Ссылка на родственные заявки
По настоящей заявке испрашивается приоритет в соответствии с китайской заявкой на выдачу патента №201710867197.2, поданной 22 сентября 2017.
Область техники, к которой относится настоящее изобретение
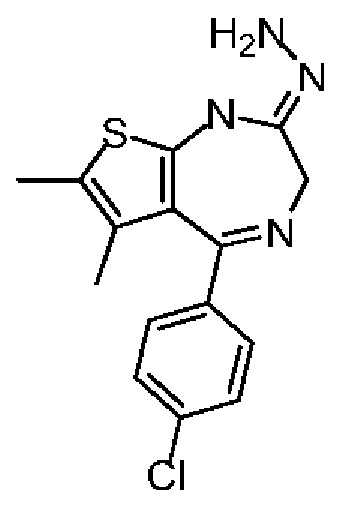
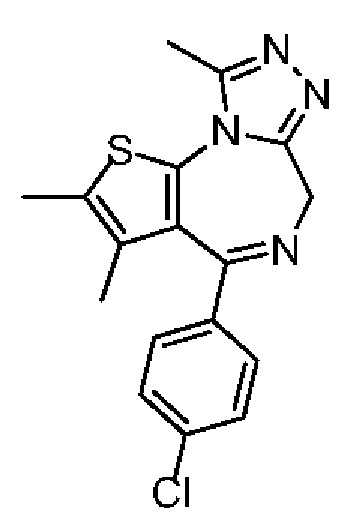
Настоящее изобретение относится к классу тиенодиазепиновых производных и их применению при получении лекарственного средства для лечения заболеваний, связанных с бромодоменовыми BET ингибиторами. В частности, настоящее изобретение относится к соединениям, представленным формулами (I) и (II), а также их фармацевтически приемлемым солям.
Предшествующий уровень техники настоящего изобретения
Распознавание ацетилирования лизина в гистоне является ключевым этапом в эпигенетической регуляции, в которой участвует ацетилирование гистонов. Ацетилированный лизин в гистоне может специфически распознаваться доменом бромодоменов (BRD) с рекрутингом тем самым регуляторных факторов хроматина в специфические области и с координацией регуляции экспрессии генов. Домен BRD, действующий на семейство бромодоменовых и экстратерминальных (BET) белков, может регулировать экспрессию ключевых онкогенов c-MYC и антиапоптотических белков. Исследование специфических ингибиторов, нацеленных на ВЕТ-бромодоменные белки, стало горячей точкой в исследованиях противораковых и противовоспалительных лекарственных средств, нацеленных на эпигенетические регуляторные механизмы. Семейство бромодоменов BET включает в себя четыре белка с тандемными бромодоменами: BRD2, BRD3, BRD4 и BRDT.
Краткое описание настоящего изобретения
Настоящее изобретение относится к соединению, представленному формулой (I) или (II), его изомеру или фармацевтически приемлемой соли
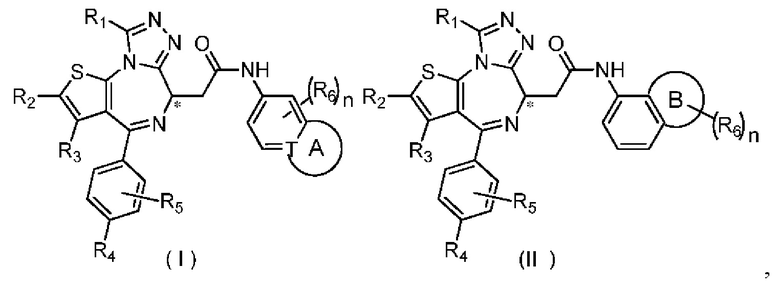
где
Т выбран из группы, состоящей из СН и N;
R1 выбран из группы, состоящей из C1-3алкила и C1-3алкоксила, оба из которых необязательно замещены 1, 2 или 3 R группой(ами);
R2, R3 и R4 отдельно и независимо выбраны из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN или отдельно и независимо выбраны из группы, состоящей из C1-6 алкила и C1-6 гетероалкила, оба из которых необязательно замещены 1, 2 или 3 R группой(ами);
R5 представляет собой Н или C1-3алкил, который необязательно замещен 1, 2 или 3 R группой(ами);
R6 отдельно и независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN, или отдельно и независимо выбраны из группы, состоящей из C1-6 алкила и C1-6 гетероалкила, оба из которых необязательно замещены 1, 2 или 3 R группой(ами); или две R6 группы, присоединенные к тому же атому углерода, образуют -С(=O) вместе с атомом углерода, присоединенным к ней;
кольцо А выбрано из группы, состоящей из С3-7 циклоалкила, 5-12-членного гетероциклоалкила и 5-6-членного гетероарила;
кольцо В выбрано из группы, состоящей из 4-7-членного гетероциклоалкила;
и структурная единица 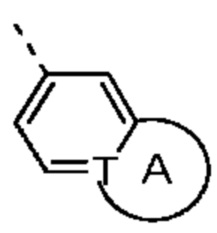 не выбрана из группы, состоящей из
не выбрана из группы, состоящей из 
n выбран из группы, состоящей из 0, 1, 2, 3, 4, 5 и 6;
R отдельно и независимо выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2 и CN, или выбран из группы, состоящей из C1-6 алкила и C1-6 гетероалкила, оба из которых необязательно замещены 1, 2 или 3 R' группой(ами);
R' отдельно и независимо выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2, CN и Me;
атом углерода, помеченный «*», представляет собой хиральный атом углерода, который присутствует в форме простого (R) или (S) энантиомера или в форме, обогащенной одним из двух энантиомеров;
термин «гетеро» в C1-6 гетероалкиле, 5-12-членном гетероциклоалкиле, 5-6-членном гетероариле и 4-7-членном гетероциклоалкиле выбран из группы, состоящей из N, -О-, -S-, -NH-, -C(=O)NH-, -С(=O)-, -С(=O)O-, -S(=O)2-, -S(=O)- и -C(=O)S-;
число вышеуказанного гетероатома или группы гетероатомов отдельно и независимо выбран из группы, состоящей из 1, 2, 3 и 4.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2 и CN, или выбран из группы, состоящей из C1-3алкила и C1-3алкокси, оба из которых необязательно замещены 1, 2 или 3 R' группой(ами), а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2 и CN, или выбран из группы, состоящей из Me, Et и 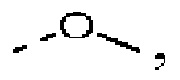 все из которых необязательно замещены 1, 2 или 3 R' группой(ами), а другие переменные определены в настоящем изобретении.
все из которых необязательно замещены 1, 2 или 3 R' группой(ами), а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2, CN, Me, CF3, Et и а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R1 выбран из группы, состоящей из Me, Et и все из которых необязательно замещены 1, 2 или 3 R группой(ами), а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R1 выбран из группы, состоящей из Me, Et, CF3 и а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R2, R3 и R4 отдельно и независимо выбраны из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN, или отдельно и независимо выбраны из группы, состоящей из C1-3алкила и C1-3алкокси, оба из которых необязательно замещены 1, 2 или 3 R группой(ами), а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R2, R3 и R4 отдельно и независимо выбраны из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, Me и а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R5 выбран из группы, состоящей из Н и Me, а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R6 отдельно и независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN, или отдельно и независимо выбраны из группы, состоящей из C1-3алкила и C1-3 гетероалкила, оба из которых необязательно замещены 1, 2 или 3 R группой(ами), а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R6 отдельно и независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN, или отдельно и независимо выбраны из группы, состоящей из Me, Et, 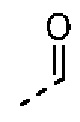 и
и 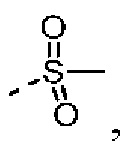 все из которых необязательно замещены 1, 2 или 3 R группой(ами), а другие переменные определены в настоящем изобретении.
все из которых необязательно замещены 1, 2 или 3 R группой(ами), а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутый R6 отдельно и независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, Me, Et, 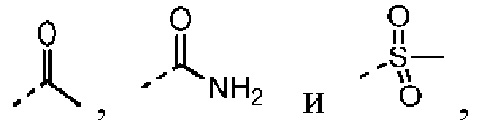 а другие переменные определены в настоящем изобретении.
а другие переменные определены в настоящем изобретении.
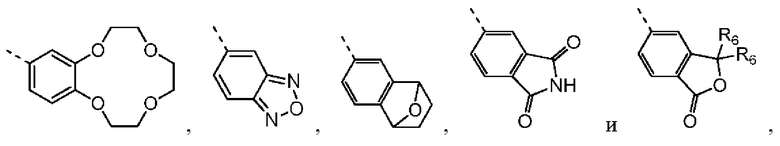
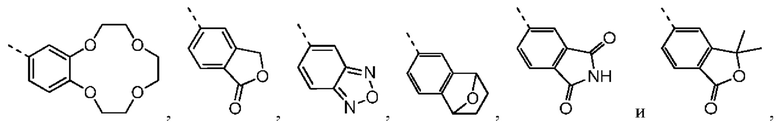
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутое кольцо А выбрано из группы, состоящей из C4-6циклоалкила, пирролидин-2-онила, пиримидин-4(3H)-онила, 5-азаспиро[2.4]гептан-4-онила, 4-азаспиро[2.4]гептан-5-онила, тетрагидротиофен-1,1-диоксидной группы, тетрагидротиофен-1-оксидной группы, тетрагидрофуранила, пирролидинила, дигидротиофен-2(3H)-онила, 2-оксаспиро[3.4]октила, дигидрофуран-2(3H)-онила, 1,4,7,10-тетраоксациклододецила, 1,2,5-оксадиазолила, 7-оксабицикло-[2.2.1]гептана, пирролидин-2,5-диона, 5,5-диметилдигидрофуран-2(3Н)-онила, а другие переменные определены в настоящем изобретении.
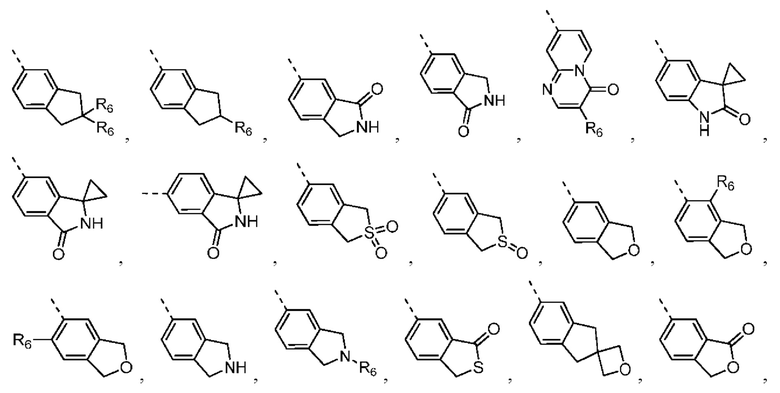
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутая структурная единица 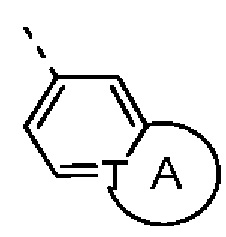 выбрана из группы, состоящей из
выбрана из группы, состоящей из
 а другие переменные определены в настоящем изобретении.
а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутая структурная единица 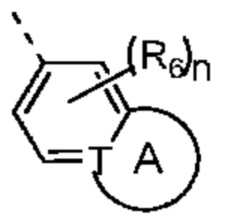 выбрана из группы, состоящей из
выбрана из группы, состоящей из
 а другие переменные определены в настоящем изобретении.
а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутая структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из
 а другие переменные определены в настоящем изобретении.
а другие переменные определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеупомянутая структурная единица 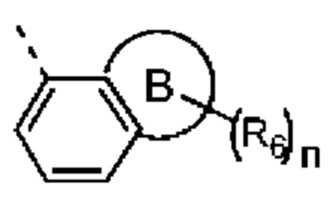 выбрана из группы, состоящей из
выбрана из группы, состоящей из  а другие переменные определены в настоящем изобретении. Настоящее изобретение также включает в себя некоторые варианты осуществления, полученные из любой комбинации вышеупомянутых переменных.
а другие переменные определены в настоящем изобретении. Настоящее изобретение также включает в себя некоторые варианты осуществления, полученные из любой комбинации вышеупомянутых переменных.
Согласно некоторым вариантам осуществления настоящего изобретения представлено вышеупомянутое соединение, его изомер или фармацевтически приемлемая соль, которые выбраны из группы, состоящей из
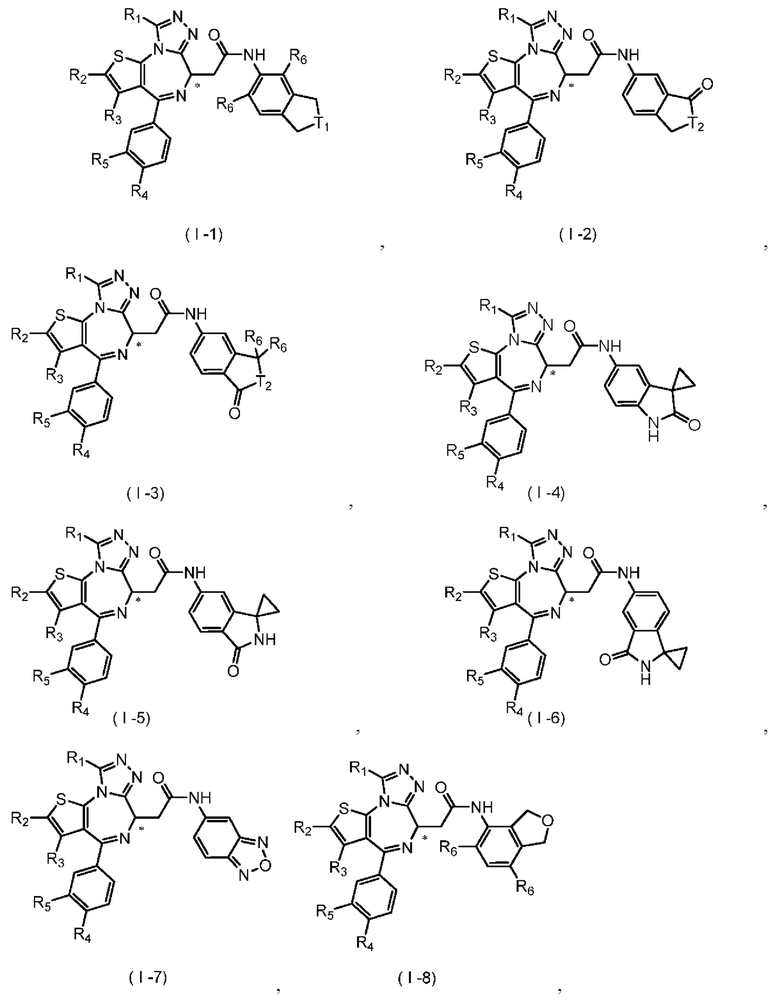
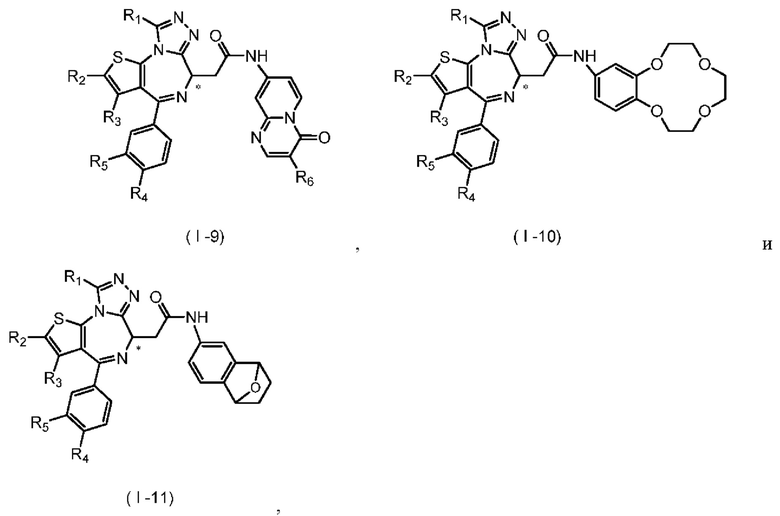
где
T1 выбран из группы, состоящей из -S(=O)-, -S(=O)2-, -N(R6)-, -О-, -C(R6)(R6)- и 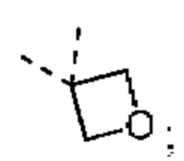
T2 отдельно и независимо выбран из группы, состоящей из -NH-, -О- и -S-; R1-R6 определены в настоящем изобретении;
атом углерода, помеченный «*», представляет собой хиральный атом углерода, который присутствует в форме простого (R) или (S) энантиомера или в форме, обогащенной одним из двух энантиомеров.
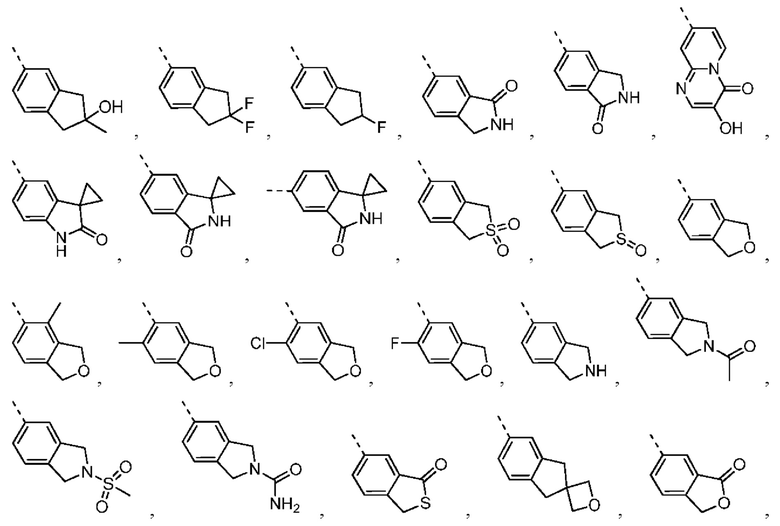
Настоящее изобретение также относится к соединению, его изомеру или фармацевтически приемлемой соли, которые выбраны из группы, состоящей из
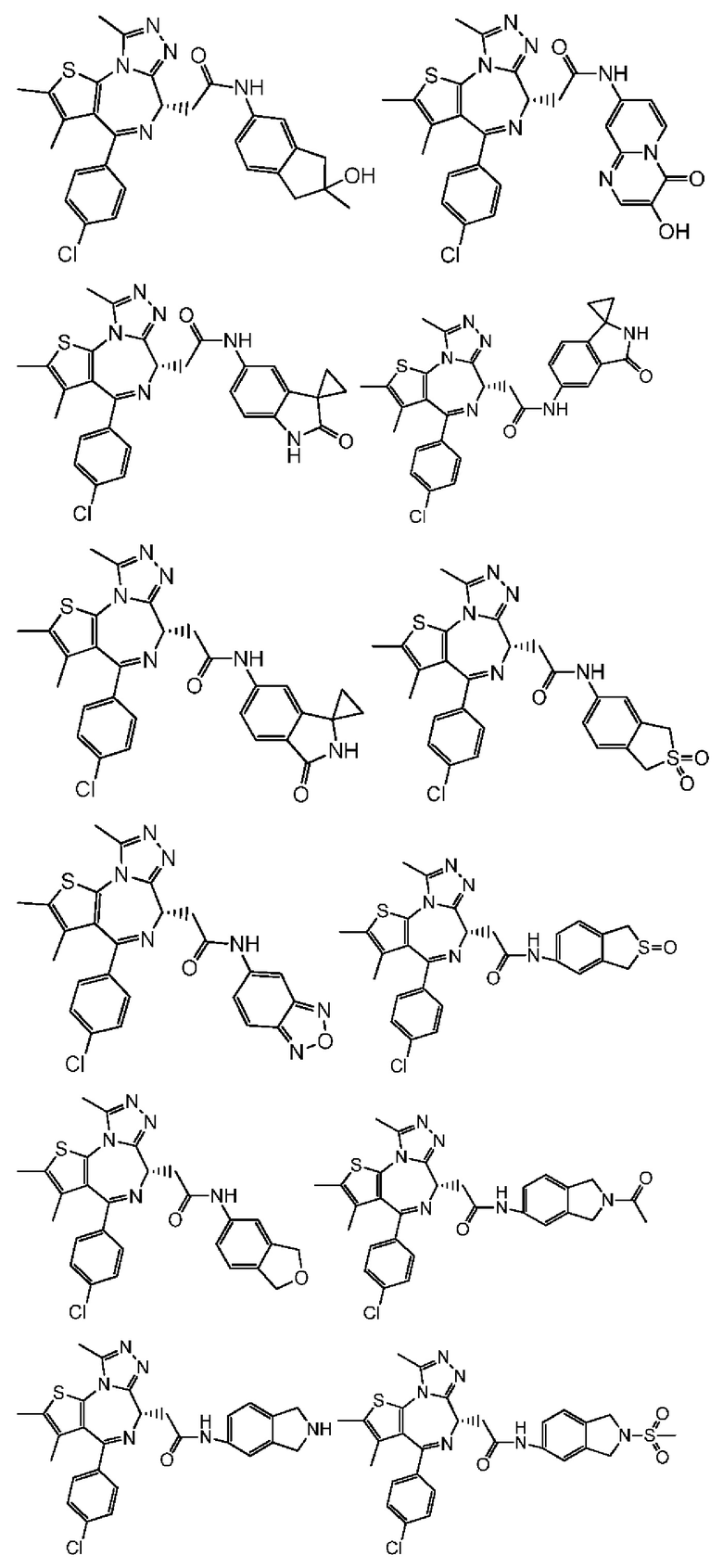
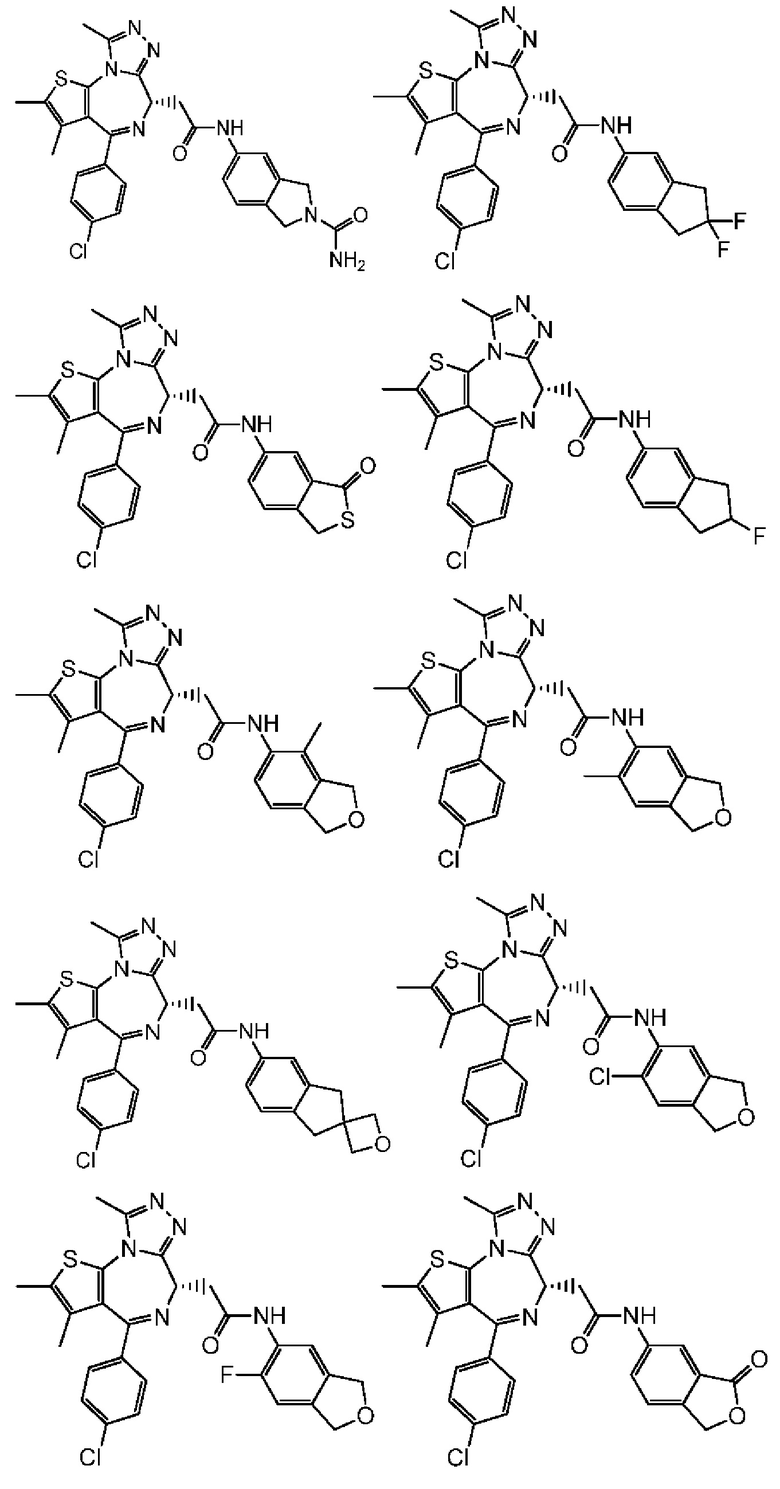
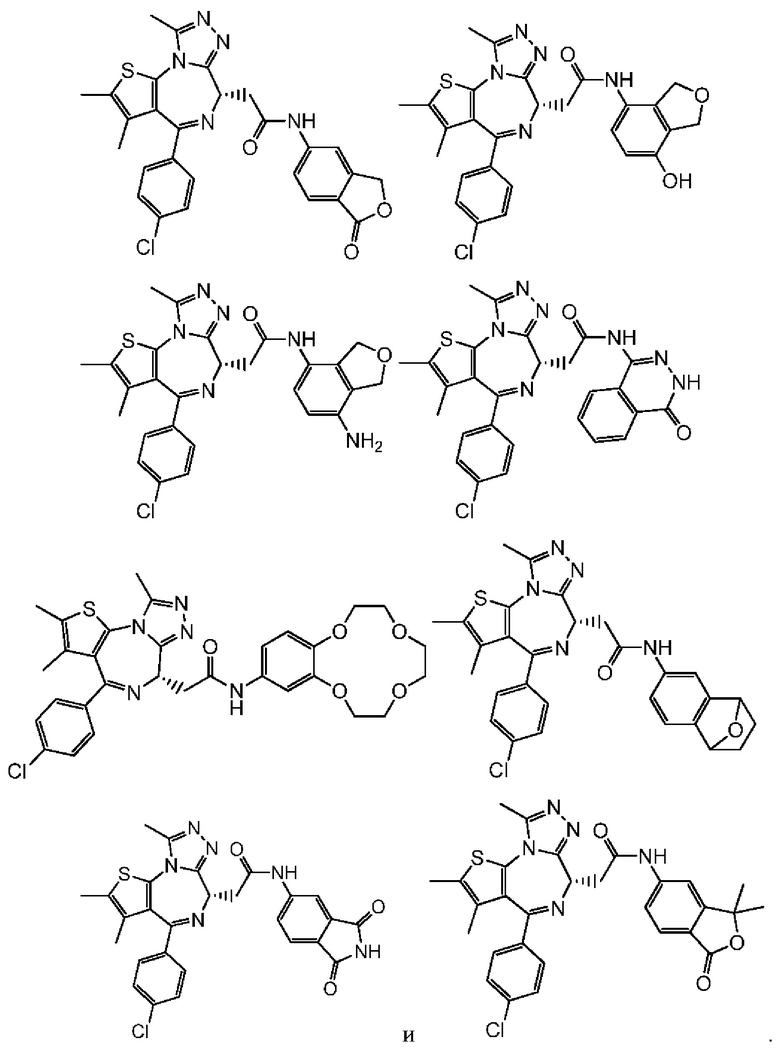
Согласно некоторым вариантам осуществления настоящего изобретения представлено соединение, его изомер или фармацевтически приемлемая соль, которые выбраны из группы, состоящей из
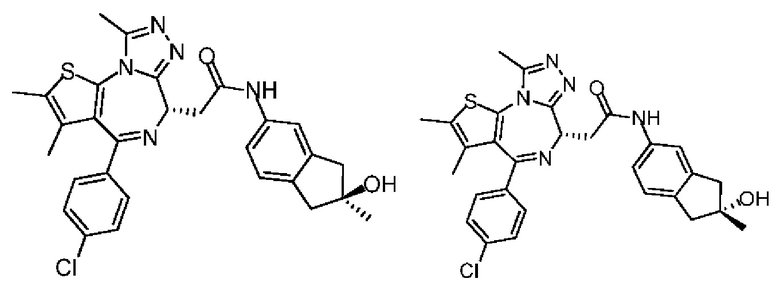
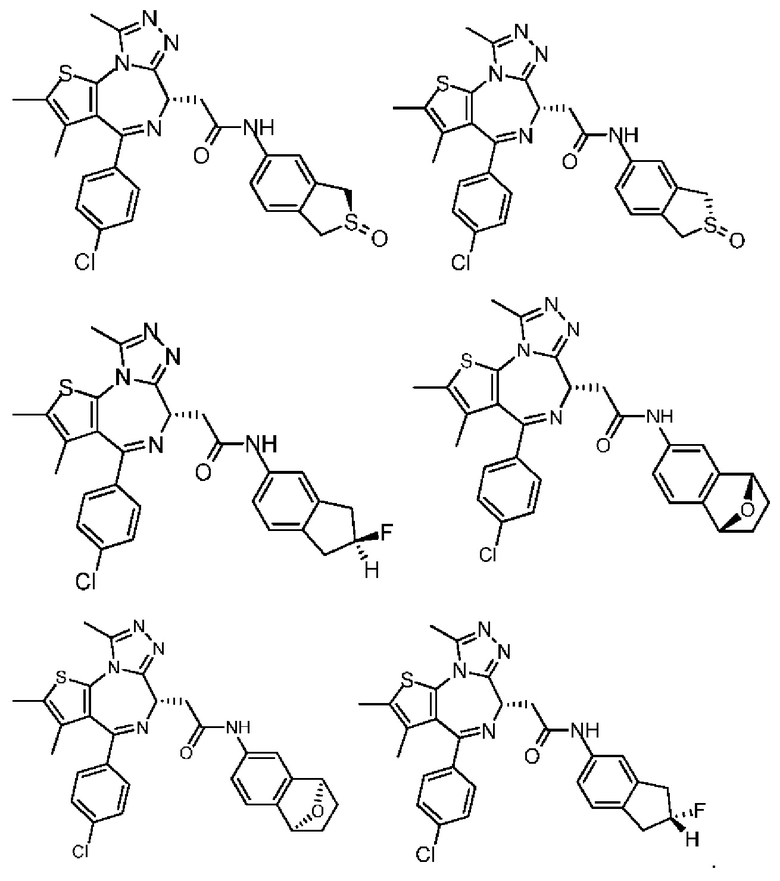
Настоящее изобретение также относится к фармацевтической композиции, которая содержит терапевтически эффективное количество вышеуказанного соединения или его фармацевтически приемлемой соли в качестве активного ингредиента и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к применению вышеуказанного соединения, его изомера или фармацевтически приемлемой соли или вышеуказанной композиции при изготовлении связанного с бромодоменовым BET ингибитором лекарственного средства.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанное связанное с бромодоменовым BET ингибитором лекарственное средство представляет собой противоопухолевое лекарственное средство.
Определение и объяснение
Если не отмечено иное, предусмотрено, что следующие термины и фразы, используемые в настоящем описании, характеризуются следующими значениями. Конкретный термин или фраза не должны рассматриваться как неопределенные или неясные без особого определения, а их следует понимать в их оригинальном значении. Если в настоящем описании встречается торговое наименование, предусмотрено, что оно относится к своему соответствующему коммерческому продукту или его активному ингредиенту. Используемый в настоящем описании термин «фармацевтически приемлемый» относится к таким соединениям, веществам, композициям и/или лекарственным формам, которые в пределах объема тщательной медицинской оценки являются подходящими для применения в контакте с тканями людей и животных без неспецифической токсичности, раздражения, аллергической реакции или других проблем или осложнений и в соответствии с подходящим соотношением польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, полученной из соединения, содержащего конкретный заместитель, который встречается в настоящем изобретении, и относительно не токсичной кислоты или основания. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, основно-аддитивная соль может быть получена приведением в контакт достаточного количества основания с нейтральной формой такого соединения в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые основно-аддитивные соли включают в себя соли натрия, калия, кальция, аммония, органического амина или магния или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, кислотно-аддитивная соль может быть получена приведением в контакт достаточного количества кислоты с нейтральной формой такого соединения в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают в себя соли неорганических кислот, включая, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, гидрокарбонат, фосфорную кислоту, вторичный кислый фосфат, первичный кислый фосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфорную кислоту и т.п.; соли органических кислот, включая, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, пара-толуолсульфоновую кислоту, лимонную кислоту, виннокаменную кислоту, метансульфоновую кислоту и т.п.; соли аминокислот (такие как аргинин); и соли органических кислот, таких как глюкуроновая кислота. Определенные конкретные соединения по настоящему изобретению содержит как основные, так и кислотные функциональные группы, и, таким образом, могут быть превращены в их любые основные или кислотные аддитивные соли.
Фармацевтически приемлемая соль настоящего изобретения может быть синтезирована из исходного соединения, содержащего кислотную группу или основную группу, традиционным химическим способом. Как правило, такие соли получали путем осуществления взаимодействия таких соединений в форме свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или органическим растворителе или их смеси.
Соединения по настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. В настоящем изобретении рассмотрены все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-пары энантиомеров, (R)- и (S)-энантиомеры, дистереоизомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и их другие смеси, такие как энантиомерно или диастереоизомерно обогащенные смеси, все из которых подпадали в пределы объема настоящего изобретения. Дополнительные асимметрические атомы углерода могут присутствовать в таком заместителе, как алкильная группа. Все такие изомеры и их смеси включены в объем настоящего изобретения.
Если не отмечено иное, термины «энантиомеры» или «оптические изомеры» относятся к стереоизомерам в отношении зеркального отражения друг к другу.
Если не отмечено иное, термин «цис-транс-изомер» или «геометрический изомер» образуется путем невозможности двойных связей или простых связей образующих кольцо атомов углерода свободно вращаться.
Если не отмечено иное, термин «диастереоизомер» относится к стереоизомеру, для которого каждая из молекул содержит два или более хиральных центров и молекулы находятся в отношении не зеркального отражения друг к другу.
Если не отмечено иное, «(D)» или «(+)» означает декстроротацию, «(L)» или «(-)» означает левостороннее вращение и «(DL)» или «(±)» означает рацемический.
Если не отмечено иное, абсолютная конфигурация стереоцентра выражена при помощи связи в виде жирной клинообразной линии  и при помощи связи в виде пунктирной клинообразной линии
и при помощи связи в виде пунктирной клинообразной линии  относительная конфигурация стереоцентра выражена при помощи связи в виде жирной прямой линии
относительная конфигурация стереоцентра выражена при помощи связи в виде жирной прямой линии 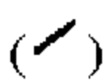 и при помощи связи в виде пунктирной прямой линии
и при помощи связи в виде пунктирной прямой линии  связь в виде жирной клинообразной линии
связь в виде жирной клинообразной линии 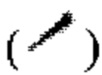 и/или связь в виде пунктирной клинообразной линии
и/или связь в виде пунктирной клинообразной линии  выражены при помощи волнистой линии
выражены при помощи волнистой линии  или связь в виде жирной прямой линии
или связь в виде жирной прямой линии 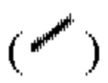 и/или связь в виде пунктирной прямой линии
и/или связь в виде пунктирной прямой линии  выражены при помощи волнистой линии
выражены при помощи волнистой линии 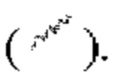
Соединения по настоящему изобретению могут существовать в конкретных формах. Если не отмечено иное, термин «таутомер» или «таутомерная форма» означает, что при комнатной температуре изомеры, содержащие разные функциональные группы, находятся в динамическом равновесии и могут быть быстро превращены друг в друга. Если таутомеры возможны (например, в растворе), может быть достигнуто химическое равновесие таутомеров. Например, протонные таутомеры (также известные как прототропные таутомеры) включают в себя взаимные превращения путем миграции протона, такие как кето-енольная изомеризация и имин-енаминовая изомеризация. Валентные таутомеры включают в себя рекомбинацию некоторых связывающих электронов для взаимного превращения. Среди прочих, конкретный пример кето-енольной таутомеризации представляет собой взаимное превращение между двумя таутомерами пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
Если не отмечено иное, термин «обогащенный в изомере», «изомерно обогащенный», «обогащенный в энантиомере» или «энантиомерно обогащенный» относится к содержимому изомера или энантиомера, что менее чем 100%, и содержимое изомера или энантиомера составляет более чем или равно 60%, или более чем или равно 70%, или более чем или равно 80%, или более чем или равно 90%, или более чем или равно 95%, или более чем или равно 96%, или более чем или равно 97%, или более чем или равно 98%, или более чем или равно 99%, или более чем или равно 99,5%, или более чем или равно 99,6%, или более чем или равно 99,7%, или более чем или равно 99,8%, или более чем или равно 99,9%.
Если не отмечено иное, термин «изомерный избыток» или «энантиомерный избыток» относится к разнице между относительным процентным содержанием двух изомеров или энантиомеров. Например, если содержимое одного изомера или энантиомера составляет 90%, а содержимое другого изомера или энантиомера составляет 10%, изомерный или энантиомерный избыток (э. и. значение) составляет 80%.
Оптически активные (R)- и (S)-изомеры, а также D и L изомеры, могут быть получены при помощи хирального синтеза, или хиральных реагентов, или другими традиционными методиками. Если необходим энантиомер соединения по настоящему изобретению, он может быть получен асимметричным синтезом или дериватизацией при помощи хирального вспомогательного элемента, при этом полученную диастереомерную смесь разделяли и вспомогательную группу отщепляли с получением чистого требуемого энантиомера. Альтернативно, если молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), диастереомерная соль образуется с соответствующей оптически активной кислотой или основанием, а затем диастереомерное расщепление проводили традиционным способом, хорошо известным из области техники, а затем чистый энантиомер восстанавливали и получали. Кроме того, разделение энантиомеров и диастереомеров обычно проводили с применением хроматографии, при которой использовали хиральную неподвижную фазу, и ее необязательно объединяли с химической дериватизацией (например, образование карбамата из амина). Соединения по настоящему изобретению могут содержать атомный изотоп в неестественной пропорции при одном или нескольких атомах, что составляют соединение. Например, соединения могут быть помечены радиоактивными изотопами, такими как тритий (3Н), йод-125 (125I) или С-14 (14С). Кроме того, например, водород может быть заменен тяжелым водородом с образованием дейтерированного лекарственного средства, и связь, образованная между дейтерием и углеродом, сильнее связи, образованной между обычным водородом и углеродом. По сравнению с недейтерированными лекарственными средствами, дейтерированные лекарственные средства обладают такими преимуществами, как уменьшенные побочные эффекты, повышенная стабильность лекарственного средства, усиленная терапевтическая эффективность и пролонгированный биологический период полураспада лекарственного средства. Превращения всех изотопных композиций соединений по настоящему изобретению, или радиоактивных, или нет, включены в объем настоящего изобретения. Термин «фармацевтически приемлемый носитель» относится к любому средству или несущей среде, которые способны доставлять эффективное количество активного вещества по настоящему изобретению, не мешают биологической активности активного вещества и не обладают токсичным побочным эффектом по отношению к хозяину или пациенту. Соответствующий носитель включает в себя воду, масло, растительное и минеральное, матричный крем, матричный лосьон, матричную мазь и т.п. Эти матрицы включают в себя суспендирующее средство, загуститель, усилитель проникновения через кожу и т.п. Их составы хорошо известны специалистам в области косметики или в области фармацевтики местного применения.
«Необязательный» или «необязательно» относится к событиям или условиям, описанным позже, которые могут, но не обязательно, происходить, и это описание включает в себя ситуации, в которых события или условия происходят, и ситуации, в которых события или условия не происходят.
Термин «замещенный» относится к замене любого одного или нескольких атомов водорода на конкретном атоме заместителем, который может включать в себя дейтерий и варианты водорода, поскольку валентность конкретного атома является нормальной и замещенное соединение является стабильным. Если заместителем является кислород (=O), это означает, что два атома водорода являются замещенными. Кислородное замещение не происходит на ароматической группе. Термин «необязательно замещенный» означает, что он может быть или не быть замещенным, и, если не отмечено иное, вид и число заместителей могут быть производными на основе химической доступности.
