Область техники
[001] Настоящее изобретение относится к новым водным композициям для антител к PD-1, и, в частности, к новым водным композициям для антитела к PD-1 пролголимаба, которые могут быть использованы в качестве лекарственного средства для лечения злокачественных новобразований.
Уровень техники
[002] Белок программируемой смерти 1 (PD-1) является ингибиторным членом семейства рецепторов CD28, которое включает в себя также CD28, CTLA-4, ICOS и BTLA. PD-1 экспрессируется активированными В-клетками, Т-клетками и миелоидными клетками (Agata et al., supra; Okazaki et al. (2002) Curr. Opin. Immunol. 14: 391779-82; Bennet et al. (2003) J Immunol 170:711-8). Первоначальные члены данного семейства, CD28 и ICOS, были обнаружены по функциональным действиям на увеличение пролиферации Т-клеток после добавления моноклональных антител (Hutloff et al. (1999) Nature 397:263-266; Hansen et al. (1980) Immunogenics 10:247-260). PD-1 был обнаружен скринингом на дифференциальную экспрессию в апоптотических клетках (Ishida et al. (1992) EMBO J 11:3887-95). Другие члены данного семейства, CTLA-4 и BTLA, были обнаружены скринингом на дифференциальную экспрессию в цитотоксических Т-лимфоцитах и TH1-клетках, соответственно. CD28, ICOS и CTLA-4, все, имеют неспаренный остаток цистеина, дающий возможность гомодимеризации. В противоположность этому, предполагается, что PD-1 существует в виде мономера, не имея неспаренного остатка цистеина, характерного для других членов семейства CD28.
[003] PD-1 является трансмембранным белком типа I 55 кДа, который является частью суперсемейства генов Ig (Agata et al. (1996) Int Immunol 8:765-72). PD-1 содержит мембранопроксимальный иммунорецепторный ингибирующий мотив на основе тирозина (ITIM) и мембранодистальный мотив переключения на основе тирозина (ITSM) (Thomas, M.L. (1995) J Exp Med 181:1953-6; Vivier, E и Daeron, M (1997) Immunol Today 18:286-91). PD-1, хотя и является структурно сходным с CTLA-4, лишен мотива MYPPPY, который является критическим для связывания В7-1 и В7-2. Были идентифицированы два лиганда для PD-1, PD-L1 и PD-L2, которые, как было показано, отрицательно регулируют активацию Т-клеток после связывания с PD-1 (Freeman et al. (2000) J Exp Med 192:1027-34; Latchman et al. (2001) Nat Immunol 2:261-8; Carter et al. (2002) Eur J Immunol 32:634-43). Как PD-L1, так и PD-L2 являются гомологами В7, которые связываются с PD-1, но не связываются с другими членами семейства CD28.
[004] Один лиганд для PD-1, PD-L1, является изобилующим в различных типах рака человека (Dong et al. (2002) Nat. Med. 8:787-9). Взаимодействие между PD-1 и PD-L1 приводит к снижению количества инфильтрирующих опухоль лимфоцитов, уменьшению опосредованной рецептором Т-клеток пролиферации и ускользанию от иммунологического надзора раковых клеток (Dong et al. (2003) J. Mol. Med. 81:281-7; Blank et al. (2005) Cancer Immunol. Immunother. 54:307-314; Konishi et al. (2004) Clin. Cancer Res. 10:5094-100). Иммуносупрессия может быть обращена ингибированием локального взаимодействия PD-1 с PD-L1, и это действие является аддитивным при блокировании взаимодействия PD-1 с PD-L2 (Iwai et al. (2002) Proc. Nat'l. Acad. Sci. USA 99:12293-7; Brown et al. (2003) J. Immunol. 170:1257-66).
[005] PD-1 является ингибирующим членом семейства CD28, экспрессируемым на активированных В-клетках, Т-клетках и миелоидных клетках (Agata et al., supra; Okazaki et al. (2002) Curr Opin Immunol 14: 391779-82; Bennett et al. (2003) J Immunol 170:711-8). PD-1-недостаточные животные развивают различные аутоиммунные фенотипы, включая аутоиммунную кардиопатию и подобный волчанке синдром с артритом и нефритом (Nishimura et al. (1999) Immunity 11:141-51; Nishimura et al. (2001) Science 291:319-22). Кроме того, было обнаружено, что PD-1 играет роль в аутоиммунном энцефаломиелите, системной красной волчанке, болезни трансплантат против хозяина (GVHD), диабете типа I и ревматоидном артрите (Salama et al. (2003) J Exp Med 198:71-78; Prokunina and Alarcon-Riquelme (2004) Hum Mol. Genet 13:R143; Nielsen et al. (2004) Lupus 13:510). Было показано, что в линии мышиных опухолевых В-клеток ITSM PD-1 является необходимым для блокирования BCR-опосредованного вхождения Са2+ и фосфорилирования тирозина, находящихся ниже по ходу процесса эффекторных молекул (Okazaki et al. (2001) PNAS 98:13866-71).
[006] В настоящее время известен ряд антител против PD-1, например, nivolumab (BMS), pembrolizumab (Merck), которые представляют собой монокональное антитело человека изотипа IgG4.
[007] Также известно новое антитело к PD-1 пролголимаб (также известное как BCD-100) представляет собой моноклональное антитело человека изотипа IgG1 с безэффекторными мутациями L234A, L235A. Пролголимаб показал повышенную аффинность к PD-1, повышенную агрегационную стабильность в сравнении с антителами изотипа IgG4. Кроме того, пролголимаб в настоящее время проходит "клинические исследования для различных типов злокачественных новообразований, включая меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого.
[008] Таким образом, в настоящее время является актуальной разработка новых улучшенных стабильных водных фармацевтических композиций для антитела к PD-1 пролголимаба.
Краткое описание чертежей
[009] Изобретение станет более понятным из последующего подробного описания вариантов осуществления настоящего изобретения со ссылкой на прилагаемые чертежи, где:
[0010] Фигура 1, Фигура 2 представляют собой графики, иллюстрирующие динамику концентраций BCD-100 в сыворотке крови пациентов на протяжении 6 введений препарата (с учетом коэффициента) в мкг/мл. (Исследование BCD-100-1).
[0011] Фигура 3 представляет собой график, иллюстрирующий дизайн исследования BCD-100-2/MIRACULUM.
[0012] Фигура 4 представляет собой график, иллюстрирующий блок-схему исследования.
[0013] Фигура 5 представляет собой график, иллюстрирующий общую выживаемость пациентов 1-ой группы (BCD-100, 1 мг/кг раз в 2 недели) по результатам исследования BCD-100-2/MIRACULUM.
[0014] Фигура 6 представляет собой график, иллюстрирующий общую выживаемость пациентов 2-ой группы (BCD-100, 3 мг/кг раз в 3 недели) по результатам исследования BCD-100-2/MIRACULUM.
[0015] Фигура 7 представляет собой график, иллюстрирующий беспрогрессивную выживаемость пациентов группы BCD-100, 1 мг/кг (в соответствии с критериями irRECIST) по результатам исследования BCD-100-2 /MIRACULUM.
[0016] Фигура 8 представляет собой график, иллюстрирующий беспрогрессивную выживаемость пациентов группы BCD-100, 3 мг/кг (в соответствии с критериями irRECIST) по результатам исследования BCD-100-2 /MIRACULUM.
[0017] Фигура 9 представляет собой график, иллюстрирующий концентрацию BCD-100 у пациентов, получавших 1 мг/кг каждые 2 недели, после введения одной дозы препарата (результаты исследования BCD-100-2 /MIRACULUM).
[0018] Фигура 10 представляет собой график, иллюстрирующий концентрацию BCD-100 у пациентов, получавших 3 мг/кг каждые 3 недели (результаты исследования BCD-100-2 /MIRACULUM).
[0019] Фигура 11 представляет собой график, иллюстрирующий долю Th9 в общей популяции Т-хелперов в подгруппах пациентов, получавших BCD-100 в дозе 1 мг/кг Q2W, с различными типами ответа на терапию (результаты исследования BCD-100-2 /MIRACULUM).
[0020] Фигура 12 представляет собой график, иллюстрирующий долю Th9 в общей популяции Т-хелперов в подгруппах пациентов, получавших BCD-100 в дозе 3 мг/кг Q3W, с различными типами ответа на терапию (результаты исследования BCD-100-2 /MIRACULUM).
Описание изобретения Определения:
[0021] Термины, используемые в настоящем описании, как правило, имеют их обычные для данной области значения в пределах контекста изобретения и в конкретном контексте, где используют каждый термин. Ниже, или в другом месте описания, определены некоторые термины, которые используют для описания изобретения, для предоставления практику дополнительного руководства в отношении описания изобретения. Приведены синонимы некоторых терминов. Изложение одного или нескольких синонимов не исключает использование других синонимов. Использование примеров в любом месте настоящего описания, включая примеры любых терминов, описываемых в настоящем документе, является исключительно иллюстративным и никоим образом не ограничивает объем и значение изобретения или любого иллюстрируемого объекта. Изобретение не ограничено различными вариантами осуществления, приведенными в настоящем описании.
[0022] "Моноклональное антитело", как используется в данной заявке, относится к гуманизированному антителу или полностью человеческому антителу, если в данной заявке не указано иное. Моноклональные антитела по изобретению могут быть получены с использованием, например, рекомбинантных технологий, технологий фагового дисплея, синтетических технологий или комбинаций таких технологий или других технологий, хорошо известных из уровня техники.
[0023] Популяция "моноклональных антител", как используется в настоящем документе, относится к гомогенной или по существу гомогенной популяции антител (т.е. по крайней мере приблизительно 96%, но более предпочтительно по крайней мере приблизительно 97 или 98% или еще более предпочтительно по крайней мере 99% антител в популяции будут конкурировать в иммунно-ферментном анализе ELISA за тот же антиген или эпитоп, или более предпочтительно антитела являются идентичными в аминокислотной последовательности).
[0024] Полноразмерное антитело, существующее в природе, представляет собой молекулу иммуноглобулина, которая состоит из четырех полипептидных цепей (две тяжелые (Н) цепи (приблизительно 50-70 кДа при полной длине) и две легкие (L) цепи (приблизительно 25 кДа при полной длине)), связанных дисульфидными мостиками. Аминоконцевая часть каждой цепи включает вариабельный домен из приблизительно 100-110 или более аминокислот, которые отвечают за связывание антигена. Карбоксиконцевая часть каждой цепи определяет константный участок, главным образом, отвечающий за функцию эффектора. Легкие, цепи классифицируют как каппа или лямбда, и они характеризуются специфичным константным участком. Каждая легкая цепь состоит из вариабельного участка N-концевой легкой цепи (в данной заявке "VL" или "VK") и константного участка легкой цепи, состоящего из одного домена (CL или СК). Тяжелые цепи классифицируют как γ (гамма), δ (дельта), альфа (α), мю (μ) или эпсилон (ε), и они определяют изотип антитела, такой как IgG, IgM, IgA, IgD и IgE, соответственно, и несколько из них могут быть дополнительно разделены на подклассы (изотипы), такие как, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2. Каждый тип тяжелой цепи характеризуется конкретным константным участком Fc. Каждая тяжелая цепь состоит из вариабельного участка N-концевой тяжелой цепи (в данной заявке "VH") и константного участка СН (тяжелой цепи). Константный участок тяжелой цепи состоит из трех доменов (CH1, СН2 и СН3) для IgG, IgD и IgA и 4 доменов (CH1, СН2, СН3 и СН4) для IgM и IgE. Вариабельные домены VH и VL могут быть дополнительно разделены на участки гипервариабельности (гипервариабельные участки, CDR), чередующиеся с более консервативными каркасными участками (FR). Каждый вариабельный домен состоит из трех CDR и четырех FR, расположенных в следующем порядке от N-конца к С-концу: FR1, CDR1, FR2, CDR2, FR3, CDR3 и FR4.
[0025] Вариабельные участки каждой из пар легкая/тяжелая цепь образуют антиген-связывающие сайты антитела. Таким образом, интактное IgG антитело имеет два сайта связывания. За исключением бифункциональных или биспецифических антител два сайта связывания являются одинаковыми. Как используется в данной заявке, "антигенсвязывающая часть", или "антигенсвязывающий участок", или "антигенсвязывающий домен" относятся, взаимозаменяемо, к такой части молекулы антитела, которая содержит аминокислотные остатки, взаимодействующие с антигеном и обуславливающие специфичность и аффинность антитела по отношению к антигену. Такая часть антитела включает "каркасные" аминокислотные остатки, необходимые для поддержания надлежащей конформации антиген-связывающих остатков.
[0026] «Фрагмент антитела» может представлять собой фрагмент антитела или фрагмент антитела, имеющий активность полноразмерного антитела. Указанный фрагмент антитела может представлять собой F(ab')2, F(ab)2, Fab', Fab Fv и scFv.
[0027] Термин "ингибировать" или "нейтрализовать", как используется в данной заявке, по отношению к функциональной активности антитела по изобретению, означает способность в значительной степени препятствовать, предотвращать, ограничивать, замедлять, прекращать, уменьшать или обращать, например, развитие или тяжесть того, что ингибируют, включая, но не ограничиваясь вышеприведенными, биологическую активность (например, активность PD-1) или свойство, заболевание или состояние. Ингибирование или нейтрализация активности PD-1 в результате связывания антитела по изобретению с PD-1 составляет предпочтительно по крайней мере приблизительно 20, 30, 40, 50, 60, 70, 80, 90, 95% или выше.
[0028] Термин "выделенный" или "изолированный" при использовании по отношению к нуклеиновой кислоте или белковому препарату (например, антителу) относится к молекуле нуклеиновой кислоты или белковой молекуле, которые идентифицируют и отделяют по крайней мере от одного контаминантного вещества, с которым она обычно связана в природном источнике. Предпочтительно "выделенное антитело" является антителом, которое по существу не содержит другие антитела, обладающие отличительной антигенной специфичностью (например, фармацевтические композиции согласно настоящему изобретению содержат выделенное антитело, которое специфически связывает PD-1 и по существу не содержит антитела, которые специфически связывают антигены, отличные от PD-1).
[0029] Полинуклеотид является "функционально связанным", если он имеет функциональные связи с другим полинуклеотидом. Например, промотор или энхансер функционально связаны с кодирующей последовательностью, если они влияют на транскрипцию последовательности. Полипептид "функционально связан" с другим полипептидом, если полинуклеотиды, кодирующие их, связаны функционально, предпочтительно, если они находятся в той же открытой рамке считывания.
[0030] Термин «Специфическое связывание» между антителом и мишенью антигена (антигеном) относится к иммунологической специфичности. Антитело может специфически связываться с мишенью антигена, если оно связывает эпитоп на антигене в большей степени, чем другие эпитопы на антигене. Специфическое связывание не исключает кросс-реактивность с другими антигенами, несущими сходные эпитопы антигенов.
[0031] VL домены в антителах, согласно изобретению, могут быть типа VL лямбда, или типа VL каппа. Термин «VL домен» относится к обоим изотипам VK лямбда и VL каппа, которые содержат одну или более аминокислотных замен, инсерций или делеций.
[0032] Термин «фармацевтическая композиция» относится к композиции и/или составу, содержащему антитело согласно изобретению в терапевтически эффективном количестве и эксцепиенты или вспомогательные вещества (носители, разбавители, наполнители, растворители и другие эксцепиенты).
[0033] В настоящей заявке, термин "буфер" или "буферный раствор" относится к водному раствору, содержащему смесь кислоты (обычно слабой кислоты, такой как, например, уксусная кислота, лимонная кислота) и ее конъюгированного основания (такой как, например, ацетатной или цитратной соли, например, ацетат натрия, цитрат натрия, а также гидраты указанных солей, например, натрия ацетат тригидрат) или альтернативно смесь основания (обычно слабого основания, например, гистидина) и его конъюгированной кислоты (например, гистидина гидрохлорида). Значение рН "буферного раствора" мало изменяется при добавлении к нему небольшого количества сильного основания или сильной кислоты, а также при разбавлении и концентрировании, благодаря "буферному эффекту", обеспечиваемому "буферным агентом".
[0034] В настоящей заявке, "буферная система" содержит один или несколько буферных агентов и/или их конъюгата (ов) с кислотой или основанием, и более подходяще содержит один или несколько буферных агентов и их конъюгата(ов) с кислотой или основанием, и наиболее подходяще содержит только один буферный агент и его кислотный/щелочной конъюгат. Если не указано иное, любые концентрации, указанные в настоящем изобретении по отношению к "буферной системе" (концентрация буфера) могут относится к объединенной концентрации буферного(ых) агента(ов) и/или его конъюгата(ов) с кислотой или основанием. Другими словами, концентрации, указанные в настоящей заявке по отношению к "буферной системе", могут относиться к объединенной концентрации релевантных буферных видов (то есть, видов в динамическом равновесии друг с другом, например, цитрат/лимонная кислота). Суммарное значение рН композиции, содержащей релевантную буферную систему, является отображением равновесной концентрация каждого из релевантных буферных видов (то есть, баланса буферного (ых) агента (ов) с его конъюгатом (ами) с кислотой или основанием).
[0035] В настоящей заявке, термин "буферный агент" относится к кислотному или щелочному компоненту (обычно слабой кислоте или слабому основанию) буфера или буферного раствора. Буферный агент помогает поддерживать значение рН данного раствора при или около заранее определенного значения, и буферные агенты обычно выбирают для дополнения заранее определенного значения. Буферный агент может представляет собой единственное соединение, которое приводит к желательному буферному эффекту, в особенности, если указанный буферный агент смешанный с (и подходяще способен к протонному обмену с) подходящим количеством (в зависимости от заранее определенного желательного значения) его соответствующего "кислотного/щелочного конъюгата".
[0036] Термин "солюбилизатор" при использовании в данном тексте означает фармацевтически приемлемое неионногенное поверхностно-активное вещество. Можно использовать один солюбилизатор, а также комбинации солюбилизаторов. Примерами солюбилизаторов являются, но не ограничиваются ими, полисорбат 20 или полисорбат 80, полоксамер 184 или полоксамер 188, или PLURONIC®.
[0037] Термины "осмотический агент" или «агент, регулирующий тоничность», а также осмолитик в том виде, как они здесь использованы, относятся к эксципиенту, который может обеспечивать требуемое осмотическое давление жидкого раствора антитела. В некоторых воплощениях агент, регулирующий тоничность, может подводить осмотическое давление жидкого препарата антитела до изотоничного так, что данный препарат антитела является физиологически совместимым с клетками ткани организма субъекта. В еще одном воплощении «агент, регулирующий тоничность», может способствовать увеличению стабильности антител. «Изотоничный» препарат представляет собой препарат, который имеет осмотическое давление, эквивалентное человеческой крови. Изотоничные препараты обычно имеют осмотическое давление от примерно 250 до 350 мОсм/кг. Термин «гипотонический» описывает препарат с осмотическим давлением, меньшим, чем осмотическое давление человеческой крови. Соответственно, термин «гипертонический» используется для описания препарата с осмотическим давлением, превышающим осмотическое давление человеческой крови. Изотоничность можно измерять с использованием, например, парового или криоскопического осмометра. Агент, регулирующий тоничность, может находиться в энантиомерной (например, L- или D-энантиомер) или рацемической форме; в форме изомеров, таких как альфа или бета, включая альфа, альфа; или бета, бета; или альфа, бета; или бета, альфа; в форме свободной кислоты или свободного основания; в форме соли; в гидратированной форме (например, моногидрат) или в безводной форме. Примерами осмотических агентов являются, но не ограничиваются ими, сахара (трегалозы дигидрат, сахароза, глюкоза), полиолы (маннитол, сорбитол), аминокислоты (пролин, аргинин, глицин), или соли (натрия хлорид, калия хлорид, магния хлорид).
[0038] Термин «длительное хранение» или «долговременная стабильность» следует понимать, как обозначение того, что фармацевтическая композиция может храниться в течение трех месяцев или более, в течение шести месяцев или более и предпочтительно в течение одного года или более, наиболее предпочтительно, с минимальным сроком хранения в стабильном состоянии по меньшей мере два года. В общем, термины «длительное хранение» и «долговременная стабильность» дополнительно включают продолжительности хранения в стабильном состоянии, которые по меньшей мере сравнимы или лучше, чем срок хранения в стабильном состоянии, как правило, необходимый для доступных в настоящее время коммерческих составов антитела к PD-1 пролголимаба без потерь в стабильности, которые могут сделать состав непригодным для определенного для него фармацевтического применения.
[0039] Термин "парентеральное введение" означает режимы введения, обычно с помощью инъекции, и включает, в частности, внутривенную, внутримышечную, внутриартериальную, внутритрахеальную, внутрикапсулярную, внутриорбитальную, внутрикардиальную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, внутрисуставную, субкапсулярную, субарахноидальную, внутриспинальную, эпидуральную и надчревную инъекцию или инфузию.
[0040] Термин «применение» относится к возможности применения антитела согласно изобретению или фармацевтической композиции, его содержащей, для лечения, облегчения течения заболеваний, для ускорения ремиссии, снижения частоты рецидивов вследствие заболеваний или нарушений, опосредуемых рецепторами, с которыми может связываться антитело согласно изобретению. Примерами заболеваний являются, но не ограничиваются ими, злокачественные новообразования, включая меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого; неплоскоклеточный немелкоклеточный рак легкого, плоскоклеточный рак легкого; мелкоклеточный рак легкого, в том числе неоперабельный или метастатический мелкоклеточный рак легкого; рак легкого ранних стадий до и после радикального лечения; рак шейки матки, в том числе метастатический рак шейки матки, ранние стадии рака шейки матки до и после радикального лечения; опухоли головы и шеи, в том числе плоскоклеточный рак головы и шеи; лимфома Ходжкина; опухоли желудка и кишечника, метастатический плоскоклеточный рак пищевода; рак мочевого пузыря, в том числе метастатическая уротелиальная карцинома, рак почки; рак эндометрия, в том числе метастатический рак эндометрия, ранние стадии рака эндометрия до и после радикального лечения; рак молочной железы, в том числе метастатический рак молочной железы, ранние стадии рака эндометрия до и после радикального лечения; рак печени, в том числе метастатический или неоперабельный рак печени, ранние стадии рака печени до и после радикального лечения; неоперабельная или метастатическая солидная опухоль, в том числе неоперабельная или метастатическая солидная опухоль с признаками микросателлитной нестабильности.
[0041] Термин «способ лечения» относится к возможности применения антитела согласно изобретению или фармацевтической композиции, его содержащей, для лечения, облегчения течения заболеваний, для ускорения ремиссии, снижения частоты рецидивов вследствие заболеваний или нарушений, связанных с активностью PD1.. "Лечить" или "лечение" заболевания, нарушения или состояния может включать предотвращение или замедление появления клинических симптомов заболевания, нарушения или состояния, развивающегося у человека, ингибирования заболевания, нарушения или состояния, то есть остановки, уменьшения или замедления развития заболевания или его рецидива (в случае поддерживающей терапии) или по меньшей мере его одного клинического или субклинического симптома, или облегчение или ослабление заболевания, то есть вызывание регресса заболевания, нарушения или состояния. Примерами заболеваний являются, но не ограничиваются ими, злокачественные новообразования, включая меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального оперативного лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого.
[0042] Термин "водная композиция" при использовании в данном документе относится к композиции на основе воды, в качестве воды могут быть использованы: вода, вода для инъекций, физиологический раствор (0,9-1,0%-ный водный раствор хлористого натрия).
[0043] В одном варианте осуществления изобретения субъект лечения или пациент является млекопитающим, предпочтительно человеческим субъектом. Вышеупомянутый субъект может быть мужского или женского пола любого возраста.
[0044] В настоящем описании и в последующей формуле изобретения, если контекстом не предусмотрено иное, слова «иметь», «включать» и «содержать» или их вариации, такие как «имеет», «имеющий», «включает», «включающий», «содержит» или «содержащий», следует понимать, как включение указанного целого или группы целых, но не исключение любого другого целого или группы целых.
Сущность изобретения
[0045] В настоящем изобретении раскрыты водные фармацевтические композиции антитела к PD-1 пролголимаба, которое имеет улучшенную агрегационную стабильность, повышенную аффинность по сравнению с известными антителами к PD-1, которые базируются на человеческом антителе изотипа IgG4.
[0046] Как описано в публикации международной заявки на изобретение WO/2018/013017, включенной в настоящий документ посредством ссылки, было показано, что антитело к PD-1 пролголимаб, которое представляет собой моноклональное антитело человека изотипа IgG1 с безэффекторными мутациями L234A, L235A, (именуемое в настоящем документе «антитело по изобретению»), обладает улучшенной агрегационной стабильностью, повышенной аффинностью и улучшенными фармакинетическими параметрами, такими как t1/2β (час) или Cmax (мкг/мл), по сравнению с известными антителами к PD-1, которые базируются на человеческом антителе изотипа IgG4, такими как ниволумаб. Пролголимаб имеет средневесовую молекулярную массу около 14 6 кДа и является специфическим по отношению к PD-1 человека. Пролголимаб имеет тяжелую цепь, которая насчитывает 459 аминокислот (SEQ ID NO: 1), и имеет легкую человека, насчитывающую 214 аминокислот (SEQ ID NO: 2), константная часть (Fc) пролголимаба содержит мутации L234A, L235A.
[0047] В этой связи было бы полезно вводить водную композицию с антителом по изобретению пациенту с злокачественным новобразованием.
[0048] Широким аспектом настоящего изобретения является водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1. Водная фармацевтическая композиция содержит фармацевтически эффективное количество антитела к PD-1 пролголимаба, эффективное количество трегалозы дигидрата, буферный агент на основе ацетата или гистидина.
[0049] В соответствии с одним широким аспектом настоящего изобретения предлагается водная фармацевтическая композиция анти-PD-1 антитела, содержащая:
(a) пролголимаб в концентрации от 15 мг/мл до 40 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 8 0 мг/мл до 110 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 мг/мл до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5.
[0050] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 15 мг/мл до 25 мг/мл.
[0051] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 20 мг/мл.
[0052] В некоторых вариантах осуществления изобретения указанная трегалозы дигидрат может находиться в концентрации от 95 мг/мл до 105 мг/мл.
[0053] В некоторых вариантах осуществления изобретения указанная трегалозы дигидрат может находиться в концентрации 100 мг/мл.
[0054] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1. 6 мг/мл до 1.9 мг/мл.
[0055] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1.7 мг/мл до 1.8 мг/мл.
[0056] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации 1.742 мг/мл.
[0057] В некоторых вариантах осуществления изобретения указанная уксусная кислота может быть добавлена до рН 5.0.
[0058] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.0 4 до 0.7 7 мг/мл.
[0059] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.40 мг/мл до 0.50 мг/мл.
[0060] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации 0.43 мг/мл.
[0061] В соответствии с одним широким аспектом настоящего изобретения предлагается водная фармацевтическая композиция анти-PD-1 антитела, содержащая:
(a) пролголимаб в концентрации от 90 мг/мл до 150 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 50 мг/мл до 110 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 мг/мл до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5.
[0062] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 90 мг/мл до 110 мг/мл.
[0063] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 100 мг/мл.
[0064] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации от 75 мг/мл до 85 мг/мл.
[0065] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации 80 мг/мл.
[0066] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1.6 мг/мл 1.9 до мг/мл.
[0067] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1.7 мг/мл до 1.8 мг/мл.
[0068] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации 1.742 мг/мл.
[0069] В некоторых вариантах осуществления изобретения указанная уксусная кислота может быть добавлена до рН от 5.0 до 5.5.
[0070] В некоторых вариантах осуществления изобретения указанная уксусная кислота может быть добавлена до рН 5.0.
[0071] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации 0.045 мг/мл до 0.77 мг/мл.
[0072] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.40 мг/мл до 0.50 мг/мл.
[0073] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.4 3 мг/мл.
[0074] В соответствии с одним широким аспектом настоящего изобретения предлагается водная фармацевтическая композиция анти-PD-1 антитела, содержащая:
(a) пролголимаб в концентрации от 5 мг/мл до 150 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 70 мг/мл до 110 мг/мл;
(c) L-гистидин в концентрации от 0,2 до 2,5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0,2 до 3,5 мг/мл.
[0075] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 15 мг/мл до 40 мг/мл.
[0076] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 15 мг/мл до 2 5 мг/мл.
[0077] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 20 мг/мл.
[0078]В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации от 95 мг/мл до 105 мг/мл.
[0079] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации 100 мг/мл.
[0080] В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации от 0,7 до 1,0 мг/мл.
[0081] В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации 0,92 мг/мл.
[0082] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации от 2,8 до 3,3 мг/мл.
[0083] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации 2,96 мг/мл.
[0084] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН от 5.5 до 6.5.
[0085] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН 5.5.
