Ссылки на родственные заявки
[1] Настоящая заявка испрашивает приоритет по заявке на патент Китая № CN201610700714.2, поданной в Национальное ведомство по интеллектуальной собственности, PRC, 22 августа 2016 г., содержание которой включено в данный документ посредством ссылки во всей своей полноте.
Область техники изобретения
[2] Настоящее изобретение относится к классу ингибитора фосфодиэстеразы-4 (PDE4) и его применению в изготовлении лекарственного препарата для лечения заболевания, связанного с PDE4. В частности, оно относится к соединению, представленному формулой (I), и его фармацевтически приемлемой соли.
Предшествующий уровень техники
[3] Фактор некроза опухолей альфа (TNF-α) представляет собой цитокин, высвобождаемый в основном мононуклеарными фагоцитами в ответ на иммуностимуляторы. TNF-α способен усиливать большинство клеточных процессов, таких как дифференциация, рекрутинг, пролиферация и протеолитический распад. При низких уровнях TNF-α обеспечивает защиту против инфекционных средств, опухолей и повреждения ткани. Однако избыточное высвобождение TNF-α также вызывает заболевания, при введении млекопитающим или людям TNF-α вызывает или усугубляет воспаление, лихорадку, сердечно-сосудистые эффекты, кровоизлияние, коагуляцию и острофазовые ответы, подобные таковым, которые наблюдаются во время острых инфекций и шоковых состояний. Усиленное или неконтролируемое продуцирование TNF-α у животных или людей часто свидетельствует о ряде заболеваний, например, эндотоксикоз и/или синдром токсического шока, кахексия, синдром дыхательной недостаточности у взрослых, виды рака (такие как виды солидной опухоли и виды гематологической опухоли), заболевание сердца (такое как застойная сердечная недостаточность), вирусные инфекции и генетические, воспалительные, аллергические или аутоиммунные заболевания.
[4] Рак является особенно тяжелым заболеванием, и повышение уровней TNF-α в крови свидетельствует о раке или риске распространения рака. Обычно, у здоровых субъектов раковые клетки не выживают в кровеносной системе, одной из причин чего является то, что внутренняя стенка кровеносных сосудов выполняет функцию барьера для проникновения опухолевой клетки. Было показано, что ELAM-1 на эндотелиальных клетках опосредует повышенную адгезию клеток рака толстой кишки к эндотелию, обработанному цитокинами.
[5] Циклический аденозинмонофосфат (cAMP) также играет роль во множестве заболеваний и симптомов. Было показано, что повышение уровня cAMP в воспалительных лейкоцитах подавляет их активацию и затем высвобождение воспалительных медиаторов, в том числе TNF-α и ядерного фактора κB (NF-κB). Повышение уровней cAMP также приводит к расслаблению гладкой мышцы дыхательных путей.
[6] Полагают, что первичным клеточным механизмом для инактивации cAMP является разрушение cAMP семейством изоферментов, называемых фосфодиэстеразами циклических нуклеотидов (PDE). Известно одиннадцать членов семейства PDE. На сегодняшний день было показано, что подавление PDE типа IV (PDE4) является особенно эффективным как в подавлении высвобождения воспалительных медиаторов, так и в расслаблении гладкой мышцы дыхательных путей. Следовательно, PDE4 стала одной из самых популярных мишеней для лекарственного средства. Семейство PDE-4 можно поделить на четыре подтипа (PDE-4A, B, C, D), исходя из различных генетических кодов, при этом PDE-4A, PDE-4B и PDE-4D более широко экспрессируются в воспалительных клетках (таких как B-клетки, T-клетки и нейтрофилы), чем PDE-4C. Подавление PDE4 приводит к повышению уровней cAMP, за счет чего обеспечивается регуляция уровней TNFα для терапевтических целей.
Содержание настоящего изобретения
[7] Настоящее изобретение предусматривает соединение, представленное формулой (I), и его фармацевтически приемлемую соль:
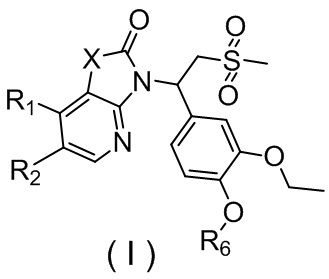 ,
,
[8] где
[9] X выбран из O, N(R3), -CH(R3)-;
[10] R3 выбран из H, F, Cl, Br, I, OH, CN, NH2, COOH, R4-L1- или выбран из группы, состоящей из C1-6алкила, C1-6гетероалкила, C3-6циклоалкила, 3-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R;
[11] R4 выбран из группы, состоящей из C3-6циклоалкила, 3-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R;
[12] L1 выбран из -CH2-, -CH2CH2-, O, S, NH, -C(=O)-;
[13] каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, CN, NH2, R5-L2- или выбран из группы, состоящей из C1-6алкила, C1-6алкокси, C1-6алкилтио, C1-6алкиламино, C2-6алкенила, C3-6циклоалкенила, 3-6-членного гетероциклоалкенила, C3-6циклоалкила, 3-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R;
[14] R5 выбран из группы, состоящей из C3-6циклоалкила, 3-6-членного гетероциклоалкила, 3-6-членного гетероциклоалкенила, фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R;
[15] L2 выбран из -CH2-, -CH2CH2-, O, S, NH;
[16] R6 выбран из C1-3алкила, который необязательно замещен 1, 2 или 3 R;
[17] R выбран из H, галогена, OH, NH2, CN или выбран из группы, состоящей из C1-6алкила и C1-6гетероалкила, каждый из которых необязательно замещен 1, 2 или 3 R';
[18] R' выбран из H, F, Cl, Br, I, OH, NH2, Me, Et, CF3, CHF2, CH2F, NHCH3, N(CH3)2;
[19] «гетеро» в C1-6гетероалкиле, 3-6-членном гетероциклоалкиле, 5-6-членном гетероариле и 3-6-членном гетероциклоалкениле выбран из -C(=O)NH-, -NH-, -C(=NH)-, -S(=O)2NH-, -S(=O)NH-, -O-, -S-, =O, =S, -O-N=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -NHC(=O)NH-;
[20] в любом из вышеуказанных случаев число гетероатомов или содержащих гетероатом групп независимо выбрано из 1, 2 или 3.
[21] В некоторых вариантах осуществления настоящего изобретения R выбран из H, F, Cl, Br, I, OH, NH2, CN или выбран из группы, состоящей из C1-3алкила, C1-3алкокси, C1-3алкилтио, C1-3алкиламино, C1-4алкил-OC(=O)- и N,N'-ди(C1-3алкил)амино, каждый из которых необязательно замещен 1, 2 или 3 R'.
[22] В некоторых вариантах осуществления настоящего изобретения R выбран из H, F, Cl, Br, I, OH, NH2, CN, Me, CF3, CHF2, CH2F, Et, 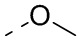 ,
, 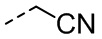 ,
, 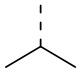 ,
,  ,
,  .
.
[23] В некоторых вариантах осуществления настоящего изобретения R4 выбран из группы, состоящей из фенила, пиридила, пиримидинила, пиразинила, тиенила, имидазолила, пиразолила, оксазолила, тиазолила, изоксазолила и изотиазолила, каждый из которых необязательно замещен 1, 2 или 3 R.
[24] В некоторых вариантах осуществления настоящего изобретения R4 выбран из группы, состоящей из  ,
,  и
и  , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
[25] В некоторых вариантах осуществления настоящего изобретения R4 выбран из  ,
,  ,
,  .
.
[26] В некоторых вариантах осуществления настоящего изобретения R4-L1- выбран из  ,
,  ,
,  .
.
[27] В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, R4-L1- или выбран из группы, состоящей из C1-3алкила, C3-6циклоалкила, -C1-3алкил-C(=O)O-C1-3алкил-, C1-3алкил-S(=O)2-C1-3алкил-, фенила, пиридила, пиримидинила и пиразинила, каждый из которых необязательно замещен 1, 2 или 3 R.
[28] В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, R4-L1- или выбран из группы, состоящей из Me, Et,  ,
, 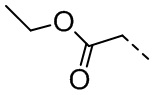 ,
, 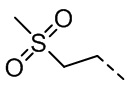 ,
, 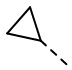 ,
, 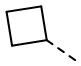 и
и 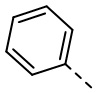 , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
[29] В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, Me, Et, 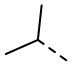 ,
, 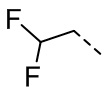 ,
, 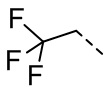 ,
, 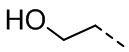 ,
, 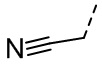 ,
, 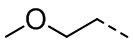 , ,
, , 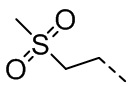 ,
, 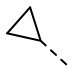 , , , , , .
, , , , , .
[30] В некоторых вариантах осуществления настоящего изобретения X выбран из 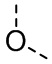 ,
, 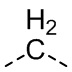 ,
, 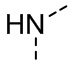 ,
, 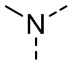 ,
, 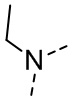 ,
, 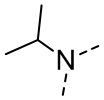 ,
, 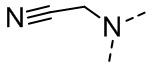 ,
, 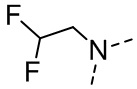 ,
, 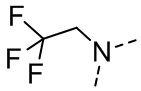 ,
, 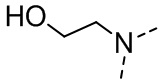 ,
, 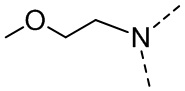 ,
, 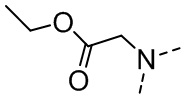 ,
, 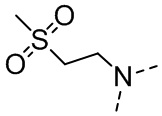 ,
, 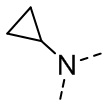 ,
, 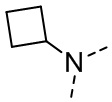 ,
, 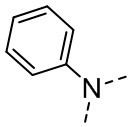 ,
, 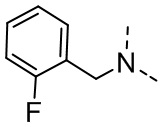 ,
, 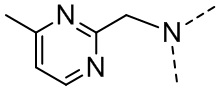 ,
, 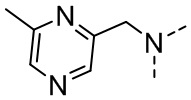 .
.
[31] В некоторых вариантах осуществления настоящего изобретения R5 выбран из группы, состоящей из фенила, пиридила, пиримидинила, пиразинила, пиридазинила, тиенила, имидазолила, пиразолила, оксазолила, тиазолила, изоксазолила и изотиазолила, каждый из которых необязательно замещен 1, 2 или 3 R.
[32] В некоторых вариантах осуществления настоящего изобретения R5 выбран из группы, состоящей из 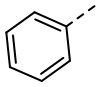 и
и 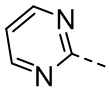 , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
[33] В некоторых вариантах осуществления настоящего изобретения R5 выбран из 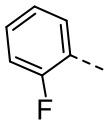 ,
, 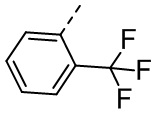 ,
,  .
.
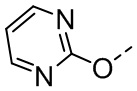
[34] В некоторых вариантах осуществления настоящего изобретения R5-L2- выбран из 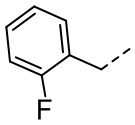 ,
, 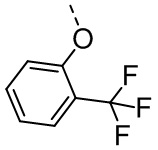 , .
, .
[35] В некоторых вариантах осуществления настоящего изобретения каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, R5-L2- или выбран из группы, состоящей из C1-3алкила, C1-3алкокси, C1-3алкилтио, C1-3алкиламино, C2-4алкенила, 1,2,3,6-тетрагидропиридила, пиридин-2(1H)-онила, фенила, пиридила, пиримидинила, пиразинила, пиридазинила, пиразолила, имидазолила, тиазолила, изотиазолила и тиенила, каждый из которых необязательно замещен 1, 2 или 3 R.
[36] В некоторых вариантах осуществления настоящего изобретения каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, R5-L2- или выбран из группы, состоящей из Me, 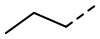 ,
,  ,
, 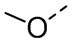 ,
,  ,
,  ,
, 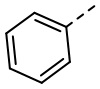 ,
, 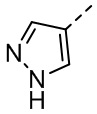 ,
, 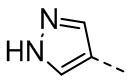 ,
, 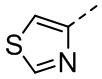 ,
, 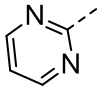 ,
, 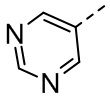 ,
, 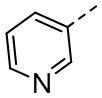 ,
, 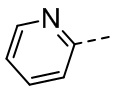 ,
, 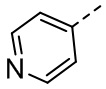 ,
, 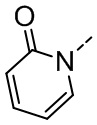 и
и 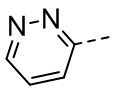 , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
[37] В некоторых вариантах осуществления настоящего изобретения каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, Me,  ,
,  ,
,  ,
,  , , ,
, , ,  , ,
, ,  ,
,  ,
,  ,
,  ,
, 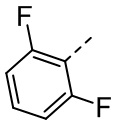 ,
, 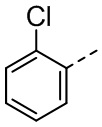 ,
, 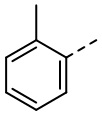 ,
, 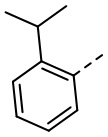 ,
, 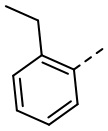 ,
, 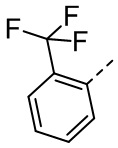 ,
, 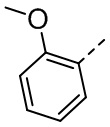 ,
, 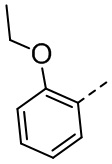 ,
, 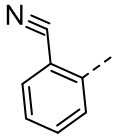 , ,
, , 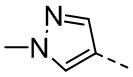 , , , , ,
, , , , , 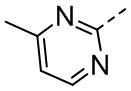 , ,
, , 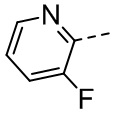 ,
,  ,
, 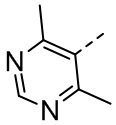 ,
, 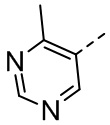 , ,
, , 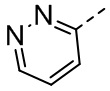 , ,
, , 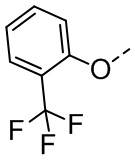 , .
, .
[38] В некоторых вариантах осуществления настоящего изобретения R6 выбран из группы, состоящей из Me и Et, каждый из которых необязательно замещен 1, 2 или 3 R.
[39] В некоторых вариантах осуществления настоящего изобретения R6 выбран из Me, CH2F, CHF2.
[40] В некоторых вариантах осуществления настоящего изобретения R выбран из H, F, Cl, Br, I, OH, NH2, CN или выбран из группы, состоящей из C1-3алкила, C1-3алкокси, C1-3алкилтио, C1-3алкиламино, C1-4алкил-OC(=O)- и N,N'-ди(C1-3алкил)амино, каждый из которых необязательно замещен 1, 2 или 3 R', и другие переменные являются такими, как определено выше.
[41] В некоторых вариантах осуществления настоящего изобретения R выбран из H, F, Cl, Br, I, OH, NH2, CN, Me, CF3, CHF2, CH2F, Et, , , , , , и другие переменные являются такими, как определено выше.
[42] В некоторых вариантах осуществления настоящего изобретения R4 выбран из группы, состоящей из фенила, пиридила, пиримидинила, пиразинила, тиенила, имидазолила, пиразолила, оксазолила, тиазолила, изоксазолила и изотиазолила, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[43] В некоторых вариантах осуществления настоящего изобретения R4 выбран из группы, состоящей из , и , каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[44] В некоторых вариантах осуществления настоящего изобретения R4 выбран из , , , каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[45] В некоторых вариантах осуществления настоящего изобретения R4-L1- выбран из , , , и другие переменные являются такими, как определено выше.
[46] В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, R4-L1- или выбран из группы, состоящей из C1-3алкила, C3-6циклоалкила, -C1-3алкил-C(=O)O-C1-3алкил-, C1-3алкил-S(=O)2-C1-3алкил-, фенила, пиридила, пиримидинила и пиразинила, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[47] В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, R4-L1- или выбран из группы, состоящей из Me, Et, , , , , и , каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[48] В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, Me, Et, , , , , , , , , , , , , , , и другие переменные являются такими, как определено выше.
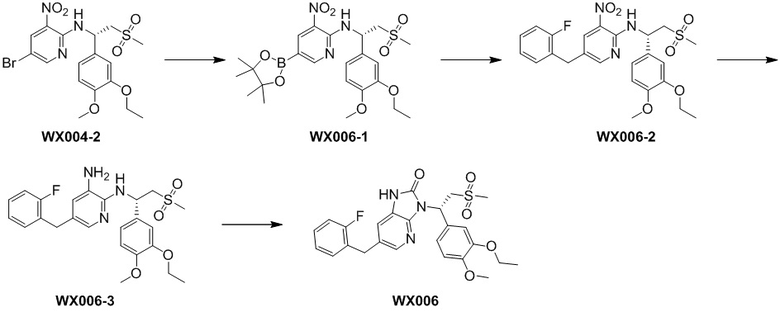
[49] В некоторых вариантах осуществления настоящего изобретения X выбран из , , , , , , , , , , , , , , , , , , , и другие переменные являются такими, как определено выше.
[50] В некоторых вариантах осуществления настоящего изобретения R5 выбран из группы, состоящей из фенила, пиридила, пиримидинила, пиразинила, пиридазинила, тиенила, имидазолила, пиразолила, оксазолила, тиазолила, изоксазолила и изотиазолила, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[51] В некоторых вариантах осуществления настоящего изобретения R5 выбран из группы, состоящей из и , каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[52] В некоторых вариантах осуществления настоящего изобретения R5 выбран из , , , и другие переменные являются такими, как определено выше.
[53] В некоторых вариантах осуществления настоящего изобретения R5-L2- выбран из , , , и другие переменные являются такими, как определено выше.
[54] В некоторых вариантах осуществления настоящего изобретения каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, R5-L2- или группы, состоящий из C1-3алкила, C1-3алкокси, C1-3алкилтио, C1-3алкиламино, C2-4алкенила, 1,2,3,6-тетрагидропиридила, пиридин-2(1H)-онила, фенила, пиридила, пиримидинила, пиразинила, пиридазинила, пиразолила, имидазолила, тиазолила, изотиазолила и тиенила, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[55] В некоторых вариантах осуществления настоящего изобретения каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, R5-L2- или выбран из группы, состоящей из Me, , , , , , , , , , , , , , , и , каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
[56] В некоторых вариантах осуществления настоящего изобретения каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, Me, , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , и , и другие переменные являются такими, как определено выше.
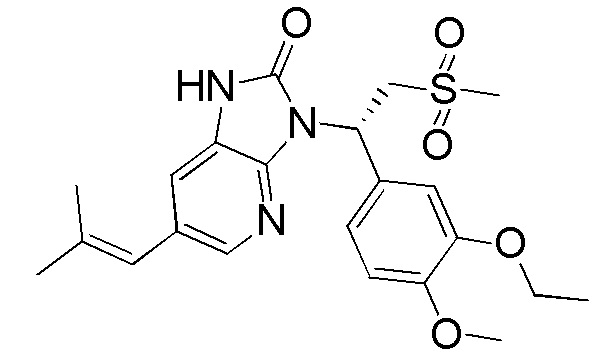
[57] В некоторых вариантах осуществления настоящего изобретения R6 выбран из группы, состоящей из Me и Et, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные являются такими, как определено выше.
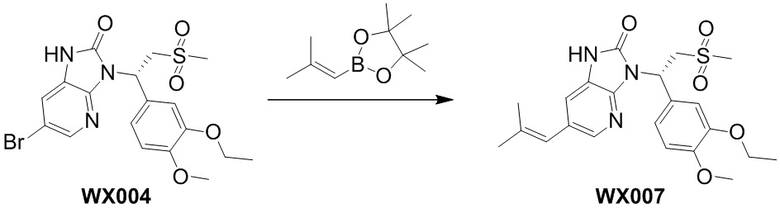
[58] В некоторых вариантах осуществления настоящего изобретения R6 выбран из Me, CH2F и CHF2, и другие переменные являются такими, как определено выше.
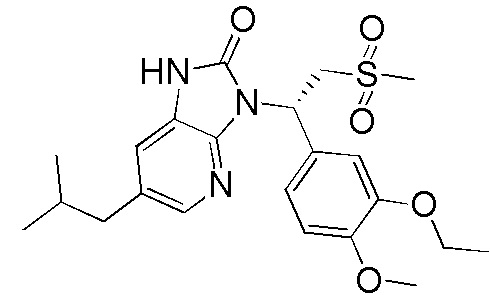
[59] В некоторых вариантах осуществления настоящего изобретения соединение или его фармацевтически приемлемая соль выбраны из 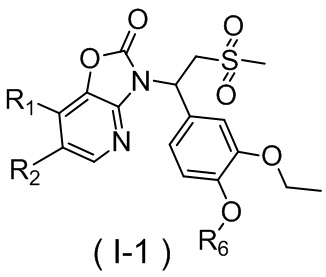 ,
, 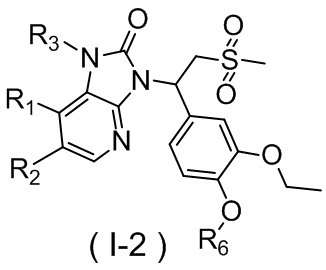 ,
, 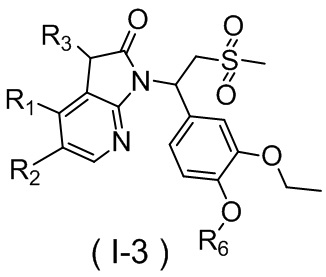 , где R1, R2, R3 и R6 являются такими, как определено выше.
, где R1, R2, R3 и R6 являются такими, как определено выше.
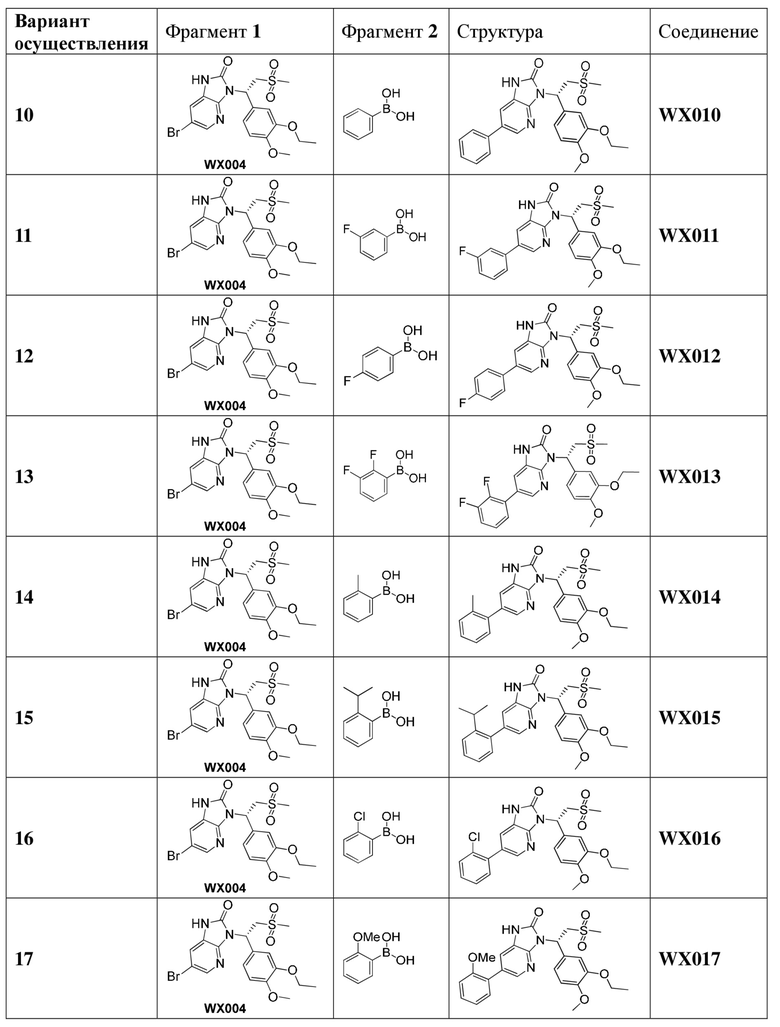
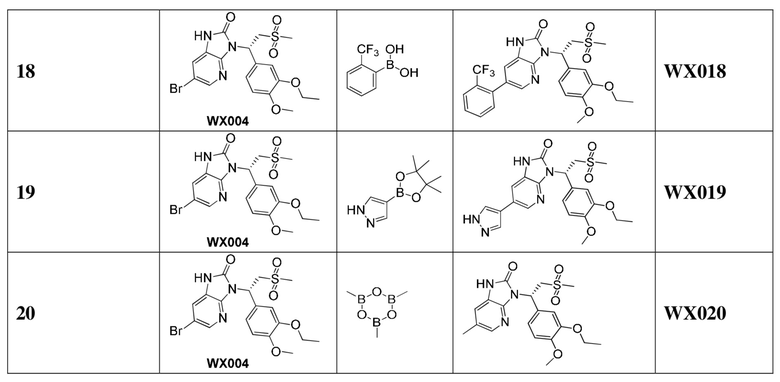
[60] В некоторых вариантах осуществления настоящего изобретения соединение или его фармацевтически приемлемая соль выбраны из 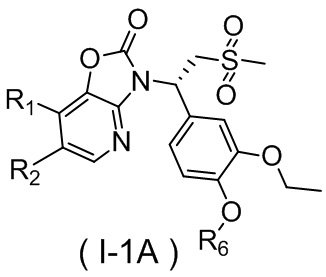 ,
, 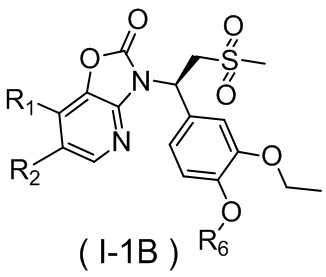 ,
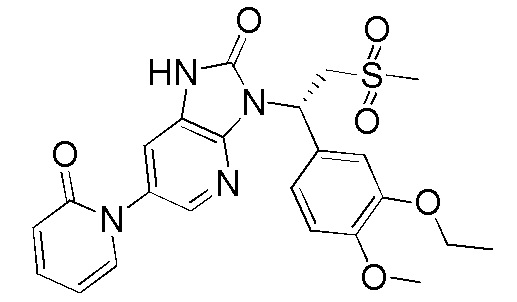
, 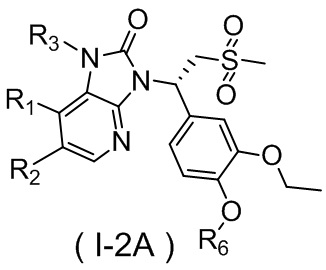 ,
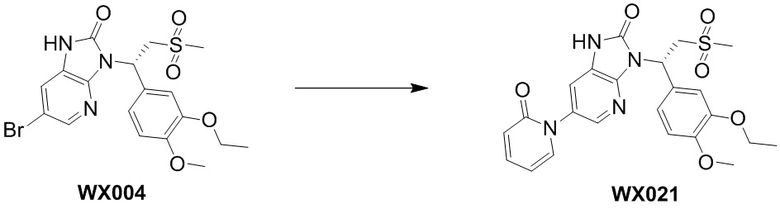
, 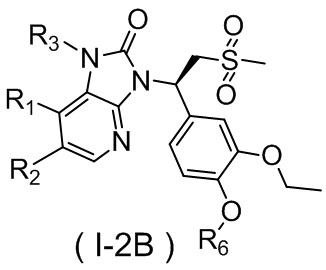 ,
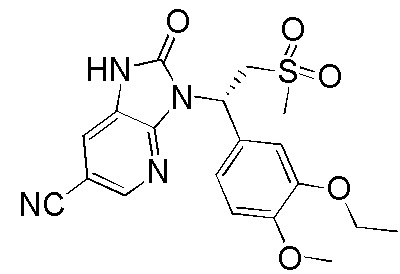
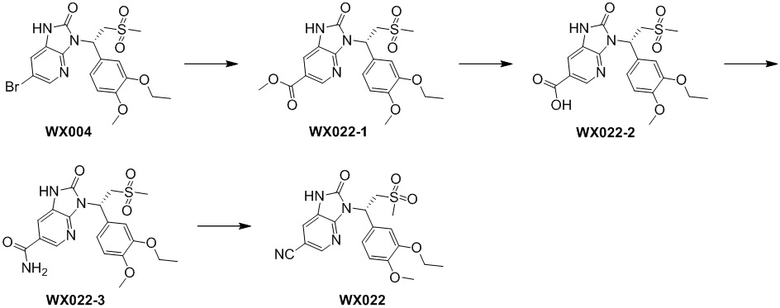
, 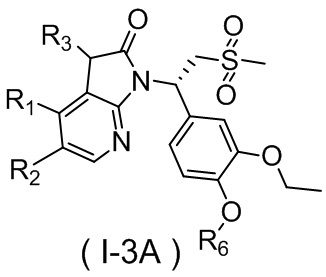 ,
, 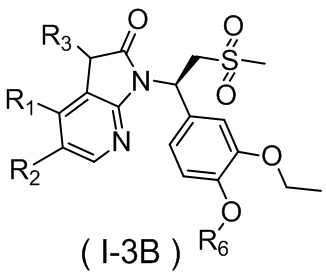 , где R1, R2, R3 и R6 являются такими, как определено выше.
, где R1, R2, R3 и R6 являются такими, как определено выше.
[61] Вышеуказанные переменные можно произвольно объединять, таким образом получают другие варианты осуществления настоящего изобретения.
