выливали в 150 мл воды и фильтровали через слой целита. Фильтрат подкисляли HCl. Образовавшийся осадок отфильтровывали и промывали последовательно водой, ацетонитрилом и простым диэтиловым эфиром.
Стадия C
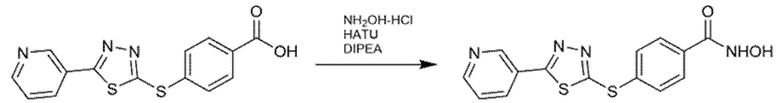
HATU (0,181 мг, 0,476 ммоль, 1,5 экв.) добавляли к раствору карбоновой кислоты, полученному на стадии B (0,1 г, 0,317 ммоль, 1 экв.), и DIPEA (0,333 мл, 1,902 ммоль, 6 экв.) в 2 мл безводного DMF. Реакционную смесь перемешивали при комнатной температуре и контролировали ЖХМС на полное превращение кислоты в промежуточное соединение с HATU: через 1 ч превращение было завершено. Добавляли NH2OH-HCl (0,066 г, 0,951 ммоль, 3 экв.) и реакционную смесь перемешивали в течение еще 2 ч. Реакцию контролировали ЖХМС. Реакционную смесь разбавляли водой до 50 мл общего объема и экстрагировали EtOAc (225 мл). После выпаривания получали 101 мг очень вязкого оранжевого масла. Затирание с ацетонитрилом (обработка ультразвуком ~ 15 мин) приводило к образованию осадка, который собирали фильтрованием, промывали ацетонитрилом и простым диэтиловым эфиром и сушили. Получали 40 мг чистого продукта (m/z 366,99 [MH+]). ЖХМС: 94,5%. ЯМР: OK.
Следующие соединения синтезировали с использованием данной методики:

Пример 13 - Синтез 3,5-дифтор-N-гидрокси-4-((5-фенил-1,3,4-тиадиазол-2-ил)метил)бензамида (соед. 198)
Стадия А

Трет-бутилмалонат (11,4 г, 52,73 ммоль, 2 экв.) добавляли по каплям в суспензию NaH (1,5 экв.) в 70 мл безводного DMF. После 5 мин перемешивания при КТ добавляли метил-3,4,5-трифторбензоат (5 г, 26,3 ммоль, 1 экв). Реакционную смесь перемешивали в течение 3 ч при КТ (наблюдали образование белого осадка), разбавляли водой и экстрагировали EtOAc. После концентрирования остаток очищали колоночной хроматографией. Получали 11,0 г неразделимой смеси продукта и трет-бутилмалоната в соотношении 1:3 (по ЯМР). Данную смесь использовали на следующей стадии без дальнейшей очистки.
Стадия B
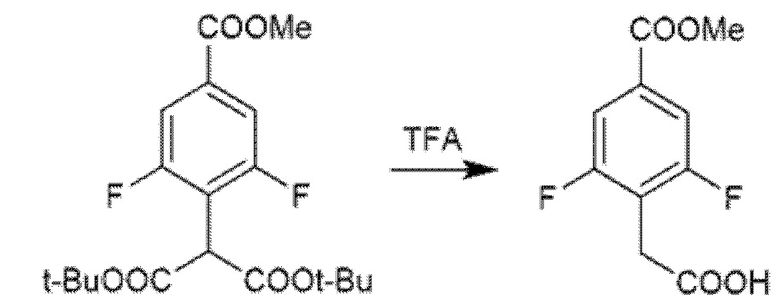
Смесь, полученную на стадии A (8,6 г, 22 ммоль, 1 экв.) и TFA (17 мл, 10 экв.), растворяли в 10 мл безводного DCE и кипятили с обратным холодильником в течение ночи. После охлаждения растворитель выпаривали, остаток обрабатывали гексаном и собирали образовавшийся осадок. ЯМР-анализ осадка и фильтрата показал, что получили смесь продукта и малоновой кислоты (примерно в том же соотношении 2:1 в пользу продукта). Собранные порции объединяли и использовали на следующей стадии.
Стадия C

Смесь, полученную на стадии B (0,5 г, 2,17 ммоль, 1 экв.), растворяли в 5 мл SOCl2, кипятили с обратным холодильником в течение 1 ч и концентрировали. Полученный неочищенный хлорангидрид смешивали с бензоилгидразином (0,643 г, 4,72 ммоль, 2 экв.) в 10 мл безводного DMF с последующим добавлением DIPEA (1,99 мл, 11,45 ммоль, 5 экв.). После перемешивания в течение ночи реакционную смесь гасили водой, экстрагировали EtOAc и концентрировали. Остаток обрабатывали DCM и фильтровали.
Стадия D
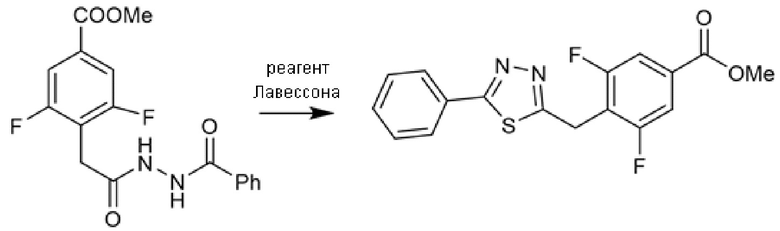
Смесь соединения, полученного на стадии С (0,277 г, 0,88 ммоль, 1 экв.), и реагента Лавессона (0,35 г, 0,86 ммоль, 0,98 экв.) в 5 мл толуола перемешивали в герметичном сосуде при 120°С в течение 15 мин. Полное превращение исходного материала контролировали с помощью СВЭЖХ. Растворитель выпаривали и остаток очищали колоночной хроматографией, сначала используя EtOAc в гексане (градиент 20-100%), затем 5% МеОН в DCM.
Стадия E
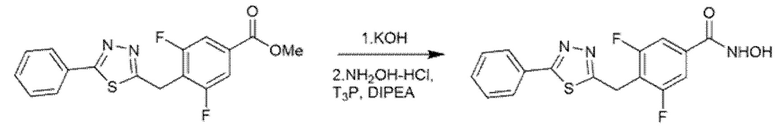
КОН (0,021 г, 0,37 ммоль, 2 экв.) добавляли к раствору циклического соединения, полученного на стадии D (0,06 г, 0,18 ммоль, 1 экв.) в 14 мл смеси THF/вода=4/1. Реакционную смесь перемешивали при КТ в течение ночи и подкисляли 1 М HCl. Полученный осадок собирали и сушили под вакуумом. Затем данное твердое вещество растворяли в THF вместе с DIPEA (0,333 мл, 1,902 ммоль, 6 экв). Добавляли HATU (0,181 мг, 0,476 ммоль, 1,5 эквив.) и реакционную смесь перемешивали при КТ, и полное превращение кислоты в промежуточное соединение с HATU контролировали с помощью ЖХМС. Добавляли NH2OH-HCl (0,066 г, 0,951 ммоль, 3 экв.) и реакционную смесь перемешивали в течение еще 2 ч. Реакционную смесь разбавляли водой до 50 мл общего объема и экстрагировали EtOAc (225 мл). После выпаривания получали 101 мг очень вязкого масла. Затирание с ацетонитрилом (обработка ультразвуком ~ 15 мин) приводило к образованию осадка, который собирали фильтрованием, промывали ацетонитрилом и простым эфиром и сушили. Получали 33 мг чистого продукта (m/z 348,09 [MH+]).
Следующие соединения синтезировали с использованием данной методики:
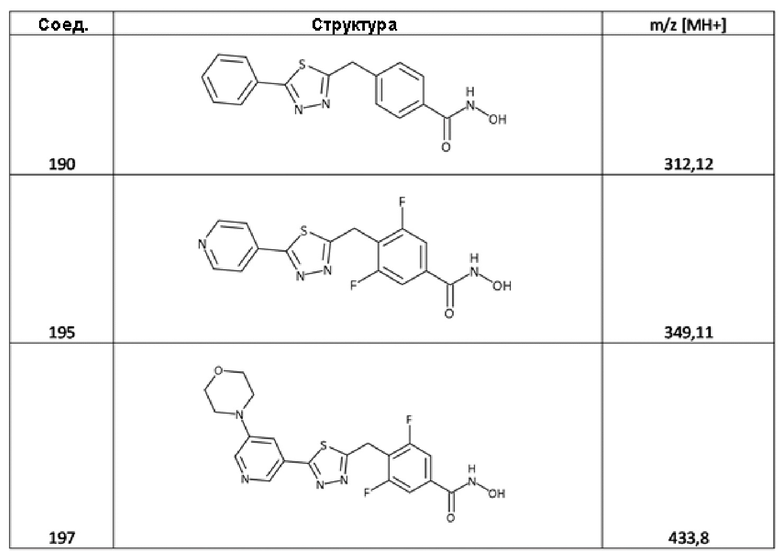
Пример 14 - Синтез 3,5-дифтор-N-гидрокси-4-((5-фенил-1,2,4-оксадиазол-3-ил)метил)бензамида (соед. 194)
Стадия А
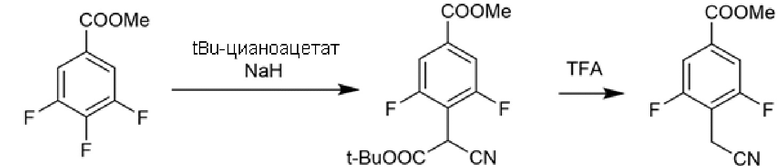
Трет-бутилцианоацетат (11,4 г, 52,73 ммоль, 2 экв.) добавляли по каплям в суспензию NaH (1,5 экв.) в 70 мл безводного DMF. После 5 мин перемешивания при КТ добавляли метил-3,4,5-трифторбензоат (5 г, 26,3 ммоль, 1 экв). Реакционную смесь перемешивали в течение 3 ч при КТ, разбавляли водой и экстрагировали EtOAc. После концентрирования остаток очищали с помощью колоночной хроматографии, затем разбавляли в 20 мл безводного DCE и обрабатывали TFA (8,6 мл, 10 экв.) при кипячении с обратным холодильником в течение ночи. Растворитель выпаривали, остаток растворяли в DCM, промывали насыщенным раствором NaHCO3, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией.
Стадия B
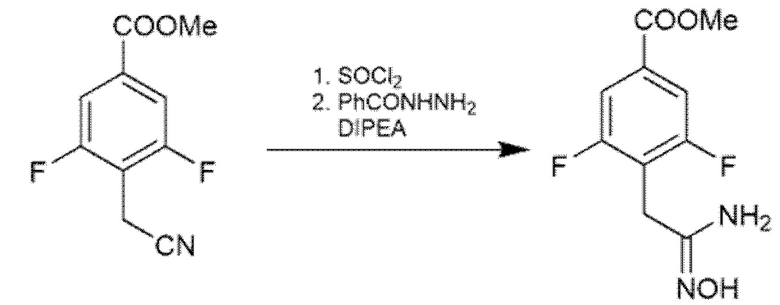
Смесь нитрильного производного, полученную на стадии A (2 г, 11 ммоль, 1 экв.), гидрохлорид NH2OH (1,5 г, 22 ммоль, 2 экв.) и NaHCO3 (1,81, 22 ммоль, 2 экв.) в 40 мл метанола кипятили с обратным холодильником в течение ночи. После фильтрования и концентрирования полученный неочищенный продукт очищали колоночной хроматографией (10% EtOAc в DCM).
Стадия C

Бензоилхлорид (0,243 г, 1,73 ммоль, 1,2 экв.) добавляли к раствору метил-(Z)-4-(2-амино-2-(гидроксиимино)этил)-3,5-дифторбензоата, полученного на стадии B (0,3 г, 1,44 ммоль, 1 экв.), и DIPEA (0,75 мл, 4,32 ммоль, 3 экв.) в 2 мл безводного DMF. После перемешивания в течение ночи реакционную смесь гасили водой и экстрагировали EtOAc. Очистка колоночной хроматографией (чистый DCM) давала 41 мг продукта.
Стадия D
KOH (0,014 г, 0,24 ммоль, 2 экв.) добавляли к раствору сложного метилового эфира, полученного на стадии C (0,04 г, 0,12 ммоль, 1 экв.), в 14 мл смеси THF/вода= 4/1. Реакционную смесь перемешивали при КТ в течение ночи и подкисляли 1 М HCl. Полученный осадок собирали и сушили под вакуумом. Полученную карбоновую кислоту растворяли в 2 мл безводного THF. Добавляли DIPEA (0,72 ммоль, 6 экв.) и HATU (0,18 ммоль, 1,5 экв.). Реакционную смесь перемешивали при КТ и контролировали ЖХМС на полное превращение кислоты в промежуточное соединение с HATU. Добавляли гидрохлорид NH2OH (0,025 г, 0,36 ммоль, 3 экв.) и реакционную смесь перемешивали еще 2 ч, затем разбавляли водой до 50 мл общего объема и экстрагировали EtOAc (225 мл). После выпаривания получали 101 мг очень вязкого масла. Затриание с ацетонитрилом (обработка ультразвуком ~ 15 мин) приводило к образованию осадка, который собирали фильтрованием, промывали ацетонитрилом и простым эфиром и сушили. Получали 20 мг чистого продукта (m/z 332,13 [MH+]).
Следующие соединения синтезировали с использованием данной методики:
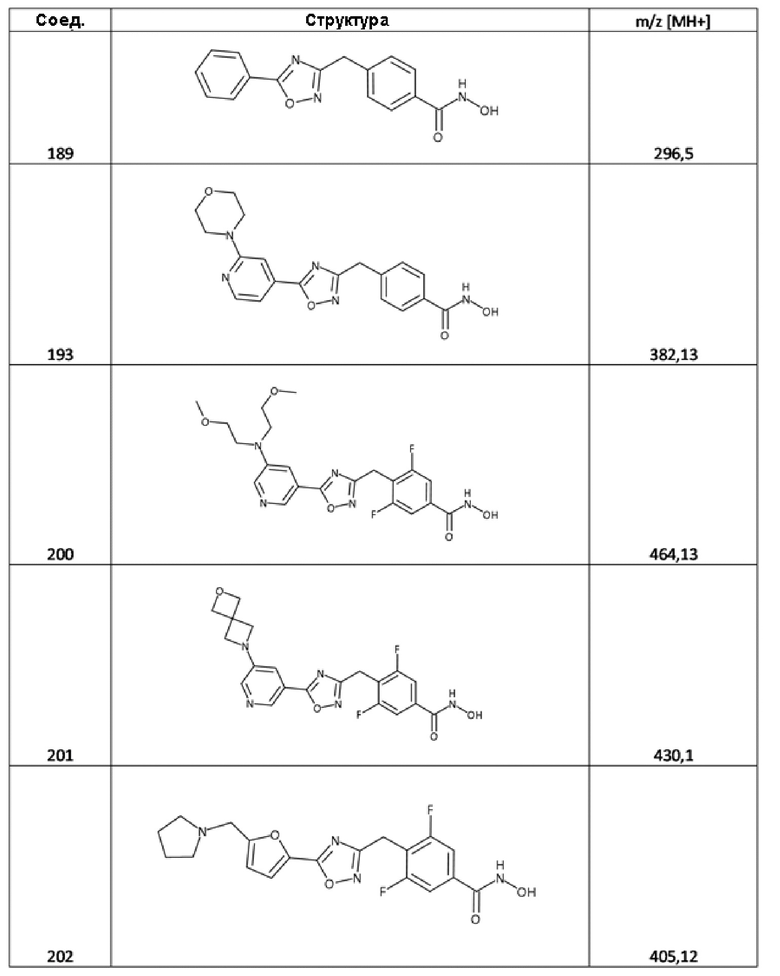
Пример 15 - Синтез N-гидрокси-4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензамида (соед. 217)
Стадия А

