ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Изобретение относится к определенным замещенным гетероциклическим конденсированным гамма-карболинам, их пролекарствам в свободной, твердой, фармацевтически приемлемой солевой и/или по существу чистой форме, как описано в данной заявке, их фармацевтическим композициям и способам применения при лечении заболеваний, включающих рецептор 5-НТ2А, транспортер серотонина (SERT), пути, включающие сигнальные системы рецепторов дофамина D1 и D2, и/или μ-опиоидный рецептор, например, заболеваний или нарушений, таких как тревожность, психоз, шизофрения, нарушения сна, сексуальные расстройства, мигрень, состояния, связанные с головной болью, социальные фобии, желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта и ожирение; депрессия и расстройства настроения, связанные с психозом или болезнью Паркинсона; психоз, такой как шизофрения, связанная с депрессией; биполярное расстройство; расстройства настроения; наркотические зависимости, такие как опиоидная зависимость и алкогольная зависимость, симптомы отмены лекарственного средства и другие психиатрические и неврологические состояния, также как комбинации с другими средствами.
УРОВЕНЬ ТЕХНИКИ
[0002] Известно, что замещенные гетероциклические конденсированные гамма-карболины являются агонистами или антагонистами 5-НТ2-рецепторов, в частности 5-НТ2А и 5-НТ2С-рецепторов для лечения расстройств центральной нервной системы. Данные соединения были раскрыты в патенте США No. №6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680 и U.S. RE39679 в качестве новых соединений, пригодных для лечения расстройств, связанных с модуляцией 5-HT2A-рецептора, таких как ожирение, тревожность, депрессия, психоз, шизофрения, нарушения сна, сексуальные расстройства, мигрень, состояния, связанные с головной болью, социальные фобии, желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта и ожирение. В PCT/US08/03340 (WO 2008/112280) и в заявке на патент США №10/786935 также раскрыты способы получения замещенных гетероциклических конденсированных гамма-карболинов и применение данных гамма-карболинов в качестве агонистов и антагонистов серотонина, пригодных для контроля и профилактики расстройств центральной нервной системы, таких как аддиктивное поведение и нарушения сна.
[0003] Кроме того, в WO/2009/145900 раскрыто применение определенных замещенных гетероциклических конденсированных гамма-карболинов для лечения комбинации психозов и депрессивных расстройств, также как нарушений сна, депрессии и/или настроения у пациентов с психозом или болезнью Паркинсона. В дополнение к расстройствам, связанным с психозом и/или депрессией, в данной патентной заявке раскрыто и заявлено применение данных соединений в низкой дозе для селективного антагонизма 5-НТ2А-рецепторов без влияния или минимального влияния на рецепторы дофамина D2, что пригодно для лечения нарушений сна без побочных эффектов путей дофамина D2 или побочных эффектов других путей (например, ГАМКA-рецепторов), связанных со стандартными седативно-гипнотическими средствами (например, бензодиазепинами), включая, но не ограничиваясь ими, развитие наркотической зависимости, мышечную гипотонию, слабость, головную боль, нечеткость зрения, головокружение, тошноту, рвоту, желудочный дискомфорт, диарею, боли в суставах и боли в груди. В WO 2009/114181 также раскрыты способы получения кристаллов аддитивной соли толуолсульфокислоты данных замещенных гетероциклических конденсированных гамма-карболинов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0004] Раскрытие предоставляет новые соединения, имеющие новый профиль рецепторного связывания, связывания 5-НТ2А, D1 и мю-опиоидных рецепторов. Уникальный фармакологический профиль данных соединений, включая антагонизм 5-НТ2А, активность D1 и антагонизм морфина, делает соединения пригодными для лечения различных нарушений применения веществ и психических расстройств, таких как депрессия, тревожность, нарушение сна, психоз или деменция, как дополнительно описано в данной заявке, включая лечение пациентов, страдающих нарушением, связанным с применением веществ, и сопутствующим психиатрическим расстройством.
[0005] Настоящее раскрытие, таким образом, предоставляет Соединения Формулы I, которые являются пригодными для лечения или профилактики расстройств центральной нервной системы. В первом аспекте настоящее раскрытие относится к соединению (Соединению I) Формулы I:
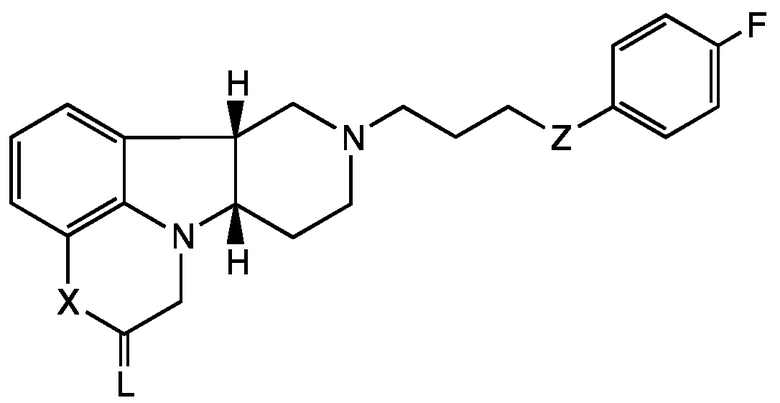 ,
,
Формула I
в которой:
X представляет собой -NH- или -N(CH3)-;
L выбран из O, NH, NRa и S;
Z представляет собой -CH(O-R1)-, -O- или -C(=O)-;
R1 представляет собой H, -C(O)-C1-21алкил (например, -C(O)-C1-5алкил, -C(O)-C6-15алкил или -C(O)-C16-21алкил), предпочтительно указанный алкил представляет собой прямую цепь, необязательно насыщенную или ненасыщенную и необязательно замещенную одной или более гидрокси или C1-22алкокси (например, этокси) группами, например, R1 представляет собой C(O)-C3алкил, -C(O)-C6алкил, -C(O)-C7алкил, -C(O)-C9алкил, -C(O)-C11алкил, -C(O)-C13алкил или -C(O)-C15алкил;
Ra представляет собой:
галоген, С1-4алкил, С2-4алкенил, С2-4алкинил или С3-6циклоалкил, каждый из которых может быть независимо замещен до трех независимо выбранных Rb-групп, например, С1-3галогеналкил или С1-3гидроксиалкил; или
арил, необязательно замещенный до пяти независимо выбранных Rb; и каждый Rb независимо выбран из H, галогена, NH2, NO2, OH, C(=O)OH, CN, SO3 и C1-4алкила;
в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме.
[0006] Настоящее раскрытие предоставляет дополнительные примерные варианты осуществления Соединения Формулы I в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме, включая:
1.1 Соединение I, в котором L представляет собой -O-;
1.2 Соединение I или 1.1, в котором Z представляет собой -CH(O-R1)-;
1.3 Соединение I или 1.1, в котором Z представляет собой -C(=O)-;
1.4 Соединение I, в котором L представляет собой NH.
1.5 Соединение I, в котором L представляет собой NRa;
1.6 Соединение I, в котором L представляет собой S;
1.7 Соединение I или любое из 1.1-1.5 в твердой форме, например, в твердой форме соли;
1.8 Соединение I или любое из 1.1-1.7, в котором Z представляет собой -CH(O-R1)-;
1.9 Соединение I или любое из 1.1-1.7, в котором Z представляет собой -C(=O)-;
1.10 Соединение I или любое из 1.1-1.7, в котором Z представляет собой -O-;
1.11 Соединение I или любое из 1.1-1.9, в котором X представляет собой -NH-;
1.12 Соединение I или любое из 1.1-1.9, в котором X представляет собой -N(CH3)-;
1.13 Соединение I или любое из 1.1-1.12, в котором L представляет собой -O- и X представляет собой -N(CH3)-;
1.14 Соединение I или любое из 1.1-1.12, в котором L представляет собой -O- и X представляет собой -NH-;
1.15 Соединение 1.13, в котором Z представляет собой -C(=O)-;
1.16 Соединение 1.14, в котором Z представляет собой -C(=O)-;
1.17 Соединение I или любое из 1.1-1.14, в котором Z представляет собой -CH(O-R1)- и R1 представляет собой H;
1.18 Соединение I или любое из 1.1-1.14, в котором Z представляет собой -CH(O-R1)- и R1 представляет собой -C(O)-C1-5алкил, -C(O)-C6-15алкил или -C(O)-C16-21алкил;
1.19 Соединение I или любое из 1.1-1.14, в котором Z представляет собой -CH(O-R1)- и R1 выбран из группы, состоящей из C(O)-C3алкила, -C(O)C6алкила, -C(O)-C7алкила, -C(O)-C9алкила, -C(O)-C11алкила, -C(O)-C13алкила или -C(O)-C15алкила; например, в котором R1 представляет собой ацетил, этилкарбонил или пропилкарбонил;
1.20 Соединение I или любое из 1.1-1.12 или 1.17-1.19, в котором L представляет собой NRa и в котором Ra представляет собой:
галоген, С1-4алкил, С2-4алкенил, С2-4алкинил или С3-6циклоалкил, каждый из которых может быть независимо замещен до трех независимо выбранных Rb-групп; или в котором Ra представляет собой арил, необязательно замещенный до пяти независимо выбранных Rb; в котором Rb независимо выбран из H, галогена, NH2, NO2, OH, C(=O)OH, CN, SO3 и C1-4алкила;
1.21 Соединение 1.20, в котором Ra представляет собой С1-4алкил или С3-6циклоалкил, необязательно замещенный до трех независимо выбранных Rb-групп;
1.22 Соединение 1.20, в котором Ra представляет собой арил, необязательно замещенный до трех независимо выбранных Rb-групп;
1.23 Соединение 1.20, в котором Ra выбран из группы, состоящей из метила, этила, пропила, бутила, изопропила, изобутила, втор-бутила или фенила;
1.24 Соединение I или любое из 1.1-1.14 или 1.17-1.23, в котором Z представляет собой -CH(O-R1)- и указанный атом углерода CH в группе -CH(O-R1)- имеет или R-конфигурацию, или S-конфигурацию, или их смесь;
1.25 Соединение 1.24, в котором атом углерода CH по существу присутствует или в R-конфигурации, или в S-конфигурации, например, в котором диастереомер, имеющий R-конфигурацию или S-конфигурацию на данном углероде, присутствует более чем в 70% диастереомерном избытке, например, более 75%, или более 80%, или более 85%, или более 90%, или более 95%, или более 97%, или более 98%, или более 99% диастереомерном избытке.
1.26 Соединение I или любое из 1.1-1.25, в котором соединение выбрано из группы, состоящей из:
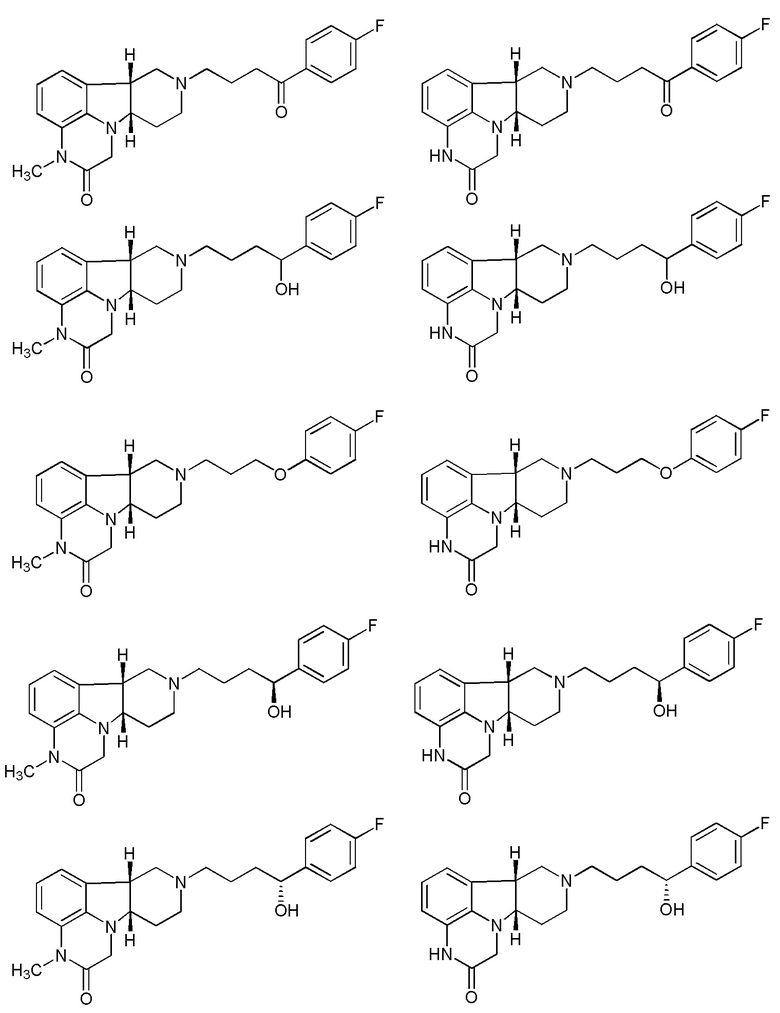 ;
;
1.27 Соединение I или любое из 1.1-1.26, в котором соединение выбрано из группы, состоящей из:
 ;
;
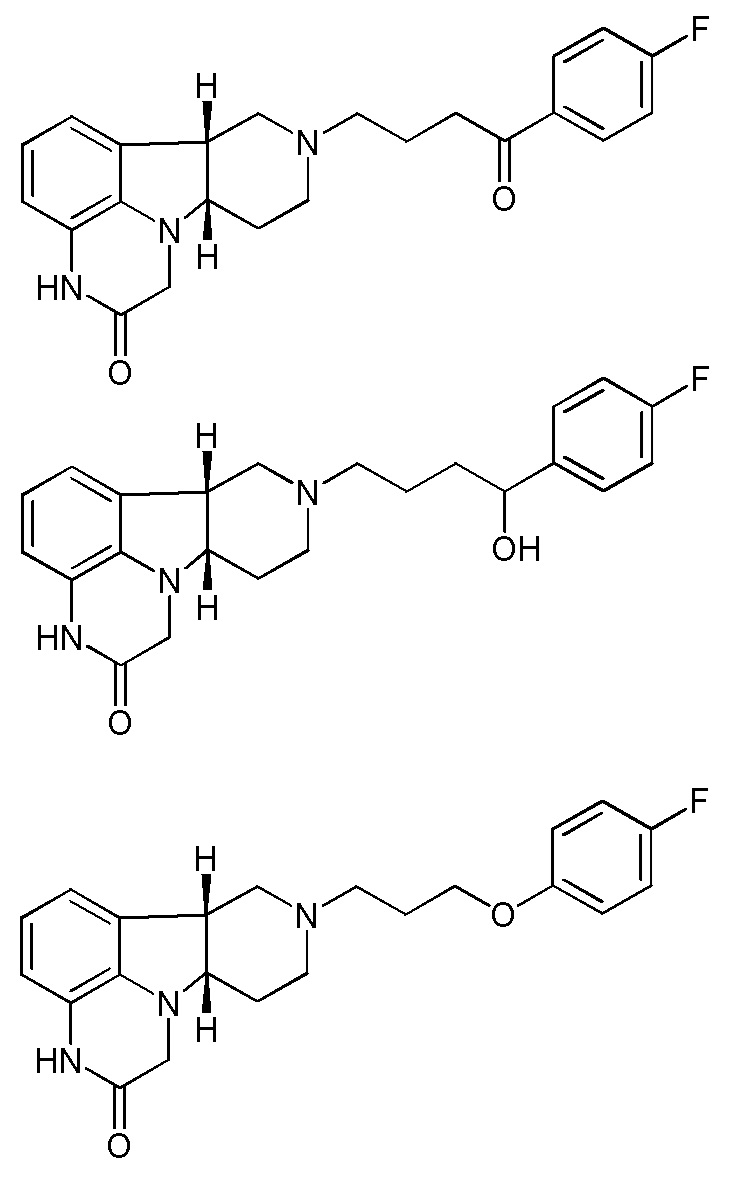
(т.е. 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фтор-фенил)бутан-1-она;
4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1Н,7Н-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фтор-фенил)бутан-1-ола; и
(6bR,10aS)-8-(3-(4-фторфенокси)пропил)-6b,7,8,9,10,10a-гексагидро-1Н-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-2(3H)-она, соответственно); например, (6bR,10aS)-8-(3-(4-фторфенокси)пропил)-6b,7,8,9,10,10а-гексагидро-1Н-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-2(3H)-она;
в свободной или фармацевтически приемлемой солевой форме.
1.28 Соединение I или любое из 1.1-1.27 в свободной форме;
1.29 Соединение I или любое из 1.1-1.28 в форме соли, например, в форме фармацевтически приемлемой соли;
1.30 Соединение I или любое из 1.1-1.29 в твердой форме;
в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме.
[0007] Во втором аспекте настоящее раскрытие относится к соединению (Соединению II) Формулы II:
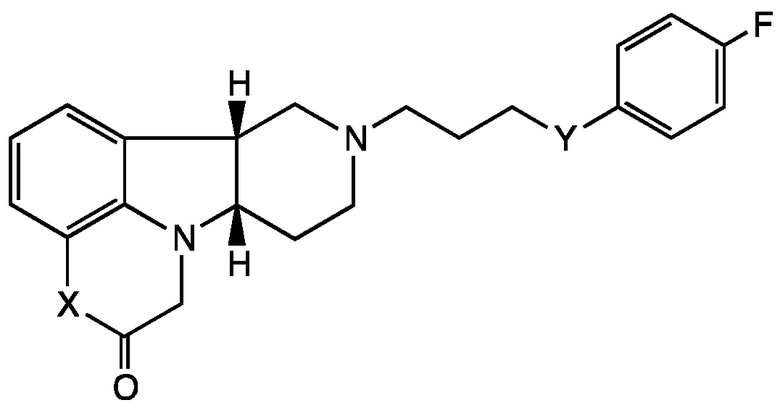
Формула II
в которой:
X представляет собой -NH- или -N(CH3)-;
Y представляет собой -CH(O-R1)- или -C(=O)-;
R1 представляет собой H, -C(O)-C1-21алкил (например, -C(O)-C1-5алкил, -C(O)-C6-15алкил или -C(O)-C16-21алкил), предпочтительно указанный алкил представляет собой прямую цепь, необязательно насыщенную или ненасыщенную и необязательно замещенную одной или более гидрокси или C1-22алкокси (например, этокси) группами, например, R1 представляет собой C(O)-C3алкил, -C(O)-C6алкил, -C(O)-C7алкил, -C(O)-C9алкил, -C(O)-C11алкил, -C(O)-C13алкил или -C(O)-C15алкил;
в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме.
