По данной заявке запрашивается приоритет в соответствии с предварительной заявкой США 61/327049, поданной 22 апреля 2010 г., и предварительной заявкой США 61/367609, поданной 26 июля 2010 г., содержание каждой из которых включено посредством ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Изобретение относится к конкретным замещенным конденсированным с гетероциклом гамма-карболинам, их пролекарствам, в свободной форме, в виде твердого вещества, в виде фармацевтически приемлемой соли и/или в по существу чистой форме, как описано здесь, их фармацевтическим композициям и способам применения при лечении заболеваний, в которые вовлечены рецептор 5-HT2A, переносчик серотонина (SERT) и/или пути, в которые вовлечены сигнальные системы рецептора допамина D2, например, заболеваний или расстройств, таких как тревога, психоз, шизофрения, нарушения сна, сексуальные расстройства, мигрень, состояния, связанные с головной болью, социальные фобии, желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта и ожирение; депрессия и расстройства настроения, связанные с психозом или болезнью Паркинсона; психоз, такой как шизофрения, связанная с депрессией; биполярное расстройство; и другие психиатрические и неврологические состояния, а также к комбинациям с другими средствами.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
[0002] Известно, что замещенные конденсированные с гетероциклом гамма-карболины являются агонистами или антагонистами рецепторов 5-HT2, особенно, рецепторов 5-HT2A и 5-HT2c, при лечении расстройств центральной нервной системы. Эти соединения были раскрыты в патентах США №№ 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680 и U.S. RE39679, в качестве новых соединений, применимых для лечения расстройств, связанных с модуляцией рецептора 5-HT2A, таких как ожирение, тревога, депрессия, психоз, шизофрения, нарушения сна, сексуальные расстройства, мигрень, состояния, связанные с головной болью, социальные фобии, желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта, и ожирение. PCT/US08/03340 (WO 2008/112280) и патентная заявка США серийный № 10/786935 также раскрывают способы получения замещенных конденсированных с гетероциклом гамма-карболинов и применения этих гамма-карболинов в качестве агонистов и антагонистов серотонина, применимых для контроля и предотвращения расстройств центральной нервной системы, таких как зависимое поведение и нарушения сна.
[0003] Кроме того, WO/2009/145900 раскрывает применение конкретных замещенных конденсированных с гетероциклом гамма-карболинов для лечения комбинации психоза и депрессивных расстройств, а также нарушений сна, депрессивных расстройств и/или расстройств настроения у пациентов с психозом или болезнью Паркинсона. В дополнение к расстройствам, связанным с психозом и/или депрессией, данная патентная заявка раскрывает и заявляет права на применение этих соединений при низкой дозе для оказания селективного антагонистического эффекта на рецепторы 5-HT2A без оказания воздействия или с минимальным воздействием на рецепторы допамина D2, что, таким образом, делает их применимыми для лечения нарушений сна без побочных эффектов путей допамина D2 или побочных эффектов других путей (например, рецепторов ГАМК), связанных с общепринятыми седативными-снотворными средствами (например, бензодиазепинами), включающими, но не ограниченными лишь ими, развитие зависимости от лекарственного средства, мышечную гипотонию, слабость, головную боль, нечеткое зрения, головокружение, тошноту, рвоту, желудочный дискомфорт, диарею, боли в суствах и боли в груди. WO 2009/114181 также раскрывает способы получения кристаллов соли присоединения толуолсульфоновой кислоты этих замещенных конденсированных с гетероциклом гамма-карболинов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
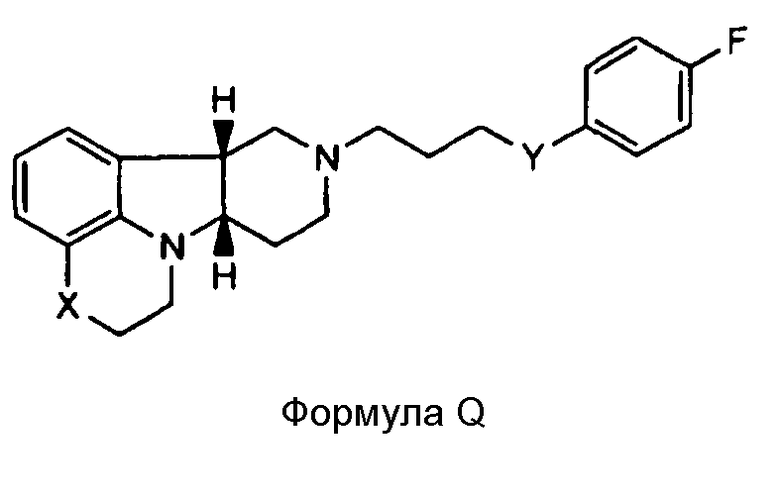
[0004] В то время как замещенные конденсированные с гетероциклом гамма-карболины и их применения являются известными, авторы изобретения неожиданно обнаружили, что конкретные замещенные конденсированные с гетероциклом гамма-карболины ("соединения формулы I", как описано здесь далее), являясь менее активными в анализах ex vivo, чем первое лекарственное средство ("соединения формулы Q", как описано здесь далее), взаимно превращаются в первое лекарственное средство и из первого лекарственного средства в плазме и мозге. Вследствие того, что соединения формулы I превращаются in vivo в соединения формулы Q и из соединений формулы Q, их можно рассматривать в качестве пролекарств для соединений формулы Q, а также метаболитов соединений формулы Q, и они могут служить в качестве депо для соединений формулы Q, расширяя его продолжительность действия. Продолжительность действия и метаболизм этих соединений формулы I могут быть модифицированы дополнительно посредством присоединения физиологически гидролизуемых и приемлемых фрагментов и/или применения готовых форм с продолжительным высвобождением. Авторы изобретения, таким образом, дополнительно предоставили пролекарства из конкретных замещенных конденсированных с гетероциклом гамма-карболинов, которые имеют измененный фармакокинетический профиль, например, измененные механизмы и/или скорость всасывания и распределения, и, следовательно, могут быть применимыми для улучшенного составления и/или для регулирования продолжительности эффекта лекарственного средства в организме (например, для замедленного или контролируемого высвобождения). Изобретение, следовательно, предоставляет соединения и их пролекарства, их фармацевтическую композицию для применения, как изложено здесь.
[0005] Дополнительно обнаружено, что соединения формулы I, кроме того, имеют интересную активность связывания с рецептором нейромедиатора, отличную от соединений формулы Q. В частности, показано, что соединения формулы I, как описано здесь далее, где Y представляет собой -CH(OH)-, имеют высокую селективность по отношению к переносчику серотонина (SERT) относительно соединений формулы Q, и могут, таким образом, усиливать эффект соединений формулы Q на SERT. Этот уникальный профиль предлагает конкретную применимость при лечении SERT-опосредованных заболеваний, таких как депрессия, тревога и психоз с депрессией или тревогой.
[0006] Не имея намерений быть связанными с теорией, полагают, что, в то время как соединения формулы I, где Y представляет собой -C(H)(OH)-, превращаются в соединения формулы Q in vivo, введение соединений формулы I могло бы иметь некоторые преимущества по сравнению с введением соединений формулы Q непосредственно, в том, что соединения формулы I будут предоставлять более длительную продолжительность действия вследствие их метаболической стабильности и непрерывного превращения в соединения формулы Q, и, кроме того, будут усиливать SERT-ингибирующую активность по сравнению с активностью к другим рецепторам, вследствие их относительно высокой активности к SERT рецептору.
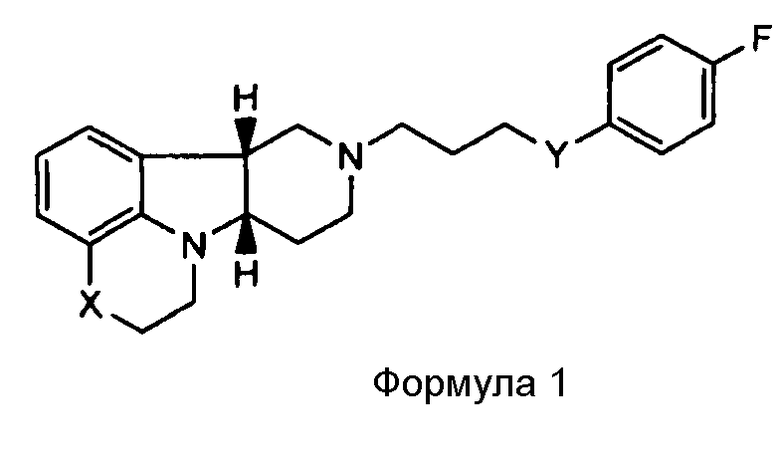
[0007] Изобретение относится к соединению формулы I:
 ,
,
где
X представляет собой -N(H)- или -N(CH3) и Y представляет собой -C(H)(OH)-;
X представляет собой -N(H), когда Y представляет собой -О-; или
X представляет собой -O- и Y представляет собой -C(H)(OH)-;
в свободной или солевой форме.
[0008] В первом аспекте изобретение предоставляет соединение формулы I, в свободной или солевой форме, как описано в следующих формулах:
1.1 соединение формулы I, при условии, что соединение не вырабатывается у млекопитающего посредством метаболизма соединения формулы Q:
 ,
,
где
X представляет собой -N(H)- или -N(CH3)- и/или Y представляет собой -C(=О);
X представляет собой -N(CH3)- и Y представляет собой -О-; или
X представляет собой -O- и Y представляет собой -C(=О)-;
1.2 соединение Формулы I или 1.1, где указанное соединение находится в твердой форме;
1.3 соединение Формулы I, 1.1 или 1.2, где указанное соединение находится в солевой форме;
1.4 соединение формулы I или любой из формул 1.1-1.3, где указанное соединение находится в фармацевтически приемлемой солевой форме;
1.5 формулы 1.4, где фармацевтически приемлемую соль выбирают из группы, состоящей из хлористоводородной, бромистоводородной, серной, сульфаминовой, фосфорной, азотной, уксусной, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памоевой, малеиновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изетионовой и т.п.;
1.6 формулы 1.5, где соль представляет собой соль присоединения фумаровой кислоты;
1.7 формулы 1.5, где соль представляет собой соль присоединения фосфорной кислоты;
1.8 формулы 1.5, где соль представляет собой соль присоединения толуолсульфоновой кислоты;
1.9 соединение Формулы I или любой из формул 1.1-1.8, где соединение представляет собой
 ;
;
1.10 соединение Формулы I или любой из 1.1-1.9, где соединение представляет собой
 ;
;
1.11 соединение формулы I или любой из 1.1-1.9, где соединение представляет собой
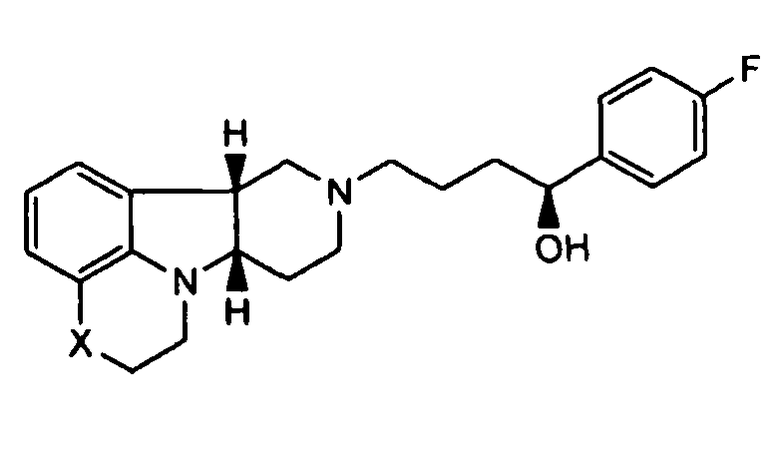 ;
;
1.12 соединение формулы I или любой из 1.1-1.11, где X представляет собой -N(CH3);
1.13 соединение формулы I или любой из 1.1-1.11, где X представляет собой -N(H)-;
1.14 соединение формулы I или любой из 1.1-1.12, где соединение представляет собой
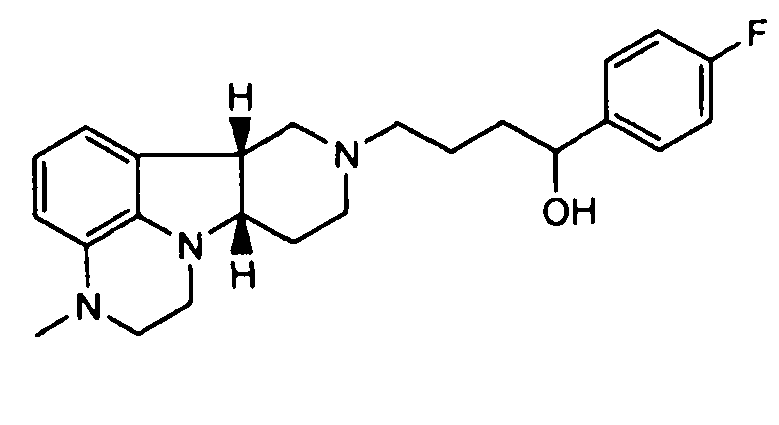 ;
;
1.15 соединение формулы I или любой из 1.1-1.11, где X представляет собой -О-;
1.16 соединение формулы I или любой из 1.9-1.15, где соединение находится в по существу чистой диастереомерной форме (т.е., не содержит по существу других диастереомеров);
1.17 соединение формулы I или любой из 1.9-1.16, где соединение имеет диастереомерный избыток, больший, чем 70%, предпочтительно больший, чем 80%, более предпочтительно больший, чем 90% и наиболее предпочтительно больший, чем 95%;
1.18 соединение формулы I или любой из формул 1.1-1.8, где соединение представляет собой
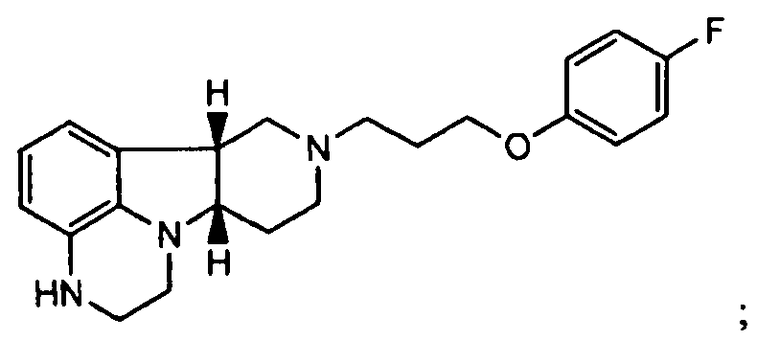
1.19 соединение формулы I или любой из формул 1.1-1.18, где указанное соединение по существу не содержит соединения формулы Q, как определено здесь прежде;
1.20 формулы 1.19, где соединение формулы I представляет собой
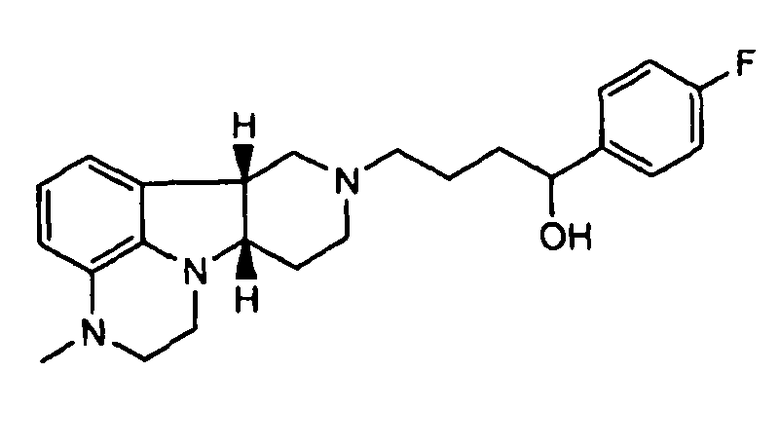 ,
,
не содержащее по существу соединения формулы Q, где Y представляет собой -C(=О) и/или X представляет собой -N(CH3)-;
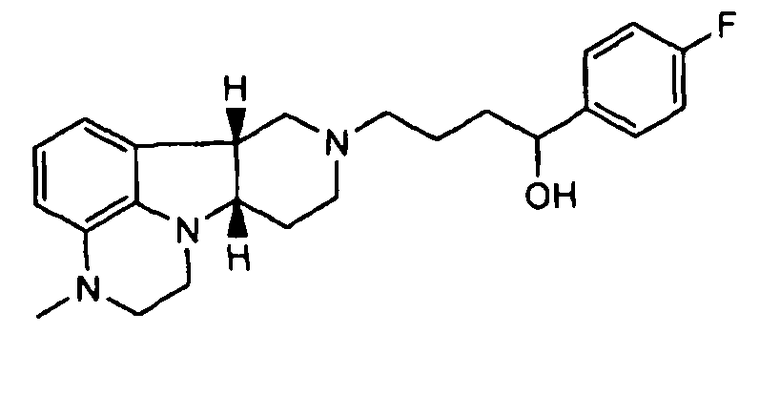
1.21, формулы 1.19, где соединение формулы I представляет собой
 ,
,
не содержащее по существу соединения формулы Q, где Y представляет собой -C(=О) и/или X представляет собой -N(CH3)- или -N(H)-;
1.22 любой из формул 1.19-1.21, где соединение формулы I больше, чем на 70%, предпочтительно больше, чем 80%, более предпочтительно больше, чем 90%, еще более предпочтительно больше, чем 95%, еще более предпочтительно больше, чем 98%, еще более предпочтительно больше, чем 99% является не содержащим соединения Формулы Q, как описано в любой из Формул 1.1-1.21,
в свободной или солевой форме.
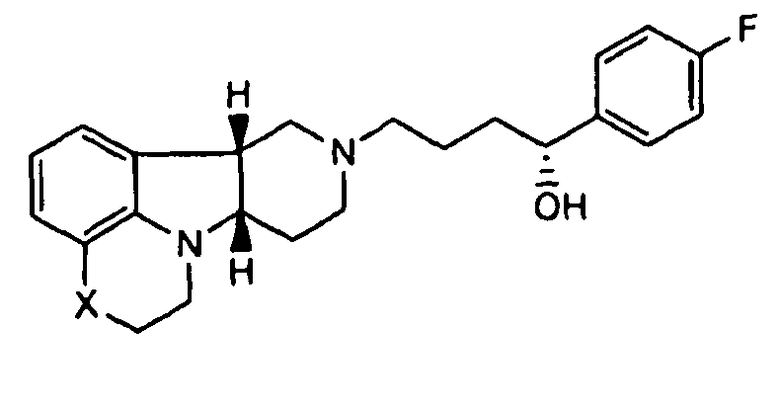
[0009] Во втором аспекте изобретение предоставляет соединение формулы II-A:
 ,
,
где X представляет собой -N(CH3)-, -N(H)- или -О-, в свободной или солевой форме. В дополнительном варианте осуществления второго аспекта, изобретение предоставляет соединение формулы II-A, где X представляет собой -N(CH3)-. В еще одном другом дополнительном варианте осуществления второго аспекта, изобретение предоставляет соединение формулы II-A, где X представляет собой -N(H)-.