Уксусную кислоту (0,3 мл) по каплям добавляли к раствору неочищенного метил-4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)бензоата (0,38 г, 1,29 ммоль, 1 экв.) в 2 М растворе MeNH2 в THF (15 мл). Реакционный сосуд герметично закрывали и реакционную смесь оставляли перемешиваться при 150°С в течение ночи. После охлаждения растворитель выпаривали; остаток обрабатывали водой и экстрагировали EtOAc. Органическую фазу сушили и выпаривали, получая 258 мг оранжевого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия B

Сложный метиловый эфир, полученный на стадии А (0,041 г, 0,139 ммоль, 1 экв), суспендировали в 8 мл метанола и полученный раствор охлаждали на ледяной бане. Добавляли 50% раствор NH2OH в воде (0,34 мл, 40 экв.) с последующим медленным добавлением 1 М раствора NaOH (1,4 мл, 10 экв.). Реакционную смесь перемешивали до достижения КТ (примерно 1 ч) и подкисляли 1 М HCl. Белый осадок собирали фильтрованием. Очистка преп. ВЭЖХ давала 24 мг чистого продукта (m/z 309,12 [MH+]).
Пример 16 - Синтез 4-((3-((1H-индол-3-ил)метил)-5-(тиофен-2-ил)-4H-1,2,4-триазол-4-ил)метил)-N-гидроксибензамида (соед. 183)
Стадия А

Гидрохлорид метил-4-(аминометил)бензоата (402 мг, 2 ммоль, 1 экв.) растворяли в дихлорметане (8 мл) в присутствии триметиламина (616 мкл, 4,4 ммоль, 2,2 экв.). Затем добавляли 2-тиофенкарбонилхлорид (236 мкл, 2,2 ммоль, 1,1 экв.) и смесь перемешивали при КТ в течение ночи.
После завершения реакционную смесь разбавляли дихлорметаном и промывали водой. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия B
Метил-4-((тиофен-2-карбоксамидо)метил)бензоат (1 ммоль, 1 экв.) суспендировали в тионилхлориде (4 мл, 5,5 экв.) в атмосфере аргона и перемешивали при температуре кипения с обратным холодильником в течение ночи. Смесь концентрировали при пониженном давлении для удаления избытка SOCl2. Полученный таким образом неочищенный имидоилхлорид суспендировали в сухом толуоле, и гидразид индол-3-уксусной кислоты (189 мг, 1 ммоль, 1 экв.) добавляли в виде твердого вещества. Полученную смесь нагревали до 120°С и перемешивали в течение выходных. Смесь концентрировали с помощью ротационного выпаривания. Продукт осаждали из EtOAc/MeOH 1% и собирали фильтрованием. Получали 113 мг продукта.
Стадия C