[0086] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 90 мг/мл до 110 мг/мл.
[0087] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 100 мг/мл.
[0088] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации от 75 мг/мл до 85 мг/мл.
[0089] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации 80 мг/мл.
[0090] В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации от 0,7 до 1,0 мг/мл.
[0091]В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации 0,92 мг/мл.
[0092] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации от 2,8 до 3,3 мг/мл.
[0093] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации 2,96 мг/мл.
[0094] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН от 5.5 до 6.5.
[0095] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН от 5.5 до 6.0.
[0096] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН 5.5.
[0097] Водная фармацевтическая композиция анти-PD-1 антитела пролголимаба по настоящему изобретению дополнительно может содержать подходящий солюбилизатор.
[0098] В некоторых вариантах осуществления изобретения указанный солюбилизатор может представлять собой полоксамер 188.
[0099] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве, которое больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00100] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве 0 мг/мл, 0.1 мг/мл, 0.2 мг/мл, 0.3 мг/мл, 04 мг/мл, 0.5 мг/мл, 0.6 мг/мл, 0.7 мг/мл, 0.8 мг/мл, 0.9 мг/мл, 1.0 мг/мл.
[00101] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 2 0 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5.
[00102] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.7 до 1.8 мг/мл; и
(d) уксусную кислоту до рН 5.0.
[00103] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации 1.742 мг/мл; и
(d) уксусную кислоту до рН 5.0.
[00104] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5.
[00105] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.7 до 1.8 мг/мл; и
(d) уксусную кислоту до рН от 5.0 до 5.5.
[00106] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.742 мг/мл; и
(d) уксусную кислоту до рН от 5.0 до 5.5.
[00107] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.742 мг/мл; и
(d) уксусную кислоту до рН 5.0.
[00108] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации от 0.2 до 2.5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0.2 до 3.5 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.5.
[00109] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН 5.5.
[00110] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) L-гистидин в концентрации от 0.2 до 2.5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0.2 до 3.5 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.5.
[00111] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.0.
[00112] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН 5.5.
[00113] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
I. Состав на 1 мл:
[00114] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
II. Состав на 1 мл:
[00115] Водная фармацевтическая композиция анти-PD-1 антитела пролголимаба по настоящему изобретению дополнительно может содержать подходящий солюбилизатор.
[00116] В некоторых вариантах осуществления изобретения указанный солюбилизатор может представлять собой полоксамер 188.
[00117] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве, которое больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00118] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве 0 мг/мл, 0.1 мг/мл, 0.2 мг/мл, 0.3 мг/мл, 0.4 мг/мл, 0.5 мг/мл, 0.6 мг/мл, 0.7 мг/мл, 0.8 мг/мл, 0.9 мг/мл, 1.0 мг/мл.
[00119] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5;
(e) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00120] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.7 до 1.8 мг/мл; и
(d) уксусную кислоту до рН 5.0;
(e) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00121] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации 1.742 мг/мл; и
(d) уксусную кислоту до рН 5.0;
(e) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00122] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5;
(е) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00123] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.7 мг/мл до 1.8 мг/мл; и
(d) уксусную кислоту до рН от 5.0 до 5.5;
(e) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00124] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации 1.742 мг/мл; и
(d) уксусную кислоту до рН от 5.0 до 5.5;
(e) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00125] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.742 мг/мл; и
(d) уксусную кислоту до рН 5.0;
(e) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00126] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации от 0.2 до 2.5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0.2 до 3.5 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.5;
(f) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00127] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН 5.5;
(f) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00128] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(а) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) L-гистидин в концентрации от 0.2 до 2.5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0.2 до 3.5 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.5;
(f) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00129] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.0;
(f) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00130] В одном варианте осуществления настоящее изобретение относится к водной фармацевтической композиции анти-PD-1 антитела, содержащей:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН 5.5;
(f) полоксамер 188 в концентрации больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00131] В некоторых вариантах осуществления настоящего изобретения указанный полоксамер 188 может находиться в количестве 0 мг/мл, 0.1 мг/мл, 0.2 мг/мл, 0.3 мг/мл, 0.4 мг/мл, 0.5 мг/мл, 0.6 мг/мл, 0.7 мг/мл, 0.8 мг/мл, 0.9 мг/мл, 1.0 мг/мл.
[00132] В некоторых вариантах осуществления водная фармацевтическая композиция анти-PD-1 антитела по настоящему изобретению может быть введена парентерально.
[00133] В некоторых вариантах осуществления водная фармацевтическая композиция анти-PD-1 антитела по настоящему изобретению может быть введена внутримышечно.
[00134] В некоторых вариантах осуществления водная фармацевтическая композиция анти-PD-1 антитела по настоящему изобретению может быть введена подкожно.
[00135] В некоторых вариантах осуществления водная фармацевтическая композиция анти-PD-1 антитела по настоящему изобретению может быть введена внутривенно.
[00136] В некоторых вариантах осуществления водная фармацевтическая композиция анти-PD-1 антитела по настоящему изобретению может быть введена внутривенно в виде инфузии.
[00137] В некоторых вариантах осуществления водная фармацевтическая композиция анти-PD-1 антитела по настоящему изобретению может быть введена внутривенно в виде инфузии длительностью 60 минут, при хорошей переносимости длительность инфузии может быть сокращена до 3 0 минут.
[00138] В одном варианте осуществления водная фармацевтическая композиция анти-PD-1 антитела пролголимаба по настоящему изобретению может находиться во флаконе.
[00139] В некоторых вариантах осуществления указанный флакон может представлять собой стеклянный флакон.
[00140] В некоторых вариантах осуществления указанный флакон может иметь объем от 1 мл до 50 мл.
[00141] В некоторых вариантах осуществления указанный флакон может иметь объем от 1 мл до 2 0 мл.
[00142] В некоторых вариантах осуществления указанный флакон может иметь объем 1 мл, 2 мл, 3 мл, 4 мл, 5 мл, 6 мл, 7 мл, 8 мл, 9 мл, 10 мл, 15 мл, 20 мл, 25 мл, 30 мл, 35 мл, 4 0 мл, 4 5 мл или 5 0 мл.
[00143] В одном варианте осуществления водная фармацевтическая композиция анти-PD-1 антитела пролголимаба по настоящему изобретению может находиться в шприце.
[00144] В некоторых вариантах осуществления указанный шприц может иметь вместимость 1 мл.
[00145] В некоторых вариантах осуществления указанный шприц может иметь вместимость 2 мл.
[00146] В одном варианте осуществления водная фармацевтическая композиция анти-PD-1 антитела пролголимаба по настоящему изобретению может находиться в преднаполненном шприце.
[00147] В некоторых вариантах осуществления указанный преднаполненный шприц может иметь вместимость 1 мл.
[00148] В некоторых вариантах осуществления указанный преднаполненный шприц может иметь вместимость 2 мл.
[00149] В соответствии с другим широким аспектом настоящего изобретения предлагается способ получения водной фармацевтической композиции, подходящей для введения субъекту для ингибирования активности белка PD-1. Способ включает комбинирование фармацевтически эффективного количества антитела к PD-1 пролголимаба с буферным агентом на основе ацетата; и эффективного количества трегалозы. Способ включает комбинирование фармацевтически эффективного количества антитела к PD-1 пролголимаба с буферным агентом на основе гистидина; и эффективного количества трегалозы.
[00150] В некоторых вариантах осуществления полоксамер 188 может быть добавлен в качестве солюбилизатора.
[00151] В соответствии с другим широким аспектом предлагается применение водной фармацевтической композиции антитела к PD-1 пролголимаба, как определено в настоящем документе, для лечения злокачественных новообразований.
[00152] В некоторых вариантах осуществления изобретения применение водной фармацевтической композиции антитела к PD-1 пролголимаба для лечения злокачественных новообразований, которое может быть выбрано из группы, содержащей меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого; неплоскоклеточный немелкоклеточный рак легкого, плоскоклеточный рак легкого; мелкоклеточный рак легкого, в том числе неоперабельный или метастатический мелкоклеточный рак легкого; рак легкого ранних стадий до и после радикального лечения; рак шейки матки, в том числе метастатический рак шейки матки, ранние стадии рака шейки матки до и после радикального лечения; опухоли головы и шеи, в том числе плоскоклеточный рак головы и шеи; лимфома Ходжкина; опухоли желудка и кишечника, метастатический плоскоклеточный рак пищевода; рак мочевого пузыря, в том числе метастатическая уротелиальная карцинома, рак почки; рак эндометрия, в том числе метастатический рак эндометрия, ранние стадии рака эндометрия до и после радикального лечения; рак молочной железы, в том числе метастатический рак молочной железы, ранние стадии рака эндометрия до и после радикального лечения; рак печени, в том числе метастатический или неоперабельный рак печени, ранние стадии рака печени до и после радикального лечения; неоперабельная или метастатическая солидная опухоль, в том числе неоперабельная или метастатическая солидная опухоль с признаками микросателлитной нестабильности.
[00153] В соответствии с другим широким аспектом предлагается применение водной фармацевтической композиции антитела к PD-1 пролголимаба для лечения злокачественного образования, включающий введение фармацевтически эффективного количества водной фармацевтической композиции, как определено в настоящем документе.
[00154] В соответствии с другим широким аспектом предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации от 15 мг/мл до 40 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 80 мг/мл до 110 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 мг/мл до 2.5 мг/мл; и
(d) уксусную кислоту до рН 4.5-5.5,
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00155] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 15 мг/мл до 2 5 мг/мл.
[00156] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 2 0 мг/мл.
[00157] В некоторых вариантах осуществления изобретения указанная трегалозы дигидрат может находиться в концентрации от 95 мг/мл до 105 мг/мл.
[00158] В некоторых вариантах осуществления изобретения указанная трегалозы дигидрат может находиться в концентрации 100 мг/мл.
[00159] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1.6 мг/мл до 1.9 мг/мл.
[00160] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1.7 мг/мл до 1.8 мг/мл.
[00161] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации 1.742 мг/мл.
[00162] В некоторых вариантах осуществления изобретения указанная уксусная кислота может быть добавлена до рН 5.0.
[00163] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.04 до 0.77 мг/мл.
[00164] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.4 0 мг/мл до 0.50 мг/мл.
[00165] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации 0.4 3 мг/мл.
[00166] В соответствии с другим широким аспектом предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации от 5 мг/мл до 4 0 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 70 мг/мл до 110 мг/мл;
(c) L-гистидин в концентрации от 0,2 до 2,5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0,2 до 3,5 мг/мл; для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00167] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 15 мг/мл до 2 5 мг/мл.
[00168] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 20 мг/мл.
[00169] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации от 95 мг/мл до 105 мг/мл.
[00170] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации 100 мг/мл.
[00171] В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации от 0,7 до 1,0 мг/мл.
[00172] В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации 0,92 мг/мл.
[00173] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации от 2,8 до 3,3 мг/мл.
[00174] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации 2.96 мг/мл.
[00175] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН от 5.5 до 6.5.
[00176] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН 5.5.
[00177] В одном варианте осуществления изобретения предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5;
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00178] В одном варианте осуществления изобретения предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.7 мг/мл до 1.8 мг/мл; и
(d) уксусную кислоту до рН 5.0;
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00179] В одном варианте осуществления изобретения предлагается применение водной фармацевтической композиции антитела к PQ-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации 1.742 мг/мл; и
(d) уксусную кислоту до рН 5.0;
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00180] В одном варианте осуществления изобретения предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации от 0.2 до 2.5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0.2 до 3.5 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.5;
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00181] В одном варианте осуществления изобретения предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН 5.5;
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00182] В одном варианте осуществления изобретения предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00183] В одном варианте осуществления изобретения предлагается применение водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
для лечения злокачественного новообразования у субъекта нуждающего в этом.
[00184] Водная фармацевтическая композиция анти-PD-1 антитела пролголимаба по настоящему изобретению дополнительно может содержать подходящий солюбилизатор.
[00185] В некоторых вариантах осуществления изобретения указанный солюбилизатор может представлять собой полоксамер 188.
[00186] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве, которое больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00187] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве 0 мг/мл, 0.1 мг/мл, 0.2 мг/мл, 0.3 мг/мл, 04 мг/мл, 0.5 мг/мл, 0.6 мг/мл, 0.7 мг/мл, 0.8 мг/мл, 0.9 мг/мл, 1.0 мг/мл.
[00188] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела.
[00189] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела.
[00190] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции каждые 2 недели.
[00191] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции 1 раз в 2 недели.
[00192] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции каждые 3 недели.
[00193] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции 1 раз в 3 недели.
[00194] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела каждые 2 недели.
[00195] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела 1 раз в 2 недели.
[00196] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела каждые 3 недели.
[00197] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела 1 раз в 3 недели.
[00198] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции парентерально.
[00199] В некоторых вариантах осуществления изобретения указанное парентеральное введение может представлять собой внутривенное, подкожное или внутримышечное введение.
[00200] В некоторых вариантах осуществления изобретения применение указанной водной фармацевтической композиции антитела к PD-1 пролголимаба может включать введение данной композиции внутривенно в виде инфузии.
[00201] В некоторых вариантах осуществления водная фармацевтическая композиция антитела к PD-1 пролголимаба по настоящему изобретению может быть введена внутривенно в виде инфузии длительностью 60 минут, при хорошей переносимости длительность инфузии может быть сокращена до 30 минут.
[00202] В некоторых вариантах осуществления изобретения злокачественное новообразование представляет собой меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого.
[00203] В одном варианте изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации от 15 мг/мл до 40 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 80 мг/мл до 110 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 мг/мл до 2.5 мг/мл; и
(d) уксусную кислоту до рН 4.5-5.5.
[00204] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 15 мг/мл до 25 мг/мл.
[00205] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 20 мг/мл.
[00206] В некоторых вариантах осуществления изобретения указанная трегалозы дигидрат может находиться в концентрации от 95 мг/мл до 105 мг/мл.
[00207] В некоторых вариантах осуществления изобретения указанная трегалозы дигидрат может находиться в концентрации 100 мг/мл.
[00208] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1.6 мг/мл до 1.9 мг/мл.
[00209] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации от 1.7 мг/мл до 1.8 мг/мл.
[00210] В некоторых вариантах осуществления изобретения указанный натрия ацетат тригидрат может находиться в концентрации 1.742 мг/мл.
[00211] В некоторых вариантах осуществления изобретения указанная уксусная кислота может быть добавлена до рН 5.0.
[00212] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.04 до 0.77 мг/мл.
[00213] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации от 0.40 мг/мл до 0.50 мг/мл.
[00214] В некоторых вариантах осуществления изобретения указанная уксусная кислота может находиться в концентрации 0.4 3 мг/мл.
[00215] В одном варианте изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации от 5 мг/мл до 40 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 7 0 мг/мл до 110 мг/мл;
(c) L-гистидин в концентрации от 0,2 до 2,5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0,2 до 3,5 мг/мл.
[00216] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации от 15 мг/мл до 2 5 мг/мл.
[00217] В некоторых вариантах осуществления изобретения указанный пролголимаб может находиться в концентрации 20 мг/мл.
[00218] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации от 95 мг/мл до 105 мг/мл.
[00219] В некоторых вариантах осуществления изобретения указанный трегалозы дигидрат может находиться в концентрации 100 мг/мл.
[00220] В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации от 0,7 до 1,0 мг/мл.
[00221] В некоторых вариантах осуществления изобретения указанный L-гистидин может находиться в концентрации 0,92 мг/мл.
[00222] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации от 2.8 до 3,3 мг/мл.
[00223] В некоторых вариантах осуществления изобретения указанный L-гистидина гидрохлорид может находиться в концентрации 2.96 мг/мл.
[00224] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН от 5.5 до 6.5.
[00225] В некоторых вариантах осуществления изобретения указанная композиция может иметь рН 5.5.
[00226] В одном варианте осуществления изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 0.2 до 2.5 мг/мл; и
(d) уксусную кислоту до рН от 4.5 до 5.5.
[00227] В одном варианте осуществления изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации от 1.7 мг/мл до 1.8 мг/мл; и
(d) уксусную кислоту до рН 5.0.
[00228] В одном варианте осуществления изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации 1.742 мг/мл; и
(d) уксусную кислоту до рН 5.0.
[00229] В одном варианте осуществления изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации от 0.2 до 2.5 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации от 0.2 до 3.5 мг/мл.;
(e) где указанная композиция имеет рН от 5.5 до 6.5.
[00230] В одном варианте осуществления изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации 0.92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2.96 мг/мл.;
(e) где указанная композиция имеет рН 5.5.
[00231] В одном варианте осуществления изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00232] В одном варианте осуществления изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00233] Водная фармацевтическая композиция анти-PD-1 антитела пролголимаба по настоящему изобретению дополнительно может содержать подходящий солюбилизатор.
[00234] В некоторых вариантах осуществления изобретения указанный солюбилизатор может представлять собой полоксамер 188.
[00235] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве, которое больше 0 мг/мл, но равно или меньше 1 мг/мл.
[00236] В некоторых вариантах осуществления изобретения указанный полоксамер 188 может находиться в количестве 0 мг/мл, 0.1 мг/мл, 0.2 мг/мл, 0.3 мг/мл, 0.4 мг/мл, 0.5 мг/мл, 0.6 мг/мл, 0.7 мг/мл, 0.8 мг/мл, 0.9 мг/мл, 1.0 мг/мл.
[00237] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела.
[00238] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела.
[00239] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба каждые 2 недели.
[00240] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба 1 раз в 2 недели.
[00241] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба каждые 3 недели.
[00242] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба 1 раз в 3 недели.
[00243] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела каждые 2 недели.
[00244] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела 1 раз в 2 недели.
[00245] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела каждые 3 недели.
[00246] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела 1 раз в 3 недели.
[00247] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба парентерально.
[00248] В некоторых вариантах осуществления изобретения указанное парентеральное введение может представлять собой внутривенное, подкожное или внутримышечное введение.
[00249] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества указанной водной фармацевтической композиции антитела к PD-1 пролголимаба внутривенно в виде инфузии.
[00250] В некоторых вариантах осуществления водная фармацевтическая композиция антитела к PD-1 пролголимаба по настоящему изобретению может быть введена внутривенно в виде инфузии длительностью 60 минут, при хорошей переносимости длительность инфузии может быть сокращена до 3 0 минут.
[00251] В некоторых вариантах осуществления изобретения злокачественное новообразование представляет собой меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого.
[00252] В одном варианте изобретения предлагается способ лечения злокачественных новообразований у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества пролголимаба.
[00253] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба в дозе 1 мг/кг.
[00254] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба в дозе 3 мг/кг.
[00255] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба каждые 2 недели.
[00256] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба 1 раз в 2 недели.
[00257] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба каждые 3 недели.
[00258] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба 1 раз в 3 недели.
[00259] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба в дозе 1 мг/кг каждые 2 недели.
[00260] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба в дозе 1 мг/кг 1 раз в 2 недели.
[00261] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба в дозе 3 мг/кг каждые 3 недели.
[00262] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба в дозе 3 мг/кг 1 раз в 3 недели.
[00263] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба парентерально.
[00264] В некоторых вариантах осуществления изобретения указанное парентеральное введение может представлять собой внутривенное, подкожное или внутримышечное введение.
[00265] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба внутривенно в виде инфузии.
[00266] В некоторых вариантах осуществления пролголимаб может быть введен внутривенно в виде инфузии длительностью 60 минут, при хорошей переносимости длительность инфузии может быть сокращена до 30 минут.
[00267] В некоторых вариантах осуществления изобретения злокачественное новообразование представляет собой меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатичесий немелкоклеточный рак легкого.
[00268] В одном варианте осуществления изобретения предлагается водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1, при этом фармацевтическая композиция содержит на каждый 1 мл фармацевтической композиции:
[00269] В одном варианте осуществления изобретения предлагается водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1, при этом фармацевтическая композиция содержит на каждый 1 мл фармацевтической композиции:
[00270] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела.
[00271] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела.
[00272] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена каждые 2 недели.
[00273] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена 1 раз в 2 недели.
[00274] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена 1 раз в 3 недели.
[00275] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела каждые 2 недели.
[00276] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела 1 раз в 2 недели.
[00277] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела каждые 3 недели.
[00278] В некоторых вариантах осуществления изобретения указанная водная фармацевтическая композиция, подходящая для введения субъекту для ингибирования активности белка PD-1 может быть введена в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела 1 раз в 3 недели.
[00279] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба парентерально.
[00280] В некоторых вариантах осуществления изобретения указанное парентеральное введение может представлять собой внутривенное, подкожное или внутримышечное введение.
[00281] В некоторых вариантах осуществления изобретения способ лечения злокачественных новообразований у нуждающегося в этом субъекта может включать введение терапевтически эффективного количества пролголимаба внутривенно в виде инфузии.
[00282] В некоторых вариантах осуществления пролголимаб может быть введен внутривенно в виде инфузии длительностью 60 минут, при хорошей переносимости длительность инфузии может быть сокращена до 30 минут.
[00283] В некоторых вариантах осуществления изобретения злокачественное новообразование представляет собой меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатичесий немелкоклеточный рак легкого.
ПРИМЕРНЫЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ:
[00284] Настоящее изобретение относится к подходящим водным фармацевтическим композициям антитела к PD-1 пролголимаба, В одном варианте осуществления изобретения водная фармацевтическая композиция пролголимаба может содержать буфер на основе ацетата и трегалозу. Полоксамер 188 может быть добавлен в качестве солюбилизатора. В еще одном варианте осуществления изобретения водная фармацевтическая композиция пролголимаба может содержать буфер на основе гистидина и трегалозу. Полоксамер 188 может быть добавлен в качестве солюбилизатора.
[00285] Буфер на основе гистидина может быть образован за счет комбинирования L-гистидина с гистидина гидрохлоридом или, дополнительно, с соляной кислотой или другими кислотами. Следует понимать, что хотя в качестве соли для буфера на основе гистидина можно использовать гистидина гидрохлорид, для буфера на основе гистидина можно использовать любую другую соль на основе гистидина, не отступая при этом от идей настоящего изобретения.
[00286] Буфер на основе ацетата может быть образован за счет комбинирования уксусной кислоты с натрия ацетат тригидратом. Следует понимать, что хотя в качестве соли для буфера на основе ацетата можно использовать натрия ацетат тригидрат, для буфера на основе ацетата можно использовать любую другую ацетатную соль, например, ацетат калия, не отступая при этом от идей настоящего изобретения.
[00287] Кроме того, композиция по настоящему изобретению может дополнительно включать один или несколько других подходящих эксципиентов, которые хорошо известны специалистам в данной области.
[00288] В некоторых вариантах осуществления жидкая фармацевтическая композиция исполнена так, что она стабильна при хранении в том смысле, что не происходит каких-либо дальнейших процессов агрегации белка или его модификаций по сравнению с показателем стабильности в нулевой временной точке.
[00289] В одном варианте осуществления авторы изобретения неожиданно получили высококонцентрированные водные фармацевтические композиции антитела к PD-1 пролголимаба, где пролголимаб может находиться в концентрации от 90 мг/мл до 150 мг/мл. В некоторых вариантах осуществления настоящего изобретения указанный пролголимаб может находиться в концентрации 90 мг/мл, 95 мг/мл, 100 мг/мл, 105 мг/мл, 110 мг/мл, 115 мг/мл, 120 мг/мл, 125 мг/мл, 130 мг/мл, 135 мг/мл, 140 мг/мл, 145 мг/мл, 150 мг/мл.
[00290] Такие высококонцентрированные водные фармацевтические композиции антитела к PD-1 пролголимаба по настоящему изобретению показали коллоидную стабильность при интенсивном перемешивании (800 об./мин) в течение 120 часов и высокую термическую стабильность при нагревании при 50°С и 37°С, а также приемлемую для парентерального введения вязкость менее 50 сР.
[00291] Указанные выше композиции подходят для парентерального введения, такого как внутривенное, подкожное, интрадермальное, внутриартериальное, интратекальное, внутрибрюшинное, внутрисуставное и/или внутримышечное введение.
[00292] Предоставляемые фармацевтические композиции можно вводить нуждающемуся в лечении индивидууму посредством системной инъекции, например, посредством внутривенной или подкожной, или внутримышечной инъекции; или посредством инъекции или нанесения на соответствующий участок, например, посредством прямой инъекции или прямого нанесения на участок, когда участок доступен при хирургическом вмешательстве; или посредством местного применения.
[00293] Указанные выше композиции можно вводить нуждающемуся в лечении индивидууму внутривенно в виде инфузии.
[00294] В некоторых вариантах осуществления водная фармацевтическая композиция антитела к PD-1 пролголимаба по настоящему изобретению может быть введена внутривенно в виде инфузии длительностью 60 минут, при хорошей переносимости длительность инфузии может быть сокращена до 30 минут.
[00295] Водная фармацевтическая композиция антитела к PD-1 пролголимаба по настоящему изобретению может быть использована после разведения. Для этого необходимое количество композиции из флакона переносят в емкость для инфузий, содержащую стерильный 0,9% раствор натрия хлорида или стерильный 5% раствор декстрозы. Концентрация указанной композиции в приготовленном растворе может составлять от 0,5 до 10 мг/мл. Приготовленный раствор перемешивают путем осторожного переворачивания емкости для инфузий во избежание пенообразования.
Способы лечения и применение водной композиции
[00296] В другом варианте осуществления настоящее изобретение относится к способу лечения млекопитающего, включающему введение млекопитающему терапевтически эффективного количества фармацевтических композиций, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по настоящему изобретению, где у млекопитающего может присутствовать заболевание или нарушение, которые можно эффективно лечить антителом к PD-1 пролголимабом по настоящему изобретению.
[00297] В другом варианте осуществления настоящее изобретение относится к способу лечения млекопитающего, включающему введение млекопитающему терапевтически эффективного количества фармацевтических композиций, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по настоящему изобретению, где у млекопитающего может присутствовать заболевание или нарушение, которые можно эффективно лечить антителом к PD-1 пролголимабом по настоящему изобретению.
[00298] В предпочтительном варианте осуществления млекопитающее представляет собой человека.
[00299] В другом варианте осуществления настоящее изобретение относится к способу лечения нуждающегося в этом субъекта, включающему введение субъекту терапевтически эффективного количества фармацевтических композиций, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по настоящему изобретению, где у субъекта может присутствовать заболевание или нарушение, которые можно эффективно лечить антителом к PD-1 пролголимабом по настоящему изобретению.
[00300] В другом варианте осуществления настоящее изобретение относится к способу лечения нуждающегося в этом субъекта, включающему введение субъекту терапевтически эффективного количества фармацевтических композиций, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по настоящему изобретению, где у субъекта может присутствовать заболевание или нарушение, которые можно эффективно лечить антителом к PD-1 пролголимабом по настоящему изобретению.
[00301] В предпочтительном варианте осуществления субъект представляет собой человека.
[00302] В одном из вариантов осуществления изобретение относится к способу лечения меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по данному изобретению.
[00303] В одном из вариантов осуществления изобретение относится к способу лечения меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по данному изобретению.
[00304] В одном из вариантов осуществления изобретение относится к способу лечения неоперабельной меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 4 0 мг/мл, по данному изобретению.
[00305] В одном из вариантов осуществления изобретение относится к способу лечения неоперабельной меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по данному изобретению.
[00306] В одном из вариантов осуществления изобретение относится к способу лечения метастатической меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по данному изобретению.
[00307] В одном из вариантов осуществления изобретение относится к способу лечения метастатической меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет 2 0 мг/мл, по данному изобретению.
[00308] В одном из вариантов осуществления изобретение относится к способу лечения ранних стадий меланомы до и после радикального лечения, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по данному изобретению.
[00309] В одном из вариантов осуществления изобретение относится к способу лечения ранних стадий меланомы до и после радикального лечения, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по данному изобретению.
[00310] В одном из вариантов осуществления изобретение относится к способу лечения рака легкого, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по данному изобретению.
[00311] В одном из вариантов осуществления изобретение относится к способу лечения рака легкого, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по данному изобретению.
[00312] В одном из вариантов осуществления изобретение относится к способу лечения немелкоклеточного рака легкого (НМРЛ), включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по данному изобретению.
[00313] В одном из вариантов осуществления изобретение относится к способу лечения немелкоклеточного рака легкого (НМРЛ), включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по данному изобретению.
[00314] В одном из вариантов осуществления изобретение относится к способу лечения неоперабельного или метастатического немелкоклеточного рака легкого, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет от 15 мг/мл до 40 мг/мл, по данному изобретению.
