[0001] Настоящая заявка испрашивает приоритет следующих заявок:
CN202110286500.6, поданной 17 марта 2021 г.;
CN202110712765.8, поданной 25 июня 2021 г.;
CN202111314330.4, поданной 8 ноября 2021 г.;
CN202210187905.9, поданной 28 февраля 2022 г.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0002] Настоящее изобретение относится к ряду глутаримидных соединений, замещенных кольцом, конденсированным с фураном, и их применению в изготовлении лекарственного средства для лечения родственных заболеваний, в частности, к соединению формулы (II) и его фармацевтически приемлемой соли.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0003] Поскольку известно, что передача сигналов андрогеновых рецепторов играет решающую роль в патогенезе рака предстательной железы и участвует в развитии других андрогенных рецептор-положительных видов рака, применяли ингибирование передачи сигналов андрогенных рецепторов с помощью антиандрогена, который является антагонистом андрогенных рецепторов, в лечении рака предстательной железы, или его применение предлагается.
[0004] Ген AR кодирует белок андрогенового рецептора, лигандами которого являются преимущественно тестостерон и дигидротестостерон. Рецептор широко представлен в большинстве органов и тканей организма, при этом он опосредует биологические эффекты андрогенов. Андрогеновый рецептор может связываться с белком теплового шока (Hsp) в цитоплазме. Когда андроген связывается с андрогеновым рецептором, рецептор активируется, а белок теплового шока диссоциирует. Андрогеновый рецептор образует димер, который проникает в ядро, связывается с элементом ответа на андрогены (ARE) на ДНК и инициирует транскрипцию и экспрессию ряда генов, расположенных ниже по ходу транскрипции относительно элемента, включая гены, такие как простат-специфический антиген (PSA), простатическая кислая фосфатаза (PAP) и ингибитор циклин-зависимой киназы (CDK) p21WAF1/CIPI, что в конечном итоге приводит к дифференцировке клеток и способствует развитию тканей и органов. Андрогены характеризуются функцией поддержания роста и развития предстательной железы. В среде, не содержащей андрогенов, клетки предстательной железы будут подвергаться спонтанному апоптозу, при этом в среде с нормальным уровнем андрогенов клетки предстательной железы могут продолжать расти и дифференцироваться. Следовательно, было высказано предположение, что избыточная секреция андрогенов и чрезмерная чувствительность андрогеновых рецепторов способствуют беспрепятственному росту клеток предстательной железы, что является факторами риска развития рака предстательной железы.
[0005] Рак предстательной железы (PCa) представляет собой один из наиболее часто диагностируемых видов рака, не связанных с кожей, среди мужчин в США. Он представляет собой вторую наиболее распространенную причину смерти от рака, при этом ежегодно в США регистрируется более 200000 новых случаев и более 30000 смертей. Андроген-депривационная терапия (ADT) представляет собой стандартный способ лечения прогрессирующего PCa. Пациентов с прогрессирующим PCa подвергают ADT с помощью агонистов лютеинизирующего гормона-рилизинг-гормона (LHRH), антагонистов LHRH или двусторонней орхиэктомии. Несмотря на первоначальный ответ на ADT, прогрессирование заболевания неизбежно, и рак развивается в виде кастрационно-резистентного рака предстательной железы (CRPC). У не более 30% пациентов с раком предстательной железы, которые проходят первичное лучевое или хирургическое лечение, будут развиваться метастатические заболевания в течение 10 лет после первичного лечения. Ежегодно приблизительно у 50000 пациентов будет развиваться метастатическое заболевание, называемое метастатический CRPC (mCRPC).
[0006] Химера, активирующая протеолиз целевого белка (PROTAC), представляет собой методику, в которой применяется система убиквитин-протеасома для нацеливания на конкретные белки и индукции их внутриклеточной деградации. Система убиквитин-протеасома представляет собой основной путь внутриклеточной деградации белка, главным образом отвечая за удаление денатурированных, мутировавших или вредных белков из клетки в качестве ее нормальных физиологических функций. 80% или более внутриклеточной деградации белка зависит от системы убиквитин-протеасома. В PROTAC используется собственный механизм разрушения белков клетки для удаления конкретных белков-мишеней из клетки.
[0007] В настоящем изобретении описаны соединения, включая содержащие их композиции, функция которых заключается в рекрутировании эндогенных белков к убиквитинлигазам Е3, таким как цереблон (CRBN) в убиквитинлигазах Е3, для убиквитинирования и последующей деградации, а также способы их применения. В частности, в настоящем изобретении предусмотрено бифункциональное или активирующее протеолиз целевого белка химерное (PROTAC) соединение, которое, как было обнаружено, выступает в качестве модулятора направленного убиквитирования и деградации андрогенового рецептора (AR).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0008] В настоящем изобретении предусмотрено соединение формулы (II) или его фармацевтически приемлемая соль,
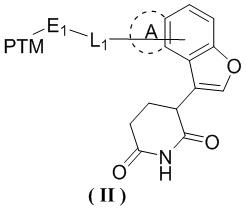 ,
,
[0009] где
[00010] PTM выбран из лекарственного средства или его производного, которое связывается с белком, нацеленным на AR;
[00011] L1 выбран из -(CH2)n-, и каждый CH2 необязательно замещен R1;
[00012] R1 выбран из C3-7циклоалкила, 6-членного гетероциклоалкила, -NRa-, -CRbRc-, -CH2CH2O- и -NHC(=O)-;
[00013] Ra выбран из H и C1-3алкила;
[00014] Rb и Rc выбраны из H, D и F;
[00015] E1 выбран из одинарной связи и O;
[00016] кольцо A отсутствует;
[00017] или кольцо A выбрано из фенила и 5-членного гетероарила;
[00018] n выбран из 1, 2, 3, 4, 5 и 6;
[00019] 6-членный гетероциклоалкил и 5-членный гетероарил содержат 1, 2 или 3 гетероатома или группы с гетероатомами, независимо выбранные из -NH-, -O-, -S- и N соответственно;
[00020] при условии, что соединение не выбрано из:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  .
.
[00021] В некоторых вариантах осуществления настоящего изобретения PTM выбран из  ,
,  и
и  , а другие переменные являются такими, как определено в настоящем изобретении.
, а другие переменные являются такими, как определено в настоящем изобретении.
[00022] В некоторых вариантах осуществления настоящего изобретения Ra выбран из H и CH3, а другие переменные являются такими, как определено в настоящем изобретении.
[00023] В некоторых вариантах осуществления настоящего изобретения R1 выбран из циклопропила, циклогексила, пиперазинила, пиперидинила, -NRa-, -CH2CH2O-, -NHC(=O)- и -C(=O)NH-, а другие переменные являются такими, как определено в настоящем изобретении.
[00024] В некоторых вариантах осуществления настоящего изобретения R1 выбран из 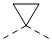 ,
, 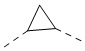 ,
,  ,
,  , -NH-, -N(CH3)-, -CH2CH2O-, -NHC(=O)- и -C(=O)NH-, а другие переменные являются такими, как определено в настоящем изобретении.
, -NH-, -N(CH3)-, -CH2CH2O-, -NHC(=O)- и -C(=O)NH-, а другие переменные являются такими, как определено в настоящем изобретении.
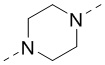
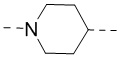
[00025] В некоторых вариантах осуществления настоящего изобретения R1 выбран из , , , , -N(CH3)-, -CH2CH2O- и -NHC(=O)-, а другие переменные являются такими, как определено в настоящем изобретении.
[00026] В некоторых вариантах осуществления настоящего изобретения L1 выбран из  ,
, 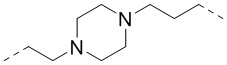 ,
, 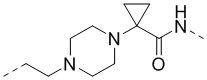 ,
, 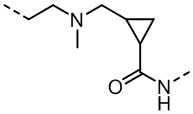 ,
, 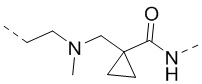 ,
, 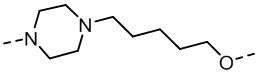 ,
, 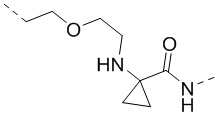 ,
, 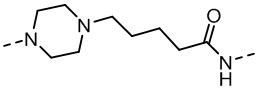 ,
, 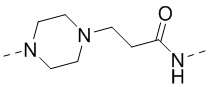 и
и 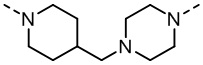 , а другие переменные являются такими, как определено в настоящем изобретении.
, а другие переменные являются такими, как определено в настоящем изобретении.
[00027] В некоторых вариантах осуществления настоящего изобретения структурный фрагмент -E1-L1- выбран из 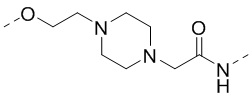 ,
, 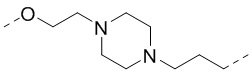 ,
, 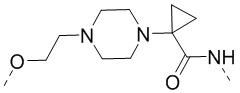 ,
, 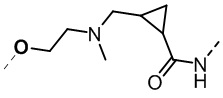 ,
, 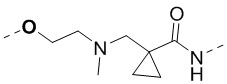 , ,
, , 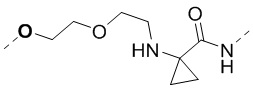 , , и , а другие переменные являются такими, как определено в настоящем изобретении.
, , и , а другие переменные являются такими, как определено в настоящем изобретении.
[00028] В некоторых вариантах осуществления настоящего изобретения структурный фрагмент 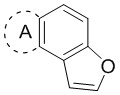 выбран из
выбран из  ,
,  и
и  , а другие переменные являются такими, как определено в настоящем изобретении.
, а другие переменные являются такими, как определено в настоящем изобретении.
[00029] В некоторых вариантах осуществления настоящего изобретения соединение выбрано из структур, представленных формулами (I-1), (I-2), (II-1), (II-2), (III-1) и (IV-1):
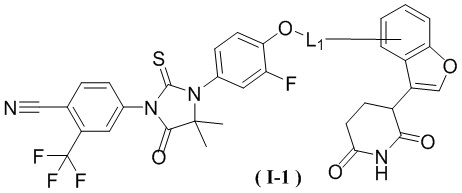
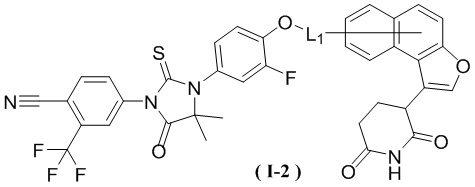
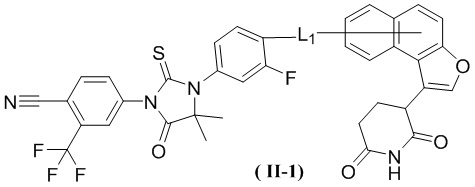
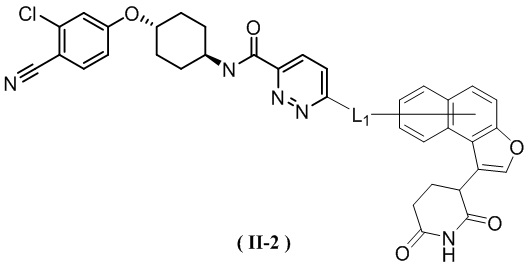
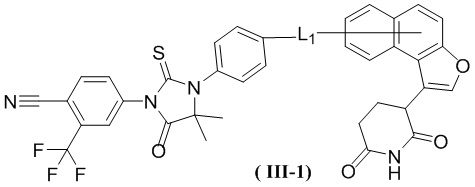
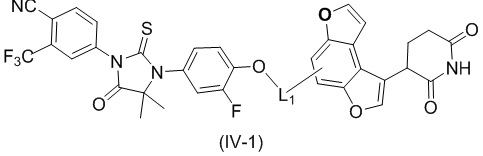 ,
,
[00030] где
[00031] L1 является таким, как определено в настоящем изобретении.
[00032] В настоящем изобретении предусмотрено соединению формулы (Ι) или его фармацевтически приемлемая соль,
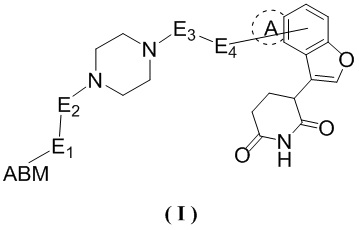 ,
,
[00033] где
[00034] E1 выбран из одинарной связи, O и NH;
[00035] E2 выбран из одинарной связи и C1-3алкила;
[00036] E3 выбран из C1-3алкила и циклопропила;
[00037] E4 выбран из одинарной связи и -C(=O)NH-;
[00038] кольцо A отсутствует;
[00039] или кольцо A выбрано из фенила;
[00040] ABM выбран из лекарственного средства или его производного, которое связывается с белком, нацеленным на AR.
[00041] В некоторых вариантах осуществления настоящего изобретения структурный фрагмент -E1-E2- выбран из одинарной связи и -OCH2CH2-, а другие переменные являются такими, как определено в настоящем изобретении.
[00042] В некоторых вариантах осуществления настоящего изобретения структурный фрагмент -E3-E4- выбран из C1-3алкила, 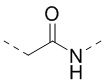 и
и 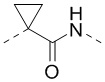 , а другие переменные являются такими, как определено в настоящем изобретении.
, а другие переменные являются такими, как определено в настоящем изобретении.
[00043] В некоторых вариантах осуществления настоящего изобретения структурный фрагмент выбран из и , а другие переменные являются такими, как определено в настоящем изобретении.
[00044] В некоторых вариантах осуществления настоящего изобретения ABM выбран из структуры, представленной формулой (ABM-1):
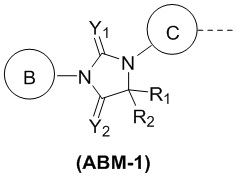 ,
,
[00045] где
[00046] R1 и R2 выбраны из метила;
[00047] или R1 и R2 вместе с атомом углерода, к которому они совместно присоединены, образуют C4-6циклоалкильное кольцо;
[00048] каждый из Y1 и Y2 независимо выбран из O и S;
[00049] кольцо B выбрано из фенила и пиридила, причем фенил и пиридил необязательно замещены 1, 2 или 3 Ra;
[00050] кольцо C выбрано из одинарной связи или фенила, причем фенил необязательно замещен 1, 2 или 3 Rb;
[00051] Ra выбран из F, Cl, Br, I, CN, CH3, CF3 и NO2;
[00052] Rb выбран из F и Cl.
[00053] В некоторых вариантах осуществления настоящего изобретения ABM выбран из структур, представленных формулами (ABM-1a) и (ABM-1b):
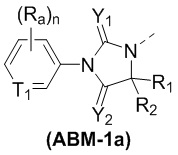
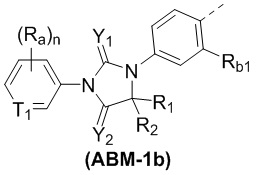 ,
,
[00054] где
[00055] T1 выбран из CH и N;
[00056] Rb1 выбран из H и F;
[00057] n выбран из 1, 2 и 3;
[00058] Y1, Y2, Ra, R1 и R2 являются такими, как определено в настоящем изобретении.
[00059] В некоторых вариантах осуществления настоящего изобретения ABM выбран из 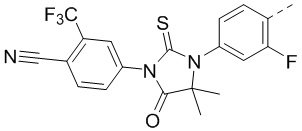 .
.
[00060] В некоторых вариантах осуществления настоящего изобретения, соединение выбрано из структур, представленных формулами (I-1a) и (I-2a):
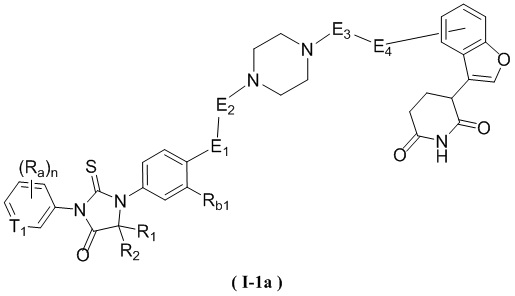 ,
, 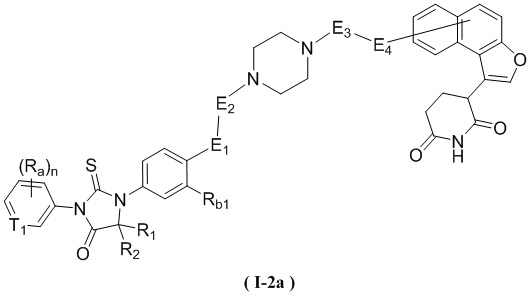 ,
,
[00061] где
[00062] T1, R1, R2, Ra, Rb1, n, E1, E2, E3 и E4 являются такими, как определено в любом из объектов настоящего изобретения.
[00063] Также существуют некоторые варианты осуществления настоящего изобретения, которые получают посредством любой комбинации вышеуказанных переменных.
[00064] В настоящем изобретении также предусмотрены следующие соединения или их фармацевтически приемлемые соли:
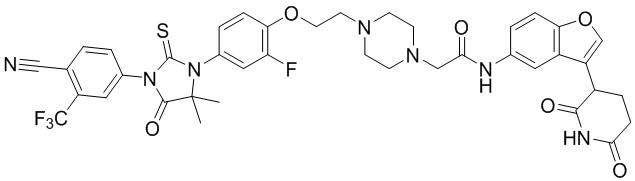 ,
, 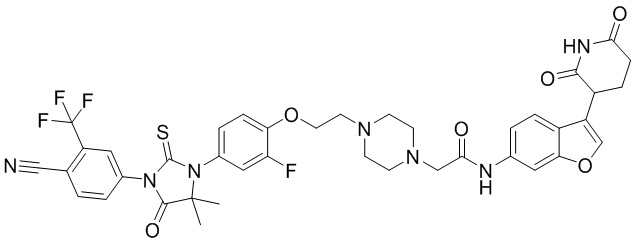 ,
, 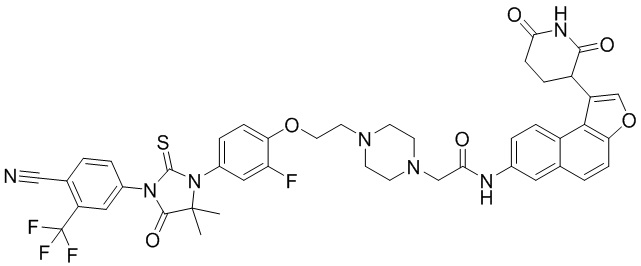 ,
, 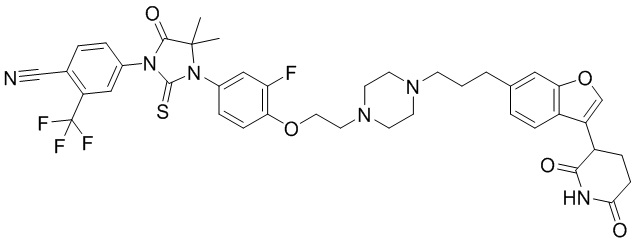 ,
, 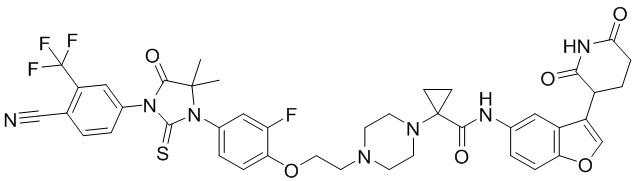 ,
, 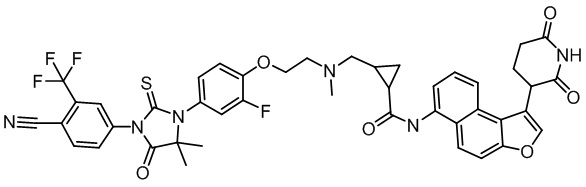 ,
, 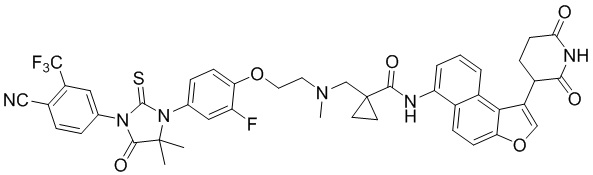 ,
, 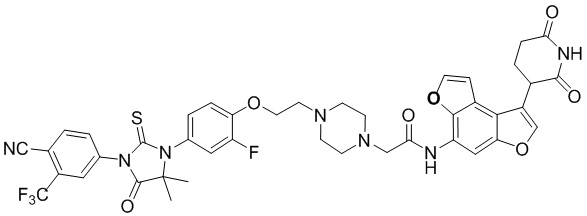 ,
, 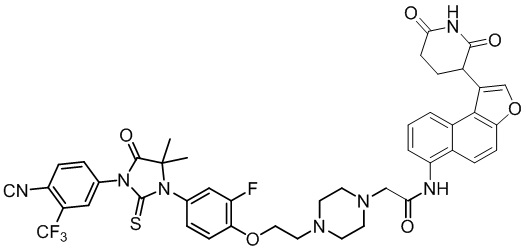 ,
, 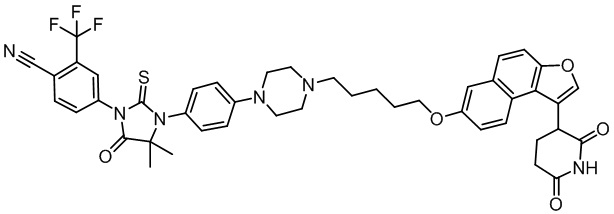 ,
,  ,
,  ,
,  ,
,  ,
, 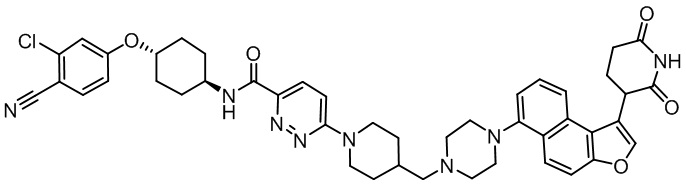 .
.
[00065] В настоящем изобретении также предусмотрены следующие соединения или их фармацевтически приемлемые соли:
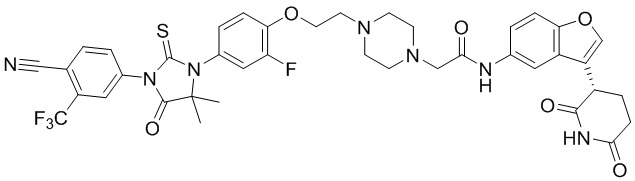
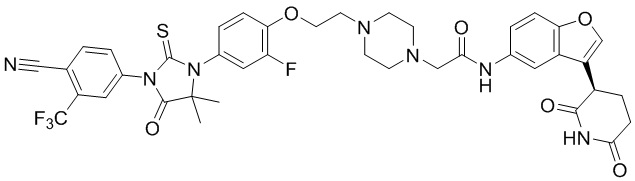
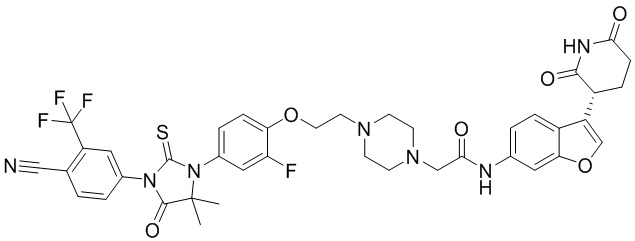
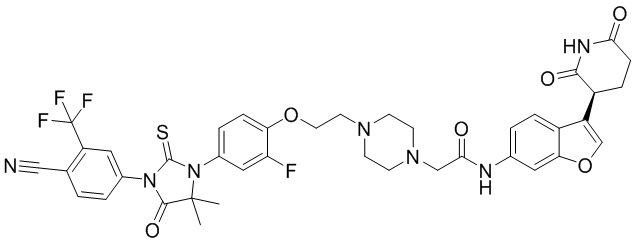
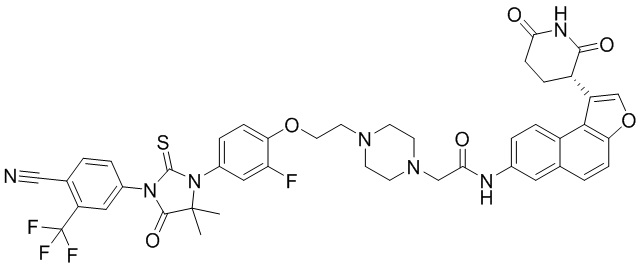
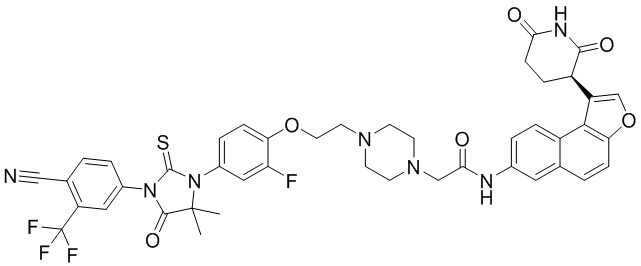
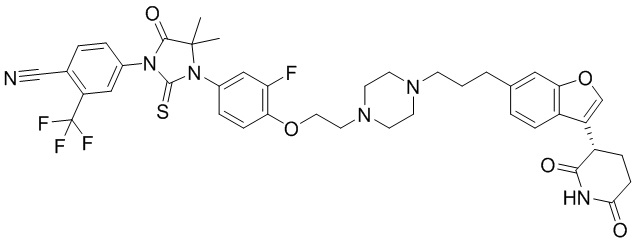
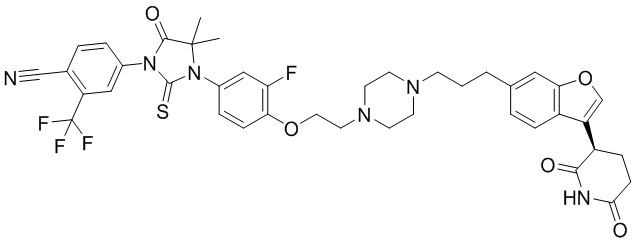
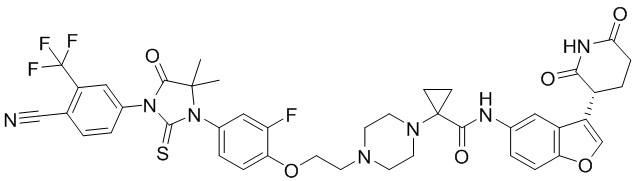 ,
, 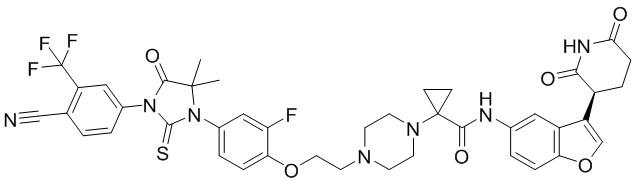 ,
, 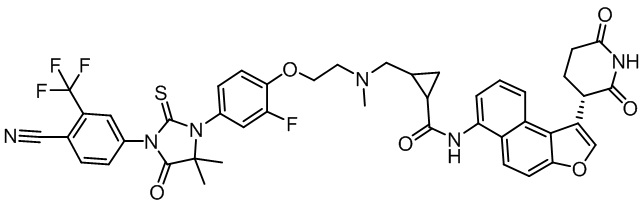 ,
, 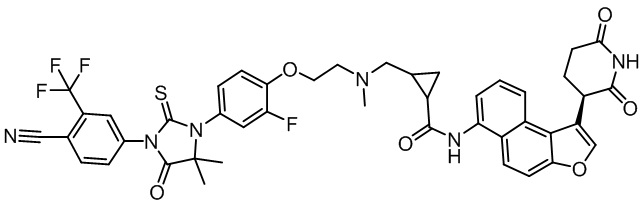 ,
, 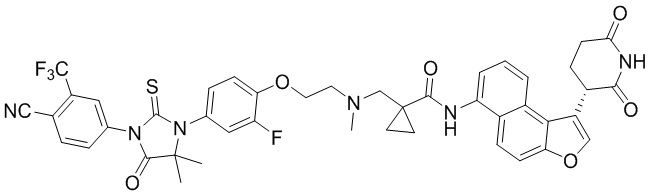 ,
, 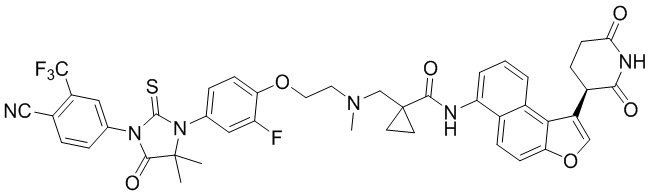 ,
, 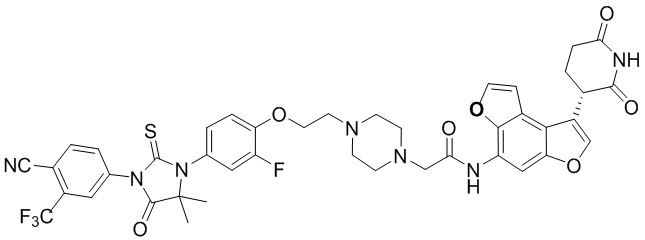 ,
, 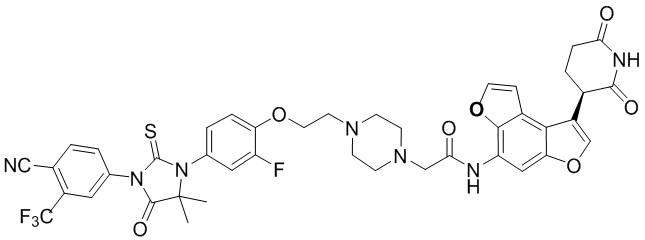 ,
, 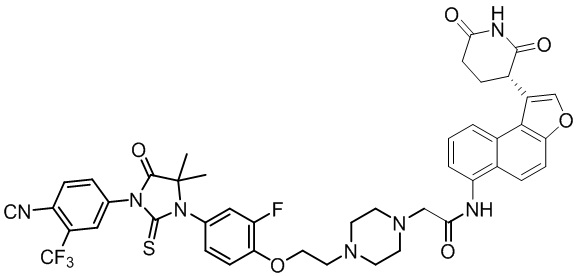 ,
, 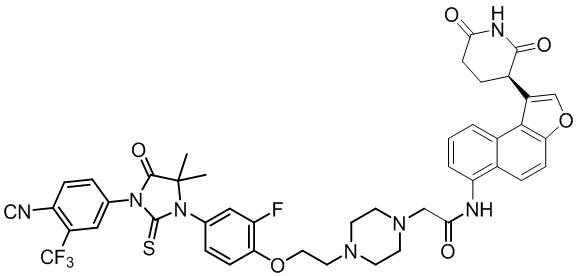 ,
, 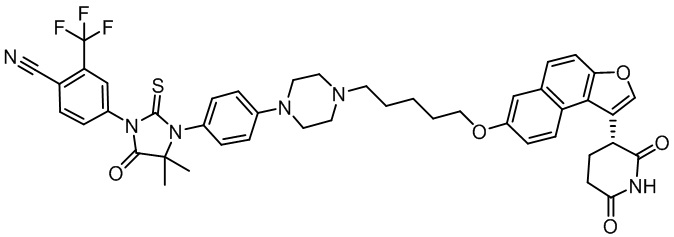 ,
, 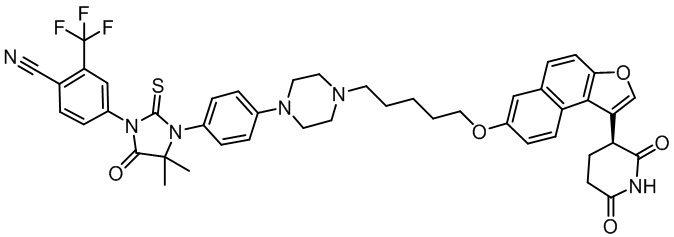 ,
, 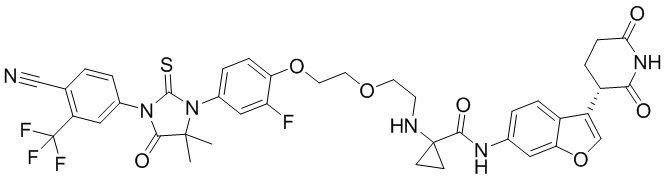 ,
, 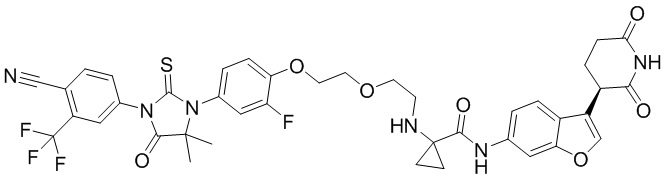 ,
, 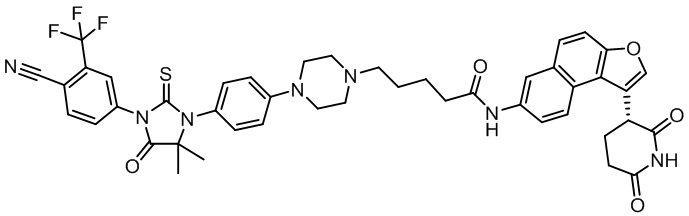 ,
, 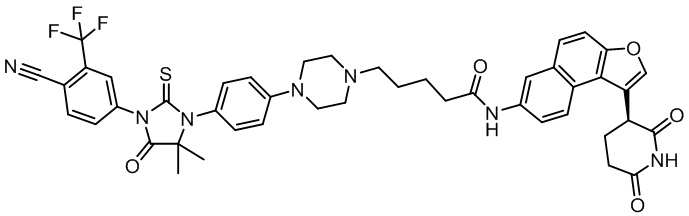 ,
, 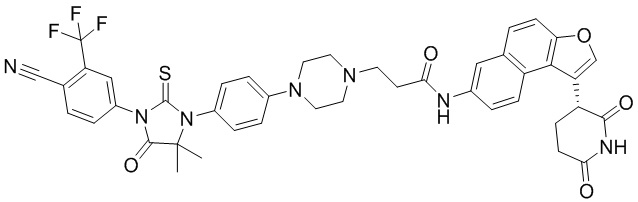 ,
, 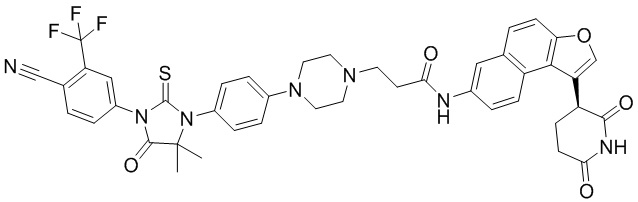 ,
, 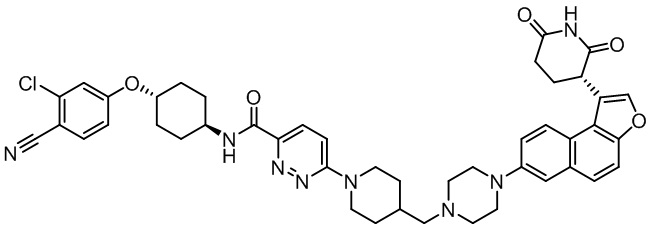 ,
, 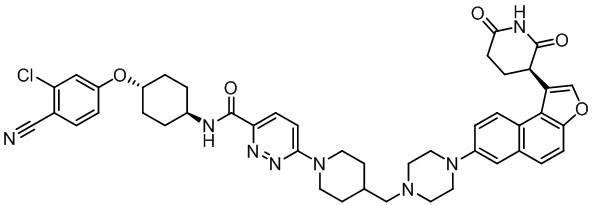 ,
, 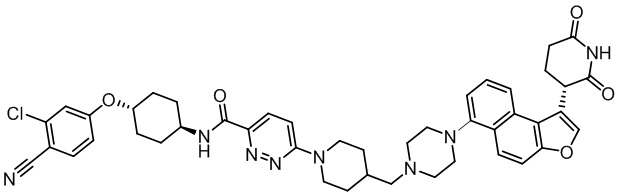 ,
, 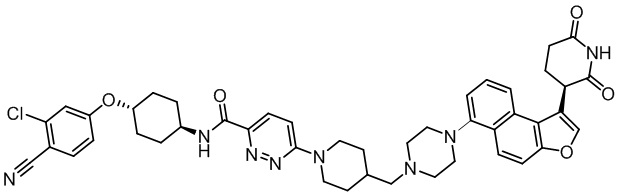 .
