Область техники
Настоящее изобретение относится к С-арил глюкозидам, которые являются ингибиторами натрий-зависимых переносчиков глюкозы, обнаруживаемых в кишечнике и почках (SGLT2), а также к способу лечения сахарного диабета, преимущественно сахарного диабета 2-го типа, гипергликемии, гиперинсулинемии, ожирения, гипертриглицеридемии, Синдрома X, осложнений сахарного диабета, атеросклероза и сопутствующих состояний, заключающемуся в использовании таких С-арил глюкозидов как в качестве монотерапии, так и в комбинации с одним, двумя или более антидиабетическими препаратами и в комбинации с одним, двумя или более другими лекарственными средствами, такими, как антигиперлипидемические препараты.
Предпосылки создания изобретения
Приблизительно 100 миллионов человек во всем мире страдают сахарным диабетом 2-го типа (NIDDM), для которого характерна гипергликемия, являющаяся следствием избыточной продукции глюкозы печенью и инсулинорезистентности периферических тканей, причина которых до сих пор неизвестна. Гипергликемия считается основным фактором риска для развития осложнений сахарного диабета, и, вероятно, вносит непосредственный вклад в ухудшение секреции инсулина, наблюдаемой при развитом сахарном диабете 2-го типа. Нормализация уровня глюкозы в плазме крови у пациентов, страдающих NIDDM, позволит улучшить секрецию инсулина и отсрочить развитие осложнений сахарного диабета.
Предположительно, ингибирование натрий-зависимых переносчиков глюкозы SGLT2 в почках будет способствовать нормализации уровня глюкозы в плазме крови, а также, возможно, нормализации массы тела за счет увеличения экскреции глюкозы.
Создание новых безопасных и пероральных антидиабетических средств также желательно для дополнения уже существующей терапии, основанной на использовании таких препаратов, как производные сульфонилмочевины, метформин и инсулин, а также для снижения возможных побочных эффектов, возникающих при использовании других указанных сахароснижающих препаратов.
Гипергликемия является маркером сахарного диабета 2-го типа (NIDDM); стойкий контроль уровня глюкозы в плазме крови может замедлить развитие осложнений и деструкции бета-клеток, которая наблюдается при развитом сахарном диабете 2-го типа. В норме глюкоза плазмы крови фильтруется в почечных клубочках и активно реабсорбируется в проксимальных почечных канальцах. Считается, что основными переносчиками, отвечающими за реабсорбцию глюкозы в проксимальных почечных канальцах, являются SGLT2. Применение специфического ингибитора SGDT - флоризина или его аналогов у грызунов и собак - моделей диабета - приводило к нормализации уровня глюкозы плазмы крови путем усиления экскреции глюкозы с мочой без развития побочных эффектов, связанных с гипогликемией. Продолжительное (в течение 6 месяцев) лечение крыс линии Zucker - моделей диабета - с помощью ингибитора SGLT2 приводило к усилению инсулинового ответа на гликемию, улучшению чувствительности тканей к инсулину и препятствовало развитию нефропатии и нейропатии у этих животных при одновременном отсутствии каких-либо патологических изменений в почках и электролитного дисбаланса в плазме крови. Предполагается, что селективное ингибирование SGrT2 у пациентов, страдающих сахарным диабетом, позволит нормализовать уровень глюкозы в плазме крови путем усиления ее экскреции с мочой, а также улучшит чувствительность тканей к инсулину и отсрочит развитие осложнений.
Девяносто процентов всей глюкозы, реабсорбирующейся в почках, реабсорбируется в эпителиальных клетках, выстилающих начальный S1 сегмент почечных кортикальных проксимальных канальцев, и, вероятно, SGLT2 являются основными переносчиками ответственными за эту реабсорбцию. SGDT2 представляет собой белок, состоящий из 67 2 аминокислот и включающий 14 перекрывающихся мембранных сегментов, который, преимущественно, экспрессируется в начальном S1 сегменте проксимальных почечных канальцев. Субстратная специфичность, зависимость от ионов натрия и локализация SGLT2 сочетаются с такими их свойствами, как большая емкость и низкая аффинность. Кроме того, исследования, основанные на истощении гибридов, показали, что SGLT2 являются преобладающими натрий-зависимыми переносчиками глюкозы в S1 сегменте проксимальных канальцев, поскольку практически вся натрий-зависимая активность транспорта глюкозы, определяемая мРНК, выделенной из коркового вещества почек крысы, ингибируется антисмысловым олигонуклеотидом, специфичным к SGLT2 крысы. Возможно, ген SGLT2 отвечает за некоторые формы семейной глюкозурии, генетически обусловленного заболевания, при котором различным образом нарушается реабсорбция глюкозы в почках. Ни один из этих синдромов, обнаруженных на сегодняшнее время, не связан с 36bT2-локусом на хромосоме 16. Однако, исследования высокогомологичных SGLT2 грызунов явно свидетельствуют о том, что SGLT2 являются основными натрий-зависимыми переносчиками глюкозы в почках, и позволяют считать, что картированный локус глюкозурии кодирует регулятор SGLT2. Можно предположить, что ингибирование SGLT2 позволит снизить уровень глюкозы в плазме крови посредством усиления ее экскреции у больных сахарным диабетом.
SGLT1, другой натрий-зависимый переносчик глюкозы, который идентичен по своей аминокислотной последовательности SGLT2 на 60%, экспрессируется в тонком кишечнике и более дистальном S3 сегменте проксимальных почечных канальцев. Несмотря на сходство аминокислотных последовательностей, SGLT1 и SGLT2 различны по своим биохимическим свойствам. Для SGLT1 молярное соотношение между ионами Na+ и транспортируемой глюкозой составляет 2:1, в то время как для SGLT2 оно равно 1:1. Km для ионов Na+ составляет 32 и 250-300 мМоль для SGLT1 и SGLT2, соответственно. Значения Km по захвату глюкозы и неметаболизируемого аналога глюкозы α-метил-D-глюкопиранозида (AMG) являются сходными для SGLT1 и SGLT2, то есть 0.8 и 1.6 мМоль (глюкозы) и 0.4 и 1.6 мМоль (AMG) для SGLT1 и SGLT2 переносчиков, соответственно. Однако эти два переносчика значительно различаются своей субстратной специфичностью по отношению к сахарам, например, галактозе, которая является субстратом только для SGLT1.