[00315] В одном из вариантов осуществления изобретение относится к способу лечения неоперабельного или метастатического немелкоклеточного рака легкого, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества одной из приведенных композиций антитела к PD-1 пролголимаба, где концентрация антитела к PD-1 пролголимаба составляет 20 мг/мл, по данному изобретению.
[00316] В одном варианте осуществления изобретения предлагается способ лечения меланомы у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00317] В одном варианте осуществления изобретения предлагается способ лечения меланомы у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00318] В одном варианте осуществления изобретения предлагается способ лечения неоперабельной меланомы у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00319] В одном варианте осуществления изобретения предлагается способ лечения неоперабельной меланомы у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00320] В одном варианте осуществления изобретения предлагается способ лечения метастатической меланомы у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00321] В одном варианте осуществления изобретения предлагается способ лечения метастатической меланомы у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00322] В одном варианте осуществления изобретения предлагается способ лечения ранних стадий меланомы до и после радикального лечения у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00323] В одном варианте осуществления изобретения предлагается способ лечения ранних стадий меланомы до и после радикального лечения у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00324] В одном варианте осуществления изобретения предлагается способ лечения рака легкого у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной
фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00325] В одном варианте осуществления изобретения предлагается способ лечения рака легкого у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00326] В одном варианте осуществления изобретения предлагается способ лечения немелкоклеточного рака легкого (НМРЛ) у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00327] В одном варианте осуществления изобретения предлагается способ лечения немелкоклеточного рака легкого (НМРЛ) у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00328] В одном варианте осуществления изобретения предлагается способ лечения неоперабельного или метастатического немелкоклеточного рака легкого у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
I. Состав на 1 мл:
[00329] В одном варианте осуществления изобретения предлагается способ лечения неоперабельного или метастатического немелкоклеточного рака легкого у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции антитела к PD-1, содержащей:
II. Состав на 1 мл:
[00330] В одном из вариантов осуществления изобретение относится к способу лечения меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества антитела к PD-1 пролголимаба.
[00331] В одном из вариантов осуществления изобретение относится к способу лечения неоперабельной меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества антитела к PD-1 пролголимаба.
[00332] В одном из вариантов осуществления изобретение относится к способу лечения метастатической меланомы, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества антитела к PD-1 пролголимаба.
[00333] В одном из вариантов осуществления изобретение относится к способу лечения ранних стадий меланомы до и после радикального лечения, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества антитела к PD-1 пролголимаба.
[00334] В одном из вариантов осуществления изобретение относится к способу лечения рака легкого, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества антитела к PD-1 пролголимаба.
[00335] В одном из вариантов осуществления изобретение относится к способу лечения немелкоклеточного рака легкого (НМРЛ), включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества антитела к PD-1 пролголимаба.
[00336] В одном из вариантов осуществления изобретение относится к способу лечения неоперабельного или метастатического немелкоклеточного рака легкого, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества антитела к PD-1 пролголимаба.
[00337] Терапевтически эффективное количество антитела к PD-1 пролголимаба по настоящему изобретению, и водных композиций, включающих антитело к PD-1 пролголимаб, по настоящему изобретению, в предлагаемых составах зависит от состояния, подлежащего лечению, тяжести состояния, предшествующей терапии и истории болезни пациента, и ответу на терапевтическое средство. Подходящую дозу можно регулировать по решению лечащего врача так, что ее можно вводить пациенту один раз или посредством нескольких введений.
[00338] В одном из вариантов осуществления эффективное количество антитела к PD-1 на дозу для пациента составляет приблизительно от 0.01 до 10 мг на килограмм массы тела, или приблизительно от 1 до 10 мг на килограмм массы тела, или приблизительно 0.05 мг на килограмм массы тела, или приблизительно 0.25 мг на килограмм массы тела, или приблизительно 0.5 мг на килограмм веса тела, или приблизительно 1 мг на килограмм массы тела, или приблизительно 2 мг на килограмм массы тела, или приблизительно 3 мг на килограмм массы тела, или приблизительно 4 мг на килограмм массы тела, или приблизительно 5 мг на килограмм массы тела, или приблизительно 6 мг на килограмм массы тела, или приблизительно 7 мг на килограмм массы тела, или приблизительно 8 мг на килограмм массы тела, или приблизительно 9 мг на килограмм массы тела, или приблизительно 10 мг на килограмм массы тела.
[00339] Частота введения доз обычно может быть примерно один раз в неделю, или примерно один раз каждые 2 недели, или примерно один раз каждые 3 недели.
[00340] В другом варианте осуществления приемлемая доза для введения посредством инфузии может содержать 5-450 мг/дозу, или может содержать 40 мг, или 50 мг, или 60 мг на одну дозу; или может содержать 70 мг, или 80 мг, или 90 мг, или 100 мг на одну дозу; или может содержать 110 мг, или 12 0 мг, или 130 мг, или 140 мг на одну дозу; или может содержать 150 мг, или 160 мг, или 170 мг, или 180 мг на одну дозу; или может содержать 190 мг, или 200 мг, или 210 мг, или 220 мг на одну дозу; или может содержать 230 мг, или 240 мг, или 250 мг, или 260 мг на одну дозу; или может содержать 270 мг, или 280 мг, или 290 мг на одну дозу, или может содержать 300 мг, или 310 мг, или 320 мг, или 330 мг, или 340 мг, или 350 мг на одну дозу; или может содержать 360 мг, или 370 мг, или 380 мг, или 390 мг, или 400 мг, или 410 мг, или 420 мг, или 430 мг, или 440 мг, или 450 мг на одну дозу.
[00341] В одном варианте осуществления изобретения доза может быть доставлена посредством одной или более одной инфузий. Доза может быть доставлена посредством одной, двух или трех инфузий. В некоторых вариантах изобретения длительность терапии может составлять от одной или нескольких инфузий. В некоторых вариантах изобретения улучшения состояния пациента можно добиться посредством лечения в течение продолжительного периода времени. В некоторых вариантах изобретения длительность терапии может быть до прогрессирования заболевания или пожизненно.
[00342] В другом варианте осуществления фармацевтические композиции по настоящему изобретению можно получать в виде нерасфасованного состава, и, по существу, компоненты фармацевтической композиции присутствуют в количествах выше, чем может требоваться для введения, и их соответствующим образом разбавляют до введения.
[00343] Альтернативно, фармацевтическая композиция может быть заморожена, высушена распылением, либо лиофилизирована и восстановлена перед применением в подходящем стерильном носителе. Лиофилизация может быть выполнена с использованием способов, известных в данной области техники, которые включают в себя различные шаги, такие как замораживание, отжиг, первичная и вторичная сушка.
[00344] Фармацевтические композиции можно вводить в виде одного терапевтического средства или в комбинации с дополнительными терапевтическими средствами по мере необходимости. Таким образом, в одном варианте осуществления предлагаемые способы лечения и/или профилактики используют в комбинации с введением терапевтически эффективного количества другого активного средства. Другое активное средство можно вводить до, в течение или после введения фармацевтических композиций по настоящему изобретению. Другое активное средство можно вводить как часть предлагаемой композиции или, альтернативно, в виде отдельного состава.
[00345] Введение предлагаемых фармацевтических композиций можно проводить различными способами, включая парентеральное, пероральное, буккальное, назальное, ректальное и местное введение. Парентеральное введение может включать, без ограничения, трансдермальное, подкожное, внутривенное, внутриартериальное, внутрибрюшинное, интрадермальное, внутрисердечное, внутрижелудочковое, внутричерепное, внутритрахеальное, интратекальное введение, внутримышечную инъекцию, внутривитреальную инъекцию.
[00346] Фармацевтические композиции по настоящему изобретению особенно пригодны для парентерального введения, т.е. подкожно, внутримышечно, внутривенно, интраперитонеально, в спинной, мозг, в суставы, интрасиновиально и/или интратекально. Парентеральное введение можно проводить посредством болюсной инъекции или непрерывной инфузии. Фармацевтические композиции для инъекций могут находиться в стандартной лекарственной форме, например, в ампулах, флаконах, преднаполненных шприцах или в контейнерах с несколькими дозами с добавленным консервантом, но не ограничиваясь этим. Кроме того, разработан ряд недавних подходов к доставке лекарственного средства, и фармацевтические композиции по настоящему изобретению подходят для введения этими новыми способами, например, BD Physioject™, Inject-ease®, Genject®, ручки-инжекторы, такие как GenPen®, и безыгольные устройства, такие как MediJector®40 и BioJector®. Фармацевтическую композицию по настоящему изобретению также можно адаптировать для еще не открытых способов введения.
[00347] Также см. Langer, 1990, Science, 249: 1527-1533.
[00348] Предлагаемые фармацевтические композиции также можно формулировать в качестве депо-препарата. Такие длительно действующие составы можно вводить посредством имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Таким образом, например, составы можно модифицировать с использованием подходящих полимерных или гидрофобных материалов (например, в виде эмульсии в приемлемом масле), или ионообменных смол, или в виде умеренно растворимых производных, например, в виде умеренно растворимой соли.
[00349] Фармацевтические композиции при желании можно предоставлять во флаконе, упаковке или в устройстве-распределителе, которые могут содержать одну или несколько стандартных лекарственных форм, содержащих активный ингредиент. В одном варианте осуществления устройство-распределитель может содержать шприц, содержащий однократную дозу жидкого состава, готового к инъекции. Шприц может сопровождаться инструкцией по введению.
[00350] В другом варианте осуществления настоящее изобретение относится к набору или контейнеру, содержащим водную фармацевтическую композицию по изобретению. Концентрация антитела в водной фармацевтической композиции может варьировать в широком диапазоне, но, как правило, в пределах диапазона приблизительно от 1 до приблизительно 200 мг/мл. Набор также может сопровождаться инструкциями по применению.
[00351] Способ получения вышеуказанных композиций включает добавление в водную фазу ацетатных буферных агентов, с последующим добавлением, в любой последовательности, следующих компонентов: трегалозы, пролголимаба, и/или солюбилизатора, выбранного из группы: полисорбат 20, полисорбат 80, полоксамер 188 или их комбинация.
[00352] Способ получения вышеуказанных композиций включает добавление в водную фазу гистидиновых буферных агентов, с последующим добавлением, в любой последовательности, следующих компонентов: трегалозы, пролголимаба, и/или солюбилизатора, выбранного из группы: полисорбат 20, полисорбат 80, полоксамер 188 или их комбинация.
ПРИМЕР ИССЛЕДОВАНИЯ:
[00353] Ниже представлены примеры исследования по определению реагентов и их концентраций для получения водной фармацевтической композиции антитела к PD-1 пролголимаба по настоящему изобретению.
[00354] Исследования и примеры, представленные в настоящем документе, предназначены исключительно для иллюстративных целей, которые демонстрируют подходимость определенных компонентов, используемых в водных фармацевтических композициях антитела к PD-1 пролголимаба. Подразумевается, что средний специалист в данной области техники может использовать и другие способы, не отступая при этом от идей настоящего изобретения.
[00355] Подходимость водных композиций по настоящему изобретению была испытана посредством иллюстративных способов, описанных в настоящем документе.
[00356] Пример 1. Получение стабильных составов антитела к PD-1 пролголимаба
[00357] Получение образцов антитела с концентрацией 5 мг/мл осуществляли в концентрационных ячейках Stirred Cell (Millipore) под давлением. Для этого антитело в исходном составе помещали в ячейку, при непрерывном перемешивании белок концентрировали под потоком сжатого воздуха до концентрации 10 мг/мл, после чего вносили в ячейку не менее чем 10 кратный объем водного раствора с целевым составом, включающим буферные, осмотические агенты и, если необходимо, дополнительные водорастворимые стабилизаторы. По завершении процесса диафильтрации антитело концентрировали до концентрации около 10 мг/мл, выгружали из ячейки, определяли точную концентрацию белка методом УФ-спектрофотометрии. Затем к образцу вносили соответствующий раствор вспомогательных веществ для получения раствора с целевой концентрацией белка 5±0.2 мг/мл.
[00358] Получение образцов белка с концентрацией более 50 мг/мл проводили в кассетах Pellicon (Millipore) в режиме тангенциального потока. Для этого антитело в исходном составе помещали в емкость для диафильтрации, концентрировали белок до концентрации 50-100 мг/мл, после чего к системе подключали подачу не менее чем 10 кратного объема раствора с целевым составом, содержащим буферные, осмотические агенты и, если необходимо, дополнительные водорастворимые стабилизаторы. Допускается внесение концентрата осмотических агентов и водорастворимых стабилизаторов после завершения диафильтрации. По завершении процесса диафильтрации антитело концентрировали до концентрации, превышающей целевую, выгружали из системы и определяли точную концентрацию белка. Затем к образцу вносили соответствующий раствор вспомогательных веществ для получения раствора с целевой концентрацией белка.
[00359] При получении составов, содержащих солюбилизаторы, концентраты поверхностно-активных веществ вносили к антителу после завершения диафильтрации и концентрирования при финальном разведении антитела раствором вспомогательных веществ до целевой концентрации.
[00360] Перед асептическим наполнением в конечный контейнер (например, стеклянный или полимерный сосуд, флакон или шприц) раствор антитела фильтровали через мембрану с размером пор 0.22 мкм.
[00361] Пример 2. Определение концентрации белка в исследуемых образцах
[00362] Концентрацию белка определяли с помощью метода УФ-спектрофотометрии при длине волны 280 нм в УФ-прозрачных планшетах.
[00363] Каждый образец разводили соответствующим раствором вспомогательных веществ до концентрации ~ 0.5 мг/мл. В лунку планшета для УФ-спектрофотометрии помещали 150 мкл разведенного образца. Измеряли оптическую плотность помещенных в планшет растворов на планшетном спектрофотометре при длине волны 280 нм. В качестве раствора сравнения использовали соответствующий раствор вспомогательных веществ.
[00364] Концентрацию белка (С) в мг/мл рассчитывали по формуле:
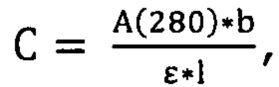 где
где
А280 - значение оптической плотности при длине волны 280 нм;
ε - коэффициент экстинкции исследуемого белка;
b - суммарный коэффициент разведения образца;
l - толщина слоя в лунке планшета; для 150 мкл l=0.42 см.
[00365] Пример 3. Исследование ПЭГ-агрегации
[00366] Готовили раствор ПЭГ 6000 с массовой концентрацией 20-25% в исследуемом составе эксципиентов. Итоговые растворы фильтровали через фильтр Durapore 0.4 5 мкм.
[00367] В 96-луночные планшеты для УФ-спектрофотометрии переносили расчетное количество образца, раствора вспомогательных веществ и 20-25% раствора ПЭГ 6000 так, чтобы в ряде лунок была концентрация ПЭГ6000 от 0 до 18%, а концентрация белка в каждой лунке составляла 1 мг/мл. Все полученные в лунках растворы хорошо перемешивали, пипетированием.
[00368] После этого оценивали степень мутности растворов визуально, а также измеряли оптическую плотность растворов при длине волны 400 нм.
[00369] Осаждение белка в присутствии ПЭГ связано с эффектом замещения объема, то есть белок стерически вытесняется из регионов растворителя полимером. Это приводит к концентрированию белка до тех пор, пока не будет превышена его растворимость и не выпадет осадок. Чем менее стабилен образец, тем при меньшей концентрации ПЭГ 6000 он будет образовывать видимые агрегаты (опалесценцию).
[00370] Пример 4. Определение коллоидной стабильности методом «шейк-тест»
[00371] Исследуемые образцы разделяли на 2 части по 200 мкл и помещали в стеклянные виалы, по 1 виале на каждый состав закладывали в холодильник на хранение при 2-8°С, остальные устанавливали в шейкер и шейкировали со скоростью 800 об./мин при температуре 2-8°С в течение указанного времени. После окончания стресса пробирки встряхивали на вортексе и передавали на анализ.
[00372] Пример 5. Определение коллоидной стабильности методом криоконцентрирования
[00373] Исследуемые образцы разделяли на 2 части и помещали в полимерные пробирки: по 1 пробирке на каждый состав откладывали в холодильник на хранение при 2-8°С, остальные устанавливали в морозильную камеру и хранили при температуре минус 16-20°С в течение указанного времени. После окончания стресса пробирки убирали из морозильной камеры, выдерживали при комнатной температуре до полного оттаивания содержимого, перемешивали растворы с помощью vortex и передавали на анализ.
[00374] Пример 6. Определение термической стабильности методом «термостресс»
[00375] Исследуемые образцы разделяли на 2 части и помещали в отдельные стеклянные виалы: по 1 виале на каждый состав откладывали в холодильник на хранение при 2-8°С, остальные устанавливали в термостат и инкубировали при необходимой температуре в течение указанного времени. После окончания прогрева виалы убирали из термостата, выдерживали при комнатной температуре около 15 мин и передавали на анализ.
[00376] Пример 7. Определение гомогенности образцов методом эксклюзионной (Э) высокоэффективной жидкостной хроматографии (ВЭЖХ)
Колонка Tosoh TSK-GelG3000SWXL 7.8 mm ID × 30 cm, cat. №08541.
Температура колонки: 25°C
Скорость потока подвижной фазы: 0.7 мл/мин.
Объем вкола: 10 мкл.
Концентрация образца: 5 мг/мл.
Длина волны детектора: 220 нм.
Продолжительность элюирования: 25 мин.
Подвижная фаза: Динатрия гидрофосфат б/в 7.1 мг/мл.
Натрия хлорид 17.54 мг/мл.
рН подвижной фазы доводили до 7.0 ортофосфорной кислотой.
Изменение чистоты после стресса рассчитывали по формуле:
Δ = (доля основного пика после стресса - доля основного пика до стресса)
[00377] Пример 8. Определение гетерогенности по заряду (профиль заряженных форм) образцов на приборе Caliper Labchip GX II
[00378] Приготовление анализируемых образцов.
[00379] Образцы разводили до концентрации 1 мг/мл. К 200 мкл полученного раствора добавляли 2 мкл раствора карбоксипептидазы В (СрВ) с концентрацией 5 мг/мл, перемешивали и инкубировали в течение 2 часов при температуре 37°С. Испытуемые образцы диализовывали против трех смен воды. Для проведения диализа поместили испытуемые растворы в центрифужные пробирки Amicon Ultra объемом 0.5 мл и центрифугировали в течение 10 мин при скорости вращения ротора 10000 об/мин в центрифуге Eppendorf Centrifuge 5417R. Измеряли интенсивность поглощения растворов при помощи спектрофотометра Сагу 50 Bio относительно воды. Готовили пробы исследуемых серий с концентрацией 2 мг/мл. В 96-луночный планшет Bio-Rad вносили по 3 мкл Labelling Buffer (из набора НТ Protein Charge Variant Labeling Kit), по 15 мкл испытуемых растворов и по 3 мкл раствора Dye Mixture (из набора НТ Protein Charge Variant Labeling Kit). Помещали планшет в темное место на 10 минут, после чего добавляли в каждую лунку по 3 6 мкл воды и перемешивали, пипетируя. Центрифугировали в течение 1 минуты при 1000 об/мин в центрифуге Eppendorf Centrifuge 5417R.
[00380] Приготовление рабочих растворов и заполнение чипа.
[00381] Приготовление рабочих растворов и подготовку чипа проводили в соответствии с методикой от производителя с использованием набора НТ Protein Charge Variant Labeling Kit. Запуск анализа является стандартной процедурой. Метод анализа НТ Protein Charge Variant 90s.
[00382] Пример 9. Определение чистоты образцов на приборе Caliper Labchip GXII в редуцирующих и нередуцирующих условиях
[00383] Приготовление анализируемых образцов.
Для приготовления денатурирующего и восстанавливающего растворов использовали по 700 мкл НТ Protein Express Sample Buffer. Для восстановления образцов вносили 24,5 мкл 1М дитиотреитола (DTT). В буфер для невосстанавливаемых образцов вносили алкилирующий агент - 24,5 мкл 1М йодацетамида (IAM).
[00384] Для каждого образца подготавливали две микропробирки - в одну вносили 35 мкл денатурирующего буфера, в другую - 3 5 мкл восстанавливающего. Образцы разводили до концентрации 2 мг/мл. Вносили в каждую пару пробирок по 5 мкл образца. Денатурировали образцы при 100°С в течение 5 минут. Перемешивали пробирки на вортексе, после чего добавляли 7 0 мкл воды в каждую пробирку и перемешивали на вортексе. Переносили по 44 мкл каждого образца в лунки 96-луночного планшета.
[00385] Приготовление рабочих растворов и заполнение чипа
Рабочие растворы и подготовку чипа проводили в соответствии с методикой от производителя с использованием набора НТ Protein Express Reagent Kit. Запуск анализа является стандартной операцией. Метод анализа НТ Protein Express 200.
[00386] Пример 10. Определение профиля заряженных форм образцов
методом ионообменной (ИО) высокоэффективной жидкостной хроматографии (ВЭЖХ)
Колонка: ProPac WCX-10 Analytical, 4×250 мм.
Предколонка: Pro Рас WCX-10G, 4×50 мм.
Температура колонки: 30°С.
Скорость потока подвижной фазы: 0.7 мл/мин.
Объем вкола: 50 мкл.
Концентрация образца: 1 мг/мл.
Длина волны детектора: 220 нм.
Продолжительность элюирования: 60 мин.
Подвижная фаза:
Элюент А: 0,03М 2-монфолиноэтансульфаоновая кислота (MES), рН 6.0.
Элюент В: 0,03 М MES, 0,5 М NaCl, рН 6.0.
Градиент элюента А: 86%-0%-86%.
[00387] Перед анализом испытуемый образец обрабатывали раствором карбоксипептидазы Б при температуре +37±1°С в течение 2 ч.
[00388] Абсолютное изменение профиля заряженных форм после стресса рассчитывали по формуле:
Δ = | содержание кислых фракций до стресса - содержание кислых фракций после стресса | + | содержание доминирующей фракции до стресса - содержание доминирующей фракции после стресса | + | содержание щелочных фракций до стресса - содержание щелочных фракций после стресса.
[00389] Пример 11. Определение низкомолекулярных примесей методом вертикального гель-электрофореза (ВЭФ) в полиакриламидном геле (ПААГ) в редуцирующих и нередуцирующих условиях
[00390] Готовили ПААГ в стеклянных пластинах в присутствии натрия додецилсульфата, состоящие из концентрирующего слоя - 4% ПААГ и разделяющего слоя: в редуцирующих условиях - 12.5% ПААГ, в нередуцирующих условиях - 8% ПААГ.
[00391] Собирали и устанавливали электрофорезную камеру в соответствии с инструкцией по эксплуатации прибора для проведения вертикального электрофореза. Пробы готовили, разводя образцы очищенной водой до конечной концентрации 1 мг/мл. Отбирали объем, эквивалентный 40 мкг и смешивали подготовленные пробы исследуемого образца в соотношении 3:1 (объем/объем) с буферным раствором для нанесения образцов 4-кратным, содержащим 2-меркаптоэтанол (редуцирующие условия) и не содержащим 2-меркаптоэтанол (нередуцирующие условия), перемешивали. Полученные растворы инкубировали при температуре (99±1)°С в течение 3 мин (образцы, содержащие 2-меркаптоэтанол) и при температуре (99±1)°С в течение 1 мин (образцы, не содержащие 2-меркаптоэтанол). Растворы охлаждали до комнатной температуры, перемешивали и наносили в лунки ПААГ под слой электродного буферного раствора.
[00392] Электрофорез проводили в режиме постоянного тока, используя систему водяного охлаждения. Задавали параметры работы источника питания: при прохождении фронта красителя через концентрирующий гель напряжение тока составляло 110 В. После вхождения фронта красителя в нижний разделяющий гель на 5-7 мм напряжение тока увеличивали до 180 В. Источник питания отключали, когда фронт красителя достиг нижней границы геля.
[00393] После окончания электрофореза гели отделяли от стекол и проводили фиксацию белков в фиксирующем растворе в течение 16-18 часов при комнатной температуре. Затем проводили окрашивание гелей (в растворе кислотном синем 83) и отмывку до получения четкой визуализации полос. Гели сканировали. Чистоту и примеси в испытуемых образцах оценивали с помощью программного обеспечения GelPro.
[00394] Пример 12. Определение относительной специфической активности
[00395] Относительную специфическую активность образцов моноклонального антитела против PD-1 оценивали по способности специфично связываться с белком PD-1 на поверхности мембраны клеток линии Jurkat PD-1 NFAT. За день до проведения анализа иммобилизировали PDL-1 в лунках культуральных планшетов. На следующий день промывали планшеты, вносили по 50 мкл/лунку раствора фитогемагглютинина-П (ПанЭко, Россия, кат. № М021). При помощи роботизированной станции Freedom Evo готовили серийные разведения стандартного и исследуемого образцов и вносили в культуральные планшеты по 10 мкл/лунку. Вносили в культуральные планшеты по 40 мкл/лунку клеточной суспензии Jurkat PD-1 NFAT. Инкубировали планшеты 4-6 часов при 37°С, 5% СО2. Все описанные выше процедуры проводили в асептических условиях.
[00396] По завершении инкубации добавляли 100 мкл/лунку раствора BioGlo (Promega, США, кат. № G7941) и определяли уровень люминесценции.
[00397] С помощью программного обеспечения Magellan строили четырехпараметровые кривые зависимости среднего значения люминесценции от концентрации белка для растворов стандартного образца и испытуемого образцов, расположенных на одном планшете.
[00398] Относительную специфическую активность испытуемого образца в процентах (RP) рассчитывали по формуле: 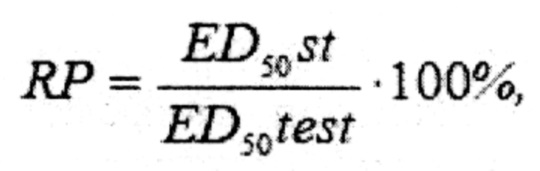 , где
, где
ED50st - значение половинной эффективной дозы стандартного образца, нг/мл;
ED50test - значение половинной эффективной дозы испытуемого образца, нг/мл.
[00399] За конечный результат принимали среднее значение относительной специфической активности, рассчитанное по трем независимым определениям (определенное на 3-х разных культуральных планшетах).
[00400] Пример 13. Определение вязкости
Динамическую вязкость исследуемых растворов определяли методом ротационной вискозиметрии на вискозиметре Брукфильда CAP2000+L.
[00401] Пример 14. Исследования природы буферного раствора
[00402] Исследуемые составы
Для настоящего исследования выбраны четыре буферных раствора. При выборе молярности и рН буфера учтены ограничения при возможном подкожном введении, а также pI антитела (рН исследуемых буферных растворов выбраны в минимальном возможном физиологичном значении).
Состав (на 1 мл):

[00403] Определение коллоидной стабильности методом ПЭГ-агрегации.
Исследование проводили в трех повторностях для каждого образа. Результаты представлены в таблице 1 и на Фиг. 1.
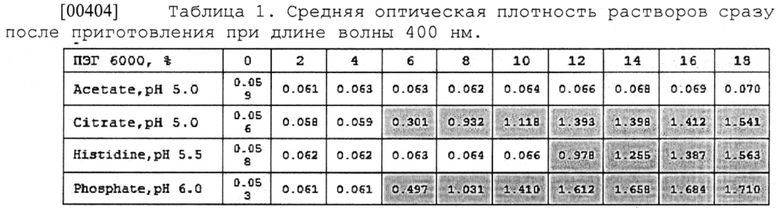
[00405] В результате исследования наиболее высокую коллоидную стабильность в присутствии ПЭГ показали образцы в гистидиновом и ацетатном буферных растворах. Композиции на основе цитрата и фосфата были исключены из дальнейшего исследования, так как агрегация при 6% ПЭГ является показателем неудовлетворительной коллоидной стабильности. На основании полученных результатов для дальнейшего выбора рН и молярности буферного раствора выбраны ацетатный и гистидиновый буферные растворы.
[00406] Пример 15. Выбор рН и буферной емкости раствора
[00407] Исследуемые составы (на 1 мл)
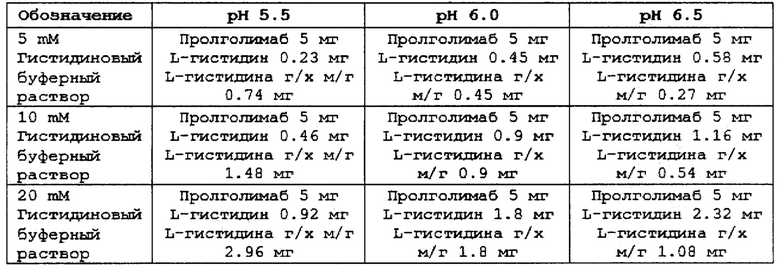

[00408] Исследование проводили методом термостресс при 50°С в течение 72 часов. Результаты представлены в таблице 2. Гомогенность образцов анализировали методом Э ВЭЖХ и электрофореза на приборе Labchip.Профиль заряженных форм образцов анализировали на приборе Labchip.