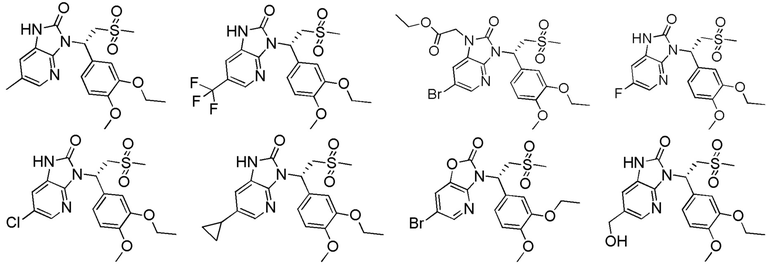
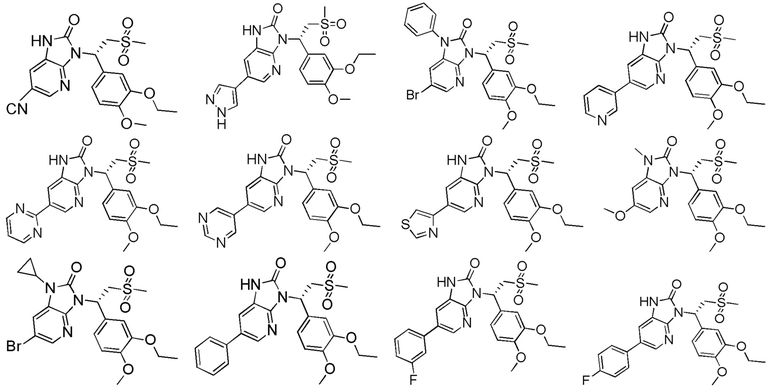
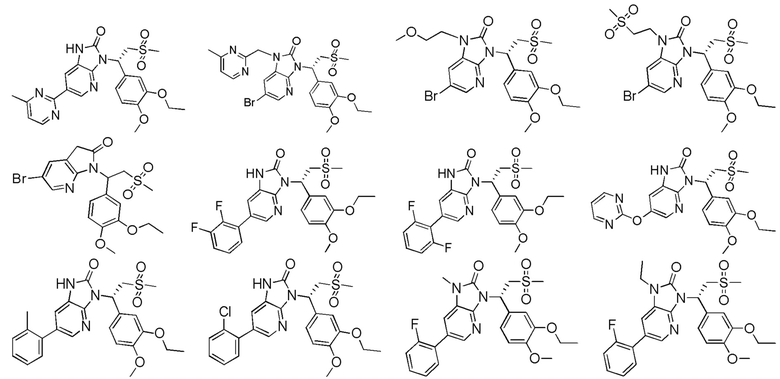
[62] Настоящее изобретение также предусматривает соединение, представленное формулой, указанной ниже, или его фармацевтически приемлемую соль, которые выбраны из группы, состоящей из 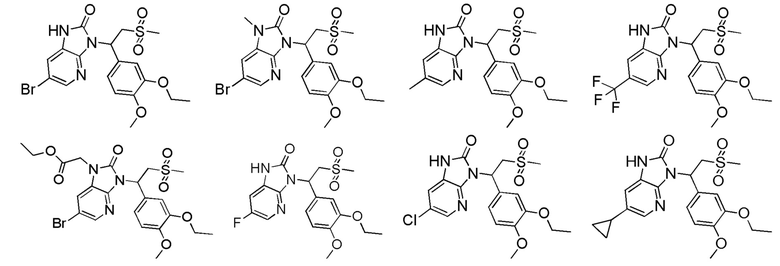
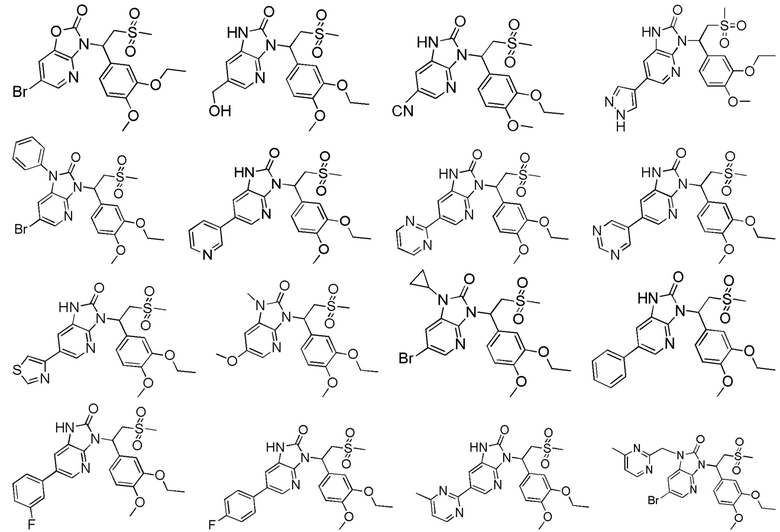
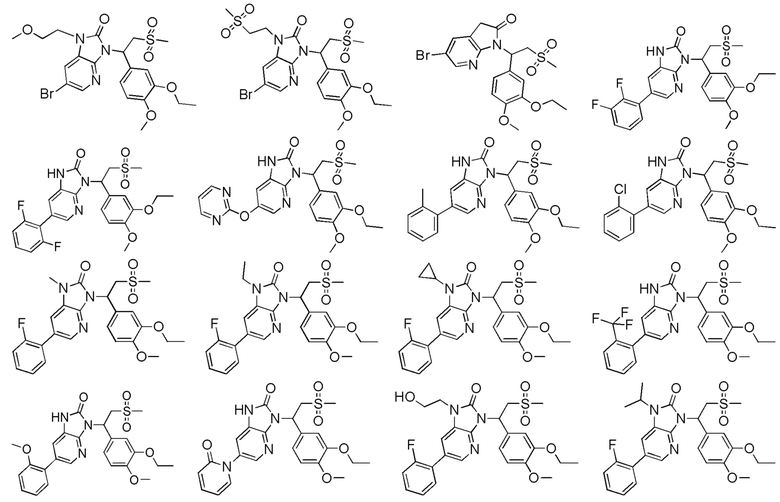
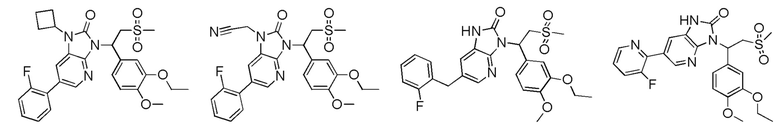
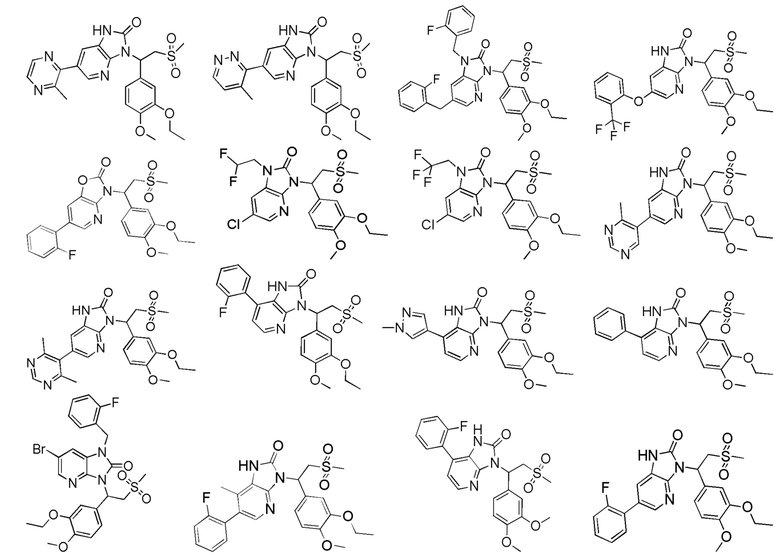
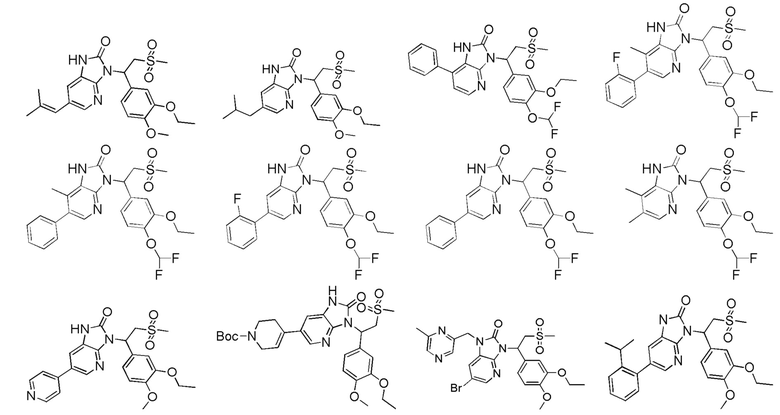
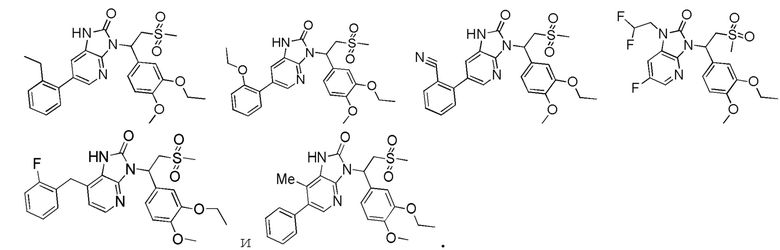
[63] В некоторых вариантах осуществления настоящего изобретения соединение или его фармацевтически приемлемая соль выбраны из группы, состоящей из 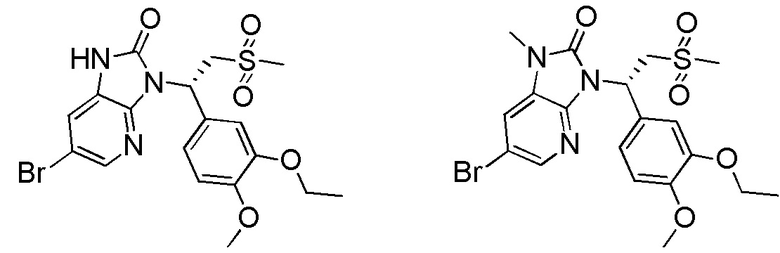
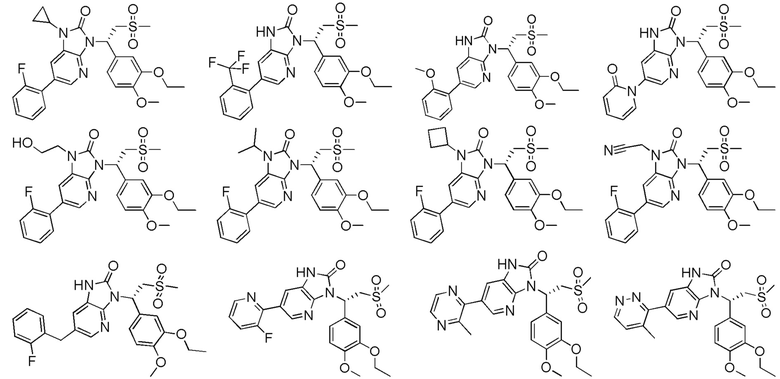
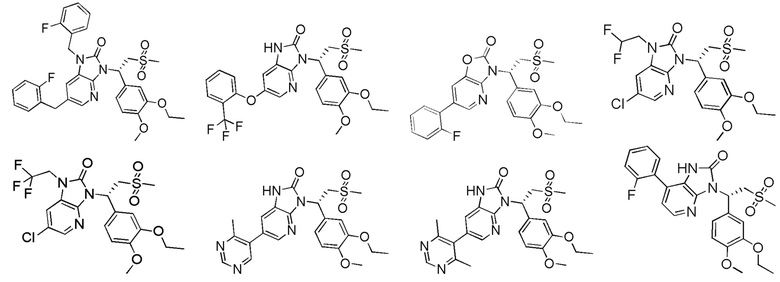
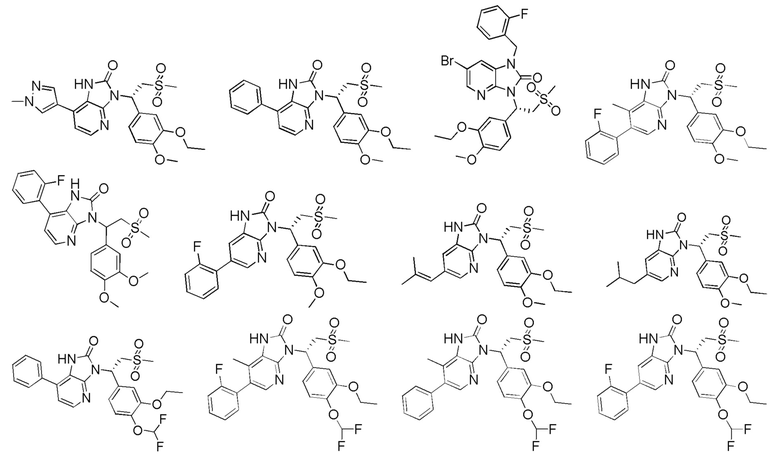
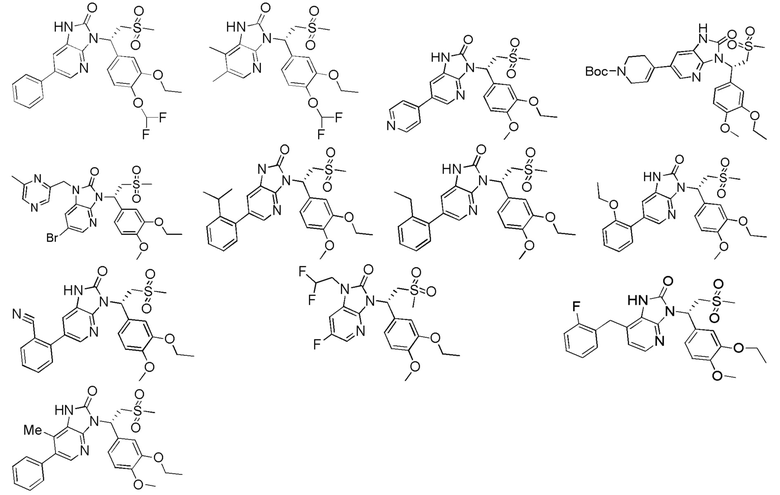
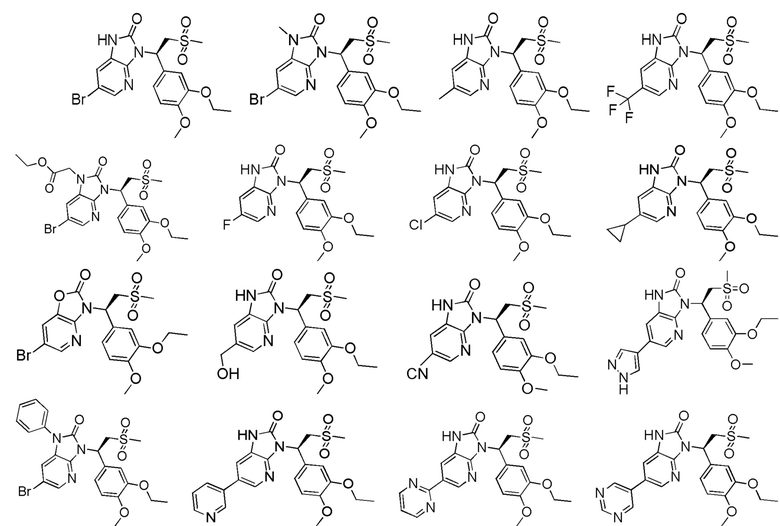
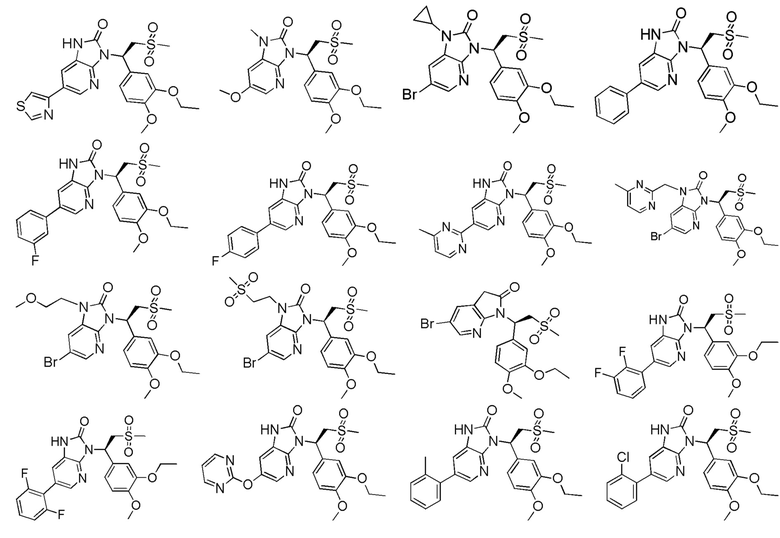
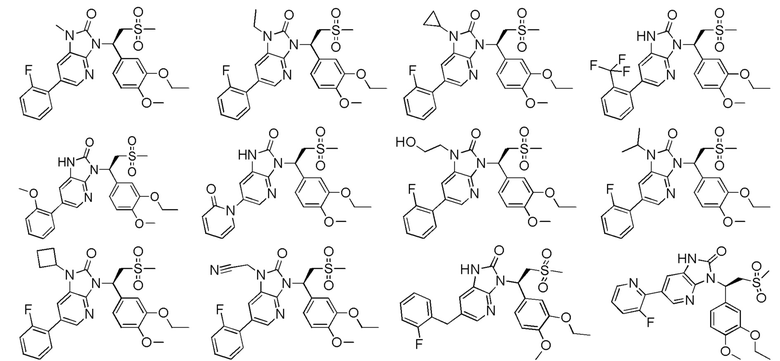
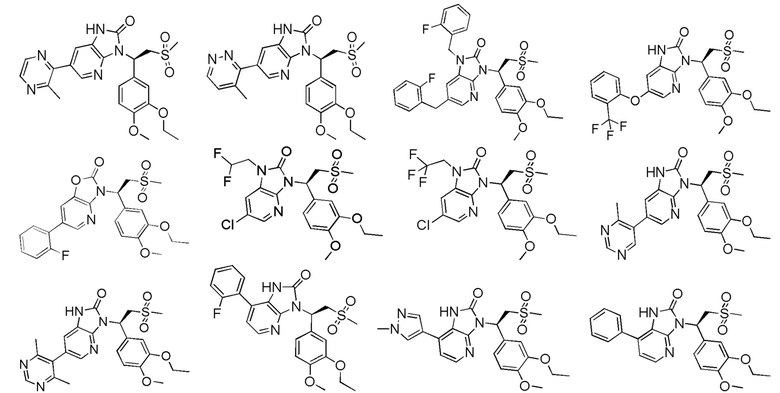
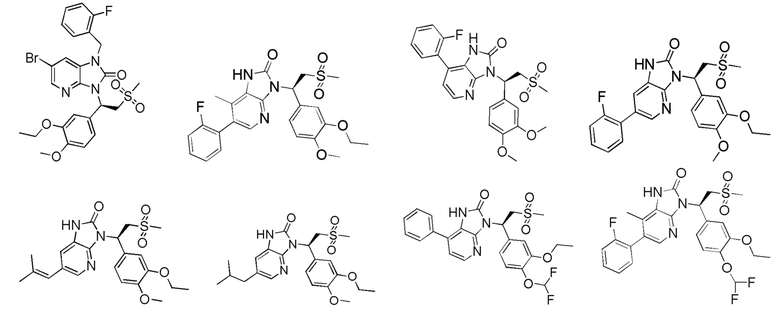
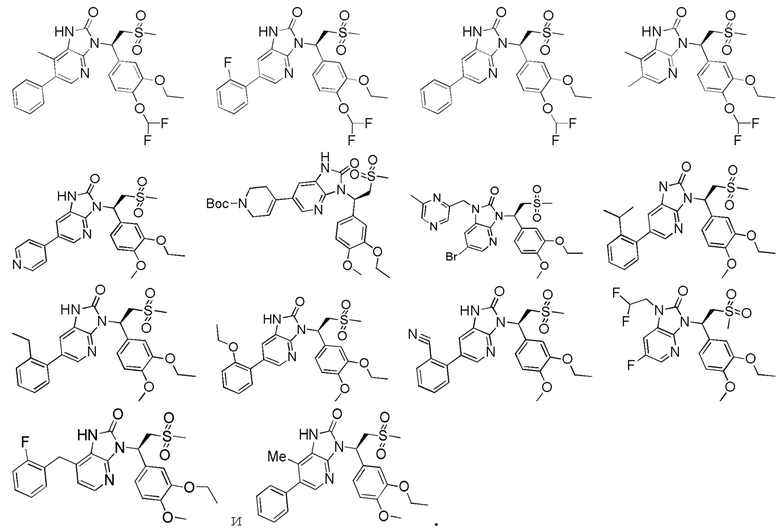
[64] Настоящее изобретение также предусматривает применение соединения или его фармацевтически приемлемой соли в изготовлении лекарственного препарата для лечения заболевания, связанного с PDE4.
[65] В некоторых вариантах осуществления настоящего изобретения заболевание, связанное с PDE4, представляет собой псориаз, псориатический артрит, хроническую обструктивную пневмонию, анкилозирующий спондилит, воспалительное заболевание кишечника.
[66] Технический эффект
[67] По сравнению с апремиластом соединение по настоящему изобретению уменьшает распространение в головном мозге, потенциально уменьшая рвоту и побочные эффекты, ассоциированные с головным мозгом. Это значительно повышает ингибирующий эффект соединения по настоящему изобретению в отношении TNFα в hPBMC и снижает терапевтически эффективную дозу в экспериментах на животных, за счет чего обеспечивается снижение терапевтически эффективной дозы для человека и повышение фактора безопасности для него. Оно обладает улучшенными характеристиками фармакокинетики и, как ожидается, подлежит введению человеку один раз в день.
[68] Определение и описание
[69] Если не указано иное, при использовании в описании и формуле настоящего изобретения следующие термины имеют следующие значения. Конкретный термин или выражение при отсутствии точного определения не стоит считать неопределенным или неясным, а следует понимать в соответствии с обычным значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту. Термин «фармацевтически приемлемый» используют в данном документе применительно к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках объективного врачебного мнения являются подходящими для применения в контакте с тканями людей и животных без избыточной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с обоснованным соотношением польза/риск.
[70] Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают путем осуществления реакции соединения, имеющего конкретный заместитель согласно настоящему изобретению, с относительно нетоксичной кислотой или основанием. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, то соль присоединения основания может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина или магния или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, то соль присоединения кислоты может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и т. п.; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту и метансульфоновую кислоту и т. п.; и соль аминокислоты (такой как аргинин и т. п.) и соль органической кислоты, такой как глюкуроновая кислота и т. п. (см. Berge et al., «Pharmaceutical Salts», Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые конкретные соединения по настоящему изобретению, которые содержат как основные, так и кислотные функциональные группы, могут быть превращены в любую соль присоединения основания или присоединения кислоты.
[71] Предпочтительно посредством приведения соли в контакт с основанием или кислотой традиционным способом, а затем отделения исходного соединения, таким образом, восстанавливают нейтральную форму соединения. Отличие между исходной формой соединения и его различными солевыми формами заключается в конкретных физических свойствах, как например различная растворимость в полярном растворителе.
[72] «Фармацевтически приемлемая соль», используемая в данном документе, относится к производному соединения по настоящему изобретению, где исходное соединение модифицировано путем образования соли с кислотой или основанием. Примеры фармацевтически приемлемой соли включают без ограничения соль неорганической кислоты или органической кислоты с основным фрагментом, такую как амин, соль щелочного металла или органическую соль с кислотным фрагментом, такую как карбоновая кислота, и т. п. Фармацевтически приемлемая соль включает традиционную нетоксичную соль или соль четвертичного аммония исходного соединения, такую как соль, образованная с помощью нетоксичной неорганической кислоты или органической кислоты. Традиционная нетоксичная соль включает без ограничения соль, полученную из неорганической кислоты и органической кислоты, где неорганическая кислота или органическая кислота выбраны из группы, состоящей из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфоновой кислоты, уксусной кислоты, аскорбиновой кислоты, бензолсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, гидройодида, гидроксила, гидроксинафталина, изэтионовой кислоты, молочной кислоты, лактозы, додецилсульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памоевой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактуроновой кислоты, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, надуксусной кислоты, янтарной кислоты, сульфаминовой кислоты, сульфаниловой кислоты, серной кислоты, танина, винной кислоты и п-толуолсульфоновой кислоты.
[73] Фармацевтически приемлемую соль по настоящему изобретению можно получать из исходного соединения, которое содержит кислотный или основный фрагмент, с помощью традиционного химического способа. Как правило, такая соль может быть получена путем осуществления реакции свободных кислотных или основных форм соединения со стехиометрическим количеством соответствующего основания или кислоты в воде, или в органическом растворителе, или в их смеси. Как правило, неводные среды, такие как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил, являются предпочтительными.
[74] В дополнение к солевой форме соединение, предусмотренное в настоящем изобретении, также находится в форме пролекарства. Пролекарственная форма соединения, описанного в данном документе, представляет собой соединение, которое легко подвергается химическим изменениям в физиологических условиях с превращением в соединение по настоящему изобретению. Кроме того, пролекарство можно превращать в соединение по настоящему изобретению посредством химического или биохимического способа в условиях in vivo.
[75] Некоторые соединения по настоящему изобретению могут находиться в несольватированной форме или сольватированной форме, в том числе в гидратированной форме. Как правило, сольватированная форма является эквивалентной несольватированной форме, и обе формы включены в объем настоящего изобретения.
[76] Некоторые соединения по настоящему изобретению могут иметь асимметрический атом углерода (оптический центр) или двойную связь. Все из рацемата, диастереомера, геометрического изомера и отдельного изомера включены в объем настоящего изобретения.
[77] Если не указано иное, абсолютная конфигурация стереогенного центра представлена клиновидной сплошной связью ( ) и клиновидной пунктирной связью (
) и клиновидной пунктирной связью (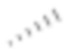 ), а относительная конфигурация стереогенного центра представлена прямой сплошной связью (
), а относительная конфигурация стереогенного центра представлена прямой сплошной связью ( ) и прямой пунктирной связью (
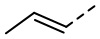
) и прямой пунктирной связью (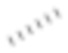 ). Если соединение, описанное в данном документе, содержит олефиновую двойную связь или другие геометрические асимметрические центры, то геометрические E- и Z-изомеры включены, если не указано иное. Подобным образом, в объем настоящего изобретения включены все таутомерные формы.
). Если соединение, описанное в данном документе, содержит олефиновую двойную связь или другие геометрические асимметрические центры, то геометрические E- и Z-изомеры включены, если не указано иное. Подобным образом, в объем настоящего изобретения включены все таутомерные формы.
[78] Соединение по настоящему изобретению может находиться в форме конкретного геометрического или стереоизомера. В настоящем изобретении подразумеваются все такие соединения, в том числе цис- и транс-изомер, (-)- и (+)-энантиомер, (R)- и (S)-энантиомер, диастереоизомер, (D)-изомер, (L)-изомер, и рацемическая смесь и другие смеси, например, энантиомерно или диастереоизомерно обогащенная смесь, все из которых включены в объем настоящего изобретения. Заместитель, такой как алкил, может иметь дополнительный асимметрический атом углерода. Все эти изомеры и их смеси включены в объем настоящего изобретения.
[79] Оптически активные (R)- и (S)-изомеры или D- и L-изомеры могут быть получены с использованием хирального синтеза, или хиральных реагентов, или других традиционных методик. Если требуется получение одного типа энантиомера конкретного соединения по настоящему изобретению, чистый необходимый энантиомер может быть получен путем асимметрического синтеза или дериватизации с помощью хирального вспомогательного вещества с последующим разделением полученной диастереомерной смеси и отщеплением вспомогательной группы. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксильная), соединение вступает в реакцию с подходящей оптически активной кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному разделению посредством традиционного способа из уровня техники с получением чистого энантиомера. Кроме того, энантиомер и диастереоизомер обычно выделяют посредством хроматографии, которую проводят с использованием хиральной неподвижной фазы и необязательно объединяют со способом химической дериватизации (как например с карбаматом, образованным из амина).
[80] Соединение по настоящему изобретению может содержать неприродное соотношение атомных изотопов при одном или более атомах, которые составляют соединение. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). Все изотопные варианты соединения по настоящему изобретению, вне зависимости от радиоактивности, включены в объем настоящего изобретения.
[81] Термин «фармацевтически приемлемый носитель» относится к любому средству или несущей среде, которые способны доставлять эффективное количество активного вещества по настоящему изобретению, не оказывают отрицательного воздействия на биологическую активность активного вещества и не вызывают какого-либо токсичного побочного эффекта у хозяина или пациента. Иллюстративный носитель включает воду, растительное и минеральное масло, кремовую основу, лосьонную основу, мазевую основу и т. п. Основа включает суспендирующее средство, загуститель, вещество, способствующее проникновению, и т. п. Их составы хорошо известны специалисту в области косметических средств или в области фармацевтических препаратов для местного применения.
[82] Термин «вспомогательное вещество» обычно относится к носителю, разбавителю и/или среде, необходимым для составления эффективной фармацевтической композиции.
[83] Применительно к лекарственному препарату или фармакологически активному средству термин «эффективное количество» или «терапевтически эффективное количество» относится к нетоксичному, но достаточному количеству для достижения необходимого эффекта лекарственного препарата или средства. В отношении лекарственной формы по настоящему изобретению для перорального применения «эффективное количество» активного вещества в композиции относится к количеству, необходимому для достижения необходимого эффекта при объединении с другим активным веществом в композиции. Эффективное количество отличается для каждого человека и определяется в зависимости от возраста и общего состояния реципиента, а также от конкретного активного вещества. Подходящее эффективное количество в каждом отдельном случае может быть определено специалистом в данной области техники на основе стандартного эксперимента.
[84] Термин «активный ингредиент», «терапевтическое средство», «активное вещество» или «активное средство» относится к химическому соединению, с помощью которого можно осуществлять эффективное лечение целевого нарушения, заболевания или состояния.
[85] «Необязательный» или «необязательно» означает, что последующее событие или условие может реализовываться, но не является необходимым, и что термин включает случаи, в которых событие или условие реализуется, и случаи, в которых событие или условие не реализуется.
[86] Термин «замещенный» означает, что один или более атомов водорода при конкретном атоме замещены заместителем, в том числе дейтерием и вариантами водорода, при условии, что валентность конкретного атома является нормальной и замещенное соединение является стабильным. Если заместитель представляет собой кетогруппу (т. е. =O), то это означает, что два атома водорода являются замещенными. Положения в ароматическом кольце не могут быть замещены кетогруппой. Термин «необязательно замещенный» означает, что атом может быть замещенным или не быть замещенным заместителем, если не указано иное, причем тип и число заместителей могут быть произвольными при условии, что это химически достижимо.
[87] Если любая переменная (такая как R) встречается более одного раза в составе или структуре соединения, то определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то данная группа необязательно может быть замещена не более чем двумя R, при этом определение R в каждом случае является независимым. Более того, комбинация заместителя и/или его варианта является допустимой, только если такая комбинация приводит к стабильному соединению.
[88] Если число линкерных групп равняется 0, как например -(CRR)0-, это означает, что линкерная группа представляет собой одинарную связь.
[89] Если одна из переменных выбрана из одинарной связи, это означает, что две группы, соединенные одинарной связью, соединены непосредственно. Например, если L в A-L-Z представляет собой одинарную связь, то структура A-L-Z фактически представляет собой A-Z.
[90] Если заместитель не указан, это означает, что заместитель отсутствует. Например, если X не указан в A-X, то структура A-X фактически представляет собой A. Если связь заместителя может обеспечивать сшивку с более чем одним атомом в кольце, то такой заместитель может быть связан с любым атомом в кольце. Если для перечисленного заместителя не указано посредством какого атома он присоединен к соединению, включенному в общую химическую формулу, но конкретно не указанному, то такой заместитель может быть связан посредством любого из его атомов. Комбинация заместителей и/или их вариантов является допустимой, только если такая комбинация может приводить к стабильному соединению. Например, структурное звено 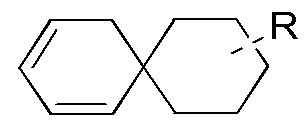 или
или 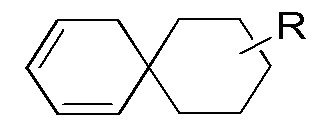 означает, что заместитель R может находиться в любом положении в циклогексиле или циклогексадиене.
означает, что заместитель R может находиться в любом положении в циклогексиле или циклогексадиене.
[91] Если не указано иное, термин «гетеро» представляет собой гетероатом или содержащую гетероатом группу (например, радикал, содержащий гетероатом), включают атом, отличный от углерода (C) и водорода (H), и при этом радикал, содержащий вышеуказанный гетероатом, например, включает кислород (O), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2- и группу, состоящую из -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- и -S(=O)N(H)-, каждый из которых необязательно замещен.
[92] Если не указано иное, термин «кольцо» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Так называемое кольцо включает одно кольцо, сборку колец, спирокольцо, конденсированное кольцо или кольцо с мостиковой связью. Число атомов в кольце обычно определено как число членов в кольце, например «5-7-членное кольцо» означает, что 5-7 атомов объединены в кольцо. Если не указано иное, кольцо необязательно содержит 1-3 гетероатома. Следовательно, «5-7-членное кольцо» включает, например, фенил, пиридинил и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо соответствует вышеуказанному определению.
[93] Если не указано иное, термин «гетероцикл» или «гетероцикло» относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или содержащую гетероатом группу, которое может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим), и может содержать атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, независимо выбранных из N, O и S, где любой из вышеуказанного гетероцикла может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы, представляющие собой азот и серу, необязательно могут быть окислены (т. е. NO и S(O)p, причем p равняется 1 или 2). Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероцикл может быть присоединен к боковой группе посредством любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является устойчивым, гетероцикл, описанный в данном документе, может быть замещен в положениях, соответствующих атому углерода или азота. Атом азота в гетероцикле необязательно является кватернизированным. В предпочтительном варианте осуществления, если общее количество атомов S и O в гетероцикле превышает 1, то гетероатомы не являются смежными друг с другом. В другом предпочтительном варианте осуществления общее количество атомов S и O в гетероцикле не превышает 1. Как используется в данном документе, термин «ароматическая гетероциклическая группа» или «гетероарил» относится к стабильному 5-, 6-, 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклическому ароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, независимо выбранных из N, O и S. Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероатомы, представляющие собой азот и серу, необязательно могут быть окислены (т. е. NO и S(O)p, причем p равняется 1 или 2). Следует отметить, что общее количество атомов S и O в ароматическом гетероцикле не превышает одного. Кольцо с мостиковой связью также включено в определение гетероцикла. Кольцо с мостиковой связью образуется, если один или более атомов (т. е. C, O, N или S) соединяют два несмежных атома углерода или азота. Предпочтительное кольцо с мостиковой связью включает без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостиковая связь всегда превращает моноциклическое кольцо в трициклическое кольцо. В кольце с мостиковой связью заместитель в кольце также может присутствовать при мостиковой связи.
[94] Примеры гетероциклического соединения включают без ограничения акридинил, азоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидрохиндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, фенолоксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазолил, пиридоимидазолил, пиридотиазолил, пиридинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Также включены соединения с конденсированными кольцами и спиросоединения.
[95] Если не указано иное, термин «гидрокарбил» или его гипонимы (например, алкил, алкенил, алкинил и арил и т. д.), сами по себе или в качестве части другого заместителя, относятся к линейной, разветвленной цепи, или циклическому углеводородному радикалу, или любой их комбинации. Они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно-, ди- или полизамещенными, могут быть одновалентными (например, метил), двухвалентными (например, метилен) или многовалентными (например, метенил), могут также включать двухвалентную или многовалентную группу, имеют конкретное число атомов углерода (например, C1-C12 означает 1-12 атомов углерода, причем C1-12 выбран из C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12; C3-12 выбран из C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12). Термин «гидрокарбил» включает без ограничения алифатический гидрокарбил и ароматический гидрокарбил. Алифатический гидрокарбил включает линейный и циклический гидрокарбил, в частности, включает без ограничения алкил, алкенил и алкинил. Ароматический гидрокарбил включает без ограничения 6-12-членный ароматический гидрокарбил, такой как фенил, нафтил и т. п. В некоторых вариантах осуществления термин «гидрокарбил» относится к линейной или разветвленной группе или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной и может включать двухвалентную или многовалентную группу. Примеры насыщенной гидрокарбильной группы включают без ограничения метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил и гомолог или изомер н-амила, н-гексила, н-гептила, н-октила и других радикалов. Ненасыщенный гидрокарбил содержит одну или более двойных или тройных связей. Примеры ненасыщенного алкила включают без ограничения винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и более высшие гомологи и изомеры.
[96] Если не указано иное, термин «гетерогидрокарбил» или его гипонимы (такие как гетероалкил, гетероалкенил, гетероалкинил и гетероарил и т. д.), сами по себе или в качестве части другого заместителя, относятся к устойчивой линейной, разветвленной или циклической углеводородной группе или любой их комбинации, которая содержит конкретное число атомов углерода и по меньшей мере один гетероатом. В некоторых вариантах осуществления термин «гетероалкил», сам по себе или в комбинации с другим термином, относится к стабильной линейной цепи, разветвленному углеводородному радикалу или их комбинации, которые содержат конкретное число атомов углерода и по меньшей мере один гетероатом. В конкретном варианте осуществления гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизирован. Гетероатом или содержащая гетероатом группа могут находиться в любом внутреннем положении гетерогидрокарбила, включая положение, в котором гидрокарбил присоединяется к остальной части молекулы. Но термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкил) применяются в их обычном значении и относятся к алкильной группе, соединенной с остальной частью молекулы посредством атома кислорода, аминогруппы или атома серы соответственно. Примеры включают без ограничения -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Могут присутствовать не более двух смежных гетероатомов, как например -CH2-NH-OCH3.
[97] Если не указано иное, термин «циклогидрокарбил», «гетероциклогидрокарбил» или их гипонимы (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т. д.), применяемые сами по себе или в комбинации с другим термином, относятся к циклизированному «гидрокарбилу» или «гетерогидрокарбилу». Кроме того, в случае гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкил и гетероциклоалкил) один гетероатом может занимать положение, в котором гетероцикл присоединяется к остальной части молекулы. Примеры циклоалкила включают без ограничения циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т. п. Неограничивающие примеры гетероциклоалкила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[98] Если не указано иное, термин «алкил» относится к линейной цепи или разветвленной насыщенной углеводородной группе, которая может быть монозамещенной (например, -CH2F) или полизамещенной (например, -CF3), может быть одновалентной (например, метил), двухвалентной (например, метилен) или многовалентной (например, метенил). Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и т. п.
[99] Если не указано иное, термин «алкенил» относится к алкильной группе, содержащей одну или более углерод-углеродных двойных связей в любом положении в цепи, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или многовалентной. Примеры алкенила включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и т. п.
[100] Если не указано иное, термин «алкилен» или «алкенилалкил» относится к алкилу, замещенному алкенилом.
[101] Если не указано иное, термин «алкинил» относится к алкильной группе, содержащей одну или более углерод-углеродных тройных связей в любом положении в цепи, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или многовалентной. Примеры алкинила включают этинил, пропинил, бутинил, пентинил, гексинил и т. п.
[102] Если не указано иное, циклоалкил включает любой устойчивый циклический или полициклический гидрокарбил, и при этом любой атом углерода является насыщенным, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным. Примеры циклоалкила включают без ограничения циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодеканил и т. п.
[103] Если не указано иное, циклоалкенил включает любой устойчивый циклический или полициклический гидрокарбил, содержащий одну или более ненасыщенных углерод-углеродных двойных связей в любом положении в кольце, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным. Примеры циклоалкенила включают без ограничения циклопентенил, циклогексенил и т. п.
[104] Если не указано иное, циклоалкинил включает любой устойчивый циклический или полициклический гидрокарбил, имеющий одну или более углерод-углеродных тройных связей в любом положении в кольце, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным.
[105] Если не указано иное, термин «галогено» или «галоген», сам по себе или как часть другого заместителя, обозначает атом фтора, хлора, брома или йода. Кроме того, подразумевается, что термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин «галоген(C1-C4)алкил» включает без ограничения трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т. п. Примеры галогеналкила включают без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
[106] Термин «алкокси» представляет собой любой алкил, определенный выше, характеризующийся конкретным числом атомов углерода, присоединенный с помощью кислородного мостика. Если не указано иное, C1-6алкокси включает C1, C2, C3, C4, C5 и C6алкокси. Примеры алкокси включают без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси. Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому заместителю, который может быть моно-, ди- или полизамещенным, может быть одновалентным, двухвалентным или многовалентным, может представлять собой одно кольцо или несколько колец (например, от одного до трех колец; где по меньшей мере одно кольцо является ароматическим), которые являются вместе конденсированными или соединенными ковалентно. Термин «гетероарил» относится к арилу (или кольцу), содержащему от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизирован. Гетероарил может присоединяться к остальной части молекулы посредством гетероатома. Неограничивающие примеры арила или гетероарила включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместители любой описанной выше арильной и гетероарильной кольцевой системы выбраны из приемлемых заместителей, описанных ниже.
[107] Если не указано иное, в случае объединения арила с другими терминами (такими как арилокси, арилтио, арилалкил), арил включает арильное и гетероарильное кольцо, как определено выше. Таким образом, подразумевается, что термин «арилалкил» включает группы (например, бензил, фенэтил, пиридилметил и т. д.), где арил присоединен к алкилу, в том числе алкилу, где атом углерода (например, метилен) был заменен на такой атом, как кислород, например, феноксиметил, 2-пиридилокси, 3-(1-нафтилокси)пропил и т. п.
[108] Термин «уходящая группа» относится к функциональной группе или атому, которые могут быть заменены на другую функциональную группу или атом посредством реакции замещения (такой как реакция замещения по аффинности). Например, иллюстративные уходящие группы включают трифлат; хлор, бром и йод; сульфонатную группу, как например мезилат, тозилат, п-бромбензолсульфонат, п-толуолсульфонаты и т. п.; ацилокси, как например ацетокси, трифторацетокси и т. п.
[109] Термин «защитная группа» включает без ограничения «защитную группу для аминогруппы», «защитную группу для гидроксигруппы» или «защитную группу для тиогруппы». Термин «защитная группу для аминогруппы» относится к защитной группе, подходящей для блокирования побочных реакций с участием азота аминогруппы. Иллюстративные защитные группы для аминогруппы включают без ограничения формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутилоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т. п. Термин «защитная группа для гидроксигруппы» относится к защитной группе, подходящей для блокирования побочных реакций с участием гидроксигруппы. Иллюстративные защитные группа для гидроксигруппы включают без ограничения алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т. п.
[110] Соединение по настоящему изобретению можно получать посредством различных способов синтеза, хорошо известных специалистам в данной области техники, в том числе следующий перечисленный вариант осуществления, вариант осуществления, образованный следующим перечисленным вариантом осуществления в комбинации с другими способами химического синтеза и эквивалентными заменами, хорошо известными специалистам в данной области техники. Предпочтительный вариант осуществления включает без ограничения вариант осуществления настоящего изобретения.
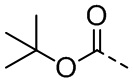
[111] Все растворители, используемые в настоящем изобретении, являются коммерчески доступными. В данном настоящем изобретении используются следующие сокращения: водн. означает водный; «HATU» означает гексафторфосфат 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; «EDC» означает гидрохлорид N-(3-диметиламинопропил)-N’-этилкарбодиимида; «m-CPBA» означает 3-хлорпероксибензойную кислоту; «экв.» означает эквивалент; «CDI» означает карбонилдиимидазол; «DCM» означает дихлорметан; «PE» означает петролейный эфир; «DIAD» означает диизопропилазодикарбоксилат; «DMF» означает N,N-диметилформамид; «DMSO» означает диметилсульфоксид; «EtOAc» означает этилацетат; «EtOH» означает этанол; «MeOH» означает метанол; «CBz» означает карбобензилокси, тип защитной группы для амина; «BOC» означает трет-бутилоксикарбонил; «HOAc» означает уксусную кислоту; «NaCNBH3» означает цианоборгидрид натрия; к. т. означает комнатную температуру; O/N означает в течение ночи; «THF» означает тетрагидрофуран; «Boc2O» означает ди-трет-бутилдикарбонат; «TFA» означает трифторуксусную кислоту; «DIPEA» означает этилдиизопропиламин; «SOCl2» означает тионилхлорид; «CS2» означает сероуглерод; TsOH означает п-толуолсульфоновую кислоту; «NFSI» означает N-фторбензолсульфонимид; «NCS» означает 1-хлорпирролидин-2,5-дикетон; «n-Bu4NF» означает фторид тетрабутиламмония; «iPrOH» означает 2-пропанол; «т. пл.» означает точку плавления; «LDA» означает диизопропиламид лития.
[112] Соединения названы вручную или с помощью программного обеспечения ChemDraw®, а для коммерчески доступных соединений используются названия в соответствии с каталогом их поставщика.
Подробное описание предпочтительных вариантов осуществления
[113] Следующие примеры дополнительно иллюстрируют настоящее изобретение, однако настоящее изобретение ими не ограничивается. Настоящее изобретение было описано подробно в данном документе, и его конкретные варианты осуществления также раскрыты. Для специалиста в данной области техники очевидно как модифицировать и улучшить варианты осуществления настоящего изобретения в пределах сущности и объема настоящего изобретения.
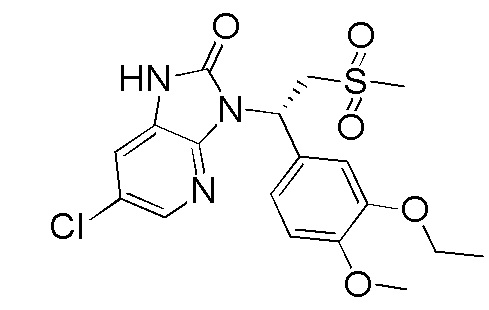
[114] Вариант осуществления 1: WX001
Путь синтеза
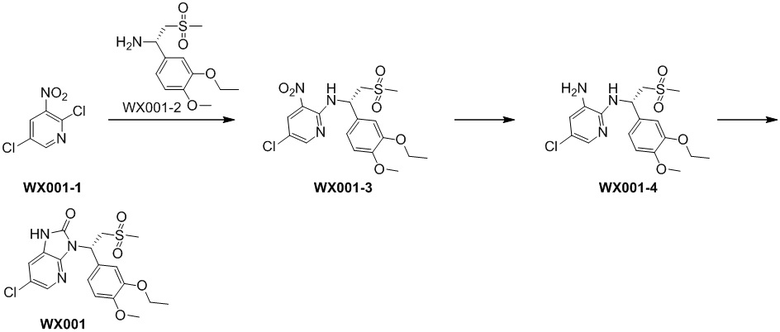
[115] Стадия 1. Получение соединения WX001-3
[116] 2,5-Дихлор-3-нитропиридин (WX001-1) (464,42 мг, 1,04 ммоль) и соединение WX001-2 (569,40 мг, 2,08 ммоль) растворяли в ацетонитриле (3,00 мл) при комнатной температуре с последующим добавлением карбоната калия (287,48 мг, 2,08 ммоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 2 часов. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали этилацетатом (5 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток очищали с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=10/1-2/1, объемное соотношение) с получением целевого продукта WX001-3. MS-ESI масса/заряд: 430,0 [M+H]+.
[117] Стадия 2. Получение соединения WX001-4
[118] Соединение WX001-3 (180,00 мг, 418,73 мкмоль) и хлорид аммония (179,18 мг, 3,35 ммоль) растворяли в этаноле (5,00 мл) и воде (500,00 мкл) при комнатной температуре с последующим добавлением порошка железа (116,93 мг, 2,09 ммоль). Реакционную смесь перемешивали при 80°C в течение 3 часов. После завершения реакции добавляли этилацетат (5 мл) с последующей фильтрацией, фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли воду (8 мл) и экстрагировали этилацетатом (5 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX001-4. MS-ESI масса/заряд: 400,0 [M+H]+.
[119] Стадия 3. Получение соединения WX001
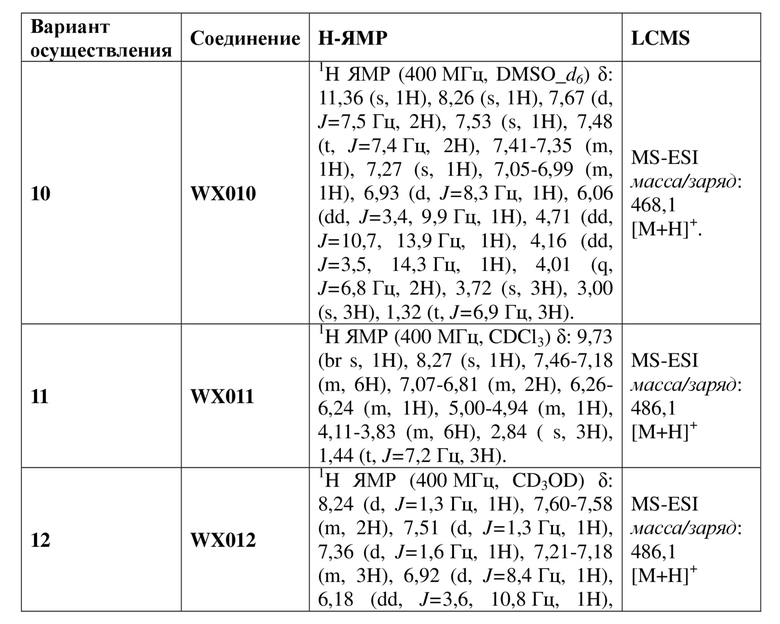
[120] Соединение WX001-4 (100,00 мг, 250,07 мкмоль) и триэтиламина (50,6 мг, 500,14 мкмоль) растворяли в тетрагидрофуране (2 мл) при комнатной температуре. Раствор охлаждали до 0°C на ледяной бане и к нему добавляли триэтилортоформиат (89,05 мг, 300,08 мкмоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа. После завершения реакции смесь гасили водой (8 мл) и экстрагировали этилацетатом (5 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX001. MS-ESI масса/заряд: 426,0 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 8,03 (d, J=2,0 Гц, 1H), 7,36 (dd, J=1,9, 19,4 Гц, 2H), 7,17 (dd, J=1,8, 8,3 Гц, 1H), 6,93 (d, J=8,3 Гц, 1H), 6,15 (dd, J=3,9, 10,7 Гц, 1H), 4,87-4,77 (m, 1H), 4,09-3,99 (m, 3H), 3,82 (s, 3H), 2,95 (s, 3H), 1,40 (t, J=7,0 Гц, 3H).