Применение флоризина, специфического ингибитора активности SGLT, позволило in vivo подтвердить данную концепцию, поскольку привело к усилению экскреции глюкозы, снижению уровней глюкозы плазмы натощак и после еды, а также к усилению утилизации глюкозы без возможных побочных эффектов, связанных с гипогликемией, у нескольких грызунов и одной собаки - моделей диабета. Такие возможные последствия применения флоризина в течение двух недель, как ионный дисбаланс в плазме крови, нарушение функции почек и изменение нормальной структуры почечной ткани, отмечены не были. Кроме того, гипогликемия и другие побочные эффекты не наблюдались даже при применении флоризина у здоровых животных, несмотря на наличие глюкозурии. Применение ингибитора почечных SGLT в течение 6 месяцев приводило к нормализации уровней глюкозы плазмы натощак и после еды, улучшению секреции инсулина и усилению утилизации глюкозы, а также препятствовало развитию нефропатии и нейропатии у крыс, являющихся моделями сахарного диабета и ожирения, при одновременном отсутствии гипогликемии и патологических изменений со стороны почек.
Флоризин не подходит для перорального применения, поскольку он представляет собой неспецифический SGLT1/ SGLT2 ингибитор, который подвергается гидролизу в кишечнике, в результате которого образуется агликоновый флоретин, являющийся сильным ингибитором облегченных переносчиков глюкозы. Конкурентное ингибирование облегченных переносчиков глюкозы (ГЛЮТов) является нежелательным, поскольку оно может привести к усилению инсулинорезистентности периферических тканей, а также способствовать гипергликемии в центральной нервной системе. Ингибирование SGLT1 также может иметь серьезные последствия, которые возможно проследить на наследственном синдроме мальабсорбции глюкозы и галактозы (GGM), при котором мутация SGLT1 переносчика приводит к снижению захвата глюкозы в кишечнике и развитию угрожающей жизни диареи и дегидратации. Различные биохимические свойства SGLT1 и SGLT2, а также процент несоответствий их аминокислотных последовательностей, позволяют идентифицировать селективные ингибиторы SGLT2.
Синдромы семейной глюкозурии представляют собой состояния, при которых кишечный транспорт глюкозы, а также почечный транспорт других ионов и аминокислот является нормальным. Пациенты, страдающие семейной глюкозурией, развиваются без отклонений, имеют нормальный уровень глюкозы плазмы крови и не страдают от каких-либо серьезных нарушений здоровья, несмотря на иногда довольно высокие значения экскретируемой глюкозы (110-140 г/сутки). Основные симптомы, наблюдаемые у таких пациентов - это полифагия, полиурия и полидипсия; при этом их почки функционируют нормально и не имеют морфологических изменений. Таким образом, из имеющихся данных можно сделать вывод о том, что нарушения почечной реабсорбции глюкозы в почках, по-видимому, имеют минимальные отсроченные негативные последствия, у в остальном нормальных индивидуумов.
Последующие ссылки относятся к С-арил глюкозидным ингибиторам SGLT2 для лечения сахарного диабета.
WO 01/27128 относится к соединениям, имеющим следующую структуру:
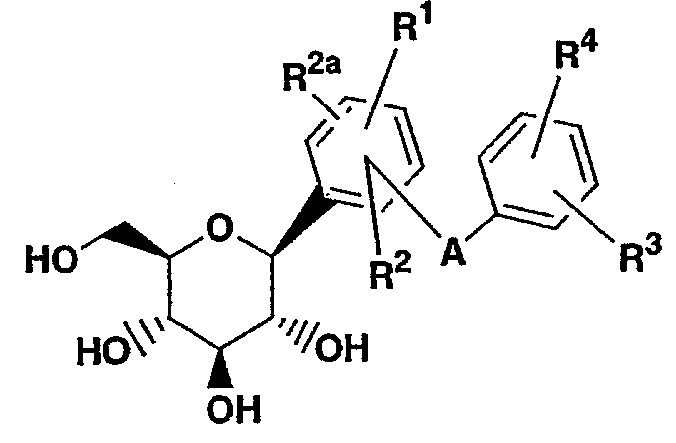
где А представляет собой О, S, NH или (СН2)n, где n - это 0-3; R1, R2, и R2a независимо друг от друга представляют собой водород, ОН, OR5, алкил, CF3, OCHF2, OCF3, SR5i или галоген, и так далее;
R3 и R4 независимо друг от друга представляют собой водород, ОН, OR5a, ОАрил, ОСН2Арил, алкил, циклоалкил, CF3, -OCHF2, OCF3, галоген, и т.д. Считается, что данные соединения являются ингибиторами SGLT2 переносчиков и, следовательно, могут представлять собой средства для лечения сахарного диабета и его осложнений.
WO 98/31697 относится к соединениям, имеющим следующую структуру:
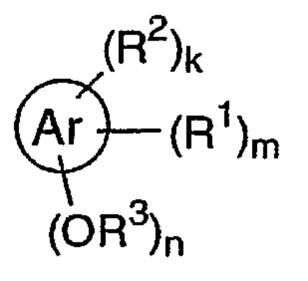
где Ar представляет собой, помимо всего прочего, фенил, бифенил, дифенилметан, дифенилэтан и дифенилэфир, R1 представляет собой гликозид, R2 представляет собой Н, ОН, амино, галоген, карбокси, алкил, циклоалкил или карбоксамидо и R3 представляет собой галоген, алкил, или ацил, а к, тип независимо друг от друга равны 1-4. Подмножество соединений, описанных в WO 98/31697, включает соединения, имеющие следующую структуру:
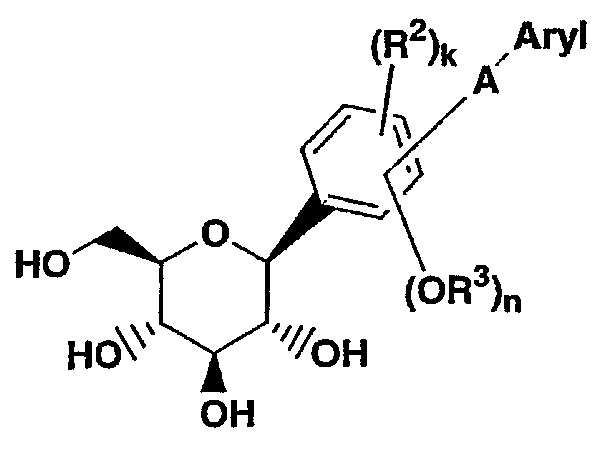
где А представляет собой О или (СН2)x, где х=0-3
R3 представляет собой водород, алкил- или ацил-группу, где n=1-4
R2 представляет собой водород, алкил, ОН, NH, галоген, СО2Н или карбоксимид, где k=1-4
Указанные соединения рассматриваются как средства для лечения и профилактики воспалительных заболеваний, аутоиммунных заболеваний, инфекций, рака и раковых метастазов, реперфузионных заболеваний, тромбозов, язв, ран, остеопороза, сахарного диабета и атеросклероза и др.