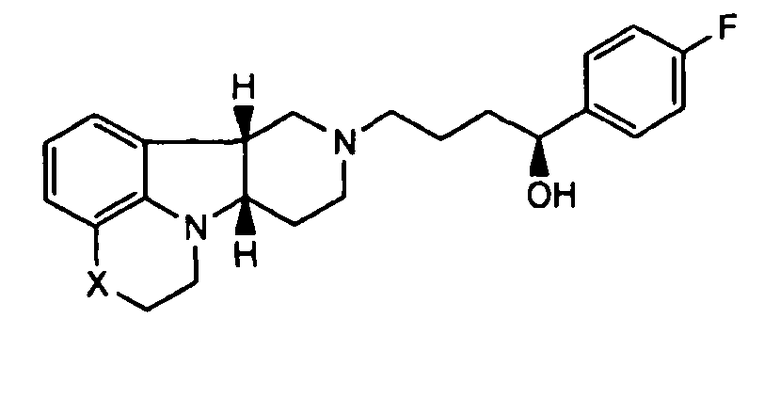
[0010] В третьем аспекте изобретение предоставляет соединение формулы II-B:
 ,
,
где X представляет собой -(CH3)-, -N(H)- или -О-, в свободной или солевой форме. В дополнительном варианте осуществления третьего аспекта, изобретение предоставляет соединение формулы II-B, где X представляет собой -N(CH3)-. В еще одном другом дополнительном варианте осуществления третьего аспекта, изобретение предоставляет соединение формулы II-B, где X представляет собой -N(H)-.
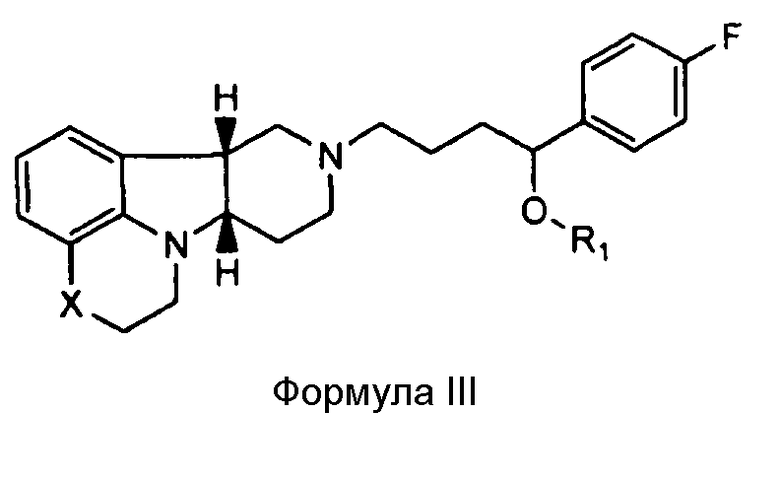
[0011] В четвертом аспекте изобретение предоставляет соединение формулы III:
 ,
,
где
X представляет собой -N(CH3)-, -N(H)- или -О-; и
R1 представляет собой -C(О)-C1-21алкил (например, -C(О)-C1-5алкил, -C(О)-C6-15алкил или -C(О)-C16-21алкил), предпочтительно, указанный алкил является прямоцепочечным, необязательно насыщенным или ненасыщенным и необязательно замещенным одной или более гидрокси или C1-22алкокси (например, этокси) группами, например, R1 представляет собой -C(О)-C6алкил, -C(О)-C7алкил, -C(О)-C9алкил, -C(О)-C11алкил, -C(О)-C13алкил или -C(О)-C15алкил, и такое соединение гидролизуется с образованием остатка природной или неприродной, насыщенной или ненасыщенной жирной кислоты, например, соединение гидролизуется с образованием гидроксисоединения, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны,
в свободной или солевой форме.
[0012] В дополнительном варианте осуществления четвертого аспекта, изобретение предоставляет соединение формулы III, в свободной или солевой форме, как описано в следующих формулах:
4.1. соединение формулы III, где соль выбирают из группы, состоящей из хлористоводородной, бромистоводородной, серной, сульфаминовой, фосфорной, азотной, уксусной, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памовой, малеиновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изетионовой и т.п.;
4.2 соединение Формулы III или формулы 4.1, где соль представляет собой соль присоединения фумаровой кислоты;
4.3 соединение формулы III или формулы 4.1, где соль представляет собой соль присоединения фосфорной кислоты;
4.4 соединение формулы III или формулы 4.1, где соль представляет собой соль присоединения толуолсульфоновой кислоты;
4.5 соединение формулы III или любой из формул 4.1-4.4, где соединение представляет собой:
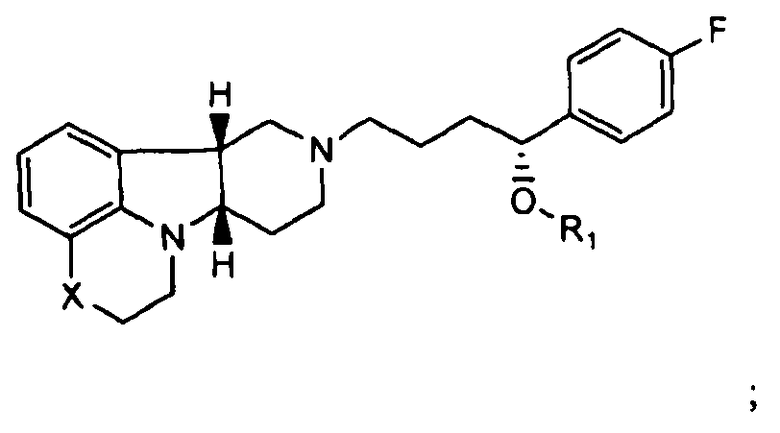
4.6 соединение формулы III или любой из Формул 4.1-4.4, где соединение представляет собой:
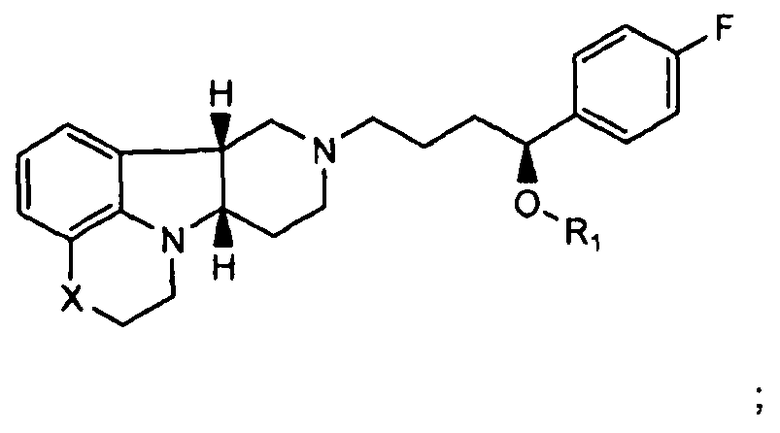
4.7 соединение формулы III или любой из формул 4.1-4.6, где соединение находится в по существу чистой диастереомерной форме (т.е., не содержит по существу других диастереомеров);
4.8 соединение формулы III или любой из 4.1-4.7, где соединение имеет диастереомерный избыток, больший, чем 70%, предпочтительно больший, чем 80%, более предпочтительно больший, чем 90% и наиболее предпочтительно больший, чем 95%;
4.9 соединение формулы III или любой из формул 4.1-4.8, где X представляет собой -N(CH3);
4.10 соединение формулы III или любой из формул 4.1-4.8, где X представляет собой -N(H)- ;
4.11 соединение формулы III или любой из формул 4.1-4.10, где R1 представляет собой -C(О)-C1-21алкил (например, -C(О)-C1-5алкил, -C(О)-C6-15алкил или -C(О)-C16-21алкил), предпочтительно указанный алкил является прямоцепочечным, необязательно насыщенным или ненасыщенным и необязательно замещенным одной или более гидрокси или C1-22алкокси (например, этокси) группами, например, R1 представляет собой -C(O)-C6алкил, -C(О)-C7алкил, -C(О)-C9алкил, -C(О)-C11алкил, -C(О)-C13алкил или -C(О)-C15алкил, и такое соединение гидролизуется с образованием остатка природной или неприродной, насыщенной или ненасыщенной жирной кислоты, например, соединение гидролизуется с образованием гидроксисоединения, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны;
4.12 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C1-21алкил (например, -C(О)-C1-5алкил, -C(О)-C6-15алкил или -C(О)-C16-21алкил), предпочтительно указанный алкил является прямоцепочечным, необязательно насыщенным или ненасыщенным;
4.13 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C1-21алкил (например, -C(О)-C1-5алкил, -C(О)-C6-15алкил или -C(О)-C16-21алкил);
4.14 соединение формулы III или любой из формул 4.1-4.11, где R1 выбирают из -C(О)-C6алкила, -C(О)-C7алкила, -C(О)-C9алкила, -C(О)-C11алкила, -C(О)-C13алкила и -C(О)-C15алкила;
4.15 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C6алкил;
4.16 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C7алкил;
4.17 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C9алкил;
4.18 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C11алкил;
4.19 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C13алкил;
4.20 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C15алкил;
4.21 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C17алкил;
4.22 соединение формулы III или любой из формул 4.1-4.11, где R1 представляет собой -C(О)-C2lалкил;
4.23 соединение формулы III или любой из формул 4.1-4.8 или 4.11-4.14, где X представляет собой -О-;
4.24 соединение формулы III или любой из формул 4.1-4.23, где указанное соединение не содержит по существу соединения формулы Q, как в любой из формул 1.1-1.21;
4.25 формулы 4.24, где соединение формулы III представляет собой
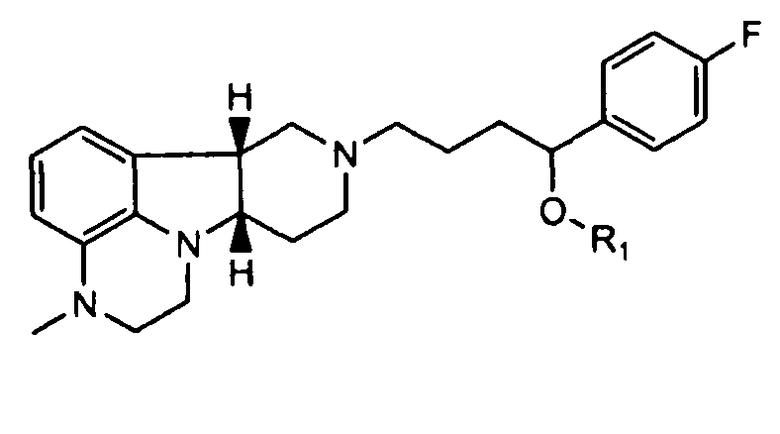 ,
,
не содержащее по существу соединения формулы Q, где Y представляет собой -C(=О) и/или X представляет собой -N(CH3)-;
4.26 формулы 4.24, где соединение формулы III представляет собой
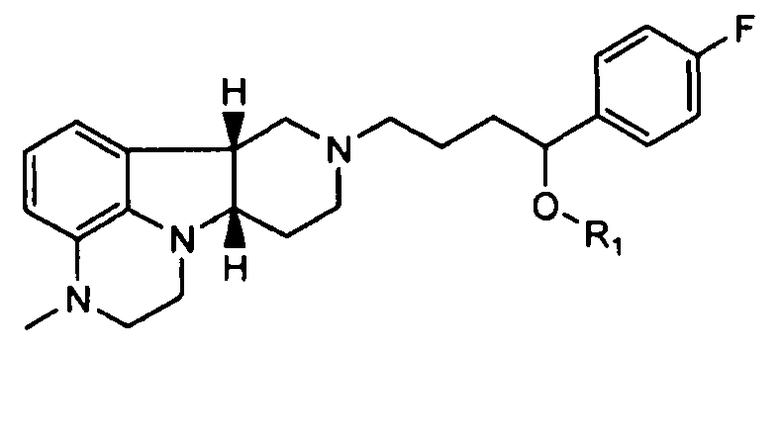 ,
,
не содержащее по существу соединения формулы Q, где Y представляет собой -C(=О) и/или X представляет собой -N(CH3)- или -N(H)-;
4.27 любой из формул 4.24-4.26, где соединение формулы III больше, чем на 70%, предпочтительно больше, чем 80%, более предпочтительно больше, чем 90%, еще более предпочтительно больше, чем 95%, еще более предпочтительно больше, чем 98%, еще более предпочтительно больше, чем 99% является не содержащим соединения формулы Q, как описано в любой из формул 1.1-1.21;
в свободной или солевой форме.
[0013] В еще одном другом дополнительном варианте осуществления четвертого аспекта, изобретение предоставляет соединение формулы III, в свободной или солевой форме, как описано в любой одной из следующих формул:
4.28. соединение формулы III или любой из 4.1-4.13 или 4.23-4.27, где R1 представляет собой -C(О)-C3алкил;
4.29. соединение формулы III или любой из 4.1-4.13 или 4.23-4.27, где R1 представляет собой -C(О)-C9алкил;
4.30. соединение формулы III, где указанное соединение выбирают из любого одного из следующих:
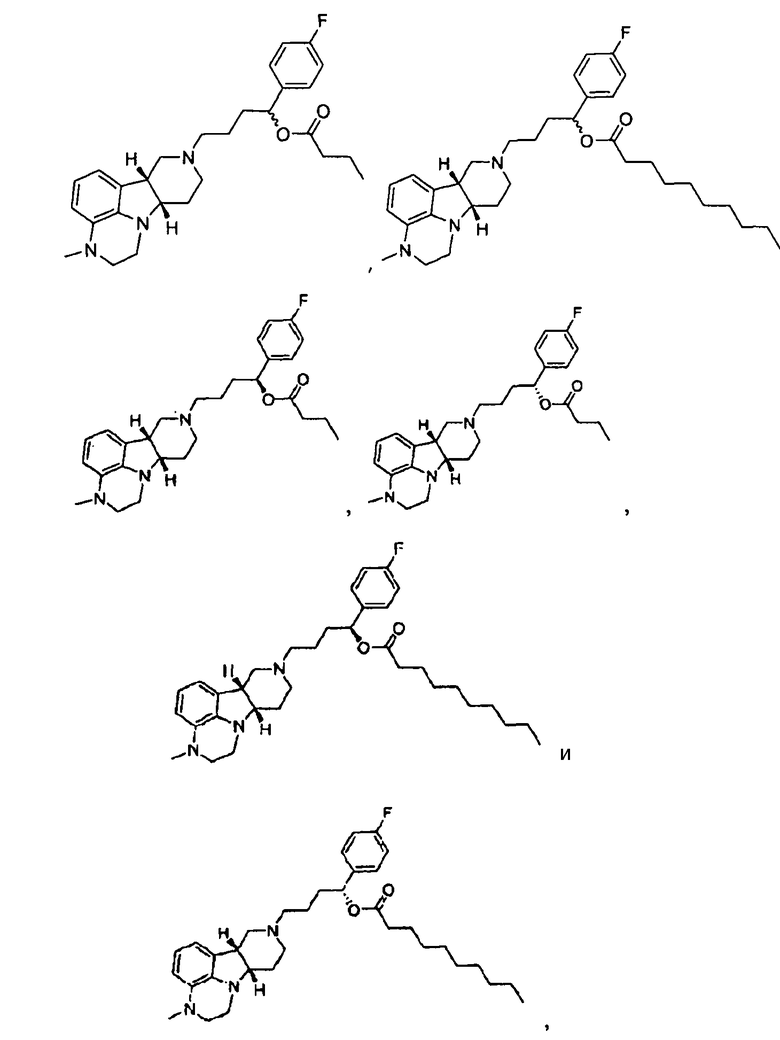
в свободной или солевой форме.
[0014] В пятом аспекте изобретение предоставляет фармацевтическую композицию, как описано в следующих формулах:
5.1 фармацевтическая композиция, содержащая соединение формулы I или любой из формул 1.1-1.22, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем (фармацевтическая композиция 5.1);
5.2 фармацевтическая композиция, содержащая соединение формулы II-A, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем (фармацевтическая композиция 5.2);
5.3 фармацевтическая композиция, содержащая соединение формулы II-B, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем (фармацевтическая композиция 5.3); или
5.4 фармацевтическая композиция, содержащая соединение формулы III или любой из формул 4.1-4.27, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем (фармацевтическая композиция 5.4);
5.4P фармацевтическая композиция, содержащая соединение формулы III или любой из формул 4.28-4.30, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем (фармацевтическая композиция 5.4P).
В предпочтительном варианте осуществления фармацевтическая композиция по изобретению содержит соединение формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)-, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем. В еще одном варианте осуществления фармацевтическая композиция по изобретению содержит соединение формулы 1.14, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем. В еще одном другом варианте осуществления фармацевтическая композиция по изобретению содержит соединение формулы 4.26, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем. В еще одном другом варианте осуществления фармацевтическая композиция по изобретению содержит соединение формулы 4.20, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем.
[0015] В дополнительном варианте осуществления пятого аспекта, фармацевтическая композиция по изобретению предназначена для замедленного или отсроченного высвобождения, является, например, готовой лекарственной формы пролонгированного действия. В одном варианте осуществления готовая лекарственная форма пролонгированного действия представляет собой фармацевтическую композицию 5.4 (композицию пролонгированного действия 5.5). В дополнительном варианте осуществления композиция пролонгированного действия 5.5 содержит соединение формулы III, где R1 представляет собой -C(О)-C6-15алкил, в свободной или фармацевтически приемлемой солевой форме (композиция пролонгированного действия 5.6). Например, готовая лекарственная форма пролонгированного действия представляет собой фармацевтическую композицию, содержащую:
5.7 1-(4-фторфенил)-4-((6bR, 10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-энантрат (т.е., соединение формулы III, где X представляет собой -N(CH3)- и R1 представляет собой -C(O)-C6лкил) (композиция пролонгированного действия 5.7);
5.8 1-(4-фторфенил)-4-((6bR, 10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-деканоат, (т.е., соединение формулы III, где X представляет собой -N(CH3)- и R1 представляет собой -C(O)-C19алкил) (композиция пролонгированного действия 5.8); или
5.9 1-(4-фторфенил)-4-((6bR, 10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-пальмитат (т.е., соединение формулы III, где X представляет собой -N(CH3)- и R1 представляет собой -C(O)-C15алкил) (композиция пролонгированного действия 5.9),
в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем.