[121] Вариант осуществления 2: WX002
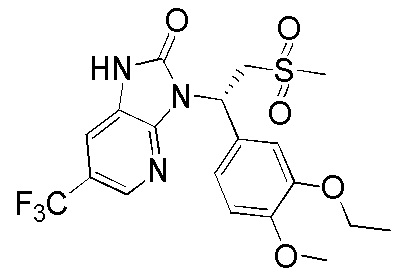
Путь синтеза
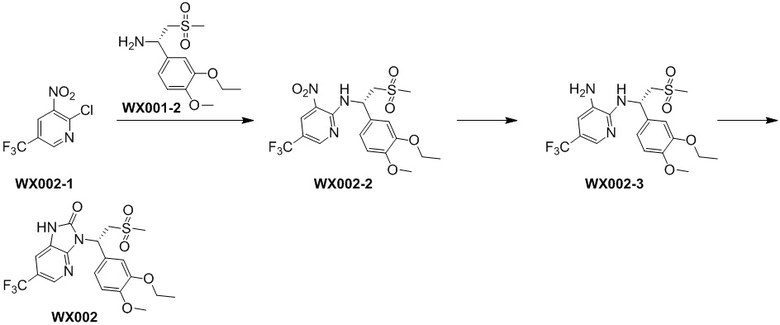
[122] Стадия 1. Получение соединения WX002-2
[123] Соединение WX002-1 (295,68 мг, 662,13 мкмоль) и соединение WX001-2 (354,9 мг, 1,30 ммоль) растворяли в ацетонитриле (3,00 мл) при комнатной температуре с последующим добавлением карбоната калия (183,03 мг, 1,32 ммоль). Реакционную смесь нагревали при комнатной температуре и перемешивали в течение 2 часов. После завершения реакции смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (5 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=10/1-2/1, объемное соотношение) с получением целевого продукта WX002-2. MS-ESI масса/заряд: 464,0 [M+H]+.
[124] Стадия 2. Получение соединения WX002-3
[125] Соединение WX002-2 (200,00 мг, 431,56 мкмоль) и хлорид аммония (184,67 мг, 3,45 ммоль) добавляли в этанол (5,00 мл) и воду (500,00 мкл) при комнатной температуре с последующим добавлением порошка железа (120,51 мг, 2,16 ммоль). Реакционную смесь перемешивали при 90°C в течение 3 часов. После завершения реакции добавляли этилацетат (8 мл) и нерастворимые вещества удаляли посредством фильтрации. Фильтрат концентрировали при пониженном давлении. К остатку добавляли воду (10 мл), экстрагировали этилацетатом (8 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX002-3. MS-ESI масса/заряд: 434,0 [M+Na]+.
[126] Стадия 3. Получение соединения WX002
[127] Соединение WX002-3 (30,00 мг, 69,21 мкмоль) и триэтиламин (14,12 мг, 0,14 ммоль) растворяли в тетрагидрофуране (2,00 мл) при комнатной температуре. Раствор охлаждали до 0°C на ледяной бане и к нему добавляли триэтилортоформиат (24,65 мг, 83,05 мкмоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. После завершения реакции добавляли воду (8 мл) и экстрагировали этилацетатом (5 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX002. MS-ESI масса/заряд: 460,0 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,97 (br s, 1H), 8,39 (s, 1H), 7,50 (s, 1H), 7,33 (s, 1H), 7,25 (d, J=8,3 Гц, 1H), 6,86 (d, J=8,3 Гц, 1H), 6,26 (dd, J=4,0, 10,0 Гц, 1H), 4,93 (dd, J=10,4, 14,2 Гц, 1H), 4,20-4,06 (m, 2H), 3,97-3,78 (m, 4H), 2,86 (s, 3H), 1,47 (t, J=6,9 Гц, 3H).
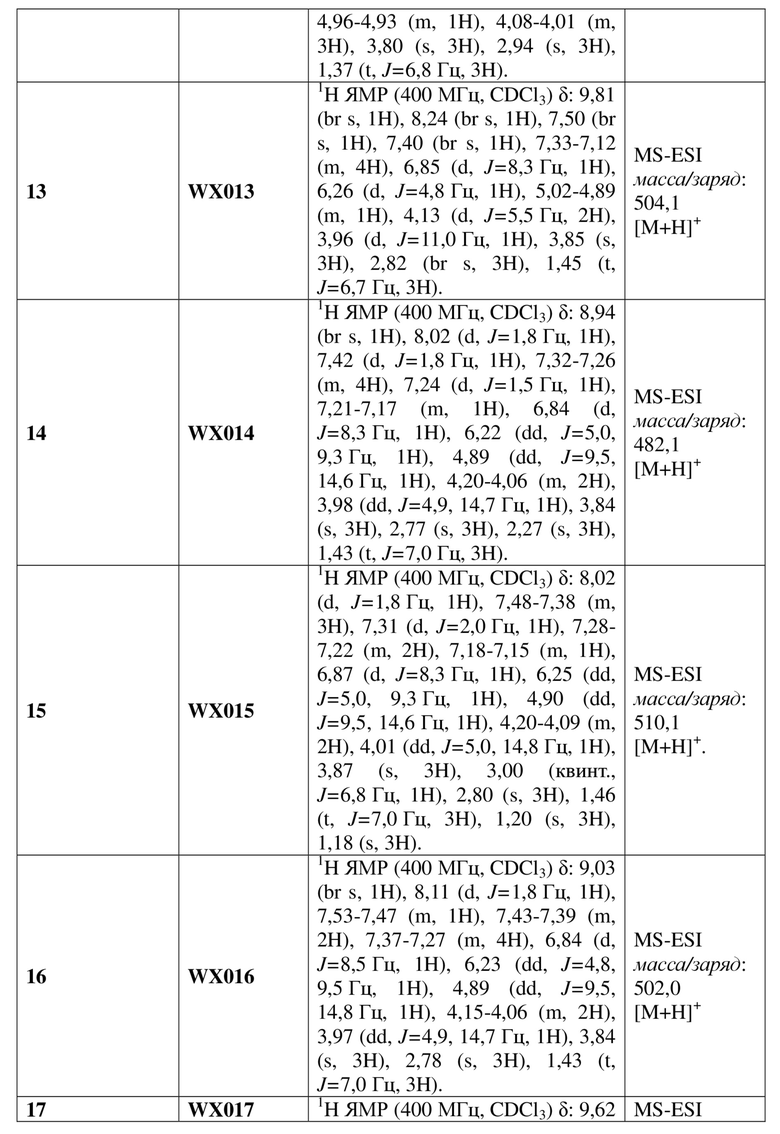
[128] Вариант осуществления 3: WX003
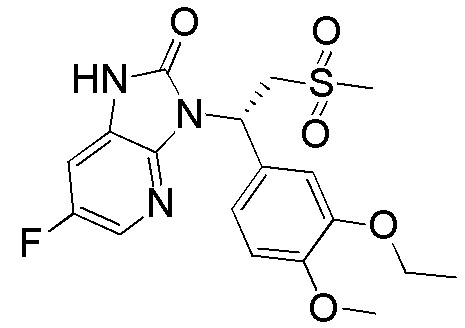
Путь синтеза
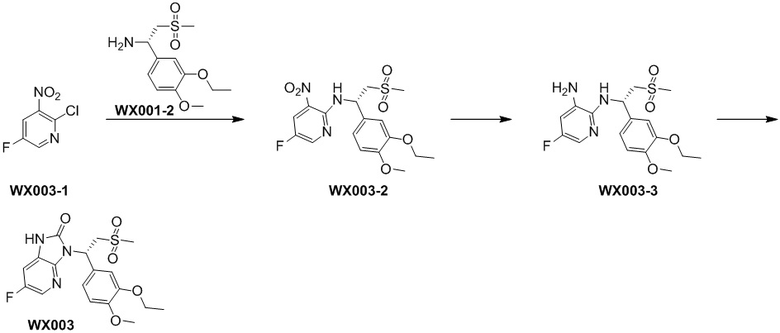
[129] Стадия 1. Получение соединения WX003-2
[130] Соединение WX003-1 (505,93 мг, 1,13 ммоль) и соединение WX001-2 (671,00 мг, 2,26 ммоль) растворяли в ацетонитриле (3,00 мл) при комнатной температуре с последующим добавлением карбоната калия (313,17 мг, 2,27 ммоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 16 часов. После завершения реакции добавляли воду (10 мл) и экстрагировали этилацетатом (5 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=10/1-2/1, объемное соотношение) с получением целевого продукта WX003-2. MS-ESI масса/заряд: 435,9 [M+Na]+.
[131] Стадия 2. Получение соединения WX003-3
[132] Соединение WX003-2 (150,00 мг, 362,83 мкмоль) и хлорид аммония (155,26 мг, 2,90 ммоль) растворяли в этаноле (5,00 мл) и воде (500,00 мкл) при комнатной температуре с последующим добавлением порошка железа (101,32 мг, 1,81 ммоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 3 часов. После завершения реакции смесь разбавляли этилацетатом (5 мл) с последующей фильтрацией и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли воду (5 мл) и экстрагировали этилацетатом (5 мл x 2). Органические фазы объединяли, промывали водой (10 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX003-3. MS-ESI масса/заряд: 384,0 [M+Na]+.
[133] Стадия 3. Получение соединения WX003
[134] Соединение WX003-3 (70,00 мг, 182,56 ммоль) и триэтиламин (36,42 мг, 0,36 ммоль) растворяли в тетрагидрофуране (2,00 мл) при комнатной температуре. Раствор охлаждали до 0°C на ледяной бане и к нему добавляли триэтилортоформиат (65,01 мг, 219,07 мкмоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа. После завершения реакции добавляли воду (8 мл) и экстрагировали этилацетатом (5 x 2 мл). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX003. MS-ESI масса/заряд: 410,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 7,95 (t, J=2,0 Гц, 1H), 7,33 (d, J=1,8 Гц, 1H), 7,24 (dd, J=2,4, 8,4 Гц, 1H), 7,17 (dd, J=1,8, 8,3 Гц, 1H), 6,93 (d, J=8,3 Гц, 1H), 6,15 (dd, J=3,8, 10,8 Гц, 1H), 4,88-4,82 (m, 1H), 4,13-3,99 (m, 3H), 3,81 (s, 3H), 2,98-2,90 (m, 3H), 1,39 (t, J=7,0 Гц, 3H).
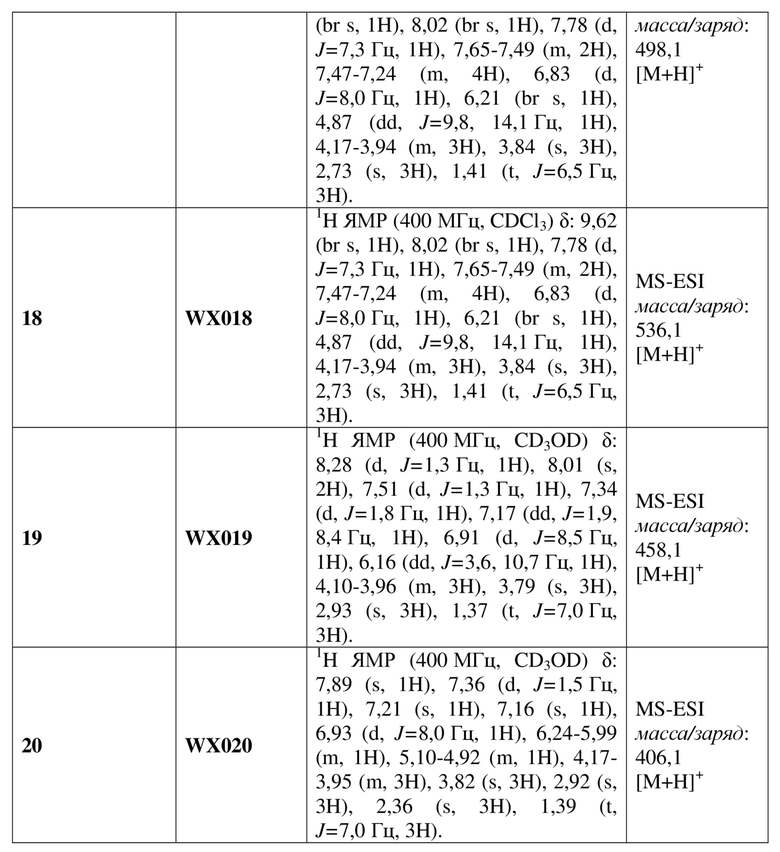
[135] Вариант осуществления 4: WX004
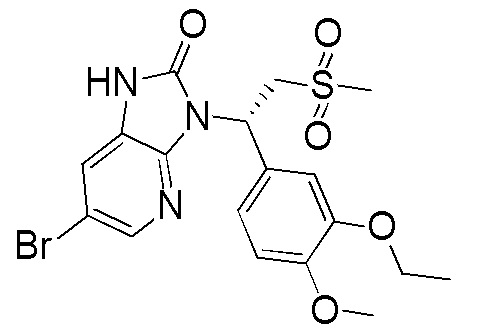
Путь синтеза
 Стадия 1. Получение соединения WX004-2
Стадия 1. Получение соединения WX004-2
[136] Соединение WX001-2 (10,00 г, 36,58 ммоль) растворяли в N,N-диметилформамиде (100,00 мл) при комнатной температуре с последующим последовательным добавлением N,N-диизопропилэтиламина (14,18 г, 109,74 ммоль, 19,16 мл) и соединение WX004-1 (17,37 г, 73,16 ммоль). Реакционную смесь нагревали до 100°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. К остатку добавляли метанол (30 мл) и перемешивали при комнатной температуре в течение 10 минут с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя с получением целевого продукта WX004-2. MS (ESI) масса/заряд: 473,7 [M+H]+, 475,7 [M+H+2]+.
[137] Стадия 2. Получение соединения WX004-3
[138] Соединение WX004-2 (5,00 г, 10,54 ммоль) и хлорид аммония (5,64 г, 105,40 ммоль) растворяли в метаноле (50,00 мл) при комнатной температуре с последующим добавлением порциями порошка цинка (3,45 г, 52,70 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа в атмосфере азота. После завершения реакции смесь фильтровали с применением диатомита. Фильтрат концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли воду (50 мл) и этилацетат (50 мл). Водную фазу экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX004-3. MS (ESI) масса/заряд: 443,7 [M+H]+, 445,7 [M+H+2]+.
[139] Стадия 3. Получение соединения WX004
[140] Соединение WX004-3 (4,20 г, 9,45 ммоль) и триэтиламин (5,26 г, 51,99 ммоль, 7,21 мл) растворяли в тетрагидрофуране (50,00 мл) при комнатной температуре. Раствор охлаждали до 0°C на ледяной бане и к нему 4 партиями добавляли триэтилортоформиат (981,73 мг, 3,31 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 часов. После завершения реакции смесь гасили насыщенным солевым раствором (15 мл), разбавляли водой (15 мл) и экстрагировали этилацетатом (10 x 3 мл). Органические фазы объединяли, промывали водой (25 x 2 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=4/1-2/3, объемное соотношение) с получением целевого продукта WX004. (3,5 г, выход: 77,17%). MS (ESI) масса/заряд: 470,0 [M+H]+, 472,0 [M+H+2]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,09 (s, 1H), 8,14 (d, J=2,0 Гц, 1H), 7,41 (d, J=2,0 Гц, 1H), 7,31 (d, J=2,0 Гц, 1H), 7,22 (dd, J=2,0, 8,3 Гц, 1H), 6,84 (d, J=8,3 Гц, 1H), 6,17 (dd, J=4,5, 10,0 Гц, 1H), 4,86 (dd, J=10,3, 14,6 Гц, 1H), 4,98-4,78 (m, 1H), 4,13-4,08 (m, 2H), 3,88 (d, J=4,5 Гц, 1H), 3,85 (s, 4H), 2,81 (s, 3H), 1,46 (t, J=6,9 Гц, 3H).
[141] Вариант осуществления 5: WX005
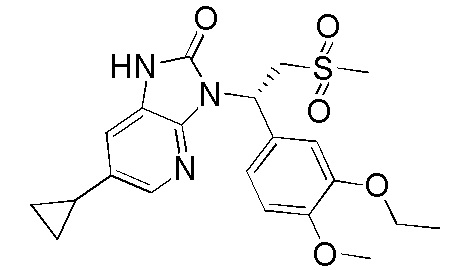
Путь синтеза

[142] Стадия 1. Получение соединения WX005-1
[143] Соединение WX004-1 (2,17 г, 25,26 ммоль) растворяли в диоксане (30,00 мл) при комнатной температуре с последующим последовательным добавлением фосфата калия (5,36 г, 25,26 ммоль), трициклогексилфосфина (3,54 г, 12,63 ммоль) и тетракис(трифенилфосфин)палладия (1,46 г, 1,26 ммоль). Реакционную смесь нагревали до 100°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и разбавляли водой (30 мл) и этилацетатом (30 мл). Водную фазу экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=40/1-10/1, объемное соотношение) с получением целевого продукта WX005-1. MS-ESI масса/заряд: 199,0 [M+H]+.
[144] Стадия 2. Получение соединения WX005-2
[145] Соединение WX005-1 (200,00 мг, 1,01 ммоль) растворяли в ацетонитриле (4,00 мл) при комнатной температуре с последующим добавлением карбоната калия (279,18 мг, 2,02 ммоль). Реакционную смесь нагревали до 90°C и перемешивали в течение 0,2 часа с последующим добавлением соединения WX001-2 (276,08 мг, 1,01 ммоль). И реакционную смесь перемешивали в течение дополнительных 11,8 часа. После завершения реакции смесь охлаждали до комнатной температуры и разбавляли водой (10 мл) и этилацетатом (10 мл), разделяли. Водную фазу экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=10/1-1/2, объемное соотношение) с получением целевого продукта WX005-2. MS-ESI масса/заряд: 436,0 [M+Na]+.
[146] Стадия 3. Получение соединения WX005-3
[147] Соединение WX005-2 (100,00 мг, 229,63 мкмоль), порошок железа (76,95 мг, 1,38 ммоль) и хлорид аммония (122,83 мг, 2,30 ммоль) растворяли в воде (300,00 мкл) и этаноле (3,00 мл) при комнатной температуре. Реакционную смесь нагревали до 90°C и перемешивали в течение 2 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении и полученный остаток разбавляли водой (10 мл) и этилацетатом (10 мл). Водную фазу экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали водой (10 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX005-3. MS-ESI масса/заряд: 406,1 [M+Na]+.
[148] Стадия 4. Получение соединения WX005
[149] Соединение WX005-3 (100,00 мг, 246,60 мкмоль) и триэтиламин (124,89 мг, 1,23 ммоль, 172,02 мкл) растворяли в тетрагидрофуране (4,00 мл) при комнатной температуре. Реакционную смесь охлаждали до 0°C на ледяной бане и перемешивали в течение 10 минут с последующим добавлением триэтилортоформиата (87,81 мг, 295,92 мкмоль). Реакцию проводили при 0°C с перемешиванием в течение 80 минут. После завершения реакции смесь разбавляли водой (10 мл) и этилацетатом (10 мл). Водную фазу экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали водой (10 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX005. MS-ESI масса/заряд: 432,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 7,92 (d, J=1,5 Гц, 1H), 7,34 (d, J=2,0 Гц, 1H), 7,20-7,15 (m, 1H), 7,14-7,10 (m, 1H), 6,93 (d, J=8,3 Гц, 1H), 6,27-5,86 (m, 1H), 4,88-4,82 (m, 1H), 4,07 (d, J=7,0 Гц, 3H), 3,82 (s, 3H), 2,95 (s, 3H), 2,16-1,80 (m, 1H), 1,39 (t, J=6,9 Гц, 3H), 1,03 (dd, J=1,5, 8,3 Гц, 2H), 0,83-0,65 (m, 2H).
[150] Вариант осуществления 6: WX006
Путь синтеза
Стадия 1. Получение соединения WX006-1
[151] Соединение WX004-2 (200,00 мг, 421,65 мкмоль), бис(пинаколато)дибор (214,15 мг, 843,30 мкмоль), ацетат калия (124,14 мг, 1,26 ммоль) и комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (34,43 мг, 42,17 мкмоль) растворяли в диметилсульфоксиде (5,00 мл) при комнатной температуре. Реакционную смесь нагревали до 90°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью воды (8 мл) и экстрагировали этилацетатом (10 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/1, объемное соотношение) с получением целевого продукта WX006-1. MS-ESI масса/заряд: 522,2 [M+H]+.
[152] Стадия 2. Получение соединения WX006-2
[153] Соединение WX006-1 (80,00 мг, 153,44 мкмоль), 2-фторбензилбромид (58,01 мг, 306,88 мкмоль), фосфат калия (65,14 мг, 306,87 мкмоль) и тетракис(трифенилфосфин)палладий (177,30 мг, 153,44 мкмоль) растворяли в диметиловом эфире этиленгликоля (2,00 мл), этаноле (500,00 мкл) и воде (500,00 мкл) при комнатной температуре. Реакционную смесь нагревали до 90°C и перемешивали в течение 2 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью воды (8 мл) и экстрагировали этилацетатом (10 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=2/1, объемное соотношение) с получением целевого продукта WX006-2. MS-ESI масса/заряд: 504,2 [M+H]+.
[154] Стадия 3. Получение соединения WX006-3
[155] Соединение WX006-2 (80,00 мг, 158,88 мкмоль), порошок цинка (83,11 мг, 1,27 ммоль) и хлорид аммония (84,98 мг, 1,59 ммоль) растворяли в метаноле (3,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения реакции добавляли этилацетат (8 мл) с последующей фильтрацией. К полученному остатку добавляли воду (8 мл) и экстрагировали этилацетатом (10 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX006-3. MS-ESI масса/заряд: 474,2 [M+H]+.
[156] Стадия 4. Получение соединения WX006
[157] Соединение WX006-3 (30,00 мг, 63,35 мкмоль) и триэтиламин (19,23 мг, 190,05 мкмоль, 26,34 мкл) растворяли в тетрагидрофуране (2,00 мл) при комнатной температуре. Реакционную смесь охлаждали до 0°C на ледяной бане и к ней добавляли триэтилортоформиат (7,52 мг, 25,34 мкмоль). Реакцию проводили при 0°C с перемешиванием в течение 0,5 часа. После завершения реакции смесь гасили водой (5 мл) и экстрагировали этилацетатом (8 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX006. MS-ESI масса/заряд: 500,2 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 7,95 (s, 1H), 7,37-7,01 (m, 7H), 6,91 (d, J=8,3 Гц, 1H), 6,12 (dd, J=4,0, 10,5 Гц, 1H), 4,86-4,85 (m, 1H), 4,09-3,98 (m, 5H), 3,79 (s, 3H), 2,91 (s, 3H), 1,35 (t, J=6,9 Гц, 3H).
[158] Вариант осуществления 7: WX007
Путь синтеза
[159] Соединение WX004 (150,00 мг, 318,92 мкмоль) и 4,4,5,5-тетраметил-2(2-метилпропан-1-алкенил)-1,3,2-диоксаборолан (69,68 мг, 382,70 мкмоль) растворяли в диоксане (20,00 мл) при комнатной температуре с последующим добавлением карбоната калия (132,23 мг, 956,75 мкмоль) и воды (2,00 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут в атмосфере азота с последующим добавлением дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия (70,01 мг, 95,68 мкмоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 16 часов. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли воду (30 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX007. MS-ESI масса/заряд: 446,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,70 (s, 1 H), 7,85 (s, 1 H), 7,39 (s, 1 H), 7,24 (d, J=7,6 Гц, 1 H), 7,16 (s, 1 H), 6,76 (d, J=8,0 Гц, 1 H), 6,31 (s, 1 H), 6,11 (s, 1 H), 4,86 (dd, J=14,2, 10,2 Гц, 1 H), 4,05 (dd, J=7,0, 3,4 Гц, 2 H), 3,81 (d, J=3,2 Гц, 1 H), 3,77 (s, 3 H), 2,81 (s, 3 H), 1,86 (s, 3 H), 1,78 (s, 3 H), 1,37 (t, J=6,8 Гц, 3 H).
[160] Вариант осуществления 8: WX008
Путь синтеза
[161] Соединение WX007 (100,00 мг, 224,45 мкмоль) растворяли в метаноле (10,00 мл) при комнатной температуре с последующим добавлением гидроксида палладия (31,52 мг, 224,45 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере водорода (40 фунтов/кв. дюйм). После завершения реакции нерастворимые катализаторы удаляли посредством фильтрации. Фильтрат выделяли с помощью препаративной HPLC с получением целевого продукта WX008. MS-ESI масса/заряд: 448,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,83 (s., 1 H), 7,78 (s, 1 H), 7,32 (s, 1 H), 7,17 (d, J=8,4 Гц, 1 H), 7,07 (s, 1 H), 6,74 (d, J=8,0 Гц, 1 H), 6,13 (dd, J=9,0, 4,6 Гц, 1 H), 4,82 (dd, J=14,6, 9,4 Гц, 1 H), 4,09-3,95 (m, 2 H), 3,88 (dd, J=14,4, 4,4 Гц, 1 H), 3,75 (s, 3 H), 2,65 (s, 3 H), 2,40 (d, J=6,8 Гц, 2 H), 1,83-1,70 (m, 1 H), 1,35 (t, J=7,0 Гц, 3 H), 0,84 (d, J=6,4 Гц, 6 H).
[162] Вариант осуществления 9: WX009
Путь синтеза
[163] Соединение WX004 (1,00 г, 2,13 ммоль), 2-фторфенилбороновую кислоту (357,6 мг, 2,56 ммоль) и карбонат калия (441,58 мг, 3,20 ммоль) растворяли в диоксане (10,00 мл) при комнатной температуре с последующим добавлением комплекса [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид и дихлорметана (173,94 мг, 213,00 мкмоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали с применением диатомита и концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли водой (50 мл) и этилацетатом (50 мл), разделяли. Водную фазу экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX009. MS-ESI масса/заряд: 486,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 10,24 (br s, 1H), 8,24 (br s, 1H), 7,73-7,03 (m, 7H), 6,98-6,08 (m, 2H), 4,94 (br s, 1H), 4,36-3,50 (m, 6H), 2,78 (br s, 3H), 1,94 (br s, 1H), 1,42 (br s, 3H).
[164] Соединения из каждого варианта осуществления в следующей таблице получали в соответствии со способом согласно варианту осуществления 9.
Таблица 1
[165] Данные LCMS и 1H ЯМР для каждого варианта осуществления
Таблица 2
[166] Вариант осуществления 21: WX021
Путь синтеза
[167] 1H-Пиридин-2-кетон (24,26 мг, 255,14 мкмоль), соединение WX004 (60,00 мг, 127,57 мкмоль), N1,N2-диметилэтилендиамин (4,50 мг, 51,03 мкмоль), карбонат калия (35,26 мг, 255,154 мкмоль) и йодид меди (4,86 мг, 25,51 мкмоль) растворяли в N,N-диметилформамиде (2,00 мл) при комнатной температуре. Реакционную смесь нагревали до 120°C в течение 15 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX021. MS-ESI масса/заряд: 485,3 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,54 (br s, 1H), 7,99 (s, 1H), 7,53-7,42 (m, 2H), 7,38 (d, J=6,5 Гц, 1H), 7,30 (s, 1H), 7,22 (d, J=8,3 Гц, 1H), 6,81 (d, J=8,3 Гц, 1H), 6,75 (d, J=8,3 Гц, 1H), 6,35 (t, J=6,8 Гц, 1H), 6,20 (dd, J=4,6, 9,2 Гц, 1H), 4,91-4,79 (m, 1H), 4,18-4,04 (m, 2H), 3,96-3,86 (m, 1H), 3,83 (s, 3H), 2,82 (s, 3H), 1,44 (t, J=6,9 Гц, 3H).
[168] Вариант осуществления 22: WX022
Путь синтеза
[169] Стадия 1. Получение соединения WX022-1
[170] Соединение WX004 (100,00 мг, 212,61 мкмоль) растворяли в метаноле (5,00 мл) при комнатной температуре с последующим последовательным добавлением триэтиламина (43,03 мг, 425,22 мкмоль) и дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия (155,57 мг, 212,61 мкмоль). Реакционную смесь нагревали до 60°C и перемешивали в течение 12 часов в атмосфере монооксида углерода (50 фунтов/кв. дюйм). После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и этилацетатом (10 мл), разделяли. Водную фазу экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX022-1. MS-ESI масса/заряд: 450,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,95 (br s, 1H), 8,79 (s, 1H), 7,86 (br s, 1H), 7,37 (br s, 1H), 6,84 (d, J=7,5 Гц, 1H), 6,27 (br s, 1H), 4,90 (br s, 1H), 4,11 (br s, 2H), 3,99-3,82 (m, 8H), 2,84 (br s, 3H), 1,46 (t, J=6,5 Гц, 3H).
[171] Стадия 2. Получение соединения WX022-2
[172] Соединение WX022-1 (30,00 мг, 66,74 мкмоль) растворяли в метаноле (1,00 мл) и тетрагидрофуране (500,00 мкл) при комнатной температуре с последующим добавлением моногидрата гидроксида лития (5,60 мг, 133,49 мкмоль) и воды (300,00 мкл). Реакционную смесь нагревали до 50°C и перемешивали в течение 3 часов. После завершения реакции смесь охлаждали до комнатной температуры, к ней добавляли разбавленную хлористоводородную кислоту (2 M, 0,1 мл) и экстрагировали этилацетатом (8 мл x 3). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX022-2. MS-ESI масса/заряд: 436,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 8,75 (s, 1H), 7,85 (s, 1H), 7,37 (s, 1H), 7,20 (d, J=8,0 Гц, 1H), 6,95 (d, J=8,3 Гц, 1H), 6,22 (dd, J=3,8, 10,5 Гц, 1H), 4,95 (br s, 1H), 4,14-3,99 (m, 3H), 3,82 (s, 3H), 2,96 (s, 3H), 1,40 (t, J=7,0 Гц, 3H).
[173] Стадия 3. Получение соединения WX022-3
[174] Соединение WX022-2 (40,00 мг, 91,86 мкмоль), хлорид аммония (19,65 мг, 367,44 ммоль) и гексафторфосфат тетраметилурония (10,48 мг, 27,56 мкмоль) растворяли в этилдиизопропиламине (47,49 мг, 367,44 мкмоль) и N,N-диметилформамиде (2,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения реакции смесь разбавляли водой (5 мл) и экстрагировали этилацетатом (8 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (5 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX022-3. MS-ESI масса/заряд: 435,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 8,61 (s, 1H), 7,79 (s, 1H), 7,36 (d, J=1,5 Гц, 1H), 7,23-7,12 (m, 1H), 6,93 (d, J=8,3 Гц, 1H), 6,21 (dd, J=3,8, 10,5 Гц, 1H), 4,94 (br s, 1H), 4,14-3,98 (m, 3H), 3,81 (s, 3H), 2,96 (s, 3H).
[175] Стадия 4. Получение соединения WX022
[176] Соединение WX022-3 (30,00 мг, 69,05 мкмоль) и триэтиламин (20,96 мг, 207,15 мкмоль) растворяли в дихлорметане (1,00 мл) с последующим добавлением по каплям трифторуксусного ангидрида (29,00 мг, 138,10 мкмоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа. После завершения реакции смесь разбавляли водой (3 мл) и экстрагировали этилацетатом (5 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (5 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX022. MS-ESI масса/заряд: 417,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 8,43 (d, J=1,8 Гц, 1H), 7,61 (d, J=1,6 Гц, 1H), 7,33 (d, J=2,0 Гц, 1H), 7,18 (dd, J=2,0, 8,2 Гц, 1H), 6,94 (d, J=8,6 Гц, 1H), 6,21 (dd, J=3,8, 10,8 Гц, 1H), 4,83 (br s, 1H), 4,13-3,99 (m, 3H), 3,82 (s, 3H), 2,98 (s, 3H), 1,40 (t, J=7,0 Гц, 3H).
[177] Вариант осуществления 23: WX023
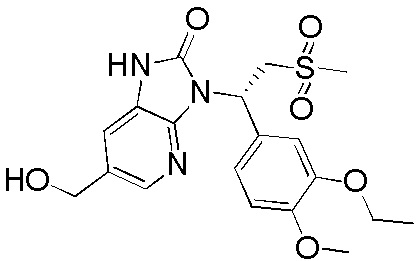
Путь синтеза
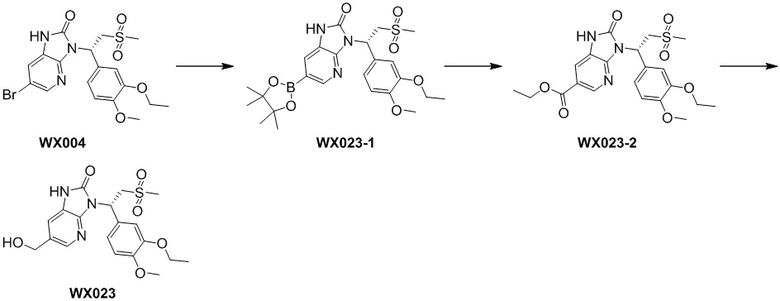
[178] Стадия 1. Получение соединения WX023-1
[179] Дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (46,67 мг, 63,78 мкмоль) добавляли в раствор бис(пинаколато)дибора (388,73 мг, 1,53 ммоль), соединения WX004 (300,00 мг, 637,84 мкмоль) и ацетата калия (375,58 мг, 3,83 ммоль) в диоксане (5,00 мл) при комнатной температуре. Реакционную смесь нагревали до 100°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью воды (10 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (15 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=3/1-1/2, объемное соотношение) с получением целевого продукта WX023-1. MS-ESI масса/заряд: 518,1 [M+H]+.
[180] Стадия 2. Получение соединения WX023-2
[181] Добавляли ацетат палладия (5,42 мг, 24,16 мкмоль) и раствор соединения WX023-1 (250,00 мг, 483,19 мкмоль) и бензофенона (88,05 мг, 483,19 мкмоль) в этаноле (1,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов в атмосфере монооксида углерода (15 фунтов/кв. дюйм). После завершения реакции смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (15 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=3/1-1/2, объемное соотношение) с получением целевого продукта WX023-2. MS-ESI масса/заряд: 464,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,60 (s, 1H), 8,79 (d, J=1,5 Гц, 1H), 7,86 (d, J=1,8 Гц, 1H), 7,34 (d, J=2,0 Гц, 1H), 7,23 (dd, J=1,9, 8,4 Гц, 1H), 6,83 (d, J=8,3 Гц, 1H), 6,23 (dd, J=4,5, 10,0 Гц, 1H), 4,90 (dd, J=10,2, 14,7 Гц, 1H), 4,40 (q, J=7,0 Гц, 2H), 4,09 (дуб. квинт., J=2,5, 7,0 Гц, 2H), 3,96-3,78 (m, 4H), 2,80 (s, 3H), 1,49-1,35 (m, 6H).
[182] Стадия 3. Получение соединения WX023
[183] LiBH4 (3,38 мг, 155,34 мкмоль) добавляли в раствор соединения WX023-2 (60,00 мг, 129,45 мкмоль) в тетрагидрофуране (4,00 мл) при комнатной температуре. Реакционную смесь нагревали до 50°C и перемешивали в течение 2 часов. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью воды (8 мл) и экстрагировали этилацетатом (10 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX023. MS-ESI масса/заряд: 422,1 [M+H]+. 1H ЯМР (400 МГц, DMSO_d6) δ: 8,06 (s, 1H), 7,53 (s, 1H), 7,36 (d, J=2,0 Гц, 1H), 7,19 (dd, J=2,0, 8,2 Гц, 1H), 6,95 (d, J=8,6 Гц, 1H), 6,19 (dd, J=3,6, 10,8 Гц, 1H), 4,87 (d, J=11,0 Гц, 1H), 4,67 (s, 2H), 4,13-4,01 (m, 3H), 3,82 (s, 3H), 2,97 (s, 3H), 1,40 (t, J=7,0 Гц, 3H).
[184] Вариант осуществления 24: WX024
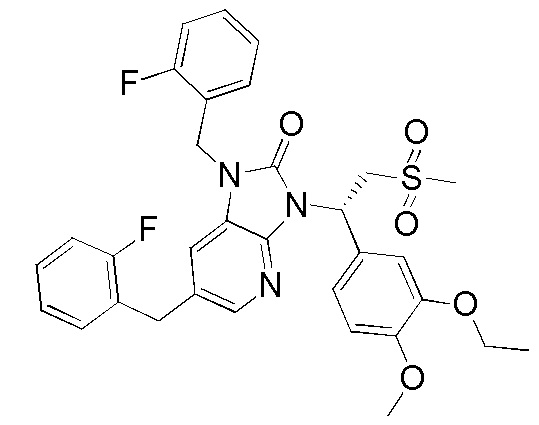
Путь синтеза
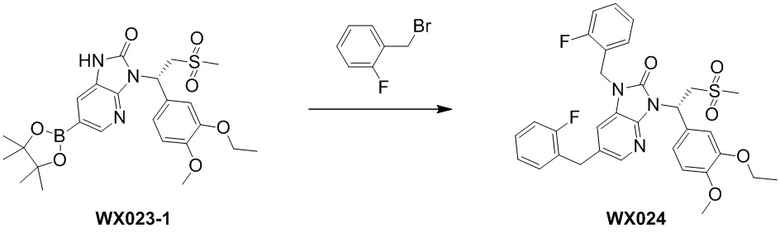
[185] Соединение WX023-1 (50,00 мг, 96,64 мкмоль), 2-фторбензилбромид (36,53 мг, 193,28 мкмоль), тетракис(трифенилфосфин)палладий (11,17 мг, 9,66 мкмоль) и фосфат калия (41,03 мг, 193,28 мкмоль) растворяли в диметиловом эфире (2,00 мл), этаноле (500,00 мкл) и воде (500,00 мкл) при комнатной температуре. Реакционную смесь нагревали до 90°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли воду (5 мл) и экстрагировали этилацетатом (5 мл x 3). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX024. MS-ESI масса/заряд: 608,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 7,86 (s, 1H), 7,29 (s, 1H), 7,19-7,12 (m, 4H), 7,05-6,87 (m, 6H), 6,74 (d, J=8,5 Гц, 1H), 6,18 (dd, J=4,5, 10,0 Гц, 1H), 4,97 (s, 2H), 4,79 (dd, J=10,0, 14,6 Гц, 1H), 4,05-3,94 (m, 2H), 3,88-3,74 (m, 6H), 2,69 (s, 3H), 1,36 (t, J=7,0 Гц, 3H).
