Область техники, к которой относится изобретение
Настоящее изобретение относится к области фармации и медицины, а именно к водным композициям анти-IL-6R антитела левилимаба, которые могут быть использованы в качестве лекарственного средства для лечения IL-6R-ассоциированных заболеваний.
Уровень техники
Интерлейкин-6 (IL-6, IL6) является одним из основных провоспалительных цитокинов. IL-6 продуцируется активированными моноцитами, макрофагами, Т-клетками, а также рядом других клеток. Наряду с другими цитокинами он участвует в процессах иммунного ответа, воспаления, ангиогенеза, костного метаболизма. Основное действие IL-6 связано с его участием в качестве кофактора в дифференцировке В-лимфоцитов, их созревании и трансформации в плазматические клетки, секретирующие иммуноглобулины. Помимо этого, IL-6 индуцирует экспрессию рецептора IL-6 на активированных клетках иммунной системы, а также индуцирует продукцию IL-2 Т-лимфоцитами. IL-6 стимулирует пролиферацию Т-лимфоцитов и реакции гемопоэза. По многообразию клеток продуцентов и мишеней биологического действия IL-6 является одним из наиболее активных цитокинов, участвующих в реализации иммунного ответа и воспалительной реакции. Установлено, что дисбаланс между про- и антивоспалительными действиями IL-6 приводит к различным аутоиммунным заболеваниям, хроническому воспалению и остеопорозу, псориазу, а избыточная продукция ассоциирована с различными формами рака.
Таким образом, подавление действия IL-6 является привлекательной терапевтической мишенью (PeterC. HeinrichBiochem. J. (2003) 374:1).
Рецептор IL-6 (IL-6R, IL6R), активируясь, запускает каскад реакций в клетке, приводящих к активному синтезу белков, участвующих в реакциях воспалительного ответа. Рецептор активируется при связывании IL-6 с альфа-субъединицей рецептора IL-6 (CD126), и двух молекул gp130, передающих сигнал внутрь клетки (SimonA.JonesTheFASEBJournal 15(1): 43-58). Существуют 2 формы альфа-рецептора: мембранная (mIL-6R) и растворимая (sIL-6R). Растворимая форма образуется в результате протеолиза трансмембранной части mIL-6R или в процессе альтернативного сплайсинга мРНК mIL-6R. Растворимая форма
sIL-6R обеспечивает реакцию на IL-6 клеток, не имеющих mIL-6R на поверхности.
Таким образом, проведение сигнала IL-6 внутрь клетки возможно 2 путями. Первый (классический сигналинг), при котором IL-6 связывается с клетками иммунной системы, экспрессирующими на своей поверхности mIL-6R ассоциированную с молекулой gp130. Второй (транс-сигналинг)– IL-6 связывается с циркулирующей sIL-6R, формируя комплекс, который связывается с клетками, имеющими на мембране только молекулы gp130, т.е. потенциально любыми клетками человеческого организма. В этом случае, на мембране клетки происходит сборка полного комплекса рецептора IL-6 и последующий запуск сигнального каскада в клетке.
Блокирования действия IL-6 и, следовательно, воспалительной реакции можно добиться, помешав сборке полного комплекса рецептора IL-6, состоящего из альфа-субъединицы, молекул gp130 и IL-6. Полипептиды, связываясь с IL-6R, способны препятствовать сборке полного комплекса, соответственно, блокируют проведение сигнала внутрь клетки.
Полипептиды, специфически связывающиеся с IL-6 (патент RU2550262), IL-6R или gp130 показали значительное ингибирующее влияние на функционирование IL-6. В настоящее время известно антитело, связывающееся с IL-6R, тоцилизумаб, которое представляет собой рекомбинантное гуманизированное моноклональное антитело подкласса иммуноглобулина IgG1κ (гамма-1, каппа), сконструированное путем переноса региона, определяющего комплементарность (CDR) мышиного анти-IL-6R антитела в человеческий IgG1.
Препараты на основе антитела (тоцилизумаб) связывающегося с IL-6R и блокирующего взаимодействие с IL-6 применяются при лечении ревматоидного артрита и системного ювенильного идиопатического артрита, как в виде монотерапии, так и в комбинации с метотрексатом и/или с другими базисными противовоспалительными препаратами.
Также известно новое антитело к IL-6R - левилимаб (также известный как BCD-089), который представляет собой моноклональное антитело изотипа IgG1 c внесенными в константную часть мутациями. Левилимаб в настоящее время проходит клинические исследования у пациентов с различными заболеваниями, включая ревматоидный артрит и (острый) респираторный дистресс-синдром у взрослых.
Известно, что применение моноклональных антител против рецептора интерлейкина-6 (ИЛ6Р, IL-6R) позволяет эффективно купировать синдром цитокинового шторма, который развивается при использовании CAR-T терапии в онкологии. Эффективность терапии (купирование синдрома в течение 14 дней от первого и единственного введения) достигает 69%
В контексте пандемии COVID-19, показано успешное применение анти- IL-6R терапии у пациентов с тяжелым или критическим течением COVID-19 пневмонии. Проведенный мета-анализ опубликованных данных эффективности ингибиторов IL-6R у пациентов с COVID-19 позволил предварительно подтвердить их эффективность (Xu, X.; Han, M.; etc., Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020; Coomes, E. A.; Haghbayan, H., Interleukin-6 in COVID-19: A Systematic Review and Meta-Analysis. medRxiv 2020, 2020.03.30.20048058).
В клиническом течении пневмонии COVID-19 существует период окна между постановкой диагноза и развитием синдрома полиорганной недостаточности, составляющий около 5-7 дней, по истечении которого у большинства пациентов наблюдается улучшение, однако у около 20% пациентов наблюдается нарастание тяжести пневмонии (СВЦ, ОРДС). Для улучшения прогноза и снижения летальности, рекомендуется применение упреждающей противовоспалительной терапии, начиная с момента постановки диагноза COVID-19 пневмонии (Sun, X.; Wang, T.; etc., Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev 2020).
Ингибиторы IL-6R включены в российские рекомендации по лечению COVID-19 в качестве препаратов упреждающей противовоспалительной терапии COVID-19 у взрослых (для пациентов со среднетяжелым и тяжелым течением: с острым респираторным дистресс-синдромом, синдромом цитокинового шторма).
Таким образом, в настоящее время является актуальной разработка новых улучшенных стабильных водных фармацевтических композиций для антитела к IL-6R левилимаба.
Краткое описание чертежей
Фигура 1 представляет собой график зависимости оптической плотности растворов при 400 нм от концентрации ПЭГ для моноклонального антитела против рецептора IL-6 левилимаба в исследуемых составах.
Фигура 2 представляет собой график, иллюстрирующий температурный тренд фармацевтической композиции 5 Acet.Buf + 300Glu (выбор осмотического агента).
Фигура 3 представляет собой график, иллюстрирующий температурный тренд фармацевтической композиции 5 Acet Buf. +Mann (выбор осмотического агента).
Фигура 4 представляет собой график, иллюстрирующий температурный тренд фармацевтической композиции 5 Acet. Buf + 100Arg + Mann (выбор осмотического агента).
Фигура 5 представляет собой график, иллюстрирующий температурный тренд фармацевтической композиции 5 Acet Buf. + 200Arg (выбор осмотического агента).
Фигура 6 представляет собой график, иллюстрирующий изменение показателей качества в зависимости от времени при ускоренном хранении в концентрации 220 мг/мл левилимаба.
Фигура 7 представляет собой график, иллюстрирующий изменение показателей качества в зависимости от времени при ускоренном хранении в концентрации 180 мг/мл левилимаба.
Фигура 8 представляет собой график, иллюстрирующий изменение показателей качества в зависимости от времени при ускоренном хранении в концентрации 20 мг/мл левилимаба.
Фигура 9 представляет собой график, иллюстрирующий долю пациентов, достигших улучшения в течение болезни, соответствующее ACR20 к 4, 8, 12, 16, 24, 36, 48 и 52 неделе.
Фигура 10 представляет собой график, иллюстрирующий долю пациентов, достигших улучшения в течение болезни, соответствующее ACR50 к 4, 8, 12, 16, 24, 36, 48 и 52 неделе.
Фигура 11 представляет собой график, иллюстрирующий долю пациентов, достигших улучшения в течение болезни, соответствующее ACR70 к 4, 8, 12, 16, 24, 36, 48 и 52 неделе.
Фигура 12 представляет собой график, иллюстрирующий изменение индекса DAS-28-CRP относительно исходного уровня на протяжение 52 недель терапии.
Фигура 13 представляет собой график, иллюстрирующий долю больных, достигших ремиссии болезни к 24, 36, 48 и 52 неделям терапии.
Фигура 14 представляет собой график, иллюстрирующий изменения СОЭ на фоне терапии.
Фигура 15 представляет собой график, иллюстрирующий динамику концентрации растворимого рецептора интерлейкина-6 у пациентов на протяжении 12 недель терапии.
Фигура 16 представляет собой график, иллюстрирующий изменение концентрации C-реактивного белка в сыворотке крови пациентов на протяжении 12 недель терапии.
Описание изобретения
Определения
Если иное не определено в настоящем документе, научные и технические термины, используемые в связи с настоящим изобретением, будут иметь значения, которые обычно понятны специалистам в данной области.
Кроме того, если по контексту не требуется иное, термины в единственном числе включают в себя термины во множественном числе, и термины во множественном числе включают в себя термины в единственном числе. Как правило, используемая классификация и методы культивирования клеток, молекулярной биологии, иммунологии, микробиологии, генетики, аналитической химии, химии органического синтеза, медицинской и фармацевтической химии, а также гибридизации и химии белка и нуклеиновых кислот, описанные в настоящем документе, хорошо известны специалистам и широко применяются в данной области. Ферментативные реакции и способы очистки осуществляют в соответствии с инструкциями производителя, как это обычно осуществляется в данной области, или как описано в настоящем документе.
Термин «антитело» или «иммуноглобулин» (Ig), как использовано в данном описании, включает полноразмерные антитела и любой антигенсвязывающий фрагмент (т.е. «антигенсвязывающую часть») или его отдельные цепи.
Термин «антигенсвязывающая часть» антитела или «антигенсвязывающий фрагмент» (или просто «часть антитела» или «фрагмент антитела»), как использовано в данном описании, относится к одному или нескольким фрагментам антитела, которые сохраняют способность специфически связываться с антигеном. Было показано, что антигенсвязывающая функция антитела может выполняться фрагментами полноразмерного антитела. Примеры связывающих фрагментов, включенных в термин “антигенсвязывающая часть" антитела включают (i) Fab-фрагмент, одновалентный фрагмент, состоящий из доменов VL, VH, CL и CH 1; (ii) F(ab’)2-фрагмент, двухвалентный фрагмент, содержащий два Fab-фрагмента, связанных дисульфидным мостиком в шарнирной области; (iii) Fd-фрагмент, состоящий из доменов VH и CH 1; (iv) Fv-фрагмент, состоящий из доменов VL и VH в едином плече антитела, (v) dAb-фрагмент (Ward et al., (1989) Nature 341:544-546), который состоит из домена VH/VHH; и (vi) выделенная определяющая комплементарность область (CDR). Кроме того, две области Fv-фрагмента, VL и VH, кодируются разными генами, они могут быть соединены при помощи рекомбинантных способов с использованием синтетического линкера, который дает возможность получать их в виде единой белковой цепи, в которой области VL и VH спариваются с образованием одновалентных молекул (известных как одноцепочечный Fv (scFv); см., например, Bird et al. (1988) Science 242:423-426; и Huston et al. (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883). Предполагается, что такие одноцепочечные молекулы также включены в термин “антигенсвязывающая часть" антитела. Такие фрагменты антител получают с использованием общепринятых способов, известных специалистам в данной области, и эти фрагменты подвергают скринингу таким же образом, как и интактные антитела.
Предпочтительно CDR антигенсвязывающего участка или весь антигенсвязывающий участок антител по изобретению имеет происхождение из мыши, ламы или донорской человеческой библиотеки или по существу человеческое происхождение с определенными аминокислотными остатками, измененными, например, замещенными разными аминокислотными остатками с тем, чтобы оптимизировать конкретные свойства антитела, например KD, koff, IC50, EC50, ED50. Предпочтительно каркасные участки антитела по изобретению имеют человеческое происхождение или по существу человеческое происхождение (по крайней мере на 80, 85, 90, 95, 96, 97, 98 или 99% человеческое происхождение).
Термин «моноклональное антитело» или «mAb» относится к антителу, которое синтезировано и выделено отдельной клональной популяцией клеток. Клональная популяция может быть клональной популяцией иммортализованных клеток. В некоторых вариантах осуществления изобретения иммортализованные клетки в клональной популяции являются гибридными клетками, гибридомами, которые обычно получают путем слияния отдельных В-лимфоцитов от иммунизированных животных с отдельными клетками лимфоцитарной опухоли. Гибридомы представляют собой тип сконструированных клеток и не встречаются в природе.
Популяция «моноклональных антител», как используется в настоящем документе, относится к гомогенной или по существу гомогенной популяции антител (т.е. по крайней мере приблизительно 96%, но более предпочтительно по крайней мере приблизительно 97 или 98% или еще более предпочтительно по крайней мере 99% антител в популяции будут конкурировать в иммунно-ферментном анализе ELISA за тот же антиген или эпитоп, или более предпочтительно антитела являются идентичными в аминокислотной последовательности).
Полноразмерное антитело, существующее в природе, представляет собой молекулу иммуноглобулина, которая состоит из четырех полипептидных цепей (две тяжелые (Н) цепи (приблизительно 50-70 кДа при полной длине) и две легкие (L) цепи (приблизительно 25 кДа при полной длине)), связанных дисульфидными мостиками. Аминоконцевая часть каждой цепи включает вариабельный домен из приблизительно 100-110 или более аминокислот, которые отвечают за связывание антигена. Карбокси-концевая часть каждой цепи определяет константный участок, главным образом, отвечающий за функцию эффектора. Легкие цепи классифицируют как каппа или лямбда, и они характеризуются специфичным константным участком. Каждая легкая цепь состоит из вариабельного участка N-концевой легкой цепи (в данной заявке «VL» или «VK») и константного участка легкой цепи, состоящего из одного домена (CL или СК). Тяжелые цепи классифицируют как γ (гамма), δ (дельта), альфа (α), мю (μ) или эпсилон (ε), и они определяют изотип антитела, такой как IgG, IgM, IgA, IgD и IgE, соответственно, и несколько из них могут быть дополнительно разделены на подклассы (изотипы), такие как, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2. Каждый тип тяжелой цепи характеризуется конкретным константным участком Fc. Каждая тяжелая цепь состоит из вариабельного участка N-концевой тяжелой цепи (в данной заявке «VH») и константного участка CH (тяжелой цепи). Константный участок тяжелой цепи состоит из трех доменов (CH1, СН2 и СН3) для IgG, IgD и IgA и 4 доменов (CH1, СН2, СН3 и СН4) для IgM и IgE. Вариабельные домены VH и VL могут быть дополнительно разделены на участки гипервариабельности (гипервариабельные участки, CDR), чередующиеся с более консервативными каркасными участками (FR). Каждый вариабельный домен состоит из трех CDR и четырех FR, расположенных в следующем порядке от N-конца к С-концу: FR1, CDR1, FR2, CDR2, FR3, CDR3 и FR4.
Вариабельные участки каждой из пар легкая/тяжелая цепь образуют антиген-связывающие сайты антитела. Таким образом, интактное IgG антитело имеет два сайта связывания. За исключением бифункциональных или биспецифических антител два сайта связывания являются одинаковыми. Как используется в данной заявке, «антигенсвязывающая часть», или «антигенсвязывающий участок», или «антигенсвязывающий домен» относятся, взаимозаменяемо, к такой части молекулы антитела, которая содержит аминокислотные остатки, взаимодействующие с антигеном и обуславливающие специфичность и аффинность антитела по отношению к антигену. Такая часть антитела включает «каркасные» аминокислотные остатки, необходимые для поддержания надлежащей конформации антиген-связывающих остатков.
«Фрагмент антитела» может представлять собой фрагмент антитела или фрагмент антитела, имеющий активность полноразмерного антитела. Указанный фрагмент антитела может представлять собой F(ab')2, F(ab)2, Fab', Fab Fv и scFv.
Термин «ингибировать» или «нейтрализовать», как используется в данной заявке, по отношению к функциональной активности антитела по изобретению, означает способность в значительной степени препятствовать, предотвращать, ограничивать, замедлять, прекращать, уменьшать или обращать, например, развитие или тяжесть того, что ингибируют, включая, но не ограничиваясь вышеприведенными, биологическую активность (например, активность IL-6R) или свойство, заболевание или состояние. Ингибирование или нейтрализация активности IL-6R в результате связывания антитела по изобретению с IL-6R составляет предпочтительно по крайней мере приблизительно 20, 30, 40, 50, 60, 70, 80, 90, 95% или выше.
Термин «выделенный» или «изолированный» при использовании по отношению к нуклеиновой кислоте или белковому препарату (например, антителу) относится к молекуле нуклеиновой кислоты или белковой молекуле, которые идентифицируют и отделяют по крайней мере от одного контаминантного вещества, с которым она обычно связана в природном источнике. Предпочтительно «выделенное антитело» является антителом, которое по существу не содержит другие антитела, обладающие отличительной антигенной специфичностью (например, фармацевтические композиции, согласно настоящему изобретению, содержат выделенное антитело, которое специфически связывает IL-6R и по существу не содержит антитела, которые специфически связывают антигены, отличные от IL-6R).
Термин «специфическое связывание», как используется в данной заявке, относится к той ситуации, при которой один участник пары специфического связывания не связывает в значительной степени молекулы, отличные от его партнера (партнеров) по специфическому связыванию. Термин также применим, когда, например, антигенсвязывающий домен антитела по изобретению является специфическим по отношению к конкретному эпитопу, который переносится рядом антигенов, в таком случае специфическое антитело, имеющее антигенсвязывающий домен, будет способно к специфическому связыванию различных антигенов, несущих эпитоп.
«Kabat номенклатура» или «номенклатура по Kabat» применяются в данной заявке к системе нумерации аминокислотных остатков, которые являются более вариабельными (т.е. гипервариабельными), чем остальные аминокислотные остатки в вариабельных участках тяжелой и легкой цепи антитела (Kabat et al. Ann. N.Y. Acad. Sci., 190:382-93 (1971); Kabat et al. Sequences of Proteins of Immunological Interest, Fifth Edition, U.S. Department of Health and Human Services, NIH Publication No. 91-3242 (1991)).
Термин «фармацевтическая композиция» относится к композиции и/или составу, содержащему антитело согласно изобретению в терапевтически эффективном количестве и эксцепиенты или вспомогательные вещества (носители, разбавители, наполнители, растворители и другие эксцепиенты).
Термин «буфер» или «буферный раствор» относится к водному раствору, содержащему смесь кислоты (обычно слабой кислоты, такой как, например, уксусная кислота, лимонная кислота) и ее конъюгированного основания (такой как, например, ацетатной или цитратной соли, например, ацетат натрия, цитрат натрия, а также гидраты указанных солей, например, натрия ацетат тригидрат) или альтернативно смесь основания (обычно слабого основания, например, гистидина) и его конъюгированной кислоты (например, гистидина гидрохлорида). Значение рН «буферного раствора» мало изменяется при добавлении к нему небольшого количества сильного основания или сильной кислоты, а также при разбавлении и концентрировании, благодаря «буферному эффекту», обеспечиваемому «буферным агентом».
В настоящей заявке, «буферная система» содержит один или несколько буферных агентов и/или их конъюгата(ов) с кислотой или основанием, и более подходяще содержит один или несколько буферных агентов и их конъюгата(ов) с кислотой или основанием, и наиболее подходяще содержит только один буферный агент и его кислотный/щелочной конъюгат. Если не указано иное, любые концентрации, указанные в настоящем изобретении по отношению к «буферной системе» (концентрация буфера) могут относиться к объединенной концентрации буферного(ых) агента(ов) и/или его конъюгата(ов) с кислотой или основанием. Другими словами, концентрации, указанные в настоящей заявке по отношению к «буферной системе», могут относиться к объединенной концентрации релевантных буферных видов (то есть, видов в динамическом равновесии друг с другом, например, цитрат/лимонная кислота). Суммарное значение рН композиции, содержащей релевантную буферную систему, является отображением равновесной концентрация каждого из релевантных буферных видов (то есть баланса буферного(ых) агента(ов) с его конъюгатом(ами) с кислотой или основанием).
Термин «буферный агент» относится к кислотному или щелочному компоненту (обычно слабой кислоте или слабому основанию) буфера или буферного раствора. Буферный агент помогает поддерживать значение рН данного раствора при или около заранее определенного значения, и буферные агенты обычно выбирают для дополнения заранее определенного значения. Буферный агент может представляет собой единственное соединение, которое приводит к желательному буферному эффекту, в особенности, если указанный буферный агент смешан с (и подходяще способен к протонному обмену с) подходящим количеством (в зависимости от заранее определенного желательного значения) его соответствующего «кислотного/щелочного конъюгата».
Термин «солюбилизатор» при использовании в данном тексте означает фармацевтически приемлемое неионногенное поверхностно-активное вещество. Можно использовать один солюбилизатор, а также комбинации солюбилизаторов. Примерами солюбилизаторов являются, но не ограничиваются ими, полисорбат 20 или полисорбат 80, полоксамер 184 или полоксамер 188, или PLURONIC®.
Термины «осмотический агент» или «агент, регулирующий тоничность», а также осмолитик в том виде, как они здесь использованы, относятся к эксципиенту, который может обеспечивать требуемое осмотическое давление жидкого раствора антитела. В некоторых воплощениях агент, регулирующий тоничность, может подводить осмотическое давление жидкого препарата антитела до изотоничного так, что данный препарат антитела является физиологически совместимым с клетками ткани организма субъекта. В еще одном воплощении «агент, регулирующий тоничность», может способствовать увеличению стабильности антител. «Изотоничный» препарат представляет собой препарат, который имеет осмотическое давление, эквивалентное человеческой крови. Изотоничные препараты обычно имеют осмотическое давление от примерно 239 до 376 мОсм/кг. Термин «гипотонический» описывает препарат с осмотическим давлением, меньшим, чем осмотическое давление человеческой крови. Соответственно, термин «гипертонический» используется для описания препарата с осмотическим давлением, превышающим осмотическое давление человеческой крови. Изотоничность можно измерять с использованием, например, парового или криоскопического осмометра. Агент, регулирующий тоничность, может находиться в энантиомерной (например, L- или D-энантиомер) или рацемической форме; в форме изомеров, таких как альфа или бета, включая альфа, альфа; или бета, бета; или альфа, бета; или бета, альфа; в форме свободной кислоты или свободного основания; в форме соли; в гидратированной форме (например, моногидрат) или в безводной форме. Примерами осмотических агентов являются, но не ограничиваются ими, сахара (трегалозы дигидрат, сахароза, глюкоза), полиолы (маннитол, сорбитол), аминокислоты (пролин, аргинин, глицин), или соли (натрия хлорид, калия хлорид, магния хлорид).