[0016] В еще одном варианте осуществления пятого аспекта, готовая лекарственная форма пролонгированного действия представляет собой Фармацевтическую композицию 5.4P (композицию пролонгированного действия 5.10). Например, готовая лекарственная форма пролонгированного действия представляет собой фармацевтическую композицию, содержащую:
5.11 1-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-бутаноат (т.е., соединение формулы III, где X представляет собой -N(CH3)- и R1 представляет собой -C(O)-C3алкил) (композиция пролонгированного действия 5.11),
в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем.
[0017] В шестом аспекте изобретение предоставляет композицию, содержащую соединение по изобретению, как описано в следующих формулах:
6.1. формула I или любая из формул 1.1-1.22, в свободной или (фармацевтически приемлемой) солевой форме (композиция 6.1);
6.2. формула II-A, в свободной или (фармацевтически приемлемой) солевой форме (композиция 6.2);
6.3. формула II-B, в свободной или (фармацевтически приемлемой) солевой форме (композиция 6.3); или
6.4. формула III или любая из формул 4.1-4.27, как описано здесь прежде, в свободной или (фармацевтически приемлемой) солевой форме (композиция 6.4);
6.5. формула III или любая из формул 4.28-4.30, как описано здесь прежде, в свободной или (фармацевтически приемлемой) солевой форме (композиция 6.5),
в полимерной матрице. В одном варианте осуществления соединение по изобретению диспергировано или растворено внутри полимерной матрицы. В дополнительном варианте осуществления полимерная матрица содержит стандартные полимеры, применяемые в готовых лекарственных формах пролонгированного действия, такие как полимеры, выбранные из сложного полиэфира гидроксижирных кислот и их производных, или полимер из алкил-альфа-цианоакрилата, полиалкиленоксалата, сложного полиортоэфира, поликарбоната, полиортокарбоната, полиаминокислоты, сложного эфира гиалуроновой кислоты и их смесей. В дополнительном варианте осуществления полимер выбирают из группы, состоящей из полилактида, поли-d,l-лактида, полигликолида, полимера PLGA 50:50, PLGA 85:15 и PLGA 90:10. В еще одном варианте осуществления полимер выбирают из поли(гликолевой кислоты), поли-D,L-молочной кислоты, поли-L-молочной кислоты, сополимеров вышеуказанных, поли(алифатических карбоновых кислот), сополиоксалатов, поликапролактона, полидиоксонона, поли(ортокарбонатов), поли(ацеталей), поли(молочной кислоты-капролактона), сложных полиортоэфиров, поли(гликолевой кислоты-капролактона), полиангидридов, и природных полимеров, включающих альбумин, казеин, и воска, такие как моно- и дистеарат глицерина и т.п. В предпочтительном варианте осуществления полимерная матрица содержит поли(d,l-лактид-co-гликолид). Например, композиция любой из формул 6.1-6.4, где соединение по изобретению представляет собой соединение формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)-, в свободной или солевой форме. В еще одном примере композиция формулы 6.5, где соединение по изобретению представляет собой соединение формулы III, где X представляет собой -N(CH3)- и R1 представляет собой-C(О)-C3алкил или -C(О)-C9алкил, в свободной или солевой форме. В еще одном варианте осуществления соединение по изобретению представляет собой соединение формулы 1.14, 4.26 или 4.20, в свободной или солевой форме, и полимерная матрица содержит поли(d,l-лактид-со-гликолид). Любая из композиций формул 6.1-6.4, как описано здесь прежде, может являться фармацевтической композицией, где указанная композиция находится в смеси с фармацевтически приемлемым разбавителем или носителем (фармацевтическая композиция 6.1-6.4). Аналогично, любая из композиций формулы 6.5, как описано здесь прежде, может являться фармацевтической композицией, где указанная композиция находится в смеси с фармацевтически приемлемым разбавителем или носителем.
(Фармацевтическая композиция 6.5).
[0018] (Фармацевтические) композиции любой из Формул 6.1-6.4 являются особенно применимыми для замедленного или отсроченного высвобождения, где соединение по изобретению высвобождается при разрушении полимерной матрицы. Эти композиции могут быть составлены для контролируемого- и/или замедленного высвобождения соединений по изобретению (например, в виде композиции пролонгированного действия) в течение периода, равного до 180 дней, например, от приблизительно 14 до приблизительно 30, до приблизительно 180 дней. Например, полимерная матрица может разрушаться и высвобождать соединения по изобретению в течение периода, равного приблизительно 30, приблизительно 60 или приблизительно 90 дней. В еще одном примере полимерная матрица может разрушаться и высвобождать соединения по изобретению в течение периода, равного приблизительно 120 или приблизительно 180 дней.
[0019] В еще одном другом дополнительном варианте осуществления, фармацевтические композиции по изобретению, конкретно, композицию пролонгированного действия по изобретению (например, композицию пролонгированного действия любой из формул 5.5-5.9 или (фармацевтическую) композицию любой из формул 6.1-6.4 или 6.5) составляют для введения посредством инъекции.
[0020] В седьмом аспекте изобретение предоставляет соединение по изобретению, как описано здесь прежде, например,
соединение формулы I или любой из формул 1.1-1.22, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме;
соединение формулы II-A, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме;
соединение формулы II-B, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме; или
соединение формулы III или любой из формул 4.1-4.30, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме;
в системе для пероральной доставки с осмотически контролируемом высвобождением (OROS), которая описана в WO 2000/35419 и EP 1539115 (публикация патентной заявки США № 2009/0202631), содержание каждой из этих заявок включено посредством ссылки во всей их полноте. Следовательно, в одном варианте осуществления седьмого аспекта, изобретение предоставляет фармацевтическую композицию или устройство, содержащее (a) желатиновую капсулу, содержащую соединение изобретения в свободной или фармацевтически приемлемой солевой форме или фармацевтическую композицию по изобретению, как описано здесь прежде; (b) многослойную стенку, наложенную на желатиновую капсулу, содержащую, в порядке снаружи от капсулы: (i) барьерный слой, (ii) расширяемый слой и (iii) полупроницаемый слой; и (c) и отверстие, образованное или образуемое через стенку. (Композиция P.1)
[0021] В еще одном варианте осуществления седьмого аспекта, изобретение предоставляет композицию, содержащую желатиновую капсулу, содержащую жидкость, соединение по изобретению в свободной или фармацевтически приемлемой солевой форме или фармацевтическую композицию по изобретению, например, любую из фармацевтических композиций 6.1-6.5; желатиновая капсула является окруженной композитной стенкой, содержащей барьерный слой, контактирующий с наружной поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или образуемое в стенке. (Композиция P.2)
[0022] В еще одном другом варианте осуществления седьмого аспекта, изобретение предоставляет композицию, содержащую желатиновую капсулу, содержащую жидкость, Соединение изобретения в свободной или фармацевтически приемлемой солевой форме или Фармацевтическую композицию изобретения, например, любую из Фармацевтических композиций 6.1-6.5, желатиновая капсула является окруженной композитной стенкой, содержащей барьерный слой, контактирующий с наружной поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или образуемое в стенке, где барьерный слой образует закрывающий слой между расширяемым слоем и окружением по выходному отверстию. (Композиция P.3)
[0023] В еще одном другом варианте осуществления седьмого аспекта, изобретение предоставляет композицию, содержащую желатиновую капсулу, содержащую жидкость, соединение по изобретению в свободной или фармацевтически приемлемой солевой форме или фармацевтическую композицию по изобретению, например, любую из фармацевтических композиций 6.1-6.5; желатиновая капсула является окруженной барьерным слоем, контактирующим с наружной поверхностью желатиновой капсулы, расширяемым слоем, контактирующим с частью барьерного слоя, полупроницаемым слоем, охватывающим, по меньшей мере, расширяемый слой, и выходным отверстием, образованным или образуемым в дозированной форме, простирающейся от наружной поверхности желатиновой капсулы до окружения применения. (Композиция P.4). Расширяемый слой может быть образован в одной или более дискретных секций, как, например, двух секциях, расположенных на противоположных сторонах или концах желатиновой капсулы.
[0024] В конкретном варианте осуществления седьмого аспекта, соединение изобретения в пероральной системе доставки с осмотически контролируемым высвобождением (т.е., в композициях P.1-P.4) находится в жидкой готовой форме, причем готовая форма может являться чистым, жидким активным средством, жидким активным средством в растворе, суспензии, эмульсии или самоэмульгирующейся композицией или т.п.
[0025] Дополнительную информацию по пероральной системе доставки с осмотически контролируемым высвобождением композиции, включающей характеристики желатиновой капсулы, барьерного слоя, расширяемого слоя, полупроницаемого слоя и отверстия, можно найти в WO 2000/35419, содержание которой включено посредством ссылки во всей полноте.
[0026] Описание другой пероральной системы доставки с осмотически контролируемым высвобождением для соединения или фармацевтической композиции изобретения можно найти в EP 1539115 (публикация патентной заявки США № 2009/0202631), содержание которой включено посредством ссылки во всей полноте. Следовательно, в еще одном варианте осуществления седьмого аспекта, изобретение предоставляет композицию или устройство, содержащие (a) два или более слоев, указанные два или более слоев, содержащие первый слой и второй слой, указанный первый слой содержит соединение по изобретению, в свободной или фармацевтически приемлемой солевой форме, или фармацевтическую композицию, как описано здесь прежде, указанный второй слой содержит полимер; (b) внешнюю стенку, окружающую указанные два или более слоев; и (c) отверстие в указанной внешней стенке. (Композиция P.5)
[0027] В композиции P.5 предпочтительно используют полупроницаемую мембрану, окружающую трехслойную сердцевину: в этих вариантах осуществления первый слой называется первым лекарственным слоем, и он содержит низкие количества лекарственного средства (например, соединение по изобретению) и осмотический агент, такой как соль; средний слой, именуемый вторым лекарственным слоем, содержит более высокие количества лекарственного средства, эксципиентов и не содержит соли; и третий слой, именуемый выдавливаемым слоем, содержит осмотические агенты и не содержит лекарственного средства. По меньшей мере, одно отверстие просверливают через мембрану на конце первого лекарственного слоя капсуловидной таблетки. (Композиция P.6)
[0028] Композиция P.5 или P.6 может содержать мембрану, определяющую отсек, мембрану, окружающую внутреннюю защитную оболочку, по меньшей мере, одно выходное отверстие, образованное или образуемое в ней и, по меньшей мере, часть мембраны является полупроницаемой; расширяемый слой, расположенный внутри отсека дистанционно от выходного отверстия и находящийся в текучем сообщении с полупроницаемой частью мембраны; первый лекарственный слой располагается вблизи от выходного отверстия; и второй лекарственный слой располагается внутри отсека между первым лекарственным слоем и расширяемым слоем, причем лекарственные слои содержат соединение по изобретению в свободной форме или форме его фармацевтически приемлемой соли. В зависимости от относительной вязкости первого лекарственного слоя и второго лекарственного слоя, получают различные профили высвобождения. Крайне важно идентифицировать оптимальную вязкость для каждого слоя. В настоящем изобретении вязкость модулируют посредством добавления соли, хлорида натрия. Профиль доставки из сердцевины является зависимым от массы, состава и толщины каждого из слоев лекарственного средства. (Композиция P.7)
[0029] В конкретном варианте осуществления изобретение предоставляет композицию P.7, где первый лекарственный слой содержит соль, а второй лекарственный слой не содержит соли. Композиции P.5-P.7 могут необязательно содержать инициирующий текучесть слой между мембраной и лекарственными слоями.
[0030] Композиции P.1-P.7 будут, как правило, именоваться как композиция пероральной системы доставки с осмотически контролируемым высвобождением.
[0031] В восьмом аспекте изобретение предоставляет способ (Способ I) лечения или профилактики расстройства центральной нервной системы, включающий введение пациенту, нуждающемуся в лечении:
7.1 соединения формулы I или любой из формул 1.1-1.22 в свободной или фармацевтически приемлемой солевой форме;
7.2 соединения формулы II-A в свободной или фармацевтически приемлемой солевой форме;
7.3 соединения формулы II-B в свободной или фармацевтически приемлемой солевой форме;
7.4 соединения формулы III или любой из формул 4.1-4.27, описано здесь выше, в свободной или фармацевтически приемлемой солевой форме;
7.5 фармацевтической композиции, как описано в формуле 5.1;
7.6 фармацевтической композиции, как описано в формуле 5.2;
7.7 Фармацевтической композиции, как описано в формуле 5.3;
7.8 фармацевтической композиции, как описано в формуле 5.4;
7.9 композиции пролонгированного действия любой из формул 5.5-5.9; или
7.10 (фармацевтической) композиции любой из Формул 6.1-6.4, как описано здесь прежде;
[0032] В дополнительном варианте осуществления восьмого аспекта, изобретение предоставляет способ I или любую из формул 7.1-7.10, где способ представляет собой дополнительно, как описано в следующих формулах:
7.11 способ I или любая из формул 7.1-7.10, где расстройство центральной нервной системы является нарушением, выбранным из группы, состоящей из ожирения, тревоги, депрессии (например, рефрактерной депрессии и MDD), психоза, шизофрении, нарушений сна (особенно, нарушений сна, связанных с шизофренией и другими психиатрическими и неврологическими заболеваниями), сексуальных расстройств, мигрени, состояний, связанных с головной болью, социальных фобий, возбуждения при деменции (например, возбуждения при болезни Альцгеймера), возбуждения при аутизме и родственных аутических расстройствах, и желудочно-кишечных расстройств, таких как дисфункция моторики желудочно-кишечного тракта;
7.12 способ I или любая из формул 7.1-7.10, где расстройство центральной нервной системы представляет собой нарушение, в которое вовлечены пути серотонина 5-HT2A, рецепторной системы допамина D2 и/или переносчик обратного захвата серотонина (SERT), как аналогично описано в WO/2009/145900, содержание которой включено здесь посредством ссылки во всей своей полноте;
7.13 способ I или любая из формул 7.1-7.12, где расстройство центральной нервной системы представляет собой нарушение, выбранное из следующих: (i) психоз, например, шизофрения у пациента, страдающего от депрессии; (2) депрессия у пациента, страдающего от психоза, например, шизофрении; (3) расстройства настроения, связанные с психозом, например, шизофренией или болезнью Паркинсона; и (4) нарушения сна, связанные с психозом, например, шизофренией или болезнью Паркинсона;
7.14 способ I или любая из формул 7.1-7.13, где расстройство центральной нервной системы представляет собой психоз, например, шизофрению, и указанный пациент является пациентом, страдающим от депрессии;
7.15 способ I или любая из формул 7.1-7.14, где указанный пациент не способен переносить побочные эффекты общепринятых антипсихотических средств, например, хлорпромазина, галоперидола дроперидола, флуфеназина, локсапина, мезоридазина, молидона, перфеназина, пимозида, прохлорперазина промазина, тиоридазина, тиотиксена, трифторперазина, клозапина, арипипаразола, оланзапина, кветиапина, рисперидона и зипрасидона;
7.16 способ I или любая из формул 7.1-7.15, где указанный пациент не способен переносить побочные эффекты общепринятых антипсихотических средств, например, галоперидола, арипипаразола, клозапина, оланзапина, кветиапина, рисперидона и зипрасидона;
7.17 способ I или любая из формул 7.1-7.16, где указанное расстройство является депрессией, и указанный пациент является пациентом, страдающим от психоза, например, шизофрении, или болезни Паркинсона;
7.18 способ I или любая из формул 7.1-7.13, где указанное расстройство представляет собой нарушение сна, и указанный пациент является страдающим от депрессии;
7.19 способ I или любая из 7.1-7.13, где указанные одно или более расстройств представляют собой нарушение сна, и указанный пациент является страдающим от психоза, например, шизофрении;
7.20 способ I или любая из 7.1-7.13, где указанные одно или более расстройств представляют собой нарушение сна, и указанный пациент является страдающим от болезни Паркинсона;
7.21 способ I или любая из 7.1 -7.13, где указанные одно или более расстройств представляют собой нарушение сна, и указанный пациент является страдающим от депрессии и психоза, например, шизофрении или болезни Паркинсона.
7.22 Любой из вышеприведенных способов, где эффективное количество составляет l мг-l000 мг, предпочтительно, 2,5 мг-50 мг;
7.23 любой из вышеприведенных способов, где эффективное количество составляет 1 мг-100 мг в день, предпочтительно 2,5 мг-50 мг в день;
7.24 любой из вышеприведенных способов, где состояние, подлежащее лечению, представляет собой дискинезию, например, у пациента, принимающего допаминергические препараты, например, препараты, выбранные из леводопа и вспомогательных средств для леводопа (карбидопа, ингибиторы СОМТ, ингибиторы MAO-B), агонисты допамина, и антихолинергические средства, например, леводопа;
7.25 любой из вышеприведенных способов, где пациент страдает от болезни Паркинсона.
[0033] В конкретном варианте осуществления восьмого аспекта, изобретение предоставляет способ (способ Ip) лечения или профилактики расстройства центральной нервной системы, включающий введение пациенту, нуждающемуся в лечении:
7.4P соединения формулы III или любой из формул 4.28-4.30, как описано здесь прежде, в свободной или (фармацевтически приемлемой) солевой форме;
7.8P фармацевтической композиции, как описано в 5.4P;
7.9P композиции пролонгированного действия любой из Формул 5.10-5.11;
7.10P (фармацевтической) композиции формулы 6.5, как описано здесь прежде;
7.11P композиции пероральной системы доставки с осмотически контролируемым высвобождением, как описано здесь прежде.
[0034] В дополнительном варианте осуществления восьмого аспекта, изобретение предоставляет способ Ip, где способ дополнительно описан в любой из формул 7.11-7.25.
[0035] В предпочтительном варианте осуществления восьмого аспекта, изобретение предоставляет способ I или любое из 7.1-7.25, где соединение представляет собой соединение Формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)-, в свободной или фармацевтически приемлемой солевой форме. В еще одном предпочтительном варианте осуществления, изобретение предоставляет способ I или любое из 7.1-7.25, где соединение представляет собой соединение формулы 1.14, 4.26 или 4.20, в свободной или фармацевтически приемлемой солевой форме. В еще одном другом предпочтительном варианте осуществления, изобретение предоставляет способ, как описано здесь прежде, где расстройство представляет собой шизофрению или нарушение сна.
