СМЕЖНЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет по предварительной заявке на патент США № 62/097,835, поданной 30 декабря 2014 г., и предварительной заявке на патент США № 62/163,150, поданной 18 мая 2015 г. Содержание этих предварительных заявок полностью включено в настоящий документ путем ссылки.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Инфекция вируса хронического гепатита B (ВГВ) представляет собой глобальную проблему здравоохранения и поражает свыше 5% населения планеты (более 350 миллионов человек по всему миру и 1,25 миллиона субъектов в США).
Несмотря на наличие профилактической вакцины против ВГВ, влияние хронической инфекции ВГВ по-прежнему остается серьезной нерешенной глобальной медицинской проблемой в связи с недостаточными возможностями лечения и стабильными частотами появления новых инфекций в большинстве развивающихся стран. Текущие способы лечения не обеспечивают выздоровление и ограничены только двумя классами агентов (интерферон-альфа и аналоги нуклеозида/ингибиторы вирусной полимеразы); их воздействие ограничивают лекарственная устойчивость, низкая эффективность и проблемы с переносимостью. Низкие частоты выздоровления от ВГВ по меньшей мере частично объясняются тем фактом, что полного подавления продукции вируса сложно достичь с помощью одного противовирусного агента. Однако постоянное подавление ДНК ВГВ замедляет прогрессирование заболевания печени и помогает предотвратить гепатоцеллюлярную карциному. Текущая терапия инфицированных ВГВ пациентов направлена на снижение сывороточной ДНК ВГВ до низких или необнаруживаемых концентраций и, в конечном счете, на замедление или предотвращение развития цирроза печени и гепатоцеллюлярной карциномы.
В данной области существует потребность в терапевтических агентах, которые могут усиливать подавление продукции вируса и которые могут обеспечивать лечение, облегчение и/или предотвращение инфекции ВГВ. Введение таких терапевтических агентов инфицированному ВГВ пациенту либо в качестве монотерапии, либо в комбинации с другими (или вспомогательными) способами лечения ВГВ приведет к существенному снижению вирусной нагрузки, улучшению прогноза, замедлению прогрессирования заболевания и повышению показателей сероконверсии.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложены соединения, используемые для лечения инфекции ВГВ у нуждающегося в этом субъекта, имеющие следующую структуру:
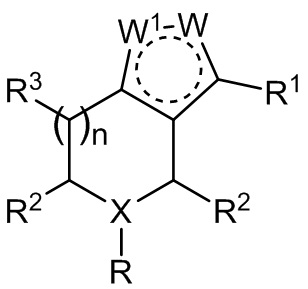 ,
,
или их фармацевтически приемлемая соль.
В одном аспекте в настоящем документе предложено соединение формулы I:
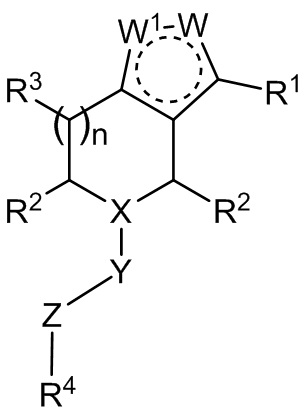
I
или его фармацевтически приемлемая соль.
В одном варианте осуществления соединение формулы I представляет собой соединение формулы II:
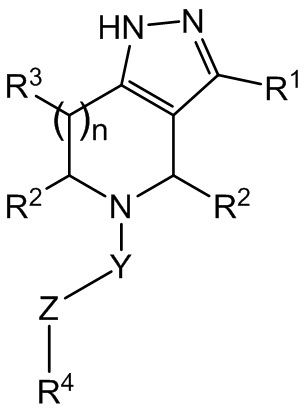
II
или его фармацевтически приемлемую соль.
В другом варианте осуществления соединение формулы I представляет собой соединение формулы III:
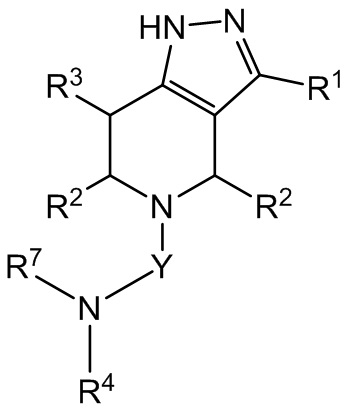
III
или его фармацевтически приемлемую соль.
В другом варианте осуществления соединение формулы I представляет собой соединение формулы IV:
IV
или его фармацевтически приемлемую соль.
В другом аспекте настоящего изобретения предложены фармацевтические композиции, содержащие соединение формулы I, II, III или IV или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
В одном аспекте настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ эрадикации инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ снижения вирусной нагрузки, связанной с инфекцией ВГВ, у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ снижения повторного возникновения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ ингибирования или снижения образования или наличия частиц, содержащих ДНК ВГВ, или частиц, содержащих РНК ВГВ, у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ снижения негативного физиологического влияния инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ индукции ремиссии поражения печени из-за инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ снижения физиологического влияния долгосрочной противовирусной терапии инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ профилактического лечения инфекции ВГВ у нуждающегося в этом субъекта, страдающего от латентной инфекции ВГВ, включающий введение субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли.
В одном варианте осуществления способы, предложенные в настоящем документе, могут дополнительно включать введение субъекту по меньшей мере одного дополнительного терапевтического агента, выбранного из группы, состоящей из ингибитора полимеразы ВГВ, иммуномодулирующих агентов, пегилированного интерферона, ингибитора проникновения вируса в клетку, ингибитора созревания вируса, описанного в литературе модулятора сборки капсида, ингибитора обратной транскриптазы, ингибитора циклофилина/ФНО, агониста толл-подобного рецептора (TLR), вакцины против ВГВ и агентов с другим или неизвестным механизмом действия и их комбинации. В дополнительном варианте осуществления предложенные в настоящем документе способы позволяют вводить по меньшей мере один дополнительный терапевтический агент в меньшей дозе или с меньшей частотой по сравнению с введением по меньшей мере одного дополнительного терапевтического агента в виде монотерапии, чем требуемая для достижения аналогичных результатов в профилактическом лечении инфекции ВГВ у нуждающегося в этом субъекта.
В другом варианте осуществления способы, предложенные в настоящем документе, снижают вирусную нагрузку у субъекта в большей степени или с большей скоростью по сравнению с введением соединения, выбранного из группы, состоящей из ингибитора полимеразы ВГВ, интерферона, ингибитора проникновения вируса в клетку, ингибитора созревания вируса, другого модулятора сборки капсида, противовирусных соединений с другим или неизвестным механизмом действия и любой их комбинации.
В другом варианте осуществления способы, предложенные в настоящем документе, приводят к снижению частоты мутации вируса и/или устойчивости вируса по сравнению с введением соединения, выбранного из группы, состоящей из ингибитора полимеразы ВГВ, интерферона, ингибитора проникновения вируса в клетку, ингибитора созревания вируса, другого модулятора сборки капсида, противовирусных соединений с другим или неизвестным механизмом действия и их комбинации.
В другом варианте осуществления способы, предложенные в настоящем документе, дополнительно включают введение субъекту по меньшей мере одной вакцины против ВГВ, ингибитора нуклеозида ВГВ, интерферона или любой их комбинации.
В одном аспекте настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий снижение вирусной нагрузки ВГВ посредством введения субъекту терапевтически эффективного количества соединения формулы I, II, III или IV или его фармацевтически приемлемой соли в виде монотерапии или в комбинации с ингибитором обратной транскриптазы; и дополнительного введения субъекту терапевтически эффективного количества вакцины против ВГВ.
В одном варианте осуществления способы, предложенные в настоящем документе, дополнительно включают контроль вирусной нагрузки ВГВ у субъекта, причем способ осуществляют в течение периода времени до тех пор, пока вирус ВГВ перестает обнаруживаться.
ПОДРОБНОЕ ОПИСАНИЕ
В настоящем документе предложены соединения, например соединения формулы I, II, III или IV или их фармацевтически приемлемые соли, которые используются при лечении и профилактике инфекции ВГВ у субъекта. В не имеющем ограничительного характера аспекте эти соединения могут модулировать или нарушать сборку ВГВ и другие функции ядерного белка ВГВ, необходимые для репликации ВГВ или образования инфекционных частиц, могут ингибировать продукцию инфекционных вирусных частиц или инфекцию или могут взаимодействовать с капсидом ВГВ с получением дефектных вирусных частиц со значительно сниженной инфективностью или способностью к репликации. Другими словами, предложенные в настоящем документе соединения могут выступать в качестве модуляторов сборки капсида. Предложенные в настоящем документе соединения обладают выраженной противовирусной активностью, показывают благоприятные метаболические свойства, распределение в ткани, профили безопасности и фармацевтические профили и являются приемлемыми для применения у людей.
Капсидный белок ВГВ выполняет важнейшие функции в течение жизненного цикла вируса. Капсидные/ядерные белки ВГВ образуют метастабильные вирусные частицы или белковые оболочки, которые защищают вирусный геном во время межклеточного перехода, а также играют центральную роль в процессах репликации вируса, включая капсидирование генома, репликацию генома и морфогенез и выход вириона. Структуры капсида также реагируют на внешние стимулы, обеспечивая декапсидацию после проникновения вируса в клетку. Таким образом, обнаружено, что соответствующее время сборки и разборки капсида, надлежащая стабильность капсида и функция ядерного белка представляют собой критические факторы для инфективности вируса.
Решающая функция белков капсида ВГВ накладывает строгие эволюционные ограничения на последовательность капсидного белка вируса, приводя к наблюдаемой низкой изменчивости последовательности и высокой консервативности. Таким образом, мутации в капсиде ВГВ, которые нарушают его сборку, являются летальными, а мутации, которые ухудшают стабильность капсида, существенно ослабляют репликацию вируса. Значительные функциональные ограничения, присущие многофункциональному ядерному/капсидному белку ВГВ, согласуются с высокой консервативностью последовательности, так как большое число мутаций является губительным для функции. Действительно, последовательности ядерного/капсидного белка являются более чем на 90% идентичными в различных генотипах ВГВ и имеют только небольшое число полиморфных остатков. Следовательно, может быть сложно выполнить отбор по устойчивости к соединениям, связывающимся с ядерным/капсидным белком ВГВ, без выраженного воздействия на пригодность вируса к репликации.
Статьи, описывающие соединения, которые связываются с вирусными капсидами и ингибируют репликацию ВИЧ, риновируса и ВГВ, обеспечивают мощное фармакологическое доказательство концепции, подразумевающей, что капсидные белки вируса являются мишенями противовирусного лекарственного средства.
В одном аспекте предложенные в настоящем документе соединения используются для лечения ВГВ посредством нарушения, ускорения, снижения, замедления и/или ингибирования нормальной сборки и/или разборки вирусного капсида, что индуцирует образование ненормальной морфологии капсида и приводит к противовирусным эффектам, таким как нарушение сборки и/или разборки вириона, созревания вириона, выхода вируса и/или инфицирование клеток-мишеней. В одном варианте осуществления соединение, нарушающее сборку капсида, взаимодействует со зрелым или незрелым вирусным капсидом для снижения стабильности капсида, таким образом оказывая воздействие на сборку и\или разборку. В другом варианте осуществления соединение, нарушающее сборку капсида, повреждает фолдинг белка и/или солевые мостики, требуемые для стабильности, функционирования и/или нормальной морфологии вирусного капсида, таким образом нарушая и/или ускоряя сборку и/или разборку капсида. В еще одном варианте осуществления соединения настоящего изобретения связываются с капсидом и изменяют метаболизм клеточных полипротеинов и предшественников, приводя к ненормальному накоплению белковых мономеров и/или олигомеров и/или ненормальных частиц, что вызывает клеточную токсичность и гибель инфицированных клеток. В другом варианте осуществления предложенные в настоящем документе соединения приводят к нарушению образования капсидов с оптимальной стабильностью, влияя на эффективную декапсидацию и/или разборку вирусов (например, во время заражения).
В одном варианте осуществления предложенные в настоящем документе соединения нарушают и/или ускоряют сборку и/или разборку капсида, когда капсидный белок является незрелым. В другом варианте осуществления предложенные в настоящем документе соединения нарушают и/или ускоряют сборку и/или разборку капсида, когда капсидный белок является зрелым. В еще одном варианте осуществления предложенные в настоящем документе соединения нарушают и/или ускоряют сборку и/или разборку капсида во время заражения вирусом. В еще одном варианте осуществления нарушение и/или ускорение сборки и/или разборки капсида ослабляет инфективность вируса ВГВ и/или снижает вирусную нагрузку. В еще одном варианте осуществления нарушение, ускорение, ингибирование, замедление и/или ослабление сборки и/или разборки капсида приводит к эрадикации вируса из организма-хозяина. В еще одном варианте осуществления эрадикация вируса ВГВ из хозяина преимущественно избавляет от необходимости хронической долгосрочной терапии и/или уменьшает длительность долгосрочной терапии.
В одном варианте осуществления описанные в настоящем документе соединения являются приемлемыми для монотерапии и эффективными против природных или нативных штаммов ВГВ и против штаммов ВГВ, устойчивых к известным в настоящее время лекарственным средствам. В другом варианте осуществления описанные в настоящем документе соединения являются приемлемыми для применения в комбинированной терапии.
В другом варианте осуществления предложенные в настоящем документе соединения можно использовать в способах модуляции (например, ингибирования или нарушения) активности, стабильности, функции и относящихся к репликации вируса свойств замкнутой ковалентно-непрерывной кольцевой ДНК ВГВ. В еще одном варианте осуществления соединения настоящего изобретения можно использовать в способах уменьшения или предотвращения образования замкнутой ковалентно-непрерывной кольцевой ДНК ВГВ.
В другом варианте осуществления предложенные в настоящем документе соединения можно использовать в способах модуляции (например, ингибирования или нарушения) активности замкнутой ковалентно-непрерывной кольцевой ДНК ВГВ. В еще одном варианте осуществления соединения настоящего изобретения можно использовать в способах уменьшения образования замкнутой ковалентно-непрерывной кольцевой ДНК ВГВ.
В другом варианте осуществления предложенные в настоящем документе соединения можно использовать в способах модуляции, ингибирования или нарушения образования или высвобождения частиц РНК ВГВ из инфицированной клетки. В дополнительном варианте осуществления выполняют модуляцию общей нагрузки (или концентрации) частиц РНК ВГВ. В предпочтительном варианте осуществления уменьшают общую нагрузку РНК ВГВ.
Определения
Ниже перечислены определения различных терминов, использующихся для описания настоящего изобретения. Эти определения относятся только к терминам, использующимся в настоящем описании и формуле изобретения, если специально не указано иное, индивидуально или как часть более крупной группы.
Если не оговорено иное, все используемые в настоящем документе технические и научные термины по существу имеют общепринятое значение, понятное любому специалисту в области, к которой имеет отношение настоящее изобретение. По существу условные обозначения, применяемые в настоящем документе, и лабораторные процедуры в клеточной культуре, методы молекулярной генетики, органической химии и химии пептидов являются хорошо известными и часто используются в данной области.
В настоящем документе существительные в единственном числе используются для обозначения одного или нескольких (т. е. по меньшей мере одного) грамматических объектов. Например, «элемент» означает «один элемент или несколько элементов». Более того, применение термина «включая», а также других форм, например «включают», «включает» и «включал», не имеет ограничительного характера.
В настоящем документе термин «около» будет понятен специалистам в данной области, и его значение будет в некоторой степени зависеть от контекста, в котором он используется. В настоящем документе термин «около» при указании измеримой величины, такой как количество, продолжительность по времени и т. п., считается охватывающим отклонения ± 20% или ± 10%, включая ± 5%, ± 1% и ± 0,1%, от указанного значения, поскольку такие отклонения приемлемы для реализации описанных способов.
В настоящем документе термин «модулятор сборки капсида» относится к соединению, которое нарушает, или ускоряет, или ингибирует, или препятствует, или замедляет, или уменьшает, или модифицирует нормальную сборку капсида (например, во время созревания) или нормальную разборку капсида (например, во время заражения), или снижает стабильность капсида, таким образом вызывая нарушение морфологии или функционирования капсида. В одном варианте осуществления модулятор сборки капсида ускоряет сборку или разборку капсида, таким образом вызывая нарушение морфологии капсида. В другом варианте осуществления модулятор сборки капсида взаимодействует (например, связывается в активном участке, связывается в аллостерическом участке, модифицирует и/или препятствует фолдингу и т. п.) с главным белком сборки капсида (CA), таким образом нарушая сборку или разборку капсида. В еще одном варианте осуществления модулятор сборки капсида вызывает нарушение структуры или функции CA (например, способность CA к сборке, разборке, связыванию с субстратом, укладке в приемлемую конформацию или т. п.), что ослабляет инфективность вируса и/или является летальным для вируса.
В настоящем документе термин «способ лечения» или «лечение» обозначает применение или введение терапевтического агента, т. е. соединения настоящего изобретения (в виде монотерапии или в композиции с другим фармацевтическим агентом), пациенту или применение или введение терапевтического агента в выделенную ткань или клеточную линию, полученную от пациента (например, для диагностики или применения ex vivo), у которого имеется инфекция ВГВ, симптом инфекции ВГВ или потенциал к развитию инфекции ВГВ, с целью излечения, устранения, ослабления, изменения, уничтожения, улучшения или воздействия на инфекцию ВГВ, симптомы инфекции ВГВ или потенциал к развитию инфекции ВГВ. Такие способы лечения могут быть специально приспособлены или модифицированы на основании знаний, полученных в области фармакогеномики.
В настоящем документе термин «предотвращать» или «предотвращение» означает отсутствие развития расстройства или заболевания, если этого не произошло, или отсутствие дополнительного развития расстройства или заболевания, если такое развитие расстройства или заболевания уже произошло. Также подразумевается способность предотвращать некоторые или все симптомы, связанные с расстройством или заболеванием.
В настоящем документе термин «пациент» или «субъект» означает человека или не относящееся к человеку млекопитающее. Не относящиеся к человеку млекопитающие включают, например, домашний скот и домашних животных, таких как овцы, коровы, свиньи, собаки, кошки и мыши. Предпочтительно пациент или субъект представляет собой человека.
В настоящем документе термин «эффективное количество», «фармацевтически эффективное количество» и «терапевтически эффективное количество» означает нетоксичное, но достаточное количество агента для достижения желаемого биологического результата. Таким результатом может быть уменьшение и/или ослабление признаков, симптомов или причин заболевания, либо любое другое желательное изменение биологической системы. Нужное терапевтическое количество в каждом отдельном случае может определяться специалистом в данной области с помощью стандартных экспериментов.
В настоящем документе термин «фармацевтически приемлемый» относится к материалу, такому как носитель или разбавитель, который не подавляет биологическую активность или свойства соединения и является относительно нетоксичным, т. е. материал можно вводить лицу, что не вызывает нежелательных биологических воздействий или взаимодействия нежелательным образом с любым из компонентов композиции, в которой он содержится.
В настоящем документе под термином «фармацевтически приемлемая соль» понимают производные описанных соединений, где исходное соединение модифицируют путем преобразования имеющегося кислотного или щелочного остатка в его солевую форму. Примеры фармацевтически приемлемых солей включают, без ограничений, неорганические или органические кислые соли щелочных остатков, например амины; щелочные или органические соли кислотных остатков, таких как карбоновые кислоты; и т. п. Фармацевтически приемлемые соли настоящего изобретения включают стандартные нетоксичные соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли настоящего изобретения можно синтезировать из исходного соединения, которое содержит щелочной или кислотный остаток, традиционными химическими способами. По существу такие соли можно получить в результате реакции свободных кислотных или щелочных форм этих соединений со стехиометрическим количеством соответствующей щелочи или кислоты в воде, или в органическом растворителе, или в смеси этих двух веществ; как правило, предпочтительна неводная среда, например эфир, этилацетат, этанол, изопропанол или ацетонитрил. Список приемлемых солей приведен в публикациях Remingtonʹs Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 и Journal of Pharmaceutical Science, 66, 2 (1977), каждая из которых полностью включена в настоящий документ путем ссылки.
В настоящем документе термин «композиция» или «фармацевтическая композиция» означает смесь по меньшей мере одного соединения, используемого в настоящем изобретении, с фармацевтически приемлемым носителем. Фармацевтическая композиция облегчает введение соединения пациенту или субъекту. В данной области существует множество способов введения соединения, включая, без ограничений, внутривенное, пероральное, аэрозольное, парентеральное, глазное, легочное и местное введение.
В настоящем документе термин «фармацевтически приемлемый носитель» означает фармацевтически приемлемый материал, композицию или носитель, такой как жидкий или твердый наполнитель, стабилизатор, диспергирующий агент, суспендирующий агент, разбавитель, эксципиент, загуститель, растворитель или инкапсулирующий материал, участвующий в переносе или транспортировке соединения, используемого в настоящем изобретении, внутрь или на пациента таким образом, что он может выполнять предусмотренную функцию. Как правило, такие конструкции переносятся или транспортируются из одного органа или участка тела к другому органу или участку тела. Каждый носитель должен быть «приемлемым» в плане наличия совместимости с другими ингредиентами состава, включая соединение, используемое в настоящем изобретении, и безвредным для пациента. Некоторые примеры материалов, которые могут выступать в качестве фармацевтически приемлемых носителей, включают сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и ацетатцеллюлоза; порошковую трагакантовую камедь; солод; желатин; тальк; эксципиенты, такие как масло какао и свечные воски; масла, такие как растительное арахисовое масло, хлопковое масло, сафлоровое масло, сезамовое масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферизующие агенты, такие как гидроксид магния и гидроксид алюминия; поверхностно-активные вещества; альгиновую кислоту; апирогенную воду; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы; и другие нетоксичные совместимые вещества, используемые в фармацевтических составах.
В настоящем документе термин «фармацевтически приемлемый носитель» также включает любые покрытия, антибактериальные и противогрибковые агенты и замедляющие всасывание агенты и т. п., которые являются совместимыми с действием соединения, используемого в настоящем изобретении, и являются физиологически приемлемыми для пациента. В композиции также можно включить вспомогательные активные соединения. Термин «фармацевтически приемлемый носитель» может дополнительно включать фармацевтически приемлемую соль соединения, используемого в настоящем изобретении. Другие дополнительные ингредиенты, которые могут быть включены в фармацевтические композиции, используемые в практике настоящего изобретения, известны в данной области и описаны, например, в публикации Remingtonʹs Pharmaceutical Sciences (Genaro, Ed., Mack Publishing Co., 1985, Easton, PA), которая включена в настоящий документ путем ссылки.
В настоящем документе термин «алкил» отдельно или как часть другого заместителя означает, при отсутствии иных указаний, прямую или разветвленную углеводородную цепь, имеющую определенное число обозначенных атомов углерода (т. е. C1-C6 алкил означает от одного до шести атомов углерода), и включает прямую, разветвленную цепь. Примеры включают метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, неопентил и гексил. Другие примеры C1-C6 алкила включают этил, метил, изопропил, изобутил, н-пентил и н-гексил.
В настоящем документе термин «алкенил» означает моновалентную группу, полученную из углеводородной функциональной группы, которая содержит по меньшей мере два атома углерода и по меньшей мере одну двойную углеродную связь. Двойная связь может быть или может не быть местом присоединения другой группы. Алкенильные группы (например, C2-C8 алкенил) включают, без ограничений, например, этенил, пропенил, проп-1-ен-2-ил, бутенил, 1-метил-2-бутен-1-ил, гептенил, октенил и т. п.
В настоящем документе термин «галоген» отдельно или как часть другого заместителя означает, при отсутствии иных указаний, атом фтора, хлора, брома или иода, предпочтительно фтора, хлора или брома, более предпочтительно фтора или хлора.
В настоящем документе термин «галогеналкил» означает алкильные радикалы, в которых один или более атомов углерода алкила замещены галогеном, как определено выше. Галогеналкил включает моногалогенаклильный, дигалогеналкильный и полигалогеналкильный радикалы. Термин «галогеналкил» включает, без ограничений, фторметил, дифторметил, трифторметил, хлорметил, дихлорметил, трихлорметил и пентафторэтил.
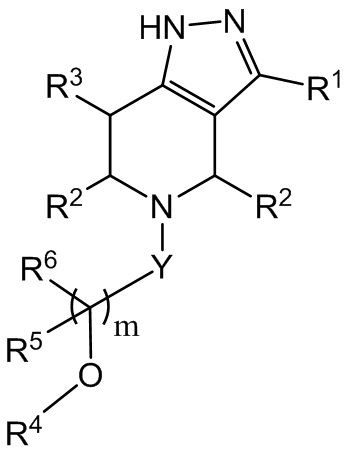
В настоящем документе термин «циклоалкил» означает моноциклический или полициклический неароматический радикал, в котором каждый из атомов, образующих кольцо (т. е. атомов каркаса), представляет собой атом углерода. В одном варианте осуществления циклоалкильная группа является насыщенной или частично ненасыщенной. В другом варианте осуществления циклоалкильная группа соединена с ароматическим кольцом. Циклоалкильные группы включают группы, имеющие от 3 до 10 атомов кольца (C3-C10 циклоалкил), группы, имеющие от 3 до 8 атомов кольца (C3-C8 циклоалкил), группы, имеющие от 3 до 7 атомов кольца (C3-C7 циклоалкил), и группы, имеющие от 3 до 6 атомов кольца (C3-C6 циклоалкил). Наглядные примеры циклоалкильных групп включают, без ограничений, следующие функциональные группы:
 .
.
Моноциклические циклоалкилы группы включают, без ограничений, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Дициклические циклоалкилы включают, без ограничений, тетрагидронафтил, инданил и тетрагидропентален. Полициклические циклоалкилы включают адамантин и норборнан. Термин циклоалкил включает группы «ненасыщенного неароматического карбоциклила» или «неароматического ненасыщенного карбоциклила», обе из которых относятся к неароматическому углеродному циклу, как определено в настоящем документе, который содержит по меньшей мере одну двойную углеродную связь или одну тройную углеродную связь.
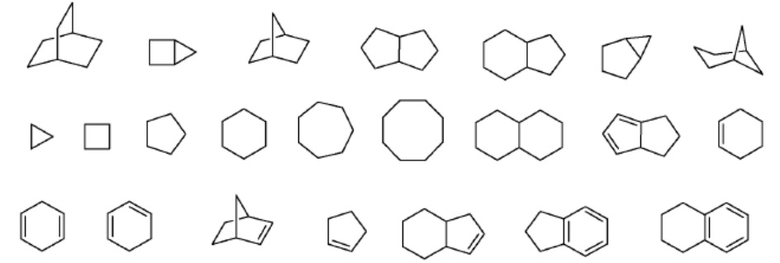
В настоящем документе термин «гетероциклоалкил» или «гетероциклил» означает гетероалициклическую группу, содержащую от одного до четырех гетероатомов кольца, каждый из которых выбран из O, S и N. В одном варианте осуществления каждая гетероциклическая группа имеет в своей кольцевой системе от 3 до 10 атомов, при условии, что кольцо указанной группы не содержит двух смежных атомов O или S. Гетероциклические заместители могут быть альтернативно образованы определенным числом атомов углерода, например, C2-C8 гетероциклил указывает на число атомов углерода, содержащихся в гетероциклической группе без включения числа гетероатомов. Например, C2-C8 гетероциклил будет включать дополнительно от одного до четырех гетероатомов. В другом варианте осуществления гетероциклоалкильная группа соединена с ароматическим кольцом. В одном варианте осуществления гетероатомы азота и серы могут быть необязательно окислены, а атом азота может быть необязательно кватернизован. Гетероциклическая система может быть присоединена, при отсутствии иных указаний, к любому гетероатому или атому углерода, который обеспечивает стабильную структуру.
Пример 3-членной гетероциклической группы включает, без ограничений, азиридин. Примеры 4-членных гетероциклических групп включают, без ограничений, азетидин и бета-лактам. Примеры 5-членных гетероциклических групп включают, без ограничений, пирролидин, оксазолидин и тиазолидиндион. Примеры 6-членных гетероциклоалкильных групп включают, без ограничений, пиперидин, морфолин и пиперазин.
Другие не имеющие ограничительного характера примеры представляют собой:
 .
.
Примеры гетероциклов включают моноциклические группы, такие как азиридин, оксиран, тииран, азетидин, оксетан, тиетан, пирролидин, пирролин, пиразолидин, имидазолин, диоксолан, сульфолан, 2,3-дигидрофуран, 2,5-дигидрофуран, тетрагидрофуран, тиофан, пиперидин, 1,2,3,6-тетрагидропиридин, 1,4-дигидропиридин, пиперазин, морфолин, тиоморфолин, пиран, 2,3-дигидропиран, тетрагидропиран, 1,4-диоксан, 1,3-диоксан, гомопиперазин, гомопиперидин, 1,3-диоксепан, 4,7-дигидро-1,3-диоксепин и гексаметиленоксид.
В настоящем документе термин «ароматический» означает углеродный цикл или гетероцикл с одним или более полиненасыщенными кольцами и имеющий ароматический характер, т. е. имеющий (4n+2) делокализованных π (пи) электронов, где n представляет собой целое число.
В настоящем документе термин «арил», используемый самостоятельно или в комбинации с другими терминами, означает, при отсутствии иных указаний, карбоциклическую ароматическую систему, содержащую одно или более колец (как правило, одно, два или три кольца), причем такие кольца могут быть соединены друг с другом по типу подвешивания, как в бифениле, или могут быть слитыми, как в нафталине. Примеры арильных групп включают фенил, антрацил и нафтил. Предпочтительные примеры представляют собой фенил (например, C6 арил) и бифенил (например, C12 арил). В некоторых вариантах осуществления арильные группы имеют от шести до шестнадцати атомов углерода. В некоторых вариантах осуществления арильные группы имеют от шести до двенадцати атомов углерода (например, C6-C12 арил). В некоторых вариантах осуществления арильные группы имеют шесть атомов углерода (например, C6 арил).
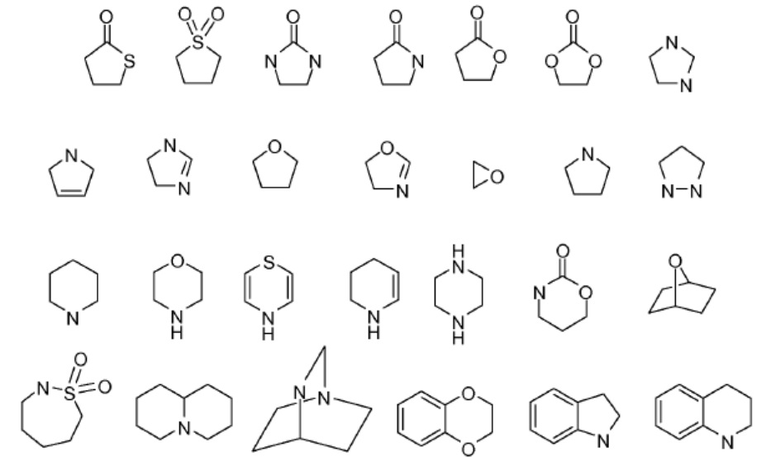
В настоящем документе термин «гетероарил» или «гетероароматический» означает гетероцикл, имеющий ароматический характер. Гетероарильные заместители могут быть образованы определенным числом атомов углерода, например, C1-C9 гетероарил указывает на число атомов углерода, содержащихся в гетероарильной группе без включения числа гетероатомов. Например, C1-C9 гетероарил будет дополнительно включать от одного до четырех гетероатомов. Полициклический гетероарил может включать одно или более колец, которые являются частично насыщенными. Не имеющие ограничительного характера примеры гетероарилов включают:
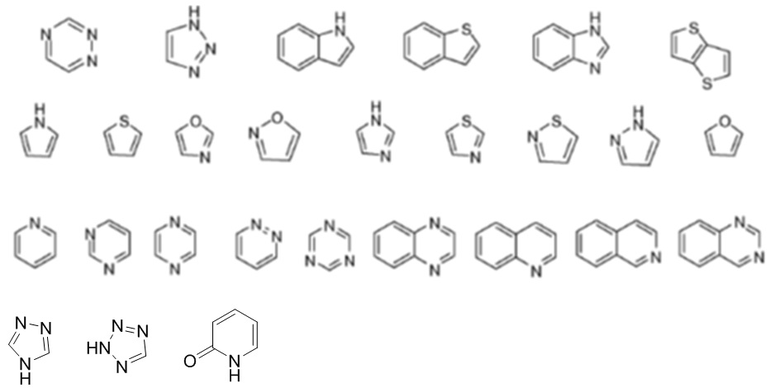 .
.
Дополнительные не имеющие ограничительного характера примеры гетероарильных групп включают пиридил, пиразинил, пиримидинил (включая, например, 2- и 4-пиримидинил), пиридазинил, тиенил, фурил, пирролил (включая, например, 2-пирролил), имидазолил, тиазолил, оксазолил, пиразолил (включая, например, 3- и 5-пиразолил), изотиазолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,3,4-триазолил, тетразолил, 1,2,3-тиадиазолил, 1,2,3-оксадиазолил, 1,3,4-тиадиазолил и 1,3,4-оксадиазолил.
Не имеющие ограничительного характера примеры полициклических гетероциклов и гетероарилов включают индолил (включая, например, 3-, 4-, 5-, 6- и 7-индолил), индолинил, хинолил, тетрагидрохинолил, изохинолил (включая, например, 1- и 5-изохинолил), 1,2,3,4-тетрагидроизохинолил, циннолинил, хиноксалинил (включая, например, 2- и 5-хиноксалинил), хиназолинил, фталазинил, 1,8-нафтиридинил, 1,4-бензодиоксанил, кумарин, дигидрокумарин, 1,5-нафтиридинил, бензофурил (включая, например, 3-, 4-, 5-, 6- и 7-бензофурил), 2,3-дигидробензофурил, 1,2-бензизоксазолил, бензотиенил (включая, например, 3-, 4-, 5-, 6- и 7-бензотиенил), бензоксазолил, бензотиазолил (включая, например, 2-бензотиазолил и 5-бензотиазолил), пуринил, бензимидазолил (включая, например, 2-бензимидазолил), бензотриазолил, тиоксантинил, карбазолил, карболинил, акридинил, пирролизидинил и хинолизидинил.
В настоящем документе термин «замещенный» означает, что атом или группа атомов заместила водород в качестве заместителя, присоединенного к другой группе.
Соединения настоящего изобретения
В настоящем изобретении предложены соединения, используемые для лечения инфекции ВГB у нуждающегося в этом субъекта, имеющие следующую структуру:
,
или их фармацевтически приемлемые соли.
В одном аспекте в настоящем документе предложено соединение формулы Ia
Ia
или его фармацевтически приемлемая соль,
где
каждый из W1 и W независимо выбран из N, NRa и CRa, причем один из W1 и W представляет собой NRa;
X представляет собой N или CRb;
Y выбран из связи, -C(O)- и -SO2-;
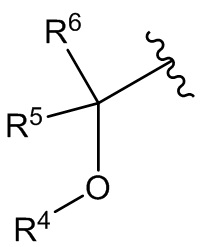
Z выбран из -(CR5R6)m-, -(CR5R6)mO-, -(CR5R6)mCR5=CR5-, -(CR5R6)m-C3-C6 циклоалкилена- и -(CR5R6)m-NR7-;
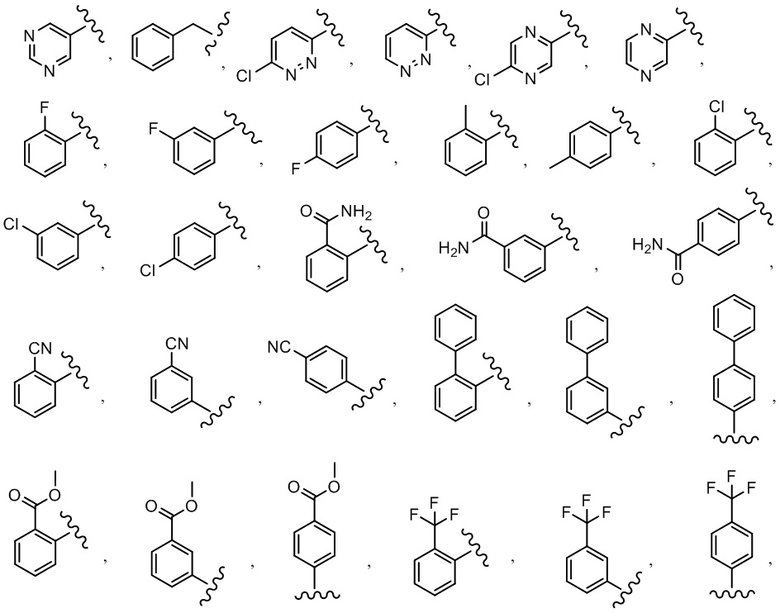
R1 выбран из C6-C12 арила, C1-C9 гетероарила, C3-C8 циклоалкила, C2-C8 гетероциклила, -ORc, C1-C6 алкила, C(O)ORc, C(O)Rc, C(O)NRdRe, NRdC(O)Rc, -OC(O)Rc, галогена и C2-C8 алкенила, причем алкил, арил, гетероарил, циклоалкил, гетероциклил и алкенил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R2 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R3 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R4 выбран из C1-C6 алкила, (CR8R9)p-C3-C8 циклоалкила, (CR8R9)p-C2-C8 гетероциклила, (CR8R9)p-C6-C12 арила и (CR8R9)p-C1-C9 гетероарила, причем алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf, C1-C6 алкил-OH и C3-C8 циклоалкила;
R5 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
альтернативно R4 и R5 необязательно соединены с образованием кольца;
R6 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R7 выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
R8 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R9 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
Ra выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
Rb выбран из H и C1-C6 алкила;
Rc выбран из H, C1-C6 алкила, C1-C6 алкил-OH, C3-C8 циклоалкила, C2-C8 гетероциклила, C6-C12 арила и C1-C9 гетероарила;
Rd выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
Re выбран из H, C1-C6 алкила, C1-C6 алкил-OH, C3-C8 циклоалкила, C2-C8 гетероциклила, C6-C12 арила, C1-C9 гетероарила и -O-C1-C6 алкила;
альтернативно Rd и Re необязательно соединены с образованием гетероциклического кольца;
Rf в каждом случае независимо выбран из H и C1-C6 алкила;
m составляет 0, 1, 2, 3 или 4;
n составляет 0, 1, 2 или 3; и
p составляет 0, 1, 2, 3 или 4.
В другом аспекте в настоящем документе предложено соединение формулы I
I
или его фармацевтически приемлемая соль,
где
каждый из W1 и W независимо выбран из N, NRa и CRa, причем один из W1 и W представляет собой NRa;
X представляет собой N или CRb;
Y выбран из связи, -C(O)- и -SO2-;
Z выбран из -(CR5R6)m-, -(CR5R6)mO-, -(CR5R6)mCR5=CR5-, -(CR5R6)m-C3-C6 циклоалкилена- и -(CR5R6)m-NR7-;
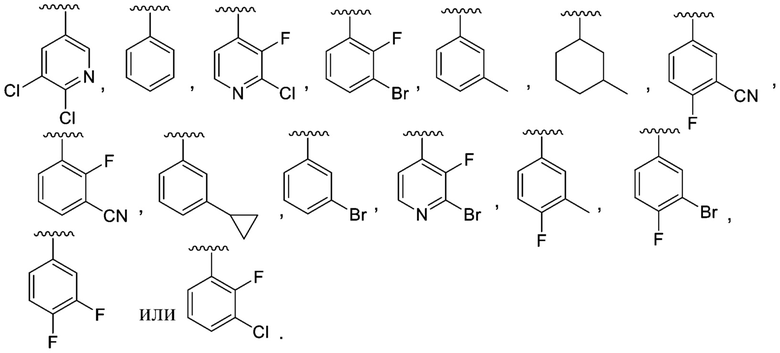
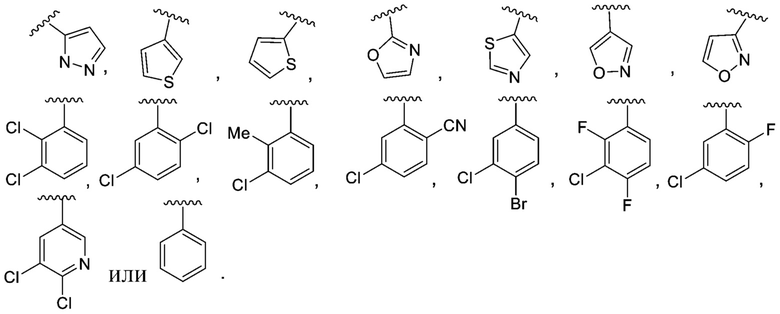
R1 выбран из C6-C12 арила и C1-C9 гетероарила, причем арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H;
R2 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R3 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R4 выбран из C1-C6 алкила, (CR8R9)p-C3-C8 циклоалкила, (CR8R9)p-C2-C8 гетероциклила, (CR8R9)p-C6-C12 арила и (CR8R9)p-C1-C9 гетероарила, причем алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf, C1-C6 алкил-OH, C3-C8 циклоалкила и C6 арила;
R5 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
альтернативно R4 и R5 необязательно соединены с образованием гетероциклического кольца;
R6 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R7 выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
R8 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R9 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
Ra выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
Rb выбран из H и C1-C6 алкила;
Rf в каждом случае независимо выбран из H и C1-C6 алкила;
m составляет 0, 1, 2, 3 или 4;
n составляет 0, 1, 2 или 3; и
p составляет 0, 1, 2, 3 или 4.
В одном варианте осуществления соединения формулы I
I
или его фармацевтически приемлемой соли
каждый из W1 и W независимо выбран из N, NRa и CRa, причем один из W1 и W представляет собой NRa;
X представляет собой N или CRb;
Y выбран из связи, -C(O)- и -SO2-;
Z выбран из -(CR5R6)m-, -(CR5R6)mO-, -(CR5R6)mCR5=CR5-, -(CR5R6)m-C3-C6 циклоалкилена- и -(CR5R6)m-NR7-;
R1 выбран из C6-C12 арила и C1-C9 гетероарила, причем арил и гетероарил необязательно замещены 1 или 2 группами, каждая из которых независимо выбрана из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)HH;
R2 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R3 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R4 выбран из C1-C6 алкила, (CR8R9)p-C3-C8 циклоалкила, (CR8R9)p-C2-C8 гетероциклила, (CR8R9)p-C6-C12 арила и (CR8R9)p-C1-C9 гетероарила, причем алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf и C1-C6 алкил-OH;
R5 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
альтернативно R4 и R5 необязательно соединены с образованием кольца;
R6 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R7 выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
R8 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R9 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
Ra выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
Rb выбран из H и C1-C6 алкила;
Rf в каждом случае независимо выбран из H и C1-C6 алкила;
m составляет 0, 1, 2, 3 или 4;
n составляет 0, 1, 2 или 3; и
p составляет 0, 1, 2, 3 или 4.
В другом варианте осуществления соединения формулы I W1 представляет собой NRa, а W представляет собой N или CRa. В дополнительном варианте осуществления W1 представляет собой NH.
В другом варианте осуществления соединения формулы I W1 представляет собой N или CRa, а W представляет собой NRa.
В другом варианте осуществления соединения формулы I X представляет собой N.
В одном варианте осуществления соединения формулы I Y представляет собой -C(O)- или -SO2-.
В дополнительном варианте осуществления соединения формулы I Z представляет собой -(CR5R6)m-, -(CR5R6)mO- или -(CR5R6)m-NR7-.
В одном варианте осуществления соединения формулы I
m составляет 0 или 1;
R5 представляет собой H, -OH или C1-C6 алкил;
R6 представляет собой H или C1-C6 алкил; и
R7 представляет собой H или C1-C6 алкил.
В одном варианте осуществления соединения формулы I R1 представляет собой C6 арил и C1-C9 гетероарил, причем арил и гетероарил необязательно замещены 1 или 2 группами, которые независимо выбраны из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H.
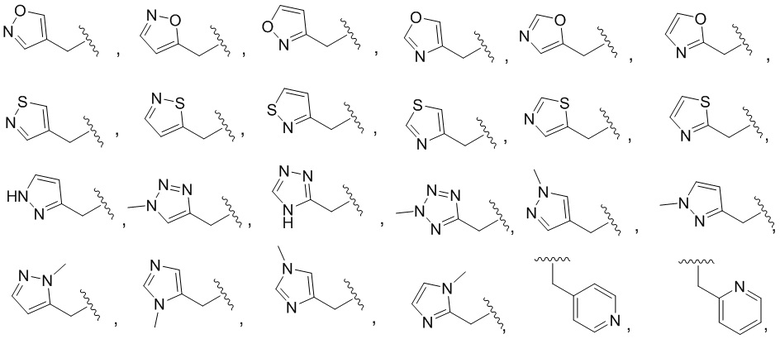
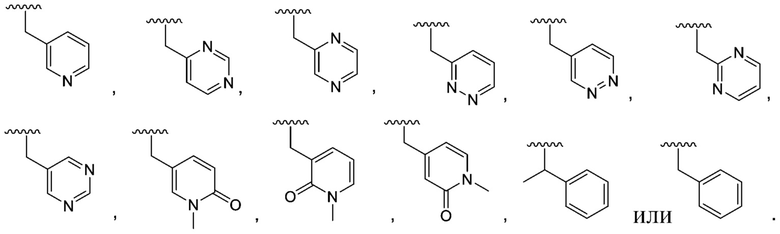
В другом варианте осуществления соединения формулы I R1 представляет собой C6 арил, пиримидинил, пиридинил, пиразолил, тиофенил, тиазолил, изотиазолил, оксазолил, пиридазинил, пиразинил или пирролил, любой из которых необязательно замещен 1 или 2 группами, независимо выбранными из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H.
В одном варианте осуществления соединения формулы I каждый R2 независимо выбран из H, C1-C6 алкила или C1-C6 алкил-OH. В дополнительном варианте осуществления соединения формулы I каждый R2 независимо выбран из C1-C6 алкила или H. В другом дополнительном варианте осуществления соединения формулы I R2 представляет собой H.
В одном варианте осуществления соединения формулы I R3 представляет собой H или C1-C6 алкил.
В одном варианте осуществления соединения формулы I R4 представляет собой (CR8R9)p-C3-C8 циклоалкил, (CR8R9)p-C2-C8 гетероциклил, (CR8R9)p-C6-C12 арил или (CR8R9)p-C1-C9 гетероарил, причем циклоалкил, гетероциклил, арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf и C1-C6 алкил-OH.
В другом варианте осуществления соединения формулы I R4 представляет собой (CR8R9)p-C6-C12 арил или (CR8R9)p-C1-C9 гетероарил, причем арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf и C1-C6 алкил-OH.
В другом варианте осуществления соединения формулы I
p составляет 0 или 1;
R8 представляет собой H, -OH или C1-C6 алкил; и
R9 представляет собой H или C1-C6 алкил.
В одном варианте осуществления соединения формулы I n составляет 1.
В другом варианте осуществления соединения формулы I
X представляет собой N;
Y представляет собой -C(O)-;
Z представляет собой NR7; и
R7 представляет собой H или C1-4 алкил.
В дополнительном варианте осуществления соединения формулы I
X представляет собой N;
Y представляет собой -C(O)-;
Z представляет собой NR7;
R7 представляет собой H или C1-4 алкил; и
n составляет 1.
Кроме того, в настоящем изобретении предложено соединение формулы I, имеющее структуру формулы II (также называется «соединением формулы II»):
II,
или его фармацевтически приемлемая соль.
В одном варианте осуществления соединения формулы II Y представляет собой -C(O)- или -SO2-.
В одном варианте осуществления соединения формулы II Z представляет собой -(CR5R6)m-, -(CR5R6)mO- или -(CR5R6)m-NR7-.
В одном варианте осуществления соединения формулы II
m составляет 0 или 1;
R5 представляет собой H, -OH или C1-C6 алкил;
R6 представляет собой H или C1-C6 алкил; и
R7 представляет собой H или C1-C6 алкил.
В одном варианте осуществления соединения формулы II R1 представляет собой C6-C12 арил или C1-C9 гетероарил, причем арил и гетероарил необязательно замещены 1 или 2 группами, каждая из которых независимо выбрана из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H.
В одном варианте осуществления соединения формулы II R1 представляет собой C6 арил, пиримидинил, пиридинил, пиразолил, тиофенил, тиазолил, изотиазолил, оксазолил, пиридазинил, пиразинил или пирролил, любой из которых необязательно замещен 1 или 2 группами, независимо выбранными из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H.
В одном варианте осуществления соединения формулы II каждый R2 независимо выбран из H, C1-C6 алкила или C1-C6 алкил-OH. В дополнительном варианте осуществления соединения формулы II каждый R2 независимо выбран из C1-C6 алкила или H. В другом дополнительном варианте осуществления соединения формулы II R2 представляет собой H.
В одном варианте осуществления соединения формулы II R3 представляет собой H или C1-C6 алкил. В дополнительном варианте осуществления R3 представляет собой H.
В одном варианте осуществления соединения формулы II n составляет 1.
В одном варианте осуществления соединения формулы II R4 представляет собой (CR8R9)p-C3-C8 циклоалкил, (CR8R9)p-C2-C8 гетероциклил, (CR8R9)p-C6-C12 арил или (CR8R9)p-C1-C9 гетероарил, причем циклоалкил, гетероциклил, арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf и C1-C6 алкил-OH.
В одном варианте осуществления соединения формулы II R4 представляет собой (CR8R9)p-C6-C12 арил или (CR8R9)p-C1-C9 гетероарил, и при этом арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf и C1-C6 алкил-OH.
В одном варианте осуществления соединения формулы II
Y представляет собой -C(O)-;
Z представляет собой -(CR5R6)m-, -(CR5R6)mO- или -(CR5R6)m-NR7-;
R1 представляет собой C6 арил, пиримидинил, пиридинил, пиразолил, тиофенил, тиазолил, изотиазолил, оксазолил, пиридазинил, пиразинил или пирролил, любой из которых необязательно замещен 1 или 2 группами, независимо выбранными из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H; каждый R2 независимо выбран из H, C1-C6 алкила или C1-C6 алкил-OH, а R3 представляет собой H;
R4 представляет собой (CR8R9)p-C6-C12 арил или (CR8R9)p-C1-C9 гетероарил, и при этом арил и гетероарил необязательно замещены 1, 2, 3 или 4 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf и C1-C6 алкил-OH;
R5 представляет собой H, -OH или C1-C6 алкил;
R6 представляет собой H или C1-C6 алкил;
R7 представляет собой H или C1-C6 алкил;
R8 в каждом случае независимо выбран из H, -OH, галогена и C1-C6 алкила;
R9 в каждом случае независимо выбран из H, -OH, галогена и C1-C6 алкила;
Rf в каждом случае независимо выбран из H и C1-C6 алкила;
m составляет 1 или 2;
n составляет 1; и
p составляет 0, 1 или 2.
В одном варианте осуществления данного варианта осуществления R1 представляет собой C6 арил, необязательно замещенный -OH или галогеном.
В одном варианте осуществления данного варианта осуществления R1 представляет собой C6 арил, необязательно замещенный галогеном.
В одном варианте осуществления данного варианта осуществления R4 представляет собой (CR8R9)p-C6-C12 арил или (CR8R9)p-C1-C9 гетероарил, и при этом арил и гетероарил необязательно замещены 1, 2 или 3 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH.
В одном варианте осуществления соединения формулы II
p составляет 0 или 1;
R8 независимо выбран из H, -OH и C1-C6 алкила; и
R9 независимо выбран из H и C1-C6 алкила.
В одном варианте осуществления соединения формулы II n составляет 1.
В одном варианте осуществления соединения формулы II
Y представляет собой -C(O)-;
Z представляет собой NR7; и
R7 представляет собой H или C1-4 алкил.
В одном варианте осуществления соединения формулы II
Y представляет собой -C(O)-;
Z представляет собой NR7;
R7 представляет собой H или C1-4 алкил; и
n составляет 1.
Кроме того, в настоящем изобретении предложено соединение формулы I, имеющее структуру формулы III (также называется «соединением формулы III»):
III,
или его фармацевтически приемлемая соль, где
Y представляет собой -C(O)- или -SO2-;
R1 представляет собой C6-C12 арил или C1-C9 гетероарил, причем арил и гетероарил необязательно замещены 1 или 2 группами, каждая из которых независимо выбрана из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H;
R2 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R3 выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
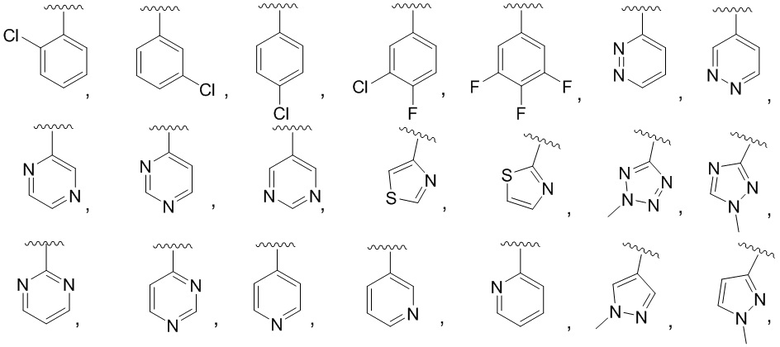
R4 выбран из (CR8R9)p-C1-C9 гетероарила, (CR8R9)p-C6-C12 арила и C3-C8 циклоалкила, причем гетероарил, арил и циклоалкил необязательно замещены 1, 2 или 3 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH и C3-C8 циклоалкила.
R7 выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
R8 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R9 в каждом случае независимо выбран из H и C1-C6 алкила; и
p составляет 0, 1, 2, 3 или 4.
В одном варианте осуществления соединения формулы III
Y представляет собой -C(O)- или -SO2-;
R1 представляет собой C6 арил, пиримидинил, пиридинил, пиразолил, тиофенил, тиазолил, изотиазолил, оксазолил, пиридазинил, пиразинил или пирролил, любой из которых необязательно замещен 1 или 2 группами, независимо выбранными из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H;
R2 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R3 выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R4 выбран из (CR8R9)p-C1-C9 гетероарила и (CR8R9)p-C6-C12 арила, причем гетероарил и арил необязательно замещены 1, 2 или 3 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH.
R7 выбран из H, C1-C6 алкила и C1-C6 алкил-OH;
R8 в каждом случае независимо выбран из H, -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила и C1-C6 алкил-OH;
R9 в каждом случае независимо выбран из H и C1-C6 алкила; и
p составляет 0, 1, 2, 3 или 4.
В одном варианте осуществления соединения формулы III Y представляет собой -C(O)-.
В одном варианте осуществления соединения формулы III R1 представляет собой C6 арил или C1-C9 гетероарил, причем арил или гетероарил необязательно замещены -OH, галогеном, C1-C6 алкилом или -O-C1-C6 алкилом.
В одном варианте осуществления соединения формулы III R1 представляет собой C6 арил, C6 арил, пиримидинил, пиридинил, пиразолил, тиофенил, тиазолил, изотиазолил, оксазолил или пиридазинил, любой из которых необязательно замещен 1 или 2 группами, независимо выбранными из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH, CN и C(O)H.
В другом варианте осуществления соединения формулы III R1 представляет собой C6 арил.