Описание изобретения
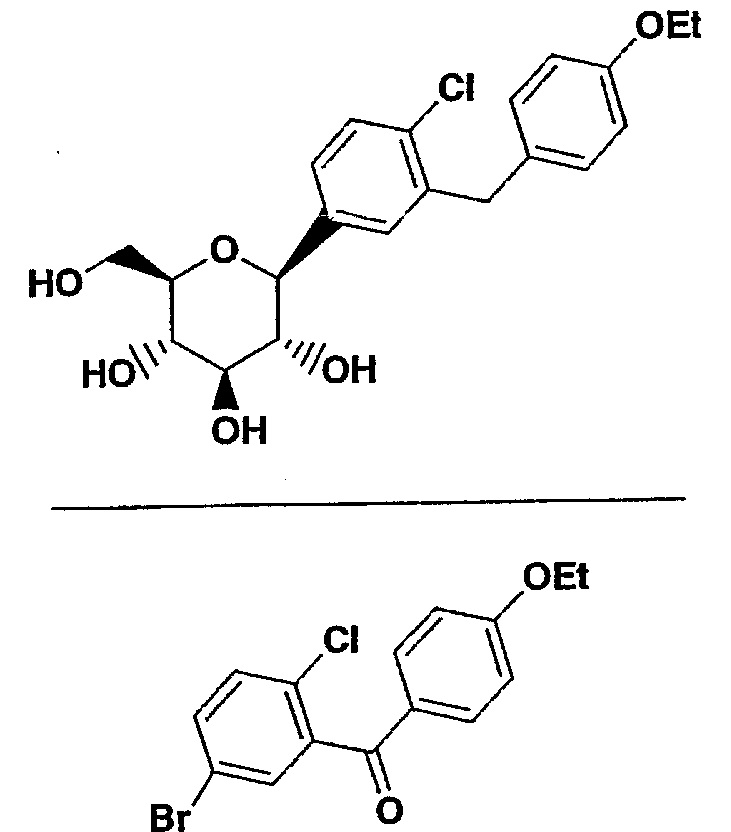
Настоящее изобретение относится к С-арил глюкозидным соединениям, имеющим формулу I:
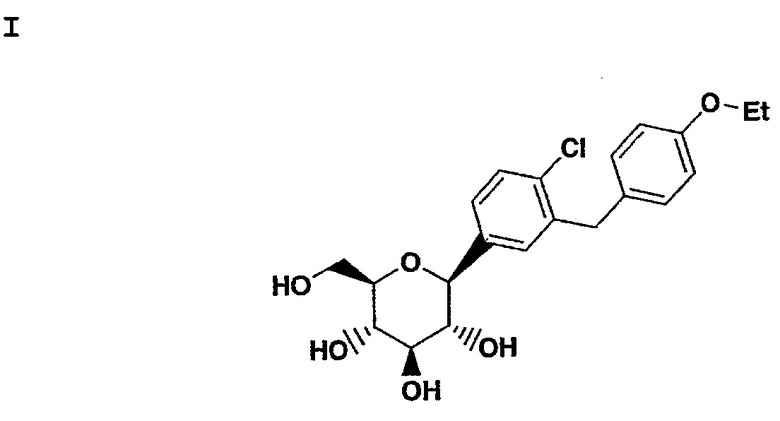
а также к их фармацевтически приемлемым солям, всем возможным их стереоизомерам и сложным эфирам этих соединений, представляющим собой пролекарства.
Соединение формулы I обладает способностью ингибировать натрий-зависимые переносчики глюкозы, обнаруживаемые в кишечнике и почках у млекопитающих, и представляет ценность для лечения сахарного диабета и его микро- и макрососудистых осложнений, таких как ретинопатия, нейропатия, нефропатия, а также для лечения ран.
Настоящее изобретение относится к соединению формулы I, фармацевтическим композициям, включающим это соединение, и к способам его применения.
Кроме того, настоящее изобретение относится к способу лечения и замедления прогрессирования или начала диабета, преимущественно сахарного диабета 1-го и 2-го типов, включая осложнения диабета, такие как ретинопатия, нейропатия, нефропатия; к способу лечения ран, а также к способу лечения сопутствующих состояний, таких как инсулиновая резистентность (нарушенный гомеостаз глюкозы), гипергликемия, гиперинсулинемия, повышение в крови уровня жирных кислот и глицерина, ожирение, гиперлипидемия, включая гипертриглицеридемию, Синдром X, атеросклероз и гипертензия, а также к способу повышения в крови уровня липопротеинов высокой плотности, заключающемуся в применении по отношению к человеку, который нуждается в лечении, соединения формулы I.
Настоящее изобретение также относится к способу лечения диабета и сопутствующих состояний, описываемому выше, заключающемуся в применении по отношению к человеку, который нуждается в лечении, терапевтически эффективного количества смеси соединения формулы I и антидиабетического средства другого типа и/или другого лекарственного средства, такого как гиполипидемическое средство.
Состояния, заболевания и расстройства, объединенные понятием "Синдром X" (также известный как метаболический синдром), детально описаны у Johannsson J. Clin. Endocrinol. Metab., 82, 121-34 (1991).
Термин "другие лекарственные средства", используемый здесь, относится к одному или более антидиабетическому средству (не к ингибитору SGLT2 формулы I), одному или более средству против ожирения, антигипертензивному средству, антитромбоцитарному средству, антиатеросклеротическому средству и/или одному или более гиполипидемическому средству (включая антиатеросклеротические средства).
Согласно способу, описанному выше, соединение формулы I, заявленное в соответствии с настоящим изобретением, будет вводиться совместно с одним или двумя, или более антидиабетическими средствами и/или совместно с одним, двумя или более другими лекарственными средствами (в зависимости от способа их введения), в соотношениях от 0.01:1 до 300:1, предпочтительно, от 0.1:1 до 10:1.
Детальное описание изобретения
Соединение формулы I, заявленное в соответствии с настоящим изобретением, может быть получено согласно схеме, приведенной ниже, и ее описанию, при этом температура выражена в значениях стоградусной шкалы.