[0008] Настоящее раскрытие предоставляет дополнительные примерные варианты осуществления Соединения Формулы II в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме, включая:
2.1 Соединение II, в котором X представляет собой -NH-;
2.2 Соединение II, в котором X представляет собой -N(CH3)-;
2.3 Соединение II или 2.1-2.4, в котором Y представляет собой -C(=O)-;
2.4 Соединение II, в котором Y представляет собой -CH(O-R1)-; т.е. имеющее Формулу II-A:
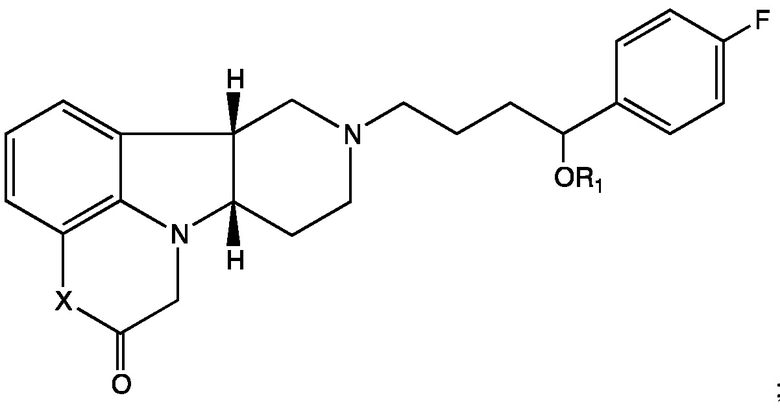 ;
;
Формула II-A
2.5 Соединение II или 2.1-2.4, в котором Y представляет собой -CH(O-R1)-;
2.6 Соединение II, в котором Х представляет собой NH и Y представляет собой -C(=O)-; т.е. имеющее Формулу II-B:
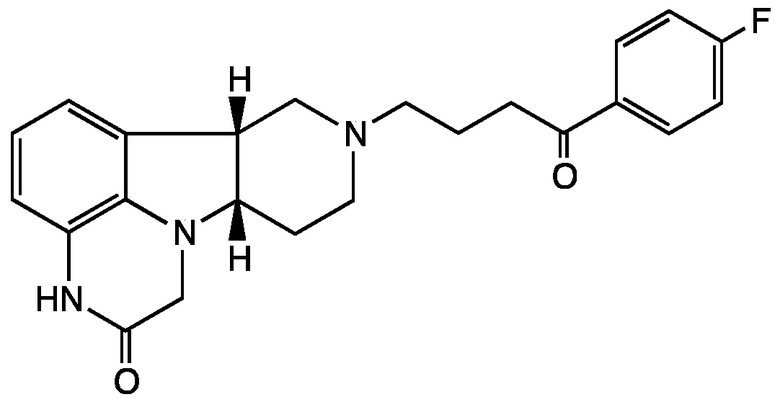 ;
;
Формула II-B
2.7 Соединение II, в котором X представляет собой -NH- и Y представляет собой -CH(O-R1)-;
2.8 Соединение II, в котором Х представляет собой -NH- и Y представляет собой -CH(O-R1)-, в котором R1 представляет собой H; т.е. имеющее Формулу II-C:
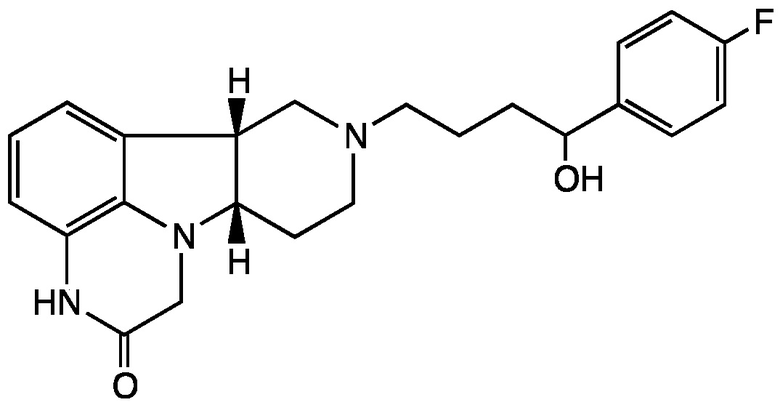 ;
;
Формула II-C
2.9 Соединение II, в котором X представляет собой -N(CH3)- и Y представляет собой -C(=O)-; т.е. имеющее Формулу II-D:
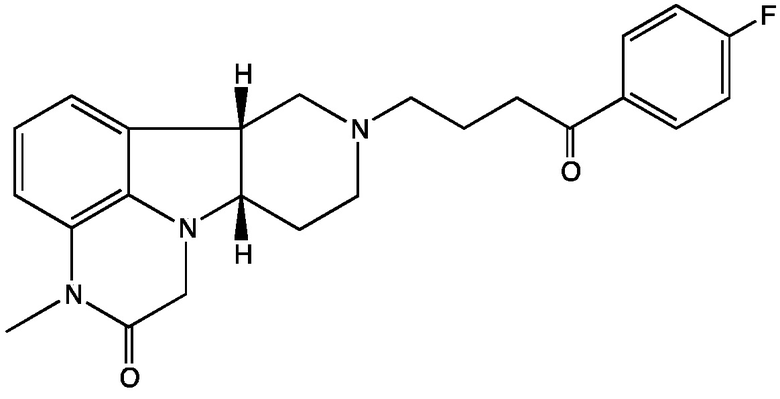 ;
;
Формула II-D
2.10 Соединение II, в котором X представляет собой -N(CH3)- и Y представляет собой -CH(O-R1)-;
2.11 Соединение II, в котором X представляет собой -N(CH3)- и Y представляет собой -CH(O-R1)-, в котором R1 представляет собой H; т.е. имеющее Формулу II-E:
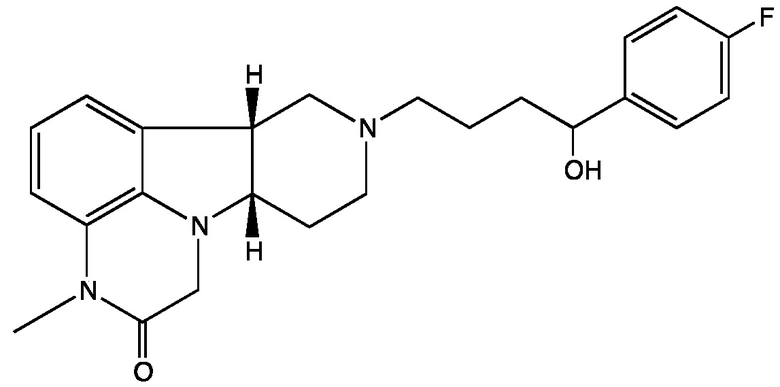
Формула II-E
2.12 Соединение II или любое из 2.1-2.11 в твердой форме, например, в твердой форме соли.
[0009] В третьем аспекте настоящее раскрытие относится к соединению (Соединению III) Формулы III:
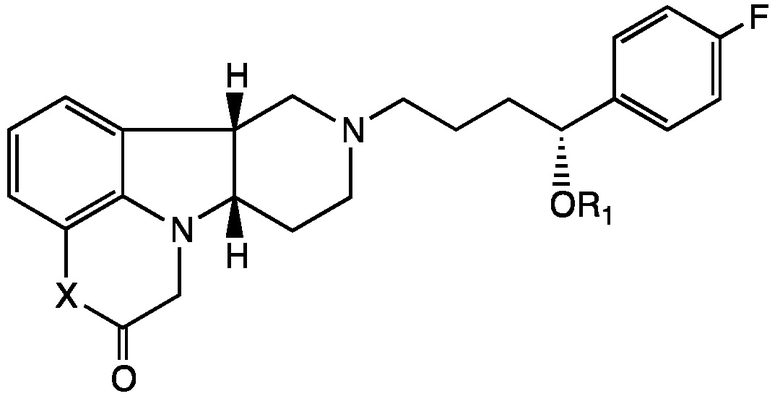
Формула III
в которой:
X представляет собой -NH- или -N(CH3)-;
R1 представляет собой H, -C(O)-C1-21алкил (например, -C(O)-C1-5алкил, -C(O)-C6-15алкил или -C(O)-C16-21алкил), предпочтительно указанный алкил представляет собой прямую цепь, необязательно насыщенную или ненасыщенную и необязательно замещенную одной или более гидрокси или C1-22алкокси (например, этокси) группами, например, R1 представляет собой C(O)-C3алкил, -C(O)-C6алкил, -C(O)-C7алкил, -C(O)-C9алкил, -C(O)-C11алкил, -C(O)-C13алкил или -C(O)-C15;
в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме.
[00010] Настоящее раскрытие предоставляет дополнительные примерные варианты осуществления Соединения Формулы III в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме, включая:
3.1 Соединение III, в котором R1 представляет собой Н; т.е. имеющее Формулу III-A:
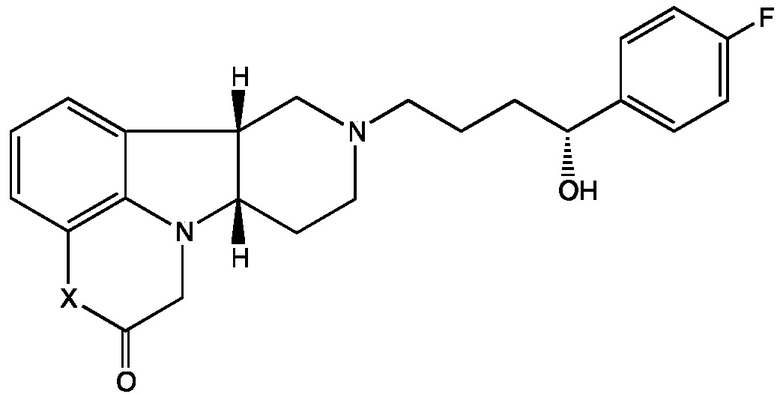
Формула III-A
в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме;
3.2 Соединение III или 3.1, в котором X представляет собой -NH-;
3.3 Соединение III или 3.1, в котором X представляет собой -N(CH3)-;
3.4 Соединение 3.1, в котором X представляет собой -NH-; т.е. имеющее Формулу III-B:
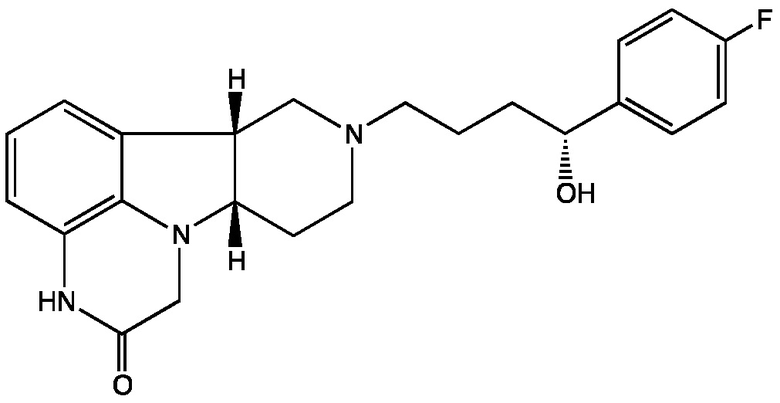
Формула III-B
3.5 Соединение 3.1, в котором X представляет собой -N(CH3)-; т.е. имеющее Формулу III-C:
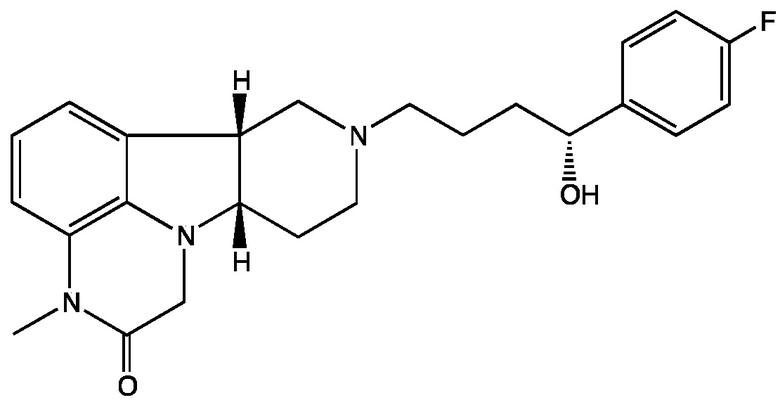 ;
;
Формула III-C
3.6 Соединение III или любое из 3.1-3.5, в котором Соединение имеет диастереомерный избыток более 70%;
3.7 Соединение III или любое из 3.1-3.6, в котором Соединение имеет диастереомерный избыток более 80%;
3.8 Соединение III или любое из 3.1-3.7, в котором Соединение имеет диастереомерный избыток более 90%;
3.9 Соединение III или любое из 3.1-3.8, в котором Соединение имеет диастереомерный избыток более 95%;
3.10 Соединение III или любое из 3.1-3.9, в котором Соединение находится по существу в чистой диастереомерной форме (т.е. по существу свободной от других диастереомеров)
3.11 Соединение III или любое из 3.1-3.10 в твердой форме, например, в твердой форме соли.
[00011] В четвертом аспекте настоящее раскрытие относится к соединению (Соединению IV) Формулы IV:
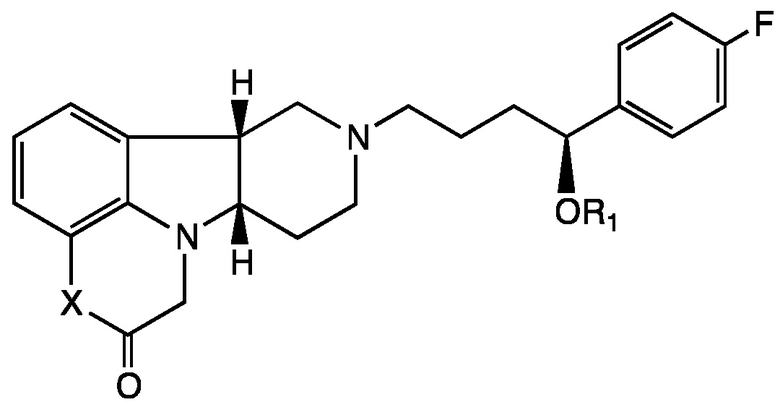
Формула IV
в которой:
X представляет собой -NH- или -N(CH3)-;
R1 представляет собой H, -C(O)-C1-21алкил (например, -C(O)-C1-5алкил, -C(O)-C6-15алкил или -C(O)-C16-21алкил), предпочтительно указанный алкил представляет собой прямую цепь, необязательно насыщенную или ненасыщенную и необязательно замещенную одной или более гидрокси или C1-22алкокси (например, этокси) группами, например, R1 представляет собой C(O)-C3алкил, -C(O)-C6алкил, -C(O)-C7алкил, -C(O)-C9алкил, -C(O)-C11алкил, -C(O)-C13алкил или -C(O)-C15алкил;
в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме.
[00012] Настоящее раскрытие предоставляет дополнительные примерные варианты осуществления Соединения Формулы IV в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме, включая:
4.1 Соединение IV, в котором R1 представляет собой Н; т.е. имеющее Формулу IV-A:
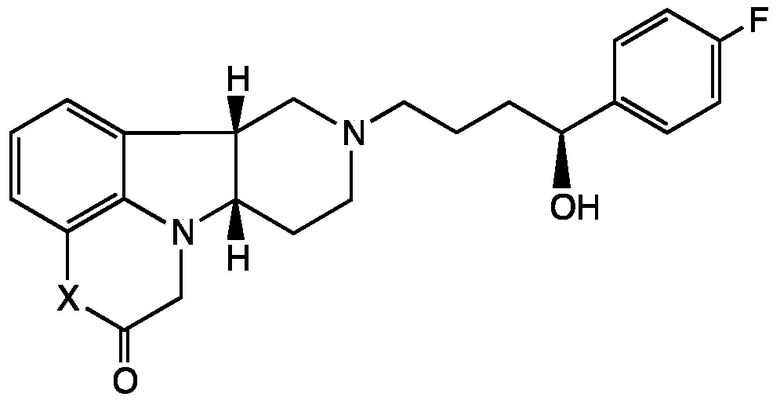
Формула IV-A
в свободной или солевой форме, например, в выделенной или очищенной свободной или солевой форме.
4.2 Соединение IV или 4.1, в котором X представляет собой -NH-;
4.3 Соединение IV или 4.1, в котором X представляет собой -N(CH3)-;
4.4 Соединение 4.1, в котором X представляет собой -NH-; т.е. имеющее Формулу IV-B:
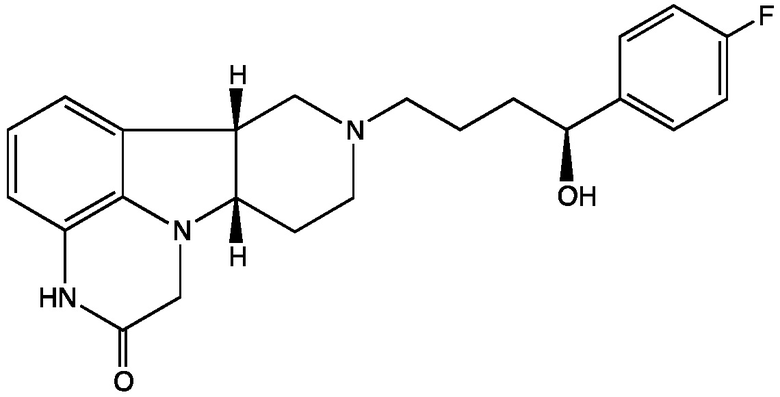
Формула IV-B
4.5 Соединение 4.1, в котором X представляет собой -N(CH3)-; т.е. имеющее Формулу IV-C:
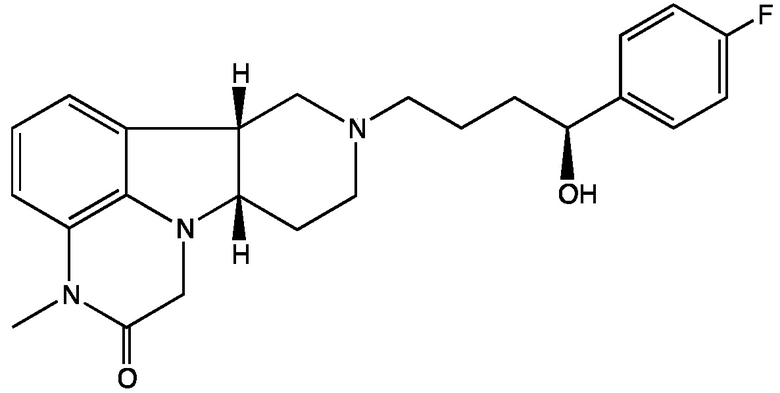
Формула IV-C
4.6 Соединение IV или любое из 4.1-4.5, в котором Соединение имеет диастереомерный избыток более 70%;
4.7 Соединение IV или любое из 4.1-4.6, в котором Соединение имеет диастереомерный избыток более 80%;
4.8 Соединение IV или любое из 4.1-4.7, в котором Соединение имеет диастереомерный избыток более 90%;
4.9 Соединение IV или любое из 4.1-4.8, в котором Соединение имеет диастереомерный избыток более 95%;
4.10 Соединение IV или любое из 4.1-4.9, в котором Соединение находится по существу в чистой диастереомерной форме (т.е. по существу свободной от других диастереомеров);
4.11 Соединение IV или любое из 4.1-4.10 в твердой форме, например, в твердой форме соли.
