Настоящее изобретение относится к способам получения соединений хлорированных C3-6 алканов высокой степени чистоты, таких как тетрахлорпропан, пентахлорпропан, пентахлорбутан и гептахлоргексан, а также к композициям, содержащим такие соединения.
Галогеноалканы находят применение в различных областях. Например, галогенуглеводороды широко используются в качестве хладагентов, вспенивающих агентов и пенообразователей. На протяжении второй половины двадцатого столетия использование хлорфторалканов экспоненциально возрастало вплоть до 1980-х годов, когда были высказаны опасения относительно их воздействия на окружающую среду, в частности в связи с истощением озонового слоя.
Впоследствии вместо хлорфторалканов стали использовать фторированные углеводороды, такие как перфторуглероды и гидрофторуглероды, хотя в последнее время были подняты экологические вопросы, связанные с использованием этого класса соединений, и в ЕС и в других странах были приняты законы, направленные на сокращение их использования.
Появляются и исследуются новые классы экологически безопасных галогенуглеводородов, которые в ряде случаев находят применение в ряде областей, особенно в качестве хладагентов в автомобильной и бытовой областях. Примеры таких соединений включают в себя 1,1,1,2-тетрафторэтан (R-134a), 2-хлор-3,3,3-трифторпропен (HFO-1233xf), 1,3,3,3-тетрафторпропен (HFO-1234ze), 3,3,3-трифторпропен (HFO-1243zf) и 2,3,3,3-тетрафторпропен (HFO-1234yf), 1,2,3,3,3-пентафторпропен (HFO-1225ye), 1-хлор-3,3,3-трифторпропен (HFO-1233zd), 3,3,4,4,4-пентафторбутен (HFO-1345zf), 1,1,1,4,4,4-гексафторбутен (HFO-1336mzz), 3,3,4,4,5,5,5-гептафторпентен (HFO1447fz), 2,4,4,4-тетрафторбут-1-ен (HFO-1354mfy) и 1,1,1,4,4,5,5,5-октафторпентен (HFO-1438mzz).
Хотя эти соединения, условно говоря, не являются сложными с химической точки зрения, их синтез в промышленном масштабе до требуемых уровней чистоты является сложной задачей. В синтезе таких соединений все чаще предлагают использование в качестве исходных материалов или промежуточных соединений хлорированных алканов или алкенов. Как правило, многие из способов, разработанных для получения таких соединений, включали добавление хлорированных алканов к фторированным олефинам. Однако было установлено, что такие способы не являются достаточно эффективными и приводят к образованию многочисленных примесей. Способы, разработанные в недавнее время, как правило, являются прямыми и включают в себя конверсию исходных материалов или исходного сырья хлорированных алканов или алкенов во фторированные целевые соединения с использованием фтористого водорода и катализаторов на основе переходных металлов, например катализаторов на основе хрома.
Было установлено, что когда хлорированное исходное сырье получают многостадийным способом, особенно если стадии такого способа связаны и непрерывно выполняются для достижения промышленно приемлемых объемов продукта, очень важна необходимость предотвращать образование недопустимых примесей при кумулятивных побочных реакциях на каждой стадии способа.
Чистота хлорированных исходных материалов имеет существенное влияние на успешность и результативность способов (особенно непрерывных способов) для получения желаемых фторированных продуктов. Присутствие определенных примесей приводит к побочным реакциям, что минимизирует выход целевого соединения. Удаление этих примесей даже при использовании стадий интенсивной дистилляции также является сложной задачей. Кроме того, присутствие определенных примесей уменьшает срок службы катализатора, например, путем загрязнения катализатора.
Соответственно, существует потребность в эффективных, надежных и высокоселективных способах получения хлорированных алканов высокой степени чистоты для применения в синтезе упомянутых выше фторированных соединений, а также других специальных соединений. В данной области техники предложено несколько способов получения очищенных хлорированных соединений, например, US6187978, US6313360, US2008/091053, US6552238, US6720466, JP2013-189402 и US2014/0171698.
Несмотря на эти достижения, проблемы в результате использования хлорированных соединений, полученных описанными выше способами, все же могут возникать. В частности, проблемы может вызывать присутствие примесей, особенно таких, которые нелегко отделить от целевых соединений (например, из-за сходных точек кипения) или которые снижают эффективность или срок службы катализаторов, используемых в последующих процессах.
Вследствие необходимости минимизации таких недостатков сохраняется спрос на хлорированные алкановые соединения высокой степени чистоты, а также на эффективные, надежные и высокоселективные способы получения таких соединений.
Таким образом, в соответствии с одним аспектом настоящего изобретения предложен способ получения хлорированного C3-6 алкана, включающий обеспечение реакционной смеси, содержащей алкен и четыреххлористый углерод в зоне основного алкилирования для образования хлорированного C3-6 алкана в реакционной смеси, и извлечение части реакционной смеси из зоны основного алкилирования, отличающийся тем, что:
а) концентрацию хлорированного C3-6 алкана в реакционной смеси в зоне основного алкилирования поддерживают на таком уровне, чтобы молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси, извлеченной из зоны алкилирования, не превышало 95:5, когда зона основного алкилирования работает в непрерывном режиме, или 99:1, когда зона основного алкилирования работает в периодическом режиме; и/или
б) реакционная смесь, извлеченная из зоны основного алкилирования, дополнительно содержит алкен, и реакционную смесь подвергают стадии деалкенирования, на которой по меньшей мере приблизительно 50% или более по массе алкена, присутствующего в реакционной смеси, выделяют из нее и по меньшей мере приблизительно 50% выделенного алкена направляют обратно в реакционную смесь, находящуюся в зоне основного алкилирования; и/или
в) реакционная смесь, присутствующая в зоне основного алкилирования и извлеченная из зоны основного алкилирования, дополнительно содержит катализатор, и реакционную смесь, извлеченную из зоны основного алкилирования, подвергают стадии водной обработки, на которой реакционную смесь приводят в контакт с водной средой в зоне водной обработки с образованием двухфазной смеси, и из двухфазной смеси выделяют органическую фазу, содержащую катализатор.
Способы в настоящем изобретении сосредоточены вокруг высокоселективной реакции теломеризации, которая происходит частично или полностью в зоне основного алкилирования. В этой реакции четыреххлористый углерод подвергают взаимодействию с алкеном с образованием хлорированного C3-6 алкана. В то время как реакции теломеризации для получения хлорированных C3-6 алканов известны в данной области техники, одной из проблем, связанных с такими способами, является получение нежелательных примесей; остается потребность в способе получения хлорированных C3-6 алканов высокой степени чистоты в промышленных объемах и в идеале на непрерывной основе.
Было неожиданно и преимущественно установлено, что проведение одной, некоторых или всех указанных выше стадий а) - в) при получении хлорированного C3-6 алкана из алкена и четыреххлористого углерода, повышает эффективность (включая снижение потребления энергии) и/или минимизирует образование примесей, которые в противном случае может быть трудно удалить из целевого хлорированного C3-6 алкана, и/или которые могут представлять собой проблему в последующих реакциях, в которых может использоваться хлорированный C3-6 алкан. Кроме того, неожиданно, способы согласно настоящему изобретению сочетают эти преимущества с высокой селективностью и высоким выходом.
Реакционную смесь получают путем приведения в контакт алкена и четыреххлористого углерода. Это может происходить в зоне основного алкилирования, например, за счет поступления алкена и четыреххлористого углерода в эту зону. Дополнительно или альтернативно, алкен могут приводить в контакт с четыреххлористым углеродом в зоне перед зоной основного алкилирования и затем подавать в зону основного алкилирования.
В вариантах осуществления изобретения молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси контролируют в определенных числовых пределах. Как будет понятно специалистам в данной области, в подобных вариантах осуществления, в то время как контроль способа характеризуется здесь с точки зрения молярного соотношения между исходным четыреххлористым углеродом и продуктом хлорированного C3-6 алкана, он также может рассматриваться как контроль конверсии исходного материала в продукт – таким образом, молярное соотношение исходного материала к продукту, составляющее 95:5, соответствует конверсии 5%. Авторы изобретения обнаружили, что ограничение конверсии исходного материала, как описано выше, сводит к минимуму образование нежелательных примесей. Кроме того, когда приводится ссылка на молярное соотношение исходного материала к продукту, превышающее заданную величину, это означает более высокую степень конверсии исходного материала в продукт, то есть такую, что пропорция продукта увеличивалась, в то время как пропорция исходного материала уменьшалась.
Например, в вариантах осуществления изобретения может быть задействована зона первичного алкилирования, предшествующая зоне основного алкилирования. Реакционная смесь может быть образована путем подачи четыреххлористого углерода и алкена в зону первичного алкилирования с образованием реакционной смеси, которую затем подают в зону основного алкилирования. В таком варианте осуществления частичная конверсия четыреххлористого углерода в целевой хлорированный C3-6 алкан может происходить в зоне первичного алкилирования так, что этот алкан образуется и содержится в реакционной смеси, подаваемой в зону основного алкилирования наряду с четыреххлористым углеродом. В дополнительных или альтернативных вариантах осуществления количество алкена, подаваемого в зону первичного алкилирования, может быть ограничено для замедления конверсии четыреххлористого углерода в целевой хлорированный C3-6 алкан в зоне первичного алкилирования так, что реакционная смесь, подаваемая в зону основного алкилирования из нее, содержит четыреххлористый углерод и хлорированный C3-6 алкан, но содержит мало или по существу не содержит алкен.
Алкен и четыреххлористый углерод, используемые в способах согласно настоящему изобретению, могут взаимодействовать в зоне (например, в зоне первичного алкилирования или в зоне основного алкилирования) путем подачи в эту зону с использованием любого метода или оборудования, известных специалистам в данной области, например, с помощью дисперсионных устройств, таких как погружная трубка/и, сопло/а, струйные насосы, устройства статического смешивания и/или разбрызгиватель/и. В таких вариантах осуществления подача алкена и/или четыреххлористого углерода может быть непрерывной или периодической. Алкен, подаваемый в качестве сырья в зону, в которой формируется реакционная смесь, может находиться в жидкой и/или газообразной форме. Аналогично, четыреххлористый углерод может находиться в жидкой и/или газообразной форме.
В вариантах осуществления настоящего изобретения реакционная смесь (содержащая четыреххлористый углерод, продукт хлорированного C3-6 алкана и необязательно катализатор и/или непрореагировавший алкен), присутствующая в зоне основного алкилирования (и/или любой другой зоне алкилирования, которая может быть задействована), может быть гомогенной, то есть находиться в одной фазе, например жидкой или газообразной. Это может быть достигнуто даже тогда, когда один из компонентов реакционной смеси вводится в систему в другой фазе, отличной от фазы других компонентов. Например, в вариантах осуществления газообразный алкен может взаимодействовать с жидким четыреххлористым углеродом, что вызывает растворение алкена, в результате чего образуется гомогенная реакционная смесь в жидкой фазе. Альтернативно, реакционная смесь может быть гетерогенной.
Таким образом, в соответствии с одним аспектом настоящего изобретения предложен способ получения хлорированного C3-6 алкана, включающий обеспечение реакционной смеси, содержащей алкен и четыреххлористый углерод в зоне первичного алкилирования для образования хлорированного C3-6 алкана в реакционной смеси, извлечение части реакционной смеси из зоны первичного алкилирования, подача извлеченной части реакционной смеси в основную реакционную зону и извлечение части реакционной смеси из основной реакционной зоны, где молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси, извлеченной из зоны основного алкилирования, превышает молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси, извлеченной из зоны первичного алкилирования.
Алкен, используемый в способах согласно настоящему изобретению, может представлять собой C2-5 алкен, например этен (то есть C2 алкен), пропен, бутен или пентен. Алкен может быть галогенированным и нет, например хлорированным и/или замещенным. В механизмах, в которых происходит хлорирование алкена, он предпочтительно содержит 1, 2, 3, 4 или 5 атомов хлора. В вариантах осуществления настоящего изобретения хлоралкен имеет общую формулу: CHaXb=R, где a равно 1 или 2, b равно 0 или 1, X представляет собой галоген (например, хлор), а R представляет собой замещенный или незамещенный C1-4 алкил.
Примеры материалов алкенов, которые могут быть использованы в способах согласно настоящему изобретению, включают этен, винилхлорид, пропен, 2-хлорпропен, 3-хлорпропен, 2,3,3,3-тетрахлорпропен, 1,1-дихлорэтен, трихлорэтен, хлорфторэтен 1,2-дихлорэтен, 1,1-дихлордифторэтен, 1-хлорпропен, 1-хлорбутен и/или любой из других алкенов, описанных в патентах US5902914 и EP131561, содержание которых включено в качестве ссылки.
Четыреххлористый углерод и алкен в качестве исходных материалов, используемых в способах согласно настоящему изобретению, могут иметь высокую степень чистоты, например, один или оба из этих материалов могут обладать чистотой по меньшей мере приблизительно 95%, чистотой по меньшей мере приблизительно 97%, чистотой по меньшей мере приблизительно 99%, чистотой по меньшей мере приблизительно 99,5%, чистотой по меньшей мере приблизительно 99,7% или чистотой по меньшей мере приблизительно 99,9%.
В вариантах осуществления настоящего изобретения исходный материал четыреххлористого углерода содержит менее чем приблизительно 2000 млн-1, менее чем приблизительно 1000 млн-1, менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1, менее чем приблизительно 100 млн-1, менее чем приблизительно 50 млн-1 или менее чем приблизительно 20 млн-1 бромидов или бромированных органических соединений.
Дополнительно или альтернативно, исходный материал четыреххлористого углерода может иметь содержание влаги приблизительно 200 млн-1 или менее, приблизительно 100 млн-1 или менее, приблизительно 50 млн-1 или менее или приблизительно 35 млн-1 или менее.
В способах согласно настоящему изобретению четыреххлористый углерод предпочтительно используют в качестве исходного галогеноалкана. Однако в альтернативных вариантах осуществления настоящего изобретения могут использоваться исходное сырье галогеналканов, отличных от четыреххлористого углерода, например 1,1-дихлорметан, 1,1,1-трихлорэтан, дихлорфторметан, 1,1,1-трихлортрифторэтан, 1,1,2-трихлортрифторэтан, тетрахлорэтан, пентахлорэтан, гексахлорэтан и/или любой другой хлоралкан, описанный в патенте US5902914, содержание которого включено в настоящее описание.