Если любая переменная (такая как R) встречается более чем один раз в композиции или структуре соединения, ее определение в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R заместителем(ями), группа может быть необязательно замещенной не более чем двумя R заместителями, и для каждого заместителя R содержит независимый вариант. Кроме того, комбинация заместителей и/или их вариантов разрешена, только если такая комбинация приводит к стабильному соединению.
Если число связывающей группы равно 0, такое как -(CRR)0-, это означает, что связывающая группа представляет собой простую связь.
Если одна из переменных выбрана из простой связи, две группы, соединенные с ней, прямо соединены. Например, если L представляет собой простую связь в A-L-Z, на самом деле структурой является A-Z.
Если заместитель свободный, это означает, что заместитель отсутствует. Например, если X в А-Х свободный, это означает, что на самом деле структурой является А. Если заместитель может быть присоединен к более чем одному атому на кольце, этот заместитель может быть связан с любым атомом на кольце. Например, структурная единица 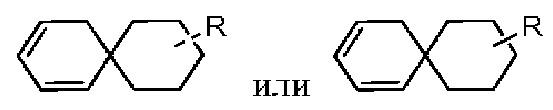 представляет, что замещение R заместителем может возникать на любом положении циклогексила или циклогексадиена. В случае отсутствия указания того, какой атом в изложенном заместителе будет присоединен к замещаемой группе, такой заместитель может быть присоединен через любой ее атом. Например, пиридильная группа в качестве замещающей группы может быть присоединена к замещаемой группе при помощи любого атома углерода на пиридиновом кольце. В случае отсутствия указания направления связывания изложенной связывающей группы, его направление связывания является произвольным. Например, в
представляет, что замещение R заместителем может возникать на любом положении циклогексила или циклогексадиена. В случае отсутствия указания того, какой атом в изложенном заместителе будет присоединен к замещаемой группе, такой заместитель может быть присоединен через любой ее атом. Например, пиридильная группа в качестве замещающей группы может быть присоединена к замещаемой группе при помощи любого атома углерода на пиридиновом кольце. В случае отсутствия указания направления связывания изложенной связывающей группы, его направление связывания является произвольным. Например, в 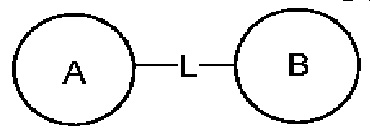 связывающая группа L представляет собой -M-W-; в это же время -M-W- может или связывать кольцо А и кольцо В в том же направлении, что и порядок чтения слева направо с образованием
связывающая группа L представляет собой -M-W-; в это же время -M-W- может или связывать кольцо А и кольцо В в том же направлении, что и порядок чтения слева направо с образованием 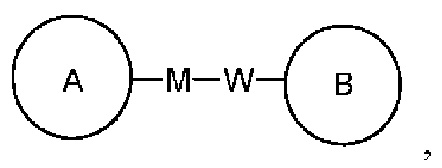 или связывать кольцо А и кольцо В в направлении, противоположном порядку чтения слева направо с образованием
или связывать кольцо А и кольцо В в направлении, противоположном порядку чтения слева направо с образованием
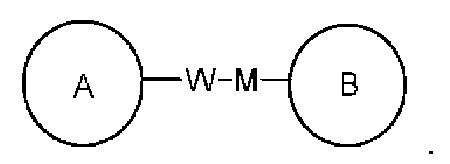
Комбинация связывающей группы, заместителей и/или их вариантов разрешена, только если такая комбинация приводит к стабильному соединению.
Если не отмечено иное, «кольцо» представляет собой замещенный или незамещенный циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил, арил или гетероарил. Так называемое кольцо включает в себя простое кольцо, соединенное кольцо, спирокольцо, конденсированное кольцо или кольцо с мостиковыми связями. Число атомов на кольце обычно определено как номер элемента колец. Например, «5-7-членное кольцо» означает 5-7 атомов, расположенных по кругу. Если не отмечено иное, кольцо необязательно содержит 1-3 гетероатома. Таким образом, «5-7-членное кольцо» включает в себя, например, фенил, пиридинил и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает в себя пиридил и пиперидил, но не включает в себя фенил. Термин «кольцо» также включает в себя кольцевую систему, содержащую по меньшей мере одно кольцо, каждое из «колец» независимо попадает под указанное определение.
Если не отмечено иное, термин «гетероцикл» или «гетероциклил» означает стабильное, содержащее гетероатом или гетерогруппу моноциклическое, бициклическое или трициклическое кольцо, которое может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим) и содержит атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, независимо выбранные из N, О и S, где любой из вышеуказанных гетероциклов может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы азота и серы необязательно могут быть окислены (т.е., NO и S(O)p, где р представляет собой 1 или 2). Атом азота может быть замещенным или незамещенным (т.е., N или NR, где R представляет собой Н или другие заместители, которые были определены в настоящем описании). Гетероцикл может быть присоединен к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, описанный в настоящем изобретении гетероцикл может подвергаться замещению в положении углерода или азота. Атом азота в гетероцикле необязательно кватернизирован. Предпочтительным вариантом осуществления является тот, в котором общее число S и О атомов в гетероцикле превышает 1, такие гетероатомы не являются смежными друг с другом. Другим предпочтительным вариантом осуществления является тот, в котором общее число S и О атомов в гетероцикле не превышает 1. Используемый в настоящем описании термин «ароматическая гетероциклическая группа» или «гетероарил» означает стабильное 5-, 6-, 7-членное моноциклическое или бициклическое или 7-, 8-, 9- или 10-членное бициклическое гетероциклическое ароматическое кольцо, которое содержит атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, которые независимо выбраны из N, О и S. Атом азота может быть замещенным или незамещенным (т.е., N или NR, если R представляет собой Н или другие заместители, которые были определены в настоящем описании). Гетероатомы азота и серы необязательно могут быть окислены (т.е., NO и S(O)p, где р представляет собой 1 или 2). Следует отметить, что общее число атомов S и О в ароматическом гетероцикле не превышает 1. Кольцо с мостиковыми связями также включено в определение гетероцикла. Кольцо с мостиковыми связями образуется, если один или несколько атомов (т.е. С, О, N или S) соединяют два не смежных атома углерода или азота. Предпочтительное кольцо с мостиковыми связями включает в себя без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостик всегда превращает простое кольцо в трициклическое кольцо. В кольце с мостиковыми связями заместитель в кольце также может располагаться на мостике.
Пример гетероциклического соединения включает в себя без ограничения акридинил, азоцинил, бензимидазолил, бензофуранил, меркаптобензофуранил, меркаптобензофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3Н-индолил, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксииндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонин, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридинооксазол, пиридиноимидазол, пиридинотиазол, пиридинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6Н-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1Н-1,2,3-триазолил, 2Н-1,2,3-триазолил, 1Н-1,2,4-триазолил, 4Н-1,2,4-триазолил и ксантенил. Также включены соединение с конденсированным кольцом и соединение со спирокольцом.
Если не отмечено иное, термин «гидрокарбил» или его родовое понятие (например, алкил, алкенил, алкинил, арил и т.п.), сам по себе или в комбинации с другим заместителем, относится к углеводородному радикалу с неразветвленной, разветвленной цепью или циклическому углеводородному радикалу или их любой комбинации. Они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно- или полизамещенными, могут быть моно валентными (например, метил), дивалентными (например, метилен) или поливалентными (например, метинил), также могут включать в себя дивалентную или поливалентную группу, содержат конкретное число атомов углерода (например, С1-С12 означает 1-12 атомов углерода, С1-12 выбран из C1, С2, С3, С4, С5, С6, С7, C8, С9, С10, С11 и С12; С3-12 выбран из С3, С4, С5, С6, С7, C8, С9, С10, С11 и С12). Термин «гидрокарбил» включает в себя без ограничения алифатический гидрокарбил и ароматический гидрокарбил. Алифатический гидрокарбил включает в себя неразветвленный и циклический гидрокарбил, особенно включая в себя без ограничения алкил, алкенил и алкинил. Ароматический гидрокарбил включает в себя без ограничения 6-12-членный ароматический гидрокарбил, такой как фенил, нафталенил и т.п. Согласно некоторым вариантам осуществления термин «гидрокарбил» относится к неразветвленной или разветвленной группе или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной и может включать в себя дивалентную или поливалентную группу. Пример насыщенной гидрокарбильной группы включает в себя без ограничения метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, изобутил, циклогексил, (циклогексил)метил, циклопропилметил и гомолог или изомер н-пентила, н-гексила, н-гептила, н-октила и подобные группы. Ненасыщенный гидрокарбил содержит одну или более чем одну двойную или тройную связь, и их пример включает в себя без ограничения этенил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и более высокие гомологи и изомеры.
Если не отмечено иное, термин «гетерогидрокарбил» или его родовое понятие (например, гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т.п.), сам по себе или в комбинации с другим термином, относится к стабильной неразветвленной, разветвленной или циклической углеводородной группе или любой их комбинации, которая состоит из конкретного числа атомов углерода и по меньшей мере одного гетероатома. Согласно некоторым вариантам осуществления термин «гетероалкил», сам по себе или в комбинации с другим термином, относится к стабильной неразветвленной или разветвленной углеводородной группе или любой их комбинации, которая состоит из конкретного числа атомов углерода и по меньшей мере одного гетероатома. Согласно конкретному варианту осуществления гетероатом выбран из В, О, N и S, где атомы азота и серы необязательно окислены, а атом азота является необязательно кватернизирован. Гетероатом или гетерогруппа могут быть расположены в любом внутреннем положении гетерогидрокарбила, включая положение, где гидрокарбил присоединен к оставшейся части молекулы. Но термины «алкокси», «алкиламино» и «алкилтио» (или алкоксил, в которой О заменен S) принадлежат к идиоматическому выражению и относятся к алкильной группе, соединенной с оставшейся частью молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Пример включает в себя без ограничения -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2-S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-O-CH3 и -СН=СН-N(СН3)-СН3. Вплоть до двух гетероатомов могут быть последовательными, например, -CH2-NH-OCH3.
Если не отмечено иное, термин «циклогидрокарбил», «гетероциклогидрокарбил» или его родовое понятие (например, арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т.п.), сам по себе или в комбинации с другим термином, относится к циклизированному «гидрокарбилу» и «гетерогидрокарбилу», соответственно. Более того, для гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкила и гетероциклоалкила) гетероатом может занимать положение, в котором гетероцикл присоединен к оставшемуся положению молекулы. Пример циклогидрокарбила включает в себя без ограничения циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т.п. Неограничивающий пример гетероциклоалкила включает в себя 1-(1,2,5,6-тетрагидропиридинил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофураниндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
Если не отмечено иное, термин «алкил» относится к неразветвленному или разветвленному насыщенному гидрокарбилу, который может быть монозамещенным (например, -CH2F) или полизамещенным (например, -CF3), и может быть моновалентным (например, метил), дивалентным (например, метилен) или поливалентным (например, метенил). Пример алкила включает в себя метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и т.п.
Если не отмечено иное, циклоалкил включает в себя любой стабильный циклический или полициклический гидрокарбил, любой атом углерода которого является насыщенным, и который может быть монозамещенным или полизамещенным и может быть моновалентным, дивалентным или поливалентным. Пример циклоалкила включает в себя без ограничения циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодекан и т.п.
Если не отмечено иное, термин «гало» или «галоген», сам по себе или как часть другого заместителя, относится к атому фтора, хлора, брома или йода. Более того, подразумевается, что термин «галогеналкил» включает в себя моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин «галоген(С1-С4)алкил» включает в себя без ограничения трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п. Если не отмечено иное, пример галогеналкила включает в себя без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
«Алкокси» представляет собой любой алкил, определенный выше, с конкретным числом атомов углерода, присоединенный кислородным мостиком. Если не отмечено иное, C1-6 алкокси включает в себя C1, С2, С3, С4, С5 и С6 алкокси. Пример алкокси включает в себя без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси.
Если не отмечено иное, термин «арил» относится к полиненасыщенному ароматическому углеводородному заместителю, который может быть моно- или полизамещенным, может быть моновалентным, двухвалентным или поливалетным, может быть моноциклическим или полициклическим (например, содержащим 1-3 кольца; причем по меньшей мере одно кольцо является ароматическим), и может быть конденсированным вместе или соединенным ковалентно. Термин «гетероарил» относится к арильной группе (или кольцу), что содержит от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбран из группы, состоящей из В, N, О и S, в которой атомы азота и серы необязательно окислены, и атомы азота необязательно кваретнизированы. Гетероарил может быть соединен с оставшейся частью молекулы при помощи гетероатома. Неограничивающие примеры арила или гетероарила включают в себя фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенилоксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидинил, бензотиазолил, пуринил, бензимидазолил, индолил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридинил, 3-пиридинил, 4-пиридинил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензоимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель любой из вышеуказанных арильных и гетероарильных кольцевых систем выбран из описанных ниже приемлемых заместителей.
Если не отмечено иное, при использовании в комбинации с другими терминами (например, арилокси, арилтио, арилалкил) термин арил включает в себя арильное и гетероарильное кольцо, как определено выше. Таким образом, предусмотрено, что термин «арилалкил» включает в себя группу (например, бензил, фенилэтил, пиридилметил и т.п.), где арил присоединен к алкилу включая алкил, где атом углерода (например, метилен) заменен атомом кислорода, например, феноксиметил, 2-пиридилоксиметил, 3-(1-нафтилокси)пропил и т.п.
Термин «уходящая группа» относится к функциональной группе или атому, который может быть заменен другой функциональной группой или атомом путем реакции замещения (такой как реакция нуклеофильного замещения). Например, приводимая в качестве примера уходящая группа включает в себя трифлат; хлор, бром и йод; сульфонатную группу, такую как мезилат, тозилат, пара-бромбензолсульфонат, пара-толуолсульфонаты и т.п.; ацилокси, такой как ацетокси, трифторацетокси и т.п.
Термин «защитная группа» включает в себя без ограничения «аминозащитную группу», «гидроксизащитную группу» или «меркаптозащитную группу». Термин «аминозащитная группа» относится к защитной группе, которая подходит для блокировки побочной реакции при положении азота на амине. Типичные аминозащитные группы включают в себя без ограничения формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилмсилил (TMS) и трет-бутилдиметилсилил (TBS) и т.п. Термин «гидроксизащитная группа» относится к защитной группе, которая подходит для блокировки побочной реакции при гидрокси. Типичные гидроксизащитные группы включают в себя без ограничения: алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), пара-метоксибензил (РМВ), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и т.п.
Соединение по настоящему изобретению может быть получено различными способами синтеза, хорошо известными специалистам настоящей области техники, включая следующие перечисленные варианты осуществления, варианты осуществления, образованные следующими перечисленными вариантами осуществления в комбинации с другими химическими способами синтеза, и эквивалентными режимами подстановки, хорошо известными специалистам настоящей области техники. Предпочтительный вариант осуществления включает в себя без ограничения примеры по настоящему изобретению.
Все используемые в настоящем изобретении растворители являются коммерчески доступными. В настоящем изобретении используют следующие аббревиатуры: водн. представляет собой воду; HATU представляет собой
O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC представляет собой N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид; мета-СРВА представляет собой 3-хлорпероксибензойную кислоту; экв. представляет собой эквивалент, эквивалентное количество; CDI представляет собой карбонилдиимидазол; DCM представляет собой метиленхлорид; РЕ представляет собой петролейный эфир; DIAD представляет собой диизопропилазодиформиат; DMF представляет собой N,N-диметилформамид; DMSO представляет собой диметилсульфоксид; EtOAc представляет собой этилацетат; EtOH представляет собой этанол; МеОН представляет собой метанол; CBz представляет собой бензилоксикарбонил, аминозащитную группу; ВОС представляет собой трет-бутоксилкарбонил, аминозащитную группу; НОАс представляет собой уксусную кислоту; NaCNBH3 представляет собой цианоборгидрид натрия; к. т. представляет собой комнатную температуру; O/N представляет собой всю ночь; THF представляет собой тетрагидрофуран; Вос2О представляет собой ди-трет-бутилдикарбонат; TFA представляет собой трифторуксусную кислоту; DIPEA представляет собой диизопропилэтиламин; SOCl2 представляет собой тионилхлорид; CS2 представляет собой бисульфид углерода; TsOH представляет собой паратолуолсульфоновую кислоту; NFSI представляет собой N-фтор-N-(бензолсульфонил)бензолсульфонамид; NCS представляет собой 1-хлорпирролидин-2,5-дион; н-Bu4NF представляет собой тетрабутиламмония фторид; iPrOH представляет собой 2-пропиловый спирт; т.пл. представляет собой точку плавления; LDA представляет собой диизопропиламид лития; EDCI представляет собой 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид; dppf представляет собой 1,1'-бис(дифенилфосфин)ферроцен; HATU представляет собой 2-(7-бензотриазолоксид)-N,N,N',N'-тетраметилмочевины гексафторфосфат; Ti(i-PrO)4 представляет собой тетраизопропоксид титана; NBS представляет собой N-бромсукцинимид; dast представляет собой (диэтиламино)серы трифторид; LiHMDS представляет собой гексаметилдисилазид лития; AIBN представляет собой азобисизобутиронитрил; POCl3 представляет собой оксихлорид фосфора; PEG400 представляет собой полиэтиленгликоль 400.
Соединения называли вручную или при помощи программного обеспечения ChemDraw®, и в коммерчески доступных соединениях использовали названия из каталогов их поставщиков.
Технический эффект: соединение по настоящему изобретению обладает значительной ингибиторной активностью в отношении бромодомена BET и значительным эффектом ингибирования опухоли, а также обладает хорошей переносимостью у животных; при этом, соединение по настоящему изобретению обладает низким фармакокинетическим клиренсом и хорошей абсорбцией.
Подробное описание предпочтительных вариантов осуществления
Настоящее изобретение конкретно будет описано ниже при помощи примеров, но оно не подразумевает никаких неприемлемых ограничений настоящего изобретения. Настоящее изобретение подробно было описано здесь и его конкретные варианты осуществления также раскрыты. Специалистам настоящей области техники будет очевидно, что различные изменения и улучшения могут быть сделаны по отношению к конкретным вариантам осуществления настоящего изобретения без отклонения от сущности и объема настоящего изобретения.
Схема 1
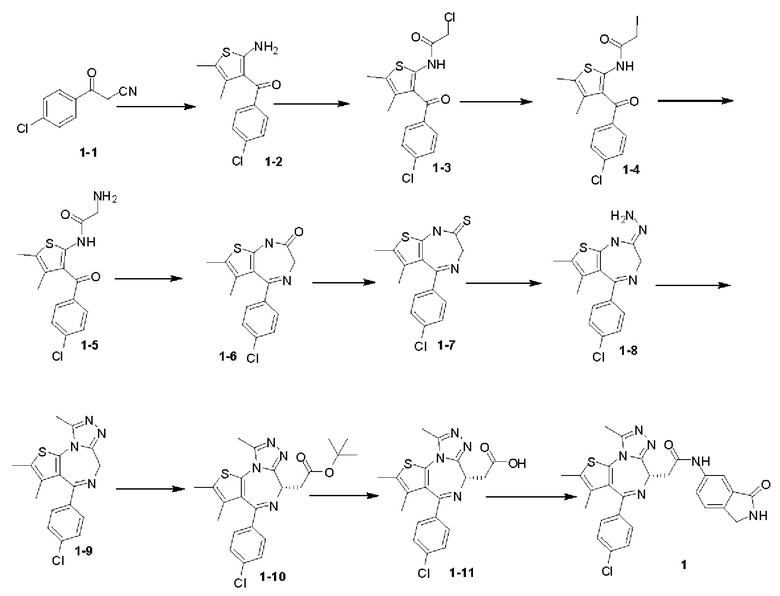
Пример 1
Синтез соединения 1-2
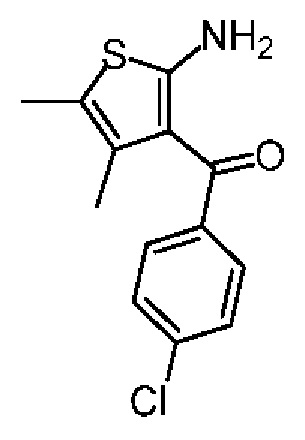
Соединение 1-1 (25,00 г, 139,20 ммоль, 1,00 экв.), 2-бутанон (11,04 г, 153,12 ммоль, 13,63 мл, 1,10 экв.) и морфолин (12,13 г, 139,20 ммоль, 12,25 мл, 1,00 экв.) растворяли в этаноле (200,00 мл) и добавляли сублимированную серу (4,46 г, 139,20 ммоль, 1,00 экв.). Суспензию нагревали до 70°С и перемешивали под защитой газообразного азота в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением желтого масла. К масляному веществу добавляли воду (500 мл) и полученную смесь экстрагировали этилацетатом (200 мл×4). Объединенные органические фазы собирали, промывали насыщенным солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали на колонке с силикагелем (петролейный эфир/этилацетат=10/1) с получением соединения 1-2. 1H ЯМР (400 МГц, CDCl3) δppm 7.47 (d, J=8.0 Гц, 2H), 7.38 (d, J=8.0 Гц, 2H), 6.43 (br s, 2H), 2.13 (s, 3Н), 1.56 (s, 3Н).
Синтез соединения 1-3
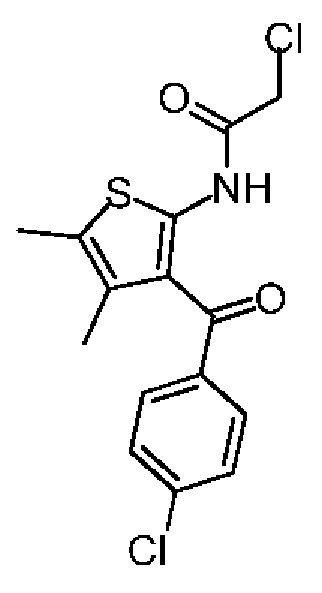
Соединение 1-2 (10,00 г, 37,63 ммоль, 1,00 экв.) растворяли в хлороформе (100,00 мл) и по каплям добавляли 2-хлорацетилхлорид (6,37 г, 56,45 ммоль, 4,49 мл, 1,50 экв.). После завершения добавления по каплям реакционную смесь перемешивали при 70°С в течение 1 часа. Реакционную смесь промывали насыщенным раствором бикарбоната натрия (100 мл) и насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Полученное соединение в виде неочищенного продукта перекристаллизовывали с метанолом (40 мл) с получением соединения 1-3. 1Н ЯМР (400 МГц, CDCl3) δppm 11.81 (br s, 1H), 7.58 (dd, J=2.0, 6.4 Гц, 2H), 7.45 (dd, J=2.2, 8.6 Гц, 2H), 4.25 (s, 2H), 2.29 (s, 3Н), 1.72 (s, 3Н).
Синтез соединения 1-4
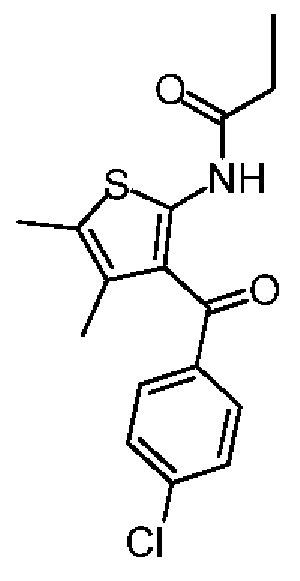
Соединение 1-3 (11,00 г, 32,14 ммоль, 1,00 экв.) и йодид натрия (9,63 г, 64,28 ммоль, 2,00 экв.) добавляли к тетрагидрофурану (50,00 мл). Полученную смесь перемешивали при 60°С в течение 2 часов. Реакционную смесь сразу концентрировали при пониженном давлении с получением соединения 1-4, которое не очищали и сразу использовали на следующей стадии реакции. LCMS (ESI) m/z: 433.9 (М+1). Синтез соединения 1-5
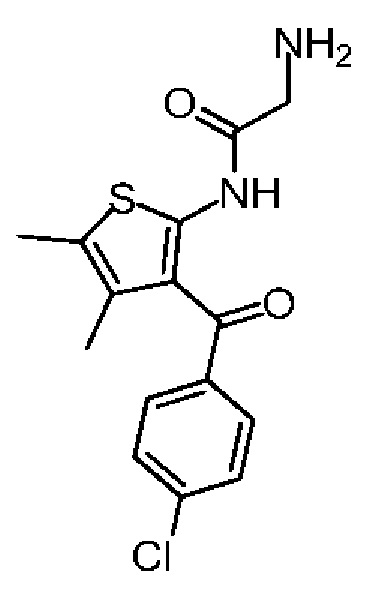
Соединение 1-4 (14,00 г, 32,28 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (100,00 мл). Полученную смесь охлаждали до -60°С и заполняли газообразным аммиаком в течение 30 минут. Полученную реакционную смесь медленно нагревали до 20°С и перемешивали в течение 3 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Полученное твердое вещество растворяли в этилацетате (150 мл) и промывали водой (50 мл×3) и насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Полученное соединение 1-5 сразу использовали на следующей стадии реакции. LCMS (ESI)m/z: 322.9 (М+1), 344.9 (M+Na).
Синтез соединения 1-6
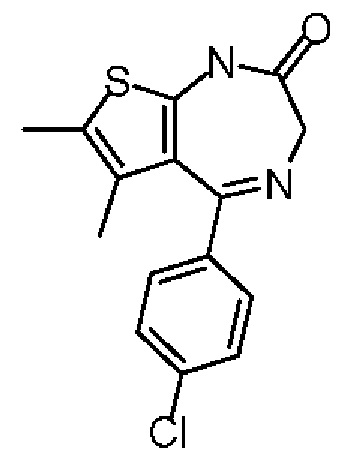
Соединение 1-5 (10,00 г, 30,98 ммоль, 1,00 экв.) растворяли в изопропаноле (150,00 мл) и ледяной уксусной кислоте (50,00 мл). Полученную смесь перемешивали при 90°С в течение 3 часов. Растворитель удаляли при пониженном давлении из реакционной смеси. Полученную смесь растворяли в хлороформе (20 мл), промывали насыщенным раствором бикарбоната натрия (20 мл) и насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Соединение в виде неочищенного продукта перекристаллизовывали с этилацетатом (50 мл) с получением соединения 1-6. 1H ЯМР (400 МГц, CDCl3) δppm 8.98 (br s, 1H), 7.46 (d, 8.4 Гц, 2H), 7.35 (d, 8.4 Гц, 2Н), 4.80 (d, 7=8.8 Гц, 1H), 3.93 (d, J=8.6 Гц, 1H), 2.28 (s, 3Н), 1.59 (s, 3Н).
Синтез соединения 1-7
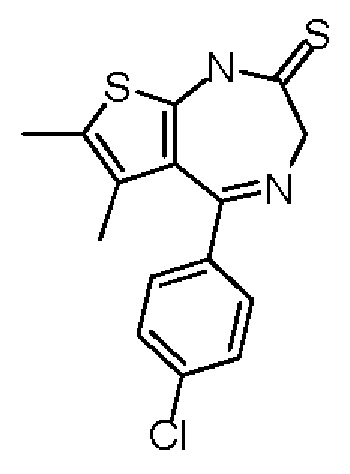
Фосфорный пентасульфид (17,07 г, 76,79 ммоль, 8,17 мл, 3,60 экв.) добавляли к постоянно перемешиваемой мутной жидкости карбоната натрия (4,07 г, 38,39 ммоль, 1,80 экв.) в 1,2-дихлорэтане (200,00 мл). Полученную смесь перемешивали при 20°С в течение 1 часа. Затем добавляли соединение 1-6 (6,50 г, 21,33 ммоль, 1,00 экв.). Полученная мутная жидкость реагировала при 65°С в течение 5 часов. Реакционную смесь охлаждали до 20°С и фильтровали. Фильтрационный кек растворяли в этилацетате (2 л) и промывали насыщенным солевым раствором (500 мл), сушили над сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали на колонке с силикагелем (петролейный эфир/этилацетат=5/1) с получением соединения 1-7.
Синтез соединения 1-8
При 0°С к мутной жидкости соединения 1-7 (3,50 г, 10,91 ммоль, 1,00 экв.) в метаноле (5,00 мл) добавляли гидразина гидрат (1,67 г, 32,72 ммоль, 1,62 мл, 98% чистоты, 3.00 экв.). Смесь реагировала при перемешивании при 0°С в течение 1 часа. Реакционную смесь фильтровали, и фильтрационный кек сушили в сушильном шкафу. Соединение 1-8 получали и сразу использовали на следующей стадии реакции. LCMS (ESI) m/z: 318.9 (М+1).
Синтез соединения 1-9
К смешанной жидкости соединения 1-8 (2,50 г, 7,84 ммоль, 1,00 экв.) в толуоле (100,00 мл) добавляли триэтилортоацетат (3,82 г, 23,52 ммоль, 4,29 мл, 3,00 экв.). Полученная смесь реагировала при перемешивании при 80°С в течение 1 часа. Реакционную смесь сразу концентрировали при пониженном давлении. Соединение в виде неочищенного продукта перекристаллизовывали с этилацетатом (10 мл) с получением соединения 1-9. LCMS (ESI) m/z: 344.9 (М+1).
Синтез соединения 1-10
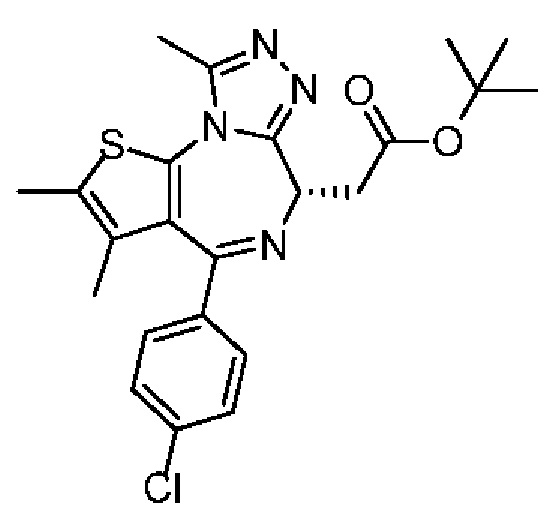
При -70°С к раствору соединения 1-9 (1,50 г, 4,38 ммоль, 1,00 экв.) в тетрагидрофуране (180 мл) добавляли по каплям LiHMDS (1 М, 8,76 мл, 2,00 экв.). Смесь реагировала при перемешивании при той же температуре в течение 1 часа, а затем раствор трет-бутил-2-бромацетата (1,28 г, 6,57 ммоль, 970,82 мкл, 1,50 экв.), растворенный в тетрагидрофуране (20 мл), добавляли по каплям. После завершения добавления по каплям реакционную смесь медленно нагревали до 20°С и перемешивали в течение 5 часов. Реакционную смесь гасили насыщенным раствором NH4Cl (50 мл), экстрагировали этилацетатом (100 мл) и промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали на колонке методом флеш-хроматографии и полученное соединение отделяли при помощи SFC с получением соединения 1-10 (основность-EtOH, хроматографическая колонка: AS(250 мм×30 мм, 5 мкм), подвижная фаза В: 30%, скорость потока (мл/мин): 55)([α]25D+54 (С 0,6, CHCl3)). LCMS (ESI)m/z: 457.0 (М+1).
Синтез соединения 1-11
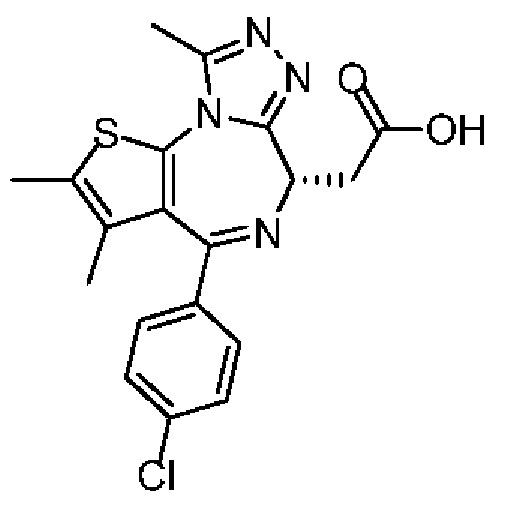
Соединение 1-10 (150,00 мг, 328,23 мкмоль, 1,00 экв.) растворяли в метиленхлориде (5,00 мл) и трифторуксусной кислоте (1,00 мл). Смесь реагировала при перемешивании при 20°С в течение 4 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Соединение 1-11 получали и сразу использовали на следующей стадии реакции. LCMS (ESI)m/z: 401.0 (М+1).
Синтез соединения 1
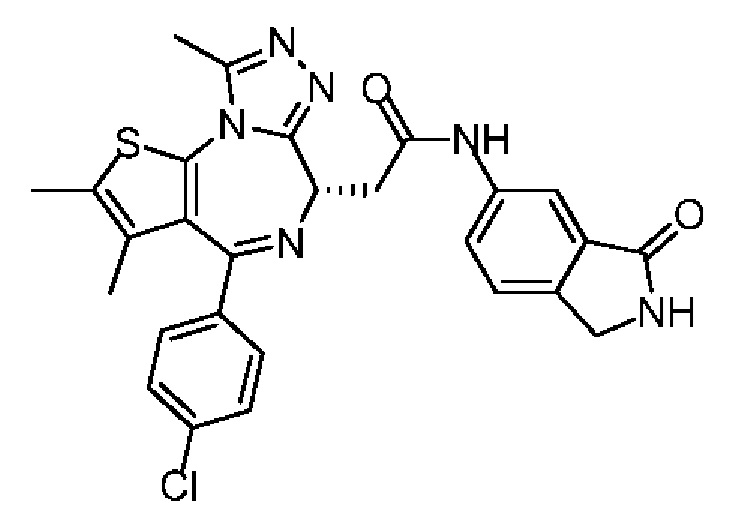
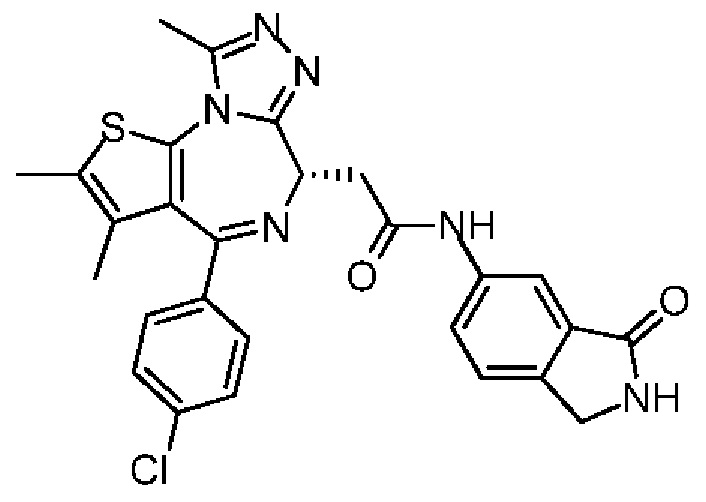
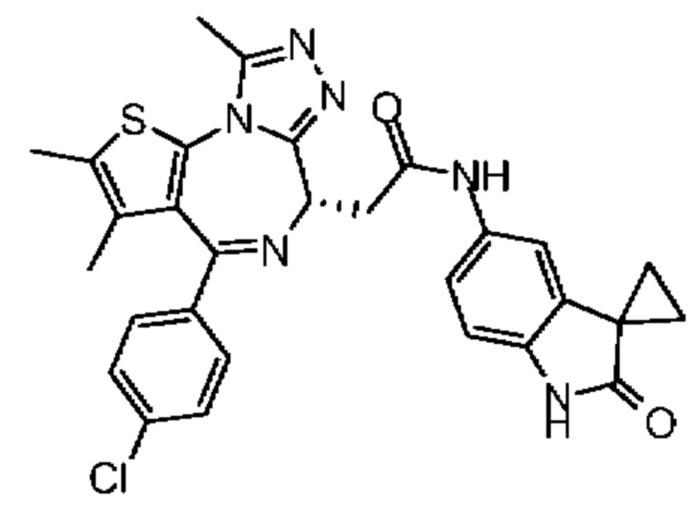
При 30°С и при защите газообразным азотом N,N-диизопропилэтиламин (48,36 мг, 374,19 мкмоль) медленно по каплям добавляли к раствору соединения 1-11 (50,00 мг, 124,73 мкмоль), 6-аминоизоиндолин-1-она (27,72 мг, 187,10 мкмоль) и HATU (71,14 мг, 187,10 мкмоль) в безводном метиленхлориде (15,00 мл). После добавления смесь реагировала при 30°С в течение 12 часов. Реакционную смесь промывали водой (20 мл×2). Водную фазу экстрагировали метиленхлоридом (20 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали, затем концентрировали при пониженном давлении и очищали при помощи препаративной хроматографии с получением соединения 1. 1Н ЯМР (400 МГц, CDCl3): δ ppm 9.46 (br s, 1H), 8.03 (br s, 1H), 7.85 (d, J=7.6 Гц, 1H), 7.41 (d, J=8.4 Гц, 2H), 7.31-7.33 (m, 3H), 6.57-6.61 (m, 1H), 4.69-4.73 (m, 1H), 4.34 (s, 2H), 3.83-3.88 (m, 1H), 3.56-3.61 (m, 1H), 2.69 (s, 3H), 2.41 (s, 3H), 1.68 (s, 3H). LCMS (ESI) m/z: 531.1 (M+1).
Схема 2
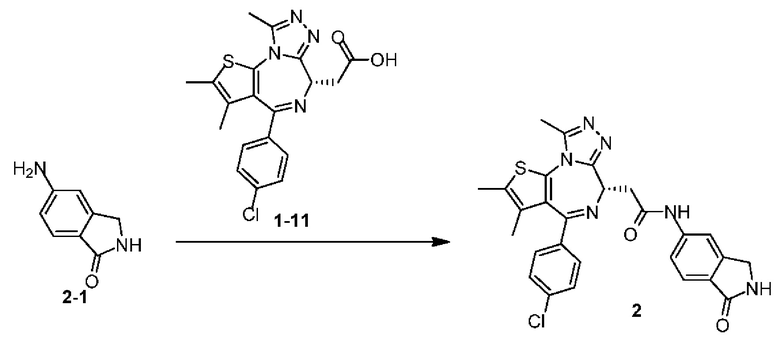
Пример 2
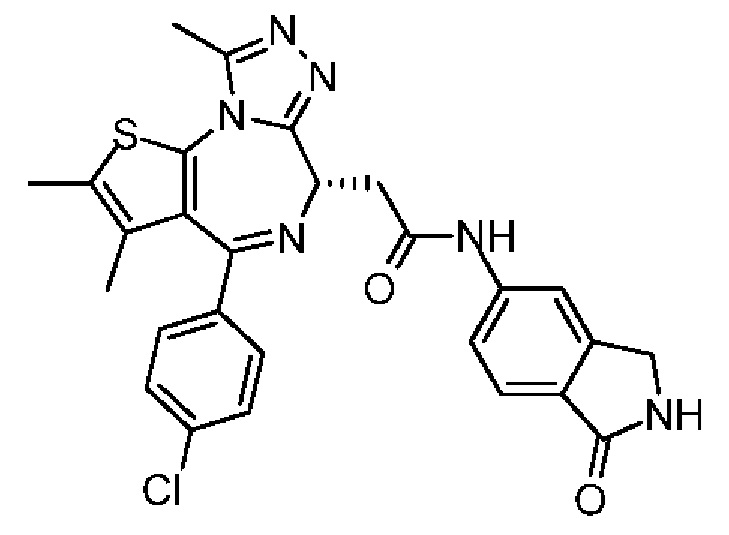
Соединение примера 2 синтезировали на основании примера 1.
1Н ЯМР (400 МГц, CDCl3): δ ppm 9.62 (br s, 1H), 8.00 (s, 1H), 7.76 (d, J=8.4 Гц 1H), 7.34-7.45 (m, 5H), 6.14-6.15 (m, 1H), 4.65-4.68 (m, 1H), 4.38 (s, 2H), 3.85-3.91 (m, 1H), 3.50-3.54 (m, 1H), 2.71 (s, 3H), 2.44 (s, 3H), 1.72 (s, 3H). LCMS (ESI)m/z: 531.1 (M+1).