В другом варианте осуществления соединения формулы III каждый R2 независимо выбран из H, C1-C6 алкила или C1-C6 алкил-OH. В дополнительном варианте осуществления соединения формулы III каждый R2 независимо выбран из C1-C6 алкила или H. В другом дополнительном варианте осуществления соединения формулы III R2 представляет собой H.
В одном варианте осуществления соединения формулы III R3 представляет собой H или C1-C6 алкил. В дополнительном варианте осуществления соединения формулы III R3 представляет собой H.
В одном варианте осуществления соединения формулы III R7 представляет собой H или C1-C4 алкил. В дополнительном варианте осуществления R7 представляет собой H или -CH3. В еще одном варианте осуществления R7 представляет собой H.
В одном варианте осуществления соединения формулы III R4 представляет собой (CR8R9)p-C1-C5 гетероарил, или (CR8R9)p-C6 арил, или C3-C8 циклоалкил, причем гетероарил, арил и циклоалкил необязательно замещены 1, 2 или 3 группами, каждая из которых независимо выбрана из -OH, галогена, CN и C1-C6 алкила;
R8 представляет собой H или C1-C6 алкил;
R9 представляет собой H или C1-C6 алкил; и
p составляет 0 или 1.
В одном варианте осуществления соединения формулы III R4 представляет собой (CR8R9)p-C1-C5 гетероарил или (CR8R9)p-C6 арил, причем гетероарил и арил необязательно замещены 1, 2 или 3 группами, каждая из которых независимо выбрана из -OH, галогена, CN и C1-C6 алкила;
R8 представляет собой H или C1-C6 алкил;
R9 представляет собой H или C1-C6 алкил; и
p составляет 0 или 1.
В конкретном варианте осуществления соединения формулы III R4 представляет собой
В другом конкретном варианте осуществления соединения формулы III R4 представляет собой
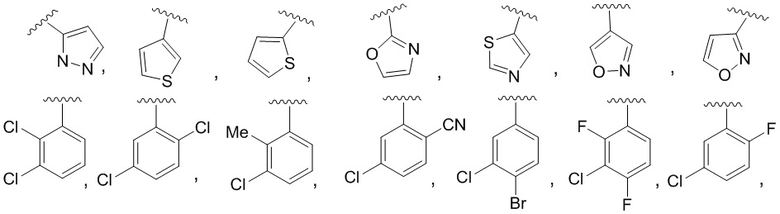
В другом конкретном варианте осуществления соединения формулы III R4 представляет собой
Кроме того, в настоящем изобретении предложено соединение формулы I, имеющее структуру формулы IV (также называется «соединением формулы IV»):
IV,
или его фармацевтически приемлемая соль, где
Y представляет собой -C(O)- или -SO2-; и
m составляет 0, 1 или 2.
В одном варианте осуществления соединения формулы IV Y представляет собой -C(O)-.
В одном варианте осуществления соединения формулы IV R1 представляет собой C6-C12 арил или C1-C9 гетероарил, причем арил и гетероарил необязательно замещены 1 или 2 группами, каждая из которых независимо выбрана из -OH, галогена, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C1-C6 алкил-OH и CN. В одном варианте осуществления соединения формулы IV R1 представляет собой C6 арил, необязательно замещенный -OH или галогеном.
В одном варианте осуществления соединения формулы IV R2 представляет собой H.
В одном варианте осуществления соединения формулы IV R3 представляет собой H.
В другом варианте осуществления соединения формулы IV m составляет 1, R5 представляет собой H или C1-C6 алкил, R6 представляет собой H или C1-C6 алкил, и при этом R5 и R4 необязательно соединены с образованием кольца. В другом варианте осуществления соединения формулы IV m составляет 1; R5 представляет собой C1-C6 алкил; R6 представляет собой H или C1-C6 алкил; и R5 и R4 необязательно соединены с образованием кольца. Например, в одном варианте осуществления
 представляет собой
представляет собой
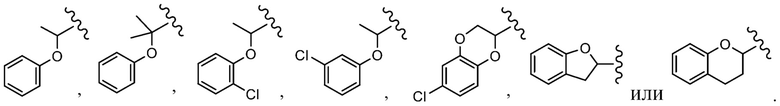
В другом варианте осуществления соединения формулы IV R4 представляет собой C1-C6 алкил или (CR8R9)p-C6-C12 арил, причем алкил и арил необязательно замещены 1, 2 или 3 группами, каждая из которых независимо выбрана из -OH, галогена, CN, C1-C6 алкила, C1-C6 галогеналкила, -O-C1-C6 алкила, C(O)N(Rf)2, C(O)ORf, -OCH2C(O)ORf, -SO2Rf и C1-C6 алкил-OH.
В дополнительных вариантах осуществления соединения формулы IV R4 представляет собой
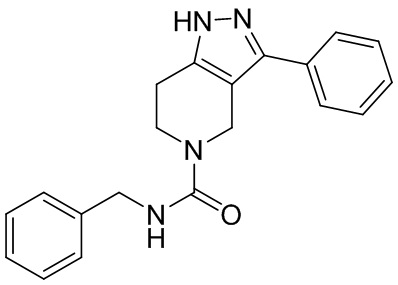
Определенные варианты осуществления соединений формул I-IV, включая их фармацевтически приемлемые соли, показаны ниже в таблице 1. Все соединения формул I, II, III и IV, а также их фармацевтически приемлемые соли и соединения из таблицы 1, а также их фармацевтически приемлемые соли представляют собой «соединения настоящего изобретения».
Таблица 1.
 001
001 002
002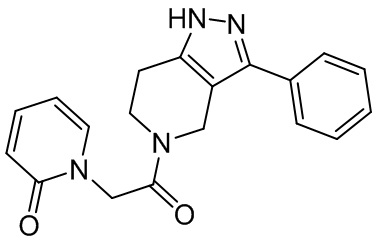
075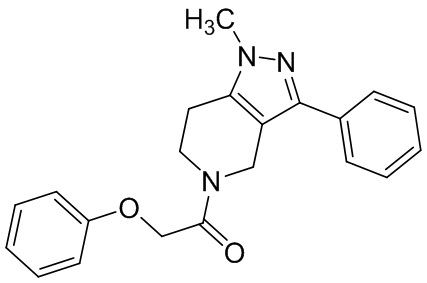
010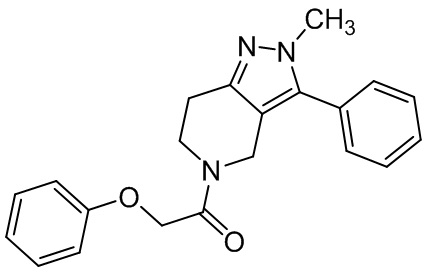 059
059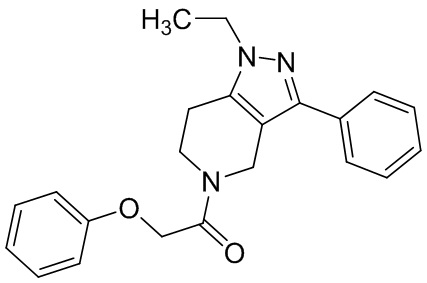
011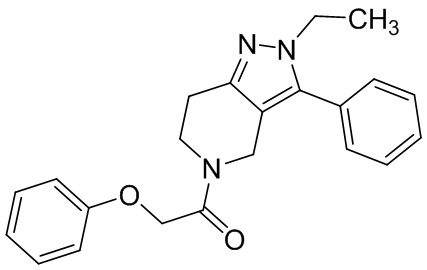
060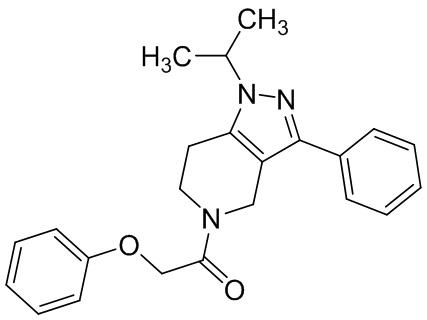
012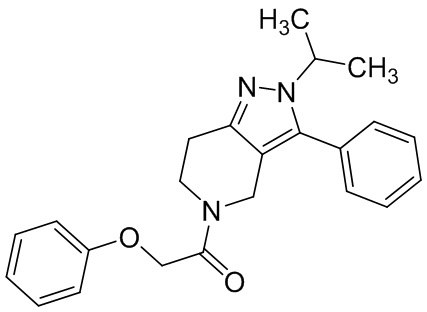
061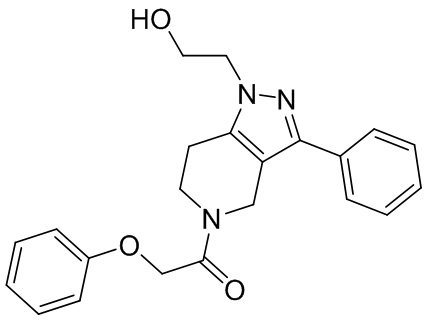
013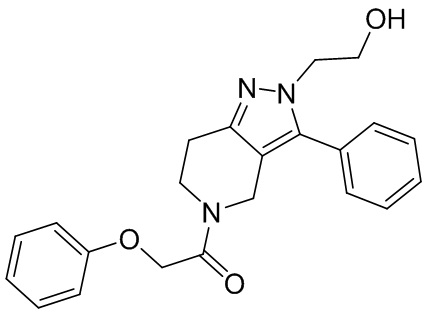
062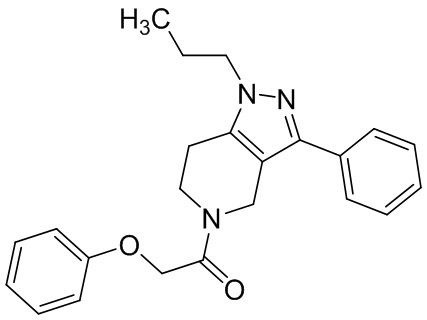
056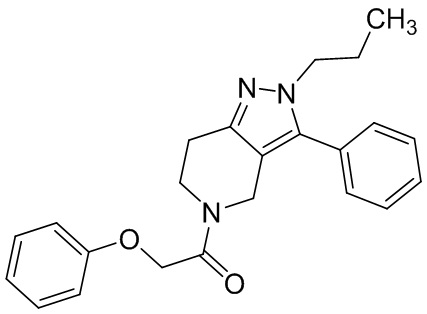
063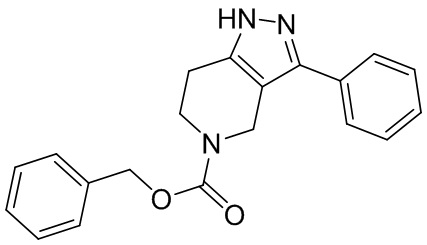
024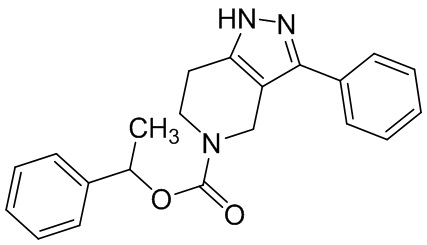
016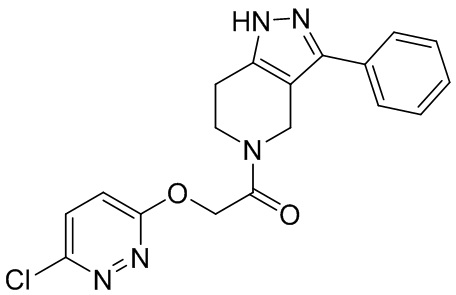
187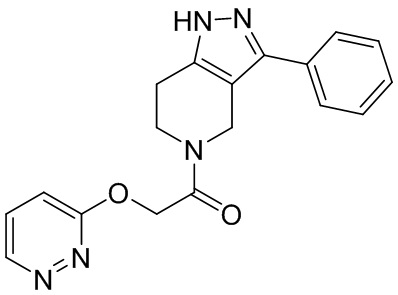
004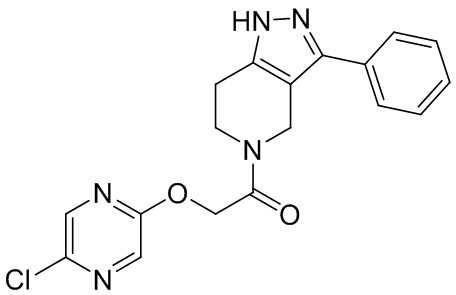
188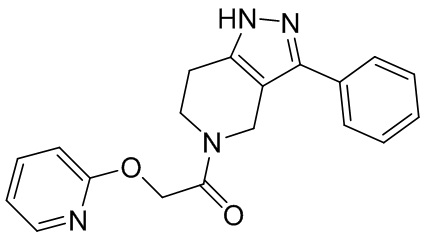
005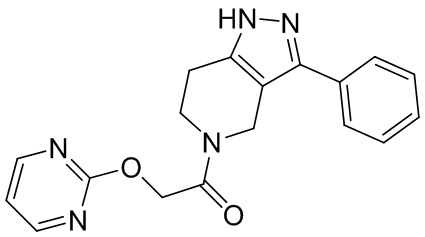
008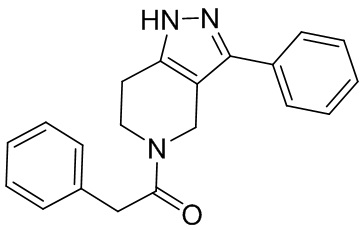
025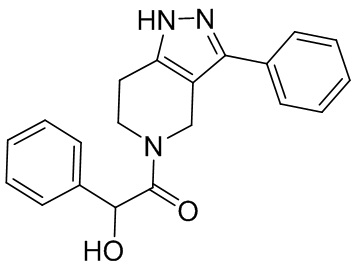
026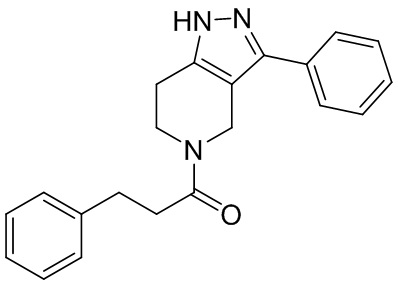
027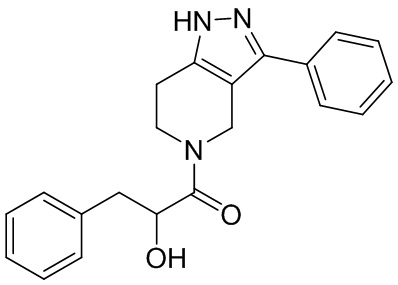
028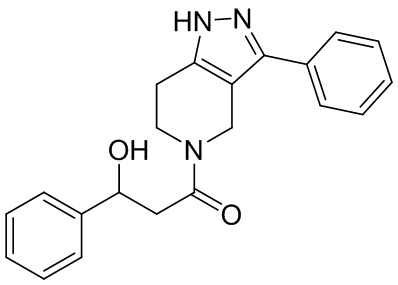
029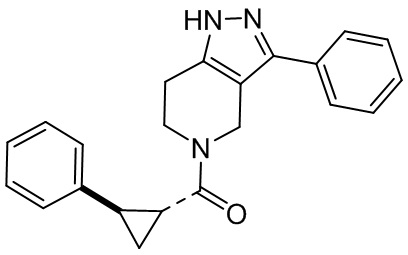 030
030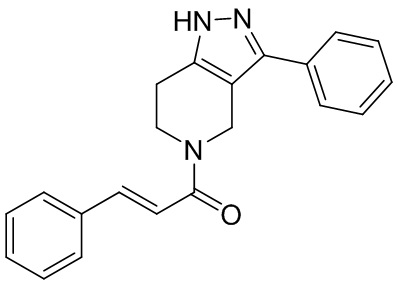
057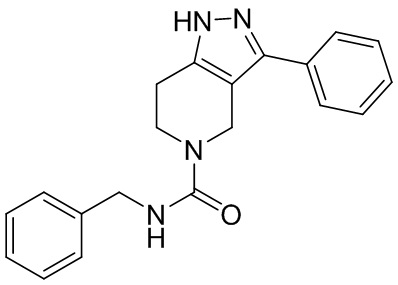
017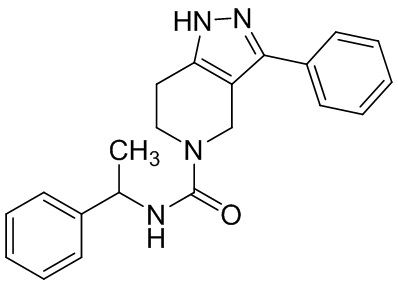
018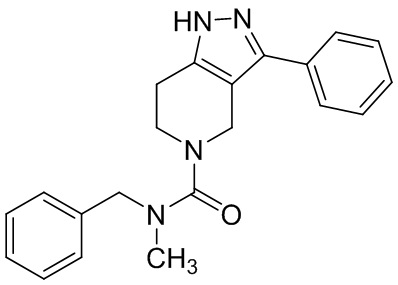
019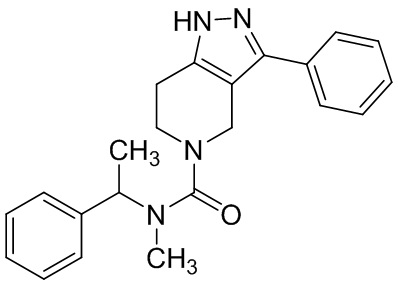
020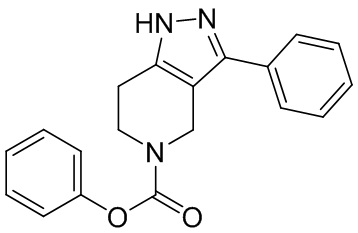
021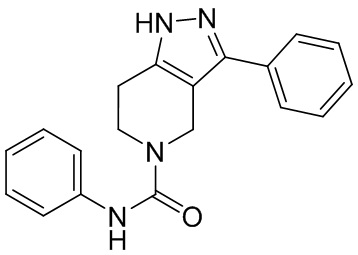
022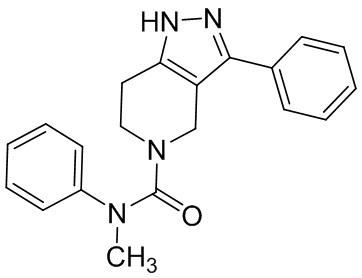
023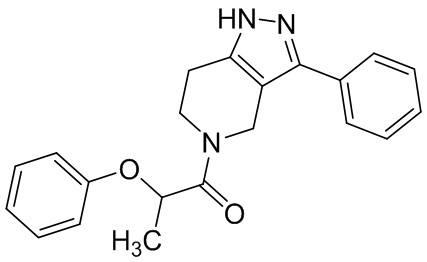
014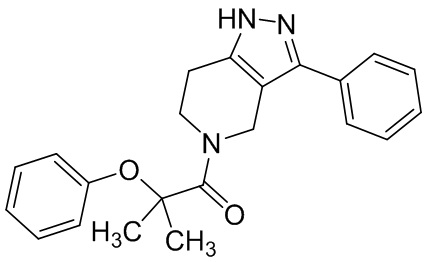
015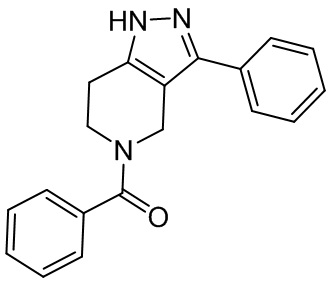
064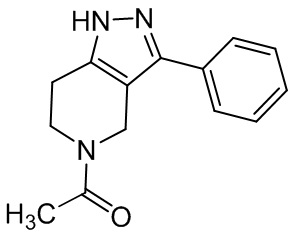
065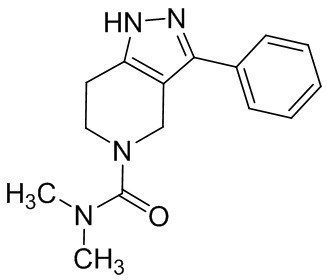
066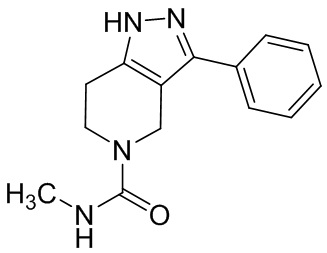
067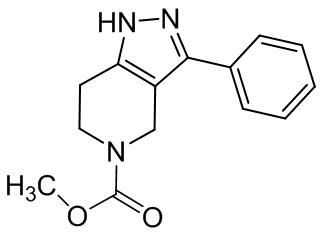
068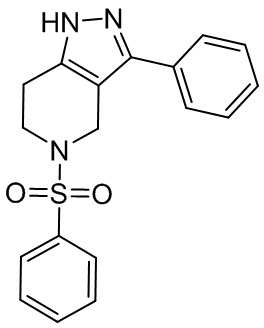
069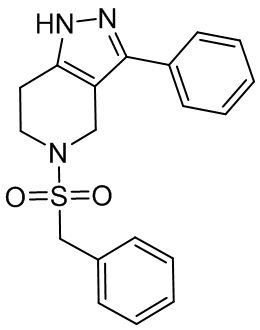
070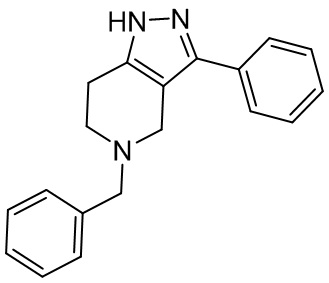
073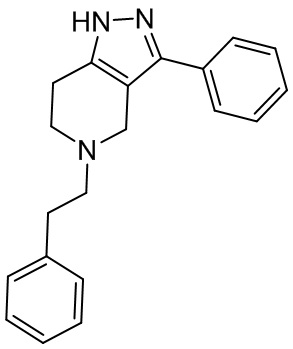
074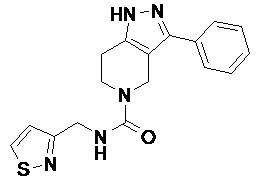
085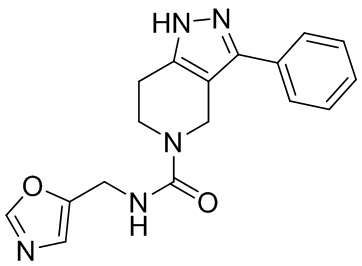
081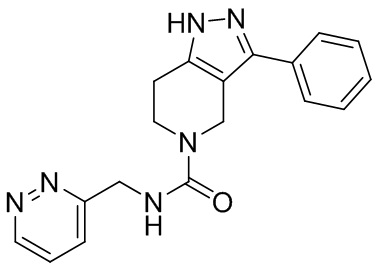
104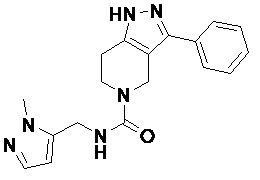
095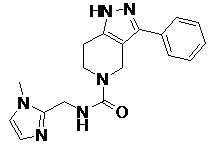
098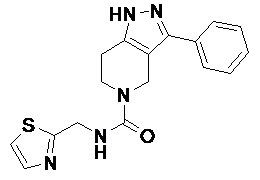
088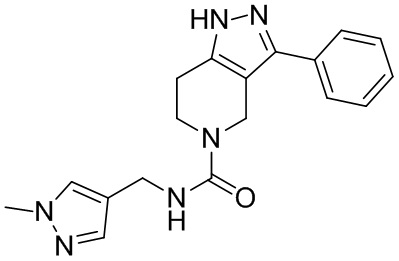
093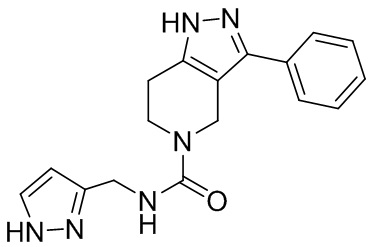
089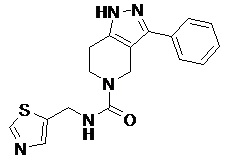
087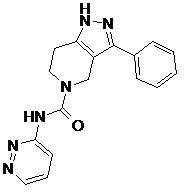
119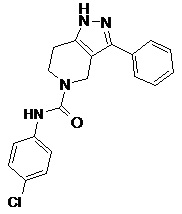
113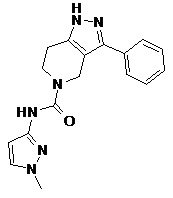
117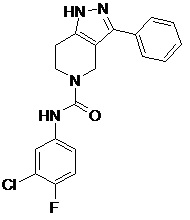
114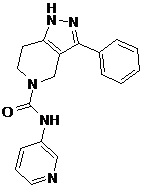
130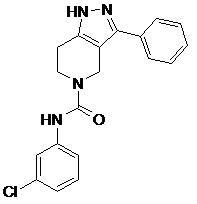
112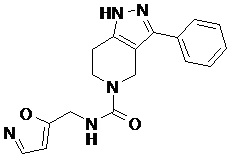
078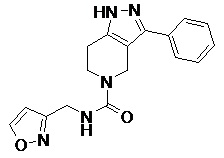
079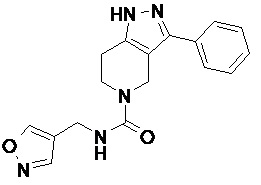
077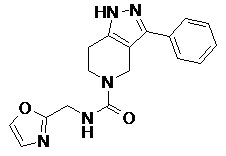
082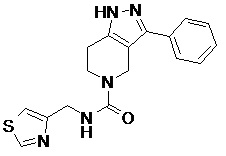
086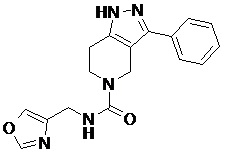
080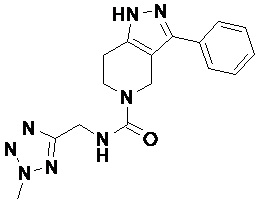
092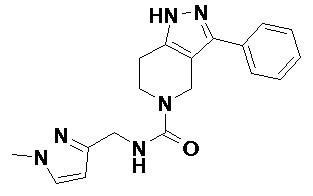
094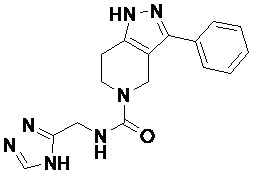
091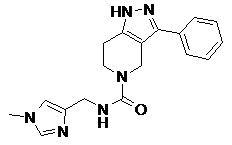
097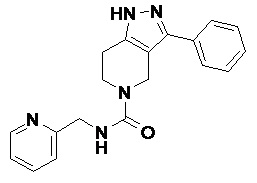
100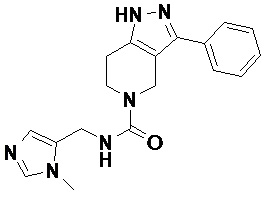
096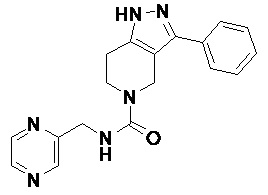
103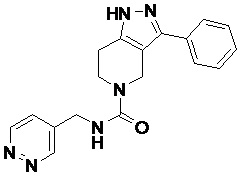
105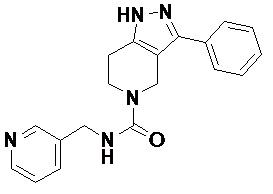
101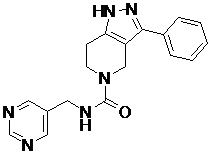
107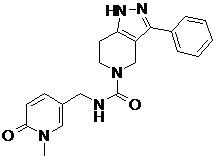
108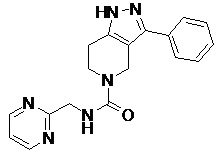
106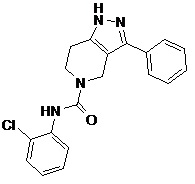
111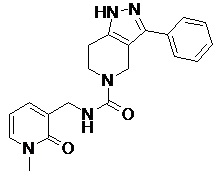
109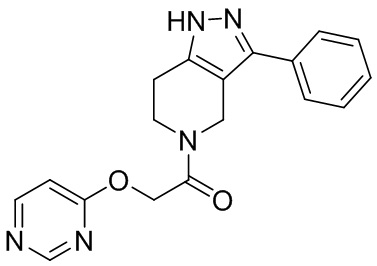
009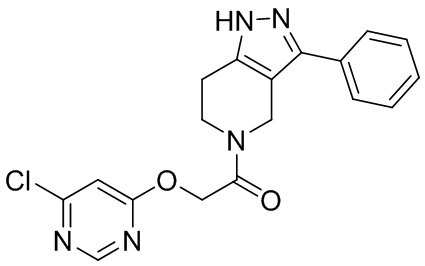
240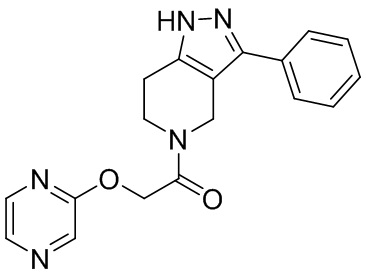
003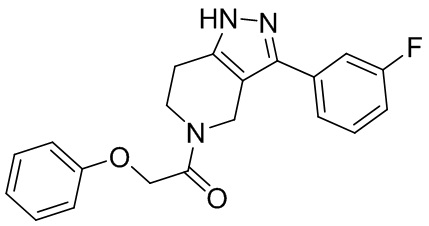
243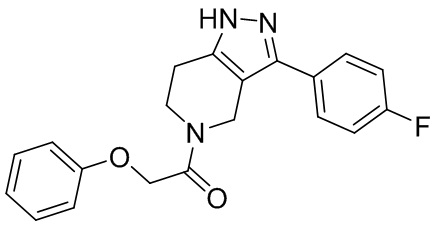
244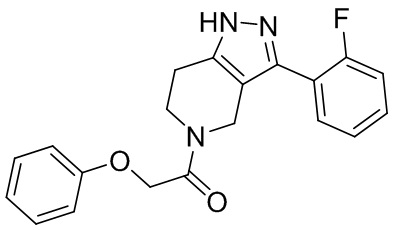
242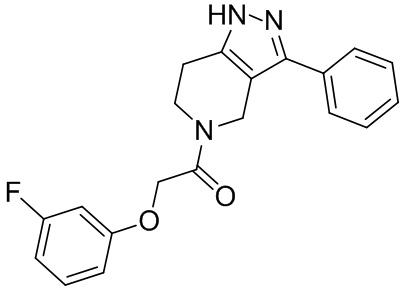
143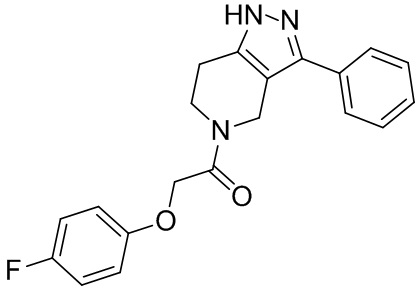
144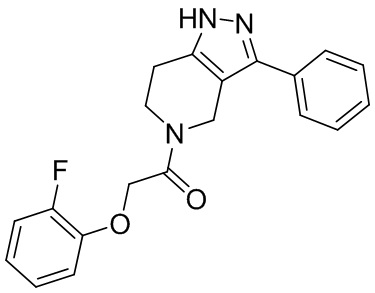
142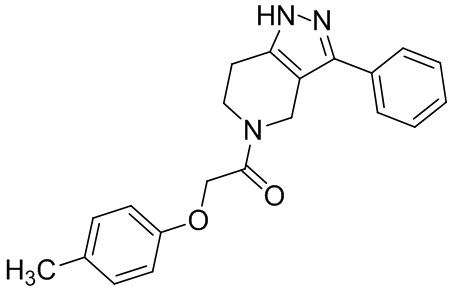
147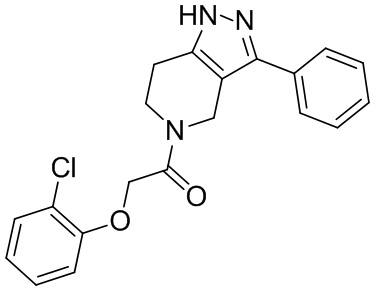
148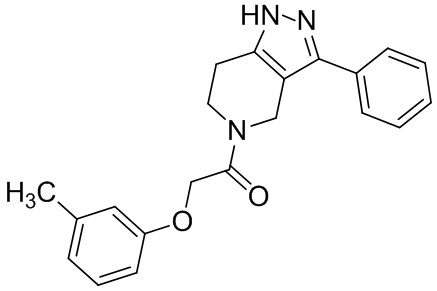
145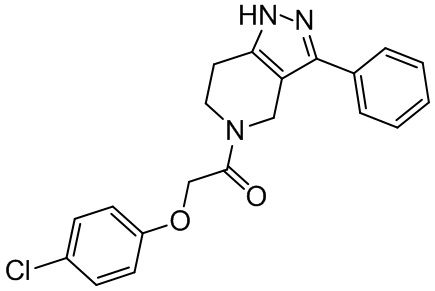
150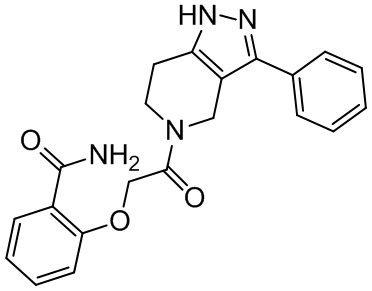
151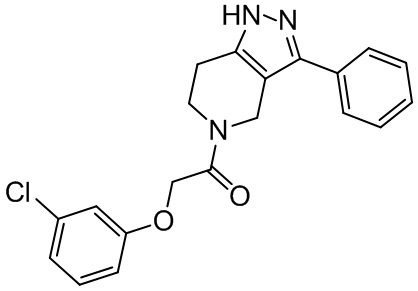
149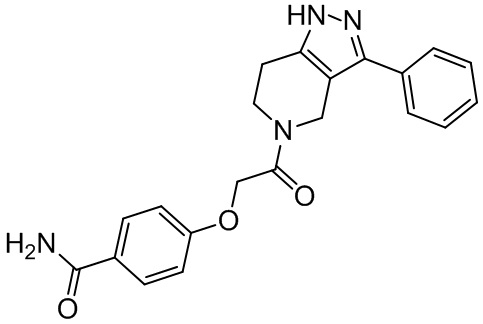
153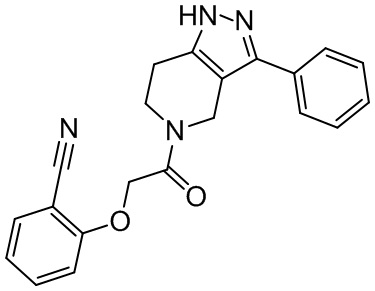
154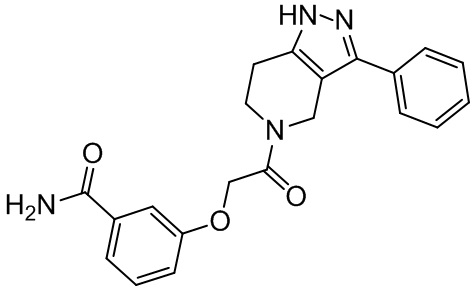
152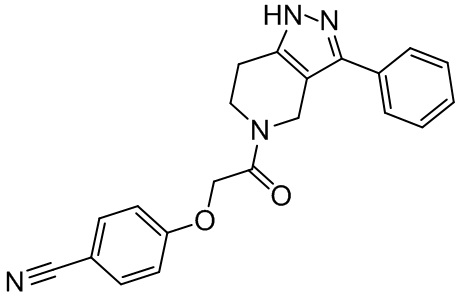
156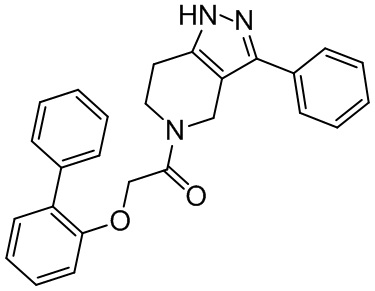
157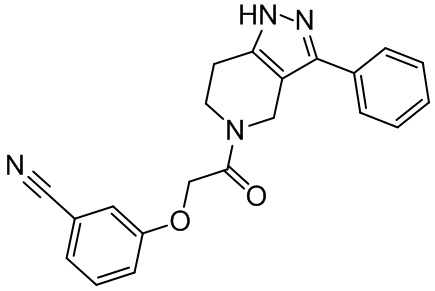
155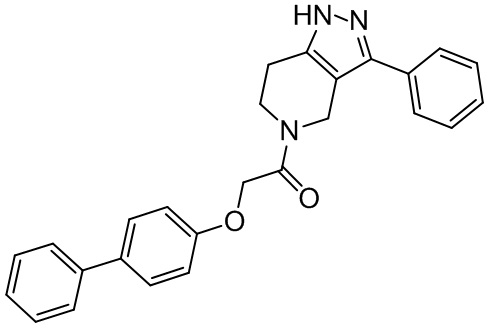
159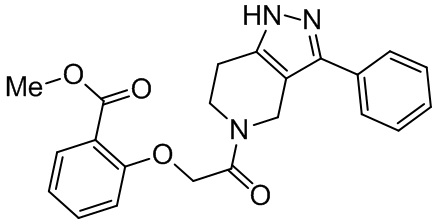
160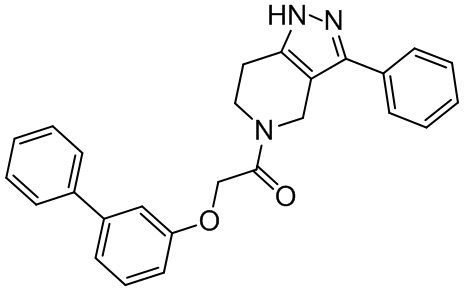
158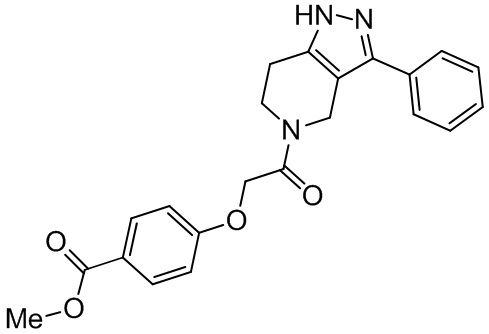
162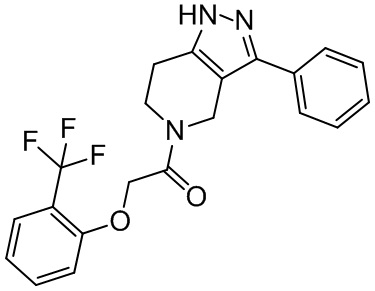
163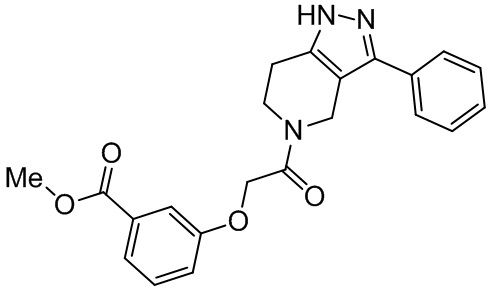
161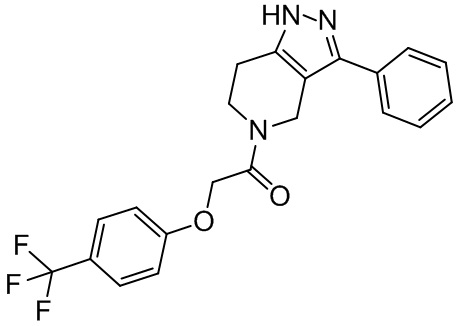
165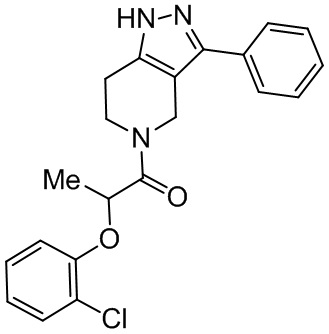
169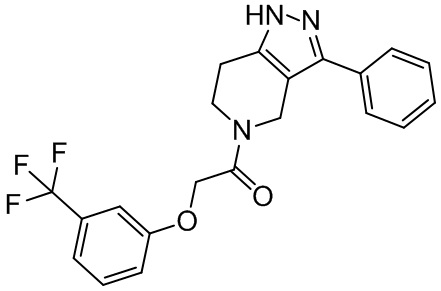
164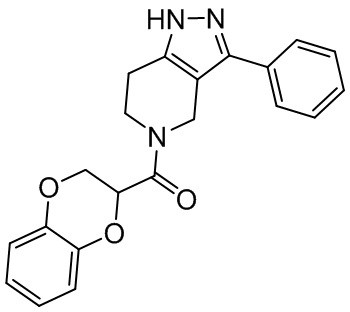
171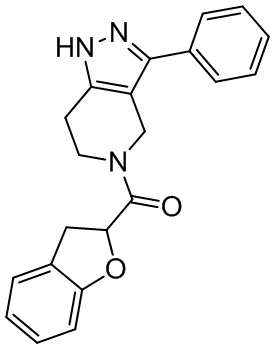
172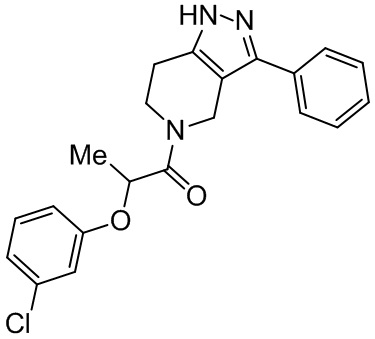
170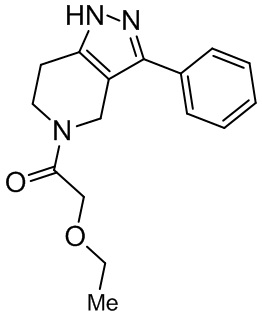
174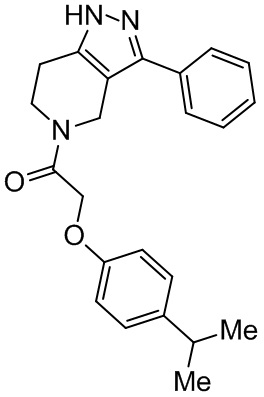
175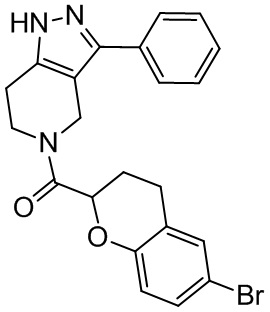
173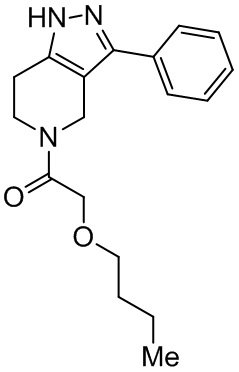
177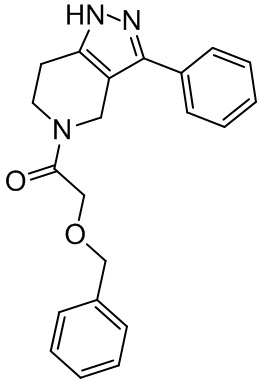
178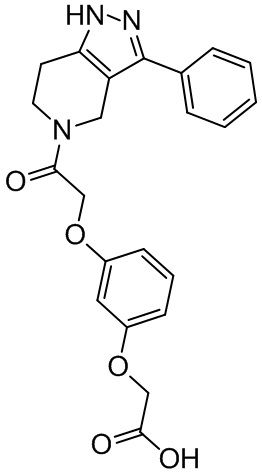
176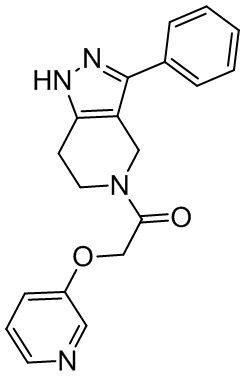
182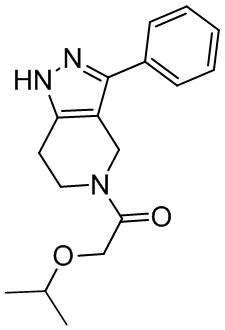
183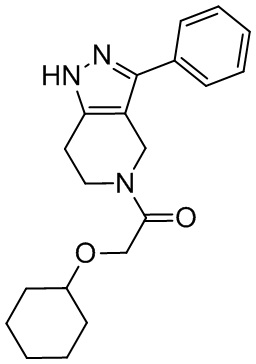
180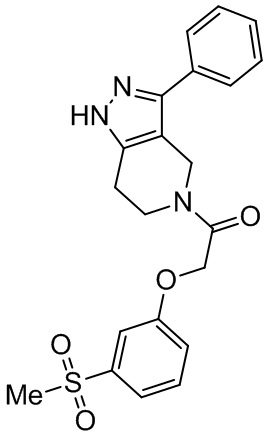
184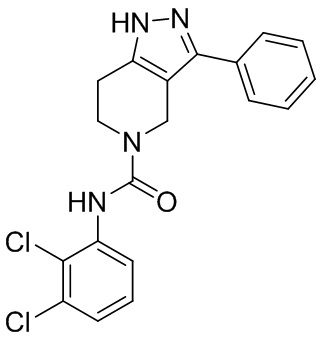
190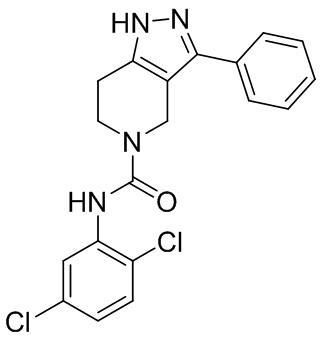
191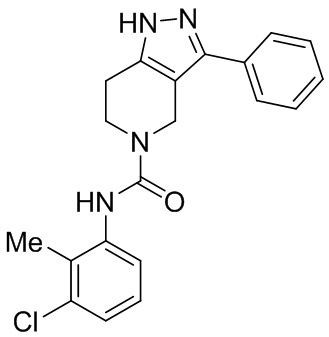
192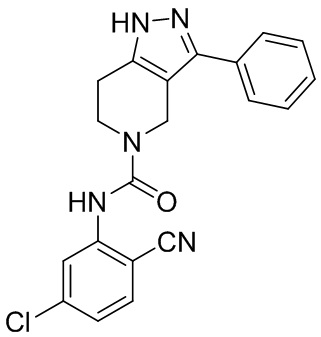
201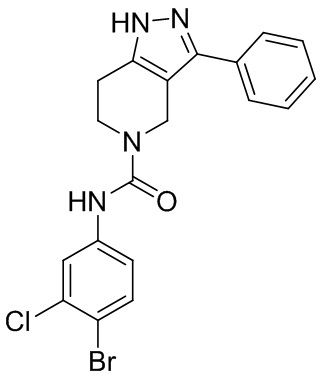
204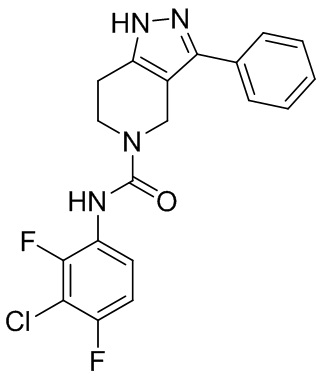
205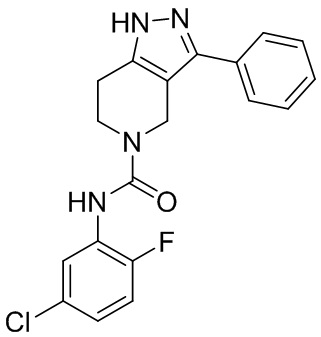
214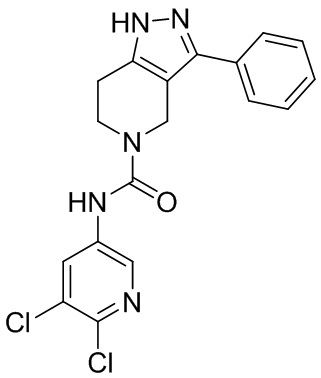
226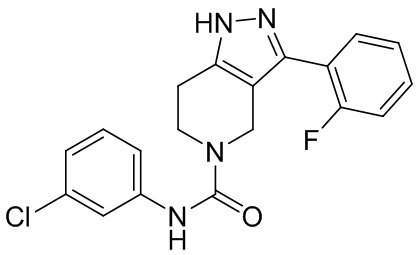
276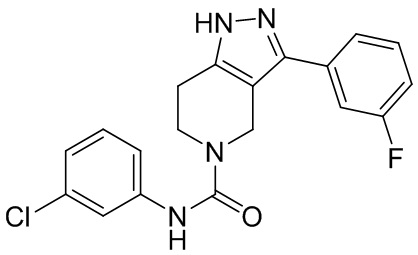
277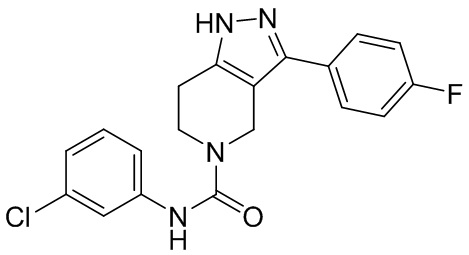
278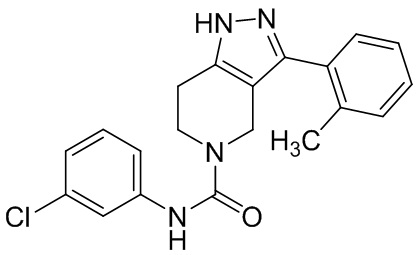
279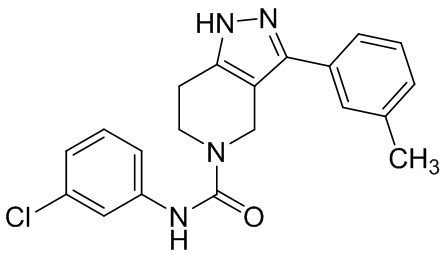
280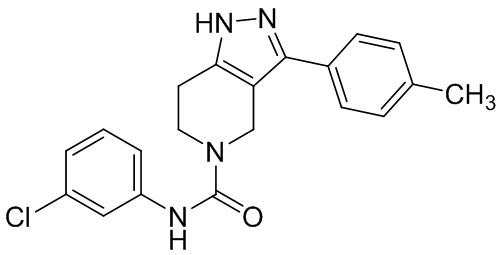
281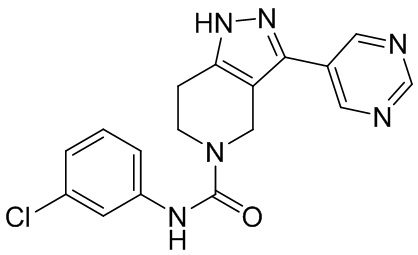
291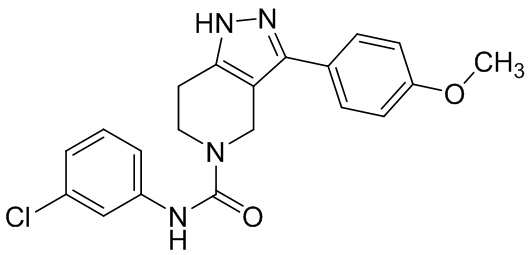
325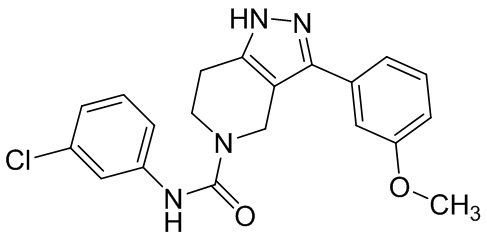 326
326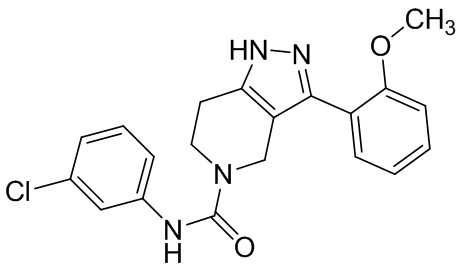 327
327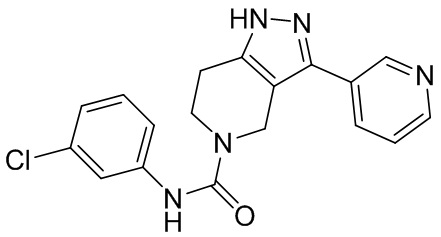
286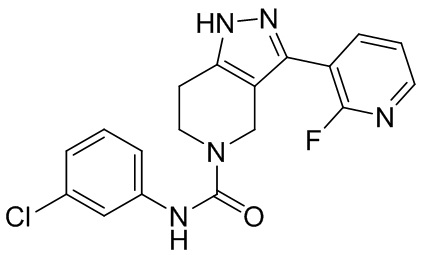
472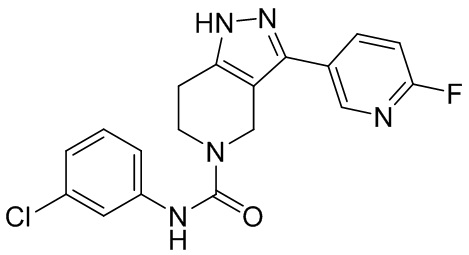
473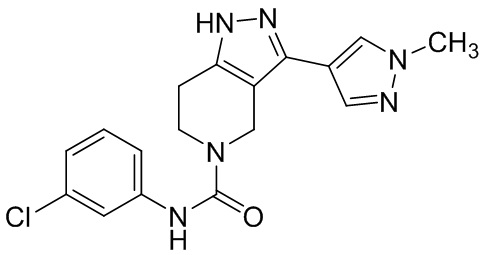
495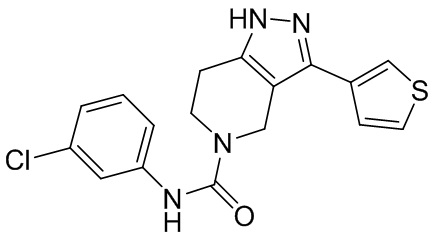 562
562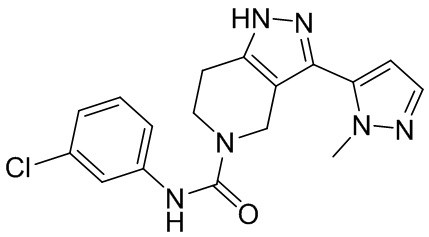
496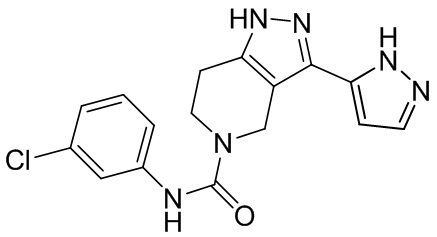
497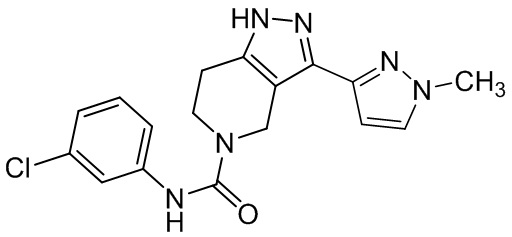
555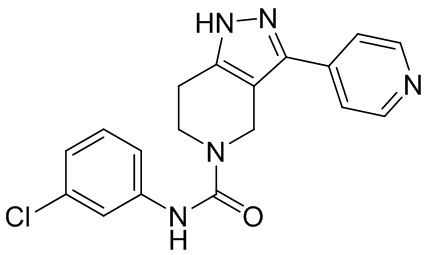
287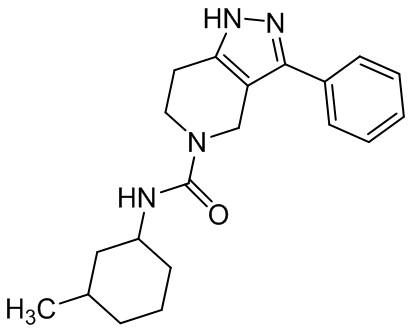
436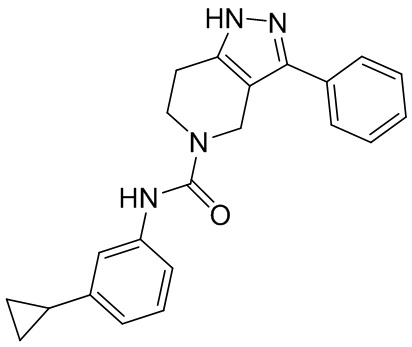
556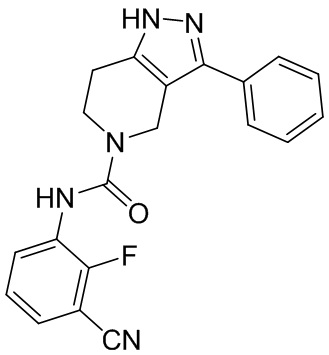
559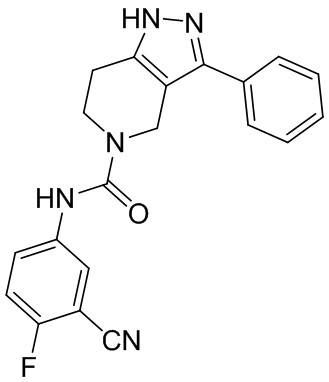
560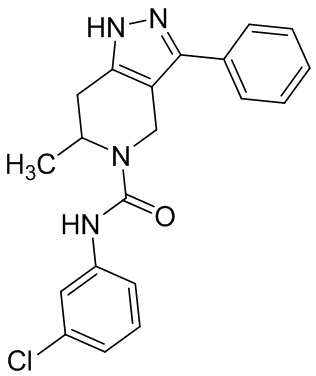
317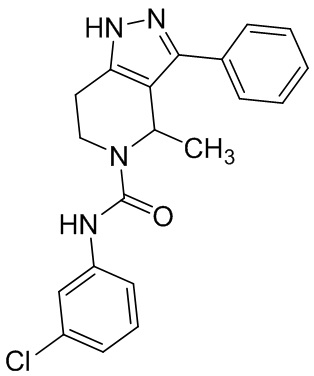
318 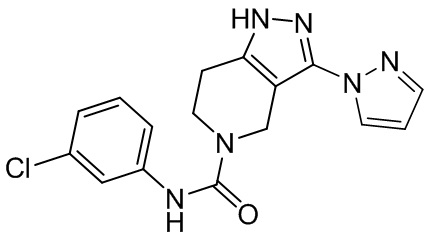
576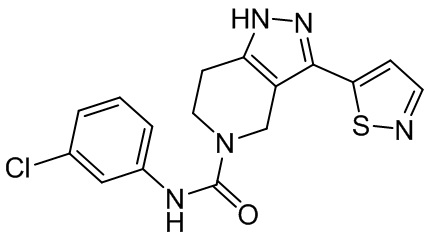
751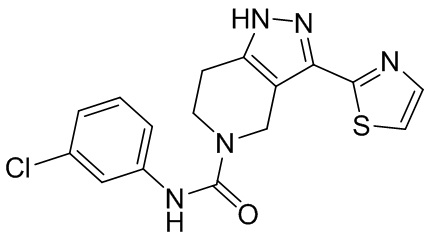 569
569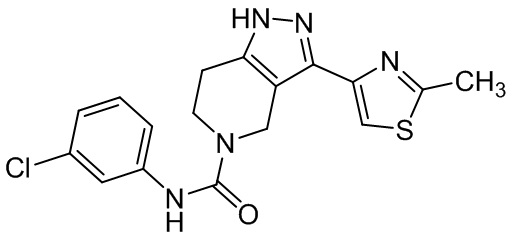
726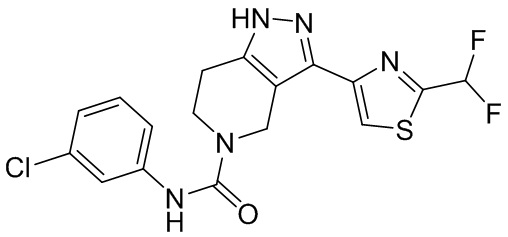
730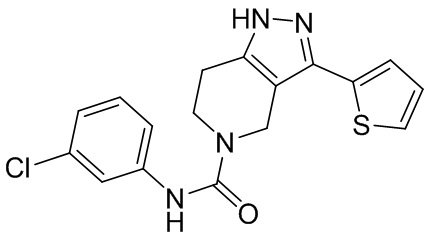
645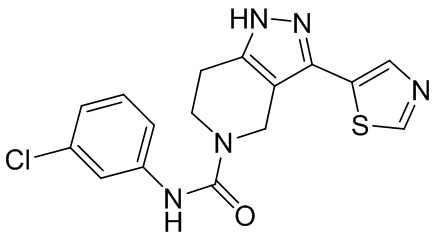
568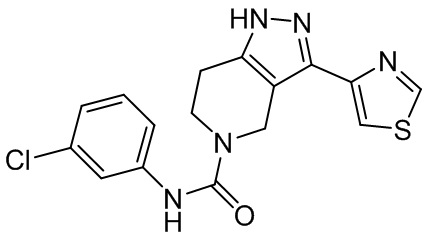
570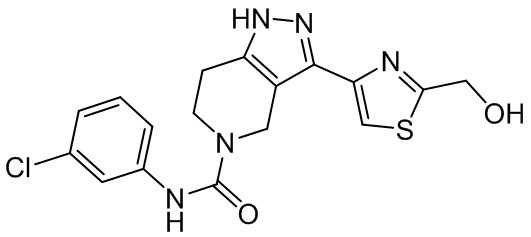
729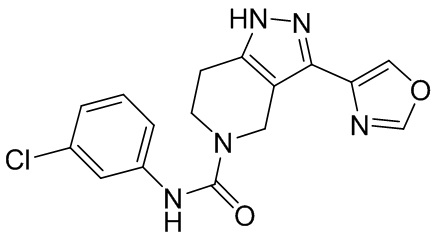
741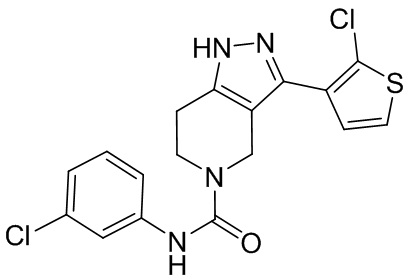
707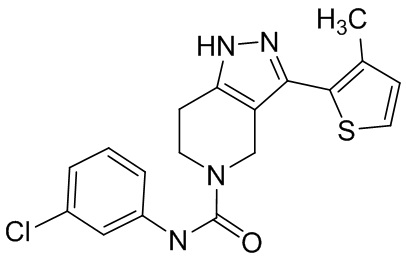
708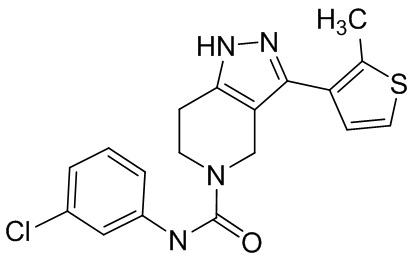
712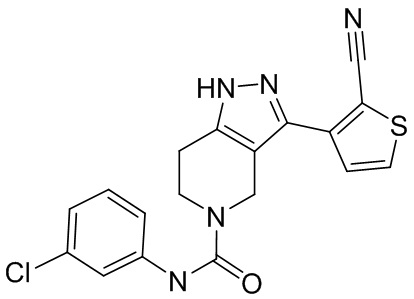
715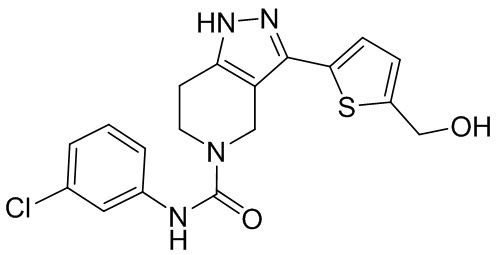
769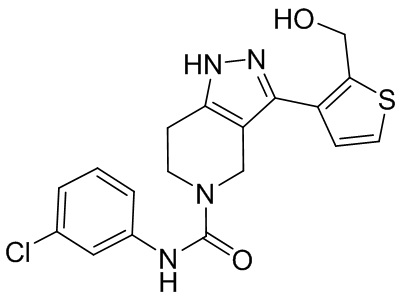 766
766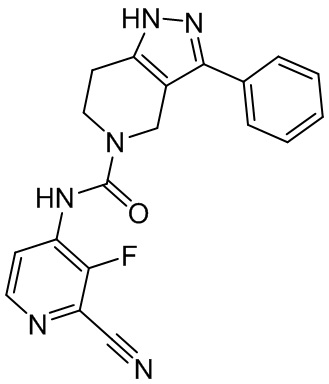
747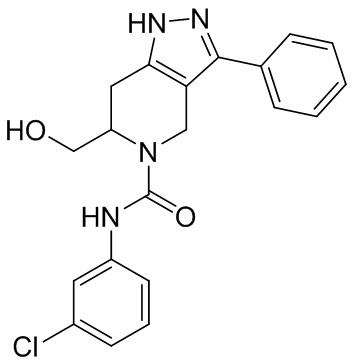
756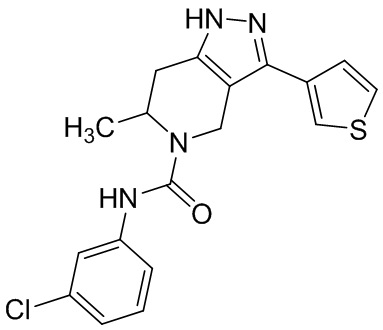
754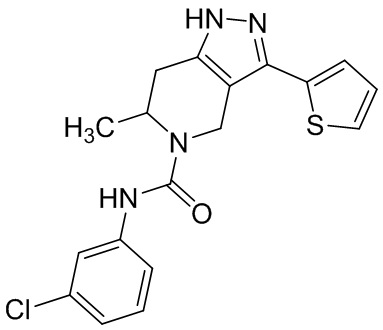
753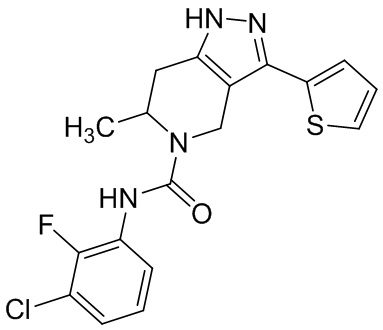
819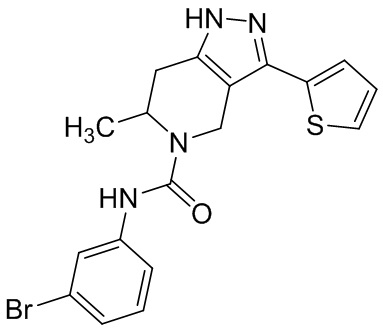
820 (851)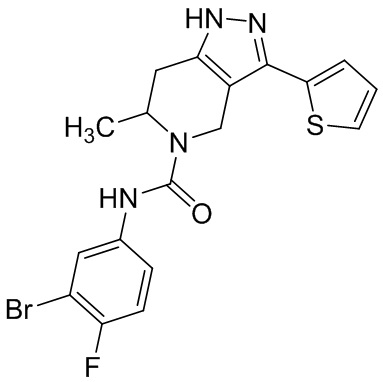
821 (852)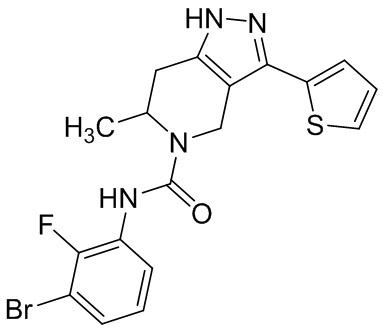
822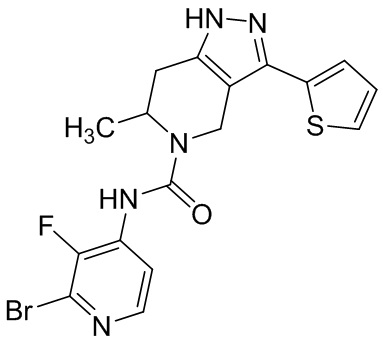
823 (853)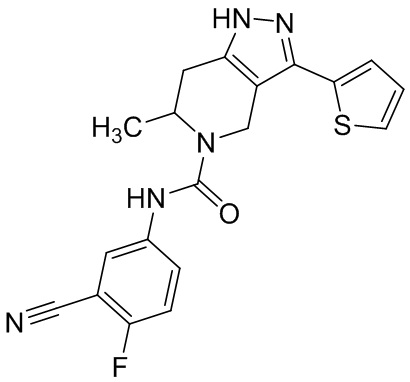
824 (854)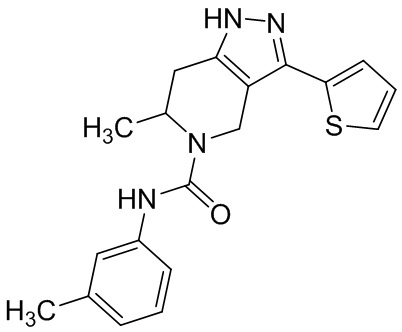
825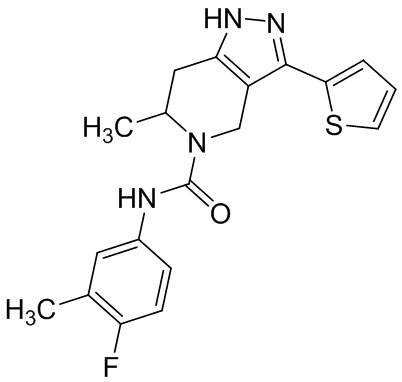 826 (855)
826 (855)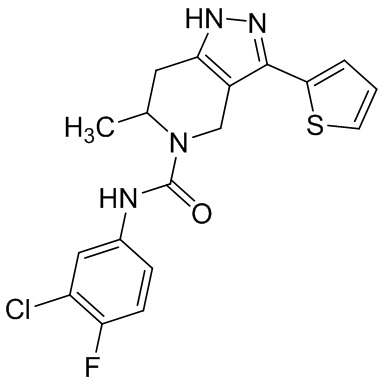
857 (856)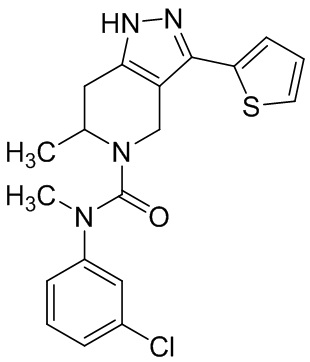
830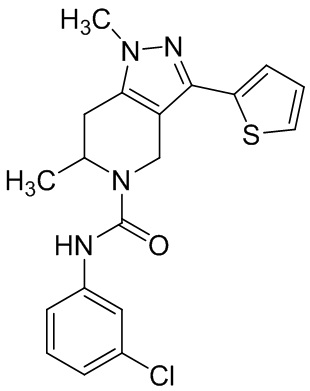
831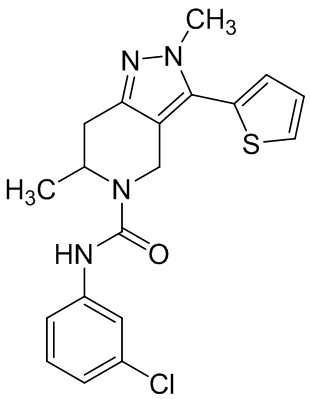
832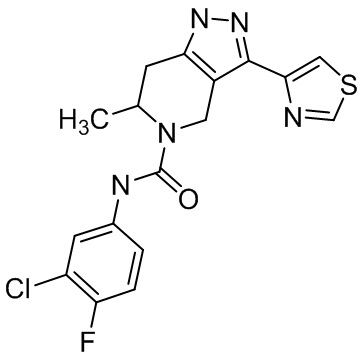
917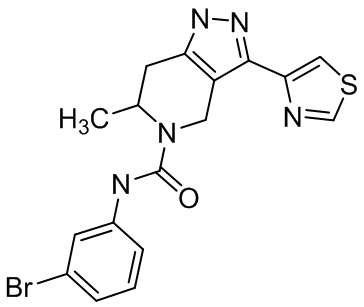
918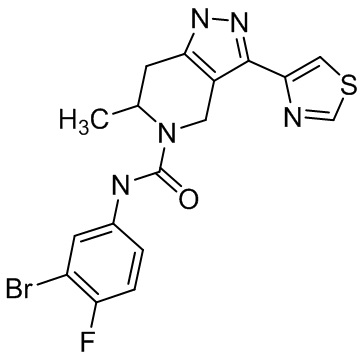
919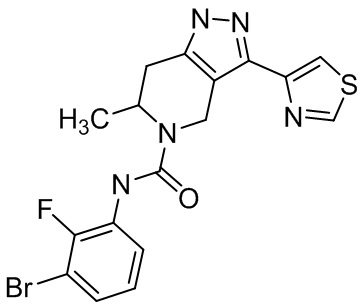
920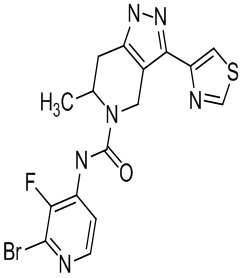
921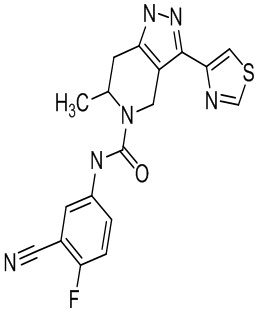
922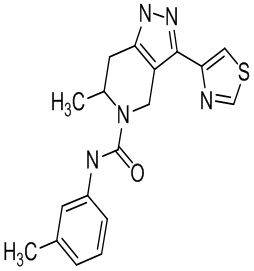
923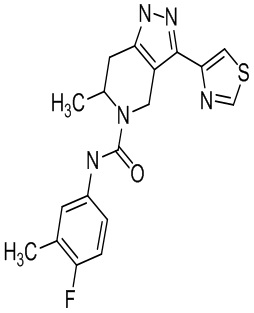
924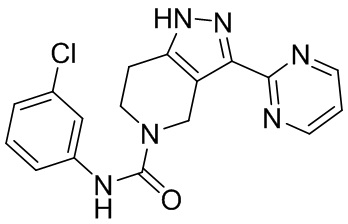
289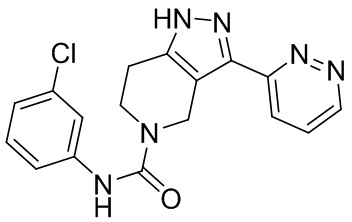
290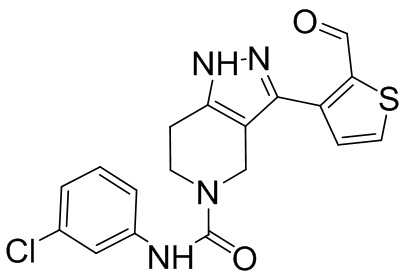
716
В еще одном варианте осуществления формулы I, предложенном в настоящем документе, соединение формулы III или его фармацевтически приемлемая соль выбраны из соединений, показанных в таблице 2, и их фармацевтически приемлемых солей.
Таблица 2.
017
018
019
020
022
023
085
081
104
095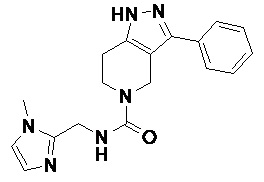
098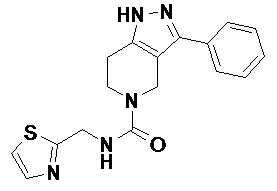
088
093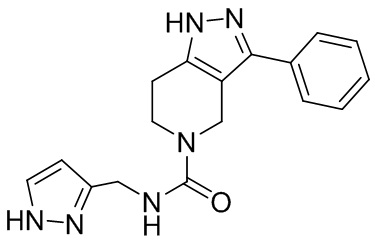
089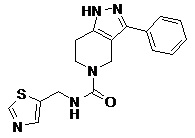
087
119
113
117
114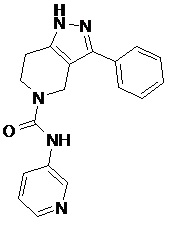
130
112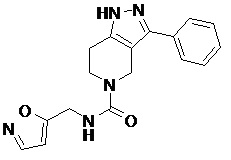
078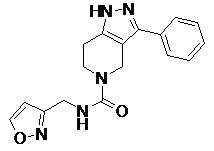
079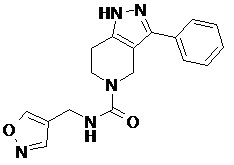
077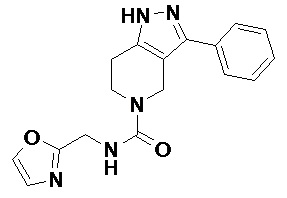
082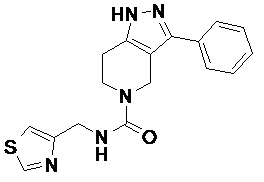
086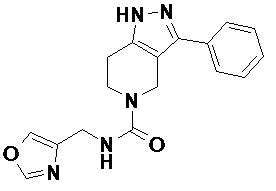
080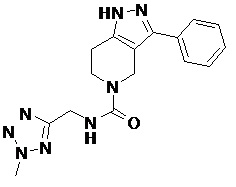
092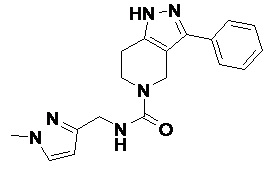
094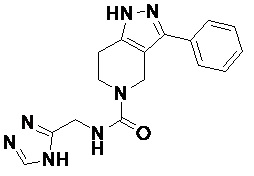
091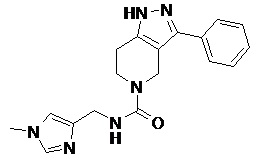
097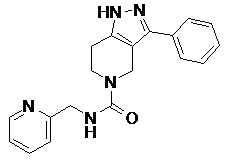
100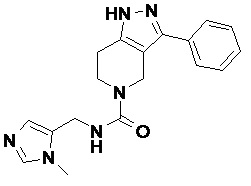
096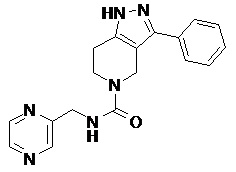
103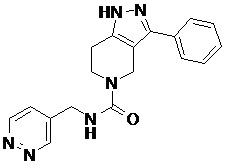
105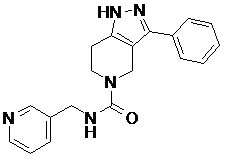
101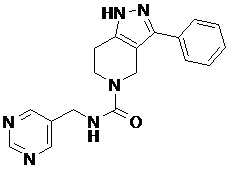
107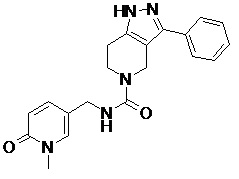
108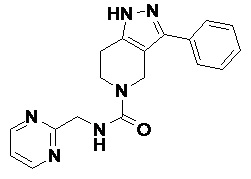
106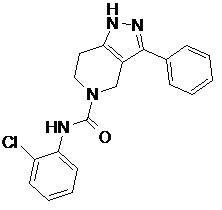
111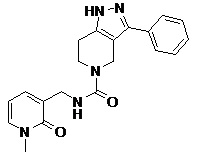
109
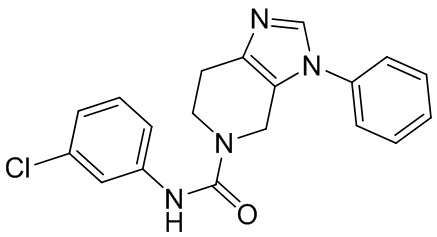
716
190
191
192
201
204
205
214
226
276
277
278
279
280
281
291
325
326327286
472
473495562
496
497555
287
436
556
559
560
317
318
576
751569726
730645
568
570729
741
707
708
712
715
769
766
747
756
754
753
819
820 (851)
821 (852)
822
823 (853)
824 (854)
825
826 (855)
857 (856)
830
831
832
917
918
919
920
921
922
923
924
289
290
и их фармацевтически приемлемые соли.
В настоящем изобретении также предложены следующие соединения:
542
583
Соединения настоящего изобретения могут иметь один или более стереоцентров, и каждый стереоцентр может существовать независимо в R- или S-конфигурации. В одном варианте осуществления соединения, описанные в настоящем документе, присутствуют в оптически активной или рацемической формах. Следует понимать, что соединения, описанные в настоящем документе, включают рацемические, оптически активные, региоизомерные и стереоизомерные формы или их комбинации, которые обладают терапевтически полезными свойствами, описанными в настоящем документе.
Получение оптически активных форм выполняют любым приемлемым образом, включая, в качестве не имеющего ограничительного характера примера, посредством разделения рацемической формы с помощью способов рекристаллизации, синтеза из оптически активных исходных материалов, хирального синтеза или хроматографического разделения с использованием хиральной неподвижной фазы. В одном варианте осуществления в качестве терапевтического соединения, описанного в настоящем документе, используется смесь одного или более изомеров. В другом варианте осуществления соединения, описанные в настоящем документе, содержат один или более хиральных центров. Эти соединения получают любыми способами, включая стереоселективный синтез, энантиоселективный синтез и/или разделение смеси энантиомеров и/или диастереомеров. Разделение соединений и их изомеров выполняют с помощью любых способов, включая, в качестве не имеющего ограничительного характера примера, химические способы, ферментативные способы, фракционную кристаллизацию, дистилляцию и хроматографию.
В одном варианте осуществления соединения настоящего изобретения могут существовать в виде таутомеров. Все таутомеры включены в объем соединений, представленных в настоящем документе.
Соединения, описанные в настоящем документе, также включают изотопно меченные соединения, в которых один или более атомов замещены атомом, имеющим такой же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, обычно обнаруживаемого в природе. Примеры изотопов, приемлемых для включения в соединения, описанные в настоящем документе, включают, без ограничений, 2H, 3H, 11C, 13C, 14C, 36Cl, 18F, 123I, 125I, 13N, 15N, 15O, 17O, 18O, 32P и 35S. В одном варианте осуществления изотопно меченные соединения используются в исследованиях распределения в тканях лекарственного средства и/или субстрата. В другом варианте осуществления замещение более тяжелыми изотопами, такими как дейтерий, может обеспечить большую метаболическую стабильность (например, увеличение периода полужизни in vivo или уменьшение требуемой дозы). В еще одном варианте осуществления замещение позитрон-излучающими изотопами, такими как 11C, 18F, 15O и 13N, используется в исследованиях позитронно-эмиссионной топографии (ПЭТ) для оценки заполненности рецепторов субстратом. Изотопно меченные соединения получают любым приемлемым способом или посредством способов, в которых используется подходящий изотопно меченный реагент вместо немеченого реагента, применяемого в других случаях.
В одном варианте осуществления соединения, описанные в настоящем документе, метят другими способами, включая, без ограничений, применение хромофоров или флуоресцентных групп, биолюминесцентных меток или хемилюминесцентных меток.
Соединения, описанные в настоящем документе, и другие схожие соединения, имеющие другие заместители, синтезируют с использованием способов и материалов, описанных в настоящем документе, и в соответствии с описанием, например, в Fieser and Fieserʹs Reagents for Organic Synthesis, Volumes 1-17 (John Wiley and Sons, 1991); Rodd's Chemistry of Carbon Compounds, Volumes 1-5 and Supplementals (Elsevier Science Publishers, 1989); Organic Reactions, Volumes 1-40 (John Wiley and Sons, 1991), Larockʹs Comprehensive Organic Transformations (VCH Publishers Inc., 1989), March, Advanced Organic Chemistry 4th Ed., (Wiley 1992); Carey and Sundberg, Advanced Organic Chemistry 4th Ed., Vols. A and B (Plenum 2000, 2001) и Green and Wuts, Protective Groups in Organic Synthesis 3rd Ed. (Wiley 1999) (все из которых включены в настоящее описание путем ссылки). Общие способы получения соединения, описанные в настоящем документе, модифицированы посредством применения соответствующих реагентов и условий для внедрения различных функциональных групп, обнаруживаемых в формуле, предложенной в настоящем документе.
Соединения, описанные в настоящем документе, синтезируют с использованием любых приемлемых способов, используя в качестве исходного материала соединения, присутствующие на рынке, или получают с использованием способов, описанных в настоящем документе.
В одном варианте осуществления реактивные функциональные группы, такие как гидроксильные, амино, имино, тио или карбоксильные группы, защищены, чтобы избежать нежелательного участия в реакциях. Защитные группы используются для блокировки некоторых или всех реактивных функциональных групп и предотвращают участие таких групп в химических реакциях до тех пор, пока защитная группа не будет удалена. В другом варианте осуществления каждую защитную группу можно удалять различными способами. Защитные группы, которые расщепляются в совершенно иных условиях реакции, удовлетворяют требованию о дифференцированном удалении.
Способы настоящего изобретения
В настоящем изобретении предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении также предложен способ эрадикации инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении также предложен способ уменьшения вирусной нагрузки, связанной с инфекцией ВГВ, у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении дополнительно предложен способ уменьшения вероятности повторного возникновения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении дополнительно предложен способ ингибирования или снижения образования или наличия частиц, содержащих ДНК ВГВ, или частиц, содержащих РНК ВГВ, у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении также предложен способ уменьшения нежелательного физиологического влияния инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении дополнительно предложен способ ослабления, замедления или подавления инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении также предложен способ индукции ремиссии поражения печени из-за инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении дополнительно предложен способ уменьшения физиологического влияния долгосрочной противовирусной терапии инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В настоящем изобретении дополнительно предложен способ профилактического лечения инфекции ВГВ у нуждающегося в этом субъекта, страдающего от латентной инфекции ВГВ, включающий введение субъекту терапевтически эффективного количества соединения настоящего изобретения.
В одном варианте осуществления способы, описанные в настоящем документе, дополнительно включают введение по меньшей мере одного дополнительного терапевтического агента, который выбран из группы, состоящей из аналогов нуклеотида/нуклеозида, ингибиторов проникновения, ингибиторов слияния и любой комбинации этих или других противовирусных механизмов. В другом варианте осуществления соединение настоящего изобретения и по меньшей мере один дополнительный терапевтический агент помещают в один состав. В еще одном варианте осуществления соединение настоящего изобретения и по меньшей мере один дополнительный терапевтический агент вводят совместно.
В одном варианте осуществления субъект является невосприимчивым к другим терапевтическим классам лекарственных средств против ВГВ (например, ингибиторам полимеразы ВГВ, интерферонам, ингибиторам проникновения вируса в клетку, ингибиторам созревания вируса, описанным в литературе модуляторам сборки капсида, противовирусным соединениям с другим или неизвестным механизмом действия и т. п. или их комбинации). В другом варианте осуществления способ настоящего изобретения уменьшает вирусную нагрузку у субъекта, страдающего от инфекции ВГВ, в большей степени или с большей скоростью по сравнению со степенью, в которой другие терапевтические классы лекарственных средств против ВГВ уменьшают вирусную нагрузку у субъекта.
В одном варианте осуществления введение соединения настоящего изобретения или его фармацевтически приемлемой соли позволяет вводить по меньшей мере один дополнительный терапевтический агент в меньшей дозе или с меньшей частотой по сравнению с введением по меньшей мере одного дополнительного терапевтического агента в виде монотерапии, чем требуемая для достижения аналогичных результатов в профилактическом лечении инфекции ВГВ у нуждающегося в этом субъекта.