Источник четыреххлористого углерода может быть расположен на том же участке, что и устройство для осуществления способов согласно настоящему изобретению. В вариантах осуществления источник четыреххлористого углерода может быть расположен рядом с хлорщелочной установкой, например, с установкой мембранного электролиза, хлор высокой степени чистоты из которой может использоваться для получения четыреххлористого углерода. Участок может также содержать установки для производства эпихлоргидрина (например, из глицеринового сырья), глицидола и/или эпоксидной смолы, так что газообразный хлористый водород, получаемый в качестве побочного продукта на любых связанных стадиях или в процессах, также эффективно используется. Таким образом, для наиболее экономически выгодного использования хлорщелочной установки предусмотрена комплексная установка с установками для проведения реакций с хлором и улавливания/повторного использования хлористого водорода.
Реакционная смесь может извлекаться из зоны основного алкилирования (и/или, если она задействована, из зоны первичного алкилирования) на непрерывной или периодической основе. Во избежание сомнений, отсылка в настоящей заявке на непрерывное извлечение материала из зон, задействованных в способе согласно настоящему изобретению, не должна восприниматься буквально. Специалист в данной области должен понимать, что в таких вариантах осуществления материал может удаляться по существу непрерывно, в то время как рассматриваемая зона находится в рабочем состоянии и, если ее целью является осуществление реакции стационарного состояния (например, алкилирования), как только реакционная смесь в ней достигнет требуемого стационарного состояния.
Одним из преимуществ настоящего изобретения является то, что присутствие определенных примесей, обычно содержащихся в коммерчески поставляемом алкене (таких как некоторые органические примеси, например в виде спиртов, простых эфиров, сложных эфиров и альдегидов), можно нивелировать и/или они могут быть удалены с использованием описанных в данном документе стадий способа. Это преимущество особенно полезно в случаях, когда алкеном является этен; этен может быть получен из биоэтанола, из этанола или из неочищенной нефти.
Дополнительным преимуществом способов согласно настоящему изобретению является то, что I) непрерывное получение хлорированного алкана и II) по существу полное использование исходного материала алкена может быть достигнуто без выхода алкена в систему отходящего газа.
Хлорированный C3-6 алкан может представлять собой хлорпропан, хлорбутан, хлорпентан или хлоргексан. Хлорированный C3-6 алкан может содержать 4, 5, 6, 7, 8 или более атомов хлора. Примеры соединений хлорированных C3-6 алканов, которые могут быть получены с высокой степенью чистоты в соответствии со способами согласно настоящему изобретению, включают тетрахлорпропан, например 1,1,1,3-тетрахлорпропан, тетрахлорбутан, гексахлорбутан, гептахлорбутан, октахлорбутан, пентахлорпропан, пентахлорбутан, например 1,1,1,3,3-пентахлорбутан, и гептахлоргексан.
Одним из преимуществ способов согласно настоящему изобретению является то, что они могут обеспечить получение целевого хлорированного C3-6 алкана с высокой степенью изомерной селективности. Таким образом, в таких вариантах осуществления изобретения продукт хлорированного C3-6 алкана получают с изомерной селективностью по меньшей мере приблизительно 95%, по меньшей мере приблизительно 97%, по меньшей мере приблизительно 98%, по меньшей мере приблизительно 99%, по меньшей мере приблизительно 99,5%, по меньшей мере приблизительно 99,7%, по меньшей мере приблизительно 99,8% или по меньшей мере приблизительно 99,9%.
Реакция алкилирования, осуществляемая в способе согласно настоящему изобретению для получения хлорированного C3-6 алкана, может быть ускорена с использованием катализатора. Используемый здесь термин «катализатор» охватывает не только применение одного соединения или материала, обладающего каталитическим действием, например твердого металла или соли металла, но каталитической системы, которая может дополнительно содержать каталитический материал и сокатализатор или промотор, такой как лиганд.
Может быть использован любой катализатор, применение которого для образования хлорированного C3-6 алкана из четыреххлористого углерода известно специалистам в данной области техники.
В вариантах осуществления изобретения катализатор является металлическим. Может быть использован любой металл, который может функционировать в качестве катализатора в реакции алкилирования согласно настоящему изобретению, включая, но не ограничиваясь указанными, медь и/или железо. Металлический катализатор может присутствовать в твердой форме (например, в случае меди или железа, в форме частиц (например, порошка или опилок), проволоки и/или сетки или тому подобного) и/или в форме соли, в которой металл может находиться в любой степени окисления (например, одновалентной соли меди, такой как хлорид меди, бромид меди, цианид меди, сульфат меди, фенил меди и/или соли железа (II) и/или (III), такой как хлорид железа (II) и хлорид железа (III)).
Если в способах согласно настоящему изобретению в качестве катализаторов используют соли металлов, они могут быть добавлены в зону/ы алкилирования и/или образовываться в ней/них in situ. В последнем случае твердый металл может быть добавлен в зону/ы алкилирования и благодаря условиям в ней/них может быть образована соль. Например, если твердое железо добавляют в реакционную смесь для хлорирования, присутствующий хлор может сочетаться с элементарным железом с образованием хлорида железа (II) и хлорида железа (III) in situ. В том случае, когда металлические соли образуются in situ, поддержание заданного уровня элементарного металлического катализатора в реакционной смеси (например, избыток элементарного металла по сравнению с уровнем соли/ей металла и/или лиганда) тем не менее может быть желательным, и, таким образом, дополнительный элементарный металлический катализатор может добавляться по мере протекания реакции либо непрерывно, либо периодически.
Как упомянуто выше, в вариантах осуществления настоящего изобретения катализатор может также содержать лиганд, предпочтительно органический лиганд, который может образовывать комплекс с металлическим катализатором. Подходящие лиганды включают в себя амины, нитриты, амиды, фосфаты и фосфиты. В вариантах осуществления изобретения используемый лиганд представляет собой алкилфосфат, такой как триметилфосфат, триэтилфосфат, трибутилфосфат и трифенилфосфат.
Дополнительные металлические катализаторы и лиганды известны специалистам в данной области и описаны в существующем уровне техники, например в патенте US6187978, содержание которого включено в настоящее описание в качестве ссылки.
Компоненты каталитической системы, если они используются, могут непрерывно или периодически подаваться в зону/ы алкилирования (например, в зону основного алкилирования и/или, если она используется, в зону первичного алкилирования). Дополнительно или альтернативно, они могут быть введены в зону/ы алкилирования (например, в зону основного алкилирования и/или, если она используется, в зону первичного алкилирования) до и/или в начале реакции алкилирования.
Дополнительно или альтернативно, катализатор (или компоненты катализатора, например лиганд) могут подаваться в зону/ы алкилирования (например, в зону основного алкилирования или, если она используется, в зону первичного алкилирования) вместе с другими компонентами реакционной смеси, например с сырьевым четыреххлористым углеродом и/или этеном.
В вариантах осуществления изобретения, в которых катализатор содержит металлический катализатор и промотор, такой как лиганд, молярное соотношение промотора к металлическому катализатору в реакционной смеси, присутствующей в зоне основного алкилирования, и/или, если она используется, в зоне первичного алкилирования, поддерживают в соотношении более 1:1, более предпочтительно в соотношении более 2:1, 5:1 или 10:1.
Твердый металлический катализатор может быть добавлен в реакционную смесь в зоне первичного алкилирования, если она используется, и/или в зоне основного алкилирования. В вариантах осуществления изобретения твердый металлический катализатор добавляют в зону первичного алкилирования, если она используется, и/или в зону основного алкилирования в количествах, поддерживающих уровень от приблизительно 0,1 до 4%, от приблизительно 0,5 до приблизительно 3% или от приблизительно 1 до 2% от массы реакционной смеси.
Дополнительно или альтернативно, в случае применения металлических катализаторов, их добавляют для установления содержания растворенного металла приблизительно 0,1%, приблизительно 0,15% или приблизительно 0,2-1,0%, приблизительно 0,5% или приблизительно 0,3% от массы реакционной смеси.
В вариантах осуществления изобретения, в которых используемая каталитическая система содержит металлический катализатор и промотор, металлический катализатор и промотор могут быть добавлены к реакционной смеси одновременно и/или в одной и той же части устройства, например в зоне первичного алкилирования (если она используется) и/или в зоне основного алкилирования.
Альтернативно, металлический катализатор и промотор могут быть добавлены в разных зонах устройства, или последовательно, или раздельно. Например, твердый металлический катализатор может быть добавлен в зону первичного алкилирования с промотором, подаваемым в эту зону из цикла рециркуляции, к которому также может быть добавлен дополнительный свежий промотор.
В вариантах осуществления изобретения зоны первичного и/или основного алкилирования работают при атмосферном или превышающем атмосферное давлении, то есть при давлении более чем приблизительно 100 кПа, более чем приблизительно 200 кПа, более чем приблизительно 300 кПа, более чем приблизительно 400 кПа, более чем приблизительно 500 кПа, более чем приблизительно 600 кПа, более чем приблизительно 700 кПа или более чем приблизительно 800 кПа. Обычно давление в зонах первичного и/или основного алкилирования составляет или поддерживается ниже чем приблизительно 2000 кПа, приблизительно 1700 кПа, приблизительно 1500 кПа, приблизительно 1300 кПа, приблизительно 1200 кПа или приблизительно 1000 кПа.
Дополнительно или альтернативно, в вариантах осуществления изобретения зоны первичного и/или основного алкилирования работают при повышенных температурах, то есть температурах, равных или превышающих приблизительно 30°С, приблизительно 40°С, приблизительно 50°С, приблизительно 60°С , приблизительно 70°С, приблизительно 80°С, приблизительно 90°С или приблизительно 100°С. Обычно зоны первичного и/или основного алкилирования работают при температурах, равных или составляющих ниже чем приблизительно 200°С, приблизительно 180°С, приблизительно 160°С, приблизительно 140°С, приблизительно 130°С, приблизительно 120°С или приблизительно 115°С.
Было обнаружено, что использование температур и давлений в этих пределах в сочетании с другими особенностями способов согласно настоящему изобретению является преимущественным для максимизации выходов и/или селективности целевого хлорированного C3-6 алкана и при этом сведения к минимуму образования проблемных побочных продуктов.
В способах согласно изобретению может быть задействовано несколько зон алкилирования. Может быть задействовано любое количество зон алкилирования, например 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 или более. В вариантах осуществления, в которых используют множество зон первичного и/или основного алкилирования, может быть любое количество (например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 или более) зон первичного и/или основного алкилирования.
Во избежание сомнений, в тех случаях, когда речь идет о свойствах зоны алкилирования (первичного и/или основного), например, ее рабочих условиях, способах ее работы, ее свойствах и т. д., в части вариантов осуществления настоящего изобретения, которые включают в себя несколько зон первичного и/или основного алкилирования, одна, некоторые или все из этих зон могут демонстрировать рассматриваемое/ые свойство/а. Например, если для краткости ссылка делается на зону основного алкилирования, имеющую заданную рабочую температуру, в части вариантов осуществления, включающих в себя несколько зон основного алкилирования, это следует рассматривать как ссылку на то, что одна, некоторые или все из этих зон основного алкилирования работают при указанной температуре.
В устройствах, в которых задействованы несколько зон первичного и/или основного алкилирования, эти зоны алкилирования могут эксплуатироваться параллельно и/или последовательно.
В устройствах, в которых задействованы зоны первичного и основного алкилирования, реакция алкилирования между алкеном и четыреххлористым углеродом может контролироваться для предотвращения ее продолжения, превышающего определенную степень завершенности в зоне первичного алкилирования, например так, чтобы молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси, извлеченной из зоны первичного алкилирования и/или подаваемой в зону основного алкилирования, не превышало 85:15, 90:10, 93:7 или 95:5, хотя это не является существенным. Дополнительно или альтернативно, реакция может протекать до относительно продвинутой стадии завершения, так что молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси, извлеченной из зоны первичного алкилирования и/или подаваемой в зону основного алкилирования, составляет больше 50:50, 60:40, 70:30, 75:25 или 80:20.
Контроль за ходом реакции алкилирования в зоне первичного алкилирования может осуществляться путем установления условий реакции, которые не способствуют полной конверсии четыреххлористого углерода в 1,1,1,3-тетрахлорпропан. Дополнительно или альтернативно, контроль за ходом реакции алкилирования в зонах первичного алкилирования может осуществляться путем тщательного выбора времени пребывания реакционной смеси в зонах первичного алкилирования, например, приблизительно от 20 до 300 минут, приблизительно от 40 до 250 минут, от приблизительно 60 до приблизительно 200 минут или от приблизительно 90 до приблизительно 180 минут. В вариантах осуществления изобретения молярное соотношение может контролироваться путем ограничения количества алкена, подаваемого в зоны первичного и/или основного алкилирования. Например, молярное соотношение четыреххлористого углерода к алкену, подаваемых в зоны первичного и/или основного алкилирования, может находиться в диапазоне от приблизительно 50:50 до приблизительно 55:45, приблизительно 60:40, приблизительно 65:35, приблизительно 70:30, приблизительно 75:25, приблизительно 80:20, приблизительно 85:15 или приблизительно 90:10.
В вариантах осуществления, где задействованы зоны первичного и основного алкилирования, основная часть хлорированного C3-6 алкана может быть получена в зоне первичного алкилирования. В таких вариантах осуществления доля C3-6 алкана, полученного в основной реакционной зоне, может быть значительно ниже, например, так, что молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси увеличивается на от 1 до 10, от 2 до 8 или от 3 до 5.
Например, если молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси, извлеченной из зоны первичного алкилирования и подаваемой в зону основного алкилирования, составляет 90:10, молярное соотношение может быть увеличено на 2, 3 или 5 в зоне основного алкилирования, так, что молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду, присутствующих в смеси, извлеченной из зоны основного алкилирования, может составлять 92:8, 93:7 или 95:5.
Однако результативность способов согласно настоящему изобретению не зависит от происхождения основной конверсии четыреххлористого углерода в целевой хлорированный C3-6 алкан в первичной реакционной зоне. Таким образом, в альтернативных вариантах осуществления степень конверсии четыреххлористого углерода в целевой хлорированный C3-6 алкан может быть сбалансирована между зонами первичного и основного алкилирования или может быть больше в зоне основного алкилирования по сравнению с зоной первичного алкилирования.