.
[00066] В настоящем изобретении также предусмотрено применение соединения или его фармацевтически приемлемой соли в изготовлении лекарственного средства, предназначенного для лечения рака предстательной железы.
[00067] Определение и описание
[00068] Если не указано иное, предполагается, что следующие термины и выражения, используемые в данном документе, имеют следующие значения. Конкретные термин или выражение при отсутствии точного определения не следует считать неопределенным или неясным, а следует понимать в соответствии с обычным значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту.
[00069] Термин «фармацевтически приемлемый» используют в данном документе для обозначения таких соединений, материалов, композиций и/или лекарственных форм, которые, в рамках здравого медицинского суждения, подходят для применения по отношению к тканям людей и животных без избыточной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с обоснованным соотношением польза/риск.
[00070] Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают путем осуществления реакции соединения, содержащего конкретный заместитель по настоящему изобретению, с относительно нетоксичными кислотой или основанием. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, соль присоединения основания можно получать путем приведения соединения в нейтральной форме в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина, магния или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, соль присоединения кислоты можно получать путем приведения соединения в нейтральной форме в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, при этом неорганическая кислота включает, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту; и соль органической кислоты, при этом органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту и метансульфоновую кислоту; и соли аминокислоты (такой как аргинин), и соль органической кислоты, такой как глюкуроновая кислота. Определенные конкретные соединения по настоящему изобретению содержат как основные, так и кислотные функциональные группы, таким образом, они могут быть превращены в любую соль присоединения основания или соль присоединения кислоты.
[00071] Термин «лекарственное средство или его производное» включает лекарственное средство или его производное, которое было разработано для связывания с белком-мишенью.
[00072] Фармацевтически приемлемая соль по настоящему изобретению может быть синтезирована из исходного соединения, которое содержит кислотный или основный фрагмент, посредством обычного химического способа. Как правило, такая соль может быть получена путем осуществления реакции свободной кислотной или основной формы соединения со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе или их смеси.
[00073] Соединения по настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. В настоящем изобретении рассматриваются все такие соединения, в том числе цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, рацемические и другие их смеси, такие как энантиомеры или смеси, обогащенные диастереомерами, все из которых находятся в пределах объема настоящего изобретения. В заместителях, таких как алкил, могут присутствовать дополнительные асимметрические атомы углерода. Все эти изомеры и их смеси включены в объем настоящего изобретения.
[00074] Если не указано иное, термин «энантиомер» или «оптический изомер» относится к стереоизомерам, которые являются зеркальными отражениями друг друга.
[00075] Если не указано иное, термин «диастереомер» относится к стереоизомеру, молекула которого содержит два или более хиральных центров, и молекулы не соотносятся друг с другом как зеркальные отражения.
[00076] Если не указано иное, «(+)» относится к правостороннему вращению, «(-)» относится к левостороннему вращению, и «(±)» относится к рацемической смеси.
[00077] Если не указано иное, абсолютная конфигурация стереогенного центра представлена клиновидной сплошной связью ( ) и клиновидной пунктирной связью (
) и клиновидной пунктирной связью (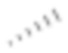 ); при отсутствии в структуре стереогенного центра относительная конфигурация представлена одновременным присутствием клиновидной сплошной связи () и клиновидной пунктирной связи (), например, транс-1,4-диметилциклогексан представлен в виде
); при отсутствии в структуре стереогенного центра относительная конфигурация представлена одновременным присутствием клиновидной сплошной связи () и клиновидной пунктирной связи (), например, транс-1,4-диметилциклогексан представлен в виде 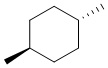 .
.
[00078] Если не указано иное, при содержании в группе одного или более соединяемых сайтов любой один или более сайтов группы могут быть соединены с другими группами посредством химических связей. Химическая связь между сайтом и другими группами может быть представлена прямой сплошной связью ( ), прямой пунктирной связью (
), прямой пунктирной связью ( ) или волнистой линией (
) или волнистой линией (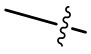 ). Например, прямая сплошная связь в -OCH3 означает, что группа присоединяется к другим группам посредством атома кислорода в группе; прямая пунктирная связь в
). Например, прямая сплошная связь в -OCH3 означает, что группа присоединяется к другим группам посредством атома кислорода в группе; прямая пунктирная связь в 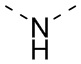 означает, что группа присоединяется к другим группам посредством двух концов от атома азота в группе; волнистая линия в
означает, что группа присоединяется к другим группам посредством двух концов от атома азота в группе; волнистая линия в 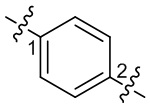 означает, что фенильная группа присоединяется к другим группам посредством атомов углерода в положении 1 и положении 2.
означает, что фенильная группа присоединяется к другим группам посредством атомов углерода в положении 1 и положении 2.
[00079] Соединения по настоящему изобретению могут существовать в конкретных формах. Если не указано иное, термин «таутомер» или «таутомерная форма» означает, что при комнатной температуре изомеры разных функциональных групп находятся в состоянии динамического равновесия и могут быстро превращаться друг в друга. Если возможно наличие таутомеров (как, например, в растворе), то может быть достигнуто химическое равновесие таутомеров. Например, протонный таутомер (также называемый прототропным таутомером) предусматривает взаимопревращение посредством протонного переноса, например, кето-енольная изомеризация и имин-енаминовая изомеризация. Валентный таутомер предусматривает некоторую перестройку связывающих электронов со взаимным преобразованием. Конкретным примером кето-енольной таутомеризации является таутомерия между двумя таутомерами - пентан-2,4-дионом и 4-гидроксипент-3-ен-2-оном.
[00080] Если не указано иное, термины «обогащенный одним изомером», «обогащенный изомерами», «обогащенный одним энантиомером» или «обогащенный энантиомерами» относятся к содержанию одного из изомеров или энантиомеров, составляющему менее 100%, и содержанию изомера или энантиомера, составляющему 60% или больше, или составляющему 70% или больше, или составляющему 80% или больше, или составляющему 90% или больше, или составляющему 95% или больше, или составляющему 96% или больше, или составляющему 97% или больше, или составляющему 98% или больше, или составляющему 99% или больше, или составляющему 99,5% или больше, или составляющему 99,6% или больше, или составляющему 99,7% или больше, или составляющему 99,8% или больше, или составляющему 99,9% или больше.
[00081] Если не указано иное, термин «изомерный избыток» или «энантиомерный избыток» относится к разности значений относительного процентного содержания двух изомеров или двух энантиомеров. Например, если содержание одного изомера или энантиомера составляет 90%, а содержание другого изомера или энантиомера составляет 10%, то изомерный или энантиомерный избыток (значение ee) составляет 80%.
[00082] Оптически активные (R)- и (S)-изомеры и D- и L-изомеры можно получать с применением хирального синтеза, или хиральных реагентов, или других традиционных методик. Если необходимо получить один тип энантиомера определенного соединения по настоящему изобретению, его можно получать путем асимметричного синтеза или производного действия хирального вспомогательного вещества, при этом полученную в результате диастереомерную смесь разделяют и вспомогательную группу отщепляют с получением чистого требуемого энантиомера. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), соль диастереоизомера образуется при использовании соответствующих оптически активных кислоты или основания, а затем проводят диастереомерное разделение общепринятыми способами, известными из уровня техники, и затем выделяют чистый энантиомер. Кроме того, энантиомер и диастереоизомер обычно разделяют путем хроматографии, в которой используется хиральная стационарная фаза и необязательно объединяется с методом химических производных (например, карбамат, полученный из амина).
[00083] Соединение по настоящему изобретению может содержать неприродную долю атомного изотопа одного или более атомов, которые составляют соединение. Например, соединение может быть меченным радиоактивным изотопом с применением радиоактивного изотопа, такого как тритий (3H), йод-125 (125I) или C-14 (14C). В качестве другого примера, дейтерированные лекарственные средства могут быть образованы посредством замещения атома водорода дейтерием, при этом связь, образованная между дейтерием и углеродом, сильнее, чем связь между обычным водородом и углеродом, при этом по сравнению с недейтерированными лекарственными средствами дейтерированные лекарственные средства обладают преимуществами, состоящими в снижении токсичности и побочных эффектов, повышении стабильности лекарственного средства, усилении эффективности, продлении периода биологического полувыведения лекарственных средств и т. п. Все изотопные варианты соединения по настоящему изобретению, радиоактивные или нет, охватываются объемом настоящего изобретения.
[00084] Термин «необязательный» или «необязательно» означает, что последующее событие или условие может быть реализовано, но не обязательно, так что термин включает случай, при котором событие или условие реализуется, и случай, при котором событие или условие не реализуется.
[00085] Термин «замещенный» означает, что один или более атомов водорода при конкретном атоме замещены заместителем, в том числе дейтерием и вариантами водорода, при условии, что валентность конкретного атома является нормальной, и замещенное соединение является стабильным. Если заместитель представляет собой атом кислорода (т. е. = O), то это означает, что два атома водорода являются замещенными. Положения в ароматическом кольце не могут быть замещены кетоном. Термин «необязательно замещенный» означает, что атом может быть замещен или не замещен заместителем, если не указано иное, причем тип и число заместителей могут быть произвольными при условии, что это химически достижимо.
[00086] Термин «замещенный» означает, что указанные атом или группа могут быть замещены другим атомом или группой, как указано. Например, CH2 в CH3CH2CH3 может быть замещен O, S и NH с получением CH3OCH3, CH3SCH3 и CH3NHCH3.
[00087] Если любая переменная (такая как R) встречается в составе или структуре соединения более одного раза, то определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то данная группа может быть необязательно замещена не более чем двумя R, при этом определение R в каждом случае является независимым. Более того, комбинация заместителя и/или его переменной является допустимой, только если данная комбинация приводит к образованию стабильного соединения.
[00088] Если число линкерной группы равняется 0, как например -(CRR)0-, то это означает, что линкерная группа представляет собой одинарную связь.
[00089] Когда заместитель вакантен, это означает, что заместитель отсутствует, например, когда X вакантен в A-X, структура A-X фактически представляет собой A. Когда перечисленный заместитель не указывает, каким атомом он связан с группой, которую необходимо замещать, такой заместитель может быть связан посредством любого его атома. Например, если пиридил выполняет функцию заместителя, он может быть присоединен к замещаемой группе посредством любого атома углерода в пиридиновом кольце.
[00090] Если не указано иное, термин «C1-3алкил» относится к прямой или разветвленной насыщенной углеводородной группе, состоящей из 1-3 атомов углерода. C1-3алкил включает C1-2-, C2-3алкил и т. п.; он может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин). Примеры C1-3алкила включают без ограничения метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т. п.
[00091] Если не указано иное, число атомов в кольце обычно определяется как число членов кольца, например, «5-7-членное кольцо» относится к «кольцу», в котором 5-7 атомов расположены по кругу.
[00092] Если не указано иное, «C3-7циклоалкил» относится к насыщенной циклической углеводородной группе, состоящей из 3-7 атомов углерода, включая моноциклические и бициклические системы, при этом бициклические системы включают спирокольцо, конденсированное кольцо и кольцо, соединенное мостиковой связью. C3-7циклоалкил включает C3-6, C3-5, C3-4, C4-7, C4-6, C4-5, C5-7, C5-6циклоалкил и т. п.; он может быть одновалентным, двухвалентным или поливалентным. Примеры C3-7циклоалкила включают без ограничения циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т. п.
[00093] Если не указано иное, термин «6-членный гетероциклоалкил» сам по себе или в комбинации с другими терминами относится к насыщенной циклической группе, состоящей из 6 атомов в кольце, причем 1, 2, 3 или 4 атома в кольце представляют собой гетероатомы, независимо выбранные из O, S и N, а остальные атомы представляют собой атомы углерода, при этом атомы азота необязательно кватернизованы, а гетероатомы, представляющие собой азот и серу, могут быть необязательно окислены (т. е. NO и S(O)p, p равняется 1 или 2). Он включает моноциклические и бициклические системы, при этом бициклические системы включают спирокольцо, конденсированное кольцо и кольцо, соединенное мостиковой связью. Примеры 6-членного гетероциклоалкила включают без ограничения тетрагидропиранил, пиперидинил (в том числе 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т. п.), пиперазинил (в том числе 1-пиперазинил, 2-пиперазинил и т. п.), морфолинил (в том числе 3-морфолинил, 4-морфолинил и т. п.) и т. п.
[00094] Если не указано иное, термины «5-членное гетероарильное кольцо» и «5-членный гетероарил» в настоящем изобретении можно использовать взаимозаменяемо, и термин «5-членный гетероарил» относится к моноциклической группе, состоящей из 5 атомов в кольце с сопряженной π-электронной системой, где 1, 2 или 3 атома в кольце представляют собой гетероатомы, независимо выбранные из O, S и N, а остальные представляют собой атомы углерода. Атомы азота необязательно кватернизованы, и гетероатомы, представляющие собой азот и серу, могут быть необязательно окислены (т. е. NO и S(O)p, p равняется 1 или 2). 5-членный гетероарил можно присоединять к остальной части молекулы посредством гетероатома или атома углерода. Примеры 5-членного гетероарила включают без ограничения пирролил (в том числе N-пирролил, 2-пирролил, 3-пирролил и т. п.), пиразолил (в том числе 2-пиразолил, 3-пиразолил и т. п.), имидазолил (в том числе N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил и т. п.), оксазолил (в том числе 2-оксазолил, 4-оксазолил, 5-оксазолил и т. п.), триазолил (1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил, 4H-1,2,4-триазолил и т. п.), тетразолил, изоксазолил (3-изоксазолил, 4-изоксазолил, 5-изоксазолил и т. п.), тиазолил (в том числе 2-тиазолил, 4-тиазолил, 5-тиазолил и т. п.), фурил (в том числе 2-фурил, 3-фурил и т. п.), тиенил (в том числе 2-тиенил, 3-тиенил и т. п.).
[00095] Если не указано иное, Cn-n+m или Cn-Cn+m включает любой конкретный случай от n до n+m атомов углерода, например, C1-12 включает C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12, и любой диапазон от n до n+m также включен, например, C1-12 включает C1-3, C1-6, C1-9, C3-6, C3-9, C3-12, C6-9, C6-12, C9-12 и т. п.; аналогично: n-членный - n+m-членный означает, что число атомов в кольце составляет от n до n+m, например, 3-12-членное кольцо включает 3-членное кольцо, 4-членное кольцо, 5-членное кольцо, 6-членное кольцо, 7-членное кольцо, 8-членное кольцо, 9-членное кольцо, 10-членное кольцо, 11-членное кольцо и 12-членное кольцо, и любой диапазон от n до n+m также включен, например, 3-12-членное кольцо включает 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо, 6-10-членное кольцо и т. п.
[00096] Если не указано иное, при содержании в группе одного или более соединяемых сайтов любой один или более сайтов группы могут быть соединены с другими группами посредством химических связей. Если сайт соединения химической связи не установлен, и в присоединяемом сайте присутствует атом H, то число атомов H в указанном сайте будет соответственно уменьшаться на число присоединяемых к нему химических связей, чтобы соответствовать соответствующей валентности. Химическая связь между сайтом и другими группами может быть представлена прямой сплошной связью (), прямой пунктирной связью () или волнистой линией (). Например, прямая сплошная связь в -OCH3 означает, что группа присоединяется к другим группам посредством атома кислорода в группе; прямая пунктирная связь в означает, что группа присоединяется к другим группам посредством двух концов от атома азота в группе; волнистая линия в означает, что фенильная группа присоединяется к другим группам посредством атомов углерода в положении 1 и положении 2; 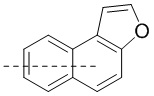 означает, что группа может присоединяться к другим группам посредством любых доступных для присоединения сайтов в нафталиновом кольце посредством одной химической связи, включая по меньшей мере шесть типов соединения, в том числе
означает, что группа может присоединяться к другим группам посредством любых доступных для присоединения сайтов в нафталиновом кольце посредством одной химической связи, включая по меньшей мере шесть типов соединения, в том числе 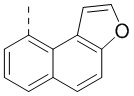 ,
, 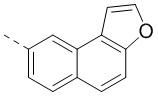 ,
, 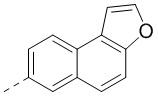 ,
, 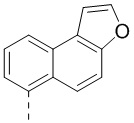 ,
, 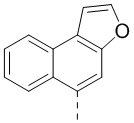 и
и 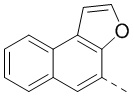 .
.
[00097] Соединения по настоящему изобретению могут быть получены посредством ряда способов синтеза, известных специалистам в данной области, в том числе посредством конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, образованных путем их комбинирования с другими способами химического синтеза, и эквивалентных альтернатив, известных специалистам в данной области, при этом предпочтительные варианты осуществления включают без ограничения примеры из настоящего изобретения.
[00098] Структура соединений по настоящему изобретению может быть подтверждена общепринятыми способами, известными специалистам в данной области техники, и если настоящее изобретение включает абсолютную конфигурацию соединения, то абсолютная конфигурация может быть подтверждена с помощью средств общепринятых методик из данной области техники. Например, в случае рентгеновской дифракции монокристаллов (SXRD) данные интенсивности дифракции собирают от выращенного монокристалла с использованием дифрактометра Bruker D8 Venture с источником излучения CuKα в качестве источника света и следующим режимом сканирования: сканирование ϕ/ω, и после сбора соответствующих данных структуру кристалла дополнительно анализируют прямым способом (Shelxs97) таким образом, чтобы подтвердить абсолютную конфигурацию.
[00099] Используемый в настоящем изобретении растворитель является коммерчески доступным. В настоящем изобретении используются следующие сокращения: вод. означает воду; экв. означает эквивалент или эквивалентный вес; CDI означает карбонилдиимидазол; DCM означает дихлорметан; PE означает петролейный эфир; DMF означает N,N-диметилформамид; DMSO означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; MeOH означает метанол; Boc означает трет-бутоксикарбонил, который представляет собой защитную группу для аминогруппы; к. т. означает комнатную температуру; O/N означает в течение ночи; THF означает тетрагидрофуран; Boc2O означает ди-трет-бутилдикарбонат; M означает моль/л; HPLC означает высокоэффективную жидкостную хроматографию.
[000100] Соединения по настоящему изобретению названы в соответствии с традиционными принципами номенклатуры в данной области техники или с помощью программного обеспечения ChemDraw®, а для коммерчески доступных соединений используют названия согласно каталогу поставщика.
[000101] Технический эффект
[000102] Соединения по настоящему изобретению характеризуются превосходным эффектом деградации белка AR, эффектом подавления пролиферации клеток и значительным противоопухолевым эффектом.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[000103] На фигуре 1 показана активность энзалутамида и WX001 в отношении деградации белка AR в клеточной линии LNCaP.
[000104] На фигуре 2 показана активность WX002 в отношении деградации белка AR в клеточной линии LNCaP.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
[000105] Настоящее изобретение подробно описано с помощью примеров, представленных ниже, но это не означает, что есть какие-либо противоположные ограничения в отношении настоящего изобретения. В данном документе подробно описано настоящее изобретение, а также раскрыты его конкретные примеры; для специалиста в данной области техники очевидно, что осуществление модификаций и улучшений по отношению к примерам настоящего изобретения происходит без отступления от сущности и объема настоящего изобретения.
[000106] Сравнительный пример 1
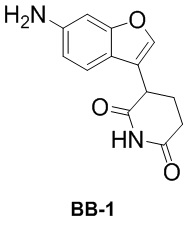
[000107] Путь синтеза:
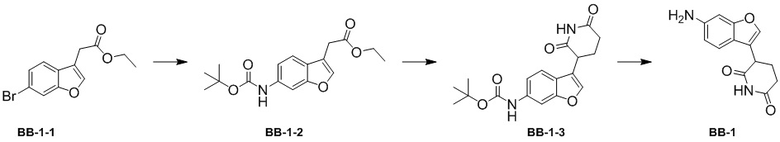
[000108] Стадия 1: синтез промежуточного соединения BB-1-2
[000109] К толуолу (15 мл) и воде (1,5 мл) добавляли соединение BB-1-1 (1 г, 3,53 ммоль) и трет-бутилкарбамат (2,07 г, 17,66 ммоль). К реакционной смеси медленно последовательно добавляли трис(дибензилиденацетон)дипалладий (226,41 мг, 247,25 мкмоль), 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (209,98 мг, 494,49 мкмоль) и фосфат калия (3,00 г, 14,13 ммоль). Смесь трижды продували азотом, затем нагревали до 100°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении для удаления большей части органического растворителя. Остаток разбавляли этилацетатом (30 мл). Органическую фазу дважды промывали водой (30 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный осадок разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 3/2, об./об.) с получением промежуточного соединения BB-1-2. 1H ЯМР (400 MГц, CDCl3) δ: 7,83-7,73 (m, 1H), 7,57 (s, 1H), 7,47-7,40 (m, 1H), 7,14-7,05 (m, 1H), 6,81 (br d, J = 1,0 Гц, 1H), 4,19 (q, J = 7,0 Гц, 2H), 3,67 (d, J = 1,3 Гц, 2H), 1,49-1,42 (m, 9H), 1,32-1,23 (m, 3H).
[000110] Стадия 2: синтез промежуточного соединения BB-1-3
[000111] К тетрагидрофурану (5 мл) добавляли соединение BB-1-2 (300 мг, 939,40 мкмоль) и акриламид (73,45 мг, 1,03 ммоль, 71,31 мкл) при 0°C. К реакционной смеси медленно добавляли трет-бутоксид калия (158,12 мг, 1,41 ммоль). Смесь трижды продували азотом, а затем перемешивали при 0°C в течение 1 часа. К реакционной смеси медленно добавляли насыщенный водный раствор хлорида аммония (20 мл) и дважды экстрагировали этилацетатом (20 мл × 2). Органическую фазу промывали один раз насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный осадок разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 3/1, об./об.) с получением промежуточного соединения BB-1-3.
[000112] Стадия 3: синтез гидрохлорида промежуточного соединения BB-1
[000113] К этилацетату (2 мл) добавляли соединение BB-1-3 (40 мг, 116,16 мкмоль), а затем к реакционной смеси медленно по каплям добавляли хлористоводородную кислоту/диоксан (4 M, 1 мл). Смесь трижды продували азотом, а затем перемешивали при 25°C в течение 12 часов. Реакционную смесь непосредственно выпаривали до сухого состояния путем ротационного выпаривания с получением гидрохлорида промежуточного соединения BB-1.
[000114] Сравнительный пример 2
[000115] Путь синтеза:
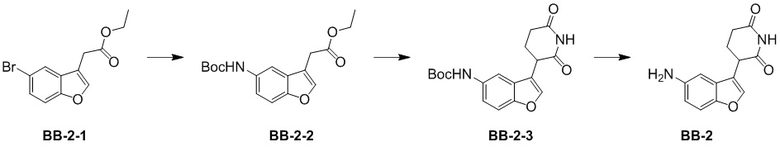
[000116] Стадия 1: синтез промежуточного соединения BB-2-2
[000117] К смеси толуола (70 мл) и воды (10 мл) добавляли промежуточное соединение BB-2-1 (4 г, 14,13 ммоль) и трет-бутилкарбамат (4,97 г, 42,39 ммоль) при комнатной температуре в атмосфере азота, а затем последовательно добавляли фосфат калия (12,00 г, 56,51 ммоль), трис(дибензилиденацетон)дипалладий (905,63 мг, 988,99 мкмоль) и 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (839,93 мг, 1,98 ммоль). Реакционную смесь перемешивали и обеспечивали ее реакцию при 110°C в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры и фильтровали через диатомит. К фильтрату добавляли воду (100 мл) и экстрагировали этилацетатом (500 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (300 мл × 2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении для удаления растворителя и полученный в результате остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 0/1 до 10/1, об./об.) с получением промежуточного соединения BB-2-2. 1H ЯМР (400 MГц, CDCl3) δ: 7,71 (br s, 1H), 7,63 (s, 1H), 7,37 (d, J = 8,8 Гц, 1H), 7,14 (dd, J = 2,0, 8,8 Гц, 1H), 6,59 (br s, 1H), 4,20 (q, J = 7,2 Гц, 2H), 3,67 (d, J = 0,8 Гц, 2H), 1,53 (s, 9H), 1,29 (t, J = 7,2 Гц, 3H).