Термин «длительное хранение» или «долговременная стабильность» следует понимать, как обозначение того, что фармацевтическая композиция может храниться в течение трех месяцев или более, в течение шести месяцев или более и предпочтительно в течение одного года или более, наиболее предпочтительно, с минимальным сроком хранения в стабильном состоянии по меньшей мере два года. В общем, термины «длительное хранение» и «долговременная стабильность» дополнительно включают продолжительности хранения в стабильном состоянии, которые по меньшей мере сравнимы или лучше, чем срок хранения в стабильном состоянии, как правило, необходимый для доступных в настоящее время коммерческих составов антитела к IL-6R левилимаба без потерь в стабильности, которые могут сделать состав непригодным для определенного для него фармацевтического применения.
Термин «парентеральное введение» означает режимы введения, обычно выполняемые с помощью инъекции (инфузии), и включает, в частности, внутривенную, внутримышечную, внутриартериальную, внутритрахеальную, внутрикапсулярную, внутриорбитальную, внутрикардиальную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, внутрисуставную, субкапсулярную, субарахноидальную, внутриспинальную, эпидуральную и надчревную инъекцию или инфузию.
Термин «лекарственное средство» или «препарат» подразумевает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, растворов, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «заболевание или нарушение, ассоциированное с IL-6R» или «заболевание или нарушение, опосредованное IL-6R» подразумевает все заболевания или нарушения, которые либо прямо, либо косвенно связаны с активацией сигнального пути IL6, включая этиологию, патогенез, прогрессирование, персистирование, или патологию заболевания или нарушения.
Термин «применение» относится к возможности применения антитела согласно изобретению или фармацевтической композиции, его содержащей, для лечения, облегчения течения заболеваний, для ускорения ремиссии, снижения частоты рецидивов заболеваний или нарушений, опосредуемых рецепторами, с которыми может связываться антитело согласно изобретению. Примерами заболеваний являются, но не ограничиваются ими, ревматоидный артрит, ювенильный хронический артрит, склеродермия, реакция "трансплантат против хозяина", отторжение трансплантата органа, острое или хроническое иммунное заболевание, связанное с трансплантацией органа, кахексия, взрослый (острый) респираторный дистресс-синдром, болезнь Стилла, системная склеродермия, синдром Шегрена, болезнь/артериит Такаясу, нарушения, связанные с цитокиновой терапией, синдром выброса цитокинов (синдром высвобождения цитокинов), иридоциклит, увеит, оптический неврит, оптический нейромиелит, ювенильный ревматоидный артрит, гигантоклеточный артериит, полиартикулярный ювенильный идиопатический артрит, системный ювенильный идиопатический артрит; рак, в частности, множественная миелома и злокачественные солидные опухоли, колоректальный рак, рак предстательной железы, рак яичника.
Термин «способ лечения» относится к возможности применения антитела согласно изобретению или фармацевтической композиции, его содержащей, для лечения, облегчения течения заболеваний, для ускорения ремиссии, снижения частоты рецидивов вследствие заболеваний или нарушений, связанных с активностью IL-6R. «Лечить» или «лечение», «профилактика» заболевания, нарушения или состояния может включать предотвращение или замедление появления клинических симптомов заболевания, нарушения или состояния, развивающегося у человека, ингибирования заболевания, нарушения или состояния, то есть остановки, уменьшения или замедления развития заболевания или его рецидива (в случае поддерживающей терапии) или по меньшей мере его одного клинического или субклинического симптома, или облегчение или ослабление заболевания, то есть вызывание регресса заболевания, нарушения или состояния. Примерами заболеваний являются, но не ограничиваются ими, ревматоидный артрит, ювенильный хронический артрит, склеродермия, реакция "трансплантат против хозяина", отторжение трансплантата органа, острое или хроническое иммунное заболевание, связанное с трансплантацией органа, кахексия, взрослый (острый) респираторный дистресс-синдром, болезнь Стилла, системная склеродермия, синдром Шегрена, болезнь/артериит Такаясу, нарушения, связанные с цитокиновой терапией, синдром выброса цитокинов (синдром высвобождения цитокинов), иридоциклит, увеит, оптический неврит, оптический нейромиелит, ювенильный ревматоидный артрит, гигантоклеточный артериит, полиартикулярный ювенильный идиопатический артрит, системный ювенильный идиопатический артрит; рак, в частности, множественная миелома и злокачественные солидные опухоли, колоректальный рак, рак предстательной железы, рак яичника.
Термин «водная композиция» при использовании в данном документе относится к композиции на основе воды, в качестве воды могут быть использованы: вода, вода для инъекций, физиологический раствор (0,9-1,0%-ный водный раствор хлористого натрия).
В одном варианте осуществления изобретения субъект лечения или пациент является млекопитающим, предпочтительно человеческим субъектом. Вышеупомянутый субъект может быть мужского или женского пола любого возраста.
В настоящем описании и в последующей формуле изобретения, если контекстом не предусмотрено иное, слова «иметь», «включать» и «содержать» или их вариации, такие как «имеет», «имеющий», «включает», «включающий», «содержит» или «содержащий», следует понимать, как включение указанного целого или группы целых, но не исключение любого другого целого или группы целых.
Сущность изобретения
В настоящем изобретении раскрыты стабильные водные фармацевтические композиции антитела к IL-6R левилимаба, которые могут быть использованы в качестве лекарственного средства для лечения IL-6R- ассоциированных заболеваний.
Антитело к IL-6R левилимаб, представляющее собой моноклональное антитело изотипа IgG1, включает тяжелую цепь (HC) с аминокислотной последовательностью SEQ ID NO: 5, где вариабельный домен тяжелой цепи (SEQ ID NO: 4) содержит HCDR1 (SEQ ID NO: 1), HCDR2 (SEQ ID NO: 2) и HCDR3 (SEQ ID NO: 3); и легкую цепь (LC) с аминокислотной последовательностью SEQ ID NO: 10, где вариабельный домен легкой цепи (SEQ ID NO: 9) содержит LCDR1 (SEQ ID NO: 6), LCDR2 (SEQ ID NO: 7) и LCDR3 (SEQ ID NO: 8).
Левилимаб – рекомбинантное моноклональное антитело к рецептору интерлейкина-6. Левилимаб связывается и блокирует как растворимые (sIL-6R), так и мембранные рецепторы IL-6 (mIL-6R). Блокада обеих форм рецептора позволяет предотвратить развитие IL-6-ассоциированного провоспалительного каскада, в том числе препятствует активации антигенпрезентирующих клеток, В- и Т-лимфоцитов, моноцитов и макрофагов, эндотелиальных клеток и фибробластов, и избыточной продукции других провоспалительных цитокинов. IL-6 участвует в активации и поддержании местных воспалительных реакций (образование паннуса в синовии, стимуляция остеокластогенеза – эрозии хрящевой ткани, остеопороз), кроме того, IL-6 непосредственно индуцируюет синтез острофазовых белков в гепатоцитах: СРБ, фибриногена, сывороточного амилоидного белка А – SAA, гипсидина, лептина.
В одном из аспектов настоящее изобретение относится к водной фармацевтической композиции левилимаба, содержащей:
(a) 5-220 мг/мл левилимаба;
(b) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(c) 20-50 мг/мл полиола и 5-10 мг/мл глицина
или
10-32 мг/мл аргинина гидрохлорида; и
(d) уксусную кислоту до pH 4,5-6,5.
В некоторых вариантах осуществления изобретения указанный полиол выбран из маннитола или сорбитола.
В одном из аспектов настоящее изобретение относится к водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 20-50 мг/мл полиола;
(iv) 5-10 мг/мл глицина; и
(v) уксусную кислоту до pH 4,5-6,5.
Концентрация левилимаба, содержащегося в фармацевтических композициях по настоящему изобретению, может варьироваться в зависимости от желаемых свойств композиций, а также от конкретных условий, способов и целей использования фармацевтических композиций.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 5-40 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 5 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 5-15 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 10 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 15-25 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 20 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 100-180 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 140-220 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 180-220 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 160-200 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 180 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 200 мг/мл.
В некоторых вариантах осуществления изобретения указанный натрия ацетата тригидрат находится в концентрации 0,4-1,0 мг/мл.
В некоторых вариантах осуществления изобретения указанный натрия ацетата тригидрат находится в концентрации 0,4-0,5 мг/мл.
В некоторых вариантах осуществления изобретения указанный натрия ацетата тригидрат находится в концентрации 0,436 мг/мл.
В некоторых вариантах осуществления изобретения указанный полиол находится в концентрации 20-26 мг/мл.
В некоторых вариантах осуществления изобретения указанный полиол находится в концентрации 22-24 мг/мл.
В некоторых вариантах осуществления изобретения указанный полиол находится в концентрации 23 мг/мл.
В некоторых вариантах осуществления изобретения указанный полиол может быть выбран из сахароспирта, такого как маннитол, сорбитол, глицерин или ксилит или их комбинаций.
В некоторых вариантах осуществления изобретения указанный маннитол находится в концентрации 20-26 мг/мл.
В некоторых вариантах осуществления изобретения указанный маннитол находится в концентрации 22-24 мг/мл.
В некоторых вариантах осуществления изобретения указанный маннитол находится в концентрации 23 мг/мл.
В некоторых вариантах осуществления изобретения указанный сорбитол находится в концентрации 20-26 мг/мл.
В некоторых вариантах осуществления изобретения указанный сорбитол находится в концентрации 22-24 мг/мл.
В некоторых вариантах осуществления изобретения указанный сорбитол находится в концентрации 23 мг/мл.
В некоторых вариантах осуществления изобретения указанная комбинация маннитола и сорбитола находится в концентрации 20-26 мг/мл.
В некоторых вариантах осуществления изобретения указанная комбинация маннитола и сорбитола находится в концентрации 22-24 мг/мл.
В некоторых вариантах осуществления изобретения указанная комбинация маннитола и сорбитола находится в концентрации 23 мг/мл.
В некоторых вариантах осуществления изобретения указанный глицин находится в концентрации 7-8 мг/мл.
В некоторых вариантах осуществления изобретения указанный глицин находится в концентрации 7,5 мг/мл.
Необходимое значение pH фармацевтической композиции по настоящему изобретению может достигаться путем добавления уксусной кислоты.
В некоторых вариантах осуществления изобретения указанная уксусная кислота добавлена до pH 4,5-5,5.
В некоторых вариантах осуществления изобретения указанная уксусная кислота добавлена до pH 4,5 5,0, 5,5, 6,0, или 6,5.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 20 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii)23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 5 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 10 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 100 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 180 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 200 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 220 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
уксусную кислоту до pH 5,0.
В одном из аспектов настоящее изобретение относится к водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 10-32 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 4,5-6,5.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 5-40 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 5 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 5-15 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 10 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 15-25 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 20 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 100-180 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 140-220 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 180-220 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 160-200 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 200 мг/мл.
В некоторых вариантах осуществления изобретения указанный левилимаб находится в концентрации 180 мг/мл.
В некоторых вариантах осуществления изобретения указанный натрия ацетата тригидрат находится в концентрации 1,7-1,8 мг/мл.
В некоторых вариантах осуществления изобретения указанный натрия ацетата тригидрат находится в концентрации 1,744 мг/мл.
В некоторых вариантах осуществления изобретения указанный аргинина гидрохлорид находится в концентрации 18-24 мг/мл.
В некоторых вариантах осуществления изобретения указанный аргинина гидрохлорид находится в концентрации 20-22 мг/мл.
В некоторых вариантах осуществления изобретения указанный аргинина гидрохлорид находится в концентрации 21,1 мг/мл.
В некоторых вариантах осуществления изобретения указанная уксусная кислота добавлена до pH 4,5-5,5.
В некоторых вариантах осуществления изобретения указанная уксусная кислота добавлена до pH 4,5, 5,0, 5,5, 6,0, или 6,5.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 20 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 5 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 10 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 100 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 180 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 200 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
В некоторых вариантах осуществления изобретения предлагается водная фармацевтическая композиция, содержащая:
(i) 220 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
В одном из аспектов настоящее изобретение относится к водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 0,392 мг натрия ацетата тригидрата;
(iii) 20,7 мг маннитола или сорбитола;
(iv) 6,75 мг глицина;
(v) уксусную кислоту до pH 5,0; и
(vi) воду для инъекций до 0,9 мл.
В одном из аспектов настоящее изобретение относится к водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 1,57 мг натрия ацетата тригидрата;
(iii) 18,99 мг аргинина гидрохлорида;
(iv) уксусную кислоту до pH 5,0; и
(v) воду для инъекций до 0,9 мл.
В одном из аспектов настоящее изобретение относится к водной фармацевтической композиции левилимаба, содержащей:
Состав на 0,9 мл:
(i) 162 мг левилимаба;
(ii) 0,392 мг натрия ацетата тригидрата;
(iii) 20,7 мг маннитола или сорбитола;
(iv) 6,75 мг глицина;
(v) уксусную кислоту до pH 5,0; и
(vi) воду для инъекций до 0,9 мл.
В одном из аспектов настоящее изобретение относится к водной фармацевтической композиции левилимаба, содержащей:
Состав на 0,9 мл:
(i) 162 мг левилимаба;
(ii) 1,57 мг натрия ацетата тригидрата;
(iii) 18,99 мг аргинина гидрохлорида;
(iv) уксусную кислоту до pH 5,0; и
(v) воду для инъекций до 0,9 мл.
В некоторых вариантах осуществления изобретения указанная уксусная кислота является ледяной уксусной кислотой.
В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция левилимаба по настоящему изобретению предназначена для парентерального введения.
В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция левилимаба по настоящему изобретению предназначена для внутримышечного, внутривенного или подкожного введения.
В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция левилимаба по настоящему изобретению может быть введена внутривенно в виде инфузии.
Фармацевтические композиции по настоящему изобретению можно хранить в любом подходящем для этого сосуде. Например, стеклянные или полимерные контейнер, флакон, ампула, шприц, картридж, автоинжектор или бутылка необходимого объема.
В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция во флаконе.
В некоторых вариантах осуществления изобретения указанный флакон представляет собой стеклянный или полимерный флакон.
В некоторых вариантах осуществления изобретения указанный флакон имеет объем 4-20 мл.
В некоторых вариантах осуществления изобретения указанный флакон имеет объем 1 мл, 2 мл, 3 мл, 4 мл, 5 мл, 6 мл, 7 мл, 8 мл, 9 мл, 10 мл, 15 мл или 20 мл.
В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция находится в шприце или автоинжекторе.
В некоторых вариантах осуществления изобретения указанный шприц или автоинжектор является стеклянным или полимерным.
В некоторых вариантах осуществления изобретения указанный шприц или автоинжектор имеет вместимость 0,9 мл.
В некоторых вариантах осуществления изобретения указанный шприц или автоинжектор имеет вместимость 1 мл.
В некоторых вариантах осуществления изобретения указанный шприц или автоинжектор имеет вместимость 2 мл.
В некоторых вариантах осуществления изобретения указанный шприц или автоинжектор может быть объёмом 1 мл с номинальным объемом 0,9 мл.
В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция находится в преднаполненном шприце или в преднаполненном автоинжекторе.
В некоторых вариантах осуществления изобретения указанный преднаполненный шприц или в преднаполненный автоинжектор является стеклянным или полимерным.
В некоторых вариантах осуществления изобретения указанный предзаполненный шприц или предзаполненный автоинжектор имеет вместимость 0,9 мл.
В некоторых вариантах осуществления изобретения указанный предзаполненный шприц или предзаполненный автоинжектор имеет вместимость 1 мл.
В некоторых вариантах осуществления изобретения указанный предзаполненный шприц или предзаполненный автоинжектор имеет вместимость 2 мл.
В некоторых вариантах осуществления изобретения указанный предзаполненный шприц или предзаполненный автоинжектор может быть объёмом 1 мл с номинальным объемом 0,9 мл.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 20-50 мг/мл полиола;
(iv) 5-10 мг/мл глицина; и
(v) уксусную кислоту до pH 4,5-6,5.
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 10-32 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 4,5-6,5
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 0,392 мг натрия ацетата тригидрата;
(iii) 20,7 мг полиола, выбранного из маннитола или сорбитола;
(iv) 6,75 мг глицина;
(v) уксусную кислоту до pH 5,0; и
(vi)воду для инъекций до 0,9 мл
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 1,57 мг натрия ацетата тригидрата;
(iii) 18,99 мг аргинина гидрохлорида;
(iv) уксусную кислоту до pH 5,0; и
(v) воду для инъекций до 0,9 мл
для лечения или профилактики IL6R-ассоциированных заболеваний или нарушений.
В некоторых вариантах осуществления изобретения указанное IL6R-ассоциированное заболевание или нарушение выбрано из: ревматоидного артрита, ювенильного хронического артрита, склеродермии, реакции "трансплантат против хозяина", отторжения трансплантата органа, острого или хронического иммунного заболевания, связанного с трансплантацией органа, кахексии, взрослого (острого) респираторного дистресс-синдрома, синдрома выброса цитокинов (синдром высвобождения цитокинов), болезни Стилла, системной склеродермии, синдрома Шегрена, болезни/артериита Такаясу, нарушений, связанных с цитокиновой терапией, иридоциклита, увеита, оптического неврита, оптического нейромиелита, ювенильного ревматоидного артрита, гигантоклеточного артериита, полиартикулярного ювенильного идиопатического артрита, системного ювенильного идиопатического артрита; рака, в частности, множественной миеломы и злокачественных солидных опухолей, колоректального рака, рака предстательной железы, рака яичника.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению может включать введение данной композиции парентерально.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению может включать введение данной композиции внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению может включать введение данной композиции внутривенно в виде инфузии.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 20-50 мг/мл полиола;
(iv) 5-10 мг/мл глицина; и
(v)уксусную кислоту до pH 4,5-6,5.
для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 10-32 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 4,5-6,5
для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0
для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 0,392 мг натрия ацетата тригидрата;
(iii) 20,7 мг полиола, выбранного из маннитола или сорбитола;
(iv) 6,75 мг глицина;
(v) уксусную кислоту до pH 5,0; и
(vi) воду для инъекций до 0,9 мл
для лечения ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 1,57 мг натрия ацетата тригидрата;
(iii) 18,99 мг аргинина гидрохлорида;
(iv) уксусную кислоту до pH 5,0; и
(v) воду для инъекций до 0,9 мл
для лечения ревматоидного артрита.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать введение данной композиции в дозе левилимаба 162 мг.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать введение данной композиции один раз в неделю или один раз каждые две недели.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать введение данной композиции в месячной дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать введение данной композиции в месячной дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать введение данной композиции парентерально.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать введение данной композиции внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать введение данной композиции внутривенно в виде инфузии.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения ревматоидного артрита может включать дополнительно применение метотрексата.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 20-50 мг/мл полиола;
(iv) 5-10 мг/мл глицина; и
(v) уксусную кислоту до pH 4,5-6,5.
для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 10-32 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 4,5-6,5
для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0
для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 0,392 мг натрия ацетата тригидрата;
(iii) 20,7 мг полиола, выбранного из маннитола или сорбитола;
(iv) 6,75 мг глицина;
(v) уксусную кислоту до pH 5,0; и
(vi)воду для инъекций до 0,9 мл
для лечения активного ревматоидного артрита.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 1,57 мг натрия ацетата тригидрата;
(iii) 18,99 мг аргинина гидрохлорида;
(iv) уксусную кислоту до pH 5,0; и
(v) воду для инъекций до 0,9 мл
для лечения активного ревматоидного артрита.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать введение данной композиции в дозе левилимаба 324 мг или 648 мг.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать введение данной композиции в дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать введение данной композиции в дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать введение данной композиции 1 раз в 2 недели, или 1 раз в 4 недели, или 1 раз в 6 недель.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать введение данной композиции парентерально.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать введение данной композиции внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать введение данной композиции внутривенно в виде инфузии.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения активного ревматоидного артрита может включать дополнительно применение метотрексата.
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 20-50 мг/мл полиола;
(iv) 5-10 мг/мл глицина; и
(v) уксусную кислоту до pH 4,5-6,5.
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 10-32 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 4,5-6,5
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 20 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 180 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 0,392 мг натрия ацетата тригидрата;
(iii) 20,7 мг полиола, выбранного из маннитола или сорбитола;
(iv) 6,75 мг глицина;
(v) уксусную кислоту до pH 5,0; и
(vi) воду для инъекций до 0,9 мл
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В одном варианте осуществления настоящее изобретение относится к применению водной фармацевтической композиции левилимаба, содержащей:
(i) 162 мг левилимаба;
(ii) 1,57 мг натрия ацетата тригидрата;
(iii) 18,99 мг аргинина гидрохлорида;
(iv) уксусную кислоту до pH 5,0; и
(v) воду для инъекций до 0,9 мл
для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может включать введение данной композиции в дозе левилимаба 324 мг или 648 мг.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может включать введение данной композиции в дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может включать введение данной композиции в дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может включать введение данной композиции однократно, или двукратно, или трехкратно, или четырехкратно с интервалом минимум 8 часов.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может включать введение данной композиции парентерально.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может включать введение данной композиции внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может включать введение данной композиции внутривенно в виде инфузии.
В одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики IL6R-ассоциированных заболевания или нарушения, включающему введение субъекту, нуждающемуся в такой профилактике или лечении, терапевтически эффективного количества водной фармацевтической композиции левилимаба по настоящему изобретению.
В некоторых вариантах осуществления изобретения указанное IL6R-ассоциированное заболевание или нарушение выбрано из: ревматоидного артрита, ювенильного хронического артрита, склеродермии, реакции "трансплантат против хозяина", отторжения трансплантата органа, острого или хронического иммунного заболевания, связанного с трансплантацией органа, кахексии, взрослого (острого) респираторного дистресс-синдрома, болезни Стилла, системной склеродермии, синдрома Шегрена, болезни/артериита Такаясу, нарушений, связанных с цитокиновой терапией, синдрома выброса цитокинов (синдром высвобождения цитокинов), иридоциклита, увеита, оптического неврита, оптического нейромиелита, ювенильного ревматоидного артрита, гигантоклеточного артериита, полиартикулярного ювенильного идиопатического артрита, системного ювенильного идиопатического артрита; рака, в частности, множественной миеломы и злокачественных солидных опухолей, колоректального рака, рака предстательной железы, рака яичника.
В некоторых вариантах осуществления изобретения способ лечения или профилактики IL6R-ассоциированного заболевания или нарушения у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества водной фармацевтической композиции левилимаба по настоящему изобретению парентерально.