Реакционная смесь затем может быть отобрана из зоны первичного алкилирования (непрерывно или периодически) и подана в зону основного алкилирования, в которой доля оставшегося четыреххлористого углерода, присутствующего в реакционной смеси, переходит в целевой хлорированный C3-6 алкан. В таких вариантах осуществления любой непрореагировавший исходный материал алкена, присутствующий в реакционной смеси, может преимущественно быть использован полностью (или по меньшей мере почти полностью).
В способах согласно изобретению зоны первичного и основного алкилирования, если они используются, могут эксплуатироваться в различных условиях. Зона основного алкилирования может эксплуатироваться при большем давлении, чем зона/ы первичного алкилирования, например, под давлением, которое составляет по меньшей мере приблизительно на 10 кПа, приблизительно на 20 кПа больше, приблизительно на 50 кПа больше, приблизительно на 100 кПа больше, приблизительно на 150 кПа больше, приблизительно на 200 кПа больше, приблизительно на 300 кПа больше или приблизительно на 500 кПа больше.
В вариантах осуществления изобретения алкен не может быть введен в зону основного алкилирования; единственный источник алкена в этой зоне/ах может находиться в реакционной смеси, подаваемой в зону основного алкилирования.
Кроме того, в вариантах осуществления, в которых реакцию алкилирования между четыреххлористым углеродом и алкеном катализируют металлическим катализатором (необязательно включающим лиганд), металлический катализатор и/или лиганд не могут подаваться в зону основного алкилирования. В таких вариантах осуществления единственным источником катализатора может быть реакционная смесь, подаваемая в зону основного алкилирования. Дополнительно или альтернативно, зона основного алкилирования может быть снабжена слоем катализатора.
В способах согласно настоящему изобретению, в которых задействованы зоны первичного и основного алкилирования, и в реакционной смеси в зоне первичного алкилирования присутствует твердый металлический катализатор (например, добавленный непосредственно в нее), когда реакционную смесь извлекают из зоны первичного алкилирования, чтобы подать в зону основного алкилирования, извлечение реакционной смеси из зоны первичного алкилирования может быть осуществлено таким образом, чтобы в реакционной смеси присутствовало очень маленькое количество, при наличии, твердого металлического катализатора, например, менее чем приблизительно 5 мг, приблизительно 2 мг, приблизительно 1 мг, приблизительно 0,5 мг, приблизительно 0,2 мг, приблизительно 0,1 мг твердого металлического катализатора на литр реакционной смеси.
Это может быть достигнуто посредством использования любого метода и/или оборудования, известных специалистам в данной области техники, например посредством трубки, проходящей в зону/ы первичного алкилирования в соответствующем месте, снабженной фильтрующей сеткой и/или имеющей соответствующий диаметр.
В случае использования зоны первичного и основного алкилирования могут находиться в одинаковых или разных реакторах, которые могут относиться к одинаковым или разным типам реакторов. Кроме того, в вариантах осуществления, подразумевающих использование нескольких зон первичного алкилирования, они могут находиться в одинаковых или разных реакторах. Аналогично, в вариантах осуществления, подразумевающих использование нескольких зон основного алкилирования, они могут находиться в одинаковых или разных реакторах.
В способах согласно настоящему изобретению могут быть задействованы любые типы реакторов, известные специалистам в данной области техники. Конкретными примерами реакторов, которые могут использоваться для обеспечения зон алкилирования, являются реакторы-колонки (например, газожидкостные реакторы-колонки), трубчатые реакторы, барботажные колонные реакторы, реакторы идеального вытеснения (например, трубчатые реакторы идеального вытеснения) и реакторы с мешалкой (например, реакторы непрерывного перемешивания).
Установки, в которых зона первичного алкилирования находится в реакторе непрерывного перемешивания (CSTR) и основная зона алкилирования находится в реакторе идеального вытеснения, обеспечили благоприятные результаты.
Одним из преимуществ способа согласно настоящему изобретению является то, что целевые результаты получены независимо от того, эксплуатируются ли зоны алкилирования (например, зона первичного алкилирования и/или зона основного алкилирования) непрерывно (стационарно) или периодически. Термины «непрерывно» и «периодически» будут понятны специалистам в данной области техники.
В вариантах осуществления зона первичного алкилирования, где она задействована, используется непрерывно или периодически. Дополнительно или альтернативно, вторая зона/ы алкилирования, где задействована, используется непрерывно или периодически.
В способах согласно настоящему изобретению концентрацию хлорированного C3-6 алкана в реакционной смеси в зоне основного алкилирования поддерживают на таком уровне, чтобы молярное соотношение хлорированного C3-6 алкана к четыреххлористому углероду в реакционной смеси, извлеченной из зоны основного алкилирования, не превышало: I) 95:5, где зона основного алкилирования работает в непрерывном режиме, или II) 99: 1, где зона основного алкилирования работает в периодическом режиме.
В некоторых вариантах осуществления изобретения, в которых зона основного алкилирования работает в непрерывном режиме, содержание хлорированного C3-6 алкана может регулироваться таким образом, чтобы соотношение этого соединения к четыреххлористому углероду в реакционной смеси, извлеченной из зоны основного алкилирования, не превышало приблизительно 94:6, приблизительно 92:8 или приблизительно 90:10
В альтернативных вариантах осуществления изобретения, где зона основного алкилирования работает в периодическом режиме, содержание хлорированного C3-6 алкана может регулироваться таким образом, чтобы соотношение этого соединения к четыреххлористому углероду в реакционной смеси, извлеченной из зоны основного алкилирования, не превышало приблизительно 97:3, приблизительно 95:5 или приблизительно 90:10.
Независимо от того, работает ли зона основного алкилирования в непрерывном или периодическом режиме, содержание хлорированного C3-6 алкана может регулироваться таким образом, чтобы соотношение этого соединения к четыреххлористому углероду в реакционной смеси, извлеченной из зоны основного алкилирования, составляло или было больше чем приблизительно 70:30, приблизительно 80:20, приблизительно 85:15 или приблизительно 90:10.
Неожиданно было установлено, что при контроле степени конверсии четыреххлористого углерода в целевой хлорированный C3-6 алкан и препятствовании завершению реакции образование примесей преимущественно снижается. Например, в вариантах осуществления, в которых алкеновым сырьем, используемым в способах согласно настоящему изобретению, является этен, образование нежелательных побочных продуктов, таких как пентаны (которые иначе образовывались бы) минимизировано.
Таким образом, в вариантах осуществления изобретения в реакционной смеси, извлеченной из основной реакционной зоны, содержание серийных продуктов реакции, т.е. соединений, содержащих большее количество атомов углерода, чем продукт хлорированного C3-6 алкана, составляет менее чем приблизительно 5%, менее чем приблизительно 2%, менее чем приблизительно 1%, менее чем приблизительно 0,5%, менее чем приблизительно 0,2%, менее чем приблизительно 0,1%, менее чем приблизительно 0,05% или менее чем приблизительно 0,02%.
Контроль содержания хлорированного C3-6 алкана может осуществляться путем замедления процесса алкилирования и/или введения дополнительного четыреххлористого углерода в зону основного алкилирования.
В вариантах осуществления, в которых содержание хлорированного C3-6 алкана контролируется путем замедления процесса алкилирования, это может быть достигнуто посредством поддержания условий реакции, которые не способствуют полной конверсии четыреххлористого углерода в 1,1,1,3- тетрахлорпропан. Например, это может быть достигнуто путем подвергания реакционной смеси или по меньшей мере ее части условиям, которые замедляют или останавливают протекание реакции алкилирования. В таких вариантах осуществления давление, воздействию которого подвергается реакционная смесь в зоне/ах алкилирования (например, в зоне/ах основного алкилирования, если они задействованы), может быть значительно снижено, например, по меньшей мере приблизительно на 500 кПа, по меньшей мере приблизительно на 700 кПа, по меньшей мере приблизительно на 1000 кПа.
Дополнительно или альтернативно, давление, воздействию которого подвергается реакционная смесь, может быть уменьшено до атмосферного или ниже атмосферного давления. Снижение давления может происходить в одной или нескольких зонах алкилирования (например, одной, некоторых или всех зонах основного алкилирования, если они задействованы). Дополнительно или альтернативно, снижение давления может происходить после извлечения реакционной смеси из зоны/зон алкилирования.
Дополнительно или альтернативно, в вариантах осуществления, в которых содержание хлорированного C3-6 алкана контролируется путем замедления процесса алкилирования, это может быть достигнуто путем ограничения уровня содержания алкена в реакционной смеси.
В вариантах осуществления изобретения контроль за ходом реакции алкилирования в зоне/ах алкилирования может осуществляться путем тщательного выбора времени пребывания реакционной смеси в зоне/ах алкилирования. Например, в вариантах осуществления, в которых задействована одна или несколько зон основного алкилирования, время пребывания реакционной смеси в этой зоне/ах может составлять, например, от приблизительно 1 до 120 минут, от приблизительно 5 до 100 минут, от приблизительно 15 до приблизительно 60 минут или от приблизительно 20 до приблизительно 40 минут.
В вариантах осуществления, в которых содержание хлорированного C3-6 алкана контролируется путем замедления процесса алкилирования, это может быть дополнительно или альтернативно достигнуто путем снижения рабочей температуры зоны основного алкилирования, например, приблизительно на 5°C или более, приблизительно на 10°С или более, приблизительно на 20°С или более, приблизительно на 50°С или более или приблизительно на 100°С или более. Дополнительно или альтернативно, рабочая температура в зоне основного алкилирования может быть уменьшена до приблизительно 20°С, приблизительно 10°С или приблизительно 0°С.
Дополнительно или альтернативно, процесс алкилирования может быть замедлен путем ограничения количества присутствующего катализатора в реакционной смеси или удаления слоя катализатора (если он имеется) из зоны основного алкилирования.
Скорость перемешивания в зоне основного алкилирования также может быть уменьшена для замедления процесса алкилирования.
Как упомянуто выше, реакционная смесь, извлеченная из зоны основного алкилирования, содержит четыреххлористый углерод и хлорированный C3-6 алкан. Однако в вариантах осуществления изобретения в зависимости от условий и задействованного оборудования реакционная смесь, извлеченная из зоны основного алкилирования, может дополнительно содержать непрореагировавший исходный материал алкена, катализатор и/или примеси (например, примеси хлорированных алканов, хлорированных алкенов и/или кислородосодержащие органические соединения).
Учитывая, что присутствие непрореагировавшего алкена рядом с хлорированным C3-6 алканом может создавать проблемы, особенно для дальнейших процессов с использованием хлорированного C3-6 алкана, в вариантах осуществления изобретения реакционную смесь, извлеченную из зоны основного алкилирования, подвергают стадии деалкенирования, на которой по меньшей мере приблизительно 50% по массе алкена, присутствующего в реакционной смеси, выделяют из нее и по меньшей мере приблизительно 50% выделенного алкена направляют обратно в реакционную смесь, находящуюся в зоне основного алкилирования.
Такие варианты осуществления являются особенно предпочтительными, поскольку они позволяют осуществлять существенное, если не полное, использование алкенового сырья, используемого в способах согласно настоящему изобретению.
В вариантах осуществления изобретения по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 95%, по меньшей мере приблизительно 97% или по меньшей мере приблизительно 99% алкена, присутствующего в реакционной смеси, извлеченной из зоны основного алкилирования, удаляют на стадии деалкенирования.
Удаление непрореагировавшего алкена из реакционной смеси может быть достигнуто за счет использования любого метода, известного специалистам в данной области техники. В вариантах осуществления изобретения выделение алкена из реакционной смеси может быть достигнуто за счет использования методов дистилляции, которые приводят к получению потока, обогащенного алкеном, например, методом мгновенного выпаривания, который может быть преимущественно использован в вариантах осуществления, где температура кипения алкена существенно ниже, чем температура кипения других соединений, присутствующих в реакционной смеси, как в случае с этеном (-103,7°С) по сравнению с четыреххлористым углеродом (76,6°С) и 1,1,1,3-тетрахлорпропаном (159°С).
Деалкенирование реакционной смеси может быть селективным. Другими словами, алкен удаляют избирательно без существенного удаления других соединений из реакционной смеси. В таких вариантах осуществления алкен, выделенный из реакционной смеси, может содержать менее чем приблизительно 10%, менее чем приблизительно 5%, менее чем приблизительно 2% или менее чем приблизительно 1% соединений, отличных от исходного материала алкена.
Дистилляция реакционной смеси может осуществляться любыми методами или с использованием любого оборудования, известного специалистам в данной области техники. Например, может быть использовано обычное дистилляционное устройство (например, дистилляционная колонка). Дополнительно или альтернативно, в вариантах осуществления изобретения, когда давление в зоне основного алкилирования, из которой извлекают реакционную смесь, является избыточным, выпаривание алкена из реакционной смеси может осуществляться путем поддержания реакционной смеси при избыточном давлении после извлечения реакционной смеси из зоны основного алкилирования и подачи ее в зону выпаривания, в которой происходит выпаривание алкена из реакционной смеси.
В вариантах осуществления изобретения выпаривание алкена из реакционной смеси в зоне выпаривания может осуществляться путем сброса давления, например путем существенного снижения давления реакционной смеси, например, по меньшей мере приблизительно на 500 кПа, по меньшей мере приблизительно на 700 кПа, по меньшей мере приблизительно на 1000 кПа и/или до атмосферного или ниже атмосферного давления. В вариантах осуществления, в которых используется либо частичный, либо полный сброс давления для замедления или остановки конверсии четыреххлористого углерода в целевой хлорированный C3-6 алкан, а также для отделения алкена от реакционной смеси эти цели могут быть одновременно достигнуты при одностадийном сбросе давления.
Зона выпаривания может находиться в любом устройстве, в котором может осуществляться выпаривание алкена, присутствующего в реакционной смеси, например, в устройстве мгновенного выпаривания, таком как испаритель.
Алкен, дистиллируемый из реакционной смеси, например, мгновенным выпариванием, предпочтительно выделяют из дистилляционного устройства в жидкой или газообразной форме.