[000118] Стадия 2: синтез промежуточного соединения BB-2-3
[000119] К тетрагидрофурану (8 мл) добавляли соединение BB-2-2 (0,75 г, 1,69 ммоль) и трет-бутоксид калия (284,42 мг, 2,53 ммоль). К реакционной смеси медленно добавляли акриламид (312,60 мг, 2,03 ммоль). Смесь трижды продували азотом, а затем перемешивали при 0-5°C в течение 2 часов. После завершения реакции к реакционной смеси медленно добавляли насыщенный водный раствор хлорида аммония (20 мл) и экстрагировали дважды этилацетатом (20 мл × 2). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный осадок разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 1/1, об./об.) с получением промежуточного соединения BB-2-3. MS-ESI масса/заряд: 245,0 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ: 8,13 (br s, 1H), 7,65 (br s, 1H), 7,46 (s, 1H), 7,32 (d, J = 8,8 Гц, 1H), 7,04 (dd, J = 2,1, 8,8 Гц, 1H), 6,57 (br s, 1H), 3,91 (t, J = 7,6 Гц, 1H), 2,80-2,53 (m, 2H), 2,35-2,20 (m, 2H), 1,45 (s, 9H).
[000120] Стадия 3: синтез гидрохлорида промежуточного соединения BB-2
[000121] Промежуточное соединение BB-2-3 (300 мг, 871,18 мкмоль) растворяли в растворе хлористоводородная кислота/этилацетат (4 M, 5 мл) при комнатной температуре и реакционную смесь перемешивали и обеспечивали ее реакцию при комнатной температуре в течение 2 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении для удаления растворителя с получением гидрохлорида промежуточного соединения BB-2.
[000122] 1H ЯМР (400 MГц, DMSO-d6) δ: 10,97 (s, 1H), 10,34 (br s, 2H), 8,05 (s, 1H), 7,72 (d, J = 8,8 Гц, 1H), 7,58 (d, J = 2,0 Гц, 1H), 7,35 (dd, J = 2,1, 8,7 Гц, 1H), 4,20 (dd, J = 4,8, 12,3 Гц, 1H), 2,88-2,73 (m, 1H), 2,68-2,58 (m, 1H), 2,30 (dq, J = 4,4, 12,6 Гц, 1H), 2,20-2,10 (m, 1H).
[000123] Сравнительный пример 3
[000124] Путь синтеза:
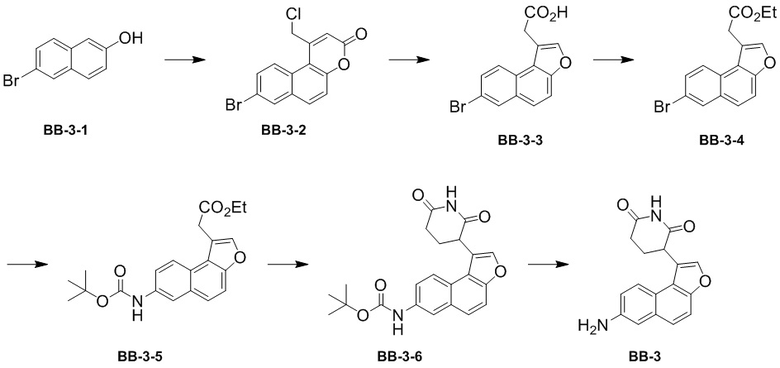
[000125] Стадия 1: синтез промежуточного соединения BB-3-2
[000126] К ледяной воде (40 мл) по каплям добавляли концентрированную серную кислоту (220,80 г, 2,21 моль, 120 мл, чистота: 98%) при комнатной температуре в атмосфере азота, затем добавляли соединение BB-3-1 (10 г, 44,83 ммоль), охлаждали до 5-10°C и к полученному медленно по каплям добавляли этил-4-хлорацетоацетат (7,38 г, 44,83 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов, затем нагревали до 50°C и продолжали перемешивать в течение 16 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду (1 л). Большое количество твердого вещества осаждали и фильтровали для сбора осадка на фильтре. К полученному твердому веществу добавляли толуол (400 мл), концентрировали при пониженном давлении для удаления растворителя, затем добавляли толуол (400 мл) и снова концентрировали при пониженном давлении для удаления растворителя с получением промежуточного соединения BB-3-2.
[000127] Стадия 2: синтез промежуточного соединения BB-3-3
[000128] Промежуточное соединение BB-3-2 (14,5 г, 44,81 ммоль) растворяли в растворе гидроксида натрия (8,70 г, 217,52 ммоль) в воде (150 мл) при комнатной температуре в атмосфере азота. Реакционную смесь нагревали до 80°C, перемешивали и обеспечивали ее реакцию в течение 5 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры и разбавляли дихлорметаном (150 мл). Органическую фазу собирали после разделения фаз, а водную фазу экстрагировали дихлорметаном (150 мл × 3). К водной фазе добавляли 2 M разбавленную хлористоводородную кислоту для доведения pH до 4 и экстрагировали этилацетатом (200 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя с получением промежуточного соединения BB-3-3.
[000129] Стадия 3: синтез промежуточного соединения BB-3-4
[000130] Промежуточное соединение BB-3-3 (11,3 г, 37,03 ммоль) растворяли в этаноле (300 мл) при комнатной температуре в атмосфере азота, затем к полученному добавляли концентрированную серную кислоту (2,08 г, 20,78 ммоль, 1,13 мл, чистота: 98%) и реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении для удаления растворителя, добавляли воду (150 мл) и экстрагировали этилацетатом (150 мл × 1, 100 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный осадок разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 97/3, об./об.) с получением промежуточного соединения BB-3-4. 1H ЯМР (400 MГц, DMSO-d6) δ: 8,35 (d, J = 2,0 Гц, 1H), 8,09 (t, J = 4,4 Гц, 2H), 7,86 (s, 2H), 7,73 (dd, J = 2,0, 8,8 Гц, 1H), 4,18-4,09 (m, 4H), 1,18 (t, J = 7,2 Гц, 3H).
[000131] Стадия 4: синтез промежуточного соединения BB-3-5
[000132] Промежуточное соединение BB-3-4 (2,00 г, 6,00 ммоль) растворяли в толуоле (60 мл) и воде (6 мл) при перемешивании при комнатной температуре и затем к полученному последовательно добавляли трет-бутилкарбамат (3,52 г, 30,01 ммоль), трис(дибензилиденацетон)дипалладий (384,78 мг, 420,20 мкмоль), 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (356,87 мг, 840,40 мкмоль) и фосфат калия (5,10 г, 20,01 ммоль). Систему реакционной смеси трижды продували азотом, медленно нагревали до 105°C и обеспечивали ее реакцию в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры. К полученной в результате реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным солевым раствором (50 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный осадок очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 10/1 до 5/1, об./об.) с получением промежуточного соединения BB-3-5.
[000133] Стадия 5: синтез промежуточного соединения BB-3-6
[000134] Соединение BB-3-5 (4,00 г, 10,83 ммоль) растворяли в растворителе N,N-диметилформамиде (60 мл) при 0°C в атмосфере азота, затем к полученному добавляли трет-бутоксид калия (1,22 г, 10,83 ммоль) и акриламид (1,67 г, 10,83 ммоль) соответственно и реакционную смесь перемешивали и обеспечивали ее реакцию при 0°C в течение 3 часов. После завершения реакции к реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным солевым раствором (30 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный осадок очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 3/1 до 1/1, об./об.) с получением промежуточного соединения BB-3-6. MS-ESI масса/заряд: 338,4 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,95 (s, 1H), 9,55 (s, 1H), 8,25 (s, 1H), 7,96 (d, J = 4,0 Гц, 2H), 7,72 (s, 2H), 2,89 (s, 3H), 2,74 (s, 2H), 1,52 (s, 9H).
[000135] Стадия 6: синтез гидрохлорида промежуточного соединения BB-3
[000136] Соединение BB-3-6 (1,0 г, 2,54 ммоль) растворяли в этилацетате (20 мл) при 0°C, затем к полученному медленно добавляли хлористоводородную кислоту/этилацетат (2,54 мл) и реакционную смесь перемешивали в течение 4 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении с получением гидрохлорида BB-3. MS-ESI масса/заряд: 294,31 [M+H]+.
[000137] Сравнительный пример 4
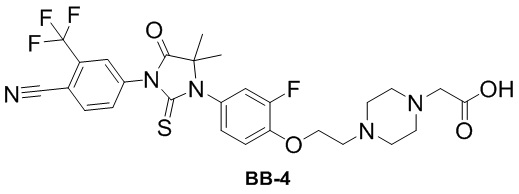
[000138] Путь синтеза:
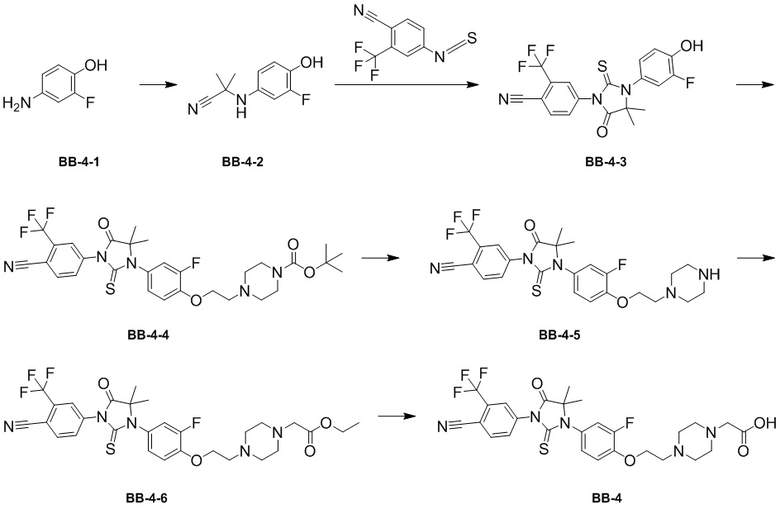
[000139] Стадия 1: синтез промежуточного соединения BB-4-2
[000140] Соединение BB-4-1 (10 г, 78,67 ммоль) растворяли в дихлорметане (120 мл) и ацетоне (60 мл) при 0-5°C в атмосфере азота, затем последовательно медленно по каплям добавляли цианотриметилсилан (12,45 г, 125,50 ммоль, 15,70 мл) и триметилсилилтрифторметансульфонат (820,00 мг, 3,69 ммоль, 666,67 мкл) и реакционную смесь перемешивали при 25°C в течение 2 часов. После завершения реакции реакционную смесь охлаждали до 0-5°C, разбавляли водой (200 мл) и экстрагировали этилацетатом (200 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 10/1 до 5/1, об./об.) с получением соединения BB-4-2. H ЯМР(400 MГц, CDCl3) δ: 6,83 (t, J = 9,0 Гц, 1H), 6,74 (dd, J = 2,7, 12,1 Гц, 1H), 6,66-6,59 (m, 1H), 1,65 (s, 6H).
[000141] Стадия 2: синтез соединения BB-4-3
[000142] Соединение BB-4-2 (8 г, 45,40 ммоль) растворяли в N,N-диметилацетамиде (150 мл) при комнатной температуре в атмосфере азота. К реакционной смеси порциями добавляли 4-изотиоцианато-2-(трифторметил)бензoнитрил (10,36 г, 45,40 ммоль). Реакционную смесь перемешивали при 25°C в течение 12 часов, затем добавляли метанол (60 мл) и разбавленную хлористоводородную кислоту (2 M, 60 мл) и перемешивали при 70°C в течение 3 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (500 мл) и экстрагировали этилацетатом (200 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 3/1 до 1/1, об./об.) с получением соединения BB-4-3. 1H ЯМР (400 MГц, CDCl3) δ: 8,03-7,94 (m, 1H), 8,02-7,94 (m, 1H), 8,05-7,92 (m, 1H), 8,04-7,92 (m, 1H), 8,15-7,89 (m, 1H), 7,86 (d, J = 1,5 Гц, 1H), 7,82-7,79 (m, 1H), 7,19-7,10 (m, 2H), 6,99-6,90 (m, 2H), 6,06 (br s, 1H), 1,58 (s, 6H).
[000143] Стадия 3: синтез соединения BB-4-4
[000144] Соединение BB-4-3 (2 г, 4,72 ммоль) растворяли в N,N-диметилформамиде (50 мл) при комнатной температуре в атмосфере азота и последовательно добавляли трет-бутил-4-(2-бромэтил)пиперазин-1-карбоксилат (1,66 г, 5,67 ммоль), карбонат калия (1,31 г, 9,45 ммоль) и йодид калия (784,17 мг, 4,72 ммоль). Реакционную смесь нагревали до 80°C, перемешивали и обеспечивали ее реакцию в течение 12 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 3/1 до 1/1, об./об.) с получением соединения BB-4-4. MS-ESI масса/заряд: 636,3 [M+H]+.
[000145] Стадия 4: синтез гидрохлорида соединения BB-4-5
[000146] Соединение BB-4-4 (2,0 г, 3,15 ммоль) растворяли в этилацетате (50 мл) при комнатной температуре в атмосфере азота. К реакционной смеси медленно по каплям добавляли хлористоводородную кислоту/этилацетат (4 M, 3,93 мл) и перемешивали при 25°C в течение 12 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении с получением гидрохлорида соединения BB-4-5. 1H ЯМР (400 MГц, CD3OD) δ: 8,23-8,13 (m, 2H), 8,00 (dd, J = 1,8, 8,3 Гц, 1H), 7,40 (t, J = 8,9 Гц, 1H), 7,32 (dd, J = 2,4, 11,4 Гц, 1H), 7,24 (br d, J = 8,8 Гц, 1H), 4,69-4,60 (m, 2H), 3,88-3,79 (m, 6H), 3,75-3,65 (m, 4H), 1,58 (s, 6H).
[000147] Стадия 5: синтез соединения BB-4-6
[000148] Соединение BB-4-5 (1,5 г, 2,62 ммоль, гидрохлорид) растворяли в ацетонитриле (50 мл) при комнатной температуре в атмосфере азота, а затем последовательно добавляли этилбромацетат (875,85 мг, 5,24 ммоль, 580,04 мкл) и карбонат калия (724,86 мг, 5,24 ммоль). Реакционную смесь нагревали до 80°C, перемешивали и обеспечивали ее реакцию в течение 5 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 3/1 до 1/1, об./об.) с получением соединения BB-4-6. MS-ESI масса/заряд: 622,2 [M+H]+.
[000149] Стадия 6: синтез соединения BB-4
[000150] Соединение BB-4-6 (1,5 г, 2,42 ммоль) растворяли в этаноле (50 мл) при комнатной температуре в атмосфере азота. К реакционной смеси медленно по каплям добавляли гидроксид лития (1 M, 7,25 мл) и перемешивали при 25°C в течение 12 часов. После завершения реакции к реакционной смеси добавляли 2 M хлористоводородную кислоту для доведения pH до 2-3 и концентрировали при пониженном давлении с получением соединения BB-4. MS-ESI масса/заряд: 594,1 [M+H]+.
[000151] Сравнительный пример 5
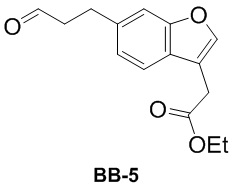
[000152] Путь синтеза:
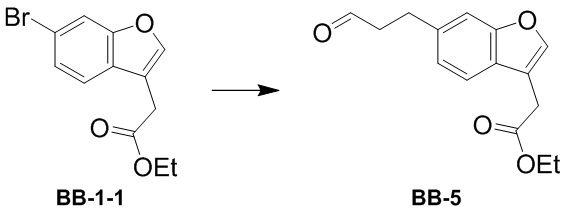
[000153] К диоксану (30 мл) добавляли соединение BB-1-1 (3 г, 10,60 ммоль) и аллиловый спирт (1,34 г, 23,07 ммоль, 1,57 мл) и к реакционной смеси медленно добавляли N-циклогексил-N-метилциклогексиламин (2,48 г, 12,72 ммоль, 2,70 мл), три-трет-бутилфосфин (4,29 г, 2,12 ммоль, 4,97 мл, содержание: 10%) и трис(дибензилиденацетон)дипалладий (970,32 мг, 1,06 ммоль). Смесь трижды продували азотом, а затем перемешивали при 30°C в течение 12 часов. К реакционной смеси медленно добавляли воду (50 мл) и трижды экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 0/1 до 3/1, об./об.) с получением соединения BB-5. 1H ЯМР (400 MГц, CDCl3) δ: 9,84 (s, 1H), 7,60 (s, 1H), 7,50 (d, J = 8,0 Гц, 1H), 7,33 (s, 1H), 7,12 (dd, J = 1,0, 8,0 Гц, 1H), 4,24-4,16 (m, 2H), 3,69 (d, J = 0,8 Гц, 2H), 3,14-3,03 (m, 2H), 2,90-2,78 (m, 2H), 1,29 (t, J = 7,2 Гц, 3H).
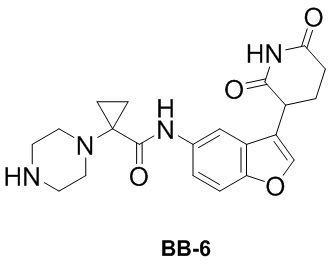
[000154] Сравнительный пример 6
[000155] Путь синтеза:
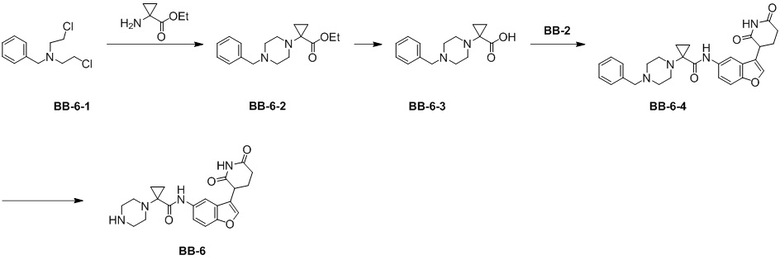
[000156] Стадия 1: синтез промежуточного соединения BB-6-2
[000157] К этанолу (10 мл) добавляли соединения, представляющее собой гидрохлорид этил-1-аминоциклопропан-1-карбоксилата (1,0 г, 6,04 ммоль), при 25°C и начинали перемешивание. К полученному последовательно добавляли соединение, представляющее собой гидрохлорид N-бензил-2-хлор-N-(2-хлорэтил)этиламина (1,78 г, 6,64 ммоль), и N,N-диизопропилэтиламин (7,88 г, 60,98 ммоль, 10,62 мл). Реакционную смесь нагревали до 80°C в атмосфере азота и перемешивали в течение дополнительных 12 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры, а затем непосредственно выпаривали до сухого состояния путем ротационного выпаривания. К неочищенному продукту добавляли воду (20 мл) и экстрагировали этилацетатом (20 мл × 2). Органические фазы объединяли, промывали полунасыщенным солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении для удаления растворителя и полученный в результате остаток очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 3/1 до 1/1, об./об.) с получением промежуточного соединения BB-6-2. 1H ЯМР (400 МГц, CDCl3) δ: 7,40-7,27 (m, 5 H), 4,18 (q, J = 7,20 Гц, 2H), 3,55 (s, 2H), 3,02 (s, 4H), 2,40 (s, 4H), 1,28-1,34 (m, 5H), 0,94 (q, J = 3,6 Гц, 2H).
[000158] Стадия 2: синтез промежуточного соединения BB-6-3
[000159] К этанолу (20 мл) добавляли промежуточное соединение BB-6-2 (0,3 г, 1,04 ммоль) при 25°C и начинали перемешивание. К полученному последовательно добавляли соединение, представляющее собой гидроксид калия (583,66 мг, 10,40 ммоль). Реакционную смесь нагревали до 120°C и перемешивали в течение дополнительных 12 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры и выпаривали до сухого состояния путем ротационного выпаривания. Затем к реакционной смеси добавляли воду (40 мл) и экстрагировали этилацетатом (40 мл × 2). К водной фазе добавляли 4 M разбавленную хлористоводородную кислоту для доведения pH до 3, а затем экстрагировали этилацетатом (40 мл). Продукт находился в водной фазе, которую собирали лиофилизацией. Получали соединение BB-6-3. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,70 (s, 1H), 7,59 (s, 2H), 7,45 (s, 3H), 4,26 (s, 2H), 3,46 (t, J = 12,4 Гц, 2H), 3,21 (d, J = 11,6 Гц, 2H), 2,94 (d, J = 12,8 Гц, 2H), 2,83 (s, 2H), 1,16 (s, 2H), 0,94 (s, 2H).
[000160] Стадия 3: синтез соединения BB-6-4
[000161] К N,N-диметилформамиду (20 мл) добавляли промежуточное соединение BB-6-3 при 25°C и начинали перемешивание. К полученному последовательно добавляли соединение BB-2 (938,21 мг, 3,84 ммоль), гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (2,19 г, 5,76 ммоль) и триэтиламин (1,55 г, 15,37 ммоль, 2,14 мл). Реакционную смесь перемешивали в течение дополнительных 12 часов в атмосфере азота. После завершения реакции к реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали полунасыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении для удаления растворителя и полученный в результате остаток очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/1 до 0/1, об./об.) с получением промежуточного соединения BB-6-4. MS-ESI масса/заряд: 487,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,91 (s, 1H), 9,92 (s, 1H), 7,95 (s, 1H), 7,88 (s, 1H), 7,53 (d, J = 8,8 Гц, 1H), 7,41 (dd, J = 8,8, 1,6 Гц, 1H), 7,35-7,28 (m, 4H), 7,27-7,22 (m, 1H), 4,12 (dd, J = 12,0, 4,8 Гц, 1H), 3,50 (s, 2H), 3,40-3,30 (m, 2H), 2,89 (s, 2H), 2,83-2,74 (m, 1H), 2,73 (s, 2H), 2,61-2,60 (m, 1H), 2,56-2,55 (m, 2H), 2,34-2,20 (m, 1H), 2,14-2,10 (m, 1H), 1,08-1,06 (m, 4 H).
[000162] Стадия 4: синтез соединения BB-6
[000163] Соединение BB-6-4 (50 мг, 97,91 мкмоль) растворяли в тетрагидрофуране (10 мл) при 25°C и добавляли влажный палладий на угле (5 мг, 10,28 мкл) в атмосфере аргона. Реакционную смесь трижды продували водородом, а давление в системе поддерживали на уровне 40 фунтов/кв. дюйм. Реакционную смесь нагревали до 30°C и перемешивали в течение 12 часов в атмосфере водорода. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя с получением соединения BB-6. MS-ESI масса/заряд: 397,1 [M+H]+.
[000164] Сравнительный пример 7
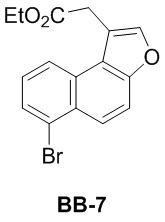
[000165] Путь синтеза:
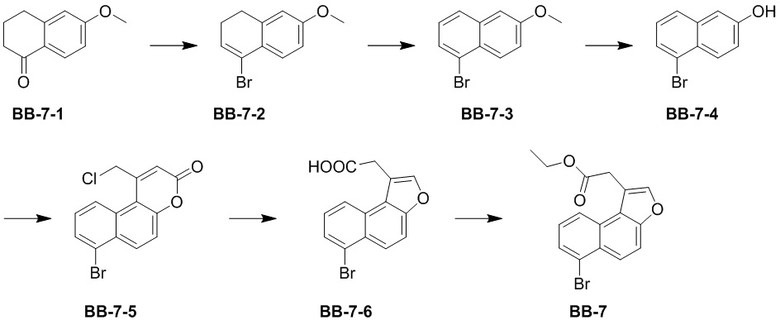
[000166] Стадия 1: синтез соединения BB-7-2
[000167] Соединение трифенилфосфит (48,42 г, 156,06 ммоль, 41,04 мл) растворяли в дихлорметане (250 мл) при 25°C, охлаждали до -70°C в атмосфере азота и по каплям добавляли жидкий бром (27,21 г, 170,25 ммоль, 8,78 мл). После завершения добавления к реакционной смеси последовательно по каплям добавляли раствор триэтиламина (18,66 г, 184,44 ммоль, 25,67 мл) и соединение BB-7-1 (25,00 г, 141,88 ммоль) в дихлорметане (60 мл). После завершения добавления реакционную смесь нагревали до 25°C и перемешивали в течение 12 часов. После завершения реакции реакционную смесь медленно выливали в насыщенный водный раствор сульфита натрия (400 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (200 мл × 3). Органическую фазу промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир). Получали соединение BB-7-2. 1H ЯМР (400 МГц, CDCl3) δ: 7,48 (d, J = 8,78 Гц, 1H), 6,75 (dd, J = 8,53, 2,51 Гц, 1H), 6,68 (d, J = 2,76 Гц, 1H), 6,30 (t, J = 4,77 Гц, 1H), 3,82 (s, 3H), 2,82 (t, J = 8,03 Гц, 2H), 2,35 (td, J = 8,03, 5,02 Гц, 2H).
[000168] Стадия 2: синтез соединения BB-7-3
[000169] Соединение BB-7-2 (10,00 г, 41,82 ммоль) растворяли в толуоле (100 мл) при 25°C, охлаждали до 0°C в атмосфере азота и к нему порциями добавляли дихлордицианобензохинон (10,44 г, 46,00 ммоль). После завершения добавления обеспечивали реакцию реакционной смеси при 25°C в течение 15 часов. К реакционной смеси для гашения по каплям добавляли насыщенный водный раствор сульфита натрия (200 мл), перемешивали в течение 10 минут, добавляли 1 н. водный раствор гидроксида натрия (100 мл) и экстрагировали этилацетатом (50 мл × 3). Органическую фазу промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Полученный в результате сырой продукт суспендировали петролейным эфиром (50 мл) в течение 10 минут, фильтровали и осадок на фильтре ополаскивали петролейным эфиром (25 мл × 2). Фильтрат выпаривали до сухого состояния путем ротационного выпаривания и неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир). Получали соединение BB-7-3. 1H ЯМР (400 МГц, CDCl3) δ: 8,13 (d, J = 9,29 Гц, 1H), 7,67 (d, J = 8,28 Гц, 1H), 7,61 (d, J = 7,28 Гц, 1H), 7,21-7,26 (m, 2H), 7,11 (d, J = 2,51 Гц, 1H), 3,92 (s, 3H).
[000170] Стадия 3: синтез соединения BB-7-4
[000171] Соединение BB-7-3 (4,90 г, 20,67 ммоль) растворяли в дихлорметане (50 мл) при 25°C, охлаждали до 0°C и медленно по каплям добавляли трибромид бора (6,21 г, 24,80 ммоль, 2,39 мл). После завершения добавления реакционную смесь перемешивали при 25°C в течение 3 часов. Реакционную смесь выливали в ледяную воду (250 мл) для гашения и экстрагировали дихлорметаном (100 мл). Органическую фазу промывали насыщенным солевым раствором (100 мл × 1), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Получали соединение BB-7-4. 1H ЯМР (400 МГц, CDCl3) δ: 8,19 (d, J = 9,03 Гц, 1H), 7,63-7,73 (m, 2H), 7,26-7,32 (m, 1H), 7,23 (dd, J = 9,03, 2,51 Гц, 1H), 7,19 (d, J = 2,51 Гц, 1H).
[000172] Стадия 4: синтез соединения BB-7-5
[000173] Соединение BB-7-4 (2,50 г, 11,21 ммоль) растворяли в метансульфоновой кислоте (25 мл) при 20°C, по каплям добавляли этил-4-хлорацетоацетат (2,77 г, 16,81 ммоль, 2,27 мл) и перемешивали при 20°C в течение 15 часов. Реакционную смесь выливали в ледяную воду (200 мл) для гашения, перемешивали в течение 10 минут и фильтровали. Осадок на фильтре промывали водой (30 мл × 3), а затем выпаривали до сухого состояния путем ротационного выпаривания. Получали соединение BB-7-5. 1H ЯМР (400 MГц, DMSO-d6) δ: 8,56 (d, J = 8,78 Гц, 1H), 8,46 (d, J = 9,29 Гц, 1 H), 7,71 (d, J = 9,29 Гц, 1H), 7,63 (dd, J = 8,66, 7,65 Гц, 1H), 6,92 (s, 1H), 5,39 (s, 2H).
[000174] Стадия 5: синтез соединения BB-7-6
[000175] К водному раствору гидроксида натрия (2 M, 1,03 мл) добавляли соединение BB-7-5 (0,10 г, 309,05 мкмоль) при 25°C и реакционную смесь перемешивали при 80°C в течение 3 часов. Реакционную смесь охлаждали до 25°C, разбавляли водой (10 мл), добавляли разбавленную хлористоводородную кислоту (2 M, водный раствор) для доведения pH до 3 и экстрагировали этилацетатом (10 мл × 3). Органическую фазу промывали насыщенным солевым раствором (10 мл × 1), высушивали над безводным сульфатом натрия, фильтровали и фильтрат выпаривали до сухого состояния путем ротационного выпаривания. Получали соединение BB-7-6. 1H ЯМР (400 MГц, DMSO-d6) δ: 12,59 (br s, 1H), 8,24 (d, J = 8,38 Гц, 1H), 7,96 (d, J = 9,26 Гц, 1H), 7,90 (d, J = 7,50 Гц, 1H), 7,54 (dd, J = 2,8 Гц, 1H), 7,53 (s, 1H), 7,41 (s, 1H), 4,08 (s, 2H).