В некоторых вариантах осуществления изобретения способ лечения или профилактики IL6R-ассоциированного заболевания или нарушения у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества водной фармацевтической композиции левилимаба по настоящему изобретению внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения способ лечения или профилактики IL6R-ассоциированного заболевания или нарушения у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества водной фармацевтической композиции левилимаба по настоящему изобретению внутривенно в виде инфузии.
В одном варианте осуществления настоящее изобретение относится к способу лечения ревматоидного артрита, включающему введение субъекту, нуждающемуся в такой профилактике или лечении, терапевтически эффективного количества водной фармацевтической композиции левилимаба по настоящему изобретению.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в дозе левилимаба 162 мг.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению один раз в неделю или один раз каждые две недели.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в месячной дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в месячной дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению парентерально.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению внутривенно в виде инфузии.
В некоторых вариантах осуществления изобретения способ лечения ревматоидного артрита у нуждающегося в этом субъекта может включать дополнительно введение метотрексата.
В одном варианте осуществления настоящее изобретение относится к способу лечения активного ревматоидного артрита, включающему введение субъекту, нуждающемуся в такой профилактике или лечении, терапевтически эффективного количества водной фармацевтической композиции левилимаба по настоящему изобретению.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в дозе левилимаба 324 мг или 648 мг.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению 1 раз в 2 недели, или 1 раз в 4 недели, или 1 раз в 6 недель.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению парентерально.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению внутривенно в виде инфузии.
В некоторых вариантах осуществления изобретения способ лечения активного ревматоидного артрита у нуждающегося в этом субъекта может включать дополнительно введение метотрексата.
В одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов), включающему введение субъекту, нуждающемуся в такой профилактике или лечении, терапевтически эффективного количества водной фармацевтической композиции левилимаба по настоящему изобретению.
В некоторых вариантах осуществления изобретения способ лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в дозе левилимаба 324 мг или 648 мг.
В некоторых вариантах осуществления изобретения способ лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения способ лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению в дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения способ лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению однократно, или двукратно, или трехкратно, или четырехкратно с интервалом минимум 8 часов.
В некоторых вариантах осуществления изобретения способ лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению парентерально.
В некоторых вариантах осуществления изобретения способ лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения способ лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) у нуждающегося в этом субъекта может включать введение водной фармацевтической композиции левилимаба по настоящему изобретению внутривенно в виде инфузии.
В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции левилимаба по настоящему изобретению для применения для лечения или профилактики IL6R-ассоциированного заболевания или нарушения.
В некоторых вариантах осуществления изобретения указанное IL6R-ассоциированное заболевание или нарушение выбрано из: ревматоидного артрита, ювенильного хронического артрита, склеродермии, реакции "трансплантат против хозяина", отторжения трансплантата органа, острого или хронического иммунного заболевания, связанного с трансплантацией органа, кахексии, взрослого (острого) респираторного дистресс-синдрома, болезни Стилла, системной склеродермии, синдрома Шегрена, болезни/артериита Такаясу, нарушений, связанных с цитокиновой терапией, синдрома выброса цитокинов (синдром высвобождения цитокинов), иридоциклита, увеита, оптического неврита, оптического нейромиелита, ювенильного ревматоидного артрита, гигантоклеточного артериита, полиартикулярного ювенильного идиопатического артрита, системного ювенильного идиопатического артрита; рака, в частности, множественной миеломы и злокачественных солидных опухолей, колоректального рака, рака предстательной железы, рака яичника.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики IL6R-ассоциированного заболевания или нарушения может вводиться парентерально.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики IL6R-ассоциированного заболевания или нарушения может вводиться внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики IL6R-ассоциированного заболевания или нарушения может вводиться внутривенно в виде инфузии.
В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может вводиться в дозе левилимаба 162 мг.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может вводиться один раз в неделю или один раз каждые две недели.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может вводиться в дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может вводиться в дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может вводиться парентерально.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может вводиться внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может вводиться внутривенно в виде инфузии.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может применяться в комбинации с метотрексатом.
В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может вводиться в дозе левилимаба 324 мг или 648 мг.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может вводиться в дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может вводиться в дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может вводиться 1 раз в 2 недели, или 1 раз в 4 недели, или 1 раз в 6 недель.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может вводиться парентерально.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может вводиться внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может вводиться внутривенно в виде инфузии.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может применяться в комбинации с метотрексатом.
В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов).
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может вводиться в дозе левилимаба 324 мг или 648 мг.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может вводиться в дозе левилимаба 4 мг/кг массы тела.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может вводиться в дозе левилимаба 8 мг/кг массы тела.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может вводиться однократно, или двукратно, или трехкратно, или четырехкратно с интервалом минимум 8 часов.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может вводиться парентерально.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может вводиться внутримышечно, внутривенно или подкожно.
В некоторых вариантах осуществления изобретения водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдрома выброса цитокинов (синдром высвобождения цитокинов) может вводиться внутривенно в виде инфузии.
В одном из аспектов настоящее изобретение относится к способу получения водной фармацевтической композиции левилимаба, включающему комбинирование 5-220 мг/мл левилимаба с
0,4-1,8 мг/мл натрия ацетата тригидрата;
20-50 мг/мл полиола;
5-10 мг/мл глицина; и
уксусной кислотой до pH 4,5-6,5.
В одном из аспектов настоящее изобретение относится к способу получения водной фармацевтической композиции левилимаба, включающему комбинирование 5-220 мг/мл левилимаба с
0,4-1,8 мг/мл натрия ацетата тригидрата;
10-32 мг/мл аргинина гидрохлорида; и
уксусной кислотой до pH 4,5-6,5.
В одном из аспектов настоящее изобретение к способу получения водной фармацевтической композиции левилимаба, где указанная уксусная кислота является ледяной уксусной кислотой.
Настоящее изобретение относится к подходящим водным фармацевтическим композициям анти-IL-6R антитела левилимаба. Одна водная фармацевтическая композиция может содержать левилимаб, буфер на основе ацетата, полиол, глицин и уксусную кислоту. Еще одна водная фармацевтическая композиция может содержать левилимаб, буфер на основе ацетата, аргинина гидрохлорид и уксусную кислоту.
Буфер на основе ацетата может быть образован за счет комбинирования уксусной кислоты с натрия ацетат тригидратом. Следует понимать, что хотя в качестве соли для буфера на основе ацетата можно использовать натрия ацетат тригидрат, для буфера на основе ацетата можно использовать любую другую ацетатную соль, например, ацетат калия, не отступая при этом от идей настоящего изобретения.
В водных фармацевтических композициях левилимаба по настоящему изобретению может быть использован аргинин, в частности, L-аргинин, или аргинина гидрохлорид.
Настоящее изобретение относится к применению водной фармацевтической композиции левилимаба по настоящему изобретению для лечения или профилактики IL6R-ассоциированного заболевания или нарушения.
Заболевания или нарушения, которые можно лечить предложенными в настоящем документе композициями состоят из, без ограничения, ревматоидного артрита, ювенильного хронического артрита, склеродермии, реакции "трансплантат против хозяина", отторжения трансплантата органа, острого или хронического иммунного заболевания, связанного с трансплантацией органа, кахексии, взрослого (острого) респираторного дистресс-синдрома, болезни Стилла, системной склеродермии, синдрома Шегрена, болезни/артериита Такаясу, нарушений, связанных с цитокиновой терапией, синдром выброса цитокинов (синдром высвобождения цитокинов), иридоциклита, увеита, оптического неврита, оптического нейромиелита, ювенильного ревматоидного артрита, гигантоклеточного артериита, полиартикулярного ювенильного идиопатического артрита, системного ювенильного идиопатического артрита; рака, в частности, множественной миеломы и злокачественных солидных опухолей, колоректального рака, рака предстательной железы, рака яичника.
Предоставляемые фармацевтические композиции можно вводить нуждающемуся в лечении индивидууму посредством системной инъекции, например, посредством внутривенной или подкожной, или внутримышечной инъекции; или посредством прямой инъекции.
Водная фармацевтическая композиция левилимаба по настоящему изобретению может быть использована после разведения. Для этого необходимое количество композиции из флакона переносят в ёмкость для инфузий, содержащую стерильный 0,9% раствор натрия хлорида или стерильный 5% раствор декстрозы. Приготовленный раствор перемешивают путем осторожного переворачивания емкости для инфузий во избежание пенообразования.
В одном варианте осуществления изобретения доза может быть доставлена посредством одной или более одной инфузий. Доза может быть доставлена посредством одной, двух или трех инфузий. В некоторых вариантах изобретения длительность терапии может составлять от одной или нескольких инфузий.
Терапевтически эффективное количество водных композиций, включающих левилимаб, по настоящему изобретению, в предлагаемых составах зависит от состояния, подлежащего лечению, тяжести состояния, предшествующей терапии и истории болезни пациента, и ответу на терапевтическое средство. Подходящую дозу можно регулировать по решению лечащего врача так, что ее можно вводить пациенту один раз или посредством нескольких введений.
В одном из вариантов осуществления эффективное количество левилимаба на дозу для пациента составляет приблизительно 4 мг/кг веса тела или 8 мг на килограмм веса тела.
Доза может быть доставлена посредством одной или более одной инъекции. Доза может быть доставлена посредством одной, двух или трех инъекций. Одна инъекция может содержать 0,9 мл, 1 мл, 1,8 мл или 2 мл раскрытой в настоящем документе композиции.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения ревматоидного артрита может быть введены в дозе левилимаба 162 мг посредством одной инъекции.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может быть введены в дозе 324 мг посредством одной инъекции.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может быть введены в дозе 324 мг посредством двух инъекции по 162 мг.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может быть введены в дозе 648 мг посредством одной инъекции.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может быть введены в дозе 648 мг посредством двух инъекции по 324 мг.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения активного ревматоидного артрита может быть введены в дозе 648 мг посредством четырех инъекции по 162 мг.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдром выброса цитокинов (синдром высвобождения цитокинов) может быть введены в дозе 324 мг посредством одной инъекции.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдром выброса цитокинов (синдром высвобождения цитокинов) может быть введены в дозе 324 мг посредством двух инъекции по 162 мг.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдром выброса цитокинов (синдром высвобождения цитокинов) может быть введены в дозе 648 мг посредством одной инъекции.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдром выброса цитокинов (синдром высвобождения цитокинов) может быть введены в дозе 648 мг посредством двух инъекции по 324 мг.
В одном варианте осуществления водная фармацевтическая композиция левилимаба по настоящему изобретению для применения для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдром выброса цитокинов (синдром высвобождения цитокинов) может быть введены в дозе 648 мг посредством четырех инъекции по 162 мг.
В другом варианте осуществления фармацевтические композиции по настоящему изобретению можно получать в виде нерасфасованного состава, и, по существу, компоненты фармацевтической композиции присутствуют в количествах выше, чем может требоваться для введения, и их соответствующим образом разбавляют до введения.
Альтернативно, фармацевтическая композиция может быть заморожена, высушена распылением, либо лиофилизирована и восстановлена перед применением в подходящем стерильном носителе. Лиофилизация может быть выполнена с использованием способов, известных в данной области техники, которые включают в себя различные шаги, такие как замораживание, отжиг, первичная и вторичная сушка.
Фармацевтические композиции можно вводить в виде одного терапевтического средства или в комбинации с дополнительными терапевтическими средствами по мере необходимости. Таким образом, в одном варианте осуществления предлагаемые способы лечения и/или профилактики используют в комбинации с введением терапевтически эффективного количества другого активного средства. Другое активное средство можно вводить до, в течение или после введения фармацевтических композиций по настоящему изобретению. Другое активное средство можно вводить как часть предлагаемой композиции или, альтернативно, в виде отдельного состава.
Фармацевтические композиции при желании можно предоставлять во флаконе, упаковке или в устройстве-распределителе, которые могут содержать одну или несколько стандартных лекарственных форм, содержащих активный ингредиент. В одном варианте осуществления устройство-распределитель может содержать шприц, содержащий однократную дозу жидкого состава, готового к инъекции. Шприц может сопровождаться инструкцией по введению.
В другом варианте осуществления настоящее изобретение относится к набору или контейнеру, содержащим водную фармацевтическую композицию по изобретению. Набор также может сопровождаться инструкциями по применению.
Методики
1. Получение образцов левилимаба
Получение образцов антитела с концентрацией 5-20 мг/мл осуществляли в концентрационных ячейках Stirred Cell (Millipore) под давлением. Для этого антитело в исходном составе помещали в ячейку, при непрерывном перемешивании белок концентрировали под потоком сжатого воздуха до концентрации 10 мг/мл, после чего вносили в ячейку не менее чем 10 кратный объем водного раствора с целевым составом, включающим буферные, осмотические агенты и, если необходимо, дополнительные водорастворимые стабилизаторы. По завершении процесса диафильтрации антитело концентрировали до концентрации около 30 мг/мл, выгружали из ячейки, определяли точную концентрацию белка методом УФ-спектрофотометрии. Затем к образцу вносили соответствующий раствор вспомогательных веществ для получения раствора с целевой концентрацией белка.
Получение образцов белка с концентрацией более 20 мг/мл проводили в кассетах Pellicon (Millipore) в режиме тангенциального потока. Для этого антитело в исходном составе помещали в емкость для диафильтрации, концентрировали белок до концентрации около 45-50 мг/мл, после чего к системе подключали подачу не менее чем 10 кратного объема раствора с целевым составом, содержащим буферные агенты и, если необходимо, дополнительные водорастворимые стабилизаторы. По завершении процесса диафильтрации антитело концентрировали до концентрации 100 мг/мл, выгружали из системы, вносили осмотические агенты и стабилизаторы, продолжали концентрирование до концентрации, превышающей целевую, выгружали из системы и определяли точную концентрацию белка. Затем к образцу вносили соответствующий раствор вспомогательных веществ для получения раствора с целевой концентрацией белка.
При получении составов, содержащих солюбилизаторы, концентраты поверхностно-активных веществ вносили к антителу после завершения диафильтрации и концентрирования при финальном разведении антитела раствором вспомогательных веществ до целевой концентрации.
При асептическом наполнении в конечный контейнер (например, стерильный стеклянный или полимерный сосуд, флакон или шприц) раствор антитела фильтровали через стерилизующую мембрану с размером пор 0.22 мкм.
2. Определение концентрации белка в исследуемых образцах
Концентрацию белка определяли с помощью метода УФ-спектрофотометрии при длине волны 280 нм в УФ-прозрачных планшетах.
Каждый образец разводили соответствующим раствором вспомогательных веществ до концентрации ~0.5 мг/мл. В лунку планшета для УФ-спектрофотометрии помещали 150 мкл разведенного образца. Измеряли оптическую плотность помещенных в планшет растворов на планшетном спектрофотометре при длине волны 280 нм. В качестве раствора сравнения использовали соответствующий раствор вспомогательных веществ.
Концентрацию белка (С) в мг/мл рассчитывали по формуле:
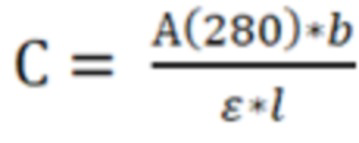 , где
, где
А280 – значение оптической плотности при длине волны 280 нм;
ε – коэффициент экстинкции исследуемого белка;
b – суммарный коэффициент разведения образца;
l – толщина слоя в лунке планшета; для 150 мкл l=0,42 см.
3. Определение температуры агрегации белка методом динамического светорассеяния
Определение точки агрегации исследуемых белков (в концентрации 1 мг/мл) осуществляли на приборе Zetasizer Nano ZSP. Для этого 0.5 мл раствора помещали в кварцевую обеспыленную кювету, которую постепенно нагревали в установке при постоянном измерении интенсивности рассеянного света.
• Аналитическая модель: Protein analysis.
• Режим Temperature trend, mod: Protein aggregation point. От 50 до 83°С при шаге нагрева 1,5°С.
• Выдерживание при температуре перед началом измерения 30 секунд.
• Интенсивность рассеянного света детектировали при угле θ = 173°.
• В каждой точке среднее значение по 13 измерениям в 1 повторности.
Температурный тренд и точка агрегации были определены с использованием ПО прибора.
4. Определение коллоидной стабильности методом «ПЭГ-агрегация»
Готовили раствор ПЭГ 6000 с массовой концентрацией 20 – 25% в исследуемом составе вспомогательных веществ. Итоговые растворы фильтровали через фильтр Durapore 0,45 мкм.
В 96-луночные планшеты для УФ-спектрофотометрии переносили расчетное количество образца, раствора вспомогательных веществ и 20 – 25 % раствора ПЭГ 6000 так, чтобы в ряде лунок была концентрация ПЭГ6000 от 0 до 18%, а концентрация белка в каждой лунке составляла 1 мг/мл. Все полученные в лунках растворы хорошо перемешивали, пипетируя.
После этого оценивали степень мутности растворов визуально, а также измеряли оптическую плотность растворов при длине волны 400 нм.
Осаждение белка в присутствии ПЭГ связано с эффектом замещения объема, то есть белок стерически вытесняется из регионов растворителя полимером (L. Li, A. Kantor, N. Warne. Application of a PEG precipitation method for solubility screening: A tool for developing high protein concentration formulations, Protein Sci. 2013 Aug; 22(8): 1118-1123). Это приводит к концентрированию белка до тех пор, пока не будет превышена его растворимость и не выпадет осадок. Чем менее стабилен образец, тем при меньшей концентрации ПЭГ 6000 он будет образовывать видимые агрегаты (опалесценцию).
5. Определение термической стабильности при термострессе при 50°С
Исследуемые образцы разделяли на две аликвоты по 150 мкл и помещали в отдельные стеклянные виалы: по одной виале на каждый состав откладывали в холодильник на хранение при 5 ± 3°С, остальные устанавливали в термостат и инкубировали при 50°С в течение указанного времени. При отборе контрольных точек или после окончания прогрева виалы убирали из термостата, выдерживали при комнатной температуре около 15 мин и передавали на анализ.
6. Определение коллоидной стабильности при шейкировании.
Исследуемые образцы разделяли на две аликвоты по 150 мкл и помещали в стеклянные виалы по одной виале на каждый состав откладывали в холодильник на хранение при температуре 5 ± 3°С, остальные устанавливали в термошейкер и шейкировали со скоростью 800 об/мин при температуре 5 ± 3°С в течение указанного времени. При отборе контрольных точек или после окончания стресса виалы убирали из термошейкера и передавали на анализ.
7. Определение коллоидной стабильности при замораживании и оттаивании
Исследуемые образцы разделяли на две аликвоты и помещали в полимерные пробирки: по одной пробирке на каждый состав откладывали в холодильник на хранение при 5 ± 3°С, остальные устанавливали в морозильную камеру и хранили при температуре минус 16 – 20°С в течение указанного времени. После окончания стресса пробирки убирали из морозильной камеры, выдерживали при комнатной температуре до полного оттаивания содержимого, перемешивали растворы с помощью вортекса и передавали на анализ.
8. Ускорение хранения
Исследуемые образцы в концентрации белка 20, 180 и 220 мг/мл разделяли на отдельные аликвоты (одна для входного контроля – допускается передавать на анализ единожды для всех исследований при старте хранения) и помещали в отдельные стерильные стеклянные флаконы и шприцы: часть флаконов и шприцов на каждый состав откладывали в холодильник на хранение при 5 ± 3°С (входной контроль), остальные устанавливали в термостат и инкубировали при 25°С в течение 6 месяцев, периодически отбирая контрольные точки согласно плану. При отборе контрольных точек и после завершения хранения флаконы и шприцы убирали из термостата и передавали на анализ.
9. Определение чистоты образцов методом эксклюзионной высокоэффективной жидкостной хроматографии (Э ВЭЖХ)
Колонка Tosoh TSK-GelG3000SWXL 7.8 mm ID × 30 cm, cat. № 08541.
Температура колонки: 25.
Скорость потока подвижной фазы: 0.7 мл/мин.
Объем вкола: 10 мкл.
Концентрация образца: 5 мг/мл.
Длина волны детектора: 220 и 280 нм.
Продолжительность элюирования: 23 мин.
Подвижная фаза: Динатрия гидрофосфат б/в 7.1 мг/мл.
Натрия хлорид 17.54 мг/мл.
рН подвижной фазы доводили до 7.0 ортофосфорной кислотой.
10. Определение профиля заряженных форм методом ионообменной высокоэффективной жидкостной хроматографии (ИО ВЭЖХ)
Колонка: TSKgel CM-STAT, 4,6 мм × 100 мм, размер частиц 7 мкм (Tosoh Bioscience LLC, Япония, 21966)
Элюент А: Раствор 10 мМ динатрия гидрофосфата безводного, pH=6,8
Элюент В: Раствор 10 мМ динатрия гидрофосфата безводного, 200 mM NaCl, pH=6,8
Скорость потока: 0,7 мл/мин.
Температура колонки: 35°С
Температура
автосэмплера: 5°С
Детектор: УФ, 280 нм
Референсная длина волны: 360 нм, ширина окна пропускания (bandwidth) 100 нм
Объем пробы: 40 мкл
Режим элюирования: Элюент А 100 → 0 → 100%
Элюент B 0 → 100 → 0%
Время хроматографирования: 60 мин.
Исследуемый образец разводили до концентрации 1,0 мг/мл и проводили обработку карбоксипептидазой B (1% от объема образца) в течение 2 ч при температуре (37 ± 1)°C.
11. Определение гомогенности методом вертикального электрофореза в полиакриламидном геле в редуцирующих и нередуцирующих условиях (ВЭФ ред. и неред.)
Готовили ПААГ в стеклянных пластинах в присутствии натрия додецилсульфата, состоящие из концентрирующего слоя - 4% ПААГ и разделяющего слоя: в редуцирующих условиях – 12.5% ПААГ, в нередуцирующих условиях – 8 % ПААГ.
Собирали и устанавливали электрофорезную камеру в соответствии с инструкцией по эксплуатации прибора для проведения вертикального электрофореза. Пробы готовили, разводя образцы очищенной водой до конечной концентрации 1 мг/мл. Отбирали объем, эквивалентный 40 мкг и смешивали подготовленные пробы исследуемого образца в соотношении 3:1 (объем/объем) с буферным раствором для нанесения образцов 4-кратным, содержащим 2-меркаптоэтанол (редуцирующие условия) и не содержащим 2-меркаптоэтанол (нередуцирующие условия), перемешивали. Полученные растворы инкубировали при температуре (99 ± 1)°С в течение 3 мин (образцы, содержащие 2-меркаптоэтанол) и при температуре (99 ± 1)°С в течение 1 мин (образцы, не содержащие 2-меркаптоэтанол). Растворы охлаждали до комнатной температуры, перемешивали и наносили в лунки ПААГ под слой электродного буферного раствора.