В способах согласно настоящему изобретению по меньшей мере приблизительно 50%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 95%, по меньшей мере приблизительно 97% или по меньшей мере приблизительно 99% по массе алкена, извлеченного из зоны выпаривания, возвращаются (т.е. рециклируются) в зону первичного и/или основного алкилирования.
Во избежание сомнений, в вариантах осуществления изобретения дистиллированный алкен, если он находится в газообразной форме, может быть переведен или не переведен обратно в жидкость до подачи в реакционную смесь, находящуюся в зоне основного алкилирования. Например, конверсия газообразного алкена в жидкий алкен может осуществляться путем пропускания через конденсатор и/или улавливания в потоке жидкого (предпочтительно охлажденного) четыреххлористого углерода, который затем может быть подан в зону/ы алкилирования. Газообразный алкен может улавливаться в жидком потоке четыреххлористого углерода с использованием любых методов или оборудования, известных специалистам в данной области техники, например, абсорбционной колонки. Такой механизм является предпочтительным, так как он способствует полному промышленному использованию соединений, используемых в процессе алкилирования.
Как упоминалось выше, в вариантах осуществления изобретения в реакционной смеси, присутствующей в зоне/ах алкилирования, может быть использован катализатор, например, каталитическая система, содержащая твердый металл и/или катализатор на основе соли металла и промотор, такой как лиганд. В таких вариантах осуществления реакционная смесь, извлеченная из зоны/зон алкилирования, может содержать катализатор. Учитывая, что присутствие катализатора может вызывать проблемы в последующих реакциях с участием хлорированного C3-6 алкана, может оказаться предпочтительным удаление катализатора из реакционной смеси.
Кроме того, для каталитических систем, в которых используются дорогостоящие катализаторы и/или промоторы, такие как упомянутые выше алкилфосфатные и алкилфосфитные лиганды, извлечение повторно используемых каталитических систем и/или их компонентов также предпочтительно для минимизации необходимого количества свежего катализатора, что снижает эксплуатационные расходы.
Хотя проблема удаления катализаторов типа, используемого в способах согласно настоящему изобретению, из реакционных смесей была рассмотрена ранее, применяемые методы и условия (обычно включающие перегонку в агрессивных условиях) могут наносить ущерб каталитическим системам и могут снизить их каталитическую способность. Это особенно важно, когда каталитическая система чувствительна к температуре, как в случае систем, включающих определенные органические лиганды в качестве промоторов, таких как алкилфосфаты и алкилфосфиты.
Таким образом, в вариантах осуществления настоящего изобретения реакционную смесь, извлеченную из зоны основного алкилирования, подвергают стадии водной обработки, на которой реакционную смесь приводят в контакт с водной средой в зоне водной обработки с получением двухфазной смеси, и органическую фазу, содержащую катализатор, выделяют из двухфазной смеси.
В вариантах осуществления изобретения, в которых реакционная смесь подвергается водной обработке, реакционная смесь может содержать непрореагировавший четыреххлористый углерод и продукт хлорированного C3-6 алкана. Кроме того, реакционная смесь может содержать катализатор (например, комплекс металлического катализатора и каталитического лиганда или свободный каталитический лиганд) и/или непрореагировавший исходный материал алкена.
Стадия водной обработки позволяет избежать повреждающих условий, описанных в предшествующем уровне техники (например, высокая температура, высокая концентрация катализатора и/или присутствие соединений железа в безводной форме), что означает, что восстановленный катализатор и/или его компоненты (например, лиганд или промотор) могут быть повторно использованы (например, могут быть возвращены обратно в реакционную смесь, находящуюся в зоне/ах алкилирования без какого-либо существенного снижения каталитической способности). В вариантах осуществления изобретения паровая десорбция двухфазной смеси после стадии водной обработки является предпочтительной, поскольку позволяет избежать температуры котла выше 100°С, а также позволяет использовать атмосферное давление.
Еще одно преимущество стадии водной обработки заключается в том, что она приводит к удалению примесей из реакционной смеси, например кислородосодержащих органических продуктов, если они присутствуют. Более конкретно, когда в способах используют алкены в качестве предшественников, например этен, пропен, 1-бутен, 2-бутен, 1-гексен, 3-гексен, 1-фенил-3-гексен, бутадиен, винилгалогениды и т.п., могут образовываться кислородосодержащие органические соединения (например, алканолы, алканоилы и хлорированные аналоги таких кислородосодержащих соединений). Преимущественно, чтобы содержание таких материалов в реакционной смеси были значительно снижено до допустимых уровней, если не устранено, на стадии водной обработки.
В вариантах осуществления изобретения, в которых проводится стадия водной обработки, реакционная смесь, находящаяся в зоне водной обработки, может содержать целевой хлорированный C3-6 алкан (например, в количествах приблизительно 50% или более), катализатор и необязательно четыреххлористый углерод и/или примеси, например кислородосодержащие органические соединения, соединения хлорированных алканов (отличные от целевого хлорированного C3-6 алкана) и/или соединения хлорированных алкенов.
Этот процесс восстановления катализатора включает в себя стадию водной обработки реакционной смеси, на которой реакционную смесь приводят в контакт с водной средой в зоне водной обработки. В вариантах осуществления водная среда представляет собой воду (в виде жидкости и/или пара). Кроме того, водная среда может дополнительно содержать другие соединения, такие как кислоты. Могут быть использованы неорганические кислоты, такие как соляная кислота, серная кислота и/или фосфорная кислота.
Когда водная среда, подаваемая в зону водной обработки, частично или полностью находится в жидкой форме, двухфазная смесь будет образовываться с жидкой водной средой, контактирующей с реакционной смесью.
Альтернативно, когда водная среда находится в газообразной форме, например в форме пара, двухфазная смесь может образоваться не сразу, а только после того, как газообразная водная среда конденсируется. Устройство, используемое на стадии водной обработки, может быть сконструировано таким образом, что конденсация водной среды с образованием двухфазной смеси происходит внутри и/или удаленно от зоны водной обработки.
В вариантах осуществления изобретения продукт хлорированного C3-6 алкана может быть выделен из смеси, образованной в зоне водной обработки. Большая часть (например, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80% или по меньшей мере приблизительно 90%) хлорированного C3-6 алкана, присутствующего в реакционной смеси, подаваемой в зону водной обработки, может быть выделена из смеси, образованной в зоне водной обработки, с использованием любых методов или оборудования, известных специалистам в данной области техники.
В вариантах осуществления изобретения дистилляцию используют для выделения продукта хлорированного C3-6 алкана из смеси, образованной в зоне водной обработки. Перегонка может привести к получению потока, обогащенного продуктом хлорированного C3-6 алкана.
Используемый в данном описании термин «поток, обогащенный» конкретным соединением (или соответствующие формулировки) используется для обозначения того, что поток содержит по меньшей мере приблизительно 90%, приблизительно 95%, приблизительно 97%, приблизительно 98% или приблизительно 99% конкретного соединения. Кроме того, термин «поток» не следует толковать узко, он охватывает композиции (включая фракции), выделенные из смеси любыми способами.
Например, C3-6 алкан может отгоняться, например, из газообразной смеси, содержащей этот алкан и водяной пар. Хлорированный C3-6 алкан может отгоняться в потоке, обогащенном хлорированным C3-6 алканом. В вариантах осуществления изобретения, в которых водная среда частично или полностью находится в жидкой форме, дистилляция хлорированного C3-6 алкана может быть достигнута путем кипячения присутствующей смеси для выпаривания хлорированного C3-6 алкана и получения газообразной смеси хлорированного C3-6 алкана и водяных паров, из которых хлорированный C3-6 алкан может быть дистиллирован, например, с использованием технологий паровой дистилляции.
Дополнительно или альтернативно, когда водная среда подводится частично или полностью в газообразной форме, это приводит к выпариванию хлорированного C3-6 алкана с образованием газообразной смеси, содержащей этот алкан и водяной пар, которые затем могут быть необязательно подвергнуты дистилляции для удаления хлорированного C3-6 алкана, например, паровой дистилляции. Хлорированный C3-6 алкан может быть получен в потоке, обогащенном этим соединением.
В вариантах осуществления, в которых хлорированный C3-6 алкан отгоняют из газообразной смеси хлорированного C3-6 алкана и водяного пара, дистилляционное устройство может быть соединено с зоной водной обработки так, чтобы смесь газообразного хлорированного алкана и водяного пара могла проходить непосредственно от зоны водной обработки до этого устройства. Альтернативно, дистилляционное устройство может быть расположено удаленно от зоны водной обработки, так, что газообразная смесь сначала извлекается из зоны водной обработки, а затем транспортируется в дистилляционный аппарат. В любой конфигурации устройства хлорированный C3-6 алкан может быть получен в потоке, обогащенном этим соединением.
В альтернативных вариантах осуществления, когда водная среда и реакционная смесь находятся в жидкой форме, хлорированный C3-6 алкан может быть выделен из этой жидкой смеси с использованием обычных методов перегонки, известных специалистам в данной области техники. Хлорированный C3-6 алкан может быть получен в потоке, обогащенном этим соединением.
Двухфазная смесь может быть образована в зоне водной обработки или удаленно от нее. Двухфазная смесь включает водную фазу (которая является результатом добавления водной среды в зону водной обработки) и органическую фазу (содержащую хлорированный C3-6 алкан, необязательно непрореагировавший четыреххлористый углерод и, что важно, катализатор).
Для максимизации объема органической фазы и, таким образом, для облегчения выделения этой фазы из двухфазной смеси в двухфазную смесь может быть добавлен агент выделения галогеналканов (например, четыреххлористый углерод и/или хлорированный C3-6 алкан) (например, путем непрерывной или периодической подачи в зону водной обработки) с использованием методов и оборудования, известных специалистам в данной области техники.
Органическая фаза может быть выделена из двухфазного остатка с использованием любого метода, известного специалистам в данной области, например, декантации. Например, выделение органической фазы может быть осуществлено путем последовательного фазового отделения из зоны водной обработки или сосуда, в котором она содержится. Альтернативно, двухфазная смесь может быть извлечена из зоны водной обработки и подвергнута стадии разделения фаз удаленно от зоны водной обработки.
В вариантах осуществления изобретения двухфазная смесь и/или выделенная органическая фаза может быть отфильтрована. В вариантах осуществления это приводит к получению фильтрационного осадка, который может быть необязательно полностью или частично использован в качестве источника железа.
Выделение продукта хлорированного C3-6 алкана из смеси, образованной на стадии водной обработки, может быть выполнено до извлечения органической фазы из нее и/или после извлечения органической фазы из этой смеси. Некоторые примерные варианты осуществления, в которых продукт хлорированного C3-6 алкана выделяют из смеси, образованной во время водной обработки, описаны выше.
В качестве еще одного примера двухфазная смесь может быть нагрета с образованием газообразной смеси, из которой может быть выделен продукт хлорированного C3-6 алкана (необязательно в виде потока, обогащенного хлорированным C3-6 алканом), например, путем дистилляции. Органическая фаза, имеющая пониженное содержание хлорированного C3-6 алкана, может быть затем выделена из двухфазной смеси.
Дополнительно или альтернативно, органическая фаза может быть выделена из двухфазной смеси, как описано выше. Хлорированный C3-6 алкан затем может быть выделен (необязательно в виде потока, обогащенного хлорированным C3-6 алканом) из этой фазы, например, путем дистилляции. В таких вариантах осуществления, когда органическая фаза содержит катализатор, условия дистилляции, выбранные для выделения хлорированного C3-6 алкана, являются мягкими для минимизации дезактивации каталитической системы, например, при температуре приблизительно 100°C или ниже, приблизительно 95°С или ниже, приблизительно 90°С или ниже, приблизительно 85°С или ниже или приблизительно 80°С или ниже и/или под давлением приблизительно от 1 до 10 кПа. Дополнительно или альтернативно, может быть использовано более низкое давление.
Выделенная органическая фаза может содержать четыреххлористый углерод и/или продукт хлорированного C3-6 алкана. Кроме того, реакционная смесь может содержать катализатор (например, комплекс металлического катализатора и каталитического лиганда или свободный лиганд) и/или непрореагировавший исходный материал алкена. Когда поток, обогащенный хлорированным C3-6 алканом, выделяют из смеси, образованной на стадии водной обработки (либо напрямую, либо после выделения органической фазы из нее), содержание хлорированного C3-6 алкана в этой фазе ниже, чем в реакционной смеси.
В установках согласно изобретению, особенно в тех, в которых органическая фаза содержит четыреххлористый углерод и/или катализатор, органическая фаза может быть возвращена в зону/ы алкилирования, например, в жидкой форме. В таких установках исходный материал алкена (например, в газообразной форме) может быть захвачен потоком органической фазы, подаваемой в зону/ы алкилирования.
В вариантах осуществления изобретения одна или несколько стадий дистилляции в дополнение к тем, которые обсуждались выше, могут быть выполнены в любой момент способа, необязательно для получения потока/ов, обогащенных конкретными продуктами. Например, перед проведением стадии водной обработки, если она проводится, реакционная смесь может быть подвергнута стадии дистилляции. В вариантах осуществления, в которых реакционная смесь содержит чувствительную к температуре каталитическую систему, например, включает органический лиганд в качестве промотора, стадию дистилляции обычно проводят в условиях, исключающих дезактивацию катализатора, например, при температуре приблизительно 100°С или ниже, приблизительно 95°С или ниже, приблизительно 90°С или ниже, приблизительно 85°С или ниже или приблизительно 80°С или ниже и/или под давлением приблизительно от 1 до 10 кПа. Дополнительно или альтернативно могут быть использованы более низкие значения давлений.
Кроме того, было обнаружено, что дезактивации чувствительных к температуре каталитических систем можно избежать, если не подвергать реакционную смесь дополнительной дистилляции. Таким образом, в вариантах осуществления изобретения, в которых дистиллируется реакционная смесь, содержащая каталитическую систему, нельзя допускать, чтобы дистилляция приводила к уменьшению объема технологической жидкости в дистилляционном устройстве так, чтобы концентрация каталитической системы в этой технологической жидкости составляла приблизительно в 2, приблизительно в 5, или приблизительно в 10 раз больше, чем уровень этой каталитической системы, присутствующей в реакционной смеси, находящейся в зоне основного алкилирования.