Сложный метиловый эфир, полученный на стадии В (0,041 г, 0,139 ммоль, 1 экв.), суспендировали в 8 мл метанола и полученный раствор охлаждали на ледяной бане. Добавляли 50% раствор NH2OH в воде (0,34 мл, 40 экв.) с последующим медленным добавлением 1 М раствора NaOH (1,4 мл, 10 экв.). Реакционную смесь перемешивали до достижения КТ (примерно 1 ч) и подкисляли 1 М HCl. Белый осадок собирали фильтрованием. Очистка преп. ВЭЖХ давала чистый продукт (m/z 430,3 [MH+]).
Следующее соединение синтезировали с использованием данной методики:
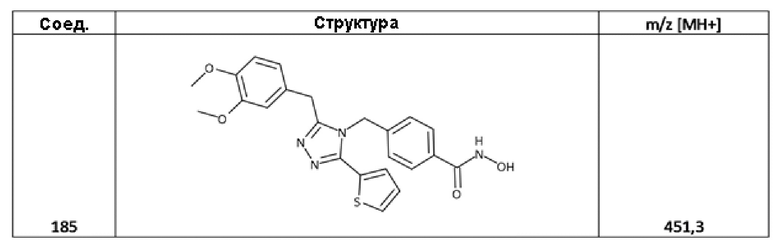
Пример 17 - Ферментативный скрининг
Ферментативную активность в отношении рекомбинантных человеческих HDAC6 и HDAC3 оценивали (таблица 2) для каждого синтезированного соединения. Соединения, которые показали хорошую селективность в отношении HDAC6, определяемую как log отношения IC50 между HDAC6 и другой изоформой менее чем -2, также подвергали скринингу на всех других изоформах для получения полного профиля (таблица 3).
Для каждого тестируемого соединения получали растворы в пяти различных концентрациях (обычно в диапазоне 3-30000 нМ), 5-кратно концентрированных в реакционном буфере (25 мМ Трис-HCl, рН 8, 130 мМ NaCl, 0,05% Tween-20, 10% глицерин) плюс ДМСО, нормализованных к количеству, присутствующему в более концентрированном растворе ингибитора, обычно 0,75% эквивалента к конечным 0,15% в планшете. 10 мкл раствора в трехкратном повторе для каждой концентрации тестируемого соединения помещали в 96-луночный планшет и в каждую лунку добавляли 15 мкл 3,33-кратно концентрированного раствора фермента в реакционном буфере (25 мМ Трис-HCl, рН 8, 130 мМ NaCl 0,05% Tween-20, 10% глицерина, 1 мг/мл BSA или 2 мг/мл для HDAC4, HDAC5 и HDAC9 - примечание: для HDAC7 использовали 50 мМ Трис-HCl, pH 8, 137 мМ NaCl, 2,7 мМ KCl и 1 мМ MgCl2). После периода инкубации при 30°С (время инкубации варьирует для разных изоформ и показано в таблице 1) добавляли 25 мкл раствора, содержащего субстрат. В качестве субстрата использовали субстрат деацетилазы FLUOR DE LYS® (Enzo Life Sciences, кат.№ BML-KI104, FdL), субстрат FLUOR DE LYS®-Green (Enzo Life Sciences, кат.№ BML-KI572 FdL_G) и трифторацетил-L-лизин (Tfal) - 2-кратно концентрированный раствор в 25 мМ Трис-HCl, рН 8, 130 мМ NaCl, 0,05% Tween-20, 10% глицерин). По истечении времени реакции при 30°C (время реакции изменяется для разных изоформ и представлено в таблице 1) добавляли 50 мкл проявляющего раствора, состоящего из концентрата проявителя I FLUOR DE LYS® (Enzo Life Sciences, кат. № BML-KI105), разбавленного в 200 раз в HAB плюс 2 мкМ TSA, и, через 25 мин при комнатной температуре в темноте с помощью прибора Victor 1420 Multilabel Counter Perkin Elmer Wallac осуществляли измерение флуоресценции.
Таблица 1 - Подробные данные ферментативного теста каждой отдельной изоформы
λ ex/λ em
(0,1 с)
Данные по ферментативному ингибированию HDAC6 и HDAC3 синтезированных соединений приведены в таблице 2. Полные профили ингибирования всех изоформ для выбранных соединений приведены в таблице 3. Вещества показали хорошую активность в отношении HDAC6 и заметную селективность в отношении других изоформ.
Таблица 2 - Анализ ингибирующей активности фермента по отношению к HDAC6 (IC50 нМ) и селективности по сравнению с HDAC3 (логарифмическое отношение IC50 для двух ферментов)
Предпочтительные соединения настоящего изобретения показывают значения IC50 HDAC6 менее 20 нМ и индекс селективности относительно HDAC3 менее -1,6.
Таблица 3 - Полный профиль ингибирования всех HDAC для некоторых предпочтительных соединений по изобретению (IC50 нМ)
Пример 18 - Цитотоксичность
Цитотоксическую активность оценивали на промиелоцитарной клеточной линии B 697 для всех синтезированных соединений и на мононуклеарных клетках периферической крови (РВМС) для соединений, демонстрирующих хороший профиль эффективности/селективности.
Клетки высевали в планшет (2×104 клеток на лунку для 697, 5×105 клеток на лунку для PBMC). Тестируемые соединения (концентрации от 1,5 нМ до 10000 нМ для РВМС и от 1 нМ до 10000 нМ для 697) добавляли через 24 ч и инкубировали 72 ч. Цитотоксическую активность соединений оценивали с использованием CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega), измеряющим функцию митохондрий, следуя инструкциям производителя.
Значения IC50 показаны в таблице 4. Большинство соединений проявляет низкую токсичность.
Таблица 4 - Цитотоксичность клеток для клеточной линии 697 и PBMC (IC50 нМ)
н.д. = нет данных
Предпочтительные соединения настоящего изобретения показывают значения IC50 для клеточной линии 697 более 1000 нМ и для PBMC более 5000 нМ.
Пример 19 - Устойчивость к фазе I метаболизма во фракции печени S9 крысы и человека
Тестируемые соединения инкубировали во фракции S9 печени крысы и человека при 37°С до 90 мин для оценки их устойчивости к фазе I метаболизма ферментами печени.
Каждое тестируемое соединение инкубировали при концентрации мкМ (50 мкМ, когда образцы анализировали с помощью УФ/ВЭЖХ; 1 или 2 мкМ, когда образцы анализировали с помощью ЖХМС/МС) с фракцией S9 (содержание белка 2 мг/мл) в 100 мМ фосфатного буфера (рН 7,4), 3,3 мМ MgCl2 и 1,3 мМ NADPH в течение 0 мин, 10 мин, 30 мин, 60 мин и 90 мин при 37°С в термостатированной бане с вибрационным устройством. Реакцию останавливали помещением образцов на ледяную баню и добавлением подкисленного ацетонитрила. После центрифугирования (10 мин при 14000 об/мин) аликвоту супернатанта разбавляли водой, фильтровали через 0,45 мкм шприцевые фильтры из регенерированной целлюлозы и вводили в ВЭЖХ-УФ или в ЖХМС/МС. Рассчитывали проценты количества, остающегося в различные периоды инкубации по отношению к исходному количеству. Также вычисляли внутренний клиренс.
Пример 20 - Устойчивость в плазме крысы и человека
Для оценки устойчивости к циркулирующим ферментам тестируемые соединения инкубировали в плазме человека и крысы при 37°С в термостатированной бане с вибрационным устройством. Каждое тестируемое соединение инкубировали при концентрации мкМ (50 мкМ, когда образцы анализировали с помощью УФ/ВЭЖХ; 1 или 2 мкМ, когда образцы анализировали с помощью ЖХМС/МС) в течение 0 мин, 15 мин, 30 мин и 1 ч, 2 ч и 4 ч. Реакцию останавливали помещением пробирок на ледяную баню и добавлением подкисленного ацетонитрила. После центрифугирования в течение 10 мин при 14000 об/мин аликвоту супернатанта разбавляли водой, фильтровали через 0,45 мкм шприцевые фильтры и вводили в ВЭЖХ-УФ или в ЖХМС/МС. Рассчитывали проценты количества, остающегося в различные периоды инкубации по отношению к исходному количеству. Рассчитывали также период полувыведения из плазмы.
Данные по устойчивости в процессе метаболизма in vitro приведены в таблицах 5 и 5'. Большинство соединений отличалось хорошей устойчивостью.
Таблица 5 - Анализ in vitro устойчивости к воздействию ферментов для предпочтительных соединений (остаточный процент в S9 через 90 мин и в плазме через 4 ч).
н.д. = нет данных
Предпочтительные соединения настоящего изобретения демонстрируют остаточное процентное содержание во фракции S9 крысы более 25%, во фракции S9 человека более 85%, в плазме крысы более 75% и в плазме человека более 90%.
Таблица 5' - Анализ in vitro устойчивости к воздействию ферментов (остаточный процент в S9 через 90 мин и в плазме через 4 ч).
н.д. = нет данных
Предпочтительные соединения настоящего изобретения демонстрируют остаточное процентное содержание во фракции S9 крысы более 25% и в плазме крысы более 75%.
Пример 21 - Ацетилирование α-тубулина и гистона Н3 в клеточной линии 697 in vitro
Определение ацетилирования α-тубулина и гистона Н3 in vitro оценивали на промиелоцитарной клеточной линии B 697.
Тестируемые вещества разбавляли из 20 мМ исходного раствора в ДМСО с RPMI 10% FCS+0,01% ДМСО при 20-кратной концентрации по сравнению с конечной концентрацией, добавляли к клеткам (15×106 клеток в общем объеме 30 мл в среде RPMI 10% FCS+0,01% ДМСО) для получения конечных концентраций 1000 нМ, 333 нМ, 111 нМ и 37 нМ и инкубировали при 37°С, 5% СО2 в течение 16 ч.
В конце периода инкубации 5×106 клеток отбирали из каждого образца, центрифугировали в течение 5 мин при 1100 об/мин и промывали в 0,9% NaCl при 4°С. Полученный осадок лизировали обработкой при 4°C в течение 30 мин 150 мкл Complete Lysis-M (Roche, кат. № 04719956051), содержащего ингибиторы протеаз и фосфатаз (таблетки из смеси ингибиторов протеиназы Complete Easy Pack, кат. № 04693116001; смесей ингибиторов фосфатазы Phostop easypack, кат. №: 01906837001- Roche), затем центрифугировали 10 мин при 14000 об/мин (20817× g). 0,150 мкг супернатанта (общий белковый экстракт) разбавляли в 100 мкл 1× PBS и иммобилизовали в планшете Maxisorp F96 NUN-IMMUNO (Nunc кат. № 5442404) при комнатной температуре в течение ночи. Планшеты дважды промывали промывочным буфером (PBS1X+0,005% Tween-20) и насыщали в течение 1 ч при комнатной температуре 300 мкл 1× PBS, содержащего 10FCS. После промывания буфером (1× PBS, содержащим 0,005% Tween) планшеты инкубировали в течение 2 ч при комнатной температуре в присутствии антитела против ацетилированного α-тубулина (клон моноклонального антиацетилированного тубулина 6-11B-1, мышиная асцитная жидкость, кат. № T6793 Sigma, 100 мкл, разбавленная 1:1000 в 1× PBS, содержащем 10% FCS) или с антителом против общего α-тубулина (моноклональное антитело против α-тубулина, продуцируемое у мыши; кат. № T6074 Sigma). После промывки добавляли 100 мкл на лунку набора субстратов TMB в течение 10 мин при комнатной температуре в темноте. Реакцию останавливали добавлением 50 мкл 2 н. H2SO4. Планшеты считывали на спектрофотометре Multiskan Spectrum при длине волны 450 нм.
Степень ацетилирования рассчитывали путем деления поглощения, полученного для ацетилированного α-тубулина, на поглощение общего α-тубулина.
Оставшиеся клетки (10×106) обрабатывали кислотной экстракцией гистонов (Kazuhiro et al., PNAS (2002), 99 (13) 8921-8926). Клетки центрифугировали 5 мин при 1100 об/мин при 4°С и однократно промывали в 0,9% NaCl. Полученный осадок лизировали лизиновым буфером (10 мМ Трис-HCl, рН 6,5/50 мМ бисульфита натрия, 1% Triton X-100/10 мМ MgCl2/8,6% сахарозы, содержащим смесь ингибиторов протеаз (Roche)) в течение 20 мин при 4°С. Полученный осадок ядер многократно промывали в буфере до осветления супернатанта (центрифугировали при 7500× g, 5 мин после каждой промывки) и, наконец, промывали в буфере для ядер (10 мМ Трис-HCl/13 мМ ЭДТА, рН 7, 4) и ресуспендировали в 250 мкл 0,2 М HCl/H2SO4. Гистоновые белки экстрагировали в кислой среде путем инкубации в течение ночи при 4°С при осторожном встряхивании. После центрифугирования при 14000 об/мин при 4°С в течение 10 мин к супернатанту добавляли 1250 мкл холодного ацетона и инкубировали в течение ночи при -20°С, что приводило к осаждению гистоновых белков. Осадок, полученный после центрифугирования в течение 10 мин при 14000 об/мин и 4°С, промывали холодным ацетоном, выпаривали досуха и ресуспендировали в 50 мкл дистиллированной воды. Определение содержания белка как общего, так и гистонового экстрактов проводили колориметрическим анализом с использованием набора BCA Protein Assay Kit (Pierce, кат.№: 23227).
Количество ацетилированных гистонов H3 и общее количество H3 определяли, применяя коммерчески доступные анализы ELISA (набор PathScan для сэндвич-анализа ацетилированного гистона H3, кат. № 7232C, и набор PathScan для сэндвич-анализа Elisa общего гистона H3, кат. № 7253C Cell Signaling) в соответствии со способом, указанным поставщиком, и путем определения поглощения при длине волны 450 нм с помощью Multiskan Spectrum. Тесты ELISA проводили путем анализа 0,250 мкг и 0,500 мкг гистонового экстракта каждого образца. Степень ацетилирования рассчитывали делением поглощения гистонов Н3 на общее поглощение гистонов.
Результаты испытаний ацетилирования тубулина и гистона Н3, выраженные в кратном увеличении соотношения ацетилированного α-тубулина/общего α-тубулина и H3Ac/H3Tot, соответственно, каждого образца относительно контрольного образца (необработанного), подытожены в таблицах 6 и 6'. Соединения показали хорошее ацетилирование тубулина и плохое ацетилирование гистона Н3.
Гивиностат, ингибитор пан-HDAC, использовали в качестве контрольного соединения. Как и ожидалось, контрольное соединение показало хорошее ацетилирование как тубулина, так и Н3 гистона. Приведенный в примере 43 WO 2012/106343 ингибитор HDAC использовали в качестве сравнительного соединения, чтобы показать неожиданные эффекты соединений изобретения по сравнению с соединением известного уровня техники, имеющим следующую формулу:
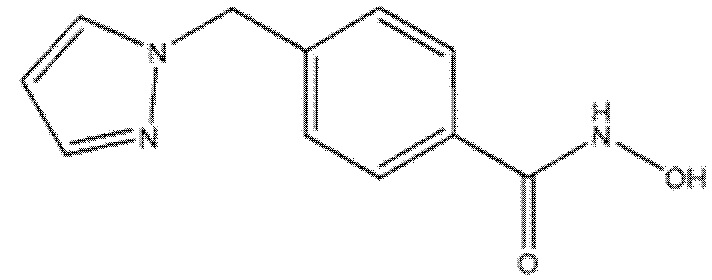
Пример 43
Таблица 6 - Ацетилирование тубулина в клеточной линии 697 (кратное увеличение отношения ацетилированного тубулина и общего тубулина к контролю).
По сравнению с примером 43 соединения по изобретению показали более высокое ацетилирование тубулина.
Таблица 6' - Ацетилирование гистона H3 в клеточной линии 697 (кратное увеличение показаний между ацетилированным H3 и общим H3 относительно контроля).
н.д. = нет данных
За исключением гивиностата, все вещества показали плохое ацетилирование гистона Н3.
Пример 22 - Фармакокинетика
Уровни в плазме и основные фармакокинетические параметры исследуемых соединений оценивали после однократного внутривенного и перорального введения мышам.
Вводимые дозы составляли 1,3-2,6 мг/кг при внутривенном введении и 2,6-5,2 мг/кг при пероральном введении. Композиции готовили в смеси ДМСО/PEG400/Н2О. Кровь отбирали в следующие моменты времени: 5 мин, 10 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч и 6 ч после введения. Образцы плазмы (100 мкл) депротеинизировали добавлением 1% муравьиной кислоты в ACN, затем перемешивали на вортексе и центрифугировали. Для каждого образца собирали аликвоту супернатанта и разбавляли водой, фильтровали через регенерированный целлюлозный фильтр 0,45 мкм и анализировали методом ЖХМС/МС. Уровни в плазме тестируемых соединений рассчитывали по калибровочной кривой, полученной в диапазоне 0,5-200 нг/мл.
Фармакокинетические параметры рассчитывали по средней кривой концентрации в плазме, используя программное обеспечение KineticaTM v. 5.1, с помощью некомпартментного метода.
Основные параметры приведены в таблице 7. Три протестированных соединения показали хорошую пероральную биодоступность.
Таблица 7 - Фармакокинетические параметры у мыши для трех предпочтительных соединений
Пример 23 - Оценка максимально переносимой дозы (MTD)
После длительного внутрибрюшинного введения C57BL/6 мышам MTD соединений оценивали по клиническим параметрам (масса тела и поведение) и параметрам крови (лейкоциты и тромбоциты). Соединения вводили после растворения в смеси H2O/PEG400 в соотношении 1:1 масс./масс., содержащей 5% ДМСО (для соединения 17 использовали 20% ДМСО).
Всех животных взвешивали за день до эксперимента (день 0) и определяли среднюю массу тела.
Животным (8 животных в группе) проводили введение один раз в день, начиная с дня 1 в течение 5 последовательных дней в неделю:
a) соединений в дозах 10, 30, 50 мг/кг ip,
b) гивиностата в дозе 100 мг/кг (внутренний контроль) и
c) растворов носителей, используемых для растворения веществ.
Объем вводимых растворов составлял 10 мл/кг. Введение повторяли в течение 2 недель, всего 10 введений на группу.
Ежедневно сообщалось о любых клинических проявлениях (внешний вид кожи, подвижность и реактивность животных, дыхание и т.д.), указывающих на возможную токсичность соединений. Вес животных оценивали на 2, 4, 9 и 11 сутки.
В 1, 3, 5, 8, 10 и 12 день из хвоста животного брали образец крови (примерно 50 мкл) для оценки влияния веществ на параметры крови. Исключения осуществляли для 4 животных на группу через день.
Образцы собирали в пробирки, содержащие ЭДТА, разбавляли соответствующим образом в физиологическом растворе и анализировали с помощью счетчика клеток.
В конце исследования (день 12) животных умерщвляли через 60 мин после последнего введения. Макроскопическую оценку при вскрытии проводили для выявления каких-либо нарушений внутренних органов. В таблице 8 обобщены данные, полученные в экспериментах по определению MTD для некоторых соединений по изобретению. Гивиностат является пан-ингибитором HDAC и использовался в качестве контроля. Тестируемые соединения хорошо переносились.
Таблица 8 - Значения параметров на 12-й день, контролируемые в эксперименте MTD на мышах для четырех предпочтительных соединений по изобретению
50 мг/кг
50 мг/кг
30 мг/кг
50 мг/кг
100 мг/кг
Пример 24 - Анализ супрессии пролиферации Т-CD4-лимфоцитов, опосредуемой регуляторными Т-клетками мыши
Для оценки способности соединений по данному патенту повышать супрессорную активность регуляторной Т-клетки (Treg, CD4+CD25+) использовали анализ супрессии пролиферации Т-клеток (отвечающие Т-клетки, Teff). Treg-клетки в различных концентрациях культивировали с Teff-клетками (CD4+CD25-) в присутствии пролиферативных стимулов. Т-клеткам необходимы два стимула для пролиферации: первый определяется распознаванием антигена, связанного с МНС, рецептором Т-клеток (TCR), и второй - из костимулирующих молекул, таких как CD28. В отсутствие специфического антигена активация TCR может происходить с помощью антитела, распознающего одну из составляющих субъединиц, CD3ε. В данном анализе в качестве стимулов-активаторов использовали моноклональное антитело против CD3ε и спленоциты с элиминированными T-клетками CD4. Соответственно, оценивали способность Treg-клеток уменьшать пролиферацию Teff-клеток в присутствии ингибиторов HDAC6 или без них.
Treg и Teff клетки разделяли с использованием набора для выделения Treg, основанного на методике разделения магнитных гранул (Miltenyi Biotec), посредством первоначального отрицательного отбора и конечного положительного отбора.
Суспензию отдельных клеток получали из селезенки мышей C57BL/6 с помощью фильтра 70 мкм.
Суспензию клеток обрабатывали буфером ACK для лизиса красных кровяных клеток и затем центрифугировали в течение 5 мин при 300 × g. После центрифугирования клетки ресуспендировали в PBS (Phosphate Bufferd Saline, Gibco) и подсчитывали. Затем спленоциты ресуспендировали в буфере, состоящем из PBS, 0,5% BSA и 2 мМ ЭДТА.
Для первой стадии разделения Treg CD4-негативные клетки подвергали непрямому магнитному мечению с использованием смеси конъюгированных с биотином антител против CD8a, CD11b, CD45R, CD49b, Ter-119 и, соответственно, получали CD4+ клетки с микрогранулами, покрытыми антителом против биотина, путем отрицательного отбора в виде фильтрата через магнитную колонку MACS.
Клетки, связанные с микрогранулами, элюировали и консервировали для их использования в качестве антигенпрезентирующих клеток (АРС) в анализе пролиферации.
На второй стадии очистки Treg предварительно обогащенные CD4+ клетки метили конъюгированным с R-фикоэритрином (PE) антителом против CD25, которое преимущественно связывается с Treg-клетками и магнитными гранулами, связанными с антителом против PE. Затем клеточную суспензию загружали в колонку. Teff-клетки проходили через колонку без связывания (отрицательный отбор), тогда как связанные с микрогранулами Treg-клетки прилипали с магнитными гранулами (положительный отбор). Затем Treg-клетки элюировали из колонки с потоком буфера с помощью плунжера. Очистка Treg и Teff клеток представлена в следующей схеме:

Полученные клетки использовали для анализа супрессии Treg следующим образом:
- CD4- клетки из первого положительного отбора в качестве APC
- Treg CD4+ CD25+ в качестве клеток-супрессоров
- Teff CD4+ CD25- в качестве отвечающих/пролиферирующих клеток
CD4- клетки обрабатывали митомицином C (50 мкг/мл, Sigma) в течение 30 мин при 37°C для предотвращения их пролиферации. Затем их ресуспендировали в концентрации 4,0 × 105/ 50 мкл в полной среде (RPMI, FBS10%, пенициллин/стрептомицин 1X, 50 мкМ бета-меркаптоэтанол). Teffs метили сложным сукцинимидиловым эфиром карбоксифлуоресцеина (CFSE) в концентрации 2 мкМ в PBS при 37°C, и после инкубации 10 мин реакцию останавливали 10% раствором FBS PBS. Мечение CFSE допускает ковалентную модификацию Teff-клеток для анализа их пролиферации путем флуоресцентного разбавления. Меченые Teffs затем центрифугировали и ресуспендировали до конечной концентрации 5,0 × 104/50 мкл в полной среде. Наконец, Treg, полученные очисткой, разбавляли до конечной концентрации 5,0 × 104/50 мкл в полной среде.
Затем туда добавляли сокультуру Teff (5,0×104), T CD4- (4,0×105) и Treg-клеток при различных соотношениях (Teff-клетки:Treg-клетки 1:1, 1:2, 1:4, 1:8). Тестируемые соединения в различных концентрациях или носитель ДМСО добавляли в клеточную суспензию. Наконец, моноклональное антитело против CD3ε (Miltenyi Biotec) добавляли в концентрации 1 мкг/мл. Клетки высевали в плоскодонные 96-луночные планшеты, и каждое условие устанавливалось в двух повторностях. Для определения того, каким образом различные вещества непосредственно влияют на пролиферацию клеток, оценивали влияние соединений на сокультуру меченных Teff и CD4- клеток в присутствии стимула, обеспечиваемого моноклональным антителом против CD3ε, в отсутствие Treg-клеток. Отрицательный контроль пролиферации клеток был определен только для меченых Teff, которые в отсутствие Т-CD4- и анти-CD3ε-моноклонального антитела не должны пролиферировать.
После 72 ч инкубации совместно культивированные клетки метили PE/Cy5-меченным анти-CD4 антителом (разведение 1:200, Biolegend) в течение 15 мин при комнатной температуре (КТ). После мечения клетки промывали и ресуспендировали в 200 мкл PBS.
Процент пролиферированных Teff определяли с помощью проточной цитометрии, наблюдая сигнал от CFSE в популяции клеток T CD4+. Мечение CFSE наследуется дочерними клетками после митоза, индуцированного активацией клеток. Анализ сигнала флуоресценции CFSE позволяет получить процент пролиферирующих клеток, которые представлены популяциями с более низкой флуоресценцией относительно непролиферирующей популяции.
В этом анализе прямая антипролиферативная активность тестируемых веществ должна быть исключена. Таким образом, был установлен порог, при котором, если наблюдается уменьшение пролиферации >10% в клетках Teff без Treg, ингибирование пролиферации не может быть полностью объяснено одной только индукцией супрессорной активности Treg.
Чтобы сравнить влияние соединений на супрессорную способность Treg, стандартизированную скорость пролиферации рассчитывали, применяя min-max стандартизацию к скоростям пролиферации для каждого образца по сравнению с контролем. Полученные значения преобразовывали в процент стандартизированной супрессии:
Стандартизированная супрессия=100 - (% стандартизированной пролиферации)
Затем рассчитывали площадь под кривой (AUC) графика значений процента стандартизированной супрессии. Относительную супрессию определяли по формуле:(AUC препарата/AUC контроля) - что представляет собой значение, которое позволяет сравнивать активность соединений. Вышеуказанные процедуры были выполнены посредством обработки данных с использованием программного обеспечения GraphPad Prism 7.
Более подробную информацию о всей процедуре можно найти в статье Akimova et al., Methods Mol Biol (2016), 1371: 43-78.
Результаты анализа супрессии Treg-клеток приведены в таблицах 9 и 10. Соединение с RS более 1,5 индуцирует хорошую супрессорную активность в Treg-клетках. Значения RS выше 2,5 указывают на высокую активность в этом анализе. Многие из протестированных соединений проявляют высокую активность.
Таблица 9 - Относительная супрессия T-reg для некоторых из предпочтительных соединений изобретения
мкМ
Таблица 10 - Относительная супрессия T-reg для других соединений по изобретению
мкМ
Пример 25 - Реакция смешанных лимфоцитов (MLR) с РВМС человека
Для изучения способности ингибиторов HDAC6 ингибировать активацию аллогенных T CD4+ клеток осуществляли анализ реакции смешанных лимфоцитов (MLR или смешанной культуры лимфоцитов, CLM). Это реакция, включающая бластную трансформацию культивированных in vitro лимфоцитов в присутствии аллогенных лимфоцитов. Существует так называемая «двусторонняя» реакция, в которой две популяции лимфоцитов стимулируют пролиферацию друг друга, и так называемая «односторонняя» реакция, при которой пролиферация одной из двух популяций ингибируется митомицином С или облучением, эти клетки обеспечивают стимул пролиферации (стимулятор) для так называемых «отвечающих» клеток.
Мононуклеарные клетки периферической крови человека (РВМС), используемые в MLR, получали разделением в градиенте Фиколла из лейкоцитарной пленки здоровых доноров.
Авторы изобретения использовали двустороннюю MLR. Клетки от двух доноров высевали в соотношении 1:1 (аллогенная стимуляция) до конечной концентрации 2 × 105 на лунку в 96-луночных планшетах с U-образным дном в среде RPMI 1640 с 10% FBS и антибиотиками. В качестве контроля авторы изобретения индивидуально высевали клетки из каждого донора (сингенный стимул). Эксперимент для каждого ингибитора устанавливали в двух повторностях для аллогенных стимулов и в пяти повторностях для сингенных стимулов. Клетки культивировали в течение 6 дней в инкубаторе при 37°С.
Через 6 дней действие тестируемых соединений оценивали путем измерения продукции провоспалительных цитокинов, признанных характеристиками данного анализа. Для этой цели собирали супернатант культуры и использовали для анализа воспалительных цитокинов IFN-γ, TNF-α и IL-6.
Результаты тестов MLR приведены в таблицах 11 и 12. Ингибитор JAK руксолитиниб использовали в качестве активного эталонного соединения в тесте.
Таблица 11 - Тест MLR для некоторых предпочтительных соединений по изобретению
мкМ
Значения в таблице указывают процент ингибирования. Отрицательные значения указывают на индукцию.
Таблица 12 - Тест MLR для других соединений по изобретению
мкМ
Пример 26 - Ингибирование экспрессии PD-L1 в дендритных клетках, полученных in vitro
В существующей на данный момент литературе описано, что селективные ингибиторы HDAC6 имеют большой потенциал в качестве иммуномодуляторов для использования в иммунотерапии рака (Tavares MT et al. ACS Med Chem Lett. 2017; 8(10):1031-1036).
Известно, что солидные опухоли имеют сильный миелоидный компонент, который вносит вклад в развитие, прогрессирование и распространение опухоли.
Дендритные клетки (DC) представляют собой профессиональные антигенпрезентирующие клетки (АРС), которые играют решающую роль в регуляции адаптивного иммунного ответа. Они могут эффективно представлять опухолевые неоантигены в контексте МНС класса I и II для стимуляции Т-клеточных ответов против опухоли. Однако в микроокружении опухоли раковые клетки могут ослаблять активацию Т-клеток через DC различными способами. Примером такой активности является индукция экспрессии ингибитора иммунной контрольной точки PD-L1 на поверхности DC. PD-L1 может взаимодействовать с PD-1, экспрессируемым на Т-клетках, и подавлять их активацию. Таким образом, уменьшение экспрессии PD-L1 на DC может представлять собой средство противодействия этому процессу.
Авторы изобретения предположили, что селективное ингибирование HDAC6 может снизить экспрессию PD-L1 на DC, тем самым увеличивая стимулирующую активность T-клеток.
Для получения in vitro DC, моноциты человека, очищенные от PBMC, обрабатывали в течение 5 дней GMCSF (50 нг/мл) и IL-4 (10 нг/мл) в присутствии двух селективных ингибиторов HDAC6, описанных в данном изобретении (соединения 10 и 19) и пример 43 ингибитора HDAC из WO 2012/106343. Контрольные клетки обрабатывали носителем ингибитора. Эта процедура вызывает образование незрелых дендритных клеток (iDC), которые экспрессируют PD-L1 (Brown JA et al. J Immunol. 2003;170:1257-66). Через 5 дней iDC анализировали на экспрессию ингибиторного маркера PD-L1.
Как показано на фиг. 1, соединения 10 и 19 данного изобретения снижали экспрессию PD-L1 статистически значимым образом. В противоположность этому, соединение примера 43 из WO 2012/106343 не смогло снизить экспрессию PD-L1, что указывает на отличающуюся биологическую активность этого соединения по сравнению с тем, что наблюдается для соединений 10 и 19.
Пример 27 - Модели опухолей мыши in vivo
Для оценки эффективности соединений 8 и 10 данного изобретения in vivo использовали четыре различные иммуноонкологические мышиные модели рака. В данном эксперименте авторы сравнивали эффективность анти-PD-1 антитела с эффективностью, проявляемой ингибиторами HDAC6. Анти-PD-1 таргетирует иммунную контрольную точку оси PD-1/PD-L1 и является признанной иммунотерапией при растущем числе злокачественных новообразований (Pardoll D.M., Nature Reviews Cancer, 2012, 12: 252-264).
Опухоли индуцировали у иммунокомпетентных мышей с использованием следующих клеточных линий:
- EMT6 (мышиный рак молочной железы)
- CT26 (мышиный рак толстой кишки)
- 4Т1 (трижды негативный мышиный рак молочной железы)
В соответствии с литературными данными, чувствительность этих мышиных опухолей к лечению анти-PD-1 резюмируется в следующей таблице:
- Терапевтическое лечение начинали, когда опухолевые узелки достигали примерно 3 мм в диаметре.
- Соединения 8 и 10 вводили через желудочный зонд один раз в день в течение 5 дней в неделю в дозе 50 мг/кг.
- Анти-PD-1 антитело вводили три раза в неделю путем интраперитонеальной инъекции в дозе 10 мг/кг.
Результаты экспериментов представлены на фиг. 2. Соединения 8, 10 и анти-PD1 антитело имели сопоставимую эффективность в снижении роста опухоли. Результаты также согласуются с ожидаемой эффективностью анти-PD-1 антитела. Селективные ингибиторы HDAC6 по данному изобретению обладают пониженной прямой противоопухолевой активностью, что подтверждается отсутствием цитотоксической активности in vitro. Следовательно, результаты in vivo на этих иммуноонкологических моделях позволяют предположить, что лечение селективными ингибиторами HDAC6 приводит к возможной активации противоопухолевого иммунного ответа.
Для демонстрации того, что противоопухолевая активность соединений 8 и 10 in vivo опосредована активацией иммунной системы, авторы изобретения провели дальнейшие эксперименты с использованием мышиной модели СТ-26.
Взрослым BALB/c мышам вводили s.c. 1×106 опухолевых клеток CT26 (разбавленных до 100 мкл фосфатно-солевым буферным раствором). Через неделю мышам ежедневно вводили соединения 8 и 10 p. o. в дозе 50 мг/кг и/или вводили анти-PD1 антитело в дозе 10 мг/кг. Во время умерщвления брали селезенку для анализа ex vivo иммунного ответа опухоли. Клетки селезенки стимулировали смесью опухолеспецифических пептидов CT-26, распознаваемых в контексте как MHC I, так и MHC II. Таким образом, с помощью этой стимуляции ex vivo можно обнаружить опухолеспецифический ответ, опосредованный Т-клетками CD4 и CD8.
Результаты, показанные на фиг. 3, подтверждают предыдущие данные об эффективности соединений по изобретению в качестве самостоятельных агентов при уменьшении роста опухоли. Это снижение снова сопоставимо с тем, что было получено с антителом против PD-1. Кроме того, комбинированное лечение анти-PD-1 антителом и ингибиторами HDAC6 приводит к дополнительному улучшению, особенно с соединением 10.
Для демонстрации специфической активации иммунной системы против опухоли выделяли селезенки животных и культивировали спленоциты в присутствии специфических пептидов CT-26, распознаваемых Т-клетками CD4 и CD8 (Kreiter S. et al. Nature, 2015, 520:692-696).
Результаты представлены на фиг. 4 и фиг. 5, на которых процентное содержание Т-клеток CD4 и CD8, которые продуцируют IFN-α и TNF-β, показано для каждой группы лечения.
Обобщая, результаты, представленные на фиг. 3 - фиг. 5, показывают, что:
- Селективные ингибиторы HDAC6 соединений 8 и 10 могут значительно уменьшить прогрессирование опухоли CT-26.
- Эффективность этих двух соединений сравнима с эффективностью анти-PD-1 антитела.
- Сочетание соединения 10 с анти-PD-1 антителом дополнительно улучшает ингибирование роста опухоли.
- Стимуляция ex vivo CT-26 специфическими пептидами указывает на то, что лечение ингибиторами HDAC6, самостоятельно и в сочетании с анти-PD-1 антителом, вызывает специфический противоопухолевый опосредованный Т-клетками иммунный ответ.
- Результаты анализа ex vivo показывают, что с соединениями 8 и 10 достигается более сильный иммунный ответ на неоантиген, чем с анти-PD-1 антителом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ САХАРИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ АДДИТИВНАЯ СОЛЬ КИСЛОТЫ ИЛИ ОСНОВАНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ ИНГИБИТОРА ЭЛАСТАЗЫ | 1992 |
|
RU2114835C1 |
| Бициклические производные пиридина, полезные в качестве ингибитора белков, связывающих жирные кислоты (FABP) 4 и/или 5 | 2013 |
|
RU2648247C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АРИЛСУЛЬФОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, КОТОРЫЕ ОТВЕЧАЮТ НА МОДУЛИРОВАНИЕ 5HT РЕЦЕПТОРОВ | 2007 |
|
RU2451012C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| ПРОИЗВОДНОЕ 2-ЗАМЕЩЕННОГО САХАРИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ ИНГИБИТОРНУЮ АКТИВНОСТЬ ПРОТИВ ЭЛАСТАЗЫ | 1992 |
|
RU2101281C1 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| ЗАМЕЩЕННЫЕ ФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ (ВАРИАНТЫ) | 1998 |
|
RU2197482C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ПРОИЗВОДНЫЕ НИТРОКАТЕХОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СОМТ | 2006 |
|
RU2441001C2 |
| ЗАМЕЩЕННЫЕ АРИЛТИОАЛКИЛТИОПИРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА. | 1994 |
|
RU2154062C2 |