Соединение формулы I может быть получено согласно схеме 1 путем обработки соединения формулы II
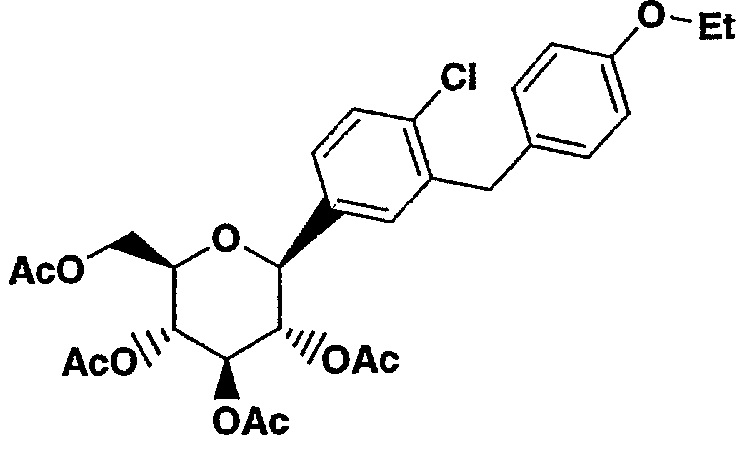
основанием, таким как LiOH или NaOH, в присутствии растворителя, такого как смесь H2O/THF/MeOH в соотношении 1:2:3 или водн. МеОН, или водн. EtOH.
Соединение формулы II (которое представляет собой легко кристаллизующееся новое промежуточное соединение) обеспечивает необходимые условия для очистки соединения формулы Ia, получаемого в виде смеси α- и β-аномеров.
Соединение формулы II может быть получено путем обработки соединения Ia с помощью Ас2О в растворителе, таком как CH2Cl2, содержащем пиридин и катализатор, такой как диметиламинопиридин (DMAP).
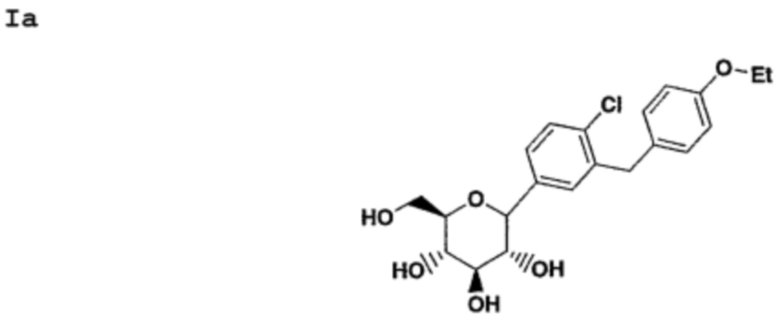
Соединения формулы Ia могут быть получены путем восстановления соединения формулы III с помощью восстанавливающего агента, такого как Et3SiH в растворителе, таком как смесь CH2Cl2/MeCN в соотношении 1:1 при температуре - 10° в присутствии кислого катализатора Льюиса, такого как BF3⋅Et2O.
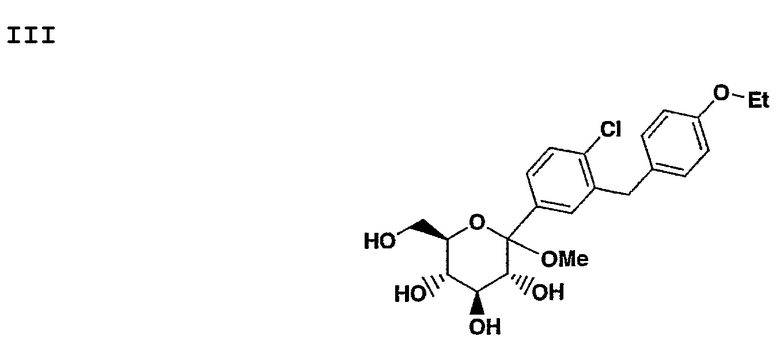
Соединение формулы II может быть альтернативно получено из соединения формулы III с помощью ацетилирования соединения формулы III при использовании Ac2O в растворителе, таком как толуол или CH2Cl2, содержащем основание, такое как основание Хьюнига или Et3N, и катализатор, такой как DMAP, для получения соединения формулы IV.
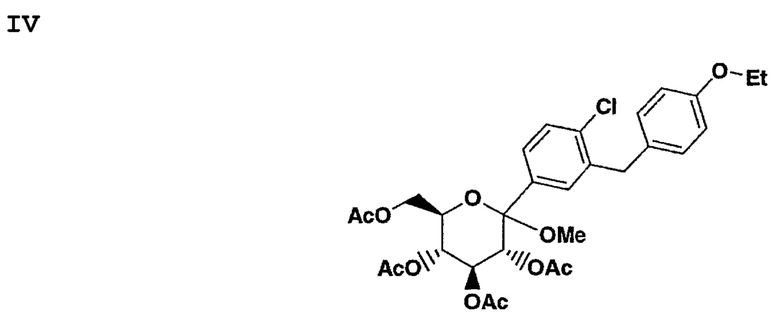
Последующее превращение соединения IV в соединение II может быть достигнуто путем обработки соединения IV при температуре 20° восстановителем, таким как Et3SiH, в растворителе, таком как МеОН, содержащем 1 экв. Н2О и кислый катализатор Льюиса, такой как BF3⋅Et2O.
Схема 1
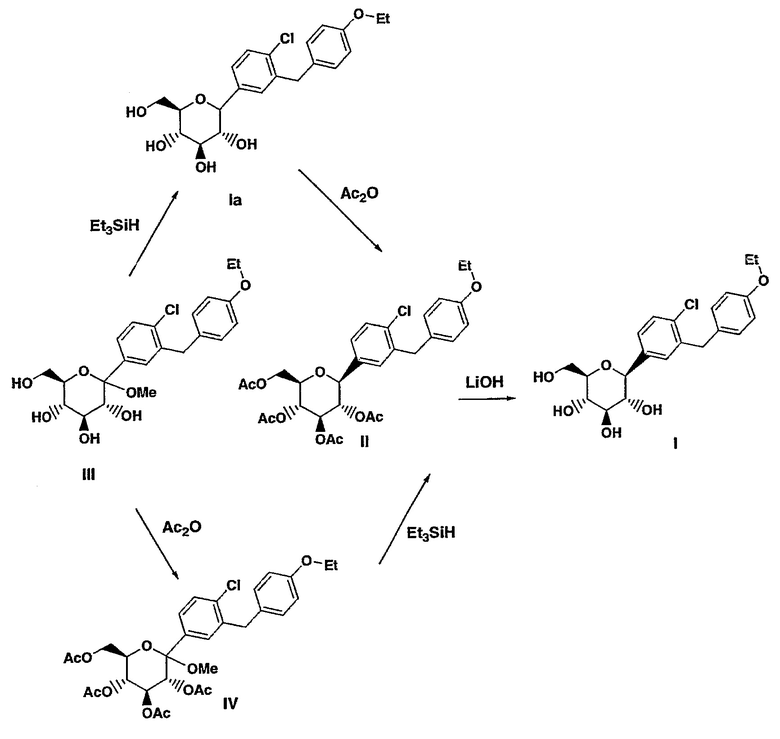
Соединение формулы III может быть получено согласно схеме 2, путем 1) добавления холодного THF раствора арил лития формулы V к персилированному глюконолактону формулы VI в растворителе, таком как толуол при температуре -75°. Затем через 30 минут добавляют раствор протонсодержащей кислоты, такой метансульфоновая кислота (MSA), в метаноле и раствор перемешивают при температуре 20° до тех пор, пока не произойдет полная трансформация промежуточного лактола в соединение III.