[00013] В пятом аспекте настоящее раскрытие предоставляет каждое из вышеуказанного Соединения I или 1.1-1.30, Соединения II или 2.1-2.12, Соединения III или 3.1-3.11 или Соединения IV или 4.1-4.11 (в дальнейшем совместно «Соединения Формул I-IV и последующие» или «соединения раскрытия») в свободной или фармацевтически приемлемой солевой форме. Настоящее раскрытие предоставляет дополнительные примерные варианты осуществления Соединений Формул I-IV и последующие, включая:
5.1 Соединения Формул I-IV и последующие, в которых соль представляет собой кислотно-аддитивную соль, выбранную из хлористоводородной, бромистоводородной, серной, сульфаминовой, фосфорной, азотной, уксусной, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, пальмитиновой, малеиновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изэтионовой и подобного;
5.2 Соединения Формул I-IV и последующие, в которых соль представляет собой аддитивную соль фумаровой кислоты;
5.3 Соединения Формул I-IV и последующие, в которых соль представляет собой аддитивную соль фосфорной кислоты;
5.4 Соединения Формул I-IV и последующие, в которых соль представляет собой аддитивную соль толуолсульфоновой кислоты;
5.5 Любой из 5.1-5.4, в котором соль находится в твердой форме.
[00014] В шестом аспекте настоящее раскрытие предоставляет фармацевтическую композицию (Фармацевтическую Композицию 6), содержащую соединение по любому одному из Соединения I или 1.1-1.30, Соединения II или 2.1-2.12, Соединения III или 3.1-3.11 или Соединения IV или 4.1-4.11 (совместно Соединений Формул I-IV и последующие или соединений раскрытия), например, в смеси с фармацевтически приемлемым разбавителем или носителем. Настоящее раскрытие предоставляет дополнительные примерные варианты осуществления Фармацевтической Композиции 6, включая:
6.1 Фармацевтическую Композицию 6, содержащую Соединение I или любое из 1.1-1.30;
6.2 Фармацевтическую Композицию 6, содержащую Соединение II или любое из 2.1-2.12;
6.3 Фармацевтическую Композицию 6, содержащую Соединение III или любое из 3.1-3.11;
6.4 Фармацевтическую Композицию 6, содержащую Соединение IV или любое из 4.1-4.11;
6.5 Фармацевтическую Композицию 6 или любую из 6.1-6.4, в которой Соединение Формулы I-IV и последующие находятся в твердой форме;
6.6 Фармацевтическую Композицию 6 или любую из 6.1-6.5, в которой Соединение Формул I-IV и последующие находятся в форме фармацевтически приемлемой соли, как описано в Соединениях 5.1-5.5;
6.7 Фармацевтическую Композицию 6 или любую из 6.1-6.6, в которой Соединение Формул I-IV и последующие находятся в смеси с фармацевтически приемлемым разбавителем или носителем.
[00015] В предпочтительном варианте осуществления Фармацевтическая Композиция настоящего раскрытия содержит Соединение Формулы II-A, II-B или II-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем. В другом предпочтительном варианте осуществления Фармацевтическая Композиция настоящего раскрытия содержит Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем. В другом предпочтительном варианте осуществления Фармацевтическая Композиция настоящего раскрытия содержит Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем.
[00016] В дополнительном варианте осуществления Фармацевтические Композиции настоящего раскрытия предназначены для замедленного или отсроченного высвобождения, например, депо, состав. В одном варианте осуществления состав депо (Состав Депо 6.8) представляет собой Фармацевтическую Композицию любую из 6.1-6.7 предпочтительно в свободной или фармацевтически приемлемой солевой форме и предпочтительно в смеси с фармацевтически приемлемым разбавителем или носителем, например, предоставляя замедленное или отсроченное высвобождение в качестве инъекционного депо.
[00017] В дополнительном варианте осуществления Композиция Депо (Композиция Депо 6.9) содержит Фармацевтическую Композицию любую из 6.1-6.7, в которой R1 представляет собой -C(O)-C6-15алкил в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем.
[00018] В другом аспекте настоящее раскрытие предоставляет Фармацевтическую Композицию 6.10, которая представляет собой Фармацевтическую Композицию 6 или любую из 6.1-6.9, в которой Соединение Формул I-IV и последующие находятся в полимерной матрице. В одном варианте осуществления Соединение настоящего раскрытия диспергируется или растворяется в полимерной матрице. В дополнительном варианте осуществления полимерная матрица содержит стандартные полимеры, используемые в составах депо, такие как полимеры, выбранные из сложного полиэфира гидроксижирных кислот и их производных, или полимер из алкил-альфа-цианоакрилата, полиалкиленоксалата, сложного полиортоэфира, поликарбоната, полиортокарбоната, полиаминокислоты, сложного эфира гиалуроновой кислоты и их смесей. В дополнительном варианте осуществления полимер выбирают из группы, состоящей из полилактида, поли-d,l-лактида, полигликолида, полимера PLGA 50:50, PLGA 85:15 и PLGA 90:10. В другом варианте осуществления полимер выбирают из поли(гликолевой кислоты), поли-D,L-молочной кислоты, поли-L-молочной кислоты, сополимеров вышеуказанных, поли(алифатических карбоновых кислот), сополиоксалатов, поликапролактона, полидиоксонона, поли(ортокарбонатов), поли(ацеталей), поли(молочной кислоты-капролактона), сложных полиортоэфиров, поли(гликолевой кислоты-капролактона), полиангидридов, и природных полимеров, включающих альбумин, казеин, и восков, таких как моно- и дистеарат глицерина, и подобного. В предпочтительном варианте осуществления полимерная матрица содержит поли(d,l-лактид-сo-гликолид).
[00019] Например, в одном варианте осуществления Фармацевтической Композиции 6.10 Соединение представляет собой Соединение Формулы I, в которой X представляет собой -NH- или -N(CH3)- и Y представляет собой -C(=O)- или -C(H)(OH)- в свободной или фармацевтически приемлемой солевой форме. В другом примере Фармацевтической Композиции 6.10 Соединение представляет собой Соединение Формулы II-A, II-B или II-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем. В другом примере Фармацевтической Композиции 6.10 Соединение представляет собой Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем. В другом примере Фармацевтической Композиции 6.10 Соединение представляет собой Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем. В другом варианте осуществления каждого из вышеприведенных примеров Фармацевтической Композиции 6.10 полимерная матрица содержит поли(d,l-лактид-со-гликолид).
[0019] (Фармацевтические) Композиции 6 и 6.1-6.10 в частности пригодны для замедленного или отсроченного высвобождения, в которых Соединение настоящего раскрытия высвобождается при разрушении полимерной матрицы. Данные Композиции могут быть составлены для контролируемого и/или замедленного высвобождения Соединений настоящего раскрытия (например, в виде композиции депо) в течение периода до 180 дней, например, от приблизительно 14 до приблизительно 30 до приблизительно 180 дней. Например, полимерная матрица может разрушаться и высвобождать Соединения настоящего раскрытия в течение периода приблизительно 30, приблизительно 60 или приблизительно 90 дней. В другом примере полимерная матрица может разрушаться и высвобождать Соединения настоящего раскрытия в течение периода приблизительно 120 или приблизительно 180 дней.
[0020] В еще одном варианте осуществления Фармацевтические Композиции настоящего раскрытия, например, композиция депо настоящего раскрытия, например, Фармацевтическая Композиция 6.10 составлены для введения путем инъекции.
[0021] В седьмом аспекте настоящее раскрытие предоставляет Соединения Формул I-IV и последующие, как описано выше, в пероральной системе доставки с осмотически контролируемым высвобождением (OROS), которая описана в WO 2000/35419 и ЕР 1539115 (US Pub. No. 2009/0202631), содержание каждой из данных заявок включено посредством ссылки в полном объеме. Следовательно, в одном варианте осуществления седьмого аспекта настоящее раскрытие предоставляет фармацевтическую композицию или устройство, содержащее (а) желатиновую капсулу, содержащую Соединение любой из Формул I-IV и последующие в свободной или фармацевтически приемлемой солевой форме или Фармацевтическую Композицию изобретения, как описано выше; (b) многослойную стенку, наложенную на желатиновую капсулу, содержащую в порядке снаружи от капсулы: (i) барьерный слой, (ii) расширяемый слой и (iii) полупроницаемый слой; и (c) и отверстие, образованное или образуемое через стенку. (Композиция P.1)
[0022] В другом варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую желатиновую капсулу, содержащую жидкость, Соединение Формул I-IV и последующие в свободной или фармацевтически приемлемой солевой форме или Фармацевтическую Композицию изобретения, например, любую из Фармацевтической Композиции 6 или 6.1-6.10, причем желатиновая капсула окружена композитной стенкой, содержащей барьерный слой, контактирующий с наружной поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или образуемое в стенке. (Композиция Р.2)
[0023] В еще одном варианте осуществления седьмого аспекта изобретение предоставляет композицию, содержащую желатиновую капсулу, содержащую жидкость, Соединение Формул I-IV и последующие в свободной или фармацевтически приемлемой солевой форме или Фармацевтическую Композицию изобретения, например, любую из Фармацевтической Композиции 6 или 6.1-6.10, причем желатиновая капсула окружена композитной стенкой, содержащей барьерный слой, контактирующий с наружной поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или образуемое в стенке, в которой барьерный слой образует уплотнение между расширяемым слоем и окружающей средой на выходном отверстии. (Композиция P.3)
[0024] В еще одном варианте осуществления седьмого аспекта изобретение предоставляет композицию, содержащую желатиновую капсулу, содержащую жидкость, Соединение Формул I-IV и последующие в свободной или фармацевтически приемлемой солевой форме или Фармацевтическую Композицию изобретения, например, любую из Фармацевтической Композиции 6 или 6.1-6.10, причем желатиновая капсула окружена барьерным слоем, контактирующим с наружной поверхностью желатиновой капсулы, расширяемым слоем, контактирующим с частью барьерного слоя, полупроницаемым слоем, охватывающим, по меньшей мере, расширяемый слой, и выходным отверстием, образованным или образуемым в дозированной форме, простирающейся от наружной поверхности желатиновой капсулы до окружения применения. (Композиция P.4). Расширяемый слой может быть образован в одной или более дискретных секций, как, например, двух секциях, расположенных на противоположных сторонах или концах желатиновой капсулы.
[0025] В определенном варианте осуществления седьмого аспекта Соединение настоящего раскрытия в пероральной системе доставки с осмотически контролируемым высвобождением (т.е. в Композиции P.1-P.4) находится в жидком составе, который может представлять собой чистое, жидкое активное средство, жидкое активное средство в растворе, суспензию, эмульсию или самоэмульгирующуюся композицию или подобное.
[0026] Дополнительная информация о композиции пероральной системы доставки с осмотически контролируемым высвобождением, включающая характеристики желатиновой капсулы, барьерного слоя, расширяемого слоя, полупроницаемого слоя и отверстия, может быть найдена в WO 2000/35419, содержание которой включено посредством ссылки в полном объеме.
[0027] Другая пероральная система доставки с осмотически контролируемым высвобождением для Соединения Формул I-IV и последующие или Фармацевтическая Композиция настоящего раскрытия может быть найдена в ЕР 1539115 (опубл. США №2009/0202631), содержание которой включено посредством ссылки в полном объеме. Следовательно, в другом варианте осуществления седьмого аспекта изобретение предоставляет композицию или устройство, содержащее (а) два или более слоев, указанные два или более слоев, содержащие первый слой и второй слой, причем указанный первый слой содержит Соединение Формул I-IV и последующие в свободной или фармацевтически приемлемой солевой форме или Фармацевтическую Композицию, как описано ранее в данной заявке, причем указанный второй слой содержит полимер; (b) наружную стенку, окружающую указанные два или более слоев; и (c) отверстие в указанной наружной стенке. (Композиция P.5)
[0028] В Композиции P.5 предпочтительно используют полупроницаемую мембрану, окружающую трехслойное ядро: в данных вариантах осуществления первый слой называется первым слоем лекарственного средства и содержит малые количества лекарственного средства (например, Соединения Формул I-IV и последующие) и осмотический агент, такой как соль, средний слой, называемый вторым слоем лекарственного средства, содержит большие количества лекарственного средства, эксципиентов и не содержит соль; и третий слой, называемый выдавливаемым слоем, содержит осмотические агенты и не содержит лекарственного средства. По меньшей мере, одно отверстие просверливают через мембрану на конце первого слоя лекарственного средства капсуловидной таблетки (Композиция P.6).
[0029] Композиция P.5 или P.6 может содержать мембрану, определяющую отсек, мембрану, окружающую внутреннюю защитную оболочку, по меньшей мере, одно выходное отверстие, образованное или образуемое в ней, и, по меньшей мере, часть мембраны является полупроницаемой; расширяемый слой, расположенный в отсеке, удаленный от выходного отверстия и сообщающийся по текучей среде с полупроницаемой частью мембраны; первый слой лекарственного средства, расположенный рядом с выходным отверстием; и второй слой лекарственного средства, расположенный внутри отсека между первым слоем лекарственного средства и расширяемым слоем, причем слои лекарственного средства содержат Соединение изобретения в свободной или его фармацевтически приемлемой солевой форме. В зависимости от относительной вязкости первого слоя лекарственного средства и второго слоя лекарственного средства получают различные профили высвобождения. Крайне необходимо определить оптимальную вязкость для каждого слоя. В настоящем изобретении вязкость модулируется добавлением соли, хлорида натрия. Профиль доставки из ядра зависит от массы, состава и толщины каждого из слоев лекарственного средства (Композиция Р.7).
[0030] В определенном варианте осуществления изобретение предоставляет Композицию P.7, в которой первый слой лекарственного средства содержит соль и второй слой лекарственного средства не содержит соль. Композиция P.5-P.7 может необязательно содержать способствующий текучести слой между мембраной и слоями лекарственного средства.
[0031] Композиции P.1-P.7, как правило, будут называться как композиция пероральной системы доставки с осмотически контролируемым высвобождением.
[0032] В восьмом аспекте изобретение предоставляет способ (Способ 1) для лечения или профилактики расстройства центральной нервной системы, включающий введение пациенту, нуждающемуся в таком лечении, Соединения Формул I-IV и последующие или Фармацевтической Композиции 6, или 6.1-6.10, или P.1-P.7, например, Способ 1, в котором вводимое соединение или композиция представляет собой:
1.1 Соединение I или любое из 1.1-1.30 в свободной или фармацевтически приемлемой солевой форме;
1.2 Соединение II или любое из 2.1-2.12 в свободной или фармацевтически приемлемой солевой форме;
1.3 Соединение III или любое из 3.1-3.11 в свободной или фармацевтически приемлемой солевой форме;
1.4 Соединение IV или любое из 4.1-4.11 в свободной или фармацевтически приемлемой солевой форме;
1.5 Соединения Варианта осуществления 5 или любое из 5.1-5.5;
1.6 Соединение Формулы II-A, II-B или II-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
1.7 Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
1.8 Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
1.9 Фармацевтическая Композиция, как описано в любой из Композиций 6 и 6.1-6.10;
1.10 Фармацевтическая Композиция, содержащая Соединение Формулы II-A, II-B или II-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
1.11 Фармацевтическая Композиция, содержащая Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
1.12 Фармацевтическая Композиция, содержащая Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
1.13 Композиция Депо, как описано в Композиции Депо 6.09 или 6.10;
1.14 Фармацевтическая Композиция P.1-P.7;
1.15 Композиция пероральной системы доставки с осмотически контролируемым высвобождением, как описано выше в данной заявке;
[0033] В дополнительном варианте осуществления восьмого аспекта настоящее раскрытие предоставляет Способ 1 или любой из Способов 1.1-1.15, в котором способ дополнительно описан ниже:
1.16 Способ 1 или любой из Способов 1.1-1.15, в котором расстройство центральной нервной системы представляет собой расстройство, выбранное из группы, состоящей из ожирения, тревожности, депрессии (например, рефрактерной депрессии и БДР), психоза (включая психоз, связанный с деменцией, такой как галлюцинации при прогрессирующей болезни Паркинсона или параноидальные идеи), шизофрении, нарушений сна (в частности нарушений сна, связанных с шизофренией и другими психиатрическими и неврологическими заболеваниями), сексуальных расстройств, мигрени, состояний, связанных с головной болью, социальных фобий, возбуждения при деменции (например, возбуждения при болезни Альцгеймера), возбуждения при аутизме и родственных аутических расстройствах, желудочно-кишечных расстройств, таких как дисфункция моторики желудочно-кишечного тракта, и деменции, например, деменции болезни Альцгеймера или болезни Паркинсона; расстройств настроения; и наркотических зависимостей, например, опиатной зависимости и/или алкогольной зависимости или отмены наркотической или алкогольной зависимости (например, опиатной зависимости); или компульсивного переедания; например, симптомов расстройств настроения и расстройства употребления веществ, например, злоупотребления опиатами, например, у пациентов с нарушением, связанным с употреблением веществ и сопутствующим расстройством центральной нервной системы, например депрессией, включая биполярную депрессию, тревожностью, нарушением сна, психозом, например, шизофренией или деменцией, включая болезнь Альцгеймера;
1.17 Способ 1 или любой из Способов 1.1-1.16, в котором нарушение центральной нервной системы представляет собой нарушение, в которое вовлечены пути серотонина 5-HT2A, рецепторной системы дофамина D2 и/или транспортера обратного захвата серотонина (SERT), как аналогично описано в WO/2009/145900, содержание которой включено в настоящее описание посредством ссылки в полном объеме;
1.18 Способ 1 или любой из Способов 1.1-1.17, в котором расстройство центральной нервной системы представляет собой расстройство, в которое вовлечен μ-опиоидный рецептор;
1.19 Способ 1 или любой из Способов 1.1-1.18, в котором расстройство центральной нервной системы представляет собой расстройство, выбранное из следующего: (i) психоз, например, шизофрения у пациента, страдающего от депрессии; (2) депрессия у пациента, страдающего от психоза, например, шизофрении; (3) расстройства настроения, связанные с психозом, например, шизофренией или болезнью Паркинсона; (4) нарушения сна, связанные с психозом, например, шизофренией или болезнью Паркинсона; и (5) наркотическая зависимость, нарушения, связанные с употреблением веществ, и/или нарушения, вызванные употреблением веществ, необязательно, когда пациент страдает от остаточных симптомов тревожности или тревожного расстройства;
1.20 Способ 1 или любой из Способов 1.1-1.18, в котором расстройство центральной нервной системы представляет собой психоз, например, шизофрению и указанный пациент является пациентом, страдающим депрессией;
1.21 Способ 1 или любой из Способов 1.1-1.20, в котором указанный пациент не способен переносить побочные эффекты стандартных нейролептических средств, например, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина, промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона и зипрасидона;
1.22 Способ 1 или любой из Способов 1.1-1.20, в котором указанный пациент не способен переносить побочные эффекты стандартных нейролептических средств, например, галоперидола, арипипразола, клозапина, оланзапина, кветиапина, рисперидона и зипрасидона;
1.23 Способ 1 или любой из Способов 1.1-1.22, в котором указанное расстройство является депрессией и указанный пациент является пациентом, страдающим психозом, например, шизофренией или болезнью Паркинсона;
1.24 Способ 1 или любой из Способов 1.1-1.22, в котором указанное расстройство является нарушением сна и указанный пациент страдает депрессией;
1.25 Способ 1 или любой из Способов 1.1-1.22, в котором указанное одно или более расстройств являются нарушением сна и указанный пациент страдает психозом, например, шизофренией;
1.26 Способ 1 или любой из Способов 1.1-1.22, в котором указанное одно или более расстройств являются нарушением сна и указанный пациент страдает болезнью Паркинсона;
1.27 Способ 1 или любой из Способов 1.1-1.22, в котором указанное одно или более расстройств являются нарушением сна и указанный пациент страдает депрессией и психозом, например, шизофренией или болезнью Паркинсона.
