Ссылка на родственные заявки
[0001] Настоящая заявка заявляет приоритет следующих заявок:
[0002] CN201711080753.8, дата подачи 06.11.2017 г.;
[0003] CN201810136962.8, дата подачи 09.02.2018 г.;
[0004] CN201810661825.6, дата подачи 25.06.2018 г.
Область техники, к которой относится изобретение
[0005] Настоящее изобретение относится к ряду пиридопиримидиновых соединений и их применению в изготовлении лекарственных препаратов, связанных с двойными ингибиторами mTORC 1/2, в частности относится к применению соединений, представленных формулой (IV), их таутомеров или их фармацевтически приемлемых солей в изготовлении лекарственных препаратов, связанных с двойными ингибиторами mTORC 1/2.
Предшествующий уровень техники
[0006] Опухоли, особенно злокачественные опухоли, в настоящее время являются одними из наиболее серьезных заболеваний, которые ставят под угрозу здоровье человека. С развитием науки и техники, а также углублением исследований, проводимых на людях, в отношении лечения опухоли был достигнут быстрый прогресс в изучении механизма возникновения и развития опухоли и лечении опухоли. Было открыто множество новых механизмов и биологических маркеров. Настоящее изобретение относится к сигнальному пути, который играет ключевую роль в пролиферации клеток опухоли, инвазивном метастазировании и антиапоптическом действии, т. е. сигнальному пути фосфатидилинозитол-3-киназа (PI3K)-AKT – мишени рапамицина млекопитающих (mTOR).
[0007] Активация PI3K в основном происходит в субстрате, вблизи внутренней части плазматической мембраны. Различные факторы роста и комплексы передачи сигнала, включающие фактор роста фибробластов (FGF), фактор роста эндотелия сосудов (VEGF), фактор роста человека (HGF), ангиопоэтин-1 (Ang1) и инсулин, могут инициировать процесс активации PI3K. Данные факторы активируют рецепторную тирозинкиназу (RTK), что вызывает аутофосфорилирование. Результатом активации PI3K является образование второго мессенджера PIP3 на плазматической мембране. PIP3 связывается с сигнальными белками AKT и PDK1 (фосфоинозитид-зависимая киназа-1), содержащими PH-домен в клетке, обуславливая фосфорилирование PDK1 Ser308 белка AKT, что приводит к активации AKT. Другие субстраты PDK1 включают PKC (протеинкиназа C), S6K (p70S6) и SGK (киназы, регулируемые сывороткой/глюкокортикоидами). AKT, также известная как протеинкиназа B (PKB), является основным нижележащим эффектором PI3K. Активированная AKT регулирует функцию клетки посредством фосфорилирования нижележащих факторов, таких как ферменты, киназы и факторы транскрипции. AKT оказывает антиапоптический эффект путем фосфорилирования целевых белков посредством множества нисходящих путей. PTEN (гомолог фосфатазы и тензина, удаленный на хромосоме 10), ген-супрессор опухоли, подвергнут мутации или делеции в широком спектре опухолей человека. PTEN представляет собой PIP3-фосфатазу, которая, в отличие от функции PI3K, может преобразовывать PIP3 в PIP2 посредством дефосфорилирования. PTEN может снизить степень активации AKT и предотвратить все последующие события передачи сигнала, регулируемые AKT. Будучи нижележащим субстратом AKT mTOR является сравнительно консервативным в развитии, может интегрировать множественные сигналы факторов питания, энергии и роста, участвовать в биологических процессах, таких как транскрипция генов, трансляция белков, синтез рибосом и апоптоз, и играет чрезвычайно важную роль в росте клетки. Существует два вида высокогомологичных комплексов, Tor и KOG01 объединяются с образованием mTORC1, mTOR с AVO1/AVO2/AVO3/ и LST8 образуют mTORC2, который нечувствителен к рапамицину. mTOR регулирует трансляцию последующего белка посредством фосфорилирования последующего рибосомного белка-мишени S6 S40S протеинкиназы, например, S6K1 и 4EBP1. mTOR связывается с eIF3, фосфорилирует S6K1, обеспечивает высвобождение S6K1 от eIF3 и активирование, с последующим фосфорилированием, субстрата клетки, такого как p70S6, что способствует трансляции и экспрессии белка. 4EBP1 связывается с эукариотическим фактором инициации транскрипции 4E и ингибирует его активность. mTOR фосфорилирует 4E-BP1 с его активацией и отделением его от eiF-4E с обеспечением транскрипции в эукариотических клетках. mTORC2 может фосфорилировать AKT, обеспечивая тем самым повышение степени регулирования ее киназной активности.
[0008] Как видно из вышесказанного, любая мутация или сверхэкспрессия более ранних этапов сигнального пути PI3K/AKT/mTOR приведет к ряду последующих каскадных реакций, что в конечном итоге приведет к возникновению, развитию и метастазированию опухоли. Пока mTOR находится по середине сигнального пути, ингибирование mTORC1 и mTORC2 может эффективно блокировать передачу сигналов, за счет чего достигается контроль развития опухоли.
[0009] В исследованиях обнаружили, что данный сигнальный путь обнаружен в различных солидных опухолях, таких как рак молочной железы, рак предстательной железы, рак легкого, рак толстой кишки, рак поджелудочной железы, рак печени, рак желудка, колоректальный рак, рак почки, рак щитовидной железы, рак, сопровождающийся менингитом, а также острый и хронический лимфоцитарный лейкоз, трабекулярный рак и т. д. И это тесно связано с переносимостью лечения и плохим прогнозом. Было видно, что разработка низкомолекулярных соединений для достижения ингибирования сигнального пути PI3K/AKT/mTOR имеет хорошие перспективы касаемо развития.
[0010] Настоящее изобретение направлено на поиск целенаправленно воздействующих лекарственных средств, представляющих собой двойные ингибиторы mTOR1/2, при этом такие соединения обладают хорошей активностью и проявляют превосходные эффекты и функции.
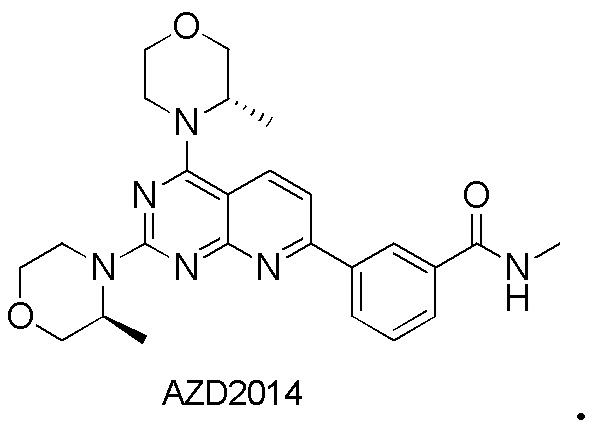
[0011] В US20170281637 раскрывается соединение AZD2014, которое принадлежит к ингибиторам киназы mTORC1 и mTORC2, и его структурная формула является следующей:
Содержание настоящего изобретения
[0012] Настоящее изобретение предусматривает соединение, представленное формулой (IV), его фармацевтически приемлемую соль или его изомер,
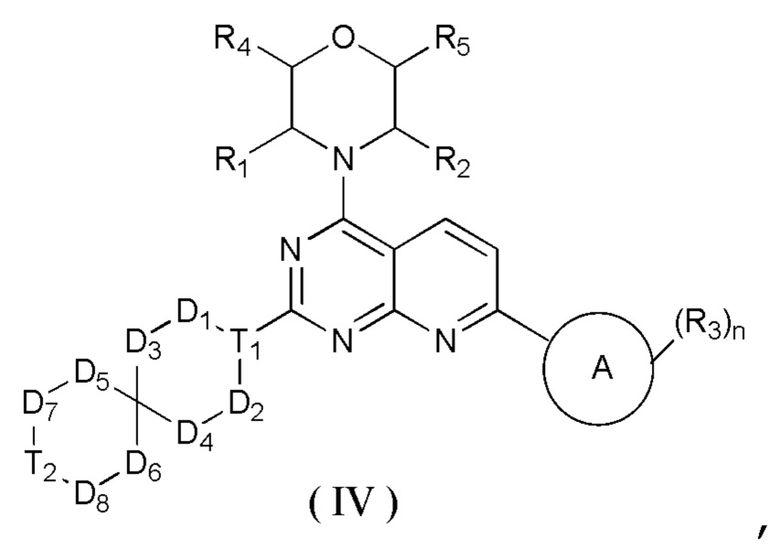
где
R1 представляет собой H;
R2 представляет собой Me;
в качестве альтернативы R1, R2 и атом N в морфолиновом кольце образуют 5-6-членный гетероциклоалкил;
R3 выбран из NH2, 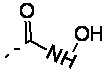 , C1-3гетероалкила, 5-6-членного гетероарила и C3-6циклоалкил-NH-C(=O)-, где C1-3гетероалкил, 5-6-членный гетероарил и C3-6циклоалкил-NH-C(=O)- необязательно замещены R, и при этом число R равняется 1, 2 или 3;
, C1-3гетероалкила, 5-6-членного гетероарила и C3-6циклоалкил-NH-C(=O)-, где C1-3гетероалкил, 5-6-членный гетероарил и C3-6циклоалкил-NH-C(=O)- необязательно замещены R, и при этом число R равняется 1, 2 или 3;
n выбран из 1 и 2;
кольцо A выбрано из фенила и 6-10-членного гетероарила;
R4 представляет собой H;
R5 представляет собой H;
в качестве альтернативы R4 и R5 соединены вместе с образованием 5-6-членного гетероциклоалкила;
D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2- и -O-, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, где -CH2- или- CH2CH2- необязательно замещен R, и при этом число R равняется 1 или 2;
D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O- и -NH-, и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
T1 выбран из CH и N;
T2 выбран из -CH2-, -NH-, -O-,  -S- и -C(=O)NH-, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
-S- и -C(=O)NH-, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
R соответственно выбран из F, Cl, Br, I, OH, NH2, C1-3алкила, C1-3алкокси и C3-6циклоалкила, где C1-3алкил, C1-3алкокси и C3-6циклоалкил необязательно замещены R', и при этом число R' равняется 1 или 2;
R' соответственно выбран из F, Cl, Br, I, OH и NH2;
C1-3гетероалкил, 5-6-членный гетероарил и 6-10-членный гетероарил соответственно содержат 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из -O-, -S-, -NH-, N, -C(=O)-, -C(=O)NH- и -C(=S)NH-.
[0013] Настоящее изобретение предусматривает соединение, его фармацевтически приемлемую соль или его изомер, описанные выше, которые выбраны из
где
R1 представляет собой H;
R2 представляет собой Me;
в качестве альтернативы R1, R2 и атом N в морфолиновом кольце образуют 5-6-членный гетероциклоалкил;
R3 выбран из NH2, , C1-3гетероалкила, 5-6-членного гетероарила и C3-6циклоалкил-NH-C(=O)-, где C1-3гетероалкил, 5-6-членный гетероарил и C3-6циклоалкил-NH-C(=O)- необязательно замещены R, и при этом число R равняется 1, 2 или 3;
кольцо A выбрано из фенила и 6-10-членного гетероарила;
R4 выбран из H;
R5 выбран из H;
в качестве альтернативы R4 и R5 соединены вместе с образованием 5-6-членного гетероциклоалкила;
D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2- и -O-, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, где -CH2- или -CH2CH2- необязательно замещен R, и при этом число R равняется 1 или 2;
D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O- и -NH-, и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
T1 выбран из CH и N;
T2 выбран из -CH2-, -NH-, -O-,  -S- и -C(=O)NH-, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
-S- и -C(=O)NH-, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
R соответственно выбран из F, Cl, Br, I, OH, NH2, C1-3алкила, C1-3алкокси и C3-6циклоалкила, где C1-3алкил, C1-3алкокси и C3-6циклоалкил необязательно замещены R’, и при этом число R’ равняется 1 или 2;
R’ соответственно выбран из F, Cl, Br, I, OH и NH2;
C1-3гетероалкил, 5-6-членный гетероарил и 6-10-членный гетероарил соответственно содержат 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из -O-, -S-, -NH-, N, -C(=O)-, -C(=O)NH- и -C(=S)NH-.
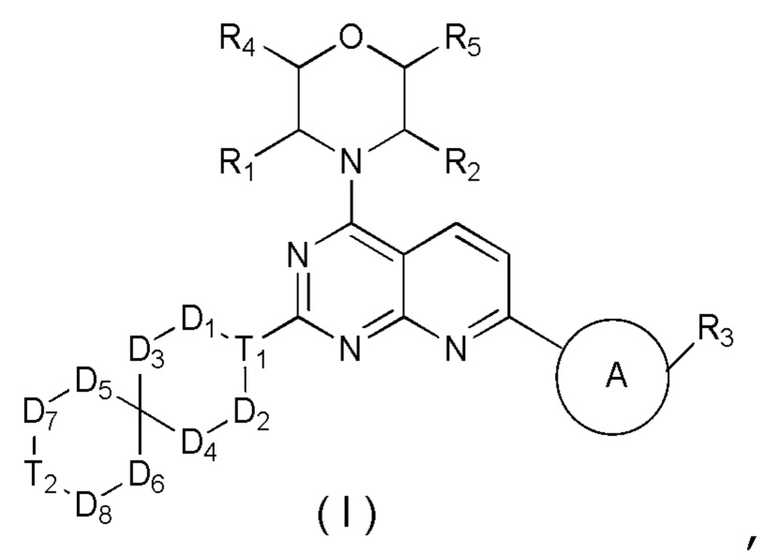
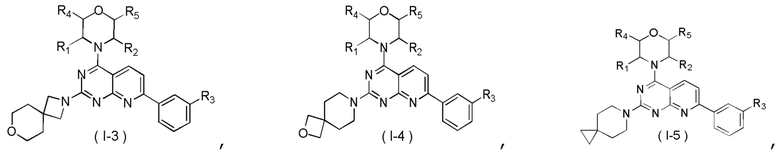
[0014] Настоящее изобретение предусматривает соединение, представленное формулой (I), его фармацевтически приемлемую соль или его изомер,
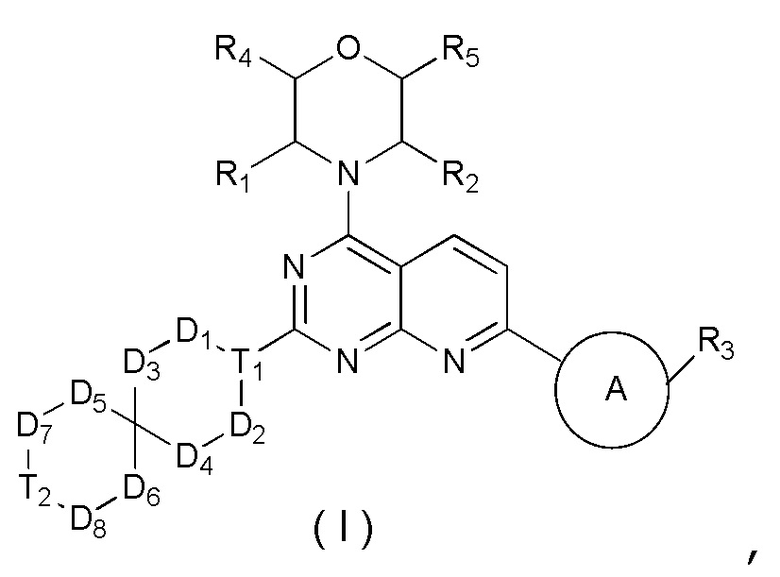
где
R1 выбран из H;
R2 выбран из Me;
в качестве альтернативы R1 и R2 соединены вместе с образованием 5-6-членного гетероциклоалкила;
R3 выбран из NH2, , C1-3гетероалкила, 5-6-членного гетероарила и C3-6циклоалкил-NH-C(=O)-, где C1-3гетероалкил, 5-6-членный гетероарил и C3-6циклоалкил-NH-C(=O)- необязательно замещены R, и при этом число R равняется 1, 2 или 3;
кольцо A выбрано из фенила и 6-10-членного гетероарила;
R4 выбран из H;
R5 выбран из H;
D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2- и -O-, где -CH2- или -CH2CH2- необязательно замещены R, и при этом число R равняется 1 или 2;
D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O- и -NH-, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, и -NH- необязательно замещен R;
T1 выбран из CH и N;
T2 выбран из одинарной связи, CH2 и -O-;
R соответственно выбран из F, Cl, Br, I, OH, NH2, C1-3алкила, C1-3алкокси и C3-6циклоалкила, где C1-3алкил, C1-3алкокси и C3-6циклоалкил необязательно замещены R’, при этом число R’ равняется 1 или 2;
R’ соответственно выбран из F, Cl, Br, I, OH и NH2;
C1-3гетероалкил, 5-6-членный гетероарил и 6-10-членный гетероарил соответственно содержат 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из -O-, -S-, -NH-, N, -C(=O)-, -C(=O)NH- и -C(=S)NH-.
[0015] В некоторых вариантах осуществления настоящего изобретения R соответственно выбран из F, Cl, Br, I, OH, NH2, Me, Et,  и
и  , где Me, Et,
, где Me, Et,  и необязательно замещены R’, и при этом число R’ равняется 1 или 2, другие переменные определены в настоящем изобретении.
и необязательно замещены R’, и при этом число R’ равняется 1 или 2, другие переменные определены в настоящем изобретении.
[0016] В некоторых вариантах осуществления настоящего изобретения R соответственно выбран из F, Cl, Br, I, OH, NH2, Me, CF3, Et,  и , другие переменные определены в настоящем изобретении.
и , другие переменные определены в настоящем изобретении.
[0017] В некоторых вариантах осуществления настоящего изобретения фрагмент 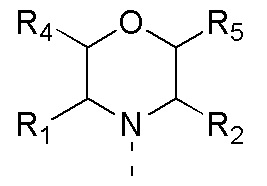 выбран из
выбран из 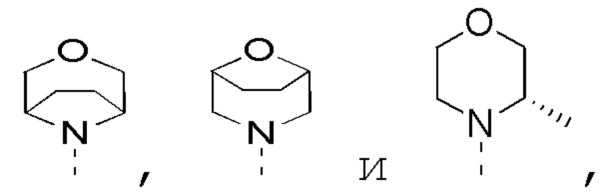 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0018] В некоторых вариантах осуществления настоящего изобретения R1 и R2 соединены вместе, и фрагмент 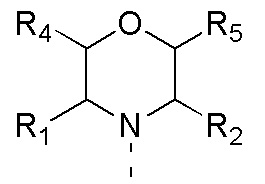 выбран из
выбран из 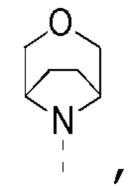 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0019] В некоторых вариантах осуществления настоящего изобретения R3 выбран из NH2, 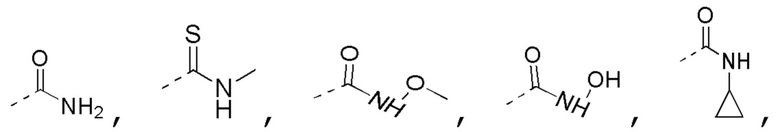
 1H-пиразолила и 1H-1,2,4-триазолила, где NH2,
1H-пиразолила и 1H-1,2,4-триазолила, где NH2, 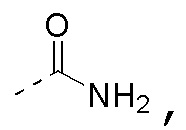
 1H-пиразолил и 1H-1,2,4-триазолил необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
1H-пиразолил и 1H-1,2,4-триазолил необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
[0020] В некоторых вариантах осуществления настоящего изобретения R3 выбран из NH2,  1H-пиразолила и 1H-1,2,4-триазолила, где NH2,
1H-пиразолила и 1H-1,2,4-триазолила, где NH2, 
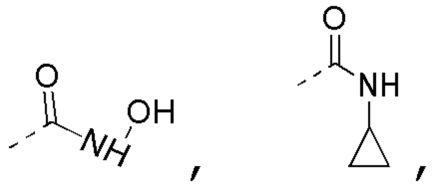 1H-пиразолил и 1H-1,2,4-триазолил необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
1H-пиразолил и 1H-1,2,4-триазолил необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
[0021] В некоторых вариантах осуществления настоящего изобретения R3 выбран из NH2, 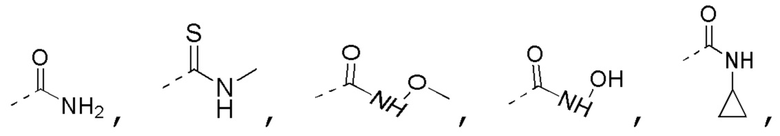
 где NH2,
где NH2, 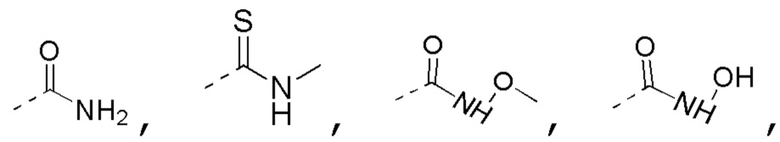
 необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
[0022] В некоторых вариантах осуществления настоящего изобретения R3 выбран из NH2, 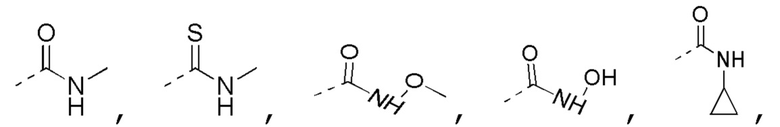
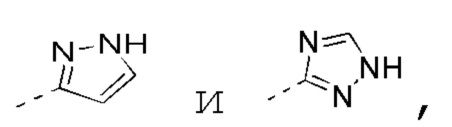 где NH2,
где NH2, 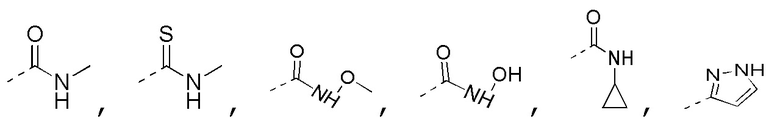 и
и  необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
необязательно замещены R, и при этом число R равняется 1, 2 или 3, другие переменные определены в настоящем изобретении.
[0023] В некоторых вариантах осуществления настоящего изобретения R3 выбран из NH2, 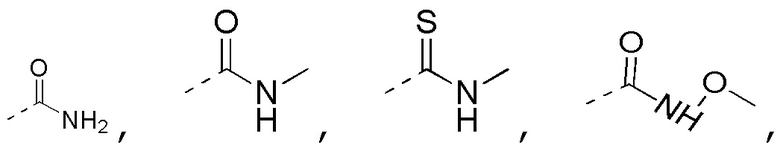
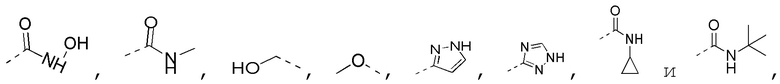 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0024] В некоторых вариантах осуществления настоящего изобретения R3 выбран из NH2, 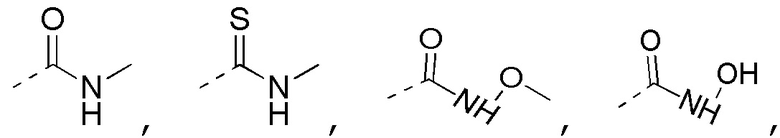
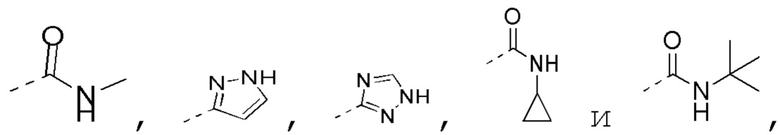 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
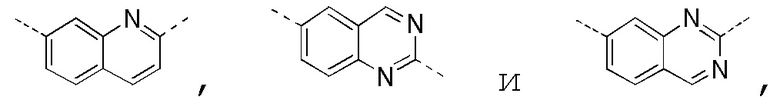
[0025] В некоторых вариантах осуществления настоящего изобретения кольцо A выбрано из фенила, бензо[d]оксазола, хинолинила и хиназолинила, другие переменные определены в настоящем изобретении.
[0026] В некоторых вариантах осуществления настоящего изобретения кольцо A выбрано из 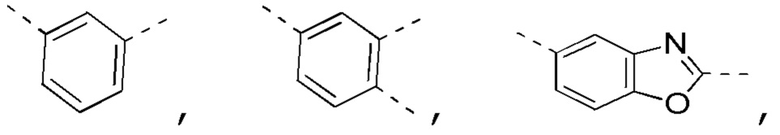
 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0027] В некоторых вариантах осуществления настоящего изобретения кольцо A выбрано из 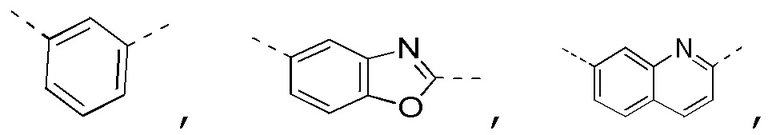
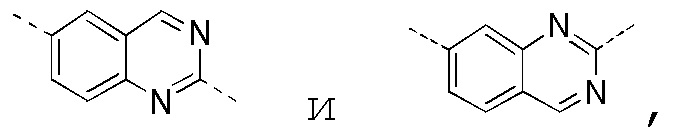 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0028] В некоторых вариантах осуществления настоящего изобретения фрагмент 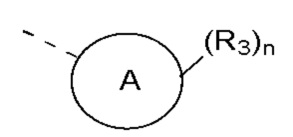 выбран из
выбран из 
другие переменные определены в настоящем изобретении.
[0029] В некоторых вариантах осуществления настоящего изобретения фрагмент  выбран из
выбран из 
 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0030] В некоторых вариантах осуществления настоящего изобретения D1, D2, D3 и D4, описанные выше, соответственно выбраны из одинарной связи, -CH2-, -CH2CH2-, -O- и 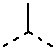 , и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, другие переменные определены в настоящем изобретении.
, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, другие переменные определены в настоящем изобретении.
[0031] В некоторых вариантах осуществления настоящего изобретения D5, D6, D7 и D8, описанные выше, соответственно выбраны из одинарной связи, -CH2-, -O-, -NH-, 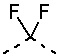 ,
,  ,
,  и
и  , и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, другие переменные определены в настоящем изобретении.
, и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, другие переменные определены в настоящем изобретении.
[0032] В некоторых вариантах осуществления настоящего изобретения фрагмент 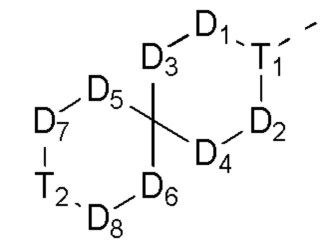 выбран из
выбран из 
 и
и 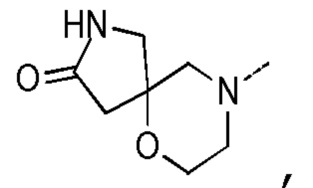 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0033] В некоторых вариантах осуществления настоящего изобретения фрагмент  выбран из
выбран из 
 другие переменные определены в настоящем изобретении.
другие переменные определены в настоящем изобретении.
[0034] В некоторых вариантах осуществления настоящего изобретения соединение, его фармацевтически приемлемая соль или его изомер выбраны из 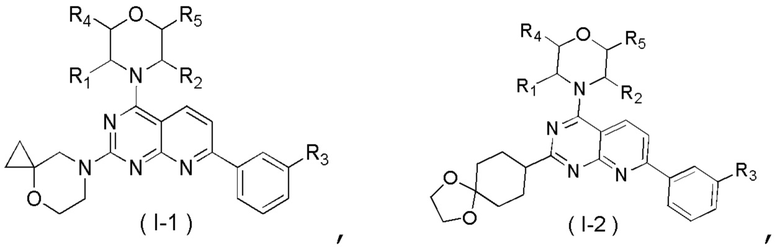
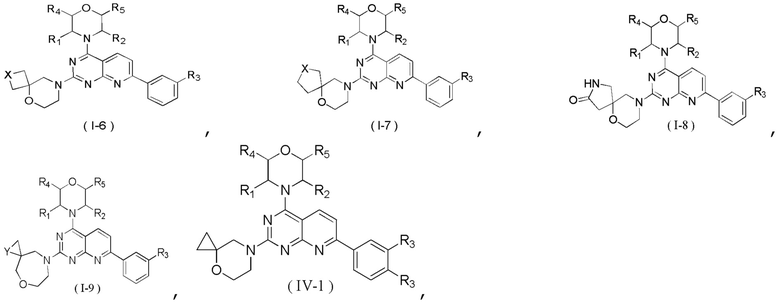
где R1, R2, R3, R4 и R5 являются такими, как определено выше, и X выбран из -CH2-, -NH-, -O-,  и -S-.
и -S-.
[0035] Настоящее изобретение также характеризуется некоторыми вариантами осуществления, полученными за счет любой комбинации переменных, описанных выше.
[0036] Настоящее изобретение также предусматривает соединение, показанное ниже, его фармацевтически приемлемую соль или его изомер:
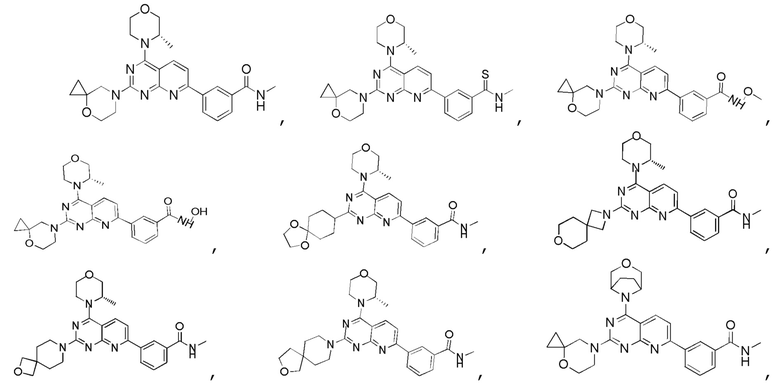
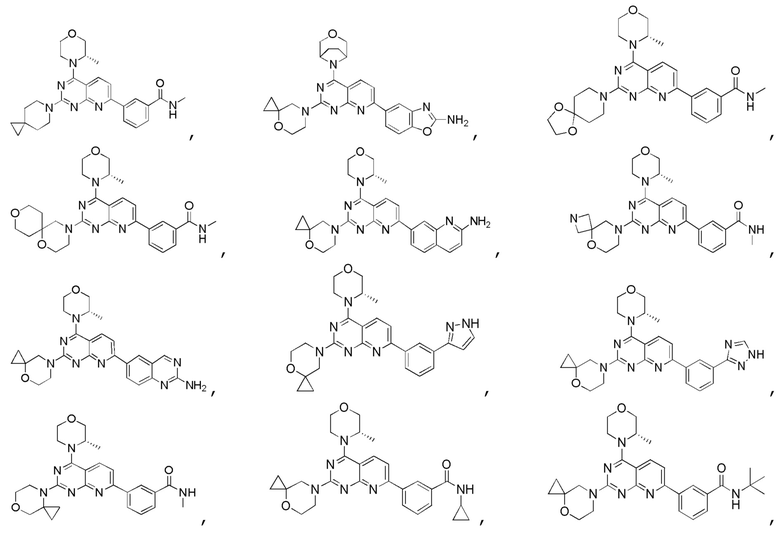
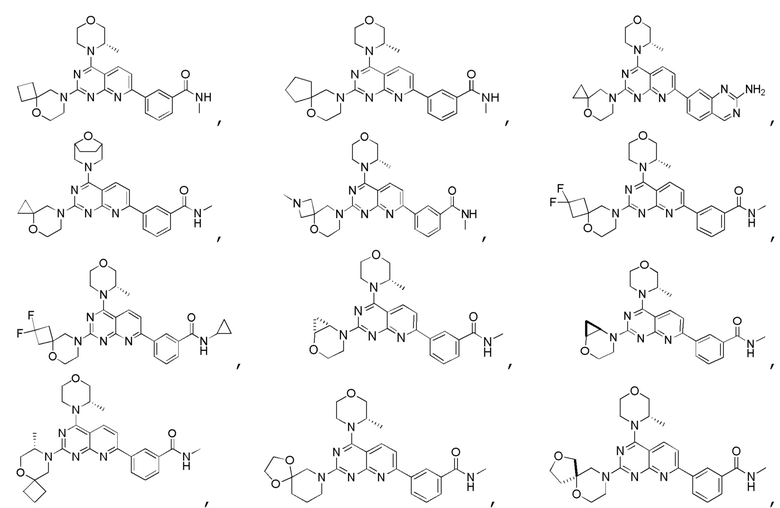
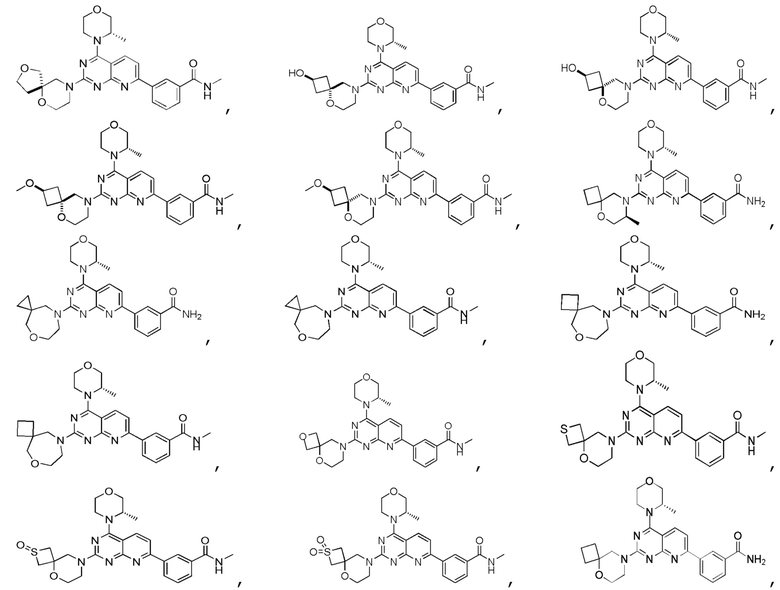
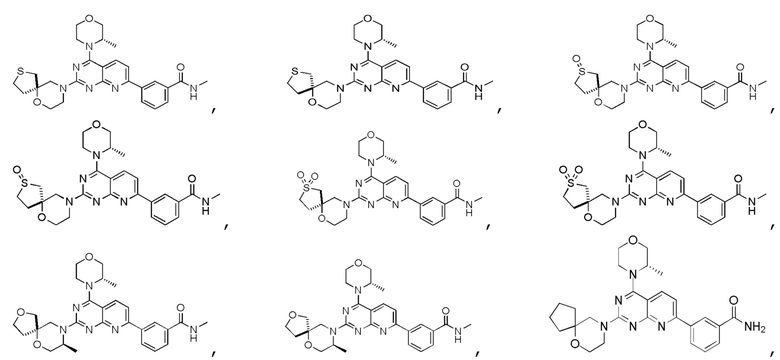
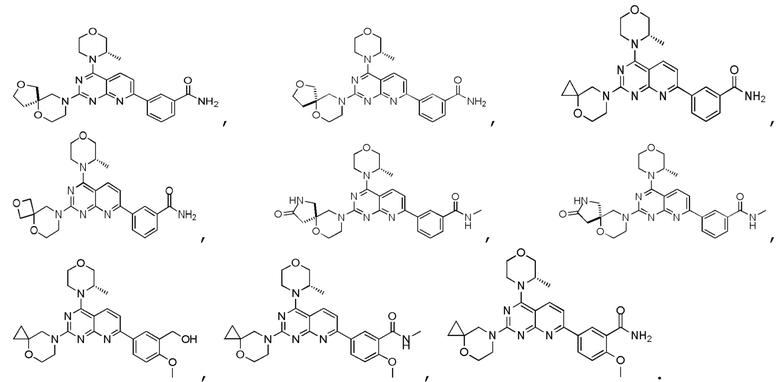
[0037] Настоящее изобретение также предусматривает фармацевтическую композицию, которая содержит терапевтически эффективное количество соединения, его фармацевтически приемлемой соли или его изомера, описанных выше, в качестве активного ингредиента и фармацевтически приемлемый носитель.
[0038] Настоящее изобретение также предусматривает применение соединения, его фармацевтически приемлемой соли или его изомера, описанных выше, или фармацевтической композиции, описанной выше, в изготовлении лекарственного препарата, предназначенного для лечения заболеваний, связанных с двумя комплексами mTORC1/2.
[0039] Настоящее изобретение также предусматривает применение соединения, его фармацевтически приемлемой соли или его изомера, описанных выше, или фармацевтической композиции, описанной выше, в изготовлении лекарственного препарата для лечения рака молочной железы, рака молочной железы, рака головы и шеи и колоректального рака.
Технический эффект
[0040] Соединения по настоящему изобретению тестировали на предмет активности в отношении киназы mTORC1/2, и данные показывают, что соединения по настоящему изобретению обладают значительной или даже неожиданной ингибирующей активностью в отношении киназы mTOR, которая превосходит таковую у известного клинического соединения AZD2014.
[0041] Соединения по настоящему изобретению обладают очевидной ингибирующей активностью в отношении пролиферации клеток MCF-7, N87 и OE-21 и обладают определенной ингибирующей активностью в отношении пролиферации клеток HT-29.
[0042] Результаты PK показывают, что соединения по настоящему изобретению характеризуются биодоступностью, составляющей примерно 100%, и являются превосходными пригодными для разработки молекулами для перорального введения.
[0043] В модели, полученной путем трансплантации опухоли из клеток MCF-7, некоторые соединения обладают такой же эффективностью, что и AZD2014. Соединения по настоящему изобретению потенциально могут стать ингибиторами различных опухолей.
Определения и описание
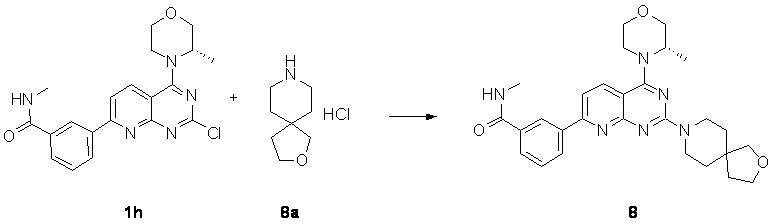
[0044] Если не указано иное, предполагается, что следующие термины и выражения, используемые в данном документе, имеют следующие значения. Конкретный термин или выражение при отсутствии точного определения не следует считать неопределенными или неясными, а следует понимать в соответствии с общепринятым значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту. Термин «фармацевтически приемлемый» используется в данном документе применительно к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках тщательной медицинской оценки являются подходящими для применения в контакте с тканями человека и животного без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с обоснованным соотношением польза/риск.
[0045] Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают путем осуществления реакции соединения, содержащего конкретный заместитель по настоящему изобретению, с относительно нетоксичными кислотой или основанием. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, то соль присоединения основания может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина или магния, или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, то соль присоединения кислоты может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и т. п.; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту, метансульфоновую кислоту и т. п.; и соль аминокислоты (такой как аргинин и т. п.), и соль органической кислоты, такой как глюкуроновая кислота и т. п. Некоторые конкретные соединения по настоящему изобретению, которые содержат как основные, так и кислотные функциональные группы, могут быть превращены в любую соль присоединения основания или кислоты.
[0046] Фармацевтически приемлемая соль по настоящему изобретению может быть получена из исходного соединения, которое содержит кислотный или основный фрагмент, с помощью общепринятого химического способа. Как правило, такая соль может быть получена путем осуществления реакции свободной кислотной или основной формы соединения со стехиометрическим количеством соответствующих основания или кислоты в воде, или в органическом растворителе, или в их смеси.
[0047] Соединения по настоящему изобретению могут находиться в конкретных геометрической или стереоизомерной формах. В настоящем изобретении подразумеваются все такие соединения, в том числе цис- и транс-изомеры, (–)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, а также рацемическая и другие их смеси, такие как энантиомерно или диастереоизомерно обогащенные смеси, все из которых охватываются объемом настоящего изобретения. Дополнительные асимметрические атомы углерода могут присутствовать в таких заместителях, как алкил. Все такие изомеры и их смеси включены в объем настоящего изобретения.
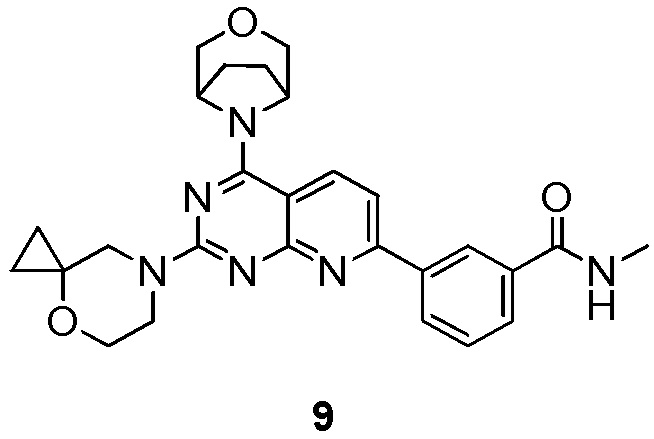
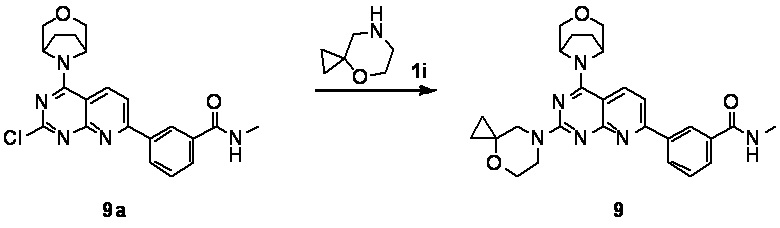
[0048] Если не указано иное, термины «энантиомеры» или «оптические изомеры» относятся к стереоизомерам, которые являются зеркальными отражениями друг друга.
[0049] Если не указано иное, термины «цис/транс-изомер» или «геометрический изомер» определяются неспособностью к свободному вращению вокруг двойной связи или одинарной связи между атомами углерода, образующими кольцо.
[0050] Если не указано иное, термин «диастереомер» относится к стереоизомеру, в молекуле которого имеется два или более хиральных центров, и молекулы не являются зеркальным отражением друг друга.
[0051] Если не указано иное, связь, обозначенная клиновидной сплошной линией ( ), и связь, обозначенная клиновидной пунктирной линией (
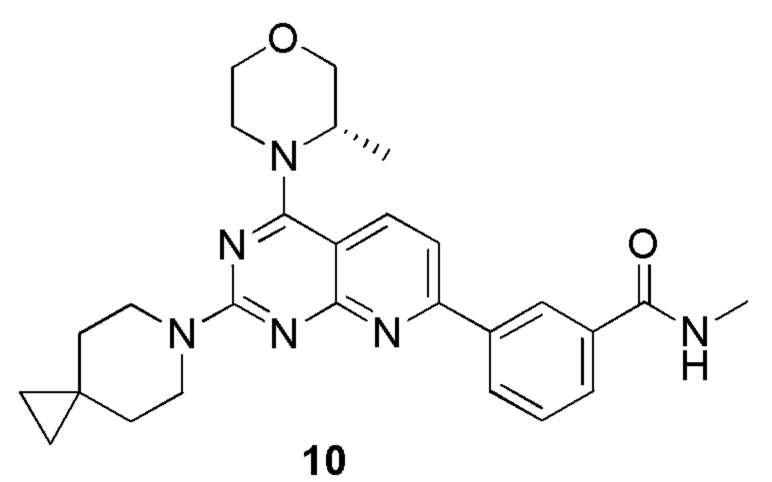
), и связь, обозначенная клиновидной пунктирной линией ( ), представляют абсолютную конфигурацию стереоцентра; связь, обозначенная прямой сплошной линией (
), представляют абсолютную конфигурацию стереоцентра; связь, обозначенная прямой сплошной линией ( ), и связь, обозначенная прямой пунктирной линией (
), и связь, обозначенная прямой пунктирной линией ( ), представляют относительную конфигурацию стереоцентра; волнистая линия (
), представляют относительную конфигурацию стереоцентра; волнистая линия ( ) представляет связь, обозначенную клиновидной сплошной линией (), или связь, обозначенную клиновидной пунктирной линией (), или волнистая линия () представляет связь, обозначенную прямой сплошной линией (), или связь, обозначенную прямой пунктирной линией ().
) представляет связь, обозначенную клиновидной сплошной линией (), или связь, обозначенную клиновидной пунктирной линией (), или волнистая линия () представляет связь, обозначенную прямой сплошной линией (), или связь, обозначенную прямой пунктирной линией ().
[0052] Соединения по настоящему изобретению могут находиться в конкретной форме. Если не указано иное, термин «таутомер» или «таутомерная форма» означает, что при комнатной температуре изомеры с разными функциональными группами находятся в состоянии динамического равновесия и могут быстро превращаться друг в друга. Если возможно наличие таутомеров (например, в растворе), то может быть достигнуто состояние химического равновесия таутомеров. Например, протонные таутомеры (также известные как прототропные таутомеры) предусматривают взаимопревращения посредством миграции протона, такие как кето-енольная изомеризация и имино-енаминовая изомеризация. Валентный таутомер предусматривает взаимное превращение, происходящее в результате рекомбинации некоторых связывающих электронов. Конкретным примером кето-енольной таутомеризации является взаимное превращение двух таутомеров – пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
[0053] Если не указано иное, термины «обогащенный конкретным изомером», «обогащенный изомером», «обогащенный конкретным энантиомером» или «обогащенный энантиомером» относятся к тому, что содержание изомера или энантиомера составляет меньше 100%, и при этом содержание изомера или энантиомера составляет 60% или больше, или 70% или больше, или 80% или больше, или 90% или больше, или 95% или больше, или 96% или больше, или 97% или больше, или 98% или больше, или 99% или больше, или 99,5% или больше, или 99,6% или больше, или 99,7% или больше, или 99,8% или больше, или 99,9% или больше.
[0054] Если не указано иное, термины «избыток изомера» или «избыток энантиомера» относится к разнице между значениями относительного процентного содержания двух изомеров или двух энантиомеров. Например, если содержание одного изомера или энантиомера составляет 90%, и содержание другого изомера или энантиомера составляет 10%, то избыток изомера или энантиомерный избыток (значение ee) составляет 80%.
[0055] Оптически активный (R)- и (S)-изомер или D- и L-изомер можно получить с применением хирального синтеза, или хиральных реагентов, или других общепринятых методик. Если требуется получение одного типа энантиомера конкретного соединения по настоящему изобретению, чистый необходимый энантиомер может быть получен путем асимметрического синтеза или дериватизации с помощью хирального вспомогательного вещества с последующим разделением полученной в результате диастереомерной смеси и отщеплением вспомогательной группы. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксильная), соединение реагирует с соответствующими оптически активными кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному разделению посредством общепринятого способа, известного из уровня техники, с получением чистого энантиомера. Кроме того, энантиомеры и диастереоизомеры обычно выделяют посредством хроматографии, в которой используется хиральная неподвижная фаза, и необязательно в комбинации со способом химической дериватизации (например, карбамат, полученный из амина). Соединения по настоящему изобретению могут содержать неприродные соотношения атомных изотопов при одном или более атомах, составляющих соединение. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). В качестве другого примера дейтерированные лекарственные средства можно получать посредством замены водорода дейтерием. Связь между дейтерием и углеродом является более прочной, чем связь между обычным водородом и углеродом. По сравнению с недейтерированными лекарственными средствами, дейтерированные лекарственные средства обладают следующими преимуществами: менее выраженными токсическими и побочными эффектами, более высокой стабильностью лекарственных средств, усиленной эффективностью, продленным биологическим периодом полувыведения лекарственных средств и т. п. Превращения всех изотопных композиций на основе соединений по настоящему описанию, вне зависимости от того, являются ли они радиоактивными или нет, включены в объем настоящего изобретения. Термин «фармацевтически приемлемый носитель» относится к любому средству или несущей среде, которые способны доставлять эффективное количество активного вещества по настоящему изобретению, не оказывают отрицательного воздействия на биологическую активность активного вещества и не вызывают какого-либо токсичного побочного эффекта у хозяина или пациента. Иллюстративный носитель включает воду, растительное и минеральное масло, основу для крема, основу для лосьона, основу для мази и т. п. Основа содержит суспендирующее средство, загуститель, вещество, способствующее проникновению, и т. п. Их составы хорошо известны специалистам в области косметических средств или в области фармацевтических препаратов для местного применения.