Схемы 3 и 4
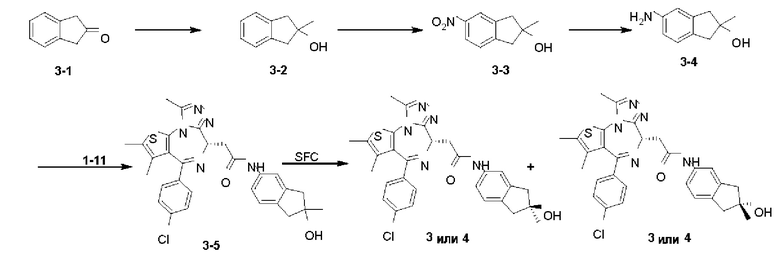
Примеры 3 и 4
Синтез соединения 3-2
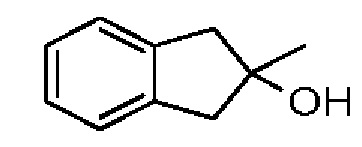
При 0°С метилбромид магния (3 М, 6,30 мл) добавляли к безводному диэтиловому эфиру (10 мл). Атмосферу заменяли газообразным азотом три раза. Раствор соединения 3-1 (2,00 г, 15,13 ммоль) в безводном диэтиловом эфире (40 мл) медленно по каплям добавляли. Смесь перемешивали при 25°С в атмосфере азота в течение 3 часов. Реакционную смесь выливали в ледяную воду (50 г) при перемешивании. Добавляли насыщенный раствор NH4Cl (50 мл) и смесь перемешивали в течение 5 минут. Смесь разделяли на две фазы. Водную фазу экстрагировали этилацетатом (50 мл). Органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (50 мл), водой (50 мл) и насыщенным солевым раствором (80 мл), каждым один раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 3-2. 1Н ЯМР (400 МГц, CDCl3): δ ppm 7.14-7.23 (m, 4Н), 2.97-3.08 (m, 4Н), 1.52 (s, 3Н).
Синтез соединения 3-3
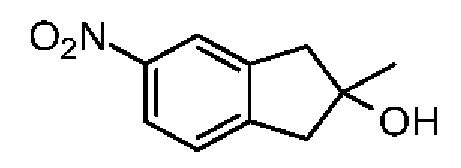
При 0°С и при перемешивании раствор соединения 3-2 (200,00 мг, 1,35 ммоль) в метиленхлориде (2 мл) медленно добавляли к раствору концентрированной азотной кислоты (4,20 г, 66,66 ммоль, 3,00 мл) и концентрированной серной кислоты (132,36 мг, 1,35 ммоль) в безводном метиленхлориде (10 мл). Смесь перемешивали при 0°С в течение 5 минут. Реакционную смесь медленно выливали в раздробленный лед (50 г) при перемешивании. Смесь перемешивали в течение 10 минут и разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (30 мл × 2). Объединенные органические фазы промывали насыщенным раствором бикарбоната натрия (80 мл) и насыщенным солевым раствором (80 мл), соответственно, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продует очищали методом тонкослойной хроматографии на пластине с получением соединения 3-3. 1Н ЯМР (400 МГц, CDCl3): δ ppm 8.04-8.14 (m, 2Н), 7.32-7.38 (m, 1H), 3.53-3.57 (m, 2Н),3.28-3.33 (m, 2Н), 1.81 (s, 3Н).
Синтез соединения 3-4
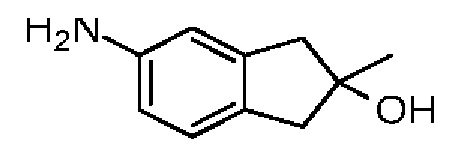
Соединение 3-3 (220,00 мг, 1,14 ммоль) и Pd/C (200,00 мг, 10% чистоты) добавляли к абсолютному метанолу (10,00 мл). Атмосферу заменяли газообразным водородом три раза. Смесь перемешивали в течение 16 часов при 25°С в атмосфере водорода. Реакционную смесь сразу фильтровали через слой целита с воронкой Бюхнера и концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 3-4.
Синтез соединения 3-5
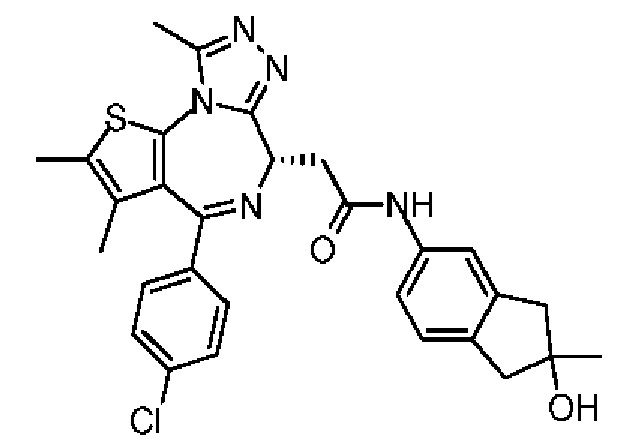
Соединение 3-4 (46,00 мг, 281,83 мкмоль, 1,20 экв.) и диизопропилэтиламин (91,06 мг, 704,58 мкмоль, 123,05 мкл, 3,00 экв.) добавляли к метиленхлориду (5,00 мл). Добавляли соединение 1-11 (94,15 мг, 234,86 мкмоль, 1,00 экв.) и HATU (89,30 мг, 234,86 мкмоль, 1,00 экв.). Атмосферу заменяли газообразным азотом три раза. Смесь перемешивали при 25°С в атмосфере азота в течение 2 часов. Реакционную смесь промывали водой при встряхивании (10 мл). Органическую фазу промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 3-5. LCMS (ESI) m/z: 546.2 (М+1).
Синтез соединений 3 и 4
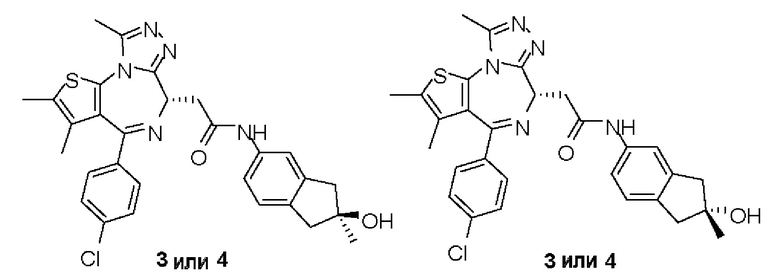
Соединение 3-5 (98 мг, 179,46 мкмоль) подвергали SFC хиральному расщеплению (хроматографическая колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% NH3H2O EtOH]; В%: 40%-40%, 60 мл/мин) с получением двух продуктов, каждый характеризуется простой конфигурацией.
Соединение примера 3 (Rt=5.311 мин).1Н ЯМР (400 МГц, CDCl3): δ ppm 8.68 (s, 1Н), 7.51 (s, 1H), 7.41 (d, J=8.4 Гц, 2Н), 7.33 (d, J=8.8 Гц, 2Н), 7.28-7.29 (m, 1Н), 7.12 (d, J=8.0 Гц, 1H), 4.60-4.64 (m, 1H), 3.74-3.80 (m, 1Н), 3.44-3.49 (m, 1H), 2.95-3.03 (m, 4Н), 2.68 (s, 3Н), 2.40 (s, 3Н), 1.68 (s, 3Н), 1.49 (s, 3Н). LCMS (ESI) m/z: 546.2 (М+1).
Соединение примера 4 (Rt=5.926 мин) 1Н ЯМР (400 МГц, CDCl3): δppm 8.72 (s, 1Н), 7.51 (s, 1H), 7.41 (d, J=8.4 Гц, 2Н), 7.33 (d, J=8.8 Гц, 2Н), 7.28-7.29 (m, 1Н), 7.12 (d, J=8.0 Гц, 1H), 4.60-4.64 (m, 1H), 3.74-3.80 (m, 1Н), 3.44-3.49 (m, 1H), 2.95-3.03 (m, 4Н), 2.68 (s, 3Н), 2.40 (s, 3Н), 1.68 (s, 3Н), 1.49 (s, 3Н). LCMS (ESI) m/z: 546.1 (М+1).
Схема 5
Пример 5
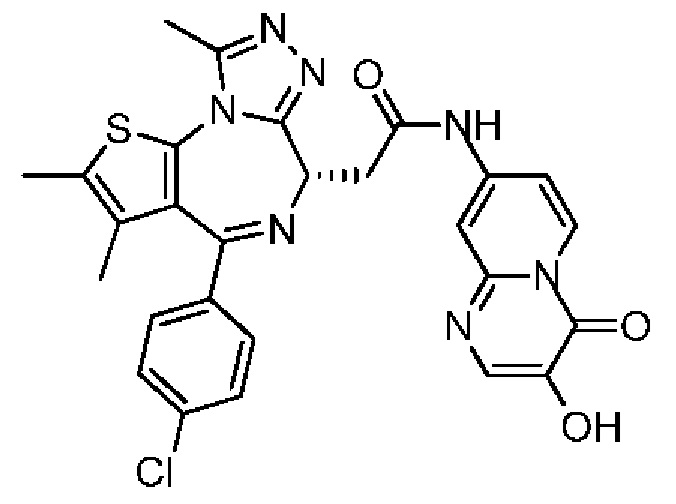
Синтез соединения 5-2
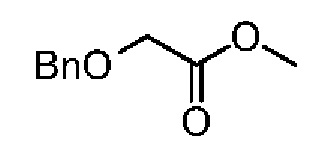
При 0°С тионилхлорид (25,77 г, 216,65 ммоль, 15,71 мл, 1,20 экв.) добавляли по каплям к метанолу (200,00 мл). Смесь перемешивали при 0°С в течение 30 минут. Раствор соединения 5-1 (30,00 г, 180,54 ммоль, 25,86 мл, 1,00 экв.) в метаноле (100,00 мл) добавляли по каплям. После завершения добавления по каплям смесь реагировала при 26°С в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в метиленхлориде. Значение рН смеси доводили при помощи насыщенного раствора карбоната натрия до 8-9, экстрагировали и разделяли на две фазы. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 5-2, которое сразу использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3): δ ppm 7.26-7.37 (m, 5Н), 4.63 (s, 2Н), 4.11 (s, 2Н), 3.76 (s, 3Н).
Синтез соединения 5-3
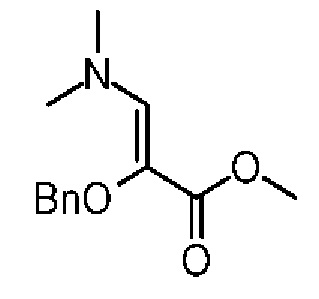
Смесь соединения 5-2 (16,00 г, 88,79 ммоль, 1,00 экв.) и трет-бутокси-ди(диметиламино)метана (17,02 г, 97,67 ммоль, 20,26 мл, 1,10 экв.) нагревали до 90°С и осуществляли взаимодействие в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в метиленхлориде (80 мл). Смесь промывали насыщенным солевым раствором (25 мл), экстрагировали и разделяли на две фазы. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 5-3, которое сразу использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3): δ ppm 7.26-7.33 (m, 5Н), 6.78 (s, 1Н), 4.63(s, 2H), 3.64 (s, 3H), 2.88(m, 6H).
Синтез соединения 5-4
Ледяную уксусную кислоту (40,00 мл) добавляли к смеси соединения 5-3 (12,00 г, 51,00 ммоль, 1,00 экв.) и 4-бром-2-аминопиридина (8,82 г, 51,00 ммоль, 1,00 экв.). Полученную смесь нагревали до 130°С и перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 4:1) с получением соединения 5-4. 1Н ЯМР (400 МГц, CDCl3): δ ppm 8.72 (d, J=7.6 Гц, 1H), 8.19 (s, 1H), 7.89 (s, 1H), 7.35-7.47 (m, 5H), 5.19 (s, 2H).
Синтез соединения 5-5
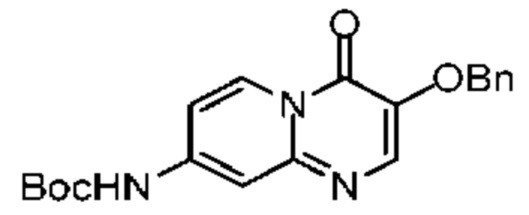
К раствору соединения 5-4 (1,00 г, 3,02 ммоль, 1,00 экв.) и трет-бутилкарбамата (459,88 мг, 3,93 ммоль, 1,30 экв.) в 1,4-диоксане (20,00 мл) последовательно добавляли 4,5-бис(дифенилфосфин)-9,9-диметилксантен (174,73 мг, 301,97 мкмоль, 0,10 экв.), трис(дибензилиденацетон)дипалладий (276,52 мг, 301,97 мкмоль, 0,10 экв.) и карбонат цезия (2,95 г, 9,06 ммоль, 3,00 экв.). Атмосферу заменяли газообразным азотом три раза. Смесь нагревали до 100°С при защите газообразным азотом, и она реагировала в течение 10 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрационный кек промывали метиленхлоридом (10 мл × 2). Полученный фильтрат концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 5:1-2:1) с получением соединения 5-5. LCMS (ESI) m/z: 368.2 (М+1).
Синтез соединения 5-6
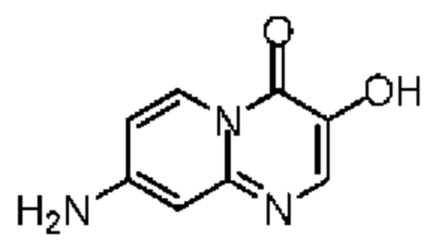
Смесь соединения 5-5 (190,00 мг, 517,15 мкмоль, 1,00 экв.) и трифторуксусной кислоты (5,00 мл) нагревали до 90°С и перемешивали в течение 20 часов. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток растворяли в метиленхлориде (10 мл). Смесь снова концентрировали при пониженном давлении с получением соединения 5-6, которое сразу использовали на следующей стадии без дополнительной очистки.
Синтез соединения 5
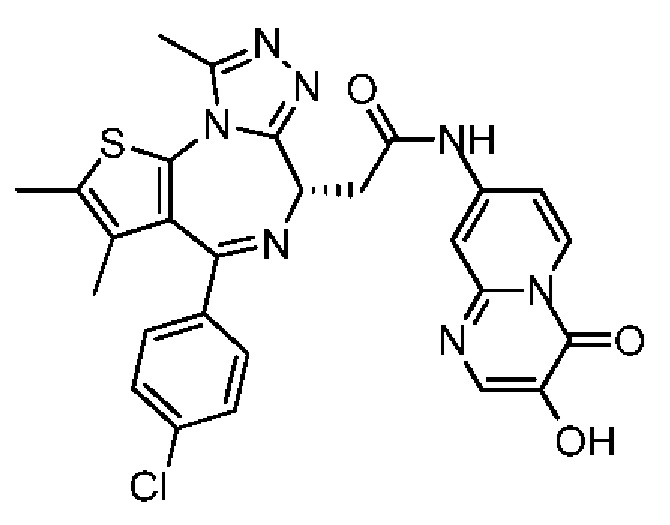
При -10°С и при защите газообразным азотом соединение 1-11 (60,00 мг, 149,67 мкмоль, 1,00 экв.) и триэтиламин (30,29 мг, 299,34 мкмоль, 41,49 мкл, 2,00 экв.) растворяли в смешанной жидкости безводного тетрагидрофурана (2,00 мл) и безводного N,N-диметилформамида (1,00 мл). Затем медленно по каплям добавляли пивалоилхлорид (18,05 мг, 149,67 мкмоль, 18,42 мкл, 1,00 экв.) к вышеуказанному раствору. Полученную смесь перемешивали при -10°С в течение 0,5 часа. Затем раствор соединения 5-6 (40,00 мг, 137,37 мкмоль, 0,92 экв., TFA) в безводном N,N-диметилформамиде (1,00 мл) добавляли по каплям к реакционной смеси. После завершения добавления по каплям смесь нагревали до 27°С и перемешивали в течение 5 часов. Реакционную смесь гасили водой (5 мл) и смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (5 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали методом препаративной хроматографии (основность) с получением соединения 5. 1H ЯМР (400 МГц, CDCl3): δ ppm 8.76 (d, J=7.28 Гц, 1H), 8.12 (s, 1H), 7.47 (d, J=8.28 Гц, 2H), 7.34 (d, J=8.53 Гц, 3Н), 6.56-6.64 (m, 2Н), 5.12 (s, 1H), 4.72 (t, J=7.15 Гц, 1H), 3.93-4.00 (m, 2H), 2.69 (s, 3Н), 2.40 (s, 3Н), 1.68 (s, 3Н). LCMS (ESI) m/z: 560.0 (M+1).
Схемы 6 и 7
Примеры 6 и 7
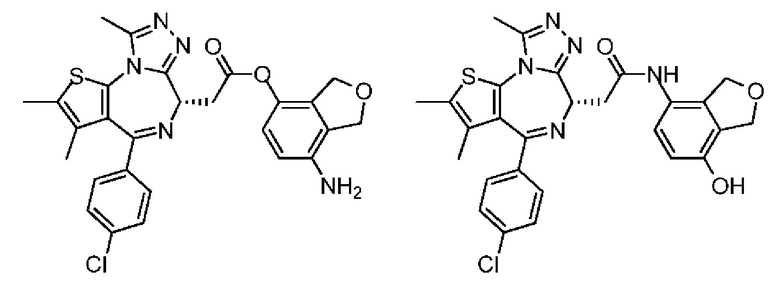
Синтез соединения 6-2
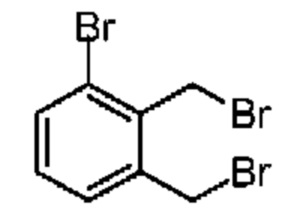
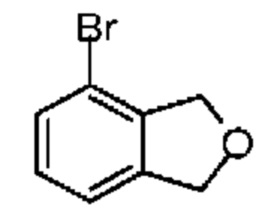
При защите газообразным азотом к раствору соединения 6-1 (5,00 г, 27,02 ммоль, 3,65 мл, 1,00 экв.) в тетрахлориде углерода (20,00 мл) добавляли NBS (10,00 г, 56,19 ммоль, 2,08 экв.) и AIBN (1,04 г, 6,33 ммоль, 0,23 экв.). Смесь реагировала при перемешивании при 65°С в течение 4 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 6-2. 1Н ЯМР (400 МГц, CDCl3) δ ppm 7.57 (d, J=8.0 Гц, 1H), 7.32 (d, J=8.0 Гц, 1H), 7.16 (t, J=7.8 Гц, 1H), 4.84 (s, 2Н), 4.63 (s, 2Н).
Синтез соединения 6-3
Нейтральный оксид алюминия (100,00 г, 980,78 ммоль, 93,41 экв.) добавляли к раствору соединения 6-2 (3,60 г, 10,50 ммоль, 1,00 экв.), растворенному в н-гексане (200,00 мл), и смесь реагировала при перемешивании при 75°С в течение 2 часов. Реакционную смесь непосредственно фильтровали. Фильтрационный кек промывали этилацетатом (200 мл). Фильтрат сразу концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали на колонке методом флеш-хроматографии с получением соединения 6-3. 1Н ЯМР (400 МГц, CDCl3): δ ppm 7.38-7.40 (m, 1H), 7.16-7.16 (m, 2Н), 5.21 (s, 2Н), 5.10 (s, 2Н).
Синтез соединения 6-4
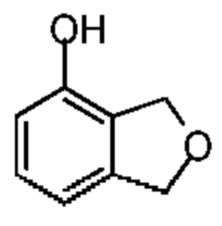
К смешанной жидкости соединения 6-3 (400,00 мг, 2,01 ммоль, 1,00 экв.), гидроксида калия (225,52 мг, 4,02 ммоль, 2,00 экв.) и 2-ди-трет-бутилфосфин-2',4',6'-триизопропилбифенила (85,34 мг, 201,00 мкмоль, 0,10 экв.) в 1,4-диоксане (10,00 мл) добавляли воду (1,00 мл) и трис(дибензилиденацетон)дипалладий (184,03 мг, 201,00 мкмоль, 0,10 экв.). Смесь реагировала при 120°С при защите газообразным азотом в микроволновом устройстве в течение 1 часа. Реакционную смесь сразу концентрировали при пониженном давлении. Остаток растворяли в этилацетате (20 мл). Смесь промывали водой (10 мл) и насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали при помощи препаративной пластины (петролейный эфир/этилацетат=5/1) с получением соединения 6-4. 1H ЯМР (400 МГц, CDCl3-d) δ ppm 7.15 (t, J=7.6 Гц, 1H), 6.80 (d, J=7.6 Гц, 1H), 6.66 (d, J=7.6 Гц, 1H), 5.15 (m, 4Н).
Синтез соединения 6-5
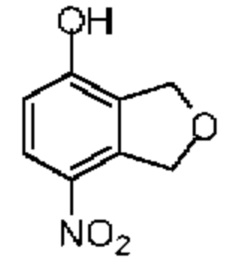
При -5°С и при защите газообразным азотом концентрированную серную кислоту (36,75 мг, 367,24 мкмоль, 19,97 мкл, 98% чистоты, 1,00 экв.) добавляли к раствору соединения 6-4 (50,00 мг, 367,24 мкмоль, 1,00 экв.), растворенного в метиленхлориде (2 мл). Затем медленно к реакционной смеси добавляли дымящуюся азотную кислоту (24,36 мг, 367,24 мкмоль, 17,40 мкл, 1,00 экв.) (чистота 95%), разбавленную в метиленхлориде (0,5 мл). Полученную смесь перемешивали в течение 0,5 часа. Реакционную смесь разбавляли метиленхлоридом (10 мл), затем промывали водой (5 мл) и насыщенным солевым раствором (5 мл), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали при помощи препаративной пластины (петролейный эфир/этилацетат = 3/1) с получением соединения 6-5. 1H ЯМР (400 МГц, CDCl3) δppm 7.94 (d, J=8.8 Гц, 1H), 6.72 (d, J=8.8 Гц, 1H), 5.32 (s, 2Н), 4.97 (s, 2Н).
Синтез соединения 6-6
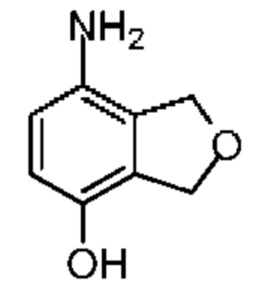
При защите газообразным азотом к раствору соединения 6-5 (40,00 мг, 220,81 мкмоль, 1,00 экв.), растворенного в метаноле (10.00 мл), добавляли Pd/C (100,00 мг) (с содержанием палладиума 20%, воды 50%). Затем атмосферу реакционной системы заменяли газообразным водородом три раза. Реакционная смесь реагировала при 20°С в атмосфере водорода (баллон) (15 фунт/кв.дюйм) в течение 1 часа. Реакционную смесь сразу фильтровали. Фильтрационный кек промывали метанолом (10 мл). Фильтрат сразу концентрировали при пониженном давлении с получением соединения 6-6, которое сразу использовали на следующей стадии реакции. LCMS (ESI) m/z: 151.9 (М+1).
Синтез соединений 6 и 7
Соединение 1-11 (50,00 мг, 124,73 мкмоль, 1,00 экв.), соединение 6-6 (30,00 мг, 198,32 мкмоль, 1,59 экв.), триэтиламин (37,86 мг, 374,19 мкмоль, 51,86 мкл, 3,00 экв.) и HATU (71,14 мг, 187,10 мкмоль, 1,50 экв.) растворяли в метиленхлориде (5,00 мл). Смесь перемешивали при 20°С при защите газообразным азотом в течение 2 часов. Реакционную смесь разбавляли метиленхлоридом (10 мл) и промывали водой (10 мл) и насыщенным солевым раствором (10 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали при помощи препаративной пластины с получением соединения 6. LCMS (ESI) m/z: 534.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δ ppm 8.47 (s, 1H), 7.36 (d, J=8.0 Гц, 2H), 7.28 (d, J=8.4 Гц, 2H), 7.07 (d, J=8.4 Гц, 1H), 6.57 (d, J=8.0 Гц, 1H), 4.85-5.00 (m, 4H), 4.53-4.56 (m, 1H), 3.63-3.65 (m, 1H), 3.34-3.38 (m, 1H), 2.62 (s, 3H), 2.34 (s, 3H), 1.62 (s, 3H).
Соединение 7. LCMS (ESI) m/z: 534.1 (M+1). 1H ЯМР (400 МГц, CDCl3) δ ppm 8.65 (s, 1H), 8.57 (br s, 1H), 7.35 (d, J=8.4 Гц, 2H), 7.26 (d, J=8.4 Гц, 2H), 6.90 (d, J=8.0 Гц, 1H), 6.53 (d, J=8.8 Гц, 1H), 4.95 (s, 2H), 4.90 (d, J=12.8 Гц, 1H), 4.80 (d, J=12.8 Гц, 1H), 4.57-4.60 (m, 1H), 3.63-3.69 (m, 1H), 3.35-3.39 (m, 1H), 2.62 (s, 3H), 2.34 (s, 3H), 1.61 (s, 3H).
Схема 8
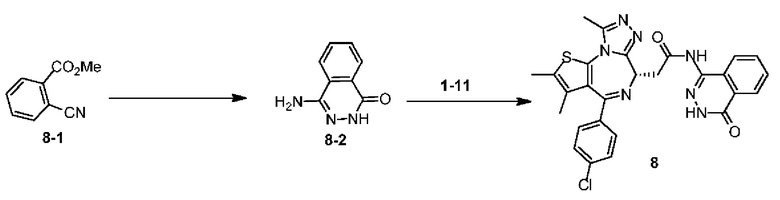
Пример 8
Синтез соединения 8-2
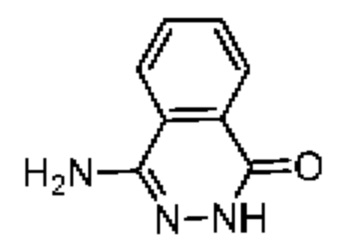
Соединение 8-1 (500,00 мг, 3,10 ммоль, 1,00 экв.) и гидразина гидрат (4,12 г, 80,66 ммоль, 4,00 мл, 26,02 экв.) добавляли в пробирку под микроволновым облучением. Смесь реагировала под микроволновым облучением при 90°С в течение 1 часа. Большое количество твердого вещества осаждалось из реакционной смеси. Реакцию заканчивали. Реакционную смесь фильтровали. Фильтрационный кек промывали водой (20 мл × 2). Затем добавляли безводный тетрагидрофуран (20 мл × 2). Смесь концентрировали при пониженном давлении с получением соединения 8-2 без дополнительной очистки. LCMS (ESI)m/z: 161.9 (М+1).
Синтез соединения 8
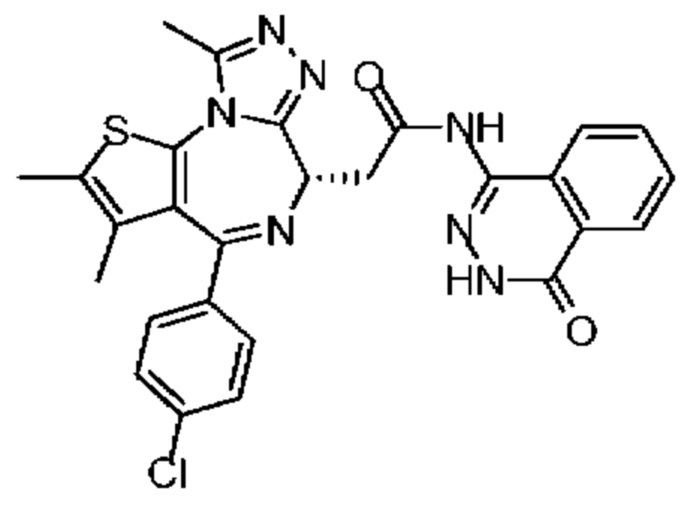
Раствор соединения 1-11 (100,00 мг, 249,45 мкмоль, 1,00 экв.) и соединения 8-2 (100,50 мг, 623,63 мкмоль, 2,50 экв.) в пиридине (5,00 мл) добавляли по каплям к POCl3 (114,74 мг, 748,35 мкмоль, 69,54 мкл, 3,00 экв.). Смесь реагировала при перемешивании при 20°С в течение 12 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и промывали водой (5 мл × 2) и насыщенным солевым раствором (5 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали методом препаративной хроматографии с получением соединения 8. 1Н ЯМР (400 МГц, CDCl3) δ ppm 10.49-10.54 (m, 1H), 9.84 (br s, 1H), 8.36-8.37 (m, 1H), 7.79-7.98 (m, 1H), 7.72-7.74 (m, 2Н), 7.46 (d, J=8.4 Гц, 2Н), 7.34 (d, J=8.4 Гц, 2Н), 4.70-4.73 (m, 1H), 3.73-3.86 (m, 2Н), 2.68 (s, 3Н), 2.40 (s, 3Н),1.67 (s, 3Н). LCMS (ESI)m/z: 544.1 (М+1).
Схема 9
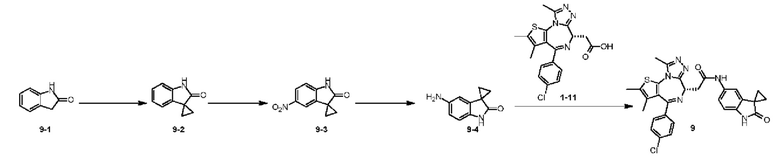
Пример 9
Синтез соединения 9-2
При -20°С и при защите газообразным азотом соединение 9-1 (2,00 г, 15,02 ммоль, 1,00 экв.) и безводный диизопропиламин (3,16 г, 31,24 ммоль, 4,39 мл, 2,08 экв.) растворяли в безводном тетрагидрофуране (30,00 мл). Затем раствор охлаждали до -20°С, медленно по каплям добавляли н-бутиллитий (2,5 М, 23,73 мл, 3,95 экв.) и полученную смесь сохраняли при температуре от -20 до -30°С. После завершения добавления по каплям смесь нагревали до 0°С и она реагировала при перемешивании в течение 1 часа. Затем медленно по каплям добавляли раствор 1,2-дибромэтана (9,62 г, 51,22 ммоль, 3,86 мл, 3,41 экв.) в безводном тетрагидрофуране (10,00 мл). После завершения добавления по каплям смесь нагревали до 27°С и она реагировала при перемешивании в течение 18 часов. При -20°С добавляли насыщенный раствор хлорида аммония (20 мл) для гашения реакции. Значение рН реакционной смеси доводили при помощи 3 н хлористоводородной кислоты (5 мл) до 2-3, а затем экстрагировали этилацетатом (3×20 мл). Вышеуказанные органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (50 мл) и насыщенным солевым раствором (50 мл) последовательно. Полученную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением пурпурного твердого вещества. Неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 9-2. 1H ЯМР (400 МГц, CDCl3): δ ppm 9.12 (s, 1Н), 7.11 (m, 1H), 6.89-6.96 (m, 2H), 6.75 (d, J=7.28 Гц, 1H), 1.70 (t, J=4.0 Гц, 2H), 1.47 (t, J=4.0 Гц, 2H).
Синтез соединения 9-3
При -15°C и при защите газообразным азотом медленно по каплям добавляли азотную кислоту (118,46 мг, 1,88 ммоль, 84,61 мкл, 1,00 экв.) к раствору соединения 9-2 (300,00 мг, 1,88 ммоль, 1,00 экв.) и концентрированной серной кислоты (184,85 мг, 1,88 ммоль, 100,46 мкл, 1,00 экв.) в метиленхлориде (4,00 мл). После завершения добавления по каплям смесь нагревали до 27°С и перемешивание поддерживали в течение 10 часов. Лед (приблизительно 2 г) добавляли к реакционной смеси для гашения реакции. Реакционную смесь экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (10 мл) и насыщенным солевым раствором (10 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 9-3. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8.32 (s, 1H), 8.12 (m, 1H), 7.67 (d, J=2.0 Гц, 1H), 6.97 (d, J=8.4 Гц, 1H), 1.81-1.84 (m, 2Н), 1.61-1.68 (m, 2Н).
Синтез соединения 9-4
Восстановленное порошковое железо (462,27 мг, 8,28 ммоль, 13,00 экв.) добавляли к раствору соединения 9-3 (130 мг, 636,69 мкмоль, 1,0 экв.) в ледяной уксусной кислоте (8,00 мл). Смесь перемешивали при 25°С при защите газообразным азотом в течение 16 часов. Реакционную смесь фильтровали. Фильтрат концентрировали и затем добавляли к воде (5 мл). Водную фазу экстрагировали этилацетатом (3×5 мл). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Реакционную смесь фильтровали при помощи диатомитовой земли. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт отделяли и очищали методом тонкослойной хроматографии на пластине с получением соединения 9-4. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8.37 (s, 1H), 6.75 (d, J=8.0 Гц, 1H), 6.48-6.63 (m, 1H), 6.23 (s, 1Н), 1.64-1.82 (m, 2H), 1.40-1.51 (m, 2H).
Синтез соединения 9
При 25°С и при защите газообразным азотом соединение 1-11 (36,82 мг, 91,85 мкмоль, 1,00 экв.) и соединение 9-4 (16,00 мг, 91,85 мкмоль, 1,00 экв.) добавляли к раствору HATU (41,91 мг, 110,22 мкмоль, 1,20 экв.) в безводном метиленхлориде (4,00 мл), а затем медленно по каплям добавляли триэтиламин (27,88 мг, 275,55 мкмоль, 38,19 мкл, 3,00 экв.). Смесь перемешивали при 25°С при защите газообразным азотом в течение 5 часов. Воду (3 мл) добавляли к реакционной смеси для гашения реакции. Смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (2 мл) и насыщенным солевым раствором (2 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали методом препаративной хроматографии (основность) с получением соединения 9. 1H ЯМР (400 МГц, CDCl3) δ ppm 9.19 (s, 1H), 8.64 (s, 1H), 7.31-7.37 (m, 2H), 7.23-7.27 (m, 2H), 7.19 (s, 1H), 7.08-7.11 (m, 1H), 6.76 (d, J=8.4 Гц, 1H), 4.60-4.64 (m, 1H), 3.73-3.79 (m, 1H), 3.39-3.44 (m, 1H), 2.61 (s, 3H), 2.34 (s, 3H), 1.62-1.64 (m, 2H), 1.61 (s, 3H), 1.42-1.43 (m, 2H). LCMS (ESI)m/z: 557.1 (M+1).
Схема 10
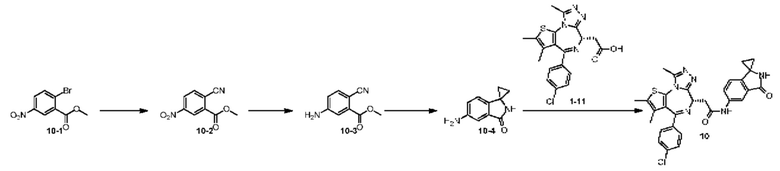
Пример 10
Синтез соединения 10-2
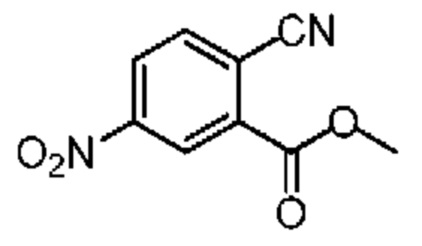
Смешанную жидкость соединения 10-1 (1,76 г, 6,77 ммоль, 1,00 экв.), цианида меди (910,00 мг, 10,16 ммоль, 2,22 мл, 1,50 экв.), dppf (375,32 мг, 677,00 мкмоль, 0,10 экв.), бис(дибензилиденацетон)палладия (389,28 мг, 677,00 мкмоль, 0,10 экв.) и N,N-диметилформамида (20,00 мл) нагревали до 110°С и перемешивали в течение 16 часов. Реакционную смесь фильтровали при пониженном давлении. Фильтрат концентрировали при пониженном давлении. Концентрированный остаток очищали на колонке с силикагелем (петролейный эфир/этилацетат = 1/0-3/1) с получением соединения 10-2. 1H ЯМР (400 МГц, CDCl3) δ ppm 8.98 (d, J=2.3 Гц, 1H), 8.52 (dd, J=2.3, 8.5 Гц, 1H), 8.05 (d, J=8.3 Гц, 1H), 4.09 (s, 3Н).
Синтез соединения 10-3
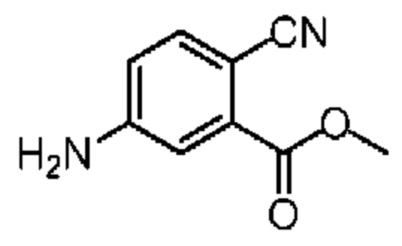
Раствор соединения 10-2 (700,00 мг, 3,40 ммоль, 1,00 экв.) в ледяной уксусной кислоте (10,00 мл) добавляли к восстановленному порошковому железу (1,90 г, 34,00 ммоль, 10,00 экв.). Полученную реакционную смесь перемешивали при 20°С в течение 16 часов. Реакционную смесь фильтровали при помощи диатомитовой земли и концентрировали при пониженном давлении. К концентрированному остатку добавляли этилацетат (100 мл) и насыщенный водный раствор бикарбоната натрия (рН 7-8). Органическую фазу промывали насыщенным солевым раствором (60 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрированный остаток очищали на колонке с силикагелем (петролейный эфир/этилацетат = 1/0-1/1) с получением соединения 10-3. 1H ЯМР (400 МГц, CDCl3) δ ppm 7.47 (d, J=8.3 Гц, 1H), 7.26 (d, J=2.5 Гц, 1Н), 6.73 (dd, J=2.5, 8.3 Гц, 1H), 4.21 (br s, 2H), 3.90 (s, 3H).
Синтез соединения 10-4
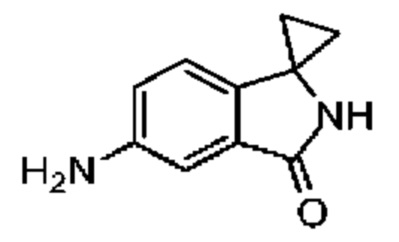
При -78°C к раствору соединения 10-3 (100,00 мг, 567,63 мкмоль, 1,00 экв.) и Ti(i-PrO)4 (643,20 мг, 2,26 ммоль, 670,00 мкл, 3,99 экв.) в тетрагидрофуране (2,00 мл) добавляли этилбромид магния (3 М, 1,50 мл, 7,93 экв.). Полученную реакционную смесь перемешивали при температуре от -78°С до 10°С (при медленном нагревании) в течение 18 часов. К реакционной смеси добавляли насыщенный раствор хлорида аммония (20 мл) с образованием вязкой взвеси. Этилацетат (20 мл) добавляли. Полученную смесь перемешивали в течение 10 минут, а затем фильтровали. Фильтрат разделяли на две фазы. Органическую фазу промывали насыщенным солевым раствором (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении.
Концентрированный остаток очищали методом тонкослойной хроматографии на пластине с получением соединения 10-4. LCMS: MS (ESI) m/z: 174.9 (М+1).