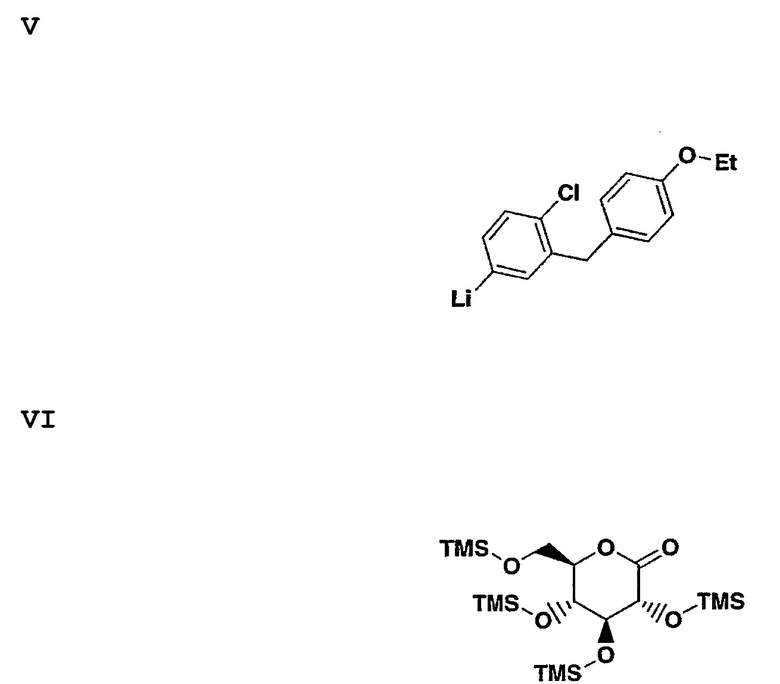
Соединение формулы VI может быть получено путем обработки коммерчески-доступного D-глюконолактона силирующим агентом, таким как хлорид триметилсилила в растворителе, таком как THF, содержащем основание, такое как N-метилморфолин.
Соединение формулы V может быть получено путем обработки соединения формулы VII алкил литием, таким как n-BuLi или t-Buli в растворителе, таком как THF, при температуре -75°.
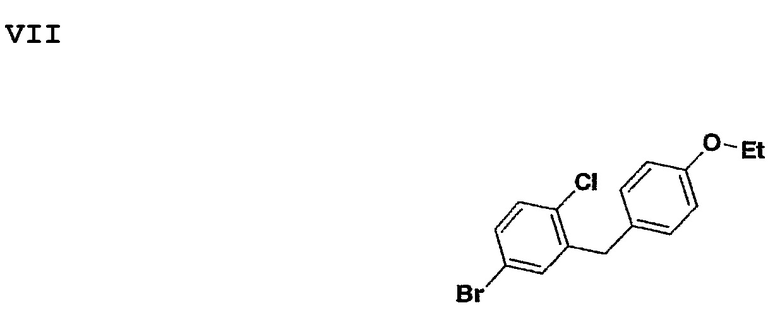
Соединение формулы VIII может быть легко получено путем обработки соединения формулы III восстанавливающим агентом, таким как Et3SiH в растворителе, таком как смесь CH2Cl3/MeCN в соотношении 1:1 при температуре 0°-20° в присутствии кислого катализатора Льюиса, такого как BF3⋅Et2O.
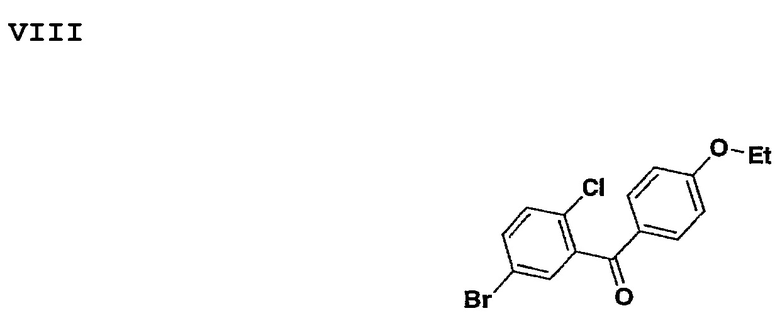
Соединение формулы VIII может быть получено с помощью реакции ацилирования Фриделя-Крафта с использованием коммерчески доступного этоксибензена (фенетола)и 2-хлор-5-бромбензоилхлорида в растворителе, таком как CH2Cl2, содержащем эквивалент кислого катализатора Льюиса, такого как AlCl3 или AlBr3.
2-хлор-5-бромбензоилхлорид можно легко получить из коммерчески доступной 2-хлор-5-бромбензойной кислоты путем обработки последней оксалилхлоридом в растворителе, таком, как CH2Cl2, содержащем DMF в каталитическом количестве.
Схема 2
Далее приводится список сокращений химических соединений и терминов, используемых для характеристики настоящего изобретения. Эти сокращения соответствуют указанным соединениям и терминам по всему тексту описания (если специальным образом не оговаривается иное)и могут быть использованы как сами по себе, так и в сочетаниях друг с другом.