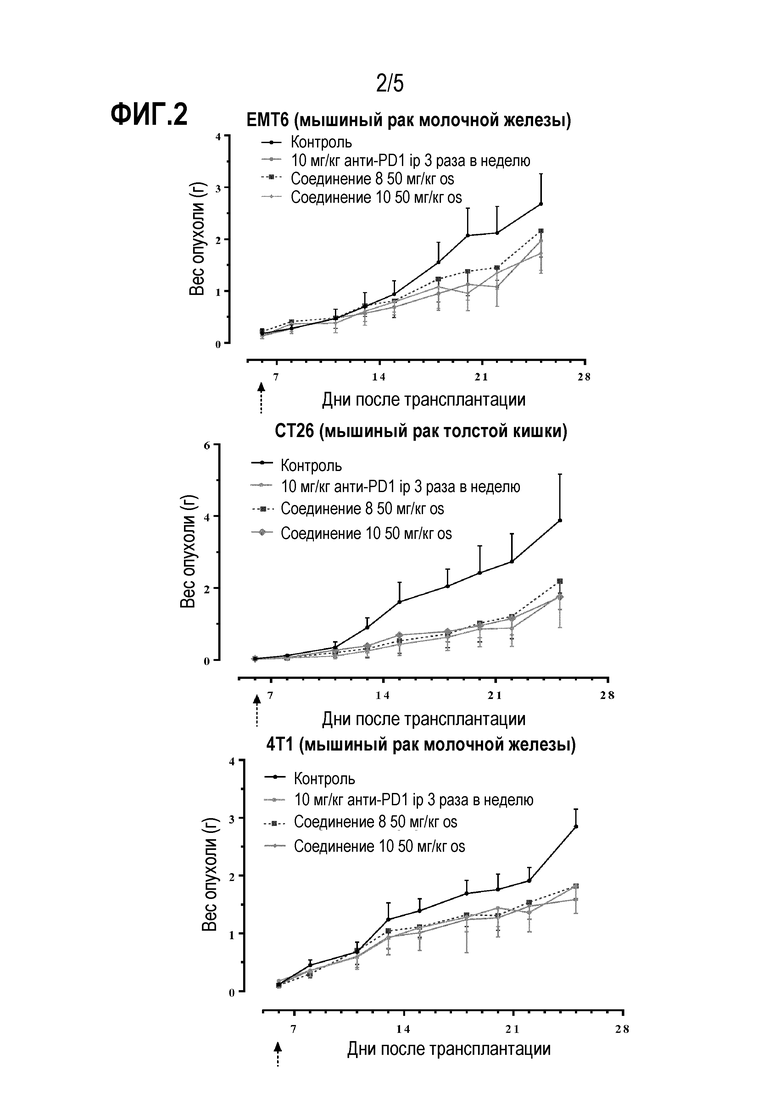

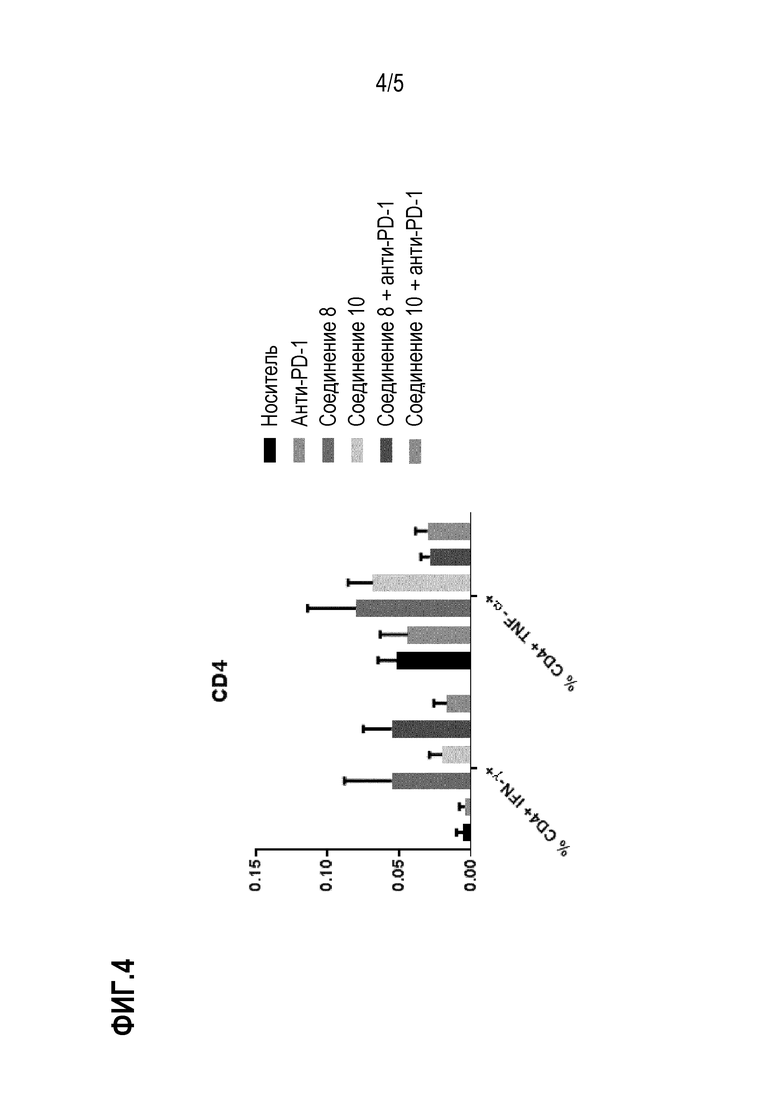
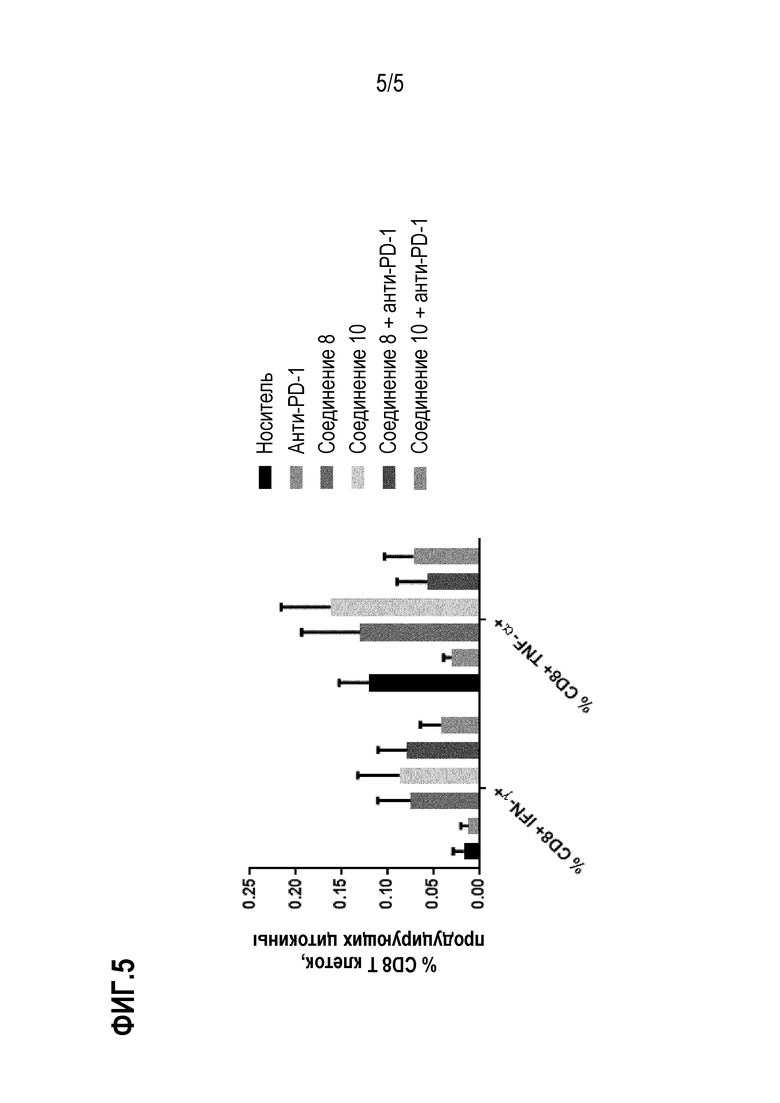
Изобретение относится к соединению формулы (I) и (II), его фармацевтически приемлемым солям и стереоизомерам, в котором А=N, O, S в формуле (I), А=N в формуле (II); B=C, N; С=N, O в формуле (I), когда С=N в формуле (II); X=CH2, S, NH; n=0, 1; когда n=1, атом углерода может быть замещен R12 и R13, независимо выбранными из группы, включающей H, –Me, –фенил, или вместе R12 и R13 могут образовывать циклопропан, циклобутан, циклопентан или циклогексан; когда n=1, R6 не отсутствует; R4=R5=H, F; R1 отсутствует или выбран из группы, включающей –H, –NH2, – C1–C4 алкил, фенил, фенил, замещенный одним или несколькими галогенами, метилфуран, метилфенил, тиофен и 2–(морфолин–4–ил)этил; R2 отсутствует или выбран из фенила; R3 отсутствует или выбран из o–метоксифенила, п–трифторметилфенила, бензила или пиридила; R6, R7 и R8 определены в формуле изобретения, R9=R10 = –Me, –Et; R11 выбран из группы, содержащей –H, –Cl и –CH3, при условии, что в соединениях формулы (I), когда пентагетероциклическое ядро представляет собой 1,3,4-оксадиазол, R6 не является нафтилом. Изобретение также относится к фармацевтической композиции для лечения заболеваний, опосредованных HDAC6, на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, проявляющие высокую селективную ингибирующую активность в отношении фермента гистондеацетилазы 6 (HDAC6), которые могут найти применение в медицине для лечения множественной миеломы, рака молочной железы и рака толстой кишки. 2 н. и 6 з.п. ф-лы, 5 ил., 15 табл., 27 пр.

(I)

(II)
1. Соединение формулы (I) и (II), его фармацевтически приемлемые соли и стереоизомеры:

(I)
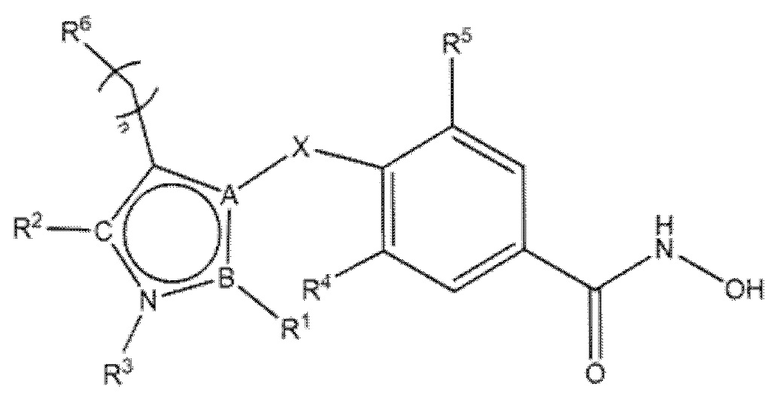
(II)
где
А=N, O, S в формуле (I), когда А=N в формуле (II);
B=C, N;
С=N, O в формуле (I), когда С=N в формуле (II);
X=CH2, S, NH;
n=0, 1;
когда n=1, атом углерода может быть замещен R12 и R13, независимо выбранными из группы, включающей H, –Me, –фенил, или вместе R12 и R13 могут образовывать циклопропан, циклобутан, циклопентан или циклогексан;
когда n=1, R6 не отсутствует;
R4=R5=H, F;
R1 отсутствует или выбран из группы, включающей –H, –NH2, – C1–C4 алкил, фенил, фенил, замещенный одним или несколькими галогенами, метилфуран, метилфенил, тиофен и 2–(морфолин–4–ил)этил;
R2 отсутствует или выбран из фенила;
R3 отсутствует или выбран из o–метоксифенила, п–трифторметилфенила, бензила или пиридила;
R6 выбран из группы, содержащей:
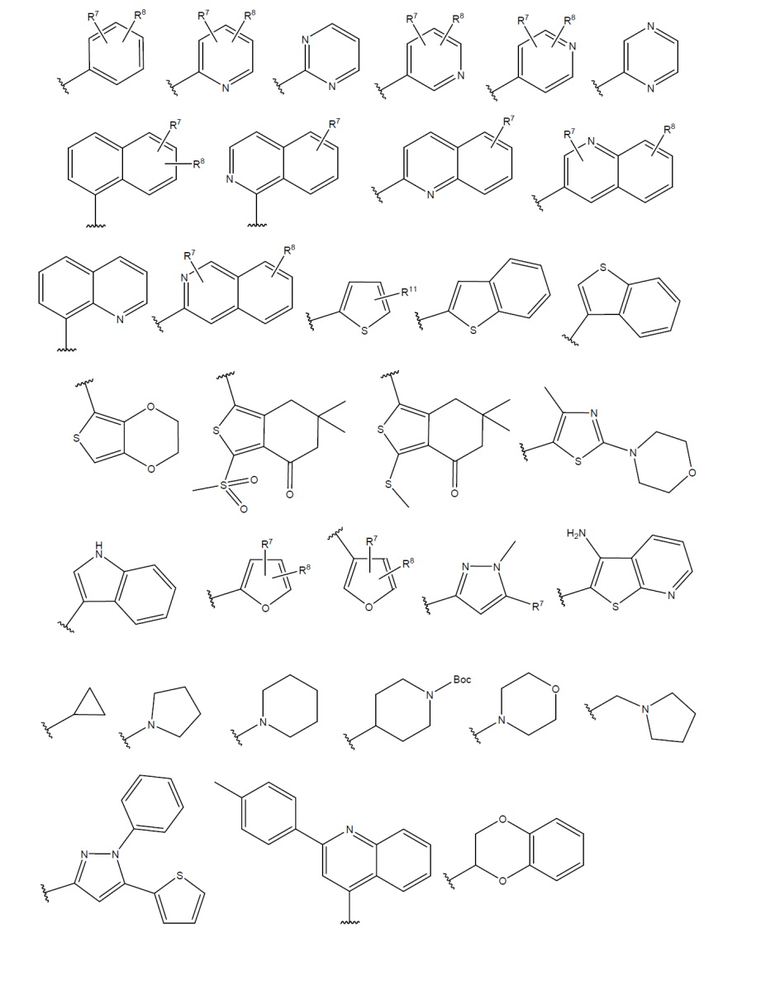