Синтез соединения 10
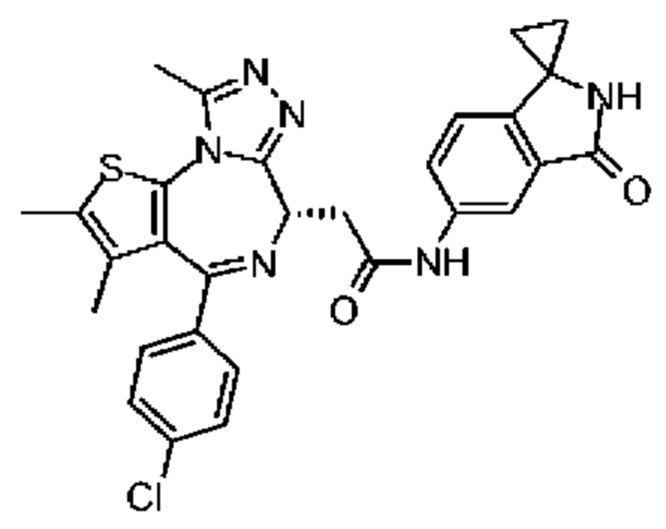
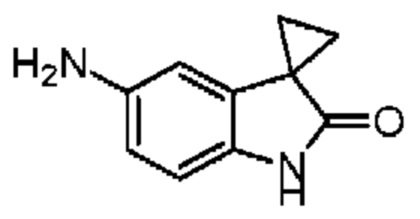
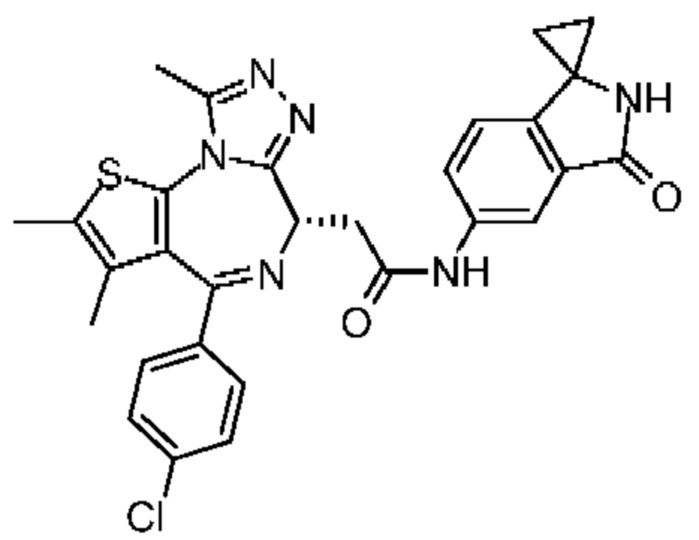
Соединение 1-11 (25,00 мг, 62,36 мкмоль, 1,00 экв.), соединение 10-4 (11,95 мг, 68,60 мкмоль, 1,10 экв.), HATU (28,45 мг, 74,84 мкмоль, 1,20 экв.) и триэтиламин (15,78 мг, 155,91 мкмоль, 21,61 мкл, 2,50 экв.) растворяли в безводном метиленхлориде (1,00 мл). Смесь перемешивали при 20°С при защите газообразным азотом в течение 2 часов. Реакционную смесь разбавляли метиленхлоридом (10 мл) и промывали водой (5 мл) и насыщенным солевым раствором (5 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали методом препаративной хроматографии с получением соединения 10. 1H ЯМР (400 МГц, CDCl3) δ ppm 8.10 (br s, 1H), 7.94 (dd, J=2.0, 8.4 Гц, 1H), 7.43 (d, J=8.4 Гц, 2H), 7.35 (d, J=8.8 Гц, 2H), 6.96 (d, J=8.0 Гц, 1H), 6.83 (s, 1H), 4.72-4.76 (m, 1H), 3.86-3.92 (m, 1H), 3.61-3.66 (m, 1H), 2.71 (s, 3H), 2.43 (s, 3H), 1.71 (s, 3H), 1.54-1.57 (m, 2H),1.35-1.50 (m, 2H). LCMS (ESI) m/z: 557.1 (M+1).
Схема 11
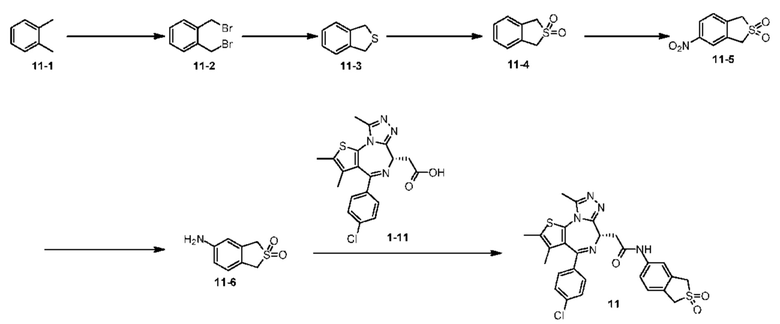
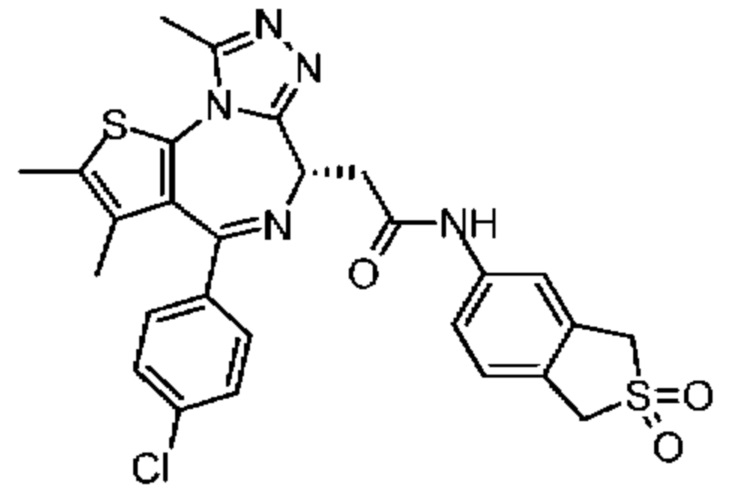
Пример 11
Синтез соединения 48-2
Смесь орто-ксилола (5,00 г, 47,09 ммоль, 5,68 мл, 1,00 экв.), NBS (17,60 г, 98,89 ммоль, 2,10 экв.), бензоилпероксида (228,13 мг, 941,80 мкмоль, 0,02 экв.) и хлороформа (50,00 мл) перемешивали при 80°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, затем разбавляли метиленхлоридом (100 мл), промывали водой (80 мл × 2) и промывали насыщенным солевым раствором (50 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученное твердое вещество переводили во взвесь (петролейный эфир/этанол = 30:1; 60 мл/2 мл) при 80°С в течение 20 минут, охлаждали до комнатной температуры и фильтровали. Фильтрационный кек промывали петролейным эфиром (20 мл). Фильтрационный кек сушили в сушильном шкафу с получением соединения 11-2. 1Н ЯМР (400МГц, CDCl3) δ ppm 7.40-7.36 (m, 2Н), 7.34-7.30 (m, 2Н), 4.68 (s, 4Н).
Синтез соединения 11-3
Смесь соединения 11-2 (6,00 г, 22,73 ммоль, 3,06 мл, 1,00 экв.), сульфида натрия нонагидрата (16,38 г, 68,19 ммоль, 11,45 мл, 3,00 экв.), бензилтриэтиламмония хлорида (258,86 мг, 1,14 ммоль, 0,05 экв.), метиленхлорида (60,00 мл) и воды (60,00 мл) перемешивали при 18°С в темноте в течение 24 часов. Смесь экстрагировали метиленхлоридом (100 мл). Органическую фазу промывали водой (80 мл × 5), промывали насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом натрия, и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 11-3, которое сразу использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ ppm 7.20-7.14 (m, 4Н), 4.21 (s, 4Н).
Синтез соединения 11-4
Соединение 11-3 (2,80 г, 20,56 ммоль, 1,00 экв.) растворяли в ледяной уксусной кислоте (15,00 мл) при 5-10°С, а затем по каплям добавляли пероксид водорода (5,90 г, 52,02 ммоль, 5,00 мл, 30% чистоты, 2,53 экв.). После завершения добавления по каплям смесь перемешивали при 20°С в течение 1 часа, а затем нагревали до 90°С и перемешивали в течение 3 часов. Реакционную смесь охлаждали и твердое вещество осаждалось. После фильтрации фильтрационный кек промывали водой (20 мл). Фильтрационный кек переводили во взвесь при помощи этанола (20 мл) при 80°С в течение 20 минут и полученную смесь фильтровали. Фильтрационный кек сушили в сушильном шкафу (1 г). Фильтрат помещали в течение 24 часов и твердое вещество осаждалось. После фильтрации фильтрационный кек сушили в сушильном шкафу (1 г). Фильтрационные кеки объединяли с получением соединения 11-4, которое сразу использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ ppm 7.41-7.35 (m, 2Н), 7.35-7.29 (m, 2Н), 4.39 (s, 4Н).
Синтез соединения 11-5
Соединение 11-4 (300,00 мг, 1,78 ммоль, 1,00 экв.) растворяли в концентрированной серной кислоте (2,00 мл) при -10°С, а затем добавляли нитрат калия (180,31 мг, 1,78 ммоль, 1.00 экв.). Смесь перемешивали при -10°С в течение 5 минут. При -10°С добавляли ледяные кубики (20 г) для гашения реакции. Ледяные кубики таяли и твердое вещество осаждалось. После фильтрации фильтрационный кек промывали водой (10 мл). Фильтрационный кек сушили в сушильном шкафу с получением соединения 11-5, которое сразу использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, DMSO-d6) δ ppm 8.30 (s, 1H), 8.24 (dd, J=2.0, 8.5 Гц, 1Н), 7.69 (d, J=8.5 Гц, 1H), 4.67 (d, J=8.3 Гц, 4H).
Синтез соединения 11-6
Соединение 11-5 (200,00 мг, 938,04 мкмоль, 1,00 экв.) и дигидрат двухлористого олова (846,68 мг, 3,75 ммоль, 312,43 мкл, 4,00 экв.) растворяли в этаноле (3,00 мл), а затем по каплям добавляли концентрированную хлористоводородную кислоту (1,32 г, 5,85 ммоль, 1,20 мл, 37% чистоты, 6,23 экв.) при 15°С. После завершения добавления по каплям смесь перемешивали при 80°С в течение 1 часа, доводили значение рН при помощи раствора NaOH (2 н) до 10. Смесь концентрировали при пониженном давлении до приблизительно 50 мл, а затем экстрагировали (метиленхлорид/метанол = 10:1) (40 мл × 6).
Объединенные органические фазы промывали насыщенным солевым раствором (50 мл × 2). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 11-6, которое сразу использовали на следующей стадии без дополнительной очистки. LCMS (ESI) m/z: 184.1, 206.0 (М+1), (М+23). 1H ЯМР (400 МГц, DMSO-d6) δ ppm 6.98 (d, J=8.3 Гц, 1H), 6.56-6.52 (m, 1H), 6.50 (s, 1H), 5.31 (br s, 2H), 4.30 (s, 2H), 4.24 (s, 2H).
Синтез соединения 11
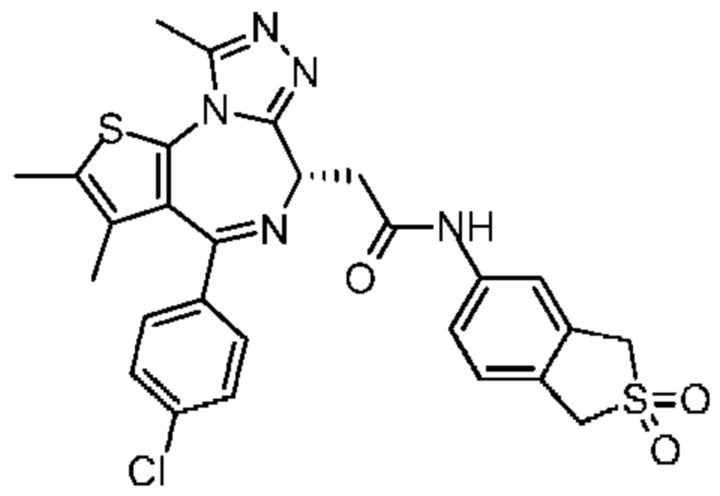
При 15°C и при защите газообразным азотом соединение 1-11 (1,00 г, 2,49 ммоль, 1,00 экв.) и соединение 11-6 (547,49 мг, 2,99 ммоль, 1,20 экв.) растворяли в безводном N,N-диметилформамиде (15,00 мл). Добавляли HATU (946,77 мг, 2,49 ммоль, 1,00 экв.) и по каплям добавляли диизопропилэтиламин (965,42 мг, 7,47 ммоль, 1,30 мл, 3,00 экв.). Смесь перемешивали при 15°С в атмосфере азота в течение 1 часа. Реакционную смесь сразу сушили. Полученное твердое вещество промывали водой при встряхивании (40 мл). Смесь экстрагировали метиленхлоридом (30 мл × 2). Органическую фазу промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 11. LCMS (ESI) m/z: 567.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δ ppm 9.68 (s, 1H), 7.57 (s, 1H), 7.22-7.37 (m, 5H), 7.02 (d, J=8.8 Гц, 1H), 4.60-4.63 (m, 1H), 4.17-4.21 (m, 4H), 3.82-3.88 (m, 1H), 3.38-3.43 (m, 1H), 2.62 (s, 3H), 2.35 (s, 3H),1.63 (s, 3H).
Схема 12
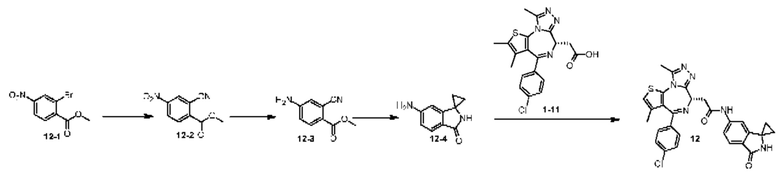
Пример 12
Синтез соединения 12-2
Смешанную жидкость соединения 12-1 (2,00 г, 7,69 ммоль, 1,00 экв.), CuCN (1,04 г, 11,61 ммоль, 2,54 мл, 1,51 экв.), dppf (426,38 мг, 769,00 мкмоль, 0,10 экв.), трис(дибензилиденацетон)дипалладия (442,25 мг, 769,00 мкмоль, 0,10 экв.) и N,N-диметилформамида (20,00 мл) нагревали до 120°С и перемешивали в течение 16 часов. Реакционную смесь фильтровали при помощи диатомитовой земли. Фильтрат концентрировали при пониженном давлении. Концентрированный остаток очищали на колонке с силикагелем (петролейный эфир/этилацетат = 1/0-1/1) с получением соединения 12-2. 1H ЯМР (400 МГц, CDCl3) δ ppm 8.65 (d, J=2.3 Гц, 1H), 8.51 (dd, J=2.3, 8.8 Гц, 1H), 8.36 (d,J=8.5 Гц, 1H), 4.08 (s, 3Н).
Синтез соединения 12-3
К раствору соединения 12-2 (900,00 мг, 4,37 ммоль, 1,00 экв.) в ледяной уксусной кислоте (20,00 мл) добавляли восстановленное порошковое железо (2,44 г, 43,70 ммоль, 10,00 экв.). Полученную реакционную смесь перемешивали при 20°С в течение 4 часов. Реакционную смесь фильтровали при помощи диатомитовой земли и концентрировали при пониженном давлении. К концентрированному остатку добавляли этилацетат (150 мл) и насыщенный водный раствор бикарбоната натрия (рН 7-8). Органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрированный остаток очищали методом тонкослойной хроматографии на пластине с получением соединения 12-3. LCMS (ESI) m/z: 177.1 (М+1).
Синтез соединения 12-4
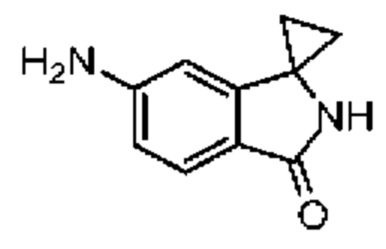
При -78°С к раствору соединения 12-3 (490,00 мг, 2,78 ммоль, 1,00 экв.) и Ti(i-PrO)4 (3,07 г, 10,81 ммоль, 3,20 мл, 3,89 экв.) в тетрагидрофуране (15,00 мл) добавляли этилбромид магния (3 М, 7,40 мл, 7,99 экв.). Полученную реакционную смесь перемешивали при температуре от -78°С до 15°С (при медленном нагревании) в течение 16 часов. Как только прошла реакция, осаждалось желтое твердое вещество, и реакционная смесь постепенно становилась земельно-желтой и вязкой. К реакционной смеси добавляли насыщенный раствор хлорида аммония (30 мл) и образовывалась вязкая взвесь. Этилацетат (30 мл) добавляли. Смесь перемешивали в течение 10 минут, а затем фильтровали при помощи диатомитовой земли. Фильтрат разделяли на две фазы. Органическую фазу промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрированный остаток очищали методом тонкослойной хроматографии на пластине с получением соединения 12-4. LCMS (ESI) m/z: 174.9 (М+1). 1Н ЯМР (400 МГц, CDCl3) δ ppm 7.65 (d, J=8.0 Гц, 1H), 6.68 (dd, J=2.0, 8.3 Гц, 1H), 6.43 (br s, 1Н), 6.23 (d, J=1.8 Гц, 1H), 4.04 (br s, 2H), 1.52-1.46 (m, 2H), 1.37-1.32 (m, 2H).
Синтез соединения 12
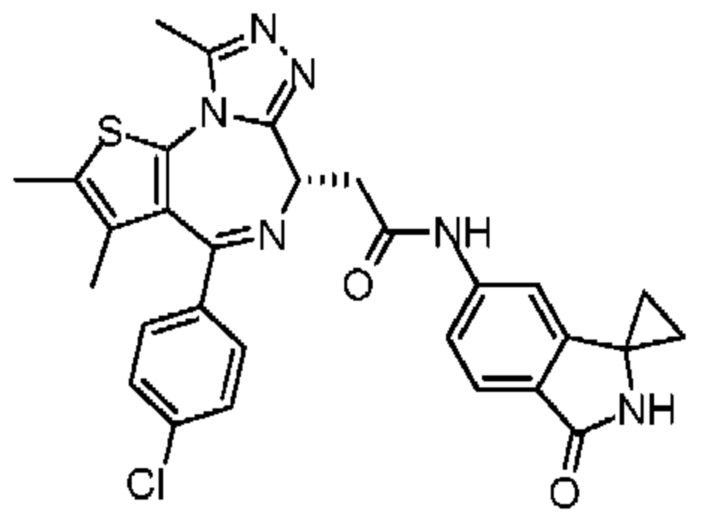
Соединение 12-4 (15,64 мг, 89,80 мкмоль, 1,20 экв.) и диизопропилэтиламин (29,02 мг, 224,51 мкмоль, 39,21 мкл, 3,00 экв.) растворяли в безводном N,N-диметилформамиде (300 мл). Добавляли соединение 1-11 (30,00 мг, 74,84 мкмоль, 1,00 экв.) и HATU (28,45 мг, 74,84 мкмоль, 1,00 экв.). Атмосферу заменяли газообразным азотом три раза. Смесь перемешивали при 15°С в атмосфере азота в течение 16 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Остаток растворяли в метиленхлориде (15 мл), а затем промывали водой при встряхивании (10 мл). Органическую фазу промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом препаративной хроматографии с получением соединения 12. LCMS (ESI) m/z: 557.2 (M+1). 1H ЯМР (400 МГц, CDCl3) δ ppm 9.91 (br s, 1H), 7.68 (d, J=8.0 Гц, 1H), 7.63 (s, 1H), 7.44 (d, J=8.8 Гц, 2H), 7.35 (d, J=8.8 Гц, 2H), 7.22 (d, J=8.0 Гц, 1H), 4.70-4.74 (m, 1H), 3.95-3.99 (m, 1H), 3.50-3.54 (m, 1H), 2.73 (s, 3H), 2.45 (s, 3H), 1.72 (s, 3H), 1.46-1.51 (m, 2H), 1.39-1.41 (m, 2H).
Схема 13
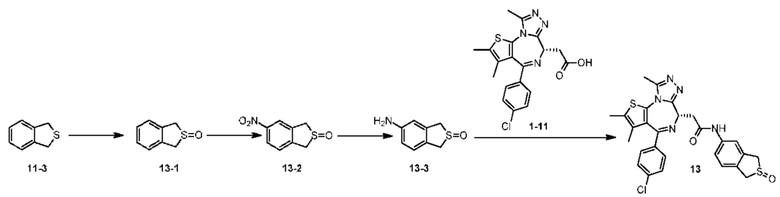
Пример 13
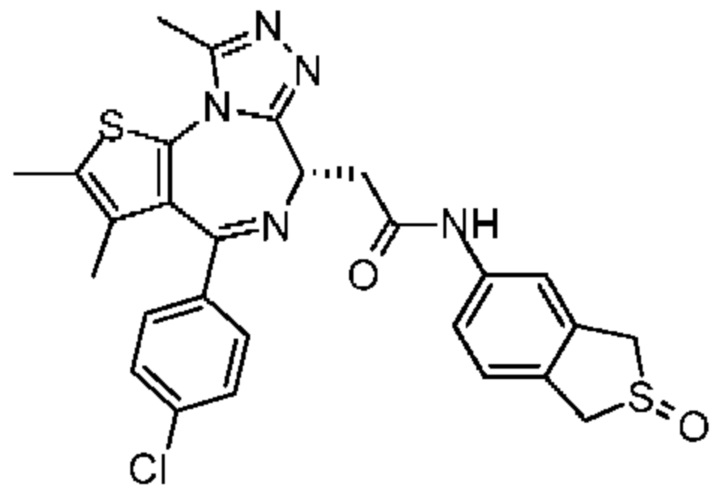
Синтез соединения 13-1
Соединение 11-3 (1,80 г, 13,21 ммоль, 1,00 экв.) растворяли в метаноле (15,00 мл), а затем по каплям добавляли раствор периодата натрия (2,83 г, 13,21 ммоль, 732,26 мкл, 1,00 экв.) в H2O (15,00 мл). После завершения добавления по каплям смесь перемешивали при 15°С в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении до приблизительно 10 мл и экстрагировали этилацетатом (20 мл × 5). Объединенные органические фазы промывали насыщенным солевым раствором (40 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 13-1, которое сразу использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ ppm 7.40-7.36 (m, 2Н), 7.36-7.31 (m, 2Н), 4.34-4.27 (m, 2Н), 4.22-4.13 (m, 2Н).
Синтез соединения 13-2
Соединение 13-1 (600,00 мг, 3,94 ммоль, 1,00 экв.) растворяли в концентрированной серной кислоте (5,00 мл) при -10°С, а затем добавляли нитрат калия (398,53 мг, 3,94 ммоль, 1,00 экв.). Смесь перемешивали при -10°С в течение 5 минут. При -10°С добавляли ледяные кубики (20 г) для гашения реакции. Ледяные кубики таяли и смесь экстрагировали (метиленхлорид/метанол = 10:1) (30 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (40 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 13-2, которое сразу использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, DMSO-d6) δ ppm 8.30 (s, 1Н), 8.24 (dd, J=2.0, 8.5 Гц, 1H), 7.69 (d, J=8.5 Гц, 1H), 4.67 (d, J=8.3 Гц, 4H).
Синтез соединения 13-3
Соединение 13-2 (300,00 мг, 1,52 ммоль, 1,00 экв.) растворяли в этаноле (8,00 мл), а затем добавляли дигидрат двухлористого олова (686,53 мг, 3,04 ммоль, 253,33 мкл, 2,00 экв.). Смесь перемешивали при 80°С в течение 1 часа. Значение рН смеси доводили при помощи раствора гидроксида натрия (1 н) до 10, концентрировали при пониженном давлении приблизительно до 50 мл, затем экстрагировали (метиленхлорид/метанол = 10:1) (40 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (60 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 13-3. LCMS (ESI)m/z: 167.8 (M+1).
Синтез соединения 13
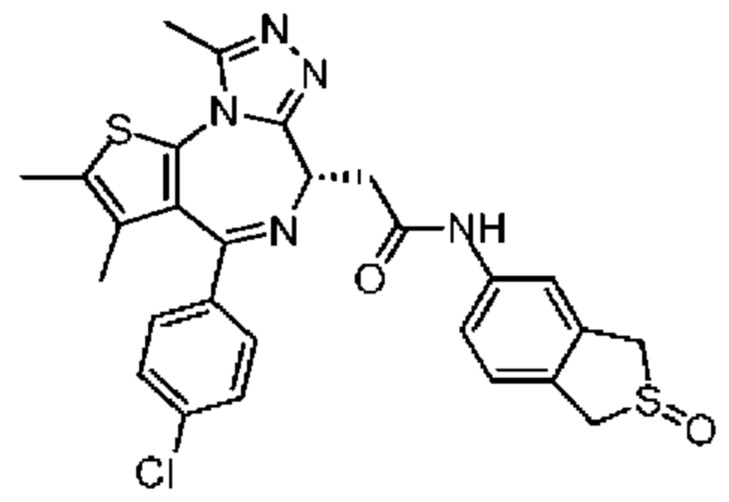
Соединение 1-11 (60,00 мг, 149,67 мкмоль, 1,00 экв.) и соединение 13-3 (30,04 мг, 179,60 мкмоль, 1,20 экв.) последовательно добавляли к раствору HATU (74,00 мг, 194,62 мкмоль, 1,30 экв.) в безводном N,N-диметилформамиде (3,00 мл), а затем медленно по каплям добавляли диизопропилэтиламин (59,20 мг, 458,06 мкмоль, 80,00 мкл, 3,06 экв.). Смесь перемешивали при 15°С при защите газообразным азотом в течение 6 часов. К реакционной смеси добавляли воду (3 мл) для гашения реакции. Смесь экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали методом препаративной хроматографии с получением соединения 13. 1H ЯМР (400 МГц, CDCl3) δ ppm 9.35-9.73 (m, 1H), 7.54-7.64 (m, 1H), 7.22-7.38 (m, 5 H), 7.09-7.23 (m, 1H), 4.62-4.64 (m, 1H), 3.94-4.18 (m, 4H), 3.81-3.93 (m, 1H), 3.40-3.50 (m, 1H), 2.62 (s, 3H), 2.34 (s, 3H), 1.62 (s, 3H). LCMS (ESI) m/z: 550.1 (M+1).
Схемы 14 и 15
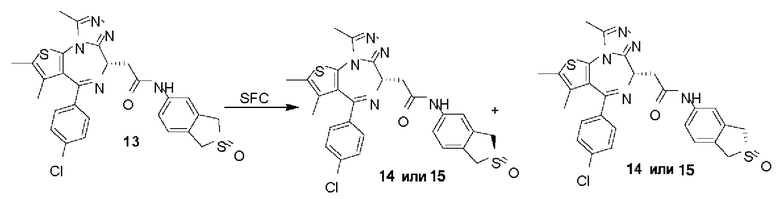
Примеры 14 и 15
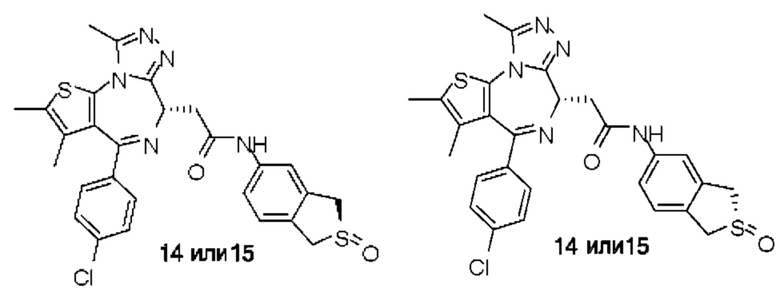
Соединение 13 (38 мг, 69,08 мкмоль) подвергали разделению методом SFC (хроматографическая колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% NH3H2O EtOH]; В%: 55%-55%, 50 мл/мин) с получением соединения 14 (Rt=0,837 мин). 1Н ЯМР (400 МГц, CDCl3) δ ppm 9.49 (s, 1H), 7.59 (s, 1H), 7.25-7.38 (m, 5Н), 7.12 (d, 7=8.0 Гц, 1Н), 4.61-4.64 (m, 1Н), 3.99-4.18 (m, 4Н), 3.74-3.77 (m, 1Н), 3.44-3.50 (m, 1Н), 2.62 (s, 3Н), 2.34 (s, 3Н), 1.61 (s, 3Н). LCMS (ESI) m/z: 550.0 (М+1).
Соединение 15 (Rt=1,666 мин). 1Н ЯМР (400 МГц, CDCl3) δ ppm 9.73 (s, 1H), 7.54 (s, 1Н), 7.22-7.38 (m, 5Н), 7.03 (d, J=8.8 Гц, 1H), 4.65-4.68 (m, 1Н), 4.06-4.13 (m, 2Н), 3.88-3.98 (m, 3Н), 3.40-3.50 (m, 1H), 2.62 (s, 3Н), 2.34 (s, 3Н), 1.62 (s, 3Н). LCMS (ESI) m/z: 550.0 (М+1).
Схема 16
Пример 16
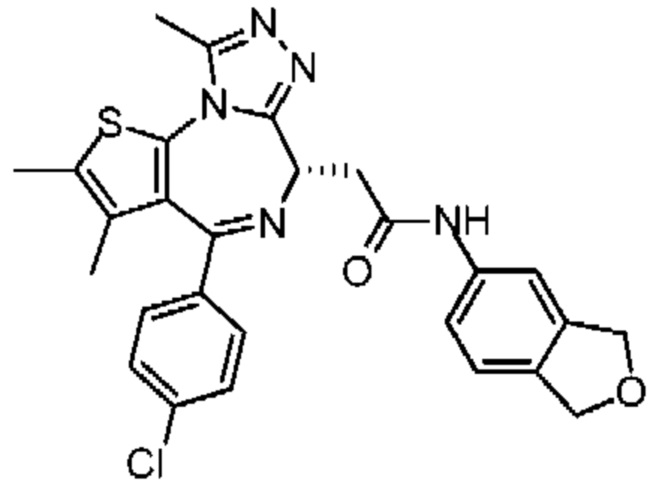
Соединение 1-11 (60,00 мг, 149,67 мкмоль, 1,00 экв.) и соединение 16-1 (24,28 мг, 179,60 мкмоль, 1,20 экв.) растворяли в безводном метиленхлориде (5,00 мл), добавляли HATU (56,91 мг, 149,67 мкмоль, 1,00 экв.) и по каплям добавляли диизопропилэтиламин (58,03 мг, 449,01 мкмоль, 78,42 мкл, 3,00 экв.). Смесь перемешивали при 15°С в атмосфере азота в течение 1 часа. Реакционную смесь промывали водой при встряхивании (10 мл). Органическую фазу промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом препаративной хроматографии с получением соединения 16.
LCMS (ESI) m/z: 518.0 (М+1). 1H ЯМР (400 МГц, CDCl3) δ ppm 8.88 (s, 1Н), 7.62 (s, 1H), 7.44 (d, J=8.8 Гц, 2H), 7.36 (d, J=8.8 Гц, 2H), 7.32-7.34 (m, 1H),7.16 (d, J=8.0 Гц, 1H), 5.08 (s, 4H), 4.62-4.65 (m, 1H), 3.79-3.85 (m, 1H), 3.48-3.53 (m, 1H), 2.71 (s, 3H), 2.43 (s, 3H), 1.71 (s, 3H).
Схема 17
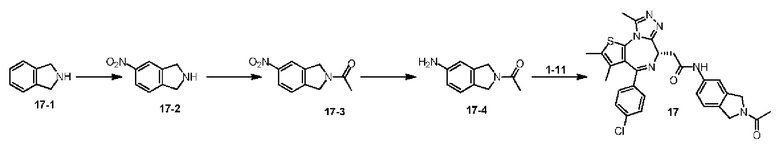
Пример 17
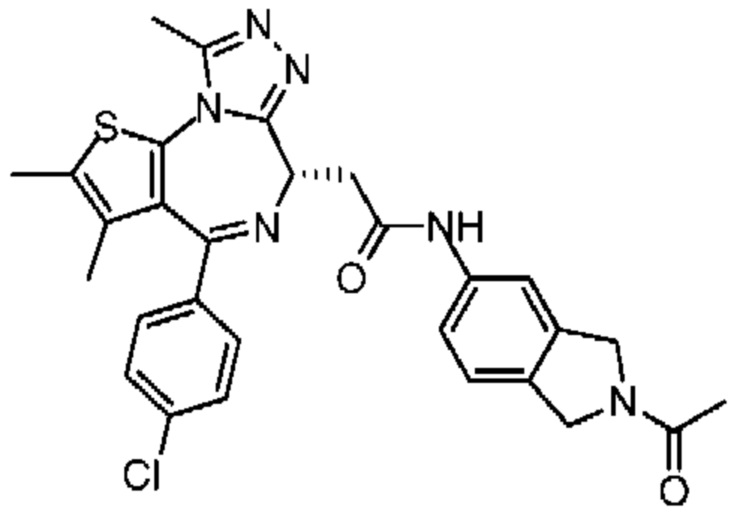
Синтез соединения 17-2
При -20°С и при защите газообразным азотом концентрированную серную кислоту (16,00 мл) медленно добавляли к раствору соединения 17-1 (3,90 г, 32,73 ммоль, 3,71 мл, 1,00 экв.), растворенного в метиленхлориде (10,00 мл). После завершения добавления по каплям смесь медленно нагревали до 20°С. Затем метиленхлорид удаляли при пониженном давлении с получением светло-коричневого раствора. Концентрированную азотную кислоту (5,60 г, 62,19 ммоль, 4,00 мл, 1,90 экв.) (содержимое приблизительно 70%) медленно по каплям добавляли к вышеуказанному светло-коричневому раствору с обеспечением внутренней температуры, которая не превышала 20°С. После завершения добавления по каплям смесь перемешивали в течение 0,5 часа. Реакционную смесь медленно добавляли к ледяной воде (300 мл), а затем значение рН при помощи твердого бикарбоната натрия доводили до 8. Добавляли метил-трет-бутиловый эфир (300 мл). Смесь перемешивали в течение 1 часа. Органическую фазу отделяли. Водную фазу экстрагировали метил-трет-бутиловым эфиром (200 мл × 2). Объединенные органические фазы промывали насыщенным солевым раствором (300 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 17-2, которое сразу использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ ppm 8.05 (dd, J=2.0, 8.0 Гц, 1Н), 8.03 (s, 1H), 7.30 (d, J=8.0 Гц, 1H), 4.24 (s, 4H).
Синтез соединения 17-3
При 0°С к раствору соединения 17-2 (150,00 мг, 913,74 мкмоль, 1,00 экв.) и триэтиламина (277,38 мг, 2,74 ммоль, 379,98 мкл, 3,00 экв.) в безводном метиле нхлориде (5,00 мл) добавляли ацетилхлорид (71,73 мг, 913,74 мкмоль, 65,21 мкл, 1,00 экв.). Смесь реагировала при перемешивании при 20°С в течение 2 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 17-3. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8.08-8.15 (m, 2Н), 7.35-7.41 (m, 1H), 4.84 (s, 2Н), 4.81 (s, 2Н), 2.13 (s, 3Н).
Синтез соединения 17-4
Влажный Pd/C (100,00 мг, 10% Pd) добавляли к раствору соединения 17-3 (140,00 мг, 678,95 мкмоль, 1,00 экв.) в метаноле (3.00 мл). Атмосферу заменяли газообразным водородом три раза. Смесь перемешивали при 15°С в водородных условиях (баллон) (15 фунт/кв.дюйм) в течение 18 часов. Реакционную смесь фильтровали при помощи диатомитовой земли. Фильтрат сразу концентрировали при пониженном давлении с получением соединения 17-4, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ ppm 7.01-7.07 (m, 1H), 6.57-6.65 (m, 2Н), 4.67-4.73 (m, 4Н), 2.15 (s, 3Н).
Синтез соединения 17
Соединение 1-11 (200,00 мг, 498,90 мкмоль, 1,00 экв.) и соединение 17-4 (100,00 мг, 567,50 мкмоль, 1,14 экв.) последовательно добавляли к раствору HATU (240,00 мг, 631,20 мкмоль, 1,27 экв.) в безводном N,N-диметилфорамиде (5,00 мл), а затем медленно по каплям добавляли триэтиламин (146,00 мг, 1,44 ммоль, 200,00 мкл, 2,89 экв.). Смесь перемешивали при 15°С при защите газообразным азотом в течение 4 часов. Воду (5 мл) добавляли к реакционной смеси для гашения реакции. Смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом препаративной хроматографии с получением соединения 17. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9.16-9.20 (m, 1H), 7.61-7.70 (m, 1Н), 7.31-7.43 (m, 5 H), 7.14-7.18 (m, 1H), 4.62-4.76 (m, 5 H), 3.81-3.84 (m, 1H), 3.47-3.52 (m, 1H), 2.69 (s, 3H), 2.41 (s, 3H), 2.15 (s, 3H), 1.69 (s, 3H). LCMS (ESI) m/z: 559.1 (M+1).
Схема 18
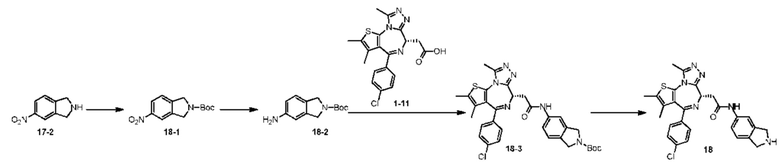
Пример 18
Синтез соединения 18-1
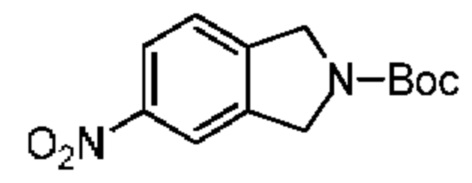
Соединение 17-2 (30,00 мг, 182,75 мкмоль, 1,00 экв.) и (Вос)2O (47,50 мг, 217,47 мкмоль, 50,00 мкл, 1,19 экв.) последовательно добавляли к раствору 4-диметиламинопиридина (1,00 мг, 8,19 мкмоль, 0,04 экв.) в безводном метиленхлориде (3,00 мл). Затем к реакционной смеси медленно по каплям добавляли триэтиламин (55,48 мг, 548,25 мкмоль, 76,00 мкл, 3,00 экв.). Смесь перемешивали при 15°С при защите газообразным азотом в течение 6 часов. Воду (3 мл) добавляли к реакционной смеси для гашения реакции. Смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 18-1, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8.00-8.15 (m, 2Н), 7.28-7.38 (m, 1Н), 4.68 (d, J=10.8 Гц, 4H), 1.45 (s, 9H).
Синтез соединения 18-2
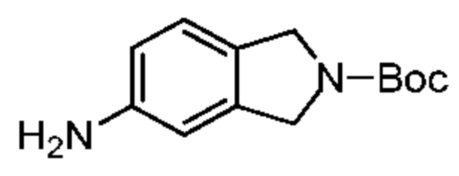
Влажный Pd/C (100,00 мг, 10% Pd) добавляли к раствору соединения 18-1 (40,00 мг, 151,35 мкмоль, 1,00 экв.) в метаноле (3,00 мл). Атмосферу заменяли газообразным водородом три раза. Смесь перемешивали при 15°С в водородных условиях (баллон) (15 фунт/кв.дюйм) в течение 3 часов. Реакционную смесь фильтровали при помощи диатомитовой земли. Фильтрат сразу концентрировали при пониженном давлении с получением соединения 18-2, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ ppm 6.86-6.99 (m, 1Н), 6.44-6.56 (m, 2H), 4.41-4.53 (m, 4H), 1.43 (s, 9H).
Синтез соединения 18-3
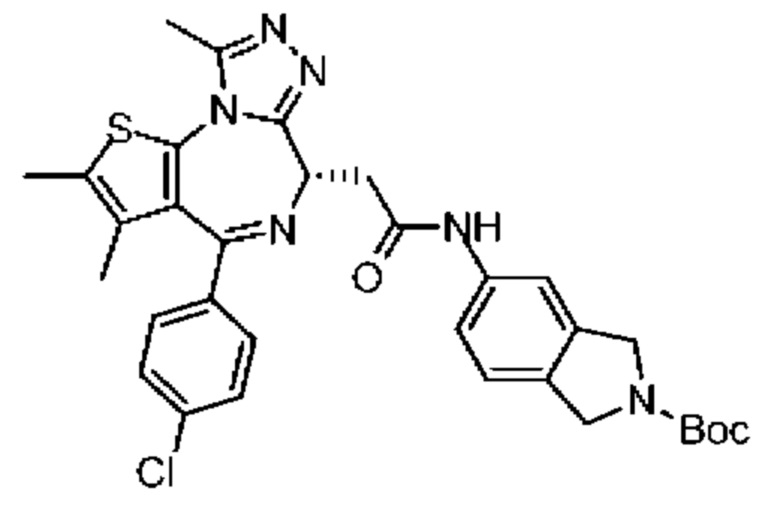
Соединение 1-11 (40,00 мг, 99,78 мкмоль, 1,00 экв.) и соединение 18-2 (30,00 мг, 128,05 мкмоль, 1,28 экв.) последовательно добавляли к раствору HATU (48,00 мг, 126,24 мкмоль, 1,27 экв.) в безводном N,N-диметилфорамиде (3,00 мл), а затем медленно по каплям добавляли триэтиламин (29,20 мг, 288,57 мкмоль, 40,00 мкл, 2,89 экв.). Смесь перемешивали при 15°С при защите газообразным азотом в течение 4 часов. Воду (3 мл) добавляли к реакционной смеси для гашения реакции. Смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом препаративной хроматографии с получением соединения 18-3. LCMS (ESI) m/z: 617.0 (M+1).