Электрофорез проводили в режиме постоянного тока, используя систему водяного охлаждения. Задавали параметры работы источника питания: при прохождении фронта красителя через концентрирующий гель напряжение тока составляло 110 В. После вхождения фронта красителя в нижний разделяющий гель на 5 – 7 мм напряжение тока увеличивали до 180 В. Источник питания отключали, когда фронт красителя достиг нижней границы геля.
После окончания электрофореза гели отделяли от стекол и проводили фиксацию белков в фиксирующем растворе в течение 16 – 18 часов при комнатной температуре. Затем проводили окрашивание гелей (в растворе кислотном синем 83) и отмывку до получения четкой визуализации полос. Гели сканировали. Чистоту и примеси в испытуемых образцах оценивали с помощью программного обеспечения GelPro.
12. Определение относительной специфической активности
Специфическую активность определяли с использованием антипролиферативного теста на культуре клеток DS-1. Пробоподготовку проводили с использованием роботизированной станции TecanEvo 200, в качестве СКО (среды для количественного определения) использовали RPMI1640, содержащая 2 мМ Gln, 10 % FBS, 1 мМ натрия пирувата, 50 мкг/мл гентамицина.
Исследуемый образец антитела разводили с использованием СКО до концентрации 5 мг/мл и помещали в роботизированную станцию. С помощью TecanEvo 200 проводили подготовку трех независимых разведений стандартного и исследуемого образца в концентрациях 1 000 000, 250 000, 100 000, 25 000, 5000, 2500, 1000, 250, 50, 5,0 нг/мл с использованием СКО. Переносили разведения и СКО в культуральные планшеты и вносили клеточную суспензию DS-1 в концентрации (1,5 ± 0,1)×105 клеток/мл и рабочий раствор IL6 7,5 нг/мл к разведениям испытуемых и стандартных образцов. Культуральные планшеты помещали в СО2-инкубатор, инкубировали при температуре (37 ± 1)°С в увлажненном воздухе с содержанием углерода диоксида 5 % в течение 70-72 ч.
По истечении срока инкубации в лунки культурального планшета вносили краситель Аламар синий и инкубировали планшеты при тех же условиях до развития градиентной окраски. Оценивали интенсивность флуоресценции при длине волны возбуждения/испускания 544/590 нм. С помощью ПО Magellan ver 7.2 проводили построение кривых зависимости интенсивности флуоресценции от концентрации белка. Относительную специфическую активность исследуемых образцов определяли как соотношение ED50 стандартного образца к ED50 испытуемого образца, выраженное в процентах.
13. Обработка результатов.
Абсолютное изменение показателей качества в ходе стрессов рассчитывали по формуле:
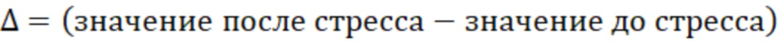
Абсолютное изменение профиля заряженных форм рассчитывали по формуле:

Примеры
Пример 1. Выбор буферной системы
В настоящем исследовании в качестве основы фармацевтической композиции выбраны 2 типовых буферных системы, пригодных для парентерального введения: ацетатная и гистидиновая.
Для оценки пригодности буферных систем в отношении технологических характеристик фармацевтической композиции было проведено исследование влияния природы буферного раствора на коллоидную стабильность белка при его концентрировании. В качестве отклика было проведено измерение времени фильтрации образца через стерилизующий фильтр 0,22 мкм. Исследуемые фармацевтические композиции представлены в таблице 1.
Таблица 1. Исследуемые составы

Исследование времени фильтрации.
Образцы концентрировали согласно методике 1. При достижении концентрации 100 мг/мл, 130 мг/мл и 180 мг/мл проводили измерение времени стерилизующей фильтрации фармацевтической композиции. Результаты исследования времени проведения стерилизующей фильтрации представлены в таблице 2.
Таблица 2. Время стерилизующей фильтрации
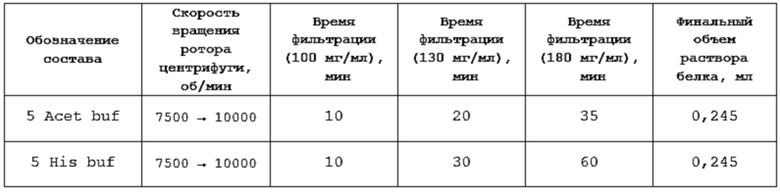
Использование ацетатной буферной системы позволяет снизить время фильтрации фармацевтической композиции, содержащей 180 мг/мл белка приблизительно в 1.7 раз по сравнению с гистидиновой буферной системой, что свидетельствует о ее лучшей растворимости и коллоидной стабильности.
Пример 2. Первичный выбор осмотического агента
Исследуемые составы
В качестве осмотических агентов исследовали пригодные для парентерального введения вспомогательные вещества. Исследуемые составы представлены в таблице 3.
Таблица 3. Исследуемые составы
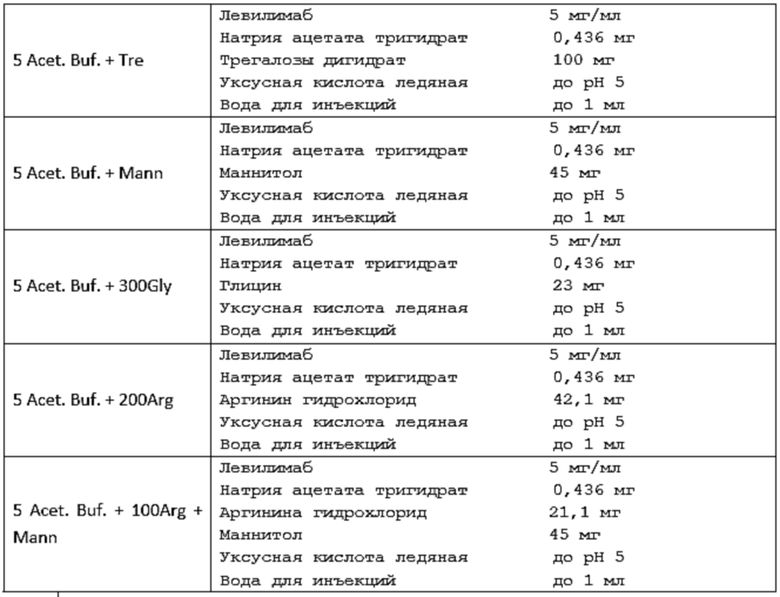
Определение коллоидной стабильности методом «ПЭГ-агрегации».
Тест «ПЭГ-агрегация» позволяет смоделировать прямое концентрирование левилимаба за счет его вытеснения инертным полимером ПЭГ 6000, а также сделать сравнительную оценку теоретической растворимости антитела в различных составах. Исследование выполнено согласно методике 4. Данные средней оптической плотности растворов представлены в таблице 4. Результаты также представлены на Фиг. 1.
Таблица 4. Средняя оптическая плотность растворов после приготовления
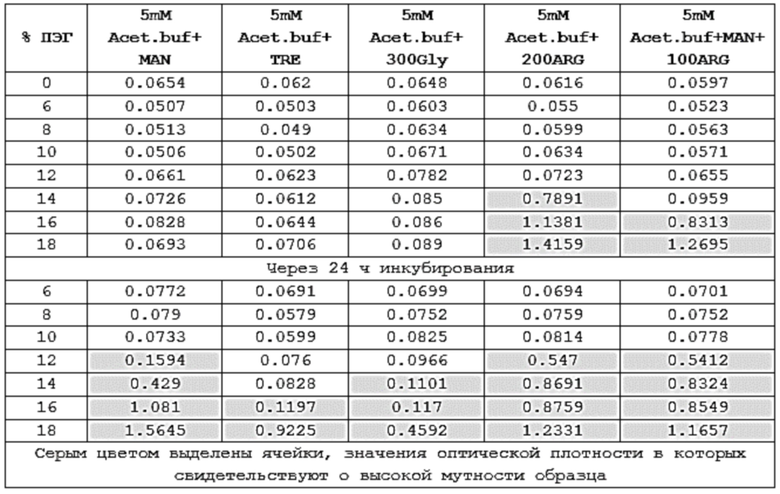
Определение термической стабильности
Оценка термической стабильности выполнена с использованием методик 3 и 5. До и после термостресса определено суммарное содержание примесей методом Э ВЭЖХ с использованием методики 9.
Результаты представлены в таблице 5 и на Фиг. 2, 3, 4 и 5.
Таблица 5. Результаты определения термической стабильности
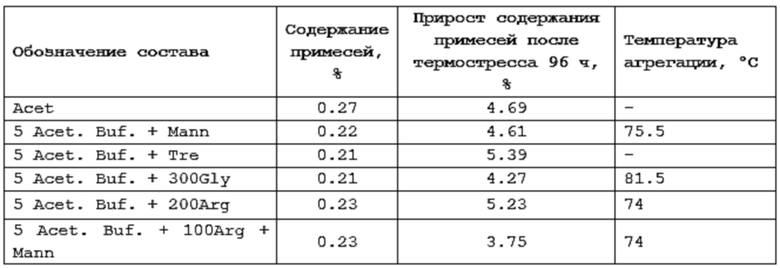
Фармацевтические композиции на основе ацетатной буферной системы, содержащие в качестве осмотических агентов маннитол, трегалозы дигидрат и глицин, продемонстрировали лучшую коллоидную стабильность при ПЭГ-агрегации.
Высокую термическую стабильность продемонстрировали составы на основе ацетатного буферного раствора, содержащие в качестве осмотических агентов глицин и маннитол.
Пример 3. Скрининг осмотических агентов и стабилизаторов
Для проведения скрининга осмотических агентов и стабилизаторов были использованы вспомогательные вещества, пригодные для парентерального введения. Исследуемые составы представлены в таблице 6. Фармацевтические композиции, содержащие левилимаб с концентрацией 10 мг/мл в исследуемых составах, были получены в соответствии с методикой 2.
Таблица 6. Исследуемые составы
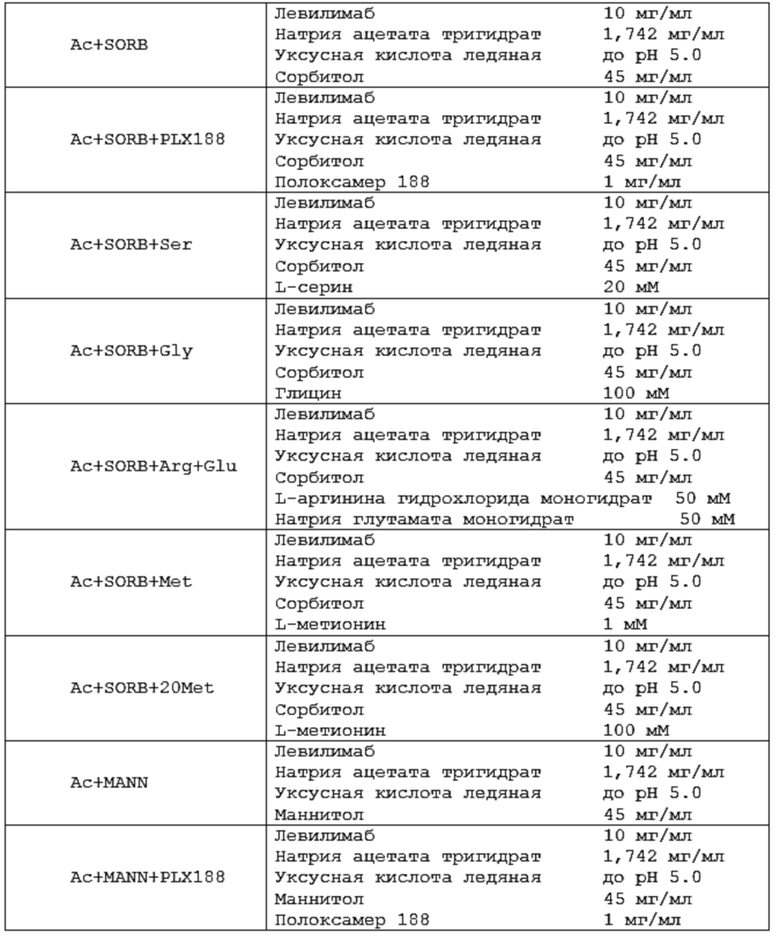
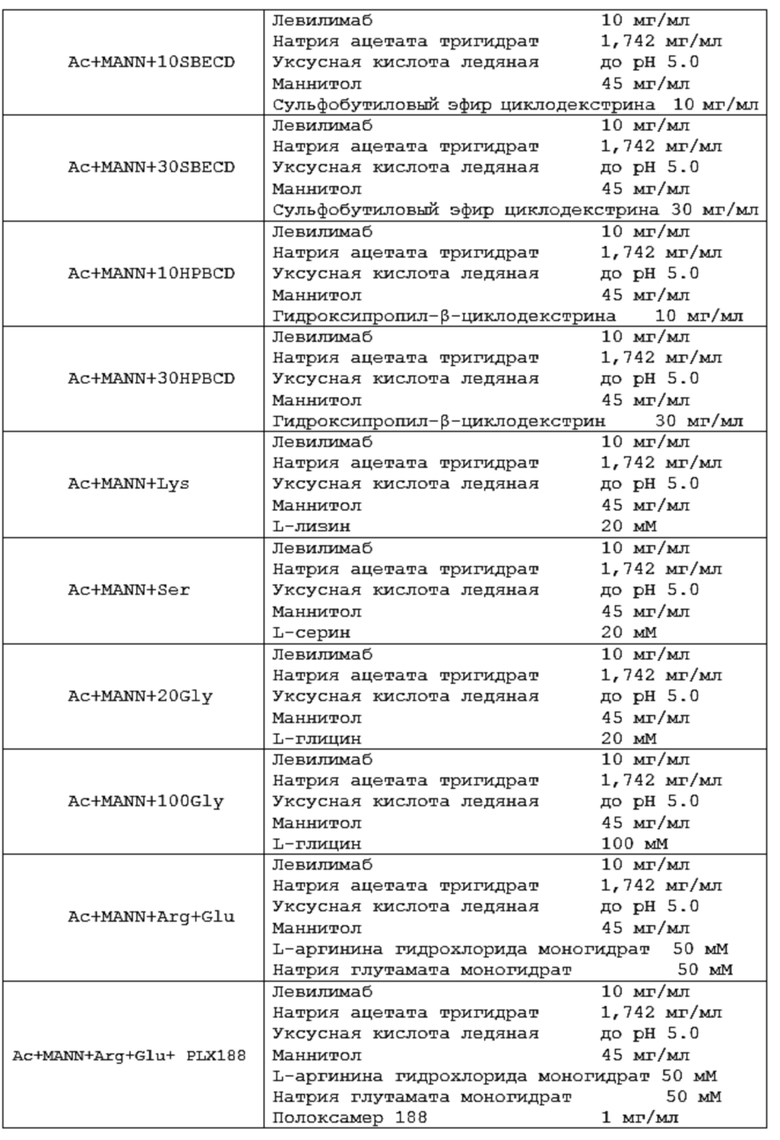
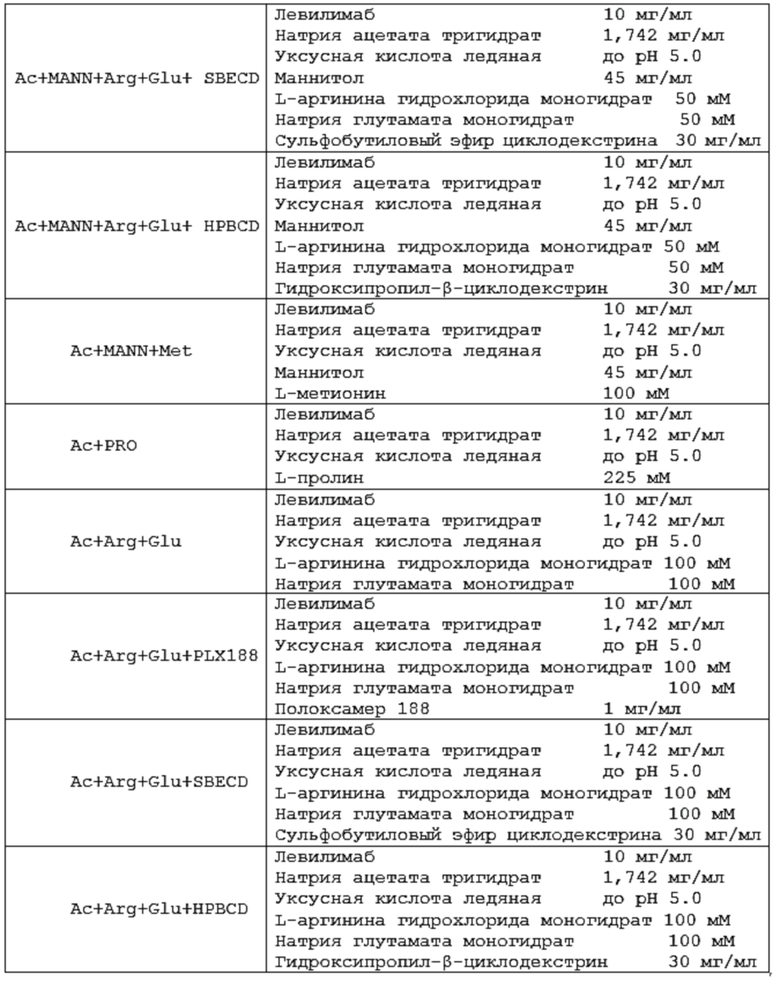
Определение термической стабильности
Исследование термической стабильности проведено в соответствии с методикой 5 в течение 96 часов. Анализ проводили согласно методикам 9-10. Результаты представлены в таблице 7. Результаты анализировали с использованием инструмента «heat-mapping» в ПО «Microsoft Excel». Лучшие результаты имеют более светлый оттенок окраски.
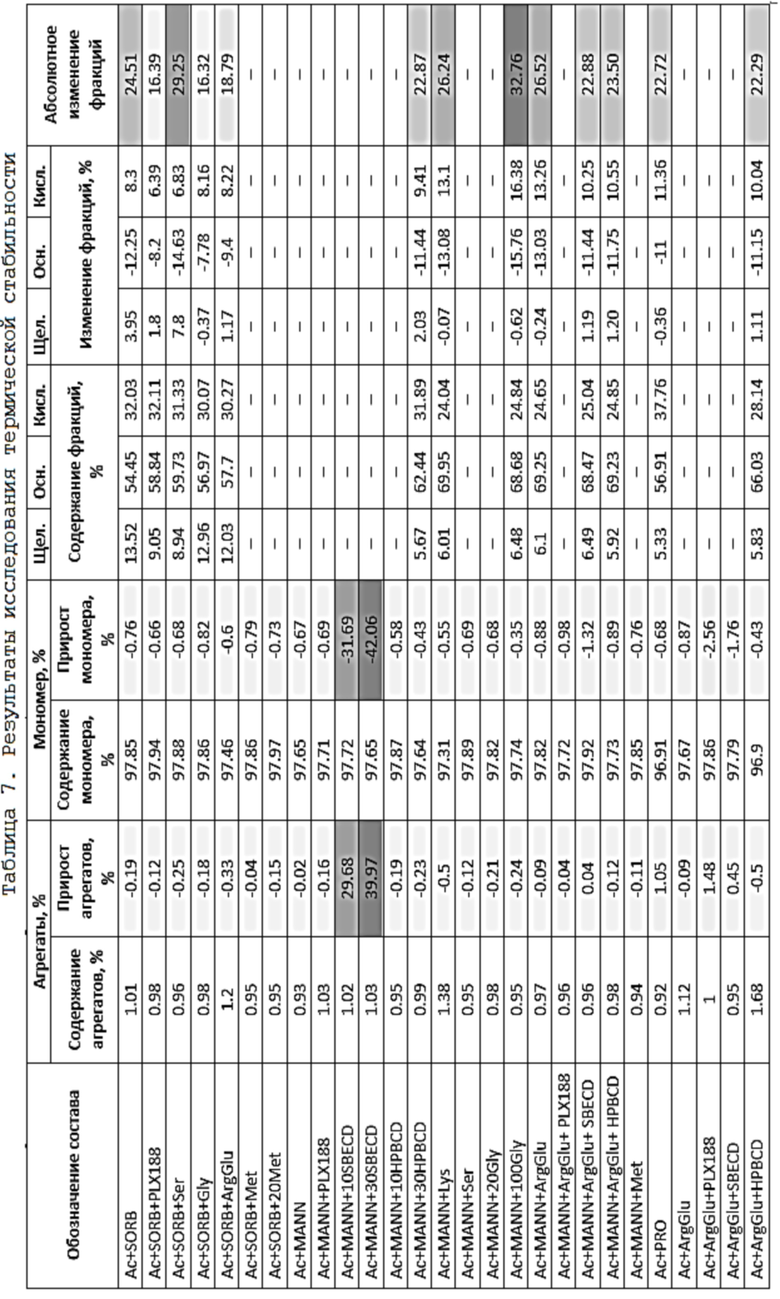
Определение коллоидной стабильности при шейкировании
Исследование коллоидной стабильности проведено согласно методике 6 в течение 96 часов. Результаты исследования представлены в таблице 8. Результаты анализировали с использованием инструмента «heat-mapping» в ПО «Microsoft Excel». Лучшие результаты имеют более светлый оттенок окраски.
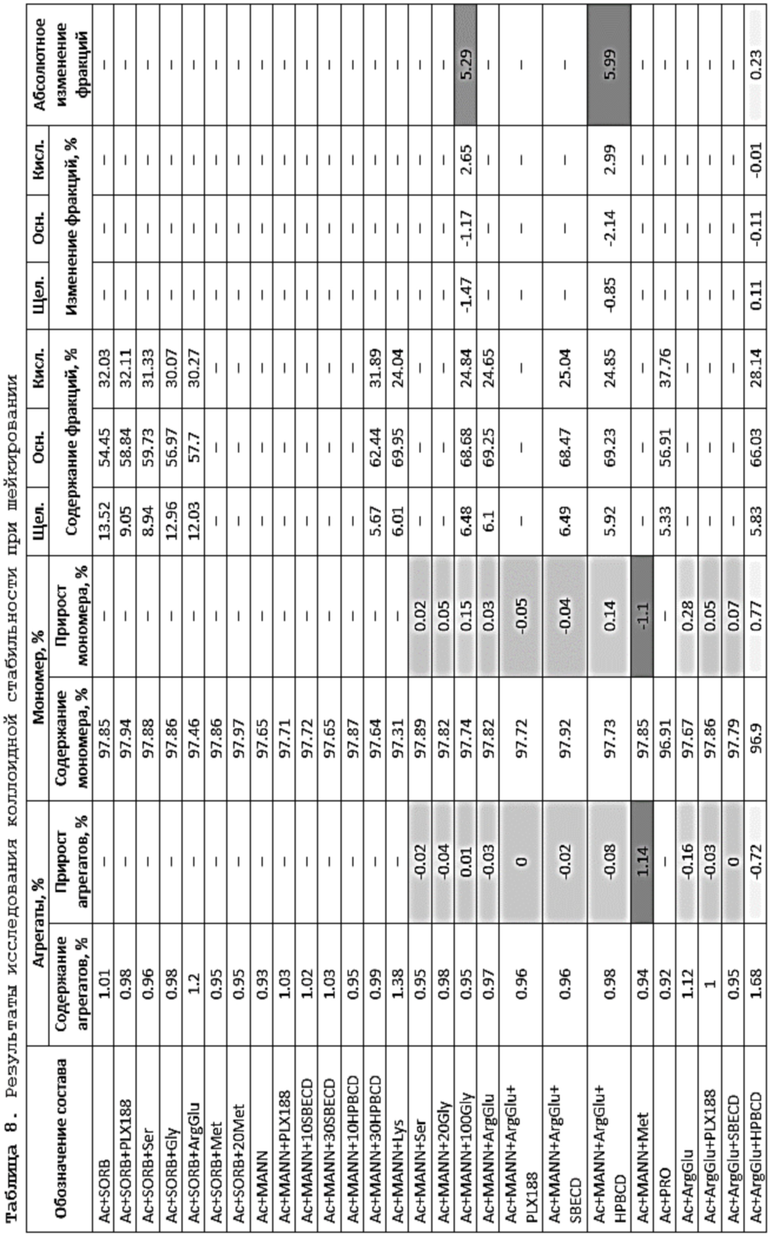
Определение коллоидной стабильности при замораживании и оттаивании.