1.28 Способ 1 или любой из 1.1-1.27, в котором указанный пациент страдает от расстройства наркотической зависимости, необязательно в сочетании с любыми предшествующими расстройствами, например, когда указанный пациент страдает от опиатной зависимости и/или алкогольной зависимости или от отмены наркотической или алкогольной зависимости, необязательно, когда пациент страдает от остаточных симптомов тревожности или тревожного расстройства;
1.29 Любой из вышеприведенных способов, в котором эффективное количество составляет 1 мг-1000 мг, предпочтительно 2,5 мг-50 мг;
1.30 Любой из вышеприведенных способов, в котором эффективное количество составляет 1 мг-100 мг в день, предпочтительно 2,5 мг-50 мг в день;
1.31 Любой из вышеприведенных способов, в котором состояние, подлежащее лечению, представляет собой дискинезию, например, у пациента, получающего дофаминергические препараты, например, препараты, выбранные из леводопы и вспомогательных средств леводопы (карбидопы, ингибиторов КОМТ, ингибиторов МАО-Б), агонистов дофамина и антихолинергических средств, например, леводопы;
1.32 Любой из вышеприведенных способов, в котором пациент страдает болезнью Паркинсона.
[00035] Расстройства, связанные с употреблением веществ, и расстройства, вызванные употреблением веществ, представляют собой две категории расстройств, связанных с веществом, которые определены в пятом издании DSM (Диагностическое и статистическое руководство по психическим расстройствам). Расстройство, связанное с употреблением веществ, представляет собой образец симптомов, возникающих вследствие употребления вещества, которое объект продолжает принимать, несмотря на то, что в результате возникают проблемы. Расстройство, вызванное веществом, представляет собой расстройство, вызванное употреблением вещества. Расстройства, вызванные употреблением веществ, включают интоксикацию, синдром отмены, вызванные употреблением веществ психические расстройства, включая вызванный употреблением веществ психоз, вызванные употреблением веществ биполярные и связанные с ними расстройства, вызванные употреблением веществ депрессивные расстройства, вызванные употреблением веществ тревожные расстройства, вызванные употреблением веществ обсессивно-компульсивные и связанные с ними расстройства, вызванные употреблением веществ нарушения сна, вызванные употреблением веществ сексуальные дисфункции, вызванный употреблением веществ делириозный синдром и вызванные употреблением веществ нейрокогнитивные расстройства.
[00036] DSM-V включает критерии классификации расстройств употребления веществ как мягкие, умеренные или тяжелые. В некоторых вариантах осуществления способов, раскрытых в настоящем описании, расстройство употребления веществ выбрано из мягкого расстройства употребления веществ, умеренного расстройства употребления веществ или тяжелого расстройства употребления веществ. В некоторых вариантах осуществления расстройство употребления веществ представляет собой мягкое расстройство употребления веществ. В некоторых вариантах осуществления расстройство употребления веществ представляет собой умеренное расстройство употребления веществ. В некоторых вариантах осуществления расстройство употребления веществ представляет собой тяжелое расстройство употребления веществ.
[00037] Тревожность представляет собой широко распространенное коморбидное расстройство у пациентов, проходящих лечение употребления веществ или злоупотребления веществами. Стандартное лечение расстройства, связанного с злоупотреблением веществами, представляет собой комбинацию частичного агониста опиоидных рецепторов бупренорфина с антагонистом опиоидных рецепторов налоксоном, но ни одно из данных лекарственных средств не оказывает какого-либо существенного влияния на тревожность, приводя к общему результату, который представляет собой третье лекарственное средство, такое как бензодиазепин - класс анксиолитического средства. Это усложняет режим лечения и соблюдение его пациентом. Напротив, Соединения настоящего раскрытия обеспечивают опиатный антагонизм наряду с антагонизмом серотонина и модуляцией дофамина. Это может привести к значительному улучшению лечения пациентов с расстройством употребления или злоупотребления веществами, связанным с тревожностью.
[00038] Таким образом, в некоторых вариантах осуществления настоящее раскрытие предоставляет способ в соответствии со Способом 1 или любым из Способов 1.1-1.32, в котором расстройство центральной нервной системы представляет собой наркотическую зависимость, расстройства употребления веществ и/или вызванные употреблением веществ расстройства или связанное с употреблением веществ расстройство, например, у пациента, страдающего от симптомов тревожности или у которого диагностирована тревожность как коморбидное расстройство или как остаточное расстройство, в котором способ не включает дополнительное введение анксиолитического средства, такого как бензодиазепин. Бензодиазепины являются ГАМК-модулирующими соединениями, включая те, которые обсуждаются со ссылкой на Способ 3.1 и 3.2 ниже.
[00039] В другом варианте осуществления настоящее раскрытие предоставляет Способ 1 или любой из Способов 1.1-1.32, в котором Соединения Формул I-IV и последующие или Фармацевтическая Композиция 6, или 6.1-6.10, или P.1-P.7 содержит:
1.33 Соединение Формулы II-A, II-B, II-C или II-D в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
1.34 Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем или
1.35 Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем.
[0036] В еще одном варианте осуществления настоящее раскрытие предоставляет любой из Способов 1 или 1.1-1.35, как описано выше в данной заявке, в котором нарушение представляет собой шизофрению или нарушение сна.
[0037] В еще одном варианте осуществления настоящее раскрытие предоставляет любой из Способов 1.1-1.35, в котором Композицию Депо Изобретения (например, Композицию Депо любую из формул 6.8-6.10), или (Фармацевтическую) Композицию 6 или 6.1-6.7, или Композицию P.1-P.7 вводят для контролируемого и/или замедленного высвобождения Соединений Изобретения в течение периода от приблизительно 14 дней, приблизительно 30 до приблизительно 180 дней, предпочтительно в течение периода приблизительно 30, приблизительно 60 или приблизительно 90 дней. Контролируемое и/или замедленное высвобождение наиболее пригодно для избежания преждевременного прекращения терапии, в частности для терапии нейролептическими средствами, где несоблюдение или невыполнение режимов приема препарата является обычным явлением.
[0038] В еще одном варианте осуществления изобретение предоставляет любой из Способов 1 или 1.1-1.35, как описано выше в данной заявке, в котором Композицию Депо настоящего раскрытия вводят для контролируемого и/или замедленного высвобождения Соединений Изобретения в течение периода времени.
[0039] В девятом аспекте изобретение предоставляет способ (Способ 2) для профилактики или лечения одного или более нарушений сна, включающий введение пациенту, нуждающемуся в таком лечении, Соединения Формул I-IV и последующие или Фармацевтической Композиции 6, или 6.1-6.10, или P.1-P.7, (Способ 2), например, Способ 2, в котором вводимым соединением или композицией является:
2.1 Соединение I или 1.1-1.30 в свободной или фармацевтически приемлемой солевой форме;
2.2 Соединение II или 2.1-2.12 в свободной или фармацевтически приемлемой солевой форме;
2.3 Соединение III или 3.1-3.11 в свободной или фармацевтически приемлемой солевой форме;
2.4 Соединение IV или 4.1-4.11 в свободной или фармацевтически приемлемой солевой форме;
2.5 Соединение 5 или 5.1-5.5;
2.6 Соединение Формулы II-A, II-B, II-C или II-D в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
2.7 Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
2.8 Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
2.9 Фармацевтическая композиция, как описано в любой из Композиций 6 и 6.1-6.10;
2.10 Фармацевтическая Композиция, содержащая Соединение Формулы II-A, II-B или II-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
2.11 Фармацевтическая Композиция, содержащая Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
2.12 Фармацевтическая Композиция, содержащая Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем;
2.13 Композиция Депо, как описано в Композиции Депо 6.09 или 6.10;
2.14 Фармацевтическая композиция P.1-P.7;
2.15 Композиция пероральной системы доставки с осмотически контролируемым высвобождением, как описано выше в данной заявке;
[0040] В дополнительном варианте осуществления девятого аспекта изобретение предоставляет Способ 2 или 2.1-2.15, в котором нарушение сна включает бессонницу поддержания сна, частые пробуждения и ощущение несвежести при пробуждении; например:
2.16 Любой из вышеприведенных способов, в котором нарушение сна представляет собой бессонницу поддержания сна;
2.17 Любой из вышеприведенных способов, в котором эффективное количество составляет 1 мг-5 мг, предпочтительно 2,5-5 мг в день;
2.18 Любой из вышеприведенных способов, в котором эффективное количество составляет 2,5 мг или 5 мг в день;
2.19 Любой из вышеприведенных способов, в котором нарушение сна наблюдается у пациента, страдающего или находящегося под угрозой дискинезии, например, пациента, получающего дофаминергические препараты, например, выбранные из леводопы и вспомогательных средств леводопы (карбидопы, ингибиторов КОМТ, ингибиторов МАО-Б), агонистов дофамина и антихолинергических средств, например, получающего леводопу;
2.20 Любой из вышеприведенных способов, в котором пациент страдает болезнью Паркинсона.
[0041] В дополнительном варианте осуществления девятого аспекта изобретение предоставляет Способ 2 или любой из 2.1-2.20, в котором нарушение сна включает бессонницу поддержания сна, частые пробуждения и ощущение несвежести при пробуждении.
[0042] Соединения настоящего раскрытия, Фармацевтические Композиции настоящего раскрытия или Композиции Депо настоящего раскрытия могут использоваться в комбинации со вторым терапевтическим средством, в частности при более низких дозировках, чем когда отдельные средства применяют в виде монотерапии, таким образом, чтобы усилить терапевтические активности комбинированных средств, не вызывая нежелательных побочных эффектов, обычно имеющих место при стандартной монотерапии. Следовательно, Соединения настоящего раскрытия могут одновременно, последовательно или параллельно вводить вместе с другими антидепрессантными, нейролептическими, другими снотворными средствами и/или средствами, применяемыми для лечения болезни Паркинсона или расстройств настроения. В другом примере побочные эффекты могут быть снижены или минимизированы путем введения Соединения настоящего раскрытия в комбинации с одним или более вторыми терапевтическими средствами в свободной или солевой форме, в котором дозировки (i) второго терапевтического средств(а) или (ii) как Соединения настоящего раскрытия, так и второго терапевтического средства являются более низкими, чем если бы средства/соединения вводили в виде монотерапии. В определенном варианте осуществления Соединения настоящего раскрытия пригодны для лечения дискинезии у пациента, получающего дофаминергические препараты, например, выбранные из леводопы и вспомогательных средств леводопы (карбидопы, ингибиторов КОМТ, ингибиторов МАО-Б), агонистов дофамина и антихолинергических средств, например, таких как используемые при лечении болезни Паркинсона.
[0043] Следовательно, в десятом аспекте настоящее раскрытие предоставляет Способ I или любой из Способов 1.1-35 или Способ 2 или любой из 2.1-20, дополнительно включающий одно или более терапевтических средств, выбранных из соединений, которые модулируют активность ГАМК (например, усиливают активность и облегчают передачу ГАМК), агониста ГАМК-B, модулятора 5-HT (например, агониста 5-HTIa, антагониста 5-HT2А, обратного агониста 5-HT2a и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста/ингибитора обратного захвата серотонина-2 (SARIs), антагониста рецепторов орексина, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, человеческого гормона роста, агониста гормона роста, эстрогена, агониста эстрогена, лекарственного средства нейрокинина-1, антидепрессанта, и опиоидного агониста и/или частичного опиатного агониста, и нейролептического средства, например, атипичного нейролептического средства в свободной или фармацевтически приемлемой солевой форме (Способ I-A и II-A соответственно; совместно «Способ 3»).
[0044] В дополнительном варианте осуществления десятого аспекта изобретение предоставляет Способ I-A или II-A, как указано, дополнительно включающий одно или более терапевтических средств.
3.1 Способ I-A или II-A, в котором терапевтическое средство(а) представляет собой соединение, которое модулирует активность ГАМК (например, усиливает активность и облегчает передачу ГАМК);
3.2 Способ I-A, или II-A, или 3.1, в котором соединение ГАМК выбрано из группы, состоящей из одного или более из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габоксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals) и эстазолама;
3.3 Способ I-A или II-A, в котором терапевтическое средство представляет собой дополнительный антагонист 5HT2a;
3.4 Способ I-A, или II-A, или 3.3, в котором указанный дополнительный антагонист 5HT2a выбран из одного или более из кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, Франция), прувансерина, MDL 100907 (Sanofi-Aventis, Франция), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, Сан-Диего, Калифорния) и AVE8488 (Sanofi-Aventis, Франция);
3.5 Способ I-A или II-A, в котором терапевтическое средство представляет собой агонист мелатонина;
3.6 Способ I-A, или II-A, или 3.5, в котором агонист мелатонина выбран из группы, состоящей из одного или более из мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Япония), VEC-162 (Vanda Pharmaceuticals, Роквилл, Мэриленд), PD-6735 (Phase II Discovery) и агомелатина;
3.7 Способ I-A или II-A, в котором терапевтическое средство представляет собой блокатор ионного канала;
3.8 Способ I-A, или II-A, или 3.7, в котором указанный блокатор ионного канала представляет собой один или более из ламотриджина, габапентина и прегабалина.
3.9 Способ I-A или II-A, в котором терапевтическое средство представляет собой антагонист рецепторов орексина;
3.10 Способ I-A, или II-A, или 3.9, в котором антагонист рецепторов орексина выбран из группы, состоящей из орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, Великобритания), GW649868 (GlaxoSmithKline) и производного бензамида;
3.11 Способ I-A или II-A, в котором терапевтическое средство представляет собой антагонист/ингибитор обратного захвата серотонина-2 (SARI);
3.12 Способ I-A, или II-A, или 3.11, в котором антагонист/ингибитор обратного захвата серотонина-2 (SARI) выбран из группы, состоящей из одного или более из Org 50081 (Organon-Нидерланды), ритансерина, нефазодона, серзона и тразодона;
3.13 Способ I-A или II-A, в котором терапевтическое средство представляет собой агонист 5HTIa;
3.14 Способ I-A, или II-A, или 3.13, в котором агонист 5HTIa выбран из группы, состоящей из одного или более из репинотана, саризотана, эптапирона, буспирона и MN-305 (MediciNova, Сан-Диего, Калифорния);
3.15 Способ I-A или II-A, в котором терапевтическое средство представляет собой лекарственное средство нейрокинина-1;
3.16 Способ I-A, или II-A, или 3.15, в котором лекарственное средство нейрокинина-1 представляет собой Казопитант (GlaxoSmithKline);
3.17 Способ I-A или II-A, в котором терапевтическое средство представляет собой нейролептическое средство;
3.18 Способ I-A, или II-A, или 3.17, в котором нейролептическое средство выбрано из группы, состоящей из хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина, промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
3.19 Способ I-A или II-A, в котором терапевтическое средство представляет собой антидепрессант;
3.20 Способ I-A, или II-A, или 3.19, в котором антидепрессант выбран из амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, сульфата фенелзина, протриптилина, сертралина, транилципромина, тразодона, тримипрамина и венлафаксина;
3.21 Способ I-A или II-A, 3.17 или 3.18, в котором нейролептическое средство представляет собой атипичное нейролептическое средство;
3.22 Способ I-A, или II-A, или любой из 3.17-3.21, в котором атипичное нейролептическое средство выбрано из группы, состоящей из клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
3.23 Способ I-A или II-A, в котором терапевтическое средство выбрано из любого из способов 3.1-3.22, например, выбрано из группы, состоящей из модафинила, армодафинила, доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габоксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама, кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, Франция), прувансерина, MDL 100907 (Sanofi-Aventis, Франция), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, Сан-Диего, Калифорния), AVE8488 (Sanofi-Aventis, Франция), репинотана, саризотана, эптапирона, буспирона, MN-305 (MediciNova, Сан-Диего, Калифорния), мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Япония), VEC-162 (Vanda Pharmaceuticals, Роквилл, Мэриленд), PD-6735 (Phase II Discovery), агомелатина, ламотриджина, габапентина, прегабалина, орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, Великобритания), GW649868 (GlaxoSmithKline), производного бензамида, Org 50081 (Organon-Нидерланды), ритансерина, нефазодона, серзона, тразодона, Казопитанта (GlaxoSmithKline), амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, сульфата фенелзина, протриптилина, сертралина, транилципромина, тразодона, тримипрамина, венлафаксина, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина, промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
3.24 Способ I-A или II-A, в котором терапевтическое средство представляет собой агонист H3;
3.25 Способ I-A или II-A, в котором терапевтическое средство представляет собой антагонист H3;
3.26 Способ I-A или II-A, в котором терапевтическое средство представляет собой норадренергический агонист или антагонист;
3.27 Способ I-A или II-A, в котором терапевтическое средство представляет собой агонист галанина;
3.28 Способ I-A или II-A, в котором терапевтическое средство представляет собой антагонист CRH;
3.29 Способ I-A или II-A, в котором терапевтическое средство представляет собой гормон роста человека;
3.30 Способ I-A или II-A, в котором терапевтическое средство представляет собой агонист гормона роста;
3.31 Способ I-A или II-A, в котором терапевтическое средство представляет собой эстроген;
3.32 Способ I-A или II-A, в котором терапевтическое средство представляет собой агонист эстрогена;
3.33 Способ I-A или II-A, в котором терапевтическое средство представляет собой лекарственное средство нейрокинин-1;
3.34 Способ I-A или II-A, в котором терапевтическое средство объединяют с соединениями Формулы (I) и терапевтическое средство представляет собой средство против болезни Паркинсона, такое как L-допа, ко-карелдопа, дуодопа, сталево, Симметрел, бензтропин, бипериден, бромокриптин, энтакапон, перголид, прамипексол, проциклидин, ропинирол, селегилин и толкапон;
3.35 Способ I-A или II-A, в котором терапевтическое средство представляет собой опиатный агонист или частичный опиатный агонист, например, мю-агонист или частичный агонист или каппа-агонист или частичный агонист, включая смешанные агонисты/антагонисты (например, средство с частичной мю-агонистической активностью и каппа-антагонистической активностью);
3.36 Способ 3.35, в котором терапевтическое средство представляет собой бупренорфин, необязательно в котором указанный способ не включает совместное лечение анксиолитическим средством, например, ГАМК-соединением или бензодиазепином;
3.37 Способ I-A или II-A, в котором соединения Формулы (I) могут быть использованы для лечения нарушений сна, депрессии, психоза или их любых комбинаций у пациентов, страдающих перечисленными заболеваниями и/или болезнью Паркинсона;
3.38 Способ I-A или II-A, в котором нарушение выбрано, по меньшей мере, из одного или более из психоза, например, шизофрении, депрессии, расстройств настроения, нарушений сна (например, поддержания сна и/или наступления сна) или их любой комбинации нарушений;
3.39 Любой из вышеприведенных способов, в котором нарушение представляет собой нарушение сна;
3.40 Любой из вышеприведенных способов, в котором нарушение представляет собой нарушение сна, связанное с психозом, например, шизофренией или болезнью Паркинсона; в свободной или фармацевтически приемлемой солевой форме.