Синтез соединения 18
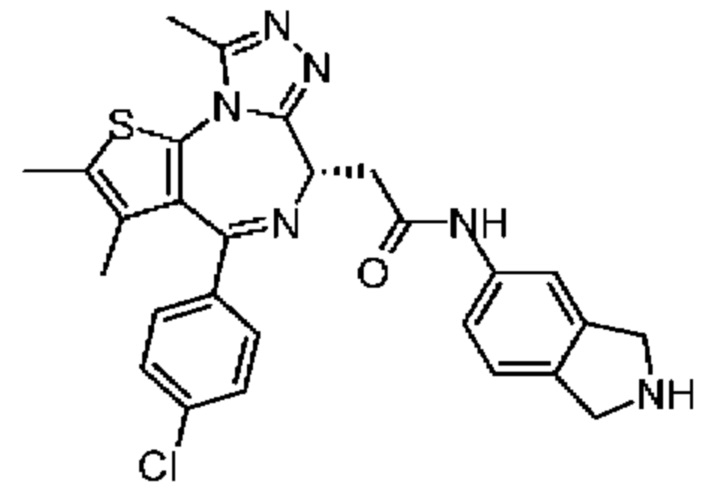
При 15°С и при защите газообразным азотом трифторуксусную кислоту (3,08 г, 27,01 ммоль, 2,00 мл, 333,41 экв.) добавляли к раствору соединения 18-3 (50,00 мг, 81,02 мкмоль, 1,00 экв.) в безводном метиленхлориде (6,00 мл). Смесь перемешивали при 15°С в течение 4 часов. Реакционную смесь сразу концентрировали при пониженном давлении, значение рН при помощи насыщенного водного раствора бикарбоната натрия доводили до 7 и экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным солевым раствором (5 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом препаративной хроматографии с получением соединения 18. 1H ЯМР (400 МГц, CDCl3) δppm 9.12 (s, 1H), 7.57 (s, 1H), 7.32-7.47 (m, 5H), 7.13-7.15 (m, 1H), 4.68-4.71 (m, 1H), 3.82-3.88 (m, 1H), 3.50-3.55 (m, 1H), 2.71 (s, 3H), 2.43 (s, 3H), 1.70 (s, 3H). LCMS (ESI) m/z: 539.1 (M+1).
Схема 19
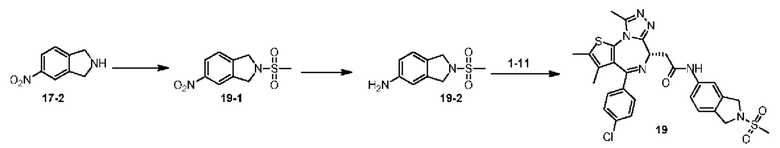
Пример 19
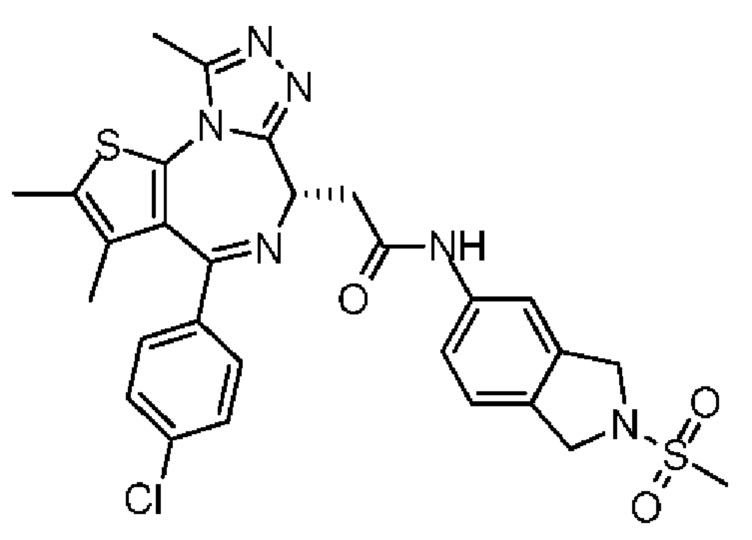
Синтез соединения 19-1
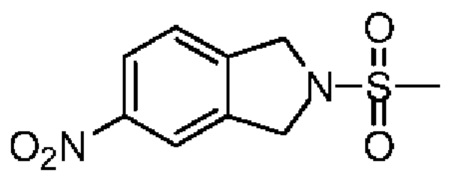
Триэтиламин (948,78 мг, 9,38 ммоль, 1,30 мл, 2,96 экв.) добавляли к раствору соединения 17-2 (520,00 мг, 3,17 ммоль, 1,00 экв.) в метиленхлориде (5,00 мл). Смесь охлаждали до 0°С при защите газообразным азотом. К реакционной смеси медленно по каплям добавляли метилсульфонилхлорид (370,00 мг, 3,23 ммоль, 250,00 мкл, 1,02 экв.). После завершения добавления по каплям смесь нагревали до 15°С и перемешивали в течение 2 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 19-1. 1H ЯМР (400 МГц, CDCl3) δppm 8.24 (d, J=8.4 Гц, 1H), 8.17 (s, 1H), 7.46 (d, J=8.4 Гц, 1H), 4.83 (s, 4H), 2.97 (s, 3H).
Синтез соединения 19-2
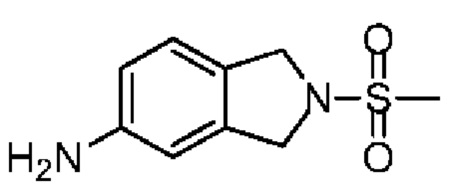
Влажный Pd/C (100,00 мг, 10% Pd) добавляли к раствору соединения 19-1 (50,00 мг, 206,40 мкмоль, 1,00 экв.) в метаноле (3,00 мл). Атмосферу заменяли газообразным водородом три раза. Смесь перемешивали при 15°С в водородных условиях (баллон) (15 фунт/кв.дюйм) в течение 3 часов. Реакционную смесь фильтровали при помощи диатомитовой земли. Фильтрат сразу концентрировали при пониженном давлении с получением соединения 19-2, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δppm 6.95 (d, J=8.28 Гц, 1H), 6.54-6.57 (m, 1H), 6.50 (s, 1H), 4.52 (s, 4H), 2.78 (s, 3H).
Синтез соединения 19
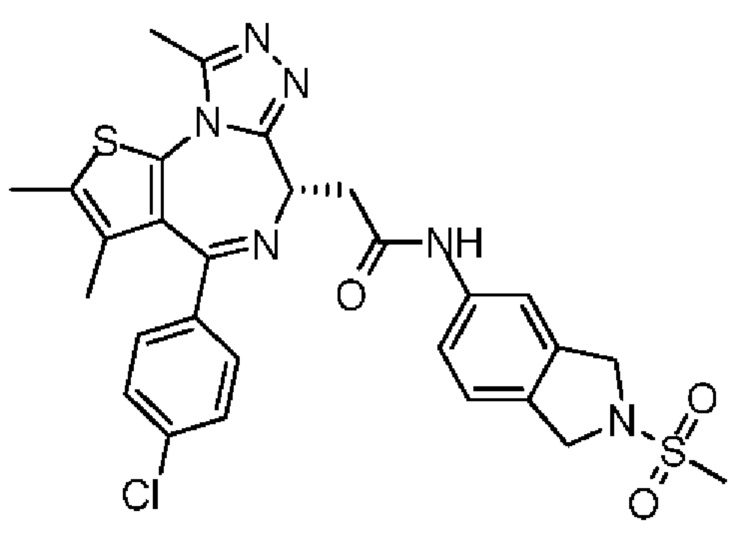
Соединение 1-11 (60,00 мг, 149,67 мкмоль, 1,00 экв.) и соединение 19-2 (40,00 мг, 188,58 мкмоль, 1,26 экв.) последовательно добавляли к раствору HATU (73,00 мг, 191,99 мкмоль, 1,28 экв.) в безводном метиленхлориде (3,00 мл), а затем медленно по каплям добавляли триэтиламин (43,80 мг, 432,55 мкмоль, 60,00 мкл, 2,89 экв.). Смесь перемешивали при 15°С при защите газообразным азотом в течение 2 часов. Воду (3 мл) добавляли к реакционной смеси для гашения реакции. Смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 19. 1H ЯМР (400 МГц, CDCl3) δppm 9.82 (s, 1Н), 7.51 (s, 1H), 7.17-7.40 (m, 5H) 6.96-6.98 (m, 1H), 4.68-4.70 (m, 1H), 4.49 (d, J=17.2 Гц, 4H), 3.83-3.89 (m, 1H),3.46-3.51 (m, 1H), 2.82 (s, 3H), 2.62 (s, 3H), 2.35 (s, 3H), 1.62 (s, 3H). LCMS (ESI) m/z: 595 (M+1).
Схема 20
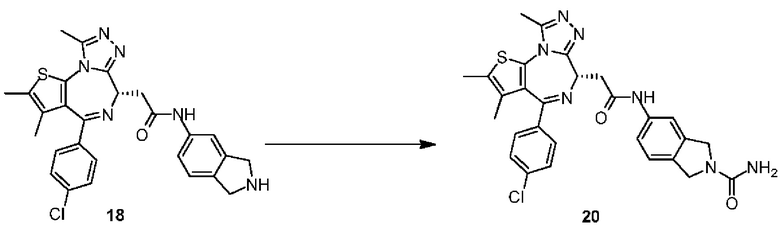
Пример 20
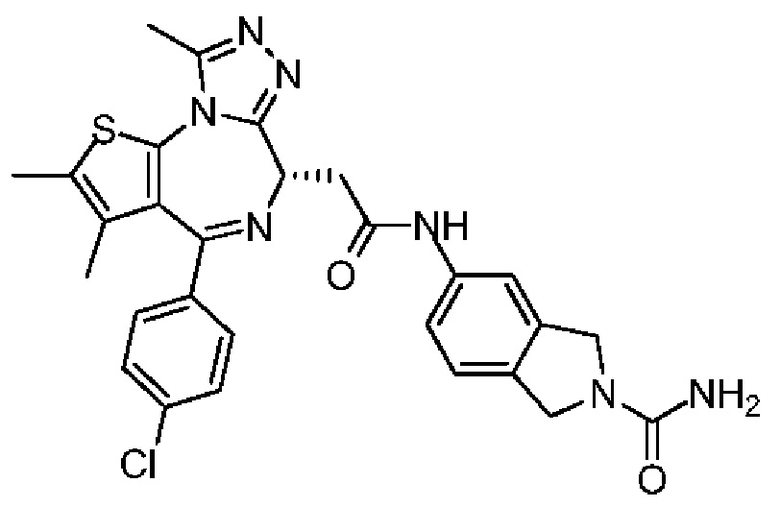
Синтез соединения 20
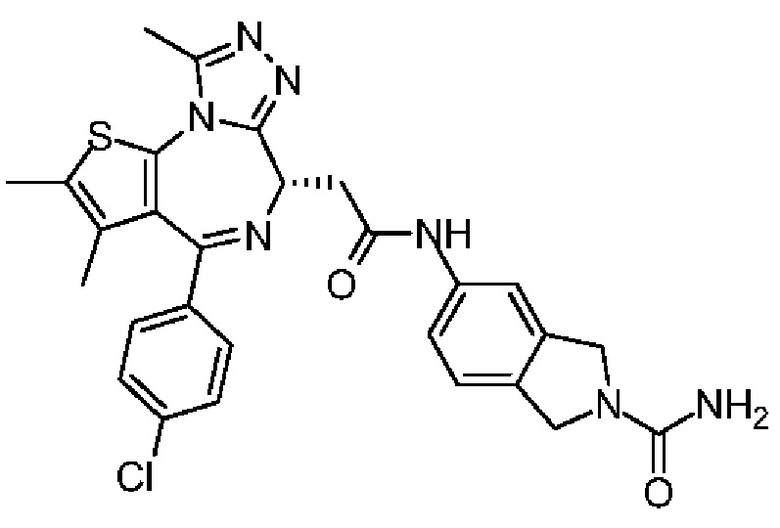
Соединение 18 (140,00 мг, 270,77 мкмоль, 1,00 экв.) добавляли к смешанной жидкости ледяной уксусной кислоты (2,00 мл) и H2O (2 мл). Затем к реакционной смеси медленно по каплям добавляли раствор цианата натрия (35,00 мг, 538,38 мкмоль, 1,99 экв.) в воде (2 мл). Смесь перемешивали при 15°С в течение 18 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Неочищенный продукт очищали методом препаративной хроматографии с получением соединения 20. 1H ЯМР (400 МГц, CDCl3) δppm 9.09 (s, 1Н), 7.58 (s, 1H), 7.24-7.37 (m, 5H), 7.08 (d, J=8.4 Гц, 1H), 4.50-4.58 (m, 5H), 4.42 (br s, 2H), 3.73-3.79 (m, 1H), 3.39-3.44 (m, 1H), 2.62 (s, 3H), 2.34 (s, 3H), 1.62 (s, 3H). LCMS (ESI) m/z: 560.0 (M+1).
Схема 21
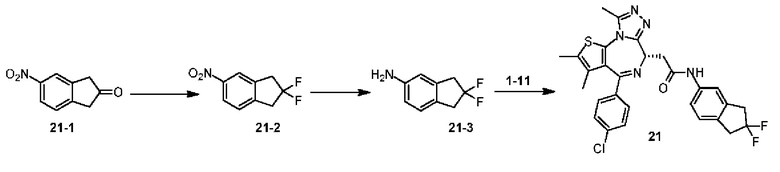
Пример 21
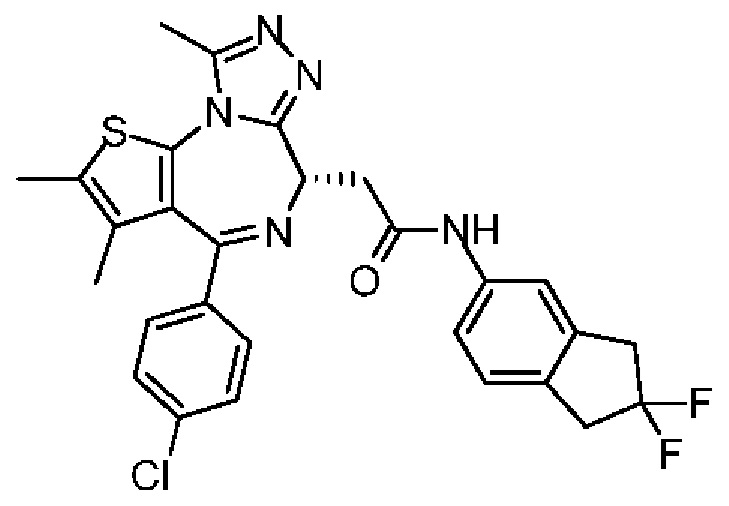
Синтез соединения 21-2
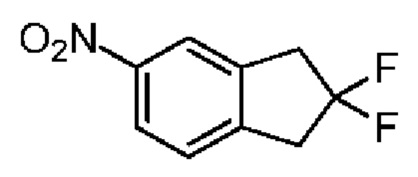
Соединение 21-1 (300,00 мг, 1,69 ммоль, 1,00 экв.) и диэтиламиносеры трифторид (545,91 мг, 3,39 ммоль, 447,47 мкл, 2,00 экв.) растворяли в безводном метиленхлориде (10,00 мл). Смесь перемешивали при 15°С в атмосфере азота в течение 16 часов. К реакционной смеси добавляли воду (30 мл). Затем смесь экстрагировали метиленхлоридом (20 мл ×2). Органическую фазу промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 21-2. 1H ЯМР (400 МГц, CDCl3) δppm 8.13-8.17 (m, 2H), 7.39-7.42 (m, 1H), 3.51-3.59 (m, 4H).
Синтез соединения 21-3
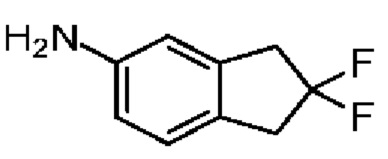
Соединение 21-2 (70,00 мг, 351,49 мкмоль, 1,00 экв.) и Pd(OH)2/C (50 мг, 10% чистоты) добавляли к метанолу (5,00 мл). Смесь перемешивали при 15°С в атмосфере водорода в течение 16 часов. Реакционную смесь фильтровали через слой целита и концентрировали при пониженном давлении с получением соединения 21-3, которое использовали сразу на следующей стадии без необходимости дополнительной очистки.
Синтез соединения 21
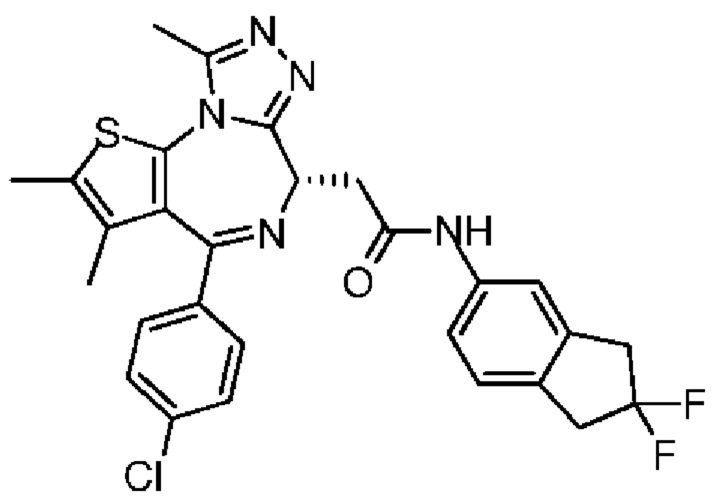
Соединение 1-11 (60,00 мг, 149,67 мкмоль, 1,00 экв.) и соединение 21-3 (30,38 мг, 179,60 мкмоль, 1,20 экв.) добавляли к безводному метиленхлориду (5,00 мл), затем добавляли HATU (56,91 мг, 149,67 мкмоль, 1,00 экв.) и по каплям добавляли диизопропилэтиламин (58,03 мг, 449,01 мкмоль, 78,42 мкл, 3,00 экв.). Атмосферу заменяли газообразным азотом три раза. Смесь перемешивали при 15°С в атмосфере азота в течение 16 часов. Реакционную смесь сразу концентрировали при пониженном давлении, затем промывали водой при встряхивании (10 мл) и экстрагировали этилацетатом (10 мл ×2). Органическую фазу промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом препаративной хроматографии с получением соединения 21. LCMS (ESI) m/z: 551.9 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 9.00 (s, 1H), 7.60 (s, 1H), 7.43 (d, J=8.4 Гц,2Н), 7.33-7.38 (m, 3H), 7.14 (d, J=8.0 Гц, 1H), 4.64-4.67(m, 1H), 3.52-3.80 (m, 1H), 3.51-3.52 (m, 1H), 3.34-3.44 (m, 4H), 2.70 (s, 3H), 2.43 (s, 3H), 1.71 (s, 3H).
Схема 22
Пример 22
Синтез соединения 22-2
К раствору соединения 22-1 (1,00 г, 5,12 ммоль, 1,00 экв.) и NBS (1,00 г, 5,63 ммоль, 1,10 экв.) в тетрахлориде углерода (10,00 мл) добавляли бензоила пероксид (124,02 мг, 512,00 мкмоль, 0,10 экв.). Смесь реагировала при перемешивании при 85°С в течение 4 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 22-2. 1H ЯМР (400 МГц, CDCl3) δppm 8.85 (d, J=2.4 Гц, 1H), 8.34-8.37 (m, 1H), 7.71 (d, J=8.8 Гц, 1H), 5.02 (s, 2H), 4.03 (s, 3H).
Синтез соединения 22-3
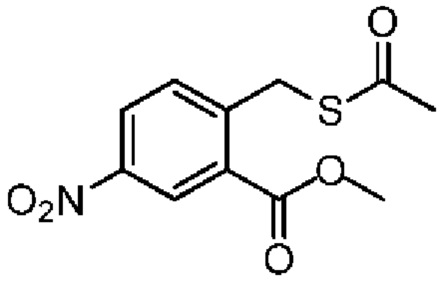
При 0°C и при защите газообразным азотом тиоуксусную кислоту медленно добавляли (288,90 мг, 3,80 ммоль, 270,00 мкл, 1,04 экв.) по каплям к раствору соединения 22-2 (1,00 г, 3,65 ммоль, 1,00 экв.) и карбоната калия (948,40 мг, 6,86 ммоль, 1,88 экв.) в ацетоне (4,00 мл). После завершения добавления по каплям смесь перемешивали при 0°С при защите газообразным азотом в течение 30 минут, а затем нагревали до 15°С и перемешивали в течение 3 часов. Реакционную смесь концентрировали с удалением ацетона. Полученную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3×10 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 22-3. 1H ЯМР (400 МГц, CDCl3) δppm 8.83 (s, 1H), 8.31-8.32 (m, 1H), 7.77 (m, J=8.4 Гц, 1H), 4.56 (s, 2Н), 4.00 (s, 3Н), 2.34 (s, 3Н).
Синтез соединения 22-4
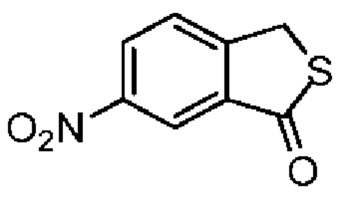
Соединение 22-3 (800,00 мг, 2,97 ммоль, 1,00 экв.) добавляли к концентрированной хлористоводородной кислоте (5,88 г, 47,76 ммоль, 4,00 мл, 16,08 экв.) и смесь перемешивали при 90°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и доводили до значения рН насыщенным раствором бикарбоната натрия до 7. Смесь экстрагировали этилацетатом (3×10 мл) и разделяли на две фазы. Вышеуказанные органические фазы объединяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 22-4. 1H ЯМР (400 МГц, CDCl3) δppm 8.59 (m, J=2.0 Гц, 1H), 8.43 (dd, J=8.4, 2.0 Гц, 1H), 7.66 (d, J=8.4 Гц, 1Н), 4.53 (s, 2Н).
Синтез соединения 22-5
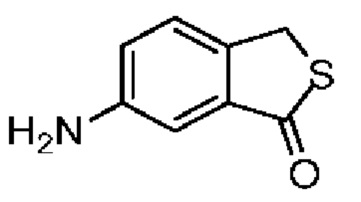
Влажный Pd/C (50,00 мг, 10% Pd) добавляли к раствору соединения 22-4 (100,00 мг, 512,30 мкмоль, 1,00 экв.) в метаноле (3,00 мл). Атмосферу заменяли газообразным водородом три раза. Смесь перемешивали при 15°С в водородных условиях (баллон) (15 фунт/кв.дюйм) в течение 18 часов. Реакционную смесь фильтровали при помощи диатомитовой земли. Фильтрат сразу концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 22-5. LCMS (ESI) m/z: 165.8 (М+1).
Синтез соединения 22
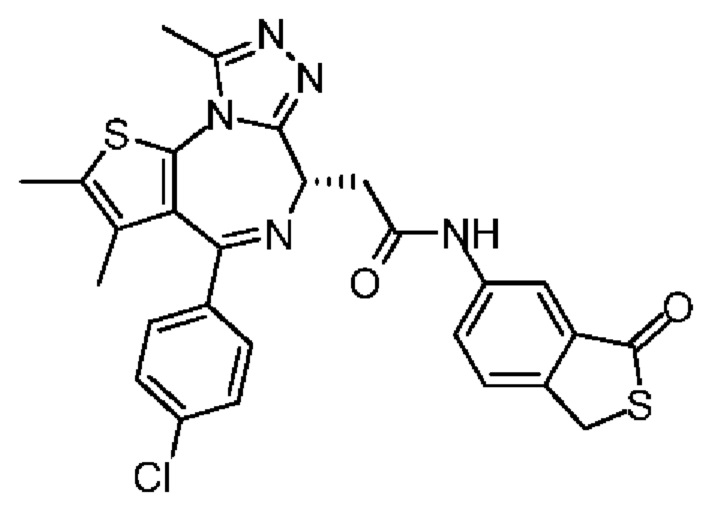
Соединение 1-11 (30 мг, 74,84 мкмоль, 1,00 экв.) и соединение 22-5 (99,218% чистоты) последовательно добавляли к раствору HATU (36 мг, 94,68 мкмоль, 1,27 экв.) в безводном N,N-диметилфорамиде (2 мл), а затем медленно по каплям добавляли триэтиламин (21,90 мг, 216,42 мкмоль, 30 мкл, 2,89 экв.). Смесь перемешивали при 15°С при защите газообразным азотом в течение 18 часов. Воду (3 мл) добавляли к реакционной смеси для гашения реакции. Смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли. Органическую фазу промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом препаративной хроматографии с получением соединения 22. LCMS (ESI) m/z: 548.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 10.08 (s, 1H), 8.01 (s, 1H), 7.66 (d, J=8.0 Гц, 1H), 7.29-7.46 (m, 5H), 4.80-4.83 (m, 1H), 4.30 (s, 2H),3.97-4.03 (m, 1H), 3.58-3.63 (m, 1H), 2.78 (s, 3H), 2.45 (s, 3H), 1.71 (s, 3H).
Схемы 23 и 24
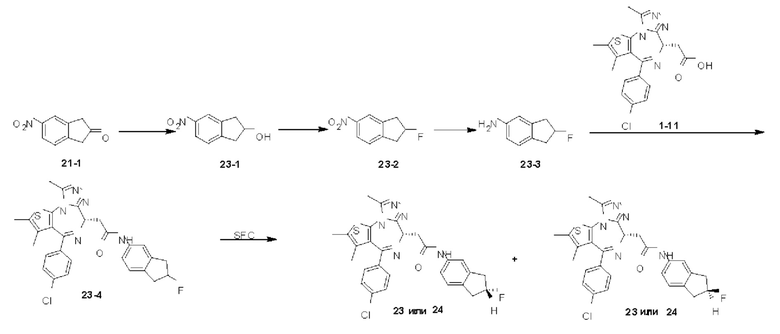
Примеры 23 и 24
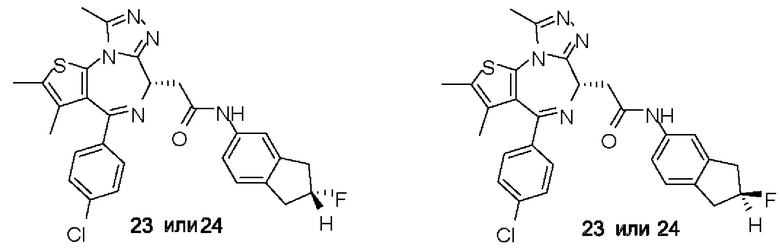
Синтез соединения 23-1
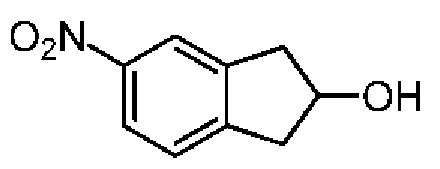
Соединение 21-1 (200,00 мг, 1,13 ммоль, 1,00 экв.) и боргидрид натрия (85,50 мг, 2,26 ммоль, 2,00 экв.) растворяли в абсолютном метаноле (5,00 мл) и смесь перемешивали при 15°С в атмосфере азота в течение 1 часа. Реакционную смесь сразу концентрировали при пониженном давлении и к ней добавляли воду (20 мл). Затем смесь экстрагировали этилацетатом (10 мл ×2). Органическую фазу промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 23-1, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δppm 8.00-8.03 (m, 2Н), 7.31 (d, J=8.0 Гц, 1H), 4.75 (br s, 1H), 3.18-3.25 (m, 2Н), 2.92-2.97 (m, 2Н), 1.61 (br s, 1Н).
Синтез соединения 23-2
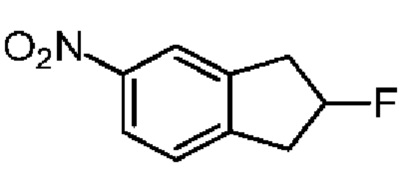
Соединение 23-1 (190 мг, 1,06 ммоль, 1,00 экв.) и диэтиламиносеры трифторид (188,02 мг, 1,17 ммоль, 154,12 мкл, 1,1 экв.) растворяли в безводном метиленхлориде (5,00 мл). Смесь перемешивали при 15°С в атмосфере азота в течение 16 часов. К реакционной смеси добавляли воду (20 мл). Затем смесь экстрагировали метиленхлоридом (20 мл ×2). Органическую фазу промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 23-2. 1H ЯМР (400 МГц, CDCl3) δppm 8.03-8.07 (m, 2Н), 7.31-7.35 (m, 1H), 5.27-5.61 (m, 1H), 3.26-3.28 (m, 2H),3.19-3.20 (m, 2H).
Синтез соединения 23-3
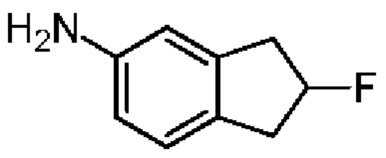
Соединение 23-2 (30 мг, 165,60 мкмоль, 1,00 экв.) и Pd/C (30 мг, 10% чистоты) добавляли к метанолу (3 мл). Смесь перемешивали при 15°С в атмосфере водорода в течение 2 часов. Реакционную смесь сразу фильтровали и концентрировали при пониженном давлении с получением соединения 23-3, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δppm 6.90-6.95 (m, 1H), 6.52 (s, 1H), 6.41-6.49 (m, 1H), 5.24-5.50 (m, 1H), 2.95-3.11 (m, 4Н).
Синтез соединения 23-4
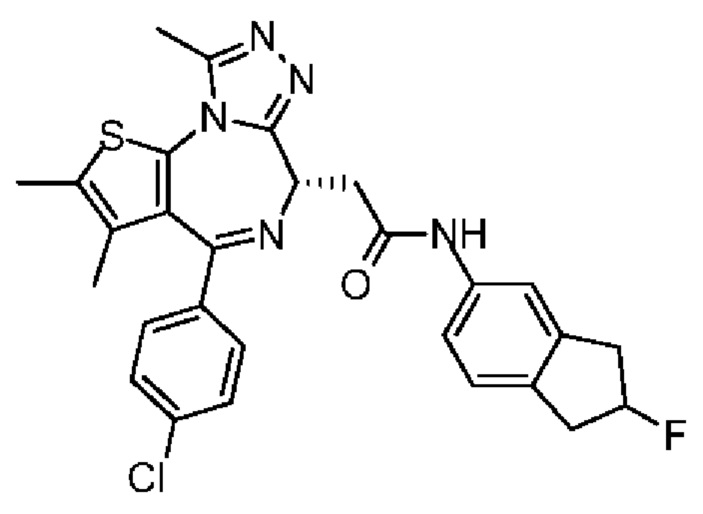
Соединение 1-11 (60 мг, 149,67 мкмоль, 1,00 экв.) и соединение 23-3 (25 мг, 165,37 мкмоль, 1,10 экв.) добавляли к безводному метиленхлориду (3 мл), затем добавляли HATU (56,91 мг, 149,67 мкмоль, 1,00 экв.) и по каплям добавляли диизопропилэтиламин (58,03 мг, 449,01 мкмоль, 78,21 мкл, 3,00 экв.). Атмосферу заменяли газообразным азотом три раза. Смесь перемешивали при 15°С в атмосфере азота в течение 1 часа. Реакционную смесь сразу концентрировали при пониженном давлении, затем промывали водой при встряхивании (10 мл), а затем экстрагировали этилацетатом (10 мл ×2). Органические фазы промывали насыщенным солевым раствором (15 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом препаративной хроматографии с получением соединения 23-4. LCMS (ESI) m/z: 534.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 8.66 (br s, 1H), 7.60 (br s, 1H), 7.43 (d, J=8.4 Гц,2Н), 7.30-7.37 (m, 3H), 7.20 (d, J=8.4 Гц, 1H), 5.33-5.64 (m, 1H), 4.61-4.65 (m, 1H), 3.79-3.80 (m, 1H), 3.46-3.51 (m, 1H), 3.06-3.30 (m, 4H), 2.70 (s, 3H), 2.42 (s, 3H), 1.70 (s, 3H).
Синтез соединений 23 и 24
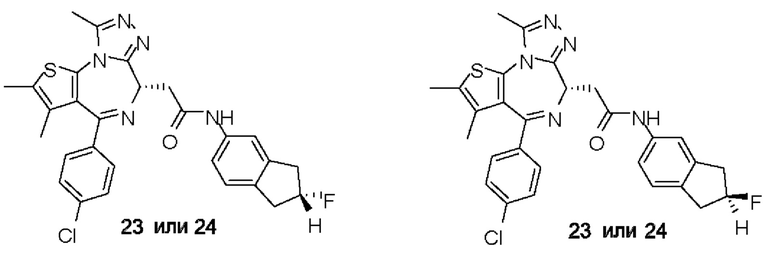
Соединение 23-4 (28 мг, 52,43 мкмоль) подвергали разделению методом SFC ((хроматографическая колонка: OJ (250 мм × 30 мм, 5 мкм); подвижная фаза: [0,1% NH3H2O EtOH]; В%: 30%-30%, 60 мл/мин)) с получением соединения 23 (Rt=5,404 мин). LCMS (ESI) m/z: 534.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 8.99 (br s, 1H), 7.50 (s, 1H), 7.23-7.34 (m, 5H), 7.06 (br d, J=8.0 Гц, 1H), 5.36 (d, J=53.2 Гц 1H), 4.58-4.61 (m, 1H), 3.71-3.75 (m, 1H), 3.43-3.47 (m, 1H), 2.78-3.20 (m, 4H), 2.60 (s, 3H), 2.33 (s, 3H), 1.60 (s, 3H).
Соединение 24 (Rt=5,702 мин). LCMS (ESI) m/z: 534.1 (M+1). 1H ЯМР (400 МГц, CDCl3) δppm 9.00 (s, 1H), 7.52 (s, 1H), 7.20-7.34 (m, 5H), 7.07 (br d, J=8.0 Гц, 1H), 5.36 (d, J=52.0 Гц, 1H), 4.58-4.61 (bm, 1H), 3.65-3.72 (m, 1H), 3.45-3.49 (m, 1H), 3.02-3.31 (m, 4H), 2.60 (s, 3H), 2.33 (s, 3H), 1.60 (s, 3H).
Схема 25
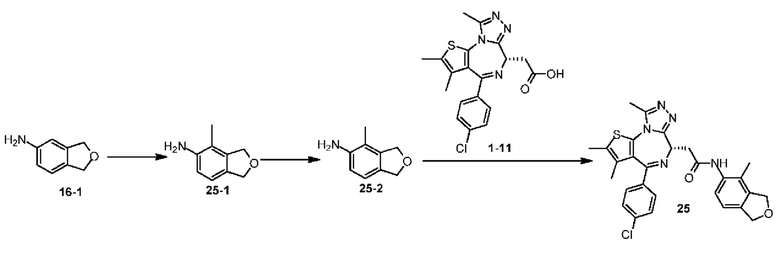
Пример 25
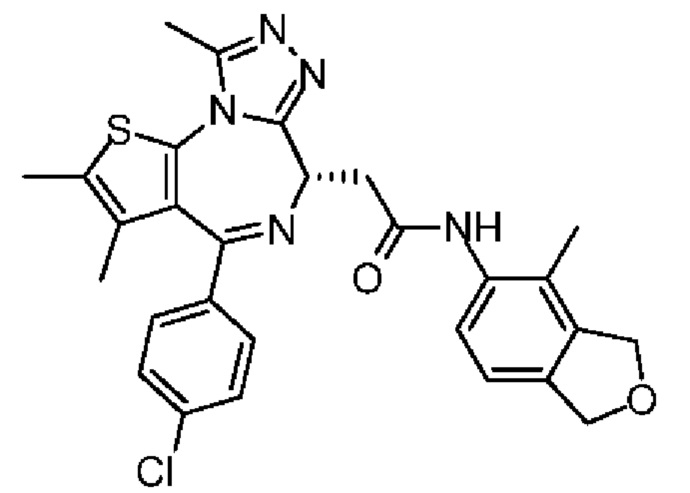
Синтез соединения 25-1
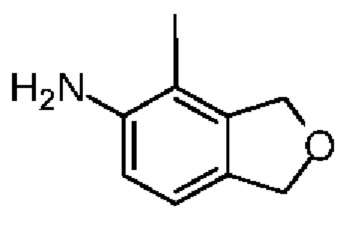
При 15°С и в атмосфере азота к смешанной жидкости соединения 16-1 (300 мг, 2,22 ммоль, 1 экв.), бикарбоната натрия (279,70 мг, 3,33 ммоль, 129,49 мкл, 1,5 экв.) в метиленхлориде (10 мл) и метаноле (2,5 мл) добавляли по каплям хлорид йода (1 М, 2,44 мл, 1,1 экв.), а затем смесь перемешивали при той же температуре в течение 4 часов. При 15°С и при перемешивании к реакционной смеси добавляли по каплям насыщенный раствор Na2SO3 (25 мл), а затем смесь экстрагировали метиленхлоридом (10 мл ×2). Органическую фазу промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 25-1. LCMS (ESI) m/z: 261.8 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 6.88 (d, J=8.0 Гц, 1H), 6.57 (d, J=8.0 Гц, 1Н), 5.12 (s, 2Н), 4.89 (s, 2Н), 4.00 (br s, 2Н).
Синтез соединения 25-2
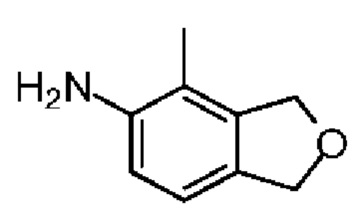
При 15°С соединение 25-1 (140 мг, 489,22 мкмоль, 1 экв.), фторид цезия (260,10 мг, 1,71 ммоль, 63,13 мкл, 3,5 экв.), [1,1'-бис(дифенилфосфин)ферроцен]палладия (II) дихлорид (35,80 мг, 48,92 мкмоль, 0,1 экв.) и метилбороновую кислоту (87,85 мг, 1,47 ммоль, 3 экв.) растворяли в диоксане (5 мл) и смесь перемешивали при 80°С в атмосфере азота в течение 4 часов. Реакционную смесь фильтровали, затем промывали водой при встряхивании (20 мл), а затем экстрагировали этилацетатом (10 мл ×2). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 25-2. 1H ЯМР (400 МГц, CDCl3) δppm 6.81 (d, J=8.0 Гц, 1H), 6.56 (d, J=8.00 Гц, 1H), 4.99 (s, 4Н), 3.52 (br s, 2Н), 1.98 (s, 3Н).
Синтез соединения 25
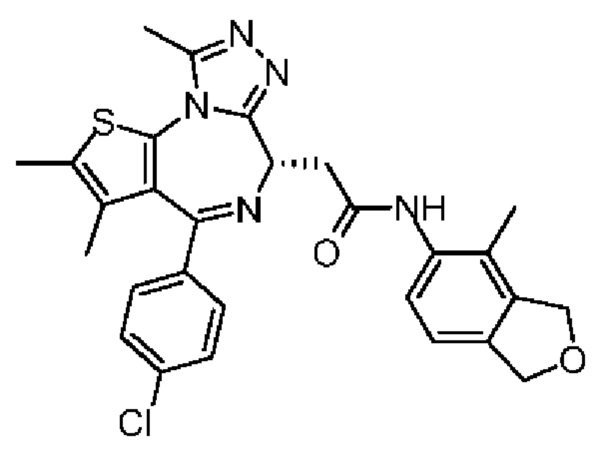
Соединение 1-11 (50 мг, 124,73 мкмоль, 1,00 экв.) и соединение 25-2 (27,91 мг, 187,09 мкмоль, 1,5 экв.) растворяли в безводном метиленхлориде (3 мл), добавляли HATU (47,42 мг, 124,73 мкмоль, 1,00 экв.) и по каплям добавляли диизопропилэтиламин (48,36 мг, 374,18 мкмоль, 65,17 мкл, 3,00 экв.). Смесь перемешивали при 15°С и в атмосфере азота в течение 2 часов. Реакционную смесь промывали водой при встряхивании (10 мл). Органическую фазу промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом препаративной хроматографии с получением соединения 25. LCMS (ESI) m/z: 532.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 8.44 (s, 1H), 7.57 (d, J=7.6 Гц, 1H), 7.35 (d, J=8.4 Гц, 2H), 7.26 (d, J=8.4 Гц, 2H), 6.96 (d, J=8.0 Гц, 1H), 4.99-5.02 (m, 4H), 4.55-4.59 (m, 1H), 3.70-3.75 (m, 1Н),3.45-3.50 (m, 1H), 2.61 (s, 3H), 2.34 (s, 3H), 2.08 (s, 3H), 1.61 (s, 3H).
