Область техники
Изобретение относится к области медицинской генетики и может быть использовано в медицинской генетике и оториноларингологии для диагностики наследственной несиндромальной тугоухости (ННТ) в популяции чувашей.
Уровень техники
Наследственная несиндромальная нейросенсорная тугоухость (ННТ) заболевание, при котором происходит снижение слуха из-за поражения органов внутреннего уха, слухового нерва или центра в головном мозге, который отвечает за восприятие звука, характеризуется широкой локусной и аллельной гетерогенностью и разными типами наследования.
Картирование генов, ответственных за возникновение нейросенсорной тугоухости, несомненно, явилось прорывом к пониманию механизмов возникновения и наследования тугоухости. За последние два десятилетия после открытия первого клонированного гена GJB2 в 1992 году [Online Mendelian Inheritance in Man] ежегодно увеличивалось число выявленных генов, ассоциированных с ННТ. На данный момент идентифицировано 99 генов, ассоциированных с аутосомно-рецессивным (АР), 67 - с аутосомно-доминантным (АД) и 7 генов - с Х-сцепленным рецессивным типом наследования ННТ. Аутосомно-рецессивные формы ННТ составляют около 70% всех форм [Meena et al., 2017].
Проведенные исследования как в Европе, так и в мире показали, что мутации гена GJB2 оказались самой частой причиной ННТ (27,1-50% случаев аутосомно-рецессивной наследственной нейросенсорной тугоухости (АР ННТ) в европейских популяциях и России [Online Mendelian Inheritance in Man, Kenneson et al, 2002]. Однако показано, что в странах Африки вклад гена GJB2 в ННТ небольшой - 5,6% [Chan et all, 2014]. Также в работах многих авторов показано, что, как и для большинства наследственных болезней, для ННТ характерна не только локусная, но и аллельная гетерогенность. В большинстве европейских стран у больных с АР ННТ самой частой среди патогенных мутаций гена GJB2 является c.35delG [Chan et all, 2014]. Вариант c.35delG превалирует и среди русского населения России с ННТ (81% мутантных аллелей GJB2) [Bliznetz et all, 2012]. Частота мутации c.35delG в европейских, американских, азиатских, африканских популяциях и популяции Океании в среднем составила 1,89%, 1,52%, 0,64%, 0,64% и 1%, соответственно [Mahdieh et all, 2009].
Менее частыми причинами, обусловливающими ННТ как в Европе, так и в мире, оказались патогенные варианты генов STRC, USH2A, SLC26A4, MY07A, OTOF, MY015A, ТЕСТА. Мутации в остальных генах наблюдались в единичных случаях [Hereditary Hearing Loss Homepage, Online Mendelian Inheritance in Man; Azaiez H., et al, 2018].
В наших предыдущих исследованиях было показано, что в популяциях и этнических группах РФ наблюдается как аллельная, так и локусная гетерогенность по ННТ [Zinchenko et al., 2003; Зинченко др., 2007; Петрина и др., 2017; Шокарев и др., 2005; Шаронова и др., 2009; Осетрова и др., 2010; Zinchenko et al., 2012; Zinchenko et al., 2012; Петрина и др, 2018; Zinchenko et al., 2018]. Но вклад гена GJB2 в развитие ННТ у представителей ряда этнических групп, кроме русских, незначителен. Так, было показано, что популяционная частота мутации c.35delG составила у башкир 0,25%, карачаевцев 0,14%, у удмуртов 0,25% при одинаковой с русскими частоте заболевания 1:2000-2500 новорожденных. Секвенирование гена GJB2 позволило дополнительно определить только 1-2 мутации с очень низкими частотами.
Чуваши являются пятым по численности народом Российской Федерации. Исследование, проведенное ранее, не выявило нарушений гена GJB2 у чувашских пациентов с ННТ [Zinchenko et al., 2003; Зинченко др., 2007]. В связи с этим необходимость выявления молекулярно-генетических основ развития данного заболевания с целью разработки эффективных методов диагностики с учетом этнической принадлежности больного является важной задачей исследования.
Изучение структурных особенностей генома, приводящих к развитию наследственных заболеваний, выявление патогенных вариантов генов, являющихся молекулярной причиной у значительного числа пациентов, позволяет разрабатывать методы, позволяющие быстро и точно тестировать эти варианты у значительного числа лиц.
Применение разработанного способа обеспечит выявление лиц с несиндромальной нейросенсорной тугоухостью (ННТ), тип 15 (DFNB15), с целью уточнения дифференциального диагноза и осуществления своевременных, целенаправленных мероприятий по профилактике развития данной патологии.
До сих пор варианты гена GIPC3, ответственного за развитие ННТ, тип 15, выявляли в ходе применения методов высокопроизводительного секвенирования (анализа полного генома или полного экзома [Charizopoulou N, et al., 2011; Rehman AU et al, 2011; Ramzan К et al., 2013; Diaz-Horta О et al., 2012; Sirmaci A et al., 2012], секвенирования панели генов [Bitarafan Fet al., 2020; Kannan-Sundhari A et al., 2020]) - высокотехнологичной и трудозатратной процедуры, требующей задействования биоинформационных ресурсов и продолжительного времени биоинформатического анализа, наличия дорогостоящего оборудования и квалифицированных биоинформатиков, что можно рассматривать как недостаток метода.
Раскрытие сущности изобретения
Задачей изобретения явилась разработка объективного, информативного, но простого, быстрого и дешевого способа молекулярно-генетической диагностики патогенного варианта, обусловливающего значительную долю заболеваемости ННТ в популяции чувашей.