[000176] Стадия 6: синтез соединения BB-7
[000177] Соединение BB-7-6 (1,40 г, 4,59 ммоль) растворяли в безводном этаноле (14 мл) при 25°C, медленно по каплям добавляли серную кислоту (413,28 мг, 4,13 ммоль, 224,61 мкл, концентрация: 98%), затем нагревали до 80°C и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, разбавляли этилацетатом (60 мл), добавляли насыщенный водный раствор бикарбоната натрия (60 мл) и экстрагировали этилацетатом (60 мл × 2). Органическую фазу промывали насыщенным солевым раствором (60 мл × 1), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Остаток суспендировали петролейным эфиром (5 мл) при комнатной температуре, перемешивали в течение 10 минут, а затем фильтровали. Осадок на фильтре промывали петролейным эфиром (5 мл × 2), собирали и выпаривали до сухого состояния путем ротационного выпаривания. Получали соединение BB-7. 1H ЯМР (400 МГц, CDCl3) δ: 8,22 (t, J = 8,41 Гц, 2H), 7,78-7,85 (m, 2H), 7,75 (d, J = 9,29 Гц, 1H), 7,41 (t, J = 7,91 Гц, 1H), 4,23 (q, J = 7,03 Гц, 2H), 4,06 (s, 2H), 1,26 (t, J = 7,15 Гц, 3H).
[000178] Сравнительный пример 8
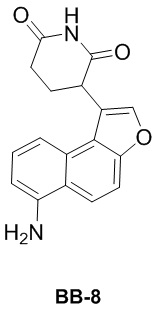
[000179] Путь синтеза:
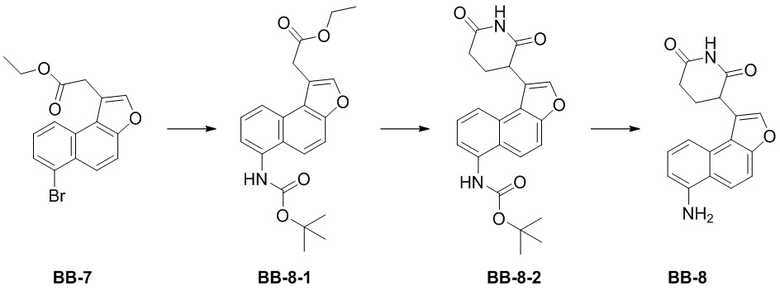
[000180] Стадия 1: синтез соединения 8-1
[000181] Соединение BB-7 (5 г, 15,01 ммоль) растворяли в смешанном растворителе из толуола (50 мл) и воды (10 мл), а затем добавляли трет-бутилкарбамат (2,64 г, 22,51 ммоль), фосфат калия (12,74 г, 60,03 ммоль), трис(дибензилиденацетон)дипалладий (961,96 мг, 1,05 ммоль) и 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (892,16 мг, 2,10 ммоль). Реакционную смесь трижды продували азотом, нагревали до 100°C и перемешивали в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и осадок на фильтре промывали этилацетатом (30 мл × 3). Фильтрат промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. К неочищенному продукту добавляли метил-трет-бутиловый эфир (50 мл), перемешивали в течение 10 минут и фильтровали. Осадок на фильтре ополаскивали метил-трет-бутиловым эфиром (10 мл × 2) и собирали с получением соединения BB-8-1.
[000182] Стадия 2: синтез соединения BB-8-2
[000183] Соединение BB-8-1 (4,2 г, 11,37 ммоль) и акриламид (888,93 мг, 12,51 ммоль, 863,04 мкл) растворяли в N,N-диметилформамиде (40 мл), охлаждали до 0°C в атмосфере азота, добавляли по каплям раствор трет-бутоксида калия (2,55 г, 22,74 ммоль) в N,N-диметилформамиде (10 мл), нагревали до 20°C и перемешивали в течение 2 часов. Реакционную смесь выливали в 0,2 н. разбавленную хлористоводородную кислоту (200 мл) и экстрагировали этилацетатом (100 мл × 3). Органическую фазу промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. К неочищенному продукту добавляли дихлорметан (20 мл), перемешивали в течение 10 минут и фильтровали. Осадок на фильтре ополаскивали дихлорметаном (10 мл) и собирали с получением соединения BB-8-2. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,94 (s, 1H), 9,28 (s, 1H), 8,04-7,96 (m, 3H), 7,79 (d, J = 9,6 Гц, 1H), 7,55-7,51 (m, 2H), 4,67 (dd, J = 4,4 Гц, 12,0 Гц, 1H), 2,93-2,84 (m, 1H), 2,66-2,60 (m, 1H), 2,45-2,36 (m, 1H), 2,33-2,26 (m, 1H), 1,49 (s, 9H).
[000184] Стадия 3: синтез гидрохлорида соединения BB-8
[000185] Соединение BB-8-2 (1 г, 2,54 ммоль) растворяли в этилацетате (10 мл), по каплям добавляли смесь хлористоводородная кислота/этилацетат (4 M, 50,00 мл) и перемешивали при 20°C в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении с получением гидрохлорида соединения BB-8.
[000186] Сравнительный пример 9
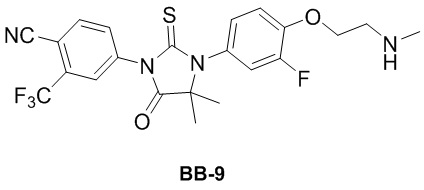
[000187] Путь синтеза:
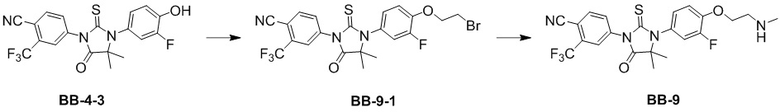
[000188] Стадия 1: синтез соединения BB-9-1
[000189] Соединение BB-4-3 (13 г, 30,71 ммоль) и карбонат калия (8,49 г, 61,41 ммоль) растворяли в N,N-диметилформамиде (300 мл), добавляли 1,2-дибромэтан (28,84 г, 153,53 ммоль, 11,58 мл), нагревали до 80°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, к ней добавляли воду (1 л) и экстрагировали этилацетатом (300 мл × 3). Органическую фазу промывали насыщенным солевым раствором (300 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = 4/1) с получением соединения BB-9-1. 1H ЯМР (400 МГц, CDCl3) δ: 8,00-7,96 (m, 2H), 7,85-7,83 (m, 1H), 7,14-7,04 (m, 3H), 4,44 (t, J = 6,0 Гц, 2H), 3,71 (t, J = 6,4 Гц, 2H), 1,60 (s, 6H).
[000190] Стадия 2: синтез соединения BB-9
[000191] Соединение BB-9-1 (3 г, 5,66 ммоль) и гидрохлорид метиламина (1,91 г, 28,28 ммоль) помещали в N,N-диметилформамид (30 мл), добавляли карбонат калия (11,73 г, 84,85 ммоль), нагревали до 50°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (70 мл) и экстрагировали этилацетатом (50 мл × 4). Органическую фазу промывали насыщенным солевым раствором (70 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт разделяли колоночной хроматографией (элюент: дихлорметан/метанол = от 50/1 до 10/1) с получением соединения BB-9.
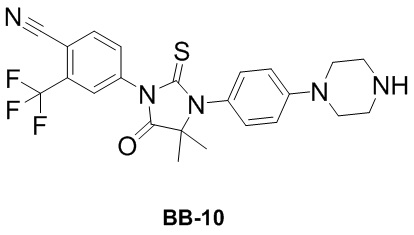
[000192] Сравнительный пример 10: фрагмент BB-10
[000193] Путь синтеза:
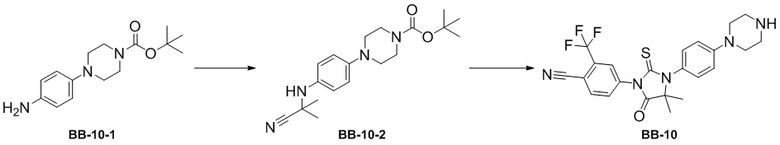
[000194] Стадия 1: синтез соединения BB-10-2
[000195] Соединение BB-10-1 (5 г, 18,03 ммоль) растворяли в дихлорметане (50 мл) и ацетоне (25 мл) при 0°C в атмосфере азота, затем медленно последовательно по каплям добавляли цианотриметилсилан (2,68 г, 27,04 ммоль, 3,38 мл) и триметилсилилтрифторметансульфонат (400,67 мг, 1,80 ммоль, 325,74 мкл) и реакционную смесь перемешивали при 20°C в течение 2 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 4/1, об./об.) с получением соединения BB-10-2.
[000196] Стадия 2: синтез соединения BB-10
[000197] Соединение BB-10-2 (10 г, 29,03 ммоль) растворяли в N,N-диметилацетамиде (100 мл) при комнатной температуре в атмосфере азота. К реакционной смеси порциями добавляли 4-изотиоцианато-2-(трифторметил)бензoнитрил (6,62 г, 29,03 ммоль). Реакционную смесь перемешивали при 20°C в течение 3 часов, затем добавляли метанол (100 мл) и разбавленную хлористоводородную кислоту (2 M, 56,32 мл) и перемешивали при 70°C в течение 2 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: дихлорметан/метанол = от 20/1 до 10/1, об./об.) с получением соединения BB-10. 1H ЯМР (400 MГц, CDCl3) δ: 7,99 (d, J = 8,8 Гц, 1H), 7,96 (d, J = 1,6 Гц, 1H), 7,84 (dd, J = 2,0, 8,4 Гц, 1H), 7,22 (d, J = 9,2 Гц, 2H), 7,04 (d, J = 8,8 Гц, 2H), 3,64-3,57 (m, 4H), 3,45-3,38 (m, 4 H), 1,58 (s, 6H).
[000198] Сравнительный пример 11: фрагмент BB-11
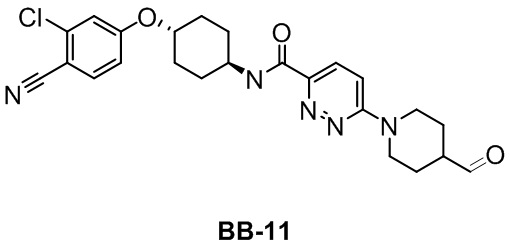
[000199] Путь синтеза:
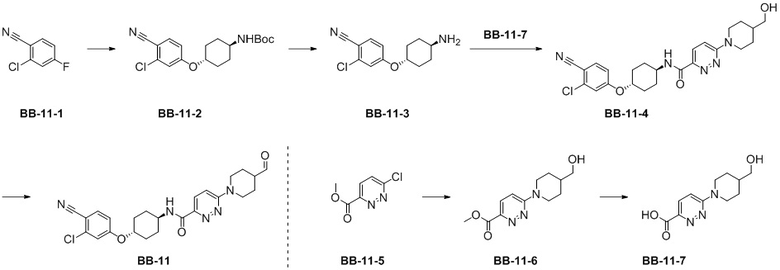
[000200] Стадия 1: синтез соединения BB-11-2
[000201] Соединение BB-11-1 (0,87 г, 5,57 ммоль) и соединение трет-бутил-[(1R,4R)-4-гидроксициклогексил]карбамат (1,00 г, 4,64 ммоль) растворяли в N,N-диметилформамиде (20 мл) при 0°C в атмосфере азота, затем добавляли гидрид натрия (278,68 мг, 6,97 ммоль, 60%), перемешивали и обеспечивали прохождение реакции при 0°C в течение 2 часов. После завершения реакции к реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, последовательно промывали водой (50 мл × 3) и насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 6/1, об./об.) с получением соединения BB-11-2. 1H ЯМР (400 МГц, CDCl3) δ: 8,00 (s, 1H), 7,56-7,47 (m, 1H), 6,97-6,92 (m, 1H), 6,84-6,72 (m, 1H), 4,29-4,17 (m, 1H), 4,13-4,08 (m, 1H), 2,14-2,03 (m, 4H), 1,47-1,38 (m, 9H), 1,26-1,20 (m, 4H).
[000202] Стадия 2: синтез гидрохлорида соединения BB-11-3
[000203] Соединение BB-11-2 (2,70 г, 7,70 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, затем добавляли 4 M раствор хлороводорода в метаноле (25 мл) и реакционную смесь перемешивали и обеспечивали ее реакцию при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении для удаления растворителя с получением гидрохлорида соединения BB-11-3, который непосредственно использовали на следующей стадии реакции.
[000204] Стадия 3: синтез соединения BB-11-6
[000205] Соединение BB-11-5 (10,00 г, 57,95 ммоль) и 4-пиперидинметанол (6,67 г, 57,95 ммоль) растворяли в диметилсульфоксиде (100 мл) при комнатной температуре, а затем добавляли триэтиламин (11,73 г, 115,90 ммоль, 16,13 мл). Реакционную смесь нагревали до 90°C, перемешивали и обеспечивали ее реакцию в течение 12 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, добавляли воду (200 мл) и экстрагировали дихлорметаном (100 мл × 4). Органические фазы объединяли, последовательно промывали водой (100 мл × 2) и насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: дихлорметан/метанол = от 1/0 до 20/1, об./об.) с получением соединения BB-11-6. 1H ЯМР (400 MГц, DMSO-d6) δ: 7,79 (d, J = 9,2 Гц, 1H), 7,27 (d, J = 9,6 Гц, 1H), 4,57-4,53 (m, 1H), 4,53-4,48 (m, 2H), 3,86 (s, 3H), 3,30-3,24 (m, 2H), 2,99 (td, J = 1,9, 12,7 Гц, 2H), 1,81-1,66 (m, 3H), 1,21-1,06 (m, 2H).
[000206] Стадия 4: синтез соединения BB-11-7
[000207] Соединение BB-11-6 (2,00 г, 7,96 ммоль) растворяли в тетрагидрофуране (15 мл) при комнатной температуре, затем добавляли 2 M водный раствор гидроксида натрия (15 мл) и реакционную смесь перемешивали и обеспечивали ее реакцию при комнатной температуре в течение 12 часов. После завершения реакции к реакционной смеси добавляли 4 M хлористоводородную кислоту для доведения pH до 4-5, концентрировали при пониженном давлении для удаления растворителя с получением неочищенного продукта соединения BB-11-7, который непосредственно применяли на следующей стадии реакции.
[000208] Стадия 5: синтез соединения BB-11-4
[000209] Гидрохлорид соединения BB-11-3 (7,65 ммоль) и соединения BB-11-7 (7,65 ммоль, неочищенный продукт) растворяли в N,N-диметилформамиде (46 мл) при комнатной температуре в атмосфере азота, затем добавляли диизопропилэтиламин (1,03 г, 7,96 ммоль, 1,39 мл) и гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (3,03 г, 7,96 ммоль) и реакционную смесь перемешивали и обеспечивали ее реакцию при комнатной температуре в течение 12 часов. После завершения реакции к реакционной смеси добавляли воду (100 мл) и экстрагировали этилацетатом (70 мл × 3). Органические фазы объединяли, последовательно промывали водой (100 мл × 2) и насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: дихлорметан/метанол = от 1/0 до 20/1, об./об.) с получением соединения BB-11-4. MS-ESI масса/заряд: 470,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 8,58 (d, J = 8,4 Гц, 1H), 7,85 (d, J = 8,4 Гц, 1H), 7,79 (d, J = 9,6 Гц, 1H), 7,38 (d, J = 2,4 Гц, 1H), 7,33 (d, J = 9,6 Гц, 1H), 7,13 (dd, J = 2,4, 8,8 Гц, 1H), 4,60-4,44 (m, 4H), 3,92-3,79 (m, 1H), 3,32-3,27 (m, 2H), 3,07-2,95 (m, 2H), 2,18-2,05 (m, 2H), 1,95-1,84 (m, 2H), 1,81-1,68 (m, 3H), 1,68-1,51 (m, 2H), 1,51-1,48 (m, 2H), 1,20-1,06 (m, 2H).
[000210] Стадия 6: синтез соединения BB-11
[000211] Соединение BB-11-4 (100,02 мг, 205,06 мкмоль) растворяли в дихлорметане (6 мл) при комнатной температуре, затем добавляли периодинан Десс-Мартина (130,46 мг, 307,59 мкмоль) и реакционную смесь перемешивали и обеспечивали ее реакцию при комнатной температуре в течение 1 часа. После завершения реакции к реакционной смеси добавляли воду (20 мл) и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 0/1, об./об.) с получением соединения BB-11. MS-ESI масса/заряд: 468,2 [M+H]+.
[000212] Сравнительный пример 12: фрагмент BB-12
[000213] Путь синтеза:
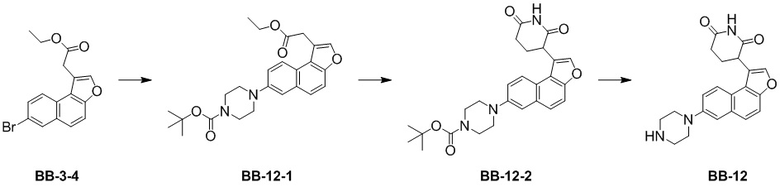
[000214] Стадия 1: синтез соединения BB-12-1
[000215] К толуолу (150 мл) и воде (20 мл) добавляли соединение BB-3-4 (10,00 г, 30,01 ммоль) и 1-(трет-бутоксикарбонил)пиперазин (8,39 г, 45,02 ммоль) при 25°C. К реакционной смеси медленно добавляли трис(дибензилиденацетон)дипалладий (1,92 г, 2,10 ммоль), фосфат калия (19,11 г, 90,04 ммоль) и 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (1,78 г, 4,20 ммоль). Смесь трижды продували азотом, затем нагревали до 100°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали до 25°С, фильтровали через диатомит и осадок на фильтре промывали этилацетатом (20 мл). К фильтрату добавляли этилацетат (300 мл) и воду (300 мл) и водную фазу экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат =от 50/1 до 5/1) с получением соединения BB-12-1.
[000216] MS-ESI масса/заряд: 439,0 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,15 (d, J = 9,0 Гц, 1H), 7,74 (s, 1H), 7,60 (s, 2H), 7,36 (dd, J = 2,5, 9,0 Гц, 1H), 7,31 (d, J = 2,5 Гц, 1H), 4,23 (q, J = 7,1 Гц, 2H), 4,04 (s, 2H), 3,71-3,62 (m, 4H), 3,29-3,20 (m, 4H), 1,53 (s, 9H), 1,28 (dt, J = 7,2 Гц, 3H).
[000217] Стадия 2: синтез соединения BB-12-2
[000218] К N,N-диметилформамиду (80 мл) добавляли соединение BB-12-1 (8,6 г, 15,86 ммоль) и акриламид (1,13 г, 15,86 ммоль, 1,09 мл) при 0°C и начинали перемешивание. К реакционной смеси добавляли порциями трет-бутоксид калия (2,67 г, 23,80 ммоль). Реакционную смесь трижды продували азотом, а затем перемешивали при 0°C в течение 1 часа. После завершения реакции к реакционной смеси медленно по каплям добавляли насыщенный водный раствор хлорида аммония (480 мл). Большое количество твердого вещества осаждали и фильтровали. Осадок на фильтре дважды промывали водой (20 мл × 2), собирали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. К полученному в результате неочищенному продукту добавляли метанол (10 мл), перемешивали в течение 30 минут и фильтровали. Осадок на фильтре промывали метанолом (5 мл), собирали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении с получением соединения BB-12-2. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,96 (br dd, J = 2,9, 6,1 Гц, 1H), 8,02 (br d, J = 8,8 Гц, 1H), 7,97-7,91 (m, 1H), 7,72-7,65 (m, 2H), 7,44-7,37 (m, 2H), 4,62 (br dd, J = 4,3, 11,8 Гц, 1H), 3,53 (br s, 4H), 3,22-3,20 (m, 4H), 2,93-2,83 (m, 1H), 2,68-2,57 (m, 1H), 2,39 (dq, J = 4,3, 12,5 Гц, 1H), 2,26 (br dd, J = 3,9, 8,7 Гц, 1H), 1,44 (s, 9H).
[000219] Стадия 3: синтез гидрохлорида соединения BB-12
[000220] К этилацетату (20 мл) добавляли соединение BB-12-2 (3,4 г, 7,34 ммоль) и начинали перемешивание. К полученному медленно добавляли раствор хлористоводородная кислота/этилацетат (4 M, 30 мл). Реакционную смесь трижды продували азотом и перемешивали при 25°C в течение 12 часов. После завершения реакции реакционную смесь фильтровали. Осадок на фильтре промывали этилацетатом (5 мл × 2), собирали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении с получением гидрохлорида соединения BB-12. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,96 (s, 1H), 9,65 (br s, 2H), 8,07 (br d, J = 9,3 Гц, 1H), 7,96 (s, 1H), 7,75-7,69 (m, 2H), 7,53 (d, J = 2,3 Гц, 2H), 7,45 (dd, J = 2,4, 9,2 Гц, 1H), 4,64 (br dd, J = 4,4, 11,9 Гц, 1H), 3,54 (br d, J = 5,0 Гц, 4H), 3,28 (br s, 4H), 2,94-2,82 (m, 1H), 2,67-2,58 (m, 1H), 2,39 (br dd, J = 4,0, 12,5 Гц, 1H), 2,30-2,17 (m, 1H).
[000221] Сравнительный пример 13: фрагмент BB-13
[000222] Путь синтеза:
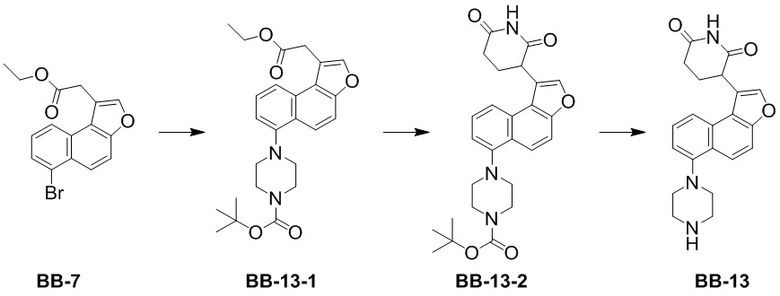
[000223] Стадия 1: синтез соединения BB-13-1
[000224] К диоксану (20 мл) и воде (4 мл) добавляли соединение BB-7 (1,4 г, 4,20 ммоль) и 1-трет-бутоксикарбонилпиперазин (1,17 г, 6,30 ммоль) при 25°C. К реакционной смеси медленно добавляли трис(дибензилиденацетон)дипалладий (269,35 мг, 294,14 мкмоль), фосфат калия (2,68 г, 12,61 ммоль) и 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (249,81 мг, 588,28 мкмоль). Смесь трижды продували азотом, затем нагревали до 100°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали до 25°С, фильтровали через диатомит и осадок на фильтре промывали этилацетатом (20 мл). К фильтрату добавляли этилацетат (100 мл) и воду (100 мл) и водную фазу экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат =от 50/1 до 10/1) с получением соединения BB-13-1. MS: ESI, масса/заряд: 439,0 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 8,17 (d, J = 9,3 Гц, 1H), 8,04 (s, 1H), 7,86 (d, J = 8,3 Гц, 1H), 7,77 (d, J = 9,3 Гц, 1H), 7,53 (t, J = 8,0 Гц, 1H), 7,19 (d, J = 7,3 Гц, 1H), 4,20-4,09 (m, 4H), 3,36-2,59 (m, 8H), 1,45 (s, 9H), 1,20 (t, J = 7,0 Гц, 3H).
[000225] Стадия 2: синтез соединения BB-13-2
[000226] К N,N-диметилформамиду (20 мл) добавляли соединение BB-13-1 (1,5 г, 3,26 ммоль) и акриламид (231,68 мг, 3,26 ммоль, 224,93 мкл) при 0°C и начинали перемешивание. К реакционной смеси медленно порциями добавляли трет-бутоксид калия (438,91 мг, 3,91 ммоль). Реакционную смесь трижды продували азотом, а затем перемешивали при 0°C в течение 1 часа. К насыщенному водному раствору хлорида аммония (75 мл) медленно по каплям добавляли смесь. Твердое вещество осаждали и фильтровали. Осадок на фильтре промывали водой (5 мл × 2), а собранное твердое вещество выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. К неочищенному продукту добавляли метанол (5 мл), перемешивали в течение 15 минут и фильтровали. Осадок на фильтре промывали метанолом (3 мл × 2), а собранное твердое вещество выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Получали соединение BB-13-2. MS-ESI масса/заряд: 464,0 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 8,19 (d, J = 9,3 Гц, 1H), 7,97 (d, J = 16,8 Гц, 2H), 7,78 (d, J = 9,0 Гц, 1H), 7,51 (br t, J = 8,0 Гц, 1H), 7,18 (br d, J = 7,5 Гц, 1H), 4,66 (br d, J = 7,8 Гц, 1H), 3,34 (m, 4H), 3,17 (m, 4H), 2,89-2,81 (m, 1H), 2,68-2,55 (m, 1H), 2,40 (dq, J = 3,8, 12,4 Гц, 1H), 2,26 (br s, 1H), 1,44 (s, 9H).
[000227] Стадия 3: синтез гидрохлорида соединения BB-13
[000228] К этилацетату (6 мл) добавляли соединение BB-13-2 (0,6 г, 1,16 ммоль) и начинали перемешивание. К реакционной смеси медленно добавляли смесь хлористоводородная кислота/этилацетат (4 M, 10 мл). Реакционную смесь трижды продували азотом и перемешивали при 25°C в течение 12 часов. Смесь выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении с получением гидрохлорида соединения BB-13. MS-ESI масса/заряд: 364,0 [M+H]+.
[000229] Пример 1
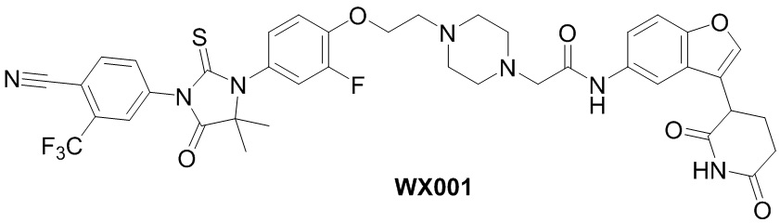
[000230] Путь синтеза:
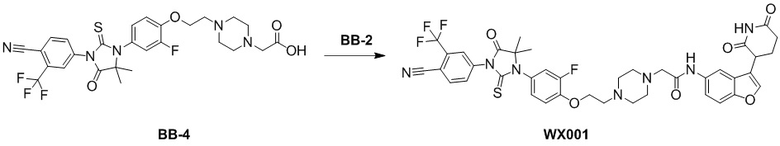
[000231] Соединение BB-4 (0,3 г, 505,40 мкмоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре в атмосфере азота, затем добавляли BB-2 (123,44 мг, 505,40 мкмоль, гидрохлорид), триэтиламин (255,70 мг, 2,53 ммоль, 351,72 мкл) и гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (288,25 мг, 758,10 мкмоль) и реакционную смесь перемешивали при 20°C в течение 2 часов. После завершения реакции к реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный в результате остаток разделяли посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% хлористоводородная кислота) и лиофилизировали с получением целевого соединения WX001. MS-ESI масса/заряд: 820,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,94 (s, 1H), 10,61 (s, 1H), 8,40 (d, J = 8,0 Гц, 1H), 8,29 (d, J = 1,6 Гц, 1H), 8,08 (m, 1H), 7,93-7,92 (m, 2H), 7,57 (d, J = 8,8 Гц, 1H), 7,49-7,43 (m, 3H), 7,40 (d, J = 2,4 Гц, 1H), 4,59 (s, 2H), 4,15-4,11 (m, 1H), 3,62-3,54 (m, 6H), 2,90-2,51 (m, 8H), 2,25-2,15 (m, 2H), 1,52 (s, 6H).
[000232] Пример 2
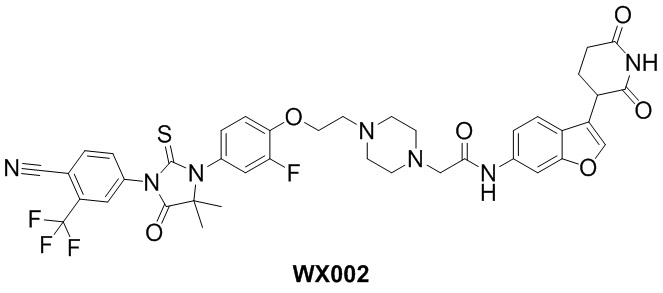
[000233] Путь синтеза:
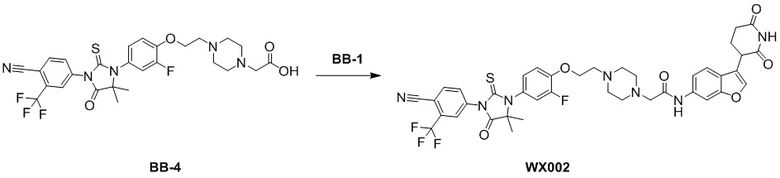
[000234] К N,N-диметилформамиду (2 мл) добавляли соединение BB-4 (100 мг, 168,47 мкмоль) и гидрохлорид промежуточного соединения BB-1 (47,29 мг, 168,47 мкмоль). К реакционной смеси медленно добавляли гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (96,08 мг, 252,70 мкмоль) и триэтиламин (34,09 мг, 336,93 мкмоль, 46,90 мкл). Смесь трижды продували азотом и перемешивали при 25°C в течение 12 часов. Реакционную смесь выливали в воду (20 мл) и этилацетат (30 мл) и отделенную органическую фазу дважды промывали водой (20 мл × 2). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный в результате остаток разделяли посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% хлористоводородная кислота) с получением соединения WX002. MS-ESI масса/заряд: 820,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,90 (s, 1H), 10,47 (br s, 1H), 8,40 (d, J = 8,3 Гц, 1H), 8,28 (d, J = 1,5 Гц, 1H), 8,13-7,97 (m, 2H), 7,87 (s, 1H), 7,54 (d, J = 8,5 Гц, 1H), 7,47-7,29 (m, 3H), 7,26-7,13 (m, 1H), 4,54 (br s, 2H), 4,12 (dd, J = 4,8, 11,8 Гц, 1H), 3,67-3,45 (m, 2H), 2,83-2,67 (m, 2H), 2,63-2,43 (m, 10H), 2,39-2,25 (m, 1H), 2,19-2,03 (m, 1H), 1,52 (s, 6H).