Схема 26
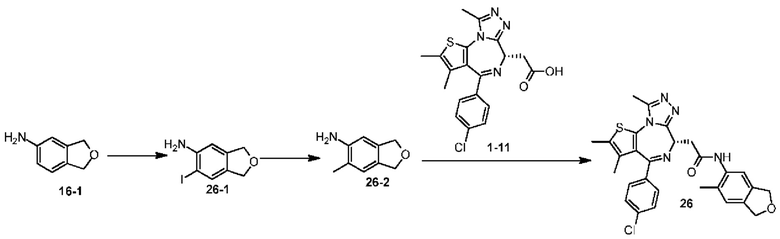
Пример 26
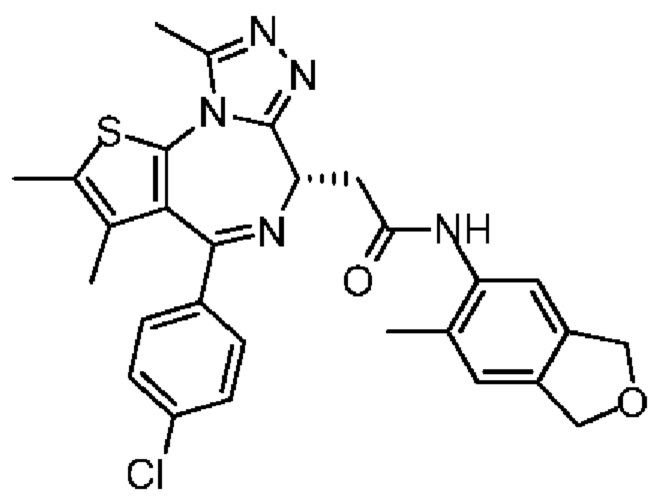
Соединение примера 26 синтезировали на основании примера 25.
LCMS (ESI) m/z: 532.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 8.39 (s, 1H), 7.75 (s, 1H), 7.36 (d, J=8.8 Гц, 2H), 7.27 (d, J=8.8 Гц, 2H), 6.97 (s, 1H), 4.98 (d, J=9.6 Гц, 4H), 4.53-4.57 (m, 1H), 3.73-3.78 (m, 1H), 3.42-3.47 (m, 1H), 2.62 (s, 3H), 2.34 (s, 3H), 2.24 (s, 3H), 1.62 (s, 3H).
Схема 27
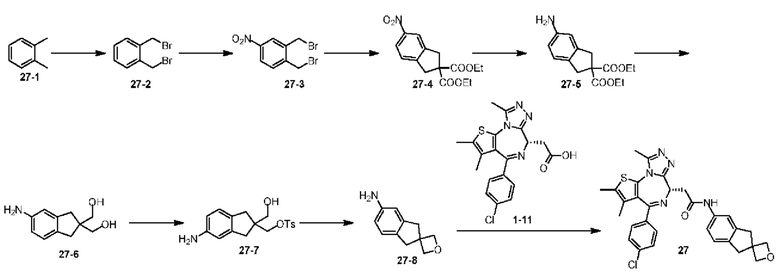
Пример 27
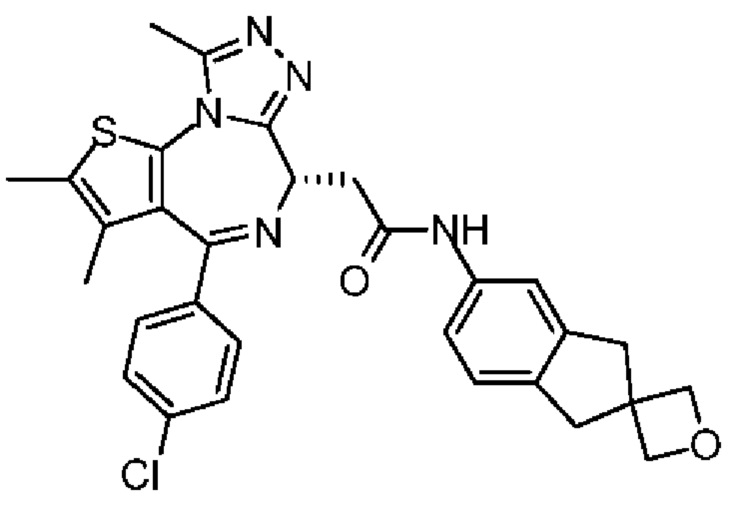
Синтез соединения 27-2
Смесь орто-ксилола (20,00 г, 188,39 ммоль, 22,73 мл, 1,00 экв.), NBS (70,41 г, 395,62 ммоль, 2,10 экв.), бензоил пероксида (912,70 мг, 3,77 ммоль, 0,02 экв.) и хлороформа (200,00 мл) перемешивали при 80°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, затем разбавляли метиленхлоридом (200 мл) и промывали водой (100 мл ×2) и промывали насыщенным солевым раствором (100 мл), соответственно. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученное твердое вещество перекристаллизовывали при 80°С (петролейный эфир/этанол=30:1; 240 мл/8 мл) и охлаждали до комнатной температуры. Твердое вещество осаждалось и его фильтровали. Фильтрационный кек промывали петролейным эфиром (50 мл). Фильтрационный кек сушили в сушильном шкафу с получением соединения 27-2. 1H ЯМР (400 МГц, CDCl3) δppm 7.41-7.35 (m, 2Н), 7.35-7.29 (m, 2Н), 4.68 (s, 4Н).
Синтез соединения 27-3
При 0°С к раствору соединения 27-2 (5 г, 18,94 ммоль, 2,55 мл, 1 экв.) и концентрированной серной кислоты (30 мл) добавляли партиями нитрат калия (2,30 г, 22,73 ммоль, 1,2 экв.). Затем смесь перемешивали при 0°С в течение 3 часов. Реакционная смесь становилась красно-коричневой. Реакционную смесь медленно по каплям добавляли в лабораторный стакан объемом 500 мл, содержащий кубики льда (200 г), и осаждалось светло-желтое твердое вещество. Затем смесь перемешивали при 0°С в течение 1 часа, а затем фильтровали. Фильтрационный кек промывали водой (100 мл). Фильтрационный кек сушили в сушильном шкафу с получением соединения 27-3, которое сразу использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δppm 8.39 (d, J=2.5 Гц, 1H), 8.19 (dd, J=2.4, 8.4 Гц, 1H), 7.78 (d, J=8.3 Гц, 1Н), 4.94 (s, 2Н), 4.90 (s, 2Н).
Синтез соединения 27-4
Металлический натрий (892,94 мг, 38,84 ммоль, 920,56 мкл, 2,4 экв.) добавляли к этанолу (20 мл). При 10°С смесь перемешивали в течение 0,5 часа до полного исчезновения Na. Затем добавляли раствор диэтилмалоната (3,00 г, 18,73 ммоль, 2,83 мл, 1,16 экв.) в тетрагидрофуране (10 мл) и быстро добавляли смешанную жидкость соединения 27-3 (5 г, 16,18 ммоль, 1 экв.) и тетрагидрофурана (10 мл). Затем полученную смесь нагревали с обратным холодильником при 80°С при защите газообразным азотом в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. Воду (60 мл) добавляли. Затем смесь экстрагировали этилацетатом (50 мл ×3). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, разделяли и очищали методом колоночной хроматографии с получением соединения 27-4. 1H ЯМР (400 МГц, DMSO-d6) δppm 8.13 (s, 1H), 8.06 (dd, J=2.3, 8.3 Гц, 1Н), 7.51 (d, J=8.3 Гц, 1H), 4.18-4.13 (m, 4H), 3.59 (s, 4H), 1.19-1.16 (m, 6H).
Синтез соединения 27-5
Соединение 27-4 (1,3 г, 4,23 ммоль, 1 экв.) растворяли в этаноле (20 мл), а затем добавляли дигидрат двухлористого олова (4,77 г, 21,15 ммоль, 1,76 мл, 5 экв.). Смесь перемешивали при 80°С в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, затем значение рН при помощи раствора NaOH (4 н) доводили до 10, а затем экстрагировали этилацетатом (40 мл ×3). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, разделяли и очищали методом колоночной хроматографии с получением соединения 27-5. LCMS (ESI) m/z: 277.9 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 6.97 (d, J=8.0 Гц, 1H, 6.56-6.48 (m, 2H), 4.20 (q, J=7.2 Гц, 4H), 3.62 (br s, 2H), 3.50 (s, 2H), 3.48 (s, 2H), 1.27-1.24 (t, J=7.2 Гц, 6H).
Синтез соединения 27-6
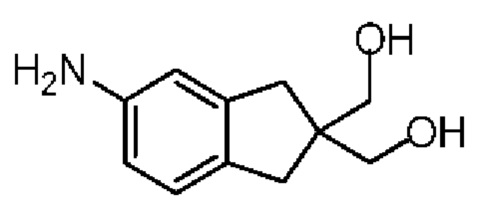
Соединение 27-5 (500 мг, 1,80 ммоль, 1 экв.) растворяли в тетрагидрофуране (10 мл) при 0°С, а затем добавляли алюмогидрид лития (157,39 мг, 4,15 ммоль, 2,3 экв.). Смесь перемешивали при 0°С в течение 1 часа. Воду (0,2 мл) добавляли для гашения реакции. Затем смесь концентрировали при пониженном давлении, затем переводили во взвесь при помощи этилацетата (100 мл) в течение 10 минут при 10°С и фильтровали. Фильтрационный кек промывали этилацетатом (50 мл). Фильтрат концентрировали при пониженном давлении с получением соединения 27-6, которое сразу использовали на следующей стадии без дополнительной очистки.
Синтез соединения 27-7
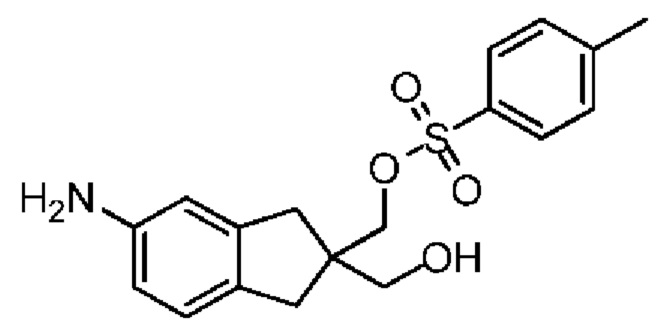
Соединение 27-6 (150 мг, 776,23 мкмоль, 1 экв.) растворяли в тетрагидрофуране (3 мл). Гидрид натрия (46,57 мг, 1,16 ммоль, 60% чистоты, 1,5 экв.) добавляли при 0°С. Смесь перемешивали 40 минут. Затем добавляли смешанную жидкость пара-толуолсульфонилхлорида (147,99 мг, 776,23 мкмоль, 1 экв.) и тетрагидрофурана (3 мл). Смесь перемешивали при 0°С в течение 40 минут, а затем концентрировали при пониженном давлении с получением масляного вещества. К масляному веществу добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл ×3). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали методом тонкослойной хроматографии на пластине с получением соединения 27-7. LCMS (ESI) m/z: 348.1 (М+1).
Синтез соединения 27-8
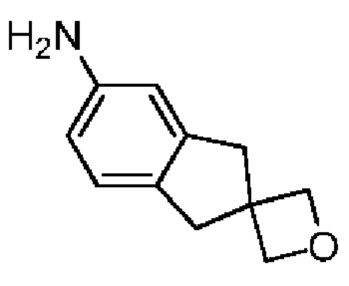
Соединение 27-7 (50 мг, 143,91 мкмоль, 1 экв.) растворяли в тетрагидрофуране (3 мл), а затем добавляли гидрид натрия (57,56 мг, 1,44 ммоль, 60% чистоты, 10 экв.). Смесь перемешивали при 70°С в течение 5 часов. Воду (0,5 мл) добавляли для гашения реакции. Реакционную смесь концентрировали при пониженном давлении с получением масляного вещества. К масляному веществу добавляли воду (30 мл). Смесь экстрагировали (метиленхлорид/метнол=10/1) (40 мл ×3). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали методом тонкослойной хроматографии на пластине с получением соединения 27-8. LCMS (ESI) m/z: 175.9 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 6.98 (d, J=8.0 Гц, 1H), 6.57 (s, 1H), 6.51 (dd, J=2.1, 7.9 Гц, 1H), 4.67 (s, 4H), 3.62 (br s, 2H), 3.16 (s, 2H), 3.14 (s, 2H).
Синтез соединения 27
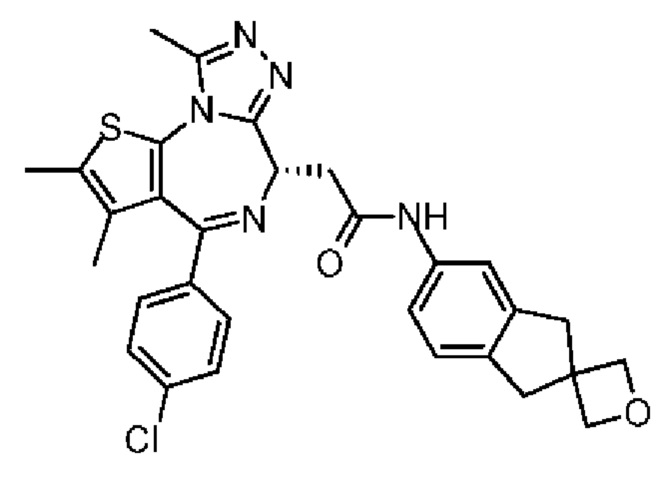
Диизопропилэтиламин (58,03 мг, 449,01 мкмоль, 78,21 мкл, 3 экв.) добавляли к раствору соединения 1-11 (0,06 г, 149,67 мкмоль, 1 экв.), соединения 27-8 (15,74 мг, 89,80 мкмоль, 388,88 мкл, 0,6 экв.) и HATU (62,60 мг, 164,64 мкмоль, 1,1 экв.) в безводном метиленхлориде (10 мл). Смесь реагировала при перемешивании при 15°С при защите газообразным азотом в течение 1 часа. Реакционную смесь разбавляли метиленхлоридом (10 мл), затем последовательно промывали 1 н хлористоводородной кислотой (5 мл), водой (5 мл) и насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Неочищенный продукт очищали методом препаративной хроматографии с получением соединения 27. LCMS (ESI) m/z: 558.1 (М+1). 1Н ЯМР (CDCl3 400 МГц) δppm 8.58 (s, 1Н), 7.52 (s, 1H), 7.42 (d, J=8.8 Гц, 2H), 7.33 (d, J=8.8 Гц, 2H), 7.23-7.26 (m, 1H), 7.11-7.23(m, 1H, 4.66 (s, 4H), 4.58-4.65 (m, 1H, 3.74-3.79 (m, 1H, 3.43-3.48 (m, 1H, 3.21 (s, 2H), 3.18 (s, 2H), 2.67 (s, 3H), 2.40 (s, 3H), 1.69 (s, 3H).
Схема 28
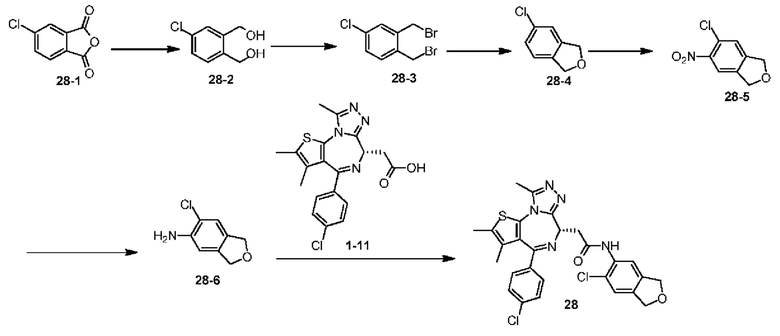
Пример 28
Синтез соединения 28-2
При 0°С и при защите газообразным азотом раствор соединения 28-1 (6 г, 32,87 ммоль, 1 экв.) в тетрагидрофуране (10 мл) медленно по каплям добавляли к суспензии алюмогидрида лития (2,50 г, 65,76 ммоль, 2 экв.) и хлорида цинка (2,69 г, 19,72 ммоль, 923,71 мкл, 0,6 экв.) в тетрагидрофуране (20 мл). После завершения добавления по каплям смесь нагревали до 10°С и перемешивали в течение 6 часов. Реакционную смесь охлаждали до 0°С. Воду (10 мл) добавляли для гашения реакции (белый осадок образовывался в процессе гашения). Значение рН смесь доводили при помощи водного раствора хлористоводородной кислоты (2М) до приблизительно 6. Вышеуказанный смешанный раствор экстрагировали этилацетатом (3×20 мл) и разделяли на две фазы. Органические фазы объединяли, затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт добавляли к этилацетату (3 мл) и петролейному эфиру (10 мл). Смесь нагревали до 75°С и перемешивали с обратным холодильником в течение 30 минут, затем охлаждали до комнатной температуры. Затем добавляли петролейный эфир (20 мл) и смесь перемешивали в течение 20 минут (осаждалось белое твердое вещество) и фильтровали. Фильтрационный кек сразу сушили в сушильном шкафу с получением соединения 28-2. 1Н ЯМР (400 МГц, CDCl3) δppm 7.30 (s, 1H, 7.21-7.22 (m, 2Н), 4.63 (s, 4Н), 2.76 (br s, 1H), 2.67 (br s, 1H).
Синтез соединения 28-3
При 0°C и при защите газообразным азотом по каплям медленно добавляли трибромид фосфора (4,70 г, 17,38 ммоль, 1,2 экв.) к раствору соединения 28-2 (2,5 г, 14,48 ммоль, 1 экв.) в метиленхлориде (20 мл). После завершения добавления по каплям смесь нагревали до 10°С, перемешивали в течение 5 часов, разбавляли и экстрагировали метиленхлоридом (3×20 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 28-3. 1H ЯМР (400 МГц, CDCl3) δppm 7.30 (d, J=2.4 Гц, 1H, 7.21-7.24 (m, 2Н), 4.53 (d, J=8.8 Гц, 4Н).
Синтез соединения 28-4
Нейтральный оксид алюминия (30 г, 294,23 ммоль, 35,12 экв.) добавляли к раствору соединения 28-3 (2,5 г, 8,38 ммоль, 1 экв.) в н-гексане (40 мл). Смесь перемешивали при 75°С в течение 3 часов. Реакционную смесь охлаждали до 10°С и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 28-4. 1H ЯМР (400 МГц, CDCl3) δppm 7.22-7.25 (m, 2Н), 7.15-7.17 (m, 1H, 5.08 (s, 4Н).
Синтез соединения 28-5

При -10°С раствор соединения 28-4 (300 мг, 1,94 ммоль, 1 экв.) в концентрированной серной кислоте (2 мл) медленно по каплям добавляли к раствору нитрата калия (195,00 мг, 1,93 ммоль, 9,94е-1 экв.) в концентрированной серной кислоте (6 мл). После завершения добавления по каплям смесь перемешивали при -10°С в течение 20 минут. Реакционную смесь выливали в лед (приблизительно 10 мл). Смесь перемешивали в течение 10 минут и экстрагировали этилацетатом (3×10 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 28-5. 1Н ЯМР (400 МГц, CDCl3) δppm 7.78 (s, 1H, 7.44 (s, 1H, 5.15 (s, 4Н).
Синтез соединения 28-6
Дигидрат двухлористого олова (900 мг, 3,99 ммоль, 332,10 мкл, 3,98 экв.) добавляли к раствору соединения 28-5 (200 мг, 1.00 ммоль, 1,00 экв.) в метаноле (4 мл). Смесь перемешивали при 20°С в течение 5 минут. Реакционную смесь сразу концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 28-6. 1Н ЯМР (400 МГц, CD3OD) δppm 7.33 (s, 1H, 7.0-7.06 (m, 1Н), 5.01 (m, 4H).
Синтез соединения 28
При 0°С POCl3 (133,87 мг, 873,08 мкмоль, 81,13 мкл, 5 экв.) медленно по каплям добавляли к раствору соединения 28-6 (40 мг, 235,84 мкмоль, 1,35 экв.) и соединения 1-11 (70 мг, 174,62 мкмоль, 1,00 экв.) в пиридине (3 мл). При 0°С и при защите газообразным азотом смесь перемешивали в течение 1 часа. Ледяную воду (3 мл) добавляли к реакционной смеси для гашения реакции. Значение рН вышеуказанного смешанного раствора доводили при помощи разбавленного водного раствора хлористоводородной кислоты (0,5 М) до приблизительно 6 и экстрагировали этилацетатом (3×5 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом препаративной хроматографии с получением соединения 28. LCMS (ESI) m/z: 552.0 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 8.71 (s, 1H, 8.22 (s, 1H, 7.36 (d, J=8.4 Гц, 2H), 7.26 (d, J=8.4 Гц, 2H), 7.17 (s, 1H), 4.98 (d, J=4.0 Гц, 4H), 4.58 (t, J=6.8 Гц, 1H), 3.66-3.72 (m, 1H), 3.51-3.57 (m, 1H), 2.62 (s, 3H), 2.34 (s, 3H), 1.62 (s, 3H).
Схема 29
Пример 29
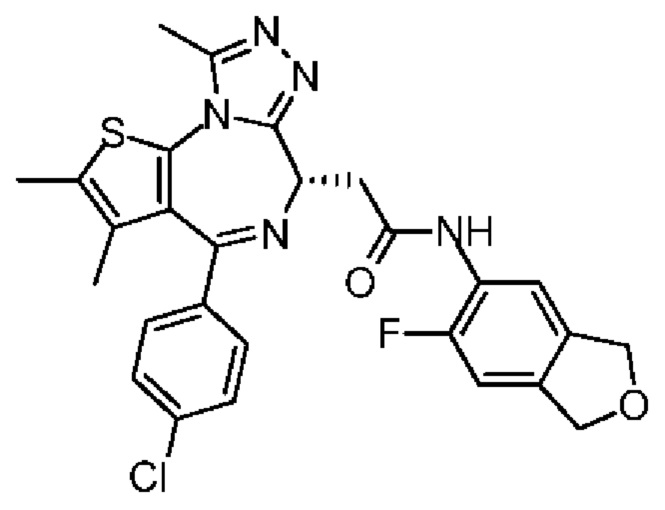
Синтез соединения 29-2
При 0°С и при защите газообразным азотом раствор соединения 29-1 (5 г, 30,10 ммоль, 1 экв.) в безводном тетрагидрофуране (40 мл) медленно добавляли к суспензии алюмогидрида лития (2,28 г, 60,20 ммоль, 2 экв.) и хлорида цинка (2,46 г, 18,06 ммоль, 845,91 мкл, 0,6 экв.) в безводном тетрагидрофуране (100 мл). После добавления смесь реагировала при 10°С в течение 16 часов. Воду (3 мл) медленно добавляли к реакционной смеси для гашения реакции, а затем к реакционной смеси добавляли воду (50 мл). Водную фазу экстрагировали этилацетатом (50 мл ×2). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении с получением соединения 29-2, которое сразу использовали на следующей стадии без очистки. 1Н ЯМР (400 МГц, CDCl3) δppm 7.21-7.23 (m, 1H, 6.93-7.03 (m, 2Н), 4.53-4.55 (m, 4Н).
Синтез соединения 29-3
При 0°С и при защите газообразным азотом трибромид фосфора (7,76 г, 28,66 ммоль, 1,2 экв.) медленно добавляли к раствору соединения 29-2 (3,73 г, 23,89 ммоль, 1 экв.) в безводном метиленхлориде (100 мл). После добавления смесь реагировала при 10°С при защите газообразным азотом в течение 6 часов. Реакционную смесь промывали водой (50 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 29-3.
Синтез соединения 29-4
Нейтральный оксид алюминия (40 г, 392,31 ммоль, 29,89 экв.) добавляли к раствору соединения 29-3 (3,7 г, 13,12 ммоль, 1 экв.) в н-гексане (80 мл). После добавления смесь реагировала при 75°С в течение 20 часов. Реакционную смесь охлаждали до 50°С и нерастворимое вещество отфильтровывали, пока оно было горячее. Затем фильтрационный кек промывали метиленхлоридом (50 мл). Фильтраты объединяли и выпаривали досуха при пониженном давлении с получением соединения 29-4, которое сразу использовали на следующей стадии без очистки. 1H ЯМР (400 МГц, CDCl3) δppm 7.07-7.09 (m, 1H, 6.84-6.90 (m, 2Н), 5.00 (s, 4Н).
Синтез соединения 29-5
Нитрат калия (878,27 мг, 8,69 ммоль, 1 экв.) добавляли к раствору концентрированной серной кислоты (10 мл). Соединение 29-4 (1,2 г, 8,69 ммоль, 1 экв.) растворяли в концентрированной серной кислоте (5 мл), которую охлаждали до -10°С в ледяной солевой бане, а затем добавляли к вышеуказанной реакционной смеси в ледяной солевой бане. После добавления смесь реагировала при -10°С в ледяной солевой бане в течение 30 минут. Реакционную смесь медленно по каплям добавляли к непрерывно перемешиваемому дробленому льду (100 мл). Смесь фильтровали с получением светло-коричневого твердого вещества в виде неочищенного продукта. Неочищенный продукт растворяли в этилацетате (50 мл). Затем смесь сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении с получением соединения 29-5, которое сразу использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δppm 8.71 (d, J=1.6 Гц, 1H, 8.51 (dd, J=2.0, 8.4 Гц, 1H, 7.65 (d, J=8.4 Гц, 1H, 5.38 (s, 4H).
Синтез соединения 29-6
Соединение 29-5 (0,183 г, 999,25 мкмоль, 1 экв.) и дигидрат двухлористого олова (901,91 мг, 4,00 ммоль, 332,81 мкл, 4 экв.) добавляли к абсолютному метанолу (5 мл). После добавления смесь реагировала при 30°С в течение 4 часов. Реакционную смесь сразу концентрировали при пониженном давлении. Остаток растворяли в этилацетате (50 мл). Нерастворимое вещество отфильтровывали. Фильтрат сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении с получением соединения 29-6, которое сразу использовали на следующей стадии без дополнительной очистки. LCMS (ESI) m/z: 153.9 (М+1).
Синтез соединения 29
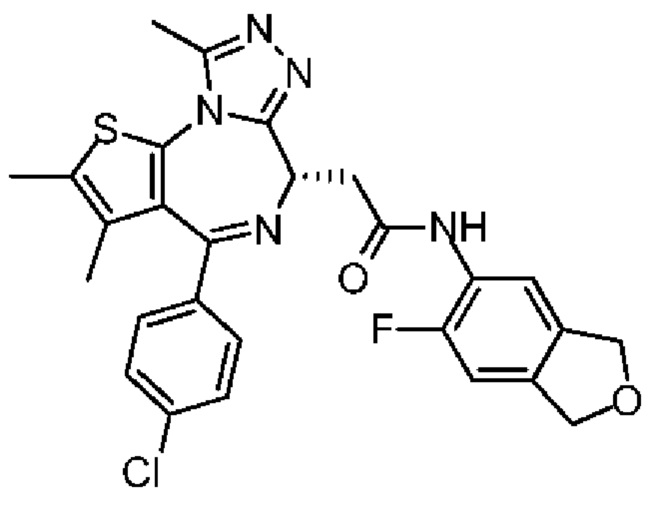
При 0°С и при защите газообразным азотом POCl3 (71,27 мг, 464,83 мкмоль, 43,20 мкл, 2 экв.) добавляли к раствору соединения 1-11 (100 мг, 232,41 мкмоль, 1 экв.) и соединения 29-6 (42,71 мг, 278,90 мкмоль, 1,2 экв.) в пиридине (2 мл). После добавления смесь реагировала при 15°С в течение 2 часов. Воду (5 мл) добавляли к реакционной смеси. Значение рН водной фазы доводили при помощи хлористоводородной кислоты до 7 и экстрагировали метиленхлоридом (5 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, затем концентрировали при пониженном давлении и очищали методом тонкослойной хроматографии на пластине с получением соединения 29. LCMS (ESI) m/z: 536.1 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 8.80 (br s, 1H, 8.13 (d, J=6.8 Гц, 1H, 7.36 (d, J=8.4 Гц, 2H), 7.26 (d, J=8.8 Гц, 2Н), 6.88 (d, J=10.0 Гц, 1Н), 4.97 (s, 4H), 4.5-4.57 (m, 1H, 3.64-3.66 (m, 1H, 3.52-3.54 (m, 1H, 2.61 (s, 3H), 2.34 (s, 3H), 1.62 (s, 3H).
Схема 30
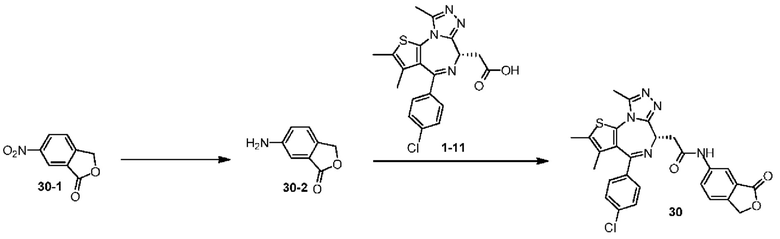
Пример 30
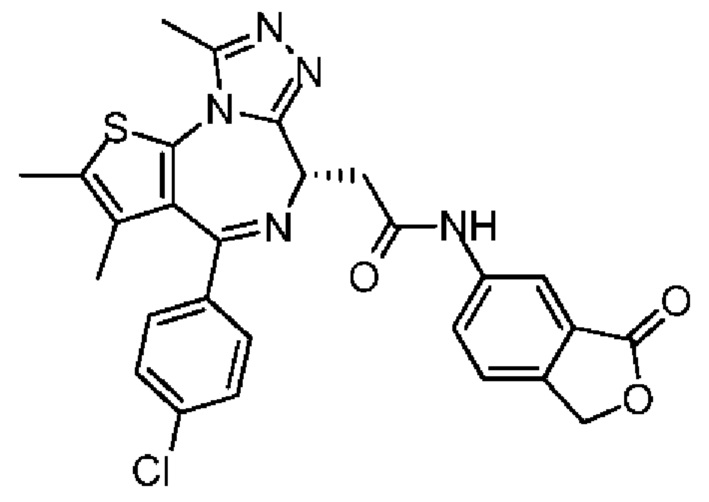
Синтез соединения 30-2
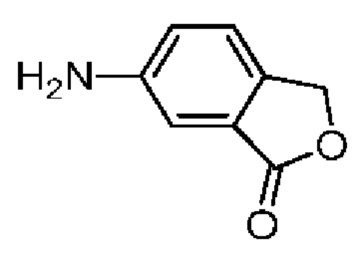
Соединение 30-1 (0,5 г, 2,79 ммоль, 1 экв.) и Pd/C (0,5 г, 10% чистоты) добавляли к метанолу (5 мл). После добавления атмосферу заменяли баллоном, заполненным газообразным водородом, 3 раза. Затем смесь реагировала при 20°С под защитой водорода под баллоном (15 фунт/кв.дюйм) в течение 12 часов. Нерастворимое вещество отфильтровывали. Фильтрат концентрировали при пониженном давлении. Остаток растворяли в метиленхлориде (50 мл). Смесь фильтровали с получением фильтрата. Затем фильтрат концентрировали при пониженном давлении с получением соединения 30-2, которое сразу использовали на следующей стадии без дополнительной обработки. 1Н ЯМР (400 МГц, CD3OD) δppm 7.19 (d, J=9.2 Гц, 1H, 6.96-6.98 (m, 2Н), 5.12 (s, 3H).
Синтез соединения 30
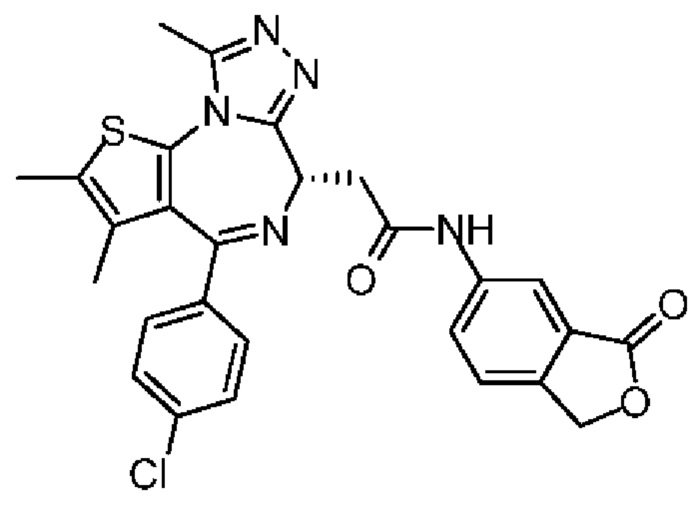
Соединение 1-11 (0,1 г, 232,41 мкмоль, 1 экв.), соединение 30-2 (52,00 мг, 348,62 мкмоль, 1,5 экв.) и HATU (106,04 мг, 278,90 мкмоль, 1,2 экв.) добавляли к безводному N,N-диметилфорамиду (2 мл). Затем добавляли диизопропилэтиламин (60,08 мг, 464,83 мкмоль, 80,96 мкл, 2 экв.) к вышеуказанному раствору при 20°С. После добавления смесь реагировала при 20°С в течение 12 часов. Воду (5 мл) добавляли к реакционной смеси. Полученную смесь экстрагировали метиленхлоридом (5 мл ×3). Органическую фазу концентрировали при пониженном давлении. Затем добавляли воду (10 мл). Смесь лиофилизировали с удалением остаточного N,N-диметилфорамида и очищали методом препаративной хроматографии с получением соединения 30. LCMS (ESI) m/z: 532.1 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 9.65 (br s, 1H, 8.09 (d, J=6.8 Гц, 1H, 7.68 (d, J=8.0 Гц, 1Н), 7.33-7.35 (m, 2H), 7.24-7.27 (m, 3H), 5.15 (s, 2H), 4.62-4.66 (m, 1H, 3.80-3.86 (m, 1H, 3.46-3.51 (m, 1H, 2.64 (s, 3H), 2.35 (s, 3H), 1.63 (s, 3H).
Схема 31
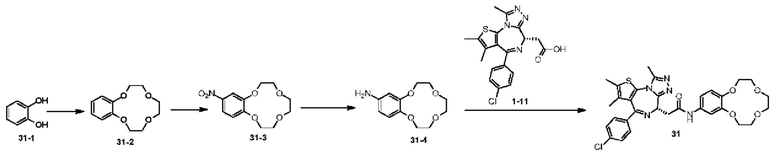
Пример 31
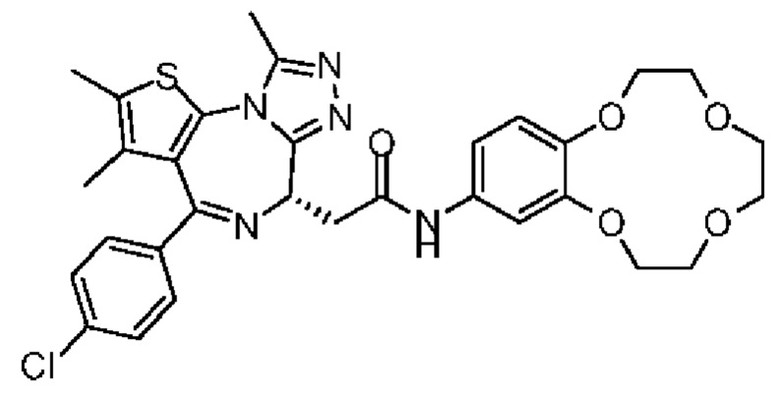
Синтез соединения 31-2
При 20°С и при защите газообразным азотом к постоянно перемешиваемому раствору соединения 31-1 (0,5 г, 4,54 ммоль, 757,58 мкл, 1 экв.), растворенному в н-бутиловом спирте (10 мл), добавляли по каплям раствор бромида лития моногидрата (1,19 г, 11,35 ммоль, 2,5 экв.) и гидроксида лития моногидрата (400,16 мг, 9,53 ммоль, 2,1 экв.), растворенного в воде (1 мл). После завершения добавления по каплям реакционную смесь нагревали до 110°С и перемешивали в течение 0,1 часов. Затем добавляли 1,2-ди(2-хлорэтокси)этан (849,27 мг, 4,54 ммоль, 532,29 мкл, 1 экв.). Перемешивание при той же температуре поддерживали в течение 5 минут. Значение рН реакционной смеси доводили при помощи HCl до 4, а затем сразу концентрировали при пониженном давлении. К остатку добавляли метиленхлорид (50 мл) и воду (30 мл). Смесь перемешивали в течение 0,5 часа и разделяли на две фазы. Органическую фазу промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении. Соединение в виде неочищенного продукта очищали на колонке методом флеш-хроматографии с получением соединения 31-2. 1Н ЯМР (400 МГц, CDCl3) δppm 6.89-6.90 (m, 4Н), 4.09-4.11 (m, 4Н), 3.73-3.79 (m, 8Н).
Синтез соединения 31-3
Раствор соединения 31-2 (50 мг, 222,96 мкмоль, 1 экв.) в ацетонитриле (2 мл) нагревали до 85°С. Концентрированную азотную кислоту (38 мг, 337,71 мкмоль, 27,14 мкл, 56% чистоты, 1,51 экв.) медленно по каплям добавляли. После завершения добавления по каплям смесь перемешивали при 85°С в течение 0,5 часа. Реакционную смесь охлаждали до комнатной температуры и выливали в лед (приблизительно 10 мл) для гашения реакции. Смесь экстрагировали этилацетатом (3×10 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 31-3, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δppm 7.86-7.89 (m, 1H, 7.81 (d, J=2.8 Гц, 1H, 6.93 (d, J=8.8 Гц, 1Н), 4.17-4.23 (m, 4H), 3.84-3.85 (m, 2H), 3.77-3.80 (m, 2H), 3.71 (s, 4H).
Синтез соединения 31-4
Влажный Pd/C (50 мг, 10% чистоты) добавляли к раствору соединения 31-3 (50 мг, 185,70 мкмоль, 1,00 экв.) в метаноле (10 мл). Атмосферу заменяли газообразным водородом три раза. Смесь перемешивали при 15°С в водородных условиях (баллон) (15 фунт/кв.дюйм) в течение 2 часов. Реакционную смесь фильтровали при помощи диатомитовой земли. Фильтрат сразу концентрировали при пониженном давлении с получением соединения 31-4, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δppm 6.75 (d, J=8.4 Гц, 1Н), 6.26 (d, J=2.4 Гц, 1H), 6.18 (dd, J=8.4, 2.4 Гц, 1Н), 3.98-4.05 (m, 4Н), 3.75-3.84 (m, 2Н), 3.71 (s, 6Н), 3.27 (br s, 2Н).
Синтез соединения 31
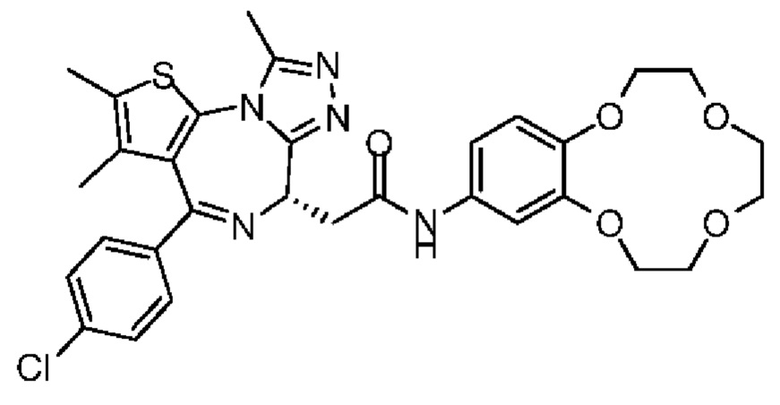
Соединение 1-11 (35 мг, 87,31 мкмоль, 1,00 экв.) и соединение 31-4 (25 мг, 104,49 мкмоль, 1,20 экв.) последовательно добавляли к раствору HATU (35,00 мг, 92,05 мкмоль, 1,05 экв.) в безводном метиленхлориде (2 мл), а затем медленно по каплям добавляли триэтиламин (29,08 мг, 287,38 мкмоль, 40 мкл, 3,29 экв.). Смесь перемешивали при 10°С при защите газообразным азотом в течение 6 часов. Воду (3 мл) добавляли к реакционной смеси для гашения реакции. Смесь экстрагировали метиленхлоридом (3×5 мл) и вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом препаративной хроматографии с получением соединения 31. LCMS (ESI) m/z: 622.1 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 8.82 (m, 1H, 7.23-7.38 (m, 5Н), 6.92-6.94 (m, 1H, 6.81-6.83 (m, 1H), 4.57-4.59 (m, 1H), 3.97-4.15 (m, 4H), 3.70-3.80 (m, 9H), 3.43-3.45 (m, 1H), 2.61 (s, 3H), 2.34 (s, 3H), 1.61 (s, 3H).