[0045] В одиннадцатом аспекте изобретения комбинация Соединения настоящего раскрытия и одного или более вторых терапевтических средств, как описано в Способах I-A, II-A или любом из Способов 3 или 3.1-3.40, может быть введена в виде Фармацевтической Композиции или Композиции депо, как описано выше в данной заявке. Комбинированные композиции могут включать смеси комбинированных лекарственных средств, также как две или более отдельных композиций лекарственных средств, причем отдельные композиции могут быть, например, совместно введены пациенту.
[0046] В определенном варианте осуществления Способы I-A, II-A, 3 или 3.1-3.40 включают введение пациенту, нуждающемуся в таком лечении, Соединения Изобретения в комбинации с атипичным нейролептическим средством, например, соединением, выбранным из клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона или палиперидона, в свободной или фармацевтически приемлемой солевой форме, например, в котором дозировка атипичного нейролептического средства снижена и/или уменьшены побочные эффекты.
[0047] В другом варианте осуществления Способы I-A, II-A, 3 или 3.1-3.40 включают введение пациенту, нуждающемуся в таком лечении, Соединения Изобретения в комбинации с антидепрессантом, например, амитриптилином, амоксапином, бупропионом, циталопрамом, кломипрамином, дезипрамином, доксепином, дулоксетином, эсциталопрамом, флуоксетином, флувоксамином, имипрамином, изокарбоксазидом, мапротилином, миртазапином, нефазодоном, нортриптилином, пароксетином, сульфатом фенелзина, протриптилином, сертралином, транилципромином, тразодоном, тримипрамином или венлафаксином в свободной или фармацевтически приемлемой солевой форме. Альтернативно, антидепрессант может быть использован в качестве вспомогательного лекарственного средства в дополнение к соединению Изобретения.
[0048] В еще одном варианте осуществления Способы I-A, II-A, 3 или 3.1-3.40 включают введение пациенту, нуждающемуся в таком лечении, Соединения Изобретения в комбинации с соединением, которое модулирует активность ГАМК, например, соединением, выбранным из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габоксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама или их любых комбинаций, в свободной или фармацевтически приемлемой солевой форме. В других вариантах осуществления способы, раскрытые в настоящем описании, дополнительно не включают введение ГАМК-соединения, бензодиазепина или любого другого анксиолитического средства.
[0049] В другом предпочтительном варианте осуществления Способы I-A, II-A, 3 или 3.1-3.40 включают введение пациенту, нуждающемуся в таком лечении, Соединения Изобретения в комбинации с доксепином в свободной или фармацевтически приемлемой солевой форме. Дозировки доксепина могут варьироваться в любом диапазоне, известном специалисту в данной области техники. В одном примере доза доксепина 10 мг может быть объединена с любой дозировкой соединения Изобретения.
[0050] В другом варианте осуществления Способы I-A, II-A, 3 или 3.1-3.40 включают введение пациенту, нуждающемуся в таком лечении, Соединения Изобретения в комбинации (включая как часть ежедневного режима дозирования) с атипичным стимулятором, например, модафинилом, адрафинилом или армодафинилом. Режим, включающий Соединение Изобретения с такими лекарственными средствами, способствует более регулярному сну и позволяет избежать побочных эффектов, таких как психоз или мания, связанных с более высокими уровнями таких лекарственных средств, например, при лечении биполярной депрессии, когнитивного нарушения, связанного с шизофренией, и избыточной сонливости и утомляемости в состояниях, таких как болезнь Паркинсона и рак.
[0051] В некоторых вариантах осуществления каждого из Соединений Формул I-IV и последующие; Фармацевтических Композиций 6 и 6.1-6.8; Композиций Депо 6.9 и 6.10; Композиций P.1-P.7; Способов 1 и 1.1-1.35 и Способов 2 и 2.1-2.20 соединение настоящего раскрытия по существу не содержит соединения Формулы A.
[0052] В двенадцатом аспекте изобретение предоставляет применение соединения, как описано далее:
11.1 Соединение I или 1.1-1.30 в свободной или фармацевтически приемлемой солевой форме;
11.2 Соединение II или 2.1-2.12 в свободной или фармацевтически приемлемой солевой форме;
11.3 Соединение III или 3.1-3.11 в свободной или фармацевтически приемлемой солевой форме;
11.4 Соединение IV или 4.1-4.11 в свободной или фармацевтически приемлемой солевой форме;
11.5 Соединение 5 или 5.1-5.5;
11.6 Соединение Формулы II-A, II-B, II-C, II-D или II-E в свободной или фармацевтически приемлемой солевой форме;
11.7 Соединение Формулы III-A, III-B или III-C в свободной или фармацевтически приемлемой солевой форме;
11.8 Соединение Формулы IV-A, IV-B или IV-C в свободной или фармацевтически приемлемой солевой форме;
11.9 Фармацевтическая Композиция 6 и 6.1-6.10;
11.10 Фармацевтическая Композиция P.1-P.7;
11.11 Композиция пероральной системы доставки с осмотически контролируемым высвобождением, как описано выше в данной заявке;
(при изготовлении лекарственного средства) для лечения или профилактики одного или более нарушений, как раскрыто выше в данной заявке, например, в любом из Способов 1 или 1.1-1.35, любом из Способов 2 и 2.1-2.20 и Способов 3 или 3.3-3.40 или любых способах, описанных в одиннадцатом аспекте изобретения.
[0053] В тринадцатом аспекте изобретение предоставляет фармацевтическую композицию, как описано выше в данной заявке, например:
12.1 Фармацевтическая Композиция 6 и 6.1-6.10;
12.2 Фармацевтическая композиция P.1-P.7;
12.3 Композиция пероральной системы доставки с осмотически контролируемым высвобождением, как описано выше в данной заявке,
для применения при лечении или профилактике одного или более нарушений, как раскрыто выше в данной заявке, например, в любом из Способов 1 и 1.1-1.35, Способов 2 и 2.1-2.20, Способов I-A, II-A, 3 или 3.1-3.40 или любых способах описанных в одиннадцатом или двенадцатом аспекте изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0054] Если не указано иначе или не ясно из контекста, следующие термины, используемые в данном описании, имеют следующие значения:
[0055] Используемый в данном описании «алкил» представляет собой насыщенный или ненасыщенный углеводородный фрагмент, например, длиной от одного до двадцати одного атома углерода, который может быть линейным или разветвленным (например, н-бутил или трет-бутил), предпочтительно линейным, если не указано иначе. Например, «C1-21алкил» обозначает алкил, содержащий от 1 до 21 атома углерода. В одном варианте осуществления алкил является необязательно замещенным одной или более гидрокси или C1-22алкокси (например, этокси) группами. В другом варианте осуществления алкил содержит от 1 до 21 атома углерода, предпочтительно имеет прямую цепь и необязательно является насыщенным или ненасыщенным, например, в некоторых вариантах осуществления, в которых R1 представляет собой алкильную цепь, содержащую от 1 до 21 атома углерода, предпочтительно 6-15 атомов углерода, 16-21 атом углерода, например, таким образом, что вместе с -C(O)-, к которому он присоединен, например, когда отщепляется от соединения Формулы I, образует остаток природной или неприродной, насыщенной или ненасыщенной жирной кислоты.
[0056] Термин «фармацевтически приемлемый разбавитель или носитель» предназначен для обозначения разбавителей и носителей, которые являются пригодными в фармацевтических препаратах и которые не содержат веществ, которые являются аллергенными, пирогенными или патогенными и которые, как известно, потенциально вызывают или способствуют развитию заболевания. Таким образом, фармацевтически приемлемые разбавители или носители исключают физиологические жидкости, такие как, например, кровь, моча, спинномозговая жидкость, слюна и подобные, также как их составляющие компоненты, такие как клетки крови и циркулирующие белки. Подходящие фармацевтически приемлемые разбавители и носители могут быть найдены в любом из нескольких хорошо известных трактов о фармацевтических составах, например, Anderson, Philip O.; Knoben, James E.; Troutman, William G, eds., Handbook of Clinical Drug Data, Tenth Edition, McGraw-Hill, 2002; Pratt and Taylor, eds., Principles of Drug Action, Third Edition, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, Ninth Edition, McGraw Hill, 20037ybg; Goodman and Gilman, eds., The Pharmacological Basis of Therapeutics, Tenth Edition, McGraw Hill, 2001; Remington's Pharmaceutical Sciences, 20th Ed., Lippincott Williams & Wilkins., 2000; и Martindale, The Extra Pharmacopoeia, Thirty-Second Edition (The Pharmaceutical Press, London, 1999); все из которых включены в настоящее описание посредством ссылки в полном объеме.
[0057] Термины «очищенный», «в очищенной форме» или «в выделенной и очищенной форме» для соединения относятся к физическому состоянию указанного соединения после выделения из синтетического процесса (например, из реакционной смеси), или природного источника, или их комбинации. Таким образом, термин «очищенный», «в очищенной форме» или «в выделенной и очищенной форме» для соединения относится к физическому состоянию указанного соединения после получения из процесса очистки или процессов, описанных в данной заявке или хорошо известных специалисту в данной области техники (например, хроматография, перекристаллизация, методы ЖХ-МС и ЖХ-МС/МС и подобные), с достаточной степенью чистоты, чтобы характеризоваться с помощью стандартных аналитических методов, описанных в данной заявке или хорошо известных специалисту в данной области техники.
[0058] Соединения Формулы I, в которой Z представляет собой -(C=O)- или -(СН(ОН))-, включая, например, Соединения Формул II-B и II-C, могут быть получены в виде метаболитов соединения Формулы A и/или в виде метаболитов соединения Формулы B:
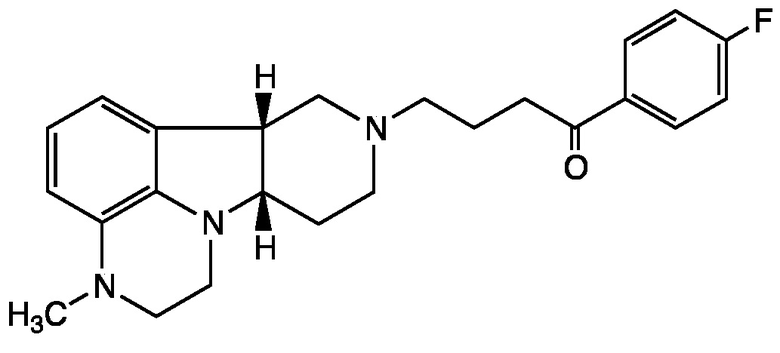
Формула А
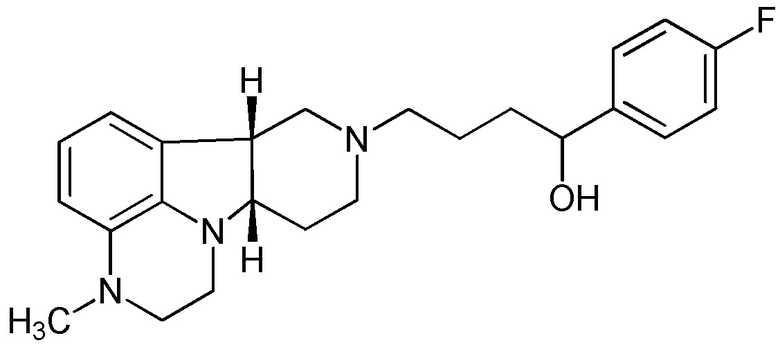
Формула B
Известно, что соединение Формулы A обеспечивает эффективное лечение нарушений, связанных с 5-HT2A, SERT и/или D2-рецепторами, без значительных экстрапирамидных побочных эффектов, как подобным образом раскрыто и заявлено в WO 2009/145900, содержание которой включено посредством ссылки в полном объеме. Уровни в плазме соединений Формул II-B и II-C, полученных из метаболизма соединения Формулы A, однако, довольно низки и, вероятно, не вносят значительного вклада в терапевтическую активность соединения Формулы A. Соединения Формул II-D и II-E также могут присутствовать в виде метаболитов, хотя это до сих пор не было обнаружено. Неожиданно было обнаружено, что Соединения Формулы I обладают активностью в качестве антагонистов μ-опиоидного рецептора. Это неожиданно, потому что соединение Формулы A не было известно, или было непонятно, имеет оно активность μ-опиоидного рецептора или связывание. Показано, что Соединения Формулы I, в которой X представляет собой -NH- и в которой L представляет собой -O-, имеют в частности хороший антагонизм μ-опиоидного рецептора. Следовательно, такие Соединения Формулы I могут быть пригодны при лечении наркотической зависимости, такой как опиатная зависимость и/или алкогольная зависимость с помощью ингибирования эндогенного опиатного ответа на введение запрещенного лекарственного средства, также как с помощью ингибирования прямого воздействия приема запрещенных опиатных средств.
[0059] Удивительно, что метаболиты соединения Формулы А имеют несколько иную относительную аффинность связывания с рецептором, чем соединения Формулы А. Например, профиль связывания с рецептором соединения Формулы II-B является уникальным, с комбинацией антагонистической активности при 5-HT2A, D1 и Мю-опиоидных рецепторах, что делает данное соединение очень интересным для лечения расстройств настроения. Соединение Формулы А, например, неактивно при Мю-опиоидном рецепторе.
[0060] Если не указано иначе, Соединения настоящего раскрытия, например, Соединение I или 1.1-1.30, Соединение II или 2.1-2.18, Соединение III или 3.1-3.13 или Соединение IV или 4.1-4.13 (совместно Соединения Формул I-IV и последующие) могут существовать в свободной или солевой форме, например, в виде кислотно-аддитивных солей. Кислотно-аддитивная соль соединения изобретения, которое является достаточно основным, например, кислотно-аддитивная соль, например, с неорганической или органической кислотой, например, хлористоводородной, бромистоводородной, серной, фосфорной кислотой, уксусной, трифторуксусной, лимонной, малеиновой кислотой, толуолсульфоновой, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памовой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изэтионовой кислотой и подобной. Кроме того, соль соединения изобретения, которое является достаточно кислотным, представляет собой соль щелочного металла, например, соль натрия или калия, соль щелочноземельного металла, например, соль кальция или магния, соль аммония или соль с органическим основанием, которое предоставляет физиологически приемлемый катион, например, соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис-(2-гидроксиэтил)амином. В определенном варианте осуществления соль Соединений Изобретения представляет собой аддитивную соль толуолсульфоновой кислоты. В другом определенном варианте осуществления соль Соединений Изобретения представляет собой аддитивную соль фумаровой кислоты. В определенном варианте осуществления соль Соединений Изобретения представляет собой аддитивную соль фосфорной кислоты.
[0061] Соединения настоящего раскрытия предназначены для применения в качестве фармацевтических препаратов, следовательно, фармацевтически приемлемые соли являются предпочтительными. Соли, которые непригодны для фармацевтического применения, могут быть пригодны, например, для выделения или очистки свободных Соединений Изобретения и поэтому также включены.
[0062] Соединения настоящего раскрытия могут содержать один или более хиральных атомов углерода. Таким образом, соединения существуют в отдельной изомерной, например, энантиомерной или диастереомерной форме или в виде смесей отдельных форм, например, рацемических/диастереомерных смесей. Любой изомер может быть представлен, в котором асимметрический центр находится в (R)-, (S)- или (R,S)-конфигурации. Изобретение следует понимать, как охватывающее как отдельные оптически активные изомеры, также их смеси (например, рацемические/диастереомерные смеси). Соответственно, Соединения Изобретения могут представлять собой рацемическую смесь или оно может находиться преимущественно, например, в чистой или по существу чистой изомерной форме, например, более 70% энантиомерном/диастереомерном избытке («ee»), предпочтительно более 80% ee, более предпочтительно более 90% ee, наиболее предпочтительно более 95% ee. Очистка указанных изомеров и разделение указанных изомерных смесей могут быть осуществлены с помощью стандартных методов, известных в данной области техники (например, колоночной хроматографии, препаративной ТСХ, препаративной ВЭЖХ, псевдодвижущегося слоя и подобного).
[0063] Геометрические изомеры по природе заместителей вокруг двойной связи или кольца могут присутствовать в цис (Z) или транс (Е)-форме, и обе изомерные формы охвачены объемом данного изобретения.
[0064] Также предполагается, что соединения настоящего раскрытия охватывают их стабильные и нестабильные изотопы. Стабильные изотопы представляют собой нерадиоактивные изотопы, которые содержат один дополнительный нейтрон по сравнению с распространенными нуклидами того же вида (т.е. элемента). Ожидается, что активность соединений, содержащих такие изотопы, будет сохранена, и такое соединение также будет пригодно для измерения фармакокинетики неизотопных аналогов. Например, атом водорода в определенном положении на соединениях раскрытия может быть заменен дейтерием (стабильный изотоп, который является нерадиоактивным). Примеры известных стабильных изотопов включают, но не ограничиваются ими, дейтерий, 13C, 15N, 18O. В качестве альтернативы, нестабильные изотопы, которые представляют собой радиоактивные изотопы, которые содержат дополнительные нейтроны по сравнению с распространенными нуклидами того же вида (т.е. элемента), например, 123I, 131I, 125I, 11C, 18F, могут заменять соответствующие распространенные виды I, C и F. Другим примером пригодного изотопа соединения изобретения является 11С-изотоп. Данные радиоактивные изотопы пригодны для радиоизображения и/или фармакокинетических исследований соединений изобретения. Кроме того, замещение атомов, имеющих природное изотопное распределение, более тяжелыми изотопами может привести к желаемому изменению фармакокинетических скоростей, когда данные замещения производятся на метаболически предрасположенных участках. Например, включение дейтерия (2Н) вместо водорода может замедлять метаболическое разложение, когда положение водорода является местом ферментативной или метаболической активности.
[0065] В дополнение к уникальной характеристике Соединений настоящего раскрытия Соединения Формулы I, в которой Y представляет собой -C(H)(OH), также могут быть этерифицированы с образованием физиологически гидролизуемых и приемлемых сложноэфирных пролекарств. Используемый в настоящем описании термин «физиологически гидролизуемые и приемлемые сложные эфиры» означает сложные эфиры Соединений настоящего раскрытия, которые гидролизуются в физиологических условиях с образованием гидрокси с одной стороны и кислоты, например, карбоновой кислоты с другой, которые сами по себе являются физиологически переносимыми в дозах, которые вводятся. Например, Соединение Формулы I или Формулы II, в которой Y представляет собой -C(H)(OH), может быть этерифицировано с образованием пролекарства, т.е. Соединения Формулы I или Формулы II, в которой R1 представляет собой -C(O)-C1-21алкил. В некоторых предпочтительных вариантах осуществления R1 представляет собой -C(O)-C1-21алкил, например, сложные эфиры ацилкислоты, например, сложный эфир гептановой, октановой, декановой, додекановой, тетрадекановой или гексадекановой кислоты.