Исследование коллоидной стабильности проведено согласно методике 7 в течение 96 часов. Результаты исследования представлены в таблице 9. Результаты анализировали с использованием инструмента «heat-mapping» в ПО «Microsoft Excel». Лучшие результаты имеют более светлый оттенок окраски.
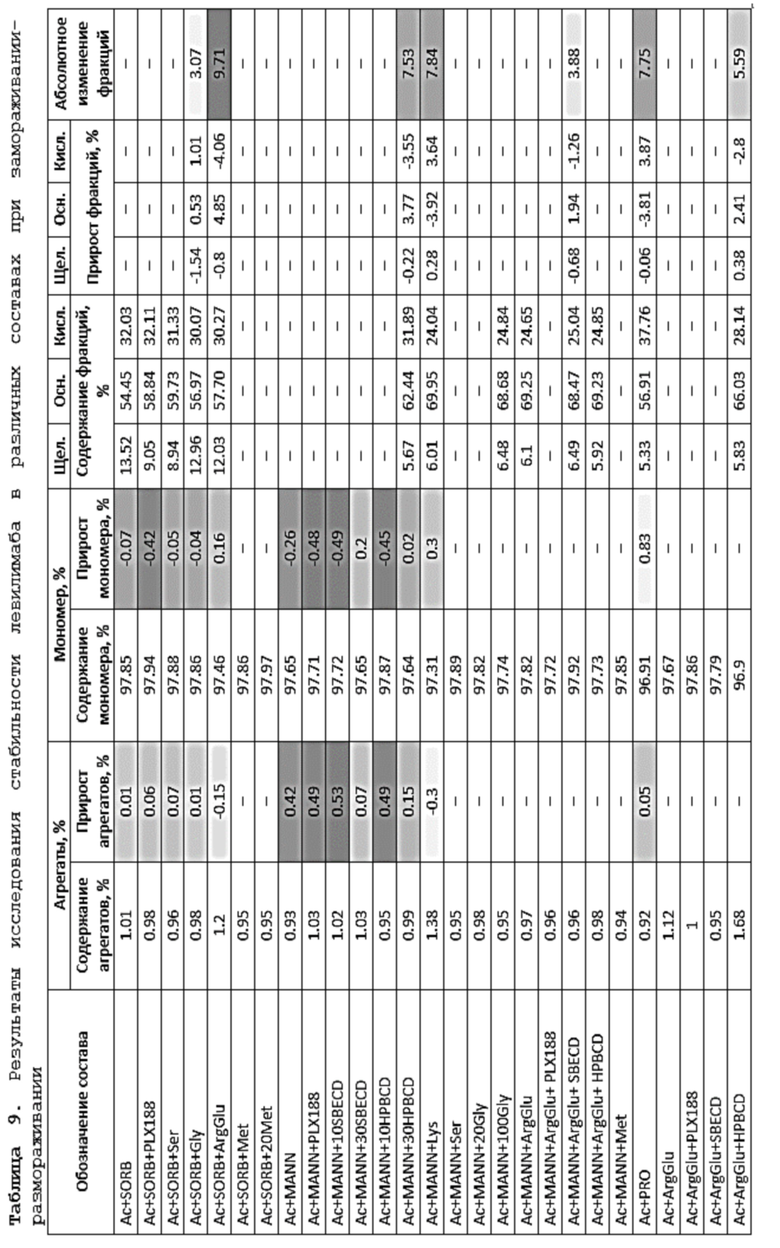
Среди исследуемых образцов можно выделить по стабильности следующий состав:
Данная фармацевтическая композиция продемонстрировала достаточные стабилизирующие свойства среди всех исследуемых образцов: низкий уровень абсолютного изменения кислотно-щелочного профиля при термострессе, а также низкое снижение содержания мономера при термострессе и замораживании.
Состав, содержащий маннитол в качестве осмотического агента и глицин в качестве стабилизатора, продемонстрировал минимальное снижение мономера в ходе термостресса.
В ходе исследования не было выявлено значимых преимуществ использования солюбилизаторов на термическую или коллоидную стабильность белка по сравнению с использованием в качестве стабилизатора аминокислот.
Пример 4. Определение стабильности при ускоренном хранении
Исследования стабильности были проведены для составов: два состава, содержащие глицин в качестве стабилизатора, сорбитол и маннитол в качестве осмотических агентов, а также состав на основе аргинин-ацетатной буферной системы. Составы, содержащие аргинин, продемонстрировали достаточно низкий уровень изменений кислотно-щелочного профиля при термострессе и шейкировании, кроме того, согласно литературным данным, использование аргинина (Hong, T., et al., Current Protein and Peptide Science, 2018, 19, 748-758) значительно снижает вязкость фармацевтической композиции. Исследуемые составы представлены в таблице 10.
Таблица 10. Исследуемые составы
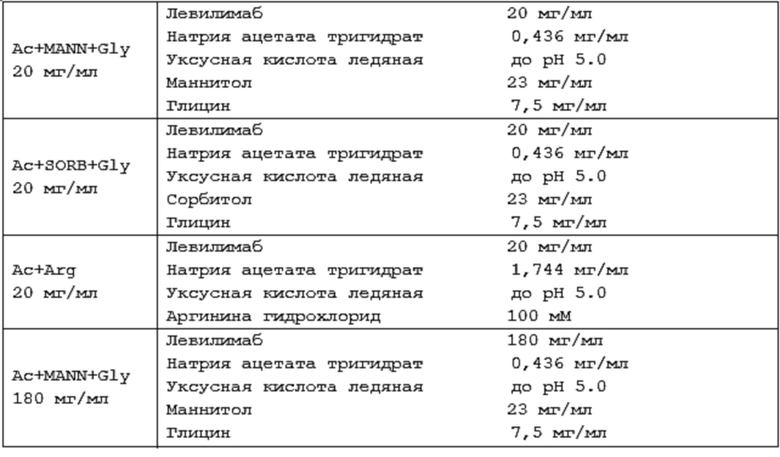
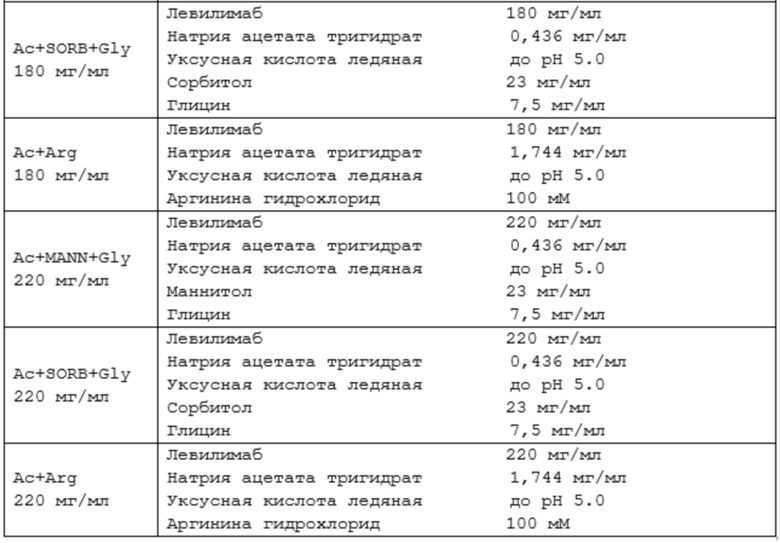
Ускоренное хранение
Фармацевтические композиции, содержащие белок в концентрации 20, 180 и 220 мг/мл приготовлены методом диафильтрации согласно методике 1 и заложены на ускоренное хранение при температуре 25 ± 2°С в соответствии с методикой 8. Результаты исследования представлены в таблице 11 и на Фиг. 6, 7 и 8.
Таблица 11. Результаты исследования стабильности
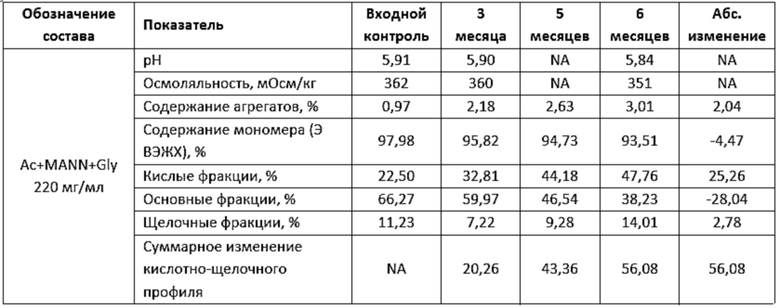
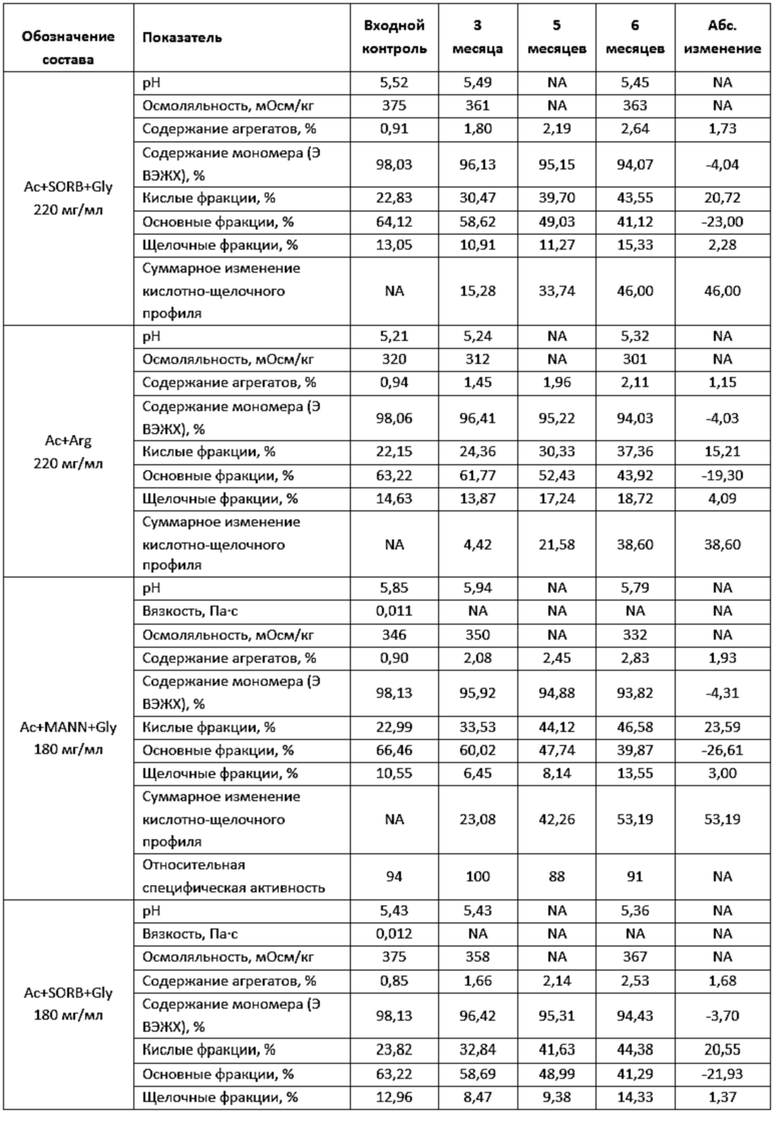
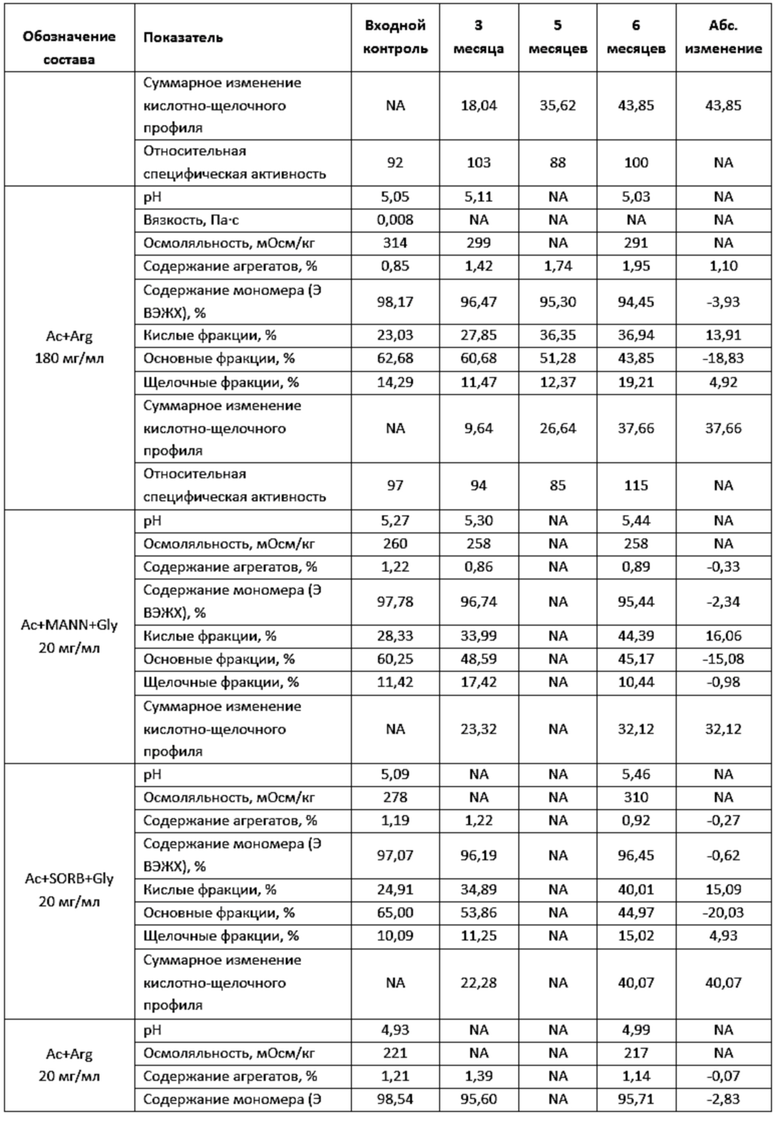
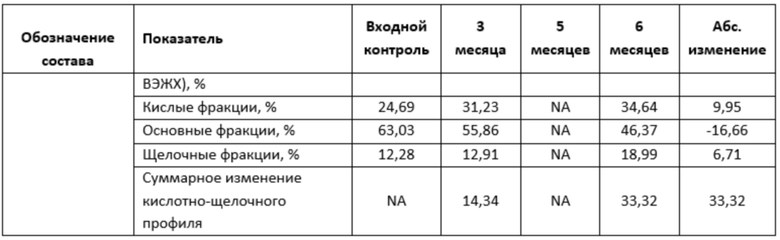
Все фармацевтические композиции продемонстрировали приемлемый уровень изменений в ходе ускоренного хранения.
Фармацевтическая композиция, содержащая ацетат-аргининовую буферную систему, продемонстрировала приемлемый уровень агрегатообразования, а также низкое изменение кислотно-щелочного профиля при ускоренном хранении как при концентрации моноклонального антитела против рецептора IL-6 20 мг/мл, так и при повышенной концентрации до 180-220 мг/мл.
Фармацевтическая композиция, содержащая в составе аргинин, продемонстрировала сниженное значение вязкости.
Фармацевтическая композиция, содержащая в качестве осмотического агента сорбитол, продемонстрировала низкий уровень снижения содержания мономера при ускоренном хранении как при концентрации моноклонального антитела против рецептора IL-6 20 мг/мл, так и при повышенной концентрации 180-220 мг/мл.
Были проведены примеры исследований с использованием водных фармацевтических композиций левилимаба для лечения IL-6-ассоциированных заболеваний. Водные фармацевтические композиции, используемые для данных исследований, охарактеризованы в таблице 11.1.
Таблица 11.1.
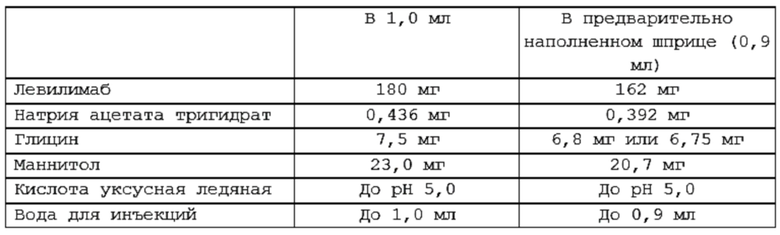
Было показано, что различные дозы фармацевтических композиций левилимаба по настоящему изобретению и/или препарата Левилимаб (также обозначенного в примерах 5 и 6 как LVL и BCD-089), включающий водные фармацевтическиу композиции левилимаба по настоящему изобретению, охарактеризованные в таблице 11.1, являются подходящими для лечения соответствующих IL-6-ассоциированных заболеваний.
Пример 5. Международное многоцентровое сравнительное рандомизированное двойное слепое плацебо-контролируемое клиническое исследование эффективности и безопасности препарата левилимаба в разных режимах дозирования у больных активным ревматоидным артритом
Исследование включало период скрининга, основной период (в ходе которого пациенты получали терапию в заслепленном режиме), открытый период (в ходе которого пациенты получали терапию в открытом режиме) и период последующего наблюдения:
• Скрининг-период (28 - 42 дня)
• Основной период исследования: Неделя 0 – Неделя 12
• Открытый период (Неделя 12 - Неделя 52)
• Период последующего наблюдения (4 календарных недели вплоть до недели 56).
В исследование включались мужчины и женщины в возрасте 18 - 80 лет включительно, с достоверным диагнозом ревматоидного артрита, соответствующим критериям ACR 2010, установленным минимум за 6 месяцев до даты подписания информированного согласия, получавших терапию метотрексатом не менее трех месяцев и не менее последних 4 недель в стабильной дозе, и при сохранении активности заболевания на момент подписания информированного согласия и сохранение активности ревматоидного артрита, несмотря на проведенную в рамках скринингового периода (4-6 недель) терапию метотрексатом без значимой сопутствующей патологии, в соответствии с критериями включения и невключения в исследование.
Итоговая выборка в данном исследовании составила 105 пациентов:
• 35 пациентов рандомизированы в группу, получавшую препарат левилимаб в дозе 162 мг подкожно 1 раз в неделю (группа LVL QW);
• 35 пациентов рандомизированы в группу, получавшую препарат левилимаб в дозе 162 мг подкожно 1 раз в 2 недели (группа LVL Q2W);
• 35 пациентов рандомизированы в группу, получавшую плацебо (группа Плацебо / LVL Q2W) на протяжении первых 12 недель лечения. Начиная с 12 недели, пациенты этой группы получали терапию левилимабом, в дозе 162 мг подкожно в режиме 1 раз в 2 недели до 52 недели исследования.
Исследуемый лекарственный препарат:
МНН: левилимаб – моноклональное антитело против рецептора интерлейкина-6, раствор для инъекций, 180 мг/мл.
Дозы: 162 мг / 0,9 мл
Путь введения: подкожный.
Длительность лечения левилимабом:
• Группа LVL QW:53 недели (Неделя 0 – Неделя 52). Пациенты данной группы могли получить максимум 53 введения препарата левилимаб.
• В группе LVL Q2W 53 недели (Неделя 0 – Неделя 52). Пациенты данной группы могли получить максимум 27 введений препарата левилимаб.
• В группе Плацебо / LVL Q2W: 41 неделя (Неделя 12 – Неделя 52). Пациенты данной группы могли получить максимум 21 введение препарата левилимаб за анализируемый период.
Конечные точки для оценки эффективности основного периода исследования:
Первичная конечная точка:
• Доля больных ревматоидным артритом в каждой группе, у которых к 12 неделе от момента первого введения BCD-089 / плацебо было достигнуто улучшение в течение болезни, соответствующее ACR20.
Дополнительные конечные точки для основного периода исследования:
• Доля больных ревматоидным артритом в каждой группе, у которых было достигнуто улучшение в течение болезни, соответствующее ACR20, к 4 и 8 неделям от момента первого введения BCD-089 / плацебо.
• Доля больных ревматоидным артритом в каждой группе, у которых было достигнуто улучшение в течение болезни, соответствующее ACR50/70, к 4, 8 и 12 неделям от момента первого введения BCD-089 / плацебо.
• Доля больных в каждой группе с низкой активностью РА согласно индексу DAS28-CRP(4) (DAS28-CRP(4)<3,2) CDAI (CDAI≤10, SDAI (SDAI≤11) к 4, 8 и 12 неделям от момента первого введения BCD-089 / плацебо.
• Изменение индексов DAS28-CRP(4), CDAI и SDAI на 12 неделе по сравнению с исходными значениями.
• Изменение скорости оседания эритроцитов на 12 неделе терапии по сравнению с исходными значениями.
Дополнительные конечные точки для открытого периода исследования
• Доля больных ревматоидным артритом, у которых было достигнуто улучшение в течение болезни, соответствующее ACR20/50/70 к 16, 24, 36, 48 и 52 неделе от момента первого введения BCD-089.
• Доля больных с низкой активностью РА согласно индексу DAS28-CRP(4) (DAS28-CRP(4)<3,2) CDAI (CDAI≤10), SDAI (SDAI≤11) к 16, 24, 36, 48 и 52 неделям от момента первого введения BCD-089.
• Изменение индексов DAS28-CRP(4), CDAI и SDAI по сравнению с исходными значениями.