где:
R7 и R8 независимо выбраны из группы, содержащей H, –Cl, –F, –Br, –CF3, –Me, –Et, –OMe, –Oбензил, –SF5, –OCH2F, –CH2NH2, –CH2NMe2, –NH2, –NMe2, –N(CH2CH2OCH3)2, –COOH, –COOMe,
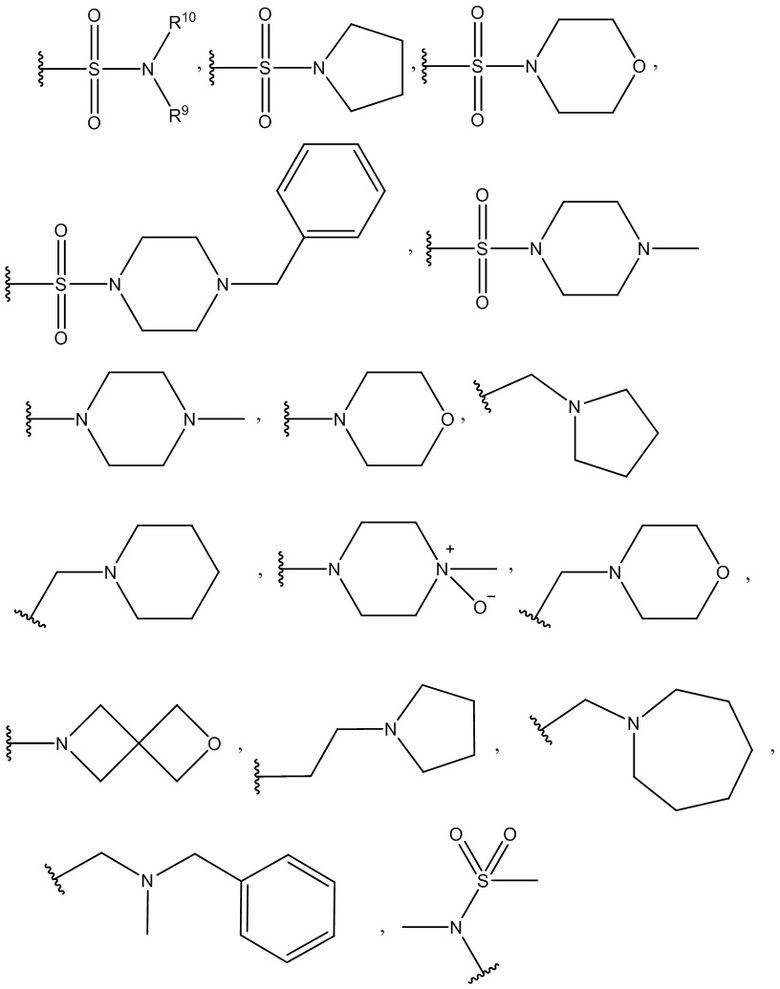
или R7 и R8 вместе могут образовывать гетеропентациклический фрагмент (–OCH2O–);
R9=R10 = –Me, –Et;
R11 выбран из группы, содержащей –H, –Cl и –CH3,
при условии, что в соединениях формулы (I), когда пентагетероциклическое ядро представляет собой 1,3,4-оксадиазол, R6 не является нафтилом.
2. Соединение по п.1, выбранное из перечисленного:
(S)–N–(1–(3–(4–(гидроксикарбамоил)бензил)–1,2,4–оксадиазол–5–ил)–2–(тиазол–4–ил)этил)–3,4–диметоксибензамид (соединение 1);
3,5–дифтор–N–гидрокси–4–((4–метил–5–(нафталин–1–ил)–4H–1,2,4–триазол–3–ил)тио)бензамид (соединение 2);
4–((5–(3–(N,N–диметилсульфамоил)фенил)–1,3,4–оксадиазол–2–ил)метил)–N–гидроксибензамид (соединение 3);
3,5–дифтор–N–гидрокси–4–((4–метил–5–(2–фенилпропан–2–ил)–4H–1,2,4–триазол–3–ил)тио)бензамид (соединение 4);
4–((5–(2,3–дигидротиено[3,4–b][1,4]диоксин–5–ил)–1H–тетразол–1–ил)метил)–3,5–дифтор–N–гидроксибензамид (соединение 5);
3,5–дифтор–N–гидрокси–4–((5–(пиридин–2–ил)–2H–тетразол–2–ил)метил)бензамид (соединение 6);
дифтор–N–гидрокси–4–((5–(пиримидин–2–ил)–2H–тетразол–2–ил)метил)бензамид (соединение 7);
N–гидрокси–4–((5–(тиофен–2–ил)–1H–тетразол–1–ил)метил)бензамид (соединение 8);
3,5–дифтор–N–гидрокси–4–((4–метил–5–(4–метил–2–морфолинoтиазол–5–ил)–4H–1,2,4–триазол–3–ил)тио)бензамид (соединение 9);
N–гидрокси–4–((4–метил–5–(тиофен–2–ил)–4H–1,2,4–триазол–3–ил)тио)бензамид (соединение 10);
4–((5–(фуран–2–ил)–2H–тетразол–2–ил)метил)–N–гидроксибензамид (соединение 12);
3,5–дифтор–N–гидрокси–4–((5–(пиридин–2–ил)–1H–тетразол–1–ил)метил)бензамид (соединение 13);
3,5–дифтор–N–гидрокси–4–((4–метил–5–(пиридин–2–ил)–4H–1,2,4–триазол–3–ил)тио)бензамид (соединение 14);
3,5–дифтор–N–гидрокси–4–((5–(тиофен–2–ил)–1H–тетразол–1–ил)метил)бензамид (соединение 15);
3,5–дифтор–N–гидрокси–4–((4–метил–5–(4–(пиперидин–1–илметил)фенил)–4H–1,2,4–триазол–3–ил)тио)бензамид (соединение 16);
3,5–дифтор–N–гидрокси–4–((4–метил–5–(тиофен–2–ил)–4H–1,2,4–триазол–3–ил)тио)бензамид (соединение 17);
3,5–дифтор–4–((5–(фуран–2–ил)–2H–тетразол–2–ил)метил)–N–гидроксибензамид (соединение 19);
N–гидрокси–4–((5–(пиридин–2–ил)–1H–тетразол–1–ил)метил)бензамид (соединение 20);
3–(3,4–диметоксифенил)–N–[(1S)–1–[3–[[4–(гидроксикарбамоил)фенил]метил]–1,2,4–оксадиазол–5–ил]–2–тиазол–4–ил–этил]пропанамид (соединение 21);
4–[[5–[4–(трифторметил)фенил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 23);
4–[(4,5–дифенил–1,2,4–триазол–3–ил)сульфанил]бензолкарбогидроксамовая кислота (соединение 24);
4–[[4–(2–фурилметил)–5–(1H–индол–3–ил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота; 2,2,2–трифторуксусная кислота (соединение 25);
4–[5–[(3,4–диметоксифенил)метил]–1,3,4–оксадиазол–2–ил]бензолкарбогидроксамовая кислота (соединение 26);
4–[[5–бензил–4–(4–фторфенил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 27);
4–[[4–амино–5–[4–(дифторметокси)фенил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 28);
4–[[5–(4–фторфенил)–4H–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 29);
4–[[4–этил–5–(4–фторфенил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 30);
4–[[5–(4–хлорфенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 31);
4–[[5–(5–хлор–2–тиенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 32);
4–[[5–(2–фторфенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 33);
4–[[5–(4–фторфенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 34);
4–[[5–(4–метоксифенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 35);
4–[(5–бензилтетразол–2–ил)метил]бензолкарбогидроксамовая кислота (соединение 36);
4–[(5–бензилтетразол–1–ил)метил]бензолкарбогидроксамовая кислота (соединение 37);
4–[[5–(2,4–дихлорфенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 38);
4–[[5–(3–метил–2–тиенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 39);
4–[[5–(5–метил–2–тиенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 41);
4–[[5–(бензoтиофен–3–ил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 42);
4–[[5–(2,3–дигидротиено[3,4–b][1,4]диоксин–5–ил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 43);
4–[[5–[(3,4–диметоксифенил)метил]–2–[4–(трифторметил)фенил]–1,2,4–триазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 44);
4–[[5–[(3,4–диметоксифенил)метил]–1,3,4–оксадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 45);
4–[[5–(2–фторфенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 46);
4–[[5–[(1S)–1–амино–2–тиазол–4–ил–этил]–1,2,4–оксадиазол–3–ил]метил]бензолкарбогидроксамовая кислота; 2,2,2–трифторуксусная кислота (соединение 48);
4–[[5–(3,4–диметоксифенил)–1,2,4–оксадиазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 49);
4–[[5–(2–тиенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 50);
4–[[2–бензил–5–(4–хлорфенил)–1,2,4–триазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 51);
4–[[2–(2–пиридил)–5–(2–тиенил)–1,2,4–триазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 52);
4–[[2–(2–метоксифенил)–5–(2–тиенил)–1,2,4–триазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 53);
4–[[5–(6,6–диметил–3–метилсульфанил–4–оксо–5,7–дигидро–2–бензoтиофен–1–ил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 54);
4–[[5–(бензoтиофен–2–ил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 55);
4–[[5–(3,4–диметоксифенил)–1,3,4–оксадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 57);
4–[[5–(2,4–дифторфенил)–1,3,4–оксадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 58);
4–[[5–[3–(диметилсульфамоил)фенил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 59);
4–[(5–фенил–1,3,4–оксадиазол–2–ил)амино]бензолкарбогидроксамовая кислота (соединение 60);
4–[[4–амино–5–[3–(диэтилсульфамоил)фенил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 61);
4–[[5–(3–пирролидин–1–илсульфонилфенил)–1,3,4–оксадиазол–2–ил]амино]бензолкарбогидроксамовая кислота (соединение 63);
4–[[5–(3–морфолинoсульфонилфенил)–1,3,4–оксадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 64);
3,5–дифтор–4–[[5–(2–тиенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 65);
4–[[5–[3–(диэтилсульфамоил)фенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 66);
4–[[4–метил–5–[2–(п–толил)–4–хинолил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 67);
4–[(5–фенил–1,3,4–оксадиазол–2–ил)метил]бензолкарбогидроксамовая кислота (соединение 68);
4–[[5–(4–пирролидин–1–илсульфонилфенил)–1,3,4–оксадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 69);
4–[[5–(3–бензилокси–4–метокси–фенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 70);
4–[[5–(3–бензилокси–4–метокси–фенил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 71);
4–[(5–циклопропил–1–фенил–1,2,4–триазол–3–ил)сульфанил]бензолкарбогидроксамовая кислота (соединение 72);
4–[[5–[4–(диметиламино)фенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 73);
4–[[5–(4–метил–2–морфолинo–тиазол–5–ил)–1,3,4–оксадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 75);
4–[[5–[3–(диметиламино)фенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 77);
4–[[5–(3–метоксифенил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 78);
4–[[5–(2,3–дигидротиено[3,4–b][1,4]диоксин–5–ил)тетразол–2–ил]метил]–3,5–дифтор–бензолкарбогидроксамовая кислота (соединение 79);
4–[[5–[3–(диметиламино)фенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифтор–бензолкарбогидроксамовая кислота (соединение 80);