[0036] В еще одном другом предпочтительном варианте осуществления восьмого аспекта, изобретение предоставляет способ I или любой из 7.1-7.25, где композицию пролонгированного действия изобретения (композицию пролонгированного действия любой из формул 5.5-5.9; или (фармацевтическую) композицию любой из формул 6.1-6.4) вводят для контролируемого и/или замедленного высвобождения соединения по изобретению в течение периода, равного от приблизительно 14 дней, от приблизительно 30 до приблизительно 180 дней, предпочтительно в течение периода, равного приблизительно 30, приблизительно 60 или приблизительно 90 дней. Контролируемое и/или замедленное высвобождение является особенно применимым для избегания преждевременного прерывания лечения, особенно, в случае лечения антипсихотическими средствами, где несоблюдение или невыполнение режимов приема препарата является обычной практикой.
[0037] В еще одном другом предпочтительном варианте осуществления восьмого аспекта, изобретение предоставляет способ Ip, как описано здесь прежде, где композицию пролонгированного действия по изобретению вводят для контролируемого и/или замедленного высвобождения соединения по изобретению в течение периода времени.
[0038] В девятом аспекте изобретение предоставляет способ (способ II) для профилактики и лечения одного или более нарушений сна, включающий введение пациенту, нуждающемуся в лечении, соединения, как описано в следующих формулах:
8.1 соединения формулы I или любой из формул 1.1-1.22 в свободной или фармацевтически приемлемой солевой форме;
8.2 соединения формулы II-A в свободной или фармацевтически приемлемой солевой форме;
8.3 соединения формулы II-B в свободной или фармацевтически приемлемой солевой форме;
8.4 соединения формулы III или любой из формул 4.1-4.27, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме;
8.5 фармацевтической композиции, как описано в формуле 5.1;
8.6 фармацевтической композиции, как описано в формуле 5.2;
8.7 фармацевтической композиции, как описано в формуле 5.3;
8.8 фармацевтической композиции, как описано в формуле 5.4;
8.9 композиции пролонгированного действия по любой из формул 5.5-5.9; или
8.10 (фармацевтической) композиции по любой из формул 6.1-6.4, как описано здесь прежде.
[0039] В дополнительном варианте осуществления девятого аспекта, изобретение предоставляет способ II, 8.1-8.10, где нарушение сна включает бессонницу при поддержании сна, частые пробуждения, и ощущение несвежести при пробуждении;
8.11 любой из вышеприведенных способов, где нарушение сна представляет собой бессонницу при поддержании сна;
8.12 любой из вышеприведенных способов, где эффективное количество составляет l мг-5 мг, предпочтительно 2,5-5 мг, в день;
8.13 любой из вышеприведенных способов, где эффективное количество составляет 2,5 мг или 5 мг в день;
8.14 любой из вышеприведенных способов где нарушение сна происходит у пациента, страдающего от или имеющего риск дискинезии, например, пациента, принимающего допаминергические препараты, например, выбранные из леводопа и вспомогательных средств для леводопа (карбидопа, ингибиторов СОМТ, ингибиторов MAO-B), агонистов допамина и антихолинергических средств, например, принимающего леводопа;
8.15 любой из вышеприведенных способов, где пациент страдает от болезни Паркинсона.
[0040] В еще одном варианте осуществления девятого аспекта, изобретение предоставляет способ (Способ IIp) для профилактики и лечения одного или более из нарушений сна, включающий введение пациенту, нуждающемуся в этом, соединения, как описано в следующих формулах:
8.4P формулы III или любой из формул 4.28-4.30, как описано здесь прежде, в свободной или (фармацевтически приемлемой) солевой форме;
8.8P фармацевтической композиции, как описано в 5.4P;
8.9P композиции пролонгированного действия любой из Формул 5.10-5.11;
8.10P (фармацевтической) композиции формулы 6.5, как описано здесь прежде;
8.11 композиции пероральной системы доставки с осмотически контролируемым высвобождением, как описано здесь прежде.
[0041] В дополнительном варианте осуществления девятого аспекта, изобретение предоставляет способ IIp, или любой из 8.4P, 8.8P-8.11P, где нарушение сна включает бессонницу при поддержании сна, частые пробуждения и ощущение несвежести при пробуждении. В еще одном другом варианте осуществления девятого аспекта, способ IIp является таким, как описано в любой из формул 8.11-8.15.
[0042] Соединения по изобретению, при превращении в соединения формулы Q, как описано здесь прежде, предоставляют эффективное лечение нарушений, относящихся к 5-HT2A, SERT и/или D2 рецепторам с минимальными экстрапирамидальными побочными эффектами или без них, как аналогично раскрыто и заявлено в WO 2009/145900, содержание которой включено посредством ссылки во всей полноте. Следовательно, соединения по изобретению, фармацевтические композиции по изобретению или композиции пролонгированного действия по изобретению могут применяться в комбинации со вторым терапевтическим средством, особенно, при более низких дозировках, чем когда индивидуальные средства применяют в виде монотерапии, таким образом, чтобы усилить терапевтические активности комбинированных агентов, не вызывая нежелательных побочных эффектов, обычно имеющих место при общепринятой монотерапии. Следовательно, соединения по изобретению могут одновременно, последовательно или параллельно вводиться вместе с другим антидепрессантом, антипсихотическим средством, другими снотворными средствами и/или агентами, применяемыми для лечения болезни Паркинсона или расстройств настроения. В еще одном примере побочные эффекты могут быть снижены или минимизированы посредством введения соединения по изобретению в комбинации с одним или более из вторых терапевтических средств в свободной или солевой форме, где дозировки (i) второго терапевтического средства (средств) или (ii) как соединения по изобретению, так и второго терапевтического средства являются более низкими, чем если бы средства/соединения вводились в виде монотерапии. В конкретном варианте осуществления соединения по изобретению являются применимыми для лечения дискинезии у пациента, принимающего допаминергические препараты, например, выбранные из леводопа и вспомогательных средств для леводопа (карбидопа, ингибиторов COMT, ингибиторов MAO-B), агонистов допамина и антихолинергических средств, например, таких как применяются при лечении болезни Паркинсона.
[0043) Следовательно, в десятом аспекте рассматриваемое изобретение предоставляет способ I, например, или любую из формул 7.1-7.25, или способ II или любой из 8.1-8.15, который дополнительно включает одно или более терапевтических средств, выбранных из соединений, которые модулируют активность ГАМК (например, усиливают активность и облегчают передачу ГАМК), агониста ГАМК-B, 5-HT модулятора (например, 5-HTIa агонист, 5- HT2a антагониста, 5-HT2a обратного агониста и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста/ингибитора обратного захвата серотонина-2 (SARIs), антагониста рецептора орексина, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, человеческого гормона роста, агониста гормона роста, эстрогена, агониста эстрогена, лекарственного средства нейрокинин-1, антидепрессанта и антипсихотического средства, например, атипичного антипсихотического средства, в свободной или фармацевтически приемлемой солевой форме (Способ I-A и II-A, соответственно).
[0044] В дополнительном варианте осуществления десятого аспекта, изобретение предоставляет следующие Способ I-A или II-A, дополнительно включающие одно или более терапевтических средств.
9.1 Способ I-A или II-A, где терапевтическое средство (средства) представляет(ют) собой соединения, которые модулируют активность ГАМК (например, усиливают активность и облегчают передачу ГАМК);
9.2 способ I-A или II-A или 9.1, где соединение ГАМК выбирают из группы, состоящей из одного или более из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals) и эстазолама;
9.3 способ I-A или II-A, где терапевтическое средство представляет собой дополнительный антагонист 5HT2a;
9.4 способ I-A или II-A или 9.3, где указанный дополнительный антагонист 5HT2a выбран из одного или более из кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, Франция), прувансерина, MDL 100907 (Sanofi-Aventis, Франция), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA) и AVE8488 (Sanofi-Aventis, Франция);
9.5 способ I-A или II-A, где терапевтическое средство представляет собой агонист мелатонина;
9.6 способ I-A или II-A или 9.5, где агонист мелатонина выбирают из группы, состоящей из одного или более из мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (Phase II Discovery) и агомелатина;
9.7 способ I-A или II-A, где терапевтическое средство представляет собой блокатор ионных каналов;
9.8 способ I-A или II-A или 9.7, где указанный блокатор ионных каналов является одним или более из ламотригина, габапентина и прегабалина.
9.9 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист рецептора орексина;
9.10 способ I-A или II-A или 9.9, где антагонист рецептора орексина выбирают из группы, состоящей из орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmith Kline, UK), GW649868 (GlaxoSmithKline) и производного бензамида;
9.11 способ I-A или II-A, где терапевтическое средство представляет собой антагонист/ингибитор обратного захвата серотонина-2 (SARI);
9.12 способ I-A или II-A или 9.11, где антагонист/ингибитор обратного захвата серотонина-2 (SARI) выбирают из группы, состоящей из одного или более из Org 50081 (Organon-Netherlands), ритансерина, нефазодона, серзона и тразодона;
9.13 способ I-A или II-A, где терапевтическое средство представляет собой агонист 5HTIa;
9.14 способ I-A или II-A или 9.13, где агонист 5HTIa выбирают из группы, состоящей из одного или более из репинотана, саризотана, эптапирона, буспирона и MN-305 (MediciNova, San Diego, CA);
9.15 способ I-A или II-A, где терапевтическое средство представляет собой лекарственное средство нейрокинин-1;
9.16 способ I-A или II-A или 9.15, где лекарственное средство нейрокинин-1 представляет собой Касопитант (GlaxoSmithKline);
9.17 способ 1-A или II-A, где терапевтическое средство представляет собой антипсихотическое средство;
9.18 способ I-A или II-A или 9.17, где антипсихотическое средство выбирают из группы, состоящей из хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молидона, перфеназина, пимозида, прохлорперазина, промазина, тиоридазина, тиотиксена, трифторперазина, клозапина, арипипаразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
9.19 способ I-A или II-A, где терапевтическое средство представляет собой антидепрессант;
9.20 способ 1-A или II-A или 9.19, где антидепрессант выбирают из амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, сульфата фенизина, протиптилина, сертралина, транилципромина, тразодона, тримипрамина и велафаксина;
9.21 способ I-A или II-A, 9.17 или 9.18, где антипсихотическое средство представляет собой атипичное антипсихотическое средство;
9.22 способ 1-A или II-A, или любое из 9.17-9.21, где атипичное антипсихотическое средство выбирают из группы, состоящей из клозапина, арипипаразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
9.23 способ I-A или II-A, где терапевтическое средство выбирают из любого из способов 9.1-9.22, например, выбирают из группы, состоящей из модафинила, армодафинила, доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама, кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, Франция), прувансерина, MDL 100907 (Sanofi-Aventis, Франция), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA), AVE8488 (Sanofi-Aventis, Франция), репинотана, саризотана, эптапирона, буспирона, MN-305 (MediciNova, San Diego, CA), мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (Phase II Discovery), агомелатина, ламотригина, габапентина, прегабалина, орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithCline, UK), GW649868 (GlaxoSmithKline), производного бензамида, Org 50081 (Organon-Netherlands), ритансерина, нефазодона, серзона, тразодона, Касопитанта (GlaxoSmithKline), амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, сульфата фенизина, протиптилина, сертралина, транилципромина, тразодона, тримипрамина, велафаксина, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молидона, перфеназина, пимозида, прохлорперазина промазина, тиоридазина, тиотиксена, трифторперазина, клозапина, арипипаразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
9.24 способ I-A или II-A, где терапевтическое средство представляет собой агонист H3;
9.25 способ I-A или II-A, где терапевтическое средство представляет собой антагонист H3;
9.26 способ I-A или II-A, где терапевтическое средство представляет собой норадренергический агонист или антагонист;
9.27 способ I-A или II-A, где терапевтическое средство представляет собой агонист галанина;
9.28 способ I-A или II-A, где терапевтическое средство представляет собой антагонист CRH;
9.29 способ I-A или II-A, где терапевтическое средство представляет собой человеческий гормон роста;
9.30 способ I-A или II-A, где терапевтическое средство представляет собой агонист гормона роста;
9.31 способ I-A или II-A, где терапевтическое средство представляет собой эстроген;
9.32 способ I-A или II-A, где терапевтическое средство представляет собой агонист эстрогена;
9.33 способ I-A или II-A, где терапевтическое средство представляет собой лекарственное средство нейрокинин-1;
9.34 способ I-A или II-A, где терапевтическое средство комбинируют с соединениями формулы (I), и терапевтическое средство представляет собой средство против болезни Паркинсона, такое как L-допа, со-карелдопа, дуодопа, сталова, Симметрел, бензотропин, бипериден, бромокриптин, энтакапон, перголид, прамипексол, проциклидин, ропинирол, селегилин и толкапон;
9.35 способ I-A или II-A, где соединения формулы (I) могут применяться для лечения нарушений сна, депрессии, психоза или любых их комбинаций, у пациентов, страдающих от перечисленных заболеваний и/или болезни Паркинсона;
9.36 способ I-A или II-A, где расстройство выбирают из, по меньшей мере, одного или более из психоза, например, шизофрении, депрессии, расстройств настроения, нарушений сна (например, поддержания сна и/или наступления сна) или любой комбинации этих расстройств;
9.37 любой из вышеприведенных способов, где расстройство представляет собой нарушение сна;
9.38 любой из вышеприведенных способов, где расстройство представляет собой нарушение сна, связанное с психозом, например, шизофренией или болезнью Паркинсона; в свободной или фармацевтически приемлемой солевой форме.
[0045] В еще одном варианте осуществления десятого аспекта, рассматриваемое изобретение предоставляет Способ Ip или Способ IIp, как описано здесь прежде, дополнительно включает одно или более терапевтических средств, выбранных из соединений, которые модулируют активность ГАМК (например, усиливают активность и облегчают передачу ГАМК), агониста ГАМК-B, модулятора 5-HT (например, агониста 5-HTIa, антагониста 5-HT2A, обратного агониста 5-HT2a и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста/ингибитора обратного захвата серотонина-2 (SARIs), антагониста рецептора орексина, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, человеческого гормона роста, агониста гормона роста, эстрогена, агониста эстрогена, лекарственного средства нейрокинин-1, антидепрессанта и антипсихотического средства, например, атипичного антипсихотического средства, в свободной или фармацевтически приемлемой солевой форме (Способ Ip-A и IIp-A, соответственно). В дополнительном варианте осуществления этого аспекта, изобретение предоставляет Способ Ip-A или IIp-A, как описано сходным образом в любой из формул 9.1-9.38.
[0046] В одиннадцатом аспекте изобретения комбинация из соединения по изобретению и одного или более вторых терапевтических средств, как описано в способах I-A, II-A или любого из 9.1-9.38, может быть введена в виде фармацевтической композиции или композиции пролонгированного действия, как описано здесь прежде. Аналогично, комбинация соединения по изобретению и одного или более второго терапевтических средств, как описано в способах Ip-A, IIp-A или любом из 9.1-9.38, может быть введена в виде фармацевтической композиции или композиции пролонгированного действия, как описано здесь прежде. Комбинированные композиции может включать смеси комбинированных лекарственных средств, а также две или более отдельных композиций лекарственных средств, причем эти индивидуальные композиции могут, например, совместно вводиться пациенту.
[0047] В конкретном варианте осуществления способы I-A, II-A, Ip-A, IIp-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с атипичным антипсихотическим средством, например, соединением, выбранным из клозапина, арипипаразола, оланзапина, кветиапина, рисперидона, зипрасидона или палиперидона, в свободной или фармацевтически приемлемой солевой форме, например, где дозировка атипичного антипсихотического средства снижена и/или снижаются побочные эффекты.
[0048] В еще одном варианте осуществления, способы I-A, II-A, способы Ip-A, IIp-A или любой из 9.1-9.38 включает введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с антидепрессантом, например, амитриптилином, амоксапином, бупропионом, циталопрамом, кломипрамином, дезипрамином, доксепином, дулоксетином, эсциталопрамом, флуоксетином, флувоксамином, имипрамином, изокарбоксазидом, мапротилином, миртазапином, нефазодоном, нортриптилином, пароксетином, сульфатом фенизина, протиптилином, сертралином, транилципромином, тразодоном, тримипрамином или велафаксином, в свободной или фармацевтически приемлемой солевой форме. Альтернативно, антидепрессант может применяться как вспомогательный препарат в дополнение к соединению по изобретению.
[0049] В еще одном другом варианте осуществления способы I-A, II-A, Ip-A, IIp-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с соединением, которое модулирует активность ГАМК, например, соединением, выбранным из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама или любых их комбинаций, в свободной или фармацевтически приемлемой солевой форме.
[0050] В еще одном предпочтительном варианте осуществления, способы I-A, II-A, Ip-A, IIp-Α или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с доксепином в свободной или фармацевтически приемлемой солевой форме. Дозировки доксепина могут изменяться в любом интервале, известном рядовому специалисту в области. В одном примере доза 10 мг доксепина может комбинироваться с любой дозировкой соединения по изобретению.
[0051] В еще одном варианте осуществления способы I-A, II-A, Ip-A, IIp-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации (включающей в качестве части режима суточной дозировки) с атипичным стимулятором, например, модафинилом, адрафинилом или армодафинилом. Режим, включающий в себя соединение по изобретению с такими лекарственными средствами, способствует более регулярному сну и позволяет избежать побочных эффектов, таких как психоз или мания, связанных с более высокими уровнями таких лекарственных средств, например, при лечении биполярной депрессии, когнитивного нарушения, связанного с шизофренией, и избыточной сонливости и утомляемости в состояниях, таких как болезнь Паркинсона и рак.