• Доля больных, достигших ремиссии согласно критериям ACR/EULAR 2011 к 24, 36, 48 и 52 неделям терапии BCD-089.
• Оценка качества жизни пациентом до лечения, через 24 и 52 недели после первого введения BCD-089 согласно опроснику SF36.
• Изменение скорости оседания эритроцитов по сравнению с исходными значениями.
• Рентгенологическая характеристика пораженных суставов через 52 недели после первого введения BCD-089.
– Изменение среднего значения общего балла согласно оценке по методу Sharp в модификации van der Heijde (1989 г.).
– Доля больных с повышением рентгенологической стадии ревматоидного артрита (при оценке по методу Штейнброкера).
Конечная точка для оценки фармакодинамики
Вторичные конечные точки
Фармакодинамика была проанализирована с помощью определения концентрации аналитов, перечисленных ниже, в сыворотке крови методом твердофазного ИФА:
• растворимого рецептора к интерлейкину-6
• С-реактивного белка
• ИЛ-6 (IL-6)
• ФНОα
Вторичные конечные точки
• Emin (минимальная концентрация C-РБ в сыворотке крови).
• ETmin (время достижения минимальной концентрации C-РБ).
• AUEC0-last (площадь под кривой «концентрация для С-РБ, sIL-6R, ФНОб и IL-6-время» (AUC — от англ. «area under curve») от момента введения препарата до последнего измерения концентрации).
• Emax (максимальная концентрация sIL-6R в сыворотке крови).
• EТmax (время достижения максимальной концентрации sIL-6R).
Конечные точки для оценки безопасности
• Доля пациентов с нежелательными явлениями, в том числе серьезными, в каждой группе.
• Доля пациентов с серьезными нежелательными явлениями в каждой группе.
• Доля пациентов с нежелательными явлениями 3-4 степени в каждой группе.
• Доля пациентов с нейтропенией 3-4 степени в каждой группе.
• Доля пациентов в каждой группе с нежелательными явлениями, характерными для использования ингибиторов рецептора IL-6:
– Повышение активности АЛТ/АСТ;
– Лейкопения/Нейтропения;
– Тромбоцитопения;
– Инфекции верхних дыхательных путей; флегмоны; пневмонии; инфекции, вызываемые Herpes Simplex 1 типа и Herpes Zoster; дивертикулит;
– Повышение общего холестерина / ЛПВП / ЛПНП / триглицеридов.
• Доля пациентов, досрочно прекративших участие в исследовании в связи с развитием НЯ/СНЯ, в каждой группе.
Конечная точка для оценки иммуногенности
Основной период исследования
• Доля пациентов с выявленными связывающими и/или нейтрализующими антителами к препарату BCD-089 на неделе 12.
Открытый период исследования
• Доля пациентов с выявленными связывающими и/или нейтрализующими антителами к препарату BCD-089 на неделях 24 и 52.
Результаты оценки эффективности:
Применение левилимаба на протяжении года характеризовалось продолжающимся ростом числа пациентов с улучшением в течение болезни. При этом наименее выраженный ответ, соответствующий ACR20, достигался большинством пациентов в течение первых 24 недель терапии, и в дальнейшем рост числа ответчиков обеспечивался, за счет достижения пациентами ACR50 и в большей степени ACR70 (фигуры 9, 10, 11).
Начиная с недели 4 терапии, доли пациентов с низкой активностью РА в группе LVL QW была численно выше по сравнению с группой LVL Q2W для индексов CDAI и SDAI, а для DAS28-CRP(4) различия достигали статистической значимости на неделе 12. Индексы DAS-28-CRP(4), CDAI и SDAI демонстрировали отчетливую положительную динамику на протяжении 52 недель терапии, отражая уменьшение выраженности клинической симптоматики РА. В течение первых 12 недель терапии изменения индексов численно были более выражены в группе LVL QW по сравнению с группой LVL Q2W, а для индекса DAS28-CRP(4) различия достигали статистической значимости также к 12 неделе (фигура 12). В целом, динамика активности РА, отражаемая как долями пациентов с низкой активностью, так и изменениями индексов, свидетельствует о более высоком темпе клинического ответа в группе LVL QW.
Частота достижения ремиссии в течение РА (по ACR/EULAR 2011) была сопоставима на 52 неделе терапии в группах LVL QW и LVL Q2W, однако несмотря на отсутствие статистической значимости различий, в группе LVL QW наблюдались численно более высокие значения показателя на неделях 24, 36 и 48 по сравнению с LVL Q2W, что, также, подтверждает более высокий темп клинического ответа в группе пациентов, применявших препарат один раз в неделю (фигура 13).
Анализ изменений СОЭ относительно исходного уровня на фоне терапии левилимабом выявил, что в группах LVL QW и LVL Q2W уже после первого введения исследуемого препарата, наблюдалось выраженное снижение скорости оседания эритроцитов, которая достигала минимальных значений в течение первых 2-4-х недель терапии и в дальнейшем существенно не изменялась, оставаясь минимальной до конца исследования (фигура 14).
Анализ результатов оценки физического (PH) и психологического (MH) компонентов качества жизни по опроснику SF-36 показал, что терапия левилимабом сопровождается улучшением оценки пациентами как физического, так и психологического компонентов качества жизни.
Оценка рентгенологических изменений в суставах (по Sharp в модификации van der Heijde) показала, что на фоне терапии левилимабом абсолютные значения показателя между скринингом и неделей 52 статистически значимо не различались. Однако анализ изменений показателя, выявил статистически значимое различие между группами LVL QW и LVL Q2W (p=0,0494) на неделе 52. В группе LVL QW не наблюдалось изменений общего бала в течение года, а в группе LVL Q2W нарастание рентгенологических изменений было выявлено у 3 пациентов. Повышение рентгенологической стадии РА по методу Штейнбокера было зарегистрировано лишь у одного пациента группы Плацебо / LVL Q2W.
Оценка показателя доля пациентов с повышением рентгенологической стадии ревматоидного артрита не выявила пациентов с прогрессированием рентгенологической стадии по методу Штейнбокера в группах LVL QW и LVL Q2W.
Полученные данные об эффективности по первичной конечной точке подтвердили гипотезу о превосходстве эффективности препарата левилимаб над плацебо во всех исследуемых популяциях (PP и ITT), как при использовании схемы 1 раз в неделю, так и 1 раз в 2 недели, и, следовательно, позволили сделать вывод об эффективности обоих исследованных режимов дозирования препарата левилимаб у пациентов с активным ревматоидным артритом и достижении исследованием своей цели. При этом, более частое введение исследуемого препарата – 1 раз в неделю в течение года, показало несколько лучшую эффективность по сравнению с режимом 1 раз в 2 недели, как по времени достижения, так и по величине ответа на терапию.
Анализ данных по безопасности применения препарата левилимаб у пациентов с активным ревматоидным артритом в течение 1 года, показал, что препарат левилимаб в дозе 162 мг независимо от режима введения обладает благоприятным профилем безопасности и низкой иммуногенностью.
На фоне терапии исследуемым препаратом происходит выраженное изменение сывороточных концентраций фармакодинамических маркеров – нарастание концентрации sIL-6R, IL-6 и снижение концентрации СРБ. Режим дозирования 1 раз в неделю обеспечивает существенно более выраженный рост концентрации sIL-6R (характерный для группы ингибиторов sIL-6R) и характеризуется тенденцией к более быстрому и выраженному снижению концентрации СРБ. В целом, динамика фармакодинамических маркеров свидетельствует о высокоэффективной нейтрализации растворимого рецептора к IL-6 препаратом левилимаб, что, в свою очередь, проявляется быстрым и выраженным снижением сывороточной концентрации СРБ, отражающим эффективное подавление воспалительного процесса у пациентов с активным ревматоидным артритом. При этом, введение препарата левилимаб в режиме 1 раз в 1 неделю обладает большей эффективностью в отношении фармакодинамических маркеров по сравнению с режимом 1 раз в 2 недели.
Пример 6. Оценка фармакодинамики
В качестве фармакодинамических маркеров в данном исследовании использовались сывороточные концентрации растворимого рецептора к интерлейкину-6 (sIL-6R), С-реактивного белка (СРБ).
В анализ фармакодинамических параметров были включены данные 104 пациентов: 35 пациентов получавших п/к введение левилимаба 1 раз в неделю (группа BCD-089 QW), 34 пациента получавших п/к введение левилимаба 1 раз в 2 недели (группа BCD-089 Q2W), и 35 пациентов группы Плацебо.
Из анализа фармакодинамики был исключен 1 пациент группы BCD-089 Q2W, - отозвавший информированное согласие на участие в исследовании на визите 1 до первого введения исследуемого препарата.
Оценка концентраций растворимого рецептора к IL-6 (sIL-6R)
Концентрация sIL-6R в сыворотке крови (отражает блокирование рецептора исследуемым препаратом, характерное для препаратов группы ингибиторов sIL6R) нарастала в сыворотке крови пациентов обеих групп исследуемого препарата и достигала наибольших значений (Emax) в группе BCD-089 QW (3240960 [1937060-4108080] пг/мл через 2016 [1344-2016] ч. В группе BCD-089 Q2W Emax составила 1835030 [1536920-3020400] пг/мл через 2016 [1344; 2016] часов. В группе Плацебо, нарастания концентрации sIL-6R не наблюдалось, Emax составила 228440 [168822-367380] пг/мл через 96 [48-504] часов. Статистически значимые различия были выявлен как между группами исследуемого препарата и плацебо (p<0,0001; критерий Краскелла-Уоллиса), так и между группами исследуемого препарата (p=0,0112; критерий Краскелла-Уоллиса).
Подробно, результаты статистического анализа концентрации sIL-6R представлены в таблице ниже.
Введенная доза BCD-089 определяла значения показателей площади под кривой концентрация/время (AUEC0-last), которые достигали значимо высоких значений в группах BCD-089 QW и с BCD-089 Q2W и Плацебо (p<0.0001; критерий Краскелла-Уоллиса). При этом различия между исследуемыми группами также имели значимые различия (p=0,0066; критерий Краскелла-Уоллиса) (фигуры 15).
Таблица 12. Фармакодинамические показатели сывороточных концентраций sIL-6 в исследуемых группах
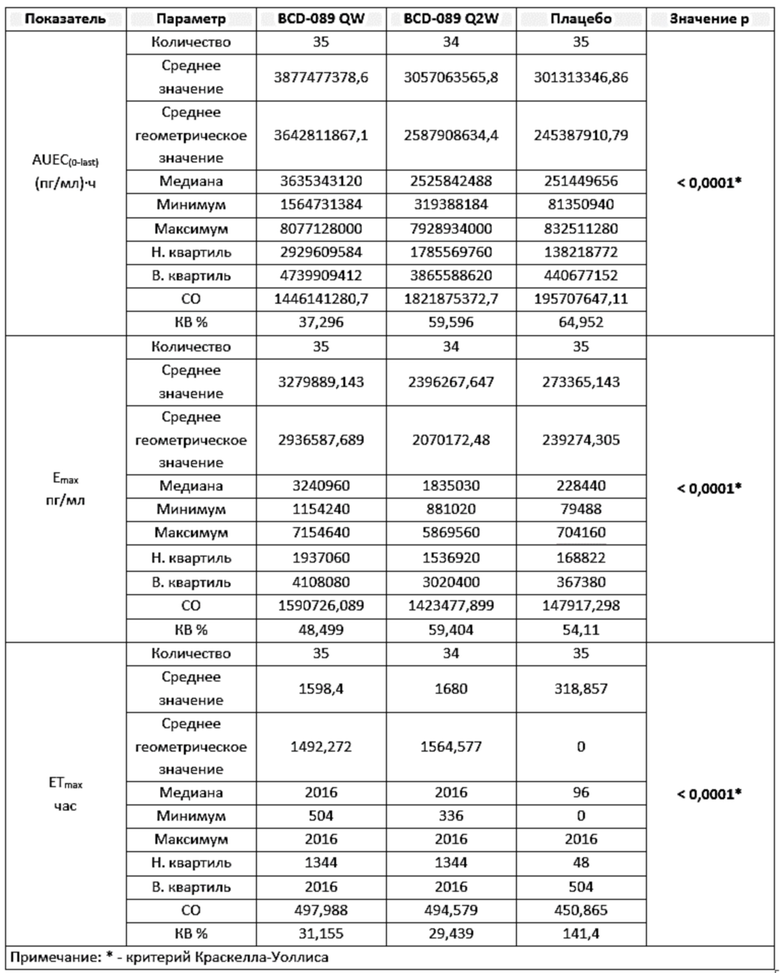
Оценка концентрации С-реактивного белка
Концентрации C-реактивного белка в сыворотке крови пациентов групп демонстрировали отчетливое снижение на фоне проводимой терапии. Максимальное снижение было выявлено в группе BCD-089 QW – Emin, составили 0 [0;404] нг/мл и достигалось через 672 [336; 1344] часа. В группе BCD-089 Q2W, соответствующие значения составили 72 [0;421] нг/мл, которые достигались через 1344 [504;2016] часов, при этом, значимые различия между группами отсутствовали (p>0,05).
В группе Плацебо минимальная концентрация СРБ составляла 1421 [1087; 2266] нг/мл, которая наблюдалась через 336 [96; 672] часа. Показатели Emin и ETmin в группе плацебо значимо отличались от соответствующих показателей в обеих группах исследуемого препарата (фигура 16).
Таблица 13. Фармакодинамические показатели C-реактивного белка в сыворотке крови пациентов
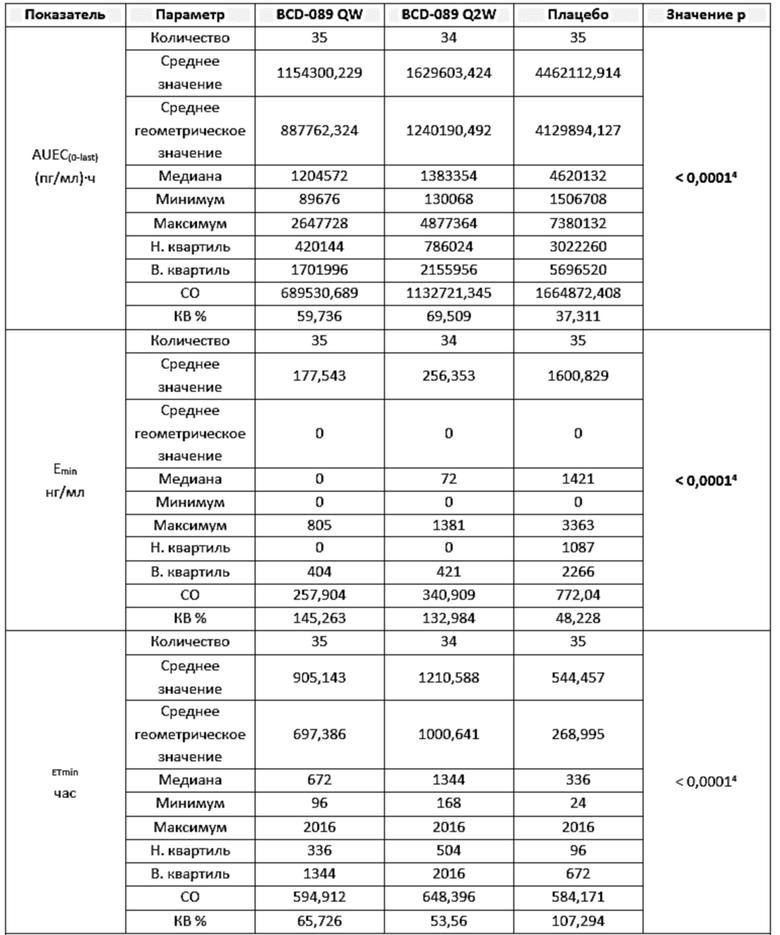
На фоне терапии исследуемым препаратом наблюдалось выраженное изменение сывороточных концентраций фармакодинамических маркеров – нарастание концентрации sIL-6R и снижение концентрации СРБ.
Полученные значения фармакодинамических показателей характеризовались статистически значимыми различиями с полученными у пациентов группы Плацебо. Кроме того, параметры, характеризующие концентрацию sIL-6R (Рост концентрации которого отражает блокирование рецептора исследуемым препаратом и характерен для препаратов группы ингибиторов sIL6R) также имели значимые различия между группами исследуемого препарата. При этом наблюдалась тенденция к более быстрому и выраженному снижению концентрации СРБ в группе BCD-089 qw по сравнению с BCD-089 Q2W, однако, данные различия были не значимыми.
В целом, динамика фармакодинамических маркеров свидетельствует о высокоэффективной нейтрализации растворимого рецептора к IL-6 препаратом BCD-089, что, в свою очередь, проявляется быстрым и выраженным снижением сывороточной концентрации СРБ, свидетельствующем об эффективном подавлении воспалительного процесса у пациентов с активным ревматоидным артритом. Введение препарата BCD-089 в режиме 1 раз в неделю продемонстрировало лучшую эффективность в отношении фармакодинамических маркеров, по сравнению с режимом 1 раз в 2 недели.
Известно, что применение анти-IL-6R терапии эффективно при синдроме высвобождения цитокинов и взрослом (остром) респираторном дистресс синдроме. Принимая во внимание полученные данные фармакодинамики левилимаба, свидетельствующие о его способности эффективно блокировать сигналинг IL-6, можно заключить, что препарат левилимаб будет эффективен в терапии синдрома высвобождения цитокинов (СВЦ) и взрослого (острого) респираторного дистресс синдрома (ОРДС).