В одном варианте осуществления введение соединения настоящего изобретения или его фармацевтически приемлемой соли снижает вирусную нагрузку у субъекта в большей степени или с большей скоростью по сравнению с введением соединения, которое выбрано из группы, состоящей из ингибитора полимеразы ВГВ, интерферона, ингибитора проникновения вируса в клетку, ингибитора созревания вируса, другого модулятора сборки капсида, противовирусных соединений с другим или неизвестным механизмом действия и любой их комбинации.
В одном варианте осуществления способ настоящего изобретения уменьшает вирусную нагрузку у субъекта, страдающего от инфекции ВГВ, что позволяет снизить дозы или изменить режимы используемой комбинированной терапии.
В одном варианте осуществления способ настоящего изобретения вызывает снижение частоты мутации вируса и/или устойчивости вируса по сравнению с другими классами лекарственных средств против ВГВ, таким образом позволяя осуществлять долгосрочную терапию и сводя к минимуму необходимость в изменениях режимов лечения.
В одном варианте осуществления введение соединения настоящего изобретения или его фармацевтически приемлемой соли приводит к снижению частоты мутации вируса и/или устойчивости вируса по сравнению с введением соединения, выбранного из группы, состоящей из ингибитора полимеразы ВГВ, интерферона, ингибитора проникновения вируса в клетку, ингибитора созревания вируса, другого модулятора сборки капсида, противовирусных соединений с другим или неизвестным механизмом действия и их комбинации.
В одном варианте осуществления способ настоящего изобретения увеличивает показатель сероконверсии по сравнению с текущими режимами лечения.
В одном варианте осуществления способ настоящего изобретения увеличивает, и/или нормализует, и/или восстанавливает нормальное здоровье, приводит к полному восстановлению нормального здоровья, увеличивает ожидаемую продолжительность жизни и/или уничтожает вирусную инфекцию у нуждающегося в этом субъекта.
В одном варианте осуществления способ настоящего изобретения удаляет или уменьшает число частиц РНК ВГВ, которые высвобождаются из инфицированных ВГВ клеток, таким образом усиливая, удлиняя или увеличивая терапевтическую пользу соединений настоящего изобретения.
В одном варианте осуществления способ настоящего изобретения обеспечивает эрадикацию ВГВ у субъекта, инфицированного ВГВ, таким образом устраняя необходимость в долгосрочном и/или пожизненном лечении, или сокращая длительность лечения, и/или позволяя уменьшить дозу других противовирусных агентов.
В другом варианте осуществления способ настоящего изобретения дополнительно включает контроль вирусной нагрузки ВГВ у субъекта, причем способ осуществляют в течение периода времени до тех пор, пока вирус ВГВ перестает обнаруживаться.
Соответственно, в одном варианте осуществления настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
В другом варианте осуществления настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы II или его фармацевтически приемлемой соли.
В другом варианте осуществления настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы III или его фармацевтически приемлемой соли.
В другом варианте осуществления настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения формулы IV или его фармацевтически приемлемой соли.
В другом варианте осуществления настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения из таблицы 1 или его фармацевтически приемлемой соли.
В другом варианте осуществления настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения из таблицы 2 или его фармацевтически приемлемой соли.
В одном варианте осуществления любого из способов, предложенных в настоящем изобретении, способ может дополнительно включать контроль вирусной нагрузки ВГВ у субъекта, причем способ осуществляют в течение периода времени до тех пор, пока вирус ВГВ перестает обнаруживаться.
Комбинированные терапии
Соединения настоящего изобретения предназначены для использования в комбинации с одним или более дополнительными соединениями, используемыми для лечения инфекции ВГВ. Эти дополнительные соединения могут содержать соединения настоящего изобретения или соединения, которые, как известно, способны лечить, предотвращать или уменьшать симптомы или эффекты инфекции ВГВ. Такие соединения включают, без ограничений, ингибиторы полимеразы ВГВ, интерфероны, ингибиторы проникновения вируса в клетку, ингибиторы созревания вируса, описанные в литературе модуляторы сборки капсида, ингибитор обратной транскриптазы, иммуномодулирующие агенты, агонист TLR и другие агенты с другими или неизвестными механизмами действия, которые влияют на жизненный цикл ВГВ и/или влияют на последствия инфекции ВГВ.
В не имеющих ограничительного характера примерах соединения настоящего изобретения можно использовать в комбинации с одним или более лекарственными средствами (или их солями), выбранными из группы, состоящей из:
ингибиторов обратной транскриптазы ВГВ и ингибиторов ДНК- и РНК-полимеразы, включая, без ограничений, ламивудин (3TC, Zeffix, Heptovir, Epivir и Epivir-HBV), энтекавир (Baraclude, Entavir), адефовир дипивоксил (Hepsara, Preveon, bis-POM PMEA), тенофовира дизопроксил фумарат (Viread, TDF или PMPA);
интерферонов, включая, без ограничений, интерферон-альфа (IFN-α), интерферон-бета (IFN-β), интерферон-лямбда (IFN-λ) и интерферон-гамма (IFN-γ);
ингибиторов проникновения вируса в клетку;
ингибиторов созревания вируса;
описанных в литературе модуляторов сборки капсида, таких как, без ограничений, BAY 41-4109;
ингибитора обратной транскриптазы;
иммуномодулирующего агента, такого как агонист TLR; и
агентов с другим или неизвестным механизмом действия, таких как, без ограничений, AT-61 ((E)-N-(1-хлор-3-оксо-1-фенил-3-(пиперидин-1-ил)проп-1-ен-2-ил)бензамид), AT-130 ((E)-N-(1-бром-1-(2-метоксифенил)-3-оксо-3-(пиперидин-1-ил)проп-1-ен-2-ил)-4-нитробензамид) и схожие аналоги.
В одном варианте осуществления дополнительный терапевтический агент представляет собой интерферон. Термин «интерферон» или IFN относится к любому члену семейства высокогомологичных видоспецифичных белков, которые ингибируют репликацию вируса и клеточную пролиферацию и модулируют иммунный ответ. Человеческие интерфероны сгруппированы в три класса: тип I, который включает интерферон-альфа (IFN-α), интерферон-бета (IFN-β) и интерферон-омега (IFN-ω); тип II, который включает интерферон-гамма (IFN-γ); и тип III, который включает интерферон-лямбда (IFN-λ). В настоящем документе термин «интерферон» включает рекомбинантные формы интерферонов, которые были разработаны и доступны на рынке. В настоящем документе термин «интерферон» также включает подтипы интерферонов, такие как химически модифицированные или мутировавшие интерфероны. Химически модифицированные интерфероны включают пегилированные интерфероны и гликозилированные интерфероны. Примеры интерферонов также включают, без ограничений, интерферон-альфа-2a, интерферон-альфа-2b, интерферон-альфа-n1, интерферон-бета-1a, интерферон-бета-1b, интерферон-лямбда-1, интерферон-лямбда-2 и интерферон-лямбда-3. Примеры пегилированных интерферонов включают пегилированный интерферон-альфа-2a и пегилированный интерферон-альфа-2b.
Соответственно, в одном варианте осуществления соединения формулы I, II, III или IV можно вводить в комбинации с интерфероном, выбранным из группы, состоящей из интерферона-альфа (IFN-α), интерферона-бета (IFN-β), интерферона-лямбда (IFN-λ) и интерферона-гамма (IFN-γ). В одном конкретном варианте осуществления интерферон представляет собой интерферон-альфа-2a, интерферон-альфа-2b или интерферон-альфа-n1. В другом конкретном варианте осуществления интерферон-альфа-2a или интерферон-альфа-2b являются пегилированными. В предпочтительном варианте осуществления интерферон-альфа-2a представляет собой пегилированный интерферон-альфа-2a (PEGASYS).
В другом варианте осуществления дополнительный терапевтический агент выбран из терапевтической группы иммуномодуляторов или иммуностимуляторов, которая включает биологические агенты, принадлежащие к классу интерферонов.
Кроме того, дополнительный терапевтический агент может представлять собой агент с другим или неизвестным механизмом действия, включая агенты, которые нарушают функцию другого (-их) основного (-ых) вирусного (-ых) белка (-ов) или белков хозяина, требуемых для репликации или персистенции ВГВ.
В другом варианте осуществления дополнительный терапевтический агент представляет собой противовирусный агент, который блокирует проникновение в клетку или созревание вируса или воздействует на полимеразу ВГВ, такой как нуклеозидные или нуклеотидные или ненуклеоз(т)идные ингибиторы полимеразы. В дополнительном варианте осуществления комбинированной терапии ингибитор обратной транскриптазы и/или ингибитор ДНК- и/или РНК-полимеразы представляет собой зидовудин, диданозин, залцитабин, диданозин (ddA), ставудин, ламивудин, абакавир, эмтрицитабин, энтекавир, априцитабин, атевирапин, рибавирин, ацикловир, фамцикловир, валацикловир, ганцикловир, валганцикловир, тенофовир, адефовир, 9-(2-фосфонилметоксипропил)аденин (PMPA), цидофовир, эфавиренз, невирапин, делавирдин или этравирин.
В одном варианте осуществления дополнительный терапевтический агент представляет собой иммуномодулирующий агент, который вызывает естественный, ограниченный иммунный ответ, приводящий к индукции иммунных ответов против неродственных вирусов. Другими словами, иммуномодулирующий агент способен вызывать созревание антигенпредставляющих клеток, пролиферацию Т-клеток и высвобождение цитокинов (например, среди прочих, IL-12, IL-18, IFN-альфа, -бета и -гамма и TNF-альфа).
В дополнительном варианте осуществления дополнительный терапевтический агент представляет собой модулятор TLR или агонист TLR, такой как агонист TLR-7 или агонист TLR-9. В дополнительном варианте осуществления комбинированной терапии агонист TLR-7 выбран из группы, состоящей из SM360320 (9-бензил-8-гидрокси-2-(2-метоксиэтокси)аденина) и AZD 8848 (метил-[3-({[3-(6-амино-2-бутокси-8-оксо-7,8-дигидро-9H-пурин-9-ил)пропил][3-(4-морфолинил)пропил]амино}метил)фенил]ацетата).
В любом из способов, предложенных в настоящем документе, способ может дополнительно включать введение субъекту по меньшей мере одной вакцины против ВГВ, ингибитора нуклеозида ВГВ, интерферона или любой их комбинации. В одном варианте осуществления вакцина против ВГВ представляет собой по меньшей мере одно из RECOMBIVAX HB, ENGERIX-B, ELOVAC B, GENEVAC-B или SHANVAC B.
В другом аспекте настоящего изобретения предложен способ лечения инфекции ВГВ у нуждающегося в этом субъекта, включающий снижение вирусной нагрузки ВГВ посредством введения субъекту терапевтически эффективного количества соединения настоящего изобретения в виде монотерапии или в комбинации с ингибитором обратной транскриптазы; и дополнительного введения субъекту терапевтически эффективного количества вакцины против ВГВ. Ингибитор обратной транскриптазы может представлять собой один из зидовудина, диданозина, залцитабина, ddA, ставудина, ламивудина, абакавира, эмтрицитабина, энтекавира, априцитабина, атевирапина, рибавирина, ацикловира, фамцикловира, валацикловира, ганцикловира, валганцикловира, тенофовира, адефовира, PMPA, цидофовира, эфавиренза, невирапина, делавирдина или этравирина.
Для любой комбинированной терапии, описанной в настоящем документе, синергетический эффект может быть рассчитан, например, с использованием приемлемых способов, таких как уравнение Sigmoid-Eмакс (Holford & Scheiner, 19981, Clin. Pharmacokinet. 6: 429-453), уравнение аддитивности Лоэва (Loewe & Muischnek, 1926, Arch. Exp. Pathol Pharmacol. 114: 313-326) и уравнение медианного эффекта (Chou & Talalay, 1984, Adv. Enzyme Regul. 22: 27-55). Каждое из упомянутых выше уравнений можно применять для обработки экспериментальных данных с построением соответствующего графика, который помогает оценить эффекты комбинации лекарственных средств. К соответствующим графикам, связанным с указанными выше уравнениями, относятся кривая зависимости эффекта от концентрации, изоболограмма и кривая показателей комбинации соответственно.
В одном варианте осуществления любого из способов введения комбинированных терапий, предложенных в настоящем изобретении, способ может дополнительно включать контроль вирусной нагрузки ВГВ у субъекта, причем способ осуществляют в течение периода времени до тех пор, пока вирус ВГВ перестает обнаруживаться.
Введение/доза/составы
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение настоящего изобретения или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
Фактические уровни дозы активных ингредиентов в фармацевтических композициях, описанных в данном изобретении, можно изменять для получения количества активного ингредиента, которое позволяет эффективно достигать требуемой терапевтической реакции у конкретного пациента, композиции или способа введения без проявления токсичности для пациента.
В частности, выбранный уровень дозы будет зависеть от множества факторов, например активности конкретного используемого соединения, времени введения, скорости выведения соединения, длительности лечения, других лекарственных средств, соединений или материалов, применяемых в комбинации с соединением, возраста, пола, веса, состояния, общего состояния здоровья и предшествующего анамнеза пациента, по отношению к которому проводится лечение, и других факторов, хорошо известных в медицине.
Медицинский работник, например врач или ветеринар, являющийся специалистом в данной области, может легко определить и назначить необходимое эффективное количество фармацевтической композиции. Например, врач или ветеринар может начинать с более низких доз соединений изобретения в фармацевтической композиции, чем необходимо для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозировку, пока не будет достигнут желаемый эффект.
В конкретных вариантах осуществления особенно предпочтительно получить композицию в единичной дозированной форме для простоты введения и однородности дозировки. Единичная дозированная форма, как описано в этом документе, относится к физически дискретным единицам, пригодным в качестве единичных доз для пациента, подлежащего лечению; причем каждая единица содержит предварительно заданное количество терапевтического соединения, рассчитанное на получение желаемого терапевтического эффекта в сочетании с требуемым фармацевтическим носителем. Единичные дозированные формы изобретения продиктованы и непосредственно зависят от: (а) уникальных характеристик терапевтического соединения и конкретного терапевтического эффекта, который должен быть достигнут; и (b) ограничений, присущих в данной области рецептуры/композиции, например, в случае терапевтического соединения для лечения инфекции ВГB у пациента.
В одном варианте осуществления композиции настоящего изобретения получают с использованием одного или более фармацевтически приемлемых эксципиентов или носителей. В одном варианте осуществления фармацевтические композиции настоящего изобретения содержат терапевтически эффективное количество соединения настоящего изобретения и фармацевтически приемлемый носитель.
В некоторых вариантах осуществления доза соединения настоящего изобретения составляет от около 1 мг до около 2500 мг. В некоторых вариантах осуществления доза соединения настоящего изобретения, используемая в композициях, описанных в настоящем документе, составляет менее около 10 000 мг, или менее около 8000 мг, или менее около 6000 мг, или менее около 5000 мг, или менее около 3000 мг, или менее около 2000 мг, или менее около 1000 мг, или менее около 500 мг, или менее около 200 мг, или менее около 50 мг. Аналогично в некоторых вариантах осуществления доза второго соединения (т. е. другого лекарственного средства для лечения ВГВ), как описано в настоящем документе, составляет менее около 1000 мг, или менее около 800 мг, или менее около 600 мг, или менее около 500 мг, или менее около 400 мг, или менее около 300 мг, или менее около 200 мг, или менее около 100 мг, или менее около 50 мг, или менее около 40 мг, или менее около 30 мг, или менее около 25 мг, или менее около 20 мг, или менее около 15 мг, или менее около 10 мг, или менее около 5 мг, или менее около 2 мг, или менее около 1 мг, или менее около 0,5 мг и любые ее целые или частичные приращения.
В одном варианте осуществления настоящее изобретение относится к укомплектованной фармацевтической композиции, содержащей контейнер, удерживающий терапевтически эффективное количество соединения настоящего изобретения, самого по себе или в комбинации со вторым фармацевтическим агентом; и инструкцию по применению соединения для лечения, предотвращения или ослабления одного или более симптомов инфекции ВГВ у пациента.
Способы введения любой из композиций настоящего изобретения включают пероральный, назальный, ректальный, интравагинальный, парентеральный, буккальный, сублингвальный или местный способ введения. Соединения для применения в настоящем изобретении могут быть составлены для введения любым приемлемым способом, таким как пероральный или парентеральный, например, трансдермальный, чресслизистый (например, сублингвальный, лингвальный, (транс)буккальный, (транс)уретральный, вагинальный (например, транс- и перивагинально), (интра)назальный и (транс)ректальный), внутрипузырный, внутрилегочный, интрадуоденальный, внутрижелудочный, интратекальный, подкожный, внутримышечный, интрадермальный, внутриартериальный, внутривенный, эндобронхиальный, ингаляционный и местный способ введения.
Приемлемые композиции и дозированные формы включают, например, таблетки, капсулы, таблетки в форме капсулы, пилюли, желатиновые капсулы, пастилки, дисперсии, суспензии, растворы, сиропы, гранулы, шарики, трансдермальные пластыри, гели, порошки, взвеси, таблетки для рассасывания, кремы, пасты, пластыри, лосьоны, диски, свечи, жидкие спреи для назального или перорального введения, сухой порошок или аэрозольные составы для ингаляции, композиции и составы для внутрипузырного введения и т. п. Следует понимать, что составы и композиции, которые можно использовать в настоящем изобретении, не ограничены конкретными составами и композициями, которые описаны в настоящем документе.
Для перорального применения особенно приемлемыми являются таблетки, драже, жидкости, капли, свечи или капсулы, таблетки в форме капсулы и желатиновые капсулы. Композиции, предназначенные для перорального применения, можно получать в соответствии с любым способом, известным в данной области, и такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из инертных, нетоксичных фармацевтических эксципиентов, которые являются приемлемыми для изготовления таблеток. Такие эксципиенты включают, например, инертный разбавитель, такой как лактоза; гранулирующие и разрыхляющие агенты, такие как кукурузный крахмал; связующие агенты, такие как крахмал; и смазывающие агенты, такие как стеарат магния. Таблетки могут не быть покрыты оболочкой, или они могут быть покрыты оболочкой с использованием известных способов для улучшения внешнего вида или замедления высвобождения активных ингредиентов. Составы для перорального применения могут также быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным разбавителем.
В случае парентерального введения соединения настоящего изобретения могут быть составлены для инъекции или инфузии, например внутривенной, внутримышечной или подкожной инъекции или инфузии, или для введения в виде болюсной дозы и/или непрерывной инфузии. Можно использовать суспензии, растворы или эмульсии в масляной или водной несущей среде, необязательно содержащие другие вспомогательные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты.
Специалисты в данной области смогут определить или будут способны установить с помощью стандартных экспериментов множество эквивалентов конкретных способов, вариантов осуществления изобретения, пунктов формулы изобретения и примеров, описанных в настоящем документе. Такие эквиваленты входят в объем настоящего изобретения и включены в формулу изобретения, приложенную к настоящему изобретению. Например, следует понимать, что в объем настоящей заявки включены модификации условий реакции, включая, без ограничений, времена реакций, размер/объем реакции и экспериментальные реагенты, такие как растворители, катализаторы, давление, атмосферные условия, например атмосфера азота, и восстанавливающие/окисляющие агенты, с использованием известных в данной области альтернативных вариантов и без выполнения чрезмерного экспериментирования.
Следует понимать, что в объем настоящего изобретения включены все значения и диапазоны, которые включены в значения и диапазоны, предложенные в настоящем документе. Более того, все значения, которые находятся внутри этих диапазонов, а также верхние и нижние пределы этих значений также учтены в настоящей заявке.
Представленные ниже примеры дополнительно иллюстрируют аспекты настоящего изобретения. Однако они никоим образом не ограничивают идеи или описание настоящего изобретения, представленного в настоящем документе.
Примеры
Изобретение ниже описано со ссылкой на следующие примеры. Эти примеры приведены только с иллюстративной целью, и настоящее изобретение не ограничено этими примерами, но включает все вариации, которые являются очевидными в результате предложенных в настоящем документе идей.
Пример 1. Получение соединений 010 и 059
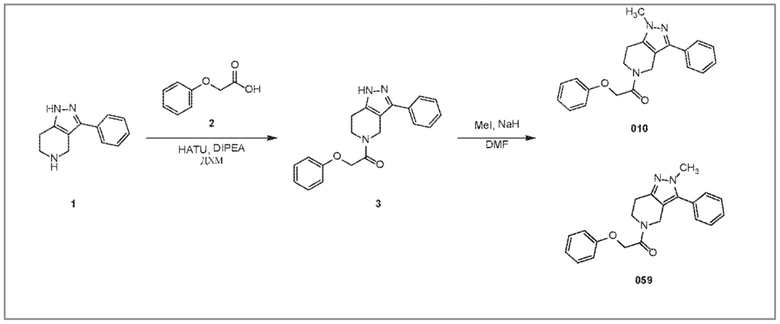
Стадия 1. Получение соединения 3
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (1,00 г, 4,24 ммоль, 1,00 экв.) и 2-феноксиуксусной кислоты (645,50 мг, 4,24 ммоль, 1,00 экв.) в ДХМ (40 мл) добавляли HATU (1,93 г, 5,09 ммоль, 1,20 экв.) и (диизопропилэтиламин) DIPEA (1,37 г, 10,60 ммоль, 2,50 экв.) за один раз при 30°C в атмосфере N2. Смесь перемешивали при температуре 30°C в течение 12 ч. Жидкостная хроматография с масс-спектрометрией (ЖХМС) показала завершение реакции. Смесь вливали в воду (50 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=3/1) с получением 2-фенокси-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)этанона (1,30 г, 3,90 ммоль, выход 91,97%) в виде желтого твердого вещества. ЖХМС: 334 [M+1].
Получение соединений 010 и 059
К смеси 2-фенокси-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)этанона (150,00 мг, 449,94 мкмоль, 1,00 экв.) в DMF (10 мл) добавляли NaH (36,00 мг, 899,88 мкмоль, 2,00 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 0,5 ч, впоследствии к смеси добавляли MeI (191,59 мг, 1,35 ммоль, 3,00 экв.) при 0 °C. Смесь нагревали до 30°C и перемешивали в течение 12 часов. ЖХМС показала завершение реакции. Смесь гасили водой. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением региоизомерной смеси (85,34 мг, 245,65 мкмоль, выход 54,60%) в виде желтого твердого вещества. Желтое твердое вещество разделяли посредством сверхкритической жидкостной хроматографии (SFC) с получением 1-(1-метил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (21,34 мг, 61,43 мкмоль) и 1-(2-метил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (32,11 мг, 92,43 мкмоль). Определение характеристик соединения 010: 1H ЯМР (400 МГц, метанол-d4) δ 7,56-7,66 (м, 2 H), 7,43 (с, 5 H), 6,84-7,04 (м, 3 H), 4,89-4,92 (м, 1 H), 4,85 (с, 1 H), 4,76-4,80 (м, 2 H), 3,87-4,00 (м, 2 H), 3,74-3,82 (м, 3 H), 2,73-2,95 (м, 2 H). ЖХМС: 348 [M+1]. Определение характеристик соединения 059: 1H ЯМР (400 МГц, метанол-d4) δ 7,39-7,67 (м, 5 H), 7,19-7,33 (м, 2 H), 6,83-7,02 (м, 3 H), 4,88-4,90 (м, 2 H), 4,54-4,61 (м, 2 H), 3,84-4,00 (м, 2 H), 3,77 (с, 3 H), 2,71-2,92 (м, 2 H). ЖХМС: 348 [M+1].