Для характеристики настоящего изобретения используются следующие сокращения:
Ph = фенил
Bn = бензил
t-Bu = третичный бутил
Me = метил
Et = этил
TMS = триметилсилил
TBS - трет-бутилдиметилсилил
THF = тетрагидрофуран
Et2O = диэтиловый эфир
EtOAc = этилацетат
DMF = диметилформамид
МеОН = метанол
EtOH = этанол
i-PrOH = изопропанол
НОАс или АсОН = уксусная кислота
TFA - трифторуксусная кислота
i-Pr2NEt = диизопропилэтиламин
Et3N = триэтиламин
DMAP = 4-диметиламинопиридин
NaBH4 = борогидрат натрия
n-BuLi = n-бутиллитий
Pd/С = палладий на углероде
KOH = гидроксид калия
NaOH = гидроксид натрия
LiOH = гидроксид лития
K2CO3 = карбонат калия
NaHCO3 = гидрокарбонат натрия
Ar = аргон
N2 = азот
мин = минута (минуты)
ч = час (часы)
л = литр
мл = миллилитр
мкл = микролитр
г = грамм
мг = миллиграмм
мол. = моль
мМол. = миллимоль
мэкв. = миллиэквивалент
RT = комнатная температура
нас. = насыщенный
водн. = водный
TLC = тонкослойная хроматография
HPLC = жидкостная хроматография высокого разрешения
LC/MS = жидкостная хроматография высокого разрешения/масс-спектрометрия
MS или Mass Spec = масс-спектрометрия
ЯМР = ядерно-магнитный резонанс
т.пл. = точка плавления
Термин "низший алкил", используемый здесь как самостоятельно, так и в составе других терминов, если дополнительно не оговаривается иное, относится к прямым и разветвленным углеводородным цепям, содержащим от 1 до 8 атомов углерода, а термин "алкил" или "алк", используемый здесь как самостоятельно, так и в составе других терминов, относится к прямым и разветвленным углеводородным цепям, содержащим от 1 до 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, еще более предпочтительно от 1 до 8 атомов углерода в нормальной цепи, таким как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил, додецил, а также к их изомерам с разветвленными цепями и подобным соединениям, а также к группам, имеющим от 1 до 4 заместителей, таких как галоген, например, F, Br, Cl или J или CF3, алкил алкокси, арил, арилокси, арил(арил)или диарил, арилалкил, арилалкилокси, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкилалкилокси, факультативно замещенный амин, гидрокси, гидроксиалкил, ацил, алканоил, гетероарил, гетероарилокси, циклогетероалкил, арилгетероарил, арилалкоксикарбонил, гетероарилалкил, гетероарилалкокси, арилоксиалкил, арилоксиарил, алкиламино, алканоиламино, арилкарбониламино, нитро, циано, тиол, галоалкил, тригалоалкил и/или алкилтио.
Термин "циклоалкил", используемый здесь как самостоятельно, так и в составе других терминов, если дополнительно не оговаривается иное, относится к насыщенным или частично насыщенным (имеющим 1 или 2 двойные связи) циклическим углеводородным группам, имеющим от 1 до 3 колец, и включающим моноциклический алкил, бициклический алкил и трициклический алкил, кольца которых состоят в целом из 3-20 атомов углерода, более предпочтительно, из 3-10 атомов углерода и могут быть объединены с 1-2 ароматическими кольцами, как описано для арила, которые включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил и циклододецил, циклогексенил,
причем любая из этих групп может быть факультативно замещена 1-4 заместителями, такими как галоген, алкил, алкокси, гидрокси, арил, арилокси, арилалкил, циклоалкил, алкиламидо, алканоиламино, оксо, ацил, арилкарбониламино, амино, нитро, циано, тиол и/или алкилтио и/или любым алкильным заместителем.
Термин "алканоил", используемый здесь как самостоятельно, так и в составе других терминов, относится к алкилу, связанному с карбонильной группой.
Термин "галоген" или "гало", используемый здесь как самостоятельно, так и в составе других терминов, относится к хлору, брому, фтору и йоду; при этом хлор и фтор являются более предпочтительными.
Термин "ион металла" относится к ионам щелочных металлов, таким как натрий, калий или литий, и к ионам щелочноземельных металлов, таким как магний и кальций, а также цинк и алюминий.
Термин "арил" или "Арил", используемый здесь как самостоятельно, так и в составе других терминов, если дополнительно не оговаривается иное, относится к моноциклическим и бициклическим ароматическим группам, имеющим от 6 до 10 атомов углерода в кольце (таким как фенил или нафтил, включая 1-нафтил и 2-нафтил),которые могут необязательно включать 1-3 дополнительных кольца, объединенных с углеродным кольцом или гетероциклическим кольцом (таким как, например, кольцо арила, циклоалкила, гетероарила или циклогетероалкила, например
и могут быть факультативно замещены по подходящим углеродным атомам 1, 2 или 3 группами, включающими водород, гало, галоалкил, алкил, галоалкил, алкокси, галоалкокси, алкенил, трифторметил, трифторметокси, алкинил, циклоалкил-алкил, циклогетероалкил, циклогетероалкилалкил, арил, гетероарил, арилалкил, арилокси, арилоксиалкил, арилалкокси, алкоксикарбонил, арилкарбонил, арилалкенил, аминокарбониларил, арилтиоарилсульфинил, арилазо, гетероарилалкил, гетероарилалкенил, гетероарилгетероарил, гетероарилокси, гидрокси, нитро, циано, амино, замещенные амины, где амин содержит 1-2 заместителя (к которым относятся алкил, арил или любое другое арильное соединение, упоминающееся в определениях), тиол, алкилтио, арилтио, гетероарилтио, арилтиоалкил, алкоксиарилтио, алкилкарбонил, арилкарбонил, алкиламинокарбонил, ариламинокарбонил, алкоксикарбонил, аминокарбонил, алкилкарбонилокси, арилкарбонилокси, алкилкарбониламино, арилкарбониламино, арилсульфинил, арилсульфинилалкил, арилсульфониламино и арилсульфонаминокарбонил и/или любой алкильный заместитель, упоминающийся здесь.
Термины "низший алкокси", "алкокси", "арилокси" или "аралкокси", используемые здесь как самостоятельно, так и в составе других терминов, если дополнительно не оговаривается иное, относятся к любым упоминающимся выше группам алкила, аралкила или арила, связанным с атомом кислорода.
Термины "низший алкилтио", "алкилтио", "арилтио" или "аралкилтио", используемые здесь как самостоятельно, так и в составе других терминов, если дополнительно не оговаривается иное, относятся к любым упоминающимся выше группам алкила, аралкила или арила, связанным с атомом серы.
Термин "полигалоалкил", используемый здесь, относится к "алкил" группе, охарактеризованной выше, имеющей от 2 до 9, предпочтительно от 2 до 5 гало-заместителей, таких как F или Cl, предпочтительно F, таких как CF3CH2, CF3 или CF3CF2CH2.
Термин "полигалоалкилокси", используемый здесь, относится к "алкокси" или "алкилокси" группе, охарактеризованной выше, имеющей от 2 до 9, предпочтительно, от 2 до 5 гало-заместителей, таких как F или Cl, предпочтительно F, таких как CF3CH2O, CF3O или CF3CF2CH2O.
Термин "сложные эфиры, представляющие собой пролекарства", используемый здесь, относится к сложным эфирам и карбонатам, полученным при взаимодействии одного или более гидроксила соединений формулы I с алкил, алкокси или арил-замещенным ацилирующим агентом при использовании методов для получения ацетатов, пивалатов, метилкарбонатов, бензоатов и им подобных соединений, хорошо известных специалистам в данной области. Кроме того, из уровня техники известны сложные эфиры, на основе карбоновых и фосфорных кислот, такие как метиловый, этиловый, бензиловый и т.д.