СВЦ относят к основной причине летальности пациентов с SARS-CoV, MERS-CoV и COVID-19, при этом наблюдающийся у этих пациентов повышенный уровень интерлейкина 6 (IL-6) коррелирует с уровнем С-реактивного белка (СРБ), дыхательной недостаточностью, ОРДС и неблагоприятными клиническими исходами.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> ЗАО «БИОКАД»
<120> ВОДНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ЛЕВИЛИМАБА И ЕЕ ПРИМЕНЕНИЕ
<160> 10
<170> BiSSAP 1.3.6
<210> 1
<211> 5
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_HCDR1
<400> 1
Ser Tyr Tyr Met Ser
1 5
<210> 2
<211> 16
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_HCDR2
<400> 2
Gly Ile Tyr Ser Asp Gly Thr Thr His Tyr Gly Asp Ser Val Lys Gly
1 5 10 15
<210> 3
<211> 12
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_HCDR3
<400> 3
Gly Ala Gly Pro Thr Trp Trp Tyr Ala Leu Asp Ala
1 5 10
<210> 4
<211> 120
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_VH
<400> 4
Gln Val Gln Leu Val Gln Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr
20 25 30
Tyr Met Ser Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Gly Ile Tyr Ser Asp Gly Thr Thr His Tyr Gly Asp Ser Val Lys
50 55 60
Gly Arg Phe Thr Ile Ser Arg Asp Asn Ala Lys Asn Thr Val Tyr Leu
65 70 75 80
Gln Leu Asn Ser Leu Arg Ala Glu Asp Thr Ala Met Tyr Tyr Cys Ala
85 90 95
Lys Gly Ala Gly Pro Thr Trp Trp Tyr Ala Leu Asp Ala Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 5
<211> 450
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_HC
<400> 5
Gln Val Gln Leu Val Gln Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr
20 25 30
Tyr Met Ser Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Gly Ile Tyr Ser Asp Gly Thr Thr His Tyr Gly Asp Ser Val Lys
50 55 60
Gly Arg Phe Thr Ile Ser Arg Asp Asn Ala Lys Asn Thr Val Tyr Leu
65 70 75 80
Gln Leu Asn Ser Leu Arg Ala Glu Asp Thr Ala Met Tyr Tyr Cys Ala
85 90 95
Lys Gly Ala Gly Pro Thr Trp Trp Tyr Ala Leu Asp Ala Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Arg Val Glu Pro Lys Ser Cys Asp
210 215 220
Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Gly
225 230 235 240
Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Tyr Ile
245 250 255
Thr Arg Glu Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
260 265 270
Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His
275 280 285
Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg
290 295 300
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320
Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr
340 345 350
Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu
355 360 365
Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp
370 375 380
Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val
385 390 395 400
Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp
405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430
Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro
435 440 445
Gly Lys
450
<210> 6
<211> 15
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_LCDR1
<400> 6
Lys Ser Ser Gln Ser Val Leu Ser Ala Ser Asn Thr Tyr Leu Asn
1 5 10 15
<210> 7
<211> 7
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_LCDR2
<400> 7
Tyr Ala Ser Thr Arg Glu Ser
1 5
<210> 8
<211> 9
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_LCDR3
<400> 8
Gln Gln Ala Tyr Arg Ala Pro Val Thr
1 5
<210> 9
<211> 111
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_VL
<400> 9
Asp Ile Gln Leu Thr Gln Ser Pro Ser Ser Val Ser Val Ser Val Gly
1 5 10 15
Glu Arg Val Thr Ile Asp Cys Lys Ser Ser Gln Ser Val Leu Ser Ala
20 25 30
Ser Asn Thr Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro
35 40 45
Gln Leu Leu Ile Tyr Tyr Ala Ser Thr Arg Glu Ser Gly Val Pro Asp
50 55 60
Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser
65 70 75 80
Ser Leu Gln Ala Glu Asp Ala Ala Val Tyr Tyr Cys Gln Gln Ala Tyr
85 90 95
Arg Ala Pro Val Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys
100 105 110
<210> 10
<211> 218
<212> PRT
<213> Искусственная последовательность
<220>
<223> Levilimab_LC
<400> 10
Asp Ile Gln Leu Thr Gln Ser Pro Ser Ser Val Ser Val Ser Val Gly
1 5 10 15
Glu Arg Val Thr Ile Asp Cys Lys Ser Ser Gln Ser Val Leu Ser Ala
20 25 30
Ser Asn Thr Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro
35 40 45
Gln Leu Leu Ile Tyr Tyr Ala Ser Thr Arg Glu Ser Gly Val Pro Asp
50 55 60
Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser
65 70 75 80
Ser Leu Gln Ala Glu Asp Ala Ala Val Tyr Tyr Cys Gln Gln Ala Tyr
85 90 95
Arg Ala Pro Val Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Arg
100 105 110
Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln
115 120 125
Leu Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr
130 135 140
Pro Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser
145 150 155 160
Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr
165 170 175
Tyr Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys
180 185 190
His Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro
195 200 205
Val Thr Lys Ser Phe Asn Arg Gly Glu Cys
210 215
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИ-TRBV9 АНТИТЕЛА И ЕЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2826886C1 |
| ВОДНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИ-IL17A АНТИТЕЛА И ЕЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2754760C2 |
| ВОДНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИТЕЛА К PD-1 ПРОЛГОЛИМАБА И ЕЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2806320C2 |
| СПОСОБЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА | 2020 |
|
RU2828027C2 |
| КОМБИНИРОВАННЫЙ ПРЕПАРАТ, ВКЛЮЧАЮЩИЙ КОРТИКОСТЕРОИД И ЭКЗОСОМЫ | 2010 |
|
RU2557897C2 |
| АНТИТЕЛА, НЕЙТРАЛИЗУЮЩИЕ GM-CSF, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РЕВМАТОИДНОГО АРТРИТА ИЛИ В КАЧЕСТВЕ АНАЛЬГЕТИКОВ | 2014 |
|
RU2714919C2 |
| СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОГО НОВООБРАЗОВАНИЯ С ИСПОЛЬЗОВАНИЕМ КОМБИНАЦИИ АНТИТЕЛА К PD-1 И ХИМИОТЕРАПЕВТИЧЕСКОГО АГЕНТА | 2021 |
|
RU2787457C2 |
| СОСТАВЫ, СОДЕРЖАЩИЕ АНТИТЕЛА | 2009 |
|
RU2745601C2 |
| НОВЫЕ ПОКАЗАНИЯ К ПРИМЕНЕНИЮ ПРИ ЛЕЧЕНИИ АНТИТЕЛАМИ ПРОТИВ IL-1-БЕТА | 2019 |
|
RU2834722C2 |
| НОВЫЕ ПОКАЗАНИЯ К ПРИМЕНЕНИЮ ПРИ ЛЕЧЕНИИ АНТИТЕЛАМИ ПРОТИВ IL-1-БЕТА | 2014 |
|
RU2698208C2 |
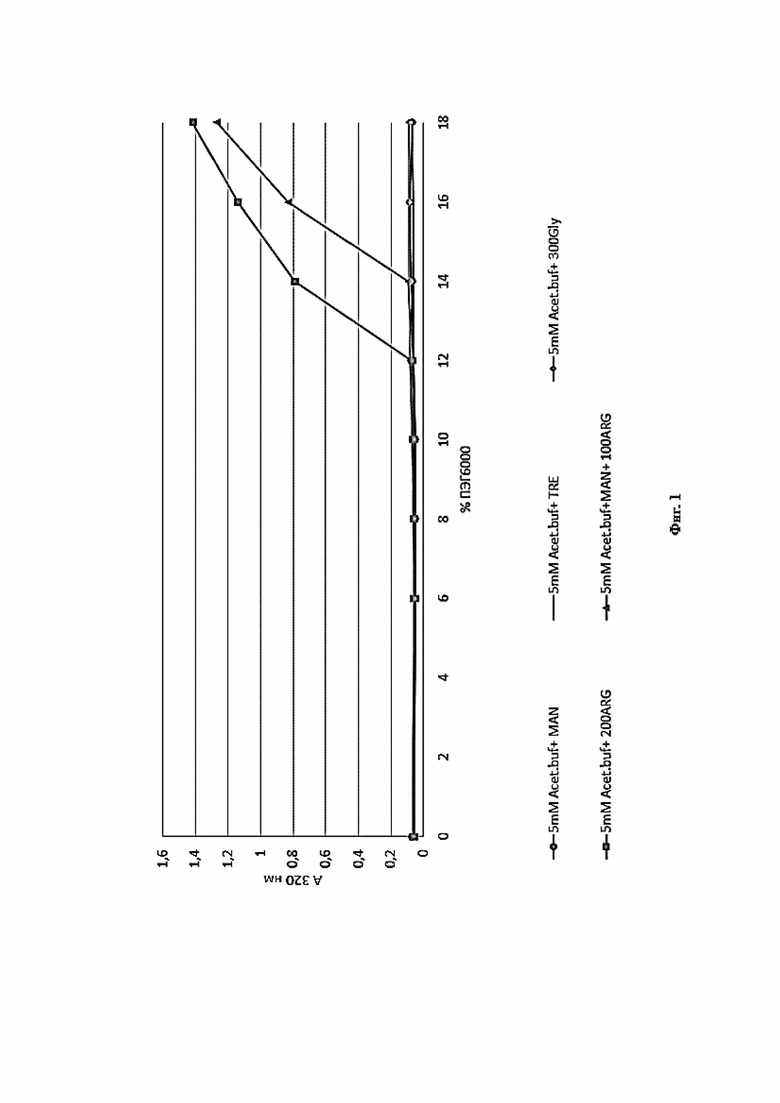
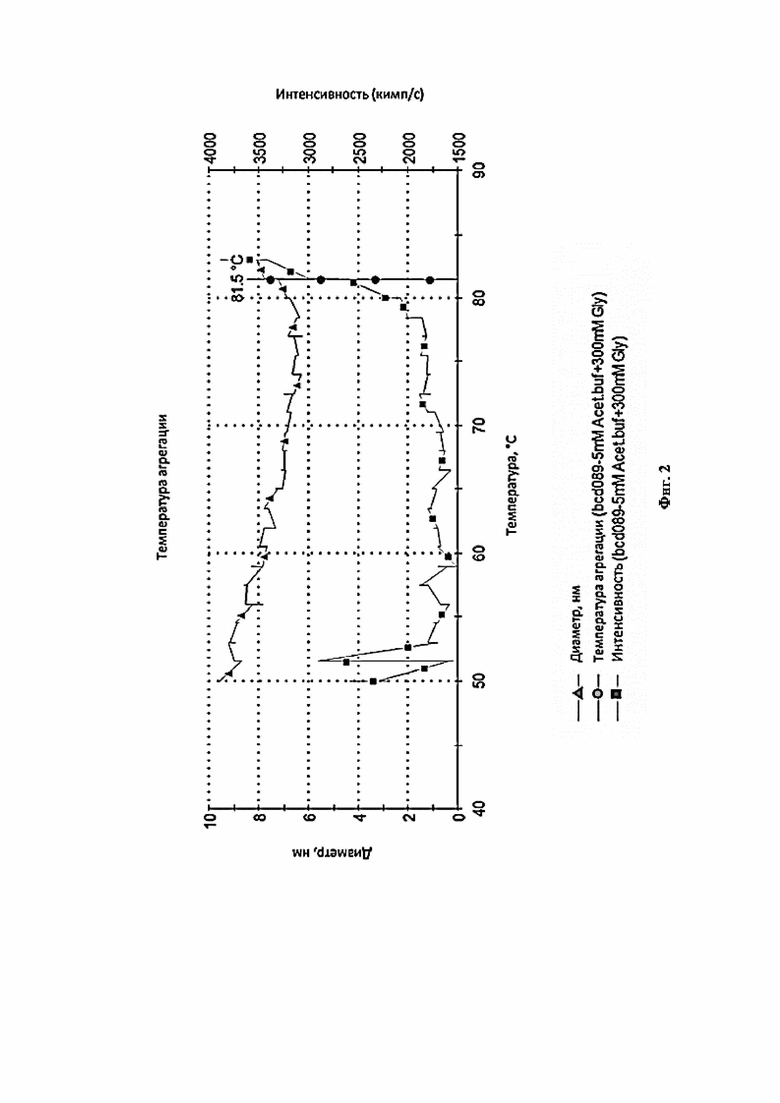
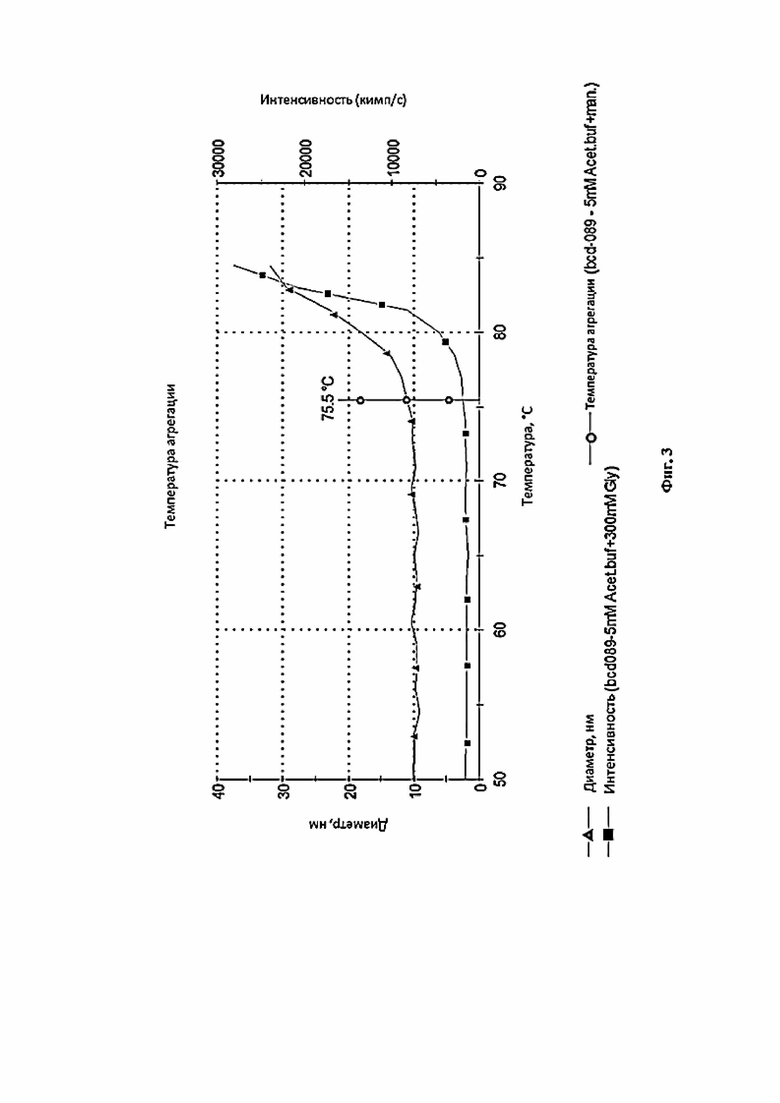
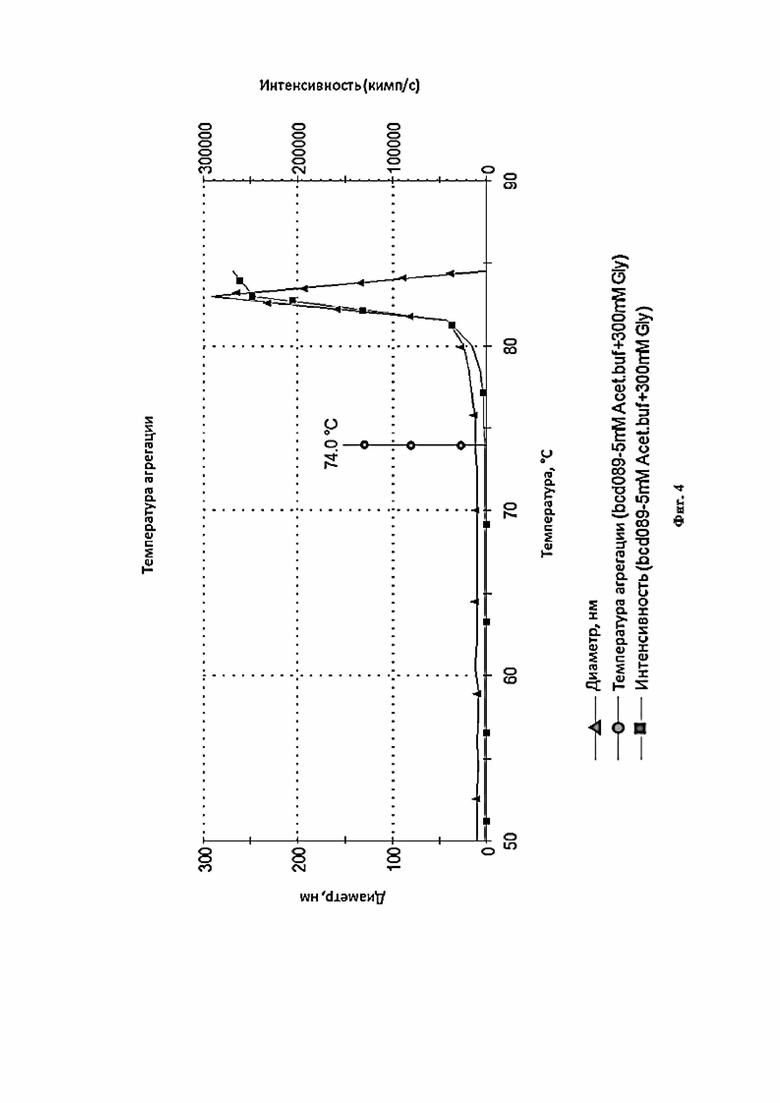
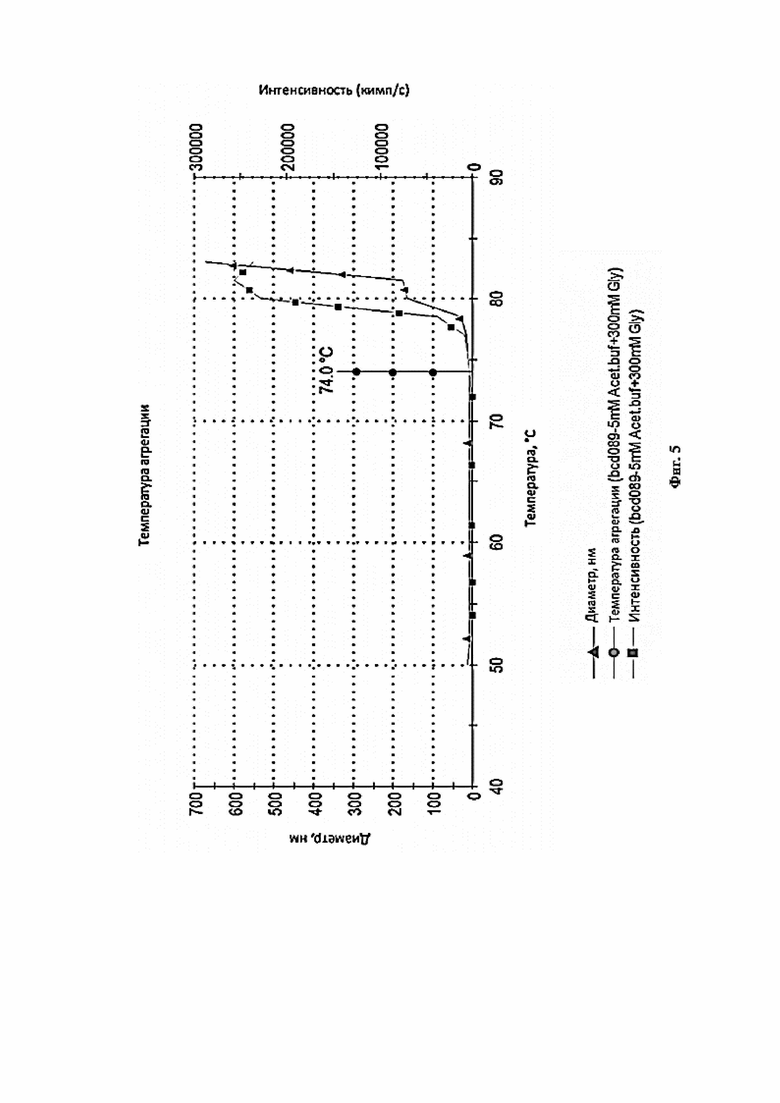
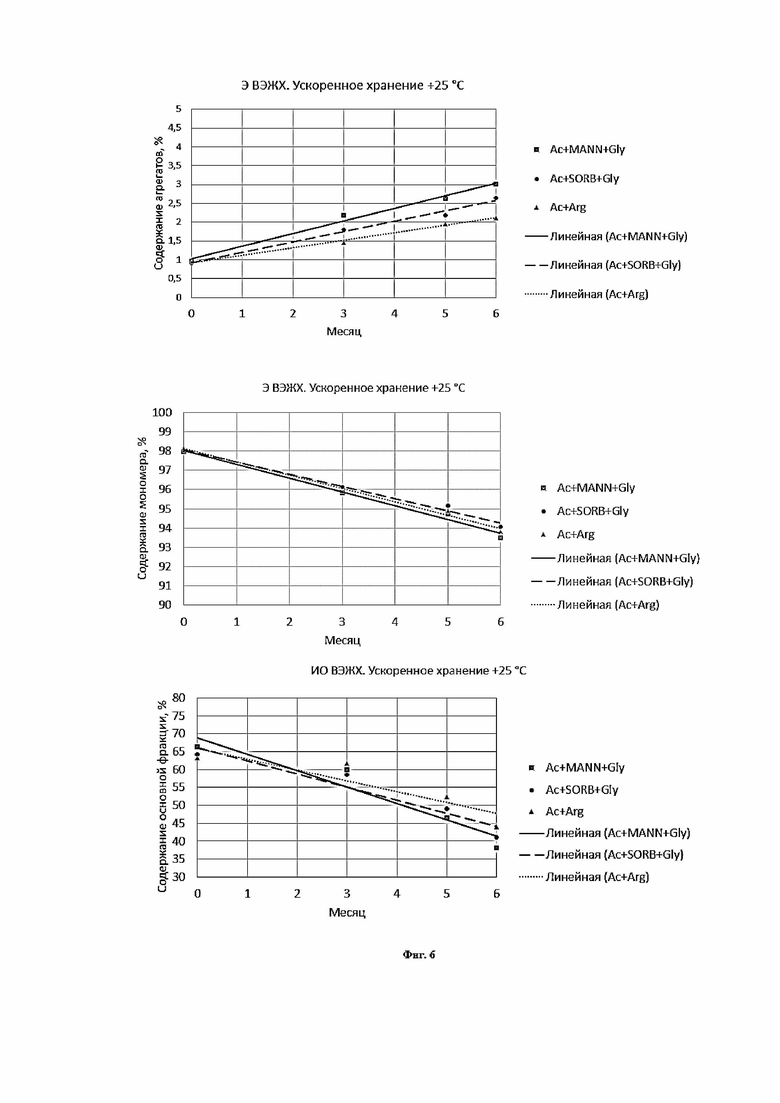
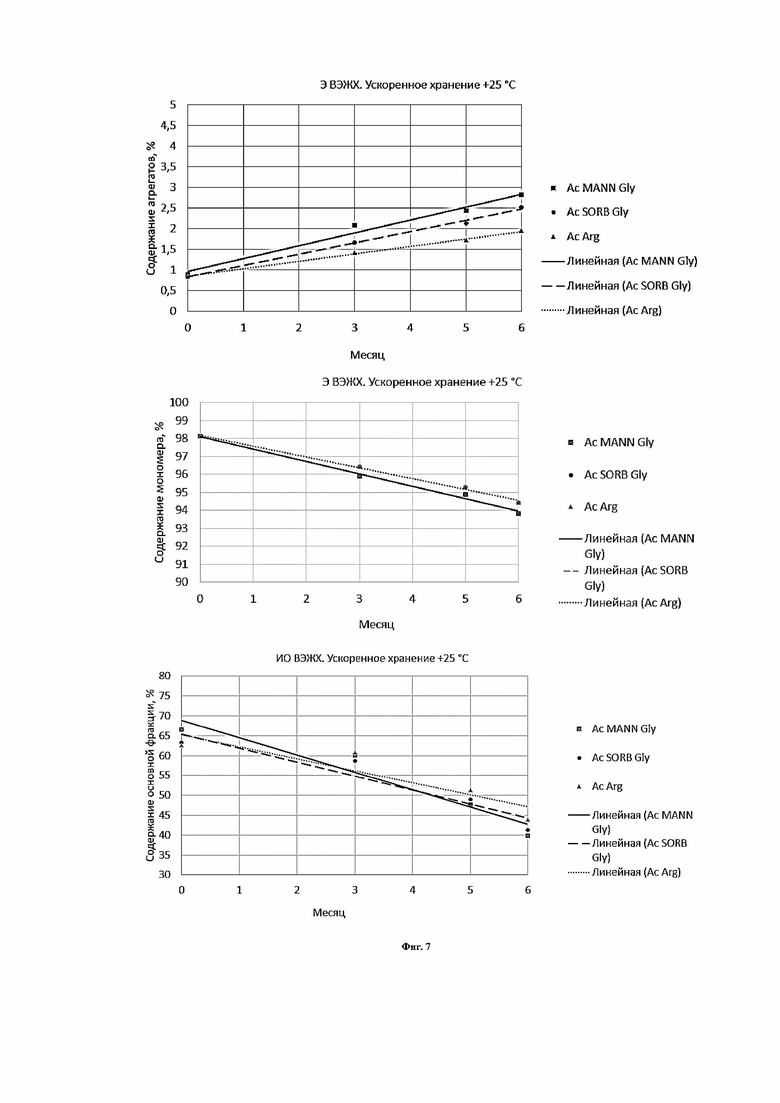
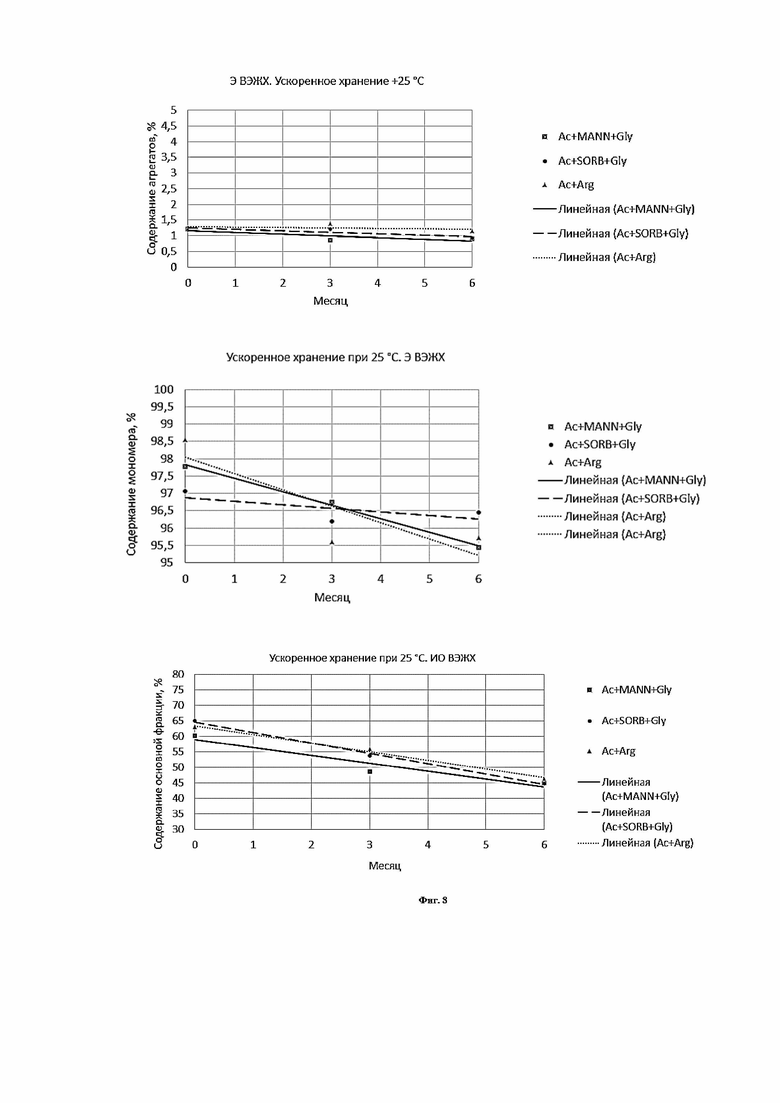
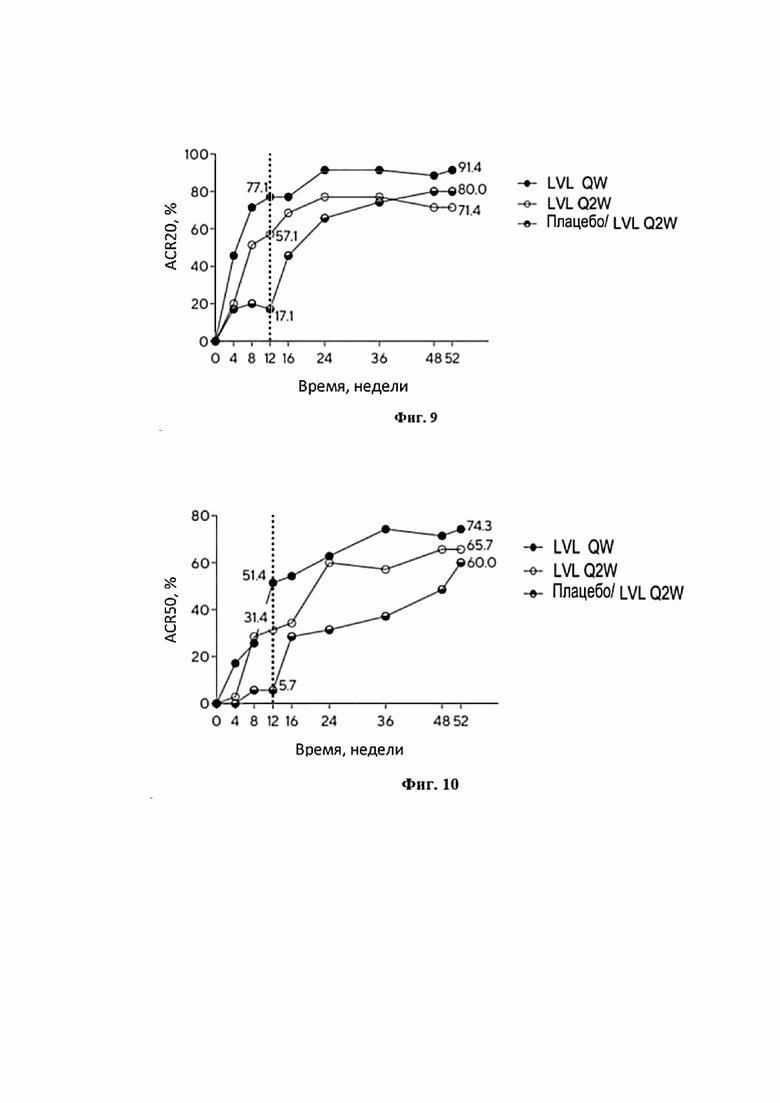
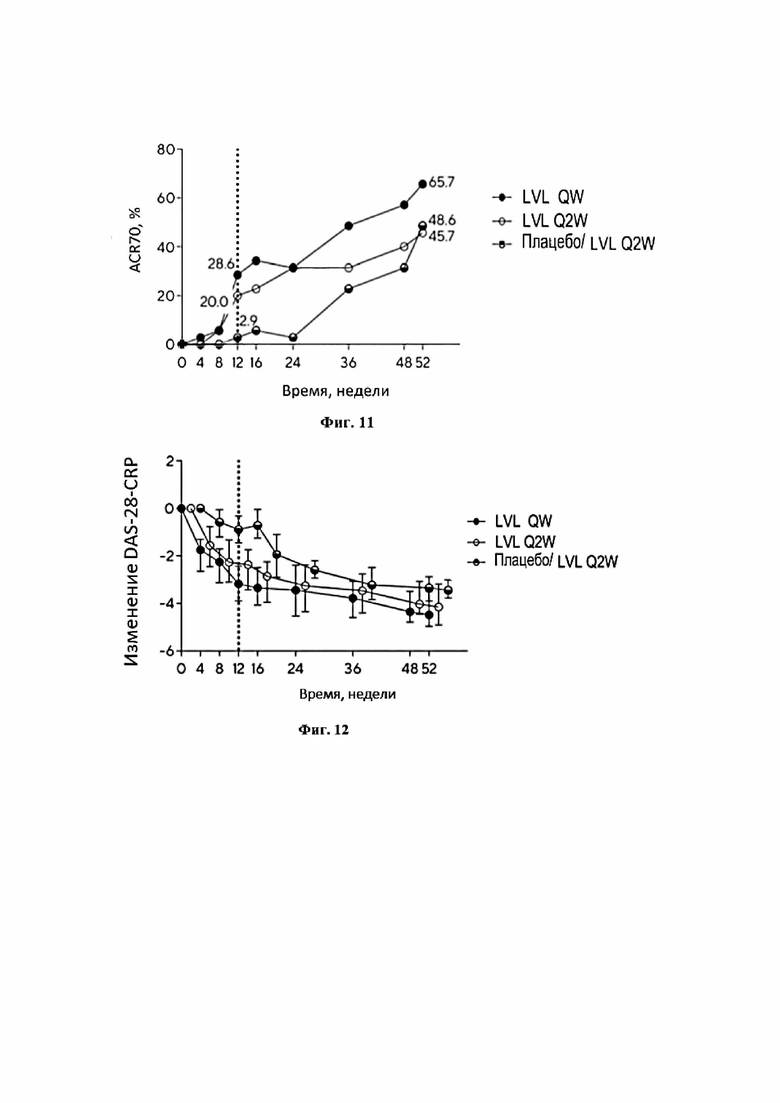
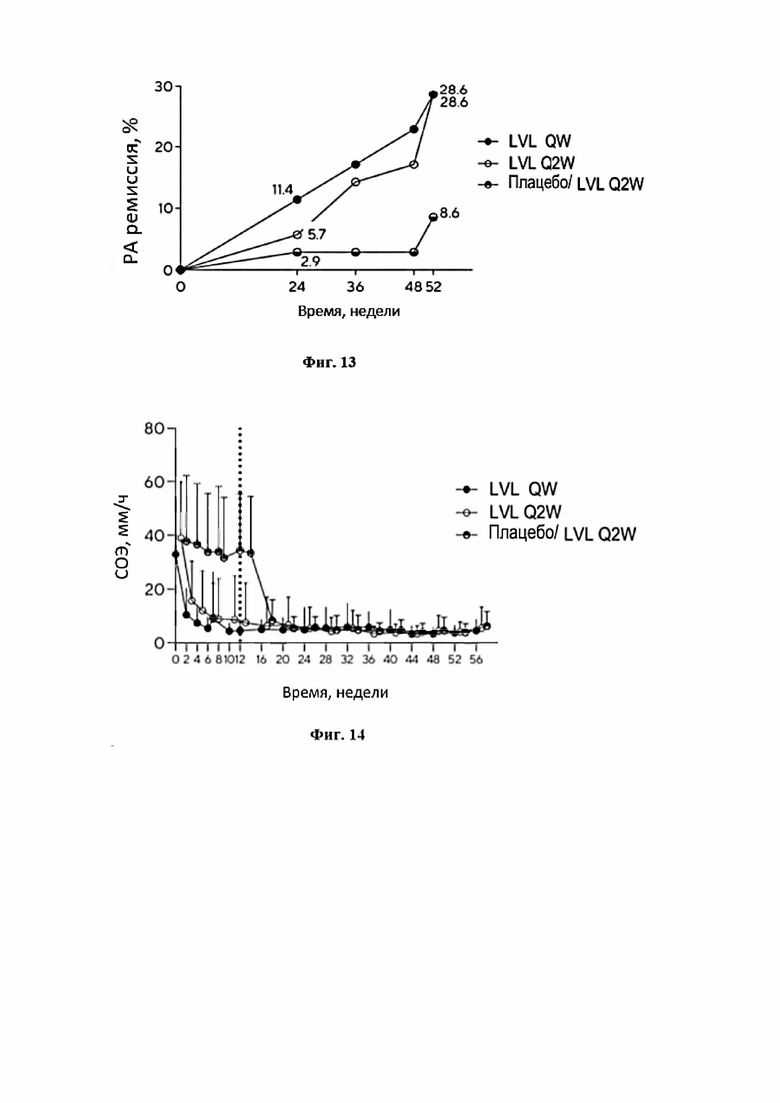
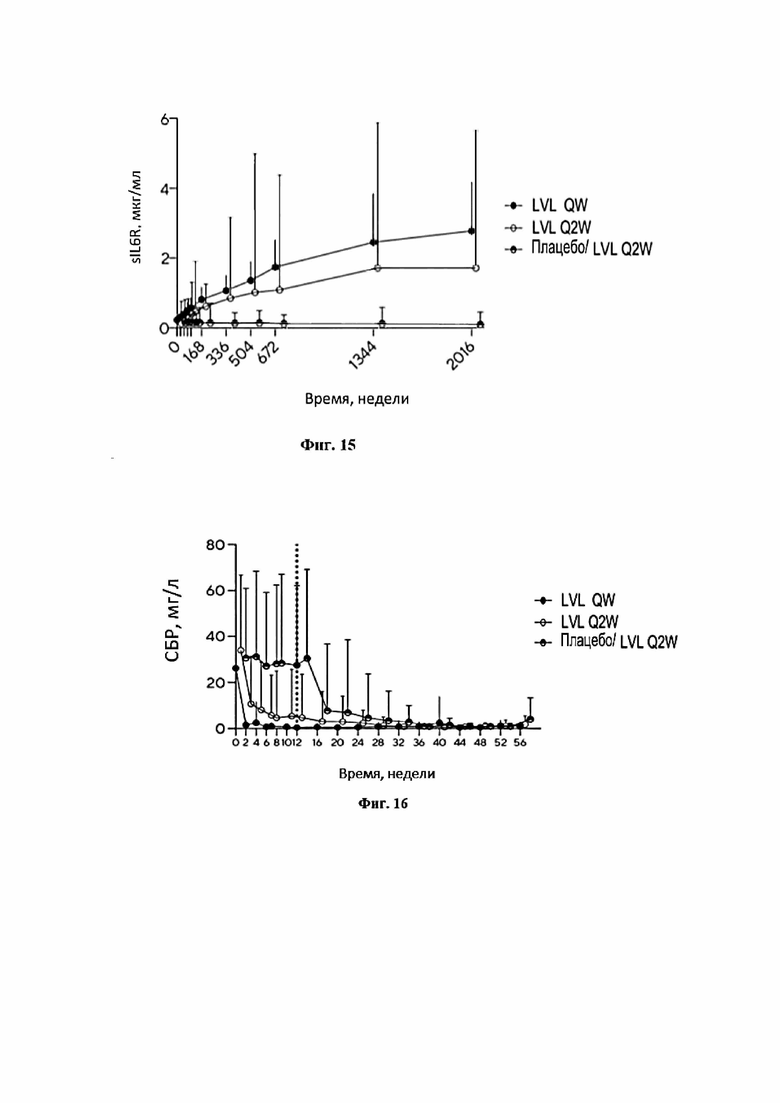
Группа изобретений относится к лечению IL-6R-ассоциированных заболеваний. Водная фармацевтическая композиция левилимаба содержит 5-220 мг/мл левилимаба; 0,4-1,8 мг/мл натрия ацетата тригидрата; 20-50 мг/мл полиола; 5-10 мг/мл глицина; и уксусную кислоту до pH 4,5-6,5. Также раскрыты другие варианты выполнения водной фармацевтической композиции левилимаба и её применение для лечения или профилактики IL6R-ассоциированного заболевания или нарушения, в частности для лечения ревматоидного артрита, активного ревматоидного артрита, острого респираторного дистресс-синдрома или синдрома выброса цитокинов. Группа изобретений обеспечивает стабильные водные композиции для антитела к IL-6R левилимаба. 8 н. и 56 з.п. ф-лы, 13 табл., 6 пр., 16 ил.
1. Водная фармацевтическая композиция левилимаба, содержащая:
(i) 5-220 мг/мл левилимаба;
(II) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 20-50 мг/мл полиола;
(iv) 5-10 мг/мл глицина; и
(v) уксусную кислоту до pH 4,5-6,5.
2. Водная фармацевтическая композиция по п. 1, где левилимаб находится в концентрации 5-40 мг/мл.
3. Водная фармацевтическая композиция по п. 1, где левилимаб находится в концентрации 20 мг/мл.
4. Водная фармацевтическая композиция по п. 1, где левилимаб находится в концентрации 180-220 мг/мл.
5. Водная фармацевтическая композиция по п. 1, где левилимаб находится в концентрации 180 мг/мл.
6. Водная фармацевтическая композиция по любому из пп. 1-5, где указанный натрия ацетата тригидрат находится в концентрации 0,4-1,0 мг/мл.
7. Водная фармацевтическая композиция по любому из пп. 1-5, где указанный натрия ацетата тригидрат находится в концентрации 0,4-0,5 мг/мл.
8. Водная фармацевтическая композиция по любому из пп. 1-5, где указанный натрия ацетата тригидрат находится в концентрации 0,436 мг/мл.