Пример 2. Получение соединений 011 и 060
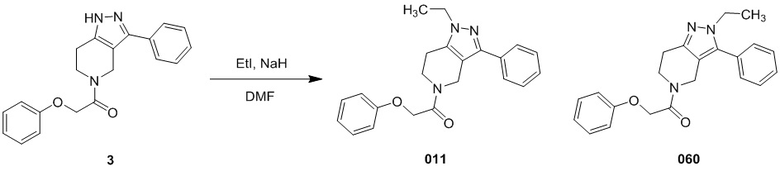
К смеси 2-фенокси-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)этанона (150,00 мг, 449,94 мкмоль, 1,00 экв.) в DMF (10 мл) добавляли NaH (36,00 мг, 899,88 мкмоль, 2,00 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 0,5 ч, впоследствии к смеси добавляли иодэтан (210,53 мг, 1,35 ммоль, 3,00 экв.) при 0 °C. Смесь нагревали до 30°C и перемешивали в течение 12 часов. ЖХМС показала завершение реакции. Смесь гасили водой (5 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (15 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-(1-этил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (71,20 мг, 196,99 мкмоль, выход 43,78%) и 1-(2-этил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (21,49 мг, 59,46 мкмоль, выход 13,21%) в виде белого твердого вещества.
Определение характеристик соединения 011: 1H ЯМР (400 МГц, метанол-d4) δ 7,57-7,66 (м, 2 H), 7,38-7,50 (м, 2 H), 7,15-7,38 (м, 3 H), 6,83-7,05 (м, 3 H), 4,91 (с, 1 H), 4,83-4,86 (м, 1 H), 4,75-4,81 (м, 2 H), 4,05-4,18 (м, 2 H), 3,85-4,03 (м, 2 H), 2,87-2,96 (м, 1 H), 2,75-2,83 (м, 1 H), 1,40 (с, 3 H). ЖХМС: 362 [M+1].
Определение характеристик соединения 060: 1H ЯМР (400 МГц, метанол-d4) δ 7,44-7,59 (м, 3 H), 7,37-7,44 (м, 2 H), 7,19-7,33 (м, 2 H), 6,84-7,02 (м, 3 H), 4,89 (уш. с, 2 H), 4,51-4,62 (м, 2 H), 4,08 (д, J=7,28 Гц, 2 H), 3,89 (с, 2 H), 2,85-2,91 (м, 1 H), 2,74-2,81 (м, 1 H), 1,31 (т, J=7,22 Гц, 3 H). ЖХМС: 362 [M+1].
Пример 3. Получение соединений 012 и 061
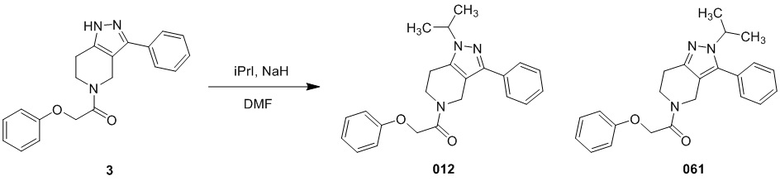
К смеси 2-фенокси-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)этанона (150,00 мг, 449,94 мкмоль, 1,00 экв.) в DMF (10 мл) добавляли NaH (36,00 мг, 899,88 мкмоль, 2,00 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 0,5 ч, впоследствии к смеси добавляли 2-иодпропан (229,46 мг, 1,35 ммоль, 3,00 экв.), смесь нагревали до 30°C и перемешивали в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-(1-изопропил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (22,18 мг, 59,07 мкмоль, выход 13,13%) и 1-(2-изопропил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (54,13 мг, 144,17 мкмоль, выход 32,04%) в виде желтого твердого вещества. Определение характеристик соединения 012: 1H ЯМР (400 МГц, метанол-d4) δ 7,57-7,69 (м, 2 H), 7,39-7,49 (м, 2 H), 7,17-7,38 (м, 3 H), 6,84-7,06 (м, 3 H), 4,91 (с, 2 H), 4,75-4,81 (м, 2 H), 4,40-4,58 (м, 1 H), 3,84-4,04 (м, 2 H), 2,75-3,00 (м, 2 H), 1,49 (д, J=6,53 Гц, 6 H). ЖХМС: 376 [M+1]. Определение характеристик соединения 061: 1H ЯМР (400 МГц, метанол-d4) δ 7,43-7,60 (м, 3 H), 7,34-7,41 (м, 2 H), 7,18-7,33 (м, 2 H), 6,82-7,02 (м, 3 H), 4,89 (уш. с, 2 H), 4,43-4,63 (м, 3 H), 3,81-4,01 (м, 2 H), 2,74-2,94 (м, 2 H), 1,41 (д, J=6,65 Гц, 6 H). ЖХМС: 376 [M+1].
Пример 4. Получение соединений 013, 056, 062 и 063
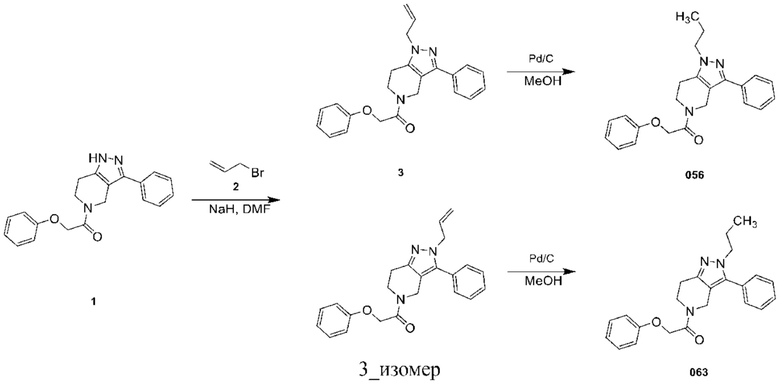
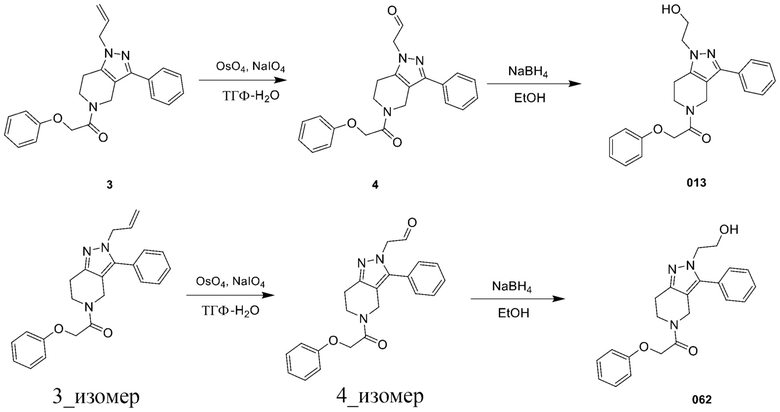
Стадия 1. Получение соединения 3 и изомера соединения 3
К смеси 2-фенокси-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)этанона (400,00 мг, 1,20 ммоль, 1,00 экв.) в DMF (10 мл) добавляли NaH (95,99 мг, 2,40 ммоль, 2,00 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 1 ч, впоследствии к смеси добавляли 3-бромпроп-1-ен (362,94 мг, 3,00 ммоль, 2,50 экв.), смесь нагревали до 30°C и перемешивали в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в ледяную воду (масс./масс.=1/1) (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (40 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (30 л*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=3/1) с получением 1-(1-аллил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (330,00 мг, 883,65 мкмоль, выход 73,64%) и 1-(2-аллил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (120,00 мг, 267,77 мкмоль, выход 22,31%) в виде желтого твердого вещества. ЖХМС: 374 [M+1].
Получение соединения 056
К раствору 1-(1-аллил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (100,00 мг, 267,77 мкмоль, 1,00 экв.) в MeOH (10 мл) добавляли Pd-C (10%, 20,00 мг) в атмосфере N2. Суспензию дегазировали в вакууме и несколько раз продували H2. Смесь перемешивали в H2 (15 фунтов/кв. дюйм) при температуре 30°C в течение 12 часов. ЖХМС показала, что исходный материал был полностью использован. Реакционную смесь фильтровали и фильтрат концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ (FA) с получением 2-фенокси-1-(3-фенил-1-пропил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)этанона (45,32 мг, 120,71 мкмоль, выход 45,08%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,56-7,70 (м, 2 H), 7,35-7,48 (м, 2 H), 7,20-7,34 (м, 3 H), 6,85-7,00 (м, 3 H), 4,87-5,01 (м, 2 H), 4,60-4,77 (м, 2 H), 3,89-4,07 (м, 2 H), 3,67-3,85 (м, 2 H), 2,80-2,95 (м, 1 H), 2,65-2,76 (м, 1 H), 1,69-1,84 (м, 2 H), 0,86 (т, J=7,34 Гц, 3 H). ЖХМС: 376 [M+1].
Получение соединения 063
К раствору 1-(2-аллил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (50,00 мг, 133,89 мкмоль, 1,00 экв.) в MeOH (10 мл) добавляли Pd-C (10%, 5 мг) в атмосфере N2. Суспензию дегазировали в вакууме и несколько раз продували H2. Смесь перемешивали в атмосфере H2 (103 кПа (15 фунтов/кв. дюйм)) при 30°C в течение 10 часов. ЖХМС показала, что исходный материал был полностью использован. Реакционную смесь фильтровали и фильтрат концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ (FA) с получением 2-фенокси-1-(3-фенил-2-пропил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)этанона (25,34 мг, 67,49 мкмоль, выход 50,41%) в виде белого твердого вещества. ЖХМС: 376 [M+1].
Получение соединения 013
К смеси 1-(1-аллил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (150,00 мг, 401,66 мкмоль, 1,00 экв.) в ТГФ (10 мл) и H2O (2 мл) добавляли NaIO4 (189,00 мг, 883,65 мкмоль, 2,20 экв.) и OsO4 (10,21 мг, 40,17 мкмоль, 0,10 экв.) за один раз в атмосфере N2. Смесь перемешивали при температуре 30°C в течение 5 ч. Смесь промывали насыщенным Na2SO3 (20 мл*2). Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток разбавляли EtOH (10 мл), к смеси добавляли NaBH4 (15,20 мг, 401,66 мкмоль, 1,00 экв.). Смесь перемешивали при температуре 30°C в течение 5 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (15 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-[1-(2-гидроксиэтил)-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил]-2-феноксиэтанона (45,32 мг, 120,07 мкмоль, выход 29,89%) в виде белого твердого вещества. ЖХМС: 378 [M+1].
Получение соединения 062
К смеси 1-(2-аллил-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (70,00 мг, 187,44 мкмоль, 1,00 экв.) в ТГФ (10 мл) и H2O (2 мл) добавляли NaIO4 (88,20 мг, 412,37 мкмоль, 2,20 экв.) и OsO4 (4,77 мг, 18,74 мкмоль, 0,10 экв.) за один раз в атмосфере N2. Смесь перемешивали при температуре 30°C в течение 5 ч. Смесь промывали насыщенным Na2SO3 (20 мл*2). Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток разбавляли EtOH (10 мл), к смеси добавляли NaBH4 (8,51 мг, 224,93 мкмоль, 1,20 экв.). Смесь перемешивали при температуре 30°C в течение 5 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (15 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-[2-(2-гидроксиэтил)-3-фенил-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-ил]-2-феноксиэтанона (18,24 мг, 48,33 мкмоль, выход 25,78%) в виде белого твердого вещества. ЖХМС: 378 [M+1].
Пример 5. Получение соединения 024
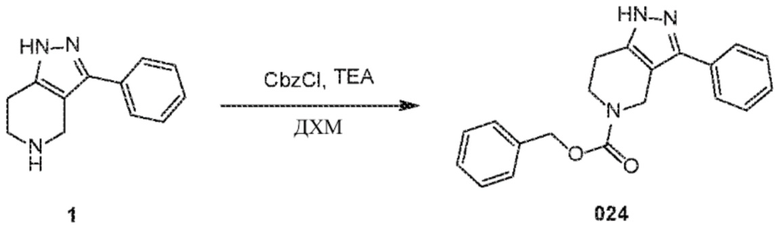
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (150,00 мг, 636,38 мкмоль, 1,00 экв.) в ДХМ (5 мл) добавляли CbzCl (130,27 мг, 763,66 мкмоль, 1,20 экв.) и TEA (193,19 мг, 1,91 ммоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при температуре 30°C в течение 12 ч. ЖХМС показала завершение реакции. Смесь промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением бензил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (48,97 мг, 146,89 мкмоль, выход 23,08%) в виде белого твердого вещества. 1H ЯМР (400 МГц, метанол-d4) 7,50-7,64 (м, 2 H), 7,41-7,50 (м, 2 H), 7,24-7,41 (м, 6 H), 5,12-5,22 (м, 2 H), 4,69-4,77 (м, 2 H), 3,78-3,89 (м, 2 H), 2,74-2,85 (м, 2 H). ЖХМС: 334 [M+1].
Пример 6. Получение соединения 016
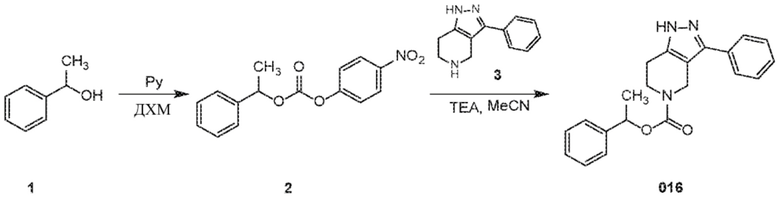
Стадия 1. Получение соединения 2
К смеси 1-фенилэтанола (2,00 г, 16,37 ммоль, 1,00 экв.) и Py (3,24 г, 40,93 ммоль, 2,50 экв.) в ДХМ (75,00 мл) по каплям добавляли (4-нитрофенил)хлорформиат (3,30 г, 16,37 ммоль, 1,00 экв.) в ДХМ (75,00 мл) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 1 часа, впоследствии нагревали до 30°C и перемешивали в течение 12 часов. Тонкослойная хроматография (ТСХ) показала завершение реакции. Смесь вливали в воду (30 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=10/1) с получением (4-нитрофенил)-1-фенилэтилкарбоната (2,90 г, 10,10 ммоль, выход 61,67%) в виде желтого твердого вещества. ЖХМС: 288 [M+1].
Получение соединения 016
К смеси (4-нитрофенил)-1-фенилэтилкарбоната (150,00 мг, 522,16 мкмоль, 1,00 экв.) и 3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (123,08 мг, 522,16 мкмоль, 1,00 экв.) в MeCN (10,00 мл) добавляли TEA (158,51 мг, 1,57 ммоль, 3,00 экв.) за один раз при 30°C в атмосфере N2. Смесь перемешивали при 30°C в течение 5 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-фенилэтил-3-фенил-1,4,6,7-
тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (54,40 мг, 156,59 мкмоль, выход 29,99%) в виде белого твердого вещества. ЖХМС: 348 [M+1].
Пример 7. Получение соединений 025-030 и 057
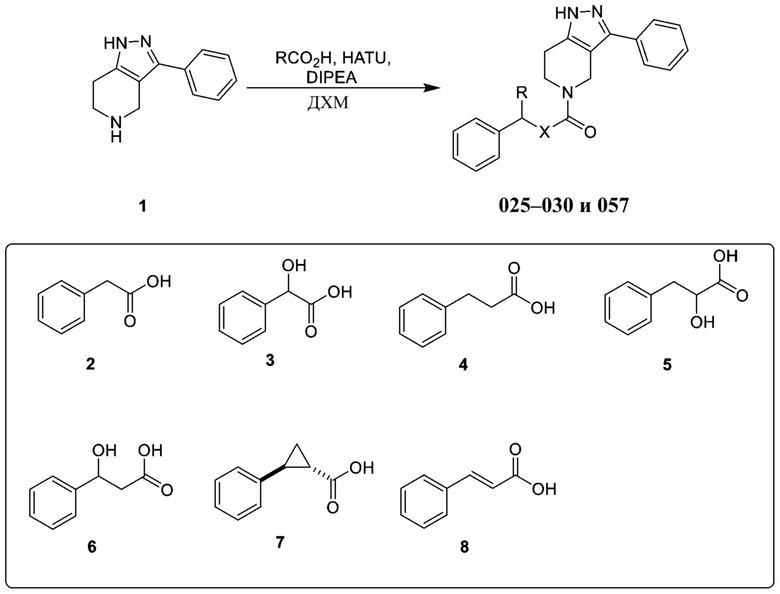
Получение соединения 025
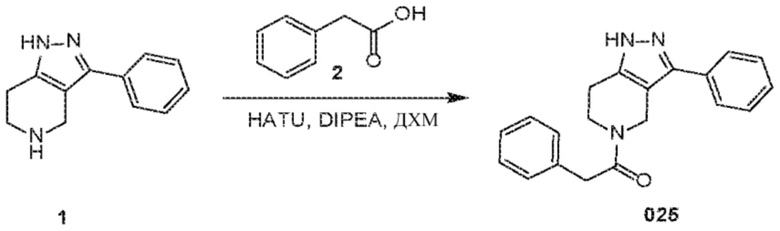
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 2-фенилуксусной кислоты (57,76 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) за один раз при 30°C в атмосфере N2. Смесь перемешивали при температуре 30°C в течение 8 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 2-фенил-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)этанона (32,58 мг, 102,65 мкмоль, выход 24,20%) в виде белого твердого вещества. ЖХМС: 318 [M+1].
Получение соединения 026
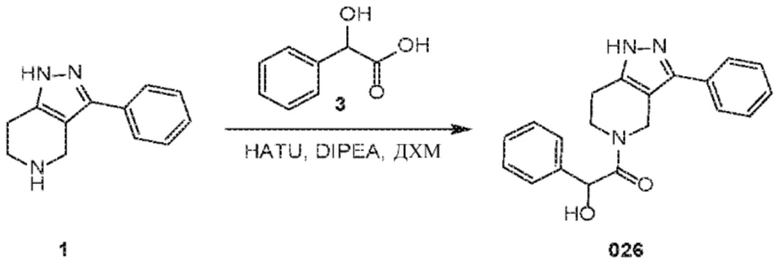
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 2-гидрокси-2-фенилуксусной кислоты (64,55 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) за один раз при 30°C в атмосфере N2. Смесь перемешивали при температуре 30°C в течение 8 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 2-гидрокси-2-фенил-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)этанона (46,19 мг, 138,55 мкмоль, выход 32,66%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) 7,13-7,69 (м, 10 H), 5,75 (д, J=6,53 Гц, 1 H), 5,51 (уш. с, 1 H), 4,62-4,83 (м, 2 H), 3,74 (уш. с, 3 H), 2,67 (уш. с, 1 H), 2,15 (д, J=15,18 Гц, 1 H). ЖХМС: 334 [M+1].
Получение соединения 027
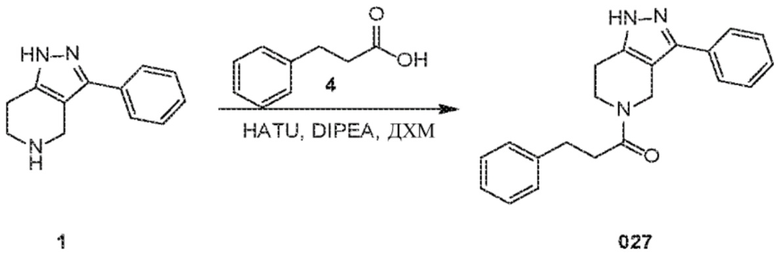
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 3-фенилпропановой кислоты (63,71 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 14 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 3-фенил-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)пропан-1-она (24,76 мг, 74,71 мкмоль, выход 17,61%) в виде белого твердого вещества. ЖХМС: 332 [M+1].
Получение соединения 028
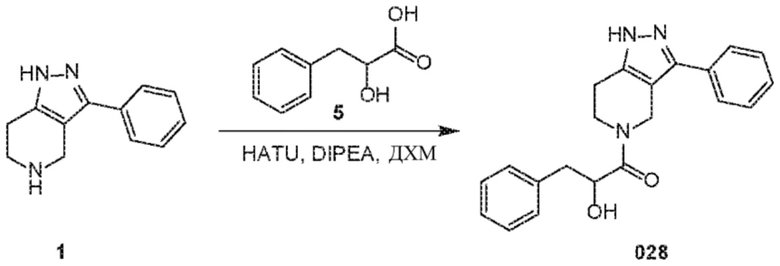
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 2-гидрокси-3-фенилпропановой кислоты (70,50 мг, 424,25 мкмоль, 1,00 экв.) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 15 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 2-гидрокси-3-фенил-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)пропан-1-она (58,00 мг, 166,95 мкмоль, выход 39,35%) в виде белого твердого вещества. ЖХМС: 348 [M+1].
Получение соединения 029
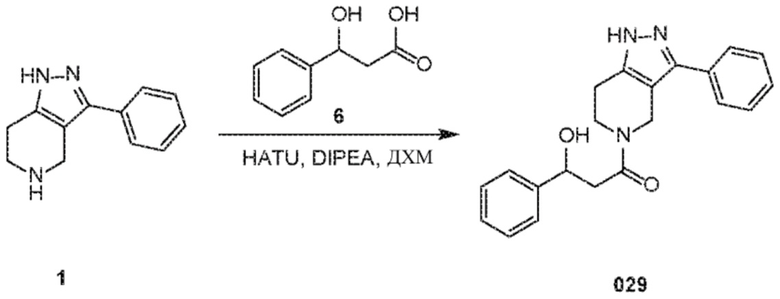
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 3-гидрокси-3-фенилпропановой кислоты (70,50 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 15 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 3-гидрокси-3-фенил-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)пропан-1-она (95,00 мг, 273,45 мкмоль, выход 64,46%) в виде белого твердого вещества. ЖХМС: 348 [M+1].
Получение соединения 030
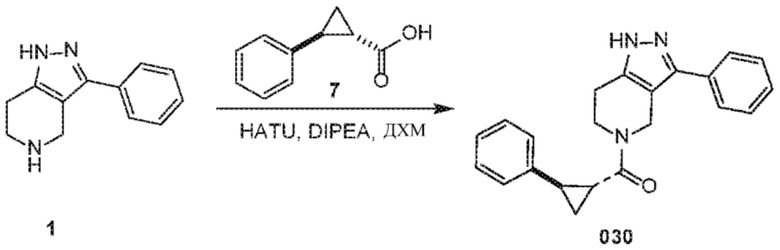
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 2-фенилциклопропанкарбоновой кислоты (68,80 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) и HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) за один раз при 30°C в атмосфере N2. Смесь перемешивали при температуре 30°C в течение 4 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением (2-фенилциклопропил)-(3-фенил-2,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)метанона (27,92 мг, 81,30 мкмоль, выход 19,16%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) 7,11-7,63 (м, 10 H), 4,88 (д, J=14,56 Гц, 1 H), 4,72 (с, 1 H), 3,67-3,99 (м, 2 H), 2,63-2,86 (м, 2 H), 2,27-2,47 (м, 2 H), 1,43 (тд, J=8,97, 4,52 Гц, 1 H), 1,18-1,28 (м, 1 H). ЖХМС: 344 [M+1].
Получение соединения 057
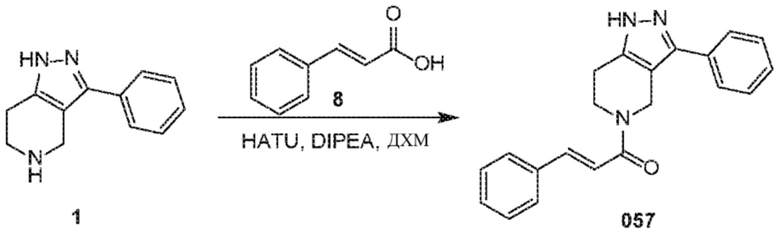
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и (Е)-3-фенилпроп-2-еновой кислоты (62,86 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 5 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением (Е)-3-фенил-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)проп-2-ен-1-она (61,85 мг, 187,77 мкмоль, выход 44,26%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) 12,63-13,14 (м, 1 H), 7,32-7,80 (м, 12 H), 4,75-5,00 (м, 2 H), 3,86-4,06 (м, 2 H),2,67-2,90 (м, 2 H). ЖХМС: 330 [M+1].
Пример 8. Получение соединений 017-023
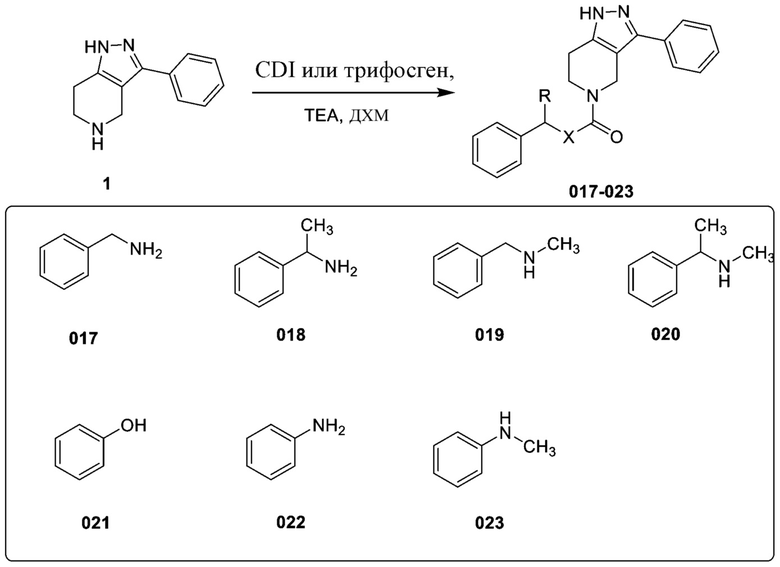
Получение соединения 017
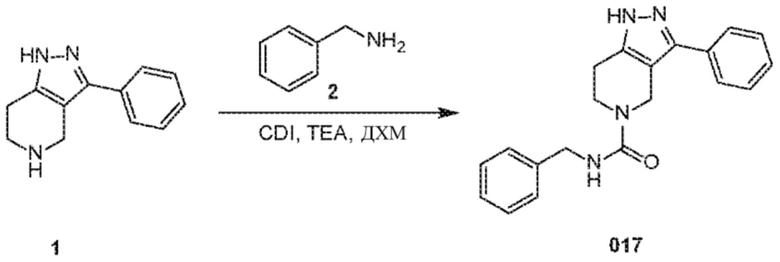
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и фенилметанамина (45,46 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли TEA (51,52 мг, 509,10 мкмоль, 1,20 экв.) и CDI (68,79 мг, 424,25 мкмоль, 1,00 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 5 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-бензил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (29,09 мг, 87,52 мкмоль, выход 20,63%) в виде белого твердого вещества. ЖХМС: 333 [M+1].
Получение соединения 018
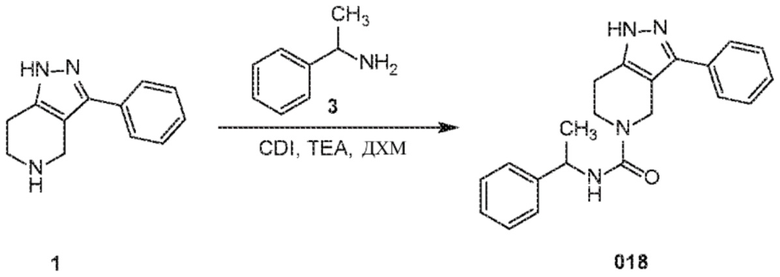
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 1-фенилэтанамина (51,41 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли TEA (51,52 мг, 509,10 мкмоль, 1,20 экв.) и CDI (68,79 мг, 424,25 мкмоль, 1,00 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 14 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 3-фенил-N-(1-фенилэтил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (39,91 мг, 115,21 мкмоль, выход 27,16%) в виде белого твердого вещества. ЖХМС: 347 [M+1].
Получение соединения 019
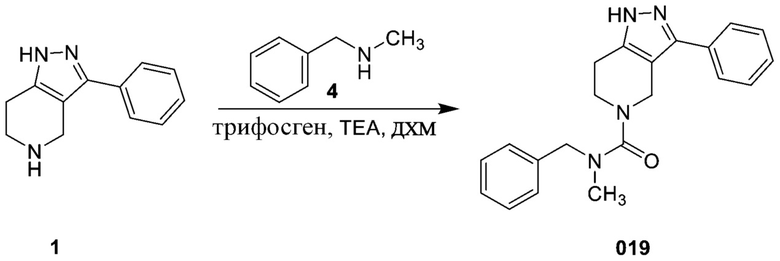
К смеси N-метил-1-фенилметанамина (51,41 мг, 424,25 мкмоль, 1,00 экв.) и TEA (214,65 мг, 2,12 ммоль, 5,00 экв.) в ДХМ (15 мл) добавляли ТРИФОСГЕН (50,36 мг, 169,70 мкмоль, 0,40 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 5 мин, впоследствии добавляли 3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин (100,00 мг, 424,25 мкмоль, 1,00 экв.) и перемешивали при 0°C в течение 2 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-бензил-N-метил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (23,51 мг, 67,87 мкмоль, выход 16,00%) в виде белого твердого вещества. ЖХМС: 347 [M+1].
Получение соединения 020
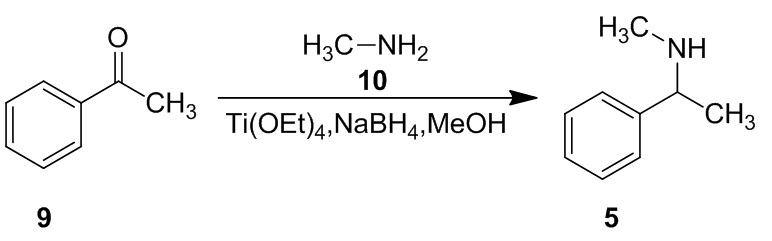
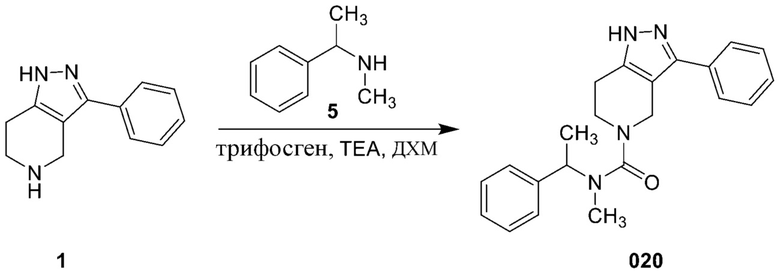
Стадия 1. Получение соединения 5. К смеси 1-фенилэтанона (1,00 г, 8,32 ммоль, 1,00 экв.) и этоксида титана (3,80 г, 16,64 ммоль, 2,00 экв.) в MeOH (15 мл) добавляли метанамин (2,58 г, 83,20 ммоль, 10,00 экв.) при 30°C в атмосфере N2. Смесь перемешивали при 30°C в течение 5 ч, впоследствии добавляли NaBH4 (472,12 мг, 12,48 ммоль, 1,50 экв.) при 0°C в атмосфере N2 и перемешивали в течение 10 мин. Смесь нагревали до 30°C и перемешивали в течение 19 часов. ЖХМС показала завершение реакции. Смесь добавляли в воду (20 мл) за один раз и перемешивали в течение 10 мин. Водную фазу экстрагировали EA (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением N-метил-1-фенилэтанамина (700,00 мг, неочищенный) в виде желтого масла. Неочищенный продукт использовали на следующей стадии непосредственно без очистки. ЖХМС: 136 [M+1].
Стадия 2. Получение соединения 020. К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и фенола (39,93 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли TEA (51,52 мг, 509,10 мкмоль, 1,20 экв.) и CDI (68,79 мг, 424,25 мкмоль, 1,00 экв.) при 30 °C. Смесь перемешивали при температуре 30°C в течение 8 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением фенил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (42,83 мг, 134,11 мкмоль, выход 31,61%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 7,11-7,66 (м, 10 H), 5,20 (д, J=7,15 Гц, 1 H), 4,42-4,58 (м, 2 H), 3,61 (к, J=5,98 Гц, 2 H), 2,89 (т, J=5,58 Гц, 2 H), 2,65 (с, 3 H), 1,57 (д, J=7,03 Гц, 3 H). ЖХМС: 361 [M+1].
Получение соединения 021
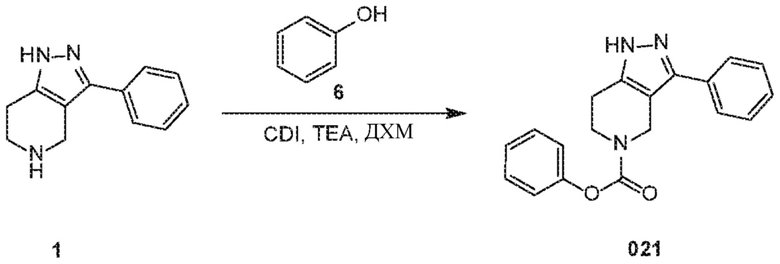
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и фенола (39,93 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли TEA (51,52 мг, 509,10 мкмоль, 1,20 экв.) и CDI (68,79 мг, 424,25 мкмоль, 1,00 экв.) при 30 °C. Смесь перемешивали при температуре 30°C в течение 8 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением фенил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (42,83 мг, 134,11 мкмоль, выход 31,61%) в виде белого твердого вещества. ЖХМС: 320 [M+1].
Получение соединения 022
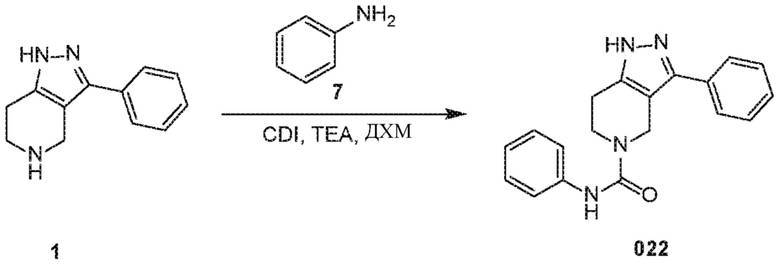
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и анилина (39,51 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли TEA (51,52 мг, 509,10 мкмоль, 1,20 экв.) и CDI (68,79 мг, 424,25 мкмоль, 1,00 экв.) при 30 °C. Смесь перемешивали при температуре 30°C в течение 8 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N,3-дифенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (20,23 мг, 63,54 мкмоль, выход 14,98%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) м. д. 12,58-13,11 (м, 1 H), 8,68 (с, 1 H), 7,65 (уш. с, 2 H), 7,44 (д, J=7,53 Гц, 5 H), 7,23 (т, J=7,97 Гц, 2 H), 6,90-6,97 (м, 1 H), 4,72 (с, 2 H), 3,78 (т, J=5,33 Гц, 2 H), 2,76 (уш. с, 2 H).
ЖХМС: 319 [M+1].
Получение соединения 023
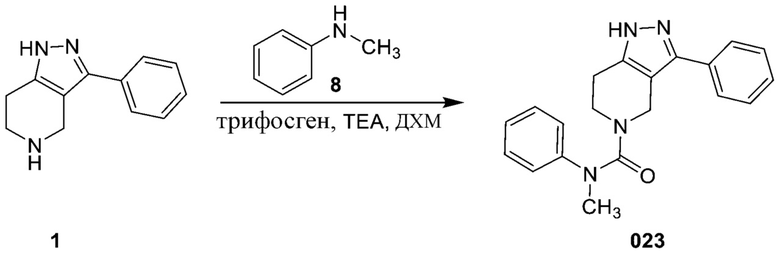
К смеси N-метиланилина (45,46 мг, 424,25 мкмоль, 1,00 экв.) и TEA (214,65 мг, 2,12 ммоль, 5,00 экв.) в ДХМ (15 мл) добавляли трифосген (50,36 мг, 169,70 мкмоль, 0,40 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 5 мин, впоследствии добавляли 3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин (100,00 мг, 424,25 мкмоль, 1,00 экв.) и перемешивали при 0°C в течение 2 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-метил-N,3-дифенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (32,10 мг, 96,57 мкмоль, выход 22,76%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) м. д. 12,47-13,04 (м, 1 H), 7,38 (дд, J=16,00, 8,09 Гц, 7 H), 7,10-7,22 (м, 3 H), 4,28 (с, 2 H), 3,45 (д, J=5,52 Гц, 2 H), 3,11 (с, 3 H), 2,38 (уш. с, 2 H). ЖХМС: 333 [M+1].
Пример 9. Получение соединения 014
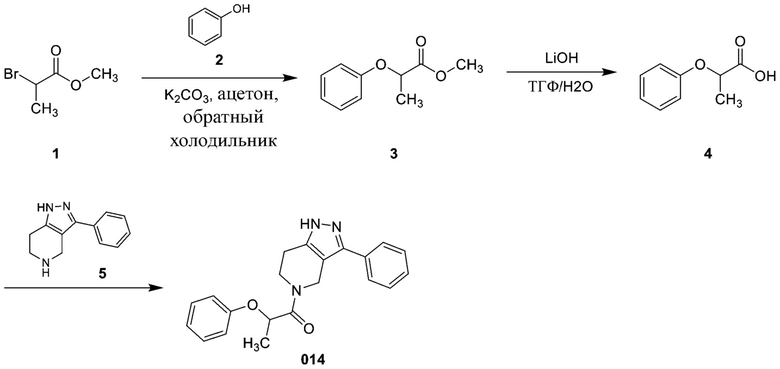
Стадия 1. Получение соединения 3
К смеси метил-2-бромпропаноата (2,00 г, 11,98 ммоль, 1,00 экв.) и фенола (1,13 г, 11,98 ммоль, 1,00 экв.) в АЦЕТОНЕ (30 мл) добавляли K2CO3 (2,48 г, 17,97 ммоль, 1,50 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 5 часов. ЖХМС показала завершение реакции. Смесь концентрировали при пониженном давлении при 45 oC. Остаток вливали в воду (50 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (50 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением метил-2-феноксипропаноата (3,30 г, неочищенный). Неочищенный продукт использовали на следующей стадии непосредственно без очистки. ЖХМС: 181 [M+1].
Стадия 2. Получение соединения 4
К смеси метил-2-феноксипропаноата (3,30 г, 18,31 ммоль, 1,00 экв.) и LiOH (877,05 мг, 36,62 ммоль, 2,00 экв.) в ТГФ (50 мл). Смесь перемешивали при температуре 30°C в течение 15 ч. ТСХ показала завершение реакции. Смесь вливали в воду (100 мл) и перемешивали в течение 10 мин и подкисляли разведенной соляной кислотой, впоследствии водную фазу экстрагировали этилацетатом (50 мл*3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл*3), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением 2-феноксипропановой кислоты (1,03 г, 6,20 ммоль, выход 33,85%).
Получение соединения 014
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 2-феноксипропановой кислоты (70,50 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 15 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 2-фенокси-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)пропан-1-она (96,00 мг, 276,33 мкмоль, выход 65,13%) в виде белого твердого вещества. ЖХМС: 348 [M+1].
Пример 10. Получение соединения 015
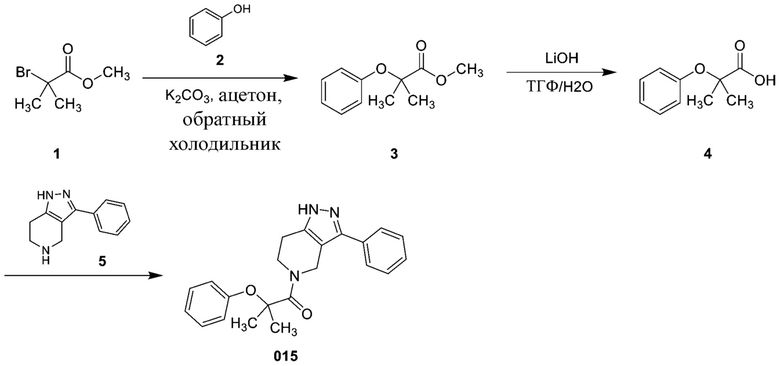
Стадия 1. Получение соединения 3
К смеси метил-2-бром-2-метилпропаноата (2,00 г, 11,05 ммоль, 1,00 экв.) и фенола (1,04 г, 11,05 ммоль, 1,00 экв.) в АЦЕТОНЕ (30 мл) добавляли K2CO3 (2,29 г, 16,58 ммоль, 1,50 экв.) при 30 °C. Впоследствии нагревали до 60°C и перемешивали в течение 5 часов. ЖХМС показала завершение реакции. Смесь охлаждали до 30°C и концентрировали под пониженным давлением при 45 °C. Остаток вливали в воду (50 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (50 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением метил-2-метил-2-феноксипропаноата (1,50 г, неочищенный) в виде желтого масла. Неочищенный продукт использовали на следующей стадии непосредственно без очистки. ЖХМС: 195 [M+1].
Стадия 2. Получение соединения 4
К смеси метил-2-метил-2-феноксипропаноата (1,50 г, 7,72 ммоль, 1,00 экв.) и LiOH (369,79 мг, 15,44 ммоль, 2,00 экв.) в ТГФ (50 мл) и H2O (10 мл). Смесь перемешивали при температуре 30°C в течение 15 ч. ТСХ показала завершение реакции. Смесь вливали в воду (100 мл) и перемешивали в течение 10 мин. Водную фазу подкисляли разведенной соляной кислотой, впоследствии водную фазу экстрагировали этилацетатом (50 мл*3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл*3), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением 2-метил-2-феноксипропановой кислоты (850,00 мг, 4,72 ммоль, выход 61,10%).
Получение соединения 015
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и 2-метил-2-феноксипропановой кислоты (76,45 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (10 мл) добавляли HATU (241,97 мг, 636,38 мкмоль, 1,50 экв.) и DIPEA (137,08 мг, 1,06 ммоль, 2,50 экв.) при 30 °C. Смесь перемешивали при 30°C в течение 18 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 2-метил-2-фенокси-1-(3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)пропан-1-она (21,30 мг, 58,93 мкмоль, выход 13,89%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) м. д. 7,59 (д, J=7,40 Гц, 1 H), 7,43-7,52 (м, 3 H), 7,34 (т, J=7,03 Гц, 1 H), 7,25 (т, J=7,53 Гц, 1 H), 7,11-7,18 (м, 1 H), 6,77-6,99 (м, 2 H), 6,68 (д, J=7,65 Гц, 1 H), 5,02 (уш. с, 1 H), 4,74 (уш. с, 1 H), 4,08 (уш. с, 2 H), 3,81 (уш. с, 2 H), 1,42-1,64 (м, 6 H). ЖХМС: 362 [M+1].
Пример 11. Получение соединения 067
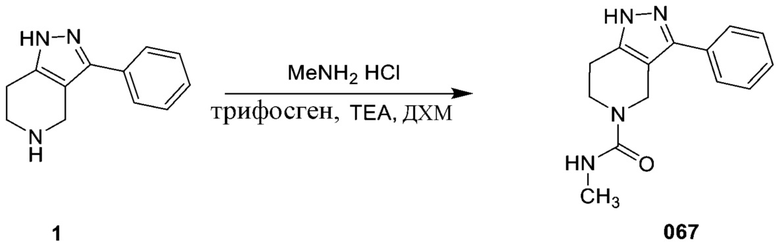
К смеси 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (100,00 мг, 424,25 мкмоль, 1,00 экв.) и гидрохлорида метанамина (28,64 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (15,00 мл) добавляли TEA (429,30 мг, 4,24 ммоль, 10,00 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 5 мин, впоследствии добавляли ТРИФОСГЕН (50,36 мг, 169,70 мкмоль, 0,40 экв.) и нагревали до 30°C в течение 15 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (20 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-метил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (11,68 мг, 45,57 мкмоль, выход 10,74%) в виде белого твердого вещества. ЖХМС: 257 [M+1].
Пример 12. Получение соединений 111, 112, 113 и 114
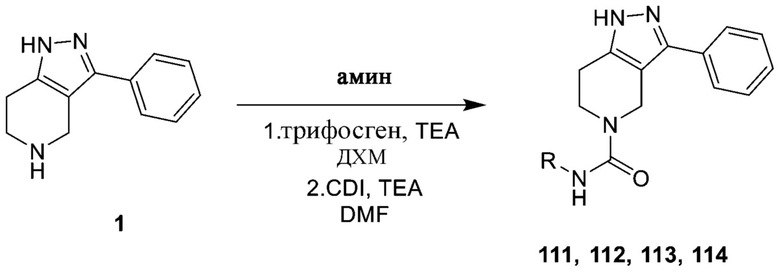
Условие 1.
К смеси трифосгена (0,6 экв.) и амина (1,05 экв.) в ДХМ (5 мл) добавляли Et3N (1,0 экв.) за один раз при 0 °C. Смесь перемешивали при 0~10°C в течение 20 мин и впоследствии добавляли промежуточный продукт 1 (1,0 экв.) и Et3N (2,2 экв.) при 0 °C. Смесь перемешивали при 0-10°C в течение 5 мин. Нужный продукт обнаруживали посредством ЖХМС. Реакционную смесь концентрировали под пониженным давлением. Остаток очищали посредством препаративной ВЭЖХ для получения нужных продуктов.
Условие 2.
К смеси амина (1,05 экв.) в DMF (5,00 мл) добавляли CDI (1,05 экв.) за один раз при 25 °C. Смесь перемешивали в течение 20 мин и впоследствии добавляли промежуточный продукт 1 (1,0 экв.) и Et3N (2,2 экв.). Смесь перемешивали в течение 16~48 часов (25 °C~40 °C). Нужный продукт обнаруживали посредством ЖХМС. Реакционную смесь концентрировали под пониженным давлением. Остаток очищали посредством препаративной ВЭЖХ для получения нужных продуктов.
1H ЯМР (CDCl3, 400 МГц): δ 8,17 (д, 1 H, J=9,6 Гц), 7,56 (д, 2 H, J=7,2 Гц), 7,48 (т, 2 H, J=8,0 Гц)
7,42-7,34 (м, 2 H), 7,12 (с, 1 H), 6,98 (т, 1 H, J=8,0 Гц), 4,82 (с, 2 H), 3,90 (т, 2 H, J=5,6 Гц), 2,98 (т, 2 H, J=5,6 Гц).
Пример 13. Получение соединений 242-244
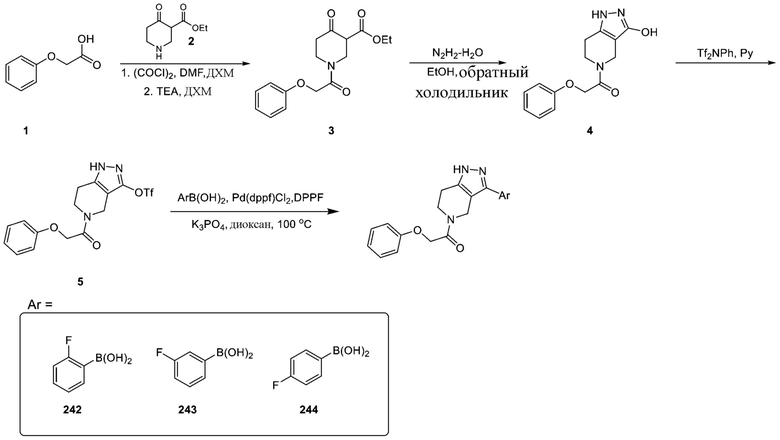
Стадия 1. Получение соединения 3
К смеси 2-феноксиуксусной кислоты (7,33 г, 48,16 ммоль, 1,00 экв.) в ДХМ (100,00 мл) по каплям добавляли оксалилхлорид (9,17 г, 72,24 ммоль, 1,50 экв.) при 0°C в атмосфере N2 с последующим добавлением каталитического количества DMF. Смесь перемешивали при 0°C в течение 2 часов. ТСХ показала завершение реакции. Смесь концентрировали в вакууме. Остаток разбавляли ДХМ (50,00 мл), добавляли к этил-4-оксопиперидин-3-карбоксилату (10,00 г, 48,16 ммоль, 1,00 экв.) и TEA (14,62 г, 144,48 ммоль, 3,00 экв.) в ДХМ (80,00 мл) при 0°C в атмосфере N2. Смесь перемешивали при 20°C в течение 10 часов. ТСХ показала завершение реакции. Смесь вливали в воду (100 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (50 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=5/1) с получением этил-4-оксо-1-(2-феноксиацетил)пиперидин-3-карбоксилата (14,20 г, 46,51 ммоль, выход 96,57%) в виде желтого твердого вещества. ЖХМС: 306 [M+1].
Стадия 2. Получение соединения 4
К смеси этил-4-оксо-1-(2-феноксиацетил)пиперидин-3-карбоксилата (12,00 г, 39,30 ммоль, 1,00 экв.) в EtOH (200,00 мл) добавляли N2H4-H2O (2,36 г, 47,16 ммоль, 1,20 экв.) за один раз в атмосфере N2. Смесь перемешивали при 80°C в течение 3 часов. ТСХ показала завершение реакции. Смесь охлаждали и концентрировали в вакууме с получением 1-(3-гидрокси-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (10,00 мг, 36,59 ммоль, выход 93,11%) в виде белого твердого вещества. ЖХМС: 274 [M+1].
Стадия 3. Получение соединения 5
К смеси 1-(3-гидрокси-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил)-2-феноксиэтанона (3,00 г, 10,98 ммоль, 1,00 экв.) в Py (30,00 мл) добавляли 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамид (5,10 г, 14,27 ммоль, 1,30 экв.) за один раз при 20°C в атмосфере N2. Смесь перемешивали при 20°C в течение 16 часов. ТСХ показала завершение реакции. Смесь концентрировали в вакууме. Остаток вливали в воду (15 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (15 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=1/1) с получением [5-(2-феноксиацетил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (4,10 г, 10,11 ммоль, выход 92,12%) в виде желтого твердого вещества. ЖХМС: 406 [M+1].
Получение соединения 242
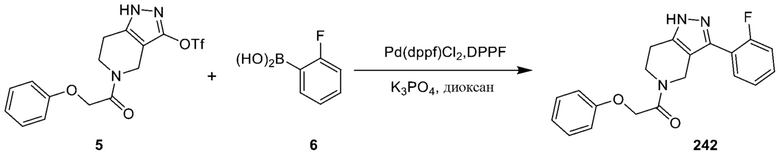
К смеси [5-(2-феноксиацетил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (200,00 мг, 493,40 мкмоль, 1,00 экв.) и (2-фторфенил)бороновой кислоты (138,07 мг, 986,80 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (36,10 мг, 49,34 мкмоль, 0,10 экв.), DPPF (13,68 мг, 24,67 мкмоль, 0,05 экв.), K3PO4 (314,20 мг, 1,48 ммоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 10 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-[3-(2-фторфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил]-2-феноксиэтанона (35,46 мг, 100,11 мкмоль, выход 20,29%, чистота 99,2%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,51-7,61 (м, 1 H), 7,39-7,49 (м, 1 H), 7,18-7,33 (м, 4 H), 6,83-7,02 (м, 3 H), 4,90 (с, 1 H), 4,80-4,82 (м, 1 H), 4,64-4,70 (м, 2 H), 3,87-4,00 (м, 2 H), 2,78-2,97 (м, 2 H). ЖХМС: 352 [M+1].
Получение соединения 243
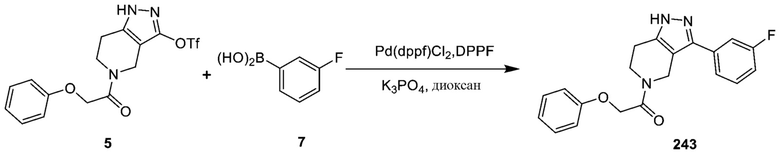
К смеси [5-(2-феноксиацетил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (200,00 мг, 493,40 мкмоль, 1,00 экв.) и (3-фторфенил)бороновой кислоты (138,07 мг, 986,80 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (36,10 мг, 49,34 мкмоль, 0,10 экв.), DPPF (13,68 мг, 24,67 мкмоль, 0,05 экв.) и K3PO4 (314,20 мг, 1,48 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 10 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-[3-(3-фторфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил]-2-феноксиэтанона (45,32 мг, 126,61 мкмоль, выход 25,66%, чистота 98,16%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,33-7,53 (м, 3 H), 7,19-7,32 (м, 2 H), 7,05-7,15 (м, 1 H), 6,99 (д, J=7,91 Гц, 3 H), 4,92 (уш. с, 2 H), 4,80-4,85 (м, 2 H), 3,86-3,99 (м, 2 H), 2,88-2,96 (м, 1 H), 2,77-2,84 (м, 1 H). ЖХМС: 352 [M+1].
Получение соединения 244
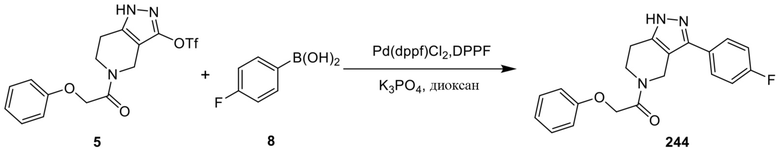
К смеси [5-(2-феноксиацетил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (100,00 мг, 246,70 мкмоль, 1,00 экв.) и (4-фторфенил)бороновой кислоты (69,04 мг, 493,40 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (18,05 мг, 24,67 мкмоль, 0,10 экв.), DPPF (13,68 мг, 24,67 мкмоль, 0,10 экв.) и K3PO4 (157,10 мг, 740,10 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 1 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 1-[3-(4-фторфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-ил]-2-феноксиэтанона (8,00 мг, 20,80 мкмоль, выход 8,43%, чистота 91,35%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,62 (уш. с, 2 H), 7,14-7,34 (м, 4 H), 6,87-7,03 (м, 3 H), 5,00-5,12 (м, 2 H), 4,80 (уш. с, 2 H), 3,90 (уш. с, 2 H), 2,75-2,95 (м, 2 H). ЖХМС: 352 [M+1].