[0052] В двенадцатом аспекте изобретение предоставляет применение соединения, как описано в следующих формулах:
11.1 соединения формулы I или любой из формул 1.1-1.22 в свободной или фармацевтически приемлемой солевой форме;
11.2 соединения формулы II-A в свободной или фармацевтически приемлемой солевой форме;
11.3 соединения формулы II-B в свободной или фармацевтически приемлемой солевой форме;
11.4 соединения формулы III или любой из формул 4.1-4.27, как описано здесь прежде, в свободной или фармацевтически приемлемой солевой форме;
11.5 фармацевтической композиции, как описано в формуле 5.1;
11.6 фармацевтической композиции, как описано в формуле 5.2;
11.7 фармацевтической композиции, как описано в формуле 5.3;
11.8 фармацевтической композиции, как описано в формуле 5.4;
11.9 композиции пролонгированного действия любой из формул 5.5-5.9; или
11.10 (фармацевтической) композиции любой из формул 6.1-6.4, как описано здесь прежде;
11.4P соединения формулы III или любой из формул 4.28-4.30, как описано здесь прежде, в свободной или (фармацевтически приемлемой) солевой форме;
11.8P фармацевтической композиции, как описано в 5.4P;
11.9P композиции пролонгированного действия любой из формул 5.10-5.11;
11.10P (фармацевтической) композиции формулы 6.5, как описано здесь прежде;
8.11P композиции пероральной системы доставки с осмотически контролируемым высвобождением, как описано здесь прежде,
(в производстве лекарственного средства) для лечения или профилактики одного или более расстройств, как раскрыто здесь прежде, например, в любом из способа I, любом из 7.1-7.25, способа II, любом из 8.1-8.15, способов I-A, II-A, любом из 9.1-9.38, способа Ip, способа IIp, способов Ip-A, IIp-A или любых способов, описанных в одиннадцатом аспекте изобретения.
[0053] В тринадцатом аспекте изобретение предоставляет фармацевтическую композицию, как описано здесь прежде, например, в следующих формулах:
12.1 фармацевтическую композицию, как описано в формуле 5.1;
12.2 фармацевтическую композицию, как описано в формуле 5.2;
12.3 фармацевтическую композицию, как описано в формуле 5.3;
12.4 фармацевтическую композицию, как описано в формуле 5.4;
12.5 композицию пролонгированного действия любой из Формул 5.5-5.9; или
12.6 (фармацевтическую) композицию любой из формул 6.1-6.4, как описано здесь прежде;
12.8P фармацевтическую композицию, как описано в 5.4P;
12.9P композицию пролонгированного действия любой из формул 5.10-5.11;
12.10P (фармацевтическую) композицию формулы 6.5, как описано здесь прежде;
12.11P композицию пероральной системы доставки с осмотически контролируемым высвобождением, как описано здесь прежде, для применения при лечении или профилактики одного или более расстройств, как раскрыто здесь прежде, например, в любом из способа I, любом из 7.1-7.25, способа II, любом из 8.1-8.15, способов I-A, II-A, любом из 9.1-9.38, способа Ip, способа IIp, способов Ip-A, IIp-A или любых способов, описанных в одиннадцатом или двенадцатом аспекте изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0054] Фигура 1 иллюстрирует ингибирование DOI-индуцированных судорожных движений головой у мышей посредством соединения Примера 1, как описано в Примере 8.
[0055] Фигура 2 иллюстрирует ингибирование латентности при спуске у мышей после перорального введения соединения Примера 1, как описано в Примере 9.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0056] Если не указано иначе или не ясно из контекста, следующие термины, как используют здесь, имеют следующие значения:
a. "Алкил", как используют здесь, представляет собой насыщенный или ненасыщенный углеводородный остаток, например, от одного до двадцати одного атома углерода по длине, который может быть линейным или разветвленным (например, н-бутил или трет-бутил), предпочтительно линейным, если не установлено иначе. Например, "C1-21алкил" обозначает алкил, имеющий от 1 до 21 атома углерода. В одном варианте осуществления алкил является необязательно замещенным одной или более гидрокси или C1-22 алкокси (например, этокси) группами. В еще одном варианте осуществления алкил содержит от 1 до 21 атома углерода, предпочтительно имеет прямую цепь и необязательно является насыщенным или ненасыщенным, например, R1 представляет собой алкильную цепь, содержащую от 1 до 21 атома углерода, предпочтительно 6-15 атомов углерода, 16-21 атома углерода, например, таким образом, что вместе с -C(O)-, к которому он присоединен, например, когда отщепляется от соединения формулы III, образует остаток природной или неприродной, насыщенной или ненасыщенной жирной кислоты.
[0057] Если не указано иначе, соединения по изобретению, например, соединения формулы I или любой из 1.1-1.22, формулы II-A, формулы II-B или формулы III или любой из формул 4.1-4.27 или 4.28-4.30, могут существовать в свободной форме или в виде соли, например, в виде солей присоединения кислоты. Кислотно-аддитивная соль соединения изобретения, которое является в достаточной степени основным, например, кислотно-аддитивная соль с, например, неорганической или органической кислотой, например хлористоводородной, бромистоводородной, серной, фосфорной кислотой, уксусной, трифторуксусной, лимонной, малеиновой кислотой, толуолсульфоновой, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памоевой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изетионовой кислотой и т.п. Кроме того, соль соединения по изобретению, которое является достаточно кислотным, представляет собой соль щелочного металла, например, натриевую или калиевую соль, соль щелочноземельного металла, например, соль кальция или магния, соль аммония или соль с органическим основанием, которое предоставляет физиологически приемлемый катион, например, соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис-(2-гидроксиэтил)амином. В конкретном варианте осуществления соль соединения по изобретению представляет собой соль присоединения толуолсульфоновой кислоты. В еще одном конкретном варианте осуществления, соль соединения по изобретению представляет собой соль присоединения фумаровой кислоты. В конкретном варианте осуществления соль соединения по изобретению представляет собой соль присоединения фосфорной кислоты.
[0058] Соединения по изобретению предназначены для применения в качестве фармацевтических средств, следовательно, фармацевтически приемлемые соли являются предпочтительными. Соли, которые являются непригодными для фармацевтических применений, могут быть применимыми, например, для выделения или очистки свободного соединения по изобретению и, следовательно, также являются включенными.
[0059] Соединения по изобретению могут содержать один или более хиральных атомов углерода. Соединения, таким образом, существуют в индивидуальной изомерной, например, энантиомерной или диастереомерной форме или в виде смесей индивидуальных форм, например, рацемических/диастереомерных смесей. Любой изомер может быть представлен, в котором асимметрический центр находится в (R)-, (S)- или (R,S)-конфигурации. Изобретение следует понимать, как охватывающее индивидуальные оптически активные изомеры, а также их смеси (например, рацемические/диастереомерные смеси). Соответственно, соединение по изобретению может представлять собой рацемическую смесь, или оно может находиться, преобладающим образом, например, в чистой или по существу чистой изомерной форме, например, с большим, чем 70% энантиомерным/диастереомерным избытком ("ee"), предпочтительно большим, чем 80% ee, более предпочтительно большим, чем 90% ee, наиболее предпочтительно большим, чем 95% ee. Очистка указанных изомеров и разделение указанных изомерных смесей могут осуществляться посредством стандартных методов, известных в данной области (например, колоночной хроматографии, препаративной ТСХ, препаративной ВЭЖХ, псевдодвижущегося слоя и т.п.).
[0060] Геометрические изомеры, по природе заместителей вокруг двойной связи или кольца, могут присутствовать в цис (Z) или транс (Е) форме, и обе изомерные формы охвачены объемом настоящего изобретения.
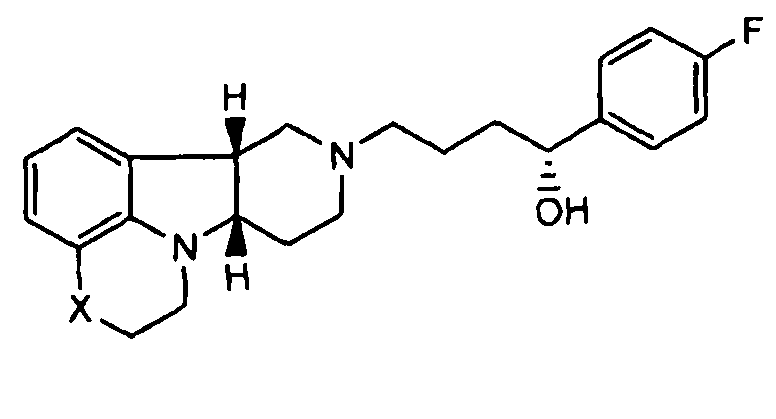
[0061] Соединения по изобретению могут в некоторых случаях также существовать в форме пролекарства. Термин "пролекарство" является признанным в данной области и относится к предшественнику лекарственного средства перед введением, но который генерирует или высвобождает активный метаболит in vivo после введения, посредством некоторого химического или физиологического процесса. В некоторых случаях соединение по изобретению может представлять собой пролекарство, а также метаболит. Авторы настоящего изобретения неожиданно обнаружили, что соединения по изобретению, особенно, соединения, несущие свободную гидроксигруппу, например, соединение формулы I, где X представляет собой -N(CH3) и Y представляет собой -C(H)(OH)-, является относительно неактивным соединением, где гидроксигруппа на указанном соединении окисляется in vivo с образованием активного l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-ил)бутан-1-она (т.е., формулы Q, где X представляет собой -N(CH3)- и Y представляет собой -C(=О)-). Это активное исходное соединение, несущее кетоновую группу, может быть также метаболизировано обратно в относительно неактивный гидроксиметаболит/пролекарство (например, формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)). Не имея намерения быть связанными с какой-либо конкретной теорией, полагают, что активное соединение формулы Q, где X представляет собой -N(CH3)- и Y представляет собой -C(=О), непрерывно образуется из соединения по изобретению, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH). Посредством введения соединения по изобретению достигают превосходящего фармакокинетического профиля (например, с более длительным временем пребывания активного соединения в организме, особенно в мозге) по отношению к той ситуации, когда вводят активное соединение формулы Q.
[0062] Там, где X представляет собой -N(CH3)-, соединения по изобретению могут дополнительно метаболизироваться in vivo с образованием дезметильного производного (т.е., где X представляет собой -N(H)-). Конкретно, соединение формулы I, где X представляет собой -N(CH3) и Y представляет собой -C(H)(OH)- или -C(=О), может метаболизироваться с образованием дезметильного производного (например, где X представляет собой -N(H)- и Y представляет собой -C(H)(OH) или -C(=О)-, соответственно), где гидроксисоединение может затем окисляться in vivo с образованием соответствующего активного дезметильного соединения формулы Q, где X представляет собой -N(H)- и Y представляет собой -C(=О).
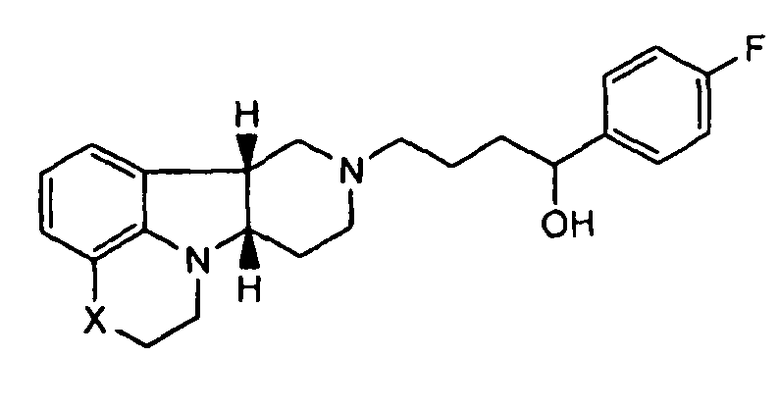
[0063] В дополнение к уникальным характеристикам соединений по изобретению, соединения формулы I, где Y представляет собой -C(H)(OH)-, могут также быть этерифицированы с образованием физиологически гидролизуемых и приемлемых сложноэфирных пролекарств. Как используют здесь, "физиологически гидролизуемые и приемлемые сложные эфиры" означает сложные эфиры соединений изобретения, которые являются гидролизуемыми при физиологических условиях с получением гидрокси, с одной стороны, и кислоты, например, карбоновой кислоты, с другой стороны, которые сами по себе являются физиологически переносимыми в дозах, подлежащих введению. Например, соединение формулы I, где Y представляет собой -C(H)(OH), может быть этерифицировано с образованием пролекарства, т.е. соединения формулы III или любой из формул 4.1-4.27 или 4.28-4.30. Например, соединение формулы I, где Y представляет собой -C(H)(OH)-, или любое из 1.1-1.17 или 1.19-1.22 может быть этерифицировано с образованием соединения формулы III, которое может быть гидролизовано in vivo с образованием соединения формулы I, где Y представляет собой -C(H)(OH)-, и затем окислено in vivo до соответствующего активного соединения формулы Q. В частности, соединение формулы III, где R1 представляет собой -C(О)-C1-21алкил, например, ацил сложных эфиров кислот, например, сложный эфир гептановой, октановой, декановой, додекановой, тетрадекановой или гексадекановой кислоты, может гидролизоваться в организме с образованием соединения формулы I, где Y представляет собой -C(H)(OH)-, с одной стороны, и HO-C(О)-C1-21алкил, с другой стороны (например, гептановой, октановой, декановой, додекановой, тетрадекановой или гексадекановой кислоты, соответственно), причем это гидроксисоединение будет затем превращаться в активное соединение формулы Q, где Y представляет собой -C(=О)-.
[0064] Аналогично, там, где соединения по изобретению содержат аминогруппу, пролекарство такого амина, например, пролекарства метиламина могут также существовать там, где пролекарство расщепляется с высвобождением аминного метаболита in vivo после введения.
[0065] Пролекарства соединений по изобретению, т.е. соединений формулы III, конкретно, когда R1 представляет собой -C(О)-C1-21алкил, предпочтительно -C6-21алкил, более предпочтительно C6-15алкил, более предпочтительно линейный, насыщенный или ненасыщенный и необязательно замещенный одной или более гидрокси или алкокси группами, являются особенно применимыми для замедленного и/или отсроченного высвобождения, таким образом, чтобы достичь эффекта длительного действия, например, где соединения по изобретению высвобождаются в течение периода, равного от приблизительно 14 до приблизительно 30, до приблизительно 180 дней, предпочтительно в течение приблизительно 30 или приблизительно 60 или приблизительно 90 дней, например, как описано в любой из композиций пролонгированного действия любой из Формул 5.5-5.9 или 5.10-5.11. Предпочтительно, готовая лекарственная форма замедленного и/или отсроченного высвобождения является инъекционной готовой лекарственной формой.
[0066] Альтернативно и/или дополнительно, соединения по изобретению (например, соединения формулы I или любое из 1.1-1.22, формулы II-A, формулы II-B или формулы III или любой из формул 4.1-4.27 или 4.28-4.30) могут быть включены в качестве готовой лекарственной формы пролонгированного действия, например, посредством диспергирования, растворения или инкапсулирования соединений по изобретению в полимерную матрицу, как описано в любой из композиций 6.1-6.4 или 6.5, таким образом, что соединение непрерывно высвобождается по мере того, как полимер разрушается с течением времени. Высвобождение соединений по изобретению из полимерной матрицы предоставляет контролируемое и/или отсроченное и/или замедленное высвобождение соединений, например, из фармацевтической композиции пролонгированного действия, в субъекте, например, теплокровном животном, таком как человек, которому вводят фармацевтическое депо. Таким образом, фармацевтическое депо доставляет соединения по изобретению субъекту при концентрациях, эффективных для лечения конкретного заболевания или медицинского состояния в течение длительного периода времени, например, 14-180 дней, предпочтительно приблизительно 30, приблизительно 60 или приблизительно 90 дней.
[0067] Полимеры, применимые для полимерной матрицы в композиции по изобретению (например, композиции пролонгированного действия по изобретению), могут включать сложный полиэфир гидроксижирной кислоты и ее производных или других средств, таких как полимолочная кислота, полигликолевая кислота, полилимонная кислота, полияблочная кислота, поли-бета-гидроксимасляная кислота, полимер с раскрытием кольца эпсилон-капролактона, сополимер молочной кислоты-гликолевой кислоты, сополимер 2-гидроксимасляной кислоты-гликолевой кислоты, сополимер полимолочной кислоты-полиэтиленгликоля или сополимер полигликолевой кислоты-полиэтиленгликоля), полимер алкил-альфа-цианоакрилата (например поли(бутил-2-цианоакрилат)), полиалкиленоксалат (например политриметиленоксалат или политетраметиленоксалат), сложный полиортоэфир, поликарбонат (например, полиэтиленкарбонат или полиэтиленпропиленкарбонат), полиортокарбонат, полиаминокислоту (например поли-гамма-L-аланин, поли-гамма-бензил-L-глутаминовую кислоту или поли-γ-метил-L-глутаминовую кислоту), сложный эфир гиалуроновой кислоты и т.п., и могут применяться один или более из этих полимеров.
[0068] Если полимеры представляют собой сополимеры, они могут представлять собой любые из статистических, блок- и/или привитых сополимеров. Когда вышеуказанные альфа-гидроксикарбоновые кислоты, гидроксидикарбоновые кислоты и гидрокситрикарбоновые кислоты имеют оптическую активность в их молекулах, может применяться любой из D-изомеров, L-изомеров и/или DL-изомеров. Среди прочих, могут применяться полимер альфа-гидроксикарбоновой кислоты (предпочтительно, полимер молочной кислоты-гликолевой кислоты), его сложный эфир, сложные эфиры поли-альфа-цианоакриловой кислоты и т.д.; и сополимер молочной кислоты-гликолевой кислоты (также называемый поли(лактид-альфа-гликолид) или поли(молочная-co-гликолевая кислота), и здесь далее именуемый PLGA) является предпочтительным. Таким образом, в одном аспекте полимер, применимый для полимерной матрицы, представляет собой PLGA. Как используют здесь, термин PLGA включает полимеры молочной кислоты (также называемые полилактидом, поли(молочной кислотой) или PLA). Наиболее предпочтительно, полимер представляет собой биодеградируемый полимер поли(d,l-лактид-co-гликолид).
[0069] В предпочтительном варианте осуществления полимерная матрица по изобретению представляет собой биосовместимый и биодеградируемый полимерный материал. Термин "биосовместимый" определяют как полимерный материал, который не является токсичным, не является канцерогенным и не индуцирует существенное воспаление в тканях тела. Материал матрицы должен быть биодеградируемым там, где полимерный материал должен разрушаться под действием процессов в организме до продуктов, легко утилизируемых организмом, и не должен накапливаться в организме. Продукты биодеградации должны быть также биосовместимыми с организмом в том смысле, что полимерная матрица является биосовместимой с организмом. Конкретные применимые примеры материалов полимерной матрицы включают поли(гликолевую кислоту), поли-D,L-молочную кислоту, поли-L-молочную кислоту, сополимеры вышеприведенных, поли(алифатические карбоновые кислоты), сополиоксалаты, поликапролактон, полидиоксонон, поли(ортокарбонаты), поли(ацетали), поли(молочная кислота-капролактон), сложные полиортоэфиры, поли(гликолевая кислота-капролактон), полиангидриды, и природные полимеры, включающие альбумин, казеин и воска, такие как моно- и дистеарат глицерина, и т.п. Предпочтительным полимером для применения при практической реализации данного изобретения является dl(полилактид-со-гликолид). Предпочтительно, чтобы молярное отношение лактида к гликолиду в таком сополимере находилось в интервале от приблизительно 75:25 до 50:50.