9. Водная фармацевтическая композиция по любому из пп. 1-8, где указанный полиол находится в концентрации 20-26 мг/мл.
10. Водная фармацевтическая композиция по любому из пп. 1-8, где указанный полиол находится в концентрации 23 мг/мл.
11. Водная фармацевтическая композиция по любому из пп. 1-8, где указанный полиол выбран из маннитола, или сорбитола, или их комбинации.
12. Водная фармацевтическая композиция по любому из пп. 1-11, где указанный глицин находится в концентрации 7-8 мг/мл.
13. Водная фармацевтическая композиция по любому из пп. 1-11, где указанный глицин находится в концентрации 7,5 мг/мл.
14. Водная фармацевтическая композиция по любому из пп. 1-13, где указанная уксусная кислота добавлена до pH 5,0.
15. Водная фармацевтическая композиция по п. 1, содержащая:
(i) 20 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
16. Водная фармацевтическая композиция по п. 1, содержащая:
(i) 180 мг/мл левилимаба;
(ii) 0,436 мг/мл натрия ацетата тригидрата;
(iii) 23 мг/мл полиола, выбранного из маннитола или сорбитола;
(iv) 7,5 мг/мл глицина; и
(v) уксусную кислоту до pH 5,0.
17 Водная фармацевтическая композиция левилимаба, содержащая:
(i) 5-220 мг/мл левилимаба;
(ii) 0,4-1,8 мг/мл натрия ацетата тригидрата;
(iii) 10-32 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 4,5-6,5.
18. Водная фармацевтическая композиция по п. 17, где левилимаб находится в концентрации 5-40 мг/мл.
19. Водная фармацевтическая композиция по п. 17, где левилимаб находится в концентрации 20 мг/мл.
20. Водная фармацевтическая композиция по п. 17, где левилимаб находится в концентрации 180-220 мг/мл.
21. Водная фармацевтическая композиция по п. 17, где указанное левилимаб находится в концентрации 180 мг/мл.
22. Водная фармацевтическая композиция по любому из пп. 17-21, где указанный натрия ацетата тригидрат находится в концентрации 1,7-1,8 мг/мл.
23. Водная фармацевтическая композиция по любому из пп. 17-21, где указанный натрия ацетата тригидрат находится в концентрации 1,744 мг/мл.
24. Водная фармацевтическая композиция по любому из пп. 17-23, где указанный аргинина гидрохлорид находится в концентрации 18-24 мг/мл.
25. Водная фармацевтическая композиция по любому из пп. 17-23, где указанный аргинина гидрохлорид находится в концентрации 21,1 мг/мл.
26. Водная фармацевтическая композиция по любому из пп. 17-25, где указанная уксусная кислота добавлена до pH 5,0.
27. Водная фармацевтическая композиция по п. 17, содержащая:
(i) 20 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
28 Водная фармацевтическая композиция по п. 17, содержащая:
(i) 180 мг/мл левилимаба;
(ii) 1,744 мг/мл натрия ацетата тригидрата;
(iii) 21,1 мг/мл аргинина гидрохлорида; и
(iv) уксусную кислоту до pH 5,0.
29. Водная фармацевтическая композиция левилимаба, содержащая:
(i) 162 мг левилимаба;
(ii) 0,392 мг натрия ацетата тригидрата;
(iii) 20,7 мг полиола, выбранного из маннитола или сорбитола;
(iv) 6,75 мг глицина;
(v) уксусную кислоту до pH 5,0; и
(vi) воду для инъекций до 0,9 мл.
30. Водная фармацевтическая композиция левилимаба, содержащая:
(i) 162 мг левилимаба;
(ii) 1,57 мг натрия ацетата тригидрата;
(iii) 18,99 мг аргинина гидрохлорида;
(iv) уксусную кислоту до pH 5,0; и
(v) воду для инъекций до 0,9 мл.
31. Водная фармацевтическая композиция по любому из пп. 1-30, где указанная уксусная кислота является ледяной уксусной кислотой.
32. Водная фармацевтическая композиция по любому из пп. 1-31, где указанная композиция предназначена для парентерального введения.
33. Водная фармацевтическая композиция по любому из пп. 1-31, где указанная композиция предназначена для внутримышечного, внутривенного или подкожного введения.
34. Водная фармацевтическая композиция по любому из пунктов пп. 1-31, где указанная композиция находится во флаконе.
35. Водная фармацевтическая композиция по п. 34, где указанный флакон представляет собой стеклянный или полимерный флакон.
36. Водная фармацевтическая композиция по любому из пп. 34, 35, где указанный флакон имеет объем 4-20 мл.
37. Водная фармацевтическая композиция по п. 34, где указанный флакон имеет объем 4 мл, 10 мл или 20 мл.
38. Водная фармацевтическая композиция по любому из пп. 1-31, где указанная композиция находится в шприце или автоинжекторе.
39. Водная фармацевтическая композиция по п. 38, где указанный шприц или автоинжектор является стеклянным или полимерным.
40. Водная фармацевтическая композиция по любому из пп. 38, 39, где указанный шприц или автоинжектор имеет вместимость 1 мл.
41. Водная фармацевтическая композиция по любому из пп. 1-31, где указанная композиция находится в преднаполненном шприце или в преднаполненном автоинжекторе.
42. Водная фармацевтическая композиция по п. 41, где указанный преднаполненный шприц или в преднаполненный автоинжектор является стеклянным или полимерным.
43. Водная фармацевтическая композиция по пп. 41, 42, где указанный предзаполненный шприц или преднаполненный автоинжектор имеет вместимость 1 мл.
44. Применение водной фармацевтической композиции левилимаба по любому из пп. 1, 17, 29, 30 для лечения или профилактики IL6R-ассоциированного заболевания или нарушения.
45. Применение по п. 44, где IL6R-ассоциированное заболевание или нарушение выбрано из:
ревматоидного артрита, ювенильного хронического артрита, склеродермии, реакции "трансплантат против хозяина", отторжения трансплантата органа, острого или хронического иммунного заболевания, связанного с трансплантацией органа, кахексии, взрослого (острого) респираторного дистресс-синдрома, болезни Стилла, системной склеродермии, синдрома Шегрена, болезни/артериита Такаясу, нарушений, связанных с цитокиновой терапией, синдром выброса цитокинов (синдром высвобождения цитокинов), иридоциклита, увеита, оптического неврита, оптического нейромиелита, ювенильного ревматоидного артрита, гигантоклеточного артериита, полиартикулярного ювенильного идиопатического артрита, системного ювенильного идиопатического артрита; рака, в частности, множественной миеломы и злокачественных солидных опухолей, колоректального рака, рака предстательной железы, рака яичника.
46. Применение по п. 44, где указанную водную фармацевтическую композицию вводят парентерально.
47. Применение по п. 46, где указанную водную фармацевтическую композицию вводят внутримышечно, внутривенно или подкожно.
48. Применение водной фармацевтической композиции левилимаба по любому из пп. 1, 17, 29, 30 для лечения ревматоидного артрита.
49. Применение по п. 48, где указанную водную фармацевтическую композицию вводят в дозе левилимаба 162 мг.
50. Применение по п. 48, где указанную водную фармацевтическую композицию вводят один раз в неделю или один раз каждые две недели.
51. Применение по п. 48, где указанную водную фармацевтическую композицию вводят парентерально.
52. Применение по п. 51, где указанную водную фармацевтическую композицию вводят внутримышечно, внутривенно или подкожно.
53. Применение по п. 48, дополнительно включающее применение метотрексата.
54. Применение водной фармацевтической композиции левилимаба по любому из пп. 1, 17, 29, 30 для лечения активного ревматоидного артрита.
55. Применение по п. 54, где указанную водную фармацевтическую композицию вводят в дозе левилимаба 324 мг или 648 мг.
56. Применение по п. 54, где указанную водную фармацевтическую композицию вводят 1 раз в 2 недели, или 1 раз в 4 недели, или 1 раз в 6 недель.
57. Применение по п. 54, где указанную водную фармацевтическую композицию вводят парентерально.
58. Применение по п. 57, где указанную водную фармацевтическую композицию вводят внутримышечно, внутривенно или подкожно.
59. Применение по п. 54, дополнительно включающее применение метотрексата.
60. Применение водной фармацевтической композиции левилимаба по любому из пп. 1, 17, 29, 30 для лечения или профилактики взрослого (острого) респираторного дистресс-синдрома или синдром выброса цитокинов (синдром высвобождения цитокинов).
61. Применение по п. 60, где указанную водную фармацевтическую композицию вводят в дозе левилимаба 324 мг или 648 мг.
62. Применение по п. 60, где указанную водную фармацевтическую композицию вводят однократно, или двукратно, или трехкратно, или четырехкратно с интервалом минимум 8 часов.
63. Применение по п. 60, где указанную водную фармацевтическую композицию вводят парентерально.
64. Применение по п. 63, где указанную водную фармацевтическую композицию вводят внутримышечно, внутривенно или подкожно.
| АНТИТЕЛО ИЛИ ЕГО АНТИГЕНСВЯЗЫВАЮЩИЙ ФРАГМЕНТ, СПОСОБНЫЙ СВЯЗЫВАТЬСЯ С РЕЦЕПТОРОМ ИНТЕРЛЕЙКИНА-6 ЧЕЛОВЕКА | 2016 |
|
RU2656160C2 |
| Водогрейный котел | 1926 |
|
SU15262A1 |
| EP 3563867 A1, 06.11.2019 | |||
| CN 111057152 A, 24.04.2020. | |||