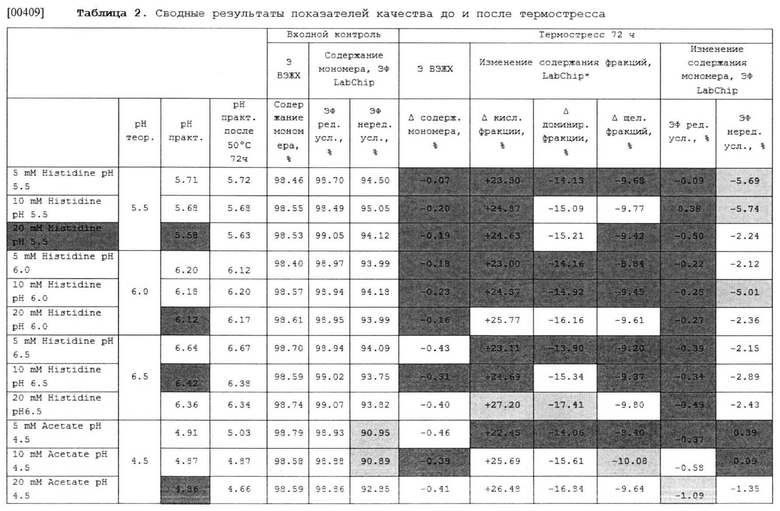
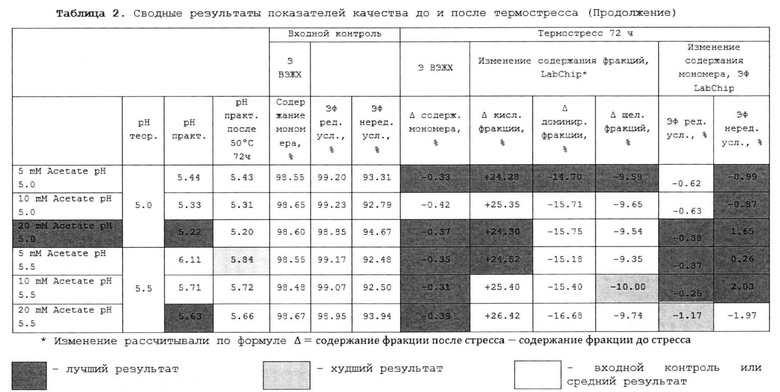
[00410] В результате исследования показано, что присутствие моноклонального антитела против PD-1 (пролголимаб) в водном растворе приводит к увеличению уровня рН. Наибольшая стабилизация рН обеспечивается 20 мМ буферными растворами.
[00411] Все исследуемые образцы продемонстрировали высокую агрегационную стабильность на Э ВЭЖХ, так как прирост примесей в ходе термостресса составил не более 0.5% для всех образцов. Наилучшую стабильность показали образцы на основе гистидина с рН 5.5-6.0, а также на основе ацетата с рН 5.0-5.5.
[00412] Все составы продемонстрировали схожую стабильность профиля заряженных форм, количественная разница изменения содержания фракций между составами находится в пределах погрешности метода.
[00413] Согласно результатам гель-электрофореза в редуцирующих условиях, более высокую стабильность продемонстрировали все составы на основе гистидина, в то время как в нередуцирующих условиях лучшие показатели были получены для ацетатных буферных растворов.
[00414] На основании полученных данных стабильности по показателям рН, чистота и профиль заряженных форм моноклонального антитела против PD-1 для дальнейшей работы рекомендованы следующие составы вспомогательных веществ:

[00415] Пример 16. Выбор фармацевтической композиции на основе гистидина
[00416] Исследуемые составы
Для скрининга стабильной фармацевтической композиции на основе 20 мМ гистидинового буферного раствора с рН 5.5 были взяты следующие вспомогательные вещества: маннитол, трегалозы дигидрат, сахароза (осмотические агенты). Все исследуемые растворы являются изотоническими.
[00417] Исследуемые составы (мг до 1 мл)
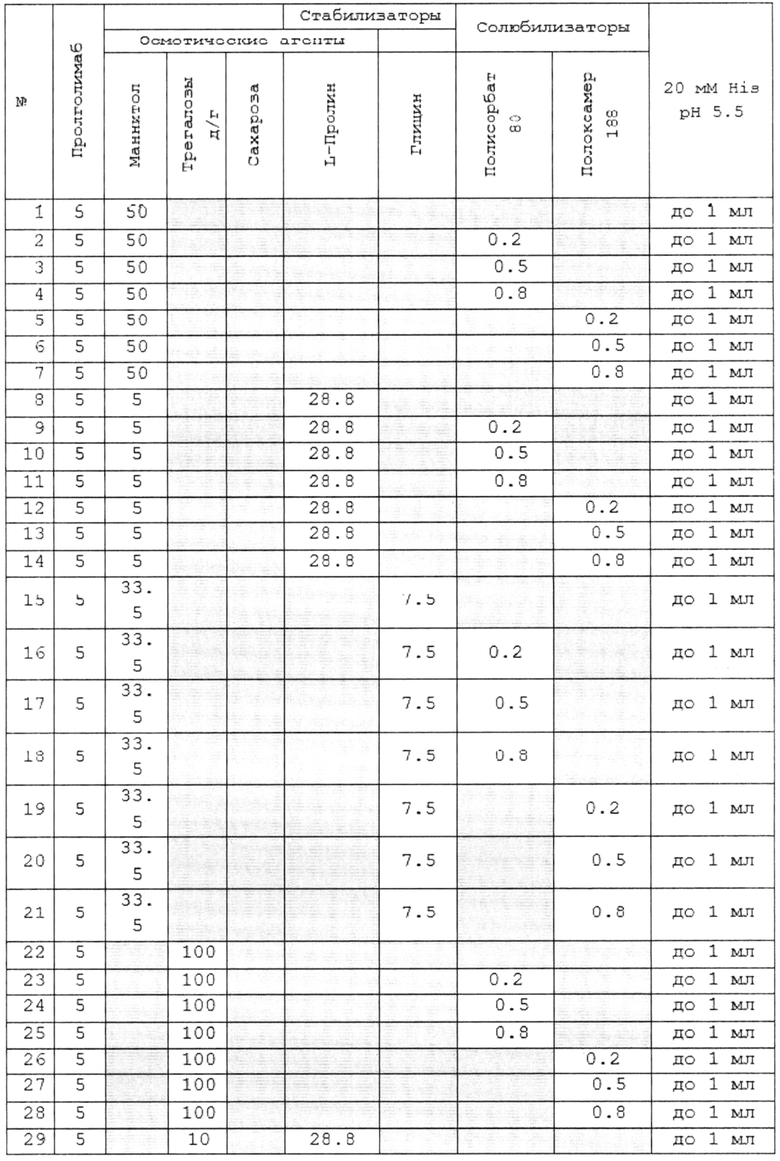
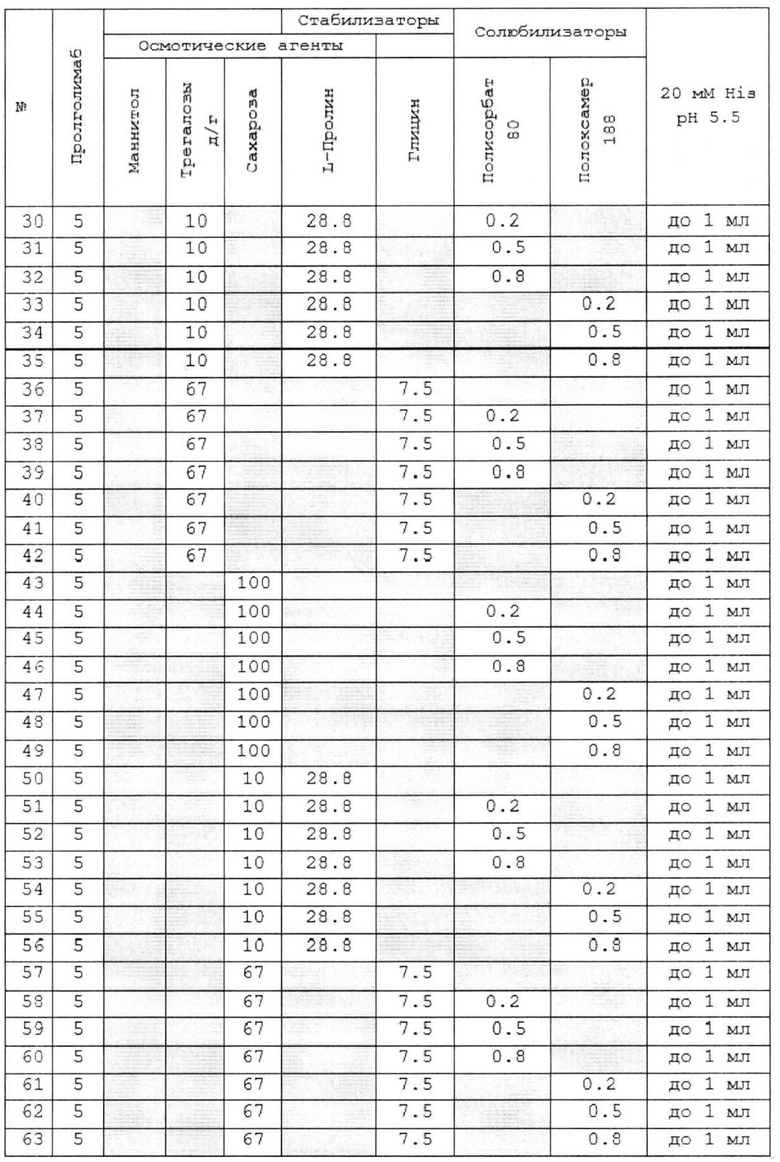
[00418]
Исследование стабильности образцов проводили методом термостресс при 50°С в течение 72 часов, шейк-тест в течение 120 ч при скорости 800 об/мин, а также однократного замораживания при температуре минус 16-20°С и размораживания при температуре +25±1°С. Мутность растворов оценивали методом спектрофотометрии при 400 нм. Гомогенность образцов анализировали методом Э ВЭЖХ и электрофореза на приборе Labchip. Профиль заряженных форм образцов анализировали на приборе Labchip.
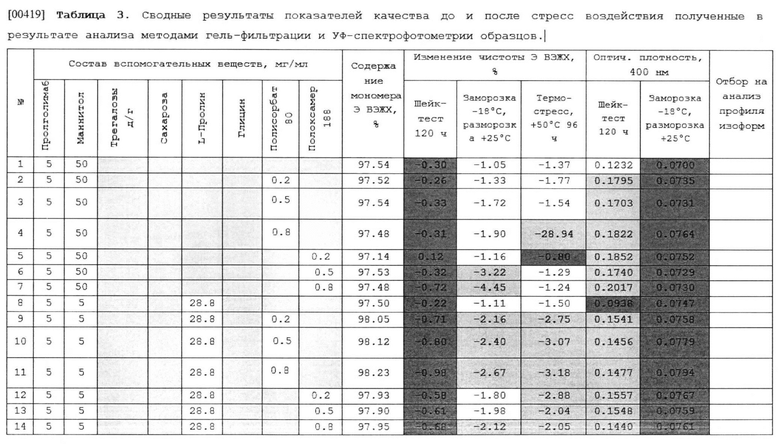
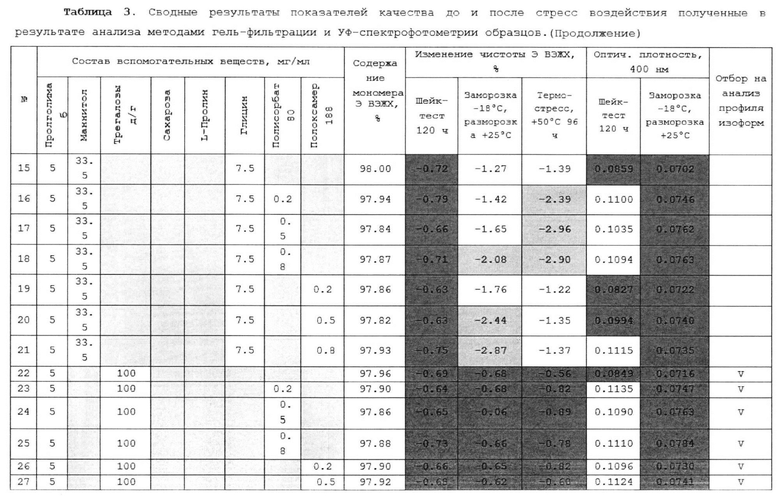
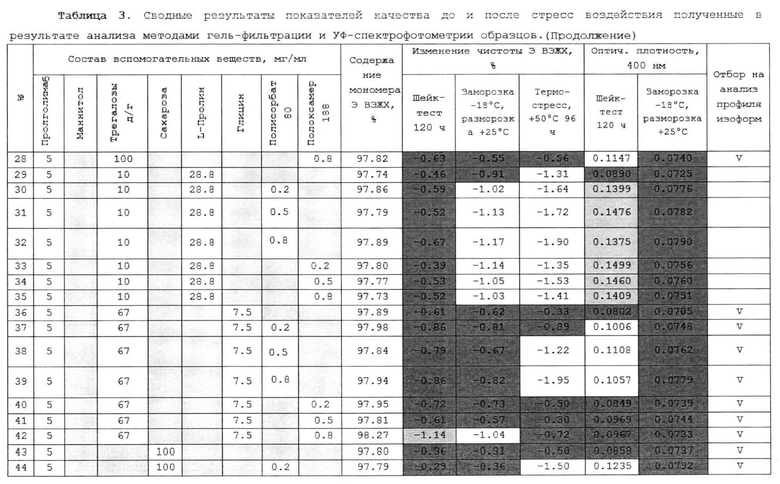
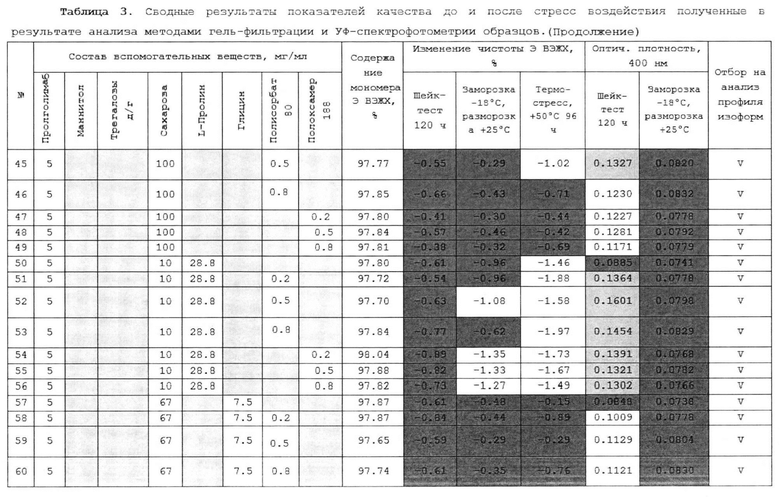
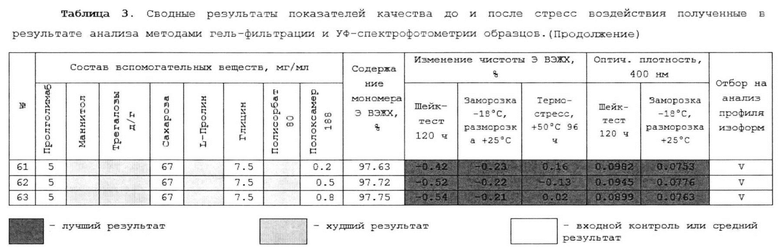
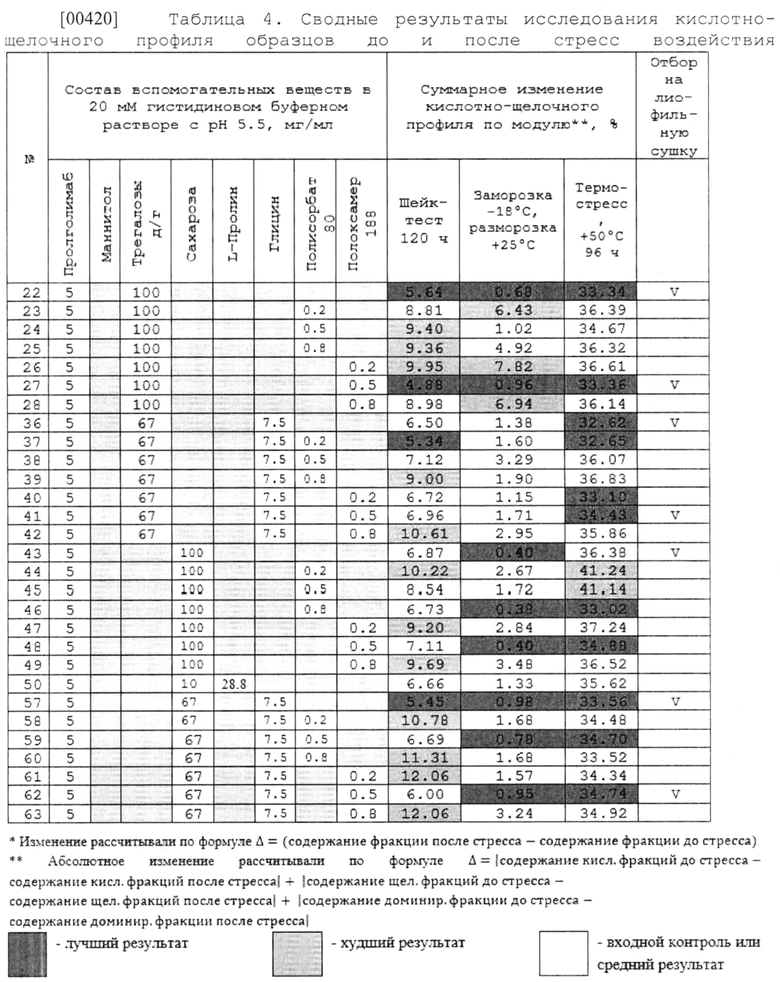
[00421] В таблице 3 показано негативное влияние маннитола на
термическую и коллоидную стабильность моноклонального антитела против PD-1 в 20 мМ гистидиновом буферном растворе с рН 5.5: в ходе термостресса в течение 96 ч прирост примесей на Э ВЭЖХ составил от 1.24 до 3.18% в ходе шейк-теста в течение 120 ч растворы приобрели видимую агрегацию. Составы с маннитолом показали также отрицательные результаты стабильности в ходе криоконцентрирования: после одного цикла заморозки-разморозки прирост примесей на Э ВЭЖХ составил от 1.05 до 4.45%, что гораздо выше данного показателя для других составов.
[00422] Составы, содержащие L-пролин, также продемонстрировали низкую коллоидную стабильность вследствие образования видимой агрегации в процессе шейк-теста. После тестов заморозки-разморозки и термостресса прирост примесей составов с L-пролином составил в среднем более 1% на Э ВЭЖХ.
[00423] В ходе эксперимента выявлена высокая термическая и коллоидная стабильность пролголимаба (отсутствие значимых изменений показателей качества на всех стресс-воздействиях) в составах с трегалозы дигидратом без наличия солюбилизаторов, таких как полисорбат 80 и полоксамер 188.
[00424] Наибольшим стабилизирующим эффектом на моноклональное антитело против PD-1 в 20 мМ гистидиновом буферном растворе обладают: трегалозы дигидрат, сахароза, а также их комбинации с глицином. Эти составы могут быль использованы для лиофильной лекарственной формы моноклонального антитела.
[00425] Оценка стабильности перспективных составов на основе гистидина при лиофильной сушке
[00426] Образцы, продемонстрировавшие наилучшую стабильность на предыдущем этапе скрининга, были исследованы на возможность получения лиофилизата для приготовления раствора для инфузий.
[00427] Для этого растворы, содержащие 20 мг/мл или 100 мг/мл моноклонального антитела к PD-1 пролголимаба, разливали в стеклянные флаконы I гидролитического класса, флаконы неплотно укупоривали резиновой пробкой с прорезью. Флаконы с раствором помещали в камеру лиофильной сушки. Проводили лиофильную сушку в автоматическом режиме. Стадию заморозки проводили при температуре минус 40°С, первичную сушку проводили при давлении (0,10±0,03) мбар, во время вторичной сушки температуру повышали, давление составляло (0,05±0,02) мбар. После завершения процесса сушки давление сбрасывали до необходимого значения вакууму 0.76±0,03 мбар, проводили укупорку флаконов резиновыми пробками и далее сбрасывали вакуум до атмосферного давления. Укупоренные флаконы передавали на дальнейшие исследования. Лиофилизированные образцы моноклонального антитела к PD-1 пролголимаба хранили при температуре 2-8°С.
[00428] Для подтверждения стабильности белка после лиофилизации образцы восстанавливали. Содержание мономера оценивали методом Э ВЭЖХ, профиль заряженных форм анализировали на приборе Labchip. Также определяли значение рН до и после лиофилизации. Результаты представлены в таблице 5.
[00429] При лиофильной сушке отобранных составов на основе гистидина и после восстановления лиофилизатов наблюдали незначительный сдвиг рН, находящийся в пределах погрешности метода. Все составы продемонстрировали высокую стабильность по чистоте на Э ВЭЖХ и профилю заряженных форм после восстановления. Однако ощутимое различие чистоты образцов было продемонстрировано методом гель-электрофорез в системе LabChip Caliper. Наиболее стабильными образцами по этому показателю являются составы №22, 27, 36, содержащие трегалозы дигидрат. Составы в L-пролином также показали значимое изменение профиля заряженных форм, поэтому не рекомендованы для использования. Была продемонстрирована возможность лиофилизации составов №22 и 27 с концентрацией белка 100 мг/мл.
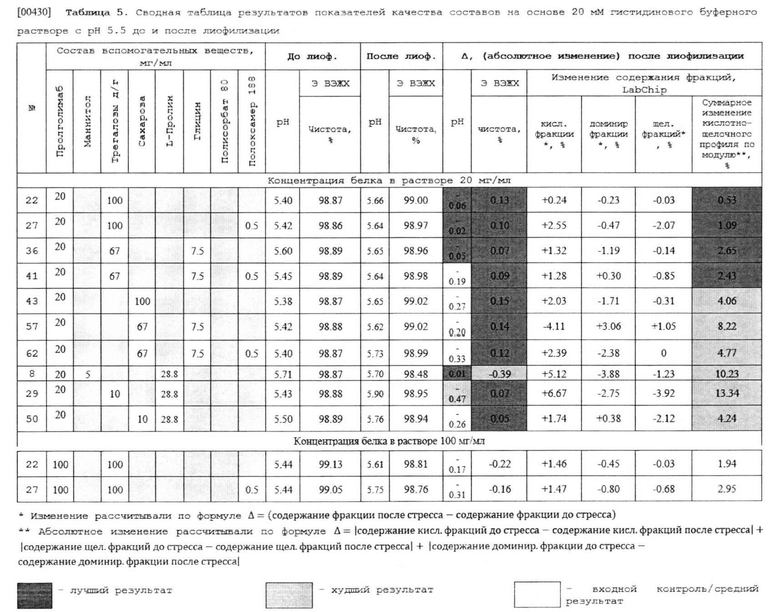
[00431] Получение высококонцентрированной формы и подтверждение стабильности при ускоренном хранении
[00432] На основании результатов скрининга в жидкой и лиофильной лекарственных форм для ускоренного исследования стабильности выбраны следующие составы:

[00433] Содержание трегалозы дигидрата было снижено для выравнивания уровня осмоляльности растворов с повышенной концентрацией моноклонального антитела против PD-1 до физиологического уровня (около 300 мОсм/кг), а также для снижения вязкости растворов до уровня менее 100 сР. Стабильность образцов подтверждали ускоренным хранением при +37°С, анализ проводили методами Э ВЭЖХ, ИО ВЭЖХ и ВЭФ, а также определяли относительную специфическую активность пролголимаба. Результаты представлены в таблицах 6 и 7.
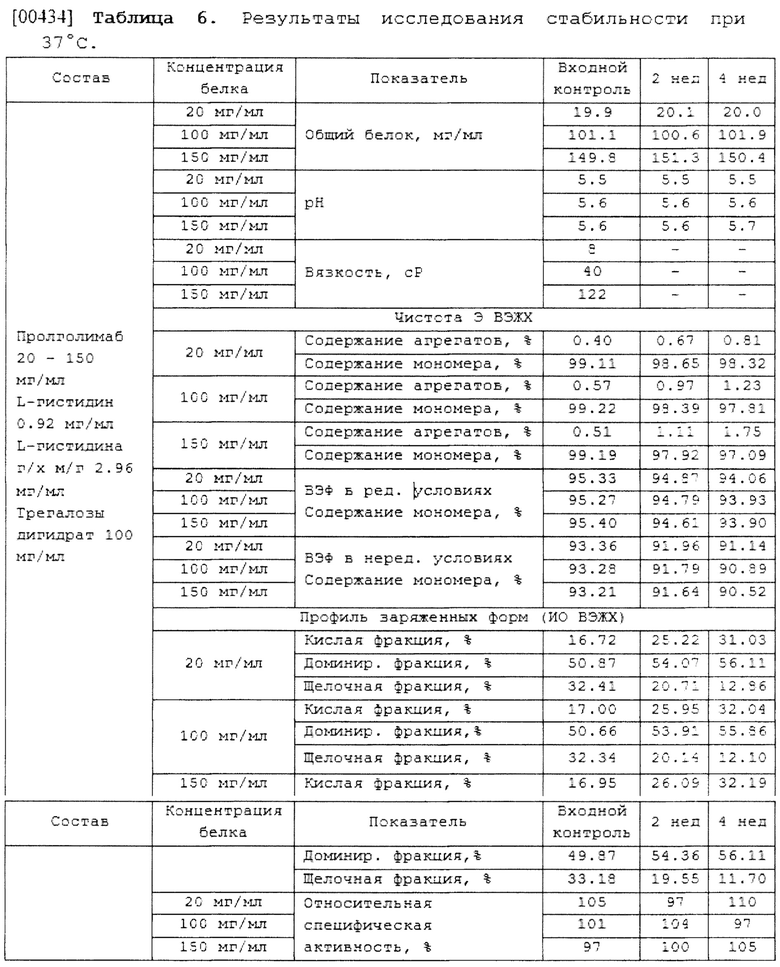
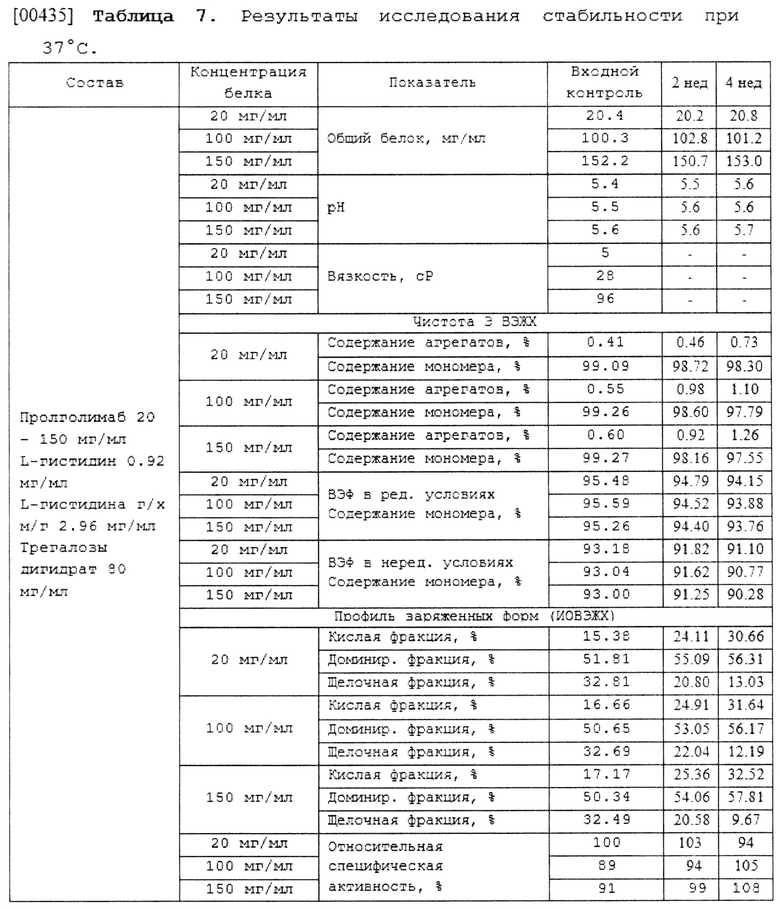
[00436] Пример 17. Выбор фармацевтической композиции на основе ацетата
[00437] Исследуемые составы
Для скрининга стабильной фармацевтической композиции на основе 20 мМ ацетатного буферного раствора с рН 5.0 были взяты следующие вспомогательные вещества: маннитол, трегалозы дигидрат, сахароза (осмотические агенты), L-пролин (осмотический агент и стабилизатор), глицин (осмотический агент и стабилизатор), полисорбат 80 и полоксамер Р18 8 (солюбилизаторы). Все исследуемые растворы являются изотоническими.