[0070] Применимые полимеры PLGA могут иметь среднемассовую молекулярную массу, равную от приблизительно 5000 до 500000 дальтонов, предпочтительно приблизительно 150000 дальтонов. В зависимости от скорости разрушения, которой нужно достичь, могут применяться полимеры с различной молекулярной массой. Для диффузионного механизма высвобождения лекарственного средства, полимер должен оставаться неповрежденным, пока все лекарственное средство не высвободится из полимерной матрицы, и затем разрушаться. Лекарственное средство может также высвобождаться из полимерной матрицы по мере того, как полимерный эксципиент биоэродирует.
[0071] PLGA может быть получен посредством любого общепринятого способа или может являться доступным для приобретения. Например, PLGA может быть получен полимеризацией с раскрытием цикла с подходящим катализатором из циклического лактида, гликолида и т.д. (см. EP-0058481B2; Эффекты переменных полимеризации на свойства PLGA: молекулярную массу, композицию и структуру цепи).
[0072] Полагают, что PLGA является биодеградируемым посредством разрушения цельной композиции твердого полимера, вследствие разрыва гидролизуемых и ферментативно расщепляемых сложноэфирных связей при биологических условиях (например, в присутствии воды и биологических ферментов, обнаруживаемых в тканях теплокровных животных, таких как люди), с образованием молочной кислоты и гликолевой кислоты. Как молочная кислота, так и гликолевая кислота являются водорастворимыми, нетоксичными продуктами нормального метаболизма, которые могут далее биодеградировать с образованием диоксида углерода и воды. Другими словами, полагают, что PLGA разрушается посредством гидролиза его сложноэфирных групп в присутствии воды, например, в организме теплокровного животного, такого как человек, с получением молочной кислоты и гликолевой кислоты и созданием кислотного микроклимата. Молочная и гликолевая кислоты являются побочными продуктами разнообразных метаболических путей в организме теплокровного животного, такого как человек, при нормальных физиологических условиях и, следовательно, являются хорошо переносимыми и продуцируют минимальную системную токсичность.
[0073] В еще одном варианте осуществления полимерная матрица, применимая для изобретения, может содержать звездообразный полимер, где структура сложного полиэфира является звездообразной. Эти сложные полиэфиры имеют единичный остаток полиола в качестве центрального фрагмента, окруженного цепями остатков кислот. Полиольный фрагмент может представлять собой, например, глюкозу или, например, маннит. Эти сложные эфиры являются известными и описаны в патенте Великобритании GB 2145422 и в патенте США № 5538739, содержание которых включено посредством ссылки.
[0074] Звездообразные полимеры могут быть получены с использованием полигидроксисоединений, например, полиола, например, глюкозы или маннитола в качестве инициатора. Полиол содержит, по меньшей мере, 3 гидроксигруппы и имеет молекулярную массу до приблизительно 20000 дальтонов, с, по меньшей мере, 1, предпочтительно, по меньшей мере, 2, например, в среднем 3 из гидроксигрупп полиола, находящихся в виде сложноэфирных групп, которые содержат полилактидные или co-полилактидные цепи. Разветвленные сложные полиэфиры, например, поли(d,l-лактид-co-гликолид), имеют центральный фрагмент глюкозы, имеющий лучи из линейных полилактидных цепей.
[0075] Композиция пролонгированного действия по изобретению (например, композиции любой из формул 6.1-6.4 или 6.5), как описано здесь прежде, может содержать полимер в форме микрочастиц или наночастиц или в жидкой форме, вместе с соединениями по изобретению, диспергированными или инкасулированными в них. "Микрочастицы" означают твердые частицы, которые содержат соединения по изобретению или в растворе, или в твердой форме, где такое соединение диспергировано или растворено в полимере, который служит в качестве матрицы частицы. Посредством соответствующего выбора полимерных материалов, может быть изготовлена готовая лекарственная форма в виде микрочастиц, в которой полученные в результате микрочастицы проявляют как диффузионное высвобождение, так и свойства высвобождения при биодеградации.
[0076] Когда полимер находится в форме микрочастиц, микрочастицы могут быть получены с использованием любого подходящего способа, такого как упаривание растворителя или способ экстракции растворителем. Например, при способе с упариванием растворителя, соединения по изобретению и полимер могут быть растворены в летучем органическом растворителе (например, кетоне, таком как ацетон, галогенированном углеводороде, таком как хлороформ или метиленхлорид, галогенированном ароматическом углеводороде, циклическом простом эфире, таком как диоксан, сложном эфире, таком как этилацетат, нитриле, таком как ацетонитрил, или спирте, таком как этанол) и диспергированы в водной фазе, содержащей подходящий стабилизатор эмульсии (например, поливиниловый спирт, PVA). Органический растворитель затем упаривают с получением микрочастиц с соединениями по изобретению, инкапсулированными в них. При способе экстракции растворителем, соединения по изобретению и полимер могут быть растворены в полярном растворителе (таком как ацетонитрил, дихлорметан, метанол, этилацетат или метилформиат) и затем диспергированы в водной фазе (такой как вода/раствор PVA). Получают эмульсию с предоставлением микрочастиц с соединениями по изобретению, инкапсулированными в них. Распылительная сушка является альтернативным технологическим методом производства для получения микрочастиц.
[0077] Еще один способ получения микрочастиц по изобретению также описан, как в патенте США № 4389330, так и патенте США № 4530840.
[0078] Микрочастица по настоящему изобретению может быть получена посредством любого способа, способного обеспечить получение микрочастиц в интервале размеров, приемлемом для применения в инъекционной композиции. Одним предпочтительным способом получения является способ, описанный в патенте США № 4389330. В данном способе активный агент растворяют или диспергируют в соответствующем растворителе. К среде, содержащей агент, добавляют материал полимерной матрицы в количестве относительно активного ингредиента, который предоставляет продукт, имеющий желательную загрузку активного агента. Необязательно, все из ингредиентов продукта в виде микрочастиц могут быть смешаны вместе в среде растворителя.
[0079] Растворители для соединений по изобретению и материала полимерной матрицы, которые могут использоваться при практической реализации настоящего изобретения, включают органические растворители, такие как ацетон; галогенированные углеводороды, такие как хлороформ, метиленхлорид и т.п.; ароматические углеводородные соединения; галогенированные ароматические углеводородные соединения; циклические простые эфиры; спирты, такие как бензиловый спирт; этилацетат; и т.п. В одном варианте осуществления растворитель для применения на практике настоящего изобретения может представлять собой смесь бензилового спирта и этилацетата. Дополнительную информацию по получению микрочастиц, применимых для изобретения, можно найти в публикации патентной заявки США № 2008/0069885, содержание которой включено здесь посредством ссылки во всей своей полноте.
[0080] Количество соединений по изобретению, включенных в микрочастицы, обычно находится в интервале от приблизительно 1 масс.% до приблизительно 90 масс.%, предпочтительно от 30 до 50 масс.%, более предпочтительно от 35 до 40 масс.%. Под массовыми % подразумевают части соединений по изобретению на общую массу микрочастицы.
[0081] Фармацевтическое депо может содержать фармацевтически приемлемый разбавитель или носитель, такой как смешивающийся с водой разбавитель или носитель.
[0082] Подробности по композиции пероральной системы доставки с осмотически контролируемым высвобождением можно найти в EP 1539115 (публикация патентной заявки США № 2009/0202631) и WO 2000/35419, содержание каждой из которых включено посредством ссылки во всей своей полноте.
[0083] "Терапевтически эффективное количество" представляет собой любое количество соединений по изобретению (например, такое, какое содержится в фармацевтическом депо), которое, когда его вводят субъекту, страдающему от заболевания или расстройства, является эффективным, чтобы вызвать снижение, ремиссию или регрессию заболевания или расстройства в течение периода времени, как предписано для лечения.
[0084] Дозировки, используемые при практической реализации настоящего изобретения, будут, несомненно, изменяться в зависимости, например, от конкретного заболевания или состояния, подлежащих лечению, конкретного применяемого соединения по изобретению, режима введения и желательной терапии. Если не указано иначе, количество соединения по изобретению для введения (либо вводимое как свободное основание или как солевая форма) относится к количеству соединения по изобретению в свободной основной форме или основано на этом количестве (т.е., расчет количества базируется на количестве свободного основания).
[0085] Соединения по изобретению могут вводиться посредством любого удовлетворительного пути, включающего пероральный, парентеральный (внутривенный, внутримышечный или подкожный) или трансдермальный, но предпочтительно их вводят перорально. В определенном варианте осуществления соединения по изобретению, например, в готовой лекарственной форме пролонгированного действия, предпочтительно вводят парентерально, например, посредством инъекции.
[0086] В целом, удовлетворительные результаты для способа I или любой из формул 7.1-7.25 или способа Ip или применения соединений по изобретению, как описано здесь прежде, например, для лечения комбинации заболеваний, такой как комбинация, по меньшей мере, депрессии, психоза, например, (1) психоза, например, шизофрении, у пациента, страдающего от депрессии; (2) депрессии у пациента, страдающего от психоза, например, шизофрении; (3) расстройств настроения, связанных с психозом, например, шизофренией или болезнью Паркинсона; и (4) нарушений сна, связанных с психозом, например, шизофренией, или болезнью Паркинсона, как изложено выше, указаны для получения при пероральном введении при дозировках порядка от приблизительно l мг до l00 мг однократно ежедневно, предпочтительно 2,5 мг-50 мг, например, 2,5 мг, 5 мг, l0 мг, 20 мг, 30 мг, 40 мг или 50 мг, однократно ежедневно, предпочтительно посредством перорального введения.
[0087] Удовлетворительные результаты для способа II или любого из 8.1-8.15, способа IIp или применения соединения по изобретению, как описано здесь прежде, например, для лечения нарушений сна, по отдельности указаны для получения при пероральном введении при дозировках порядка от приблизительно 2,5 мг-5 мг, например, 2,5 мг, 3 мг, 4 мг или 5 мг соединения по изобретению, в свободной или фармацевтически приемлемой солевой форме, однократно ежедневно, предпочтительно посредством перорального введения.
[0088] Удовлетворительные результаты для способа I-A или любого из 9.1-9.38 или способ Ip-A указаны для получения при менее чем l00 мг, предпочтительно менее чем 50 мг, например, менее чем 40 мг, менее чем 30 мг, менее чем 20 мг, менее чем l0 мг, менее чем 5 мг, менее чем 2,5 мг, однократно ежедневно. Удовлетворительные результаты для способа II-A или любого из 9.1-9.38 указаны для получения при менее чем 5 мг, предпочтительно менее чем 2,5 мг.
[0089] Для лечения расстройств, раскрытых здесь, где композицию пролонгированного действия применяют для достижения более длительной продолжительности действия, дозировки будут выше относительно композиции с более коротким действием, например, больше, чем 1-l00 мг, например, 25 мг, 50 мг, l00 мг, 500 мг, l000 мг, или больше, чем 1000 мг. Продолжительность действия соединений по изобретению может регулироваться посредством манипуляции с полимерной композицией, т.е. отношением полимер:лекарственное средство и размером микрочастиц. Там, где композиция по изобретению является композицией пролонгированного действия, введение посредством инъекции является предпочтительным.
[0090] Фармацевтически приемлемые соли соединений по изобретению могут быть синтезированы из исходного соединения, которое содержит основный или кислотный фрагмент, посредством общепринятых химических способов. Как правило, такие соли могут быть получены посредством взаимодействия свободных основных форм этих соединений со стехиометрическим количеством соответствующей кислоты в воде или в органическом растворителе, или в смеси двух; в целом, неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил, являются предпочтительными. Дополнительные подробности получения этих солей, например, толуолсульфоновой соли в аморфной или кристаллической форме, можно найти в PCT/US08/03340 и/или предварительной патентной заявке США № 61/036069.
[0091] Фармацевтические композиции, содержащие соединения по изобретению, могут быть получены с использованием общепринятых разбавителей или эксципиентов (пример включает, но не ограничен им, кунжутное масло) и методами, известными в области галеновых форм. Таким образом, пероральные дозированные формы могут включать таблетки, капсулы, растворы, суспензии и т.п.
Способы получения соединений по изобретению:
[0092] Соединения по изобретению, где Y представляет собой -C(H)(OH)-, могут быть получены посредством взаимодействия восстанавливающего агента с соединением формулы Q, где Y представляет собой -C(=О), как описано здесь прежде, причем исходное соединение формулы Q может быть получено, как описано с дополнительными подробностями в WO PCT/US08/03340 (WO 2008/112280); патентной заявке США серийный № 10/786935; патентах США №№ 6548493; 7238690; 6552017; 6713471; 7183282; U.S.RE39680 и U.S.RE39679, содержание которых включено посредством ссылки во всей полноте. Восстанавливающий агент может представлять собой гидрид металла, например, боргидрид натрия, цианоборгидрид натрия, алюмогидрид лития, гидрид алюминия, гидрид диизобутилалюминия, предпочтительно боргидрид натрия. Дополнительные реагенты для восстановления кетонов можно найти в Jerry March, Advanced Organic Chemistry, Reactions Mechanisms and Structures, p. 910-911, (1992, John Wiley & Sons, Inc.), Fourth Edition, содержание которой включено посредством ссылки.
[0093] Там, где X представляет собой -N(H)- и Y представляет собой -C(H)(OH), соединения по изобретению (например, формулы I) альтернативно получают посредством взаимодействия соединения формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)-, с восстанавливающими агентами, как описано выше, для восстановления кетоновой группы формулы Q, например, при использовании боргидрида натрия.
[0094] Выделения или очистки диастереомеров соединений по изобретению можно достичь посредством общепринятых способов, известных в области, например, посредством колоночной очистки, препаративной тонкослойной хроматографии, препаративной ВЭЖХ, кристаллизации, растирания, псевдодвижущихся слоев и т.п.
[0095] Соединения формулы III или любых из 4.1-4.27 или 4.28-4.30 могут быть получены посредством нескольких обычно применяемых способов этерификации, таких как алкоголиз ацилгалогенидов, ангидридов или активных сложных эфиров. Например, соединения формулы III, где R1 представляет собой -C(О)-алкил, могут быть получены посредством взаимодействия:
(a) L-C(О)-C1-21алкила, где L является уходящей группой, такой как группа галогена (например, хлора или брома), трифторметилсульфонилокси (-OSO2CF3), тозилокси (-О-S(О)2-C6H4-CH3), метилсульфонилокси (-О-S(О)2-CH3), 1H-бензо[d][l,2,3]триазол-l-илокси или сукцинимидилокси группа,
с
(b) соединением формулы I, где Y представляет собой -C(H)(OH),
предпочтительно в присутствии основания (например, диизопропиламина, триэтиламина или пиридина). Например, L-C(О)-C1-21алкил представляет собой ацетилгалогенид, деканоилгалогенид или гептаноилгалогенид, которые могут быть получены посредством взаимодействия HO-C(О)-C1-21алкила, например, с тионилхлоридом, P(X')3 или P(X')5, где X' представляет собой Cl или Br. Там, где L представляет собой тозилокси-C(О)-C1-21алкил или метилсульфонилокси-C(O)-C1-21алкил, эти соединения могут быть получены посредством взаимодействия HO-C(О)-C1-21алкила с тозилхлоридом или мезилхлоридом, предпочтительно в присутствии основания, такого как пиридин. Синтез соединения формулы II может быть обобщен в схеме реакции ниже:
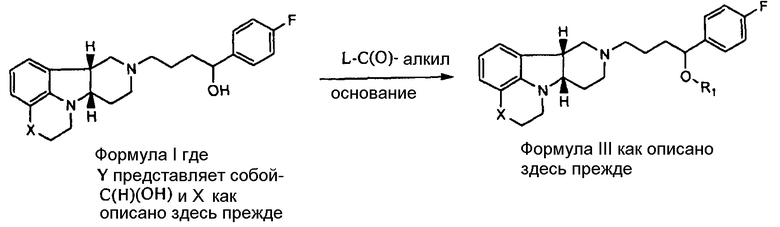
[0096] Альтернативно, синтез соединения формулы III может достигаться посредством взаимодействия HO-C(О)-C1-21алкила с (i) соединением формулы 1, где Y представляет собой -C(H)(OH), в присутствии основания, такого как DIEPA и NEt3, и (ii) дегидратирующим реагентом или реагентом сочетания, таким как тетрафторборат 2-фтор-l-этилпиридиния (FEP), гексафторфосфат тетрафторамидиния (TFFH) или гексафторфосфат l,l,3,3-бис(тетраметилен)хлорурония (PyCIU).
[0097] Соли соединений по изобретению могут быть получены, как описано сходным образом в патентах США №№ 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680; U.S. RE39679 и WO 2009/114181, содержание каждого из которых включено посредством ссылки во всей своей полноте.
ПРИМЕР 1
Синтез l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-ил)бутан-l-ола
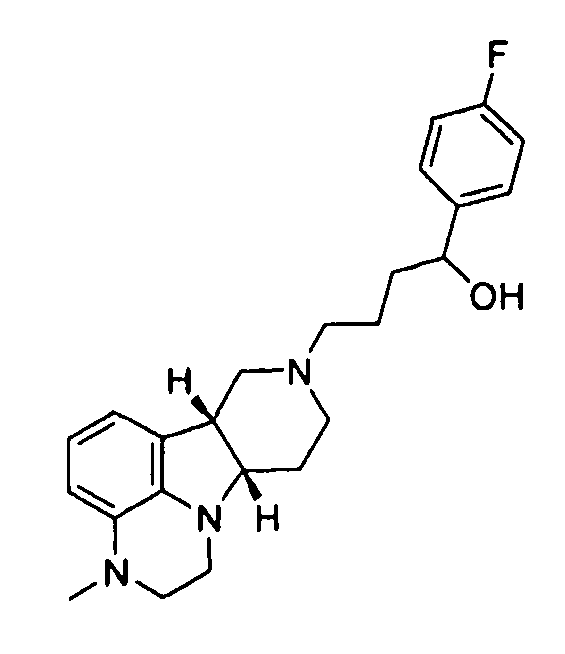
[0098] Тозилатную соль 1-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-она (8,58 г, 15,2 ммоль) растворяют в 150 мл метанола. Полученный коричневый раствор охлаждают до -10°C, и затем медленно добавляют NaBH4 (1,72 г, 45,5 ммоль). После завершения добавления, реакционную смесь перемешивают при комнатной температуре в течение часа, и затем реакцию гасят посредством добавления 20 мл воды. После удаления растворителя роторным испарением, остаток обрабатывают 50 мл водного раствора 1N NaOH и затем экстрагируют метиленхлоридом четыре раза. Объединенную органическую фазу промывают водой, сушат безводным сульфатом натрия и затем упаривают досуха в вакууме с получением 6 г белого пенообразного твердого вещества 97,8% чистотой и 99% выходом. МС (ЭРИ) m/z 396,1 [M+H]+.