Пример 14. Получение соединений 142, 143, 144, 145, 147, 148, 150, 163, 164 и 165
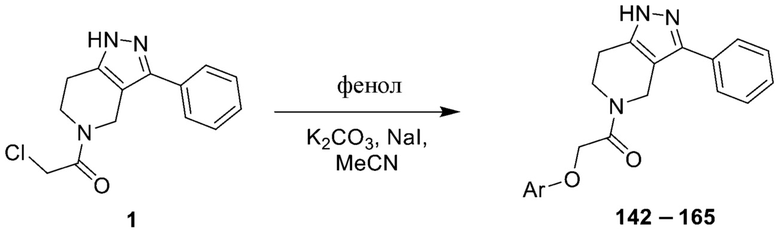
Общая процедура
К смеси фенола (0,35 ммоль, 1,2 экв.) в MeCN (5 мл) добавляли K2CO3 (60 мг, 0,435 ммоль, 1,5 экв.) и NaI (2,17 мг, 14,51 мкмоль, 0,05 экв.) при 25°C с последующим добавлением соединения 1 (80 мг, 0,29 ммоль, 1 экв.). Смесь нагревали до 80°C и перемешивали в течение 18 часов. ТСХ показала завершение реакции. Смесь охлаждали до 25 °C. Смесь добавляли в 10 мл воды. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ для получения нужного продукта.
1H ЯМР (400 МГц, ДМСО-d6) d 12,66-13,16 (м, 1 H), 7,21-7,76 (м, 7 H), 6,87-7,09 (м, 2 H), 4,94-5,05 (м, 2 H), 4,71 (уш. с, 2 H), 3,77 (д, J=15,18 Гц, 2 H), 2,85 (уш. с, 1 H), 2,68 (д, J=1,63 Гц, 1 H)
Пример 15. Получение соединения 169

Общая процедура
К смеси соединения 1 (80 мг, 0,40 ммоль, 1 экв.) и карбоновой кислоты (0,48 ммоль, 1,2 экв.) в ДХМ (5,00 мл) добавляли DIPEA (62,27 мг, 0,48 ммоль, 1,2 экв.) и HATU (305,3 мг, 0,80 ммоль, 2 экв.) при 25 ℃. Смесь перемешивали при 25°C в течение 5 ч. ТСХ показала завершение реакции. Смесь добавляли в 10 мл воды. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ для получения нужного продукта.
Пример 16. Получение соединения 276
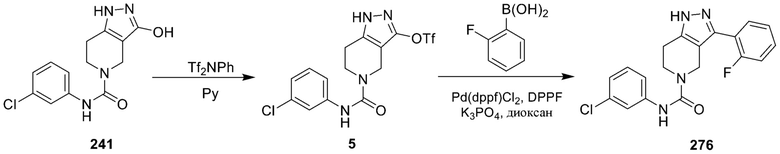
Стадия 1. Получение соединения 5
К смеси N-(3-хлорфенил)-3-гидрокси-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (3,60 г, 12,30 ммоль, 1,00 экв.) в Py (30,00 мл) добавляли 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамид (6,15 г, 17,22 ммоль, 1,40 экв.) за один раз в атмосфере N2. Смесь перемешивали при 10°C в течение 16 часов. ТСХ показала завершение реакции. Смесь концентрировали в вакууме. Остаток разбавляли этилацетатом (50 мл), и вливали в 1 Н HCl (50 мл), и перемешивали в течение 3 мин. Водную фазу экстрагировали этилацетатом (50 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (30 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=4/1, 1/1) с получением [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (3,20 г, 7,19 ммоль, выход 58,49%, чистота 95,5%) в виде белого твердого вещества. ЖХМС: 425 [M+1].
Стадия 2. Получение соединения 276
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (100,00 мг, 235,42 мкмоль, 1,00 экв.) и (2-фторфенил)бороновой кислоты (65,88 мг, 470,84 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (17,23 мг, 23,54 мкмоль, 0,10 экв.), DPPF (13,05 мг, 23,54 мкмоль, 0,10 экв.) и K3PO4 (149,92 мг, 706,26 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 120°C в течение 2 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 3 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(2-фторфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (13,56 мг, 35,69 мкмоль, выход 15,16%, чистота 97,59%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,54-7,62 (м, 1 H), 7,50 (с, 1 H), 7,40-7,48 (м, 1 H), 7,18-7,34 (м, 4 H), 6,96-7,03 (м, 1 H), 4,58-4,66 (м, 2 H), 3,83-3,92 (м, 2 H), 2,86-2,95 (м, 2 H). ЖХМС: 371 [M+1].
Пример 17. Получение соединения 277
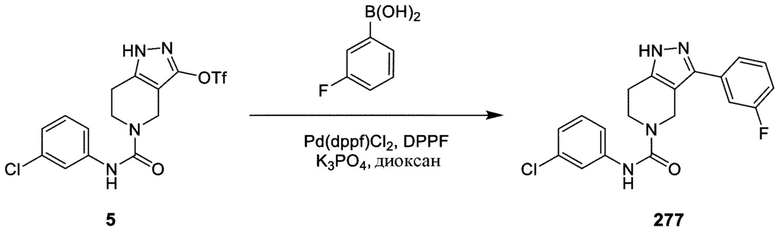
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (100,00 мг, 235,42 мкмоль, 1,00 экв.) и (3-фторфенил)бороновой кислоты (65,88 мг, 470,84 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (17,23 мг, 23,54 мкмоль, 0,10 экв.), DPPF (13,05 мг, 23,54 мкмоль, 0,10 экв.) и K3PO4 (149,92 мг, 706,26 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(3-фторфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (15,36 мг, 40,97 мкмоль, выход 17,40%, чистота 98,9%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,38-7,56 (м, 1 H), 7,26-7,33 (м, 1 H), 7,19-7,26 (м, 1 H), 7,06-7,15 (м, 1 H), 6,97-7,03 (м, 1 H), 4,76-4,81 (м, 2 H), 3,84-3,92 (м, 2 H), 2,84-2,92 (м, 2 H). ЖХМС: 371 [M+1].
Пример 18. Получение соединения 278
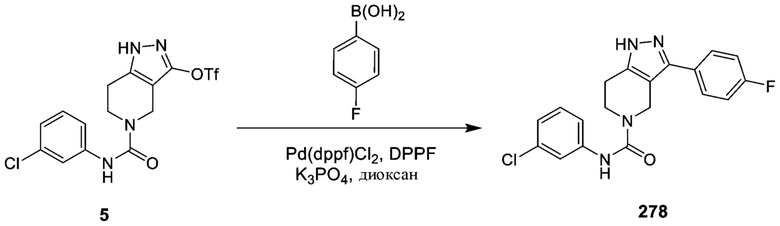
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (100,00 мг, 235,42 мкмоль, 1,00 экв.) и (4-фторфенил)бороновой кислоты (65,88 мг, 470,84 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (17,23 мг, 23,54 мкмоль, 0,10 экв.), DPPF (13,05 мг, 23,54 мкмоль, 0,10 экв.) и K3PO4 (149,92 мг, 706,26 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(4-фторфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (35,42 мг, 92,53 мкмоль, выход 39,30%, чистота 96,87%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ7,62-7,73 (м, 1 H), 7,50-7,56 (м, 1 H), 7,26-7,32 (м, 1 H), 7,16-7,26 (м, 3 H), 6,97-7,03 (м, 1 H), 4,73-4,78 (м, 2 H), 3,83-3,91 (м, 2 H), 2,83-2,91 (м, 2 H). ЖХМС: 371 [M+1].
Пример 19. Получение соединения 279
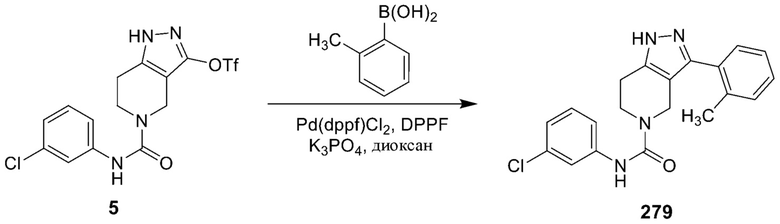
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (120,00 мг, 282,50 мкмоль, 1,00 экв.) и о-толилбороновой кислоты (76,82 мг, 565,00 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (20,67 мг, 28,25 мкмоль, 0,10 экв.), DPPF (15,66 мг, 28,25 мкмоль, 0,10 экв.) и K3PO4 (179,90 мг, 847,50 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь герметизировали и выдерживали в микроволновой печи при 130°C в течение 2 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 3 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(о-толил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (25,25 мг, 68,18 мкмоль, выход 24,14%, чистота 99,06%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,47-7,52 (м, 1 H), 7,16-7,36 (м, 6 H), 6,94-7,03 (м, 1 H), 4,46 (с, 2 H), 3,82-3,94 (м, 2 H), 2,84-2,95 (м, 2 H), 2,29 (с, 3 H). ЖХМС: 367 [M+1].
Пример 20. Получение соединения 280
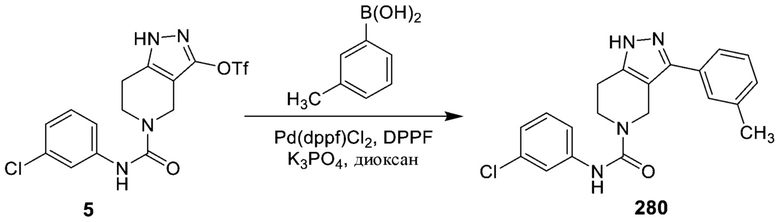
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (120,00 мг, 282,50 мкмоль, 1,00 экв.) и м-толилбороновой кислоты (76,82 мг, 565,00 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (20,67 мг, 28,25 мкмоль, 0,10 экв.), DPPF (15,66 мг, 28,25 мкмоль, 0,10 экв.) и K3PO4 (179,90 мг, 847,50 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 130°C в течение 2 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 1 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(м-толил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (15,32 мг, 41,05 мкмоль, выход 14,53%, чистота 98,3%) в виде белого твердого вещества. ЖХМС: 367 [M+1].
Пример 21. Получение соединения 281
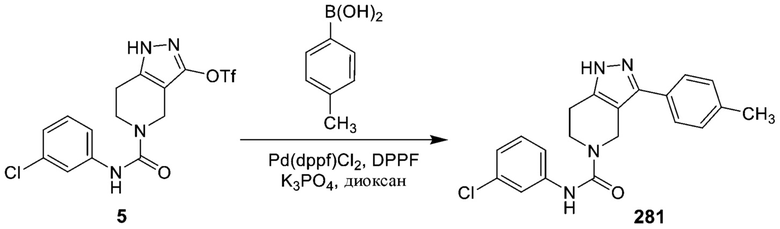
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (120,00 мг, 282,50 мкмоль, 1,00 экв.) и п-толилбороновой кислоты (76,82 мг, 565,00 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (20,67 мг, 28,25 мкмоль, 0,10 экв.), DPPF (15,66 мг, 28,25 мкмоль, 0,10 экв.) и K3PO4 (179,90 мг, 847,50 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 130°C в течение 2 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 1 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(п-толил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (20,35 мг, 54,25 мкмоль, выход 19,20%, чистота 97,8%) в виде белого твердого вещества. ЖХМС: 367 [M+1].
Пример 22. Получение соединения 291
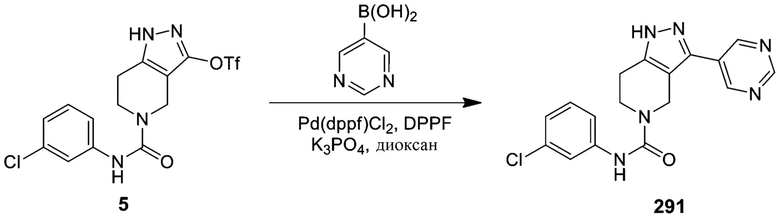
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (100,00 мг, 235,42 мкмоль, 1,00 экв.) и пиримидин-5-илбороновой кислоты (58,34 мг, 470,84 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (17,23 мг, 23,54 мкмоль, 0,10 экв.), DPPF (13,05 мг, 23,54 мкмоль, 0,10 экв.) и K3PO4 (149,92 мг, 706,26 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 145°C в течение 2 ч. ЖХМС показала показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-пиримидин-5-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (25,63 мг, 69,47 мкмоль, выход 29,51%, чистота 96,16%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ9,12 (д, J=5,02 Гц, 1 H), 7,48-7,58 (м, 1 H), 7,29 (с, 1 H), 7,19-7,26 (м, 1 H), 6,98-7,05 (м, 1 H), 4,83 (с, 2 H), 3,85-3,93 (м, 2 H), 2,86-2,95 (м, 2 H). ЖХМС: 355 [M+1].
Пример 23. Получение соединения 325
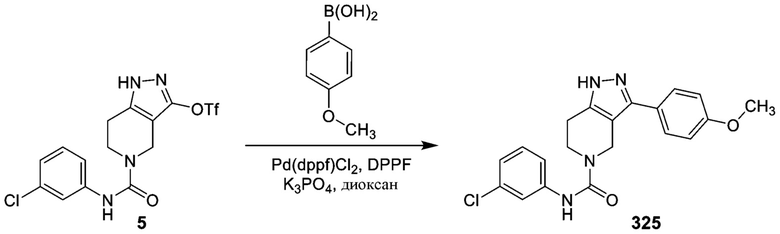
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (120,00 мг, 282,50 мкмоль, 1,00 экв.) и (4-метоксифенил)бороновой кислоты (64,39 мг, 423,75 мкмоль, 1,50 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (20,67 мг, 28,25 мкмоль, 0,10 экв.), DPPF (15,66 мг, 28,25 мкмоль, 0,10 экв.) и K3PO4 (179,90 мг, 847,50 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 145°C в течение 3 ч. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(4-метоксифенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (34,26 мг, 83,85 мкмоль, выход 31,68%, чистота 93,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ7,55 (с, 3 H), 7,19-7,32 (м, 2 H), 7,03 (д, J=8,78 Гц, 3 H), 4,73-4,77 (м, 2 H), 3,85-3,89 (м, 2 H), 3,84 (с, 3 H), 2,83-2,89 (м, 2 H). ЖХМС: 383 [M+1].
Пример 24. Получение соединения 326
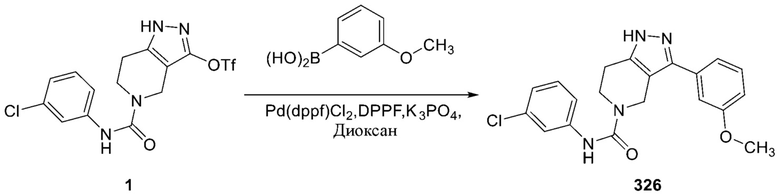
Смесь [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (120,00 мг, 282,50 мкмоль, 1,00 экв.), (3-метоксифенил)бороновой кислоты (85,86 мг, 565,00 мкмоль, 2,00 экв.), Pd(dppf)Cl2 (20,67 мг, 28,25 мкмоль, 0,10 экв.), DPPF (15,66 мг, 28,25 мкмоль, 0,10 экв.) и K3PO4 (179,90 мг, 847,50 мкмоль, 3,00 экв.) переносили в пробирку для микроволновой обработки с диоксаном (5,00 мл). Герметично закрытую пробирку выдерживали при температуре 145°C в течение 3 ч под воздействием микроволн. ЖХМС показала завершение реакции. Смесь фильтровали и концентрировали под пониженным давлением с получением осадка. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(3-метоксифенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (20,62 мг, 53,75 мкмоль, выход 19,03%, чистота 99,8%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 7,52 (с, 1 H), 7,39 (т, J=7,84 Гц, 1 H), 7,28 (с, 1 H), 7,17-7,26 (м, 3 H), 7,00 (д, J=7,91 Гц, 1 H), 6,92-6,97 (м, 1 H), 4,77 (с, 2 H), 3,83-3,92 (м, 5 H), 2,88 (т, J=5,65 Гц, 2 H). ЖХМС: 383 [M+1].
Пример 25. Получение соединения 327
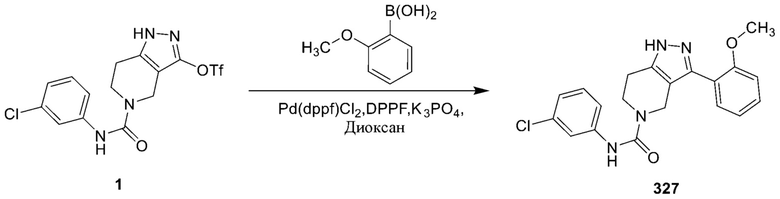
Смесь [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (120,00 мг, 282,50 мкмоль, 1,00 экв.), (2-метоксифенил)бороновой кислоты (85,86 мг, 565,00 мкмоль, 2,00 экв.), Pd(dppf)Cl2 (20,67 мг, 28,25 мкмоль, 0,10 экв.), DPPF (15,66 мг, 28,25 мкмоль, 0,10 экв.) и K3PO4 (179,90 мг, 847,51 мкмоль, 3,00 экв.) переносили в пробирку для микроволновой обработки с диоксаном (5,00 мл). Герметично закрытую пробирку выдерживали при температуре 145°C в течение 3 ч под воздействием микроволн. ЖХМС показала, что исходный материал израсходован и нужный продукт обнаружен. Смесь фильтровали и концентрировали под пониженным давлением с получением осадка. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(2-метоксифенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (4,62 мг, 11,98 мкмоль, выход 4,24%, чистота 99,3%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 7,50 (с, 1 H), 7,37-7,44 (м, 2 H), 7,25-7,30 (м, 1 H), 7,18-7,24 (м, 1 H), 7,12 (д, J=8,03 Гц, 1 H), 7,05 (т, J=7,59 Гц, 1 H), 6,99 (д, J=7,40 Гц, 1 H), 4,60 (с, 2 H), 3,82-3,96 (м, 5 H), 2,88 (т, J=5,58 Гц, 2 H). ЖХМС: 383 [M+1].
Пример 26. Получение соединения 286
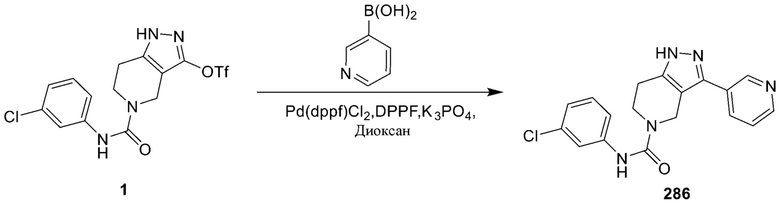
Смесь [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (120,00 мг, 282,50 мкмоль, 1,00 экв.), 3-пиридилбороновой кислоты (69,45 мг, 565,00 мкмоль, 2,00 экв.), Pd(dppf)Cl2 (20,67 мг, 28,25 мкмоль, 0,10 экв.), DPPF (15,66 мг, 28,25 мкмоль, 0,10 экв.) и K3PO4 (179,90 мг, 847,50 мкмоль, 3,00 экв.) переносили в пробирку для микроволновой обработки с диоксаном (5,00 мл). Герметично закрытую пробирку выдерживали при температуре 145°C в течение 3 ч под воздействием микроволн. ЖХМС показала, что соотношение исходного материала/нужного продукта составляет 1/1. Смесь фильтровали и концентрировали под пониженным давлением с получением осадка. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(3-пиридил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (13,00 мг, 34,02 мкмоль, выход 12,04%, чистота 92,6%) в виде белого твердого вещества.
1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 8,87 (д, J=1,51 Гц, 1 H), 8,50-8,55 (м, 1 H), 8,12 (тд, J=1,77, 8,00 Гц, 1 H), 7,51-7,57 (м, 2 H), 7,27-7,32 (м, 1 H), 7,20-7,26 (м, 1 H), 6,98-7,03 (м, 1 H), 4,81 (с, 2 H), 3,89 (т, J=5,77 Гц, 2 H), 2,90 (т, J=5,71 Гц, 2 H). ЖХМС: 354 [M+1].
Пример 27. Получение соединения 472
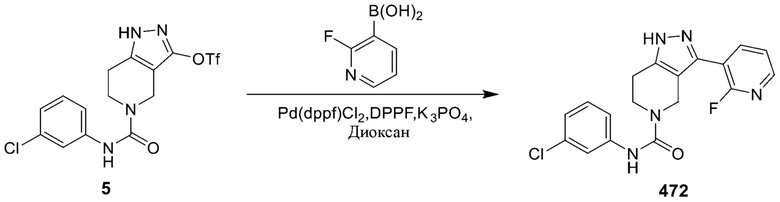
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (100,00 мг, 235,42 мкмоль, 1,00 экв.) и (2-фтор-3-пиридил)бороновой кислоты (66,35 мг, 470,84 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (17,23 мг, 23,54 мкмоль, 0,10 экв.), DPPF (13,05 мг, 23,54 мкмоль, 0,10 экв.), KBr (2,80 мг, 23,54 мкмоль, 0,10 экв.) и K3PO4 (149,92 мг, 706,26 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 145°C в течение 2 часов. ЖХМС показала, что нужное соединение обнаружено. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(2-фтор-3-пиридил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (17,56 мг, 46,71 мкмоль, выход 19,84%, чистота 98,9%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 8,22-8,28 (м, 1 H), 8,10-8,19 (м, 1 H), 7,48-7,52 (м, 1 H), 7,40-7,46 (м, 1 H), 7,18-7,30 (м, 2 H), 6,96-7,02 (м, 1 H), 4,62-4,69 (м, 2 H), 3,85-3,93 (м, 2 H), 2,86-2,94 (м, 2 H). ЖХМС: 372 [M+1].
Пример 28. Получение соединения 473
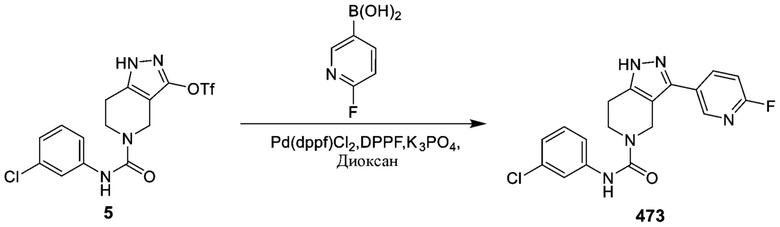
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (100,00 мг, 235,42 мкмоль, 1,00 экв.) и (6-фтор-3-пиридил)бороновой кислоты (66,35 мг, 470,84 мкмоль, 2,00 экв.) в диоксане (5,00 мл) добавляли Pd(dppf)Cl2 (17,23 мг, 23,54 мкмоль, 0,10 экв.), DPPF (13,05 мг, 23,54 мкмоль, 0,10 экв.) и K3PO4 (149,92 мг, 706,26 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 145°C в течение 2 ч. ЖХМС показала показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(6-фтор-3-пиридил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (21,36 мг, 57,05 мкмоль, выход 24,23%, чистота 99,3%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) 8,46-8,54 (м, 1 H), 8,18-8,28 (м, 1 H), 7,49-7,57 (м, 1 H), 7,16-7,32 (м, 3 H), 6,98-7,04 (м, 1 H), 4,78 (с, 2 H), 3,88 (с, 2 H), 2,84-2,95 (м, 2 H). ЖХМС: 372 [M+1].
Пример 29. Получение соединения 495
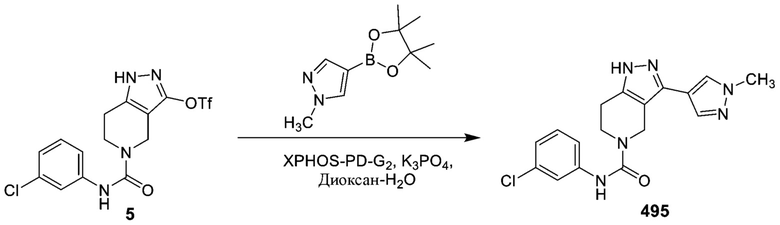
К смеси [5-[(3-хлорфенил)карбамоил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-3-ил]трифторметансульфоната (30,00 мг, 70,62 мкмоль, 1,00 экв.) и 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразола (22,04 мг, 105,93 мкмоль, 1,50 экв.) в диоксане (2,00 мл) и H2O (200,00 мкл) добавляли K3PO4 (29,98 мг, 141,24 мкмоль, 2,00 экв.), XPHOS-PD-G2 (5,56 мг, 7,06 мкмоль, 0,10 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 10 часов. ЖХМС показала, что нужный продукт обнаружен. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(1-метилпиразол-4-ил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (5,20 мг, 14,25 мкмоль, выход 20,18%, чистота 97,8%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) 7,90-7,95 (м, 1 H), 7,79-7,83 (м, 1 H), 7,52-7,56 (м, 1 H), 7,29-7,34 (м, 1 H), 7,20-7,28 (м, 1 H), 6,99-7,05 (м, 1 H), 4,63-4,69 (м, 2 H), 3,93-4,00 (м, 3 H), 3,81-3,89 (м, 2 H), 2,79-2,89 (м, 2 H). ЖХМС: 357 [M+1].
Пример 30. Получение соединения 562
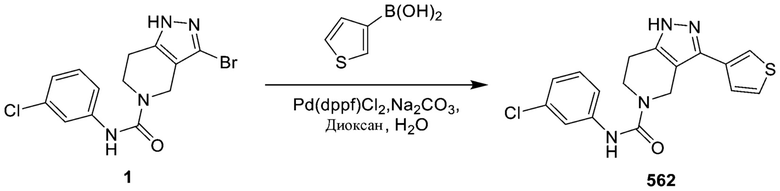
К смеси 3-бром-N-(3-хлорфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (80,00 мг, 224,96 мкмоль, 1,00 экв.) и 3-тиенилбороновой кислоты (57,57 мг, 449,92 мкмоль, 2,00 экв.) в диоксане (2,00 мл) и H2O (200,00 мкл) добавляли Pd(dppf)Cl2 (16,46 мг, 22,50 мкмоль, 0,10 экв.), Na2CO3 (47,69 мг, 449,92 мкмоль, 2,00 экв.) за один раз в атмосфере N2. Реакционный сосуд герметизировали и выдерживали в микроволновой печи при 110°C в течение 2 часов. ЖХМС показала, что исходный материал был полностью израсходован. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(3-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (16,00 мг, 44,05 мкмоль, выход 19,58%, чистота 98,8%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,61-7,66 (м, 1 H), 7,51-7,57 (м, 2 H), 7,46-7,51 (м, 1 H), 7,28-7,35 (м, 1 H), 7,20-7,27 (м, 1 H), 6,97-7,05 (м, 1 H), 4,72-4,78 (м, 2 H), 3,83-3,92 (м, 2 H), 2,82-2,90 (м, 2 H). ЖХМС: 359 [M+1].
Пример 31. Получение соединений 496 и 497
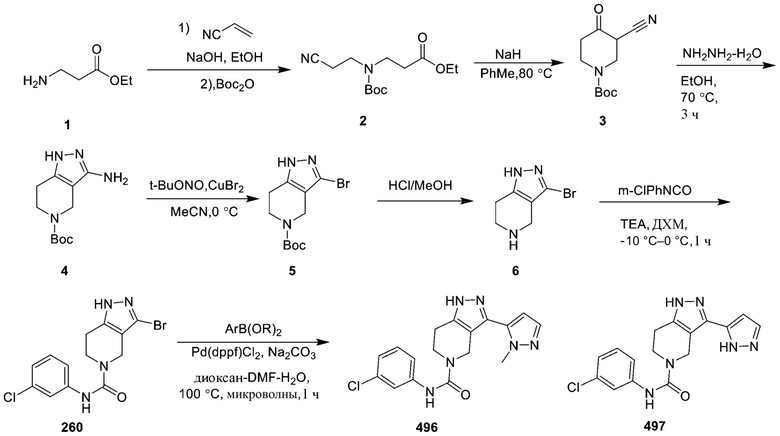
Стадия 1. Получение соединения 2
К смеси этил-3-аминопропаноата (50,00 г, 320,55 ммоль, 1,00 экв. соль HCl) в MeOH (150,00 мл) добавляли NaOH (13 г, 320,55 ммоль, 1,00 экв.). Смесь нагревали до 70 °C. В вышеупомянутую смесь по каплям добавляли акрилонитрил (21,8 г, 410,1 ммоль, 1,26 экв.). И смесь перемешивали при 70°C в течение 4 ч. Ее охлаждали до 25 °C, добавляли Boc2O (6,39 г, 29,30 ммоль, 0,90 экв.). Впоследствии смесь перемешивали при 25°C в течение 16 ч. ТСХ показала завершение реакции. Смесь фильтровали, фильтрат промывали водой (500 мл), экстрагировали EtOAc (500 мл*3), фильтрат сушили Na2SO4 и концентрировали с получением соединения 2A (6,70 г, 24,79 ммоль, выход 76,15%), которое использовали непосредственно. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=3,71 (с, 3 H), 3,50-3,63 (м, 4 H), 2,56-2,70 (м, 4 H), 1,49 (с, 9 H).
Стадия 2. Получение соединения 3
К смеси этил-3-[трет-бутоксикарбонил(2-цианоэтил)амино]пропаноата (70,00 г, 258,95 ммоль, 1,00 экв.) в PhMe (150,00 мл) добавляли NaH (10,46 г, 261,54 ммоль, 1,01 экв.) за три раза. Смесь перемешивали при 110°C в течение 4 ч. ТСХ показала завершение реакции. Реакцию гасили водным насыщенным раствором NH4Cl (200 мл), водную фазу подкисляли HCl (2 Н) до рН=6, впоследствии смесь экстрагировали EtOAc (150 мл*3), органический слой промывали солевым раствором (100 мл), сушили Na2SO4 и концентрировали с получением соединения 3, которое использовали непосредственно. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=4,40 (уш. с, 1 H), 4,16-4,26 (м, 1 H), 3,58 (уш. с, 2 H), 3,41 (д, J=7,28 Гц, 1 H), 2,67 (д, J=14,31 Гц, 1 H), 2,53 (дд, J=5,77, 9,54 Гц, 1 H), 1,52 (с, 9 H).
Стадия 3. Получение соединения 4
К смеси трет-бутил-3-циано-4-оксопиперидин-1-карбоксилата (20,00 г, 89,18 ммоль, 1,00 экв.) в EtOH (200,00 мл) добавляли NH2NH2·H2O (8,93 г, 178,36 ммоль, 2,00 экв.) за один раз. Смесь перемешивали при 80°C в течение 2 ч. ТСХ показала, что реакция прошла успешно. Смесь концентрировали с получением соединения 4 (19,70 г, 82,67 ммоль, выход 92,70%).
Стадия 4. Получение соединения 5
К суспензии соединения 4 (40,00 г, 0,47 ммоль, 1,00 экв.) и CuBr2 (44 г, 0,58 моль, 1,20 экв.) в 500 мл ацетонитрила по каплям добавляли t-BuONO (20,2 г, 0,58 моль, 1,20 экв.) при 0 °C. Содержимое выдерживали при 50°C в течение 4 ч с перемешиванием. ТСХ показала завершение реакции. Впоследствии реакцию гасили HCl (1 М, 300 мл), экстрагировали EtOAc (200 мл*3), органический слой промывали солевым раствором (300 мл), сушили Na2SO4 и концентрировали с получением соединения 5 (11,00 г, 36,40 ммоль, выход 21,69%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=4,33 (уш. с, 2 H), 3,72 (уш. с, 2 H), 2,83 (т, J=5,27 Гц, 2 H), 1,50 (с, 9 H).
Стадия 5. Получение соединения 6
К смеси соединения 5 (11,00 г, 36,40 ммоль, 1,00 экв.) в ДХМ (10,00 мл) добавляли HCl/диоксан (4 М, 20,02 мл) за один раз при 0 °C. Смесь перемешивали при 0°C в течение 1 ч. Смесь концентрировали с получением соединения 5 (HCl).
Получение соединения 260
К смеси 3-бром-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (10,50 г, 38,19 ммоль, 1,00 экв., 2HCl) в MeOH (350,00 мл) добавляли K2CO3 (13,20 г, 95,48 ммоль, 2,50 экв.). Впоследствии смесь фильтровали, фильтрат использовали непосредственно. И в вышеупомянутый фильтрат медленно добавляли 1-хлор-3-изоцианатбензол (5,86 г, 38,19 ммоль, 1,00 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 1 ч. ЖХМС показала, что реакция прошла успешно. Смесь концентрировали. Остаток промывали смешанным раствором PE/EA (10/1, 20 мл). Смесь фильтровали и собирали фильтрат с получением соединения 260 (11,00 г, 30,93 ммоль, выход 80,99%). 1H ЯМР (400 МГц, ДМСО-d6) δ=12,95 (уш. с, 1 H), 8,88 (с, 1 H), 7,64 (с, 1 H), 7,41 (д, J=8,03 Гц, 1 H), 7,26 (т, J=8,16 Гц, 1 H), 6,99 (д, J=7,78 Гц, 1 H), 4,34 (с, 2 H), 3,72 (уш. с, 2 H), 2,72 (уш. с, 2 H). ЖХМС: 355 [M+1].
Получение соединения 496
К смеси соединения 260 (100,00 мг, 281,20 мкмоль, 1,00 экв.), 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразола (70,21 мг, 337,44 мкмоль, 1,20 экв.) в диоксане (1,50 мл) добавляли Pd(dppf)Cl2 (20,58 мг, 28,12 мкмоль, 0,10 экв.) и Na2CO3 (65,57 мг, 618,64 мкмоль, 2,20 экв.) за один раз. Смесь перемешивали при 100°C в течение 1 ч посредством микроволн. ЖХМС показала, что основное соединение представляло собой нужный продукт. Смесь фильтровали и фильтрат концентрировали. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением соединения 496 (22,00 мг, 59,19 мкмоль, выход 21,05%). 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ=7,47-7,56 (м, 2 H), 7,26-7,33 (м, 1 H), 7,18-7,26 (м, 1 H), 7,00 (д, J=7,53 Гц, 1 H), 6,51 (д, J=1,76 Гц, 1 H), 4,60 (с, 2 H), 4,04 (уш. с, 3 H), 3,87 (т, J=5,65 Гц, 2 H), 2,89 (т, J=5,65 Гц, 2 H). ЖХМС: 357 [M+1].
Получение соединения 497
К смеси соединения 260 (80,00 мг, 224,96 мкмоль, 1,00 экв.), 1H-пиразол-5-илбороновой кислоты (30,20 мг, 269,95 мкмоль, 1,20 экв.) в диоксане (1,50 мл) добавляли Pd(dppf)Cl2 (16,46 мг, 22,50 мкмоль, 0,10 экв.) и Na2CO3 (65,57 мг, 618,64 мкмоль, 2,20 экв.) за один раз. Смесь перемешивали при 100°C в течение 1 ч посредством микроволн. ЖХМС показала, что отношение DP: SM=1: 1. Смесь фильтровали и фильтрат концентрировали. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением соединения 497 (3,50 мг, 10,01 мкмоль, выход 4,45%, чистота 98,00%). 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ=7,71 (с, 1 H), 7,55 (с, 1 H), 7,29-7,35 (м, 1 H), 7,23 (т, J=8,03 Гц, 1 H), 7,00 (д, J=7,78 Гц, 1 H), 6,66 (д, J=1,76 Гц, 1 H), 4,77 (с, 2 H), 3,87 (т, J=5,65 Гц, 2 H), 2,86 (т, J=5,52 Гц, 2 H). ЖХМС: 343 [M+1].
Пример 32. Получение соединения 555
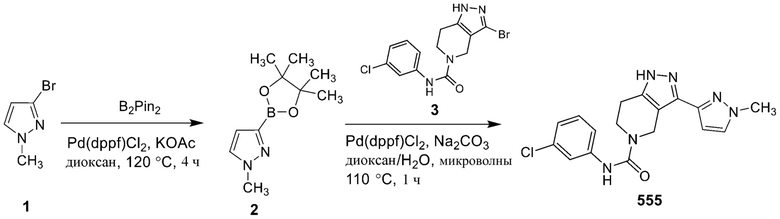
Стадия 1. Получение соединения 2
Смесь 3-бром-1-метилпиразола (100,00 мг, 621,12 мкмоль, 1,00 экв.), 4,4,4ʹ,4ʹ,5,5,5ʹ,5ʹ-октаметил-2,2ʹ-би(1,3,2-диоксаборолан) (236,59 мг, 931,68 мкмоль, 1,50 экв.), AcOK (152,39 мг, 1,55 ммоль, 2,50 экв.) и Pd(dppf)Cl2 (22,72 мг, 31,06 мкмоль, 0,05 экв.) в диоксане (3,00 мл) выдерживали при 120°C в течение 4 ч. Смесь фильтровали, а фильтрат использовали непосредственно на следующей стадии.
Стадия 2. Получение соединения 555
Смесь соединения 3 (60,00 мг, 168,72 мкмоль, 1,00 экв.), соединения 2 (70,21 мг, 337,44 мкмоль, 2,00 экв.), Na2CO3 (35,77 мг, 337,44 мкмоль, 2,00 экв.) и Pd(dppf)Cl2 (6,17 мг, 8,44 мкмоль, 0,05 экв.) в диоксане (2,00 мл)/H2O (400,00 мкл) выдерживали при 110°C в микроволновой печи в течение 1 ч. Реакционную смесь разбавляли солевым раствором (60 мл) и экстрагировали EA (80 мл). Органический слой концентрировали под пониженным давлением с получением желтого остатка. Остаток очищали посредством препаративной ТСХ с получением неочищенного продукта (20 мг). Неочищенный продукт очищали посредством препаративной ВЭЖХ (FA) с получением нужного продукта, соединения 555 (6,00 мг, 16,65 мкмоль, выход 9,87%, чистота 99,00%), в виде белого твердого вещества. ЖХМС: 357/359 [M+1]. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ=7,65 (с, 1 H), 7,55 (т, J=1,94 Гц, 1 H), 7,30-7,38 (м, 1 H), 7,18-7,29 (м, 1 H), 6,92-7,08 (м, 1 H), 6,62 (д, J=2,26 Гц, 1 H), 4,78 (с, 2 H), 3,97 (с, 3 H), 3,87 (т, J=5,71 Гц, 2 H), 2,81-2,92 (м, 2 H).
Пример 33. Получение соединения 287
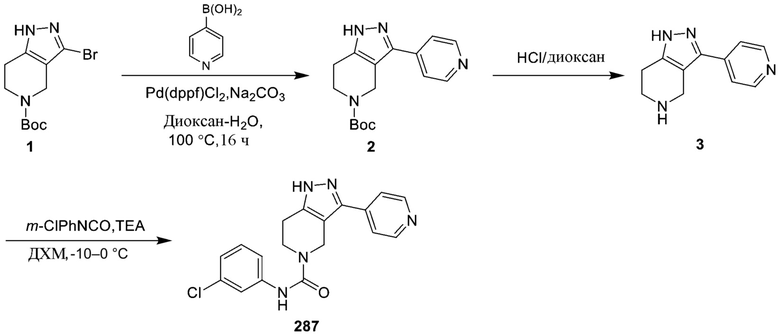
Стадия 1. Получение соединения 2
К смеси соединения 1 (100,00 мг, 330,94 мкмоль, 1,00 экв.) в диоксане (2,00 мл) добавляли Pd(dppf)Cl2 (24,21 мг, 33,09 мкмоль, 0,10 экв.) и Na2CO3 (70,15 мг, 661,88 мкмоль, 2,00 экв.) за один раз. Смесь перемешивали при 100°C в течение 1 ч посредством микроволн. ЖХМС показала, что основное соединение представляло собой нужный продукт. Смесь фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле (PE: EA=1: 1) с получением соединения 2 (70,00 мг, 233,06 мкмоль, выход 70,42%). ЖХМС: 301 [M+1].
Стадия 2. Получение соединения 3
К смеси соединения 2 (70,00 мг, 233,06 мкмоль, 1,00 экв.) в ДХМ (5,00 мл) добавляли HCl/MeOH (4 М, 6,21 мл, 106,65 экв.) за один раз. Смесь перемешивали при 0°C в течение 0,5 часа. ТСХ показала завершение реакции. Смесь концентрировали с получением соединения 3 (65,00 мг, неочищенное).
Стадия 3. Получение соединения 287
К смеси соединения 3 (50,00 мг, неочищенное) в ДХМ (5,00 мл) добавляли TEA (64,83 мг, 640,64 мкмоль, 3,50 экв.) и 1-хлор-3-изоцианатбензол (28,11 мг, 183,04 мкмоль, 1,00 экв.) за один раз. Смесь перемешивали при 0°C в течение 0,5 часа. ЖХМС показала, что продукт представляет собой основное соединение. Смесь концентрировали. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением соединения 287 (32,00 мг, 88,64 мкмоль, выход 48,43%, чистота 98%). 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ=8,59 (д, J=5,52 Гц, 2 H), 7,72 (д, J=5,27 Гц, 2 H), 7,53 (с, 1 H), 7,27-7,34 (м, 1 H), 7,19-7,26 (м, 1 H), 7,01 (д, J=8,03 Гц, 1 H), 4,84 (с, 2 H), 3,88 (т, J=5,65 Гц, 2 H), 2,89 (т, J=5,52 Гц, 2 H). ЖХМС: 354 [M+1].
Пример 34. Получение соединения 436
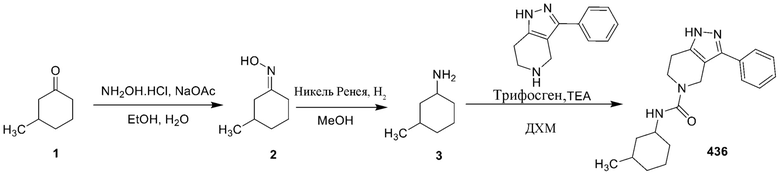
Стадия 1. Получение соединения 2
К раствору 3-метилциклогексанона (2,00 г, 17,83 ммоль, 1,00 экв.) в EtOH (15,00 мл) и H2O (15,00 мл) последовательно добавляли NaOAc (4,39 г, 53,49 ммоль, 3,00 экв.) и NH2OH·HCl (6,20 г, 89,15 ммоль, 5,00 экв.), впоследствии смесь перемешивали при температуре 20°C в течение 16 ч. ТСХ показала, что реакция завершена и появилась одна новая зона. Смесь концентрировали для удаления EtOH, а впоследствии добавляли в воду (30 мл), экстрагировали EA (30 мл*3), органические слои сушили Na2SO4, фильтровали и концентрировали с получением соединения 2 (2,00 г, 15,73 ммоль, выход 88,20%).
Стадия 2. Получение соединения 3
К раствору 3-метилциклогексаноноксима (1,00 г, 7,86 ммоль, 1,00 экв.) в MeOH (10,00 мл) добавляли никель Ренея (67,34 мг, 786,00 мкмоль, 0,10 экв.) в атмосфере аргона, а впоследствии перемешивали при 30°C в течение 16 ч в атмосфере H2 (45 фунтов/кв. дюйм). ТСХ показала, что реакция завершена и появилась одна новая зона. Смесь фильтровали посредством диатомита, впоследствии фильтрат добавляли в HCl/MeOH (4 M, 30 мл), смесь перемешивали при 10°C в течение 5 мин и концентрировали с получением соединения 3 (800,00 мг, 5,35 ммоль, выход 68,01%, HCl) в виде белого твердого вещества.
Стадия 3. Получение соединения 436
К смеси 3-метилциклогексанамина (63,49 мг, 424,25 мкмоль, 1,00 экв., HCl) и TEA (429,30 мг, 4,24 ммоль, 10,00 экв.) в ДХМ (20,00 мл) добавляли ТРИФОСГЕН (50,36 мг, 169,70 мкмоль, 0,40 экв.) при 0°C и перешивали в течение 10 мин, в реакционную смесь добавляли 3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин (100,00 мг, 424,25 мкмоль, 1,00 экв., HCl) и перемешивали при 0°C в течение 2 ч. ЖХМС показала, что реакция завершена и обнаружен один основной пик с нужным масс-спектром. Реакционную смесь гасили посредством добавления воды (20 мл) при 10 °C, экстрагировали ДХМ (20 мл*3). Объединенные органические слои сушили Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали посредством препаративной ТСХ с получением соединения 436 (40,00 мг, 118,19 мкмоль, выход 27,86%) в виде светло-желтого твердого вещества.
ЖХМС: 339 [M+H]. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ м. д. 7,30-7,77 (м, 5 H) 4,65 (д, J=4,27 Гц, 2 H) 3,88-3,97 (м, 1 H) 3,72-3,82 (м, 2 H) 3,55-3,65 (м, 1 H) 2,75-2,87 (м, 2 H) 1,62-1,96 (м, 4 H) 1,13-1,61 (м, 5 H) 0,92-1,00 (м, 3 H).
Пример 35. Получение соединения 559
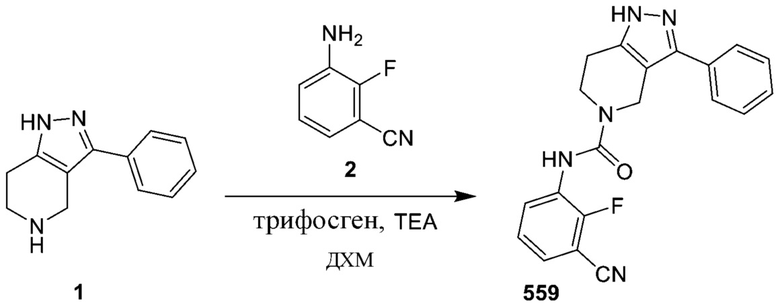
К раствору 3-амино-2-фторбензонитрила (57,75 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (3 мл) добавляли TEA (100 мг) и ТРИФОСГЕН (50,36 мг, 169,70 мкмоль, 0,40 экв.) при 0°C в атмосфере N2, смесь перемешивали при этой температуре в течение 10 мин, впоследствии добавляли в раствор 3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин (100,00 мг, 424,25 мкмоль, 1,00 экв., HCl), в который был добавлен TEA (71 мг) в ДХМ (3 мл), перемешивали при 10°C в течение 2 ч. ЖХМС показала, что реакция завершена и обнаружен один основной пик с нужным масс-спектром. Смесь добавляли в HCl (0,5 М, 10 мл), экстрагировали ДХМ (20 мл*3). Органические слои концентрировали. Остаток очищали посредством препаративной ВЭЖХ (условие FA) с получением соединения 559 (45,00 мг, 119,55 мкмоль, выход 28,18%, чистота 96%) в виде белого твердого вещества. ЖХМС: 262 [M+H]. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ м. д. 7,72-7,80 (м, 1 H) 7,57-7,70 (м, 2 H) 7,43-7,53 (м, 3 H) 7,33-7,40 (м, 1 H) 7,29 (с, 1 H) 4,81 (с, 2 H) 3,91 (т, J=5,77 Гц, 2 H) 2,90 (т, J=5,65 Гц, 2 H).
Пример 36. Получение соединения 560
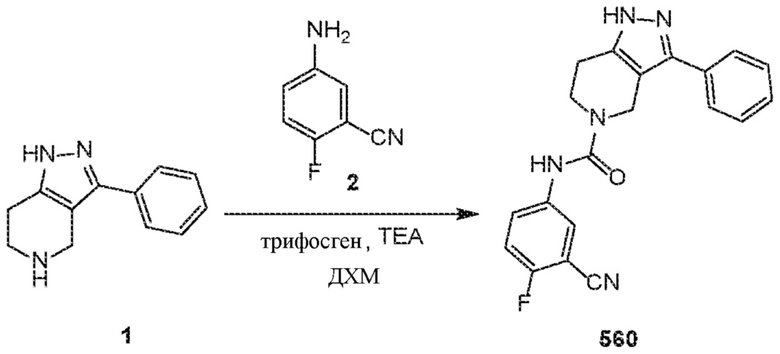
К раствору 5-амино-2-фторбензонитрила (57,75 мг, 424,25 мкмоль, 1,00 экв.) в ДХМ (3,00 мл) добавляли ТРИФОСГЕН (125,90 мг, 424,25 мкмоль, 1,00 экв.) и TEA (42,93 мг, 424,25 мкмоль, 1,00 экв.) при 0 °C, смесь перемешивали в течение 10 мин, впоследствии в нее добавляли 3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин (100,00 мг, 424,25 мкмоль, 1,00 экв., HCl), реакционную смесь перемешивали при 10°C в течение 2 ч. ЖХМС показала, что реакция завершена и обнаружен один основной пик с нужным масс-спектром. Смесь концентрировали и разбавляли 10 мл HCl (0,5 М), впоследствии экстрагировали EA (20 мл*3), органические слои сушили Na2SO4, впоследствии фильтровали и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (условие FA) с получением соединения 560 (50,00 мг, 135,60 мкмоль, выход 31,96%, чистота 98%) в виде белого твердого вещества. ЖХМС: 362 [M+H]. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ м. д. 7,80 (дд, J=5,52, 2,76 Гц, 1 H) 7,57-7,72 (м, 3 H) 7,47 (т, J=7,40 Гц, 2 H) 7,38 (д, J=7,28 Гц, 1 H) 7,26 (т, J=9,03 Гц, 1 H) 4,79 (с, 2 H) 3,88 (т, J=5,77 Гц, 2 H) 2,88 (т, J=5,65 Гц, 2 H).
Пример 37. Получение соединения 556
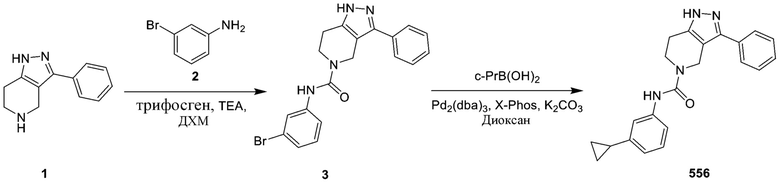
Стадия 1. Получение соединения 3
К раствору 3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (500,00 мг, 2,12 ммоль, 1,00 экв., HCl) в ДХМ (5,00 мл) добавляли TEA (1,07 г, 10,61 ммоль, 5,00 экв.), смесь перемешивали в течение 10 мин, и в реакционную смесь добавляли 3-броманилин (2,12 ммоль, 1,00 экв., HCl), и перемешивали при 10°C в течение 16 ч. ТСХ показала, что реакция завершена и появилась одна новая основная зона. Смесь добавляли в HCl (0,5 М, 20 мл) и экстрагировали ДХМ (30 мл*3). Органические слои сушили Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии с получением соединения 3 (450,00 мг, 1,04 ммоль, выход 49,16%, чистота 92%) в виде белого твердого вещества.
Стадия 2. Получение соединения 556
Смесь N-(3-бромфенил)-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (130,00 мг, 327,23 мкмоль, 1,00 экв.), циклопропилбороновой кислоты (140,55 мг, 1,64 ммоль, 5,00 экв.), K2CO3 (135,68 мг, 981,69 мкмоль, 3,00 экв.), Pd2(dba)3 (26,97 мг, 29,45 мкмоль, 0,09 экв.) и дициклогексил-[2-(2,4,6-триизопропилфенил)фенил]фосфана (12,48 мг, 26,18 мкмоль, 0,08 экв.) в диоксане (2,00 мл) перемешивали при 110°C в течение 16 часов в атмосфере N2. ЖХМС показала, что реакция завершена и обнаружен один основной пик с нужным масс-спектром. Смесь добавляли в воду (10 мл) и экстрагировали ДХМ (20 мл*3). Органические слои сушили Na2SO4, фильтровали и концентрировали. Остаток очищали посредством препаративной ТСХ с получением нужного соединения в виде светло-желтого твердого вещества, которое дополнительно очищали посредством препаративной ВЭЖХ (условие FA) с получением соединения 556 (20,00 мг, 55,24 мкмоль, выход 16,88%, чистота 99%) в виде белого твердого вещества. ЖХМС: 359 [M+H]. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) d м. д. 7,60-7,67 (м, 1 H) 7,43-7,51 (м, 1 H) 7,33-7,40 (м, 1 H) 7,05-7,16 (м, 3 H) 6,74-6,80 (м, 1 H) 4,77 (с, 2 H) 3,84-3,91 (м, 2 H) 2,84-2,91 (м, 2 H) 1,80-1,91 (м, 1 H) 0,88-0,96 (м, 2 H) 0,63-0,70 (м, 2 H).
Пример 38. Получение соединений 317 и 318
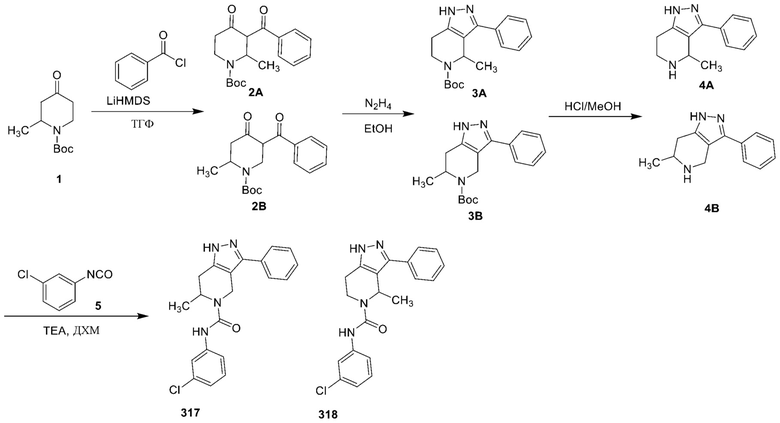
Стадия 1. Получение соединений 2A и 2B
К раствору LiHMDS (1 M, 15,47 мл, 2,20 экв.) в ТГФ (10,00 мл) по каплям добавляли трет-бутил-2-метил-4-оксопиперидин-1-карбоксилат (1,50 г, 7,03 ммоль, 1,00 экв.) при -70°C и перемешивали в течение 0,5 ч, впоследствии по каплям добавляли PhCOCl (988,68 мг, 7,03 ммоль, 1,00 экв.) в ТГФ (2,00 мл) при -70 °C. Реакционную смесь перемешивали при температуре от -70°C до 16°C в течение 3 ч. Реакцию гасили насыщенным NH4Cl (20 мл) и впоследствии экстрагировали EA (20 мл*2). Объединенную органическую фазу промывали солевым раствором (15 мл), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством флэш-хроматографии на силикагеле (градиент соотношения этилацетата/петролейного эфира ~ 5%) с получением смеси 2A и 2B (1,75 г) в виде желтого масла. ЖХМС: 218 [M+1].
Стадия 2. Получение соединений 3A и 3B
К раствору 2A и 2B (1,75 г смеси 2A и 2B, 5,51 ммоль, 1,00 экв.) в EtOH (15,00 мл) добавляли N2H4·H2O (324,51 мг, 5,51 ммоль, 1,00 экв.), раствор перемешивали при 90°C в течение 3 ч. Раствор концентрировали с получением смеси 3A и 3B (1,75 г, 4,47 ммоль, выход 81,08%, чистота 80%) в виде светло-желтого твердого вещества. ЖХМС: 314 [M+1].
Стадия 3. Получение соединений 4A и 4B
К смеси 3A и 3B (330,00 мг, 842,40 мкмоль, 1,00 экв.) в диоксане (3,00 мл) добавляли HCl/диоксан (4 М, 10,00 мл, 47,48 экв.). Смесь перемешивали при температуре 15°C в течение 1 ч. Было образовано твердое вещество, и растворитель выпаривали с получением смеси 4A и 4B (210,00 мг, неочищ., соль HCl) в виде светло-желтого твердого вещества.