[000235] Пример 3
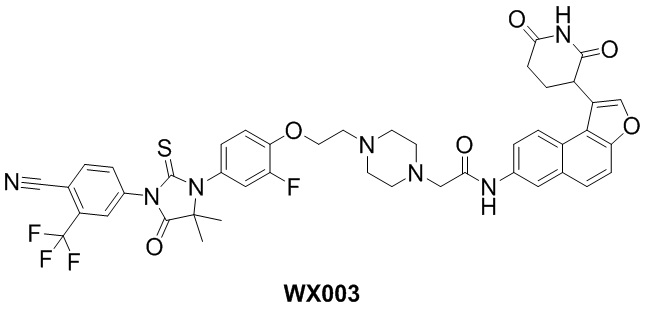
[000236] Путь синтеза:
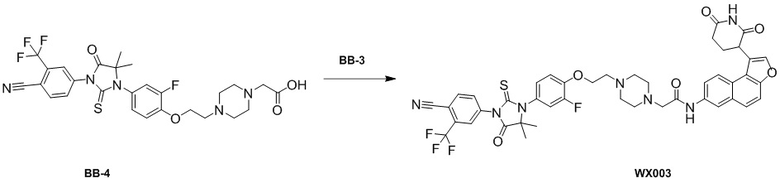
[000237] Гидрохлорид BB-3 (378 мг, 1,08 ммоль) и BB-4 (767,86 мг, 1,29 ммоль) растворяли в растворителе N,N-диметилформамиде (5 мл) при комнатной температуре в атмосфере азота, затем добавляли гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (614,82 мг, 1,62 ммоль) и триэтиламин (272,70 мг, 2,69 ммоль) соответственно и реакционную смесь перемешивали при 25°C в течение 3 часов. После завершения реакции к реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный в результате остаток разделяли посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% хлористоводородная кислота) с получением целевого соединения WX003. MS-ESI масса/заряд: 870,1 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,96 (s, 1H), 10,73 (s, 1H), 8,41-8,40 (m, 2H), 8,39 (d, J = 1,6 Гц, 1H), 8,08 (d, J = 8,8 Гц, 1H), 8,07 (dd, J = 1,6, 8,4 Гц, 1H), 8,01 (s, 1H), 7,78 (s, 3H), 7,75 (d, J = 2,0 Гц, 1H), 7,46-7,37 (m, 2H), 7,23 (d, J = 8,8 Гц, 1H), 4,68 (dd, J = 4,0, 11,2 Гц, 1H), 4,58 (s, 2H), 3,98 (m, 1H), 3,47 (s, 9H), 2,95-2,58 (m, 3H), 2,49-2,21 (m, 3H), 1,52 (s, 6H).
[000238] Пример 4
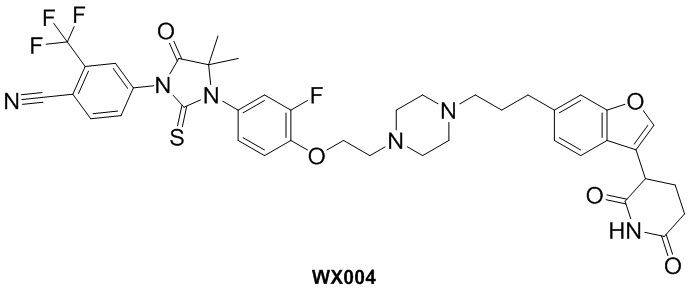
[000239] Путь синтеза:
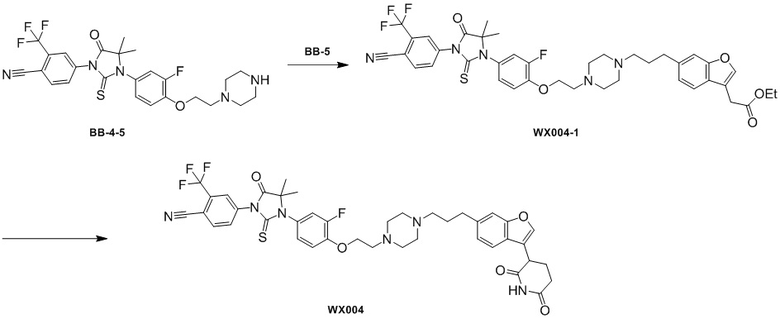
[000240] Стадия 1: синтез WX004-1
[000241] К дихлорметану (10 мл) добавляли соединение BB-5 (102,38 мг, 393,35 мкмоль) и BB-4-5 (150 мг, 262,23 мкмоль, 145,82 мкл, гидрохлорид). К смеси медленно добавляли триэтиламин (53,07 мг, 524,46 мкмоль, 73,00 мкл), перемешивали при 25°C в течение 0,5 часа, медленно добавляли триацетоксиборгидрид натрия (138,94 мг, 655,58 мкмоль), трижды продували азотом, а затем перемешивали при 25°C в течение 11,5 часа. К реакционной смеси медленно добавляли воду (50 мл), трижды экстрагировали дихлорметаном (50 мл × 3) и объединенные органические фазы выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный остаток очищали колоночной хроматографией (элюент: дихлорметан/метанол = от 1/0 до 5,7/1, об./об.) с получением соединения WX004-1. MS-ESI масса/заряд: 780,1 [M+H]+.
[000242] Стадия 2: синтез WX004
[000243] К N,N-диметилформамиду (5 мл) добавляли соединение WX004-1 (280 мг, 309,14 мкмоль) и акриламид (26,37 мг, 370,97 мкмоль, 25,60 мкл) при 0°C. К реакционной смеси медленно порциями добавляли трет-бутоксид калия (173,45 мг, 1,55 ммоль). Смесь трижды продували азотом, а затем перемешивали при 0°C в течение 1 часа. К реакционной смеси медленно добавляли насыщенный раствор хлорида аммония, дважды экстрагировали этилацетатом (20 мл × 2) и объединенные органические фазы дважды промывали насыщенным солевым раствором (20 мл × 2). Собранные органические фазы высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный в результате остаток разделяли посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением целевого соединения WX004. MS-ESI масса/заряд: 805,4 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,89 (s, 1H), 8,40 (d, J = 8,3 Гц, 1H), 8,28 (d, J = 1,0 Гц, 1H), 8,07 (dd, J = 1,4, 8,2 Гц, 1H), 7,85 (d, J = 6,8 Гц, 1H), 7,53 (dd, J = 4,0, 7,8 Гц, 1H), 7,49-7,32 (m, 3H), 7,21 (br d, J = 8,8 Гц, 1H), 7,15 (dd, J = 5,5, 7,3 Гц, 1H), 4,53 (br d, J = 1,0 Гц, 2H), 4,12 (dd, J = 4,9, 11,9 Гц, 1H), 3,83-3,62 (m, 5H), 3,27-3,06 (m, 2H), 2,84-2,65 (m, 3H), 2,57 (br dd, J = 4,1, 17,2 Гц, 4H), 2,49-2,42 (m, 2H), 2,40-2,26 (m, 1H), 2,19-2,01 (m, 3H), 1,52 (s, 6H).
[000244] Пример 5
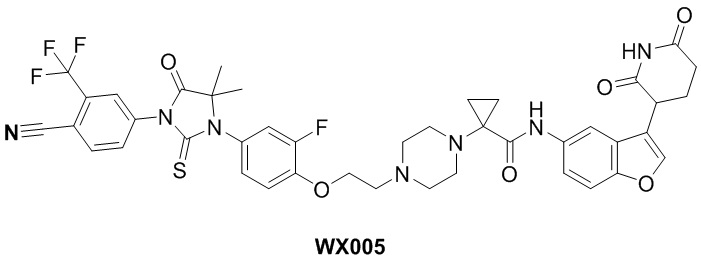
[000245] Путь синтеза:
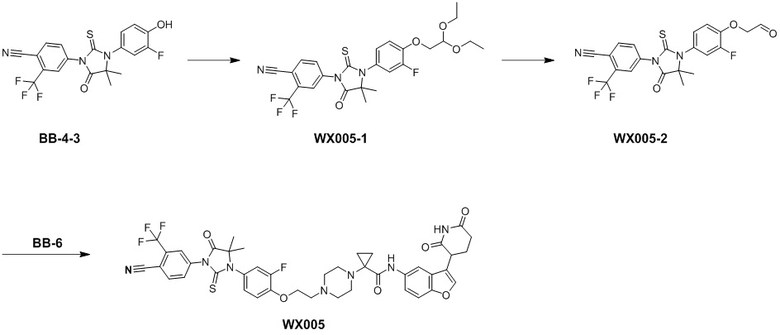
[000246] Стадия 1: синтез промежуточного соединения WX005-1
[000247] Соединение BB-4-3 (1,00 г, 2,36 ммоль) и бромацетальдегид диэтилацеталь (512,01 мг, 2,60 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре, затем к полученному добавляли карбонат калия (391,73 мг, 2,83 ммоль) и реакционную смесь перемешивали и обеспечивали ее реакцию при 80°C в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры, добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным солевым раствором (50 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный осадок разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 3/1, об./об.) с получением промежуточного соединения WX005-1. 1H ЯМР (400 МГц, CDCl3) δ: 8,01-7,94 (m, 2H), 7,84 (dd, J = 2,0, 8,4 Гц, 1H), 7,16-7,10 (m, 1H), 7,09-7,00 (m, 2H), 4,88 (t, J = 5,2 Гц, 1H), 4,13 (d, J = 5,2 Гц, 2H), 3,86-3,76 (m, 2H), 3,72-3,62 (m, 2H), 1,59 (s, 6H), 1,27 (t, J = 7,0 Гц, 6H).
[000248] Стадия 2: синтез промежуточного соединения WX005-2
[000249] Соединение WX005-1 (500,00 мг, 926,71 мкмоль) растворяли в этаноле (20 мл) при комнатной температуре, затем к полученному добавляли уксусную кислоту (5,25 г, 87,42 ммоль) и хлористоводородную кислоту (1 M, 2,5 мл) и реакционную смесь перемешивали и обеспечивали ее реакцию при 80°C в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении для удаления растворителя (этанола), добавляли воду (30 мл) и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным солевым раствором (30 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении для удаления растворителя. Полученный осадок разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = от 1/0 до 1/1, об./об.) с получением промежуточного соединения WX005-2. 1H ЯМР (400 МГц, CDCl3) δ: 9,92 (s, 1H), 8,02-7,92 (m, 2H), 7,84 (dd, J = 2,0, 8,0 Гц, 1H), 7,19-7,10 (m, 1H), 7,10-7,01 (m, 2H), 4,74 (s, 2H), 1,60 (s, 6H).
[000250] Стадия 3: синтез соединения WX005
[000251] К дихлорметану (10 мл) добавляли соединение WX005-2 (40 мг, 85,94 мкмоль) при 25°C и начинали перемешивание. К полученному добавляли соединение BB-6 (36,80 мг, 77,35 мкмоль) и триацетоксиборгидрид натрия (54,65 мг, 257,83 мкмоль). Реакционную смесь перемешивали в течение дополнительных 12 часов в атмосфере азота. После завершения реакции к реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали с помощью полунасыщенного солевого раствора (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Полученный в результате остаток разделяли посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением целевого соединения WX005. ESI масса/заряд: 846,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,89 (s, 2H), 9,77 (s, 1H), 8,39 (d, J = 8,4 Гц, 1H), 8,27 (d, J = 1,25 Гц, 1H), 8,07 (d, J = 8,4 Гц, 1 H), 7,96 (d, J = 1,2 Гц, 1H), 7,88 (s, 1H), 7,51-7,56 (m, 1H), 7,44-7,50 (m, 1H), 7,35-7,44 (m, 2H), 7,21-7,23 (m, 1H), 4,61 (s, 2H), 4,11 (dd, J = 12,0, 4,8 Гц, 1H), 3,5-3,60 (m, 4H), 3,50-3,51 (m, 2H), 2,93-3,02 (m, 2H), 2,80-2,90 (m, 2H), 2,70-2,79 (m, 1H), 2,55-2,68 (m, 1H), 2,22-2,35 (m, 1H), 2,06-2,19 (m, 1H), 1,51 (s, 6H), 1,14-1,21 (m, 2H), 1,08-1,10 (m, 2H).
[000252] Пример 6
[000253] Путь синтеза:
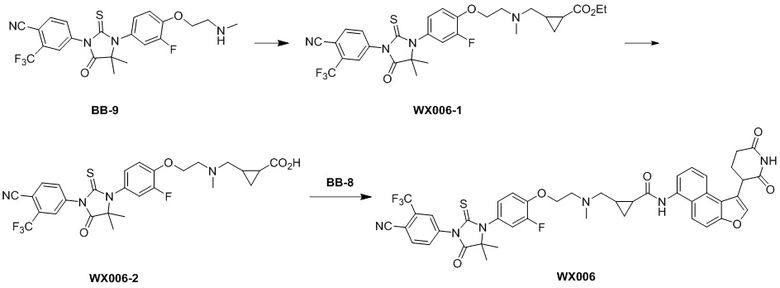
[000254] Стадия 1: синтез соединения WX006-1
[000255] Соединение BB-9 (71,00 мг, 499,50 мкмоль, 66,36 мкл) и соединение этил-2-формил-1-циклопропанкарбоксилат (0,2 г, 416,25 мкмоль) растворяли в смешанном растворителе дихлорметана (4 мл) и ледяной уксусной кислоты (0,1 мл), затем добавляли триацетоксиборгидрид натрия (132,33 мг, 624,38 мкмоль) при 20°C и перемешивали при 20°C в течение 15 часов. К реакционной смеси добавляли воду (2 мл), перемешивали в течение 10 минут, добавляли насыщенный водный раствор бикарбоната натрия (5 мл) и экстрагировали дихлорметаном (5 мл × 4). Органическую фазу промывали насыщенным солевым раствором (10 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Полученный остаток очищали препаративной тонкослойной хроматографией (проявляющий растворитель: дихлорметан/метанол = 10/1) с получением соединения WX006-1.
[000256] Стадия 2: синтез соединения WX006-2
[000257] Соединение WX006-1 (0,11 г, 181,33 мкмоль) растворяли в смешанном растворе тетрагидрофурана (2 мл) и воды (0,4 мл), добавляли моногидрат гидроксида лития (38,05 мг, 906,65 мкмоль) и перемешивали при 20°C в течение 15 часов. К реакционной смеси добавляли этилацетат (5 мл) и промывали 1 н. водным раствором гидроксида лития (2 мл × 5). К водной фазе добавляли 1 н. разбавленную хлористоводородную кислоту для доведения pH до 4-5 и экстрагировали дихлорметаном (5 мл × 3). Органическую фазу непосредственно высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания с получением соединения WX006-2.
[000258] Стадия 3: синтез соединения WX006
[000259] Соединение WX006-2 (90 мг, 155,55 мкмоль) растворяли в N,N-диметилформамиде (2 мл), добавляли гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (88,72 мг, 233,33 мкмоль) и N,N-диизопропилэтиламин (100,52 мг, 777,77 мкмоль, 135,47 мкл), перемешивали в течение 10 минут, затем добавляли гидрохлорид соединения BB-8 (54,94 мг, 186,66 мкмоль) и перемешивали при 20°C в течение 60 часов. Реакционную смесь очищали посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; нейтральная система) с получением соединения WX006. 1H ЯМР (400 MГц, CD3OD) δ: 8,15-8,13 (m, 2H), 8,06-8,05 (m, 1H), 7,98 (t, J = 10 Гц, 2H), 7,84 (s, 1H), 7,69 (d, J = 9,2 Гц, 1H), 7,58-7,55 (m, 2H), 7,29-7,19 (m, 2H), 7,10-7,08 (m, 1H), 4,65-4,61 (m, 1H), 4,33 (t, J = 5,2 Гц, 2H), 3,07 (t, J = 4,8 Гц, 2H), 2,92-2,84 (m, 1H), 2,77-2,70 (m, 2H), 2,61-2,58 (t, 1H), 2,55 (s, 3H), 2,46-2,42 (m, 2H), 1,94-1,91 (m, 1H), 1,67-1,65 (m, 1H), 1,52 (s, 6H), 1,35-1,31 (m, 1H), 0,95-0,90 (m, 1H).
[000260] Пример 7
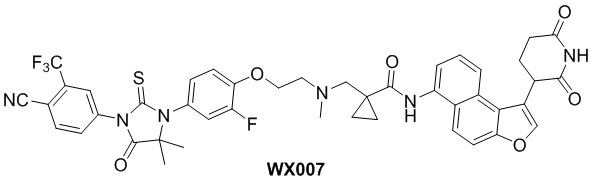
[000261] Путь синтеза:
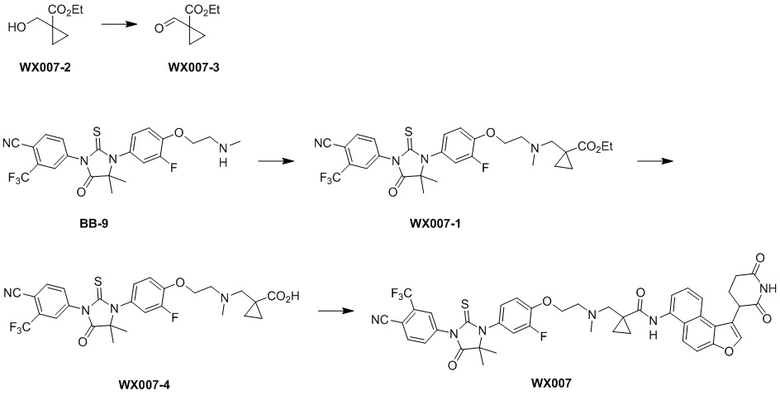
[000262] Стадия 1: синтез соединения WX007-1
[000263] Соединение WX007-2 (0,5 г, 3,47 ммоль) растворяли в дихлорметане (5 мл) и реакционную смесь охлаждали до 0°C, порциями добавляли периодинан Десс-Мартина (2,94 г, 6,94 ммоль, 2,15 мл), медленно нагревали до 20°C и перемешивали в течение 2 часов. Реакционную смесь выливали в насыщенный водный раствор бикарбоната натрия (10 мл) и 10% водный раствор тиосульфата натрия (30 мл) при 0-10°C и экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным солевым раствором (10 мл), высушивали над безводным сульфатом натрия и фильтровали с получением раствора соединения WX007-3. Раствор соединения WX007-3 и соединения BB-9 (1,4 г, 2,91 ммоль) растворяли в дихлорметане (5 мл) и ледяной уксусной кислоте (0,1 мл), затем добавляли триацетоксиборгидрид натрия (1,24 г, 5,83 ммоль) при 20°C и перемешивали в течение 12 часов. К реакционной смеси добавляли воду (50 мл) и экстрагировали дихлорметаном (30 мл × 3). Органическую фазу промывали насыщенным солевым раствором (20 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат =от 1/1 до 0/1) с получением соединения WX007-1. 1H ЯМР (400 МГц, CDCl3) δ: 8,00-7,96 (m, 2H), 7,85 (dd, J = 1,6 Гц, 8,4 Гц, 1H), 7,14-7,02 (m, 3H), 4,21-4,18 (m, 2H), 4,13 (q, J = 7,2 Гц, 2H), 2,98-2,90 (m, 2H), 2,78 (s, 2H), 2,41 (s, 3H), 1,59 (s, 6H), 1,29-1,23 (m, 5H), 0,88-0,85 (m, 2H).
[000264] Стадия 2: синтез соединения WX007-4
[000265] Соединение WX007-1 (0,53 г, 873,68 мкмоль) растворяли в тетрагидрофуране (5 мл) и воде (1 мл), затем добавляли моногидрат гидроксида лития (183,30 мг, 4,37 ммоль) и перемешивали при 20°C в течение 24 часов. Реакционную смесь нагревали до 40°C и перемешивали в течение 24 часов. К реакционной смеси добавляли 1 н. хлористоводородную кислоту для доведения pH до 6-7, выпаривали до сухого состояния путем ротационного выпаривания для удаления большей части тетрагидрофурана, добавляли метил-трет-бутиловый эфир (30 мл) и 10% водный раствор карбоната калия (20 мл × 3) и фазы разделяли. К водной фазе добавляли 1 н. хлористоводородную кислоту для доведения pH до 5-6 и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении с получением соединения WX007-4.
[000266] Стадия 3: синтез соединения WX007
[000267] Соединение WX007-4 (50 мг, 86,42 мкмоль) и N,N-диизопропилэтиламин (44,68 мг, 345,68 мкмоль, 60,21 мкл) растворяли в N,N-диметилформамиде (2 мл), добавляли гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (39,43 мг, 103,70 мкмоль) при 20°C, перемешивали в течение 5 минут, добавляли гидрохлорид соединения BB-8 (34,30 мг, 103,70 мкмоль) и перемешивали при 20°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя с получением неочищенного продукта, который очищали посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением соединения WX007. 1H ЯМР (400 MГц, CD3OD) δ: 8,17-8,15 (m, 3H), 7,98 (d, J = 8,4 Гц, 1H), 7,87-7,84 (m, 2H), 7,74 (d, J = 9,6 Гц, 1H), 7,62 (t, J = 7,6 Гц, 1H), 7,45 (d, J = 7,2 Гц, 1H), 7,28-7,25 (m, 2H), 7,12 (d, J = 8,8 Гц, 1H), 4,65 (dd, J = 5,6 Гц, 11,2 Гц, 1H), 4,52 (t, J = 4,4 Гц, 2H), 3,90 (dd, J = 4,0 Гц, 13,2 Гц, 1H), 3,83-3,79 (m, 1H), 3,65-3,61 (m, 1H), 3,32-3,29 (m, 1H), 3,08 (s, 3H), 2,94-2,85 (m, 1H), 2,77-2,72 (m, 1H), 2,53-2,43 (m, 2H), 1,92-1,84 (m, 2H), 1,53 (s, 6H), 1,48-1,45 (m, 1H), 1,39-1,35 (m, 1H).
[000268] Пример 8
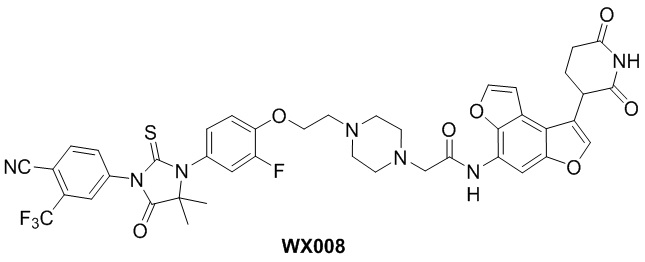
[000269] Путь синтеза:
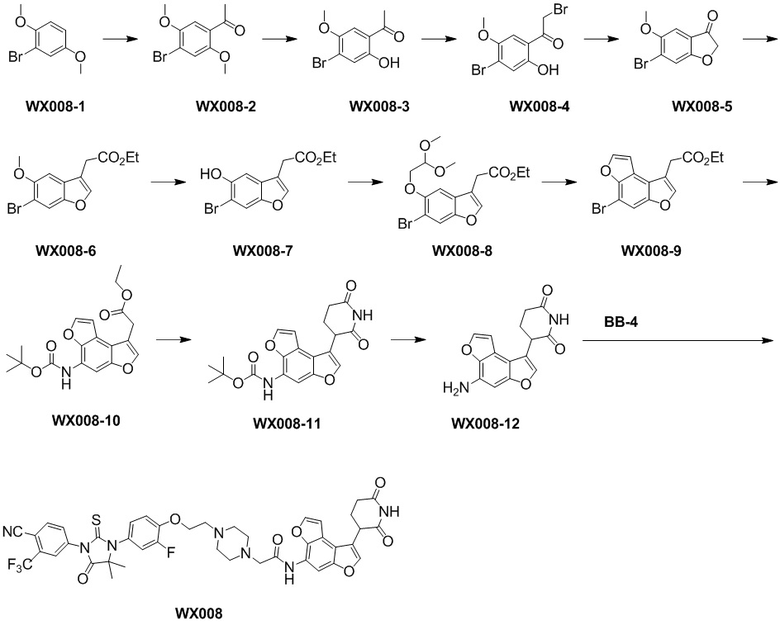
[000270] Стадия 1: синтез соединения WX008-2
[000271] Соединение WX008-1 (100 г, 460,70 ммоль) и ацетилхлорид (54,25 г, 691,05 ммоль, 49,31 мл) растворяли в дихлорметане (1000 мл), затем реакционную смесь замещали азотом, порциями добавляли хлорид алюминия (92,15 г, 691,05 ммоль, 37,76 мл) при 20°C в атмосфере азота и перемешивали в течение 2 часов. Реакционную смесь выливали в 6 н. ледяную хлористоводородную кислоту (400 мл) и экстрагировали дихлорметаном (700 мл × 3). Органическую фазу промывали насыщенным солевым раствором (700 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Полученный в результате неочищенный продукт суспендировали в метаноле (500 мл), перемешивали в течение 15 минут и фильтровали. Осадок на фильтре ополаскивали метанолом (100 мл × 2) и собирали с получением соединения WX008-2.
[000272] Стадия 2: синтез соединения WX008-3
[000273] Соединение WX008-2 (20 г, 77,19 ммоль) растворяли в дихлорметане (200 мл), охлаждали до -70°C, по каплям добавляли трибромид бора (17,40 г, 69,47 ммоль, 6,69 мл), медленно нагревали до 20°C и перемешивали в течение 1 часа. Реакционную смесь выливали в 4 н. разбавленную хлористоводородную кислоту (100 мл) и экстрагировали этилацетатом (100 мл × 3). Органическую фазу промывали насыщенным солевым раствором (100 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Полученный в результате остаток разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = 7/1) с получением соединения WX008-3.
[000274] Стадия 3: синтез соединения WX008-4
[000275] Соединение WX008-3 (13,84 г, 56,47 ммоль) растворяли в хлороформе (70 мл) и этилацетате (70 мл), добавляли бромид меди (25,23 г, 112,95 ммоль, 5,29 мл), нагревали до 90°C и перемешивали в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и осадок на фильтре промывали дихлорметаном (110 мл) с получением раствора соединения WX008-4 в дихлорметане.
[000276] Стадия 4: синтез соединения WX008-5
[000277] К раствору соединения WX008-4 в дихлорметане медленно по каплям добавляли триэтиламин (11,43 г, 112,97 ммоль, 15,72 мл) при 0°C. После завершения добавления по каплям реакционную смесь нагревали до 20°C и перемешивали в течение 2 часов. Реакционную смесь промывали насыщенным водным раствором хлорида аммония (100 мл × 3), затем органическую фазу высушивали над безводным сульфатом натрия и фильтровали с получением раствора соединения WX008-5 в дихлорметане.
[000278] Стадия 5: синтез соединения WX008-6
[000279] К раствору WX008-5 в дихлорметане добавляли толуол (150 мл) и (этоксикарбонилметилен)трифенилфосфоран (23,62 г, 67,79 ммоль) и нагревали до 130°C. Растворитель с низкой температурой кипения разделяли с помощью водоотделителя и реакционную смесь перемешивали в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и непосредственно концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = 10/1) с получением соединения WX008-6. 1H ЯМР (400 МГц, CDCl3) δ: 7,70 (s, 1H), 7,60 (s, 1H), 7,05 (s, 1H), 4,20 (q, J = 7,2 Гц, 2H), 3,95 (s, 3H), 3,67 (s, 2H), 1,28 (t, J = 7,2 Гц, 3H).
[000280] Стадия 6: синтез соединения WX008-7
[000281] Соединение WX008-6 (4 г, 12,77 ммоль) растворяли в дихлорметане (40 мл) и смесь охлаждали до -70°C, по каплям добавляли трибромид бора (3,36 г, 13,41 ммоль, 1,29 мл), перемешивали в течение 0,5 часа при той же температуре, медленно нагревали до 0°C и перемешивали в течение 1 часа. Реакционную смесь охлаждали до -70°C, добавляли дополнительное количество трибромида бора (3,36 г), медленно нагревали до 0°C и перемешивали в течение 1 часа. Реакционную смесь выливали в 1 н. хлористоводородную кислоту (50 мл) и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу промывали насыщенным солевым раствором (50 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = 5/1) с получением соединения WX008-7.
[000282] Стадия 7: синтез соединения WX008-8
[000283] Соединение WX008-7 (1,8 г, 6,02 ммоль) и карбонат калия (1,83 г, 13,24 ммоль) помещали в N,N-диметилформамид (10 мл), добавляли 2-бром-1,1-диметоксиэтан (1,22 г, 7,22 ммоль, 847,58 мкл), нагревали до 100°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (50 мл) и экстрагировали этилацетатом (30 мл × 3). Органическую фазу промывали насыщенным солевым раствором (30 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = 10/1) с получением соединения WX008-8.
[000284] Стадия 8: синтез соединения WX008-9
[000285] Соединение WX008-8 (1,84 г, 4,75 ммоль) помещали в толуол (20 мл), добавляли полифосфорную кислоту (1,84 г, 8,74 ммоль), нагревали до 100°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (30 мл) и экстрагировали этилацетатом (30 мл × 3). Органическую фазу промывали насыщенным водным раствором бикарбоната натрия (30 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = 10/1) с получением соединения WX008-9. 1H ЯМР (400 МГц, CDCl3) δ: 7,81 (d, J = 2,4 Гц, 1H), 7,71 (m, 1H), 7,66 (s, 1H), 7,13 (d, J = 2,0 Гц, 1H), 4,20 (q, J = 7,2 Гц, 2H), 3,85 (d, J = 0,8 Гц, 2H), 1,25 (t, J = 7,2 Гц, 3H).