[0099] Диастереомеры этого соединения разделяют посредством ВЭЖХ, используя CHIRALPAK® AY-H, 5 мк, 30×250 мм при комнатной температуре и элюируют смесью 10% этанола/90% гексана/0,1% диметилэтиламина. Пики детектируют при 230 нм с получением 98-99,9% ee диастереомера.
Пример 2:
Синтез l-(4-фторфенил)-4-((6bR,10aS)-2,3,6b,9,10,10a-гексагидро-1Н,7Н-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-ола
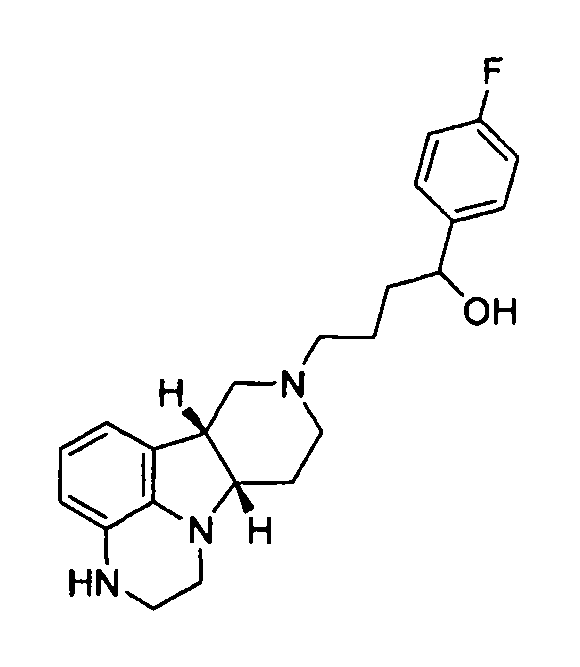
[0100] HCl соль l-(4-фторфенил)-4-((6bR,10aS)-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-она (500 мг, 1,20 ммоль) суспендируют в 20 мл метанола. NaBH4 (136 мг, 3,60 ммоль) медленно добавляют в суспензию при комнатной температуре. Суспензия изменяется на прозрачный раствор во время добавления NaBH4. Через час дополнительные 90 мг NaBH4 добавляют в смесь для сдвига реакции в сторону завершения. Реакционную смесь гасят посредством добавления 10 мл воды. После удаления растворителя роторным испарением, остаток обрабатывают 10 мл 1N NaOH водного раствора и затем экстрагируют метиленхлоридом три раза. Объединенную органическую фазу промывают водой, сушат безводным сульфатом натрия и затем упаривают досуха в вакууме с получением 434 мг грязно-белого пенообразного твердого вещества с 98% чистотой и 95% выходом. МС (ЭРИ) m/z 382,2 [M+H]+.
Пример 3:
Синтез l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-деканоата
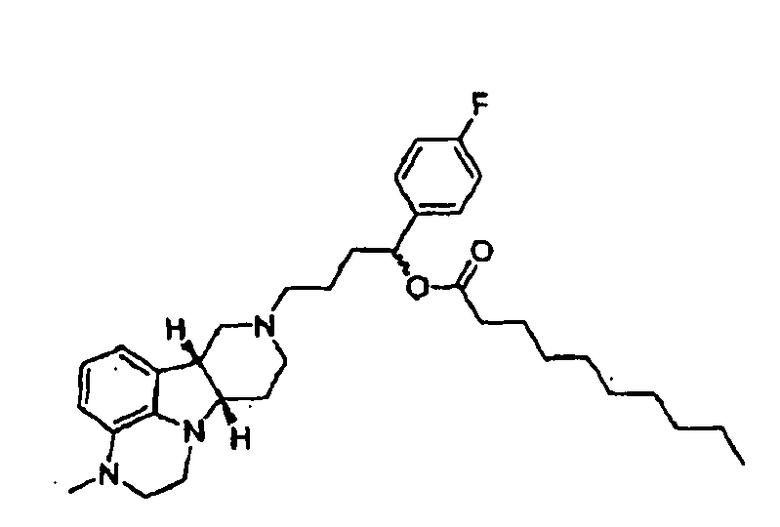
[0101] Сто семьдесят два мг декановой кислоты (1,0 эквивалент) растворяют в 1,0 мл дихлорметана (ДХМ) при комнатной температуре, и по каплям добавляют оксалилхлорид (1,0 эквивалент) с последующим добавлением 2 капель N,N-диметилформамида (ДМФА). Прозрачный раствор перемешивают в течение 1,0 ч с получением хлорида декановой кислоты, который используют непосредственно для следующей стадии.
[0102] Триста девяносто шесть мг 1-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-ил)бутан-l-ола (1,0 эквив.), полученного аналогично примеру 1, и 1,5 эквив. N,N-диметил-4-аминопиридина (DMAP) растворяют в 3,0 мл ДХМ при перемешивании. Свежеприготовленный раствор хлорида декановой кислоты медленно добавляют по каплям при комнатной температуре, и смесь перемешивают в течение дополнительных 2,0 часов до тех пор, пока тонкослойная хроматография (ТСХ) не покажет израсходование всего исходного материала. По завершении реакции, по каплям добавляют два миллилитра воды для гашения реакции и pH регулируют до >9 30% NaOH. Органический слой отделяют и концентрируют. Неочищенный продукт очищают колоночной хроматографией (этилацетат:метанол=5:1) с получением 160 мг свободного основания сложного эфира декановой кислоты в виде коричневого масла. Rt (UPLC-МС)=3,42 мин, ВРМС для C34H48FN3O2 (M++l) 550,1024.
Пример 4:
Синтез l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,l0,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-бутаноата
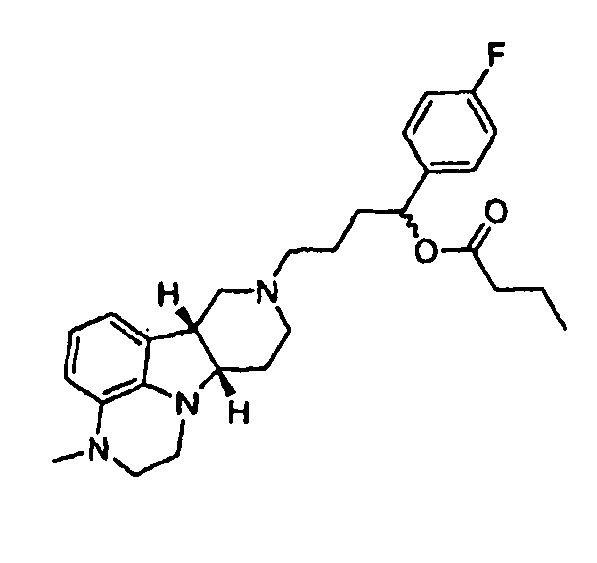
[0103] Это соединение синтезируют с использованием аналогичной методики, как описано в Примере 3 с получением свободного основания l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-бутаноата в виде коричневого масла. Rt (UPLC-МС)=2,54 мин, ВРМС для C28H36FN3O2 (M++1) 466,0489.
Пример 5: Исследование пероральной биодоступности и фармакокинетики разовой дозы на крысах Спрага-Доули
Животные, применяемые для исследования:
Штамм/Вид: Sprague Dawley (Crl:CD®(SD)BR) Крыса
Пол: Самцы и самки
Возраст при поступлении: 7-8 недель
Диапазон массы: 225-300 г
Количество животных в исследовании: 3 самца и 3 самки
Получение дозированной готовой лекарственной формы
[0104] Получают суспензию l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-ил)бутан-l-ола (т.е., соединение примера 1) с использованием деионизированной воды, содержащей 0,5% метилцеллюлозы. Суспензию получают, помещая требуемое количество метилцеллюлозы в деионизированной воде в стеклянный контейнер. Суспензию поддерживают при перемешивании с помощью магнитной мешалки до завершения суточного дозирования. Суспензии дозы получают в день дозирования для каждой группы тестируемого изделия.
[0105] Всего 3 самца и 3 самки крыс получают дозу в 30 мг/кг. Образцы крови отбирают при 0,30 мин, 1, 4 и 8 часов после дозирования. Образцы плазмы анализируют для определения плазменных концентраций соединения примера 1, соединения формулы Q, где X представляет собой -N(CH3)- и Y представляет собой -C(=О), и соединения формулы Q, где X представляет собой -N(H)- и Y представляет собой -C(=О). Фармакокинетический профиль определяют, используя WinNonlin.
[0106] Животных подвергают анестезии изофлураном, и цельную кровь (~0,5 мл/временная отметка) отбирают из глазного синуса, используя стеклянную капиллярную трубку в первый день обработки при следующих временных отметках: 0,30 мин, 1, 4 и 8 часов после дозирования. Цельную кровь (~0,5 мл) отбирают в охлажденные пластиковые пробирки, содержащие гепарин натрия, поставленные производителем. Пробирки для сбора крови перемешивают несколько раз, переворачивая вручную или осторожно встряхивая, и хранят на льду с водой до центрифугирования. Кровь центрифугируют (~2700×g, ~10 мин, ~5°C). Плазму переносят с помощью одноразовых пластиковых пипеток в пластиковые пузырьки и хранят при ~70°C до анализа. Результаты обобщены в таблице 2:
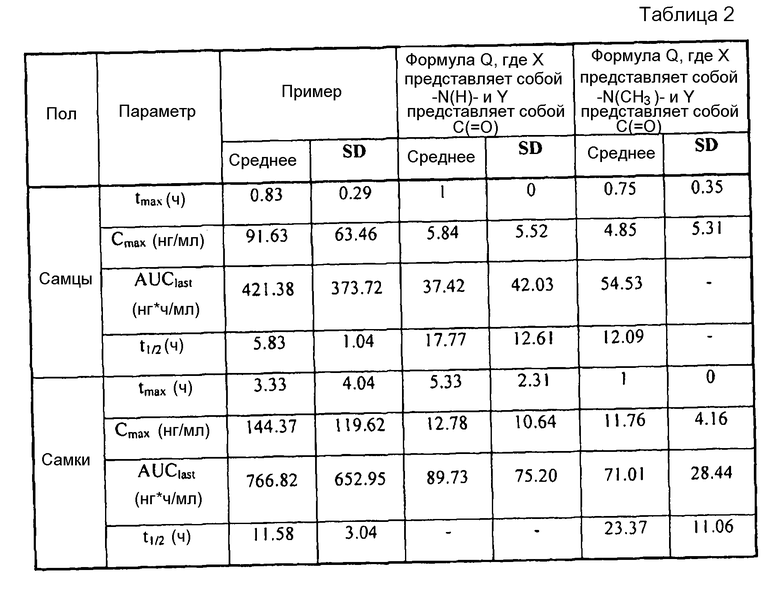
[0107] Как наблюдается в таблице 2, концентрация соединения примера 1 в плазме в целом снижается с течением времени, в то время как концентрация соединения формулы Q, где X представляет собой -N(CH3)- и Y представляет собой -C(=О), или X представляет собой -N(H)- и Y представляет собой -C(=О)-, поддерживается в течение 8 часов.
Пример 6: Активность SERT
[0108] Ингибирование активности транспорта обратного захвата серотонина (SERT) и связывания с SERT посредством l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-ил)бутан-l-ола измеряют, используя имипрамин в качестве стандарта, применяя способы, описанные в WO/2009/145900. Обнаружено, что соединение обладает сильной ингибирующей активностью по отношению к SERT, аналогично соответствующему кето-соединению Формулы Q.
[0109] Ингибирование обратного захвата серотонина измеряют в анализе на человеческих тромбоцитах, используя меченный тритием серотонин. Тромбоциты разбавляют в не содержащем Ca буфере KRH, содержащем 0,5 мМ ЭДТА, pH 7,4, и добавляют в контрольный и тестируемый образцы на 15 минут при комнатной температуре. Пробирки затем уравновешивают в водяной бане при 37°C перед добавлением субстрата. Реакцию останавливают посредством быстрой вакуумной фильтрации после 15-минутной инкубации. Определяют радиоактивность, осажденную на фильтрах, и сравнивают с контрольными значениями, чтобы подтвердить какие-либо взаимодействия тестируемого соединения с захватом серотонина. КТ (активность связывания меченного тритием серотонина) в данном анализе составляет 970 нМ. Vmax (скорость транспорта) составляет 25 пмоль/мин/мг белка. Тестируемое соединение имеет Ki, равное 1,5 микромолей. Положительный контроль, имипрамин, имеет Ki, равный 0,1 микромолей в данном анализе.
[0110] Ингибирование связывания с SERT измеряют с использованием меченного тритием циталопрама в качестве конкурента. Источником рецептора в данном анализе являются мембраны человеческих тромбоцитов. Радиолиганд представляет собой [3H] Циталопрам, N-Метил (70-87 Ки/ммоль). Конечная концентрация лиганда составляет [0,7 нМ]. Кломипрамин [1,0 мкМ] является неспецифическим детерминантом и положительным контролем у имипрамина. Реакции осуществляют в 50 мМ TRIS-HCl (pH 7,4), содержащем 120 мМ NaCl и 5 мМ KCl, при 25°C в течение 60 минут. Реакцию останавливают посредством быстрой вакуумной фильтрации через стекловолоконные фильтры. Определяют радиоактивность, уловленную фильтрами, используя жидкостную сцинтилляционную спектрометрию, и сравнивают ее с контрольными значениями, чтобы подтвердить какие-либо взаимодействия тестируемого соединения с участком связывания переносчика серотонина. KD (сродство связывания циталопрама) в данном анализе составляет 2,5 нМ и Вмакс (число рецептора) составляет 425 фмоль/мг белка. Тестируемое соединение имеет Ki, равное 71 нМ, по сравнению с Ki, равным 3 нМ для имипраминового положительного контроля.
Пример 7: Гидролиз примера 4 в цельной крови крысы
[00100] Скорость гидролиза l-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,9,l0,10a-гексагидро-lH,7H-пиридо[3',4':4,5]пирроло[l,2,3-de]хиноксалин-8-ил)бутан-l-бутаноата (или соединения примера 4) в ЭДТА-содержащей цельной крови крысы измеряют при инкубации образца с цельной кровью крысы при 37 градусах температуры в течение различного времени, с отбором образцов после каждого пятиминутного интервала, остающаяся концентрация исходного лекарственного средства и гидролизованного сложного эфира измеряют посредством ВЭЖХ-МС после экстракции крови ацетонитрилом. Результаты показаны в таблице 3 ниже:

Как можно видеть из таблицы 3, соединение примера 4 неуклонно расщепляется обратно в соединение примера 1 с очевидностью увеличения концентрации соединения примера 1 в крови в течение 90 минут.
Пример 8: Тест на DOI(DOI (R(-)-2,5-диметокси-4-йодамфетамин))-индуцированные судорожные движения головой у мышей
[0111] DOI является агонистом семейства 5-HT2 рецепторов серотонина. При введении мышам он продуцирует поведенческий профиль, ассоциированный с частыми судорожными движениями головой. Частота этих судорожных движений головой во время предварительно определенного периода времени может быть взята в качестве оценки агонизма по отношению к 5-HT2 рецептору в мозге. Напротив, этот поведенческий анализ может применяться для определения антагонизма к 5-HT2 рецептору в мозге посредством введения DOI вместе с антагонистом или без него и регистрации снижения DOI-индуцированных судорожных движений головой после введения антагониста.
[0112] Способ Darmani et al., Pharmacol Biochem Behav. (1990) 36:901-906 (содержание которой включено посредством ссылки во всей полноте) применяют с некоторыми модификациями. (±)-DOI HCl вводят инъекционно подкожно, и мышей немедленно помещают в общепринятую пластиковую клетку. Считают количество судорожных движений головой в течение 6 мин, начиная с 1 мин после введения DOI. Соединение примера 1 вводят перорально через 0,5 час перед инъекцией DOI. Результаты показаны на фигуре 1.
[0113] Как можно видеть на фигуре 1, пероральное введение соединения Примера 1 перед DOI значительно и дозозависимо снижало судорожные движения головой, что указывает на активность антагониста рецептора 5-HT2. IC50 соединения примера 1 для снижения DOI-индуцированных судорожных движений головой в данном анализе составляет 0,31 мг/кг.
Пример 9: Тест на латентность при спуске
[0114] Лекарственные средства, которые антагонизируют допаминовые рецепторы в мозге, замедляют моторное поведение и могут индуцировать каталептическое состояние. Эту активность можно оценить у мышей, используя простой тест на латентность при спуске. В этом тесте мышей держат за хвост, и передние лапки грызунов помещают на стержень, а их задние лапы помещают на рабочую поверхность стола. Время, затраченное для спуска передних лапок со стержня, затем измеряют. Максимально дают 2 минуты, и в течение этого времени животное забирают со стержня и возвращают в клетку для содержания. Соединение примера 1 или галоперидол (используемый в качестве положительного контроля) вводят перорально за 120 минут перед первым тестом. Тесты на латентность при спуске проводят при 120, 180, 240 и 360 минутах после введения соединения примера 1 или галоперидола. После каждого теста мышей возвращают в их клетки. Результаты показаны на Фигуре 2.