[0066] Подобным образом, в тех случаях, когда Соединения настоящего раскрытия содержат аминогруппу, может также существовать пролекарство такого амина, например, пролекарства метиламина, в котором пролекарство расщепляется для высвобождения метаболита амина in vivo после введения.
[0067] Пролекарства Соединений настоящего раскрытия, в которых R1 представляет собой -С(O)-С1-21алкил, предпочтительно -С6-21алкил, более предпочтительно С6-15алкил, более предпочтительно линейный, насыщенный или ненасыщенный и необязательно замещенный одной или более гидрокси или алкокси группами, являются наиболее пригодными для замедленного и/или отсроченного высвобождения с целью достижения эффекта длительного действия, например, в которых Соединения настоящего раскрытия высвобождаются в течение периода от приблизительно 14 до приблизительно 30, до приблизительно 180 дней, предпочтительно в течение приблизительно 30, или приблизительно 60, или приблизительно 90 дней, например, как описано в любой из композиций депо, как описано в данной заявке. Предпочтительно, состав с замедленным и/или отсроченным высвобождением представляет собой инъекционный состав.
[0068] Альтернативно и/или дополнительно Соединения настоящего раскрытия могут быть включены в качестве состава депо, например, с помощью диспергирования, растворения или инкапсулирования Соединений Изобретения в полимерную матрицу, как описано в любой из Композиций 6 и 6.1-6.10, так что Соединение непрерывно высвобождается по мере того, как полимер разрушается с течением времени. Высвобождение Соединений Изобретения из полимерной матрицы предоставляет контролируемое, и/или отсроченное, и/или замедленное высвобождение Соединений, например, из фармацевтической композиции депо объекту, например, теплокровному животному, такому как человек, которому вводится фармацевтическое депо. Таким образом, фармацевтическое депо доставляет Соединения Изобретения объекту в концентрациях, эффективных для лечения определенного заболевания или медицинского состояния в течение длительного периода времени, например, 14-180 дней, предпочтительно приблизительно 30, приблизительно 60 или приблизительно 90 дней.
[0069] Полимеры, пригодные для полимерной матрицы в Композиции Изобретения (например, композиции Депо Изобретения), могут включать сложный полиэфир гидроксижирной кислоты и ее производных или других средств, таких как полимолочная кислота, полигликолевая кислота, полилимонная кислота, полияблочная кислота, поли-бета-гидроксимасляная кислота, полимер с раскрытием кольца эпсилон-капролактона, сополимер молочной кислоты-гликолевой кислоты, сополимер 2-гидроксимасляной кислоты-гликолевой кислоты, сополимер полимолочной кислоты-полиэтиленгликоля или сополимер полигликолевой кислоты-полиэтиленгликоля, полимер алкил-альфа-цианоакрилата (например, поли(бутил-2-цианоакрилат)), полиалкиленоксалат (например, политриметиленоксалат или политетраметиленоксалат), сложный полиортоэфир, поликарбонат (например, полиэтиленкарбонат или полиэтиленпропиленкарбонат), полиортокарбонат, полиаминокислоту (например, поли-гамма-L-аланин, поли-гамма-бензил-L-глутаминовую кислоту или поли-γ-метил-L-глутаминовую кислоту), сложный эфир гиалуроновой кислоты и подобные, и один или более из данных полимеров можно использовать.
[0070] Если полимеры являются сополимерами, они могут представлять собой любые из статистических, блок и/или привитых сополимеров. Когда вышеуказанные альфа-гидроксикарбоновые кислоты, гидроксидикарбоновые кислоты и гидрокситрикарбоновые кислоты обладают оптической активностью в их молекулах, можно использовать любой один из D-изомеров, L-изомеров и/или DL-изомеров. В том числе может быть использован полимер альфа-гидроксикарбоновой кислоты (предпочтительно, полимер молочной кислоты-гликолевой кислоты), его сложный эфир, сложные эфиры поли-альфа-цианоакриловой кислоты и т.д., и предпочтительным является сополимер молочной кислоты-гликолевой кислоты (также называемый как поли(лактид-альфа-гликолид) или поли(молочная-co-гликолевая кислота) и далее в настоящем описании называемый PLGA). Таким образом, в одном аспекте полимер, применяемый для полимерной матрицы, представляет собой PLGA. Используемый в настоящем описании термин PLGA включает полимеры молочной кислоты (также называемые как полилактид, поли(молочная кислота) или PLA). Наиболее предпочтительно, полимер представляет собой биоразлагаемый полимер поли(d,l-лактид-со-гликолид).
[0071] В предпочтительном варианте осуществления полимерная матрица изобретения представляет собой биосовместимый и биоразлагаемый полимерный материал. Термин «биосовместимый» определяется как полимерный материал, который не является токсичным, не является канцерогенным и не вызывает в значительной степени воспаление в тканях тела. Материал матрицы должен быть биоразлагаемым там, где полимерный материал должен разлагаться под действием процессов в организме до продуктов, легко утилизируемых организмом, и не должен накапливаться в организме. Продукты биоразложения также должны быть биосовместимы с организмом в том смысле, что полимерная матрица биосовместима с организмом. Определенные пригодные примеры материалов полимерной матрицы включают поли(гликолевую кислоту), поли-D,L-молочную кислоту, поли-L-молочную кислоту, сополимеры вышеприведенных, поли(алифатические карбоновые кислоты), сополиоксалаты, поликапролактон, полидиоксонон, поли(ортокарбонаты), поли(ацетали), поли(молочная кислота-капролактон), сложные полиортоэфиры, поли(гликолевая кислота-капролактон), полиангидриды и природные полимеры, включающие альбумин, казеин и воски, такие как моно- и дистеарат глицерина и подобные. Предпочтительным полимером для применения в практике данного изобретения является dl(полилактид-со-гликолид). Предпочтительно, чтобы молярное соотношение лактида и гликолида в таком сополимере находилось в диапазоне от приблизительно 75:25 до 50:50.
[0072] Пригодные PLGA-полимеры могут иметь средневесовую молекулярную массу от приблизительно 5000 до 500000 дальтон, предпочтительно приблизительно 150000 дальтон. В зависимости от скорости разложения, которая должна быть достигнута, могут быть использованы полимеры с различной молекулярной массой. Для диффузионного механизма высвобождения лекарственного средства полимер должен оставаться неповрежденным, пока все лекарственное средство не высвободится из полимерной матрицы, и затем разлагаться. Лекарственное средство может также высвобождаться из полимерной матрицы по мере того, как полимерный эксципиент биоразлагается.
[0073] PLGA может быть получен с помощью любого стандартного способа или может быть коммерчески доступным. Например, PLGA может быть получен путем полимеризации с открытием кольца с подходящим катализатором из циклического лактида, гликолида и т.д. (см. EP-0058481B2; Эффекты переменных полимеризации на свойства PLGA: молекулярную массу, композицию и структуру цепи).
[0074] Полагают, что PLGA является биоразлагаемым посредством разложения цельной композиции твердого полимера вследствие разрыва гидролизуемых и ферментативно расщепляемых сложноэфирных связей при биологических условиях (например, в присутствии воды и биологических ферментов, обнаруживаемых в тканях теплокровных животных, таких как люди) с образованием молочной кислоты и гликолевой кислоты. Как молочная кислота, так и гликолевая кислота являются водорастворимыми, нетоксичными продуктами нормального метаболизма, которые могут дополнительно биоразлагаться с образованием диоксида углерода и воды. Другими словами, полагают, что PLGA разлагается посредством гидролиза его сложноэфирных групп в присутствии воды, например, в организме теплокровного животного, такого как человек для получения молочной кислоты и гликолевой кислоты и создания кислого микроклимата. Молочная и гликолевая кислоты являются побочными продуктами разнообразных метаболических путей в организме теплокровного животного, такого как человек при нормальных физиологических условиях и, следовательно, являются хорошо переносимыми и продуцируют минимальную системную токсичность.
[0075] В другом варианте осуществления полимерная матрица, пригодная для изобретения, может содержать звездообразный полимер, в котором структура сложного полиэфира является звездообразной. Данные сложные полиэфиры имеют единичный остаток полиола в качестве центрального фрагмента, окруженного цепями остатков кислот. Полиольный фрагмент может представлять собой, например, глюкозу или, например, маннит. Данные сложные эфиры известны и описаны в патенте Великобритании 2145422 и в патенте США № 5538739, содержание которых включено посредством ссылки.
[0076] Звездообразные полимеры могут быть получены с использованием полигидроксисоединений, например, полиола, например, глюкозы или маннита в качестве инициатора. Полиол содержит, по меньшей мере, 3 гидроксигруппы и имеет молекулярную массу до приблизительно 20000 дальтон, по меньшей мере, с 1, предпочтительно, по меньшей мере, 2, например, в среднем 3 из гидроксигрупп полиола, находящихся в виде сложноэфирных групп, которые содержат полилактидные или co-полилактидные цепи. Разветвленные сложные полиэфиры, например, поли(d,l-лактид-со-гликолид), имеют центральный фрагмент глюкозы, имеющий лучи из линейных полилактидных цепей.
[0077] Композиции депо изобретения (например, Композиции 6 и 6.1-6.10 в полимерной матрице), как описано выше в данной заявке, могут содержать полимер в форме микрочастиц или наночастиц или в жидкой форме с Соединениями Изобретения, диспергированными или инкапсулированными в них. «Микрочастицы» означают твердые частицы, которые содержат Соединения Изобретения или в растворе, или в твердой форме, в которой такое соединение диспергировано или растворено в полимере, который служит в качестве матрицы частицы. С помощью соответствующего выбора полимерных материалов может быть получен состав микрочастиц, в котором полученные микрочастицы обладают как свойствами диффузионного высвобождения, так и высвобождения при биоразложении.
[0078] Когда полимер находится в форме микрочастиц, микрочастицы могут быть получены с использованием любого подходящего способа, такого как испарение растворителя или экстракция растворителем. Например, в способе испарения растворителя Соединения Изобретения и полимер могут быть растворены в летучем органическом растворителе (например, кетоне, таком как ацетон, галогенированном углеводороде, таком как хлороформ или метиленхлорид, галогенированном ароматическом углеводороде, циклическом простом эфире, таком как диоксан, сложном эфире, таком как этилацетат, нитриле, таком как ацетонитрил или спирте, таком как этанол) и диспергированы в водной фазе, содержащей подходящий стабилизатор эмульсии (например, поливиниловый спирт, ПВС). Затем органический растворитель испаряют для получения микрочастиц с Соединениями Изобретения, инкапсулированными в них. В способе экстракции растворителем Соединения Изобретения и полимер могут быть растворены в полярном растворителе (таком как ацетонитрил, дихлорметан, метанол, этилацетат или метилформиат) и затем диспергированы в водной фазе (такой как вода/раствор ПВС). Эмульсию получают с предоставлением микрочастиц с Соединениями Изобретения, инкапсулированными в них. Сушка распылением является альтернативной технологией изготовления для получения микрочастиц.
[0079] Другой способ получения микрочастиц изобретения также описан как в патенте США № 4389330, так и патенте США. № 4530840.
[0080] Микрочастица настоящего изобретения может быть получена с помощью любого способа, способного обеспечить получение микрочастиц в интервале размеров, приемлемом для применения в инъекционной композиции. Одним из предпочтительных способов получения является способ, описанный в патенте США № 4389330. В данном способе активный агент растворяют или диспергируют в соответствующем растворителе. К среде, содержащей агент, добавляют материал полимерной матрицы в количестве относительно активного ингредиента, который предоставляет продукт, имеющий желательную загрузку активного агента. Необязательно, все из ингредиентов продукта в виде микрочастиц могут быть смешаны вместе в среде растворителя.
[0081] Растворители для Соединений Изобретения и материала полимерной матрицы, которые могут использоваться в практике осуществления настоящего изобретения, включают органические растворители, такие как ацетон; галогенированные углеводороды, такие как хлороформ, метиленхлорид и подобные; ароматические углеводородные соединения; галогенированные ароматические углеводородные соединения; циклические простые эфиры; спирты, такие как бензиловый спирт; этилацетат и подобные. В одном варианте осуществления растворитель для применения в практике осуществления настоящего изобретения может представлять собой смесь бензилового спирта и этилацетата. Дополнительную информацию по получению микрочастиц, пригодных для изобретения, можно найти в публикации патентной заявки США № 2008/0069885, содержание которой включено в настоящее описание посредством ссылки в полном объеме.
[0082] Количество Соединений настоящего раскрытия, включенных в микрочастицы, обычно находится в диапазоне от приблизительно 1 мас.% до приблизительно 90 мас.%, предпочтительно от 30 до 50 мас.%, более предпочтительно от 35 до 40 мас.%. Под массовыми % подразумевают части Соединений настоящего раскрытия на общую массу микрочастицы.
[0083] Композиции фармацевтического депо могут содержать фармацевтически приемлемый разбавитель или носитель, такой как смешивающийся с водой разбавитель или носитель.
[0084] Дополнительные сведения о композиции пероральной системы доставки с осмотически контролируемым высвобождением могут быть найдены в ЕР 1539115 (публикация патентной заявки США №2009/0202631) и WO 2000/35419, содержание каждой из которых включено посредством ссылки в полном объеме.
[0085] «Терапевтически эффективное количество» представляет собой любое количество Соединений изобретения (например, такое, какое содержится в фармацевтическом депо), которое, когда его вводят объекту, страдающему от заболевания или нарушения, является эффективным, чтобы вызвать снижение, ремиссию или регрессию заболевания или нарушения в течение периода времени, как предписано для лечения.
[0086] Дозировки, применяемые в практике осуществления настоящего изобретения, будут, разумеется, варьироваться в зависимости, например, от определенного заболевания или состояния, подлежащего лечению, определенного применяемого Соединения Изобретения, способа введения и желательной терапии. Если не указано иначе, количество Соединения Изобретения для введения (независимо от того, что вводится в виде свободного основания или в виде солевой формы) относится или основывается на количестве Соединения Изобретения в форме свободного основания (т.е. расчет количества основывается на количестве свободного основания).
[0087] Соединения Изобретения могут быть введены с помощью любого приемлемого пути, включая перорально, парентерально (внутривенно, внутримышечно или подкожно) или трансдермально, но предпочтительно их вводят перорально. В определенном варианте осуществления Соединения Изобретения, например, в составе депо предпочтительно вводят парентерально, например, с помощью инъекции.
[0088] В целом, удовлетворительные результаты для Способа 1 и 1.1-1.35, Способа 2 и 2.1-2.20 и Способа 3 и 3.1-3.40 или применения Соединений настоящего раскрытия, как описано выше в данной заявке, например, для лечения комбинации заболеваний, такой как комбинация, по меньшей мере, депрессии, психоза, например, (1) психоза, например, шизофрении, у пациента, страдающего депрессией; (2) депрессии у пациента, страдающего психозом, например, шизофренией; (3) расстройств настроения, связанных с психозом, например, шизофренией или болезнью Паркинсона; (4) нарушений сна, связанных с психозом, например, шизофренией или болезнью Паркинсона; и (5) наркотической зависимости, нарушений, связанных с употреблением веществ, и/или нарушений, вызванных употреблением веществ, как изложено выше, указаны для получения при пероральном введении при дозировках порядка от приблизительно 1 мг до 100 мг один раз в день, предпочтительно 2,5 мг-50 мг, например, 2,5 мг, 5 мг, 10 мг, 20 мг, 30 мг, 40 мг или 50 мг один раз в день, предпочтительно посредством перорального введения.
[0089] Удовлетворительные результаты для Способа 2 или 2.1-2.20 или применения Соединений настоящего раскрытия, как описано выше в данной заявке, например, для лечения нарушения сна по отдельности указаны для получения при пероральном введении при дозировках порядка от приблизительно 2,5 мг-5 мг, например, 2,5 мг, 3 мг, 4 мг или 5 мг Соединения Изобретения в свободной или фармацевтически приемлемой солевой форме один раз в день, предпочтительно посредством перорального введения.
[0090] Удовлетворительные результаты для Способа I-A, или Способа II-A, или любого из 3.1-3.40 указаны для получения при менее 100 мг, предпочтительно менее 50 мг, например, менее 40 мг, менее 30 мг, менее 20 мг, менее 10 мг, менее 5 мг, менее 2,5 мг один раз в день. Удовлетворительные результаты для Способа II-A или любого из 3.1-3.40 указаны для получения при менее 5 мг, предпочтительно менее 2,5 мг.
[0091] Для лечения нарушений, раскрытых в данном описании, в которых композицию депо применяют для достижения более длительной продолжительности действия, дозировки будут выше относительно композиции с более коротким действием, например, более 1-100 мг, например, 25 мг, 50 мг, 100 мг, 500 мг, 1000 мг или более 1000 мг. Продолжительность действия Соединений настоящего раскрытия может контролироваться путем воздействия на полимерную композицию, т.е. соотношение полимер:лекарственное средство и размер микрочастиц. В том случае, когда композиция изобретения представляет собой композицию депо, предпочтительным является введение с помощью инъекции.
[0092] Фармацевтически приемлемые соли Соединений настоящего раскрытия могут быть синтезированы из исходного соединения, которое содержит основный или кислотный фрагмент с помощью стандартных химических способов. Как правило, такие соли могут быть получены путем взаимодействия форм свободного основания данных соединений со стехиометрическим количеством подходящей кислоты в воде, или в органическом растворителе, или в смеси двух; обычно неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил являются предпочтительными. Дополнительные подробности получения данных солей, например, толуолсульфоновой соли в аморфной или кристаллической форме можно найти в PCT/US08/03340 и/или предварительной патентной заявке США №61/036069.
[0093] Фармацевтические композиции, содержащие Соединения настоящего раскрытия, могут быть получены с использованием стандартных разбавителей или эксципиентов (пример включает, но не ограничивается им, кунжутное масло) и методов, известных в галеновой области техники. Таким образом, пероральные лекарственные формы могут включать таблетки, капсулы, растворы, суспензии и подобное.
Способы получения Соединений Изобретения:
[0094] Соединения настоящего раскрытия, в которых X представляет собой -NH- или -N(CH3)- и Y представляет собой -C(=O), могут быть получены с помощью взаимодействия (6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалина или его 1-метилового аналога с 4-хлор-4'-фторбутирофеноном в соответствии со Схемой 1 ниже:
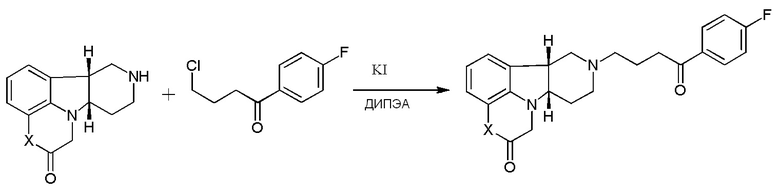
X=-NH- или -N(CH3)-
Схема 1
[0095] Соединения настоящего раскрытия, в которых Х представляет собой -NH- или -N(CH3)- и Y представляет собой -CH(OH)-, могут быть получены с помощью взаимодействия 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1Н,7Н-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фторфенил)-бутан-1-она, полученного в Схеме 1 (или его 1-метилового аналога) с восстановителем в соответствии со Схемой 2 ниже:
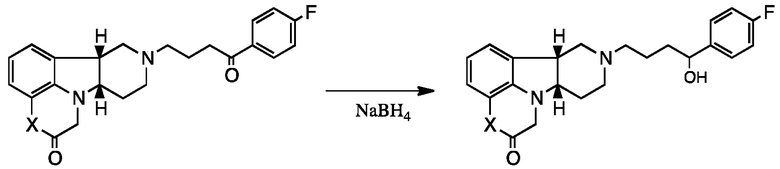
X=-NH- или -N(CH3)-
Схема 2
[0096] Восстановитель может представлять собой гидрид металла, например, боргидрид натрия, цианоборгидрид натрия, алюмогидрид лития, гидрид алюминия, гидрид диизобутилалюминия, предпочтительно боргидрид натрия. Дополнительные реагенты для восстановления кетонов могут быть найдены в Jerry March, Advanced Organic Chemistry, Reactions Mechanisms and Structures, p. 910-911, (1992, John Wiley & Sons, Inc.), Fourth Edition, содержание которой включено посредством ссылки.