[00438] Исследуемые составы (на 1 мл).
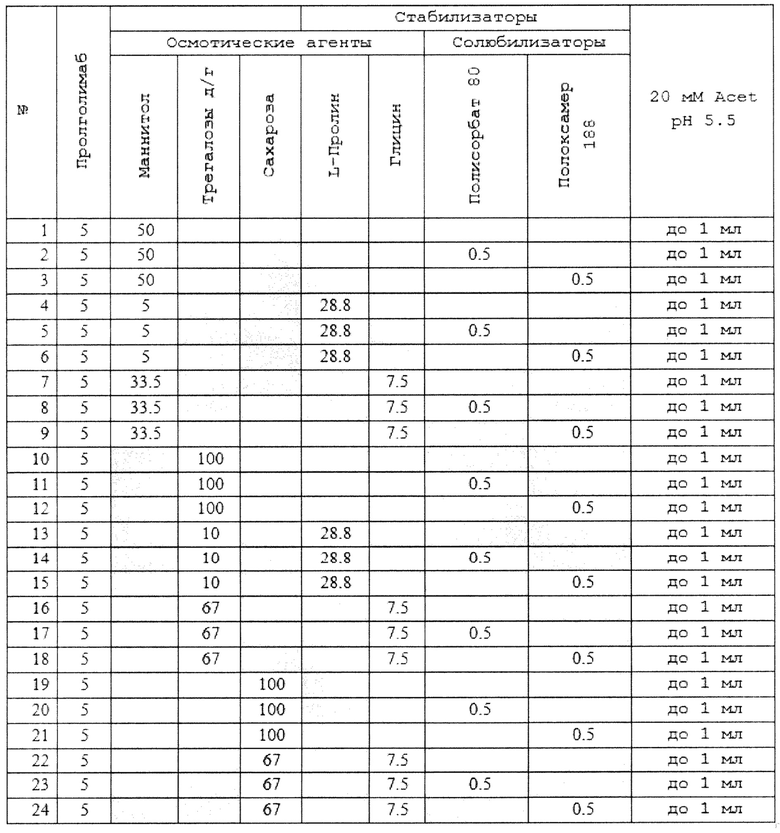
[00439] Исследование стабильности образцов проводили методом термостресс при 50°С в течение 72 часов, шейк-тест в течение 120 ч при скорости 800 об/мин, а также однократного замораживания минус 16 - 20°С и размораживания при температуре +25±1°С. Мутность растворов оценивали методом спектрофотометрии при 400 нм. Гомогенность образцов анализировали методом Э ВЭЖХ и электрофореза на приборе Labchip. Профиль заряженных форм образцов анализировали на приборе Labchip.
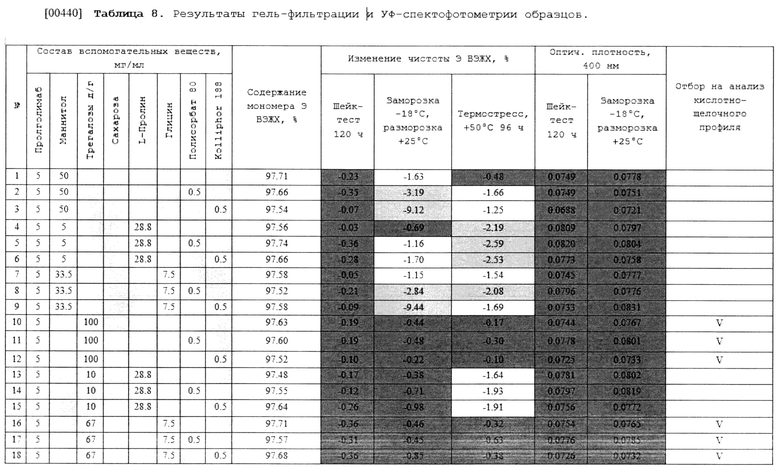
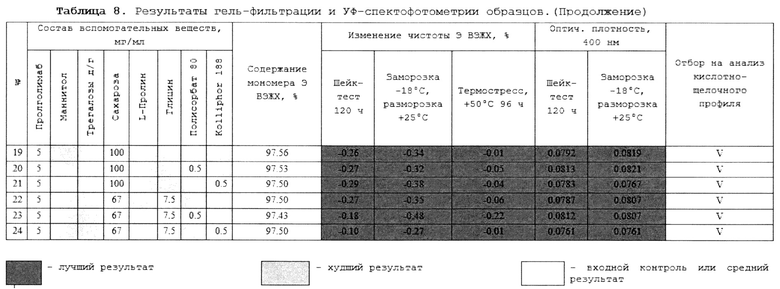
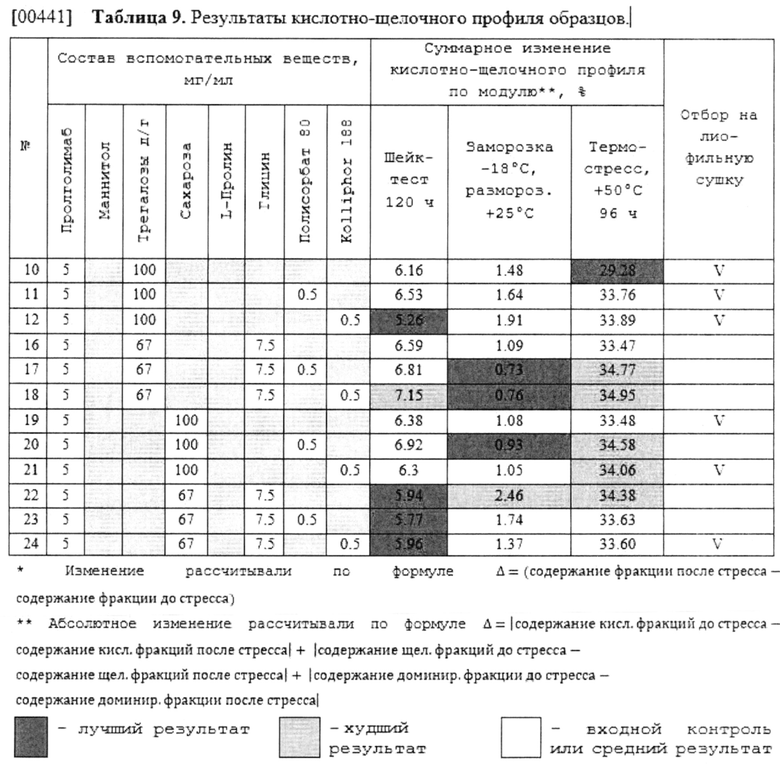
[00442] В результате работы показано негативное влияние маннитола на термическую и коллоидную стабильность моноклонального антитела против PD-1 в 20 мМ ацетатном буферном растворе с рН 5.0: в ходе термостресса в течение 96 ч прирост примесей на Э ВЭЖХ составил от 0.48 до 2.59%, в ходе шейк-теста в течение 120 ч все исследуемые растворы не приобрели видимой агрегации (см. результаты УФ-спектрофотометрии). Составы с маннитолом показали также отрицательные результаты стабильности в ходе криоконцентрирования: после одного цикла заморозки-разморозки прирост примесей на Э ВЭЖХ составил от 0.69 до 9.44%, что гораздо выше данного показателя для других составов.
[00443] Составы, содержащие L-пролин, также продемонстрировали низкую термическую стабильность по показателю чистота при исследовании методом Э ВЭЖХ: прирост примесей через 96 ч стресса составил 1.64-1.93%. После заморозки-разморозки прирост примесей составов с L-пролином не превысил среднее значение наиболее стабильных составов в группе. В ходе эксперимента не было выявлено влияния солюбилизаторов на термическую или коллоидную стабильность белка. Пролголимаб в составах с трегалозы дигидратом проявил высокую стабильность на всех стресс-воздействиях в отсутствии поверхностно-активных веществ, таких как полисорбат 80 и полоксамер 188.
[00444] Наибольшим стабилизирующим эффектом на моноклональное антитело против PD-1 в 20 мМ ацетатном буферном растворе обладают: трегалозы дигидрат, сахароза, а также их комбинации с глицином. Эти составы могут быть использованы для получения лиофильной лекарственной формы моноклонального антитела.
[00445] Определение стабильности перспективных составов на основе ацетата при лиофильной сушке
[00446] Образцы, продемонстрировавшие наилучшую стабильность на предыдущем этапе скрининга, были исследованы на возможность получения лиофилизата для приготовления раствора для инфузий.
[00447] Для этого растворы, содержащие 20 мг/мл или 100 мг/мл моноклонального антитела против PD-1, разливали в стеклянные флаконы I гидролитического класса, флаконы неплотно укупоривали резиновой пробкой с прорезью. Флаконы с раствором помещали в камеру лиофильной сушки. Проводили лиофильную сушку в автоматическом режиме. Стадию заморозки проводили при температуре минус 40°С, первичную сушку проводили при давлении (0,10±0,03) мбар, во время вторичной сушки температуру повышали, давление составляло (0,05±0,02) мбар. После завершения процесса сушки давление сбрасывали до необходимого значения вакууму 0.76±0,03 мбар, проводили укупорку флаконов резиновыми пробками и далее сбрасывали вакуум до атмосферного давления. Укупоренные флаконы передавали на дальнейшие исследования. Лиофилизированные образцы моноклонального антитела против PD-1 хранили при температуре 2-8°С.
[00448] Для подтверждения стабильности белка после лиофилизации образцы восстанавливали. Содержание мономера оценивали методом Э ВЭЖХ, профиль заряженных форм анализировали на приборе Labchip. Также определяли значение рН до и после лиофилизации. Результаты представлены в таблице 8.
[00449] При лиофильной сушке отобранных составов на основе ацетата и после восстановления лиофилизатов наблюдается значительный сдвиг рН в диапазоне 0.26-0.45. Данный сдвиг является может быть обусловлен потерей ацетат-ионов в процессе сушки. Таким образом, составы на основе ацетата могут быть рекомендованы к лиофильной сушке при условии учета данного явления.
[00450] Все составы продемонстрировали высокую стабильность по чистоте на Э ВЭЖХ и профилю заряженных форм после восстановления. В ходе работы была продемонстрирована возможность лиофилизации составов №10 и 12 с концентрацией белка 100 мг/мл.
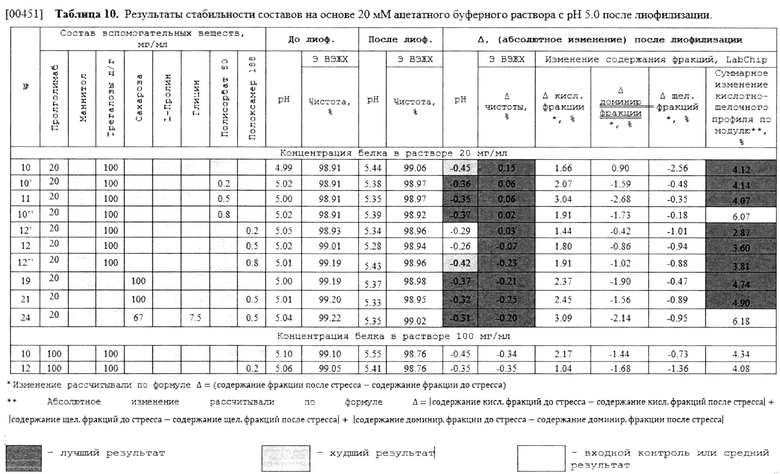
[00452] Получение высококонцентрированной формы и подтверждение стабильности при ускоренном хранении
[00453] На основании результатов скрининга в жидкой и лиофильной лекарственных форм для ускоренного исследования стабильности выбраны следующие составы:

[00454] Содержание трегалозы дигидрата было снижено для выравнивания уровня осмоляльности растворов с повышенной концентрацией моноклонального антитела против PD-1 до физиологического уровня (около 300 мОсм/кг), а также снижения вязкости растворов до уровня менее 100 сР. Образцы закладывали на хранение при +37°С и анализировали методами Э ВЭЖХ, ИО ВЭЖХ и ВЭФ, а также определяли специфическую активность белка. Результаты представлены в таблицах 9 и 10.
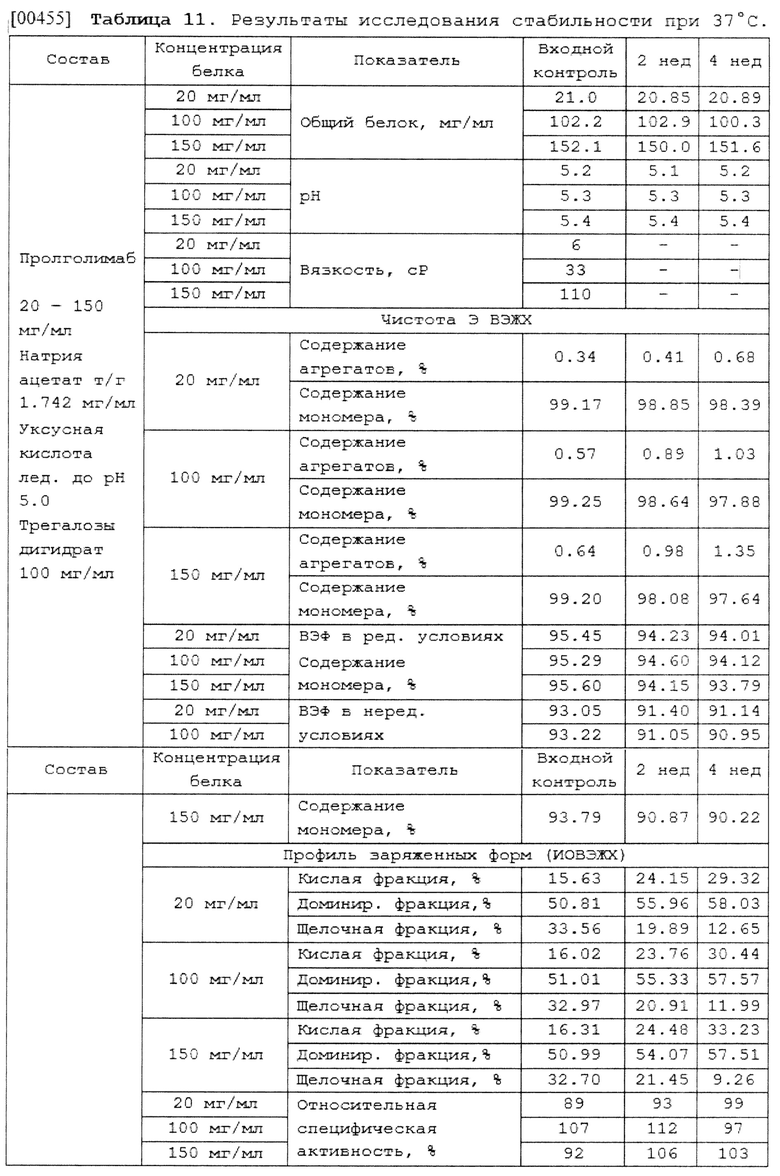
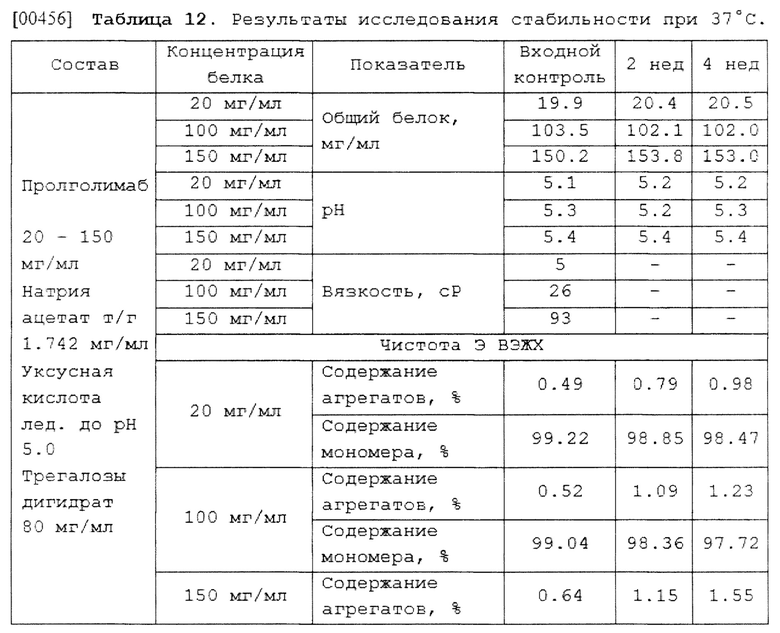
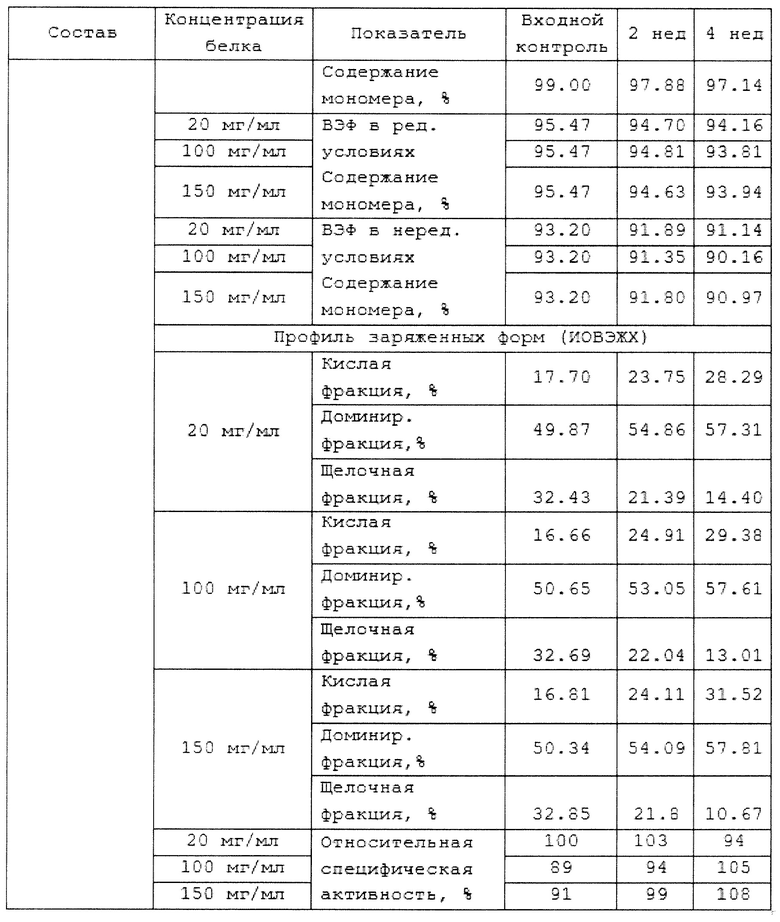
[00457] При том, что изобретение было описано со ссылкой на предпочтительные варианты осуществления, следует понимать, что можно прибегнуть к изменениям и модификациям, как очевидно для специалистов в данной области техники. Подобные изменения и модификации следует рассматривать в рамках и объеме притязаний настоящего изобретения.
[00458] Выше были подробно описаны иллюстративные, неограничивающие примеры осуществления настоящего изобретения. Данное детальное описание приведено просто для того, чтобы ознакомить специалиста в данной области техники с дополнительными деталями практической реализации предпочтительных аспектов настоящих идей, и не ограничивает объема данного изобретения. Кроме того, каждый из дополнительных признаков и идей, раскрытых выше по тексту и ниже по тексту, могут быть использованы по отдельности или в совокупности с другими признаками или идеями.
[00459] Более того, комбинации отличительных признаков и шагов, раскрытых в нижеприведенном детальном описании, также, как и эксперементальные примеры, могут быть необязательными для практической реализации изобретения в самом широком смысле, и приведены только для того, чтобы подробно описать конкретные примеры для настоящего изобретения. Более того, отличительные признаки описанных выше конкретных примеров и независимые и зависимые пункты формулы изобретения, приведенные в данном изобретении, можно комбинировать способами, которые конкретно и непосредственно не указаны, с целью реализации дополнительных целесообразных вариантов осуществления настоящего изобретения.
[00460] Были проведены дополнительные примеры исследований с использованием различных водных фармацевтических композиций, содержащих антитело к PD-1 пролголимаб, для лечения злокачественных новобразований, таких как меланома, в том числе неоперабельная или метастатическая меланома, ранние стадии меланомы до и после радикального лечения, рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого. Типовые фармацевтические композиции, используемые для данных исследований, охарактеризованы следующими составами ниже (таблица 12.1).

[00461] Основные сведения о клинической разработке исследуемого препарата
Фармакокинетика, фармакодинамика, безопасность и иммуногенность BCD-100 (пролголимаб) в монотерапии при внутривенном введении изучены в клиническом исследовании фазы I с эскалацией дозы. В данном исследовании были продемонстрированы безопасность и преимущества исследуемого продукта у пациентов с распространенными формами злокачественных новообразований различных локализаций (меланома, НМРЛ,) и по результатам исследования два режима введения BCD-100 выбраны для дальнейшей клинической разработки, 3 мг/кг в/в 1 раз в 3 недели и 1 мг/кг в/в 1 раз в 2 недели.
[00462] Целью исследования № BCD-100-2/MIRACULUM применения BCD-100 у пациентов с нерезектабельной или метастатической меланомой была оценка фармакокинетики, эффективности, безопасности и иммуногенности в монотерапии в двух режимах введения: 3 мг/кг в/в 1 раз в 3 недели и 1 мг/кг в/в 1 раз в 2 недели. Первичная конечная точка (ОЧО) оценивалась по результатам промежуточного анализа через 6 месяцев. BCD-100 продемонстрировал благоприятный профиль безопасности на всех дозовых режимах. Оба дозовых режима продемонстрировали эффективность около 60% КЗ и 30% ОЧО у пациентов с нерезектабельной или метастатической меланомой.
[00463] Пример 18. Применение пролголимаба (BCD-100) для терапии пациентов с распространенными формами злокачественных новообразований различных локализаций, исследование I фазы, BCD-100-1
[00464] Дизайн исследования
В исследовании № BCD-100-1 были изучены фармакокинетика и переносимость препарата BCD-100 (пролголимаба). Исследование № BCD-100-1 (NCT03050047) являлось многоцентровым открытым исследованием I фазы у пациентов с солидными опухолями, проведенным на территории Российской Федерации. Первичной задачей исследования была оценка фармакокинетики и клинических фармакологических параметров BCD-100. Основные характеристики исследования BCD-100-1 приведены в таблице 13.
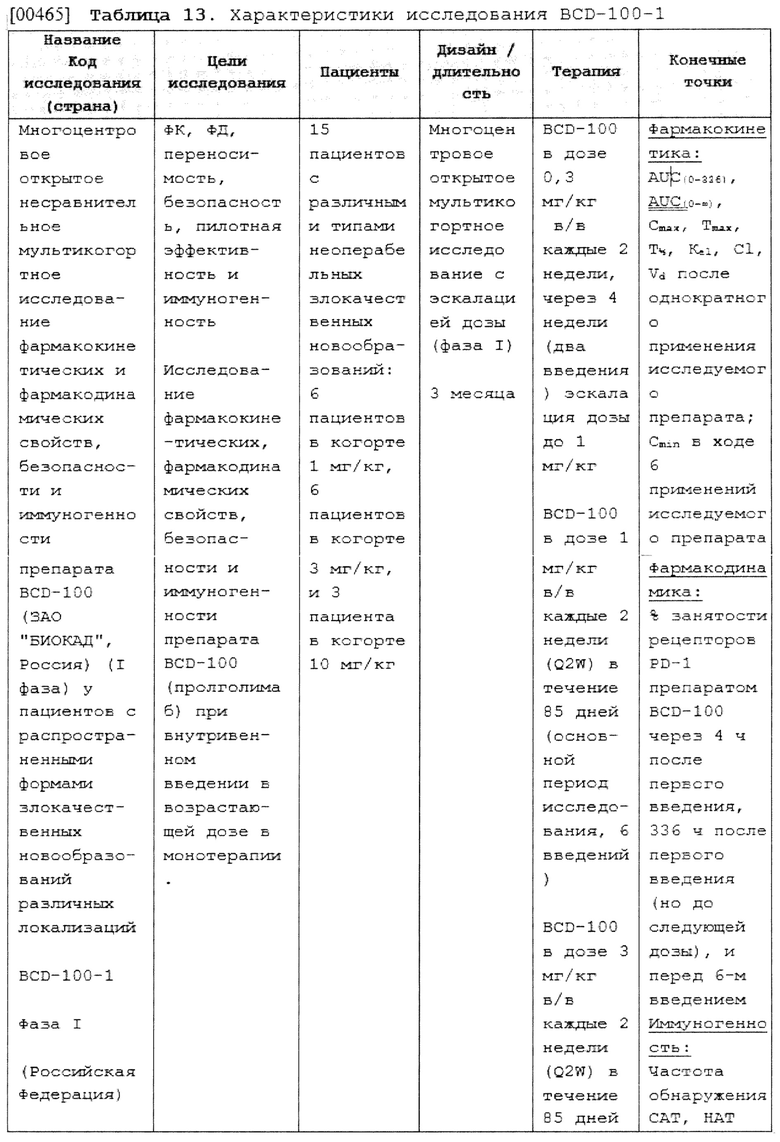
[00466] Первому пациенту назначалась стартовая доза препарата BCD-100 - 0,3 мг/кг каждые две недели. В случае отсутствия явлений дозолимитирующей токсичности в течение 4-х недель, пациенту проводилось увеличение дозы до 1 мг/кг раз в две недели. После первого пациента исследование продолжалось по классическому дизайну с эскалацией дозы 3+3, т.е. при отсутствии ДЛТ каждые 4 недели в исследование последовательно включалась когорта из 3-х пациентов на следующем дозовом уровне. На каждом дозовом уровне BCD-100 назначался на 85 дней (около 3 месяцев) либо до появления признаков ДЛТ или прогрессирования заболевания.
[00467] Если явления ДЛТ на текущем дозовом уровне отмечались у одного пациента, следующая когорта из трех пациентов получала препарат на этом же уровне, то есть всего шесть пациентов начинало получать данную дозу BCD-100. Если явления ДЛТ отмечались у двух и более пациентов на одном уровне дозы, набор дозы и назначение препарата новым пациентам прекращались.
[00468] Длительность последующего наблюдения за пациентами после терапии препаратом BCD-100 составляла 126 дней. На протяжении этого периода производился мониторинг нежелательных явлений, медицинские осмотры, осуществлялись анализы крови (1 раз в 2 недели - первые 28 дней, далее 1 раз в 28 дней).
[00469] Результаты исследования
Сводная информация о характеристиках популяции пациентов
В исследование были включены 15 пациентов с распространенными формами злокачественных солидных новообразований различных локализаций (меланома, включая меланому хориоидеи, НМРЛ, почечно-клеточный рак, мезотелиома), в возрасте 18 лет и старше, обоих полов. Для включения в исследование пациенты должны были соответствовать 0-2 баллам по шкале ECOG и иметь как минимум один измеряемый целевой очаг согласно RECIST 1.1 (не считая костных метастазов). Описание характеристик заболевания у пациентов и распределение пациентов представлено в таблиц 14, таблице 15.