[000286] Стадия 9: синтез соединения WX008-10
[000287] Соединение WX008-9 (700 мг, 2,17 ммоль), 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (128,78 мг, 303,28 мкмоль), трет-бутилкарбамат (304,53 мг, 2,60 ммоль), фосфат калия (1,84 г, 8,67 ммоль) и трис(дибензилиденацетон)дипалладий (138,86 мг, 151,64 мкмоль) помещали в толуол (10 мл) и воду (2 мл), затем реакционную смесь продували азотом, нагревали до 100°C и перемешивали в течение 6 часов в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (20 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фазу промывали насыщенным солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. К неочищенному продукту добавляли н-гептан (10 мл), перемешивали в течение 15 минут и фильтровали. Осадок на фильтре ополаскивали н-гептаном (3 мл × 3) и собирали с получением соединения WX008-10.
[000288] Стадия 10: синтез соединения WX008-11
[000289] Соединение WX008-10 (760 мг, 2,11 ммоль) и акриламид (165,35 мг, 2,33 ммоль, 160,53 мкл) растворяли в N,N-диметилформамиде (20 мл), добавляли трет-бутоксид калия (498,34 мг, 4,44 ммоль) при 20°C и перемешивали в течение 1 часа при 20°C. Реакционную смесь выливали в 1 н. хлористоводородную кислоту (20 мл) при 0-10°C и экстрагировали этилацетатом (20 мл × 3). Органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт разделяли колоночной хроматографией (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения WX008-11.
[000290] Стадия 11: синтез гидрохлорида соединения WX008-12
[000291] Соединение WX008-11 (240 мг, 624,38 мкмоль) помещали в этилацетат (1 мл), добавляли смесь хлористоводородная кислота/этилацетат (4 M, 10 мл) и перемешивали при 20°C в течение 3 часов. Реакционную смесь непосредственно концентрировали при пониженном давлении с получением гидрохлорида соединения WX008-12. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,94 (s, 1H), 8,16 (d, J = 2,0 Гц, 1H), 7,87 (s, 1H), 7,26 (s, 1H), 7,09 (d, J = 2,0 Гц, 1H), 4,29 (dd, J = 4,8 Гц, 12,0 Гц, 1H), 2,86-2,77 (m, 1H), 2,68-2,58 (m, 1H), 2,34-2,22 (m, 1H), 2,19-2,14 (m, 1H).
[000292] Стадия 12: синтез соединения WX008
[000293] Соединение BB-4 (125,29 мг, 211,07 мкмоль) и N,N-диизопропилэтиламин (90,93 мг, 703,57 мкмоль) растворяли в N,N-диметилформамиде (2 мл), добавляли гексафторфосфат 2-(азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (86,94 мг, 228,66 мкмоль) при 20°C и перемешивали в течение 5 минут. К реакционной смеси добавляли гидрохлорид соединения WX008-12 (50 мг, 175,89 мкмоль) при 20°C и перемешивали в течение 12 часов. Реакционную смесь непосредственно концентрировали при пониженном давлении. Неочищенный продукт очищали посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением соединения WX008. 1H ЯМР (400 MГц, CD3OD) δ: 8,19 (s, 1H), 8,17-8,15 (m, 2H), 7,98 (dd, J = 1,6 Гц, 8,0 Гц, 1H), 7,91 (d, J = 2,0 Гц, 1H), 7,81 (s, 1H), 7,40-7,35 (m, 1H), 7,32 (dd, J = 2,4 Гц, 11,6 Гц, 1H), 7,25-7,22 (m, 1H), 7,02 (d, J = 2,0 Гц, 1H), 4,60-4,58 (m, 2H), 4,29 (dd, J = 5,2 Гц, 11,6 Гц, 1H), 3,92 (s, 2H), 3,77-3,69 (m, 6H), 3,42 (m, 4H), 2,92-2,83 (m, 1H), 2,79-2,73 (m, 1H), 2,44-2,30 (m, 2H), 1,57 (s, 6H).
[000294] Пример 9
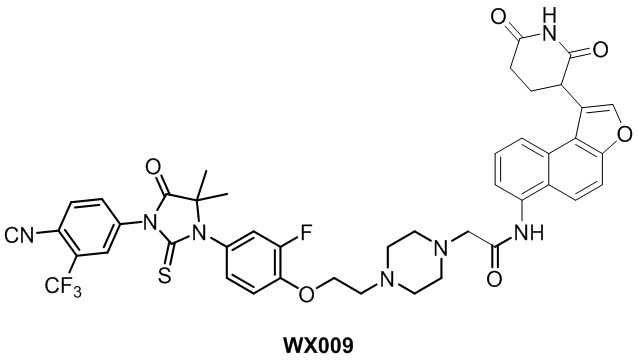
[000295] Путь синтеза:
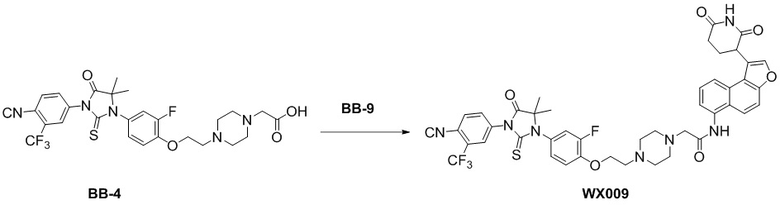
[000296] Синтез соединения WX009
[000297] К N,N-диметилформамиду (10 мл) добавляли соединение BB-4 (200 мг, 336,93 мкмоль) и гидрохлорид BB-8 (133,73 мг, 404,32 мкмоль). К реакционной смеси медленно добавляли гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (256,22 мг, 673,86 мкмоль) и N,N-диизопропилэтиламин (130,64 мг, 1,01 ммоль). Смесь трижды продували азотом и перемешивали при 20°C в течение 12 часов. Реакционную смесь выливали в воду (10 мл) и этилацетат (10 мл) и отделенную органическую фазу трижды промывали водой (10 мл × 3). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Неочищенный продукт очищали посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением соединения WX009. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,95 (s, 1H), 10,69-10,37 (m, 1H), 8,39 (d, J = 8,0 Гц, 1H), 8,27 (d, J = 1,5 Гц, 1H), 8,17-7,97 (m, 4H), 7,86 (d, J = 9,3 Гц, 1H), 7,68-7,54 (m, 2H), 7,49-7,33 (m, 2H), 7,22 (br d, J = 8,0 Гц, 1H), 4,70 (br dd, J = 4,3, 11,8 Гц, 1H), 4,66-4,46 (m, 2H), 4,28-3,98 (m, 2H), 3,81-3,61 (m, 10H), 3,00-2,81 (m, 1H), 2,72-2,60 (m, 1H), 2,47-2,37 (m, 1H), 2,35-2,21 (m, 1H), 1,51 (s, 6H).
[000298] Пример 10
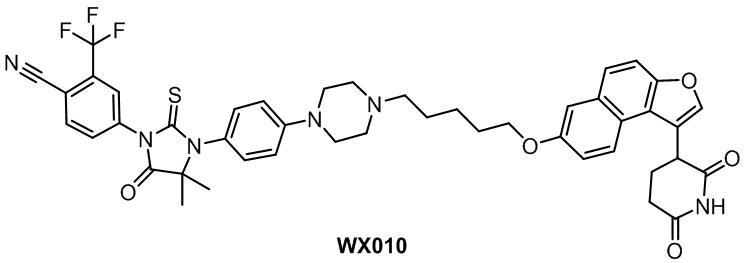
[000299] Путь синтеза:
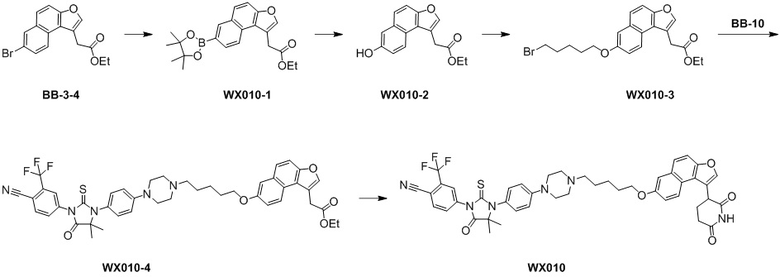
[000300] Стадия 1: синтез соединения WX010-1
[000301] К диоксановому (20 мл) растворителю добавляли соединение BB-3-4 (2 г, 6,00 ммоль) при комнатной температуре и начинали перемешивание. К полученному добавляли соединение бис(пинаколато)дибор (2,29 г, 9,00 ммоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (439,23 мг, 600,28 мкмоль) и ацетат калия (1,18 г, 12,01 ммоль) и реакционную смесь нагревали до 100°C и перемешивали в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры, добавляли воду (30 мл) и экстрагировали этилацетатом (30 мл × 2). Органические фазы объединяли, промывали с помощью полунасыщенного солевого раствора (30 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Полученный в результате неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат =от 1/0 до 4/1) с получением соединения WX010-1. MS-ESI масса/заряд: 380,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ: 8,47 (s, 1H), 8,19 (d, J = 8,5 Гц, 1H), 7,94 (d, J = 8,3 Гц, 1H), 7,83-7,75 (m, 2H), 7,65 (d, J = 9,0 Гц, 1H), 4,23 (q, J = 7,1 Гц, 2H), 4,09-4,09 (m, 1H), 4,08 (s, 1H), 1,41 (s, 12H), 1,30-1,25 (m, 3H).
[000302] Стадия 2: синтез соединения WX010-2
[000303] К смешанному растворителю из тетрагидрофурана (40 мл) и воды (20 мл) добавляли соединение WX010-1 (2,3 г, 6,05 ммоль) при комнатной температуре и начинали перемешивание. К полученному добавляли соединение бикарбонат натрия (1,02 г, 12,10 ммоль, 470,52 мкл) и реакционную смесь охлаждали до 0°C, медленно по каплям добавляли пероксид водорода (5,49 г, 48,39 ммоль, 4,65 мл, концентрация: 30%) и перемешивали в течение дополнительных 2 часов в атмосфере азота. К реакционной смеси медленно по каплям добавляли насыщенный водный раствор сульфита натрия (50 мл) на ледяной бане. После завершения добавления по каплям реакционную смесь перемешивали в течение дополнительных 10 минут. Реакционную смесь экстрагировали этилацетатом (100 мл × 2). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат =от 8/1 до 4/1) с получением соединения WX010-2. 1H ЯМР (400 МГц, CDCl3) δ: 8,13 (d, J = 8,9 Гц, 1H), 7,74 (s, 1H), 7,64-7,51 (m, 2H), 7,29 (d, J = 2,6 Гц, 1H), 7,18 (dd, J = 2,6, 8,9 Гц, 1H), 4,23 (q, J = 7,1 Гц, 2H), 4,05 (d, J = 0,9 Гц, 2H), 1,31-1,22 (m, 3H).
[000304] Стадия 3: синтез соединения WX010-3
[000305] К N,N-диметилформамиду (10 мл) добавляли соединение WX010-2 (400 мг, 1,33 ммоль) и 1,5-дибромпентан (1,53 г, 6,64 ммоль, 897,79 мкл) и начинали перемешивание. К реакционной смеси медленно добавляли карбонат калия (550,41 мг, 3,98 ммоль) и йодид натрия (198,99 мг, 1,33 ммоль). Смесь трижды продували азотом, а затем перемешивали при 80°C в течение 10 часов. Реакционную смесь охлаждали до 25°C, затем к полученному добавляли этилацетат (30 мл) и воду (30 мл) и отделенную органическую фазу дважды промывали насыщенным солевым раствором (30 мл × 2). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный в результате неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат =от 50/1 до 10/1) с получением соединения WX010-3. MS-ESI масса/заряд: 418,9 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ: 8,06 (d, J = 9,0 Гц, 1H), 7,66 (s, 1H), 7,53 (s, 2H), 7,25-7,10 (m, 3H), 4,17-4,08 (m, 3H), 4,03 (t, J = 6,3 Гц, 2H), 3,96 (s, 2H), 3,39 (t, J = 6,8 Гц, 2H), 1,97-1,86 (m, 2H), 1,85-1,77 (m, 2H), 1,69-1,57 (m, 3H), 1,18 (t, J = 7,2 Гц, 3H).
[000306] Стадия 4: синтез соединения WX010-4
[000307] К N,N-диметилформамиду (5 мл) добавляли соединение BB-10 (200 мг, 422,37 мкмоль) и соединение WX010-3 (177,11 мг, 422,37 мкмоль, 72,61 мкл) и начинали перемешивание. К реакционной смеси медленно добавляли карбонат калия (175,12 мг, 1,27 ммоль) и йодид калия (70,12 мг, 422,37 мкмоль). Реакционную смесь трижды продували азотом, а затем перемешивали при 80°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (30 мл) и органическую фазу дважды промывали водой (20 мл × 2). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный в результате неочищенный продукт очищали колоночной хроматографией (элюент: дихлорметан/метанол = от 50/1 до 10/1) с получением соединения WX010-4.
[000308] MS-ESI масса/заряд: 812,3 [M+H]+.
[000309] Стадия 5: синтез соединения WX010
[000310] К N,N-диметилформамиду (10 мл) добавляли соединение WX010-4 (300 мг, 369,50 мкмоль) и акриламид (26,26 мг, 369,50 мкмоль, 25,50 мкл) при 0°C. К реакционной смеси медленно порциями добавляли трет-бутоксид калия (82,92 мг, 739,00 мкмоль). Смесь трижды продували азотом, а затем перемешивали при 0°C в течение 1 часа. К реакционной смеси медленно добавляли насыщенный раствор хлорида аммония (50 мл) и дважды экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, трижды промывали насыщенным солевым раствором (30 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Неочищенный продукт разделяли путем препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением соединения WX010. MS-ESI масса/заряд: 837,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 11,23 (br s, 1H), 10,95 (s, 1H), 8,39 (d, J = 8,3 Гц, 1H), 8,30 (d, J = 1,3 Гц, 1H), 8,15-8,04 (m, 2H), 7,98 (d, J = 6,8 Гц, 1H), 7,82-7,68 (m, 2H), 7,53 (d, J = 2,3 Гц, 1H), 7,34-7,20 (m, 3H), 7,14 (br d, J = 9,0 Гц, 2H), 4,65 (br dd, J = 4,1, 11,9 Гц, 1H), 4,18-4,09 (m, 2H), 4,03 (s, 1H), 3,93 (br s, 2H), 3,60 (br d, J = 11,3 Гц, 2H), 3,38-3,23 (m, 2H), 3,22-3,04 (m, 4H), 2,98-2,79 (m, 1H), 2,71-2,53 (m, 1H), 2,41 (br dd, J = 3,9, 12,7 Гц, 1H), 2,31-2,17 (m, 1H), 1,95-1,76 (m, 4H), 1,64-1,43 (m, 8H).
[000311] Пример 11
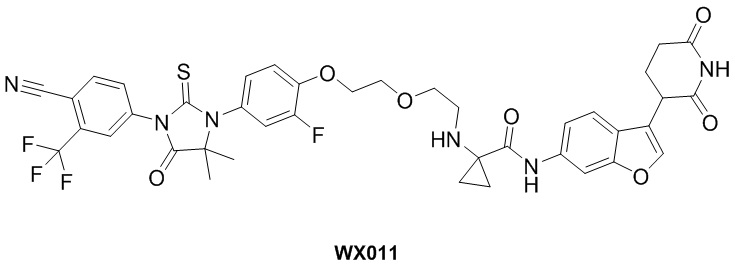
[000312] Путь синтеза:
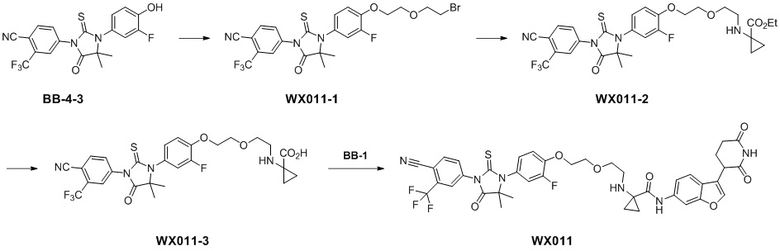
[000313] Стадия 1: синтез соединения WX011-1
[000314] Соединение BB-4-3 (1 г, 2,36 ммоль) растворяли в N,N-диметилформамиде (30 мл), добавляли карбонат калия (652,86 мг, 4,72 ммоль) и 2,2'-дибромдиэтиловый эфир (2,74 г, 11,81 ммоль, 1,48 мл), нагревали до 80°C и перемешивали в течение 30 часов. К реакционной смеси добавляли воду (150 мл) и экстрагировали этилацетатом (30 мл × 3). Органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат = 3/1) с получением соединения WX011-1.
[000315] Стадия 2: синтез соединения WX011-2
[000316] Гидрохлорид этил-1-аминоциклопропанкарбоксилата (346,01 мг, 2,09 ммоль) и карбонат калия (577,48 мг, 4,18 ммоль) растворяли в N,N-диметилформамиде (10 мл), добавляли соединение WX011-1 (0,6 г, 1,04 ммоль), нагревали до 75°C и перемешивали в течение 15 часов. К реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (15 мл × 2). Органическую фазу промывали насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания. Неочищенный продукт очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат = 3/1) с получением соединения WX011-2.
[000317] Стадия 3: синтез соединения WX011-3
[000318] Соединение WX011-2 (0,2 г, 321,22 мкмоль) растворяли в смешанном растворе тетрагидрофурана (4 мл) и воды (1 мл), добавляли моногидрат гидроксида лития (67,40 мг, 1,61 ммоль) при 20°C и перемешивали в течение 30 часов. К реакционной смеси добавляли воду (5 мл), затем добавляли 1 н. разбавленную хлористоводородную кислоту для доведения pH до 5 и экстрагировали этилацетатом (10 мл × 4). Органическую фазу промывали насыщенным солевым раствором (10 мл), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания с получением соединения WX011-3.
[000319] Стадия 4: синтез соединения WX011
[000320] Соединение WX011-3 (80 мг, 134,55 мкмоль) растворяли в N,N-диметилформамиде (1 мл), добавляли гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (76,74 мг, 201,82 мкмоль) и N,N-диизопропилэтиламин (69,56 мг, 538,20 мкмоль, 93,74 мкл), перемешивали в течение 10 минут, затем добавляли гидрохлорид соединения BB-1 (49,10 мг, 174,91 мкмоль) и перемешивали при 20°C в течение 15 часов. Реакционную смесь очищали посредством препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением соединения WX011. 1H ЯМР (400 MГц, CD3OD) δ: 8,16-8,14 (m, 2H), 7,95 (dd, J = 1,6 Гц, 8,4 Гц, 1H), 7,91 (d, J = 1,6 Гц, 1H), 7,71 (s, 1H), 7,50 (d, J = 8,8 Гц, 1H), 7,30-7,22 (m, 3H), 7,16-7,14 (m, 1H), 4,35 (t, J = 4,0 Гц, 2H), 4,10 (dd, J = 4,2 Гц, 11,6 Гц, 1H), 3,99-3,97 (m, 2H), 3,89 (t, J = 2,8 Гц, 2H), 3,47 (s, 2H), 2,83-2,66 (m, 2H), 2,42-2,32 (m, 1H), 2,29-2,22 (m, 1H), 1,81-1,78 (m, 2H), 1,65-1,63 (m, 2H), 1,54 (s, 9H).
[000321] Пример 12
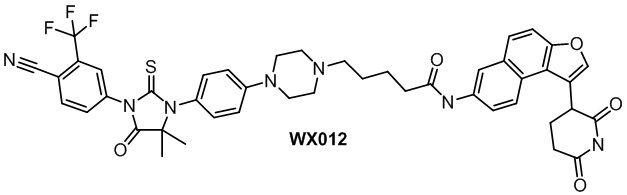
[000322] Путь синтеза:
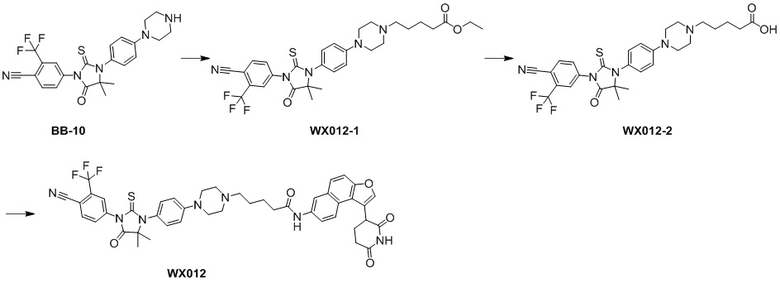
[000323] Стадия 1: синтез соединения WX012-1
[000324] К N,N-диметилформамиду (5 мл) добавляли соединение BB-10 и этил-5-бромвалерат (662,33 мг, 3,17 ммоль, 505,59 мкл) и начинали перемешивание. К реакционной смеси медленно добавляли карбонат калия (262,69 мг, 1,90 ммоль) и йодид натрия (94,97 мг, 633,56 мкмоль). Реакционную смесь трижды продували азотом, а затем перемешивали при 80°C в течение 12 часов. После завершения реакции реакционную смесь охлаждали до 25°C, разбавляли этилацетатом (20 мл). Органическую фазу дважды промывали водой (20 мл × 2) и промывали один раз насыщенным раствором соли (20 мл). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (элюент: дихлорметан/метанол = от 50/1 до 10/1) с получением соединения WX012-1. MS-ESI масса/заряд: 602,1 [M+H]+.
[000325] Стадия 2: синтез соединения WX012-2
[000326] К безводному этанолу (2,5 мл) и воде (0,5 мл) добавляли соединение WX012-1 (170 мг, 282,54 мкмоль) и начинали перемешивание. К реакционной смеси медленно добавляли моногидрат гидроксида лития (29,64 мг, 706,35 мкмоль). Реакционную смесь трижды продували азотом и перемешивали при 25°C в течение 12 часов. После завершения реакции к реакционной смеси добавляли воду (10 мл) и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении для удаления большей части органического растворителя. К водной фазе добавляли 2 н. хлористоводородную кислоту для доведения pH до 3 и непосредственно лиофилизировали. Получали соединение WX012-2. MS-ESI масса/заряд: 574,0 [M+H]+.
[000327] Стадия 3: синтез гидрохлорида соединения WX012
[000328] К N,N-диметилформамиду (5 мл) добавляли гидрохлорид соединения BB-3 (54,30 мг, 139,46 мкмоль). К реакционной смеси медленно добавляли гексафторфосфат 2-(азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (165,71 мг, 435,82 мкмоль) и триэтиламин (52,92 мг, 522,99 мкмоль, 72,79 мкл). Смесь трижды продували азотом и перемешивали при 25°C в течение 3 часов. Реакционную смесь разбавляли этилацетатом (30 мл) и органическую фазу дважды промывали водой (20 мл × 2). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный в результате неочищенный продукт разделяли путем препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением гидрохлорида соединения WX012. MS-ESI масса/заряд: 850,4 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,95 (s, 1H), 10,54 (br d, J = 4,5 Гц, 1H), 10,29 (s, 1H), 8,48-8,35 (m, 2H), 8,29 (d, J = 1,3 Гц, 1H), 8,18-8,04 (m, 2H), 7,99 (s, 1H), 7,79-7,69 (m, 3H), 7,28-7,21 (m, 2H), 7,14 (d, J = 9,0 Гц, 2H), 4,65 (br dd, J = 4,4, 12,2 Гц, 1H), 3,94 (br d, J = 11,3 Гц, 2H), 3,62 (br d, J = 10,8 Гц, 2H), 3,28-3,07 (m, 6H), 2,95-2,80 (m, 1H), 2,69-2,59 (m, 1H), 2,49-2,36 (m, 3H), 2,33-2,22 (m, 1H), 1,92-1,77 (m, 2H), 1,76-1,65 (m, 2H), 1,49 (s, 6H).
[000329] Пример 13
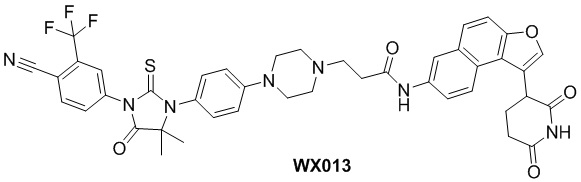
[000330] Путь синтеза:
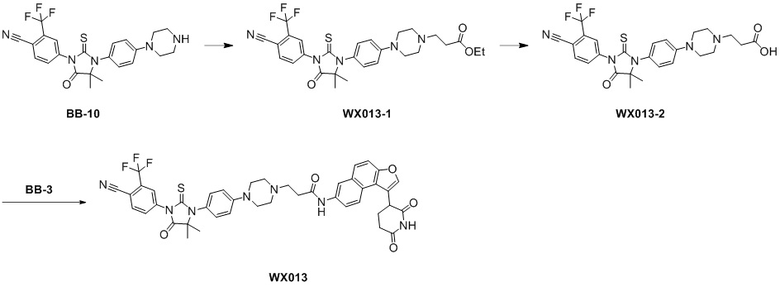
[000331] Стадия 1: синтез соединения WX013-1
[000332] К ацетонитрилу (5 мл) добавляли соединение BB-10 (0,15 г, 284,79 мкмоль) и этилбромпропионат (103,11 мг, 569,57 мкмоль, 72,61 мкл) и начинали перемешивание. К реакционной смеси медленно добавляли карбонат калия (78,72 мг, 569,57 мкмоль) и йодид калия (47,28 мг, 284,79 мкмоль). Реакционную смесь трижды продували азотом, а затем перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, затем к полученному добавляли этилацетат (50 мл) и воду (50 мл) и отделенную органическую фазу дважды промывали насыщенным солевым раствором (30 мл × 2). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (элюент: дихлорметан/метанол = от 50/1 до 10/1, об./об.) с получением соединения WX013-1. MS-ESI масса/заряд: 574,0 [M+H]+.
[000333] Стадия 2: синтез соединения WX013-2
[000334] К безводному этанолу (4 мл) и воде (1 мл) добавляли соединение WX013-1 (0,2 г, 348,66 мкмоль) и начинали перемешивание. К реакционной смеси медленно добавляли моногидрат гидроксида лития (43,89 мг, 1,05 ммоль). Реакционную смесь трижды продували азотом и перемешивали при 25°C в течение 12 часов. Реакционную смесь выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении для удаления большей части этанола и к полученному добавляли воду (20 мл). К водной фазе добавляли 2 н. хлористоводородную кислоту для доведения pH до 3-4 и непосредственно лиофилизировали. Получали соединение WX013-2. MS-ESI масса/заряд: 546,0 [M+H]+.
[000335] Стадия 3: синтез гидрохлорида соединения WX013
[000336] К N,N-диметилформамиду (3 мл) добавляли соединение WX013-2 (60 мг, 109,98 мкмоль) и гидрохлорид соединения BB-3. К реакционной смеси медленно добавляли гексафторфосфат 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония (83,63 мг, 219,95 мкмоль) и N,N-диизопропилэтиламин (42,64 мг, 329,93 мкмоль, 57,47 мкл). Смесь трижды продували азотом, а затем перемешивали при 20°C в течение 12 часов. После завершения реакции реакционную смесь выливали в смешанный растворитель из воды и этилацетата (10 мл : 10 мл) и отделенную органическую фазу трижды промывали водой (10 мл × 3). Собранную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния путем ротационного выпаривания при пониженном давлении. Полученный в результате неочищенный продукт очищали путем препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением гидрохлорида соединения WX013. MS-ESI масса/заряд: 822,3 [M+H]+.
[000337] 1H ЯМР (400 MГц, DMSO-d6) δ: 10,95 (s, 1H), 10,54 (br s, 1H), 10,46 (br s, 1H), 8,47-8,35 (m, 2H), 8,29 (br s, 1H), 8,16 (br s, 1H), 8,08 (br d, J = 8,5 Гц, 1H), 8,00 (s, 1H), 7,80-7,67 (m, 3H), 7,34-7,20 (m, 2H), 7,16 (br d, J = 8,8 Гц, 2H), 4,68 (br s, 1H), 3,98 (m, 2H), 3,64 (m, 2H), 3,55 (m, 4H), 3,23 (m, 2H), 3,05 (br s, 2H), 2,86 (m, 1H), 2,67 (m, 1H), 2,63-2,62 (m, 1H), 2,41-2,33 (m, 1H), 1,49 (br s, 6H).
[000338] Пример 14
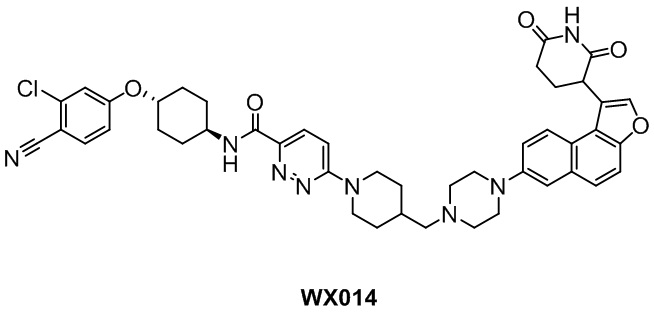
[000339] Путь синтеза:
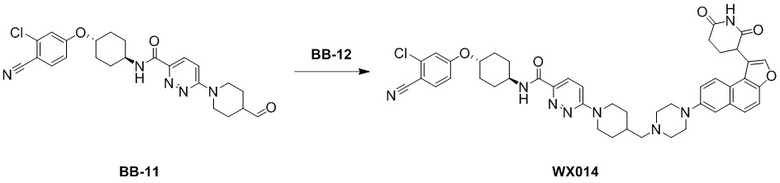
[000340] Синтез гидрохлорида соединения WX014
[000341] Соединение BB-11 (400 мг, 854,80 мкмоль) и гидрохлорид соединения BB-12 (403,83 мг, 1,11 ммоль) растворяли в дихлорметане (30 мл), затем добавляли триацетоксиборгидрид натрия (543,50 мг, 2,56 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь разбавляли дихлорметаном (100 мл) и водой (100 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали путем препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением гидрохлорида соединения WX014. MS-ESI масса/заряд: 815,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,97 (s, 1H), 10,83 (br s, 1H), 8,63 (d, J = 8,0 Гц, 1H), 8,05 (br d, J = 8,5 Гц, 1H), 7,96 (s, 1H), 7,91-7,84 (m, 2H), 7,73 (s, 2H), 7,54-7,43 (m, 3H), 7,41-7,38 (m, 1H), 7,16-7,11 (m, 1H), 4,63 (br dd, J = 4,0, 12,0 Гц, 1H), 4,58-4,46 (m, 3H), 3,98-3,81 (m, 3H), 3,66 (br d, J = 11,0 Гц, 2H), 3,43 (br t, J = 12,2 Гц, 2H), 3,28-3,05 (m, 6H), 2,96-2,83 (m, 1H), 2,76-2,58 (m, 2H), 2,56-2,53 (m, 1H), 2,46-2,37 (m, 1H), 2,31-2,21 (m, 2H), 2,11 (br d, J = 9,0 Гц, 2H), 2,02 (br d, J = 12,0 Гц, 2H), 1,90 (br d, J = 10,5 Гц, 2H), 1,72-1,58 (m, 2H), 1,58-1,45 (m, 2H).