Стадия 4. Получение соединений 317 и 318
Смесь 6-метил-3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина и 4-метил-3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (210,00 мг, 840,87 мкмоль, 1,00 экв., HCl) в ДХМ (10,00 мл) добавляли TEA (170,18 мг, 1,68 ммоль, 2,00 экв.) и 1-хлор-3-изоцианатбензол (129,13 мг, 840,87 мкмоль, 1,00 экв.). Смесь перемешивали при температуре 15°C в течение 1 ч. ЖХМС показала, что материал полностью израсходован и обнаружен основной пик с нужным продуктом. Смесь промывали водой (10 мл), а водный слой экстрагировали ДХМ (15 мл*3). Объединенный органический слой сушили безводным Na2SO4 и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением смеси N-(3-хлорфенил)-4-метил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида и N-(3-хлорфенил)-6-метил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (160,00 мг, 1: 1 по данным H ЯМР, 436,16 мкмоль, выход 51,87%, 4 изомера обнаружено посредством SFC) в виде белого твердого вещества. Смесь разделяли посредством SFC (прибор: SFC 80, колонка: OD-10 мкм, подвижная фаза: А для CO2 и В для MeOH (0,1% NH3H2O), градиент: В 35%, скорость потока: 65 мл/мин, противодавление: 100 бар, температура колонки: 35 ℃, длина волны: 220 нм) с получением соединения 318, энантиомер 1 (пик 1, Rt=3,003 мин, 22,76 мг, чистота: 98,6%), соединения 317, энантиомер 1 (пик 2, Rt=3,219 мин, 26,27 мг, чистота: 99,9%), соединения 318, энантиомер 2 (пик 3, Rt=3,509 мин, 24,3 мг, чистота: 99,1%), соединения 317, энантиомер 2 (пик 4, Rt=3,930 мин, 27,21 мг, чистота: 99,0%), все в виде белого твердого вещества.
Соединение 318, энантиомер 1: 1H ЯМР (400 МГц, МЕТАНОЛ-d4) d м. д. 7,68 (д, J=7,53 Гц, 2 H), 7,56 (д, J=1,76 Гц, 1 H), 7,50 (т, J=7,65 Гц, 2 H), 7,42 (д, J=7,28 Гц, 1 H), 7,32-7,38 (м, 1 H), 7,27 (т, J=8,03 Гц, 1 H), 7,05 (д, J=7,78 Гц, 1 H), 5,77 (к, J=6,27 Гц, 1 H), 4,40 (дд, J=13,80, 4,52 Гц, 1 H), 3,37 (уш. с, 1 H), 2,75-2,95 (м, 2 H), 1,31 (д, J=6,53 Гц, 3 H). ЖХМС: 367/369 [M+1].
Соединение 317, энантиомер 1: 1H ЯМР (400 МГц, МЕТАНОЛ-d4) d м. д. 7,69 (д, J=7,53 Гц, 2 H), 7,46-7,59 (м, 3 H), 7,39 (с, 1 H), 7,29-7,34 (м, 1 H), 7,22-7,28 (м, 1 H), 7,03 (д, J=7,78 Гц, 1 H), 5,07 (д, J=15,31 Гц, 1 H), 4,99 (с, 1 H), 4,48 (д, J=15,31 Гц, 1 H), 3,09 (дд, J=15,81, 5,77 Гц, 1 H), 2,73 (д, J=15,81 Гц, 1 H), 1,28 (д, J=6,78 Гц, 3 H). ЖХМС: 367/369 [M+1]. ЖХМС: 367/369 [M+1].
Соединение 318, энантиомер 2: 1H ЯМР (400 МГц, МЕТАНОЛ-d4) d м. д. 7,68 (д, J=7,53 Гц, 2 H), 7,57 (с, 1 H), 7,50 (т, J=7,65 Гц, 2 H), 7,33-7,44 (м, 2 H), 7,24-7,30 (м, 1 H), 7,05 (д, J=7,53 Гц, 1 H), 5,72-5,83 (м, 1 H), 4,35-4,47 (м, 1 H), 3,37 (уш. с, 1 H), 2,76-2,94 (м, 2 H), 1,31 (д, J=6,53 Гц, 3 H). ЖХМС: 367/369 [M+1].
Соединение 317, энантиомер 2: 1H ЯМР (400 МГц, МЕТАНОЛ-d4) d м. д. 7,69 (д, J=7,78 Гц, 2 H), 7,47-7,58 (м, 3 H), 7,37-7,44 (м, 1 H), 7,30-7,34 (м, 1 H), 7,21-7,28 (м, 1 H), 7,04 (с, 1 H), 5,08 (д, J=15,31 Гц, 1 H), 4,99-5,01 (м, 1 H), 4,48 (д, J=15,31 Гц, 1 H), 3,10 (дд, J=15,81, 5,77 Гц, 1 H), 2,73 (д, J=15,81 Гц, 1 H), 1,28 (д, J=6,78 Гц, 3 H). ЖХМС: 367/369 [M+1].
Пример 39. Получение соединений 542 и 583
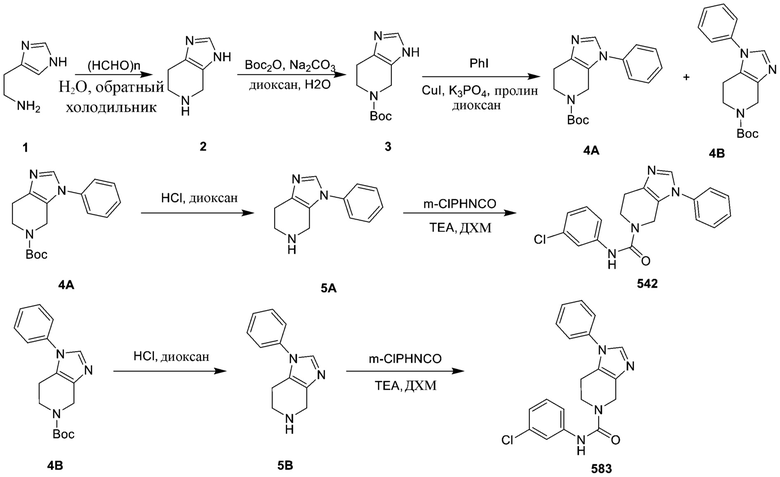
Стадия 1. Получение соединения 2
К смеси 2-(1Н-имидазол-4-ил)этанамина (1,00 г, 5,43 ммоль, 1,00 экв., 2HCl) в H2O (8,00 мл) добавляли HCHO (244,59 г, 8,15 ммоль, 1,50 экв.) за один раз в атмосфере N2. Смесь перемешивали при 100°C в течение 10 часов. ЖХМС показала завершение реакции. Смесь концентрировали в вакууме с получением 4,5,6,7-тетрагидро-3Н-имидазо[4,5-c]пиридина (1,25 г, неочищенный, 2HCl) в виде желтого твердого вещества.
Стадия 2. Получение соединения 3
К смеси 4,5,6,7-тетрагидро-3Н-имидазо[4,5-c]пиридина (669,00 мг, 3,41 ммоль, 1,00 экв., 2HCl) в диоксане (5,00 мл) и H2O (3,00 мл) добавляли Na2CO3 (904,06 мг, 8,53 ммоль, 2,50 экв.) и Boc2O (819,11 мг, 3,75 ммоль, 1,10 экв.) за один раз при 15°C в атмосфере N2. Смесь перемешивали при 15°C в течение 10 часов. ЖХМС показала завершение реакции. Осадок экстрагировали этилацетатом (40 мл*2). Объединенную органическую фазу промывали солевым раствором (40 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (этилацетат) с получением трет-бутил-3,4,6,7-тетрагидроимидазо[4,5-c]пиридин-5-карбоксилата (740,00 мг, 2,85 ммоль, выход 83,58%, чистота 86%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) 9,08-9,41 (м, 1 H), 7,51 (с, 1 H), 4,48 (с, 2 H), 3,63-3,83 (м, 2 H), 2,59-2,76 (м, 2 H), 1,47 (с, 9 H). ЖХМС: 224 [M+1].
Стадия 3. Получение соединений 4A и 4B
К смеси трет-бутил-3,4,6,7-тетрагидроимидазо[4,5-c]пиридин-5-карбоксилата (300,00 мг, 1,34 ммоль, 1,00 экв.) и иодбензола (356,36 мг, 1,75 ммоль, 1,30 экв.) в диоксане (3,00 мл) добавляли K3PO4 (570,44 мг, 2,69 ммоль, 2,00 экв.), CuI (51,18 мг, 268,73 мкмоль, 0,20 экв.) и N,Nʹ-диметилэтан-1,2-диамин (236,89 мг, 2,69 ммоль, 2,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 16 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (20 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (20 мл*2). Объединенную органическую фазу промывали солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=4/1) с получением трет-бутил-3-фенил-6,7-дигидро-4H-имидазо[4,5-c]пиридин-5-карбоксилата (32,00 мг, 106,89 мкмоль, выход 7,98%) в виде желтого масла и трет-бутил-1-фенил-6,7-дигидро-4H-имидазо[4,5-c]пиридин-5-карбоксилата (46,00 мг, 153,66 мкмоль, выход 11,47%) в виде желтого масла.
Соединение 4A: 1H ЯМР (400 МГц, МЕТАНОЛ-d4) 7,80 (с, 1 H), 7,57 (с, 2 H), 7,48-7,52 (м, 1 H), 7,44 (д, J=1,25 Гц, 2 H), 4,47 (с, 2 H), 3,74-3,80 (м, 2 H), 2,66-2,74 (м, 2 H), 1,39-1,49 (м, 9 H). ЖХМС: 300 [M+1].
Соединение 4B: 1H ЯМР (400 МГц, МЕТАНОЛ-d4) 7,82 (с, 1 H), 7,55 (с, 2 H), 7,43 (с, 3 H), 4,44-4,53 (м, 2 H), 3,69-3,77 (м, 2 H), 2,62-2,73 (м, 2 H), 1,50 (с, 9 H). ЖХМС: 300 [M+1].
Стадия 4a. Получение соединения 5A
К смеси трет-бутил-3-фенил-6,7-дигидро-4H-имидазо[4,5-c]пиридин-5-карбоксилата (30,00 мг, 100,21 мкмоль, 1,00 экв.) в диоксане (2,00 мл) добавляли HCl/диоксан (4 М, 4,00 мл, 159,66 экв.) за один раз в атмосфере N2. Смесь перемешивали при температуре 15°C в течение 1 часа. ТСХ (этилацетат: петролейный эфир=1: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением 3-фенил-4,5,6,7-тетрагидроимидазо[4,5-c]пиридина (23,62 мг, 100,21 мкмоль, выход 100,00%, HCl) в виде желтого твердого вещества.
Стадия 5a. Получение соединения 542
К смеси 3-фенил-4,5,6,7-тетрагидроимидазо[4,5-c]пиридина (23,62 мг, 100,21 мкмоль, 1,00 экв., HCl) и TEA (30,42 мг, 300,62 мкмоль, 3,00 экв.) в ДХМ (4,00 мл) добавляли 1-хлор-3-изоцианатбензол (15,39 мг, 100,21 мкмоль, 1,00 экв.) за один раз при 15°C в атмосфере N2. Смесь перемешивали при 15°C в течение 1 часа. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-фенил-6,7-дигидро-4Н-имидазо[4,5-c]пиридин-5-карбоксамида (17,00 мг, 47,36 мкмоль, выход 47,26%, чистота 98,3%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,82-7,91 (м, 1 H), 7,55-7,62 (м, 3 H), 7,49 (д, J=1,63 Гц, 6 H), 7,18-7,29 (м, 3 H), 6,96-7,03 (м, 1 H), 4,61 (с, 2 H), 3,89 (с, 2 H), 2,81 (уш. с, 2 H). ЖХМС: 353 [M+1].
Стадия 4b. Получение соединения 5B
К смеси трет-бутил-1-фенил-6,7-дигидро-4H-имидазо[4,5-c]пиридин-5-карбоксилата (36,00 мг, 120,25 мкмоль, 1,00 экв.) в диоксане (2,00 мл) добавляли HCl/диоксан (4 М, 2,00 мл, 66,53 экв.) за один раз в атмосфере N2. Смесь перемешивали при температуре 15°C в течение 30 мин. ТСХ (этилацетат: петролейный эфир=2: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением 1-фенил-4,5,6,7-тетрагидроимидазо[4,5-c]пиридина (28,34 мг, 120,23 мкмоль, выход 100,00%, HCl) в виде желтого твердого вещества.
Стадия 5b. Получение соединения 583
К смеси 1-фенил-4,5,6,7-тетрагидроимидазо[4,5-c]пиридина (28,34 мг, 120,23 мкмоль, 1,00 экв., HCl) и TEA (36,50 мг, 360,69 мкмоль, 3,00 экв.) в ДХМ (2,00 мл) добавляли 1-хлор-3-изоцианатбензол (18,46 мг, 120,23 мкмоль, 1,00 экв.) за один раз при 15°C в атмосфере N2. Смесь перемешивали при 15°C в течение 30 мин. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-1-фенил-6,7-дигидро-4Н-имидазо[4,5-c]пиридин-5-карбоксамида (24,00 мг, 64,35 мкмоль, выход 53,52%, чистота 94,6%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) 8,34-8,38 (м, 1 H), 7,60 (д, J=7,65 Гц, 1 H), 7,48-7,57 (м, 4 H), 7,25 (с, 1 H), 7,00-7,05 (м, 1 H), 4,68 (с, 2 H), 3,87 (т, J=5,52 Гц, 2 H), 2,80 (т, J=5,21 Гц, 2 H). ЖХМС: 353 [M+1].
Пример 40. Получение соединения 576
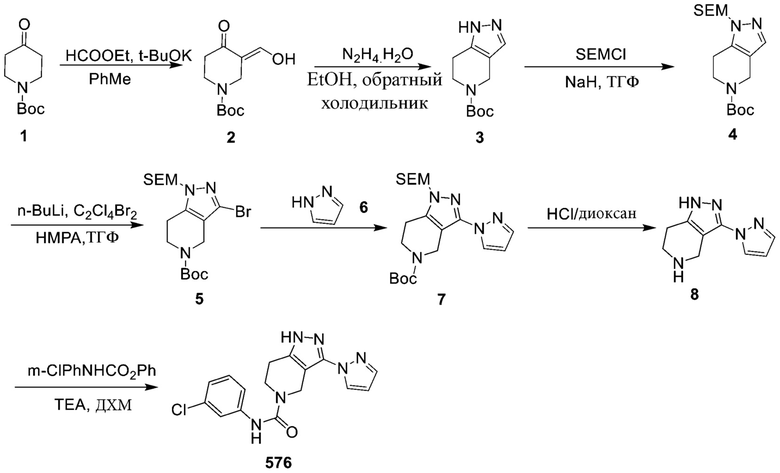
Стадия 1. Получение соединения 2
К смеси трет-бутил-4-оксопиперидин-1-карбоксилата (60,00 г, 301,13 ммоль, 1,00 экв.) в толуоле (600,00 мл) добавляли t-BuOK (50,68 г, 451,70 ммоль, 1,50 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 30 мин. Впоследствии добавляли этилформиат (33,46 г, 451,70 ммоль, 1,50 экв.) при 0°C и смесь перемешивали при 15°C в течение 16 ч в атмосфере N2. ТСХ показала завершение реакции. Смесь вливали в ледяную воду (600 мл), экстрагировали EA (300 мл*2), объединенный органический слой промывали NaOH 10% (300 мл), рН объединенного водного слоя доводили до 4 посредством 1 Н HCl, впоследствии водный слой экстрагировали EA (600*3), объединенный органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением трет-бутил-(3Е)-3-(гидроксиметилен)-4-оксопиперидин-1-карбоксилата (70,60 г, неочищенный) в виде желтого масла и использовали непосредственно на следующей стадии.
Стадия 2. Получение соединения 3
Смесь трет-бутил-(3Е)-3-(гидроксиметилен)-4-оксопиперидин-1-карбоксилата (70,00 г, 308,02 ммоль, 1,00 экв.), NH2NH2·H2O (36,28 г, 616,04 ммоль, 2,00 экв.) в EtOH (700,00 мл) дегазировали и 3 раза продували N2, а впоследствии смесь перемешивали при 90°C в течение 5 часов в атмосфере N2. ЖХМС показала завершение реакции. Смесь вливали в HCl (0,5 Н, 700 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (300 мл*3). Объединенную органическую фазу промывали солевым раствором (700 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением трет-бутил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (65,00 г, 291,13 ммоль, выход 94,52%) в виде желтого масла, которое использовали непосредственно на следующей стадии. ЖХМС: 224 [M+1].
Стадия 3. Получение соединения 4
К смеси NaH (16,12 г, 403,10 ммоль, чистота 60%, 1,50 экв.) в ТГФ (750 мл) по каплям добавляли раствор трет-бутил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (60,00 г, 268,73 ммоль, 1,00 экв.) в ТГФ (50 мл) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 30 мин. Впоследствии к смеси по каплям добавляли SEM-Cl (58,24 г, 349,35 ммоль, 61,96 мл, 1,30 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 15°C в течение 2,5 часа в атмосфере N2. ТСХ показала завершение реакции. Смесь вливали в ледяную воду (800 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (500 мл*3). Объединенную органическую фазу промывали солевым раствором (800 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 100/1 до 20/1) с получением трет-бутил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (72,00 г, 203,66 ммоль, выход 75,79%) в виде белого масла. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) м. д. 7,32 (с, 1 H), 5,32-5,41 (м, 2 H), 4,40-4,53 (м, 2 H), 3,67-3,78 (м, 2 H), 3,52-3,65 (м, 2 H), 2,72-2,81 (м, 2 H), 1,48 (с, 9 H), 0,87-0,92 (м, 2 H), -0,02 (д, J=5,52 Гц, 9 H).
Стадия 4. Получение соединения 5
Смесь трет-бутил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (20,00 г, 56,57 ммоль, 1,00 экв.) и HMPA (25,34 г, 141,42 ммоль, 24,84 мл, 2,50 экв.) в ТГФ (200,00 мл) при -78 °C, впоследствии за один раз добавляли n-BuLi (2,5 М, 33,94 мл, 1,50 экв.) при -78°C в атмосфере N2. Смесь перемешивали при -78°C в течение 0,5 ч в атмосфере N2. Впоследствии за один раз добавляли 1,2-дибром-1,1,2,2-тетрахлорэтан (36,84 г, 113,14 ммоль, 13,59 мл, 2,00 экв.) при -78°C в атмосфере N2. Смесь перемешивали при 15°C в течение 2,5 ч в атмосфере N2. ТСХ показала завершение реакции. Смесь вливали в ледяную воду (300 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (100 мл*3). Объединенную органическую фазу промывали солевым раствором (300 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 100/1 до 30/1) с получением трет-бутил-3-бром-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (8,30 г, 19,19 ммоль, выход 33,93%) в виде желтого масла. 1H ЯМР (300 МГц, ХЛОРОФОРМ-d) м. д. 5,40-5,42 (м, 1 H), 5,38 (с, 1 H), 4,27-4,42 (м, 2 H), 3,55-3,71 (м, 4 H), 2,66-2,78 (м, 2 H), 1,47 (с, 9 H), 0,91 (с, 2 H), -0,03 (с, 9 H).
Стадия 5. Получение соединения 7
К смеси трет-бутил-3-бром-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (100,00 мг, 231,25 мкмоль, 1,00 экв.) и 1Н-пиразола (23,62 мг, 346,88 мкмоль, 1,50 экв.) в DMF (2,00 мл) добавляли Bu4NCuI2 (25,89 мг, 46,25 мкмоль, 0,20 экв.), (1S,2S)-N1,N2-диметилциклогексан-1,2-диамин (6,58 мг, 46,25 мкмоль, 0,20 экв.) и t-BuOK (77,85 мг, 693,75 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 100°C в течение 16 часов. ТСХ (петролейный эфир: этилацетат=3: 1) показала, что нужный продукт обнаружен. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (15 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением трет-бутил-3-пиразол-1-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (60,00 мг, неочищенный) в виде желтого масла. ЖХМС: 420 [M+1].
Стадия 6. Получение соединения 8
К смеси трет-бутил-3-пиразол-1-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (20,00 мг, 47,67 мкмоль, 1,00 экв.) в диоксане (1,00 мл) добавляли HCl/диоксан (4 М, 4,00 мл, 335,64 экв.). Смесь перемешивали при 20°C в течение 2 часов. ТСХ (петролейный эфир: этилацетат=3: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением 3-пиразол-1-ил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (10,76 мг, 47,68 мкмоль, выход 100,00%, HCl) в виде желтого твердого вещества.
Получение соединения 576
К смеси 3-(1-бицикло[3.1.0]гексанил)-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (15,80 мг, 65,90 мкмоль, 1,00 экв., HCl) и TEA (20,01 мг, 197,70 мкмоль, 3,00 экв.) в ДХМ (2,00 мл) добавляли 1-хлор-3-изоцианатбензол (9,11 мг, 59,31 мкмоль, 0,90 экв.) за один раз при 15°C в атмосфере N2. Смесь перемешивали при 15°C в течение 30 мин. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением 3-(1-бицикло[3.1.0]гексанил)-N-(3-хлорфенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (10,00 мг, 26,90 мкмоль, выход 40,82%, чистота 96,0%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 8,08-8,18 (м, 1 H), 7,70-7,75 (м, 1 H), 7,50-7,56 (м, 1 H), 7,18-7,34 (м, 2 H), 6,96-7,04 (м, 1 H), 6,44-6,52 (м, 1 H), 4,77 (с, 2 H), 3,86 (т, J=5,71 Гц, 2 H), 2,86 (т, J=5,65 Гц, 2 H). ЖХМС: 343/345 [M+1].
Пример 41. Получение соединения 751
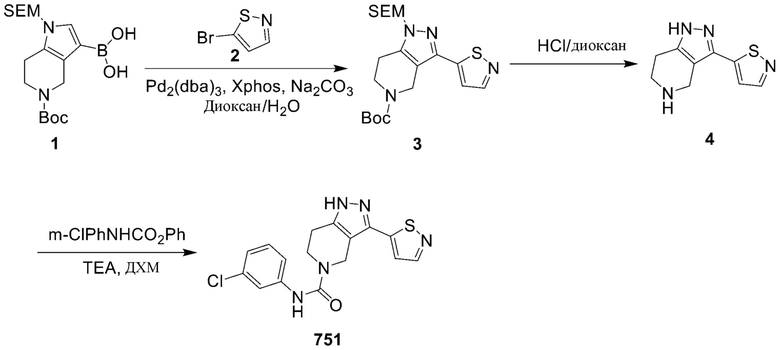
Стадия 1. Получение соединения 3
Смесь [5-трет-бутоксикарбонил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-3-ил]бороновой кислоты (120,00 мг, 302,00 мкмоль, 1,00 экв.), 5-бромизотиазола (59,44 мг, 362,40 мкмоль, 1,20 экв.), XPhos (14,40 мг, 30,20 мкмоль, 0,10 экв.), Pd2(dba)3 (13,83 мг, 15,10 мкмоль, 0,05 экв.) и Na2CO3 (80,02 мг, 755,00 мкмоль, 2,50 экв.) в диоксане (4,00 мл) и H2O (500,00 мкл) дегазировали и 3 раза продували N2, а впоследствии смесь перемешивали при 105°C в течение 1 часа в атмосфере N2. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (5 мл*3). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (PE/EA=5/1) с получением трет-бутил-3-изотиазол-5-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилата (29,00 мг, 46,49 мкмоль, выход 15,39%, чистота 70%) в виде белого твердого вещества. ЖХМС: 437 [M+1].
Стадия 2. Получение соединения 4
Смесь трет-бутил-3-изотиазол-5-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилата (29,00 мг, 66,42 мкмоль, 1,00 экв.) в HCl/диоксане (4М, 5,00 мл, 301,11 экв.), а впоследствии смесь перемешивали при 15°C в течение 0,5 часа. ТСХ показала завершение реакции. Смесь концентрировали в вакууме с получением 5-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)изотиазола (14,00 мг, 57,68 мкмоль, выход 86,84%, HCl) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии.
Получение соединения 751
Смесь 5-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)изотиазола (14,00 мг, 57,68 мкмоль, 1,00 экв., HCl), фенил-N-(3-хлорфенил)карбамата (12,86 мг, 51,91 мкмоль, 0,90 экв.), TEA (8,75 мг, 86,52 мкмоль, 11,99 мкл, 1,50 экв.) в ДХМ (3,00 мл) дегазировали и 3 раза продували N2, а впоследствии смесь перемешивали при 15°C в течение 16 часов в атмосфере N2. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (5 мл*3). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-изотиазол-5-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (11,10 мг, 30,85 мкмоль, выход 53,48%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 8,50-8,54 (м, 1 H), 7,51-7,57 (м, 2 H), 7,30 (дд, J=1,00, 2,01 Гц, 1 H), 7,21-7,27 (м, 1 H), 7,03 (с, 1 H), 4,74 (с, 2 H), 3,87 (т, J=5,71 Гц, 2 H), 2,89 (т, J=5,71 Гц, 2 H). ЖХМС: 360/362 [M+1].
Пример 42. Получение соединения 569
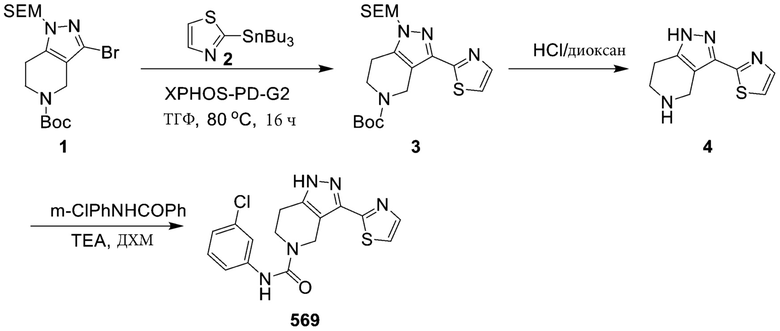
Стадия 1. Получение соединения 3
Раствор трет-бутил-3-бром-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (200,00 мг, 462,50 мкмоль, 1,00 экв.) и трибутил(тиазол-2-ил)станнана (259,58 мг, 693,75 мкмоль, 1,50 экв.) в ТГФ (10,00 мл) добавляли XPHOS-PD-G2 (36,39 мг, 46,25 мкмоль, 0,10 экв.). Смесь перемешивали при 80°C в течение 16 часов под защитой N2. ТСХ (петролейный эфир/этилацетат=5: 1) показала, что материал сохранился и обнаружена новая зона. Растворитель выпаривали. Остаток очищали посредством препаративной ТСХ (петролейный эфир/этилацетат=5: 1) с получением неочищенного продукта. Неочищенный продукт повторно очищали посредством препаративной ВЭЖХ (FA) с получением трет-бутил-3-тиазол-2-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (40,00 мг, 43,97 мкмоль, выход 9,51%, чистота 48%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,90-8,00 (м, 1 H), 7,48 (д, J=3,01 Гц, 1 H), 5,95 (с, 1 H), 4,70 (уш. с, 2 H), 3,77 (уш. с, 2 H), 3,63-3,68 (м, 2 H), 2,83 (д, J=5,27 Гц, 2 H), 1,51 (с, 9 H), 0,93 (д, J=8,28 Гц, 2 H), -0,05 (с, 9 H). ЖХМС: 437 [M+1].
Стадия 2. Получение соединения 4
Смесь трет-бутил-3-тиазол-2-ил-1-(2-триметилсилилэтоксиметил)-
6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилата (40,00 мг, 91,61 мкмоль, 1,00 экв.) и HCl/диоксана (4М, 5,00 мл, 218,32 экв.) перемешивали при 25°C в течение 2 ч. Растворитель выпаривали с получением 2-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]
пиридин-3-ил)тиазола (22,00 мг, 90,64 мкмоль, выход 98,94%, HCl) в виде белого твердого вещества, которое не очищали и использовали непосредственно на следующей стадии.
Получение соединения 569
К смеси 2-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)тиазола (22,00 мг, 90,64 мкмоль, 1,00 экв., HCl) и фенил-N-(3-хлорфенил)карбамата (22,45 мг, 90,64 мкмоль, 1,00 экв.) в ДХМ (3,00 мл) добавляли TEA (18,34 мг, 181,28 мкмоль, 25,12 мкл, 2,00 экв.). Смесь перемешивали при температуре 25°C в течение 16 ч. ЖХМС показала, что материал полностью израсходован и обнаружен основной нужный масс-спектр. Растворитель выпаривали. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-тиазол-2-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (14,41 мг, 40,05 мкмоль, выход 44,18%, чистота 100%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ 7,90 (д, J=2,51 Гц, 1 H), 7,52-7,60 (м, 2 H), 7,31-7,36 (м, 1 H), 7,21-7,29 (м, 1 H), 7,02 (д, J=8,03 Гц, 1 H), 4,89-4,97 (м, 2 H), 3,89 (т, J=5,65 Гц, 2 H), 2,91 (т, J=5,65 Гц, 2 H). ЖХМС: 360/362 [M+1].
Пример 43. Получение соединения 726
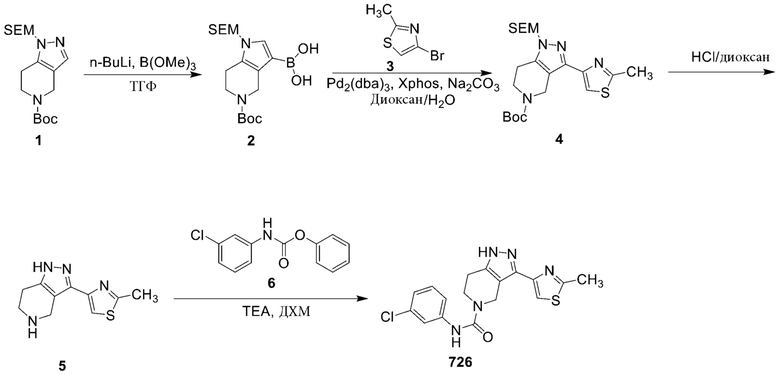
Стадия 1. Получение соединения 2
Смесь трет-бутил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (5,70 г, 16,12 ммоль, 1,00 экв.) в ТГФ (60,00 мл) при -78°C в атмосфере N2. Добавляли N-BuLi (2,5 М, 7,74 мл, 1,20 экв.) за один раз при -78°C в атмосфере N2. Смесь перемешивали при -78°C в течение 30 мин. Впоследствии добавляли B(OMe)3 (5,03 г, 48,36 ммоль, 5,47 мл, 3,00 экв.) за один раз при -78°C в атмосфере N2. Смесь перемешивали при 15°C в течение 1,5 ч в атмосфере N2. ЖХМС показала, что соотношение исходного материала/нужного продукта составляет 1/3. Смесь вливали в ледяной NH4Cl (водн. раствор, 80 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (50 мл*3). Объединенную органическую фазу промывали солевым раствором (80 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением [5-трет-бутоксикарбонил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-3-ил]бороновой кислоты (6,80 г, неочищенная) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии. ЖХМС: 398 [M+1].
Стадия 2. Получение соединения 4
[5-трет-бутоксикарбонил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-3-ил]бороновую кислоту (120,00 мг, 302,00 мкмоль, 1,00 экв.), 4-бром-2-метилтиазол (64,53 мг, 362,40 мкмоль, 1,20 экв.), Pd2(dba)3 (13,83 мг, 15,10 мкмоль, 0,05 экв.), XPhos (14,40 мг, 30,20 мкмоль, 0,10 экв.) и Na2CO3 (80,02 мг, 755,00 мкмоль, 2,50 экв.) помещали в пробирку для микроволновой обработки с диоксаном (4,00 мл) и H2O (500,00 мкл), дегазировали и 3 раза продували N2. Герметично закрытую пробирку выдерживали при температуре 105°C в течение 1 ч под воздействием микроволн. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (5 мл*3). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (PE/EA=5/1) с получением трет-бутил-3-(2-метилтиазол-4-ил)-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (34,00 мг, 75,44 мкмоль, выход 24,98%) в виде светло-желтого твердого вещества. ЖХМС: 451 [M+1].
Стадия 3. Получение соединения 5
Смесь трет-бутил-3-(2-метилтиазол-4-ил)-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (34,00 мг, 75,44 мкмоль, 1,00 экв.) в HCl/диоксане (4 М, 2,00 мл, 106,04 экв.), впоследствии смесь перемешивали при 20°C в течение 15 мин. ТСХ показала завершение реакции. Смесь концентрировали в вакууме с получением 2-метил-4-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)тиазола (15,00 мг, 58,42 мкмоль, выход 77,44%, HCl) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии.
Получение соединения 726
Смесь 2-метил-4-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)изотиазола (15,00 мг, 58,42 мкмоль, 1,00 экв., HCl), фенил-N-(3-хлорфенил)карбамата (13,02 мг, 52,58 мкмоль, 0,90 экв.), TEA (8,87 мг, 87,63 мкмоль, 12,15 мкл, 1,50 экв.) в ДХМ (3,00 мл) дегазировали и 3 раза продували N2, а впоследствии смесь перемешивали при 20°C в течение 16 часов в атмосфере N2. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали ДХМ (5 мл*3). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-(2-метилтиазол-4-ил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (12,00 мг, 31,84 мкмоль, выход 54,50%, чистота 99,2%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 7,54 (с, 1 H), 7,28-7,33 (м, 1 H), 7,20-7,26 (м, 1 H), 6,98-7,03 (м, 1 H), 4,80 (с, 2 H), 3,83-3,88 (м, 2 H), 2,84-2,89 (м, 2 H), 2,76 (с, 3 H). ЖХМС: 374/376 [M+1].
Пример 44. Получение соединения 730
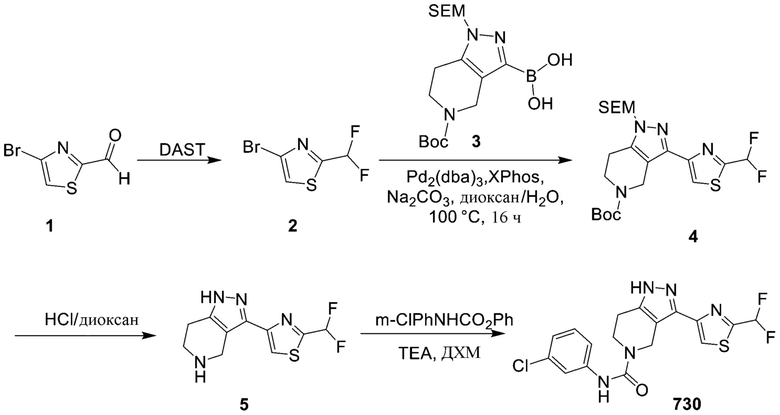
Стадия 1. Получение соединения 2
К раствору 4-бромтиазол-2-карбальдегида (500,00 мг, 2,60 ммоль, 1,00 экв.) в ДХМ (10,00 мл) по каплям добавляли DAST (838,19 мг, 5,20 ммоль, 687,04 мкл, 2,00 экв.) при -78°C под защитой N2. Смесь перемешивали при температуре 25°C в течение 16 ч. ТСХ (петролейный эфир/этилацетат=10: 1) показала, что материал полностью израсходован и обнаружена новая зона. Смесь вливали в воду (10 мл), экстрагировали этилацетатом (10 мл*2), объединенный органический слой сушили безводным Na2SO4, концентрировали. Остаток очищали посредством хроматографии (на силикагеле, элюирование с помощью смеси петролейного эфира/этилацетата=100: 1) с получением 4-бром-2-(дифторметил)тиазола (300,00 мг, 1,40 ммоль, выход 53,91%) в виде светло-желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,38 (с, 1 H), 6,75 (т, J=52,00 Гц, 1 H).
Стадия 2. Получение соединения 4
К раствору 4-бром-2-(дифторметил)тиазола (300,00 мг, 1,40 ммоль, 1,00 экв.) и [5-трет-бутоксикарбонил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-H-пиразол[4,3-c]пиридин-3-ил]бороновой кислоты (556,29 мг, 1,40 ммоль, 1,00 экв.) в диоксане (10,00 мл) добавляли Pd2(dba)3 (64,10 мг, 70,00 мкмоль, 0,05 экв.), XPhos (66,74 мг, 140,00 мкмоль, 0,10 экв.) и Na2CO3 (296,77 мг, 2,80 ммоль, 2,00 экв.). Смесь перемешивали при температуре 100°C в течение 16 ч под защитой N2. ЖХМС показала, что материал полностью израсходован и обнаружен нужный масс-спектр. ТСХ (петролейный эфир: этилацетат=10: 1) показала наличие основного продукта. Растворитель выпаривали. Остаток очищали посредством хроматографии (на силикагеле, элюирование с помощью смеси петролейного эфира/этилацетата=10: 1) с получением трет-бутил-3-[2-(дифторметил)тиазол-4-ил]-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (196,00 мг, 237,62 мкмоль, выход 16,97%, чистота 59%) в виде светло-желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,98 (с, 1 H), 7,34 (с, 1 H), 6,74-7,06 (м, 1 H), 5,62 (с, 1 H), 5,38 (с, 1 H), 4,70 (уш. с, 1 H), 4,45 (уш. с, 1 H), 3,66-3,78 (м, 3 H), 3,52-3,61 (м, 1 H), 2,80 (д, J=17,07 Гц, 2 H), 1,45-1,56 (м, 9 H), 0,86-1,00 (м, 2 H), -0,04-0,06 (м, 9 H). ЖХМС: 487 [M+1].
Стадия 3. Получение соединения 5
Смесь трет-бутил-3-[2-(дифторметил)тиазол-4-ил]-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилата (190,00 мг, 390,42 мкмоль, 1,00 экв.) и HCl/диоксана (4М, 5,00 мл, 51,23 экв.) перемешивали при 25°C в течение 1 ч. Растворитель выпаривали с получением 2-(дифторметил)-4-(4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин-3-ил)тиазола (114,00 мг, 389,42 мкмоль, выход 99,74%, HCl) в виде белого твердого вещества, которое не очищали и использовали непосредственно на следующей стадии.
Получение соединения 730
К раствору 2-(дифторметил)-4-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)тиазола (114,00 мг, 389,42 мкмоль, 1,00 экв., HCl) в ДХМ (4,00 мл) добавляли TEA (78,81 мг, 778,84 мкмоль, 107,96 мкл, 2,00 экв.) и фенил-N-(3-хлорфенил)карбамат (96,45 мг, 389,42 мкмоль, 1,00 экв.). Смесь перемешивали при температуре 25°C в течение 16 ч. ЖХМС показала, что материал полностью израсходован и обнаружен основной нужный масс-спектр. Растворитель выпаривали. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-[2-(дифторметил)тиазол-4-ил]-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (29,83 мг, 72,09 мкмоль, выход 18,51%, чистота 99,04%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ 7,94 (с, 1 H), 7,49 (с, 1 H), 7,26 (с, 1 H), 6,92-7,23 (м, 3 H), 4,74-4,81 (м, 2 H), 3,83 (т, J=5,65 Гц, 2 H), 2,85 (т, J=5,52 Гц, 2 H). ЖХМС: 410/412 [M+1].
Пример 45. Получение соединения 645
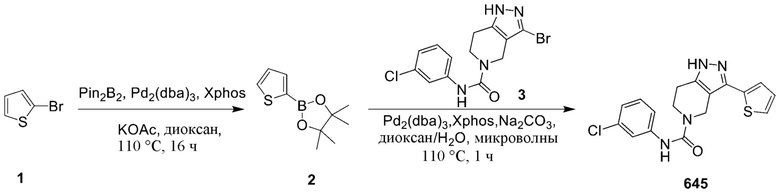
Стадия 1. Получение соединения 2
Смесь 2-бромтиофена (400,00 мг, 2,45 ммоль, 1,00 экв.), 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолана (933,23 мг, 3,68 ммоль, 1,50 экв.), AcOK (601,11 мг, 6,13 ммоль, 2,50 экв.), XPhos (116,80 мг, 245,00 мкмоль, 0,10 экв.) и Pd2(dba)3 (112,18 мг, 122,50 мкмоль, 0,05 экв.) в диоксане (10,00 мл) выдерживали при 110°C в атмосфере N2 в течение 16 ч. Реакционную смесь разбавляли солевым раствором (60 мл) и экстрагировали EA (80 мл). Органический слой сушили Na2SO4, фильтровали и концентрировали под пониженным давлением с получением коричневого остатка. Остаток очищали на колонке с силикагелем (PE/EA=100/1) с получением нужного продукта (420,00 мг, неочищенный).
Получение соединения 645
Смесь соединения 3 (60,00 мг, 168,72 мкмоль, 1,00 экв.), 4,4,5,5-тетраметил-2-(2-тиенил)-1,3,2-диоксаборолана (120,00 мг, 571,16 мкмоль, 3,39 экв.), Na2CO3 (44,71 мг, 421,80 мкмоль, 2,50 экв.), XPhos (8,04 мг, 16,87 мкмоль, 0,10 экв.) и Pd2(dba)3 (7,72 мг, 8,44 мкмоль, 0,05 экв.) в диоксане (2,50 мл)/H2O (300,00 мкл) выдерживали при 110°C в микроволновой печи в течение 1 ч. Реакционную смесь разбавляли солевым раствором (40 мл) и экстрагировали EA (40 мл). Органический слой сушили Na2SO4, фильтровали и концентрировали под пониженным давлением с получением коричневого остатка. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением нужного продукта (13,00 мг, 34,92 мкмоль, выход 20,70%, чистота 96,4%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,54-7,56 (м, 1 H), 7,34-7,36 (м, 1 H), 7,30-7,32 (м, 2 H), 7,25-7,27 (м, 1 H), 7,13-7,16 (м, 1 H), 7,00-7,05 (м, 1 H), 4,75 (с, 2 H), 3,86-3,89 (м, 2 H), 2,86-2,89 (м, 2 H). ЖХМС: 359/361 [M+1].
Пример 46. Получение соединения 568
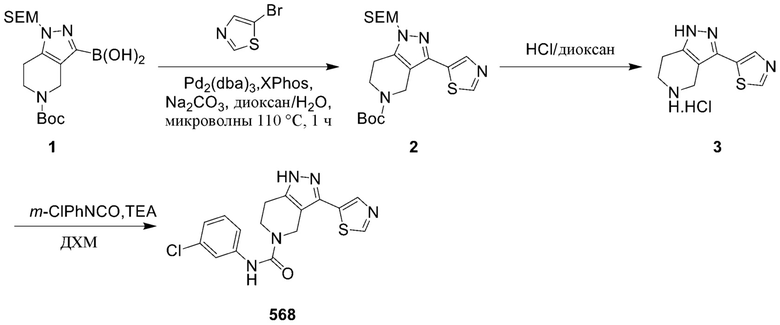
Стадия 1. Получение соединения 2
Смесь соединения 1 (120,00 мг, 181,20 мкмоль, 1,00 экв.), 5-бромтиазола (35,66 мг, 217,44 мкмоль, 1,20 экв.), Na2CO3 (48,01 мг, 453,00 мкмоль, 2,50 экв.), XPhos (8,64 мг, 18,12 мкмоль, 0,10 экв.) и Pd2(dba)3 (8,30 мг, 9,06 мкмоль, 0,05 экв.) в диоксане (2,50 мл)/H2O (500,00 мкл) выдерживали при 110°C в микроволновой печи в течение 1 ч. Смесь разбавляли EA (50 мл) и промывали солевым раствором (40 мл). Органический слой сушили Na2SO4, фильтровали и концентрировали под пониженным давлением с получением желтого масла. Желтое масло очищали посредством препаративной ТСХ (EA/PE=2/3) с получением нужного продукта (24,00 мг, 46,72 мкмоль, выход 25,78%, чистота 85%) в виде бесцветного масла. ЖХМС: 437 [M+1].
Стадия 2. Получение соединения 3
Смесь соединения 2 (24,00 мг, 54,97 мкмоль, 1,00 экв.) в HCl/диоксане (4 М, 3,84 мл, 279,43 экв.) перемешивали при 18°C в течение 0,5 ч. Смесь концентрировали под пониженным давлением с получением желтого остатка, содержащего нужный продукт (14,00 мг, неочищенный, HCl) в виде желтого твердого вещества, который использовали непосредственно на следующей стадии.
Получение соединения 568
К раствору 5-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)тиазола (14,00 мг, 57,68 мкмоль, 1,00 экв., HCl) и TEA (23,35 мг, 230,71 мкмоль, 4,00 экв.) в ДХМ (5,00 мл)/MeOH (500,00 мкл) добавляли 1-хлор-3-изоцианатбензол (7,97 мг, 51,91 мкмоль, 0,90 экв.) в атмосфере N2 и смесь перемешивали при 18°C в течение 0,5 ч. Реакционную смесь разбавляли ДХМ (30 мл) и промывали солевым раствором (30 мл). Органический слой концентрировали под пониженным давлением с получением желтого твердого вещества. Желтое твердое вещество очищали посредством препаративной ВЭЖХ (FA) с получением нужного продукта (8,00 мг, 21,57 мкмоль, выход 37,39%, чистота 97%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 9,00 (с, 1 H), 8,12 (с, 1 H), 7,55-7,56 (м, 1 H), 7,23-7,34 (м, 2 H), 7,02-7,04 (м, 1 H), 4,76 (с, 2 H), 3,88-3,90 (м, 2 H), 2,88-2,91 (м, 2 H). ЖХМС: 360/362 [M+1].
Пример 47. Получение соединения 570
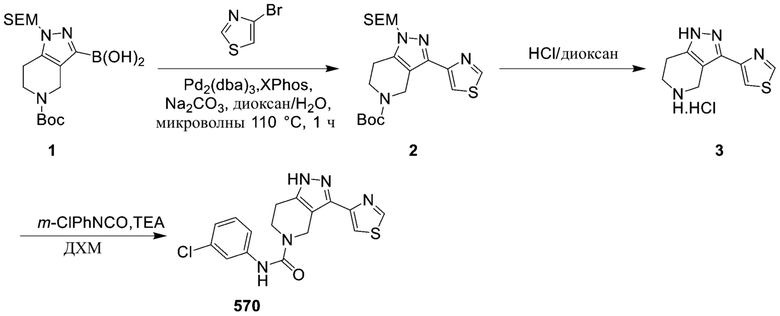
Стадия 1. Получение соединения 2
Смесь соединения 1 (120,00 мг, 181,20 мкмоль, 1,00 экв.), 4-бромтиазола (35,66 мг, 217,44 мкмоль, 1,20 экв.), Na2CO3 (38,41 мг, 362,40 мкмоль, 2,00 экв.), XPhos (8,64 мг, 18,12 мкмоль, 0,10 экв.) и Pd2(dba)3 (8,30 мг, 9,06 мкмоль, 0,05 экв.) в диоксане (2,50 мл)/H2O (500,00 мкл) выдерживали при 110°C в микроволновой печи в течение 1 ч. Смесь разбавляли EA (50 мл) и промывали солевым раствором (40 мл). Органический слой сушили Na2SO4, фильтровали и концентрировали под пониженным давлением с получением желтого масла. Желтое масло очищали посредством препаративной ТСХ (EA/PE=2/3) с получением нужного продукта (50,00 мг, 68,71 мкмоль, выход 37,92%, чистота 60%) в виде бесцветного масла. ЖХМС: 437 [M+1].
Стадия 2. Получение соединения 3
Смесь соединения 2 (50,00 мг, 114,51 мкмоль, 1,00 экв.) в HCl/диоксане (4 М, 6,67 мл, 232,86 экв.) перемешивали при 18°C в течение 0,5 ч. Смесь концентрировали под пониженным давлением с получением желтого остатка, содержащего нужный продукт (28,00 мг, неочищенный, HCl) в виде желтого твердого вещества, который использовали непосредственно на следующей стадии.
Получение соединения 570
К раствору 4-(4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин-3-ил)тиазола (28,00 мг, 115,35 мкмоль, 1,00 экв., HCl) и TEA (46,69 мг, 461,42 мкмоль, 4,00 экв.) в ДХМ (5,00 мл) добавляли 1-хлор-3-изоцианатбензол (15,94 мг, 103,82 мкмоль, 0,90 экв.) в атмосфере N2 и смесь перемешивали при 18°C в течение 0,5 ч. Реакционную смесь разбавляли ДХМ (30 мл) и промывали солевым раствором (30 мл). Органический слой концентрировали под пониженным давлением с получением желтого остатка. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением нужного продукта (13,00 мг, 35,41 мкмоль, выход 30,69%, чистота 98%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ=9,09 (с, 1 H), 7,82 (с, 1 H), 7,55-7,56 (м, 1 H), 7,32-7,33 (м, 1 H), 7,23-7,27 (м, 1 H), 7,03-7,04 (м, 1 H), 4,86 (с, 2 H), 3,87-3,90 (м, 2 H), 2,88-2,91 (м, 2 H). ЖХМС: 360/362 [M+1].
Пример 48. Получение соединения 729
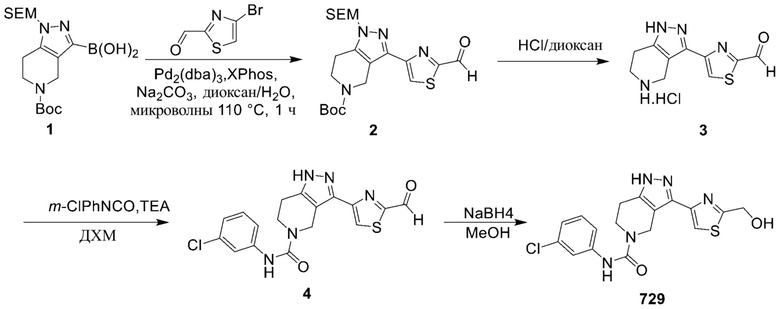
Стадия 1. Получение соединения 2
Смесь соединения 1 (1,20 г, 1,81 ммоль, 1,00 экв.), 4-бромтиазол-2-карбальдегида (347,57 мг, 1,81 ммоль, 1,00 экв.), Na2CO3 (479,60 мг, 4,52 ммоль, 2,50 экв.), XPhos (86,29 мг, 181,00 мкмоль, 0,10 экв.) и Pd2(dba)3 (82,87 мг, 90,50 мкмоль, 0,05 экв.) в диоксане (15,00 мл)/H2O (2,00 мл) выдерживали при 110°C в микроволновой печи в течение 1 ч. Смесь фильтровали и фильтрат разбавляли EA (80 мл) и промывали солевым раствором (80 мл). Органический слой сушили Na2SO4, фильтровали и концентрировали под пониженным давлением с получением коричневого масла. Коричневое масло очищали на колонке с силикагелем (PE/EA=15/1) с получением нужного продукта (724,00 мг, 856,99 мкмоль, выход 47,35%, чистота 55%) в виде бесцветного масла. ЖХМС: 465 [M+1].
Стадия 2. Получение соединения 3
Смесь соединения 2 (100,00 мг, 118,37 мкмоль, 1,00 экв.) в HCl/диоксане (4 М, 4,00 мл, 135,17 экв.) перемешивали при 25°C в течение 0,5 ч. Смесь концентрировали под пониженным давлением с получением нужного продукта (32,00 мг, 65,01 мкмоль, выход 54,92%, чистота 55%, HCl) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии.
Стадия 3. Получение соединения 4
К раствору соединения 3 (32,00 мг, 65,01 мкмоль, 1,00 экв., HCl) и TEA (19,73 мг, 195,03 мкмоль, 27,03 мкл, 3,00 экв.) в ДХМ (5,00 мл)/MeOH (500,00 мкл) добавляли 1-хлор-3-изоцианатбензол (9,48 мг, 61,76 мкмоль, 7,46 мкл, 0,95 экв.) в атмосфере N2 и смесь перемешивали при 25°C в течение 0,5 ч. Остаток разбавляли ДХМ (40 мл) и промывали солевым раствором (40 мл). Органический слой концентрировали под пониженным давлением с получением желтого остатка. Остаток очищали посредством препаративной ТСХ (ДХМ/MeOH=12/1) с получением нужного продукта (31,00 мг, 31,97 мкмоль, выход 49,18%, чистота 40%) в виде белого твердого вещества. ЖХМС: 388/390 [M+1].
Получение соединения 729
К раствору соединения 4 (31,00 мг, 43,96 мкмоль, 1,00 экв.) в MeOH (2,00 мл) добавляли NaBH4 (3,33 мг, 87,92 мкмоль, 2,00 экв.) и смесь перемешивали при 25°C в течение 0,5 ч. Реакционную смесь гасили солевым раствором (30 мл) и экстрагировали EA (30 мл). Органический слой сушили Na2SO4, фильтровали и фильтрат концентрировали под пониженным давлением с получением желтого остатка. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением нужного продукта (10,00 мг, 25,14 мкмоль, выход 57,19%, чистота 98%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,68 (с, 1 H), 7,54-7,56 (м, 1 H), 7,27-7,33 (м, 1 H), 7,22-7,25 (м, 1 H), 7,01-7,03 (м, 1 H), 4,92 (с, 2 H), 4,82 (с, 2 H), 3,86-3,89 (м, 2 H), 2,87-2,90 (м, 2 H). ЖХМС: 390/392 [M+1].
Пример 49. Получение соединения 741
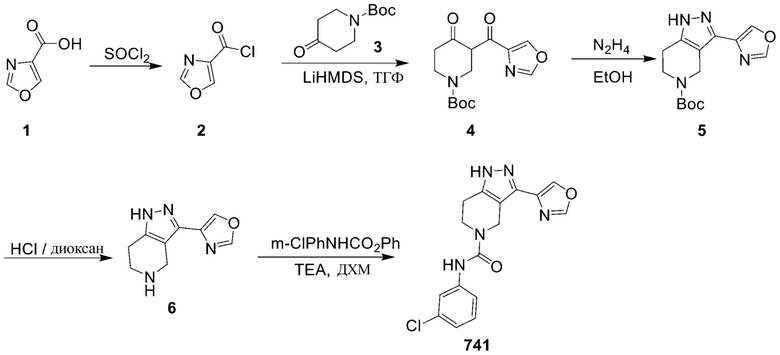
Стадия 1. Получение соединения 2
Оксазол-4-карбоновую кислоту (500,00 мг, 4,42 ммоль, 1,00 экв.) добавляли к SOCl2 (10,00 мл), реакционную смесь нагревали до 70°C и перемешивали при 70°C в течение 2 часов. ТСХ показала, что исходный материал был полностью израсходован. Растворитель выпаривали. Было получено соединение оксазол-4-карбонилхлорид (570,00 мг, неочищенный) в виде желтого масла. Неочищенный продукт использовали непосредственно на следующей стадии без очистки.
Стадия 2. Получение соединения 4
В охлажденную до -78°C трехгорлую круглодонную колбу в атмосфере N2 добавляли LiHMDS (1 M, 5,20 мл, 1,20 экв.), впоследствии по каплям добавляли раствор трет-бутил-4-оксопиперидин-1-карбоксилата (690,83 мг, 3,46 ммоль, 0,80 экв.) в ТГФ (10,00 мл). Реакционную смесь перемешивали при -78°C в течение одного часа в атмосфере N2. К смеси добавляли раствор оксазол-4-карбонилхлорида (570,00 мг, 4,33 ммоль, 1,00 экв.) в ТГФ (10,00 мл). После добавления реакционную смесь нагревали до 30°C и перемешивали при 30°C в течение еще 2 ч. При ЖХМС выявили несколько новых пиков и обнаружили только 15% нужного соединения. Реакционную смесь добавляли в водный раствор NH4Cl (30 мл) и экстрагировали EA (40 мл*3). Объединенную органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением трет-бутил-3-(оксазол-4-карбонил)-4-оксопиперидин-1-карбоксилата (930,00 мг, неочищенный) в виде желтого масла. Неочищенный продукт использовали непосредственно на следующей стадии без очистки. ЖХМС: 295 [M+1].
