Настоящая заявка относится к применению новых замещенных индазолов для лечения и/или профилактики аллергических и/или воспалительных заболеваний у животных и к их применению для изготовления лекарственных средств для лечения и/или профилактики аллергических и/или воспалительных заболеваний у животных, в особенности атопического дерматита и/или аллергического блошиного дерматита, и в особенности у домашних животных, особенно у собак.
Настоящее изобретение относится к применению новых замещенных индазолов общей формулы (I), которые ингибируют ассоциированную с рецептором интерлейкин-1 киназу 4 (IRAK4).
Человеческая IRAK4 (ассоциированная с рецептором интерлейкин-1 киназа 4) играет ключевую роль в активации иммунной системы. Поэтому, эта киназа является важной терапевтической молекулой-мишенью для разработки ингибирующих воспаление веществ. IRAK4 экспрессируется множеством клеток и опосредует сигнальную трансдукцию толл-подобных рецепторов (TLR), за исключением TLR3, и рецепторов семейства интерлейкина (IL)-1β, состоящего из IL-1R (рецептор), IL-18R, IL-33R и IL-36R (Janeway and Medzhitov, Annu. Rev. Immunol., 2002; Dinarello, Annu. Rev. Immunol., 2009; Flannery and Bowie, Biochemical Pharmacology, 2010).
Ни IRAK4-нокаутные мыши, ни человеческие клетки пациентов с отсутствующей IRAK4 не реагируют на стимуляцию посредством TLR (за исключением TLR3) и семейства IL-1β (Suzuki, Suzuki, et al., Nature, 2002; Davidson, Currie, et al., The Journal of Immunology, 2006; Ku, von Bernuth, et al., JEM, 2007; Kim, Staschke, et al., JEM, 2007).
Связывание лигандов TLR или лигандов семейства IL-1β с соответствующим рецептором приводит к рекрутменту и связыванию MyD88 [ген первичного иммунного ответа миелоидной дифференцировки (88)] с рецептором. В результате этого MyD88 взаимодействует с IRAK4, что приводит к образованию активного комплекса, который взаимодействует с и активирует киназы IRAK1 или IRAK2 (Kollewe, Mackensen, et al., Journal of Biological Chemistry, 2004; Precious et al., J. Biol. Chem., 2009). Вследствие чего активируется сигнальный путь NF (ядерный фактор)-kB и сигнальный путь MAPK (митоген-активируемая протеинкиназа) (Wang, Deng, et al., Nature, 2001). Активация как сигнального пути NF-kB, так и сигнального пути MAPK приводит к процессам, связанным с различными иммунными процессами. Например, увеличивается экспрессия различных воспалительных сигнальных молекул и ферментов, таких как цитокины, хемокины и СОХ-2 (циклооксигеназа-2), и повышается стабильность мРНК связанных с воспалением генов, например, СОХ-2, IL-6 (интерлейкин-6), IL-8 (Holtmann, Enninga, et al., Journal of Biological Chemistry, 2001; Datta, Novotny, et al., The Journal of Immunology, 2004). К тому же, эти процессы могут быть связаны с пролиферацией и дифференциацией конкретных типов клеток, например, моноцитов, макрофагов, дендритных клеток, Т-клеток и В-клеток (Wan, Chi, et al., Nat Immunol, 2006; McGettrick and J.  British Journal of Haematology, 2007).
British Journal of Haematology, 2007).
Главная роль IRAK4 в патологии различных воспалительных нарушений была показана путем непосредственного сравнения мышей дикого типа (WT) с генетически модифицированными животными, имеющими киназа-инактивированную форму IRAK4 (IRAK4 KDKI). Животные IRAK4 KDKI имеют улучшенную клиническую картину в животной модели рассеянного склероза, атеросклероза, инфаркта миокарда и болезни Альцгеймера (Rekhter, Staschke, et al., Biochemical and Biophysical Research Communication, 2008; Maekawa, Mizue, et al., Circulation, 2009; Staschke, Dong, et al., The Journal of Immunology, 2009; Kim, Febbraio, et al., The Journal of Immunology, 2011; Cameron, Tse, et al., The Journal of Neuroscience, 2012). Кроме того, было обнаружено, что делеция IRAK4 в животной модели защищает от вызванного вирусами миокардита посредством улучшенной противовирусной реакции с одновременно сниженным системным воспалением (Valaperti, Nishii, et al., Circulation, 2013). Также было доказано, что экспрессия IRAK4 коррелирует со степенью синдрома Фогта-Коянаги-Харада (Sun, Yang, et al., PLoS ONE, 2014). Кроме того, была продемонстрирована высокая значимость IRAK4 для продуцирования опосредованного иммунным комплексом IFNα (интерферон-альфа) с помощью плазмоцитоидных дендритных клеток, ключевого процесса в патогенезе системной красной волчанки (СКВ) (Chiang et al., J Immunol, 2010). Кроме того, сигнальный путь связан с ожирением (Ahmad, R., P. Shihab, et al., Diabetology & Metabolic Syndrome, 2015).
Помимо основной роли IRAK4 во врожденном иммунитете, также имеются указания на то, что IRAK4 влияет на дифференциацию Th17 Т-клеток, компонентов приобретенного иммунитета. При отсутствии киназной активности IRAK4, генерируется меньше IL-17-продуцирующих Т-клеток (Т-клеток Th17) по сравнению с мышами WT. Ингибирование IRAK4 позволяет проводить профилактику и/или лечение атеросклероза, сахарного диабета 1 типа, ревматоидного артрита, спондилоартрита (в особенности псориатического спондилоартрита и болезни Бехтерева), красной волчанки, псориаза, витилиго, гигантоклеточного артериита, хронического воспалительного заболевания кишечника и вирусных заболеваний, например, ВИЧ (вирус иммунодефицита человека), вирусного гепатита (Staschke, et al., The Journal of Immunology, 2009; Marquez, et al., Ann Rheum Dis, 2014; Zambrano-Zaragoza, et al., International Journal of Inflammation, 2014; Wang, et al., Experimental and Therapeutic Medicine, 2015; Ciccia, et al., Rheumatology, 2015).
Вследствие центральной роли IRAK4 в MyD88-опосредованном сигнальном каскаде TLR (за исключением TLR3) и семейства рецепторов IL-1, ингибирование IRAK4 может быть использовано для профилактики и/или лечения нарушений, опосредованных указанными рецепторами.
В уровне техники раскрыто множество ингибиторов IRAK4 (см., например, Annual Reports in Medicinal Chemistry (2014), 49, 117-133).
В US 8293923 и US 20130274241 описаны ингибиторы IRAK4, имеющие 3-замещенную индазольную структуру. Описание 2-замещенных индазолов отсутствует.
В заявках WO 2013106254 и WO 2011153588 раскрыты производные 2,3-дизамещенного индазола.
В заявке WO 2007091107 описано производное 2-замещенного индазола для лечения мышечной дистрофии Дюшенна. Раскрытые соединения не имеют 6-гидроксиалкильного замещения.
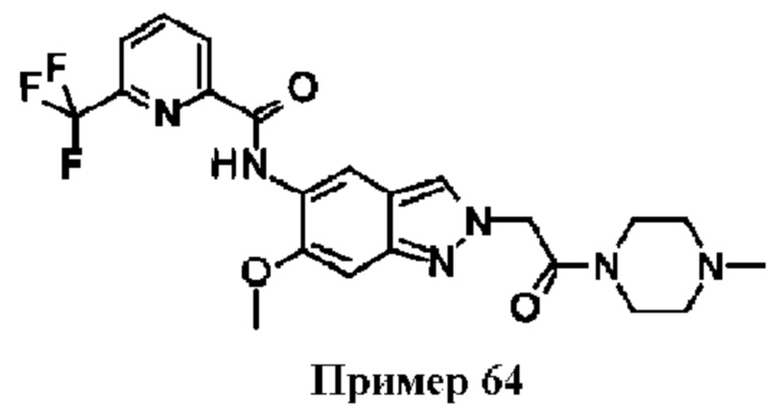
В WO 2015091426 описаны индазолы, такие как Пример 64, замещенные во 2 положении карбоксамидной боковой цепью.
В заявке WO 2015104662 описаны 2-замещенные индазолы следующей общей формулы:
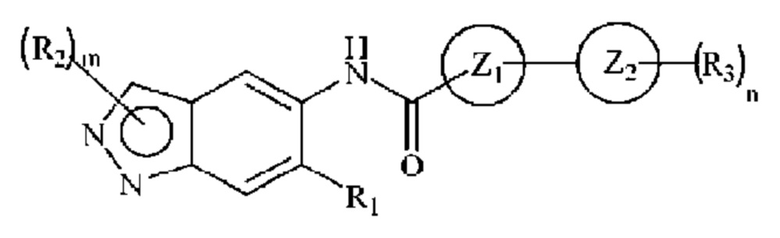
в которой R2 представляет собой алкильную или циклоалкильную группу. Имеются ясные описания 2-замещенных индазолов, имеющих метильную, 2-метоксиэтильную и циклопентильную группу в положении 2 (Примеры 1, 4 и 76). В Примере 117 также описано производное индазола, имеющее гидроксиэтильный заместитель в положении 1. Однако не описаны производные индазола, имеющие 3-гидрокси-3-метилбутильный заместитель в положении 1 или положении 2.
Индазолы, имеющие гидроксил-замещенную алкильную группу в положении 2, в общем, охватываются общей формулой, но явно не раскрыты в WO 2015104662.
Индазолы, имеющие алкильную группу в положении 2, где алкильная группа дополнительно замещена метилсульфонильной группой не охватываются общей формулой и определениями заместителей R2 в заявке WO 2015104662.
В дополнение к описанной выше схеме замещения для индазола в положениях 1 и 2 в заявке WO 2015104662 описаны индазолы, имеющие замещение в положении 6, для которого R1 определен следующим образом: алкил, циано, -NRaRb или необязательно замещенные группы, выбранные из циклоалкила, арила или гетероциклила, где заместителями являются независимо алкил, алкокси, галоген, гидроксил, гидроксиалкил, амино, аминоалкил, нитро, циано, галогеналкил, галогеналкокси, -ОСОСН2-O-алкил, -ОР(O)(O-алкил)2 или -СН2-ОР(O)(O-алкил)2. Для соединений индазола, в которых R1 представляет собой алкильную группу, действительная дата подачи заявки составляет 7 января 2015 (дата подачи международной заявки WO 2015104662). В заявках Индии 146/СНЕ/2014 и 3018/СНЕ/2014, приоритет которых заявлен, не описаны никакие соединения индазола, для которых R1 представляет собой алкильную группу.
Таким образом, соединения индазола следующей общей формулы:

в которой R1 представляет собой необязательно замещенную алкильную группу описаны впервые 7 января 2015 года а, следовательно, после даты приоритета настоящей заявки.
Примерами заместителей в положении 6, описанных в WO 2015104662 для R1 являются циклопропильный, циклогексильный, циано, 3-фторфенильный и насыщенный гетероциклический заместители. Индазолы, имеющие гидроксил-замещенную алкильную группу в положении 6, явно не описаны в WO 2015104662.
Используемые в настоящем изобретении соединения, также описаны в находящейся на рассмотрении патентной заявке РСТ/ЕР2015/077596, опубликованной как WO 2016083433 2 июня 2016 года.
Современные варианты лечения аллергических и/или воспалительных заболеваний у животных, например аллергических кожных заболеваний, обычно включают применение стероидов и циклоспорина - оба связаны с побочными эффектами. Недавно ингибитор янус-киназы (JAK) был одобрен для использования при атопическом дерматите у собак (АДС), который симптоматически обеспечивает избавление от зуда, однако режим дозирования снова может быть ограничен побочными эффектами. Лечение АДС с помощью модифицирующего заболевание средства и без побочных эффектов, связанных с лечением, остается неудовлетворенной медицинской потребностью.
Задача, которую решают с помощью настоящего изобретения, заключается в том, чтобы обеспечить лучший вариант лечения воспалительных и/или аллергических заболеваний у животных.
Данные ингибиторы IRAK4 в особенности пригодны для лечения и профилактики воспалительных заболеваний у животных, для которых характерна чрезмерная реакция иммунной системы. Особо следует упомянуть собачий атопический дерматит, аллергический блошиный дерматит у собак и кошек, воспалительные заболевания кишечника у собак и кошек, остеоартрит и воспалительные боли у собак, кошек, лошадей и крупного рогатого скота, неинфекционные рецидивирующие заболевания дыхательных путей у лошадей (также известная как хроническая обструктивная болезнь легких, запал), гиперчувствительность к насекомым у лошадей (также известная как сладкая чесотка, летняя экзема), астма у кошек, респираторные заболевания крупного рогатого скота, мастит и эндометрит у крупного рогатого скота и респираторные заболевания у свиней.
Например, атопический дерматит является распространенным заболеванием у домашних животных, особенно у кошек и собак.
В качестве одного конкретного примера, собачий атопический дерматит (АДС) является одним из наиболее распространенных заболеваний собак. АДС может поражать пациентов с раннего возраста, повторяться в течение всей жизни. В исследовании Lund et al. 1999, в ходе которого было исследовано 31484 собаки в 52 частных практиках в США, распространенность АДС составила 8,7%. АДС является второй наиболее распространенной причиной зуда у собак после аллергического блошиного дерматита (АБД).
Атопический дерматит у собак можно определить как «генетически предрасположенное воспалительное и зудящее аллергическое заболевание кожи с характерными клиническими признаками, связанными с IgE, чаще всего направленными против аллергенов окружающей среды (Halliwell, Veterinary Immunology and Immunopathology, 2006), таких как пылевые клещи и пыльца, которых домашним животным невероятно трудно избежать, так как пылевые клещи практически повсюду, а пыльца проникает в воздух снаружи.
Атопический дерматит у собак является сложным и многофакторным заболеванием, включающим иммунную дисрегуляцию, аллергическую сенсибилизацию, дефекты кожного барьера, микробную колонизацию и факторы окружающей среды.
IgE не является обязательным условием для развития клинических признаков во всех случаях, и отдельная клиническая единица, известная как атопический дерматит, была определена как «воспалительное и зудящее заболевание кожи с клиническими признаками, идентичными тем, которые наблюдаются при атопическом дерматите у собак, при котором ответ IgE на внешние или другие аллергены не может быть задокументирован» (Nuttall et al., Vet. Record, 2013).
Наиболее распространенные симптомы собачьего атопического дерматита включают зуд, чрезмерное расчесывание, истирание ковра, выпадение шерсти, жирную или шелушащуюся кожу с неприятным запахом, чрезмерное кусание лап и таких областей, как пах и подмышки. Со временем на поцарапанной коже могут образоваться горячие очаги - влажные воспаленные участки, которые могут стать инфицированными.
В настоящее время лечение острых вспышек атопического дерматита (АД) должно включать в себя поиск, а затем устранение причины обострений, купание в мягких шампунях и контроль зуда и поражений кожи с помощью вмешательств, которые включают местные и/или пероральные глюкокортикоиды или оклацитиниб. Для хронического АДС первыми шагами в терапии являются выявление и предотвращение вспышечных факторов, а также обеспечение надлежащей гигиены кожи и шерсти, и ухода за ними; это может включать более частое купание и, возможно, увеличение потребления незаменимых жирных кислот. В настоящее время наиболее эффективными препаратами для уменьшения хронического зуда и поражений кожи являются местные и пероральные глюкокортикоиды, пероральный циклоспорин, пероральный оклацитиниб и, где это доступно, инъекционные рекомбинантные интерфероны. Аллергенспецифическая иммунотерапия и проактивное прерывистое местное применение глюкокортикоидов являются единственными вмешательствами, которые могут предотвратить или отсрочить рецидив вспышек АД. (Olivry et al., ВМС Veterinary Research, 2015).
В качестве другого конкретного примера аллергический блошиный дерматит (АБД) или гиперчувствительность к укусу блох является наиболее распространенным дерматологическим заболеванием домашних собак (Scott et al., In: Muller and  Small Animal Dermatology, 2001), вызванным наиболее распространенной блохой на собаках и кошках: Ctenocephalides felis (Beresford-Jones, J Small Animal Practice, 1981; Chesney, Veterinary Record, 1995). У кошек также развивается АБД, который является одной из основных причин милиарного дерматита у кошек.
Small Animal Dermatology, 2001), вызванным наиболее распространенной блохой на собаках и кошках: Ctenocephalides felis (Beresford-Jones, J Small Animal Practice, 1981; Chesney, Veterinary Record, 1995). У кошек также развивается АБД, который является одной из основных причин милиарного дерматита у кошек.
АБД наиболее распространен летом, хотя в теплых климатах заражение блохами может сохраняться в течение всего года. В северных умеренных регионах тесная связь домашних животных и их блох с жилищами людей создает условия, которые обеспечивают круглогодичную проблему. Экстремальные температуры и низкая влажность препятствуют распространению блох.
Во время укуса блохи впрыскивают слюну, которая содержит различные гистаминоподобные соединения, ферменты, полипептиды и аминокислоты, которые охватывают широкий диапазон размеров (40-60 кДа) и вызывают гиперчувствительность типа I, типа IV и базофильную гиперчувствительность. У незараженных блохами собак, периодически подвергающихся укусам блох, развиваются немедленные (15 мин.) или отсроченные (24-48 ч.) реакции, или обе, и обнаруживаемые уровни как циркулирующих антител против IgE, так и против IgG. Собаки, постоянно подвергающиеся укусам блох, имеют низкий уровень этих циркулирующих антител и либо не проявляют кожных реакций, либо развивают их позже и в значительно сниженной степени. Это может указывать на то, что иммунологическая толерантность может естественно развиваться у собак, постоянно подвергающихся укусам блох. Хотя патофизиология АБД у кошек плохо изучена, подобные механизмы могут существовать.
Кошачья блоха (Ctencephalides felis) вызывает сильное раздражение у животных и людей и отвечает за аллергический блошиный дерматит. Типичными симптомами являются: зуд, воспаление кожи и поражения кожи (эритема, чешуйки, папулы, корки и лихенификация). Эти поражения чаще всего наблюдаются вдоль спины и у основания хвоста.
По мере прогрессирования состояния может произойти выпадение шерсти, ломкость шерсти, сочащиеся или покрытые коркой язвы, пупырышки и общее покраснение и воспаление кожи. Раны могут быть очень болезненными. В тяжелых случаях кожа становится утолщенной и темной, преимущественно в области спины у основания хвоста собаки. Собака сама наносит себе вред нанесением себе увечий из-за сильного зуда.
В целом, предпочтительным вариантом лечения является профилактика и лечение заражения блохами. Чаще всего используют неоникотиноиды, такие как имидаклоприд, или блокаторы хлоридных каналов, контролируемые гамма-аминомасляной кислотой (ГАМК), такие как фипронил. В случаях, когда симптомы кожного аллергического дерматита не проходят, используют современные методы лечения, упомянутые в разделе АДС, такие как местные и пероральные глюкокортикоиды, пероральный циклоспорин, пероральный оклацитиниб.
Настоящее изобретение обеспечивает соединения общей формулы (I)
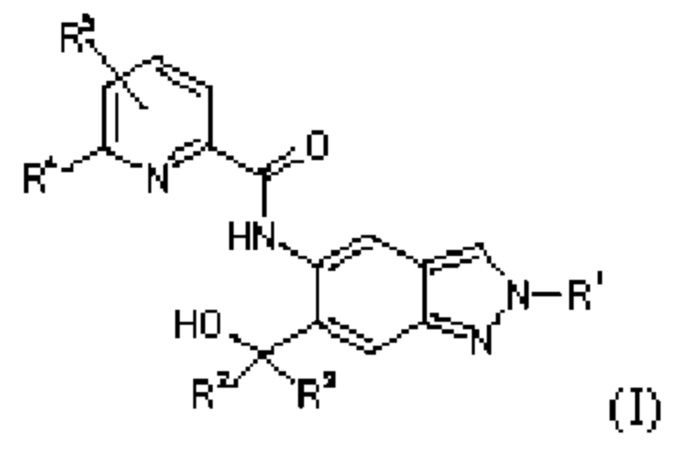
в которой:
R1 представляет собой C1-C6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством
галогена, гидроксила, незамещенного или моно- или поли-галоген-замещенного С3-С6-циклоалкила или группы R6, R7SO2, R7SO или R8O,
или группу, выбранную из:

где * представляет собой место связывания группы с остальной частью молекулы;
R2 и R3 всегда имеют одно и то же определение и оба являются или водородом, или С1-С6-алкилом;
R4 представляет собой галоген, циано, незамещенный или однократно или многократно одинаково или по-разному замещенный C1-C6-алкил или незамещенный или однократно или многократно одинаково или по-разному замещенный С3-С6-циклоалкил, и заместители выбраны из группы галогена и гидроксила;
R5 представляет собой водород, галоген или незамещенный или моно- или поли-галоген-замещенный С1-С6-алкил;
R6 представляет собой незамещенный или моно- или ди-метил-замещенный моноциклический насыщенный гетероцикл с 4-6 кольцевыми атомами, который содержит гетероатом или гетерогруппу из группы О, S, SO и SO2;
R7 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена, гидроксила или С3-С6-циклоалкила,
или R7 представляет собой С3-С6-циклоалкил;
R8 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена;
и их диастереомеры, энантиомеры, метаболиты, соли, сольваты или сольваты солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
В случае описанных ниже промежуточных продуктов синтеза и демонстрационных примеров изобретения любое соединение, указанное в форме соли соответствующего основания или кислоты, обычно представляет собой соль неизвестного точного стехиометрического состава, полученного соответствующим способом получения и/или очистки. Поэтому, если не указано более подробно, дополнения к названиям и структурным формулам, таким как «гидрохлорид», «трифторацетат», «натриевая соль» или "х HCl", "х CF3COOH", "х Na+" не следует понимать в стехиометрическом смысле в случае таких солей, но они носят описательный характер в отношении присутствующих в них солеобразующих компонентов.
Это же соответствующим образом относится и к случаю, когда промежуточные продукты синтеза или демонстрационные примеры или их соли были получены в форме сольватов, например, гидратов, неизвестного стехиометрического состава (если они определенного типа) описанными способами получения и/или очистки.
Настоящие соединения представляют собой соединениями формулы (I) и их соли, сольваты и сольваты солей, соединения, охватываемые формулой (I) и имеющие формулы, указанные ниже и их соли, сольваты и сольваты солей и соединения, которые охватываются формулой (I) и которые упоминаются ниже в качестве вариантов осуществления и их соли, сольваты и сольваты солей, если соединения, которые охватываются формулой (I) и указаны ниже, еще не являются солями, сольватами и сольватами солей.
Предпочтительными солями в контексте настоящего изобретения являются физиологически приемлемые соли соединений настоящего изобретения. Тем не менее, изобретение также охватывает соли, которые сами непригодны для фармацевтических применений, но которые можно применять, например, для выделения или очистки соединений настоящего изобретения.
Физиологически приемлемые соли соединений настоящего изобретения включают кислотно-аддитивные соли минеральных кислот, карбоновых кислот и сульфоновых кислот, например, соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, нафталиндисульфоновой кислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты и бензойной кислоты.
Физиологически приемлемые соли соединений настоящего изобретения также включают соли обычных оснований, в качестве примера и с предпочтением - соли щелочных металлов (например, соли натрия и калия), соли щелочноземельных металлов (например, соли кальция и магния) и соли аммония - производные аммиака или органических аминов, содержащих от 1 до 16 атомов углерода, в качестве примера и с предпочтением - этиламина, диэтиламина, триэтиламина, этилдиизопропиламина, моноэтаноламина, диэтаноламина, триэтаноламина, дициклогексиламина, диметиламиноэтанола, прокаина, дибензиламина, N-метилморфолина, аргинина, лизина, этилендиамина и N-метилпиперидина.
Сольваты в контексте изобретения описаны в тех формах соединений в соответствии с изобретением, которые в твердом или жидком состоянии образуют комплекс путем координации с молекулами растворителя. Гидраты являются особой формой сольватов, в которых координация происходит с водой.
Соединения настоящего изобретения в зависимости от их структуры, могут существовать в различных стереоизомерных формах, т.е. в форме конфигурационных изомеров или же необязательно в виде конформационных изомеров (энантиомеров и/или диастереомеров, включая таковые в случае атропоизомеров). Следовательно, настоящее изобретение охватывает энантиомеры и диастереомеры, и их соответствующие смеси. Стереоизомерно однородные составные части можно выделить из таких смесей энантиомеров и/или диастереомеров известным способом; для этой цели предпочтительно применяют хроматографические способы, особенно ВЭЖХ хроматографию на ахиральной или хиральной фазе.
Если соединения в соответствии с настоящим изобретением могут встречаться в таутомерных формах, то настоящее изобретение охватывает все таутомерные формы.
Настоящее изобретение также охватывает применение всех пригодных изотопных вариантов данных соединений. Изотопный вариант соединения в соответствии с изобретением в данном случае понимают в значении соединения, в котором по меньшей мере один атом в рамках такого соединения в соответствии с изобретением заменен на другой атом того же атомного номера, но с атомной массой, отличающейся от атомной массы, которая обычно или преимущественно встречается в природе. Примерами изотопов, которые можно включить в соединение в соответствии с изобретением являются изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора, брома и йода, такие как 2Н (дейтерий), 3Н (тритий), 13С, 14С, 15N, 17O, 18O, 32Р, 33Р, 33S, 34S, 35S, 36S, 18F, 36Cl, 82Br, 123I, 124I, 129I и 131I. Особые изотопные варианты соединения в соответствии с изобретением, особенно варианты, в которые включены один или несколько радиоактивных изотопов, могут быть полезными, например, для исследования механизма действия или распределения активного ингредиента в организме; вследствие сравнительной легкости получения и обнаружения, для этой цели пригодны особенно соединения, меченые изотопами 3Н или 14С. Кроме того, введение изотопов, например, дейтерия, может приводить к особенным терапевтическим преимуществам в результате большей метаболической стабильности соединения, например, к удлинению периода полураспада в организме или к снижению требуемой активной дозы; следовательно, такие модификации соединений в соответствии с настоящим изобретением могут в некоторых случаях также составлять предпочтительный вариант осуществления настоящего изобретения. Изотопные варианты данных соединений могут быть получены способами, известными специалисту в данной области техники, например, с помощью методов, описанных ниже, и методик, описанных в демонстрационных примерах, путем использования соответствующих изотопных модификаций соответствующих реагентов и/или исходных соединений.
Настоящее изобретение также предусматривает применение всех возможных кристаллических и полиморфных форм данных соединений, где полиморфы могут присутствовать либо в виде отдельных полиморфов, либо в виде смеси множества полиморфов во всех диапазонах концентраций.
Настоящее изобретение дополнительно также охватывает применение пролекарств настоящих соединений. Термин «пролекарства» в данном контексте относится к соединениям, которые сами могут быть биологически активными или неактивными, но превращаются (например, метаболически или гидролитически) в соединения, присутствующие в организме в течение времени их удержания в организме.
В контексте настоящего изобретения, если не указано иное, заместители имеют следующие значения:
Алкил в контексте изобретения представляет собой алкильную группу с прямой или разветвленной цепью, содержащую указанное конкретное число атомов углерода. Примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, 1-метилпропил, 2-метилпропил, трет-бутил, н-пентил, 1-этилпропил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 2,2-диметилпропил, н-гексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1-этилбутил и 2-этилбутил. Предпочтение отдают метилу, этилу, н-пропилу, н-бутилу, 2-метилбутилу, 3-метилбутилу и 2,2-диметилпропилу.
Циклоалкил в контексте изобретения представляет собой моноциклическую насыщенную алкильную группу, содержащую указанное в каждом случае число атомов углерода. Предпочтительные примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
Алкокси в контексте изобретения представляет собой алкокси-группу с прямой или разветвленной цепью, содержащий указанное конкретное число атомов углерода. Предпочтительны от 1 до 6 атомов углерода. Примеры включают метокси, этокси, н-пропокси, изопропокси, 1-метилпропокси, н-бутокси, изобутокси, трет-бутокси, н-пентокси, изопентокси, 1-этилпропокси, 1-метилбутокси, 2-метилбутокси, 3-метилбутокси и н-гексокси. Особое предпочтение отдают линейному или разветвленному алкокси-радикалу, содержащему от 1 до 4 атомов углерода. Примеры, которые могут быть упомянуты как предпочтительные, представляют собой метокси, этокси, н-пропокси, 1-метилпропокси, н-бутокси и изобутокси.
Циклоалкил в контексте изобретения представляет собой моноциклическую насыщенную алкильную группу, содержащую указанное в каждом случае число атомов углерода. Предпочтительные примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
Алкокси в контексте изобретения представляет собой алкокси-группу с прямой или разветвленной цепью, содержащую указанное конкретное число атомов углерода. Предпочтительны от 1 до 6 атомов углерода. Примеры включают метокси, этокси, н-пропокси, изопропокси, 1-метилпропокси, н-бутокси, изобутокси, трет-бутокси, н-пентокси, изопентокси, 1-этилпропокси, 1-метилбутокси, 2-метилбутокси, 3-метилбутокси и н-гексокси. Особое предпочтение отдают линейному или разветвленному алкокси-радикалу, содержащему от 1 до 4 атомов углерода. Примеры, которые могут быть упомянуты как предпочтительные, представляют собой метокси, этокси, н-пропокси, 1-метилпропокси, н-бутокси и изобутокси.
Галоген в контексте изобретения означает фтор, хлор и бром. Предпочтение отдают фтору.
Гидроксил в контексте изобретения означает ОН.
Моноциклический насыщенный гетероцикл представляет собой моноциклический насыщенный гетероцикл, который имеет от 4 до 6 кольцевых атомов и содержит гетероатом или гетерогруппу из группы О, S, SO и SO2. Предпочтительным является гетероцикл, содержащий гетероатом или гетерогруппу из группы О, SO и SO2. Примеры включают: оксетан, тетрагидрофуран, тетрагидро-2Н-пиран-4-ил, 1,1-диоксидотетрагидро-2Н-тиопиран-3-ил, 1,1-диоксидотетрагидро-2Н-тиопиран-2-ил, 1,1-диоксидотетрагидро-2Н-тиопиран-4-ил, 1,1-диоксидотетрагидротиофен-3-ил, 1,1-диоксидотетрагидротиофен-2-ил, 1,1-диоксидотиэтан-2-ил или 1,1-диоксидотиэтан-3-ил. В данном случае особое предпочтение отдают оксетану и тетрагидрофурану. Наибольшее предпочтение отдают оксетан-3-илу.
Символ * на связи обозначает точку присоединения в молекуле.
Когда группы в соединениях в соответствии с настоящим изобретением замещены, то такие радикалы могут быть моно- или полизамещенными, если не указано иное. В контексте настоящего изобретения, все радикалы, встречающиеся более одного раза, определены независимо друг от друга. Предпочтительным является замещение одним, двумя или тремя одинаковыми или различными заместителями.
Предпочтительный вариант осуществления R1 представляет собой С2-С6-алкильную группу, замещенную 1, 2 или 3 атомами фтора. Особое предпочтение отдают 2,2,2-трифторэтилу, 3,3,3-трифторпропилу и 4,4,4-трифторбутилу. Наиболее предпочтительной является 4,4,4-трифторбутильная группа.
Дополнительным предпочтительным вариантом осуществления R1 является С2-С6-алкильная группа, замещенная одним или двумя гидроксильными группами или одним С1-С3-алкокси или три-фтор-замещенным C1-С3-алкокси. Особое предпочтение отдают С2-С5-алкильной группе, замещенной гидроксилом или C1-С3-алкокси или трифторметокси или 2,2,2-трифторэтокси. Наиболее предпочтительным является 3-гидрокси-3-метилбутил, 3-метоксипропил, 3-гидроксипропил, 3-трифторметоксипропил, 2-метоксиэтил или 2-гидроксиэтил. В особенности предпочтительной является 3-гидрокси-3-метилбутильная группа.
Кроме того, предпочтительно, R1 представляет собой С2-С6-алкильную группу замещенную посредством С1-С6-алкил-SO2 группы. Особенно предпочтительной является метил-SO2-замещенная С2-С4-алкильная группа. В особенности предпочтительными для R1 являются 2-(метилсульфонил)этил или 3-(метилсульфонил)пропил. Из последней группы особенно предпочтительным является 2-(метилсульфонил)этил.
Дополнительно предпочтительно, R1 представляет собой C1-С3-алкильную группу, замещенную оксетанилом, тетрагидрофуранилом, тетрагидро-2Н-пиран-4-илом, 1,1-диоксидотетрагидро-2Н-тиопиран-3-илом, 1,1-диоксидотетрагидро-2Н-тиопиран-2-илом, 1,1-диоксидотетрагидро-2Н-тиопиран-4-илом, 1,1-диоксидотетрагидротиофен-3-илом, 1,1-диоксидотетрагидротиофен-2-илом, 1,1-диоксидотиэтан-2-илом или 1,1-диоксидотиэтан-3-илом. Особое предпочтение отдают C1-С3-алкильной группе, замещенной оксетановой группой. В особенности предпочтительно для R1 является оксетан-3-илметильная группа.
Для R2 и R3, которые всегда имеют одно и то же определение, предпочтительными являются водород или метил. Особенно предпочтительным является метил.
В случае с R4, предпочтительной является незамещенная или моно- или поли-галоген-замещенная C1-С3-алкильная группа или C1-С3-алкильная группа, замещенная одной гидроксильной группой или C1-С3-алкильная группа, замещенная одной гидроксильной группой и тремя атомами фтора.
Для R4, особое предпочтение отдают следующим группам: метил, этил, трифтор-С1-С3-алкил, дифтор-С1-С3-алкил, гидроксиметил, 1-гидроксиэтил, 2-гидроксипропан-2-ил и 2,2,2-трифтор-1-гидроксиэтил. Для R4, особое предпочтение отдают метальной, трифторметильной и дифторметильной группам. При этом особое предпочтение отдают трифторметильной группе.
Предпочтительный вариант осуществления R5 представляет собой водород, фтор, хлор или C1-С3-алкил. Более предпочтительно, R5 представляет собой водород, фтор или метил. Наиболее предпочтительно, R5 представляет собой водород или фтор.
Также особое предпочтение отдают соединениям, в которых R4 представляет собой метил или трифторметил и R5 представляет собой фтор. Наиболее предпочтительными являются соединения, в которых R4 представляет собой метил и R5 представляет собой фтор, где R5 находится в орто-положении к R4.
Для R6, предпочтительные варианты осуществления включают оксетанил, тетрагидрофуранил, тетрагидро-2Н-пиран-4-ил, 1,1-диоксидотетрагидро-2Н-тиопиран-3-ил, 1,1-диоксидотетрагидро-2Н-тиопиран-2-ил, 1,1-диоксидотетрагидро-2Н-тиопиран-4-ил, 1,1-диоксидотетрагидротиофен-3-ил, 1,1-диоксидотетрагидротиофен-2-ил, 1,1-диоксидотиэтан-2-ил или 1,1-диоксидотиэтан-3-ил. При этом особое предпочтение отдают оксетанилу. Наиболее предпочтительным является оксетан-3-ил.
R7 исключительно связан с функциональными группами -SO2- и -SO-, т.е. представляет собой R7-замещенную -SO2- или SO группу. В этой связи, R7 представляет собой предпочтительно С1-С4-алкил, где С1-С4-алкильная группа является незамещенной или монозамещенной гидроксилом или циклопропилом или замещенной тремя атомами фтора. Дополнительно предпочтительно R7 представляет собой циклопропильную группу. В особенности предпочтительно R7 означает метил, этил или гидроксиэтил. Наиболее предпочтительным для R7 является метил.
Это означает, что в случае С1-С6-алкильной группы, замещенной посредством R7SO2- или R7SO-, в контексте R1, предпочтение отдают C1-C6-алкилу, замещенному посредством С1-С6-алкил-SO2 или а С1-С6-алкил-SO. При этом для R1, предпочтение отдают в особенности метилсульфонилэтилу и метилсульфонилпропилу. Причем наибольшее предпочтение отдают метилсульфонилэтилу.
Для R8, предпочтение отдают незамещенной С1-С4-алкильной группе или три-фтор-замещенной С1-С4-алкильной группе. Особое предпочтение отдают метилу, этилу, трифторметилу или 2,2,2-трифторэтилу. Наиболее предпочтительным является метил, трифторметил или 2,2,2-трифторэтил.
Как описано ранее, внутриклеточный фермент ассоциированная с рецептором интерлейкин-1 киназа 4 (IRAK4) играет неотъемлемую роль в сигнальном пути рецепторов, активируемых цитокинами и лигандами TLR, которые участвуют в воспалительных процессах. Помимо воспаления, IRAK4 также участвует в передаче сигналов аллергических процессов. Такие аллергические процессы играют важную роль в патогенезе аллергических заболеваний кожи, таких как атопический дерматит.
Например, IL-33, недавнее дополнение к семейству цитокинов IL-1, которое также включает IL-18 и IL-1, связывается и активирует рецепторы IL-33 (IL-33R), которые затем связываются с MyD88, IRAK4 и TRF6 (Schmitz et al, Immunity, 2005). IRAK4 является основным компонентом этого сигнального пути. IL-33R сильно экспрессируются на хелперных Т-клетках 2 типа (Th2), тучных клетках и эозинофилах. IL-33 активирует эти клетки и стимулирует иммунные ответы Th2 (Schmitz et al, Immunity, 2005). Каждый из этих типов клеток участвует в патогенезе атопического дерматита. Уровни IL-33 в сыворотке коррелируют с тяжестью атопического дерматита у человека и снижаются при лечении местными стероидами и ингибитором кальциневрина (Tamagawa-Mineoka et al, J American Academy Dermatology, 2014). На моделях острого собачьего атопического дерматита было показано, что экспрессия гена IL-33 была значительно повышена при поражениях кожи (Schamber et al., G3 (Bethesda), 2014; Olivry et al, Journal of Investigative Dermatology, 2016).
Кроме того, в атопический дерматит был вовлечен второй член семейства цитокинов IL-1, IL-18. Уровни IL-18 в сыворотке крови повышаются с тяжестью атопического дерматита у детей (Sohn et al, Allergy and Asthma Proceedings, 2004). На моделях острого собачьего атопического дерматита было показано, что ген IL-18 был значительно активирован при поражениях кожи (Schamber et al., G3 (Bethesda), 2014; Olivry et al, Journal of Investigative Dermatology, 2016 Кроме того, подобное атопическому дерматиту воспаление и зуд были вызваны чрезмерным высвобождением IL-18 и ускорены посредством IL-1 у мышей (Konishi et al, Proceedings of the National Academy of Sciences, 2002). И снова было доказано, что IRAK4 является важным компонентом сигнального каскада IL-18 (Suzuki et al, J Immunology, 2003). Аналогичным образом IRAK4 является решающей для передачи сигналов IL-1 и TLR лигандов (Suzuki et al, Nature, 2002). Известно, что агонисты TLR вызывают зуд (Liu et al, Neuroscience bulletin, 2012), важный симптом атопического дерматита, и для лечения атопического дерматита анти-IL-1 терапии применяют не по показаниям. Кроме того, полиморфизмы в IRAK4 связаны с повышенным общим IgE при аллергических заболеваниях, таких как астма и хронический риносинусит (Tewfik et al, Allergy, 2009). Уровни IgE также повышены при атопическом дерматите.
Поэтому, поскольку IRAK4 является важным элементом сигнальных путей, которые активируются рядом цитокинов, лигандов TLR и IRAK4 имеет полиморфизмы, связанные с повышенным уровнем IgE, то ингибирование IRAK4 является важной терапевтической стратегией для лечения аллергических заболеваний кожи, таких как атопический дерматит. Кроме того, у домашних животных (в особенности собак и кошек) как атопический дерматит, так и аллергический блошиный дерматит являются соответствующими показаниями, поскольку оба заболевания заключают в себе гиперчувствительности I типа, которая включает антитела IgE, клетки Th2, тучные клетки и эозинофилы. К тому же, АБД может включать гиперчувствительность IV типа, в которую вовлечены IL-1 и IL-18.
Соединения в соответствии с настоящим изобретением действуют как ингибиторы киназы IRAK4 и, следовательно, обладают непредвиденным спектром полезной фармакологической активности при лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Предпочтение отдают соединениям формулы (I), в которой
R1 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством фтора, гидроксила или группы R6, R7SO2, R7SO или R8O;
R2 и R3 всегда имеют одно и то же определение и оба являются или водородом, или C1-С3-алкилом;
R4 представляет собой галоген, циано или C1-С3-алкил, где С1-С3-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена или гидроксила;
R5 представляет собой водород, фтор, хлор или C1-С3-алкил;
R6 представляет собой оксетанил или тетрагидрофуранил;
R7 представляет собой С1-С4-алкил, где С1-С4-алкильная группа является незамещенной или монозамещенной гидроксилом или циклопропилом или замещенной тремя атомами фтора;
R8 представляет собой незамещенный С1-С4-алкил или три-фтор-замещенный С1-С4-алкил;
и их диастереомерам, энантиомерам, метаболитам, солям, сольватам или сольватам солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Дополнительно предпочтение отдают соединениям формулы (I), в которой
R1 представляет собой С2-С6-алкил, где С2-С6-алкил является незамещенным, или
С2-С6-алкил является моно-, ди- или три-фтор-замещенным или
С2-С6-алкил является монозамещенным посредством гидроксила, R6, R7SO2, или R8O,
или в которой R1 представляет собой оксетанил-замещенный C1-С3-алкил;
R2 и R3 всегда имеют одно и то же определение и оба являются или водородом, или метилом;
R4 представляет собой незамещенную или моно- или поли-галоген-замещенную C1-С3-алкильную группу или C1-С3-алкильную группу, замещенную одной гидроксильной группой или C1-С3-алкильную группу, замещенную одной гидроксильной группой и тремя атомами фтора;
R5 представляет собой водород, фтор или C1-С3-алкил;
R7 представляет собой C1-С3-алкил;
R8 представляет собой С1-С4-алкил, где С1-С4-алкильная группа является незамещенной или моно-, ди- или три-фтор-замещенной;
и их диастереомеры, энантиомеры, метаболиты, соли, сольваты или сольваты солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Особое предпочтение также отдают соединениям общей формулы (I), в которой
R1 представляет собой С2-С5-алкильную группу, замещенную посредством гидроксила или C1-С3-алкокси или трифторметокси или 2,2,2-трифторэтокси или трифторметила или
представляет собой метил-SO2-замещенную С2-С4-алкильную группу или
представляет собой оксетан-3-ил-замещенную С1-С2-алкильную группу;
R2 и R3 всегда имеют одно и то же определение и оба представляют собой водород или метил;
R4 представляет собой метил, этил, трифтор-С1-С3-алкил, дифтор-C1-С3-алкил, гидроксиметил, 1-гидроксиэтил, 2-гидроксипропан-2-ил и 2,2,2-трифтор-1-гидроксиэтил и
R5 представляет собой водород, фтор или метил;
и их диастереомерам, энантиомерам, метаболитам, солям, сольватам или сольватам солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Наиболее предпочтительными являются соединения, в которых
R1 представляет собой 4,4,4-трифторбутил, 3-гидрокси-3-метилбутил, 3-гидроксибутил, 3-метоксипропил, 3-гидроксипропил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-трифторметоксипропил, 2-метоксиэтил, 2-гидроксиэтил, 2-(метилсульфонил)этил или 3-(метилсульфонил)пропил;
R2 и R3 оба представляют собой метил или водород и
R4 представляет собой дифторметил, трифторметил или метил и
R5 представляет собой водород или фтор;
и их диастереомеры, энантиомеры, метаболиты, соли, сольваты или сольваты солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Также особое предпочтение отдают соединениям, в которых
R1 представляет собой 3-гидрокси-3-метилбутил, 3-гидроксибутил, 3-гидрокси-2-метилпропил,
3-гидрокси-2,2-диметилпропил, 3-(метилсульфонил)пропил или 2-(метилсульфонил)этил;
R2 и R3 оба представляют собой метил;
R4 представляет собой дифторметил или трифторметил; и
R5 представляет собой водород;
и их диастереомерам, энантиомерам, метаболитам, солям, сольватам или сольватам солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Также особое предпочтение дополнительно отдают соединениям, в которых
R1 представляет собой 3-гидрокси-3-метилбутил, 3-гидроксибутил, 3-гидрокси-2-метилпропил,
3-гидрокси-2,2-диметилпропил, 3-(метилсульфонил)пропил или 2-(метилсульфонил)этил;
R2 и R3 оба представляют собой метил;
R4 представляет собой метил и
R5 представляет собой фтор, где R5 находится в орто-положении к R4;
и их диастереомерам, энантиомерам, метаболитам, солям, сольватам или сольватам солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Настоящее изобретение в особенности обеспечивает следующие соединения:
1) N-[6-(2-Гидроксипропан-2-ил)-2-(2-метоксиэтил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
2) N-[6-(Гидроксиметил)-2-(2-метоксиэтил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
3) N-[6-(2-Гидроксипропан-2-ил)-2-(3-метоксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
4) N-[6-(Гидроксиметил)-2-(3-метоксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
5) N-[2-(2-Гидроксиэтил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
6) N-[6-(2-Гидроксипропан-2-ил)-2-(3-гидроксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
7) N-[2-(2-Гидроксиэтил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
8) N-[6-(2-Гидроксипропан-2-ил)-2-(оксетан-3-илметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
9) N-[6-(Гидроксиметил)-2-(оксетан-3-илметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
10) N-{6-(2-Гидроксипропан-2-ил)-2-[3-(метилсульфонил)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид
11) N-[2-(3-Гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
12) N-{6-(2-Гидроксипропан-2-ил)-2-[2-(метилсульфонил)этил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид
13) 6-(Дифторметил)-N-[2-(3-гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]пиридин-2-карбоксамид
14) 6-(Дифторметил)-N-{6-(2-гидроксипропан-2-ил)-2-[2-(метилсульфонил)этил]-2Н-индазол-5-ил}пиридин-2-карбоксамид
15) 6-(Дифторметил)-N-[6-(2-гидроксипропан-2-ил)-2-(3-гидроксипропил)-2Н-индазол-5-ил]пиридин-2-карбоксамид
16) N-[6-(2-Гидроксипропан-2-ил)-2-(4,4,4-трифторбутил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
17) N-{6-(2-Гидроксипропан-2-ил)-2-[3-(трифторметокси)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид
18) N-{6-(2-Гидроксипропан-2-ил)-2-[3-(2,2,2-трифторэтокси)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид
19) 5-Фтор-N-[2-(3-гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-метилпиридин-2-карбоксамид
20) N-[2-(3-Гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-метилпиридин-2-карбоксамид
21) 6-(2-Гидроксипропан-2-ил)-N-[6-(2-гидроксипропан-2-ил)-2-(4,4,4-трифторбутил)-2Н-индазол-5-ил]пиридин-2-карбоксамид
22) N-{2-[2-(1-Гидроксициклопропил)этил]-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Кроме того, изобретение обеспечивает соединения общей формулы (III)
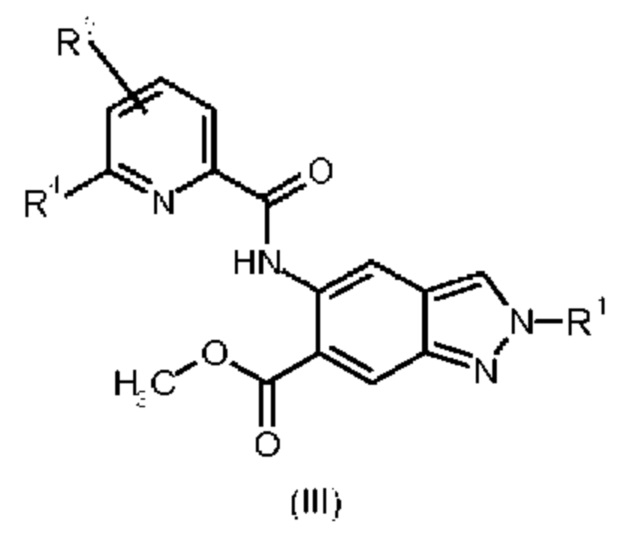
в которой
R1 представляет собой 4,4,4-трифторбутил, 3-гидрокси-3-метилбутил, 3-метоксипропил, 3-гидроксипропил, 3-гидроксибутил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-трифторметоксипропил, 2-метоксиэтил, 2-гидроксиэтил, 2-(метилсульфонил)этил, 3-(метилсульфонил)пропил или 2-(1-гидроксициклопропил)этил;
R4 представляет собой дифторметил, трифторметил или метил; и
R5 представляет собой водород или фтор;
и их диастереомеры, энантиомеры, метаболиты, соли, сольваты или сольваты солей,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
В особенности предпочтение отдают нижеследующим соединениям общей формулы (III):
метил 5-{[(5-фтор-6-метилпиридин-2-ил)карбонил]амино}-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилат и
метил 2-(3-гидрокси-3-метилбутил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат,
для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у животных.
Соединения общей формулы (III) пригодны для получения части соединений общей формулы (I).
К тому же, соединения общей формулы (III) представляют собой ингибиторы ассоциированной с рецептором интерлейкин-1 киназы-4 (IRAK4).
Соединения общей формулы (III) могут быть получены из соединений общей формулы (II)
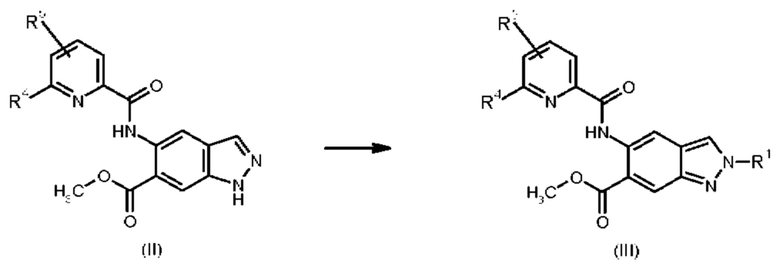
в которых
R1 представляет собой 4,4,4-трифторбутил, 3-гидрокси-3-метилбутил, 3-метоксипропил, 3-гидроксипропил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-трифторметоксипропил, 2-метоксиэтил, 2-гидроксиэтил, 2-(метилсульфонил)этил, 3-(метилсульфонил)пропил или 2-(1-гидроксициклопропил)этил;
R4 представляет собой дифторметил, трифторметил или метил; и
R5 представляет собой водород или фтор;
при помощи реакции (II) с соответственно замещенными алкилгалогенидами или алкил 4-метилбензолсульфонатами в присутствии карбоната калия.
Далее, соединения общей формулы (I) могут быть получены из соединений формулы (III)
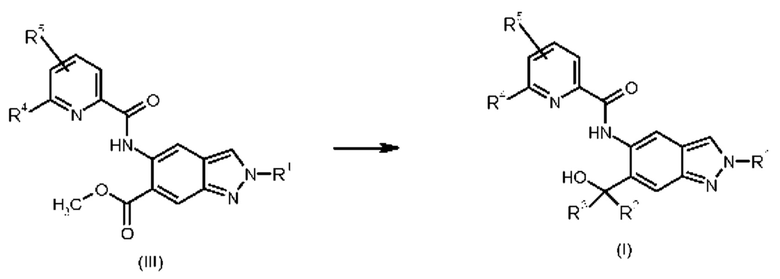
в которой
R1 представляет собой 4,4,4-трифторбутил, 3-гидрокси-3-метилбутил, 3-гидроксибутил, 3-метоксипропил, 3-гидроксипропил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-трифторметоксипропил, 2-метоксиэтил, 2-гидроксиэтил, 3-(метилсульфонил)пропил 2-(1-гидроксициклопропил)этил;
R2 и R3 представляют собой метил;
R4 представляет собой дифторметил, трифторметил или метил; и
R5 представляет собой водород или фтор;
путем реакции Гриньяра с бромидом метилмагния.
Предлагаемые в настоящем изобретении соединения действуют как ингибиторы киназы IRAK4 и обладают непредсказуемым спектром полезной фармакологической активности.
Кроме того, предпочтение отдают соединениям формулы (I), или соединениям в особенности упомянутым выше, для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у домашних животных, особенно у кошек и собак, и особенно у собак.
Понятие «домашние животные» в данном контексте включает, например, млекопитающих, таких как хомяки, морские свинки, крысы, мыши, шиншиллы, хорьки или, в частности, собаки, кошки; клеточных птиц; рептилий; земноводных или аквариумных рыбок.
Кроме того, предпочтение отдают соединениям формулы (I), или соединениям в особенности упомянутым выше, для применения в лечении и/или профилактике аллергического дерматита у домашних животных, в частности собачьего и кошачьего аллергического дерматита и, в частности, собачьего аллергического дерматита. Кроме того, предпочтение отдают соединениям формулы (I), или соединениям в особенности упомянутым выше, для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у сельскохозяйственных животных, особенно у овец, коз, лошадей, крупного рогатого скота и свиней, и в особенности у крупного рогатого скота и свиней.
Понятие «сельскохозяйственные животные» в данном контексте включает, например, млекопитающих, таких как лошади, овцы, козы, буйволы, северные олени, лани или, в частности, крупный рогатый скот или свиньи.
Кроме того, предпочтение отдают соединениям формулы (I), или в особенности упомянутым выше соединениям, для применения в способе лечения и/или профилактики атопического дерматита, аллергического блошиного дерматита, воспалительного заболевания кишечника, остеоартрита и воспалительной боли, неинфекционного рецидивирующего заболевания дыхательных путей, гиперчувствительности к насекомым, астмы, респираторных заболеваний, мастита и эндометрита у животных, в частности атопического дерматита и аллергического блошиного дерматита.
Особое предпочтение отдают соединениям формулы (I), или в особенности упомянутым выше соединениям, для применения в способе лечения и/или профилактики атопического дерматита у собак, аллергического блошиного дерматита у собак или кошек, воспалительного заболевания кишечника у собак или кошек, остеоартрита и воспалительных болей у собак, кошек, лошадей или крупного рогатого скота, неинфекционного рецидивирующего заболевания дыхательных путей у лошадей, гиперчувствительности к насекомым у лошадей, кошачьей астмы, респираторных заболеваний у крупного рогатого скота, мастита, эндометрита у крупного рогатого скота, и респираторных заболеваний у свиней.
Наиболее предпочтительными являются соединения формулы (I), или в особенности упомянутые выше соединения, для применения в способе лечения и/или профилактики атопического дерматита у собак и аллергического блошиного дерматита у собак или кошек, более конкретно у собак.
Кроме того, наиболее предпочтительными являются соединения формулы (I), или соединения в особенности упомянутые выше, для применения в способе лечения и/или профилактики остеоартрита и воспалительной боли у крупного рогатого скота, респираторных заболеваний у крупного рогатого скота, мастита у крупного рогатого скота, эндометрита у крупного рогатого скота, и респираторных заболеваний у свиней.
Что касается соединений формулы (III), кроме того, предпочтение отдают соединениям формулы (III) для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у домашних животных, особенно у кошек и собак, и более конкретно у собак.
Кроме того, предпочтение отдают соединениям формулы (III) для применения в лечении и/или профилактике аллергического дерматита у домашних животных, в частности аллергического дерматита у собак и кошек и, в особенности, аллергического дерматита у собак.
Кроме того, предпочтение отдают соединениям формулы (III) для применения в лечении и/или профилактике аллергических и/или воспалительных заболеваний у сельскохозяйственных животных, особенно у овец, коз, лошадей, крупного рогатого скота и свиней, и более конкретно у крупного рогатого скота и свиней.
Кроме того, предпочтение отдают соединениям формулы (III) для применения в способе лечения и/или профилактики атопического дерматита, аллергического блошиного дерматита, воспалительного заболевания кишечника, остеоартрита и воспалительной боли, неинфекционного рецидивирующего заболевания дыхательных путей, гиперчувствительности к насекомым, астмы, респираторных заболеваний, мастита и эндометрита у животных, в частности атопического дерматита и аллергического блошиного дерматита.
Особое предпочтение отдают соединениям формулы (III) для применения в способе лечения и/или профилактики атопического дерматита у собак, аллергического блошиного дерматита у собак или кошек, воспалительного заболевания кишечника у собак или кошек, остеоартрита и воспалительной боли у собак, кошек, лошадей или крупного рогатого скота, неинфекционного рецидивирующего заболевания дыхательных путей у лошадей, гиперчувствительности к насекомым у лошадей, астмы у кошек, респираторных заболеваний у крупного рогатого скота, мастита у крупного рогатого скота, эндометрита у крупного рогатого скота, и респираторных заболеваний у свиней.
Наиболее предпочтительными являются соединения формулы (III) для применения в способе лечения и/или профилактики атопического дерматита у собак и аллергического блошиного дерматита у собак или кошек, более конкретно у собак.
Кроме того, наиболее предпочтительными являются соединения формулы (III) для применения в способе лечения и/или профилактики остеоартрита и воспалительной боли у крупного рогатого скота, респираторных заболеваний у крупного рогатого скота, мастита у крупного рогатого скота, эндометрита у крупного рогатого скота, и респираторных заболеваний у свиней.
В качестве примера, соединения примеров 11, 12, 13, 19 (как показано ниже) были оценены в анализе TR-FRET IRAK4 in vitro, подробно описанном ниже с использованием рекомбинантного фермента IRAK4 собаки. Значения IC50 для каждого соединения были рассчитаны для ингибирования IRAK4 собаки. Примерные соединения (11, 12, 13, 19) были определены как пригодные в лечении аллергических кожных заболеваний у животных, в частности собак и кошек, таких как атопический дерматит и аллергический блошиный дерматит. Примерные соединения 11, 12, 13, 19, каждое, являлись сильнодействующими ингибиторами IRAK4 собаки со значениями IC50 в 1.7, 9.2, 2.2, 7.6 нм, соответственно. Значения IC50 для каждого из этих примерных соединений также были аналогичны значениям IC50, рассчитанным для ингибирования IRAK4 человека.
В качестве дополнительного примера Примерное соединение 12 также оценивали в анализе in vitro, чтобы установить воздействия соединений на вызванную липополисахаридами (ЛПС) выработку цитокинов мононуклеарными клетками периферической крови собак (МКПК). Примерное соединение 12 ингибировало выработку провоспалительного цитокина фактора некроза опухолей альфа (TNFα) посредством МКПК собак, вызванную при помощи ЛПС, в зависимости от концентрации. МКПК включают типы клеток, такие как дендритные клетки, Т- и В-лимфоциты, а также моноциты, каждый из которых вовлечен в атопический дерматит, а TNFα повышен у пациентов с атопический дерматитом (Sumimoto et al, Archives of Disease in Childhood, 1992). Этот пример также представлен на Фигуре 7.
Следовательно, соединения в соответствии с настоящим изобретением демонстрируют ингибирование рекомбинантного фермента собачьей IRAK4 и выработки цитокинов собачьими МКПК, что указывает на потенциальную терапевтическую пользу таких примерных соединений при атопическом дерматите у собак и аллергическом блошином дерматите.
Кроме того, примерное соединение 12 также был оценено in vivo в дальнейшем исследовании для установления воздействий соединений при лечении клинических признаков, связанных с аллергическим дерматитом у собак, в частности с атопическим дерматитом у собак (АДС), в модели домашнего пылевого клеща. Примерное соединение 12 значительно уменьшало клинические признаки АДС, такие как отек кожи и эритема. Этот пример также представлен на Фигурах 11 и 12.
Следовательно, соединения в соответствии с настоящим изобретением демонстрируют уменьшение характерных клинических признаков аллергического дерматита у собак, что указывает на терапевтическую пользу таких примерных соединений при аллергическом дерматите у собак, в частности атопическом дерматите у собак (АДС). К тому же, примерное соединение 12 было оценено в модели in vivo аллергического блошиного дерматита у собак (АБД) для установления противозудного действия соединений. Лечение при помощи Примерного соединения 12 существенно уменьшило зуд, связанный с аллергическими заболеваниями, такими как аллергический блошиный дерматита. Этот пример также представлен на Фигуре 13.
Следовательно, соединения в соответствии с настоящим изобретением демонстрируют уменьшение сопутствующих патогномоничных клинических признаков аллергического дерматита, таких как воспаление кожи и зуд, что указывает на терапевтическую пользу таких примерных соединений при аллергическом дерматите у собак, в частности при аллергическом блошином дерматите (АБД) и атопическом дерматите у собак (АДС).
Термин «аллергический дерматит у собак» в данном контексте включает, в частности, атопический дерматит у собак (АДС) и аллергический блошиный дерматит (АБД).
В качестве дополнительного примера, Примерное соединение 12 также оценивали в анализе ex vivo для установления воздействий соединений на вызванную липополисахаридами (ЛПС) выработку цитокинов мононуклеарными клетками периферической крови крупного рогатого скота (МКПК). Примерное соединение 12 ингибировало выработку провоспалительного цитокина фактора некроза опухолей альфа (TNFα) посредством МКПК крупного рогатого скота, вызванного при помощи ЛПС, в зависимости от концентрации. МКПК включают типы клеток, такие как дендритные клетки, Т- и В-лимфоциты, а также моноциты, каждый из которых вовлечен в воспалительные и инфекционные заболевания с подавлением провоспалительного иммунного ответа, такие как респираторные заболевания (Sterner-Kock, Haider, et al., Tropical Animal Health and Production, 2016), кишечные заболевания (Pan, Rostagnio, et al., Veterinary Immunology and Immunopathology, 2015), и мастит (Zheng, Xu, et al., Free Radical Biology and Medicine, 2016), при которых TNFα повышен у этих пациентов. Этот пример также представлен на Фигурах 8 и 9.
Следовательно, соединения в соответствии с настоящим изобретением демонстрируют ингибирование выработки цитокинов посредством МКПК крупного рогатого скота, что указывает на потенциальную терапевтическую пользу таких примерных соединений при воспалительных и/или инфекционных заболеваниях, таких как респираторные заболевания, кишечные заболевания и мастит.
В качестве дополнительного примера, Примерное соединение 12 также оценивали в анализе ex vivo для установления воздействий соединений на вызванную липополисахаридами (ЛПС) выработку цитокинов мононуклеарными клетками периферической крови свиньи (МКПК). Примерное соединение 12 ингибировало выработку провоспалительного цитокина фактора некроза опухоли альфа (TNFα) МКПК свиньи, вызванную при помощи ЛПС. МКПК включают типы клеток, такие как дендритные клетки, Т- и В-лимфоциты, а также моноциты, каждый из которых вовлечен в воспалительные и инфекционные заболевания с чрезмерным провоспалительным иммунным ответом, такие как респираторные заболевания и кишечные заболевания, при которых у этих пациентов повышен TNFα. Этот пример также представлен на Фигуре 10.
Поэтому, соединения в соответствии с настоящим изобретением демонстрируют ингибирование выработки цитокинов МКПК свиньи, что указывает на потенциальную терапевтическую пользу таких примерных соединений при воспалительных и/или инфекционных заболеваниях, таких как респираторные заболевания и кишечные заболевания.
Профилактика и/или лечение зуда и боли, в особенности острой, хронической, воспалительной и невропатической боли у животных, также обеспечивается соединениями в соответствии с настоящим изобретением.
Кроме того, соединения в соответствии с настоящим изобретением пригодны для лечения и/или профилактики болевых расстройств, в особенности острой, хронической, воспалительной и невропатической боли у животных. Предпочтительно это включает в себя гипералгезию, аллодинию, боль от артрита (такого как остеоартрит, ревматоидный артрит и спондилоартрит), предменструальную боль, боль, связанную с эндометриозом, послеоперационную боль, боль от интерстициального цистита, CRPS (комплексный региональный болевой синдром), невралгию тройничного нерва, боль от простатита, боль, вызванная повреждениями спинного мозга, боль, вызванная воспалением, боль в пояснице, боль при раке, боль, связанную с химиотерапией, невропатию, вызванную лечением ВИЧ, боль, вызванную ожогом и хроническую боль.
Кроме того, в настоящем изобретении предлагается способ лечения и/или профилактики заболеваний у животных, в особенности упомянутых выше заболеваний, с применением эффективного количества по меньшей мере одного из представленных соединений.
Предпочтение отдают способу лечения и/или профилактики аллергических и/или воспалительных заболеваний у животных путем введения эффективного количества по меньшей мере одного соединения представленной формулы (I), как определено выше, нуждающемуся в этом животному.
В контексте настоящего изобретения термин «лечение» или «проведение лечения» включает ингибирование, замедление, контроль, облегчение, ослабление, ограничение, уменьшение, подавление, преодоление или излечение заболевания, состояния, расстройства, повреждения или проблемы со здоровьем, или развития, течения или прогрессирования таких состояний и/или симптомов таких состояний. При этом термин «терапия» следует понимать как синоним термина «лечение».
Термины «предупреждение», «профилактика» и «предотвращение» в контексте настоящего изобретения использованы как синонимы и относятся к предотвращению или снижению риска заразиться, перенести, страдать от или иметь заболевание, состояние, расстройство, повреждение или проблему со здоровьем, или развитие или прогрессирование таких состояний и/или симптомов таких состояний.
Лечение или предупреждение заболевания, состояния, расстройства, повреждения или проблемы со здоровьем может быть частичным или полным.
Соединения в соответствии с настоящим изобретением можно применять отдельно или, если требуется, в сочетании с другими активными ингредиентами. Настоящее изобретение также относится к лекарственным средствам, содержащим, по меньшей мере, одно из соединений в соответствии с настоящим изобретением и один или несколько дополнительных активных ингредиентов, для лечения и/или предупреждения аллергических и/или воспалительных заболеваний у животных. Предпочтительные примеры активных ингредиентов, пригодных для комбинаций, включают:
В общем, можно упомянуть такие активные ингредиенты, как антибактериальные (например, пенициллины, ванкомицин, ципрофлоксацин), противовирусные (например, ацикловир, осельтамивир) и противогрибковые (например, нафтифин, нистатин) вещества, гамма-глобулины, иммуномодулирующие и иммуносупрессорные соединения, такие как циклоспорин, метотрексат®, антагонисты ФНО (например, Humira®, этанерцепт, инфликсимаб), ингибиторы IL-1 (например, анакинра, канакинумаб, рилонацепт), ингибиторы фосфодиэстеразы (например, апремиласт), ингибиторы Jak/STAT (например, тофацитиниб, барицитиниб, GLPG0634), лефлуномид, циклофосфамид, ритуксимаб, белимумаб, такролимус, рапамицин, микофенолат мофетила, интерфероны, кортикостероиды (например, преднизон, преднизолон, метилпреднизолон, гидрокортизон, бетаметазон), циклофосфамид, азатиоприн и сульфасалазин; парацетамол, нестероидные противовоспалительные вещества (НПВВ) (аспирин, ибупрофен, напроксен, этодолак, целекоксиб, колхицин).
В дополнение к приведенным выше, ингибиторы IRAK4 в соответствии с изобретением можно также комбинировать со следующими активными ингредиентами:
вещества для лечения заболеваний легких, например, бета-2-симпатомиметики (например, сальбутамол), антихолинергические средства (например, гликопирроний), метилксантины (например, теофиллин), антагонисты лейкотриеновых рецепторов (например, монтелукаст), ингибиторы PDE-4 (фосфодиэстераза типа 4) (например, рофлумиласт), метотрексат, антитела IgE, азатиоприн и циклофосфамид, препараты, содержащие кортизол; вещества для лечения остеоартрита, такие как нестероидные противовоспалительные вещества (НПВВ). В дополнение к двум указанным видам лечения следует упомянуть метотрексат и биологические препараты для В-клеточной и Т-клеточной терапии (например, ритуксимаб, абатацепт) при ревматоидных расстройствах, например, ревматоидного артрита, ревматоидном артрите, спондилоартрите и юношеском идиопатическом артрите. Нейротрофические вещества, такие как ингибиторы ацетилхолинэстеразы (например, донепезил), ингибиторы МАО (моноаминооксидазы) (например, селегилин), интерфероны и противосудорожные средства (например, габапентин); активные ингредиенты для лечения сердечно-сосудистых заболеваний, такие как бета-блокаторы (например, метопролол), ингибиторы АПФ (например, беназеприл), блокаторы рецепторов ангиотензина (например, лозартан, валсартан), диуретики (например, гидрохлоротиазид), блокаторы кальциевых каналов (например, нифедипин), статины (например, симвастатин, флувастатин); противодиабетические средства, например, метформин, глиниды (например, натеглинид), ингибиторы DPP-4 (дипептидилпептидаза-4) (например, линаглиптин, саксаглиптин, ситаглиптин, вилдаглиптин), ингибиторы SGLT2 (натрий-глюкозный котранспортер 2)/глифлозин (например, дапаглифлозин, эмпаглифлозин), миметики инкретина (аналоги/агонисты гормон глюкозо-зависимого инсулинотропного пептида (GIP) и глюкагоноподобного пептида 1 (GLP-1)) (например, эксенатид, лираглутид, ликсисенатид), ингибиторы α-глюкозидазы (например, акарбоза, миглитол, воглибиоза) и сульфонилмочевины (например, глибенкламид, толбутамид), сенсибилизаторы к инсулину (например, пиоглитазон) и терапия инсулином (например, инсулин NPH, инсулин лизпро). Активные ингредиенты, такие как мезалазин, сульфасалазин, азатиоприн, 6-меркаптопурин или метотрексат, пробиотические бактерии (Mutaflor, VSL#3®, Lactobacillus GG, Lactobacillus plantarum, L. acidophilus, L. casei, Bifidobacterium infantis 35624, Enterococcus fecium SF68, Bifidobacterium longum, Escherichia coli Nissle 1917), антибиотики, например, ципрофлоксацин и метронидазол, противодиарейные препараты, например, лоперамид или слабительные средства (бисакодил) для лечения хронических воспалительных заболеваний кишечника. Иммунодепрессанты, такие как глюкокортикоиды и нестероидные противовоспалительные вещества (НПВВ), кортизон, хлорохин, циклоспорин, азатиоприн, белимумаб, ритуксимаб, циклофосфамид для лечения красной волчанки. Аналоги витамина D3, например, кальципотриол, такальцитол или кальцитриол, салициловая кислота, мочевина, циклоспорин, метотрексат, эфализумаб при дерматологических нарушениях.
Следует также упомянуть лекарственные средства, содержащие по меньшей мере одно из соединений в соответствии с настоящим изобретением и один или несколько других активных ингредиентов для применения в соответствии с изобретением, в особенности ингибиторы ЕР4 (ингибиторы рецептора 4 простагландина Е2), ингибиторы Р2Х3 (Р2Х пуриноцептор 3), ингибиторы PTGES (ингибиторы простагландин Е синтазы) или ингибиторы AKR1C3 (ингибиторы альдокеторедуктазы 1-го семейства, член С3) для лечения и/или предупреждения вышеупомянутых заболеваний.
Соединения в соответствии с настоящим изобретением могут действовать системно и/или местно. Для этой цели их можно вводить пригодным способом, например, пероральным, парентеральным, внутрилегочным, назальным, сублингвальным, лингвальным, буккальным, ректальным, дермальным, трансдермальным, конъюнктивальным путем или в ухо, или в виде имплантата или стента.
Соединения в соответствии с настоящим изобретением можно вводить в лекарственных формах, пригодных для этих путей введения.
Пригодными лекарственными формами для перорального введения являются формы, которые функционируют в соответствии с известным уровнем техники и высвобождают соединения в соответствии с настоящим изобретением быстро и/или модифицированным образом и которые содержат соединения в соответствии с настоящим изобретением в кристаллической и/или аморфной и/или растворенной форме, например, таблетки (непокрытые или покрытые оболочкой таблетки, например, имеющие устойчивые к желудочному соку покрытия или покрытия, которые растворяются с задержкой, или нерастворимые покрытия, которые контролируют высвобождение соединения в соответствии с изобретением), таблетки или пленки/облатки, которые быстро распадаются в ротовой полости, пленки/лиофилизаты, капсулы (например, твердые или мягкие желатиновые капсулы), таблетки с сахарным покрытием, жевательные таблетки (например, мягкие жевательные таблетки), гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное введение можно осуществить, избегая стадии всасывания (например, внутривенным, внутриартериальным, внутрисуставным внутрисердечным, интраспинальным или интралюмбальным путем) или с включением стадии всасывания (например, внутримышечным, подкожным, внутрикожным, чрескожным или внутрибрюшинным путем). Пригодные лекарственные формы для парентерального введения включают составы для инъекций и инфузий в форме растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для других путей введения, пригодными примерами являются ингаляционные формы лекарственных средств (включающие применение порошковых ингаляторов, небулайзеров), капли в нос, растворы или спреи, таблетки, пленки/облатки или капсулы для лингвального, сублингвального или буккального введения, суппозитории, препараты для введения в уши или в глаза, водные суспензии (лосьоны, взбалтываемые смеси), липофильные суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластыри), молочко, пасты, пены, присыпки, имплантаты или стенты.
Предпочтение отдают пероральному или парентеральному введению, особенно пероральному введению.
Соединения в соответствии с настоящим изобретением могут быть преобразованы в упомянутые лекарственные формы. Это можно осуществить по сути известным способом смешивания с инертными, нетоксичными, фармацевтически пригодными наполнителями. Такие наполнители включают носители (например, микрокристаллическую целлюлозу, лактозу, маннит), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергирующие или смачивающие средства (например, додецилсульфат натрия, полиоксисорбитанолеат), связующие вещества (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты, например, аскорбиновую кислоту), красители (например, неорганические пигменты, например, оксиды железа) и добавки для коррекции вкуса и/или запаха.
Настоящее изобретение также обеспечивает лекарственные средства, которые содержат, по меньшей мере, одно соединение в соответствии с настоящим изобретением, как правило, вместе с одним или несколькими инертными, нетоксичными, фармацевтически приемлемыми наполнителями для применения в способе лечения и/или профилактики аллергических и/или воспалительных заболеваний у животных.
В общем, было обнаружено, что в случае парентерального введения для достижения эффективных результатов выгодно вводить соединения в количестве приблизительно от 0,001 до 1 мг/кг, приблизительно от 0,01 до 0,5 мг/кг массы тела. В случае перорального введения дозировка составляет приблизительно от 0,01 до 100 мг/кг, предпочтительно приблизительно от 0,01 до 20 мг/кг и наиболее предпочтительно от 0,1 до 10 мг/кг массы тела.
Тем не менее, в некоторых случаях может оказаться необходимым отклониться от указанных количеств, а именно в зависимости от массы тела, пути введения, индивидуального ответа на активный ингредиент, природы препарата и времени введения или интервала, в течение которого его осуществляют. Таким образом, в некоторых случаях может быть достаточно количеств, меньших упомянутого выше минимального количества, в то время как в других случаях упомянутый верхний предел должен быть превышен. В случае введения более значительных количеств, может оказаться целесообразным разделить их на несколько отдельных доз, вводимых в течение суток.
Демонстрационные примеры, которые приведены ниже, иллюстрируют изобретение. Изобретение не ограничено примерами.
Если не указано иное, процентные значения, упомянутые в тестах и примерах, которые приведены ниже, являются процентными значениями по массе; части являются частями по массе. Соотношения растворителей, степени разбавлений и данные концентраций для растворов жидкость/жидкость в каждом случае пересчитаны на объем.
Получение соединений
Получение соединений в соответствии с настоящим изобретением показано при помощи схем синтеза, которые следуют ниже.
Исходные вещества, используемые для синтеза соединений в соответствии с настоящим изобретением представляют собой карбоновые кислоты (Промежуточное соединение V3), которые являются коммерчески доступными или их можно получить известными из литературных источников способами или аналогично известным из литературных источников способам (см., например, European Journal of Organic Chemistry 2003, 8, 1559-1568, Chemical and Pharmaceutical Bulletin, 1990, 38, 9, 2446 2458, Synthetic Communications 2012, 42, 658-666, Tetrahedron, 2004, 60, 51, 11869-11874) (см., например, Схему синтеза 1). Некоторые карбоновые кислоты V3 могут быть получены из сложных эфиров карбоновых кислот (Промежуточное соединение V2) путем гидролиза (см., например, реакцию этил 6-(гидроксиметил)пиридин-2-карбоксилата с водным раствором гидроксида натрия в метаноле, WO 200411328) или - в случае сложного трет-бутилового эфира - путем реакции с кислотой, например, хлористоводородной или трифторуксусной кислотой (см., например, Dalton Transactions, 2014, 43, 19, 7176-7190). Карбоновые кислоты V3 также можно использовать в виде их солей щелочных металлов. Промежуточные соединения V2 также необязательно можно получить из промежуточных соединений V1, которые несут атом хлора, брома или йода в качестве заместителя X1 по реакции в атмосфере монооксида углерода, необязательно при повышенном давлении, в присутствии фосфинового лиганда, например, 1,3-бис(дифенилфосфино)пропана, соединения палладия, например, ацетата палладия(II), и основания, например триэтиламина, с добавлением этанола или метанола в растворителе, например диметилсульфоксиде (для способов получения см., например, WO 2012112743, WO 2005082866, Chemical Communications (Cambridge, Англия), 2003, 15, 1948-1949, WO 200661715). Промежуточные соединения V1 являются или коммерчески доступными, или их можно получить известными из литературных источников способами. Иллюстративные способы получения подробно описаны в WO 2012061926, European Journal of Organic Chemistry, 2002, 2, 327-330, Synthesis, 2004, 10, 1619-1624, Journal of the American Chemical Society, 2013, 135, 32, 12122-12134, Bioorganic and Medicinal Chemistry Letters, 2014, 24, 16, 4039-4043, US 2007185058, WO 2009117421.
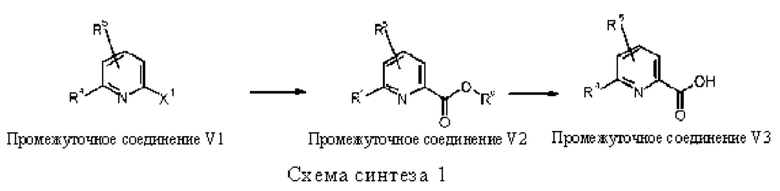
X1 представляет собой хлор, бром или йод.
Rd представляет собой метил, этил, бензил или трет-бутил.
R4, R5 имеет значения, определенные в общей формуле (I).
Метил 5-амино-1Н-индазол-6-карбоксилат (Промежуточное соединение 2) может быть получен, исходя из метил 1Н-индазол-6-карбоксилата (Промежуточное соединение 0) согласно Схеме синтеза 2 в соответствии со схемой синтеза 2 путем нитрирования и восстановления нитрогруппы промежуточного соединения 1 водородом в присутствии палладия на угле, аналогично WO 2008/001883. Для получения промежуточных соединений 3, исходя из промежуточного соединения 2, можно использовать различные реагенты сочетания, известные из литературных источников (Amino Acids, Peptides and Proteins in Organic Chemistry, том 3 - Building Blocks, Catalysis and Coupling Chemistry, Andrew B. Hughes, Wiley, глава 12 - Peptide-Coupling Reagents, 407-442; Chem. Soc. Rev., 2009, 38, 606). Например, можно использовать гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида в комбинации с гидратом 1-гидрокси-1Н-бензотриазола (HOBt, WO 2012107475, Bioorg. Med. Chem. Lett., 2008, 18, 2093), тетрафторборатом (1Н-бензотриазол-1-илокси)(диметиламино)-N,N-диметилметанаминия (TBTU, CAS 125700-67-6), гексафторфосфатом (диметиламино)-N,N-диметил(3Н-[1,2,3]триазол[4,5-b]пиридин-3-илокси) метанаминия (HATU, CAS 148893-10-1), пропанфосфоновым ангидридом (в виде раствора в этилацетате или ДМФА, CAS68957-94-8) или ди-1Н-имидазол-1-илметаноном (CDI) в качестве реагентов сочетания, с добавлением к реакционной смеси в каждом случае основания, такого как триэтиламин или N-этил-N-изопропилпропан-2-амин. Предпочтение отдают применению TBTU и N-этил-N-изопропилпропан-2-амина в ТГФ.

Каждый из заместителей R4, R5 является таким, как определено для общей формулы (I).
Исходя из промежуточных соединений 3, можно получить производные 2-замещенного индазола (Промежуточное соединение 4) (см. Схему синтеза 3). Применимые реакции для этой цели включают в себя реакции с необязательно замещенными алкилхлоридами, алкилбромидами, алкилйодидами или алкил-4-метилбензолсульфонатами. Используемые алкилгалогениды или алкил-4-метилбензолсульфонаты коммерчески доступны или могут быть получены аналогично известным из литературных источников способам (для получения алкил-4-метилбензолсульфонатов одним примером является реакция соответствующего спирта с 4-метилбензолсульфонилхлоридом в присутствии триэтиламина или пиридина; см., например, Bioorganic and Medicinal Chemistry, 2006, 14, 12, 4277-4294). Необязательно, в случае применения алкилхлоридов или алкилбромидов, также возможно добавлять йодид щелочного металла, такой как йодид калия или йодид натрия. Применимые основания могут представлять собой, например, карбонат калия, карбонат цезия или гидрид натрия. В случае реакционноспособных алкилгалогенидов также возможно в некоторых случаях использовать N-циклогексил-N-метилциклогексанамин. Применимые растворители включают, например, 1-метилпирролидин-2-он, ДМФА, ДМСО или ТГФ. Необязательно, используемые алкилгалогениды или алкил-4-метилбензолсульфонаты могут иметь функциональные группы, которые необязательно заранее защищены защитной группой (см. также P.G.М. Wilts, Т.W. Greene, Greene's Protective Groups in Organic Synthesis, Fourth Edition, ISBN: 9780471697541). При применении, например, алкилгалогенидов или алкил-4-метилбензолсульфонатов, имеющих одну или несколько гидроксильных групп, эти гидроксильные группы могут быть необязательно защищены трет-бутил(диметил)силильной группой или подобной кремнийсодержащей защитной группой, известной специалистам в данной области. Альтернативно, гидроксильные группы также могут быть защищены тетрагидро-2Н-пирановой группой (ТНР) или ацетильной или бензоильной группой. Используемые защитные группы затем можно отщепить до синтеза промежуточного соединения 4, или так же после синтеза (I). Если, например трет-бутил(диметилсилильная) группа используется в качестве защитной группы, то ее можно отщепить, с использованием тетрабутилфторида аммония в растворителе, таком как, например, ТГФ. ТНР защитную группу можно отщепить, например, с использованием 4-метилбензолсульфоновой кислоты (необязательно в форме моногидрата). Ацетильные группы или бензоильные группы можно отщепить обработкой водным раствором гидроксида натрия.
Используемые алкилгалогениды или алкил-4-метилбензолсульфонаты могут необязательно содержать функциональные группы, которые можно подвергнуть реакциям окисления или восстановления, известным специалистам в данной области (см., например, Science of Synthesis, Georg Thieme Verlag). Если, например, функциональная группа представляет собой сульфидную группу, то ее можно окислить способами, известными в литературных источниках, до сульфоксидной или сульфоновой группы. В случае сульфоксидной группы ее также можно окислить до сульфоновой группы. Для этих стадий окисления можно использовать, например, 3-хлорпербензойную кислоту (CAS 937-14-4) (в этом отношении см. также, например, US 201094000 для окисления производного 2-(метилсульфанил)этил-1Н-пиразола до производного 2-(метилсульфинил)этил-1H-пиразола и окисления дополнительного производного 2-(метилсульфанил)этил-1Н-пиразола до производного 2-(метилсульфонил)этил-1H-пиразола). Если используемые алкилгалогениды или тозилаты содержат кетогруппу, то ее можно восстановить с помощью способов восстановления, известных специалистам в данной области, до спиртовой группы (см., например, Chemische Berichte, 1980, 113, 1907-1920 для использования боргидрида натрия). Эти стадии окисления или восстановления можно впоследствии осуществить до синтеза промежуточного соединения 4, или же после синтеза соединения в соответствии с настоящим изобретением общей формулы (I). Альтернативно, промежуточное соединение 4 можно получить по реакции Мицунобу (см., например, K.С.K. Swamy и др. Chem. Rev. 2009, 109, 2551-2651) из промежуточного соединения 3 с необязательно замещенными алкиловыми спиртами. Можно применять различные фосфины, такие как трифенилфосфин, трибутилфосфин или 1,2-дифенилфосфиноэтан в комбинации с диизопропилазодикарбоксилатом (CAS 2446-83-5) или другими производными диазола, упомянутыми в литературных источниках (K.С.K. Swamy и др. Chem. Rev. 2009, 109, 2551-2651). Предпочтение отдают применению трифенилфосфина и диизопропилазодикарбоксилата. Если алкиловый спирт несет функциональную группу, то возможно - как в случае вышеупомянутых реакций с алкилгалогенидами - осуществить известные приемы с защитными группами (дальнейшие указания можно найти в P.G.М. Wuts, Т.W. Greene, Greene's Protective Groups in Organic Synthesis, четвертое издание, ISBN: 9780471697541) и - как в случае вышеупомянутых реакций с алкилгалогенидами - осуществить стадии окисления или восстановления соответственно до синтеза промежуточного соединения 4, или же после синтеза соединений в соответствии с настоящим изобретением общей формулы (I). Исходя из промежуточного соединения 4, соединения в соответствии с настоящим изобретением общей формулы (I) где R2 и R3 определены как C1-C6-алкил (где R2 и R3 имеют одинаковое определение) можно получить с помощью реакции Гриньяра (см., например, реакцию производного метил-1H-индазол-6-карбоксилата с метилмагнийбромидом в ЕР 2489663). Для реакции Гриньяра можно использовать галогениды алкилмагния. Особое предпочтение отдают метилмагнийхлориду или метилмагнийбромиду в ТГФ или диэтиловом эфире, или так же в смесях ТГФ и диэтилового эфира. Альтернативно, исходя из промежуточного соединения 4, соединения в соответствии с изобретением общей формулы (I), где R2 и R3 определены как C1-C6-алкил (где R2 и R3 имеют одинаковое определение), можно получить с помощью реакции с алкиллитиевым реагентом (см., например, реакцию производного метил-2-амино-4-хлор-1-метил-1H-бензимидазол-7-карбоксилата с изопропиллитием или трет-бутиллитием в WO 2006116412). Исходя из промежуточного соединения 4, можно получить соединения в соответствии с настоящим изобретением общей формулы (I), где R2 и R3 определены как Н, путем восстановления алюмогидридом лития в ТГФ, боргидридом лития в ТГФ или боргидридом натрия в ТГФ, необязательно с добавлением метанола, или смесями боргидрида лития и боргидрида натрия.
Каждый из заместителей R1, R2, R3, R4, R5 имеет значения, определенные в общей формуле (I).
Исходя из промежуточного соединения 3, промежуточное соединение 5, где R2 и R3 определены как C1-C6-алкил (где R2 и R3 имеют одинаковое определение) можно получить с помощью реакции Гриньяра (см., например, Схему синтеза 4). Для этой цели можно применять пригодные галогениды алкилмагния, например метилмагнийхлорид или метилмагнийбромид в ТГФ или в диэтиловом эфире или же в смесях ТГФ и диэтилового эфира.
Исходя из промежуточного соединения 5, затем можно получить часть (I-а) соединений в соответствии с настоящим изобретением формулы (I), где R2 и R3 определены как C1-C6-алкил (где R2 и R3 имеют одинаковое определение). Для этой цели, аналогично Схеме синтеза 3 (получение промежуточного соединения 3), применимыми являются реакции промежуточного соединения 5 с необязательно замещенными алкилхлоридами, алкилбромидами, алкилйодидами или алкил-4-метилбензолсульфонатами. Можно использовать приемы с защитными группами, аналогично описанными в Схеме синтеза 3.
Альтернативно, для получения части (I-а) соединений в соответствии с настоящим изобретением формулы (I), где R2 и R3 определены как C1-С6-алкил (где R2 и R3 имеют одинаковое определение), можно использовать реакцию Мицунобу промежуточного соединения 5 с необязательно замещенными алкиловыми спиртами (аналогично Схеме синтеза 3).
Если R1 в соединениях формулы (I-а) включает пригодную функциональную группу, затем необязательно возможно, по аналогии со Схемой синтеза 3, использовать реакции окисления или реакции восстановления для получения дополнительных соединений в соответствии с настоящим изобретением.

Каждый из заместителей R1, R4, R5 имеет значения, определенные в общей формуле (I). R2 и R3 всегда имеют одно и то же определение и оба представляют собой С1-С6-алкил.
Исходя из промежуточного соединения 1, можно получить промежуточное соединение 4 альтернативным способом (см. Схему синтеза 5). Прежде всего, промежуточное соединение 1 превращают в промежуточное соединение 6 способами, как в Схеме синтеза 3 (получение промежуточного соединения 4 из промежуточного соединения 3).
Промежуточное соединение 6 затем можно превратить в промежуточное соединение 7 путем восстановления нитрогруппы. Например, нитрогруппу можно восстановить палладием на угле в атмосфере водорода (см, например, WO 2013174744 для восстановления 6-изопропокси-5-нитро-1Н-индазола в 6-изопропокси-1H-индазол-5-амине) или используя железо и хлорид аммония в воде и этаноле (см., например, также Journal of the Chemical Society, 1955, 2412-2419), или используя хлорид олова(II) (CAS 7772-99-8). Предпочтительным является применение железа и хлорида аммония в воде и этаноле. Получение промежуточного соединения 4 из промежуточного соединения 7 можно осуществить аналогично Схеме синтеза 2 (получение промежуточного соединения 3 из промежуточного соединения 2).
Как описано для Схемы синтеза 3, необязательно можно использовать приемы с защитными группами и в случае Схемы синтеза 5. Необязательно, дополнительно возможно, исходя из промежуточного соединения 6 или промежуточного соединения 7, как описано для схемы синтеза 3, провести реакции окисления или восстановления, известные специалистам в данной области (см., например, Science of Synthesis, Georg Thieme Verlag).
Каждый из заместителей R1, R4, R5 имеет значения, определенные в общей формуле (I).
Синтез примерных соединений
Сокращения и пояснения
Термин раствор хлорида натрия всегда означает насыщенный водный раствор хлорида натрия.
Химические названия промежуточных соединений и примеров генерировали с использованием программного обеспечения ACD/LABS (Batch Version 12.01.).
Методы
В некоторых случаях данные соединения и предшественники и/или их промежуточные продукты были проанализированы при помощи ЖХ-МС.
Метод А1: СЭЖХ (MeCN-HCOOH):
Прибор: Waters Acquity СЭЖХ-МС SQD 3001; колонка: Acquity СЭЖХ ВЕН С18 1.7 50×2.1 мм; элюент А: вода + 0.1 объемн. % муравьиной кислоты (99%), элюент В: ацетонитрил; градиент: 0-1.6 мин 1-99% В, 1.6-2.0 мин 99% В; скорость потока 0.8 мл/мин; температура: 60°С; объем вводимой пробы: 2 мкл; DAD скан: 210-400 нм; PC ESI+, ESI-, диапазон сканирования 160-1000 m/z; ELSD.
Метод А2: СЭЖХ (MeCN-NH3):
Прибор: Waters Acquity СЭЖХ-МС SQD 3001; колонка: Acquity СЭЖХ ВЕН С18 1.7 50×2.1 мм; элюент А: вода + 0.2 объемн. % аммиака (32%), элюент В: ацетонитрил; градиент: 0-1.6 мин 1-99% В, 1.6-2.0 мин 99% В; скорость потока 0.8 мл/мин; температура: 60°С; объем вводимой пробы: 2 мкл; DAD скан: 210-400 нм; PC ESI+, ESI-, диапазон сканирования 160-1000 m/z; ELSD.
Метод A3: (ЖХ-МС)
Прибор: Agilent 1290 Infinity ЖХ; колонка: Acquity СЭЖХ ВЕН С18 1.7 50×2.1 мм; элюент А: вода + 0.05 объемн. % муравьиной кислоты, элюент В: ацетонитрил + 0.05 объемн. % муравьиной кислоты; градиент: 0-1.7 мин 2-90% В, 1.7-2.0 мин 90% В; скорость потока 1.2 мл/мин; температура: 60°С; объем вводимой пробы: 2 мкл; DAD скан: 190-390 нм; МС: Agilent TOF 6230.
Метод А4: (ЖХ-МС)
Прибор: Waters Acquity; колонка: Kinetex (Phenomenex), 50×2 мм; элюент А: вода + 0.05 объемн. % муравьиной кислоты, элюент В: ацетонитрил + 0.05 объемн. % муравьиной кислоты; градиент: 0-1.9 мин 1-99% В, 1.9-2.1 мин 99% В; скорость потока 1.5 мл/мин; температура: 60°С; объем вводимой пробы: 0.5 мкл; DAD скан: 200-400 нм.
В некоторых случаях данные соединения и предшественники и/или их промежуточные продукты очищали следующими иллюстративными препаративными методами ВЭЖХ:
Метод Р1: система: Waters Autopurification system: Pump 2545, Sample Manager 2767, CFO, DAD 2996, ELSD 2424, SQD; колонка: XBridge C18 5 мкм 100×30 мм; элюент А: вода + 0.1 объемн. % муравьиной кислоты, элюент В: ацетонитрил; градиент: 0-8 мин 10-100% В, 8-10 мин 100% В; поток: 50 мл/мин; температура: комнатная температура; раствор: макс. 250 мг/макс. 2.5 мл ДМСО или DMF; объем вводимой пробы: 1×2.5 мл; детектирование: DAD диапазон сканирования 210-400 нм; PC ESI+, ESI-, диапазон сканирования 160-1000 m/z.
Метод Р2: система: Waters Autopurification system: Pump 254, Sample Manager 2767, CFO, DAD 2996, ELSD 2424, SQD 3100; колонка: XBridge C18 5 мкм 10×30 мм; элюент А: вода + 0.2 объемн. % аммиака (32%), элюент В: метанол; градиент: 0-8 мин 30-70% В; поток: 50 мл/мин; температура: комнатная температура; детектирование: DAD диапазон сканирования 210-400 нм; PC ESI+, ESI-, диапазон сканирования 160-1000 m/z; ELSD.
Метод Р3: система: Labomatic, насос: HD-5000, отборник фракций: LABOCOL Vario-4000, УФ детектор: Knauer UVD 2.1S; колонка: XBridge С18 5 мкм 100×30 мм; элюент А: вода + 0,2 объемн. % аммиака (25%), элюент В: ацетонитрил; градиент: 0-1 мин 15% В, 1-6.3 мин 15-55% В, 6.3-6.4 мин 55-100% В, 6.4-7.4 мин 100% В; поток: 60 мл/мин; температура: комнатная температура; раствор: макс. 250 мг/2 мл ДМСО; объем вводимой пробы: 2×2 мл; детектирование: УФ 218 нм; программное обеспечение: SCPA PrepCon5.
Метод Р4: система: Labomatic, насос: HD-5000, отборник фракций: LABOCOL Vario-4000, УФ детектор: Knauer UVD 2.1S; колонка: Chromatorex RP C18 10 мкм 125×30 мм; элюент А: вода + 0,1 объемн. % муравьиной кислоты, элюент В: ацетонитрил; градиент: 0-15 мин. 65-100% В; поток: 60 мл/мин; температура: комнатная температура; раствор: макс. 250 мг/2 мл ДМСО; объем вводимой пробы: 2×2 мл; детектирование: УФ 254 нм; программное обеспечение: SCPA PrepCon5.
Метод Р5: система: Sepiatec: Преп. SFC100, колонка: Chiralpak IA 5 мкм 250×20 мм; элюент А: двуокись углерода, элюент В: этанол; градиент: изократический 20% В; поток: 80 мл/мин; температура: 40°С; раствор: макс. 250 мг/2 мл ДМСО; объем вводимой пробы: 5×0.4 мл; детектирование: УФ 254 нм.
Метод Р6: система: Agilent: Преп. 1200, 2 х преп. насос, DLA, MWD, Gilson: Liquid Handler 215; колонка: Chiralcel OJ-H 5 мкм 250×20 мм; элюент А: гексан, элюент В: этанол; градиент: изократический 30% В; поток: 25 мл/мин; температура: 25°С; раствор: 187 мг/8 мл этанол/метанол; объем вводимой пробы: 8×1.0 мл; детектирование: УФ 280 нм.
Метод Р7: система: Labomatic, насос: HD-5000, отборник фракций: LABOCOL Vario-4000, УФ детектор: Knauer UVD 2.1S; колонка: XBridge С18 5 мкм 100×30 мм; элюент А: вода + 0.1 объемн. % муравьиной кислоты, элюент В: ацетонитрил; градиент: 0-3 мин: 65% В изократический, 3-13 мин: 65-100% В; поток: 60 мл/мин; температура: комнатная температура; раствор: макс. 250 мг/2 мл ДМСО; объем вводимой пробы: 2×2 мл; детектирование: УФ 254 нм.
Метод Р8: система: Agilent: Преп. 1200, 2 х преп. насос, DLA, MWD, Gilson: Liquid Handler 215; колонка: Chiralpak IF 5 мкм 250×20 мм; элюент А: этанол, элюент В: метанол; градиент: изократический 50% В; поток: 25 мл/мин; температура: 25°С; раствор: 600 мг/7 мл N,N-диметилформамид; объем вводимой пробы: 10×0.7 мл; детектирование: УФ 254 нм.
В некоторых случаях смеси веществ очищали колоночной хроматографией на силикагеле.
Для получения некоторых соединений в соответствии с настоящим изобретением и их предшественников и/или промежуточных соединений очистку с помощью колоночной хроматографии («флеш-хроматографии») проводили на силикагеле с использованием устройств Isolera® от Biotage. Это осуществляли с использованием картриджей от Biotage, например, картриджа "SNAP Cartridge, KP_SIL" разного размера и картриджей "Interchim Puriflash Silica HP 15UM flash column" от Interchim разного размера.
Исходные вещества
Промежуточное соединение V2-1
Метил 6-(2-гидроксипропан-2-ил)пиридин-2-карбоксилат

2.00 г (9.26 ммоль) 2-(6-бромпиридин-2-ил)пропан-2-ола (CAS 638218-78-7) растворяли в 20 мл метанола и 20 мл ДМСО. Затем добавляли 250 мг 1,3-бис(фенилфосфино)пропана, 130 мг ацетата палладия(II) и 3 мл триэтиламина. Реакционную смесь три раза продували окисью углерода при комнатной температуре и перемешивали в атмосфере монооксида углерода 13 бар в течение 30 мин. Атмосферу окиси углерода удаляли путем применения вакуума, и смесь перемешивали под 14 бар атмосферы окиси углерода при 100°С в течение 24 ч. Автоклав декомпрессировали, к реакционной смеси добавляли воду, и реакционную смесь экстрагировали три раза этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия и раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Это обеспечило 1,60 г сырого продукта.
СЭЖХ-МС (Метод A1): By=0.76 мин (УФ детектор: TIC), обнаруженная масса 195.00.
Промежуточное соединение V3-1
6-(2-Гидроксипропан-2-ил)пиридин-2-карбоксилат калия
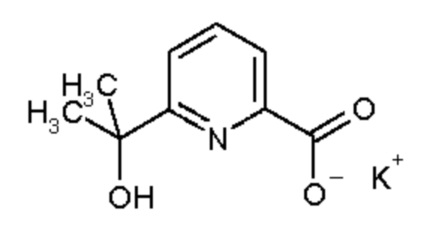
1.60 г сырого продукта промежуточного соединения 0-1 сначала загружали в 15 мл метанола, добавляли 0,74 г гидроксида калия, и смесь перемешивали при 50°С в течение 16,5 ч. После концентрирования это обеспечило 2.1 г остатка, который использовали без дополнительной очистки.
СЭЖХ-МС (Метод A1): By=0.47 мин (УФ детектор: TIC), обнаруженная масса 181.00.
Промежуточное соединение 1-1
Метил 5-нитро-1Н-индазол-6-карбоксилат
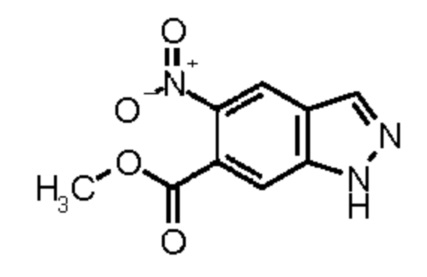
4.60 г (26.1 ммоль) метил 1Н-индазол-6-карбоксилата (CAS №: 170487-40-8) растворяли в 120 мл серной кислоты (96%) и охлаждали до -15°С в трехгорлой колбе, содержащей CPG-мешалку, капельную воронку и внутренний термометр. В течение 15 мин нитрирующую кислоту (10 мл 96% серной кислоты в 5 мл 65% азотной кислоты), которую предварительно получали и охлаждали, добавляли по каплям к этому раствору. По окончании добавления по каплям смесь перемешивали в течение еще 1 часа (внутренняя температура -13°С). Реакционную смесь добавляли ко льду и осадок отфильтровывали путем отсасывания, промывали водой и сушили в сушильном шкафу при температуре 50°С под сниженным давлением. Получали 5,49 г указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=0.75 мин
МС (ЭРИпоз): m/z=222 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=3.87 (s, 3Н), 7.96 (s, 1Н), 8.44 (s, 1H), 8.70 (s, 1H), 13.98 (br. s., 1H).
Промежуточное соединение 2-1
Метил 5-амино-1Н-индазол-6-карбоксилат
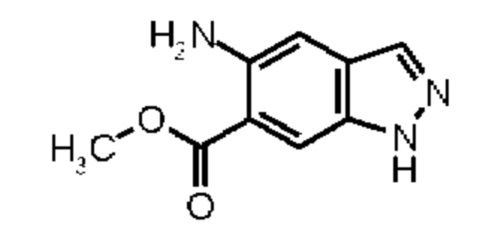
4,40 г (19.8 ммоль) метил 5-нитро-1Н-индазол-6-карбоксилата (Промежуточное соединение 1-1) растворяли в 236 мл метанола и гидрировали с 1,06 г (0,99 ммоль) палладия на активированном угле под стандартным давлением водорода при 25°С в течение 3 ч. Реакционную смесь фильтровали через целит, фильтр промывали метанолом, и фильтрат концентрировали. Получали 3,53 г указанного в заголовке соединения.
1H ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=3.85 (s, 3H) 6.01 (s, 2Н) 6.98 (s, 1H) 7.79-7.91 (m, 1H) 7.99 (s, 1H) 12.84 (br. s., 1H).
Промежуточное соединение 3-1
Метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилат
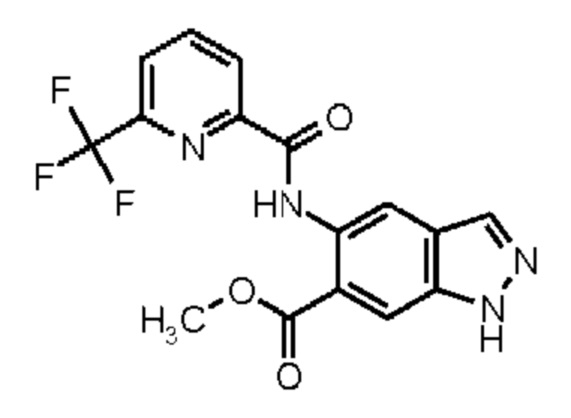
4,95 г (25.9 ммоль) 6-(трифторметил)пиридин-2-карбоновой кислоты сначала загружали в 45 мл ТГФ. Добавляли 9.07 г (28.2 ммоль) тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и 4.92 мл (28.2 ммоль) N-этил-N-изопропилпропан-2-амин и смесь перемешивали при 25°С в течение 30 мин. Затем добавляли 4.50 г (23.5 ммоль) метил 5-амино-1Н-индазол-6-карбоксилата (Промежуточное соединение 2-1) и смесь перемешивали при 25°С в течение 24 ч. Реакционную смесь фильтровали путем отсасывания через мембранный фильтр, твердые вещества промывали посредством ТГФ и водой, и сушили в сушильном шкафу в течение ночи. Получали 7,60 г указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=1.16 мин
МС (ЭРИпоз): m/z=365 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=3.97 (s, 3H), 8.13-8.27 (m, 2Н), 8.30 (s, 1H), 8.33-8.45 (m, 1H), 8.45-8.51 (m, 1H), 9.15 (s, 1H), 12.57 (s, 1H), 13.44 (s, 1H).
Промежуточное соединение 3-2
Метил 5-({[6-(дифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилат

2,85 г (23.5 ммоль) 6-(дифторметил)пиридин-2-карбоновой кислоты сначала загружали в 30 мл ТГФ. Добавляли 6.05 г (18.8 ммоль) тетрафторбората О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и 3,3 мл N-этил-N-изопропилпропан-2-амина и смесь перемешивали при комнатной температуре в течение 10 минут. Затем добавляли 3.00 г (15.7 ммоль) метил 5-амино-1H-индазол-6-карбоксилата и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси подмешивали воду и осадок отфильтровывали путем отсасывания и многократно промывали водой и дихлорметаном. Это обеспечило 1.53 г (27% от теории) указанного в заголовке соединения. Фазы фильтрата разделяли, органическую фазу концентрировали, подмешивали небольшое количество дихлорметана и суспендировали в ультразвуковой бане, и осадок отфильтровывали путем отсасывания. Это обеспечило еще 1.03 г указанного в заголовке соединения.
1Н-ЯМР (фракция первого продукта, 300 МГц, ДМСО-d6): δ [част. на млн.]=3.99 (s, 3Н), 7.09 (t, 1Н), 8.00 (d, 1H), 8.21-8.40 (m, 4Н), 9.14 (s, 1H), 12.53 (s, 1Н), 13.44 (s, 1H).
Промежуточное соединение 3-3
Метил 5-({[6-(2-гидроксипропан-2-ил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилат
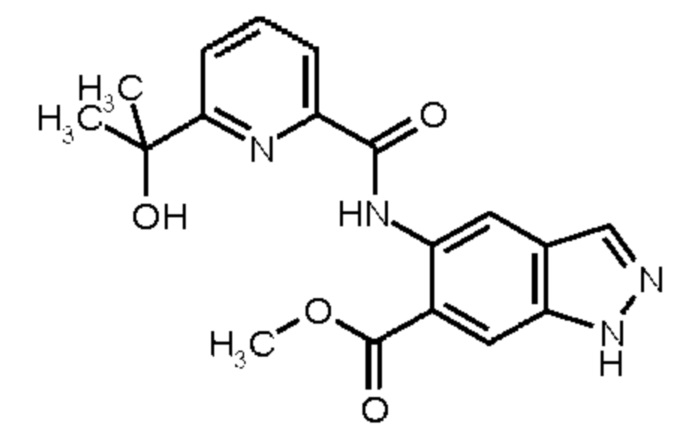
2,10 г 6-(2-гидроксипропан-2-ил)пиридин-2-карбоксилата калия (Промежуточное соединение V3-1) сначала загружали в 15 мл ТГФ. Добавляли 3.69 г (11.5 ммоль) тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и 2.00 мл N-этил-N-изопропилпропан-2-амина, и смесь перемешивали при комнатной температуре в течение 15 мин. Затем добавляли 1.83 г (9.58 ммоль) метил 5-амино-1Н-индазол-6-карбоксилата (Промежуточное соединение 2-1), и смесь перемешивали при комнатной температуре в течение 19 ч. К смеси подмешивали воду и этилацетат, нерастворенные твердые вещества отфильтровывали, фазы фильтрата разделяли, и водную фазу экстрагировали дважды этилацетатом, промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр, концентрировали и очищали колоночной хроматографией на силикагеле (гексан/этилацетат). После удаления растворителей получали 1.56 г указанного в заголовке соединения в виде желтой пены.
СЭЖХ-МС (Метод A1): By=1.00 мин (УФ детектор: TIC Smooth), обнаруженная масса 354.00.
1H-ЯМР (500 МГц, ДМСО-d6): δ [част. на млн.]=1.63 (s, 6Н), 3.97 (s, 3Н), 5.37 (s, 1Н), 7.90-7.95 (m, 1H), 8.03-8.07 (m, 2Н), 8.23 (s, 1Н), 8.29 (s, 1Н), 9.19 (s, 1Н), 12.79 (s, 1H), 13.41 (br.s., 1H).
Промежуточное соединение 4-1
Метил 2-(оксетан-3-илметил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат

1.00 г (2.66 ммоль) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1) растворяли в 10 мл ДМФА и, после добавления 1.10 г (7.99 ммоль) карбоната калия и 221 мг (1.33 ммоль) йодида калия, смесь перемешивали при 25°С в течение 30 мин. Добавляли 603 мг (3.99 ммоль) 3-бромметилоксетана, и смесь перемешивали при 25°С в течение 24 ч. Реакционную смесь распределяли между водой и этилацетатом. Смесь экстрагировали дважды этилацетатом, и объединенные органические фазы фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат). Получали 260 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=1.24 мин
МС (ЭРИпоз): m/z=435 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=3.49-3.64 (m, 1H), 3.95 (s, 3H), 4.49 (t, 2Н), 4.68 (dd, 2Н), 4.81 (d, 2Н), 8.20 (dd, 1H), 8.35-8.41 (m, 1H), 8.43-8.49 (m, 2Н), 8.55-8.58 (m, 1H), 9.06 (s, 1H), 12.53 (s, 1H).
Промежуточное соединение 4-2
Метил 2-(2-метоксиэтил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат

1.00 г (2.75 ммоль) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1) растворяли в 5 мл ДМФА, и при перемешивании добавляли 387 мкл (4,12 ммолей) простого 2-бромэтил-метилового эфира, 1.14 г (8.23 ммоль) карбоната калия и 228 мг (1.37 ммоль) йодида калия. Реакционную смесь перемешивали при 25°С в течение 24 ч., разбавляли с водой и экстрагировали два раза этилацетатом. Объединенные органические фазы фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат). Получали 12 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.24 мин
МС (ЭРИпоз): m/z=423 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=3.24 (s, 3H), 3.86 (t, 2Н), 3.96 (s, 3H), 4.65 (t, 2Н), 8.21 (dd, 1H), 8.35-8.42 (m, 1H), 8.43-8.51 (m, 2Н), 8.52 (d, 1H), 9.06 (s, 1H), 12.53 (s, 1H).
Промежуточное соединение 4-3
Метил 2-(3-метоксипропил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат
1.00 г (2.75 ммоль) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1) растворяли в 5 мл ДМФА, и при перемешивании добавляли 460 мкл (4.12 ммоль) 1-бром-3-метоксипропана, 1.14 г (8.23 ммоль) карбоната калия и 228 мг (1.37 ммоль) йодида калия. Реакционную смесь перемешивали при 25°С в течение 72 ч., разбавляли с водой и экстрагировали два раза этилацетатом. Объединенные органические фазы фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат). Получали 28 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.29 мин
МС (ЭРИпоз): m/z=437 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=2.17 (quin, 2Н), 3.24 (s, 3H), 3.33-3.36 (m, 2Н), 3.96 (s, 3H), 4.53 (t, 2Н), 8.21 (dd, 1H), 8.35-8.42 (m, 1H), 8.45-8.49 (m, 2Н), 8.54 (d, 1H), 9.06 (s, 1H), 12.54 (s, 1H).
Промежуточное соединение 4-4
Метил 2-(3-гидрокси-3-метилбутил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат
Способ получения 1
930 мг (2.55 ммоль) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1), 1.06 г карбоната калия и 212 мг йодида калия сначала загружали в 9 мл ДМФА и смесь перемешивали в течение 15 мин. Затем добавляли 0.62 мл 4-бром-2-метилбутан-2-ола, и смесь перемешивали при 60°С в течение 16 ч. К смеси подмешивали воду и экстрагировали два раза этилацетатом, и экстракт промывали три раза насыщенным раствором хлорида натрия, фильтровали и концентрировали. Очистка с помощью колоночной хроматографии на силикагеле (гексан/этилацетат) обеспечивала 424 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=1.21 мин (УФ детектор: TIC), обнаруженная масса 450.00.
1Н-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.16 (s, 6Н) 2.02-2.11 (m, 2Н) 3.96 (s, 3H) 4.51-4.60 (m, 3H) 8.20 (dd, J=7.83, 1.01 Гц, 1H) 8.39 (s, 1H) 8.45 (s, 2Н) 8.55 (d, J=0.76 Гц, 1H) 9.05 (s, 1H) 12.52 (s, 1H)
Способ получения 2
1.95 г (7.03 ммоль) метил 5-амино-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилата (Промежуточное соединение 7-1) сначала загружали в 30 мл ТГФ. Добавляли 1.48 г (7.73 ммоль) 6-(трифторметил)пиридин-2-карбоновой кислоты, 2.71 г (8.44 ммоль) тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и 1.47 мл (8.44 ммоль) N-этил-N-изопропилпропан-2-амина, и смесь перемешивали при 25°С в течение 20,5 ч. Добавляли воду, смесь экстрагировали три раза этилацетатом и экстракты промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток разделяли колоночной хроматографией на силикагеле (гексан/этилацетат градиент). Получали 2.79 г указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1,23 мин (УФ детектор: TIC), обнаруженная масса 450.00.
Промежуточное соединение 4-5
Метил 2-(2-{[трет-бутил(диметил)силил]окси}этил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат

1.00 г (2.66 ммоль, 97%) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1) сначала загружали в 50 мл ДМФА, при перемешивании добавляли 1.10 г (7.99 ммоль) карбоната калия и 221 мг (1.33 ммоль) йодида калия, и смесь перемешивали при 25°С в течение 30 мин. Затем добавляли 857 мкл (3.99 ммоль) (2-бромэтокси)(трет-бутил)диметилсилана, и смесь перемешивали при 25°С в течение 24 ч. Реакционную смесь разбавляли с водой и экстрагировали этилацетатом. Объединенные органические фазы фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат). Получали 400 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.58 мин
МС (ЭРИпоз): m/z=523 (М+Н)+
1H ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=-0.18--0.13 (m, 6Н), 0.74 (s, 9Н), 3.96 (s, 3H), 4.08 (t, 2Н), 4.57 (t, 2Н), 8.15-8.25 (m, 1H), 8.32-8.43 (m, 1H), 8.43-8.52 (m, 3H), 9.07 (s, 1H), 12.53 (s, 1H).
Промежуточное соединение 4-6
Метил 2-(3-{[трет-бутил(диметил)силил]окси}пропил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат
Аналогично промежуточному соединению 4-5, 1.00 г (2.75 ммоль) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1) растворяли в 10 мл ДМФА, 1.14 г (8.24 ммоль) карбоната калия и добавляли 228 мг (1.37 ммоль) йодида калия при перемешивании, и смесь перемешивали при 25°С в течение 30 мин. Затем добавляли 1.04 г (4.12 ммоль) (3-бромпропокси)(трет-бутил)диметилсилана и смесь перемешивали при 25°С в течение 24 ч. Реакционную смесь фильтровали и осадок на фильтре промывали этилацетатом. Реакционную смесь распределяли между водой и этилацетатом, и водную фазу экстрагировали дважды этилацетатом. Объединенные органические фазы фильтровали через гидрофобный фильтр и концентрировали. Очистка остатка при помощи препаративной ВЭЖХ обеспечивала 428 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.63 мин
МС (ЭРИпоз): m/z=537 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=-0.02-0.06 (m, 6Н), 0.87 (s, 9Н), 2.14 (quin, 2Н), 3.62 (t, 2Н), 3.96 (s, 3H), 4.54 (t, 2Н), 8.20 (d, 1H), 8.35-8.42 (m, 1H), 8.43-8.48 (m, 3H), 8.49-8.53 (m, 1H), 9.06 (s, 1H).
Промежуточное соединение 4-7
Метил 5-({[6-(2-гидроксипропан-2-ил)пиридин-2-ил]карбонил}амино)-2-(4,4,4-трифторбутил)-2Н-индазол-6-карбоксилат

300 мг (0.80 ммоль) метил 5-({[6-(2-гидроксипропан-2-ил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-3) сначала загружали в 4.5 мл ДМФА. 287 мг (1.21 ммоль) 1,1,1-трифтор-4-йодбутана и 333 мг карбоната калия добавляли и смесь перемешивали при 100°С в течение 23 ч. Добавляли воду, и смесь экстрагировали три раза этилацетатом. Смесь концентрировали продукт очищали при помощи препаративной ВЭЖХ. Это обеспечило 72 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.26 мин (УФ детектор: TIC), обнаруженная масса 464.17.
Промежуточное соединение 4-8
Метил 5-{[(5-фтор-6-метилпиридин-2-ил)карбонил]амино}-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилат

195 мг (0.46 ммоль) метил 5-амино-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилата (Промежуточное соединение 7-1) вводили в реакцию с 78 мг (0.50 ммоль) 5-фтор-6-метилпиридин-2-карбоновой кислоты аналогично промежуточному соединению 4-4 (Способ получения 2) в течение 19,5 ч. Получали 228 мг сырого продукта после аналогичной водной обработки.
СЭЖХ-МС (Метод A1): By=1.20 мин (УФ детектор: TIC), обнаруженная масса 414.00.
Промежуточное соединение 4-9
Метил 2-(3-гидрокси-3-метилбутил)-5-{[(6-метилпиридин-2-ил)карбонил]амино}-2Н-индазол-6-карбоксилат
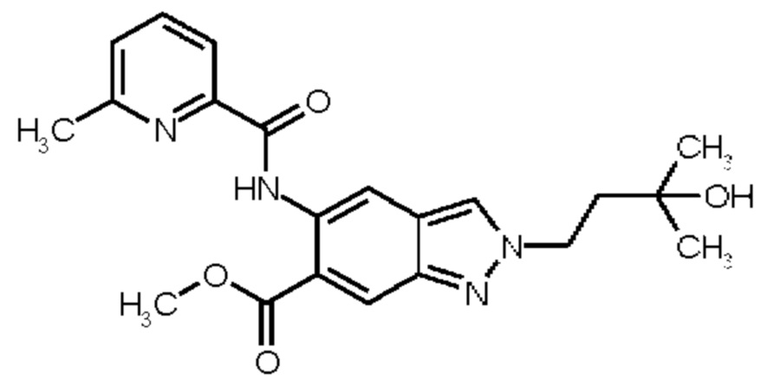
195 мг (0.45 ммоль) метил 5-амино-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилата (Промежуточное соединение 7-1) вводили в реакцию с 70 мг (0.50 ммоль) 6-метилпиридин-2-карбоновой кислоты аналогично с получением промежуточного соединения 4-4 (Способ получения 2) в течение 19.5 ч. Получали 278 мг указанного в заголовке соединения в виде сырого продукта после аналогичной водной обработки.
СЭЖХ-МС (Метод A1): By=1.14 мин (УФ детектор: TIC), обнаруженная масса 396.00.
Промежуточное соединение 4-10
Метил 2-[3-(2,2,2-трифторэтокси)пропил]-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат
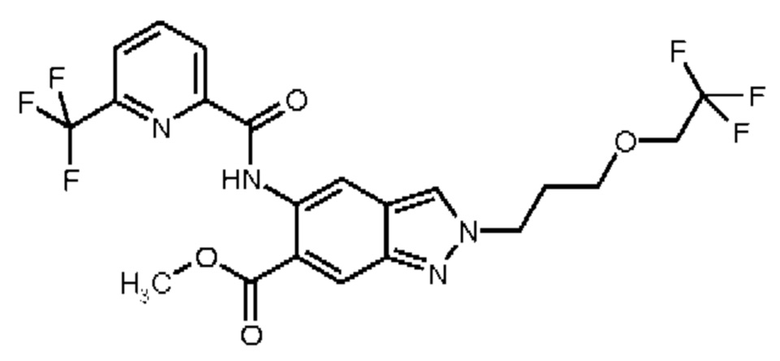
Смесь из 250 мг (0.58 ммоль) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1), 193 мг (0.88 ммоль) 3-бромпропил 2,2,2-трифторэтилового эфира, 242 мг карбоната калия и 145 мг йодида калия в 3 мл ДМФА перемешивали при 100°С в течение 20 ч. Добавляли воду, смесь экстрагировали этилацетатом и экстракт промывали раствором хлорида натрия и концентрировали. Очистка при помощи препаративной ВЭЖХ обеспечивала 52 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.39 мин (УФ детектор: TIC), обнаруженная масса 504.12.
Промежуточное соединение 4-11
Метил 5-({[6-(дифторметил)пиридин-2-ил]карбонил}амино)-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилат
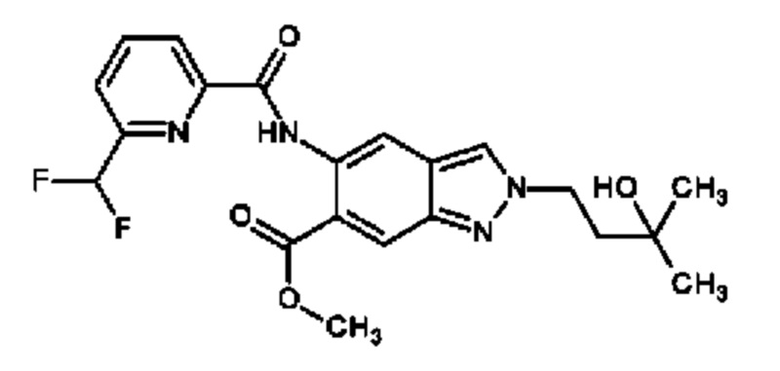
2.00 г метил 5-амино-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилата (Промежуточное соединение 7-1) сначала загружали в 40 мл ТГФ. Добавляли 1.50 г 6-(дифторметил)пиридин-2-карбоновой кислоты, 2.78 г тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU, Номер CAS 125700-67-6) и 1.5 мл N-этил-N-изопропилпропан-2-амина, и смесь перемешивали при КТ в течение 24 ч. Добавляли воду, смесь экстрагировали три раза этилацетатом, и объединенные органические фазы промывали раствором хлорида натрия и фильтровали через гидрофобный фильтр. Смесь концентрировали и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат). Это обеспечило 3.05 г указанного в заголовке соединения в виде желтого твердого вещества.
СЭЖХ-МС (Метод A1): By=1.15 мин (УФ детектор TIC), обнаруженная масса 432.00.
1Н-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.17 (s, 6Н), 2.04-2.11 (m, 2Н), 3.99 (s, 3Н), 4.52-4.60 (m, 3Н), 7.10 (t, 1H), 8.00 (dd, 1H), 8.28-8.38 (m, 2Н), 8.44-8.47 (m, 1H), 8.56 (d, 1H), 9.05 (s, 1Н), 12.49 (s, 1H).
Промежуточное соединение 5-1
N-[6-(2-Гидроксипропан-2-ил)-1Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид

К раствору, охлажденному в охлаждающей бане с ледяной водой, 1.50 г (4.12 ммоль) метил 5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-1) в 20 мл ТГФ осторожно добавляли 6.9 мл (5 эквивалентов) 3М раствора бромида метилмагния в диэтиловом эфире. Смесь перемешивали при охлаждении на ледяной бане в течение 1 ч. и при комнатной температуре в течение 19,5 ч. Добавляли еще 2 эквивалента раствора бромида метилмагния, и смесь перемешивали при комнатной температуре в течение еще 24 ч. Добавляли насыщенный водный раствор хлорида аммония и смесь перемешивали и три раза экстрагировали этилацетатом. Объединенные органические фазы промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат). Получали 763 мг указанного в заголовке соединения.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.63 (s, 6Н), 5.99 (s, 1H), 7.49 (s, 1Н), 8.06 (s, 1H), 8.14-8.19 (m, 1H), 8.37 (t, 1H), 8.46 (d, 1H), 8.78 (s, 1Н), 12.32 (s, 1Н), 12.97 (s, 1H).
Промежуточное соединение 5-2
6-(Дифторметил)-N-[6-(2-гидроксипропан-2-ил)-1H-индазол-5-ил]пиридин-2-карбоксамид

Аналогично получению промежуточного соединения 5-1, 2.40 г (6.93 ммоль) метил 5-({[6-(дифторметил)пиридин-2-ил]карбонил}амино)-1Н-индазол-6-карбоксилата (Промежуточное соединение 3-2) в 10 мл ТГФ вводили в реакцию с тремя частями 3М раствора бромида метилмагния в диэтиловом эфире (6.9 мл, затем перемешивали при комнатной температуре в течение 45 мин; 11.6 мл, затем перемешивали при комнатной температуре в течение 2 ч; 6.9 мл, затем перемешивали при комнатной температуре в течение 2 ч). После обработки, как для промежуточного соединения 5-1, получали 2,39 г сырого продукта, которые использовали далее без дальнейшей очистки.
Промежуточное соединение 6-1
Метил 2-(3-гидрокси-3-метилбутил)-5-нитро-2Н-индазол-6-карбоксилат
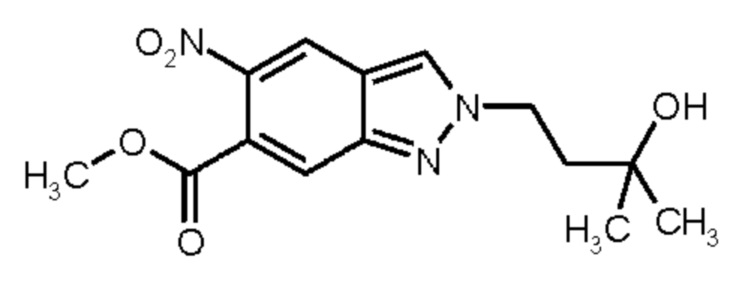
5.00 г (22.6 ммоль) метил 5-нитро-1Н-индазол-6-карбоксилата (Промежуточное соединение 1-1) сначала загружали в 40 мл ДМФА. Добавляли 5.65 г (33.9 ммоль) 4-бром-2-метилбутан-2-ола, 9.37 г (67.8 ммоль) карбоната калия и 5.63 г (33.9 ммоль) йодида калия и смесь перемешивали при 100°С в течение 20 ч. Добавляли воду, смесь экстрагировали три раза этилацетатом и экстракты промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат). Полученные твердые вещества экстрагировали перемешиванием с диэтиловым эфиром, отфильтровывали путем отсасывания, промывали диэтиловым эфиром и сушили. Получали 2,49 г указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=0.93 мин (УФ детектор: TIC), обнаруженная масса 307.00.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.15 (s, 6Н), 2.02-2.11 (m, 2Н), 3.84 (s, 3Н), 4.54 (s, 1Н), 4.58-4.65 (m, 2Н), 8.05 (s, 1H), 8.69 (s, 1Н), 8.86 (s, 1Н).
Промежуточное соединение 7-1
Метил 5-амино-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилат

4.53 г железа и 217 мг хлорида аммония добавляли к 2.49 г (8.10 ммоль) метил 2-(3-гидрокси-3-метилбутил)-5-нитро-2Н-индазол-6-карбоксилата (Промежуточное соединение 6-1) в 30 мл этанола и 10 мл воды, и смесь перемешивали при 90°С в течение 21,5 ч. Смесь фильтровали через целит и три раза промывали этанолом, и фильтрат концентрировали, а к остатку подмешивали воду. Экстракцию осуществляли три раза этилацетатом (добавляли раствор хлорида натрия для улучшения разделения фаз). Объединенные органические фазы промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Это обеспечило 1.95 г (85% от теории) указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=0.67 мин (УФ детектор: TIC), обнаруженная масса 277.00.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.14 (s, 6Н), 1.96-2.08 (m, 2Н), 3.85 (s, 3Н), 4.39-4.51 (m, 3Н), 5.81 (s, 2Н), 6.80 (s, 1Н), 8.05 (s, 1H), 8.18 (s, 1Н).
Демонстрационные примеры
Пример 1
N-[6-(2-Гидроксипропан-2-ил)-2-(2-метоксиэтил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид

75 мг (0.18 ммоль) метил 2-(2-метоксиэтил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-2) растворяли в 500 мкл ТГФ и смешивали с 887 мкл (0.89 ммоль) 1 М раствора бромида метилмагния в ТГФ. Реакционную смесь перемешивали при 25°С в течение 60 мин. Затем, 1 мл насыщенного водного раствора хлорида аммония осторожно добавляли и смесь фильтровали. Водную фазу экстрагировали дважды этилацетатом, и органические фазы объединяли, фильтровали через гидрофобный фильтр и концентрировали. Остаток растворяли в 3 мл ДМСО и очищали при помощи препаративной ВЭЖХ. Содержащие продукт фракции подвергали лиофилизации. Получали 20 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.08 мин
МС (ЭРИпоз): m/z=423 (М+Н)+
1H ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=1.62 (s, 6Н), 3.22 (s, 3H), 3.82 (t, 2Н), 4.55 (t, 2H), 5.96 (s, 1H), 7.57 (s, 1H), 8.16 (d1 1Н), 8.29-8.42 (m, 2Н), 8.42-8.50 (m, 1H), 8.71 (s, 1H), 12.36 (s, 1H)
Пример 2
N-[6-(Гидроксиметил)-2-(2-метоксиэтил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид

13 мг (0.36 ммоль) алюмогидрида лития суспендировали в 1 мл ТГФ и смесь охлаждали до 0°С. Добавляли по каплям 75 мг (0.17 ммоль) метил 2-(2-метоксиэтил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-2), растворенного в 500 мкл ТГФ и смесь перемешивали при 25°С в течение 60 мин. Смесь разбавляли с водой и экстрагировали два раза этилацетатом, и объединенные органические фазы промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр, концентрировали и сушили под сниженным давлением. Это обеспечило 13 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=0.99 мин
МС (ЭРИпоз): m/z=394 (М+Н)+
1H ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=3.23 (s, 3H), 3.83 (t, 2Н), 4.56 (t, 2Н), 4.69 (d, 2Н), 5.77 (t, 1H), 7.57 (s, 1H), 8.19 (d, 1H), 8.33-8.41 (m, 2Н), 8.43-8.47 (m, 1H), 8.51 (s, 1H), 11.20 (s, 1H)
Пример 3
N-[6-(2-Гидроксипропан-2-ил)-2-(3-метоксипропил)-2H-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид

75 мг (0.17 ммоль) метил 2-(3-метоксипропил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-3) растворяли в 500 мкл ТГФ и смешивали с 859 мкл (0.86 ммоль) 1 М раствора бромида метилмагния в ТГФ. Реакционную смесь перемешивали при 25°С в течение 60 мин. Затем, 1 мл насыщенного раствора хлорида аммония осторожно добавляли и смесь фильтровали. Водную фазу экстрагировали дважды этилацетатом, и органические фазы объединяли, фильтровали через гидрофобный фильтр и концентрировали. Остаток растворяли в 3 мл ДМСО и очищали при помощи препаративной ВЭЖХ. Содержащие продукт фракции подвергали лиофилизации. 25 мг указанного в заголовке соединения получали.
СЭЖХ-МС (Метод A1): By=1.13 мин
МС (ЭРИпоз): m/z=437 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.62 (s, 6Н), 2.14 (quin, 2Н), 3.23 (s, 3H), 3.26-3.32 (m, 2Н), 4.44 (t, 2Н), 5.95 (s, 1H), 7.58 (s, 1H), 8.16 (d, 1H), 8.31-8.40 (m, 2Н), 8.43-8.48 (m, 1H), 8.72 (s, 1H), 12.36 (s, 1H).
Пример 4
N-[6-(Гидроксиметил)-2-(3-метоксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
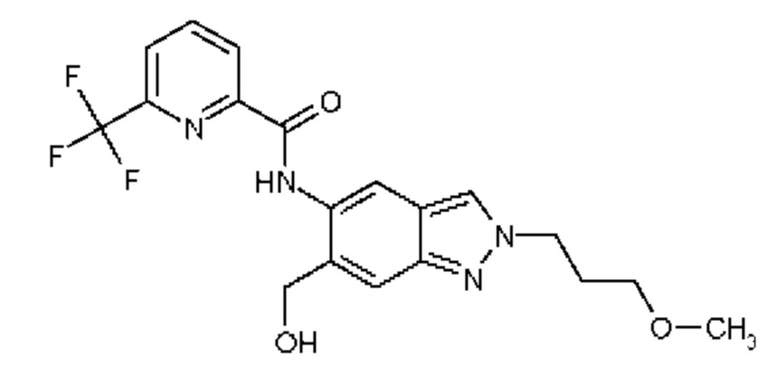
13 мг алюмогидрида лития суспендировали в ТГФ, и смесь охлаждали до 0°С. Добавляли по каплям 75 мг (0.17 ммоль) метил 2-(3-метоксипропил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-3) в ТГФ, и смесь оставляли достичь комнатной температуры в течение 30 мин. Смесь разбавляли водой и фильтровали, остаток промывали этилацетатом и фильтрат экстрагировали этилацетатом. Объединенные фазы этилацетата промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ.
1H ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=2.14 (quin, 2Н), 3.23 (s, 3H), 3.29 (t, 2Н), 4.45 (t, 2Н), 4.68 (d, 2Н), 5.77 (t, 1H), 7.58 (s, 1H), 8.18 (d, 1H), 8.32-8.48 (m, 3H), 8.51 (s, 1H), 11.21 (s, 1H).
Пример 5
N-[2-(2-Гидроксиэтил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
Стадия А:
Получение N-[2-(2-{[трет-бутил(диметил)силил]окси}этил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида

100 мг (0.19 ммоль) метил 2-(2-{[трет-бутил(диметил)силил]окси}этил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-5) растворяли в 1 мл ТГФ и смешивали с 669 мкл (0.67 ммоль) 1 М раствора бромида метилмагния в ТГФ. Реакционную смесь перемешивали при 25°С в течение 60 мин. Добавляли еще 287 мкл (0.29 ммоль) 1 М раствора бромида метилмагния в ТГФ и смесь перемешивали при 25°С в течение 3 ч. Затем осторожно добавляли 20 мл насыщенного раствора хлорида аммония и смесь фильтровали. Водную фазу экстрагировали дважды этилацетатом, и органические фазы объединяли, сушили над сульфатом магния, фильтровали, концентрировали и сушили под сниженным давлением. Это обеспечило 50 мг N-[2-(2-{[трет-бутил(диметил)силил]окси}этил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида.
СЭЖХ-МС (Метод А2): By=1.51 мин
МС (ЭРИпоз): m/z=523 (М+Н)+
1Н ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=-0.17--0.09 (m, 6Н), 0.78 (s, 9Н), 1.62 (s, 6Н), 4.04 (t, 2Н), 4.47 (t, 2Н), 5.98 (s, 1H), 7.57 (s, 1H), 8.16 (d, 1H), 8.29 (s, 1H), 8.37 (t, 1H), 8.45 (d, 1H), 8.73 (s, 1H), 12.38 (s, 1H).
Стадия В:
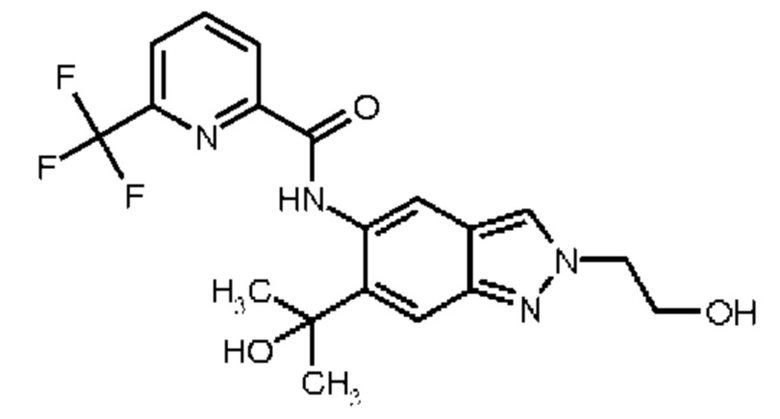
50 мг (96 мкмолей) N-[2-(2-{[трет-бутил(диметил)силил]окси}этил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида растворяли в 1.0 мл ТГФ и смешивали с 144 мкл (0.14 ммоль) 1 М раствора фторида тетрабутиламмония в ТГФ. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разбавляли с водой и экстрагировали два раза этилацетатом, и объединенные органические фазы промывали насыщенным раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Это обеспечило 36 мг N-[2-(2-гидроксиэтил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Пример 5).
1H-ЯМР (400 МГц, ДМСО-d6): d [част. на млн.]=1.62 (s, 6Н), 3.86 (q, 2Н), 4.43 (t, 2Н), 4.95 (t, 1H), 5.94 (s, 1Н), 7.57 (s, 1Н), 8.16 (dd, 1Н), 8.30 (s, 1H), 8.37 (t, 1Н), 8.45 (d, 1Н), 8.72 (s, 1H), 12.36 (s, 1Н).
СЭЖХ-МС (Метод А2): By=0.97 мин (УФ детектор: TIC), обнаруженная масса 408.00.
Пример 6
N-[6-(2-Гидроксипропан-2-ил)-2-(3-гидроксипропил)-2H-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
Стадия А:
Получение N-[2-(3-{[трет-бутил(диметил)силил]окси}пропил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида

50 мг (0.09 ммоль) метил 2-(3-{[трет-бутил(диметил)силил]окси}пропил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-6) растворяли в 500 мкл ТГФ и смешивали с 326 мкл (0.33 ммоль) 1 М раствора бромида метилмагния в ТГФ. Реакционную смесь перемешивали при 25°С в течение 60 мин. Затем осторожно добавляли 20 мл насыщенного раствора хлорида аммония и смесь экстрагировали дважды этилацетатом. Объединенные органические фазы фильтровали через гидрофобный фильтр, концентрировали и сушили под сниженным давлением. Остаток очищали при помощи препаративной ВЭЖХ. Получали 40 мг N-[2-(3-{[трет-бутил(диметил)силил]окси}пропил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида.
СЭЖХ-МС (Метод A1): By=1.58 мин
МС (ЭРИпоз): m/z=537 (М+Н)+
1H ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=0.02-0.05 (m, 6Н), 0.84-0.91 (m, 9Н), 1.62 (s, 6Н), 2.02-2.18 (m, 2Н), 3.55-3.62 (m, 2Н), 4.45 (t, 2Н), 5.96 (s, 1H), 7.57 (s, 1H), 8.16 (d, 1H), 8.31 (s, 1H), 8.33-8.42 (m, 1H), 8.45 (d, 1H), 8.72 (s, 1H), 12.37 (s, 1H).
Стадия В:
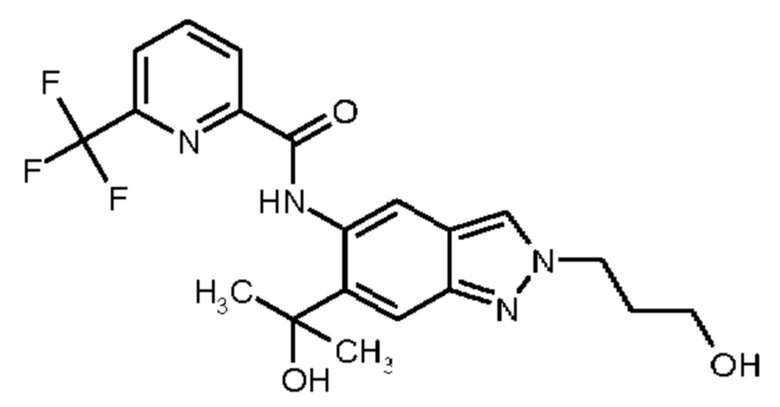
37 мг (0.07 ммоль) N-[2-(3-{[трет-бутил(диметил)силил]окси}пропил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида растворяли в 500 мкл ТГФ и смешивали с 207 мкл (0.21 ммоль) 1 М раствора фторида тетрабутиламмония в ТГФ. Реакционную смесь перемешивали при 25°С в течение 2 ч. Смесь разбавляли с водой и экстрагировали два раза этилацетатом, и объединенные органические фазы промывали насыщенным раствором хлорида натрия, фильтровали и концентрировали. После очистки при помощи препаративной ВЭЖХ, получали 10 мг N-[6-(2-гидроксипропан-2-ил)-2-(3-гидроксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Пример 6, содержащий вторичный компонент).
СЭЖХ-МС (Метод А2): By=1.00 мин
МС (ЭРИпоз): m/z=423 (М+Н)+
1H ЯМР выбранные сигналы (400 МГц, ДМСО-d6): δ [част. на млн.]=1.61 (s), 2.00-2.12 (m), 3.38 (t, 2Н), 4.44 (t, 2Н), 4.62 (br. s., 1H), 5.93 (br. s., 1H), 7.55 (s, 1H), 8.13 (d, 1H), 8.27-8.38 (m, 2H), 8.43 (d, 1H), 8.71 (s, 1H), 12.30 (br. s., 1H).
Пример 7
N-[2-(2-Гидроксиэтил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
Стадия A:
N-[2-(2-{[трет-Бутил(диметил)силил]окси}этил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид

100 мг (0.19 ммоль) метил 2-(2-{[трет-бутил(диметил)силил]окси}этил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-5) растворяли в 1 мл ТГФ и смешивали с 191 мкл (0.38 ммоль) 2 М раствора боргидрида лития. Смесь оставляли перемешиваться при 25°С в течение 24 ч. Добавляли 14 мг (0.38 ммоль) боргидрида натрия и 500 мкл метанола, и смесь перемешивали при 25°С в течение 4 ч. Добавляли еще 14 мг (0.38 ммоль) боргидрида натрия, и смесь перемешивали при 25°С в течение 24 ч. К реакционной смеси осторожно добавляли воду и органическую фазу удаляли. Затем смесь экстрагировали два раза этилацетатом, и объединенные органические фазы промывали насыщенным раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток ресуспендировали в 2 мл ДМСО и очищали при помощи препаративной ВЭЖХ. Это обеспечило 30 мг N-[2-(2-{[трет-бутил(диметил)силил]окси}этил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида.
СЭЖХ-МС (Метод А2): By=1.44 мин
МС (ЭРИпоз): m/z=495 (М+Н)+
1H ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=-0.16--0.12 (m, 6Н), 0.75-0.79 (m, 9Н), 4.05 (t, 2Н), 4.48 (t, 2Н), 4.69 (d, 2Н), 5.75-5.77 (m, 1H), 7.57 (s, 1H), 8.18 (dd, 1H), 8.30-8.33 (m, 1H), 8.38 (t, 1H), 8.45 (d, 1H), 8.51 (s, 1H), 11.20 (s, 1H).
Стадия В:

33 мг (0.07 ммоль) N-[2-(2-{[трет-бутил(диметил)силил]окси}этил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида растворяли в 1 мл ТГФ и смешивали с 100 мкл (0.10 ммоль) 1 М раствора фторида тетрабутиламмония в ТГФ. Реакционную смесь перемешивали при 25°С в течение 1 ч. Смесь разбавляли с водой и экстрагировали два раза этилацетатом, и объединенные органические фазы промывали насыщенным раствором хлорида натрия, фильтровали через гидрофобный фильтр, концентрировали и сушили под сниженным давлением. Получали 25 мг N-[2-(2-гидроксиэтил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Пример 7).
СЭЖХ-МС (Метод А2): By=0.87 мин
МС (ЭРИпоз): m/z=381 (М+Н)+
1H ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=3.87 (q, 2Н), 4.44 (t, 2Н), 4.69 (d, 2Н), 4.98 (t, 1H), 5.70-5.81 (m, 1H), 7.57 (s, 1H), 8.11-8.23 (m, 1H), 8.31-8.42 (m, 2Н), 8.43-8.49 (m, 1H), 8.51 (s, 1H), 11.20 (s, 1H).
Пример 8
N-[6-(2-Гидроксипропан-2-ил)-2-(оксетан-3-илметил)-2H-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
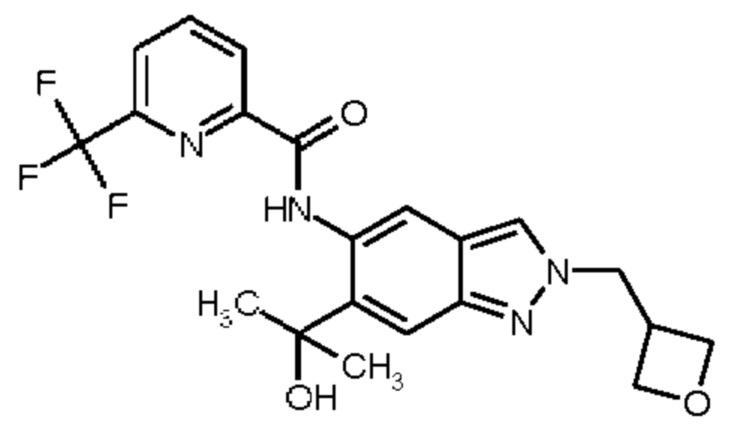
50 мг (0.12 ммоль) метил 2-(оксетан-3-илметил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-1) растворяли в 500 мкл ТГФ и смешивали с 576 мкл (0.58 ммоль) 1 М раствора бромида метилмагния в ТГФ. Реакционную смесь перемешивали при 25°С в течение 60 мин. Затем осторожно добавляли 20 мл насыщенного водного раствора хлорида аммония и смесь концентрировали. Водную фазу экстрагировали дважды этилацетатом, и органические фазы объединяли, сушили над сульфатом магния, фильтровали и концентрировали. Остаток растворяли в 2.0 мл ДМСО и очищали при помощи препаративной ВЭЖХ. Содержащие продукт фракции подвергали лиофилизации. Получали 30 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=1.03 мин
МС (ЭРИпоз): m/z=435 (М+Н)+
1Н ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.62 (s, 6Н), 3.45-3.61 (m, 1H), 4.48 (t, 2Н), 4.66 (dd, 2Н), 4.72 (d, 2Н), 5.94 (s, 1H), 7.57 (s, 1H), 8.16 (d, 1H), 8.33-8.42 (m, 2Н), 8.42-8.47 (m, 1H), 8.72 (s, 1H), 12.36 (s, 1H).
Пример 9
N-[6-(Гидроксиметил)-2-(оксетан-3-илметил)-2H-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид

75 мг (0.17 ммоль) метил 2-(оксетан-3-илметил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-1) растворяли в 1 мл смеси ТГФ/метанол (1:1), и добавляли 8 мг (0.21 ммоль) боргидрида натрия. Смесь оставляли перемешиваться при 25°С в течение 60 мин. Реакционную смесь концентрировали, и остаток смешивали с водой. Суспензию энергично перемешивали в течение 15 мин., и твердые вещества отфильтровывали путем отсасывания, промывали два раза водой и два раза диэтиловым эфиром, и сушили под сниженным давлением. Получали 48 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=0.94 мин
МС (ЭРИпоз): m/z=407 (М+Н)+
1Н ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=3.55 (s, 1H), 4.48 (t, 2Н), 4.61-4.77 (m, 6Н), 7.57 (s, 1H), 8.18 (dd, 1H), 8.33-8.49 (m, 3H), 8.51 (s, 1H), 11.21 (s, 1H).
Пример 10
N-{6-(2-Гидроксипропан-2-ил)-2-[3-(метилсульфонил)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид
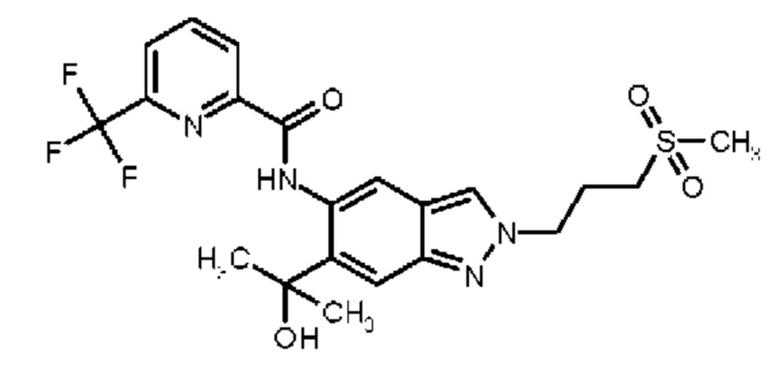
Смесь из 500 мг (1.32 ммоль) N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Промежуточное соединение 5-1), 569 мг карбоната калия и 114 мг йодида калия в 5.0 мл ДМФА перемешивали при комнатной температуре в течение 15 мин. Добавляли 414 мг 1-бром-3-(метилсульфонил)пропана, и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду, смесь два раза экстрагировали этилацетатом и экстракты промывали раствором хлорида натрия и концентрировали. Остаток очищали колоночной хроматографией (дихлорметан/метанол градиент). Экстракция фракции продукта путем перемешивания с диэтиловым эфиром обеспечивала 59 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By=1.02 мин
МС (ЭРИпоз): m/z=485 (М+Н)+
1Н-ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=1.63 (s, 6Н), 2.26-2.42 (m, 2Н), 2.99 (s, 3Н), 3.06-3.16 (m, 2Н), 4.55 (t, 2Н), 5.96 (s, 1H), 7.60 (s, 1H), 8.16 (d, 1Н), 8.33-8.48 (m, 3Н), 8.73 (s, 1Н), 12.37 (s, 1H).
Пример 11
N-[2-(3-Гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид
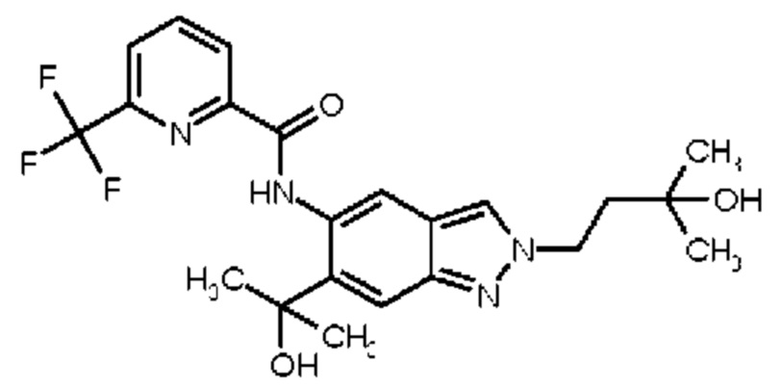
Способ получения 1
705 мг (1.57 ммоль) метил 2-(3-гидрокси-3-метилбутил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-4) сначала загружали в 10 мл ТГФ и охлаждали в охлаждающей бане с ледяной водой. Добавляли 2.6 мл (5.0 эквивалентов) 3М раствора бромида метилмагния (в диэтиловом эфире), и смесь оставляли перемешиваться при охлаждении на ледяной бане в течение 1 ч. и при комнатной температуре в течение 4,5 ч. Добавляли еще 1 эквивалент раствора бромида метилмагния, и смесь оставляли перемешиваться при комнатной температуре в течение 20,5 ч. Снова добавляли еще 1 эквивалент раствора бромида метилмагния, и смесь оставляли перемешиваться при комнатной температуре в течение 22 ч. Реакционную смесь смешивали с насыщенным водным раствором хлорида аммония, перемешивали и экстрагировали три раза этилацетатом. Объединенные органические фазы промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Это обеспечило 790 мг остатка, который очищали при помощи препаративной ВЭЖХ. Это обеспечило 234 мг указанного в заголовке соединения и 164 мг фракции продукта, который перемешивали с диэтиловым эфиром. После фильтрации путем отсасывания с последующей сушкой, получали дополнительные 146 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.10 мин (УФ детектор: TIC), обнаруженная масса 450.00.
1Н-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.14 (s, 6Н), 1.61 (s, 6Н), 1.99-2.08 (m, 2Н), 4.42-4.55 (m, 3Н), 5.93 (s, 1H), 7.56 (s, 1Н), 8.15 (dd, 1Н), 8.32-8.39 (m, 2Н), 8.41-8.47 (m, 1H), 8.70 (s, 1H), 12.34 (s, 1Н).
Способ получения 2
Смесь из 500 мг (1.37 ммоль) N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Промежуточное соединение 5-1), 569 мг карбоната калия и 114 мг йодида калия в 5 мл ДМФА перемешивали при комнатной температуре в течение 15 мин. Добавляли 344 мг (1.5 эквивалентов) 4-бром-2-метилбутан-2-ола и смесь нагревали до 100°С в течение 2 ч. Добавляли еще 0.5 эквивалента 4-бром-2-метилбутан-2-ола, и смесь перемешивали при комнатной температуре в течение 16 ч. К смеси подмешивали воду и экстрагировали два раза этилацетатом, и объединенные органические фазы промывали насыщенным раствором хлорида натрия и фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией очистка на силикагеле (гексан/этилацетат). Это обеспечило 100 мг фракции продукта, который перемешивали с диэтиловым эфиром. Твердое вещество фильтровали и сушили. Получали 60 мг указанного в заголовке соединения.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.14 (s, 6Н), 1.61 (s, 6Н), 1.99-2.07 (m, 2Н), 4.43-4.52 (m, 3H) 5.94 (s, 1H) 7.57 (s, 1H) 8.15 (dd, 1Н) 8.33-8.40 (m, 2Н), 8.42-8.48 (m, 1H), 8.71 (s, 1H), 12.35 (s, 1H)
Пример 12
N-{6-(2-Гидроксипропан-2-ил)-2-[2-(метилсульфонил)этил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид

160 мг (0.44 ммоль) N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Промежуточное соединение 5-1) суспендировали вместе с 182 мг карбоната калия и 36 мг йодида калия в 1.0 мл ДМФА, и смесь перемешивали при комнатной температуре в течение 15 мин. Затем добавляли 123 мг 2-бромэтилметилсульфона (0.66 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду, смесь экстрагировали дважды этилацетатом и экстракты промывали насыщенным водным раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Очистка остатка при помощи препаративной ВЭЖХ обеспечивала 20 мг указанного в заголовке соединения.
СЭЖХ (Метод А2): By=1.01 мин;
МС (ЭРИпоз): m/z=471 (М+Н)+
1H ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.63 (s, 6Н), 2.90 (s, 3H), 3.85 (t, 2Н), 4.86 (t, 2Н), 5.97 (s, 1H), 7.59 (s, 1H), 8.13-8.19 (m, 1H), 8.37 (s, 1H), 8.41-8.48 (m, 2Н), 8.74 (s, 1H), 12.37 (s, 1H).
Пример 13
6-(Дифторметил)-N-[2-(3-гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]пиридин-2-карбоксамид
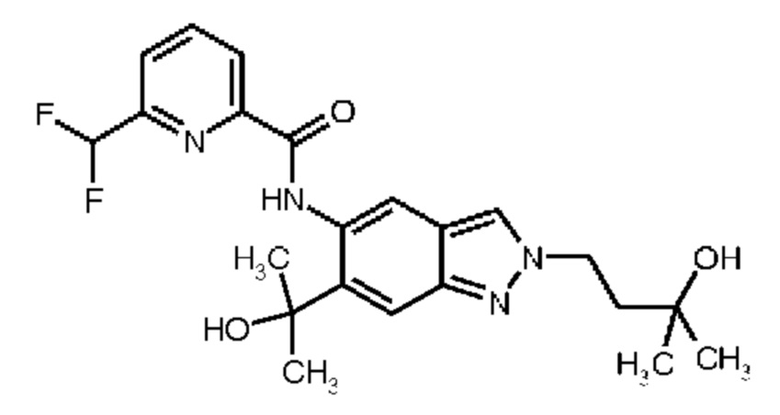
Способ получения 1
Смесь из 250 мг 6-(дифторметил)-N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]пиридин-2-карбоксамида (сырой продукт промежуточного соединения 5-2), 144 мг йодида калия и 239 мг карбоната калия в 2.5 мл ДМФА перемешивали при комнатной температуре в течение 15 мин. Добавляли 145 мг (0.87 ммоль) 4-бром-2-метилбутан-2-ола, смесь перемешивали при 110°С в течение 3 ч., добавляли еще 96 мг 4-бром-2-метилбутан-2-ола, и смесь перемешивали при 110°С в течение 4 ч. Добавляли воду, смесь экстрагировали дважды этилацетатом и экстракт промывали полунасыщенным водным раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Очистку осуществляли колоночной хроматографией на силикагеле (гексан/этилацетат). Получали 61 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.00 мин (УФ детектор: TIC), обнаруженная масса 432.00.
1Н-ЯМР (300 МГц, ДМСО-d6): δ [част. на млн.]=1.14 (s, 6Н), 1.63 (s, 6Н), 1.97-2.08 (m, 2Н), 4.41-4.55 (m, 3Н), 5.99 (s, 1H), 7.03 (t, 1Н), 7.56 (s, 1H), 7.94-8.00 (m, 1H), 8.24-8.38 (m, 3Н), 8.71 (s, 1H), 12.49 (s, 1Н).
Способ получения 2
Аналогично получению Примера 11 (Способ получения 1), 3.00 г метил 5-({[6-(дифторметил)пиридин-2-ил]карбонил}амино)-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-11) вводили в реакцию с 3М раствором бромида метилмагния (в диэтиловом эфире). После очистки сырого продукта при перемешивании с диэтиловым эфиром, фильтрации с последующей препаративной ВЭЖХ, получали 1,37 г указанного в заголовке соединения.
Пример 14
6-(Дифторметил)-N-{6-(2-гидроксипропан-2-ил)-2-[2-(метилсульфонил)этил]-2Н-индазол-5-ил}пиридин-2-карбоксамид

Смесь из 250 мг 6-(дифторметил)-N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]пиридин-2-карбоксамида (сырой продукт промежуточного соединения 5-2), 144 мг йодида калия и 239 мг карбоната калия в 2,5 мл ДМФА перемешивали при комнатной температуре в течение 15 мин. Добавляли 162 мг 2-бромэтилметилсульфона (0.87 ммоль), и смесь перемешивали при 110°С в течение 3 ч. Добавляли воду, смесь экстрагировали дважды этилацетатом и экстракт промывали полунасыщенным водным раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ и фракции продукта дополнительно очищали путем очистки колоночной хроматографией на силикагеле (гексан/этилацетат). 40 мг указанного в заголовке соединения получали.
1Н-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.65 (s, 6Н), 2.90 (s, 3Н), 3.85 (t, 2Н), 4.85 (t, 2Н), 6.03 (s, 1Н), 7.04 (t, 1Н), 7.59 (s, 1H), 7.98 (d, 1Н), 8.25-8.36 (m, 2Н), 8.43 (s, 1Н), 8.75 (s, 1H), 12.52 (s, 1H).
Пример 15
6-(Дифторметил)-N-[6-(2-гидроксипропан-2-ил)-2-(3-гидроксипропил)-2Н-индазол-5-ил]пиридин-2-карбоксамид
Стадия А:
Получение N-[2-(3-{[трет-бутил(диметил)силил]окси}пропил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(дифторметил)пиридин-2-карбоксамида

Смесь из 250 мг 6-(дифторметил)-N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]пиридин-2-карбоксамида (Промежуточное соединение 5-2), 48 мг йодида калия и 239 мг карбоната калия в 2.5 мл ДМФА перемешивали при комнатной температуре в течение 15 мин. Добавляли 219 мг (0.87 ммоль, 1.5 эквивалентов) (3-бромпропокси)(трет-бутил)диметилсилана и смесь перемешивали при 110°С в течение 3 ч. Добавляли еще 1 эквивалент (3-бромпропокси)(трет-бутил)диметилсилана, и смесь перемешивали при 100°С в течение 4 ч. Добавляли воду, смесь экстрагировали этилацетатом и экстракт промывали водным раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали колоночной хроматографией (гексан/этилацетат). Получали 92 мг указанного в заголовке соединения.
Стадия В:
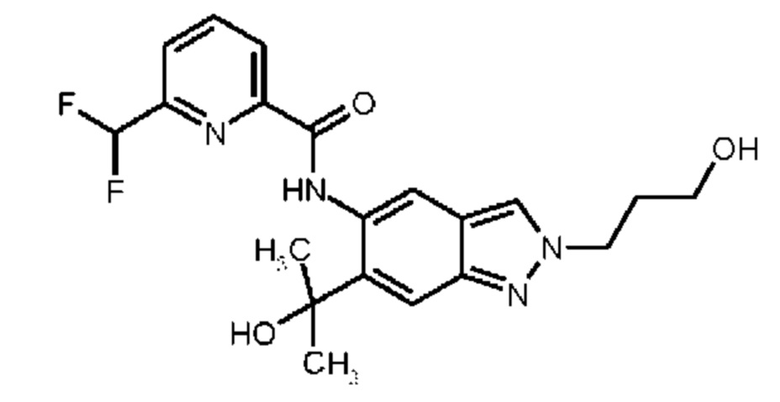
Аналогично получению Примера 6, Стадия В, 92 мг N-[2-(3-{[трет-бутил(диметил)силил]окси}пропил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(дифторметил)пиридин-2-карбоксамида вводили в реакцию с 0.53 мл 1 М раствора фторида тетрабутиламмония в ТГФ в течение 1 ч. Водная обработка как в Примере 6 и очистка при помощи препаративной ВЭЖХ обеспечивала 46 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=0.92 мин (УФ детектор: TIC), обнаруженная масса 404.00.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.64 (s, 6Н), 2.05 (quin, 2Н), 3.35-3.46 (m, 2Н), 4.45 (t, 2Н), 4.64 (t, 1Н), 5.99 (s, 1H), 7.04 (t, 1Н), 7.57 (s, 1Н), 7.95-7.99 (m, 1H), 8.25-8.36 (m, 3Н), 8.73 (s, 1H), 12.50 (s, 1Н).
Пример 16
N-[6-(2-Гидроксипропан-2-ил)-2-(4,4,4-трифторбутил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид

Смесь из 210 мг (0.58 ммоль) N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Промежуточное соединение 5-1) в 3 мл ДМФА смешивали с 0.11 мл (0.87 ммоль) 1,1,1-трифтор-4-йодбутана и 239 мг карбоната калия, и смесь перемешивали при 80°С в течение 6 ч. После добавления воды, смесь экстрагировали три раза этилацетатом, и объединенные органические фазы промывали насыщенным раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Сырой продукт очищали при помощи препаративной ВЭЖХ. Получали 19 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By=1.27 мин (УФ детектор: TIC), обнаруженная масса 474.15.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.]=1.62 (s, 6Н), 2.10-2.33 (m), 4.49 (t, 2Н), 5.94 (s, 1H), 7.59 (s, 1Н), 8.13-8.18 (m, 1H), 8.32-8.41 (m, 2Н), 8.41-8.47 (m, 1H), 8.72 (s, 1Н), 12.35 (s, 1Н).
Пример 17
N-{6-(2-Гидроксипропан-2-ил)-2-[3-(трифторметокси)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид
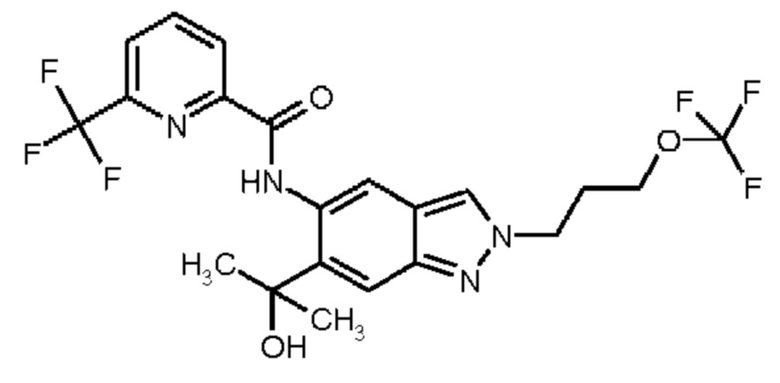
150 мг (0.33 ммоль) N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Промежуточное соединение 5-1) сначала загружали в 2 мл ТГФ. Добавляли 58 мг (0.40 ммоль) 3-(трифторметокси)пропан-1-ола, 131 мг трифенилфосфина и 71 мкл диизопропил азодикарбоксилата (DIAD, CAS 2446-83-5), и смесь перемешивали при комнатной температуре в течение 19 ч. Добавляли 0.83 мл раствора гидроксида натрия (2М) и смесь перемешивали при 40°С в течение 5 ч. Смесь разбавляли с водой и экстрагировали три раза этилацетатом, и объединенные органические фазы концентрировали и очищали при помощи препаративной ВЭЖХ. 16 мг указанного в заголовке соединения получали в виде сырого продукта.
СЭЖХ-МС (Метод А2): By=1.26 мин (УФ детектор: TIC), обнаруженная масса 490.14.
1H-ЯМР (400 МГц, ДМСО-d6, выбранные сигналы): δ [част. на млн.]=1.61 (s, 6Н), 1.84 (d, 1Н), 2.32 (quint., 2Н), 4.08 (t, 2Н), 4.51 (t, 2Н), 7.58 (s, 1Н), 8.15 (d, 1H), 8.31-8.39 (m, 2Н), 8.44 (d, 1Н), 8.72 (s, 1Н), 12.35 (s, 1H).
Пример 18
N-{6-(2-Гидроксипропан-2-ил)-2-[3-(2,2,2-трифторэтокси)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид
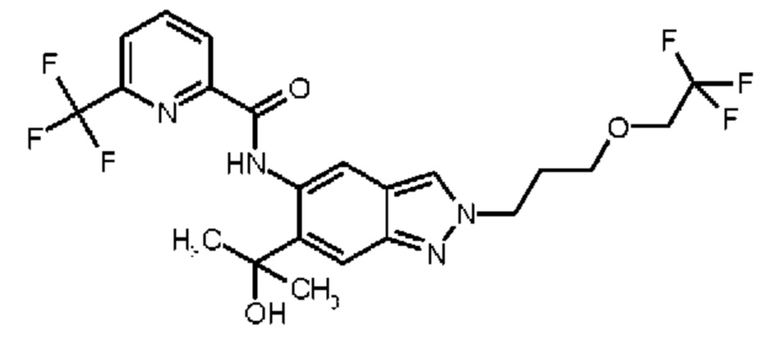
Аналогично получению Примера 11 (Способ получения 1), 52 мг (0.10 ммоль) метил 2-[3-(2,2,2-трифторэтокси)пропил]-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-10) в 3 мл ТГФ вводили в реакцию с 2×171 мкл 3М бромида магния раствор в диэтиловом эфире. Очистка при помощи препаративной ВЭЖХ обеспечивала 12 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод A1): By = 1.25 мин (УФ детектор: TIC), обнаруженная масса 504.16.
1H-ЯМР (500 МГц, ДМСО-d6): δ [част. на млн.] = 1.63 (s, 6H), 2.20 (quin, 2Н), 3.58 (t, 2H), 4.05 (q, 2Н), 4.47 (t, 2H), 5.94 (s, 1H), 7.58 (s, 1H), 8.15 (dd, 1H), 8.32 (s, 1H), 8.36 (t, 1H), 8.45(d, 1H), 8.73 (s, 1H), 12.36 (s, 1H).
Пример 19
5-Фтор-N-[2-(3-гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2H-индазол-5-ил]-6-метилпиридин-2-карбоксамид
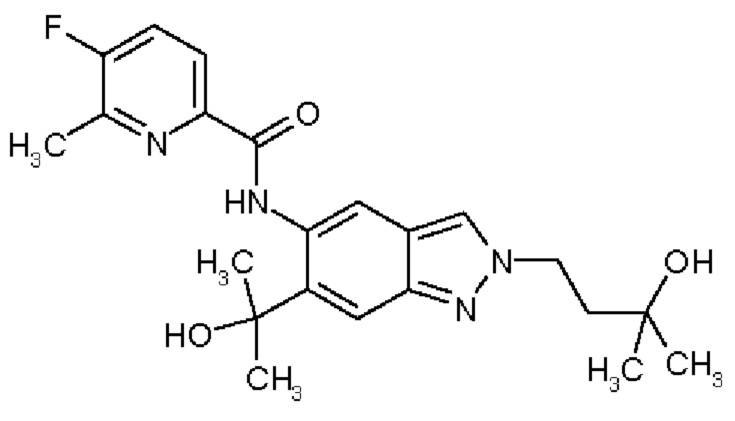
228 мг (0.31 ммоль) метил 5-{[(5-фтор-6-метилпиридин-2-ил)карбонил]амино}-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-8) сначала загружали в 4.5 мл ТГФ и охлаждали на ледяной бане. Добавляли 0,63 мл 3М раствор бромида метилмагния (в диэтиловом эфире), и смесь оставляли перемешиваться при охлаждении ледяной баней в течение 2 ч. и при комнатной температуре в течение 21 ч. Реакционную смесь смешивали с насыщенным водным раствором хлорида аммония и экстрагировали три раза этилацетатом. Объединенные органические фазы концентрировали. Остаток очищали при помощи препаративной ВЭЖХ. 82 мг указанного в заголовке соединения получали.
СЭЖХ-МС (Метод А2): By = 1.03 мин (УФ детектор: TIC), обнаруженная масса 414.21.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.] = 1.13 (s, 6H), 1.63 (s, 6H), 1.99-2.05 (m, 2H), 2.55-2.59 (m, 3H), 4.42-4.50 (m, 3H), 5.95 (s, 1H), 7.54 (s, 1H), 7.83 (t, 1H), 8.05 (dd, 1H), 8.31 (s, 1H), 8.68 (s, 1H), 12.33 (s, 1H).
Пример 20
N-[2-(3-Гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-метилпиридин-2-карбоксамид
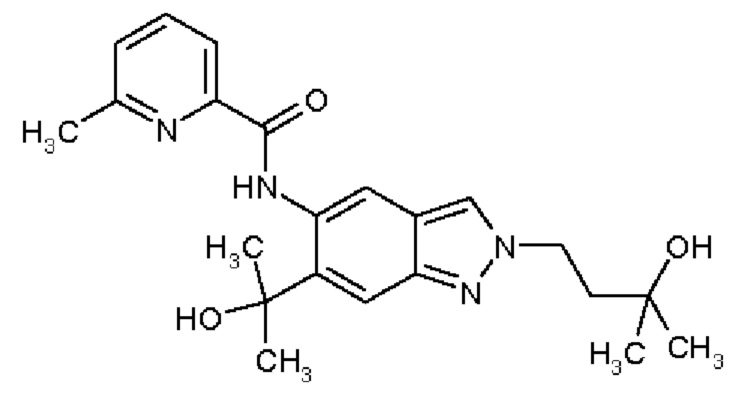
278 мг (0.48 ммоль) метил 2-(3-гидрокси-3-метилбутил)-5-{[(6-метилпиридин-2-ил)карбонил]амино}-2Н-индазол-6-карбоксилата
(Промежуточное соединение 4-9) сначала загружали в 5.0 мл ТГФ и охлаждали на ледяной бане. Добавляли 0.97 мл 3М раствор бромида метилмагния (в диэтиловом эфире) и смесь оставляли перемешиваться при охлаждении ледяной баней в течение 2 ч. и при комнатной температуре в течение 20,5 ч. Добавляли еще 0.48 мл 3М раствора бромида метилмагния и смесь оставляли перемешиваться при комнатной температуре в течение 67 ч. Смесь смешивали с насыщенным водным раствором хлорида аммония и экстрагировали три раза этилацетатом, и экстракты промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ. Получали 111 мг указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By = 0.97 мин (УФ детектор: TIC), обнаруженная масса 396.22.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.] = 1.15 (s, 6H), 1.64 (s, 6H), 2.00-2.08 (m, 2H), 2.61 (s, 3Н), 4.41-4.59 (m, 3Н), 5.92 (s, 1H), 7.50 (dd, 1H), 7.56 (s, 1H), 7.90-7.99 (m, 2H), 8.33 (s, 1H), 8.70 (s, 1H), 12.39 (s, 1H).
Пример 21
6-(2-Гидроксипропан-2-ил)-N-[6-(2-гидроксипропан-2-ил)-2-(4,4,4-трифторбутил)-2Н-индазол-5-ил]пиридин-2-карбоксамид
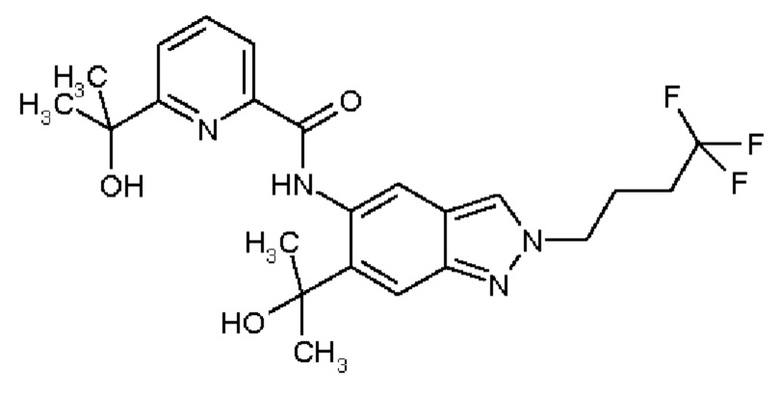
Раствор 72 мг (0.16 ммоль) метил 5-({[6-(2-гидроксипропан-2-ил)пиридин-2-ил]карбонил}амино)-2-(4,4,4-трифторбутил)-2Н-индазол-6-карбоксилата (Промежуточное соединение 4-7) в 10 мл ТГФ охлаждали в охлаждающей бане со льдом/водой. Добавляли 0,26 мл 3М раствора бромида метилмагния в диэтиловом эфире и смесь перемешивали в течение 2 ч. и затем при комнатной температуре в течение 20 ч. Добавляли еще 1 эквивалент 3М раствора бромида метилмагния, и смесь перемешивали при комнатной температуре в течение 24 ч. Добавляли насыщенный водный раствор хлорида аммония, смесь три раза экстрагировали этилацетатом и экстракты промывали раствором хлорида натрия и концентрировали. Препаративная ВЭЖХ обеспечивала 22 мг (31% от теории) указанного в заголовке соединения.
СЭЖХ-МС (Метод А2): By = 1.15 мин (УФ детектор: TIC), обнаруженная масса 464.20.
1H-ЯМР (400 МГц, ДМСО-d6): δ [част. на млн.] = 1.56 (s, 6H), 1.64 (s, 6H), 2.07-2.34 (m, 4H), 4.49 (t, 2H), 5.32 (s, 1H), 6.05 (s, 1H), 7.60 (s, 1H), 7.87 (dd, 1H), 7.99-8.05 (m, 2H), 8.35 (s, 1H), 8.79 (s, 1H), 12.45 (s, 1H).
Пример 22
N-{2[2-(1-Гидроксициклопропил)этил]-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид

250 мг (0.69 ммоль) N-[6-(2-гидроксипропан-2-ил)-1Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамида (Промежуточное соединение 5-1) сначала загружали в 5 мл ДМСО. Добавляли 159 мг (0.96 ммоль) 1-(2-бромэтил)циклопропанола, 285 мг карбоната калия и 171 мг йодида калия и смесь перемешивали при 100°С в течение 5 ч. Добавляли воду и смесь экстрагировали три раза этилацетатом. Объединенные органические фазы промывали раствором хлорида натрия, фильтровали через гидрофобный фильтр и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (колонка: Waters XBridge C18 5 мкм 100×30 мм, элюент А: вода + 0.1 объемн. % муравьиной кислоты (99%), элюент В: ацетонитрил). Лиофилизация обеспечивала 45 мг указанного в заголовке соединения.
1H-ЯМР (500 МГц. ДМСО-d6): δ [част. на млн.] = 0.18-0.22 (m, 2Н), 0.48-0.52 (m, 2Н), 1.62 (s, 6H), 2.08 (t, 2Н), 4.54-4.60 (m, 2Н), 5.36 (s, 1H), 5.96 (s, 1H), 7.57 (s, 1H), 8.16 (dd, 1H), 8.34-8.39 (m, 2Н), 8.45 (d, 1H), 8.72 (s, 1H), 12.36 (s, 1H).
Оценка физиологической эффективности при различных аутоиммунных заболеваниях
Анализ IRAK4 киназы
IRAK4-ингибирующую активность веществ в соответствии с настоящим изобретением измеряли в анализе IRAK4 TR-FRET (TR-FRET = времяразрешенный флуоресцентный индуктивно-резонансный перенос энергии), описанном ниже в настоящей заявке.
В качестве фермента использовали рекомбинантный белок слияния из N-концевой GST (глутатион S-трансфераза) и IRAK4 человека, экспрессированный в клетках насекомых, инфицированных бакуловирусом (Hi5, BTI-TN-5B1-4, клеточную линию приобретали у Invitrogen, каталожный № В855-02) и очищенных с помощью аффинной хроматографии. Применяемый для киназной реакции субстрат представлял собой биотинилированный пептид биотин-Ahx-KKARFSRFAGSSPSQASFAEPG (С-конец в амидной форме), который можно приобрести, например, у Biosyntan GmbH (Berlin-Buch).
Для анализа получали 11 различных концентраций в диапазоне от 20 мкМ до 0,073 нМ из 2 мМ раствора тестируемого вещества в ДМСО. 50 нл соответствующего раствора пипетировали в черный низкообъемный 384-луночный микротитрационный планшет (Greiner Bio-One, Frickenhausen, Германия), добавляли 2 мкл раствора IRAK4 в буфере для анализа [50 мм HEPES рН 7.5, 5 мм MgCl2, 1.0 мм дитиотреитола, 30 мкм активированного ортованадата натрия, 0.1% (мас./объемн.) бычьего гамма-глобулина (BGG), 0.04% (объемн./объемн.) нонидет-Р40 (Sigma)], и смесь инкубировали в течение 15 мин. для обеспечения предварительного связывания веществ с ферментом перед киназной реакцией. Затем инициировали киназную реакцию путем добавления 3 мкл раствора аденозинтрифосфата (АТР, 1.67 мм = конечная концентрация в 5 мкл анализируемого объема: 1 мм) и пептидного субстрата (0.83 мкм = конечная концентрация в 5 мкл анализируемого объема: 0.5 мкм) в буфере для анализа, и полученную в результате смесь инкубировали при 22°С в течение времени реакции 45 мин. Концентрацию IRAK4 регулировали до соответствующей активности фермента и устанавливали такой, которая обеспечивала проведение анализа в линейном диапазоне. Типичные концентрации составляли около 0.2 нм. Реакцию останавливали добавлением 5 мкл раствора детектирующих реагентов TR-FRET [0.1 мкм стрептавидин-XL665 (Cisbio Bioassays; Франция, каталожный №610SAXLG)] и 1.5 нм анти-фосфосеринового антитела [Merck Millipore, "STK Antibody", каталожный №35-002] и 0.6 нм LANCE EU-W1024-меченного анти-мышиного-IgG антитела (Perkin-Elmer, № продукта AD0077; альтернативно можно использовать меченое криптатом тербия анти-мышиное-IgG антитело от Cisbio Bioassays) в водном растворе ЭДТУ (100 мм EDTA, 0.4% [мас./объемн.] бычьего сывороточного альбумина [BSA] в 25 мм HEPES рН 7.5).
Полученную в результате смесь инкубировали при 22°С в течение 1 ч. для обеспечения образования комплекса биотинилированного фосфорилированного субстрата и детектирующих реагентов. Количество фосфорилированного субстрата затем оценивали путем измерения резонансного переноса энергии от меченного хелатом европия анти-мышиного-IgG антитела к стрептавидин-XL665. Для этой цели испускания флуоресценции на длинах волн 620 нм и 665 нм измеряли после возбуждения на длине волны 350 нм в измерительном приборе TR-FRET, например, Rubystar (BMG Labtechnologies, Оффенбург, Германия) или Viewlux (Perkin-Elmer). Соотношение испусканий на длинах волн 665 нм и 622 нм принимали в качестве меры количества фосфорилированного субстрата. Данные нормализовали (ферментативная реакция без тестируемого вещества = 0% ингибирования; все остальные компоненты анализируемой среды, но без фермента = 100% ингибирования). Обычно, тестируемые вещества тестировали на одних и тех же микротитрационных планшетах в 11 различных концентрациях в диапазоне от 20 мкМ до 0.073 нм (20 мкм, 5.7 мкм, 1.6 мкм, 0.47 мкм, 0.13 мкм, 38 нм, 11 нм, 3.1 нм, 0.89 нм, 0.25 нм и 0.073 нм). Серии разбавлений готовили перед анализом (от 2 мм до 7.3 нм в 100% ДМСО) путем серийных разбавлений. Значения IC50 были рассчитаны путем подбора по 4 параметрам.

Ингибирующую активность данных веществ общей формулы (III) по отношению к IRAK4 равным образом измеряли в описанном выше анализе IRAK4 TR-FRET. В качестве примера упоминаются следующие: промежуточное соединение 4-2 с IC50=21.7 нм, промежуточное соединение 4-3 с IC50=13.0 нм и промежуточное соединение 4-4 с IC50=6.2 нм.
Секреция TNF-α в клетках ТНР-1
При помощи этого теста можно протестировать вещества на их способность ингибировать секрецию TNF-α (фактор некроза опухоли-альфа) в клетках ТНР-1 (линия клеток острого моноцитарного лейкоза человека). TNF-α является цитокином, участвующим в воспалительных процессах. В этом тесте секрецию TNF-α запускают путем инкубирования с бактериальным липополисахаридом (ЛПС).
Клетки ТНР-1 выдерживали в непрерывной суспензионной клеточной культуре [среда RPMI 1460 с L-глутамаксом (Gibco, кат. №.61870-044) дополненной 10% фетальной бычьей сывороткой (FCS) (Invitrogen, кат. №.10082-147), 1% смеси пенициллин/стрептомицин (Gibco BRL, кат. №.15140-114)] и концентрация клеток не должна превышать 1×106 клеток/мл. Анализ осуществляли в среде для культивирования клеток (среда RPMI 1460 с L-глутамаксом, дополненная FCS 10%).
В каждом случае 2-2,5 мкл клеточной суспензии (соответствует 4000 клеткам) на лунку распределяли в 384-луночный планшет для тестирования (Greiner, кат. №784076), в каждой из которых 40-50 нл вещества было растворено в 100% ДМСО. Это было сделано с использованием 10 различных концентраций в диапазоне от 20 мкм до 0.073 нм для каждого вещества. Клетки инкубировали при комнатной температуре в течение 15 мин. Затем в каждую лунку распределяли 2-2.5 мкл 0.1 мкг/мл ЛПС (Sigma, Escherichia coli 055:B5, кат. № L5418), растворенного в среде для культивирования клеток (конечная концентрация 0.05 мкг/мл). В качестве нейтрального контроля клетки обрабатывали концентрацией ЛПС в 0.05 мкг/мл и 1% ДМСО и, в качестве контроля ингибитора только посредством 1% ДМСО.
Планшеты центрифугировали при 80 g в течение 30 с и инкубировали при 37°С, 5% CO2 и 95% атмосферной влажности в течение 17 ч. Количество TNF-α определяли с использованием набора для детектирования TNF-альфа HTRF (Cisbio, кат. №62TNFPEB/C). Для этой цели в каждом случае 2 мкл детектирующего раствора, состоящего из конъюгата анти-TNF-α-XL665 и конъюгата анти-TNF-α-криптата, растворенных в восстанавливающем буфере в соответствии с инструкциями изготовителя, добавляли для проведения теста HTRF (гомогенная флуоресценция с временным разрешением). После добавления смесь инкубировали или при комнатной температуре в течение 3 ч., или при 4°С в течение ночи. Затем сигналы считывали на длинах волн 620/665 нм, с использованием поддерживаемого HTRF измерительного прибора, такого как BMG PheraStar.
Активность веществ выражали в процентах в виде соотношения между нейтральным контролем и контрольным ингибитором. Значения IC50 были рассчитаны путем подбора по 4 параметрам.

Вызванная посредством ЛПС (липополисахаридов) выработка цитокинов in vitro в МКПК (мононуклеарные клетки периферической крови) человека)
Исследовали воздействие соединений общей формулы (I) на вызванную выработку цитокинов в МКПК человека. В данном случае выработку цитокинов вызывали с помощью ЛПС, лиганда TLR4, который приводит к активации IRAK4-опосредованного сигнального пути.
МКПК человека получали из антикоагулированной цельной крови человека. Для этой цели 15 мл среды фиколл-пак (Biochrom, кат. № L6115) изначально пипетировали в пробирки Leucosep и добавляли 20 мл крови человека. После центрифугирования крови при 800 g в течение 15 мин. при комнатной температуре плазму, включая тромбоциты, удаляли и отбрасывали. МКПК переносили в центрифужные пробирки и доливали PBS (фосфатно-солевой буферный раствор) (Gibco, кат. №14190). Суспензию клеток центрифугировали при комнатной температуре при 250 g в течение 10 мин. и супернатант отбрасывали. МКПК ресуспендировали в полной среде (RPMI 1640, без L-глутамина (РАА, кат. № Е15-039), 10% FCS; 50 Ед/мл пенициллина, 50 мкг/мл стрептомицина (РАА, кат. № Р11-010) и 1% L-глутамина (Sigma, кат. № G7513)).
Анализ также проводили в полной среде. РВМС высевали в 96-луночные планшеты при плотности клеток 2,5×105 клеток/лунку. Соединения в соответствии с настоящим изобретением подвергали серийному разбавлению в постоянном объеме 100% ДМСО и использовали в анализе в 8 различных концентрациях в диапазоне от 10 мкМ до 3 нМ таким образом, что конечная концентрация ДМСО составляла 0,4% ДМСО. Затем перед фактической стимуляцией клетки предварительно инкубировали с ними в течение 30 мин. Для индуцирования секреции цитокинов клетки стимулировали посредством 0,1 мкг/мл ЛПС (Sigma, Escherichia coli 0128:B12, кат. № L2887) в течение 24 часов. Жизнеспособность клеток определяли с использованием люминесцентного анализа CellTiter-Glo (Promega, кат. № G7571 (G755/G756A)) в соответствии с инструкциями изготовителя. Количество секретированного TNF-α в супернатанте культуры клеток определяли с использованием набора Human ProInflammatory 9-Plex Tissue Culture Kit (MSD, кат. № K15007B) в соответствии с инструкциями изготовителя. В качестве примера, примерное соединение 11 и примерное соединение 12 обладают активностью ≤1 мкм.
In vitro TLR-4/TLR-7-индуцированная секреция интерлейкина (IL)-23 дендритных клеток (ДК) человека
Исследовали воздействие соединений в соответствии с настоящим изобретением общей формулы (I) на индуцированное продуцирование провоспалительного цитокина IL-23, который играет значительную роль для генерации клеток ТН-17 в ДК человека. Установлено, что клетки ТН-17 играют решающую роль в патогенезе нарушений, таких как ревматоидный артрит, псориатический артрит, болезнь Бехтерева (анкилозирующий спондилоартрит) или рассеянный склероз (Lubberts, Nat. Rev. Rheumatol., 2015; Marinoni et al., Auto. Immun. Highlights, 2014; Isailovic et al., J. Autoimmun., 2015; Staschke et al., J Immunol., 2009). Для определения воздействия соединений в соответствии с настоящим изобретением на продуцирование IL-23, первичные человеческие моноциты (выделенные из РВМС человека с использованием магнитного разделения [Miltenyi Biotech, Monocyte Isolation Kit, кат. №130-091-153] с помощью добавления факторов роста (рекомбинантный человеческий GM-CSF [PeproTech, кат. №300-03] и IL-4 [PeproTech, кат. №200-04]) в полной среде (VLE (сверх-низкий эндотоксин) RPMI 1640 [Biochrom AG, кат. №FG1415], 10% фетальной бычьей сыворотки (FBS) [Gibco, кат. №10493-106]; 50 мкМ β-меркаптоэтанола (Gibco, кат. №31350], 50 Ед/мл пенициллина и стрептомицина [Gibco, кат. №15140-114]) были дифференцированы в культуре в течение 6 дней, чтобы получить ДК. После сбора ДК их ресуспендировали в полной среде и высевали с плотностью клеток 2×107 клеток/лунку в 96-луночный планшете (Costar, кат. №3599). Соединения в соответствии с настоящим изобретением подвергали серийному разведению в постоянном объеме 100% ДМСО и использовали в анализе в 9 различных концентрациях в диапазоне от 10 мкМ до 1 нМ. В данном случае обеспечивали, чтобы присутствующая концентрация ДМСО всегда составляла 0.1% ДМСО для каждой из 9 используемых концентраций. Проводили 30-минутную предварительную инкубацию ДК с соединениями в соответствии с настоящим изобретением. После этого, ДК стимулировали для продуцирования IL-23 при помощи 10 нг/мл ЛПС (Sigma, Escherichia coli серотип 0127:В8, кат. № L3129) (лиганд TLR4) и 2.5 мкг/мл лиганда TLR7/8 R848 (Invivogen, кат. № tlrl-r848-5), оба из которых приводят к активации IRAK4-опосредованного сигнального пути, в инкубаторе (37°С, 95%rH, 5% СО2) в течение 24 часов. По истечении этого времени инкубации в течение 24 часов, супернатанты собирали и анализировали с использованием коммерчески доступного hIL-23 ELISA (eBiosciences, кат. №88-7237-88), что проводили в соответствии с инструкциями изготовителя. Результаты ингибирования IL-23 в ДК человека показаны в качестве примера для примерного Соединения 12 на Фигуре 1.
In vitro TLR7/8- или TLR9-индуцированная выработка IFN-α плазмоцитоидных дендритных клеток человека (pDCs)
С помощью этого теста можно изучить в человеческих pDCs воздействие соединений в соответствии с настоящим изобретением общей формулы (I) на выработку IFN-α (интерферон-альфа), ключевого цитокина в патогенезе системной красной волчанки (Mathian et al., Arthritis Rheum, 2009; Crow M.K., Rheum Dis Clin N Am, 2010). Для этой цели, как описано выше, МКПК человека выделяли из цельной крови и плазмоцитоидные ДК (pDCs) выделяли из них при помощи коммерчески доступного набора для разделения клеток (Miltenyi Biotech, Plasmacytoid Dendritic Cell Isolation Kit II, кат. №130-097-415). Полученные таким образом pDC ресуспендировали в полной среде (RPMI 1640 + GlutaMax [Gibco, кат. №61870-010], дополненной посредством 10% FBS [Gibco, кат. №10493-106] и 50 ед. пенициллина/стрептомицина [Gibco, кат. №15140-114]) и высевали с плотностью клеток в 5×104 клеток/лунку в 96-луночный микротитрационный планшет (Costar, кат. №3599). Соединения в соответствии с настоящим изобретением подвергали серийному разбавлению в постоянном объеме 100% ДМСО и использовали в анализе в 9 различных концентрациях в диапазоне от 10 мкМ до 1 нМ. При этом было гарантировано, что присутствующая концентрация ДМСО всегда составляла 0,1% ДМСО для каждой из 9 применяемых концентраций. Проводили 30-минутную предварительную инкубацию pDC с соединениями в соответствии с настоящим изобретением. pDC стимулировали или с лигандом TLR7/8 (имиквимод, R837, Invivogen, кат. № tlrl-imq), или с лигандом TLR9 (CPG-A, ODN2216, Invivogen, кат. № tlrl-2216-1), и это приводило к активации IRAK-4-опосредованных сигнальных путей. После инкубации в течение 24 часов супернатанты клеточной культуры удаляли и анализировали с использованием коммерчески доступного человеческого IFN-α ELISA (IFNalpha Multi-Subtype ELISA Kit, pbl Assay Science, кат. №41105-1). Результаты ингибирования IFN-α в плазмоцитоидных ДК человека показаны в качестве примера для Примерного соединения 12 на Фигуре 2.
Модель in vivo воспаления, опосредованного TLR
Соединения в соответствии с настоящим изобретением общей формулы (I) исследовали в отношении их эффективности in vivo в модели in vivo опосредованного TLR воспаления. Эта механистическая модель в частности показывает потенциальное воздействие соединений общей формулы (I) на опосредованные TLR4 нарушения, так как была использована опосредованная ЛПС модель воспаления. В этой модели самки мышей NMRI (в возрасте приблизительно 6 недель; Charles River Laboratories, Германия) были распределены на группы по 5 животных в каждой. Здоровой контрольной группе давали лекарственную основу (этанол-арахисовое масло 10:90 объемн./объемн.), в которой было растворено вещество (наполнитель для вещества), а также лекарственную основу, в которой был растворен ЛПС. Наряду с группами лечения субстратом, группе положительного контроля также вводили внутрибрюшинно (в.б.) 0,2 мг в каждом случае ЛПС/кг массы тела (Sigma, кат. № L4391) (липополисахариды из Е. coli 0111:В4). Кроме того, группе положительного контроля вводили наполнитель для вещества, описанный выше. Вещество вводили перорально за 16 часов до индукции воспаления путем введения ЛПС. Чтобы исследовать воздействие соединений в соответствии с настоящим изобретением на воспаление, у животных брали конечный образец крови через 1,5 часа. Концентрацию цитокинов TNF-α и IL-6 в плазме определяли с использованием набора Mouse Pro Inflammatory 7-Plex Tissue Culture Kit (MSD, кат. № K15012B) в соответствии с инструкциями производителя. Ингибиторы IRAK4 эффективны в модели опосредованного TLR воспаления. На Фигуре 3 показано количество TNF-α в плазме крови, которое уменьшается в зависимости от дозы при введении Примерного соединения 11 в сравнении с вызванной при помощи ЛПС концентрацией.
Модель in vivo воспаления, опосредованного IL-1β
Чтобы оценить потенциальную эффективность соединений в соответствии с настоящим изобретением общей формулы (I) в IL-1β-опосредованных заболеваниях, IL-1β вводили в.б. самкам Balb/с (в возрасте приблизительно 8 недель, Charles River Laboratories, Германия) и исследовали действие соединений в соответствии с настоящим изобретением на IL-1β-опосредованную секрецию цитокинов. В каждой группе было по 5 животных. Контрольная группа получала лекарственные основы, используемые для растворения вещества и IL-1β. Группам, получавшим вещество, и группе положительного контроля в каждом случае вводили в.б. 90 мкг IL-1β/кг массы тела (R&D, кат. №401-МЛ/CF). Вещество или его лекарственную основу в группе положительного контроля вводили за 6 часов до введения IL-1β. Через 2 часа после введения IL-1β, определяли TNF-α в плазме, выделенной из крови с применением набора Mouse ProInflammatory 7-Plex Tissue Culture Kit (MSD, кат. № K15012B) в соответствии с инструкциями изготовителя. Введение IL-1β приводило к повышению концентрации TNF-α в плазме, что ингибировалось введением Примерных Соединений 11 и 12. Это представлено на Фигуре 4.
Модель in vivo вызванного адъювантом артрита
Для определения противовоспалительной активности соединений в соответствии с настоящим изобретением общей формулы (I), они были исследованы в отношении их эффективности in vivo в модели артрита. Для этой цели каждому самцу крыс Льюиса (приблизительно 100-125 г, Charles River Laboratories, Германия) в основание хвоста в день 0 вводили подкожно 100 мкл полного раствора адъюванта Фрейнда (CFA) (M. tuberculosis H37Ra [Difo Lab, кат. №-231141], растворенного в неполном адъюванте Фрейнда [Difco Lab, кат. №-263910]). Каждая группа насчитывала n=8 крыс. В исследование включали как здоровую контрольную группу, так и контрольную группу заболевания. Каждая контрольная группа получала п.о. в качестве лечения только лекарственную основу исследуемого вещества. Лечение различными дозами исследуемого вещества проводили в профилактическом порядке, т.е. начиная с 0 дня, путем перорального введения. В день 0 дополнительно определяли начальное состояние животных с точки зрения показателей активности заболевания (оценка тяжести артрита на основе системы баллов). В данном случае, баллы присваивали и суммировали в зависимости от степени воспаления суставов от 0 до 4 при наличии эритемы, включая опухание суставов (0 = нет; 1 = незначительное; 2 = умеренное; 3 = выраженное; 4 = тяжелое) для обеих задних лап. Чтобы определить противовоспалительную эффективность соединений активность заболевания животных оценивали при помощи оценки активности заболевания, начиная с 8-го дня, когда у животных сначала проявляются признаки артрита, а затем 3 раза в неделю до конца (20-й день). Статистический анализ осуществляли с использованием однофакторного дисперсионного анализа (ANOVA) и путем сравнения с контрольной группой с помощью многократного сравнительного анализа (тест Даннетта).
Подкожное введение CFA крысам приводит к острому артриту с отчетливым воспалением суставов у крыс. Этот индуцированный артрит был ингибирован путем лечения при помощи Примерного Соединения 11. Это представлено на Фигуре 5.
Модель in vivo, вызванного коллагеновым антителом артрита у мышей
Исследовали противовоспалительное воздействие соединений в соответствии с настоящим изобретением общей формулы (I) в еще одной мышиной модели артрита. Для этой цели самкам мышей Balb/c (в возрасте приблизительно 9 недель, Charles River Laboratories, Kingston, Канада) в 0 день каждой вводили внутривенно в хвостовую вену 200 мкл коктейля коллагенового антитела (10 мг/мл; ArthritoMab, MD Bioproducts) (за исключением здоровой контрольной группы, включенной в исследование). Затем на 6-й день каждая из этих мышей получала дополнительную внутрибрюшинную инъекцию 200 мкл ЛПС. Каждая группа насчитывала n=10 мышей. В исследование включали как здоровую контрольную группу, так и контрольную группу заболевания. Каждая контрольная группа получала п.о. в качестве лечения только лекарственную основу тестируемого вещества в профилактическом порядке, т.е. начиная с 0 дня. Лечение различными дозами тестируемого вещества осуществляли в профилактическом порядке, т.е. начиная с 0 дня путем перорального введения. В течение эксперимента, степень заболевания оценивали на основе системы баллов для оценки активности заболевания на всех четырех лапах. В такой системе начисления баллов за здоровую лапу баллы не начисляли, тогда как баллы с 1 [легкое воспаление, например, пальца(ев) лапы) до 4 [сильное воспаление, распространяющееся по всей лапе) начисляли в каждом случае за определенную степень воспаления суставов, возникшего от пальцев лап через плюсневый сустав к голеностопному суставу, как описано ниже:
0 = норма
1 = эритема или слабое опухание, ограниченное предплюсневым или голеностопным суставом или пальцами лапы
2 = эритема или слабое опухание, простирающееся от голеностопного сустава до плюсневой кости (2 сегмента)
3 = эритема и умеренное опухание, простирающееся от голеностопного сустава до плюсневых суставов
4 = эритема и сильное опухание, охватывающее плюсневые суставы, стопы и пальцы лап.
Для этого параметра начальное условие было определено заранее за один день до начала эксперимента (день -1), и этот показатель активности заболевания впоследствии оценивали три раза в неделю, начиная с 8 дня. Статистический анализ проводили с использованием однофакторного дисперсионного анализа (ANOVA) и путем сравнения с контрольной группой с помощью многократного сравнительного анализа (тест Даннетта).
Внутривенное введение коктейля из коллагеновых антител, включая последующее внутрибрюшинное введение ЛПС мышам приводит к острому артриту с выраженным воспалением суставов. Этот вызванный артрит был ингибирован вследствие лечения при помощи Примерного Соединения 12. Это представлено на Фигуре 6.
Мышиная модель НАСГ in vivo
Чтобы экспериментально индуцировать НАСГ; 200 мкг стрептозотоцина (STZ; Sigma-Aldrich, США) вводят подкожно каждому из 45 самцов 2-дневных мышей C57BL/6. Начиная с 4-недельного возраста, эти животные получают неограниченно рацион с высоким содержанием жиров (HFD; 57 ккал % жира, #HFD32 от CLEA, Япония). В возрасте 6 недель животных рандомизируют на 3 группы (15 животных на группу). В то время как одна из групп не получает никакого лечения, другим двум группам ежедневно в течение 4 недель вводят перорально или лекарственную основу, или исследуемое вещество. После 4-недельного лечения всех животных безболезненно умерщвляют, а печень удаляют и фиксируют для гистологического исследования в растворе Буина (Н. Denk, "Fixierung histologischer Praparate" [Фиксация гистологических препаратов], в: Р.  (ed.): "Romeis Mikroskopische Technik" [Romei's Microscopy Techniques], Urban & Schwarzenberg, Munich-Vienna-Baltimore 1989, 17-е издание, стр. 97, ISBN 3-541-11227-1). После этого образцы печени помещают в парафин и получают срезы парафина толщиной 5 мкм. Гистологические срезы каждой печени окрашивают а) для определения показателя активности НЖБП (НАСГ) с помощью гематоксилин-эозина (НС) и б) для определения фиброза печени с помощью Picro-Sirius red (Waldeck, Германия). Оценку активности НЖБП определяют в срезах гематоксилин-эозина на основе критериев, рекомендованных D.E. Kleiner et al., Hepatology 41 (2005), 1313-1321 (Таблица 1). Для гистологического количественного определения фиброзных зон делают 5 цифровых фотографий (DFC280; Leica, Германия) для каждого среза при 200-кратном увеличении микроскопа, и процент фиброза определяют с использованием программного обеспечения ImageJ (Национальный институт здравоохранения, США).
(ed.): "Romeis Mikroskopische Technik" [Romei's Microscopy Techniques], Urban & Schwarzenberg, Munich-Vienna-Baltimore 1989, 17-е издание, стр. 97, ISBN 3-541-11227-1). После этого образцы печени помещают в парафин и получают срезы парафина толщиной 5 мкм. Гистологические срезы каждой печени окрашивают а) для определения показателя активности НЖБП (НАСГ) с помощью гематоксилин-эозина (НС) и б) для определения фиброза печени с помощью Picro-Sirius red (Waldeck, Германия). Оценку активности НЖБП определяют в срезах гематоксилин-эозина на основе критериев, рекомендованных D.E. Kleiner et al., Hepatology 41 (2005), 1313-1321 (Таблица 1). Для гистологического количественного определения фиброзных зон делают 5 цифровых фотографий (DFC280; Leica, Германия) для каждого среза при 200-кратном увеличении микроскопа, и процент фиброза определяют с использованием программного обеспечения ImageJ (Национальный институт здравоохранения, США).
Мышиная модель in vivo db/db
Использовали 30 самцов мышей db/db в возрасте 8 недель. Эта модель является общепринятой моделью для ожирения, резистентности к инсулину и диабету 2 типа (Aileen JF King; The use of animal models in diabetes research; British Journal of Pharmacology 166 (2012), 877 894). Во время эксперимента животные получали неограниченно стандартный рацион питания (RM1(E) 801492, SDS) и водопроводную воду. Животных распределяли рандомизировано на 3 группы (по 10 животных в группе) и лечили перорально тестируемым веществом в течение 6 недель. В течение периода исследования у животных брали кровь в разные моменты времени (до начала лечения; через 3 недели после начала лечения и за 2 дня до окончания лечения) для определения параметров чувствительности к инсулину (например, HbA1c, содержание глюкозы, содержание инсулина). Кроме того, проводили OGTT (тест на толерантность к глюкозе) в качестве параметра для определения чувствительности к инсулину за 1 день до начала лечения и через 2 дня после окончания лечения. Кроме того, рассчитывали индекс HOMA-IR (уровень глюкозы натощак (мЕд/л) * уровень инсулина натощак (ммоль/л) / 22,5).
In vivo ксенотрансплантантная модель, ассоциированная с В-клеточной лимфомой
Изучали противоопухолевую активность соединений в соответствии с настоящим изобретением общей формулы (I) в мышиных ксенотрансплантантных моделях. Для этой цели, самкам мышей С.В-17 SCID имплантировали подкожно клеточные линии В-клеточной лимфомы человека, например, TMD-8. При среднем размере опухоли 20-30 мм2 пероральное монотерапевтическое лечение начинали соединением в соответствии с настоящим изобретением или путем введения соединения в соответствии с настоящим изобретением в комбинации со стандартной терапией, каждое из которых вводят перорально. Тем не менее, животные были рандомизированы заранее. Лечение заканчивали, как только у необработанной контрольной группы были большие опухоли. Размер опухоли и массу тела определяли три раза в неделю. Снижение массы тела является мерой токсичности, связанной с лечением (>10% = критическая, остановка в лечении до восстановления, >20% = токсическая, прекращение). Площадь опухоли определяли электронным штангенциркулем [длина (мм) × ширина (мм)]. В конце исследования также определяли массу опухоли. Противоопухолевую эффективность определяют соотношением массы опухоли, подвергавшейся лечению, по сравнению с контролем (Т/С) [масса опухоли группы, получавшей лечение в день х/масса опухоли контрольной группы в день х] или соотношением площади опухоли, подвергавшейся лечению по сравнению с контролем [площадь опухоли группы, получавшей лечение в день х/площадь опухоли контрольной группы в день x]. Соединение, имеющее Т/С выше чем 0,5 определяют как активное (эффективное). Статистический анализ осуществляют с использованием однофакторного дисперсионного анализа ANOVA и путем сравнения с контрольной группой с помощью сравнительного анализа «пары к паре» (тест Даннетта).
Анализ киназы IRAK4 собаки
Ингибирующую IRAK4 активность соединений в соответствии с настоящим изобретением в отношении IRAK4 собаки измеряли в анализе Irak4 TR-FRET (TR-FRET = флуоресцентный резонансный перенос энергии с временным разрешением), описанном ниже.
В качестве фермента использовали рекомбинантный белок слияния из N-концевого HIS (полигистидин) и Irak4 собаки, экспрессированный в инфицированных бакуловирусом клетках насекомых (Hi5, BTI-TN-5B1-4, клеточная линия, приобретенная у Invitrogen, № по каталогу В855-02) и очищенный с помощью аффинной хроматографии. Применямым для киназной реакции субстратом был биотинилированный пептид биотип-Ahx-KKARFSRFAGSSPSQASFAEPG (С-конец в амидной форме), который можно приобрести, например, у Biosyntan GmbH (Berlin-Buch).
Для анализа были приготовлены 11 разных концентраций в диапазоне от 20 мкМ до 0,073 нМ из 2 мМ раствора исследуемого вещества в ДМСО. 50 нл соответствующего раствора пипетировали в черный низкообъемный 384-луночный микротитрационный планшет (Greiner Bio-One, Frickenhausen, Германия), добавляли 2 мкл раствора Irak4 в буфере для анализа [50 мм HEPES рН 7.5, 5 мм MgCl2, 1.0 мм дитиотреитола, 30 мкм активированного ортованадата натрия, 0.1% (мае./объем.) бычьего гамма-глобулина (BGG) 0.04% (объемн./объемн.) нонидет-Р40 (Sigma)], и смесь инкубировали в течение 15 мин., чтобы обеспечить предварительное связывание веществ с ферментом до киназной реакции. Затем киназную реакцию начинали посредством добавления 3 мкл раствора аденозинтрифосфата (АТФ, 1.67 мм = конечная концентрация в 5 мкл объема анализа: 1 мм) и пептидного субстрата (0.83 мкм = конечная концентрация в 5 мкл объема анализа: 0.5 мкм) в буфер для анализа, и полученную смесь инкубировали при 22°С в течение времени реакции 45 мин.
Концентрацию Irak4 подбирали в соответствии с соответствующей активностью фермента и устанавливали так, чтобы анализ проводился в линейном диапазоне. Типичные концентрации составляли порядка 0,1 нМ. Реакцию останавливали путем добавления 5 мкл раствора реагентов для детектирования TR-FRET [0,1 мкм стрептавидин-XL665 (Cisbio Bioassays; France, № по каталогу 610SAXLG)] и 1,5 нм антифосфосеринового антитела [Merck Millipore, "STK Antibody", № по каталогу 35-002] и 0,6 нм LANCE EU-W1024-меченного антимышиного-IgG антитела (Perkin-Elmer, № продукта AD0077; в качестве альтернативы, можно использовать меченое криптатом тербия анти-мышиное антитело IgG от Cisbio Bioassays) в водном растворе EDTA (100 мм EDTA, 0,4% [мас./объем.] бычьего сывороточного альбумина [BSA] в 25 мм HEPES pH 7,5).
Полученную смесь инкубировали при 22°С в течение 1 ч., чтобы обеспечить образование комплекса биотинилированного фосфорилированного субстрата и реагентов для детектирования. Количество фосфорилированного субстрата затем оценивали путем измерения резонансной передачи энергии от меченного хелатом европия антимышиного-IgG антитела к стрептавидину-XL665. Для этой цели флуоресцентные излучения при 620 нм и 665 нм измеряли после возбуждения при 350 нм в измерительном приборе TR-FRET, например, Rubystar (BMG Labtechnologies, Offenburg, Германия) или Viewlux (Perkin-Elmer). Соотношение излучений при 665 нм и 622 нм было принято в качестве меры количества фосфорилированного субстрата. Данные были нормализованы (реакция фермента без тестируемого вещества = 0% ингибирования; все другие компоненты анализа, но без фермента = 100% ингибирования). Обычно тестируемые вещества тестировали на одних и тех же микротитрационных планшетах в 11 различных концентрациях в диапазоне от 20 мкм до 0.073 нм (20 мкм, 5.7 мкм, 1.6 мкм, 0.47 мкм, 0.13 мкм, 38 нм, 11 нм, 3.1 нм, 0.89 нм, 0.25 нм и 0.073 нм). Серии разведении готовили перед анализом (2 мм to 7.3 нм в 100% ДМСО) путем серийных разведении. Значения IC50 рассчитывали путем подбора по 4 параметрам.
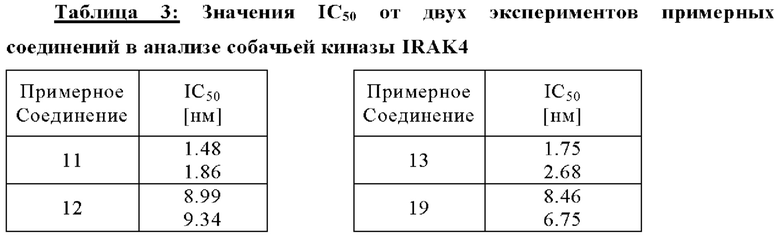
Вызванная липополисахаридом (ЛПС) in vitro выработка цитокинов мононуклеарными клетками периферической крови собак (МКПК)
Исследовали воздействие соединений в соответствии с настоящим изобретением общей формулы (I) на вызванную выработку цитокинов в МКПК собак. В данном случае выработку цитокинов вызывали при помощи ЛПС, лиганд TLR4, что приводит к активации сигнального пути, опосредованного IRAK4.
МКПК собак были получены из антикоагулированной цельной крови собаки. Для этой цели обогащенная лейкоцитами собаки плазма была приготовлена из 15 мл крови собаки путем центрифугирования при 400g в течение 15 мин. при 4°С, с последующим сбором и затем суспендированием лейкоцитарной оболочки МКПК собак в плазме. Семь (7) мл Ficoll-Paque Plus (Fischer Scientific, кат. №11778538) пипетировали в пробирку для центрифугирования и 7 мл обогащенной лейкоцитами собаки плазмы затем наслаивали поверх Ficoll-Paque Plus. После центрифугирования пробирки при 400 g в течение 20 мин. при 4°С, МКПК собаки собирали с поверхности раздела плазмы собак и Ficoll-Paque Plus. МКПК переносили в новую пробирку для центрифугирования и дополняли сбалансированным солевым раствором Хэнкса 1× (HBSS) без Ca2+/Mg2+ (Sigma-Aldrich, кат. № Н9394). Суспензию клеток центрифугировали при 400g в течение 5 мин. при 4°С и супернатант отбрасывали. Затем клеточный осадок повторно суспендировали в 0,2% гипотоническом солевом растворе, чтобы лизировать любые оставшиеся эритроциты. Через 30 секунд суспензию клеток делали изотонической и центрифугировали при 400 g в течение 5 мин. при 4°С. Затем клеточный осадок ресуспендировали в HBSS без Ca2+/Mg2+ для окончательной промывки и центрифугировали при 400 g в течение 5 мин. при 4°С. Затем МКПК ресуспендировали в полной среде (RPMI 1640 с GlutaMAX (Sigma-Aldrich, кат. № R0883), 10% FCS; 50 ед./мл пенициллина, 50 мкг/мл стрептомицина (Sigma-Aldrich, кат. № Р4333)).
Анализ также осуществляли в полной среде. МКПК высевали в 96-луночные планшеты при плотности клеток 2,5×105 клеток/лунку. Соединения в соответствии с настоящим изобретением растворяли в ДМСО и подвергали серийному разбавлению в полной среде. Примерные соединения использовали в анализе в 8 различных концентрациях в диапазоне от 3 нМ до 10 мкМ, так что конечная концентрация ДМСО составляла 0,0003-0,4%. Для индукции секреции цитокинов клетки стимулировали посредством 0,1 мкг/мл ЛПС (Sigma-Aldrich, Escherichia coli 0111:B4, кат. № L3024) в течение 24 часов. Жизнеспособность клеток определяли с использованием 0,2% трипанового синего (Sigma-Aldrich, кат. № Т8154). Количество секретируемого TNF-α в супернатанте клеточной культуры определяли с использованием собачьего TNFα DuoSet Elisa (R&D Systems, кат. № DY1507) в соответствии с инструкциями производителя. В качестве примера, Примерное соединение 12 ингибировало выработку TNFα собачьими МКПК, стимулированными посредством ЛПС. Это представлено на Фигуре 7.
In vitro вызванная липополисахаридом (ЛПС) выработка цитокинов бычьими мононуклеарными клетками периферической крови (МКПК)
Изучали воздействие соединений в соответствии с настоящим изобретением общей формулы (I) на вызванную выработку цитокинов в бычьих МКПК. В данном случае выработку цитокинов вызывали посредством ЛПС, лиганд TLR4, что приводит к активации сигнального пути, опосредованного IRAK4.
Бычьи МКПК были получены из антикоагулированной цельной крови крупного рогатого скота. Для этой цели обогащенную бычьими лейкоцитами плазму готовили из 500 мл крови крупного рогатого скота центрифугированием при 1000 g в течение 20 мин. при комнатной температуре (КТ), с последующим сбором и затем суспендированием бычьей лейкоцитарной пленки МКПК в равном объеме PBS/5 мм EDTA (КТ). Тридцать (30) мл Ficoll-Paque Plus (Fischer Scientific, кат. №11778538) пипетировали в пробирку Leucosep и 30 мл смеси бычья лейкоцитарная пленка МКПК/PBS/EDTA затем наслаивали поверх Ficoll-Paque Plus. После центрифугирования пробирки при 800 g в течение 25 мин. при RT, бычьи МКПК собирали с поверхности раздела бычьей плазмы и Ficoll-Paque Plus. МКПК переносили в новую пробирку для центрифугирования и добавляли холодным PBS/ 5 мм EDTA. Суспензию клеток центрифугировали при 350 g в течение 10 мин. при 4°С и супернатант отбрасывали. Затем клеточный осадок повторно суспендировали в 0,2% гипотоническом солевом растворе, чтобы лизировать любые оставшиеся эритроциты. Через 30 секунд суспензию клеток делали изотонической и центрифугировали при 500 g в течение 5 мин. при 4°С. Затем осадок клеток МКПК ресуспендировали в полной среде (DMEM с GlutaMAX (ThermoFisher, кат. №32430100), 10% лошадиной сыворотки (АТСС® 30-2040™), 20 мкм β- меркаптоэтаноле (ThermoFisher кат. №31350010 [исходный раствор: 50 мм]).
Анализ также осуществляли в полной среде. МКПК высевали в 24-луночные планшеты при плотности клеток 1×106 клеток/лунку. Соединения в соответствии с настоящим изобретением растворяли в ДМСО и подвергали серийному разбавлению в полной среде. Примерные соединения использовали в анализе в 8 различных концентрациях в диапазоне от 0,003 мкМ до 10 мкМ, так что конечная концентрация ДМСО составляла 0,5%. Для индукции секреции цитокинов клетки стимулировали посредством 1 мкг/мл (Фиг. 8) и 0,1 мкг/мл ЛПС (Фиг. 9) (ЛПС из Е. coli K12; Invivogen # tlrl-eklps) в течение 24 часов. Жизнеспособность клеток определяли с использованием раствора Тюрка (Merck Millipore # 1092770100).
Количество секретируемого TNFα в супернатанте клеточной культуры подвергшихся воздействию ЛПС бычьих МКПК определяли с использованием кроличьего антибычьего антитела TNFα на основе считывания данных ELISA. Анализ ELISA проводили в 384-луночных планшетах ELISA, которые были покрыты 5 мкг/мл кроличьего антибычьего антитела TNFα (BioRad, AHP2383) в 50 мм Na2CO3/NaHCO3 pH 9,6 буфера в 10 мкл/лунку в течение ночи при 4°С. После удаления антитела и промывки лунок три раза посредством 50 мкл промывочного буфера (PBS, 0,05% (объемн./объемн.) Tween 20), лунки инкубировали в течение 90 мин. при 37°С с 40 мкл блокирующего буфера (PBS, 0,05% (объемн./объемн.) Tween 20, 1% (мас./объем.) бычьего сывороточного альбумина). После этого блокирующий буфер удаляли и добавляли образцы культурального супернатанта (20 мкл/лунку). После инкубации в течение 90 мин. при 37°С, образцы удаляли, и лунки три раза промывали посредством 50 мкл промывочного буфера. 1 мкг/мл конъюгированного с биотином кроличьего антибычьего антитела TNFα (BioRad, AHP2383B) в блокирующем буфере добавляли в планшеты (20 мкл/лунку), которые инкубировали в течение 60 мин. при 37°С. После удаления биотинилированного антитела и промывания лунок три раза посредством 50 мкл промывочного буфера, 20 мкл/лунку ExtrAvidin™-щелочной фосфатазы (Sigma, E2636), 1:10.000 разведенной в блокирующем буфере, добавляли в течение 1 ч. при 37°С. После удаления ExtrAvidin™-щелочной фосфатазы и промывки лунок три раза посредством 50 мкл промывочного буфера, инициировали ферментативную реакцию/проявление цвета путем добавления 50 мкл/лунку буфера для проявления (5 мм пара-нитрофенилфосфат (pNPP) в 50 мм Na2CO3/NaHCO3 pH 9.6, 2 мм MgCl2). Оптическую плотность регистрировали при длине волны 405 нм. Для кинетических измерений точки данных регистрировали каждые 5 минут в течение 1 часа, измерения конечных точек проводили через 2 часа. В качестве примера, Примерное соединение 12 ингибировало выработку TNFα бычьими МКПК, стимулированными посредством ЛПС. Это представлено на Фигурах 8 и 9.
In vitro вызванная липополисахаридом (ЛПС) выработка цитокинов мононуклеарными клетками периферической крови свиней (МКПК)
В качестве еще одного примера, было исследовано воздействие соединений в соответствии с настоящим изобретением общей формулы (I) на вызванную выработку цитокинов в МКПК свиней. В данном случае выработку цитокинов индуцировали посредством ЛПС, лиганд TLR4, что приводит к активации сигнального пути, опосредованного IRAK4.
МКПК свиней были получены из антикоагулированной цельной крови свиней. Для этой цели обогащенную лейкоцитами свиней плазму готовили из 36 мл свиной крови центрифугированием при 1000 g в течение 20 мин. при комнатной температуре (КТ), с последующим сбором, а затем суспендированием лейкоцитарной пленки МКПК свиней в равном объеме PBS/5 мм EDTA (КТ). Тридцать (30) мл Ficoll-Paque Plus (Fischer Scientific, кат. №11778538) пипетировали в пробирку Leucosep и 30 мл смеси лейкоцитарная пленка МКПК свиней/PBS/EDTA затем наслаивали поверх Ficoll-Paque Plus. После центрифугирования пробирки при 800 g в течение 25 мин. при КТ, МКПК свиней собирали с поверхности раздела свиной плазмы и Ficoll-Paque Plus. МКПК переносили в новую пробирку для центрифугирования и дополняли посредством холодного PBS/ 5 мм EDTA. Суспензию клеток центрифугировали при 350 g в течение 10 мин. при 4°С и супернатант отбрасывали. Затем клеточный осадок повторно суспендировали в 0,2% гипотоническом солевом растворе, чтобы лизировать любые оставшиеся эритроциты. Через 30 секунд суспензию клеток делали изотонической и центрифугировали при 500 g в течение 5 мин. при 4°С. Затем осадок клеток МКПК ресуспендировали в полной среде (DMEM с GlutaMAX (ThermoFisher, кат. №32430100). 10% лошадиной сыворотке (АТСС® 30-2040™), 20 мкм β-меркаптоэтанола (ThermoFisher кат. №31350010 [исходный раствор: 50 мм]).
Анализ также проводили в полной среде. МКПК высевали в 24-луночные планшеты при плотности клеток 1×106 клеток/лунку. Соединения в соответствии с настоящим изобретением растворяли в ДМСО и подвергали серийному разбавлению в полной среде. Примерные соединения использовали в анализе в 8 различных концентрациях в диапазоне от 0,003 мкМ до 10 мкМ, так что конечная концентрация ДМСО составляла 0,5%. Для индукции секреции цитокинов клетки стимулировали посредством ЛПС (ЛПС из Е. coli K12; Invivogen # tlrl-eklps) в диапазоне концентраций от 0,01 до 1 нг/мл в течение 24 часов. Жизнеспособность клеток определяли с использованием раствора Тюрка (Merck Millipore # 1092770100).
Количество секретируемого TNFα в супернатанте клеточной культуры подвергшихся воздействию ЛПС МКПК свиней определяли с использованием кроличьего антисвиного антитела TNFα на основе считывания данных ELISA. Анализ ELISA проводили в 384-луночных планшетах ELISA, которые были покрыты 3 мкг/мл кроличьего антисвиного антитела TNFα (BioRad, AHP2397) в 50 мм Na2CO3/NaHCO3 pH 9,6 буфера в 10 мкл/лунку в течение 48 ч. при 4°С. После удаления антитела и промывки лунок три раза посредством 50 мкл промывочного буфера (PBS, 0,05% (объемн./объемн.) Tween 20), лунки инкубировали в течение 60 мин. при 37°С с 50 мкл блокирующего буфера (PBS, 0,05% (объемн./объемн.) Tween 20, 1% (мас./объем.) бычьего сывороточного альбумина). После этого блокирующий буфер удаляли и добавляли образцы культурального супернатанта (20 мкл/лунку). После инкубации в течение 90 мин. при 37°С, образцы удаляли, и лунки три раза промывали посредством 50 мкл промывочного буфера. 0,25 мкг/мл конъюгированного с биотином кроличьего антисвиного антитела TNFα (BioRad, AHP2397B) в блокирующем буфере добавляли в планшеты (20 мкл/лунку), которые инкубировали в течение 60 мин. при 37°С. После удаления биотинилированного антитела и промывания лунок три раза посредством 50 мкл промывочного буфера, 20 мкл/лунку ExtrAvidin™-щелочной фосфатазы (Sigma, E2636), 1:10.000 разведенной в блокирующем буфере, добавляли в течение 1 ч. при 37°С. После удаления ExtrAvidin™-щелочной фосфатазы и промывки лунок три раза посредством 50 мкл промывочного буфера, инициировали ферментативную реакцию/проявление цвета путем добавления 50 мкл/лунку буфера для проявления (5 мм пара-нитрофенилфосфат (pNPP) в 50 мм Na2CO3/NaHCO3 рН 9.6, 2 мм MgCl2). Оптическую плотность регистрировали при длине волны 405 нм. Для кинетических измерений точки данных регистрировали каждые 5 минут в течение 1 часа, измерения конечных точек проводили через 2 часа. В качестве примера, при 10 мкм Примерное соединение 12 ингибировало выработку TNFα бычьими МКПК, стимулированными посредством 0,1 нг/мл ЛПС. Это представлено на Фигуре 10.
Модель in vivo вызванного домашним пылевым клещом аллергического дерматита у собак
Чтобы оценить потенциальную противоаллергическую/противовоспалительную эффективность соединений в соответствии с настоящим изобретением общей формулы (I) использовали модель собак породы бигль, сенсибилизированных домашним пылевым клещом (HDM). При этом сенсибилизация HDM состояла из серии подкожных инъекций антигена HDM (10 мкг, Greer Laboratories, Lenoir, NC, США) и Alhydrogel® (0.2 мл, InvivoGen, San Diego, CA 921221, США) в качестве адъюванта с временными интервалами примерно две недели. Процесс сенсибилизации контролировали и подтверждали внутрикожным тестированием кожи. После того, как собаки дали положительный результат на внутрикожное тестирование кожи HDM, через месяц после последней сенсибилизации местно наносили антиген (135 мкг) и вкалывали в кожу (посредством микроиголок длиной 2 мм) взрослых собак породы бигль во внутреннюю часть задних лап и исследовали воздействие соединений в соответствии с настоящим изобретением на признаки аллергического дерматита, например, эритема и отек. Было 2 группы по 4 животных в каждой: 1 1 контрольная группа плацебо и 1 группа, получавшая Примерное соединение 12. Контрольной группе вводили перорально желатиновые капсулы, содержащие микроцеллюлозу, в то время как группе, которую лечили при помощи Примерного соединения 12 вводили перорально желатиновые капсулы, содержащие Примерное соединение 12 и микроцеллюлозу. Введение Примерного соединения 12 или плацебо начинали за 5 дней до заражения антигеном HDM и продолжали до 2 дней после заражения. Частота введения составляла один раз в день, с дозой 10 мг/кг массы тела в случае Примерного соединения 12. Начиная с 30 мин после заражения и в разные моменты времени в течение 48 ч., эритему и отек оценивали с использованием VAS (Visual Analogue Scale) в 2 группах. Образцы плазмы были проанализированы для определения воздействия соединения в связи с клиническими оценками. Отек и эритема были значительно уменьшены после лечения при помощи примерного Соединения 12. Это представлено в Таблицах 4 и 5, и на Фигурах 11 и 12.


Модель зуда in vivo аллергического блошиного дерматита
Для оценки потенциального противозудного эффекта соединений в соответствии с настоящим изобретением общей формулы (I) использовали модель аллергического блошиного дерматита (АБД). В исследование были включены только взрослые собаки с историей АБД. Жизненная фаза исследования состояла из двух фаз: фазы индукции зуда (2 недели) с последующей фазой лечения (2 недели). Собак заражали блохами Ctenocephalides (первое контрольное заражение посредством 100 блох/собаку, все последующие заражения посредством 30 блох/собаку) два раза в неделю в течение обеих фаз исследования. Было 2 группы по 12 животных в каждой: 1 контрольная группа плацебо и 1 группа, которую лечили при помощи Примерного Соединения 12. Контрольной группе вводили перорально желатиновые капсулы, содержащие микроцеллюлозу, тогда как группе, которую лечили при помощи Примерного Соединения 12, перорально вводили желатиновые капсулы, содержащие Примерное соединение 12 и микроцеллюлозу. Частота введения составляла один раз в сутки, с дозой 20 мг/кг массы тела в случае Примерного соединения 12. Начиная с 1 дня после лечения и каждый третий день, собак записывали в течение 4 часов и время, проведенное в состоянии зуда, определяли как секунды, потраченные на царапанье, лизание, кусание. Образцы плазмы были проанализированы для определения воздействия соединения в связи с клиническими оценками. Зуд значительно уменьшился после 10 дней лечения при помощи Примерного Соединения 12. Это представлено в Таблице 6 и Фигуре 13.
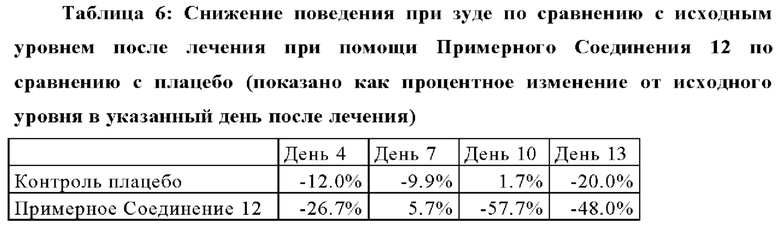
Фигура 1: Ингибирование IL-23 в человеческих, образованных моноцитами ДК для Примерного соединения 12. Данные представлены в виде средних значений со стандартными отклонениями.
Фигура 2: Ингибирование INF-α в стимулированных (А) имиквимодом (R837)- или (В) CpG-A-стимулированных человеческих плазмоцитоидных ДК для Примерного соединения 12. Данные представлены в виде средних значений со стандартными отклонениями.
Фигура 3: Лечение вызванного посредством ЛПС воспаления при помощи Примерного Соединения 11 приводит к сниженному количеству секретируемого TNF-α. Данные представлены в виде средних значений со стандартными отклонениями.
Фигура 4: Лечение вызванного посредством IL-1β воспаления при помощи Примерных Соединений 11 (слева) и 12 (справа) приводит к дозозависимому уменьшению количества секретируемого TNF-α. Данные представлены в виде средних значений со стандартными отклонениями.
Фигура 5: Противовоспалительные воздействия Примерного соединения 11 в животной модели ревматоидного артрита (вызванная адъювантом крысиная модель). Значительное и дозозависимое ингибирование ревматического воспаления суставов, измеренное на основании оценки активности заболевания. Данные соответствуют средним значениям + стандартные отклонения. Однофакторный дисперсионный анализ ANOVA с последующим многократным сравнительным анализом с CFA контрольной группой при помощи теста Даннетта; *р<0.05; **р<0.01; ***p<0.001; ****р<0.0001.
Фигура 6: Противовоспалительные воздействия Примерного соединения 12 в животной модели ревматоидного артрита (мышиная модель, индуцированная коллагеновыми антителами). Значительное и дозозависимое ингибирование ревматического воспаления суставов, измеренное на основании оценки активности заболевания. Данные соответствуют средним значениям + стандартные отклонения. Статистические значения между контролем коллагеновых антител (AK) и терапевтическими группами подсчитывали при помощи однофакторного дисперсионного анализа ANOVA с последующим многократным сравнительным анализом (тест Даннетта) (*р<0.05; **р<0.01; ***p<0.001; ****р<0.0001).
Фигура 7: Ингибирование вызванной посредством ЛПС выработки TNFα собачьими МКПК для Примерного соединения 12. Данные представлены в виде средних значений со стандартными отклонениями.
Фигура 8: Дозозависимое ингибирование посредством Примерного соединения 12 выработки TNFα бычьими МКПК, вызванной посредством 1 мкг/мл ЛПС (кинетическое измерение). Данные показывают средние значения со стандартными отклонениями биологических трипликатов, каждый из которых измеряется в двух экземплярах. Значение IC50, определенное из этой кривой составляет 120 нм.
Фигура 9: Дозозависимое ингибирование посредством Примерного соединения 12 выработки TNFα бычьими МКПК, вызванной посредством 0,1 мкг/мл ЛПС (кинетическое измерение). Данные показывают средние значения со стандартными отклонениями биологических трипликатов, каждый из которых измеряется в двух экземплярах. Значение IC50, определенное из этой кривой составляет 70,5 нм.
Фигура 10: Ингибирование посредством 10 мкм Примерного соединения 12 выработки TNFα свиными МКПК, вызванной при помощи 0,1 нг/мл ЛПС (кинетическое измерение). Данные показывают средние значения со стандартными отклонениями биологических трипликатов, каждый из которых измеряется в двух экземплярах.
Фигура 11: Лечение модели аллергического дерматита у собак, вызванного домашним пылевым клещом, при помощи Примерного Соединения 12 приводит к уменьшению эритемы (а). Данные представлены в виде средних значений со стандартными отклонениями.
Фигура 12: Лечение модели аллергического дерматита у собак, вызванного домашним пылевым клещом, при помощи Примерного Соединения 12 приводит к уменьшению отека (б). Данные представлены в виде средних значений со стандартными отклонениями.
Фигура 13: Противозудный эффект Примерного соединения 12 в животной модели аллергического блошиного дерматита. Данные выражены в процентах изменения от базовой линии, соответствуя срединным значениям.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ MAS-СВЯЗАННОГО РЕЦЕПТОРА G-БЕЛКА X2 И СВЯЗАННЫЕ С НИМИ ПРОДУКТЫ И СПОСОБЫ | 2021 |
|
RU2841536C1 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2797295C1 |
| 5-ЗАМЕЩЕННЫЕ ИНДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2487873C2 |
| ТЕРПИРИДИН-ДИКЕТОНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2022 |
|
RU2825875C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
| СОЕДИНЕНИЕ, НАЦЕЛЕННОЕ НА БЕЛОК И ЕГО ДЕГРАДАЦИЮ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2829459C1 |
| Бициклические конденсированные гетероарильные или арильные соединения в качестве модуляторов IRAK4 | 2016 |
|
RU2684324C1 |
| ЗАМЕЩЕННЫЕ N-(1H-ИНДАЗОЛ-4-ИЛ) ИМИДАЗОЛ [1,2-А]ПИРИДИН-3- КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ III ТИПА | 2011 |
|
RU2591195C2 |
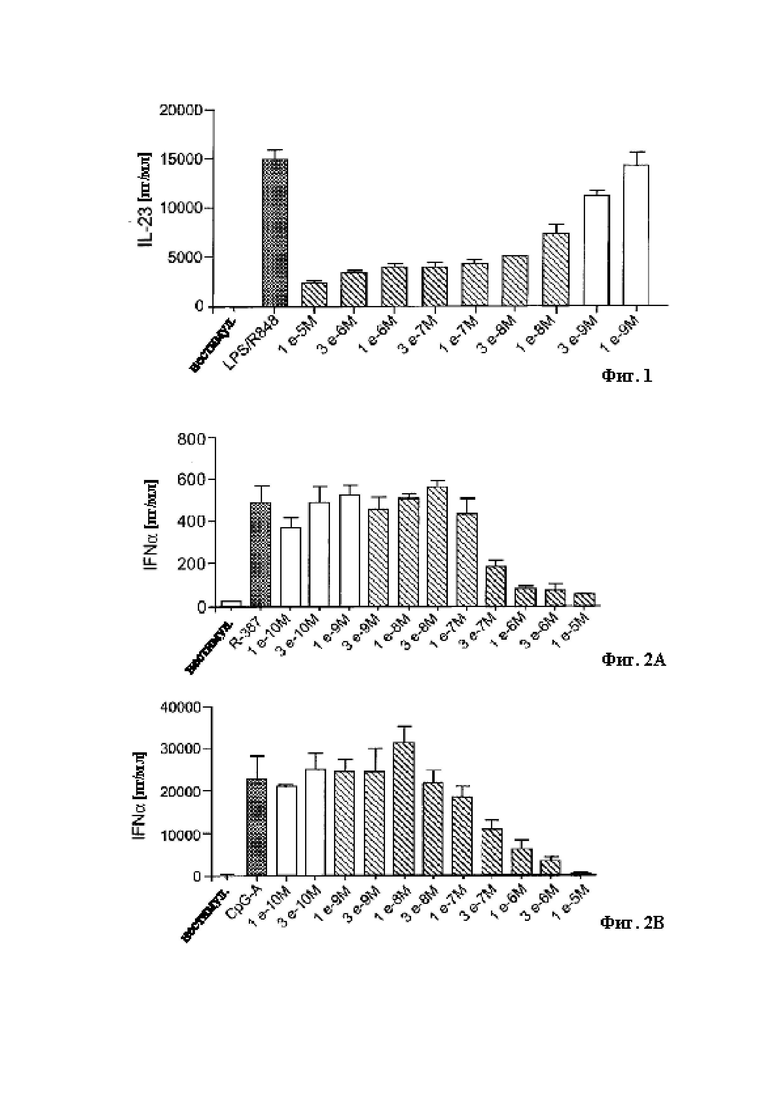

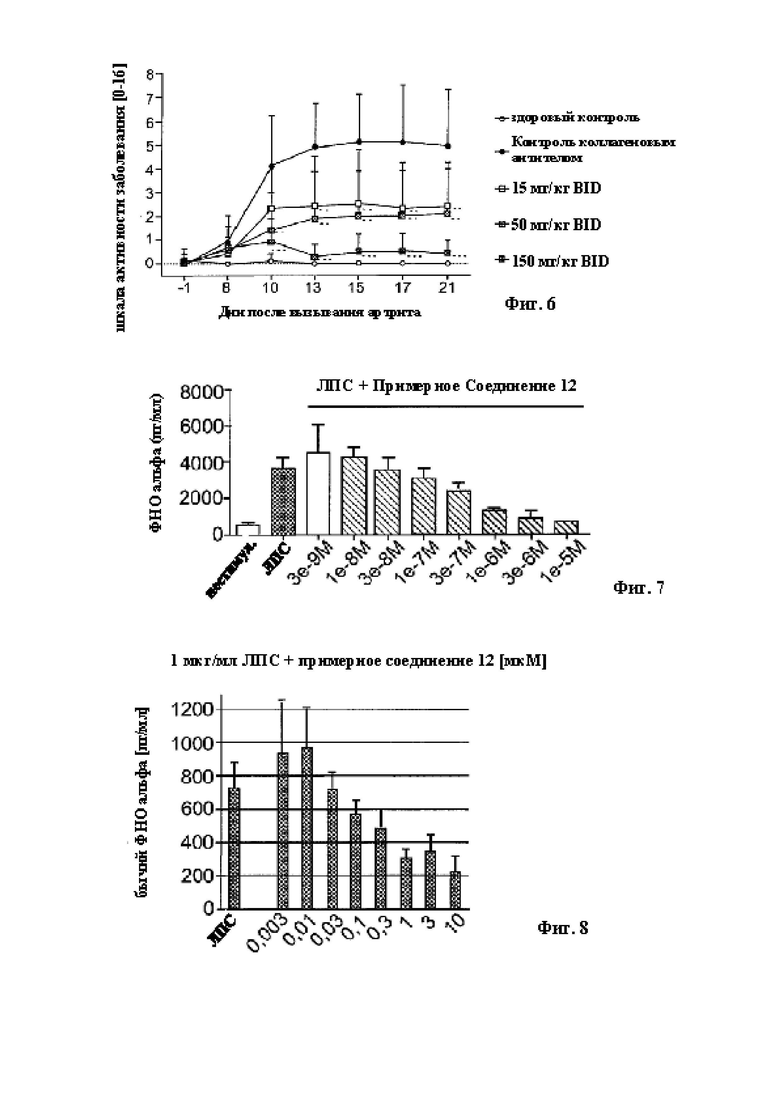

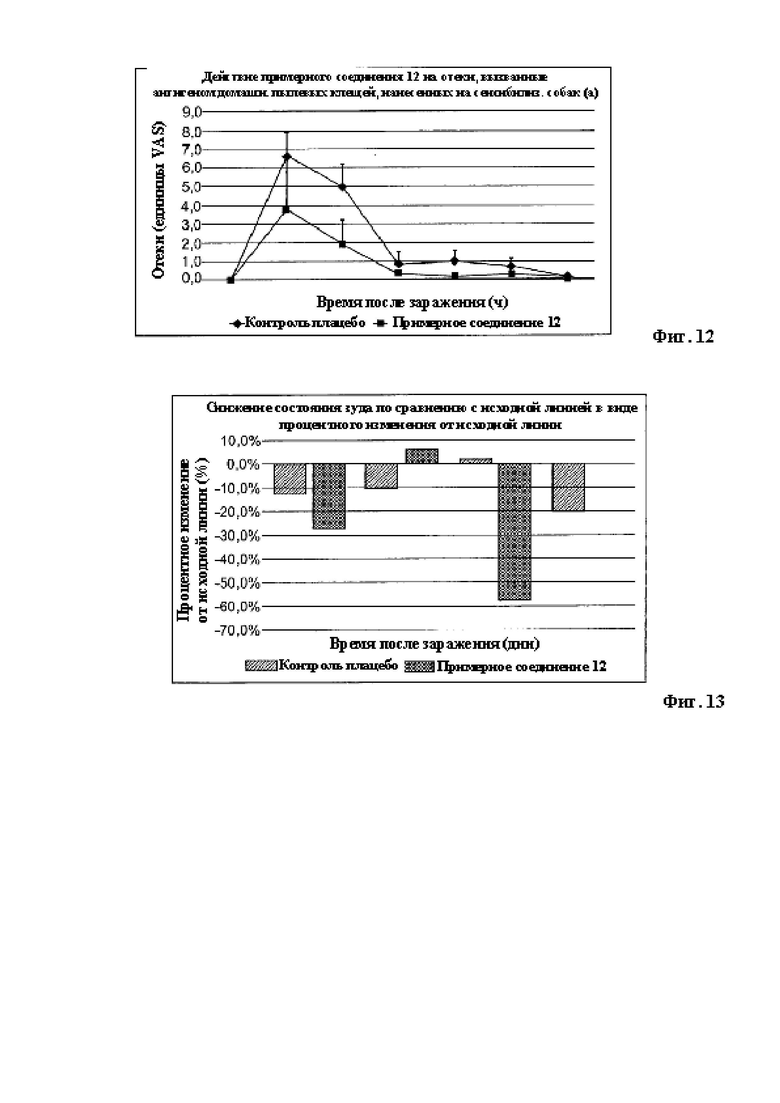
Изобретение относится к применению соединения формулы (I) или его диастереомера, энантиомера, соли, сольвата или сольвата солей, где R1 представляет собой C1-C6-алкил, где C1-C6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена, гидроксила, незамещенного или моно- или поли-галоген-замещенного С3-С6-циклоалкила, или группы R6, R7SO2, R7 SO или R8O, или группу, выбранную из:  , R2 и R3 всегда имеют одно и то же определение и оба представляют собой или водород, или C1-С6-алкил; R4 представляет собой галоген, циано, незамещенный или однократно или многократно одинаково или по-разному замещенный C1-С6-алкил или незамещенный или однократно или многократно одинаково или по-разному замещенный С3-С6-циклоалкил, и заместители выбраны из группы галогена и гидроксила; R5 представляет собой водород, галоген или незамещенный или моно- или поли-галоген-замещенный C1-С6-алкил; R6 представляет собой незамещенный или моно- или ди-метил-замещенный моноциклический насыщенный гетероцикл с 4-6 кольцевыми атомами, который содержит гетероатом или гетерогруппу из группы О, S, SO и SO2; R7 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена, гидроксила или С3-С6-циклоалкила; или R7 представляет собой С3-С6-циклоалкил; R8 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена. Изобретение также относится к лекарственному средству, содержащему соединение формулы (I), которое ингибирует ассоциированную с рецептором интерлейкин-1 киназу 4 (IRAK4). Технический результат - найдено новое применение соединения формулы (I), которое может использоваться в ветеринарии для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у животных. 5 н. и 22 з.п. ф-лы, 13 ил., 6 табл., 22 пр.
, R2 и R3 всегда имеют одно и то же определение и оба представляют собой или водород, или C1-С6-алкил; R4 представляет собой галоген, циано, незамещенный или однократно или многократно одинаково или по-разному замещенный C1-С6-алкил или незамещенный или однократно или многократно одинаково или по-разному замещенный С3-С6-циклоалкил, и заместители выбраны из группы галогена и гидроксила; R5 представляет собой водород, галоген или незамещенный или моно- или поли-галоген-замещенный C1-С6-алкил; R6 представляет собой незамещенный или моно- или ди-метил-замещенный моноциклический насыщенный гетероцикл с 4-6 кольцевыми атомами, который содержит гетероатом или гетерогруппу из группы О, S, SO и SO2; R7 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена, гидроксила или С3-С6-циклоалкила; или R7 представляет собой С3-С6-циклоалкил; R8 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена. Изобретение также относится к лекарственному средству, содержащему соединение формулы (I), которое ингибирует ассоциированную с рецептором интерлейкин-1 киназу 4 (IRAK4). Технический результат - найдено новое применение соединения формулы (I), которое может использоваться в ветеринарии для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у животных. 5 н. и 22 з.п. ф-лы, 13 ил., 6 табл., 22 пр.

1. Применение соединений общей формулы (I)
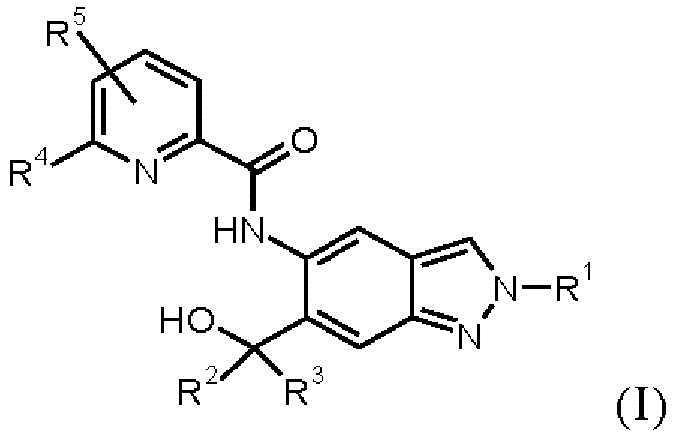
в которой:
R1 представляет собой C1-C6-алкил, где C1-C6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена, гидроксила, незамещенного или моно- или поли-галоген-замещенного С3-С6-циклоалкила, или группы R6, R7SO2, R7SO или R8O,
или группу, выбранную из:

R2 и R3 всегда имеют одно и то же определение и оба представляют собой или водород, или C1-С6-алкил;
R4 представляет собой галоген, циано, незамещенный или однократно или многократно одинаково или по-разному замещенный C1-С6-алкил или незамещенный или однократно или многократно одинаково или по-разному замещенный С3-С6-циклоалкил, и заместители выбраны из группы галогена и гидроксила;
R5 представляет собой водород, галоген или незамещенный или моно- или поли-галоген-замещенный C1-С6-алкил;
R6 представляет собой незамещенный или моно- или ди-метил-замещенный моноциклический насыщенный гетероцикл с 4-6 кольцевыми атомами, который содержит гетероатом или гетерогруппу из группы О, S, SO и SO2;
R7 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена, гидроксила или С3-С6-циклоалкила;
или R7 представляет собой С3-С6-циклоалкил;
R8 представляет собой С1-С6-алкил, где С1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена;
и их диастереомеров, энантиомеров, солей, сольватов или сольватов солей,
для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у животных.
2. Применение соединений по п. 1, в которых
R1 представляет собой C1-С6-алкил, где C1-С6-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством фтора, гидроксила или группы R6, R7SO2, R7SO или R8O;
R2 и R3 всегда имеют одно и то же определение и оба являются или водородом, или C1-С3-алкилом;
R4 представляет собой галоген, циано или C1-С3-алкил, где С1-С3-алкильная группа является незамещенной или моно- или полизамещенной одинаково или по-разному посредством галогена или гидроксила;
R5 представляет собой водород, фтор, хлор или C1-С3-алкил;
R6 представляет собой оксетанил или тетрагидрофуранил;
R7 представляет собой С1-С4-алкил, где С1-С4-алкильная группа является незамещенной или монозамещенной гидроксилом или циклопропилом или замещенной тремя атомами фтора;
R8 представляет собой незамещенную С1-С4-алкильную группу или три-фтор-замещенную С1-С4-алкильную группу.
3. Применение соединений по п. 1 или 2, в которых R4 представляет собой дифторметил, трифторметил или метил.
4. Применение соединений по пп. 1, 2 или 3, в которых R5 представляет собой водород или фтор.
5. Применение соединений по пп. 1-3 или 4, в которых R2 и R3 оба представляют собой водород или метил.
6. Применение соединений по п. 2, в которых
R1 представляет собой С2-С6-алкил, где С2-С6-алкильная группа является незамещенной, или С2-С6-алкильная группа является моно-, ди- или три-фтор-замещенной, или С2-С6-алкильная группа является монозамещенной посредством гидроксила, R6, R7 SO2 или R8O,
или R1 представляет собой оксетанил-замещенную C1-С3-алкильную группу;
R2 и R3 всегда имеют одно и то же определение и оба являются или водородом, или метилом;
R4 представляет собой незамещенную или моно- или поли-галоген-замещенную C1-С3-алкильную группу или C1-С3-алкильную группу, замещенную одной гидроксильной группой, или C1-С3-алкильную группу, замещенную одной гидроксильной группой и тремя атомами фтора;
R5 представляет собой водород, фтор или C1-С3-алкил;
R7 представляет собой C1-С3-алкил;
R8 представляет собой С1-С4-алкил, где С1-С4-алкильная группа является незамещенной или моно-, ди- или три-фтор-замещенной.
7. Применение соединений по п. 6, в которых
R1 представляет собой С2-С5-алкильную группу, замещенную гидроксилом или C1-С3-алкокси или трифторметокси или 2,2,2-трифторэтокси или трифторметилом, или представляет собой метил-SO2-замещенную С2-С4-алкильную группу, или представляет собой оксетан-3-ил-замещенную С1-С2-алкильную группу;
R2 и R3 всегда имеют одно и то же определение и оба представляют собой водород или метил;
R4 представляет собой метил, этил, трифтор-С1-С3-алкил, дифтор-C1-С3-алкил, гидроксиметил, 1-гидроксиэтил, 2-гидроксипропан-2-ил и 2,2,2-трифтор-1-гидроксиэтил;
R5 представляет собой водород, фтор или метил.
8. Применение соединений по п. 7, в которых
R1 представляет собой 4,4,4-трифторбутил, 3-гидрокси-3-метилбутил, 3-гидроксибутил, 3-метоксипропил, 3-гидроксипропил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-трифторметоксипропил, 2-метоксиэтил, 2-гидроксиэтил, 2-(метилсульфонил)этил или 3-(метилсульфонил)пропил;
R2 и R3 оба представляют собой метил или водород;
R4 представляет собой дифторметил, трифторметил или метил;
R5 представляет собой водород или фтор.
9. Применение соединений по п. 8, в которых
R1 представляет собой 3-гидрокси-3-метилбутил, 3-гидроксибутил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-(метилсульфонил)-пропил или 2-(метилсульфонил)этил;
R2 и R3 оба представляют собой метил;
R4 представляет собой дифторметил или трифторметил;
R5 представляет собой водород.
10. Применение соединений по п. 8, в которых
R1 представляет собой 3-гидрокси-3-метилбутил, 3-гидроксибутил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-(метилсульфонил)-пропил или 2-(метилсульфонил)этил;
R2 и R3 оба представляют собой метил;
R4 представляет собой метил;
R5 представляет собой фтор, где R5 находится в орто-положении к R4.
11. Применение соединений по пп. 1-10, которые представляют собой:
1) N-[6-(2-Гидроксипропан-2-ил)-2-(2-метоксиэтил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
2) N-[6-(Гидроксиметил)-2-(2-метоксиэтил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
3) N-[6-(2-Гидроксипропан-2-ил)-2-(3-метоксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
4) N-[6-(Гидроксиметил)-2-(3-метоксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
5) N-[2-(2-Гидроксиэтил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
6) N-[6-(2-Гидроксипропан-2-ил)-2-(3-гидроксипропил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
7) N-[2-(2-Гидроксиэтил)-6-(гидроксиметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
8) N-[6-(2-Гидроксипропан-2-ил)-2-(оксетан-3-илметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
9) N-[6-(Гидроксиметил)-2-(оксетан-3-илметил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
10) N-{6-(2-Гидроксипропан-2-ил)-2-[3-(метилсульфонил)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид;
11) N-[2-(3-Гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
12) N-{6-(2-Гидроксипропан-2-ил)-2-[2-(метилсульфонил)этил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид;
13) 6-(Дифторметил)-N-[2-(3-гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]пиридин-2-карбоксамид;
14) 6-(Дифторметил)-N-{6-(2-гидроксипропан-2-ил)-2-[2-(метилсульфонил)этил]-2Н-индазол-5-ил}пиридин-2-карбоксамид;
15) 6-(Дифторметил)-N-[6-(2-гидроксипропан-2-ил)-2-(3-гидроксипропил)-2Н-индазол-5-ил]пиридин-2-карбоксамид;
16) N-[6-(2-Гидроксипропан-2-ил)-2-(4,4,4-трифторбутил)-2Н-индазол-5-ил]-6-(трифторметил)пиридин-2-карбоксамид;
17) N-{6-(2-Гидроксипропан-2-ил)-2-[3-(трифторметокси)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид;
18) N-{6-(2-Гидроксипропан-2-ил)-2-[3-(2,2,2-трифторэтокси)пропил]-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид;
19) 5-Фтор-N-[2-(3-гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-метилпиридин-2-карбоксамид;
20) N-[2-(3-Гидрокси-3-метилбутил)-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил]-6-метилпиридин-2-карбоксамид;
21) 6-(2-Гидроксипропан-2-ил)-N-[6-(2-гидроксипропан-2-ил)-2-(4,4,4-трифторбутил)-2Н-индазол-5-ил]пиридин-2-карбоксамид;
22) N-{2-[2-(1-Гидроксициклопропил)этил]-6-(2-гидроксипропан-2-ил)-2Н-индазол-5-ил}-6-(трифторметил)пиридин-2-карбоксамид.
12. Применение соединения общей формулы (I) по любому из пп. 1-11 для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у домашних животных.
13. Применение соединения общей формулы (I) по любому из пп. 1-11 для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у сельскохозяйственных животных.
14. Применение соединения общей формулы (I) по любому из пп. 1-12 или 13 для лечения и/или профилактики атопического дерматита, аллергического блошиного дерматита, воспалительного заболевания кишечника, остеоартрита и воспалительной боли, неинфекционного рецидивирующего заболевания дыхательных путей, гиперчувствительности к насекомым, астмы, респираторных заболеваний, мастита и эндометрита у животных.
15. Применение соединения общей формулы (I) по любому из пп. 1-11 для лечения и/или профилактики атопического дерматита у собак, аллергического блошиного дерматита у собак или кошек, воспалительного заболевания кишечника у собак или кошек, остеоартрита и воспалительной боли у собак, кошек, лошадей или крупного рогатого скота, неинфекционного рецидивирующего заболевания дыхательных путей у лошадей, гиперчувствительности к насекомым у лошадей, астмы у кошек, респираторных заболеваний у крупного рогатого скота, мастита у крупного рогатого скота, эндометрита у крупного рогатого скота и респираторных заболеваний у свиней.
16. Применение соединения общей формулы (I) по любому из пп. 1-11 для лечения и/или профилактики атопического дерматита у собак и аллергического блошиного дерматита у собак или кошек.
17. Применение cоединения общей формулы (I) по любому из пп. 1-11 для лечения и/или профилактики остеоартрита и воспалительной боли у крупного рогатого скота, респираторных заболеваний у крупного рогатого скота, мастита у крупного рогатого скота, эндометрита у крупного рогатого скота и респираторных заболеваний у свиней.
18. Применение соединения общей формулы (I), как определено в любом из пп. 1-11, для производства лекарственного средства для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у животных.
19. Применение по п. 18, где лекарственное средство используют для лечения и/или профилактики атопического дерматита, аллергического блошиного дерматита, воспалительного заболевания кишечника, остеоартрита и воспалительной боли, неинфекционного рецидивирующего заболевания дыхательных путей, гиперчувствительности к насекомым, астмы, респираторных заболеваний, мастита и эндометрита у животных.
20. Применение по пп. 18 и 19, где животное является домашним животным.
21. Применение по пп. 18 и 19, где животное является сельскохозяйственным животным.
22. Применение по пп. 18 и 19 для лечения и/или профилактики атопического дерматита у собак, аллергического блошиного дерматита у собак или кошек, воспалительного заболевания кишечника у собак или кошек, остеоартрита и воспалительной боли у собак, кошек, лошадей или крупного рогатого скота, неинфекционного рецидивирующего заболевания дыхательных путей у лошадей, гиперчувствительности к насекомым у лошадей, астмы у кошек, респираторных заболеваний у крупного рогатого скота, мастита у крупного рогатого скота, эндометрита у крупного рогатого скота и респираторных заболеваний у свиней.
23. Применение по пп. 18 и 19 для лечения и/или профилактики атопического дерматита у собак и аллергического блошиного дерматита у собак или кошек.
24. Лекарственное средство, содержащее соединение формулы (I), как определено в любом из пп. 1-11, в эффективном количестве в комбинации с инертным, нетоксическим, фармацевтически приемлемым наполнителем для применения для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у животных.
25. Способ лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у животных путем введения эффективного количества по меньшей мере одного соединения формулы (I), как определено в любом из пп. 1-11, нуждающемуся в этом животному.
26. Применение соединений общей формулы (III)
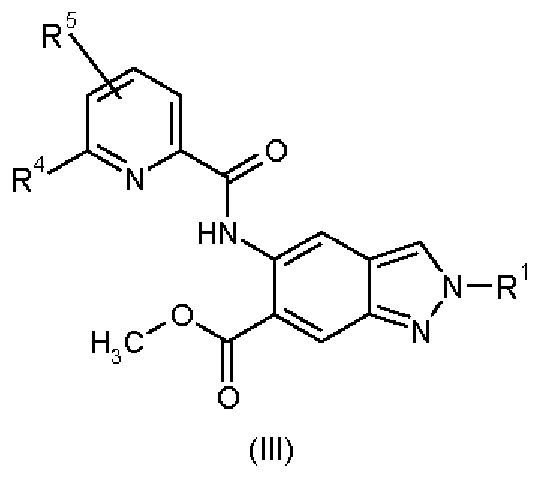
в которой
R1 представляет собой 4,4,4-трифторбутил, 3-гидрокси-3-метилбутил, 3-метоксипропил, 3-гидроксипропил, 3-гидроксибутил, 3-гидрокси-2-метилпропил, 3-гидрокси-2,2-диметилпропил, 3-трифторметоксипропил, 2-метоксиэтил, 2-гидроксиэтил, 2-(метилсульфонил)этил, 3-(метилсульфонил)пропил или 2-(1-гидроксициклопропил)этил;
R4 представляет собой дифторметил, трифторметил или метил; и
R5 представляет собой водород или фтор;
и их диастереомеров, энантиомеров, солей, сольватов или сольватов солей, для лечения и/или профилактики аллергических и/или воспалительных IRAK4-связанных заболеваний у животных.
27. Применение соединений по п. 26, которые представляют собой:
метил 5-{[(5-фтор-6-метилпиридин-2-ил)карбонил]амино}-2-(3-гидрокси-3-метилбутил)-2Н-индазол-6-карбоксилат и
метил 2-(3-гидрокси-3-метилбутил)-5-({[6-(трифторметил)пиридин-2-ил]карбонил}амино)-2Н-индазол-6-карбоксилат.
| WO 2015104662 A1, 16.07.2015 | |||
| WO 2004013102 A1, 12.02.2004. |