Стадия дистилляции, проводимая перед стадией водной обработки (если она проводится), может быть проведена с использованием методов и оборудования, известных специалистам в данной области, например, дистилляционного котла (для периодической или непрерывной дистилляции), связанного с вакуумной дистилляционной колонкой. В таком варианте осуществления изобретения реакционная смесь, подвергаемая дистилляции, может содержать более приблизительно 50% по массе целевого хлорированного C3-6 алкана, катализатор и необязательно четыреххлористый углерод и/или примеси, например кислородосодержащие органические соединения, соединения хлорированных алканов (за исключением целевого хлорированного C3-6 алкана) и/или соединения хлорированных алкенов.
Стадия дистилляции обычно приводит к удалению потока/ов дистиллята хлорированных алканов, например потока/ов (и необязательно обогащенного) непрореагировавшего четыреххлористого углерода, целевого C3-6 алкана и/или хлорированных органических примесей (т.е. хлорированных органических соединений, отличных от целевого хлорированного C3-6 алкана и четыреххлористого углерода) из реакционной смеси. Четыреххлористый углерод может быть возвращен обратно в зону/ы алкилирования. Остаток после такой стадии, который обычно содержит хлорированный C3-6 алкан, четыреххлористый углерод и/или катализатор, может быть подвергнут дополнительным стадиям обработки, например, стадии водной обработки и/или дополнительной стадии/ям дистилляции.
В вариантах осуществления изобретения, где реакционную смесь подвергают дистилляции до стадии водной обработки (если она проводится), по меньшей мере приблизительно 30%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60% или по меньшей мере приблизительно 70%, вплоть до по большей мере приблизительно 95%, по большей мере приблизительно 90%, по большей мере приблизительно 85% или по большей мере приблизительно 80% по массе хлорированного C3-6 алкана удаляют из реакционной смеси на этой стадии дистилляции.
Одна или несколько стадий дистилляции могут быть дополнительно или альтернативно проведены после стадии водной обработки (если она проводится). Например, хлорированный C3-6 алкан, выделенный из реакционной смеси, подаваемой в зону водной обработки, может присутствовать в виде смеси, содержащей в качестве основного компонента хлорированный C3-6 алкан, агент выделения галогеналканов, а также хлорированные органические примеси (т.е. хлорированные органические соединения, отличные от целевого хлорированного C3-6 алкана и четыреххлористого углерода). Эта смесь может быть подвергнута одной или нескольким стадиям дистилляции для удаления хлорированных органических примесей с получением потока, обогащенного хлорированным C3-6 алканом, и/или удаления агента выделения галогеналканов. Опять же любое оборудование или условия, известные специалистам в данной области техники, могут быть использованы на такой стадии дистилляции, например, дистилляционный котел (для периодической или непрерывной дистилляции), связанный с вакуумной дистилляционной колонкой.
На такой стадии дистилляции хлорированный C3-6 алкан, выделенный из реакционной смеси, находящейся в зоне водной обработки, может быть подвергнут дистилляции для отделения целевого хлорированного C3-6 алкана от хлоралкановых примесей. Например, в вариантах осуществления, в которых целевой хлорированный C3-6 алкан представляет собой 1,1,1,3-тетрахлорпропан, было обнаружено, что стадия дистилляции для очистки хлорированного C3-6 алкана, выделенного из реакционной смеси, находящейся в зоне водной обработки, является особенно эффективной при удалении примесей хлорпентана/хлорпентена.
Хлорированные органические примеси, отделенные от смесей, содержащих целевой хлорированный C3-6 алкан, на стадиях дистилляции, осуществляемых на любой стадии способа согласно настоящему изобретению, могут быть извлечены и повторно использованы при производстве четыреххлористого углерода. Это может быть осуществлено путем введения хлорированных органических примесей в процесс высокотемпературного хлоринолиза. В таком процессе любые присутствующие хлорированные органические соединения перерабатываются в основном обратно в чистый четыреххлористый углерод с высокими выходами. Таким образом, использование стадии хлоринолиза в способах согласно настоящему изобретению является полезным для максимизации общего выхода синтеза и чистоты целевого хлоралкана при минимизации образования отходов.
Независимо от целевого хлорированного C3-6 алкана, в вариантах осуществления изобретения в дистилляционном котле после водной обработки может быть образован остаток «тяжелых фракций». Остатки «тяжелых фракций» обычно выделяют из системы и обрабатывают, например, высокотемпературным хлоринолизом, что предпочтительно приводит к получению хлорметана.
Способы согласно настоящему изобретению особенно преимущественны, поскольку они позволяют получать хлорированные алканы высокой степени чистоты, хотя задействованы простые и прямые методы и оборудование, с которыми знаком специалист в данной области техники.
Как можно видеть из описания, представленного здесь, предлагаемые в настоящем изобретении способы могут осуществляться комплексно в полностью непрерывном режиме, необязательно в сочетании с другими процессами. В стадиях способа согласно настоящему изобретению могут быть использованы исходные соединения, которые превращаются в промежуточные соединения высокой степени чистоты, которые затем обрабатываются с получением требуемых целевых хлорированных соединений. Эти соединения имеют требуемую степень чистоты для использования в качестве исходного сырья в ряде последующих процессов, например, при гидрофторировании.
Способы согласно настоящему изобретению позволяют регулировать уровни чистоты продукта для достижения высоких степеней чистоты соединений на каждой стадии. Способы преимущественно сочетают высокие выходы, высокую селективность и высокую эффективность, что является особенно сложным, особенно в непрерывных процессах. Способы согласно настоящему изобретению позволяют экономично получать соединения хлоралканов высокой степени чистоты в промышленном масштабе, причем такие соединения имеют очень низкие уровни содержания ряда примесей, таких как галогеналканы или галогеналкены (в частности, таких, которые изомерны целевым соединениям и/или которые имеют более высокую молекулярную массу, таких как продукты последовательных реакций) и/или кислородосодержащие и/или бромированные аналоги, а также вода и металлические соединения.
В вариантах осуществления изобретения способы согласно изобретению могут быть использованы для получения композиций хлорированных алканов высокой степени чистоты, которые содержат:
Приблизительно 99,0% или более, приблизительно 99,5% или более, приблизительно 99,7% или более, приблизительно 99,8% или более или приблизительно 99,9% или более хлорированного алкана,
менее чем приблизительно 2000 млн-1, менее чем приблизительно 1000 млн-1, менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1 или менее чем приблизительно 100 млн-1 примесей хлорированных алканов (то есть хлорированных алканов, отличных от целевого хлорированного C3-6 алкана),
менее чем приблизительно 2000 млн-1, менее чем приблизительно 1000 млн-1, менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1 или менее чем приблизительно 100 млн-1 хлорированных алкенов,
менее чем приблизительно 2000 млн-1, менее чем приблизительно 1000 млн-1, менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1 или менее чем приблизительно 100 млн-1 кислородосодержащих органических соединений,
менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1, менее чем приблизительно 100 млн-1, менее чем приблизительно 50 млн-1 или менее чем приблизительно 20 млн-1 металлического катализатора,
менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1, менее чем приблизительно 100 млн-1, менее чем приблизительно 50 млн-1 или менее чем приблизительно 20 млн-1 промотора катализатора,
менее чем приблизительно 2000 млн-1, менее чем приблизительно 1000 млн-1, менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1 или менее чем приблизительно 100 млн-1 бромидов или бромированных органических соединений и/или
менее чем приблизительно 1000 млн-1, менее чем приблизительно 500 млн-1, менее чем приблизительно 200 млн-1, менее чем приблизительно 100 млн-1, менее чем приблизительно 50 млн-1 или менее чем приблизительно 20 млн-1 воды.
Для вариантов осуществления, в которых целевой хлорированный C3-6 алкан представляет собой 1,1,1,3-тетрахлорпропан, ниже приведены примеры конкретных примесей, содержание которых минимизировано, которые не образовались и/или были удалены в способах согласно настоящему изобретению: трихлорметан, 1,2-дихлорэтан, 1-хлорбутан, 1,1,1-трихлорпропан, тетрахлорэтен, 1,1,3-трихлорпропен-1,1,1,1,3,3-пентахлорпропан, 1,1,1,2,3-пентахлорпропан, гексахлорметан, 1,1,1,5-тетрахлорпентан, 1,3,3,5-тетрахлорпентан, трибутилфосфат, хлорированный алканол и соединения хлорированных алканоилов.
Таким образом, в вариантах осуществления настоящего изобретения продукт 1,1,1,3-тетрахлорпропан содержит приблизительно 500 млн-1 или менее, приблизительно 200 млн-1 или менее, приблизительно 100 млн-1 или менее, приблизительно 50 млн-1 или менее, приблизительно 20 млн-1 или менее или приблизительно 10 млн-1 или менее или менее одного или нескольких из этих соединений.
Во избежание сомнений, когда чистота композиции или материала представлена в % или млн-1, если не указано иное, имеются в виду массовые проценты/массовые миллионные доли.
Как упоминалось ранее, в уровне техники не раскрыты или не описаны способы получения хлорированных алканов с такой высокой степенью чистоты, в ходе которых осуществляется контроль содержания большого ряда возможных примесей. Таким образом, согласно дополнительным аспектам настоящего изобретения, предложены композиции хлорированного алкана высокой степени чистоты, как указано выше.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1. Стадия алкилирования
Фиг. 2. Первая стадия дистилляции
Фиг. 3. Стадия водного восстановления катализатора
Фиг. 4. Вторая стадия дистилляции
ПРИМЕРЫ
Далее настоящее изобретение дополнительно будет проиллюстрировано с помощью следующих примеров.
ПРИМЕР 1. ДЕМОНСТРАЦИЯ КАТАЛИТИЧЕСКОЙ СПОСОБНОСТИ КАТАЛИЗАТОРА, ВОССТАНОВЛЕННОГО С ПОМОЩЬЮ ВОДНОЙ ОБРАБОТКИ
Этен и четыреххлористый углерод подвергали взаимодействию с получением 1,1,1,3-тетрахлорпропана в присутствии катализатора, который либо I) восстанавливали из реакционной смеси с использованием обычных методов дистилляции, либо II) восстанавливали из реакционной смеси с использованием стадии водной обработки согласно настоящему изобретению, описанной здесь. Реакционная смесь дополнительно содержала 1,1,1,3-тетрахлорпропан (присутствующий в рециркуляционном потоке) и тетрахлорпентан (примесь хлорированного алкана, обычно образующаяся в качестве побочного продукта в ходе реакций теломеризации между четыреххлористым углеродом и этеном).
Эти примеры экспериментов демонстрируют, что проведение стадии водной обработки для восстановления катализатора значительно увеличивает производительность катализатора по сравнению с производительностью катализатора, восстановленного с использованием обычных методов дистилляции.
Для краткости в приведенных ниже примерах используются следующие термины:
TeCM: четыреххлористый углерод,
TeCPa: 1,1,1,3-тетрахлорпропан,
TeCPna: тетрахлорпентан,
Bu3PO4: трибутилфосфат.
За ходом реакции следили с помощью газовой хроматографии.
Периодическая организация эксперимента:
Автоклав из нержавеющей стали объемом 405 мл, снабженный мешалкой, термогильзой для измерения температуры и пробоотборной трубкой (с клапаном), заполняли описанной ниже реакционной смесью и закрывали. Нагревание осуществляли с помощью масляной бани, помещенной на магнитную (нагревательную) мешалку. Этен подавали через медную капиллярную трубку из 10-литрового цилиндра, помещенного на весы. Газовую среду в автоклаве заменяли этеновой промывкой. После герметизации этеном до 5 бар автоклав нагревали до 105°С, после чего подавали этен в автоклав. Подачу этена контролировали вручную в течение первых десяти минут (для поддержания температуры реакции до 112°C), а затем поддерживали при постоянном давлении 9 бар. Реакции давали возможность протекать в течение определенного периода времени. Затем реактор охлаждали и реакционную смесь отводили после открытия разгерметизованного реактора.
Сравнительные примеры 1-1 и 1-3 и Примеры 1-2, 1-4 и 1-5
В первом примере дистилляционный остаток непосредственно использовали в качестве восстановленного катализатора (Сравнительный пример 1-1). Во втором примере дистилляционный остаток выделяли с помощью 5% соляной кислоты и в качестве катализатора использовали фильтрованную органическую фракцию (Пример 1-2).
Сравнительный пример 1-1
90,1 г дистилляционного остатка, содержащего 63,7% TeCPa, 22,8% TeCPna и 7,49% Bu3PO4, смешивали с 400 г TeCM. Смесь затем вводили в автоклав, куда добавляли 5,0 г железа. После продувки этеном смесь нагревали в автоклаве до 110°С. При этой температуре и при давлении этена в 9 бар реакционную смесь оставляли реагировать в течение 4,5 часов. Первый образец был взят через 3 часа. Концентрация остаточного TeCM в конце эксперимента составляла 19,7% (33,0% через 3 часа).
Пример 1-2
90,1 г дистилляционного остатка, содержащего 63,7% TeCPa, 22,8% TeCPna и 7,49% Bu3PO4, выделяли с помощью 370 г 5% HCl. Нижний органический слой фильтровали и смешивали с 400 г TeCM. Смесь затем вводили в автоклав, куда добавляли 5,0 г железа. После продувки этеном смесь нагревали в автоклаве до 110°С. При этой температуре и под давлением этена в 9 бар реакционную смесь оставляли реагировать в течение 4,5 часов. Первый образец был взят через 3 часа. Концентрация остаточного TeCM в конце эксперимента составляла 5,5% (24,6% через 3 часа).
Сравнительный пример 1-3
Сравнительный пример 1-3 проводили в условиях, идентичных условиям Сравнительного примера 1-1, за исключением того, что использовали другие концентрации четыреххлористого углерода и трибутилфосфата.
Примеры 1-4 и 1-5
Примеры 1-4 и 1-5 проводили в условиях, идентичных условиям Примера 1-2, за исключением того, что использовали другие концентрации четыреххлористого углерода и трибутилфосфата.