Ввиду высокой генетической гетерогенности наследственной патологии слуха, а молекулярно-генетические причины ННТ в чувашской популяции неизвестны, обследование пациентов чувашской национальности проводили в несколько этапов. На первом этапе исследования, в качестве генов-кандидатов, выбраны гены коннексинов, наиболее часто ассоциированных с заболеванием (GJB2, GJB6, GJB3), следует проанализировать наличие как точковых мутаций (секвенирование по Сенгеру), так и вариаций числа копий локусов путем мультиплексной реакции лигазо-зависимой амплификации зондов (MLPA). На следующем этапе, ввиду высокой генетической гетерогенности ННТ и необнаружения патогенных вариантов в генах GJB2, GJB6, GJB3, проведено полноэкзомное секвенирование у пациента, родители которого также поражены. Следующим этапом выполнено секвенирование гена, в котором выявлены вероятно патогенные варианты, у протестированного пациента (ген GIPC3) и в выборке пациентов с ННТ, а далее определение популяционной частоты выявленного варианта.
Обследовано 26 пациентов с наследственной нейросенсорной тугоухостью (ННТ) из 21 неродственных семей. Все пациенты и их родители - чуваши из Республики Чувашия. Средний возраст 18,57±2,10 лет (SD=10,72; 1,00-42,00). У всех обследованных произведен забор биоматериала для молекулярно-генетического исследования при проведении комплексного генетико-эпидемиологического исследования чувашской популяции. В Таблице 1 представлены клинические портреты пациентов обследуемой выборки.
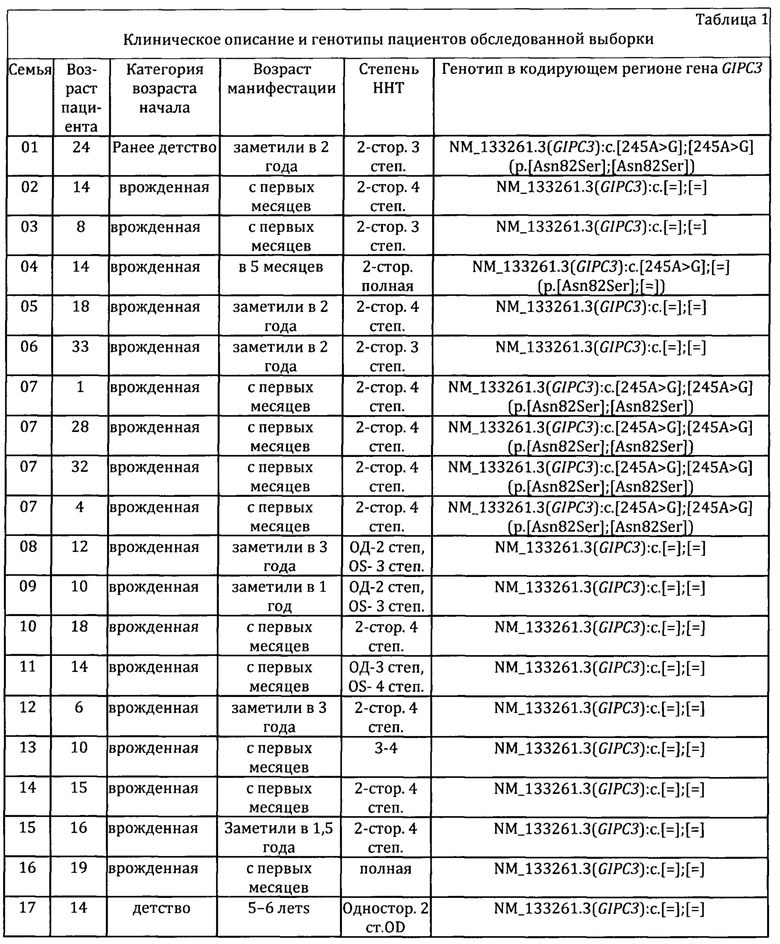
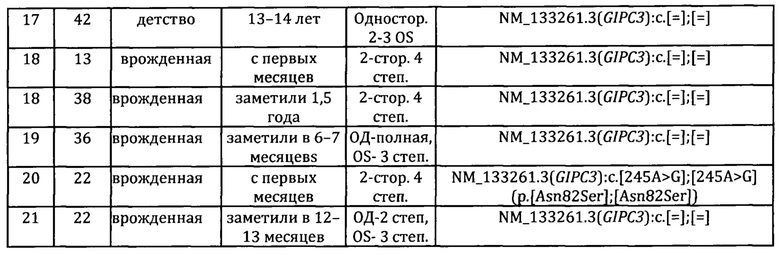
На первом этапе проведено секвенирование кодирующих последовательностей и фланкирующих их интронных зон генов GJB2, GJB6, GJB3, поиск варианта c.-23+1G>A в гене GJB2 и определение числа копий локусов GJB3 (1р34.3), WFS1 (4р16.1), PSPC1 (13q12.11), ZMYM2 (13q12.11), GJB2 (13q12.11), GJB6 (13q12.11), LATS2 (13q12.11), P0U3F4 (Xq21.1). Ни у одного пациента патогенных изменений не выявлено.