[0056] «Необязательный» или «необязательно» означает, что последующее событие или условие может реализовываться, но не является необходимым, и что термин включает случаи, в которых событие или условие реализуется, и случаи, в которых событие или условие не реализуется.
[0057] Термин «замещенный» означает, что один или более атомов водорода при конкретном атоме замещены заместителем, в том числе дейтерием и вариантами водорода, при условии, что валентность конкретного атома является нормальной, и замещенное соединение является стабильным. Если заместитель представляет собой атом кислорода (т. е. =O), то это означает, что два атома водорода являются замещенными. Положения в ароматическом кольце не могут быть замещены кетоном. Термин «необязательно замещенный» означает, что атом может быть замещен или не замещен заместителем, если не указано иное, причем тип и число заместителей могут быть произвольными при условии, что это химически достижимо.
[0058] Если любая переменная (такая как R) встречается более одного раза в составе или структуре соединения, то определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то данная группа может быть необязательно замещена не более чем двумя R, при этом определение R в каждом случае является независимым. Более того, комбинация заместителя и/или его варианта является допустимой, только если такая комбинация приводит к образованию стабильного соединения.
[0059] Если число линкерных групп равно 0, например -(CRR)0-, это означает, что линкерная группа представляет собой одинарную связь.
[0060] Если одна из переменных представляет собой одинарную связь, это означает, что две группы, соединенные одинарной связью, соединены непосредственно. Например, если L в A-L-Z представляет собой одинарную связь, то структура A-L-Z фактически представляет собой A-Z.
[0061] Если заместитель не указан, это означает, что заместитель отсутствует. Например, если X не указан в A-X, то структура A-X фактически представляет собой A. Если заместитель может быть присоединен к более чем одному атому в кольце, такой заместитель может быть связан с любым атомом кольца. Например, структурное звено  или
или  означает, что заместитель R может находиться в любом положении в циклогексиле или циклогексадиене. Если в перечисленном заместителе не указано, посредством какого атома он присоединен к замещаемой группе, такой заместитель может быть связан посредством любого его атома. Например, если пиридил выполняет функцию заместителя, он может быть присоединен к замещаемой группе посредством любого атома углерода в пиридиновом кольце. Если в перечисленной линкерной группе не указано направление связывания, то направление связывания является произвольным; например, если линкерная группа L, содержащаяся в
означает, что заместитель R может находиться в любом положении в циклогексиле или циклогексадиене. Если в перечисленном заместителе не указано, посредством какого атома он присоединен к замещаемой группе, такой заместитель может быть связан посредством любого его атома. Например, если пиридил выполняет функцию заместителя, он может быть присоединен к замещаемой группе посредством любого атома углерода в пиридиновом кольце. Если в перечисленной линкерной группе не указано направление связывания, то направление связывания является произвольным; например, если линкерная группа L, содержащаяся в 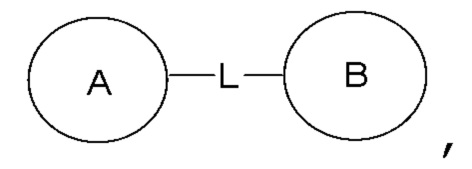 представляет собой –M-W, то –M-W- может связывать кольцо A и кольцо B с образованием
представляет собой –M-W, то –M-W- может связывать кольцо A и кольцо B с образованием 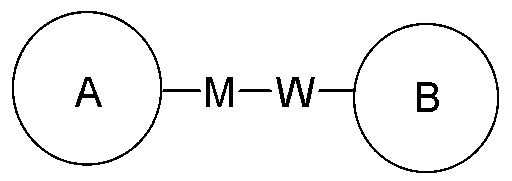 в направлении, соответствующем порядку чтения слева направо, и с образованием
в направлении, соответствующем порядку чтения слева направо, и с образованием 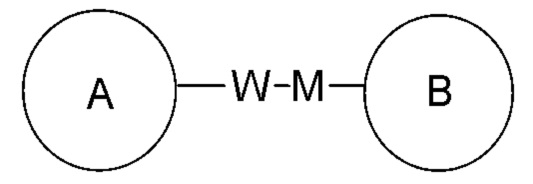 в направлении, противоположном порядку чтения слева направо. Комбинация линкерной группы, заместителя и/или их вариантов является допустимой, только если такая комбинация может приводить к стабильному соединению.
в направлении, противоположном порядку чтения слева направо. Комбинация линкерной группы, заместителя и/или их вариантов является допустимой, только если такая комбинация может приводить к стабильному соединению.
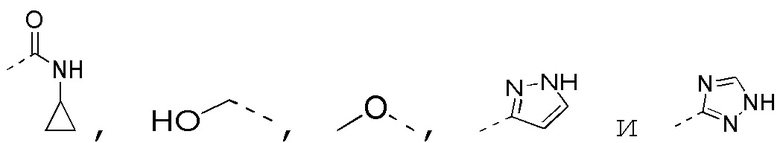
[0062] Если не указано иное, термин «гетероцикло» относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или гетероатомную группу, которое является насыщенным и содержит атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, соответственно выбранных из N, O и S. Гетероатомы азота и серы необязательно могут являться окисленными (т. е. NO и S(O)p, p равняется 1 или 2). Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероцикл может быть присоединен к боковой группе посредством любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное в результате соединение является стабильным, гетероцикл, описанный в данном документе, может иметь замещение в положении, соответствующему атому углерода или азота. Атом азота в гетероцикле необязательно кватернизирован. В предпочтительном варианте осуществления, если общее число атомов S и O в гетероцикле превышает 1, то гетероатомы не являются смежными друг с другом. В другом предпочтительном варианте осуществления общее число атомов S и O в гетероцикле не превышает 1. Используемый в данном документе термин «гетероарил» относится к стабильному 5-, 6- или 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклическому ароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, соответственно выбранных из N, O и S. Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероатомы, представляющие собой азот и серу, необязательно могут быть окислены (т. е. NO и S(O)p, p равняется 1 или 2). Следует отметить, что общее число атомов S и O в ароматическом гетероцикле не превышает один. Кольцо с мостиковой связью образуется, если один или более атомов (т. е. C, O, N или S) соединяют два несмежных атома углерода или азота. Предпочтительное кольцо с мостиковой связью включает без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостиковая связь всегда превращает моноциклическое кольцо в трициклическое кольцо. В кольце с мостиковой связью заместитель в кольце также может присутствовать при мостиковой связи.
[0063] Если не указано иное, термин «5-6-членный гетероциклоалкил» сам по себе или в комбинации с другими терминами, относится к насыщенной циклической группе, состоящей из 5-6 атомов кольца соответственно, из которых 1, 2, 3 или 4 атома кольца независимо выбраны из гетероатомов O, S и N, остальные представляют собой атомы углерода, где атом азота необязательно кватернизирован, и гетероатомы, представляющие собой азот и серу, необязательно окислены (т. е. NO и S(O)P, p равняется 1 или 2). Он включает как моноциклическую, так и бициклическую системы, где бициклическая система включает спирокольцо, конденсированное кольцо или кольцо с мостиковой связью. Кроме того, в случае «5-6-членного гетероциклоалкила», гетероатом может занимать положение присоединения гетероциклоалкила к остальной части молекулы. 5-6-Членный гетероциклоалкил включает 5-членный и 6-членный гетероциклоалкил. Примеры 5-6-членного гетероциклоалкила включают без ограничения пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиофенил (в том числе тетрагидротиофен-2-ил, тетрагидротиофен-3-ил и т. д.), тетрагидрофуранил (в том числе тетрагидрофуран-2-ил и т. д.), тетрагидропиранил, пиперидинил (в том числе 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т. д.), пиперазинил (в том числе 1-пиперазинил, 2-пиперазинил и т. д.), морфолинил (в том числе 3-морфолинил, 4-морфолинил и т. д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил или гомопиперидинил и т. п.
[0064] Если не указано иное, термины «6-10-членное гетероарильное кольцо» и «6-10-членный гетероарил» по настоящему изобретению можно применять взаимозаменяемо. Термин «6-10-членный гетероарил» относится к циклической группе, состоящей из 6-10 атомов кольца с сопряженной системой π-электронов, где 1, 2, 3 или 4 атома кольца являются гетероатомами, соответственно выбранными из O, S и N, остальные представляют собой атомы углерода. Это может быть моноциклическая, конденсированная бициклическая или конденсированная трициклическая система, где каждое кольцо является ароматическим. Атом азота необязательно кватернизирован, а гетероатомы, представляющие собой атомы азота и серы, необязательно окислены (т. е. NO и S(O)P, p равняется 1 или 2). 6-10-Членный гетероарил может быть присоединен к остальной части молекулы посредством гетероатома или атома углерода. 6-10-Членный гетероарил включает 6-8-членный, 6-7-членный, 6-9-членный, 6-членный и 10-членный гетероарил. Примеры 6-10-членного гетероарила включают без ограничения фуранил (в том числе 2-фуранил, 3-фуранил и т. д.), пиридил (в том числе 2-пиридил, 3-пиридил, 4-пиридил и т. д.), пиразинил, пиримидинил (в том числе 2-пиримидинил, 4-пиримидинил и т. д.), бензтиазолил (в том числе 5-бензотиазолил и т. д.), пуринил, бензимидазолил (в том числе 2-бензимидазолил и т. д.), бензоксазолил, индолил (в том числе 5-индолил и т. д.), изохинолинил (в том числе 1-изохинолинил, 5-изохинолинил и т. д.), хиноксалинил (в том числе 2-хиноксалинил, 5-хиноксалинил и т. д.) или хинолинил (в том числе 3-хинолинил, 6-хинолинил и т. д.).
[0065] Если не указано иное, термины «5-6-членное гетероарильное кольцо» и «5-6-членный гетероарил» по настоящему изобретению можно применять взаимозаменяемо. Термин «5-6-членный гетероарил» относится к циклической группе, состоящей из 5-6 атомов кольца с сопряженной системой π-электронов, где 1, 2, 3 или 4 атома кольца являются гетероатомами, соответственно выбранными из O, S и N, остальные представляют собой атомы углерода. Атом азота необязательно кватернизирован, а гетероатомы, представляющие собой атомы азота и серы, необязательно окислены (т. е. NO и S(O)P, p равняется 1 или 2). 5-6-Членный гетероарил может быть присоединен к остальной части молекулы посредством гетероатома или атома углерода. 5-6-Членный гетероарил включает 5-членный и 6-членный гетероарил. Примеры 5-6-членного гетероарила включают без ограничения пирролил (в том числе N-пирролил, 2-пирролил, 3-пирролил и т. д.), пиразолил (в том числе 2-пиразолил, 3-пиразолил и т. д.), имидазолил (в том числе N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил и т. д.), оксазолил (в том числе 2-оксазолил, 4-оксазолил, 5-оксазолил и т. д.), триазолил (1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил, 4H-1,2,4-триазолил и т. д.), тетразолил, изоксазолил (3-изоксазолил, 4-изоксазолил, 5-изоксазолил и т. д.), тиазолил (в том числе 2-тиазолил, 4-тиазолил, 5-тиазолил и т. д.), фуранил (в том числе 2-фуранил, 3-фуранил и т. д.), тиенил (в том числе 2-тиенил, 3-тиенил и т. д.), пиридил (в том числе 2-пиридил, 3-пиридил, 4-пиридил и т. д.), пиразинил или пиримидил (в том числе 2-пиримидил, 4-пиримидил и т. д.).
[0066] Если не указано иное, «C3-6циклоалкил» относится к насыщенному циклическому гидрокарбилу, состоящему из 3-6 атомов углерода, который представляет собой моноциклическую и бициклическую систему, и C3-6циклоалкил предусматривает C3-5-, C4-5-, C5-6циклоалкил и т. д.; при этом он может являться одновалентным, двухвалентным или поливалентным. Примеры C3-6циклоалкила включают без ограничения циклопропил, циклобутил, циклопентил, циклогексил и т. п.
[0067] В некоторых вариантах осуществления термин «гетероалкил» сам по себе или в комбинации с другим термином относится к стабильной линейной цепи, разветвленному углеводородному радикалу или их комбинации, которые содержат конкретное число атомов углерода и по меньшей мере один гетероатом. В конкретном варианте осуществления гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены, и гетероатом, представляющий собой атом азота, необязательно кватернизирован. Гетероатом или гетероатомная группа могут находиться в любом внутреннем положении гетерогидрокарбила, в том числе в положении, где гидрокарбил соединяется с остальной частью молекулы. Однако термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкокси) применяют в соответствии с их обычными значениями, и они относятся к алкильной группе, которая присоединена к остальной части молекулы посредством атома кислорода, аминогруппы или атома серы соответственно. Примеры включают без ограничения -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3 и -CH2-CH2-S(O)2-CH3. Могут присутствовать не более двух последовательно расположенных гетероатомов, например -CH2-NH-OCH3.
[0068] Если не указано иное, термин «алкил» относится к линейной цепи или разветвленной насыщенной углеводородной группе, которая может быть однозамещенной (например, -CH2F) или полизамещенной (например, -CF3), может быть одновалентной (например, метил), двухвалентной (например, метилен) или многовалентной (например, метенил). Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и т. п.
[0069] Если не указано иное, термин «C1-3алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-3 атомов углерода. C1-3алкил включает C1-2- и C2-3алкил, и пр.; при этом он может являться одновалентным (таким как метил), двухвалентным (таким как метилен) или многовалентным (таким как метин). Примеры C1-3алкила включают без ограничения метил (Me), этил (Et), пропил (в том числе н-пропил и изопропил) и т. п.
[0070] Если не указано иное, термин «C1-3алкокси» относится к такому алкилу, который содержит 1-3 атома углерода, присоединенные к остальной части молекулы посредством одного атома кислорода. C1-3алкоксигруппа включает C1-2-, C2-3-, C3- и C2алкокси. Примеры C1-3алкокси включают без ограничения метокси, этокси, пропокси (в том числе н-пропокси и изопропокси) и т. п.
[0071] Если не указано иное, циклоалкил включает любой стабильный циклический или полициклический гидрокарбил, и любой атом углерода, который является насыщенным, может быть однозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным. Примеры циклоалкила включают без ограничения циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодеканил и т. п.
[0072] Если не указано иное, термины «галогено» или «галоген», сами по себе или как часть другого заместителя, относятся к атому фтора, хлора, брома или йода. Кроме того, подразумевается, что термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин «галоген(C1-C4)алкил» включает без ограничения трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т. п. Примеры галогеналкила включают без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
[0073] Термин «алкокси» означает алкил, определенный выше, содержащий конкретное число атомов углерода, присоединенных посредством кислородного мостика. Если не указано иное, C1-6алкокси включает C1-, C2-, C3-, C4-, C5- и C6алкокси. Примеры алкокси включают без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси.
[0074] Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому заместителю, который может быть моно-, ди- или полизамещенным, может быть одновалентным, двухвалентным или многовалентным, может представлять собой одно кольцо или несколько колец (например, от одного до трех колец; где по меньшей мере одно кольцо является ароматическим), которые являются конденсированными совместно или соединенными ковалентно. Термин «гетероарил» относится к арилу (или кольцу), содержащему от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбран из B, N, O и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизирован. Гетероарил может быть присоединен к остальной части молекулы посредством гетероатома. Неограничивающие примеры арила или гетероарила включают фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенилоксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидинил, бензотиазолил, пуринил, бензимидазолил, индолил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель в любой из указанных выше арильных и гетероарильных кольцевых систем выбран из приемлемых заместителей, описанных ниже.
[0075] Если не указано иное, Cn-n+m или Cn-Cn+m включает любой конкретный случай в отношении от n до n+m атомов углерода, например, C1-12 включает C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12, также включает любой диапазон от n до n+m, например, C1-12 включает C1-3, C1-6, C1-9, C3-6, C3-9, C3-12, C6-9, C6-12, C9-12 и т. д.; подобным образом «от n до n+m» означает, что число атомов в кольце составляет от n до n+m, например, 3-12-членные кольца включают 3-членное кольцо, 4-членное кольцо, 5-членное кольцо, 6-членное кольцо, 7-членное кольцо, 8-членное кольцо, 9-членное кольцо, 10-членное кольцо, 11-членное кольцо и 12-членное кольцо, также включает любой диапазон от n до n+m, например, 3-12-членные кольца включают 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо, 6-10-членное кольцо и т. п.
[0076] Соединения по настоящему изобретению можно получать посредством различных способов синтеза, хорошо известных специалистам в данной области техники, в том числе посредством конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, образованных посредством объединения конкретных вариантов осуществления, перечисленных ниже, с другими способами химического синтеза, и эквивалентных вариантов осуществления, хорошо известных специалистам в данной области техники, при этом предпочтительные варианты осуществления включают без ограничения варианты осуществления настоящего изобретения.
[0077] Соединения по настоящему изобретению могут иметь различные применения или предназначения, в том числе без ограничения конкретные применения или предназначения, перечисленные в данной заявке.
[0078] Используемый в настоящем изобретении растворитель является коммерчески доступным.
[0079] Каждый вариант осуществления настоящего изобретения включает нейтральную очистку для очистки посредством высокоэффективной жидкостной хроматографии.
[0080] В настоящем изобретении используются следующие сокращения: водн. означает водный; HATU означает O-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC означает N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид; m-CPBA означает 3-хлорпероксибензойную кислоту; экв. означает эквивалентный или эквивалент; CDI означает карбонилдиимидазол; DCM означает дихлорметан; PE означает петролейный эфир; DIAD означает диизопропилазодикарбоксилат; DMF означает N,N-диметилформамид; DMSO означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; MeOH означает метанол; CBz означает бензилоксикарбонил, который является защитной группой для аминогруппы; BOC означает трет-бутилкарбонил, который является защитной группой для аминогруппы; HOAc означает уксусную кислоту; NaCNBH3 означает цианоборогидрид натрия; к. т. означает комнатную температуру; O/N означает в течение ночи; THF означает тетрагидрофуран; Boc2O означает ди-трет-бутилдикарбонат; TFA означает трифторуксусную кислоту; DIPEA означает диизопропилэтиламин; SOCl2 означает тионилхлорид; CS2 означает сероуглерод; TsOH означает п-толуолсульфоновую кислоту; NFSI означает N-фтор-N-(фенилсульфонил)бензолсульфонамид; n-Bu4NF означает тетрабутиламмония фторид; iPrOH означает 2-пропанол; т. пл. означает точку плавления; LDA означает диизопропиламид лития; Pd(PPh3)4 означает тетракис(трифенилфосфин)палладий; IV означает внутривенную инъекцию; PO означает пероральное введение.
[0081] Названия соединениям даны в соответствии с традиционными принципами номенклатуры в данной области техники или с помощью программного обеспечения ChemDraw®, для коммерчески доступных соединений используются их названия в соответствии с каталогом поставщика.
Подробное описание предпочтительного варианта осуществления
[0082] Настоящее изобретение будет конкретно описано ниже с помощью вариантов осуществления, но объем настоящего изобретения ими не ограничивается. В данном документе было подробно описано настоящее изобретение и в данном документе был раскрыт вариант осуществления настоящего изобретения. В отношении варианта осуществления настоящего изобретения могут быть выполнены различные модификации и изменения без отступления от сущности и объема настоящего изобретения, которые будут очевидны для специалиста в данной области техники.
[0083] Вариант осуществления 1
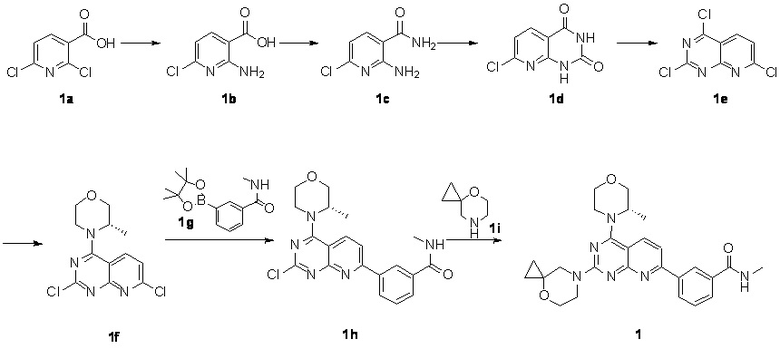
[0084] Первая стадия
[0085] Соединение 1a (20,0 г, 104 ммоль, 1,00 экв.) и концентрированный гидроксид аммония (200 мл, 1,45 моль, 14,0 экв.) закрывали в автоклаве и перемешивали при 130°C в течение 24 часов при давлении приблизительно 0,9 МПа. Реакционный раствор концентрировали с получением соединения 1b.
[0086] MS-ESI: расчетное значение [M+H]+: 173 и 175, найденное значение: 173 и 175.
[0087] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,03 (d, J=8,0 Гц, 1H), 7,56 (br s, 2H), 6,61 (d, J=8,0 Гц, 1H).
[0088] Вторая стадия
[0089] Соединение 1b (17,0 г, 98,5 ммоль, 1,00 экв.), хлорид аммония (10,5 г, 197 ммоль, 2,00 экв.), 1-гидроксибензотриазол (13,3 г, 98,5 ммоль, 1,00 экв.), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (18,9 г, 98,5 ммоль, 1,00 экв.) и диизопропилэтиламин (38,2 г, 296 ммоль, 3,00 экв.) растворяли в N,N-диметилформамиде (200,0 мл). Смесь перемешивали при 20°C в течение 16 часов. После завершения реакции растворитель удаляли путем выпаривания на роторном испарителе при пониженном давлении с последующим добавлением воды (200 мл) и экстрагированием этилацетатом (200 мл × 3). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и подвергали колоночной хроматографии (1:1 петролейный эфир/этилацетат, Rf = 0,4) с получением соединения, которое суспендировали с помощью этилацетата (50 мл) в течение десяти минут с получением соединения 1c.
[0090] 1H ЯМР (400 MГц, DMSO-d6) δ: 7,96 (d, J=8,0 Гц, 2H), 7,62 (br s, 2H), 7,40 (br s, 1H), 6,61 (d, J=8,0 Гц, 1H).
[0091] Третья стадия
[0092] Соединение 1c (8,00 г, 46,6 ммоль, 1,00 экв.) и оксалилхлорид (7,1 г, 56,0 ммоль, 4,9 мл, 1,00 экв.) последовательно добавляли к толуолу (200 мл). Смесь перемешивали при 110°C в течение 15 часов, затем охлаждали до комнатной температуры, фильтровали и высушивали с получением соединения 1d.
[0093] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,24 (d, J=8,0 Гц, 1H), 7,30 (d, J=8,0 Гц, 1H).
[0094] Четвертая стадия
[0095] Соединение 1d (6,00 г, 30,4 ммоль, 1,00 экв.) и диизопропилэтиламин (11,8 г, 91,1 ммоль, 15,9 мл, 3,00 экв.) последовательно добавляли к толуолу (100 мл). Смесь перемешивали при 70°C в течение получаса и охлаждали до комнатной температуры. В полученную смесь по каплям добавляли оксихлорид фосфора (14,0 г, 91,1 ммоль, 8,5 мл, 3,00 экв.). Смесь перемешивали при 100°C в течение 2 часов, охлаждали до комнатной температуры, концентрировали и очищали посредством колоночной хроматографии (3:1 петролейный эфир/этилацетат, Rf = 0,4) с получением соединения 1e.
[0096] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,45 (d, J=8,0 Гц, 1H), 7,63 (d, J=8,3 Гц, 1H).
[0097] Пятая стадия
[0098] Соединение 1e (1,90 г, 8,10 ммоль, 1,00 экв.), (S)-2-метилморфолин (819 мг, 8,10 ммоль, 1,00 экв.) и диизопропилэтиламин (2,09 г, 16,2 ммоль, 2,83 мл, 2,00 экв.) растворяли в дихлорметане (50 мл) и обеспечивали осуществление реакции в полученном в результате растворе при 25°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали и подвергали колоночной хроматографии (3:1 петролейный эфир/этилацетат) с получением соединения 1f.
[0099] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,47 (d, J=8,8 Гц, 1H), 7,55 (d, J=8,8 Гц, 1H), 4,71-4,72 (m, 1H), 4,12-4,09 (m, 1H), 3,92-3,91 (m, 1H), 3,84-3,74 (m, 1H), 3,73-3,64 (m, 2H), 3,54-3,53 (m, 1H), 1,46 (d, J=6,8 Гц, 3H).
[0100] Шестая стадия
[0101] Соединение 1f (1,2 г, 4,01 ммоль, 1,00 экв.), соединение 1g (1,15 г, 4,41 ммоль, 1,10 экв.), тетракис(трифенилфосфин)палладий (232 мг, 200 мкмоль, 0,05 экв.) и карбонат калия (1,66 г, 12,0 ммоль, 3,00 экв.) растворяли в воде (24 мл) и 1,4-диоксане (120 мл) и обеспечивали осуществление реакции в смеси при 60°C в течение 5 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (30 мл) и проводили экстрагирование с помощью этилацетата (50 мл × 2). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (100% этилацетат) с получением соединения 1h.
[0102] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,71 (s, 1H), 8,67 (d, J=4,8 Гц, 1H), 8,55 (d, J=8,8 Гц, 1H), 8,39 (d, J=8,0 Гц, 1H), 8,14 (d, J=8,8 Гц, 1H), 8,01 (d, J=8,0 Гц, 1H), 7,68 (t, J=7,6 Гц, 1H), 4,75 (d, J=6,4 Гц, 1H), 4,17-4,15 (m, 1H), 3,94-3,92 (m, 1H), 3,87-3,77 (m, 1H), 3,72 (s, 2H), 3,59-3,57 (m, 1H), 2,86-2,84 (m, 3H), 1,49 (d, J=6,8 Гц, 3H).
[0103] Седьмая стадия
[0104] Соединение 1h (50 мг, 126 мкмоль, 1 экв.), 1i (18,8 мг, 126 мкмоль, 1 экв.) и DIPEA (16,2 мг, 126 мкмоль, 21,89 мкл, 1 экв.) растворяли в DMSO (3 мл). Обеспечивали осуществление реакции в реакционном растворе при 70°C в течение 20 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 1.
[0105] MS-ESI: расчетное значение [M+H]+: 475, найденное значение: 475.
[0106] 1H ЯМР (400 MГц, CD3OD) δ: 8,62 (s, 1H), 8,32-8,30 (m, 2H), 7,96 (d, J=7,2 Гц, 1H), 7,72-7,61 (m, 2H), 4,52 (br s, 1H), 4,09-3,92 (m, 6H), 3,91-3,82(m, 3H), 3,76-3,74 (m, 3H), 2,99 (s, 3H), 1,49 (d, J=6,8 Гц, 3H), 0,81 (s, 2H), 0,71 (s, 2H).
[0107] Вариант осуществления 2
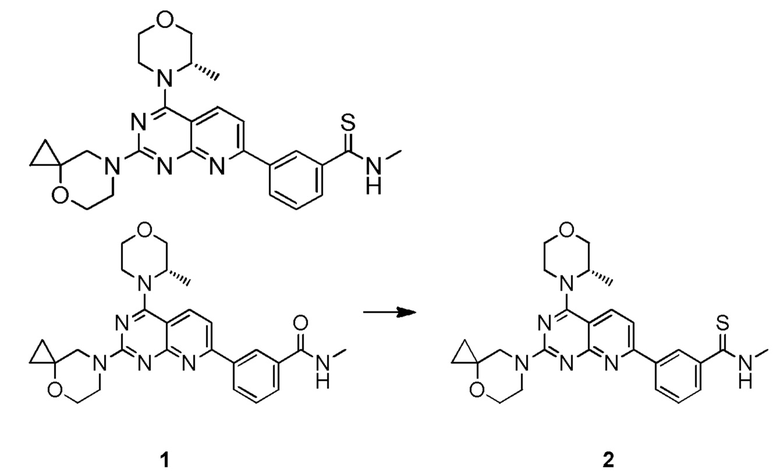
[0108] Соединение 1 (40 мг, 84,3 мкмоль, 1,00 экв.) растворяли в ксилоле (2 мл) при 130°C и затем к вышеуказанному реакционному раствору порциями добавляли реагент Лоуссона (37,5 мг, 92,7 мкмоль, 1,10 экв.). Три раза проводили замену азота и затем обеспечивали осуществление реакции при 130°C в защитной атмосфере азота в течение 17 часов. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении с удалением растворителя. К неочищенному продукту добавляли воду (10 мл) и проводили экстрагирование с помощью дихлорметана (10 мл × 3). Органические фазы объединяли и высушивали с помощью безводного сульфата натрия, фильтровали и подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния. Остаток разделяли посредством препаративной тонкослойной хроматографии (100% этилацетат). Неочищенный продукт разделяли посредством препаративной жидкостной хроматографии с получением соединения 2.
[0109] MS-ESI: расчетное значение [M+H]+: 491, найденное значение: 491.
[0110] 1H ЯМР (400 МГц, CDCl3) δ: 8,47 (s, 1H), 8,15 (s, 1H), 8,05 (d, J=7,8 Гц, 1H), 8,02-7,95 (m, 2H), 7,47-7,36 (m, 2H), 4,30 (s, 1H), 4,04-3,83 (m, 5H), 3,80-3,78 (m, 4H), 3,73-3,60 (m, 3H), 3,33 (d, J=4,8 Гц, 3H), 1,41 (d, J=6,8 Гц, 3H), 0,80-0,74 (m, 2H), 0,60-0,59 (m, 2H).
[0111] Вариант осуществления 3
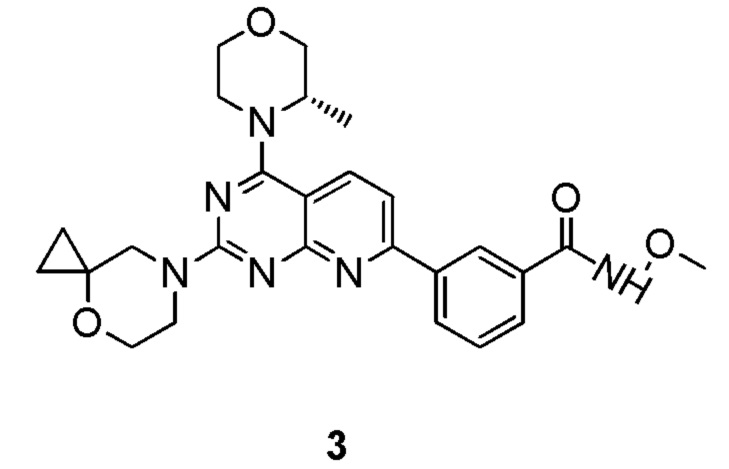
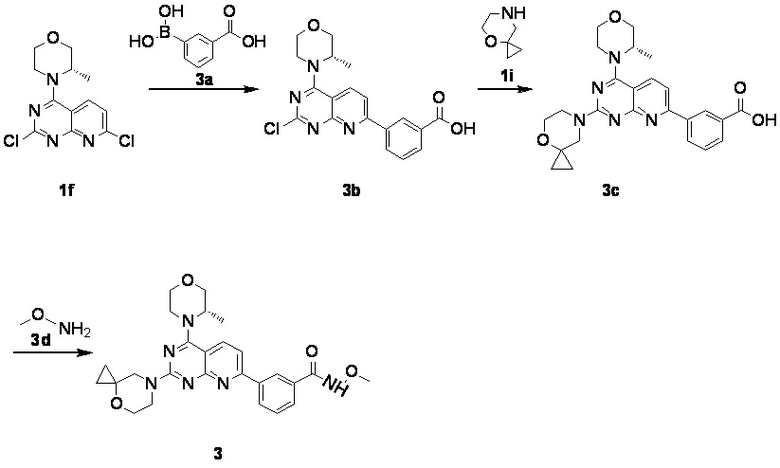
[0112] Первая стадия
[0113] Соединение 1f (0,5 г, 1,37 ммоль, 1 экв.), 3a (246 мг, 1,37 ммоль, 1 экв.), тетракис(трифенилфосфин)палладий (79,2 мг, 68,53 мкмоль, 0,05 экв.) и безводный карбонат натрия (436 мг, 4,11 ммоль, 3 экв.) растворяли в диоксане (6,0 мл) и воде (2,0 мл). Три раза проводили замену азота. Обеспечивали осуществление реакции в защитной атмосфере азота при 70°C в течение 3 часов. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении с удалением растворителя. Оставшееся твердое вещество разбавляли водой (20 мл) и этилацетатом (10 мл × 3), фильтровали и разделяли. Полученные органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе до сухого состояния и очищали посредством колоночной хроматографии (3:1 петролейный эфир/этилацетат) с получением соединения 3b.
[0114] Вторая стадия
[0115] Соединение 3b (180 мг, 375 мкмоль, 1 экв.), 1i (56,0 мг, 375 мкмоль, 1 экв., HCl) и DIPEA (145 мг, 1,12 ммоль, 196 мкл, 3 экв.) растворяли в DMSO (3 мл) и затем обеспечивали осуществление реакции в смеси при 70°C в течение 15 часов. После завершения реакции 3c непосредственно использовали в реакции на следующей стадии без обработки.
[0116] Третья стадия
[0117] Соединение 3c (30,0 мг, 65,0 мкмоль, 1 мл, 1 экв.), 3d (10,9 мг, 130 мкмоль, 2 экв., HCl), DIPEA (25,2 мг, 195 мкмоль, 34,0 мкл, 3 экв.) и HATU (49,4 мг, 130 мкмоль, 2 экв.) растворяли в DMSO (2 мл) и затем обеспечивали осуществление реакции в смеси при 27°C в течение 20 часов. После завершения реакции реакционный раствор подвергали высокоэффективной жидкостной хроматографии с получением соединения 3.
[0118] MS-ESI: расчетное значение [M+H]+: 491, найденное значение: 491.
[0119] 1H ЯМР: (400 MГц, CDCl3) δ: 9,29 (s, 1H), 8,49 (s, 1H), 8,13 (br d, J=7,6 Гц, 1H), 7,98 (d, J=8,4 Гц, 1H), 7,8 (d, J=7,6 Гц, 1H), 7,49 (t, J=7,6 Гц, 1H), 7,37-7,46 (m, 1H), 4,22-4,37 (m, 1H), 3,89-4,06 (m, 3H), 3,75-3,89 (m, 9H), 3,60-3,73 (m, 3H), 1,41 (d, J=6,4 Гц, 3H), 0,74-0,81 (m, 2H), 0,61 (br s, 2H).
Вариант осуществления 4
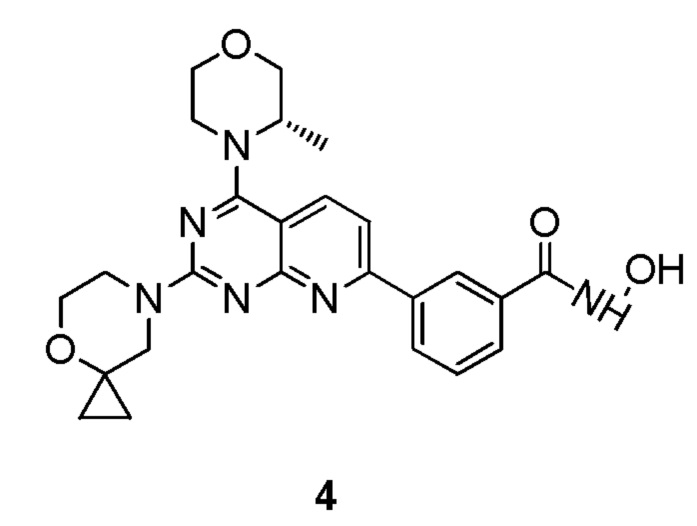
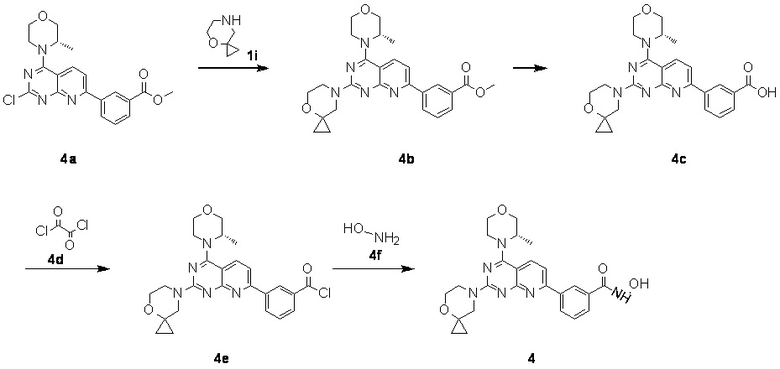
[0120] Первая стадия
[0121] Соединение 4a (180 мг, 375 мкмоль, 1 экв.), 1i (56,0 мг, 375 мкмоль, 1 экв., HCl) и DIPEA (145 мг, 1,12 ммоль, 196 мкл, 3 экв.) растворяли в DMSO (3 мл) и затем обеспечивали осуществление реакции в смеси при 70°C в течение 15 часов. После завершения реакции получали соединение 4b, которое непосредственно использовали в реакции на следующей стадии.
[0122] MS-ESI: расчетное значение [M+H]+: 476, найденное значение: 476.
[0123] Вторая стадия
[0124] Соединение 4b (178 мг, 374,31 мкмоль, 1 экв.) и LiOH (23,56 мг, 561,47 мкмоль, 1,5 экв.) растворяли в диметилсульфоксиде (3 мл) и обеспечивали осуществление реакции в смеси при комнатной температуре в течение 24 часов. Посредством обнаружения с помощью LC-MS определили, что соединение 3 не было получено. В реакционный раствор добавляли гидроксид натрия (29,95 мг, 748,63 мкмоль, 2 экв.) и обеспечивали продолжение реакции в течение 20 часов. После завершения реакции полученную в результате смесь непосредственно очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 4c.
[0125] Третья стадия
[0126] Соединение 4c (20 мг, 42,9 мкмоль, 1 экв.), 4d (5,99 мг, 47,2 мкмоль, 4,13 мкл, 1,1 экв.) и DMF (314 мкг, 4,29 мкмоль, 0,33 мкл, 0,1 экв.) растворяли в дихлорметане (2 мл) и обеспечивали осуществление реакции в полученном растворе при комнатной температуре в течение часа. После завершения реакции получали соединение 4e, которое непосредственно использовали в реакции на следующей стадии без обработки.
[0127] Четвертая стадия
[0128] Соединение 4f (60 мг, 125,01 мкмоль, 1 экв.) добавляли к раствору 4e (86,9 мг, 1,25 ммоль, 10 экв., HCl) и DIPEA (194 мг, 1,50 ммоль, 261 мкл, 12 экв.) в дихлорметане (2 мл), затем обеспечивали осуществление реакции в смеси при комнатной температуре в течение 2 часов. Полученный в результате раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 4.
[0129] MS-ESI: расчетное значение [M+H]+: 477, найденное значение: 477.
[0130] 1H ЯМР (400 MГц, CD3OD) δ: 8,57 (s, 1H), 8,34-8,31 (m, 2H), 7,90 (d, J=7,6 Гц, 1H), 7,57-7,78 (m, 2H), 4,62-4,60 (m, 3H), 4,04-4,02 (m, 2H), 4,01-3,92 (m, 3H), 3,87-3,85 (m, 3H), 3,78-3,76 (m, 2H), 1,51 (d, J=6,8 Гц, 3H), 0,93-0,61 (m, 4H).
[0131] Вариант осуществления 5
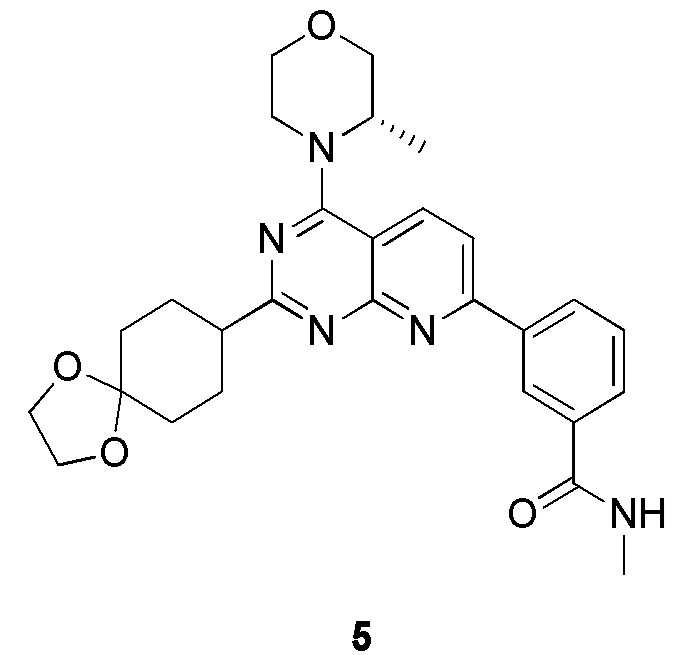
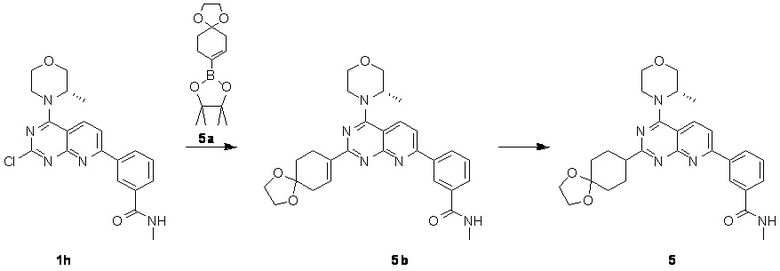
[0132] Первая стадия
[0133] 1h (300 мг, 754 мкмоль, 1,00 экв.), 5a (301 мг, 1,13 ммоль, 1,50 экв.), тетракис(трифенилфосфин)палладий (43,6 мг, 37,7 мкмоль, 0,05 экв.) и карбонат натрия (240 мг, 2,26 ммоль, 3,00 экв.) растворяли в воде (3 мл) и 1,4-диоксане (10 мл). Обеспечивали осуществление реакции при 90°C в течение 16 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (10 мл) и проводили экстрагирование с помощью этилацетата (20 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (100% этилацетат, Rf=0,4) с получением желтого твердого вещества (200 мг) с чистотой 63,8% и выходом 27%. 40 мг полученного разделяли и очищали посредством препаративной высокоэффективной жидкостной хроматографии с получением соединения 5b.
[0134] MS-ESI: расчетное значение [M+H]+: 502, найденное значение: 502.
[0135] 1H ЯМР (400 MГц, CD3OD) δ: 8,70 (s, 1H), 8,44 (d, J = 8,8 Гц, 1H), 8,41 (br d, J = 8,0 Гц, 1H), 8,02-7,92 (m, 2H), 7,66 (t, J = 8,0 Гц, 1H), 7,35-7,25 (m, 1H), 4,68-4,66 (m, 1H), 4,21-4,210 (m, 1H), 4,05-3,96 (m, 5H), 3,90-3,72 (m, 4H), 3,00 (s, 3H), 2,92 (br s, 2H), 2,56 (s, 2H), 1,94 (t, J = 6,4 Гц, 2H), 1,53 (d, J = 6,4 Гц, 3H).
[0136] Вторая стадия
[0137] К раствору 5b (80 мг, 160 мкмоль, 1,00 экв.) в метаноле (10 мл) добавляли палладий на угле (10 мг, содержание 10%, влажность: 50%) и три раза проводили замену водорода. Обеспечивали осуществление реакции в полученной смеси в атмосфере водорода (15 фунтов/кв. дюйм) при 20°C в течение 16 часов и фильтровали. Осадок на фильтре промывали с помощью 10 мл метанола и фильтрат концентрировали. Неочищенный продукт разделяли и очищали посредством препаративной высокоэффективной жидкостной хроматографии с получением соединения 5.
[0138] MS-ESI: расчетное значение [M+H]+: 504, найденное значение: 504.
[0139] 1H ЯМР (400 MГц, CD3OD) δ: 8,73 (s, 1H), 8,48 (d, J=8,4 Гц, 1H), 8,45 (br d, J=8,0 Гц, 1H), 8,02 (d, J=8,4 Гц, 1H), 7,98 (br d, J=8,0 Гц, 1H), 7,66 (t, J=8,0 Гц, 1H), 4,78-4,76 (m, 1H), 4,26-4,24 (m, 1H), 4,05-3,94 (m, 5H), 3,89-3,70 (m, 4H), 3,00 (s, 3H), 2,94-2,82 (m, 1H), 2,12-1,99 (m, 4H), 1,92-1,90 (m, 2H), 1,78-1,67 (m, 2H), 1,57 (d, J=6,8 Гц, 3H).
[0140] Вариант осуществления 6
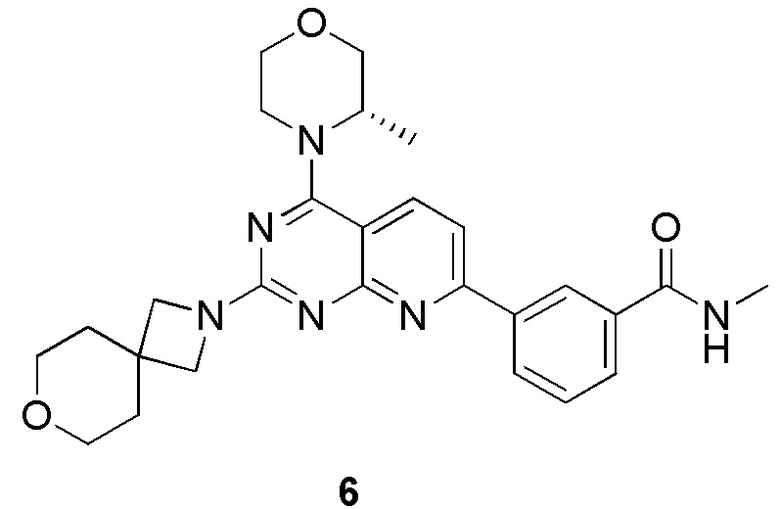
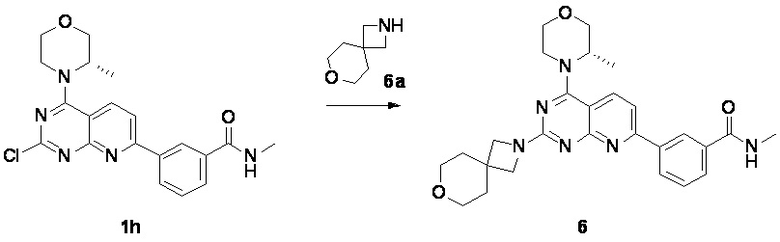
[0141] Первая стадия
[0142] Соединение 1h (50,0 мг, 126 мкмоль, 1,00 экв.), соединение 6a (32,5 мг, 189 мкмоль, 1,50 экв., 0,5 эквивалента оксалата) и N,N-диизопропилэтиламин (48,7 мг, 377 мкмоль, 3,00 экв.) растворяли в диметилсульфоксиде (1,00 мл) и затем реакционный раствор перемешивали при 70°C в течение 16 часов. После завершения реакции реакционный раствор очищали посредством препаративной высокоэффективной жидкостной хроматографии с получением соединения 6.
[0143] MS-ESI: расчетное значение [M+H]+: 489, найденное значение: 489.