[0115] Как можно видеть на фигуре 2, пероральное введение соединения Примера 1 значительно и дозозависимо увеличивало латентность при спуске, что указывает на активность антагониста допаминового рецептора.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2743513C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ РАССТРОЙСТВА ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2017 |
|
RU2776800C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2780002C2 |
| НОВЫЕ СПОСОБЫ | 2014 |
|
RU2682658C1 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2733975C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2777366C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2785871C2 |
| НОВЫЕ КОМПОЗИЦИИ И СПОСОБЫ | 2018 |
|
RU2767410C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2728787C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2679142C2 |
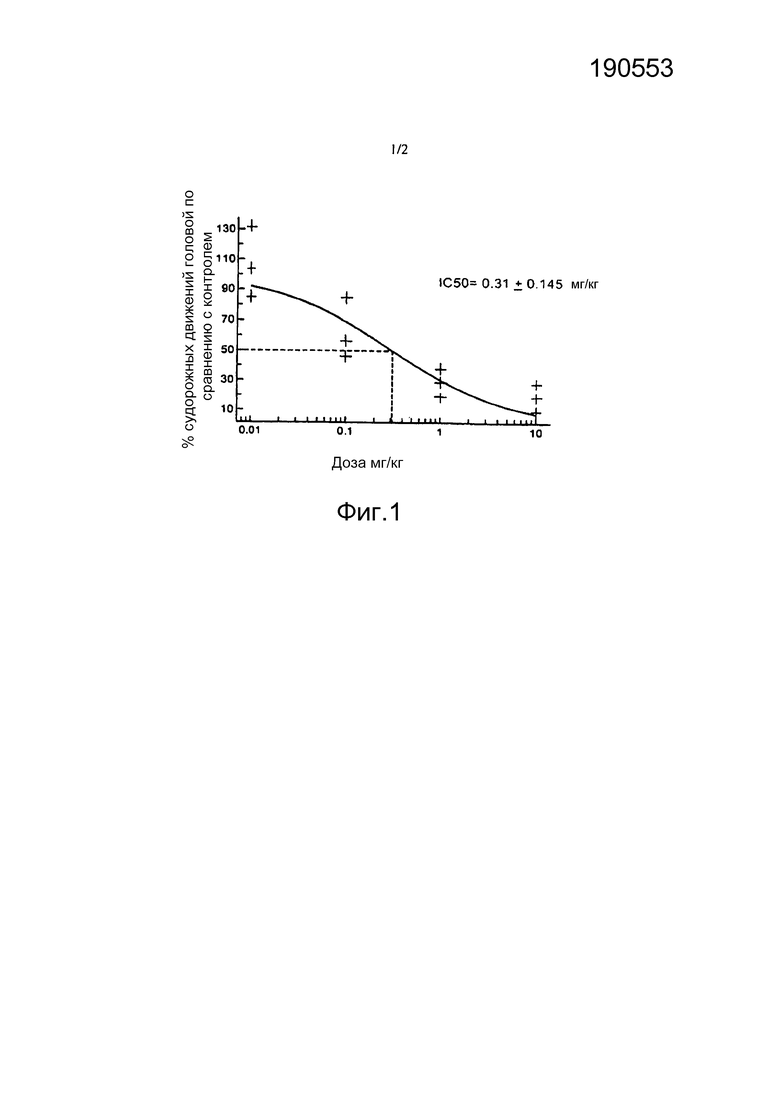
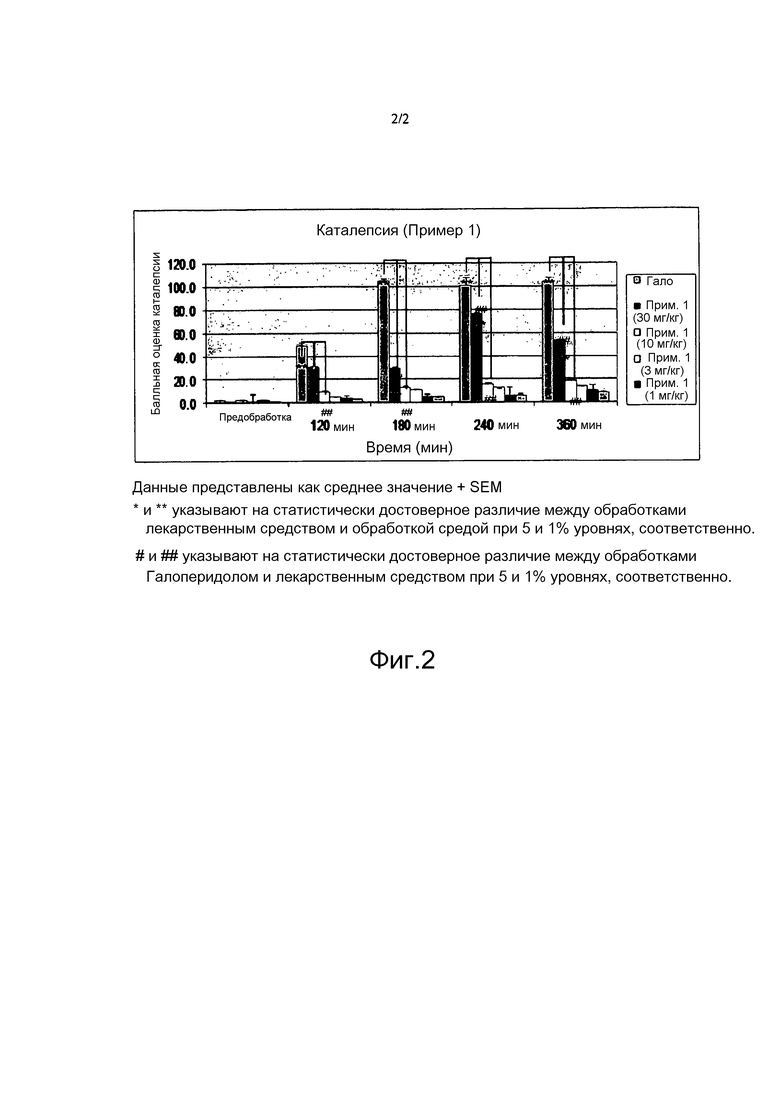
Изобретение относится к конкретным замещенным конденсированным с гетероциклом гамма-карболинам формулы I, где X представляет собой -N(H)- или -N(CH3) и Y представляет собой -С(Н)(ОН)-; и формулы III, где X представляет собой -N(CH3)-, -N(H)-; и R1 выбран из -С(О)-С1-5алкила, -С(O)-С6алкил, -С(О)-С7алкил и -С(О)-С9алкила, в свободной форме, в виде твердого вещества, в виде фармацевтически приемлемой соли и/или в по существу чистой форме, а также к фармацевтическим композициям на основе этих соединений и применению их при лечении заболеваний, в которые вовлечены рецептор 5-HT2A, переносчик серотонина (SERT) и/или пути, в которые вовлечены сигнальные системы рецептора допамина D2. 8 н. и 22 з.п. ф-лы, 2 ил., 2 табл., 9 пр.
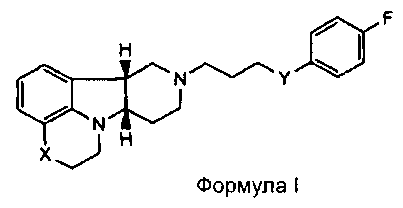
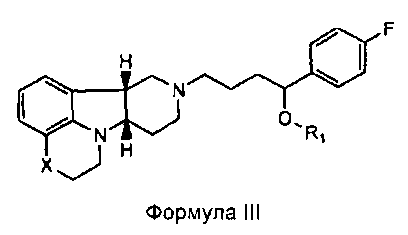
1. Соединение формулы I:
,
где
X представляет собой -N(H)- или -N(CH3) и Y представляет собой -С(Н)(ОН)-;
в свободной или солевой форме, при условии, что соединение не вырабатывается у млекопитающего посредством метаболизма соединения формулы Q:
 ,
,
где
X представляет собой -N(Н)- или -N(CH3)- и Y представляет собой -С(=O).
2. Соединение формулы I:
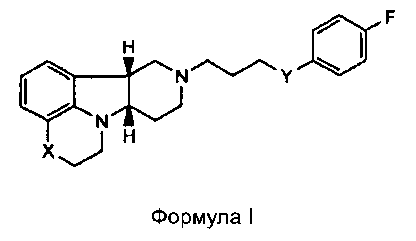 ,
,
в свободной или солевой форме, где
X представляет собой -N(Н)- или -N(CH3) и Y представляет собой -С(Н)(ОН)-;
в твердой форме.
3. Соединение по п. 1 или 2, где указанное соединение находится в солевой форме.
4. Соединение по п. 1 или 2, где соль выбирают из группы, состоящей из солей присоединения толуолосульфоновой, фумаровой и фосфорной кислоты.
5. Соединение по п. 1 или 2, где указанное соединение представляет собой
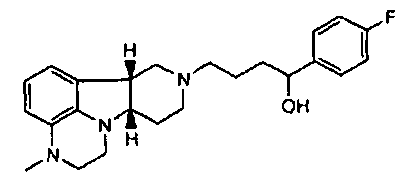
в свободной или солевой форме.
6. Соединение по п. 1 или 2, где указанное соединение не содержит по существу соединения формулы Q:
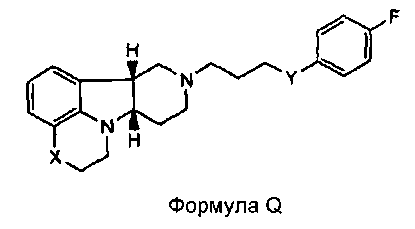 ,
,
где
X представляет собой -N(H)- или -N(CH3)- и Y представляет собой -С(=O).
7. Соединение по п. 1 или 2, где указанное соединение не
содержит по существу соединения формулы Q:
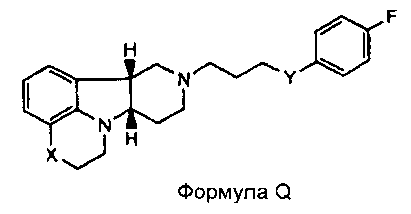 ,
,
где Y представляет собой -С(=O) и/или X представляет собой -N(CH3)-.
8. Соединение формулы III:
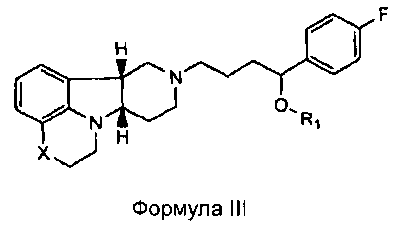 ,
,
где
X представляет собой -N(CH3)-, -N(Н)-; и
R1 выбран из -С(О)-С1-5алкила, -С(О)-С6алкила, -С(О)-С7алкила и -С(О)-С9алкила, и такое соединение гидролизуется с образованием остатка природной или неприродной, насыщенной или ненасыщенной жирной кислоты, например, соединение гидролизуется с образованием гидроксисоединения, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны,
в свободной или солевой форме.
9. Соединение по п. 8, где указанное соединение представляет собой соединение формулы III:
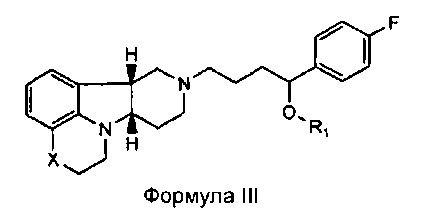 ,
,
где X представляет собой -N(CH3)- или -N(Н)- и R1 представляет собой -С(О)-С3алкил,
в свободной или солевой форме.
10. Соединение по п. 8, где соединение формулы III представляет собой
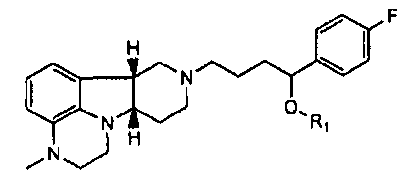
в свободной или солевой форме, где R1 выбран из -C(O)-C1-5алкила, -С(О)-С6алкила, -С(О)-С7алкила и -С(О)-С9алкила.
11. Соединение по п. 8, где соединение выбрано из группы, состоящей из
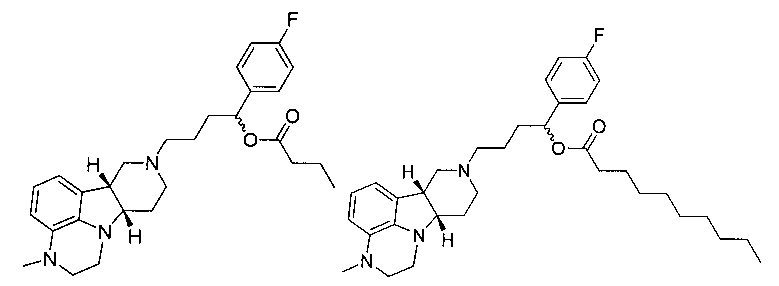
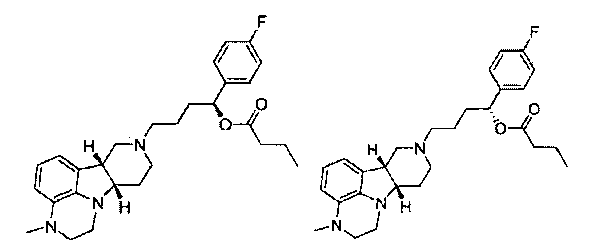
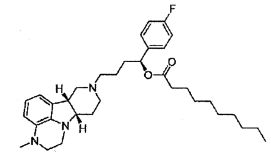 и
и 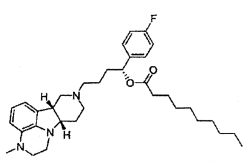
в свободной или солевой форме.
12. Фармацевтическая композиция, содержащая соединение по любому из пп. 1-11, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем для применения для лечения заболевания или нарушения опосредованного SERT.
13. Фармацевтическая композиция по п. 12, где соединение представляет собой соединение формулы III:
,
где
X представляет собой -N(CH3)-, -N(H)- или -O-; и
R1 выбран из -С(О)-С1-5алкила, -С(О)-С3алкила, -С(O)-С6алкил, -С(О)-С7алкил и -С(О)-С9алкила, и такое соединение
гидролизуется с образованием остатка природной или неприродной, насыщенной или ненасыщенной жирной кислоты, например, соединение гидролизуется с образованием гидроксисоединения, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны,
в свободной или солевой форме.
14. Фармацевтическая композиция, содержащая соединение по любому из пп. 1-11, в свободной или фармацевтически приемлемой солевой форме в полимерной матрице, где указанная композиция пригодна для лечения заболевания или нарушения опосредованного SERT.
15. Фармацевтическая композиция по п. 14, где полимерная матрица представляет собой биодеградируемую микросферу поли(d,l-лактид-со-гликолида).
16. Фармацевтическая композиция по п. 14 или 15 в смеси с фармацевтически приемлемым разбавителем или носителем.
17. Фармацевтическая композиция по любому из пп. 13-16, где указанную композицию составляют для контролируемого и/или замедленного высвобождения соединения по любому из пп. 1-11 в течение периода, равного приблизительно 30 дням, примерно 60 дням или примерно 90 дням.
18. Фармацевтическая композиция по любому из пп. 13-17, где указанную композицию составляют для введения посредством инъекции.
19. Композиция, содержащая (а) желатиновую капсулу, содержащую соединение по любому из пп. 1-11 или
фармацевтическую композицию по п. 12 или 13; (b) многослойную стенку, наложенную на желатиновую капсулу, содержащую, в порядке снаружи от капсулы: (i) барьерный слой, (ii) расширяемый слой и (iii) полупроницаемый слой; и (с) отверстие, образованное или образуемое через стенку, где указанная композиция пригодна для лечения заболевания или нарушения опосредованного SERT.
20. Композиция, содержащая (а) два или более слоя, указанные два или более слоя, содержащие первый слой и, второй слой, указанный первый слой содержит соединение по любому из пп. 1-11, в свободной или фармацевтически приемлемой солевой форме, или фармацевтическую композицию по любому из пп. 12-13, указанный второй слой содержит полимер; (b) внешнюю стенку, окружающую указанные два или более слоев; и (с) отверстие в указанной внешней стенке, где указанная композиция пригодна для лечения заболевания или нарушения опосредованного SERT.
21. Композиция по п. 19 или 20, где указанную композицию составляют для перорального введения.
22. Применение соединения по любому из пп. 1-11, в свободной или фармацевтически приемлемой солевой форме, или фармацевтической композиции по любому из пп. 12-21 в производстве лекарственного средства для лечения или профилактики расстройства центральной нервной системы.
23. Применение по п. 22, где указанное расстройство выбирают из группы, состоящей из ожирения, тревоги, депрессии (например, рефрактерной депрессии и MDD), психоза, шизофрении, нарушений сна (особенно, нарушений сна, связанных с шизофренией и другими психиатрическими и неврологическими заболеваниями), сексуальных
расстройств, мигрени, состояний, связанных с головной болью, социальных фобий, возбуждения при деменции (например, возбуждения при болезни Альцгеймера), возбуждения при аутизме и родственных аутических расстройствах, и желудочно-кишечных расстройств, таких как дисфункция моторики желудочно-кишечного тракта.
24. Применение по п. 22, где указанное расстройство представляет собой расстройство, в которое вовлечен путь серотонина 5-НТ2А, допамина D2 и/или переносчика обратного захвата серотонина (SERT).
25. Применение по п. 24, где указанное расстройство представляет собой расстройство, выбранное из следующих: (i) психоз, например, шизофрения, у пациента, страдающего от депрессии; (2) депрессия у пациента, страдающего от психоза, например, шизофрении; (3) расстройства настроения, связанные с психозом, например, шизофренией или болезнью Паркинсона; и (4) нарушения сна, связанные с психозом, например, шизофренией или болезнью Паркинсона.
26. Применение по п. 22, где расстройство представляет собой психоз.
27. Применение по п. 22, где расстройство представляет собой шизофрению.
28. Применение по п. 22, где расстройство представляет собой депрессию.
29. Применение по любому из пп. 22-28, где соединение представляет собой
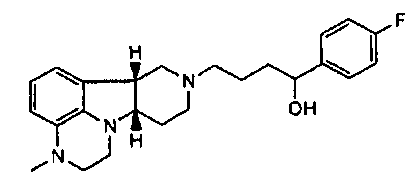
в свободной или фармацевтически приемлемой солевой форме.
30. Применение по любому из пп. 22-28, где соединение представляет собой
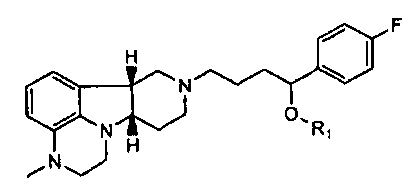 ,
,
где R1 выбран из -С(О)-С1-5алкила, -С(О)-С6алкила, -С(О)-С7алкила и -С(О)-С9алкила, и такое соединение гидролизуется с образованием остатка природной или неприродной, насыщенной или ненасыщенной жирной кислоты, например, соединение гидролизуется с образованием гидроксисоединения, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны,
в свободной или фармацевтически приемлемой солевой форме.
| WO 2009145900 A1 03.12.2009 | |||
| US2008280941 A1 13.11.2008 | |||
| Nantaka Khorana et al, y-Carbolines:Binding at 5-HT5a Serotonin Reseptors,Bioorganic & Medicinal Chemistry,2003,v.11,no.5-6,p.717-722 | |||
| US2004180875 A1 16.09.2004 | |||
| RU 2004104454 A 10.08.2005 | |||
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННЫХ ПОЛИЦИКЛИЧЕСКИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2167877C2 |