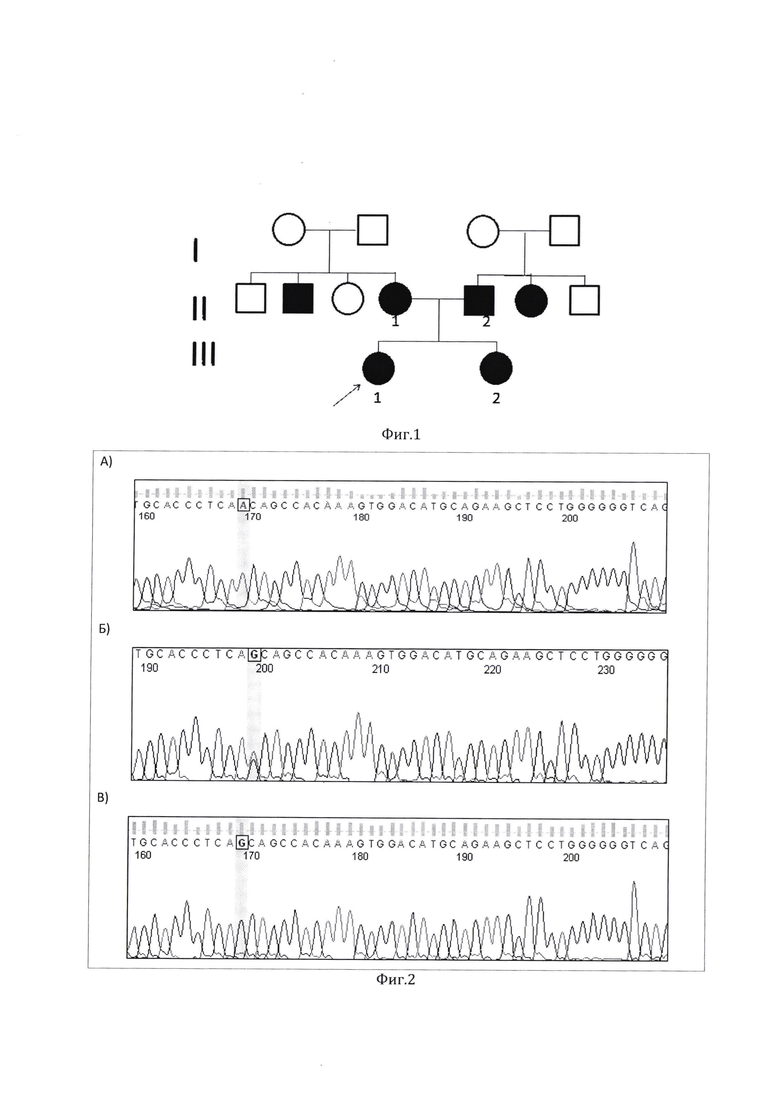
На втором этапе провели полноэкзомное секвенирование ДНК одного пациента из семьи 07, в которой оба родителя и сибс были поражены (НСТ 4 степени - глухонемота) (фигура 1). Родители - по национальности этнические чуваши, не состоявшие в кровнородственном браке. Бабушки и дедушки со стороны как матери, так и отца без нарушений слуха.
Пациент III-1 (семья 07) был отправлен на анализ нуклеотидной последовательности ДНК высокопроизводительного секвенирования. Анализ кодирующих последовательностей генов методом полноэкзомного секвенирования проведен с использованием таргетного обогащения геномной ДНК системой Agilent SureSelect V4 (51М), расчетное среднее покрытие 75×. Секвенирование выполнено на приборе Illumina HiSeq 2000 с использованием парно-концевых прочтений длиной 2×100 п. о. Выявленные варианты были проанализированы с помощью комплекса программных алгоритмов, позволяющих аннотировать каждую выявленную мутацию и полиморфизм в масштабах всего генома с учетом следующих параметров: эволюционная консервативность; потенциальное влияние мутации на структуру гена и белка, в том числе на экзон-интронную структуру (нарушения сайтов сплайсинга); предсказанное влияние мутации на функцию белка, в том числе по алгоритмам PolyPhen2 и SIFT; наличие мутации в базах данных dbSNP и ClinVar и ее клинический статус; популяционные частоты встречаемости вариантов в проектах "1000 геномов" и "6500 экзомов (ESP6500)". Для анализа используются все известные варианты транскрипта каждого гена из базы NCBI RefSeq. При поиске клинически значимых мутаций для моногенных заболеваний не выводятся полиморфизмы, не влияющие на структуру белка и при этом не отмеченные как патогенные в ClinVar, а также любые мутации/полиморфизмы с частотой встречаемости в популяциях более 2%. Метод предназначен для поиска однонуклеотидных замен в кодирующих участках генов человека. С помощью этой технологии может быть определена последовательность 90-95% кодирующих участков генов человека с покрытием не ниже 10х.
Выявлен вариант нуклеотидной последовательности g.chr19:3586512A>G, NM_133261.3(GIPC3):c.245A>G; p.(Asn82Ser), в гомозиготном состоянии, приводящий к замене аспарагина на серии p.(Asn82Ser) в эволюционно консервативной позиции белка, кодируемого геном GIPC3. Мутация находится во втором экзоне гена. Вариант NM_133261.3(GIPC3):c.245A>G в гене GIPC3 идентифицирован на 9 из 30778 хромосомах из южноазиатской популяции (9/30778) в базе Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org; dbSNP rs747242422). Хотя вариант отмечен в общей популяции, его частота недостаточна, чтобы исключить его патогенную роль. Инструменты компьютерного прогнозирования и анализ консервативности предполагают, что вариант может нарушать функцию белка. Ген GIPC3 кодирует белок длиной 312 аминокислотных остатков, участвующий в функционировании чувствительных клеток внутреннего уха и нейронов спирального ганглия. Мутации в данном гене ассоциированы с несиндромальной нейросенсорной тугоухостью, тип 15 [Charizopoulou et al., 2011], с аутосомно-рецессивным наследованием; у мышей мутации данного гена ассоциированы с несиндромальной потерей слуха либо с синдромом ювенильных аудиогенных судорог.У пациентов с нейросенсорной тугоухостью описаны нонсенс-, миссенс- и фреймшифт-мутации гена GIPC3 в гомозиготной или компаунд-гетерозиготной форме [Rehman et al., 2011].