Примерами таких эфиров являются:
В том случае, когда соединения формулы I находятся в форме кислот, они могут формировать фармацевтически приемлемые соли, такие как соли щелочных металлов, таких как литий, натрий или калий; соли щелочноземельных металлов, таких как кальций или магний, а также цинк и алюминий, и другие катионы, такие как аммоний, хлор, диэтаноламин, лизин (D или L), этилендиамин, трет-бутиламин, трет-октиламин, три-(гидроксиметил)аминометан (TRIS), N-метил глюкозамин (NMG), триэтаноламин и дегидроабиетиламин.
Настоящее изобретение также рассматривает все стереоизомеры соединений, как в смеси, так в очищенной или частично очищенной форме. Соединение, заявленное в соответствии с настоящим изобретением, может иметь асимметричные центры в любом из атомов углерода, включая любой R-заместитель. Следовательно, соединение формулы I может существовать в формах энантиомеров или диастереомеров или их смеси. При получении соединений можно использовать рацематы, энантиомеры или диастереомеры в качестве начальных соединений. После получения, энантиомеры и диастереомеры могут быть разделены с помощью традиционных методов, таких как хроматография или фракционная кристаллизация.
При необходимости соединения формулы I могут быть использованы в комбинации с одним или более другими антидиабетическими средствами или одним или более другими лекарственными средствами, которые могут вводиться как перорально в одинаковой дозированной форме, а также в отдельной оральной дозированной форме или путем инъекции.
К таким антидиабетическим средствам, которые могут применяться совместно с ингибиторами SGLT2 формулы I, относятся 1,2,3 или более антидиабетических средств или антигипергликемические средства, включая стимуляторы секреции инсулина или сенсибилизаторы инсулина, или другие антидиабетические средства, предпочтительно имеющие отличный от ингибиторов SGLT2 механизм действия и включающие бигуаниды, производные сульфонилмочевины, ингибиторы глюкозидазы, PPAR у агонисты, такие как тиазолидиндионы, аР2 ингибиторы, PPAR α/γ двойные агонисты, ингибиторы дипептидил пептидазы IV (DP4) и/или меглитиниды, а также инсулин, глюкагоноподобный пептид 1, ингибиторы РТР1 В, ингибиторы гликоген-фосфорилазы и/или ингибиторы глюкозо-6-фосфатазы.
К другим лекарственным средствам, которые могут применяться совместно с ингибиторами SGLT2 формулы I, относятся средства для лечения ожирения, антигипертензивные средства, антитромбоцитарные средства, антиатеросклеротические средства и/или гиполипидемические средства.
Ингибитор SGLT2 формулы I также может применяться совместно с лекарственными средствами для лечения осложнений диабета. К таким лекарственным средствам относятся ингибиторы PKC и/или ингибиторы AGE.
Считается, что применение соединений формулы I в комбинации с 1,2,3 или более другими антидиабетическими средствами приводит к более значительному антигипергликемическому эффекту, чем при использовании этих же препаратов по отдельности и большему, чем общий аддитивный антигипергликемический эффект, обусловливаемый этими препаратами.
Таким антидиабетическим средством может быть оральный антигипергликемический препарат, предпочтительно из группы бигуанида, такой как метформин или фенформин или их соли, предпочтительно метформин HCl.
В том случае, когда в качестве дополнительного антидиабетического средства используется препарат из группы бигуанида, соединение формулы I вводится в весовом соотношении к этому препарату, равном 0.01:1-100:1, предпочтительно, 0.1:1-5:1.
Также дополнительным антидиабетическим средством может, предпочтительно, быть средство из группы производных сульфонилмочевины, такое как глубирид (также известный как глибенкламид), глимепирид (описанный в патенте СШ7А №4,379,785), глипизид, гликлазид или хлорпропамид, или любое известное средство из этой группы или другое антигипергликемическое средство, которое воздействует на АТФ-зависимые каналы р-клеток; глубирид и глипизид являются предпочтительными и могут применяться в одинаковых или различных оральных дозированных формах.
Соединение формулы I в таком случае вводится в весовом соотношении к производному сульфонилмочевины, равном 0.01:1-100:1, предпочтительно, 0.2-10:1.
Кроме того, дополнительным антидиабетическим средством может быть средство из группы ингибиторов глюкозидазы, такое как акарбоза (описанная в патенте США №4,904,769) или миглитол (описанный в патенте США №4,639,436), которые могут применяться в одинаковых или различных оральных дозированных формах.
Соединение формулы I в таком случае вводится в весовом соотношении к ингибитору глюкозидазы, равном 0.01:1-100:1, предпочтительно, 0.5:1-50:1.
Соединение формулы I может применяться в комбинации с PPAR у агонистом, таким как оральный антигипогликемический препарат тиазолидиндион или другой сенсибилизатор иснулина (который обладает инсулин-сенсибилизирующим эффектом у пациентов с NIDDM), такой как троглитазон (Rezulin® фирмы Warner-Lambert, описанный в патенте США №4,572,912), розиглитазон (SKB), пиоглитазон (Takeda), МСС-555 фирмы Mitsubishi (описанный в патенте США №5,594,016), GL-262570 фирмы Glaxo-Welcome, энглитазон (СР-68722 фирмы Pfizer) или дарглитазон (СР-86325 фирмы Pfizer, изаглитазон (MIT/J&J), JTT-501 (JPNT/P&U), L-895645 (Merk), R-119702 (Sakyo/WL), NN-2344 (Dr. Reddy/NN)или YM-440 (Yamanouchi), предпочтительно розиглитазон и пиоглитазон.
Соединение формулы I в таком случае вводится в весовом соотношении к тиазолидиндиону равном 0.01:1-100:1, предпочтительно, 0.2:1-10:1.
Производные сульфонилмочевины и тиазолидиндион в количествах менее чем 150 мг орального антидиабетического агента могут быть объединены в одну таблетку с соединением формулы I.
Соединение формулы I может также применяться в комбинации с антигипергликемическим средством, таким как инсулин или глюкагоноподобный пептид 1(GLP-1), такой как GLP-1(1-36)амид, GLP-1(7-36)амид, GLP-1(7-37) (как описано в патенте США №5,614,492 Habener, приведенном здесь в качестве ссылки), а также АС2993 (Amylen) и LY-315902 (Lilly), которые могут вводиться инъекционно, интраназально, трансдермально или буккально.