[0144] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,64 (s, 2H), 8,31 (d, J=7,6 Гц, 1H), 8,24 (d, J=8,4 Гц, 1H), 7,95 (d, J=7,6 Гц, 1H), 7,69 (d, J=8,4 Гц, 1H), 7,62 (t, J=7,6 Гц, 1H), 4,47-4,45 (m, 1H), 3,95-3,82 (m, 6H), 3,79-3,70 (m, 1H), 3,69-3,50 (m, 7H), 2,84 (d, J=4,8 Гц, 3H), 1,76 (br t, J=4,8 Гц, 4H), 1,38 (d, J=6,8 Гц, 3H).
[0145] Вариант осуществления 7
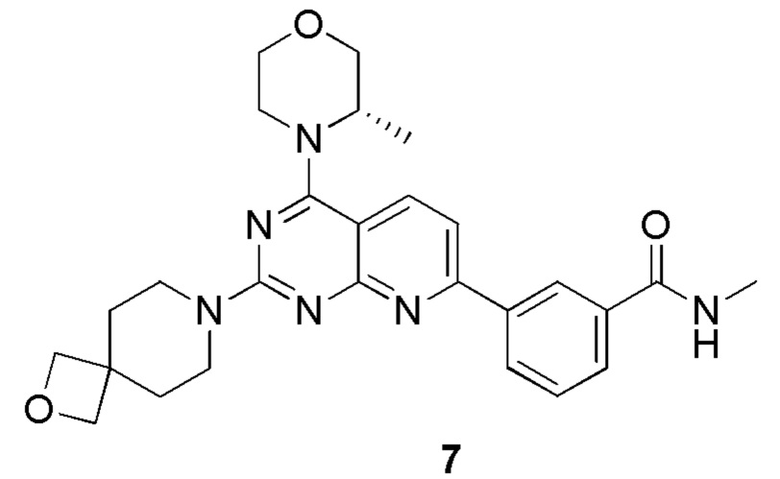
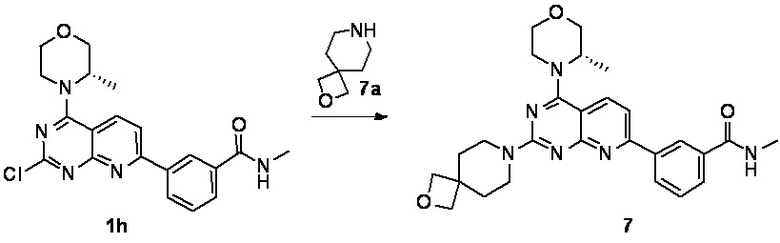
[0146] Соединение 1h (50,0 мг, 126 мкмоль, 1,00 экв.), соединение 7a (32,5 мг, 189 мкмоль, 1,50 экв., 0,5 эквивалента оксалата) и диизопропилэтиламин (48,7 мг, 377 мкмоль, 3,00 экв.) растворяли в диметилсульфоксиде (1,00 мл). Реакционный раствор затем перемешивали при 70°C в течение 16 часов. После завершения реакции реакционный раствор очищали посредством препаративной высокоэффективной жидкостной хроматографии с получением соединения 7.
[0147] MS-ESI: расчетное значение [M+H]+: 489, найденное значение: 489.
[0148] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,70-8,60 (m, 2H), 8,30 (br d, J=8,0 Гц, 1H), 8,22 (d, J=8,4 Гц, 1H), 7,95 (d, J=8,0 Гц, 1H), 7,68 (d, J=8,4 Гц, 1H), 7,62 (t, J=8,0 Гц, 1H), 4,47-4,27 (m, 5H), 3,96-3,56 (m, 10H), 2,84 (d, J=4,4 Гц, 3H), 1,83 (br t, J=5,2 Гц, 4H), 1,37 (d, J=6,8 Гц, 3H).
[0149] Вариант осуществления 8
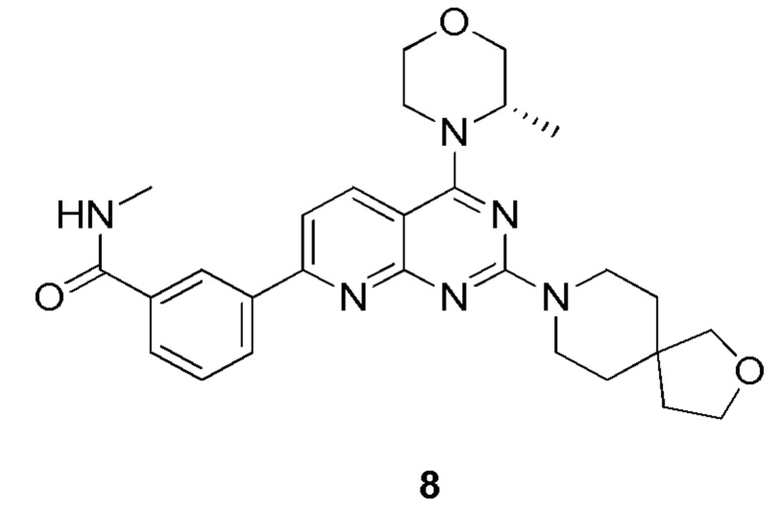
[0150] Соединение 1h (20 мг, 50,3 мкмоль, 1,00 экв.), 8a (13,4 мг, 75,4 мкмоль, 1,50 экв., HCl) и N,N-диизопропилэтиламин (19,5 мг, 151 мкмоль, 26,3 мкл, 3,00 экв.) растворяли в диметилсульфоксиде (1 мл) и обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 40 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 8.
[0151] MS-ESI: расчетное значение [M+H]+: 503, найденное значение: 503.
[0152] 1H ЯМР (400 MГц, CD3OD) δ: 8,62 (s, 1H), 8,33 (d, J=8,0 Гц, 1H), 8,26 (d, J=8,0 Гц, 1H), 7,95 (d, J=8,0 Гц, 1H), 7,67-7,61 (m, 2H), 4,51 (br d, J=7,03 Гц, 1H), 4,13-4,05 (m, 2H), 4,02-3,85 (m, 7H), 3,82-3,69 (m, 3H), 3,65 (s, 2H), 2,99 (s, 3H), 1,92 (t, J=7,2 Гц, 2H), 1,69 (t, J=5,6 Гц, 4H), 1,48 (d, J=6,8 Гц, 3H).
[0153] Вариант осуществления 9
[0154] Соединение 9a (100 мг, 175 мкмоль, 1 экв.), 1i (26,28 мг, 175,67 мкмоль, 1 экв., HCl) и DIPEA (68,1 мг, 527 мкмоль, 91,8 мкл, 3 экв.) растворяли в DMSO (5 мл) и затем обеспечивали осуществление реакции в смеси при 70°C в течение 18 часов. После завершения реакции реакционный раствор разбавляли водой (10 мл), затем экстрагировали с помощью этилацетата (15 мл × 5) и разделяли. Объединенные органические фазы высушивали над безводным сульфатом натрия, фильтровали и подвергали выпариванию на роторном испарителе до сухого состояния. Остаток очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 9.
[0155] MS-ESI: расчетное значение [M+H]+: 487, найденное значение: 487.
[0156] 1H ЯМР (400 МГц, CDCl3) δ: 8,63 (s, 1H), 8,22 (d, J=8,0 Гц, 1H), 8,14 (d, J=8,0 Гц, 1H), 7,98 (d, J=8,0 Гц, 1H), 7,50-7,61 (m, 2H), 6,56 (s, 1H), 4,59 (s, 2H), 3,69-4,23 (m, 10H), 3,07 (d, J=4,8 Гц, 3H), 2,06-2,20 (m, 2H), 1,98-2,06 (m, 2H), 0,90(s, 2H), 0,69 (s, 2H).
[0157] Вариант осуществления 10
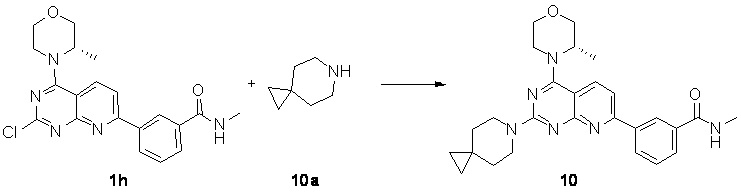
[0158] Соединение 1h (50,0 мг, 126 мкмоль, 1,00 экв.), 10a (20,9 мг, 188 мкмоль, 1,50 экв.) и триэтиламин (38,2 мг, 377 мкмоль, 52,3 мкл, 3,00 экв.) растворяли в диметилсульфоксиде (1,00 мл). Три раза проводили замену азота и реакционный раствор перемешивали при 70°C в течение 12 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 10.
[0159] MS-ESI: расчетное значение [M+H]+: 473, найденное значение: 473.
[0160] 1H ЯМР (400 MГц, CD3OD) δ: 8,61 (s, 1H), 8,32 (d, J=8,0 Гц, 1H), 8,23 (d, J=8,4 Гц, 1H), 7,95 (d, J=8,0 Гц, 1H), 7,59-7,66 (m, 2H), 4,49-4,47 (m, 1H), 4,00-4,07 (m, 4H), 4,00-3,92 (m, 2H), 3,89-3,87 (m, 1H), 3,82-3,67 (m, 3H), 2,99 (s, 3H), 1,49-1,46 (m, 7H), 0,43 (s, 4H).
[0161] Вариант осуществления 11
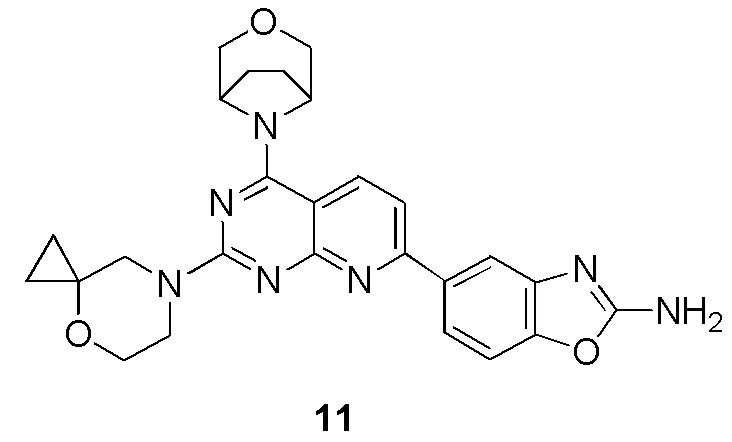
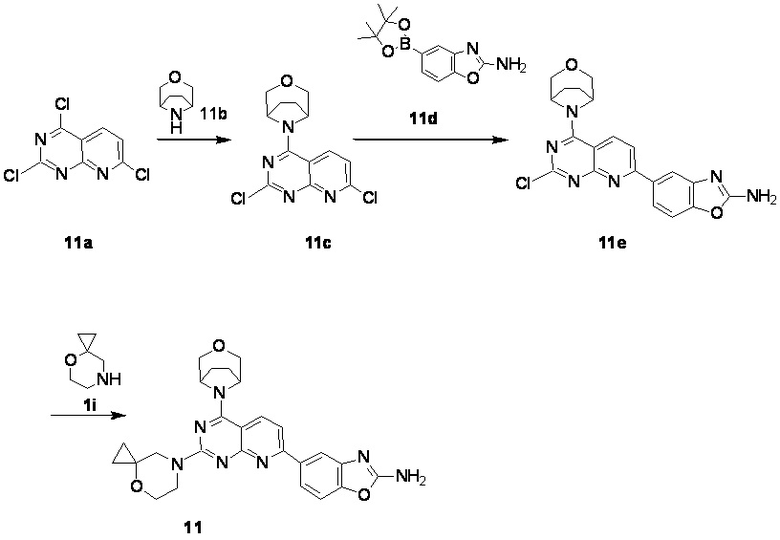
[0162] Первая стадия
[0163] Соединение 11a (1,0 г, 3,41 ммоль, 1 экв.) и 11b (510 мг, 3,41 ммоль, 1 экв., HCl) растворяли в безводном дихлорметане (80 мл) с последующим добавлением DIPEA (441 мг, 3,41 ммоль, 594 мкл, 1 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 18 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и остаток на фильтре очищали посредством препаративной тонкослойной хроматографии (1:1 петролейный эфир/этилацетат) с получением соединения 11c.
[0164] MS-ESI: расчетное значение [M+H]+: 311, 312 и 313, найденное значение: 311, 312 и 313.
[0165] 1H ЯМР (400 MГц, CDCl3) δ: 8,14 (d, J=8,8 Гц, 1H), 7,36 (d, J=8,8 Гц, 1H), 4,84 (s, 2H), 3,97 (d, J=11,2 Гц, 2H), 3,83 (d, J=11,2 Гц, 2H), 2,24-2,16 (m, 2H), 2,10-2,01 (m, 2H).
Вторая стадия
[0166] Соединение 11c (200 мг, 643 мкмоль, 1 экв.), 11d (167 мг, 643 мкмоль, 1 экв.), K2CO3 (266 мг, 1,93 ммоль, 3 экв.) и Pd (PPh3)4 (37,1 мг, 32,1 мкмоль, 0,05 экв.) растворяли в безводном диоксане (30 мл) и воде (6 мл). Три раза проводили замену азота и обеспечивали осуществление реакции в смешанном растворе при 90°C в течение 2 часов в атмосфере азота. После завершения реакции реакционный раствор концентрировали при пониженном давлении, затем добавляли в него 20 мл воды и проводили экстрагирование с помощью этилацетата (20 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали, фильтрат концентрировали при пониженном давлении и остаток на фильтре очищали посредством хроматографии на пластине (100% этилацетат) с получением соединения 11e.
[0167] MS-ESI: расчетное значение [M+H]+: 409 и 411, найденное значение: 409 и 411.
[0168] Третья стадия
[0169] Соединение 11e (120 мг, 294 мкмоль, 1 экв.), 1i (33,2 мг, 222 мкмоль, HCl) и DIPEA (37,9 мг, 294 мкмоль, 51,1 мкл, 1 экв.) растворяли в DMSO (6 мл) и обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 17 часов в атмосфере азота. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 11.
[0170] MS-ESI: расчетное значение [M+H]+: 486, найденное значение: 486.
[0171] 1H ЯМР (400 MГц, CD3OD) δ: 8,38 (d, J=8,4 Гц, 1H), 8,24 (br s, 1H), 8,03 (d, J=1,2 Гц, 1H), 7,94-7,89 (m, 1H), 7,71 (d, J=8,4 Гц, 1H), 7,43 (d, J=8,4 Гц, 1H), 4,83 (s, 1H), 4,06-3,97 (m, 4H), 3,94 (s, 2H), 3,87 (t, J=4,8 Гц, 2H), 3,80 (br d, J=10,4 Гц, 2H), 2,17-2,00 (m, 4H), 0,86-0,80 (m, 2H), 0,76-0,70 (m, 2H).
[0172] Вариант осуществления 12
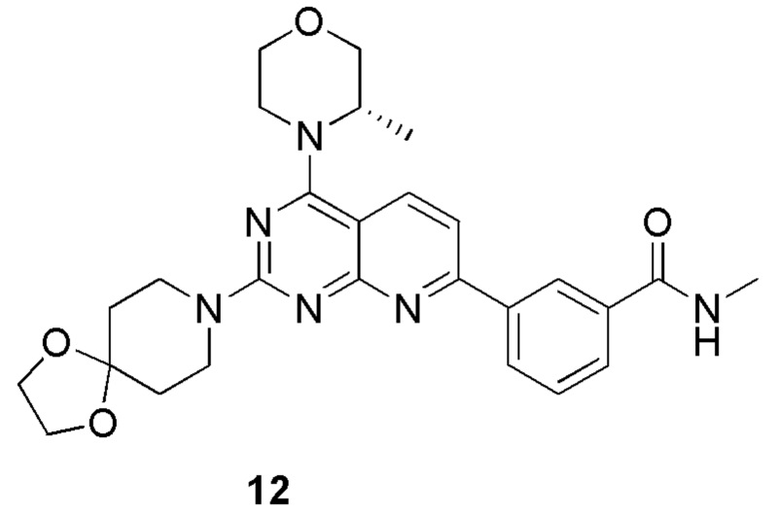
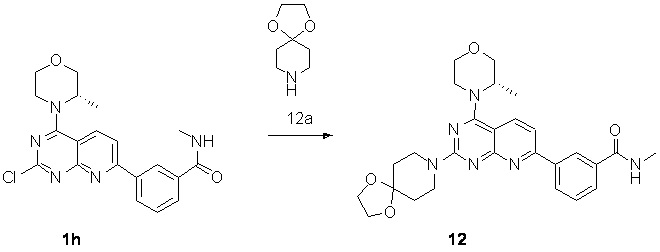
[0173] Соединение 1h (80 мг, 201 мкмоль, 1 экв.), 12a (28,8 мг, 201 мкмоль, 25,7 мкл, 1 экв.) и диизопропилэтиламин (26,0 мг, 201 мкмоль, 35,0 мкл, 1 экв.) растворяли в диметилсульфоксиде (4,00 мл), обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 1,5 часа. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 12.
[0174] MS-ESI: расчетное значение [M+H]+: 505, найденное значение: 505.
[0175] 1H ЯМР (400 MГц, CD3OD) δ: 8,62-8,60 (m, 1H), 8,31 (d, J=8,0 Гц, 1H), 8,24 (d, J=8,4 Гц, 1H), 7,95 (d, J=8,0 Гц, 1H), 7,65-7,60 (m, 2H), 4,50 (br d, J=6,8 Гц, 1H), 4,09-4,02 (m, 8H), 4,00-3,93 (m, 2H), 3,90-3,84 (m, 1H), 3,81-3,68 (m, 3H), 2,99 (s, 3H), 1,80-1,74 (m, 4H), 1,48 (d, J=6,8 Гц, 3H).
[0176] Вариант осуществления 13
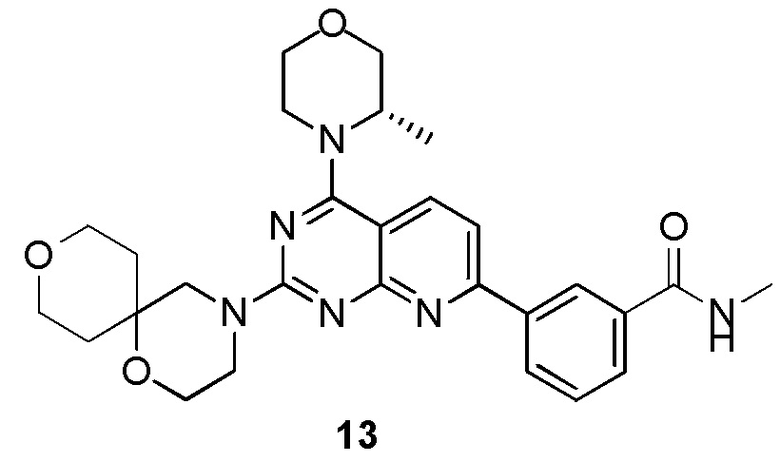
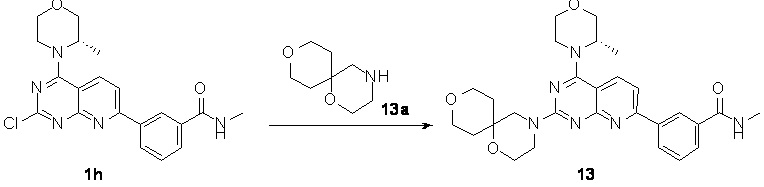
[0177] Соединение 1h (50,0 мг, 126 мкмоль, 1 экв.), 13a (19,8 мг, 126 мкмоль, 1 экв.) и N,N-диизопропилэтиламин (48,7 мг, 377 мкмоль, 65,7 мкл, 3 экв.) растворяли в диметилсульфоксиде (2 мл) и обеспечивали осуществление реакции в смеси при 70°C в течение 16 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 13.
[0178] MS-ESI: расчетное значение [M+H]: 519, найденное значение: 519.
[0179] 1H ЯМР (400 МГц, CDCl3) δ: 8,63 (s, 1H), 8,24 (d, J=8,00 Гц, 1H), 8,06 (d, J=8,00 Гц, 1H), 7,98 (d, J=8,00 Гц, 1H), 7,49-7,64 (m, 2H), 6,52 (br s, 1H), 4,40 (br d, J=6,8 Гц, 1H), 4,02 (br d, J=10,8 Гц, 3H), 3,83-3,94 (m, 6H), 3,66-3,83 (m, 7H), 3,08 (d, J=4,8 Гц, 3H), 1,80-1,93 (m, 2H), 1,73 (br s, 2H), 1,50 (d, J=6,8 Гц, 3H).
[0180] Вариант осуществления 14
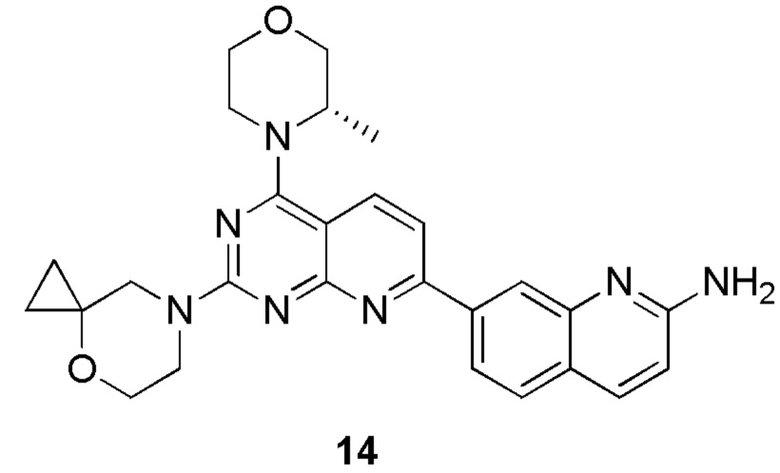
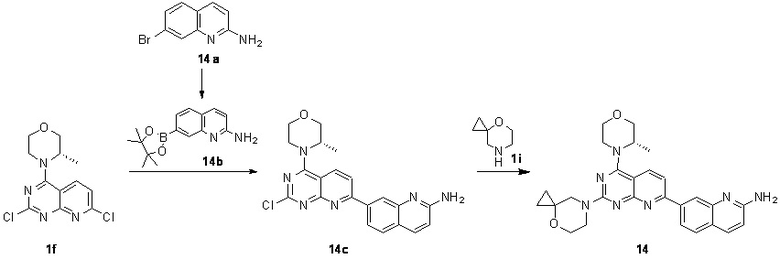
[0181] Первая стадия
[0182] Соединение 14a (0,08 г, 359 мкмоль, 1 экв.), пинаколдиборат (109 мг, 430 мкмоль, 1,2 экв.), Pd(dppf)Cl2 (10,5 мг, 14,4 мкмоль, 0,04 экв.) и ацетат калия (106 мг, 1,08 ммоль, 3 экв.) растворяли в 20 мл безводного диоксана и обеспечивали осуществление реакции в смешанном растворе при 90°C в течение 22 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с получением 14b.
[0183] MS-ESI: расчетное значение [M+H]+: 270, найденное значение: 270.
[0184] Вторая стадия
[0185] Соединение 1f (0,1 г, 334 мкмоль, 1 экв.), 14b (99,3 мг, 367 мкмоль, 1,1 экв.), тетракис(трифенилфосфин)палладий (19,3 мг, 16,7 мкмоль, 0,05 экв.) и карбонат калия (139 мг, 1,00 ммоль, 3 экв.) растворяли в безводном диоксане (20 мл) и воде (4 мл) и обеспечивали осуществление реакции в смешанном растворе в защитной атмосфере азота при 70°C в течение 3,5 часа. После завершения реакции и охлаждения реакционного раствора в реакционный раствор добавляли 10 мл воды и 60 мл (20 мл × 3) этилацетата для экстрагирования. Затем органическую фазу высушивали над безводным сульфатом натрия, концентрировали и очищали посредством колоночной хроматографии с получением 14c.
[0186] MS-ESI: расчетное значение [M+H]+: 407 и 409, найденное значение: 407 и 409.
[0187] Третья стадия
[0188] Соединение 14c (0,04 г, 93,4 мкмоль, 1 экв.), 1i (13,9 мг, 93,4 мкмоль, 1 экв., HCl) и диизопропиламин (12,1 мг, 93,4 мкмоль, 16,3 мкл, 1 экв.) растворяли в диметилсульфоксиде (3 мл). Обеспечивали осуществление реакции в реакционном растворе при 70°C в течение 17 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 14.
[0189] MS-ESI: расчетное значение [M+H]+: 484, найденное значение: 484.
[0190] 1H ЯМР (400 MГц, CD3OD) δ: 8,30 (d, J=8,4 Гц, 1H), 8,21 (s, 1H), 8,08 (dd, J=1,6, 8,4 Гц, 1H), 8,01 (d, J=9,2 Гц, 1H), 7,80 (d, J=8,4 Гц, 1H), 7,71 (d, J=8,4 Гц, 1H), 6,90 (d, J=8,8 Гц, 1H), 4,52 (br s, 1H), 4,07-3,94 (m, 6H), 3,91-3,84 (m, 3H), 3,80-3,72 (m, 3H), 1,49 (d, J=6,8 Гц, 3H), 0,84-0,79 (m, 2H), 0,72 (s, 2H).
[0191] Вариант осуществления 15
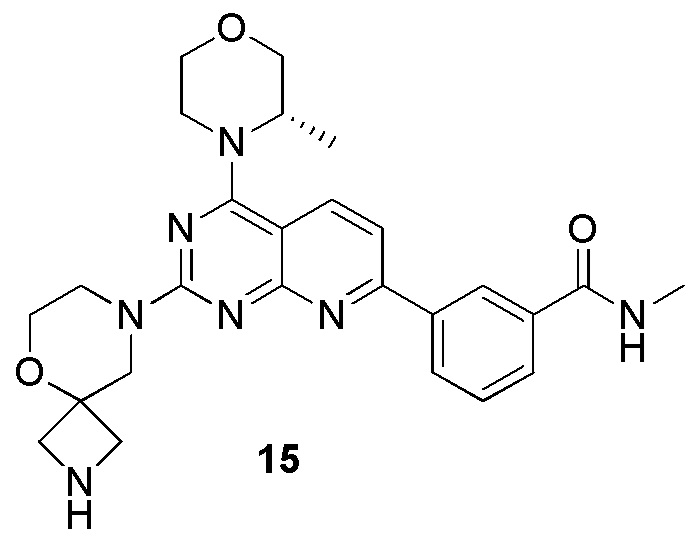
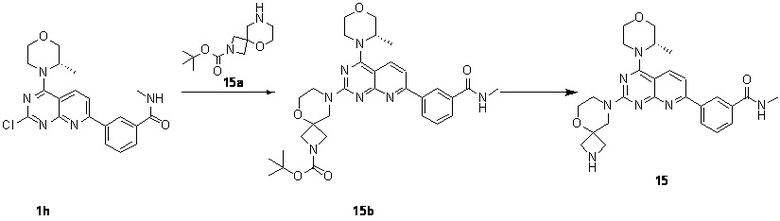
[0192] Первая стадия
[0193] Соединение 1h (70 мг, 176 мкмоль, 1 экв.), 15a (40,2 мг, 176 мкмоль, 1 экв.) и диизопропилэтиламин (22,7 мг, 176 мкмоль, 30,7 мкл, 1 экв.) растворяли в диметилсульфоксиде (5 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 17 часов. После завершения реакции и охлаждения реакционного раствора в реакционный раствор добавляли 10 мл воды и 30 мл этилацетата для экстрагирования. Затем к органической фазе добавляли воду для экстракции избыточного диметилсульфоксида. Органическую фазу высушивали над безводным сульфатом натрия, концентрировали и подвергали плоскостной хроматографии (0/1 петролейный эфир/этилацетат) с получением соединения 15b.
[0194] MS-ESI: расчетное значение [M+H]+: 590, найденное значение: 590.
[0195] Вторая стадия
[0196] Растворяли соединение 15b (100 мг, 169 мкмоль, 1 экв.) в этилацетате (3 мл). К полученному раствору добавляли хлористоводородную кислоту/этилацетат (4 M, 3 мл, 70,8 экв.) и обеспечивали осуществление реакции при 20°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали и проводили экстрагирование с помощью 10 мл воды и 45 мл этилацетата (15 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия и концентрировали. Небольшое количество реакционного раствора очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 15.
[0197] MS-ESI: расчетное значение [M+H]+: 490, найденное значение: 490.
[0198] 1H ЯМР (400 MГц, CD3OD) δ: 8,62 (s, 1H), 8,36-8,29 (m, 2H), 7,96 (d, J=7,8 Гц, 1H), 7,69 (d, J=8,4 Гц, 1H), 7,64 (t, J=7,8 Гц, 1H), 4,62 (br d, J=6,4 Гц, 1H), 4,19-3,86 (m, 7H), 3,81-3,72 (m, 5H), 3,65 (br d, J=9,2 Гц, 2H), 3,53 (br d, J=8,0 Гц, 2H), 2,99 (s, 3H), 1,51 (d, J=6,8 Гц, 3H).
[0199] Вариант осуществления 16
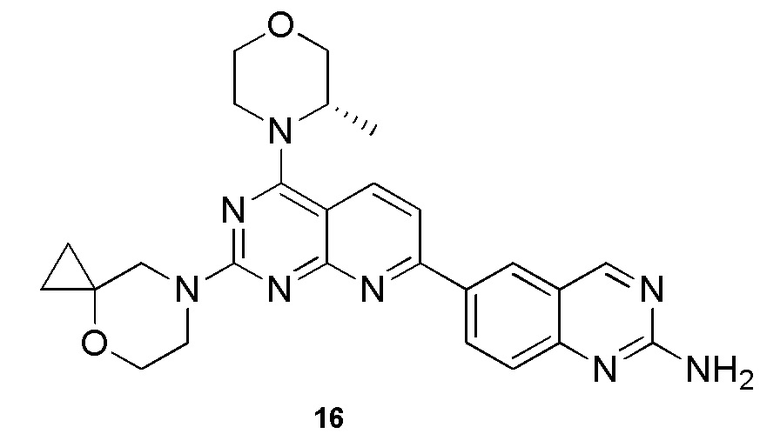
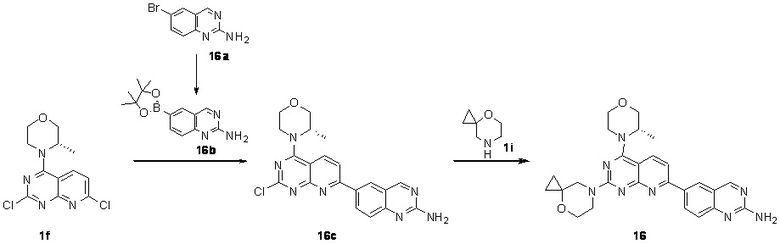
[0200] Первая стадия
[0201] Соединение 16a (0,1 г, 446 мкмоль, 1 экв.), пинаколборат (136 мг, 536 мкмоль, 1,2 экв.), Pd(dppf)Cl2 (13,1 мг, 17,9 мкмоль, 0,04 экв.) и ацетат калия (131 мг, 1,34 ммоль, 3 экв.) растворяли в 10 мл безводного диоксана и обеспечивали осуществление реакции в смешанном растворе при 90°C в течение 2,5 часа в защитной атмосфере азота. Реакцию останавливали, когда посредством масс-спектрометрии обнаружили, что было получено 30% продукта. Затем реакционный раствор концентрировали с получением соединения 16b.
[0202] MS-ESI: расчетное значение [M+H]+: 272, найденное значение: 272.
[0203] Вторая стадия
[0204] Соединение 1f (0,1 г, 334 мкмоль, 1 экв.), 16b (90,6 мг, 334 мкмоль, 1 экв.), тетракис(трифенилфосфин)палладий (19,3 мг, 16,7 мкмоль, 0,05 экв.) и карбонат калия (139 мг, 1,00 ммоль, 3 экв.) растворяли в безводном диоксане (20 мл) и воде (4 мл) и обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 15 часов в защитной атмосфере азота. После завершения реакции и охлаждения реакционного раствора в реакционный раствор добавляли 10 мл воды и 60 мл (20 мл × 3) этилацетата для экстрагирования. Затем органическую фазу высушивали над безводным сульфатом натрия, концентрировали и разделяли посредством колоночной хроматографии (метанол/этилацетат) с получением соединения 16c.
[0205] MS-ESI: расчетное значение [M+H]+: 408 и 410, найденное значение: 408 и 410.
[0206] Третья стадия
[0207] Соединение 16c (0,1 г, 153 мкмоль, 1 экв.), 1i (22,9 мг, 153 мкмоль, 1 экв., HCl) и диизопропиламин (19,7 мг, 153 мкмоль, 26,6 мкл, 1 экв.) растворяли в диметилсульфоксиде (4 мл), обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 15 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 16.
[0208] MS-ESI: расчетное значение [M+H]+: 485, найденное значение: 485.
[0209] 1H ЯМР (400 MГц, CD3OD) δ: 9,21 (s, 1H), 8,61-8,55 (m, 2H), 8,23 (d, J=8,4 Гц, 1H), 7,69 (d, J=8,4 Гц, 1H), 7,61 (d, J=9,6 Гц, 1H), 4,50 (br d, J=6,8 Гц, 1H), 4,04 (q, J=4,4 Гц, 2H), 4,00-3,92 (m, 4H), 3,90-3,84 (m, 3H), 3,80-3,69 (m, 3H), 1,48 (d, J=6,8 Гц, 3H), 0,86-0,78 (m, 2H), 0,75-0,69 (m, 2H).
[0210] Вариант осуществления 17
[0211] Первая стадия
[0212] Соединение 17a (100 мг, 448 мкмоль, 1,0 экв.), бис(пинаколато)дибор (125 мг, 493 мкмоль, 1,1 экв.), ацетат калия (132 мг, 1,34 ммоль, 3,0 экв.) и Pd(dppf)Cl2 (16,4 мг, 22,4 мкмоль, 0,05 экв.) растворяли в безводном диоксане (5 мл) с последующей вентиляцией три раза. Обеспечивали осуществление реакции в смешанном растворе при 100°C в течение 17 часов в атмосфере азота. После завершения реакции реакционный раствор непосредственно использовали на следующей стадии.
[0213] MS-ESI: расчетное значение [M+H]+: 271, найденное значение: 271.
[0214] Вторая стадия
[0215] В реакционный раствор, содержащий соединение 17b, добавляли соединение 17c (134 мг, 448 мкмоль, 1,0 экв.), тетракис(трифенилфосфин)палладий (25,9 мг, 22,4 мкмоль, 0,05 экв.) и карбонат натрия (143 мг, 1,34 ммоль, 3,0 экв.), смесь растворяли в воде (2 мл) и 1,4-диоксане (5 мл). Обеспечивали осуществление реакции при 70°C в течение 4 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (10 мл), экстрагировали с помощью этилацетата (10 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством плоскостной хроматографии (чистый этилацетат) с получением 17d.
[0216] MS-ESI: расчетное значение [M+H]+: 407 и 409, найденное значение: 407 и 409.
[0217] 1H ЯМР (400 МГц, CDCl3) δ: 8,60 (s, 1H), 8,14 (d, J=8,8 Гц, 1H), 8,10 (br d, J=7,8 Гц, 1H), 7,85 (d, J=7,8 Гц, 1H), 7,79 (d, J=8,8 Гц, 1H), 7,62 (d, J=2,0 Гц, 1H), 7,54-7,45 (m, 1H), 6,68 (d, J=2,0 Гц, 1H), 4,62 (br d, J=4,3 Гц, 1H), 4,15 (br d, J=12,5 Гц, 1H), 3,98-3,92 (m, 1H), 3,81-3,67 (m, 4H), 1,53 (d, J=6,8 Гц, 3H).
[0218] Третья стадия
[0219] Соединение 17d (30 мг, 58,3 мкмоль, 1,00 экв.), 1i (13,1 мг, 87,5 мкмоль, 1,50 экв.) и DIPEA (22,6 мг, 175 мкмоль, 3,00 экв.) растворяли в DMSO (3 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 17 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 17.
[0220] MS-ESI: расчетное значение [M+H]+: 484, найденное значение: 484.
[0221] 1H ЯМР (400 МГц, CDCl3) δ: 8,48 (s, 1H), 8,02 (d, J=8,0 Гц, 1H), 7,97 (d, J=8,3 Гц, 1H), 7,79 (br d, J=7,8 Гц, 1H), 7,59 (d, J=2,3 Гц, 1H), 7,49-7,42 (m, 2H), 6,67 (d, J=2,0 Гц, 1H), 4,30 (br s, 1H), 4,01-3,56 (m, 12H), 1,40 (d, J=6,8 Гц, 3H), 0,79-0,73 (m, 2H), 0,61 (br s, 2H).
[0222] Варианты осуществления 18-1 и 18-2
[0223] Соединение 1h (100 мг, 251 мкмоль, 1 экв.), 18a (34,1 мг, 251 мкмоль, 1 экв., HCl) и N,N-диизопропилэтиламин (97,5 мг, 754 мкмоль, 131 мкл, 3 экв.) растворяли в диметилсульфоксиде (2 мл) и обеспечивали осуществление реакции в смеси при 70°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением рацемата, 50 мг которого подвергали хиральному разделению с получением соединений 18-1 и 18-2.
[0224] MS-ESI: расчетное значение [M+H]+: 461, найденное значение: 461.
[0225] Положение пика соединения 18-1: 3,032 мин. (хиральная колонка: AD-3 150 × 4,6 мм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,5 мл/мин., температура колонки: 40°C).
[0226] ЯМР 18-1: 1H ЯМР (400 МГц, CDCl3) δ: 8,58 (br s, 1H), 7,84-8,23 (m, 3H), 7,41-7,59 (m, 2H), 6,52 (s, 1H), 4,39 (br s, 1H), 3,63-4,02 (m, 10H), 3,04-3,63 (m, 2H), 2,99 (d, J=4,8 Гц, 3H), 1,46 (s, 3H), 0,57-0,92 (m, 2H).
[0227] Положение пика соединения 18-2: 3,587 мин. (хиральная колонка: AD-3 150 × 4,6 мм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,5 мл/мин., температура колонки: 40°C).
[0228] ЯМР 18-2: 1H ЯМР (400 МГц, CDCl3) δ: 8,49-8,82 (m, 1H), 7,82-8,23 (m, 3H), 7,42-7,58 (m, 2H), 6,53 (br s, 1H), 4,39 (br s, 1H), 3,60-4,06 (m, 10H), 3,03-3,60 (m, 2H), 2,99 (d, J=4,8 Гц, 3H), 1,46 (s, 3H), 0,54-0,99 (m, 2H).
[0229] Вариант осуществления 19
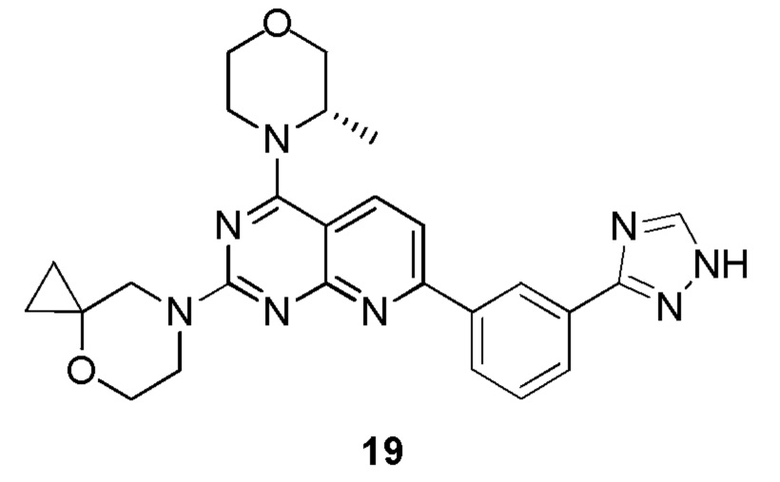
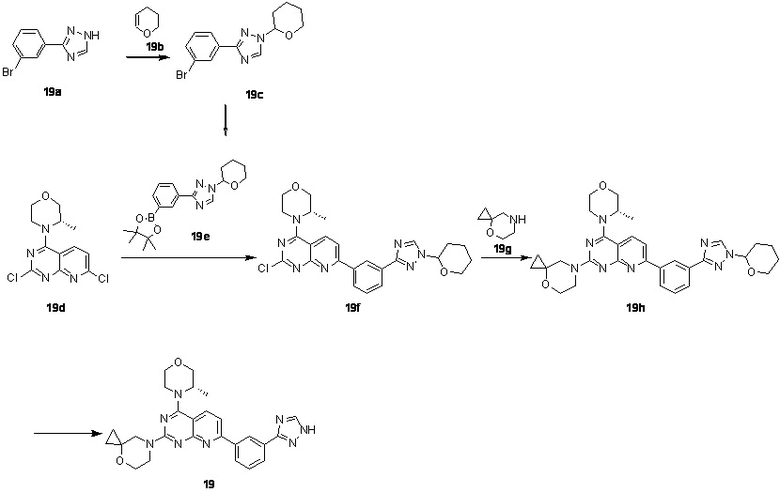
[0230] Первая стадия
[0231] Соединение 19a (50 мг, 223 мкмоль, 1 экв.), соединение 19b (37,5 мг, 446 мкмоль, 40,8 мкл, 2 экв.) и п-толуолсульфоновую кислоту (2,14 мг, 22,3 мкмоль, 1,59 мкл, 0,1 экв.) растворяли в тетрагидрофуране (2 мл) и обеспечивали осуществление реакции в смеси при 75°C в течение 3 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (40 мл), добавляли насыщенный раствор бикарбоната натрия (10 мл), экстрагировали с помощью этилацетата (20 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали соединение 19c.
[0232] MS-ESI: расчетное значение [M+H]+: 308 и 310, найденное значение: 308 и 310.
[0233] Вторая стадия
[0234] Соединение 19c (70 мг, 227 мкмоль, 1 экв.), бис(пинаколато)дибор (57,7 мг, 227 мкмоль, 1 экв.), Pd(dppf)Cl2 (8,31 мг, 11,4 мкмоль, 0,05 экв.) и ацетат натрия (66,9 мг, 681 мкмоль, 3 экв.) растворяли в 1,4-диоксане (3 мл) и обеспечивали осуществление реакции в смеси при 100°C в течение 16 часов в защитной атмосфере азота. После завершения реакции получали 19e, которое непосредственно использовали на следующей стадии без обработки.
[0235] MS-ESI: расчетное значение [M+H]+: 356, найденное значение: 356.
[0236] Третья стадия
[0237] Соединение 19d (80,7 мг, 227 мкмоль, 1 экв.), соединение 19e (68,0 мг, 227 мкмоль, 1 экв.), тетракис(трифенилфосфин)палладий (13,1 мг, 11,4 мкмоль, 0,05 экв.) и карбонат натрия (72,2 мг, 681 мкмоль, 3 экв.) растворяли в воде (1 мл) и 1,4-диоксане (3 мл) и обеспечивали осуществление реакции в смеси при 90°C в течение 4 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (30 мл) и проводили экстрагирование с помощью этилацетата (20 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (100% этилацетат) с получением 19f.
[0238] MS-ESI: расчетное значение [M+H]+: 492 и 494, найденное значение: 492 и 494.
[0239] Четвертая стадия
[0240] Соединение 19f (90 мг, 166 мкмоль, 1 экв.), 19g (24,9 мг, 166 мкмоль, 1 экв., HCl) и DIPEA (64,6 мг, 499 мкмоль, 87,0 мкл, 3 экв.) растворяли в DMSO (2 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 16 часов. После завершения реакции получали 19h, которое непосредственно использовали в реакции на следующей стадии без обработки.
[0241] MS-ESI: расчетное значение [M+H]+: 569, найденное значение: 569.
[0242] Пятая стадия
[0243] К раствору соединения 19h (94,7 мг, 166 мкмоль, 1 экв.) в DMSO (2 мл) добавляли HCl (6 M, 166,48 мкл, 6 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 66 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 19.
[0244] MS-ESI: расчетное значение [M+H]+: 485, найденное значение: 485.
[0245] 1H ЯМР (400 МГц, CDCl3) δ: 8,88 (s, 1H), 8,02-8,35 (m, 4H), 7,42-7,77 (m, 3H), 4,40 (br s, 1H), 4,01 (br d, J=10,4 Гц, 3H), 3,82-3,95 (m, 6H), 3,65-3,82 (m, 3H), 1,50 (d, J=6,8 Гц, 3H), 0,83 (s, 2H), 0,59-0,69 (m, 2H).
[0246] Вариант осуществления 20
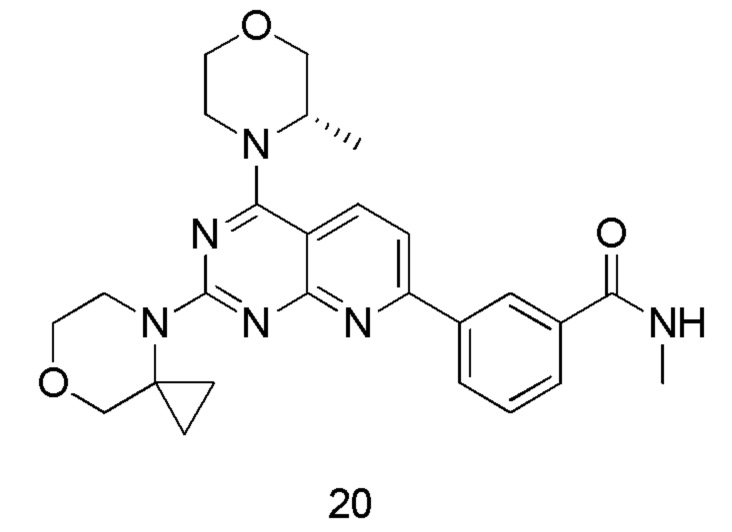
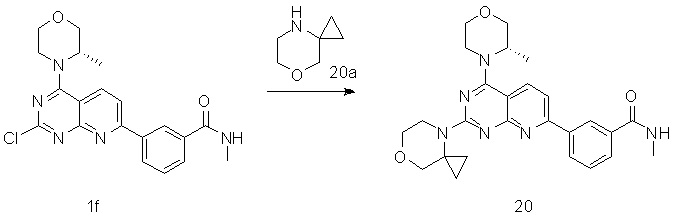
[0247] Соединение 1f (60 мг, 151 мкмоль, 1 экв.) и 20a (22,6 мг, 151 мкмоль, 1 экв.) растворяли в 0,8 мл тетрагидрофурана с последующим добавлением трет-бутоксида калия (29,0 мг, 302 мкмоль, 2 экв.) и Ruphos-Pd-G3 (12,6 мг, 15,1 мкмоль, 0,1 экв.). Реакционный раствор подвергали замене азота в течение 2 минут и обеспечивали осуществление реакции при 80°C в течение часа. После завершения реакции к реакционному раствору добавляли 1 мл воды и реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 20.
[0248] 1H ЯМР (400 МГц, CDCl3) δ: 8,18 (s, 1H), 8,09 (d, J=8,0 Гц, 2H), 7,62-7,53 (m, 2H), 7,21 (s, 1H), 4,51-4,18 (m, 3H), 4,08-3,95 (m, 2H), 3,90-3,75 (m, 6H), 3,64 (s, 2H), 3,08 (d, J=5,0 Гц, 3H), 1,31 (s, 3H), 1,09-1,02 (m, 2H), 0,90 (s, 2H).