трет–бутил–4–[5–[4–(гидроксикарбамоил)фенил]сульфанил–4–метил–1,2,4–триазол–3–ил]пиперидин–1–карбоксилат (соединение 82);
4–[[5–(2,3–дигидро–1,4–бензодиоксин–3–ил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 83);
4–[[5–(1,3–бензoдиоксол–5–ил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 84);
4–[[5–(1,5–диметилпиразол–3–ил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 85);
4–[[5–(2–фурил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 86);
4–[[5–(1–изохинолил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 87);
4–[[5–(1–изохинолил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 88);
4–[[5–(2–пиридил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 89);
4–[[5–(2–хинолил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 90);
4–[[5–(2–хинолил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 91);
3,5–дифтор–4–[[5–(2–фурил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 92);
3,5–дифтор–4–[[5–(1–изохинолил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 93);
3,5–дифтор–4–[[5–(1–изохинолил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 94);
3,5–дифтор–4–[[5–(2–хинолил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 95);
3,5–дифтор–4–[[5–(2–хинолил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 96);
3,5–дифтор–4–[[5–(2–тиенил)–4H–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 97);
4–[(5–бензгидрил–4–метил–1,2,4–триазол–3–ил)сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 98);
4–[[5–(3–аминотиено[2,3–b]пиридин–2–ил)–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 99);
4–[[5–(1,5–диметилпиразол–3–ил)–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 100);
3,5–дифтор–4–[[4–метил–5–(1–фенилциклобутил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 101);
3,5–дифтор–4–[[5–[1–(3–фторфенил)циклопентил]–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 102);
3,5–дифтор–4–[[5–[1–(4–метоксифенил)циклогексил]–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 103);
3,5–дифтор–4–[[5–[1–(4–метоксифенил)циклопропил]–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 104);
4–[[5–[3–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 106);
4–[[5–[3–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 107);
3,5–дифтор–4–[[5–[3–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 108);
3,5–дифтор–4–[[5–[3–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 109);
4–[[5–[4–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 110);
4–[[5–[4–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 111);
3,5–дифтор–4–[[5–[4–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 112);
3,5–дифтор–4–[[5–[4–(пентaфтор–лямбда–6–сульфанил)фенил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 113);
3,5–дифтор–4–[[4–метил–5–[3–(4–метил–4– оксидопиперазин–4–ий–1–ил)фенил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 114);
3,5–дифтор–4–[[4–(4–фторфенил)–5–(1–пиперидилметил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 115);
3,5–дифтор–4–[[4–(2–фурилметил)–5–пирролидин–1–ил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 116);
4–[(4–бензил–5–морфолинo–1,2,4–триазол–3–ил)сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 117);
4–[[5–(2,3–дигидротиено[3,4–b][1,4]диоксин–5–ил)–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 118);
3,5–дифтор–4–[[5–(1–изохинолил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 121);
3,5–дифтор–4–[[4–метил–5–(2–хинолил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 122);
4–[(5–пиримидин–2–илтетразол–2–ил)метил]бензолкарбогидроксамовая кислота (соединение 123);
4–[(5–пиримидин–2–илтетразол–1–ил)метил]бензолкарбогидроксамовая кислота (соединение 124);
3,5–дифтор–4–[(5–пиримидин–2–илтетразол–1–ил)метил]бензолкарбогидроксамовая кислота (соединение 125);
4–[[5–[5–(трифторметил)–2–пиридил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 126);
4–[[5–[5–(трифторметил)–2–пиридил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 127);
3,5–дифтор–4–[[5–[5–(трифторметил)–2–пиридил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 128);
3,5–дифтор–4–[[5–[5–(трифторметил)–2–пиридил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 129);
4–[[5–[3–морфолинo–5–(трифторметил)–2–пиридил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 130);
4–[[5–[3–морфолинo–5–(трифторметил)–2–пиридил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 131);
4–[[5–(2–пиридилметил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота; 2,2,2–трифторуксусная кислота (соединение 132);
4–[[5–(2–пиридилметил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота; 2,2,2–трифторуксусная кислота (соединение 133);
3,5–дифтор–4–[[5–(2–пиридилметил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота; 2,2,2–трифторуксусная кислота (соединение 134);
3,5–дифтор–4–[[5–(2–пиридилметил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота; 2,2,2–трифторуксусная кислота (соединение 135);
3,5–дифтор–4–[[4–метил–5–[1–фенил– 5–(2– тиенил)пиразол–3–ил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 136);
3,5–дифтор–4–[[5–(6–фтор–2–метил–3–хинолил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 137);
3,5–дифтор–4–[[5–(4–фторфенил)–4–(2–морфолинoэтил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 138);
3,5–дифтор–4–[[4–(2–фурилметил)–5–пиразин–2–ил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 139);
3,5–дифтор–4–[[4–(2–фурилметил)–5–(2–пиридил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 140);
4–[[4–бензил–5–(пирролидин–1–илметил)–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 141);
4–[[4–бензил–5–(2–фурил)–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 142);
4–[[4–бензил–5–(2–тиенил)–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 143);
3,5–дифтор–4–[[4–(2–фурилметил)–5–(2–тиенил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 144);
3,5–дифтор–4–[[5–(2–фторфенил)–4–(2–фурилметил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 145);
3,5–дифтор–4–[[4–(2–фурилметил)–5–(4–пиридил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 146);
3,5–дифтор–4–[[4–(2–фурилметил)–5–(3–пиридил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 147);
3,5–дифтор–4–[[5–(3–изохинолил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 148);
3,5–дифтор–4–[(5–имидазо[1,2–a]пиридин–3–ил–4–метил–1,2,4–триазол–3–ил)сульфанил]бензолкарбогидроксамовая кислота (соединение 149);
4–[[5–(1–бензил–4–фенил–4–пиперидил)–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 150);
3,5–дифтор–4–[[4–метил–5–[3–(4–метилпиперазин–1–ил)сульфонилфенил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 151);
4–[[5–[3–(4–бензилпиперазин–1–ил)сульфонилфенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 152);
3,5–дифтор–4–[[4–метил–5–(3–пиридил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 153);
метил–4–[[2–[[2,6–дифтор–4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]метил]бензоат (соединение 154);
метил–4–[[1–[[2,6–дифтор–4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]метил]бензоат (соединение 155);
метил–6–[2–[[4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]пиридин–3–карбоксилат (соединение 156);
метил–6–[1–[[4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]пиридин–3–карбоксилат (соединение 157);
4–[[2–[[4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]метил]бензойная кислота (соединение 158);
4–[[1–[[4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]метил]бензойная кислота (соединение 159);
4–[[2–[[2,6–дифтор–4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]метил]бензойная кислота (соединение 160);