[000342] Пример 15
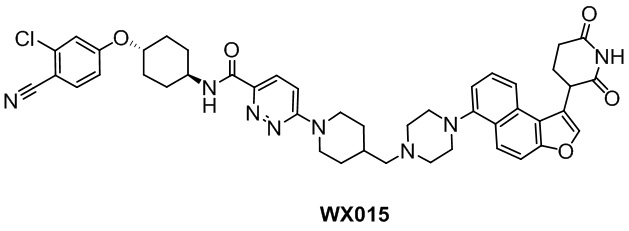
[000343] Путь синтеза:
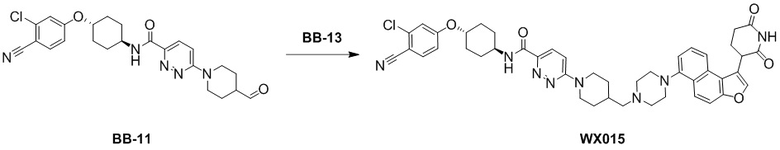
[000344] синтез гидрохлорида соединения WX015
[000345] Соединение BB-11 (400 мг, 854,80 мкмоль) и гидрохлорид соединения BB-13 (403,83 мг, 1,11 ммоль) растворяли в дихлорметане (30 мл), затем добавляли триацетоксиборгидрид натрия (181,17 мг, 854,80 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь разбавляли дихлорметаном (100 мл) и водой (100 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали путем препаративной HPLC (подвижная фаза: ацетонитрил/вода; кислотная система: 0,05% HCl) с получением гидрохлорида соединения WX015. MS-ESI масса/заряд: 815,2 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,96 (s, 1H), 10,82 (br s, 1H), 8,63 (d, J = 8,3 Гц, 1H), 8,17 (d, J = 9,3 Гц, 1H), 8,03 (s, 1H), 7,96 (br d, J = 5,8 Гц, 1H), 7,86 (dt, J = 9,4, 15,6 Гц, 3H), 7,60-7,51 (m, 2H), 7,39 (d, J = 2,5 Гц, 1H), 7,27-7,22 (m, 1H), 7,14 (dd, J = 2,3, 8,8 Гц, 1H), 4,68 (br dd, J = 3,9, 11,9 Гц, 1H), 4,58-4,47 (m, 3H), 3,92-3,81 (m, 1H), 3,69 (br s, 3H), 3,50-3,38 (m, 3H), 3,23-3,06 (m, 4H), 2,98-2,81 (m, 1H), 2,70-2,58 (m, 1H), 2,45-2,20 (m, 4H), 2,16-1,99 (m, 5H), 1,91 (br d, J = 10,3 Гц, 2H), 1,71-1,58 (m, 2H), 1,57-1,42 (m, 2H), 1,40-1,27 (m, 2H).
[000346] Пример 16 и пример 17
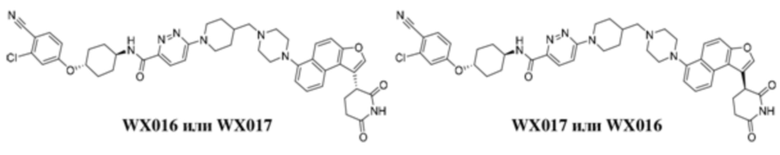
[000347] Путь синтеза:
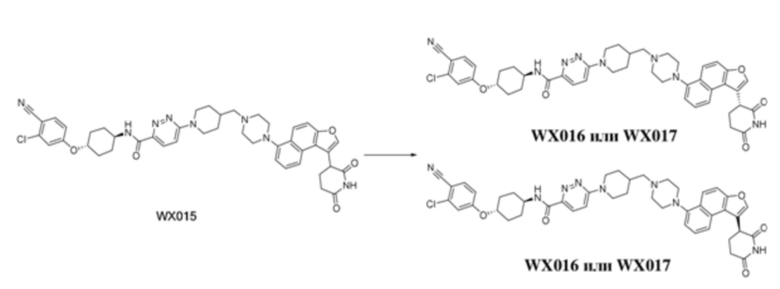
[000348] синтез соединения 16 и соединения 17
[000349] Соединение BB-11 (7 г, 14,96 ммоль) и гидрохлорид соединения BB-13 (7,18 г, 17,95 ммоль) растворяли в дихлорметане (500 мл), перемешивали при комнатной температуре в течение 12 часов, затем добавляли триацетоксиборгидрид натрия (4,76 г, 22,44 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов в атмосфере азота. После завершения реакции реакционную смесь разбавляли водой (200 мл). Органическую фазу отделяли, промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией (элюент: дихлорметан/метанол = 10/1) с получением соединения WX015, которое подвергали хиральному разделению с помощью HPLC (тип колонки: DAICEL CHIRALPAK IE (250 мм * 30 мм, 10 мкм); подвижная фаза: A (IPA), B (ACN), B%: 50% - 100%; скорость потока: 80 мл/мин) с получением соединений WX016 и WX017.
[000350] Способ анализа: хроматографическая колонка: Chiralpak IA, 100 * 4,6 мм, 3 мкм; подвижная фаза: A: н-гексан (0,1% диэтиламин); B: изопропанол: ацетонитрил = 2:1, A:B = 40:60; температура колонки: 35°C; длина волны: 220 нм.
[000351] WX016: время удерживания составляло 4,679 мин., ee %: 98,77%; MS-ESI масса/заряд: 815,4 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ: 10,97 (s, 1H), 8,60 (d, J = 8,3 Гц, 1H), 8,16 (d, J = 9,3 Гц, 1H), 7,99 (s, 1H), 7,91-7,74 (m, 4H), 7,51 (t, J = 7,9 Гц, 1H), 7,41-7,31 (m, 2H), 7,21-7,10 (m, 2H), 4,66 (br dd, J = 4,1, 11,7 Гц, 1H), 4,58-4,45 (m, 3H), 3,94-3,79 (m, 1H), 3,22-3,00 (m, 7H), 2,89 (ddd, J = 5,3, 12,2, 17,4 Гц, 2H), 2,69-2,56 (m, 2H), 2,47-2,33 (m, 2H), 2,29 (br d, J = 7,0 Гц, 3H), 2,11 (br d, J = 9,3 Гц, 2H), 2,00-1,81 (m, 5H), 1,72-1,58 (m, 2H), 1,58-1,44 (m, 2H), 1,26-1,09 (m, 2H).
[000352] WX017: время удерживания составляло 5,797 мин., ee %: 99,86%; MS-ESI масса/заряд: 815,4 [M+H]+. 1H ЯМР (400 MГц, DMSO-d6) δ = 11,00 (br s, 1H), 8,60 (br d, J = 8,3 Гц, 1H), 8,22-8,13 (m, 1H), 8,04-7,95 (m, 1H), 7,94-7,71 (m, 4H), 7,57-7,47 (m, 1H), 7,41-7,31 (m, 2H), 7,16 (br dd, J = 8,0, 19,1 Гц, 2H), 4,66 (br d, J = 7,3 Гц, 1H), 4,58-4,40 (m, 3H), 3,87 (br d, J = 7,8 Гц, 1H), 3,28-2,97 (m, 7H), 2,88 (br t, J = 12,3 Гц, 2H), 2,69-2,56 (m, 2H), 2,49-2,35 (m, 2H), 2,28 (br d, J = 6,0 Гц, 3H), 2,17-2,00 (m, 2H), 2,00-1,79 (m, 5H), 1,71-1,58 (m, 2H), 1,51 (q, J = 10,6 Гц, 2H), 1,26-1,01 (m, 2H).
[000353] Биологические данные
[000354] Пример исследования 1. Тест in vitro в отношении регуляции уровня белка AR и нижележащего эффекторного белка простат-специфического антигена (PSA) в клетках LNCaP рака предстательной железы человека.
[000355] Экспериментальная цель: изучить регуляцию уровня белка AR и нижележащего эффекторного белка простат-специфического антигена (PSA) в клетках LNCaP рака предстательной железы человека под воздействием соединений в различных концентрациях с использованием метода WB (вестерн блоттинг).
[000356] Схема проведения эксперимента:
1) клетки LNCaP размораживали и выполняли их пассаж по меньшей мере дважды;
2) Клетки LNCaP инокулировали в 6-луночный планшет по 3 × 105 клеток на лунку, культивировали адгезивным способом в течение ночи и обрабатывали исследуемым соединением при определенной концентрации;
3) После 24 часов обработки супернатант образцов культивируемых клеток удаляли, а остаток дважды промывали с помощью DPBS (фосфатно-солевого буфера Дульбекко); затем клетки лизировали определенным количеством 2% лизирующего буфера SDS, предварительно нагретого до 100°C, собирали и денатурировали при 100°C в течение 15 минут;
4) Вышеуказанный буфер для лизиса денатурировали и охлаждали для количественного определения белка (набор для анализа белка Pierce BCA, Thermo), затем объем фиксировали с помощью 5-кратного загрузочного буфера (содержащего дитиотреитол (DDT), Beyotime) в соответствии с той же концентрацией белка и восстанавливали и денатурировали при 100°C в течение 10 минут;
5) Вышеуказанные образцы (от 10 до 20 мкг белка) разделяли с помощью SDS-PAGE и переносили на мембрану PVDF (Biorad);
6) Полоску разрезали согласно молекулярной массе белка-мишени и блокировали блокирующим раствором (3% раствором бычьего сывороточного альбумина TBS-T, причем TBS-T раствор представлял собой буфер Трис-HCl, содержащий 0,2% Tween-20) в течение 1 часа, затем инкубировали в течение ночи при 4°C с первичными антителами (антителами к AR (№ 5153, CST), антителами к PSA/KLK3 (№ 5365, CST) и антителами к β-актину (№ 4970, CST), разбавленными 1:1000, 1:1000 и 1:2000 соответственно в блокирующем растворе);
7) Наконец, ее инкубировали при комнатной температуре в течение 1 часа с конъюгированным с HRP вторичным антителом (антителом к IgG кролика (№ 7074, CST), разбавленным 1:2000 в блокирующем растворе) и полоски на мембране подвергали детекции с использованием хемилюминесцентного субстрата (Clarity ECL, Biorad).
[000357] Результаты эксперимента
[000358] Результаты испытания показаны на фигуре 1 и фигуре 2.
[000359] Заключение: соединения по настоящему изобретению демонстрируют надлежащую активность в отношении деградации белка AR, при этом их способность к деградации демонстрирует надлежащую зависимость от концентрации. Соединения по настоящему изобретению могут в значительной степени влиять на соответствующие изменения нижележащего эффектора PSA, при этом степень их эффекта демонстрирует надлежащую зависимость от концентрации.
[000360] Пример исследования 2. Оценка антипролиферативных эффектов в клетках LNCaP рака предстательной железы человека
[000361] Экспериментальная цель
[000362] В данном эксперименте обнаружили ингибирующий эффект исследуемого соединения на пролиферацию клеток LNCaP рака предстательной железы человека.
[000363] Материалы для экспериментов:
[000364] 1. Клеточная линия и способ культивирования
[000365] Таблица 1. Клеточная линия и способ культивирования

[000366] 2. Культуральная среда и реагенты
[000367] Таблица 2. Культуральная среда и реагенты
[000368] 3. Многолуночный планшет
[000369] 96-луночный планшет Greiner CELLSTAR®, черный планшет с плоским дном (с крышкой и прозрачным дном), № 655090.
[000370] 4. Реагенты и инструменты для анализа жизнеспособности клеток
(1) Набор для люминесцентного анализа жизнеспособности клеток Promega CellTiter-Glo (Promega-G7573).
(2) Планшет-ридер 2104 EnVision®, PerkinElmer.
[000371] Схема проведения эксперимента:
[000372] 1. Культура клеток
[000373] Линии опухолевых клеток культивировали в инкубаторе при 37°C, 5% CO2 при вышеуказанных условиях культивирования. Для посева отбирали периодические пассажи и клетки в логарифмической фазе роста.
[000374] 2. Посев клеток
(1) Клетки окрашивали трипановым синим и подсчитывали жизнеспособные клетки;
(2) Концентрацию клеток доводили до соответствующей концентрации;
[000375] Таблица 3. Количество высеянных клеток

(3) В культуральный планшет добавляли 90 мкл клеточной суспензии на лунку, как показано в таблице выше, а в лунку для холостого контроля добавляли бесклеточную культуральную среду;
(4) Осуществляли культивирование в культуральном планшете в течение ночи в инкубаторе при 37°C, 5% CO2 и относительной влажности 100%.
[000376] 3. Подготовка планшета для хранения соединения
[000377] Получение исходного раствора для планшета для хранения с 400-кратной исходной концентрацией соединения: соединение серийно разбавляли с помощью DMSO от самой высокой концентрации до самой низкой концентрации. Раствор получали каждый раз по мере необходимости.
[000378] Получение рабочего раствора с 10-кратной исходной концентрацией соединений и обработка клеток соединениями:
(1) В 96-луночный планшет с V-образным дном добавляли 78 мкл среды для культивирования клеток и 2 мкл соединения отбирали пипеткой из планшета для хранения исходного раствора с 400-кратной начальной концентрацией соединения и добавляли к среде для культивирования клеток в 96-луночном планшете. К контролю в виде среды-носителя и холостому контролю добавляли 2 мкл DMSO. После добавления соединения или DMSO проводили продувку пипеткой и тщательное перемешивание.
(2) Введение: в планшет для клеточных культур добавляли 10 мкл рабочего раствора с 10-кратной исходной концентрацией соединения. К контролю в виде среды-носителя и холостому контролю добавляли 10 мкл смеси DMSO-среда для культивирования клеток.
(3) 96-луночный планшет возвращали в инкубатор и культивировали в течение 6 дней.
[000379] 5. Люминесцентный анализ жизнеспособности клеток CellTiter-Glo
[000380] Следующие шаги выполняли в соответствии с инструкциями набора для люминесцентного анализа жизнеспособности клеток Promega CellTiter-Glo (Promega-G7573).
(1) Буфер CellTiter-Glo расплавляли и доводили до комнатной температуры;
(2) Субстрат CellTiter-Glo доводили до комнатной температуры;
(3) Рабочий раствор CellTiter-Glo получали путем добавления 100 мл буфера CellTiter-Glo во флакон с субстратом CellTiter-Glo для растворения субстрата;
(4) Смесь медленно перемешивали с помощью вортекса и встряхивали до полного растворения;
(5) Планшет для клеточных культур вынимали и оставляли на 30 минут для достижения равновесия до комнатной температуры;
(6) В каждую лунку добавляли по 50 мкл рабочего раствора CellTiter-Glo (эквивалентно половине объема среды для культивирования клеток на лунку). Планшет для клеток оборачивали алюминиевой фольгой для защиты от света;
(7) Культуральный планшет встряхивали на орбитальном шейкере в течение 2 минут для индукции лизиса клеток;
(8) Культуральный планшет оставляли при комнатной температуре на 10 минут для стабилизации люминесцентного сигнала;
(9) Люминесцентный сигнал регистрировали с помощью планшет-ридера 2104 EnVision.
[000381] 6. Анализ данных
[000382] Степень ингибирования (IR) исследуемого соединения рассчитывали с применением следующей формулы: IR (%) = (RLU контроля в виде среды-носителя - RLU соединения) / (RLU контроля в виде среды-носителя - RLU холостого контроля) × 100%. Значения степени ингибирования соединений в различных концентрациях рассчитывали в Excel, а затем использовали программное обеспечение GraphPad Prism для построения кривых ингибирования и расчета соответствующих параметров, включая минимальную степень ингибирования, максимальную степень ингибирования и IC50.
[000383] Результаты эксперимента
[000384] Результаты исследования показаны в таблице 4.
[000385] Таблица 4. Ингибирующий эффект соединений по настоящему описанию на пролиферацию клеток в клеточных линиях LNCaP
[000386] Заключение: соединения по настоящему изобретению проявляют превосходный ингибирующий эффект в отношении пролиферации клеток LNCaP рака предстательной железы человека.
[000387] Пример исследования 3: фармакодинамическое исследование подкожного ксенотрансплантата опухоли CB-17 SCID на модели рака предстательной железы человека LNCaP in vivo.
[000388] Культура клеток
[000389] Клетки LNCaP рака предстательной железы человека (ECACC-89110211) культивировали in vitro в монослое в культуральных условиях среды RPMI-1640 с 10% фетальной бычьей сывороткой, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина и культивировали в инкубаторе при 37°C, 5% CO2. Рутинное расщепление трипсином-EDTA проводили дважды в неделю для выполнения пассажа. Если плотность насыщения клеток составляла 80% - 90% и количество клеток достигало необходимого уровня, клетки собирали, подсчитывали и инокулировали.
[000390] Экспериментальные животные
[000391] Мыши CB-17 SCID в возрасте 6-8 недель, самцы весом 18-22 г.
[000392] Схема проведения эксперимента
[000393] 0,2 мл (1 × 107) клеток LNCaP (с матричным гелем, объемное соотношение 1:1) инокулировали подкожно в правую часть спины каждой мыши. Мышей подвергали хирургической кастрации когда средний объем опухоли достигал примерно 80 мм3, а группировку и введение начинали, когда средний объем опухоли достигал 166 мм3. Экспериментальная группировка животных и схема введения показаны в таблице 5.
[000394] Таблица 5. Экспериментальная группировка животных и схема введения
[000395] Примечание:
[000396] 1. N: Количество мышей на группу
[000397] 2. Объем введения: 10 мкл/г в зависимости от массы тела мыши. Если потеря массы превышает 15%, режим введения следует корректировать соответствующим образом.
[000398] 3. Интервал «Дважды в сутки» составлял 8 часов.
[000399] Измерение параметров опухоли и экспериментальные показатели
[000400] Диаметры опухолей измеряли дважды в неделю с помощью штангенциркуля. Формула расчета объема опухоли представляла собой: V = 0,5a × b2, где a и b представляют собой длинный и короткий диаметры опухоли соответственно.
[000401] Противоопухолевую эффективность соединений оценивали по TGI (%). TGI (%) отражал степень ингибирования роста опухоли. TGI (%) = [(1 - (средний объем опухоли в конце введения в группе с обработкой - средний объем опухоли в начале введения в данной группе с обработкой)) / (средний объем опухоли в конце обработки в группе контроля в виде среды-носителя - средний объем опухоли в начале обработки в группе контроля в виде среды-носителем)] × 100%.
[000402] Результаты эксперимента
[000403] Результаты исследования показаны в таблице 6.
[000404] Таблица 6. Противоопухолевый эффект соединений в модели ксенотрансплантата опухоли LNCaP
[000405] Заключение: соединения по настоящему изобретению проявляют значительный противоопухолевый эффект в модели ксенотрансплантата опухоли LNCaP рака предстательной железы человека.
[000406] Пример исследования 4: тест in vitro в отношении регуляции уровня белка AR в клетках LNCaP рака предстательной железы человека.
[000407] Экспериментальная цель: проверить эффект деградации соединений в отношении андрогенового рецептора AR в клетках LNCaP способом ICW.
[000408] Экспериментальный способ:
[000409] День 1
1. Среду для культивирования клеток, 0,025% трипсин и фосфатно-солевой буфер предварительно нагревали на водяной бане до 37°C.
2. Клетки, расщепленные трипсином, центрифугировали при 1000 об/мин в течение 5 минут. Супернатант удаляли, а затем клетки ресуспендировали в свежей культуральной среде.
3. Клетки инокулировали в покрытый COL планшет CellCarrier-384 при плотности 4 тыс./36 мкл/лунка.
4. 384-луночный планшет для клеток, инокулированный клетками, помещали в ультрачистый бокс на 10 минут.
5. Планшет для клеток культивировали в течение ночи в клеточном инкубаторе с CO2.
[000410] День 2
1. Соединение при концентрации 10 мМ разбавляли до 3 мМ (3 мкл + 7 мкл диметилсульфоксида) перед проведением последовательного разведения.
2. Соединение серийно разбавляли в 3 раза (4 мкл + 8 мкл) в планшете Echo с использованием Bravo.
3. Были установлены контрольные лунки ZPE и HPE, при этом контрольные лунки ZPE содержали диметилсульфоксид.
4. Планшет Echo центрифугировали при 1000 об/мин в течение 1 минуты, а затем разбавленное соединение переносили из планшета Echo в промежуточный планшет с использованием Echo в количестве 200 нл/лунку.
5. В промежуточный планшет добавляли среду для культивирования клеток с использованием Multidrop combi в количестве 20 мкл/лунка.
6. Промежуточный планшет встряхивали на горизонтальном шейкере в течение 1 минуты для смешивания соединения с культуральной средой.
7. Соединение переносили из планшета с промежуточными разведениями в планшет для клеток с использованием Bravo в количестве 4 мкл/лунку.
8. Планшет для клеток встряхивали на горизонтальном шейкере в течение 1 минуты и центрифугировали при 800 об/мин в течение 30 секунд.
9. Планшет для клеток возвращали в инкубатор с CO2 и культивировали в течение дополнительных 24 часов.
[000411] День 3
1. 4% параформальдегид извлекали из холодильника и приводили к комнатной температуре.
2. Планшет для клеток извлекали из инкубатора.
3. В планшет для клеток непосредственно добавляли 4% параформальдегид в количестве 40 мкл/лунку.
4. Планшет инкубировали при комнатной температуре в течение 20 минут.
5. Культуральную среду вручную стряхивали с планшета для клеток, помещали в центрифугу в перевернутом положении и центрифугировали при 800 об/мин в течение 7 секунд для полного удаления остатков культуральной среды.
6. В планшет с клетками добавляли 4% параформальдегид в количестве 25 мкл/лунка.
7. Планшет инкубировали при комнатной температуре в течение 30 минут.
8. Раствор параформальдегида вручную стряхивали с планшета для клеток, помещали в центрифугу в перевернутом положении и центрифугировали при 800 об/мин в течение 7 секунд.
9. Добавляли 0,1% тритион в количестве 25 мкл/лунка.
10. Планшет инкубировали при комнатной температуре в течение 10 минут.
11. 0,1% раствор тритиона вручную стряхивали с планшета для клеток, помещали в центрифугу в перевернутом положении и центрифугировали при 800 об/мин в течение 7 секунд.
12. Добавляли блокирующий буфер Intercept® (PBS) в количестве 25 мкл/лунка.
13. Планшет инкубировали при комнатной температуре в течение 1 часа.
14. Раствор блокирующего буфера Intercept® (PBS) вручную стряхивали с планшета для клеток, помещали в центрифугу в перевернутом положении и центрифугировали при 800 об/мин в течение 7 секунд.
15. Добавляли раствор первичного антитела в количестве 20 мкл/лунка. Планшет для клеток помещали в центрифугу и центрифугировали при 800 об/мин в течение 7 секунд.
16. Планшет для клеток герметизировали, а затем инкубировали в течение ночи при 4°C.
[000412] День 4
1. Раствор первичного антитела вручную стряхивали с планшета для клеток, помещали в центрифугу в перевернутом положении и центрифугировали при 800 об/мин в течение 7 секунд.
2. Планшет трижды промывали с помощью 110 мкл PBS.
3. Планшет для клеток помещали в центрифугу в перевернутом положении и центрифугировали при 800 об/мин в течение 7 секунд.
4. Добавляли раствор вторичного антитела и DAPI в количестве 20 мкл/лунка. Планшет для клеток помещали в центрифугу и центрифугировали при 800 об/мин в течение 7 секунд.
5. Планшет инкубировали при комнатной температуре в течение 1 часа.
6. Раствор вторичного антитела и DAPI вручную стряхивали с планшета для клеток, помещали в центрифугу в перевернутом положении и центрифугировали при 800 об/мин в течение 7 секунд.
7. Планшет для клеток трижды промывали с помощью 110 мкл PBS, при этом в планшете оставалось 30 мкл PBS.
8. Планшет для клеток помещали в центрифугу и центрифугировали при 800 об/мин в течение 7 секунд.
9. Планшет для клеток сканировали с использованием Operetta.
[000413] Результаты эксперимента
[000414] Результаты исследования показаны в таблице 4.
[000415] Таблица 7. Эффект деградации соединений в отношении андрогенового рецептора AR в клетках LNCaP
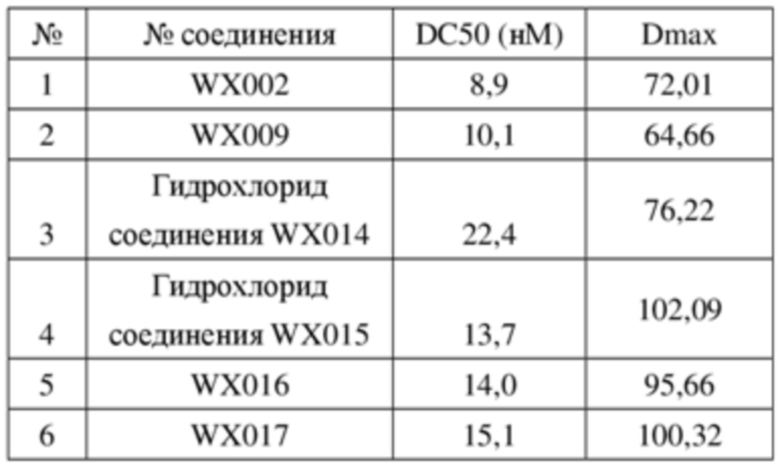
[000416] Заключение: соединения по настоящему изобретению демонстрируют надлежащую активность в отношении деградации белка AR.
[000417] Пример исследования 5: Внутриклеточный вестерн-анализ уровня экспрессии белка AR в клетках 293T AR F876L
[000418] Экспериментальная цель: в данном эксперименте эффекты соединений WX015, WX016 и WX017 на уровень экспрессии белка AR с точечной мутацией в клеточной линии 293T AR F876L определяли с помощью внутриклеточного вестерн-анализа для оценки эффекта деградации соединений на белки с точечной мутацией AR F876L.
[000419] Материалы для экспериментов:
[000420] 1. Клеточная линия и способ культивирования
[000421] Таблица 8. Клеточная линия и способ культивирования

[000422] 2. Культуральная среда и реагенты
[000423] Таблица 9. Культуральная среда и реагенты
[000424] 3. Приборы
[000426] 4. Антитела

[000428] Экспериментальный способ:
[000429] 1. Культура клеток:
[000430] Клеточную линию культивировали в инкубаторе при 37°C, 5% CO2. Для посева отбирали периодические пассажи и клетки в логарифмической фазе роста.
[000431] 2. Посев клеток:
1) Клетки окрашивали трипановым синим и подсчитывали жизнеспособные клетки.
2) Концентрацию клеток доводили до соответствующей концентрации с плотностью 20000/лунка.
3) 90 мкл клеточной суспензии на лунку добавляли в 96-луночный культуральный планшет, а в лунку холостого контроля добавляли бесклеточную культуральную среду.
4) Осуществляли культивирование культурального планшета в течение ночи в инкубаторе при 37°C, 5% CO2 и относительной влажности 100%.
[000432] 3. Подготовка планшета для хранения соединения и внутриклеточное вестерн-детектирование.
1) Подготовка планшета для хранения соединения в концентрации 400X: соединение серийно разбавляли с помощью DMSO от самой высокой концентрации до самой низкой концентрации.
2) Получение 10-кратного рабочего раствора соединения: в 96-луночный планшет с V-образным дном добавляли 78 мкл среды для культивирования клеток, при этом 2 мкл соединения отбирали пипеткой из планшета для хранения соединения в концентрации 400X и добавляли в среду для культивирования клеток в 96-луночном планшете. 2 мкл DMSO добавляли к контролю в виде среды-носителя и холостому контролю. После добавления соединения или DMSO проводили продувку пипеткой и тщательное перемешивание.
3) Введение: в планшет для культуры клеток добавляли 10 мкл 10-кратного рабочего раствора соединения. К контролю в виде среды-носителя и холостому контролю добавляли 10 мкл смеси DMSO-среда для культивирования клеток. Конечная концентрация DMSO составляла 0,25%.
4) 96-луночный планшет для клеток возвращали в инкубатор и культивировали в течение 48 часов.
5) Культуральную среду удаляли, а планшет один раз промывали с помощью PBS;
6) В планшет добавляли 8% раствор формалина (разбавленный в PBS) и инкубировали в течение ночи при 4°C.
7) 8% раствор формалина удаляли. В планшет добавляли 150 мкл на лунку блокирующего буфера Licor и блокировали на шейкере при комнатной температуре в течение 1,5 часов.
8) Блокирующий раствор выливали. В планшет добавляли 50 мкл на лунку первичного антитела, разведенного блокирующим буфером Licor (первичное антитело не добавляли в лунки с DMSO), инкубировали в течение ночи при 4°C, а затем пять раз промывали с помощью PBST (содержащим 0,1% Triton-X) каждый раз в течение 5 минут.