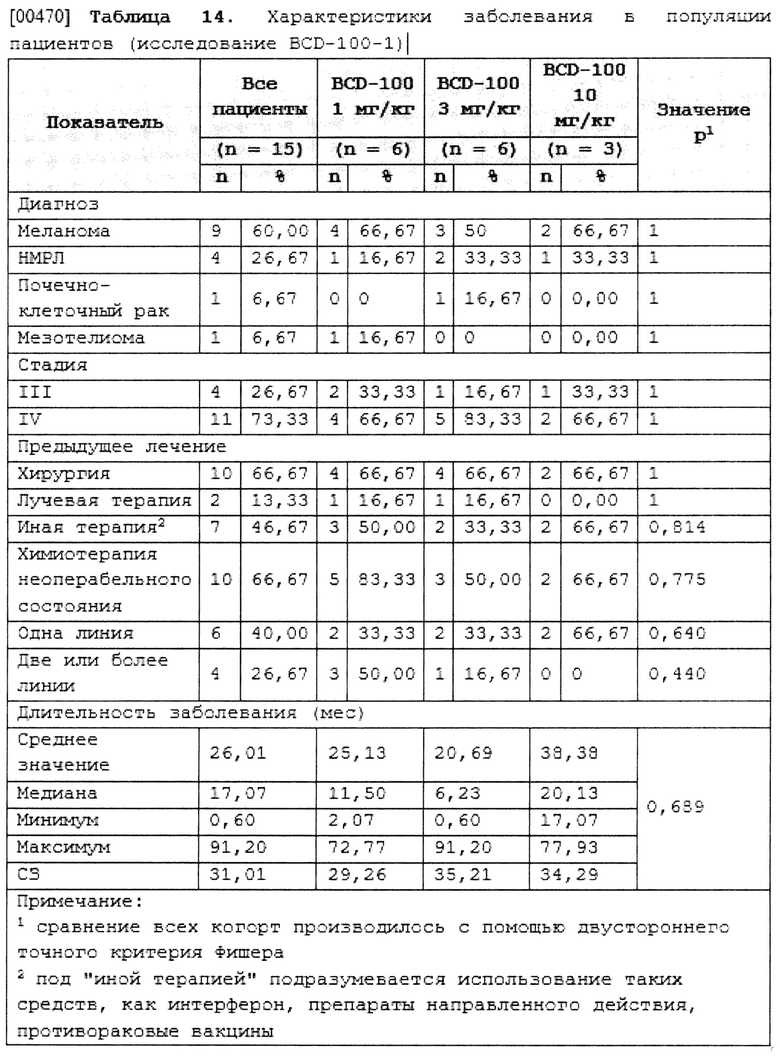
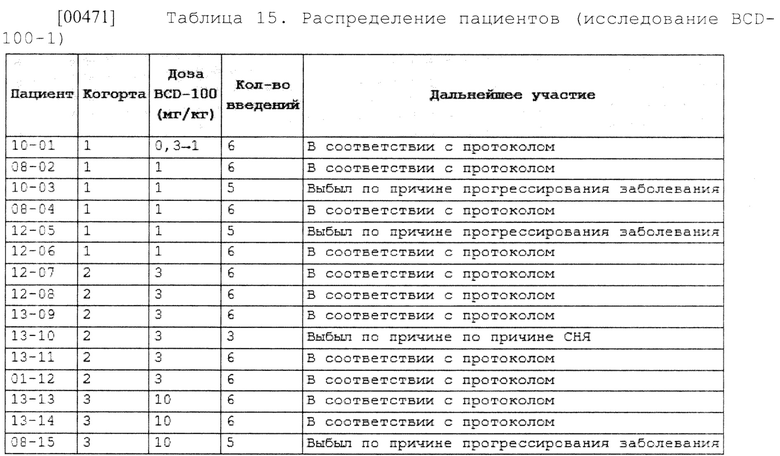
[00472] В итоговый анализ были включены данные 15 пациентов (6 пациентов из когорты BCD-100 1 мг/кг, включая 1 пациента после эскалации дозы, 6 пациентов из когорты BCD-100 3 мг/кг и 3 пациента из когорты BCD-100 10 мг/кг). Все пациенты получили терапию в рамках основного этапа исследования (85 дней).
[00473] Все когорты пациентов были сбалансированы по основным демографическим характеристикам. Распределение всех пациентов по половой принадлежности было равномерным: женщины - 53,33% (8 пациентов), мужчины - 46,67% (7 пациентов). Медиана возраста пациентов составила 56 лет (минимум - 35 лет, максимум - 77 лет).
[00474] По характеристикам основного заболевания в популяции преобладали пациенты с меланомой (9/15 пациентов). Популяция также включала 4 пациентов с НМРЛ, 1 пациента с мезотелиомой плевры и 1 больного почечно-клеточным раком. Длительность заболевания на момент скрининга во всей популяции составила (медиана) 17,07 месяцев, минимум - 0,6 месяца, максимум - 91,2 месяца.
[00475] Сводная информация о клинической безопасности, полученная в ходе I фазы исследования
Безопасность была исследована на популяции, в которую были включены все пациенты, получившие как минимум одну дозу исследуемого препарата (n=15). У каждого из 15 пациентов были отмечены НЯ и/или СНЯ, общее число явлений составило 247. Статистический анализ не выявил различий между когортами по количеству НЯ (р=0,567, критерий Краскела - Уоллиса). Сводная таблица 16 с данными по безопасности препарата приведена ниже.
[00476] НЯ, соответствующие 3-й степени тяжести по шкале СТСАЕ 4.03, были отмечены у 40,00% (6 из 15) пациентов - у двух пациентов в каждой когорте. У 4 из 6 данных пациентов НЯ со степенью тяжести 3 и выше были сочтены исследователем связанными с терапией: у пациента, получавшего BCD-100 в дозировке 1 мг/кг, у пациента, получавшего BCD-100 в дозировке 3 мг/кг, и у двух пациентов, получавших BCD-100 в дозировке 10 мг/кг. Многие из этих нежелательных явлений носили гематологический характер.
[00477] СНЯ были отмечены у трех пациентов, получавших BCD-100 в дозировке 1 мг/кг, и у одного пациента, получавшего 3 мг/кг BCD-100.
[00478] В ходе исследования был выявлен один случай дозолимитирующей токсичности (ДЛТ). У пациента 13-09, получившего две дозы BCD-100 в дозировке 3 мг/кг, нарушился гормональный фон (снизился ТТГ) и развился аутоиммунный тиреоидит (иммуноопосредованное нежелательное явление 2-й степени тяжести по критериям СТСАЕ 4.03). Комиссия по мониторингу данных и безопасности сочла данный случай явлением ДЛТ. Пациент 13-0 9 продолжил получать исследуемую терапию, течение его заболевания было охарактеризовано как стабильное по критериям RECIST 1.1 и irRC.
[00479] Помимо аутоиммунного тиреоидита, иммуноопосредованные НЯ были отмечены у 33,33% (5 из 15) пациентов: у 33,33% (2 из б) пациентов, получавших 1 мг/кг BCD-100, у 50,00% (3 из 6) пациентов, получавших 3 мг/кг BCD-100, но ни у одного из пациентов, получавших 10 мг/кг BCD-100. Все эти НЯ были явлениями 1-й степени тяжести по шкале СТСАЕ 4.03, и не были сочтены явлениями ДЛТ.
[00480] НЯ/СНЯ привели к временному прекращению исследуемой терапии у трех пациентов: у 16,67% (1 из 6) пациентов, получавших 1 мг/кг BCD-100 и у 33,33% (2 из 6) пациентов, получавших 3 мг/кг BCD-100. Все НЯ, из-за которых была временно прекращена терапия, носили иммунный характер (гипертиреоз 1-й степени тяжести у пациента 10-03, получавшего 1 мг/кг BCD-100; аутоиммунный тиреоидит 1-й степени тяжести у пациента 13-10, и аутоиммунный тиреоидит 2-й степени тяжести у пациента 13-09 - оба пациента получали 3 мг/кг BCD-100). Получение исследуемой терапии пациентом 10-03 было приостановлено. Пациентам 13-09 и 13-10 были назначены глюкокортикоиды по поводу заболевания, после чего исследуемая терапия была возобновлена. Ни один из перерывов в терапии не превысил норм (2-4 недели), установленных в протоколе исследования.
[00481] У трех пациентов исследуемый препарат был отменен из-за прогрессирования заболевания (у двух из них - после 5 введений в дозировке 1 мг/кг и у одного - после 5 введений в дозировке 10 мг/кг). Причиной отмены во всех случаях было прогрессирование заболевания, а не прием BCD-100.
[00482] СНЯ привело к отмене терапии, а затем к смерти одного из испытуемых. У пациента 13-10, перенесшего три введения препарата в дозировке 3 мг/кг, произошло кровоизлияние 5-й степени тяжести в правом полушарии. Исследователь счел, что данное НЯ было, вероятно, связано с исследуемой терапией.
[00483] Не было отмечено различий между группами пациентов в плане изменения лабораторных и физиологических параметров с течением времени. Хотя анализ данных параметров и выявил изолированные примеры статистически значимых различий, было сочтено, что эти различия отражают естественную вариабельность в рамках нормы. Отклонения, обнаруженные по результатам биохимического анализа крови и анализа крови на свертываемость (повышение уровней трансаминаз, билирубина, отклонения в показателях свертываемости крови и т.п.) в большинстве случаев были типичны для исследуемой популяции (пациентов с неоперабельными новообразованиями), ожидаемыми, и преходящими; нормализация параметров прошла без последствий, дополнительной терапии не потребовалось.
[00484] Описанные результаты демонстрируют благоприятный профиль безопасности препарата BCD-100 в любой из изученных дозировок при внутривенном применении.
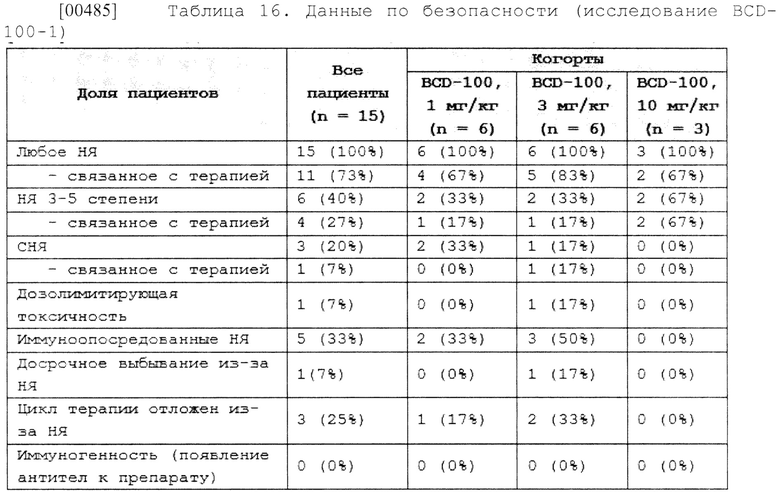
[00486] Иммуногенность
Оценка иммуногенности производилась во время скрининга, на 28-й день исследуемой терапии, по завершению основного периода (на 85-й день), а среди пациентов, продолжавших получать терапию препаратом BCD-100, также и каждые 42 дня после этого. Оценка иммуногенности осуществлялась в два этапа: на первом этапе производился скрининг на содержание связывающих антител к препарату (CAT) в образцах сыворотки пациентов, на втором - скрининг образцов с положительным титром CAT на содержание нейтрализующих антител (HAT).
[00487] В анализ иммуногенности были включены все пациенты, образцы сыворотки которых были доступны для анализа на момент скрининга и в последующие дни анализов (n=15). Не было обнаружено ни одного образца, содержащего CAT. Ввиду отсутствия образцов с CAT анализ на содержание HAT не осуществлялся.
[00488] Сводная информация по фармакокинетике и метаболизму, полученная в ходе I фазы исследования
В анализ фармакокинетики (ФК) включены данные пациентов, у которых не было пропущено/утеряно/испорчено более 3-х образцов сыворотки крови для определения концентрации BCD-100 после первого введения препарата (n=15).
[00489] Исследование фармакокинетики (как после первого введения препарата, так и на протяжении всех введений) проводилось по следующим группам:
• у первого пациента (доза BCD-100 0,3 мг/кг) (n=1);
• в первой когорте пациентов (доза BCD-100 1 мг/кг) (n=5);
• во второй когорте пациентов (доза BCD-100 3 мг/кг) (n=6).
• в третьей когорте пациентов (доза BCD-100 10 мг/кг) (n=3).
[00490] В фармакокинетический анализ входила оценка стандартных ФК характеристик распределения и выведения исследуемого препарата после неоднократного внутривенного введения. Результаты анализа представлены в таблице 17.
[00491] Образцы крови для анализа фармакокинетики (содержания BCD-100 в сыворотке) собирались у всех пациентов, включенных в исследование, на каждом дозовом уровне. Использовались следующие временные точки забора образцов: до первого введения препарата; через 30 минут, 2 ч (±15 мин), 4 ч (±15 мин), 6 ч (±15 мин), 24 ч (±1 ч), 48 ч (±2 ч), 192 ч (±8 ч), и 336 ч (±8 ч) после первого введения препарата (перед вторым введением), и перед каждым последующим введением препарата раз в две недели. Забор образцов крови для анализа концентрации BCD-100 в сыворотке производился одновременно с забором образцов для анализа иммуногенности (для того, чтобы оценить возможное воздействие антител к препарату на фармакокинетические показатели BCD-100). Концентрация BCD-100 в сыворотке крови определялась с помощью валидированной методики ИФА.
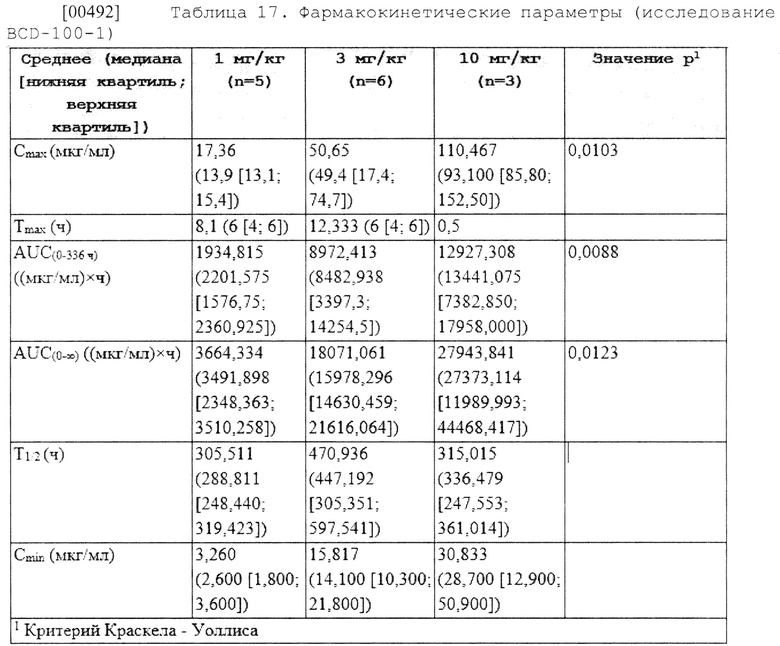
[00493] Измерение всех параметров происходило после единичного введения препарата, за исключением показателя Cmin, который рассчитывался для 6 введений препарата (12 недель).
[00494] Статистически значимые отличия между тремя когортами, получавшими разные дозы, были отмечены для показателя AUC(0-336 ч) (р=0,0088, критерий Краскела - Уоллиса); AUMC(0-336 ч) (р=0,0088, критерий Краскела - Уоллиса); AUC(o-∞) (р=0,0123, критерий Краскела - Уоллиса); Cmax (р=0,0103, критерий Краскела - Уоллиса). На дозах 3 мг/кг и 10 мг/кг были отмечены более благоприятные профили фармакокинетики по сравнению с дозой 1 мг/кг; однако при этом показатели ФК, эффективности и безопасности оказались не связаны с дозировкой.
[00495] Профили концентрации препарата BCD-100 в период с первого введения до 10-й недели во всех дозовых когортах (когорта 1 мг/кг [n=5], когорта 3 мг/кг [n=6] и когорта 10 мг/кг [n=3]) показаны на фигуре 1 и фигуре 2.
[00496] Расчет параметров ФК показал, что концентрация BCD-100 изменяется прямо пропорционально введенной дозе, достигая пика между 30 мин и 6 ч после введения и постепенно снижаясь после этого. Не было обнаружено зависимости между периодом полувыведения и количеством введенного препарата. Значение показателя 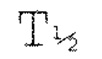 оказалось типичным для моноклональных антител (12-18 дней).
оказалось типичным для моноклональных антител (12-18 дней).
[00497] Сводная информация о фармакодинамике, полученная в ходе I фазы исследования
В анализ фармакодинамики были включены все пациенты, образцы сыворотки которых были доступны для анализа на момент скрининга и в последующие дни анализов (n=15).
[00498] Исследование фармакодинамики проводилось по следующим группам:
• у первого пациента (доза BCD-100 0,3 мг/кг) (n=1);
• в первой когорте пациентов (доза BCD-100 1 мг/кг) (n=5);
• во второй когорте пациентов (доза BCD-100 3 мг/кг) (n=6).
• в третьей когорте пациентов (доза BCD-100 10 мг/кг) (n=3).
[00499] Показатель насыщенности рецептора PD-1 препаратом BCD-100 помогает оценить взаимодействие исследуемого препарата с его мишенью и является наиболее важным фармакодинамическим маркером активности моноклонального антитела к PD-1, поскольку блокада рецептора PD-1 инициирует антиопухолевый иммунный ответ. Забор крови для оценки фармакодинамики (% насыщенности рецептора PD-1 препаратом BCD-100) производился у всех пациентов. Забор образцов для оценки ФД осуществлялся до введения препарата, через 4 ч и 336 ч после введения (но до второго введения), и перед шестым введением препарата.
[00500] Результаты демонстрируют высокую насыщенность рецептора PD-1 препаратом BCD-100 (от 95% до 100%) на всех дозовых уровнях и во всех популяциях клеток (таблица 18).
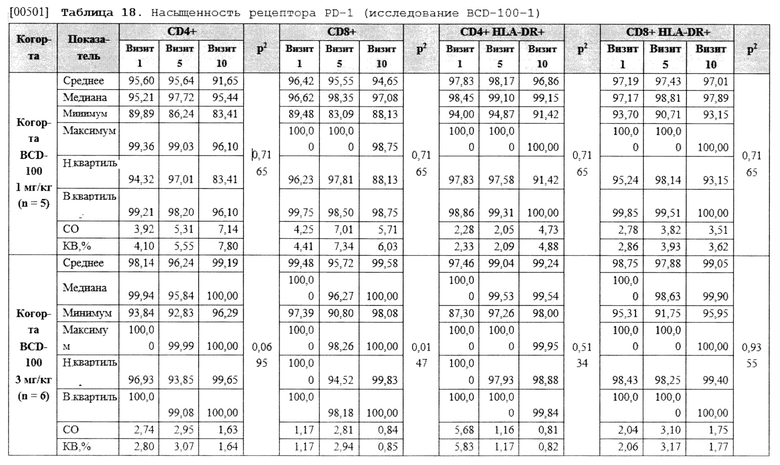
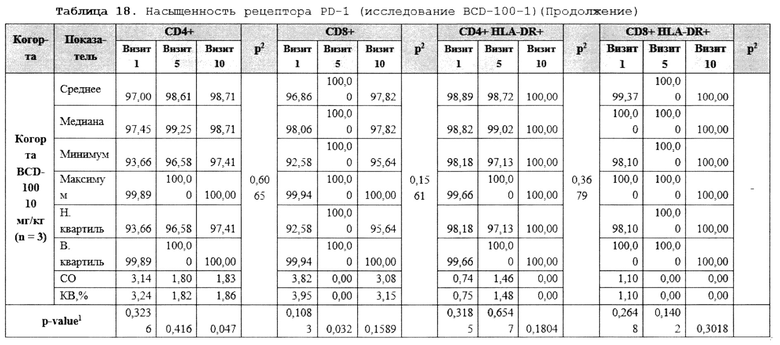
[00502] Сводная информация о клинической эффективности, полученная в ходе I фазы исследования
В популяцию для оценки эффективности было включено 14 из 15 пациентов, получавших BCD-100. Терапия одного из пациентов была прекращена из-за СНЯ (смерть); к моменту смерти пациента оценка ответа опухоли на терапию с помощью КТ не была проведена.
[00503] Определение общей частоты ответов (частота частичного ответа + частота полного ответа) и контроля заболевания (стабильное заболевание + частичный ответ + полный ответ) производилось через 85 дней терапии препаратом BCD-100. Оценка эффективности, производившаяся через 85 дней терапии препаратом BCD-I00, была основана на анализе результатов КТ-сканирования. Ответ опухоли оценивался по критериям RECIST 1.1 и Immune-Related RECIST (irRECIST).
[00504] Согласно критериям irRECIST, которые были разработаны с учетом иммунотерапии, контроль заболевания был достигнут у 28,57% (4 из 14) пациентов. Общая частота ответа составила 7,14% (1 из 14). Не было обнаружено значимых различий по частоте ответа между разными когортами пациентов (таблица 19).
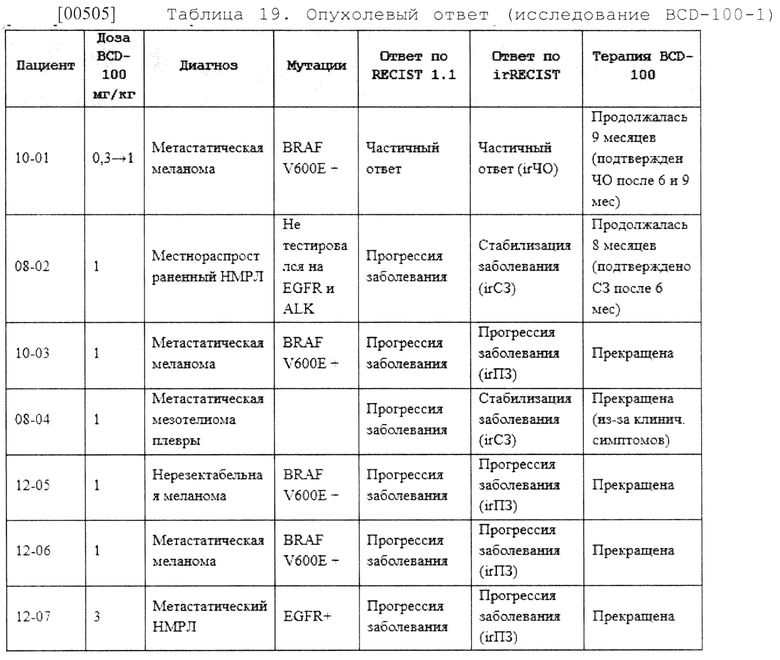
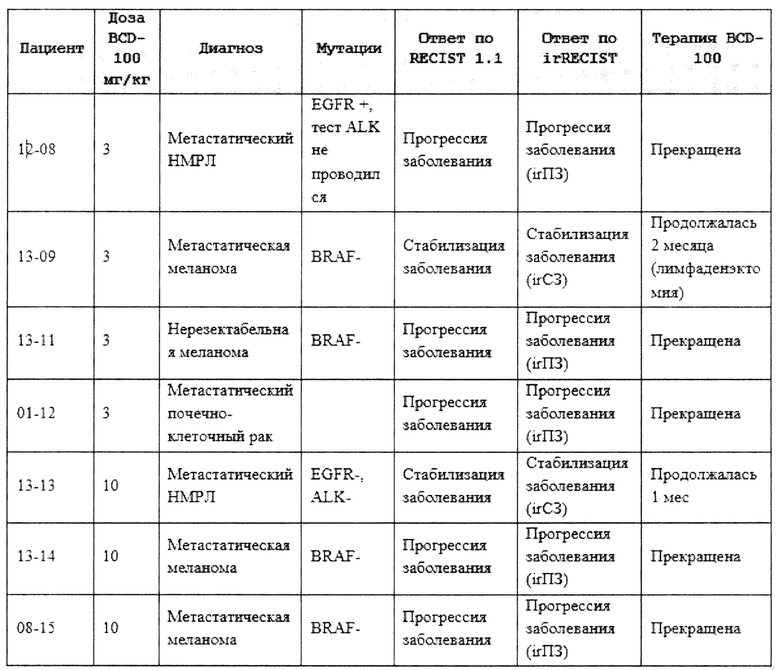
[00506] Вывод
Проведенный анализ полученных данных наглядно демонстрирует благоприятный профиль безопасности препарата BCD-100 при его использовании в широком диапазоне доз от 0,03 мг/кг до 10,0 мг/кг. Для дальнейшего клинического изучения представляется оптимальным выбор 2-х режимов введения препарата BCD-100: 1 мг/кг 1 раз в 2 недели и 3 мг/кг раз в 3 недели.
[00507] Пример 19. Применение пролголимаба (BCD-100) для терапии пациентов с нерезектабельной/метастатической меланомой, исследование II фазы, BCD-100-2/MIRACULUM
[00508] Дизайн исследования
Исследование № BCD-100-2/MIRACULUM (MIRACULUM, NCT03269565) - многоцентровое открытое рандомизированное исследование фармакокинетики, эффективности, безопасности и иммуногенности препарата BCD-100 (пролголимаб) в качестве монотерапии у пациентов с нерезектабельной/метастатической меланомой, ранее не получавших либо получавших лечение. Сравнение осуществлялось между препаратом BCD-100, вводимым внутривенно в дозе 3 мг/кг раз в три недели, и BCD-100, вводимым внутривенно в дозе 1 мг/кг раз в две недели. Исследование в настоящий момент продолжается в Российской Федерации и Белоруссии. Были получены и проанализированы данные за 1 год терапии.
[00509] Оценка каждого параметра эффективности проводилась в отдельности для каждой группы. Первичной конечной точкой для оценки эффективности являлась общая частота ответа (частота достижения частичного ответа + полного ответа) по irRECIST у участников исследования на фоне терапии препаратом BCD-100. К расчету ОЧО (общей частоты ответа) и контроля над заболеванием принимался лучший ответ на терапию за вышеуказанный период. Вторичными конечными точками для оценки эффективности являлись беспрогрессивная и общая выживаемость пациентов через 12 месяцев от начала терапии, частота достижения контроля над заболеванием (частота достижения стабилизации + частичного ответа + полного ответа), время достижения ответа на терапию, длительность ответа на терапию (фигура 3, таблица 20).
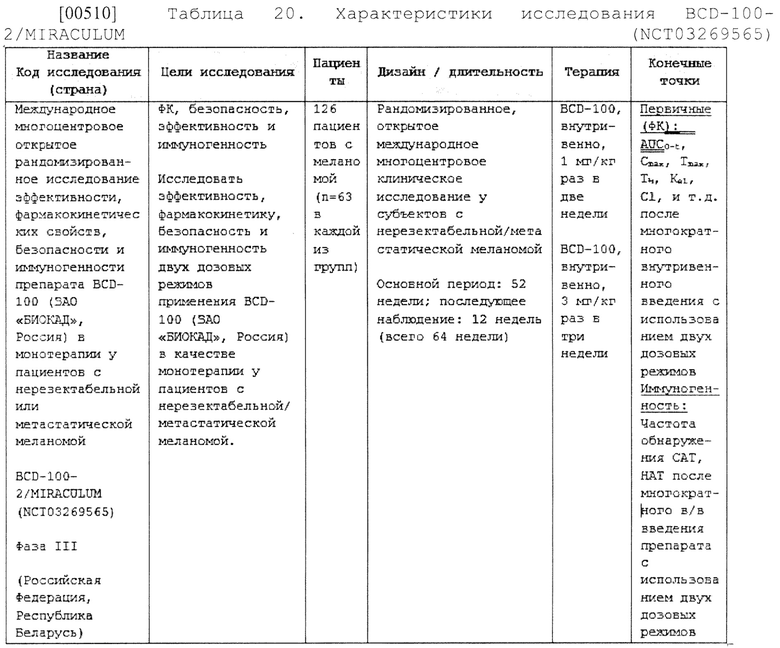
[00511] Результаты исследования
Сводная информация о характеристиках популяции пациентов
[00512] В исследование был включен 131 пациент в возрасте от 18 лет. Популяция пациентов включала как мужчин, так и женщин, страдавших распространенной меланомой, в том числе меланомой хориоидеи. Данная популяция включала пациентов, которые заняли место тех, кто выбыл до первого введения BCD-100 либо вследствие серьезных отклонений от Протокола. Образованная в итоге модифицированная ITT-популяция, состоящая из пациентов, получивших хотя бы одну дозу BCD-100, насчитывала 126 пациентов. Для включения в исследование пациенты должны были соответствовать 0-1 баллам по шкале EC0G и иметь как минимум один измеряемый целевой очаг согласно RECIST 1.1 (не считая метастазов в костях) (фигура 4, таблица 21).