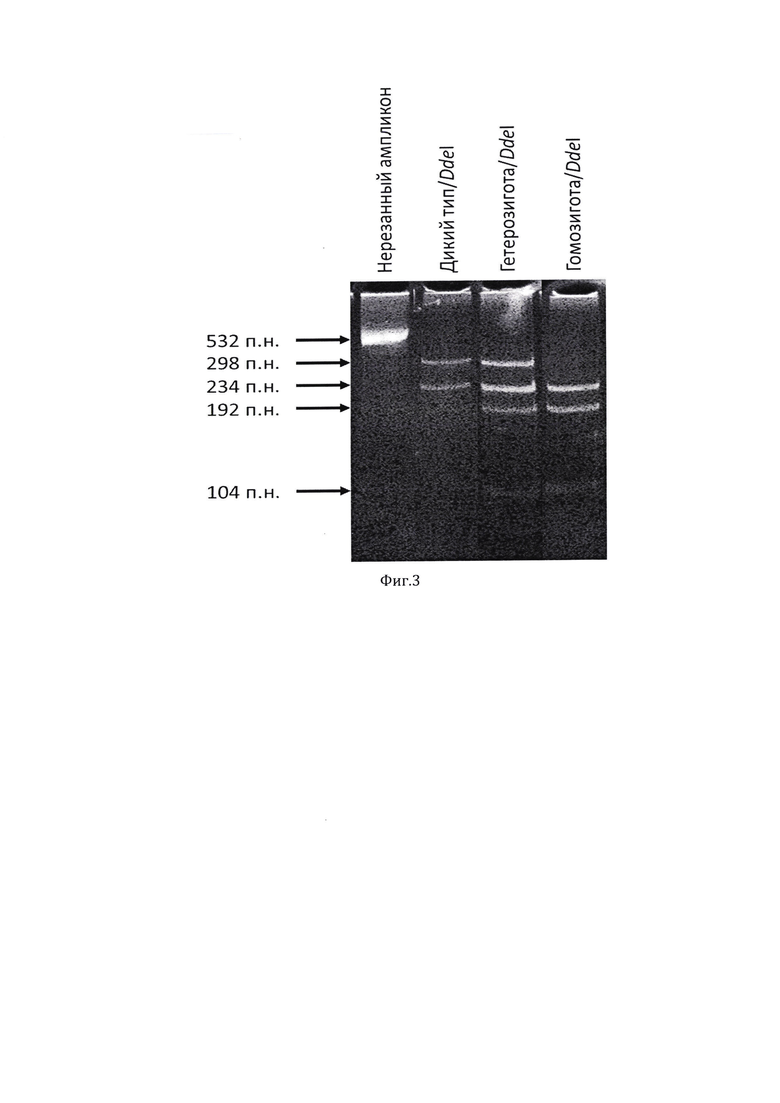
Для подтверждения наличия выявленных мутаций у пациента использован метод секвенирования ДНК по Сэнгеру. На фигуре 2 представлены результаты секвенирования. У пациентов (III-1 и III-2) и родителей (II-1 и II-2) проведен сегрегационный анализ, который позволил установить, что все пораженные члены семьи имеют данный вариант в гомозиготном состоянии.
На третьем этапе проведено двунаправленное секвенирование шести экзонов гена GIPC3 на выборке 22 чувашских пациентов с ННТ. Для этого разработана система праймеров на все экзоны гена GIPC3 (Таблица 2). Дополнительно выявлены два пациента, гомозиготные по варианту c.245A>G, и один пациент, гетерозиготный по варианту c.245A>G в гене GIPC3. Т.е. всего выявлено шесть пациентов, гомозиготных по варианту c.245A>G, и один гетерозиготный носитель этой замены. Таким образом, частота варианта c.245A>G у пациентов с ННТ - чувашей составила 25,0% (13/52).
В настоящее время в гене GIPC3 описан ряд различных мутаций, ассоциированых с АР ННТ, типом 15 (DFNB15) [Rehman et al., 2011, Charizopoulou et al., 2011, Ramzan K., et al., 2013, Diaz-Horta et al., 2012; Sirmaci et al., 2012]. Большинство описанных случаев встретилось в Пакистане; в Индии и в Голландии по единичному случаю. У всех описанных пациентов диагностировали двустороннюю прелингвальную тугоухость с различной степенью нарушений слуха от средней до полной. Вариант NM_133261.3(GIPC3):c.245A>G обнаружен двумя группами исследователей [Bitarafan F.,2020; Kannan-Sundhari А., 2020] в двух иранских близкородственных семьях у трех пациентов с ННТ в гомозиготном состоянии. В Российской Федерации, в частности в Республике Чувашия, о случаях ННТ тип 15 (DFNB15) ранее не сообщалось.
Таким образом, вариант NM_133261.3(GIPC3):c.245A>G является молекулярной причиной заболевания ННТ у 23% пациентов чувашской национальности (6/26), а выявлен в 25,0% мутантных хромосом (13/52).
Технический результат
Технический результат при использовании изобретения - оптимизация способа молекулярной диагностики патогенных вариантов, приводящих к ННТ, ассоциированной с геном GIPC3, в чувашской популяции.
Указанный технический результат достигается тем, что выделяют ДНК методом солевой экстракции, проводят генотипирование на наличие варианта NM_133261.3(GIPC3):c.245A>G в гене GIPC3 у пациентов с ННТ и у здоровых родственников для поддверждения сегрегации варианта в семье; обнаружение варианта NM_133261.3(GIPC3):c.245A>G у пациента с ННТ в гомозиготном состоянии является подтверждением диагноза наследственной нейросенсорной тугоухости тип 15, обнаружение этого варианта в гетерозиготном состоянии указывает, по крайней мере, на статус гетерозиготного носителя индивида и предполагает дальнейшее секвенирование экзонов гена GIPC3 с целью обнаружения второго генетического варианта в транс-положении. В качестве биологического материала можно использовать образцы из группы: венозной или капиллярной крови, слюны, буккального соскоба, осадка эпителия нижних мочевыводящих путей.
Осуществление изобретения
Основным материалом для достижения технического результата является ДНК, выделенная из венозной крови.
ДНК выделяют из крови методом солевой экстракции с использованием набора для выделения ДНК «Wizard Genomic DNA Purification Kit» фирмы «Promega» (USA), либо альтернативных наборов для выделения геномной ДНК. Венозную кровь собирают в пробирку-вакутейнер, содержащую антикоагулянт ЗмМ ЭДТА (двунатриевая соль диаминтетрауксусной кислоты), тщательно перемешивают, хранят в холодильнике при +4°С не более недели. Для выделения ДНК используют «набор для выделения ДНК»: к 200 мкл крови добавляют 600 мкл лизирующего буфера А. Полученную смесь перемешивают и центрифугируют при 4°С, 12000 об./мин в течение 20 секунд. Надосадочную жидкость сливают, к осадку добавляют 200 мкл солевой буфера Saline и инкубируют при 37°С в течение 15 минут. После этого к лизату добавляют 100 мкл преципитационный буфер, перемешивают, встряхивают на вортексе до выпадения осадка; центрифугируют при 4°С, 12000 об./мин в течение 1 минуты. Надосадочную жидкость переносят в чистый эппендорф, добавляют 300 мкл изопропанола, перемешивают, центрифугируют при 4°С, 12000 об./мин в течение 20 секунд. Надосадочную жидкость отсасывают пипеткой, добавляют 300 мкл 70% этанола, промывают, перемешивают, центрифугируют при 4°С, 12000 об./мин в течение 20 секунд. Осадок подсушивают в термостате при 65°С в течение 5 минут, растворяют в 100 мкл дистиллированной воды и хранят при температуре -20°С. Выделенная ДНК используется для проведения полимеразной цепной реакции синтеза ДНК (ПЦР).
Амплификацию фрагмента ДНК 2 экзона гена GIPC3, содержащего сайт расположения варианта NM_133261.3(GIPC3):c.245A>G, проводят методом ПЦР синтеза ДНК в 20 мкл общего объема смеси, содержащей 2 мкл 10×Taq-буфера (67 мМ трис-HCl (рН 8,8), 16,6 мМ (NH4)2SO4, 2 мМ MgCl2, 0,01% Tween-20), 0,1 мкг геномной ДНК, смесь dNTP (dATP, dGTP, dCTP, dTTP no 200 мкМ каждого), 1,5 ед. SmarTaq ДНК полимеразы (производства фирмы «ДиаЛат», г. Москва) и 5-10 рМ специально разработанных локусспецифичных олигонуклеотидных праймеров.
Последовательности праймеров, размеры амплифицируемых фрагментов и температурные режимы ПЦР представлены в таблице 2.
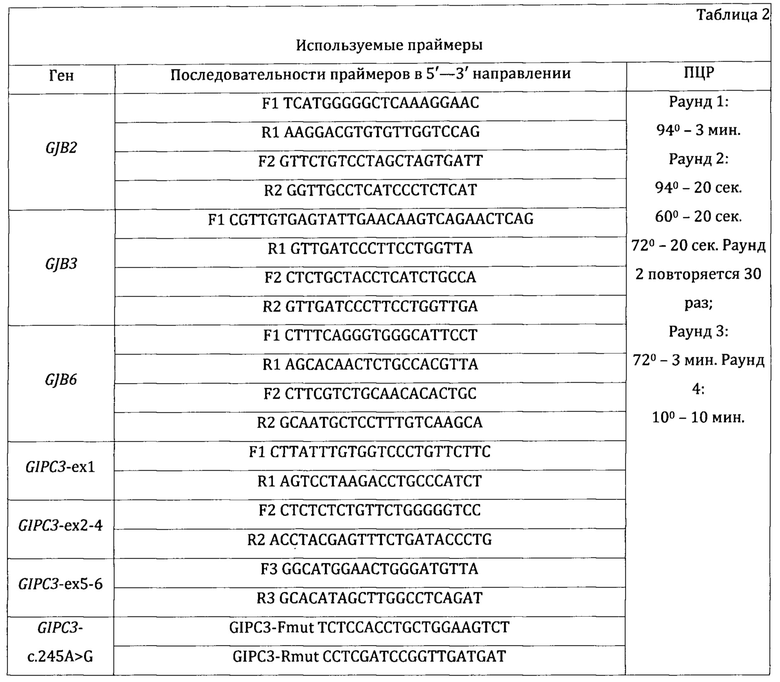
Для определения нуклеотидной замены NM_133261.3(GIPC3):c.245A>G проводят гидролиз амплифицированных фрагментов рестриктазой BstDEI (прототип Ddel) в соответствии с рекомендациями фирмы-производителя (ООО «Сибэнзим», Новосибирск). Данные о длине амплифицированного фрагмента, название рестриктазы и длины продуктов расщепления представлены в таблице 3.
Разделение фрагментов ДНК после амплификации и рестрикции проводят при помощи электрофореза в 8% полиакриламидном геле (ПААГ), приготовленном из 30% раствора ПААГ (соотношение акриламид: N,N'-метиленбисакриламид - 29:1). Электрофорез проводят в 1×ТБЕ буфере (0,089 М трис-HCl; 0,089 М борная кислота; 0,002 М ЭДТА, рН=8,0) в течение 1,5 часов. Перед нанесением на гель пробы смешивают в соотношении 5:1 с краской, содержащей 0,25% бромфенолового синего, 0,25% ксиленцианола и 40% сахарозы. После окончания электрофореза гель окрашивают раствором бромистого этидия в течение 5 секунд и визуализируют при УФ-освещении на трансиллюминаторе. Результаты электрофореза фрагментов ДНК после амплификации и рестрикции BstDEI (прототип Ddel) представлены на фигуре 3.
Частоты аллелей определяют по формуле: pi=Ni/N, где Ni - число i-гo аллеля, N - объем выборки [Животовский, 1991]. Минимальное и максимальное значения 95% доверительного интервала рассчитывали согласно методу Вальда [Животовский, 1991].
Валидация способа молекулярно-генетической диагностики патогенного варианта NM_133261.3(GIPC3):c.245A>G с использованием ПЦР-амплификации с последующей рестрикцией проведена на группе 26 чувашских пациентов с ННТ, исследованных ранее методом секвенирования по Сэнгеру, и тестированием здоровых индивидов, относящимся к разным этническим группам (175 чувашей, 93 русских из Кировской области, 320 марийцев, 42 удмурта, 183 татар и 283 башкир).
Результаты анализа ДНК 26 чувашских пациентов с ННТ, проведенного разработанным способом: ПЦР-амплификация с последующей рестрикцией, не различались от результатов, полученных при генотипировании методом секвенирования: шесть пациентов, имеющих гомозиготные по варианту NM_133261.3(GIPC3):c.245A>G в гене GIPC3 генотипы, и один - гетерозиготный. Частота варианта у чувашских пациентов составила 0,2500 (95%ДИ 0,1403-0,3895). Результат достигнут в течение одного рабочего дня: выделение ДНК - 2 часа; пробоподготовка - 30 мин; амплификация - 1,5 часа; пробоподготовка к рестрикции - 30 мин; рестрикция - 3 часа; электрофорез с документацией результата - один час 40 минут.
В результате тестирования 175 здоровых чувашских индивидов выявлены 4 гетерозиготных носителя варианта. Частота носительства варианта NM_133261.3(GIPC3):c.245A>G в чувашской популяции составила 1: 44 (175/4), а популяционная частота - 0,01142 (95%ДИ 0,0031-0,290) (4/350 хромосом), т.е. более 1%. Это очень высокое значение для патогенного аллеля, поэтому было проведено исследование гетерозиготного носительства варианта NM_133261.3(GIPC3):c.245A>G в российских популяциях с другим этническим составом.
Протестированы здоровые индивиды из других, соседних с чувашской, популяций Волго-Уральского региона (93 русских из Кировской области, 320 марийцев, 42 удмурта, 183 татар и 283 башкир) на носительство варианта NM_133261.3(GIPC3):c.245A>G. В протестированных выборках не выявлено ни одного носителя варианта NM_133261.3(GIPC3):c.245A>G. Т.е. можно заключить, что вариант c.245A>G в гене GIPC3 в этнических группах, помимо чувашей, является очень редким (0 из 1842 хромосом, частота 0,0000; 95% ДИ 0,0000-0,0016).
Вариант NM_133261.3(GIPC3):c.245A>G является молекулярной причиной наследственной тугоухости у 23% пациентов-чувашей, а его высокая частота в популяции может быть обусловлена рядом последовательных событий произошедших в историческом прошлом чувашской популяции: эффектом основателя, «бутылочного горлышка» и/или дрейфом генов.
Использование данного способа идентификации патогенного варианта NM_133261.3(GIPC3):c.245A>G позволяет с высокой точностью подтвердить ННТ, обусловленную этим вариантом, и при этом ускоряет и удешевляет процесс молекулярно-генетической диагностики ННТ для популяции чувашей.
Краткое описание чертежей
Фигура 1. Родословная семьи 07 с несиндромальной нейросенсорной тугоухостью.
Фигура 2. Хроматограмма секвенирования фрагмента экзона 2 гена GIPC3:
а) гомозигота по мутации NM_133261.3(GIPC3):c.[245A>G];[245A>G];
б) гетерозиготный носитель мутации NM_133261.3(GIPC3):c.[245A>G];[=];
в) гомозигота по дикому типу NM_133261.3(GIPC3):c.[=];[=].
Фигура 3. Электрофореграмма продуктов рестрикции, демонстрирующая тест-систему для определения варианта NM_133261.3(GIPC3):c.245A>G.
Слева направо:
нерезаный - продукт амплификации - 532 п. н.,
продукты рестрикции эндонуклеазой BstDEI (Ddel):
дикий тип NM_133261.3(GIPC3):c.[=];[=]- 298 п. н. и 234 п. н.;
гетерозигота NM_133261.3(GIPC3):c.[245A>G];[=] - 298 п. н., 234 п. н., 192 п. н. и 104 п. н.;
гомозигота NM_133261.3(GIPC3):c.[245A>G];[245A>G] - 234 п. н., 192 п. н. и 104 п. н. Список литературы
1. Online Mendelian Inheritance in Man. URL: http://www.ncbi.nlm.nih.gov/OMIM
2. Meena, R.; Ayub, M. Genetics of human hereditary hearing impairment. J. Ayub. Med. Coll. Abbottabad 2017, 29, 671-676.
3. Kenneson A., Van Naarden Braun, K. & Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet. Med. 2002; 4: 258-274. https://doi.org/10.1097/00125817-200207000-00004
4. Hereditary Hearing Loss Homepage https://hereditaryhearingloss.org/
5. Azaiez H., Booth K.T., Ephraim S.S., Crone В., Black-Ziegelbein E.A., Marini R.J., Eliot Shearer A., Sloan-Heggen С.М., Kolbe D., Casavant Т., Schnieders M.J., Nishimura C, Braun Т., Smith R.J.H. Genomic landscape and mutational signatures of deafness-associated genes. Am. J. Hum. Genet. 2018;103,484-497. https://doi.Org/10.1016/j.ajhg.2018.08.006.
6. Chan D.K., & Chang K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. The Laryngoscope, 2014;124(2):E34-E53. https://doi.org/10.1002/lary.24332
7. Bliznetz E.A., Galkina V.A., Matyushchenko G.N., Kisina A.G., Markova T.G., Polyakov A.V. Changes in the connexin 26 gene (GJB2) in Russian patients with hearing loss: Results of long-term molecular diagnostics of hereditary nonsyndromic hearing loss. Rus J of Genetics. 2012; 48(1): 101-112. doi:10.1134/sl022795412010036
8. Mahdieh N., Rabbani B. Statistical study of 35delG mutation of GJB2 gene: a metaanalysis of carrier frequency // Int J of Audiology. - 2009. - Vol. 48. - P. 363-70
9. Zinchenko R.A., Zinchenko S.P., Galkina V.A., Elchinova G.I., Nurbaev S.D., Polyakov A.V., Nekrasova N.Yu., Ginter E.K. Prevalence and molecular genetic typing of nonsyndromic sensorineural deafness in Chuvash Republic // Rus J of Genetics. 2003. T. 39. №9. C. 1076-1084. doi: 10.1023/A:1025739521156
10. Зинченко С.П., Кириллов А.Г., Абрукова А.В., Сорокина Т.В., Шаронова Е.И., Хидиятова И.М., Джемилева Л.У., Шокарев Р.А., Близнец Е.А., Хуснутдинова Э.К., Зинченко Р.А., Гинтер Е.К. Генетико-эпидемиологическое исследование наследственных (изолированных и синдромальных) нарушений слуха в Республике Чувашия. Медицинская генетика. 2007. Т. 6. №5. С. 18-28.
11. Петрина Н.В., Близнец Е.А., Зинченко Р.А., Макаов А.Х-М., Петрова Н.В., Васильева ТА, Чудакова Л.В., Петрин А.Н., Поляков А.В., Гинтер Е.К., Частота мутаций гена GJB2 у больных наследственной несиндромальной нейросенсорной тугоухостью в девяти популяциях Карачаево-Черкесской Республики // Медицинская генетика. 2017. Т. 16. №2. С. 19-25.
12. Шокарев Р.А., Амелина С.С., Кривенцова Н.В., Хлебникова О.В., Близнец Е.А., Поляков А.В., Зинченко Р.А. Генетико-эпидемиологическое и молекулярно-генетическое исследование наследственной тугоухости в Ростовской области// Медицинская генетика. - 2005. - Т. 4, №12. - С. 556-567.
13. Шаронова Е.И., Осетрова А.А., Зинченко Р.А. Наследственные нарушения слуха в Кировской области // Якутский медицинский журнал. - 2009. №2(29). - С. 28-31.
14. Осетрова А.А., Шаронова Е.И., Российская Т.Г., Галкина В.А., Зинченко Р.А. Изучение генетических причин врожденной и ранней детской тугоухости в специализированных школах для детей с нарушением слуха в Кировской области // Медицинская генетика. - 2010. - Т.9, №9 (99). - С. 30-40.
15. Zinchenko R.A., Sharonova Е.I., El'chinova G.I, Osetrova A.A. Hereditary deafness in Kirov oblast: a genetic epidemiological study // Rus J of Genetics s. 2012. T. 48. №3. C. 329-335. doi: 10.1134/S102279541203012X.
16. Zinchenko R.A., Sharonova E.I, Osetrova A.A. Hereditary deafness in Kirov oblast: Estimation of the incidence rate and DNA diagnosis in children // Rus J of Genetics. 2012. T. 48. №4. C. 455-462 doi: 10.1134/S1022795412030131.
17. Петрина H.E., Макаов A.X.-M., Зинченко P.A., Ижевская В.Л., Марахонов А.В., Близнец Е.А., Петрова Н.В., Васильева ТА, Поляков А.В., Гинтер Е.К. Особенности медико-генетического консультирования семей с несиндромальной нейросенсорной тугоухости при ассортативных браках // Медицинская генетика. 2018. Т. 17. №5. С. 47-50.
18. Zinchenko R.A., Kadyshev V.V., El'chinova G.I., Marakhonov A.V. et al. Study of the genetic load and diversity of hereditary diseases in the Russian population of the Karachay-Cherkess Republic // International Journal of Molecular Epidemiology and Genetics. 2018. T. 9. №4. C. 34-42. /IJMEG0081697.
19. Charizopoulou N, Lelli A, Schraders M, Ray K, Hildebrand MS, Ramesh A, Srisailapathy CR, Oostrik J, Admiraal RJ, Neely HR, Latoche JR, Smith RJ, Northup JK, Kremer H, Holt JR, Noben-Trauth K. Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat Commun. 2011 Feb 15;2:201. doi:10.1038/ncommsl200. PMID: 21326233; PMCID: PMC3105340.
20. Rehman AU, Gul K, Morell RJ, Lee K, Ahmed ZM, Riazuddin S, Ali RA, Shahzad M, Jaleel AU, Andrade PB, Khan SN, Khan S, Brewer CC, Ahmad W, Leal SM, Riazuddin S, Friedman ТВ. Mutations of GIPC3 cause nonsyndromic hearing loss DFNB72 but not DFNB81 that also maps to chromosome 19p.Hum Genet. 2011 Dec;130(6):759-65. doi: 10.1007/s00439-011-1018-5. Epub 2011 Jun 10. PMID: 21660509; PMCID: PMC3303183.
21. Ramzan K, Al-Owain M, Allam R, Berhan A, Abuharb G, Taibah K, Imtiaz F. Homozygosity mapping identifies a novel GIPC3 mutation causing congenital nonsyndromic hearing loss in a Saudi family. Gene. 2013 May 25;521(l):195-9. doi: 10.1016/j.gene.2013.03.042. Epub 2013 Mar 16. PMID: 23510777.
22. Diaz-Horta 0, Duman D, Foster J 2nd, Sirmaci A, Gonzalez M, Mahdieh N, Fotouhi N, Bonyadi M, Cengiz FB, Menendez I, Ulloa RH, Edwards YJ, Zuchner S, Blanton S, Tekin M. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One. 2012;7(11):e50628. doi: 10.1371/journal.pone.0050628. Epub 2012 Nov 30. PMID: 23226338; PMCID: PMC3511533.
23. Sirmaci A, Edwards YJ, Akay H, Tekin M. Challenges in whole exome sequencing: an example from hereditary deafness. PLoS One. 2012;7(2):e32000. doi: 10.1371/journal.pone.0032000. Epub 2012 Feb 21. PMID: 22363784; PMCID: PMC3283682.
24. Bitarafan F, Seyedena SY, Mahmoudi M, Garshasbi M. Identification of novel variants in Iranian consanguineous pedigrees with nonsyndromic hearing loss by next-generation sequencing. J Clin Lab Anal. 2020 Dec;34(12):e23544. doi: 10.1002/jcla.23544. Epub 2020 Aug 30. PMID: 32864763; PMCID: PMC7755797.
25. Kannan-Sundhari A, Yan D, Saeidi K, Sahebalzamani A, Blanton SH, Liu XZ. Screening Consanguineous Families for Hearing Loss Using the MiamiOtoGenes Panel. Genet Test Mol Biomarkers. 2020 Oct;24(10):674-680. doi: 10.1089/gtmb.2020.0153. Epub 2020 Sep 29. PMID: 32991204; PMCID: PMC7585618.
26. gnomAD, http://gnomad.broadinstitute.org; dbSNP rs747242422
27. https://www.ncbi.nlm.nih.gov/clinvar/variation/506266/, обращение от 10.02.2021.
28. Животовский, Л.А. Популяционная биометрия // М.: Наука, 1991. - 272 с.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ диагностики мутации 35delG (rs80338939) гена GJB2 | 2020 |
|
RU2739889C1 |
| Способ диагностики мутации 167delT (rs80338942) гена GJB2 | 2020 |
|
RU2739943C1 |
| Способ диагностики мутации c.-23+1G>A (rs80338940) гена GJB2 | 2020 |
|
RU2746055C1 |
| Способ ДНК-диагностики аутосомно-рецессивной глухоты-103 | 2019 |
|
RU2727684C1 |
| СПОСОБ ДЕТЕКЦИИ 17 МУТАЦИЙ ГЕНОВ GJB2 И GJB6 ПРИ НАСЛЕДСТВЕННОЙ НЕСИНДРОМАЛЬНОЙ ГЛУХОТЕ | 2010 |
|
RU2448163C2 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИИ с.-53-2А>G В ГЕНЕ ПРЕСТИНА (SLC26A5), ВЫЗЫВАЮЩЕЙ РАЗВИТИЕ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ | 2012 |
|
RU2505608C1 |
| Способ преимплантационного генетического тестирования несиндромальной нейросенсорной тугоухости | 2022 |
|
RU2791878C1 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ GJB2, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ | 2006 |
|
RU2317547C1 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ MYO7A, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ И СИНДРОМОМ УШЕРА | 2013 |
|
RU2555755C1 |
| Способ выявления мутаций гена GJB2, обуславливающих аутосомно-рецессивную глухоту 1А типа | 2017 |
|
RU2688180C1 |
Изобретение относится к биотехнологии, а именно к медицинской генетике, и может быть использовано в медицинской генетике и оториноларингологии для диагностики наследственной несиндромальной тугоухости (ННТ) в популяции чувашей. Предложенный способ включает в себя метод полимеразной цепной реакции с последующим анализом полиморфизма длин рестрикционных фрагментов (ПЦР-ПДРФ) с использованием праймеров, фланкирующих локус варианта нуклеотидной последовательности NM_133261.2(GIPC3):c.245A>G: F-TCTCCACCTGCTGGAAGTCT, R-CCTCGATCCGGTTGATGAT, и эндонуклеазы рестрикции BstDEI. Предлагаемый способ позволяет с высокой точностью подтвердить ННТ, обусловленную вариантом NM_133261.3(GIPC3):c.245A>G, и при этом ускоряет и удешевляет процесс молекулярно-генетической диагностики ННТ для популяции чувашей. 1 з.п. ф-лы, 3 ил., 3 табл., 3 пр.
1. Способ подтверждающей молекулярно-генетической диагностики наследственной нейросенсорной тугоухости (ННТ), при котором у пациента из чувашской популяции, имеющего клинические признаки ННТ, производят забор образца биологического материала для проведения ДНК-диагноститки, в отношении которого первоначально осуществляют поиск варианта нуклеотидной последовательности NM_133261.2(GIPC3):c.245A>G методом полимеразной цепной реакции с последующим анализом полиморфизма длин рестрикционных фрагментов (ПЦР-ПДРФ) с использованием системы праймеров, фланкирующих локус варианта нуклеотидной последовательности NM_133261.2(GIPC3):c.245A>G: F-TCTCCACCTGCTGGAAGTCT, R-CCTCGATCCGGTTGATGAT, и эндонуклеазы рестрикции BstDEII (прототип Ddel), и на основании результатов этого поиска производят дальнейшие мероприятия, выбранные из следующей группы диагностических инструментов:
- в случае обнаружения варианта NM_133261.2(GIPC3):c.245A>G в гомозиготном состоянии пациенту подтверждается диагноз ННТ и проводится медико-генетическое консультирование семьи,
- в случае отсутствия варианта NM_133261.2(GIPC3):c.245A>G или его наличия в гетерозиготной форме проводят секвенирование 6 экзонов гена GIPC3.
2. Способ по п. 1, отличающийся тем, что производят забор образца биологического материала из группы венозной или капиллярной крови, слюны, буккального соскоба, осадка эпителия нижних мочевыводящих путей.
| СПОСОБ ДИФФЕРЕНЦИАЛЬНОЙ ДИАГНОСТИКИ ПРОФЕССИОНАЛЬНОЙ НЕЙРОСЕНСОРНОЙ ТУГОУХОСТИ И НЕЙРОСЕНСОРНОЙ ТУГОУХОСТИ НЕПРОФЕССИОНАЛЬНОГО ГЕНЕЗА | 2008 |
|
RU2367347C1 |
| ЗИНЧЕНКО С.П, и др | |||
| Генетико-эпидемиологическое исследование наследственных (изолированных и синдромальных) нарушений слуха в Республике Чувашия | |||
| Медицинская генетика, 2007, т | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| ZINCHENKO R.A., et al | |||
| Study of the genetic load and diversity of hereditary diseases in the Russian population of the | |||