[186] Вариант осуществления 25: WX025
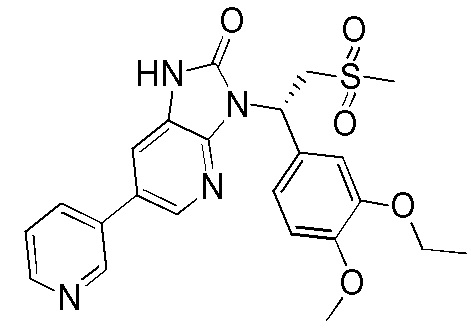
Путь синтеза
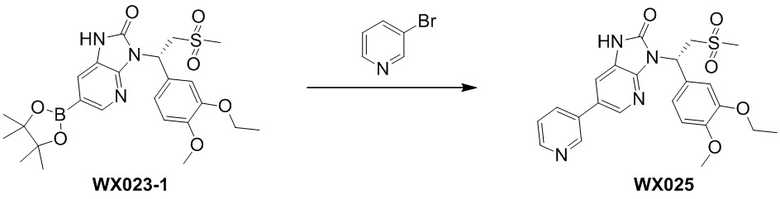
[187] Комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (3,95 мг, 4,83 мкмоль) добавляли в раствор 3-бромпиридина (15,27 мг, 96,64 мкмоль), соединения WX023-1 (50,00 мг, 96,64 мкмоль) и карбоната калия (40,07 мг, 289,92 мкмоль) в диоксане (3,00 мл) и воде (1,00 мл) при комнатной температуре. Реакционную смесь нагревали до 70-80°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и к ней добавляли воду (3 мл) и этилацетат (3 мл) с последующей фильтрацией. Фильтрат экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX025. MS-ESI масса/заряд: 469,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,18 (br s, 1H), 8,78 (br s, 1H), 8,40 (d, J=6,8 Гц, 1H), 8,31 (br s, 1H), 7,87 (br s, 1H), 7,62 (br s, 1H), 7,33 (br s, 1H), 7,28 (s, 1H), 7,25 (d, J=8,3 Гц, 1H), 6,84 (d, J=8,5 Гц, 1H), 6,29 (d, J=10,5 Гц, 1H), 5,05 (t, J=12,4 Гц, 1H), 4,11 (d, J=7,0 Гц, 2H), 3,85 (s, 4H), 2,93 (s, 3H), 1,46 (t, J=6,8 Гц, 3H).
[188] Соединения из каждого варианта осуществления в следующей таблице получали в соответствии со способом согласно варианту осуществления 25.
Таблица 3
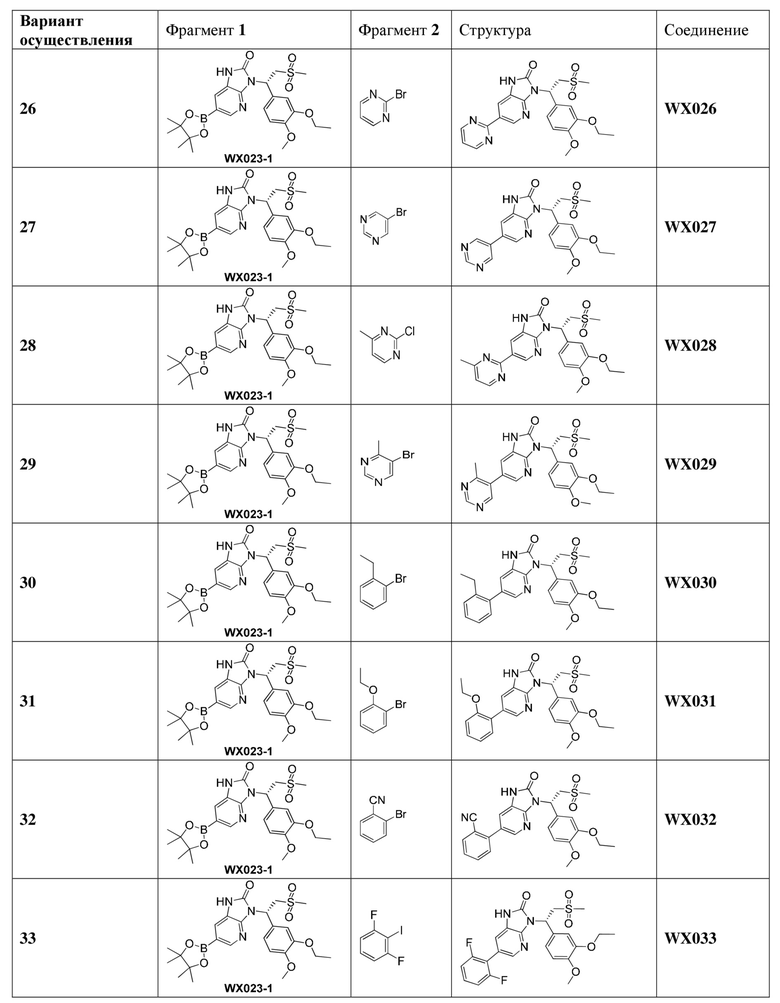
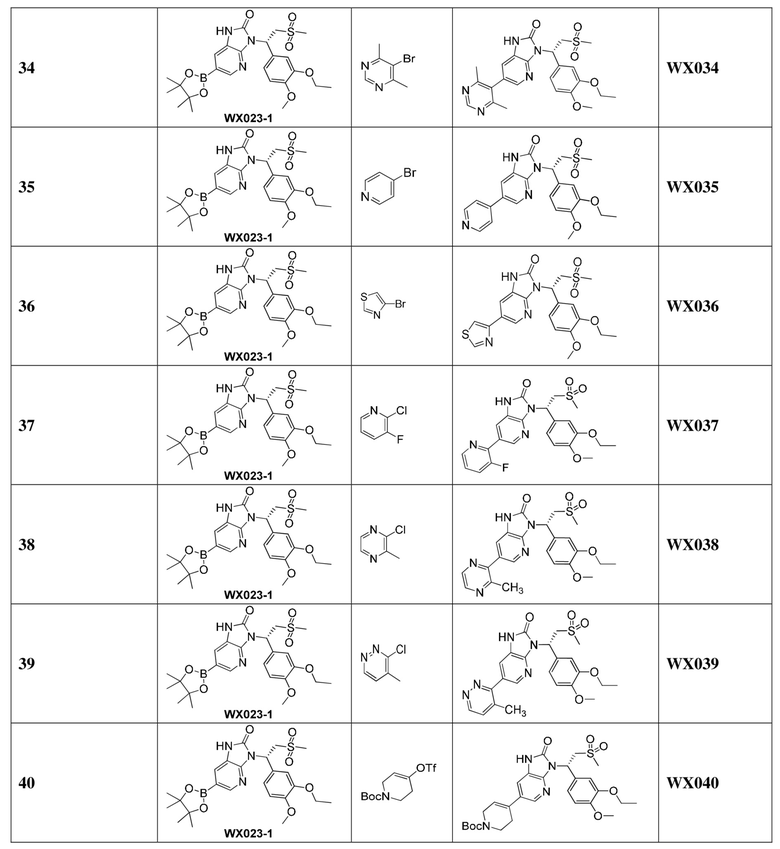
[189] Результаты LCMS и 1H ЯМР для каждого варианта осуществления
Таблица 4
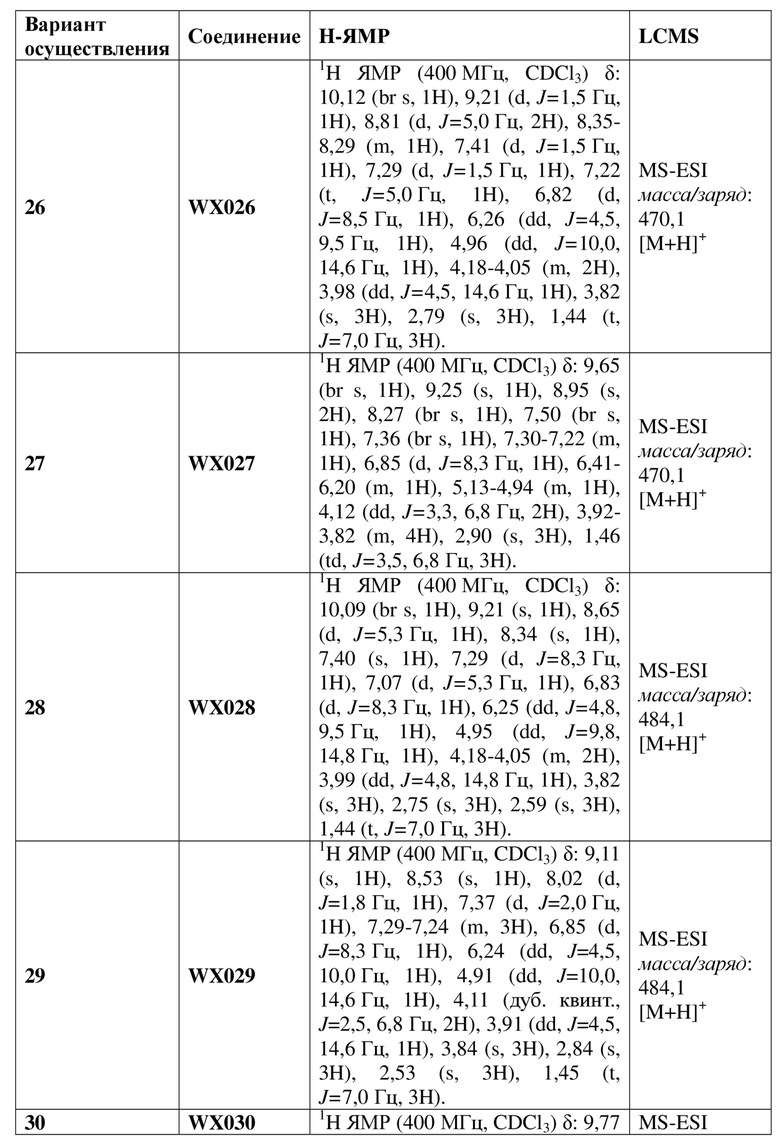
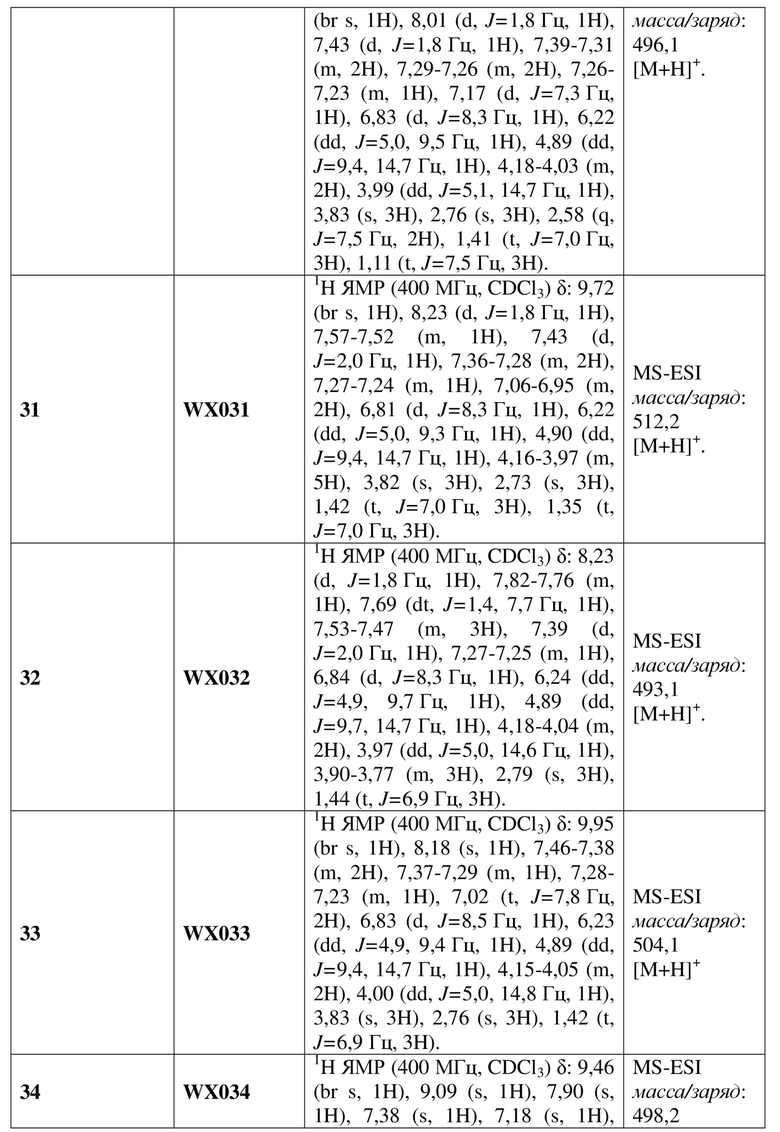
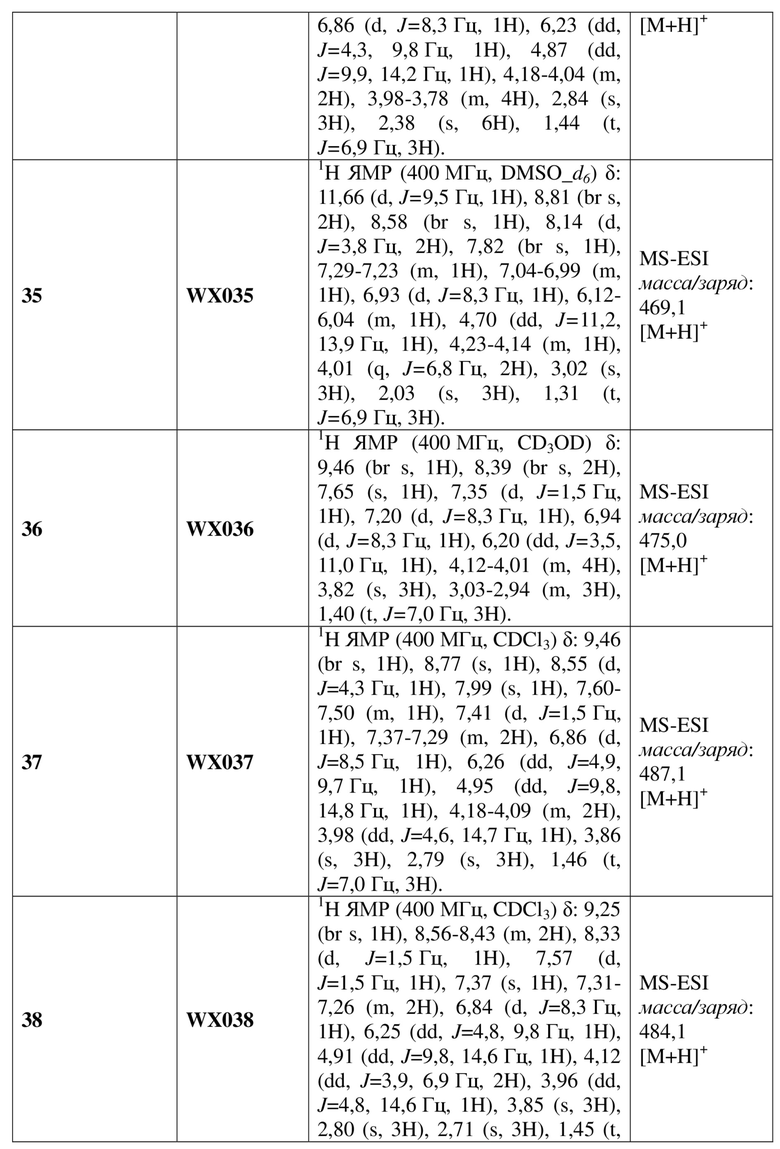
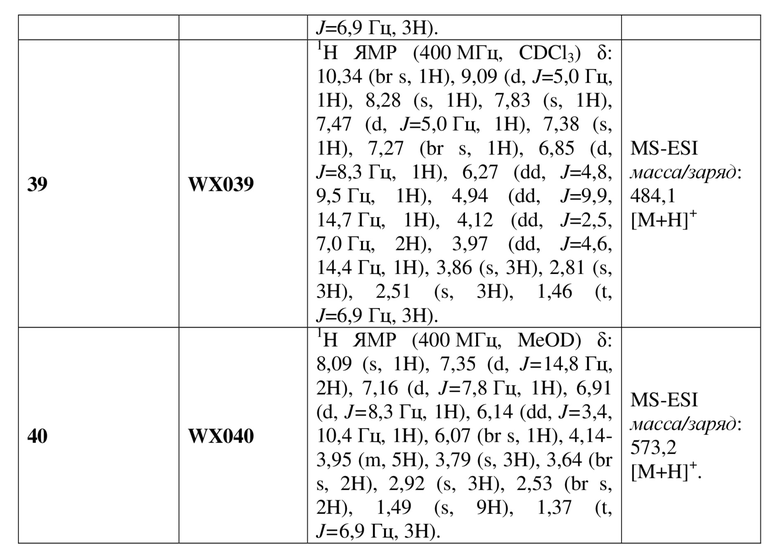
[190] Вариант осуществления 41: WX041
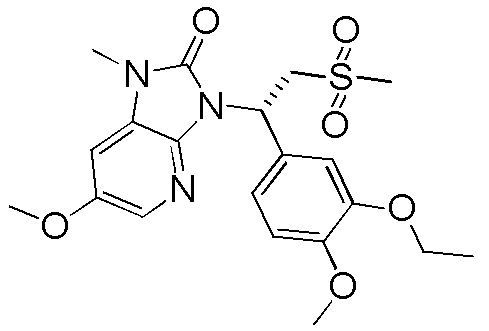
Путь синтеза
[191] Стадия 1. Получение WX041-1
[192] H2O2 (21,91 мг, 193,28 мкмоль, 30%) и карбонат калия (26,71 мг, 193,28 мкмоль) добавляли к раствору соединения WX023-1 (50,00 мг, 96,64 мкмоль) в дихлорметане (1,00 мл) при 0°C. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 10 часов. После завершения реакции смесь концентрировали при пониженном давлении для удаления дихлорметана. Полученный остаток разбавляли водой (5 мл), подкисляли с помощью насыщенного раствора лимонной кислоты до pH 3-4 и экстрагировали смесью дихлорметан/метанол = 4/1 (5 мл x 5). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX041-1. 1H ЯМР (400 МГц, CD3OD) δ: 7,65 (d, J=2,3 Гц, 1H), 7,34 (d, J=1,5 Гц, 1H), 7,23-7,11 (m, 1H), 6,98-6,85 (m, 2H), 6,09 (dd, J=3,9, 10,4 Гц, 1H), 4,86-4,79 (m, 1H), 4,14-3,95 (m, 3H), 3,82 (s, 3H), 2,91 (s, 3H), 1,39 (t, J=7,0 Гц, 3H).
[193] Стадия 2. Получение WX041-1
[194] Соединение WX041-1 (30,00 мг, 73,63 мкмоль), 2-йодпиримидин (104,51 мг, 736,30 мкмоль) и карбонат калия (10,18 мг, 73,63 мкмоль) растворяли в N,N-диметилформамиде (1,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов в атмосфере азота. После завершения реакции воду (8 мл) и этилацетат (10 мл) добавляли в реакционную смесь, разделяли. Органическую фазу промывали насыщенным солевым раствором (5 мл x 2), высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX041. MS-ESI масса/заряд: 436,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 7,76 (d, J=1,5 Гц, 1H), 7,34 (s, 1H), 7,23 (d, J=8,0 Гц, 1H), 6,84-6,78 (m, 2H), 6,15 (dd, J=5,0, 9,5 Гц, 1H), 4,85 (dd, J=10,0, 14,6 Гц, 1H), 4,10 (dd, J=3,5, 7,0 Гц, 2H), 3,94-3,80 (m, 7H), 3,37 (s, 3H), 2,74 (s, 3H), 1,45 (t, J=7,0 Гц, 3H).
[195] Вариант осуществления 42: WX042
Путь синтеза
[196] Карбонат калия (67,84 мг, 490,86 мкмоль) добавляли к раствору соединения WX041-1 (100,00 мг) и 2-хлорпиримидина (28,11 мг, 245,43 мкмоль) в N,N-диметилформамиде (3,00 мл) при комнатной температуре. Реакционную смесь нагревали до 50°C и перемешивали в течение 10 часов. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя с последующей фильтрацией. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX042. MS-ESI масса/заряд: 486,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 8,61 (d, J=4,8 Гц, 1H), 7,97 (d, J=2,3 Гц, 1H), 7,37 (d, J=1,8 Гц, 1H), 7,30 (d, J=2,3 Гц, 1H), 7,27-7,18 (m, 2H), 6,96 (d, J=8,3 Гц, 1H), 6,19 (dd, J=4,0, 10,5 Гц, 1H), 4,95 (br s, 1H), 4,15-4,01 (m, 3H), 3,83 (s, 3H), 2,96 (s, 3H), 1,40 (t, J=7,0 Гц, 3H).
[197] Вариант осуществления 43: WX043
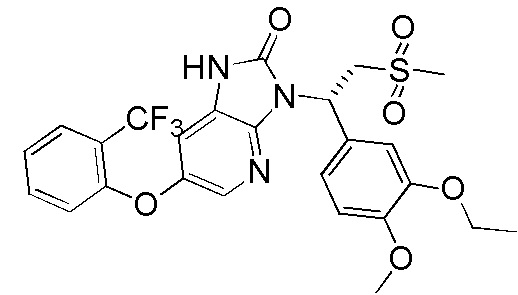
Путь синтеза
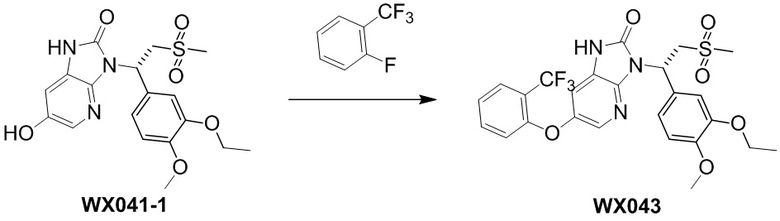
[198] Соединение WX041-1 (90,00 мг, 220,89 мкмоль), 1-фтор-2-трифторметилбензол (54,37 мг, 331,34 мкмоль) и карбонат калия (91,59 мг, 662,67 мкмоль) растворяли в метилпирролидоне (2,00 мл) при комнатной температуре. Реакционную смесь нагревали до 160°C с помощью микроволнового излучения и перемешивали в течение 50 минут. После завершения реакции смесь охлаждали до комнатной температуры. Воду (8 мл) добавляли в смесь и экстрагировали этилацетатом (10 мл x 2). Органические фазы объединяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX043. MS-ESI масса/заряд: 552,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ: 7,87 (d, J=2,3 Гц, 1H), 7,74 (d, J=7,5 Гц, 1H), 7,63-7,51 (m, 1H), 7,37 (s, 1H), 7,30-7,17 (m, 2H), 7,13 (d, J=2,3 Гц, 1H), 6,97 (dd, J=8,5, 13,8 Гц, 2H), 6,18 (dd, J=3,8, 10,5 Гц, 1H), 4,83-4,81 (m, 1H), 4,12-3,99 (m, 3H), 3,83 (s, 3H), 2,97 (s, 3H), 1,40 (t, J=7,0 Гц, 3H).
[199] Вариант осуществления 44: WX044
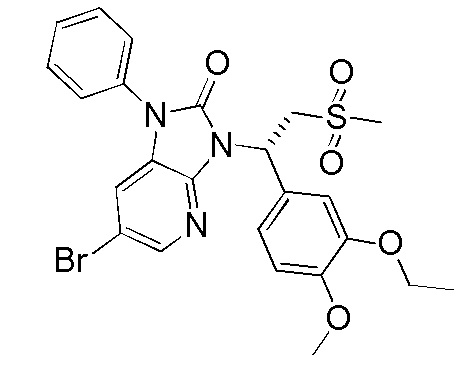
Путь синтеза
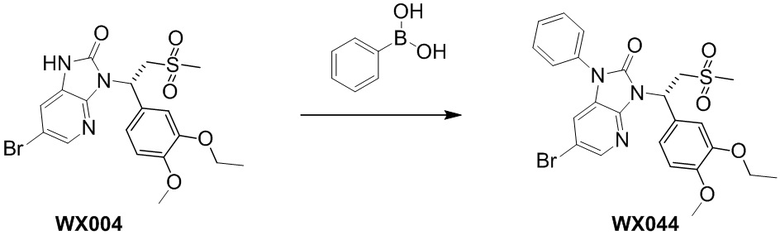
[200] Ацетат меди (19,31 мг, 106,31 мкмоль) и 2-(2-пиридил)пиридин (16,60 мг, 106,31 мкмоль) добавляли к раствору соединения WX004 (50,00 мг, 106,31 мкмоль) и фенилбороновой кислоты (19,44 мг, 159,47 мкмоль) в дихлорметане (2,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов в атмосфере азота. После завершения реакции смесь фильтровали и концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX044. MS-ESI масса/заряд: 546,0 [M+H]+, 548,0 [M+H+2]+. 1H ЯМР (400 МГц, CD3OD) δ: 8,23 (d, J=2,0 Гц, 1H), 7,65-7,58 (m, 2H), 7,56-7,46 (m, 4H), 7,40 (d, J=2,0 Гц, 1H), 7,24 (dd, J=2,1, 8,4 Гц, 1H), 6,97 (d, J=8,5 Гц, 1H), 6,27 (dd, J=4,0, 10,8 Гц, 1H), 4,17-4,02 (m, 3H), 3,84 (s, 3H), 3,27-3,22 (m, 1H), 3,00 (s, 3H), 1,41 (t, J=6,9 Гц, 3H).
[201] Вариант осуществления 45: WX045

Путь синтеза
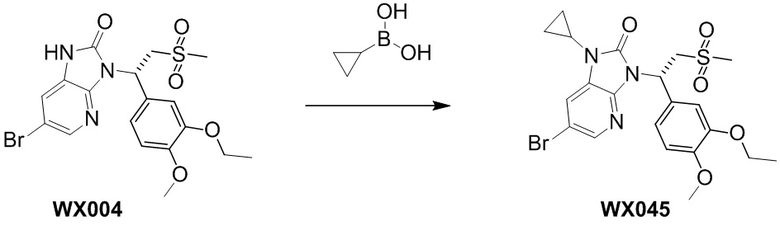
[202] Ацетат меди (19,31 мг, 106,31 мкмоль) и 2-(2-пиридил)пиридин (16,60 мг, 106,31 мкмоль) добавляли к раствору соединения WX004 (50,00 мг, 106,31 мкмоль) и циклопропилбороновой кислоты (9,13 мг, 106,31 мкмоль) в дихлорметане (2,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 10 часов в атмосфере азота. После завершения реакции смесь фильтровали и концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX045. MS-ESI масса/заряд: 510,0 [M+H]+, 512,0 [M+H+2]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,12 (s, 1H), 7,48 (s, 1H), 7,34 (s, 1H), 7,24 (d, J=8,3 Гц, 1H), 6,84 (d, J=8,5 Гц, 1H), 6,14 (dd, J=4,6, 9,9 Гц, 1H), 4,79 (dd, J=10,0, 14,3 Гц, 1H), 4,19-4,05 (m, 2H), 3,93-3,83 (m, 4H), 2,88 (d, J=3,3 Гц, 1H), 2,80 (s, 3H), 2,20 (s, 1H), 1,48 (t, J=6,9 Гц, 3H), 1,13 (d, J=6,8 Гц, 2H), 0,99 (br s, 2H).
[203] Вариант осуществления 46: WX046
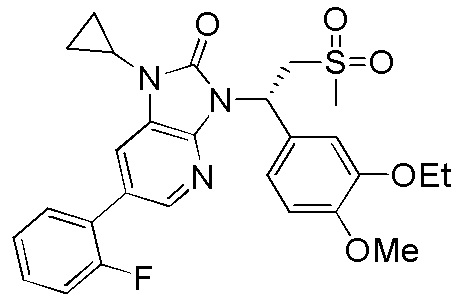
Путь синтеза
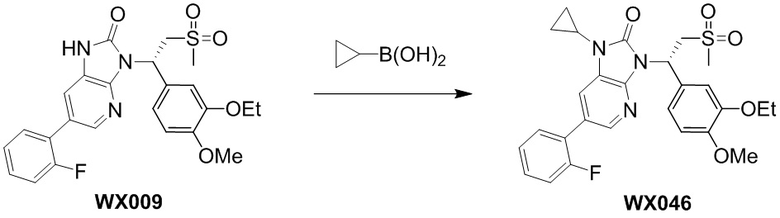
[204] Ацетат меди (18,70 мг, 102,98 мкмоль) и 2-(2-пиридил)пиридин (16,08 мг, 102,98 мкмоль) добавляли к раствору циклопропилбороновой кислоты (13,27 мг, 154,47 мкмоль) и соединения WX009 (50,00 мг, 102,98 мкмоль) в дихлорметане (2,00 мл) при комнатной температуре. Реакционную смесь перемешивали при 20-40°C в течение 85 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью воды (10 мл) и экстрагировали дихлорметаном (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX046. MS-ESI масса/заряд: 526,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,19 (s, 1H), 7,52 (t, J=1,8 Гц, 1H), 7,46-7,32 (m, 3H), 7,29-7,26 (m, 1H), 7,26-7,15 (m, 2H), 6,83 (d, J=8,3 Гц, 1H), 6,19 (dd, J=5,0, 9,8 Гц, 1H), 4,83 (dd, J=9,5, 14,6 Гц, 1H), 4,17-4,07 (m, 2H), 3,95 (dd, J=5,0, 14,8 Гц, 1H), 3,84 (s, 3H), 2,94-2,86 (m, 1H), 2,77 (s, 3H), 1,45 (t, J=7,0 Гц, 3H), 1,14-0,97 (m, 4H).
[205] Вариант осуществления 47: WX047
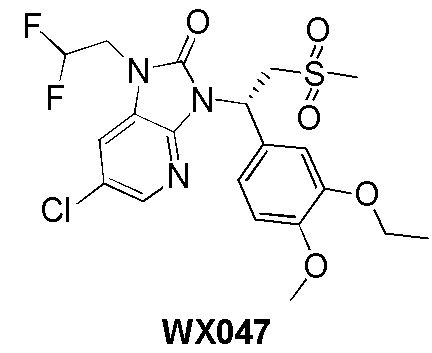
Путь синтеза
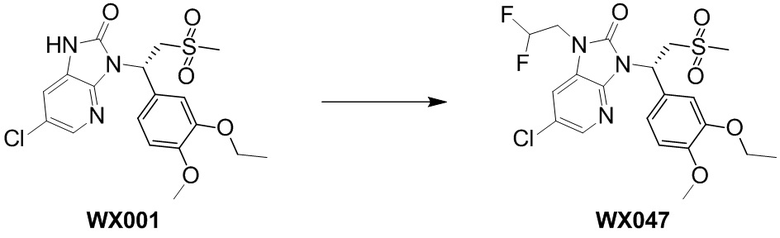
[206] Соединение WX001 (300,00 мг, 704,41 мкмоль), 2,2-дифторэтил-4-метилбензолсульфонат (498,72 мг, 2,11 ммоль) и карбонат калия (292,07 мг, 2,11 ммоль) растворяли в N,N-диметилформамиде (10,00 мл) при комнатной температуре. Реакционную смесь нагревали до 30°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции к смеси добавляли этилацетат (100 мл) и воду (30 мл). Органические фазы отделяли, промывали насыщенным солевым раствором и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток очищали с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=5/1-1/1, объемное соотношение) с получением целевого продукта WX047. MS (ESI) масса/заряд 490,0 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,06 (d, J=2,0 Гц, 1H), 7,31-7,20 (m, 3H) 6,84 (d, J=8,4 Гц, 1H), 6,19-6,15 (m, 2H), 4,82 (m, 1H), 4,20-4,09 (m, 4H), 3,86-3,81 (m, 4H), 2,80 (s, 3H), 1,46 (t, J=6,8 Гц, 3H).
[207] Соединения из каждого варианта осуществления в следующей таблице получали в соответствии со способом согласно варианту осуществления 47.
Таблица 5
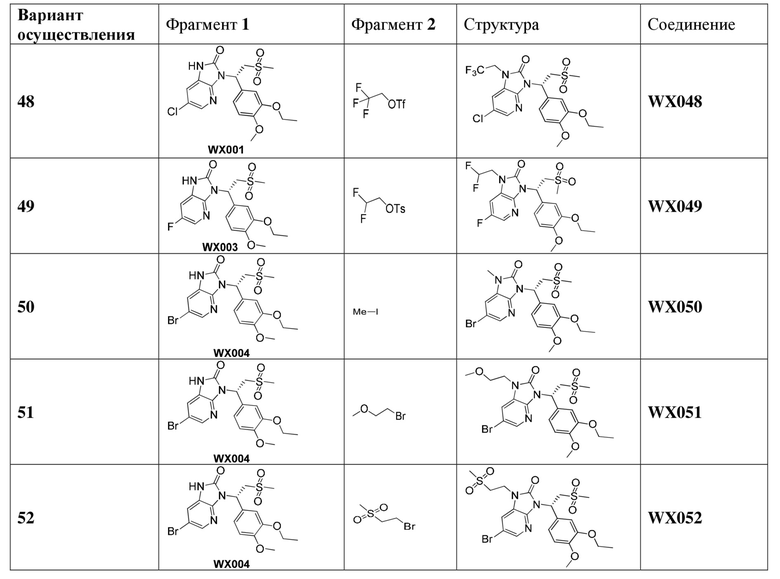
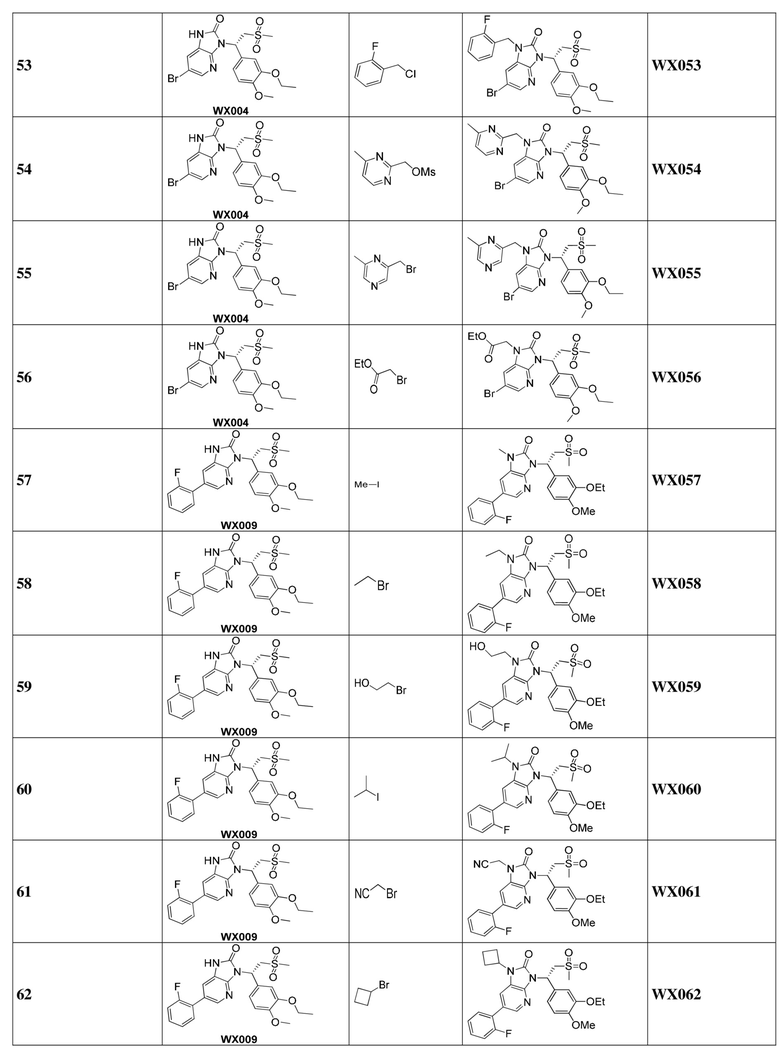
[208] Данные LCMS и 1H ЯМР для каждого варианта осуществления
Таблица 6
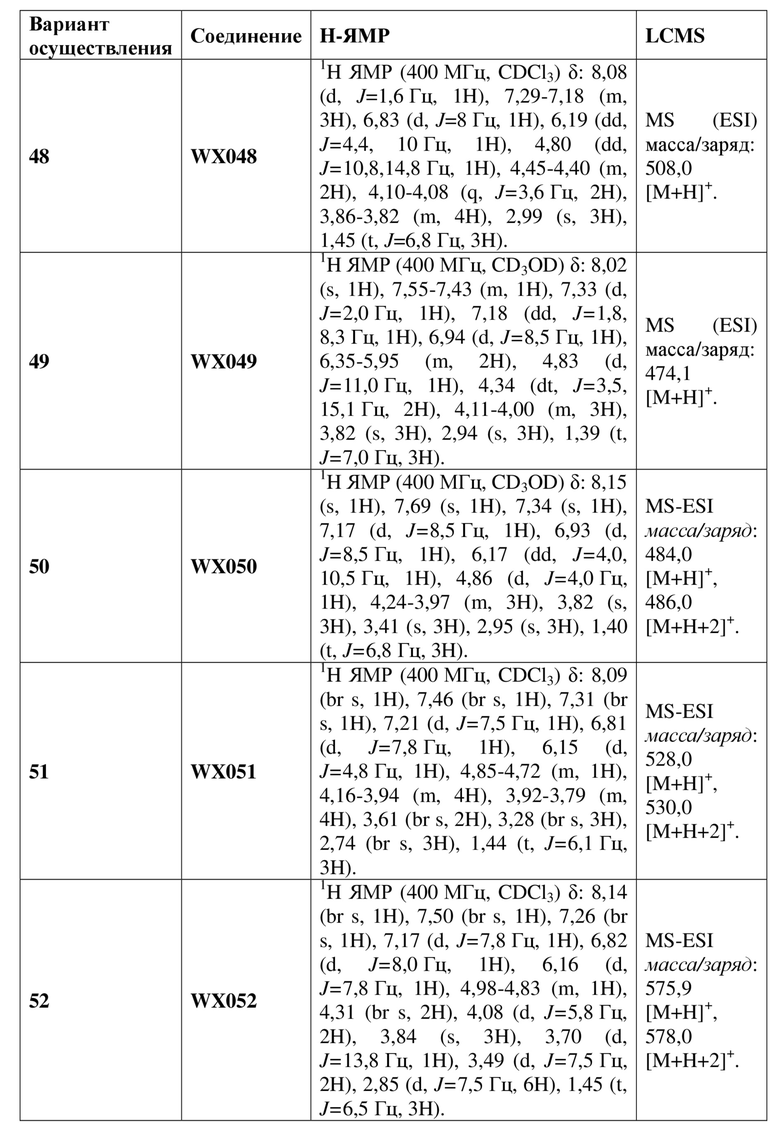
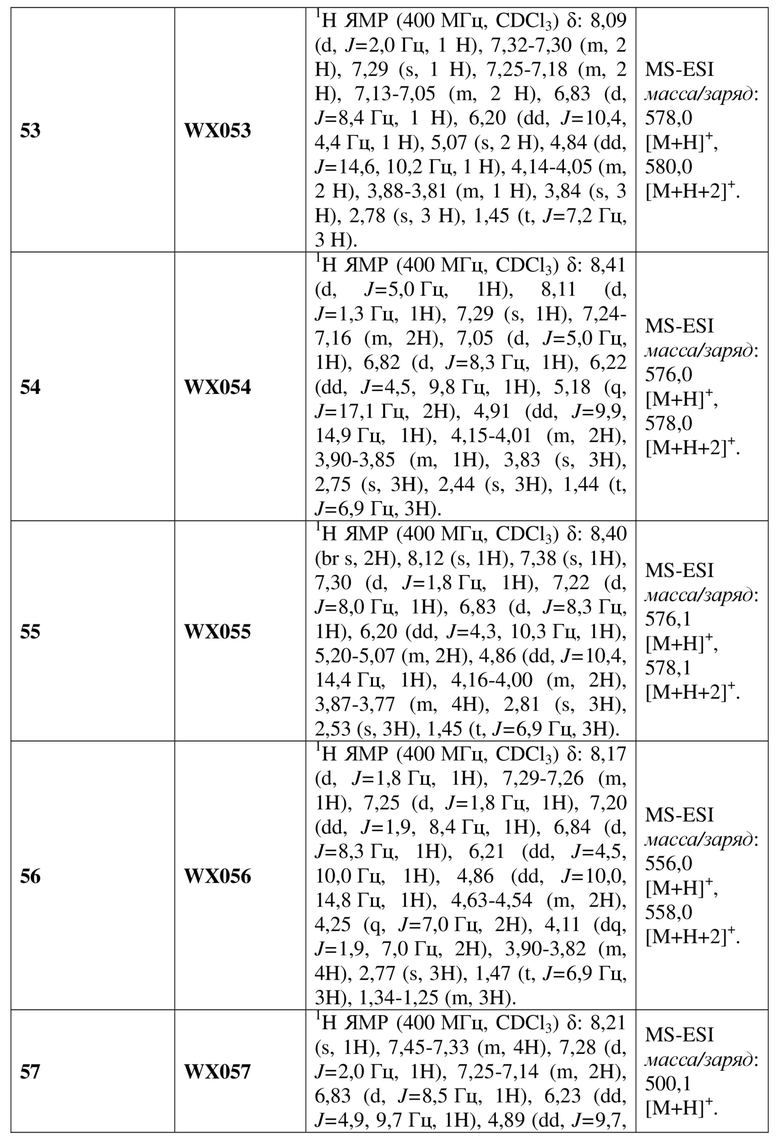

[209] Вариант осуществления 63: WX063
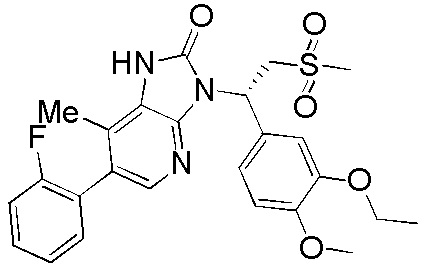
Путь синтеза
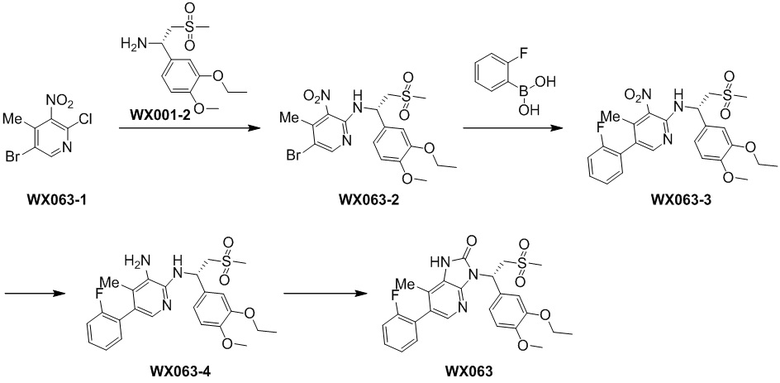
[210] Стадия 1. Получение соединения WX063-2
[211] Соединение WX063-1 (500,00 мг, 1,99 ммоль), соединение WX001-2 (489,15 мг, 1,79 ммоль) и диизопропиламин (513,94 мг, 3,98 ммоль) растворяли в N,N-диметилформамиде (10,00 мл) при комнатной температуре. Реакционную смесь нагревали до 120°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл x 2). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл x 3) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=1/0-1/1, объемное соотношение) с получением целевого продукта WX063-2. 1H ЯМР (400 МГц, CDCl3) δ: 8,34 (s, 1H), 7,33 (d, J=6,8 Гц, 1H), 6,99-6,94 (m, 1H), 6,92 (d, J=2,0 Гц, 1H), 6,90-6,86 (m, 1H), 5,78 (q, J=6,7 Гц, 1H), 4,15-4,06 (m, 2H), 3,86 (s, 3H), 3,75 (dd, J=6,5, 14,6 Гц, 1H), 3,46 (dd, J=6,4, 14,7 Гц, 1H), 2,59 (s, 3H), 2,52 (s, 3H), 1,47 (t, J=7,0 Гц, 3H).