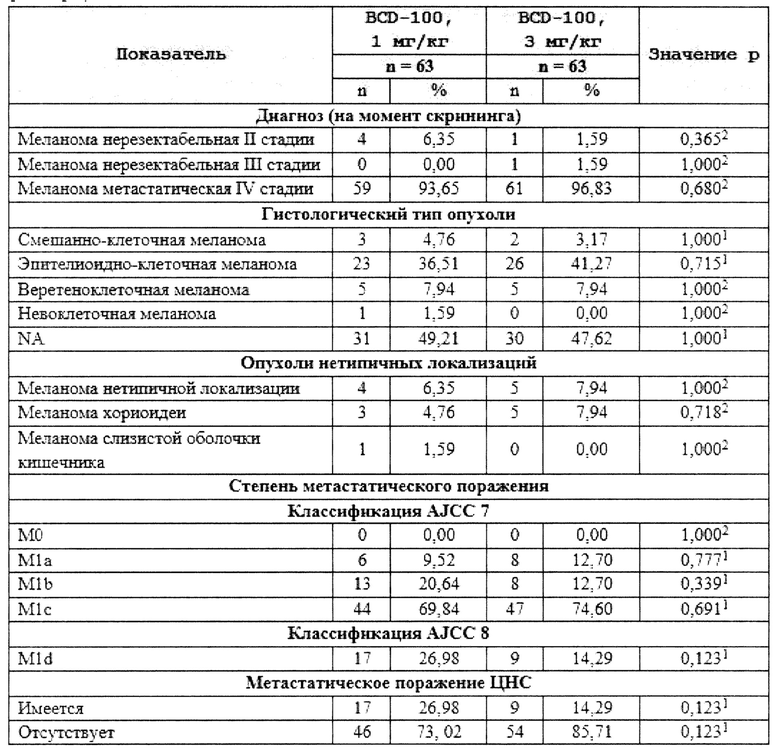
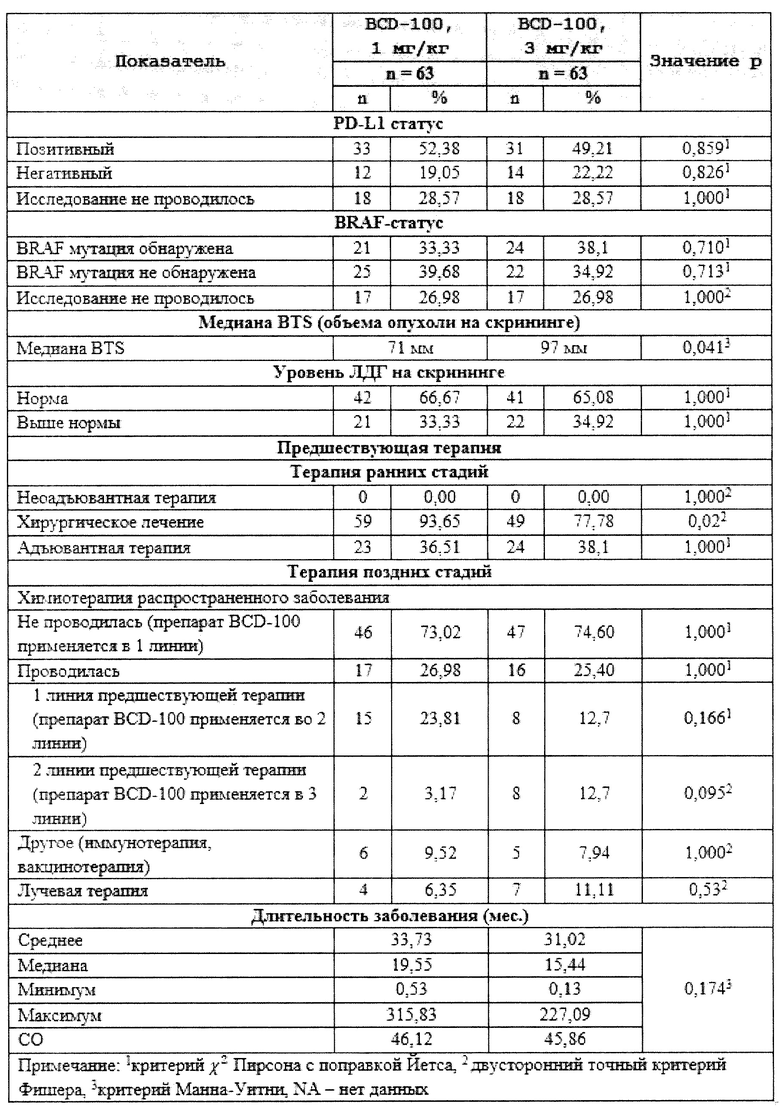
[00514] В анализ безопасности вошли все пациенты, получившие как минимум одно введение исследуемого препарата («modified Intent-to-treat» (mITT) популяция, n=126).
[00515] Анализ эффективности проведен в двух популяциях пациентов:
• все пациенты, получившие как минимум одно введение исследуемого препарата (mITT популяция, n=126);
• пациенты, получившие, как минимум, одно введение исследуемого препарата и прошедшие, как минимум, I плановое КТ-исследование в динамике для оценки ответа (популяция «рег protocol» (РР), n=114).
[00516] В анализ фармакокинетики были включены данные пациентов, получивших, как минимум, одно введение препарата BCD-100, у которых не было пропущено/утеряно/испорчено необходимых образцов, согласно протоколу, действующей версии, на момент определения (n=125).
[00517] Сводная информация о клинической эффективности, полученная в ходе исследования № BCD-100-2/MIRACULUM
Результаты оценки эффективности по первичной конечной точке (ОЧО) свидетельствуют о достаточной эффективности препарата BCD-100 в обоих дозовых режимах, во всех исследуемых популяциях пациентов. Так, ОЧО и контроль над заболеванием в популяции РР составили 40,68% и 67,80% в 1-ой группе и 32,73% и 52,73% - во 2-ой группе терапии. В популяции mITT наблюдались схожие показатели эффективности. Таким образом, ожидаемая целевая частота ответа (28%) и критическое значение r (11 ответов) были достигнуты в обеих группах терапии.
[00518] Подгрупповой анализ по линиям терапии продемонстрировал высокую эффективность препарата BCD-100 в минимальной дозе 1 мг/кг в популяции не леченных прежде пациентов (ОЧО составила 50,00%).
[00519] Результаты анализа эффективности по вторичным конечным точкам подтвердили заключение о достаточной эффективности препарата BCD-100 в обеих дозах. Показатели БПВ, ОВ, длительности ответа были сопоставимы с таковыми лучших в классе ингибиторов PD-1.
[00520] 12-месячная БПВ составила 41,27% для пациентов группы BCD-100, 1 мг/кг и 34,92% - для пациентов группы BCD-100, 3 мг/кг. Медиана БПВ составила 5,78 месяца (95% ДИ 3,52 - -) для пациентов группы BCD-100, 1 мг/кг и 2,33 месяца (95% ДИ 2,07-10,25) - для пациентов группы BCD-10C, 3 мг/кг (р=0,4 00, лог-ранговый критерий). При детальной оценке беспрогрессивной выживаемости в каждой из групп, по линиям терапии (по критериям irRECIST, в mITT популяции), статистически значимых различий выявлено не было. Препарат BCD-100 был равно эффективен как при применении у не леченных прежде пациентов, так и при применении у ранее получавших терапию пациентов.
[00521] При медиане наблюдения 13,8 месяца (95% ДИ 13,2-14,7) медиана общей выживаемости для пациентов группы BCD-100, 1 мг/кг не была достигнута (95% ДИ -1 В данном случае и далее по тексту "-" означает "не достигнута" - -). 12-месячная ОВ составила 74,60% для пациентов группы BCD-100, 1 мг/кг. Медиана ОВ для пациентов группы BCD-100, 3 мг/кг составила 15 месяцев (95% ДИ 9,99 - -) при медиане наблюдения 14,5 месяца (95% ДИ 13,9-15,2). 12-месячная ОВ составила 53,97% для пациентов группы BCD-100, 3 мг/кг. При детальной оценке общей выживаемости в каждой из групп, по линиям терапии, статистически значимых различий выявлено не было. Препарат BCD-100 был равноэффективен как при применении у не леченных прежде пациентов, так и при применении у ранее получавших терапию пациентов. Медианы ОВ не были достигнуты ни в одной из подгрупп у пациентов, получавших препарат BCD-100 в дозе 1 мг/кг (р=0,800; лог-ранговый критерий). Медиана ОВ составила 16,8 месяца (95% ДИ 9,33 - -) для не леченных прежде пациентов и 15 месяцев (95% ДИ 7,4 6 - -) для получавших ранее терапию, в группе препарата BCD-100, 3 мг/кг (р=0,900; лог-ранговый критерий) (таблица 22, таблица 23, таблица 24, таблица 25, фигура 5, фигура 6, фигура 7, фигура 8, ).
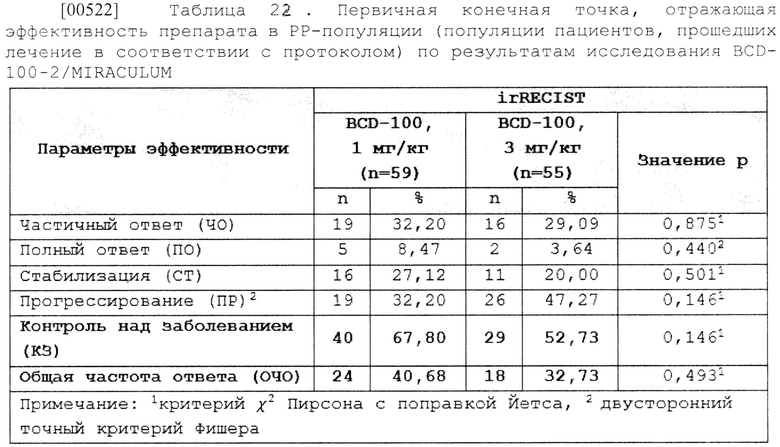
[00523] Сводная информация о клинической безопасности, полученная в ходе исследования № BCD-100-2/MIRACULUM
BCD-100 продемонстрировал благоприятный профиль безопасности в обоих дозовых режимах (таблица 23).
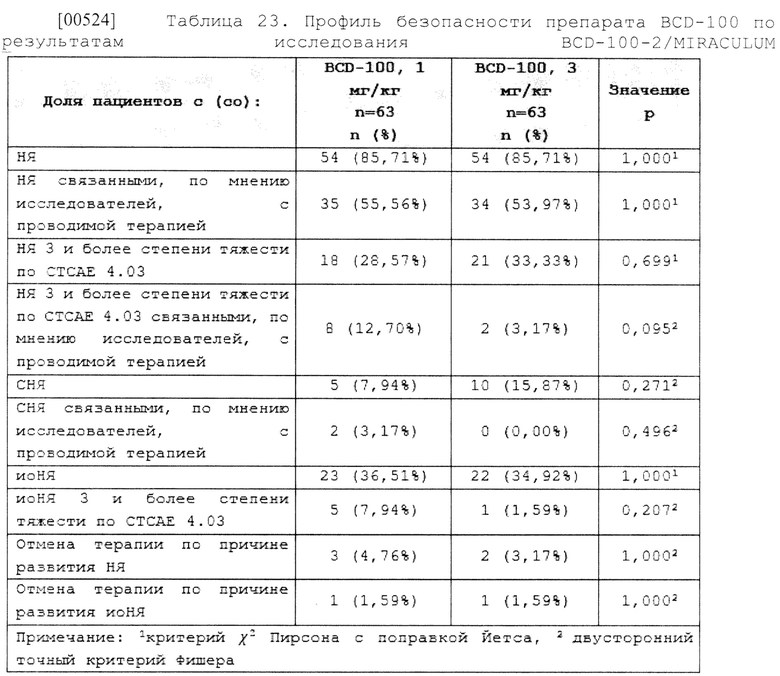
[00525] На основании приведенных выше данных можно сделать вывод о том, что результаты, полученные в настоящем исследовании, не противоречат известным данным о профиле безопасности препаратов моноклональных антител к рецептору PD-1. Статистически значимых различий в профиле безопасности препарата в изученных дозах выявлено не было, за исключением количества СНЯ с последующим летальным исходом (значимо большее количество отмечалось среди пациентов группы BCD-100, 3 мг/кг).
[00526] Иммуногенность
В анализ иммуногенности были включены все пациенты, образцы сыворотки которых были доступны для анализа на момент скрининга и в последующие дни анализов (n=121). Ни у одного из пациентов не было обнаружено CAT к BCD-100.
[00527] Сводная информация по фармакокинетике и метаболизму, полученная в ходе исследования № BCD-100-2/MIRACULUM
Проведенный анализ показал, что однократное внутривенное введение препарата BCD-100 в дозе от 1 до 3 мг/кг характеризуется линейным нарастанием концентрации исследуемого вещества в плазме крови, с последующим равномерным снижением, при этом период полувыведения препарата, независимо от введенной в организм дозы, характеризуется обычной для иммуноглобулинов класса IgG продолжительностью - от 11,5 до 17 дней (фигура 9, фигура 10).
[00528] Тенденция к дозозависимому нарастанию концентрации исследуемого вещества в плазме крови (по параметрам Cmax и AUC0-t,ss) сохраняется также и после многократного введения препарата BCD-100, что соответствует литературным данным о ФК профиле других препаратов анти-PD-1 антител.
[00529] Продемонстрированное в рамках клинического исследования № BCD-100-2/MIRACULUM стойкое сохранение Cmin на достаточном уровне на фоне проведения курса лечения свидетельствует о том, что терапевтические концентрации препарата сохраняются как при применении препарата BCD-100 в дозе 1 мг/кг 1 раз в 2 недели, так и при применении препарата в дозе 3 мг/кг в режиме 1 раз в 3 недели. Таким образом, применение BCD-100 в обоих дозовых режимах является оправданным с позиции фармакокинетических свойств.
[00530] Оценка параметра Т1/2, в том числе и сравнительная оценка между группами терапии, была затруднена ввиду отсутствия у ряда пациентов типичного терминального периода фазы элиминации препарата после однократного введения.
[00531] В целом, препарат BCD-100 как в дозе 1 мг/кг раз в 2 недели, так и в дозе 3 мг/кг раз в 3 недели характеризовался фармакокинетическими показателями, обеспечивающими длительное пребывание препарата в организме, достаточное для поддержания стабильной терапевтической концентрации на фоне многократного введения. Дозозависимости в отношении показателей эффективности и безопасности препарата отмечено не было.
[00532] Сводная информация о фармакодинамике, полученная в ходе I фазы исследования
[00533] Процент насыщенности PD-1 рецепторов препаратом BCD-100 во всех изученных субпопуляциях лейкоцитов не отличался между группами терапии. Насыщенность PD-1 на уровне >99% в популяции активированных Т-хелперов и цитотоксических лейкоцитов была зарегистрирована у 33,33% пациентов, включенных в анализ данного показателя (14 из 42). При этом статистически значимой разницы между группами терапии отмечено не было: 8 пациентов получали BCD-100 в дозе 1 мг/кг, 6 пациентов - в дозе 3 мг/кг.
[00534] Ki-67 является универсальным маркером клеточной пролиферации, что обусловлено его присутствием в клетке только во время процесса деления и разрушением в течение 1,5-2 ч. после окончания митоза. Маркер Ki-67 выявляется в области теломер, центромер и ядрышках. Процент Ki-67 положительных цитотоксических Т-лимфоцитов в ходе терапии возрастал в обеих группах, но достоверных различий при этом обнаружено не было. Анализ доли Ki-67 положительных цитотоксических Т-лимфоцитов также не выявил статистически значимых различий между группами.
[00535] Процент Th9 в общей популяции Т-хелперов в ходе терапии возрастал преимущественно в группе 2 (BCD-100, 3 мг/кг Q3W), но достоверных различий при этом обнаружено не было. Достоверные различия отсутствовали и при межгрупповом сравнении.
[00536] По имеющимся литературным данным, раннее повышение уровня Th9 ассоциировано с лучшим ответом на терапию ниволумабом при меланоме3 ( ). Следует обратить внимание, что эти результаты были получены на ограниченной выборке пациентов (n=42), гетерогенной по половой принадлежности и характеристикам основного заболевания.
). Следует обратить внимание, что эти результаты были получены на ограниченной выборке пациентов (n=42), гетерогенной по половой принадлежности и характеристикам основного заболевания.
[00537] Сравнительный анализ доли Th9 в общей популяции Т-хелперов в подгруппах с различными типами ответа на терапию препаратом BCD-100 не выявил статистически значимых различий в обеих дозовых когортах. Тем не менее, на представленных графиках видна тенденция к лучшему ответу на проводимую терапию у пациентов с исходно повышенным уровнем Тп9 (фигура 11, фигура 12).
[00538] Вывод
[00539] Полученные данные об эффективности, безопасности и фармакокинетических свойствах BCD-100 являются достаточными для обоснования применения препарата как в дозовом режиме 1 мг/кг 1 раз в 2 недели, так и в дозовом режиме 3 мг/кг 1 раз в 3 недели.
[00540] В рамках международного многоцентрового рандомизированного открытого исследования II фазы № BCD-100-2/MIRACULUM в популяции пациентов с нерезектабельной и метастатической меланомой были продемонстрированы значимые терапевтические преимущества по сравнению с известными данными об эффективности химиотерапии, и результаты сопоставимые с лучшими существующими методами лечения. С учетом полученных в рамках исследования показателей эффективности при благоприятном профиле безопасности, применение препарата BCD-100 в рутинной клинической практике отвечает балансу риска и пользы в данной популяции пациентов.
--->
Перечень последовательностей
<110> ЗАКРЫТОЕ АКЦИОНЕРНОЕ ОБЩЕСТВО "БИОКАД"
<120> ВОДНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИТЕЛА К PD-1 ПРОЛГОЛИМАБА И ЕЕ ПРИМЕНЕНИЕ
<160> 2
<170> BiSSAP 1.3.6
<210> 1
<211> 459
<212> PRT
<213> Homo sapiens
<400> 1
Gln Val Gln Leu Val Gln Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr
20 25 30
Trp Met Tyr Trp Val Arg Gln Val Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Ala Ile Asp Thr Gly Gly Gly Arg Thr Tyr Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Ala Ile Ser Arg Val Asn Ala Lys Asn Thr Met Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asp Glu Gly Gly Gly Thr Gly Trp Gly Val Leu Lys Asp Trp
100 105 110
Pro Tyr Gly Leu Asp Ala Trp Gly Gln Gly Thr Leu Val Thr Val Ser
115 120 125
Ser Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser
130 135 140
Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu Val Lys Asp
145 150 155 160
Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr
165 170 175
Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr
180 185 190
Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly Thr Gln
195 200 205
Thr Tyr Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys Val Asp
210 215 220
Lys Arg Val Glu Pro Lys Ser Cys Asp Lys Thr His Thr Cys Pro Pro
225 230 235 240
Cys Pro Ala Pro Glu Ala Ala Gly Gly Pro Ser Val Phe Leu Phe Pro
245 250 255
Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr
260 265 270
Cys Val Val Val Asp Val Ser His Glu Asp Pro Glu Val Lys Phe Asn
275 280 285
Trp Tyr Val Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro Arg
290 295 300
Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val
305 310 315 320
Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser
325 330 335
Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser Lys Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Glu Glu Met Ala Lys Gly Gln Pro Arg Glu Pro
355 360 365
Gln Val Thr Lys Asn Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe
370 375 380
Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu
385 390 395 400
Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe
405 410 415
Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly
420 425 430
Asn Val Phe Ser Cys Ser Val Met His Glu Ala Leu His Asn His Tyr
435 440 445
Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys
450 455
<210> 2
<211> 214
<212> PRT
<213> Homo sapiens
<400> 2
Gln Pro Val Leu Thr Gln Pro Leu Ser Val Ser Val Ala Leu Gly Gln
1 5 10 15
Thr Ala Arg Ile Thr Cys Gly Gly Asn Asn Ile Gly Ser Lys Asn Val
20 25 30
His Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Val Leu Val Ile Tyr
35 40 45
Arg Asp Ser Asn Arg Pro Ser Gly Ile Pro Glu Arg Phe Ser Gly Ser
50 55 60
Asn Ser Gly Asn Thr Ala Thr Leu Thr Ile Ser Arg Ala Gln Ala Gly
65 70 75 80
Asp Glu Ala Asp Tyr Tyr Cys Gln Val Trp Asp Ser Ser Thr Ala Val
85 90 95
Phe Gly Thr Gly Thr Lys Leu Thr Val Leu Gln Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОГО НОВООБРАЗОВАНИЯ С ИСПОЛЬЗОВАНИЕМ КОМБИНАЦИИ АНТИТЕЛА К PD-1 И ХИМИОТЕРАПЕВТИЧЕСКОГО АГЕНТА | 2021 |
|
RU2787457C2 |
| ВОДНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИ-IL17A АНТИТЕЛА И ЕЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2754760C2 |
| ВОДНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ЛЕВИЛИМАБА И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2745814C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ПЕМБРОЛИЗУМАБА И ЕЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2791857C2 |
| КОМПОЗИЦИИ АНТИ-PD-1 АНТИТЕЛ | 2019 |
|
RU2772781C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИ-CD20 АНТИТЕЛА И ЕЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2824627C2 |
| Водная фармацевтическая композиция рекомбинантного моноклонального антитела к ФНОа | 2017 |
|
RU2764521C2 |
| Водная фармацевтическая композиция рекомбинантного моноклонального антитела к ФНОα | 2016 |
|
RU2665966C2 |
| КОМБИНИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ | 2020 |
|
RU2829637C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИ-TRBV9 АНТИТЕЛА И ЕЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2826886C1 |
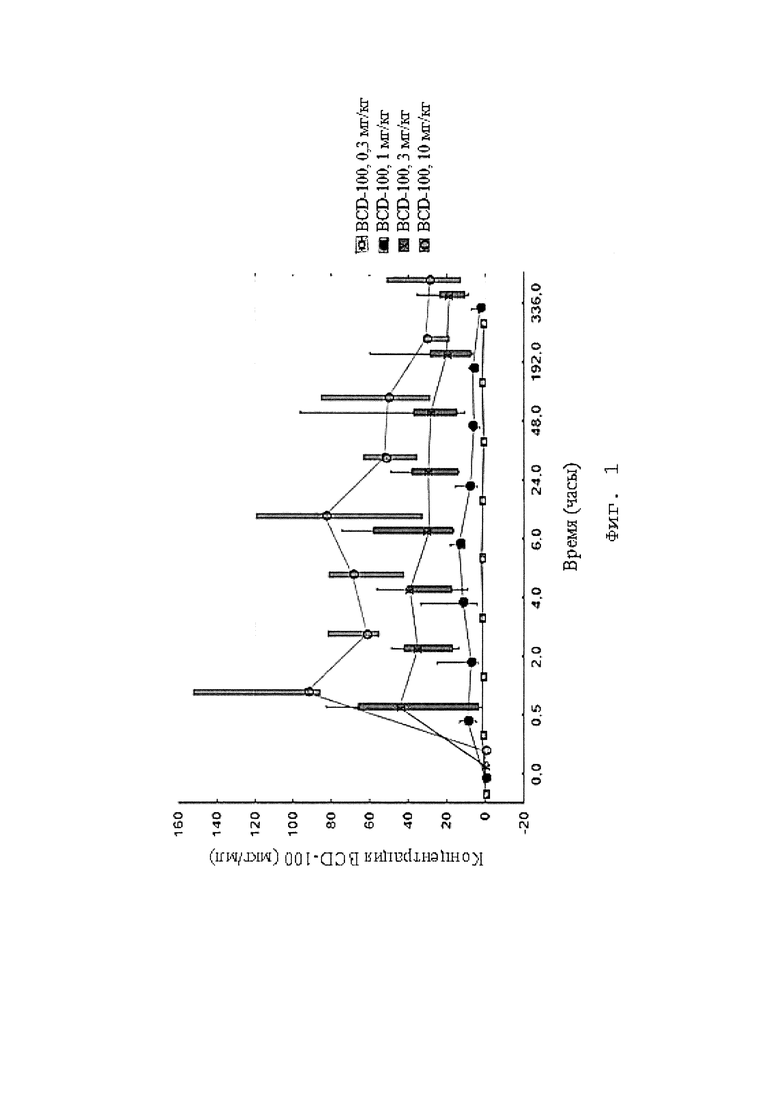
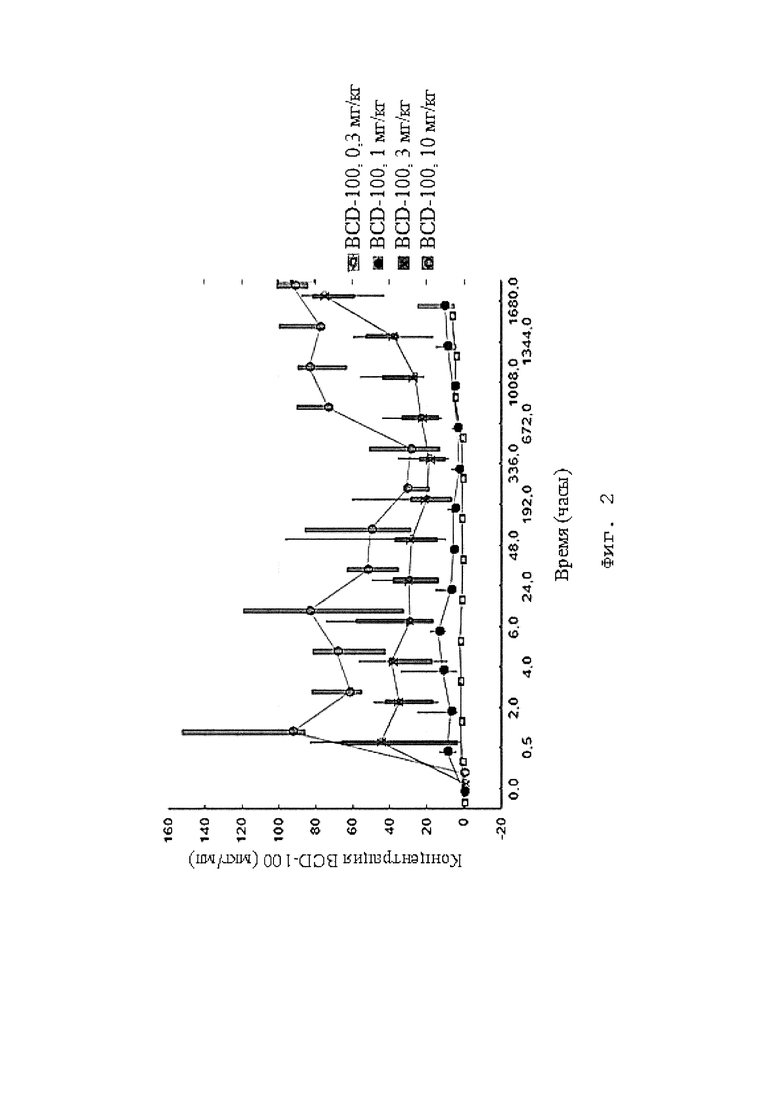

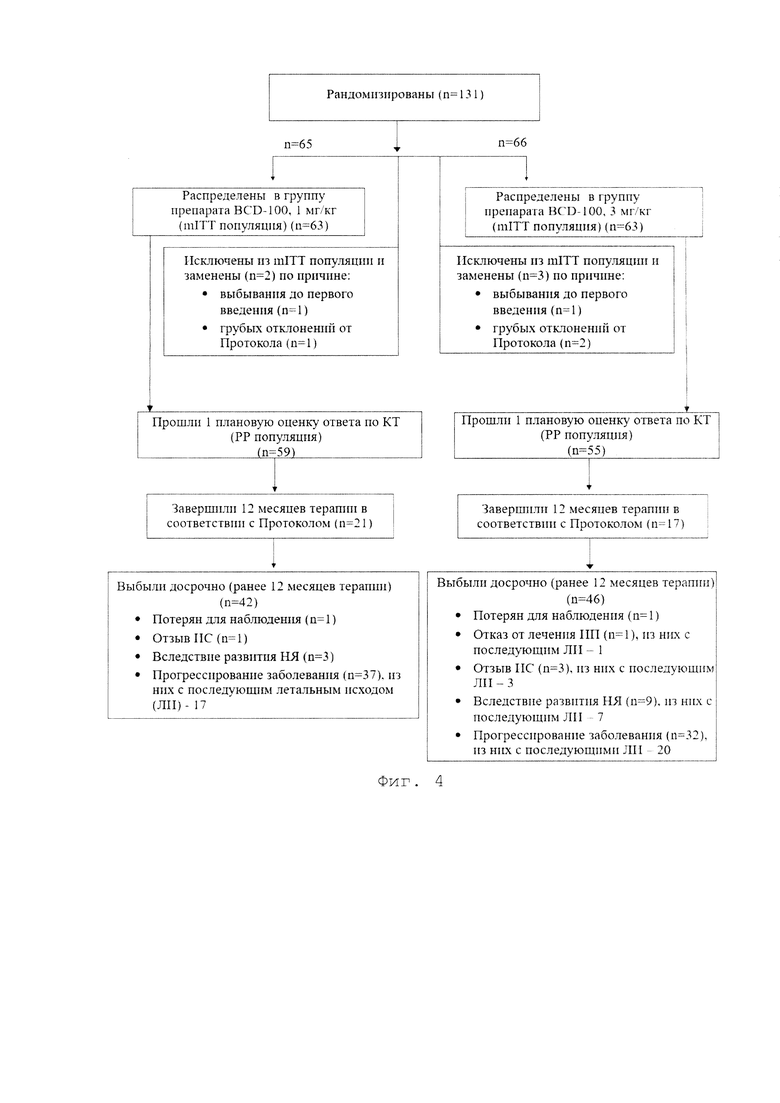
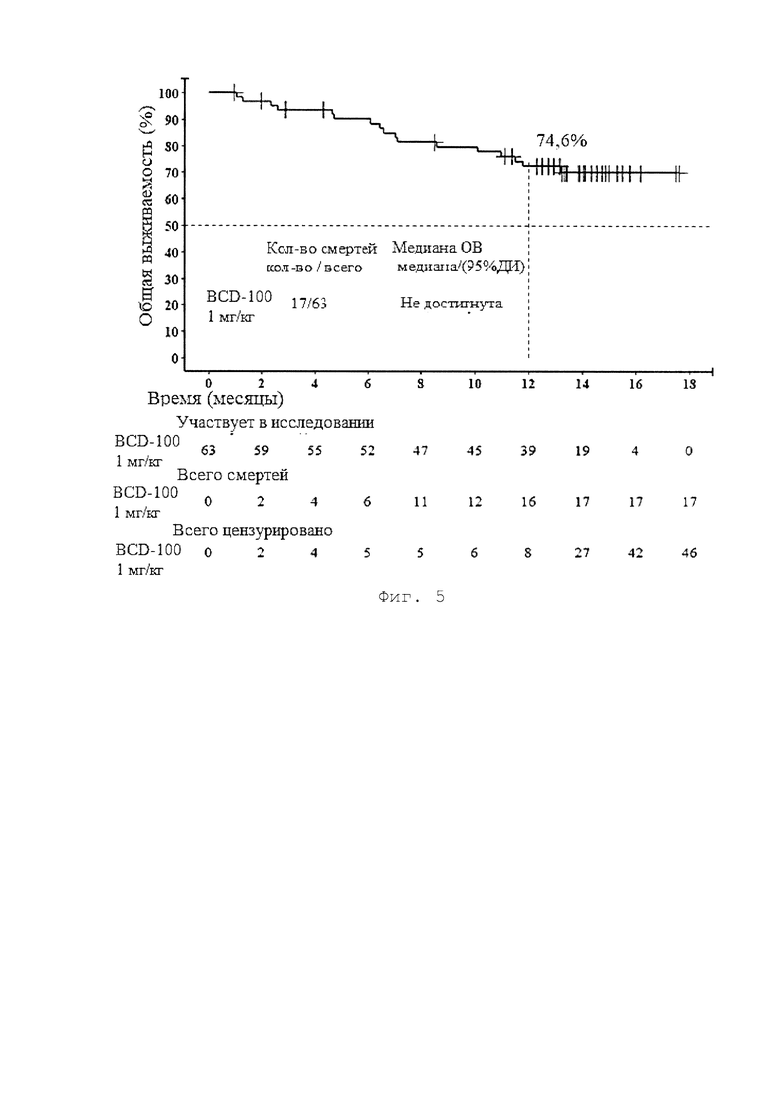
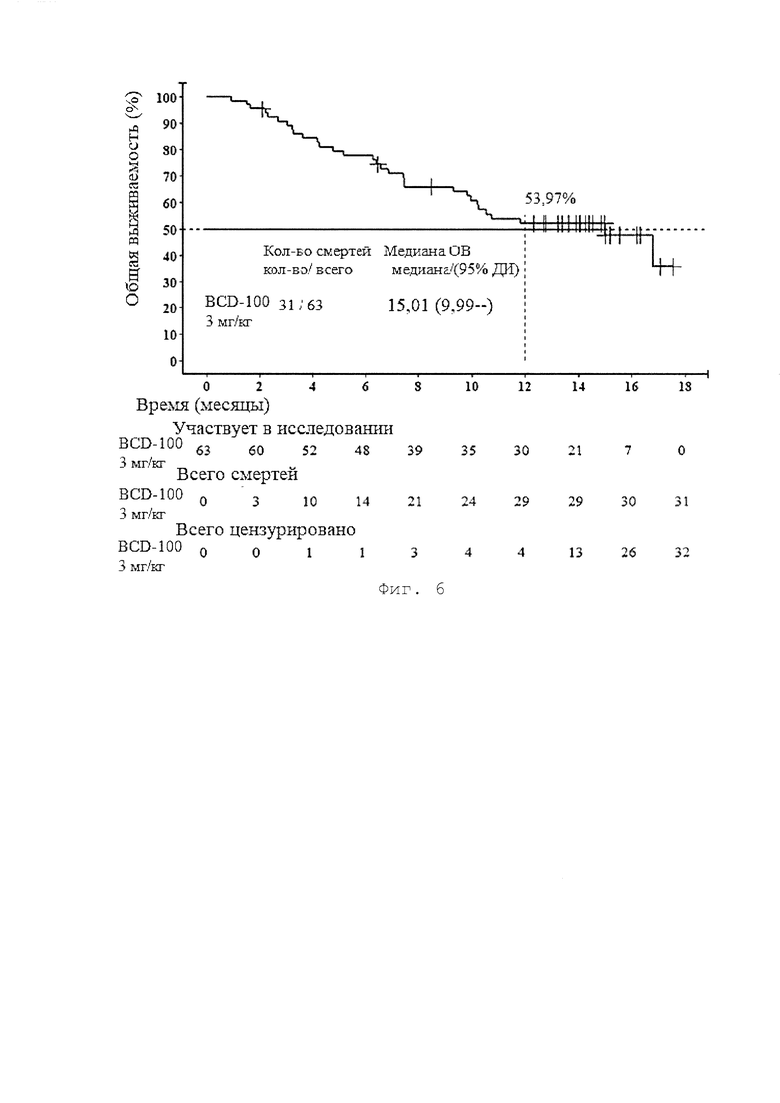
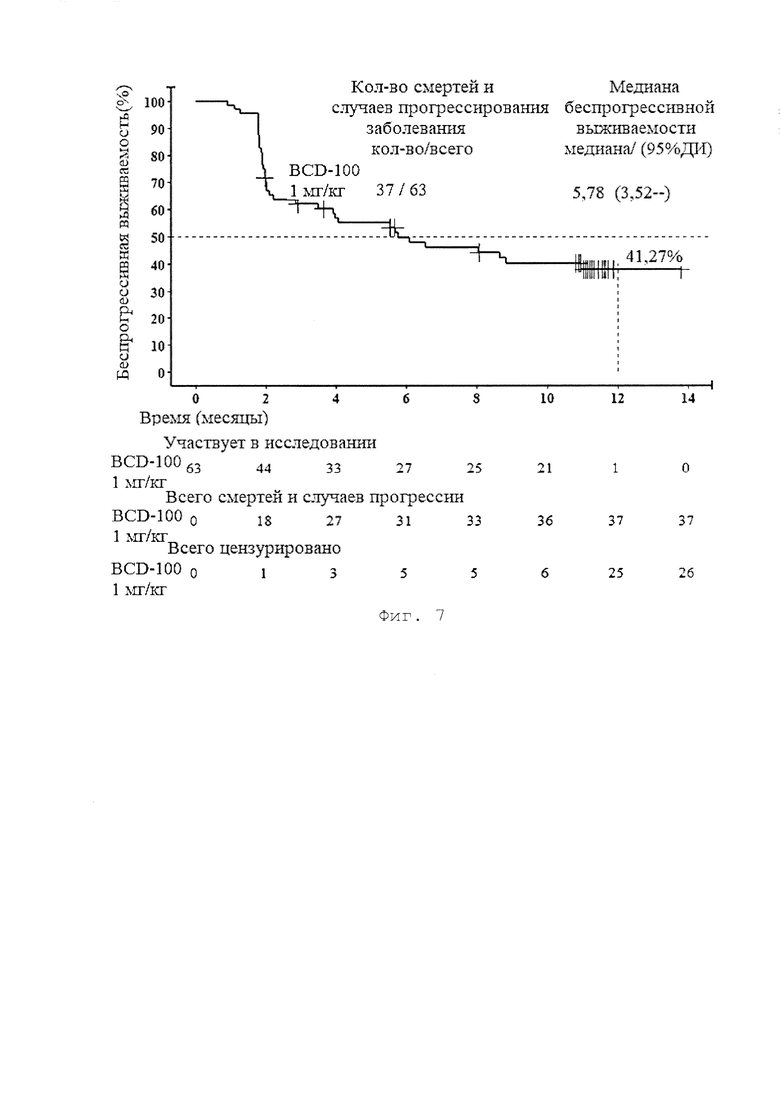
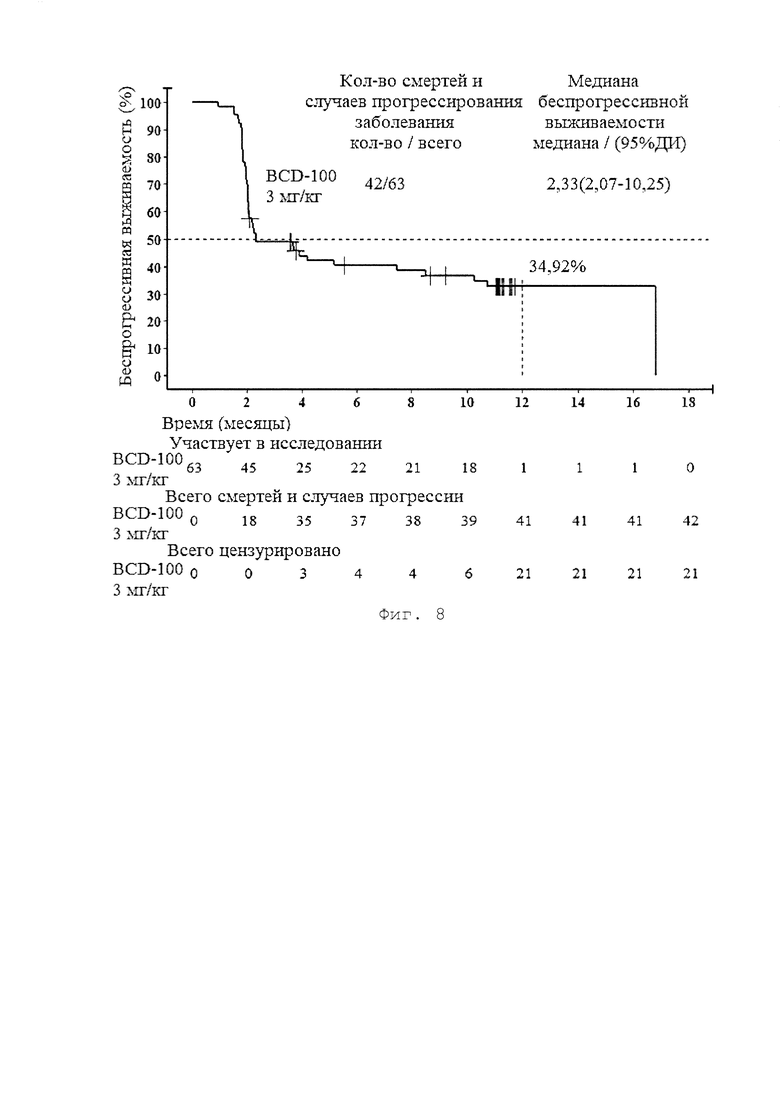
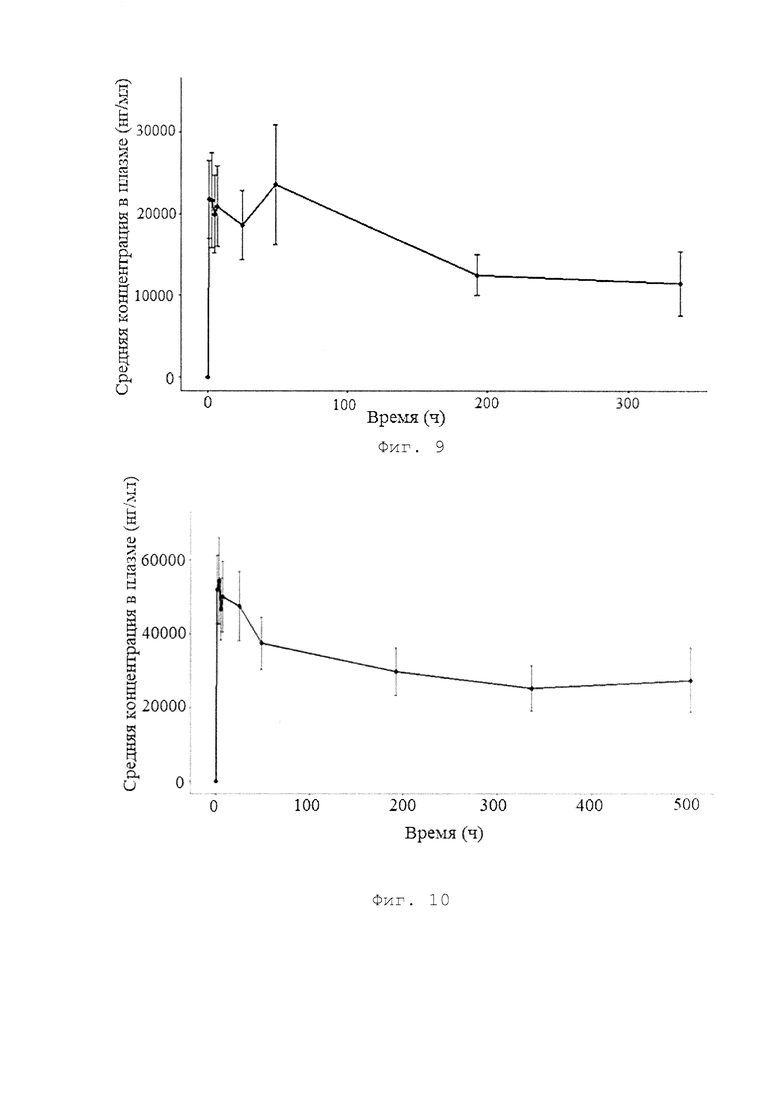
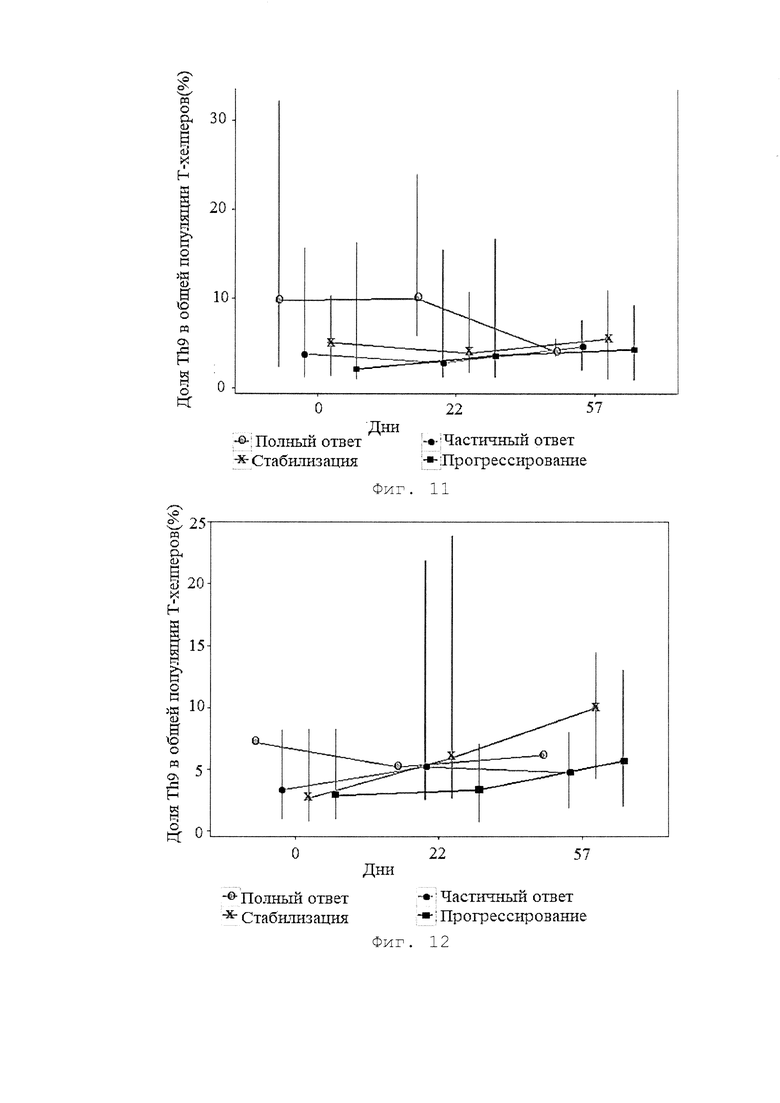
Группа изобретений относится к водным фармацевтическим композициям для антитела к PD-1 пролголимаба, к применению таких фармацевтических композиций и способу лечения с их использованием. Предлагаются варианты водной фармацевтической композиции анти-PD-1 антитела, состоящие из: пролголимаба в качестве антитела, трегалозы дигидрата, натрия ацетата тригидрата в определенных количествах и уксусной кислоты для обеспечения рН в диапазоне 5,0-5,5; а также варианты водной фармацевтической композиции анти-PD-1 антитела, состоящие из: пролголимаба в качестве антитела, трегалозы дигидрата, L-гистидина и L-гистидина гидрохлорида в определенных количествах, при рН в диапазоне от 5,5 до 6,0. Качественный и количественный состав вспомогательных веществ, используемый в вариантах композиции, обеспечивает стабильность водных композиций пролголимаба в широком диапазоне его концентраций. Предлагается также применение указанных выше вариантов водной фармацевтической композиции антитела к PD-1 пролголимаба для лечения злокачественного новообразования у субъекта, нуждающегося в этом, и способ лечения злокачественного новообразования у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества вариантов указанной выше водной фармацевтической композиции. 10 н. и 48 з.п. ф-лы, 23 табл.. 19 пр., 12 ил.
1. Водная фармацевтическая композиция анти-PD-1 антитела, состоящая из:
(a) пролголимаба в концентрации от 15 мг/мл до 40 мг/мл в качестве антитела;
(b) трегалозы дигидрата в концентрации от 80 мг/мл до 110 мг/мл;
(c) натрия ацетата тригидрата в концентрации от 1,6 мг/мл до 1,9 мг/мл; и
(d) уксусной кислоты до рН 5,0-5,5.
2. Водная фармацевтическая композиция по п. 1, где указанный пролголимаб находится в концентрации 15 мг/мл до 25 мг/мл.
3. Водная фармацевтическая композиция по п. 2, где указанный пролголимаб находится в концентрации 20 мг/мл.
4. Водная фармацевтическая композиция по любому из пп. 1-3, где указанный трегалозы дигидрат находится в концентрации от 95 мг/мл до 105 мг/мл.
5. Водная фармацевтическая композиция по п. 4, где указанный трегалозы дигидрат находится в концентрации 100 мг/мл.
6. Водная фармацевтическая композиция по п. 1, где указанный натрия ацетат тригидрат находится в концентрации от 1,7 мг/мл до 1,8 мг/мл.
7. Водная фармацевтическая композиция по п. 6, где указанный натрия ацетат тригидрат находится в концентрации 1,742 мг/мл.
8. Водная фармацевтическая композиция по любому из пп. 1-7, где указанная уксусная кислота добавлена до рН 5,0.
9. Водная фармацевтическая композиция анти-PD-1 антитела, состоящая из:
(a) пролголимаба в концентрации от 90 мг/мл до 150 мг/мл в качестве антитела;
(b) трегалозы дигидрата в концентрации от 75 мг/мл до 85 мг/мл;
(c) натрия ацетата тригидрата в концентрации от 1,6 мг/мл до 1,9 мг/мл; и
(d) уксусной кислоты до рН от 5,0 до 5,5.
10. Водная фармацевтическая композиция по п. 9, где указанный пролголимаб находится в концентрации от 90 мг/мл до 110 мг/мл.
11. Водная фармацевтическая композиция любому из пп. 9, 10, где указанный пролголимаб находится в концентрации 100 мг/мл.
12. Водная фармацевтическая композиция по п. 9, где указанный трегалозы дигидрат находится в концентрации 80 мг/мл.
13. Водная фармацевтическая композиция по п. 9, где указанный натрия ацетат тригидрат находится в концентрации от 1,7 мг/мл до 1,8 мг/мл.
14. Водная фармацевтическая композиция по п. 13, где указанный натрия ацетат тригидрат находится в концентрации 1,742 мг/мл.
15. Водная фармацевтическая композиция анти-PD-1 антитела, содержащая:
(a) пролголимаб в концентрации от 5 мг/мл до 150 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации от 75 мг/мл до 85 мг/мл или в концентрации от 95 мг/мл до 105 мг/мл;
(c) L-гистидин в концентрации от 0,7 до 1,0 мг/мл;
(d) L-гистидина гидрохлорид в концентрации от 2,8 до 3,3 мг/мл; и
композиция имеет рН от 5,5 до 6,0.
16. Водная фармацевтическая композиция по п. 15, где указанный пролголимаб находится в концентрации от 15 мг/мл до 40 мг/мл.
17. Водная фармацевтическая композиция по п. 16, где указанный пролголимаб находится в концентрации от 15 мг/мл до 25 мг/мл.
18. Водная фармацевтическая композиция по п. 17, где указанный пролголимаб находится в концентрации 20 мг/мл.
19. Водная фармацевтическая композиция по любому из пп. 16-18, где указанный трегалозы дигидрат находится в концентрации от 95 мг/мл до 105 мг/мл.
20. Водная фармацевтическая композиция по п. 19, где указанный трегалозы дигидрат находится в концентрации 100 мг/мл.
21. Водная фармацевтическая композиция по п. 15, где указанный пролголимаб находится в концентрации от 90 мг/мл до 110 мг/мл.
22. Водная фармацевтическая композиция по п. 21, где указанный пролголимаб находится в концентрации 100 мг/мл.
23. Водная фармацевтическая композиция по любому из пп. 21, 22, где указанный трегалозы дигидрат находится в концентрации от 75 мг/мл до 85 мг/мл.
24. Водная фармацевтическая композиция по п. 23, где указанный трегалозы дигидрат находится в концентрации 80 мг/мл.
25. Водная фармацевтическая композиция по п. 15, где указанный L-гистидин находится в концентрации 0,92 мг/мл.
26. Водная фармацевтическая композиция по п. 15, где указанный L-гистидина гидрохлорид находится в концентрации 2,96 мг/мл.
27. Водная фармацевтическая композиция по п. 15, где указанная композиция имеет рН 5,5.
28. Водная фармацевтическая композиция анти-PD-1 антитела, состоящая из:
(a) пролголимаба в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрата в концентрации 100 мг/мл;
(c) натрия ацетата тригидрата в концентрации от 1,7 до 1,8 мг/мл; и
(d) уксусной кислоты до рН 5,0.
29. Водная фармацевтическая композиция анти-PD-1 антитела по п. 28, содержащая:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) натрия ацетат тригидрат в концентрации 1,742 мг/мл; и
(d) уксусную кислоту до рН 5,0.
30. Водная фармацевтическая композиция анти-PD-1 антитела, состоящая из:
(a) пролголимаба в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрата в концентрации 80 мг/мл;
(c) натрия ацетата тригидрата в концентрации от 1,7 мг/мл до 1,8 мг/мл; и
(d) уксусной кислоты до рН от 5,0 до 5,5.
31. Водная фармацевтическая композиция анти-PD-1 антитела по п. 30, содержащая:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) натрия ацетат тригидрат в концентрации 1,742 мг/мл; и
(d) уксусную кислоту до рН от 5,0 до 5,5.
32. Водная фармацевтическая композиция анти-PD-1 антитела, состоящая из:
(a) пролголимаба в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрата в концентрации 100 мг/мл;
(c) L-гистидина в концентрации от 0,7 до 1,0 мг/мл; и
(d) L-гистидина гидрохлорида в концентрации от 2,8 до 3,3 мг/мл;
(e) где указанная композиция имеет рН 5,5.
33. Водная фармацевтическая композиция анти-PD-1 антитела по п. 32, содержащая:
(a) пролголимаб в концентрации 20 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 100 мг/мл;
(c) L-гистидин в концентрации 0,92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2,96 мг/мл;
(e) где указанная композиция имеет рН 5,5.
34. Водная фармацевтическая композиция анти-PD-1 антитела, состоящая из:
(a) пролголимаба в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрата в концентрации 80 мг/мл;
(c) L-гистидина в концентрации от 0,7 до 1,0 мг/мл; и
(d) L-гистидина гидрохлорида в концентрации от 2,8 до 3,3 мг/мл;
(e) где указанная композиция имеет рН от 5,5 до 6,0.
35. Водная фармацевтическая композиция анти-PD-1 антитела по п. 34, содержащая:
(a) пролголимаб в концентрации 100 мг/мл в качестве антитела;
(b) трегалозы дигидрат в концентрации 80 мг/мл;
(c) L-гистидин в концентрации 0,92 мг/мл; и
(d) L-гистидина гидрохлорид в концентрации 2,96 мг/мл;
(e) где указанная композиция имеет рН от 5,5 до 6,0.
36. Водная фармацевтическая композиция анти-PD-1 антитела, состоящая из:
(a) пролголимаба в концентрации 20 мг в качестве антитела;
(b) трегалозы дигидрата в концентрации 100 мг;
(c) натрия ацетата тригидрата в концентрации 1,742 мг;
(d) уксусной кислоты до рН 5,0, и
(e) воды для инъекций до 1 мл.
37. Применение водной фармацевтической композиции антитела к PD-1 пролголимаба по любому из пп. 1-36 для лечения злокачественного новообразования у субъекта, нуждающегося в этом.
38. Применение по п. 37, где злокачественное новообразование выбрано из группы, содержащей меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого; неплоскоклеточный немелкоклеточный рак легкого, плоскоклеточный рак легкого; мелкоклеточный рак легкого, в том числе неоперабельный или метастатический мелкоклеточный рак легкого; рак легкого ранних стадий до и после радикального лечения; рак шейки матки, в том числе метастатический рак шейки матки, ранние стадии рака шейки матки до и после радикального лечения; опухоли головы и шеи, в том числе плоскоклеточный рак головы и шеи; лимфома Ходжкина; опухоли желудка и кишечника, метастатический плоскоклеточный рак пищевода; рак мочевого пузыря, в том числе метастатическая уротелиальная карцинома, рак почки; рак эндометрия, в том числе метастатический рак эндометрия, ранние стадии рака эндометрия до и после радикального лечения; рак молочной железы, в том числе метастатический рак молочной железы, ранние стадии рака эндометрия до и после радикального лечения; рак печени, в том числе метастатический или неоперабельный рак печени, ранние стадии рака печени до и после радикального лечения; неоперабельная или метастатическая солидная опухоль, в том числе неоперабельная или метастатическая солидная опухоль с признаками микросателлитной нестабильности.
39. Применение по п. 37, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела.
40. Применение по п. 37, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела.
41. Применение по п. 37, где указанную водную фармацевтическую композицию вводят каждые 2 недели.
42. Применение по п. 37, где указанную водную фармацевтическую композицию вводят каждые 3 недели.
43. Применение по п. 37, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела каждые 2 недели.
44. Применение по п. 37, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела каждые 3 недели.
45. Применение по п. 37, где указанную водную фармацевтическую композицию вводят парентерально.
46. Применение по п. 37, где указанную парентеральное введение представляет собой внутривенное, подкожное или внутримышечное введение.
47. Применение по п. 37, где указанную водную фармацевтическую композицию вводят внутривенно в виде инфузии.
48. Способ лечения злокачественного новообразования у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества водной фармацевтической композиции по любому из пп. 1-8, 16-19, 28, 29, 32, 33.
49. Способ по п. 48, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела.
50. Способ по п. 48, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела.
51. Способ по п. 48, где указанную водную фармацевтическую композицию вводят каждые 2 недели.
52. Способ по п. 48, где указанную водную фармацевтическую композицию вводят каждые 3 недели.
53. Способ по п. 48, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 1 мг/кг массы тела каждые 2 недели.
54. Способ по п. 48, где указанную водную фармацевтическую композицию вводят в дозе антитела к PD-1 пролголимаба 3 мг/кг массы тела каждые 3 недели.
55. Способ по п. 48, где указанную водную фармацевтическую композицию вводят парентерально.
56. Способ по п. 55, где указанное парентеральное введение представляет собой внутривенное, подкожное или внутримышечное введение.
57. Способ по п. 56, где указанную водную фармацевтическую композицию вводят внутривенно в виде инфузии.
58. Способ по п. 48, где злокачественное новообразование представляет собой меланому, в том числе неоперабельную или метастатическую меланому, ранние стадии меланомы до и после радикального лечения; рак легкого, немелкоклеточный рак легкого (НМРЛ), в том числе неоперабельный или метастатический немелкоклеточный рак легкого.
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Способ получения соединения висмута с винной или лимонной кислотой | 1932 |
|
SU31013A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |

