Область изобретения
Настоящее изобретение относится к области изобретения химических лекарственных средств и относится к терпиридин-дикетоновому соединению или его рацемату, изомеру или фармацевтически приемлемой соли и способу его получения и его применению.
Предшествующий уровень техники
Митоген-активируемая протеинкиназа (MAPK - от англ. Mitogen-Activated Protein Kinase) представляет собой консервативное семейство ферментов, которыми используется каскад фосфорилирования для передачи и доставки внешних стимулов с получением координированного клеточного ответа на окружающую среду. MAPK представляет собой пролин-индуцируемую серин/треониновую протеинкиназу, которая регулирует активности клетки, как например, экспрессию генов, митоз, дифференцировку и выживаемость/апоптоз клеток. До настоящего времени идентифицированы MAPK млекопитающих четырех разных категорий, которые представляют собой киназу передачи внеклеточного сигнала (ERK1 и ERK2), c-jun N-концевую киназу-1 (JNK1-3), p38MAPK (р38α, р38β, р38γ и р38δ) и ERK5.
Научное исследование такого пути согласно биологическим, клеточным и in vivo представлениям в основном осуществляется за счет доступности высокоэффективного, селективного низкомолекулярного ингибитора p38MAPK. Низкомолекулярный ингибитор нацелен на подтип α p38MAPK и в меньшей степени нацелен на подтип β. p38αMAPK представляет собой основной подтип, участвующий в иммунной реакции и воспалительном ответе. Таким образом, функции p38MAPK имеют важнейшее значение для образования и активности разных провоспалительных цитокинов в клетках, таких как макрофаги, моноциты, синовиоциты и эндотелиальные клетки. Провоспалительные цитокины включают TNFα (от англ. Tumor necrosis factor alpha - фактор некроза опухоли-альфа), IL-1 (от англ. interleukin-1 - интерлейкин-1), IL-6 (от англ. interleukin-6 - интерлейкин-6) и IL-8 (от англ. interleukin-8 - интерлейкин-8). p38MAPK также отвечает за индукцию ключевых провоспалительных ферментов, таких как СОХ2 и iNOS, которые, соответственно, являются основными источниками эйкозаноида и оксида азота в участках воспаления. Кроме того, путь p38MAPK регулирует экспрессию матриксной металлопротеиназы (ММР - от англ. matrix metalloproteinase). ММР включает ММР2, ММР9 и ММР13.
Применение селективных и эффективных ингибиторов облегчило открытие множества семейств субстратов p38MAPK. Субстраты p38MAPK включают транскрипционные факторы, MAPKAP-киназы и другие ферменты. MAPKAP-киназы (MK2, MK-3 и PRAK) селективно фосфорилируются под действием p38MAPK, и фосфорилирование MSK1/2, MNK1/2 и RSKb катализируется p38MAPK и ERK. Несмотря на то, что идентификация субстратов является сложной вследствие отсутствия специфичных ингибиторов, считается, что активация RSKb играет роль в выживаемости клеток.
Сразу после фосфорилирования и активации под действием p38MAPK, MK-2, MK-3 и PRAK разделяют похожие специфичности в отношении субстрата. Все из данных киназ могут фосфорилировать малые белки теплового шока Hsp27. Исследования показали, что мыши с дефицитом PRAK и MK3 не демонстрируют какой-либо устойчивости к эндотоксическому шоку или липополисахарид (LPS - от англ. lipopolysaccharide) - индуцированного уменьшения продукции цитокинов. Напротив, мыши с дефицитом MK-2 демонстрируют устойчивость к эндотоксическому шоку и нарушенный воспалительный ответ, а также значительно уменьшенную продукцию цитокинов, таких как TNFα, IFNγ и IL-6. Таким образом, в частности, ось р38/MK2 является необходимой и достаточной для того, чтобы опосредовать провоспалительные ответы.
Посредством взаимодействия р38:MK2 и посредством применения MK2 в качестве субстрата р38 открыт новый ингибитор р38α, демонстрирующий интересующие свойства (Davidson et al.). Ингибитор демонстрирует селективность в отношении субстрата за счет предотвращения р38α-зависимого фосфорилирования MK2 (Ki приблизительно 300 нМ) и одновременно поддержания р38α-зависимого фосфорилирования ATF2 (Ki приблизительно больше 20 мкМ). По сравнению с традиционным конкурентным ингибитором р38АТР, который блокирует р38-зависомое фосфорилирование всех из субстратов р38, новый ингибитор является уникальным по функции. Во втором независимом исследовании также описывается ингибитор р38, имеющий уникальную эффективность механизма. В данной работе показаны новые механизмы селективного ингибирования р38-зависимого фосфорилирования MK2. В отличие от предыдущих исследований Davidson et al., уникальные соединения данных механизмов конкурируют с АТР (от англ. adenosine triphosphate - аденозинтрифосфат) и стабилизируют соединения р38/MK2.
Подводя итоги, два исследования явным образом доказывают концепцию, согласно которой блокада оси р38/MK2 может селективно осуществляться посредством использования низкомолекулярных ингибиторов. По сравнению с известными ингибиторами p38MAPK, данные ингибиторы р38/MK2 будут сохранять или улучшать эффективность и демонстрировать улучшенные характеристики безопасности на животных моделях заболевания или в клинических средах организма человека.
Краткое изложение сущности изобретения
Ввиду проблем в предшествующем уровне техники, в данной заявке предложено соединение в качестве ингибитора р38/MK2. Указанное соединение может ингибировать продукцию цитокина TNFα таким образом, чтобы осуществлять регуляцию соответствующих заболеваний, таких как воспалительная реакция.
Согласно первому аспекту, согласно данной заявке предложено соединение, показанное в общей формуле (I), или его рацемат, изомер или фармацевтически приемлемая соль:

Согласно второму аспекту настоящего изобретения предложена фармацевтическая композиция. Фармацевтическая композиция включает любое терапевтически эффективное количество соединения, описанного выше, или его фармацевтической приемлемой соли и фармацевтически приемлемый носитель.
Согласно третьему аспекту настоящего изобретения предложено применение терапевтически эффективного количества соединения, описанного выше, или его фармацевтически приемлемой соли в получении лекарственных средств для лечения медицинских состояний. Заболевание представляет собой заболевание, связанное с р38/MK2. Данное соединение может ингибировать продукцию цитокина TNFα, таким образом, регулируя соответствующие заболевания, такие как воспалительная реакция. В частности, медицинские состояния выбраны из хронического воспалительного заболевания, острого воспалительного заболевания или аутовоспалительного заболевания.
В частности, настоящее изобретение достигается посредством следующих технических решений.
Предложено соединение, показанное в общей формуле (I), или его рацемат или изомер или фармацевтически приемлемая соль.
R1 и R1' независимо выбраны из атома водорода, галогена, алкила, циклоалкила, гетероциклоалкила, алкил циклоалкила, алкилгетероциклила, фенила, гетероарила, галогеналкила или циано, или R1 и R1', взятые вместе, циклизированы с образованием циклоалкила или гетероциклоалкила.
R2 выбран из атома водорода, галогена, гидроксила, циано, алкила, алкокси, алкоксиалкила или галогеналкила.
R3 независимо выбран из атома водорода, алкила, циклоалкила, гетероциклоалкила, галогена, алкокси, циано, галогеналкокси или сульфонила.
m равно 0, 1, 2 или 3.
R4 выбран из атома водорода, алкила, циклоалкила, алкокси или галогеналкила.
R5 выбран из атома водорода, алкила, циклоалкила, алкокси или галогеналкила.
R6 выбран из атома водорода, галогена, циано, алкила, циклоалкила или галогеналкила.
кольцо А выбрано из ароматического кольца или гетероароматического кольца.
R7 независимо выбран из атома водорода, галогена, циано, алкила, циклоалкила, галогеналкила, алкокси, галогеналкокси или сульфонила.
n равно 0, 1, 2, 3, 4 или 5.
X представляет собой О, СН2 или NH.
R8 и R9 независимо выбраны из атома водорода, алкила, циклоалкила, алкилциклоалкила или галогеналкила, или R8 и R9 совместно циклизированы с образованием циклоалкила или гетероциклоалкила.
В качестве предпочтительного технического решения настоящего изобретения алкил выбран из C1-6алкила, и С1-6алкил выбран из метила, этила, пропила, изопропила, н-бутила, изобутила, втор-бутила, трет-бутила, н-пентила, втор-пентила, 1-этилпропила, 2-метилбутила, трет-пентила, 1,2-диметилпропила, изопентила, неопентила, н-гексила, изогексила, втор-гексила, трет-гексила, неогексила, 2-метилпентила, 1,2-диметилбутила или 1-этилбутила.
Алкокси выбран из C1-6алкокси, и C1-6алкокси выбран из метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, втор-пентилокси, 1-этилпропокси, 2-метилбутокси, трет-пентилокси, 1,2-диметилпропокси, изопентилокси, неопентилокси, н-гексилокси, изогексилокси, втор-гексилокси, трет-гексилокси, неогексилокси, 2-метилпентилокси, 1,2-диметилбутокси или 1-этилбутокси. Алкоксиалкил выбран из С1-4алкокси - С1-4алкила, и, кроме того, С1-4алкокси - С1-4алкил выбран из метоксиметила, метоксиэтила, метоксипропила, метоксибутила, этоксиметила, этоксиэтила, этоксипропила, этоксибутила, пропоксиметила, пропоксиэтила, пропоксипропила, пропоксибутила, бутоксиметила, бутоксиэтила, бутоксипропила или бутоксибутила.
В качестве предпочтительного технического решения настоящего изобретения циклоалкил выбран из циклоалкила С3-6, и циклоалкил С3-6 выбран из циклопропила, циклобутила, циклопентила или циклогексила.
В качестве предпочтительного технического решения настоящего изобретения ароматическое кольцо выбрано из четырехчленного кольца, конденсированного кольца, содержащего данное четырехчленное кольцо, пятичленного кольца, конденсированного кольца, содержащего данное пятичленное кольцо, шестичленного кольца, конденсированного кольца, содержащего данное шестичленное кольцо, ароматического кольца бифенильного типа. Гетероароматическое кольцо означает, что один или более атомов углерода на ароматическом кольце замещены гетероатомом.
Ароматическое кольцо включает бензольное кольцо и нафталиновое кольцо.
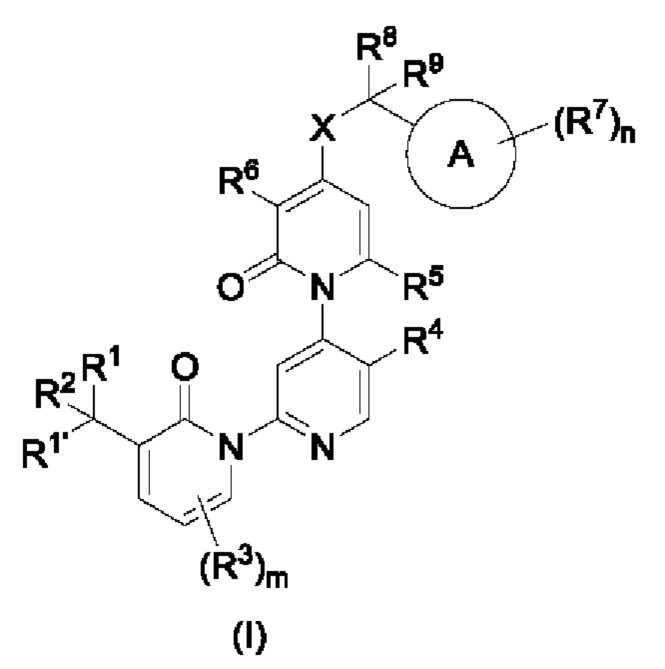
Гетероароматическое кольцо включает индазол, хинолин, изохинолин, хиноксалин, индол, изоиндол, циннолин, хиназолин, фталазин, пурин, нафтиридин, птеридин, бензофуран, бензотиофен, бензоксазол, бензотиазол, бензисоксазол, бензисотиазол, бензоксадиазол, бензотиадиазол, бензотриазол, бензотриазин, бензимидазол, пиразинопиразол, пиразинопиримидин, пиразинопиридазин, пиразинотриазин, пиримидопиразол, пиримидазол, пиримидотриазол, пиримидотриазин, пиримидопиридазин, пиридазиноимидазол, пиридазинопиразол, пиридазинотриазол, пиридазинотриазин, триазиноимидазол, триазинопиразол, триазинотриазол, пиридооксазол, пиридотиазол, пиридоизоксазол, пиридоизотиазол, пиридооксадиазол, пиридотиадиазол, пиридофуран, пиридопиррол, пиразинооксазол, пиразинотиазол, пиразиноизоксазол, пиразиноизотиазол, пиразинооксадиазол, пиразинотиадиазол, пиразинофуран, пиразинопиррол, пиримидооксазол, пиримидотиазол, пиримидоизоксазол, пиримидоизотиазол, пиримидооксадиазол, пиримидотиадиазол, пиримидофуран, пиримидопиррол, пиридазинооксазол, пиридазинотиазол, пиридазиноизоксазол, пиридазиноизотиазол, пиридазинооксадиазол, пиридазинотиадиазол, пиридазинофуран, пиридазинопиррол, триазинооксазол, триазинотиазол, триазиноизоксазол, триазиноизотиазол, триазинооксадиазол, триазинотиадиазол, триазинофуран и триазинопиррол.
В частности, например, нафтидин выбран из 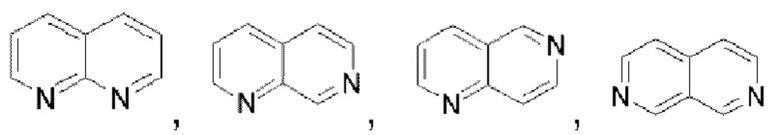 или
или  . Пиридоимидазол выбран из
. Пиридоимидазол выбран из  или
или  . Пиразиноимидазол выбран из
. Пиразиноимидазол выбран из  или
или 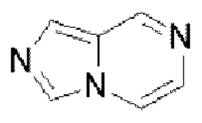 . Пиразинотриазол выбран из
. Пиразинотриазол выбран из 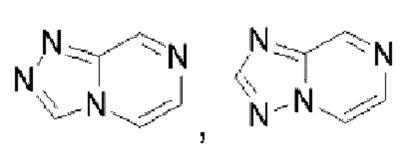 или
или  . Пиримидопиразол выбран из
. Пиримидопиразол выбран из  или
или 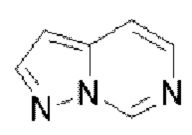 . Пиримидоимидазол выбран из
. Пиримидоимидазол выбран из 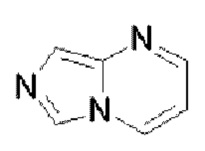 или
или 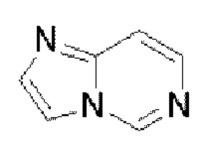 . Пиримидотриазол выбран из
. Пиримидотриазол выбран из 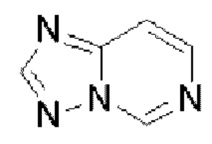 ,
,  или
или 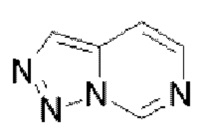 . Пиридазиноимидазол выбран из
. Пиридазиноимидазол выбран из 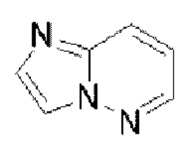 или
или 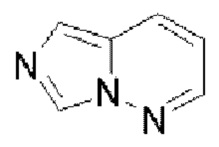 . Пиридазинотриазол выбран из
. Пиридазинотриазол выбран из 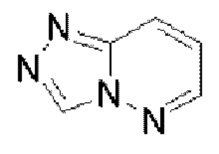 или
или 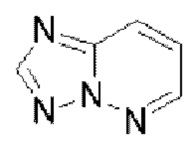 .
.
Триазиноимидазол выбран из 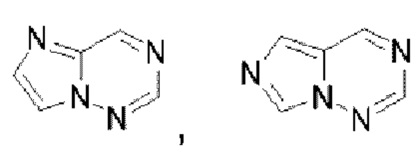 или
или  . Пиридопиридазин выбран из
. Пиридопиридазин выбран из  или
или  . Пиридопиразол выбран из
. Пиридопиразол выбран из  ,
,  или
или  . Пиридопиримидин выбран из
. Пиридопиримидин выбран из 
 или
или  . Пиридотриазин выбран из
. Пиридотриазин выбран из 
 или
или  . Пиримидотриазин выбран из
. Пиримидотриазин выбран из 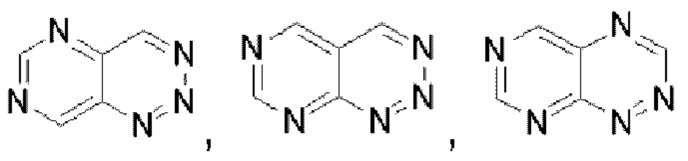 или
или  .
.
В частности, например, гетероциклический алкил выбран из 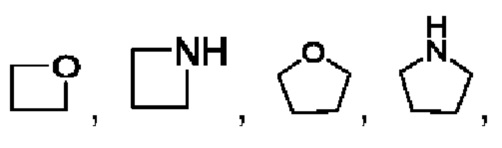
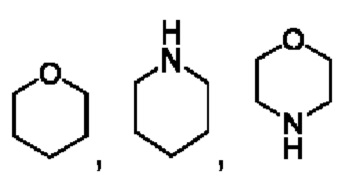 или
или  .
.
Гетероарил выбран из 

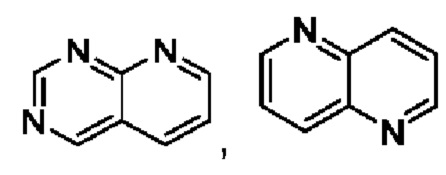 или
или  .
.
В качестве предпочтительного технического решения настоящего изобретения галоген выбран из фтора, хлора, брома или йода.
Галогеналкил означает, что один или более атомов водорода на алкиле замещены галогеном. Галогеналкокси означает, что один или более атомов водорода на алкокси замещены галогеном.
Гетероциклоалкил означает, что один или более атомов углерода на циклоалкиле замещены гетероатомом.
Алкилциклоалкил означает, что один или более атомов водорода на циклоалкиле замещены алкилом, и алкилгетероциклоалкил означает, что один или более атомов водорода на гетероциклоалкиле замещены алкилом.
В качестве предпочтительного технического решения настоящего изобретения гетероатом выбран из азота, кислорода или серы, и имеется один или более гетероатомов.
В качестве предпочтительного технического решения настоящего изобретения кольцо А выбрано из 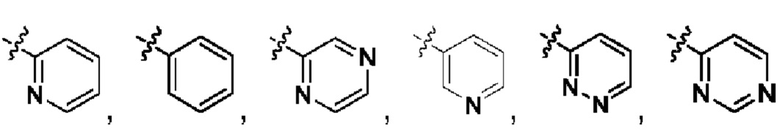 или
или 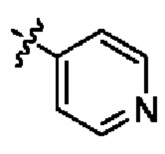 .
.
В качестве предпочтительного технического решения настоящего изобретения соединение или его рацемат, изомер или фармацевтически приемлемая соль имеет структуру формулы (Ia), формулы (Ib), формулы (Ic), формулы (Id), формулы (Ie), формулы (If) или формулы (Ig).

Где m равно 0, 1, 2 или 3; n равно 0, 1, 2, 3, 4 или 5; и R1, R1', R2, R3, R4, R5, R6, R7, R8 и R9 определены, как указано выше.
В качестве предпочтительного технического решения настоящего изобретения m равно 0 или 1; и n равно 0, 1 или 2.
Кольцо А выбрано из  или
или  .
.
R1 и R1' выбраны из атома водорода, метила или этила, и R1 и R1' циклизированы с образованием  .
.
R2 выбран из атома водорода, гидроксила, дифторметила или циано.
R3 выбран из атома водорода, метила, метокси, хлора или брома.
R4 выбран из метила, циклопропила, метокси, этила, фторметила или трифторметила.
R5 выбран из метила.
R6 выбран из хлора, атома водорода или брома.
R7 выбран из атома водорода, фтора, хлора, брома, циклопропила, метокси, циано, метила или трифторметила.
R8 и R9 представляют собой атом водорода.
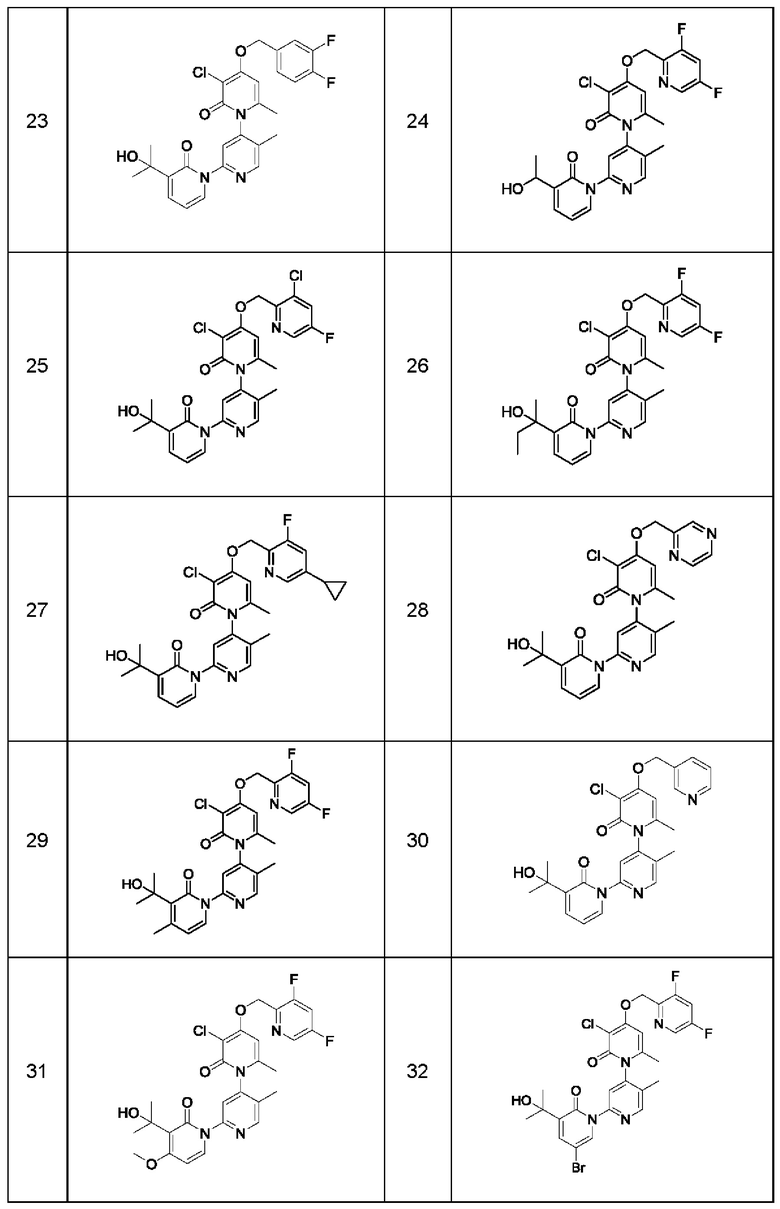
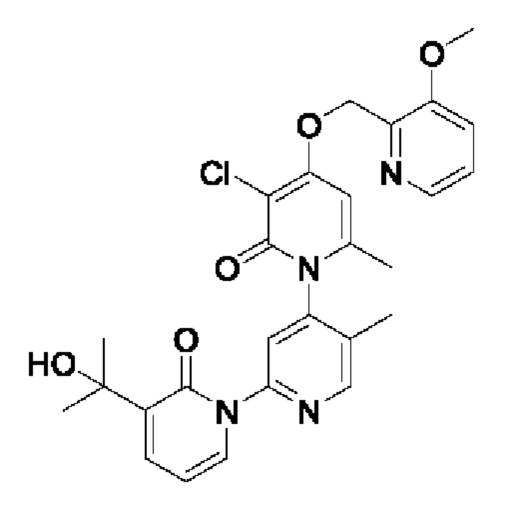
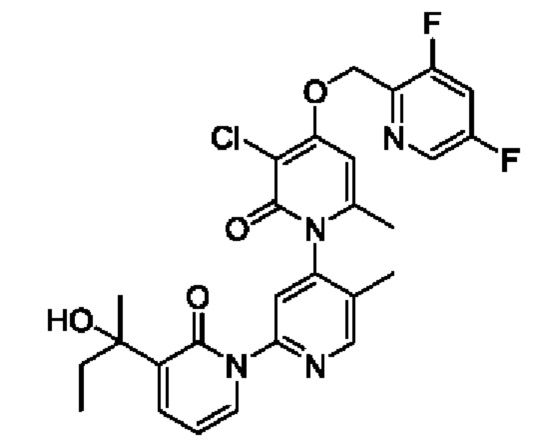
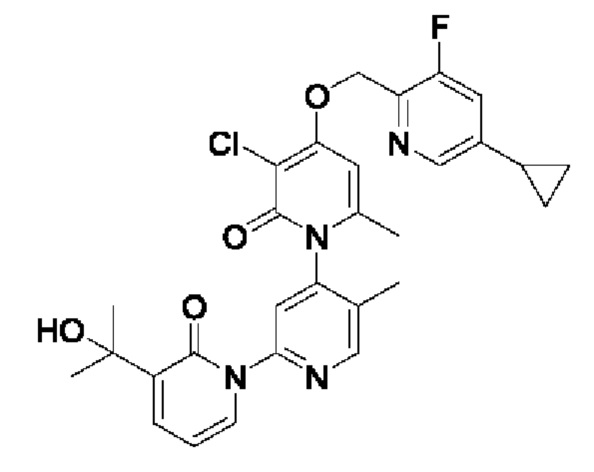
В качестве предпочтительного технического решения настоящего изобретения соединение или его рацемат, изомер или фармацевтически приемлемая соль выбраны из нижеследующего:





В качестве предпочтительного технического решения настоящего изобретения фармацевтически приемлемая соль относится к соли, полученной посредством соединения или его рацемата или изомера и фармацевтически приемлемой кислоты или основания.
В качестве предпочтительного технического решения настоящего изобретения более чем один атом водорода соединения или его рацемата, изомера или фармацевтически приемлемой соли замещен изотопом - дейтерием.
Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция. Фармацевтическая композиция включает терапевтически эффективное количество соединения или его рацемата, изомера или фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
Кроме того, согласно настоящему изобретению предложено применение соединения или его рацемата, изомера или фармацевтически приемлемой соли в получении лекарственных средств для лечения заболеваний. Заболевания представляют собой заболевания, связанные с р38/MK2, и, в частности, выбраны из хронического воспалительного заболевания и острого воспалительного заболевания. Предпочтительно, хроническое воспалительное заболевание представляет собой ревматоидный артрит.
По сравнению с соединениями в предшествующем уровне техники, соединение по настоящему изобретению обладает улучшенной активностью в отношении TNFα, растворимостью и эффектами PK in vivo.
Для ясности, в данном документе определены непатентованные термины, используемые в описании соединения.
Если не указано иное, предполагают, что следующие термины и фразы, используемые в данном документе, имеют следующие значения. Конкретный термин или фразу не следует считать неопределенными или неясными без конкретных определений, а следуют понимать в своем обычном значении. Когда торговое наименование появляется в данном документе, предполагают, что данное торговое наименование относится к соответствующему товару или активному ингредиенту. В контексте данного документа, термин «фармацевтически приемлемый» нацелен на соединения, вещества, композиции и/или лекарственные формы, которые, в рамках здравого медицинского суждения, подходят для применения в контакте с тканью человека и животного без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений и соответствуют разумному отношению польза/риск.
Термин «фармацевтически приемлемая соль» представляет собой соль соединения по настоящему изобретению, и ее получают из соединения по настоящему изобретению, имеющего конкретно указанный заместитель, и фармацевтически приемлемой кислоты или основания.
Помимо формы соли соединение, предложенное в настоящем изобретении, также имеет форму пролекарства. Пролекарство соединения, описанного в данном документе, легко химически изменяется в физиологических условиях, таким образом, что превращается в соединение по настоящему изобретению. Кроме того, пролекарство может химически или биохимически превращаться в соединение по настоящему изобретению в окружающей среде in vivo.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных или сольватированных формах, включая гидратные формы. Обычно, сольватированные формы и несольватированные формы эквивалентны и, как предполагают, включены в пределах объема настоящего изобретения.
Соединение по настоящему изобретению может существовать в конкретной геометрической или стереоизомерной форме. Все такие соединения, предполагаемые в настоящем изобретении, включают цис- и транс- изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, изомеры диастереомеры, (D)-изомеры, (L)-изомеры, атропизомеры и их рацемические смеси и другие смеси. Смеси, например, представляют собой смеси, обогащенные энантиомерами или диастереомерами. Все из данных смесей находятся в пределах объема настоящего изобретения. Заместители, такие как алкил, могут иметь дополнительные ассиметрические атомы углерода. Все из данных изомеров и их смесей включены в объем настоящего изобретения.
Оптически активные (R)- и (S)-изомеры, а также D- и L-изомеры, атропизомеры и т.п., могут быть получены посредством хирального синтеза или хиральных реагентов или других общепринятых технологий. Энантиомер определенного соединения в настоящем изобретении может быть получен посредством ассиметрического синтеза или дериватизации с хиральными вспомогательными веществами. Полученные диастереомерные смеси разделяют, и вспомогательную группу отщепляют с получением чистого требуемого энантиомера. В качестве альтернативы, когда молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксильная), диастереомерная соль образуется соответствующими оптически активными кислотами или основаниями. Затем, диастереоизомеры разбивают посредством традиционных способов, известных в данной области, и чистые энантиомеры используются повторно. Кроме того, разделение энантиомеров и диастереоизомеров обычно завершают посредством использования хроматографии. В хроматографии вводится в использование хиральная неподвижная фаза, и она, возможно, объединяется с химической дериватизацией (например, образование карбамата из аминов).
Атомы молекулы соединения по настоящему изобретению представляют собой изотопы, и обычно могут быть достигнуты эффекты увеличения периода полураспада, уменьшения скорости клиренса, стабилизации метаболизма и улучшения активностей in vivo посредством дериватизации изотопов. Кроме того, включено решение осуществления. По меньшей мере один атом замещен атомом с таким же атомным номером (количеством протонов) и отличными массовыми числами (сумма протонов и нейтронов). Примеры изотопов, включенных в соединение по настоящему изобретению, включают атомы водорода, атомы углерода, атомы азота, атомы кислорода, атомы фосфора, атомы серы, атомы фтора и атомы хлора, которые, соответственно, включают 2Н, 3Н, 13С, 14С, 15N, 17O, 18O, 31Р, 32Р, 35S, 18F, 36Cl. В частности, радиоизотопы, которые испускают излучение с распадом радиоизотопов, распадаются, как например, 3Н или 14С, могут быть использованы в исследовании топографической анатомии фармацевтических препаратов или соединений in vivo. Стабильные изотопы не распадаются, не изменяются по количеству изотопов, не являются радиоактивными изотопами, таким образом, данные изотопы безопасны для применения. Когда атомы, составляющие молекулу соединения по настоящему изобретению, представляют собой изотопы, можно осуществлять превращение данных изотопов в соответствии с общими способами посредством замены реагента, используемого в синтезе, реагентом, содержащим соответствующие изотопы.
Соединение по настоящему изобретению может включать неестественные количества атомных изотопов одного или более атомов, которые составляют данное соединение. Например, соединение может быть мечено радиоизотопами, такими как дейтерий (2Н), йод-125 (125I) или С-14 (14С). Превращения всех данных изотопных составов соединения по настоящему изобретению, вне зависимости от радиоактивности, включены в объем настоящего изобретения.
Кроме того, один или более атомов водорода соединения по настоящему изобретению замещены изотопом - дейтерием (2Н). После дейтеризации соединение по настоящему изобретению обладает эффектами увеличения периода полураспада, уменьшения скорости клиренса, стабилизации метаболизма и улучшения активности in vivo.
Способ получения изотопных производных обычно включает способ межфазного катализа. Например, в предпочтительном способе дейтеризации используется способ межфазного катализа (например, тетраалкиламмониевая соль или NBu4HSO4). Метиленовые протоны соединения дифенилметана заменяются посредством использования межфазного катализатора, что приводит к получению более высокого уровня дейтерия, вводимого посредством восстановления посредством дейтерированного бората натрия, чем посредством дейтерированного силана (такого как триэтилдейтерированный силан) или кислоты Льюиса, такой как хлорид алюминия, в присутствии кислоты (такой как метансульфоновая кислота).
Термин ''фармацевтически приемлемый носитель'' относится к любому носителю препарата или среде, которые могут доставлять эффективное количество активного вещества по настоящему изобретению без нанесения вреда биологической активности активного вещества и не обладают токсичными побочными эффектами в отношении хозяев или пациентов. Репрезентативные носители включают воду, масло, растительные продукты, минеральные вещества, основу для крема, основу для лосьона, основу для мази и тому подобное. Данные основы включают суспендирующие агенты, загустители, усилители проникновения внутрь и тому подобное. Препараты данных основ хорошо известны специалистам в области косметических средств или региональных лекарственных средств. Для получения другой информации о носителе см. Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание данного документе включено в данный документ посредством ссылки.
Термин ''вспомогательное вещество'' обычно относится к носителю, разбавителю и/или среде, требуемой для получения эффективной фармацевтической композиции.
Термин ''эффективное количество'' или ''терапевтически эффективное количество'' в отношении лекарственного средства или фармакологически активного агента относится к нетоксичному, но достаточному количеству лекарственного средства или агента для достижения желательных эффектов. Для пероральных лекарственных форм в настоящем изобретении ''эффективное количество'' одного активного вещества в композиции относится к количеству, требуемому для достижения желательного эффекта, когда активное вещество используется в комбинации с другим активным веществом в композиции. Определение эффективного количества варьирует от индивида к индивиду, которое зависит от возраста и общего состояния реципиента и также зависит от конкретного активного вещества. Соответствующее эффективное количество в отдельных случаях может быть определено специалистами в данной области на основе рутинных экспериментов.
Термин ''активный ингредиент'', ''терапевтический агент'', ''активное вещество'' или ''активный агент'' относится к химическому структурному компоненту, который является эффективным в лечении целевых расстройств, заболеваний или состояний.
Термин ''возможный'' или ''возможно'' означает, что описанное впоследствии событие или состояние может происходить, но не обязательно происходит, и описание включает ситуации, в которых событие или состояние происходит, и ситуации, в которых событие или состояние не происходит.
 указывает на линкерную связь.
указывает на линкерную связь.
Соединение по настоящему изобретению может быть получено разными способами синтеза, хорошо известными специалистам в данной области, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления, образованные в комбинации с другими способами химического синтеза, и эквивалентные способы замещения, хорошо известные специалистам в данной области, и предпочтительные варианты осуществления включают примеры осуществления настоящего изобретения, но не ограничиваются ими.
Краткое описание графических материалов
Фиг. 1 представляет собой схематичное представление кристаллической структуры одной молекулы Соединения 1А.
Фиг. 2 представляет собой параметры кристалла кристаллической структуры Соединения 1А.
Фиг. 3 представляет собой атомные координаты и параметры изотропного смещения кристаллической структуры Соединения 1А.
Фиг. 4 представляет собой длины связей в кристаллической структуре Соединения 1А.
Фиг. 5 представляет собой углы связи в кристаллической структуре Соединения 1А
Фиг. 6 представляет собой двугранные углы в кристаллической структуре Соединения 1А.
Подробное описание примеров осуществления
Данная заявка дополнительно подробно описана ниже со ссылкой на примеры осуществления, но варианты осуществления данной заявки не ограничиваются ими.
Пример осуществления 1

Путь синтеза соединения с порядковым номером, равным 1, показан ниже.
На стадии А синтезировали 2-(бромметил)-3,5-дифторпиридин.
При нуле градусов Цельсия трифенилфосфин (PPh3, 813,5 мг, 3,11 ммоль) и тетрабромметан (CBr4, 823,8 мг, 2,48 ммоль) последовательно добавляли к тетрагидрофурану (ТГФ, 5,0 мл), содержащему (3,5-дифторпиридин-2-ил)метанол (300,0 мг, 2,07 ммоль), для взаимодействия в течение 1 ч при комнатной температуре (к.т.).
После завершения реакции сразу же проводили вакуумное концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: н-гексан/этилацетат=10/1). Получали 372,4 мг бесцветного масляного 2-(бромметил)-3,5-дифторпиридина (выход: 86,5%). ЖХ-МС (жидкостная хроматография/масс-спектрометрия): RT (от англ. retention time - время удерживания)=1,89 мин, [М+Н]+=208,01.
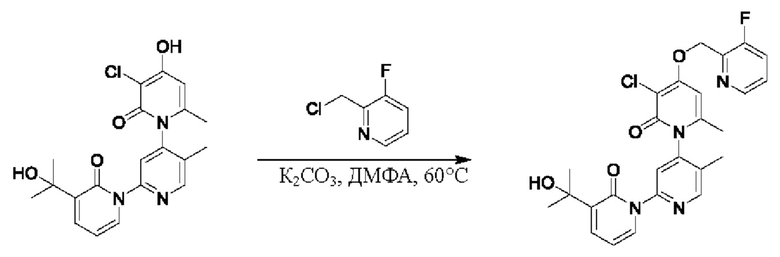
На стадии В синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
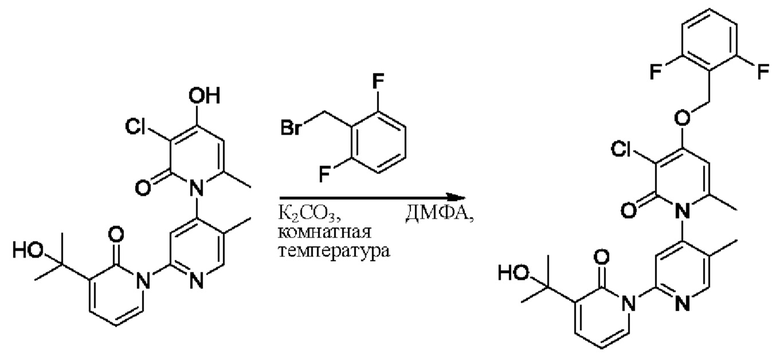
При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (37,4 мг, 0,18 ммоль) и карбонат калия (K2CO3, 33,12 мг, 0,24 ммоль) добавляли к N,N-диметилформамиду (ДМФ, 2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (20 мл × 3 раза); органическую фазу объединяли; промывку проводили раствором, насыщенным солью (20 мл × 2 раза); и проводили сушку посредством безводного сульфата натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 28,0 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 53,7%).
ЖХ-МС: RT=1,86 мин, [М+Н]+=529,22. 1Н ЯМР(400 МГц, ДМСО) δ 8.68 (s, 1Н), 8.60 (d, J=2,3 Гц, 1Н), 8.13-8.05 (m, 1Н), 7.85 (dd, J=6,9, 2,1 Гц, 1Н), 7.78 (s, 1Н), 7.69 (dd, J=7,0, 2,1 Гц, 1Н), 6.79 (s, 1Н), 6.42 (t, J=7,0 Гц, 1Н), 5.47 (d, J=1,6 Гц, 2Н), 5.22 (s, 1Н), 2.07 (s, 3Н), 2.00 (s, 3Н), 1.47 (s, 3Н), 1.46 (s, 3Н).
Сверхкритическую жидкостную хроматографию (колонка AS-H) использовали для разделения рацемического соединения Примера осуществления 1 посредством подвижной фазы диоксида углерода и изопропанола, и последовательно элюировали атропизомерные соединения Примера осуществления 1А и 1В.

Соединение Примера осуществления 1А:RT=4,21 мин (SFC, колонка AS-H, 0,46 см I.D. (от англ. Inside Diameter - внутренний диаметр) × 15 см L (от англ. length - длина), метод изоградиента с 30% изопропанолом, причем скорость потока составляет 2,5 мл/мин и период повторения цикла составляет 10 мин). [a]D25=-1,68° (МеОН, прибор для определения удельного вращения Rudolph Autopol I); и 1Н ЯМР (400 МГц, ДМСО) δ 8.68 (s, 1Н), 8.59 (d, J=2,3 Гц, 1Н), 8.11-8.05 (m, 1Н), 7.85 (dd, J=6,9, 2,0 Гц, 1Н), 7.79 (s, 1Н), 7.69 (dd, J=7,0, 2,0 Гц, 1Н), 6.80 (s, 1Н), 6.42 (t, J=6,9 Гц, 1Н), 5.48 (d, J=1,2 Гц, 2Н), 5.23 (s, 1Н), 2.07 (s, 3Н), 2.00 (s, 3Н), 1.47 (s, 3Н), 1.46 (s, 3Н). Кристаллическая структура одной молекулы показана на Фиг. 1. Главные параметры кристалла показаны на Фиг. 2. Атомные координаты и параметры изотропного смещения кристаллической структуры показаны на Фиг. 3. Длины связей в кристаллической структуре показаны на Фиг. 4. Углы связи в кристаллической структуре показаны на Фиг. 5. Двугранные углы в кристаллической структуре показаны на Фиг. 6.
Соединение Примера осуществления 1 В: RT=4,68 мин (SFC, колонка AS-H, 0,46 см I.D. × 15 см L, метод изоградиента с 30% изопропанолом, причем скорость потока составляет 2,5 мл/мин, и период повторения цикла составляет 10 мин), [a]D25=+1,64° (МеОН, прибор для определения удельного вращения Rudolph Autopol I); и 1Н ЯМР (500 МГц, ДМСО) δ 8.68 (s, 1Н), 8.59 (d, J=2,3 Гц, 1Н), 8.11-8.06 (m, 1Н), 7.85 (dd, J=6,9, 2,1 Гц, 1Н), 7.78 (s, 1Н), 7.69 (dd, J=7,0, 2,1 Гц, 1H), 6.80 (s, 1Н), 6.42 (t, J=6,9 Гц, 1H), 5.47 (d, J=1,3 Гц, 2H), 5.24 (s, 1Н), 2.07 (s, 3Н), 2.00 (s, 3Н), 1.47 (s, 3Н), 1.46 (s, 3Н).
Пример осуществления 2

Способ синтеза соединения с порядковым номером, равным 2, показан ниже. На стадии А синтезировали 2'-хлор-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он.
При комнатной температуре 2-хлор-5-метилпиридин-4-амин (2,3 г, 1,61 ммоль) растворяли в диоксане (40,0 мл), добавляли 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (2,0 г, 1,07 ммоль), температуру повышали вплоть до 90 градусов Цельсия для взаимодействия в течение 5 ч, по каплям добавляли 10 н. водного раствора серной кислоты, твердого вещества не образовалось, и реакция длилась в течение 2 ч.
После завершения реакции растворитель удаляли центрифугированием; желтоватый осадок осаждали посредством добавления соответствующего количества воды; фильтрование проводили после 0,5 ч перемешивания, и промывку проводили водой два раза; и собирали 1,25 г продукта в виде желто-землистого твердого вещества 2'-хлор-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (выход: 30,9%). ЖХ-МС: RT=1,65 мин, [М+Н]+=251,09.
На стадии В синтезировали 2'-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (1,55 г, 7,47 ммоль) и K2CO3 (1,37 г, 9,96 ммоль) добавляли к N,N-ДМФА (15,0 мл), содержащему 2'-хлор-4-гидрокси-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (1,25 г, 4,98 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (30 ил × 3 раза); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (20 мл × 2 раза); и сушку проводили посредством безводного сульфата натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 1,08 г желтоватого твердого вещества 2'-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (выход: 57,4%). ЖХ-МС: RT=1,91 мин, [М+Н]+=378,24.
На стадии С синтезировали 4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1, 2':4',1''-терпиридин]-2,2''-дикетон.
При комнатной температуре 2'-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (100,0 мг, 0,26 ммоль), 3-(2-гидроксипропан-2-ил)пиридин-2(1 Н)-он (60,1 мг, 0,39 ммоль), йодид меди (Cul, 9,5 мг, 0,05 ммоль) и K2CO3 (71,8 мг, 0,52 ммоль) растворяли в диоксане (2,0 мл), добавляли каплю N,N-диметилэтилендиамина и затем температуру повышали вплоть до 110 градусов Цельсия для микроволновой реакции в течение 1 ч.
После завершения реакции проводили фильтрование с отсасыванием, проводили капельную промывку дихлорметаном и затем концентрировали фильтрат. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 26,0 мг белого твердого вещества 4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 20,2%).
ЖХ-МС: RT=1,82 мин, [М+Н]+=495,24. 1Н ЯМР (400 МГц, ДМСО) δ 8.64 (s, 1Н), 8.59 (d, J=2,4 Гц, 1Н), 8.07 (ddd, J=10,0, 9,3, 2,4 Гц, 1Н), 7.84 (dd, J=6,9, 2,1 Гц, 1Н), 7.70 (s, 1Н), 7.70-7.66 (m, 1Н), 6.41 (t, J=6,9 Гц, 1Н), 6.13 (d, J=1,7 Гц, 1Н), 6.04 (d, J=2,5 Гц, 1Н), 5.25 (s, 2Н), 5.22 (s, 1Н), 2.08 (s, 3Н), 1.90 (s, 3Н), 1.47 (s, 3Н), 1.46 (s, 3Н).
Пример осуществления 3
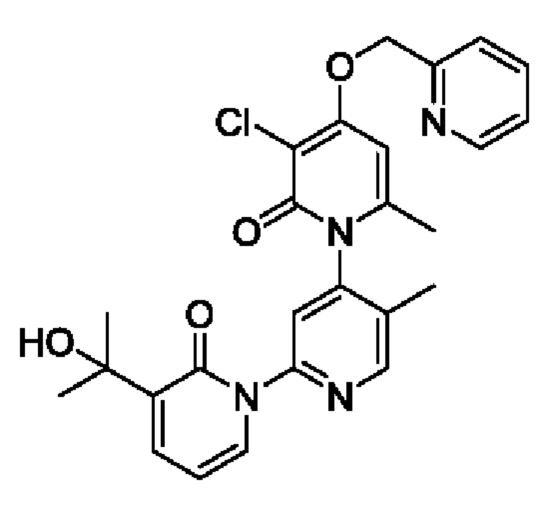
Путь синтеза соединения с порядковым номером, равным 3, показан ниже.
На стадии А синтезировали 2-(бромметил)пиридин.
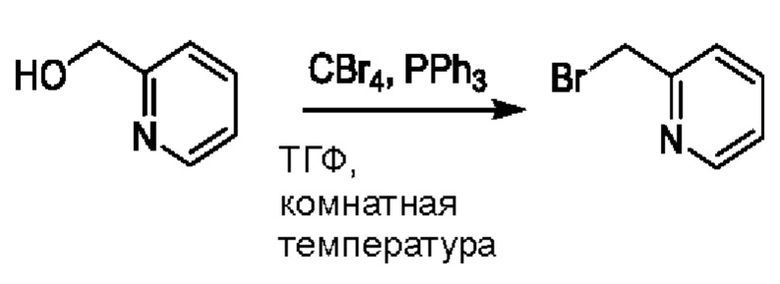
При нуле градусов Цельсия PPh3 (813,5 мг, 3,11 ммоль) и CBr4 (823,8 мг, 2,48 ммоль) добавляли к ТГФ (5,0 мл), содержащему 2-(бромметил)пиридин (225,6 мг, 2,07 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: н-гексан/этилацетат=10/1). Получали 322,3 мг бесцветного масляного 2-(бромметил)пиридина (выход: 90,5%).
На стадии В синтезировали 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиридин-2-ил)метокси-2Н,2''Н-[1,2':4', 1''-терпиридин]-2,2''-дикетон.
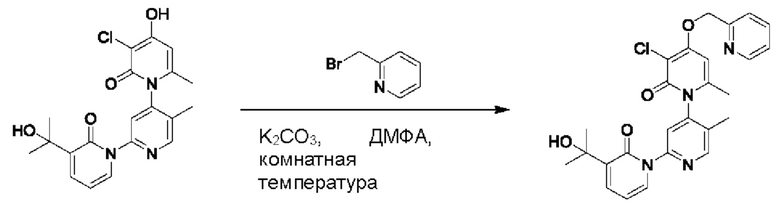
При комнатной температуре 2-(бромметил)пиридин (31,0 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гилроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (20 мл × 3 раза); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (20 мл × 2 раза); и сушку проводили безводным сульфатом натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 29,0 мг белого твердого вещества 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиридин-2-ил)метокси-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 49,0%).
ЖХ-МС: RT=1,77 мин, [М+Н]+=493,34. Данные по ядерному магнитному резонансу: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.67 (s, 1Н), 8.61 -8.58 (m, 1Н), 7.91 (td, J=7,7, 1,8 Гц, 1Н), 7.84 (dt, J=5,5, 2,8 Гц, 1Н), 7.77 (s, 1Н), 7.68 (dd, J=7,0, 2,1 Гц, 1Н), 7.58 (d, J=7,8 Гц, 1Н), 7.39 (dd, J=7,0, 5,3 Гц, 1Н), 6.76 (s, 1Н), 6.41 (t, J=6,9 Гц, 1Н), 5.42 (s, 2Н), 5.21 (s, 1Н), 2.06 (s, 3Н), 1.99 (s, 3Н), 1.45 (d, J=4,1 Гц, 6Н).
Пример осуществления 4

Путь синтеза соединения с порядковым номером, равным 4, показан ниже. На стадии А синтезировали 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он.

При комнатной температуре 2,2,6-триметил-4Н-1,3-диоксин-4-он (30 г, 211,27 ммоль) растворяли в ТГФ (100 мл), и температуру снижали до минус 70 градусов Цельсия в атмосфере азота; по каплям добавляли бис(триметилсилил)амид лития (LiHMDS, 290 мл, 1 М) для взаимодействия в течение 2 ч; диэтилцинк (ZnEt2, 290 мл, 1 М) добавляли для взаимодействия в течение 0,5 ч; и температуру медленно повышали до минус 20 градусов Цельсия в пределах 1,5 ч, и 1-ацетилимидазол (28 г, 253,52 ммоль) добавляли для взаимодействия в течение 3 ч.
После завершения реакции реакцию останавливали посредством добавления в ледяную воду; рН=3-4 доводили посредством 6 н. соляной кислоты; экстракцию проводили посредством этилацетата (50 мл × 3 раза), и органическую фазу объединяли для выполнения сушки; очистку проводили посредством хроматографии на колонке (элюент: этилацетат/н-гексан=4/1) с получением 19 г желтого твердого вещества 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (выход: 49%). ЖХ-МС: RT=1,65 мин, [М+Н]+=185,13.
На стадии В синтезировали 2'-бром-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он.
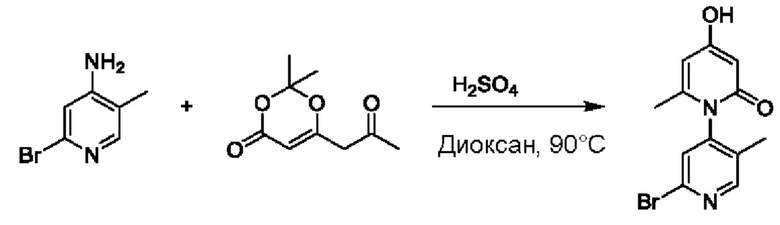
При комнатной температуре 2-бром-5-метилпиридин-4-амин (5 г, 26,74 ммоль) растворяли в диоксане (100 мл), добавляли 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (5,9 г, 32,09 ммоль), температуру повышали вплоть до 90 градусов Цельсия для взаимодействия в течение 5 ч, по каплям добавляли 10 н. водный раствор серной кислоты до тех пор, пока не образовалось твердое вещество, и реакция продолжалась в течение 2 ч.
После завершения реакции проводили концентрирование с удалением растворителя; желтоватый осадок осаждали посредством добавления воды; проводили фильтрование после 0,5 ч перемешивания, и проводили промывку водой два раза; и собирали продукт, 6 г землисто-желтого твердого вещества 2'-бром-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (выход: 76%). ЖХ-МС: RT=1,66 мин, [М+Н]+=295,11.
На стадии С синтезировали 2'-бром-3-хлор-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2'-бром-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (200 мг, 0,68 ммоль) растворяли в изопропаноле (Пропан-2-ол, 5 мл); добавляли N-хлорсукцинимид (NCS (от англ. N-Chlorosuccinimide), 62 мг, 0,37 ммоль) и каплю 2,2-дихлоруксусной кислоты; и температуру повышали вплоть до 60 градусов Цельсия для взаимодействия в течение 6 ч.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом; органическую фазу объединяли; проводили центробежную сушку; и проводили очистку (этилацетатом) посредством хроматографии на колонке с получением 100 мг белого твердого вещества 2'-бром-3-хлор-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (выход: 83%). ЖХ-МС: RT=1,68 мин, [М+Н]+=329,06.
На стадии D синтезировали 3-(2-гидроксипропан-2-ил)пиридин-2(1Н)-он.
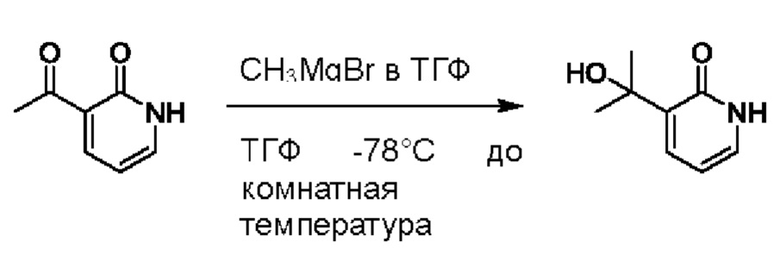
При температуре минус 78 градусов Цельсия раствор ТГФ (5,47 мл, 1 моль/мл) метилмагнийбромида (CH3MgBr) по каплям добавляли в ТГФ (5,0 мл), содержащий 3-ацетил-2(1Н)-пиридон (500,0 мг, 3,65 ммоль); и температуру медленно повышали вплоть до комнатной температуры для взаимодействия в течение 3 ч.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили смешанным раствором дихлорметана и изопропанола (дихлорметан/изопропанол=3/1, 30 мл × 5 раз); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (30 мл × 2 раза); и сушку проводили безводным сульфатом натрия, и затем проводили концентрацию. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: дихлорметан/метанол=10/1). Получали 496,0 мг белого твердого вещества 3-(2-гидроксипропан-2-ил)пиридин-2(1Н)-он (выход: 19,0%). ЖХ-МС: RT=1,33 мин, [М+Н]+=154,14.
На стадии Е синтезировали 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
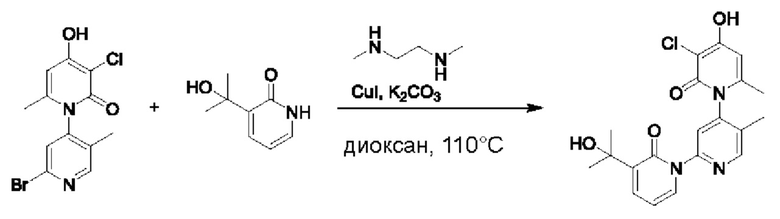
При комнатной температуре 2'-бром-3-хлор-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (100 мг, 0,30 ммоль), 3-(2-гидроксипропан-2-ил)пиридин-2(1Н)-он (93 мг, 0,61 ммоль), Cul (12 мг, 0,06 ммоль) и K2CO3 (84 мг, 0,61 ммоль) растворяли в диоксане (4 мл), добавляли каплю диметилэтилендиамина и затем температуру повышали вплоть до 100 градусов Цельсия для микроволновой реакции в течение 1 ч.
После завершения реакции проводили фильтрацию; промывку проводили этилацетатом; и проводили центробежную сушку на фильтрате, и смешанный образец очищали (этилацетатом) посредством колонок с получением 67 мг белого твердого вещества 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 55%). ЖХ-МС: RT=1,70 мин, [М+Н]+=402,20.
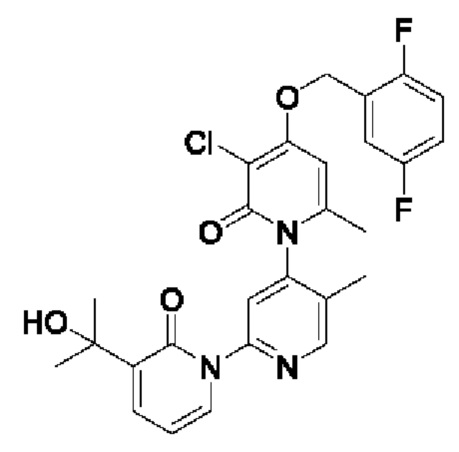
На стадии F синтезировали 3''-хлор-4''-((2,4-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2': 4',1''-терпиридин]-2,2''-дикетон.
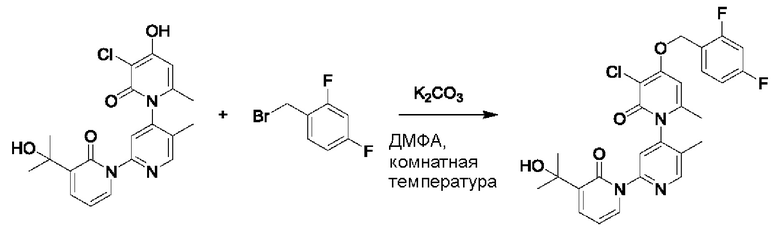
При комнатной температуре 2-фтор-4-хлорбензилбромид (36 мг, 0,17 ммоль) и K2CO3 (24 мг, 0,17 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (35 мг, 0,09 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (20 мл × 3 раза); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (20 мл × 2 раза); и сушку проводили посредством безводного сульфата натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 11,0 мг белого твердого вещества 3''-хлор-4''-((2,4-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 24%). ЖХ-МС: RT=1,95 мин, [М+Н]+=528,13.
Пример осуществления 5

Путь синтеза соединения с порядковым номером, равным 5, показан ниже.
На стадии 2 синтезировали А2-(бромметил)-3-фтор-5-хлорпиридин.

При нуле градусов Цельсия PPh3 (325 мг, 1,24 ммоль) и CBr4 (406 мг, 1,24 ммоль) добавляли к ТГФ (5,0 мл), содержащему (3-фтор-5-хлорпиридин-2-пиридин)метанол (200 мг, 1,24 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: н-гексан/этилацетат=10/1). Получали 142 мг бесцветного масляного 2-(бромметил)-3-фтор-5-хлорпиридина (выход: 51%).
На стадии В синтезировали 3''-хлор-4''-((3-фтор-5-хлорпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2-(бромметил)-3-фтор-5-хлорпиридин (38 мг, 0,17 ммоль) и K2CO3 (24 мг, 0,17 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (35 мг, 0,09 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили посредством этилацетата (20 мл × 3 раза); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (20 мл × 2 раза); и сушку проводили посредством безводного сульфата натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 14 мг белого твердого вещества 3''-хлор-4''-((3-фтор-5-хлорпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 53,7%).
ЖХ-МС: RT=1,91 мин, [М+Н]+=545,17.
Пример осуществления 6
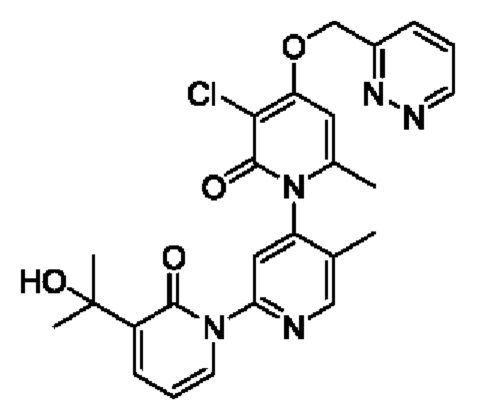
Путь синтеза соединения с порядковым номером, равным 6, показан ниже. На стадии А синтезировали 2-(бромметил)пиридазин.
При нуле градусов Цельсия PPh3 (813,5 мг, 3,11 ммоль) и CBr4 (823,8 мг, 2,48 ммоль) добавляли к ТГФ (5,0 мл), содержащему 2-пиридазинметанол (227,7 мг, 2,07 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: н-гексан/этилацетат=10/1). Получали 328,5 мг бесцветного масляного 2-(бромметил)пиридазина (выход: 91,7%).
На стадии В синтезировали 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиридазин-3-ил)метокси-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2-(бромметил)пиридазин (31,1 мг, 0,18 ммоль) и K2CO3 (33,1 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (20 мл × 3 раза); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (20 мл × 2 раза); и сушку проводили безводным сульфатом натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 30,5 мг белого твердого вещества 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиридазин-3-ил)метокси-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 51,4%).
ЖХ-МС: RT=1,68 мин, [М+Н]+=494,26. Данные по ядерному магнитному резонансу: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9.26 (dd, J=4,7, 1,8 Гц, 1Н), 8.68 (s, 1Н), 7.88-7.82 (m, 3Н), 7.77 (s, 1Н), 7.68 (dd, J=7,0, 2,0 Гц, 1Н), 6.82 (s, 1Н), 6.41 (t, J=6,9 Гц, 1Н), 5.65 (s, 2Н), 5.21 (s, 1Н), 2.07 (s, 3Н), 2.00 (s, 3Н), 1.45 (d, J=4,0 Гц, 6Н).
Пример осуществления 7
Путь синтеза соединения с порядковым номером, равным 7, показан ниже.
На стадии А синтезировали 3''-хлор-3-(2-гидроксипропан-2-ил)-4''-((3-метоксипиридин-2-ил)метокси)-5',6''-диметил-2Н,2''Н-[1,2':4',1''терпиридин]-2,2''-дикетон.

При комнатной температуре 2-(бромметил)-3-метоксипиридин (36,4 мг, 0,18 ммоль) и K2CO3 (33,1 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (20 мл × 3 раза); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (20 мл × 2 раза); и сушку проводили посредством безводного сульфата натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 24 мг белого твердого вещества 3''-хлор-4''-(пиридин-4-илметокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 38,3%).
ЖХ-МС: RT=1,79 мин, [М+Н]+=523,28. Данные по ядерному магнитному резонансу: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.67 (s, 1Н), 8.20-8.16 (m, 1Н), 7.84 (dd, J=6,9, 2,0 Гц, 1Н), 7.77 (s, 1Н), 7.68 (dd, J=7,0, 2,1 Гц, 1Н), 7.56 (d, J=8,4 Гц, 1Н), 7.44 (dd, J=8,4, 4,6 Гц, 1Н), 6.76 (s, 1Н), 6.41 (t, J=6,9 Гц, 1Н), 5.36 (s, 2Н), 5.21 (s, 1Н), 3.88 (s, 3Н), 2.06 (s, 3Н), 1.98 (s, 3Н), 1.45 (d, J=4,2 Гц, 6Н).
Пример осуществления 8

Путь синтеза соединения с порядковым номером, равным 8, показан ниже.
На стадии А синтезировали 4-(((3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2,2''-диоксо-2Н,2''Н-[1,22':4',1''-терпиридин]-4''-ил)окси)метил)-3-фторбензонинтрил.
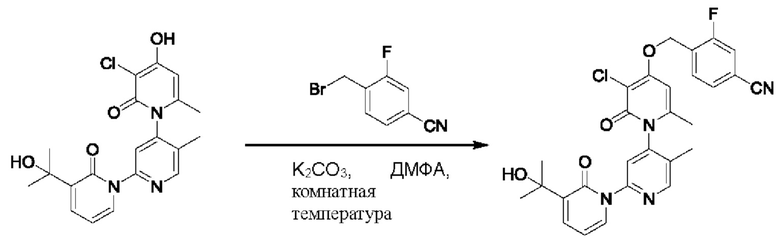
При комнатной температуре 4-(бромметил)-3-фторбензонитрил (38,16 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (20 мл × 3 раза); органическую фазу объединяли; промывку проводили насыщенным солевым раствором (20 мл × 2 раза); и сушку проводили посредством безводного сульфата натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 26,5 мг белого твердого вещества 4-(((3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2,2''-диоксо-2Н,2''Н-[1,22':4',1''-терпиридин]-4''-ил)окис)метил)-3-фторбензонитрил (выход: 41,3%). ЖХ-МС: RT=1,79 мин, [М+Н]+=535,23. Данные по ядерному магнитному резонансу: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.68 (s, 1Н), 7.97 (d, J=9,6 Гц, 1Н), 7.83 (dt, J=10,8, 3,4 Гц, 3Н), 7.77 (s, 1Н), 7.68 (dd, J=7,0, 2,0 Гц, 1Н), 6.80 (s, 1Н), 6.41 (t, J=6,9 Гц, 1Н), 5.49 (s, 2Н), 5.21 (s, 1Н), 2.07 (d, J=8,7 Гц, 3Н), 2.00 (s, 3Н), 1.45 (d, J=3,9 Гц, 6Н).
Пример осуществления 9
Путь синтеза соединения с порядковым номером, равным 9, показан ниже.
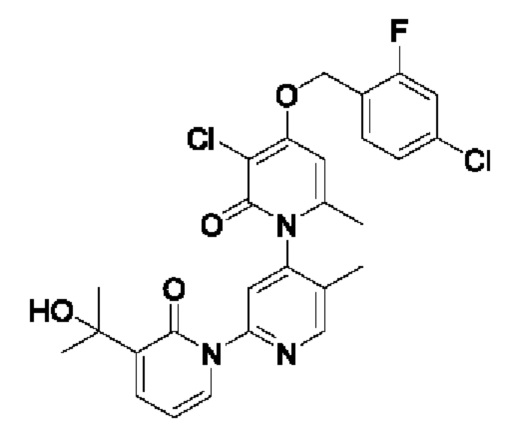
На стадии А синтезировали 3''-хлор-4''-((4-хлор-2-фторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терперидин]-2,2''-дикетон.
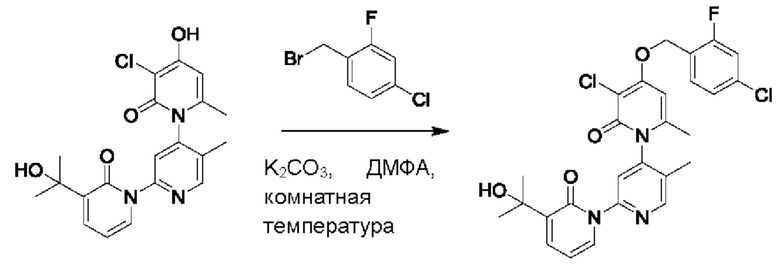
При комнатной температуре 2-фтор-4-хлорбензилбромид (122 мг, 0,50 ммоль) и K2CO3 (69 мг, 0,50 ммоль) добавляли к N,N-ДМФА (2,5 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (100 мг, 0,25 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (20 мл × 3 раза); органическую фазу объединяли; промывку проводили насыщенным водным раствором (20 мл × 2 раза); и сушку проводили посредством безводного сульфата натрия, и затем проводили концентрирование. Полученные остатки очищали посредством хроматографии на колонке с силикагелем (элюент: этилацетат/н-гексан=1/0). Получали 50 мг белого твердого вещества 3''-хлор-4''-((4-хлор-2-фторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (выход: 36%).
ЖХ-МС: RT=2,02 мин, [М+Н]+=544,18. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.67 (s, 1Н), 7.84 (dd, J=6,9, 2,1 Гц, 1Н), 7.77 (s, 1Н), 7.72-7.61 (m, 2Н), 7.55 (dd, J=10,0, 2,0 Гц, 1Н), 7.40 (dd, J=8,3, 1,8 Гц, 1 Н), 6.79 (s, 1Н), 6.41 (t, J=7,0 Гц, 1Н), 5.37 (s, 2Н), 5.21 (s, 1Н), 2.06 (s, 3Н), 2.00 (s, 3Н), 1.45 (d, J=3,9 Гц, 6Н).
Пример осуществления 10

Путь синтеза соединения с порядковым номером 10 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((5-(трифторметил)пиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
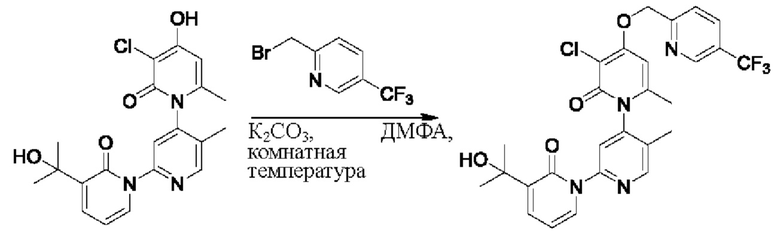
При комнатной температуре 2-(бромметил)-5-(трифторметил)пиридин (43,2 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 11,0 мг белого твердого вещества 3''-хлор-4''-((5-(трифторметил)пиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 16,3%).
ЖХ-МС: RT=1,93 мин, [М+Н]+=561,23.
Пример осуществления 11
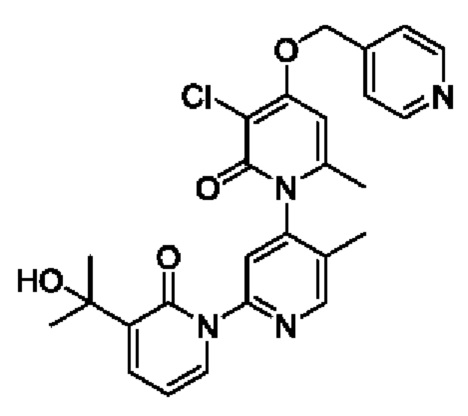
Путь синтеза соединения с порядковым номером 11 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((пиридин-4-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
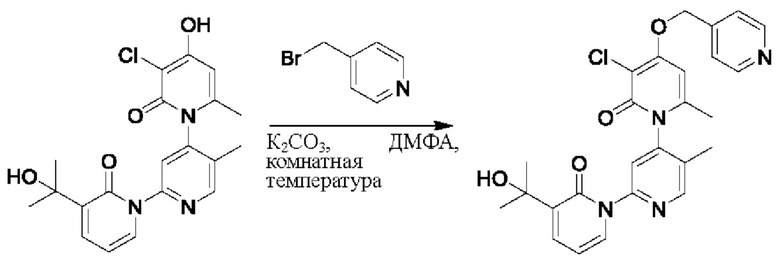
При комнатной температуре 4-(бромметил)пиридин (45,5 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 10,0 мг белого твердого вещества 3''-хлор-4''-((пиридин-4-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 16,9%). ЖХ-МС: RT=1,61 мин, [М+Н]+=493,24.
Пример осуществления 12
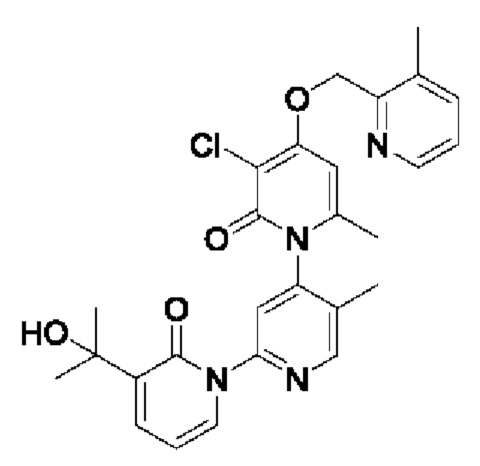
Путь синтеза соединения с порядковым номером 12 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((3-метилпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
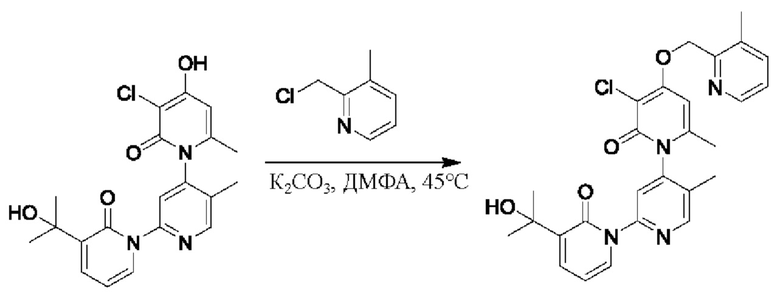
При комнатной температуре 2-(хлорметил)-3-метилпиридин (71 мг, 0,50 ммоль) и K2CO3 (69 мг, 0,50 ммоль) добавляли к N,N-ДМФА (2,5 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпириди н]-2,2''-дикетон (100 мг, 0,25 ммоль) для взаимодействия в течение 4 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 21 мг белого твердого вещества 3''-хлор-4''-((3-метилпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2'' Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 16%).
ЖХ-МС: RT=1,75 мин, [М+Н]+=507,21.
Пример осуществления 13
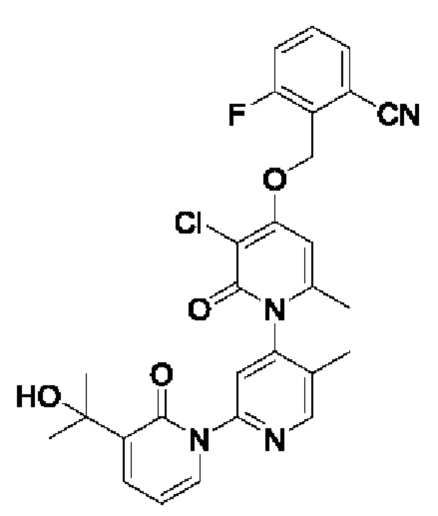
Путь синтеза соединения с порядковым номером 13 показан следующим образом.
На стадии А синтезировали 2-(((3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2,2''-дион-2Н,2''Н-[1,2':4',1''-терпиридин]-4''-ил)окси)метил)-3-фторбензонитрил.

При комнатной температуре 2-(бромметил)-3-фторбензонитрил (38,16 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 29,5 мг белого твердого вещества 2-(((3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2,2''-дион-2Н,2''Н-[1,2':4',1''-терпиридин]-4''-ил)окси)метил)-3-фторбензонитрила (выход: 45,9%).
ЖХ-МС: RT=1,88 мин, [М+Н]+=535,21. 1Н ЯМР (400 МГц, ДМСО) δ 8.70 (s, 1Н), 7.92-7.84 (m, 2Н), 7.82 (s, 1Н), 7.80-7.73 (т, 2Н), 7.70 (dd, J=7.0, 2.1 Гц, 1Н), 6.88 (s, 1Н), 6.43 (t, J=6.9 Гц, 1Н), 5.46 (s, 2Н), 5.23 (s, 1Н), 2.09 (s, 3Н), 2.05 (s, 3Н), 1.48 (s, 3Н), 1.47 (s, 3Н).
Пример осуществления 14

Путь синтеза соединения с порядковым номером 14 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((5-хлорпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2-(бромметил)-5-хлорпиридин (36,9 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 14,6 мг белого твердого вещества 3''-хлор-4''-((5-хлорпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 23,1%).
ЖХ-МС: RT=1,89 мин, [М+Н]+=527,20.
Пример осуществления 15
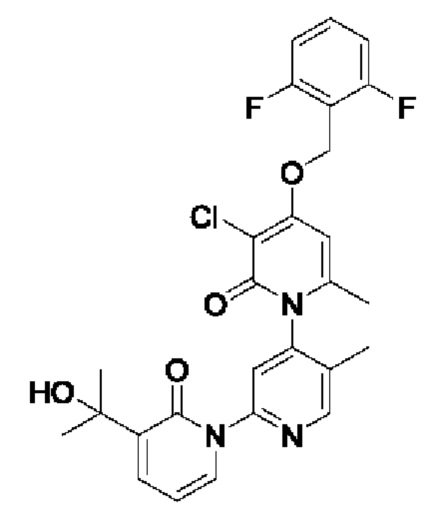
Путь синтеза соединения с порядковым номером 15 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((2,6-дифторфенил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
При комнатной температуре 2-(бромметил)-1,3-дифторбензол (37,26 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 15,31 мг белого твердого вещества 3''-хлор-4''-((2,6-дифторфенил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 24,1%).
ЖХ-МС: RT=1,93 мин, [М+Н]+=528,21.
Пример осуществления 16
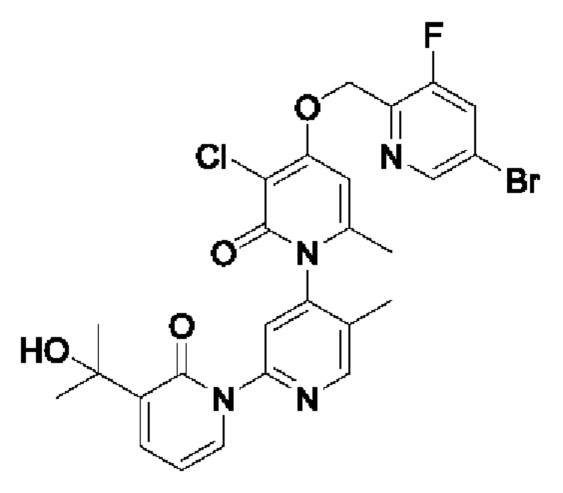
Путь синтеза соединения с порядковым номером 16 показан следующим образом. На стадии А синтезировали 2-(бромметил)-3-фтор-5-бромпиридин.
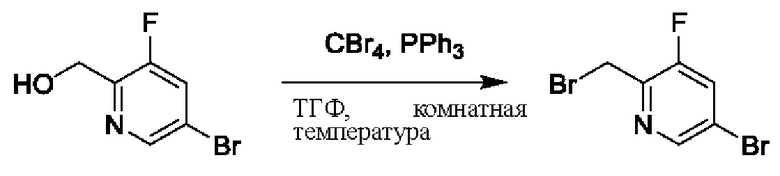
При 0°С PPh3 (267 мг, 1,02 ммоль) и CBr4 (334 мг, 1,02 ммоль) успешно добавляли к ТГФ (10 мл), содержащему (3-фтор-5-бромпиридин-2-пиридин)метанол (210 мг, 1,02 ммоль) для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции осуществляли концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: н-гексан/этилацетат=10/1). Получали 200 мг бесцветного масла 2-(бромметил)-3-фтор-5-бромпиридина (выход: 73%).
На стадии В синтезировали 3''-хлор-4''-((3-фтор-5-бромпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил -2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
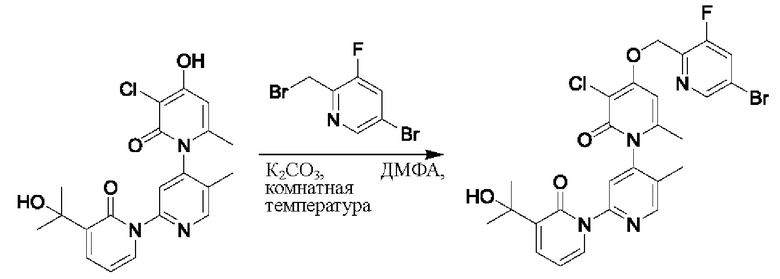
При комнатной температуре 2-(бромметил)-3-бром-5-фторпиридин (134 мг, 0,50 ммоль) и K2CO3 (39 мг, 0,50 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (100 мг, 0,25 ммоль) для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 14 мг белого твердого вещества 3''-хлор-4''-((3-фтор-5-бромпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил -2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 53,7%).
ЖХ-МС: RT=1,91 мин, [М+Н]+=589,13.
Пример осуществления 17

Путь синтеза соединения с порядковым номером 17 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((2,3-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2': 4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2,3-дифторбензил бромид (31 мг, 0,149 ммоль) и K2CO3 (35 мг, 0,50 ммоль) добавляли к N,N-ДМФА (2,5 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпириди н]-2,2''-дикетон (50 мг, 0,124 ммоль) для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 6,8 мг белого твердого вещества 3''-хлор-4''-((2,3-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 10%).
ЖХ-МС: RT=1,97 мин, [М+Н]+=528,18.
Пример осуществления 18
Путь синтеза соединения с порядковым номером 18 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((2,5-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2,5-дифторбензил бромид (31 мг, 0,149 ммоль) и K2CO3 (35 мг, 0,50 ммоль) добавляли к N,N-ДМФА (2,5 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпириди н]-2,2''-дикетон (50 мг, 0,124 ммоль) для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 7,8 мг белого твердого вещества 3''-хлор-4''-((2,5-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 12%).
ЖХ-МС: RT=1,93 мин, [М+Н]+=528,21.
Пример осуществления 19
Путь синтеза соединения с порядковым номером 19 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((3-хлорпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 3-хлор-2-(хлорметил)пиридин (29,2 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N.N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 4 ч при температуре 60°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 19,8 мг белого твердого вещества 3''-хлор-4''-((3-хлорпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 31,3%).
ЖХ-МС: RT=1,84 мин, [М+Н]+=527,20. Данные ядерного магнитного резонанса: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.67 (s, 1Н), 8.59 (dd, J=4.7, 1.4 Гц, 1Н), 8.05 (dd, J=8.1, 1.4 Гц, 1Н), 7.84 (dd, J=6.9, 2.1 Гц, 1H), 7.78 (s, 1H), 7.68 (dt, J=9.0, 2.0 Гц, 1H), 7.51 (dd, J=8.1, 4.7 Гц, 1H), 6.76 (s, 1H), 6.41 (t, J=7.0 Гц, 1H), 5.48 (d, J=12.4 Гц, 2H), 5.21 (s, 1H), 2.07 (s, 3H), 1.98 (s, 3H), 1.46 (d, J=4.1 Гц, 6H).
Пример осуществления 20

Путь синтеза соединения с порядковым номером 20 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((3-фторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
При комнатной температуре 3-фтор-2-(хлорметил)пиридин (26,2 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 4 ч при температуре 60°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 23,0 мг белого твердого вещества 3''-хлор-4''-((3-фторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 37,5%).
ЖХ-МС: RT=1,79 мин, [М+Н]+=511,20. Данные ядерного магнитного резонанса: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.67 (s, 1Н), 8.51-8.47 (m, 1Н), 7.86-7.82 (т, 2Н), 7.78 (s, 1Н), 7.68 (dd, J=7.0, 2.1 Гц, 1Н), 7.57 (dt, J=8.6, 4.4 Гц, 1Н), 6.80 (s, 1Н), 6.41 (t, J=6.9 Гц, 1Н), 5.48 (d, J=1.6 Гц, 2Н), 5.21 (s, 1Н), 2.07 (s, 3Н), 1.99 (s, 3Н), 1.46 (d, J=4.1 Гц, 6Н).
Пример осуществления 21
Путь синтеза соединения с порядковым номером 21 показан следующим образом.
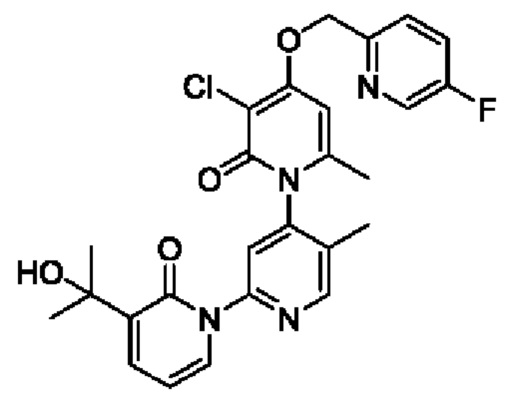
На стадии А синтезировали 3''-хлор-4''-((5-фторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н -[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2-(бромметил)-5-фторпиридин (34,38 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 11,0 мг белого твердого вещества 3''-хлор-4''-((5-фторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 18,0%).
ЖХ-МС: RT=1,82 мин, [М+Н]+=511,23.
Пример осуществления 22

Путь синтеза соединения с порядковым номером 22 показан следующим образом.
На стадии А синтезировали 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиримидин-4-ил)метокси)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 4-(бромметил)пиридин (31,1 мг, 0,18 ммоль) и K2CO3 (33,1 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 31,5 мг белого твердого вещества 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиримидин-4-ил)метокси-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 53,2%).
ЖХ-МС: RT=1,70 мин, [М+Н]+=494,19. Данные ядерного магнитного резонанса: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9.22 (s, 1Н), 8.92 (s, 1Н), 8.68 (s, 1Н), 7.85 (dd, J=7.0, 2.1 Гц, 1Н), 7.77 (s, 1H), 7.71-7.63 (m, 2H), 6.73 (s, 1H), 6.41 (t, J=7.0 Гц, 1H), 5.48 (s, 2H), 5.21 (s, 1H), 2.06 (s, 2H), 1.98 (s, 3H), 1.45 (d, J=4.1 Гц, 6H).
Пример осуществления 23

Путь синтеза соединения с порядковым номером 23 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((3,4-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 3,4-дифторбензил бромид (31 мг, 0,149 ммоль) и K2CO3 (35 мг, 0,50 ммоль) добавляли к N,N-ДМФА (2,5 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпириди н]-2,2''-дикетон (50 мг, 0,124 ммоль) для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 6,5 мг белого твердого вещества 3''-хлор-4''-((3,4-дифторбензил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 10%).
ЖХ-МС: RT=1,95 мин, [М+Н]+=528,22.
Пример осуществления 24
Путь синтеза соединения с порядковым номером 24 показан следующим образом.
На стадии А синтезировали 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (160 мг, 0,77 ммоль) и K2CO3 (177 мг, 1,28 ммоль) добавляли к раствору N,N-ДМФА (2,0 мл), содержащему 2'-бром-3-хлор-4-гидроксил-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (211 мг, 0,64 ммоль) для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции добавляли воду (10,0 мл), и затем твердое вещество осаждали и фильтровали; полученный остаток на фильтре промывали водой два раза; и осуществляли декомпрессионную сушку с получением 290 мг желтоватого твердого вещества 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-она (выход: 99%). ЖХ-МС: RT=1,91 мин, [М+Н]+=458,03.
На стадии В синтезировали 3-ацетил-3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-те рпиридин]-2,2''-дикетон.
При комнатной температуре раствор 1,4-диоксана (5,0 мл), содержащий 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (100 мг, 0,22 ммоль), 3-ацетилпиридин-2(1Н)-он (36 мг, 0,26 ммоль), K2CO3 (60 мг, 0,44 ммоль), Cul (8 мг, 0,044 ммоль) и диметилэтилендиамин (8 мг, 0,087 ммоль) дегазировали для проведения реакции с помощью микроволн в течение 1 часа путем нагревания до 100 градусов Цельсия в атмосфере азота.
После завершения реакции проводили фильтрацию; фильтрат концентрировали; и полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 105 мг белого твердого вещества 3-ацетил-3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 93,7%). ЖХ-МС: RT=1,84 мин, [М+Н]+=513,16.
На стадии С синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(1-гидроксиэтил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
При 0°С борогидрид натрия (6 мг, 0,15 ммоль) добавляли к раствору метанола (МеОН, 2,0 мл), содержащему 3-ацетил-3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50 мг, 0,097 ммоль), для взаимодействия в течение 0,5 ч путем повышения температуры до комнатной температуры.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 3 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (5 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0), и затем получали 36 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(1-гидроксиэтил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 72%).
ЖХ-МС: RT=1,84 мин, [М+Н]+=513,16. 1Н ЯМР (400 МГц, ДМСО) δ 8.67 (s, 1Н), 8.59 (d, J=2.3 Гц, 1Н), 8.12-8.04 (m, 1Н), 7.89-7.83 (m, 1Н), 7.79 (d, J=11.5 Гц, 1Н), 7.60-7.53 (m, 1Н), 6.79 (s, 1Н),6.43 (t, J=6.9 Гц, 1Н), 5.49 (t, J=9.4 Гц, 2Н), 5.15-5.08 (m, 1Н), 4.73 (dd, J=10.7, 5.6 Гц, 1Н), 2.06 (s, 3Н), 2.00 (s, 3Н), 1.26 (dd, J=8.8, 6.3 Гц, 3Н).
Пример осуществления 25
Путь синтеза соединения с порядковым номером 25 показан следующим образом. На стадии А синтезировали 2-(бромметил)-3-хлор-5-фторпиридин.

При 0°С, PPh3 (813,5 мг, 3,11 ммоль) и CBr4 (823,8 мг, 2,48 ммоль) добавляли к ТГФ (5,0 мл), содержащему (3-хлор-5-фторпиридин-2-ил)метанол (334,4 мг, 2,07 ммоль), для взаимодействия в течение 1 ч путем повышения температуры до комнатной температуры.
После завершения реакции осуществляли концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: н-гексан/этилацетат=10/1). Получали 380,6 мг бесцветного масла 2-(бромметил)-3-хлор-5-фторпиридин (выход: 81,9%).
На стадии В синтезировали 3''-хлор-4''-((3-хлор-5-фторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2-(бромметил)-3-хлор-5-фторпиридин (40,4 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 15,31 мг белого твердого вещества 3''-хлор-4''-((3-хлор-5-фторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 25,7%).
ЖХ-МС: RT=1,89 мин, [М+Н]+=545,17.
Пример осуществления 26
Путь синтеза соединения с порядковым номером 26 показан следующим образом.
На стадии А синтезировали 3-(2-гидроксибутан-2-ил)пиридин-2-(1Н)-он.

При комнатной температуре 3-ацетилпиридин-2(1Н)-он (300,0 мг, 2,2 ммоль) растворяли в ТГФ (10,0 мл), и замену азота осуществляли 3 раза; температуру понижали до минус 20°С; и медленно по каплям добавляли этилмагния бромид (EtMgBr, 6,6 мл, 1,0 моль/л), и затем добавляли EtMgBr, взаимодействие проводили в течение 1 ч путем медленного повышения температуры до комнатной температуры.
После завершения реакции, реакцию гасили добавлением насыщенного раствора хлорида аммония (25,0 мл) при 0°С; проводили экстракцию этилацетатом (15 мл × 5 раз); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл × 1 раз); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: н-гексан/этилацетат=1/3). Получали 200,0 мг бесцветного масла 3-(2-гидроксибутан-2-ил)пиридин-2(1Н)-она (выход: 54,1%). ЖХ-МС: RT=1,57 мин, [М+Н]+=168,17.
На стадии В синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксибутан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (200,0 мг, 0,5 ммоль), 3-(2-гидроксибутан-2-ил)пиридин-2-(1 Н)-он (109,7 мг, 0,6 ммоль) и K2CO3 (138,0 мг, 1,0 ммоль) растворяли в 1,4-диоксане (5,0 мл); добавляли Cul (18,2 мг, 1,0 ммоль) и диметилэтилендиамин (0,01 мл); и 3 раза осуществляли замену азота, для проведения реакции с помощью микроволн в течение 1 ч путем повышения температуры до 100°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 4 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл × 1 раз); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=1/20). Получали 90,0 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксибутан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 34,6%).
ЖХ-МС: RT=1,89 мин, [М+Н]+=543,27.
Пример осуществления 27
Путь синтеза соединения с порядковым номером 27 показан следующим образом.
На стадии А синтезировали (3-фтор-5-циклопропилпиридин-2-ил)метанол.

При комнатной температуре циклопропилбороновую кислоту (177 мг, 2,24 ммоль), палладиевый катализатор (Pd(dppf)Cl2, 82 мг, 0,11 ммоль) и K2CO3 (309 мг, 2,24 ммоль) добавляли к смеси растворителей (диоксан/воды=5/1, 18 мл), содержащей (3-фтор-5-бромпиридин-2-ил)метанол (230 мг, 1,12 ммоль), для взаимодействия в течение 8 ч путем повышения температуры до 100°С.
После завершения реакции осуществляли фильтрацию; фильтрат концентрировали и разбавляли путем добавления воды; проводили экстракцию этилацетатом (10 мл × 3 раза); органическую фазу объединяли; и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/10), с получением 80 мг бесцветной жидкости (3-фтор-5-циклопропилпиридин-2-ил)метанола (выход: 43%).
На стадии В синтезировали 2-(бромметил)-3-фтор-5-циклопропилпиридин.

При 0°С, PPh3 (126 мг, 0,48 ммоль) и CBr4 (157 мг, 0,48 ммоль) добавляли к ТГФ (5 мл), содержащему (3-фтор-5-циклопропилпиридин-2-ил)метанол (80 мг, 0,48 ммоль), для осуществления взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции осуществляли концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: н-гексан/этилацетат=10/1), с получением 40 мг бесцветного масла 2-(бромметил)-3-фтор-5-циклопропилпиридина (выход: 36%).
На стадии С синтезировали 3''-хлор-4''-((3-фтор-5-циклопропилпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
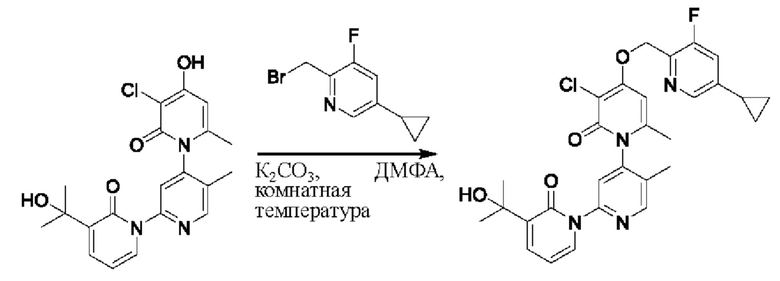
При комнатной температуре 2-(бромметил)-3-фтор-5-циклопропилпиридин (40 мг, 0,17 ммоль) и K2CO3 (48 мг, 0,35 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (68 мг, 0,17 ммоль) для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 10 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0) с получением 1,07 мг белого твердого вещества 3''-хлор-4''-((3-фтор-5-циклопропилпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 1%).
ЖХ-МС: RT=1,91 мин, [М+Н]+=551,21.
Пример осуществления 28

Путь синтеза соединения с порядковым номером 28 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((пиразин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2-(бромметил)пиразин (31,14 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 12,0 мг белого твердого вещества 3''-хлор-4''-((пиразин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 20,3%).
ЖХ-МС: RT=1,72 мин, [М+Н]+=494,23.
Пример осуществления 29

Путь синтеза соединения с порядковым номером 29 показан следующим образом.
На стадии А синтезировали 3-ацетил-4-метилпиридин-2(1Н)-он.

При комнатной температуре 4-метил-2-оксо-1,2-дигидропиридин-3-карбонитрил (350,0 мг, 2,6 ммоль) растворяли в ТГФ (10,0 мл), и замену азота осуществляли 3 раза; температуру понижали до минус 20°С; по каплям добавляли MeMgBr (2,2 мл, 3,0 моль/л), и после добавления MeMgBr взаимодействие проводили в течение 2 ч путем медленного повышения температуры до комнатной температуры; взаимодействие проводили в течение 1 ч путем повышения температуры до 40°С; и добавляли 10 мг, 1 моль/л хлористоводородной кислоты, и взаимодействие проводили в течение 1 ч путем понижения температуры до комнатной температуры.
После завершения реакции, реакционную смесь гасили добавлением 25,0 мл насыщенного раствора бикарбоната натрия; экстракцию проводили этилацетатом (15 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл × 1 раз); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование с получением 350,0 мг желтоватого твердого вещества 3-ацетил-4-метилпиридин-2(1Н)-она (выход: 89,1%). ЖХ-МС: RT=1,21 мин, [М+Н]+=152,10.
На стадии В синтезировали 3-(2-гидроксипропан-2-ил)-4-метилпиридин-2(1Н)-он.

При комнатной температуре 3-ацетил-4-метилпиридин-2(1Н)-он (350,0 мг, 2,3 ммоль) растворяли в ТГФ (10,0 мл), и замену азота осуществляли 3 раза; температуру понижали до минус 20°С; и по каплям добавляли MeMgBr (2,3 мл, 3,0 моль/л), и после добавления MeMgBr взаимодействие проводили в течение 2 ч путем медленного повышения температуры до комнатной температуры.
После завершения реакции, реакционную смесь гасили добавлением насыщенного раствора хлорида аммония (30,0 мл); экстракцию проводили этилацетатом (15 мл × 5 раз); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл × 1 раз); проводили сушку безводным сульфатом натрия, а затем проводили концентрирование; и полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=1/20) с получением 106,0 мг желтоватого твердого вещества 3-(2-гидроксипропан-2-ил)-4-метилпиридин-2(1 Н)-она (выход: 27,5%). ЖХ-МС: RT=1,57 мин, [М+Н]+=168,17.
На стадии С синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-4,5',6''-триметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (100,0 мг, 0,2 ммоль), 3-(2-гидроксибутан-2-ил)-4-метилпиридин-2-(1Н)-он (54,8 мг, 0,3 ммоль) и K2CO3 (58,0 мг, 0,4 ммоль) растворяли в 1,4-диоксане (5,0 мл); добавляли Cul (8,0 мг, 0,4 ммоль) и диметилэтилендиамин (0,01 мл); и 3 раза осуществляли замену азота, для проведения реакции с помощью микроволн в течение 1 ч путем повышения температуры до 100°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 4 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл × 1 раз); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=1/20). Получали 50,0 мг желтоватого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-4,5',6''-триметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 43,9%).
ЖХ-МС: RT=1,89 мин, [М+Н]+=543,24. 1Н ЯМР (400 МГц, ДМСО): δ=8.66 (s, 1Н), 8.59 (d, J=2.4 Гц, 1Н), 8.10-8.05 (m, 1Н), 7.74 (d, J=1.7 Гц, 1Н), 6.79 (s, 1Н), 6.21 (d, J=7.3 Гц, 1Н), 5.90 (s, 1Н), 5.47 (d, J=1.5 Гц, 2Н), 3.57 (s, 1Н), 2.44 (s, 3Н), 2.06 (s, 3Н), 1.99 (s, 3Н), 1.53 (d, J=3.3 Гц, 6Н).
Пример осуществления 30
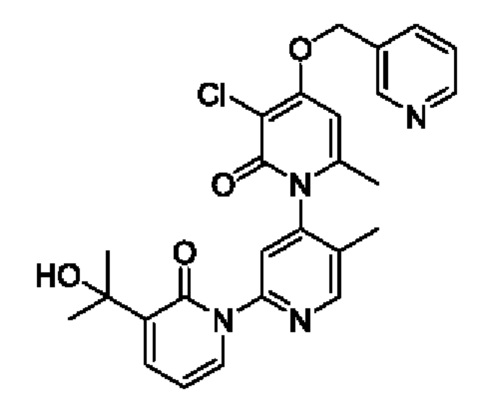
Конкретный путь синтеза соединения с порядковым номером 30 показан следующим образом.
На стадии А синтезировали 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиридин-3-ил)метокси)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 3-(бромметил)пиридин (31,0 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль) для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 29,6 мг белого твердого вещества 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((пиридин-3-ил)метокси-2Н,2''Н-[1,2':4', 1''-терпиридин]-2,2''-дикетона (выход: 40,1%).
ЖХ-МС: RT=1,64 мин, [М+Н]+=493,22. Данные ядерного магнитного резонанса: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.71 (d, J=1.7 Гц, 1Н), 8.67 (s, 1Н), 8.59 (dd, J=4.8, 1.5 Гц, 1Н), 7.91 (d, J=7.9 Гц, 1Н), 7.84 (dd, J=6.9, 2.0 Гц, 1Н), 7.77 (s, 1Н), 7.68 (dd, J=7.0, 2.1 Гц, 1Н), 7.48 (dd, J=7.8, 4.8 Гц, 1Н), 6.77 (s, 1Н), 6.41 (t, J=7.0 Гц, 1Н), 5.40 (s, 2Н), 5.21 (s, 1Н), 2.06 (s, 3Н), 2.00 (s, 3Н), 1.45 (d, J=3.8 Гц, 6Н).
Пример осуществления 31

Путь синтеза соединения с порядковым номером 31 показан следующим образом.
На стадии А синтезировали 3-ацетил-4-метоксипиридин-2(1Н)-он.
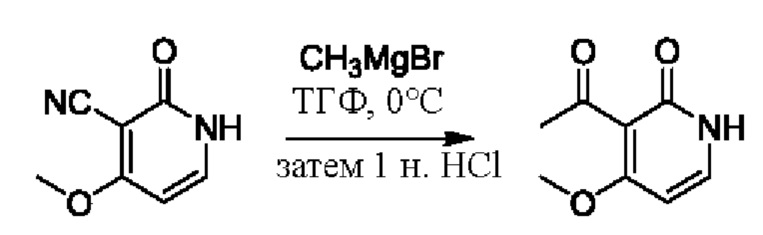
При 0°С, MeMgBr (1,33 мл, 4,00 ммоль, 3 моль/мл) добавляли к раствору ТГФ (4,0 мл), содержащему 4-метокси-2-оксо-1,2-дигидропиридин-3-карбонитрил (200 мг, 1,33 ммоль), для взаимодействия в течение 1 ч путем повышения температуры до комнатной температуры; и добавляли водный раствор хлористоводородной кислоты (1 мл, 1 моль/мл), и взаимодействие продолжали в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 5 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (5 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0) с получением 150 мг желтоватого твердого вещества 3-ацетил-4-метоксипиридин-2(1Н)-она (выход: 67,6%). ЖХ-МС: RT=1,93 мин, [М+Н]+=168,01.
На стадии В синтезировали 3-(2-гидроксипропан-2-ил)-4-метоксипиридин-2(1Н)-он.
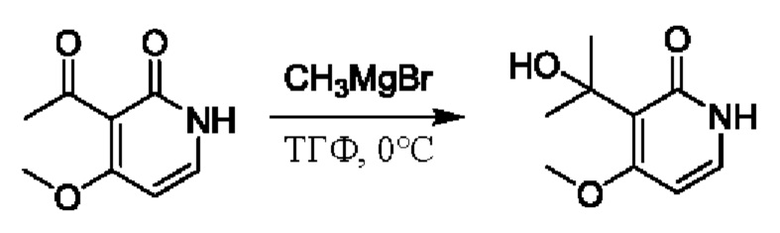
При 0°С, MeMgBr (0,90 мл, 2,69 ммоль, 3 моль/мл) добавляли к раствору ТГФ (4,0 мл), содержащему 3-ацетил-4-метоксипиридин-2(1Н)-он (150 мг, 0,90 ммоль), для взаимодействия в течение 1 ч путем повышения температуры до комнатной температуры.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 5 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (5 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование.
Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0) с получением 65 мг желтоватого твердого вещества 3-(2-гидроксипропан-2-ил)-4-метоксипиридин-2(1Н)-она (выход: 39,6%). ЖХ-МС: RT=1,95 мин, [М+Н]+=184,12.
На стадии С синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-4-метокси-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
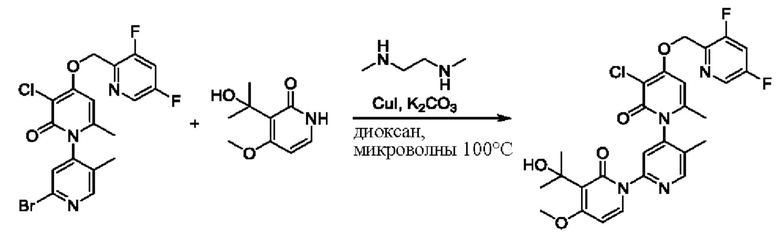
При комнатной температуре раствор 1,4-диоксана (4,0 мл), содержащий 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (82 мг, 0,18 ммоль), 3-(2-гидроксипропан-2-ил)-4-метоксипиридин-2(1Н)-он (58 мг, 0,32 ммоль), K2CO3 (50 мг, 0,36 ммоль), Cul (7 мг, 0,036 ммоль) и диметилэтилендиамин (6 мг, 0,072 ммоль) дегазировали для проведения реакции с помощью микроволн в течение 1 ч путем повышения температуры до 100°С в атмосфере азота.
После завершения реакции, осуществляли фильтрацию; фильтрат концентрировали. Остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 75 мг желтоватого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-4-метокси-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 75%).
ЖХ-МС: RT=1,88 мин, [М+Н]+=559,19. 1Н ЯМР (400 МГц, ДМСО) δ 8.67 (s, 1Н), 8.59 (d, J=2.4 Гц, 1Н),8.10-8.06 (m, 1Н),8.00 (d, J=7.9 Гц, 1Н),7.75 (s, 1Н),7.59 (s, 1Н), 6.78 (s, 1Н), 6.60 (d, J=8.0 Гц, 1Н), 5.47 (d, J=2.0 Гц, 2Н), 3.91 (s, 3Н), 2.07 (s, 3Н), 1.99 (s, 3Н), 1.46 (s, 3Н), 1.43 (s, 3Н).
Пример осуществления 32

Путь синтеза соединения с порядковым номером 32 показан следующим образом.
На стадии А синтезировали 3-ацетил-5-бромпиридин-2(1Н)-он.

При минус 40°С, раствор MeMgBr в ТГФ (1,0 мл, 2,0 моль/л) медленно по каплям добавляли к ТГФ (4,0 мл), содержащему 5-бром-2-оксо-1,2-дигидропиридин-3-карбонитрил (199,0 мг, 1,0 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до комнатной температуры.
После завершения реакции, реакционную смесь гасили добавлением разбавленного водного раствора хлористоводородной кислоты (4,0 мл, 1,0 моль/л); рН доводили до нейтрального с применением насыщенного раствора бикарбоната натрия; проводили экстракцию дихлорметаном (20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); проводили сушку безводным сульфатом натрия, а затем проводили концентрирование; и полученный остаток непосредственно применяли для следующей реакции. ЖХ-МС: RT=1,57 мин, [М+Н]+=216,02.
На стадии В синтезировали 5-бром-3-(2-гидроксипропан-2-ил)пиридин-2-(1Н)-он.
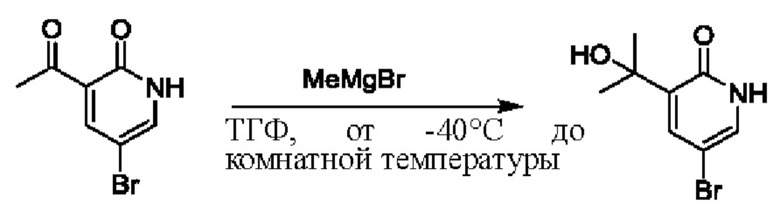
При минус 40°С, раствор MeMgBr в ТГФ (1,0 мл, 2,0 моль/л) медленно по каплям добавляли к ТГФ (4,0 мл), содержащему 3-ацетил-5-бромпиридин-2(1Н)-он (216,0 мг, 1,0 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до комнатной температуры.
После завершения реакции, реакционную смесь гасили добавлением разбавленного водного раствора хлористоводородной кислоты (4,0 мл, 1,0 моль/л); рН доводили до нейтрального с применением насыщенного раствора бикарбоната натрия; проводили экстракцию дихлорметаном (20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=2/1). Получали 150 мг белого твердого вещества 5-бром-3-(2-гидроксипропан-2-ил)пиридин-2-(1Н)-она (выход: 64,7%). ЖХ-МС: RT=1,63 мин, [М+Н]+=234,08.
На стадии С синтезировали 5-бром-3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (137 мг, 0,30 ммоль), 5-бром-3-(2-гидроксипропан-2-ил)пиридин-2(1Н)-он (141,5 мг, 0,61 ммоль), Cul (12 мг, 0,06 ммоль), и K2CO3 (84 мг, 0,61 ммоль) растворяли в диоксане (4 мл), добавляли каплю диметилэтилендиамина, и затем температуру повышали до 100°С для проведения реакции с помощью микроволн в течение 1 ч в атмосфере азота.
После завершения реакции проводили фильтрацию; фильтрат концентрировали; и полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 129,6 мг белого твердого вещества 5-бром-3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 71,1%).
ЖХ-МС: RT=1,97 мин, [М+Н]+=609,14. Данные ядерного магнитного резонанса: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.67 (s, 1Н), 8.59 (d, J=2.3 Гц, 1Н), 8.14-8.04 (m, 2Н), 7.77 (s, 1Н), 7.72 (d, J=2.8 Гц, 1Н), 6.78 (s, 1Н), 5.47 (d, J=1.5 Гц, 2Н), 5.28 (s, 1Н), 2.07 (s, 3Н), 1.98 (s, 3Н), 1.45 (d, J=4.0 Гц, 6Н).
Пример осуществления 33

Путь синтеза соединения с порядковым номером 33 показан следующим образом.
На стадии А синтезировали 3-ацетил-5-хлорпиридин-2(1Н)-он.

При комнатной температуре NCS (500 мг, 3,67 ммоль) добавляли к N,N-ДМФА (5 мл), содержащему 3-ацетилпиридин-2(1Н)-он (500 мг, 3,67 ммоль), для взаимодействия в течение 3 ч путем повышения температуры до 80°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (30 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=10/1). Получали 300 мг желтоватого твердого вещества 3-ацетил-5-хлорпиридин-2(1Н)-она (выход: 54%). ЖХ-МС: RT=1,68 мин, [М+Н]+=172,07.
На стадии В синтезировали 3-(2-гидроксипропан-2-ил)-5-хлорпиридин-2(1Н)-он.
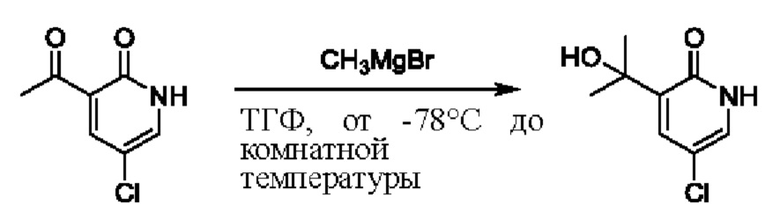
При минус 78°С, раствор MeMgBr в ТГФ (3 мл, 1 моль/мл) по каплям добавляли к ТГФ (20 мл), содержащему 3-ацетил-5-хлорпиридин-2(1 Н)-он (300 мг, 1,75 ммоль), для взаимодействия в течение 3 ч путем повышения температуры до комнатной температуры.
После завершения реакции реакцию гасили водой; экстракцию осуществляли смесью дихлорметана и изопропанола (дихлорметан/изопропанол=3/1, 30 мл × 5 раз); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (30 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=10/1). Получали 150 мг белого твердого вещества 3-(2-гидроксипропан-2-ил)-5-хлорпиридин-2(1Н)-она (выход: 46%). ЖХ-МС: RT=1,59 мин, [М+Н]+=188,10.
На стадии С синтезировали 3'',5-дихлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
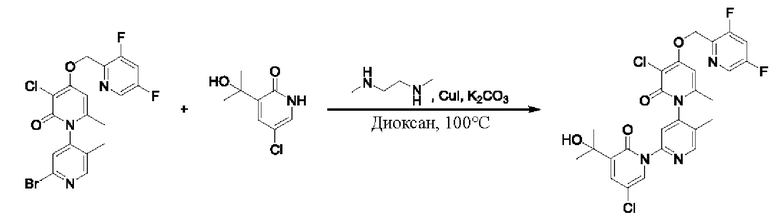
При комнатной температуре 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (80 мг, 0,18 ммоль), 3-(2-гидроксипропан-2-ил)-5-хлорпиридин-2(1Н)-он (33 мг, 0,18 ммоль), Cul (7 мг, 0,04 ммоль), и K2CO3 (48 мг, 0,35 ммоль) растворяли в диоксане (4 мл), и добавляли каплю диметилэтилендиамина, для проведения реакции с помощью микроволн в течение 1 ч путем повышения температуры до 100°С.
После завершения реакции, осуществляли фильтрацию; капельную промывку проводили этилацетатом, и фильтрат концентрировали; и полученный неочищенный продукт получали высокоэффективной жидкостной фазой с получением 50 мг белого твердого вещества 3'',5-дихлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 51%).
ЖХ-МС: RT=1,93 мин, [М+Н]+=563,18. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.67 (s, 1Н), 8.59 (d, J=2.3 Гц, 1Н), 8.14-8.01 (m, 2Н), 7.78 (s, 1Н), 7.65 (d, J=3.0 Гц, 1Н), 6.78 (s, 1Н), 5.47 (d, J=1.4 Гц, 2Н), 5.29 (s, 1Н), 2.07 (s, 3Н), 1.98 (s, 3Н), 1.50-1.42 (m, 6Н).
Пример осуществления 34

Путь синтеза соединения с порядковым номером 34 показан следующим образом.
На стадии А синтезировали 3-ацетил-6-метилпиридин-2(1Н)-он.
При минус 78°С, раствор MeMgBr в ТГФ (5,47 мл, 1 моль/мл) по каплям добавляли к ТГФ (20 мл), содержащему 6-метил-2-оксо-1,2-дигидропиридин-3-карбонитрил (500 мг, 3,73 ммоль), для взаимодействия в течение 3 ч путем медленного повышения температуры до комнатной температуры.
После завершения реакции реакцию гасили водой; экстракцию осуществляли смесью дихлорметана и изопропанола (дихлорметан/изопропанол=3/1, 30 мл × 5 раз); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (30 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=10/1) с получением 300 мг желтоватого твердого вещества 3-ацетил-6-метилпиридин-2(1Н)-она (выход: 53%). ЖХ-МС: RT=1,29 мин, [М+Н]+=152,15.
На стадии В синтезировали 3-(2-гидроксипропан-2-ил)-6-метилпиридин-2(1Н)-он.
При минус 78°С, раствор MeMgBr в ТГФ (3 мл, 1 моль/мл) по каплям добавляли к ТГФ (20 мл), содержащему 3-ацетил-6-метилпиридин-2(1Н)-он (300 мг, 1,98 ммоль), для взаимодействия в течение 3 ч путем повышения температуры до комнатной температуры.
После завершения реакции реакцию гасили водой; экстракцию осуществляли смесью дихлорметана и изопропанола (дихлорметан/изопропанол=3/1, 30 мл × 5 раз); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (30 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=10/1). Получали 200 мг белого твердого вещества 3-(2-гидроксипропан-2-ил)-6-метилпиридин-2(1Н)-она (выход: 60%). ЖХ-МС: RT=1,52 мин, [М+Н]+=168,20.
На стадии С синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6,6''-триметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
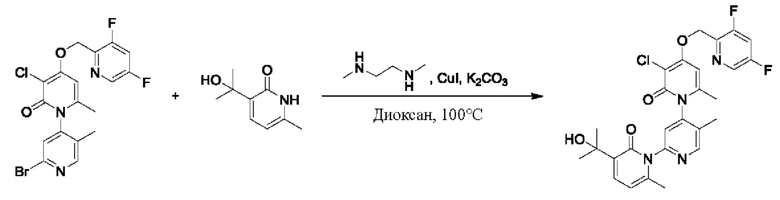
При комнатной температуре 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (80 мг, 0,18 ммоль), 3-(2-гидроксипропан-2-ил)-6-метилпиридин-2(1Н)-он (30 мг, 0,18 ммоль), Cul (7 мг, 0,04 ммоль), и K2CO3 (48 мг, 0,35 ммоль) растворяли в диоксане (4 мл), и добавляли каплю диметилэтилендиамина, для проведения реакции с помощью микроволн в течение 1 ч путем повышения температуры до 100°С.
После завершения реакции, осуществляли фильтрацию; капельную промывку осуществляли этилацетатом, и фильтрат концентрировали; и полученный неочищенный продукт получали высокоэффективной жидкостной фазой с получением 2,49 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6,6''-триметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 3%).
ЖХ-МС: RT=1,88 мин, [М+Н]+=543,18.
Пример осуществления 35
Путь синтеза соединения с порядковым номером 35 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5,5',6''-триметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре триметилбороксин (0,02 мл, 0,16 ммоль), [1,1'-Бис(дифенилфосфино)ферроцен]дихлорпалладий (1,17 мг, 0,0016 ммоль) и Na2CO3 (33,9 мг, 0,32 ммоль) добавляли к смеси растворителей (2,0 мл, толуол/вода=3/1), содержащей 5-бром-3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (100 мг, 0,16 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до 90°С в атмосфере азота.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 15,4 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5,5',6''-триметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 17,8%).
ЖХ-МС: RT=1,89 мин, [М+Н]+=543,25.
Пример осуществления 36
Путь синтеза соединения с порядковым номером 36 показан следующим образом. На стадии А синтезировали 3-(бромметил)-2-метоксипиридин.
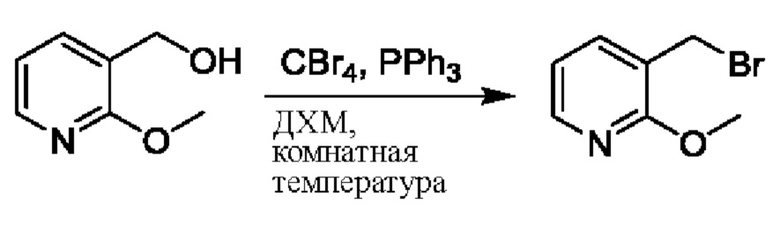
При 0°С, PPh3 (5,65 г, 21,56 ммоль) и CBr4 (5,72 г, 17,24 ммоль) добавляли к дихлорметану (30,0 мл), содержащему (2-метоксипиридин-3-ил)метанол (2,0 г, 14,37 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции осуществляли концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: Получали 2,95 г желтого бесцветного масла 3-(бромметил)-2-метоксипиридина (выход: 97,7%). ЖХ-МС: RT=1,97 мин, [М+Н]+=202,03.
На стадии В синтезировали 2-(2-метоксипиридин-3-ил)ацетонитрил.
При комнатной температуре триметилсилил цианид (3,60 мл, 28,68 ммоль) и раствор тетрабутиламмония фторида в ТГФ (28,7 мл, 1,0 моль/л) добавляли к ацетонитрилу (30,0 мл), содержащему 3-(бромметил)-2-метоксипиридин (2,9 г, 14,34 ммоль), для взаимодействия в течение 1 ч при 80°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: Получали 1,90 г белого твердого вещества 2-(2-метоксипиридин-3-ил)ацетонитрила (выход: 88,82%). ЖХ-МС: RT=1,73 мин, [М+Н]+=149,17.
На стадии С синтезировали 2-(2-метоксипиридин-3-ил)-2-метилпропионитрил.

При комнатной температуре ТГФ (5,0 мл), содержащий 2-(2-метоксипиридин-3-ил)ацетонитрил (1,90 г, 0,013 ммоль), добавляли к ТГФ (15,0 мл), содержащему гидрид натрия (2,0 г, 0,05 ммоль, 60% которого диспергированы в парафиновом масле), для взаимодействия в течение 2 ч при комнатной температуре; и добавляли CH3I для продолжения взаимодействия в течение ночи.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом.
Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: Получали 2,17 г бесцветной жидкости 2-(2-метоксипиридин-3-ил)-2-метилпропионитрила (выход: 94,2%). ЖХ-МС: RT=1,89 мин, [М+Н]+=177,11.
На стадии D синтезировали 2-(2-метоксипиридин-3-ил)-2-метилпропаналь.

При минус 78°С, раствор диизобутилалюминия гидрида в толуоле (12,15 мл, 1,5 моль/л) добавляли к дихлорметану (200,0 мл), содержащему 2-(2-метоксипиридин-3-ил)-2-метилпропионитрил (2,17 г, 12,23 ммоль), для осуществления взаимодействия путем повышения температуры до комнатной температуры.
После завершения реакции реакционную смесь гасили водой; дихлорметан выпаривали; добавляли разбавленную хлористоводородную кислоту при перемешивании в течение 30 мин; экстракцию проводили этилацетатом (по 30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: Получали 1,56 г бесцветной жидкости 2-(2-метоксипиридин-3-ил)-2-метилпропаналя (выход: 70,9%). ЖХ-МС: RT=1,91 мин, [М+Н]+=180,16.
На стадии Е синтезировали 3-(1,1-дифтор-2-метилпропан-2-ил)-2-метоксипиридин.
При 0°С, диэтиламиносератрифторид (2,13 мл, 17,34 моль/л) добавляли к дихлорметану (20,0 мл), содержащему 2-(2-метоксипиридин-3-ил)-2-метилпропаналь (1,56 г, 8,67 ммоль), для осуществления взаимодействия при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили дихлорметаном (по 30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: Получали 700,0 мг бесцветной жидкости 3-(1,1-дифтор-2-метилпропан-2-ил)-2-метоксипиридина (выход: 40,0%). ЖХ-МС: RT=2,05 мин, [М+Н]+=202,15.
На стадии F синтезировали 3-(1,1-дифтор-2-метилпропан-2-ил)пиридин-2(1Н)-он.
При комнатной температуре йодид натрия (780,7 мг, 5,2 ммоль) и хлортриметилсилан (660,0 мкл, 5,2 ммоль) добавляли к ацетонитрилу (7,0 мл), содержащему 3-(1,1-дифтор-2-метилпропан-2-ил)-2-метоксипиридин (700,0 мг, 3,47 ммоль), для осуществления взаимодействия при 65°С.
После завершения реакции реакционную смесь гасили водой; рН доводили до 8 с помощью насыщенного карбоната натрия; экстракцию проводили этилацетатом (по 30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: метанол/дихлорметан=1/20). Получали 533,0 мг желтого бесцветного масла 3-(1,1-дифтор-2-метилпропан-2-ил)пиридин-2(1Н)-она (выход: 81,6%). ЖХ-МС: RT=1,68 мин, [М+Н]+=188,16.
На стадии G синтезировали 3''-хлор-3-(1,1-дифтор-2-метилпропан-2-ил)-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (100,0 мг, 0,22 ммоль), 3-(1,1-дифтор-2-метилпропан-2-ил)пиридин-2(1Н)-он (49,6 мг, 0,26 ммоль), Cul (9,5 мг, 0,05 ммоль), и K2CO3 (60,7 мг, 0,44 ммоль) растворяли в диоксане (2,0 мл), и добавляли каплю N,N-диметилэтилендиамин, для проведения реакции с помощью микроволн в течение 1 ч при 80°С.
После завершения реакции осуществляли фильтрацию с отсасыванием; остаток на фильтре промывали дихлорметаном; и для фильтрата осуществляли концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат). Получали 28,0 мг белого твердого вещества 3''-хлор-3-(1,1-дифтор-2-метилпропан-2-ил)-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 22,6%). ЖХ-МС: RT=1,99 мин, [М+Н]+=563,17.
Пример осуществления 37

Путь синтеза соединения с порядковым номером 37 показан следующим образом.
На стадии А синтезировали 2-хлор-5-циклопропилпиридин-4-амин.
При комнатной температуре раствор 1,4-диоксана (20,0 мл), содержащий 2-хлор-5-йод-4-пиридинамин (1,0 г, 3,93 ммоль), циклопропилбороновую кислоту (0,68 г, 7,86 ммоль), [1,1'-Бис(дифенилфосфино)ферроцен]дихлорпалладий (290 мг, 0,39 ммоль) и K2CO3 (1,09 г, 7,86 ммоль), нагревали до 100°С для взаимодействия в течение 24 ч.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/5) с получением 580 мг 2-хлор-5-циклопропилпиридин-4-амина (выход: 87,6%). ЖХ-МС: RT=0,7 мин, [М+Н]+=169,09.
На стадии В синтезировали
2'-хлор-5'-циклопропил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он.
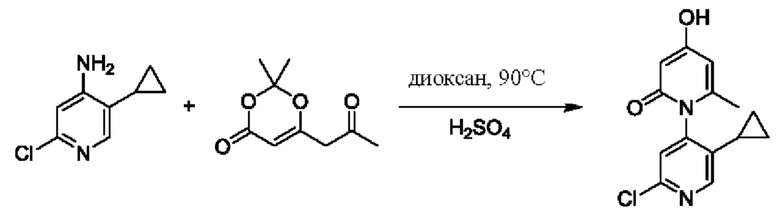
При комнатной температуре 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (760 мг, 4,13 ммоль) добавляли к 1,4-диоксану (10,0 мл), содержащему 2-хлор-5-циклопропилпиридин-4-амин (580 мг, 3,44 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до 90°С. Затем по каплям добавляли раствор серной кислоты (1,5 мл, 10 моль/л) для продолжения взаимодействия в течение 2 ч при 90°С.
После завершения реакции температуру понижали до комнатной температуры; твердое вещество осаждали путем добавления воды, и осуществляли фильтрацию; капельную промывку водой осуществляли 2 раза; и затем проводили декомпрессионную сушку с получением 550 мг желтоватого твердого вещества 2'-хлор-5'-циклопропил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 57,8%). ЖХ-МС: RT=1,70 мин, [М+Н]+=277,12.
На стадии С синтезировали
2',3-дихлор-5'-циклопропил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 1,2-дихлорэтан (2,5 мл), NCS (159 мг, 1,19 ммоль) и дихлоруксусную кислоту (1 капля) добавляли к изопропанолу (5,0 мл), содержащему 2'-хлор-5'-циклопропил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он (300 мг, 1,08 ммоль); и перемешанный раствор нагревали до 65°С для взаимодействия в течение 1 ч.
После завершения реакции реакционную смесь гасили добавлением воды (1,0 мл) растворитель удаляли при пониженном давлении; этилацетат добавляли к полученному остатку для получения кашицы, и затем осуществляли фильтрацию; и осуществляли декомпрессионную сушку с получением 230 мг желтоватого твердого вещества 2',3-дихлор-5'-циклопропил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 68,2%). ЖХ-МС: RT=1,73 мин, [М+Н]+=311,10.
На стадии D синтезировали 2',3-дихлор-5'-циклопропил-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (136 мг, 0,66 ммоль) и K2CO3 (151 мг, 1,01 ммоль) добавляли к раствору N,N-ДМФА (2,0 мл), содержащему 2',3-дихлор-5'-циклопропил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он (170 мг, 0,55 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции добавляли воду (10,0 мл), и затем осаждали твердое вещество; осуществляли фильтрацию, и два раза осуществляли капельную промывку водой; и осуществляли декомпрессионную сушку с получением 180 мг желтоватого твердого вещества 2',3-дихлор-5'-циклопропил-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-2Н-[1,4'-бипири дил]-2-она (выход: 75%). ЖХ-МС: RT=1,96 мин, [М+Н]+=438,13.
На стадии Е синтезировали 3''-хлор-5'-циклопропил-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 1,4-диоксан (5,0 мл), содержащий 2',3-дихлор-5'-циклопропил-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-2Н-[1,4'-бипири дил]-2-он (180 мг, 0,41 ммоль), 3-(2-гидроксипропан-2-ил)-пиридин-2(1Н)-он (75 мг, 0,49 ммоль), K2CO3(114 мг, 0,82 ммоль), Cul (16 мг, 0,082 ммоль) и диметилэтилендиамин (15 мг, 0,16 ммоль), дегазировали для проведения реакции с помощью микроволн в течение 2 ч при 110°С в атмосфере азота.
После завершения реакции, осуществляли фильтрацию; и проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат). Получали 16 мг желтоватого твердого вещества 3''-хлор-5'-циклопропил-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 7%). ЖХ-МС: RT=1,89 мин, [М+Н]+=555,22. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.59 (d, J=2.4 Гц, 1Н), 8.40 (s, 1Н), 8.14-8.01 (m, 1Н), 7.82 (dd, J=6.9, 2.0 Гц, 1Н), 7.75 (s, 1Н), 7.68 (dd, J=7.1, 2.1 Гц, 1Н), 6.80 (s, 1Н), 6.40 (t, J=6.9 Гц, 1Н), 5.47 (s, 2Н), 5.22 (s, 1Н), 2.02 (s, 3Н), 1.54-1.47 (m, 1Н), 1.46 (s, 3Н), 1.44 (s, 3Н), 1.06-1.00 (m, 1Н), 0.89 (m, 2Н), 0.72-0.67 (m, 1Н).
Пример осуществления 38
Путь синтеза соединения с порядковым номером 38 показан следующим образом.
На стадии А синтезировали 2-метил-2-(2-оксо-1,2-дигидропиридин-3-ил)пропионитрил.

При комнатной температуре йодид натрия (255,0 мг, 1,7 ммоль) и хлортриметилсилан (215,0 мкл, 1,7 ммоль) добавляли к ацетонитрилу (3,0 мл), содержащему 2-(2-метоксипиридин-3-ил)-2-метилпропионитрил (200,0 мг, 1,13 ммоль), для осуществления взаимодействия путем повышения температуры до 65°С.
После завершения реакции реакционную смесь гасили водой; рН доводили до 8 с помощью насыщенного карбоната натрия; экстракцию проводили этилацетатом (по 30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: метанол/дихлорметан=1/20). Получали 170,0 мг желтого бесцветного масла 2-метил-2-(2-оксо-1,2-дигидропиридин-3-ил)пропионитрила (92,3%). ЖХ-МС: RT=1,56 мин, [М+Н]+=163,13.
На стадии В синтезировали 2-(3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-2,2''-дикетон-2Н,2''Н-[1,2':4',1''-терпиридин]-3-ил)-2-метилпропионитрил.
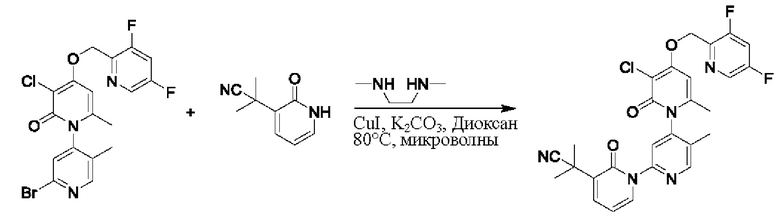
При комнатной температуре
2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (100,0 мг, 0,22 ммоль), 2-метил-2-(2-оксо-1,2-дигидропиридин-3-ил)пропионитрил (42,6 мг, 0,26 ммоль), Cul (9,5 мг, 0,05 ммоль), и K2CO3 (60,7 мг, 0,44 ммоль) растворяли в диоксане (2,0 мл), и добавляли каплю N,N-диметилэтилендиамина для проведения реакции с помощью микроволн в течение 1 ч при 80°С.
После завершения реакции, осуществляли фильтрацию с отсасыванием; проводили капельную промывку дихлорметаном; и проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат). Получали 37,0 мг белого твердого вещества 2-(3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-2,2''-дикетон-2Н,2''Н-[1,2':4',1''-терпиридин]-3-ил)-2-метилпропионитрила (выход: 31,2%). ЖХ-МС: RT=1,91 мин, [М+Н]+=538,20.
Пример осуществления 39

Путь синтеза соединения с порядковым номером 39 показан следующим образом.
На стадии А синтезировали 2'-хлор-4-гидроксил-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (700 мг, 3,78 ммоль) добавляли к 1,4-диоксану (10,0 мл), содержащему 2-хлор-4-амино-5-метоксипиридин (500 мг, 3,15 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до 90°С. Затем по каплям добавляли раствор серной кислоты (1,2 мл, 10 моль/л) для продолжения взаимодействия в течение 2 ч при температуре 90°С.
После завершения реакции температуру понижали до комнатной температуры; твердое вещество осаждали путем добавления воды, и осуществляли фильтрацию; проводили капельную промывку водой 2 раза; и затем осуществляли декомпрессионную сушку с получением 540 мг желтоватого твердого вещества 2'-хлор-4-гидроксил-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 64,3%). ЖХ-МС: RT=1,66 мин, [М+Н]+=267,04.
На стадии В синтезировали 2',3-дихлор-4-гидроксил-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 1,2-дихлорэтан (3,0 мл), NCS (297 мг, 2,23 ммоль) и дихлоруксусную кислоту (3 капли) добавляли к изопропанолу (6,0 мл), содержащему 2'-хлор-4-гидроксил-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-он (540 мг, 2,02 ммоль), для взаимодействия в течение 1 ч путем повышения температуры до 65°С.
После завершения реакции реакционную смесь гасили добавлением воды (1,0 мл); растворитель удаляли при пониженном давлении; кашицу получали путем добавления этилацетата, и затем осуществляли фильтрацию; и осуществляли декомпрессионную сушку с получением 450 мг желтоватого твердого вещества 2',3-дихлор-4-гидроксил-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 73,9%). ЖХ-МС: RT=1,68 мин, [М+Н]+=301,09.
На стадии С синтезировали 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-он.
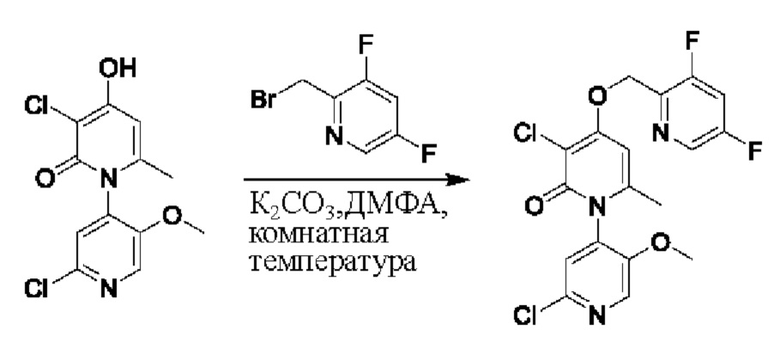
При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (180 мг, 0,87 ммоль) и K2CO3(200 мг, 1,44 ммоль) добавляли к раствору N,N-ДМФА (3,0 мл), содержащему 2',3-дихлор-4-гидроксил-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-он (217 мг, 0,72 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции, добавляли воду (10,0 мл), и затем осаждали твердое вещество; осуществляли фильтрацию, и проводили капельную промывку водой два раза; и осуществляли декомпрессионную сушку с получением 220 мг желтоватого твердого вещества 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 71,4%). ЖХ-МС: RT=1,89 мин, [М+Н]+=428,07.
На стадии D синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5'-метокси-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 1,4-диоксан (5,0 мл), содержащий 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-5'-метокси-6-метил-2Н-[1,4'-бипиридил]-2-он (220 мг, 0,51 ммоль), 3-(2-гидроксипропан-2-ил)-пиридин-2(1Н)-он (118 мг, 0,77 ммоль), K2CO3 (132 мг, 1,02 ммоль), Cul (49 мг, 0,26 ммоль) и диметилэтилендиамин (46 мг, 0,51 ммоль), дегазировали для проведения реакции с помощью микроволн в течение 2 ч при 110°С в атмосфере азота.
После завершения реакции, осуществляли фильтрацию; и проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат). Получали 22 мг желтоватого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5'-метокси-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 7,8%). ЖХ-МС: RT=1,86 мин, [М+Н]+=545,21.
Пример осуществления 40
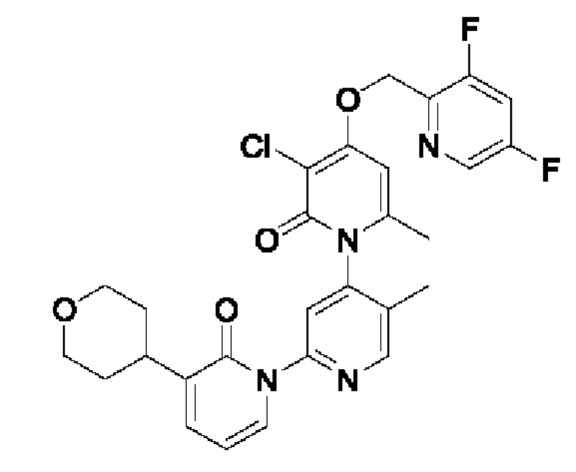
Путь синтеза соединения с порядковым номером 40 показан следующим образом.
На стадии А синтезировали 3-бром-2-((4-метоксибензил)окси)пиридин.
При комнатной температуре 4-метоксибензиловый спирт (1,5 г, 11,0 ммоль) и трет-бутоксид калия (1,34 г, 12,0 ммоль) добавляли к 1,4-диоксану (30,0 мл), содержащему 3-бром-2-хлорпиридин (1,92 г, 10,0 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до 100°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 50 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (50 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/8). Получали 2,85 г желтого бесцветного масла 3-бром-2-((4-метоксибензил)окси)пиридина (выход: 97,3%). ЖХ-МС: RT=2,18 мин, [М+Н]+=294,05.
На стадии В синтезировали 3-(3,6-дигидро-2Н-пиран-4-ил)-2-((4-метоксибензил)окси)пиридин.
При комнатной температуре тетракис(трифенилфосфин)палладий (115,6 мг, 0,1 ммоль) и Na2CO3(270 мг, 2,5 ммоль) добавляли к перемешанному раствору растворителей (4,0 мл, 1,4-диоксан/вода=3/1), содержащему 3-бром-2-((4-метоксибензил)окси)пиридин (293,0 мг, 1,0 ммоль) и пинаколовый сложный эфир 3,6-дигидро-2Н-пиран-4-бороновой кислоты (252,0 мг, 1,2 ммоль), для взаимодействия в течение 12 ч путем повышения температуры до 100°С в атмосфере азота.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/8). Получали 194 мг желтого твердого вещества 3-(3,6-дигидро-2Н-пиран-4-ил)-2-((4-метоксибензил)окси)пиридина (выход: 65,3%). ЖХ-МС: RT=2,14 мин, [М+Н]+=298,24.
На стадии С синтезировали 3-тетрагидро-2Н-пиран-4-ил)пиридин-2(1Н)-он.

При комнатной температуре влажный палладий на угле (125,8 мг, 0,065 ммоль) добавляли к перемешанному раствору растворителей (4,0 мл, метанол/этилацетат=2/1), содержащему 3-(3,6-дигидро-2Н-пиран-4-ил)-2-((4-метоксибензил)окси)пиридин (194,0 мг, 0,65 ммоль); и три раза осуществляли замену водорода, для взаимодействия в течение 12 ч путем повышения температуры до 40°С в атмосфере водорода.
После завершения реакции, осуществляли фильтрацию; и проводили концентрирование. Полученное желтоватое твердое вещество 3-(тетрагидро-2Н-пиран-4-ил)пиридин-2(1Н)-он непосредственно применяли на следующей стадии. ЖХ-МС: RT=1,50 мин, [М+Н]+=180,15.
На стадии С синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-3-тетрагидро-2Н-пиран-4-ил)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
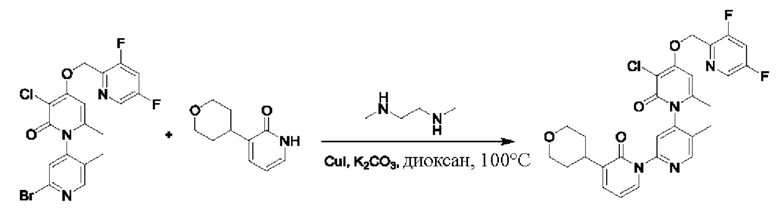
При комнатной температуре 2'-бром-3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (137 мг, 0,30 ммоль), 3-(тетрагидро-2Н-пиран-4-ил)пиридин-2(1Н)-он (109,2 мг, 0,61 ммоль), Cul (12 мг, 0,06 ммоль), и K2CO3 (84 мг, 0,61 ммоль) растворяли в диоксане (4,0 мл); и добавляли диметилэтилендиамин (одну каплю), для проведения реакции с помощью микроволн в течение 40 мин при 80°С в атмосфере азота.
После завершения реакции осуществляли фильтрацию; проводили концентрирование; и проводили очистку с помощью колоночной хроматографии на силикагеле (элюент: этилацетат). Получали 115,3 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5',6''-диметил-3-(тетрагидро-2Н-пиран-4-ил)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 69,4%). ЖХ-МС: RT=1,86 мин, [М+Н]+=555,23.
Пример осуществления 41
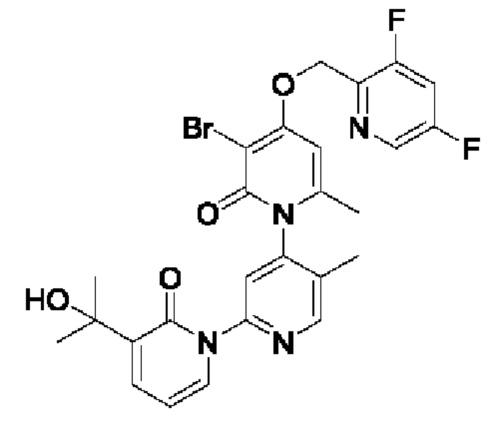
Путь синтеза соединения с порядковым номером 41 показан следующим образом.
На стадии А синтезировали 2',3-дибром-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре N-бромсукцинимид (77 мг, 0,65 ммоль) и дихлоруксусную кислоту (1 капля) добавляли к изопропанолу (3,0 мл), содержащему 2'-бром-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (250 мг, 0,59 ммоль), для взаимодействия в течение 1 ч путем повышения температуры до 65°С.
После завершения реакции, температуру понижали до комнатной температуры; осуществляли фильтрацию; и осуществляли декомпрессионную сушку с получением 230 мг белого твердого вещества 2',3-дибром-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-она (выход: 77,2%). ЖХ-МС: RT=1,93 мин, [М+Н]+=501,99.
На стадии В синтезировали 3''-бром-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 1,4-диоксан (2,0 мл), содержащий 2',3-дибром-4-((3,5-дифторпиридин-2-ил)метокси)-5',6-диметил-2Н-[1,4'-бипиридил]-2-он (100 мг, 0,20 ммоль), 3-(2-гидроксипропан-2-ил)-пиридин-2(1Н)-он (37 мг, 0,24 ммоль), K2CO3 (55 мг, 0,40 ммоль), Cul (8 мг, 0,04 ммоль) и диметилэтилендиамин (7 мг, 0,08 ммоль), дегазировали для проведения реакции с помощью микроволн в течение 1 ч при 80°С в атмосфере азота.
После завершения реакции, осуществляли фильтрацию; и проводили концентрирование. Проводили очистку с помощью колоночной хроматографии на силикагеле (элюент: этилацетат). Получали 47 мг белого твердого вещества 3''-бром-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 41,2%). ЖХ-МС: RT=1,87 мин, [М+Н]+=575,11.
Пример осуществления 42

Путь синтеза соединения с порядковым номером 42 показан следующим образом.
На стадии А синтезировали 2-хлор-5-этилпиридин-4-амин.
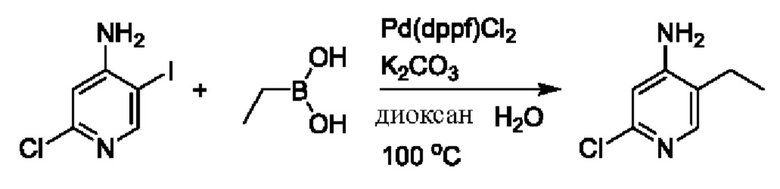
Перемешанный раствор (20,0 мл, 1,4-диоксан:вода=4:1) 1,4-диоксана, содержащий 2-хлор-5-йод-4-пиридинамин (1,5 г, 5,9 ммоль), этилбороновую кислоту (871,1 мг, 11,8 ммоль), [1,1'-Бис(дифенилфосфино)ферроцен]дихлорпалладий (431,2 мг, 0,6 ммоль) и K2CO3 (1,6 г, 11,8 ммоль) и воду нагревали до 100°С для взаимодействия в течение 24 ч.
После завершения реакции, осуществляли капельную промывку путем добавления воды; экстракцию проводили этилацетатом (20 мл × 4 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл); и осуществляли сушку безводным сульфатом натрия, и затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/5) с получением 410,0 мг желтоватого твердого вещества 2-хлор-5-этилпиридин-4-амина (выход: 44,5%). ЖХ-МС: RT=0,47 мин, [М+Н]+=157,09.
На стадии В синтезировали 2'-хлор-5'-этил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (574,6 мг, 3,1 ммоль) добавляли к 1,4-диоксану (10,0 мл), содержащему 2-хлор-5-этилпиридин-4-амин (410,0 мг, 2,6 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до 90°С. Затем по каплям добавляли раствор серной кислоты (1,5 мл, 10 моль/л) для продолжения взаимодействия в течение 2 ч при 90°С.
После завершения реакции, температуру понижали до комнатной температуры; твердое вещество осаждали путем добавления воды, и осуществляли фильтрацию; проводили капельную промывку водой 2 раза; и затем осуществляли декомпрессионную сушку с получением 180,0 мг желтоватого твердого вещества 2'-хлор-5'-этил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 26,2%). ЖХ-МС: RT=1,70 мин, [М+Н]+=265,13.
На стадии С синтезировали 2',3-дихлор-5'-этил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он.
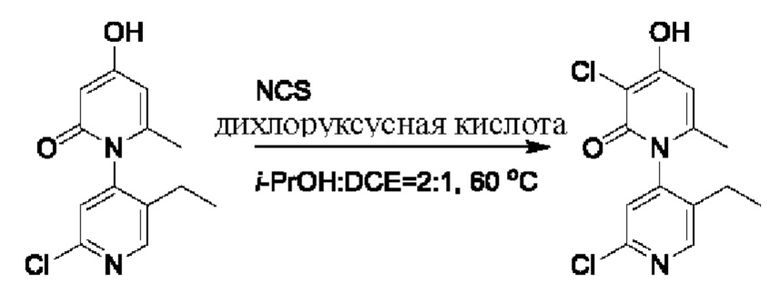
При комнатной температуре 1,2-дихлорэтан (2,5 мл), NCS (100,1 мг, 0,7 ммоль) и дихлоруксусную кислоту (1 капля) добавляли к изопропанолу (5,0 мл), содержащему 2'-хлор-5'-этил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он (180,0 мг, 0,7 ммоль); и перемешанный раствор нагревали до 65°С для взаимодействия в течение 1 ч.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 4 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=4/1) с получением 230,0 мг желтоватого твердого вещества (неочищенного) 2',3-дихлор-5'-этил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-она. ЖХ-МС: RT=1,74 мин, [М+Н]+=299,12.
На стадии D синтезировали 2',3-дихлор-5'-этил-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-2Н-[1,4'-бипиридил]-2-он
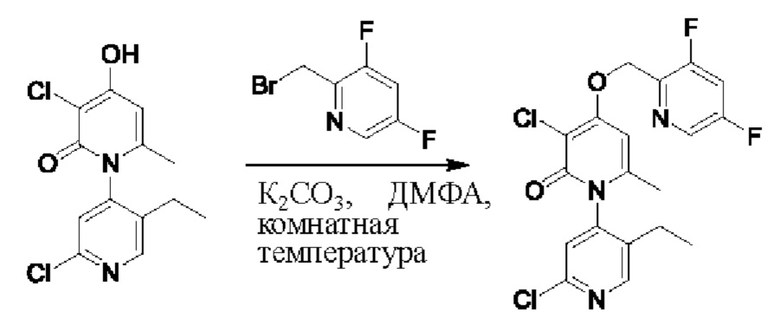
При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (176,2 мг, 0,8 ммоль) и K2CO3 (212,5 мг, 1,5 ммоль) добавляли к раствору N,N-ДМФА (2,0 мл), содержащему 2',3-дихлор-5'-этил-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он (230,0 мг, 0,8 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=4/1) с получением 91,0 мг желтоватого твердого вещества 2',3-дихлор-5'-этил-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 27,7%). ЖХ-МС: RT=1,95 мин, [М+Н]+=426,12.
На стадии Е синтезировали 3''-хлор-5'-этил-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 1,4-диоксан (5,0 мл), содержащий 2',3-дихлор-5'-этил-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-2Н-[1,4'-бипиридил]-2-он (71,0 мг, 0,61 ммоль), 3-(2-гидроксипропан-2-ил)-пиридин-2(1Н)-он (38,3 мг, 0,25 ммоль), K2CO3 (146,1 мг, 0,3 ммоль), Cul (15,8 мг, 0,08 ммоль) и диметилэтилендиамин (15 мг, 0,16 ммоль), дегазировали для проведения реакции с помощью микроволн в течение 2 ч при 110°С в атмосфере азота.
После завершения реакции, осуществляли фильтрацию; и проводили концентрирование. Получение и разделение проводили с помощью высокоэффективной жидкой фазы с получением 3,0 мг белого твердого вещества 3''-хлор-5'-этил-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 3,3%). ЖХ-МС: RT=1,91 мин, [М+Н]+=543,20.
Пример осуществления 43
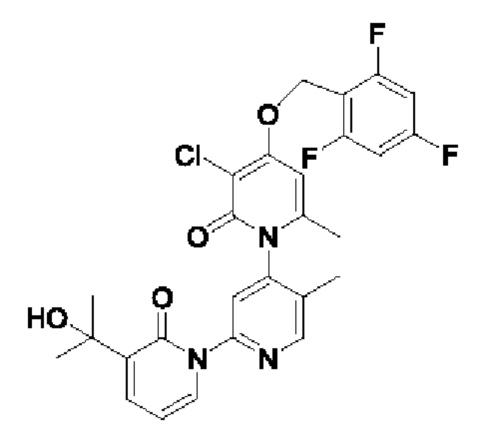
Путь синтеза соединения с порядковым номером 43 показан следующим образом.
На стадии А синтезировали 3''-хлор-4''-((2,4,6-трифторфенил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
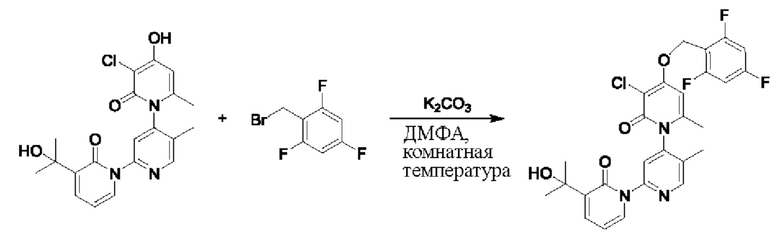
При комнатной температуре 2,4,6-трифторбензил бромид (45 мг, 0,20 ммоль) и K2CO3 (55 мг, 0,40 ммоль) добавляли к N,N-ДМФА (3 мл), содержащему 3''-хлор-4''-((2,4,6-трифторфенил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (80 мг, 0,20 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат). Получали 21 мг белого твердого вещества 3''-хлор-4''-((2,4,6-трифторфенил)окси)-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 25%). ЖХ-МС: RT=1,97 мин, [М+Н]+=546,21.
Пример осуществления 44
Путь синтеза соединения с порядковым номером 44 показан следующим образом.
На стадии А синтезировали 1-(бромметил)-2,3,4-трифторбензол.
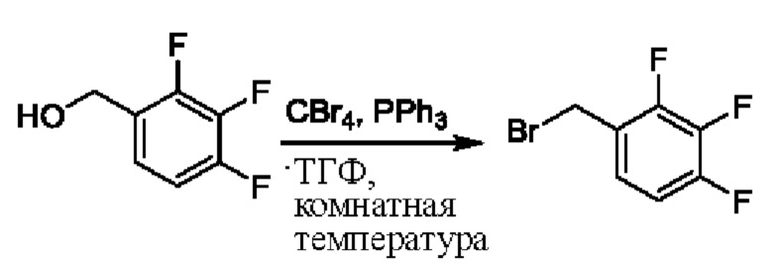
При 0°С, PPh3 (813,5 мг, 3,11 ммоль) и CBr4 (823,8 мг, 2,48 ммоль) добавляли к ТГФ (5,0 мл), содержащему (2,3,4-трифторфенил)метанол (335,4 мг, 2,07 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции осуществляли концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: н-гексан/этилацетат=10/1). Получали 388,0 мг бесцветного масла 1-(бромметил)-2,3,4-трифторбензола (выход: 73,2%). ЖХ-МС: RT=2,11 мин.
На стадии В синтезировали 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((2,3,4-трифторфенил)окси)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
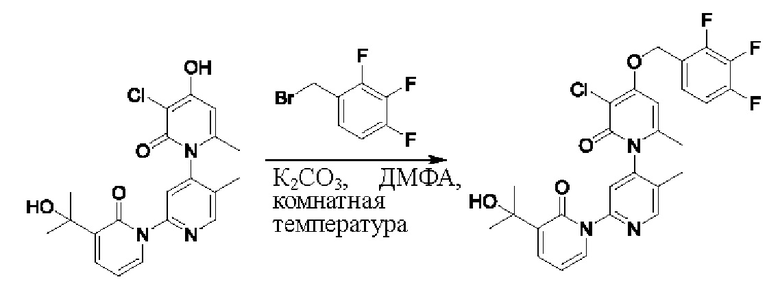
При комнатной температуре 1-(бромметил)-2,3,4-трифторбензол (40,2 мг, 0,18 ммоль) и K2CO3 (33,12 мг, 0,24 ммоль) добавляли к N,N-ДМФА (2,0 мл), содержащему 3''-хлор-4''-гидроксил-3-(2-гидроксипропан-2-ил)-5',6''-диметил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон (50,0 мг, 0,12 ммоль), для взаимодействия в течение 2 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/0). Получали 35,2 мг белого твердого вещества 3''-хлор-3-(2-гидроксипропан-2-ил)-5',6''-диметил-4''-((2,3,4-трифторфенил)окси)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 53,7%). ЖХ-МС: RT=1,98 мин, [М+Н]+=546,20.
Пример осуществления 45
Путь синтеза соединения с порядковым номером 45 показан следующим образом.
На стадии А синтезировали 6-хлор-4-((4-метоксибензил)амино)метил никотинат.

При комнатной температуре метил 4,6-дихлорникотинат (7,0 г, 33,9 ммоль), 4-метоксибензиламин (5,6 г, 40,8 ммоль) и триэтиламин (4,1 г, 40,8 ммоль) растворяли в ацетонитриле (60,0 мл), для взаимодействия в течение 12 ч при комнатной температуре.
После завершения реакции осуществляли концентрирование, и добавляли воду; экстракцию проводили этилацетатом (20 мл × 4 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл); и осуществляли сушку безводным сульфатом натрия, и затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/3) с получением 9,3 г белого твердого вещества 6-хлор-4-((4-метоксибензил)амино)метил никотината (выход: 89,6%). ЖХ-МС: RT=2,07 мин, [М+Н]+=307,13.
На стадии В синтезировали (6-хлор-4-((4-метоксибензил)амино)пиридин-3-ил)метанол.

При комнатной температуре 6-хлор-4-((4-метоксибензил)амино)метил никотинат (9,3 г, 30,3 ммоль) растворяли в ТГФ (90,0 мл); при 0°С, частями добавляли алюмогидрид лития (1,7 г, 45,5 ммоль), для взаимодействия в течение 30 мин путем повышения температуры до комнатной температуры.
После завершения реакции, 1,7 мл воды и 1,7 мл 15% водного раствора гидроксида натрия добавляли на ледяной бане; проводили фильтрацию с отсасыванием; и проводили концентрирование с получением 5,0 г белого твердого вещества (6-хлор-4-((4-метоксибензил)амино)пиридин-3-ил)метанола (выход: 60,0%). ЖХ-МС: RT=1,73 мин, [М+Н]+=1279,16.
На стадии С синтезировали 2-хлор-5-(фторметил)-N-(4-метоксибензил)пиридин-4-амине.
При комнатной температуре (6-хлор-4-((4-метоксибензил)амино)пиридин-3-ил)метанол (5,0 г, 18,0 ммоль) растворяли в дихлорметане (100,0 мл); температуру понижали до температуры ниже нуля °С; по каплям добавляли диэтиламиносератрифторид (4,3 г, 27,0 ммоль); затем добавляли диэтиламиносератрифторид, взаимодействие проводили в течение 30 мин при 0°С.
После завершения реакции реагент выливали в насыщенный водный раствор бикарбоната натрия; осуществляли экстракцию дихлорметаном (30 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл); и осуществляли сушку безводным сульфатом натрия, и затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: дихлорметан),с получением 930,0 мг желтоватой жидкости 2-хлор-5-(фторметил)-N-(4-метоксибензил)пиридин-4-амина (выход: 18,4%). ЖХ-МС: RT=1,93 мин, [М+Н]+=281,17.
На стадии D синтезировали 2-хлор-5-(фторметил)пиридин-4-амин.

При комнатной температуре 2-хлор-5-(фторметил)-N-(4-метоксибензил)пиридин-4-амин (930,0 мг, 3,3 ммоль) растворяли в дихлорметане (10,0 мл); температуру понижали до температуры ниже нуля °С; по каплям добавляли метансульфоновую кислоту (1,0 мл); после добавления метансульфоновой кислоты взаимодействие проводили в течение 2 ч при комнатной температуре.
После завершения реакции, реакционный раствор медленно выливали в насыщенный водный раствор бикарбоната натрия; проводили экстракцию дихлорметаном (10 мл × 3 раза); органическую фазу объединяли; осуществляли сушку безводным сульфатом натрия; и проводили концентрирование под вакуумом с получением 393,0 мг желтоватого твердого вещества 2-хлор-5-(фторметил)пиридин-4-амина (выход: 44,5%). ЖХ-МС: RT=0,47 мин, [М+Н]+=161,08.
На стадии Е синтезировали 2'-хлор-5'-(фторметил)-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (542,9 мг, 2,9 ммоль) добавляли к 1,4-диоксану (10,0 мл), содержащему 2-хлор-5-(фторметил)пиридин-4-амин (393,0 мг, 2,4 ммоль), для взаимодействия в течение 2 ч путем повышения температуры до 90°С. Затем по каплям добавляли раствор серной кислоты (1,0 мл, 10 моль/л) для продолжения взаимодействия в течение 2 ч при 90°С.
После завершения реакции, температуру понижали до комнатной температуры; твердое вещество осаждали путем добавления воды, и осуществляли фильтрацию; проводили капельную промывку водой 2 раза; и затем осуществляли декомпрессионную сушку с получением 163,0 мг желтоватого твердого вещества 2'-хлор-5'-(фторметил)-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 25,3%). ЖХ-МС: RT=1,68 мин, [М+Н]+=269,12.
На стадии F синтезировали 2',3-дихлор-5'-(фторметил)-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он.
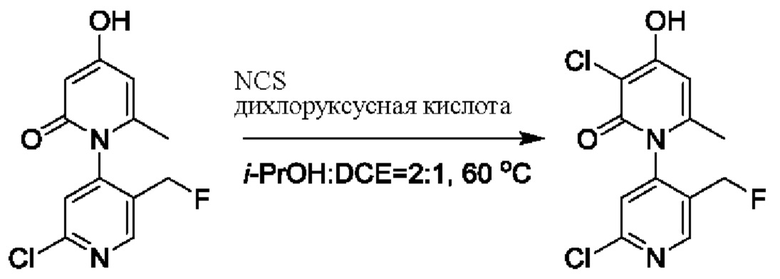
При комнатной температуре 1,2-дихлорэтан (2,5 мл), NCS (78,4 мг, 0,6 ммоль) и дихлоруксусную кислоту (1 капля) добавляли к изопропанолу (5,0 мл), содержащему 2'-хлор-5'-(фторметил)-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он (143,0 мг, 0,5 ммоль), для взаимодействия в течение 1 ч путем повышения температуры до 65°С.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 10 мл × 4 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=4/1) с получением 23,0 мг желтоватого твердого вещества 2',3-дихлор-5'-(фторметил)-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 15,2%). ЖХ-МС: RT=1,68 мин, [М+Н]+=303,07.
На стадии G синтезировали 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-5'-(фторметил)-6-метил-2Н-[1,4'-бипирид ил]-2-он.

При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (19,0 мг, 0,09 ммоль) и K2CO3 (20,9 мг, 0,15 ммоль) добавляли к раствору N,N-ДМФА (5,0 мл), содержащему 2',3-дихлор-5'-(фторметил)-4-гидроксил-6-метил-2Н-[1,4'-бипиридил]-2-он (23,0 мг, 0,07 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 10 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (10 мл); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Полученные остатки очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=6/1) с получением 15,0 мг желтого твердого вещества 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-5'-(фторметил)-6-метил-2Н-[1,4'-бипиридил]-2-она (выход: 49,9%). ЖХ-МС: RT=1,91 мин, [М+Н]+=430,09.
На стадии Н синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5'-(фторметил)-3-(2-гидроксипропан-2-ил)-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.
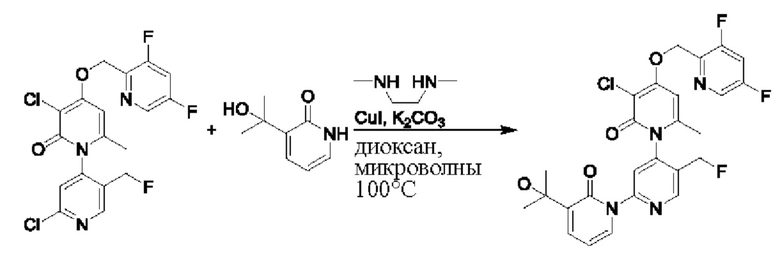
При комнатной температуре 1,4-диоксан (5,0 мл), содержащий 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-5'-(фторметил)-6-метил-2Н-[1,4'-бипирид ил]-2-он (15,0 мг, 0,035 ммоль), 3-(2-гидроксипропан-2-ил)-пиридин-2(1Н)-он (8,0 мг, 0,052 ммоль), K2CO3 (9,6 мг, 0,07 ммоль), Cul (3,3 мг, 0,0175 ммоль) и диметилэтилендиамин (15 мг, 0,16 ммоль), дегазировали для проведения реакции с помощью микроволн в течение 1 ч при 100°С в атмосфере азота.
После завершения реакции, осуществляли фильтрацию; и проводили концентрирование. Получение и разделение проводили с помощью высокоэффективной жидкой фазы с получением 0,5 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-5'-(фторметил)-3-(2-гидроксипропан-2-ил)-6''-метил-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона (выход: 2,6%). ЖХ-МС: RT=1,86 мин, [М+Н]+=547,20.
Пример осуществления 46
Путь синтеза соединения с порядковым номером 46 показан следующим образом.
На стадии А синтезировали 2'-хлор-4-гидроксил-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2-хлор-5-трифторметилпиридин-4-амин (1,55 г, 7,6 ммоль) растворяли в диоксане (20 мл), далее добавляли 2,2-диметил-6-(2-оксопропил)-4Н-1,3-диоксин-4-он (1,4 г, 7,6 ммоль) для взаимодействия в течение 5 ч путем повышения температуры до 90°С; и затем дополнительно по каплям добавляли 10 н. водный раствор серной кислоты до окончания образования твердого вещества, и взаимодействие продолжали в течение 2 ч.
После завершения реакции осуществляли концентрирование; желтоватый осадок осаждали добавлением воды; фильтрацию проводили после 0,5 ч перемешивания, и проводили капельную промывку водой два раза; и получали 1 г землисто-желтого твердого вещества 2'-хлор-4-гидроксил-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-она (выход: 43%). ЖХ-МС: RT=1,75 мин, [М+Н]+=305,04.
На стадии С синтезировали 2',3-дихлор-4-гидроксил-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2'-хлор-4-гидроксил-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-он (1 г, 3,28 ммоль) растворяли в изопропаноле (30 мл); дополнительно добавляли NCS (540 мг, 3,28 ммоль); и затем добавляли дихлоруксусную кислоту (1 капля), для взаимодействия в течение 6 ч путем повышения температуры до 60°С.
После завершения реакции, реакционную смесь гасили водой; затем проводили экстракцию этилацетатом; органическую фазу объединяли; осуществляли сушку безводным сульфатом натрия; осуществляли фильтрацию, и затем проводили концентрирование; и очистку (этилацетат) проводили с помощью колоночной хроматографии с получением 900 мг белого твердого вещества 2',3-дихлор-4-гидроксил-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-она (выход: 82%). ЖХ-МС: RT=1,78 мин, [М+Н]+=338,99.
На стадии В синтезировали 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-он.

При комнатной температуре 2-(бромметил)-3,5-дифторпиридин (185 мг, 0,89 ммоль) и K2CO3 (246 мг, 1,78 ммоль) добавляли к раствору N,N-ДМФА (3 мл), содержащему 2',3-дихлор-4-гидроксил-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-он (300 мг, 0,89 ммоль), для взаимодействия в течение 1 ч при комнатной температуре.
После завершения реакции реакционную смесь гасили водой; экстракцию проводили этилацетатом (по 20 мл × 3 раза); органическую фазу объединяли; промывание производили насыщенным водным раствором соли (20 мл × 2 раза); и проводили сушку безводным сульфатом натрия, а затем проводили концентрирование под вакуумом. Проводили очистку с помощью колоночной хроматографии на силикагеле (элюент: этилацетат). Получали 150 мг белого твердого вещества 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-5'-(трифторметил)-2Н-[1,4'-бипиридил]-2-она (выход: 36%). ЖХ-МС: RT=1,98 мин, [М+Н]+=466,04.
На стадии Е синтезировали 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-6''-метил-5'-(трифторметил)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетон.

При комнатной температуре 2',3-дихлор-4-((3,5-дифторпиридин-2-ил)метокси)-6-метил-5'-(трифторметил)-2Н-[1,4'-бипи ридил]-2-он (50 мг, 0,11 ммоль) Cul (6 мг, 0,02 ммоль) и K2CO3 (61 мг, 0,22 ммоль) растворяли в диоксане (4 мл), и затем добавляли диметилэтилендиамин (одна капля), для проведения реакции с помощью микроволн в течение 1 ч при 80°С.
После завершения реакции, осуществляли фильтрацию; капельную промывку осуществляли этилацетатом; проводили концентрирование; и очистку осуществляли с помощью колоночной хроматографии с получением 1 мг белого твердого вещества 3''-хлор-4''-((3,5-дифторпиридин-2-ил)метокси)-3-(2-гидроксипропан-2-ил)-6''-метил-5'-(трифторметил)-2Н,2''Н-[1,2':4',1''-терпиридин]-2,2''-дикетона. ЖХ-МС: RT=1,97 мин, [М+Н]+=583,20.
Сравнительный пример 1
Структура соединения 3-хлор-4-((3,5-дифторпиридин-2-ил)метокси)-2'-(2-(2-гидроксипропан-2-ил)пиримидин-4-ил -5',6-диметил-2Н-[1,4'-бипиридил]-2-она представлена следующим образом.
Для способа синтеза соединения в Сравнительном примере 1 см. путь синтеза соединения, для которого № заявки на патент Китая - CN201480032278.5, а серийный номер спецификации - 49.
Сверхкритическую жидкостную хроматографию (колонка OD-H) применяли для разделения рацемического соединения сравнительного примера 1 с помощью подвижной фазы диоксида углерода и этанола, и последовательно элюировали атропоизомерные соединения сравнительного примера 1А и 1В.

Соединение сравнительного примера 1A:RT=4,47 мин (SFC, OD-H, колонка с внутренним диаметром 0,46 см и длиной 15 см, изоградиентный метод с 40% этанолом, скорость потока 2,5 мл/мин и время цикла 10 мин); и [a]D25-0,66° (МеОН, прибор Rudolph Autopol I для специфического вращения).
Соединение сравнительного примера 1 В:RT=4,68 мин (SFC, OD-H, колонка с внутренним диаметром 0,46 см и длиной 15 см, изоградиентный метод с 40% этанолом, скорость потока 2,5 мл/мин и время цикла 10 мин). [a]D25+0,68° (МеОН, прибор Rudolph Autopol I для специфического вращения).
Пример осуществления 47: эксперимент по высвобождению TNFα из U937, индуцированного LPS
Регуляция цитокинов в моноцитах человека показала, что путь р38 является ключом к биосинтезу множества провоспалительных цитокинов, включая TNFα, IL-1β и IL-6. Следовательно, ингибирование пути p38 MAPK должно уменьшить воспалительную реакцию за счет снижения биосинтеза провоспалительных цитокинов. Это исследование показывает половину количества соединения по настоящему изобретению, необходимого для ингибирования биосинтеза TNFα (провоспалительного цитокина). Это является отражением действия соединения по настоящему изобретению в отношении обеспечения уменьшения воспаления, что полезно при лечении различных заболеваний, включая хронические воспалительные заболевания, острые воспалительные заболевания или аутовоспалительные заболевания. Потенциал и эффективность ингибитора р38 в отношении блокирования продукции цитокинов оценивают с использованием клеточной линии человека U937.
Реактивы и инструменты:
Носитель 1640 с артикулом А10491-01, Gibco; пенициллин-стрептомицин с артикулом 15140-122, Gibco; эмбриональная бычья сыворотка с артикулом 10099-141С, Gibco; PBS с артикулом 10010-031, Gibco; LPS с артикулом L2880, Sigma; РМА с артикулом Р1585, Sigma; диметилсульфоксид с артикулом D8418-1L, Sigma; и набор TNFα с артикулом K151QWD-4, MSD.
96-луночный планшет с артикулом 3599, Corning; вибрационный шейкер для планшетов с артикулом QB-9002, QILINBEIER; центрифуга с артикулом 5810R, Eppendorf; инкубатор углекислотный с артикулом 371, Thermo; счетчик с артикулом С10281, Gibco; микроскоп с артикулом CKX41, OLYMPUS; и считыватель планшетов MSD с артикулом 1201MESO SECTOR 600, MSD.
Экспериментальные клетки:
U937, АТСС с артикулом CRL-1593.2.
Приготовление лекарственного средства:
Взвешивали примерно 2 мг лекарственного средства; готовили маточный раствор 10 мМ (в пересчете на свободную щелочь) с помощью ДМСО; и маточный раствор разбавляли в 10 раз до 1 мМ, а затем последовательно разбавляли в 4 раза до 250 мкМ, 62,5 мкМ, 15,6 мкМ, 3,9 мкМ, 0,97 мкМ, 0,24 мкМ и 0,061 мкМ. Вышеупомянутую серию разведенных препаратов ДМСО разбавляли в 20 раз в качестве рабочих растворов с использованием среды.
Экспериментальный метод:
В день 0 проводили инокуляцию 10000 клеток на лунку, проводили стимуляцию в течение 48 часов с помощью 20 нг/мл РМА и культивировали при 37°С, 5% CO2.
На 2-й день, 1. Супернатант удаляли из дифференцированного U937, проводили однократную очистку с помощью PBS и добавляли 96 мкл среды 1640.
2. Добавляли 2 мкл соединения, содержащего (ДМСО с конечной концентрацией 0,1%), и проводили культивирование в течение 30 мин при 37°С, 5% CO2.
3. Добавляли 2 мкл LPS (конечная концентрация 100 нг/мл), стимулировали клетки, и проводили культивирование в течение 4 часов при 37°С, 5% CO2.
4. Проводили центрифугирование, отбирали надосадочную жидкость, и определяли содержание TNFα в надосадочной жидкости с помощью ИФА.
Статистический метод:
Содержание TNFα, соответствующее каждой лунке, рассчитывали, используя стандартную кривую из набора.
IC50 соединения рассчитывали с использованием формулы нелинейной аппроксимации GraphPad, а результаты эксперимента см. в Таблице 1.

Из экспериментальных результатов Таблицы 1 можно сделать вывод, что соединение по настоящему изобретению обладает очевидной ингибирующей активностью в отношении продукции TNFα, так что можно регулировать связанные с этим заболевания, такие как воспалительная реакция.
Пример осуществления 48 Определение растворимости.
1. Приготовление контрольного раствора
Взвешивали примерно 0,5 мг испытуемого образца в центрифужной пробирке, добавляли соответствующее количество ДМСО для полного растворения образца, а затем добавляли метанол до 1 мл. Образец, подлежащий тестированию, представляет собой соединение с серийным номером 1 и соединение в Сравнительном примере 1.
2. Приготовление испытуемого раствора
Примерно 1,0 мг каждого соединения в Сравнительном примере 1 и соединения с серийным номером 1 взвешивали в две центрифужные пробирки, и в эти две пробирки добавляли по 1 мл буферных растворов PBS=2,0 и 7,4 соответственно. (Если нужно исследовать больше значений рН, способ приготовления можно вывести по аналогии).
3. Приготовленные контрольный раствор и испытуемый раствор одновременно помещали на водяную баню с температурой 37°С и нагревали в течение 1 ч, через 1 ч растворы вынимали и охлаждали до комнатной температуры, после чего растворы фильтровали через мембранный фильтр 0,22 мкм для получения образца инъекции.
4. Концентрацию образца в испытуемом растворе рассчитывали по формуле C=(A*(ms/vs))/AS, и результаты эксперимента представлены в Таблице 2.
Примечание: ms, vs и AS представляют собой массу, объем и площадь пика образца в контрольном растворе соответственно.
А - площадь пика испытуемого раствора.
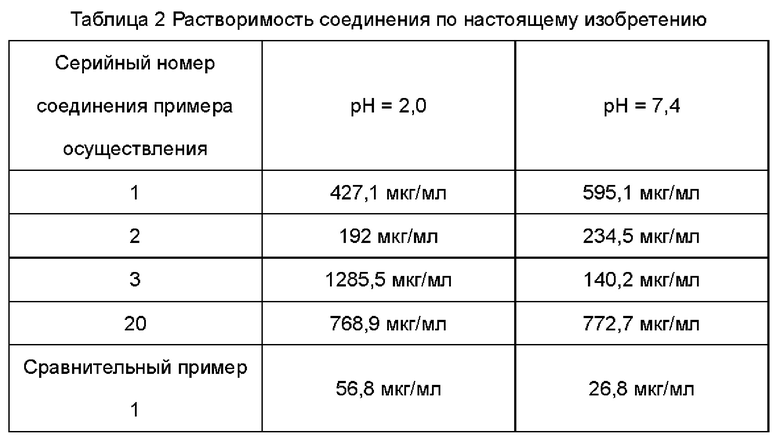
Из результатов экспериментов в Таблице 2 можно сделать вывод, что соединения с порядковыми номерами 1, 2, 3 и 20 в данной заявке обладают желаемой растворимостью и превосходят растворимость соединения в Сравнительном примере 1.
Пример осуществления 49: Эксперимент фармакокинетики
1. Реактивы и приборы
Полиэтиленгликоль 400 (номер партии: GORKREUT, Saen Chemical Technology (Shanghai) Co., Ltd.); ДМСО (номер партии: 20200319, GHTECH (Guangdong) Co., Ltd.); и нормальный физиологический раствор (номер партии: С20052604, Jiangxi Kelun Pharmaceutical Co., Ltd.). Прибор ЖХ-МС (Thermo Fisher Ultimate 3000 UPLC, TSQ QUANTUM ULTRA TSQ Quantum GC).
2. Экспериментальные животные
5 кобелей биглей массой от 5 до 7 кг, приобретенных у компании Beijing Mars Biotechnology Co., Ltd.
3. Подготовка
Тестируемый порошок точно взвешивают и полностью растворяли в ДМСО, затем добавляли ПЭГ-400; осуществляли вихревое и ультразвуковое перемешивание; затем добавляли нормальный физиологический раствор для вортекса и ультразвукового перемешивания, чтобы довести смесь до 0,5 мг/мл (ДМСО:ПЭГ-400:NS=5:60:35, об./об./об.); 5 мл/кг вводили внутрижелудочно, а 1 мл/кг вводили внутривенно.
4. Сбор образцов крови
После введения вышеуказанной смеси собакам путем внутрижелудочного введения (n=3) или внутривенного введения (n=2) из вены передней или задней конечности с помощью иглы для внутривенной инъекции отбирали примерно 1 мл крови соответственно, через 5 мин (только внутривенное введение), 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 24 ч, для введения в пробирки для забора крови, содержащие антикоагулянт ЭДТА-К2; центрифугирование проводили в течение 10 мин при 4000 об/мин для отделения плазмы; а затем пробирки хранили при температуре -80°С для проведения тестов.
5. Биоанализ
Определенное количество испытуемого образца точно взвешивали и растворяли в ДМСО до концентрации 2 мг/мл в виде маточного раствора. Исходный раствор разбавляли до 30000, 10000, 3000, 1000, 300, 100, 50, 30 и 10 нг/мл с использованием смеси ацетонитрил:воды (1:1) для получения рабочих растворов стандартной кривой. 5 мкл рабочего раствора добавляли к 45 мкл холостой плазмы собаки; и перемешивание лунок осуществляли с помощью вортекса для приготовления образцов стандартной кривой, эквивалентных концентрациям в плазме 3000, 1000, 300, 100, 30, 10, 5, 3 и 1 нг/мл. 30 мкл образца стандартной кривой и собранного образца плазмы (плазму, собранную через 5, 15 и 30 минут внутривенного введения, разводили в 5 раз) добавляли к 150 мкл раствору пропранолол-ацетонитрил (внутренний стандарт, 50 нг/мл) для осаждения белка, затем к хорошо перемешанному с помощью вортекса раствору добавляли 100 мкл воды; центрифугирование проводили 5 мин при 4000 об/мин; и супернатант отбирали для анализа ЖХ-МС. Условия обнаружения ЖХ-МС включают следующие.
Хроматографическая колонка: Waters ACQUITY™ PREMIER HSS Т3, 50*2,1 мм, 1,8 мкм.
Подвижная фаза А: вода (0,1% муравьиной кислоты); подвижная фаза В: ацетонитрил; скорость потока: 0,5 мл/мин; и градиентное элюирование показано в Таблице 3 ниже.

6. Обработка данных
После определения концентрации лекарственного средства в крови с помощью ЖХ-МС, некомпартментную модель программного обеспечения WinNonlin 6.1 использовали для расчета фармакокинетических параметров собак породы бигль после введения. Результаты показаны в Таблице 4 ниже.