9) В планшет добавляли 50 мкл на лунку вторичного антитела, разведенного блокирующим буфером Licor, инкубировали при комнатной температуре в течение 1 часа, трижды промывали PBST (содержащим 0,1% Triton-X), дважды промывали с помощью ddH2O, а затем добавляли 100 мкл PBS на лунку для выявления.
10) Выявление: PBS выливали из луночного планшета, а флуоресцентный сигнал при 700 нм на лунку определяли с помощью двухцветного устройства формирования изображений ближнего инфракрасного диапазона Li-COR odyssey.
[000433] 4. Анализ данных
[000434] Относительную количественную оценку интенсивности плотности полос хемилюминесценции иммуноблота проводили с использованием программного обеспечения Image Studio вер. 5.2.
[000435] Результаты эксперимента
[000436] Результаты исследования показаны в таблице 12.
[000437] Таблица 12. Эффект деградации соединений в отношении андрогенового рецептора AR в клетках 293T AR F876L
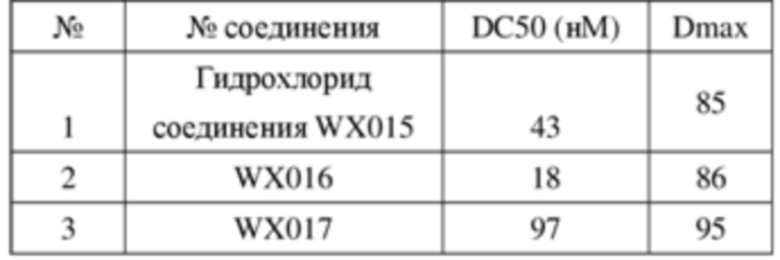
[000438] Заключение: соединения по настоящему изобретению характеризуются сильной способностью разрушать белок AR с точечной мутацией F876L.
[000439] Пример исследования 6: фармакокинетическая оценка соединений на мышах
[000440] Экспериментальная цель:
[000441] В данном исследовании самцов мышей CB-17 SCID выбирали в качестве экспериментальных животных для исследуемых соединений, при этом применяли способ LCMS/MS для количественного определения значений концентрации исследуемого соединения и референсного соединения в плазме крови, вводимых внутривенно или перорально мышам в разные моменты времени таким образом, чтобы оценить фармакокинетический профиль исследуемого лекарственного средства у мышей.
[000442] Материалы для экспериментов:
[000443] Мыши CB-17 SCID (самцы, 20-30 г, возраст 7-10 недель, Beijing Vital River или Shanghai SLAC).
[000444] Процедура эксперимента:
[000445] Осветленный раствор или суспензию исследуемого соединения вводили мышам через хвостовую вену (без голодания) или вводили мышам внутрижелудочно (без голодания). В случае проведения внутривенной инъекции кровь собирали посредством пункции яремной вены в момент 0 ч (до введения), 0,083 ч, 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 8 ч и 24 ч после введения и помещали в пробирку с антикоагулянтом с добавлением EDTA-K2. Смесь тщательно встряхивали с помощью вортекса, и перемешивали, и центрифугировали при 13000 об/мин в течение 10 минут при 4°C. В случае проведения перорального введения кровь собирали посредством пункции яремной вены в момент 0 ч (до введения), 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч после введения и помещали в пробирку с антикоагулянтом с добавлением EDTA-K2. Смесь тщательно встряхивали с помощью вортекса, и перемешивали, и центрифугировали при 13000 об/мин в течение 10 минут. Значения концентрации в плазме крови определяли с помощью метода LC-MS/MS и соответствующие фармакокинетические параметры рассчитывали с использованием фармакокинетического программного обеспечения WinNonlin™ версии 6.3 (Pharsight, Маунтин-Вью, штат Калифорния) с помощью линейного логарифмического метода трапеций в соответствии с некомпартментной моделью.
[000446] Результаты эксперимента: результаты исследования показаны в таблице 13.
[000447] Таблица 13. Фармакокинетические параметры соединений по настоящему изобретению у мышей
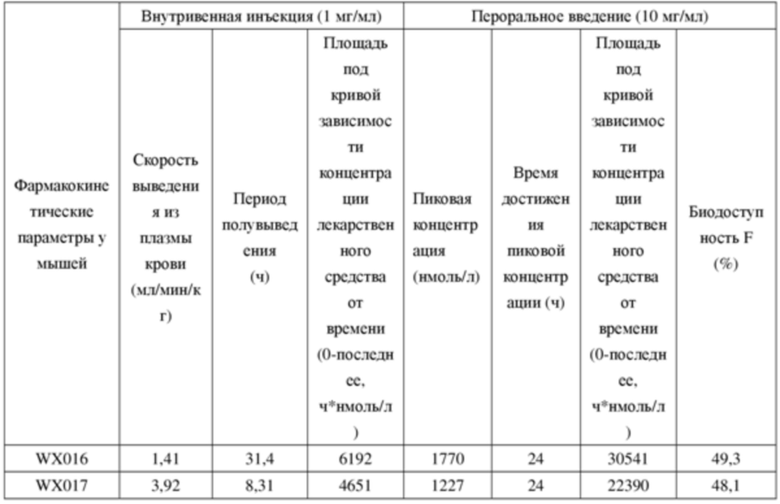
[000448] Заключение: соединения по настоящему изобретению демонстрируют надлежащую применимость в качестве лекарственных средств у мышей.
[000449] Пример исследования 7: фармакокинетическая оценка соединений на крысах
[000450] Экспериментальная цель:
[000451] В данном исследовании самцов крыс SD выбирали в качестве экспериментальных животных для исследуемых соединений, при этом применяли метод LCMS/MS для количественного определения значений концентрации исследуемого соединения и референсного соединения в плазме крови, вводимых внутривенно или перорально крысам в разные моменты времени таким образом, чтобы оценить фармакокинетический профиль исследуемого лекарственного средства у крыс.
[000452] Материалы для экспериментов:
[000453] Крысы SD (самцы, Beijing Vital River).
[000454] Процедура эксперимента:
[000455] Осветленный раствор или суспензию исследуемого соединения вводили крысам через хвостовую вену (без голодания) или вводили крысам внутрижелудочно (без голодания). В случае проведения внутривенной инъекции кровь собирали посредством пункции яремной вены в момент 0 ч (до введения), 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 24 ч и 48 ч после введения и помещали в пробирку с антикоагулянтом с добавлением гепарина натрия. Собранный образец крови помещали на лед и центрифугировали для отделения плазмы в течение 1 часа (условия центрифугирования: 6000 g, 3 минуты, 2-8°C). В случае проведения перорального введения кровь собирали посредством пункции яремной вены в момент 0 ч (до введения), 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 24 ч и 48 ч после введения и помещали в пробирку с антикоагулянтом с добавлением гепарина натрия. Собранный образец крови помещали на лед и центрифугировали для отделения плазмы в течение 1 часа (условия центрифугирования: 6000 g, 3 минуты, 2-8°C). Значения концентрации в плазме крови определяли с помощью метода LC-MS/MS и соответствующие фармакокинетические параметры рассчитывали с использованием фармакокинетического программного обеспечения WinNonlin™ версии 8.2.0 (Pharsight, Маунтин-Вью, штат Калифорния) с помощью линейного логарифмического метода трапеций в соответствии с некомпартментной моделью.
[000456] Результаты эксперимента: результаты исследования показаны в таблице 14.
[000457] Таблица 14. Фармакокинетические параметры соединений по настоящему изобретению у крыс
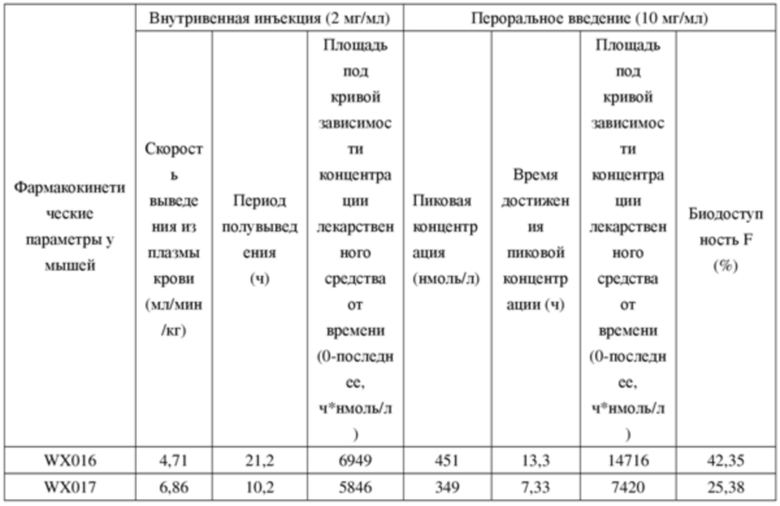
[000458] Заключение: соединения по настоящему изобретению демонстрируют надлежащую применимость в качестве лекарственных средств у крыс.
[000459] Пример исследования 8: фармакокинетическая оценка соединений на собаках породы бигль.
[000460] Экспериментальная цель:
[000461] В данном исследовании собак породы бигль выбирали в качестве экспериментальных животных для исследуемых соединений, при этом применяли метод LCMS/MS для количественного определения значений концентрации исследуемого соединения и референсного соединения в плазме крови, вводимых внутривенно или перорально собакам породы бигль в разные моменты времени таким образом, чтобы оценить фармакокинетический профиль исследуемого лекарственного средства у собак породы бигль.
[000462] Материалы для экспериментов:
[000463] Собаки породы бигль (самцы, Jiangsu Yadong Institute of Experimental Animals Co., Ltd.).
[000464] Процедура эксперимента:
[000465] Осветленный раствор или суспензию исследуемого соединения вводили собакам породы бигль через хвостовую вену (нормальное кормление) или вводили собакам породы бигль внутрижелудочно (нормальное кормление). В случае проведения внутривенной инъекции кровь собирали посредством пункции вены передней конечности в момент 0 ч (до введения), 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 24 ч и 48 ч после введения и помещали в пробирку с антикоагулянтом с добавлением EDTA-K2. Собранный образец крови помещали на лед и центрифугировали для отделения плазмы в течение 1 часа (условия центрифугирования: 6000 g, 3 минуты, 2-8°C). В случае проведения перорального введения кровь собирали посредством пункции вены передней конечности в момент 0 ч (до введения), 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 24 ч, 48 ч после введения и помещали в пробирку с антикоагулянтом с добавлением EDTA-K2. Собранный образец крови помещали на лед и центрифугировали для отделения плазмы в течение 1 часа (условия центрифугирования: 6000 g, 3 минуты, 2-8°C). Значения концентрации в плазме крови определяли с помощью метода LC-MS/MS и соответствующие фармакокинетические параметры рассчитывали с использованием фармакокинетического программного обеспечения WinNonlin™ версии 8.2.0 (Pharsight, Маунтин-Вью, штат Калифорния) с помощью линейного логарифмического метода трапеций в соответствии с некомпартментной моделью.
[000466] Результаты эксперимента: результаты исследования показаны в таблице 15.
[000467] Таблица 15. Фармакокинетические параметры соединений по настоящему изобретению у собак породы бигль
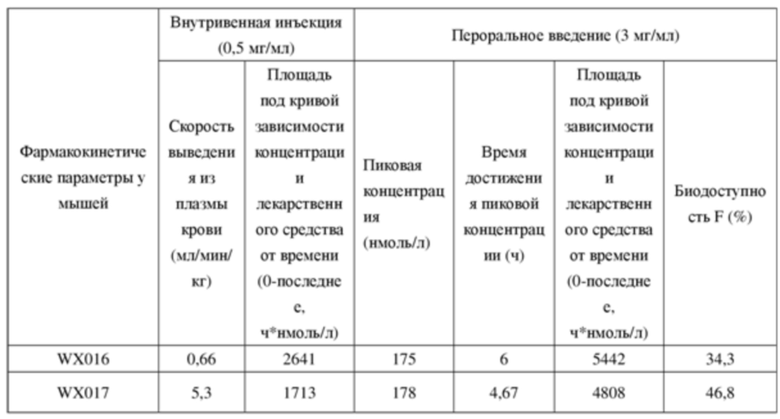
[000468] Заключение: соединения по настоящему изобретению демонстрируют хорошую применимость в качестве лекарственных средств у собак породы бигль.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ДЕЙСТВУЮЩИЕ НА БЕЛКИ CRBN | 2019 |
|
RU2801068C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
| Тиазололактамные соединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2805569C1 |
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ВЫПОЛНЯЮЩЕЕ ФУНКЦИИ ИНГИБИТОРА WEE1, И ВАРИАНТЫ ЕГО ПРИМЕНЕНИЯ | 2018 |
|
RU2783243C2 |
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
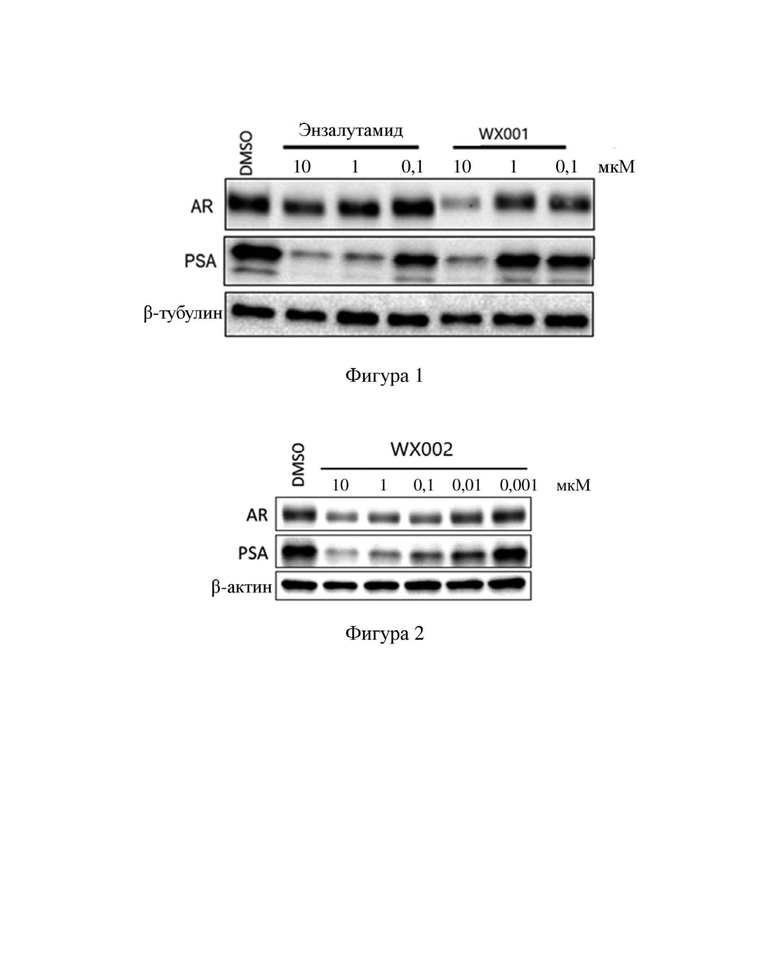
Изобретение относится к соединению формулы (II) или его фармацевтически приемлемой соли, которые являются модуляторами направленного убиквитирования и деградации андрогенового рецептора (AR) и могут найти применение для лечения рака предстательной железы. В формуле (II) PTM выбран из указанных ниже фрагментов; L1 выбран из -(CH2)n- и каждый CH2 необязательно замещен R1; R1 выбран из C3-7циклоалкила, 6-членного гетероциклоалкила, -NRa-, -CRbRc-, -CH2CH2O-, -NHC(=O)- и -C(=O)NH-; Ra выбран из H и C1-3алкила; каждый из Rb и Rc независимо выбран из H, D и F; E1 выбран из одинарной связи и O; кольцо A отсутствует; или кольцо A выбрано из фенила и 5-членного гетероарила; n выбран из 1, 2, 3, 4, 5 и 6; 6-членный гетероциклоалкил и 5-членный гетероарил содержат 1 или 2 гетероатома, независимо выбранных из -O- и N соответственно; при условии, что исключен ряд соединений. Изобретение относится также к конкретным соединениям и к применению предлагаемых соединений в изготовлении лекарственного средства для лечения рака предстательной железы. 3 н. и 7 з.п. ф-лы, 2 ил., 15 табл., 17 пр.
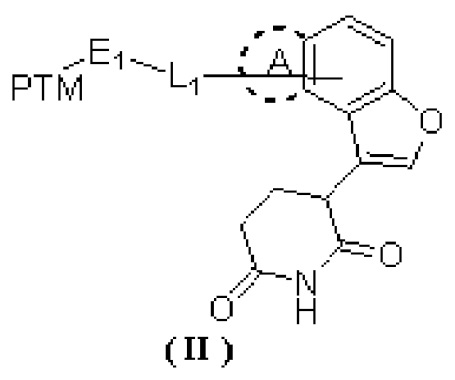
PTM:  ,
,
 и
и
1. Соединение формулы (II) или его фармацевтически приемлемая соль
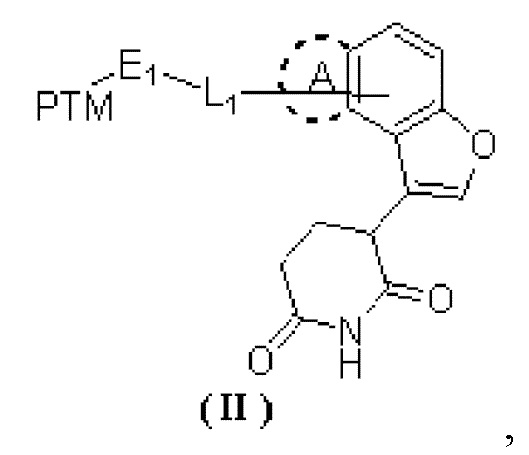
где PTM выбран из
L1 выбран из -(CH2)n- и каждый CH2 необязательно замещен R1;
R1 выбран из C3-7циклоалкила, 6-членного гетероциклоалкила, -NRa-, -CRbRc-, -CH2CH2O-, -NHC(=O)- и -C(=O)NH-;
Ra выбран из H и C1-3алкила;
каждый из Rb и Rc независимо выбран из H, D и F;
E1 выбран из одинарной связи и O;
кольцо A отсутствует;
или кольцо A выбрано из фенила и 5-членного гетероарила;
n выбран из 1, 2, 3, 4, 5 и 6;
6-членный гетероциклоалкил и 5-членный гетероарил содержат 1 или 2 гетероатома, независимо выбранных из -O- и N соответственно;
при условии, что соединение не выбрано из:
2. Соединение или его фармацевтически приемлемая соль по п. 1, где Ra выбран из H и CH3.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 выбран из
 -NH-, -N(CH3)-, -CH2CH2O-, -NHC(=O)- и -C(=O)NH-.
-NH-, -N(CH3)-, -CH2CH2O-, -NHC(=O)- и -C(=O)NH-.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где L1 выбран из 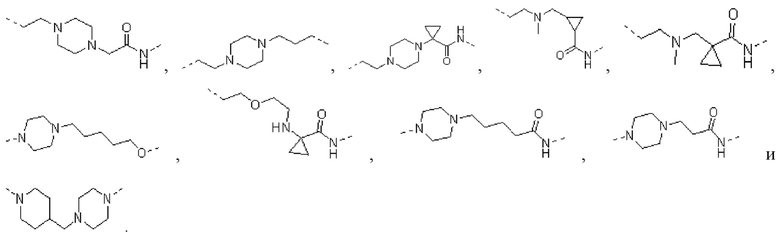
5. Соединение или его фармацевтически приемлемая соль по п. 1, где структурный фрагмент -E1-L1- выбран из
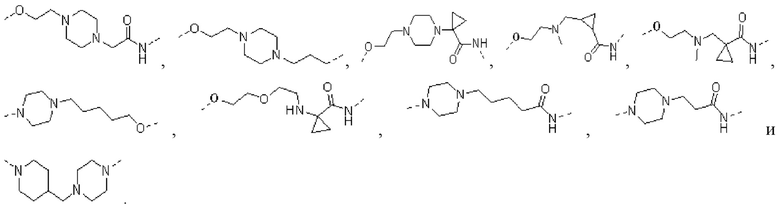
6. Соединение или его фармацевтически приемлемая соль по п. 1, где структурный фрагмент  выбран из
выбран из 
7. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-6, где соединение выбрано из структур, представленных формулами (I-1), (I-2), (II-1), (II-2), (III-1) и (IV-1),
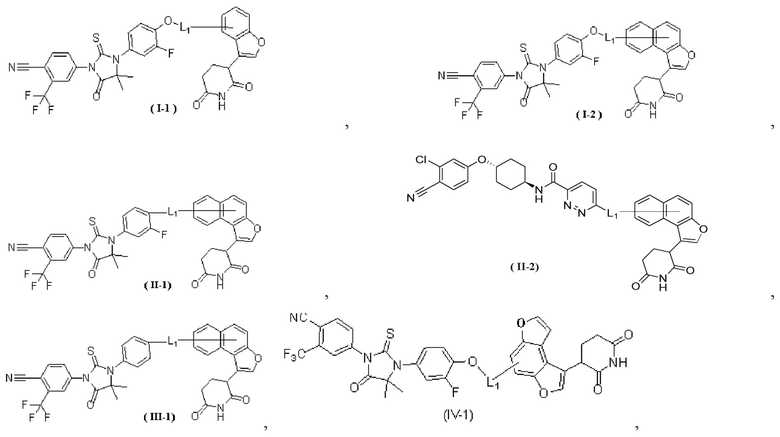
где L1 является таким, как определено в любом из пп. 1-6.
8. Соединение, представленное следующими формулами, или его фармацевтически приемлемая соль, которые выбраны из:
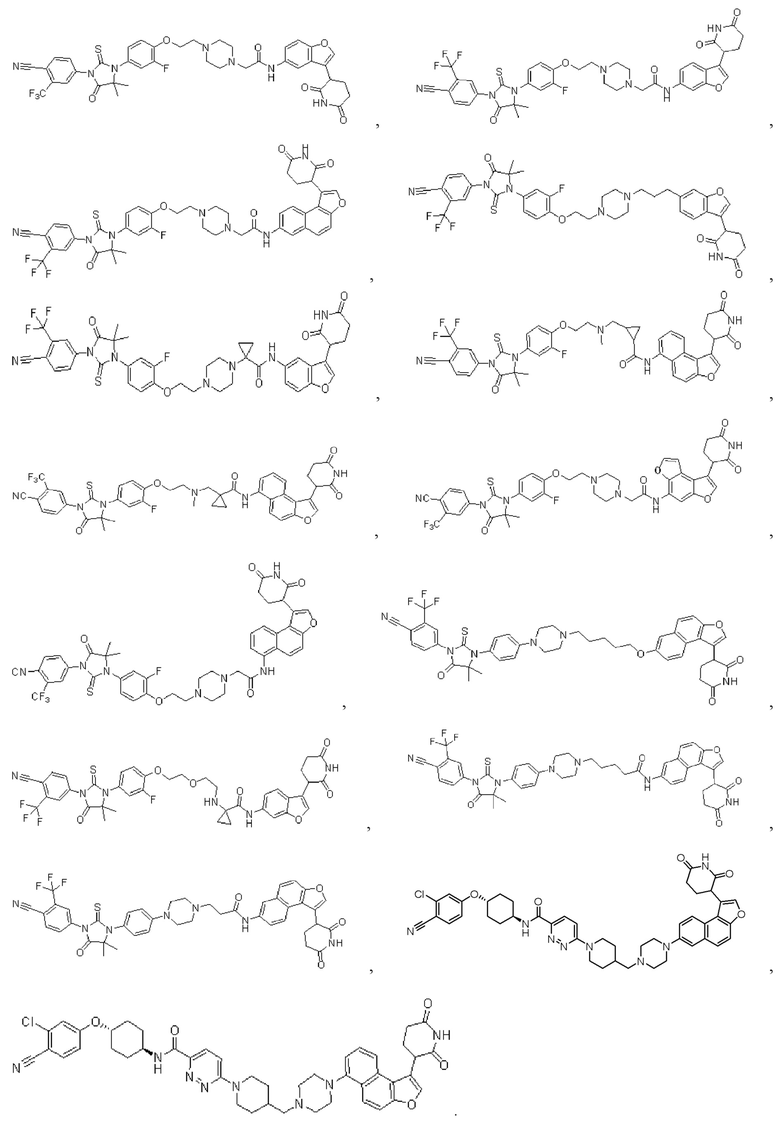
9. Соединение или его фармацевтически приемлемая соль по п. 8, которые выбраны из:
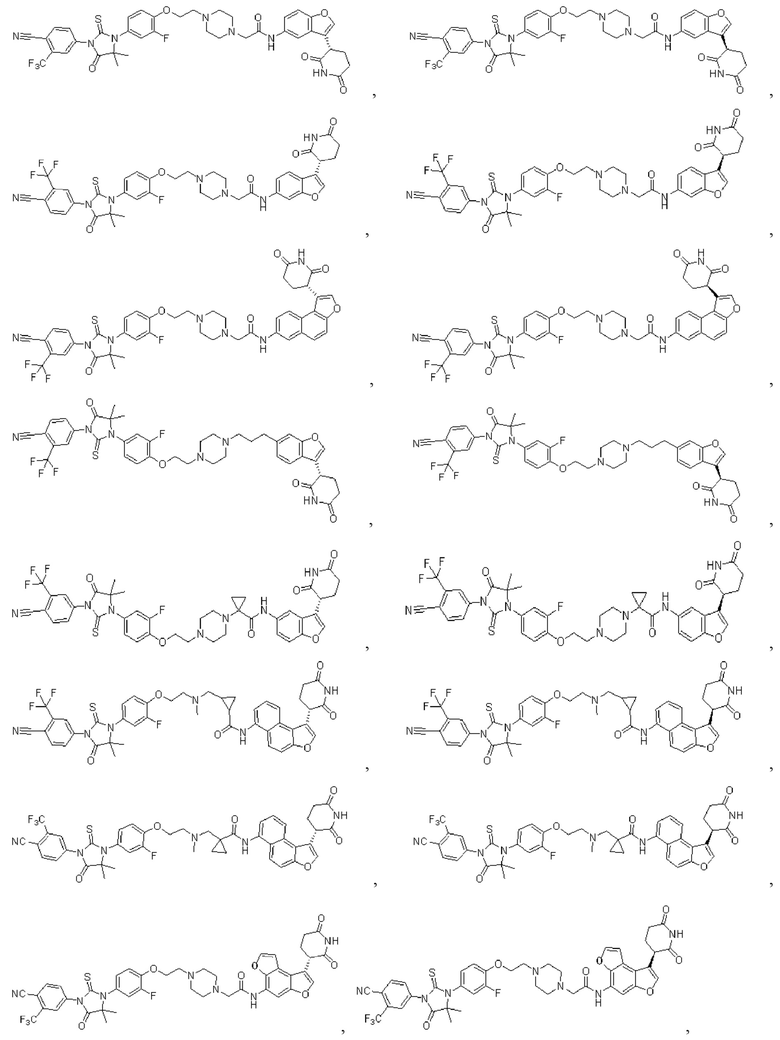
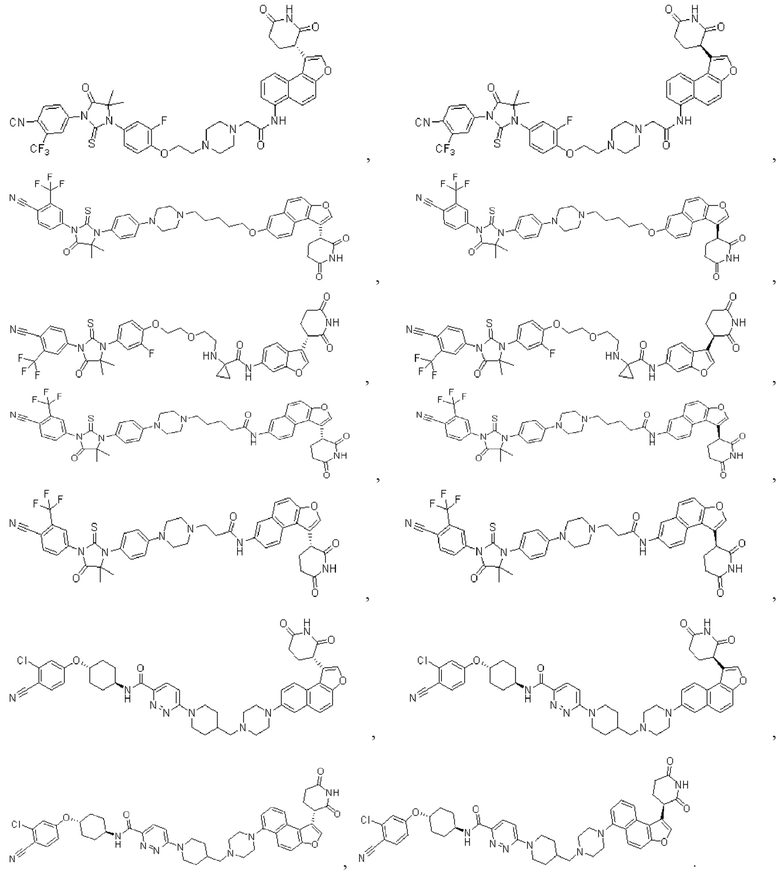
10. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-9 в изготовлении лекарственного средства для лечения рака предстательной железы.
| Способ восстановления спиралей из вольфрамовой проволоки для электрических ламп накаливания, наполненных газом | 1924 |
|
SU2020A1 |
| Способ восстановления спиралей из вольфрамовой проволоки для электрических ламп накаливания, наполненных газом | 1924 |
|
SU2020A1 |
| Способ регенерирования сульфо-кислот, употребленных при гидролизе жиров | 1924 |
|
SU2021A1 |
| RU 2019113229 A, 02.11.2020. | |||