[212] Стадия 2. Получение соединения WX063-3
[213] Соединение WX063-2 (200,00 мг, 409,54 мкмоль), 2-фторфенилбороновую кислоту (85,95 мг, 614,31 мкмоль), комплекс дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (33,44 мг, 40,95 мкмоль) и карбонат калия (113,21 мг, 819,08 мкмоль) растворяли в диоксане (3,00 мл) и воде (1,00 мл) при комнатной температуре. Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали этилацетатом (30 мл x 2). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл x 3) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX063-3.
[214] Стадия 3. Получение соединения WX063-4
[215] Соединение WX063-3 (220,00 мг, 436,91 мкмоль), порошок цинка (285,69 мг, 4,37 ммоль) и хлорид аммония (233,70 мг, 4,37 ммоль) растворяли в метаноле (5,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов в атмосфере азота. После завершения реакции смесь фильтровали и концентрировали при пониженном давлении. Дихлорметан (30 мл) добавляли в полученный остаток с перемешиванием в течение 0,5 часа при комнатной температуре с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX063-4. 1H ЯМР (400 МГц, CDCl3) δ: 7,51-7,29 (m, 3H), 7,24-6,94 (m, 7H), 6,82 (d, J=7,8 Гц, 1H), 4,10 (dd, J=6,8, 11,5 Гц, 2H), 3,99-3,89 (m, 1H), 3,86-3,78 (m, 4H), 3,55 (d, J=11,5 Гц, 1H), 2,87 (br s, 3H), 2,02 (s, 3H), 1,44-1,40 (m, 3H).
[216] Стадия 4. Получение соединения WX063
[217] Соединение WX063-4 (200,00 мг, 422,33 мкмоль), триэтиламин (213,68 мг, 2,11 ммоль) и трифосген (50,13 мг, 168,93 мкмоль) растворяли в тетрагидрофуране (10,00 мл) при комнатной температуре. Реакционную смесь перемешивали при 0-5°C в течение 2 часов в атмосфере азота. После завершения реакции смесь нагревали до комнатной температуры, гасили с помощью воды (50 мл) и экстрагировали с помощью этилацетата (50 мл x 2). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл x 3) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX063. MS-ESI масса/заряд: 500,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 10,12 (br s, 1H), 7,87 (br s, 1H), 7,32 (br s, 1H), 7,15 (br s, 4H), 7,10-7,01 (m, 1H), 6,71 (d, J=7,8 Гц, 1H), 6,13 (d, J=5,0 Гц, 1H), 4,92-4,75 (m, 1H), 3,98 (d, J=4,5 Гц, 2H), 3,82 (d, J=11,5 Гц, 1H), 3,72 (br s, 3H), 2,70 (br s, 3H), 2,11 (br s, 3H), 1,39-1,25 (m, 3H).
[218] Вариант осуществления 64: WX064
Путь синтеза
[219] Стадия 1. Получение соединения WX064-1
[220] Соединение WX063-2 (1,00 г, 2,05 ммоль), фенилбороновую кислоту (374,93 мг, 3,08 ммоль), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (167,41 мг, 205,00 мкмоль) и карбонат калия (849,99 мг, 6,15 ммоль) растворяли в диоксане (40,00 мл) и воде (10,00 мл) при комнатной температуре. Реакционную смесь нагревали до 80-90°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и этилацетатом (100 мл). Органические фазы отделяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=5/1-1/2, объемное соотношение) с получением целевого продукта WX064-1. MS-ESI масса/заряд: 486,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,16 (s, 1H), 7,48-7,35 (m, 3H), 7,29-7,21 (m, 3H), 7,05-6,96 (m, 2H), 6,90 (d, J=8,3 Гц, 1H), 5,90 (q, J=6,5 Гц, 1H), 4,13 (q, J=7,0 Гц, 2H), 3,91-3,79 (m, 4H), 3,50 (dd, J=6,5, 14,8 Гц, 1H), 2,63 (s, 3H), 2,34 (s, 3H), 1,48 (t, J=7,0 Гц, 3H).
[221] Стадия 2. Получение соединения WX064-2
[222] Соединение WX064-1 (800,00 мг, 1,65 ммоль) растворяли в метаноле (50,00 мл) при комнатной температуре с последующим добавлением порошка цинка (1,08 г, 16,50 ммоль) и хлорида аммония (1,08 г, 16,50 ммоль). Реакционную смесь перемешивали при 0-5°C в течение 1,5 часа в атмосфере азота. После завершения реакции смесь фильтровали с применением диатомита и фильтрат концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли дихлорметан (100 мл) и воду (30 мл). Органические фазы отделяли и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX064-2. MS-ESI масса/заряд: 456,1 [M+H]+.
[223] Стадия 3. Получение соединения WX064
[224] Соединение WX064-2 (700,00 мг, 1,54 ммоль), трифосген (274,20 мг, 924,00 мкмоль) и триэтиламин (935,00 мг, 9,24 ммоль, 1,28 мл) растворяли в тетрагидрофуране (30,00 мл) при комнатной температуре. Реакционную смесь перемешивали при 0-5°C в течение 2 часов в атмосфере азота. После завершения реакции смесь разбавляли водой (20 мл) и этилацетатом (100 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (30 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=10/1-1/2, объемное соотношение) с получением целевого продукта WX064. MS-ESI масса/заряд: 482,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 10,16 (br s, 1H), 7,89 (s, 1H), 7,43-7,29 (m, 3H), 7,28-7,13 (m, 4H), 6,74 (d, J = 8,3 Гц, 1H), 6,15 (dd, J = 4,4, 9,7 Гц, 1H), 4,89 (dd, J = 9,8, 14,6 Гц, 1H), 4,10-3,96 (m, 2H), 3,86 (dd, J = 4,4, 14,7 Гц, 1H), 3,75 (s, 3H), 2,70 (s, 3H), 2,22 (s, 3H), 1,35 (t, J = 6,9 Гц, 3H).
[225] Вариант осуществления 65: WX065
Путь синтеза
[226] Стадия 1. Получение соединения WX065-2
[227] Соединение WX065-1 (2,00 г, 10,36 ммоль), 2-фторфенилбороновую кислоту (1,45 г, 10,36 ммоль), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (846,30 мг, 1,04 ммоль) и карбонат калия (2,15 г, 15,54 ммоль) растворяли в диоксане (20,00 мл) и воде (7,00 мл) при комнатной температуре. Реакционную смесь нагревали до 60°C и перемешивали в течение 4 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (25 мл), разбавляли водой (25 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали водой (30 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/0-10/1, объемное соотношение) с получением целевого продукта WX065-2. 1H ЯМР (400 МГц, CDCl3) δ: 8,58 (d, J=5,0 Гц, 1H), 7,54-7,45 (m, 1H), 7,41 (d, J=4,8 Гц, 1H), 7,34-7,17 (m, 3H).
[228] Стадия 2. Получение соединения WX065-3
[229] Соединение WX065-2 (997,38 мг, 3,95 ммоль) и соединение WX001-2 (900,00 мг, 3,29 ммоль) растворяли в N,N-диметилформамиде (20,00 мл) при комнатной температуре с последующим добавлением диизопропилэтиламина (851,04 мг, 6,58 ммоль, 1,15 мл). Реакционную смесь нагревали до 80°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (20 мл), разбавляли водой (60 мл) и экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали водой (100 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/0-2/1, объемное соотношение) с получением целевого продукта WX065-3. 1H ЯМР (400 МГц, CDCl3) δ: 8,40 (d, J=4,8 Гц, 1H), 7,86 (d, J=7,0 Гц, 1H), 7,49-7,39 (m, 1H), 7,38-7,30 (m, 1H), 7,19-7,10 (m, 1H), 7,09-6,98 (m, 2H), 6,94 (d, J=8,3 Гц, 1H), 6,68 (d, J=4,8 Гц, 1H), 5,92 (q, J=6,8 Гц, 1H), 4,16 (q, J=7,0 Гц, 2H), 4,01-3,85 (m, 4H), 3,53 (dd, J=7,0, 14,6 Гц, 1H), 2,62 (s, 3H), 1,50 (t, J=7,0 Гц, 3H).
[230] Стадия 3. Получение соединения WX065-4
[231] Соединение WX065-3 (350,00 мг, 714,99 мкмоль) и хлорид аммония (573,67 мг, 10,72 ммоль, 374,95 мкл) растворяли в метаноле (10,00 мл) при комнатной температуре с последующим добавлением 5 партиями порошка цинка (467,53 мг, 7,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере азота. После завершения реакции смесь фильтровали с применением диатомита. Фильтрат концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли дихлорметан (20 мл) с перемешиванием в течение 5 минут с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX065-4.
[232] Стадия 4. Получение соединения WX065
[233] Соединение WX065-4 (300,00 мг, 652,84 мкмоль) и триэтиламин (264,24 мг, 2,61 ммоль, 361,97 мкл) растворяли в тетрагидрофуране (30,00 мл) при комнатной температуре. Раствор затем охлаждали до 0°C с последующим добавлением трифосгена (67,81 мг, 228,49 мкмоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 5 часов. После завершения реакции нерастворимые вещества удаляли посредством фильтрации и фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX065. MS-ESI масса/заряд: 486,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,16 (d, J=5,3 Гц, 1H), 7,61-7,47 (m, 2H), 7,44 (s, 1H), 7,40-7,28 (m, 3H), 7,16 (d, J=5,0 Гц, 1H), 6,82 (d, J=8,3 Гц, 1H), 6,39 (d, J=4,8 Гц, 1H), 4,92-4,79 (m, 1H), 4,09 (q, J=6,8 Гц, 2H), 3,90 (d, J=11,8 Гц, 1H), 3,84 (s, 3H), 2,85 (s, 3H), 1,44 (t, J=6,9 Гц, 3H).
[234] Вариант осуществления 66: WX066
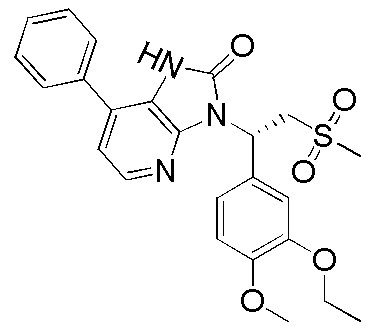
Путь синтеза
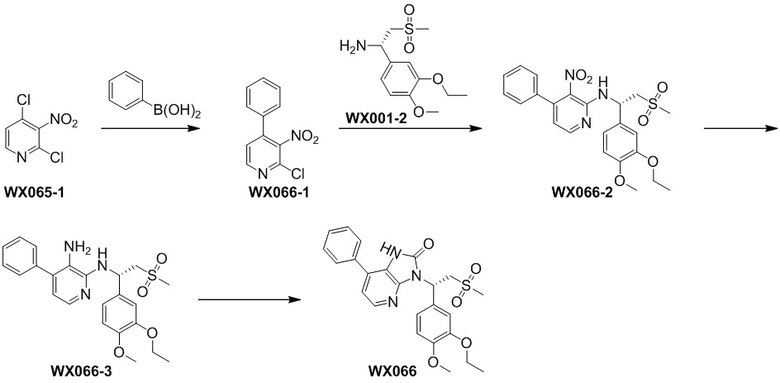
[235] Стадия 1. Получение соединения WX066-1
[236] Соединение WX065-1 (1,00 г, 5,18 ммоль), фенилбороновую кислоту (758,15 мг, 6,22 ммоль), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (189,51 мг, 259,00 мкмоль) и карбонат калия (2,15 г, 15,54 ммоль) растворяли в диоксане (18,00 мл) и воде (6,00 мл) при комнатной температуре. Реакционную смесь нагревали до 80°C и перемешивали в течение 2 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=40/1-20/1, объемное соотношение) с получением целевого продукта WX066-1. 1H ЯМР (400 МГц, DMSO_d6) δ: 8,75 (d, J=5,0 Гц, 1H), 7,80 (d, J=5,0 Гц, 1H), 7,58-7,55 (m, 3H), 7,50-7,39 (m, 2H).
[237] Стадия 2. Получение соединения WX066-2
[238] Соединение WX066-1 (300,00 мг, 1,28 ммоль) растворяли в ацетонитриле (20,00 мл) при комнатной температуре с последующим добавлением соединения WX001-2 (291,57 мг, 1,07 ммоль) и карбоната калия (442,27 мг, 3,20 ммоль). Реакционную смесь нагревали до 90°C и перемешивали в течение 72 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной тонкослойной хроматографии (элюент: петролейный эфир/этилацетат=1/1, объемное соотношение) с получением целевого продукта WX066-2. 1H ЯМР (400 МГц, CDCl3) δ: 8,30 (d, J=5,0 Гц, 1 H), 7,46-7,40 (m, 3 H), 7,37 (d, J=6,8 Гц, 1 H), 7,31-7,27 (m, 2 H), 7,05-6,97 (m, 2 H), 6,92-6,87 (m, 1 H), 6,67 (d, J=4,8 Гц, 1 H), 5,88 (d, J=6,8 Гц, 1 H), 4,14-4,11 (m, 2 H), 3,90-3,82 (m, 4 H), 3,50 (dd, J=14,6, 6,8 Гц, 1 H), 2,61 (s, 3 H), 1,51-1,45 (m, 3 H).
[239] Стадия 3. Получение соединения WX066-3
[240] Соединение WX066-2 (110,00 мг, 233,28 мкмоль) и хлорид аммония (124,78 мг, 2,33 ммоль, 81,56 мкл) растворяли в метаноле (10,00 мл) при комнатной температуре с последующим добавлением порошка цинка (152,54 мг, 2,33 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,1 часа в атмосфере азота. После завершения реакции смесь фильтровали с применением диатомита. Осадок на фильтре промывали дихлорметаном (20 мл x 2). Фильтрат объединяли и концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли дихлорметан (20 мл) с перемешиванием в течение 5 минут с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением продукта WX066-3. MS-ESI масса/заряд: 442,1 [M+H]+.
[241] Стадия 4. Получение соединения WX066
[242] Соединение WX066-3 (100,00 мг, 226,48 мкмоль) и триэтиламин (114,59 мг, 1,13 ммоль, 156,97 мкл) растворяли в тетрагидрофуране (25,00 мл) при комнатной температуре. Затем раствор охлаждали до 0°C с последующим добавлением трифосгена (26,88 мг, 90,59 мкмоль). Реакционную смесь перемешивали при 0°C в течение 5 часов. После завершения реакции смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX066. MS-ESI масса/заряд: 468,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 10,13 (s, 1 H), 8,15 (s, 1 H), 7,68-7,42 (m, 6 H), 7,30 (s, 1 H), 7,20 (s, 1 H), 6,80 (d, J=7,5 Гц, 1 H), 6,49 (s, 1 H), 4,91 (s, 1 H), 4,18-3,98 (m, 2 H), 3,93-3,70 (m, 4 H), 2,92 (s, 3 H), 1,41 (t, J=6,3 Гц, 3 H).
[243] Вариант осуществления 67: WX067
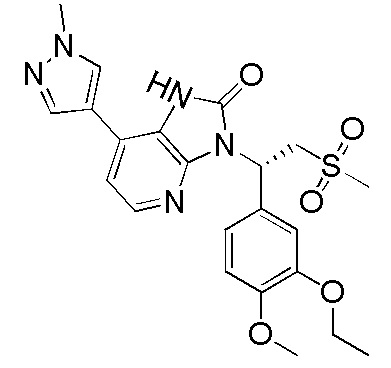
Путь синтеза
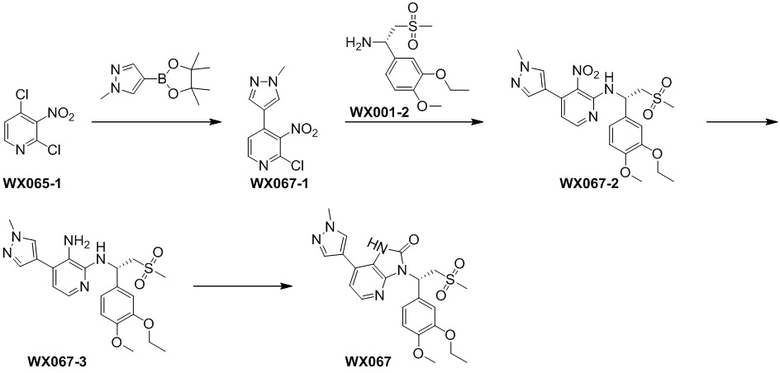
[244] Стадия 1. Получение соединения WX067-1
[245] Соединение WX065-1 (5,00 г, 25,91 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-илпиразол (5,39 г, 25,91 ммоль), карбонат калия (10,74 г, 77,73 ммоль) и комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (4,23 г, 5,18 ммоль) растворяли в диоксане (50,00 мл) и воде (10,00 мл) при комнатной температуре. Реакционную смесь нагревали до 80-90°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли этилацетатом (100 мл) и фильтровали с применением диатомита. К фильтрату добавляли воду (30 мл) и перемешивали в течение 10 минут. Органические фазы отделяли, промывали насыщенным солевым раствором (30 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=5/1-1/1, объемное соотношение) с получением целевого продукта WX067-1. MS-ESI масса/заряд: 239,0 [M+H]+.
[246] Стадия 2. Получение соединения WX067-2
[247] Соединение WX067-1 (2,00 г, 8,38 ммоль), соединение WX001-2 (2,29 г, 8,38 ммоль) и карбонат калия (3,47 г, 25,14 ммоль) растворяли в ацетонитриле (50,00 мл) при комнатной температуре. Реакционную смесь нагревали до 90-100°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и этилацетатом (200 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (30 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=3/1-1/2, объемное соотношение) с получением целевого продукта WX067-2. MS-ESI масса/заряд: 476,1 [M+H]+.
[248] Стадия 3. Получение соединения WX067-3
[249] Соединение WX067-2 (500,00 мг, 1,05 ммоль), порошок цинка (686,60 мг, 10,50 ммоль) и хлорид аммония (561,65 мг, 10,50 ммоль) растворяли в метаноле (20,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 15 часов в атмосфере азота. После завершения реакции смесь фильтровали с применением диатомита. Фильтрат концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли дихлорметан (20 мл) с перемешиванием в течение 20 минут с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта WX067-3. MS-ESI масса/заряд: 446,1 [M+H]+.
[250] Стадия 4. Получение соединения WX067
[251] Соединение WX067-3 (200,00 мг, 448,90 мкмоль), трифосген (79,93 мг, 269,34 мкмоль) и триэтиламин (136,27 мг, 1,35 ммоль, 186,68 мкл) растворяли в тетрагидрофуране (20,00 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0-5°C и перемешивали в течение 5 часов. После завершения реакции реакционный раствор выливали в ледяную воду (20,00 мл) с последующим добавлением этилацетата (50,00 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX067. MS-ESI масса/заряд: 472,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3): δ: 11,18 (s, 1H), 8,45 (s, 1H), 8,10 (s, 1H), 7,95 (d, J=5,6 Гц, 1H), 7,28-7,25 (m, 2H), 7,02 (d, J=8,0 Гц, 1H), 6,92 (d, J=8,8 Гц, 1H), 6,07 (dd, J=3,6, 9,6 Гц, 1H), 4,73-4,69 (m, 1H), 4,18-4,14 (m, 1H), 4,03-3,97 (m, 2H), 3,90 (s, 3H), 3,72 (s, 3H), 2,98 (s, 3H), 1,32 (t, J=7,2 Гц, 3H).
[252] Вариант осуществления 68: WX068
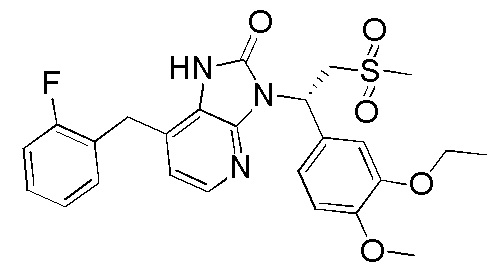
Путь синтеза
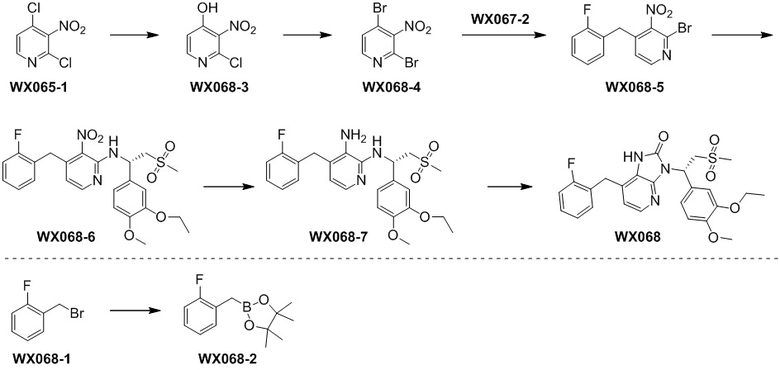
[253] Стадия 1. Получение соединения WX068-2
[254] Соединение WX068-1 (5,00 г, 26,45 ммоль, 3,18 мл), бис(пинаколато)дибор (10,08 г, 39,67 ммоль), тетракис(трифенилфосфин)палладий (3,06 г, 2,64 ммоль) и карбонат калия (10,97 г, 79,35 ммоль) растворяли в диоксане (100,00 мл) при комнатной температуре. Реакционную смесь нагревали до 80-90°C в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой (200 мл) и этилацетатом (500 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (100 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=100/1-10/1, объемное соотношение) с получением целевого продукта WX068-2. 1H ЯМР (400 МГц, CDCl3) δ: 7,28-6,93 (m, 4H), 2,26 (s, 2H), 1,25 (s, 12H).
[255] Стадия 2. Получение соединения WX068-3
[256] Соединение WX065-1 (35,00 г, 181,36 ммоль) и ацетат натрия (44,63 г, 544,08 ммоль) растворяли в N,N-диметилформамиде (600,00 мл) при комнатной температуре. Реакционную смесь нагревали до 120°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли этилацетат (500 мл) и воду (200 мл). Органические фазы отделяли и водные фазы экстрагировали этилацетатом (200 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (200 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта. Полученный неочищенный продукт суспендировали в течение 30 минут со смесью петролейного эфира и этилацетата с последующей фильтрацией. Осадок на фильтре высушивали в вакууме в течение 30 минут с получением целевого продукта WX068-3. 1H ЯМР (400 МГц, DMSO_d6) δ: 8,19 (d, J=6,0 Гц, 1H), 7,03 (d, J=6,0 Гц, 1H).
[257] Стадия 3. Получение соединения WX068-4
[258] Соединение WX068-3 (10,00 г, 57,29 ммоль) и оксибромид фосфора (49,28 г, 171,87 ммоль) растворяли в ацетонитриле (600,00 мл) при комнатной температуре. Реакционную смесь нагревали до 90-100°C и перемешивали в течение 5 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли этилацетатом (500 мл) и его pH регулировали с помощью насыщенного раствора бикарбоната натрия до 7-8 при 0-5°C. Органические фазы отделяли, промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=10/1-3/1, объемное соотношение) с получением целевого продукта WX068-4. 1H ЯМР (400 МГц, CDCl3) δ: 8,33 (d, J=5,6 Гц, 1H), 7,64 (d, J=5,6 Гц, 1H).
[259] Стадия 4. Получение соединения WX068-5
[260] Соединение WX068-4 (3,50 г, 12,42 ммоль), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (1,38 г, 1,69 ммоль) и карбонат калия (4,68 г, 33,89 ммоль) растворяли в диоксане (100,00 мл) и воде (30,00 мл) при комнатной температуре. Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали с применением диатомита и разбавляли этилацетатом (500 мл) и водой (100 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (100 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX068-5. MS-ESI масса/заряд: 310,8 [M+H]+, 312,8 [M+H+2]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,28 (d, J=5,0 Гц, 1H), 7,28-7,17 (m, 1H), 7,11-6,97 (m, 4H), 3,92 (s, 2H).
[261] Стадия 5. Получение соединения WX068-6
[262] Трифторацетатную соль соединения WX068-5 (800,00 мг, 1,88 ммоль), соединение WX001-2 (771,58 мг, 2,82 ммоль) и диизопропилэтиламин (729,60 мг, 5,65 ммоль, 985,95 мкл) растворяли в N,N-диметилформамиде (30,00 мл) при комнатной температуре. Реакционную смесь нагревали до 120-130°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и разбавляли этилацетатом (200 мл) и водой (100 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=10/1-1/2, объемное соотношение) с получением целевого продукта WX068-6. MS-ESI масса/заряд: 504,1 [M+H]+.
[263] Стадия 6. Получение соединения WX068-7
[264] Соединение WX068-6 (300,00 мг, 595,78 мкмоль), порошок цинка (389,58 мг, 5,96 ммоль) и хлорид аммония (318,68 мг, 5,96 ммоль, 208,29 мкл) растворяли в метаноле (30,00 мл) при комнатной температуре. Реакционную смесь охлаждали до 0-5°C и перемешивали в течение 1 часа в атмосфере азота. После завершения реакции смесь фильтровали с применением диатомита и осадок на фильтре промывали дихлорметаном (20 мл x 2). Объединенный фильтрат концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли дихлорметан (50 мл) и воду (20 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя с получением целевого продукта WX068-7. MS-ESI масса/заряд: 474,2 [M+H]+.
[265] Стадия 7. Получение соединения WX068
[266] Соединение WX068-7 (200,00 мг, 422,33 мкмоль), трифосген (75,20 мг, 253,40 мкмоль) и триэтиламин (256,41 мг, 2,53 ммоль, 351,25 мкл) растворяли в тетрагидрофуране (30,00 мл) при комнатной температуре. Реакционную смесь перемешивали при 0-5°C в течение 2 часов в атмосфере азота. После завершения реакции смесь разбавляли водой (20 мл) и этилацетатом (100 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX068. MS-ESI масса/заряд: 500,0 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 10,32 (br s, 1H), 8,16 (d, J=5,2 Гц, 1H), 7,52 (d, J=1,6 Гц, 1H), 7,43-7,38 (m, 3H), 7,27-7,25 (m, 2H), 7,03 (d, J=5,2 Гц, 1H), 6,94 (d, J=8,4 Гц, 1H), 6,37 (br dd, J=4,8, 9,0 Гц, 1H), 5,01 (br dd, J=9,4, 14,7 Гц, 1H), 4,24-4,19(m, 4H), 4,13-4,09 (m, 1H), 3,98 (s, 3H), 2,87 (s, 3H), 1,56 (t, J= 6,8 Гц, 3H).
[267] Вариант осуществления 69: WX069
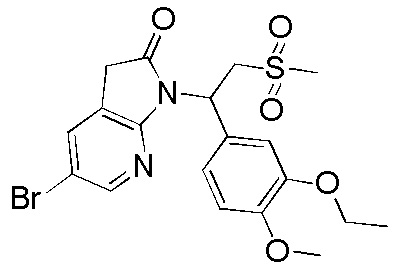
Путь синтеза
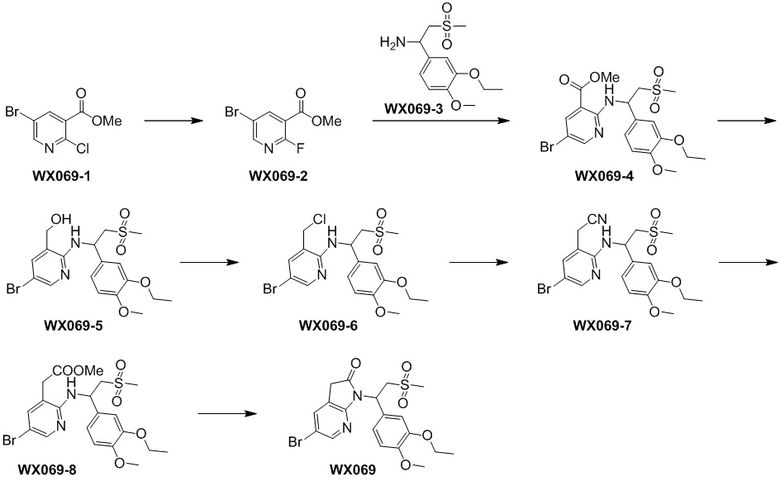
[268] Стадия 1. Получение соединения WX069-2
[269] Соединение WX069-1 (7,00 г, 27,95 ммоль) растворяли в растворе диметилсульфоксида (50,00 мл) при комнатной температуре с последующим добавлением фторида цезия (8,00 г, 52,67 ммоль) при комнатной температуре. Реакционную смесь нагревали до 55°C и перемешивали в течение 14 часов. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (10 мл), разбавляли водой (50 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы отделяли, промывали водой (50 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=100/1-100/15, объемное соотношение) с получением целевого продукта WX069-2. 1H ЯМР (400 МГц, CDCl3) δ: 8,64-8,31 (m, 2H), 3,98 (s, 3H).
[270] Стадия 2. Получение соединения WX069-4
[271] Соединение WX069-2 (2,70 г, 3,46 ммоль) и соединение WX069-3 (946,13 мг, 3,46 ммоль) растворяли в ацетонитриле (20,00 мл) при комнатной температуре с последующим добавлением карбоната калия (559,70 мг, 4,05 ммоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 5 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью воды (20 мл), разбавляли водой (40 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=100/0-100/70, объемное соотношение) с получением целевого продукта WX069-4. 1H ЯМР (400 МГц, CDCl3) δ: 8,57 (d, J=7,5 Гц, 1H), 8,32 (d, J=2,5 Гц, 1H), 8,24 (d, J=2,5 Гц, 1H), 7,06-6,95 (m, 2H), 6,89 (d, J=8,3 Гц, 1H), 5,82 (q, J=6,6 Гц, 1H), 4,13 (q, J=7,0 Гц, 2H), 3,89 (d, J=13,1 Гц, 6H), 3,81 (dd, J=6,5, 14,8 Гц, 1H), 3,47 (dd, J=6,4, 14,7 Гц, 1H), 2,64 (s, 3H), 1,48 (t, J=6,9 Гц, 3H).
[272] Стадия 3. Получение соединения WX069-5
[273] Соединение WX069-4 (700,00 мг, 1,44 ммоль) растворяли в тетрагидрофуране (10,00 мл) при комнатной температуре с последующим добавлением борогидрида лития (94,09 мг, 4,32 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 0,5 часа и затем нагревали до 30°C с перемешиванием в течение еще 2,5 часов в атмосфере азота. После завершения реакции смесь гасили с помощью насыщенного раствора хлорида аммония до прекращения образования пузырьков, разбавляли водой (40 мл) и экстрагировали дихлорметаном (20 мл x 3). Органические фазы объединяли, промывали водой (50 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/1-1/3, объемное соотношение) с получением целевого продукта WX069-5. 1H ЯМР (400 МГц, CDCl3) δ: 8,01 (br s, 1H), 7,34 (br s, 1H), 6,96 (br s, 2H), 6,82 (d, J=6,5 Гц, 1H), 6,26 (d, J=5,8 Гц, 1H), 5,72 (d, J=4,0 Гц, 1H), 4,55 (q, J=11,1 Гц, 2H), 4,07 (d, J=5,8 Гц, 2H), 3,94-3,60 (m, 4H), 3,44 (d, J=11,0 Гц, 1H), 2,63 (br s, 3H), 2,02 (br s, 1H), 1,42 (br s, 3H).
[274] Стадия 4. Получение соединения WX069-6
[275] Соединение WX069-5 (491,50 мг, 1,07 ммоль) и триэтиламин (108,27 мг, 1,07 ммоль) растворяли в дихлорметане (5,00 мл) при комнатной температуре. Раствор затем охлаждали до 0°C с последующим добавлением MsCl (122,57 мг, 1,07 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов в атмосфере азота. После завершения реакции смесь гасили с помощью насыщенного солевого раствора (20 мл), разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали водой (70 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/0-1/1, объемное соотношение) с получением неочищенного продукта WX069-6. 1H ЯМР (400 МГц, CDCl3) δ: 8,13 (d, J=1,8 Гц, 1H), 7,52 (d, J=2,0 Гц, 1H), 7,05-6,94 (m, 2H), 6,89 (d, J=7,8 Гц, 1H), 5,92-5,74 (m, 2H), 4,63-4,43 (m, 2H), 4,18-4,03 (m, 2H), 3,87 (s, 3H), 3,79 (dd, J=5,9, 14,7 Гц, 1H), 3,53 (dd, J=5,1, 14,7 Гц, 1H), 2,58 (s, 3H), 1,45 (t, J=7,0 Гц, 3H).
[276] Стадия 5. Получение соединения WX069-7
[277] Соединение WX069-6 (175 мг, неочищенный продукт) растворяли в N,N-диметилформамиде (3,00 мл) при комнатной температуре с последующим последовательным добавлением цианида натрия (17,95 мг, 366,26 мкмоль) и триэтиламина (74,12 мг, 732,52 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения реакции смесь гасили с помощью насыщенного солевого раствора (20 мл), разбавляли водой (40 мл) и экстрагировали этилацетатом (15 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (25 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной флэш-хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/0-2/3, объемное соотношение) с получением целевого продукта WX069-7. 1H ЯМР (400 МГц, CDCl3) δ: 8,13 (d, J=2,3 Гц, 1H), 7,62 (d, J=2,0 Гц, 1H), 7,06-6,94 (m, 2H), 6,93-6,82 (m, 1H), 5,82-5,71 (m, 1H), 5,61 (d, J=6,3 Гц, 1H), 4,11 (dq, J=4,3, 6,9 Гц, 2H), 3,87 (s, 3H), 3,71 (dd, J=6,9, 14,7 Гц, 1H), 3,59 (d, J=3,8 Гц, 2H), 3,51 (dd, J=4,9, 14,7 Гц, 1H), 2,62 (s, 3H), 1,46 (t, J=6,9 Гц, 3H).
[278] Стадия 6. Получение соединения WX069-8
[279] Соединение WX069-7 (60,00 мг, 128,11 мкмоль) растворяли в растворе хлористоводородной кислоты в метаноле (4 M, 5 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 5 часов. После завершения реакции pH смеси регулировали с помощью насыщенного раствора карбоната натрия до 7-8. Раствор разбавляли насыщенным солевым раствором (30 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали водой (30 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной флэш-хроматографии (элюент: петролейный эфир/этилацетат=1/0-2/3, объемное соотношение) с получением целевого продукта WX069-8.
[280] Стадия 7. Получение соединения WX069
[281] Соединение WX069-8 (60,00 мг, 119,67 мкмоль) растворяли в уксусной кислоте (2,00 мл) при комнатной температуре. Реакционную смесь нагревали до 150°C с помощью микроволнового излучения и перемешивали в течение 20 минут. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл) и концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX069. MS-ESI масса/заряд: 469,2 [M+H]+, 471,2 [M+H+2]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,26 (d, J=1,8 Гц, 1H), 7,58 (s, 1H), 7,26 (d, J=2,0 Гц, 1H), 7,19 (dd, J=1,9, 8,2 Гц, 1H), 6,83 (d, J=8,3 Гц, 1H), 6,11 (dd, J=4,5, 10,3 Гц, 1H), 4,78 (dd, J=10,3, 14,3 Гц, 1H), 4,10 (q, J=7,1 Гц, 2H), 3,85 (s, 3H), 3,79 (dd, J=4,3, 14,6 Гц, 1H), 3,55 (s, 2H), 2,84 (s, 3H), 1,47 (t, J=6,9 Гц, 3H).
[282] Вариант осуществления 70: WX070
Путь синтеза
[283] Стадия 1. Получение соединения WX070-2
[284] Диметилсульфон (5,22 г, 55,50 ммоль) растворяли в тетрагидрофуране (50,00 мл) и при 0°C добавляли по каплям н-бутиллитий (2,5 M, 22,20 мл). Реакционную смесь перемешивали при 0°C в течение 1 часа в атмосфере азота с последующим добавлением по каплям раствора соединения WX070-1 (5,00 г, 27,75 ммоль) в тетрагидрофуране (20,00 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение еще 1,5 часа в атмосфере азота. После завершения реакции смесь нагревали до комнатной температуры, гасили с помощью насыщенного раствора хлорида аммония (50 мл), разбавляли водой (150 мл) и экстрагировали этилацетатом (50 мл x 3). Органические фазы объединяли, промывали водой (100 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток перекристаллизовывали с применением этилацетата (20 мл) с получением целевого продукта WX070-2. 1H ЯМР (400 МГц, DMSO_d6) δ: 6,99 (s, 1H), 6,91 (s, 2H), 5,83 (d, J=4,0 Гц, 1H), 4,96 (ddd, J=2,8, 4,3, 10,0 Гц, 1H), 4,02 (q, J=7,0 Гц, 2H), 3,73 (s, 3H), 3,57 (dd, J=10,0, 14,6 Гц, 1H), 3,15 (d, J=14,6 Гц, 1H), 3,01 (s, 3H), 1,33 (t, J=7,0 Гц, 3H).
[285] Стадия 2. Получение соединения WX070-4
[286] Соединение WX070-3 (200,00 мг, 913,28 мкмоль) растворяли в уксусной кислоте (5,00 мл) и метаноле (5,00 мл) с последующим добавлением порошка железа (153,02 мг, 2,74 ммоль) за один раз. Реакционную смесь перемешивали при 0-5°C в течение 1,5 часа в атмосфере азота. После завершения реакции смесь разбавляли водой (50 мл) и этилацетатом (50 мл) и ее pH регулировали до 9-10 с помощью водного раствора гидроксида натрия (1 M). Отделенную водную фазу экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=5/1-1/4, объемное соотношение) с получением целевого продукта WX070-4. 1H ЯМР (400 МГц, MeOD) δ: 7,46 (d, J=2,0 Гц, 1H), 6,99 (d, J=2,0 Гц, 1H).
[287] Стадия 3. Получение соединения WX070-5
[288] Соединение WX070-4 (300,00 мг, 1,59 ммоль) растворяли в дихлорметане (20,00 мл) и пиридине (10,00 мл) с последующим добавлением трифосгена (471,83 мг, 1,59 ммоль) при 0°C. Реакционную смесь нагревали до 40°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь разбавляли водой (20 мл). Отделенную водную фазу экстрагировали дихлорметаном (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=10/1-1/1, объемное соотношение) с получением целевого продукта WX070-5. 1H ЯМР (400 МГц, MeOD) δ: 8,14 (d, J=2,0 Гц, 1H), 7,77 (d, J=2,0 Гц, 1H).
[289] Стадия 4. Получение соединения WX070
[290] Соединение WX070-5 (230,00 мг, 1,07 ммоль), трифенилфосфин (336,78 мг, 1,28 ммоль) и соединение WX070-2 (293,53 мг, 1,07 ммоль) растворяли в тетрагидрофуране (4,00 мл) при комнатной температуре с последующим добавлением диизопропилазодикарбоксилата (259,64 мг, 1,28 ммоль) при 0°C. Реакционную смесь перемешивали при 0-20°C в течение 12 часов в атмосфере азота. После завершения реакции смесь разбавляли водой (20 мл). Отделенную водную фазу экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/1, объемное соотношение) с получением целевого продукта WX070. MS-ESI масса/заряд: 470,9 [M+H]+, 473,0 [M+H+2]+. 1H ЯМР (400 МГц, CDCl3) δ: 1,47 (t, J=6,9 Гц, 3H), 2,85 (s, 3H), 3,81 (dd, J=14,6, 4,52 Гц, 1H), 3,85 (s, 3H), 4,07-4,13 (m, 2H), 4,73 (dd, J=14,3, 10,3 Гц, 1H), 6,02 (dd, J=10,2, 4,4 Гц, 1H), 6,84 (d, J=8,5 Гц, 1H), 7,19 (dd, J=8,3, 2,0 Гц, 1H), 7,24 (d, J=2,0 Гц, 1H), 7,56 (d, J=2,0 Гц, 1 H), 8,22 (d, J=2,0 Гц, 1 H).
[291] Вариант осуществления 71: WX071
Путь синтеза
[292] Стадия 1. Получение соединения WX071-1
[293] Соединение WX070-5 (1,80 г, 8,37 ммоль), 2-фторфенилбороновую кислоту (1,41 г, 10,04 ммоль) растворяли в диоксане (30,00 мл) при комнатной температуре с последующим добавлением комплекса дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (341,76 мг, 418,50 мкмоль), карбоната калия (4,68 г, 33,89 ммоль) и воды (10,00 мл). Реакционную смесь нагревали до 80°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (10 мл), разбавляли водой (30 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали водой (30 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=3/1-1/1, объемное соотношение) с получением целевого продукта WX071-1. MS-ESI масса/заряд: 231,0 [M+H]+.
[294] Стадия 2. Получение соединения WX071-2
[295] Соединение WX071-1 (800,00 мг, 3,48 ммоль), соединение WX070-2 и трифенилфосфин (1,09 г, 4,17 ммоль) растворяли в тетрагидрофуране (5,00 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением по каплям диизопропилазодикарбоксилата (843,31 мг, 4,17 ммоль, 810,88 мкл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 20 часов в атмосфере азота. После завершения реакции смесь концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=1/1-1/2, объемное соотношение) с получением целевого продукта WX071-2. 1H ЯМР (400 МГц, CDCl3) δ: 7,47-7,31 (m, 4H), 7,26-7,13 (m, 4H), 7,10 (dd, J=2,3, 8,3 Гц, 1H), 6,88 (d, J=8,3 Гц, 1H), 5,72 (dd, J=4,8, 9,3 Гц, 1H), 4,66 (dd, J=9,3, 14,8 Гц, 1H), 4,12 (q, J=7,0 Гц, 2H), 3,95-3,84 (m, 4H), 2,86 (s, 3H), 1,49 (t, J=6,9 Гц, 3H).
[296] Стадия 3. Получение соединения WX071
[297] Соединение WX071-2 (550 мг) выделяли с помощью сверхкритической жидкостной хроматографии (условия: колонка: Chiralpak AS-H 150 x 4,6 мм I.D., 5 мкм; подвижная фаза: A: диоксид углерода, B: этанол (0,05% диэтиламин); приблизительно 40%; расход: 3 мл/мин.; температура колонки: 40°C; длина волны: 220 нм) и собирали образец со временем удерживания 4,103 мин. с получением WX071 (ee%: 98,93%).
[298] Вариант осуществления 72: WX072
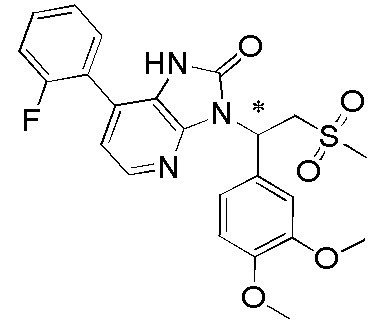
Путь синтеза
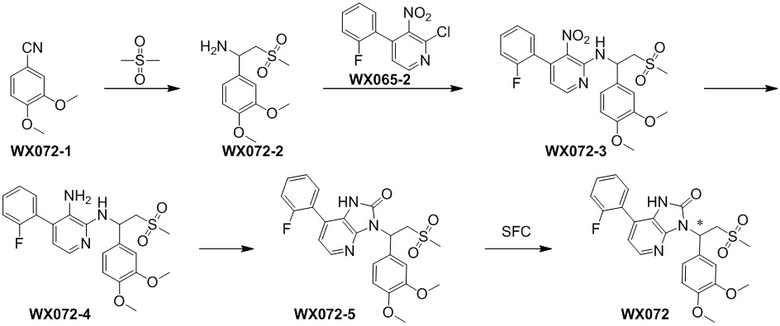
[299] Стадия 1. Получение соединения WX072-2
[300] Диметилсульфон (1,38 г, 14,71 ммоль, 1,19 мл) растворяли в тетрагидрофуране (15,00 мл) при комнатной температуре. Реакционную смесь охлаждали до -5°C в атмосфере азота с последующим добавлением по каплям н-бутиллития (2,5 M, 5,39 мл). Реакционную смесь перемешивали при 0°C в течение 1 часа с последующим добавлением по каплям раствора соединения WX072-1 (2,00 г, 12,26 ммоль) в тетрагидрофуране (20,00 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 15 минут и затем нагревали до комнатной температуры и перемешивали в течение 1,5 часа. Затем смесь охлаждали до 0°C с последующим добавлением борогидрида натрия (602,93 мг, 15,94 ммоль) с перемешиванием в течение 10 минут, уксусной кислоты (3,39 г, 56,40 ммоль, 3,23 мл) с перемешиванием в течение 2 часов и водного раствора гидроксида натрия (2,5 M, 16,18 мл) с перемешиванием в течение 15 минут. Реакционную смесь затем медленно нагревали до комнатной температуры, нагревали до 60°C и перемешивали при нагревании с обратным холодильником в течение 12 часов. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (50 мл) и экстрагировали этилацетатом (15 мл x 3). Органические фазы объединяли, промывали водой (50 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=5/1-1/1, объемное соотношение) с получением целевого продукта WX072-2. 1H ЯМР (400 МГц, CDCl3) δ: 6,95-6,88 (m, 2H), 6,87-6,80 (m, 1H), 4,61 (dd, J=3,0, 9,5 Гц, 1H), 3,89 (s, 3H), 3,87 (s, 3H), 3,42-3,15 (m, 2H), 2,92 (s, 3H).
[301] Стадия 2. Получение соединения WX072-3
[302] Соединение WX065-2 (369,85 мг, 1,46 ммоль) и соединение WX072-2 (316,00 мг, 1,22 ммоль) растворяли в N,N-диметилформамиде (3,00 мл) при комнатной температуре с последующим добавлением диизопропилэтиламина (315,35 мг, 2,44 ммоль, 426,14 мкл). Реакционную смесь нагревали до 65°C и перемешивали в течение 24 часов. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (10 мл), разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=2/1-1/1, объемное соотношение) с получением целевого продукта WX072-3. 1H ЯМР (400 МГц, CDCl3) δ: 8,38 (d, J=4,8 Гц, 1H), 7,85 (d, J=6,8 Гц, 1H), 7,47-7,38 (m, 1H), 7,36-7,28 (m, 1H), 7,26-7,22 (m, 1H), 7,12 (t, J=9,2 Гц, 1H), 7,06 (dd, J=1,9, 8,2 Гц, 1H), 7,00 (d, J=2,0 Гц, 1H), 6,92 (d, J=8,3 Гц, 1H), 6,66 (d, J=4,8 Гц, 1H), 5,93 (q, J=6,5 Гц, 1H), 3,93 (s, 3H), 3,90 (s, 3H), 3,53 (dd, J=6,9, 14,7 Гц, 1H), 2,64 (s, 3H).
[303] Стадия 3. Получение соединения WX072-4
[304] Соединение WX072-3 и хлорид аммония (151,87 мг, 2,84 ммоль) растворяли в метаноле (3 мл) при комнатной температуре с последующим добавлением порошка цинка (92,83 мг, 1,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 минут в атмосфере азота. После завершения реакции смесь разбавляли дихлорметаном (5 мл) и фильтровали с применением диатомита. Фильтрат концентрировали при пониженном давлении и остаток снова добавляли в дихлорметан (5 мл), фильтровали, концентрировали с получением неочищенного продукта WX072-4.
[305] Стадия 4. Получение соединения WX072-5
[306] Соединение WX072-4 (35,00 мг, 303,70 мкмоль) и триэтиламин (153,66 мг, 1,52 ммоль, 210,49 мкл) растворяли в тетрагидрофуране (20,00 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением трифосгена (40,56 мг, 136,66 мкмоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции нерастворимые вещества удаляли посредством фильтрации и фильтрат концентрировали при пониженном давлении. Полученный остаток выделяли с помощью препаративной HPLC с получением неочищенного продукта WX072-5. 1H ЯМР (400 МГц, CDCl3) δ: 9,49 (br s, 1H), 8,08 (d, J=5,3 Гц, 1H), 7,47-7,35 (m, 2H), 7,28 (br s, 1H), 7,26-7,12 (m, 3H), 7,02 (d, J=5,3 Гц, 1H), 6,73 (d, J=8,3 Гц, 1H), 6,16 (dd, J=4,4, 8,4 Гц, 1H), 4,80 (dd, J=9,7, 14,2 Гц, 1H), 3,87 (dd, J=4,0, 14,6 Гц, 1H), 3,77 (s, 6H), 2,67 (s, 3H).
[307] Стадия 5. Получение соединения WX072
[308] Соединение WX072-5 (39,00 мг, 82,71 мкмоль) выделяли с помощью сверхкритической жидкостной хроматографии (условия: колонка: Chiralpak AD 350 x 4,6 мм I.D., 3 мкм; подвижная фаза: A: диоксид углерода, B: этанол (0,05% диэтиламин); приблизительно 40%; температура колонки: 40°C; длина волны: 220 нм) и собирали образец со временем удерживания 0,790 мин. с получением WX072 (ee%: 100%).
[309] Вариант осуществления 73: WX073
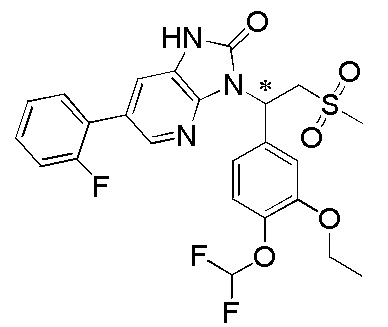
Путь синтеза
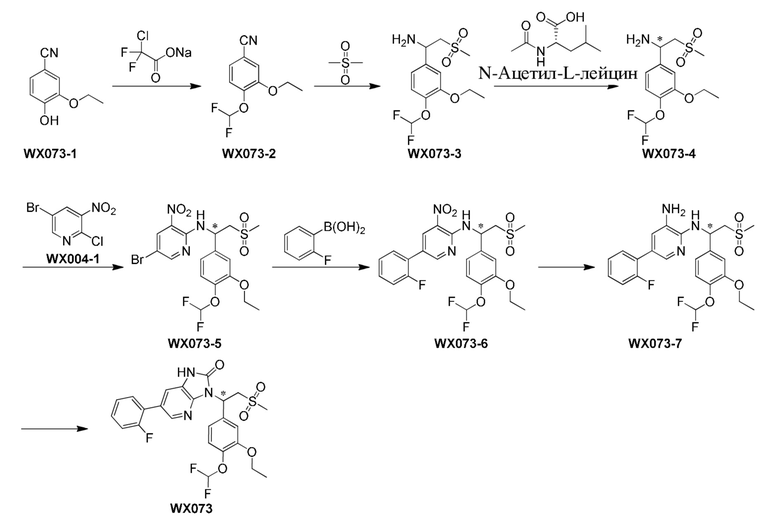
[310] Стадия 1. Получение соединения WX073-2
[311] Соединение WX073-1 (29,00 г, 177,73 ммоль), 2-хлор-2,2-дифторацетат натрия (62,32 г, 408,78 ммоль) и карбонат цезия (86,86 г, 266,60 ммоль) растворяли в N,N-диметилформамиде (600,00 мл) и воде (150,00 мл) при комнатной температуре. Реакционную смесь нагревали до 100°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции полученное охлаждали до комнатной температуры и разбавляли этилацетатом (1000 мл) и водой (200 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (200 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=10/1-3/1, объемное соотношение) с получением целевого продукта WX073-2. 1H ЯМР (400 МГц, CDCl3) δ: 7,20-7,13 (m, 3H), 6,58 (t, J=74,4 Гц, 1H), 4,05 (q, J=6,8 Гц, 2H), 1,41 (t, J=6,8 Гц, 3H).
[312] Стадия 2. Получение соединения WX073-3
[313] Диметилсульфон (9,71 г, 103,20 ммоль, 8,37 мл) растворяли в тетрагидрофуране (300 мл) при комнатной температуре. Реакционную смесь охлаждали до 0°C в атмосфере азота с последующим добавлением по каплям н-бутиллития (2,5 M, 41,28 мл). Реакционную смесь перемешивали при 0-5°C в течение 1 часа с последующим добавлением по каплям раствора соединения WX073-2 (20,00 г, 93,82 ммоль) в тетрагидрофуране (200,00 мл). Реакционную смесь перемешивали при 0-5°C в течение дополнительных 30 минут в атмосфере азота и затем нагревали до комнатной температуры с перемешиванием в течение 1,5 часа с последующим добавлением борогидрида натрия (4,61 г, 121,97 ммоль) с перемешиванием в течение дополнительных 30 минут. Реакционную смесь затем охлаждали до 0°C с последующим добавлением уксусной кислоты (25,92 г, 431,57 ммоль, 24,69 мл) с перемешиванием при 0-5°C в течение дополнительных 2 часов и водного раствора гидроксида натрия (2,5 M, 123,84 мл) с перемешиванием при 0-5°C в течение дополнительных 30 минут. Реакционную смесь нагревали до 60°C и перемешивали при нагревании с обратным холодильником в течение 12 часов. После завершения реакции смесь охлаждали до комнатной температуры и разбавляли этилацетатом (1000 мл) и водой (200 мл). Органические фазы отделяли, промывали насыщенным солевым раствором (200 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=100/1-10/1, объемное соотношение) с получением целевого продукта WX073-3. 1H ЯМР (400 МГц, CDCl3) δ: 7,08-6,84 (m, 3H), 6,50 (t, J=75,2 Гц, 1H), 4,58 (dd, J=2,8, 9,6 Гц, 1H), 4,04 (dd, J=6,8, 13,2 Гц, 2H), 3,27-3,12 (m, 2H), 2,90 (s, 3H), 1,38 (t, J=7,2 Гц, 3H).
[314] Стадия 3. Получение соединения WX073-4
[315] Соединение WX073-3 (9,00 г, 29,10 ммоль) и N-ацетил-L-лейцин (3,02 г, 17,46 ммоль) растворяли в метаноле (100,00 мл) при комнатной температуре. Реакционную смесь нагревали до 80°C и перемешивали в течение 2 часов в атмосфере азота. Затем реакционную смесь охлаждали до комнатной температуры, при этом осаждались большие количества твердого вещества, с последующей фильтрацией. Осадок на фильтре высушивали в вакууме при 30-40°C. Полученный неочищенный продукт растворяли в метаноле (100,00 мл), нагревали до 80°C и перемешивали в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, при этом осаждались большие количества твердого вещества, с последующей фильтрацией. Осадок на фильтре промывали метанолом (30,00 мл) и высушивали в вакууме при 30°C. Полученное белое твердое вещество растворяли в этилацетате (100 мл) и насыщенный раствор гидросульфата натрия (30 мл) и перемешивали при комнатной температуре в течение 15 минут. Органические фазы отделяли, промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением целевого продукта WX073-4. (время удерживания = 2,853 мин.; колонка: Chiralpak AD_3 100 X 4,6 мм, 3 мкм; время прогона: 10,0 минут).
[316] Стадия 4. Получение соединения WX073-5
[317] Соединение WX073-4 (600,00 мг, 1,94 ммоль) и соединение WX004-1 (690,84 мг, 2,91 ммоль) растворяли в ацетонитриле (6,00 мл) при комнатной температуре с последующим добавлением карбоната калия (402,19 мг, 2,91 ммоль). Реакционную смесь нагревали до 85°C и перемешивали в течение 5 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и разбавляли дихлорметаном (15 мл) с последующей фильтрацией. Фильтрат концентрировали с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=10/1-1/1, объемное соотношение) с получением неочищенного продукта WX073-5. 1H ЯМР (400 МГц, CDCl3) δ: 8,81 (d, J=7,3 Гц, 1H), 8,58 (d, J=2,3 Гц, 1H), 8,44 (s, 1H), 7,20 (d, J=8,3 Гц, 1H), 7,08-6,97 (m, 2H), 6,58 (t, J=75,0 Гц, 1H), 6,05-5,94 (m, 1H), 4,17-4,06 (m, 2H), 3,76 (dd, J=7,7, 14,7 Гц, 1H), 3,53 (dd, J=5,5, 14,6 Гц, 1H), 2,76 (s, 3H), 1,46 (t, J=7,2 Гц, 3H).
[318] Стадия 5. Получение соединения WX073-6
[319] Соединение WX073-5 (250,00 мг, 489,90 мкмоль) и 2-фторфенилбороновую кислоту (102,82 мг, 734,85 мкмоль) растворяли в диоксане (3 мл) и воде (1 мл) при комнатной температуре с последующим добавлением комплекса дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (20,00 мг, 24,50 мкмоль) и карбоната калия (101,56 мг, 734,85 мкмоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 4 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (20 мл), разбавляли водой (20 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали водой (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=10/1-2/1, объемное соотношение) с получением целевого продукта WX073-6. 1H ЯМР (400 МГц, CDCl3) δ: 8,85 (d, J=7,3 Гц, 1H), 8,71-8,62 (m, 2H), 7,46-7,33 (m, 2H), 7,26-7,16 (m, 3H), 7,10 (d, J=2,0 Гц, 1H), 7,05 (dd, J=2,0, 8,0 Гц, 1H), 6,59 (t, J=75,2 Гц, 1H), 6,18-6,03 (m, 1H), 4,17-4,17 (m, 1H), 4,14 (q, J=7,0 Гц, 1H), 3,84 (dd, J=7,4, 14,9 Гц, 1H), 3,57 (dd, J=5,8, 14,6 Гц, 1H), 2,78 (s, 3H), 1,47 (t, J=7,0 Гц, 3H).
[320] Стадия 6. Получение соединения WX073-7
[321] Соединение WX073-6 (191,00 мг, 363,46 мкмоль) и хлорид аммония (194,42 мг, 3,63 ммоль, 127,07 мкл) растворяли в метаноле (10 мл) при комнатной температуре с последующим добавлением порошка цинка (118,83 мг, 1,82 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут в атмосфере азота. После завершения реакции порошок цинка удаляли посредством фильтрации и фильтрат концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли дихлорметан (30 мл) и перемешивали в течение 15 минут с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX073-7.
[322] Стадия 7. Получение соединения WX073
[323] Соединение WX073-7 (120,00 мг, 242,17 мкмоль) и триэтиламин (122,53 мг, 1,21 ммоль, 167,85 мкл) растворяли в тетрагидрофуране (40,00 мл) при комнатной температуре. Реакционную смесь охлаждали до 0°C с последующим добавлением трифосгена (40,56 мг, 136,66 мкмоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа в атмосфере азота и затем нагревали до комнатной температуры и перемешивали в течение 12 часов. После завершения реакции смесь гасили насыщенным раствором бикарбоната натрия (20 мл), разбавляли водой (20 мл) и экстрагировали этилацетатом (15 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX073. MS-ESI масса/заряд: 521,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,79 (br s, 1H), 8,24 (s, 1H), 7,55 (s, 1H), 7,46 (s, 1H), 7,45-7,35 (m, 2H), 7,28 (d, J=17,6 Гц, 2H), 7,24-7,12 (m, 2H), 6,54 (t, J=75,2 Гц, 1H), 6,31 (dd, J=3,0, 9,5 Гц, 1H), 5,04 (dd, J=10,3, 14,3 Гц, 1H), 4,17-4,05 (m, 2H), 3,86 (dd, J=3,1, 14,4 Гц, 1H), 2,86 (s, 3H), 1,42 (t, J=6,8 Гц, 3H).
[324] Вариант осуществления 74: WX074
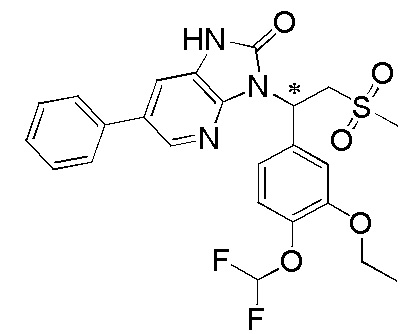
Путь синтеза
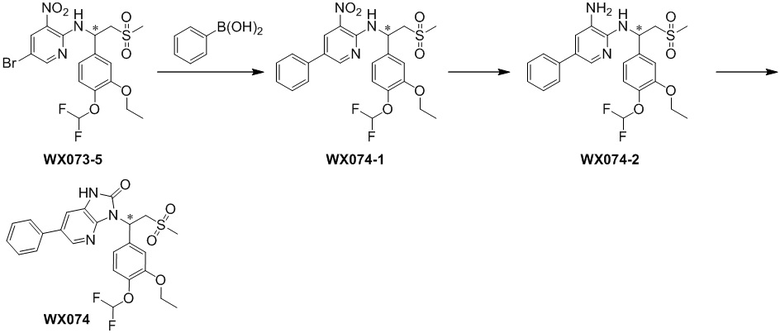
[325] Стадия 1. Получение соединения WX074-1
[326] Соединение WX073-5 (250,00 мг, 489,90 мкмоль) и фенилбороновую кислоту (89,60 мг, 734,85 мкмоль) растворяли в диоксане (3,00 мл) и воде (1,00 мл) при комнатной температуре с последующим добавлением комплекса дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (20,00 мг, 24,50 мкмоль) и карбоната калия (101,56 мг, 734,85 мкмоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 4 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, гасили с помощью насыщенного солевого раствора (20 мл), разбавляли водой (20 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали водой (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=10/1-2/1, объемное соотношение) с получением целевого продукта WX074-1. 1H ЯМР (400 МГц, CDCl3) δ: 8,81 (d, J=7,3 Гц, 1H), 8,70 (d, J=2,5 Гц, 1H), 8,67 (d, J=2,3 Гц, 1H), 7,58-7,45 (m, 5H), 7,24-7,18 (m, 1H), 7,13-7,03 (m, 2H), 6,58 (t, J=75,2 Гц, 1H), 6,17-6,07 (m, 1H), 4,13 (q, J=7,0 Гц, 2H), 3,84 (dd, J=7,4, 14,7 Гц, 1H), 3,56 (dd, J=5,8, 14,6 Гц, 1H), 2,78 (s, 3H), 1,47 (t, J=6,9 Гц, 3H).
[327] Стадия 2. Получение соединения WX074-2
[328] Соединение WX074-1 (140,00 мг, 275,86 мкмоль) и хлорид аммония (147,56 мг, 2,76 ммоль) растворяли в метаноле (10 мл) при комнатной температуре с последующим добавлением порошка цинка (90,19 мг, 1,38 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут в атмосфере азота. После завершения реакции порошок цинка удаляли посредством фильтрации и фильтрат концентрировали при пониженном давлении с удалением растворителя. К полученному остатку добавляли дихлорметан (30 мл) и перемешивали в течение 15 минут с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX074-2.
[329] Стадия 7. Получение соединения WX074
[330] Соединение WX074-2 (100,00 мг, 209,42 мкмоль) и триэтиламин (105,95 мг, 1,05 ммоль, 145,14 мкл) растворяли в тетрагидрофуране (30,00 мл) при комнатной температуре. Реакционную смесь охлаждали до 0°C с последующим добавлением трифосгена (24,86 мг, 83,77 мкмоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа в атмосфере азота и затем нагревали до комнатной температуры и перемешивали в течение 12 часов. После завершения реакции смесь гасили насыщенным раствором бикарбоната натрия (20 мл), разбавляли водой (20 мл) и экстрагировали этилацетатом (15 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX074. MS-ESI масса/заряд: 503,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,39 (br s, 1H), 8,29 (d, J=1,5 Гц, 1H), 7,56-7,51 (m, 3H), 7,51-7,44 (m, 3H), 7,44-7,38 (m, 1H), 7,30 (dd, J=1,9, 8,4 Гц, 1H), 7,14 (d, J=8,3 Гц, 1H), 6,55 (t, J=75,2 Гц, 1H), 6,33 (dd, J=4,0, 10,3 Гц, 1H), 5,05 (dd, J=10,3, 14,6 Гц, 1H), 4,11 (дуб. квинт., J=2,4, 6,9 Гц, 2H), 3,84 (dd, J=4,0, 14,6 Гц, 1H), 2,87 (s, 3H), 1,43 (t, J=7,0 Гц, 3H).
[331] Вариант осуществления 75: WX075
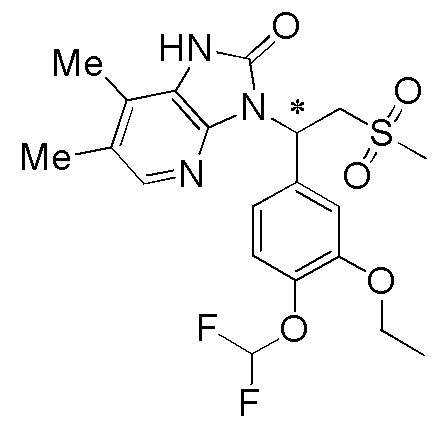
Путь синтеза
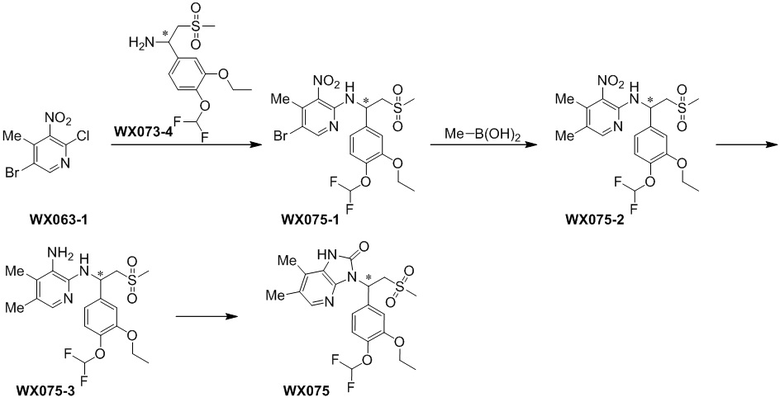
[332] Стадия 1. Получение соединения WX075-1
[333] Соединение WX063-1 (1,00 г, 3,98 ммоль) и соединение WX073-4 (1,35 г, 4,38 ммоль) растворяли в N,N-диметилформамиде (15,00 мл) при комнатной температуре с последующим добавлением диизопропилэтиламина (1,54 г, 11,93 ммоль, 2,08 мл). Реакционную смесь нагревали до 120°C и перемешивали в течение 15 часов в атмосфере азота. После завершения реакции смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (50 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат=3/1, объемное соотношение) с получением целевого продукта WX075-1. 1H ЯМР (400 МГц, CDCl3) δ: 8,30 (s, 1H), 7,40 (d, J=8,0 Гц, 1H), 7,16 (d, J=8,5 Гц, 1H), 7,02 (d, J=2,0 Гц, 1H), 6,96 (dd, J=2,0, 8,5 Гц, 1H), 6,55 (t, J=76,0 Гц 1H), 5,85 (dt, J=5,3, 7,4 Гц, 1H), 4,22-4,00 (m, 2H), 3,73 (dd, J=7,8, 14,8 Гц, 1H), 3,46 (dd, J=5,3, 14,8 Гц, 1H), 2,75 (s, 3H), 2,54-2,46 (m, 3H), 1,50-1,39 (m, 3H).
[334] Стадия 2. Получение соединения WX075-2
[335] Соединение WX075-1 (200,00 мг, 381,44 мкмоль), метилбороновую кислоту (68,50 мг, 1,14 ммоль) и карбонат калия (158,16 мг, 1,14 ммоль) растворяли в диоксане (20,00 мл) и воде (2,00 мл) при комнатной температуре с последующим добавлением комплекса дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (27,91 мг, 38,14 мкмоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 10 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли водой (30 мл) и экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=3/1, объемное соотношение) с получением целевого продукта WX075-2.MS-ESI масса/заряд: 460,1 [M+H]+.
[336] Стадия 3. Получение соединения WX075-3
[337] Соединение WX075-2 (90,00 мг, 195,88 мкмоль) растворяли в метаноле (15 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением порошка цинка (128,09 мг, 1,96 ммоль) и хлорида аммония (31,43 мг, 587,64 мкмоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов в атмосфере азота. После завершения реакции нерастворимые вещества удаляли посредством фильтрации и фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX075-3. MS-ESI масса/заряд: 430,1 [M+H]+.
[338] Стадия 4. Получение соединения WX075
[339] Соединение WX075-3 (70,00 мг, 162,99 мкмоль) и триэтиламин (105,95 мг, 1,05 ммоль, 145,14 мкл) растворяли в тетрагидрофуране (30,00 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением трифосгена (24,86 мг, 83,77 мкмоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа в атмосфере азота и затем нагревали до комнатной температуры и перемешивали в течение 12 часов. После завершения реакции смесь гасили насыщенным раствором бикарбоната натрия (20 мл), разбавляли водой (20 мл) и экстрагировали этилацетатом (15 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX075. MS-ESI масса/заряд: 456,0 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 10,59 (s, 1H), 7,81 (s, 1H), 7,49 (s, 1H), 7,30-7,12 (m, 1H), 7,04 (d, J=8,5 Гц, 1H), 6,65-6,28 (m, 2H), 4,97 (t, J=11,0 Гц, 1H), 4,03 (d, J=6,0 Гц, 2H), 3,68 (d, J=13,1 Гц, 1H), 2,86 (br s, 3H), 2,25 (m, 6H), 1,35 (t, J=6,5 Гц, 3H).
[340] Вариант осуществления 76: WX076
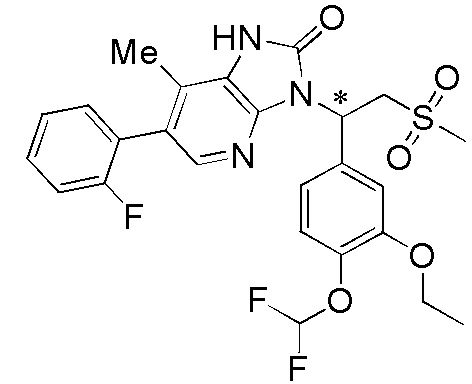
Путь синтеза
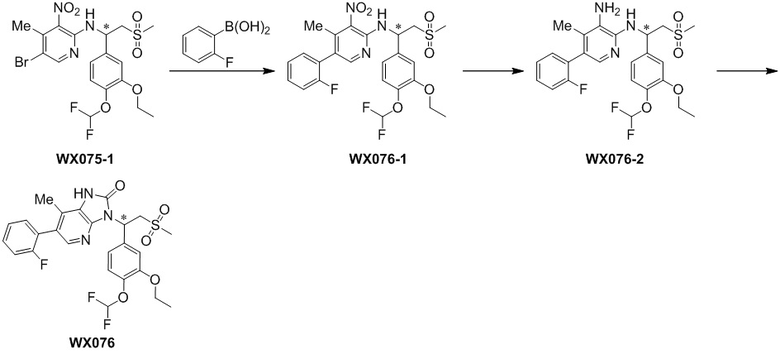
[341] Стадия 1. Получение соединения WX076-1
[342] Соединение WX075-1 (200,00 мг, 381,44 мкмоль), 2-фторбензолбороновую кислоту (58,71 мг, 419,58 мкмоль) и карбонат калия (105,44 мг, 762,88 мкмоль) растворяли в диоксане (20,00 мл) и воде (1,00 мл) при комнатной температуре с последующим добавлением комплекса дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (27,91 мг, 38,14 мкмоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 10 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали с удалением растворителя. Остаток разбавляли водой (30 мл) и экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=3/1, объемное соотношение) с получением целевого продукта WX076-1. MS-ESI масса/заряд: 540,1 [M+H]+.
[343] Стадия 2. Получение соединения WX076-2
[344] Соединение WX076-1 (160,00 мг, 296,56 мкмоль) растворяли в метаноле (15 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением порошка цинка (193,92 мг, 2,97 ммоль, 10,00 экв.) и хлорида аммония (47,59 мг, 889,68 мкмоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов в атмосфере азота. После завершения реакции нерастворимые вещества удаляли посредством фильтрации и фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX076-2. MS-ESI масса/заряд: 510,5 [M+H]+.
[345] Стадия 3. Получение соединения WX076
[346] Соединение WX076-2 (100,00 мг, 196,26 мкмоль) и триэтиламин (119,16 мг, 1,18 ммоль, 163,23 мкл) растворяли в тетрагидрофуране (10,00 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением трифосгена (34,94 мг, 117,76 мкмоль). Реакционную смесь перемешивали при 5°C в течение 2 часов в атмосфере азота. После завершения реакции смесь концентрировали с удалением растворителя, разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX076. MS-ESI масса/заряд: 535,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,73 (br s, 1H), 7,97 (s, 1H), 7,53 (s, 1H), 7,45 (dd, J=3,5, 8,5 Гц, 1H), 7,34-7,28 (m, 2H), 7,27-7,25 (m, 1H), 7,24-7,05 (m, 2H), 6,54 (t, J=76,0 Гц 1H), 6,45-6,41 (m, 1H), 5,04 (dd, J=10,5, 14,6 Гц, 1H), 4,26-4,01 (m, 2H), 3,90-3,71 (m, 1H), 2,93 (s, 3H), 2,25 (s, 3H), 1,42 (t, J=7,0 Гц, 3H).
[347] Вариант осуществления 77: WX077
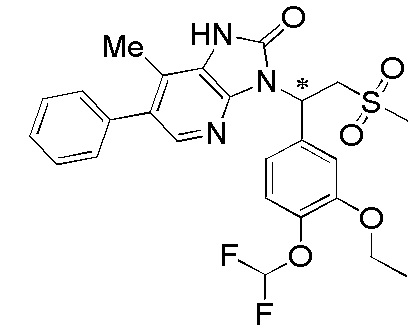
Путь синтеза
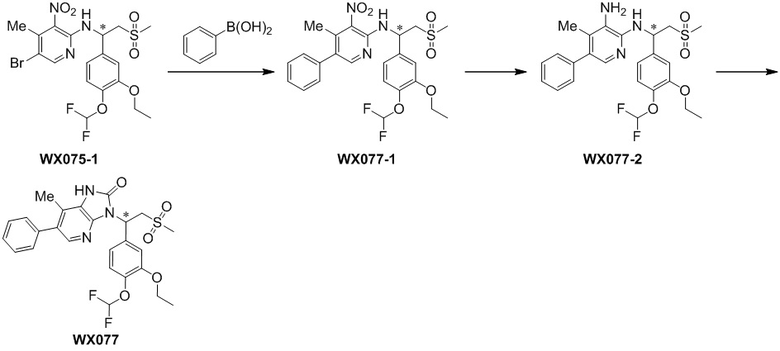
[348] Стадия 1. Получение соединения WX077-1
[349] Соединение WX075-1 (200,00 мг, 381,44 мкмоль), фенилбороновую кислоту (55,81 мг, 457,73 мкмоль) и карбонат калия (52,72 мг, 381,44 мкмоль) растворяли в диоксане (20,00 мл) и воде (2,00 мл) при комнатной температуре с последующим добавлением комплекса дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (279,1 мг, 381,44 мкмоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 10 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением растворителя. Остаток разбавляли водой (30 мл) и экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=3/1, объемное соотношение) с получением целевого продукта WX077-1. MS-ESI масса/заряд: 522,4 [M+H]+.
[350] Стадия 2. Получение соединения WX077-2
[351] Соединение WX077-1 (180,00 мг, 345,14 мкмоль) растворяли в метаноле (15 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением порошка цинка (225,69 мг, 3,45 ммоль) и хлорида аммония (55,38 мг, 1,04 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов в атмосфере азота. После завершения реакции нерастворимые вещества удаляли посредством фильтрации и фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя, с получением неочищенного продукта WX077-2. MS-ESI масса/заряд: 492,5 [M+H]+.
[352] Стадия 3. Получение соединения WX077
[353] Соединение WX077-2 (110,00 мг, 223,78 мкмоль) растворяли в тетрагидрофуране (10,00 мл) при комнатной температуре. Реакционную смесь затем охлаждали до 0°C с последующим добавлением трифосгена (34,94 мг, 117,76 мкмоль) и триэтиламина (135,87 мг, 1,34 ммоль, 186,12 мкл). Реакционную смесь перемешивали при 5°C в течение 2 часов в атмосфере азота. После завершения реакции смесь концентрировали при пониженном давлении с удалением растворителя, разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью препаративной HPLC с получением целевого продукта WX077. MS-ESI масса/заряд: 517,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 10,47 (br s, 1H), 7,90 (s, 1H), 7,43-7,30 (m, 4H), 7,23 (d, J=7,0 Гц, 2H), 7,20-7,16 (m, 1H), 7,04 (d, J=8,0 Гц, 1H), 6,46 (t, J=76,0 Гц 1H) , 6,25-6,17 (m, 1H), 4,99 (dd, J=10,5, 14,6 Гц, 1H), 4,06-3,95 (m, 2H), 3,76 (dd, J=4,0, 14,6 Гц, 1H), 2,77 (s, 3H), 2,23 (s, 3H), 1,33 (t, J=7,0 Гц, 3H).
[354] Вариант осуществления 78: WX078
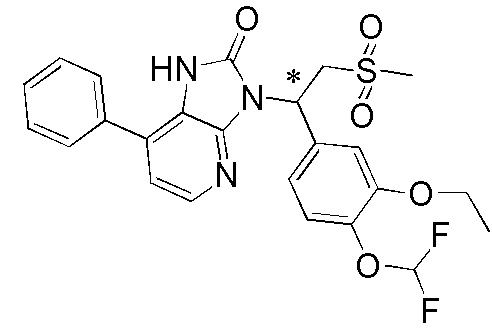
Путь синтеза
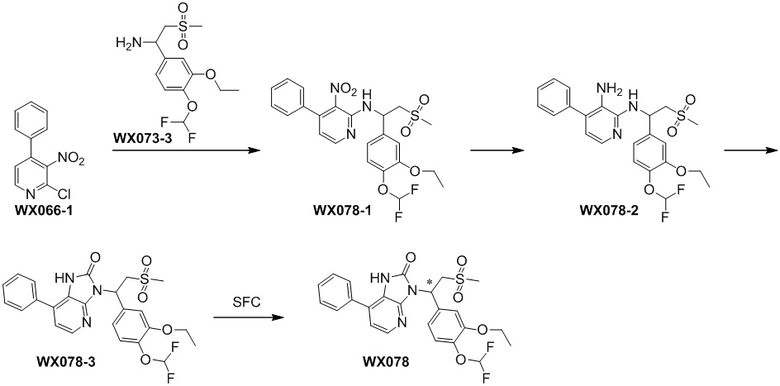
[355] Стадия 1. Получение соединения WX078-1
[356] Соединение WX066-1 (5,00 г, 21,31 ммоль), соединение WX073-3 (6,59 г, 21,31 ммоль) и фторид цезия (6,47 г, 42,62 ммоль) растворяли в диметилсульфоксиде (200,00 мл) при комнатной температуре. Реакционную смесь нагревали до 50°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (800 мл) и водой (200 мл), разделяли. Органическую фазу отделяли и промывали насыщенным солевым раствором (100 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с удалением растворителя. Полученный остаток выделяли с помощью колоночной хроматографии (элюент: петролейный эфир) с получением неочищенного продукта WX078-1. MS-ESI масса/заряд: 508,2 [M+H]+.
[357] Стадия 2. Получение соединения WX078-2
[358] Соединение WX078-1 (7,00 г, 13,79 ммоль), порошок цинка (9,02 г, 137,90 ммоль) и хлорид аммония (7,38 г, 137,90 ммоль, 4,82 мл) растворяли в метаноле (200,00 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов в атмосфере азота. После завершения реакции смесь фильтровали с применением диатомита и фильтрат концентрировали при пониженном давлении с удалением растворителя. Дихлорметан (100 мл) и воду (20 мл) добавляли в полученный остаток, разделяли. Органическую фазу отделяли и промывали насыщенным солевым раствором (20 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении с получением целевого продукта WX078-2. MS-ESI масса/заряд: 478,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 7,60-7,54 (m, 1H), 7,44-7,37 (m, 2H), 7,37-7,29 (m, 3H), 7,11-7,05 (m, 2H), 7,00-6,94 (m, 1H), 6,58 (t, J=76 Гц, 1H ),5,68 (q, J=6,4 Гц, 1H), 4,07-3,98 (m, 2H), 3,81 (br dd, J=7,2, 14,7 Гц, 1H), 3,47 (br dd, J=5,4, 14,7 Гц, 2H), 2,78 (s, 3H), 1,36 (t, J=6,9 Гц, 3H).
[359] Стадия 3. Получение соединения WX078-3
[360] Соединение WX078-2 (5,00 г, 10,47 ммоль), трифосген (1,86 г, 6,28 ммоль) и триэтиламин (6,36 г, 62,82 ммоль, 8,71 мл) растворяли в тетрагидрофуране (150,00 мл) при комнатной температуре. Реакционную смесь охлаждали до 0-5°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь разбавляли этилацетатом (300 мл) и водой (100 мл), разделяли. Органическую фазу отделяли и промывали насыщенным солевым раствором (100 мл x 2) и высушивали над безводным сульфатом натрия с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат=5/1-1/2, объемное соотношение) с получением целевого продукта WX078-3. MS-ESI масса/заряд: 504,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 9,91 (br s, 1H), 8,07 (d, J=5,5 Гц, 1H), 7,56-7,45 (m, 4H), 7,44-7,37 (m, 1H), 7,33 (d, J=1,8 Гц, 1H), 7,13 (dd, J=1,8, 8,3 Гц, 1H), 7,07-6,99 (m, 2H), 6,45(t, J=76,0 Гц, 1H), 6,18 (dd, J=4,4, 9,9 Гц, 1H), 4,89 (dd, J=9,9, 14,7 Гц, 1H), 3,99-3,91 (m, 2H), 3,78 (dd, J=4,4, 14,7 Гц, 1H), 2,70 (s, 3H), 1,32 (t, J=6,9 Гц, 3H).
[361] Стадия 4. Получение соединения WX078
[362] Соединение WX078-3 (2,50 г, 4,92 ммоль) выделяли с помощью сверхкритической жидкостной хроматографии (условия: колонка: Chiralpak AD_3 100 x 4,6 мм, 3 мкм; подвижная фаза: A: диоксид углерода, B: этанол (0,05% диэтиламин); приблизительно 40%; расход: 2,8 мл/мин.; температура колонки: 40°C; длина волны: 220 нм) и собирали образец со временем удерживания 4,842 мин. с получением WX078.
[363] Экспериментальный вариант осуществления 1. Оценка ингибирующего эффекта в отношении фермента in vitro
[364] Ингибирующая активность соединений в отношении PDE 4B
[365] Активность фермента определяли посредством измерения экспрессии AMP/GMP и обнаружения связывания антитела AMP/GMP на основании флуоресцентной поляризация в биологическом анализе.
[366] Реагенты
[367] Экспериментальный буфер раствор: 10 мМ Tris-HCl (pH 7,5), 5 мМ MgCl2, 0,01% Brij 35, 1 мМ DTT и 1% DMSO.
[368] Фермент: рекомбинантная PDE4B человека (номер доступа в Gen: NM_002600; аминокислота 305 конец) экспрессировалась бакуловирусом в клетках Sf9 насекомых с применением N-концевой метки GST. MW=78 кДа.
[369] Субстрат фермента: 1 мкМ cAMP
[370] Обнаружение: Антитело AMP2/GMP2 Transcreener® и меченое вещество AlexaFluor633 AMP2/GMP2
[371] Последовательность операций
[372] i) Рекомбинантную PDE4B человека и субстрат фермента (1 мкМ cAMP) растворяли в только что полученном буферном растворе соответственно.
[373] ii) Буферный раствор для PDE4B, определенный как указано выше, переносили в лунки для проведения реакции.
[374] iii) Соединение, растворенное в 100% DMSO, добавляли в лунки для проведения реакции с буферным раствором для PDE4B посредством генератора ультразвуковых волн (Echo 550; диапазон в нанолитрах) и инкубировали при комнатной температуре в течение 10 минут.
[375] iv) Ферментативный буферный раствор затем добавляли в лунки для проведения реакции, определенные выше, для начала реакции.
[376] v) Реакционную смесь инкубировали при комнатной температуре в течение 1 часа.
[377] vi) Реакцию останавливали посредством добавления смеси для обнаружения (антитело AMP2/GMP2 Transcreener® и меченое вещество AlexaFluor633 AMP2/GMP2) и инкубировали в течение 90 минут при медленном перемешивании. Диапазон измерения флуоресцентной поляризации составляет Ex/Em=620/688.
[378] Анализ данных. Сигнал флуоресцентной поляризации преобразовывали в нМ в соответствии со стандартной кривой AMP/GMP и процент активности фермента относительно DMSO рассчитывали с помощью Excel. GraphPad Prism применяли для построения кривых (Drawing Medicine Icon).
Таблица 7. Результаты тестирования ингибирующей активности соединения по настоящему изобретению в отношении PDE4B in vitro
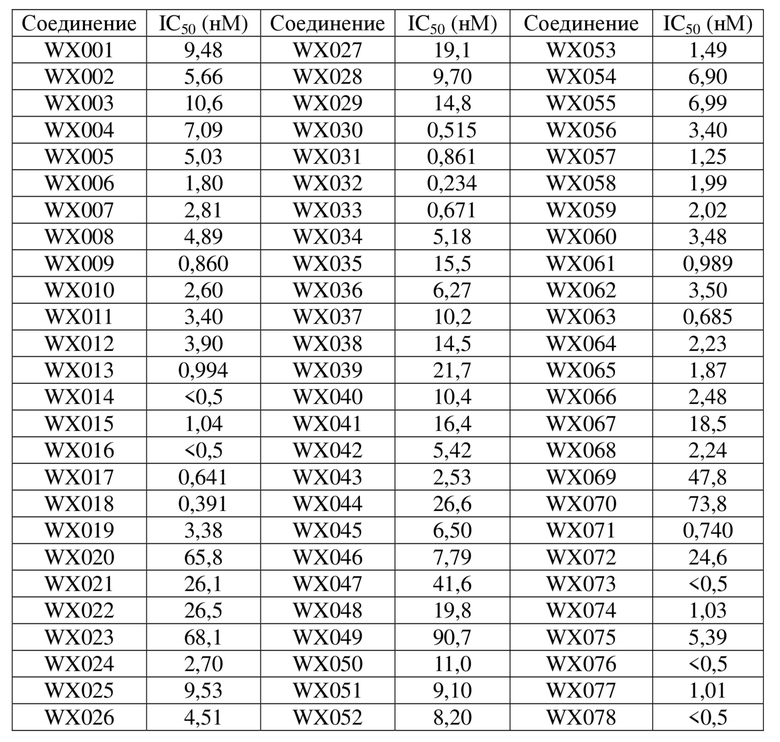
[379] Заключение. Все соединения по настоящему изобретению проявляют превосходную ингибирующую активность в отношении PDE4B in vitro.
[380] Экспериментальный вариант осуществления 2. Оценка ингибирующего эффекта в отношении TNFα в hPBMC in vitro
[381] Ингибирующая активность соединений в отношении LPS-индуцированного TNFα из периферических моноцитов человека.
[382] Экспериментальная процедура
[383] 1. У здорового человека отбирали цельную кровь и в качестве антикоагулирующего средства использовали EDTA-K2.
[384] 2. PBMC выделяли посредством центрифугирования в градиенте плотности фиколла. Концентрацию клеток регулировали до 2 x 106/мл.
[385] 3. 2 x 105 клеток высевали в лунку 96-луночного планшета с U-образным дном, после чего следовало добавление 200 мкл/лунка LPS с концентрацией 1 нг/мл и 200 мкл/на лунку соединения в различной концентрации: 100 мкМ, 10 мкМ, 1 мкМ, 100 мкМ, 10 мкМ, 1 мкМ, 100 пМ и 10 пМ, с двумя повторностями каждой концентрации.
[386] 4. Реакционную смесь инкубировали в течение 24 часов и собирали супернатант.
[387] 5. Для определения уровня TNFα в супернатанте применяли ELISA, и программное обеспечение Graphpad Prism применяли для построения кривой ингибирования и расчета значения IC50.
Таблица 8
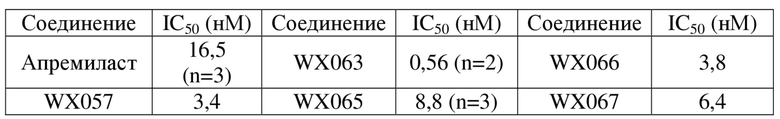
[388] Заключение. Все соединения по настоящему изобретению проявляют превосходную ингибирующую активность в отношении TNFα в hPBMC in vitro, которая превосходит апремиласт.
[389] Экспериментальный вариант осуществления 3. Модель CIA in vivo
[390] Цель эксперимента
[391] Модель коллаген-индуцированного артрита у мыши представляет собой животную модель для оценки терапевтического эффекта лечения посредством лекарственного средства псориатического артрита, патогенез и симптомы которого очевидно связаны с псориатическим артритом. В модели активировали реакционную способность B-клеток и T-клеток в отношении коллагена посредством инъекции коллагена типа II. Активированные В-клетки и T-клетки привлекались к суставам, чтобы вызвать воспаление, за счет чего обеспечивалась инициация ряда симптомов, связанных с псориатическим артритом человека, таких как покраснение и опухание суставов, суставного хряща, повреждение суставной капсулы и другие симптомы. Коллаген-индуцированный артрит мыши часто применяют для оценки терапевтического эффекта во время доклинической оценки соединений-кандидатов для применения в лекарственных препаратах для лечения псориатического артрита.
[392] Целью эксперимента было исследование терапевтического эффекта соединения WX063 согласно варианту осуществления 63 в отношении коллаген-индуцированного артрита у мыши, за счет чего обеспечивается информация о доклинической фармакодинамике для последующих клинических исследований.
[393] Экспериментальная процедура
[394] 1. Иммунизация с помощью коллагена типа II и полного адъюванта Фрейнда
[395] Получение уксусной кислоты. Уксусную кислоту разбавляли до 100 мМ, фильтровали посредством 0,22 мкм мембранный фильтр и хранили при 4ºC.
[396] Получение раствора бычьего коллагена типа II. Бычий коллаген типа II растворяли в растворе уксусной кислоты и хранили при 4ºC.
[397] Получение эмульсии. Хранившийся в течение ночи раствор CII смешивали с равным объемом полного адъюванта Фрейнда и гомогенизировали посредством высокоскоростного гомогенизатора с образованием устойчивой эмульсии.
[398] Получение липополисахарида (LPS). Взвешивание LPS, добавление нормального солевого раствора, смешивание до образования устойчивого раствора, концентрация которого составляет 0,3 мг/кг.
[399] 2. Индуцирование артрита
[400] Мышей произвольным образом разделяли на различные группы обработки. Первый день иммунизации регистрировали как день 0, а следующие дни записывали по порядку.
[401] После анестизирования мышей DBA/1 изофлураном полученную эмульсию коллагена подкожно вводили в хвост.
[402] В день 23 100 мкл раствора LPS вводили внутрибрюшинно.
[403] Мышам в нормальной группе не требовалась иммунизация.
[404] 3. Введение и доза
[405] В день 27 так как средний клинический показатель достиг приблизительно 1 балла, 60 мышей с заболеванием средней степени выбирали и повторно рандомизировали в соответствии с весом тела и показателем на 10 мышей в группе.
[406] Дексаметазон в качестве положительного контроля лекарственного средства вводили в дозе 0,3 мг/кг, которая является типичной для модели CIA. Кроме того, подходящую схему дозирования примера тестируемого соединения WX063 и контрольного соединения, представляющего собой апремиласт, определяли на основе результатов предыдущих предварительных экспериментов. Группа 1 представляла собой здоровых мышей без какой-либо обработки; группу 2 обрабатывали средой-носителем; группе 3 вводили дексаметазон в дозе 0,3 мг/кг; группе 4 вводили апремиласт в дозе 5 мг/кг; группе 5, группе 6 и группе 7 вводили соединение WX063 согласно варианту осуществления 63 в дозе 0,3, 1 и 3 мг/кг соответственно. Его вводили один раз в день в течение всего 11 дней. Объем внутрижелудочного введения составлял 10 мл/кг.
Таблица 9. Группа и доза
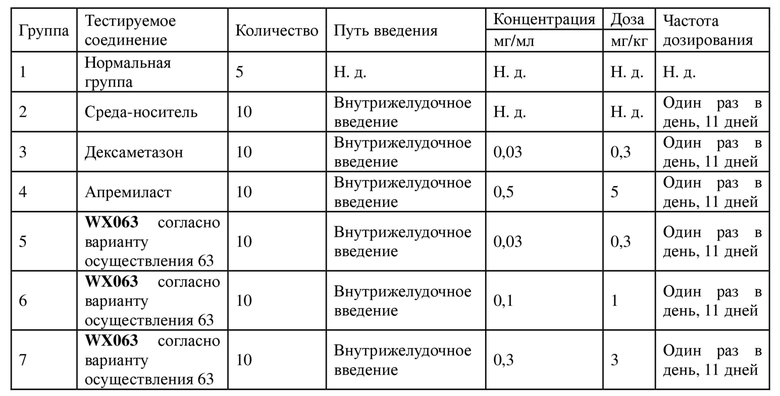
[407] 4. Определение индекса частоты возникновения артрита
[408] Клиническое наблюдение. Со дня 7 перед иммунизацией по день 23 после иммунизации ежедневно наблюдали за изменениями базового состояния здоровья и веса тела мышей DBA/1 (регистрировали раз в неделю). После дня 23 за изменениями состояния здоровья, частоты возникновения артрита и веса тела мышей наблюдали ежедневно (регистрировали по меньшей мере три раза в неделю) до конца эксперимента.
[409] Клинический показатель. После инъекции LPS за частотой возникновения артрита у мышей наблюдали ежедневно. При возникновении клинических симптомов артрита степень заболевания регистрировали в соответствии со стандартами 0-4 (покраснение, деформация сустава) по меньшей мере три раза в неделю, при этом самый высокий показатель для каждой конечности составлял 4 балла, а самый высокий показатель для каждого животного составлял 16 баллов. Стандарты оценивания являлись такими, как показано в таблице 10. Оценивали по меньшей мере три раза в неделю.
Таблица 10. Клинический стандарт оценивания артрита

[410] Патологическое исследование. В день 38 мышей умерщвляли. Задние конечности мышей отделяли, замачивали в 10% растворе формалина, декальцифицировали посредством раствора муравьиной кислоты, заключали в парафин, делали срезы и окрашивали гематоксилин-эозином и фотографировали посредством микроскопа. Степень повреждения сустава оценивали согласно четырем аспектам: инфильтрация воспалительных клеток, образование вазоспазма, повреждение хряща и резорбция кости, и оценивали в соответствии со стандартами 0-4. Различные стандарты оценивания показаны в таблице 11.
Таблица 11. Патологический стандарт оценивания артрита
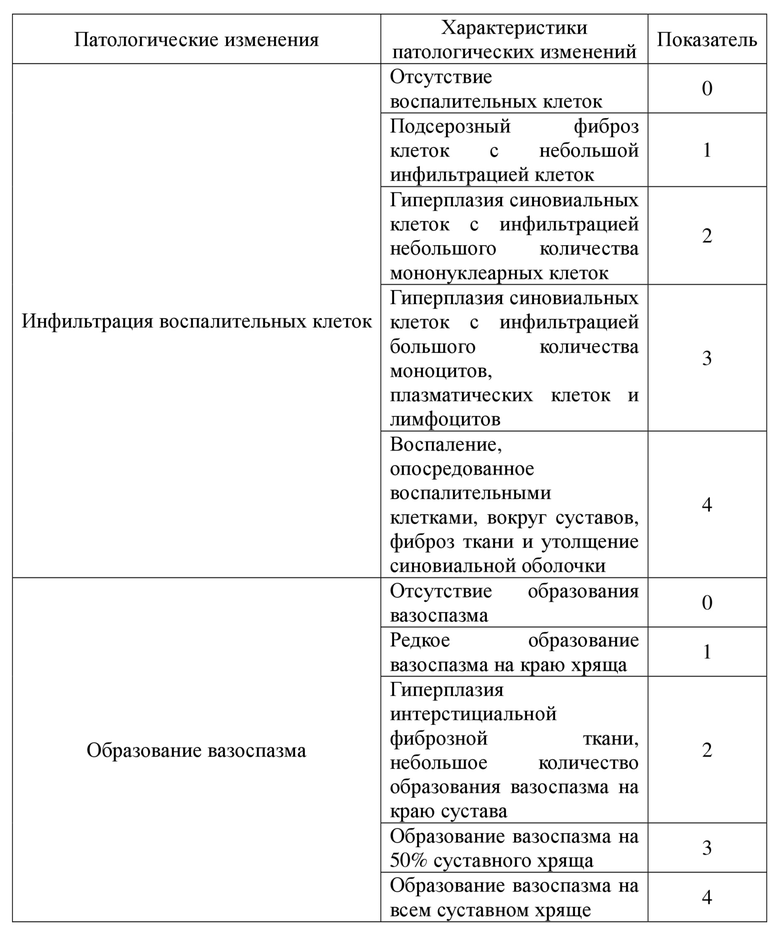
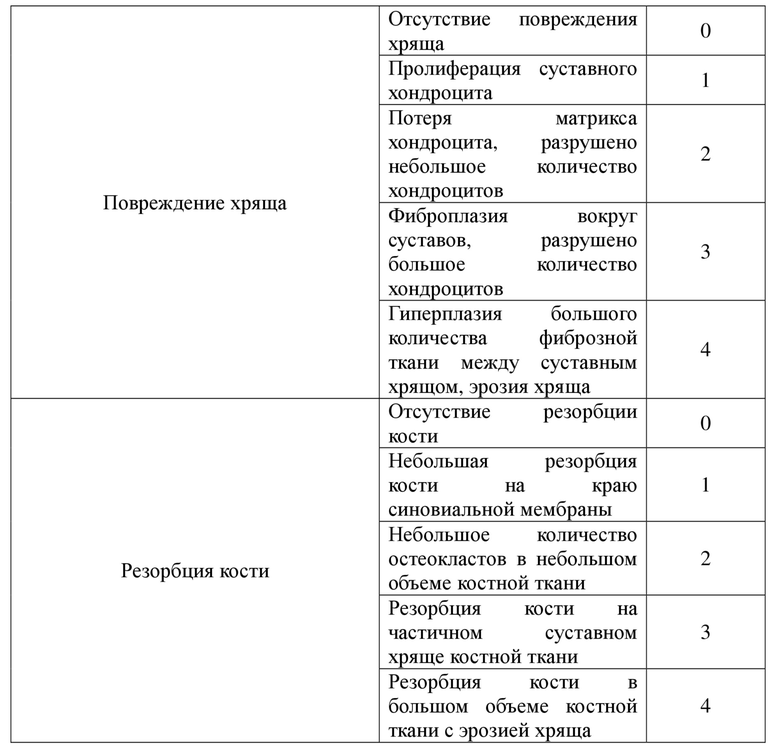
[411] 5. Статистическая обработка
[412] Экспериментальные данные выражали в виде среднего значения ± стандартная ошибка среднего значения (среднее значение ± SEM). Вес тела и клинические показатели анализировали посредством двухфакторной дисперсии (двухфакторный ANOVA). Патологические показатели и AUC тестировали посредством T-критерия. p < 0,05 считали значительным отличием.
[413] Экспериментальные результаты
[414] 1. Клинический показатель
[415] В день 25 после первой иммунизации (день 2 после второй иммунизации) у мышей начали развиваться клинические симптомы артрита. Введение начинали в день 27. Средний клинический показатель в группе со средой-носителем в качестве контроля постепенно повышался до 8,3 баллов в день 36, что указывает на успешное установление модели коллаген-индуцированного артрита.
[416] По сравнению с группой со средой-носителем в качестве контроля соединение WX063 согласно варианту осуществления 63 в дозе 0,3, 1 и 3 мг/кг в значительной степени снижало клинический показатель у мышей с артритом под конец эксперимента (день 37), и средний клинический показатель для трех доз снижался до 3,6 (p < 0,0001), 4,3 (p < 0,001) и 3,5 (p < 0,0001) соответственно. Следовательно, соединение WX063 согласно варианту осуществления 63 эффективно ослабляет коллаген-индуцированный артрит при дозе всего лишь 0,3 мг/кг. Дексаметазон в дозе 0,3 мг/кг в значительной степени подавлял повышение клинического показателя коллаген-индуцированного артрита. Клинический показатель составлял 0 в день 30 и поддерживался до конца испытания, что в значительной степени отличалось от группы со средой-носителем в качестве контроля (p < 0,0001). В группе с апремиластом в дозе 5 мг/кг также подавлялось повышение клинического показателя и проявлялось значительное отличие от группы со средой-носителем в качестве контроля со дня 33 до конца испытания. В день 37 средний клинический показатель составлял 4,2, что составляло на 3,7 балла меньше (p < 0,001) по сравнению с группой со средой-носителем в качестве контроля.
[417] Площадь под кривой (AUC) рассчитывали посредством анализа кривой клинического показателя для каждого животного в каждой группе, и процент ингибирования в каждой группе введения относительно группы со средой-носителем в качестве контроля рассчитывали посредством среднего значения площади под кривой среди групп. По сравнению с группой со средой-носителем в качестве контроля клинические показатели у животных с артритом в значительной степени снижались в группе с дексаметазоном и группе с апремиластом, и процент ингибирования составлял 96,4% (p < 0,0001) и 41,3% (p < 0,05) соответственно. Соединение WX063 согласно варианту осуществления 63 в дозе 0,3, 1 и 3 мг/кг в значительной степени снижало площадь под кривой клинического показателя у животных с артритом. Уровень ингибирования составлял 43,9% (p < 0,05), 39,4% (p < 0,05) и 51,7% (p < 0,01) соответственно. Процент ингибирования в группе с 1 мг/кг WX063 был сопоставим с таковым в группе с 5 мг/кг апремиласта (p < 0,05 для обеих групп), тогда как процент ингибирования в группе с 3 мг/кг WX063 превышал таковой в группе с 5 мг/кг апремиласта (p < 0,01 и < 0,05 соответственно).
[418] 2. Гистопатологический показатель
[419] Задние конечности мышей из каждой группы отделяли для окрашивания посредством HE, две задние конечности принимали за общий показатель. Общий патологический показатель для мышей с артритом в группе со средой-носителем в качестве контроля составлял 20,20 ± 1,15. По сравнению с группой со средой-носителем в качестве контроля патологический показатель у мышей с артритом в контрольной группе с 5 мг/кг апремиласта также в значительной степени снижался до 13,90 ± 1,89 (p < 0,05). Соединение WX063 согласно варианту осуществления 63 в дозе 1 и 3 мг/кг в значительной степени снижало патологические показатели у животных с артритом до 14,00 ± 2,43 (p < 0,05) и 9,20 ± 1,83 (p < 0,0001). Патологический показатель артрита в группе с 1 мг/кг WX063 был сопоставим с таковым в группе с 5 мг/кг апремиласта (p < 0,05), тогда как патологический показатель артрита в группе с 3 мг/кг WX063 превышал таковой в группе с 5 мг/кг апремиласта (p < 0,0001 и < 0,05 соответственно).
[420] 3. Заключение
[421] 1) Соединение WX063 согласно варианту осуществления 63 характеризовалось значительным улучшением в отношении лечения коллаген-индуцированного артрита и в отношении патологических изменений при артрите в дозах 0,3, 1 и 3 мг/кг. В группах с тремя дозами проявлялась значительная зависимость доза-эффект в отношении патологического показателя артрита.
[422] 2) Терапевтический эффект WX063 в дозе 3 мг/кг (клинический показатель и патологический показатель артрита) лучше, чем таковой апремиласта в дозе 5 мг/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ДЕЙСТВУЮЩИЕ НА БЕЛКИ CRBN | 2019 |
|
RU2801068C2 |
| ГЛУТАРИМИДНОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ КОЛЬЦОМ, КОНДЕНСИРОВАННЫМ С ФУРАНОМ | 2022 |
|
RU2836921C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| ПЕНТАЦИКЛИЧЕСКОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2815382C2 |
| ТЕТРАГИДРОИМИДАЗО[1,5-d][1,4]ОКСАЗЕПИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2659219C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
| ИНГИБИТОР LSD1, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2763898C2 |
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли:
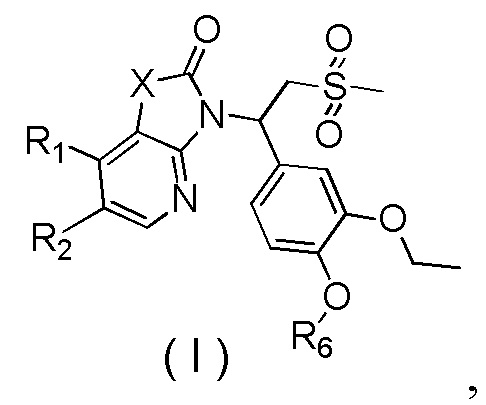
где X выбран из O, N(R3), -CH(R3)-; R3 выбран из H, COOH, R4-L1- или выбран из группы, состоящей из C1-6алкила, C1-6гетероалкила, C3-6циклоалкила и фенила, каждый из которых необязательно замещен 1, 2 или 3 R; «гетеро» в C1-6гетероалкиле выбран из -O-, -C(=O)O- и -S(=O)2; R4 выбран из группы, состоящей из фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1 или 2 R; «гетеро» в 5-6-членном гетероариле выбран из -NH-; L1 выбран из -CH2-; каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, CN, NH2, R5-L2- или выбран из группы, состоящей из C1-6алкила, C1-6алкокси, C2-6алкенила, 3-6-членного гетероциклоалкенила, C3-6циклоалкила, фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R; «гетеро» в 3-6-членном гетероциклоалкениле выбран из -NH- и -C(=O)-; «гетеро» в 5-6-членном гетероариле выбран из -NH- и -S-; R5 выбран из группы, состоящей из фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R; «гетеро» в 5-6-членном гетероариле выбран из -NH-; L2 выбран из -CH2- и O; R6 выбран из C1-3алкила, который необязательно замещен 1, 2 или 3 R; R выбран из H, галогена, OH, NH2, CN или выбран из группы, состоящей из C1-6алкила и C1-6гетероалкила, каждый из которых необязательно замещен 1, 2 или 3 R'; «гетеро» в C1-6гетероалкиле выбран из -O- и -C(=O)O-; R' выбран из F, Me, которое представляет собой ингибитор PDE4, а также к его применению в получении лекарственного препарата для лечения связанных с PDE4 заболеваний. 8 н. и 22 з.п. ф-лы, 11 табл., 78 пр.
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль:
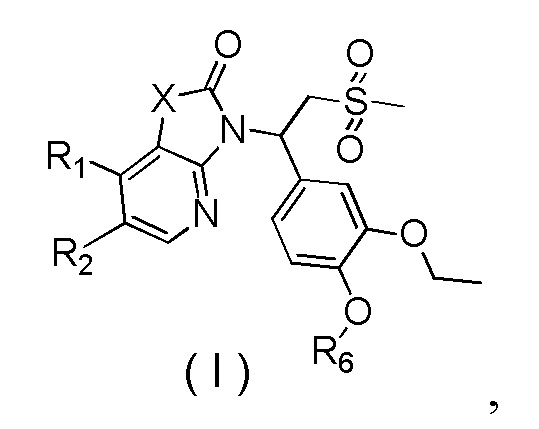
где
X выбран из O, N(R3), -CH(R3)-;
R3 выбран из H, COOH, R4-L1- или выбран из группы, состоящей из C1-6алкила, C1-6гетероалкила, C3-6циклоалкила и фенила, каждый из которых необязательно замещен 1, 2 или 3 R; «гетеро» в C1-6гетероалкиле выбран из -O-, -C(=O)O- и -S(=O)2;
R4 выбран из группы, состоящей из фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1 или 2 R; «гетеро» в 5-6-членном гетероариле выбран из -NH-;
L1 выбран из -CH2-;
каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, CN, NH2, R5-L2- или выбран из группы, состоящей из C1-6алкила, C1-6алкокси, C2-6алкенила, 3-6-членного гетероциклоалкенила, C3-6циклоалкила, фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R; «гетеро» в 3-6-членном гетероциклоалкениле выбран из -NH- и -C(=O)-; «гетеро» в 5-6-членном гетероариле выбран из -NH- и -S-;
R5 выбран из группы, состоящей из фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R; «гетеро» в 5-6-членном гетероариле выбран из -NH-;
L2 выбран из -CH2- и O;
R6 выбран из C1-3алкила, который необязательно замещен 1, 2 или 3 R;
R выбран из H, галогена, OH, NH2, CN или выбран из группы, состоящей из C1-6алкила и C1-6гетероалкила, каждый из которых необязательно замещен 1, 2 или 3 R'; «гетеро» в C1-6гетероалкиле выбран из -O- и -C(=O)O-;
R' выбран из F, Me;
в любом из вышеуказанных случаев число гетероатомов или содержащих гетероатом групп независимо выбрано из 1, 2, 3.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R выбран из H, F, Cl, Br, I, OH, NH2, CN или выбран из группы, состоящей из C1-3алкила, C1-3алкокси и C1-4алкил-OC(=O)-, каждый из которых необязательно замещен 1, 2 или 3 R'.
3. Соединение или его фармацевтически приемлемая соль по п. 2, где R выбран из H, F, Cl, Br, I, OH, NH2, CN, Me, CF3, CHF2, CH2F, Et, 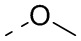 ,
,  ,
, 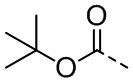 ,
, 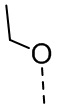 .
.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где R4 выбран из группы, состоящей из фенила, пиридила, пиримидинила, пиразинила, имидазолила и пиразолила, каждый из которых необязательно замещен 1 или 2 R.
5. Соединение или его фармацевтически приемлемая соль по п. 4, где R4 выбран из группы, состоящей из  ,
,  и
и  , каждый из которых необязательно замещен 1 или 2 R.
, каждый из которых необязательно замещен 1 или 2 R.
6. Соединение или его фармацевтически приемлемая соль по п. 5, где R4 выбран из  ,
,  ,
,  .
.
7. Соединение или его фармацевтически приемлемая соль по п. 1, где R4-L1- выбран из  ,
,  ,
,  .
.
8. Соединение или его фармацевтически приемлемая соль по п. 1, где R3 выбран из H, R4-L1- или выбран из группы, состоящей из C1-3алкила, C3-6циклоалкила, -C1-3алкил-C(=O)O-C1-3алкил-, C1-3алкил-S(=O)2-C1-3алкил- и фенила, каждый из которых необязательно замещен 1, 2 или 3 R.
9. Соединение или его фармацевтически приемлемая соль по п. 8, где R3 выбран из H, R4-L1- или выбран из группы, состоящей из Me, Et,  ,
,  ,
,  ,
,  ,
,  и
и  , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
10. Соединение или его фармацевтически приемлемая соль по п. 7, где R3 выбран из H, Me, Et, 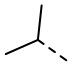 ,
, 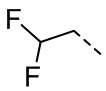 ,
, 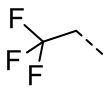 ,
, 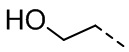 ,
, 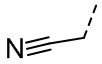 ,
, 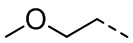 , ,
, , 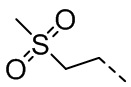 ,
, 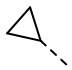 , , , , , .
, , , , , .
11. Соединение или его фармацевтически приемлемая соль по п. 1, где X выбран из  ,
,  ,
,  ,
,  ,
, 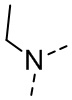 ,
, 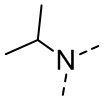 ,
, 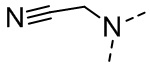 ,
, 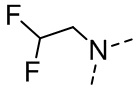 ,
, 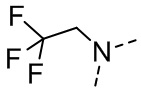 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 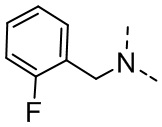 ,
, 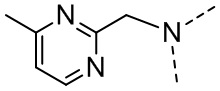 ,
, 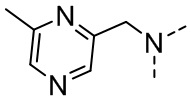 .
.
12. Соединение или его фармацевтически приемлемая соль по п. 1, где R5 выбран из группы, состоящей из фенила, пиридила, пиримидинила, пиразинила, пиридазинила, имидазолила и пиразолила, каждый из которых необязательно замещен 1, 2 или 3 R.
13. Соединение или его фармацевтически приемлемая соль по п. 12, где R5 выбран из группы, состоящей из  и
и  , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
14. Соединение или его фармацевтически приемлемая соль по п. 13, где R5 выбран из  ,
,  , .
, .
15. Соединение или его фармацевтически приемлемая соль по п. 1, где R5-L2- выбран из  ,
,  ,
, 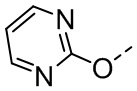 .
.
16. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, R5-L2- или выбран из группы, состоящей из C1-3алкила, C1-3алкокси, C2-4алкенила, 1,2,3,6-тетрагидропиридила, пиридин-2(1H)-онила, фенила, пиридила, пиримидинила, пиразинила, пиридазинила, пиразолила, имидазолила, тиазолила, изотиазолила и тиенила, каждый из которых необязательно замещен 1, 2 или 3 R.
17. Соединение или его фармацевтически приемлемая соль по п. 16, где каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, R5-L2- или выбран из группы, состоящей из Me, 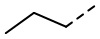 ,
, 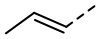 ,
, 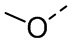 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 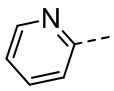 ,
, 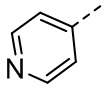 ,
,  и
и 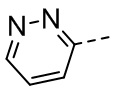 , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
18. Соединение или его фармацевтически приемлемая соль по п. 15, где каждый из R1 и R2 независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, Me, 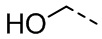 ,
,  ,
,  ,
,  , , ,
, , ,  , ,
, ,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  , ,
, ,  , , , , ,
, , , , ,  , ,
, ,  ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  , .
, .
19. Соединение или его фармацевтически приемлемая соль по п. 1, где R6 выбран из группы, состоящей из Me и Et, каждый из которых необязательно замещен 1, 2 или 3 R.
20. Соединение или его фармацевтически приемлемая соль по п. 19, где R6 выбран из Me, CH2F, CHF2.
21. Соединение или его фармацевтически приемлемая соль по п. 1, которые выбраны из 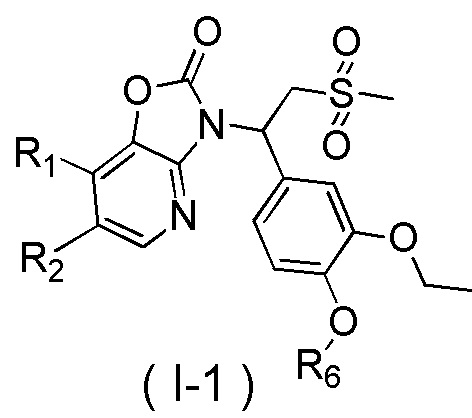 ,
, 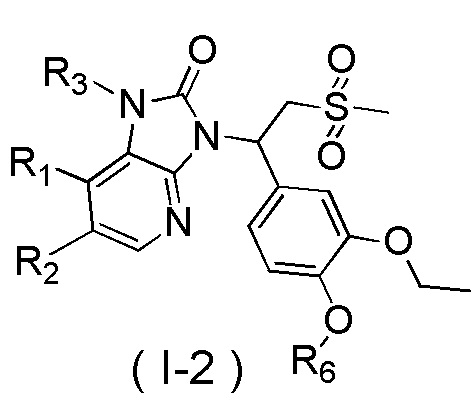 ,
, 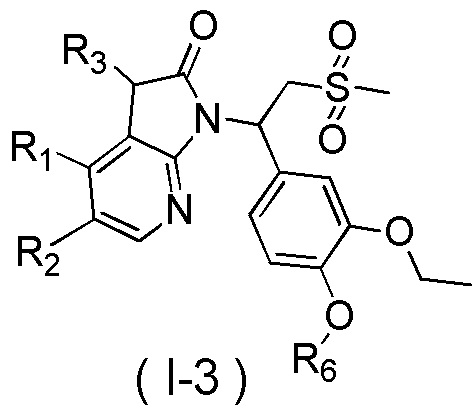 , где R1, R2, R3 и R6 определены в п. 1.
, где R1, R2, R3 и R6 определены в п. 1.
22. Соединение или его фармацевтически приемлемая соль по п. 21, которые выбраны из 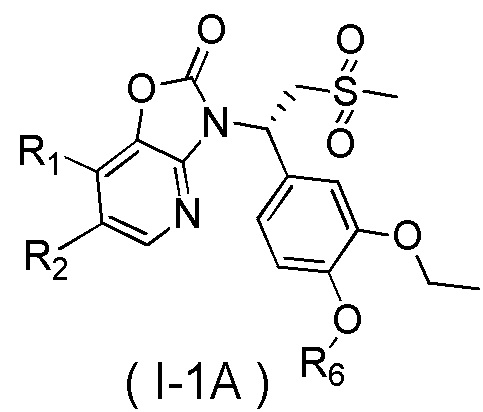 ,
, 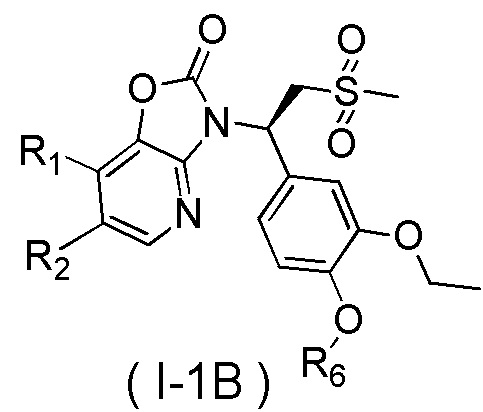 ,
, 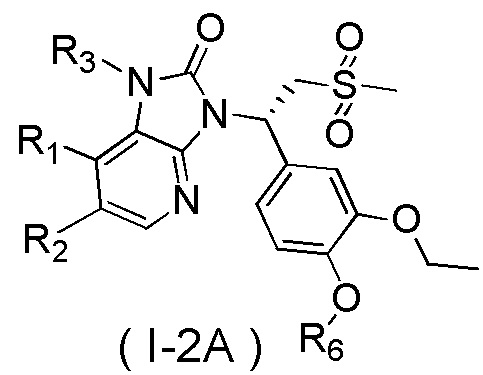 ,
, 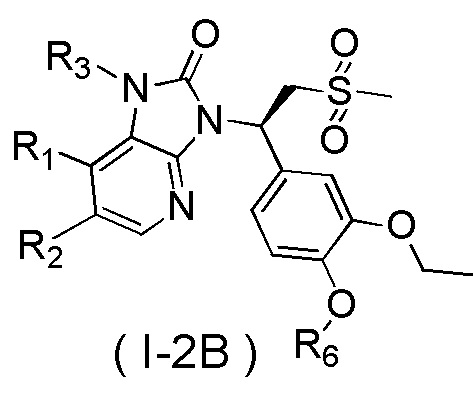 ,
, 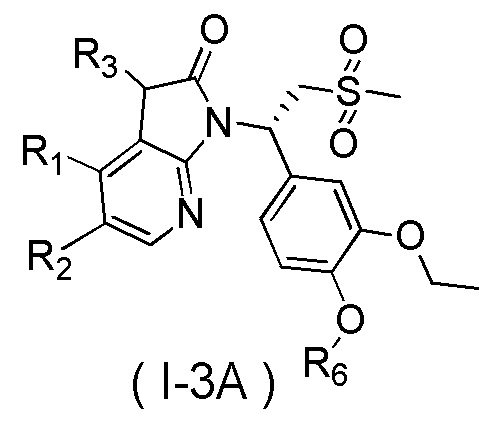 ,
, 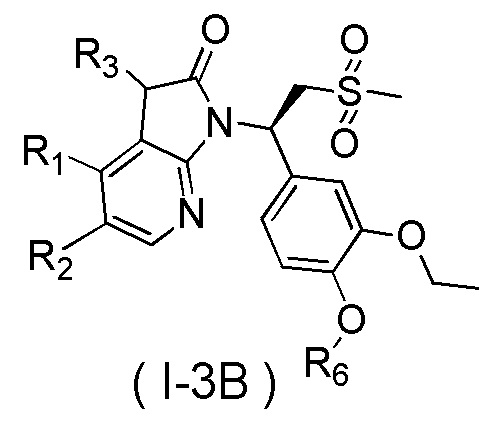 , где R1, R2, R3 и R6 определены в п. 1.
, где R1, R2, R3 и R6 определены в п. 1.
23. Соединение или его фармацевтически приемлемая соль, которые выбраны из группы, состоящей из
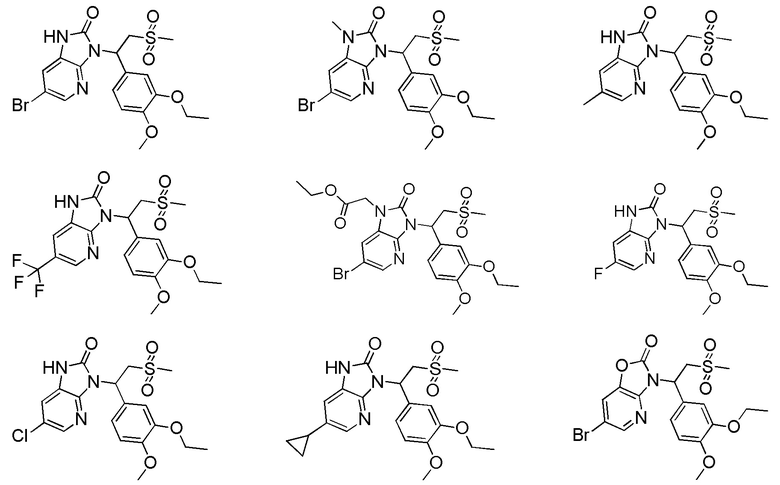
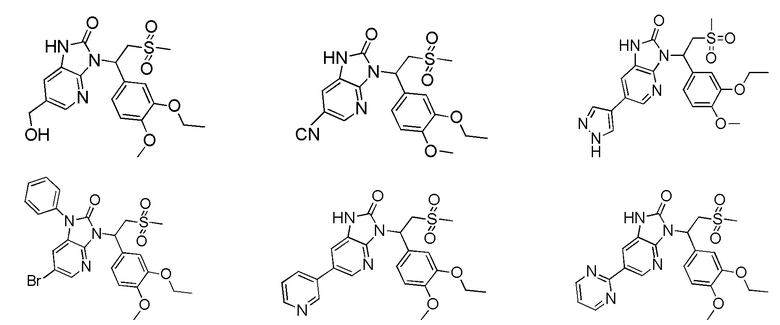
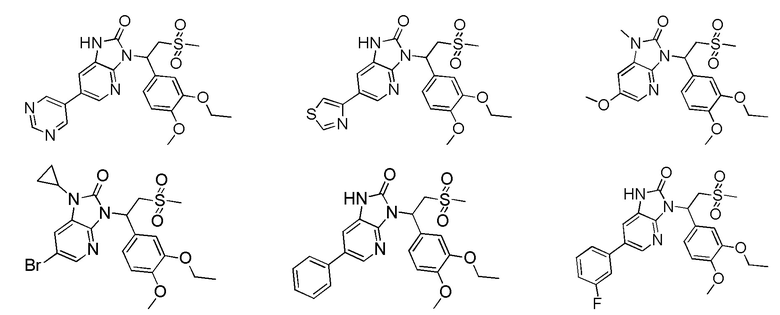
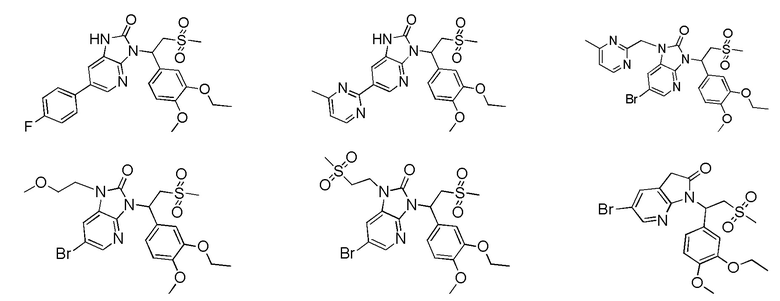
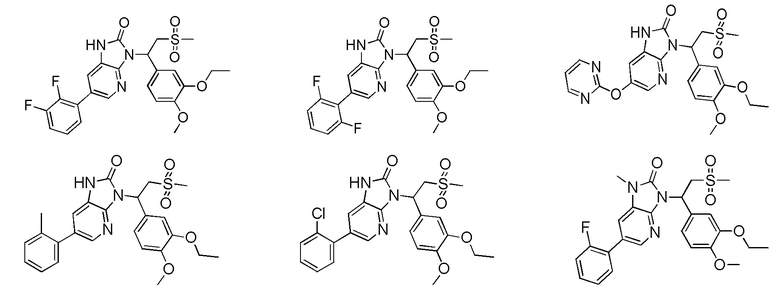
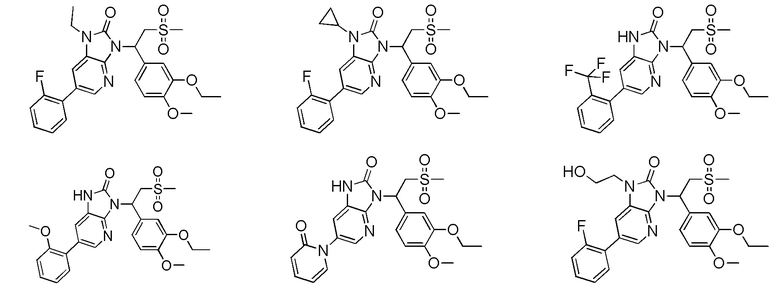
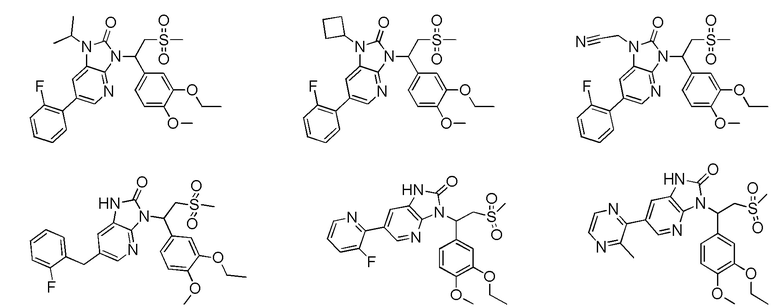
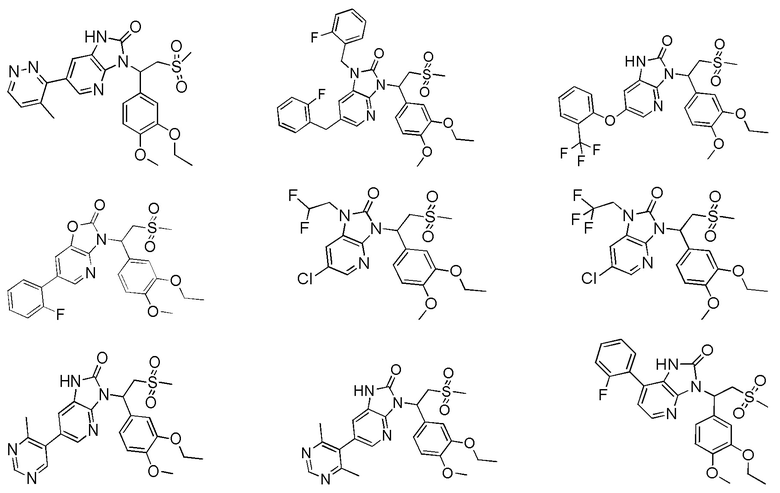
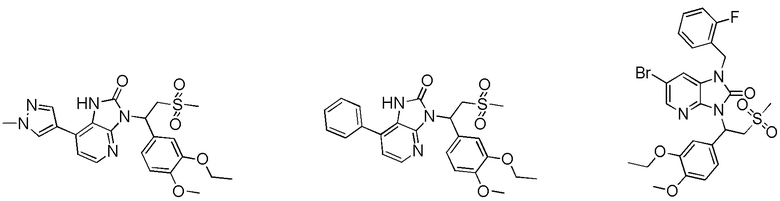
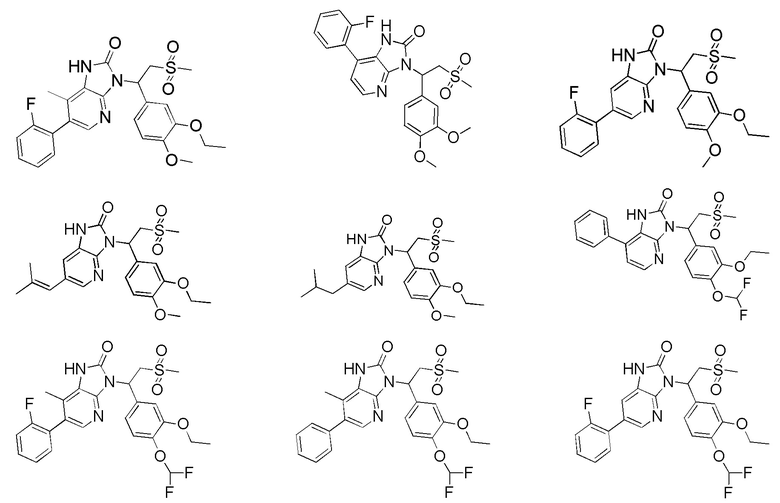
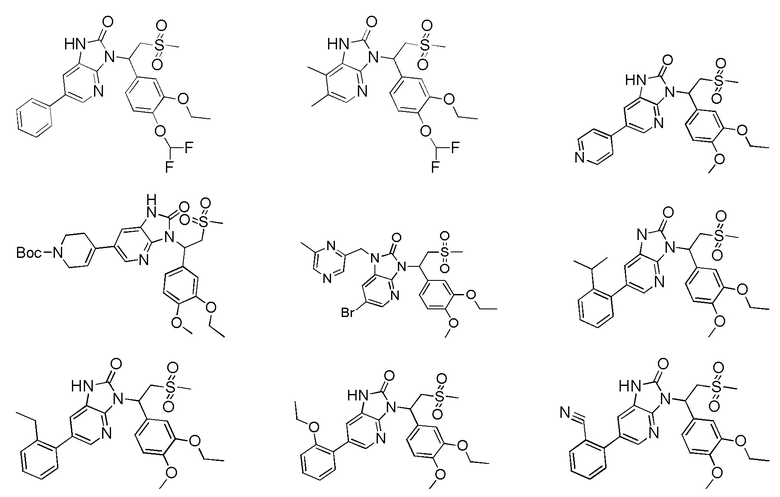
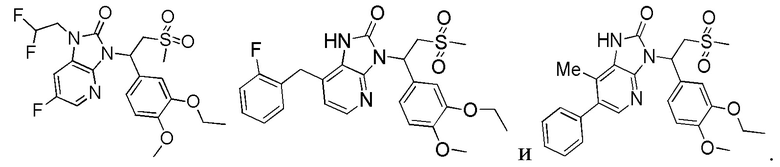
24. Соединение или его фармацевтически приемлемая соль по п. 23, которые выбраны из группы, состоящей из
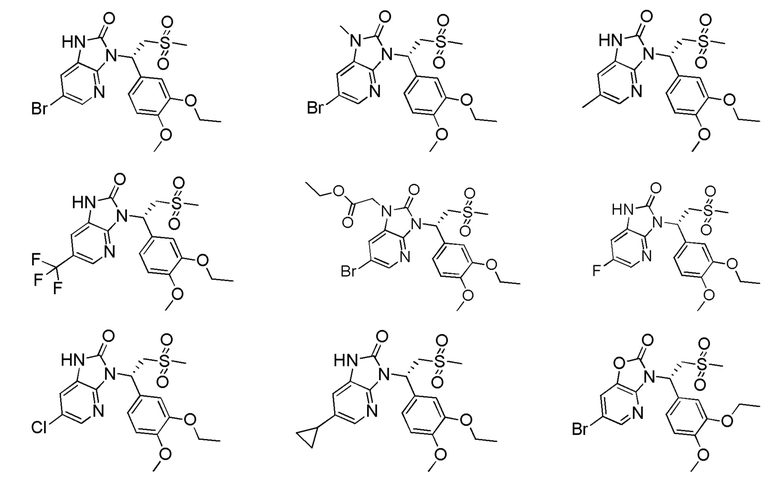
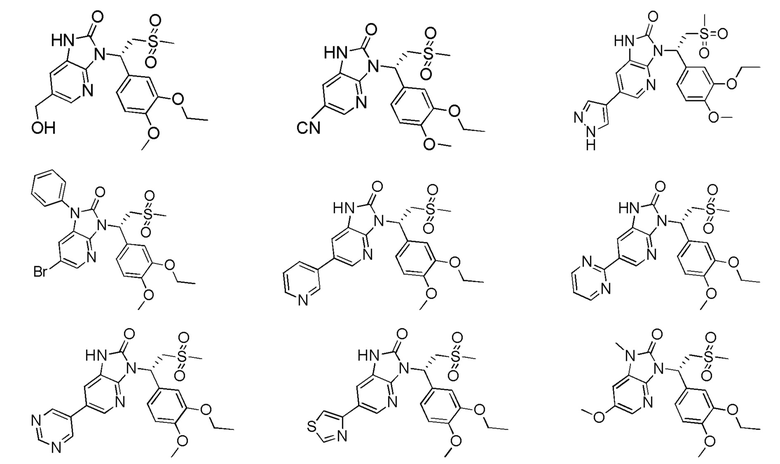
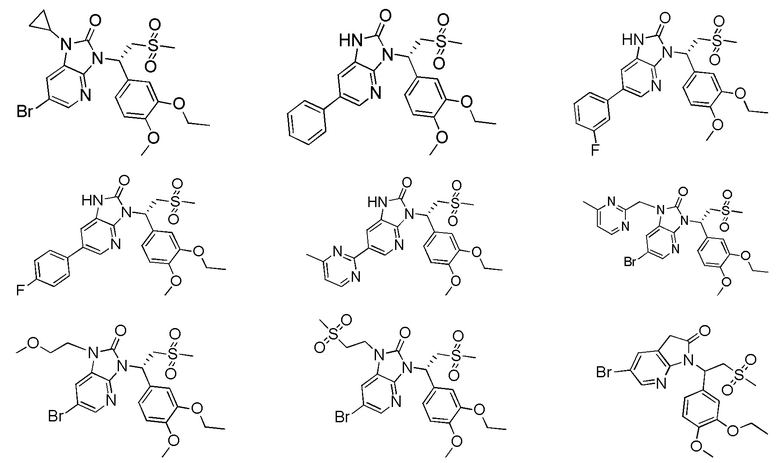
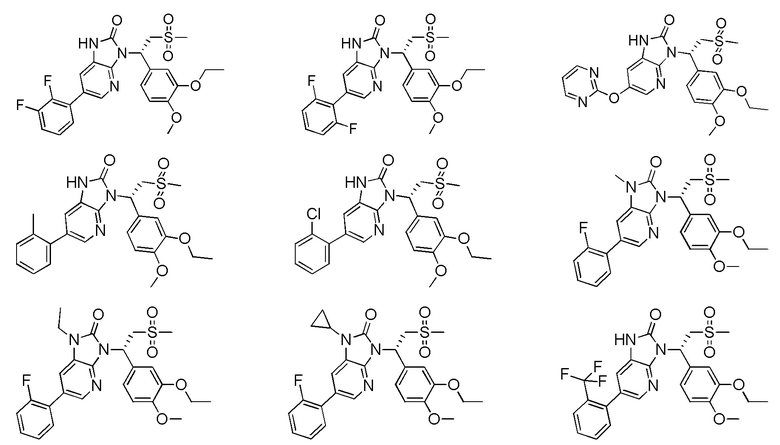
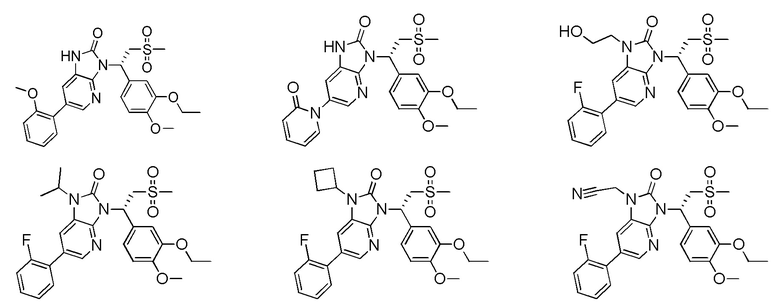
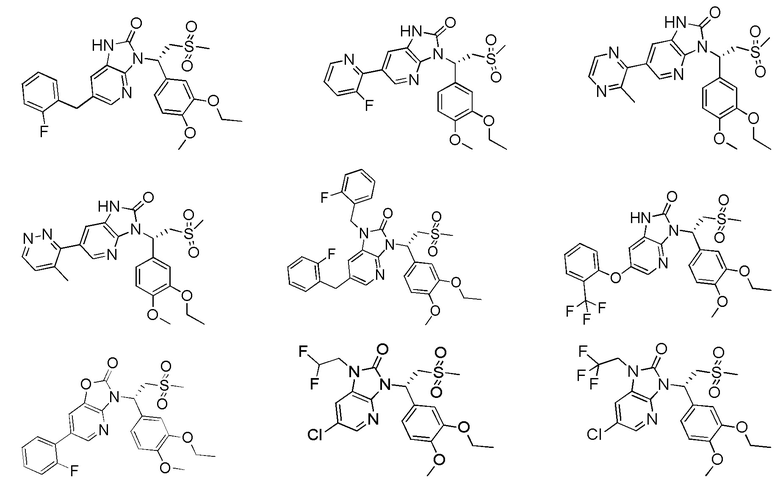
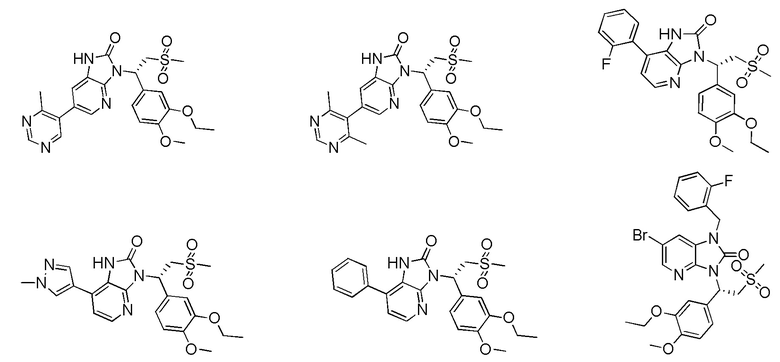
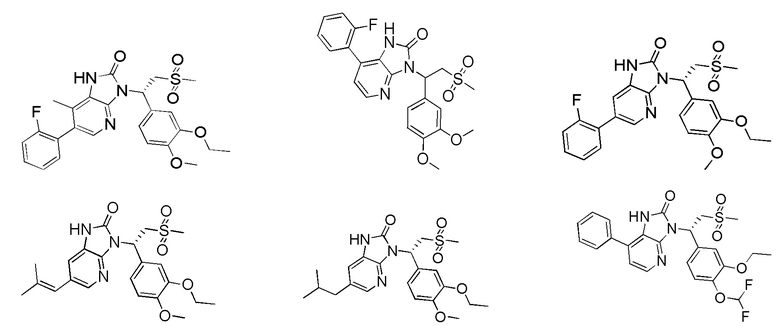
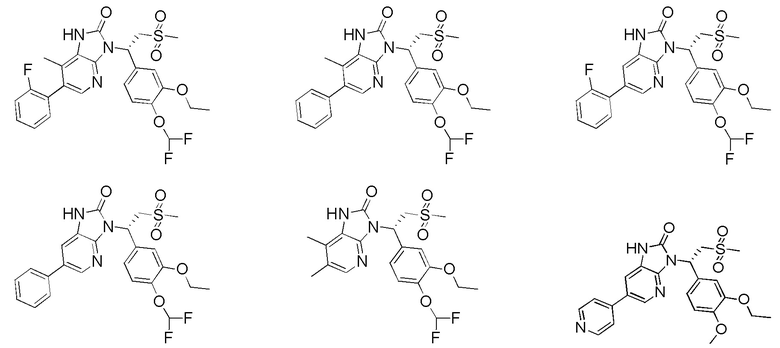
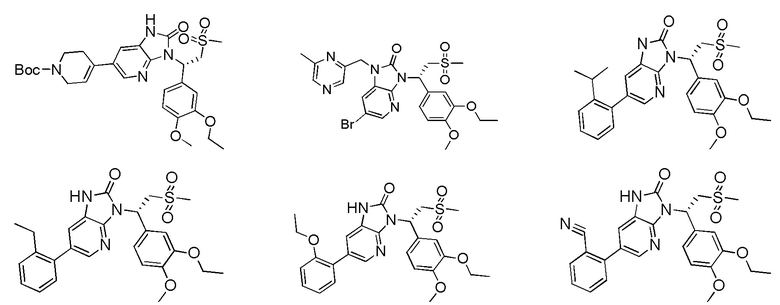
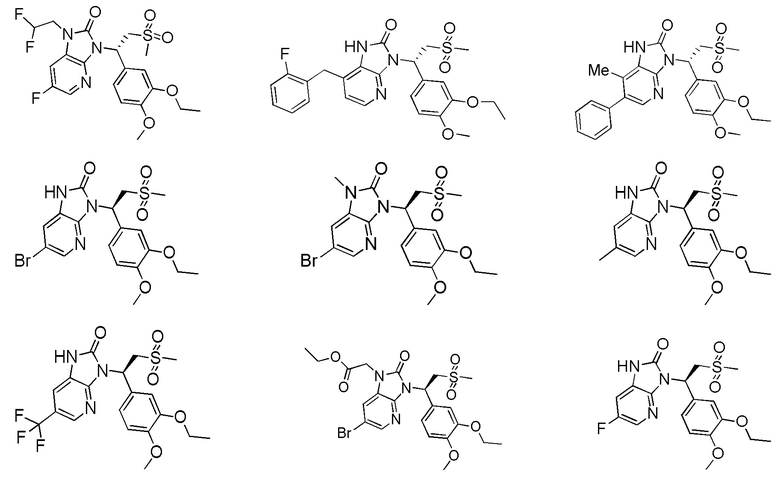
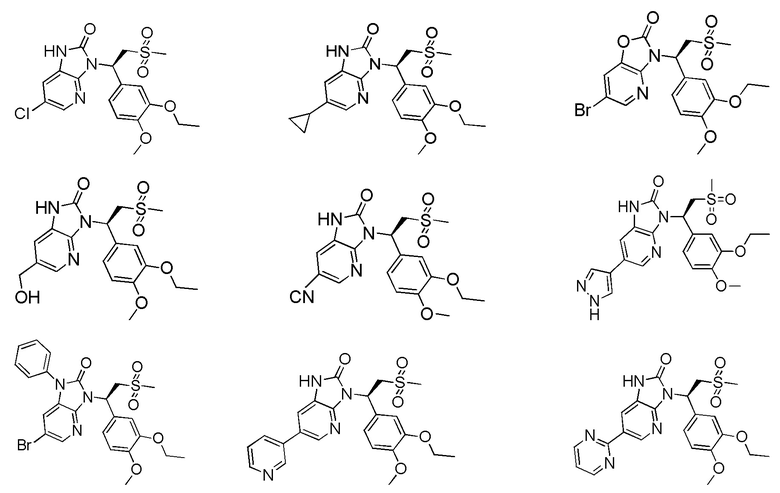
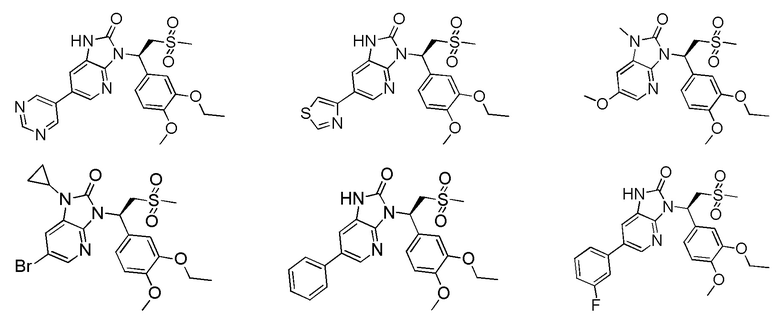
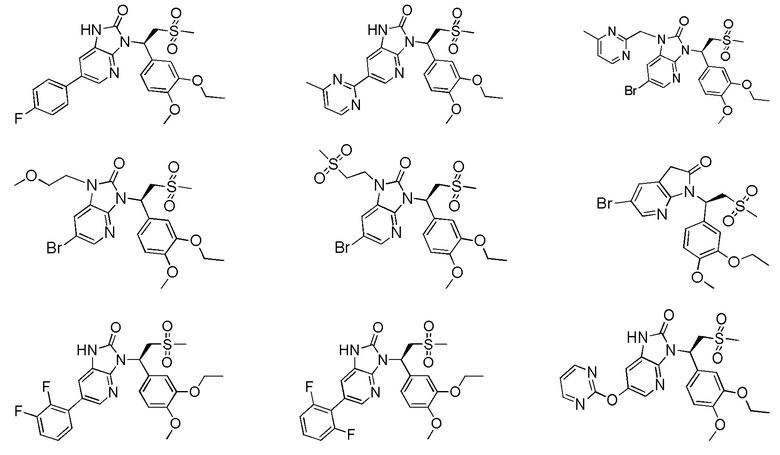
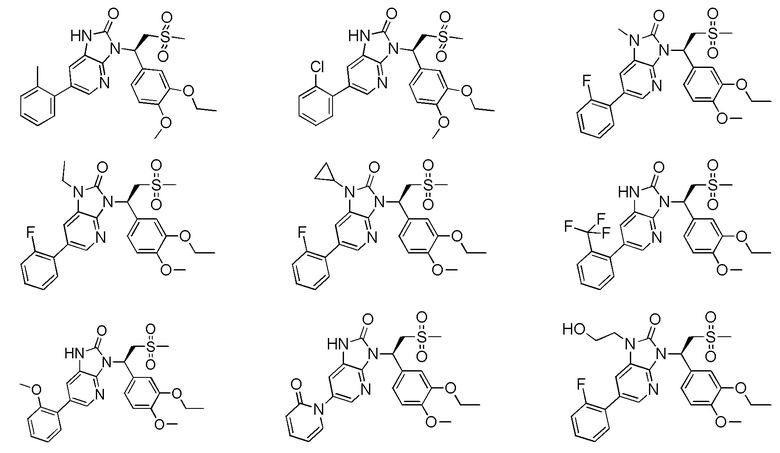
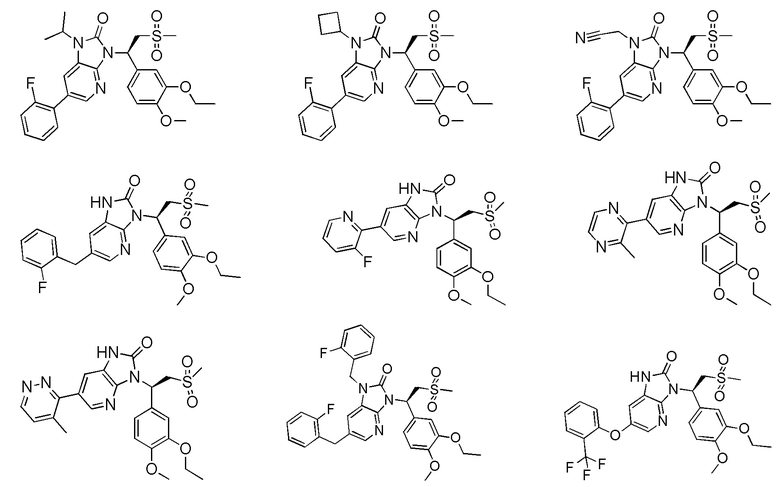
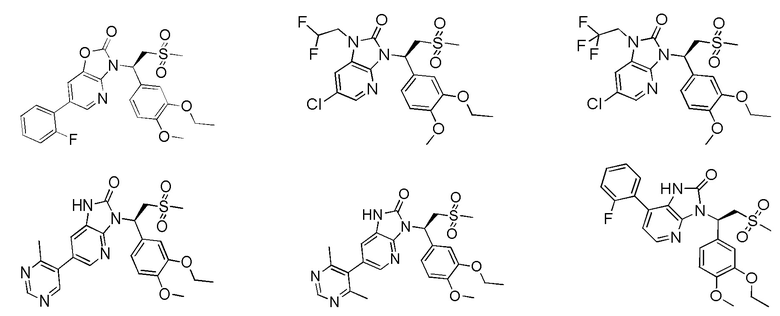
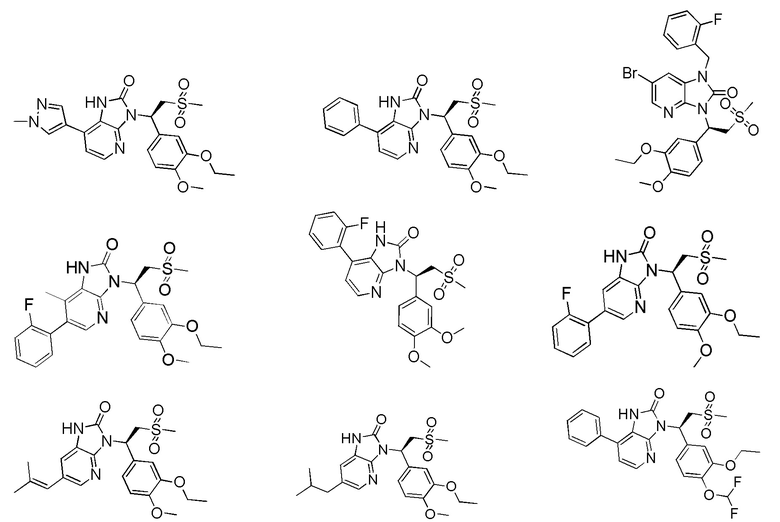
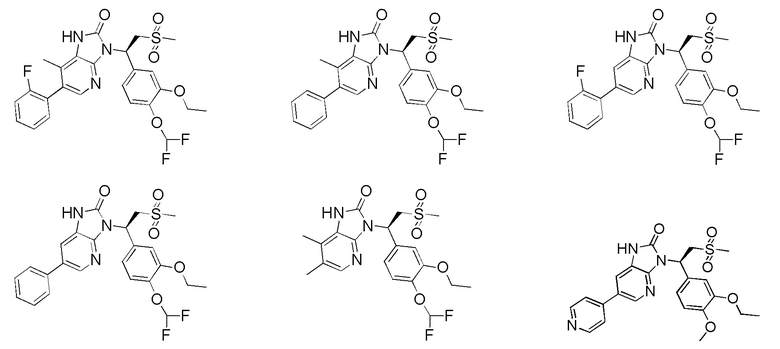
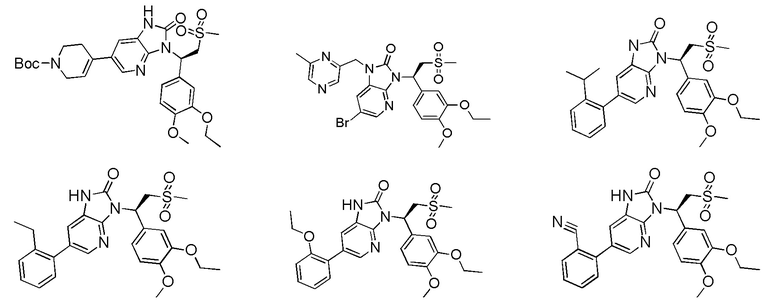
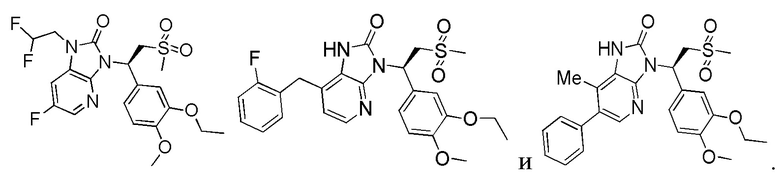
25. Применение соединения или его фармацевтически приемлемой соли по п. 1 в качестве лекарственного препарата для лечения заболеваний, связанных с PDE4, при этом необязательно заболевание, связанное с PDE4, относится к псориазу, псориатическому артриту, хронической обструктивной пневмонии, анкилозирующему спондилиту, воспалительному заболеванию кишечника.
26. Применение соединения или его фармацевтически приемлемой соли по п. 24 в качестве лекарственного препарата для лечения заболеваний, связанных с PDE4, при этом необязательно заболевание, связанное с PDE4, относится к псориазу, псориатическому артриту, хронической обструктивной пневмонии, анкилозирующему спондилиту, воспалительному заболеванию кишечника.
27. Фармацевтическая композиция для лечения заболеваний, связанных с PDE4, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п. 1 в качестве активного ингредиента и фармацевтический приемлемый носитель.
28. Фармацевтическая композиция для лечения заболеваний, связанных с PDE4, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п. 24 в качестве активного ингредиента и фармацевтический приемлемый носитель.
29. Способ лечения заболеваний, связанных с PDE4, у субъекта, нуждающегося в этом, включающий введение эффективного количества соединения или его фармацевтически приемлемой соли по п. 1 субъекту.
30. Способ лечения заболеваний, связанных с PDE4, у субъекта, нуждающегося в этом, включающий введение эффективного количества соединения или его фармацевтически приемлемой соли по п. 24 субъекту.
| WO 2011021678 A1, 24.02.2011 | |||
| WO 2010030500 A1, 18.03.2010 | |||
| RU 2009118623 A, 27.11.2010 | |||
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2006 |
|
RU2426733C2 |