Стадия 3. Получение соединения 5
К раствору трет-бутил-3-(оксазол-4-карбонил)-4-оксопиперидин-1-карбоксилата (500,00 мг, 1,70 ммоль, 1,00 экв.) в EtOH (10,00 мл) добавляли NH2NH2.H2O (120,14 мг, 2,04 ммоль, 116,64 мкл, чистота 85%, 1,20 экв.). Реакционную смесь нагревали до температуры 60°C и перемешивали ее при 60°C в течение одного часа. ЖХМС показала, что исходный материал полностью израсходован и обнаружен один основной пик с нужным масс-спектром. Смесь экстрагировали EA (20 мл*3) и водой (15 мл), органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле с получением трет-бутил-3-оксазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (180,00 мг, 620,01 мкмоль, выход 36,47%) в виде желтого масла. ЖХМС: 291 [M+1].
Стадия 4. Получение соединения 6
К раствору трет-бутил-3-оксазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (150,00 мг, 516,67 мкмоль, 1,00 экв.) в диоксане (1,00 мл) добавляли HCl/диоксан (4 М, 3,00 мл, 23,23 экв.), реакционную смесь перемешивали при 30°C в течение 30 минут. ТСХ показала, что исходный материал был полностью израсходован. Выпаривали раствор на водяной бане под пониженным давлением с использованием роторного испарителя с получением 4-(4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин-3-ил)оксазола (95,00 мг, 419,13 мкмоль, выход 81,12%, HCl) в виде желтого твердого вещества. Неочищенный продукт использовали непосредственно на следующей стадии без очистки.
Получение соединения 741
К смеси 4-(4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин-3-ил)оксазола (40,00 мг, 176,48 мкмоль, 1,20 экв., HCl) в ДХМ (3,00 мл) добавляли TEA (44,64 мг, 441,19 мкмоль, 61,16 мкл, 3,00 экв.) с последующим добавлением фенил-N-(3-хлорфенил)карбамата (36,42 мг, 147,06 мкмоль, 1,00 экв.), реакционную смесь перемешивали при 30°C в течение 16 часов. ЖХМС показала, что m-ClPhNHCO2Ph полностью израсходован и обнаружен один основной пик с нужным масс-спектром. Смесь экстрагировали ДХМ (15 мл*3) и водой (15 мл), органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Выполняли дополнительную очистку посредством препаративной ВЭЖХ (FA) с получением соединения 741 (39,00 мг, 112,77 мкмоль, выход 76,68%, чистота 99,4%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 8,26 (с, 1 H), 8,21 (с, 1 H), 7,53-7,54 (т, J=2,01 Гц, 1 H), 7,30-7,32 (м, 1 H), 7,21-7,25 (м, 1 H), 7,00-7,01 (дд, J=7,91, 1,00 Гц, 1 H), 4,75 (с, 2 H), 3,84-3,87 (т, J=5,71 Гц, 2 H), 2,84-2,87 (т, J=5,71 Гц, 2 H). ЖХМС: 344/346 [M+1].
Пример 50. Получение соединения 707
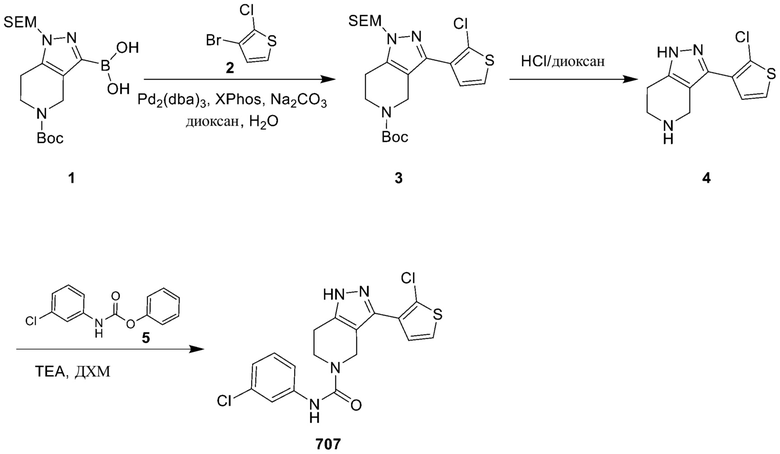
Стадия 1. Получение соединения 3
Смесь соединения 1 (200,00 мг, 503,33 мкмоль, 1,00 экв.), соединения 2 (99,40 мг, 503,33 мкмоль, 1,00 экв.), Na2CO3 (133,37 мг, 1,26 ммоль, 2,50 экв.), XPhos (23,99 мг, 50,33 мкмоль, 0,10 экв.) и Pd2(dba)3 (23,05 мг, 25,17 мкмоль, 0,05 экв.) в диоксане (5,00 мл)/H2O (500,00 мкл) выдерживали при 100°C и перемешивали в течение 16 ч. ТСХ (петролейный эфир: этилацетат=5: 1) показала, что обнаружен нужный продукт (Rf=0,7). В смесь добавляли воду (15 мл) и экстрагировали EA (3*20 мл). Объединенные органические слои сушили Na2SO4, фильтровали и концентрировали под пониженным давлением с получением остатка. Остаток очищали посредством препаративной ТСХ (PE: EA=5: 1) с получением соединения 3 (45,00 мг, неочищенное) в виде светло-желтой жидкости.
Стадия 2. Получение соединения 4
Раствор соединения 3 (45,00 мг, 95,72 мкмоль, 1,00 экв.) в HCl/диоксане (228,84 мкмоль, 3,00 мл, 2,00 экв.) перемешивали при 30°C в течение 20 мин. Впоследствии реакционную смесь концентрировали для удаления растворителя. Впоследствии к остатку добавляли другую партию HCl/диоксана (228,84 мкмоль, 3,00 мл, 2,00 экв.). Полученную смесь перемешивали при 30°C в течение 20 мин. ЖХМС показала, что обнаружено около 72% нужного продукта. Смесь концентрировали под пониженным давлением для удаления растворителя с получением соединения 4 (30,00 мг, неочищенное, HCl) в виде желтого масла. Остаток не очищали и использовали непосредственно на следующей стадии.
Получение соединения 707
Смесь соединения 4 (30,00 мг, 125,15 мкмоль, 1,00 экв.) и TEA (75,98 мг, 750,90 мкмоль, 104,08 мкл, 6,00 экв.) в ДХМ (3,00 мл) дегазировали и 3 раза продували N2 и впоследствии смесь перемешивали при 30°C в течение 16 часов в атмосфере N2. ЖХМС показала, что обнаружен нужный продукт. Показатель pH смеси доводили до 3 и концентрировали под пониженным давлением для удаления растворителя. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением соединения 707 в виде белого твердого вещества. ЖХМС: 393/395 [M+1].
Пример 51. Получение соединений 708, 712, 715 и 769
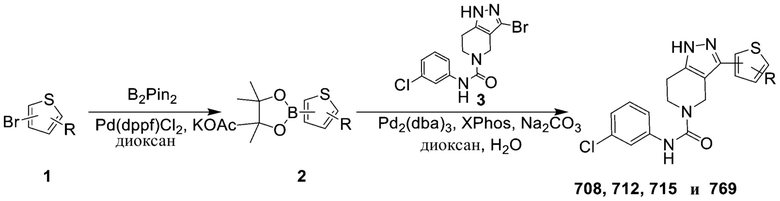
Стадия 1. Получение соединения 2
Смесь соединения 1 (1,00 экв.), B2Pin2 (1,50 экв.), KOAc (2,50 экв.) и Pd(dppf)Cl2 (0,05 экв.) в диоксане (5,00 мл) выдерживали при температуре 100°C в атмосфере N2 в течение 16 ч. ТСХ показала, что исходный материал был полностью израсходован. Смесь концентрировали. Остаток очищали на колонке с силикагелем с получением соединения 2.
Получение соединений 708, 712, 715 или 769
Смесь соединения 2 (2,50 экв.), соединения 3 (1,00 экв.), Na2CO3 (2,50 экв.), XPhos (0,10 экв.) и Pd2(dba)3 (0,05 экв.) в диоксане/H2O(10: 1) перемешивали при 100°C в атмосфере N2 в течение 16 ч. ЖХМС показала, что исходный материал полностью израсходован, а нужный продукт обнаружен. Смесь разбавляли EA и промывали солевым раствором. Органический слой сушили Na2SO4, фильтровали и концентрировали. Остаток очищали на силикагеле и повторно очищали посредством препаративной ВЭЖХ (FA) с получением соединений 708, 712, 715 или 769.
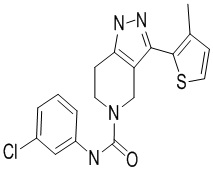
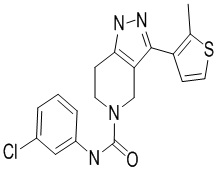
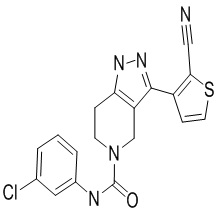
1H ЯМР (400 МГц, МЕТАНОЛ-d4) м. д. 7,94 (уш. с, 1 H), 7,51 (с, 1 H), 7,42 (д, J=5,02 Гц, 1 H), 7,26-7,33 (м, 1 H), 7,19-7,25 (м, 1 H), 7,00 (д, J=8,03 Гц, 1 H), 4,72 (с, 2 H), 3,89 (т, J=5,77 Гц, 2 H), 2,91 (т, J=5,77 Гц, 2 H).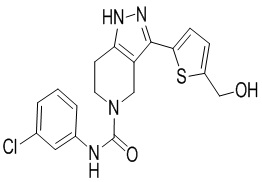
Пример 52. Получение соединений 716 и 766
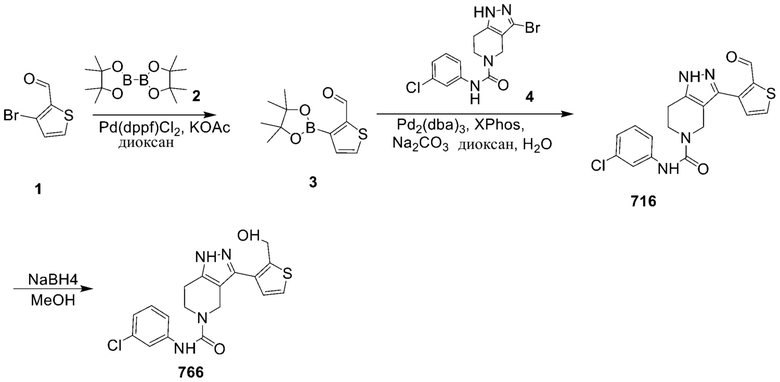
Стадия 1. Получение соединения 3
К смеси соединения 1 (500,00 мг, 2,62 ммоль, 1,00 экв.) и соединения 2 (997,98 мг, 3,93 ммоль, 1,50 экв.) в диоксане (10,00 мл) добавляли Pd(dppf)Cl2 (95,85 мг, 131,00 мкмоль, 0,05 экв.) и KOAc (514,25 мг, 5,24 ммоль, 2,00 экв.), реакционную смесь нагревали до 100°C и перемешивали при 100°C в течение 3 часов в атмосфере N2. ТСХ (петролейный эфир: этилацетат=10: 1) показала, что соединение 1 полностью израсходовано и обнаружена одна основная новая зона с более большой полярностью. Раствор выпаривали под пониженным давлением с использованием роторного испарителя. Остаток очищали посредством хроматографии на силикагеле (силикагель 200-300 меш, петролейный эфир/этилацетат=от 10/1 до 3/1) с получением соединения 3 (1,00 г, неочищенное) в виде желтого масла.
Получение соединения 716
Смесь соединения 4 (150,00 мг, 421,80 мкмоль, 1,00 экв.), соединения 3 (110,48 мг, 463,98 мкмоль, 1,10 экв.) и XPhos (20,11 мг, 42,18 мкмоль, 0,10 экв.) в диоксане (5,00 мл) и H2O (750,00 мкл) дегазировали и 3 раза продували N2. К смеси добавляли Pd2(dba)3 (19,31 мг, 21,09 мкмоль, 0,05 экв.). Полученную смесь перемешивали при 80°C в течение 15 ч. ЖХМС показала, что нужное соединение обнаружено, а соединение 4 сохранилось. Впоследствии к реакционной смеси добавляли соединение 3 из другой партии (110,48 мг, 463,98 мкмоль, 1,10 экв.). Полученную смесь перемешивали при 80°C в течение 5 ч. ТСХ показала, что обнаружен нужный продукт. Смесь разбавляли водой (15 мл) и экстрагировали EtOAc (3*15 мл). Объединенные органические фазы сушили Na2SO4, фильтровали и концентрировали. Остаток очищали посредством препаративной ТСХ (дихлорметан: метанол=20: 1) с получением соединения 716 (113,00 мг, неочищенное) в виде желтого твердого вещества. ЖХМС: 387/389 [M+1].
Получение соединения 766
Смесь соединения 716 (113,00 мг, 292,10 мкмоль, 1,00 экв.) и NaBH4 (22,10 мг, 584,20 мкмоль, 2,00 экв.) в MeOH (4,00 мл) перемешивали при 30°C в течение 1 ч. ЖХМС показала, что обнаружено около 88% нужного соединения. В реакционную смесь добавляли 10 капель воды и перемешивали 10 мин. Реакционную смесь фильтровали. Реакционную смесь очищали посредством препаративной ВЭЖХ (FA) и повторно очищали посредством препаративной ТСХ (дихлорметан: метанол=20: 1, Rf=0,2) с получением соединения 766 в виде белого твердого вещества. ЖХМС: 411/413 [M+23].
Пример 53. Получение соединения 747
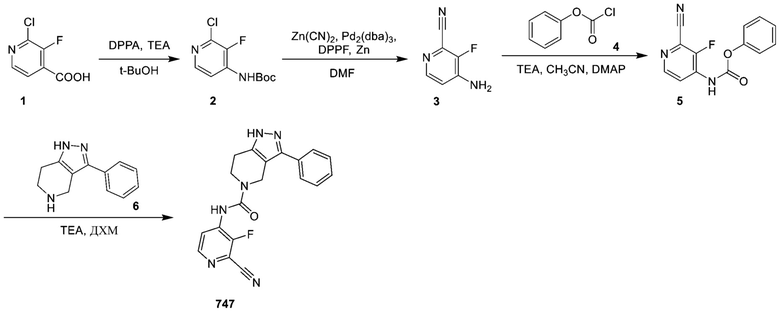
Стадия 1. Получение соединения 2
К смеси 2-хлор-3-фторпиридин-4-карбоновой кислоты (3,00 г, 17,09 ммоль, 1,00 экв.) в t-BuOH (20,00 мл) добавляли DPPA (7,05 г, 25,64 ммоль, 1,50 экв.) и TEA (3,46 г, 34,18 ммоль, 2,00 экв.), реакционную смесь нагревали до 80°C и перемешивали при 80°C в течение 16 часов. При ЖХМС выявили несколько новых пиков и обнаружили 20% нужного соединения. Смесь экстрагировали EA (80 мл*3) и водой (50 мл*2), органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (силикагель 200-300 меш, петролейный эфир/этилацетат=от 20/1 до 5/1) с получением трет-бутил-N-(2-хлор-3-фтор-4-пиридил)карбамата (1,70 г, 5,86 ммоль, выход 34,28%, чистота 85%) в виде желтого масла. ЖХМС: 247/249 [M+1].
Стадия 2. Получение соединения 3
К смеси трет-бутил-N-(2-хлор-3-фтор-4-пиридил)карбамата (1,00 г, 4,05 ммоль, 1,00 экв.) в DMF (8,00 мл) добавляли Zn(CN)2 (952,04 мг, 8,10 ммоль, 514,62 мкл, 2,00 экв.), Zn (52,97 мг, 810,00 мкмоль, 0,20 экв.), DPPF (898,98 мг, 1,62 ммоль, 0,40 экв.) и Pd2(dba)3 (742,47 мг, 810,00 мкмоль, 0,20 экв.) и реакционную смесь перемешивали при 120°C в течение 16 часов в атмосфере N2. ТСХ показала, что исходный материал полностью израсходован и образовалось множество новых зон. Смесь экстрагировали EA (20 мл*3) и водой (10 мл*2), органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (силикагель 200-300 меш, петролейный эфир/этилацетат=от 10/1 до 1/1) с получением 4-амино-3-фторпиридин-2-карбонитрила (75,00 мг, 547,01 мкмоль, выход 13,51%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,96-7,97 (д, J=5,52 Гц, 1 H), 6,91-6,94 (дд, J=7,53, 5,40 Гц, 1 H), 6,86 (уш. с, 2 H). ЖХМС: 138 [M+1].
Стадия 3. Получение соединения 5
К смеси 4-амино-3-фторпиридин-2-карбонитрила (25,00 мг, 182,34 мкмоль, 1,00 экв.) в CH3CN (2,00 мл) добавляли TEA (36,90 мг, 364,68 мкмоль, 50,55 мкл, 2,00 экв.), соединение 4 (28,55 мг, 1,00 экв.) и DMAP (2,23 мг, 18,23 мкмоль, 0,10 экв.), реакционную смесь перемешивали при 30°C в течение 16 часов. ТСХ показала, что соединение 3 полностью израсходовано и обнаружена одна основная новая зона с меньшей полярностью. Смесь экстрагировали EA (10 мл*3) и водой (10 мл), органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Выполняли дополнительную очистку посредством препаративной ТСХ с получением фенил-N-(2-циано-3-фтор-4-пиридил)карбамата (23,00 мг, 74,22 мкмоль, выход 40,70%, чистота 83%) в виде белого твердого вещества. ЖХМС: 258 [M+1].
Получение соединения 747
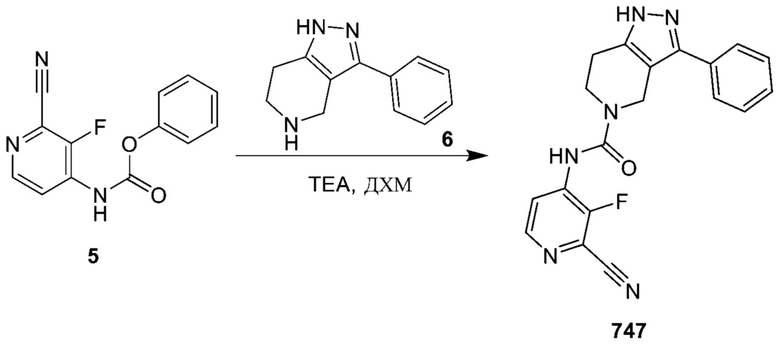
К раствору фенил-N-(2-циано-3-фтор-4-пиридил)карбамата (20,00 мг, 77,75 мкмоль, 1,00 экв.) в ДХМ (2,00 мл) добавляли TEA (23,60 мг, 233,25 мкмоль, 32,33 мкл, 3,00 экв.) с последующим добавлением 3-фенил-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридина (18,33 мг, 77,75 мкмоль, 1,00 экв.), реакционную смесь перемешивали при 25°C в течение 2 часов. ЖХМС показала, что соединение 5 полностью израсходовано и обнаружен один основной пик с нужным масс-спектром. Смесь экстрагировали ДХМ (10 мл*3) и водой (10 мл), органическую фазу сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Выполняли дополнительную очистку посредством препаративной ВЭЖХ (FA) с получением соединения 747 (8,00 мг, 21,92 мкмоль, выход 28,19%, чистота 99,3%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,31-8,32 (д, J=5,40 Гц, 1 H), 7,95-7,98 (т, J=6,02 Гц, 1 H), 7,61-7,63 (д, J=7,28 Гц, 2 H), 7,45-7,48 (т, J=6,90 Гц, 2 H), 7,34-7,36 (д, J=7,03 Гц, 1 H), 4,76 (с, 2 H), 3,79-3,82 (т, J=5,65 Гц, 2 H), 2,78 (уш. с, 2 H). ЖХМС: 363 [M+1].
Пример 54. Получение соединения 756
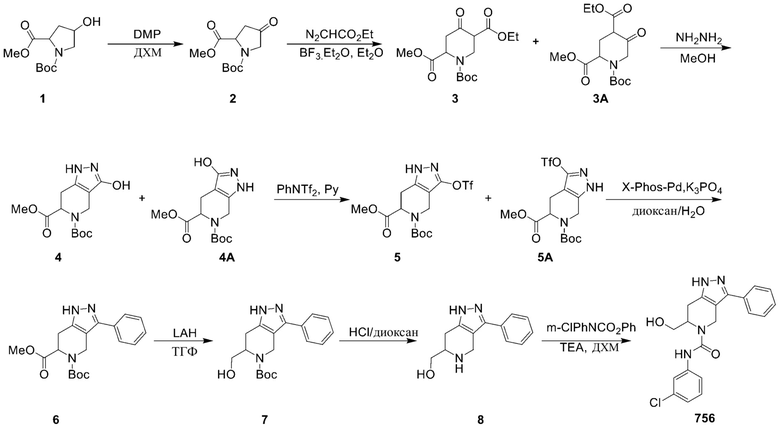 Стадия 1. Получение соединения 2
Стадия 1. Получение соединения 2
К смеси 1-трет-бутил-2-метил-4-гидроксипирролидин-1,2-дикарбоксилата (4,00 г, 16,31 ммоль, 1,00 экв.) в ДХМ (20,00 мл) добавляли реагент Десса-Мартина (8,30 г, 19,57 ммоль, 1,20 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 20°C в течение 5 часов. ТСХ (петролейный эфир: этилацетат=3: 1) показала завершение реакции. Реакцию гасили медленным добавлением Na2S2O3 и впоследствии экстрагировали ДХМ (50 мл*2). Объединенную органическую фазу промывали солевым раствором (40 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=10/1) с получением 1-трет-бутил-2-метил-4-оксопирролидин-1,2-дикарбоксилата (3,70 г, 14,60 ммоль, выход 89,53%, чистота 96%) в виде желтого масла. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 4,66-4,89 (м, 1 H), 3,85-3,97 (м, 2 H), 3,77 (с, 3 H), 2,85-3,06 (м, 1 H), 2,53-2,66 (м, 1 H), 1,48 (д, J=7,78 Гц, 9 H). ЖХМС: 244 [M+1].
Стадия 2. Получение соединений 3 и 3A
К смеси 1-трет-бутил-2-метил-4-оксопирролидин-1,2-дикарбоксилата (1,00 г, 4,11 ммоль, 1,00 экв.) и этил-2-диазоацетата (1,41 г, 12,33 ммоль, 1,29 мл, 3,00 экв.) в Et2O (10,00 мл) по каплям добавляли BF3·Et2O (1,75 г, 12,33 ммоль, 1,52 мл, 3,00 экв.) при -45°C в атмосфере N2. Смесь перемешивали при -45°C в течение 30 мин, впоследствии нагревали до 20°C и перемешивали в течение 10 часов. ТСХ (петролейный эфир: этилацетат=3: 1) показала, что нужный продукт обнаружен. Смесь вливали в насыщенный раствор NaHCO3 (20 мл) и перемешивали в течение 3 мин. Водную фазу экстрагировали этилацетатом (20 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=15/1) с получением смеси 1-трет-бутил-4-этил-2-метил-5-оксопиперидин-1,2,4-трикарбоксилата и 1-трет-бутил-5-этил-2-метил-4-оксопиперидин-1,2,5-трикарбоксилата (400,00 мг, 1,21 ммоль, выход 29,44%) в виде желтого масла. ЖХМС: 330 [M+1].
Стадия 3. Получение соединений 4 и 4A
К смеси 1-трет-бутил-4-этил-2-метил-5-оксопиперидин-1,2,4-трикарбоксилата и 1-трет-бутил-5-этил-2-метил-4-оксопиперидин-1,2,5-трикарбоксилата (670,00 мг, 2,03 ммоль, 1,00 экв.) в MeOH (10,00 мл) добавляли N2H4·H2O (131,79 мг, 2,24 ммоль, 127,96 мкл, чистота 85%, 1,10 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь концентрировали в вакууме с получением смеси 6-трет-бутил-5-метил-3-гидрокси-1,4,5,7-тетрагидропиразол[3,4-c]пиридин-5,6-дикарбоксилата и 5-трет-бутил-6-метил-3-гидрокси-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5,6-дикарбоксилата (600,00 мг, 2,02 ммоль, выход 99,41%) в виде желтого твердого вещества. ЖХМС: 298 [M+1].
Стадия 4. Получение соединений 5 и 5A
К смеси 6-трет-бутил-5-метил-3-гидрокси-1,4,5,7-тетрагидропиразол
[3,4-c]пиридин-5,6-дикарбоксилата и 5-трет-бутил-6-метил-3-гидрокси-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5,6-дикарбоксилата (600,00 мг, 2,02 ммоль, 1,00 экв.) в Py (10,00 мл) добавляли 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамид (1,08 г, 3,03 ммоль, 1,50 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ТСХ (петролейный эфир: этилацетат=3: 1) показала завершение реакции. Смесь концентрировали в вакууме. Остаток вливали в 1 Н HCl (30 мл) и перемешивали в течение 1 мин. Водную фазу экстрагировали этилацетатом (20 мл*2). Объединенную органическую фазу промывали солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=15/1, 10/1) с получением 5-трет-бутил-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5,6-дикарбоксилата (120,00 мг, 279,48 мкмоль, выход 13,84%) в виде желтого твердого вещества и 6-трет-бутил-5-метил-3-(трифторметилсульфонилокси)-1,4,5,7-тетрагидропиразол[3,4-c]пиридин-5,6-дикарбоксилата (240,00 мг, 558,96 мкмоль, выход 27,67%) в виде желтого твердого вещества.
ЖХМС: 430 [M+1].
Стадия 5. Получение соединения 6
К смеси 5-трет-бутил-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5,6-дикарбоксилата (100,00 мг, 232,90 мкмоль, 1,00 экв.) и фенилбороновой кислоты (42,60 мг, 349,35 мкмоль, 1,50 экв.) в диоксане (5,00 мл) и H2O (500,00 мкл) добавляли XPHOS-PD-G2 (18,32 мг, 23,29 мкмоль, 0,10 экв.), K3PO4 (98,88 мг, 465,80 мкмоль, 2,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 10 часов. ТСХ (этилацетат: петролейный эфир=2: 1) показала, что реакция завершена и нужный продукт обнаружен. Смесь вливали в воду (30 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (30 мл*2). Объединенную органическую фазу промывали солевым раствором (30 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=5/1) с получением 5-трет-бутил-6-метил-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5,6-дикарбоксилата (40,00 мг, 105,20 мкмоль, выход 45,17%, чистота 94%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,51-7,62 (м, 2 H), 7,42-7,50 (м, 2 H), 7,33-7,42 (м, 1 H), 5,27-5,43 (м, 1 H), 4,47-4,55 (м, 1 H), 4,36-4,44 (м, 1 H), 4,06-4,14 (м, 1 H), 3,65-3,71 (м, 3 H), 3,37 (д, J=4,39 Гц, 1 H), 2,99-3,14 (м, 1 H), 1,51 (д, J=6,15 Гц, 9 H). ЖХМС: 358 [M+1].
Стадия 6. Получение соединения 7
К смеси 5-трет-бутил-6-метил-3-фенил-1,4,6,7-
тетрагидропиразол[4,3-c]пиридин-5,6-дикарбоксилата (40,00 мг, 111,92 мкмоль, 1,00 экв.) в ТГФ (5,00 мл) добавляли LiAlH4 (21,24 мг, 559,60 мкмоль, 5,00 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 5 часов. ЖХМС и ТСХ (этилацетат: петролейный эфир=3: 1) показали завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (петролейный эфир/этилацетат=1/4) с получением трет-бутил-6-(гидроксиметил)-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (20,00 мг, 60,72 мкмоль, выход 54,25%) в виде желтого твердого вещества. ЖХМС: 330 [M+1].
Стадия 7. Получение соединения 8
К смеси трет-бутил-6-(гидроксиметил)-3-фенил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (10,00 мг, 30,36 мкмоль, 1,00 экв.) в диоксане (2,00 мл) добавляли HCl/диоксан (4 М, 4,00 мл, 527,01 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 2 часов. ТСХ (этилацетат: петролейный эфир=3: 1) показала завершение реакции. Смесь концентрировали с получением (3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин-6-ил)метанола (8,07 мг, 30,37 мкмоль, выход 100,00%, HCl) в виде желтого твердого вещества.
Получение соединения 756
К смеси (3-фенил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин-6-ил)метанола (8,07 мг, 30,37 мкмоль, 1,00 экв., HCl) и фенил-N-(3-хлорфенил)карбамата (7,52 мг, 30,37 мкмоль, 1,00 экв.) в ДХМ (5,00 мл) добавляли TEA (7,68 мг, 75,92 мкмоль, 10,52 мкл, 2,50 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-6-(гидроксиметил)-3-фенил-1,4,6,7-
тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (9,33 мг, 24,25 мкмоль, выход 79,84%, чистота 99,5%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,62-7,70 (м, 1 H), 7,42-7,56 (м, 2 H), 7,27-7,41 (м, 2 H), 7,19-7,26 (м, 1 H), 6,98-7,03 (м, 1 H), 5,05 (д, J=15,31 Гц, 2 H), 4,47 (д, J=15,43 Гц, 1 H), 3,54-3,69 (м, 2 H), 2,99-3,08 (м, 1 H), 2,86-2,94 (м, 1 H). ЖХМС: 383/385 [M+1].
Пример 55. Получение соединения 754
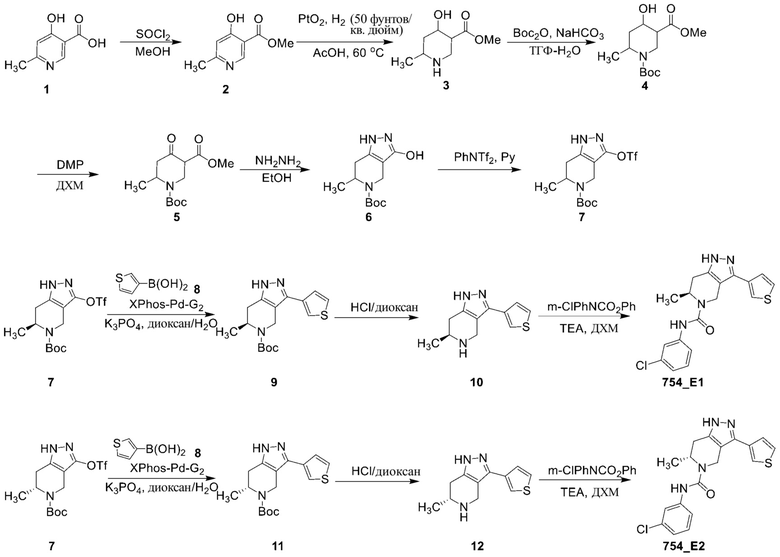
Стадия 1. Получение соединения 2
К смеси 4-гидрокси-6-метилпиридин-3-карбоновой кислоты (5,00 г, 32,65 ммоль, 1,00 экв.) в MeOH (60,00 мл) добавляли SOCl2 (23,31 г, 195,90 ммоль, 6,00 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 30 мин, впоследствии нагревали до 80°C и перемешивали в течение 10 часов. ТСХ (дихлорметан: метанол=5: 1) показала завершение реакции. Смесь охлаждали до 20°C и концентрировали в вакууме с получением метил-4-гидрокси-6-метилпиридин-3-карбоксилата (5,50 г, неочищенный, HCl) в виде желтого твердого вещества. ЖХМС: 168 [M+1].
Стадия 2. Получение соединения 3
К раствору метил-4-гидрокси-6-метилпиридин-3-карбоксилата (8,00 г, 39,29 ммоль, 1,00 экв., HCl) в AcOH (50,00 мл) добавляли PtO2 (1,34 г, 5,89 ммоль, 0,15 экв.) в атмосфере N2. Суспензию дегазировали в вакууме и несколько раз продували H2. Смесь перемешивали в атмосфере H2 (345 кПа (50 фунтов/кв. дюйм)) при 60°C в течение 24 часов. ЖХМС показала, что обнаружено нужное соединение. Реакционную смесь фильтровали и фильтрат концентрировали с получением метил-4-гидрокси-6-метилпиперидин-3-карбоксилата (9,50 г, неочищенный, соль AcOH) в виде желтого масла. ЖХМС: 174 [M+1].
Стадия 3. Получение соединения 4
К смеси метил-4-гидрокси-6-метилпиперидин-3-карбоксилата (3,00 г, 12,86 ммоль, 1,00 экв., HOAC) и NaHCO3 (1,62 г, 19,29 ммоль, 750,00 мкл, 1,50 экв.) в ТГФ (10,00 мл) и H2O (10,00 мл) добавляли Boc2O (3,37 г, 15,43 ммоль, 3,55 мл, 1,20 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 25°C в течение 5 часов. ЖХМС и ТСХ (петролейный эфир: этилацетат=3: 1) показали завершение реакции. Смесь экстрагировали этилацетатом (30 мл*2). Объединенную органическую фазу промывали солевым раствором (20 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=15/1) с получением 1-трет-бутил-3-метил-4-гидрокси-6-метилпиперидин-1,3-дикарбоксилата (1,50 г, 5,49 ммоль, выход 42,68%) в виде желтого масла. ЖХМС: 274 [M+1].
Стадия 4. Получение соединения 5
К смеси 1-трет-бутил-3-метил-4-гидрокси-6-метилпиперидин-1,3-дикарбоксилата (1,50 г, 5,49 ммоль, 1,00 экв.) в ДХМ (50,00 мл) добавляли реагент Десса-Мартина (2,79 г, 6,59 ммоль, 2,04 мл, 1,20 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 5 часов. ТСХ (петролейный эфир: этилацетат=5: 1) показала завершение реакции. Реакцию гасили медленным добавлением Na2S2O3, а впоследствии выполняли экстракцию ДХМ (30 мл*2). Объединенную органическую фазу промывали солевым раствором (30 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=20/1) с получением 1-трет-бутил-3-метил-6-метил-4-оксопиперидин-1,3-дикарбоксилата (1,00 г, 3,69 ммоль, выход 67,14%) в виде желтого масла. 1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ 11,99 (с, 1 H), 4,53-4,69 (м, 1 H), 4,46 (д, J=16,77 Гц, 1 H), 3,80 (с, 3 H), 3,54-3,72 (м, 1 H), 2,60-2,78 (м, 1 H), 2,26-2,46 (м, 1 H), 1,44-1,52 (м, 11 H), 1,15 (д, J=6,78 Гц, 3 H). ЖХМС: 272 [M+1].
Стадия 5. Получение соединения 6
К смеси 1-трет-бутил-3-метил-6-метил-4-оксопиперидин-1,3-дикарбоксилата (2,50 г, 9,21 ммоль, 1,00 экв.) в MeOH (10,00 мл) добавляли N2H4-H2O (705,14 мг, 11,97 ммоль, 684,60 мкл, чистота 85%, 1,30 экв.) за один раз в атмосфере N2. Смесь перемешивали при 75°C в течение 2 часов. ТСХ (петролейный эфир: этилацетат=4: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением трет-бутил-3-гидрокси-6-метил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (2,40 г, неочищенный) в виде желтого твердого вещества, которое разделяли посредством SFC (колонка: IC (250 мм*30 мм, 10 мкм), условие: основание-MeOH) с получением двух энантиомеров (E1: 1,0 г; E2: 1,1 г). 1H ЯМР (300 МГц, МЕТАНОЛ-d4) δ 4,72-4,81 (м, 1 H), 4,58-4,68 (м, 1 H), 3,75-3,88 (м, 1 H), 2,75-2,87 (м, 1 H), 2,37-2,49 (м, 1 H), 1,48 (д, J=1,13 Гц, 10 H), 1,13 (д, J=6,78 Гц, 3 H). ЖХМС: 254 [M+1].
Получение соединения 7(S)
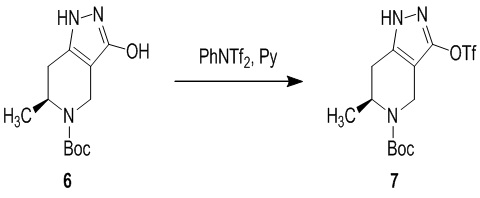
К смеси трет-бутил-3-гидрокси-6-метил-1,4,6,7-тетрагидропиразол
[4,3-c]пиридин-5-карбоксилата (1,00 г, 3,95 ммоль, 1,00 экв.) в Py (15,00 мл) добавляли 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамид (1,98 г, 5,53 ммоль, 1,40 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ТСХ показала завершение реакции. Смесь концентрировали в вакууме. Остаток разбавляли этилацетатом (50 мл), и вливали в 0,5 Н HCl (20 мл), и перемешивали в течение 1 мин. Водную фазу экстрагировали этилацетатом (50 мл*2). Объединенную органическую фазу промывали солевым раствором (40 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=10/1) с получением трет-бутил-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (850,00 мг, 2,21 ммоль, выход 55,84%) в виде желтого твердого вещества. ЖХМС: 386 [M+1].
Получение соединения 7(R)
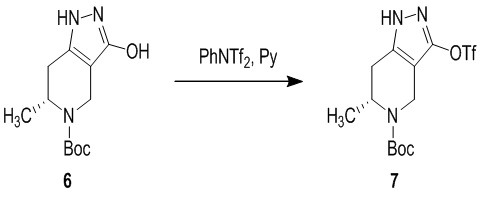
К смеси трет-бутил-3-гидрокси-6-метил-1,4,6,7-
тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (1,10 г, 4,34 ммоль, 1,00 экв.) в Py (15,00 мл) добавляли 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)
метансульфонамид (2,17 г, 6,08 ммоль, 1,40 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ТСХ показала завершение реакции. Смесь концентрировали в вакууме. Остаток разбавляли этилацетатом (60 мл), и вливали в 0,5 Н HCl (20 мл), и перемешивали в течение 1 мин. Водную фазу экстрагировали этилацетатом (50 мл*2). Объединенную органическую фазу промывали солевым раствором (50 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=10/1) с получением трет-бутил-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (920,00 мг, 2,39 ммоль, выход 55,01%) в виде желтого твердого вещества. ЖХМС: 386 [M+1].
Получение соединения 9
К смеси трет-бутил-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (60,00 мг, 155,70 мкмоль, 1,00 экв.) и 3-тиенилбороновой кислоты (29,88 мг, 233,55 мкмоль, 1,50 экв.) в диоксане (2,00 мл) и H2O (200,00 мкл) добавляли [2-(2-аминофенил)фенил]-хлорпалладий, дициклогексил-[3-(2,4,6-триизопропилфенил)фенил]фосфан (12,25 мг, 15,57 мкмоль, 0,10 экв.) и K3PO4 (99,15 мг, 467,10 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 12 часов. ТСХ (петролейный эфир: этилацетат=1: 1) показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (петролейный эфир/этилацетат=1/1) с получением трет-бутил-6-метил-3-(3-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (43,80 мг, 137,12 мкмоль, выход 88,07%, чистота 100%) в виде желтого твердого вещества. ЖХМС: 320 [M+1].
Получение соединения 10
К смеси трет-бутил-6-метил-3-(3-тиенил)-1,4,6,7-тетрагидропиразол
[4,3-c]пиридин-5-карбоксилата (43,80 мг, 137,12 мкмоль, 1,00 экв.) в диоксане (3,00 мл) добавляли HCl/диоксан (4 М, 8,00 мл, 233,37 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 2 часов. ТСХ (петролейный эфир: этилацетат=1: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением 6-метил-3-(3-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (35,07 мг, 137,12 мкмоль, выход 100,00%, HCl) в виде желтого твердого вещества.
Получение соединения 754 (E1)
К смеси 6-метил-3-(3-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (35,07 мг, 137,12 мкмоль, 1,00 экв., HCl) и фенил-N-(3-хлорфенил)карбамата (33,96 мг, 137,12 мкмоль, 1,00 экв.) в ДХМ (5,00 мл) добавляли TEA (41,62 мг, 411,35 мкмоль, 57,02 мкл, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-6-метил-3-(3-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (23,00 мг, 60,39 мкмоль, выход 44,04%, чистота 97,9%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,65-7,70 (м, 1 H), 7,51-7,57 (м, 3 H), 7,29-7,34 (м, 1 H), 7,20-7,28 (м, 1 H), 6,99-7,05 (м, 1 H), 4,99-5,06 (м, 1 H), 4,42 (д, J=15,18 Гц, 1 H), 3,05 (с, 1 H), 2,65-2,74 (м, 1 H), 1,23 (д, J=6,90 Гц, 3 H). ЖХМС: 373/375 [M+1].
Получение соединения 11
К смеси трет-бутил-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (60,00 мг, 155,70 мкмоль, 1,00 экв.) и 3-тиенилбороновой кислоты (29,88 мг, 233,55 мкмоль, 1,50 экв.) в диоксане (2,00 мл) и H2O (200,00 мкл) добавляли [2-(2-аминофенил)фенил]-хлорпалладий, дициклогексил-[3-(2,4,6-триизопропилфенил)фенил]фосфан (12,25 мг, 15,57 мкмоль, 0,10 экв.) и K3PO4 (99,15 мг, 467,10 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 12 часов. ТСХ (петролейный эфир: этилацетат=1: 1) показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (петролейный эфир/этилацетат=1/1) с получением трет-бутил-6-метил-3-(3-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (42,20 мг, 132,11 мкмоль, выход 84,85%, чистота 100%) в виде желтого твердого вещества. ЖХМС: 320 [M+1].
Получение соединения 12
К смеси трет-бутил-6-метил-3-(3-тиенил)-1,4,6,7-тетрагидропиразол
[4,3-c]пиридин-5-карбоксилата (42,00 мг, 131,49 мкмоль, 1,00 экв.) в диоксане (3,00 мл) добавляли HCl/диоксан (4 М, 8,00 мл, 243,36 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 2 часов. ТСХ (петролейный эфир: этилацетат=1: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением 6-метил-3-(3-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (33,63 мг, 131,49 мкмоль, выход 100,00%, HCl) в виде желтого твердого вещества.
Получение соединения 754 (E2)
К смеси 6-метил-3-(3-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (33,63 мг, 131,49 мкмоль, 1,00 экв., HCl) и фенил-N-(3-хлорфенил)карбамата (32,57 мг, 131,49 мкмоль, 1,00 экв.) в ДХМ (5,00 мл) добавляли TEA (39,91 мг, 394,46 мкмоль, 54,68 мкл, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водный слой экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-6-метил-3-(3-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (25,00 мг, 66,58 мкмоль, выход 50,63%, чистота 99,3%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,65-7,70 (м, 1 H), 7,51-7,57 (м, 3 H), 7,29-7,34 (м, 1 H), 7,20-7,28 (м, 1 H), 6,99-7,05 (м, 1 H), 4,99-5,06 (м, 1 H), 4,42 (д, J=15,18 Гц, 1 H), 3,05 (с, 1 H), 2,65-2,74 (м, 1 H), 1,23 (д, J=6,90 Гц, 3 H). ЖХМС: 373/375 [M+1].
Пример 56. Получение соединений 753, 819, 820, 821, 822, 823, 824, 825, 826, 851, 852, 853, 854, 856 и 857
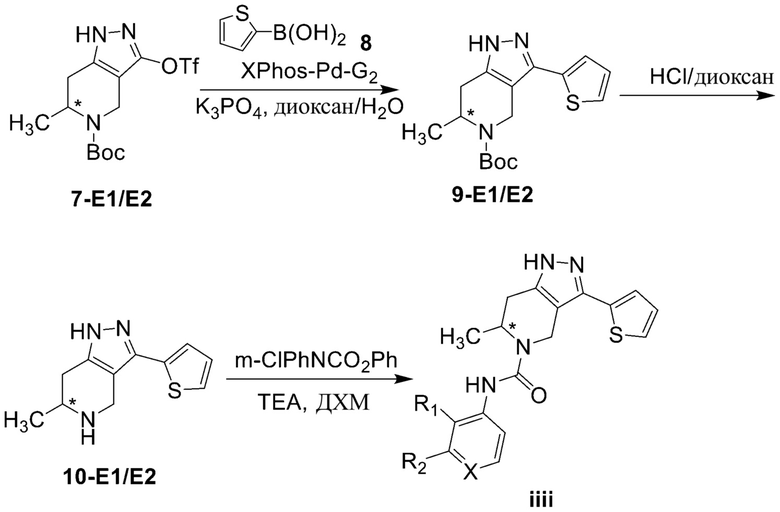
Стадия 1. Получение соединения 9
К смеси трет-бутил-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (70,00 мг, 181,65 мкмоль, 1,00 экв.) и 2-тиенилбороновой кислоты (34,87 мг, 272,47 мкмоль, 1,50 экв.) в диоксане (1,00 мл) и H2O (100,00 мкл) добавляли [2-(2-аминофенил)фенил]-хлорпалладий, дициклогексил-[3-(2,4,6-триизопропилфенил)фенил]фосфан (14,29 мг, 18,16 мкмоль, 0,10 экв.) и K3PO4 (115,68 мг, 544,94 мкмоль, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 110°C в течение 12 часов. ТСХ (петролейный эфир: этилацетат=1: 1) показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (петролейный эфир/этилацетат=1/1) с получением трет-бутил-6-метил-3-(2-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (10,00 мг, 31,31 мкмоль, выход 17,23%, чистота 100%) в виде желтого твердого вещества. ЖХМС: 320 [M+1].
Стадия 2. Получение соединения 10
К смеси трет-бутил-6-метил-3-(2-тиенил)-1,4,6,7-тетрагидропиразол
[4,3-c]пиридин-5-карбоксилата (10,00 мг, 31,31 мкмоль, 1,00 экв.) в диоксане (2,00 мл) добавляли HCl/диоксан (4 М, 5,00 мл, 638,77 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 2 часов. ТСХ (петролейный эфир: этилацетат=1: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением 6-метил-3-(2-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (8,00 мг, 31,28 мкмоль, выход 99,90%, HCl) в виде белого твердого вещества.
Получение соединения 753 (E1)
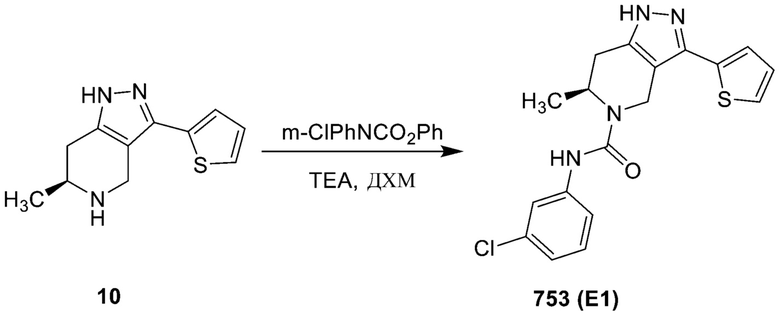
К смеси 6-метил-3-(2-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (8,01 мг, 31,32 мкмоль, 1,00 экв., HCl) и фенил-N-(3-хлорфенил)карбамата (7,76 мг, 31,32 мкмоль, 1,00 экв.) в ДХМ (5,00 мл) добавляли TEA (12,68 мг, 125,28 мкмоль, 17,37 мкл, 4,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 20°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*1), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-6-метил-3-(2-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (7,65 мг, 19,84 мкмоль, выход 63,34%, чистота 96,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,53 (с, 1 H), 7,29-7,46 (м, 3 H), 7,21-7,27 (м, 1 H), 7,12-7,17 (м, 1 H), 7,02 (д, J=8,41 Гц, 1 H), 4,99-5,06 (м, 2 H), 4,33-4,44 (м, 1 H), 3,02-3,13 (м, 1 H), 2,65-2,75 (м, 1 H), 1,22-1,26 (м, 1 H). ЖХМС: 373/375 [M+1].
Соединения 753 (E2), 819, 820, 821-826 и 851-857 получали аналогичным способом.
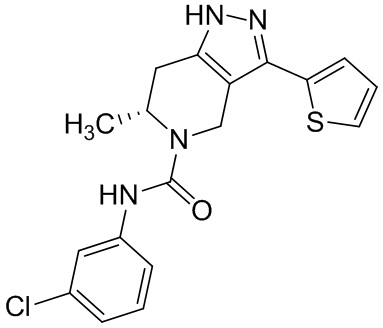
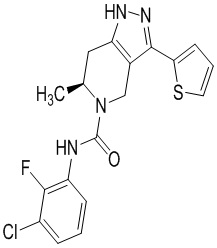
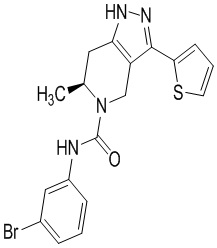
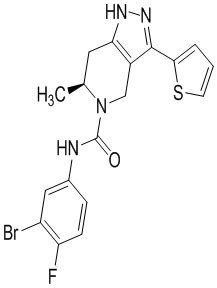
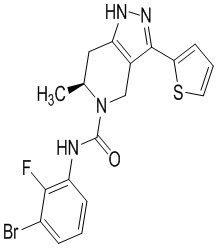
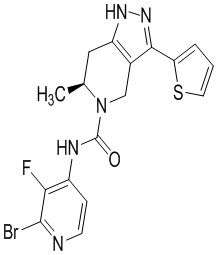
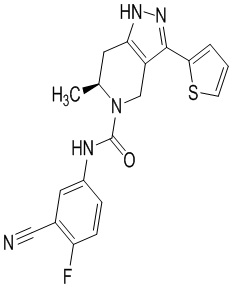
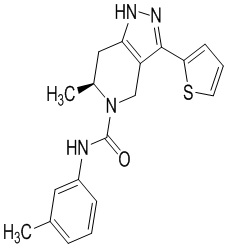
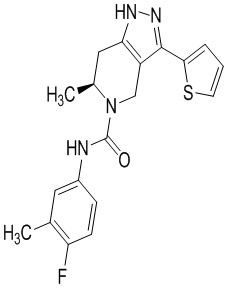
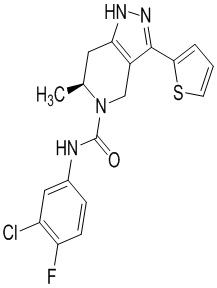
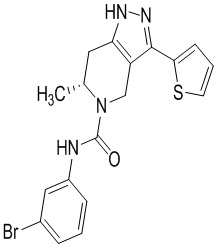
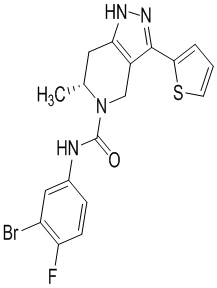
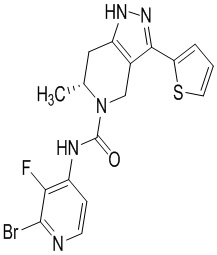
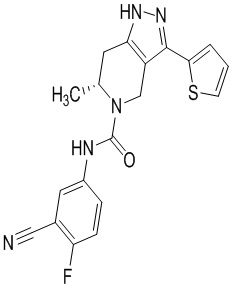
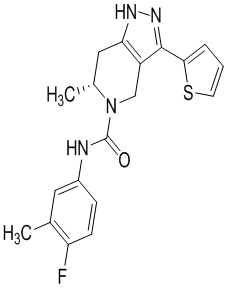
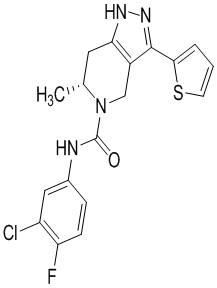
Пример 57. Получение соединения 830
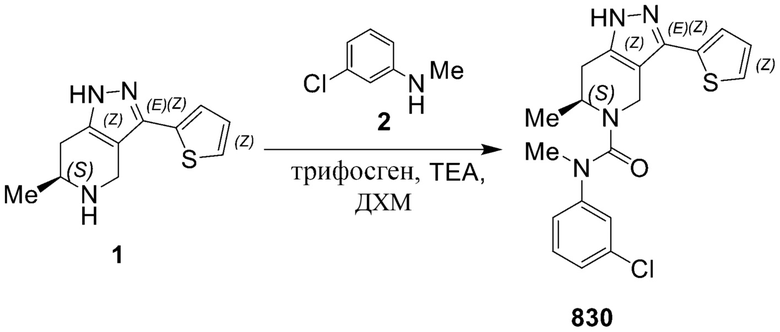
К смеси 3-хлор-N-метиланилина (18,27 мг, 129,02 мкмоль, 15,75 мкл, 1,10 экв.) и TEA (35,61 мг, 351,87 мкмоль, 48,78 мкл, 3,00 экв.) в ДХМ (2,00 мл) добавляли ТРИФОСГЕН (27,85 мг, 93,83 мкмоль, 0,80 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 5 мин. Впоследствии к смеси добавляли (6S)-6-метил-3-(2-тиенил)-4,5,6,7-тетрагидро-1H-пиразол[4,3-c]пиридин (30,00 мг, 117,29 мкмоль, 1,00 экв., HCl), смесь перемешивали при 0°C в течение 30 мин. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением (6S)-N-(3-хлорфенил)-N,6-диметил-3-(2-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (10,55 мг, 26,99 мкмоль, выход 23,01%, чистота 98,99%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,32-7,45 (м, 2 H), 7,27-7,31 (м, 1 H), 7,02-7,24 (м, 4 H), 4,67-4,74 (м, 1 H), 4,50-4,57 (м, 1 H), 3,77-3,85 (м, 1 H), 3,23 (с, 3 H), 2,78 (д, J=5,27 Гц, 1 H), 2,45-2,55 (м, 1 H), 1,07-1,17 (м, 3 H). ЖХМС: 387/389 [M+1].
