ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым производным резорцина, в частности, к соединению, представленному формулой (I), и относится к способам их получения, к фармацевтическим композициям и к их применению в получении противоопухолевого лекарственного средства и для лечения нейродегенеративных заболеваний.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
В настоящее время разновидности целевой терапии для видов лечения рака основаны на определении конкретного белка, способствующего прогрессированию опухоли, и определении конкретного средства, которое способно противодействовать влиянию вышеуказанных белков. В фармацевтической промышленности усилия в основном сконцентрированы на очень ограниченном числе хорошо исследованных белков-мишеней. Общий недостаток заключается в том, что часто наблюдают случаи мутаций, связанных с устойчивостью к лекарственному средству, у пациентов с раком, которых лечили с помощью данных конкретных ингибиторов. С недавних пор общее мнение заключается в том, что одновременное блокирование сигнальных путей, вовлеченных в прогрессирование рака, как ожидается, может способствовать лучшему противоопухолевому эффекту и снижать вероятность развития устойчивости к лекарственному средству. HSP90 принадлежит к небольшому семейству белков, которые, как правило, содержат очень специфичный связывающий аденозинтрифосфат C-концевой домен («складка Бержера») (GHKL, название, происходящее от ДНК-гиразы, HSP90, гистидинкиназы, mutL). HSP90 является одним из наиболее распространенных белков в клетках и является незаменимым для жизнеспособности эукариотных организмов. Клетки человека содержат четыре изоформы HSP90: цитозольную конститутивно экспрессируемую β-изоформу, индуцибельную α-форму, GRP94/gp96 в эндоплазматическом ретикулуме и TRAP1/HSP75 в митохондриях. α-форма и β-форма демонстрируют 85% гомологию последовательностей.
HSP90 является ключевым компонентом структуры, выполняющей функции шаперона, которая катализирует укладку белков, называемых клиентами HSP90, и контролирует их качество в нормальных клетках и в условиях стресса. Активность молекулярного шаперона, которая сильно зависит от активности аденозинтрифосфатазы, точно модулируется связыванием других регуляторных кошаперонов.
Существует убедительное доказательство, что в случае, например, рака или других пролиферативных заболеваний присутствие HSP90 становится критичным, вследствие мутаций или сверхэкспрессии конкретных онкогенов или также вследствие того, что опухоли зачастую содержат накопленные, неправильно свернутые белки (что приводит в результате к повышенной потребности в функции молекулярного шаперона).
HSP90 представляет собой гомодимер, состоящий из трех основных доменов в структуре: высококонсервативного N-концевого домена, промежуточного домена, необходимого для аденозинтрифосфатазной активности, и C-концевого домена. N-концевой и C-концевой домены могут связываться с аденозинтрифосфатом. Большинство известных ингибиторов, таких как гелданамицин, радицикол, производные диарилпиразола и пурина, проявляет конкурентное связывание c N-концевым сайтом связывания аденозинтрифосфата с аденозинтрифосфатом, тогда как новобиоцин представляет собой прототип ингибитора, который связывается с C-концевым карманом.
В настоящее время увеличивается количество сообщений о белках-клиентах HSP90 (Jolly et al., J.Natl.Cancer Inst.92; 1564-1572(2000)), принадлежащих к семейству киназ (Her2, B-RAF V600E, bcr-Abl, Flt3, NPM-ALK, Akt, Npm-Alk, ZAP-70), факторов транскрипции (p53, HIF), теломераз и другим молекулярным шаперонам, большинство из которых тесно связаны с развитием рака. Способность HSP90 ингибировать поврежденную укладку или стабилизировать его белки-клиенты приводит к деградации данных несвернутых белков с помощью протеаз. Деградацию данных белков-клиентов зачастую используют как признак ингибирования HSP90, и типичное применение заключается в том, что в сверхэксперссирующих Her2 клетках, таких как клетки рака молочной железы BT474, Her2 деградирует после обработки соединениями.
Было показано, что природное соединение гелданамицин действительно может блокировать пролиферацию многих опухолевых клеток с помощью способности конкурентного связывания с расположенным в N-концевом домене сайтом связывания аденозинтрифосфата и ингибирования аденозинтрифосфатазной активности HSP90, что изначально вызвало значительное количество исследований относительно HSP90. Неожиданно, данное соединение является неактивным в нормальных клетках, и это может быть вызвано тем, что HSP90 присутствует в виде активного комплекса (с высокой аффинностью к гелданамицину) только в опухолевых клетках (Kamal et al., Nature 425, 407-410 (2003)). Другая возможная причина для селективной чувствительности к опухолям заключается в задержании в опухоли, проявляемой многими ингибиторами HSP90.
Большое количество клинических исследований танеспимицина (17-AAG), полусинтетического производного гелданамицина (GDA), и других родственных производных (алвеспимицина, 17-DMAG, IPI-504) продолжается в данное время, но их эффекты, по-видимому, ограничены рядом факторов: сложным получением, зависимостью от метаболизма для получения активных метаболитов, отсутствием накопления у пациентов и гепатотоксичностью, возможно связанной с хиноновым фрагментом. Это приводит к большому числу усилий для определения ингибиторов HSP90 второго поколения с лучшими подобными лекарственному средству характеристиками и с лучшей переносимостью. Это приводит в результате к определению производных пурина и производных арилрезорцина.
Основной причиной нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона и прионная болезнь, является накопление неправильно свернутых белков, что приводит к образованию бляшек. Эти неправильно свернутые белки зависят от молекулярных шаперонов (HSP70, HSP40 и т. д.) относительно повторного созревания, деполимеризации и повторной солюбилизации белкового агрегата. Было показано, что белки теплового шока обеспечивают данную функцию в различных моделях клеточных культур. HSF1 может индуцировать HSP, и HSF1 точно регулируется с помощью HSP90 в нормальных клетках. Было показано, что ингибиторы HSP90, такие как гелданамицин и производные 17-AAG, могут нарушать данное взаимодействие и приводить к индукции HSP, что вызывает в результате нейропротекторную активность, а также повторную солюбилизацию и деполимеризацию неправильно свернутых белков. Сверхэкспрессия HSP90 может значительно снизить накопление неправильно свернутых белков, а накопление неправильно свернутых белков является причиной болезни Альцгеймера. По сути, было показано, что существует обратная корреляция между уровнями агрегированного тау-белка и HSP70/90. Чрезмерное агрегирование тау-белка можно снизить посредством сверхэкспрессии HSP70, HSP27 и HSP40 (путем деградации), что вызвано ингибированием HSP90. На основе in vivo эффекта GDA на нейротоксичность, вызванную 1-метил-4-фенил-1,2,3,6-тетрагидропиридином (MPTP), в мышиных моделях болезни Паркинсона для лечения болезни Паркинсона применяли ингибиторы HSP90. GDA защищает нейроны от MPTP-индуцированной токсичности, что тесно связано с повышенным уровнями HSP70. Кроме того, было показано, что сверхэкспрессия HSP90 может значительно снижать накопление неправильно свернутых белков, что является причиной повреждения двигательной системы, рассеянного склероза, спинальной и бульбарной мышечной атрофии и других заболеваний.
В GB 1406345 раскрыто соединение 4,6-дизамещенного резорцина, обладающее фармакологической активностью. В других заявках на патент описаны фенил-гетероциклические соединения в качестве ингибиторов HSP90, все из которых характеризуются наличием конкретного вида замещения пятичленных гетероциклов, например, в WO 2006/101052 от Nippon Kayaku Kabushiki Kaisha; WO 2005/000300, WO 2004/072051 и WO 2004/056782 от Vernalis; WO 2003/055860 от Ribotargets; WO 2008/097640 от Synta Pharmaceuticals и WO 2005/063222 от Kyowa Hakko Kogyo.
WO 2004072051 относится к классу ингибиторов HSP90, в том числе к луминеспибу:
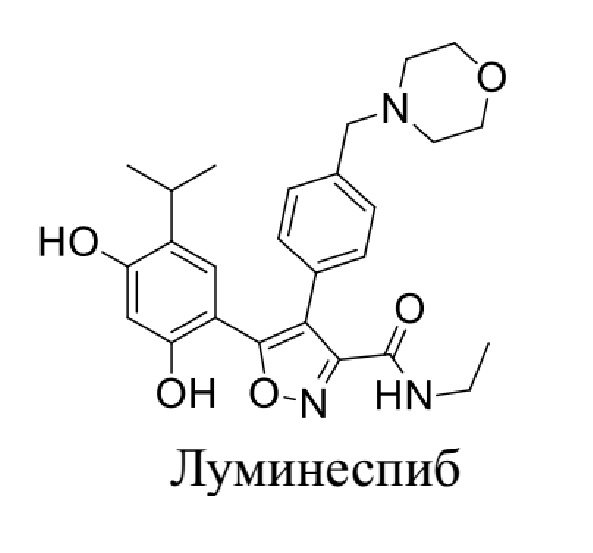 .
.
В WO 2006055760A1 сообщается от некоторых соединениях, таких как 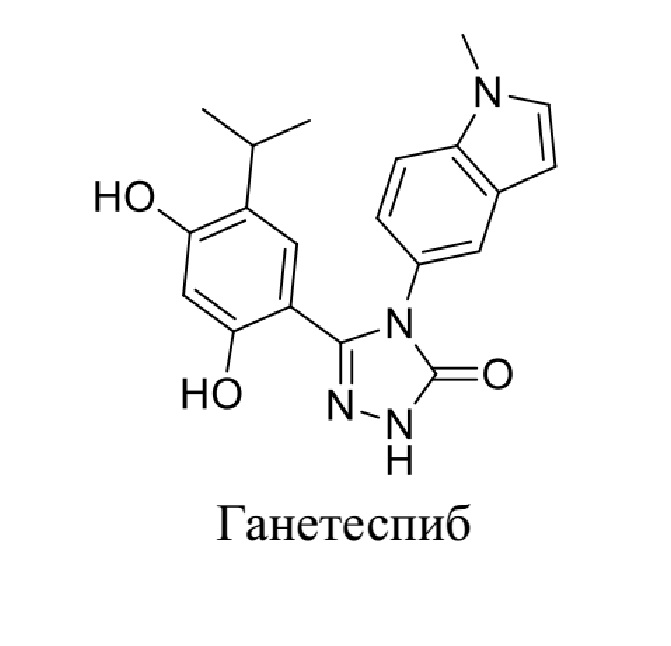 .
.
В CN 1771235A раскрыты некоторые соединения, такие как 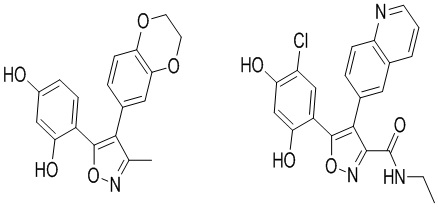 .
.
Данные соединения не являются желательными в отношении эффективности, фармакокинетики, водорастворимости, фармацевтических свойств и т. д.
Несмотря на вышеуказанные разработки, все еще существует потребность в разработке более эффективных ингибиторов HSP90 с небольшими побочными эффектами.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения заключается в обеспечении соединения, представленного формулой (I), или его фармацевтически приемлемых соли или гидрата,
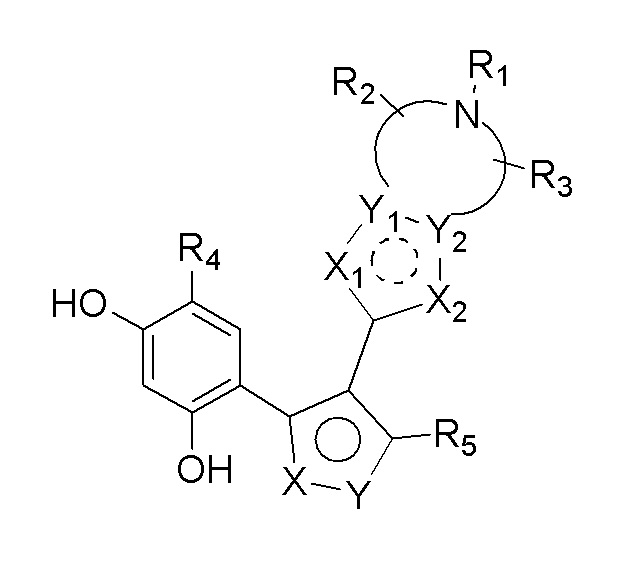
(I),
где
каждый из X и Y независимо выбран из N, O или S; предпочтительно X выбран из N, O или S, и Y выбран из N или O;
X1, X2, Y1, Y2 и атом углерода, связывающий X1 и X2, вместе образуют 5-7-членное ароматическое кольцо, алифатическое насыщенное кольцо или алифатическое ненасыщенное кольцо; предпочтительно X1, X2, Y1, Y2 и атом углерода, связывающий X1 и X2, вместе образуют 5-6-членное ароматическое кольцо, алифатическое насыщенное кольцо или алифатическое ненасыщенное кольцо;
каждый из X1 и X2 независимо выбран из C, O, S, N, -C=C-, -C=N-; и C в X1 или X2 может быть незамещенным или может быть замещен R01 или R02.
Каждый из R01 и R02 независимо выбран из галогена, CN, OH, SH, NH2, CHO, COOH, C1-10алкила, N-C1-10алкиламино, N,N-ди(C1-10алкил)амино, C1-10алкокси, C1-10алканоила, C1-10алкоксикарбонила, C1-10алкилсульфонила, C1-10алкилсульфинила, C3-10циклоалкила, C3-10циклоалкиламино, C3-10циклоалкокси, C3-10циклоалкилацила, C3-10циклоалкоксикарбонила, C3-10циклоалкилсульфонила, C3-10циклоалкилсульфинила или C1-10алкила, замещенного C3-10циклоалкилом;
каждый из Y1 и Y2 независимо выбран из C или N, и как Y1, так и Y2 предпочтительно представляют собой C; два заместителя при Y1 и Y2 связаны вместе с образованием пяти-, шести- или семичленного содержащего азот насыщенного гетерокольца или ароматического гетерокольца, содержащего заместители R1, R2 и R3;
каждый из R1, R2 и R3 независимо выбран из водорода, C1-10алкила, гидрокси-C1-10алкила, C1-6алкокси-C1-6алкила, галоген-C1-10алкила, C1-6алкилсульфонил-C1-6алкила, C1-6алкиламидо-C1-6алкила, N,N-ди(C1-6алкил)аминоацил-C1-6алкила, N,N-ди(C1-6алкил)амино-C1-6алканоила, морфолинил-C1-6алканоила, N-C1-10алкиламино, N,N-ди(C1-10алкил)амино, C1-10алкокси, C1-10алканоила, C1-10алкоксикарбонила, C1-10алкилсульфонила, C1-10алкилсульфинила, C3-10циклоалкила, C3-10циклоалкиламино, C3-10циклоалкилокси, C3-10циклоалкилацила, C3-10циклоалкоксикарбонила, C3-10циклоалкилсульфонила, C3-10циклоалкилсульфинила или C3-10циклоалкил-C1-5алкила, или заместители R2 и R3 связаны друг с другом с помощью ковалентной связи с образованием пяти-, шести- или семичленного насыщенного кольца с заместителем R03 или без заместителей; предпочтительно каждый из R1, R2 и R3 независимо выбран из водорода, C1-10алкила, гидрокси-C1-10алкила, C3-10циклоалкила, C1-6алкокси-C1-6алкила, галоген-C1-10алкила, C1-6алкилсульфонил-C1-6алкила, C1-6алкиламидо-C1-6алкила, N,N-ди(C1-6алкил)аминоацил-C1-6алкила, N,N-ди(C1-6алкил)амино-C1-6алканоила, морфолинил-C1-6алканоила или C3-10циклоалкил-C1-5алкила, или заместители R2 и R3 связаны друг с другом с помощью ковалентной связи с образованием пяти-, шести- или семичленного насыщенного кольца с заместителем R03 или без заместителей; более предпочтительно каждый из R1, R2 и R3 независимо выбран из водорода, C1-6алкила, гидрокси-C1-6алкила, C3-10циклоалкила, C1-4алкокси-C1-4алкила, галоген-C1-4алкила, C1-4алкилсульфонил-C1-4алкила, C1-4алкиламидо-C1-4алкила, N,N-ди(C1-4алкил)аминоацил-C1-4алкила, N,N-ди(C1-4алкил)амино-C1-4алканоила, морфолинил-C1-4алканоила или C3-6циклоалкил-C1-4алкила, или заместители R2 и R3 связаны друг с другом с помощью ковалентной связи с образованием пяти-, шести- или семичленного насыщенного кольца с заместителем R03 или без заместителей; наиболее предпочтительно R1 выбран из водорода, C1-6алкила, гидрокси-C1-6алкила, C3-10циклоалкила, C1-4алкокси-C1-4алкила, галоген-C1-4алкила, C1-4алкилсульфонил-C1-4алкила, C1-4алкиламидо-C1-4алкила, N,N-ди(C1-4алкил)аминоацил-C1-4алкила, N,N-ди(C1-4алкил)амино-C1-4алканоила или морфолинил-C1-4алканоила, R2 и R3 выбраны из водорода или метила, или заместители R2 и R3 связаны друг с другом с помощью ковалентной связи с образованием пяти-, шести- или семичленного насыщенного кольца без заместителей;
R03 выбран из C1-6алкила или галогена;
R4 выбран из H, галогена, C1-6алкила, C3-10циклоалкокси, замещенного фенилом C1-6алкила, замещенного фенилом C2-6алкенила, фенила, замещенного C1-6алкилом фенила или C3-6циклоалкила; предпочтительно R4 выбран из H, C1-6алкила, замещенного фенилом C1-6алкила, галогена или C3-6циклоалкила; более предпочтительно R4 выбран из C1-4алкила, Cl, Br или циклопропила; наиболее предпочтительно R4 выбран из изопропила.
R5 выбран из H, циано, карбокси, C1-6алкоксиацила, C1-7алкиламинокарбонила, галоген-C1-6алкиламинокарбонила, C1-6алкокси-C1-6алкиламинокарбонила, N,N-ди(C1-6алкил)амино-C1-6алкиламинокарбонила, аминокарбонила, гидрокси-C1-6алкиламинокарбонила, замещенного гидроксильной группой галоген-C1-6алкила или замещенного нитрильной группой амидино или выбран из C3-10циклоалкила, C3-10циклоалкенила или 5-10-членного ароматического кольца, которое необязательно замещено одним или более R05; где R05 выбран из C1-6алкила или C3-10циклоалкила.
R5 предпочтительно выбран из циано, C1-6алкиламинокарбонила, галоген-C1-4алкиламинокарбонила, C1-4алкокси-C1-4алкиламинокарбонила, N,N-ди(C1-4алкил)амино-C1-4алкиламинокарбонила, аминокарбонила, гидрокси-C1-4алкиламинокарбонила, замещенного гидроксильной группой галоген-C1-4алкила или замещенного нитрильной группой амидино или выбран из 5-6-членного содержащего азот гетероароматического кольца, которое необязательно замещено одним или более R05; где R05 выбран из C1-6алкила.
В одном варианте осуществления настоящего изобретения вышеуказанный R5 выбран из 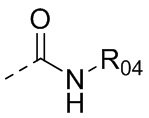 ,
, 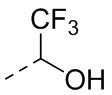 ,
, 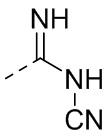 ,
, 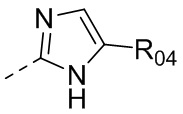 или
или 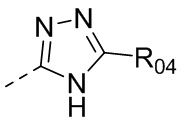 , где R04 выбран из H, C1-6алкила, галоген-C1-4алкила, C1-4алкокси-C1-4алкила, N,N-ди(C1-4алкил)амино-C1-4алкила или гидрокси-C1-4алкила.
, где R04 выбран из H, C1-6алкила, галоген-C1-4алкила, C1-4алкокси-C1-4алкила, N,N-ди(C1-4алкил)амино-C1-4алкила или гидрокси-C1-4алкила.
В одном варианте осуществления настоящего изобретения вышеуказанный 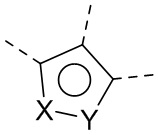 выбран из
выбран из  ,
,  или
или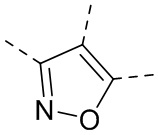 ; и другие переменные определены, как указано в формуле (I).
; и другие переменные определены, как указано в формуле (I).
В одном варианте осуществления настоящего изобретения вышеуказанный 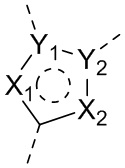 выбран из
выбран из 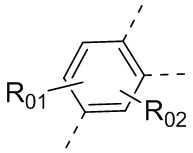 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  или
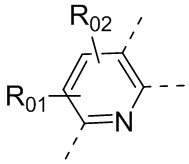
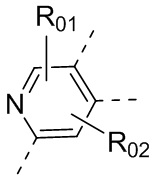
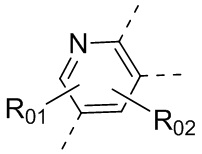
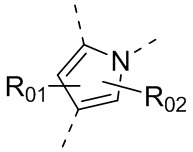
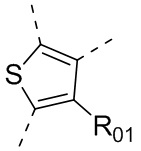
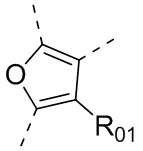
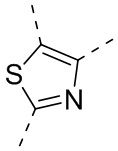
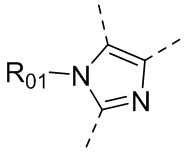
или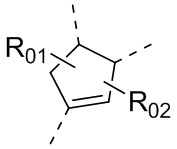 , где каждый из R01 и R02 независимо выбран из H, галогена, C1-10алкила, C3-10циклоалкила, C1-10алкила, замещенного C3-10циклоалкилом; и другие переменные определены, как указано в формуле (I).
, где каждый из R01 и R02 независимо выбран из H, галогена, C1-10алкила, C3-10циклоалкила, C1-10алкила, замещенного C3-10циклоалкилом; и другие переменные определены, как указано в формуле (I).
В одном варианте осуществления настоящего изобретения вышеуказанный выбран из 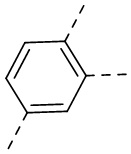 ,
, 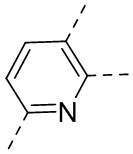 ,
, 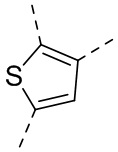 ,
, 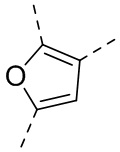 , ,
, , 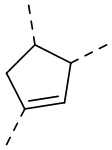 ,
, 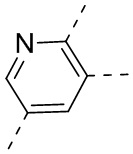 ,
, 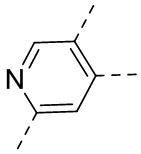 ,
, 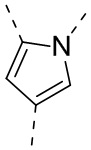 или
или 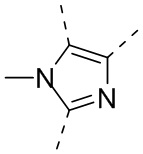 , и другие переменные определены, как указано в формуле (I).
, и другие переменные определены, как указано в формуле (I).
В одном варианте осуществления настоящего изобретения вышеуказанный 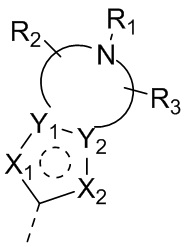 выбран из
выбран из 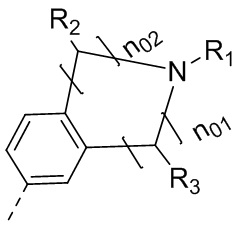 , где n01 и n02 выбраны из 0, 1, 2 или 3, и сумма n01 и n02 равняется 2, 3 или 4; и другие переменные определены, как указано в формуле (I), и R2 и R3 не связаны с образованием кольца.
, где n01 и n02 выбраны из 0, 1, 2 или 3, и сумма n01 и n02 равняется 2, 3 или 4; и другие переменные определены, как указано в формуле (I), и R2 и R3 не связаны с образованием кольца.
В одном варианте осуществления настоящего изобретения вышеуказанный выбран из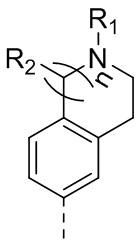 , где n равняется 1 или 2; и где другие переменные определены, как указано в формуле (I).
, где n равняется 1 или 2; и где другие переменные определены, как указано в формуле (I).
В одном варианте осуществления настоящего изобретения вышеуказанный выбран из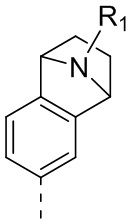 , и другие переменные определены, как указано в формуле (I).
, и другие переменные определены, как указано в формуле (I).
В одном варианте осуществления настоящего изобретения вышеуказанный выбран из 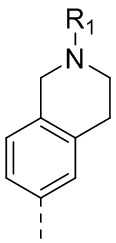 ,
,  ,
,  ,
, 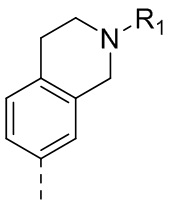 ,
, 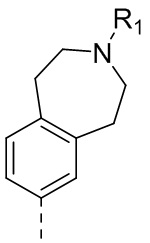 ,
, 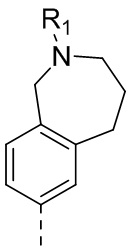 ,
, 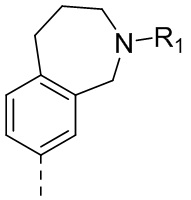 или
или 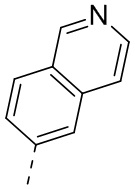 , и другие переменные определены, как указано в формуле (I).
, и другие переменные определены, как указано в формуле (I).
В одном варианте осуществления настоящего изобретения вышеуказанный выбран из 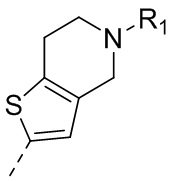 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  или
или  , и другие переменные определены, как указано в формуле (I).
, и другие переменные определены, как указано в формуле (I).
В одном варианте осуществления настоящего изобретения вышеуказанное соединение или его фармацевтически приемлемая соль или гидрат выбраны из соединения или его фармацевтически приемлемых соли или гидрата, характеризующихся следующими структурными формулами:
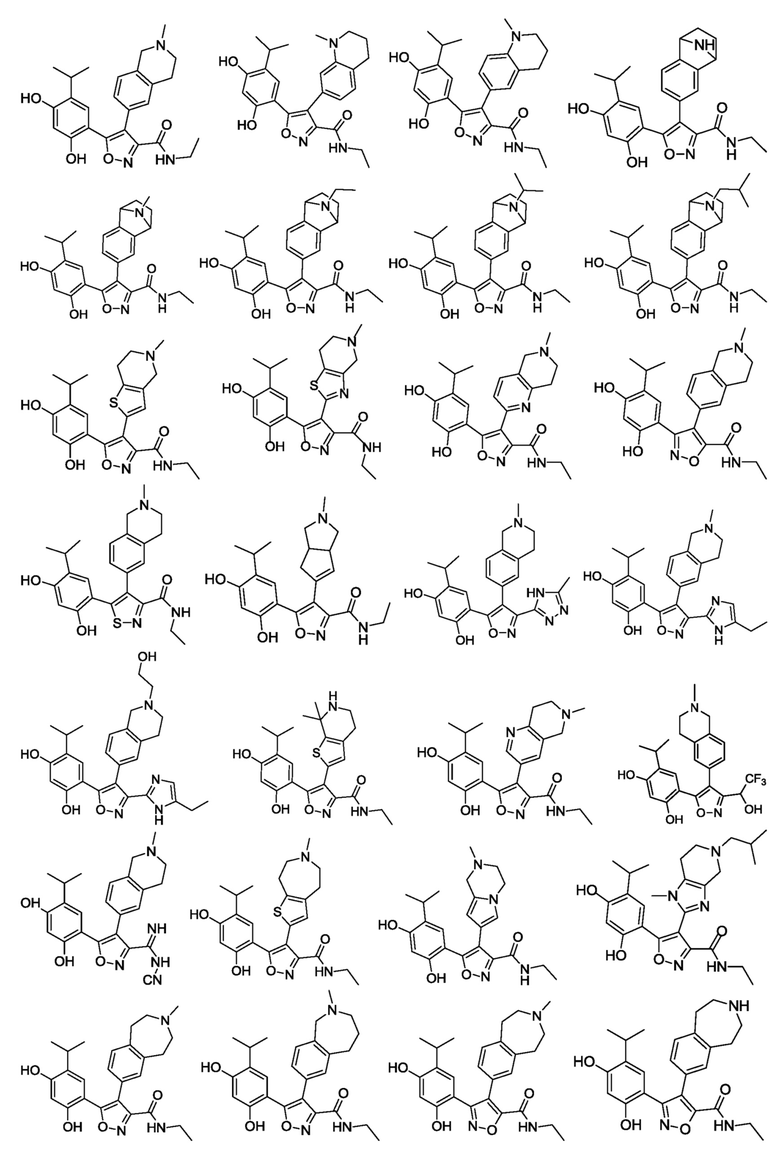
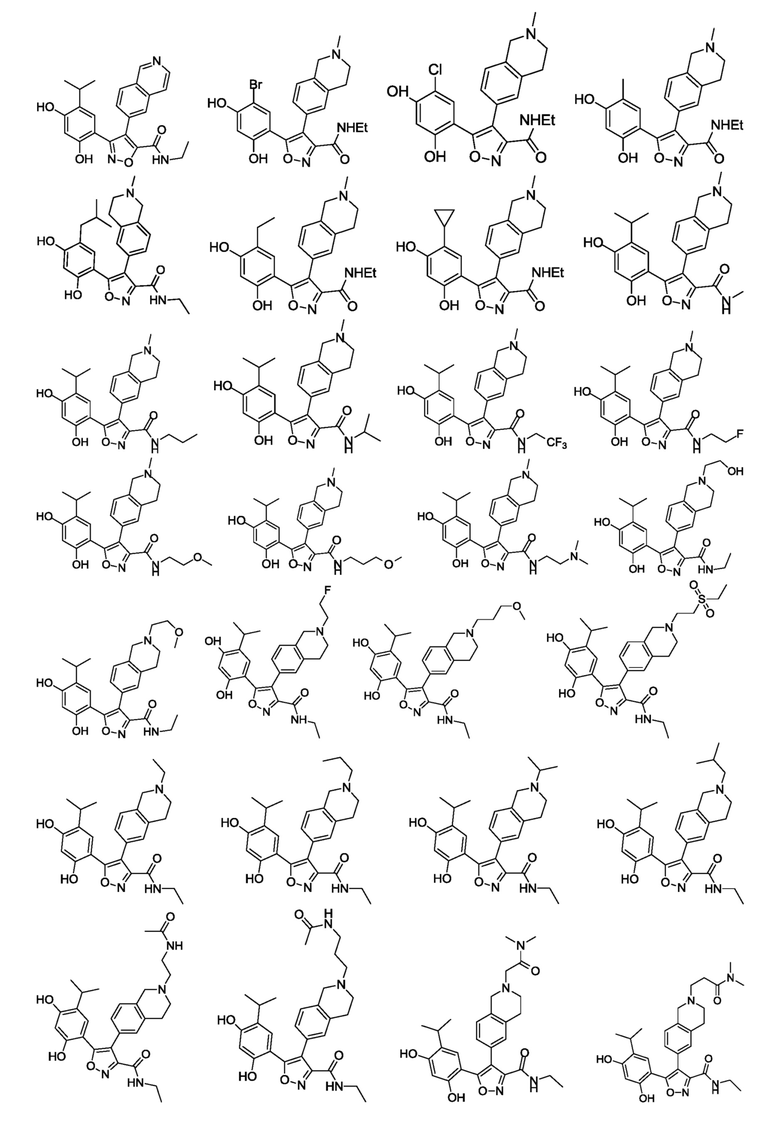
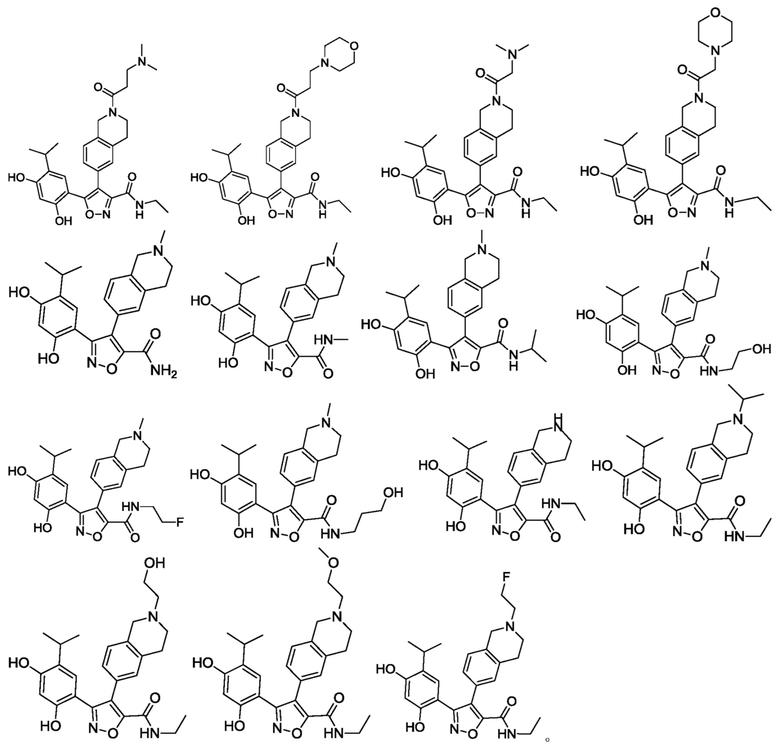
В одном варианте осуществления настоящего изобретения вышеуказанное соединение или его фармацевтически приемлемая соль выбраны из:
1) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
2) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(1-метил-1,2,3,4-тетрагидрохинолин-7-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
3) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
4) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
5) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(9-метил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
6) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(9-этил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
7) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(9-изопропил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
8) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(9-изобутил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
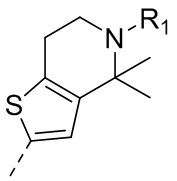
9) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
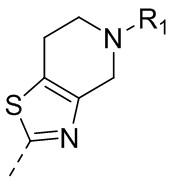
10) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(5-метил-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-2-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
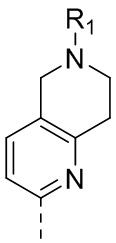
11) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
12) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
13) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изотиазол-3-карбоксамида или его фармацевтически приемлемой соли;
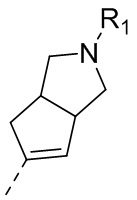
14) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,3a,4,6a-гексагидроциклопента[c]пиррол-5-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
15) 4-изопропил-6-[4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(5-метил-4H-1,2,4-триазол-3-ил)изоксазол-5-ил]бензол-1,3-диола или его фармацевтически приемлемой соли;
16) 4-(3-(5-этил-1H-имидазол-2-ил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-ил)-6-изопропилбензол-1,3-диола или его фармацевтически приемлемой соли;
17) 4-(3-(5-этил-1H-имидазол-2-ил)-4-(2-(2-гидроксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-ил)-6-изопропилбензол-1,3-диола или его фармацевтически приемлемой соли;
18) 5-(2,4-дигидрокси-5-изопропилфенил)-4-(7,7-диметил-4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-N-этилизоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
19) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
20) 4-изопропил-6-(4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(2,2,2-трифтор-1-гидроксиэтил)изоксазол-5-ил)бензол-1,3-диола или его фармацевтически приемлемой соли;
21) N-циано-5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамидина или его фармацевтически приемлемой соли;
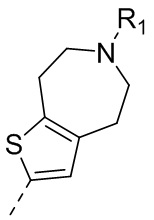
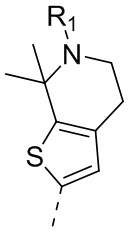
22) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
23) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
24) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(5-изобутил-1-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-2-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
25) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
26) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[c]азепин-7-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
27) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
28) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
29) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(изохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
30) 5-(2,4-дигидрокси-5-бромфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
31) 5-(2,4-дигидрокси-5-хлор-фенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
32) 5-(2,4-дигидрокси-5-метилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
33) 5-(5-изобутил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
34) 5-(5-этил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
35) 5-(5-циклопропил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
36) 5-(2,4-дигидрокси-5-изопропилфенил)-N-метил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
37) 5-(2,4-дигидрокси-5-изопропилфенил)-N-пропил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
38) 5-(2,4-дигидрокси-5-изопропилфенил)-N-изопропил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
39) 5-(2,4-дигидрокси-5-изопропилфенил)-N-(2,2,2-трифторэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
40) 5-(2,4-дигидрокси-5-изопропилфенил)-N-(2-фторэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
41) 5-(2,4-дигидрокси-5-изопропилфенил)-N-(2-метоксиэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
42) 5-(2,4-дигидрокси-5-изопропилфенил)-N-(3-метоксипропил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
43) 5-(2,4-дигидрокси-5-изопропилфенил)-N-[2-(диметиламино)этил]-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
44) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-гидроксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
45) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-метоксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
46) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-фторэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
47) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(3-метоксипропил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
48) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-этилсульфонил)этил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
49) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
50) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-пропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
51) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-изопропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
52) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-изобутил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
53) 4-(2-(2-ацетиламиноэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)-5-(2,4-дигидрокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
54) 4-(2-(2-ацетиламинопропил)-1,2,3,4-тетрагидроизохинолин-6-ил)-5-(2,4-дигидрокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
55) 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-2-оксоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
56) 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-3-оксопропил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
57) 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-(3-(диметиламино)пропионил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
58) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(3-морфолино-4-ил-пропионил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
59) 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)ацетил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
60) 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-морфолино-4-ил-ацетил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида или его фармацевтически приемлемой соли;
61) 3-(2,4-дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
62) 3-(2,4-дигидрокси-5-изопропилфенил)-N-метил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
63) 3-(2,4-дигидрокси-5-изопропилфенил)-N-изопропил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
64) 3-(2,4-дигидрокси-5-изопропилфенил)-N-(2-гидроксиэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
65) 3-(2,4-дигидрокси-5-изопропилфенил)-N-(2-фторэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
66) 3-(2,4-дигидрокси-5-изопропилфенил)-N-(3-гидроксипропил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
67) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
68) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-изопропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
69) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
70) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-метоксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли;
71) 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-фторэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида или его фармацевтически приемлемой соли.
Другая цель настоящего изобретения заключается в обеспечении фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемых соли или гидрата и фармацевтически приемлемые носители.
Другая цель настоящего изобретения заключается в обеспечении применения вышеуказанного соединения в получении лекарственного препарата для лечения опосредованных белком HSP90 заболеваний.
Опосредованные белком HSP90 заболевания согласно настоящему изобретению предпочтительно выбраны из рака и нейродегенеративных расстройств.
Другая цель настоящего изобретения заключается в обеспечении применения вышеуказанного соединения в получении лекарственного препарата для лечения конкретных типов карциномы, которые включают без ограничения рак, такой как рак мочевого пузыря, рак молочной железы, рак толстой кишки, рак почки, рак печени, рак легкого, включая мелкоклеточный рак легкого, рак пищевода, рак желчного пузыря, рак яичника, рак поджелудочной железы, рак желудка, рак шейки матки, рак щитовидной железы, рак предстательной железы и рак кожи, включая плоскоклеточный рак; гемобластоз лимфоидного происхождения, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, B-клеточную лимфому, T-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому и лимфому Беркитта; гемобластоз миелоидного происхождения, включая острый и хронический миелодный лейкоз, миелодиспластический синдром и промиелоцитарный лейкоз; рак мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; рак центральной и периферической нервной системы, включая астроцитомы, нейроцитомы, глиомы и невриномы; а также другие виды рака, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярную тиреоидную карциному и саркому Капоши.
Другая цель настоящего изобретения заключается в обеспечении применения вышеуказанного соединения в получении лекарственного препарата для лечения конкретных типов нейродегенеративных расстройств, при этом нейродегенеративные расстройства включают без ограничения болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, рассеянный склероз и спинальную и бульбарную мышечную атрофию.
Другая цель настоящего изобретения заключается в обеспечении применения соединения формулы (I) в получении лекарственного препарата для противораковой терапии, при этом лекарственный препарат применяют одновременно, отдельно или последовательно в схемах лечения с применением лучевой терапии или химиотерапии.
Кроме того, в настоящем изобретении предусмотрен способ ингибирования активности белка HSP90 in vitro, включающий приведение в контакт белка с эффективным количеством соединения формулы (I).
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая одно или более соединений формулы (I) или их фармацевтически приемлемых солей и фармацевтически приемлемые наполнители, носители или разбавители.
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) и следующие вещества: известные ингибиторы клеточного роста или цитотоксические лекарственные средства, антибиотические средства, алкилирующие средства, антиметаболиты, гормональные средства, иммунологические средства, средства на основе интерферона, ингибиторы циклооксигеназы (такие как ингибиторы COX-2), ингибиторы матриксной металлопротеиназы, ингибиторы теломеразы, ингибиторы тирозинкиназы, средства на основе рецептора фактора подавления роста, анти-HER средства, анти-EGFR средства, антиангиогенные средства (такие как ингибиторы ангиогенеза), ингибиторы фарнезилтрансферазы, ингибиторы пути передачи сигнала Ras-Raf, ингибиторы клеточного цикла, другие ингибиторы cdk, тубулин-связывающие средства, ингибиторы топоизомеразы I, ингибиторы топоизомеразы II или подобные.
Кроме того, в настоящем изобретении предусмотрен продукт или набор, содержащий соединение формулы (I), определенное выше, или его фармацевтически приемлемую соль или фармацевтическую композицию на его основе и одно или более химиотерапевтических лекарственных средств, при этом указанный продукт или набор находится в виде комбинированного препарата, и соединение формулы (I), определенное выше, или его фармацевтически приемлемую соль или фармацевтическую композицию на его основе и одно или более химиотерапевтических лекарственных средств применяют одновременно, отдельно или последовательно для противораковой терапии.
В еще одном аспекте в настоящем изобретении предусмотрено соединение формулы (I), определенное выше, или его фармацевтически приемлемая соль в качестве лекарственного средства.
Кроме того, в настоящем изобретении предусмотрено применение соединения формулы (I), описанного выше, или его фармацевтически приемлемой соли в получении лекарственного препарата, характеризующегося противоопухолевой активностью.
Наконец, в настоящем изобретении предусмотрено соединение формулы (I), описанное выше, или его фармацевтически приемлемая соль, которые применяют в получении лекарственного препарата для лечения заболеваний, вызванных и/или связанных с изменениями активности HSP90, в частности, заболеваний, представляющих собой рак или нейродегенеративные расстройства.
Если не указано иное, при ссылке на соединения формулы (I) per se и любые фармацевтические композиции на их основе или на любые терапевтические способы, предусматривающие применение соединений, настоящее изобретение охватывает все изомеры, таутомеры, гидраты, сольваты, комплексы, N-оксиды и фармацевтически приемлемые соли соединений по настоящему изобретению.
Фармацевтически приемлемые соли соединений формулы (I) включают соли присоединения кислоты, образованные с помощью неорганических или органических кислот, таких как азотная кислота, хлористоводородная кислота, бромистоводородная кислота, серная кислота, перхлорная кислота, фосфорная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, гидроксиуксусная кислота, фумаровая кислота, молочная кислота, щавелевая кислота, малоновая кислота, яблочная кислота, малеиновая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, изетионовая кислота и салициловая кислота. Фармацевтически приемлемые соли соединений формулы (I) также включают соли, образованные с помощью неорганических или органических оснований, таких как гидроксиды, карбонаты или бикарбонаты щелочных металлов или щелочноземельных металлов (в частности, натрия, калия, кальция, аммония или магния) и нециклические или циклические амины, предпочтительно метиламин, этиламин, диэтиламин, триэтиламин, пиперидин и т. д.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «алкил» означает любые насыщенные углеводороды или насыщенные углеводороды, замещенные 1-3 гетероатомами, при этом углеводороды могут быть линейными или разветвленными. «Алкил» включает алкановые группы и гетероалкил. Например, «C1-6алкил» относится к алкану, содержащему 1-6 атомов углерода или к алкану с 1-6 атомами углерода, в котором 1-3 атома углерода замещены гетероатомами. Например, «C1-10алкил» относится к алкану, содержащему 1-10 атомов углерода, или к алкану с 1-10 атомами углерода, в котором 1-3 атома углерода замещены гетероатомами. Неограничивающие примеры «алкила» включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, н-гексил и т. д.
Термин «C2-7алкенил» означает алифатические C2-7-углеводородные цепи, содержащие по меньшей мере одну двойную связь, и которые могут быть линейными или разветвленными, которые могут быть замещены одним или двумя гетероатомами. Иллюстративные примеры включают без ограничения винил, 1-пропенил, 2-пропенил, 1- или 2-бутенил и т. д.
Термин «C2-7алкинил» означает алифатические C2-7-углеводородные цепи, содержащие по меньшей мере одну углерод-углеродную тройную связь, и которые могут быть линейными или разветвленными. Иллюстративные примеры включают без ограничения этинил, 1-пропинил, 2-пропинил, 1- или 2-бутинил и т. д.
Если не указано иное, термин «циклоалкил» означает насыщенные циклические углеводороды или насыщенные циклические углеводороды, замещенные одним или более гетероатомами. «Циклоалкил» включает циклоалкановые группы и гетероциклоалкил. Неограничивающими примерами циклоалкила являются циклопропан, циклобутан, циклопентан, циклогексан, тетрагидрофуран, тетрагидротиофен и т. д. Например, термин «C3-10циклоалкил» означает насыщенные циклические углеводороды, содержащие 3-10 атомов углерода, или насыщенные циклические углеводороды, содержащие 3-10 атомов углерода, замещенные одним или более гетероатомами. Неограничивающими примерами циклоалкила являются циклопропан, циклобутан, циклопентан, циклогексан, тетрагидрофуран, тетрагидротиофен, пиран, пирролидин, имидазолидин, пиразолидин, тиазолидин, 1,3-диоксолан, пиперидин, пиперазин, морфолин и т. д.
Если не указано иное, термин «циклоалкенил» означает циклические углеводороды, содержащие двойную связь, или циклические углеводороды, содержащие двойную связь, которые замещены одним или более гетероатомами, за исключением тех, которые содержат полностью конъюгированную π-электронную систему. «Циклоалкенил» включает циклоалкеновые группы и гетероциклоалкеновые группы. Неограничивающими примерами циклоалкенила являются циклопентен, циклогексен, циклогексадиен, пирролин, имидазолин, пиразолин, тиазолин, дигидрофуран и т. д.
Термин «арил» или «ароматическое кольцо» включает ароматический гидрокарбил и гетероарил.
Термин «ароматический гидрокарбил» означает моно-, ди- или поликарбоциклический углеводород, содержащий 1-4 кольцевых системы, причем указанные кольцевые системы необязательно дополнительно конденсированы друг с другом или связаны посредством одинарной связи, при этом по меньшей мере одно карбоциклическое кольцо является «ароматическим», и при этом термин «ароматический» означает полностью конъюгированную π-электронную систему. Неограничивающими примерами арила являются фенильная группа, альфа- или бета-нафтильная группа или бифенильная группа.
Термин «гетероарил» означает ароматическое гетероциклическое кольцо и обычно представляет собой 5-10-членное гетероциклическое кольцо, содержащее 1-3 гетероатома, выбранные из N, O или S; и гетероарильные кольца необязательно могут быть дополнительно конденсированы или связаны с ароматическими и неароматическими карбоциклическими кольцами и гетероциклическими кольцами. Неограничивающими примерами гетероарила являются, например, пиридил, пиразинил, пиримидинил, пиридазинил, индолил, имидазолил, тиазолил, изотиазолил, пирролил, фенилпирролил, фуранил, фенилфурил, оксазолил, изоксазолил, пиразолил, тиенил, бензотиенил, изоиндолил, бензимидазолил, хинолинил, изохинолил, 1,2,3-триазолил, 1-фенил-1,2,3-триазолил, 2,3-дигидроиндолил, 2,3-дигидробензофуранил, 2,3-дигидробензотиенил; бензопиранил, 2,3-дигидробензоксазинил, 2,3-дигидрохиноксалинил и т. д.
Если не указано иное, согласно настоящему изобретению любая из вышеуказанных групп R1-R5 может быть необязательно замещена по любым их незанятым положениям одной или более группами, например, замещена 1-6 группами, где группы независимо выбраны из галогена, нитро, оксогруппы (=O), циано, C1-C6алкила, полифторалкила, полифторалкокси, C2-C6алкенила, C2-C6алкинила, гидроксиалкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклила, гетероциклоалкила, C3-C7циклоалкила, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкиленаминоокси, карбокси, алкоксикарбонила, арилоксикарбонила, циклоалкилоксикарбонила, гетероциклилалкилоксилкарбониламино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формамидо, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонила, алкиламинокарбонила, диалкиламинокарбонила, ариламинокарбонила, гетероциклиламинокарбонила, алкоксилкарбониламино, гидроксиаминокарбонилалкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формила, алкилкарбонила, арилкарбонила, циклоалкилкарбонила, гетероциклилкарбонила, алкилсульфонила, арилсульфонила, аминосульфонила, алкиламиносульфонила, диалкиламиносульфонила, ариламиносульфонила, гетероциклиламиносульфонила, арилтио, алкилтио, фосфонатов, фосфоновой кислоты, фосфонатного радикала и алкилфосфонатов. Каждый из вышеуказанных заместителей в свою очередь может быть дополнительно замещен одной или более из вышеупомянутых групп, таких как R1, R2, R3, R4 и R5, при необходимости.
Термин «полифторалкил» или «полифторалкокси» означает любой из вышеуказанных линейных или разветвленных C1-C8алкилов или алкокси, который замещен более чем одним атомом фтора, например, трифторметил, трифторэтил, 1,1,1,3,3,3-гексафторпропил, трифторметокси и т. д.
Термин «гидроксиалкил» означает любой из вышеуказанных C1-C8алкилов, содержащий гидроксильную группу, например, гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил и т. д.
Специалисту в данной области техники будет понятно, что по всему описанию, как указано выше, любые группы, названия которых представляют собой составные названия, должны согласно правилу означать таковые, составленные из фрагментов, образующих группу, например, ариламино представляет собой аминогруппу, которая дополнительно замещена арильной группой, где арил определен выше.
Подобным образом, алкильные, алкокси-, арильные, C3-10циклоалкильные и гетероциклильные фрагменты, содержащиеся в любых терминах, например, алкилтио, алкиламино, диалкиламино, алкоксикарбонил, алкоксикарбониламино, гетероциклилкарбонил, гетероциклилкарбониламино, циклоалкилоксикарбонил и т. д., представляют собой группы, определенные выше.
Соединения по настоящему изобретению можно получать с помощью различных способов синтеза, известных специалисту в данной области техники, в том числе с помощью конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, образованных комбинированием вышеуказанных конкретных вариантов осуществления с другими химическими способами синтеза, и эквивалентными альтернативами, известными специалисту в данной области техники, и предпочтительные варианты осуществления включают без ограничения примеры настоящего изобретения.
Все растворители, применяемые в настоящем изобретении, являются коммерчески доступными и их можно применять без дополнительной очистки. Как правило, реакции проводят в безводном растворителе в атмосфере инертного газообразного азота. Данные протонного магнитного резонанса записывали на спектрометре Bruker Avance III 400 (400 МГц) и значения химического сдвига представлены в виде δ (ppm) в сторону слабого поля от тетраметилсилана. Масс-спектры измеряли на Agilent 1200 Series plus 6110 (& 1956A). LC/MS или Shimadzu MS содержит детектор DAD: SPD-M20A (LC) и детектор Shimadzu Micromass 2020. Масс-спектрометр оснащен источником ионизации путем электрораспыления (ESI), работающим в режиме образования положительных или отрицательных ионов.
В настоящем изобретении применяют следующие сокращения: PG представляет собой защитную группу; DMF представляет собой N,N-диметилкарбоксамид; PE представляет собой петролейный эфир; EA представляет собой этилацетат; NCS представляет собой 1-хлорпирролидин-2,5-дион; NBS представляет собой 1-бромпирролидин-2,5-дион; NIS представляет собой 1-иодпирролидин-2,5-дион; экв. представляет собой эквивалентное количество или эквивалент; DCM представляет собой дихлорметан; DMSO представляет собой диметилсульфоксид; EtOH представляет собой этанол; MeOH представляет собой метанол; CBz представляет собой бензилоксикарбонил и является защитной группой для аминов; BOC представляет собой трет-бутилоксикарбонил и является защитной группой для аминов; HOAc представляет собой уксусную кислоту; NaCNBH3 представляет собой цианоборогидрид натрия; THF представляет собой тетрагидрофуран; Boc2O представляет собой ди-трет-бутилдикарбонат; TFA представляет собой трифторуксусную кислоту; TFAA представляет собой трифторуксусный ангидрид; DIEA представляет собой диизопропилэтиламин; DMAP представляет собой N,N-диметиламинопиридин; TEA представляет собой триэтиламин; TMSCl представляет собой триметилхлорсилан; MTBE представляет собой метил-трет-бутиловый эфир; AIBN представляет собой азобисизобутиронитрил; DME представляет собой диметиловый эфир; DCE представляет собой дихлорэтан; LDA представляет собой N,N-диизопропиламид лития; CAN представляет собой нитрат аммония-церия; mp представляет собой температуру плавления.
Соединения называли вручную или с помощью программного обеспечения ChemDraw®, и для коммерчески доступных соединений применяли названия из каталога поставщиков.
Высокоэффективную жидкостную хроматографию (HPLC) осуществляли с помощью колонки Xtimate C18 (диаметр наполнителя 3 мкм, с параметрами 2,1 × 300 мм) с системой Shimadzu LC20AB, оснащенной автоматическим дозатором Shimadzu SIL-20A и детектором Shimadzu DAD: SPD-M20A. Способ 0-60AB_6 минут: начинают элюирование с помощью 100% A (A представляет собой 0,0675% раствор TFA в воде) и заканчивают его с помощью 60% B (B представляет собой 0,0625% раствор TFA в MeCN) путем использования линейного градиента, при этом данный процесс занимает 4,2 мин.; затем элюируют с помощью 60% B в течение 1 мин.; и восстанавливают равновесие хроматографической колонки в течении 0,8 мин. до 100:0, и общее время анализа составляет 6 мин. Способ 10-80AB_6 минут: начинают элюирование с помощью 90% A (A представляет собой 0,0675% раствор TFA в воде) и заканчивают его с помощью 80% B (B представляет собой 0,0625% раствор TFA в ацетонитриле), при этом данный процесс занимает 4,2 мин.; затем элюируют с помощью 80% B в течении 1 мин.; и восстанавливают равновесие хроматографической колонки в течении 0,8 мин. до 90:10, и общее время анализа составляет 6 мин. Температура колонки составляет 50°C и расход составляет 0,8 мл/ мин. Детектор на диодной матрице имеет диапазон сканирования длин волн 200-400 нм.
Тонкослойную хроматографию (TLC) осуществляли на силикагеле GF254 от Sanpont-group. Пятна, как правило, детектировали с применением излучения УФ-лампы. В некоторых случаях пятна также детектировали с помощью других способов и в этих случаях пластинки для тонкослойной хроматографии проявляли с помощью йода (полученного путем добавления приблизительно 1 г йода на 10 г силикагеля и тщательно смешивания), ванилина (полученного путем растворения приблизительно 1 г ванилина в 100 мл 10% H2SO4), нингидрина (приобретенного от Aldrich) или специального проявителя (полученного путем тщательного смешивания (NH4)6Mo7O24·4H2O, 5 г (NH4)2Ce(IV)(NO3)6, 450 мл H2O и 50 мл концентрированной H2SO4) для детектирования соединений. Колоночную флэш хроматографию осуществляли на силикагеле Silicycle 40-63 мкм (230-400 меш) путем применения способа, аналогичного технологии, раскрытой в Still, W. C., Kahn, M., and Mitra, M., Journal of Organic Chemistry, 1978, 43, 2923-2925. Растворители, обычно применяемые в колоночной флэш-хроматографии или в тонкослойной хроматографии, представляют собой смесь дихлорметана/метанола, этилацетата/метанола и гексана/этилацетата.
Препаративный хроматографический анализ осуществляли с применением детектора Gilson UV/VIS-156 на системе Gilson-281 Prep LC 322 с хроматографической колонкой Agella Venusil ASB Prep C18, 5 мкм, 150 × 21,2 мм; Phenomenex Gemini C18, 5 мкм, 150 × 30 мм; Boston Symmetrix C18, 5 мкм, 150 × 30 мм или Phenomenex Synergi C18, 4 мкм, 150 × 30 мм. Когда расход составлял приблизительно 25 мл/мин., соединения элюировали ацетонитрилом/водой при малом градиенте, при этом вода содержала 0,05% HCl, 0,25% HCOOH или 0,5% NH3·H2O, и общее время анализа составляло 8-15 мин.
Анализ с помощью SFC осуществляли на системе Agilent 1260 Infinity SFC с автоматическим дозатором Agilent 1260 и детектором Agilent DAD: 1260. Используемая хроматографическая колонка представляет собой Chiralcel OD-H 250 × 4,6 мм I.D., 5 мкм; или Chiralpak AS-H 250 × 4,6 мм I.D., 5 мкм; или Chiralpak AD-H 250 × 4,6 мм I.D., 5 мкм. Хроматографическими условиями OD-H_5_40_2,35ML являются: хроматографическая колонка Chiralcel OD-H (с параметрами 250 × 4,6 мм I.D., диаметр наполнителя 5 мкм), подвижная фаза - 40% этанол (0,05% DEA)/CO2, расход - 2,35 мл/мин. и длина волны детектирования - 220 нм. Хроматографическими условиями AS-H_3_40_2,35ML являются: хроматографическая колонка Chiralpak AS-H (с параметрами 250 × 4,6 мм I.D., диаметр наполнителя 5 мкм), подвижная фаза - 40% метанол (0,05% DEA)/CO2, расход - 2,35 мл/мин. и длина волны детектирования - 220 нм. Хроматографическими условиями OD-H_3_40_2,35ML являются: хроматографическая колонка Chiralcel OD-H (с параметрами 250 × 4,6 мм I.D., диаметр наполнителя 5 мкм), подвижная фаза - 40% метанол (0,05% DEA)/CO2, расход - 2,35 мл/мин. и длина волны детектирования - 220 нм. Хроматографическими условиями AD-H_2_50_2,35ML являются: хроматографическая колонка Chiralpak AD-H (с параметрами 250 × 4,6 мм I.D., диаметр наполнителя 5 мкм), подвижная фаза - 50% метанол (0,1% MEA)/CO2, расход 2,35 мл/мин. и длина волны детектирования - 220 нм.
Анализ с помощью препаративной SFC осуществляли на системе Waters Thar 80 Pre-SFC с применением УФ-детектора Gilson, и используемая хроматографическая колонка представляет собой Chiralcel OD-H (с параметрами 250 × 4,6 мм I.D., диаметр наполнителя 5 мкм) или Chiralpak AD-H (с параметрами 250 × 4,6 мм I.D., диаметр наполнителя 5 мкм). При расходе примерно 40-80 мл/мин. соединения элюируют смесью этанол/диоксид углерода или метанол/диоксид углерода при малом градиенте, при этом метанол или этанол содержит 0,05% NH3·H2O, 0,05% DEA или 0,1% MEA, и общее время анализа составляет 20-30 мин.
Способы получения некоторых соединений по настоящему изобретению
Соединения по настоящему изобретению можно получать с помощью различных способов синтеза, известных специалисту в данной области техники, в том числе с помощью конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, образованных комбинированием конкретных вариантов осуществления с другими химическими способами синтеза, и эквивалентными замещениями, известными специалисту в данной области техники, и предпочтительные варианты осуществления включают без ограничения примеры настоящего изобретения.
Химические реакции в конкретных вариантах осуществления настоящего изобретения осуществляют в соответствующих растворителях, которые должны быть подходящими для химических превращений согласно настоящему изобретению, а также с реагентами и материалами, необходимыми для химических превращений. С целью получения соединений по настоящему изобретению иногда специалисту в данной области техники необходимо осуществить модификации или альтернативные варианты стадий синтеза или способов проведения реакции на основе существующих вариантов осуществления.
Ключевым аспектом в любом планировании пути синтеза в данной области является выбор соответствующих защитных групп для реакционноспособных функциональных групп (таких как аминогруппа в настоящем изобретении). Protective Groups In Organic Synthesis, Wiley and Sons, 1991, by Greene and Wuts, является справочным материалом в этом отношении для квалифицированных специалистов-практиков. Все ссылки, цитируемые в данном документе, включены посредством ссылки во всей своей полноте.
Соединения, представленные формулой (I) в настоящем изобретении, можно получать посредством реакционной схемы 1 и стандартных способов, известных специалисту в данной области техники. В качестве исходного материала (1-1) берут резорцин с защитной группой, например, его вводят в реакцию (3+2) циклизации с гидроксиламином в качестве субстрата и происходит элиминирование молекулы воды с образованием пятичленной ароматической гетероциклической кольцевой системы (1-2), а затем проводят электрофильное галогенирование пятичленного ароматического гетероциклического кольца. Образованный галогенид может непосредственно взаимодействовать с гетероциклическим конденсированным ароматическим боратом посредством реакции Сузуки в условиях катализа палладием, или образованный галогенид можно сначала преобразовать в борат, а затем подвергнуть реакции Сузуки с гетероциклическим конденсированным ароматическим галогенидом. С помощью двух путей можно ввести различные конденсированные ароматические кольцевые группы в пятичленный ароматический гетероцикл с получением соединения (1-5). Наконец, резорциновое соединение (1-6), т. е. ингибитор HSP90 по настоящему изобретению, образуют посредством удаления защитной группы.
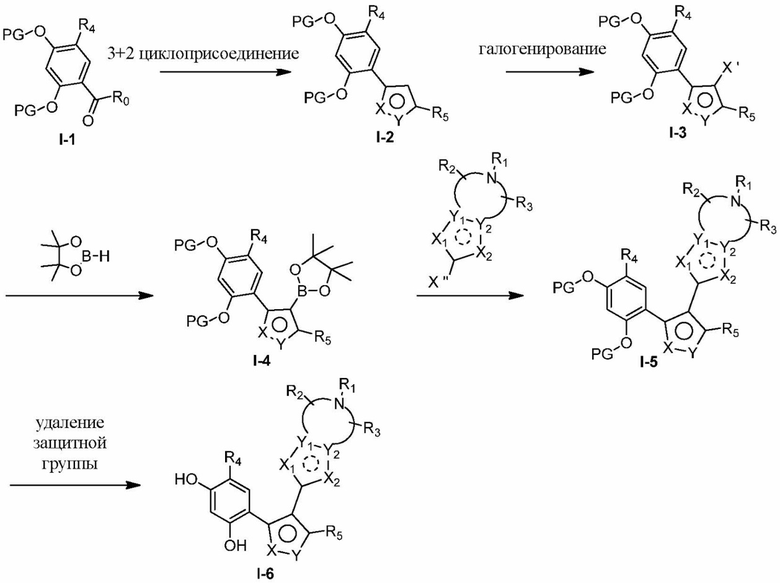
Реакционная схема 1
Более конкретно, соединения формулы (I), предусмотренные в настоящем изобретении, можно получать посредством реакционной схемы 1 и стандартных способов, известных специалисту в данной области техники. Начинают с производных валеролактама (1-1), которые являются коммерчески доступными, или можно начинать с других подобных производных, содержащих различные модификации функциональных групп, при этом R0 выбран из H, галогена, алкила, замещенного гетероатомом алкила, карбоновой кислоты, сложных алкиловых эфиров карбоновой кислоты; PG представляет собой защитную группу, выбранную из метила (Me), бензила (Bn), 4-метоксибензила (PMB), 3,4-диметоксибензила (DMB), метоксиметила (MOM), 2-метоксиэтоксиметила (MEM), 2-тетрагидрофурила (THP), триметилсилила (TMS), триэтилсилила (TES), триизопропилсилила (TIPS), трет-бутилдиметилсилила (TBDMS), трет-бутилдифенилсилила (TBDPS), ацетила, бензоила, пивалоила; X' представляет собой галоген; X'' представляет собой галоген, трифторметансульфоновую кислоту и т. д.
Другие заместители являются такими же, как и таковые в формуле (I). Все данные варианты, альтернативы будут более подробно описаны в разделе «Подробное описание». Специалисту в данной области техники будет понятно, что порядок стадий реакции в реакционной схеме 1 может изменятся для получения соединений по настоящему изобретению, что также находится в пределах объема настоящего изобретения.
Ряд новых резорциновых соединений, представленных формулой (I), по настоящему изобретению являющихся ингибиторами белка HSP90, можно применять для лечения рака и нейродегенеративных расстройств. По сравнению с предыдущим уровнем техники соединения по настоящему изобретению характеризуются улучшенной активностью и повышенной эффективностью. Следовательно, соединения формулы (I) могут представлять собой терапевтические средства от рака и нейродегенеративных расстройств.
ПОДРОБНОЕ ОПИСАНИЕ
Далее в данном документе настоящее изобретение будет подробно описано с помощью примеров, но они не предназначены для какого-либо невыгодного ограничения настоящего изобретения.
Пример 1
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
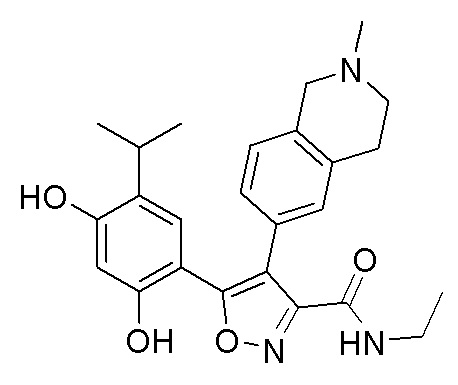
Реакционная схема
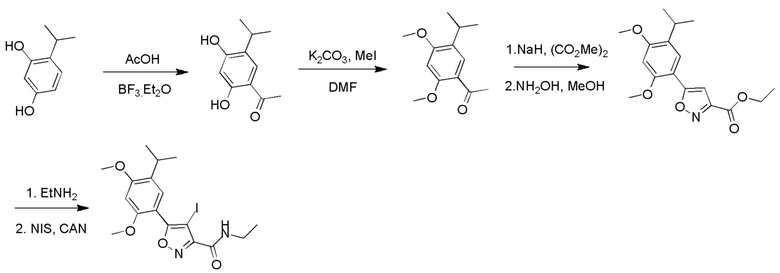
Стадия A. В защитной атмосфере газообразного азота при 20°C к раствору 4-изопропилбензол-1,3-диола (7,5 г, 49,3 ммоль, 1,0 экв.), растворенного в BF3·Et2O (60 мл), по каплям добавляли уксусную кислоту (3,26 г, 54,2 ммоль, 1,1 экв.). После завершения добавления реакционную смесь перемешивали при 80°C в течении 3 часов. Реакционный раствор гасили водным раствором ацетата калия (120 мл), а затем экстрагировали этилацетатом (150 мл × 3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, затем промывали насыщенным солевым раствором (100 мл), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = 50:1) с получением продукта, 1-(2,4-дигидрокси-5-изопропилфенил)этанона, (6,0 г, 30,9 ммоль, выход 62,7%) в виде желтого твердого вещества.
Стадия B. При 20°C в защитной атмосфере газообразного азота к смеси 1-(2,4-дигидрокси-5-изопропилфенил)этанона (6,00 г, 30,8989 ммоль, 1,00 экв.) и MeI (52,6 г, 370,789 ммоль, 12,0 экв.) в DMF (80 мл) добавляли карбонат цезия (25,2 г, 77,289 ммоль, 2,5 экв.). Смесь перемешивали при 20°C в течение 16 часов, а затем выливали в воду (80 мл). Водную фазу экстрагировали этилацетатом (100 мл × 2). Объединенную органическую фазу высушивали над безводным сульфатом натрия, затем промывали насыщенным солевым раствором (100 мл), фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = от 10/1 до 5/1) с получением 1-(5-изопропил-2,4-диметоксифенил)этанона (3,40 г, 15,389 ммоль, выход 49,5%) в виде желтого твердого вещества.
Стадия C. При 20°C в защитной атмосфере газообразного азота к раствору 1-(5-изопропил-2,4-диметоксифенил)этанона (3,40 г, 15,3089 ммоль, 1,00 экв.), растворенного в THF (50 мл), добавляли NaH (1,22 г, 30,689 ммоль, 2,0 экв.) и диметилоксалат (5,42 г, 45,989 ммоль, 3,0 экв.). После перемешивания при 60°C в течение дополнительного часа реакционную смесь выливали в водный раствор хлорида аммония (1000 мл) для подвергания гашению, а затем экстрагировали с помощью EA (80 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (80 мл) и высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением продукта, метил-2-гидрокси-4-(5-изопропил-2,4-диметоксифенил)-4-оксобутирата (4,80 г, неочищенный продукт) в виде желтого твердого вещества, которое непосредственно применяли на следующей стадии.
Стадия D. При комнатной температуре в защитной атмосфере газообразного азота к раствору метил-2-гидрокси-4-(5-изопропил-2,4-диметоксифенил)-4-оксобутирата (4,80 г, 15,5789 ммоль, 1,00 экв.) в MeOH (60 мл) добавляли NH2OH·HCl (2,16 г, 31,1489 ммоль, 2,00 экв.). Смесь нагревали до 60°C и перемешивали в течение 1 часа. Смесь охлаждали до комнатной температуры и выливали в воду (50 мл). Водную фазу экстрагировали с помощью EA (50 мл × 2). После промывания насыщенным солевым раствором (30 мл) объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = от 6/1 до 3/1) с получением этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксилата (4,80 г, 15,089 ммоль, выход 96,5%) в виде желтого твердого вещества.
Стадия E. При комнатной температуре к раствору этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксилата (4,80 г, 15,789 ммоль, 1,00 экв.) в MeOH (50 мл) добавляли этиламин (3,54 г, 78,689 ммоль, 5,0 экв.). Реакционную смесь перемешивали при 60°C в течение 2 часов. Реакционную смесь концентрировали, и неочищенный продукт промывали с помощью PE/EA = 50/1 (100 мл) с получением продукта, N-этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (3,70 г, 11,689 ммоль, выход 73,9%) в виде желтого твердого вещества.
Стадия F. При комнатной температуре в защитной атмосфере N2 к раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (3,50 г, 1189 ммоль, 1,0 экв.) в MeCN (50 мл) добавляли CAN (301 мг, 549 мкмоль, 0,05 экв.) и NIS (4,95 г, 2289 ммоль, 2,00 экв.). Смесь нагревали до 80°C и перемешивали в течение 16 часов. Смесь охлаждали до комнатной температуры и выливали в воду (30 мл) и водную фазу экстрагировали с помощью EA (40 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = от 10/1 до 4/1) с получением N-этил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (4,10 г, 9,289 ммоль, выход 84,0%) в виде желтого масла.
Стадия G. При 0°C к раствору 6-бромизохинолина (17,00 г, 81,789 ммоль), растворенного в MeOH (170 мл), одной порцией добавляли MeI (197,2 г, 1,4 моль). Смесь перемешивали при 0°C в течение 20 мин., а затем нагревали до 25°C и перемешивали в течение 16 часов. Смесь концентрировали при пониженном давлении с получением 6-бром-2-метилизохинолин-2-ия иодида (28,8 г, неочищенный продукт) в виде желтого твердого вещества. 1H ЯМР (400 МГц, метанол-d4): δ 9,90 (s, 1H), 8,65-8,61 (m, 2H), 8,45-8,39 (m, 2H), 8,22 (d, J = 8,0 Гц, 1H), 4,55 (s, 1H).
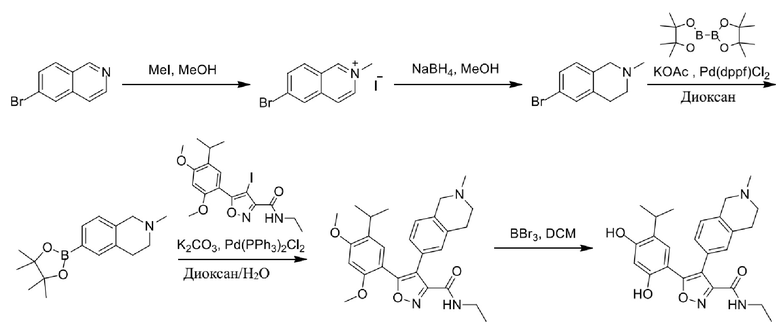
Стадия H. При 0°C к раствору 6-бром-2-метилизохинолин-2-ия йодида (28,80 г, 82,189 ммоль) в MeOH (350 мл) одной порцией добавляли NaBH4 (9,31 г, 246,289 ммоль). После перемешивания при 25°C в течение 2 часов смесь доводили с помощью водного раствора NaHCO₃ до pH = 9 и экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (100 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, дихлорметан/метанол = от 50/1 до 20/1) с получением 6-бром-2-метил-1,2,3,4-тетрагидроизохинолина (16,65 г, выход 80,8%, чистота 90%) в виде желтого твердого вещества.
Стадия I. При 25°C в защитной атмосфере газообразного азота к раствору 6-бром-2-метил-1,2,3,4-тетрагидроизохинолина (16,65 г, 73,6489 ммоль) в диоксане (150 мл) добавляли бис(пинаколато)дибор (28,05 г, 110,4689 ммоль) и KOAc (14,45 г, 147,2789 ммоль) с последующим добавлением катализатора Pd(dppf)Cl2.CH2Cl2 (6,01 г, 7,3689 ммоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 90°C при перемешивании в течение 14 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, дихлорметан/метанол = от 10/0 до 1/1) с получением указанного в заголовке продукта (31,0 г, неочищенный продукт) в виде черного твердого вещества, которое непосредственно применяли на следующей стадии. MS (ESI) масса/заряд: 274 (M+1).
Стадия J. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (6,15 г, 13,589 ммоль) и N-этил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (4,00 г, 9,089 ммоль) в диоксане (50 мл) добавляли K2CO3 (2,49 г, 18,089 ммоль), H2O (10,0 мл) и Pd(PPh3)4 (1,26 г, 1,8089 ммоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 110°C при перемешивании в течение 18 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат, дихлорметан/метанол = 5/1, 10/1) с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (2,87 г, выход 68,8%) в виде коричневого твердого вещества. MS (ESI) масса/заряд: 464 (M+1).
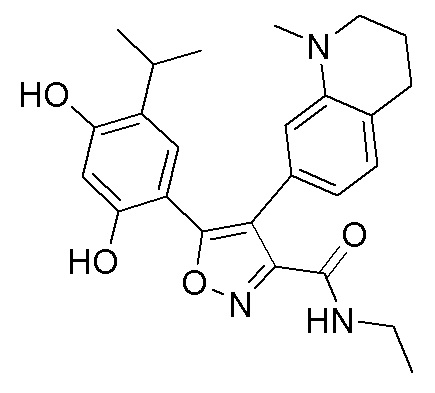
Стадия K. К раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (1,70 г, 3,6789 ммоль) в DCM (16,00 мл) медленно добавляли BBr3 (9,19 г, 36,6789 ммоль) при -78°C в защитной атмосфере газообразного азота, которую поддерживали в течение 2 часов. Обеспечивали нагревание реакционной смеси до 0°C на протяжении 1 часа и реакционную смесь перемешивали при 25°C в течение дополнительных 16 часов. Реакционную смесь гасили путем добавления MeOH (10 мл) при перемешивании при 25°C в течение 1 часа, и смесь медленно по каплям добавляли к насыщенному водному раствору NaHCO3 и фильтровали при 0°C. Водную фазу экстрагировали с помощью смеси дихлорметан:метанол = 10:1 (10 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, дихлорметан/метанол = от 10/0 до 1/1) с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (566,0 мг, выход 35,4%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,08 (s, 1H), 9,92-9,78 (m, J = 4,0 Гц, 1H), 7,16-7,12 (m, 3H), 6,89 (s, 1H), 6,53 (s, 1H), 4,6-4,42 (d, J = 16,0 Гц, 1H), 4,27-4,21 (m, 1H), 3,26-3,22 (m, 4H), 3,05-3,02 (m, 1H), 2,88-2,87 (m, 4H), 1,11-1,08 (t, J = 8,0 Гц, 3H), 1,03-0,98 (m, 6H). MS (ESI) масса/заряд: 436 (M+1).
Пример 2
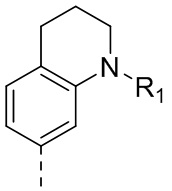
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(1-метил-1,2,3,4-тетрагидрохинолин-7-ил)изоксазол-3-карбоксамид
Реакционная схема
Стадия A. При 25°C к смеси 7-бромхинолина (100,00 мг, 480,65 мкмоль) и параформальдегида (433 мг, 4,889 ммоль), растворенных в AcOH (3 мл), добавляли NaBH3CN (151 мг, 2,489 ммоль). Смесь перемешивали при 25°C в течение 40 мин. и нейтрализовали с помощью NaOH. Весь реакционный раствор экстрагировали с помощью DCM (3 мл × 3) и органические фазы объединяли и промывали насыщенным солевым раствором (3 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Полученный в результате неочищенный продукт, 7-бром-1-метил-1,2,3,4-тетрагидрохинолин (155 мг), получали в виде коричневого масла, которое непосредственно применяли на следующей стадии без дополнительной очистки. MS (ESI) масса/заряд: 226 (M+1).
Стадия B. При 25°C в защитной атмосфере газообразного азота к раствору 7-бром-1-метил-1,2,3,4-тетрагидрохинолина (250 мг, 1,1189 ммоль) в диоксане (7 мл) добавляли бис(пинаколато)дибор (318 мг, 1,2589 ммоль) и KOAc (144 мг, 1,4789 ммоль) с последующим добавлением катализатора Pd(dppf)Cl2.CH2Cl2 (272 мг, 333 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 90°C при перемешивании в течение 17 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = 50/1) с получением неочищенного продукта, 1-метил-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолина (316 мг) в виде желтого масла. MS (ESI) масса/заряд: 274 (M+1).
Стадия C. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 1-метил-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолина (221 мг, 810,3 мкмоль) и N-этил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (360 мг, 810 мкмоль), растворенных в диоксане (9,9 мл) и H2O (2,1 мл), добавляли K2CO3 (224 мг, 1,689 ммоль) и Pd(dppf)Cl2.CH2Cl2 (66 мг, 81 мкмоль). Смесь перемешивали при 25°C в течение 20 мин., а затем нагревали до 90°C при перемешивании в течение 17 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток выливали в воду (5 мл) и перемешивали в течение 10 мин. Водную фазу экстрагировали этилацетатом (5 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (5 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с применением пластинки для тонкослойной хроматографии (дихлорметан:этилацетат = 5/1) с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(1-метил-1,2,3,4-тетрагидрохинолин-7-ил)изоксазол-3-карбоксамида (72 мг, выход 19,2%) в виде коричневого масла. MS (ESI) масса/заряд: 464 (M+1).
Стадия D. К раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(1-метил-1,2,3,4-тетрагидрохинолин-7-ил)изоксазол-3-карбоксамида (83 мг, 184,6 мкмоль) в DCM (5 мл) добавляли BBr3 (462 мг, 1,8589 ммоль) при -78°C на протяжении 15 мин. Во время данного периода поддерживали температуру на уровне -78°C. После добавления реакционную смесь нагревали до 0°C и перемешивали в течение 30 мин. Затем реакционную смесь перемешивали при 25°C в течение дополнительных 16 часов. Реакционную смесь медленно гасили насыщенным водным раствором NaHCO3 и фильтровали. Фильтрат удаляли посредством дистилляции в вакууме. Неочищенный продукт дополнительно очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(1-метил-1,2,3,4-тетрагидрохинолин-7-ил)изоксазол-3-карбоксамида (23 мг, выход 29,8%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,73 (s, 1H), 9,63 (s, 1H), 8,83 (t, J = 4,0 Гц, 1H), 6,84 (s, 1H), 6,79 (d, J = 8,0 Гц,1H), 6,49 (s, 1H), 6,43-6,42 (m, 2H), 3,27-3,21 (m, 2H), 3,06-2,99 (m, 1H), 2,65-2,63 (m, 5H), 1,88-1,82 (m, 2H), 1,09 (t, J = 4,0 Гц, 3H), 1,01 (d, J = 4,0 Гц, 6H). MS (ESI) масса/заряд: 436 (M+1).
Пример 3
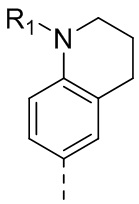
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)изоксазол-3-карбоксамид
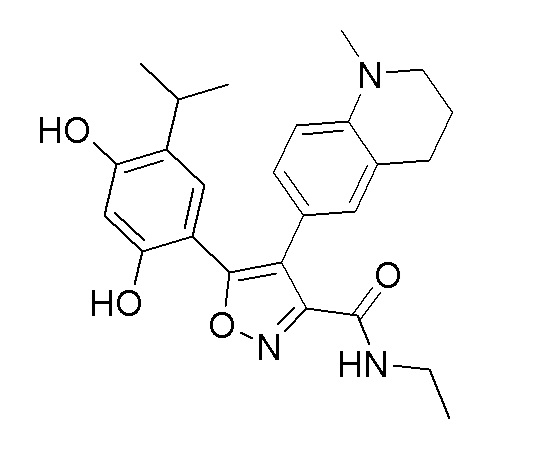
Реакционная схема
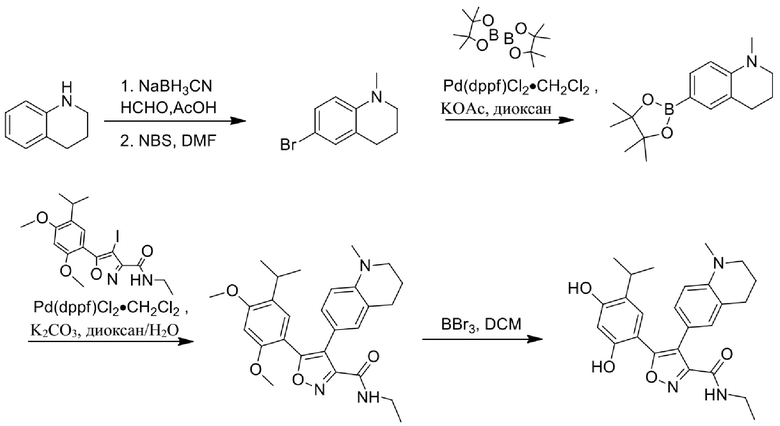
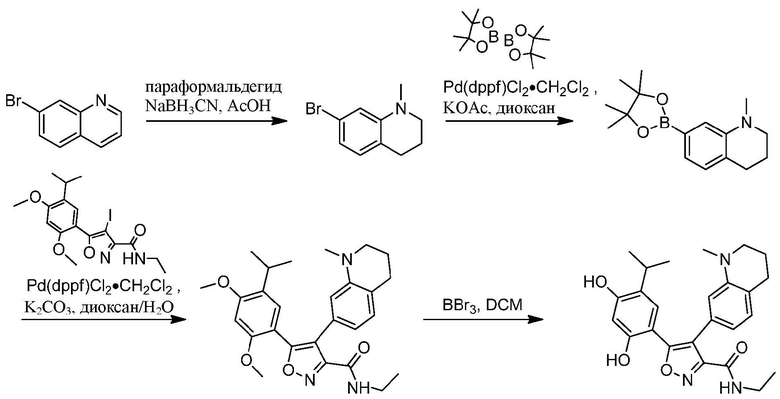
Стадия A. При 25°C к смеси 1,2,3,4-тетрагидрохинолина (2,0 г, 15,089 ммоль) и параформальдегида (6,77 г, 75,189 ммоль), растворенных в MeOH (20 мл), одной порцией добавляли AcOH (210 мг, 3,589 ммоль). Смесь перемешивали при 25°C в течение 1 часа с последующим добавлением NaBH3CN (1,89 г, 30,089 ммоль) и непрерывным перемешиванием в течение 16 часов. Смесь концентрировали при пониженном давлении. Остаток выливали в воду (15 мл) и перемешивали в течение 10 мин. Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением 1-метил-1,2,3,4-тетрагидрохинолина (1,17 г, 52,9%) в виде желтого масла. MS (ESI) масса/заряд: 148 (M+1).
Стадия B. Раствор 1-метил-1,2,3,4-тетрагидрохинолина (300 мг, 2,0489 ммоль) в DMF (5 мл) охлаждали до 0°C, а затем добавляли NBS (363,1 мг, 2,089 ммоль). Реакционный раствор перемешивали при 0°C в течение 2 часов, а затем нагревали до 25°C при перемешивании в течение 16 часов. Затем реакционную смесь выливали в 5 мл воды, и суспензию экстрагировали этилацетатом (5 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (5 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта, 6-бром-1-метил-1,2,3,4-тетрагидрохинолина (485 мг) в виде коричневого твердого вещества, которое применяли на следующей стадии без дополнительной очистки. MS (ESI) масса/заряд: 226 (M+1).
Стадия C. При 25°C в защитной атмосфере газообразного азота к раствору 6-бром-1-метил-1,2,3,4-тетрагидрохинолина (250 мг, 1,189 ммоль), бис(пинаколато)дибора (318 мг, 1,289 ммоль) и KOAc (325 мг, 3,389 ммоль), растворенных в диоксане (7 мл), добавляли Pd(dppf)Cl2.CH2Cl2 (271 мг, 331,7 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 90°C при перемешивании в течение 17 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = 50/1) с получением 1-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолина (187 мг) в виде желтого масла. MS (ESI) масса/заряд: 274 (М+1).
Стадия D. При 25°С в защитной атмосфере газообразного азота к перемешанному раствору 1-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолина (92 мг, 337,6 мкмоль) и N-этил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (100 мг, 225,1 мкмоль), растворенных в диоксане (6,6 мл) и H2O (1,4 мл), добавляли Pd(PPh3)2Cl г (15,8 мг, 22,5 мкмоль) и NaHCO3 (37,8 мг, 450,2 мкмоль). Смесь перемешивали при 25°С в течение 10 мин., а затем нагревали до 90°С при перемешивании в течение 17 часов. Смесь охлаждали до 25°С и концентрировали при пониженном давлении. Остаток выливали в воду (8 мл) и перемешивали в течение 10 мин. Водную фазу экстрагировали этилацетатом (5 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (5 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с применением пластинки для тонкослойной хроматографии (дихлорметан : этилацетат = 3/1) с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)изоксазол-3-карбоксамида (51 мг, выход 48,9%) в виде коричневого масла. MS (ESI) масса/заряд: 464 (М+1).
Стадия Е. К раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4- (1-метил-1,2,3,4-тетрагидрохинолин-6-ил)изоксазол-3-карбоксамида (50 мг, 107,9 мкмоль, 1,00 экв.) в DCM (5 мл) добавляли BBr3 (270 мг, 1,0889 ммоль, 10,00 экв.) при -78°С на протяжении 15 мин. Во время данного периода поддерживали температуру на уровне -78°С. После добавления реакционную смесь нагревали до 0°С и перемешивали в течение 30 мин. Затем реакционную смесь перемешивали при 25°С в течение дополнительных 16 часов. Реакционную смесь медленно гасили насыщенным водным раствором NaHCO3 и фильтровали. Фильтрат удаляли посредством дистилляции под вакуумом. Неочищенный продукт очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-1H-этил-4-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)изоксазол-3-карбоксамида (26,8 мг, 57,0%). 1H ЯМР (400 МГц, DMSO-d6): δ 9, 73 (s, 1Н), 9,60 (s, 1H), 8,78 (t, J=8,0 Гц, 1H), 6,87-6,89 (m, 1H), 6, 84-6,80 (m, 1H), 6,49 (d, J=8,0 Гц, 1H), 6,43 (s, 1H), 3,27-3,20 (m, 2H), 3,17-3,14 (m, 2H), 3,06-2,97 (m, 1H), 2,80 (s, 3H), 2,56 (t, J=4,0 Гц, 2H), 1,87-1,83 (m, 2H), 1,09 (t, J=4,0 Гц, 3H), 1,02-0,99 (m, 6H). MS (ESI) масса/заряд: 356 (M+1).
Пример 4
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамид
Реакционная схема
Стадия A. К раствору пиррола (4,00 г, 5989 ммоль, 1,00 экв.) и (Boc)2O (15,60 г, 71,589 ммоль, 1,20 экв.), растворенных в ацетонитриле (200 мл), добавляли DMAP (1,00 г, 8,289 ммоль, 0,14 экв.). Смесь перемешивали при 25°C в течение 2 часов. Смесь концентрировали и остаток очищали на колонке с силикагелем (элюентом являлся PE) с получением трет-бутилпиррол-1-формиата (7,80 г, 46,789 ммоль, выход 78,3%) в виде бесцветной жидкости.
Стадия B. Перемешанный раствор трет-бутилпиррол-1-формиата (4,60 г, 27,589 ммоль, 1,00 экв.) и порошка магния (720 мг, 27,589 ммоль, 1,0 экв.) в THF (20 мл) нагревали до 66°C на масляной бане. Медленно добавляли 1-бром-2-фторбензол (4,88 г, 27,8989 ммоль, 1,0 экв.) в течение 20 мин. и после добавления смесь перемешивали при 66°C в течение 8 часов. Растворитель удаляли при пониженном давлении и добавляли 0,5 н. водный раствор HCl с последующей экстракцией с помощью DCM. Органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате остаток очищали с помощью колоночной хроматографии (PE/EA = 20/1) с получением трет-бутил-1,4-дигидро-1,4-эпиминонафтил-9-формиата (2,43 г, 1089 ммоль, выход 36,3%) в виде желтой жидкости.
Стадия C. Раствор трет-бутил-1,4-дигидро-1,4-эпиминонафтил-9-формиата (2,43 г, 1089 ммоль, 1,00 экв.) и сухого Pd/C (200 мг) в MeOH (50 мл) перемешивали при 25°C в течение 2 часов в атмосфере H2. Смесь фильтровали через подушку из диатомовой земли и фильтрат концентрировали с получением трет-бутил-1,2,3,4-тетрагидро-1,4-эпиминонафтил-9-формиата (2,33 г, 9,589 ммоль, выход 95,1%) в виде желтого масла.
Стадия D. При 0°C к перемешанному раствору DCM (1,2 мл) и TFA (4,5 мл) добавляли трет-бутил-1,2,3,4-тетрагидро-1,4-эпиминонафтил-9-формиат (2,33 г, 9,589 ммоль, 1,0 экв.). Смесь перемешивали при 0°C в течение 0,5 часа, а затем перемешивали при 25°C в течение 4,5 часа. После удаления растворителя при пониженном давлении добавляли 2 н. водный раствор NaOH и водную фазу экстрагировали с помощью DCM. Органический слой высушивали над безводным сульфатом натрия и концентрировали с получением 1,2,3,4-тетрагидро-1,4-эпиминонафталина (1,38 г, 9,589 ммоль, выход 100%) в виде желтого твердого вещества.
Стадия E. Смесь 1,2,3,4-тетрагидро-1,4-эпиминонафталина (1,38 г, 9,589 ммоль, 1,00 экв.) и DIEA (1,37 г, 10,689 ммоль, 1,12 экв.) в безводном DCM (20,00 мл) охлаждали до 0°C и добавляли TFAA (2,27 г, 10,889 ммоль, 1,14 экв.). Реакционную смесь медленно нагревали до 25°C в защитной атмосфере газообразного азота и перемешивали в течение 5 часов. Полученную в результате реакционную смесь охлаждали до 0°C и добавляли воду (2 мл) для гашения оставшегося ангидрида. Водную фазу доводили до нейтрального значения pH с применением водного раствора NaOH (1 н.), а затем органическую фазу отделяли. Водную фазу дважды экстрагировали с помощью DCM, и объединенный органический слой высушивали с помощью безводного сульфата натрия и концентрировали с получением 2,2,2-трифтор-1-(1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (2,24 г, 9,2989 ммоль, выход 97,8%) в виде коричневого масла.
Стадия F. Раствор (5,00 мл) 2,2,2-трифтор-1-(1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (2,24 г, 9,2989 ммоль, 1,00 экв.) в TFA охлаждали до 0°C и по каплям добавляли дымящую азотную кислоту (1 мл). Полученную в результате реакционную смесь перемешивали при 0°C в течение 1 часа, а затем перемешивали при 25°C в течение 1 часа. Смесь выливали в 300 мл ледяной воды и экстрагировали три раза с помощью DCM. Объединенную органическую фазу последовательно промывали насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl. Органическую фазу высушивали над безводным сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюирование с помощью DCM) с получением целевого продукта, 2,2,2-трифтор-1-(6-нитро-1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (1,86 г, 6,5089 ммоль, выход 67,0%) в виде желтого твердого вещества.
Стадия G. К перемешанному раствору 2,2,2-трифтор-1-(6-нитро-1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (3,5 г, 12,2389 ммоль, 1,00 экв), растворенного в диоксане (20,00 мл)/этаноле (16,00 мл)/H2O (12,00 мл), последовательно добавляли NH4Cl (2,63 г, 49,289 ммоль, 4,02 экв.) и порошок железа (3,43 г, 61,489 ммоль, 5,02 экв.). Полученную в результате смесь нагревали до 80°C в защитной атмосфере газообразного азота и перемешивали в течение 3 часов. Реакционную смесь охлаждали до 25°C, разбавляли с помощью EtOAc и H2O и фильтровали через подушку из диатомовой земли. Органическую фазу собирали посредством отделения жидкости, высушивали над сульфатом натрия и концентрировали при пониженном давлении с получением 2,2,2-трифтор-1-(6-амино-1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (2,7 г, 10,589 ммоль, выход 86,2%), который непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия H. Раствор 2,2,2-трифтор-1-(6-амино-1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (2,7 г, 10,589 ммоль, 1,0 экв.), бис(пинаколато)дибора (2,68 г, 10,589 ммоль, 1,0 экв.), BPO (76 мг, 316 мкмоль, 0,03 экв.) и трет-бутилнитрита (1,63 г, 15,889 ммоль, 1,5 экв.), растворенных в MeCN (15,0 мл), перемешивали при 25°C в течение 16 часов. Смесь концентрировали при пониженном давлении и неочищенный остаток очищали с помощью колоночной хроматографии (петролейный эфир:этилацетат = от 40:1 до 5:1) с получением 2,2,2-трифтор-1-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (2,2 г, 6,089 ммоль, выход 56,9%) в виде желтого масла.
Стадия I. Обеспечивали прохождение реакции замещения в растворе N-этил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (500 мг, 1,1389 ммоль, 1,00 экв.), 2,2,2-трифтор-1-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидро-1,4-эпиминонафт-9-ил)этанона (539 мг, 1,4789 ммоль, 1,3 экв.), Pd(PPh3)2Cl2 (158 мг, 225,1 мкмоль, 0,20 экв.) и NaHCO3 (283 мг, 3,3889 ммоль, 3,0 экв.) в THF (5,00 мл) в атмосфере азота, затем его нагревали до 120°C с помощью облучения микроволнами и обеспечивали взаимодействие в течение 30 мин. Реакционную смесь выливали в воду (15 мл). Смесь экстрагировали этилацетатом (10 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат = от 50/1 до 5/1) с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(9-(2,2,2-трифторацетил)-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (500 мг, 807 мкмоль, выход 71,4%, чистота 90%).
Стадия J. При 30°C к перемешанному раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(9-(2,2,2-трифторацетил)-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (500 мг, 762,3 мкмоль, 1,0 экв.) в MeOH (4,2 мл) и H2O (1,8 мл) добавляли K2CO3 (526 мг, 3,889 ммоль, 5,0 экв.). Реакционный раствор перемешивали при 30°C в течение 18 часов и выливали в воду (10 мл). Водную фазу экстрагировали с помощью EA (10 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с применением пластинки для тонкослойной хроматографии (DCM/метанол = 15:1) с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (220 мг, 476,7 мкмоль, выход 62,5%) в виде белого твердого вещества.
Стадия K. При -78°C к раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (50 мг, 108,3 мкмоль, 1,0 экв.) в безводном DCM (2,0 мл) медленно по каплям добавляли BBr3 (271 мг, 1,0889 ммоль, 10,00 экв.). После добавления раствор нагревали до 30°C и перемешивали в течение 18 часов. Раствор охлаждали до -78°C и добавляли MeOH (1 мл) с последующим добавлением насыщенного водного раствора NaHCO3 (3 мл). Смесь перемешивали при 30°C в течение 5 мин. Смесь экстрагировали с помощью DCM (10 мл × 3) и органические слои объединяли, промывали солевым раствором (10 мл × 2), высушивали с помощью Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали с помощью высокоэффективной жидкостной хроматографии (система на основе муравьиной кислоты) с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (30 мг, 69,2 мкмоль, выход 63,9%). 1H ЯМР (400 МГц, DMSO-d6): δ 8,83 (t, J = 5,4 Гц, 1H), 8,31 (brs, 1H), 7,25-7,14 (m, 2H), 7,01 (d, J = 7,0 Гц, 1H), 6,71 (s, 1H), 6,45 (s, 1H), 4,72-4,57 (m, 2H), 3,28-3,17 (m, 2H), 3,03-2,90 (m, 1H), 1,95 (brs, 2H), 1,18-1,04 (m, 5H), 0,91 (t, J = 6,0 Гц, 6H).
Пример 5
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(9-метил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамид
Реакционная схема
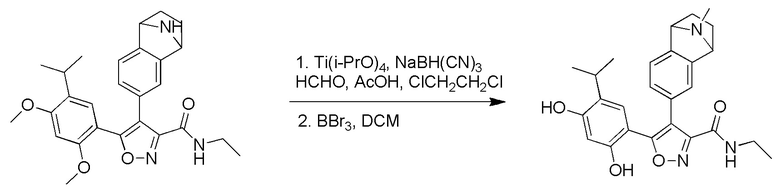
Стадия A. К раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (20 мг, 43,3 мкмоль, 1,0 экв.) в 1,2-дихлорэтане (3 мл) добавляли параформальдегид (39 мг, 433,3 мкмоль, 10,00 экв.), уксусную кислоту (1,3 мг, 21,7 мкмоль, 0,50 экв.) и тетраизопропанолят титана (6,2 мг, 21,7 мкмоль, 0,50 экв.). Перемешанный раствор перемешивали при 30°C в течение 18 часов, а затем добавляли NaBH3CN (8,2 мг, 130 мкмоль, 3,00 экв.) и смесь перемешивали в течение дополнительных 2 часов. К реакционному раствору добавляли воду (10 мл) и его фильтровали и фильтрат экстрагировали с помощью DCM (10 мл × 3). Органические слои объединяли, промывали солевым раствором (5 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(9-метил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (20 мг, 42,1 мкмоль, выход 97,1%) в виде желтого масла.
Стадия B. При -78°C к раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(9-метил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (30 мг, 63,1 мкмоль, 1,0 экв.) в безводном DCM (2 мл) медленно по каплям добавляли BBr3 (158 мг, 630,8 мкмоль, 10 экв.). После добавления раствор нагревали до 30°C и перемешивали в течение 18 часов. Раствор охлаждали до -78°C и медленно добавляли MeOH (1 мл) с последующим добавлением насыщенного водного раствора NaHCO3 (3 мл). Затем смесь перемешивали при 30°C в течение 5 мин. Смесь экстрагировали с помощью DCM (10 мл × 3) и органические слои объединяли, промывали солевым раствором (10 мл × 2), высушивали над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный в результате остаток очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(9-метил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамида (10 мг, 22,3 мкмоль, выход 35,4%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,79 (t, J = 5,5 Гц, 1H), 7,22-7,12 (m, 2H), 6,99 (d, J = 7,3 Гц, 1H), 6,68 (s, 1H), 6,46 (s, 1H), 4,04 (d, J = 19,8 Гц, 2H), 3,27-3,16 (m, 2H), 3,01-2,89 (m, 1H), 2,05-1,80 (m, 5H), 1,10-0,97 (m, 6H), 0,89 (d, J = 6,8 Гц, 6H).
Пример 6
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(9-этил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамид
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 5, при этом параформальдегид на стадии A заменяли ацетальдегидом. 1H ЯМР (400 МГц, DMSO-d6) δ 8,80 (t, J = 5,5 Гц, 1H), 8,24 (brs, 1H), 7,23-7,11 (m, 2H), 6,98 (d, J = 7,5 Гц, 1H), 6,67 (s, 1H), 6,46 (s, 1H), 4,22 (d, J = 19,8 Гц, 2H), 3,25-3,15 (m, 2H), 2,95 (td, J = 6,8, 13,7 Гц, 1H), 2,17-1,86 (m, 4H), 1,06 (t, J = 7,2 Гц, 5H), 0,95-0,84 (m, 9H).
Пример 7
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(9-изопропил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамид
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 5, при этом параформальдегид на стадии A заменяли ацетоном. 1H ЯМР (400 МГц, DMSO-d6) δ 8,80 (t, J = 5,5 Гц, 1H), 8,21 (s, 1H), 7,23-7,11 (m, 2H), 6,98 (d, J = 7,3 Гц, 1H), 6,66 (s, 1H), 6,45 (s, 1H), 4,40 (d, J = 16,6 Гц, 2H), 3,22 (q, J = 6,7 Гц, 2H), 2,95 (td, J = 6,9, 13,6 Гц, 1H), 1,98 (brs, 3H), 1,06 (t, J = 7,2 Гц, 5H), 0,96-0,79 (m, 12H).
Пример 8
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(9-изобутил-1,2,3,4-тетрагидро-1,4-эпиминонафт-6-ил)изоксазол-3-карбоксамид
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 5, при этом параформальдегид на стадии A заменяли изобутиральдегидом. 1H ЯМР (400 МГц, DMSO-d6) δ 8,78 (t, J = 5,5 Гц, 1H), 7,19-7,10 (m, 2H), 6,96 (d, J = 7,3 Гц, 1H), 6,65 (s, 1H), 6,46 (s, 1H), 4,18-4,04 (m, 2H), 3,26-3,17 (m, 3H), 3,01-2,88 (m, 1H), 1,96 (brs, 2H), 1,83 (d, J = 7,0 Гц, 2H), 1,53 (td, J = 6,7, 13,3 Гц, 1H), 1,09-0,95 (m, 5H), 0,87 (d, J = 6,0 Гц, 6H), 0,80 (dd, J = 1,8, 6,3 Гц, 6H).
Пример 9
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)изоксазол-3-карбоксамид
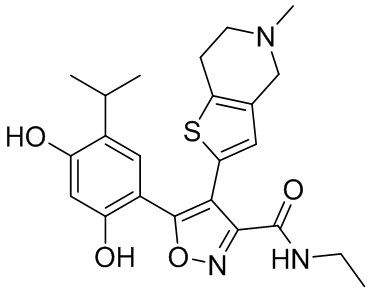
Реакционная схема
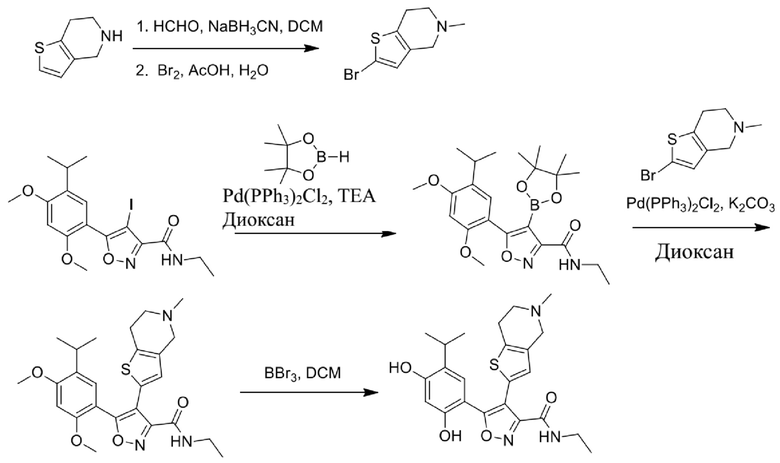
Стадия A. К раствору 4,5,6,7-тетрагидротиено[3,2-c]пиридина (10,0 г, 71,889 ммоль, 1,0 экв.) в DCM (120,0 мл) добавляли молекулярное сито с размером пор 4Å (10,00 г), тетраизопропанолят титана (1,02 г, 3,5989 ммоль, 0,05 экв.) и AcOH (215,67 мг, 3,5989 ммоль, 0,05 экв.). Смесь перемешивали при 25°C в течение 16 часов, затем добавляли NaBH3CN (9,03 г, 143,6689 ммоль, 2,00 экв.) и перемешивали при 25°C в течение дополнительных 3 часов. Смесь выливали в воду (300 мл) и экстрагировали с помощью EA (100 мл × 2). Объединенный органический слой высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (DCM/метанол = от 50/1 до 10/1) с получением 5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридина (2,50 г, 16,3189 ммоль, выход 22,71%) в виде желтого масла.
Стадия B. При 0°C к перемешанному раствору 5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридина (2,00 г, 13,0589 ммоль, 1,0 экв.), растворенного в AcOH (20,00 мл) и воде (20,00 мл), добавляли Br2 (1,88 г, 11,7589 ммоль, 0,90 экв.). После перемешивания при 0°C в течение 1 часа, смесь выливали в воду (100 мл), повышали ее основность с помощью NaOH (10 н.) до pH = 8-9, а затем экстрагировали с помощью EA (100 мл × 2). Объединенный органический слой концентрировали с получением 2-бром-5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридина (2,70 г, 11,689 ммоль, выход 89,1%), который непосредственно применяли на следующей стадии.
Стадия C. В защитной атмосфере газообразного азота к раствору N-этил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (900 мг, 2,0389 ммоль, 0,10 экв.) в диоксане (20,00 мл) добавляли Pd(PPh3)2Cl2 (142,5 мг, 203,0 мкмоль, 0,10 экв.), TEA (616 мг, 6,189 ммоль, 3,0 экв.) и 4,4,5,5-тетраметил-1,3,2-диоксаборолан (779 мг, 6,189 ммоль, 3,0 экв.). Смесь перемешивали при 80°C в течение 16 часов. После охлаждения смесь выливали в воду (60 мл) и экстрагировали с помощью EA (60 мл × 2), а затем органические слои объединяли. Органический слой концентрировали с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (1,0 г, неочищенный продукт) в виде черно-коричневого масла, которое непосредственно применяли на следующей стадии.
Стадия D. В защитной атмосфере газообразного азота к перемешанному раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (500 мг, 1,1389 ммоль, 1,00 экв.) и 2-бром-5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридина (268 мг, 1,1389 ммоль, 1,00 экв.) в диоксане (10,00 мл) и воде (2,00 мл) добавляли Pd(dppf)Cl2·CH2Cl2 (91,9 мг, 112,5 мкмоль, 0,10 экв.) и K2CO3 (466 мг, 3,489 ммоль, 3,00 экв.). Смесь перемешивали при 80°C в течение 16 часов. После охлаждения смесь выливали в воду (80 мл) и экстрагировали с помощью EA (80 мл × 2). Органические слои объединяли и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = от 1/1 до 0/1). Получали N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)изоксазол-3-карбоксамид (430 мг, 915,7 мкмоль, выход 81,0%) в виде черно-коричневого твердого вещества.
Стадия E. При -78°C к раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)изоксазол-3-карбоксамида (560 мг, 1,1989 ммоль, 1,00 экв.) в DCM (15 мл) медленно добавляли BBr3 (1,49 г, 5,9689 ммоль, 5,0 экв.). Смесь перемешивали при 25°C в течение 16 часов, добавляли MeOH (20 мл) и концентрировали. Остаток очищали с помощью препаративной HPLC (система на основе муравьиной кислоты). Получали продукт, 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)изоксазол-3-карбоксамид (243 мг, 550,3 мкмоль, выход 46,1%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,73 (m, 2H), 8,91 (t, J = 2,0 Гц, 1H), 8,15 (s, 1H), 6,91 (s, 1H), 6,80 (s, 1H), 6,45 (s, 1H), 3,27-3,24 (m, 2H), 3,07-3,04 (m, 1H), 2,72-2,70 (m, 2H), 2,65-2,63 (m, 2H), 2,34 (s, 3H), 1,16-1,04 (s, 9H).
Пример 10
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(5-метил-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-2-ил)изоксазол-3-карбоксамид
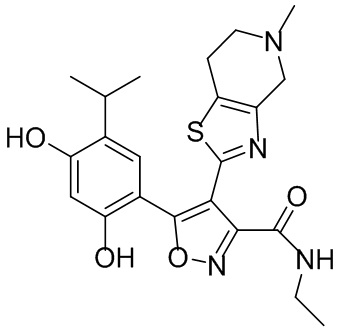
Реакционная схема
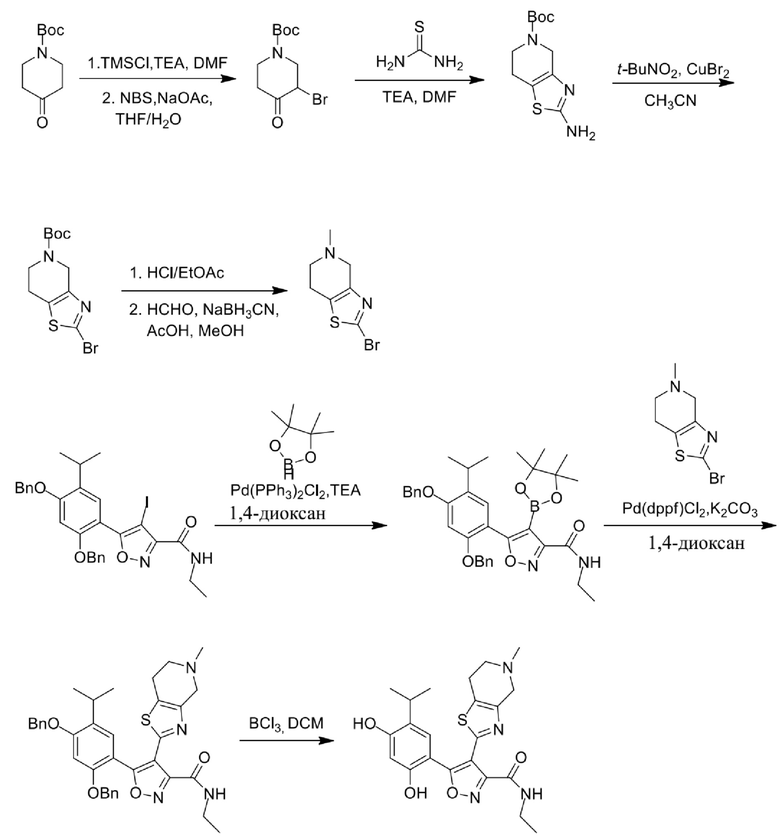
Стадия A. К раствору трет-бутил-4-оксопиперидин-1-карбоксилата (10,0 г, 50,289 ммоль, 1,0 экв.) в DMF (100,00 мл) добавляли TEA (10,2 г, 100,489 ммоль, 2,0 экв.). Затем к раствору по каплям добавляли TMSCl (6,27 г, 57,789 ммоль, 1,15 экв.) при 20°C и атмосферу меняли на атмосферу азота. После перемешивания смеси при 80°C в течение 16 часов добавляли 300 мл насыщенного водного раствора NaHCO3. Смесь экстрагировали с помощью EA (150 мл × 3), а затем объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением трет-бутил-4-триметилсилил-3,6-дигидро-2H-пиридин-1-карбоксилата (9,2 г, выход 67%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 4,77 (brs, 1H), 3,85 (brs, 2H), 3,5 (t, J = 5,6 Гц, 2H), 2,08 (brs, 2H), 1,45 (s, 9H), 0,17 (s, 9H).
Стадия B. При 0°C к перемешанному раствору трет-бутил-4-триметилсилил-3,6-дигидро-2H-пиридин-1-карбоксилата (13,9 г, 51,289 ммоль, 1,0 экв.) в THF (150 мл) и воде (150 мл) добавляли NaOAc (420 мг, 5,189 ммоль, 0,1 экв.) и NBS (13,67 г, 76,889 ммоль, 1,5 экв.). Смесь перемешивали при 20°C в течение 12 часов. Реакционную смесь гасили с помощью 100 мл насыщенного водного раствора тиосульфата натрия, а затем промывали и нейтрализовали с помощью 200 мл насыщенного водного раствора NaHCO3. Смесь экстрагировали с помощью EA (2 × 500 мл), и объединенную органическую фазу высушивали с помощью безводного Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением трет-бутил-3-бром-4-оксопиперидин-1-карбоксилата (7 г, выход 49%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 4,28-4,31 (m, 1H), 3,96 (m, 2H), 3,57-3,77 (m, 2H), 2,97-3,00 (m, 1H), 2,40-2,46 (m, 1H), 1,48 (s, 9H).
Стадия C. К раствору трет-бутил-3-бром-4-оксопиперидин-1-карбоксилата (7,0 г, 25,289 ммоль, 1,0 экв.) в DMF (70 мл) добавляли TEA (7,64 г, 75,589 ммоль, 3,0 экв.) и тиомочевину (2,11 г, 27,789 ммоль, 1,1 экв.). Смесь перемешивали при 110°C в течение 12 часов, а затем выливали в 300 мл воды с последующей фильтрацией твердого вещества. Твердое вещество промывали с помощью 100 мл воды и получали 4,5 г красного неочищенного продукта, трет-бутил-2-амино-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-5-формиата. Неочищенный продукт непосредственно применяли на следующей стадии. MS (ESI) масса/заряд: 256,0 (M+1).
Стадия D. При 0°C к раствору трет-бутил-2-амино-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-5-формиата (4,70 г, 18,489 ммоль, 1,0 экв.) в CH3CN (50 мл) добавляли CuBr2 (4,52 г, 20,389 ммоль, 1,1 экв.) и трет-бутилнитрит (2,09 г, 20,389 ммоль, 1,1 экв.). Смесь перемешивали при 0°C в течение 1 часа, гасили путем добавления 3 мл 0,5 моль/л раствора HCl, а затем нейтрализовали путем добавления 50 мл насыщенного водного раствора NaHCO₃. Смесь фильтровали и фильтрат экстрагировали с помощью EA (50 мл × 2), высушивали с помощью безводного сульфата натрия и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (PE:EA = 5:1) с получением трет-бутил-2-бром-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-5-карбоксилата (1,7 г, выход 29%) в виде белого твердого продукта. MS (ESI) масса/заряд: 318,9, 320,9 (M+1, M+3).
Стадия E. К хлористоводородной кислоте/этилацетату (4 н., 30 мл) добавляли трет-бутил-2-бром-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-5-карбоксилат (1,70 г, 5,3389 ммоль, 1,00 экв.), и смесь перемешивали при 20°C в течение 2 часов. Смесь концентрировали в вакууме с получением 1,35 г неочищенного продукта, 2-бром-4,5,6,7-тетрагидротиазоло[4,5-c]пиридина в виде белого твердого вещества, которое непосредственно применяли на следующей стадии. 1H ЯМР (400 МГц, DMSO-d6) δ 9,88 (brs, 2H), 4,31 (s, 2H), 3,39-3,41 (m, 2H), 2,99 (t, J = 6,0 Гц, 2H).
Стадия F. К раствору 2-бром-4,5,6,7-тетрагидротиазоло[4,5-c]пиридина (550,00 мг, 2,1589 ммоль, 1,00 экв.) в MeOH (10,00 мл) добавляли формальдегид (194 мг, 6,4689 ммоль, 3,00 экв.) и AcOH (258 мг, 4,389 ммоль, 2,0экв.). Смесь перемешивали при 20°C в течение 1 часа, а затем добавляли NaBH3CN (406 мг, 6,589 ммоль, 3,0 экв.), и смесь дополнительно перемешивали при 20°C в течение 4 часов. Реакционную смесь гасили с помощью 0,5 моль/л раствора HCl (2 мл) с последующим добавлением насыщенного раствора NaHCO3 (20 мл) для нейтрализации. Затем смесь экстрагировали с помощью EA (20 мл × 2), и объединенную органическую фазу высушивали с помощью безводного Na2SO4, фильтровали и концентрировали в вакууме с получением продукта, 2-бром-5-метил-4,5,6,7-тетрагидротиазоло[4,5-c]пиридина (490 мг, выход 97%) в виде белого твердого вещества. MS (ESI) масса/заряд: 232,8, 234,8 (M+1, M+3).
Стадия G. В защитной атмосфере газообразного азота к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-йодизоксазол-3-карбоксамида (1,00 г, 1,6889 ммоль, 1,0 экв.) в 1,4-диоксане (15 мл) добавляли TEA (510 мг, 5,0489 ммоль, 3,0 экв.) и Pd(PPh3)2Cl2 (117,9 мг, 168,00 мкмоль, 0,1 экв.). Наконец добавляли раствор 4,4,5,5-тетраметил-1,3,2-диоксаборолана (1,08 г, 8,489 ммоль, 5,0 экв.) и смесь перемешивали при 90°C в течение 12 часов. Реакционную смесь разбавляли с помощью EA (20 мл) и фильтровали через диатомовую землю. Фильтрат промывали с помощью 30 мл воды и органическую фазу высушивали с помощью безводного Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта, 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (1,7 г, чистота 60% согласно LCMS) в виде оранжевого геля, и неочищенный продукт непосредственно применяли на следующей стадии. MS (ESI) масса/заряд: 597,3 (M+1).
Стадия H. К перемешанному раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (800 мг, 1,3489 ммоль, 1,0 экв.) в 1,4-диоксане (20 мл) и воде (4 мл) добавляли 2-бром-5-метил-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин (313 мг, 1,3489 ммоль, 1,0 экв.) и K2CO3 (371 мг, 2,789 ммоль, 2,0 экв.). Обеспечивали прохождение реакции замещения в указанной смеси в атмосфере азота, а затем добавляли Pd(dppf)Cl2 (196 мг, 268,2 мкмоль, 0,2 экв.). Смесь перемешивали при 95°C в течение 8 часов и реакционную смесь разбавляли с помощью 20 мл EA и фильтровали через диатомовую землю. Фильтрат экстрагировали с помощью EA (20 мл × 2) и объединенную органическую фазу высушивали с помощью безводного раствора Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (DCM:метанол = 20:1) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(5-метил-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-2-ил)изоксазол-3-карбоксамида (60 мг, выход 7,2%), который представлял собой твердый продукт. MS (ESI) масса/заряд: 623,3 (M+1).
Стадия I. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-диэтил-4-(5-метил-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-2-ил)изоксазол-3-карбоксамида (60 мг, 96,3 мкмоль, 1,0 экв.) в DCM (2 мл) добавляли BCl3 (34 мг, 289 мкмоль, 3,0 экв.). После перемешивания смеси при 0°C в течение 1 часа для гашения добавляли 2 мл MeOH и смесь выливали в 5 мл насыщенного водного раствора NaHCO3. Затем смесь экстрагировали с помощью DCM (5 мл × 3) и объединенную органическую фазу высушивали с помощью безводного Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (муравьиная кислота, колонка: Welch Ultimate AQ-C18, 150 × 30 мм × 5 мкм, условия: 0,225% FA-ACN, расход: 25) с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(5-метил-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин-2-ил)изоксазол-3-карбоксамида (17 мг, выход 34%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,93 (brs, 2H), 9,09 (brs, 1H), 7,07 (s, 1H), 6,47 (s, 1H), 3,54 (s, 2H), 3,26 (m, 2H), 3,07 (m, 1H), 2,71 (brs, 4H), 2,35 (s, 3H), 1,07-1,12 (m, 9H).
Пример 11
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)изоксазол-3-карбоксамид
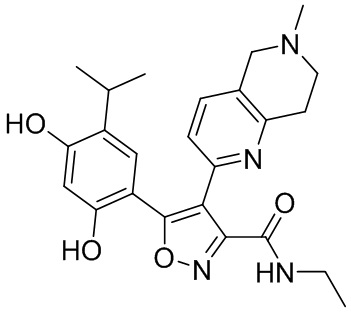
Реакционная схема
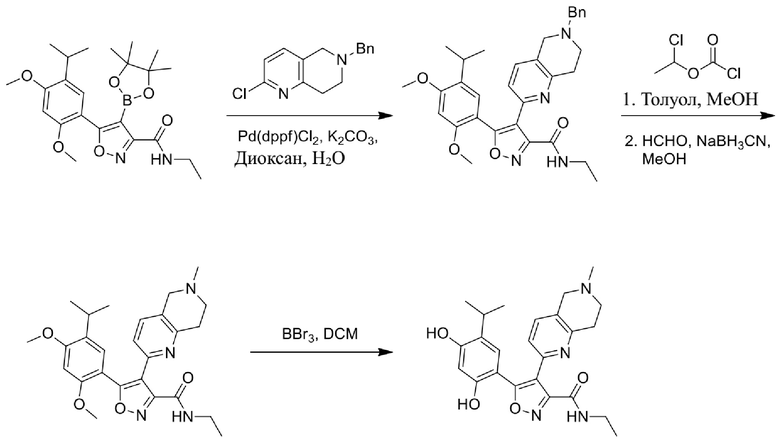
Стадия A. К перемешанному раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (700 мг, 1,5889 ммоль, 1,0 экв.) и 6-бензил-2-хлор-5,6,7,8-тетрагидро-1,6-нафтиридина (409 мг, 1,5889 ммоль, 1,0 экв.) в диоксане (3 мл) и воде (500 мкл) добавляли Pd(dppf)Cl2 (115 мг, 157,5 мкмоль, 0,1 экв.) и K2CO3 (435 мг, 3,289 ммоль, 2,0 экв.). После перемешивания смеси при 80°C в течение 16 часов смесь выливали в воду (80 мл) и экстрагировали с помощью EA (60 мл × 2). Объединенный органический слой высушивали с помощью сульфата натрия, фильтровали и концентрировали и полученный в результате неочищенный продукт очищали с помощью хроматографии на силикагеле (PE/EA = от 1:1 до EA) с получением 4-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-N-этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (250 мг, 462,4 мкмоль, выход 29,3%) в виде желтого твердого вещества.
Стадия B. При комнатной температуре к раствору 4-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-N-этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (250 мг, 462,4 мкмоль, 1,00 экв.) в толуоле (8 мл) добавляли 1-хлорэтилкарбонилхлорид (264 мг, 1,8589 ммоль, 4,0 экв.), а затем смесь перемешивали при 110°C в течение 8 часов. Смесь концентрировали и остаток растворяли в MeOH (8 мл). Смесь нагревали до 80°C и перемешивали в течение 12 часов. Смесь концентрировали с получением неочищенного продукта, N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)изоксазол-3-карбоксамида (200,00 мг, неочищенный продукт) в виде желтого твердого вещества, которое непосредственно применяли на следующей стадии.
Стадия C. К раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)изоксазол-3-карбоксамида (200 мг, 443,9 мкмоль, 1,0 экв.) в DCE (8 мл) добавляли параформальдегид (200 мг, 2,289 ммоль, 5,0 экв.), тетраизопропоксид титана (126 мг, 443,9 мкмоль, 1,0 экв.), уксусную кислоту (27 мг, 444 мкмоль, 1,0 экв.) и молекулярные сита с размером пор 4Å (300 мг, 444 мкмоль, 1,0 экв.). Смесь перемешивали при 25°C в течение 8 часов, а затем добавляли NaBH3CN (56 мг, 887,8 мкмоль, 2,0 экв.). Смесь перемешивали при 25°C в течение дополнительных 12 часов. Смесь фильтровали, фильтрат концентрировали, и неочищенный продукт очищали с применением пластинки для тонкослойной хроматографии (DCM/MeOH = 10:1) с получением N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(6-метил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)изоксазол-3-карбоксамида (120 мг, 258,3 мкмоль, выход 58,2%) в виде желтого твердого вещества.
Стадия D. При 0°C к раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)изоксазол-3-карбоксамида (60 мг, 129,2 мкмоль, 1,0 экв.) в DCM (8 мл) медленно добавляли BBr3 (323,6 мг, 1,389 ммоль, 10,0 экв.). Смесь перемешивали при 25°C в течение 16 часов и смесь медленно добавляли к MeOH (15 мл) с последующим концентрированием смеси. Неочищенный продукт очищали с помощью препаративной HPLC (Phenomenex Synergi Max-RP 250 × 80, 10 мкм, 0,225% FA-ACN) с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)изоксазол-3-карбоксамида (3,1 мг, 6,4 мкмоль, выход 5,0%). 1H ЯМР (300 МГц, DMSO-d6) δ 8,99-8,95 (m, 1H), 8,24 (s, 1H), 7,44 (d, J = 8,1 Гц, 1H), 7,03 (d, J = 7,8 Гц, 1H), 6,98 (s, 1H), 6,43 (s, 1H), 3,26-3,19 (m, 4H), 3,09-3,05 (m, 1H), 2,84-2,81 (m, 2H), 2,72-2,68 (m, 2H), 2,35 (s, 3H), 1,13-1,03 (m, 9H).
Пример 12
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
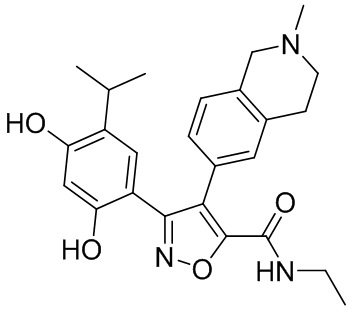
Реакционная схема
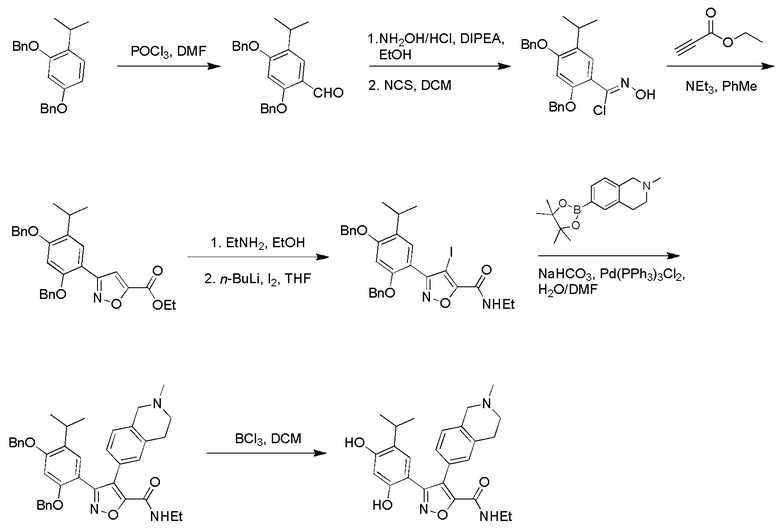
Стадия A. При 0°C к DMF (60 мл) добавляли POCl3 (6,25 г, 40,889 ммоль, 1,5 экв.) и перемешивали при 0°C в течение 10 мин. Затем при 0°C добавляли 2,4-дибензилокси-1-изопропилбензол (9,0 г, 27,189 ммоль, 1,0 экв.) и после добавления продолжали перемешивание при 0°C в течение 10 мин. Температуру повышали до 15-25°C и после перемешивания в течение 10 мин. температуру дополнительно повышали до 100°C при перемешивании в течение 2,5 часов. Реакционную смесь охлаждали до 15-25°C, выливали в ледяную воду (120 мл), доводили до pH = 6 путем добавления 10% водного раствора NaOAc, а затем экстрагировали с помощью EA (120 мл × 3). Объединенный органический слой высушивали, фильтровали и концентрировали с получением 2,4-дибензилокси-5-изопропилбензальдегида (9,70 г, неочищенный продукт) в виде желтого твердого вещества, которое непосредственно применяли на следующей стадии.
Стадия B. К раствору 2,4-дибензил-5-изопропилбензальдегида (9,5 г, 26,489 ммоль, 1,0 экв.), растворенному в EtOH (100 мл), добавляли NH2OH·HCl (3,66 г, 52,789 ммоль, 2,0 экв.) при 15-25°C с последующим добавлением DIEA (5,11 г, 39,589 ммоль, 1,5 экв.), а затем температуру повышали до 80°C при перемешивании в течение 16 часов. Реакционную смесь охлаждали до 15-25°C и концентрировали в вакууме с получением остатка. Остаток растворяли в EA (80 мл) и промывали водой (80 мл). Органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле (PE:EA = от 10:1 до 5:1). Получали оксим (1E)-2,4-дибензил-5-изопропилбензальдегида (7,00 г, 18,689 ммоль, выход 70,7%) в виде желтого твердого вещества.
Стадия C. При 0-5°C к раствору оксима (1E)-2,4-дибензил-5-изопропилбензальдегида (3,00 г, 7,9989 ммоль, 1,0 экв.) в DCM (30,00 мл) добавляли NCS (1,28 г, 9,5989 ммоль, 1,2 экв.) при перемешивании при 0-5°C в течение 2 часов, а затем перемешивали при 15-25°C в течение дополнительных 16 часов. Реакционную смесь концентрировали с получением 2,4-дибензилокси-N-гидрокси-5-изопропилбензимидоилхлорида (4,0 г, неочищенный продукт) в виде желтого масла. Неочищенный продукт непосредственно применяли на следующей стадии.
Стадия D. При 15-25°C к раствору 2,4-дибензилокси-N-гидрокси-5-изопропилбензимидоилхлорида (1,64 г, 4,0089 ммоль, 1,0 экв.) в толуоле (15,00 мл) добавляли этил-2-пропиолат (588,60 мг, 6,0089 ммоль, 1,50 экв.) с последующим добавлением по каплям TEA (445,24 мг, 4,4089 ммоль, 1,10 экв.) при 15-25°C в течение 0,5 часа. После добавления смесь перемешивали при 15-25°C в течение 0,5 часа, а затем перемешивали при 80°C в течение 3 часов. Реакционную смесь охлаждали до 15-25°C, выливали в воду (20 мл) и экстрагировали с помощью EA (20 мл × 2). Объединенный органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле (PE:EA = от 20:1 до 5:1). Получали этил-3-(2,4-дибензилокси-5-изопропилфенил)изоксазол-5-карбоксилат (1,20 г, неочищенный продукт) в виде желтого твердого вещества.
Стадия E. При 15-25°C к раствору этил-3-(2,4-дибензилокси-5-изопропилфенил)изоксазол-5-карбоксилата (600 мг, 1,2789 ммоль, 1,0 экв.), растворенного в EtOH (10,00 мл), добавляли этиламин (573,6 мг, 12,789 ммоль, 10,0 экв.) при перемешивании при 80°C в течение 16 часов. Реакционную смесь охлаждали до 15-25°C и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле (PE:EA = от 10:1 до 3:1), получали 3-(2,4-дибензилокси-5-изопропилфенил)-N-этилизоксазол-5-карбоксамид (150 мг, 318,8 мкмоль, выход 25,1%) в виде желтого твердого вещества.
Стадия F. При -78°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этилизоксазол-5-карбоксамида (50 мг, 106,3 мкмоль, 1,0 экв.) в THF (2 мл) добавляли н-бутиллитий (2 M, 132 мкл, 2,5 экв.) при перемешивании при -78°C в течение 1 часа после добавления с последующим добавлением раствора I2 (40,5 мг, 159,4 мкмоль, 1,5 экв.) в THF (1 мл) при -78°C и непрерывном перемешивании при -78°C в течение 1 часа. Реакционный раствор нагревали до 15-25°C при перемешивании в течение 16 часов. Реакционную смесь выливали в насыщенный водный раствор NH4Cl (10 мл) и экстрагировали с помощью EA (10 мл × 3). Объединенный органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной TLC (PE:EA = 3:1) с получением 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-йодизоксазол-5-карбоксамида (15 мг, 25,2 мкмоль, выход 23,7%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 597 (M+1).
Стадия G. При 15-25°C в защитной атмосфере газообразного азота к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-йодизоксазол-5-карбоксамида (15 мг, 25,15 мкмоль, 1,00 экв.) в DMF (2,5 мл) добавляли 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин (13,7 мг, 50,3 мкмоль, 2,00 экв.), воду (500,00 мкл) и бикарбонат натрия (6,34 мг, 75,5 мкмоль, 3,0 экв.) с последующим добавлением Pd(PPh3)2Cl2 (3,5 мг, 5,0 мкмоль, 0,2 экв.). Смесь перемешивали при 80°C в течение 16 часов. Реакционную смесь охлаждали до 15-25°C, выливали в воду (10 мл) и экстрагировали с помощью EA (10 мл × 3). Объединенный органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной TLC (DCM:метанол = 15:1). Получали 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид (10,00 мг, 16,24 мкмоль, выход 64,57%) в виде желтого твердого вещества. (ESI) масса/заряд: 616 (M+1).
Стадия H. При 0°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (10 мг, 16,2 мкмоль, 1,0 экв.) в DCM (1 мл) добавляли раствор BCl3 (1 M, 324,8 мкл, 20,0 экв.) в DCM и после перемешивания при 0°C в течение 2 часов температуру повышали до 15-25°C при перемешивании в течение 1 часа. Реакционную смесь охлаждали до 0°C, добавляли MeOH (2 мл) и перемешивали при 0°C в течение 0,5 часа, а затем при 15-25°C в течение дополнительного 0,5 часа. Смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (0,225% FA-ACN; Phenomenex Synergi Max-RP 250 × 80, 10 мкм) с получением 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (3,5 мг, 8,0 мкмоль, выход 49,5%). 1H ЯМР (400 МГц, DMSO) δ 8,87 (t, J = 5,6 Гц, 1H), 7,00-6,98 (m, 1H), 6,95-6,90 (m, 2H), 6,79 (s, 1H), 6,33 (s, 1H), 3,26-3,19 (m, 2H), 3,06-2,98 (m, 1H), 2,69-2,66 (m, 2H), 2,55-2,50 (m, 4H), 2,31 (s, 3H), 1,07 (t, J = 7,2 Гц, 3H), 1,01 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 436 (M+1).
Пример 13
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изотиазол-3-карбоксамид
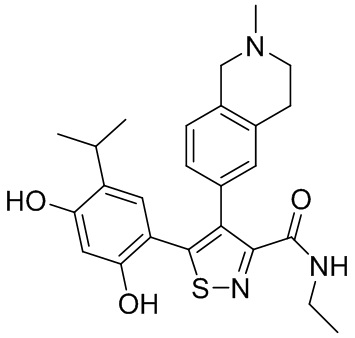
Реакционная схема
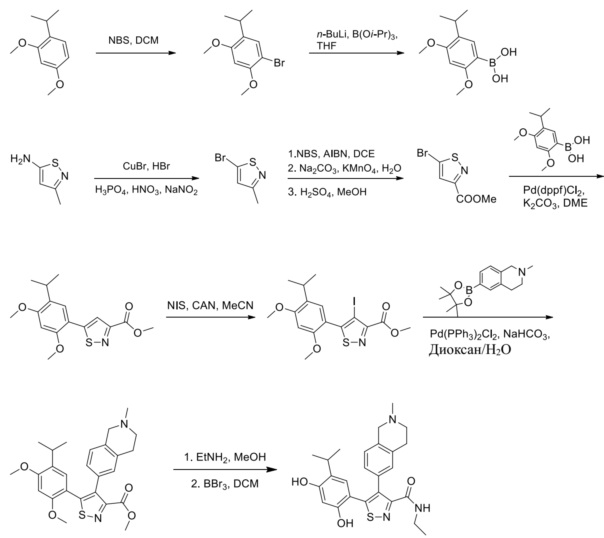
Стадия A. При 0°C к раствору 1-изопропил-2,4-диметоксибензола (2,0 г, 11,189 ммоль, 1,0 экв.) в DCM (40 мл) добавляли NBS (2,21 г, 12,4289 ммоль, 1,12 экв.). Смесь перемешивали при 0°C в течение 3 часов, а затем перемешивали при 25°C в течение 1 часа. Смесь концентрировали и остаток очищали с помощью колоночной хроматографии (PE/EA = 10/1) с получением 1-бром-5-изопропил-2,4-диметоксибензола (2,77 г, 10,6989 ммоль, выход 96,30%) в виде желтого твердого вещества.
Стадия B. При -78°C к раствору 1-бром-5-изопропил-2,4-диметоксибензола (2,77 г, 10,6989 ммоль, 1,0 экв.) в безводном THF (100 мл) добавляли n-BuLi (2,5 M, 6,00 мл, 1,40 экв.). Смесь перемешивали при -78°C в течение 1 часа. Затем медленно добавляли раствор триизопропилбората (6,03 г, 32,0789 ммоль, 3,0 экв.) в безводном THF (10 мл) и температуру поддерживали на уровне -78°C. После добавления реакционную смесь перемешивали при 25°C в течение 4 часов. Смесь выливали в ледяную воду и доводили до pH 3-4 с помощью 1 н. хлористоводородной кислоты. Полученную в результате смесь экстрагировали три раза с помощью EA и объединенный органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток перекристаллизовывали в PE с получением целевого продукта, (5-изопропил-2,4-диметоксифенил)бороновой кислоты (1,60 г, 7,1489 ммоль, выход 66,8%) в виде белого твердого вещества. MS (ESI) масса/заряд: 225,2 (M+H).
Стадия C. К EA (100 мл) добавляли гидрохлорид 3-метилизохинолин-5-амина (8,00 г, 53,1189 ммоль, 1,00 экв.) и промывали 10% водным раствором карбоната натрия. Органическую фазу высушивали и концентрировали с получением свободного основания и свободное основание растворяли в фосфорной кислоте (20 мл) и охлаждали до 0°C. Смесь по каплям добавляли к азотной кислоте (10 мл) с последующим добавлением по каплям насыщенного водного раствора нитрита натрия (4,17 г, 60,489 ммоль, 1,14 экв.), во время которого температуру поддерживали на уровне 0-5°C. Реакционную смесь перемешивали при 10°C в течение 30 мин., а затем по каплям добавляли в 48% водный раствор HBr (100 мл) с растворенным бромидом меди (9,50 г, 66,289 ммоль, 1,25 экв.) и полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор доводили до pH 6-7 с помощью 4 н. водного раствора NaOH и воды (200 мл). Водную фазу экстрагировали с помощью MTBE и органический слой высушивали над безводным сульфатом натрия и концентрировали с получением 5-бром-3-метилизотиазола (8,0 г, неочищенный продукт), который непосредственно применяли на следующей стадии.
Стадия D. Смесь 5-бром-3-метилизотиазола (8,0 г, 44,989 ммоль, 1,0 экв.), NBS (16,0 г, 89,989 ммоль, 2,0 экв.) и AIBN (1,40 г, 8,5389 ммоль, 0,19 экв.) в DCE (150 мл) нагревали до 90°C и подвергали воздействию облучения путем помещения под галогенную лампу на 150 Ватт при перемешивании в течение 48 часов. Смесь промывали насыщенным водным раствором NaHSO3 и экстрагировали три раза с помощью DCM. Объединенный органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии (PE/DCM = от 100/0 до 1/2) с получением целевого продукта, 5-бром-3-(бромметил)изотиазола (4,60 г, 17,989 ммоль, выход 39,8%).
Стадия E. Смесь 5-бром-3-(бромметил)изотиазола (4,10 г, 15,9689 ммоль, 1,0 экв.) и карбоната натрия (1,92 г, 18,1189 ммоль, 1,14 экв.) в воде (90 мл) нагревали с обратным холодильником, а затем несколькими небольшими порциями добавляли перманганат калия (3,28 г, 20,7689 ммоль, 1,3 экв.). Реакционную смесь перемешивали с применением обратного холодильника в течение 1 часа, охлаждали и фильтровали. Кислотность фильтрата повышали с помощью 1 н. HCl и его экстрагировали три раза с помощью EA. Объединенный органический слой высушивали над безводным сульфатом натрия и концентрировали с получением 5-бромизотиазол-3-карбоновой кислоты (1,26 г, 6,0689 ммоль, выход 38,0%) в виде белого твердого вещества.
Стадия F. К раствору 5-бромизотиазол-3-карбоновой кислоты (1,26 г, 6,0689 ммоль, 1,0 экв.) в MeOH (50 мл) добавляли серную кислоту (0,5 мл). Реакционную смесь нагревали с обратным холодильником при 65°C в течение 16 часов. Смесь охлаждали до комнатной температуры и гасили насыщенным водным раствором NaHCO3 и водный слой экстрагировали с помощью EA. Органический слой высушивали над безводным сульфатом натрия и концентрировали с получением метил-5-бромизотиазол-3-карбоксилата (1,30 г, 5,8589 ммоль, выход 96,6%) в виде желтого масла.
Стадия G. К перемешанному раствору DME (30 мл) и воды (0,12 мл) добавляли метил-5-бромизотиазол-3-карбоксилат (1,00 г, 4,5089 ммоль), (5-изопропил-2,4-диметоксифенил)бороновую кислоту (1,20 г, 5,3689 ммоль, 1,19 экв.), Pd(dppf)Cl2 (340,00 мг, 464,67 мкмоль, 0,10 экв.) и K2CO3 (1,29 г, 9,3389 ммоль, 2,07 экв.). В атмосфере азота реакционный раствор перемешивали при 100°C в течение 16 часов. Смесь фильтровали через диатомовую землю и фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии (PE/EA = от 6/1 до 3/1) с получением целевого продукта, метил-5-(5-изопропил-2,4-диметоксифенил)изотиазол-3-карбоксилата (1,10 г, 3,4289 ммоль, выход 76,1%) в виде белого твердого вещества. MS (ESI) масса/заряд: 322,1 (M + H).
Стадия H. К MeCN (20 мл) добавляли метил-5-(5-изопропил-2,4-диметоксифенил)изотиазол-3-карбоксилат (300 мг, 933 мкмоль, 1,0 экв.), NIS (24 мг, 1,0789 ммоль, 1,14 экв.) и CAN (55 мг, 100,3 мкмоль, 0,11 экв.). Смесь перемешивали при 82°C в течение 16 часов. Смесь концентрировали. Остаток растворяли в DCM и промывали водой и солевым раствором. Органическую фазу высушивали над безводным сульфатом натрия и концентрировали. Остаток очищали с помощью препаративной TLC (PE/EA = 3/1) с получением метил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изотиазол-3-карбоксилата (300 мг, 670,7 мкмоль, выход 71,9%) в виде желтого масла. MS (ESI) масса/заряд: 448,0 (M+H).
Стадия I. В защитной атмосфере газообразного азота к перемешанному раствору диоксана (10 мл) и H2O (1 мл) добавляли метил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изотиазол-3-карбоксилат (120 мг, 268,3 мкмоль, 1,00 экв.), 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетраизохинолин (100 мг, 366,1 мкмоль, 1,36 экв.), Pd(PPh3)2Cl2 (30 мг, 57 мкмоль, 0,21 экв.) и NaHCO3 (50 мг, 595,2 мкмоль, 2,22 экв.). Смесь перемешивали при 80°C в течение 16 часов. После охлаждения смесь фильтровали через подушку из диатомовой земли и фильтрат концентрировали. Остаток очищали с помощью препаративной TLC (DCM/метанол = 20/1) с получением целевого продукта, метил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изотиазол-3-карбоксилата (80 мг, 171,5 мкмоль, выход 63,9%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 467,2 (M+H).
Стадия J. К MeOH (10 мл) добавляли метил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изотиазол-3-карбоксилат (80 мг, 171,5 мкмоль, 1,0 экв.) и этиламин (1 мл, 15,389 ммоль, 89 экв.). Смесь перемешивали при 65°C в течение 16 часов. Смесь концентрировали в вакууме с получением целевого продукта, N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изотиазол-3-карбоксамида (82 мг, 171 мкмоль, выход 99,7%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 480,2 (M+H).
Стадия K. При -78°C к раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изотиазол-3-карбоксамида (80 мг, 166,8 мкмоль, 1,0 экв.) в DCM (1 мл) добавляли BBr3 (1 мл, 10,489 ммоль, 62 экв.). Смесь перемешивали при 25°C в течение 2 часов. К смеси добавляли 0,1 мл воды и 1 г NaHCO3 в виде твердого вещества и смесь перемешивали при 25°C в течение 10 мин. Смесь фильтровали и остаток очищали с помощью препаративной HPLC с получением целевого продукта, 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изотиазол-3-карбоксамида (16 мг, 35,4 мкмоль, выход 21,2%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,20 (brs, 1H), 7,06 (d, J = 7,6 Гц, 1H), 6,94-6,91 (m, 2H), 6,44 (s, 1H), 6,06 (brs, 1H), 3,52 (s, 2H), 3,13-3,07 (m, 3H), 2,82-2,78 (m, 4H), 2,59 (brs, 2H), 2,36 (s, 3H), 1,00 (t, J = 7,2 Гц, 3H), 0,67 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 452,2 (M+H).
Пример 14
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,3a,4,6a-гексагидроциклопента[c]пиррол-5-ил)изоксазол-3-карбоксамид
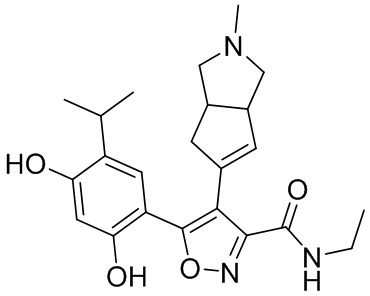
Реакционная схема
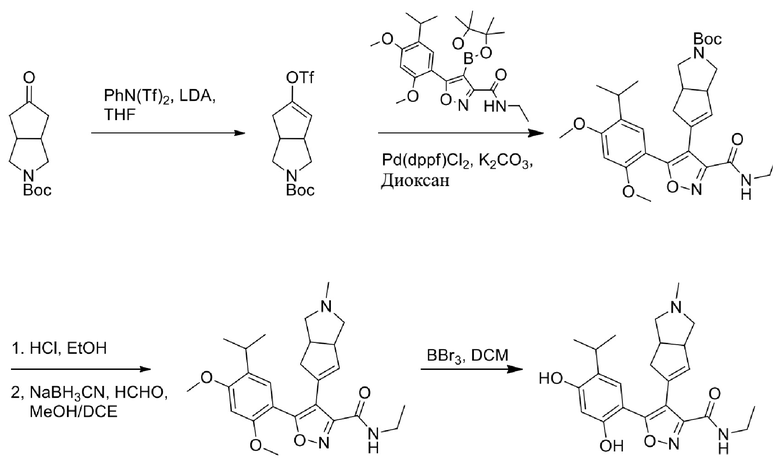
Стадия A. При -78°C к раствору трет-бутил-5-оксо-1,3,3a,4,6,6a-гексагидроциклопента[c]пиррол-2-карбоксилата (226 мг, 1,089 ммоль, 1,0 экв.) в безводном THF (20 мл) добавляли LDA (2 M, 1,0 мл, 2,0 экв.). Полученную в результате смесь перемешивали при -78°C в течение 1 часа и дополнительно добавляли раствор N,N-бис(трифторметансульфонил)анилина (467 мг, 1,289 ммоль, 1,2 экв.) в безводном THF (20 мл). Затем смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Смесь концентрировали до сухого состояния и остаток растворяли в DCM, затем промывали насыщенным водным раствором NaHCO3, высушивали над безводным MgSO4 и концентрировали с получением трет-бутил-5-(трифторметилсульфонилокси)-3,3a,6,6-тетрагидро-1H-циклопента[c]пиррол-2-карбоксилата (360 мг, неочищенный продукт) в виде коричневого масла, которое непосредственно применяли на следующей стадии.
Стадия B. В защитной атмосфере газообразного азота к перемешанному раствору диоксана (20 мл) и воды (2 мл) добавляли трет-бутил-5-(трифторметилсульфонилокси)-3,3a,6,6-тетрагидро-1H-циклопента[c]пиррол-2-карбоксилат (350 мг, 979,43 мкмоль, 1,0 экв.), N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамид (440 мг, 990,3 мкмоль, 1,01 экв.), Pd(dppf)Cl2 (140 мг, 191,3 мкмоль, 0,2 экв.) и K2CO3 (280 мг, 2,0389 ммоль, 2,07 экв.). Смесь перемешивали при 80°C в течение 16 часов. После охлаждения смесь фильтровали через диатомовую землю и фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии (PE/EA = от 5/1 до 1/1) с получением целевого продукта, трет-бутил-5-[3-(этилкарбамоил)-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-3,3a,6,6a-тетрагидро-1H-циклопента[c]пиррол-2-карбоксилата (290 мг, 551,7 мкмоль, выход 56,3%) в виде грязно-белого твердого вещества. MS (ESI) масса/заряд: 426,2 (M-99).
Стадия C. К раствору трет-бутил-5-[3-(этилкарбамоил)-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-3,3a,6,6a-тетрагидро-1H-циклопента[c]пиррол-2-карбоксилата (290 мг, 551,7 мкмоль, 1,0 экв.) в EA (10 мл) добавляли хлористоводородную кислоту/сложный этиловый эфир (4 н., 1 мл). Смесь перемешивали при 25°C в течение 2 часов. Смесь концентрировали с получением целевого продукта, 4-(1,2,3,3a,4,6a-гексагидроциклопента[c]пиррол-5-ил)-N-этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (234 мг, 549,9 мкмоль, выход 99,7%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 426,2 (M+H).
Стадия D. К перемешанному раствору 4-(1,2,3,3a,4,6a-гексагидроциклопента[c]пиррол-5-ил)-N-этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (234 мг, 549,9 мкмоль, 1,00 экв.) в DCE (12 мл) и MeOH (4 мл) добавляли формальдегид (500 мкл, 18,289 ммоль, 33,00 экв.). Смесь перемешивали при 25°C в течение 16 часов, а затем добавляли NaBH(OAc)3 (500 мг, 2,3689 ммоль, 4,3 экв.) и смесь дополнительно перемешивали при 25°C в течение 2 часов. Реакционную смесь гасили насыщенным водным раствором NaHCO3 и экстрагировали три раза с помощью DCM. Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной TLC (DCM/метанол = 10/1) с получением целевого продукта, N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,3a,4,6a-гексагидроциклопента[c]пиррол-5-ил)изоксазол-3-карбоксамида (160 мг, 364,0 мкмоль, выход 66,2%). MS (ESI) масса/заряд: 440,2 (M+H).
Стадия E. При -78°C к раствору N-этил-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,3a,4,6a-гексагидроциклопента[c]пиррол-5-ил)изоксазол-3-карбоксамида (160 мг, 364,0 мкмоль, 1,0 экв.) в DCM (1 мл) добавляли BBr3 (1 мл, 10,489 ммоль, 28,5 экв.). Смесь перемешивали при 25°C в течение 2 часов, к смеси добавляли 0,1 мл воды и 1 г NaHCO3 и полученную в результате смесь перемешивали при 25°C в течение 10 мин. Смесь фильтровали и остаточную жидкость очищали с помощью препаративной HPLC (система на основе муравьиной кислоты) с получением целевого продукта, 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,3a,4,6a-гексагидроциклопента[c]пиррол-5-ил)изоксазол-3-карбоксамида (75,5 мг, 165,0 мкмоль, выход 45,3%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,82 (t, J = 5,2 Гц, 1H), 8,20 (s, 1H), 6,91 (s, 1H), 6,47 (s, 1H), 5,61 (d, J = 1,6 Гц, 1H), 3,28-3,23 (m, 4H), 3,11-3,09 (m, 1H), 2,75 (br, 1H), 2,63-2,59 (m, 3H), 2,31-2,26 (m, 5H), 2,15-2,13 (m, 1H), 1,11 (dd, J =7,2, 1,6 Гц, 9H). MS (ESI) масса/заряд: 412,2 (M+H).
Пример 15
4-Изопропил-6-[4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(5-метил-4H-1,2,4-триазол-3-ил)изоксазол-5-ил]бензол-1,3-диол
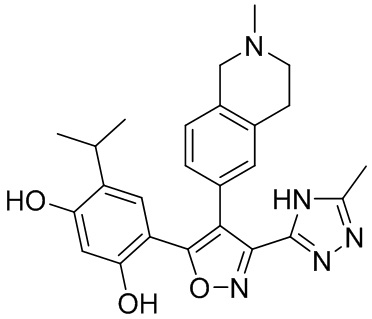
Реакционная схема
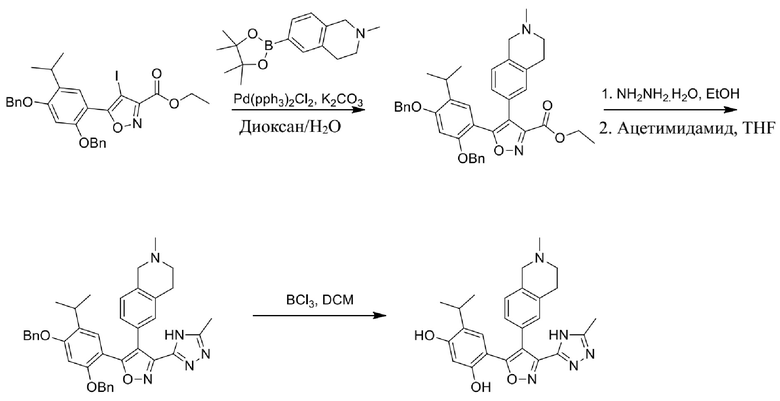
Стадия A. В защитной атмосфере газообразного азота к перемешанному раствору диоксана (30 мл) и воды (6 мл) добавляли 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин (3,66 г, 8,789 ммоль, 1,3 экв.), этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-3-карбоксилат (4,00 г, 6,789 ммоль, 1,0 экв.), Pd(PPh3)2Cl2 (470 мг, 670,0 мкмоль, 0,1 экв.) и K2CO3 (1,85 г, 13,489 ммоль, 2,0 экв.). Смесь нагревали до 80°C в защитной атмосфере N2 и перемешивали в течение 18 часов. После охлаждения реакционную смесь выливали в воду (15 мл) и экстрагировали этилацетатом (10 мл × 2). Органическую фазу промывали насыщенным солевым раствором (15 мл), высушивали над безводным Na2SO4 и концентрировали в вакууме с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат = от 50/1 до 5/1) с получением этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксилата (2,90 г, 3,589 ммоль, выход 52,6%, чистота 75%) в виде желтого масла.
Стадия B. К раствору этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксилата (2,90 г, 4,189 ммоль, 1,0 экв.) в EtOH (30 мл) добавляли гидрат гидразина (1,20 г, 24,189 ммоль, 5 экв.). Раствор нагревали до 90°C и перемешивали в течение 18 часов. Раствор концентрировали в вакууме, а затем добавляли воду (20 мл) и смесь экстрагировали с помощью EA (15 мл × 3). Органические слои объединяли, промывали солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (DCM/метанол = 60/1, 10/1) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбогидразида (1,50 г, 2,589 ммоль, выход 51,7%) в виде желтого твердого вещества.
Стадия C. К раствору 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбогидразида (1,50 г, 2,4989 ммоль, 1,00 экв.) и гидрохлорида ацетамидина (352,93 мг, 3,7389 ммоль, 1,50 экв.) в THF (4,00 мл) добавляли NaOH (149,32 мг, 3,7389 ммоль, 1,50 экв.). Раствор нагревали до 80°C и перемешивали в течение 18 часов. Раствор охлаждали, концентрировали и добавляли этиленгликоль (4,00 мл). Полученную смесь нагревали до 120°C и перемешивали в течение 2 часов. После охлаждения смеси добавляли воду (10 мл) и раствор экстрагировали с помощью EA (10 мл × 3). Органические слои объединяли, промывали солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (DCM/метанол = 60/1, 1/10) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(5-метил-4H-1,2,4-триазол-3-ил)изоксазола (1,10 г, 1,7689 ммоль, выход 70,6%) в виде желтого твердого вещества.
Стадия D. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(5-метил-4H-1,2,4-триазол-3-ил)изоксазола (120 мг, 191,77 мкмоль, 1,0 экв.) в DCM (2,0 мл) по каплям добавляли раствор BCl3 в DCM (1 M, 1,92 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., затем нагревали до 25°C и перемешивали в течение 2 часов. Смесь охлаждали до -78°C, гасили путем медленного добавления MeOH (2 мл), повышали ее основность до pH=8 с помощью насыщенного водного раствора NaHCO3, а затем экстрагировали с помощью DCM (10 мл × 3). Органические фазы объединяли, промывали солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной TLC (DCM/метанол = 5/1) с получением 4-изопропил-6-[4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(5-метил-4H-1,2,4-триазол-3-ил)изоксазол-5-ил]бензол-1,3-диола (30 мг, 67,3 мкмоль, выход 35,1%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,74 (s, 1H), 9,63 (s, 1H), 7,06-6,95 (m, 3H), 6,86 (s, 1H), 6,44 (s, 1H), 3,17 (d, J = 5,0 Гц, 1H), 3,06-2,95 (m, 1H), 2,77 (brs, 4H), 2,38 (s, 3H), 0,97 (d, J = 7,0 Гц, 7H).
Пример 16
4-(3-(5-Этил-1H-имидазол-2-ил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-ил)-6-изопропилбензол-1,3-диол
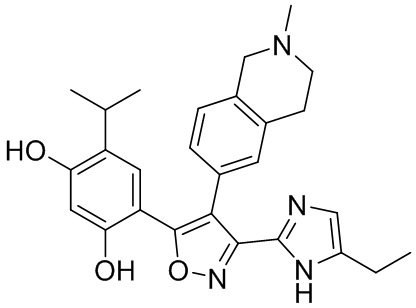
Реакционная схема
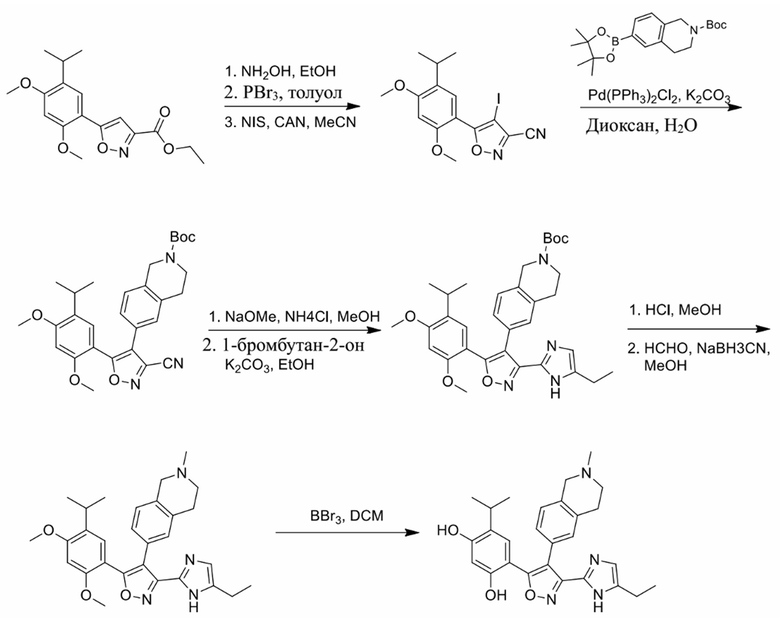
Стадия A. При 0°C к раствору NH2OH·HCl (1,16 г, 16,7489 ммоль, 100 экв.), растворенного в MeOH (6 мл), по каплям добавляли раствор KOH (1,41 г, 25,189 ммоль, 150 экв.) в MeOH (3 мл). Смесь перемешивали при 0°C в течение 30 мин., а затем твердое вещество отфильтровывали. К оставшемуся метанольному раствору добавляли этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-формиат (100,00 мг, 333,38 мкмоль, 1,0 экв.) и перемешивали при 5°C в течение 30 мин. Смесь доводили до pH 4 с помощью 1,2 M разбавленной хлористоводородной кислоты, концентрировали при пониженном давлении, добавляли воду и экстрагировали с помощью EA (10 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, N-этил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (97 мг, 330 мкмоль, выход 98%) в виде белого твердого вещества, которое непосредственно применяли на следующей стадии.
Стадия B. При 29°C в защитной атмосфере газообразного азота к раствору N-гидрокси-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (3,40 г, 11,189 ммоль, 1,0 экв.) в толуоле (80 мл) одной порцией добавляли PBr3 (6,01 г, 22,289 ммоль, 2,0 экв.). Смесь перемешивали при 29°C в течение 10 мин., а затем нагревали до 110°C при перемешивании в течение 8 часов. Смесь охлаждали до 29°C, а затем выливали в насыщенный водный раствор NaHCO3 (60 мл) при перемешивании в течение 5 мин. Водную фазу экстрагировали с помощью EA (100 мл × 3) и органические фазы объединяли, промывали солевым раствором (30 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA = от 10/1 до 5/1) с получением 5-(изопропил-2,4-диметоксифенил)изоксазол-3-карбонитрила (1,0 г, 3,6789 ммоль, выход 33,1%) в виде белого твердого вещества.
Стадия C. При 28°C в защитной атмосфере газообразного азота к раствору 5-(изопропил-2,4-диметоксифенил)изоксазол-3-карбонитрила (1,80 г, 6,689 ммоль, 1,0 экв.) в MeCN (32 мл) добавляли NIS (1,93 г, 8,5989 ммоль, 1,3 экв.) и CAN (362 мг, 661 мкмоль, 0,1 экв.). Смесь перемешивали при 28°C в течение 10 мин., а затем нагревали до 80°C при перемешивании в течение 4 часов. Смесь охлаждали до 28°C, а затем концентрировали при пониженном давлении. Остаток выливали в насыщенный водный раствор NaHCO3 (50 мл) и перемешивали в течение 5 мин. Водную фазу экстрагировали этилацетатом (5 мл × 3) и объединенную органическую фазу промывали насыщенным солевым раствором (10 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Полученный в результате неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA = от 20/1 до 10/1) с получением 4-йод-5-(изопропил-2,4-диметоксифенил)изоксазол-3-нитрила (1,30 г, 3,2689 ммоль, выход 49,4%) в виде бледно-желтого твердого вещества.
Стадия D. В защитной атмосфере газообразного азота к раствору трет-бутил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин-2-карбоксилата (2,30 г, 6,4089 ммоль, 1,0 экв.) в диоксане (30 мл) добавляли 4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-нитрил (2,04 г, 5,1289 ммоль, 0,80 экв.), Pd(PPh3)2Cl2 (314,45 мг, 448,00 мкмоль, 0,07 экв.) и K2CO3 (1,77 г, 12,8089 ммоль, 2,0 экв.). Смесь перемешивали при 80°C в течение 16 часов. Смесь выливали в воду (100 мл) и экстрагировали с помощью EA (100 мл × 2). Объединенный органический слой концентрировали и остаток очищали с помощью хроматографии на силикагеле (PE/EA = 5/1, 2/1) с получением трет-бутил-6-[3-циано-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата (2,00 г, 3,9789 ммоль, выход 62,1%) в виде черно-коричневого твердого вещества.
Стадия E. При 25°C к раствору трет-бутил-6-[3-циано-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата (2,00 г, 3,9789 ммоль, 1,0 экв.) в MeOH (15 мл) добавляли метоксид натрия (2,14 г, 39,789 ммоль, 10 экв.). Смесь перемешивали при 25°C в течение 2 часов, а затем добавляли хлорид аммония (2,12 г, 39,7189 ммоль, 10 экв.). Реакционную смесь перемешивали при 25°C в течение 14 часов. Смесь выливали в воду (80 мл) и экстрагировали с помощью EA (60 мл × 2) и органические слои объединяли и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA = 30/1) с получением трет-бутил-6-[3-формамидинил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата (1,50 г, 2,8889 ммоль, выход 72,6%) в виде белого твердого вещества.
Стадия F. К раствору трет-бутил-6-[3-формамидинил-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата (1,00 г, 1,9289 ммоль, 1,0 экв.) в EtOH (15 мл) добавляли K2CO3 (265 мг, 1,9289 ммоль, 1,0 экв.) и 1-бром-2-бутанон (290 мг, 1,9289 ммоль, 1,0 экв.). Смесь перемешивали при 80°C в течение 10 часов, выливали в воду (80 мл) и экстрагировали с помощью EA (60 мл × 2), и органические слои объединяли и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = 5/1, 2/1) с получением трет-бутил-6-[3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата (850 мг, 1,4889 ммоль, выход 77,3%) в виде белого твердого вещества.
Стадия G. Смесь трет-бутил-6-[3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата (600 мг, 1,0589 ммоль, 1,00 экв.) в HCl/MeOH (4 M, 15,00 мл) перемешивали при 25°C в течение 30 мин. Смесь концентрировали при 40°C с получением 3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазола (50 мг, 982,2 мкмоль, выход 93,6%) в виде желтого твердого вещества.
Стадия H. К раствору 3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазола (500 мг, 1,0689 ммоль, 1,0 экв.) в MeOH (8 мл) добавляли параформальдегид (476 мг, 5,2989 ммоль, 5,0 экв.) и AcOH (63,7 мг, 1,0689 ммоль, 1,0 экв.). Смесь перемешивали при 25°C в течение 2 часов, а затем добавляли NaBH3CN (133 мг, 2,1289 ммоль, 2,0 экв.). Реакционную смесь непрерывно перемешивали при 25°C в течение 14 часов, смесь выливали в воду (80 мл) и экстрагировали с помощью EA (80 мл × 2) и органические слои объединяли и концентрировали с получением 3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазола (450 мг, 924,8 мкмоль, выход 87,2%) в виде желтого твердого вещества.
Стадия I. При -78°C к раствору 3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазола (600 мг, 1,2389 ммоль, 1,0 экв.) в DCM (12 мл) добавляли BBr3 (1,54 г, 6,1589 ммоль, 5,0 экв.). Смесь перемешивали при 25°C в течение 16 часов и гасили путем медленного добавления MeOH (20 мл). Смесь концентрировали и остаток очищали с помощью препаративной HPLC (система на основе муравьиной кислоты) с получением продукта, 4-[3-(5-этил-1H-имидазол-2-ил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-ил]-6-изопропилбензол-1,3-диола (262 мг, 571,4 мкмоль, выход 46,5%). 1H ЯМР (400 МГц, DMSO-d6) δ 12,55 (m, 1H), 9,65 (m, 1H), 8,16 (s, 1H), 7,07-6,81 (brs, 5H), 6,41 (d, 1H), 3,52-3,50 (m, 4H), 3,11-2,97 (m, 1H), 2,70-2,61 (m, 4H), 2,36 (s, 3H), 1,15 (t, J = 3,6 Гц, 3H), 0,96-0,94 (m, 6H).
Пример 17
4-(3-(5-Этил-1H-имидазол-2-ил)-4-(2-(2-гидроксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-ил)-6-изопропилбензол-1,3-диол
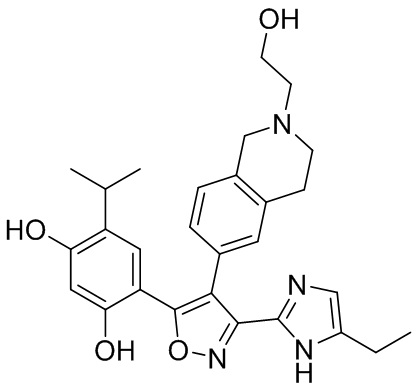
Реакционная схема
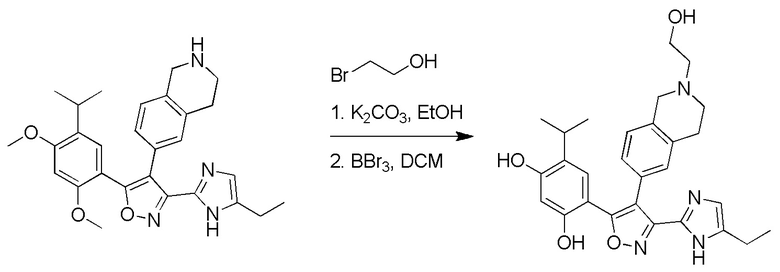
Стадия A. К раствору 3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазола (130 мг, 275,1 мкмоль, 1,0 экв.) в EtOH (6 мл) добавляли 2-бромэтанол (137,5 мг, 1,189 ммоль, 4,0 экв.) и K2CO3 (114 мг, 825,3 мкмоль, 3,0 экв.). Смесь перемешивали при 50°C в течение 16 часов, затем выливали в воду (30 мл) и экстрагировали с помощью EA (30 мл × 2). Органические слои объединяли и концентрировали. Неочищенный продукт очищали с помощью препаративной TLC (DCM/MeOH = 10:1) с получением 2-[6-[3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-ил]этанола (80 мг, 154,9 мкмоль, выход 56,3%) в виде желтого твердого вещества.
Стадия B. При 0°C к раствору 2-[6-[3-(5-этил-1H-имидазол-2-ил)-5-(5-изопропил-2,4-диметоксифенил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-ил]этанола (80 мг, 154,9 мкмоль, 1,0 экв.) в DCM (8 мл) добавляли BBr3 (387,9 мг, 1,5589 ммоль, 10,0 экв.). Смесь перемешивали при 25°C в течение 16 часов и гасили путем добавления MeOH (15 мл) и смесь концентрировали. Неочищенный продукт очищали с помощью препаративной HPLC (Phenomenex Synergi C18 250 × 21,2 мм × 4 мкм, 0,05% HCl-ACN) с получением 4-(3-(5-этил-1H-имидазол-2-ил)-4-(2-(2-гидроксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-ил)-6-изопропилбензол-1,3-диола (24,5 мг, 50,2 мкмоль, выход 32,4%). 1H ЯМР (300 МГц, DMSO-d6) δ 11,01 (brs, 1H), 10,05-9,99 (m, 2H), 7,62 (s, 1H), 7,18-7,03 (m, 3H), 6,91 (s, 1H), 6,57 (s, 1H), 4,55-4,34 (m, 2H), 4,30-4,28 (m, 2H), 3,27-3,01 (m, 7H), 2,88-2,82 (m, 2H), 1,23 (t, J = 4,5 Гц, 3H), 1,00 (d, J = 6,9 Гц, 6H).
Пример 18
5-(2,4-Дигидрокси-5-изопропилфенил)-4-(7,7-диметил-4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-N-этилизоксазол-3-карбоксамид
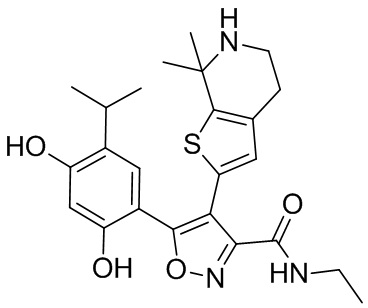
Реакционная схема
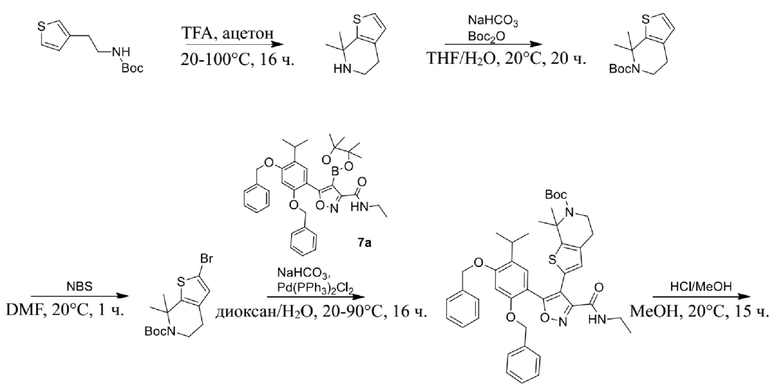
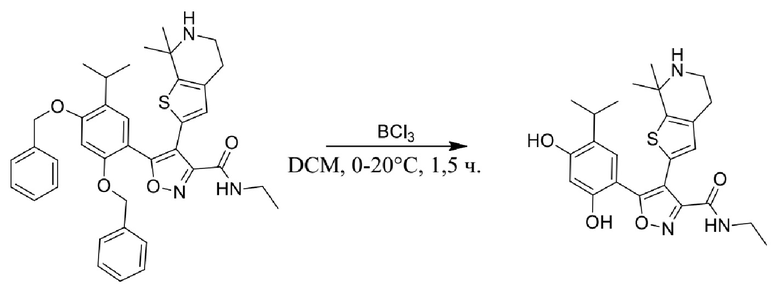
Стадия A. При 20°C в защитной атмосфере газообразного азота к раствору трет-бутил-[2-(тиофен-3-ил)этил]карбамата (2,40 г, 10,5689 ммоль, 1,0 экв.), растворенного в ацетоне (9,48 г, 163,2289 ммоль, 15,46 экв.), по каплям добавляли трифторуксусную кислоту (18,36 г, 161,0189 ммоль, 15,25 экв.). После добавления реакционную смесь перемешивали при 100°C в течение 16 часов. Реакционный раствор охлаждали до 20°C, а затем концентрировали при 50°C с получением 7,7-диметил-4,5,6,7-тетрагидротиено[2,3-c]пиридина (10,05 г, неочищенный продукт). Полученный в результате неочищенный продукт доводили до pH = 10 с помощью 2 M раствора NaOH и после обработки щелочью раствор применяли на следующей стадии реакции.
Стадия B. При 20°C в защитной атмосфере газообразного азота к раствору 7,7-диметил-4,5,6,7-тетрагидротиено[2,3-c]пиридина (10,05 г, неочищенный продукт), растворенного в тетрагидрофуране (10 мл), по каплям добавляли Boc2O (2,31 г, 10,5789 ммоль, 1 экв.). После добавления реакционную смесь перемешивали при 20°C в течение 20 часов. Реакционный раствор выливали в 20 мл воды и перемешивали в течение 5 мин. Водную фазу экстрагировали три раза, каждый раз с помощью 10 мл этилацетата, и органическую фазу промывали три раза, каждый раз с помощью 10 мл солевого раствора, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = от 400/1 до 90/1) с получением трет-бутил-7,7-диметил-4,5-дигидротиено[2,3-c]пиридин-6(7H)-карбамата (733 мг, 2,7489 ммоль, выход 25,93%) в виде желтого масла.
Стадия C. При 20°C в защитной атмосфере газообразного азота к раствору трет-бутил-7,7-диметил-4,5-дигидротиено[2,3-c]пиридин-6(7H)-карбамата (733 мг, 2,7489 ммоль, 1 экв.), растворенного в N,N-диметилкарбоксамиде (7 мл), отдельными порциями добавляли NBS (487,67 мг, 2,7489 ммоль, 1 экв.). Обеспечивали протекание реакции в реакционном растворе при 20°C в течение одного часа. Реакционный раствор выливали в 20 мл воды и перемешивали в течение 5 мин. Водную фазу экстрагировали три раза, каждый раз с помощью 10 мл этилацетата, и органическую фазу промывали три раза, каждый раз с помощью 10 мл солевого раствора. Затем органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением трет-бутил-2-бром-7,7-диметил-4,5-дигидротиено[2,3-c]пиридин-6(7H)-карбамата (840 мг, 2,4389 ммоль, выход 88,53%) в виде желтого масла.
Стадия D. При 25°C в защитной атмосфере газообразного азота к раствору трет-бутил-2-бром-7,7-диметил-4,5-дигидротиено[2,3-c]пиридин-6(7H)-карбамата (1,02 г, 2,9589 ммоль, 1 экв.) и 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (1,76 г, 2,9589 ммоль, 1 экв.) в диоксане (20 мл) и воде (4 мл) добавляли бикарбонат натрия (743,49 мг, 8,8589 ммоль, 3 экв.) и Pd(PPh3)2Cl2 (414,12 мг, 590,00 мкмоль, 0,20 экв.). Обеспечивали протекание реакции в реакционном растворе при 90°C в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры, а затем концентрировали с получением неочищенного продукта. Неочищенный остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = от 100/1 до 3/1) с получением трет-бутил-2-[5-(2,4-дибензилокси-5-изопропилфенил)-3-(этилкарбоксамид)изоксазол-4-ил]-7,7-диметил-4,5-дигидротиено[2,3-c]пиридин-6(7H)-карбамата (841,00 мг, 719,95 мкмоль, выход 24,40%, чистота 63%) в виде коричневого твердого вещества.
Стадия D. При 20°C к раствору трет-бутил-2-[5-(2,4-дибензилокси-5-изопропилфенил)-3-(этилкарбоксамид)изоксазол-4-ил]-7,7-диметил-4,5-дигидротиено[2,3-c]пиридин-6(7H)-карбамата (840 мг, 719,09 мкмоль, 1 экв.) в метаноле (8 мл) по каплям добавляли HCl/MeOH (4 моль/л, 8 мл, 44,50 экв.). Реакционный раствор перемешивали при 20°C в течение 15 часов. Реакционный раствор концентрировали с получением неочищенного продукта, а затем неочищенный продукт очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, дихлорметан/метанол = от 1/0 до 10/1) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-4-(7,7-диметил-4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-N-этилизоксазол-3-карбоксамида (442,00 мг, 695,18 мкмоль, выход 96,67%) в виде светло-коричневого твердого вещества.
Стадия E. При 0°C в атмосфере газообразного азота к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-4-(7,7-диметил-4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-N-этилизоксазол-3-карбоксамида (300 мг, 471,84 мкмоль, 1 экв.) в безводном дихлорметане (20 мл) медленно по каплям добавляли трихлорид бора (1 моль/л, 4,72 мл, 10 экв.). Обеспечивали протекание реакции в реакционном растворе при 0°C в течение одного часа, а затем его нагревали до 20°C в течение 0,5 часа. После завершения реакции реакционный раствор охлаждали до 0°C и медленно по каплям добавляли метанол (30 мл) с последующим перемешиванием в течение 0,5 часа. Органическую фазу концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (Phenomenex Synergi Max-RP 250 × 80, 10 мкм, 0,225% FA-ACN) с получением 5-(2,4-дигидрокси-5-изопропилфенил)-4-(7,7-диметил-4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-N-этилизоксазол-3-карбоксамида (150,00 мг, 311,43 мкмоль, выход 66,0%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6): δ 8,94 (t, J = 5,5 Гц, 1H), 8,28 (s, 1H), 6,90 (s, 1H), 6,82 (s, 1H), 6,50 (s, 1H), 3,31-3,24 (m, 2H), 3,09-3,03 (m, 3H), 2,56-2,54 (m, 2H), 1,38 (s, 6H), 1,11 (t, J = 7,2 Гц, 3H), 1,04 (d, J = 7 Гц, 6 H).
Пример 19
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)изоксазол-3-карбоксамид
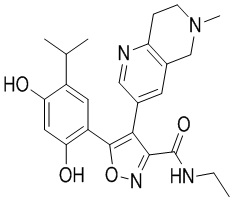
Реакционная схема
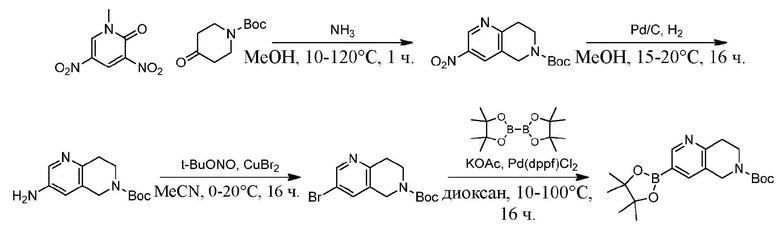
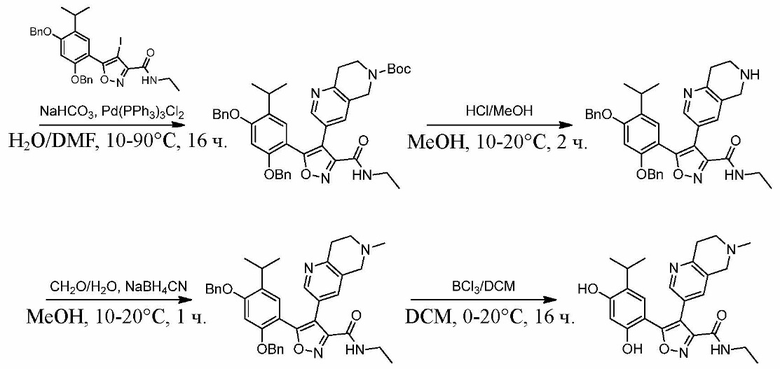
Стадия A. При комнатной температуре к раствору 1-метил-3,5-динитропиридин-2-она (2 г, 10,0489 ммоль, 1 экв.) и трет-бутил-4-пиперидон-1-карбоксилата в метаноле (50 мл) добавляли газообразный аммиак (1,37 г, 80,3289 ммоль, 8 экв.). Реакционный раствор перемешивали в герметичной емкости при 120°C в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (PE/EA = от 10/1 до 3/1) с получением трет-бутил-3-нитро-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (1,9 г, 6,889 ммоль, выход 67,76%) в виде серо-белого твердого вещества.
Стадия B. При комнатной температуре в атмосфере газообразного водорода (40 фунтов/кв. дюйм) к раствору трет-бутил-3-нитро-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (3 г, 10,7489 ммоль, 1 экв.) в метаноле (100 мл) добавляли Pd/C (1,00 г). Реакционный раствор перемешивали при комнатной температуре в течение 16 часов. После завершения реакции реакционный раствор фильтровали и концентрировали с получением трет-бутил-3-амино-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (2,5 г, 10,0389 ммоль, выход 93,37%) в виде белого твердого вещества.
Стадия C. При комнатной температуре к раствору трет-бутил-3-амино-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (300 мг, 1,289 ммоль, 1 экв.) в ацетонитриле (6 мл) добавляли CuBr2 (402,03 мг, 1,889 ммоль, 1,5 экв.) с последующим добавлением по каплям трет-бутилнитрита (148,49 мг, 1,4489 ммоль, 1,2 экв.) при 0-5°C для взаимодействия в течение 1 часа. Температуру повышали до комнатной температуры при перемешивании в течение 15 часов. Реакционный раствор выливали в воду (20 мл), фильтровали и экстрагировали три раза, каждый раз с помощью 20 мл этилацетата. Органическую фазу промывали дважды, каждый раз с помощью 30 мл воды, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (PE/EA = от 10/1 до 3/1) с получением трет-бутил-3-бром-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (180 мг, 534,5 мкмоль, выход 44,54%, чистота 93%) в виде бесцветного и прозрачного масла.
Стадия D. При 25°C в защитной атмосфере газообразного азота к раствору трет-бутил-3-бром-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (160 мг, 510,87 мкмоль, 1 экв.) в диоксане (3 мл) добавляли бис(пинаколато)дибор (194,60 мг, 766,31 мкмоль, 1,50 экв.) и KOAc (150,41 мг, 1,5389 ммоль, 3 экв.) с последующим добавлением катализатора Pd(dppf)Cl2.CH2Cl2 (74,76 мг, 102,17 мкмоль, 0,20 экв.). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 100°C при перемешивании в течение 16 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении с получением неочищенного продукта, который непосредственно применяли на следующей стадии.
Стадия E. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору трет-бутил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (181,20 мг, 502,98 мкмоль, 2,00 экв.) и N-этил-4-йод-5-(5-изопропил-2,4-диметоксифенил)изоксазол-3-карбоксамида (150,00 мг, 251,49 мкмоль, 1,00 экв.) в диоксане (5 мл) добавляли NaHCO3 (63,38 мг, 754,46 мкмоль, 3,00 экв.), H2O (1 мл) и Pd(PPh3)2Cl2 (35,30 мг, 50,30 мкмоль, 0,2 экв.). Смесь перемешивали при 25°C в течение 10 мин., затем нагревали до 90°C и перемешивали в течение 16 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат, дихлорметан/метанол = 30/1, 1/15) с получением трет-бутил-3-[5-(2,4-дибензилокси-5-изопропилфенил)-3-(этилкарбоксамид)изоксазол-4-ил]-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (90,00 мг, неочищенный продукт) в виде желтого твердого вещества.
Стадия F. При 20°C к раствору трет-бутил-3-[5-(2,4-дибензилокси-5-изопропилфенил)-3-(этилкарбоксамид)изоксазол-4-ил]-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (90 мг, 128,05 мкмоль, 1 экв.) в метаноле (2 мл) по каплям добавляли HCl/MeOH (4 моль/л, 2,00 мл, 62,4 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта, и неочищенный продукт растворяли в дихлорметане (10 мл) при комнатной температуре, а затем добавляли бикарбонат натрия (1 г). Смесь перемешивали при комнатной температуре в течение 1 часа, фильтровали и концентрировали. Неочищенный продукт отделяли и очищали с помощью больших пластин для TLC (дихлорметан/метанол = 20/1) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)изоксазол-3-карбоксилата (50,00 мг, 82,96 мкмоль, выход 64,79%) в виде желтого твердого вещества.
Стадия G. При 20°C в защитной атмосфере газообразного азота к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)изоксазол-3-карбоксилата (50,00 мг, 82,96 мкмоль, 1,00 экв.) в метаноле (5 мл) по каплям добавляли раствор формальдегида в воде (62,28 мг, 829,6 мкмоль, 10 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 10 мин., а затем добавляли NaBH3CN (15,64 мг, 248,88 мкмоль, 3,00 экв.) при комнатной температуре при перемешивании в течение 50 мин. Реакционный раствор непосредственно концентрировали с получением неочищенного продукта, и неочищенный продукт отделяли и очищали с помощью больших пластин для TLC (дихлорметан/метанол = 10/1) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)изоксазол-3-карбоксилата (40,00 мг, 64,86 мкмоль, выход 78,18%) в виде белого твердого вещества.
Стадия H. При 0°C в атмосфере газообразного азота к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)изоксазол-3-карбоксилата (40 мг, 64,86 мкмоль, 1 экв.) в безводном дихлорметане (2,5 мл) медленно по каплям добавляли трихлорид бора (1 моль/л, 0,648 мл, 10 экв.). Обеспечивали протекание реакции в реакционном растворе при 0°C в течение одного часа, а затем его нагревали до 20°C и обеспечивали протекание реакции в течение 14 часов. После завершения реакции реакционный раствор охлаждали до 0°C и медленно по каплям добавляли метанол (1 мл) с последующим перемешиванием в течение 0,5 часа. Органическую фазу концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (Phenomenex Synergi Max-RP 250 × 80, 10 мкм, 0,225% FA-ACN) с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)изоксазол-3-карбоксамида формиата (18 мг, 37,3 мкмоль, выход 57,00%). 1H ЯМР (400 МГц, DMSO): δ 8,88 (t, J = 5,6 Гц, 1H), 8,12 (d, J = 1,6 Гц, 1H), 7,32 (d, J = 1,6 Гц, 1H), 6,91 (s, 1H), 6,41 (s, 1H), 3,42 (s, 2H), 3,27-3,19 (m, 2H), 3,07-2,99 (m, 1H), 2,85 (t, J = 6,0 Гц, 2H), 2,67 (t, J = 6,0 Гц, 2H), 2,34 (s, 3H), 1,10 (t, J = 7,2 Гц, 3H), 1,01 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 437 (M+1).
Пример 20
4-Изопропил-6-(4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(2,2,2-трифтор-1-гидроксиэтил)изоксазол-5-ил)бензол-1,3-диол
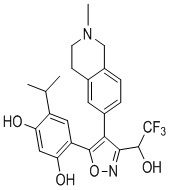
Реакционная схема
Стадия A. При 25°C в защитной атмосфере газообразного азота к раствору 6-бром-2-метил-1,2,3,4-тетрагидроизохинолина (1,00 г, 4,4289 ммоль, 1,0 экв.) в диоксане (10 мл) добавляли бис(пинаколато)дибор (1,68 г, 6,6389 ммоль, 1,5 экв.) и KOAc (1,30 г, 13,2689 ммоль, 3,0 экв.) с последующим добавлением катализатора Pd(dppf)Cl2 (323,41 мг, 442,00 мкмоль). Смесь нагревали до 90°C при перемешивании в течение 2 часов. Смесь фильтровали через диатомовую землю и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат = от 1/1 до 0/1) с получением 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (1,0 г, 3,6689 ммоль, выход 82,82%) в виде желтого масла.
Стадия B. При комнатной температуре в защитной атмосфере N2к раствору этил-5-(2,4-дибензилокси-5-изопропилфенил)изоксазол-3-карбоксилата (2,00 г, 4,2489 ммоль, 1,0 экв.) в MeCN (25 мл) добавляли CAN (232,52 мг, 549 мкмоль, 0,10 экв.) и NIS (2,83 г, 8,4889 ммоль, 2,00 экв.). Смесь нагревали до 80°C при перемешивании в течение 16 часов. Смесь охлаждали до комнатной температуры и выливали в воду (40 мл) и водную фазу экстрагировали с помощью EA (40 мл × 3). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме. Получали этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-3-карбоксилат (2,00 г, 3,3589 ммоль, выход 78,95%) в виде желтого твердого вещества.
Стадия C. При 25°C в защитной атмосфере газообразного азота к раствору 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (6,15 г, 13,589 ммоль) и этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-3-карбоксилата (1,50 г, 2,5189 ммоль, 1,0 экв.) в диоксане (15 мл) добавляли NaHCO3 (632,60 мг, 7,5389 ммоль, 3,0 экв.), H2O (3,00 мл) и Pd(PPh3)2Cl2 (176,18 мг, 251,0089 ммоль, 0,10 экв.). Смесь нагревали до 90°C при перемешивании в течение 1,5 часа. Смесь фильтровали через диатомовую землю и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (дихлорметан/метанол = 20/1) с получением этил-5-(2,4-дибензилокси-5-изопропилбензол-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксилата (600 мг, 972,86 мкмоль, выход 38,76%) в виде желтого твердого вещества, которое непосредственно применяли на следующей стадии. MS (ESI) масса/заряд: 617,2 (M+1).
Стадия D. При -20°C в защитной атмосфере газообразного азота к раствору этил-5-(2,4-дибензилокси-5-изопропилбензол-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксилата (500,00 мг, 810,71 мкмоль, 1,0 экв.) в тетрагидрофуране (2,0 мл) добавляли тетрагидроалюминат лития (153,83 мг, 4,0589 ммоль, 5,0 экв.). Смесь охлаждали до -20°C при перемешивании в течение 0,5 часа. К смеси по каплям добавляли 15% раствор NaOH (0,3 мл), а затем смесь фильтровали с получением органической фазы. Органическую фазу концентрировали при пониженном давлении. Остаток очищали с помощью препаративной TLC (дихлорметан/метанол = 10/1) с получением (5-(2,4-дибензилокси)-5-изопропилбензол)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-метанола (300 мг, 522,00 мкмоль, выход 64,39%) в виде желтого твердого вещества, которое непосредственно применяли на следующей стадии. MS (ESI) масса/заряд: 575,2 (M+1).
Стадия E. При 5°C в защитной атмосфере N2 к раствору (5-(2,4-дибензилокси)-5-изопропилбензол)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-метанола (200,00 мг, 348,00 мкмоль, 1,0 экв.) в дихлорметане (5,0 мл) добавляли реактив Десса-Мартина (221,40 мг, 522,00 мкмоль, 1,5 экв.). Смесь перемешивали при 5°C в течение 1 часа. К смеси добавляли насыщенный раствор NaHCO3 (1,0 мл) и насыщенный водный раствор Na2SO3 (1,0 мл) при перемешивании в течение 5 мин. Для разбавления добавляли 15 мл воды и водную фазу экстрагировали дихлорметаном (15 мл × 2). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Получали (5-(2,4-дибензилокси)-5-изопропилбензол)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбальдегид (200 мг, неочищенный продукт) в виде желтого твердого вещества. MS (ESI) масса/заряд: 573,2 (M+1).
Стадия F. При 5°C в защитной атмосфере N2 к раствору (5-(2,4-дибензилокси)-5-изопропилбензол)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбальдегида (200,00 мг, 349,23 мкмоль, 1,0 экв.) в тетрагидрофуране (5,0 мл) добавляли карбонат цезия (5,69 мг, 17,465 мкмоль, 0,05 экв.) и триметил(трифторметил)силан (60 мг, 419 мкмоль, 1,2 экв.). Смесь перемешивали при 5°C в течение 4 часов. К смеси добавляли фторид тетрабутиламмония (136,96 мг, 523,85 мкмоль, 1,50 экв.) при 5°C и перемешивали в течение 12 часов. Смесь выливали в воду (15 мл) и водную фазу экстрагировали дихлорметаном (15 мл × 2). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью тонкослойной хроматографии (DCM/MeOH = 8/1) с получением 1-(5-(2,4-дибензилокси)-5-изопропилбензол)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-ил)-2,2,2-трифторэтанола (100,0 мг, 155,5989 ммоль, выход 44,55%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 643,2 (M+1).
Стадия G. При -20°C к раствору 1-(5-(2,4-дибензилокси)-5-изопропилбензол)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-ил)-2,2,2,2-трифторэтанола (100,00 мг, 155,59 мкмоль, 1,0 экв.) в DCM (8 мл) добавляли 1 моль/л BCl3.DCM (2 мл). Смесь перемешивали при -20°C в течение 1 часа, гасили путем добавления 1 мл MeOH и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали дважды с помощью тонкослойной хроматографии (DCM/MeOH = 8/1) с получением 4-изопропил-6-(4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-3-(2,2,2-трифтор-1-гидроксиэтил)изоксазол-5-ил)бензол-1,3-диола (20,10 мг, выход 27,93%). 1H ЯМР B000139948 EW2407-16-P1A (400 МГц, DMSO-d6): δ 9,77 (s, 1H), 9,65 (s, 1H), 7,28 (d, J = 7,2 Гц, 1H), 7,05-7,15 (m, 3H), 6,82 (s, 1H), 6,42 (s, 1H), 5,15 (t, J = 7,2 Гц, 2H), 3,87 (brs, 2H), 2,85-3,05 (m, 5H), 2,59 (s, 3H), 0,95 (d, J = 6,8 Гц, 9H). MS (ESI) масса/заряд: 623 (M+1).
Пример 21
N-циано-5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамидин
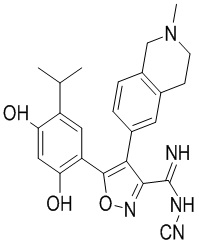
Реакционная схема
Стадия A. При 15-25°C в защитной атмосфере газообразного азота к раствору 5-(2,4-дигидрокси-5-изопропилфенил)-4-йодизоксазол-3-карбонитрила (300 мг, 545 мкмоль, 1,00 экв.) в диоксане (4,5 мл) добавляли 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин (372 мг, 818 мкмоль, 1,5 экв), воду (900,00 мкл) и бикарбонат натрия (224 мг, 2,789 ммоль, 4,9 экв.) с последующим добавлением Pd(PPh3)2Cl2 (77 мг, 109 мкмоль, 0,2 экв). Смесь перемешивали при 90°C в течение 15 часов. Реакционную смесь охлаждали до 15-25°C, выливали в воду (50 мл) и экстрагировали с помощью EA (50 мл × л). Объединенный органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (DCM:метанол = 20:1). Получали 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбонитрил (306 мг, 376 мкмоль, выход 70%) в виде желтого твердого вещества. (ESI) масса/заряд: 570 (M+1).
Стадия B. К раствору 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбонитрила (150 мг, 263 мкмоль, 1,0 экв.) в MeOH (4 мл) добавляли NaHCO3 (221 мг, 2,689 ммоль, 10,0 экв.). Смесь перемешивали при 40°C в течение 6,5 часа. Затем добавляли аминонитрил (221 мг, 589 ммоль, 20,0 экв.) при перемешивании при 40°C в течение 4 часов. Реакционный раствор выливали в воду (20 мл) и экстрагировали с помощью EA (10 мл × 3). Объединенный органический слой высушивали с помощью Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной TLC (DCM/MeOH = 15:1) с получением N-циано-5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамидина (125 мг, 204 мкмоль, выход 77%) в виде белого твердого вещества. MS (ESI) масса/заряд: 612 (M+1).
Стадия C. При 0°C к раствору N-циано-5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамидина (78 мг, 127 мкмоль, 1,0 экв.) в DCM (6 мл) добавляли раствор BCl3 (1 M, 1,3 мл, 10,0 экв.) в DCM. После перемешивания при 0°C в течение 2 часов температуру повышали до 15-25°C при перемешивании в течение 1 часа. Реакционную смесь охлаждали до 0°C и добавляли MeOH (3 мл) с последующим перемешиванием при 0°C в течение 0,5 часа и перемешиванием при 15-25°C в течение дополнительного 0,5 часа. Смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (0,225% FA-ACN; Phenomenex Synergi Max-RP 250 × 80, 10 мкм) с получением N-циано-5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамидина (17 мг, 39 мкмоль, выход 30%). 1H ЯМР (400 МГц, DMSO-d6): δ 8,26 (s, 1H), 7,05-6,83 (m, 4H), 6,44 (s, 1H), 3,02-2,98 (m, 1H), 2,72-2,55 (m, 6H), 2,33 (s, 1H), 0,96 (d, J = 6,5 Гц, 6H). MS (ESI) масса/заряд: 432 (M+1).
Пример 22
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)изоксазол-3-карбоксамид
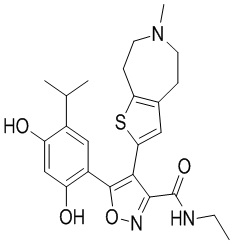
Реакционная схема
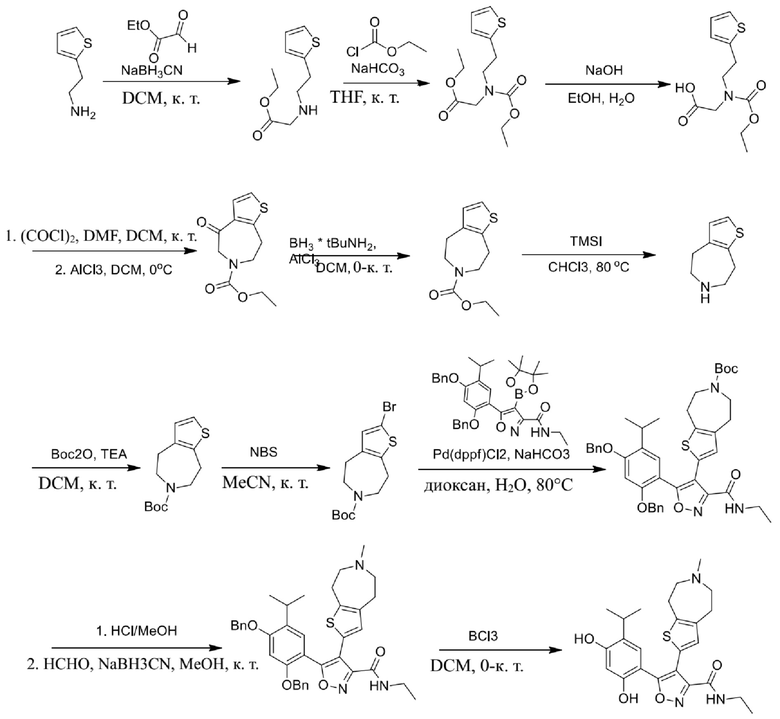
Стадия A. При 15-25°C к раствору 2-(тиофен-2-ил)этиламина (3,0 г, 23,689 ммоль, 1,00 экв.) и формиата этил-2-карбальдегида (4,8 г, 23,689 ммоль, 1,00 экв.) в DCM (40,0 мл) добавляли уксусную кислоту (1,4 г, 23,689 ммоль, 1,0 экв.) и NaBH(OAc)3 (10,0 г, 47,289 ммоль, 2,0 экв.). Смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь выливали в воду (60 мл) и экстрагировали с помощью EA (50 мл × 2). Объединенный органический слой высушивали, фильтровали и концентрировали с получением этил-2-((2-(тиофен-2-ил)этил)амино)ацетата (5,0 г, неочищенный продукт), который непосредственно применяли на следующей стадии.
Стадия B. При 0°C к раствору этил-2-((2-(тиофен-2-ил)этил)амино)ацетата (3,2 г, 15,089 ммоль, 1,00 экв.) и NaHCO3 (3,8 г, 45,089 ммоль, 3,00 экв.) в THF (40,0 мл) по каплям добавляли этилхлорформиат (1,6 г, 15,089 ммоль, 1,00 экв.). Смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь выливали в насыщенный водный раствор NaHCO3 (80 мл) и экстрагировали с помощью EA (60 мл × л). Объединенный органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = от 5:1 до 1:1) с получением этил-2-((этоксиформил)(2-(тиофен-2-ил)этил)амино)ацетата (2,9 г, 10,289 ммоль, выход 68%) в виде желтого масла.
Стадия C. При 0°C к раствору этил-2-((этоксиформил)(2-(тиофен-2-ил)этил)амино)ацетата (2,9 г, 10,289 ммоль, 1,00 экв.) в EtOH (20,0 мл) по каплям добавляли NaOH/H2O (20,3 мл, 20,489 ммоль, 2,00 экв., 1 M). Смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь выливали в воду (80 мл), доводили до pH = 1 с помощью 1 M водного раствора хлористоводородной кислоты и экстрагировали с помощью EA (80 мл × л). Объединенный органический слой высушивали, фильтровали и концентрировали с получением 2-((этоксиформил)(2-(тиофен-2-ил)этил)амино)уксусной кислоты (2,0 г, неочищенный продукт) в виде белого твердого вещества.
Стадия D. При 0°C к раствору 2-((этоксиформил)(2-(тиофен-2-ил)этил)амино)уксусной кислоты (1,0 г, 3,989 ммоль, 1,00 экв.) в DCM (15,0 мл) добавляли DMF (14 мг, 195 мкмоль, 0,05 экв.) и (COCl)2 (987 мл, 7,889 ммоль, 2,00 экв.). Смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь концентрировали с получением этил-(2-хлор-2-этокси)(2-(тиофен-2-ил)этил)карбамата (1,1 г, неочищенный продукт) в виде желтого масла.
Стадия E. При 0°C к раствору этил-(2-хлор-2-этокси)(2-(тиофен-2-ил)этил)карбамата (4,8 г, 1789 ммоль, 1,00 экв.) в DCM (50,0 мл) добавляли AlCl3 (5,8 г, 4389 ммоль, 2,5 экв). Смесь перемешивали при 25°C в течение 1 часа. К реакционному раствору добавляли EtOH (5,0 мл) и реакционную смесь выливали на лед и перемешивали в течение 1 часа. Смесь экстрагировали с помощью DCM (60 мл × л). Объединенный органический слой высушивали с помощью безводного Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = от 5:1 до 1:1) с получением этил-4-оксо-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (1,1 г, 4,689 ммоль, выход 26%) в виде желтого масла.
Стадия F. При 0°C к раствору AlCl3 (2,0 г, 1589 ммоль, 3,0 экв.) в DCM (20,0 мл) добавляли боран/2-метилпропан-2-амин (2,6 г, 3089 ммоль, 6,0 экв.). Смесь перемешивали при 0°C в течение 5 мин. с последующим добавлением раствора этил-4-оксо-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (1,2 г, 5,089 ммоль, 1,0 экв.) в DCM (10,0 мл). Реакционную смесь перемешивали при 25°C в течение 12 часов. Реакционную смесь выливали в водный раствор HCl (60 мл, 1 M) и перемешанный раствор экстрагировали с помощью DCM (50 мл × 3). Объединенный органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = от 10:1 до 5:1) с получением этил-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (700 мг, 3,189 ммоль, выход 62%) в виде желтого масла.
Стадия G. TMSI (12,4 г, 6289 ммоль, 20,0 экв.) добавляли к раствору этил-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (700 мг, 3,189 ммоль, 1,00 экв. в CHCl3 (15,0 мл) при 25°C. Смесь перемешивали при 80°C в течение 12 часов. К реакционной смеси добавляли NaOH (10,0 мл, 2 M) и выливали в насыщенный водный раствор NaHCO3 (100 мл) и перемешанный раствор экстрагировали с помощью DCM (60 мл × л). Объединенный органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали с получением продукта, 5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепина (550 мг, неочищенный продукт) в виде желтого твердого вещества.
Стадия H. При 25°C к раствору 5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепина (550 мг, 3,689 ммоль, 1,00 экв.) в DCM (10,0 мл) добавляли Et3N (1,1 г, 1189 ммоль, 3,0 экв.) и Boc2O (1,2 г, 5,489 ммоль, 1,5 экв.). Смесь перемешивали при 25°C в течение 12 часов. Реакционную смесь выливали в воду (60 мл) и перемешанный раствор экстрагировали с помощью DCM (60 мл × 2). Объединенный органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = 5:1) с получением продукта, трет-бутил-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (500 мг, 2,089 ммоль, выход 55%) в виде желтого масла.
Стадия I. При 25°C к раствору трет-бутил-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (500 мг, 2,089 ммоль, 1,00 экв.) в MeCN (5,0 мл) добавляли NBS (245 мг, 1,489 ммоль, 0,7 экв.). Смесь перемешивали при 25°C в течение 2 часов. Реакционную смесь выливали в насыщенный водный раствор NaHSO3 (30 мл) и перемешанный раствор экстрагировали с помощью DCM (30 мл × л). Объединенный органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = от 10:1 до 6:1) с получением продукта, трет-бутил-2-бром-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (470 мг, 1,489 ммоль, выход 72%) в виде желтого масла.
Стадия J. В защитной атмосфере газообразного азота к перемешанному раствору N-этил-5-(5-изопропил-2,4-дибензилоксифенил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (1,1 г, 1,889 ммоль, 1,40 экв.) и трет-бутил-2-бром-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиата (420 мг, 1,389 ммоль) в диоксане (20,00 мл) и воде (4,00 мл) добавляли Pd(PPh3)2Cl2 (88 мг, 126 мкмоль, 0,10 экв.) и NaHCO3 (212 мг, 2,589 ммоль, 2,00 экв.). Смесь перемешивали при 80°C в течение 12 часов. Смесь охлаждали, затем выливали в воду (100 мл) и экстрагировали с помощью EA (80 мл × л). Органические слои объединяли и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = от 10/1 до 3/1). Получали трет-бутил-2-(5-(2,4-дибензилокси-5-изопропилфенил)-3-(этилформамил)изоксазол-4-ил)-4,5,7,8-тетрагидротиено[2,3-d]азепинформиат (800 мг, 1,189 ммоль, выход 88,0%) в виде желтого твердого вещества.
Стадия K. К раствору трет-бутил-2-(5-(2,4-дибензилокси-5-изопропилфенил)-3-(этилформамил)изоксазол-4-ил)-4,5,7,8-тетрагидротиено[2,3-d]азепинформиата (880 мг, 1,289 ммоль, 1,00 экв.) в MeOH (10,00 мл) добавляли HCl/MeOH (4 M, 10,00 мл) при перемешивании при 25°C в течение 1 часа. Смесь концентрировали при 40°C с получением продукта 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)изоксазол-3-формиата (800 мг, 1,2 мкмоль, выход 99%) в виде желтого твердого вещества.
Стадия L. К раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)изоксазол-3-карбоксамида (730 мг, 1,289 ммоль, 1,0 экв.) в MeOH (15 мл) добавляли параформальдегид (7,93 г, 9889 ммоль, 83,5 экв.) и AcOH (70 мг, 1,289 ммоль, 1,0 экв.). Смесь перемешивали при 25°C в течение 2 часов, а затем добавляли NaBH3CN (147 мг, 2,389 ммоль, 2,0 экв.). Реакционную смесь дополнительно перемешивали при 25°C в течение 12 часов. Смесь выливали в воду (80 мл) и экстрагировали с помощью EA (80 мл × 2). Органические слои объединяли и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA = 1/1, 0/1) с получением продукта, 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(6-метил-4,5,7,8-тетрагидротиено[2,3-d]азепин-2-ил)изоксазол-3-карбоксамида (340 мг, 535 мкмоль, выход 46%) в виде желтого твердого вещества.
Стадия M. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(6-метил-4,5,7,8-тетрагидротиено[2,3-d]азепин-2-ил)изоксазол-3-карбоксамида (100 мг, 157 мкмоль, 1,0 экв.) в DCM (8 мл) добавляли раствор BCl3 (1 M, 0,8 мл, 5,0 экв.) в DCM. После перемешивания при 0°C в течение 2 часов температуру повышали до 15-25°C при перемешивании в течение 1 часа. Реакционную смесь охлаждали до 0°C и добавляли MeOH (3 мл) с последующим перемешиванием при 0°C в течение 0,5 часа и перемешиванием при 15-25°C в течение дополнительного 0,5 часа. Смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (0,225% FA-ACN; Phenomenex Synergi Max-RP 250 × 80, 10 мкм) с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(6-метил-4,5,7,8-тетрагидротиено[2,3-d]азепин-2-ил)изоксазол-3-карбоксамида (20 мг, 44 мкмоль, выход 28%). 1H ЯМР (400 МГц, DMSO-d6): δ 11,05 (m, 1H), 9,92 (s, 1H), 9,81 (s, 1H), 8,97-8,94 (m, 1H), 6,92-6,90 (m, 2H), 6,52 (s, 1H), 6,43 (s, 1H), 3,48-3,36 (m, 2H), 3,28-3,23 (m, 3H), 3,08-3,03 (m, 1H), 2,82-2,81 (m, 3H), 1,13-1,05 (m, 9H). MS (ESI) масса/заряд: 456 (M+1).
Пример 23
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-ил)изоксазол-3-карбоксамид
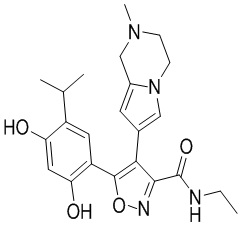
Реакционная схема
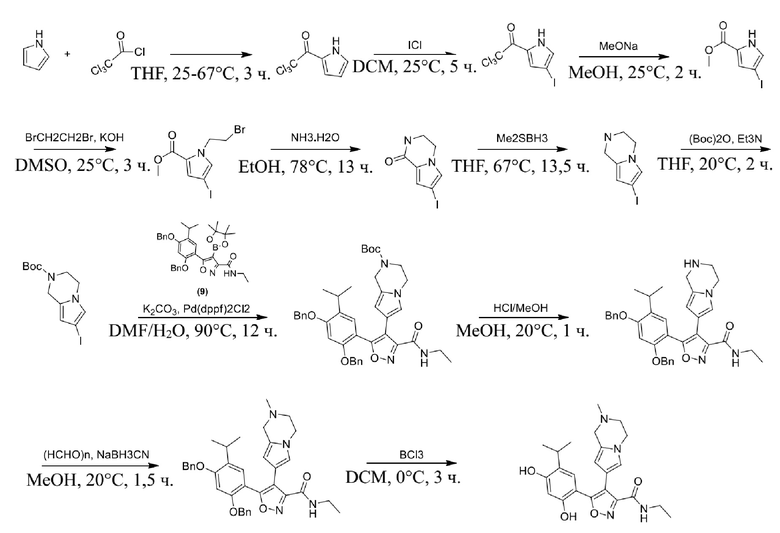
Стадия A. При 25°C в защитной атмосфере газообразного азота к раствору 2,2,2-трихлорацетилхлорида (19,51 г, 107,3289 ммоль, 1,20 экв.) в тетрагидрофуране (36 мл) медленно по каплям добавляли раствор пиррола (6,00 г, 89,4389 ммоль, 1,00 экв.), растворенного в тетрагидрофуране (120 мл). Реакционный раствор перемешивали при 25°C в течение 5 мин., а затем нагревали до 67°C при перемешивании в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и к реакционному раствору медленно по каплям добавляли раствор бикарбоната натрия (10 г) в воде (50 мл). Водную фазу экстрагировали три раза этилацетатом (каждый раз 50 мл). Органические фазы объединяли, промывали три раза водой (80 мл), промывали дважды насыщенным солевым раствором (40 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением 2,2,2-трихлор-1-(1H-пиррол-2-ил)этанона (18,00 г, 84,7289 ммоль, выход 94,74%).
Стадия B. При 20°C в защитной атмосфере газообразного азота к раствору 2,2,2-трихлор-1-(1H-пиррол-2-ил)этанона (22,00 г, 103,5589 ммоль, 1,00 экв.) в дихлорметане (250 мл) по каплям добавляли хлорид йода (16,81 г, 103,5589 ммоль, 1,00 экв.) и реакционный раствор перемешивали при 20°C в течение 5 часов. Реакционный раствор промывали 10% раствором K2CO3 (100 мл) и экстрагировали три раза дихлорметаном (300 мл). Органические фазы объединяли, промывали водой (50 мл), 1 моль/л тиосульфатом натрия (100 мл) и насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением 2,2,2-трихлор-1-(4-йод-1H-пиррол-2-ил)этанона (33,00 г, 97,5389 ммоль, выход 94,19%) в виде белого твердого вещества.
Стадия C. При 25°C в защитной атмосфере газообразного азота к раствору 2,2,2-трихлор-1-(4-йод-1H-пиррол-2-ил)этанона (34,60 г, 102,2689 ммоль, 1,0 экв.) в метаноле (200 мл) по каплям добавляли раствор метоксида натрия (6,63 г) в метаноле (50мл). Обеспечивали протекание реакции в реакционном растворе при 25°C в течение 2 часов. После завершения реакции реакционный раствор выливали в воду (150 мл) и водную фазу экстрагировали три раза этилацетатом (600 мл). Органические фазы объединяли, промывали дважды насыщенным солевым раствором (200 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением сложного метилового эфира метил-4-йод-1H-пиррол-2-карбоновой кислоты (24,00 г, 95,6189 ммоль, выход 93,50%) в виде серо-белого твердого вещества.
Стадия D. При 20°C в защитной атмосфере газообразного азота к раствору сложного метилового эфира метил-4-йод-1H-пиррол-2-карбоновой кислоты (9,00 г, 35,8589 ммоль, 1,00 экв.) в диметилсульфоксиде (80 мл) добавляли гидроксид калия (14,08 г, 250,9589 ммоль, 7,00 экв.). Обеспечивали протекание реакции в реакционном растворе при 20°C в течение 3 часов, а затем к реакционному раствор по каплям добавляли дибромэтан (53,88 г, 286,8089 ммоль, 8,00 экв.). Обеспечивали протекание реакции в реакционном растворе при 20°C в течение 10 часов. После завершения реакции реакционный раствор выливали в воду (100 мл) и экстрагировали три раза этилацетатом (450 мл). Органические фазы объединяли, промывали три раза водой (150 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = от 30/1 до 20/1) с получением метил-1-(2-бромэтил)-4-йод-1H-пиррол-2-карбоксилата (9,50 г, 26,5489 ммоль, выход 74,03%) в виде желтого масла.
Стадия E. При 20°C в защитной атмосфере газообразного азота к раствору метил-1-(2-бромэтил)-4-йод-1H-пиррол-2-карбоксилата (4,00 г, 11,1789 ммоль, 1,00 экв.) в этаноле (20 мл) по каплям добавляли водный раствор аммиака (14,56 г, 124,5589 ммоль, 11,15 экв.). Реакционный раствор перемешивали при 20°C в течение 1 часа. Затем реакционный раствор нагревали до 78°C при перемешивании в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и неочищенный продукт разбавляли водой (20 мл), а затем экстрагировали три раза с помощью этилацетата (150 мл). Объединенную органическую фазу промывали дважды насыщенным солевым раствором (40 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, дихлорметан/метанол = 10/1) с получением 7-йод-3,4-дигидропирроло[1,2-a]-пиразин-1(2H)-она (1,00 г, 3,8289 ммоль, выход 34,16%) в виде белого твердого вещества.
Стадия F. BH3-Me2S (10 M, 7,25 мл, 10,00 экв.) по каплям добавляли к раствору 7-йод-3,4-дигидропирроло[1,2-a]пиразин-1(2H)-она (1,90 г, 7,2589 ммоль, 1,00 экв.) в тетрагидрофуране (40 мл) при 0°C в защитной атмосфере газообразного азота. Реакционный раствор перемешивали при 0°C в течение 30 мин., а затем нагревали до 20°C при перемешивании в течение 1 часа. Наконец, температуру повышали до 67°C при перемешивании в течение 12 часов. После завершения реакции реакционный раствор охлаждали до 0°C, гасили метанолом (5 мл), а затем нагревали с обратным холодильником при 67°C в течение 4 часов. Реакционный раствор концентрировали при пониженном давлении с получением 7-йод-1,2,3,4-тетрагидропирроло[1,2-a]пиразина (1,80 г, неочищенный продукт) в виде желтого твердого вещества.
Стадия G. При 20°C в защитной атмосфере газообразного азота к раствору 7-йод-1,2,3,4-тетрагидропирроло[1,2-a]пиразина (1,80 г, 7,2689 ммоль, 1,00 экв.) в тетрагидрофуране (20 мл) добавляли триэтиламин (3,67 г, 36,3089 ммоль, 5,0 экв.) и (Boc)2O (3,17 г, 14,5289 ммоль, 2,00 экв.). Реакционный раствор перемешивали при 20°C в течение 2 часов. После завершения реакции реакционный раствор выливали в воду (30 мл) и экстрагировали три раза этилацетатом (150 мл). Объединенную органическую фазу дважды промывали насыщенным солевым раствором (20 мл). Органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = от 20/1 до 10/1) с получением трет-бутил-7-йод-3,4-дигидропирроло[1,2-a]пиразин-2(1H)-формиата (800,00 мг, 2,3089 ммоль, выход 31,65%) в виде желтого масла.
Стадия H. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий J, K, L и M в примере 22е, при этом трет-бутил-2-бром-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиат на стадии J заменяли трет-бутил-7-йод-3,4-дигидропирроло[1,2-a]пиразин-2(1H)-формиатом. 1H ЯМР (400 МГц, DMSO-d6): δ 1,01-1,15 (m, 9H); 2,29 (s, 3H); 2,64 (t, J = 5,40 Гц, 2H); 3,19-3,30 (m, 5H); 3,83 (t, J = 5,46 Гц, 2H); 5,61 (s, 1H);6,47 (s, 1H); 6,69 (d, J = 1,51 Гц, 1H); 6,90 (s, 1H); 8,79 (t, J = 5,52 Гц, 1H); 9,56 (s, 1H); 9,70 (s, 1H). Масса/заряд: 425,1 [M+1].
Пример 24
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(5-изобутил-1-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-2-ил)изоксазол-3-карбоксамид
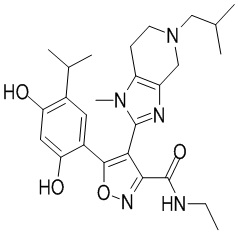
Реакционная схема
Стадия A. При 25°C в защитной атмосфере газообразного азота к раствору 4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридина (4,5 г, 22,9589 ммоль, 1,00 экв.), растворенного в метаноле (50,00 мл), добавляли DIPEA (7,42 г, 57,3789 ммоль, 2,50 экв.) и (Boc)2O (12,52 г, 57,3789 ммоль, 2,50 экв.). После добавления реакционную смесь перемешивали при 25°C в течение 18 часов. Реакционный раствор разбавляли водой (20 мл), а затем экстрагировали этилацетатом (20 мл × л). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = 2:1) с получением продукта, бис-трет-бутил-6,7-дигидро-1H-имидазо[4,5-c]пиридин-1,5(4H)-диформиата (5,4 г, 16,7089 ммоль, выход 72,76%) в виде белого твердого вещества.
Стадия B. При 25°C в защитной атмосфере газообразного азота к раствору бис-трет-бутил-6,7-дигидро-1H-имидазо[4,5-c]пиридин-1,5(4H)-диформиата (5,40 г, 16,7089 ммоль, 1,00 экв.), растворенного в метаноле (40,00 мл), добавляли 1 моль/л NaOH (20 мл). После добавления реакционную смесь перемешивали при 25°C в течение 1 часа. Реакционный раствор разбавляли водой (50 мл), а затем экстрагировали этилацетатом (50 мл × л). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Получали продукт, трет-бутил-1,4,6,7-тетрагидроимидазо[4,5-c]пиридин-5-формиат (3,30 г, 14,7889 ммоль, выход 88,50%) в виде желтого твердого вещества.
Стадия C. При 10°C в защитной атмосфере газообразного азота к раствору 1,4,6,7-тетрагидроимидазо[4,5-c]пиридин-5-формиата (3,30 г, 14,7889 ммоль, 1,00 экв.), растворенного в тетрагидрофуране (30,00 мл), добавляли NIS (4,99 г, 22,1789 ммоль, 1,50 экв.). После добавления реакционную смесь перемешивали при 10°C в течение 1 часа. Реакционный раствор разбавляли насыщенным водным раствором бикарбоната натрия (15 мл), а затем экстрагировали этилацетатом (15 мл × л). Объединенную органическую фазу промывали водой (10 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Получали продукт, трет-бутил-2-йод-1,4,6,7-тетрагидроимидазо[4,5-c]пиридин-5-формиат (4,30 г, 12,3189 ммоль, выход 83,32%) в виде желтого твердого вещества.
Стадия D. При 10°C в защитной атмосфере газообразного азота к раствору трет-бутил-2-йод-1,4,6,7-тетрагидроимидазо[4,5-c]пиридин-5-формиата (150 мг, 429,5989 ммоль, 1,00 экв.), растворенного в тетрагидрофуране (3,00 мл), добавляли гидрид натрия (34,37 мг, 859,18 мкмоль, 2,00 экв.) и метилиодид (630,00 мг, 4,4489 ммоль, 10,33 экв.). После добавления реакционную смесь перемешивали при 10°C в течение 16 часов. Реакционный раствор разбавляли водой (20 мл), а затем экстрагировали этилацетатом (20 мл × л). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме. Получали продукт, трет-бутил-2-йод-1-метил-6,7-дигидро-4H-имидазо[4,5-c]пиридин-5-формиат (150,00 мг, неочищенный продукт) в виде желтого твердого вещества. Неочищенный продукт непосредственно применяли на следующей стадии. MS (ESI) масса/заряд: 363,9 (M+1).
Стадия E. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий J, K, L и M в примере 22, при этом трет-бутил-2-бром-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиат на стадии J заменяли трет-бутил-2-йод-1-метил-6,7-дигидро-4H-имидазо[4,5-c]пиридин-5-формиатом. 1H ЯМР (400 МГц, DMSO-d6) δ 10,26 (brs, 2H), 9,96 (s, 1H), 9,96-10,03 (m, 1H), 6,87 (s, 1H), 6,48 (s, 1H), 3,23-3,26 (m, 2H), 3,15-3,23 (m, 5H), 3,00 (t, J = 6,8 Гц, 1H), 2,60-2,70(m, 2H), 2,50-2,55 (m, 2H), 2,20-2,35 (m, 2H), 1,75-1,90 (brs, 1H), 1,08 (t, J = 7,2 Гц, 1H), 0,99 (d, J = 3,6 Гц, 6H), 0,83 (d, J = 3,6 Гц, 6H). Масса/заряд: 482 [M+1].
Пример 25
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ил)изоксазол-3-карбоксамид
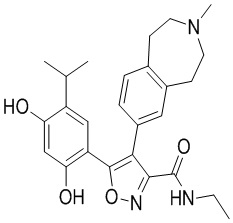
Реакционная схема
Стадия A. К раствору 6-бром-3,4-дигидронафталин-2(1H)-она (2,25 г, 1089 ммоль) в метансульфоновой кислоте (20,00 мл) медленно отдельными порциями добавляли NaN3 (1,06 г, 1689 ммоль) при 0°C на протяжении 0,5 часа. Реакционную смесь перемешивали при 0°C в течение 3,5 часов, а затем реакционную смесь перемешивали при 25°C в течение дополнительных 16 часов. Затем к насыщенному водному раствору NaHCO3 (100 мл) медленно по каплям добавляли смесь с последующей экстракцией этилацетатом (100 мл × 3). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, дихлорметан/метанол = от 50/1 до 20/1) с получением 7-бром-4,5-дигидро-1H-3-бензо[d]азепин-2(3H)-она (800 мг, выход 33%) в виде желтого твердого вещества.
Стадия B. При 0°C к раствору 7-бром-4,5-дигидро-1H-3-бензо[d]азепин-2(3H)-она (800 мг, 3,389 ммоль) в THF (8,00 мл) по каплям добавляли BH3-Me2S (3,3 мл, 10 M). Реакционную смесь перемешивали при 0°C в течение 1 часа и перемешивали при 25°C в течение 3 часов, а затем реакционную смесь перемешивали при 70°C в течение дополнительных 16 часов. Затем смесь охлаждали до 0°C и к реакционному раствору медленно по каплям добавляли MeOH (4 мл) при 0°C и перемешивали при 25°C в течение 0,5 часа. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, 7-бром-2,3,4,5-тетрагидро-1H-3-бензо[d]азепина (800 мг, неочищенный продукт) в виде желтого масла.
Стадия C. При 25°C к раствору 7-бром-2,3,4,5-тетрагидро-1H-3-бензо[d]азепина (800 мг, 3,5489 ммоль) в THF (16,00 мл) добавляли TEA (1,07 г, 10,689 ммоль) с последующим добавлением Boc2O (1,16 г, 10,689 ммоль). Реакционную смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат = от 30/1 до 10/1) с получением трет-бутил-7-бром-4,5-дигидро-1H-бензо[d]азепин-3-(2H)-формиата (500 мг, выход 43%) в виде желтого масла. MS (ESI) масса/заряд: 270, 272 (M-56+1).
Стадия D. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий J, K, L и M в примере 22, при этом трет-бутил-2-бром-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиат на стадии J заменяли трет-бутил-7-бром-4,5-дигидро-1H-бензо[d]азепин-3(2H)формиатом, и продукт представлял собой белое твердое вещество. 1H ЯМР (400 МГц, DMSO): δ 8,82 (t, J = 5,6 Гц, 1H), 7,04 (d, J = 8,0 Гц, 1H), 7,01 (s, 1H), 7,04 (dd, J = 1,6, 8,0 Гц, 1H), 6,76 (s, 1H), 6,42 (s, 1H), 3,25-3,18 (m, 2H), 3,02-2,94 (m, 1H), 2,86-2,76 (m, 2H), 2,76-2,66 (m, 2H), 2,49-2,40 (m, 4H), 2,26 (s, 3H), 1,07 (t, J = 7,2 Гц, 3H), 0,94 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 450 (M+1).
Пример 26
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-метил-2,3,4,5-тетрагидро-1H-бензо[c]азепин-7-ил)изоксазол-3-карбоксамид
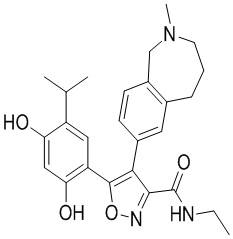
Реакционная схема
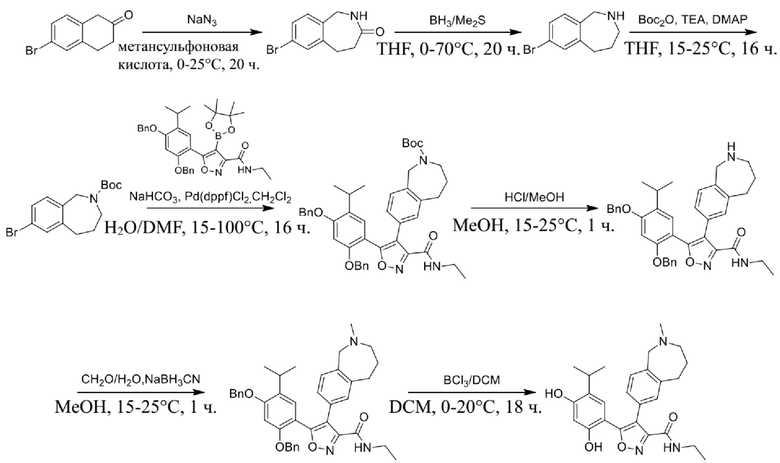
Стадия A. К раствору 6-бром-3,4-дигидронафталин-2(1H)-она (2,25 г, 1089 ммоль) в метансульфоновой кислоте (20,00 мл) медленно отдельными порциями добавляли NaN3 (1,06 г, 1689 ммоль) при 0°C на протяжении 0,5 часа. Реакционную смесь перемешивали при 0°C в течение 3,5 часов, а затем реакционную смесь перемешивали при 25°C в течение дополнительных 16 часов. Затем к насыщенному водному раствору NaHCO3 (100 мл) медленно по каплям добавляли смесь с последующей экстракцией этилацетатом (100 мл × 3). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, дихлорметан/метанол = от 50/1 до 20/1) с получением 7-бром-4,5-дигидро-1H-бензо[c]азепин-3(2H)-она (400 мг, выход 17%) в виде желтого твердого вещества.
Стадия B. При 0°C к раствору 7-бром-4,5-дигидро-1H-бензо[c]азепин-3(2H)-она (400 мг, 1,789 ммоль) в THF (4,00 мл) по каплям добавляли BH3-Me2S (1,7 мл, 10 M). Реакционную смесь перемешивали при 0°C в течение 1 часа и перемешивали при 25°C в течение 3 часов, а затем реакционную смесь перемешивали при 70°C в течение дополнительных 16 часов. Затем смесь охлаждали до 0°C и к реакционному раствору медленно по каплям добавляли MeOH (2 мл) при 0°C с последующим перемешиванием при 25°C в течение 0,5 часа. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, 7-бром-2,3,4,5-тетрагидро-1H-бензо[c]азепина (400 мг, неочищенный продукт) в виде желтого масла.
Стадия C. При 25°C к раствору 7-бром-2,3,4,5-тетрагидро-1H-бензо[c]азепина (400 мг, 1,789 ммоль) в THF (8,00 мл) добавляли TEA (537 мг, 5,389 ммоль) с последующим добавлением Boc2O (579 мг, 5,389 ммоль). Реакционную смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат = от 30/1 до 10/1) с получением трет-бутил-7-бром-4,5-дигидро-1H-бензо[c]азепин-2(3H)-формиата (210 мг, выход 36%) в виде желтого масла. MS (ESI) масса/заряд: 270, 272 (M-56+1).
Стадия D. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий J, K, L и M в примере 22, при этом трет-бутил-2-бром-7,8-дигидро-4H-тиено[2,3-d]азепин-6(5H)-формиат на стадии J заменяли трет-бутил-7-бром-4,5-дигидро-1H-бензо[c]азепин-2(3H)-формиатом. 1H ЯМР (400 МГц, DMSO): δ 8,83 (t, J = 4,8 Гц, 1H), 7,08-7,00 (m, 2H), 6,97-6,97 (m, 1H), 6,80-6,75 (m, 1H), 6,44-6,42 (m, 1H), 3,71 (s, 2H), 3,26-3,16 (m, 2H), 3,02-2,95 (m, 1H), 2,93-2,87 (m, 1,4H), 2,85-2,80 (m, 0,7H), 2,77-2,68 (m, 2H), 2,68-2,66 (m, 0,5H), 2,26-2,16 (m, 3H), 1,65-1,55 (m, 1,4H), 1,07 (t, J = 7,2 Гц, 3H), 0,96-0,92 (m, 6H). MS (ESI) масса/заряд: 450 (M+1).
Пример 27
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ил)изоксазол-5-карбоксамид
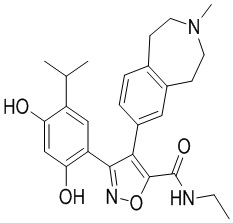
Реакционная схема
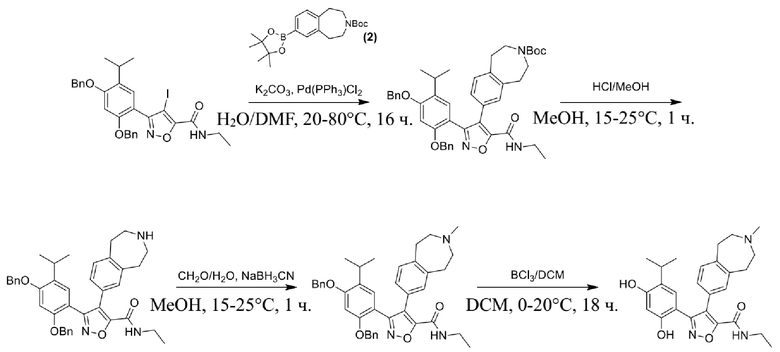
Стадия A. При 15-25°C в защитной атмосфере газообразного азота к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-йодизоксазол-5-карбоксамида (300 мг, 503 мкмоль, 1,00 экв.) в DMF (7,5 мл) добавляли трет-бутил-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,4,5-тетрагидро-бензазепин-3-формиат (282 мг, 754 мкмоль, 1,5 экв.), воду (1,5 мл) и карбонат калия (348 мг, 2,589 ммоль, 5,0 экв.) с последующим добавлением Pd(PPh3)2Cl2 (71 мг, 101 мкмоль, 0,2 экв.). Смесь перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до 15-25°C и выливали в воду (30 мл) с последующей экстракцией с помощью EA (30 мл × л). Объединенный органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = от 10:1 до 3:1) с получением трет-бутил-7(3-(2,4-дибензилокси-5-изопропилфенил)-5-(этилкарбамоил)изоксазол-4-ил)-1,2,4,5-тетрагидро-3-бензазепин-3-формиата (200 мг, 179 мкмоль, выход 36%) в виде желтого масла. (ESI) масса/заряд: 716 (M+1), 660 (M-56+1).
Стадия B. При 25°C к раствору трет-бутил-7(3-(2,4-дибензилокси-5-изопропилфенил)-5-этилкарбамоил)изоксазол-4-ил)-1,2,4,5-тетрагидро-3-бензазепин-3-формиата (200 мг, 279 мкмоль, 1,0 экв.) в MeOH (2,00 мл) добавляли HCl/MeOH (4 M, 2,00 мл). Смесь перемешивали при 25°C в течение 1 часа. Смесь концентрировали при 40°C с получением неочищенного продукта. Неочищенный продукт растворяли в DCM (10 мл) и к раствору добавляли NaHCO3 (1 г) при перемешивании в течение 1 часа. Смесь фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной TLC (DCM:MeOH = 10:1) с получением продукта, 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)изоксазол-5-карбоксамида (100 мг, 162 мкмоль, выход 58%) в виде желтого масла. MS (ESI) масса/заряд: 616 (M+1).
Стадия C. К раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)изоксазол-5-карбоксамида (100 мг, 162 мкмоль, 1,0 экв.) в MeOH (5 мл) добавляли водный раствор формальдегида (211 мг, 162 мкмоль, 1,0 экв., содержание 40%) и AcOH (10 мг, 16 мкмоль, 1,0 экв.). Смесь перемешивали при 25°C в течение 10 мин., а затем добавляли NaBH3CN (31 мг, 487 мкмоль, 3,0 экв.). Реакционную смесь дополнительно перемешивали при 25°C в течение 50 мин. и смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной TLC (DCM:MeOH = 10:1) с получением продукта, 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ил)изоксазол-5-карбоксамида (80 мг, 127 мкмоль, выход 78%) в виде бесцветного масла. MS (ESI) масса/заряд: 630 (M+1).
Стадия D. При 0°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ил)изоксазол-5-карбоксамид (60 мг, 95 мкмоль, 1,0 экв.) в DCM (5 мл) добавляли раствор BCl3 (1 M, 952 мкл, 10,0 экв.) в DCM. После перемешивания при 0°C в течение 2 часов температуру повышали до 15-25°C при перемешивании в течение 1 часа. Реакционную смесь охлаждали до 0°C и добавляли MeOH (2 мл) с последующим перемешиванием при 0°C в течение 0,5 часа и при 15-25°C в течение дополнительного 0,5 часа. Смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (0,225% FA-ACN; Phenomenex Synergi Max-RP 250 × 80, 10 мкм) с получением 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(3-метил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ил)изоксазол-5-карбоксамида (20 мг, 41 мкмоль, выход 43%). 1H ЯМР (400 МГц, DMSO): δ 10,84 (brs, 1H), 9,59 (s, 1H), 9,27 (s, 1H), 8,91-8,89 (m, 1H), 7,14-7,10 (m, 2H), 7,03 (d, J = 7,2 Гц, 1H), 6,79 (s, 2H), 6,37 (s, 1H), 3,71 (s, 2H), 3,25-3,16 (m, 5H), 3,04-2,80 (m, 5H), 2,78 (s, 3H), 1,08 (t, J = 6,8 Гц, 3H), 1,01 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 450 (M+1).
Пример 28
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)изоксазол-5-карбоксамид
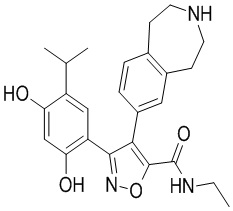
Реакционная схема
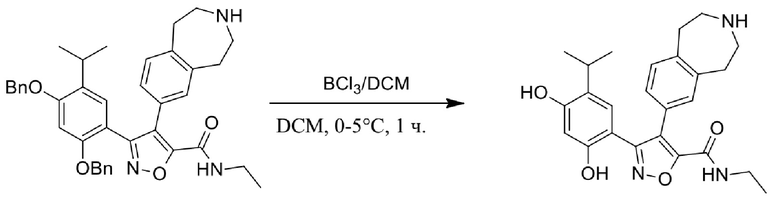
Стадия A. При 0°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)изоксазол-5-карбоксамида (130 мг, 211 мкмоль, 1,0 экв.) в DCM (10 мл) добавляли раствор BCl3 (1 M, 2,1 мл, 10,0 экв.) в DCM. После перемешивания при 0°C в течение 2 часов температуру повышали до 15-25°C при перемешивании в течение 1 часа. Реакционную смесь охлаждали до 0°C и добавляли MeOH (4 мл) при перемешивании при 0°C в течение 0,5 часа и при 15-25°C в течение дополнительного 0,5 часа. Смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC (0,225% FA-ACN; Phenomenex Synergi Max-RP 250 × 80, 10 мкм) с получением 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2,3,4,5-тетрагидро-1H-3-бензо[d]азепин-7-ил)изоксазол-5-карбоксамида (54 мг, 113 мкмоль, выход 54%). 1H ЯМР (400 МГц, DMSO): δ 9,58 (s, 1H), 9,29 (brs, 2H), 9,26 (s, 1H), 8,90 (t, J = 5,6 Гц, 1H), 7,12-7,10 (m, 2H), 7,03 (d, J = 7,6 Гц, 1H), 6,76 (s, 1H), 6,38 (s, 1H), 3,26-3,21 (m, 2H), 3,12-3,00 (m, 9H), 1,09 (t, J = 7,2 Гц, 3H), 1,00 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 436 (M+1).
Пример 29
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(изохинолин-6-ил)изоксазол-5-карбоксамид
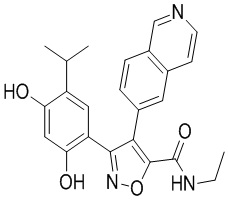
Реакционная схема
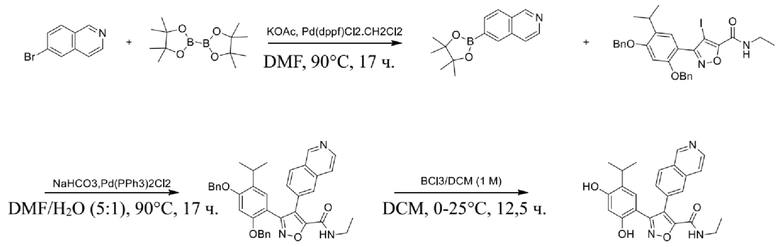
Стадия A. При 25°C в защитной атмосфере газообразного азота к раствору 6-бромизохинолина (250 мг, 1,289 ммоль) в диоксане (150 мл) добавляли бис(пинаколато)дибор (366 мг, 1,489 ммоль) и KOAc (354 мг, 3,689 ммоль) с последующим добавлением катализатора Pd(dppf)Cl2.CH2Cl2 (294 мг, 360 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 90°C при перемешивании в течение 17 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении с получением 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолина (300 мг, неочищенный продукт) в виде черного твердого вещества, которое непосредственно применяли на следующей стадии. MS (ESI) масса/заряд: 274 (M+1).
Стадия B. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолина (128 мг, 503 мкмоль) и 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-йодизоксазол-5-карбоксамида (200 мг, 335 мкмоль) в DMF (20 мл) добавляли K2CO3 (231 мг, 1,789 ммоль), H2O (5,0 мл) и Pd(PPh3)2Cl2 (71 мг, 101 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 90°C при перемешивании в течение 17 часов. Смесь охлаждали до 25°C, выливали в воду (50 мл) и экстрагировали с помощью EA (100 мл × л). Объединенный органический слой высушивали, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE:EA = от 5:1 до 2:1) с получением 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(изохинолин-6-ил)изоксазол-5-карбоксамида (180 мг, выход 90%) в виде коричневого твердого вещества.
Стадия C. При 0°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(изохинолин-6-ил)изоксазол-5-карбоксамида (180 мг, 301 мкмоль, 1,0 экв.) в DCM (10 мл) добавляли раствор BCl3 (1 M, 3,1 мл, 10,0 экв.) в DCM. После перемешивания при 0°C в течение 2 часов температуру повышали до 15-25°C при перемешивании в течение 12 часов. Реакционную смесь охлаждали до 0°C и добавляли MeOH (6 мл) с последующим перемешиванием при 0°C в течение 0,5 часа и при 15-25°C в течение дополнительного 0,5 часа. Смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной TLC (PE:EA = 1:1) с получением 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(изохинолин-6-ил)изоксазол-5-карбоксамида (15 мг, 32 мкмоль, выход 10%). 1H ЯМР (400 МГц, DMSO-d6) δ ppm: 0,83 (d, J = 6,90 Гц, 10H); 2,95-3,06 (m, 1H); 3,36 (q, J = 7,28 Гц, 2H); 6,27 (s, 1H); 6,79 (s, 1H); 7,58-7,64 (m, 1H); 7,74 (d, J = 5,77 Гц, 1H); 7,87 (s, 1H); 8,07 (d, J =8,53 Гц, 1H); 8,41 (d, J = 5,90 Гц, 1H); 9,23 (s, 1H). MS (ESI) масса/заряд: 418 (M+1).
Пример 30
5-(2,4-Дигидрокси-5-бромфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
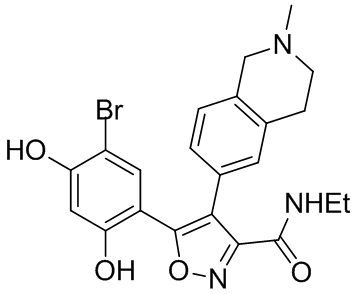
Реакционная схема
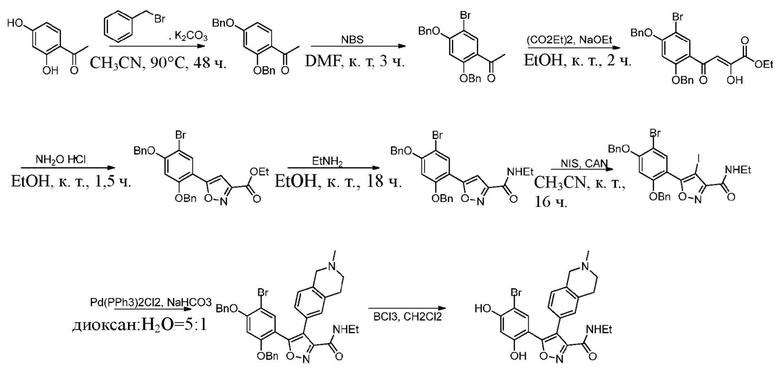
Стадия A. При 20°C в защитной атмосфере газообразного азота к смеси 1-(2,4-дигидроксифенил)этанона (50,00 г, 32989 ммоль, 1,00 экв.) и бензилбромида (124 г, 72389 ммоль, 2,2 экв.) в CH3CN (500 мл) добавляли карбонат калия (100 г, 72389 ммоль, 2,2 экв.). Смесь перемешивали при 80°C в течение 18 часов. Реакционный раствор охлаждали до комнатной температуры, затем фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт измельчали с помощью петролейного эфира в течение 30 мин. Твердое вещество собирали фильтрацией и высушивали с получением 1-(2,4-дибензилоксифенил)этанона (105 г, 31689 ммоль, выход 96%) в виде белого твердого вещества. (MS: (M+1) = 333,0).
Стадия B. При 0°C в защитной атмосфере газообразного азота к смеси 1-(2,4-дибензилоксифенил)этанона (50,00 г, 15089 ммоль, 1,00 экв.) в DMF (150 мл) добавляли NBS (27 г, 15089 ммоль, 1,0 экв.). Смесь перемешивали при 25°C в течение 3 часов. Реакционный раствор выливали в воду (200 мл) и твердое вещество собирали фильтрацией и высушивали с получением 1-(2,4-дибензилокси-5-бромфенил)этанона (61 г, 14889 ммоль, выход 98%) в виде белого твердого вещества. (MS: (M+1) = 411,1, 413,1).
Стадия C. При 20°C в защитной атмосфере газообразного азота к этанолу (400 мл) отдельными порциями добавляли металлический натрий (10,2 г, 44589 ммоль, 3,0 экв.) при перемешивании в течение 1 часа. Затем отдельными порциями добавляли 1-(2,4-дибензилокси-5-бромфенил)этанон (61,0 г, 14889 ммоль, 1,0 экв.) с последующим добавлением диметилоксалата (32,5 г, 22289 ммоль, 1,5 экв.). После перемешивания при 80°C в течение 2 часов реакционную смесь охлаждали до 65°C и добавляли ледяную уксусную кислоту (30 мл) и реакционный раствор выливали в ледяную воду (800 мл) при перемешивании для охлаждения. Твердое вещество собирали фильтрацией, промывали один раз водой (500 мл) и высушивали в вакууме с получением продукта, этил-2-гидрокси-4-(5-бром-2,4-диметоксифенил)-4-оксобутирата (32 г, 6389 ммоль, выход 42%) в виде желтого твердого вещества.
Стадия D. При комнатной температуре в защитной атмосфере газообразного азота к раствору этил-2-гидрокси-4-(5-бром-2,4-диметоксифенил)-4-оксобутирата (32 г, 6389 ммоль, 1,00 экв.) в EtOH (320 мл) добавляли NH2OH.HCl (5,2 г, 7589 ммоль, 1,2 экв.). Смесь нагревали до 80°C и перемешивали в течение 3 часов. Смесь охлаждали до комнатной температуры, и твердое вещество собирали фильтрацией, промывали водой (150 мл × 2) и этанолом (150 мл × 2) и высушивали в вакууме с получением этил-5-(5-бром-2,4-диметоксифенил)изоксазол-3-карбоксилата (23 г, 4589 ммоль, выход 72%) в виде желтого твердого вещества. (MS: (M+1) = 508,1, 510,2).
Стадия E. При комнатной температуре к раствору этил-5-(5-бром-2,4-диметоксифенил)изоксазол-3-карбоксилата (23 г, 4589 ммоль, 1,00 экв.) в EtOH (200 мл) добавляли этиламин (17 г, 38489 ммоль, 8,5 экв.). Реакционную смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и твердое вещество осаждали. Твердое вещество собирали фильтрацией, промывали этанолом (50 мл × 2) и высушивали в вакууме с получением продукта, N-этил-5-(5-бром-2,4-диметоксифенил)изоксазол-3-карбоксамида (13 г, 2689 ммоль, выход 58%) в виде желтого твердого вещества.
Стадия F. При комнатной температуре в защитной атмосфере N2 к раствору 5-(2,4-дибензилокси-5-бромфенил)-N-этилизоксазол-3-карбоксамида (2,0 г, 3,989 ммоль, 1,0 экв.) в MeCN (20 мл) добавляли CAN (432 мг, 788 мкмоль, 0,1 экв.) и NIS (1,3 г, 2,989 ммоль, 1,5 экв.). Смесь нагревали до 50°C и перемешивали в течение 12 часов. Смесь охлаждали до комнатной температуры и выливали в насыщенный водный раствор Na2S2O3 (40 мл) и водную фазу экстрагировали с помощью EA (20 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл ×), высушивали над безводным сульфатом натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = 3/1) с получением 5-(2,4-дибензилокси-5-бромфенил)-N-этил-4-йодизоксазол-3-карбоксамида (2,4 г, 3,889 ммоль, выход 98%) в виде коричневого масла. MS: [M+1] = 633,0, 635,0).
Стадия G. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (1,8 г, 489 ммоль) и 5-(2,4-дибензилокси-5-бромфенил)-N-этил-4-йодизоксазол-3-карбоксамида (2,5 г, 3,989 ммоль) в DMF (25,0 мл) добавляли NaHCO3 (1,3 г, 15,589 ммоль), H2O (2,5 мл) и Pd(PPh3)2Cl2 (407 мг, 580 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 80°C при перемешивании в течение 12 часов. Смесь охлаждали до 25°C и выливали в воду (50 мл) и водную фазу экстрагировали с помощью EA (30 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, DCM/MeOH = от 100:1 до 20:1) с получением 5-(2,4-дибензилокси-5-бромфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (2,0 г, выход 60%) в виде желтого твердого вещества.
Стадия H. При 0°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(2,4-дибензилокси-5-бромфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид (300 мг, 345 мкмоль) в дихлорметане (20,00 мл) по каплям добавляли BCl3/DCM (1,3 мл, 1,0 моль/л). Смесь перемешивали при 0°C в течение 1 часа. К смеси по каплям добавляли метанол (3 мл) и ее концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC (муравьиная кислота, колонка: Phenomenex Synergi Max-RP 250 × 80, 10 мкм, условия: 0,225% FA-ACN) с получением 5-(2,4-дигидрокси-5-бромфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (96 мг, выход 59%). 1H ЯМР (DMSO-d6, 300 МГц) δ: 1H ЯМР (400 МГц, DMSO-d6) δ =10,81 (s, 1H), 10,73 (br.s., 1H), 10,29 (s, 1H), 8,94 (t, J =5,65 Гц, 1H), 7,29 (s, 1H), 7,06-7,19 (m, 3H), 6,70 (s, 1H), 4,38-4,50 (m, 1H), 4,25 (dd, J = 15,43, 8,16 Гц, 1H), 3,59 (br.s., 1H), 3,19-3,32 (m, 3H), 3,07-3,19 (m, 1H), 2,83-2,97 (m, 4H), 1,09 (t, J = 7,15 Гц, 3H). MS (ESI) масса/заряд: 472, 474 (M+1).
Пример 31
5-(2,4-Дигидрокси-5-хлорфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
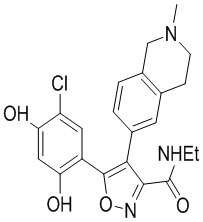
Реакционная схема
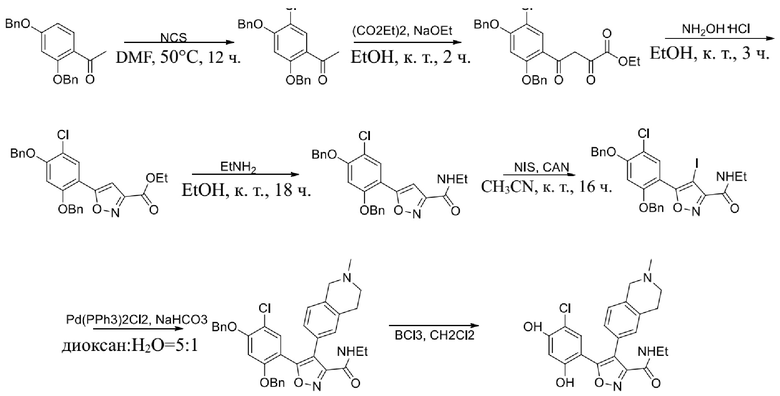
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий B, C, D, E, F, G и H в примере 30, при этом NBS на стадии B заменяли NCS, и продукт представлял собой белое твердое вещество. 1H ЯМР (400 МГц, DMSO-d6) δ = 10,72 (br.s., 1H), 10,23 (br.s., 1H), 8,95 (t, J = 5,65 Гц, 1H), 7,05-7,18 (m, 4H), 6,67 (br.s., 1H), 4,45 (d, J = 14,31 Гц, 1H), 4,25 (dd, J = 15,43, 8,41 Гц, 1H), 3,60 (br.s., 1H), 3,18-3,34 (m, 3H), 3,10 (br.s., 1H), 2,89 (br.s., 4H), 1,09 (t, J = 7,15 Гц, 3H). MS: [M+1] = 428.
Пример 32
5-(2,4-Дигидрокси-5-метилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
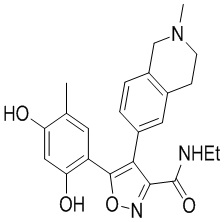
Реакционная схема
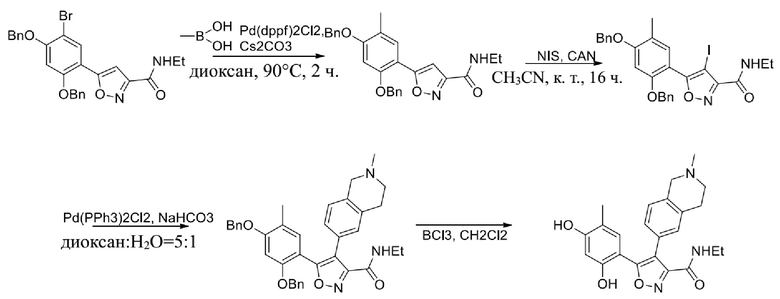
Стадия A. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору N-этил-5-(5-бром-2,4-диметоксифенил)изоксазол-3-карбоксамида (1,0 г, 2,0 мкмоль) и метилбороновой кислоты (177 мг, 3,089 ммоль) в диоксане (10,0 мл) добавляли Cs2CO3 (1,3 г, 489 ммоль) и Pd(dppf)2Cl2 (288 мг, 400 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 90°C при перемешивании в течение 2 часов. Смесь охлаждали до 25°C и выливали в воду (10 мл) и водную фазу экстрагировали с помощью EA (5 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат = 3/1) с получением 5-(5-метил-2,4-дибензилоксифенил)-N-этилизоксазол-3-карбоксамида (740 мг, выход 85%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 443 (M+1).
Стадия B. При комнатной температуре в защитной атмосфере N2 к раствору 5-(5-метил-2,4-дибензилоксифенил)-N-этилизоксазол-3-карбоксамида (740 мг, 1,789 ммоль, 1,0 экв.) в MeCN (30 мл) добавляли CAN (92 мг, 167 мкмоль, 0,1 экв.) и NIS (488 мг, 2,289 ммоль, 1,3 экв.). Смесь нагревали до 80°C при перемешивании в течение 12 часов. Смесь охлаждали до комнатной температуры и выливали в воду (30 мл) и водную фазу экстрагировали с помощью EA (20 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл × 3), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = 3/1) с получением 5-(5-метил-2,4-дибензилоксифенил)-N-этил-4-йодизоксазол-3-карбоксамида (450 мг, 79289 ммоль, выход 47,0%) в виде желтого масла.
Стадия C. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (309 мг, 679 мкмоль) и 5-(5-метил-2,4-дибензилоксифенил)-N-этил-4-йодизоксазол-3-карбоксамида (450 мг, 721 мкмоль) в диоксане (5,0 мл) добавляли NaHCO3 (228 мг, 2,789 ммоль), H2O (1,0 мл) и Pd(PPh3)2Cl2 (71 мг, 102 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 80°C при перемешивании в течение 12 часов. Смесь охлаждали до 25°C и выливали в воду (50 мл) и водную фазу экстрагировали с помощью EA (40 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат = 50/1, 3/1) с получением 5-(5-метил-2,4-дибензилоксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (280 мг, выход 70%) в виде желтого масла. MS (ESI) масса/заряд: 588 (M+1).
Стадия D. При 0°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(5-метил-2,4-дибензилоксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (280 мг, 476 мкмоль) в дихлорметане (10,00 мл) по каплям добавляли BCl3/DCM (1,8 мл, 1,0 моль/л). Смесь перемешивали при 0°C в течение 1 часа. К смеси по каплям добавляли метанол (4 мл) и ее концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC (муравьиная кислота, колонка: Phenomenex Synergi Max-RP 250 × 80, 10 мкм, условия: 0,225% FA-ACN) с получением 5-(5-метил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (57 мг, выход 29%). 1H ЯМР (DMSO-d6, 400 МГц) δ: 1H ЯМР (400 МГц, DMSO-d6) δ = 8,90 (t, J = 5,52 Гц, 1H), 8,17 (s, 1H), 7,01-7,10 (m, 1H), 6,95 (s, 2H), 6,90 (s, 1H), 6,42 (s, 1H), 3,47 (s, 2H), 3,23 (квин., J = 6,78 Гц, 2H), 2,67-2,75 (m, 2H), 2,57-2,63 (m, 2H), 2,34 (s, 3H), 1,98 (s, 3H), 1,09 (t, J = 7,15 Гц, 3H). MS (ESI) масса/заряд: 408 (M+1).
Пример 33
5-(5-Изобутил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
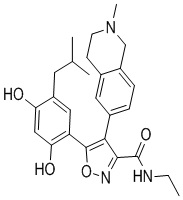
Реакционная схема
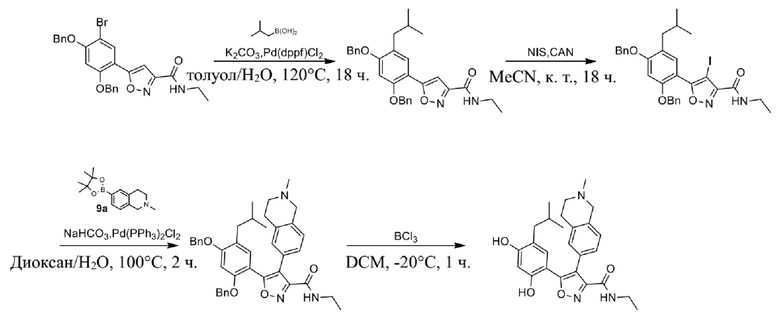
Стадия A. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(2,4-дибензилокси-5-бромфенил)-N-этилизоксазол-3-карбоксамида (2,00 г, 3,9489 ммоль, 1 экв.) и изобутилбороновой кислоты (803,67 мг, 7,8889 ммоль, 2 экв.) в толуоле (50 мл) добавляли Na2CO3 (835,60 мг, 7,8889 ммоль, 2 экв.), H2O (2,0 мл) и Pd(dppf)2Cl2 (576,86 мг, 788,0089 ммоль, 0,2 экв). Смесь нагревали до 120°C при перемешивании в течение 18 часов. Смесь фильтровали через диатомовую землю и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат = 6/1) с получением 5-(2,4-дибензилокси-5-изобутилфенил)-N-этилизоксазол-3-карбоксамида (500,00 мг, выход 26,19%) в виде белого твердого вещества.
Стадия B. При комнатной температуре в защитной атмосфере N2 к раствору 5-(2,4-дибензилокси-5-изобутилфенил)-N-этилизоксазол-3-карбоксамида (500,00 мг, 1,0389 ммоль, 1,0 экв.) в MeCN (50 мл) добавляли CAN (54,67 мг, 103,00 мкмоль, 0,10 экв.) и NIS (463,46 мг, 2,0689 ммоль, 2,00 экв.). Смесь нагревали до 10°C при перемешивании в течение 18 часов. Смесь охлаждали до комнатной температуры и выливали в водный раствор Na2SO3 (40 мл) и водную фазу экстрагировали с помощью EA (30 мл × 3). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат = 3/1) с получением 5-(2,4-дибензилокси-5-изобутилфенил)-N-этил-4-йодизоксазол-3-карбоксамида (400,00 мг, 655,22 мкмоль, выход 63,61%) в виде белого твердого вещества.
Стадия C. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(2,4-дибензилокси-5-изобутилфенил)-N-этил-4-йодизоксазол-3-карбоксамида (400 мг, 655,2289 ммоль, 1 экв.) и 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (268,49 мг, 982,83 мкмоль, 1,5 экв.) в диоксане (2,50 мл) добавляли NaHCO3 (165,14 мг, 7,8889 ммоль, 2 экв), H2O (0,5 мл) и Pd(PPh3)2Cl2 (45,99 мг, 65,52 мкмоль, 0,10 экв.). Смесь нагревали до 90°C при перемешивании в течение 3 часов. Смесь фильтровали через диатомовую землю и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (дихлорметан/метанол = 6/1) с получением 5-(5-изобутил-2,4-дибензилоксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (300,00 мг, неочищенный продукт) в виде желтого твердого вещества.
Стадия D. При -20°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(5-изобутил-2,4-дибензилоксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (150,00 мг, 238,17 мкмоль) в дихлорметане (10,00 мл) по каплям добавляли BCl3/DCM (1 мл, 1,0 моль/л). Смесь перемешивали при -20°C в течение 1 часа. К смеси по каплям добавляли метанол (2 мл) и ее концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC (муравьиная кислота, колонка: Phenomenex Synergi Max-RP 250 × 80, 10 мкм, условия: 0,225% FA-ACN) с получением 5-(5-изобутил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (28,20 мг, выход 26,34%). 1H ЯМР (DMSO-d6, 400 МГц) δ 8,87 (t, J = 5,6 Гц, 1H), 6,95 (d, J = 4,4 Гц, 1H), 6,71 (s,1H), 6,42 (s, 1H), 6,82 (s, 1H), 6,42 (s, 1H), 3,45 (s, 2H), 3,20-3,25 (m, 2H), 2,68 (d, J = 5,2 Гц, 2H), 2,53-2,60 (m, 2H), 2,50 (s, 3H), 2,18-2,35 (m, 2H), 1,68 (dd, J = 6,8 Гц, 2H), 1,07 (t, J = 7,2 Гц, 3H), 0,72 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 450,3 (M+1)
Пример 34
5-(5-Этил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
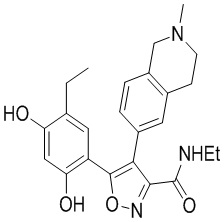
Реакционная схема
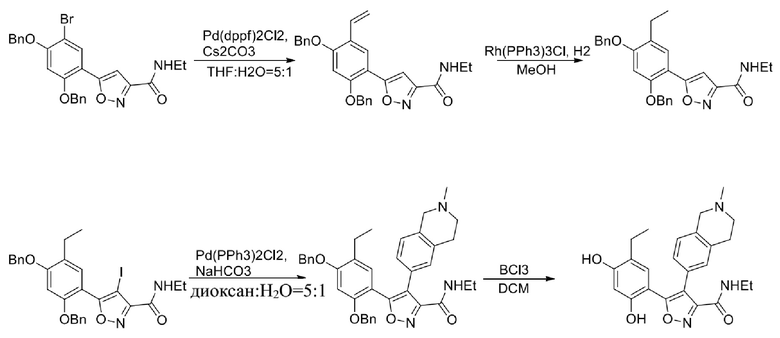
Стадия A. При 25°C в защитной атмосфере газообразного азота к раствору 5-(2,4-дибензилокси-5-бромфенил)-N-этилизоксазол-3-карбоксамида (1 г, 1,9789 ммоль) в тетрагидрофуране (40 мл) и воде (4 мл) добавляли винилтрифторборат калия (316,81 мг, 2,3689 ммоль) и карбонат цезия (1,93 г, 5,9189 ммоль). Смесь нагревали до 80°C при перемешивании в течение 12 часов. Смесь охлаждали до 25°C и выливали в воду (30 мл) и водную фазу экстрагировали с помощью EA (30 мл × 2). Объединенную органическую фазу промывали водой (50 мл) и насыщенным солевым раствором (40 мл), высушивали над безводным сульфатом натрия, фильтровали, и концентрировали в вакууме до сухого состояния, и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = 3/1) с получением целевого продукта (700,00 мг, выход 78,17%) в виде желтого твердого вещества. (MS: [M+1] = 455,1).
Стадия B. При 25°C в защитной атмосфере газообразного водорода к раствору 5-(2,4-дибензилокси-5-винилфенил)-N-этилизоксазол-3-карбоксамида (0,7 г, 1,5489 ммоль) в метаноле (20 мл) добавляли хлорид трис(трифенилфосфин)родия (71,25 мг, 77 мкмоль). Смесь нагревали до 50°C и перемешивали при 50 фунтов/кв. дюйм в течение 12 часов. Смесь охлаждали до 25°C, фильтровали и концентрировали при пониженном давлении. Остаток разбавляли водой (20 мл) и водную фазу экстрагировали с помощью EA (10 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (30 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = 2/1) с получением целевого продукта (600,00 мг, выход 85,06%) в виде желтого твердого вещества. (MS: [M+1] = 457,1)
Стадия C. При 25°C в защитной атмосфере газообразного азота к раствору 5-(2,4-дибензилокси-5-этилфенил)-N-этилизоксазол-3-карбоксамида (0,6 г, 1,3189 ммоль) в ацетонитриле (20 мл) добавляли йодсукцинимид (442,09 мг, 1,9689 ммоль). Смесь перемешивали при 25°C в течение 12 часов. Смесь охлаждали до 25°C. После фильтрации и концентрирования смеси при пониженном давлении добавляли тиосульфат натрия (30 мл). Водную фазу экстрагировали с помощью EA (20 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (40 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 100-200 меш, петролейный эфир/этилацетат = 3/1) с получением целевого продукта (420,00 мг, выход 55,05%) в виде желтого твердого вещества. (MS: [M+1] = 583,1)
Стадия D. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(2,4-дибензилокси-5-этилфенил)-N-этил-4-йодизоксазол-3-карбоксамида (320 мг, 549 мкмоль, 1 экв.) и 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (250 мг, 549 мкмоль, 1,0 экв.) в диоксане (5,0 мл) добавляли NaHCO3 (184 мг, 220 мкмоль, 4,0 экв.), H2O (1,0 мл) и Pd(PPh3)2Cl2 (39 мг, 55 мкмоль, 0,10 экв.). Смесь нагревали до 80°C при перемешивании в течение 12 часов. Смесь фильтровали через диатомовую землю и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной TLC (дихлорметан/метанол = 10/1) с получением 5-(5-этил-2,4-дибензилоксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (67,00 мг, 111 мкмоль, выход 20%) в виде коричневого твердого вещества. MS: [M+1] = 602,3.
Стадия E. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(5-этил-2,4-дибензилоксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (100,00 мг, 166 мкмоль) в дихлорметане (10,00 мл) по каплям добавляли BCl3/DCM (5 мл, 1,0 моль/л). Смесь перемешивали при 25°C в течение 0,5 часа. К смеси по каплям добавляли метанол (10 мл) и ее концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с получением 5-(5-этил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (15 мг, 36 мкмоль, выход 21%). 1H ЯМР (400 МГц, DMSO-d6) δ =8,90 (t, J = 5,52 Гц, 1H), 7,14 (s, 1H), 7,09 (br.s., 2H), 6,85 (s, 1H), 6,43 (s, 1H), 3,50 (br.s., 2H), 3,18-3,25 (m, 2H), 2,89 (s, 3H), 2,46 (br.s., 2H), 2,30-2,41 (m, 4H), 1,08 (t, J = 7,15 Гц, 3H), 0,98 (t, J = 7,40 Гц, 3H). MS (ESI) масса/заряд: 422 (M+1).
Пример 35
5-(5-Циклопропил-2,4-дигидроксифенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
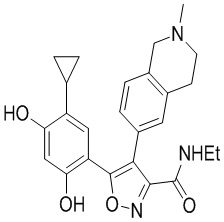
Реакционная схема
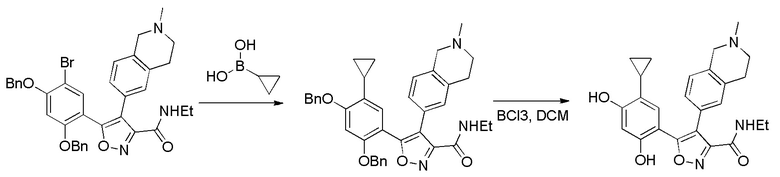
Стадия A. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(2,4-бензилокси-5-бромфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-изоксазол-3-карбоксамида (1,10 г, 1,6989 ммоль, 1,00 экв.), растворенного в PhMe (10,00 мл) и H2O (2,1 мл), добавляли циклопропилбороновую кислоту (217,76 мг, 2,5489 ммоль, 1,50 экв.), K3PO4 (717,47 мг, 3,3889 ммоль, 2,00 экв.) и Pd(OAc)2 (75,88 мг, 338,00 мкмоль, 0,20 экв.). Смесь перемешивали при 25°C в течение 20 мин., а затем нагревали до 90°C при перемешивании в течение 12 часов. Смесь охлаждали до 25°C и реакционный раствор выливали в раствор хлорида аммония (20 мл) при перемешивании в течение 10 мин. Водную фазу экстрагировали этилацетатом (10 мл × л). Объединенную органическую фазу промывали насыщенным солевым раствором (10 мл × л), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме. Остаток очищали с применением пластинки для тонкослойной хроматографии (дихлорметан:этилацетат = от 100/1 до 10/1) с получением 5-(2,4-бензилокси-5-циклопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (300,00 мг, 488,81 мкмоль, выход 28,92%) в виде черного масла.
Стадия B. При 0°C в защитной атмосфере газообразного азота к перемешанному раствору 5-(2,4-бензилокси-5-циклопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-изоксазол-3-карбоксамида (300 мг, 489 мкмоль) в дихлорметане (30,00 мл) по каплям добавляли BCl3/DCM (1,9 мл,1 моль/л). Смесь перемешивали при 0°C в течение 1 часа. К смеси по каплям добавляли метанол (4 мл) и ее концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC (муравьиная кислота, колонка: Phenomenex Synergi Max-RP 250 × 80, 10 мкм, условия: 0,225% FA-ACN) с получением 5-(2,4-дигидрокси-5-циклопропилфенил)-N-этил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-изоксазол-3-карбоксамида (22 мг, выход 10%). 1H ЯМР (DMSO-d6, 400 МГц) δ: 1H ЯМР (400 МГц, DMSO-d6) δ =8,80-8,86 (m, 1H), 6,90-7,01 (m, 3H), 6,51 (s, 1H), 6,42 (s, 1H), 3,44 (s, 2H), 3,16-3,27 (m, 2H), 2,67-2,76 (m, 3H), 2,55-2,60 (m, 3H), 2,32 (s, 3H), 2,27 (br.s., 1H), 1,08 (t, J = 7,16 Гц, 3H), 0,70 (d, J = 8,29 Гц, 2H), 0,28 (d, J = 4,14 Гц, 2H). MS (ESI) масса/заряд: 434 (M+1).
Пример 36
5-(2,4-Дигидрокси-5-изопропилфенил)-N-метил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
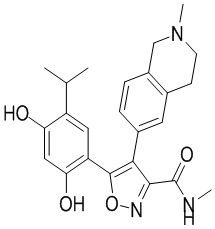
Реакционная схема
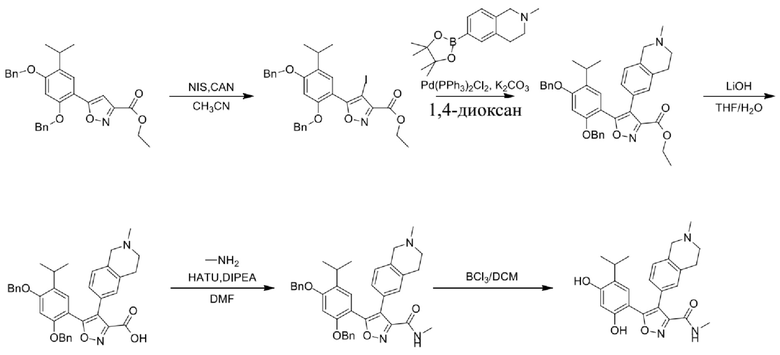
Стадия A. При комнатной температуре в защитной атмосфере N2 к раствору этил-5-(2,4-дибензилокси-5-изопропилфенил)изоксазол-3-формиата (4,00 г, 8,589 ммоль, 1,0 экв.) в MeCN (50 мл) добавляли CAN (465 мг, 848 мкмоль, 0,1 экв.) и NIS (7,63 г, 34,089 ммоль, 4,0 экв.). Смесь нагревали до 90°C при перемешивании в течение 12 часов. Смесь охлаждали до комнатной температуры и выливали в воду (40 мл) и водную фазу экстрагировали с помощью EA (50 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл × 2), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (PE/EA = от 10/1 до 4/1) с получением этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-3-карбоксилата (3,40 г) в виде желтого твердого вещества. MS (ESI) масса/заряд: (M+1).
Стадия B. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (2,47 г, 9,089 ммоль) и этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-3-карбоксилата (2,70 г, 4,589 ммоль) в диоксане (30 мл) добавляли NaHCO3 (1,52 г, 18,089 ммоль), H2O (6,0 мл) и Pd(PPh3)4 (1,26 г, 1,8089 ммоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 110°C при перемешивании в течение 18 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат, дихлорметан/метанол = 5/1, 10/1) с получением этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-формиата (1,4 г) в виде желтого масла. MS (ESI) масса/заряд: 617 (M+1).
Стадия C. При 20°C к перемешанному раствору этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксилата в THF (15 мл)/H2O (15 мл) добавляли LiOH (101 мг, 4,289 ммоль). Смесь перемешивали при 20°C в течение 2 часов. Смесь доводили до pH 6 с помощью HCl и экстрагировали с помощью EA (30 мл × 3). Объединенную органическую фазу высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме до сухого состояния. Получали неочищенный продукт, 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-муравьиную кислоту (1 г) в виде коричневого твердого вещества. MS (ESI) масса/заряд: 589 (M+1).
Стадия D. При 20°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-муравьиной кислоты (200 мг, 340 мкмоль) в DMF (5 мл) добавляли DIPEA (220 мг, 1,789 ммоль) и HATU (194 мг, 510 мкмоль). Смесь перемешивали при 20°C в течение 0,5 часа. Затем добавляли гидрохлорид метиламина (30 мг, 441 мкмоль) и реакционный раствор перемешивали при 20°C в течение 1 часа. К реакционному раствору добавляли H2O (20 мл) для осаждения твердого вещества. Твердое вещество собирали фильтрацией, промывали водой (5 мл) и высушивали с получением 5-(2,4-дибензилокси-5-изопропилфенил)-N-метил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (120 г) в виде желтого твердого вещества. MS (ESI) масса/заряд: 602 (M+1).
Стадия E. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-метил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (120 мг, 199 мкмоль, 1,0 экв.) в DCM (5 мл) добавляли BCl3 (1 M, 2 мл, 10,0 экв.). После перемешивания смеси при 20°C в течение 12 часов добавляли 5 мл MeOH для гашения и смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC с получением 5-(2,4-гидрокси-5-изопропилфенил)-N-метил-4-(2-метил-1,2,3,4-тетраизохинолин-6-ил)изоксазол-3-карбоксамида (38 мг, выход 34%). 1H ЯМР (400 МГц, DMSO-d6) δ 10,75 (s, 1H), 9,85 (s, 1H), 9,72 (s, 1H), 8,81 (s, 1H), 7,14-7,08 (m, 3H), 6,86 (s, 1H), 6,46 (s, 1H), 4,41-4,23 (m, 2H), 3,59 (brs, 1H), 3,04-2,99 (m, 4H), 2,87 (s, 3H), 2,73 (s, 3H), 1,00 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 422 (M+1).
Пример 37
5-(2,4-Дигидрокси-5-изопропилфенил)-N-пропил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
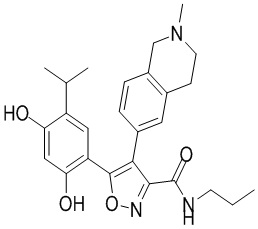
Реакционная схема
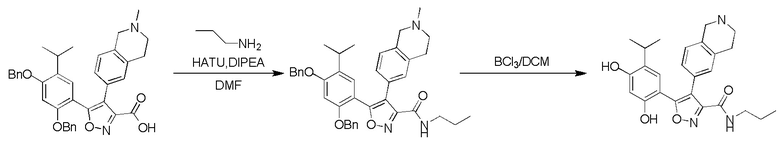
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 36, при этом гидрохлорид метиламина на стадии D заменяли пропиламином, и продукт представлял собой бледно-желтое твердое вещество. 1H ЯМР (400 МГц, DMSO-d6) δ 10,87 (s, 1H), 9,84 (s, 1H), 9,71 (s, 1H), 8,89 (t, J = 6,0 Гц, 1H), 7,14-7,08 (m, 3H), 6,88 (s, 1H), 6,48 (s, 1H), 4,45-4,21 (m, 2H), 3,58 (brs, 1H), 3,19-3,14 (m, 4H), 3,05-3,01 (m, 2H), 2,86 (s, 3H), 1,52-1,46 (m, 2H), 1,01 (d, J = 6,8 Гц, 6H), 0,86 (t, J = 7,2 Гц, 3H). MS (ESI) масса/заряд: 450 (M+1).
Пример 38
5-(2,4-Дигидрокси-5-изопропилфенил)-N-изопропил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
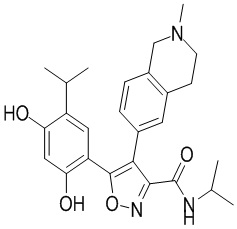
Реакционная схема
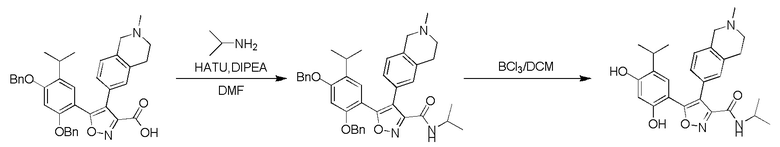
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 36, при этом гидрохлорид метиламина на стадии D заменяли пропан-2-амином, и продукт представлял собой бледно-желтое твердое вещество. 1H ЯМР (400 МГц, DMSO-d6) δ 10,41 (s, 1H), 9,80 (s, 1H), 9,66 (s, 1H), 8,81 (d, J = 7,6 Гц, 1H), 7,14-7,08 (m, 3H), 6,87 (s, 1H), 6,45 (s, 1H), 4,46-4,24 (m, 2H), 4,01 (q, J = 6,8 Гц, 1H), 3,60 (brs, 1H), 3,06-2,99 (m, 2H), 2,89 (brs, 5H), 1,13 (d, J = 6,4 Гц, 6H), 1,01 (d, J = 7,2 Гц, 6H). MS (ESI) масса/заряд: 450 (M+1).
Пример 39
5-(2,4-Дигидрокси-5-изопропилфенил)-N-(2,2,2-трифторэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
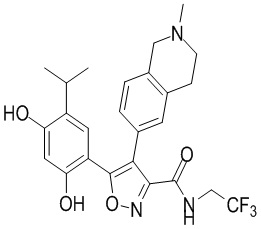
Реакционная схема
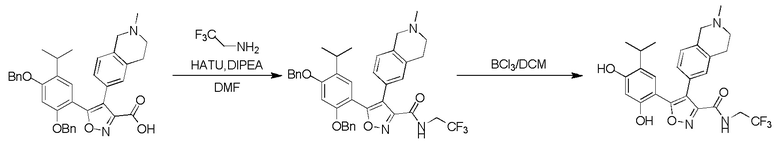
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 36, при этом гидрохлорид метиламина на стадии D заменяли 2,2,2-трифторэтиламином. 1H ЯМР (400 МГц, DMSO-d6) δδ 10,85 (brs, 1H), 9,88 (s, 1H), 9,76 (s, 1H), 9,64 (t, J = 6,4 Гц, 1H), 7,11-7,06 (m, 3H), 6,89 (s, 1H), 6,48 (s, 1H), 4,23 (brs, 2H), 4,09-4,05 (m, 2H), 3,04-2,96 (m, 3H), 2,81 (s, 3H), 1,01 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 490 (M+1).
Пример 40
5-(2,4-Дигидрокси-5-изопропилфенил)-N-(2-фторэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
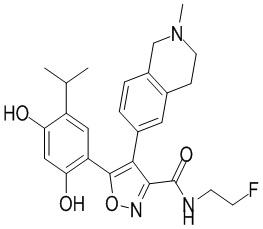
Реакционная схема
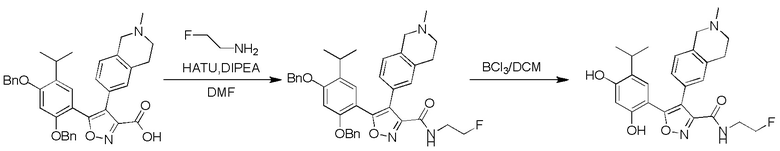
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 36, при этом гидрохлорид метиламина на стадии D заменяли 2-фторэтиламином. 1H ЯМР (400 МГц, DMSO-d6) δ 10,37 (brs, 1H), 9,83 (s, 1H), 9,70 (s, 1H), 9,13 (t, J = 5,6 Гц, 1H), 7,15-7,08 (m, 3H), 6,87 (s, 1H), 6,45 (s, 1H), 4,58-4,44 (m, 2H), 4,22 (brs, 1H), 3,56-3,36 (m, 2H), 3,20-3,00 (m, 4H), 2,90 (brs, 5H), 1,00 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 454 (M+1).
Пример 41
5-(2,4-Дигидрокси-5-изопропилфенил)-N-(2-метоксиэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
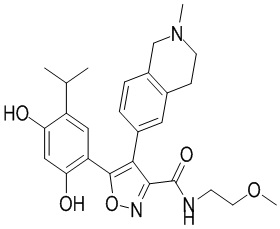
Реакционная схема
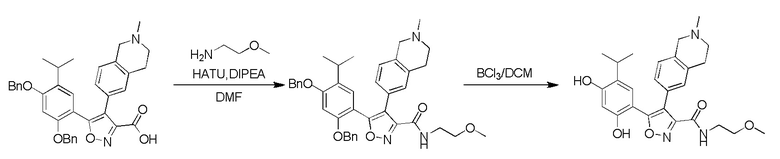
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 36, при этом гидрохлорид метиламина на стадии D заменяли 2-метоксиэтиламином. 1H ЯМР (400 МГц, DMSO-d6) δ 10,55 (brs, 1H), 9,84 (s, 1H), 9,71 (s, 1H), 8,92 (t, J = 5,6 Гц, 1H), 7,14-7,10 (m, 3H), 6,87 (s, 1H), 6,46 (s, 1H), 4,28 (brs, 3H), 3,44-3,37 (m, 6H), 3,26 (s, 3H), 3,03-2,98 (m, 2H), 2,87 (s, 3H), 1,00 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 466 (M+1).
Пример 42
5-(2,4-Дигидрокси-5-изопропилфенил)-N-(3-метоксипропил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
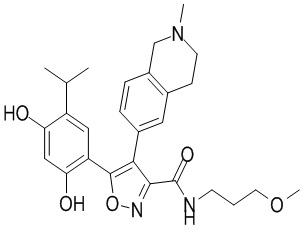
Реакционная схема
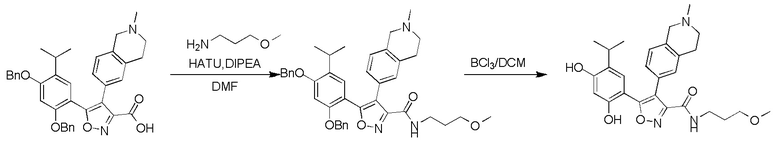
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 36, при этом гидрохлорид метиламина на стадии D заменяли 3-метоксипропан-1-амином. 1H ЯМР (400 МГц, DMSO-d6) δ 10,77 (brs, 1H), 9,83 (s, 1H), 9,71 (s, 1H), 8,89 (t, J = 5,6 Гц, 1H), 7,14-7,08 (m, 3H), 6,87 (s, 1H), 6,47 (s, 1H), 4,45-4,20 (m, 2H), 3,58 (brs, 1H), 3,32-3,21 (m, 7H), 3,19-2,99 (m, 3H), 2,87 (brs, 4H), 1,72-1,69 (m, 2H), 1,01 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 480 (M+1).
Пример 43
5-(2,4-Дигидрокси-5-изопропилфенил)-N-[2-(диметиламино)этил]-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
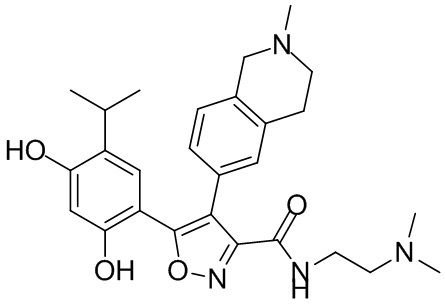
Реакционная схема
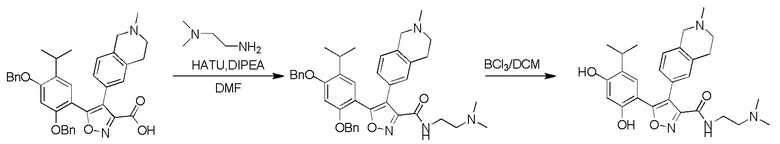
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 36, при этом гидрохлорид метиламина на стадии D заменяли N',N'-диметилэтан-1,2-диамином. 1H ЯМР (400 МГц, DMSO-d6) δ 9,83 (s, 1H), 9,72 (s, 1H), 9,05 (t, J = 5,6 Гц, 1H), 7,17-7,09 (m, 3H), 6,84 (s, 1H), 6,46 (s, 1H), 4,24 (brs, 2H), 3,59-3,58 (m, 3H), 3,19 (brs, 4H), 3,02-2,99 (m, 2H), 2,85 (s, 3H), 2,77 (s, 6H), 0,99 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 479 (M+1), 240 (M/2+1).
Пример 44
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-гидроксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
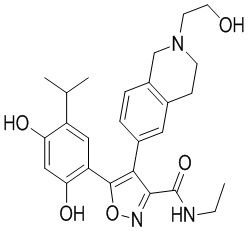
Реакционная схема
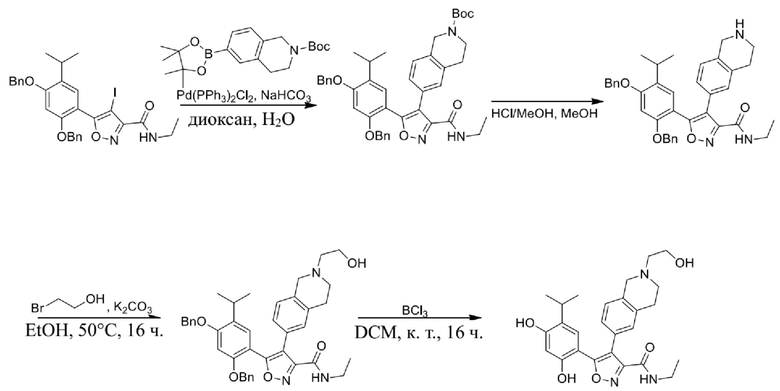
Стадия A. В защитной атмосфере газообразного азота к перемешанному раствору диоксана (40 мл) и воды (8 мл) добавляли трет-бутил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин-2-формиат (4,00 г, 11,189 ммоль, 1,6 экв.), этил-5-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-3-карбоксилат (4,10 г, 6,989 ммоль, 1,0 экв.), Pd(PPh3)2Cl2 (723 мг, 1,089 ммоль, 0,15 экв.) и NaHCO3 (2,31 г, 27,589 ммоль, 4,0 экв.). Смесь нагревали до 80°C при перемешивании в течение 16 часов в защитной атмосфере N2. После охлаждения реакционную смесь выливали в воду (50 мл) и экстрагировали этилацетатом (40 мл × 3). Органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали в вакууме с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат = от 50/1 до 3/1) с получением трет-бутил-6-[5-(2,4-дибензилокси-5-изопропилфенил)-3(этилкарбамоил)изоксазол-4-ил]-(3,4-дигидроизохинолин)-2(1H)-формиата (4,8 г, 6,889 ммоль, выход 99%) в виде желтого твердого вещества. MS: [M-56] = 646.
Стадия B. Смесь трет-бутил-6-[5-(2,4-дибензилокси-5-изопропилфенил)-3-(этилкарбамоил)изоксазол-4-ил]-(3,4-дигидроизохинолин)-2(1H)-формиата (750 мг, 1,0789 ммоль, 1,00 экв.) в HCl/MeOH (4 M, 6,00 мл) перемешивали при 25°C в течение 30 мин. Смесь концентрировали при 40°C с получением продукта, 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (630 мг, 1,0789 ммоль, выход 98%) в виде желтого твердого вещества.
Стадия C. К этанолу (5 мл) добавляли 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид (4,00 г, 382 мкмоль, 1,0 экв.), K2CO3 (158 мг, 1,289 ммоль, 3,0 экв.) и 2-бромэтанол (72 мг, 570 мкмоль, 1,5 экв.). Смесь нагревали до 50°C при перемешивании в течение 12 часов. После охлаждения реакционную смесь концентрировали до сухого состояния, добавляли воду (10 мл) и экстрагировали этилацетатом (10 мл × 2). Органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным Na2SO4 и концентрировали в вакууме с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (дихлорметан/метанол = от 50/1 до 15/1) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-[2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)]изоксазол-3-карбоксамида (132 мг, 204 мкмоль, выход 53%) в виде белого твердого вещества. MS: [M+1] = 646,3.
Стадия D. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-[2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)]изоксазол-3-карбоксамида (132 мг, 204 мкмоль, 1,0 экв.) в DCM (5,0 мл) по каплям добавляли раствор BCl3 в DCM (1 M, 2,0 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (1 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-[2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)]изоксазол-3-карбоксамида (51 мг, 110 мкмоль, выход 53,6%). 1H ЯМР (400 МГц, DMSO-d6) δ = 8,85 (t, J = 5,52 Гц, 1H), 8,21 (s, 1H), 6,94-7,01 (m, 3H), 6,86 (s, 1H), 6,42 (s, 1H), 3,54-3,60 (m, 4 H), 3,24 (dd, J = 13,18, 6,90 Гц, 5H), 3,02 (dt, J = 13,80, 6,90 Гц, 2H), 2,68 (br.s., 4H), 2,34 (br.s., 1H), 1,09 (t, J = 7,15 Гц, 3H), 1,00 (d, J = 6,78 Гц, 6H). MS (ESI) масса/заряд: 466 (M+1).
Пример 45
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-метоксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
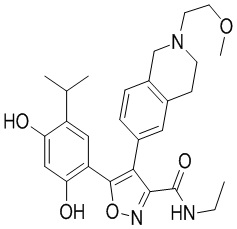
Реакционная схема
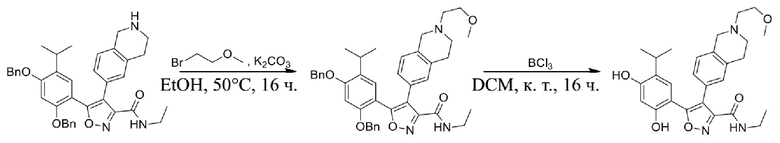
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий C и D в примере 44, при этом 2-бромэтанол на стадии C заменяли 1-бром-2-метоксиэтаном. 1H ЯМР (400 МГц, DMSO-d6) δ = 10,59 (br.s., 1H), 9,85 (s, 1H), 9,71 (s, 1H), 8,91 (t, J = 5,65 Гц, 1H), 7,05-7,20 (m, 3H), 6,83-6,93 (m, 1H), 6,48 (s, 1H), 4,43-4,52 (m, 1H), 4,26-4,36 (m, 1H), 3,63-3,83 (m, 3H), 3,38-3,44 (m, 2H), 3,32 (s, 4H), 3,23 (квин., J = 6,78 Гц, 2H), 3,08-3,18 (m, 1H), 3,02 (dt, J = 13,74, 6,81 Гц, 1H), 2,84-2,95 (m, 1H), 1,09 (t, J = 7,15 Гц, 3H), 1,01 (d, J = 6,78 Гц, 6H). MS (ESI) масса/заряд: 480 (M+1).
Пример 46
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-фторэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
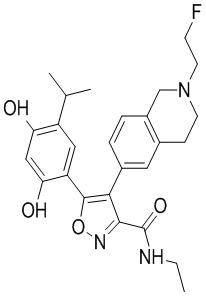
Реакционная схема
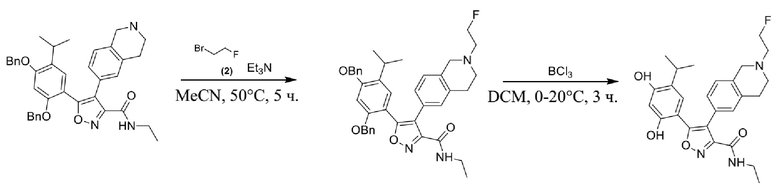
Стадия A. К ацетонитрилу (3 мл) добавляли 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид (150 мг, 250 мкмоль, 1,0 экв.), триэтиламин (63 мг, 623 мкмоль, 2,5 экв.) и 1-бром-2-фторэтан (44 мг, 350 мкмоль, 1,4 экв). Смесь нагревали до 50°C при перемешивании в течение 5 часов. После охлаждения реакционную смесь концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной TLC с получением 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-[2-(2-фторэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)]изоксазол-3-карбоксамида (120 мг, 185 мкмоль, выход 74%) в виде желтого твердого вещества. MS: [M+1] = 646,3.
Стадия B. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-[2-(2-фторэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)]изоксазол-3-карбоксамида (120 мг, 185 мкмоль, 1,0 экв.) в DCM (8,0 мл) по каплям добавляли раствор BCl3 в DCM (1 M, 1,9 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (4 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-[2-(2-фторэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)]изоксазол-3-карбоксамида (48 мг, 96 мкмоль, выход 52%). 1H ЯМР (400 МГц, DMSO-d6) ppm: 1,01 (d, J = 6,90 Гц, 6H); 1,08 (t, J = 7,22 Гц, 3H); 2,91 (d, J = 15,69 Гц, 1H); 3,01 (dt, J = 13,77, 6,85 Гц, 1H); 3,08-3,28 (m, 3H); 3,37-3,41 (m, 1H); 3,49-3,82 (m, 3H); 4,24-4,62 (m, 2H); 4,79-5,10 (m, 2H); 6,46 (s, 1H); 6,88 (s, 1H); 7,05-7,19 (m, 3H); 8,91 (t, J = 5,65 Гц, 1H); 9,70 (s, 1H); 9,83 (s, 1H); 11,01 (br.s., 1H). MS (ESI) масса/заряд: 468 (M+1).
Пример 47
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(3-метоксипропил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
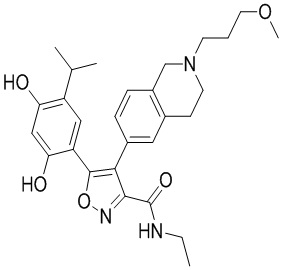
Реакционная схема
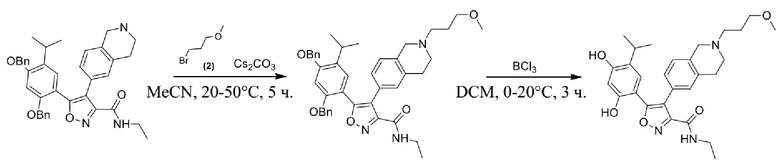
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 46, при этом 1-бром-2-фторэтан заменяли 1-бром-3-метоксипропаном, а триэтиламин заменяли карбонатом цезия на стадии A. 1H ЯМР (400 МГц, DMSO-d6) ppm: 1,01 (d, J = 6,90 Гц, 6H); 1,09 (t, J = 7,15 Гц, 3H); 1,96-2,08 (m, 2H); 2,89 (d, J = 17,32 Гц, 1H); 2,97-3,17 (m, 2H); 3,19-3,27 (m, 8H); 3,41 (t, J = 5,90 Гц, 3H); 3,67 (d, J = 10,67 Гц, 1H); 4,25 (dd, J = 15,56, 8,03 Гц, 1H); 4,50 (d, J = 15,18 Гц, 1H); 6,47 (s, 1H); 6,88 (s, 1H); 7,07-7,18 (m, 3H); 8,91 (t, J = 5,65 Гц, 1H); 9,70 (s, 1H); 9,83 (s, 1H); 10,53 (br.s., 1H). MS (ESI) масса/заряд: 494 (M+1).
Пример 48
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-этилсульфонил)этил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
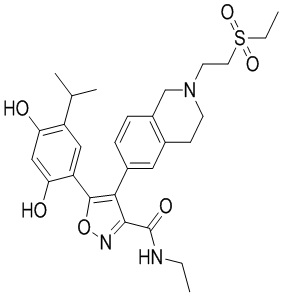
Реакционная схема
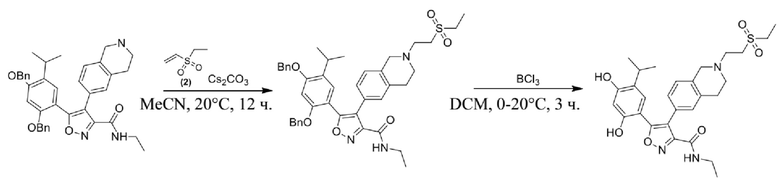
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 46, при этом 1-бром-2-фторэтан заменяли 1-винил-сульфонилэтаном на стадии A. 1H ЯМР (400 МГц, DMSO-d6) ppm: 0,96-1,13 (m, 9H); 1,28 (t, J = 7,44 Гц, 3H); 2,86-3,16 (m, 3H); 3,16-3,31 (m, 5H); 3,52-3,97 (m, 5H); 4,18-4,72 (m, 2H); 6,46 (s, 1H); 6,89 (s, 1H); 7,04-7,20 (m, 3H); 8,91 (t, J = 5,65 Гц, 1H); 9,60-9,94 (m, 2H); 11,20 (br.s., 1H). MS (ESI) масса/заряд: 542 (M+1).
Пример 49
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
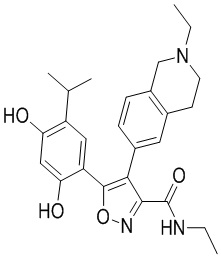
Реакционная схема
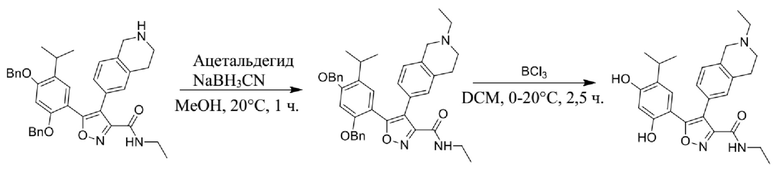
Стадия A. К раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (150 мг, 250 мкмоль, 1,0 экв.) в метаноле (3 мл) добавляли ацетальдегид (274 мг, 433,3 мкмоль, 10,00 экв.), уксусную кислоту (1,3 мг, 21,7 мкмоль, 0,50 экв.) и тетраизопропоксид титана (6,2 мг, 2,589 ммоль, 10 экв.). После перемешивания смеси при 30°C в течение 0,5 часа добавляли NaBH3CN (78 мг, 1,389 ммоль, 5,0 экв.) и смесь перемешивали в течение дополнительных 12 часов. К реакционному раствору добавляли воду (10 мл) и его фильтровали и фильтрат экстрагировали с помощью DCM (10 мл × 3). Органические слои объединяли, промывали солевым раствором (5 мл × 2), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью препаративной TLC с получением 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (60 мг, 95 мкмоль, выход 38%) в виде бесцветного масла.
Стадия B. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (60 мг, 95 мкмоль, 1,0 экв.) в безводном DCM (8 мл) по каплям добавляли раствор BCl3 в DCM (1 M, 1,0 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (2 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (15 мг, 30 мкмоль, выход 32%). 1H ЯМР (400 МГц, DMSO-d6) ppm: 1,02 (d, J = 6,90 Гц, 6H); 1,09 (t, J = 7,15 Гц, 3H); 1,32 (t, J = 7,22 Гц, 3H); 2,89 (d, J = 17,07 Гц, 1H); 3,03 (dt, J = 13,71, 6,89 Гц, 1H); 3,08-3,29 (m, 7H); 3,65 (d, J = 11,17 Гц, 1H); 4,22 (dd, J = 15,31, 8,16 Гц, 1H); 4,48 (d, J = 15,06 Гц, 1H); 6,48 (s, 1H); 6,89 (s, 1H); 7,06-7,18 (m, 3H); 8,91 (t, J = 5,71 Гц, 1H); 9,71 (s, 1H); 9,85 (s, 1H); 10,61 (br.s., 1H). MS (ESI) масса/заряд: 450 (M+1).
Пример 50
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-пропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
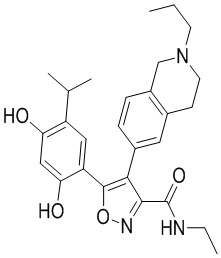
Реакционная схема
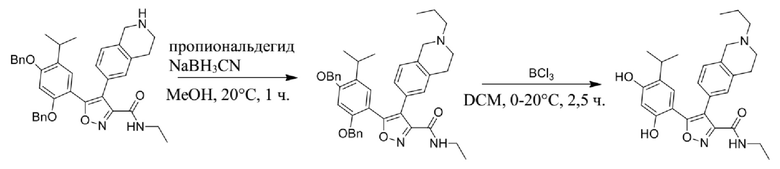
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 49, при этом ацетальдегид на стадии B заменяли пропиональдегидом. 1H ЯМР (400 МГц, DMSO-d6) δ 0,88-1,13 (m, 12H); 1,77 (d, J = 8,16 Гц, 2H); 2,90 (d, J = 16,94 Гц, 1H); 2,97-3,05 (m, 1H); 3,11 (br.s., 3H); 3,19-3,30 (m, 4H); 3,64 (br.s., 1H); 4,13-4,56 (m, 2H); 6,46 (s, 1H); 6,88 (s, 1H); 7,09-7,19 (m, 3H); 8,90 (t, J = 5,46 Гц, 1H); 9,68 (s, 1H); 9,82 (s, 1H); 10,37 (br.s., 1H). MS (ESI) масса/заряд: 464 (M+1).
Пример 51
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-изопропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
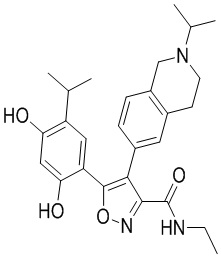
Реакционная схема
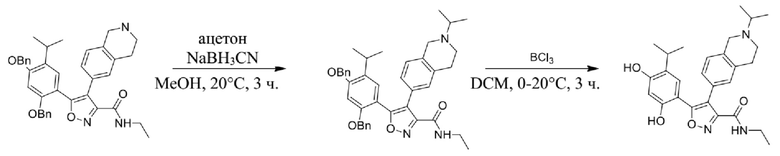
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 49, при этом ацетальдегид на стадии B заменяли ацетоном. 1H ЯМР (400 МГц, DMSO-d6) δ 0,97-1,13 (m, 9H); 1,25-1,41 (m, 6H); 2,82-2,93 (m, 1H); 2,97-3,08 (m, 1H); 3,13-3,28 (m, 4H); 3,60 (br.s., 2H); 4,23-4,43 (m, 2H); 6,49 (s, 1H); 6,91 (s, 1H); 7,06-7,24 (m, 3H); 8,92 (t, J = 5,58 Гц, 1H); 9,58-9,95 (m, 2H); 10,53 (br.s., 1H). MS (ESI) масса/заряд: 464 (M+1).
Пример 52
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-изобутил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
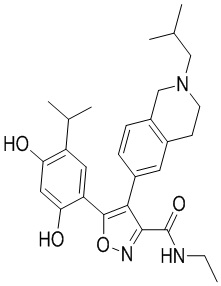
Реакционная схема
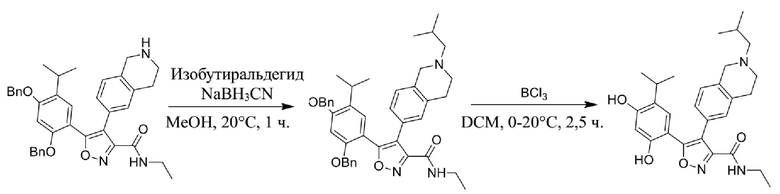
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 49, при этом ацетальдегид на стадии B заменяли 2-метилпропиональдегидом. 1H ЯМР (400 МГц, DMSO-d6) δ 0,94-1,13 (m, 15H); 2,05-2,26 (m,1H); 2,91 (d, J = 17,57 Гц, 1 H); 2,99-3,08 (m, 3H); 3,19-3,27 (m, 3H); 3,66 (br.s., 1H); 4,26 (dd, J = 15,75, 7,47 Гц, 1H); 4,51 (d, J = 15,43 Гц, 1H); 6,46 (s, 1H); 6,88 (s, 1H); 7,05-7,21 (m, 3H); 8,89 (t, J = 5,14 Гц, 1H); 9,61-9,87 (m, 3H). MS (ESI) масса/заряд: 478 (M+1).
Пример 53
4-(2-(2-Ацетиламиноэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)-5-(2,4-дигидрокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамид
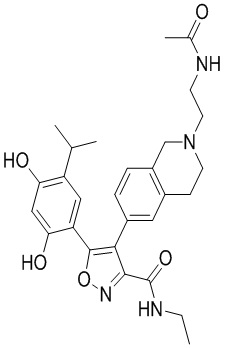
Реакционная схема
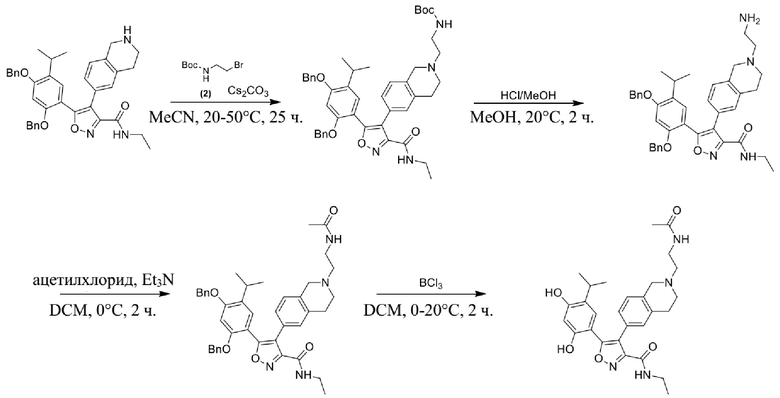
Стадия A. К ацетонитрилу (5мл) добавляли 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид (300 мг, 499 мкмоль, 1,0 экв.), карбонат цезия (244 мг, 748 мкмоль, 1,5 экв.) и трет-бутил-N-(2-бромэтил)карбамат (168 мг, 748 мкмоль, 1,5 экв). Смесь нагревали до 50°C при перемешивании в течение 25 часов. После охлаждения реакционную смесь концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной TLC с получением трет-бутил-N-[2-[6-[5-(2,4-бензилокси-5-изопропилфенил)-3-(этилкарбамоил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-ил]этил]формиата (250 мг, 336 мкмоль, выход 67%) в виде коричневого твердого вещества.
Стадия B. Смесь трет-бутил-N-[2-[6-[5-(2,4-бензилокси-5-изопропилфенил)-3-(этилкарбамоил)изоксазол-4-ил]-1,2,3,4-тетрагидроизохинолин-2-ил]этил]формиата (250 мг, 336 мкмоль, 1,00 экв.) в HCl/MeOH (4 M, 6,00 мл) перемешивали при 25°C в течение 2 часов. Смесь концентрировали при 40°C с получением продукта, 4-(2-(2-аминоэтил)-1,2,3,4-тетрагидроизохинолин-2-ил)-5-(2,4-бензилокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамида (250 мг, неочищенный продукт) в виде коричневого твердого вещества.
Стадия C. При 0°C к раствору 4-(2-(2-аминоэтил)-1,2,3,4-тетрагидроизохинолин-2-ил)-5-(2,4-бензилокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамида (170 мг, 248 мкмоль, 1,0 экв.) и триэтиламина (100 мг, 991 мкмоль, 4,0 экв.) в DCM (6 мл) добавляли ацетилхлорид (39 мг, 496 мкмоль, 2,0 экв.). Смесь перемешивали при 0°C в течение 2 часов. Реакционную смесь концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной TLC с получением 4-(2-(2-ацетиламиноэтил)-1,2,3,4-тетрагидроизохинолин-2-ил)-5-(2,4-бензилокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамида (150 мг, 218 мкмоль, выход 88%) в виде желтого твердого вещества.
Стадия D. При 0°C к раствору 4-(2-(2-ацетиламиноэтил)-1,2,3,4-тетрагидроизохинолин-2-ил)-5-(2,4-бензилокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамида (100 мг, 146 мкмоль, 1,0 экв.) в DCM (5,0 мл) добавляли раствор BCl3 в DCM (1 M, 1,5 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (3 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 4-(2-(2-ацетиламиноэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)-5-(2,4-дигидрокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамида (60 мг, 118 мкмоль, выход 81%). 1H ЯМР (400 МГц, DMSO-d6) ppm: 0,95-1,14 (m, 9H); 1,79-1,88 (m, 3H); 2,90 (d, J = 16,94 Гц, 1H); 2,97-3,07 (m, 1H); 3,10-3,34 (m, 6H); 3,51 (d, J = 5,65 Гц, 2H); 3,71 (d, J = 10,67 Гц, 1H); 4,28 (dd, J = 15,75, 7,72 Гц, 1H); 4,55 (d, J = 15,31 Гц, 1H); 6,43-6,52 (m, 1H); 6,84-6,92 (m, 1H); 7,06-7,20 (m, 3H); 8,30 (t, J = 5,58 Гц, 1H); 8,90 (t, J = 5,71 Гц, 1H); 9,56-9,95 (m, 2H); 10,52 (br.s., 1H). MS (ESI) масса/заряд: 507 (M+1).
Пример 54
4-(2-(2-Ацетиламинопропил)-1,2,3,4-тетрагидроизохинолин-6-ил)-5-(2,4-дигидрокси-5-изопропилфенил)-N-этилизоксазол-3-карбоксамид
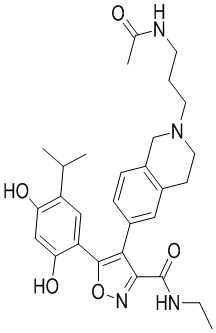
Реакционная схема
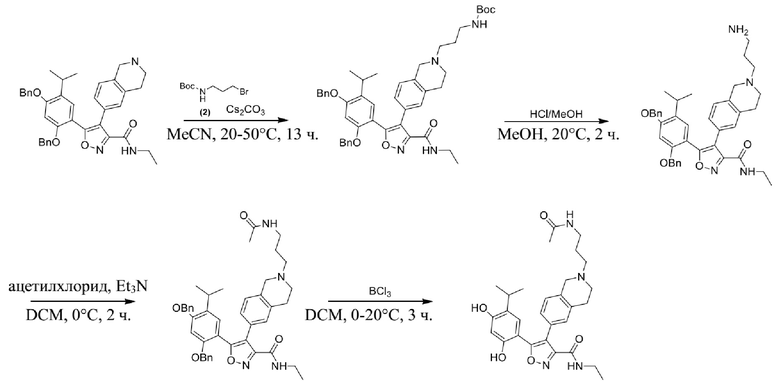
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A, B и C в примере 53, при этом трет-бутил-N-(2-бромэтил)карбамат на стадии A заменяли трет-бутил-N-(3-бромпропил)карбаматом. 1H ЯМР (400 МГц, DMSO-d6) δ ppm: 1,01 (d, J = 6,90 Гц, 6H); 1,08 (t, J = 7,22 Гц, 3H); 1,81 (s, 3H); 1,86-1,96 (m, 2H); 2,88 (d, J = 17,19 Гц, 1H); 2,96-3,34 (m, 10H); 4,23 (dd, J = 15,62, 7,97 Гц, 1H); 4,47 (d, J = 14,18 Гц, 1H); 6,48 (s, 1H); 6,88 (s, 1H); 7,07 - 7,17 (m, 3H); 8,10 (t, J = 5,71 Гц, 1H); 8,90 (t, J = 5,65 Гц, 1H); 9,52 - 10,02 (m, 2H); 10,69 (br.s., 1H). MS (ESI) масса/заряд: 521 (M+1).
Пример 55
5-(2,4-Дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-2-оксоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамид
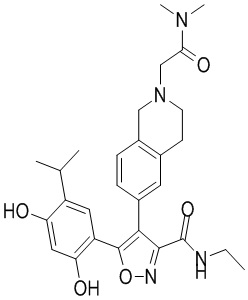
Реакционная схема
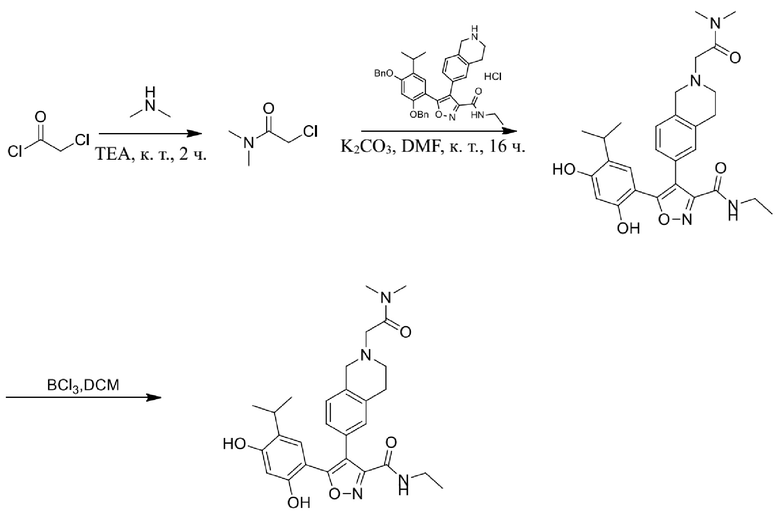
Стадия A. К DCM (30 мл) добавляли 2-хлорацетилхлорид (3,00 г, 26,689 ммоль, 1,6 экв.), гидрохлорид диметиламина (2,38 г, 29,289 ммоль, 1,1 экв.) и триэтиламин (8,0 г, 79,789 ммоль, 3,0 экв.). Смесь перемешивали при 15°C в течение 3 часов. Реакционную смесь выливали в воду (30 мл) и экстрагировали с помощью DCM (50 мл × 2). Органическую фазу промывали разведенной хлористоводородной кислотой (30 мл × 2, 1 M), высушивали над безводным Na2SO4 и концентрировали в вакууме с получением 2-хлор-N,N-диметилацетамида (2,5 г, 20,689 ммоль, выход 77%) в виде желтого масла.
Стадия B. К DMF (4 мл) добавляли 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид (200 мг, 313 мкмоль, 1,0 экв.), K2CO3 (130 мг, 940 мкмоль, 3,0 экв.) и 2-хлор-N,N-диметилацетамид (114 мг, 940 мкмоль, 3,0 экв.). Смесь перемешивали при 15°C в течение 16 часов. Реакционную смесь добавляли к воде (30 мл) и фильтровали со сбором твердого вещества с получением 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-2-оксоэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (195 мг, неочищенный продукт, чистота 74%) в виде белого твердого вещества.
Стадия C. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-2-оксоэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (190 мг, 277 мкмоль, 1,0 экв.) в DCM (10,0 мл) по каплям добавляли раствор BCl3 в DCM (1 M, 3,0 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (6 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-2-оксоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (78 мг, 138 мкмоль, выход 49,8%). 1H ЯМР (400 МГц, DMSO-d6): δ 10,22 (s, 1H), 9,86 (s, 1H), 9,72 (s, 1H), 8,92 (t, J = 5,2 Гц,1H), 7,10-7,16 (m, 3H), 6,86 (s, 1H), 6,48 (s, 1H), 4,32-4,55 (m, 4H), 3,63 (brs, 2H), 3,20-3,25 (m, 2H), 3,10-3,14 (m, 1H), 2,67-3,00 (m,7 H), 0,99-1,15 (m, 9 H). MS (ESI) масса/заряд: 507 (M+1).
Пример 56
5-(2,4-Дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-3-оксопропил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамид
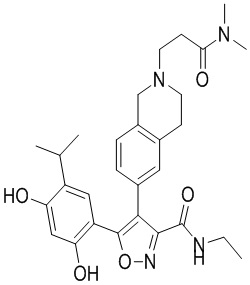
Реакционная схема
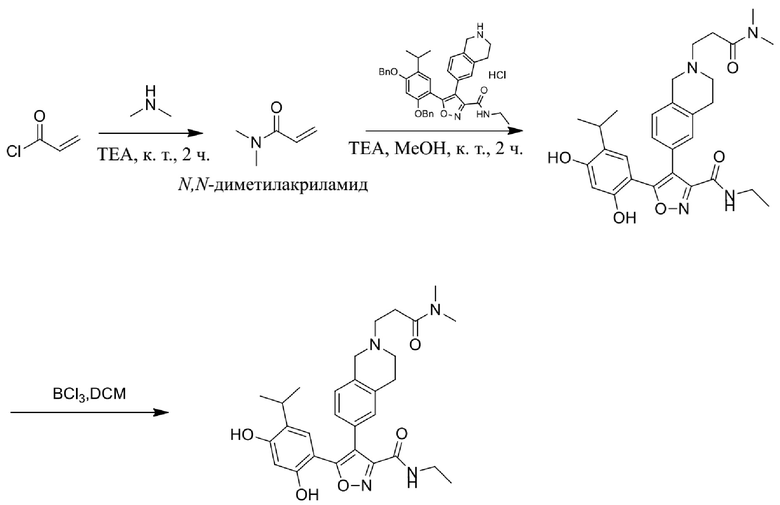
Стадия A. К DCM (50 мл) добавляли акрилоилхлорид (5,0 г, 5589 ммоль, 1,0 экв.), гидрохлорид диметиламина (4,95 г, 60,889 ммоль, 1,1 экв.) и триэтиламин (16,8 г, 16689 ммоль, 3,0 экв.). Смесь перемешивали при 15°C в течение 3 часов. Реакционную смесь выливали в воду (30 мл) и экстрагировали с помощью DCM (50 мл × 2). Органическую фазу промывали разведенной хлористоводородной кислотой (30 мл × 2, 1 M), высушивали над безводным Na2SO4 и концентрировали в вакууме с получением N,N-диметилакриламида (3,0 г, 3089 ммоль, выход 55%) в виде желтого масла.
Стадия B. К этанолу (2 мл) добавляли 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид (100 мг, 157 мкмоль, 1,0 экв.), триэтиламин (48 мг, 470 мкмоль, 3,0 экв.) и N,N-диметилакриламид (155 мг, 1,5789 ммоль, 10,0 экв.). Смесь перемешивали при 15°C в течение 2 часов. Реакционную смесь добавляли к воде (30 мл) и экстрагировали с помощью EA (30 мл × 2). Органическую фазу высушивали над безводным Na2SO4 и концентрировали в вакууме с получением 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-3-оксопропил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (195 мг, неочищенный продукт, чистота 74%) в виде желтого масла.
Стадия C. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-3-оксопропил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (105 мг, 149 мкмоль, 1,0 экв.) в DCM (15,0 мл) по каплям добавляли раствор BCl3 в DCM (1 M, 3,0 мл, 20,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (6 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)-3-оксопропил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (28 мг, 46 мкмоль, выход 31%). 1H ЯМР (400 МГц, DMSO-d6): δ 10,49 (s, 1H), 9,85 (s, 1H), 9,71 (s, 1H), 8,91 (t, J = 5,6 Гц, 1H), 7,10-7,15 (m, 3H), 6,88 (s, 1H), 6,47 (s, 1H), 4,51-4,55 (m, 1H), 4,31-4,34 (m, 2H), 3,68-3,72 (m, 1H), 3,30-3,42 (m, 4H), 3,21-3,30 (m, 1H), 3,00-3,20 (m, 4H), 2,93-3,00 (m,3H), 2,85 (s, 4H), 3,00-3,20 (m, 4H), 1,09 (t, J = 7,6 Гц, 3H), 1,01(d, J = 3,6 Гц, 6H). MS (ESI) масса/заряд: 521 (M+1).
Пример 57
5-(2,4-Дигидрокси-5-изопропилфенил)-4-(2-(3-(диметиламино)пропионил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамид
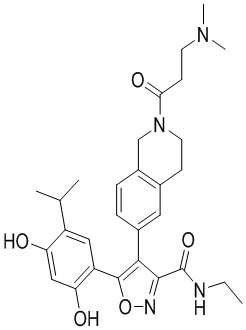
Реакционная схема
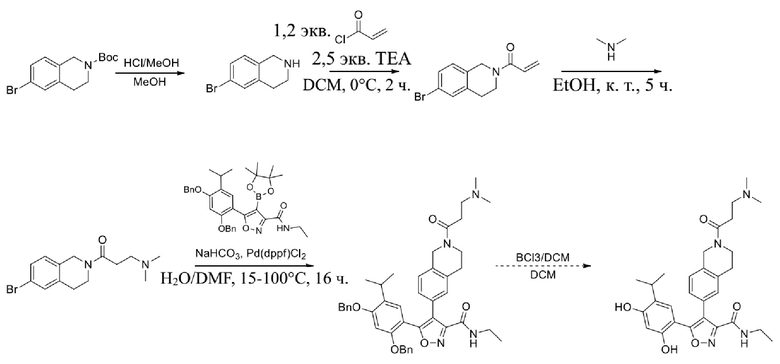
Стадия A. При 20°C к раствору трет-бутил-6-бром-1,2,3,4-тетрагидроизохинолин-2-формиата (5,0 г, 1689 ммоль, 1,00 экв.) в MeOH (20,0 мл) добавляли HCl/MeOH (4 M, 30,00 мл) и смесь перемешивали при 25°C в течение 2 часов. Смесь концентрировали при 40°C с получением продукта, гидрохлорида 6-бром-1,2,3,4-тетрагидроизохинолина (4,5 г, неочищенный продукт).
Стадия B. При 0°C к раствору гидрохлорида 6-бром-1,2,3,4-тетрагидроизохинолина (2,0 г, 989 ммоль, 1 экв.) и триэтиламина (2,9 г, 2889 ммоль, 3,0 экв.) в DCM (40 мл) добавляли акрилоилхлорид (1,0 г, 1189 ммоль, 1,2 экв.). Смесь перемешивали при 15°C в течение 14 часов. Реакционную смесь выливали в воду (30 мл) и экстрагировали с помощью DCM (50 мл × 2). Органическую фазу высушивали с помощью безводного Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии с получением 1-(6-бром-1,2,3,4-тетрагидроизохинолинил)пропил-2-ен-1-она (1,4 г, 589 ммоль, выход 56%) в виде желтого масла. MS (ESI) масса/заряд: 266 (M+1).
Стадия C. К этанолу (10 мл) добавляли 1-(6-бром-1,2,3,4-тетрагидроизохинолин-ил)пропил-2-ен-1-он (700 мг, 2,689 ммоль, 1,0 экв.) и диметиламин (1,1 г, 7,989 ммоль, 3,0 экв). Смесь перемешивали при 15°C в течение 12 часов. Реакционную смесь концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии с получением 1-(6-бром-1,2,3,4-тетрагидроизохинолинил)-3-(диметиламино)пропан-1-она (700 мг, 2,389 ммоль, выход 86%) в виде желтого масла.
Стадия D. К перемешанному раствору 5-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазол-3-карбоксамида (767 мг, 771 мкмоль, 1,0 экв.) в DMF (2,5 мл) и воды (0,5 мл) добавляли 1-(6-бром-1,2,3,4-тетрагидроизохинолинил)-3-(диметиламино)пропан-1-он (200 мг, 643 мкмоль, 1,0 экв.) и NaHCO3 (216 мг, 2,689 ммоль, 4,0 экв.). Атмосферу смеси меняли на атмосферу азота с последующим добавлением Pd(dppf)Cl2 (47 мг, 64 мкмоль, 0,1 экв.). Смесь перемешивали при 100°C в течение 16 часов, и реакционную смесь разбавляли с помощью 20 мл воды и фильтровали через диатомовую землю. Фильтрат экстрагировали с помощью EA (20 мл × 3) и объединенную органическую фазу промывали водой (20 мл × л), высушивали над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной TLC (DCM:метанол = 10:1) с получением 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-(3-(диметиламино)пропионил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (220 мг, 314 мкмоль, выход 49%) в виде белого твердого вещества. MS (ESI) масса/заряд: 701 (M+1).
Стадия E. При 0°C к раствору 5-(2,4-дибензилокси-5-изопропилфенил)-4-(2-(3-(диметиламино)пропионил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (170 мг, 243 мкмоль, 1,0 экв.) в DCM (12,0 мл) по каплям добавляли раствор BCl3 в DCM (1 M, 2,4 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (6 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 5-(2,4-дигидрокси-5-изопропилфенил)-4-(2-(3-(диметиламино)пропионил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамида (65 мг, 117 мкмоль, выход 48%). 1H ЯМР (400 МГц, DMSO-d6) ppm 8,89 (t, J = 5,65 Гц, 1H) 7,11-7,20 (m, 1H) 6,99-7,10 (m, 2H) 6,84 (s, 1H) 6,48 (d, J = 1,88 Гц, 1H) 4,54-4,72 (m, 2H) 3,65 (t, J = 5,65 Гц, 2H) 3,16-3,37 (m, 4H) 2,98-3,07 (m, 1H) 2,94 (t, J = 7,15 Гц, 2H) 2,63-2,83 (m, 8H) 1,08 (t, J = 7,22 Гц, 3H) 0,98 (d, J = 6,78 Гц, 6H). MS (ESI) масса/заряд: 521 (M+1).
Пример 58
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(3-морфолино-4-ил-пропионил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
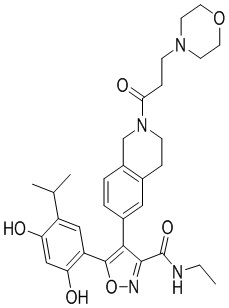
Реакционная схема
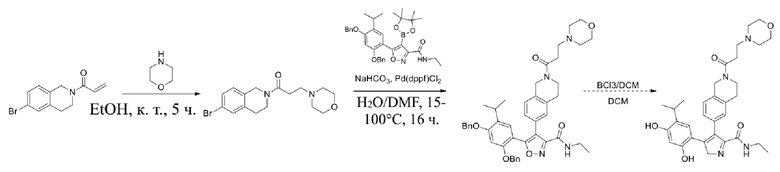
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий C, D и E в примере 57, при этом диметиламин на стадии C заменяли морфолином. 1H ЯМР (400 МГц, DMSO-d6) ppm: 8,89 (t, J = 5,65 Гц, 1H), 7,13 (t, J = 7,78 Гц, 1H), 7,00-7,10 (m, 2H), 6,85 (s, 1H), 6,44 (d, J = 1,76 Гц, 1H), 4,62 (d, J = 19,58 Гц, 2H), 3,95 (d, J = 11,92 Гц, 2H), 3,74 (t, J = 12,11 Гц, 2H), 3,62-3,69 (m, 2H), 3,45 (d, J = 12,17 Гц, 3H), 3,28-3,38 (m, 2H), 3,16-3,27 (m, 2H), 2,92-3,15 (m, 5H), 2,79 (t, J = 5,65 Гц, 1H), 2,63-2,71 (m, 1H), 1,08 (t, J = 7,15 Гц, 3H), 0,98 (d, J = 6,90 Гц, 6H). MS (ESI) масса/заряд: 563 (M+1).
Пример 59
5-(2,4-Дигидрокси-5-изопропилфенил)-4-(2-(2-(диметиламино)ацетил)-1,2,3,4-тетрагидроизохинолин-6-ил)-N-этилизоксазол-3-карбоксамид
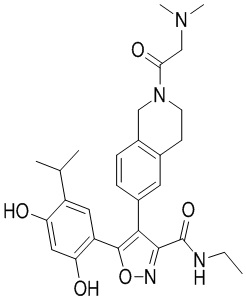
Реакционная схема
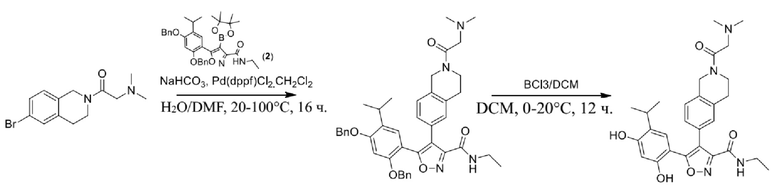
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 57, при этом 1-(6-бром-1,2,3,4-тетрагидроизохинолинил)-3-(диметиламино)пропан-1-он на стадии D заменяли 1-(6-бром-1,2,3,4-тетрагидроизохинолинил)-2-(диметиламино)этаноном. 1H ЯМР (400 МГц, DMSO-d6) ppm: 0,98 (d, J = 6,90 Гц, 6H); 1,04-1,14 (m, 3H); 2,52 (br.s., 1H); 2,71 (t, J = 5,77 Гц, 1H); 2,81 (d, J = 4,64 Гц, 7H); 3,00 (dt, J = 13,65, 6,79 Гц, 1H); 3,22 (квин., J = 6,71 Гц, 2H); 3,53 (t, J = 5,83 Гц, 1H); 3,69 (t, J = 5,77 Гц, 1H); 4,37 (d, J = 4,52 Гц, 2H); 4,50-4,69 (m, 2H); 6,48 (d, J = 2,64 Гц, 1H); 6,84 (s, 1H); 7,01-7,13 (m, 2H); 7,17 (d, J = 8,03 Гц, 1H); 8,89 (t, J = 5,65 Гц, 1H); 9,47-10,00 (m, 3H). MS (ESI) масса/заряд: 507 (M+1).
Пример 60
5-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-морфолино-4-ил-ацетил)-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамид
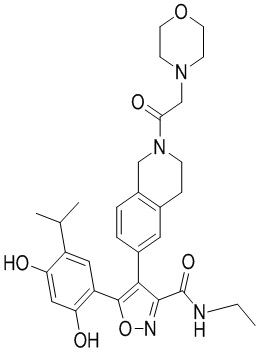
Реакционная схема
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий D и E в примере 57, при этом 1-(6-бром-1,2,3,4-тетрагидроизохинолинил)-3-(диметиламино)пропан-1-он на стадии D заменяли 1-(6-бром-1,2,3,4-тетрагидроизохинолинил)-2-морфолинилэтаноном. 1H ЯМР (400 МГц, DMSO-d6) ppm: 0,94-1,01 (m, 6H); 1,08 (td, J = 7,15, 1,38 Гц, 3H); 2,69-2,87 (m, 2H); 3,01 (dt, J = 13,71, 6,76 Гц, 1H); 3,09-3,27 (m, 4H); 3,42 (d, J = 11,67 Гц, 2H); 3,55-3,58 (m, 1H); 3,70 (t, J = 5,96 Гц, 1H); 3,75-3,86 (m, 2H); 3,87-4,01 (m, 2H); 4,45 (br.s., 2H); 4,53-4,67 (m, 2H); 6,48 (d, J = 2,76 Гц, 1H); 6,84 (s, 1H); 7,03-7,14 (m, 2H); 7,17 (d, J = 8,03 Гц, 1H); 8,90 (t, J = 5,52 Гц, 1H); 9,63-9,95 (m, 2H); 10,27 (br.s., 1H). MS (ESI) масса/заряд: 549 (M+1).
Пример 61
3-(2,4-Дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
Реакционная схема
Стадия A. При 0°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-5-муравьиной кислоты (300 мг, 527 мкмоль, 1,0 экв.) в DCM (10 мл) добавляли (COCl)2 (134 мг, 1050 мкмоль, 2 экв.) и DMF (100,00 мкл). Смесь перемешивали при 0°C в течение 1 часа, а затем реакционный раствор концентрировали в вакууме с получением промежуточного соединения. Промежуточное соединение растворяли в DCM (10 мл) при 0°C и к раствору добавляли NH3/THF (4 M, 1,32 мл, 10,00 экв). Смесь перемешивали при 0°C в течение 1 часа и выливали в 15 мл воды. Затем смесь экстрагировали с помощью DCM (15 мл × 2) и объединенную органическую фазу высушивали с помощью безводного Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, петролейный эфир/этилацетат = от 20/1 до 5/1) с получением 3-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-5-карбоксамида (140 мг, выход 47%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 569 (M+1).
Стадия B. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолина (103 мг, 377 мкмоль) и 3-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-5-карбоксамида (134 мг, 236 мкмоль) в DMF (4 мл) добавляли NaHCO3 (40 мг, 472 мкмоль), H2O (1,0 мл) и Pd(PPh3)2Cl2 (17 мг, 24 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 80°C при перемешивании в течение 12 часов. Смесь охлаждали до 25°C и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, дихлорметан/метанол = от 20/1 до 10/1) с получением 3-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (80 мг, выход 51%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 588 (M+1).
Стадия C. При 0°C в защитной атмосфере газообразного азота к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-3-карбоксамида (80 мг, 136 мкмоль) в DCM (15,00 мл) медленно добавляли BCl3 (2 мл, 1 моль в растворе DCM). Реакционную смесь перемешивали при 0°C в течение 1 часа, а затем нагревали до комнатной температуры при перемешивании в течение 2 часов. Реакционный раствор охлаждали до 0°C, а затем реакционную смесь гасили путем добавления MeOH (20 мл). Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Остаток очищали с помощью препаративной HPLC (способ с применением HCl: Phenomenex Synergi C18 150 × 30 мм × 4 мкм, вода (0,05% HCl)-ACN) с получением гидрохлорида 3-(2,4-дигидрокси-5-изопропилфенил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (9 мг, выход 15%). MS (ESI) масса/заряд: 408 (M+1). 1H ЯМР (400 МГц, DMSO-d6) δ ЯМР (400 МГц, DMSO-dnomenex Synergi C18 150 x 30 мм x 4 мкм, вода(0,05%HCl)-ACN)2 H); 4,53-4,67 (m, 2H); 6,48 (d, ), 3,30-3,42 (m, 4H), 3,21-3,30 (m,1H), 3,00-3,20 (m,4H), 2,93-3,00 (m,3H), 2,85 (s, 4H), 3,00-3,20 (m,4 H), 1,09 (t, = 8,29 Гц, 2H) J = 6,8 Гц, 6H).
Пример 62
3-(2,4-Дигидрокси-5-изопропилфенил)-N-метил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
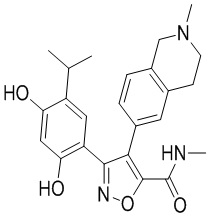
Реакционная схема
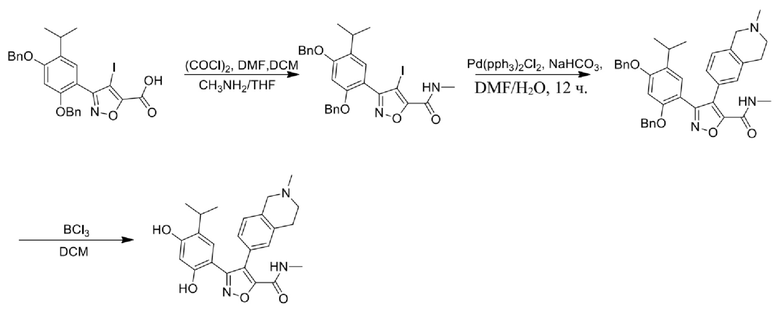
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A, B и C в примере 61, при этом NH3/THF на стадии A заменяли метиламином. 1H ЯМР (400 МГц, DMSO-d6) δ 9,64 (s, 1H), 9,36 (s, 1H), 8,91 (d, J = 4,8 Гц, 1H), 7,19 (s, 1H), 7,09 (s, 2H), 6,89 (s, 1H), 6,44 (s, 1H), 4,08 (br.s., 2H), 3,19 (br.s., 2H), 3,14-3,04 (m, 1H), 2,95 (br.s., 2H), 2,81 (d, J = 4,5 Гц, 3H), 2,75 (br.s., 3H), 1,15-1,06 (m, 6H). MS (ESI) масса/заряд: 422 (M+1).
Пример 63
3-(2,4-Дигидрокси-5-изопропилфенил)-N-изопропил-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
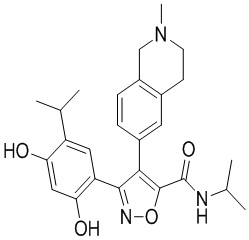
Реакционная схема
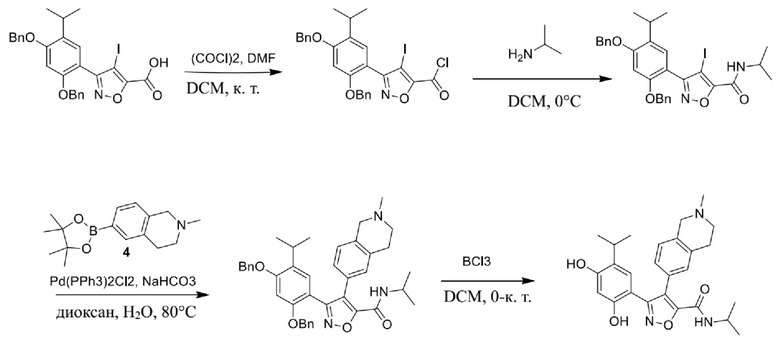
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A, B и C в примере 61, при этом NH3/THF на стадии A заменяли пропил-2-амином. 1H ЯМР (400 МГц, DMSO-d6) δ 8,80 (t, J = 5,5 Гц, 1H), 8,24 (brs, 1H), 7,23-7,11 (m, 2H), 6,98 (d, J = 7,5 Гц, 1H), 6,67 (s, 1H), 6,46 (s, 1H), 4,22 (d, J = 19,8 Гц, 2H), 3,25-3,15 (m, 2H), 2,95 (td, J = 6,8, 13,7 Гц, 1H), 2,17-1,86 (m, 4H), 1,06 (t, J = 7,2 Гц, 5H), 0,95-0,84 (m, 9H). MS (ESI) масса/заряд: 450 (M+1).
Пример 64
3-(2,4-Дигидрокси-5-изопропилфенил)-N-(2-гидроксиэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
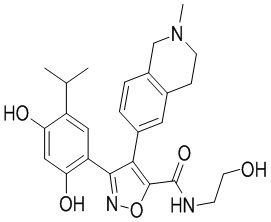
Реакционная схема
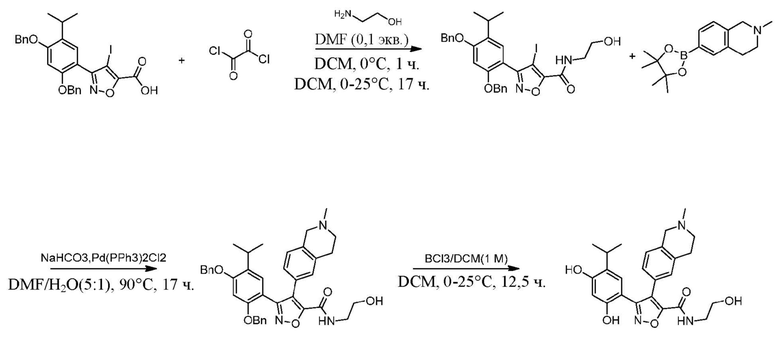
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A, B и C в примере 61, при этом NH3/THF на стадии A заменяли 2-аминоэтанолом. 1H ЯМР (400 МГц, DMSO-d6) δ 1,05 (d, J = 6,90 Гц, 6H); 2,87 (s, 3H); 3,04 (dt, J = 13,71, 6,76 Гц, 2H); 3,28 (q, J = 5,86 Гц, 3H); 3,45-3,50 (m, 2H); 3,58 (br.s., 1H); 4,10-4,52 (m, 2H); 4,80 (br.s., 1H); 6,39 (s, 1H); 6,86 (s, 1H); 7,07 (s, 2H); 7,16-7,26 (m, 1H); 8,83 (t, J = 5,58 Гц, 1H); 9,36 (s, 1H); 9,62 (s, 1H); 10,54-11,07 (m, 1H). MS (ESI) масса/заряд: 452 (M+1).
Пример 65
3-(2,4-Дигидрокси-5-изопропилфенил)-N-(2-фторэтил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
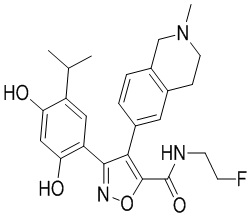
Реакционная схема
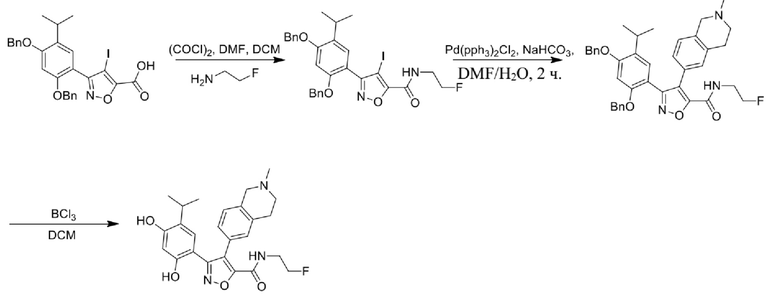
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A, B и C в примере 61, при этом NH3/THF на стадии A заменяли 2-фторэтиламином. 1H ЯМР (400 МГц, DMSO-d6) δ 9,63 (s, 1H), 9,37 (s, 1H), 9,15 (t, J = 5,4 Гц, 1H), 7,19 (s, 1H), 7,08 (s, 2H), 6,87 (s, 1H), 6,39 (s, 1H), 4,58 (t, J = 4,8 Гц, 1H), 4,46 (t, J = 4,9 Гц, 1H), 4,31 (br.s., 1H), 3,60-3,52 (m, 2H), 3,51 (br.s., 4H), 3,10-2,96 (m, 2H), 2,87 (s, 3H), 2,11-2,05 (m, 1H), 1,06 (d, J = 6,8 Гц, 6H). MS (ESI) масса/заряд: 454 (M+1).
Пример 66
3-(2,4-Дигидрокси-5-изопропилфенил)-N-(3-гидроксипропил)-4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
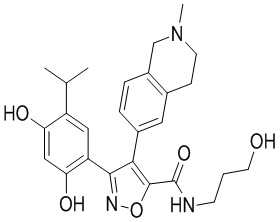
Реакционная схема
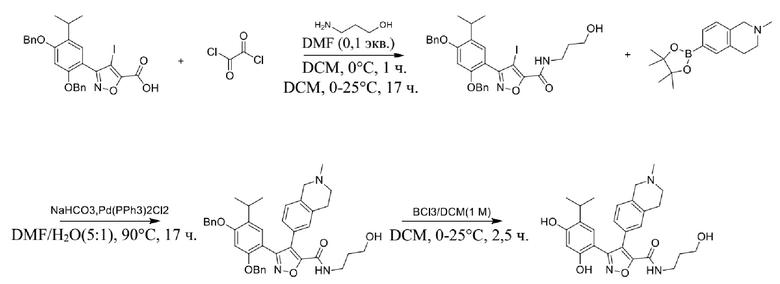
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A, B и C в примере 61, при этом NH3/THF на стадии A заменяли 3-аминопропан-1-олом. 1H ЯМР (400 МГц, DMSO-d6) δ 1,05 (d, J =6,78 Гц, 6H); 1,64 (квин., J = 6,62 Гц, 2H); 2,87 (d, J = 4,52 Гц, 3H); 2,97-3,19 (m, 3H); 3,20-3,32 (m, 4H); 3,58 (br.s., 2H); 4,22 (dd, J = 15,81, 8,03 Гц, 1H); 4,42 (d, J = 15,06 Гц, 1H); 6,39 (s, 1H); 6,86 (s, 1H); 7,03-7,11 (m, 2H); 7,19 (s, 1H); 8,89 (t, J = 5,58 Гц, 1H); 9,34 (br.s., 1H); 9,59 (br.s., 1H); 10,80 (br.s., 1H). MS (ESI) масса/заряд: 466 (M+1).
Пример 67
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
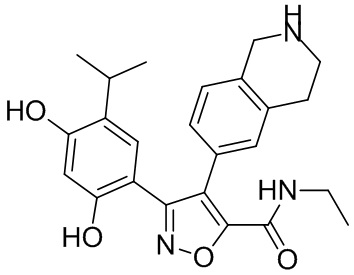
Реакционная схема
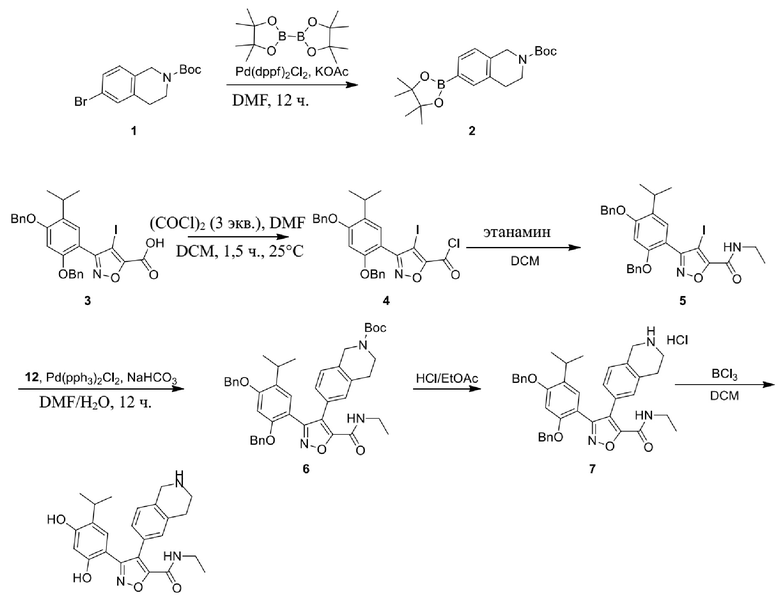
Стадия A. При 25°C в защитной атмосфере газообразного азота к раствору трет-бутил-6-бром-1,2,3,4-тетрагидроизохинолин-2-формиата (7,5 г, 2489 ммоль) в DMF (100 мл) добавляли бис(пинаколато)дибор (7,3 г, 2989 ммоль) и KOAc (7,1 г, 7289 ммоль) с последующим добавлением катализатора Pd(dppf)Cl2.CH2Cl2 (5,3 г, 789 ммоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 80°C при перемешивании в течение 12 часов. Смесь охлаждали до 25°C и выливали в воду (600 мл). Затем смесь экстрагировали с помощью EA (200 мл × 2). Объединенную органическую фазу высушивали с помощью безводного Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат = от 20/1 до 10/1) с получением трет-бутил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин-2-формиата (7,4 г, 2189 ммоль, выход 86%) в виде белого твердого вещества.
Стадия B. При 0°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-4-йодизоксазол-5-муравьиной кислоты (5 г, 989 ммоль, 1,0 экв.) в DCM (60 мл) добавляли (COCl)2 (2,2 г, 1889 ммоль, 1,5 экв.) и DMF (100,00 мкл). Смесь перемешивали при 0°C в течение 1 часа, а затем реакционный раствор концентрировали в вакууме с получением промежуточного соединения. Промежуточное соединение растворяли в DCM (80 мл) и при 0°C к раствору добавляли этиламин (1,9 г, 2,8 мл, 5,00 экв.). Смесь перемешивали при 0°C в течение 1 часа и выливали в 100 мл воды. Затем смесь экстрагировали с помощью DCM (80 мл × 2) и объединенную органическую фазу высушивали с помощью безводного Na2SO4, фильтровали и концентрировали в вакууме с получением 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-йодизоксазол-5-карбоксамида (3,8 г, выход 75%) в виде желтого твердого вещества.
Стадия C. При 25°C в защитной атмосфере газообразного азота к перемешанному раствору трет-бутил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин-2-формиата (3,2 г, 989 ммоль) и 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-йодизоксазол-5-карбоксамида (3,8 г, 689 ммоль) в DMF (50 мл) добавляли NaHCO3 (1,6 г, 1989 ммоль), H2O (17,0 мл) и Pd(PPh3)2Cl2 (447 мг, 637 мкмоль). Смесь перемешивали при 25°C в течение 10 мин., а затем нагревали до 80°C при перемешивании в течение 12 часов. Смесь охлаждали до 25°C и выливали в воду (100 мл). Затем смесь экстрагировали с помощью EA (150 мл × 2) и объединенную органическую фазу промывали водой (100 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (силикагель - 200-300 меш, PE/EA = от 10/1 до 5/1) с получением трет-бутил-6-(3-(2,4-дибензилокси-5-изопропилфенил)-5-(этилформамил)изоксазол-4-ил)-1,2,3,4-тетрагидроизохинолин-2-формиата (2,6 г, 3,789 ммоль, выход 58%) в виде желтого твердого вещества. MS (ESI) масса/заряд: 588 (M+1).
Стадия D. При 25°C к раствору трет-бутил-6-(3-(2,4-дибензилокси-5-изопропилфенил)-5-(этилформамил)изоксазол-4-ил)-1,2,3,4-тетрагидроизохинолин-2-формиата (2,6 г, 3,789 ммоль, 1,00 экв.) в MeOH (20,00 мл) добавляли HCl/MeOH (4 M, 20,00 мл) и смесь перемешивали при 25°C в течение 30 мин. Смесь концентрировали при 40°C с получением продукта, 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (2,5 г, неочищенный продукт) в виде желтого твердого вещества.
Стадия E. При 0°C в защитной атмосфере газообразного азота к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (1,0 г, 1,689 ммоль) в DCM (50,00 мл) медленно добавляли BCl3 (9,5 мл, 1 моль в растворе DCM). Реакционную смесь перемешивали при 0°C в течение 1 часа, а затем нагревали до комнатной температуры при перемешивании в течение 2 часов. Реакционный раствор охлаждали до 0°C, а затем реакционную смесь гасили путем добавления MeOH (20 мл). Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Остаток очищали с помощью препаративной HPLC (способ с применением HCl: Phenomenex Synergi C18 150 × 30 мм × 4 мкм, вода (0,05% HCl)-ACN) с получением гидрохлорида 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (315 мг, 685 мкмоль, выход 44%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,57 (s, 1H), 9,38 (s, 2H), 9,31 (s, 1H), 8,94 (t, J = 5,6 Гц,1H),7,15 (s,1H), 7,05-7,12 (m, 2 H), 6,86 (s, 1H), 6,38 (s, 1H), 4,21 (s, 2H), 3,21-3,33 (m, 4H), 3,03-3,10 (m, 1H), 2,56-2,90 (m, 2H), 1,05-1,11 (m,9H). MS (ESI) масса/заряд: 422 (M+1).
Пример 68
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-изопропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
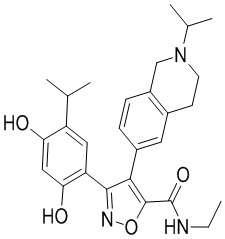
Реакционная схема
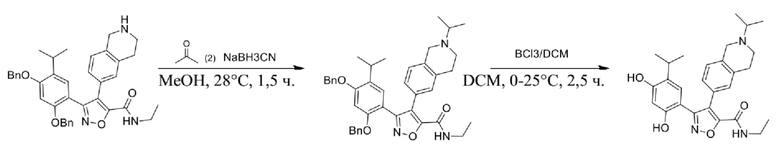
Стадия A. К раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (100 мг, 166 мкмоль, 1,0 экв.) в метаноле (3 мл) добавляли ацетон (97 мг, 289 ммоль, 10,00 экв.) и уксусную кислоту (5 мг, 8 мкмоль, 0,50 экв.). После перемешивания смеси при 30°C в течение 0,5 часа добавляли NaBH3CN (31 мг, 499 мкмоль, 3,0 экв.) и смесь перемешивали в течение дополнительного часа. Реакционный раствор концентрировали в вакууме. Остаток очищали с помощью препаративной TLC с получением 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2-изопропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (80 мг, 124 мкмоль, выход 75%) в виде желтого масла.
Стадия B. При 0°C к раствору 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(2-изопропил-1,2,3,4-тетрагидроксихинолин-6-ил)изоксазол-5-карбоксамида (80 мг, 124 мкмоль, 1,0 экв.) в безводном DCM (8 мл) медленно по каплям добавляли раствор BCl3 в DCM (1 M, 1,2 мл, 10,0 экв.) на протяжении 5 мин. Суспензию перемешивали при 0°C в течение 30 мин., а затем нагревали до 25°C при перемешивании в течение 2 часов. Смесь охлаждали до 0°C, гасили путем медленного добавления MeOH (3 мл) и концентрировали в вакууме с получением остатка. Остаток очищали с помощью препаративной HPLC с получением 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-изопропил-1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (25 мг, 48 мкмоль, выход 39%). 1H ЯМР (400 МГц, DMSO-d6) ppm: 1,02-1,11 (m, 15H); 2,64-2,69 (m, 4H); 2,85 (dt, J = 13,05, 6,53 Гц, 1H); 3,04 (dt, J = 13,71, 6,89 Гц, 1H); 3,18-3,29 (m, 4H); 3,61 (s, 2H); 6,33 (s, 1H); 6,81 (s, 1H); 6,93-6,96 (m, 2H); 6,98 (s, 1H); 8,20 (s, 1H); 8,85 (t, J = 5,71 Гц, 1H). MS (ESI) масса/заряд: 464 (M+1).
Пример 69
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
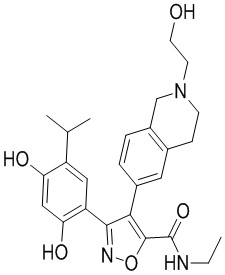
Реакционная схема
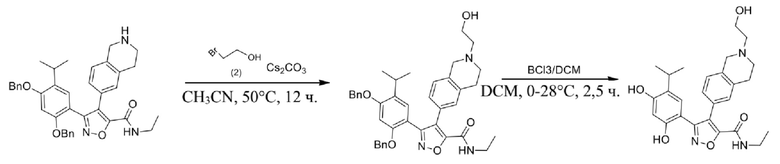
Стадия A. К ацетонитрилу (3 мл) добавляли 3-(2,4-дибензилокси-5-изопропилфенил)-N-этил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид (150 мг, 249 мкмоль, 1,0 экв.), Cs2CO3 (122 мг, 374 мкмоль, 1,5 экв.) и 2-бромэтанол (32 мг, 249 мкмоль, 1,0 экв.). Смесь нагревали до 50°C при перемешивании в течение 12 часов. После охлаждения реакционную смесь концентрировали до сухого состояния. Остаток очищали с помощью препаративной TLC (дихлорметан/метанол = 15:1) с получением 3-(2,4-дибензилоксил-5-изопропилфенил)-N-этил-4-(2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (120 мг, 185 мкмоль, выход 74%) в виде бесцветного масла.
Стадия B. При 0°C в защитной атмосфере газообразного азота к раствору 3-(2,4-дибензилоксил-5-изопропилфенил)-N-этил-4-(2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (120 мг, 186 мкмоль) в DCM (8,00 мл) медленно добавляли BCl3 (1,9 мл, 1 моль в растворе DCM). Реакционную смесь перемешивали при 0°C в течение 1 часа, а затем нагревали до комнатной температуры при перемешивании в течение двух часов. Реакционный раствор охлаждали до 0°C, а затем реакционную смесь гасили путем добавления MeOH (4 мл). Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Остаток очищали с помощью препаративной HPLC с получением гидрохлорида 3-(2,4-дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-гидроксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамида (39 мг, 77 мкмоль, выход 41%). 1H ЯМР (400 МГц, DMSO-d6) ppm: 1,03-1,12 (m, 9H); 2,88 (d, J = 17,32 Гц, 1H); 3,00-3,18 (m, 2H); 3,21-3,31 (m, 5H); 3,71 (d, J = 11,67 Гц, 1H); 3,84 (t, J = 4,96 Гц, 2H); 4,32 (dd, J = 15,69, 7,91 Гц, 1H); 4,51 (d, J = 15,18 Гц, 1H); 6,39 (s, 1H); 6,85 (s, 1H); 7,10 (s, 2H); 7,18 (s, 1H); 8,95 (t, J = 5,71 Гц, 1H); 9,32 (br.s., 1H); 9,58 (br.s., 1H); 10,43 (br.s., 1H). MS (ESI) масса/заряд: 466 (M+1).
Пример 70
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-метоксиэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
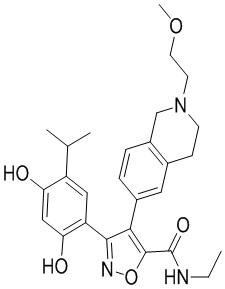
Реакционная схема
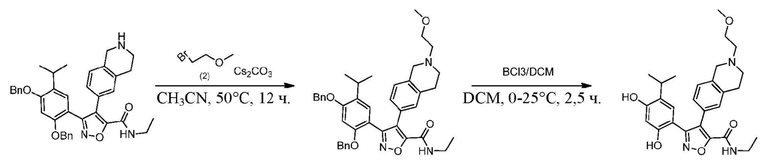
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 69, где 2-бромэтанол на стадии A заменяли 1-бром-2-метоксиэтаном. 1H ЯМР (400 МГц, DMSO-d6) ppm: 0,99-1,12 (m, 9H); 2,61-2,68 (m, 6H); 3,04 (dt, J = 13,77, 6,85 Гц, 1H); 3,19-3,24 (m, 2H); 3,47-3,60 (m, 7H); 6,33 (s, 1H); 6,81 (s, 1H); 6,90-6,96 (m, 2H); 6,98 (s, 1H); 8,18 (s, 1H); 8,85 (t, J = 5,71 Гц, 1H). MS (ESI) масса/заряд: 480 (M+1).
Пример 71
3-(2,4-Дигидрокси-5-изопропилфенил)-N-этил-4-(2-(2-фторэтил)-(1,2,3,4-тетрагидроизохинолин-6-ил)изоксазол-5-карбоксамид
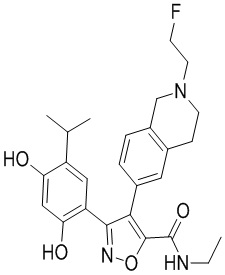
Реакционная схема
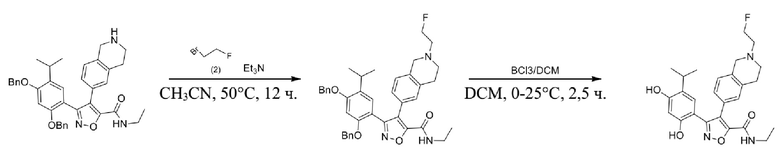
Стадия A. Указанное в заголовке данного примера соединение получали в соответствии с последовательностью стадий A и B в примере 69, где 2-бромэтанол на стадии A заменяли 1-бром-2-фторэтаном. 1H ЯМР (400 МГц, DMSO-d6) ppm: 1,02-1,11 (m, 9H); 2,89 (d, J = 16,56 Гц, 1H); 2,99-3,18 (m, 2H); 3,19-3,28 (m, 2H); 3,30-3,44 (m, 1H); 3,66-3,75 (m, 2H); 4,28-4,41 (m, 1H); 4,45-4,57 (m, 1H); 4,88 (t, J = 4,14 Гц, 1H); 5,00 (t, J = 4,27 Гц, 1H); 6,37 (s, 1H); 6,85 (s, 1H); 7,10 (s, 2H); 7,19 (s, 1H); 8,94 (t, J = 5,71 Гц, 1H); 9,18-9,76 (m, 2H); 10,97 (br.s., 1H). MS (ESI) масса/заряд: 468 (M+1).
Пример 72. Определение активности соединения на уровне фермента
В настоящем изобретении применяли способ флуоресцентной поляризации (FP) для создания платформы для скрининга ингибиторов HSP90-альфа. Принцип основан на расчете значений поляризации флуоресценции в горизонтальном и вертикальном направлениях путем определения значений изменения молекулярной массы до и после взаимодействия флуоресцентно-меченых малых молекул с другими молекулами с целью применения в корреляционном анализе. Если между флуоресцентно-меченой малой молекулой и макромолекулой установлено равновесное связывание, то при возбуждении она будет двигаться медленно и измеренные значения поляризации флуоресценции будут увеличиваться. Если флуоресцентно-меченая малая молекула, связанная с макромолекулой, замещена другими лигандами, то скорость ее вращения или обращения в свободном состоянии будет большей, испускаемый свет будет деполяризованным относительно плоскости возбуждающего света и, следовательно, измеренное значение поляризации света будет уменьшатся, таким образом рассчитывают значение поляризации (единицей поляризации является mP).
Применяемой в настоящем изобретении флуоресцентно-меченой малой молекулой являлась VER-00051001 (синтезированная в соответствии с методом синтеза, описанным в JMC, 2008, 51, 196-218). Реакцию проводили в черном 384-луночном планшете с применением HFB-буфера для проведения реакции с гидрофобным белком: 100 мМ Tris·Cl pH 7,4, 20 мМ KCl, 6 мМ MgCl2, 5 мкг/мл BSA, 25 нМ полноразмерный Hsp90U, 10 нМ VER-51001. Объем реакционной системы составлял 50 мл, состоящий из 5 нМ GM-BODIPY (гелданамицин), 30 нМ HSP90 и тестируемого малого молекулярного соединения или DMSO (диметилсульфоксида), при этом содержание DMSO составляло 2‰. Подготавливали дополнительные две группы лунок, при этом лунки лишь с добавленным HFB-буфером применяли в качестве холостых контролей, и лунки с дополнительно добавленным 5 нМ GM-BODIPY применяли в качестве отрицательных контролей. Реакцию проводили при 4°C в течение 12-16 часов. Значение mP измеряли с помощью ридера для микропланшетов Biotek при длине волны возбуждения 485 нм и длине волны испускания 535 нм. Степень ингибирования рассчитывали с применением следующего уравнения.
(mP без соединения - mP с соединением)/(mP без соединения - mP отрицательного контроля) × 100%
После расчета значений степени ингибирования для соединения при разных концентрациях можно рассчитать IC50 для соединения, и значения IC50 для каждого соединения показаны в таблице 1 ниже.
Пример 73. Измерение активности соединения на клеточном уровне
Анализ ингибирования роста опухолевых клеток проводили с помощью метода окрашивания с применением сульфородамина В (SRB). Конкретные стадии являлись следующими: инокулировали клеточную линию рака толстой кишки человека HCT116 и клеточную линию рака легкого человека A549 (приобретенные у Американской коллекции типовых культур, ATCC) в логарифмической фазе роста в 96-луночный микропланшет для культивирования при соответствующей концентрации, составляющей 4000 клеток/лунку, 100 мкл в каждой лунке; после культивирования в течение ночи добавляли соединение в разных концентрациях (100, 10, 1, 0,1, 0,01 мкМ) с обеспечением взаимодействия в течение 72 ч., при этом для каждой концентрации подготавливали лунки в трех повторностях, а также готовили лунки для контроля в виде растворителя с соответствующей концентрацией и холостые лунки без клеток. После завершения реакции культуральную среду отводили от адгезивных клеток, к последним добавляли 10% (вес/объем) трихлоруксусной кислоты (100 мкл/лунка) при 4°C с фиксированием в течение 1 ч., а затем промывали пять раз дистиллированной водой. После высушивания при комнатной температуре в каждую лунку добавляли 100 мкл SRB-раствора (4 мг/мл, растворенного в 1% ледяной уксусной кислоте) с последующим инкубированием и окрашиванием в течение 15 мин. при комнатной температуре. Несвязанный SRB удаляли путем промывания пять раз с помощью 1% ледяной уксусной кислоты. После высушивания при комнатной температуре в каждую лунку добавляли 100 мкл 10 мМ раствора Tris и измеряли оптическую плотность (OD) при 515 нм с помощью ридера VERSMax для микропланшетов. Степень ингибирования лекарственного средства по отношению к росту опухолевых клеток рассчитывали в соответствии со следующим уравнением.
Степень ингибирования (%) = (OD лунки с контролем - OD лунки с лекарственным средством)/OD лунки с контролем × 100%.
Эксперимент повторяли дважды.
Степень ингибирования (%) соединений по отношению к клеткам HCT116 и A549 показана в таблице 1 ниже.
Таблица 1. Результаты скринингового теста in vitro соединений по настоящему изобретению
 Примечания. Вышеуказанные соединения в тестах на активность в отношении фермента разделены на три группы A-C в соответствии с диапазоном активности, при этом A ≤ 20 нМ; 20 нМ < B ≤ 100 нМ; C > 100 нМ; н. о. указывает на то, что данные не определяли.
Примечания. Вышеуказанные соединения в тестах на активность в отношении фермента разделены на три группы A-C в соответствии с диапазоном активности, при этом A ≤ 20 нМ; 20 нМ < B ≤ 100 нМ; C > 100 нМ; н. о. указывает на то, что данные не определяли.
Заключения. Некоторые соединения по настоящему изобретению характеризуются хорошими значениями активности в отношении связывания с белком-мишенью HSP90 и ингибирования роста опухолевых клеток HCT116 или A549 по сравнению с известным ингибитором HSP90 луминеспибом.
Пример 74. Оценивание эффективности соединения у животных
Опухолевые клетки A549 подкожно инокулировали в правую подмышечную впадину бестимусным мышам в количестве 5x106 клеток/мышь с образованием трансплантированной опухоли. Когда объем достигал 100-200 мм3, животных случайным образом разделяли в соответствии с объемом опухоли на группу отрицательного контроля (n = 5), группу положительного контроля (n = 5 в каждой группе) и экспериментальные группы (n = 5 в каждой группе). Экспериментальным группам соответственно вводили внутривенно лекарственное средство луминеспиб (50 мг/кг) в качестве положительного контроля и соединение 1 (50 мг/кг) при разных концентрациях, дважды в неделю, и при этом группе отрицательного контроля вводили равное количество растворителя. Длину (A) и ширину (B) опухоли измеряли дважды в неделю с помощью штангенциркуля, и объем опухоли рассчитывали согласно выражению V = A × B2/2. Относительный объем опухоли (RTV) рассчитывали следующим образом. RTV = Vt/V0, где Vt представляет собой объем опухоли в конце введения, и V0 представляет собой объем опухоли, измеренный до разделения и введения. Индекс фармакодинамического оценивания противоопухолевой активности представляет собой относительную скорость роста опухоли T/C (%), и формула для расчета является следующей. T/C (%) = TRTV/CRTV × 100%, где TRTV: RTV для группы с обработкой; CRTV: RTV для группы отрицательного контроля. Критерий оценивания эффективности: T/C% > 60% неэффективное; T/C% ≤ 60% и P <0,05 с точки зрения статистики лечение эффективно. Степень ингибирования роста опухоли (TGI) рассчитывали следующим образом.
TGI (%) = {[(CVt - CV0) - (TVt - TV0)]/(CVt - CV0)} × 100%,
где CVt представляет собой объем опухоли контрольной группы в конце введения; CV0 представляет собой объем опухоли контрольной группы перед разделением и введением; TVt представляет собой объем опухоли группы с введением в конце введения; и TV0 представляет собой объем опухоли группы с введением перед разделением и введением. Разность в объеме опухоли между группой с введением и контрольной группой определяли с помощью t-критерия. При этом значения веса тела бестимусных мышей в каждой группе определяли взвешиванием дважды в неделю для предварительного оценивания побочных эффектов лекарственного средства в виде токсичности. Результаты в отношении эффективности соединений в данной модели показаны в таблице 2 ниже.
Таблица 2. Результаты тестов in vivo в отношении эффективности соединений по настоящему изобретению
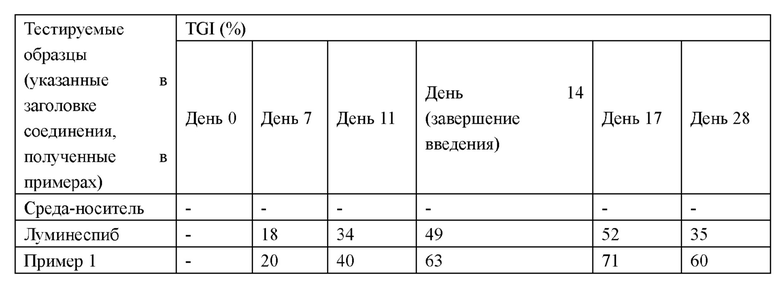
Заключения. Соединение в примере 1 по настоящему изобретению характеризуется слегка лучшей противоопухолевой эффективностью в мышиной модели с трансплантированными клетками рака легкого A549 по сравнению с известным ингибитором HSP90 луминеспибом.
Пример 75. Исследования распределения соединения в ткани
Исследование распределения соединения из примера 67 по настоящему изобретению в ткани
1. Краткое описание
В качестве субъектов-животных применяли крыс SD. Концентрации лекарственного средства в плазме крови, тканях легкого и молочной железы измеряли с помощью способа LC/MS/MS в разные моменты времени после внутривенной инъекции соединения из примера 67 в хвостовые части крыс. Исследовали распределение соединений по настоящему изобретению в ткани крыс и оценивали их фармакокинетические характеристики.
2. Протокол теста
2.1 Экспериментальные лекарственные средства: луминеспиб, ганетеспиб и соединение из примера 67.
2.2 Тестируемые животные:
Здоровые взрослые самцы крыс SD, 100-200 г, в сумме 9 крыс, приобретенные у Shanghai Slack Laboratory Animal Co., Ltd.
2.3 Получение лекарственного средства:
Взвешивали соответствующие количества образцов. Все из луминеспиба, ганетеспиба и соединения 67 по настоящему изобретению составляли в прозрачные растворы в 1 мг/мл в 0,5 мг/мл в 10% DMSO/14% Cremophor RH40/76% D5W для внутривенной инъекции.
2.4 Введение
9 самцов крыс SD разделяли на три группы, в каждой группе по три крысы. После обычного кормления осуществляли введение в хвостовые части путем внутривенной инъекции в дозе 1 мг/кг.
3. Порядок выполнения действий
Образцы собирали из крови, тканей легкого и молочной железы в трех группах крыс через 0,5 ч., 6 ч. и 24 ч. после введения. Образцы крови центрифугировали при 4°C, 3000 об./мин. в течение 15 мин. в пределах получаса после сбора с отделением и получением образцов плазмы крови. Полученные в результате образцы плазмы крови помещали в полипропиленовые пробирки и хранили при -80°C для анализа LC/MS/MS. После сбора образцов крови животных умерщвляли с помощью CO2 с последующим сбором образцов тканей. Каждый из образцов ткани промывали дважды холодной деионизированной водой и прикрепляли к фильтровальной бумаге. Затем каждый из образцов тканей несколько раз гомогенизировали в MeOH/15 мМ PBS (1: 2) (5 мл MeOH/15 мМ PBS (1: 2) на грамм ткани, с фактором разведения 6) с обеспечением образования гомогенной фазы. Весь процесс осуществляли при температуре ниже точки замерзания. Гомогенизированные образцы быстро замораживали путем применения сухого льда и хранили при -80°C для биоанализа.
Значения содержания тестируемых соединений в плазме, ткани легкого и молочной железы крыс после введения путем инъекции измеряли с помощью LC/MS/MS.
4. Результаты фармакокинетических параметров
Таблица 3. Результаты распределения соединения по настоящему изобретению в ткани
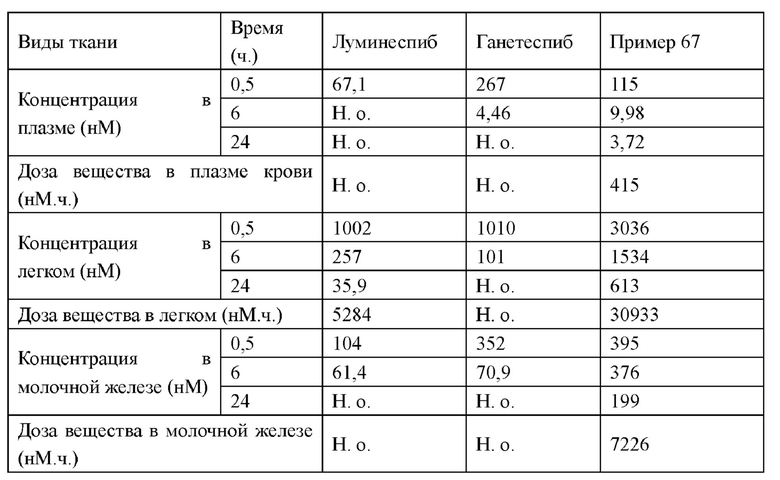
Примечания. Н. о. указывает, что был превышен предел определения и не было определено никаких данных.
Заключения. Когда доза инъекции предпочтительного соединения по настоящему изобретению составляла 1 мг/кг, значения концентрации в крови, в тканях легкого и молочной железы крыс в то же время были значительно выше, чем таковые для луминеспиба и ганетеспиба при той же дозе, и значение AUC также было значительно большим, указывая на то, что предпочтительное соединение по настоящему изобретению имеет лучшую перспективу в применении при раке, таком как рак легкого, рак молочной железы и т. д.
Пример 76. Оценивание эффективности соединения у животных с раком молочной железы
Опухолевые клетки BT-474 подкожно инокулировали в правую подмышечную впадину бестимусным мышам в количестве 5x106 клеток/мышь с образованием трансплантированной опухоли. Когда объем достигал 100-200 мм3, животных случайным образом разделяли в соответствии с объемом опухоли на группу отрицательного контроля (n = 6), группу положительного контроля (n = 6 в каждой группе) и экспериментальные группы (n = 6 в каждой группе). Экспериментальным группам соответственно вводили внутривенно лекарственное средство ганетеспиб (20 мг/кг) в качестве положительного контроля и соединение 67 (10 мг/кг и 20 мг/кг) при разных концентрациях, три раза в неделю, и при этом группе отрицательного контроля вводили равное количество растворителя. Длину (A) и ширину (B) опухоли измеряли дважды в неделю с помощью штангенциркуля, и объем опухоли рассчитывали согласно выражению V = A × B2/2. Относительный объем опухоли (RTV) рассчитывали следующим образом. RTV = Vt/V0, где Vt представляет собой объем опухоли в конце введения, и V0 представляет собой объем опухоли, измеренный до разделения и введения. Индекс фармакодинамического оценивания противоопухолевой активности представляет собой относительную скорость роста опухоли T/C (%), и формула для расчета является следующей. T/C (%) = TRTV/CRTV × 100%, где TRTV: RTV для группы с обработкой; CRTV: RTV для группы отрицательного контроля. Критерий оценивания эффективности: T/C% > 60% неэффективное; T/C% ≤ 60% и P <0,05 с точки зрения статистики лечение эффективно. Степень ингибирования роста опухоли (TGI) рассчитывали следующим образом.
TGI (%) = {[(CVt - CV0) - (TVt - TV0)]/(CVt - CV0)} × 100%,
где CVt представляет собой объем опухоли контрольной группы в конце введения; CV0 представляет собой объем опухоли контрольной группы перед разделением и введением; TVt представляет собой объем опухоли группы с введением в конце введения; и TV0 представляет собой объем опухоли группы с введением перед разделением и введением. Разность в объеме опухоли между группой с введением и контрольной группой определяли с помощью t-критерия. При этом значения веса тела бестимусных мышей в каждой группе определяли взвешиванием дважды в неделю для предварительного оценивания побочных эффектов лекарственного средства в виде токсичности. Результаты в отношении эффективности соединений в данной модели показаны в таблице 4 ниже.
Таблица 4. Результаты тестов in vivo в отношении эффективности соединений по настоящему изобретению
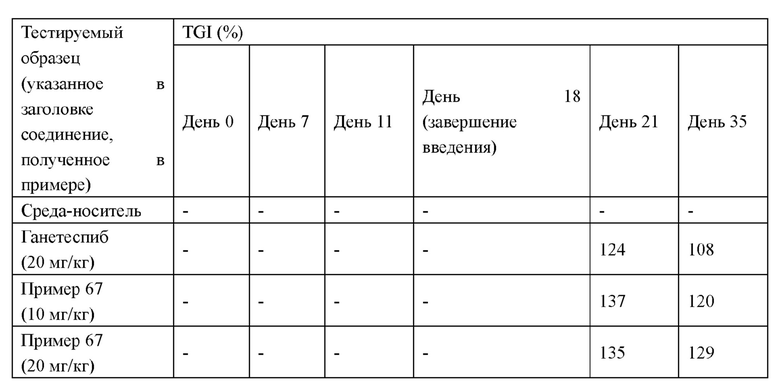
Заключения. Предпочтительное соединение по настоящему изобретению при 10 мг/кг характеризуется значительно лучшей противоопухолевой эффективностью в мышиной модели с трансплантированными клетками рака молочной железы BT-474 по сравнению с известным ингибитором HSP90 ганетеспибом при 20 мг/кг.
Пример 77. Сравнение водорастворимости
Соединения по настоящему изобретению, ганетеспиб и луминеспиб применяют в клинической практике в виде инъекций и, следовательно, необходимо, чтобы они характеризовались хорошей водорастворимостью для облегчения разработки связанных препаратов для клинической практики. Все уникальные конденсированные кольцевые структуры в соединениях по настоящему изобретению содержат основный атом азота, и соединения легко образуют соли и имеют значительное преимущество в отношении водорастворимости. В CN1771235A раскрыты ингибиторы HSP90, содержащие конденсированные кольцевые структуры, но они структурно отличаются от соединений по настоящему изобретению и не содержат групп, которые облегчают формирование соли, которые предположительно проявляют слабую водорастворимость.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ПАРАЗИТАРНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2793122C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ НАПРАВЛЕННОЙ ДЕГРАДАЦИИ БЕЛКА | 2020 |
|
RU2820673C2 |
| БЕНЗОТИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2018 |
|
RU2778370C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕКОТОРЫХ ЛЕЙКОЗОВ | 2019 |
|
RU2804709C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСИБЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Hsp90 | 2006 |
|
RU2458919C2 |
| ИНГИБИТОРЫ БЕЛКОВЫХ ТИРОЗИНФОСФАТАЗ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2833220C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
Изобретение относится к производному резорцина формулы (I) в качестве ингибитора HSP90 и к его фармацевтически приемлемым солям, способу его получения, фармацевтической композиции на его основе, его применению для получения лекарственного препарата лечения пролиферативных заболеваний, таких как рак, и нейродегенеративных заболеваний, опосредованных активностью белка теплового шока HSP90. В общей формуле (I) каждый из X и Y независимо выбран из N, O или S; X1, X2, Y1, Y2 и атом углерода, связывающий X1 и X2, вместе образуют 5-7-членное ароматическое кольцо, алифатическое насыщенное кольцо или алифатическое ненасыщенное кольцо; каждый из X1 и X2 независимо выбран из C, O, S, N, -C=C-, -C=N-; C в X1 или X2 может быть незамещенным или может быть замещен R01 или R02; каждый из R01 и R02 независимо выбран из C1-10алкила; каждый из Y1 и Y2 независимо выбран из C или N; два заместителя при Y1 и Y2 связаны вместе с образованием пяти-, шести- или семичленного содержащего азот насыщенного кольца, содержащего заместители R1, R2 и R3; R1 выбран из водорода, C1-6алкила, гидрокси-C1-6алкила, C1-4алкокси-C1-4алкила, галоген-C1-4алкила, C1-4алкилсульфонил-C1-4алкила, C1-4алкиламидо-C1-4алкила, N,N-ди(C1-4алкил)аминоацил-C1-4алкила, N,N-ди(C1-4алкил)амино-C1-4алканоила, морфолинил-C1-4алканоила; R2 и R3 выбраны из водорода или метила, или заместители R2 и R3 связаны друг с другом с помощью ковалентной связи с образованием пяти-, шести- или семичленного насыщенного кольца без заместителей; R4 выбран из галогена, C1-6алкила, C3-6циклоалкила; R5 выбран из C1-6алкоксиацила, C1-7алкиламинокарбонила, галоген-C1-6алкиламинокарбонила, C1-6алкокси-C1-6алкиламинокарбонила, N,N-ди(C1-6алкил)амино-C1-6алкиламинокарбонила, аминокарбонила, гидрокси-C1-6алкиламинокарбонила, замещенного гидроксильной группой галоген-C1-6алкила, замещенного нитрильной группой амидино или выбран из 5-6-членного ароматического кольца, необязательно замещенного одним или более R05; R05 выбран из C1-6алкила. 9 н. и 14 з.п. ф-лы, 4 табл., 77 пр.
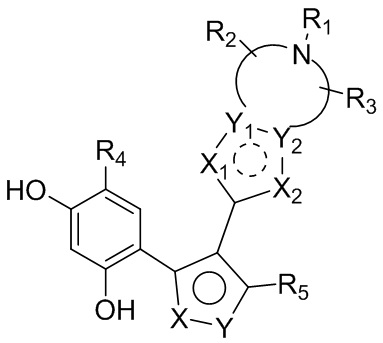
(I)
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
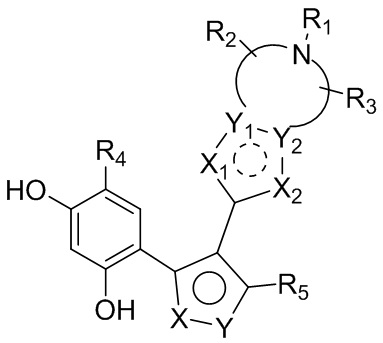
(I),
где
каждый из X и Y независимо выбран из N, O или S;
X1, X2, Y1, Y2 и атом углерода, связывающий X1 и X2, вместе образуют 5-7-членное ароматическое кольцо, алифатическое насыщенное кольцо или алифатическое ненасыщенное кольцо;
каждый из X1 и X2 независимо выбран из C, O, S, N, -C=C-, -C=N-;
C в X1 или X2 может быть незамещенным или может быть замещен R01 или R02;
каждый из R01 и R02 независимо выбран из C1-10алкила;
каждый из Y1 и Y2 независимо выбран из C или N;
два заместителя при Y1 и Y2 связаны вместе с образованием пяти-, шести- или семичленного содержащего азот насыщенного кольца, содержащего заместители R1, R2 и R3;
R1 выбран из водорода, C1-6алкила, гидрокси-C1-6алкила, C1-4алкокси-C1-4алкила, галоген-C1-4алкила, C1-4алкилсульфонил-C1-4алкила, C1-4алкиламидо-C1-4алкила, N,N-ди(C1-4алкил)аминоацил-C1-4алкила, N,N-ди(C1-4алкил)амино-C1-4алканоила, морфолинил-C1-4алканоила;
R2 и R3 выбраны из водорода или метила, или заместители R2 и R3 связаны друг с другом с помощью ковалентной связи с образованием пяти-, шести- или семичленного насыщенного кольца без заместителей; R4 выбран из галогена, C1-6алкила, C3-6циклоалкила;
R5 выбран из C1-6алкоксиацила, C1-7алкиламинокарбонила, галоген-C1-6алкиламинокарбонила, C1-6алкокси-C1-6алкиламинокарбонила, N,N-ди(C1-6алкил)амино-C1-6алкиламинокарбонила, аминокарбонила, гидрокси-C1-6алкиламинокарбонила, замещенного гидроксильной группой галоген-C1-6алкила, замещенного нитрильной группой амидино или выбран из 5-6-членного ароматического кольца, необязательно замещенного одним или более R05;
R05 выбран из C1-6алкила.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где Y1 и Y2 представляют собой C.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R4 выбран из C1-4алкила, Cl, Br или циклопропила.
4. Соединение или его фармацевтически приемлемая соль по п. 3, где R4 выбран из изопропила.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где R5 выбран из C1-6алкиламинокарбонила, галоген-C1-4алкиламинокарбонила, C1-4алкокси-C1-4алкиламинокарбонила, N,N-ди(C1-4алкил)амино-C1-4алкиламинокарбонила, аминокарбонила, гидрокси-C1-4алкиламинокарбонила, замещенного гидроксильной группой галоген-C1-4алкила, замещенного нитрильной группой амидино или выбран из 5-6-членного содержащего азот гетероароматического кольца, необязательно замещенного одним или более R05;
где R05 выбран из C1-6алкила.
6. Соединение или его фармацевтически приемлемая соль по п. 1, где R5 выбран из 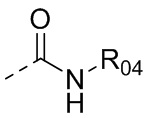 ,
,  ,
, 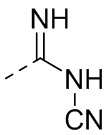 ,
, 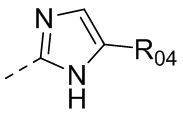 или
или 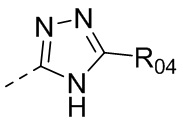 ;
;
при этом R04 выбран из H, C1-6алкила, галоген-C1-4алкила, C1-4алкокси-C1-4алкила, N,N-ди(C1-4алкил)амино-C1-4алкила, гидрокси-C1-4алкила.
7. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-6, где структурный элемент 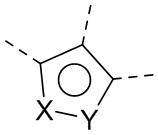 выбран из
выбран из  ,
,  или
или .
.
8. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-7, где структурный элемент 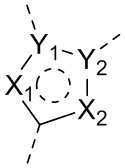 выбран из
выбран из 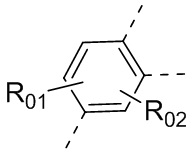 ,
, 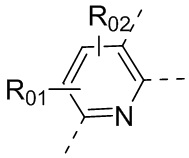 ,
, 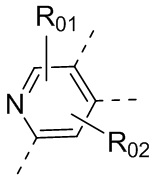 ,
, 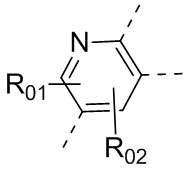 ,
, 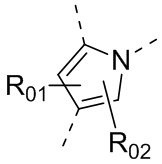 ,
, 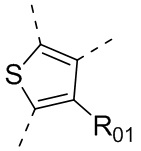 ,
, 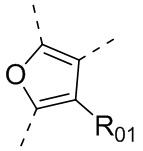 ,
, 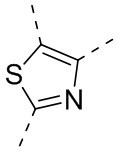 ,
, 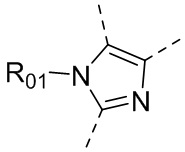 или
или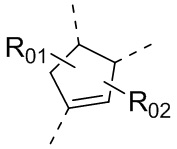 ;
;
при этом каждый из R01 и R02 независимо выбран из C1-6алкила.
9. Соединение или его фармацевтически приемлемая соль по п. 8, где выбран из  ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  или
или  .
.
10. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-7, где структурный элемент 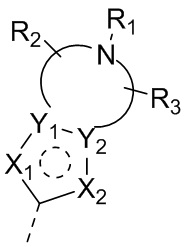 выбран из
выбран из 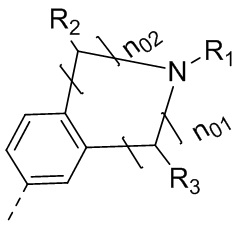 ;
;
причем n01 и n02 выбраны из 0, 1, 2 или 3, при этом сумма n01 и n02 равняется 2, 3 или 4, и R2 и R3 не связаны с образованием кольца.
11. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-7, где структурный элемент выбран из 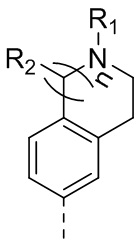 , при этом n = 1 или 2.
, при этом n = 1 или 2.
12. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-7, где структурный элемент выбран из  .
.
13. Соединение или его фармацевтически приемлемая соль по п. 10, где структурный элемент выбран из  ,
, 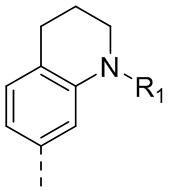 ,
, 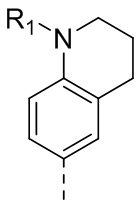 ,
, 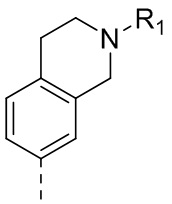 ,
, 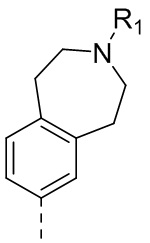 ,
, 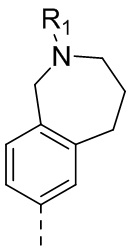 или
или 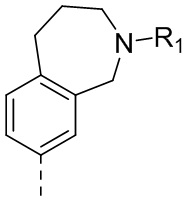 .
.
14. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-7, где структурный элемент выбран из 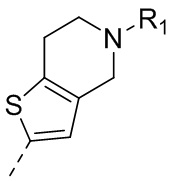 ,
, 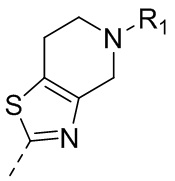 ,
, 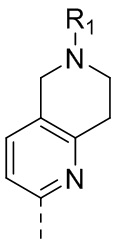 ,
, 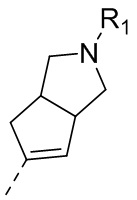 ,
, 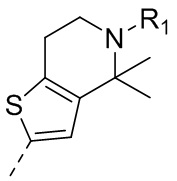 ,
, 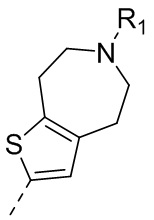 ,
,  ,
,  ,
,  или
или .
.
15. Соединение или его фармацевтически приемлемая соль, характеризующиеся следующими структурными формулами:
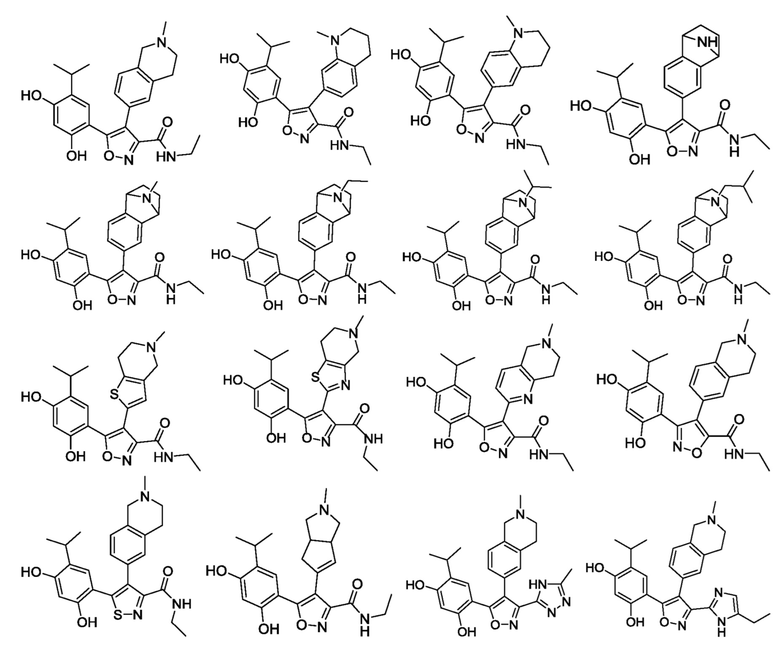
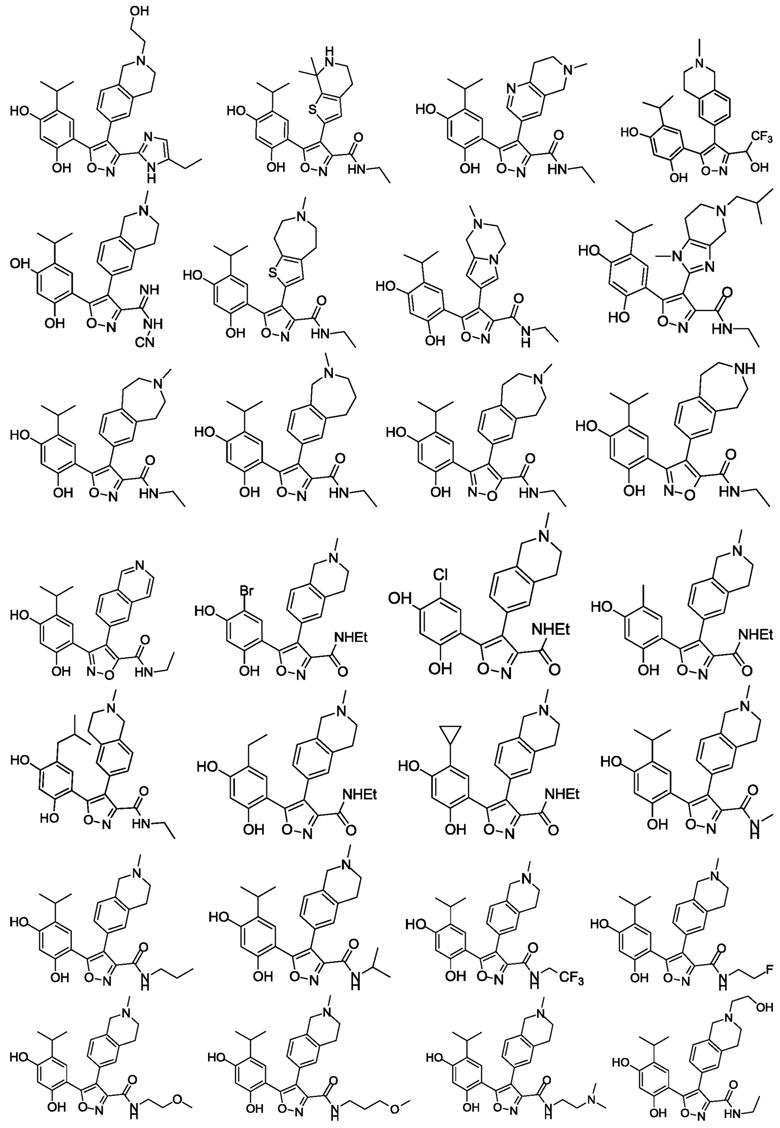
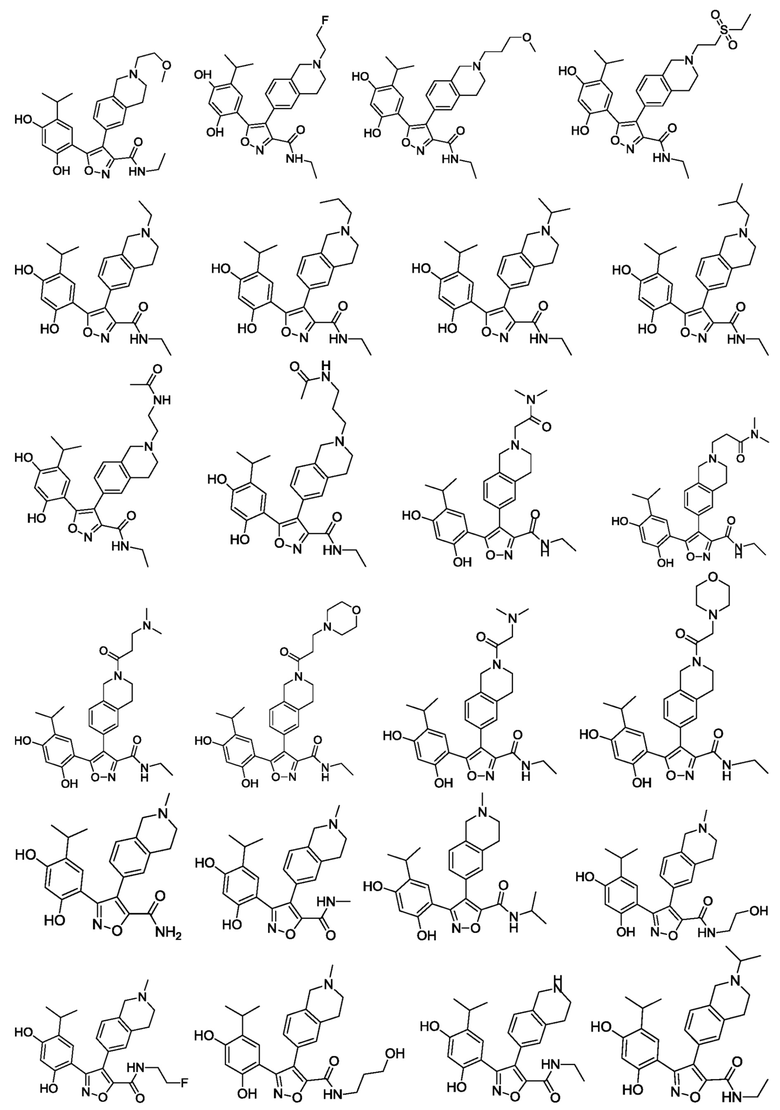
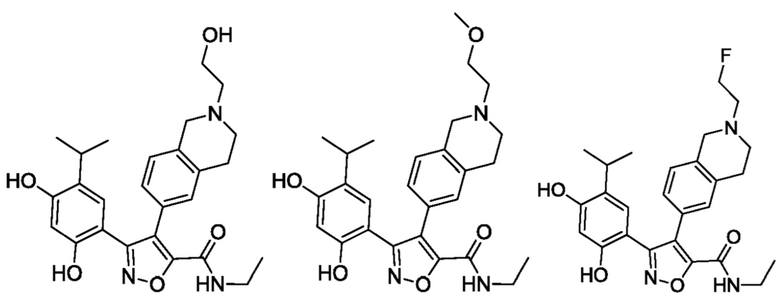
16. Способ получения соединения формулы (I) по любому из пп. 1-14, предусматривающий следующий путь:
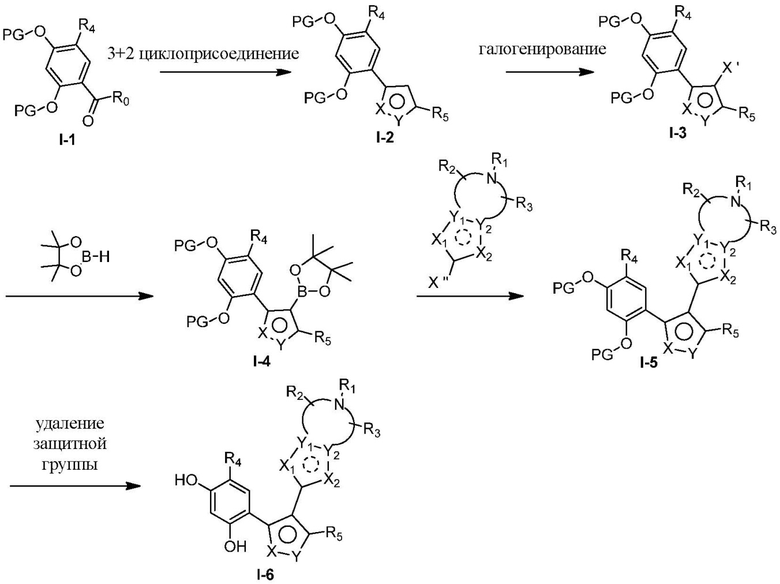 , где R0 выбран из H, алкила;
, где R0 выбран из H, алкила;
PG представляет собой защитную группу для гидроксильной группы, выбранную из метила, бензила, 4-метоксибензила, 3,4-диметоксибензила, метоксиметила, 2-метоксиэтоксиметила, 2-тетрагидрофурила, триметилсилила, триэтилсилила, триизопропилсилила, трет-бутилдиметилсилила, трет-бутилдифенилсилила, ацетила, бензоила или пивалоила;
X' представляет собой галоген;
X'' представляет собой галоген или трифторметансульфоновую кислоту;
другие переменные определены в п. 1.
17. Фармацевтическая композиция, обладающая свойством ингибирования HSP90, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-15 и фармацевтически приемлемые носители.
18. Применение соединения по любому из пп. 1-15 или композиции по п. 17 в получении лекарственного препарата для лечения опосредованных белком HSP90 заболеваний.
19. Применение по п. 18, где опосредованные белком HSP90 заболевания выбраны из рака и нейродегенеративных расстройств.
20. Применение соединения по любому из пп. 1-15 или композиции по п. 17 в получении лекарственного препарата для лечения конкретных типов карциномы, включая рак, такой как рак мочевого пузыря, рак молочной железы, рак толстой кишки, рак почки, рак печени, рак легкого, включая мелкоклеточный рак легкого, рак пищевода, рак желчного пузыря, рак яичника, рак поджелудочной железы, рак желудка, рак шейки матки, рак щитовидной железы, рак предстательной железы и рак кожи, включая плоскоклеточный рак; гемобластоз лимфоидного происхождения, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, B-клеточную лимфому, T-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому и лимфому Беркитта; гемобластоз миелоидного происхождения, включая острый и хронический миелодный лейкоз, миелодиспластический синдром и промиелоцитарный лейкоз; рак мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; рак центральной и периферической нервной системы, включая астроцитомы, нейроцитомы, глиомы и невриномы; а также другие виды рака, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярную тиреоидную карциному и саркому Капоши.
21. Применение соединения по любому из пп. 1-15 или композиции по п. 17 в получении лекарственного препарата для лечения конкретных типов нейродегенеративных расстройств, в том числе болезни Альцгеймера, болезни Паркинсона, болезни Хантингтона, рассеянного склероза и спинальной и бульбарной мышечной атрофии.
22. Применение соединения по любому из пп. 1-15 или композиции по п. 17 в получении лекарственного препарата для противораковой терапии.
23. Применение соединения по любому из пп. 1-15 или композиции по п. 17 для ингибирования активности белка HSP90 in vitro, предусматривающее приведение в контакт белка с эффективным количеством соединения формулы (I).
| CN 1771235 B, 28.04.2010 | |||
| WO 2004072051 A1, 26.08.2004 | |||
| WO 2007021966 A1, 22.02.2007 | |||
| WO 2008097640 A2, 14.08.2008 | |||
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ, ВКЛЮЧАЮЩИЕ ПРОИЗВОДНЫЕ ПИРИДО [4,3-d]ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 И ИНГИБИТОРА HER2 | 2009 |
|
RU2532375C2 |