Результаты Сравнительного примера 1-1 и Примера 1-2 и Сравнительного примера 1-3, а также Примеров 1-4 и 1-5 показаны в следующей таблице. Как можно видеть, процент четыреххлористого углерода, преобразованного в 1,1,1,3-тетрахлорпропан, значительно выше в Примерах 1-2, 1-4 и 1-5, чем в Сравнительных примерах 1-1 и 1-3, что демонстрирует, что осуществление стадии водной обработки при восстановлении катализатора оказывает сильное положительное влияние на систему. Это происходит из-за высокой эффективности катализатора, восстановленного из остатка дистиллята, а также из-за удаления примесей (например, кислородосодержащих примесей) из реакционной смеси, которые в противном случае могут замедлять реакцию.
Непрерывная организация эксперимента:
В качестве реактора с мешалкой непрерывного действия использовали тот же автоклав из нержавеющей стали, который описан выше для периодических экспериментов. Реактор изначально заполняли приблизительно 455 г реакционной смеси. После герметизации этеном до 5 бар автоклав нагревали до 105°С, затем открывали подачу этена в автоклав с непрерывной подачей сырья и начинали непрерывный отвод реакционной смеси.
Раствор исходного сырья с растворенным катализатором подавали в автоклав из резервуара из нержавеющей стали. Резервуар размещали над реактором, и, таким образом, насос для подачи в реактор не использовали. Реактор и резервуар находились в атмосфере этена, распределяемой медными капиллярами из цилиндра, причем условия в цилиндре выбирали так, чтобы предотвратить начало реакции. Отбор проб реакционной смеси осуществляли пробоотборной трубкой каждые пять минут. Для контроля за протеканием реакции взвешивали контейнер с исходным сырьем и растворенным катализатором, цилиндр с этеном и отведенную реакционную смесь. Реакционную смесь всегда собирали в течение часа, после чего заменяли сосуд.
Сравнительный пример 1-6 и 1-8 и Примеры 1-7 и 1-9
Непрерывные эксперименты, в которых сравнивали активность восстановленного катализатора (то есть дистилляционного остатка) проводили с и без осуществления водной обработки. В первом случае дистилляционный остаток использовали непосредственно в качестве восстановленного катализатора (Сравнительный пример 1-6). Во втором случае реакционную смесь после водной обработки дистилляционного остатка 5% HCl использовали в качестве исходного материала, содержащего восстановленный катализатор (Примеры 1-4 и 1-5).
Сравнительный пример 1-6
587,5 г дистилляционного остатка, содержащего 63,7% TeCPa, 22,8% TeCPna и 7,49% Bu3PO4, смешивали с 2200 г TeCM. Эта смесь содержала 78,7% TeCM, 11,8% TeCPa, 5,8% TeCPna и использовалась в качестве потока сырья для непрерывной организации эксперимента. Реакционный сосуд, представляющий собой автоклав, заполняли реакционной смесью и 8 г свежего железа. Реакцию проводили при 110°С под давлением этена 9 бар. Время пребывания составляло 2,7 часа. В ходе реакции количество прореагировавшего TeCM составляло от 75 до 76%.
Пример 1-7
587,5 г дистилляционного остатка, содержащего 63,7% TeCPa, 22,8% TeCPna, 7,49% Bu3PO4 добавляли по каплям в течение 1,5 часа в 1001,5 г кипящей 5% HCl. Эту смесь затем перегоняли. Из основного продукта перегонки собирали органическую фазу и возвращали водную фазу в виде рефлюкса. Дистилляцию прекращали через час, когда весь дистилляционный остаток был добавлен. Остаток после отгонки разбавляли 200 г TeCM и затем разделяли в делительной воронке. Нижнюю органическую фазу фильтровали и вместе с дистилляционным остатком смешивали с 2000 г TeCM. Эта смесь содержала 81,2% TeCM, 10,8% TeCPa и 5,3% TeCPna. Она использовалась в качестве потока сырья для непрерывной организации эксперимента. Реакционный сосуд (автоклав) заполняли более старой реакционной смесью и 8 г свежего железа. Реакцию проводили при 110°С и давлении этена в 9 бар. Время пребывания в реакторе составляло 2,7 ч/расход. За время реакции количество прореагировавшего TeCM составляло от 83 до 85%.
Сравнительный пример 1-8
Сравнительный пример 1-8 проводили в условиях, идентичных условиям Сравнительного примера 1-6, за исключением того, что использовали другие концентрации четыреххлористого углерода и трибутилфосфата.
Пример 1-9
Пример 1-9 проводили в условиях, идентичных условиям Примера 1-7, за исключением того, что использовали другие концентрации четыреххлористого углерода и трибутилфосфата.
ПРИМЕР 2. ПОЛУЧЕНИЕ 1,1,1,3-ТЕТРАХЛОРПРОПАНА ВЫСОКОЙ СТЕПЕНИ ЧИСТОТЫ
1,1,1,3-Тетрахлорпропан высокой степени чистоты может быть получен в соответствии со способом согласно настоящему изобретению, включающим стадию алкилирования (Фиг. 1), первую стадию дистилляции (Фиг. 2), стадию водного восстановления катализатора (Фиг. 3) и вторую стадию дистилляции (Фиг. 4). Однако следует понимать, что не все эти стадии необходимы для получения C3-6 алкана высокой степени чистоты в соответствии со способами согласно настоящему изобретению.
На стадии алкилирования, показанной на Фиг. 1, этен и порошкообразное железо вводят по линиям 1 и 2 в реактор непрерывного перемешивания 3. Этен вводят в реактор непрерывного перемешивания 3 в газообразной форме через погружную трубку, снабженную соплом. Контролируемую подачу порошкообразного железа осуществляют в реакторе непрерывного перемешивания 3.
Порошкообразное железо периодически подают в реактор непрерывного перемешивания 3 с использованием контролируемой подачи. Постоянное добавление порошкообразного железа необходимо, поскольку при протекании реакции алкилирования порошкообразное железо растворяется в реакционной смеси. Было обнаружено, что оптимальные результаты достигаются путем поддержания присутствия в реакционной смеси порошкообразного железа в концентрации 1-2% относительно массы реакционной смеси в зоне первичного алкилирования. Это приводит к получению реакционной смеси, извлеченной из зоны первичного алкилирования, с содержанием растворенного железа от 0,2 до 0,3% относительно массы реакционной смеси.
Четыреххлористый углерод подают в реактор непрерывного перемешивания 3 по линии 12 в жидкой форме. В проиллюстрированном варианте осуществления поток четыреххлористого углерода используют для улавливания газообразного этена, выделенного из реакционной смеси. Однако использование четыреххлористого углерода таким образом не является существенным для настоящего изобретения; в реактор 3 может подаваться поток свежего четыреххлористого углерода в качестве единственного или основного источника четыреххлористого углерода.
Трибутилфосфат/хлорид железа (III) также подают в реактор непрерывного перемешивания 3 по линии 12. Трибутилфосфат, присутствующий в этом потоке, частично получают в процессе водной обработки, проиллюстрированной на Фиг.3 (и обсуждаемой ниже более подробно), причем равновесие обеспечивают путем добавления свежего трибутилфосфата в систему. Поток на линии 12 дополнительно содержит агент выделения галогеналканов.
В проиллюстрированном варианте осуществления задействована одна зона первичного алкилирования, расположенная в реакторе непрерывного перемешивания 3. Конечно, при необходимости могут быть задействованы несколько зон первичного алкилирования, например, в одном или более реакторах непрерывного перемешивания, которые могут эксплуатироваться параллельно и/или последовательно.
Зона первичного алкилирования работает при давлении, превышающем атмосферное (от 5 до 8 бар) и при повышенной температуре (от 105 до 110°С) и при времени реакции 100-120 минут. Эти условия выбирают так, чтобы четыреххлористый углерод и этен образовывали 1,1,1,3-тетрахлорпропан в реакции алкилирования. Однако было обнаружено, что полная конверсия четыреххлористого углерода в 1,1,1,3-тетрахлорпропан нежелательна, так как это также приводит к образованию нежелательных примесей. Таким образом, уровень конверсии четыреххлористого углерода в целевой хлорированный C3-6 алкан контролируется и в этом варианте осуществления изобретения не допускается превышение им концентрации в 95%. Контроль за ходом реакции алкилирования достигается частично за счет использования условий реакции, которые не способствуют полной конверсии четыреххлористого углерода в 1,1,1,3-тетрахлорпропан, за счет контроля времени пребывания реакционной смеси в реакторе непрерывного перемешивания.
Реакционную смесь, содержащую I) непрореагировавший четыреххлористый углерод и этен, II) 1,1,1,3-тетрахлорпропан (целевой хлорированный C3-6 алкан в этом варианте осуществления) и III) катализатор трибутилфосфат/хлорид железа, извлекают из зоны первичного алкилирования (реактор непрерывного перемешивания 3) и подают в реактор идеального вытеснения 4 (в котором расположена зона основного алкилирования).
Реакционную смесь извлекают таким образом, чтобы порошкообразный железный катализатор, присутствующий в зоне первичного алкилирования 3, не извлекался и, таким образом, реакционная смесь по существу не содержала макрочастиц. Кроме того, в проиллюстрированном варианте осуществления никакой дополнительный катализатор не добавляют в реактор идеального вытеснения 4, хотя в реакторе идеального вытеснения 4 может быть обеспечен каталитический слой. Кроме того, дополнительный этен не добавляют в реактор идеального вытеснения 4.
В проиллюстрированном варианте рабочее давление в зоне основного алкилирования 4 такое же, как и в зоне первичного алкилирования 3. Время пребывания реакционной смеси в зоне осуществления реакции составляет приблизительно 30 минут, что в проиллюстрированном варианте является достаточным для израсходования практически всего присутствующего этена в реакции. Разумеется, должно быть понятно, что для разных типов реактора и условий эксплуатации может являться оптимальным разное время пребывания реакционной смеси в зоне осуществления реакции.
При достижении оптимального времени пребывания реакционной смеси в зоне основного алкилирования реакционную смесь извлекают из нее по линии 5, поддерживая повышенное давление и температуру, и подают в испарительный сосуд 6. В этом сосуде извлеченную реакционную смесь подвергают сбросу давления до атмосферного. Этот перепад давления вызывает выпаривание этена, содержащегося в реакционной смеси. Смесь, обогащенную 1,1,1,3-тетрахлорпропаном, которая по существу не содержит этена, выводят из испарительного сосуда по линии 7 и подвергают стадии дистилляции, показанной на Фиг. 2, что обсуждается ниже более подробно.
Выпаренный этен выводят из испарительного сосуда 6 по линии 8 и подают через конденсатор 9. Этен затем подают по линии 10 в абсорбционную колонку 11, где его приводят в контакт с потоком четыреххлористого углерода и катализатора трибутилфосфата/хлорида железа, восстановленного из реакционной смеси на стадии водной обработки, показанной на Фиг. 3, и описанной ниже более подробно. Поток восстановленного катализатора/четыреххлористого углерода 13 пропускают через охладитель 14 и затем подают по линии 15 в абсорбционную колонку 11.
Поток охлажденного четыреххлористого углерода/катализатора, проходящий через абсорбционную колонку 11, обладает эффектом улавливания этена. Полученный поток жидкого четыреххлористого углерода/катализатора/этена затем подают обратно в реактор непрерывного перемешивания 3.
Как видно из Фиг. 1, проиллюстрированный вариант осуществления включает в себя рециклирование этена, обладающее преимуществами по нескольким причинам. Во-первых, достигается почти полное использование этена, и, таким образом, количество этена, потерянного из системы, является очень низким. Кроме того, энергозатраты компонентов системы рециклирования этена также являются низкими. Кроме того, количество этена, потерянного из системы, также очень низкое, что означает уменьшение загрязнения окружающей среды.
На Фиг. 2 обогащенную 1,1,1,3-тетрахлорпропаном смесь, извлеченную из испарительного сосуда, обозначенного как 6 на Фиг. 1, подают по линии 101 в дистилляционный котел периодического действия 102, который приводят в действие совместно с вакуумной дистилляционной колонкой 104. Дистилляционный котел работает при температуре от 90°С до 95°С. Хлоралканы, присутствующие в смеси, подаваемой в котел 102, выпаривают и разделяют с помощью дистилляционной колонки 104 (и нижестоящего конденсатора 106 и делителя рефлюкса 108) на поток легких фракций 110.1, поток четыреххлористого углерода 110.2, поток тетрахлорэтена 110.3 и поток очищенного продукта 1,1,1,3-тетрахлорпропана 110.4.
Потоки легких фракций и тетрахлорэтена 110.1, 110.3 могут быть использованы для производства четыреххлористого углерода, что является преимущественным с точки зрения минимизации образования отходов. Это может быть достигнуто за счет осуществления процесса высокотемпературного хлоринолиза.
Поток четыреххлористого углерода 110.2 возвращают обратно в реактор непрерывного перемешивания 103. Поток очищенного продукта 1,1,1,3-тетрахлорпропана 110.4 извлекают из системы и он может храниться для отгрузки или использоваться в последующих процессах, для которых необходим чистый 1,1,1,3-тетрахлорпропан в качестве исходного материала.
Смесь, обогащенную 1,1,1,3-тетрахлорпропаном, которая также содержит катализатор, извлекают в виде остатка из котла 102 по линии 103 и подвергают стадии восстановления катализатора, показанной на Фиг. 3.
На этой стадии смесь, обогащенную 1,1,1,3-тетрахлорпропаном, подают в дистилляционный котел периодического действия 204 по линии 202 вместе со слабым (1-5%) раствором соляной кислоты по линии 201.
Преимущественно, вода, присутствующая в кислом растворе, дезактивирует каталитическую систему и защищает ее от термического повреждения. Это позволяет восстанавливать каталитическую систему из обогащенной 1,1,1,3-тетрахлорпропаном смеси, которая может легко быть реактивирована, восстановлена и повторно использована в процессе алкилирования без каких-либо значительных потерь каталитической активности.
Дистилляционный котел периодического действия работает при температуре приблизительно 100°C для создания газообразной смеси, содержащей 1,1,1,3-тетрахлорпропан и водяной пар.