Пример 58. Получение соединений 831 и 832
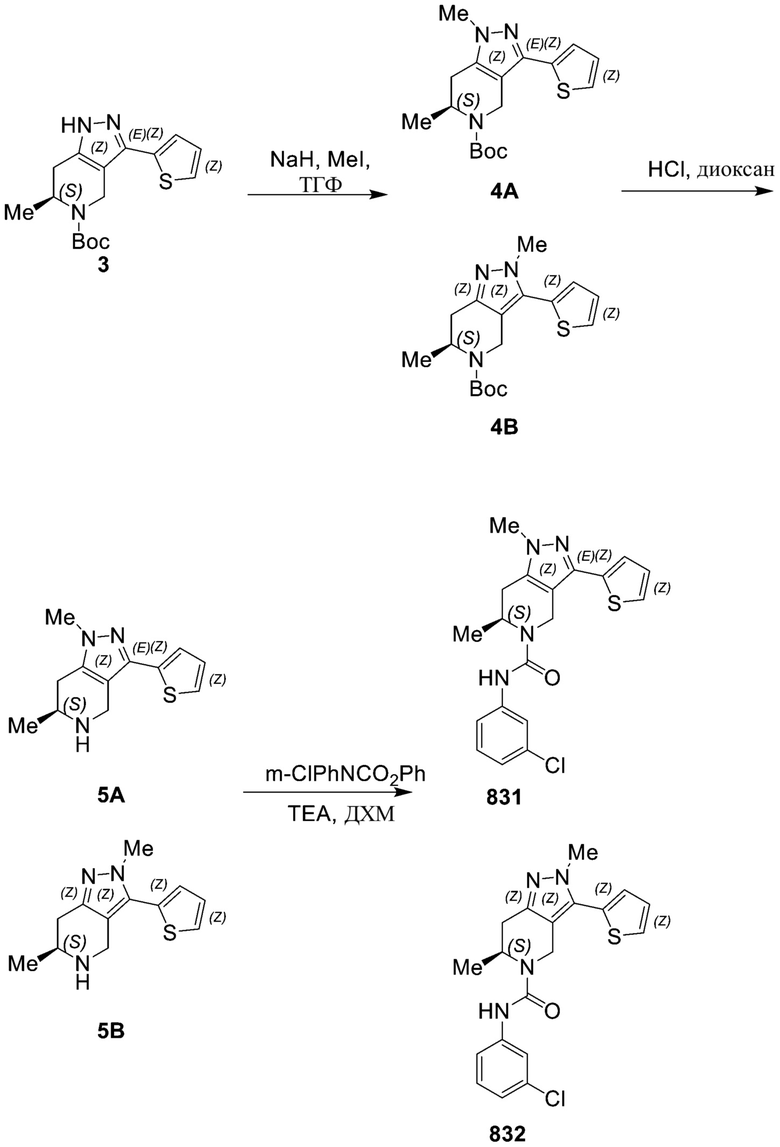
Стадия 1. Получение соединений 4A и 4B
К смеси трет-бутил-(6S)-6-метил-3-(2-тиенил)-1,4,6,7-тетрагидро
пиразол[4,3-c]пиридин-5-карбоксилата (50,00 мг, 156,53 мкмоль, 1,00 экв.) в ТГФ (2,00 мл) добавляли NaH (9,39 мг, 234,80 мкмоль, чистота 60%, 1,50 экв.) за один раз при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 30 мин. Впоследствии к смеси добавляли MeI (66,66 мг, 469,60 мкмоль, 29,23 мкл, 3,00 экв.). Смесь перемешивали при 0°C в течение 1 часа. ТСХ (петролейный эфир: этилацетат=2: 1) показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (петролейный эфир/этилацетат=5/1, 10 раз) с получением трет-бутил-(6S)-1,6-диметил-3-(2-тиенил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилата (20,00 мг, 59,98 мкмоль, выход 38,32%) в виде желтого твердого вещества и трет-бутил-(6S)-2,6-диметил-3-(2-тиенил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилата (15,00 мг, 44,98 мкмоль, выход 28,74%) в виде желтого твердого вещества.
Соединение 4A: 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δδ.26-7,29 (м, 1 H), 7,16-7,21 (м, 1 H), 7,06-7,11 (м, 1 H), 4,76-5,12 (м, 2 H), 4,08-4,22 (м, 1 H), 3,78 (с, 3 H), 2,88-3,01 (м, 1 H), 2,39-2,53 (м, 1 H), 1,51 (с, 9 H), 1,18 (д, J=6,90 Гц, 3 H). ЖХМС: 334 [M+1].
Соединение 4B: 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δδ.43-7,48 (м, 1 H), 7,14-7,18 (м, 1 H), 7,13 (с, 1 H), 4,72-4,99 (м, 2 H), 3,99-4,09 (м, 1 H), 3,93 (с, 3 H), 2,91-3,03 (м, 1 H), 2,52-2,65 (м, 1 H), 1,48 (с, 9 H), 1,16 (д, J=7,03 Гц, 3 H). ЖХМС: 334 [M+1].
Стадия 2А. Получение соединения 5A
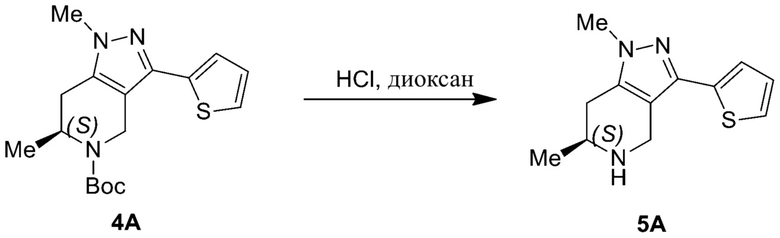
К смеси трет-бутил-(6S)-1,6-диметил-3-(2-тиенил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (20,00 мг, 59,98 мкмоль, 1,00 экв.) в диоксане (2,00 мл) добавляли HCl/диоксан (4 М, 729,81 мл, 48,67 экв.) за один раз в атмосфере N2. Смесь перемешивали при 30°C в течение 2 часов. ТСХ (петролейный эфир: этилацетат=3: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением (6S)-1,6-диметил-3-(2-тиенил)-4,5,6,7-тетрагидропиразол[4,3-c]пиридина (16,18 мг, 59,97 мкмоль, выход 100,00%, HCl) в виде белого твердого вещества.
Получение соединения 831
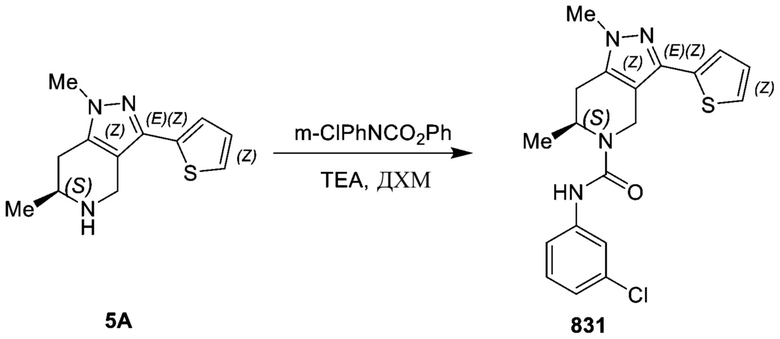
К смеси (6S)-1,6-диметил-3-(2-тиенил)-4,5,6,7-тетрагидропиразол[4,3-c]пиридина (16,18 мг, 59,97 мкмоль, 1,00 экв., HCl) и фенил-N-(3-хлорфенил)карбамата (14,85 мг, 59,97 мкмоль, 1,00 экв.) в ДХМ (5,00 мл) добавляли TEA (18,21 мг, 179,92 мкмоль, 24,94 мкл, 3,00 экв.) за один раз в атмосфере N2. Смесь перемешивали при 30°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 2 мин. Водную фазу экстрагировали ДХМ (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением (6S)-N-(3-хлорфенил)-1,6-диметил-3-(2-тиенил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксамида (10,00 мг, 25,56 мкмоль, выход 42,62%, чистота 98,9%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,51-7,54 (м, 1 H), 7,36-7,39 (м, 1 H), 7,27-7,34 (м, 2 H), 7,20-7,27 (м, 1 H), 7,09-7,14 (м, 1 H), 6,99-7,04 (м, 1 H), 4,98 (с, 2 H), 4,31-4,38 (м, 1 H), 3,78 (с, 3 H), 2,97-3,06 (м, 1 H), 2,67-2,76 (м, 1 H), 1,25 (д, J=6,90 Гц, 3 H). ЖХМС: 387/389 [M+1].
Стадия 2B. Получение соединения 5B
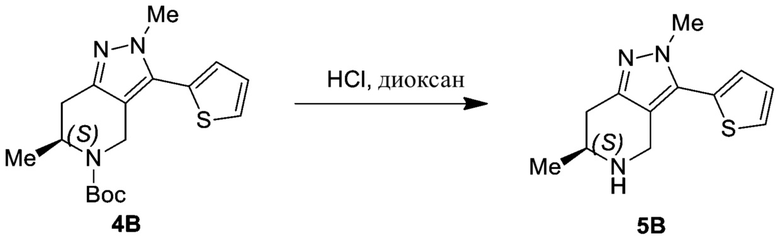
К смеси трет-бутил-(6S)-2,6-диметил-3-(2-тиенил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (15,00 мг, 44,98 мкмоль, 1,00 экв.) в диоксане (1,00 мл) добавляли HCl/диоксан (4 М, 4,00 мл, 355,71 экв.) за один раз при 30°C в атмосфере N2. Смесь перемешивали при 30°C в течение 2 часов. ТСХ (петролейный эфир: этилацетат=3: 1) показала завершение реакции. Смесь концентрировали в вакууме с получением (6S)-2,6-диметил-3-(2-тиенил)-4,5,6,7-тетрагидропиразол[4,3-c]пиридина (12,14 мг, 45,00 мкмоль, выход 100,00%, HCl) в виде желтого твердого вещества.
Получение соединения 832
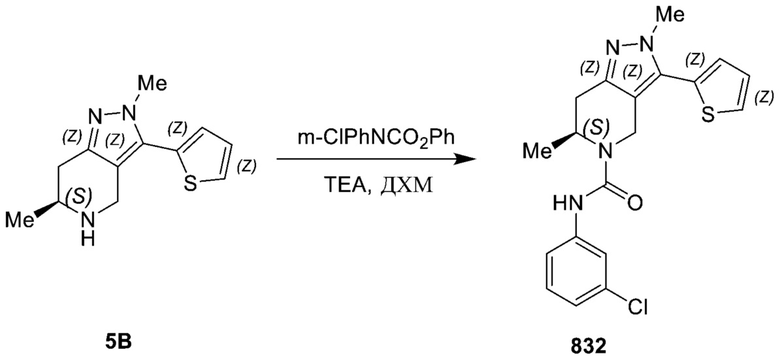
К смеси (6S)-2,6-диметил-3-(2-тиенил)-4,5,6,7-тетрагидропиразол[4,3-c]пиридина (12,14 мг, 45,00 мкмоль, 1,00 экв., HCl) и фенил-N-(3-хлорфенил)карбамата (11,15 мг, 45,00 мкмоль, 1,00 экв.) в ДХМ (3,00 мл) добавляли TEA (13,66 мг, 134,99 мкмоль, 18,71 мкл, 3,00 экв.) за один раз при 30°C в атмосфере N2. Смесь перемешивали при 30°C в течение 12 часов. ЖХМС показала завершение реакции. Смесь вливали в воду (10 мл) и перемешивали в течение 3 мин. Водную фазу экстрагировали этилацетатом (10 мл*2). Объединенную органическую фазу промывали солевым раствором (10 мл*2), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением (6S)-N-(3-хлорфенил)-2,6-диметил-3-(2-тиенил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксамида (16,38 мг, 42,34 мкмоль, выход 94,08%, чистота 100%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,64-7,69 (м, 1 H), 7,48-7,51 (м, 1 H), 7,30-7,34 (м, 1 H), 7,18-7,29 (м, 1 H), 6,97-7,03 (м, 1 H), 4,93 (д, J=6,15 Гц, 2 H), 4,22-4,30 (м, 1 H), 3,91 (с, 3 H), 2,98-3,07 (м, 1 H), 2,60-2,69 (м, 1 H), 1,23 (д, J=6,78 Гц, 3 H). ЖХМС: 387/389 [M+1].
Пример 59. Получение соединения 753 (S)
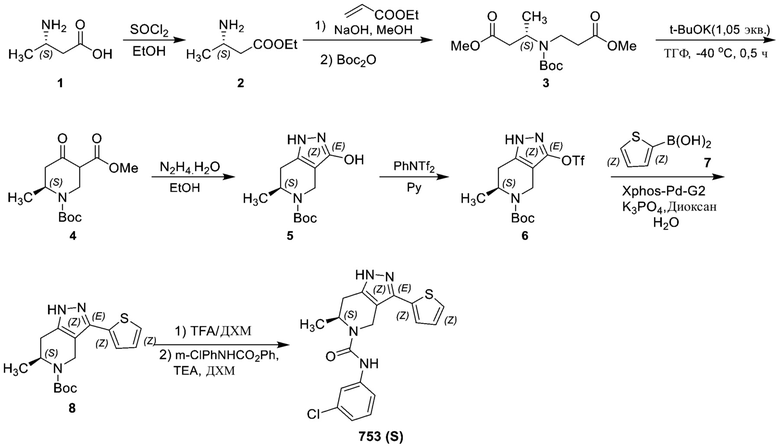
Стадия 1. Получение соединения 2
К раствору (3S)-3-аминобутановой кислоты (13,00 г, 93,14 ммоль, 1,00 экв., HCl) в EtOH (130,00 мл) добавляли SOCl2 (16,62 г, 139,70 ммоль, 10,13 мл, 1,50 экв.). Смесь нагревали при 80°C в течение 3 ч. Смесь концентрировали в вакууме с получением этил-(3S)-3-аминобутаноата (15,60 г, 93,06 ммоль, выход 99,92%, HCl) в виде коричневого масла.
Стадия 2. Получение соединения 3
К раствору этил-(3S)-3-аминобутаноата (15,60 г, 93,06 ммоль, 1,00 экв., HCl) в MeOH (150,00 мл) добавляли NaOH (4,47 г, 111,67 ммоль, 1,20 экв.) с последующим добавлением этилпроп-2-еноата (10,25 г, 102,37 ммоль, 11,14 мл, 1,10 экв.). Смесь нагревали при 80°C в течение 16 ч. Смесь охлаждали до 0°C и добавляли Boc2O (20,31 г, 93,06 ммоль, 21,38 мл, 1,00 экв.) с последующим добавлением TEA (9,42 г, 93,06 ммоль, 12,90 мл, 1,00 экв.). Смесь перемешивали при температуре 25°C в течение 2 ч. Смесь концентрировали в вакууме. Остаток экстрагировали ДХМ (100 мл*2) и H2O (100 мл). Объединенный органический слой промывали 1 Н HCl (100 мл), сушили Na2SO4, фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали посредством флэш-хроматографии (PE: EA=0%~15%) с получением метил-(3S)-3-[трет-бутоксикарбонил-(3-метокси-3-оксопропил)амино]бутаноата (13,00 г, 42,85 ммоль, выход 46,05%) в виде бесцветного масла. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 4,13 (к, J=7,2 Гц, 2 H), 3,65-3,72 (м, 5 H), 3,29-3,54 (м, 2 H), 2,51-2,73 (м, 3 H), 2,47 (дд, J=6,8, 14,9 Гц, 1 H), 1,43-1,49 (м, 9 H), 1,22-1,29 (м, 5 H).
Стадия 3. Получение соединения 4
К раствору метил-(3S)-3-[трет-бутоксикарбонил-(3-метокси-3-оксопропил)амино]бутаноата (7,00 г, 23,08 ммоль, 1,00 экв.) в ТГФ (200,00 мл) добавляли t-BuOK (2,85 г, 25,38 ммоль, 1,10 экв.) при -40 °C. Смесь перемешивали при -40°C в течение 0,5 ч. Смесь гасили NH4Cl (100 мл) и EA (200 мл). Органический слой сушили Na2SO4, фильтровали. Фильтрат концентрировали с получением O1-трет-бутил-O3-метил-(6S)-6-метил-4-оксопиперидин-1,3-дикарбоксилата (5,80 г, 21,38 ммоль, выход 92,62%) в виде коричневого масла.
Стадия 4. Получение соединения 5
К раствору O1-трет-бутил-O3-метил-(6S)-6-метил-4-оксопиперидин-1,3-дикарбоксилата (5,80 г, 21,38 ммоль, 1,00 экв.) в EtOH (60,00 мл) добавляли NH2NH2·H2O (2,52 г, 42,76 ммоль, 2,44 мл, чистота 85%, 2,00 экв.). Смесь нагревали при 80°C в течение 2 ч. Смесь концентрировали в вакууме. Остаток экстрагировали EA (100 мл*2) и H2O (80 мл). Объединенный органический слой сушили Na2SO4, фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением трет-бутил-(6S)-3-гидрокси-6-метил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (1,50 г, 5,80 ммоль, выход 27,14%, чистота 98%) в виде белого твердого вещества.
Стадия 5. Получение соединения 6
К раствору трет-бутил-(6S)-3-гидрокси-6-метил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (1,00 г, 3,95 ммоль, 1,00 экв.) в Py (10,00 мл) добавляли 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)
метансульфонамид (1,48 г, 4,15 ммоль, 1,05 экв.). Смесь перемешивали при температуре 25°C в течение 16 ч. Смесь концентрировали в вакууме и экстрагировали EA (100 мл*2) и 1 Н HCl (50 мл). Объединенный органический слой сушили Na2SO4, фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (PE: EA=10%~50%) с получением трет-бутил-(6S)-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (700,00 мг, 1,82 ммоль, выход 45,99%) в виде белого твердого вещества.
Стадия 6. Получение соединения 8
К раствору трет-бутил-(6S)-6-метил-3-(трифторметилсульфонилокси)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (50,00 мг, 129,75 мкмоль, 1,00 экв.) и 2-тиенилбороновой кислоты (24,90 мг, 194,63 мкмоль, 1,50 экв.) в диоксане (2,00 мл) и H2O (200,00 мкл) добавляли XPHOS-PD-G2 (10,21 мг, 12,98 мкмоль, 0,10 экв.) и K3PO4 (82,63 мг, 389,25 мкмоль, 3,00 экв.). Смесь нагревали при 100°C в течение 16 ч. Смесь экстрагировали EA (30 мл*2) и H2O (30 мл). Объединенный органический слой сушили Na2SO4, фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали посредством препаративной ТСХ (PE: EA=3: 1) с получением трет-бутил-(6S)-6-метил-3-(2-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата (30,00 мг, 93,92 ммоль, выход 72,39%) в виде белого твердого вещества.
Стадия 7. Получение соединения 9
К трет-бутил-(6S)-6-метил-3-(2-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилату (30,00 мг, 93,92 мкмоль, 1,00 экв.) в ДХМ (2,00 мл) добавляли TFA (27,01 ммоль, 2,00 мл, 287,62 экв.). Смесь перемешивали при температуре 25°C в течение 1 ч. Смесь концентрировали в вакууме с получением (6S)-6-метил-3-(2-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (45,00 мг, неочищенный, 2TFA) в виде коричневого масла.
Получение соединения 753(S)
К раствору (6S)-6-метил-3-(2-тиенил)-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (42,00 мг, 93,89 мкмоль, 1,00 экв., 2TFA) в ДХМ (5,00 мл) добавляли TEA (38,00 мг, 375,56 мкмоль, 52,05 мкл, 4,00 экв.) с последующим добавлением фенил-N-(3-хлорфенил)карбамата (23,25 мг, 93,89 мкмоль, 1,00 экв.). Смесь перемешивали при температуре 25°C в течение 16 ч. Смесь концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением (6S)-N-(3-хлорфенил)-6-метил-3-(2-тиенил)-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (16,00 мг, 42,65 мкмоль, выход 45,43%, чистота 99,4%) в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,55 (т, J=2,0 Гц, 1 H), 7,36-7,50 (м, 2 H), 7,30-7,36 (м, 1 H), 7,22-7,29 (м, 1 H), 7,16 (т, J=4,1 Гц, 1 H), 7,01-7,07 (м, 1 H), 4,95-5,11 (м, 2 H), 4,39 (д, J=15,4 Гц, 1 H), 3,09 (дд, J=5,8, 16,3 Гц, 1 H), 2,71 (д, J=15,9 Гц, 1 H), 1,26 (д, J=6,9 Гц, 3 H). ЖХМС: 373/375 [M+1].
Пример 60. Получение соединений 917, 918, 919, 920, 921, 922, 923 и 924 (E1 и E2)
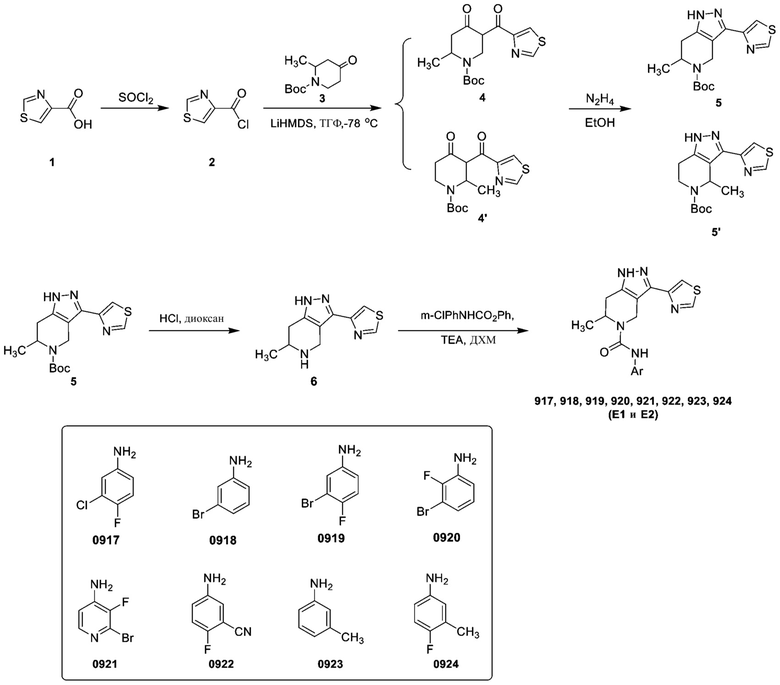
Стадия 1. Получение соединения 2
Тиазол-4-карбоновую кислоту (6,00 г, 46,46 ммоль, 1,00 экв.) растворяли в SOCl2 (100,00 мл) и выдерживали при 80°C в течение 2 ч. Смесь концентрировали в вакууме с получением тиазол-4-карбонилхлорида (7,00 г, неочищенный) в виде желтого твердого вещества.
Стадия 2. Получение соединения 4
К раствору трет-бутил-2-метил-4-оксопиперидин-1-карбоксилата (9,60 г, 45,01 ммоль, 1,00 экв.) в ТГФ (100,00 мл) по каплям добавляли LiHMDS (1 M, 67,52 мл, 1,50 экв.) при -70 °C. Смесь перемешивали при -70°C в течение 0,5 ч. По каплям добавляли тиазол-4-карбонилхлорид (6,64 г, 45,01 ммоль, 1,00 экв.) в ТГФ (60,00 мл) при -70 °C. Смесь перемешивали при -60°C в течение 2 ч. Смесь гасили NH4Cl (200 мл) и экстрагировали EA (500 мл*4). Объединенный органический слой сушили Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением смеси трет-бутил-2-метил-4-оксо-3-(тиазол-4-карбонил)пиперидин-1-карбоксилата и трет-бутил-2-метил-4-оксо-5-(тиазол-4-карбонил)пиперидин-1-карбоксилата (13,00 г, неочищенный) в виде черного масла.
Стадия 3. Получение соединения 5
К раствору смеси (13,00 г, 40,08 ммоль, 1,00 экв.) трет-бутил-2-метил-4-оксо-3-(тиазол-4-карбонил)пиперидин-1-карбоксилата и трет-бутил-2-метил-4-оксо-5-(тиазол-4-карбонил)пиперидин-1-карбоксилата в EtOH (150,00 мл) добавляли NH2NH2•H2O (3,54 г, 60,12 ммоль, 3,44 мл, чистота 85%, 1,50 экв.). Смесь перемешивали при 25°C в течение 8 ч. Смесь концентрировали в вакууме и экстрагировали EA (500 мл*5) и водой (300 мл). Объединенный органический слой сушили Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали посредством флэш-хроматографии (PE: EA=30%~100%) с получением смеси (5,00 г, 15,61 ммоль, выход 38,93%) трет-бутил-4-метил-3-тиазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата и трет-бутил-6-метил-3-тиазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата в виде желтого твердого вещества.
2,65 г смеси разделяли посредством SFC (колонка: AD-10 мкм; подвижная фаза: А для CO2 и В для изопропанола (0,1% NH3H2O); изократический: B 50%; скорость потока: 70 мл/мин; противодавление: 100 бар; температура колонки: 35 ℃; длина волны: 220 нм) для получения смеси (1,2 г) пика один, пика два и пика три (683 мг); пика четыре (559 мг). Полученную смесь (1,2 г) пика один и пика два дополнительно разделяли посредством SFC (прибор: SFC 80; колонка: AD-10 мкм; подвижная фаза: А для CO2 и В для метанола (0,1% NH3H2O); изократический: B 35%; скорость потока: 65 мл/мин.
Противодавление: 100 бар; температура колонки: 35 ℃; длина волны: 220 нм) для получения пика один (500 мг) и пика два (650 мг).
Посредством H ЯМР подтвердили, что пик 1 и пик 4 представляют собой энантиомеры трет-бутил-4-метил-3-тиазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата 5ʹ, а пик 2 и пик 3 представляют собой энантиомеры трет-бутил-4-метил-3-тиазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилата 5.
5ʹ_E1 (пик 1): 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м. д. 8,91 (уш. с, 1 H), 7,50 (д, J=1,6 Гц, 1 H), 5,76-5,43 (м, 1 H), 4,55-4,24 (м, 1 H), 3,34-3,09 (м, 1 H), 2,94-2,70 (м, 2 H), 1,63-1,36 (м, 12 H).
5_E1 (пик 2): 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м. д. 8,96 (д, J=1,8 Гц, 1 H), 7,44 (д, J=1,9 Гц, 1 H), 5,20-5,06 (м, 1 H), 4,89 (уш. с, 1 H), 4,26 (д, J=15,8 Гц, 1 H), 3,05 (дд, J=5,9, 15,7 Гц, 1 H), 2,68 (д, J=15,7 Гц, 1 H), 1,53 (с, 9 H), 1,19 (д, J=6,9 Гц, 3 H).
5_E2 (пик 3): 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м. д. 8,96 (д, J=1,6 Гц, 1 H), 7,44 (д, J=1,5 Гц, 1 H), 5,13 (д, J=15,1 Гц, 1 H), 4,90 (уш. с, 1 H), 4,26 (д, J=15,9 Гц, 1 H), 3,05 (дд, J=5,9, 15,7 Гц, 1 H), 2,68 (д, J=15,7 Гц, 1 H), 1,53 (с, 9 H), 1,19 (д, J=6,9 Гц, 3 H).
5ʹ_E2 (пик 4): 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м. д. 8,93 (уш. с, 1 H), 7,51 (с, 1 H), 5,75-5,49 (м, 1 H), 4,54-4,24 (м, 1 H), 3,31-3,10 (м, 1 H), 2,91-2,73 (м, 2 H), 1,53 (с, 9 H), 1,43 (д, J=6,5 Гц, 3 H).
Стадия 4. Получение соединения 6 (E1)
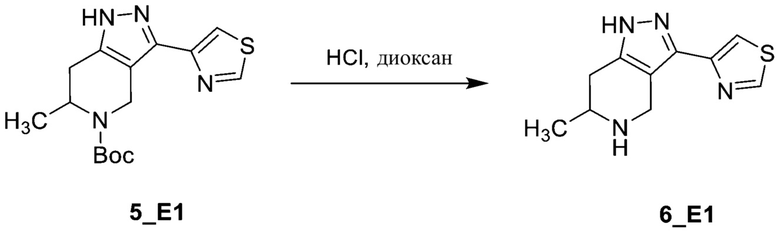
Трет-бутил-6-метил-3-тиазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксилат 5_E1 (650,00 мг, 2,03 ммоль, 1,00 экв.) растворяли в HCl/диоксане (4 М, 15,00 мл, 29,56 экв.) и перемешивали при 25°C в течение 2 ч. Смесь концентрировали в вакууме с получением 4-(6-метил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин-3-ил)тиазола (602,00 мг, неочищенный, 2HCl) в виде белого твердого вещества.
Общий способ получения соединений 917-924 (E1 и E2)
(Соединение 917 (E1) в качестве примера)
К смеси 4-(6-метил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридин-3-ил)тиазола (40,00 мг, 136,42 мкмоль, 1,00 экв., 2HCl) в ДХМ (2,00 мл) добавляли TEA (41,41 мг, 409,26 мкмоль, 56,73 мкл, 3,00 экв.) с последующим добавлением фенил-N-(3-хлор-4-фторфенил)карбамата (36,24 мг, 136,42 мкмоль, 1,00 экв.), реакционную смесь перемешивали при 25°C в течение 16 часов. ЖХМС показала, что исходный материал полностью израсходован и нужный продукт обнаружен. Растворитель удаляли в роторном испарителе, а остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлор-4-фторфенил)-6-метил-3-тиазол-4-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (43,00 мг, 109,30 мкмоль, выход 80,12%, чистота 99,6%) в виде белого твердого вещества.
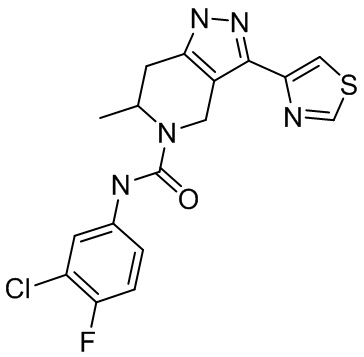
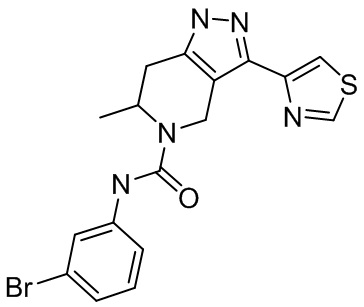
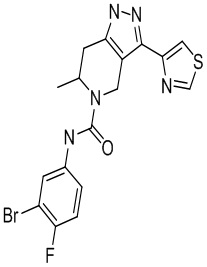
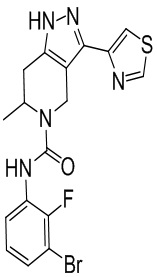
ЖХМС: 354 [M+1].
Пример 61. Получение соединения 289
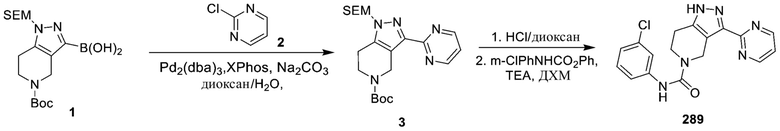
Стадия 1. Получение соединения 3
К раствору [5-трет-бутоксикарбонил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-3-ил]бороновой кислоты (300,00 мг, 755,00 мкмоль, 1,00 экв.) и 2-хлорпиримидина (129,71 мг, 1,13 ммоль, 1,50 экв.) в диоксане (2,00 мл) добавляли Pd2(dba)3 (69,14 мг, 75,50 мкмоль, 0,10 экв.), xphos (71,98 мг, 151,00 мкмоль, 0,20 экв.) с последующим добавлением раствора Na2CO3 (240,07 мг, 2,27 ммоль, 3,00 экв.) в H2O (400,00 мкл). Смесь выдерживали при 90°C в течение 16 ч. Смесь экстрагировали EA (50 мл*2) и H2O (10 мл). Объединенный органический слой сушили Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали посредством флэш-хроматографии (PE: EA: 0%~30%) с получением 220 мг продукта с чистотой 30%. Продукт очищали посредством препаративной ВЭЖХ (FA) с получением трет-бутил-3-пиримидин-2-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (46,50 мг, 107,74 мкмоль, выход 14,27%) в виде бесцветного масла. ЖХМС (M+1): 432.
Получение соединения 289
Трет-бутил-3-пиримидин-2-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилат (60,00 мг, 139,02 мкмоль, 1,00 экв.) растворяли в TFA (54,03 ммоль, 4,00 мл, 388,62 экв.) и перемешивали при 20°C в течение 16 ч. Смесь концентрировали в вакууме с получением 3-пиримидин-2-ил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (44,00 мг, неочищенный, TFA) в виде желтого масла.
К раствору 3-пиримидин-2-ил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (30,00 мг, 95,16 мкмоль, 1,00 экв., TFA) и фенил-N-(3-хлорфенил)карбамата (18,86 мг, 76,13 мкмоль, 0,80 экв.) в ДХМ (4,00 мл) добавляли TEA (48,15 мг, 475,80 мкмоль, 65,96 мкл, 5,00 экв.). Смесь перемешивали при температуре 20°C в течение 16 ч. Смесь концентрировали в вакууме. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-пиримидин-2-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (22,39 мг, 62,60 мкмоль, выход 65,79%, чистота 99,2%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ м. д. 8,86-8,97 (м, 3 H), 7,64 (с, 1 H), 7,41 (д, J=5,8 Гц, 2 H), 7,26 (т, J=8,1 Гц, 1 H), 6,94-7,02 (м, 1 H), 4,83 (уш. с, 2 H), 3,78 (уш. с, 2 H), 2,79 (уш. с, 2 H). ЖХМС: 355 [M+1].
Пример 62. Получение соединения 290
 Стадия 1. Получение соединения 3
Стадия 1. Получение соединения 3
К раствору [5-трет-бутоксикарбонил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-3-ил]бороновой кислоты (300,00 мг, 755,00 мкмоль, 1,00 экв.) и 3-хлорпиридазина (129,71 мг, 1,13 ммоль, 1,50 экв.) в диоксане (8,00 мл) добавляли Pd2(dba)3 (69,14 мг, 75,50 мкмоль, 0,10 экв.), XPhos (71,98 мг, 151,00 мкмоль, 0,20 экв.) с последующим добавлением раствора Na2CO3 (240,07 мг, 2,27 ммоль, 3,00 экв.) в H2O (2,00 мл). Смесь нагревали при 85°C в течение 16 ч. Смесь экстрагировали EA (50 мл*2) и H2O (20 мл). Объединенный органический слой сушили Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (PE: EA=20%~50%) с получением трет-бутил-3-пиридазин-3-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4Н-пиразол[4,3-c]пиридин-5-карбоксилата (75,00 мг, 147,71 мкмоль, выход 19,56%, чистота 85%) в виде желтого масла. ЖХМС (M+1): 432.
Получение соединения 290
Трет-бутил-3-пиридазин-3-ил-1-(2-триметилсилилэтоксиметил)-6,7-дигидро-4H-пиразол[4,3-c]пиридин-5-карбоксилат (75,00 мг, 137,46 мкмоль, 1,00 экв., TFA) растворяли в TFA (6,85 г, 60,09 ммоль, 4,45 мл, 437,19 экв.) и перемешивали при 20°C в течение 16 ч. Смесь концентрировали в вакууме с получением 3-пиридазин-3-ил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (59,00 мг, неочищенный, TFA) в виде желтого масла.
К раствору 3-пиридазин-3-ил-4,5,6,7-тетрагидро-1Н-пиразол[4,3-c]пиридина (59,00 мг, 187,15 мкмоль, 1,00 экв., TFA) в ДХМ (4,00 мл) добавляли фенил-N-(3-хлорфенил)карбамат (46,35 мг, 187,15 мкмоль, 1,00 экв.) с последующим добавлением TEA (94,69 мг, 935,75 мкмоль, 129,71 мкл, 5,00 экв.). Смесь перемешивали при 20°C в течение 16 ч. ЖХМС показала, что соединение 3 сохранилось. Добавляли другую партию фенил-N-(3-хлорфенил)карбамата (30 мг). Смесь перемешивали при 20°C в течение еще 16 ч. Смесь концентрировали в вакууме. ЖХМС показала, что выход нужного продукта составляет 30%. Остаток очищали посредством препаративной ВЭЖХ (FA) с получением N-(3-хлорфенил)-3-пиридазин-3-ил-1,4,6,7-тетрагидропиразол[4,3-c]пиридин-5-карбоксамида (25,00 мг, 70,32 мкмоль, выход 37,58%, чистота 99,8%) в виде белого твердого вещества.
1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ м. д. 2,93 (т, J=5,71 Гц, 2 H) 3,90 (т, J=5,71 Гц, 2 H) 5,00 (с, 2 H) 6,99-7,04 (м, 1 H) 7,21-7,29 (м, 1 H) 7,31-7,37 (м, 1 H) 7,56 (т, J=2,01 Гц, 1 H) 7,77 (дд, J=8,41, 4,77 Гц, 1 H) 8,28 (д, J=8,91 Гц, 1 H) 9,11 (д, J=4,64 Гц, 1 H). ЖХМС: 355 [M+1].
Пример 63. Анализ сборки ВГВ
Воздействие соединений настоящего изобретения на сборку капсида ВГВ можно измерить с помощью анализа сборки in vitro, основанном на гашении флуоресценции, который был разработан в соответствии со способом, описанным Zlotnick и соавторами (Nature Biotechnology 2006, 24:358). В типичном анализе мутантный белок ВГВ C150 (аминокислоты 1-150, C49A, C61A, C107A, 150C) клонируют в РНК-полимеразу Т7 на основании экспрессионного вектора, экспрессируемого в E.coli и очищенного до гомогенности в виде димера. Очищенный ядерный белок ВГВ обессоливают и метят красителем BODIPY-FL.
В не имеющем ограничительного характера варианте осуществления анализ сборки выполняют в 96-луночном планшете. Реакции сборки выполняют в 50 мМ буферном растворе HEPES, рН 7,5 и 150 мМ NaCl. Соединения предварительно инкубируют с белком CA ВГВ в течение 15 мин и инициируют реакции сборки добавлением NaCl. Реакции позволяют протекать в течение 1 часа при комнатной температуре. Изменения флуоресценции между пробами, обработанными ДМСО и обработанными соединением, фиксируют и анализируют в отношении модуляции сборки.
Пример 64. Анализ ингибирования репликации ВГВ
Ингибирование репликации ВГВ соединениями настоящего изобретения можно определять в клетках, инфицированных или трансфицированных ВГВ, или клетках со стабильно интегрированным ВГВ, таких как клетки HepG2.2.15 (Sells et al. 1987). В этом примере клетки HepG2.2.15 находились в среде для выращивания клеточной культуры, содержащей эмбриональную бычью сыворотку (FBS) 10%, генетицин, L-глутамин, пенициллин и стрептомицин. Клетки HepG2.2.15 могут быть посеяны в 96-луночные планшеты с плотностью 40 000 клеток/лунка и обработаны серийно разведенными соединениями в конечной концентрации ДМСО 0,5% либо без использования других компонентов, либо в комбинации с другими лекарственными средствами, добавленными в шахматном порядке. Клетки инкубировали с соединениями в течение трех суток, после чего среду удаляли, и к клеткам добавляли свежую среду, содержащую соединения, и инкубировали в течение еще трех суток. На 6 сутки супернатант удаляли и обрабатывали DNase при 37 ⁰C в течение 60 минут с последующей инактивацией фермента при 75 ⁰C в течение 15 минут. Заключенную в капсид ДНК ВГВ высвобождали из вирионов и ковалентно связывали с полимеразой ВГВ посредством инкубирования в лизирующем буферном растворе (Affymetrix QS0010), содержащем 2,5 мкг протеиназы K, при 50°C в течение 40 минут. ДНК ВГВ денатурировали путем добавления 0,2 М NaOH и обнаруживали с использованием набора для анализа разветвленной ДНК (РДНК) QuantiGene в соответствии с рекомендацией изготовителя (Affymetrix). Концентрации ДНК ВГВ также можно было количественно определить с использованием количественной ПЦР, основанной на амплификации капсидированной ДНК ВГВ, экстракции с помощью раствора QuickExtraction Solution (Epicentre Biotechnologies) и амплификации ДНК ВГВ с использованием ПЦР-зондов, специфичных для ВГВ, которые способны гибридизировать с ДНК ВГВ, и зонда с флуоресцентной меткой для количественного определения. Кроме того, определяли жизнеспособность клеток HepG2.2.15, инкубированных с опытными соединениями, отдельно или в комбинации с другими компонентами, с использованием реагента CellTitre-Glo в соответствии с протоколом изготовителя (Promega). Средний фоновый сигнал от лунок, содержащих только питательную среду, вычитали из всех других проб и рассчитывали процент ингибирования при каждой концентрации соединения путем нормализации по сигналам от клеток HepG2.2.15, обработанных ДМСО 0,5%, с использованием уравнения E1.
E1: % ингибирования=(ДМСОср - Xi)/ДМСОср x 100%,
где ДМСОср представляет собой средний сигнал, рассчитанный в лунках, которые обрабатывали ДМСО в качестве контроля (контроль с ингибированием 0%), а Xi представляет собой сигнал, измеренный в отдельных лунках. Значения EC50, т. е. эффективные концентрации, которые обеспечивали ингибирующий эффект в 50% случаев, определяли с помощью нелинейного подбора, используя программу Graphpad Prism (г. Сан-Диего, штат Калифорния) и уравнение E2
E2: Y=Yмин+(Yмакс - Yмин)/(1+10(LogEC50-X) x HillSlope),
где Y означает значения процента ингибирования, а X означает логарифм концентраций соединения.
Выбранные соединения настоящего изобретения оценивали в анализе репликации ВГВ (анализ РДНК), как описано выше, а репрезентативная группа этих активных соединений показана в таблице 3. В таблице 3 А означает, что 0,01 < EC50<0,10; В означает, что 0,10 ≤ EC50<0,50; С означает, что 0,50 ≤ EC50<1,0; D означает, что 1,0 ≤ EC50<1,5; а E означает, что 1,5 ≤ EC50<5.
Таблица 3. Активность в анализе РДНК:+означает, что активность составляет > 50% при 10 мкМ, или числовое значение означает EC50 (мкM).
Описания каждого патента, патентной заявки и публикации, цитируемой в настоящем документе, полностью включены в настоящий документ путем ссылки.
Хотя настоящее изобретение описано на примере конкретных вариантов осуществления, понятно, что специалисты в данной области могут найти другие варианты осуществления и модификации настоящего изобретения без отступления от сущности и объема настоящего изобретения. Предполагается, что приложенная формула изобретения включает все такие варианты осуществления и равнозначные модификации.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 2001 |
|
RU2286343C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2277909C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРИМЕНЕНИЕ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2317988C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИЙ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2290179C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕКОТОРЫХ ЛЕЙКОЗОВ | 2019 |
|
RU2804709C2 |
| N1-(4-(5-(ЦИКЛОПРОПИЛМЕТИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-ИЛ)ПИРИДИН-2-ИЛ)ЦИКЛОГЕКСАН-1,4-ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ СK1 И/ИЛИ IRAK1 ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2761457C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ПИРИМИДИНЫ И ИХ ВАРИАНТЫ, И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2760733C2 |
| ПИРИМИДИНЫ, ИХ ВАРИАНТЫ И ПРИМЕНЕНИЕ | 2017 |
|
RU2761439C2 |
| НОВЫЕ СУЛЬФОНАМИДКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2808572C2 |
Изобретение относится к соединению формулы III, в которой Y представляет собой -C(O)-; R1 представляет собой C6 арил, тиофенил, тиазолил; R2 в каждом случае независимо выбран из H и C1-C6 алкила, при условии, что по меньшей мере один из R2 представляет собой C1-C6 алкил; R3 выбран из H; R4 выбран из 
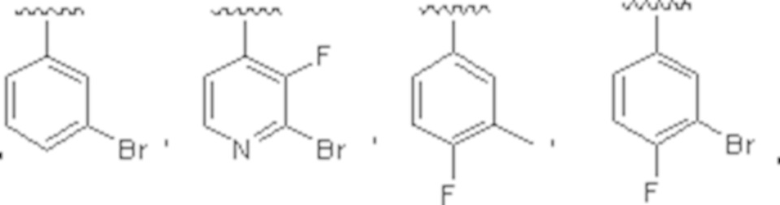
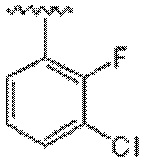 ; и R7 выбран из H, C1-C6 алкила. Изобретение также относится к индивидуальным соединениям, к фармацевтической композиции и к применению фармацевтической композиции. Технический результат: получены новые соединения формулы III, которые могут использоваться для лечения инфекции вируса гепатита В. 5 н.п. ф-лы, 3 табл., 64 пр.
; и R7 выбран из H, C1-C6 алкила. Изобретение также относится к индивидуальным соединениям, к фармацевтической композиции и к применению фармацевтической композиции. Технический результат: получены новые соединения формулы III, которые могут использоваться для лечения инфекции вируса гепатита В. 5 н.п. ф-лы, 3 табл., 64 пр.
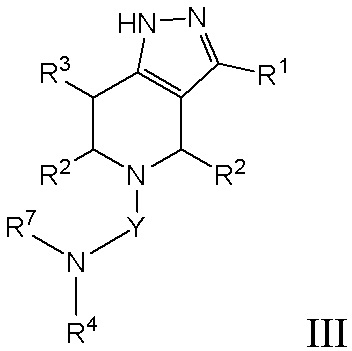
1. Соединение формулы III:
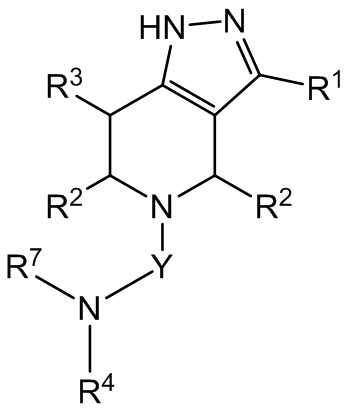
III,
где
Y представляет собой -C(O)-;
R1 представляет собой C6 арил, тиофенил, тиазолил;
R2 в каждом случае независимо выбран из H и C1-C6 алкила, при условии, что по меньшей мере один из R2 представляет собой C1-C6 алкил;
R3 выбран из H;
R4 выбран из
и
R7 выбран из H, C1-C6 алкила.
2. Соединение, выбранное из:
317 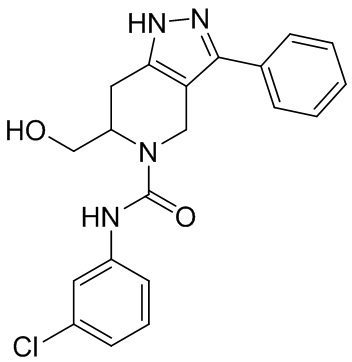
756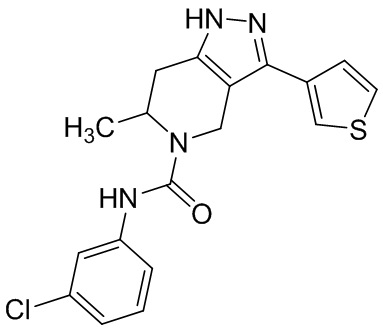
754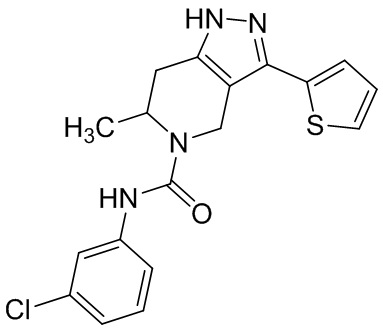
753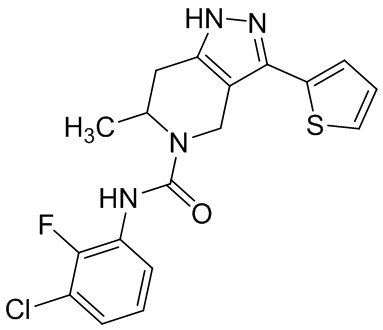
819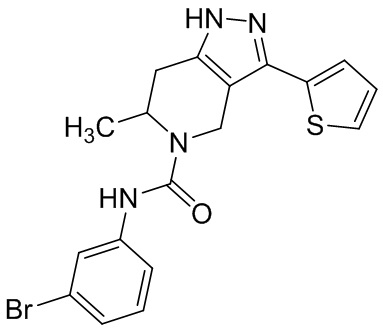
820 (851)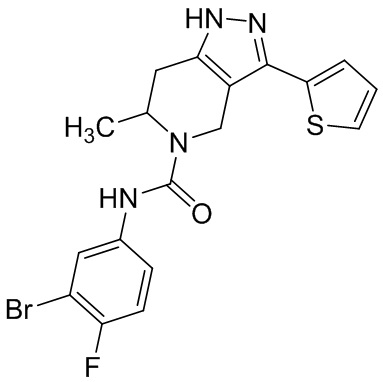
821 (852)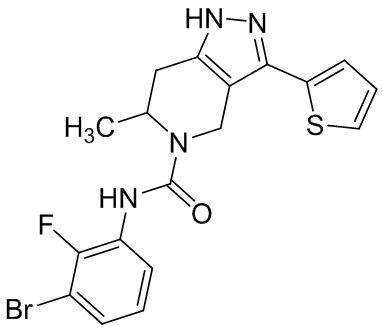
822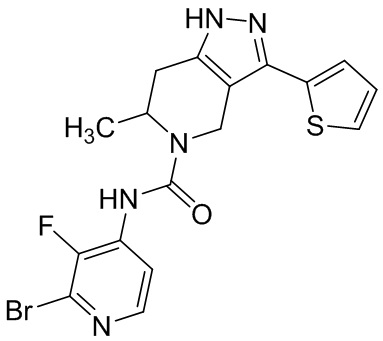
823 (853)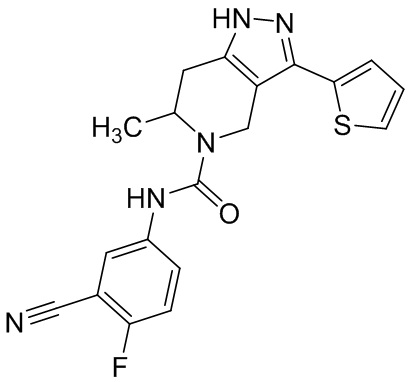
824 (854)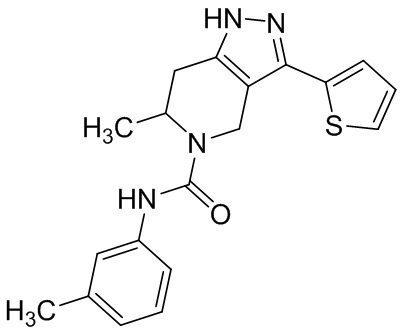
825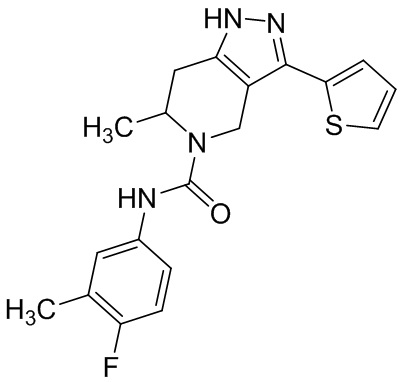
826 (855)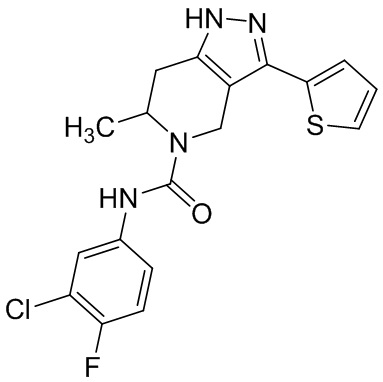
857 (856)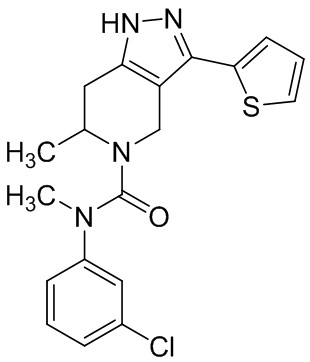
830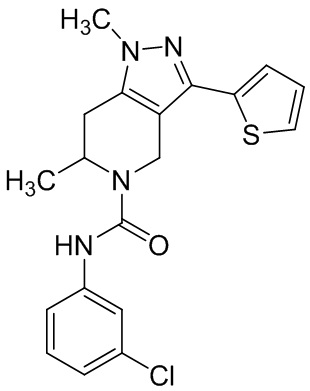
831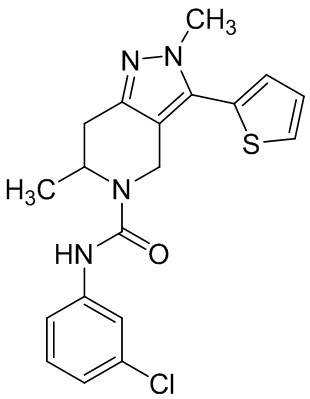
832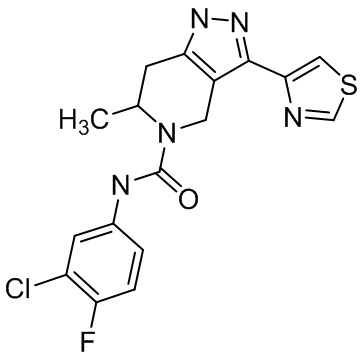
917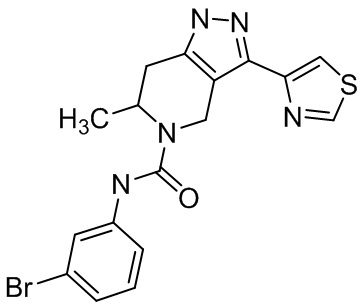
918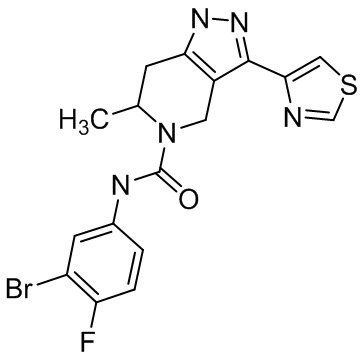
919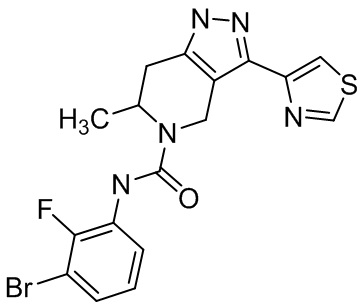
920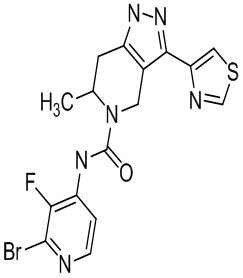
921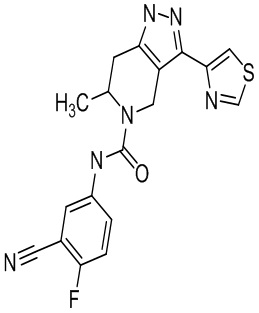
922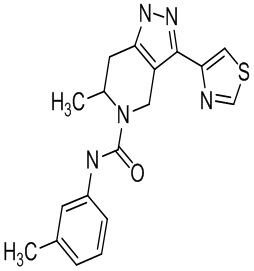
923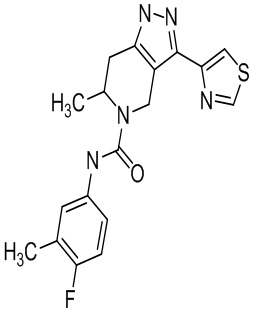
924
3. Фармацевтическая композиция для лечения инфекции вируса гепатита В, содержащая эффективное количество соединения по любому одному из пп. 1 или 2 вместе с фармацевтически приемлемым носителем.
4. Применение фармацевтической композиции, содержащей соединение по любому из пп. 1 или 2 для лечения инфекции вируса гепатита В (ВГВ).
5. Применение фармацевтической композиции, содержащей соединение по любому из пп. 1 или 2 для ингибирования или снижения образования или наличия частиц, содержащих ДНК ВГВ, у нуждающегося в этом субъекта.
| R | |||
| L | |||
| Thurmond et al | |||
| "Nonpeptidic, Noncovalent Inhibitors of the Cysteine Protease Cathepsin S" Journal of Medicinal Chemistry, vol.47, N20, 2004, 4799-4801 | |||
| C | |||
| A | |||
| Grice et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| M | |||
| K | |||
| Ameriks et al | |||