[0249] Вариант осуществления 21
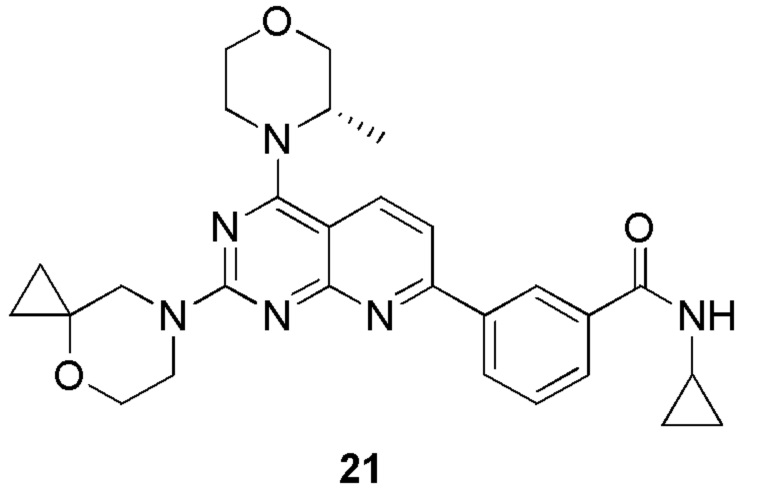
[0250] Первая стадия
[0251] Соединение 21a (5,46 г, 24,9 ммоль, 3,27 мл, 1 экв.) и бикарбонат натрия (6,27 г, 74,6 ммоль, 2,90 мл, 3 экв.) растворяли в безводном тетрагидрофуране (80 мл) и затем по каплям добавляли 21b (4,26 г, 74,6 ммоль, 5,17 мл, 3 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 16 часов в защитной атмосфере азота. После завершения реакции реакционный раствор фильтровали и затем фильтрат концентрировали с получением 21c.
[0252] Вторая стадия
[0253] Соединение 21c (0,3 г, 1,25 ммоль, 1 экв.), 21d (476 мг, 1,87 ммоль, 1,5 экв.), Pd(dppf)Cl2 (36,6 мг, 50,0 мкмоль, 0,04 экв.) и ацетат калия (368 мг, 3,75 ммоль, 3 экв.) растворяли в безводном диоксане (25 мл) и обеспечивали осуществление реакции в смешанном растворе при 90°C в течение 2 часов в защитной атмосфере азота. После завершения реакции и охлаждения реакционного раствора реакционный раствор концентрировали при пониженном давлении с последующим добавлением 15 мл воды и 60 мл (20 мл × 3) этилацетата для экстрагирования. Затем органическую фазу высушивали над безводным сульфатом натрия, концентрировали и очищали посредством колоночной хроматографии (42,5%-петролейный эфир/этилацетат) с получением 21e.
[0254] MS-ESI: расчетное значение [M+H]+: 288, найденное значение: 288.
[0255] 1H ЯМР (400 МГц, CDCl3) δ: 7,95-7,89 (m, 2H), 7,85 (d, J=7,6 Гц, 1H), 7,42-7,33 (m, 1H), 2,83 (qt, J=3,6, 7,07 Гц, 1H), 1,28 (s, 12H), 0,83-0,76 (m, 2H), 0,54-0,59 (m, 2H).
[0256] Третья стадия
[0257] Соединение 1f (0,1 г, 334 мкмоль, 1 экв.), 21e (125 мг, 435 мкмоль, 1,3 экв.), диизопропиламин тетракис(трифенилфосфин)палладий (23,2 мг, 20,1 мкмоль, 0,06 экв.) и карбонат калия (139 мг, 1,00 ммоль, 3 экв.) растворяли в безводном диоксане (10 мл) и воде (2 мл) и обеспечивали осуществление реакции в реакционном растворе при 70°C в течение 4 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и затем очищали посредством колоночной хроматографии (20,2%-метанол/этилацетат) с получением 21f.
[0258] MS-ESI: расчетное значение [M+H]+: 424 и 426, найденное значение: 424 и 426.
[0259] Четвертая стадия
[0260] Соединение 21f (0,09 г, 212 мкмоль, 1 экв.), 1i (31,8 мг, 212 мкмоль, 1 экв., HCl) и диизопропиламин (27,4 мг, 212 мкмоль, 37,0 мкл, 1 экв.) растворяли в диметилсульфоксиде (4 мл), обеспечивали осуществление реакции в реакционном растворе при 70°C в течение 15 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 21.
[0261] MS-ESI: расчетное значение [M+H]+: 501, найденное значение: 501.
[0262] 1H ЯМР (400 MГц, CD3OD) δ: 8,60 (s, 1H), 8,31 (d, J=8 Гц, 1H), 8,28 (d, J=8,4 Гц, 1H), 4,52 (br d, J=6,0 Гц, 1H), 4,07-3,93 (m, 6H), 3,89-3,83 (m, 3H), 3,80-3,71 (m, 3H), 2,92-2,91 (m, 1H), 1,48 (d, J=6,8 Гц, 3H), 0,90-0,79 (m, 4H), 0,74-0,68 (m, 4H).
[0263] Вариант осуществления 22
[0264] Первая стадия
[0265] Соединение 22a (5 г, 24,9 ммоль, 1 экв.) и соединение 22b (3,79 г, 29,9 ммоль, 2,61 мл, 1,2 экв.) растворяли в дихлорметане (50 мл) и по каплям добавляли DMF (18,2 мг, 249 мкмоль, 19,1 мкл, 0,01 экв.). Обеспечивали осуществление реакции в реакционном растворе при комнатной температуре в течение 2 часов. После завершения реакции получали соединение 22c.
[0266] Вторая стадия
[0267] Соединение 22c (5,46 г, 29 ммоль, 1 экв.), соединение 22d (7,28 г, 99,5 ммоль, 10,5 мл, 4 экв.) и DIPEA (12,9 г, 99,5 ммоль, 17,3 мл, 4 экв.) растворяли в дихлорметане (50 мл) и обеспечивали осуществление реакции в смеси при комнатной температуре в течение 5 часов. После завершения реакции реакционный раствор промывали водой (50 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния с получением 22e.
[0268] MS-ESI: расчетное значение [M+H]+: 256 и 258, найденное значение: 256 и 258.
[0269] Третья стадия
[0270] Соединение 22e (300 мг, 1,17 ммоль, 1 экв.), соединение 22f (356,9 мг, 1,41 ммоль, 1,2 экв.), ацетат калия (344 мг, 3,51 ммоль, 3 экв.) и Pd(dppf)Cl2 (42,9 мг, 58,6 мкмоль, 0,05 экв.) растворяли в 1,4-диоксане (10 мл). Обеспечивали осуществление реакции в реакционном растворе при 90°C в течение 4 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (30 мл), экстрагировали с помощью этилацетата (20 мл × 3) и органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (40% этилацетат) с получением 22g.
[0271] MS-ESI: расчетное значение [M+H]+: 304, найденное значение: 304.
[0272] Четвертая стадия
[0273] Соединение 1f (98,7 мг, 330 мкмоль, 1 экв.), 22g (100 мг, 330 мкмоль, 1 экв.), карбонат натрия (105 мг, 989 мкмоль, 3 экв.) и тетракис(трифенилфосфин)палладий (19,1 мг, 16,5 мкмоль, 0,05 экв.) растворяли в 1,4-диоксане (3 мл) и воде (1 мл). Обеспечивали осуществление реакции в реакционном растворе при 90°C в течение 5 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (20 мл) и проводили экстрагирование с помощью этилацетата (20 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (100% этилацетат) с получением 22h.
[0274] MS-ESI: расчетное значение [M+H]+: 440 и 442, найденное значение: 440 и 442.
[0275] Пятая стадия
[0276] Соединение 22h (60,0 мг, 136 мкмоль, 1 экв.), 1i (20,4 мг, 136 мкмоль, 1 экв., HCl) и DIPEA (52,9 мг, 409 мкмоль, 71,3 мкл, 3 экв.) растворяли в DMSO (3 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 15 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 22.
[0277] MS-ESI: расчетное значение [M+H]+: 517, найденное значение: 517.
[0278] 1H ЯМР (400 МГц, CDCl3) δ: 8,53 (s, 1H), 8,19 (d, J=8,0 Гц, 1H), 8,06 (d, J=8,6 Гц, 1H), 7,94 (d, J=7,8 Гц, 1H), 7,45-7,64 (m, 2H), 6,28 (br s, 1H), 4,32-4,48 (m, 1H), 3,62-4,24 (m, 12H), 1,43-1,61 (m, 12H), 0,58-0,95 (m, 4H).
[0279] Вариант осуществления 23
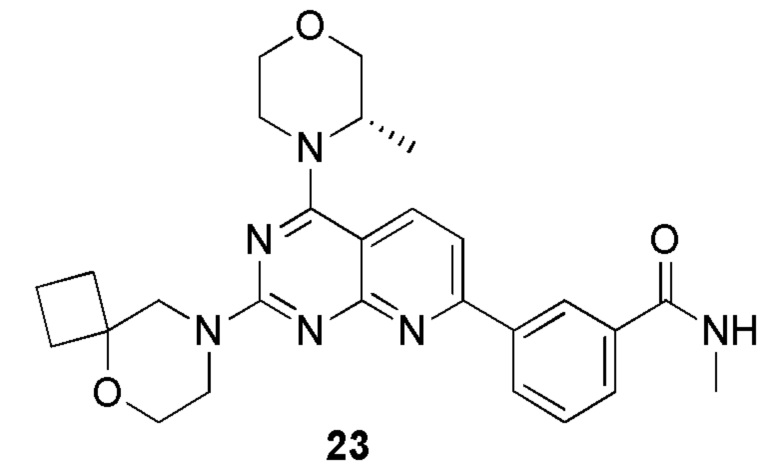
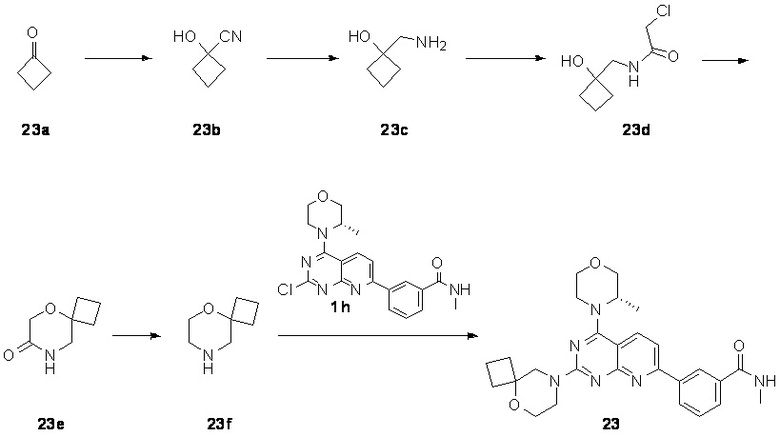
[0280] Первая стадия
[0281] Соединение 23a (23,0 г, 328 ммоль, 24,5 мл, 1,0 экв.) и дийодид цинка (5,20 г, 16,4 ммоль, 0,05 экв.) растворяли в 200 мл дихлорметана. Внутреннюю температуру полученного понижали до 0°C. Добавляли триметилсилилцианид (39,1 г, 393 ммоль, 49 мл, 1,2 экв.). Обеспечивали осуществление реакции в реакционном растворе при 25°C в течение 18 часов. После завершения реакции реакционный раствор концентрировали с последующим добавлением 50 мл ацетонитрила и 50 мл водного раствора хлористоводородной кислоты (1 M). Реакционный раствор перемешивали в течение 5 минут при 25°C и проводили экстрагирование с помощью этилацетата. Органическую фазу высушивали, фильтровали и концентрировали посредством выпаривания. Полученный остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=100:1-1:1) с получением 23b.
[0282] 1H ЯМР (400 МГц, CDCl3) δ: 4,30 (s, 1H), 2,62-2,59 (m, 2H), 2,33-2,30 (m, 2H), 1,95-1,79 (m, 2H).
[0283] Вторая стадия
[0284] 23b (16,0 г, 164 ммоль, 1 экв.) добавляли в раствор алюмогидрида лития (9,38 г, 247 ммоль, 1,5 экв.) в тетрагидрофуране (300 мл) при 0°C и обеспечивали осуществление реакции в смеси при 20°C в течение 18 часов. После завершения реакции к реакционному раствору последовательно добавляли воду (9,38 мл), 15% гидроксид натрия (9,38 мл) и воду (28,1 мл), перемешивали в течение 15 минут, фильтровали и концентрировали с получением 23c.
[0285] 1H ЯМР (400 МГц, CDCl3) δ: 2,72 (s, 2H), 2,12-1,76 (m, 7H), 1,7-1,63 (m, 1H), 1,49-1,33 (m, 1H).
[0286] Третья стадия
[0287] В раствор 23c (2,0 г, 19,8 ммоль, 1 экв.) и диизопропилэтиламина (4,0 г, 31,0 ммоль, 5,4 мл, 1,6 экв.) в дихлорметане (20,0 мл) добавляли хлорацетилхлорид (2,23 г, 19,8 ммоль, 1,6 мл, 1,0 экв.) при 0°C. Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали и остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 23d.
[0288] 1H ЯМР (400 МГц, CDCl3) δ: 6,99 (s, 1H), 4,17-4,02 (m, 2H), 3,50 (d, J = 6,0 Гц, 2H), 2,71 (s, 1H), 2,14-1,98 (m, 4H), 1,81-1,71 (m, 1H), 1,64-1,49 (m, 1H).
[0289] Четвертая стадия
[0290] Соединение 23d (2,2 г, 12,4 ммоль, 1 экв.) добавляли к безводному тетрагидрофурану (100 мл). Внутреннюю температуру полученного понижали до 0°C и добавляли гидрид натрия (1,49 г, 37,2 ммоль, чистота 60%, 3 экв.). Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 18 часов. После завершения реакции к реакционному раствору добавляли воду (15,0 мл). Реакционный раствор экстрагировали с помощью этилацетата (20 мл × 3) и органические фазы объединяли, высушивали, фильтровали и концентрировали с получением 23e.
[0291] 1H ЯМР (400 МГц, CDCl3) δ: 6,96 (s, 1H), 4,15 (s, 2H), 3,44-3,35 (m, 2H), 2,27-2,16 (m, 2H), 2,09-2,01 (m, 2H), 1,95-1,86 (m, 1H), 1,74-1,63 (m, 1H).
[0292] Пятая стадия
[0293] 23e (1,2 г, 8,5 ммоль, 1 экв.) добавляли к раствору алюмогидрида лития (645 мг, 17 ммоль, 2 экв.) в тетрагидрофуране (30 мл) при 0°C и обеспечивали осуществление реакции в смеси при 20°C в течение 18 часов. После завершения реакции к реакционному раствору последовательно добавляли воду (0,7 мл), 15% гидроксид натрия (0,7 мл) и воду (2,1 мл), перемешивали в течение 15 минут, фильтровали и концентрировали с получением 23f.
[0294] 1H ЯМР (400 МГц, CDCl3) δ: 3,61-3,51 (m, 2H), 2,87-2,76 (m, 4H), 2,03-1,97 (m, 4H), 1,86-1,82 (m, 1H), 1,62-1,54 (m, 1H).
[0295] Шестая стадия
[0296] Соединение 23f (53 мг, 414 мкмоль, 1,1 экв.), 1h (150 мг, 377 мкмоль, 1 экв.) и DIPEA (48 мг, 377 мкмоль, 66 мкл, 1 экв.) растворяли в DMSO (4 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 23.
[0297] MS-ESI: расчетное значение [M+H]+: 489, найденное значение: 489.
[0298] 1H ЯМР (400 МГц, CDCl3) δ: 8,63 (s, 1H), 8,22 (d, J=7,6 Гц, 1H), 8,05 (d, J=8,4 Гц, 1H), 7,97 (d, J=7,6 Гц, 1H), 7,65-7,41 (m, 2H), 6,53 (br s, 1H), 4,40 (d, J=6,8 Гц, 1H), 4,12-3,95 (m, 3H), 3,93-3,83 (m, 4H), 3,83-3,68 (m, 5H), 3,06 (d, J=4,8 Гц, 3H), 2,07-2,04 (m, 4H), 1,91-1,80 (m, 1H), 1,77-1,70 (m, 1H), 1,50 (d, J=6,8 Гц, 3H).
[0299] Вариант осуществления 24
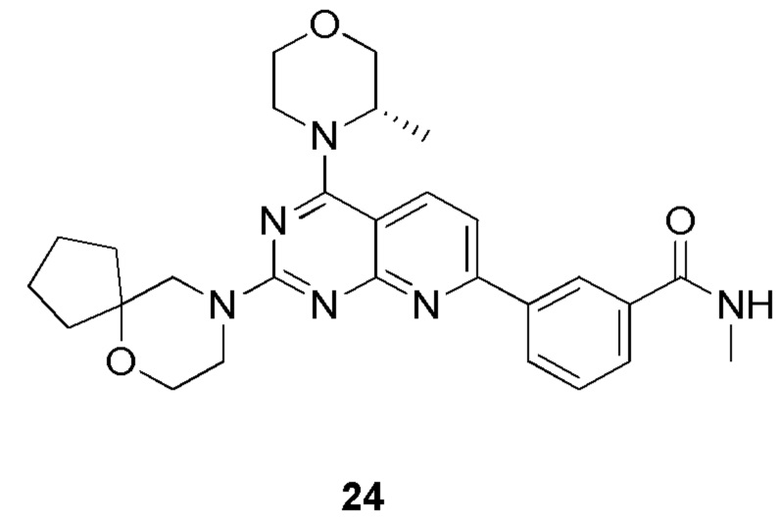
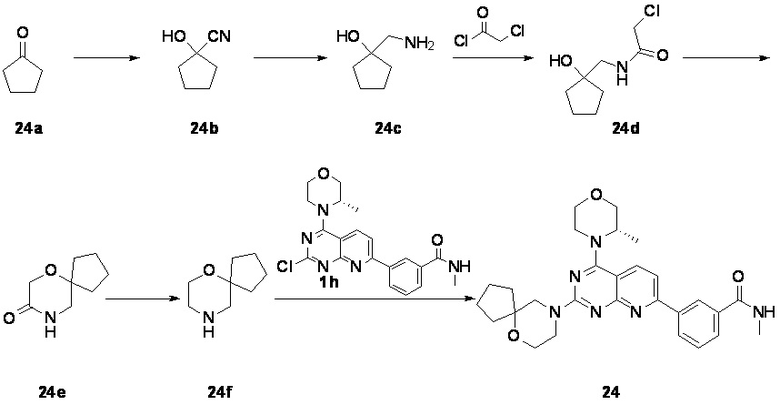
[0300] Первая стадия
[0301] Соединение 24a (23,0 г, 273 ммоль, 24,2 мл, 1 экв.) и дийодид цинка (4,36 г, 13,7 ммоль, 0,05 экв.) растворяли в 200 мл дихлорметана. Внутреннюю температуру полученного понижали до 0°C. Добавляли триметилсилилцианид (32,6 г, 328 ммоль, 41,1 мл, 1,2 экв.). Обеспечивали осуществление реакции в реакционном растворе при 25°C в течение 18 часов. После завершения реакции реакционный раствор концентрировали с последующим добавлением 50 мл ацетонитрила и 50 мл водного раствора хлористоводородной кислоты (1 M). Реакционный раствор перемешивали в течение 5 минут при 25°C и проводили экстрагирование с помощью этилацетата. Органическую фазу высушивали, фильтровали, концентрировали посредством выпаривания. Полученный остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=100:1-1:1) с получением 24b.
[0302] 1H ЯМР (400 МГц, CDCl3) δ: 3,27-3,20 (m, 1H), 2,10-2,06 (m, 4H), 1,87-1,80 (m, 4H).
[0303] Вторая стадия
[0304] 24b (16,0 г, 164 ммоль, 1 экв.) добавляли в раствор алюмогидрида лития (8,19 г, 216 ммоль, 1,5 экв.) в тетрагидрофуране (300 мл) при 0°C и обеспечивали осуществление реакции в смеси при 20°C в течение 18 часов. После завершения реакции к реакционному раствору последовательно добавляли воду (8,19 мл), 15% гидроксид натрия (8,19 мл) и воду (25 мл), перемешивали в течение 15 минут, фильтровали и концентрировали с получением 24c.
[0305] 1H ЯМР (400 МГц, CDCl3) δ: 2,74 (s, 2H), 1,90-1,55 (m, 9H), 1,54-1,47 (m, 2H).
[0306] Третья стадия
[0307] В раствор 24c (2,0 г, 17,4 ммоль, 1 экв.) и диизопропилэтиламина (3,37 г, 26,1 ммоль, 4,54 мл, 1,5 экв.) в дихлорметане (20 мл) добавляли хлорацетилхлорид (1,96 г, 17,4 ммоль, 1,38 мл, 1,0 экв.) при 0°C. Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали и остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 24d.
[0308] 1H ЯМР (400 МГц, CDCl3) δ: 7,03 (s, 1H), 4,09 (s, 2H), 3,44 (d, J = 5,6 Гц, 2H), 2,00 (s, 1H), 1,89-1,77 (m, 2H), 1,67-1,64 (m, 6H).
[0309] Четвертая стадия
[0310] Соединение 24d (2,6 г, 13,6 ммоль, 1 экв.) добавляли к безводному тетрагидрофурану (100 мл). Внутреннюю температуру полученного понижали до 0°C и добавляли гидрид натрия (1,63 г, 40,7 ммоль, 60%, 3 экв.). Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 18 часов. После завершения реакции к реакционному раствору добавляли воду (15,0 мл). Реакционный раствор экстрагировали с помощью этилацетата (20 мл × 3) и органические фазы объединяли, высушивали, фильтровали и концентрировали с получением 24e.
[0311] 1H ЯМР (400 МГц, CDCl3) δ: 6,79 (s, 1H), 4,19 (s, 2H), 3,38-3,33 (m, 2H), 1,74-1,64 (m, 8H).
[0312] Пятая стадия
[0313] 24e (1,8 г, 11,6 ммоль, 1 экв.) добавляли к раствору алюмогидрида лития (880,32 мг, 23,2 ммоль, 2 экв.) в тетрагидрофуране (50 мл) при 0°C и обеспечивали осуществление реакции в смеси при 20°C в течение 18 часов. После завершения реакции к реакционному раствору последовательно добавляли воду (0,9 мл), 15% гидроксид натрия (0,9 мл) и воду (2,7 мл), перемешивали в течение 15 минут, фильтровали и концентрировали с получением неочищенного продукта 24f.
[0314] Шестая стадия
[0315] Соединение 24f (159 мг, 1,13 ммоль, 3 экв.), 1h (150 мг, 377 мкмоль, 1 экв.) и DIPEA (97,5 мг, 754 мкмоль, 131 мкл, 2 экв.) растворяли в DMSO (4 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 24.
[0316] MS-ESI: расчетное значение [M+H]+: 503, найденное значение: 503.
[0317] 1H ЯМР (400 МГц, CDCl3) δ: 8,62 (s, 1H), 8,22 (d, J=8,0 Гц, 1H), 8,05 (d, J=8,4 Гц, 1H), 7,97 (d, J=8,0 Гц, 1H), 7,57 (t, J=8,0 Гц, 1H), 7,51 (d, J=8,4 Гц, 1H), 6,46 (s, 1H), 4,38 (d, J=5,6 Гц, 1H), 4,00 (d, J=10,8 Гц, 2H), 3,93-3,64 (m, 10H), 3,06 (d, J=4,8 Гц, 3H), 1,85-1,70 (m, 6H), 1,64 (s, 2H), 1,49 (d, J=6,8 Гц, 3H).
[0318] Вариант осуществления 25
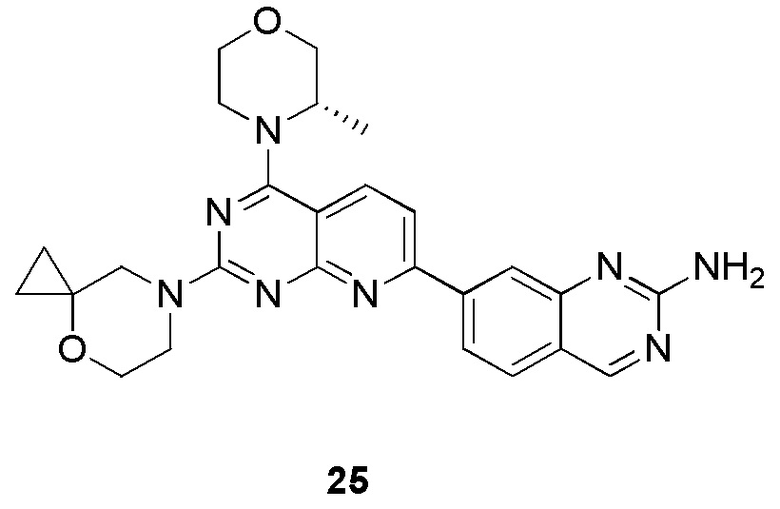
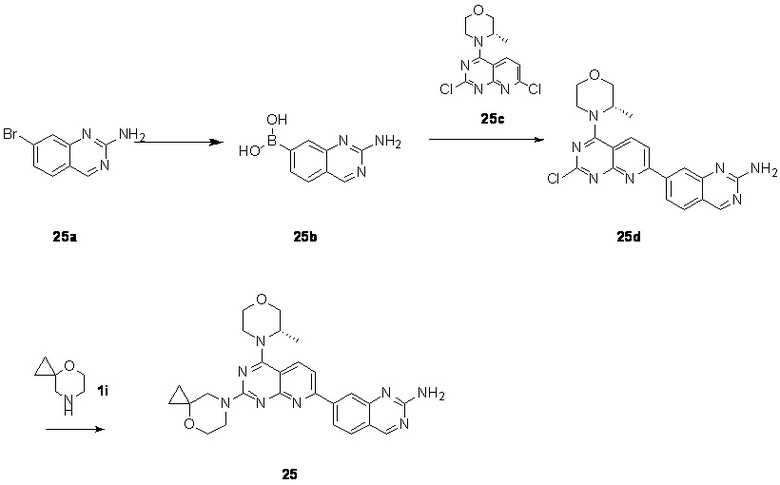
[0319] Первая стадия
[0320] Соединение 25a (100 мг, 446 мкмоль, 1,0 экв.), бис(пинаколато)дибор (136 мг, 536 мкмоль, 1,2 экв.), ацетат калия (131 мг, 1,34 ммоль, 3,0 экв.) и Pd(dppf)Cl2 (16,4 мг, 22,4 мкмоль, 0,05 экв.) растворяли в безводном диоксане (5 мл) с последующей вентиляцией три раза. Обеспечивали осуществление реакции в смешанном растворе при 90°C в течение 16 часов в атмосфере азота. После завершения реакции получали реакционный раствор, содержащий 25b, который непосредственно использовали на следующей стадии.
[0321] MS-ESI: расчетное значение [M+H]+: 190, найденное значение: 190.
[0322] Вторая стадия
[0323] К раствору, содержащему соединение 25b, добавляли соединение 25c (133 мг, 446 мкмоль, 1,0 экв.), тетракис(трифенилфосфин)палладий (25,8 мг, 22,3 мкмоль, 0,05 экв.) и карбонат натрия (142 мг, 1,34 ммоль, 3,0 экв.), растворенный в воде (2 мл) и 1,4-диоксане (5 мл). Обеспечивали осуществление реакции в реакционном растворе при 80°C в течение 17 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (10 мл), экстрагировали с помощью этилацетата (20 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (1:10 метанол/дихлорметан) с получением 25d.
[0324] MS-ESI: расчетное значение [M+H]+ 408 и 410, найденное значение: 408 и 410.
[0325] Третья стадия
[0326] Соединение 25d (30 мг, 73,5 мкмоль, 1,00 экв.), 1i (10,0 мг, 66,8 мкмоль, 1,00 экв.) и DIPEA (28,5 мг, 221 мкмоль, 3,00 экв.) растворяли в DMSO (2 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 17 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением соединения 25.
[0327] MS-ESI: расчетное значение [M+H]+: 485, найденное значение: 485.
[0328] 1H ЯМР (400 MГц, CDCl3) δ: 9,01 (s, 1H), 8,22 (br d, J=8,0 Гц, 1H), 8,09 (s, 1H), 8,01 (d, J=8,4 Гц, 1H), 7,75 (d, J=8,4 Гц, 1H), 7,50 (d, J=8,4 Гц, 1H), 5,18 (s, 2H), 4,31 (br s, 1H), 4,02-3,54 (m, 12H), 1,41 (d, J=6,4 Гц, 3H), 0,77 (s, 2H), 0,61 (br s, 2H).
[0329] Вариант осуществления 26
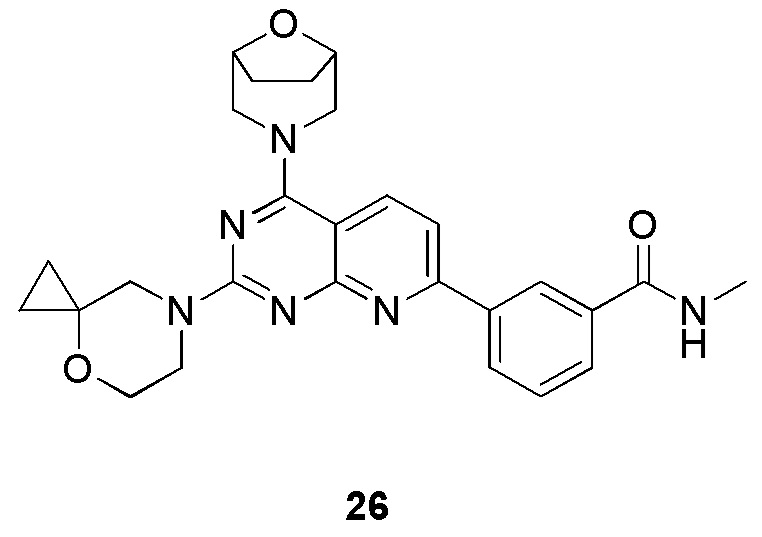
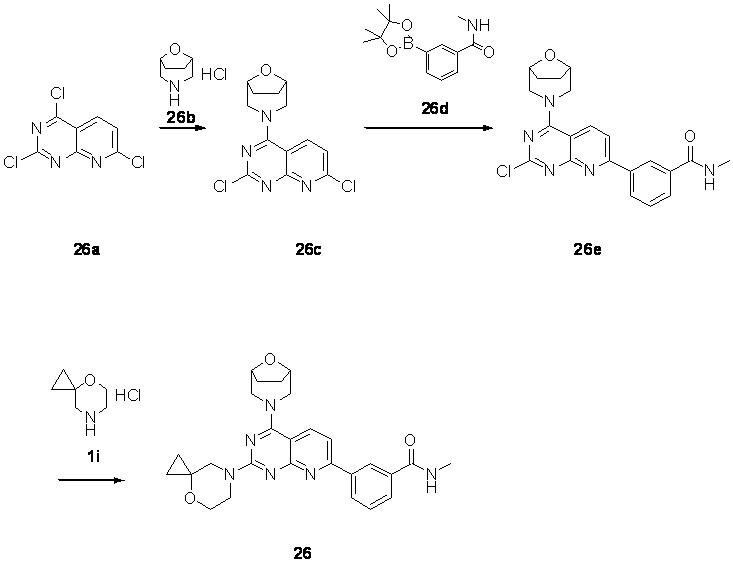
[0330] Первая стадия
[0331] Соединение 26a (150 мг, 639 мкмоль, 1,00 экв.), соединение 26b (57,4 мг, 639 мкмоль, 1,00 экв.) и диизопропилэтиламин (248 мг, 1,92 ммоль, 2,83 мл, 3,00 экв.) растворяли в дихлорметане (10 мл) и обеспечивали осуществление реакции в смеси при 20°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали и очищали посредством колоночной хроматографии (1:5 метанол/дихлорметан) с получением 26c.
[0332] Вторая стадия
[0333] Соединение 26c (0,12 г, 386 мкмоль, 1,00 экв.), соединение 26d (90,6 мг, 347 мкмоль, 0,90 экв.), тетракис(трифенилфосфин)палладий (22,3 мг, 19,3 мкмоль, 0,05 экв.) и карбонат натрия (123 мг, 1,16 ммоль, 3,00 экв.) растворяли в воде (3 мл) и 1,4-диоксане (10 мл) и обеспечивали осуществление реакции в смеси при 80°C в течение 16 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (10 мл), экстрагировали с помощью этилацетата (20 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении с удалением растворителя и очищали посредством колоночной хроматографии (1:5 метанол/дихлорметан) с получением 26e.
[0334] Третья стадия
[0335] Соединение 26e (70 мг, 171 мкмоль, 1,00 экв.), 1i (30,7 мг, 205 мкмоль, 1,20 экв.) и DIPEA (66,2 мг, 512 мкмоль, 89 мкл, 3,00 экв.) растворяли в DMSO (3 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 17 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 26.
[0336] MS-ESI: расчетное значение [M+H]+: 487, найденное значение: 487.
[0337] 1H ЯМР (400 MГц, CDCl3) δ: 8,62 (s, 1H), 8,21 (d, J=7,8 Гц, 1H), 8,06 (d, J=8,4 Гц, 1H), 7,98 (d, J=7,8 Гц, 1H), 7,56 (t, J=7,8 Гц, 1H), 7,49 (d, J=8,4 Гц, 1H), 6,59 (br s, 1H), 4,48 (br s, 2H), 4,13 (br d, J=12,8 Гц, 2H), 4,06 (br s, 2H), 3,94 (br s, 2H), 3,90-3,84 (m, 2H), 3,62 (br d, J=11,6 Гц, 2H), 3,06 (d, J=4,8 Гц, 3H), 2,06-1,90 (m, 4H), 0,89-0,82 (m, 2H), 0,69 (s, 2H).
[0338] Вариант осуществления 27
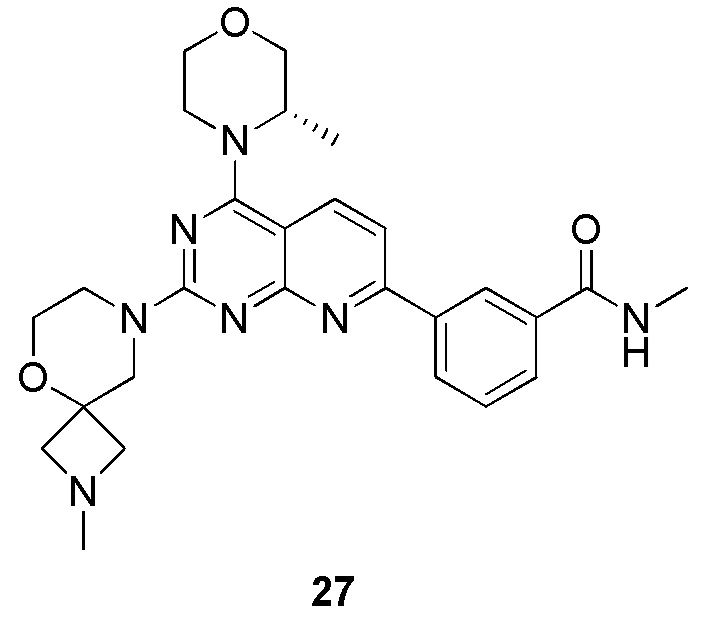
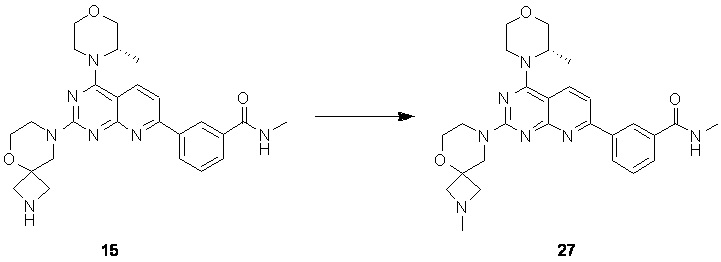
[0339] Соединение 15 (150 мг, 306 мкмоль, 1 экв.) и формальдегид (11,96 мг, 398 мкмоль, 11,0 мкл, 1,3 экв.) растворяли в дихлорэтане (10 мл) и уксусной кислоте (2 мл) с последующим добавлением цианоборогидрида натрия (38,5 мг, 613 мкмоль, 2 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 18 часов. После завершения реакции и охлаждения реакционного раствора реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали посредством высокоэффективной жидкостной хроматографии с получением 27.
[0340] MS-ESI: расчетное значение [M+H]+: 504, найденное значение: 504.
[0341] 1H ЯМР (400 MГц, CD3OD) δ: 8,63 (s, 1H), 8,36-8,30 (m, 2H), 7,96 (d, J=8,0 Гц, 1H), 7,71 (d, J=8,6 Гц, 1H), 7,65 (t, J=7,8 Гц, 1H), 4,63 (br s, 2H), 4,17-3,85 (m, 7H), 3,83-3,67 (m, 8H), 2,99 (s, 3H), 2,61 (s, 3H), 1,51 (d, J=6,8 Гц, 3H).
[0342] Вариант осуществления 28
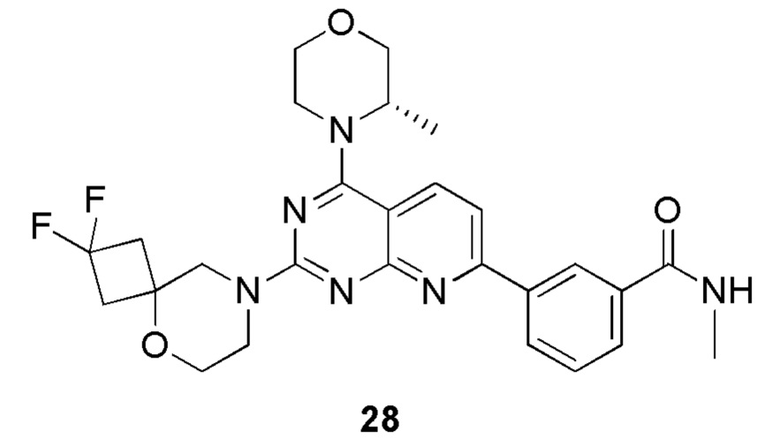
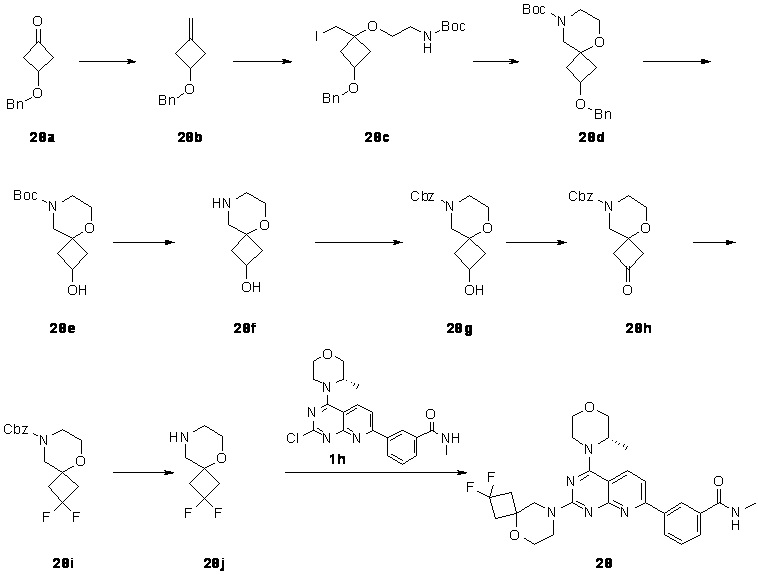
[0343] Первая стадия
[0344] Соединение метилтрифенилфосфония бромид (25,9 г, 72,6 ммоль, 1,6 экв.) растворяли в 350 мл тетрагидрофурана с последующим добавлением трет-бутоксида калия (1 M, 81,7 мл, 1,8 экв.) при 20°C. Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 3 часов. 28a (8,0 г, 45,4 ммоль, 1 экв.) добавляли к реакционному раствору и обеспечивали осуществление реакции в течение 18 часов. После завершения реакции к полученному добавляли воду (200 мл) и этилацетат (300 мл) для экстрагирования. Органическую фазу промывали с помощью насыщенного солевого раствора (100 мл × 3), высушивали, фильтровали и концентрировали. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 28b.
[0345] 1H ЯМР (400 MГц, CDCl3) δ: 7,36-7,31 (m, 5H), 4,87-4,85 (m, 2H), 4,46 (s, 2H), 4,45-4,08 (m, 1H), 2,89-2,86 (m, 2H), 2,78-2,73 (m, 2H).
[0346] Вторая стадия
[0347] К раствору 28b (1,5 г, 8,61 ммоль, 640 мкл, 1 экв.) и трет-бутил-N-(2-гидроксиэтил)формиата (1,67 г, 10,3 ммоль, 1,60 мл, 1,2 экв.) в ацетонитриле (14 мл) добавляли NIS (2,32 г, 10,3 ммоль, 1,2 экв.) и обеспечивали осуществление реакции в смеси при 20°C в течение 4 часов. После завершения реакции в реакционный раствор последовательно добавляли воду (20 мл) и этилацетат (30 мл) для экстрагирования, высушивали, фильтровали и концентрировали. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 28c.
[0348] 1H ЯМР (400 МГц, CDCl3) δ: 7,33-7,30 (m, 5H), 5,01-4,97 (m, 1H), 4,44-4,43 (m, 2H), 3,79-3,71 (m, 1H), 3,33-3,32 (m, 3H), 2,45-2,40 (m, 5H), 2,03-2,02 (m, 1H), 1,46 (s, 9H).
[0349] Третья стадия
[0350] Соединение 28c (1,8 г, 3,90 ммоль, 1 экв.) добавляли к безводному тетрагидрофурану (50 мл). Внутреннюю температуру понижали до 0°C с последующим добавлением гидрида натрия (312 мг, 7,80 ммоль, 60%, 2 экв.). Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 18 часов. После завершения реакции реакционный раствор экстрагировали с помощью воды (50,0 мл) и этилацетата (50 мл). Органические фазы объединяли, высушивали, фильтровали и концентрировали. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 28d.
[0351] 1H ЯМР (400 MГц, CDCl3) δ: 7,33-7,20 (m, 5H), 4,63-4,09 (m, 3H), 3,52-3,50 (m, 2H), 3,37 (s, 1H), 3,30 (s, 2H), 3,18 (s, 1H), 2,35 (s, 1H), 2,28-2,15 (m, 1H), 1,97-1,85 (m, 2H), 1,43-1,28 (m, 9H).
[0352] Четвертая стадия
[0353] Соединение 28d (2,6 г, 13,6 ммоль, 1 экв.) добавляли к этилацетату (15 мл) и к полученному добавляли влажный палладий на угле (0,1 г, 10%). Три раза проводили замену азота. Обеспечивали осуществление реакции в реакционном растворе при 25°C в течение 2 часов в данной атмосфере (15 фунтов/кв. дюйм). После завершения реакции реакционный раствор фильтровали и концентрировали с получением 28e. Неочищенный продукт непосредственно использовали на следующей стадии.
[0354] Пятая стадия
[0355] В реакционную колбу, содержащую 28e (360 мг, 1,48 ммоль, 1 экв.) и этилацетат (4 мл), добавляли HCl/EtOAc (4 M, 10 мл, 27 экв.) и обеспечивали осуществление реакции в смеси при 20°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали с получением неочищенного продукта 28f, который непосредственно использовали на следующей стадии.
[0356] Шестая стадия
[0357] Соединение 28f (210 мг, 1,47 ммоль, 1 экв.) и DIPEA (381 мг, 2,95 ммоль, 513 мкл, 2 экв.) растворяли в дихлорметане (5 мл) и к реакционному раствору добавляли бензилхлорформиат (302 мг, 1,77 ммоль, 251 мкл, 1,2 экв.). Обеспечивали осуществление реакции в смеси при 20°C в течение 18 часов. После завершения реакции в реакционный раствор добавляли воду (30,0 мл) и этилацетат (20 мл × 3) для экстрагирования. Органические фазы объединяли, высушивали, фильтровали, концентрировали и остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 28g.
[0358] 1H ЯМР (400 МГц, CDCl3) δ: 7,43-7,29 (m, 5H), 5,23-5,10 (m, 2H), 4,55-4,10 (m, 1H), 3,61 (s, 2H), 3,55 (s, 1H), 3,47-3,45 (m, 2H), 3,34 (s, 1H), 2,60-2,30 (m, 2H), 1,95-1,93 (m, 2H).
[0359] Седьмая стадия
[0360] Соединение 28g (320 мг, 1,15 ммоль, 1 экв.) растворяли в дихлорметане (5 мл) и к реакционному раствору добавляли DMP (636 мг, 1,50 ммоль, 1,3 экв.). Обеспечивали осуществление реакции при 20°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали и остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 28h.
[0361] 1H ЯМР (400 MГц, CDCl3) δ: 7,46-7,30 (m, 5H), 5,16 (s, 2H), 3,71 (s, 2H), 3,62 (s, 2H), 3,58-3,52 (m, 2H), 3,15-3,07 (m, 2H), 2,98 (s, 2H).
[0362] Восьмая стадия
[0363] Соединение 28h (200 мг, 727 мкмоль, 1 экв.) растворяли в дихлорметане (1 мл) и в реакционный раствор добавляли трифторид N,N-диэтилсеры (703 мг, 4,36 ммоль, 576 мкл, 6 экв.). Обеспечивали осуществление реакции при 20°C в течение 18 часов. После завершения реакции к реакционному раствору добавляли воду (40,0 мл) и дихлорметан (30 мл × 3). Органические фазы объединяли, высушивали, фильтровали, концентрировали и остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100:1-1:1) с получением 28i.
[0364] Девятая стадия
[0365] Соединение 28i (180 мг, 606 мкмоль, 1 экв.) добавляли к метанолу (5 мл) и к полученному добавляли влажный палладий на угле (0,01 г, 10%). Три раза проводили замену водорода и обеспечивали осуществление реакции при 20°C в течение 2 часов в данной атмосфере (15 фунтов/кв. дюйм). После завершения реакции реакционный раствор фильтровали и концентрировали с получением 28j. Неочищенный продукт непосредственно использовали на следующей стадии.
[0366] Десятая стадия
[0367] Соединение 28j (42 мг, 257 мкмоль, 1 экв.), 1h (102 мг, 257 мкмоль, 1 экв.) и N,N-диизопропилэтиламин (66,5 мг, 514 мкмоль, 89,7 мкл, 2 экв.) растворяли в DMSO (1 мл) и обеспечивали осуществление реакции в полученном смешанном растворе при 70°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 28.
[0368] MS-ESI: расчетное значение [M+H]+: 525, найденное значение: 525.
[0369] 1H ЯМР (400 МГц, CDCl3) δ: 8,54 (s, 1H), 8,15 (d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,89 (d, J=8,0 Гц, 1H), 7,54-7,41 (m, 2H), 6,38 (br s, 1H), 4,36 (br s, 1H), 4,06-3,84 (m, 6H), 3,79-3,53 (m, 6H), 2,99 (d, J=4,8 Гц, 3H), 2,73-2,46 (m, 4H), 1,44 (d, J=6,8 Гц, 3H).
[0370] Вариант осуществления 29
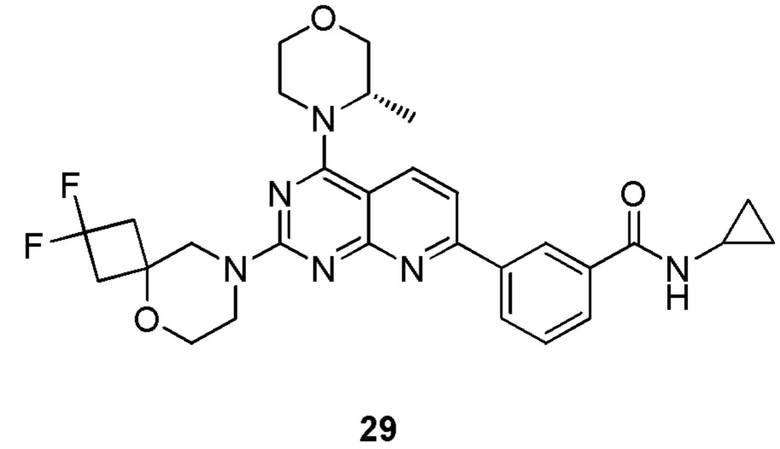
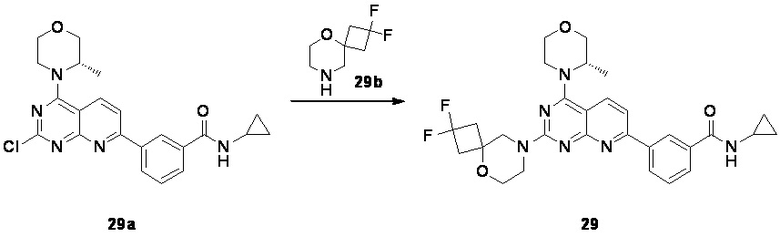
[0371] Соединение 29a (109 мг, 257 мкмоль, 1 экв.), 29b (42 мг, 257 мкмоль, 1 экв.) и N,N-диизопропилэтиламин (66,5 мг, 514,82 мкмоль, 89,7 мкл, 2 экв.) растворяли в DMSO (1 мл), обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 29.