[0097] Выделение или очистка диастереомеров Соединений Изобретения может быть достигнута с помощью стандартных способов, известных в данной области техники, например, посредством колоночной очистки, препаративной тонкослойной хроматографии, препаративной ВЭЖХ, кристаллизации, растирания, псевдодвижущихся слоев и подобного.
[0098] Соединения Формулы I, в которой Y представляет собой -CH(O-R1)- и R1 отличен от H, могут быть получены посредством нескольких широко используемых способов этерификации, таких как алкоголиз ацилгалогенидов, ангидридов или активных сложных эфиров. Например, Соединение Формулы I, в которой R1 представляет собой -C(O)-алкил, может быть получено с помощью взаимодействия:
(a) L-C(O)-C1-21алкила, в котором L представляет собой уходящую группу, такую как группа галогена (например, хлора или брома), трифторметилсульфонилокси (-OSO2CF3), тозилокси (-O-S(O)2-C6H4-CH3), метилсульфонилокси (-OS(O)2-CH3), 1H-бензо[d][1,2,3]триазол-1-илокси или сукцинимидилоксигруппа,
с
(b) Соединением Формулы I, в которой Y представляет собой -C(H)(OH),
предпочтительно в присутствии основания (например, диизопропиламина, триэтиламина или пиридина). Например, L-C(O)-C1-21алкил представляет собой ацетилгалогенид, деканоилгалогенид или гептаноилгалогенид, который может быть получен с помощью взаимодействия HO-C(O)-C1-21алкила, например, с тионилхлоридом, P(X')3 или P(X')5, в которой X' представляет собой Cl или Br. В том случае, если L представляет собой тозилокси-C(O)-C1-21алкил или метилсульфонилокси-C(O)-C1-21алкил, данные соединения могут быть получены с помощью взаимодействия HO-C(O)-C1-21алкила с тозилхлоридом или мезилхлоридом, предпочтительно в присутствии основания, такого как пиридин. Синтез Соединения Формулы II-A, в которой R1 отличен от H, может быть суммирован в Схеме 3 ниже:
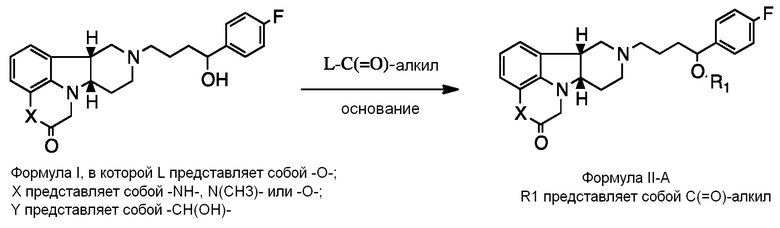
Схема 3
[0099] Альтернативно, синтез соединения Формулы II-A, в которой R1 отличен от H, может быть достигнут с помощью взаимодействия HO-C(O)-C1-21 алкила с (i) соединением Формулы I, в которой Y представляет собой -C(H)(OH), в присутствии основания, такого как ДИПЭА и NEt3 и (ii) дегидратирующим или связывающим реагентом, таким как тетрафторборат 2-фтор-1-этилпиридиния (FEP), гексафторфосфат тетраметилфторформамидиния (TFFH) или гексафторфосфат 1,1,3,3-бис(тетраметилен)хлорурония (PyClU).
[00100] Соли Соединений настоящего раскрытия могут быть получены аналогично описанным в патентах США №6548493; 7238690; 6552017; 6713471; 7183282; RE39680; RE39679 и WO 2009/114181, содержание каждого из которых включено посредством ссылки в полном объеме.
[00101] Диастереомеры полученных соединений могут быть разделены, например, с помощью ВЭЖХ с использованием CHIRALPAK® AY-H, 5μ, 30×250 мм при комнатной температуре и элюированы смесью 10% этанола/90% гексана/0,1% диметилэтиламина. Пики могут быть обнаружены при 230 нм для получения 98-99,9% ее диастереомера.
Пример 1: Синтез 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фторфенил)бутан-1-она
[00102] Этиловый эфир (6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-карбоновой кислоты (6,4 г, 21,2 ммоль) суспендируют в растворе уксусной кислоты HBr (64 мл, 33% масс./масс.) при комнатной температуре. Смесь нагревают при 50°C в течение 16 ч. После охлаждения и обработки этилацетатом (300 мл) смесь отфильтровывают. Осадок на фильтре промывают этилацетатом (300 мл) и затем высушивают под вакуумом. Полученную соль HBr затем суспендируют в метаноле (200 мл) и охлаждают сухим льдом в изопропаноле. При энергичном перемешивании медленно добавляют раствор аммиака (10 мл, 7N в метаноле) к суспензии для доведения pH смеси до 10. Полученную смесь высушивают под вакуумом без дополнительной очистки с получением неочищенного (6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалина (8,0 г), который используют непосредственно в следующей стадии. МС (ESI) m/z 230,2 [М+Н]+.
[00103] Неочищенный (6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин (1,4 г) растворяют в ДМФ (14 мл) и затем последовательно добавляют KI (2,15 г) и 4-хлор-4'-фторбутирофенон (2 мл). Смесь дегазируют аргоном с последующим добавлением N,N-диизопропилэтиламина (ДИПЭА, 2 мл). Смесь нагревают при 78°C в течение 2 ч. После охлаждения растворители удаляют при пониженном давлении. Темно-коричневый остаток суспендируют в дихлорметане (100 мл) и затем экстрагируют водой (30 мл). Органический слой отделяют и высушивают над K2CO3. После фильтрования растворители удаляют при пониженном давлении. Полученный неочищенный продукт очищают с помощью колоночной хроматографии на силикагеле, элюируя 0-10% метанола в этилацетате, содержащем 0,1% 7N аммония в метаноле с получением 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1Н,7Н-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фторфенил)-бутан-1-она в виде светло-желтого твердого вещества (767 мг). 1H ЯМР (500 МГц, ДМСО-d6) δ 10,3 (с, 1H), 8,1-8,0 (м, 2H), 7,3 (дд, J= 8,86 Гц, 2H), 6,8 (д, J= 7,25 Гц, 1H), 6,6 (дд, J= 7,55 Гц, 1H), 6,6 (д, J= 7,74 Гц, 1H), 3,8 (д, J= 14,49 Гц, 1H), 3,3-3,3 (м, 1H), 3,2-3,2 (м, 1H), 3,1-3,0 (м, 1H), 3,0 (т, J= 6,88 Гц, 2H), 2,8-2,8 (м, 1H), 2,6-2,5 (м, 1H), 2,3-2,2 (м, 2H), 2,1-2,0 (м, 1H), 1,9-1,8 (м, 1H), 1,8 (т, J= 6,99 Гц, 2H), 1,6 (т, J= 11,25 Гц, 2H). МС (ESI) m/z 394,2 [М+Н]+.
Пример 2: Синтез 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фторфенил)бутан-1-ола
[0101] 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1Н,7Н-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фторфенил)-бутан-1-он (50 мг, 0,127 ммоль) растворяют в метаноле (5 мл). При перемешивании порциями добавляют NaBH4 (31 мг, 0,82 ммоль). После завершения добавления смесь перемешивают при комнатной температуре в течение 30 мин. Метанол испаряют при пониженном давлении. Остаток растворяют в дихлорметане (10 мл) и затем экстрагируют водой (2×0,5 мл). Объединенную органическую фазу высушивают над K2CO3. После фильтрования фильтрат концентрируют при пониженном давлении и затем дополнительно высушивают под вакуумом с получением 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-8-ил)-1-(4-фторфенил)-бутан-1-ола в виде бледно-желтого пенистого твердого вещества (45 мг, выход 90%). 1H ЯМР (500 МГц, ДМСО-d6) δ 10,3 (с, 1H), 7,4-7,3 (м, 2H), 7,2-7,1 (м, 2H), 6,7 (д, J= 7,29 Гц, 1H) 6,7-6,6 (м, 1H), 6,6 (д, J= 7,74 Гц, 1H), 5,4 (с, 1H), 4,7-4,4 (м, 1H), 3,8 (д, J= 14,49 Гц, 1H), 3,3-3,3 (м, 1H), 3,3-3,2 (м, 1H), 3,2-3,1 (м, 1H), 2,8-2,7 (м, 1H), 2,6-2,5 (м, 1H), 2,3-2,1 (м, 2H), 2,1-2,0 (м, 1H), 2,0-1,9 (м, 1H), 1,8-1,7 (м, 1H), 1,7-1,5 (м, 3H), 1,5-1,4 (м, 1H), 1,4-1,3 (м, 1H). МС (ESI) m/z 396,2 [М+Н]+.
Пример 3: Синтез (6bR,10aS)-8-(3-(4-фторфенокси)пропил)-6b,7,8,9,10,10а-гексагидро-1Н-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-2(3Н)-она
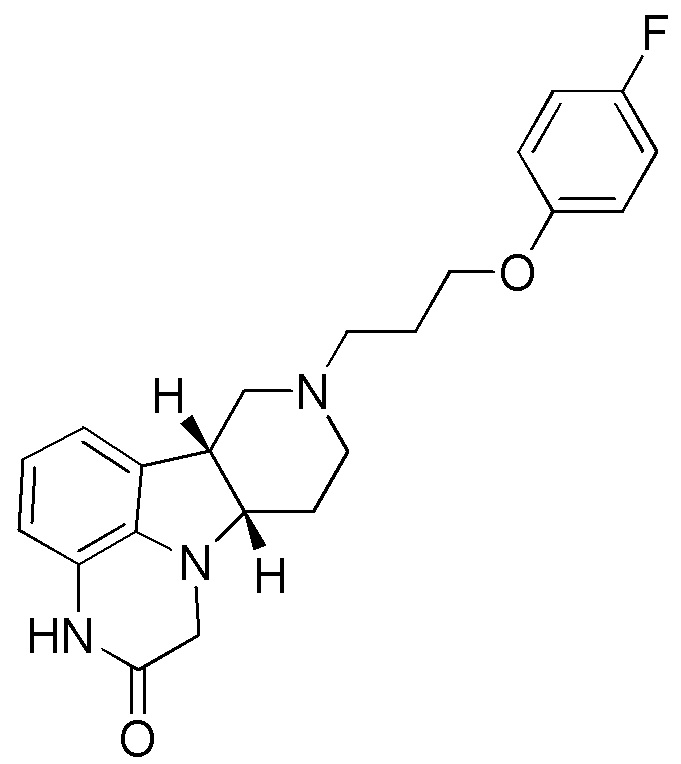
[0102] Смесь (6bR,10aS)-6b,7,8,9,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-де]хиноксалин-2(3H)-она (100 мг, 0,436 ммоль), 1-(3-хлоропрокси)-4-фторбензола (100 мкл, 0,65 ммоль) и KI (144 мг, 0,87 ммоль) в ДМФ (2 мл) дегазируют аргоном в течение 3 минут и добавляют ДИПЭА (150 мкл, 0,87 ммоль). Полученную смесь нагревают до 78°C и перемешивают при данной температуре в течение 2 ч. Смесь охлаждают до комнатной температуры и затем отфильтровывают. Фильтровальный осадок очищают с помощью колоночной хроматографией на силикагеле с использованием градиента 0-100% этилацетата в смеси метанола/7N NH3 в метаноле (1:0,1 об./об.) в качестве элюента с получением частично очищенного продукта, который далее очищают с помощью полупрепаративной системы ВЭЖХ с использованием градиента 0-60% ацетонитрила в воде, содержащей 0,1% муравьиной кислоты, в течение 16 мин с получением указанного в заголовке продукта в виде твердого вещества (50 мг, выход 30%). МС (ESI) m/z 406,2 [М+1]+. 1H ЯМР (500 МГц, ДМСО-d6) δ 10,3 (с, 1H), 7,2-7,1 (м, 2H), 7,0-6,9 (м, 2H), 6,8 (дд, J= 1,03, 7,25 Гц, 1H) 6,6 (т, J= 7,55 Гц, 1H), 6,6 (дд, J= 1,07, 7,79 Гц, 1H), 4,0 (т, J= 6,35 Гц, 2H), 3,8 (д, J= 14,74 Гц, 1H) 3,3-3,2 (м, 3H), 2,9 (дд, J= 6,35, 11,13 Гц, 1H), 2,7-2,6 (м, 1H), 2,5-2,3 (м, 2H), 2,1 (т, J= 11,66 Гц, 1H), 2,0 (д, J= 14,50 Гц, 1H), 1,9-1,8 (м, 3H), 1,7 (т, J= 11,04 Гц, 1H).
Фармакологический профиль
[0103] Как отмечалось выше, Соединения Раскрытия имеют уникальный фармакологический профиль со связыванием с 5-НТ2А, D1 и мю-опиоидными рецепторами.
[0104] Например, соединение Примера 3 обладает низкой наномолярной аффинностью к 5-НТ2А, D1 и мю-опиоидным рецепторам с величинами Ki 8,3 нМ, 50 нМ и 11 нМ соответственно.
[0105] Фармакологический профиль соединения Примера 3 исследуют с использованием связывания рецептора in vitro и функциональных анализов на основе клеток и исследований in vivo функциональной активности при 5-HT2A, D1, D2 и мю-опиоидных рецепторах, включая DOI-индуцированное судорожное движение головы, морфин-индуцированную гиперактивность, измерения с помощью вестерн-блоттинга уровней фосфопротеина в головном мозге (тирозингидроксилазы и GluN2B) и блокаду активности морфина в классическом анализе боли при подергивании хвоста у мышей.
[0106] Соединение Примера 3 проявляет активность при пероральном применении и отличную метаболическую стабильность у животных. Пероральное введение соединения Примера 3 эффективно блокирует DOI-индуцированное судорожное движение головы у мышей (EC50=0,23 мг/кг, п/о), что указывает на выраженную функциональную активность в качестве антагониста 5-HT2A. Соединение Примера 3 не нарушает нейротрансмиссию стриарного дофамина при исследуемых дозах, о чем свидетельствует отсутствие влияния на стриарную тирозингидроксилазу, скорость-лимитирующий фермент синтеза дофамина. Соединение Примера 3 (0,3 мг/кг, п/о) проявляет выраженный антагонизм морфина in vivo, блокируя морфин-индуцированную гиперактивность и морфин-индуцированную анальгезию у мышей при уровнях доз, сравнимых с его эффектами на рецептор 5-НТ2А (т.е. 0,1 мг/кг и выше, п/о).
[0107] Дополнительные сведения о фармакологических профилях Соединений Раскрытия описаны в следующих примерах.
Пример 4: Функциональные анализы клеточного и ядерного рецепторов
[0108] Функциональные анализы клеточного и ядерного рецепторов проводят на соединениях Формулы II-B и II-C в соответствии с методикой Wang, J.B. et al. (1994), FEBS Lett., 338:217-222. Соединения исследуют при нескольких концентрациях для определения их IC50 или EC50. Влияние клеточного агониста рассчитывают как процент контрольного ответа к известному стандартному агонисту для каждой мишени, и влияние клеточного антагониста рассчитывают как процент ингибирования ответа контрольного стандартного агониста для каждой мишени.
[0109] Следующий анализ проводят для определения влияния Соединения Формулы II-B на μ (МОР) (h)-рецептор:
(Рецептор)
(влияние агониста)
(0,3 мкМ DAMGO
для контроля)
37°C
(влияние антагониста)
(20 нМ)
37°C
[0110] Для антагонистов очевидные константы диссоциации (KB) рассчитывают с использованием модифицированного уравнения Ченга-Прусова:
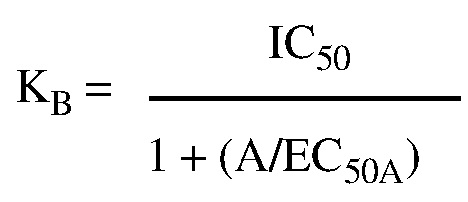
в котором A=концентрация стандартного агониста в анализе и значение EC50A=EC50 стандартного агониста.
[0111] Установлено, что соединение Формулы II-B имеет μ (MOP) (h) (влияние антагониста) с IC50 1,3×10-6 М; и KB 1,4×10-7 М; и установлено, что соединение Формулы II-C имеет IC50 выше 1×10-5, что являлось самой высокой исследуемой концентрацией.
[0112] Результаты выражены как процент контрольного ответа агониста:
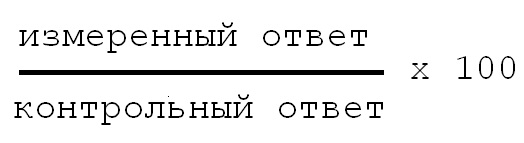
и как процентное ингибирование контрольного ответа агониста:
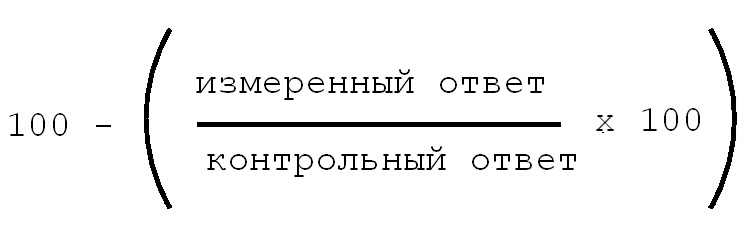
полученного в присутствии Соединения Формулы II-B или II-C.
[0113] Значения EC50 (концентрация, получающая полумаксимальный ответ) и значения IC50 (концентрация, вызывающая полумаксимальное ингибирование контрольного ответа агониста) определяют с помощью нелинейного регрессионного анализа кривых концентрация-ответ, образованных со средними значениями параллельного анализа с использованием аппроксимации кривой уравнением Хилла:
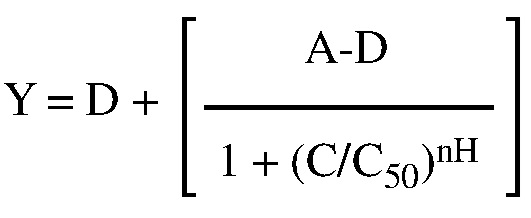
в котором Y=ответ, A=левая асимптота кривой, D=правая асимптота кривой, C=концентрация соединения и C50=EC50 или IC50 и nH=угловой коэффициент. Анализ проводят с использованием программного обеспечения, разработанного внутри компании и подтвержденного по сравнению с данными, полученными с помощью коммерческого программного обеспечения SigmaPlot® 4.0 для Windows® (© 1997 SPSS Inc.).