Из результатов эксперимента в Таблице 4 можно сделать вывод, что по сравнению с соединением Сравнительного примера 1 ФК соединения по настоящему изобретению с порядковым номером 1 у собак имеет более высокую продолжительность воздействия при пероральном введении и биодоступность.
Пример осуществления 50: Эксперимент по скринингу комплекса киназы р38α/MK2

Приготовление лекарственного средства:
Определенное количество лекарственного средства взвешивали и готовили в виде 10 мМ маточного раствора с использованием ДМСО; маточный раствор разбавляли до 100 мкМ (100-кратный рабочий раствор) в два этапа; и затем раствор последовательно разбавляли в 3 раза до 33,33 мкМ, 11,11 мкМ, 3,70 мкМ, 1,23 мкМ, 0,41 мкМ, 0,14 мкМ, 0,046 мкМ, 0,015 мкМ и 0,005 мкМ. Также готовили контроль ДМСО.
Затем ряд разбавленных в ДМСО лекарственных средств разбавляли в 25 раз реакционным буфером 1Х IMAP в качестве четырехкратного рабочего раствора лекарственного средства.
Экспериментальный метод:
a) Реакционный буфер 1Х IMAP использовали для приготовления фермента (MEK6, активный), р38α (неактивный), MK2 (неактивный), субстрата (HSP27) и АТФ.
b) 10 мкл двухкратного рабочего раствора фермента и субстрата и 5 мкл четырехкратного рабочего раствора лекарственного средства последовательно добавляли в черный 384-луночный планшет; после быстрого центрифугирования добавляли 5 мкл рабочего раствора АТФ для запуска ферментативной реакции. Для каждой группы соединений задавали 10 градиентных концентраций и 2 дублирующие лунки; и дополнительно устанавливали контрольную группу ДМСО и группу отрицательного контроля (ДМСО, без АТФ). Конечная концентрация каждого компонента показана в Таблице 7 ниже.

c) после 1 часа инкубации при комнатной температуре добавляли 1Х связующий раствор для завершения ферментативной реакции; после 30 мин инкубации для обнаружения сигнала поляризации флуоресценции (FP) [FP(Ex485/Em520/Em520nm)] использовали многофункциональный считыватель микропланшетов BMGPHERASTER FSX.
Статистический метод:
Формулу нелинейной подгонки Graph Pad Prism 7 использовали для расчета IC50 соединения, и результаты показаны в Таблице 8.

Из экспериментальных результатов Таблицы 8 можно сделать вывод, что соединение по настоящему изобретению обладает очевидной ингибирующей активностью в отношении комплекса р38α/MK2, поэтому можно регулировать связанные заболевания, такие как воспалительная реакция.
Пример осуществления 51: эксперимент по скринингу киназы р38α


Приготовление лекарственного средства:
Взвешивали определенное количество соединения, готовили в виде 10/50 мМ маточного раствора с ДМСО, разбавляли ДМСО в 3 раза и разбавляли до 10 точек концентрации в качестве рабочего раствора соединения.
Экспериментальный метод:
50 нл разведенного рабочего раствора соединения переносили в каждую лунку реакционного планшета (784075, Greiner) с Echo 655, затем добавляли 2,5 мкл (4 нг/мкл) раствора киназы р38α; через 10 мин при комнатной температуре добавляли 2,5 мкл смешанного раствора субстрата киназы (0,2 мг/мл) и АТФ (150 мкМ) для реакции в течение 60 мин при комнатной температуре; и добавляли 4 мкл реагента ADP Glo для инкубации в течение 40 мин при комнатной температуре. Добавляли 8 мкл реагента для обнаружения киназы для инкубации в течение 40 мин при комнатной температуре; и наконец на Envision 2104 считывали светоизлучающий сигнал.
Анализ данных
Формулу нелинейной подгонки Graph Pad Prism 8 использовали для расчета IC50 соединения, и результаты тестов показаны в Таблице 11.

Из результатов экспериментов, приведенных в Таблице 11, можно сделать вывод, что по сравнению с предшествующим уровнем техники соединение по настоящему изобретению обладает более высокой активностью, многократно превышающей активность р38α/(комплекс р38α/MK2), и лучшей селективностью.
Примечание: данные комплекса киназы р38α/MK2 взяты из Примера осуществления 50.
Пример осуществления 52: эксперимент по скринингу киназы р38β



Приготовление лекарственного средства:
Взвешивали определенное количество соединения, готовили в виде 10/50 мМ маточного раствора с ДМСО, разбавляли ДМСО в 3 раза и разбавляли до 10 точек концентрации в качестве рабочего раствора соединения.
Экспериментальный метод:
50 нл разведенного рабочего раствора соединения переносили в каждую лунку реакционного планшета (784075, Greiner) с Echo 655, затем добавляли 2,5 мкл (2 нг/мкл) раствора киназы р38β; через 10 мин при комнатной температуре добавляли 2,5 мкл смешанного раствора субстрата киназы (0,4 мг/мл) и АТФ (100 мкМ) для реакции в течение 60 мин при комнатной температуре; и добавляли 4 мкл реагента ADP Glo для инкубации в течение 40 мин при комнатной температуре. Добавляли 8 мкл реагента для обнаружения киназы для инкубации в течение 40 мин при комнатной температуре; и наконец на Envision 2104 считывали светоизлучающий сигнал.
Анализ данных
Формулу нелинейной подгонки Graph Pad Prism 8 использовали для расчета IC50 соединения, и результаты тестов показаны в Таблице 14.
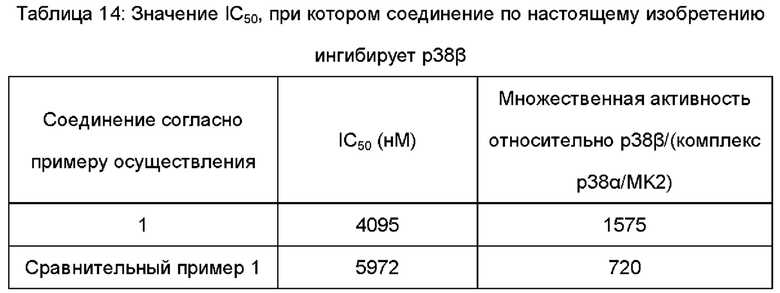
Из результатов экспериментов, приведенных в Таблице 14, можно сделать вывод, что по сравнению с предшествующим уровнем техники соединение по настоящему изобретению обладает более высокой активностью, многократно превышающей активность р38β/(комплекс р38α/MK2), и улучшенной селективностью.
Примечание: данные комплекса киназы р38α/MK2 взяты из Примера осуществления 50.
Вышеуказанные примеры осуществления являются предпочтительными примерами осуществления настоящего изобретения, но примеры осуществления настоящего изобретения не ограничиваются вышеприведенными примерами осуществления и любыми другими изменениями, модификациями, заменами, комбинациями и упрощениями, которые не отклоняются от духа и принципа настоящего изобретения и должны быть эквивалентными заменами и включены в объем охраны настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| ИНГИБИТОР PDE9 И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2793732C2 |
| ПИРИДИН-2-ИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИММУНОМОДУЛИРУЮЩИХ АГЕНТОВ | 2009 |
|
RU2494099C2 |
| СОЕДИНЕНИЕ ТРИАЗИНОНА И ИНГИБИТОР КАЛЬЦИЕВЫХ КАНАЛОВ Т-ТИПА | 2013 |
|
RU2645158C2 |
| ЗАМЕЩЕННЫЕ 3-ФЕНИЛПРОПИОНОВЫЕ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2553263C2 |
| ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-с]ПИРИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА ГИСТАМИНА 4 (hH4R) | 2012 |
|
RU2628074C2 |
| МОРФОЛИНО-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БИЦИКЛИЧЕСКИХ ПИРИМИДИНМОЧЕВИНЫ ИЛИ КАРБАМАТА В КАЧЕСТВЕ ИНГИБИТОРОВ mTOR | 2012 |
|
RU2609208C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
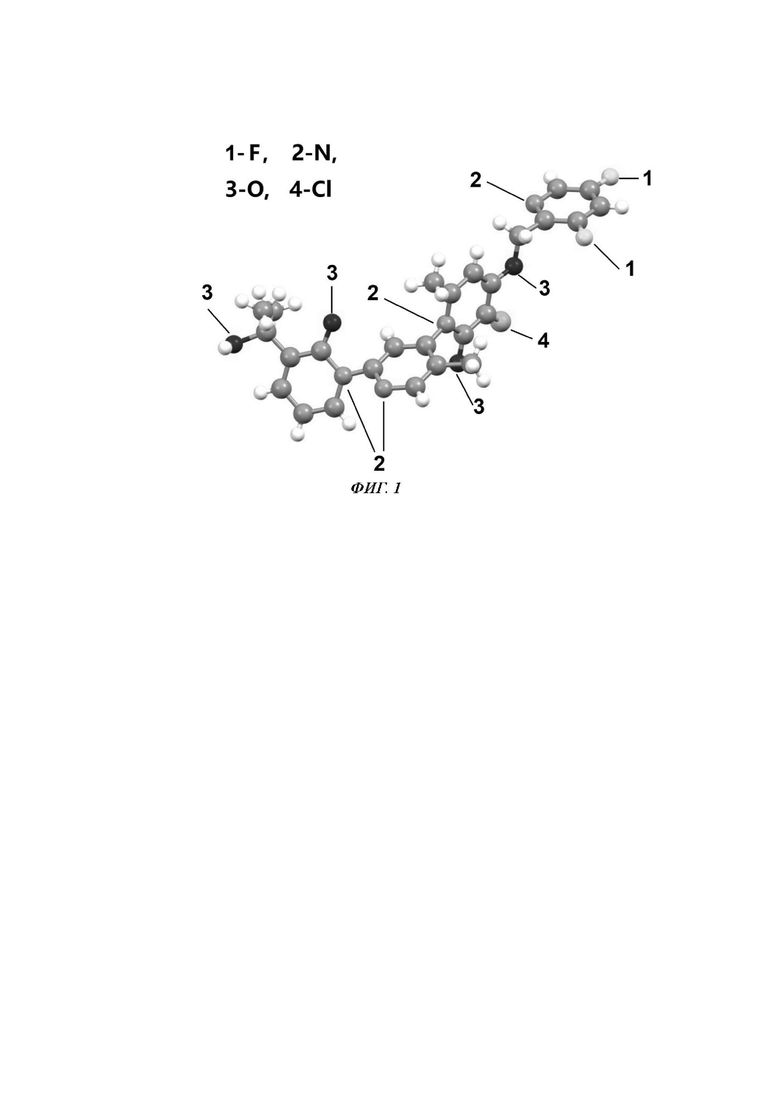



Изобретение относится к соединению, представленному общей формулой (I), где R1’, R1-R9, A, n, m определены в формуле изобретения, его рацемату в качестве ингибитора p38/MK2, способного ингибировать продукцию цитокина TNFα, и используемому для лечения заболеваний, например, такого как артрит. 3 н. и 8 з.п. ф-лы, 6 ил., 14 табл., 52 пр.
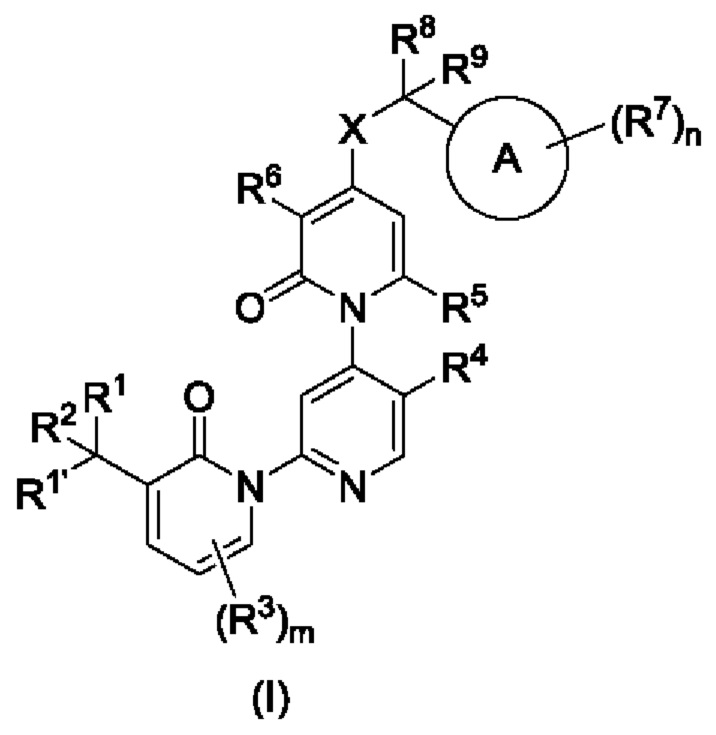
1. Соединение, представленное общей формулой (I), или его рацемат, или стереоизомер
 ,
,
где
R1 и R1' независимо выбраны из водорода или С1-6алкила, или R1 и R1', взятые вместе, циклизированы с образованием  ;
;
R2 выбран из водорода, гидроксила или С1-6галогеналкила;
R3 независимо выбран из водорода, С1-6алкила, галогена или С1-6алкокси;
m равно 0, 1, 2 или 3;
R4 выбран из C1-6алкила, С3-6циклоалкила, С1-6алкокси и C1-6галогеналкила;
R5 выбран из C1-6алкила;
R6 выбран из водорода или галогена;
кольцо А выбрано из фенила или гетероароматического шестичленного кольца, в котором один или несколько атомов углерода ароматического кольца заменены на гетероатом, представляющий собой азот;
R7 независимо выбран из водорода, галогена, циано, С1-6алкила, С3-6циклоалкила, C1-6галогеналкила и С1-6алкокси;
n равно 0, 1, 2, 3, 4 или 5;
X представляет собой О; и
R8 и R9 независимо представляют собой водород.
2. Соединение по п. 1 или его рацемат или стереоизомер, где:
C1-6алкил выбран из метила, этила, пропила, изопропила, н-бутила, изобутила, втор-бутила, трет-бутила, н-пентила, втор-пентила, 1-этилпропила, 2-метилбутила, трет-пентила, 1,2-диметилпропила, изопентила, неопентила, н-гексила, изогексила, втор-гексила, трет-гексила, неогексила, 2-метилпентила, 1,2-диметилбутила или 1-этилбутила; и/или
C1-6алкокси выбран из метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, втор-пентилокси, 1-этилпропокси, 2-метилбутокси, трет-пентилокси, 1,2-диметилпропокси, изопентилокси, неопентилокси, н-гексилокси, изогексилокси, втор-гексилокси, трет-гексилокси, неогексилокси, 2-метилпентилокси, 1,2-диметилбутокси или 1-этилбутокси; и алкоксиалкил выбран из С1-4алкокси-С1-4алкила, и, кроме того, указанный С1-4алкокси-С1-4алкил выбран из метоксиметила, метоксиэтила, метоксипропила, метоксибутила, этоксиметила, этоксиэтила, этоксипропила, этоксибутила, пропоксиметила, пропоксиэтила, пропоксипропила, пропоксибутила, бутоксиметила, бутоксиэтила, бутоксипропила или бутоксибутила.
3. Соединение по п. 1 или его рацемат или стереоизомер, где С3-6циклоалкил выбран из циклопропила, циклобутила, циклопентила или циклогексила.
4. Соединение по п. 1 или его рацемат или стереоизомер, где галоген выбран из фтора, хлора, брома или йода; галогеналкил означает, что один или более атомов водорода на алкиле замещены галогеном.
5. Соединение по п. 1 или его рацемат или стереоизомер, где кольцо А выбрано из  или
или  .
.
6. Соединение по п. 1 или его рацемат или стереоизомер, выбранные из следующего:
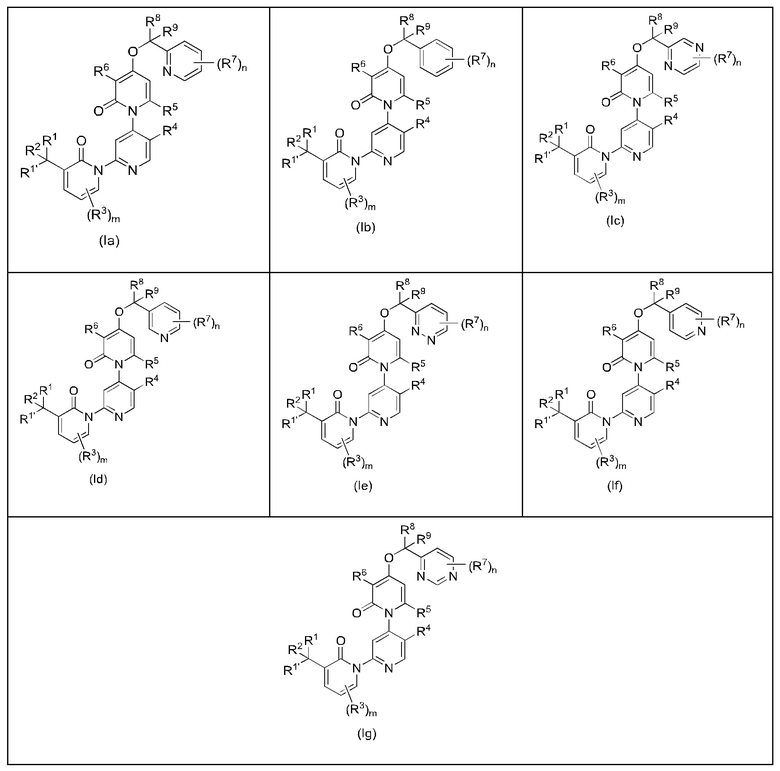 ,
,
где m равно 0, 1, 2 или 3;
n равно 0, 1, 2, 3, 4 или 5;
R1, R1', R2, R3, R4, R5, R6, R7, R8 и R9 определены выше.
7. Соединение по п. 1 или его рацемат или стереоизомер, где:
m равно 0 или 1;
n равно 0, 1, 2 или 3;
кольцо А выбрано из  или
или  ;
;
R1 и R1' выбраны из атома водорода, метила или этила, или R1 и R1', взятые вместе, циклизированы с образованием  ;
;
R2 выбран из атома водорода или гидроксила;
R3 выбран из атома водорода, метила, метокси, хлора или брома;
R4 выбран из метила, циклопропила, метокси или этила;
R5 выбран из метила;
R6 выбран из хлора, водорода или брома;
R7 выбран из водорода, фтора, хлора, брома, циклопропила, метокси, циано, метила или трифторметила; и
R8 и R9 представляют собой водород.
8. Соединение по п. 1 или его рацемат или стереоизомер, выбранные из следующего:

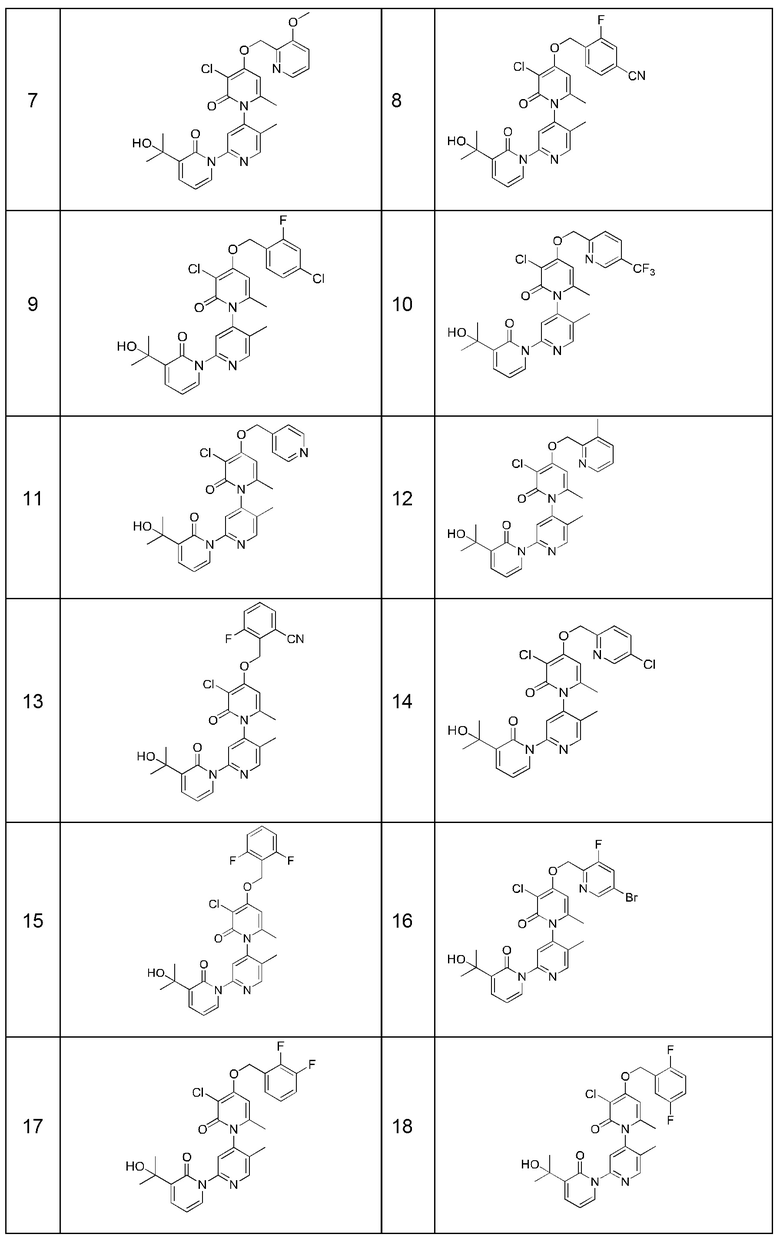
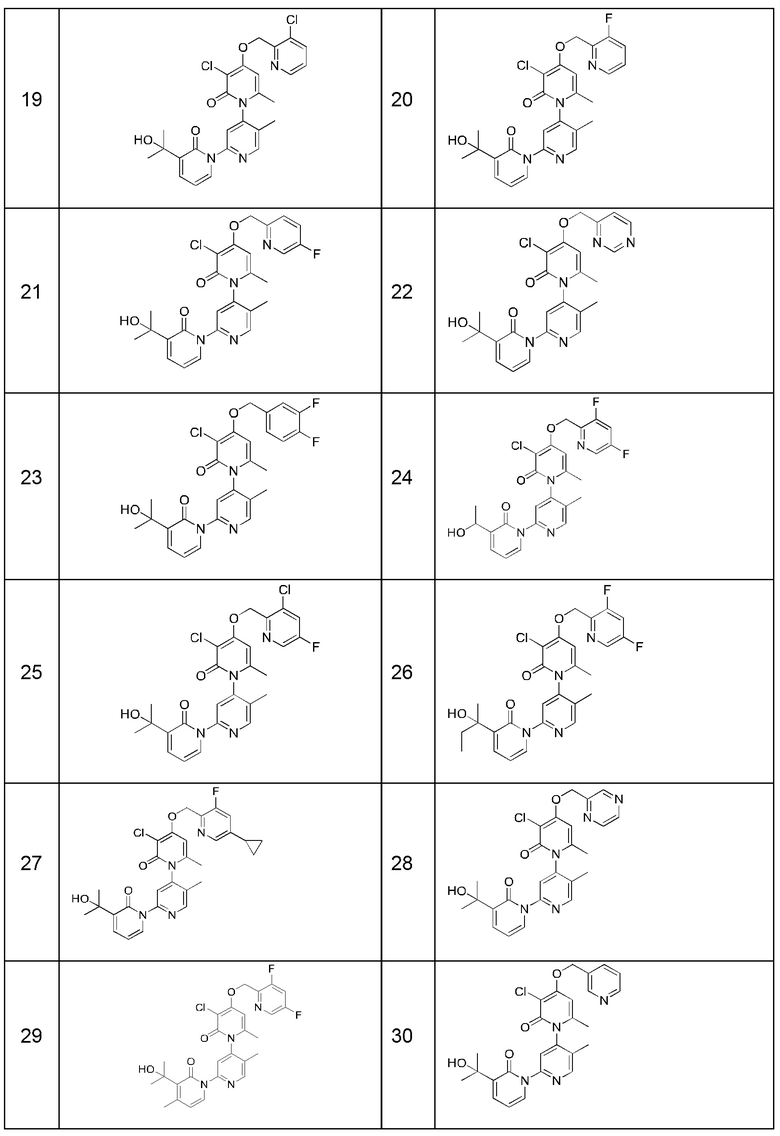

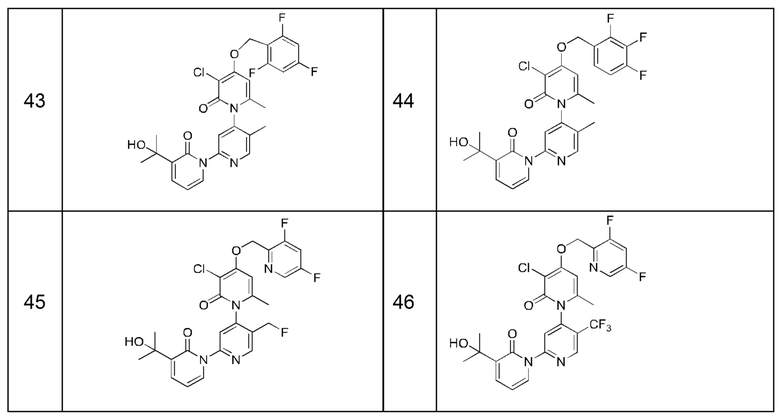
9. Фармацевтическая композиция для лечения заболеваний, связанных с р38/MK2, содержащая терапевтически эффективное количество соединения по любому из пп. 1-8, или его рацемата, или стереоизомера и фармацевтически приемлемый носитель.
10. Применение соединения по любому из пп. 1-8, или его рацемата, или стереоизомера для изготовления лекарственного средства для лечения заболеваний, где указанные заболевания представляют собой заболевания, связанные с р38/MK2, и выбраны из хронического воспалительного заболевания и острого воспалительного заболевания.
11. Применение по п. 10, где хроническое воспалительное заболевание представляет собой ревматоидный артрит.
| WO 2014197846 A1, 11.12.2014 | |||
| WO 2012078684 A1, 14.06.2012 | |||
| WO 2013086208 A1, 13.06.2013 | |||
| Устройство для присоединения бесколесных почвообрабатывающих орудий к трактору | 1926 |
|
SU15252A1 |