Схема 32
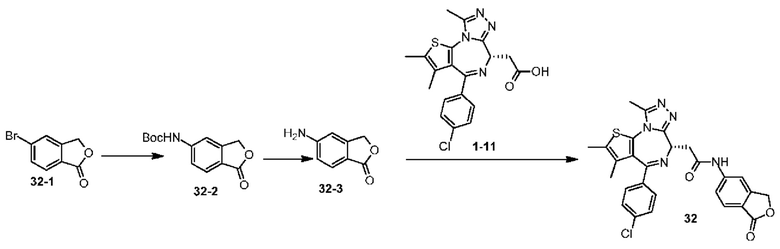
Пример 32
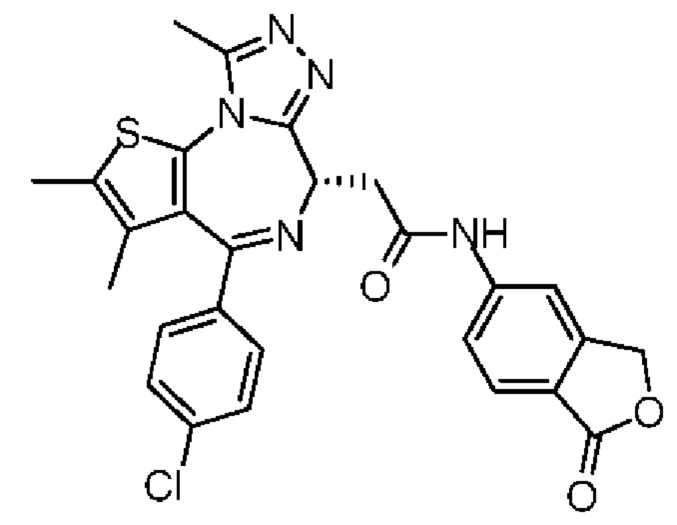
Синтез соединения 32-2
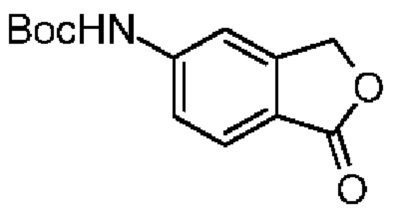
Соединение 32-1 (0,78 г, 3,66 ммоль, 1 экв.), трет-бутилкарбамат (643,39 мг, 5,49 ммоль, 1,5 экв.), трис(дибензилиденацетон)дипалладий (335,29 мг, 366,15 мкмоль, 0,1 экв.), карбонат цезия (2,39 г, 7,32 ммоль, 2 экв.) и 4,5-бис(дифенилфосфин)-9,9-диметилксантен (211,86 мг, 366,15 мкмоль, 0,1 экв.) добавляли к 1,4-диоксану (10 мл). После добавления смесь реагировала при 100°С при защите газообразным азотом в течение 12 часов. К реакционной смеси добавляли воду (20 мл), а затем этилацетат (20 мл). Нерастворимое вещество отфильтровывали. Затем водную фазу экстрагировали этилацетатом (10 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, затем концентрировали при пониженном давлении и очищали на колонке при помощи флеш-хроматографии с получением соединения 32-2. LCMS (ESI) m/z: 250.1 (М+1).
Синтез соединения 32-3
Трифторуксусную кислоту (3,85 г, 33,77 ммоль, 2,5 мл, 18,30 экв.) добавляли к соединению 32-2 (0,46 г, 1,85 ммоль, 1 экв.) в безводном метиленхлориде (20 мл). После добавления смесь реагировала при 20°С в течение 12 часов. Реакционную смесь промывали водой (20 мл). Значение рН водной фазы доводили при помощи насыщенного раствора бикарбоната натрия до 7, а затем экстрагировали метиленхлоридом (10 мл ×2). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении с получением соединения 32-3, которое сразу использовали на следующей стадии без дополнительной очистки. LCMS (ESI) m/z: 149.8 (М+1).
Синтез соединения 32
При 0°С POCl3 (76,50 мг, 498,90 мкмоль, 46,36 мкл, 2 экв.) добавляли к раствору соединения 1-11 (100 мг, 249,45 мкмоль, 1 экв.) и соединения 32-3 (44,65 мг, 299,34 мкмоль, 1,2 экв.) в пиридине (2 мл). После добавления смесь нагревали до 20°С и она реагировала в течение 1,5 часа. Воду (3 мл) добавляли к реакционной смеси для гашения реакции. Затем значение рН водной фазы доводили при помощи хлористоводородной кислоты (2 н) до 7. Водную фазу экстрагировали метиленхлоридом (5 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали, затем концентрировали при пониженном давлении и очищали на пластине методом препаративной тонкослойной хроматографии с получением соединения 32. LCMS (ESI) m/z: 532.1 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 9.98 (br s, 1H, 7.94 (d, J=6.8 Гц, 1Н), 7.67 (d, J=8.0 Гц, 1H, 7.32-7.36 (m, 3H), 7.24-7.27 (m, 2H), 5.08-5.16 (m, 2H), 4.57-4.61 (m, 1H), 3.78-3.84 (m, 1H), 3.45-3.50 (m, 1H), 2.63 (s, 3H), 2.36 (s, 3H), 1.63 (s, 3H).
Схема 33
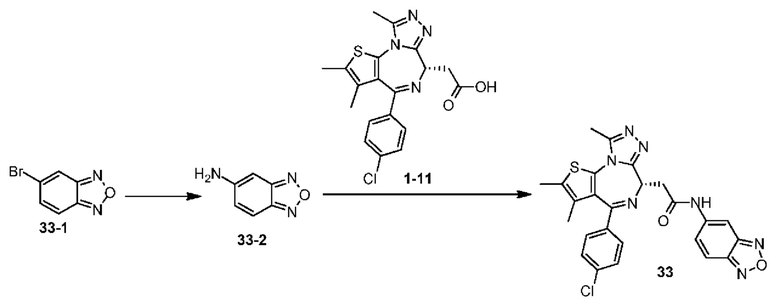
Пример 33
Синтез соединения 33-2
Соединение 33-1 (0,5 г, 2,51 ммоль, 1 экв.), CuO (19,99 мг, 251,25 мкмоль, 3,16 мкл, 0,1 экв.) и йодид меди (478,51 мг, 2,51 ммоль, 1 экв.) добавляли к концентрированной аммиачной воде (5,46 г, 155,80 ммоль, 6 мл, 62,01 экв.). К полученному раствору добавляли несколько капель N-метилпирролидина. В микроволновом приборе для синтеза реакцию проводили при 140°С в течение 0,5 часа, а затем при 150°С в течение 1 часа. К реакционной смеси добавляли воду (10 мл) для разбавления реакционной смеси, а затем добавляли метиленхлорид (10 мл). Полученную смесь фильтровали с удалением нерастворимого вещества. Водную фазу экстрагировали метиленхлоридом (10 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении с получением соединения 33-2, которое сразу использовали на следующей стадии без дополнительной очистки. LCMS (ESI) m/z: 136.0 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 7.55 (d, J=9.6 Гц, 1H, 7.67 (dd, J=2.0, 9.6 Гц, 1H, 6.50 (s, 1H, 4.63 (br s, 2Н).
Синтез соединения 33
При 0°С и при защите газообразным азотом POCl3 (35,64 мг, 232,41 мкмоль, 21,60 мкл, 2 экв.) добавляли к раствору соединения 1-11 (50 мг, 116,21 мкмоль, 1 экв.) и соединения 33-2 (23,55 мг, 174,31 мкмоль, 1,5 экв.) в пиридине (1 мл). После добавления смесь реагировала при 20°С в течение 2 часов. Воду (2 мл) добавляли к реакционной смеси для гашения реакции. Затем значение рН водной фазы доводили при помощи 2 н HCl до 7. Водную фазу экстрагировали метиленхлоридом (5 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали, затем концентрировали при пониженном давлении, очищали методом препаративной хроматографии с получением соединения 33. LCMS (ESI) m/z: 518.1 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 9.87 (br s, 1H, 8.33 (s, 1H), 7.60 (d, J=9.2 Гц, 1H), 7.37 (d, J=8.8 Гц, 2H), 7.27 (d, J=8.8 Гц, 2H), 7.16-7.17 (m, 1H), 4.55-4.58 (m, 1H), 3.76-3.82 (m, 1H), 3.44-3.48 (m, 1H), 2.64 (s, 3H), 2.36 (s, 3H), 1.64 (s, 3H).
Схемы 34 и 35
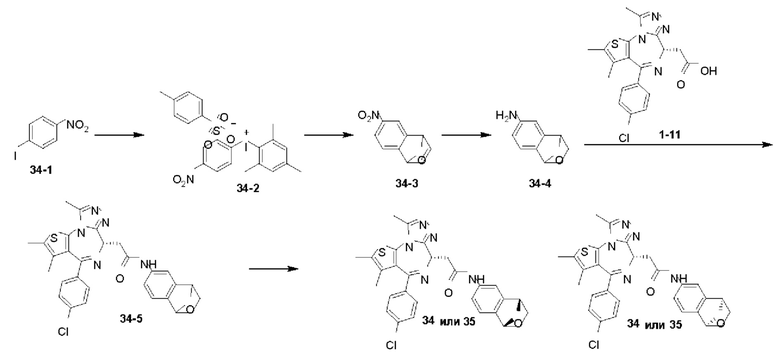
Примеры 34 и 35
Синтез соединения 34-2
3-Хлорпербензойную кислоту (3,09 г, 15,23 ммоль, 85% чистоты) добавляли к раствору соединения 34-1 (4,45 г, 17,89 ммоль, 1 экв.) и пара-толуолсульфоновой кислоты моногидрата (3,43 г, 18,01 ммоль, 1,01 экв.) в ацетонитриле (25 мл). Полученную смесь нагревали до 77°С и перемешивали в течение 30 минут. Затем медленно по каплям добавляли 1,3,5-триметилбензол (2,16 г, 17,99 ммоль, 2,5 мл, 1,01 экв.). После завершения добавления по каплям полученную смесь перемешивали при 77°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток промывали петролейным эфиром (20 мл) и фильтровали. Фильтрационный кек сушили в сушильном шкафу с получением соединения 34-2, которое сразу использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, DMSO-d6) δppm 8.24 (d, J=9.2 Гц, 1H, 8.17 (d, J=8.8 Гц, 1H, 8.06 (d, J=8.8 Гц, 2Н), 7.97 (d, J=8.8 Гц, 2Н), 7.49 (d, J=8.0 Гц, 2Н), 7.12 (d, J=8.8 Гц, 2Н), 2.58 (s, 3H), 2.51 (s, 9Н).
Синтез соединения 34-3
При 0°С и при защите газообразным азотом фуран (5,60 г, 82,26 ммоль, 5,98 мл, 5,55 экв.) добавляли по каплям к раствору соединения 34-2 (8 г, 14.83 ммоль, 1 экв.) в толуоле (20 мл), а затем медленно по каплям добавляли лития бис(триметилсилил)амид (1 М, 14,82 мл). После завершения добавления по каплям смесь перемешивали при 0°С при защите газообразным азотом в течение 3 часов. Реакционную смесь охлаждали до 0°С и добавляли насыщенный раствор хлорида аммония (4 мл) для гашения реакции. Смесь экстрагировали этилацетатом (3×5 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали на колонке методом флеш-хроматографии с получением соединения 34-3. 1Н ЯМР (400 МГц, CDCl3) δppm 8.21 (s, 1H), 8.19 (s, 1H), 7.48-7.50 (m, 1H), 7.11-7.13 (m, 1H), 7.06-7.08 (m, 1H), 5.82-5.84 (m, 2H).
Синтез соединения 34-4
При защите газообразным азотом раствор боргидрида натрия (12,00 мг, 317,18 мкмоль, 1 экв.) в воде (1 мл) медленно по каплям добавляли к суспензии соединения 34-3 (60 мг, 317,18 мкмоль, 1 экв.) и Pd/C (5 мг, 10% чистоты) в метаноле (2 мл). Смесь перемешивали при 25°С при защите газообразным азотом в течение 1 часа. Реакционную смесь фильтровали через воронку, содержащую диатомитовую землю. Фильтрат сразу концентрировали при пониженном давлении с получением соединения 34-4, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. LCMS (ESI)m/z: 162.1 (М+1).
Синтез соединения 34-5
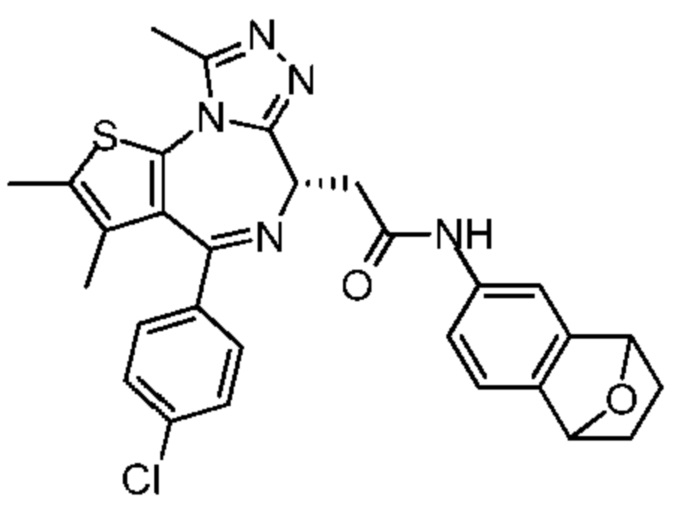
Соединение 1-11 (35 мг, 87,31 мкмоль, 1,00 экв.) и соединение 34-4 (17 мг, 105,46 мкмоль, 1,21 экв.) последовательно добавляли к раствору HATU (35,00 мг, 92,05 мкмоль, 1,05 экв.) в безводном N,N-диметилформамиде (2 мл), а затем медленно по каплям добавляли триэтилмин (28,00 мг, 276,71 мкмоль, 38.51 мкл, 3,17 экв.). Смесь перемешивали при 25°С при защите газообразным азотом в течение 1 часа. Воду (3 мл) добавляли к реакционной смеси для гашения реакции, смесь разделяли на две фазы. Водную фазу экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 34-5. LCMS (ESI) m/z: 544.1 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 8.73 (s, 1H), 7.52 (d, J=46.4 Гц, 1H), 7.34 (d, J=8.4 Гц, 2H), 7.26 (d, J=7.6 Гц, 2H), 7.05-7.22 (m, 2H), 5.28 (s, 2H), 4.52-4.56 (m, 1H), 3.68-3.74 (m, 1H), 3.37-3.41 (m, 1H), 2.60 (s, 3H), 2.33 (s, 3H), 1.61 (s, 3H), 1.27 (d, J=6.8 Гц, 2H), 1.19 (d, J=6.8 Гц, 2H).
Синтез соединений 34 и 35
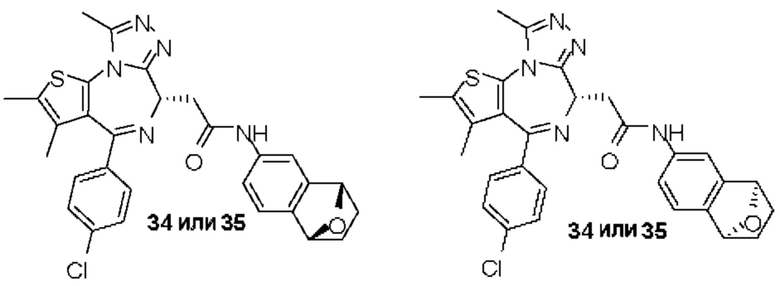
Соединение 34-5 (20 мг, 36,73 мкмоль) очищали при помощи хирального разделения (хроматографическая колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% NH3H2O ЕtOН]; В%: 50%-50%) с получением соединения 34 (Rt=0,735 минуты). LCMS (ESI) m/z: 544.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 8.79 (s, 1H), 7.58 (s, 1H), 7.34 (d, J=8.4 Гц, 2H), 7.26 (d, J=8.4 Гц, 2H), 7.11-7.15 (m, 1H), 7.05-7.07 (m, 1H), 5.29(t, J=3.8 Гц, 2H), 4.53-4.57 (m, 1H), 3.67-3.73 (m, 1H), 3.38-3.42 (m, 1H), 2.60 (s, 3H), 2.33 (s, 3H), 1.91-1.96 (m, 2H), 1.61 (s, 3H), 1.25-1.29 (m, 2H).
Соединение 35 (Rt=1,300 минуты). LCMS (ESI) m/z: 544.1 (М+1). 1H ЯМР (400 МГц, CDCl3) δppm 8.76 (s, 1H), 7.46 (s, 1H), 7.34 (d, J=8.8 Гц, 2H), 7.26 (d, J=8.8 Гц, 2H), 7.21-7.22 (m, 1H), 7.05-7.07 (m, 1H), 5.28(s, 2H), 4.53-4.57 (m, 1H), 3.68-3.74 (m, 1H), 3.37-3.42 (m, 1H), 2.60 (s, 3H), 2.33 (s, 3H), 1.94-1.96 (m, 2H), 1.61 (s, 3H), 1.25-1.29 (m, 2H).
Схема 36
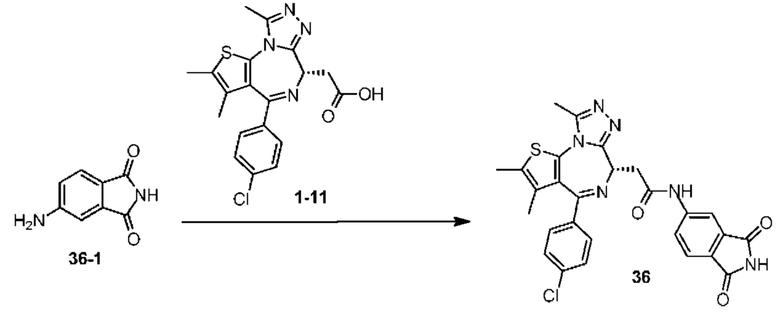
Пример 36
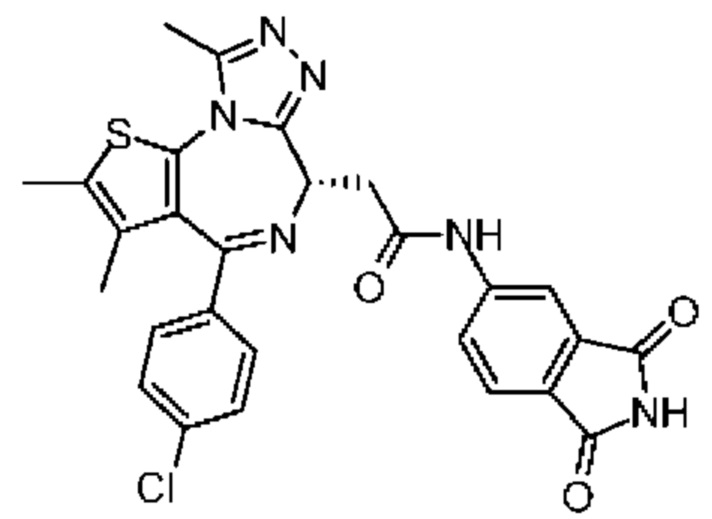
Соединение пример 36 синтезировали на основании примера 1.
LCMS (ESI) m/z: 545.1 (М+1). 1Н ЯМР (400 МГц, CDCl3) δррm 10.10 (s, 1H), 8.08 (s, 1H), 7.73 (d, J=8.0 Гц, 1H), 7.60 (d, J=8.0 Гц, 2Н), 7.34 (d, J=8.8 Гц, 2Н), 7.25 (d, J=8.8 Гц, 2Н), 4.61-4.65 (m, 2Н), 3.80-3.86 (m, 1H), 3.48-3.53 (m, 1Н), 2.65 (s, 3Н), 2.36 (s, 3Н), 1.63 (s, 3Н).
Схема 37
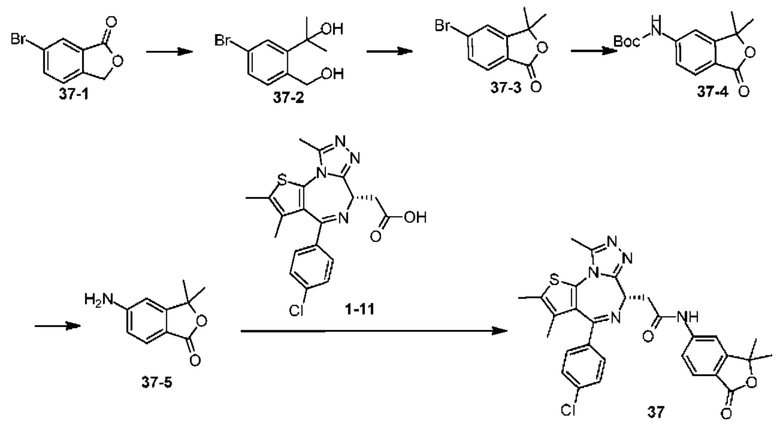
Пример 37
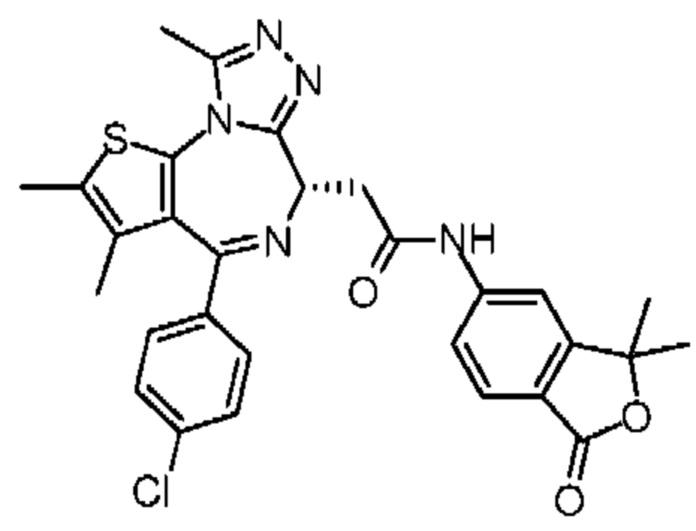
Синтез соединения 37-2
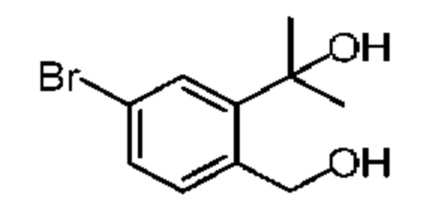
При 0°С и при защите газообразным азотом метилбромид магния (3 М, 9,39 мл, 3 экв.) медленно по каплям добавляли к раствору соединения 37-1 (2 г, 9,39 ммоль, 1 экв.) в безводном тетрагидрофуране (20 мл). После завершения добавления по каплям смесь нагревали до 30°С и перемешивали под защитой газообразного азота в течение 3 часов. Реакционную смесь охлаждали до 0°С и насыщенный раствор хлорида аммония (10 мл) медленно по каплям добавляли для гашения реакции. Смесь экстрагировали метиленхлоридом (3×10 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 37-2, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δppm 7.44 (d, J=2.0 Гц, 1H), 7.39 (dd, J=8.4, 2.0 Гц, 1Н), 7.22 (d, J=8.4, 1Н), 4.81 (s, 2Н), 1.70 (s, 6 Н).
Синтез соединения 37-3
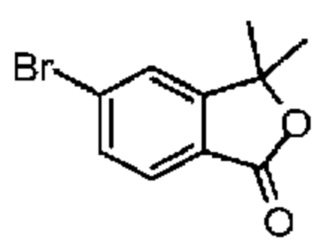
Соединение 37-2 (2,1 г, 8,57 ммоль, 1 экв.) добавляли к суспензии активного пероксида марганца (7,35 г, 84,54 ммоль, 9,87 экв.) в безводном тетрагидрофуране (15 мл). Смесь перемешивали при 70°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 37-3, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) 7.65 (d, J=8.0 Гц, 1H), 7.57 (dd, J=8.0, 1.2 Гц, 1Н), 7.49 (d, J=1.2, 1H), 1.59 (s, 6 Н).
Синтез соединения 37-4
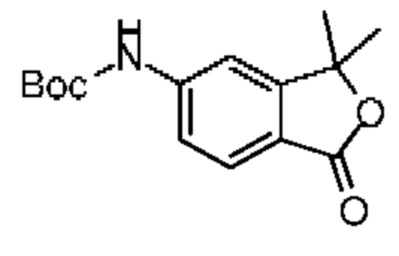
Трис(дибензилиденацетон)дипалладий (380,00 мг, 414,98 мкмоль, 0,1 экв.), 4,5-бис(дифенилфосфин)-9,9-диметилксантен (240,0 мг, 414,78 мкмоль, 0,1 экв.) и карбонат цезия (2,70 г, 8,29 ммоль, 2 экв.) последовательно добавляли к раствору соединения 37-3 (1 г, 4,15 ммоль, 1 экв.) и трет-бутилкарбамата (700,00 мг, 5,98 ммоль, 1,44 экв.) в безводном 1,4-диоксане (10 мл). Смесь перемешивали при 100°С при защите газообразным азотом в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. Воду (10 мл) добавляли к реакционной смеси. Смесь экстрагировали метиленхлоридом (3×10 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали на колонке с силикагелем (условие элюирования: петролейный эфир:этилацетат =1:1) с получением соединения 37-4. 1Н ЯМР (400 МГц, CDCl3) δppm 7.75 (s, 1H), 7.67 (d, J=8.0 Гц, 1H), 7.04 (dd, J=8.4, 1.6 Гц, 1H), 6.79 (br s, 1H), 1.58 (s, 6H), 1.47 (s, 9H).
Синтез соединения 37-5
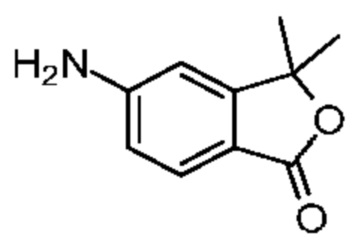
Трифторуксусную кислоту (7,70 г, 67,53 ммоль, 5 мл, 18,73 экв.) медленно по каплям добавляли к раствору соединения 37-4 (1 г, 3,61 ммоль, 1 экв.) в безводном метиленхлориде (10 мл). Смесь перемешивали при 30°С в течение 1 часа. Воду (5 мл) добавляли к реакционной смеси для гашения реакции. Значение рН смеси доводили при помощи насыщенного раствора бикарбоната натрия до 7, а затем экстрагировали метиленхлоридом (3×10 мл). Вышеуказанные органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 37-5, которое использовали сразу на следующей стадии без необходимости дополнительной очистки. LCMS (ESI) m/z: 177.8 (М+1).
Синтез соединения 37
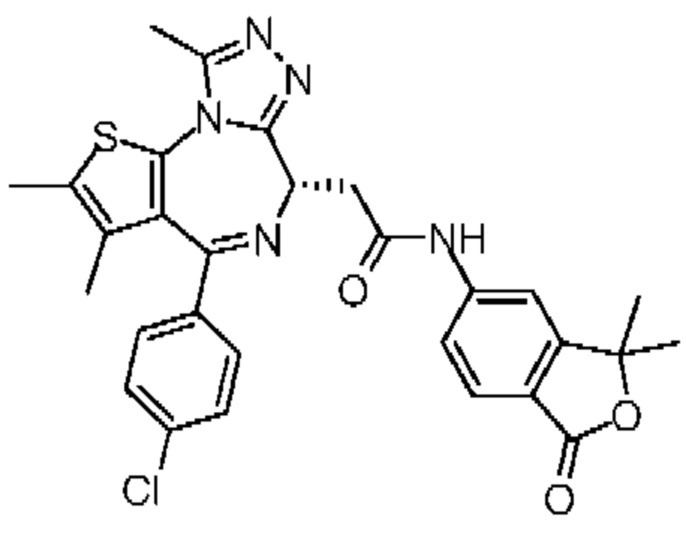
При 0°С оксихлорид фосфора (140,00 мг, 913,05 мкмоль, 84,85 мкл, 5,23 экв.) медленно по каплям добавляли к раствору соединения 1-11 (70 мг, 174,62 мкмоль, 1,00 экв.) и соединения 37-5 (40 мг, 225,73 мкмоль, 1,29 экв.) в пиридине (2 мл). Смесь перемешивали при 0°С при защите газообразным азотом в течение 1 часа. К реакционной смеси добавляли воду (3 мл) для гашения реакции. Значение рН вышеуказанной реакционной смеси доводили при помощи раствора хлористоводородной кислоты (1 моль/л) до 7 и экстрагировали метиленхлоридом (3×5 мл). Вышеуказанные органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (5 мл) и насыщенным солевым раствором (5 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Вышеуказанный неочищенный продукт очищали методом тонкослойной хроматографии на пластине с получением соединения 37. LCMS (ESI) m/z: 560.0 (М+1). 1Н ЯМР (400 МГц, CDCl3) δppm 9.89 (br s, 1H), 7.92 (s, 1H), 7.62 (d, J=8.4 Гц, 1H), 7.36 (d, J=8.4 Гц, 2H), 7.25-7.30 (m, 3Н), 4.57-4.61 (m, 1H), 3.78-3.81 (m, 1H), 3.45-3.50 (m, 1H), 2.62 (s, 3H), 2.35 (s, 3H), 1.64 (s, 3H), 1.54 (s, 3H), 1.53 (s, 3H).
Тест биохимической активности BRD4 Экспериментальное получение
1) В экспериментах использовали белки BRD4-BD1 и BRD4-BD2 от компании BPS Company, полипептид от компании ANASPEC Company и тестовый реагент от компании PerkinElmer Company.
2) В экспериментах использовали эмпирический принцип TR-FRET для скрининга соединений.
3) Соответствующие контрольные соединения. Стадии эксперимента
1) Получение планшета с соединением
Получение планшета с соединением в эксперименте осуществляли с помощью Echo.
Разбавление соединения совершали с помощью Echo и выполняли трехкратное серийное разбавление до 10 концентраций: 20000, 6666,67, 2222,22, 740,74, 246,91, 82,305, 27,435, 9,145, 3,048 и 1,016 нМ.
2) Получение реакционных реагентов
Соответствующие реакционные реагенты следует готовить в день эксперимента.
a) Составление 1× аналитического буфера.
b) Составление 3× компонентного раствора для эксперимента:
1. брали реагент и помещали его на лед для оттаивания в естественных условиях для последующего использования;
2. «раствор А» (раствор белка), «раствор В» (раствор полипептида) и «раствор С» (раствор тестового реагента), используемые в эксперименте, составляли с 1× аналитического буфера, и в ходе составления 3× раствора из компонентов, используемых в экспериментальной реакционной система, количества растворов А, В и С должны быть достаточными для требуемых в эксперименте количеств.
3) Экспериментальные операционные стадии
Экспериментальный планшет представлял собой планшет, который содержал градиентную концентрацию соединения и соответствующий раствор DMSO и готовился с ECHO перед экспериментом:
a) брали экспериментальный планшет и добавляли 5 мкл/лунка «раствора А» (раствора белка) в колонки 2-23 экспериментального планшета, затем добавляли 5 мкл/лунка 1× аналитического буфера в колонки 1 и 24 экспериментального планшета и колонки 1 и 24 использовали в качестве минимального контроля в экспериментальной системе;
b) выполняли центрифугирование при 1000 оборотах в минуту в течение 30 секунд;
c) планшет инкубировали при 23°С в течение 20 минут;
d) после 20 минут инкубации в экспериментальный планшет добавляли 5 мкл/лунка «раствора В» (раствора полипептида) в колонки 1-24 экспериментального планшета;
e) выполняли центрифугирование при 1000 оборотах в минуту в течение 30 секунд;
f) планшет инкубировали при 23°С в течение 20 минут;
g) после 20 минут инкубации в экспериментальный планшет добавляли 5 мкл/лунка «раствора В» (раствора тестового реагента) в колонки 1-24 экспериментального планшета;
h) выполняли центрифугирование при 1000 оборотах в минуту в течение 30 секунд;
i) планшет инкубировали при 23°С в течение 40 минут;
j) экспериментальный планшет помещали в EnVision для считывания планшета.
4) Данные анализа
a) Использовали соответствующие максимальный контроль и минимальный контроль каждого экспериментального планшета для преобразования в значение Z' экспериментального планшета и обеспечивали значение Z' каждого планшета >0,5.
b) Вычисляли значение IC50 из сигнала контрольного соединения с помощью XLFIT5 и обеспечивали его поддержание в пределах 3-кратного среднего значения ранее полученных данных. Результаты показаны в таблице 1.


Заключение. Соединения в соответствии с настоящим изобретением обладают значительными ингибиторными активностями в отношении бромодомена BET.
In vivo исследование фармакодинамических показателей соединения 32 на модели подкожной ксенотрансплантатной опухоли клеток MDA-MB-231_luc рака молочной железы человека
1. Схема эксперимента
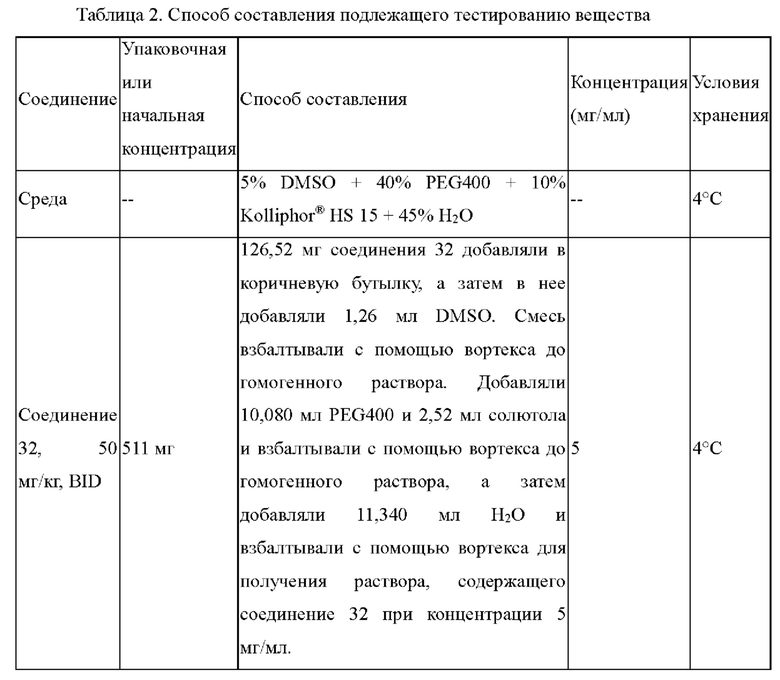
фармакодинамических экспериментов
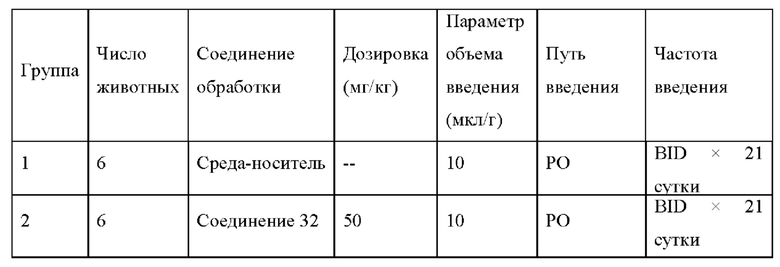
2. Экспериментальный материал 2.1.
Экспериментальные животные
Вид: мышь.
Штамм: голая мышь BALB/c.
Возраст и масса: возраст 6-8 недель, масса тела 18-22 грамма.
Пол: женский.
Поставщик: компания Shanghai Sippr-BK Laboratory Animal Co. Ltd.
3. Способы и стадии эксперимента
3.1. Клеточная культура
Клетки MDA-MB-231_luc рака молочной железы человека культивировали в монослое in vitro, и условия культивирования предусматривали культуральную среду RPMI-1640 (поставщик: компания Gibco; номер изделия: 22400-089; номер партии изготовителя: 4868546) с 10% эмбриональной бычьей сыворотки, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина. Культивирование осуществляли при 37°С в 5% CO2. Дважды в неделю выполняли традиционную обработку расщеплением панкреатином-EDTA для пересева. Когда клетки находились в экспоненциальной фазе роста, их собирали, подсчитывали и инокулировали.
3.2. Инокулирование опухолевых клеток 0,2 мл 10×106 клеток MDA-MB-231_luc подкожно инокулировали в правую часть спины каждой голой мыши (PBS: Matrigel=1:1). Распределение по группам и введение начинали, когда средний объем опухоли достигал 100-150 мм3.
3.3. Измерение опухоли и показатели эксперимента
Показатель эксперимента служил для того, чтобы исследовать, был ли рост опухоли ингибирован, отсрочен или устранен. Диаметр опухоли измеряли два раза в неделю с помощью штангенциркуля. Уравнение для расчета объема опухоли представляло собой V=0,5 × а × b2, при этом а и b представляли собой большой и меньший диаметры опухоли, соответственно.
Эффект ингибирования опухоли для соединения оценивали по TGI (%) или относительной скорости пролиферации опухоли Т/С (%). TGI (%) отражает степень ингибирования роста опухоли. Расчет TGI (%) был следующим: TGI (%)=[(1 - (средний объем опухоли в конце введения в получавшей лечение группе - средний объем опухоли в начале введения в этой получавшей лечение группе))/(средний объем опухоли в конце лечения в получавшей растворитель контрольной группе - средний объем опухоли в начале лечения в получавшей растворитель контрольной группе)] × 100%.
Относительную скорость пролиферации опухоли Т/С (%) рассчитывали по приведенному ниже уравнению: % Т/С=TRTV/CRTV × 100% (TRTV: RTV в получавшей лечение группе; CRTV: RTV в группе отрицательного контроля). Относительный объем опухоли (RTV) рассчитывали по результатам измерения опухоли. Вычислительное уравнение представляло собой RTV=Vt/V0, при этом V0 представлял собой средний объем опухоли, измеренный при распределении на группы и введении (т.е. d0), a Vt представлял собой средний объем опухоли на момент времени определенного измерения. TRTV и CRTV получали из данных в один и тот же день.
В конце эксперимента измеряли массу опухоли и рассчитывали процент Тмассы/Смассы. Тмассы и Смассы представляли собой массы опухоли в получавшей введение группе и в получавшей среду контрольной группе, соответственно.
3.4. Статистический анализ
Статистический анализ предусматривал среднее значение и стандартную ошибку (SEM) объема опухоли каждой группы в каждый момент времени. Получавшая лечение группа демонстрировала лучший эффект лечения на 21-ые сутки после введения в конце эксперимента, поэтому статистический анализ выполняли на основании этих данных для оценки различий между группами. Сравнение двух групп анализировали с помощью Т-теста, а сравнение трех или более групп анализировали с помощью однофакторного ANOVA. Если значение F существенно отличалось, применяли тест Геймса-Ховелла. Если значение F существенно не отличалось, для анализа использовали тест Даннета (2-сторонний). Анализ всех данных выполняли с SPSS 17,0. р<0,05 считали достоверной разницей.
4. Заключение эксперимента
На 21-е сутки после введения для тестируемого соединения 32 степень ингибирования роста опухоли составляла TGI=54,85%, Т/С=52,99%, р<0,05; отсутствовало значительное изменение массы тела животных, и они его хорошо переносили.
In vivo исследование фармакодинамических показателей соединения 32 на модели подкожной ксенотрансплантатной опухоли клеток РС-3 клеток рака предстательной железы человека
1. Схема эксперимента
Способ составления тестируемого вещества был таким же, как в таблице 2, а распределение животных по группам и режим введения дозы были такими же, как в таблице 3.
2. Экспериментальный материал 2.1. Экспериментальные животные
Вид: мышь.
Штамм: голая мышь BALB/c.
Возраст и масса: возраст 6-8 недель, масса тела 18-22 грамма.
Пол: мужской.
Поставщик: компания Shanghai Sippr-BK Laboratory Animal Co. Ltd.
3. Способы и стадии эксперимента 3.1.
Клеточная культура
Клетки РС-3 рака предстательной железы человека культивировали в монослое in vitro, и условия культивирования предусматривали культуральную среду F-12K (поставщик: компания Gibco; номер изделия: 21127-022; номер партии изготовителя: 1868870) с 10% эмбриональной бычьей сыворотки, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина. Культивирование осуществляли при 37°С в 5% СО2. Дважды в неделю выполняли традиционную обработку расщеплением панкреатином-EDTA для пересева. Когда клетки находились в экспоненциальной фазе роста, их собирали, подсчитывали и инокулировали.