Пример 5: Профиль связывания рецептора Соединения Формул II-B и II-C
[0114] Связывание рецептора определяют для Соединений Формул II-A и II-B и Примера 3 с использованием тозилатной соли соединения Формулы A в качестве контроля. Используют следующие литературные методики, каждая из которых включена в настоящее описание посредством ссылки в полном объеме: 5-HT2A: Bryant, H.U. et al. (1996), Life Sci., 15:1259-1268; D2: Hall, D.A. and Strange, P.G. (1997), Brit. J. Pharmacol., 121:731-736; D1: Zhou, Q.Y. et al. (1990), Nature, 347:76-80; SERT: Park, Y.M. et al. (1999), Anal. Biochem., 269:94-104; Мю-опиоидный рецептор: Wang, J.B. et al. (1994), FEBS Lett., 338:217-222.
[0115] Обычно результаты выражают как процент контрольного специфического связывания:
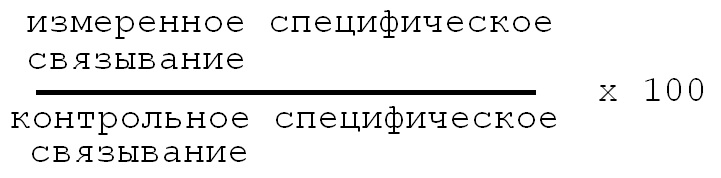
и как процент ингибирования контрольного специфического связывания:
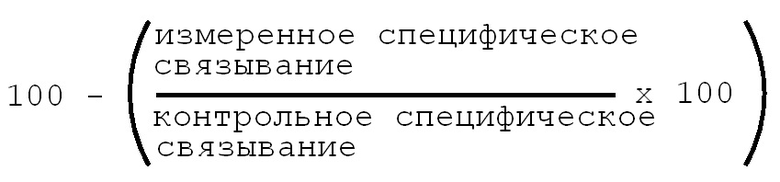
полученного в присутствии исследуемых соединений.
[0116] Значения IC50 (концентрация, вызывающая полумаксимальное ингибирование контрольного специфического связывания) и коэффициенты Хилла (nH) определяют с помощью нелинейного регрессионного анализа конкурирующих кривых, образованных со средними значениями параллельного анализа с использованием аппроксимации кривой уравнением Хилла:
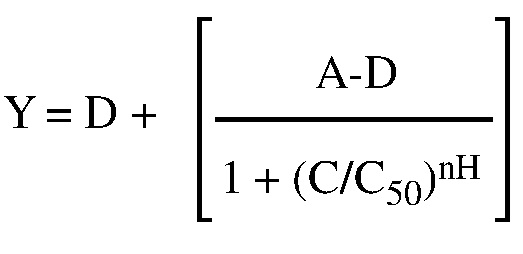
в котором Y=специфическое связывание, A=левая асимптота кривой, D=правая асимптота кривой, C=концентрация соединения, C50=IC50 и nH=угловой коэффициент. Данный анализ проводили с использованием программного обеспечения для внутреннего использования и подтверждали по сравнению с данными, полученными с помощью коммерческого программного обеспечения SigmaPlot® 4.0 для Windows® (© 1997 SPSS Inc.). Константы ингибирования (Ki) рассчитывали с использованием уравнения Ченга-Прусова:

в котором L=концентрация радиолиганда в анализе и KD=аффинность радиолиганда для рецептора. График Скэтчарда используют для определения KD.
[0117] Получают следующие результаты аффинности рецептора с использованием тозилатной соли Соединения Формулы А в качестве контроля:
(Пр. 1)
(Пр. 2)
(тозилатная соль)
Пример 6: DOI-индуцированная модель судорожного движения головы у мышей
[0118] R-(-)-2,5-диметокси-4-иодамфетамин (DOI) является агонистом семейства рецепторов серотонина 5-НТ2. При введении мышам он создает поведенческий профиль, связанный с частыми судорожными движениями головы. Частоту данных судорожных движений головы в течение заданного периода времени можно принять за оценку агонизма 5-НТ2-рецептора в головном мозге. В свою очередь, данный поведенческий анализ может быть использован для определения антагонизма 5-НТ2-рецептора в головном мозге с помощью введения DOI с или без антагониста и регистрации сокращения DOI-индуцированных судорожных движений головы после введения антагониста.
[0119] Способ Darmani et al., Pharmacol Biochem Behav. (1990) 36:901-906 (1990) 36:901-906 (содержание которого включено посредством ссылки в полном объеме) используют с некоторыми изменениями. (±)-DOI HCl вводят подкожно и мышей немедленно помещают в стандартную пластиковую клетку. Количество судорожных движений головы подсчитывают в течение 6 мин, начиная с 1 мин после введения DOI. Исследуемое соединение вводят перорально за 0,5 часа до инъекции DOI. Область результатов рассчитывают как EC50 для сокращения DOI-индуцированных судорожных движений головы. Результаты показаны в следующей Таблице:
Результаты показывают, что соединения Примера 1 и 3 значительно блокируют судорожные движения головы DOI, сравнимые с эталонными соединениями Формулы А и С, и согласуются с результатами in vitro 5-HT2A, показанными в Примере 5. Напротив, соединение Примера 2 относительно слабее в данном функциональном анализе, согласующемся с данными in vitro в Примере 5, показывая, что данное соединение относительно слабее в своем антагонизме рецептора серотонина (5-НТ2А) по сравнению с другими структурно подобными соединениями.
Пример 7: Анализ подергивания хвоста у мышей
[0120] Анализ подергивания хвоста у мышей является мерой анальгезии, обозначенной порогом болевого рефлекса сдерживаемых мышей. Хвосты мышей-самцов CD-1 располагают под фокусированным пучком высокоинтенсивного инфракрасного источника тепла, приводя к нагреванию хвоста. Регистрируют количество времени (латентность) между включением нагревательного прибора и подергиванием хвоста мыши от пути источника тепла. Введение морфина приводит к анальгезии, и это вызывает задержку реакции мыши на тепло (повышенная латентность). Предварительное введение антагониста морфина, т.е. налоксона отменяет эффект и приводит к нормальному латентному периоду. Данное исследование используют в качестве функционального анализа для определения антагонизма мю-опиоидных рецепторов.
[0121] Десяти мышам-самцам CD-1 (приблизительно 8 недель) присваивают каждую из пяти групп лечения. Группы обрабатывают следующим образом: Группа (1) [отрицательный контроль]: вводят 0,25% метилцеллюлозного наполнителя п/о за 60 минут до исследования подергивания хвоста и солевой наполнитель за 30 минут до исследования подергивания хвоста; Группа (2) [положительный контроль]: вводят 0,25% метилцеллюлозного наполнителя п/о за 60 минут до исследования и 5 мг/кг морфина в солевом растворе за 30 минут до исследования; Группа (3) [положительный контроль]: вводят 3 мг/кг налоксона в солевом растворе за 50 минут до исследования и 5 мг/кг морфина в солевом растворе за 30 минут до исследования; Группы (4)-(6): вводят или 0,1 мг/кг, 0,3 мг/кг, или 1 мг/кг исследуемого соединения в 0,25% метилцеллюлозном наполнителе п/о за 60 минут до исследования и 5 мг/кг морфина за 30 минут до исследования. Эксперимент повторяют для соединений Примера 1 и Примера 3. Результаты показаны в следующей таблице как средняя латентность, измеренная в секундах:
Нап/Нап
Соед/Мор
(0,1 мг/кг)
(0,3 мг/кг)
(1 мг/кг)
[0122] Результаты показывают, что соединения Примера 1 и Примера 3 проявляют дозозависимую блокаду морфин-индуцированной активности мю-опиоидного рецептора.
Пример 8: Профиль фосфопротеина ЦНС
[0123] Кроме того, проводят всестороннее исследование молекулярного фосфорилирования для изучения профиля центральной нервной системы (ЦНС) соединений Примера 1 и Примера 3. Степень фосфорилирования белка для выбранных ключевых белков центральной нервной системы измеряют в прилежащем ядре у мышей. Исследуемые белки включают ERK1, ERK2, Glu1, NR2B и ТГ (тирозингидроксилазу), и соединения Примера 1 и 3 сравнивали с нейролептическими средствами рисперидоном и галоперидолом.
[0124] Мышей обрабатывали соединением Примера 1 или 3 при 3 мг/кг или галоперидолом при 2 мг/кг. Мышей убивали через от 30 минут до 2 часов после инъекции путем фокусированного микроволнового черепного облучения, которое сохраняет фосфопротеин мозга, поскольку он существует в момент смерти. Затем прилежащее ядро рассекали из каждого мозга мыши, нарезали и замораживали в жидком азоте. Образцы далее готовили для анализа фосфопротеинов с помощью электрофореза SDS-PAGE с последующим фосфопротеин-специфичным иммуноблоттингом, как описано в Zhu H, et al., Brain Res. 2010 Jun 25; 1342:11-23. Фосфорилирование на каждом участке определяли количественно, нормировали к общим уровням белка (нефосфорилированного) и выражали в процентах уровня фосфорилирования у контрольных мышей, обработанных наполнителем.
[0125] Результаты показывают, что ни соединение Примера 1, ни Примера 3 не оказывают существенного влияния на фосфорилирование тирозингидроксилазы Ser40 через 30 минут или 60 минут в отличие от галоперидола, который обеспечивает увеличение более чем на 400%, и рисперидона, который обеспечивает увеличение более чем на 500% при фосфорилировании ТГ. Это демонстрирует, что соединения изобретения не нарушают метаболизм дофамина.
[0126] Результаты дополнительно показывают, что ни соединение Примера 1, ни Примера 3 не оказывают существенного влияния на фосфорилирование NR2B Tyr1472 через 30-60 минут. Соединения обеспечивают небольшое увеличение фосфорилирования GluR1 Ser845 и небольшое снижение фосфорилирования ERK2 Thr183 и Tyr185.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ РАССТРОЙСТВА ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2017 |
|
RU2776800C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2780002C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2733975C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2777366C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2011 |
|
RU2591194C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ | 2015 |
|
RU2711442C2 |
| НОВЫЕ СПОСОБЫ | 2014 |
|
RU2682658C1 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2679142C2 |
| 3-АЗАБИЦИКЛО[3.1.0]ГЕКСИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2510396C2 |
| НОВЫЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2836384C2 |
Изобретение относится к соединению формулы I в свободной или фармацевтически приемлемой солевой форме, которое может найти применение в качестве модулятора серотониновых (5-НТ2А), дофаминовых (D1) и мю-опиоидных рецепторов. В соединении формулы I X представляет собой -NH-; L представляет собой O; Z представляет собой -CH(O-R1)-, -O- или -C(=O)-; R1 представляет собой H. Изобретение также относится к фармацевтической композиции для лечения расстройства центральной нервной системы, в которое вовлечены пути серотонина 5-НТ2А, транспортера обратного захвата серотонина (SERT), дофамина D1 или мю-опиоидного рецептора, содержащей терапевтически эффективное количество соединения формулы I. Описывается также cпособ лечения или профилактики расстройств центральной нервной системы, в которые вовлечены пути серотонина 5-НТ2А, транспортера обратного захвата серотонина (SERT), дофамина D1 или мю-опиоидный рецептор. Предложенный способ включает введение пациенту, нуждающемуся в этом, соединения или фармацевтической композиции изобретения. Соединения и фармацевтические композиции изобретения могут найти применение для изготовления лекарственного средства для лечения или профилактики указанных выше расстройств центральной нервной системы. 4 н. и 16 з.п. ф-лы, 4 табл., 8 пр.
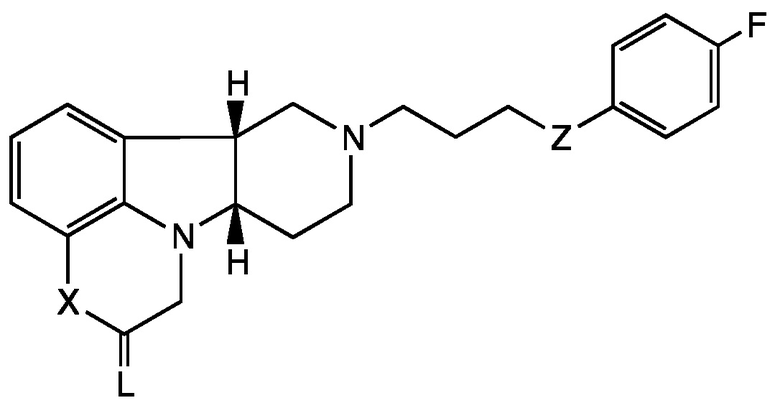
формула I
1. Соединение формулы I
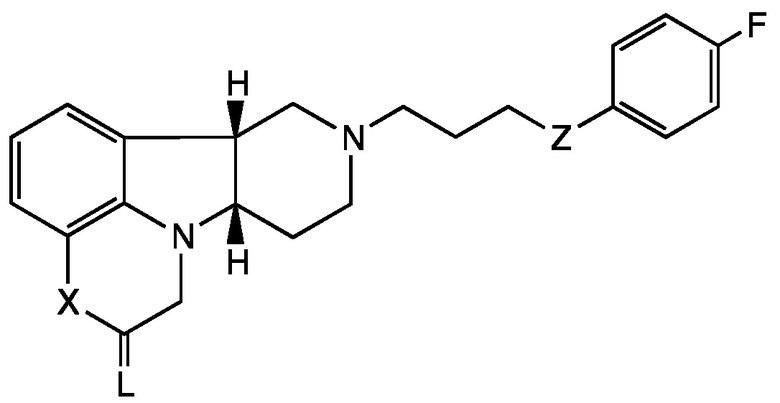 ,
,
формула I,
в которой
X представляет собой -NH-;
L представляет собой O;
Z представляет собой -CH(O-R1)-, -O- или -C(=O)-;
R1 представляет собой H;
в свободной или фармацевтически приемлемой солевой форме.
2. Соединение по п.1, в котором Z представляет собой -CH(OН)-.
3. Соединение по п.1, в котором Z представляет собой -C(=O)-.
4. Соединение по п.1, в котором Z представляет собой -O-.
5. Соединение по любому из пп.1-4 в форме фармацевтически приемлемой соли.
6. Соединение по п.5, где соединение находится в кислотно-аддитивной солевой форме, причем кислота выбрана из глутаминовой кислоты, винной кислоты, яблочной кислоты или аскорбиновой кислоты.
7. Соединение по любому из пп.1-4 в свободной форме.
8. Фармацевтическая композиция для лечения расстройства центральной нервной системы, в которое вовлечены пути серотонина 5-НТ2А, транспортера обратного захвата серотонина (SERT), дофамина D1 и/или μ-опиоидный рецептор, содержащая терапевтически эффективное количество соединения по любому одному из пп.1-7 в свободной или фармацевтически приемлемой солевой форме в смеси с фармацевтически приемлемым разбавителем или носителем.
9. Фармацевтическая композиция по п.8, в которой фармацевтически приемлемый разбавитель или носитель содержит полимерную матрицу.
10. Фармацевтическая композиция по п.9, в которой полимерная матрица представляет собой биоразлагаемую поли(d,l-лактид-со-гликолид) микросферу.
11. Способ лечения или профилактики расстройства центральной нервной системы, где указанное расстройство представляет собой расстройство, в которое вовлечены пути серотонина 5-НТ2А, транспортера обратного захвата серотонина (SERT), дофамина D1 и/или μ-опиоидный рецептор, включающий введение пациенту, нуждающемуся в таком лечении, соединения по любому одному из пп.1-7 в свободной или фармацевтически приемлемой солевой форме или фармацевтической композиции по любому одному из пп.8-10.
12. Способ по п.11, в котором указанное расстройство выбрано из группы, состоящей из расстройства, выбранного из группы, состоящей из ожирения, тревожности, депрессии (например, рефрактерной депрессии, БДР или биполярной депрессии), психоза (включая психоз, связанный с деменцией, такой как галлюцинации при прогрессирующей болезни Паркинсона или параноидальные идеи), шизофрении, нарушений сна (в частности нарушений сна, связанных с шизофренией и другими психиатрическими и неврологическими заболеваниями), сексуальных расстройств, мигрени, состояний, связанных с головной болью, социальных фобий, возбуждения при деменции (например, возбуждения при болезни Альцгеймера), возбуждения при аутизме и родственных аутических расстройствах, желудочно-кишечных расстройств, таких как дисфункция моторики желудочно-кишечного тракта, и деменции, например, деменции болезни Альцгеймера или болезни Паркинсона; расстройств настроения; и наркотических зависимостей, например, опиатной зависимости и/или алкогольной зависимости или отмены наркотической или алкогольной зависимости (например, опиатной зависимости); или компульсивного переедания.
13. Способ по п.12, в котором указанное расстройство выбрано из тревожности, депрессии и шизофрении.
14. Способ по п.12, в котором указанное расстройство представляет собой наркотическую зависимость или отмену наркотической зависимости.
15. Способ по п.14, в котором указанная наркотическая зависимость представляет собой опиатную зависимость.
16. Способ по п.14, в котором указанная наркотическая зависимость представляет собой алкогольную зависимость.
17. Способ по п.11, в котором указанное расстройство представляет собой расстройство, выбранное из следующего: (1) психоз, например, шизофрения у пациента, страдающего депрессией; (2) депрессия у пациента, страдающего психозом, например, шизофренией; (3) расстройства настроения, связанные с психозом, например, шизофренией или болезнью Паркинсона; (4) нарушения сна, связанные с психозом, например, шизофренией или болезнью Паркинсона; и (5) наркотическая зависимость, нарушения, связанные с употреблением веществ, и/или нарушения, вызванные употреблением веществ.
18. Способ по любому из пп.11-17, в котором пациент страдает расстройством, связанным с употреблением веществ вместе со вторым расстройством центральной нервной системы, где указанное расстройство представляет собой расстройство, в которое вовлечены пути серотонина 5-НТ2А, транспортера обратного захвата серотонина (SERT), дофамина D1 и/или μ-опиоидный рецептор.
19. Соединение по любому из пп.1-7 в свободной или фармацевтически приемлемой солевой форме для применения в качестве фармацевтического средства в лечении расстройства центральной нервной системы, в которое вовлечены пути серотонина 5-НТ2А, транспортера обратного захвата серотонина (SERT), дофамина D1 и/или μ-опиоидный рецептор.
20. Применение соединения по любому из пп.1-7 в свободной или фармацевтически приемлемой солевой форме или фармацевтической композиции по любому одному из пп.8-10 в свободной или фармацевтически приемлемой солевой форме при изготовлении лекарственного средства для лечения или профилактики расстройства центральной нервной системы, где указанное расстройство представляет собой расстройство, в которое вовлечены пути серотонина 5-НТ2А, транспортера обратного захвата серотонина (SERT), дофамина D1 и/или μ-опиоидный рецептор.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| US 7183282 B2, 27.02.2007 | |||
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2557059C2 |