Газообразную смесь, полученную в котле 204, затем подвергают паровой дистилляции (или отгонке паром) неочищенного 1,1,1,3-тетрахлорпропана в колонке 210, которая соединена с котлом 204. Неочищенный 1,1,1,3-тетрахлорпропан отделяют из дистилляционной колонки 210 по линии 211, конденсируют в конденсаторе 212, подают по линии 213 в жидкостно-жидкостный сепаратор рефлюкса 214. Вода из газовой смеси подается обратно в дистилляционную колонку 210 по линии 215, и часть отводится по линии 216 для дополнительной стадии дистилляции, которая более детально показана на Фиг. 4 и обсуждается ниже более подробно.
Рабочую температуру котла 204 затем снижают для остановки отгонки водяным паром, в результате чего в нем происходит конденсация водяного пара. Это приводит к образованию двухфазной смеси, содержащей водную фазу и органическую фазу, содержащую каталитическую систему, которая не подвергалась отгонке водяным паром. Для облегчения выделения органической фазы в котел 204 по линии 203 добавляют агент выделения галогеналканов (в данном случае 1,1,1,3-тетрахлорпропан) для увеличения объема этой фазы.
Выделение органической фазы из двухфазной смеси достигается последовательным выделением фаз из котла 204 по линии 205. Органическую фазу отбирают из котла 204 по линии 205 и фильтруют на фильтре 206. Фильтрационный осадок удаляют из фильтра 206 по линии 207. Органическую фазу отделяют по линии 208 и в этом варианте осуществления направляют обратно в зону первичного алкилирования, как показано на Фиг. 1, в частности, по линии 13 на Фиг. 1. Водную фазу отделяют по линии 205, фильтруют на фильтре 206 и удаляют по линии 209.
Отогнанный неочищенный продукт 1,1,1,3-тетрахлорпропана подвергают дополнительной стадии дистилляции, показанной на Фиг. 4. На этой стадии неочищенный продукт подают в дистилляционный котел 302 по линии 301. Котел 302 сообщается с дистилляционной колонной 304. Выпаренные хлорированные органические соединения, присутствующие в неочищенном 1,1,1,3-тетрахлорпропане, разделяют в дистилляционной колонке 304 (конденсаторе 306 и делителе рефлюкса 308) на поток очищенного продукта 1,1,1,3-тетрахлорпропана 310.1 и хлорированного пентана/пентена 310.2.
Поток хлорированного пентана/пентена 310.2 может быть использован в производстве четыреххлористого углерода, с преимущественной минимизацией образования отходов. Это может быть достигнуто путем использования процесса высокотемпературного хлоринолиза.
Поток очищенного продукта 1,1,1,3-тетрахлорпропана 310.1 извлекают из системы и могут объединить с потоком основного продукта (обозначенным ссылкой 110.4 на Фиг. 2). Продукт может храниться для отгрузки или использоваться в последующих процессах, требующих чистого 1,1,1,3-тетрахлорпропана в качестве исходного материала.
Остатки тяжелых фракций, извлеченные из котла 302 по линии 303, либо удаляются, либо подвергаются дальнейшей переработке.
Используя устройство и условия, описанные выше, непрерывно обрабатывали 2635 кг четыреххлористого углерода (CTC, чистота 99,97%) со средней почасовой загрузкой 78,2 кг/ч для получения 1,1,1,3-тетрахлорпропена (1113TeCPa). Основные параметры описанного способа, проведенного в соответствии с Примером 2, являются следующими.
Полный состав примесей очищенного продукта вышеописанного варианта осуществления представлен в следующей таблице. Обратите внимание, что цифры приведены в виде средневзвешенного примесей для продукта, полученного по линии 110.4 на Фиг. 2 и линии 310.1 на Фиг. 4.
ПРИМЕР 3. ВЛИЯНИЕ НА СЕЛЕКТИВНОСТЬ МОЛЯРНОГО СООТНОШЕНИЯ ИСХОДНОГО МАТЕРИАЛА К ПРОДУКТУ В РЕАКЦИОННОЙ СМЕСИ
Эти примеры были выполнены с использованием оборудования и методов, описанных выше в разделе «Непрерывная организация эксперимента» в Примере 1, за исключением случаев, когда указано иное. Молярное соотношение хлорированного C3-6 алканового продукта (в данном случае 1,1,1,3-тетрахлорпропана) к четыреххлористому углероду в реакционной смеси контролировали до различных степеней, главным образом, за счет времени пребывания реакционной смеси в зоне алкилирования. Температуру поддерживали при 110°С и поддерживали давление 9 бар. Селективность по отношению к целевому продукту, представлена в следующей таблице.
Как можно видеть из этого примера, когда молярное соотношение продукта к исходному материалу превышает 95:5, при эксплуатации способа на непрерывной основе, наблюдается заметное снижение селективности в отношении целевого продукта.
ПРИМЕР 4. ВЛИЯНИЕ НА СЕЛЕКТИВНОСТЬ МОЛЯРНОГО СООТНОШЕНИЯ ИСХОДНОГО МАТЕРИАЛА К ПРОДУКТУ В РЕАКЦИОННОЙ СМЕСИ
Эти примеры были выполнены с использованием оборудования и методов, как показано на Фиг. 1, со ссылкой на сопроводительный текст в Примере 2 выше, за исключением случаев, когда указано иное. Молярное соотношение продукта хлорированного C3-6 алкана (в данном случае 1,1,1,3-тетрахлорпропана) к четыреххлористому углероду в реакционной смеси регулировали до различных степеней, главным образом, путем регулирования скорости подачи исходного материала этена. Температура постоянно составляла 110°C. Селективность по отношению к целевому продукту, представлена в следующей таблице.
Как можно видеть из этого примера, когда молярное соотношение продукта к исходному материалу превышает 95:5, при эксплуатации способа на непрерывной основе, наблюдается заметное снижение селективности в отношении целевого продукта.
ПРИМЕР 5. ВЛИЯНИЕ ЧИСТОТЫ СЫРЬЯ
Эти примеры были выполнены с использованием оборудования и методов, как показано на Фиг. 2, со ссылкой на сопроводительный текст в Примере 2 выше, за исключением случаев, где указано иное. Испытание 5-1 представляет собой поток, полученный из потока 110.4 на Фиг. 2. Испытания 5-2 - 5-5 представляют собой альтернативные примеры, полученные с использованием тех же устройств и методов, но с использованием сырья различной чистоты. Приведенные ниже данные демонстрируют, что хотя исходное сырье более низкой чистоты было использовано в испытаниях 5-2 - 5-5, это предпочтительно не оказывает существенного влияния на чистоту продукта, если его получают способами согласно настоящему изобретению.
ПРИМЕР 6. КОМБИНАЦИЯ CSTR И РЕАКТОРА ИДЕАЛЬНОГО ВЫТЕСНЕНИЯ
Эти примеры были выполнены с использованием оборудования и методов, как показано на Фиг. 1, со ссылкой на сопроводительный текст в Примере 2 выше, за исключением случаев, когда указано иное. Была оценена эффективность реакции во втором реакторе идеального вытеснения (ссылка 4 на Фиг. 1). Было проведено два испытания с различным количеством растворенного этена на входе в реактор идеального вытеснения, который работал при той же температуре 110°C как и основной реактор CSTR (ссылка 3 на Фиг.1). Результаты показаны в следующей таблице.
Как можно видеть из этого примера, в реакторе идеального вытеснения происходит конверсия от 75 до 93% этена. Таким образом, использование реактора идеального вытеснения способствует более эффективному использованию этена в реакционной секции. Серийный реактор идеального вытеснения делает возможной эксплуатацию реактора CSTR при оптимальном давлении, что не требует сложных и неэкономичных процессов восстановления этена. Серийный реактор идеального вытеснения таким образом контролирует использование этена в эффективном замкнутом контуре.
ПРИМЕР 7. ПРОБЛЕМАТИЧНОЕ РАЗЛОЖЕНИЕ КАТАЛИТИЧЕСКОГО ЛИГАНДА ПРИ ОБЫЧНОЙ ДИСТИЛЛЯЦИИ
Устанавливали оборудование для фракционной дистилляции, состоящее из двухлитровой стеклянной четырехгорлой дистилляционной колбы, оснащенной конденсатором, термометром, нагревательной ванной и вакуумным насосом. Вначале дистилляционную колбу наполняли 1976 г реакционной смеси, полученной с использованием аппарата и методик, проиллюстрированного на Фиг. 1 и описанных в сопроводительном тексте в Примере 2 выше.
Во время дистилляции давление постепенно уменьшали от начального давления 100 мм рт.ст. до конечного давления 6 мм рт.ст. В течение этого периода было извлечено 1792 г дистиллята (в разных фракциях). В ходе дистилляции наблюдали видимое образование газообразного HCl, а также в значительных количествах, а именно от 1 до 19% для фракций дистиллята, образовывался хлорбутан (продукт распада трибутилфосфатного лиганда). После этого дистилляцию прерывали, дистилляционный остаток взвешивали и анализировали, было установлено, что содержание тетрахлорпропана составляет 16%. Продолжать перегонку без существенного разложения трибутилфосфатного лиганда было невозможно.