[0372] MS-ESI: расчетное значение [M+H]+: 551, найденное значение: 551.
[0373] 1H ЯМР (400 МГц, CDCl3) δ: 8,57 (s, 1H), 8,20 (d, J=8,0 Гц, 1H), 8,07 (d, J=8,4 Гц, 1H), 7,98 (d, J=8,0 Гц, 1H), 7,61-7,47 (m, 2H), 6,56 (s, 1H), 4,43 (d, J=6,4 Гц, 1H), 4,12-4,05 (m, 2H), 4,03-3,92 (m, 4H), 3,88-3,83 (m, 1H), 3,80-3,70 (m, 5H), 3,01-2,92 (m, 1H), 2,76-2,57 (m, 4H), 1,53-1,50 (m, 3H), 0,98-0,87 (m, 2H), 0,76-0,60 (m, 2H).
[0374] Вариант осуществления 30
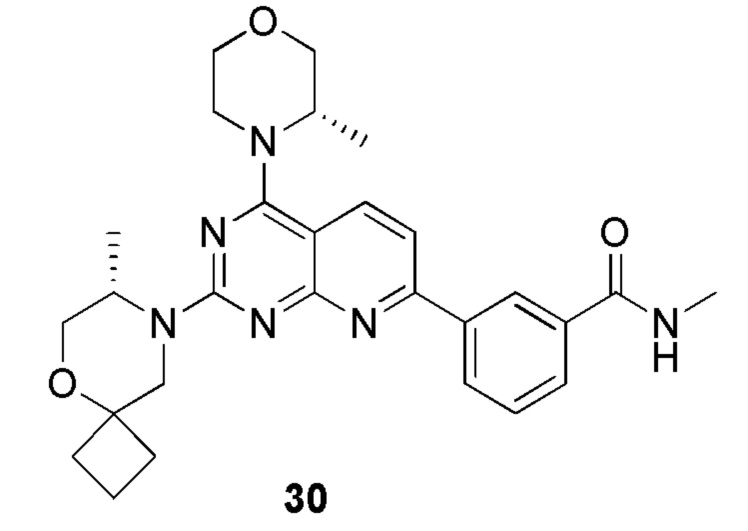
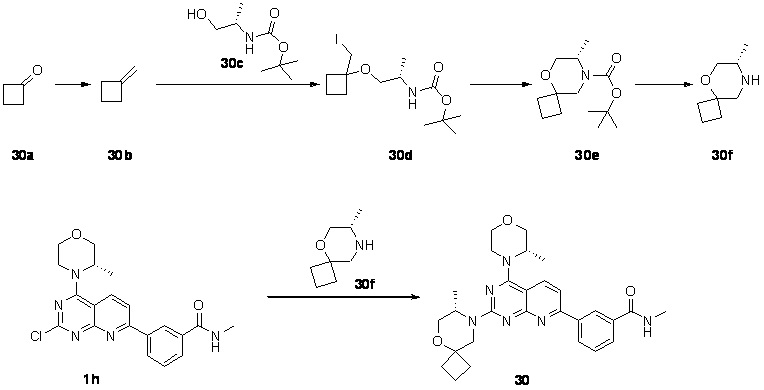
[0375] Первая стадия
[0376] Метилтрифенилфосфония бромид (102 г, 285 ммоль, 2 экв.) и трет-бутоксид калия (1 M, 257 мл, 1,8 экв.) растворяли в тетрагидрофуране (500 мл), обеспечивали осуществление реакции в смеси при комнатной температуре в течение часа с последующим добавлением соединения 30a (10,0 г, 143 ммоль, 10,7 мл, 1 экв.). После осуществления реакции при комнатной температуре в течение 17 часов реакционную смесь подвергали перегонке при 80°C с получением дистиллята, представляющего собой соединение 30b.
[0377] 1H ЯМР (400 МГц, CDCl3) δ: 4,51 (dt, J = 1,13, 2,32 Гц, 2H), 3,71-3,48 (m, 2H), 2,61-2,46 (m, 2H), 1,81-1,74 (m, 2H).
[0378] Вторая стадия
[0379] Соединение 30b (30,0 г, 17,6 ммоль, 1 экв.), соединение 30c (3,70 г, 21,1 ммоль, 1,2 экв.), N-йодсукцинимид (4,76 г, 21,1 ммоль, 1,2 экв.) растворяли в тетрагидрофуране (10,0 мл), обеспечивали осуществление реакции в смеси при комнатной температуре в течение 24 часов. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (100 мл) и проводили экстрагирование с помощью этилацетата (50,0 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (20% этилацетат) с получением соединения 30d.
[0380] MS-ESI: расчетное значение [M+H]+: 370, найденное значение: 270.
[0381] Третья стадия
[0382] Соединение 30d (30,9 г, 2,44 ммоль, 1 экв.), гидрид натрия (195 мг, 4,87 ммоль, чистота 60%, 2 экв.) растворяли в тетрагидрофуране (10,0 мл) и обеспечивали осуществление реакции в смеси при 65°C в течение 16 часов. После завершения реакции реакцию гасили посредством добавления воды (50,0 мл), проводили экстрагирование с помощью этилацетата (50,0 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали соединение 30e.
[0383] MS-ESI: расчетное значение [M+H]+: 242, найденное значение: 142.
[0384] Четвертая стадия
[0385] Соединение 30e (0,18 г, 746 мкмоль, 1 экв.) растворяли в хлористоводородной кислоте/этилацетате (20,0 мл), обеспечивали осуществление реакции в смеси и перемешивали ее в течение 5 часов. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе с удалением растворителя и получали 30f.
[0386] MS-ESI: расчетное значение [M+H]+: 142, найденное значение: 142.
[0387] Пятая стадия
[0388] Соединение 1h (141 мг, 354 мкмоль, 0,25 экв.), 30f (200 мг, 1,42 ммоль, 1 экв., HCl) и DIPEA (549 мг, 4,25 ммоль, 740 мкл, 3 экв.) растворяли в DMSO (5,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 16 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 30.
[0389] MS-ESI: расчетное значение [M+H]+: 503, найденное значение: 503.
[0390] 1H ЯМР (400 МГц, CDCl3) δ: 8,65 (s, 1H), 8,22 (br d, J=7,8 Гц, 1H), 8,07 (d, J=8,4 Гц, 1H), 7,99 (br d, J=7,8 Гц, 1H), 7,63-7,48 (m, 2H), 6,56 (br s, 1H), 4,81 (br d, J=13,6 Гц, 2H), 4,42 (br d, J=5,6 Гц, 1H), 4,08-3,97 (m, 1H), 3,96-3,67 (m, 6H), 3,58 (d, J=11,6 Гц, 1H), 3,18 (br d, J=13,2 Гц, 1H), 3,08 (d, J=4,8 Гц, 3H), 2,26-1,93 (m, 4H), 1,93-1,77 (m, 2H), 1,52 (d, J=6,8 Гц, 3H), 1,36 (br d, J=6,8 Гц, 3H).
[0391] Вариант осуществления 31
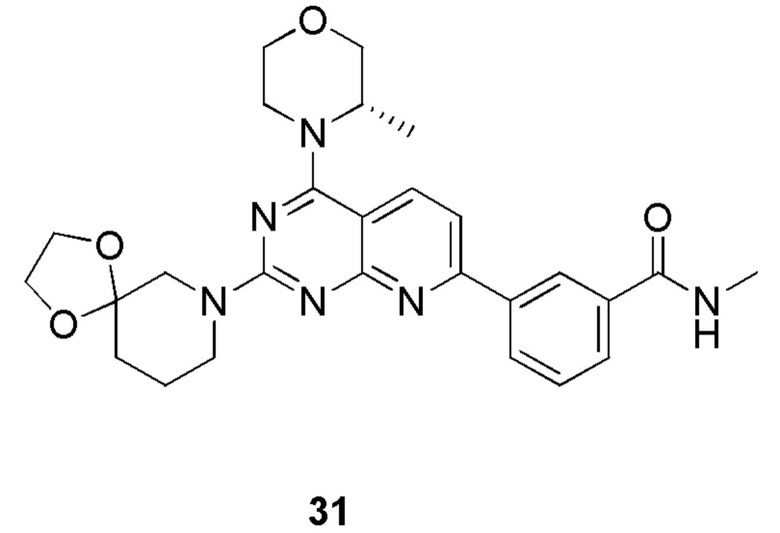
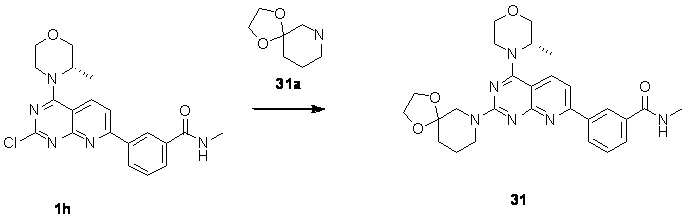
[0392] Первая стадия
[0393] Соединение 1h (100 мг, 251 мкмоль, 1,00 экв.), 31a (36,0 мг, 251 мкмоль, 1,00 экв.) и DIPEA (97,5 мг, 754 мкмоль, 131 мкл, 3,00 экв.) растворяли в DMSO (3,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 90°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 31.
[0394] MS-ESI: расчетное значение [M+H]+: 505, найденное значение: 505.
[0395] 1H ЯМР (400 MГц, CDCl3) δ: 8,57 (br s, 1H), 8,11 (br d, J=7,2 Гц, 1H), 7,95 (br d, J=8,4 Гц, 1H), 7,91 (br d, J=8,0, 1H), 7,48 (br t, J=8,0, 1H), 7,41 (br d, J=8,4 Гц, 1H), 6,53 (br s, 1H), 4,31 (br s, 1H), 4,01-3,52 (m,14H), 2,99 (d, J=4,8 Гц, 3H), 1,67 (br s, 4H), 1,41 (br d, J=6,8 Гц, 3H).
[0396] Вариант осуществления 32-1 и 32-2
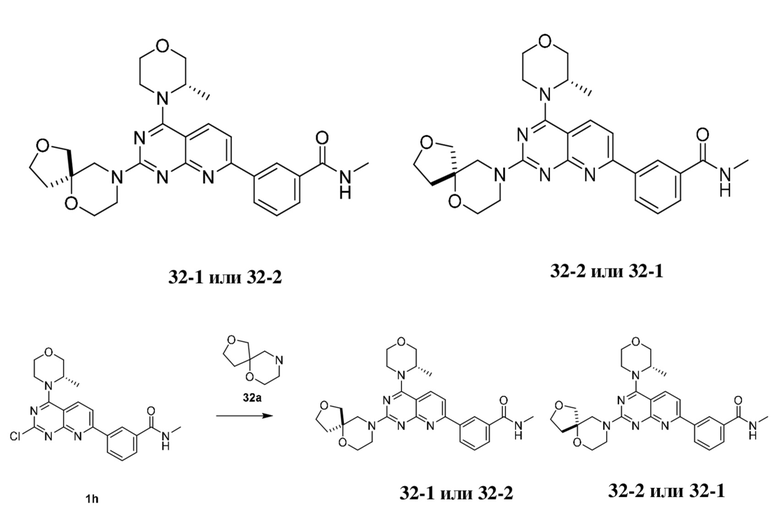
[0397] Соединение 1h (280 мг, 704 мкмоль, 1,00 экв.), 32a (101 мг, 703,8 мкмоль, 1,00 экв.) и DIPEA (273 мг, 2,11 ммоль, 131 мкл, 3,00 экв.) растворяли в DMSO (5 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии, дополнительно подвергали хиральному разделению посредством SFC с получением соединений 32-1 и 32-2.
[0398] Положение пика соединения 32-1: 2,105 мин. (хиральная колонка: AD-3 100 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,8 мл/мин., температура колонки: 40°C).
[0399] Соединение 32-1: MS-ESI: расчетное значение [M+H]+: 505, найденное значение: 505.
[0400] Соединение 32-1 1H ЯМР (400 MГц, CDCl3) δ: 0,56 (s, 1H), 8,13 (br d, J=8,0 Гц, 1H), 7,98 (d, J=8,0 Гц, 1H), 7,90 (d, J=7,8 Гц, 1H), 7,54-7,39 (m, 2H), 6,50 (br s, 1H), 4,34 (br d, J=7,2 Гц, 1H), 4,03-3,55 (m, 16H), 2,99 (d, J=4,8 Гц, 3H), 2,09-1,87 (m, 2H), 1,42 (d, J=6,8 Гц, 3H).
[0401] Положение пика соединения 32-2: 2,632 мин. (хиральная колонка: AD-3 100 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,8 мл/мин., температура колонки: 40°C).
[0402] Соединение 32-2: MS-ESI: расчетное значение [M+H]+: 505, найденное значение: 505.
[0403] Соединение 32-2: 1H ЯМР (400 MГц, CDCl3) δ: 0,56 (s, 1H), 8,13 (d, J=8,0 Гц, 1H), 7,98 (d, J=8,4 Гц, 1H), 7,90 (d, J=7,8 Гц, 1H), 7,53-7,42 (m, 2H), 6,52 (br s, 1H), 4,34 (br d, J=6,4 Гц, 1H), 4,04-3,54 (m, 16H), 2,98 (d, J=4,8 Гц, 3H), 2,08-1,88 (m, 2H), 1,42 (d, J=6,8 Гц, 3H).
[0404] Вариант осуществления 33-1 и 33-2
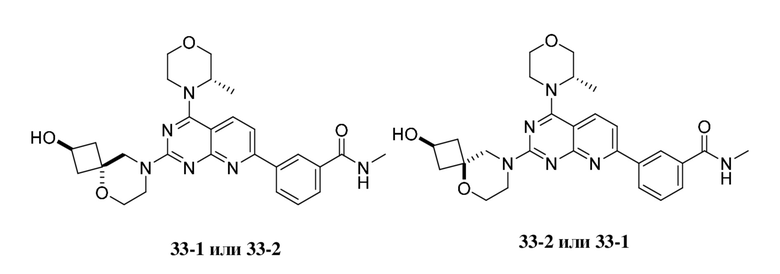
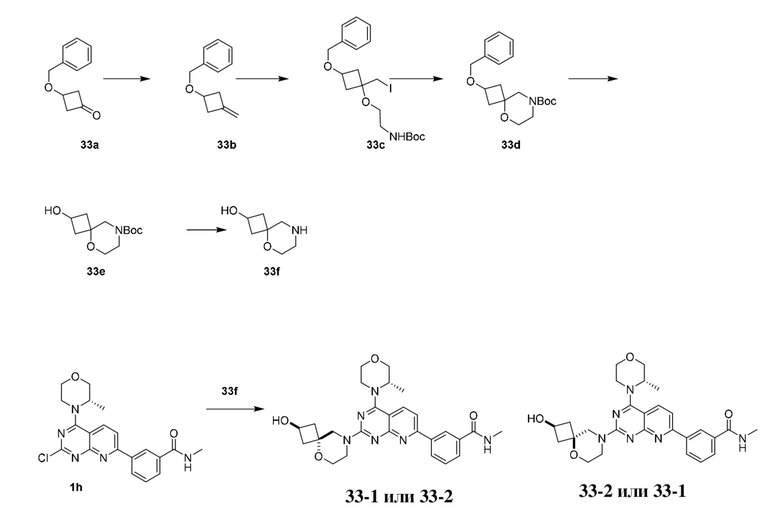
[0405] Первая стадия
[0406] Соединение метилтрифенилфосфония бромид (32,4 г, 90,8 ммоль, 1,6 экв.) растворяли в растворе трет-бутоксида калия (11,5 г, 102 ммоль, 1,8 экв.) в безводном тетрагидрофуране (400 мл) при комнатной температуре (20°C). Обеспечивали осуществление реакции в смешанном растворе при комнатной температуре в течение 3 часов, затем к раствору добавляли по каплям 33a (10,0 г, 56,8 ммоль, 1 экв.) и обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 17 часов. После завершения реакции проводили обнаружение посредством TLC, которое указывало на появление пятна нового соединения. К реакционному раствору добавляли воду (100 мл) и проводили экстрагирование с помощью этилацетата (200 мл × 3). Органическую фазу обрабатывали безводным сульфатом натрия, концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (50%-этилацетат/петролейный эфир) с получением 33b.
[0407] 1H ЯМР (400 МГц, CDCl3) δ: 7,23-7,13 (m, 5H), 4,74 (br s, 2H), 4,34-4,27 (m, 2H), 4,00-3,92 (m, 1H), 2,80-2,71 (m, 2H), 2,68-2,58 (m, 2H).
[0408] Вторая стадия
[0409] Соединение 33b (8,7 г, 49,9 ммоль, 1 экв.) и N-Boс-этаноламин (9,66 г, 59,9 ммоль, 9,29 мл, 1,2 экв.) растворяли в ацетонитриле (90,0 мл) с последующим добавлением N-йодсукцинимида (13,5 г, 59,9 ммоль, 1,2 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 18 часов. После завершения реакции проводили обнаружение посредством TLC, которое указывало на появление пятна нового соединения. В реакционный раствор добавляли воду (30,0 мл), проводили экстрагирование с помощью этилацетата (30 мл × 2). Органическую фазу обрабатывали безводным сульфатом натрия, концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (35,5%-этилацетат/петролейный эфир) с получением 33c.
[0410] 1H ЯМР (400 МГц, CDCl3) δ: 7,37-7,32 (m, 5H), 5,03 (br d, J = 10,8 Гц, 1H), 4,44 (d, J = 9,6 Гц, 2H), 3,79-3,67 (m, 1H), 3,34 (br d, J = 5,6 Гц, 3H), 2,59-2,37 (m, 3H), 2,31-2,11 (m, 2H), 2,08-1,98 (m, 1H), 1,52-1,42 (m, 9H).
[0411] Третья стадия
[0412] Соединение 33c (4 г, 8,67 ммоль, 1 экв.) растворяли в N,N-диметилацетамиде (160 мл) и затем к полученному раствору на ледяной бане добавляли гидрид натрия (694 мг, чистота 60%, 2 экв.). Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 17 часов. После завершения реакции реакционный раствор выливали в насыщенный водный раствор хлорида аммония (30 мл), добавляли воду (60,0 мл), экстрагировали с помощью этилацетата (50 мл × 3). Органическую фазу обрабатывали безводным сульфатом натрия, концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (30%-этилацетат/петролейный эфир) с получением 33d.
[0413] 1H ЯМР (400 МГц, CDCl3) δ: 7,38-7,32 (m, 5H), 4,47-4,40 (m, 2H), 4,23-4,15 (m, 1H), 3,62-3,53 (m, 2H), 3,45 (s, 1H), 3,41-3,32 (m, 2H), 3,25 (s, 1H), 2,43 (br s, 1H), 2,35-2,27 (m, 1H), 2,04 (s, 2H), 1,46 (d, J = 5,2 Гц, 9H).
[0414] Четвертая стадия
[0415] Соединение 33d (1,00 г, 3,00 ммоль, 1 экв.) растворяли в метаноле (30,0 мл) и затем к раствору добавляли гидроксид палладия (421 мг, чистота 20%, 0,2 экв.) в атмосфере азота. Несколько раз проводили замену водорода. Обеспечивали осуществление реакции в реакционном растворе при 50°C в течение 50 часов в атмосфере водорода при 50 фунтах/кв. дюйм. После завершения реакции реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении с получением 33e.
[0416] 1H ЯМР (400 МГц, CDCl3) δ: 4,06-3,87 (m, 1H), 3,51 (br s, 2H), 3,42-3,36 (m, 2H), 3,34-3,26 (m, 2H), 3,21-3,14 (m, 1H), 2,47-2,38 (m, 1H), 2,35-2,25 (m, 1H), 1,92-1,79 (m, 2H), 1,39 (s, 9H).
[0417] Пятая стадия
[0418] Соединение 33e (0,28 г, 1,15 ммоль, 1 экв.) растворяли в этилацетате (10,0 мл) и затем к раствору добавляли хлористоводородную кислоту/этилацетат (4 M, 10 мл, 34,8 экв.). Обеспечивали осуществление реакции в реакционном растворе при 20°C в течение 6 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением 33f.
[0419] Шестая стадия
[0420] Соединение 1h (0,3 г, 754 мкмоль, 1 экв.) растворяли в диметилсульфоксиде (10,0 мл) и затем к раствору добавляли N,N-диизопропилэтиламин (292 мг, 2,26 ммоль, 3 экв.) и 33f (162 мг, 902 мкмоль, 1,20 экв., HCl). Обеспечивали осуществление реакции в реакционном растворе при 70°C в течение 25 часов. После завершения реакции к реакционному раствору добавляли воду (12 мл) и проводили экстрагирование с помощью этилацетата (10,0 мл × 3). Органическую фазу обрабатывали безводным сульфатом натрия, концентрировали при пониженном давлении, очищали посредством колоночной хроматографии (12,8%-метанол/дихлорметан) и подвергали хиральному разделению с получением 33-1 и 33-2.
[0421] Положение пика соединения 33-1: 1,672 мин. (хиральная колонка: AD-3 100 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% изопропанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,8 мл/мин., температура колонки: 40°C).
[0422] Соединение 33-1: MS-ESI: расчетное значение [M+H]+: 505, найденное значение: 505.
[0423] Соединение 33-1: 1H ЯМР (400 MГц, CD3OD) δ: 8,62 (s, 1H), 8,34 (d, J=8,4 Гц, 1H), 8,29 (d, J=8,4 Гц, 1H), 7,96 (d, J=8,4 Гц, 1H), 7,68 (d, J=8,4 Гц, 1H), 7,66-7,62 (m, 1H), 4,54 (br d, J=7,2 Гц, 1H), 4,12 (br s, 1H), 4,04-3,93 (m, 3H), 3,93-3,70 (m, 10H), 3,02-2,97 (m, 3H), 2,56-2,52 (m, 2H), 1,96-1,92 (m, 2H), 1,51 (d, J=6,8 Гц, 3H).
[0424] Положение пика соединения 33-2: 3,338 мин. (хиральная колонка: AD-3 100 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% изопропанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,8 мл/мин., температура колонки: 40°C).
[0425] Соединение 33-2: MS-ESI: расчетное значение [M+H]+: 505, найденное значение: 505.
[0426] Соединение 33-2: 1H ЯМР (400 MГц, CD3OD) δ: 8,62 (s, 1H), 8,33 (d, J=7,8 Гц, 1H), 8,28 (d, J=8,4 Гц, 1H), 7,96 (d, J=7,8 Гц, 1H), 7,67 (d, J=8,4 Гц, 1H), 7,66-7,62 (m, 1H), 4,57 (br d, J=7,2 Гц, 1H), 4,44-4,36 (m, 1H), 4,08-3,95 (m, 4H), 3,95-3,85 (m, 3H), 3,82-3,70 (m, 5H), 2,99 (s, 3H), 2,43-2,40 (m, 2H), 2,04-1,95 (m, 2H), 1,51 (d, J=6,8 Гц, 3H).
[0427] Вариант осуществления 34-1 и 34-2
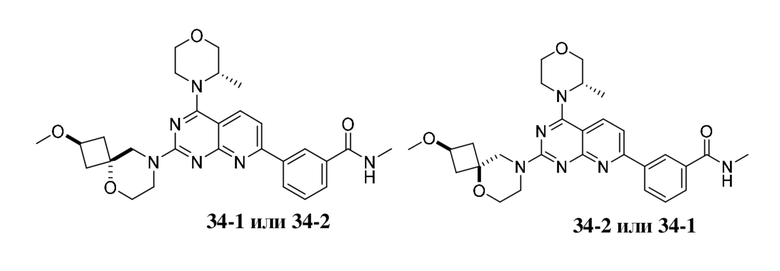
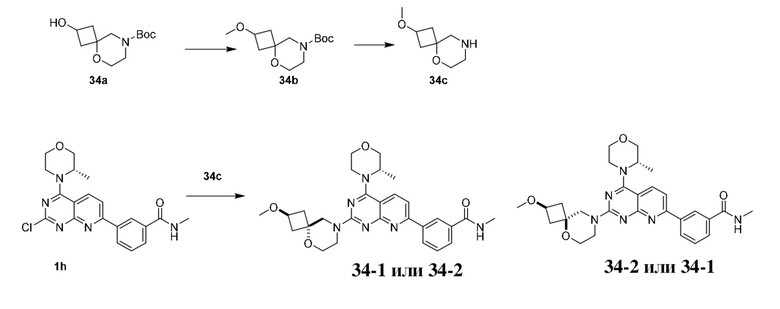
[0428] Первая стадия
[0429] Соединение 34a (0,27 г, 1,11 ммоль, 1 экв.) растворяли в безводном тетрагидрофуране (20,0 мл). К полученному смешанному раствору осторожно добавляли гидрид натрия (133 мг, чистота 60%, 3 экв.) и йодметан (1,19 г, 522 мкл, 7,55 экв.) при 0°C. Реакционный раствор постепенно нагревали до комнатной температуры и обеспечивали осуществление реакции в смеси при комнатной температуре в течение 3 часов. После завершения реакции проводили обнаружение посредством TLC, которое указывало на появление пятна нового соединения. Реакционный раствор концентрировали при пониженном давлении с получением 34b.
[0430] Вторая стадия
[0431] Соединение 34b (0,10 г, 389 мкмоль, 1 экв.) растворяли в этилацетате (8 мл). К полученному раствору добавляли хлористоводородную кислоту/этилацетат (4 M, 8,0 мл, 82,34 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 18 часов. После завершения реакции проводили обнаружение посредством TLC, которое указывало на появление пятна нового соединения. Реакционный раствор концентрировали при пониженном давлении с получением 34c.
[0432] Третья стадия
[0433] Соединение 1h (0,1 г, 251 мкмоль, 1 экв.) растворяли в диметилсульфоксиде (5,00 мл). К полученному раствору добавляли N,N-диизопропилэтиламин (65,0 мг, 87,6 мкл, 2 экв.) и 34c (73,0 мг, 377 мкмоль, 1,5 экв., HCl). Обеспечивали осуществление реакции в реакционном растворе при 70°C в течение 22 часов. После завершения реакции к реакционному раствору добавляли воду (20,0 мл) и проводили экстрагирование с помощью этилацетата (15,0 мл × 3). Органическую фазу обрабатывали безводным сульфатом натрия, концентрировали при пониженном давлении, очищали посредством колоночной хроматографии (100%-этилацетат/петролейный эфир) и подвергали хиральному разделению с получением 34-1 и 34-2.
[0434] Положение пика соединения 34-1: 2,407 минуты (хиральная колонка: OD-3 50 × 4,6 мм, I. D., 3 мкм, подвижная фаза: A: CO2, B: этанол (0,05% диэтиламин), содержание B поддерживали на уровне 5% в течение 0,2 минуты, затем увеличивали с градиентом от 5% до 40% в течение 1,4 мин., затем поддерживали на уровне 40% в течение 1,05 мин. и наконец поддерживали на уровне 5% в течение 0,35 мин., скорость потока: 4 мл/мин., температура колонки: 40°C).
[0435] Соединение 34-1: MS-ESI: расчетное значение [M+H]+: 519, найденное значение: 519.
[0436] Соединение 34-1: 1H ЯМР (400 MГц, CD3OD) δ: 8,64-8,60 (m, 1H), 8,34 (d, J=8,4 Гц, 1H), 8,29 (d, J=8,4 Гц, 1H), 7,96 (dd, J=1,2, 8,0 Гц, 1H), 7,68 (d, J=8,4 Гц, 1H), 7,66-7,61 (m, 1H), 4,55 (br d, J=7,2 Гц, 1H), 4,06-3,93 (m, 3H), 3,92-3,70 (m, 10H), 3,28 (s, 3H), 2,99 (s, 3H), 2,56-2,48 (m, 2H), 1,96-1,86 (m, 2H), 1,51 (d, J=6,8 Гц, 3H).
[0437] Положение пика соединения 34-2: 1,807 минуты (хиральная колонка: OD-3 50 × 4,6 мм, I. D., 3 мкм, подвижная фаза: A: CO2, B: этанол (0,05% диэтиламин), содержание B поддерживали на уровне 5% в течение 0,2 минуты, затем увеличивали с градиентом от 5% до 40% в течение 1,4 мин., затем поддерживали на уровне 40% в течение 1,05 мин. и вконце поддерживали на уровне 5% в течение 0,35 мин., скорость потока: 4 мл/мин., температура колонки: 40°C).
[0438] Соединение 34-2: MS-ESI: расчетное значение [M+H]+: 519, найденное значение: 519.
[0439] Соединение 34-2: 1H ЯМР (400 MГц, CD3OD) δ: 8,62 (s, 1H), 8,33 (d, J=8,03 Гц, 1H), 8,29 (d, J=8,4 Гц, 1H), 7,96 (d, J=8,4 Гц, 1H), 7,68 (d, J=8,4 Гц, 1H), 7,66-7,62 (m, 1H), 4,57 (br d, J=6,0 Гц, 1H), 4,08-3,97 (m, 5H), 3,94-3,2 (m, 8H), 3,27 (s, 3H), 2,99 (s, 3H), 2,39-2,27 (m, 2H), 2,09-2,01 (m, 2H), 1,51 (d, J=6,8 Гц, 3H).
[0440] Вариант осуществления 35
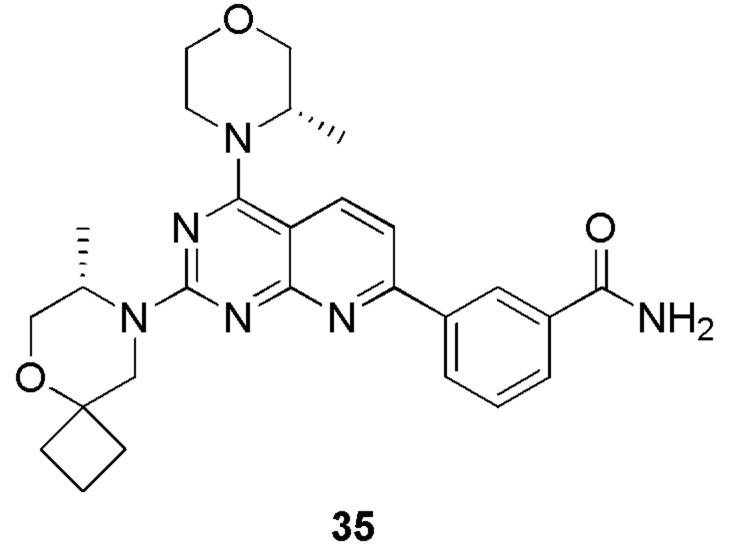
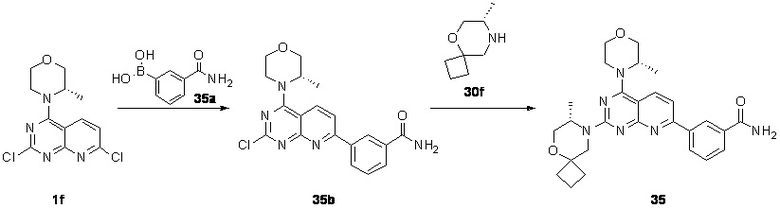
[0441] Первая стадия
[0442] Соединение 1f (2 г, 6,0 ммоль, 1 экв.), 35a (993 мг, 6,0 ммоль, 1 экв.), карбонат натрия (1,9 г, 18,1 ммоль, 3 экв.), дихлорбис(трифенилфосфин)палладий (211 мг, 301 мкмоль, 0,05 экв.) растворяли в безводном диоксане (35 мл) и воде (7,0 мл) и обеспечивали осуществление реакции в полученном растворе при 70°C в течение 19 часов в атмосфере азота. После завершения реакции реакционный раствор концентрировали при пониженном давлении, затем добавляли в него воду (15 мл) и проводили экстрагирование с помощью этилацетата (20 мл × 3). Органическую фазу обрабатывали безводным сульфатом натрия, концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии (метанол/этилацетат; Rf = 0,28) с получением 35b.
[0443] MS-ESI: расчетное значение [M+H]+: 384 и 386, найденное значение: 384 и 386.
[0444] Вторая стадия
[0445] Соединение 35b (0,1 г, 261 мкмоль, 1 экв.), 30f (73,6 мг, 414 мкмоль, 1,6 экв.), N,N-диизопропилэтиламин (33,7 мг, 261 мкмоль, 1 экв.) растворяли в диметилсульфоксиде (5,00 мл). Обеспечивали осуществление реакции в реакционном растворе при 70°C в течение 40 часов. После завершения реакции реакционный раствор фильтровали и очищали посредством высокоэффективной жидкостной хроматографии с получением 35.
[0446] MS-ESI: расчетное значение [M+H]+: 489, найденное значение: 489.
[0447] 1H ЯМР (400 MГц, CD3OD) δ: 8,70 (s, 1H), 8,36 (d, J=8,0 Гц, 1H), 8,30 (d, J=8,8 Гц, 1H), 8,04 (d, J=8,0 Гц, 1H), 7,68 (d, J=8,8 Гц, 1H), 7,68-7,64 (m, 1H), 4,88 (s, 2H), 4,56 (d, J=6,4 Гц, 1H), 4,04-3,96 (m, 2H), 3,90 (dd, J=2,8, 11,6 Гц, 1H), 3,84 (dd, J=3,2, 11,6 Гц, 1H), 3,77-3,75 (m, 3H), 3,59 (d, J=11,6 Гц, 1H), 3,14 (dd, J=1,6, 13,6 Гц, 1H), 2,20-2,08 (m, 2H), 2,08-1,96 (m, 2H), 1,92-1,76 (m, 2H), 1,50 (d, J=6,8 Гц, 3H), 1,36 (d, J=6,8 Гц, 3H).
[0448] Вариант осуществления 36
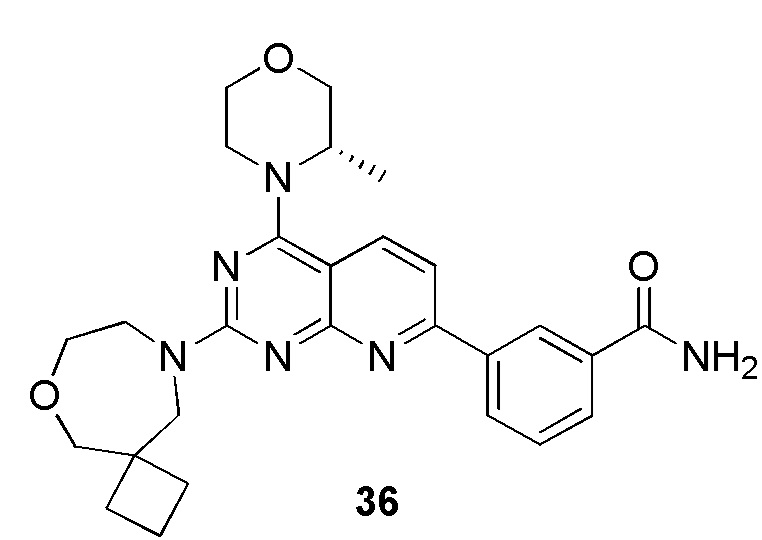
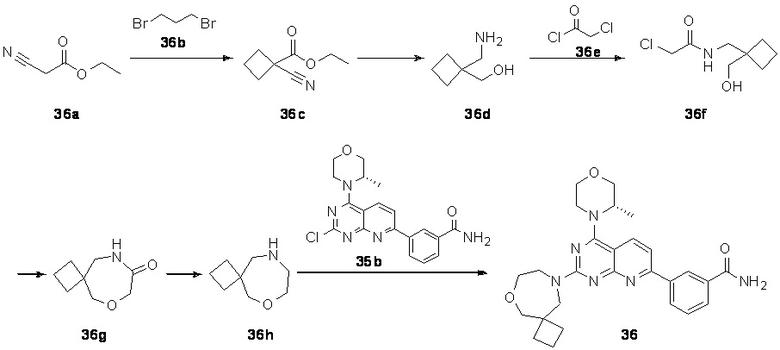
[0449] Первая стадия
[0450] Натрий (3,66 г, 159 ммоль, 3,77 мл, 2 экв.) растворяли в этаноле (147 мл). Добавляли 36a (9 г, 79,6 ммоль, 8,49 мл, 1 экв.) с последующим добавлением соединения 36b (16,1 г, 79,6 ммоль, 8,11 мл, 1 экв.). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 3 часов, затем подвергали выпариванию на роторном испарителе с удалением этанола, выливали в воду (500 мл), проводили экстрагирование с помощью этилацетата (100 мл), высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (20% этилацетат) с получением соединения 36c.
[0451] 1H ЯМР (400 МГц, CDCl3) δ: 4,26-4,14 (m, 2H), 2,67-2,56 (m, 4H), 2,20-2,04 (m, 2H), 1,28-1,25 (m, 3H).
[0452] Вторая стадия
[0453] Алюмогидрид лития (1,98 г, 52,2 ммоль, 2 экв.) растворяли в тетрагидрофуране (100 мл), добавляли соединение 36c (4,00 г, 26,1 ммоль, 1 экв.) и обеспечивали осуществление реакции в смешанном растворе при 45°C в течение 18 часов. После завершения реакции последовательно добавляли воду (2,00 мл), 15% NaOH (2,00 мл) и воду (6,00 мл) при 0-20°C для гашения реакции. Затем смесь перемешивали в течение 0,5 часа, фильтровали и подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния с получением соединения 36d.
[0454] 1H ЯМР (400 МГц, CDCl3) δ: 3,75 (s, 2H), 2,97 (s, 2H), 1,93-1,73 (m, 6H).
[0455] Третья стадия
[0456] Соединение 36d (0,5 г, 4,34 ммоль, 1 экв.) растворяли в дихлорметане (10,0 мл), добавляли карбонат натрия (920 мг, 8,68 ммоль, 2 экв.) и соединение 36e (539 мг, 4,77 ммоль, 380 мкл, 1,1 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение часа. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали с помощью колоночной хроматографии (20% метанол/дихлорметан) с получением соединения 36f.
[0457] 1H ЯМР (400 МГц, CDCl3) δ: 7,00 (s, 1H), 4,11-4,08 (m, 3H), 3,54-4,54 (m, 5H), 3,04-3,01 (m, 1H), 1,98-1,94 (m, 2H), 1,82-1,75 (m, 4H).
[0458] Четвертая стадия
[0459] Соединение 36f (0,22 г, 1,15 ммоль, 1 экв.) растворяли в тетрагидрофуране (22,0 мл) и добавляли гидрид натрия (138 мг, 3,44 ммоль, 60%, 3 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение часа. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (20% метанол/дихлорметан) с получением соединения 36g.
[0460] MS-ESI: расчетное значение [M+H]+: 156, найденное значение: 156.
[0461] Пятая стадия
[0462] Соединение 36g (0,35 г, 2,26 ммоль, 1 экв.) растворяли в тетрагидрофуране (10,0 мл), добавляли алюмогидрид лития (257 мг, 6,77 ммоль, 3 экв.). Обеспечивали осуществление реакции в смешанном растворе при 25°C в течение 18 часов. После завершения реакции последовательно добавляли воду (0,26 мл), 15% NaOH (0,26 мл) и воду (0,78 мл) при 0-20°C для гашения реакции. Реакционный раствор перемешивали в течение 0,5 часа, фильтровали и подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния с получением соединения 36h.
[0463] MS-ESI: расчетное значение [M+H]+: 142, найденное значение: 142.
[0464] Шестая стадия
[0465] Соединение 36h (0,1 г, 261 мкмоль, 1 экв.), 35b (55,18 мг, 391 мкмоль, 1,5 экв.), DIPEA (101 мг, 782 мкмоль, 136 мкл, 3 экв.) растворяли в DMSO (3,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 16 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 36.
[0466] MS-ESI: расчетное значение [M+H]+: 489, найденное значение: 489.
[0467] 1H ЯМР (400 МГц, CDCl3) δ: 8,70 (br s, 1H), 8,24 (br d, J=7,6 Гц, 1H), 8,08-7,03 (m, 2H), 7,59 (t, J=7,6 Гц, 1H), 7,49 (d, J=8,4 Гц, 1H), 6,70 (br s, 1H), 5,76 (br s, 1H), 4,40 (br s, 1H), 4,16 (br d, J=12,4 Гц, 2H), 4,02 (br s, 2H), 3,99-3,81 (m, 5H), 3,81-3,61 (m, 5H), 1,92 (br s, 6H), 1,49 (d, J=6,8 Гц, 3H).
[0468] Вариант осуществления 37
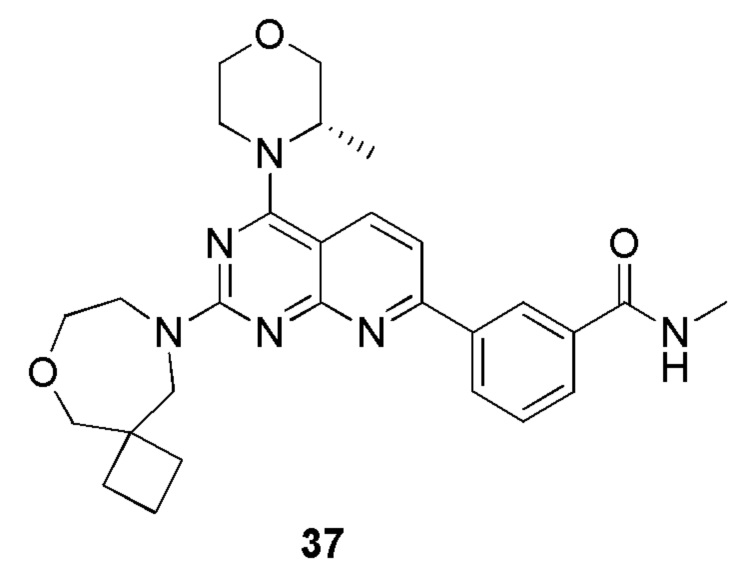
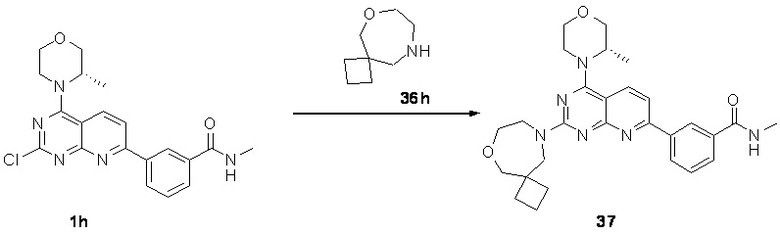
[0469] Соединение 1h (0,1 г, 251 мкмоль, 1 экв.), 36h (53,2 мг, 377 мкмоль, 1,5 экв.), DIPEA (97,5 мг, 754 мкмоль, 131 мкл, 3 экв.) растворяли в DMSO (5,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 16 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 37.
[0470] MS-ESI: расчетное значение [M+H]+: 503, найденное значение: 503.
[0471] 1H ЯМР (400 МГц, CDCl3) δ: 8,64 (br s, 1H), 8,19 (br d, J=7,2 Гц, 1H), 8,06 (br d, J=8,4 Гц, 1H), 7,98 (br d, J=7,6 Гц, 1H), 7,57 (t, J=7,6 Гц, 1H), 7,49 (d, J=8,4 Гц, 1H), 6,70 (br s, 1H), 4,40 (br s, 1H), 4,16 (br d, J=12,0 Гц, 2H), 4,02 (br s, 2H), 3,96-3,84 (m, 4H), 3,84-3,59 (m, 6H), 3,07 (d, J=4,8 Гц, 3H), 2,11-1,84 (m, 6H), 1,49 (d, J=6,8 Гц, 3H).
[0472] Вариант осуществления 38
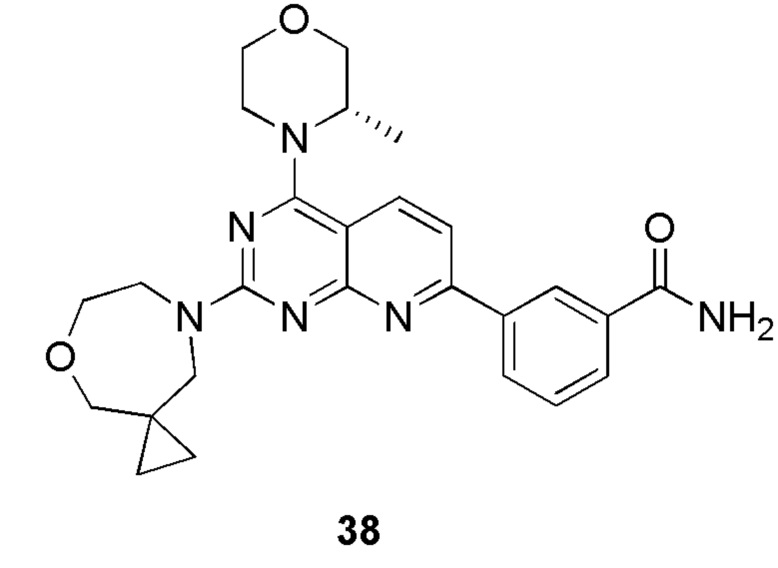
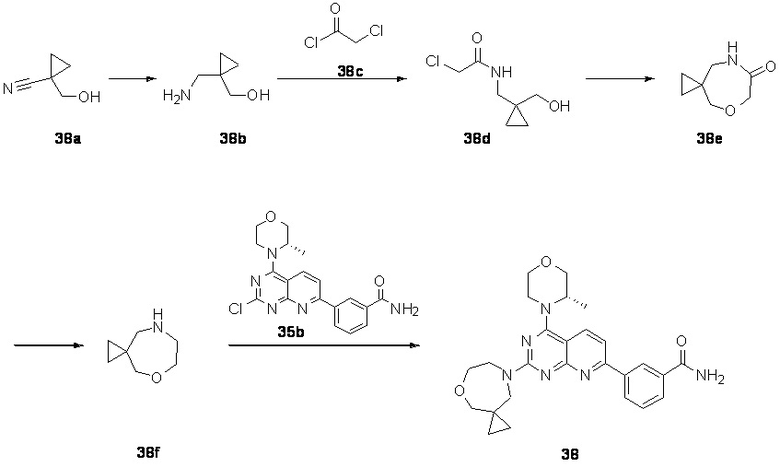
[0473] Первая стадия
[0474] Растворяли алюмогидрид лития (3,91 г, 103 ммоль, 2 экв.) в тетрагидрофуране (100 мл) и добавляли соединение 38a (5,00 г, 51,5 ммоль, 3,37 мл, 1 экв.). Обеспечивали осуществление реакции в смешанном растворе при 45°C в течение 18 часов. После завершения реакции последовательно добавляли воду (4,00 мл), 15% NaOH (4,00 мл) и воду (8,00 мл) при 0-20°C для гашения реакции. Смесь перемешивали в течение 0,5 часа, фильтровали и подвергали выпариванию на роторном испарителе при пониженном давлении с получением 38b.
[0475] MS-ESI: расчетное значение [M+H]+: 102, найденное значение: 102.
[0476] Вторая стадия
[0477] Соединение 38b (4 г, 39,6 ммоль, 1 экв.) растворяли в дихлорметане (10,0 мл), добавляли карбонат натрия (8,38 г, 79,1 ммоль, 2 экв.) и соединение 38c (4,47 г, 39,6 ммоль, 3,15 мл, 1,0 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение часа. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (20% метанол/дихлорметан) с получением соединения 38d.
[0478] MS-ESI: расчетное значение [M+H]+: 178, найденное значение: 178.
[0479] 1H ЯМР (400 МГц, CDCl3) δ: 7,27 (s, 1H), 4,09 (s, 2H), 3,44 (d, J=5,2 Гц, 2H), 3,32 (d, J=5,6 Гц, 2H), 2,83-2,81 (m, 1H), 0,52 (d, J=3,6 Гц, 4H).
[0480] Третья стадия
[0481] Соединение 38d (4,70 г, 26,5 ммоль, 1 экв.) растворяли в тетрагидрофуране (470 мл) и добавляли гидрид натрия (3,18 г, 79,4 ммоль, чистота 60%, 3 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение часа. После завершения реакции к реакционному раствору добавляли хлористоводородную кислоту для регулирования pH до 7, высушивали его над безводным сульфатом натрия с получением соединения 38e.
[0482] MS-ESI: расчетное значение [M+H]+: 142, найденное значение: 142.
[0483] Четвертая стадия
[0484] Соединение 38e (0,5 г, 3,54 ммоль, 1 экв.) растворяли в тетрагидрофуране (10 мл) и добавляли алюмогидрид лития (269 мг, 7,08 ммоль, 2 экв.). Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение часа. После завершения реакции последовательно добавляли воду (0,26 мл), 15% NaOH (0,26 мл) и воду (0,78 мл) при 0-20°C для гашения реакции. Реакционный раствор перемешивали в течение 0,5 часа, фильтровали и подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния с получением соединения 38f.
[0485] MS-ESI: расчетное значение [M+H]+: 128, найденное значение: 128.
[0486] Пятая стадия
[0487] Соединение 38f (48,2 мг, 126 мкмоль, 1 экв.), 35b (159,84 мг, 1,26 ммоль, 10 экв.), DIPEA (48,7 мг, 377 мкмоль, 65,7 мкл, 3 экв.) растворяли в DMSO (5,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 16 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 38.
[0488] MS-ESI: расчетное значение [M+H]+: 475, найденное значение: 475.
[0489] 1H ЯМР (400 МГц, CDCl3) δ: 8,69 (br s, 1H), 8,25 (br d, J=7,2 Гц, 1H), 8,06-8,03 (m, 2H), 7,71-7,40 (m, 2H), 6,63 (br s, 1H), 5,80 (br s, 1H), 4,48-4,17 (m, 3H), 4,07-3,94 (m, 2H), 3,87 (br s, 4H), 3,83-3,62 (m, 4H), 3,55 (s, 2H), 1,48 (br d, J=6,4 Гц, 3H), 0,85-0,31 (m, 4H).
[0490] Вариант осуществления 39
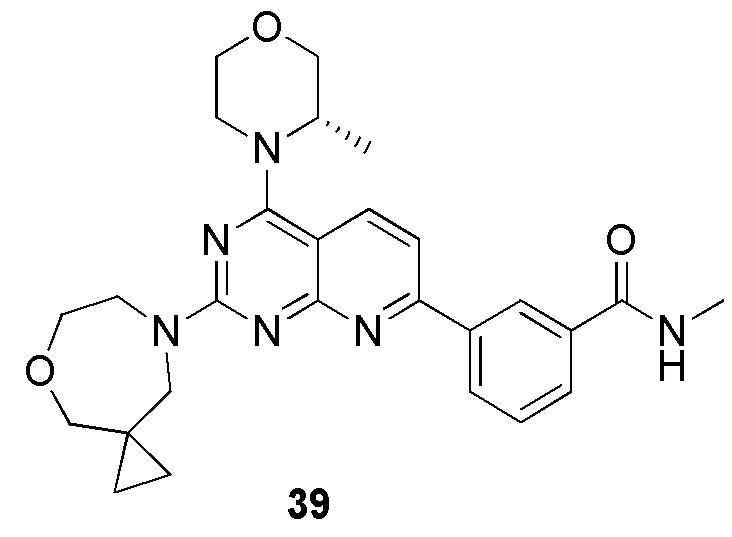
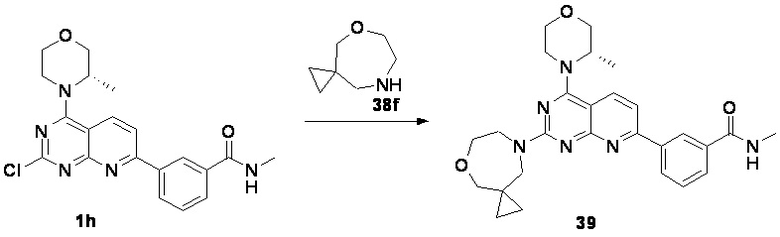
[0491] Первая стадия
[0492] Соединение 38f (141 мг, 354 мкмоль, 0,25 экв.), 1h (200 мг, 1,42 ммоль, 1 экв, HCl), DIPEA (549 мг, 4,25 ммоль, 740 мкл, 3 экв.) растворяли в DMSO (5,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 70°C в течение 16 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 39.
[0493] MS-ESI: расчетное значение [M+H]+: 489, найденное значение: 489.
[0494] 1H ЯМР (400 МГц, CDCl3) δ: 8,64 (br s, 1H), 8,21 (br d, J=7,6 Гц, 1H), 8,06 (d, J=8,4 Гц, 1H), 7,99 (br d, J=7,6 Гц, 1H), 7,58 (t, J=7,6 Гц, 1H), 7,50 (br d, J=8,4 Гц, 1H), 6,59 (br s, 1H), 4,38 (br d, J=6,8 Гц, 1H), 4,34-4,12 (m, 2H), 4,07-3,96 (m, 2H), 3,96-3,81 (m, 5H), 3,81-3,61 (m, 3H), 3,56 (s, 2H), 3,08 (d, J=4,8 Гц, 3H), 1,49 (br d, J=6,8 Гц, 3H), 0,80-0,54 (m, 4H).
[0495] Вариант осуществления 40
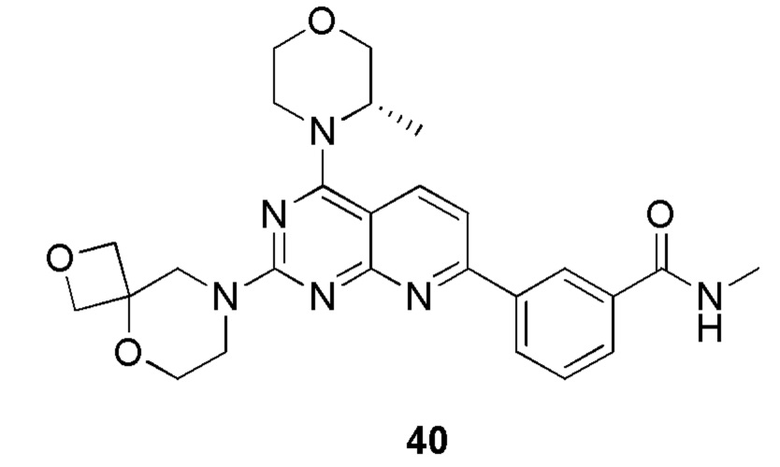
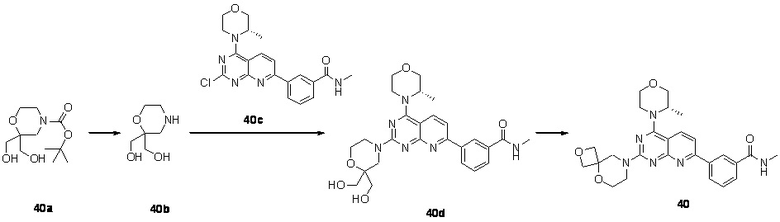
[0496] Первая стадия
[0497] Соединение 40a (0,28 г, 1,13 ммоль, 1 экв.) растворяли в трифторуксусной кислоте (5,00 мл) и дихлорметане (10,0 мл). Реакционный раствор перемешивали и обеспечивали осуществление реакции в нем при комнатной температуре в течение 2 часов. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали 40b.
[0498] MS-ESI: расчетное значение [M+H]+: 148, найденное значение: 148.
[0499] Вторая стадия
[0500] Соединение 40b (300 мг, 1,15 ммоль, 1 экв., TFA), соединение 40c (320 мг, 804 мкмоль, 0,7 экв.), DIPEA (445 мг, 3,45 ммоль, 600 мкл, 4 экв.) растворяли в диметилсульфоксиде (5,00 мл) и обеспечивали осуществление реакции в смеси при 70°C в течение 16 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 40d.
[0501] MS-ESI: расчетное значение [M+H]+: 509, найденное значение: 509.
[0502] Третья стадия
[0503] Соединение 40d (100 мг, 197 мкмоль, 1 экв.), п-толуолсульфонилхлорид (37,5 мг, 197 мкмоль, 1 экв.) и гидрид натрия (15,7 мг, 393 мкмоль, 60%, 2 экв.) растворяли в DMF (10,0 мл), обеспечивали осуществление реакции в смеси при комнатной температуре в течение 16 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 40.
[0504] MS-ESI: расчетное значение [M+H]+: 491, найденное значение: 491.
[0505] 1H ЯМР (400 МГц, CDCl3) δ: 8,65 (s, 1H), 8,23 (br d, J=7,8 Гц, 1H), 8,09 (d, J=8,4 Гц, 1H), 7,99 (br d, J=7,8 Гц, 1H), 7,62-7,53 (m, 2H), 6,60 (br s, 1H), 4,66 (d, J=6,4 Гц, 2H), 4,57-4,41 (m, 3H), 4,32-4,14 (m, 2H), 4,08-3,85 (m, 5H), 3,83-3,75 (m, 5H), 3,08 (d, J=4,8 Гц, 3H), 1,52 (d, J=6,8 Гц, 3H).
[0506] Вариант осуществления 41
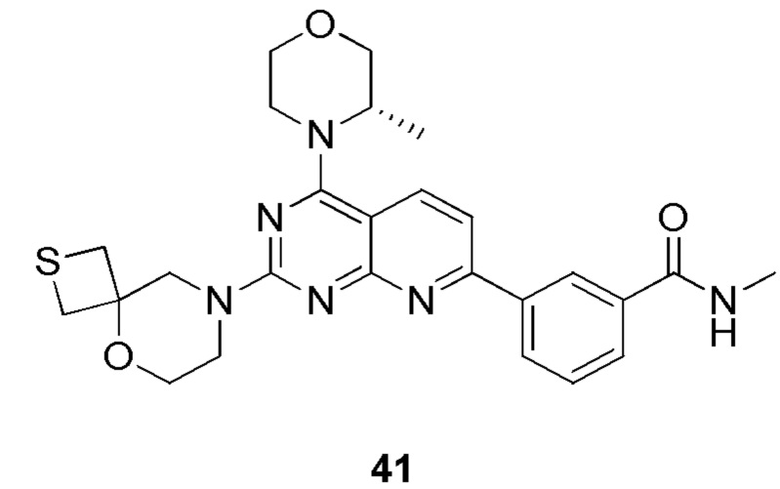
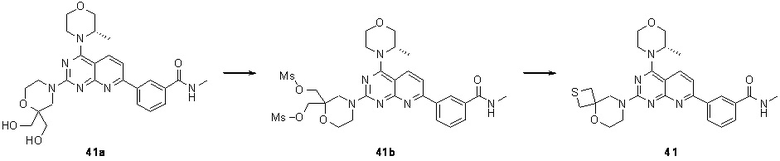
[0507] Первая стадия
[0508] Соединение 41a (70,0 мг, 138 мкмоль, 1 экв.), метансульфонилхлорид (0,8 г, 6,98 ммоль, 541 мкл, 50,7 экв.) и триэтиламин (27,9 мг, 275 мкмоль, 38,3 мкл, 2 экв.) растворяли в дихлорметане (10,0 мл) и обеспечивали осуществление реакции в смеси при комнатной температуре в течение 16 часов. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали соединение 41b.
[0509] MS-ESI: расчетное значение [M+H]+: 665, найденное значение: 665.
[0510] Вторая стадия
[0511] Соединение 41b (59,9 мг, 90,26 мкмоль, 1 экв.), тетра-н-бутиламмония йодид (3,33 мг, 9,03 мкмоль, 0,1 экв.), сульфид натрия (21,1 мг, 271 мкмоль, 11,4 мкл, 3 экв.) растворяли в N,N'-диметилформамиде (5,00 мл) и обеспечивали осуществление реакции в смеси при 70°C в течение 18 часов в защитной атмосфере азота. После завершения реакции реакционный раствор промывали водой (50,0 мл × 3) и очищали посредством высокоэффективной жидкостной хроматографии с получением 41.
[0512] MS-ESI: расчетное значение [M+H]+: 507, найденное значение: 507.
[0513] 1H ЯМР (400 МГц, CDCl3) δ: 8,67 (br s, 1H), 8,20 (br d, J=8,0 Гц, 1H), 8,10-8,03 (m, 1H), 7,99 (br d, J=8,0 Гц, 1H), 7,60-7,52 (m, 2H), 6,64 (br s, 1H), 4,46 (br d, J=5,6 Гц, 1H), 4,42-4,33 (m, 1H), 4,19 (br d, J=13,2 Гц, 1H), 4,09-3,91 (m, 3H), 3,91-3,69 (m, 7H), 3,42 (br d, J=10,0 Гц, 2H), 3,06 (d, J=4,8 Гц, 3H), 3,00 (br d, J=8,0 Гц, 2H), 1,53 (br d, J=6,8 Гц, 3H).
[0514] Вариант осуществления 42
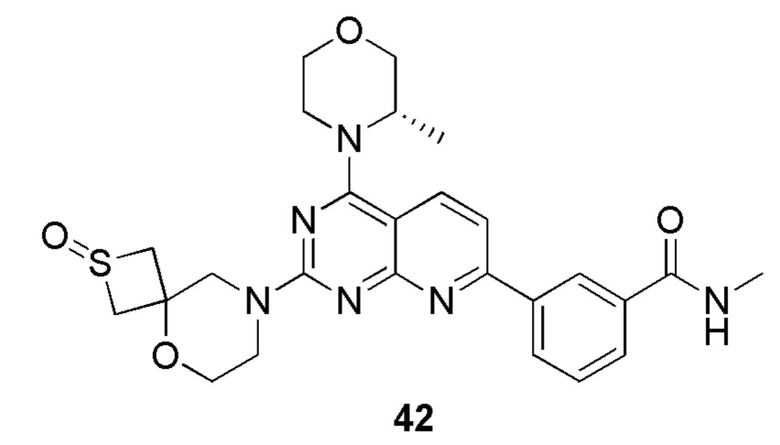
[0515] Соединение 41 (70,0 мг, 138 мкмоль, 1 экв.) растворяли в метаноле (20,0 мл), добавляли по каплям водный раствор (10,0 мл) моноперсульфата калия (84,9 мг, 138 мкмоль, 1 экв.). Реакцию проводили при комнатной температуре в течение часа. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 42.
[0516] MS-ESI: расчетное значение [M+H]+: 523, найденное значение: 523.
[0517] 1H ЯМР (400 МГц, CDCl3) δ: 8,72-7,51 (m, 1H), 8,11 (br d, J=7,2 Гц, 1H), 8,00 (d, J=8,4 Гц, 1H), 7,93 (br d, J=7,8 Гц, 1H), 7,55-7,43 (m, 2H), 4,49-4,33 (m, 1H), 4,20 (s, 2H), 3,94 (br d, J=8,4 Гц, 3H), 3,82-3,74 (m, 1H), 3,74-3,60 (m, 5H), 3,52 (br d, J=13,6 Гц, 2H), 3,28-3,06 (m, 2H), 3,00-2,94 (m, 3H), 1,44 (d, J=6,8 Гц, 3H).
[0518] Вариант осуществления 43
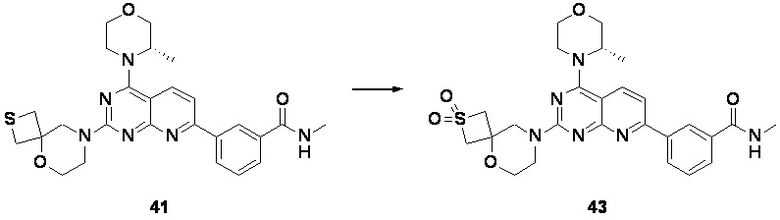
[0519] Соединение 41 (120 мг, 237 мкмоль, 1 экв.) растворяли в метаноле (5,00 мл), добавляли по каплям водный раствор (5,00 мл) моноперсульфата калия (291 мг, 474 мкмоль, 2 экв.). Реакцию проводили при комнатной температуре в течение 30 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 43.
[0520] MS-ESI: расчетное значение [M+H]+: 539, найденное значение: 539.
[0521] 1H ЯМР (400 МГц, CDCl3) δ: 8,58 (br s, 1H), 8,14 (br d, J=8,0 Гц, 1H), 8,02 (d, J=8,4 Гц, 1H), 7,91 (br d, J=7,6 Гц, 1H), 7,60-7,44 (m, 2H), 6,57 (br s, 1H), 4,42 (br d, J=6,0 Гц, 1H), 4,27-3,85 (m, 10H), 3,82-3,60 (m, 6H), 2,99 (d, J=4,8 Гц, 3H), 1,46 (d, J=6,8 Гц, 3H).
[0522] Вариант осуществления 44
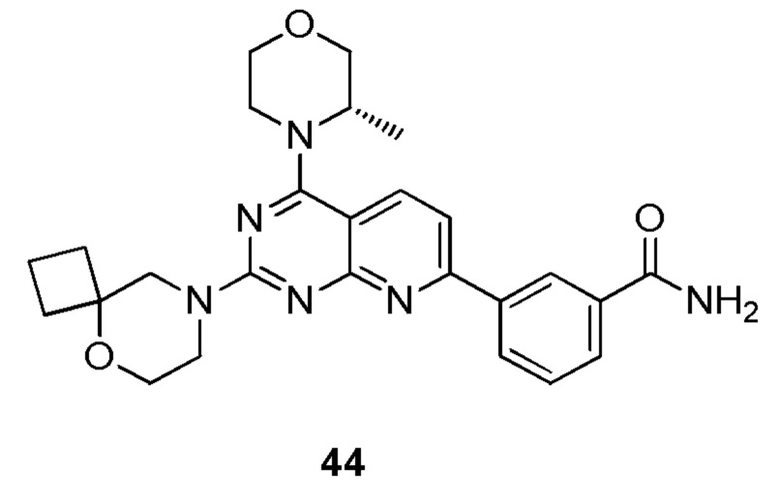
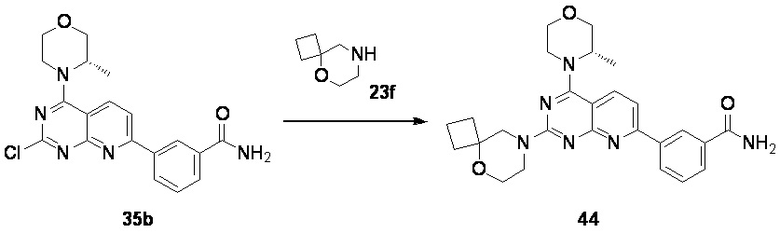
[0523] Соединение 35b (150 мг, 391 мкмоль, 1 экв.), 23f (128 мг, 782 мкмоль, 1 экв.) и DIPEA (152 мг, 1,17 ммоль, 3 экв.) растворяли в DMSO (3,00 мл) и обеспечивали осуществление реакции в смешанном растворе при 90°C в течение 20 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 44.
[0524] MS-ESI: расчетное значение [M+H]+: 475, найденное значение: 475.
[0525] 1H ЯМР (400 МГц, CDCl3) δ: 8,66 (br s, 1H), 8,18 (br d, J=7,6 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,95 (br d, J=7,6 Гц, 1H), 7,51 (t, J=7,6 Гц, 1H), 7,46 (br d, J=8,4 Гц, 1H), 6,72-6,11 (m, 1H), 5,64 (br s, 1H), 4,35 (br s, 1H), 3,95-3,57 (m, 11H), 2,07-1,77 (m, 7H), 1,45 (br d, J=6,8 Гц, 3H).
[0526] Вариант осуществления 45-1 и 45-2
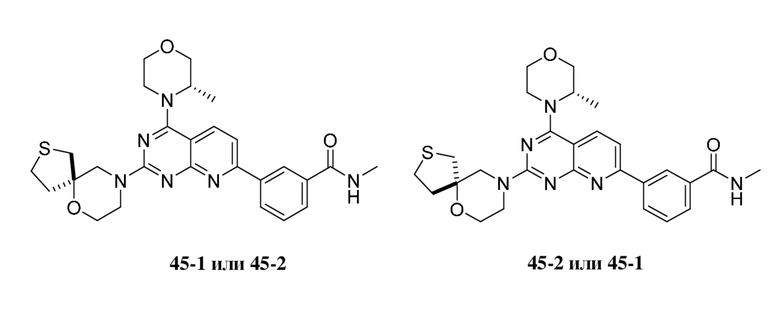
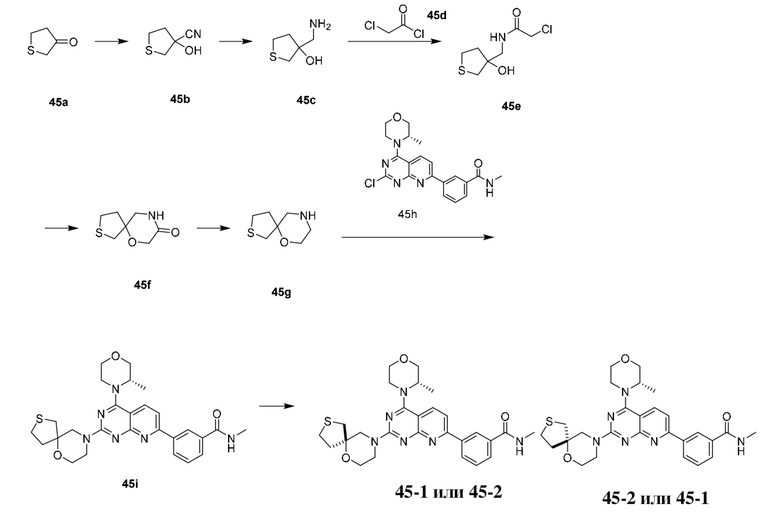
[0527] Первая стадия
[0528] К раствору соединения 45a (13,0 г, 127 ммоль, 1,0 экв.) в дихлорметане (250 мл) последовательно добавляли TMSCN (18,9 г, 191 ммоль, 1,5 экв.) и комплекс эфира с трифторидом бора (18,1 г, 127 ммоль, 1,0 экв.) и обеспечивали осуществление реакции в смеси при 20°C в течение 15 часов. После завершения реакции реакционный раствор концентрировали, очищали посредством колоночной хроматографии (2:1 петролейный эфир/этилацетат) с получением 45b.
[0529] 1H ЯМР (400 МГц, CDCl3) δ: 3,34 (d, J=12,0 Гц, 1H), 3,19 (s, 1H), 3,16 (dd, J=1,6, 12,0 Гц, 1H), 3,13-3,01 (m, 2H), 2,59-2,47 (m, 1H), 2,32 (td, J=8,8, 12,8 Гц, 1H).
[0530] Вторая стадия
[0531] Комплекс боран-тетрагидрофуран (1 M, 157 мл, 1,5 экв.) добавляли по каплям к раствору соединения 45b в тетрагидрофуране (200 мл) при 0°C. Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 17 часов. К реакционному раствору добавляли 10 мл метанола для гашения реакции. Реакционный раствор концентрировали с получением 45c и неочищенный продукт непосредственно использовали на следующей стадии.
[0532] Третья стадия
[0533] Соединение 45d (8,48 г, 75,1 ммоль, 1,0 экв.) добавляли по каплям к раствору соединения 45c (10,0 г, 75,1 ммоль, 1,0 экв.) и диизопропилэтиламина (19,4 г, 150 ммоль, 2,0 экв.) в дихлорметане (150 мл) при 0°C. Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 3 часов. К реакционному раствору добавляли 5 мл воды для гашения реакции. Реакционный раствор концентрировали и очищали посредством колоночной хроматографии (100% этилацетат) с получением 45e.
[0534] 1H ЯМР (400 МГц, CDCl3) δ: 7,12-6,88 (m, 1H), 4,06-3,99 (m, 2H), 3,55 (d, J =6,0 Гц, 2H), 3,01-2,84 (m, 3H), 2,82 (d, J=11,2 Гц, 1H), 2,68 (dd, J=1,6, 11,6 Гц, 1H), 2,08-1,99 (m, 1H), 1,81 (td, J=9,2, 13,2 Гц, 1H).
[0535] Четвертая стадия
[0536] К раствору соединения 45e (6,0 г, 28,6 ммоль, 1,0 экв.) в тетрагидрофуране (500 мл) добавляли трет-бутоксид калия (9,63 г, 85,8 ммоль, 3,0 экв.) при 0°C. Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение 2 часов. Реакционный раствор концентрировали и очищали посредством колоночной хроматографии (1:10 метанол/этилацетат) с получением 45f.
[0537] 1H ЯМР (400 МГц, CDCl3) δ: 6,43 (br s, 1H), 4,26 (s, 2H), 3,59-3,39 (m, 2H), 3,13-3,00 (m, 2H), 2,96-2,78 (m, 2H), 2,47-2,32 (m, 1H), 2,02-1,88 (m, 1H).
[0538] Пятая стадия
[0539] К раствору соединения 45f (1,0 г, 5,77 ммоль, 1,0 экв.) в безводном тетрагидрофуране (30,0 мл) добавляли алюмогидрид лития (329 мг, 8,66 ммоль, 1,5 экв.) при 0°C. Обеспечивали осуществление реакции в смешанном растворе при 20°C в течение часа. К реакционному раствору последовательно добавляли 0,3 мл воды, 0,3 мл 15% водного раствора гидроксида натрия и 1 мл воды и фильтровали. Осадок на фильтре ополаскивали с помощью 10 мл этилацетата. Фильтрат концентрировали с получением 45g. Неочищенный продукт непосредственно использовали на следующей стадии.
[0540] Шестая стадия
[0541] Соединение 45g (92,1 мг, 578 мкмоль, 2,3 экв.), 45h (100 мг, 251 мкмоль, 1,00 экв.), DIPEA (97,5 мг, 754 мкмоль, 3,00 экв.) растворяли в DMSO (3,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 18 часов. После завершения реакции реакционный раствор разбавляли водой (10,0 мл) и экстрагировали с помощью этилацетата (20,0 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством препаративной тонкослойной хроматографии (100% этилацетат) с получением 45i, который дополнительно подвергали хиральному разделению с помощью SFC с получением соединений 45-1 и 45-2.
[0542] Соединение 45-1: MS-ESI: расчетное значение [M+H]+: 521, найденное значение: 521.
[0543] Положение пика соединения 45-1: 0,950 мин. (хиральная колонка: AD-3 50 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 4 мл/мин., температура колонки: 40°C).
[0544] Соединение 45-1: 1H ЯМР (400 MГц, CDCl3) δ: 8,57 (s, 1H), 8,13 (br d, J=7,6 Гц, 1H), 7,98 (d, J=8,4 Гц, 1H), 7,90 (d, J=8,0 Гц, 1H), 7,52-7,42 (m, 2H), 6,57 (br s, 1H), 4,35 (br d, J=6,8 Гц, 1H), 4,22 (d, J=13,2 Гц, 1H), 4,13 (br s, 1H), 3,95-3,61 (m, 10H), 2,98 (d, J=4,8 Гц, 3H), 2,97-2,78 (m, 4H), 2,21 (td, J=6,0, 12,4 Гц, 1H), 1,99-1,88 (m, 1H), 1,43 (d, J=6,8 Гц, 3H).
[0545] Соединение 45-2: MS-ESI: расчетное значение [M+H]+: 521, найденное значение: 521.
[0546] Положение пика соединения 45-2: 1,168 мин. (хиральная колонка: AD-3 50 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 4 мл/мин., температура колонки: 40°C).
[0547] Соединение 45-2: 1H ЯМР (400 MГц, CDCl3) δ: 8,57 (s, 1H), 8,13 (br d, J=7,6 Гц, 1H), 7,98 (d, J=8,4 Гц, 1H), 7,90 (d, J=8,0 Гц, 1H), 7,52-7,43 (m, 2H), 6,54 (br s, 1H), 4,34 (br d, J=6,8 Гц, 1H), 4,23 (br d, J=13,2 Гц, 1H), 4,17 (br s, 1H), 3,95-3,60 (m, 10H), 2,99 (d, J=4,8 Гц, 3H), 2,97-2,76 (m, 4H), 2,20 (td, J=6,0, 12,4 Гц, 1H), 2,00-1,89 (m, 1H), 1,42 (d, J=6,8 Гц, 3H).
[0548] Вариант осуществления 46-1 и 46-2
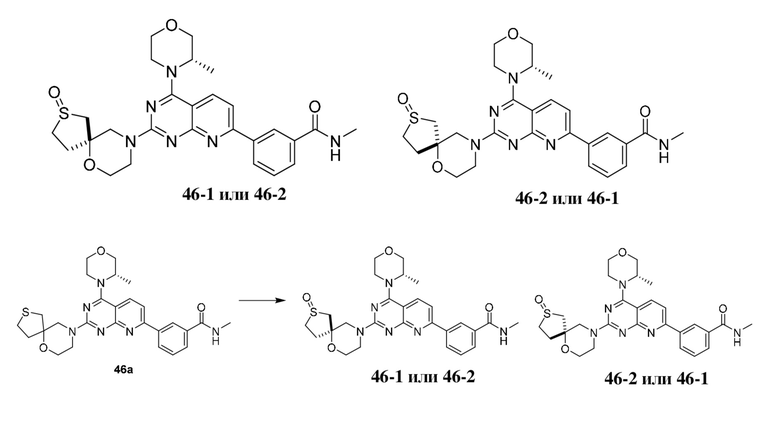
[0549] К раствору соединения 46a (400 мг, 768 мкмоль, 1,0 экв.) в метаноле (60,0 мл) добавляли по каплям водный раствор (40,0 мл) моноперсульфата калия (402 мг, 653 мкмоль, 0,85 экв.). Обеспечивали осуществление реакции при 25°C в течение 1,5 часа и гасили ее посредством добавления 10 мл насыщенного раствора тиосульфата натрия. Реакционный раствор концентрировали до объема, составляющего приблизительно 50 мл, и экстрагировали с помощью этилацетата (30 мл × 3). Объединенные органические фазы высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (1:10 метанол/этилацетат) с получением рацемата, который дополнительно подвергали хиральному разделению с помощью SFC с получением соединений 46-1 и 46-2.
[0550] Положение пика соединения 46-1: 5,297 минуты (хиральная колонка: AD-3 150 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% метанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,5 мл/мин., температура колонки: 40°C).
[0551] Соединение 46-1: MS-ESI: расчетное значение [M+H]+ 537, найденное значение: 537.
[0552] Соединение 46-1: 1H ЯМР (400 MГц, CDCl3) δ: 8,69 (br s, 1H), 8,13-7,91 (m, 3H), 7,53-7,40 (m, 2H), 4,46-4,22 (br d, J=6,8 Гц, 2H), 4,14 (br d, J=12,2 Гц, 1H), 3,97-3,62 (m, 10H), 3,17 (br s, 1H), 2,97 (d, J=4,8 Гц, 6H), 2,68 (br d, J=6,4 Гц, 1H), 2,49-2,38 (m, 1H), 1,43 (d, J=6,8 Гц, 3H).
[0553] Положение пика соединения 46-2: 6,265 минут (хиральная колонка: AD-3 150×4,6 мм, I. D., 3 мкм, подвижная фаза: 40% метанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 2,5 мл/мин., температура колонки: 40°C).
[0554] Соединение 46-2: MS-ESI: расчетное значение [M+H]+: 537, найденное значение: 537.
[0555] Соединение 46-2: 1H ЯМР (400 MГц, CDCl3) δ: 8,70 (br s, 1H), 8,13-7,91 (m, 3H), 7,55-7,39 (m, 2H), 4,45-4,22 (m, 2H), 4,20-4,08 (m, 1H), 3,94-3,63 (m, 10H), 3,17 (br s, 1H), 2,97 (d, J=4,8 Гц, 6H), 2,78-2,61 (m, 1H), 2,44 (br d, J=14,1 Гц, 1H), 1,41 (d, J=6,8 Гц, 3H).
[0556] Варианты осуществления 47-1 и 47-2
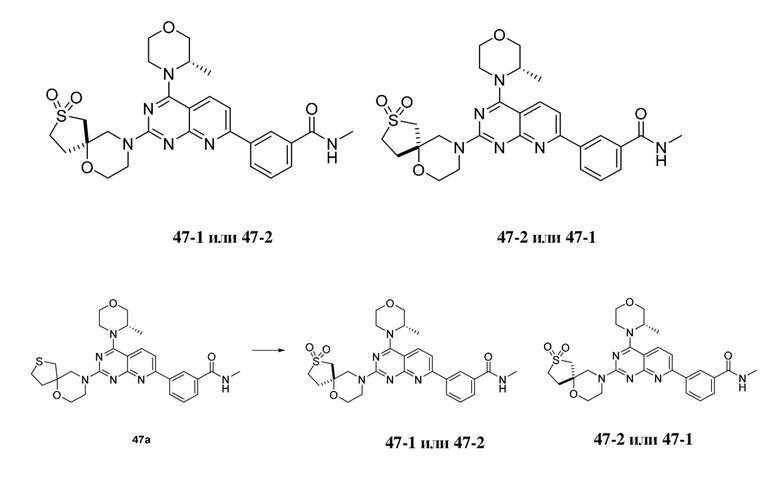
[0557] К раствору соединения 47a (200 мг, 384 мкмоль, 1,0 экв.) в метаноле (30,0 мл) добавляли по каплям водный раствор (30,0 мл) моноперсульфата калия (779 мг, 1,27 ммоль, 3,3 экв.). Обеспечивали осуществление реакции при 25°C в течение 5 часа и гасили ее посредством добавления 10 мл насыщенного раствора тиосульфата натрия. Реакционный раствор концентрировали до объема, составляющего приблизительно 40 мл, и экстрагировали с помощью дихлорметана (50,0 мл × 4). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством препаративной тонкослойной хроматографии (1:20 метанол/дихлорметан) с получением рацемического промежуточного соединения, которое дополнительно подвергали хиральному разделению с помощью SFC с получением соединений 47-1 и 47-2.
[0558] Соединение 47-1: MS-ESI: расчетное значение [M+H]+: 553, найденное значение: 553.
[0559] Положение пика соединения 47-1: 1,617 мин. (хиральная колонка: AD-3 50 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 4 мл/мин., температура колонки: 40°C).
[0560] Соединение 47-1: 1H ЯМР (400 MГц, CDCl3) δ: 8,65 (s, 1H), 8,23 (d, J=8,0 Гц, 1H), 8,10 (d, J=8,4 Гц, 1H), 7,99 (d, J=8,0 Гц, 1H), 7,64-7,55 (m, 2H), 6,58 (br s, 1H), 4,66-4,34 (m, 3H), 4,07-3,55 (m, 10H), 3,45-3,18 (m, 4H), 3,08 (d, J=4,8 Гц, 3H), 2,57-2,45 (m, 1H), 2,38-2,26 (m, 1H), 1,53 (d, J=6,8 Гц, 3H).
[0561] Соединение 47-2: MS-ESI: расчетное значение [M+H]+: 553, найденное значение: 553.
[0562] Положение пика соединения 47-2: 2,000 мин. (хиральная колонка: AD-3 50 × 4,6 мм, I. D., 3 мкм, подвижная фаза: 40% этанол (0,05% диэтиламин) + диоксид углерода, скорость потока: 4 мл/мин., температура колонки: 40°C).
[0563] Соединение 47-2: 1H ЯМР (400 MГц, CDCl3) δ: 8,65 (s, 1H), 8,23 (d, J=8,0 Гц, 1H), 8,10 (d, J=8,4 Гц, 1H), 7,99 (d, J=8,0 Гц, 1H), 7,64-7,55 (m, 2H), 6,58 (br s, 1H), 4,66-4,34 (m, 3H), 4,07-3,55 (m, 10H), 3,45-3,18 (m, 4H), 3,08 (d, J=4,8 Гц, 3H), 2,57-2,45 (m, 1H), 2,38-2,26 (m, 1H), 1,53 (d, J=6,8 Гц, 3H).
[0564] Варианты осуществления 48-1 и 48-2
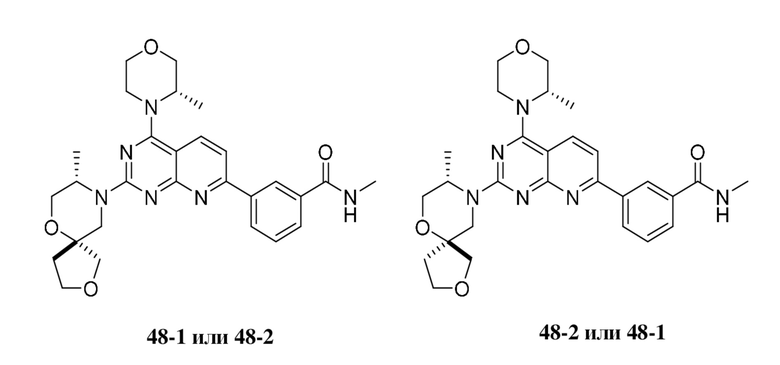
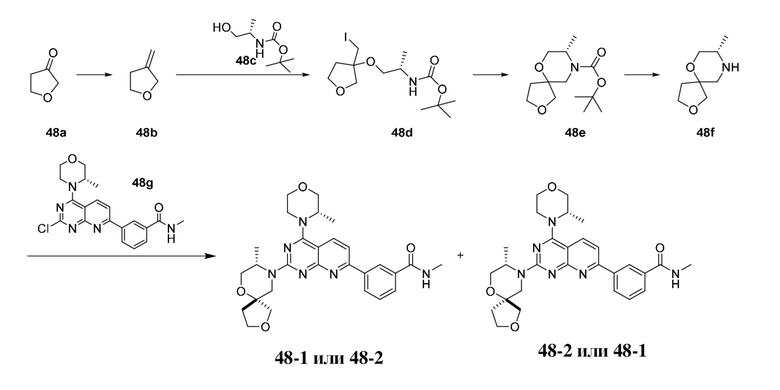
[0565] Первая стадия
[0566] Метилтрифенилфосфония бромид (83,0 г, 232 ммоль, 2,0 экв.) и трет-бутоксид калия (26,01, 232 ммоль, 2,0 экв.) растворяли в тетрагидрофуране (1,00 л) и обеспечивали осуществление реакции в смеси при 20°C в течение часа. Соединение 48a (10,0 г, 116,16 ммоль, 1,0 экв.) добавляли к реакционному раствору и обеспечивали осуществление реакции в смеси при 20°C в течение 17 часов. Реакционный раствор подвергали перегонке при 80°C при нормальном давлении с получением дистиллята, представляющего собой соединение 48b.
[0567] 1H ЯМР (400 МГц, CDCl3) δ: 5,05-4,91 (m, 2H), 4,26 (s, 2H), 3,90 (t, J=6,8 Гц, 2H), 2,56 (br t, J=6,8 Гц, 2H).
[0568] Вторая стадия
[0569] Соединение 48b (6,00 г, 28,5 ммоль, чистота 4%, 1 экв.), соединение 48c (5,0 г, 28,5 ммоль, 1,0 экв.) и N-йодсукцинимид (6,42 г, 28,5 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (80,0 мл) и обеспечивали осуществление реакции в смеси при 60°C в течение 17 часов. После завершения реакции реакцию гасили с помощью 100 мл насыщенного водного раствора тиосульфата натрия. Реакционный раствор экстрагировали с помощью этилацетата (150 мл × 3) и органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (1: 1 этилацетат/петролейный эфир) с получением соединения 48d.
[0570] MS-ESI: расчетное значение [M+H]+: 386, найденное значение: 386.
[0571] Третья стадия
[0572] Соединение 48d (0,2 г, 519 мкмоль, 1,0 экв.) и гидрид натрия (41,5 мг, 1,04 ммоль, чистота 60%, 2,0 экв.) растворяли в N,N-диметилформамиде (10,0 мл) и обеспечивали осуществление реакции в смеси при 25°C в течение 16 часов. После завершения реакции реакцию гасили посредством добавления воды (1,00 мл). Реакционный раствор концентрировали, разбавляли с помощью 10,0 мл воды, экстрагировали с помощью этилацетата (10,0 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали и подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния с получением 48e.
[0573] Четвертая стадия
[0574] Соединение 48e (0,15 г, 583 мкмоль, 1 экв.) растворяли в хлористоводородной кислоте/этилацетате (10,0 мл, 2 M) и обеспечивали осуществление реакции в смеси при 25°C в течение 17 часов при перемешивании. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния с получением 48f.
[0575] Пятая стадия
[0576] Соединение 48f (90,0 мг, 465 мкмоль, 1,85 экв.), соединение 48g (100 мг, 251 мкмоль, 1,0 экв.) и DIPEA (97,5 мг, 754 мкмоль, 3,0 экв.) растворяли в DMSO (5,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 18 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением рацемата, который дополнительно подвергали хиральному разделению с помощью SFC с получением соединений 48-1 и 48-2.
[0577] Положение пика соединения 48-1: 3,301 минуты (хиральная колонка: AS-H 150 × 4,6 мм, I. D., 5 мкм, подвижная фаза: A: CO2, B: этанол (0,05% диэтиламин), содержание B поддерживали на уровне 5% в течение 0,5 минуты, затем увеличивали с градиентом от 5% до 40% в течение 3,5 мин., затем поддерживали на уровне 40% в течение 2,5 мин. и наконец поддерживали на уровне 5% в течение 1,5 мин., скорость потока: 3 мл/мин., температура колонки: 40°C).
[0578] Соединение 48-1: MS-ESI: расчетное значение [M+H]+: 519, найденное значение: 519.
[0579] Соединение 48-1: 1H ЯМР (400 MГц, CDCl3) δ: 8,54 (s, 1H), 8,13 (d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,89 (d, J=8,0 Гц, 1H), 7,49 (t, J=8,0 Гц, 1H), 7,45 (d, J=8,4 Гц, 1H), 6,50 (br s, 1H), 4,93 (br s, 1H), 4,59 (br d, J=13,2 Гц, 1H), 4,33 (br d, J=6,4 Гц, 1H), 3,94-3,54 (m, 12H), 3,23 (d, J=13,6 Гц, 1H), 2,99 (d, J=5,2 Гц, 3H), 2,14-2,00 (m, 1H), 1,98-1,88 (m, 1H), 1,42 (d, J=6,8 Гц, 3H), 1,29 (br d, J=6,4 Гц, 3H).
[0580] Положение пика соединения 48-2: 3,608 мин. (хиральная колонка: AS-H 150 × 4,6 мм, I. D., 5 мкм, подвижная фаза: A: CO2, B: этанол (0,05% диэтиламин), содержание B поддерживали на уровне 5% в течение 0,5 минуты, затем увеличивали с градиентом от 5% до 40% в течение 3,5 мин., затем поддерживали на уровне 40% в течение 2,5 мин. и наконец поддерживали на уровне 5% в течение 1,5 мин., скорость потока: 3 мл/мин., температура колонки: 40°C).
[0581] Соединение 48-2: MS-ESI: расчетное значение [M+H]+: 519, найденное значение: 519.
[0582] Соединение 48-2: 1H ЯМР (400 MГц, CDCl3) δ: 8,54 (s, 1H), 8,13 (d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,89 (d, J=8,0 Гц, 1H), 7,49 (t, J=8,0 Гц, 1H), 7,45 (d, J=8,4 Гц, 1H), 6,50 (br s, 1H), 4,93 (br s, 1H), 4,59 (br d, J=13,2 Гц, 1H), 4,33 (br d, J=6,4 Гц, 1H), 3,94-3,54 (m, 12H), 3,23 (d, J=13,6 Гц, 1H), 2,99 (d, J=5,2 Гц, 3H), 2,14-2,00 (m, 1H), 1,98-1,88 (m, 1H), 1,42 (d, J=6,8 Гц, 3H), 1,29 (br d, J=6,4 Гц, 3H).
[0583] Вариант осуществления 49
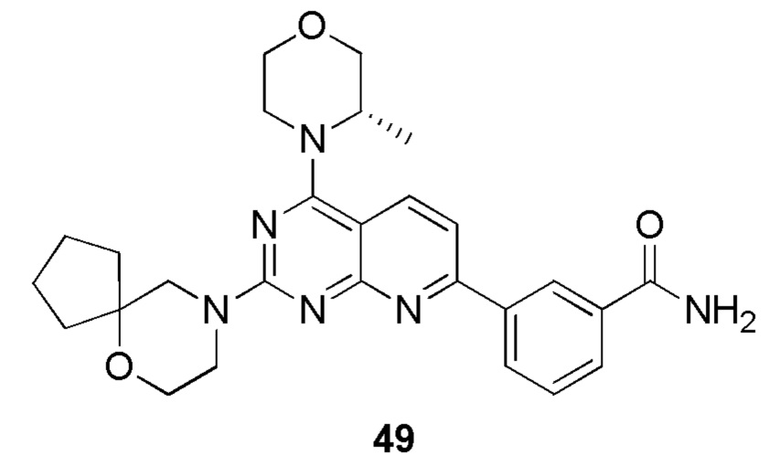
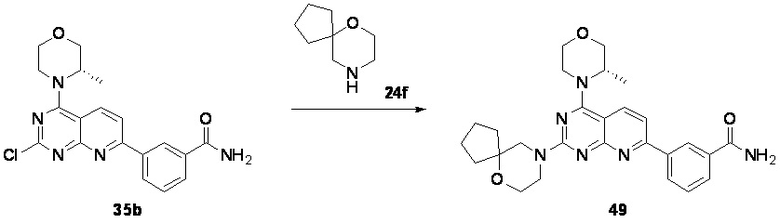
[0584] Соединение 35b (100 мг, 261 мкмоль, 1,00 экв.), 24f (92,6 мг, 522 мкмоль, 2,00 экв.), DIPEA (101 мг, 782 мкмоль, 136 мкл, 3,00 экв.) растворяли в DMSO (2,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 40 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 49.
[0585] MS-ESI: расчетное значение [M+H]+: 489, найденное значение: 489.
[0586] 1H ЯМР (400 МГц, CDCl3) δ: 8,62 (s, 1H), 8,18 (br d, J=7,6 Гц, 1H), 7,97 (d, J=8,4 Гц, 1H), 7,93 (br d, J=8,0 Гц, 1H), 7,51 (t, J=8,0 Гц, 1H), 7,43 (d, J=8,4 Гц, 1H), 6,52 (br s, 1H), 5,63 (br s, 1H), 4,29 (br s, 1H), 3,95-3,57 (m, 12H), 1,70 (br d, J=8,4 Гц, 8H), 1,41 (d, J=6,8 Гц, 3H).
[0587] Варианты осуществления 50-1 и 50-2
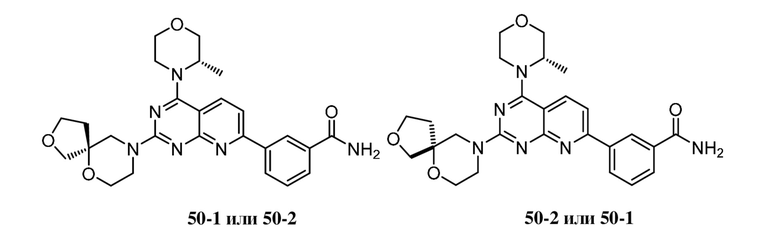
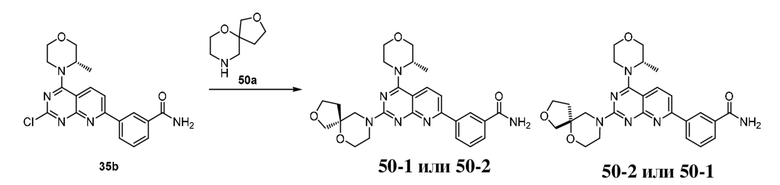
[0588] Соединение 35b (80,0 мг, 208 мкмоль, 1,00 экв.), 50a (44,9 мг, 250 мкмоль, 1,20 экв.) и DIPEA (80,8 мг, 625 мкмоль, 109 мкл, 3 экв.) растворяли в DMSO (2,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 22 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением рацемата, который дополнительно подвергали хиральному разделению с помощью SFC с получением 50-1 и 50-2.
[0589] Положение пика соединения 50-1: 3,491 мин. (хиральная колонка: OJ-3 100 × 4,6 мм, I. D., 3 мкм, подвижная фаза: A: CO2, B: метанол (0,05% диэтиламин), содержание B увеличивали с градиентом от 5% до 40% в течение 4,5 мин., затем поддерживали на уровне 5% в течение 1,0 мин., скорость потока: 2,8 мл/мин., температура колонки: 40°C).
[0590] 50-1: MS-ESI: расчетное значение [M+H]+: 491, найденное значение: 491.
[0591] 50-1: 1H ЯМР (400 MГц, CDCl3) δ: 8,63 (s, 1H), 8,18 (br d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,94 (d, J=80 Гц, 1H), 7,51 (t, J=7,2 Гц, 1H), 7,46 (d, J=8,0 Гц, 1H), 6,61 (br s, 1H), 5,69 (br s, 1H), 4,33 (br d, J=6,4 Гц, 1H), 3,97-3,59 (m, 16H), 2,07-1,85 (m, 2H), 1,41 (d, J=6,8 Гц, 3H).
[0592] Положение пика соединения 50-2: 3,846 мин. (хиральная колонка: OJ-3 100 × 4,6 мм, I. D., 3 мкм, подвижная фаза: A: CO2, B: метанол (0,05% диэтиламин), содержание B увеличивали с градиентом от 5% до 40% в течение 4,5 мин. и затем поддерживали на уровне 5% в течение 1,0 мин., скорость потока: 2,8 мл/мин., температура колонки: 40°C).
[0593] 50-2: MS-ESI: расчетное значение [M+H]+: 491, найденное значение: 491.
[0594] 50-2: 1H ЯМР (400 MГц, CDCl3) δ: 8,63 (s, 1H), 8,18 (br d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,94 (d, J=80 Гц, 1H), 7,51 (t, J=7,2 Гц, 1H), 7,46 (d, J=8,0 Гц, 1H), 6,61 (br s, 1H), 5,69 (br s, 1H), 4,33 (br d, J=6,4 Гц, 1H), 3,97-3,59 (m, 16H), 2,07-1,85 (m, 2H), 1,41 (d, J=6,8 Гц, 3H).
[0595] Вариант осуществления 51
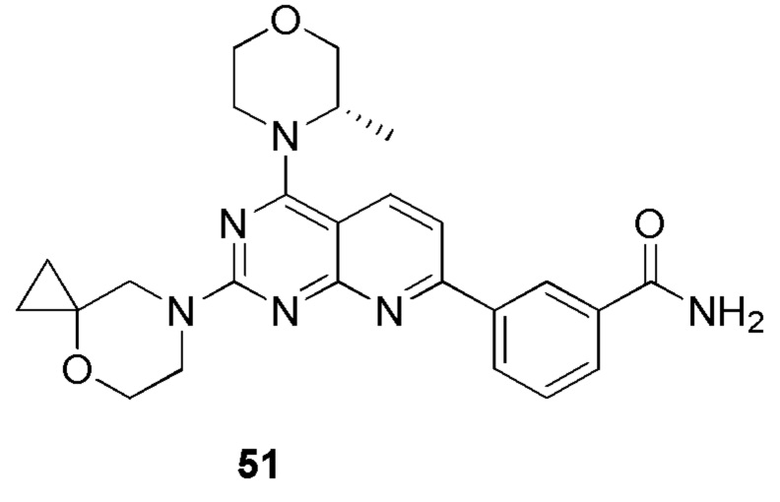
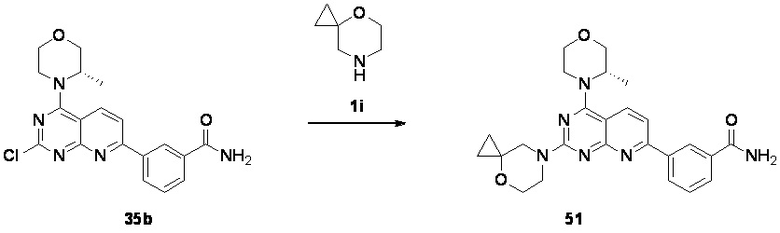
[0596] Соединение 35b (100 мг, 261 мкмоль, 1,00 экв.), 1i (46,8 мг, 313 мкмоль, 1,20 экв.) и DIPEA (101 мг, 782 мкмоль, 136 мкл, 3,00 экв.) растворяли в DMSO (2,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 18 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 51.
[0597] MS-ESI: расчетное значение [M+H]+: 461, найденное значение: 461.
[0598] 1H ЯМР (400 МГц, CDCl3) δ: 8,60 (s, 1H), 8,18 (br d, J=8,0 Гц, 1H), 7,98 (d, J=8,4 Гц, 1H), 7,92 (br d, J=8,0 Гц, 1H), 7,51 (t, J=8,0 Гц, 1H), 7,43 (d, J=8,4 Гц, 1H), 6,46 (br s, 1H), 5,62 (br s, 1H), 4,30 (br s, 1H), 4,05-3,59 (m, 12H), 1,40 (d, J=6,8 Гц, 3H), 0,80-0,73 (m, 2H), 0,61 (br s, 2H).
[0599] Вариант осуществления 52
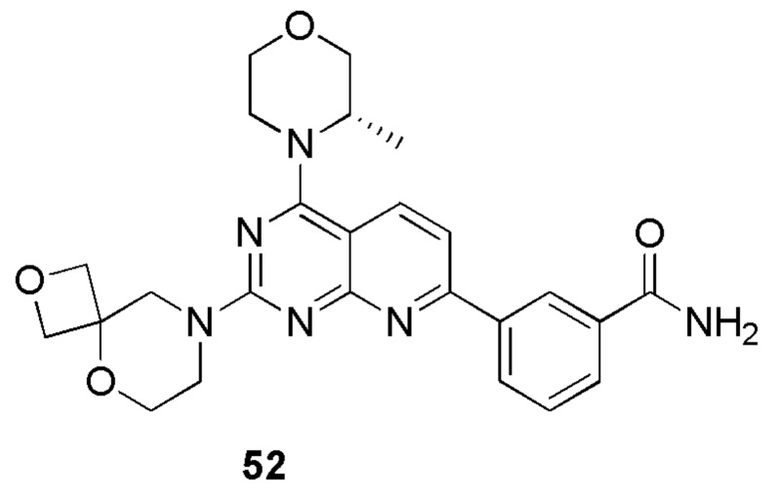
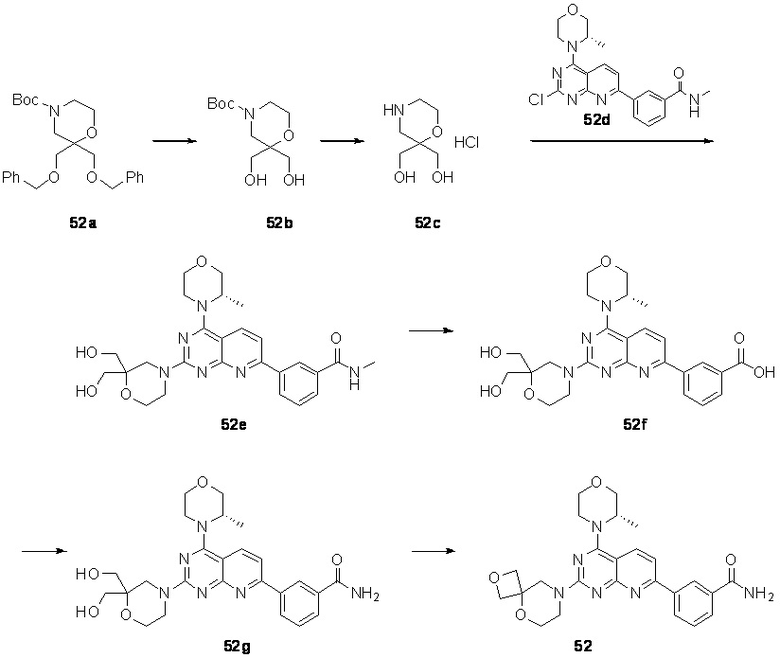
[0600] Первая стадия
[0601] Соединение 52a (900 мг, 2,11 ммоль, 1 экв.) растворяли в метаноле (20,0 мл) и к нему добавляли гидроксид палладия (591 мг, 421 мкмоль, 0,2 экв.). Обеспечивали осуществление реакции в смешанном растворе при 50°C в течение 42 часов в атмосфере водорода, 50 фунтов/кв. дюйм, затем фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния с получением соединения 52b.
[0602] 1H ЯМР (400 МГц, CDCl3) δ: 3,80-3,58 (m, 6H), 3,52-3,50 (m, 1H), 3,51 (s, 1H), 3,50-3,38 (m, 4H), 1,49 (s, 9H)
[0603] Вторая стадия
[0604] Соединение 52b (500 мг, 2,02 ммоль, 1 экв.) растворяли в растворе хлористоводородная кислота-этилацетат (20,0 мл, 2 M), обеспечивали осуществление реакции в смеси при 25°C в течение часа. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали неочищенный продукт, представляющий собой соединение 52c.
[0605] 1H ЯМР (400 MГц, CD3OD) δ: 4,15-4,10 (m, 2H), 3,71-3,63 (m, 4H), 3,31 (s, 2H), 3,23-3,17 (m, 2H).
[0606] Третья стадия
[0607] Соединение 52c (350 мг, 1,91 ммоль, 1 экв.), 52d (531 мг, 1,33 ммоль, 0,7 экв.) и DIPEA (668 мг, 5,17 ммоль, 0,9 мл, 2,71 экв.) растворяли в DMSO (7,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 23 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 52e.
[0608] MS-ESI: расчетное значение [M+H]+ 509, найденное значение: 509.
[0609] 1H ЯМР (400 МГц, CDCl3) δ: 8,61 (s, 1H), 8,21 (br d, J=8,0 Гц, 1H), 8,08 (d, J=8,4 Гц, 1H), 7,99 (d, J=8,0 Гц, 1H), 7,60-7,54 (m, 2H), 4,46 (br d, J=4,8 Гц, 1H), 4,34-3,45 (m, 19H), 3,07 (d, J=4,8 Гц, 3H), 1,52 (d, J=6,8 Гц, 3H).
[0610] Четвертая стадия
[0611] Соединение 52e (300 мг, 590 мкмоль, 1 экв.) растворяли в этаноле (15,0 мл) и воде (15,0 мл), к полученному добавляли гидроксид натрия (118 мг, 2,95 ммоль, 5 экв.). Обеспечивали осуществление реакции в смешанном растворе при 105°C в течение 285 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением желтого твердого вещества 52f.
[0612] MS-ESI: расчетное значение [M+H]+: 496, найденное значение: 496.
[0613] 1H ЯМР (400 MГц, DMSO-d6) δ: 8,78 (s, 1H), 8,28 (br d, J=8,0 Гц, 1H), 8,23 (d, J=8,4 Гц, 1H), 8,05 (br d, J=8,0 Гц, 1H), 7,68 (d, J=8,4 Гц, 1H), 7,58 (t, J=8,0 Гц, 1H), 4,66-4,39 (m, 3H), 3,96-3,41 (m, 14H), 1,38 (d, J=6,8 Гц, 3H).
[0614] Пятая стадия
[0615] Соединение 52f (78 мг, 157 мкмоль, 1 экв.), хлорид аммония (33,7 мг, 630 мкмоль, 4 экв.), DIPEA (81,4 мг, 630 мкмоль, 110 мкл, 4 экв.), EDCI (60,4 мг, 315 мкмоль, 2 экв.) и HOBt (42,5 мг, 315 мкмоль, 2 экв.) растворяли в дихлорметане (10,0 мл). Обеспечивали осуществление реакции в смешанном растворе при 30°C в течение 16 часов. После завершения реакции добавляли воду (3 мл) для гашения реакции. Реакционный раствор концентрировали с удалением растворителя, разбавляли водой (20,0 мл) и экстрагировали с помощью этилацетата (20,0 мл × 3) и метанола (5,00 мл). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали 52g.
[0616] MS-ESI: расчетное значение [M+H]+: 495, найденное значение: 495.
[0617] Шестая стадия
[0618] Соединение 52g (87,4 мг, 177 мкмоль, 1 экв.), TosCl (50,5 мг, 265 мкмоль, 1,5 экв.) и гидрид натрия (35,3 мг, 884 мкмоль, чистота 60%, 5 экв.) растворяли в DMF (4,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 30°C в течение 24 часов и очищали его посредством высокоэффективной жидкостной хроматографии с получением 52.
[0619] MS-ESI: расчетное значение [M+H]+: 477, найденное значение: 477.
[0620] 1H ЯМР (400 МГц, CDCl3) δ: 8,62 (s, 1H), 8,19 (br d, J=8,0 Гц, 1H), 8,00 (d, J=8,4 Гц, 1H), 7,94 (br d, J=8,0 Гц, 1H), 7,54-7,45 (m, 2H), 6,54 (br s, 1H), 5,73 (br s, 1H), 4,57 (d, J=6,8 Гц, 2H), 4,47-4,35 (m, 3H), 4,21-4,07 (m, 2H), 3,96-3,65 (m, 10H), 1,43 (d, J=6,8 Гц, 3H).
[0621] Варианты осуществления 53-1 и 53-2
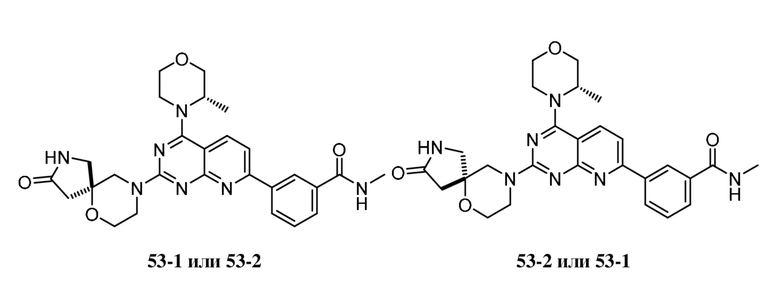
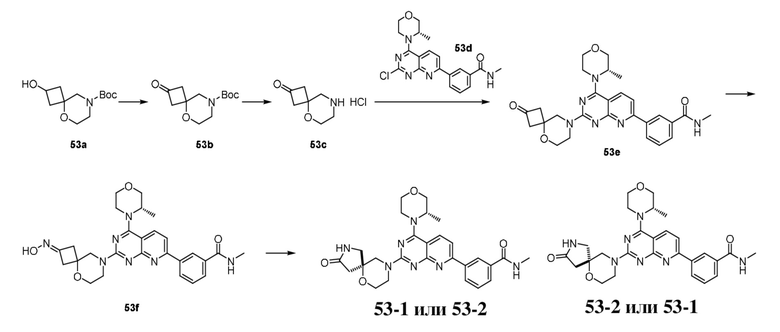
[0622] Первая стадия
[0623] К раствору соединения 53a (0,5 г, 2,06 ммоль, 1,0 экв.) в дихлорметане (5 мл) добавляли периодинан Десса-Мартина (1,31 г, 3,08 ммоль, 1,5 экв.) при 0°C и обеспечивали осуществление реакции в смеси при 25°C в течение 4 часов. Реакционный раствор концентрировали. Неочищенный продукт разбавляли с помощью 10,0 мл 1 M раствора гидроксида натрия, экстрагировали с помощью этилацетата (20,0 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (1:4 этилацетат/петролейный эфир) с получением 53b.
[0624] 1H ЯМР (400 МГц, CDCl3) δ: 3,71 (br t, J=4,8 Гц, 2H), 3,55 (s, 2H), 3,50 (br d, J=4,8 Гц, 2H), 3,21-3,08 (m, 2H), 3,06-2,91 (m, 2H), 1,49 (s, 9H).
[0625] Вторая стадия
[0626] Соединение 53b (0,25 г, 1,04 ммоль, 1 экв.) растворяли в хлористоводородной кислоте/этилацетате (10,0 мл, 2 M) и обеспечивали осуществление реакции в смеси при 30°C в течение 15 часов при перемешивании. После завершения реакции реакционный раствор подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали соединение 53c. Неочищенный продукт непосредственно использовали на следующей стадии.
[0627] Третья стадия
[0628] Соединение 53d (400 мг, 1,01 ммоль, 1,0 экв.), соединение 53c (200 мг, 1,13 ммоль, 1,12 экв.) и DIPEA (390 мг, 302 мкмоль, 3,0 экв.) растворяли в DMSO (10,0 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 17 часов, разбавляли с помощью 20,0 мл воды, экстрагировали с помощью этилацетата (20 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством колоночной хроматографии (100% этилацетат) с получением 53e.
[0629] 1H ЯМР (400 МГц, CDCl3) δ: 8,53 (s, 1H), 8,15 (d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,89 (d, J=8,0 Гц, 1H), 7,52-7,48 (m, 1H), 7,48-7,44 (m, 1H), 6,41 (br s, 1H), 4,34 (br d, J=7,2 Гц, 1H), 4,03-3,90 (m, 4H), 3,82-3,59 (m, 8H), 3,13-3,00 (m, 4H), 2,99 (d, J=4,8 Гц, 3H), 1,42 (d, J=6,8 Гц, 3H).
[0630] Четвертая стадия
[0631] К раствору соединения 53e (100 мг, 199 мкмоль, 1,0 экв.) в метаноле (3,00 мл) добавляли гидроксиламина гидрохлорид (41,5 мг, 596,9 мкмоль, 3 экв.) и смесь перемешивали при 70°C в течение часа. Реакционный раствор концентрировали. Неочищенный продукт разбавляли с помощью 5 мл воды и экстрагировали с помощью этилацетата (10,0 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и очищали посредством препаративной тонкослойной хроматографии (100% этилацетат) с получением 53f.
[0632] Пятая стадия
[0633] К раствору соединения 53f (130 мг, 251 мкмоль, 1,0 экв.) в ацетоне (40 мл) и воде (4 мл) последовательно добавляли гидроксид натрия (101 мг, 2,51 ммоль, 10,0 экв.), п-толуолсульфонилхлорид (479 мг, 2,51 ммоль, 10,0 экв.), обеспечивали осуществление реакции в смеси при 30°C в течение 16 часов. Реакционный раствор концентрировали. Неочищенный продукт разбавляли с помощью 10 мл воды и промывали этилацетатом (20 мл × 3). Органическую фазу удаляли. Водную фазу концентрировали и очищали с помощью препаративной тонкослойной хроматографии (1:10 метанол/этилацетат) с получением рацемата, который дополнительно подвергали хиральному разделению с помощью SFC с получением соединений 53-1 и 53-2.
[0634] Положение пика соединения 53-1: 2,589 мин. (хиральная колонка: AS-3 150 × 4,6 мм, подвижная фаза: A: CO2, B: метанол (0,05% диэтиламин), скорость потока: 25 мл/мин., температура колонки: 40°C).
[0635] Соединение 53-1: MS-ESI: расчетное значение [M+H]+: 518, найденное значение: 518.
[0636] Соединение 53-1: 1H ЯМР (400 MГц, CDCl3) δ: 8,55 (s, 1H), 8,13 (d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,90 (d, J=8,0 Гц, 1H), 7,53-7,43 (m, 2H), 6,52 (br s, 1H), 5,55 (br s, 1H), 4,34 (br d, J=6,4 Гц, 1H), 3,97-3,58 (m, 12H), 3,45-3,37 (m, 2H), 2,98 (d, J=5,2 Гц, 3H), 2,56-2,41 (m, 2H), 1,42 (d, J=6,8 Гц, 3H).
[0637] Положение пика соединения 53-2: 5,877 мин. (хиральная колонка: AS-3 150 × 4,6 мм, подвижная фаза: A: CO2, B: метанол (0,05% диэтиламин), скорость потока: 25 мл/мин., температура колонки: 40°C).
[0638] Соединение 53-2: MS-ESI: расчетное значение [M+H]+: 518, найденное значение: 518.
Соединение 53-2: 1H ЯМР (400 MГц, CDCl3) δ: 8,55 (s, 1H), 8,13 (d, J=8,0 Гц, 1H), 7,99 (d, J=8,4 Гц, 1H), 7,90 (d, J=8,0 Гц, 1H), 7,53-7,43 (m, 2H), 6,52 (br s, 1H), 5,55 (br s, 1H), 4,34 (br d, J=6,4 Гц, 1H), 3,97-3,58 (m, 12H), 3,45-3,37 (m, 2H), 2,98 (d, J=5,2 Гц, 3H), 2,56-2,41 (m, 2H), 1,42 (d, J=6,8 Гц, 3H).
[0639] Вариант осуществления 54
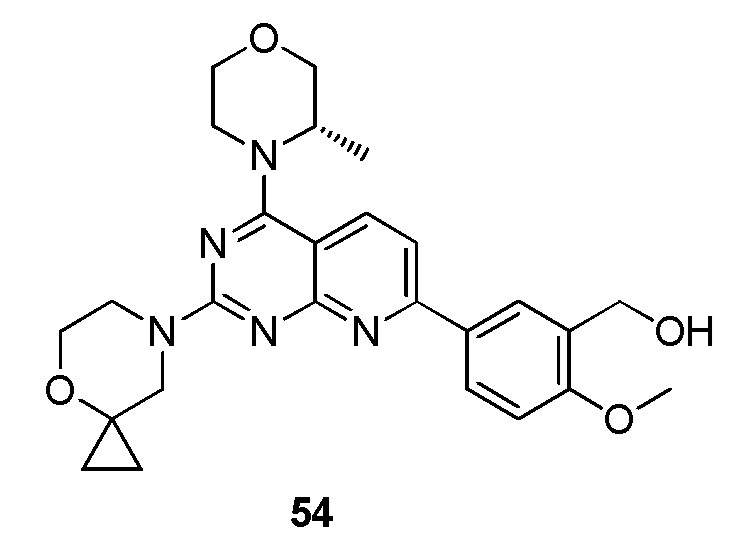
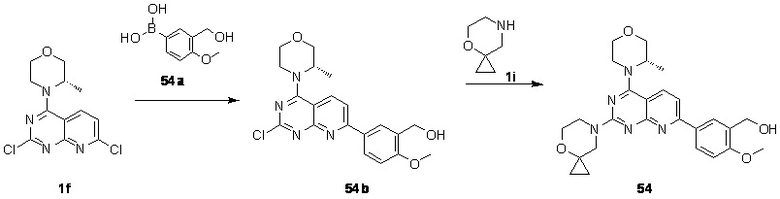
[0640] Первая стадия
[0641] Соединение 1f (150 мг, 501 мкмоль, 1,00 экв.), 54a (82,1 мг, 451 мкмоль, 0,90 экв.), дихлорид бис(трифенилфосфин)палладия (17,6 мг, 25,1 мкмоль, 0,05 экв.) и карбонат натрия (106 мг, 1,0 ммоль, 2,00 экв.) растворяли в воде (2,00 мл) и 1,4-диоксане (4,00 мл) и обеспечивали осуществление реакции в смеси при 70°C в течение 15,5 часа в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (5 мл) и экстрагировали с помощью этилацетата (10,0 мл × 3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали 54b.
[0642] Вторая стадия
[0643] Соединение 54b (100 мг, 249 мкмоль, 1,00 экв.), 1i (33,9 мг, 299 мкмоль, 1,2 экв.) и DIPEA (96,7 мг, 748 мкмоль, 130 мкл, 3 экв.) растворяли в DMSO (3,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 17 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 54.
[0644] MS-ESI: расчетное значение [M+H]+: 478, найденное значение: 478.
[0645] 1H ЯМР (400 МГц, CDCl3) δ: 8,20-8,13 (m, 2H), 8,00 (d, J=8,4 Гц, 1H), 7,45 (d, J=8,4 Гц, 1H), 7,01 (d, J=8,4 Гц, 1H), 4,79 (s, 2H), 4,36 (br s, 1H), 4,08-3,63 (m, 15H), 1,47 (d, J=6,4 Гц, 3H), 0,85 (s, 2H), 0,70 (br s, 2H).
[0646] Вариант осуществления 55
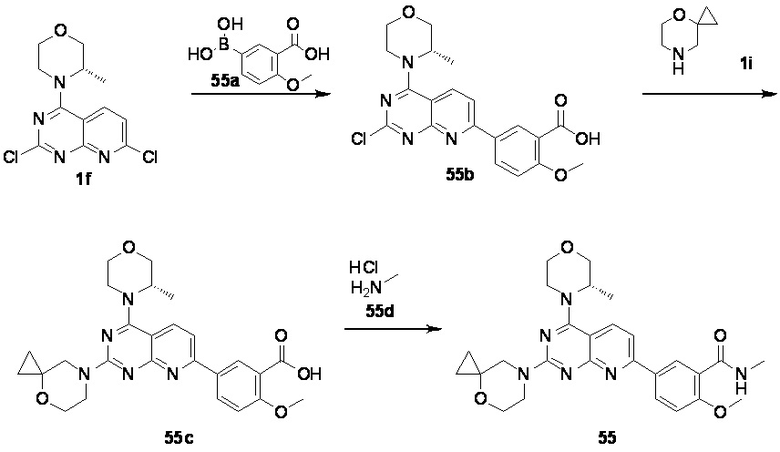
[0647] Первая стадия
[0648] Соединение 1f (100 мг, 334 мкмоль, 1,00 экв.), соединение 55a (65,5 мг, 334 мкмоль, 1,00 экв.), дихлорид бис(трифенилфосфин)палладия (11,7 мг, 16,7 мкмоль, 0,05 экв.) и карбонат натрия (70,9 мг, 669 мкмоль, 2,00 экв.) растворяли в воде (1,00 мл) и 1,4-диоксане (3,00 мл) и обеспечивали осуществление реакции в смеси при 80°C в течение 15 часов в защитной атмосфере азота. После завершения реакции реакционный раствор концентрировали с удалением растворителя, разбавляли водой (15,0 мл) и экстрагировали с помощью этилацетата (50,0 мл × 3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали, подвергали выпариванию на роторном испарителе при пониженном давлении до сухого состояния и получали 55b.
[0649] Вторая стадия
[0650] Соединение 55b (168 мг, 404 мкмоль, 1,00 экв.), 1i (54,4 мг, 364 мкмоль, 0,90 экв.) и DIPEA (157 мг, 1,21 ммоль, 211 мкл, 3,00 экв.) растворяли в DMSO (3,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 80°C в течение 15 часов. После завершения реакции реакционный раствор концентрировали с удалением растворителя с получением 55c. Неочищенный продукт непосредственно использовали на следующей стадии.
[0651] Третья стадия
[0652] Соединение 55c (99,0 мг, 201 мкмоль, 1,00 экв.), 55d (24,5 мг, 363 мкмоль, 1,80 экв.), DIPEA (104 мг, 806 мкмоль, 140 мкл, 4,00 экв.), EDCI (57,9 мг, 302 мкмоль, 1,50 экв.) и HOBt (40,8 мг, 302 мкмоль, 1,50 экв.) растворяли в DMSO (4,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 30°C в течение 22 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 55.
[0653] MS-ESI: расчетное значение [M+H]+: 505, найденное значение: 505.
[0654] 1H ЯМР (400 МГц, CDCl3) δ: 8,75 (br s, 1H), 8,51 (br d, J=8,0 Гц, 1H), 7,94 (br d, J=8,4 Гц, 1H), 7,76 (br s, 1H), 7,50 (br d, J=8,4 Гц, 1H), 7,03 (br d, J=8,4 Гц, 1H), 4,29 (br s, 1H), 3,97 (s, 3H), 3,96-3,51 (m, 12H), 2,98 (br d, J=4,4 Гц, 3H), 1,38 (br d, J=6,8 Гц, 3H), 0,76 (br s, 2H), 0,61 (br s, 2H).
[0655] Вариант осуществления 56
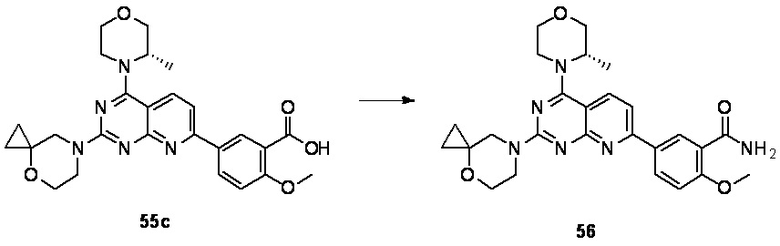
[0656] Соединение 56c (99,0 мг, 201 мкмоль, 1,00 экв.), хлорид аммония (43,1 мг, 806 мкмоль, 4,00 экв.), DIPEA (104 мг, 806 мкмоль, 140 мкл, 4,00 экв.), EDCI (57,9 мг, 302 мкмоль, 1,50 экв.) и HOBt (40,8 мг, 302 мкмоль, 1,50 экв.) растворяли в DMSO (4,00 мл). Обеспечивали осуществление реакции в смешанном растворе при 30°C в течение 22 часов. После завершения реакции реакционный раствор очищали посредством высокоэффективной жидкостной хроматографии с получением 56.
[0657] MS-ESI: расчетное значение [M+H]+: 491, найденное значение: 491.
[0658] 1H ЯМР (400 МГц, CDCl3) δ: 8,76 (d, J=2,4 Гц, 1H), 8,55 (dd, J=2,4, 8,8 Гц, 1H), 7,93 (d, J=8,8 Гц, 1H), 7,67 (br s, 1H), 7,50 (d, J=8,8 Гц, 1H), 7,06 (d, J=8,8 Гц, 1H), 5,75 (br s, 1H), 4,28 (br d, J=5,6 Гц, 1H), 3,99 (s, 3H), 3,97-3,36 (m, 12H), 1,38 (d, J=6,8 Гц, 3H), 0,79-0,73 (m, 2H), 0,61 (br s, 2H).
[0659] Экспериментальный вариант осуществления 1. Оценивание ингибирующей активности в отношении киназы mTOR in vitro
[0660] Экспериментальные материалы
[0661] Данный эксперимент проводился DiscoverX. Все материалы и способы были получены от DiscoverX.
[0662] Процедура эксперимента.
[0663] Анализ киназной активности.
[0664] 1. Меченная киназа mTOR стабильно экспрессировалась в клетках HEK-293.
[0665] 2. Магнитные гранулы, покрытые стрептавидином, обрабатывали биотинилированными низкомолекулярными лигандами при комнатной температуре в течение 30 минут с получением аффинной смолы для анализа киназы.
[0666] 3. Гранулы, обработанные лигандами, блокировали с помощью избытка биотина и промывали буфером (1% бычий сывороточный альбумин, 20 мл 0,05% Tween, 1 мл дитиотреитола) для удаления несвязанного лиганда и неспецифически связанного лиганда.
[0667] 4. Сборку киназы, обработанных лигандами гранул и аффинных гранул, а также реакцию связывания между соединением и тремя компонентами проводили в буфере (20% блокирующий буфер, 0,17x фосфатный буфер, 0,05% Tween 20, 6 мл дитиотреитола).
[0668] 5. Тестируемые соединения растворяли в диметилсульфоксиде.
[0669] 6. Все тестируемые соединения растворяли в DMSO и затем непосредственно разбавляли до концентрации, составляющей 0,9%.
[0670] 7. Раствор помещали в 384-луночный полипропиленовый планшет, где объем каждой лунки составлял 0,02 мл.
[0671] 8. Планшет встряхивали в течение часа при комнатной температуре.
[0672] 9. Планшет промывали с помощью буфера (1 x PBS, 0,05% Tween 20).
[0673] 10. Аффинные гранулы ресуспендировали в буфере (1 x PBS, 0,05% Tween 20, 0,5 мкм небиотинилированный аффинный лиганд) и инкубировали при комнатной температуре в течение 30 минут.
[0674] 11. Концентрацию киназы в элюате определяли посредством qPCR.
[0675] Результаты эксперимента.
[0676] Таблица 1. Результаты теста на активность киназного комплекса mTORC1 и mTORC2
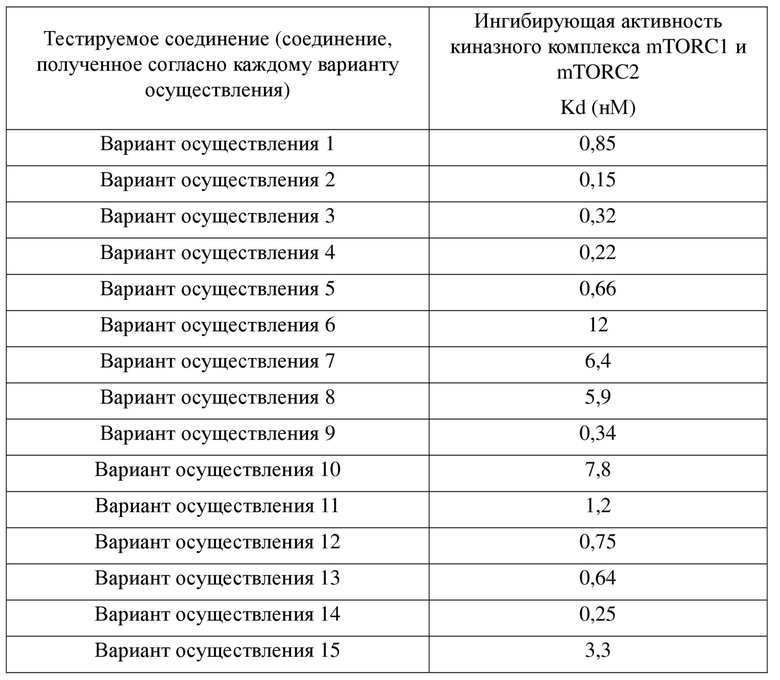
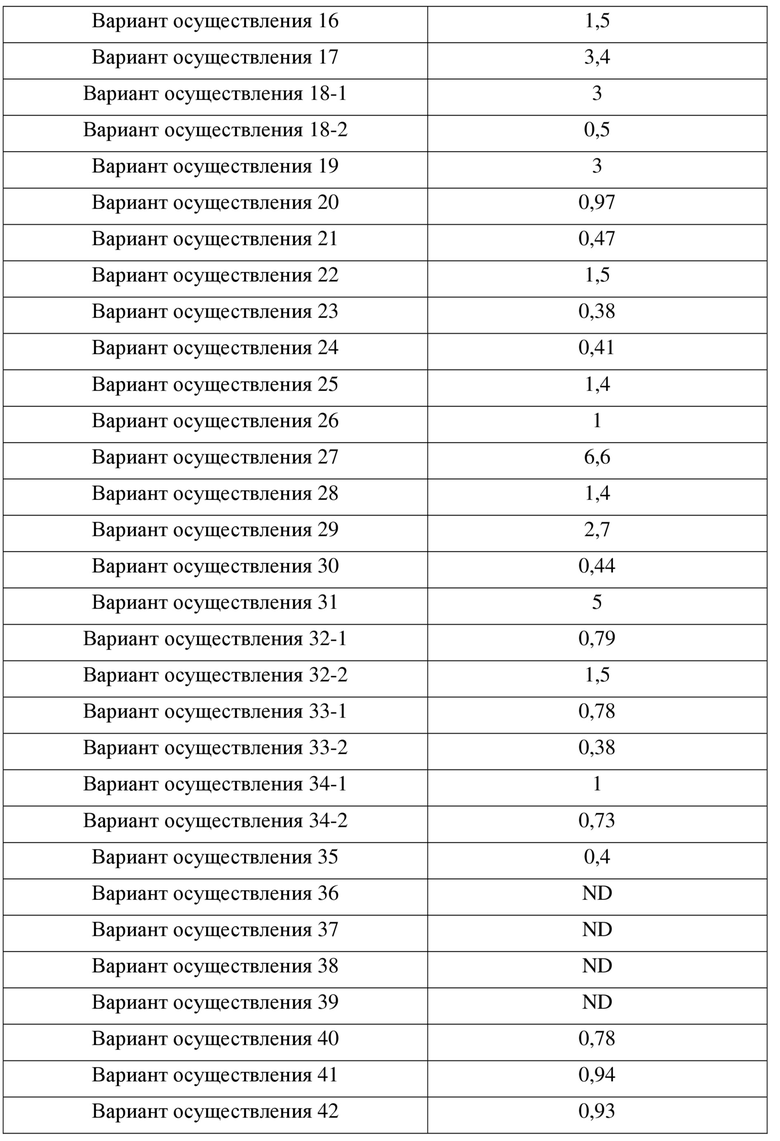
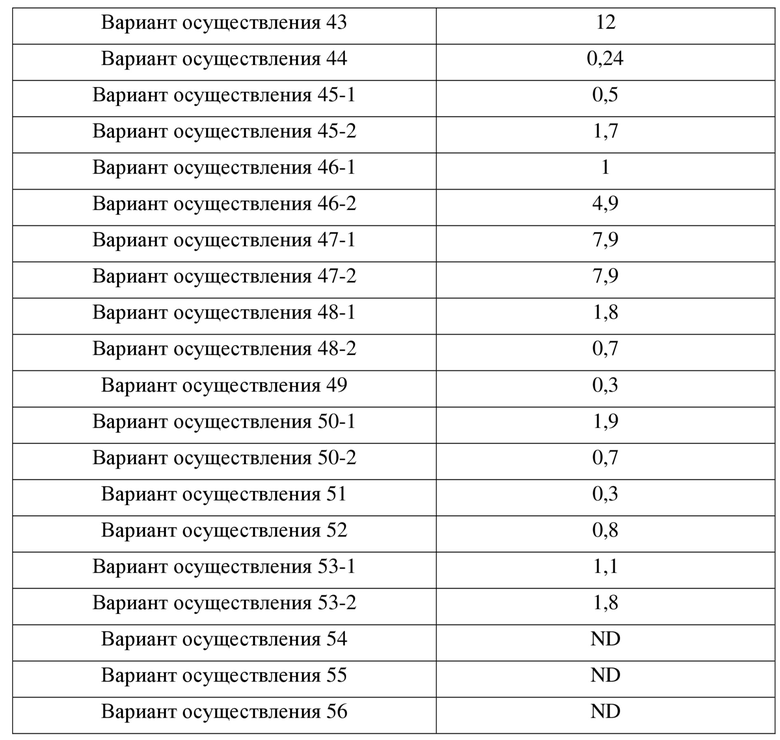
[0677] «ND» относится к необнаруженному.
[0678] Заключение: соединения по настоящему изобретению обладают значительной или неожиданной ингибирующей активностью в отношении киназы mTOR.
[0679] Экспериментальный вариант осуществления 2. Оценивание ингибирующей активности в отношении пролиферации клеток
[0680] Цель эксперимента: определить ингибирующую активность тестируемых соединений в отношении пролиферации клеток.
[0681] Принцип эксперимента: Люцифераза в реагенте Cell-Titer-Glo обеспечивает продуцирование оксилюциферина с применением люциферина, кислорода и ATP в качестве субстратов реакции, и обеспечивает высвобождение энергии в виде света. Поскольку для реакции с участием люциферазы требуется ATP, общее количество света, генерируемого вследствие реакции, пропорционально общему количеству ATP, которое отображает жизнеспособность клеток.
[0682] Экспериментальные материалы
[0683] Клеточные линии: клеточная линия MCF-7 (ATCC-CRL-22), клеточная линия HT-29 (ATCC-HTB-38), OE21 (ECACC-96062201), клеточная линия NCI-N87 (ATCC-CRL-5822).
[0684] Среда для культивирования клеток: (культуральная среда RPMI 1640 (Invitrogen № 1868546; 10% сыворотка Invitrogen № 1804958; L-глутамин 1 ×, Invitrogen № 1830863; среда, содержащая два антибиотика, Hyclone № J170012)).
[0685] Набор для анализа жизнеспособности клеток посредством люминесценции Cell Titer-Glo® (Promega № G7573).
[0686] 384-Луночный планшет для культивирования клеток (Greiner № E15103MA).
[0687] Планшет для соединений (LABCYTE № 0006346665).
[0688] CO2-инкубатор (Thermo № 371).
[0689] Устройство для подсчета клеток Vi-cell (Beckman Coulter).
[0690] Пипеточный дозатор (Eppendorf).
[0691] Пипетка для переноса (Greiner).
[0692] Держатель для пипеток (Eppendorf).
[0693] Многофункциональный ферментный маркер (Envision Reader).
[0694] Лабораторная станция ручного управления для работы с жидкостями ECHO (Labcyte-ECHO555).
[0695] Экспериментальные стадии и способы
[0696] 2.1. Первый день
[0697] Клетки инокулировали в 384- или 96-луночном планшете с плотностью 1000 клеток на лунку и 25 мкл на лунку в соответствии с инструкциями к планшету для инокуляции клеток. Краевые лунки не инокулировали клетками и дополняли 25 мкл PBS.
[0698] 2.2. День «ноль»
[0699] (i) Концентрация маточного раствора соединения составляла 10 мМ, и его разбавляли DMSO до исходной концентрации, составляющей 4 мМ. Соединение добавляли к маточному раствору соединения, 9 мкл на лунку.
[0700] (ii) Соединение разбавляли с помощью лабораторной станции ручного управления для работы с жидкостями ECHO. В каждую лунку планшета для клеток добавляли 125 нл соединения. В каждую лунку из тех, что представляли собой лунки с клетками, во 2-й колонке и 23-ей колонке добавляли 125 нл DMSO, и в каждую из лунок с PBS в 1-й колонке и 24-й колонке добавляли 125 нл DMSO.
[0701] (iii) Каждую лунку планшета для клеток дополняли 25 мкл культуральной среды, при этом конечный объем в каждой лунке планшета для клеток составлял 50 мкл, концентрация соединения составляла 1 мкМ, проводили 3 раза разбавление, получали 10 значений концентрации, каждую концентрацию дублировали в левых и правых лунках, конечная концентрация DMSO составляла 0,25%.
[0702] 2.3. После добавления соединения проводили центрифугирование при 1000 об./мин. в течение 1 мин. и планшет для клеток помещали на 3 дня в инкубатор при 37°C, 5% CO2.
[0703] 2.4. Третий день
[0704] Планшет для клеток вынимали из инкубатора и уравновешивали при комнатной температуре в течение 30 минут. В каждую лунку добавляли 25 мкл реагента Cell-Titer-Glo, встряхивали в течение одной минуты, чтобы тщательно перемешать, центрифугировали при 1000 об./мин. в течение 1 минуты. Через 10 минут планшет подвергали считыванию на PerkinElmer Envision, при этом время считывания флуоресценции выставляли на 0,2 секунды.
[0705] Результаты испытания: результаты испытания показаны в таблице 2.
[0706] Таблица 2. Результаты скринингового теста in vitro ингибирующей активности соединений по настоящему изобретению в отношении пролиферации клеток
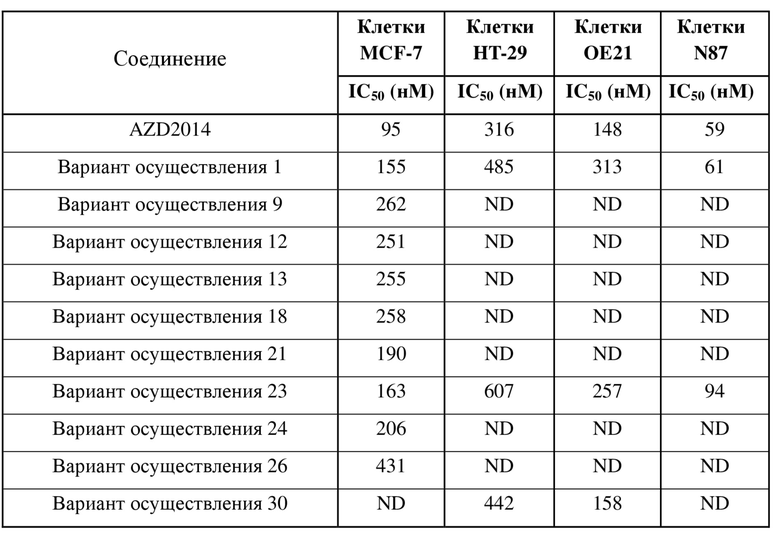

[0707] «ND» относится к необнаруженному.
[0708] Заключение: Соединения по настоящему изобретению обладают значительной ингибирующей активностью в отношении пролиферации клеток MCF-7, N87 и OE-21 и обладают определенной ингибирующей активностью в отношении пролиферации клеток HT-29.
[0709] Экспериментальный вариант осуществления 3. Фармакокинетическая оценка
[0710] 1. Экспериментальный способ
[0711] Тестируемое соединение смешивали с 5% DMSO/95% 10% полиоксиэтиленового касторового масла (Cremophor EL), перемешивали вихревым способом и подвергали воздействию ультразвука с получением 1 мг/мл практически прозрачного раствора, который фильтровали через микропористую фильтрующую мембрану для применения в будущем. Выбирали самок мышей Balb/c весом 18-20 грамм и вводили им внутривенно раствор тестируемого соединения в дозе, составляющей 1 или 2 мг/кг. Тестируемое соединение смешивали с водным раствором 1% Tween 80, 9% полиэтиленгликоля 400 и 90% воды, перемешивали вихревым способом и подвергали воздействию ультразвука с получением 1 мг/мл практически прозрачного раствора, который фильтровали через микропористую фильтрующую мембрану для применения в будущем. Выбирали самок мышей Balb/c весом 18-20 грамм и вводили им перорально раствор тестируемого соединения в дозе 2 или 10 мг/кг. В определенные моменты времени собирали цельную кровь и получали из нее плазму. Концентрацию лекарственного средства анализировали с помощью LC-MS/MS и рассчитывали фармакокинетические параметры с применением программного обеспечения Phoenix WinNonlin (Pharsight Corporation, США).
[0712] Результаты испытания:
[0713] результаты испытания показаны в таблице 3 - таблице 8.