3.2. Инокулирование опухолевых клеток
0,1 мл 10×106 клеток РС-3 подкожно инокулировали в правую часть спины каждой голой мыши. Распределение по группам и введение начинали, когда средний объем опухоли достигал 100-150 мм3.
3.3. Измерение опухоли, показатели эксперимента и статистический анализ были такими же, как и для модели MDA-MB-231
4. Заключение эксперимента
На 21- сутки после введения по сравнению с получавшей растворитель контрольной группой тестируемое соединение 32 демонстрировало значительный эффект ингибирования опухоли (Т/С=44,63%, TGI=58,4%, р=0,033); животные его хорошо переносили.
In vivo тестирование фармакокинетических показателей соединения 32 у мышей
Использовали самок мышей Balb/c в качестве тестируемых животных. Соединение 32 вводили мышам внутривенно и внутрижелудочно, затем определяли концентрации лекарственного средства в плазме в разные моменты времени с помощью способа LC/MS/MS. Исследовали поведение in vivo фармакокинетических показателей соединения 32 у мышей и оценивали его фармакокинетические характеристики.
1. Протокол эксперимента
1.1. Экспериментальное лекарственное средство: соединение 32.
1.2. Экспериментальные животные: шестнадцать здоровых взрослых самок мышей Balb/c распределяли на четыре группы по принципу подобной массы тела по четыре мыши на каждую группу. Животных приобретали у компании Shanghai Lingchang BioTech Co., Ltd. из Shanghai SLAC Laboratory Animal Co., Ltd. с номером лицензии на производство животных SCXK (Shanghai) 2013-0018.
1.3. Составление лекарственного средства
Брали соответствующее количество образца, добавляли 5% конечного объема DMSO, а затем добавляли 95% конечного объема 20% HP-β-CD. Смесь взбалтывали с помощью ультразвука с получением 0,5 мг/мл прозрачного раствора. После фильтрования его использовали для внутривенного введения.
Брали соответствующее количество образца и растворяли в 0,5% растворе карбоксиметилцеллюлозы натрия. Смесь взбалтывали с помощью ультразвука с получением 0,5 мг/мл гомогенной суспензии, которую использовали для внутрижелудочного введения. 1.4. Введение
Восемь самок мышей Balb/c распределяли в две группы. После голодной выдержки на протяжении ночи первой группе осуществляли внутривенное введение при объеме введения 2,5 мл/кг и дозировке 1 мг/кг. Второй группе осуществляли внутрижелудочное введение при объеме введения 5 мл/кг и дозировке 2 мг/кг.
2. Операция
После осуществления самкам мышей Balb/c внутривенного введения забирали 30 мкл крови у разных мышей в каждый момент времени через 0,0833, 0,25, 0,5, 1, 2, 4, 8 и 24 часа и помещали в тестовые пробирки, содержащие 2 мкл EDTA-K2; а после осуществления самкам мышей Balb/c внутрижелудочного введения забирали 30 мкл крови в альтернативном месте в каждый момент времени через 0,25, 0,5, 1, 2, 4, 8 и 24 часа и помещали в тестовые пробирки, содержащие 2 мкл EDTA-K2. Пробирку центрифугировали при 3000 g в течение 15 минут для отделения плазмы и отделенную плазму хранили при -60°С. Животные могли принимать пищу через 2 часа после введения.
Использовали способ LC/MS/MS для измерения содержания подлежащего тестированию соединения в плазме после внутривенного и внутрижелудочного введения мышам. Линейный диапазон способа составлял 2,00-6000 нмоль/л; образцы плазмы анализировали после осаждения белка обработкой ацетонитрилом. Результаты фармакокинетических параметров показаны в таблице 4.
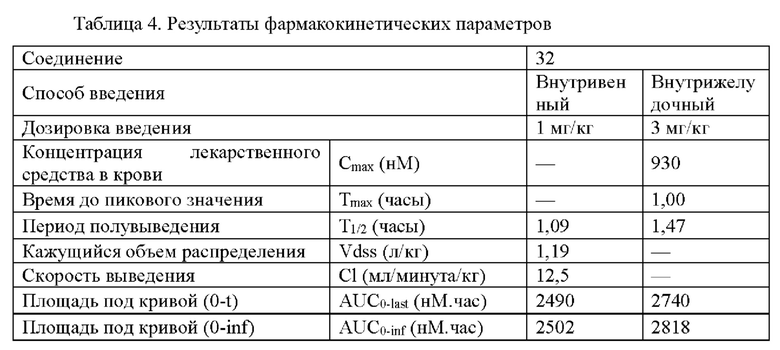
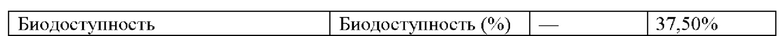
«-»: отсутствие данных.
Заключение эксперимента. Соединение 32 обладало низкими фармакокинетическими показателями выведения и хорошей абсорбцией.
In vivo противоопухолевый эффект соединения 32 на животной модели с трансплантацией опухоли в виде клеток МС38 рака толстой кишки мыши
1. Схема эксперимента
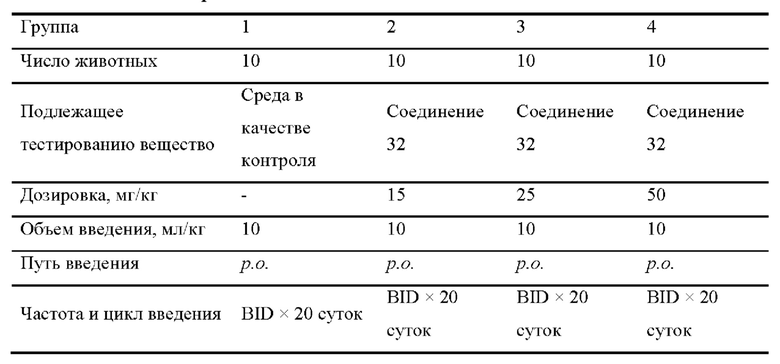
2. Экспериментальный материал 2.1.
Экспериментальные животные
Вид: мышь.
Штамм: C57BL6.
Возраст и масса: возраст 6-7 недель, масса тела 16-20 граммов.
Пол: женский.
Поставщик: компания Shanghai Sippr-BK Laboratory Animal Co. Ltd.
3. Способы и стадии эксперимента 3.1.
Клеточная культура
Клетки МС38 рака толстой кишки мыши (OBiO Technology (Shanghai) Corp., Ltd.) культивировали в монослое in vitro, и условия культивирования предусматривали культуральную среду DMEM (Gibco; номер изделия: 12100) с 10% эмбриональной бычьей сыворотки. Культивирование осуществляли при 37°С в 5% CO2 в инкубаторе. Выполняли традиционную обработку расщеплением 0,25% панкреатином-EDTA для пересева. Когда клетки находились в экспоненциальной фазе роста, и насыщение составляло 80%-90%, клетки собирали, подсчитывали и инокулировали.
3.2. Инокулирование опухолевых клеток 0,1 мл 2×105 клеток МС38 подкожно инокулировали в правую часть спины каждой голой мыши. Рандомное распределение по группам и введение выполняли согласно объему опухоли, когда средний объем опухоли достигал приблизительно 70 мм3.
3.3. Измерение опухоли
Диаметр опухоли измеряли два раза в неделю с помощью штангенциркуля. Уравнение для расчета объема опухоли представляло собой V=0,5 × а × b1, при этом а и b представляли собой большой и меньший диаметры опухоли, соответственно.
Эффект ингибирования опухоли для соединения оценивали по TGI (%) или относительной скорости пролиферации опухоли Т/С (%). Относительная скорость пролиферации опухоли Т/С (%)=TRTV/CRTV × 100% (TRTV: RTV в получавшей лечение группе; CRTV: RTV в группе отрицательного контроля). Относительный объем опухоли (RTV) рассчитывали по результатам измерения опухоли. Вычислительное уравнение представляло собой RTV=Vt/V0, при этом V0 представлял собой средний объем опухоли, измеренный при распределении на группы и введении (т.е. D0), a Vt представлял собой средний объем опухоли на момент времени определенного измерения. TRTV и CRTV получали из данных в один и тот же день.
TGI (%) отражает степень ингибирования роста опухоли. TGI (%)=[(1 - (средний объем опухоли в конце введения в получавшей лечение группе - средний объем опухоли в начале введения в этой получавшей лечение группе))/(средний объем опухоли в конце лечения в получавшей растворитель контрольной группе - средний объем опухоли в начале лечения в получавшей растворитель контрольной группе)] × 100%.
В конце эксперимента измеряли массу опухоли и рассчитывали процент Тмассы/Смассы. Тмассы и Смассы представляли собой массы опухоли в получавшей введение группе и в получавшей среду контрольной группе, соответственно.
3.4. Статистический анализ
Статистический анализ выполняли с использованием программного обеспечения SPSS на основании объема опухоли и массы опухоли в конце эксперимента. Сравнение двух групп анализировали с помощью t-теста, а сравнение трех или более групп анализировали с помощью однофакторного ANOVA. Если дисперсия была однородной (значение F существенно не отличалось), то для анализа применяли способ LSD Если дисперсия не была однородной (значение F существенно отличалось), то для теста применяли способ Геймса-Ховелла. р<0,05 считали достоверной разницей.
4. Заключение эксперимента
На 21-е сутки после введения для тестируемого соединения 32 для получавшей введение 15 мг/кг группы: относительная скорость пролиферации опухоли Т/С=33,68%, степень ингибирования роста опухоли TGI=68,81%, р<0,0001; для получавшей введение 25 мг/кг группы: относительная скорость пролиферации опухоли Т/С=27,59%, TGI=75,21%, р<0,0001; и для получавшей введение 50 мг/кг группы: Т/С=10,04%, TGI=93,46%, р<0,0001. Значительные эффекты ингибирования опухоли были продемонстрированы в каждой получавшей введение группе животных с хорошей переносимостью.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| ДИАЗАБЕНЗОФТОРАНТРЕНОВЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2697513C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИМЕЮЩИЕ СРОДСТВО К МУСКАРИНОВЫМ РЕЦЕПТОРАМ | 2008 |
|
RU2446166C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
Изобретение относится к соединению формулы (I) или (II), его стереоизомеру или фармацевтически приемлемой соли, обладающим активностью ингибирования BRD4, фармацевтической композиции на их основе и их применению при изготовлении лекарственного средства для лечения или предотвращения заболевания, связанного с BRD4. В общей формуле (I) или (II) T выбран из группы, состоящей из C и N; R1 представляет собой C1-3алкил; R2, R3 и R4 отдельно и независимо выбраны из группы, состоящей из H, F, Cl, Br и C1-6 алкила; R5 представляет собой H; R6 отдельно и независимо выбран из группы, состоящей из H, F, Cl, Br, OH, NH2, C1-6 алкила и C1-6гетероалкила, который необязательно замещен 1R группой; кольцо A выбрано из группы, состоящей из C3-7 циклоалкила, 5-12-членного гетероциклоалкила и 5-6-членного гетероарила; кольцо B представляет собой 4-7-членный гетероциклоалкил; n выбран из группы, состоящей из 0, 1, 2; R отдельно и независимо выбран из группы, состоящей из NH2 и C1-6 алкила. Технический результат: разработка соединений, обладающих значительной ингибирующей активностью в отношении бромодомена BRD4. 4 н. и 16 з.п. ф-лы, 4 табл., 37 пр.
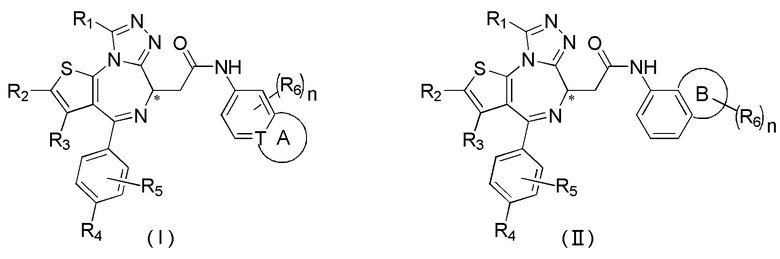
1. Соединение, представленное формулой (I) или (II), его стереоизомер или фармацевтически приемлемая соль
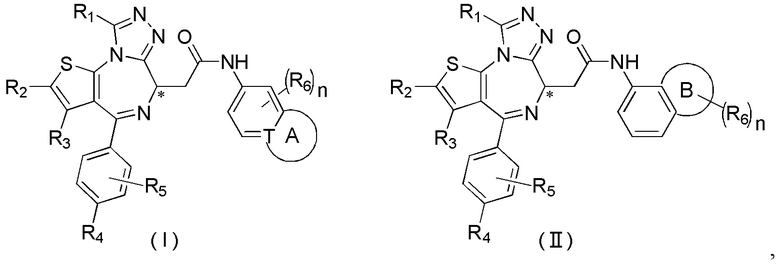
где
T выбран из группы, состоящей из C и N;
R1 представляет собой C1-3алкил;
R2, R3 и R4 отдельно и независимо выбраны из группы, состоящей из H, F, Cl, Br и C1-6 алкила;
R5 представляет собой H;
R6 отдельно и независимо выбран из группы, состоящей из H, F, Cl, Br, OH, NH2, C1-6 алкила и C1-6гетероалкила, который необязательно замещен 1R группой;
кольцо A выбрано из группы, состоящей из C3-7 циклоалкила, 5-12-членного гетероциклоалкила и 5-6-членного гетероарила;
кольцо B представляет собой 4-7-членный гетероциклоалкил;
и структурная единица  не выбрана из группы, состоящей из
не выбрана из группы, состоящей из  ,
,  и
и  ;
;
n выбран из группы, состоящей из 0, 1, 2;
R отдельно и независимо выбран из группы, состоящей из NH2 и C1-6 алкила;
атом углерода, помеченный «*», представляет собой хиральный атом углерода, который присутствует в форме простого (R) или (S) энантиомера, или в форме, обогащенной одним из двух энантиомеров;
термин «гетеро» в C1-6гетероалкиле, 5-12-членном гетероциклоалкиле, 5-6-членном гетероариле и 4-7-членном гетероциклоалкиле выбран из группы, состоящей из N, -O-, -S-, -NH-, -C(=O)NH-, -C(=O)-, -C(=O)O-, -S(=O)2-, -S(=O)- и -C(=O)S-;
число вышеуказанного гетероатома или группы гетероатомов отдельно и независимо выбрано из группы, состоящей из 1, 2, 3 и 4; при условии, что ни соединение формулы (I), ни соединение формулы (II) не содержат ни одно из следующих соединений:
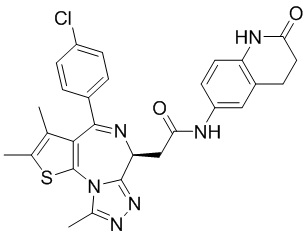 ,
,
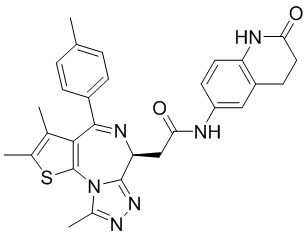 ,
,
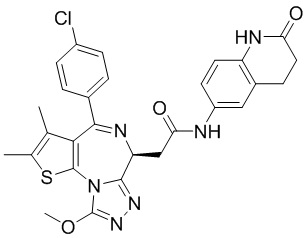 , и
, и
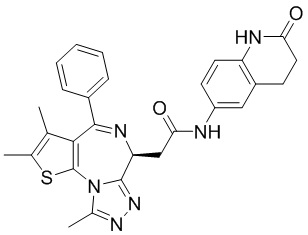 .
.
2. Соединение по п. 1, его стереоизомер или фармацевтически приемлемая соль, где R выбран из группы, состоящей из NH2 и C1-3 алкила.
3. Соединение по п. 2, его стереоизомер или фармацевтически приемлемая соль, где R выбран из группы, состоящей из NH2 и Ме.
4. Соединение по любому из пп. 1-3, его стереоизомер или фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из Me и Et.
5. Соединение по любому из пп. 1-3, его стереоизомер или фармацевтически приемлемая соль, где R2, R3 и R4 отдельно и независимо выбраны из группы, состоящей из H, F, Cl, Br, и C1-3алкила.
6. Соединение по п. 5, его стереоизомер или фармацевтически приемлемая соль, где R2, R3 и R4 отдельно и независимо выбраны из группы, состоящей из H, F, Cl, Br и Me.
7. Соединение по любому из пп. 1-3, его стереоизомер или фармацевтически приемлемая соль, где R6 отдельно и независимо выбран из группы, состоящей из H, F, Cl, Br, OH, NH2 C1-3алкила и C1-3гетероалкила, который необязательно замещен 1 R группой.
8. Соединение по п. 7, его стереоизомер или фармацевтически приемлемая соль, где R6 отдельно и независимо выбран из группы, состоящей из H, F, Cl, Br, OH, NH2 Me, Et, 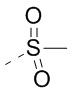 и
и  , который необязательно замещен 1 R группой.
, который необязательно замещен 1 R группой.
9. Соединение по п. 8, его стереоизомер или фармацевтически приемлемая соль, где R6 отдельно и независимо выбран из группы, состоящей из H, F, Cl, Br, OH, NH2, Me,  ,
, 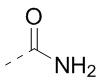 и .
и .
10. Соединение по любому из пп. 1-3, его стереоизомер или фармацевтически приемлемая соль, где кольцо A выбрано из группы, состоящей из C4-6циклоалкила, пирролидин-2-онила, пиримидин-4(3H)-онила, 5-азаспиро[2.4]гептан-4-онила, 4-азаспиро[2.4]гептан-5-онила, тетрагидротиофен-1,1-диоксидной группы, тетрагидротиофен-1-оксидной группы, тетрагидрофуранила, пирролидинила, дигидротиофен-2(3H)-онила, 2-оксаспиро[3.4]октила, дигидрофуран-2(3H)-онила, 1,4,7,10-тетраоксациклододецила, 1,2,5-оксадиазолила, 7-оксабицикло-[2.2.1]гептана, пирролидин-2,5-диона и 5,5-диметилдигидрофуран-2(3H)-онила.
11. Соединение по любому из пп. 1-3, его стереоизомер или фармацевтически приемлемая соль, где структурная единица 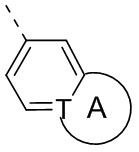 выбрана из группы, состоящей из
выбрана из группы, состоящей из  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 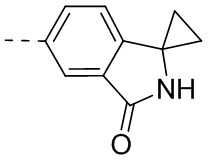 ,
, 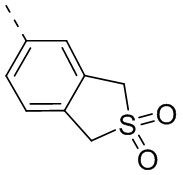 ,
, 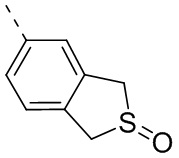 ,
, 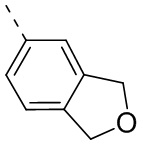 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и 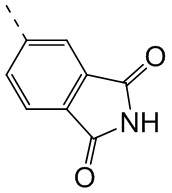 .
.
12. Соединение по п. 11, его стереоизомер или фармацевтически приемлемая соль, где структурная единица 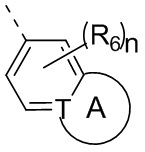 выбрана из группы, состоящей из
выбрана из группы, состоящей из 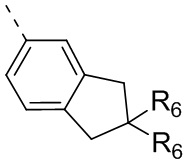 ,
,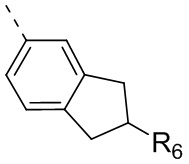 ,,,
,,,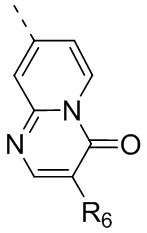 ,,,,,,,
,,,,,,,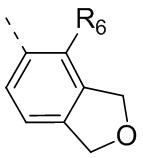 ,
,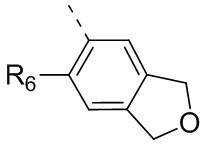 ,,
,,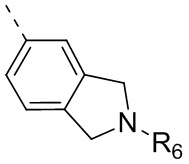 ,,,,,,
,,,,,,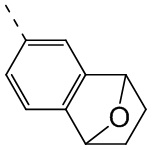 ,
,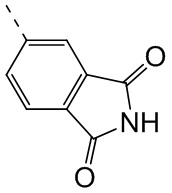 и
и 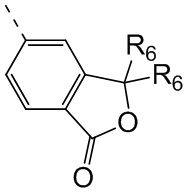 .
.
13. Соединение по п. 9 или 12, его стереоизомер или фармацевтически приемлемая соль, где структурная единица выбрана из группы, состоящей из 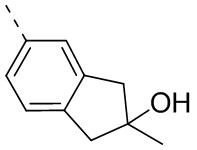 ,
,  ,
,  , , ,
, , ,  , , , , , , ,
, , , , , , ,  ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  , , , , , , , , и
, , , , , , , , и  .
.
14. Соединение по любому из пп. 1-3, его стереоизомер или фармацевтически приемлемая соль, где структурная единица 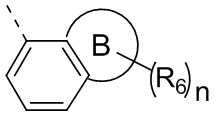 выбрана из группы, состоящей из
выбрана из группы, состоящей из  и
и  .
.
15. Соединение по любому из пп. 1-11, его стереоизомер или фармацевтически приемлемая соль, которые выбраны из группы, состоящей из
 ,
, 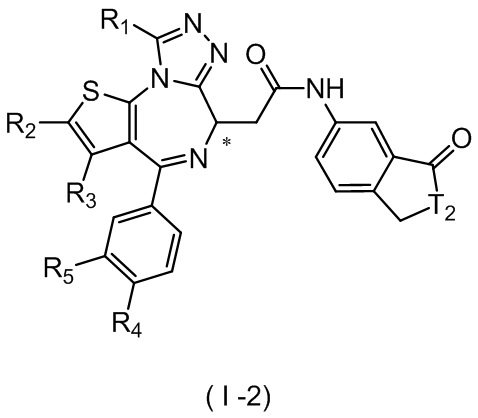 ,
,
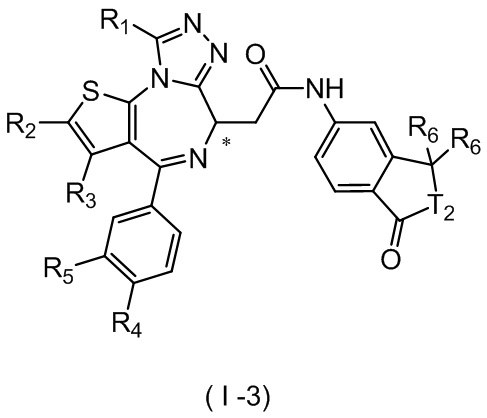 ,
, 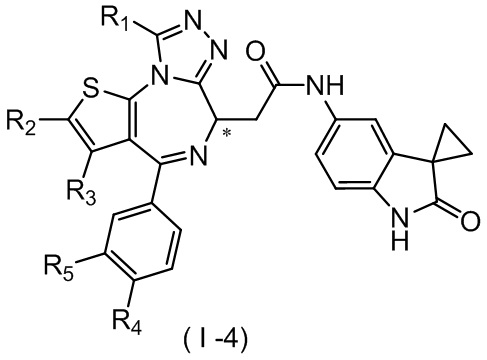 ,
,
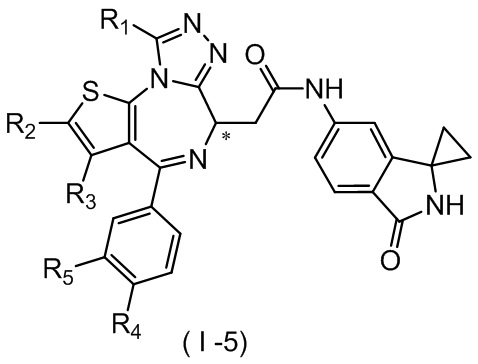 ,
, 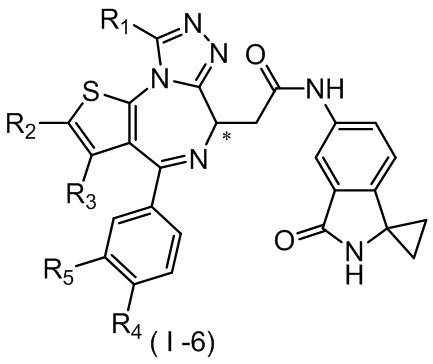 ,
,
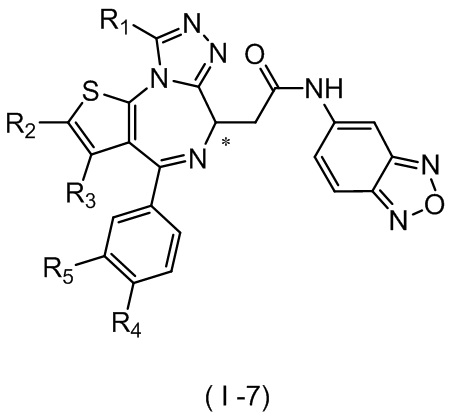 ,
, 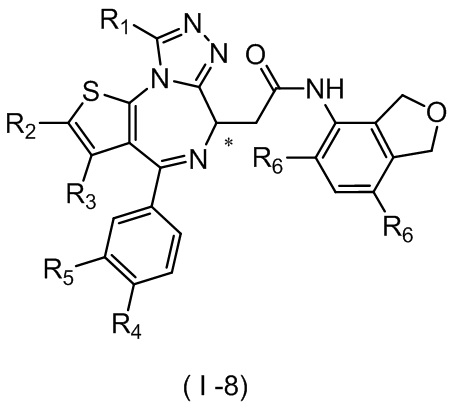 ,
,
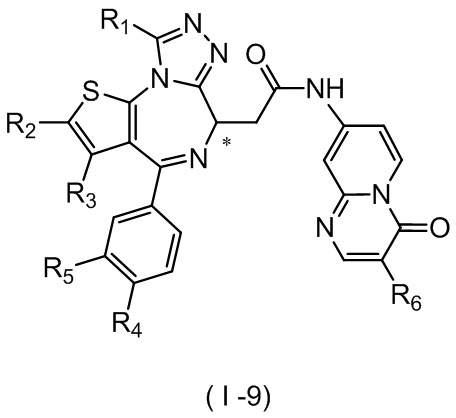 ,
, 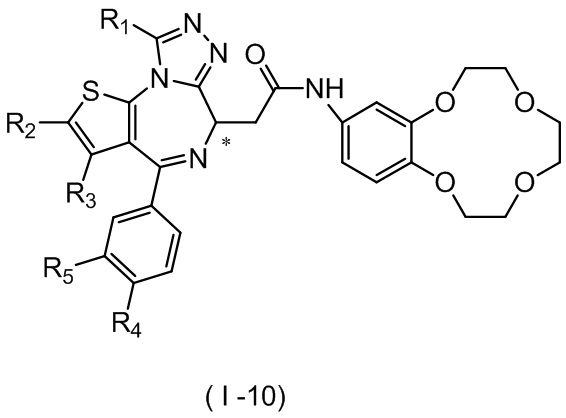 ,
,
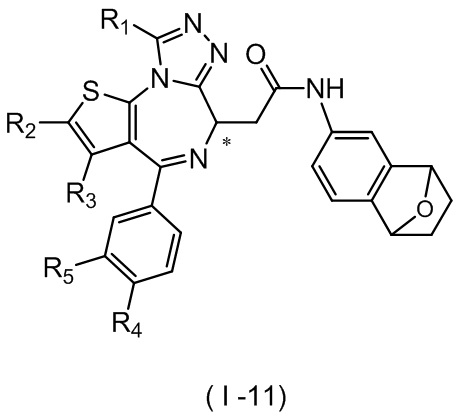 ,
,
где
T1 выбран из группы, состоящей из -S(=O)-, -S(=O)2-, -N(R6)-, -O-, -C(R6)(R6)- и  ;
;
T2 отдельно и независимо выбран из группы, состоящей из -NH-, -O- и -S-;
R1-R6 определены в любом из пп. 1-9;
атом углерода, помеченный «*», представляет собой хиральный атом углерода, который присутствует в форме простого (R) или (S) энантиомера или в форме, обогащенной одним из двух энантиомеров.
16. Соединение, его стереоизомер или фармацевтически приемлемая соль, которые выбраны из группы, состоящей из
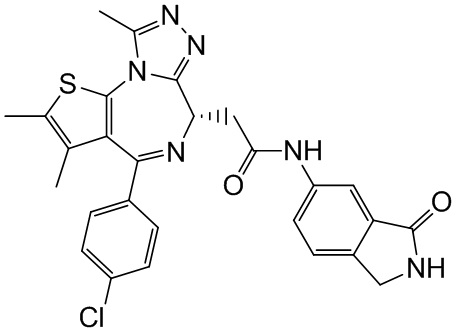
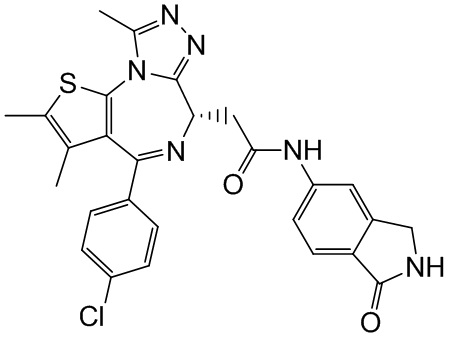
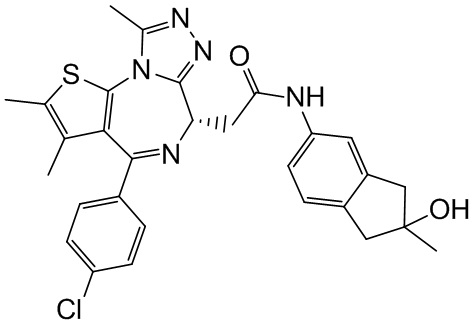
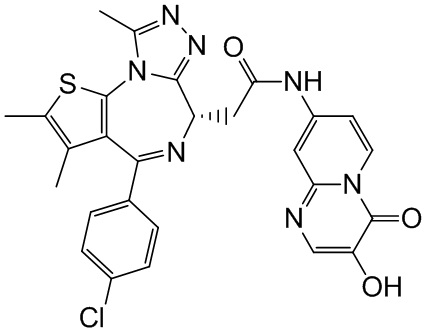
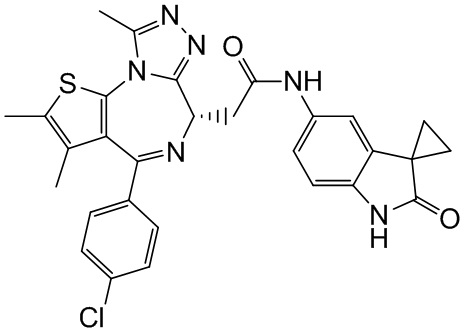
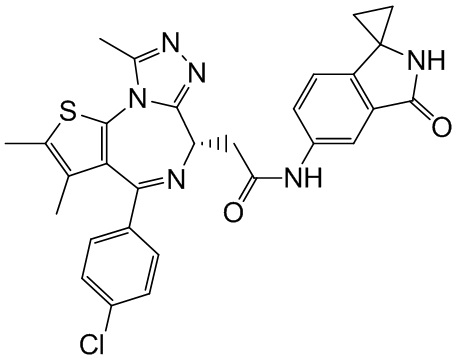
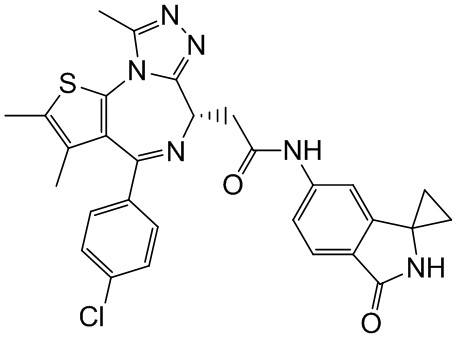
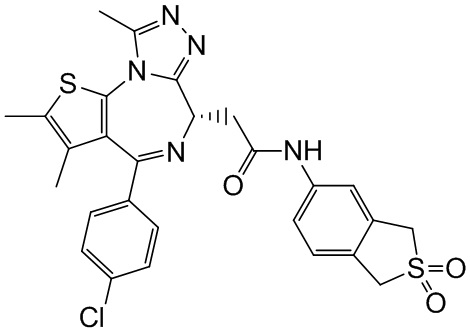
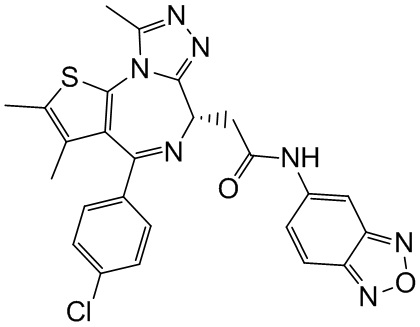
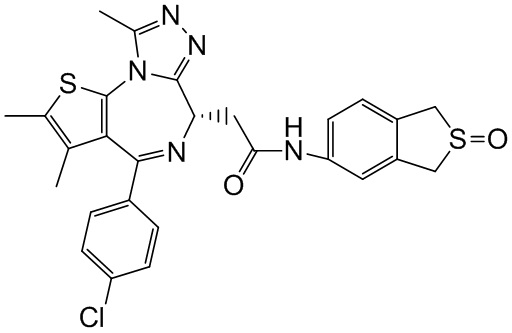
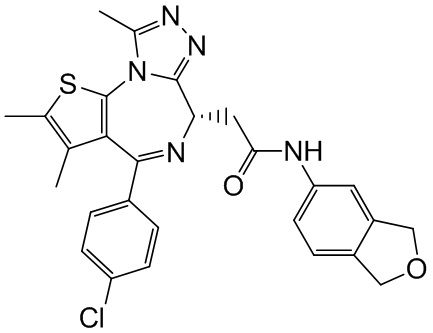
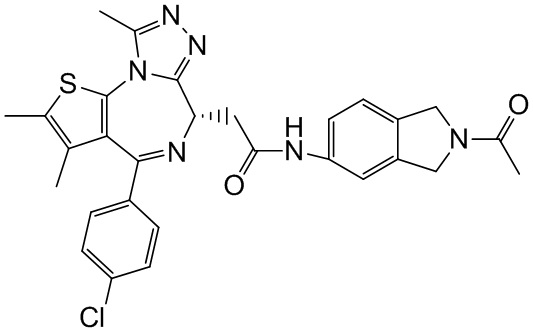
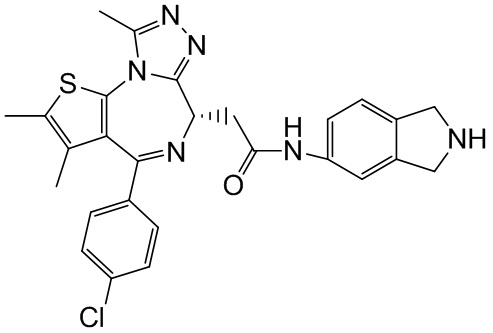
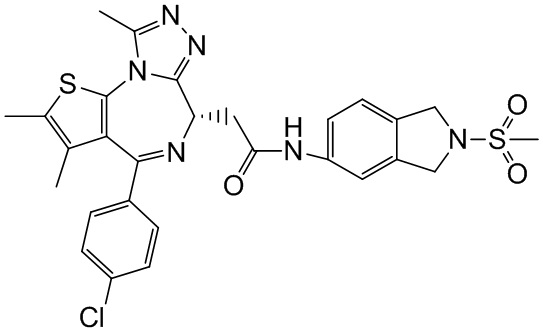
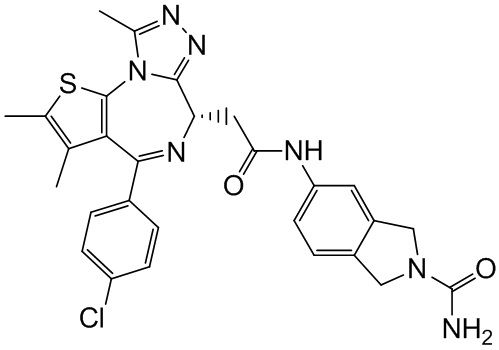
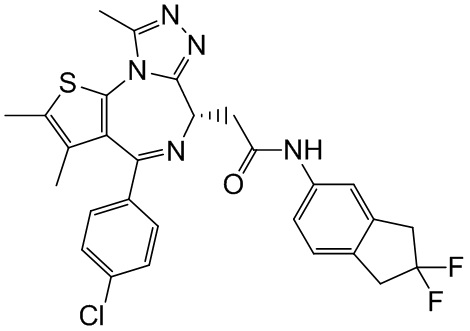
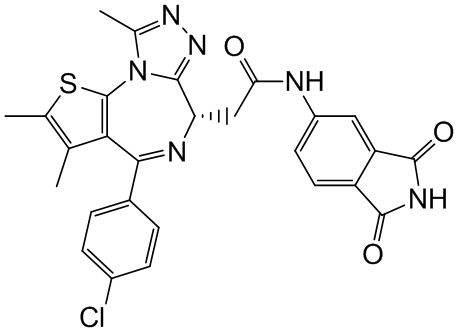 и
и  .
.
17. Соединение по п. 16, его стереоизомер или фармацевтически приемлемая соль, которые выбраны из группы, состоящей из
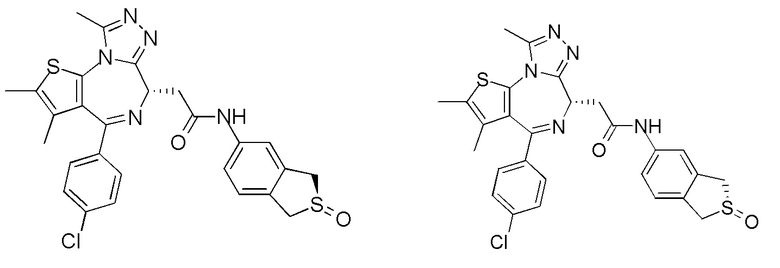
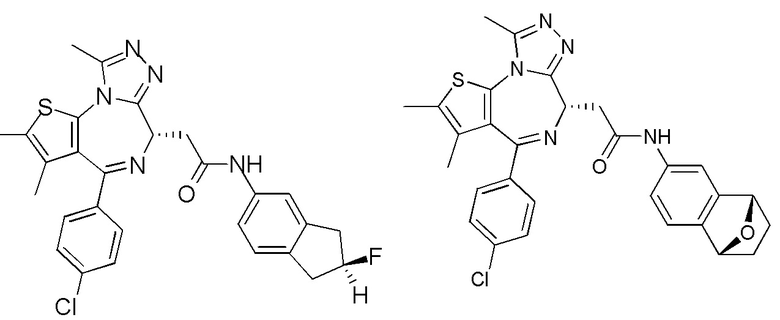
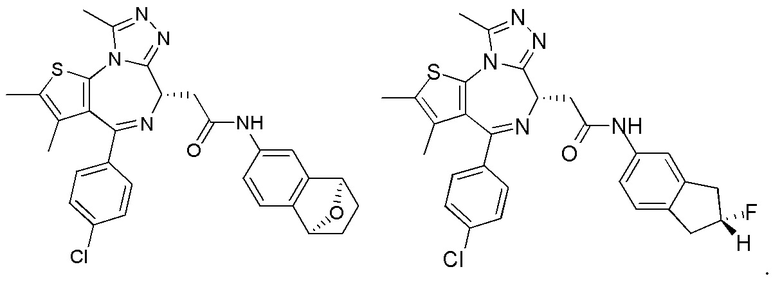
18. Фармацевтическая композиция, обладающая активностью ингибирования BRD4, содержащая терапевтически эффективное количество соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли в качестве активного ингредиента и фармацевтически приемлемый носитель.
19. Применение соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли или композиции по п. 18 при изготовлении лекарственного средства для лечения или предотвращения заболевания, связанного с BRD4.
20. Применение по пункту 19, где заболевание представляет собой опухоль, предпочтительно солидную опухоль, более предпочтительно рак груди, опухоль желудочно-кишечного тракта или рак предстательной железы, наиболее предпочтительно трижды негативный рак молочной железы, рак толстой кишки, рак прямой кишки или колоректальный рак.
| US 5712274 A1, 27.01.1998 | |||
| WO 2017044849 A8, 28.09.2017 | |||
| US 9125915 B2, 08.09.2015 | |||
| WO 2013030150 A1, 07.03.2013 | |||
| WO 2017044792 A1, 16.03.2017 | |||
| КОНЪЮГАТЫ И МАЛЫЕ МОЛЕКУЛЫ, ВЗАИМОДЕЙСТВУЮЩИЕ С РЕЦЕПТОРОМ CD16а | 2013 |
|
RU2519546C1 |