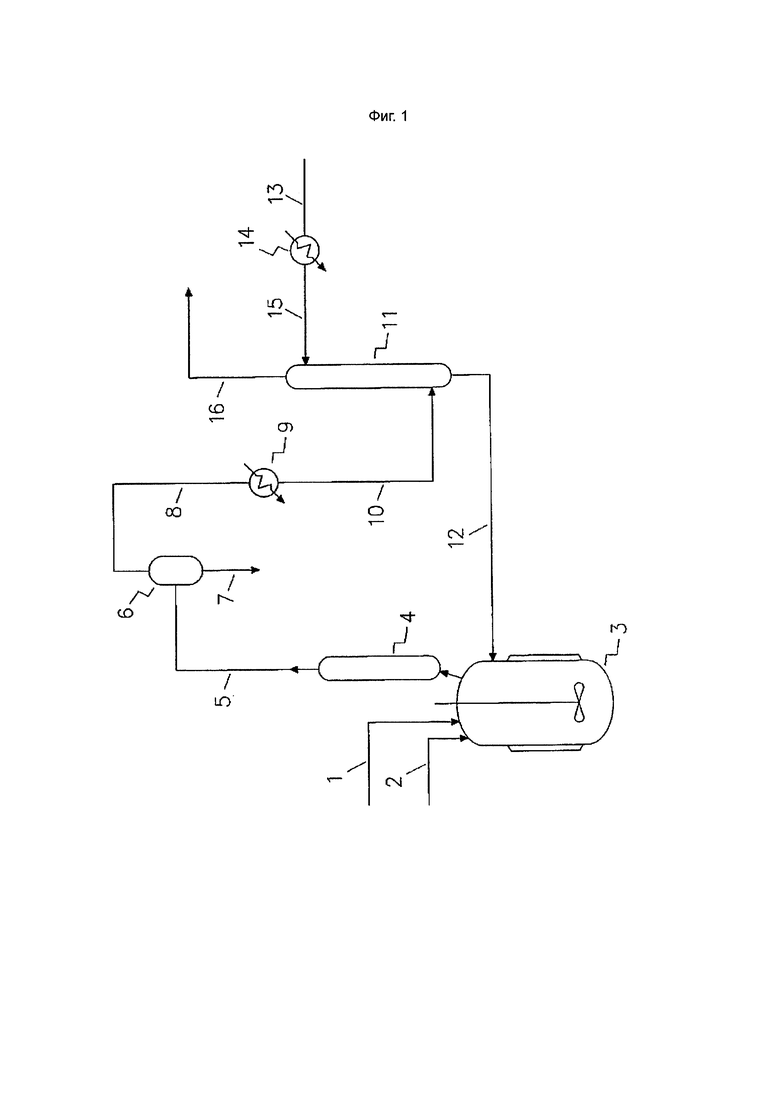
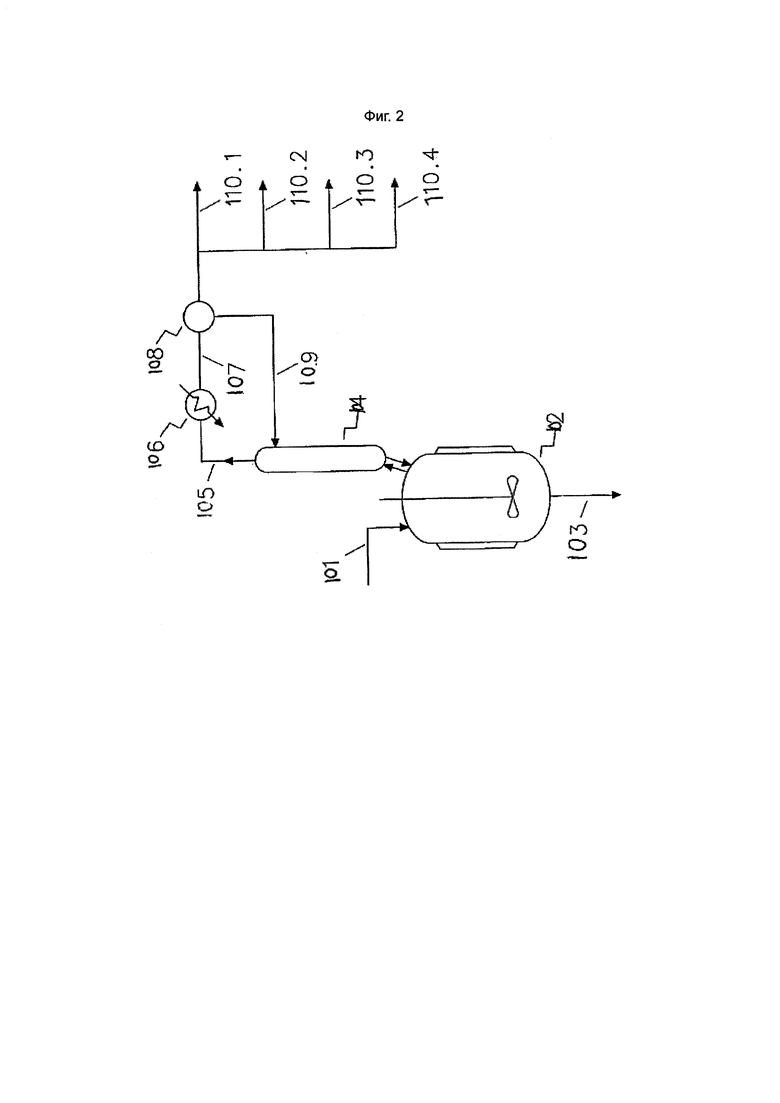
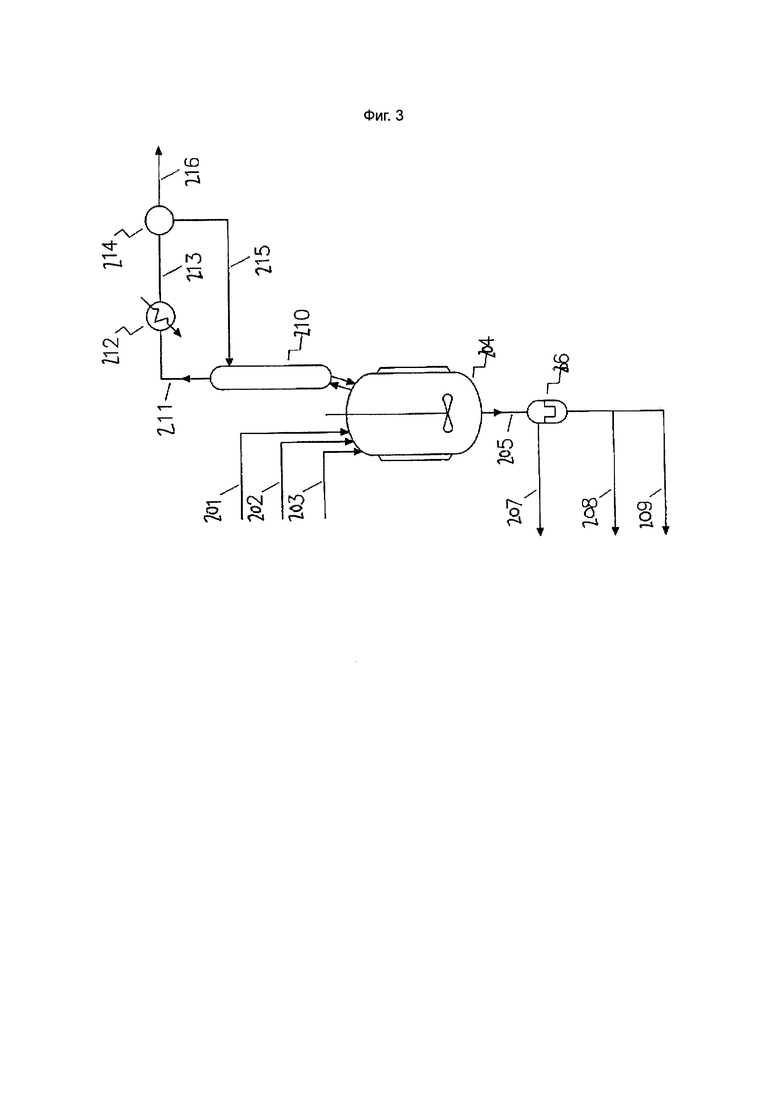
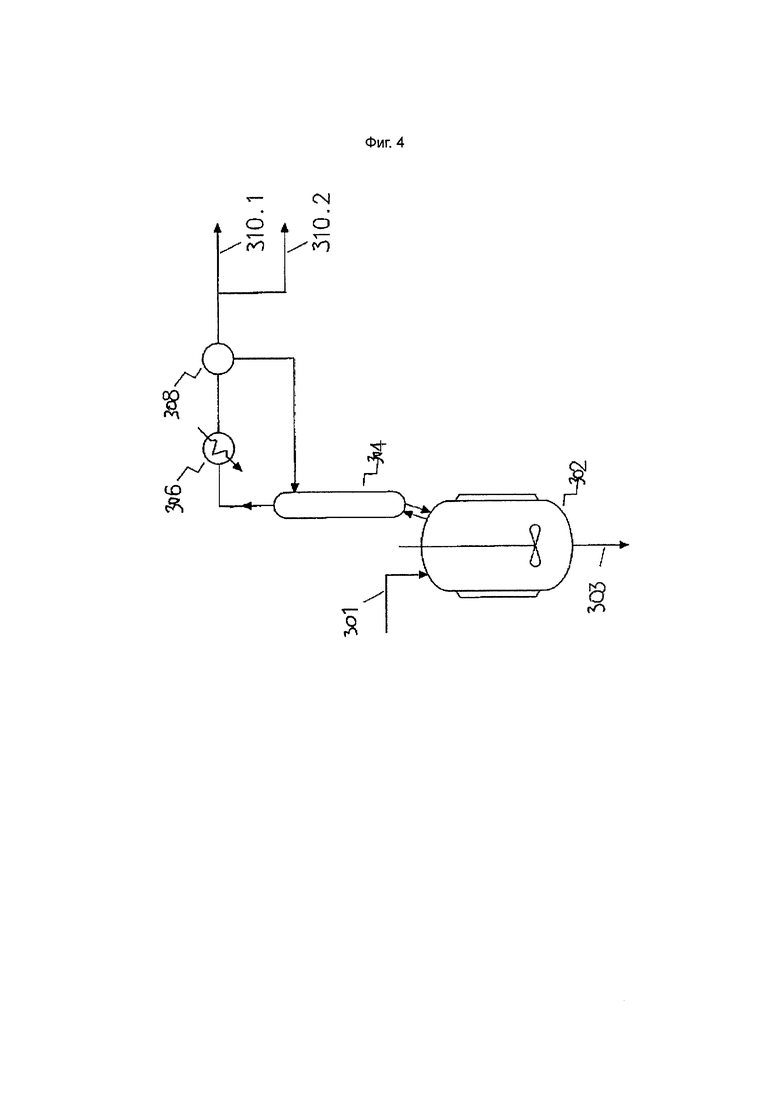
Изобретение относится к высокоселективному способу получения 1,1,1,3-тетрахлорпропана, включающему обеспечение реакционной смеси, содержащей этен, четыреххлористый углерод и металлический катализатор, содержащий алкилфосфат, в зоне основного алкилирования для образования 1,1,1,3-тетрахлорпропана в реакционной смеси, и извлечение части реакционной смеси из зоны основного алкилирования. Согласно изобретению конверсию в реакции поддерживают на уровне менее 95% путем непрерывной дистилляции смеси 1,1,1,3-тетрахлорпропана и катализатора из реакционной смеси; и указанную смесь направляют обратно в реакционную смесь, находящуюся в зоне основного алкилирования; и реакционную смесь, извлеченную из зоны основного алкилирования, подвергают стадии водной обработки, на которой реакционную смесь приводят в контакт с водной средой в зоне водной обработки с образованием двухфазной смеси; и из двухфазной смеси выделяют органическую фазу, содержащую катализатор. Изобретение также относится к композиции 1,1,1,3-тетрахлорпропана, пригодной в качестве исходного сырья в синтезе галогенированного алкена или галогенированного алкана. 3 н. и 21 з.п. ф-лы, 5 табл., 7 пр., 4 ил.
1. Способ получения 1,1,1,3-тетрахлорпропана, включающий обеспечение реакционной смеси, содержащей этен, четыреххлористый углерод и металлический катализатор, содержащий алкилфосфат, в зоне основного алкилирования для образования 1,1,1,3-тетрахлорпропана в реакционной смеси, и извлечение части реакционной смеси из зоны основного алкилирования, отличающийся тем, что:
конверсию в реакции поддерживают на уровне менее 95% путем непрерывной дистилляции смеси 1,1,1,3-тетрахлорпропана и катализатора из реакционной смеси; и
указанную смесь направляют обратно в реакционную смесь, находящуюся в зоне основного алкилирования; и
реакционную смесь, извлеченную из зоны основного алкилирования, подвергают стадии водной обработки, на которой реакционную смесь приводят в контакт с водной средой в зоне водной обработки с образованием двухфазной смеси, и из двухфазной смеси выделяют органическую фазу, содержащую катализатор.
2. Способ по п. 1, отличающийся тем, что указанный катализатор содержит триэтилфосфат и/или трибутилфосфат.
3. Способ по любому из пп. 1 или 2, отличающийся тем, что зона основного алкилирования работает при температуре от 30 до 160°С.
4. Способ по любому из пп. 1-3, отличающийся тем, что зона основного алкилирования работает при температуре от 80 до 120°С.
5. Способ по любому из пп. 1-4, отличающийся тем, что реакционную смесь формируют в зоне основного алкилирования.
6. Способ по любому из пп. 1-5, отличающийся тем, что этен и четыреххлористый углерод приводят в контакт в зоне первичного алкилирования с образованием реакционной смеси, затем реакционную смесь извлекают из зоны первичного алкилирования и подают в зону основного алкилирования, где соотношение 1,1,1,3-тетрахлорпропана к четыреххлористому углероду, присутствующих в реакционной смеси, извлеченной из зоны основного алкилирования, превышает соотношение 1,1,1,3-тетрахлорпропана к четыреххлористому углероду, присутствующих в реакционной смеси, взятой из зоны первичного алкилирования.
7. Способ по п. 6, отличающийся тем, что зона основного алкилирования работает при таком же давлении, что и зона первичного алкилирования, или отличающемся.
8. Способ по п. 6 или 7, отличающийся тем, что зона первичного алкилирования работает в непрерывном режиме.
9. Способ по любому из пп. 6-8, отличающийся тем, что в зону основного алкилирования не подают алкен или твердый металлический катализатор, отличающиеся от присутствующих в реакционной смеси, подаваемой в зону основного алкилирования.
10. Способ по любому из пп. 6-9, отличающийся тем, что реакционная смесь, взятая из зоны первичного алкилирования и подаваемая в зону основного алкилирования, по существу не содержит твердого металлического катализатора.
11. Способ по п. 1, отличающийся тем, что реакционная смесь, извлеченная из зоны основного алкилирования, дополнительно содержит этен, и реакционную смесь подвергают стадии деалкенирования, на которой по меньшей мере 50% по массе или более этена, присутствующего в реакционной смеси, выделяют из нее и по меньшей мере 50% выделенного этена направляют обратно в реакционную смесь, находящуюся в зоне основного алкилирования.
12. Способ по п. 11, отличающийся тем, что стадия деалкенирования включает подачу реакционной смеси, извлеченной из зоны основного алкилирования, в зону дистилляции, в которой этен отгоняют из реакционной смеси.
13. Способ по п. 12, отличающийся тем, что этен, отогнанный из реакционной смеси, получают в потоке, обогащенном указанным этеном.
14. Способ по п. 12 или 13, отличающийся тем, что зона дистилляции представляет собой зону выпаривания, в которой этен выпаривают из реакционной смеси.
15. Способ по любому из пп. 1-14, отличающийся тем, что от 30 до 85% по массе 1,1,1,3-тетрахлорпропана, присутствующего в реакционной смеси, извлеченной из зоны основного алкилирования, удаляют из реакционной смеси до стадии водной обработки, необязательно дистилляцией.
16. Способ по любому из пп. 1-15, отличающийся тем, что водная среда содержит разбавленную кислоту, необязательно разбавленную соляную кислоту.
17. Способ по любому из пп. 1-16, отличающийся тем, что большую часть 1,1,1,3-тетрахлорпропана, присутствующего в реакционной смеси, подаваемой в зону водной обработки, выделяют из смеси, образованной в зоне водной обработки, необязательно дистилляцией.
18. Способ по любому из пп. 1-17, дополнительно включающий стадию подачи четыреххлористого углерода и/или 1,1,1,3-тетрахлорпропана в зону водной обработки.
19. Способ по любому из пп. 1-18, отличающийся тем, что двухфазная смесь образуется в зоне водной обработки.
20. Способ по любому из пп. 1-19, отличающийся тем, что органическую фазу выделяют из двухфазной смеси путем последовательного разделения фаз.
21. Способ по любому из пп. 1-20, отличающийся тем, что пар подается в зону водной обработки и/или образуется in situ кипящей водой, присутствующей в зоне водной обработки, с образованием газообразной смеси, содержащей 1,1,1,3-тетрахлорпропан и водяной пар, из которой 1,1,1,3-тетрахлорпропан может быть дистиллирован.
22. Композиция 1,1,1,3-тетрахлорпропана, пригодная в качестве исходного сырья в синтезе галогенированного алкена или галогенированного алкана, содержащая по массе:
99,0% или более, 99,5% или более, 99,7% или более, 99,8% или более или 99,9% или более 1,1,1,3-тетрахлорпропана,
менее чем 2000 млн-1, менее чем 1000 млн-1, менее чем 500 млн-1, менее чем 200 млн-1 или менее чем 100 млн-1 примесей хлорированных алканов (то есть соединений хлорированных алканов, отличных от 1,1,1,3-тетрахлорпропана),
менее чем 2000 млн-1, менее чем 1000 млн-1, менее чем 500 млн-1, менее чем 200 млн-1 или менее чем 100 млн-1 соединений хлорированных алкенов,
менее чем 2000 млн-1, менее чем 1000 млн-1, менее чем 500 млн-1, менее чем 200 млн-1 или менее чем 100 млн-1 кислородосодержащих органических соединений,
менее чем 2000 млн-1, менее чем 1000 млн-1, менее чем 500 млн-1, менее чем 200 млн-1 или менее чем 100 млн-1 бромированных соединений,
менее чем 1000 млн-1, менее чем 500 млн-1, менее чем 200 млн-1, менее чем 100 млн-1, менее чем 50 млн-1 или менее чем 20 млн-1 воды,
менее чем 500 млн-1, менее чем 200 млн-1, менее чем 100 млн-1, менее чем 50 млн-1 или менее чем 20 млн-1 металлического катализатора и/или
менее чем 500 млн-1, менее чем 200 млн-1, менее чем 100 млн-1, менее чем 50 млн-1 или менее чем 20 млн-1 промотора катализатора.
23. Применение композиции по п. 22 в качестве исходного сырья в синтезе галогенированного алкена или галогенированного алкана.
24. Применение по п. 23, отличающееся тем, что галогенированный алкен или галогенированный алкан представляет собой фторированный и/или хлорированный алкен или фторированный и/или хлорированный алкан.
| US 2012053374 A1, 01.03.2012 | |||
| US 2004225166 A1, 11.11.2004 | |||
| WO 2010131766 A2, 18.11.2010 | |||
| Способ получения низших тетрахлоралканов | 1968 |
|
SU255229A1 |