4–[[1–[[2,6–дифтор–4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]метил]бензойная кислота (соединение 161);
6–[2–[[4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]пиридин–3–карбоновая кислота (соединение 162);
3–[2–[[4–(гидроксикарбамоил)фенил]метил]тетразол–5–ил]бензойная кислота (соединение 163);
3,5–дифтор–4–[[4–метил–5–(8–хинолилметил)–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 164);
4–[[5–(2,6–дифторфенил)–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифтор–бензолкарбогидроксамовая кислота (соединение 165);
3,5–дифтор–4–[[4–метил–5–[3–(4–метилпиперазин–1–ил)фенил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 166);
4–[[5–[3–(азепан–1–илметил)фенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 167);
4–[[5–[4–(азепан–1–илметил)фенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 168);
4–[[5–(4–аминофенил)тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 169);
4–[[5–(4–аминофенил)тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 170);
4–[[5–(4–аминофенил)тетразол–2–ил]метил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 171);
4–[[5–(4–аминофенил)тетразол–1–ил]метил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 172);
4–[[5–[4–(аминометил)фенил]тетразол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 173);
4–[[5–[4–(аминометил)фенил]тетразол–1–ил]метил]бензолкарбогидроксамовая кислота (соединение 174);
4–[[5–[4–(аминометил)фенил]тетразол–2–ил]метил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 175);
4–[[5–[4–(аминометил)фенил]тетразол–1–ил]метил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 176);
3,5–дифтор–4–[[4–метил–5–[1–(2–пиридил)циклопропил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 177);
3,5–дифтор–4–[[4–метил–5–[1–(3–пиридил)циклопропил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 178);
3,5–дифтор–4–[[5–(3–фтор–2–пиридил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 180);
3,5–дифтор–4–[[4–метил–5–[3–(1–пиперидилметил)фенил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 181);
3,5–дифтор–4–[[4–метил–5–[3–(морфолинoметил)фенил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 182);
4–((3–((1H–индол–3–ил)метил)–5–(тиофен–2–ил)–4H–1,2,4–триазол–4–ил)метил)–N–гидроксибензамид (соединение 183);
4–[[5–[3–[[бензил(метил)амино]метил]фенил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 184);
4–[[3–[(3,4–диметоксифенил)метил]–5–(2–тиенил)–1,2,4–триазол–4–ил]метил]бензолкарбогидроксамовая кислота (соединение 185);
3,5–дифтор–4–[[4–метил–5–[1–метил–1–(3–пиридил)этил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 186);
3,5–дифтор–4–[[5–[4–[метил(метилсульфонил)амино]фенил]–1,3,4–тиадиазол–2–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 187);
4–[(5–фенил–1,3,4–оксадиазол–2–ил)сульфанил]бензолкарбогидроксамовая кислота (соединение 188);
4–[(5–фенил–1,2,4–оксадиазол–3–ил)метил]бензолкарбогидроксамовая кислота (соединение 189);
4–[(5–фенил–1,3,4–тиадиазол–2–ил)метил]бензолкарбогидроксамовая кислота (соединение 190);
3,5–дифтор–N–гидрокси–4–((5–(пиридин–3–ил)–1,3,4–тиадиазол–2–ил)тио)бензамид (соединение 191);
3,5–дифтор–4–[(5–фенил–1,3,4–оксадиазол–2–ил)сульфанил]бензолкарбогидроксамовая кислота (соединение 192);
4–[[5–(2–морфолинo–4–пиридил)–1,2,4–оксадиазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 193);
3,5–дифтор–N–гидрокси–4–((5–фенил–1,2,4–оксадиазол–3–ил)метил)бензамид (соединение 194);
3,5–дифтор–4–[[5–(4–пиридил)–1,3,4–тиадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 195);
4–[[5–(5–бром–3–пиридил)–1,3,4–тиадиазол–2–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 196);
3,5–дифтор–4–[[5–(5–морфолинo–3–пиридил)–1,3,4–тиадиазол–2–ил]метил]бензолкарбогидроксамовая кислота (соединение 197);
3,5–дифтор–N–гидрокси–4–((5–фенил–1,3,4–тиадиазол–2–ил)метил)бензамид (соединение 198);
3,5–дифтор–4–[[5–(2–фурил)–4–метил–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 199);
4–[[5–[5–[бис(2–метоксиэтил)амино]–3–пиридил]–1,2,4–оксадиазол–3–ил]метил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 200);
3,5–дифтор–4–[[5–[5–(2–окса–6–азаспиро[3.3]гептан–6–ил)–3–пиридил]–1,2,4–оксадиазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 201);
3,5–дифтор–4–[[5–[5–(пирролидин–1–илметил)–2–фурил]–1,2,4–оксадиазол–3–ил]метил]бензолкарбогидроксамовая кислота (соединение 202);
3,5–дифтор–4–[[4–метил–5–[5–(морфолинoметил)–3–фурил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 203);
3,5–дифтор–4–[[4–метил–5–[5–(морфолинoметил)–2–фурил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 204);
3,5–дифтор–4–[[4–метил–5–[5–[(4–метилпиперазин–1–ил)метил]–2–фурил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 205);
4–[[5–[5–[(диметиламино)метил]–2–фурил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 206);
3,5–дифтор–4–[[4–метил–5–[5–(пирролидин–1–илметил)–2–фурил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 207);
4–[[5–[5–этил–4–(пирролидин–1–илметил)–2–фурил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 208);
4–[[4–метил–5–[5–[(4–метилпиперазин–1–ил)метил]–2–фурил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 209);
3,5–дифтор–4–[[4–метил–5–[6–(2–пирролидин–1–илэтил)–3–пиридил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 210);
4–[[5–[5–(диэтиламинометил)–2–фурил]–4–метил–1,2,4–триазол–3–ил]сульфанил]–3,5–дифторбензолкарбогидроксамовая кислота (соединение 211);
3,5–дифтор–4–[[4–метил–5–[5–(1–пиперидилметил)–2–фурил]–1,2,4–триазол–3–ил]сульфанил]бензолкарбогидроксамовая кислота (соединение 212);
4–[(5–фенилтетразол–2–ил)метил]бензолкарбогидроксамовая кислота (соединение 214);
4–[(5–фенилтетразол–1–ил)метил]бензолкарбогидроксамовая кислота (соединение 215);
4–[(5–фенил–4H–1,2,4–триазол–3–ил)метил]бензолкарбогидроксамовая кислота (соединение 216);
N–гидрокси–4–((4–метил–5–фенил–4H–1,2,4–триазол–3–ил)метил)бензамид (соединение 217).
3. Соединение по п. 2, выбранное из:
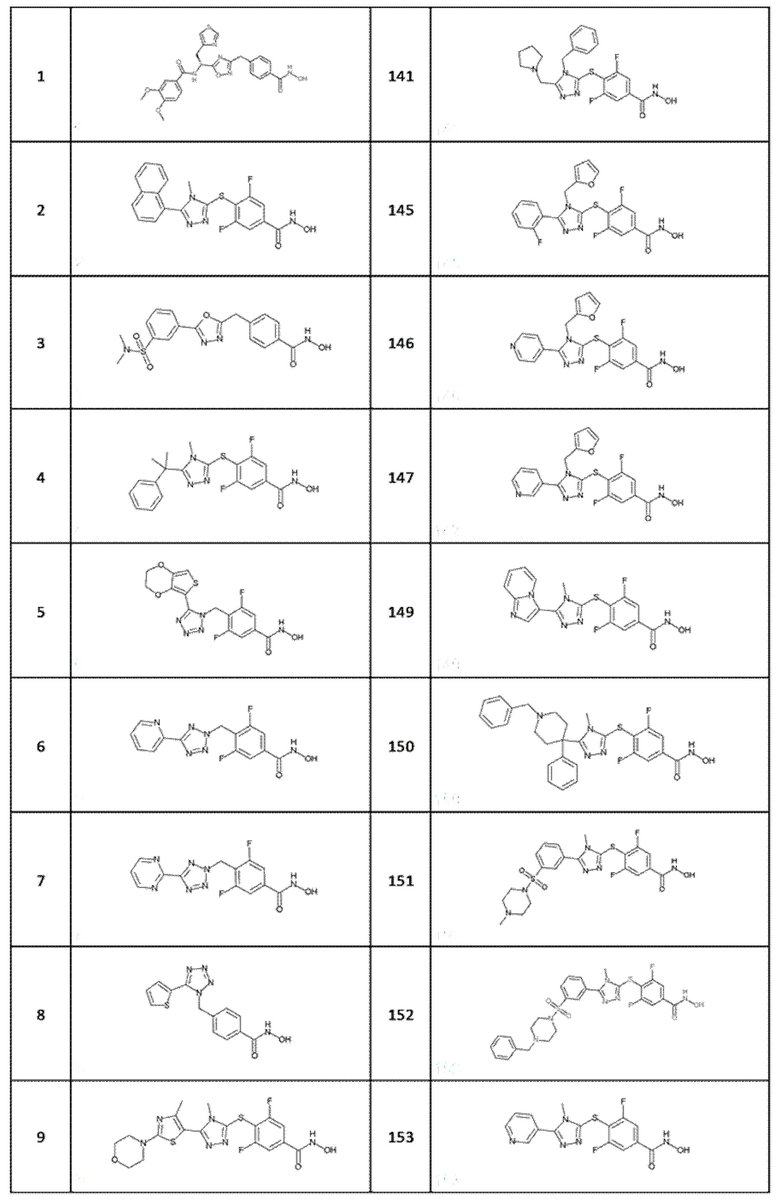
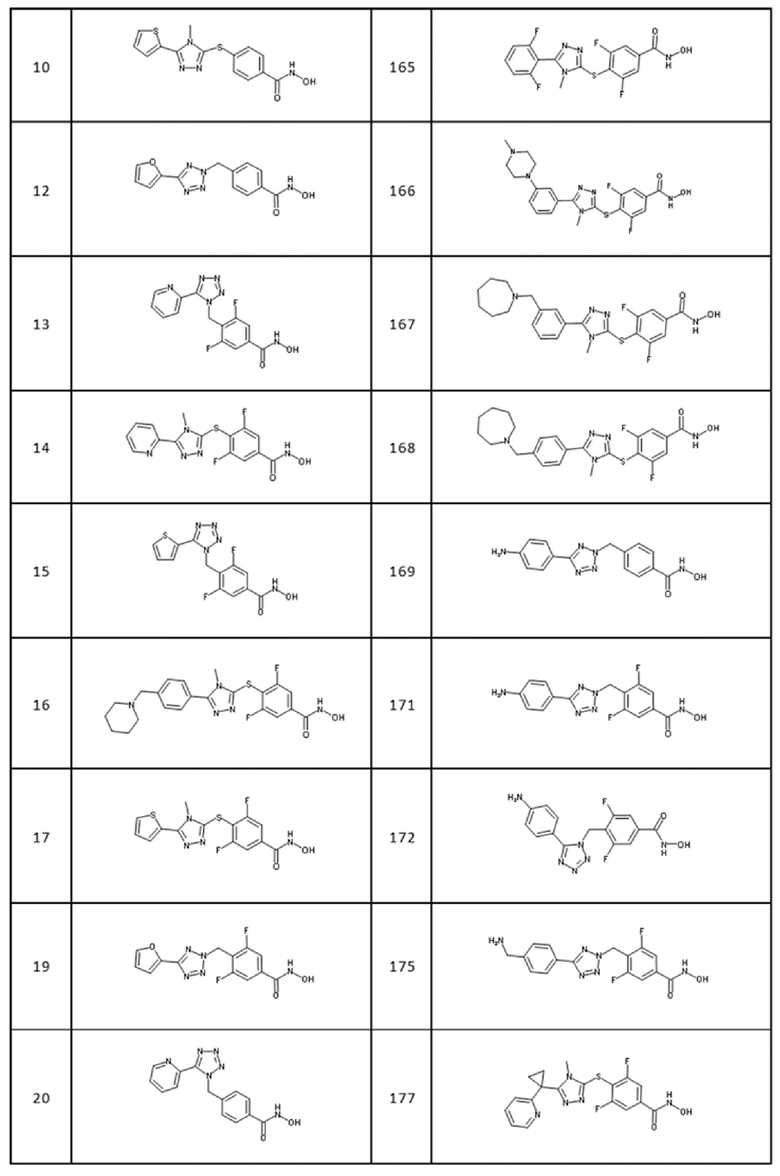
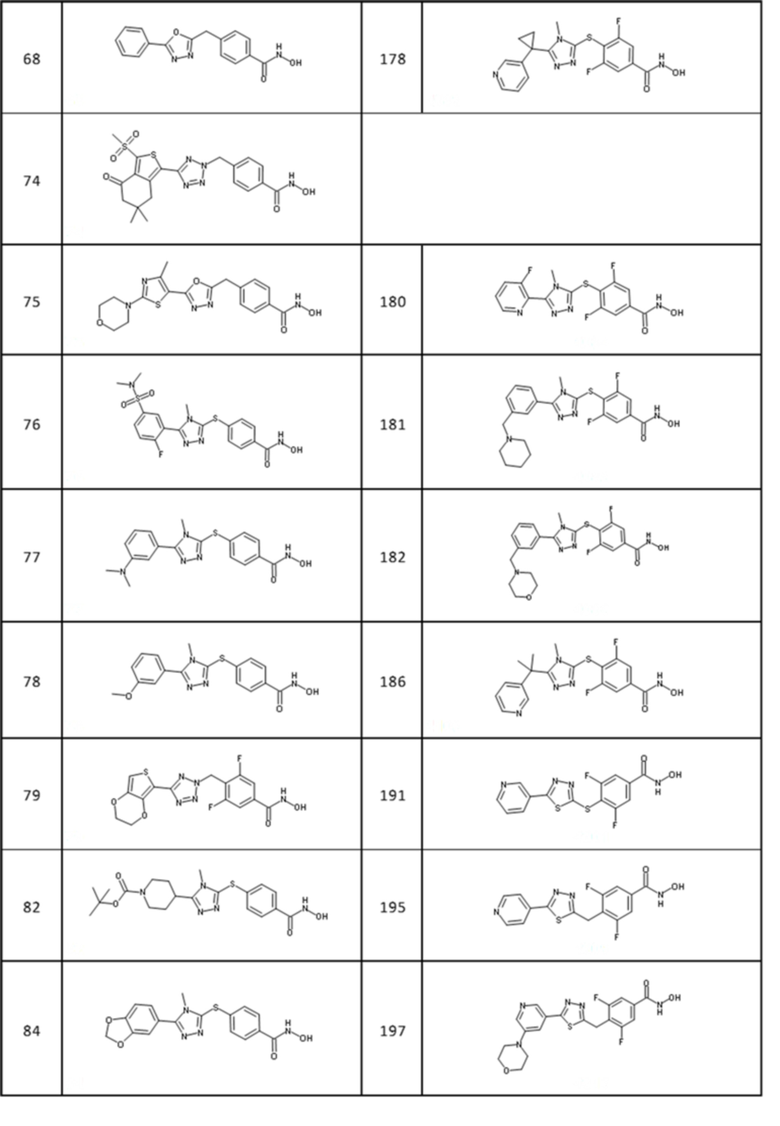
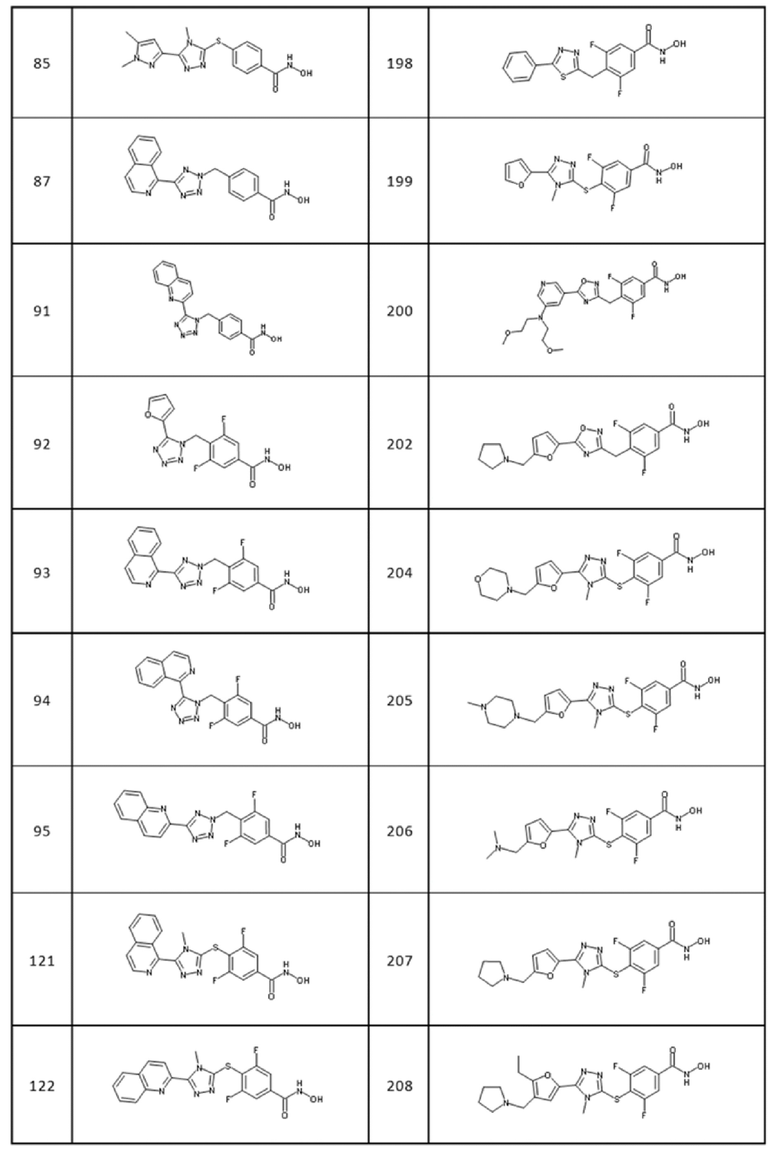
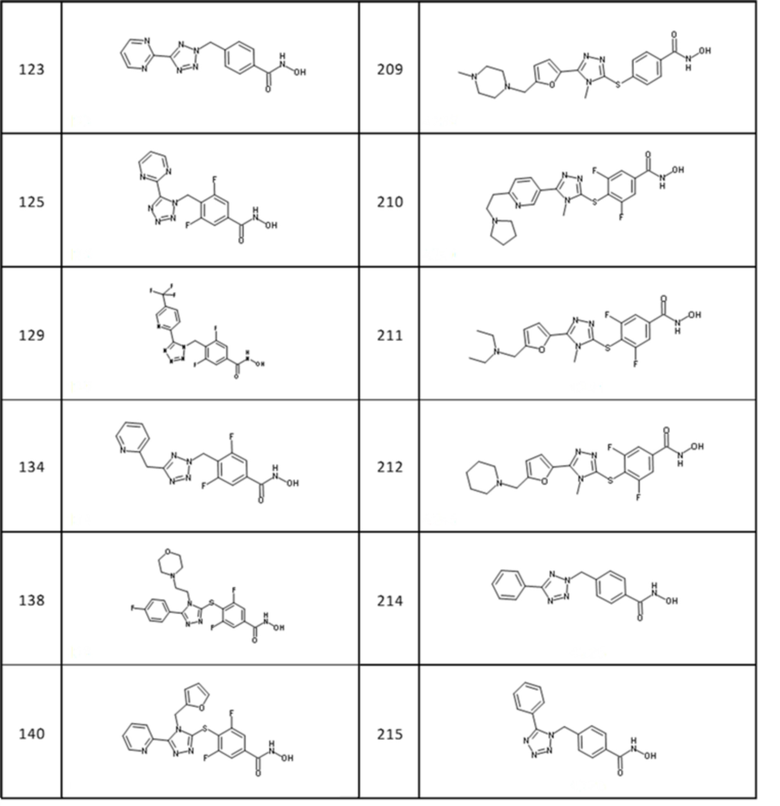
4. Соединение по любому из пп. 1–3, в комбинации с ингибитором контрольной точки PD1.
5. Соединение по любому из пп. 1-4 для применения при лечении одного или нескольких заболеваний, опосредованных HDAC6, выбранных из множественной миеломы, рака молочной железы и рака толстой кишки.
6. Фармацевтическая композиция для лечения заболеваний, опосредованных HDAC6, содержащая терапевтически эффективное количество по меньшей мере одного из соединений формулы (I) или (II) или их фармацевтически приемлемых солей и стереоизомеров по любому из пп. 1–4 вместе с по меньшей мере одним фармацевтически приемлемым эксципиентом.
7. Фармацевтическая композиция по п. 6, подходящая для введения энтеральным путем, парентеральным путем, пероральным путем, местным путем или ингаляционным путем.
8. Фармацевтическая композиция по п. 6 или 7 в форме жидкости или твердого вещества, предпочтительно в форме капсул, таблеток, таблеток с покрытием, порошков, гранул, кремов или мазей.
| WO 2011021209 A1, 24.02.2011 | |||
| WO 2012106343 A2, 09.08.2012 | |||
| WO 2012178208 A2, 27.12.2012 | |||
| SERGIO VALENTE ET AL, Journal of Medicinal Chemistry, vol | |||
| Способ получения на волокне оливково-зеленой окраски путем образования никелевого лака азокрасителя | 1920 |
|
SU57A1 |
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| ИСПАРИТЕЛЬ ДЛЯ ДВИГАТЕЛЕЙ ВНУТРЕННЕГО ГОРЕНИЯ | 1925 |
|
SU6259A1 |
| Посадочная гильза | 1930 |
|
SU25345A1 |