[0714] Таблица 3. Фармакокинетические (PK) параметры в плазме крови для соединений согласно вариантам осуществления
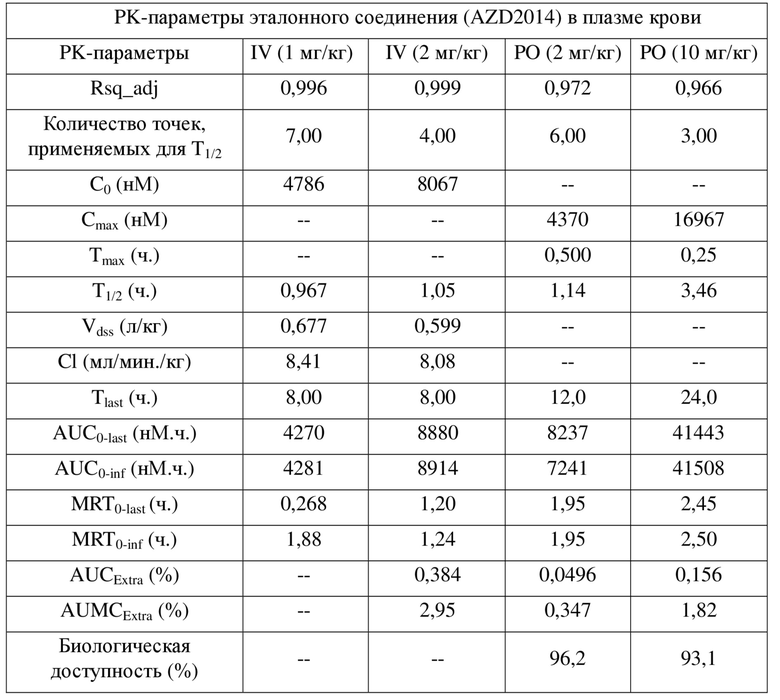
[0715] Таблица 4. PK-параметры в плазме крови для соединений согласно вариантам осуществления
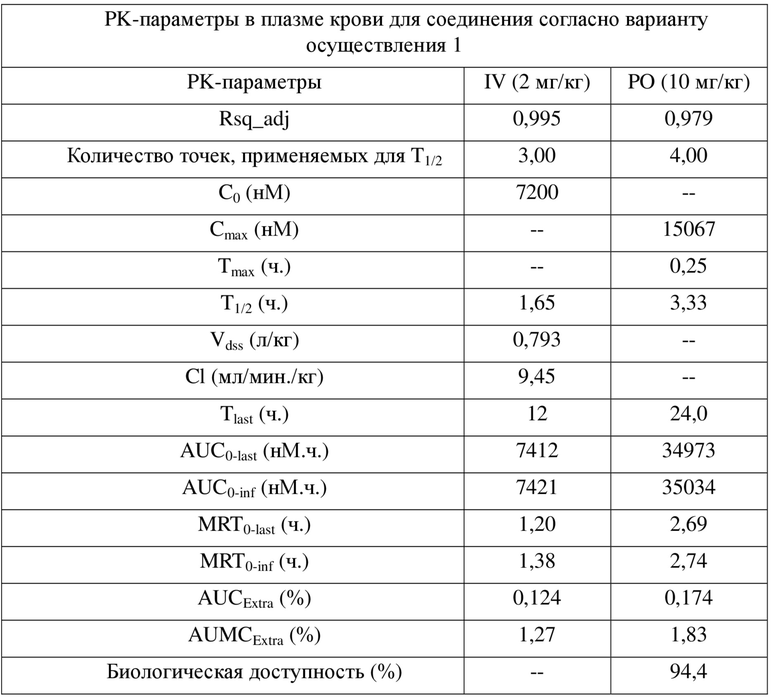
[0716] Таблица 5. PK-параметры в плазме крови для соединений согласно вариантам осуществления
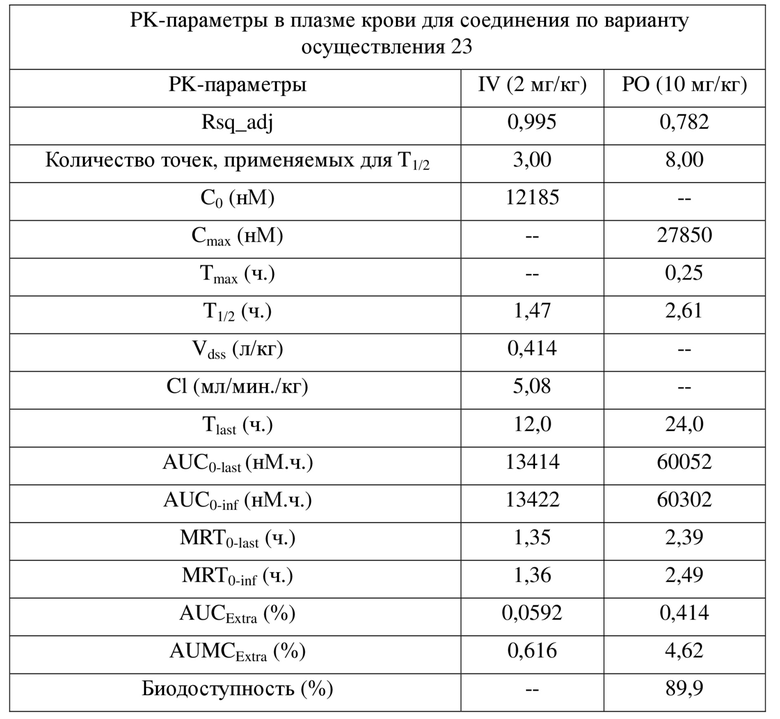
[0717] Таблица 6. PK-параметры в плазме крови для соединений согласно вариантам осуществления
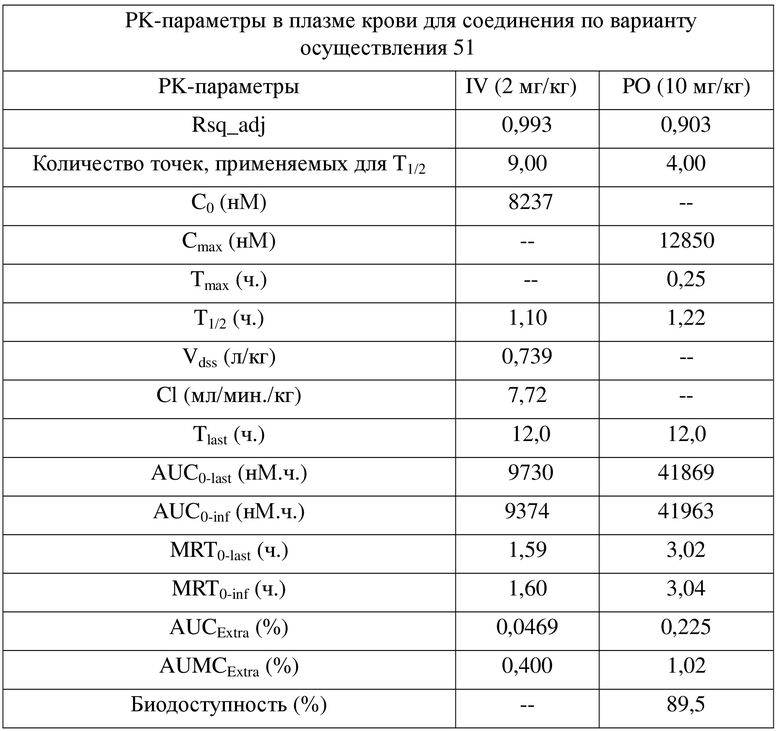
[0718] Таблица 7. PK-параметры в плазме крови для соединений согласно вариантам осуществления
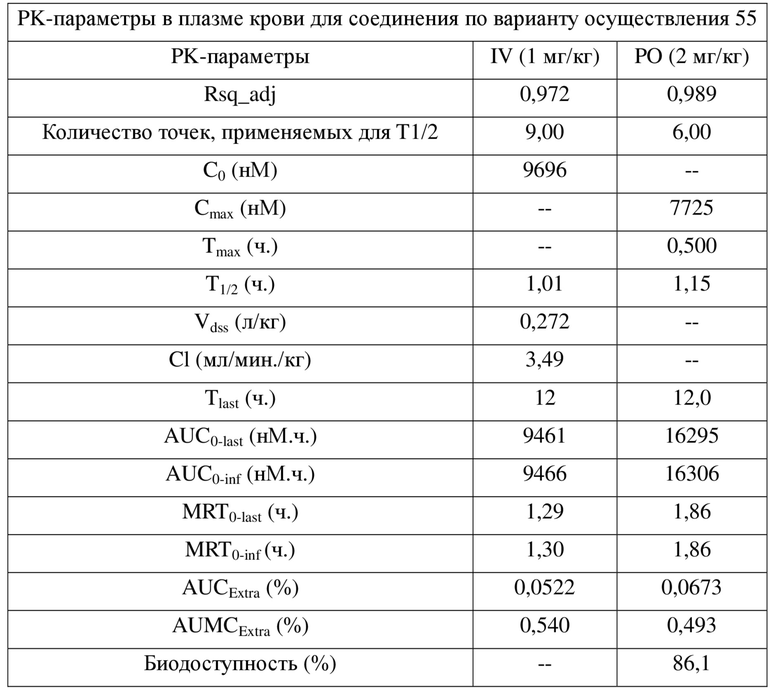
[0719] Таблица 8. PK-параметры в плазме крови для соединений согласно вариантам осуществления
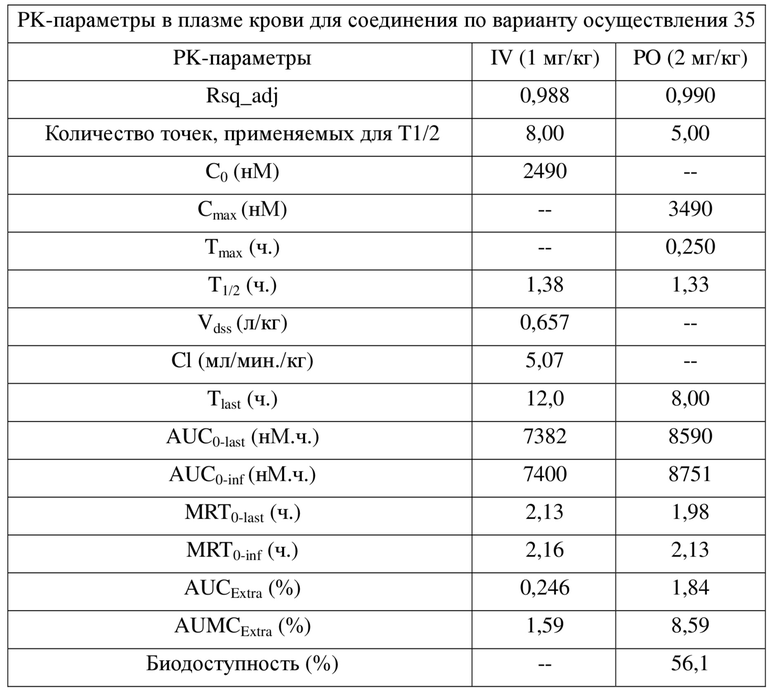
[0720] «-» относится к необнаруженному или отсутствию данных.
[0721] Заключение эксперимента: тестируемые соединения проявляют такие же фармакокинетические свойства, что и эталонное соединение, или даже лучшие; соединения по настоящему изобретению характеризуются биодоступностью, близкой к 100%, и являются превосходными пригодными для разработки молекулами для перорального введения.
[0722] Экспериментальный вариант осуществления 4. Фармакодинамическое исследование in vivo на модели рака молочной железы человека, полученной на основе подкожной ксенотрансплантатной опухоли из клеток MCF-7 у голых мышей BALB/c
[0723] Цель эксперимента. Исследование эффективности тестируемых соединений на модели рака молочной железы человека, полученной на основе подкожной ксенотрансплантатной опухоли из клеток MCF-7 у голых мышей BALB/c.
[0724] Экспериментальные животные. Самки голых мышей BALB/c возрастом 6-8 недель, весом 18-22 грамма; поставщик: Shanghai Xipuer-Bikai Experimental Animal Co., Ltd.
[0725] Экспериментальный способы и стадии
[0726] 4.1. Культивирование клеток
[0727] Клетки MCF-7 рака молочной железы человека (ECACC, номер товара: 86012803) подвергали культивированию в монослое in vitro в условиях EMEM (EBSS) + 2 мМ глутаминовая кислота + среда с 1% заменимых аминокислот (NEAA), содержащая 10% эмбриональной бычьей сыворотки, 100 Ед./мл пенициллина и 100 мкг/мл стрептомицина, и помещали в инкубатор при 37°C, 5% CO2. Проводили традиционную обработку для открепления клеток с помощью трипсин-EDTA два раза в неделю для пассирования. Когда насыщение клеток составляло 80% - 90% и количество достигало требуемого уровня, клетки собирали, подсчитывали и инокулировали.
[0728] 4.2. Инокуляция клеток опухоли (инокуляция опухоли)
[0729] Таблетки, содержащие эстроген (0,18 мг), вводили в левую часть спины каждой мыши за три дня до инокуляции клеток, 0,2 мл (1 × 107) клеток MCF-7 (добавляли с matrigel, объемное соотношение 1:1) подкожно инокулировали в правую часть спины каждой мыши. Когда средний объем опухоли достигал 142 мм3, мышей разделяли на группы и начинали введение.
[0730] 4.3. Приготовление тестируемых соединений
[0731] Тестируемые соединения готовили в виде прозрачных растворов 0,75 мг/мл, 1,5 мг/мл и 3 мг/мл. Среды-носители представляли собой следующее: 5% DMSO + 30% полиэтиленгликоль 300 + 65% вода.
[0732] 4.4. Измерение опухоли и экспериментальные показатели
[0733] Экспериментальные показатели применяли для исследования того, происходило ли подавление, замедление или устранение роста опухолей. Диаметры опухолей измеряли с помощью штангенциркуля с нониусом два раза в неделю. Формула для расчета объема опухоли была следующей: V = 0,5a × b2, где a и b представляют соответственно длинный и короткий диаметры опухоли.
[0734] Противоопухолевую эффективность соединения оценивали с помощью TGI (%) или относительной скорости разрастания опухоли T/C (%). TGI (%) отображает скорость подавления роста опухоли. Расчет TGI (%): TGI (%) = [1 − (средний объем опухоли в конце введения в группе обработки − средний объем опухоли в начале введения в группе обработки)/(средний объем опухоли в конце обработки в контрольной группе, получающей среду-носитель − средний объем опухоли в начале обработки в контрольной группе, получающей среду-носитель)] × контрольная группа.
[0735] Относительная скорость разрастания опухоли T/C (%). Формула для расчета была следующей: T/C, % = TRTV/CRTV в начале обработки (TRTV: группа обработки RTV; CRTV: группа отрицательного контроля RTV). Относительный объем опухоли (RTV) рассчитывали в соответствии с результатами измерения опухоли. Формула для расчета была следующей: RTV = Vt/V0, где V0 – средний объем опухоли, измеренный в ходе введения группам (т. е. d0), и Vt – средний объем опухоли в ходе конкретного измерения, при этом показатели TRTV и CRTV были взяты в один и тот же день.
[0736] После эксперимента можно определить вес опухоли и можно рассчитать T/вес в процентах. T вес и C вес представляют соответственно вес опухоли в группе введения и контрольной группе, получающей среду-носитель.
[0737] 4.5. Статистический анализ
[0738] Статистический анализ включал среднее значение и стандартную ошибку (SEM) объема опухоли в каждый момент времени в каждой группе. Группа обработки показала лучший эффект от обработки на 21-й день после введения в конце испытания, поэтому статистический анализ проводили на основе этих данных для оценки отличий между группами. Сравнение между двумя группами анализировали с помощью T-критерия, и сравнение между тремя или более группами анализировали с помощью однофакторного ANOVA. Если было значительное отличие в показателях F-значения, для проверки применяли способ Геймса-Ховелла. Если не было значительного отличия в показателях F-значения, для анализа применяли способ Даннета (2-сторонний). Для анализа всех данных применяли SPSS 17.0. p <0,05 считали значимым отличием.
[0739] 4.6. Результаты эксперимента
[0740] В модели ксенотрансплантатной опухоли MCF-7 некоторые соединения характеризуются такой же эффективностью, что и AZD2014.
[0741] Таблица 9. Эффект ингибирования опухоли соединениями на примере модели рака молочной железы человека, полученной на основе подкожной ксенотрансплантатной опухоли из клеток MCF-7
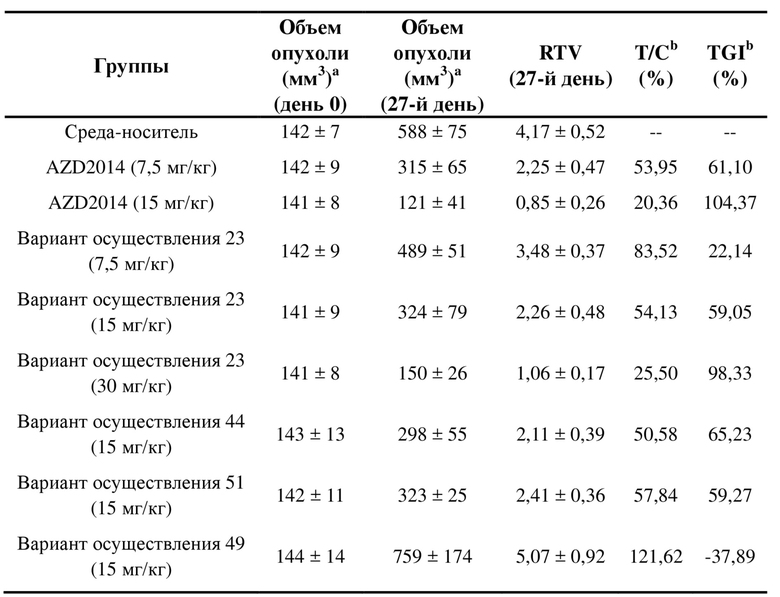
[0742] Примечание:
[0743] "--" относится к отсутствию расчета
[0744] a. Среднее значение ± SEM.
[0745] b. Ингибирование роста опухоли рассчитывали посредством T/C и TGI (TGI (%) = [1 – (T21-T0)/(V21-V0)] × 100).
[0746] c. p-значение получали на основе объема опухоли.
[0747] 4.7. Заключение эксперимента
[0748] По сравнению с группой, получавшей среду-носитель, в модели рака молочной железы человека, полученной на основе ксенотрансплантатной опухоли у голых мышей, AZD2014 имел значительное отличие при дозе 15 мг/кг, а вариант осуществления 23 имел значительное отличие при дозе 30 мг/кг по сравнению с контрольной группой, получавшей среду-носитель. Их TGI составляли 104% и 98%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДОИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДНК-PK | 2020 |
|
RU2796163C1 |
| АНТАГОНИСТ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2019 |
|
RU2832735C2 |
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ГЛУТАРИМИДНОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ КОЛЬЦОМ, КОНДЕНСИРОВАННЫМ С ФУРАНОМ | 2022 |
|
RU2836921C1 |
Изобретение относится к области органических соединений. Предложено соединение формулы (IV), его фармацевтически приемлемая соль или его стереоизомер, где R1 представляет собой H; R2 представляет собой Me; в качестве альтернативы R1, R2 и атом N в морфолиновом кольце образуют 5-6-членный гетероциклоалкил; R3 выбран из NH2, 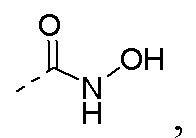
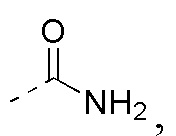
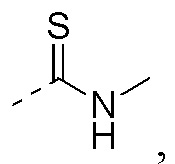
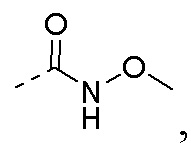
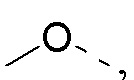
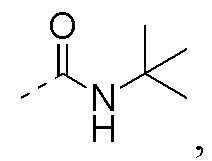
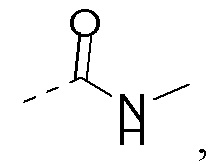 5-6-членного гетероарила и
5-6-членного гетероарила и 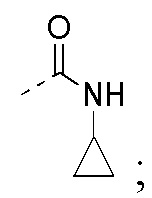 n выбран из 1 и 2; кольцо A выбрано из фенила, 9- и 10-членного гетероарила; R4 и R5 представляют собой H; в качестве альтернативы R4 и R5 соединены вместе с образованием 5-6-членного гетероциклоалкила; D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2- и -O-, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, где -CH2- или -CH2CH2- необязательно замещен R и при этом число R равняется 1; D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O- и -NH-, и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R; T1 выбран из CH и N; T2 выбран из -CH2-, -NH-, -O-,
n выбран из 1 и 2; кольцо A выбрано из фенила, 9- и 10-членного гетероарила; R4 и R5 представляют собой H; в качестве альтернативы R4 и R5 соединены вместе с образованием 5-6-членного гетероциклоалкила; D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2- и -O-, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, где -CH2- или -CH2CH2- необязательно замещен R и при этом число R равняется 1; D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O- и -NH-, и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R; T1 выбран из CH и N; T2 выбран из -CH2-, -NH-, -O-,  -S- и -C(=O)NH-, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R; где R соответственно выбран из CF3, F, OH, C1-3алкила и C1-3алкокси; 5-6-членный гетероарил, 9-членный гетероарил и 10-членный гетероарил соответственно содержат 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из -O-, -NH- и N. Также предложены конкретные соединения, фармацевтическая композиция и применение соединения формулы (IV) для получения лекарственного средства для лечения рака молочной железы. Технический результат - получение пиридопиримидиновых соединений, которые обладают двойной ингибирующей активностью в отношении mTOR1/2. 4 н. и 13 з.п. ф-лы, 9 табл., 56 пр.
-S- и -C(=O)NH-, где -CH2- необязательно замещен R, и при этом число R равняется 1 или 2, -NH- необязательно замещен R; где R соответственно выбран из CF3, F, OH, C1-3алкила и C1-3алкокси; 5-6-членный гетероарил, 9-членный гетероарил и 10-членный гетероарил соответственно содержат 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из -O-, -NH- и N. Также предложены конкретные соединения, фармацевтическая композиция и применение соединения формулы (IV) для получения лекарственного средства для лечения рака молочной железы. Технический результат - получение пиридопиримидиновых соединений, которые обладают двойной ингибирующей активностью в отношении mTOR1/2. 4 н. и 13 з.п. ф-лы, 9 табл., 56 пр.
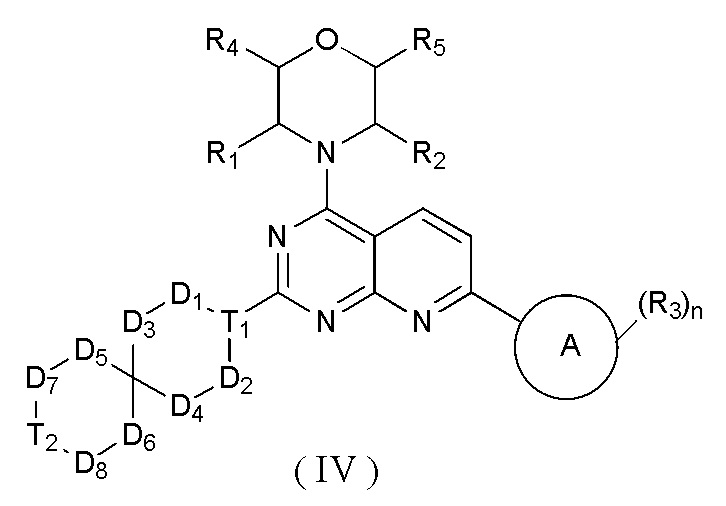
1. Соединение, представленное формулой (IV), его фармацевтически приемлемая соль или его стереоизомер,
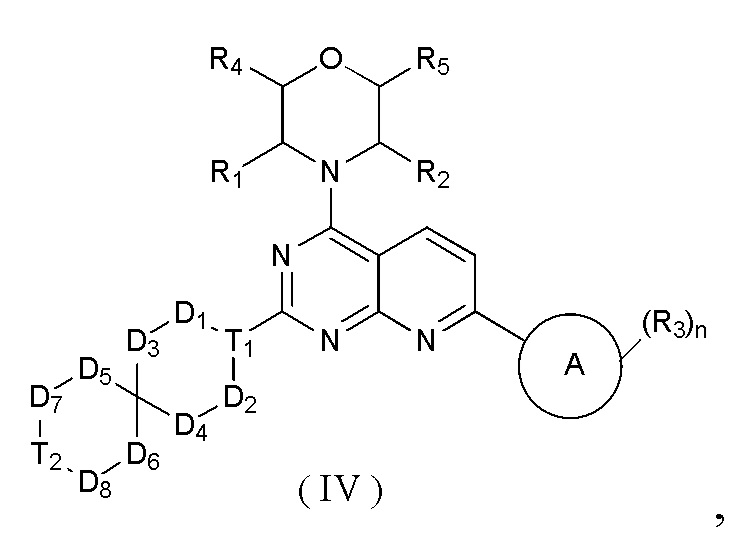
где
R1 представляет собой H;
R2 представляет собой Me;
в качестве альтернативы R1, R2 и атом N в морфолиновом кольце образуют 5-6-членный гетероциклоалкил;
R3 выбран из NH2,  5-6-членного гетероарила и
5-6-членного гетероарила и  ;
;
n выбран из 1 и 2;
кольцо A выбрано из фенила, 9-членного гетероарила и 10-членного гетероарила;
R4 представляет собой H;
R5 представляет собой H;
в качестве альтернативы R4 и R5 соединены вместе с образованием 5-6-членного гетероциклоалкила;
D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2- и -O-, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, где -CH2- или -CH2CH2- необязательно замещен R и при этом число R равняется 1;
D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O- и -NH-, и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
T1 выбран из CH и N;
T2 выбран из -CH2-, -NH-, -O-,  ,
,  , -S- и -C(=O)NH-, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
, -S- и -C(=O)NH-, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
R соответственно выбран из CF3, F, OH, C1-3алкила и C1-3алкокси;
5-6-членный гетероарил, 9-членный гетероарил и 10-членный гетероарил соответственно содержат 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из -O-, -NH- и N.
2. Соединение, его фармацевтически приемлемая соль или его стереоизомер по п. 1, которые выбраны из
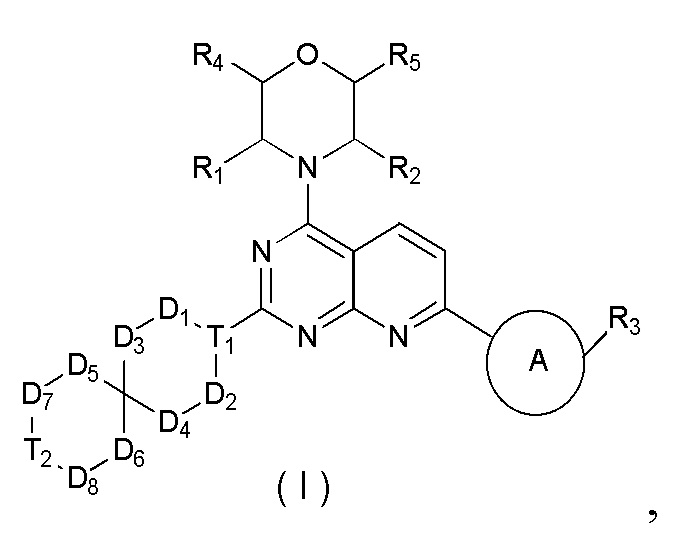
где
R1 представляет собой H;
R2 представляет собой Me;
в качестве альтернативы R1, R2 и атом N в морфолиновом кольце образуют 5-6-членный гетероциклоалкил;
R3 выбран из NH2,  5-6-членного гетероарила и
5-6-членного гетероарила и 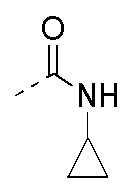 ;
;
кольцо A выбрано из фенила, 9-членного гетероарила и 10-членного гетероарила;
R4 выбран из H;
R5 выбран из H;
в качестве альтернативы R4 и R5 соединены вместе с образованием 5-6-членного гетероциклоалкила;
D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2- и -O-, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь, где -CH2- или -CH2CH2- необязательно замещен R и при этом число R равняется 1;
D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O- и -NH-, и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
T1 выбран из CH и N;
T2 выбран из -CH2-, -NH-, -O-, 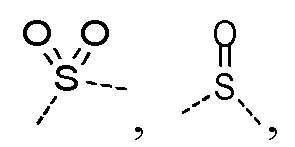 -S- и -C(=O)NH-, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
-S- и -C(=O)NH-, где -CH2- необязательно замещен R и при этом число R равняется 1 или 2, -NH- необязательно замещен R;
R соответственно выбран из CF3, F, OH, C1-3алкила и C1-3алкокси;
5-6-членный гетероарил, 9-членный гетероарил и 10-членный гетероарил соответственно содержат 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из -O-, -NH- и N.
3. Соединение, его фармацевтически приемлемая соль или его стереоизомер по п. 2, где R соответственно выбран из CF3, F, OH, Me, Et и  .
.
4. Соединение, его фармацевтически приемлемая соль или его стереоизомер по любому из пп. 1-3, где фрагмент 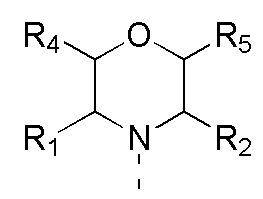 выбран из
выбран из 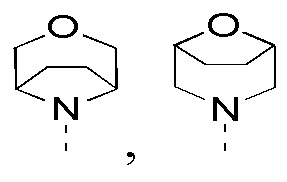 и
и 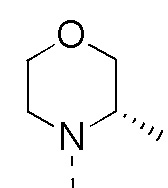 .
.
5. Соединение, его фармацевтически приемлемая соль или его стереоизомер по любому из пп. 1-3, где R3 выбран из NH2, 
 1H-пиразолила и 1H-1,2,4-триазолила.
1H-пиразолила и 1H-1,2,4-триазолила.
6. Соединение, его фармацевтически приемлемая соль или его стереоизомер по п. 5, где R3 выбран из NH2, 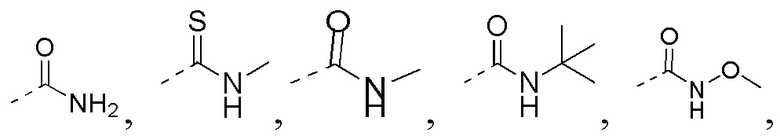
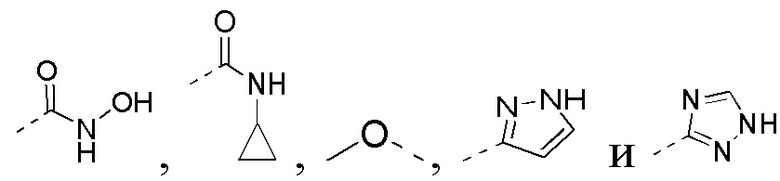 .
.
7. Соединение, его фармацевтически приемлемая соль или его стереоизомер по любому из пп. 1-3, где кольцо A выбрано из фенила, бензо[d]оксазола, хинолинила и хиназолинила.
8. Соединение, его фармацевтически приемлемая соль или его стереоизомер по п. 7, где кольцо A выбрано из 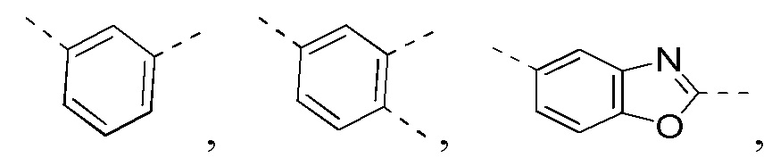
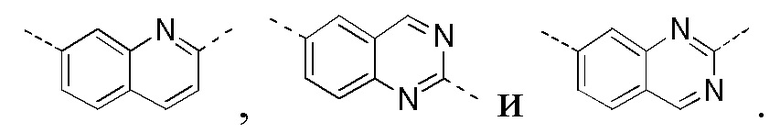
9. Соединение, его фармацевтически приемлемая соль или его стереоизомер по п. 1 или 8, где фрагмент  выбран из
выбран из 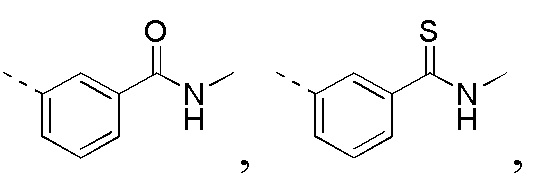
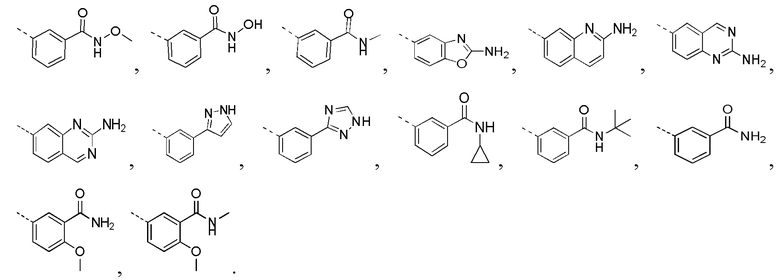
10. Соединение, его фармацевтически приемлемая соль или его стереоизомер по п. 2 или 8, где фрагмент  выбран из
выбран из 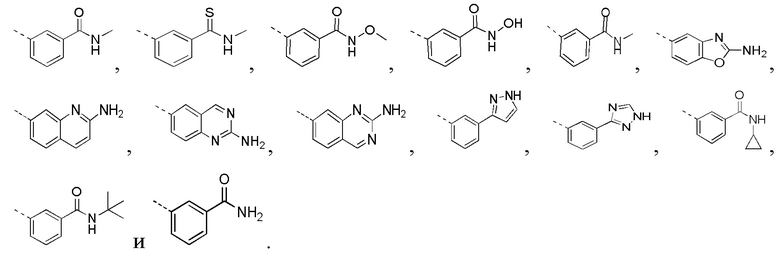
11. Соединение, его фармацевтически приемлемая соль или его стереоизомер по любому из пп. 1-3, где D1, D2, D3 и D4 соответственно выбраны из одинарной связи, -CH2-, -CH2CH2-, -O- и  , и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь;
, и по меньшей мере один из D1, D2, D3 и D4 не представляет собой одинарную связь;
или D5, D6, D7 и D8 соответственно выбраны из одинарной связи, -CH2-, -O-, -NH-, 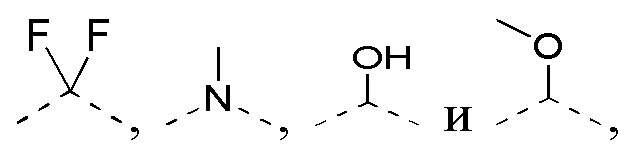 и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь.
и по меньшей мере один из D5, D6, D7 и D8 не представляет собой одинарную связь.
12. Соединение, его фармацевтически приемлемая соль или его стереоизомер по любому из пп. 1-3, где фрагмент 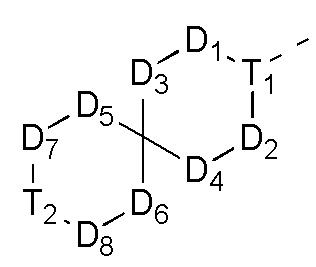 выбран из
выбран из 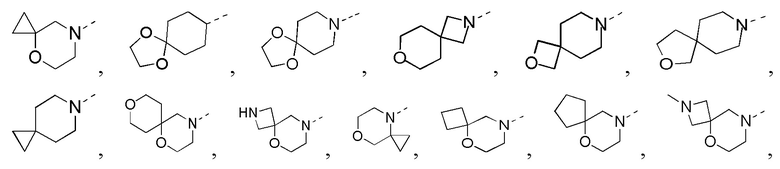
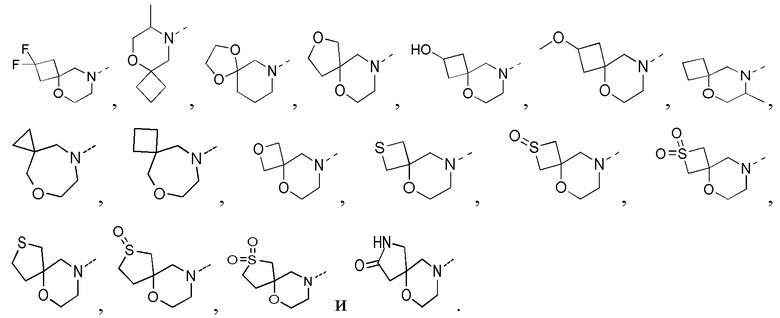
13. Соединение, его фармацевтически приемлемая соль или его стереоизомер по любому из пп. 1-7, которые выбраны из:
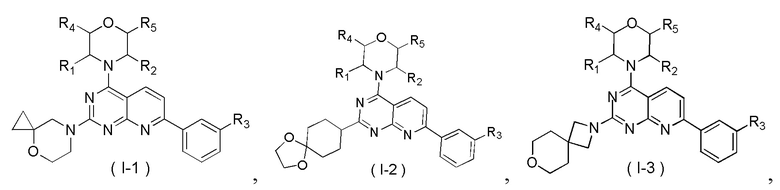
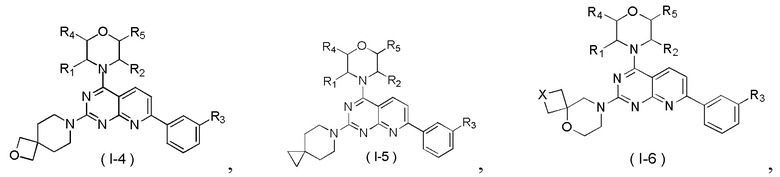

где
R1, R2, R3, R4 и R5 определены в любом из пп. 1-7;
X выбран из -CH2-, -CF2-, 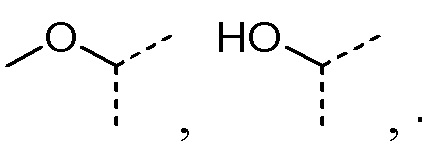 -NH-,
-NH-, 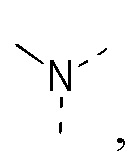 -O-,
-O-, 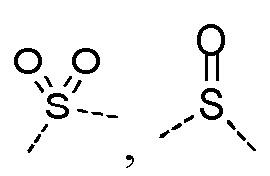 и -S-;
и -S-;
Y выбран из -CH2- и -CH2CH2-.
14. Соединение, его фармацевтически приемлемая соль или его стереоизомер по п. 1 или по любому из пп. 3-7, которые выбраны из:
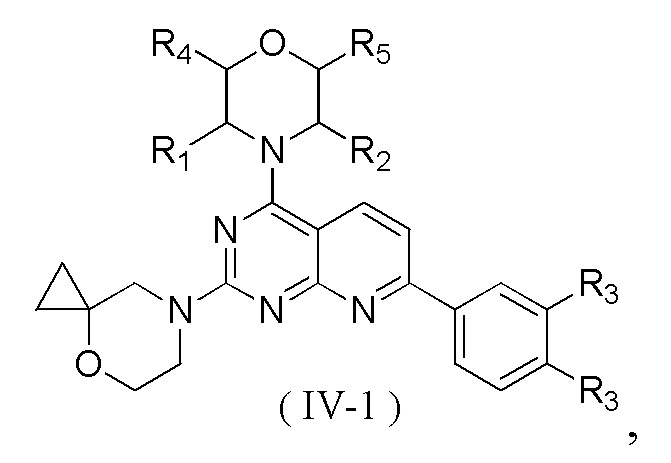
где
R1, R2, R3, R4 и R5 определены в п. 1 или в любом из пп. 3-7.
15. Соединение, показанное ниже, его фармацевтически приемлемая соль или его стереоизомер:
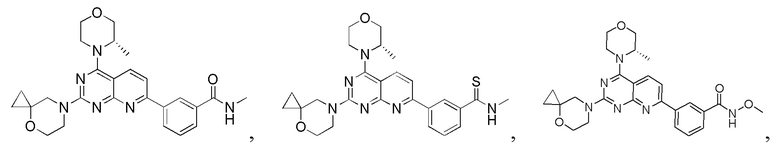

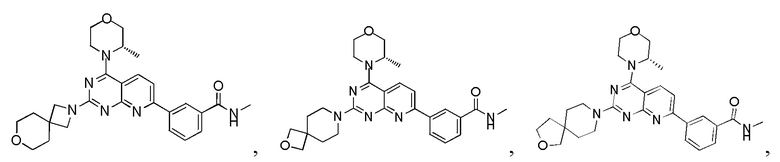
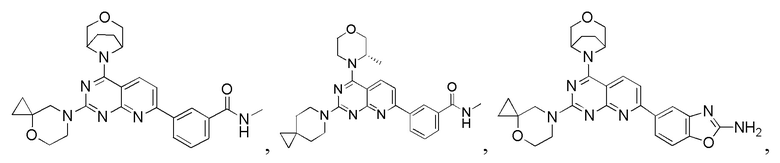
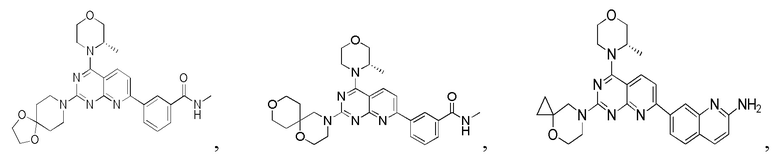
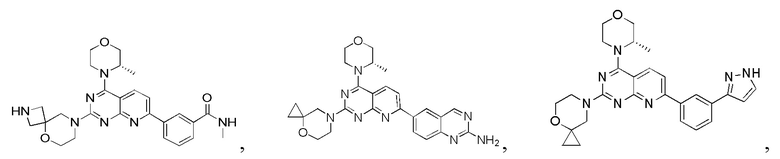
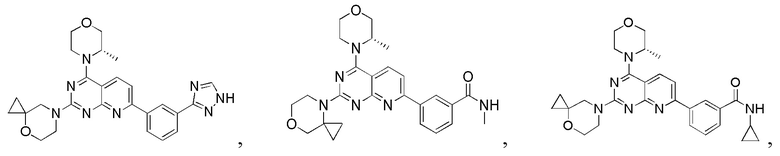
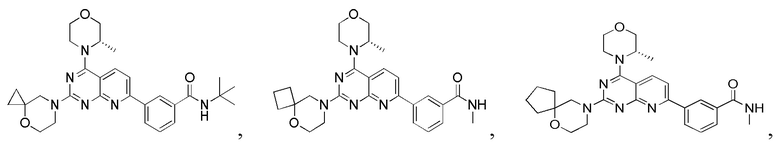
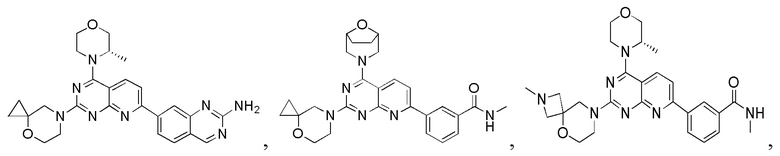

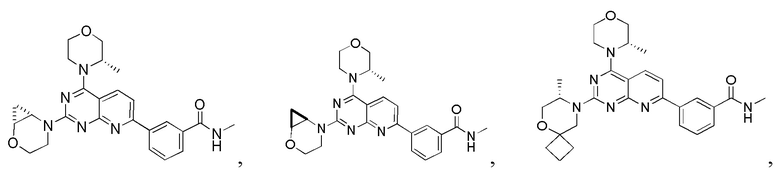
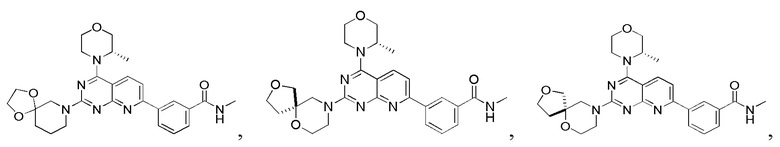
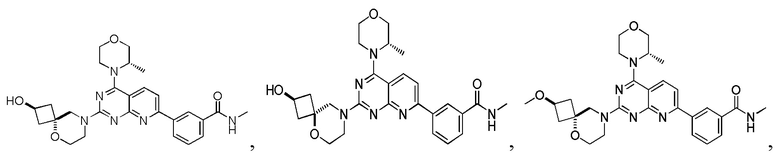
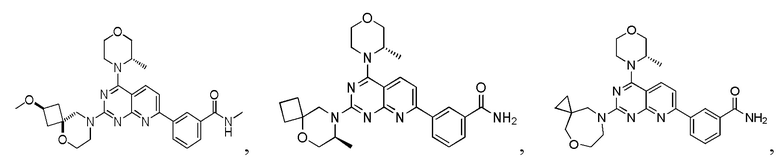
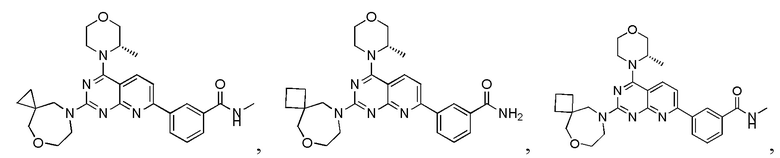
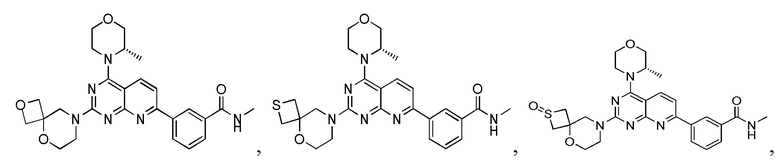
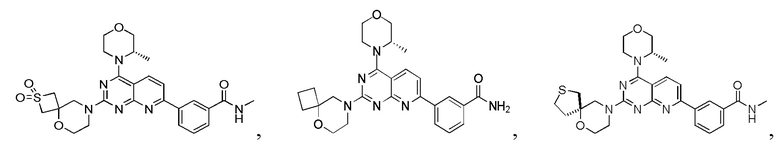
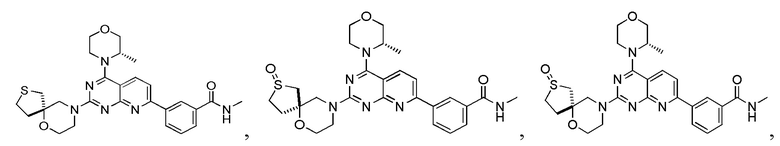
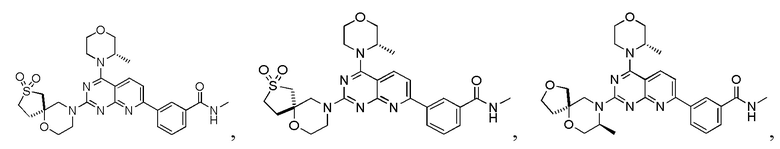
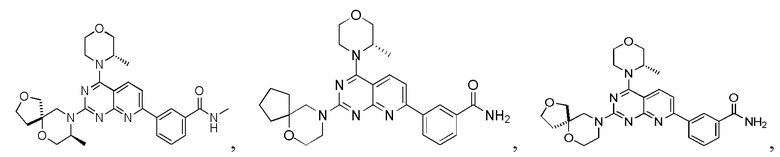
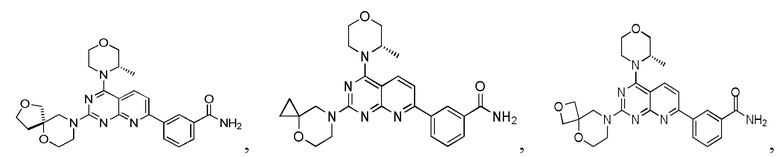
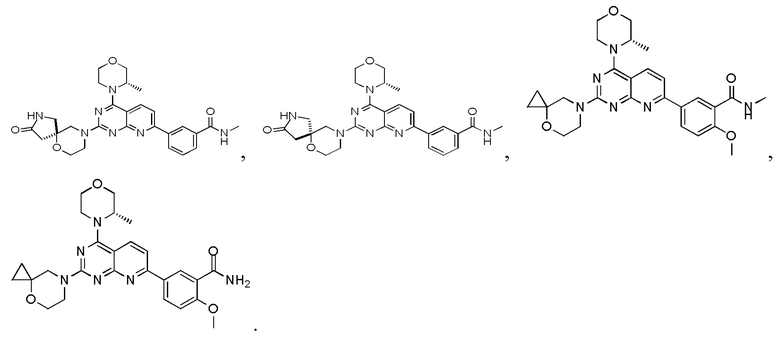
16. Фармацевтическая композиция для ингибирования активности в отношении киназы mTORC1/2 с двойной специфичностью, содержащая терапевтически эффективное количество соединения, его фармацевтически приемлемой соли или его стереоизомера по любому из пп. 1-15 в качестве активного ингредиента и фармацевтически приемлемый носитель.
17. Применение соединения, его фармацевтически приемлемой соли или его стереоизомера по любому из пп. 1-15 или фармацевтической композиции по п. 16 в изготовлении лекарственного препарата, предназначенного для лечения рака молочной железы.
| CN 103030653 A, 10.04.2013 | |||
| WO 2008023161 A1, 28.02.2008 | |||
| WO 2007060404 A1, 31.05.2007 | |||
| US 20170281637 A1, 05.10.2017 | |||
| ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2473549C2 |