В случае их использования метформин, производные сульфонилмочевины, такие как глубирид, глимепирид, глипирид, глипизид, хлорпромамид и гликлазид, и ингибиторы глюкозидазы, такие как акарбоза и миглитол, или инсулин (инъекционный, пульмонарный, буккальный или оральный), могут применяться в соответствии с рецептурами, приведенными выше, и в количествах и дозах, указанных в настольном справочнике врача.
В случае их использования, метформин и его соли могут применяться в количествах, находящихся в пределах от 500 до 2000 мг в сутки, которые могут вводиться как в единичных, так и в разделенных дозах от одного до четырех раз в сутки.
В случае их использования, антидиабетические средства из группы тиазолидиндионов могут применяться в количествах, находящихся в пределах от 0.01 до 2000 мг в сутки, которые могут вводиться как в единичных, так и в разделенных дозах от одного до четырех раз в сутки.
В случае его использования, инсулин может применяться в соответствии с рецептурами, количествами и дозировками, указанными в настольном справочнике врача.
В случае их использования, GLP-1 пептиды могут применяться в виде оральных щечных лекарственных форм, вводиться интраназально или парентерально, как описано в патентах США №5,346,701 (TheraTech), 5,614,492 и 5,631,224, приведенных здесь в качестве ссылки.
Помимо всего прочего, антидиабетическим средством, которое может применяться совместно с соединением формулы I может быть PPAR α/γ двойной агонист, такой как AR-H039242 (Astra/Zeneca), GW-409544 (Glaxo-Wel1come), KRP297 (Kyorin Merck), а также средства, описанные у Мураками и соавт."Новый сенсибилизатор инсулина работает как колиганд для альфа рецептора, активируемого пероксисомной пролиферацией (PPAR альфа), и для гамма PPAR. Влияние PPAR активизации на неправильный метаболизм липидов в печени у крыс линии Zucker, страдающих ожирением", Диабет 47, 1841-1847 (1998), а также средства раскрытые в заявке №60/155,400 от 22 сентября 1999, упомянутой здесь в качестве ссылки, и вводимые в указанных там дозировках; предпочтительными являются соединения, упомянутые в вышеуказанной заявке как наиболее пригодные.
Также другим антидиабетическим средством может быть ингибитор аР2, такой как описан в заявке на патент США №09/391,053 от 7 сентября 1999 и в заявке на патент США №60/127,745 от 5 апреля 1999, применяемый в дозировках, указанных там. Предпочтительными являются соединения, упомянутые в вышеуказанных заявках как наиболее подходящие.
Кроме того, другим антидиабетическим средством может быть ингибитор DP4, такой, как описан в WO 99/38501, WO 99/46272, WO 99/67279 (PROBIODRUG), WO 99/67278 (PROBIODRUG), WO 99/61431 (PROBIODRUG), NVP-DPP728A (1-[[2-[(5-цианопиридин-2-ил)амино]этил]амино]ацетил]-2-циано-(S)- пирролидин)(Novartis) (предпочтительно), как описано у Hunges et al, Biochemistry, 38 (36), 11597-11603, 1999, TSL-225 (триптофил-1,2,3,4-тетрагидроизохинолин-3-карбоксильная кислота (описано у Yamada et al, Biorg.& Med. Chem. Lett. 8 (1998) 1537-1540, 2-цианопирролидиды и 4-цианопирролидиды, как описано у Ashworth et al., Bioorg.& Med. Chem. Lett., Vol.6, No. 22, стр. 1163-1166 и 2745-2748 (1996), применяемый в дозировках, указанных в вышеупомянутых ссылках.
Соединением из группы меглитинида, которое может применяться совместно с соединением формулы I, может быть репаглинид, натеглинид (Novartis) или KAD1229 (PF/Kissei); рапаглинид является предпочтительным.
Ингибитор SGLT2 формулы I в таком случае вводится в весовом соотношении к меглитиниду, PPAR у агонисту, PPAR α/γ двойному агонисту, аР2 ингибитору или DP4 ингибитору, равном 0.01:1-100:1, предпочтительно 0.2:1-10:1.
Гиполипидемические средства, которые могут применяться совместно с соединениями формулы I, могут включать 1,2,3 или более средств из группы МРТ ингибиторов, ингибиторов ГМГ-КоА редуктазы, ингибиторов сквален-синтетазы, производных фибровой кислоты, ингибиторов АЛАТ, ингибиторов липоксигеназы, ингибиторов абсорбции холестерина, ингибиторов кишечного Na+/желчная кислота совместного переносчика, регуляторов активности рецепторов ЛПНП, секвестрантов желчных кислот и/или никотиновую кислоту и ее производные.
К МТР ингибиторам, которые могут применяться в соответствии с настоящим изобретением, относятся МТР ингибиторы, описанные в патенте США №5,595,872, патенте США №5,739,135, патенте США №5,712,279, патенте США №5,760,246, патенте США №5,827,875, патенте США №5,885,983, а также в патенте США №5,962,440. Предпочтительным является каждый из ингибиторов МТР, описанный в вышеупомянутых документах. Все вышеупомянутые патенты США и заявки приведены здесь в качестве ссылки.
Гиполипидемическим средством может быть средство из группы ингибиторов ГМГ-КоА редуктазы, которая включает (без ограничений указанным) мевастатин и родственные соединения, описанные в патенте США №3,983,140; ловастатин (мевинолин)и родственные соединения, описанные в патенте США №4,231,938; правастатин и родственные соединения, описанные в патенте США №4,346,227; симвастатин и родственные соединения, описанные в патентах США №4,448,784 и 4,450,171. Гиполипидемическими средствами также могут быть соединения, описанные в заявках США на патент №60/211,594 и 60/211,595. К другим ингибиторам ГМГ-КоА редуктазы, которые могут применяться в соответствии с настоящим изобретением, относятся (без ограничений указанным) флувастатин, описанный в патенте США №5,354,772; церивастатин, описанный в патентах США №5,006,530 и 5,177,080; аторвастатин, описанный в патентах США №4,681,893, 5,273,995, 5,385,929 и 5,686,104; атавастатин (нивастатин фирмы Nissan/Sankyo (NK-104)), описанный в патенте США №5,011,930; визастатин фирмы Shionogi-Astra/Zeneca (ZD-4522), описанный в патенте США №5,260,440, и родственные статинам соединения, описанные в патенте США №5,753,675; пиразоловые аналоги производных мевалонолактона, описанные в патенте США №4,613,610, инденовые аналоги производных мевалонолактона, описанные в заявке РСТ WO 86/03488; 6-[2-(замещенный-пиррол-1-ил)-алкил)пуран-2-оны и их производные, описанные в патенте США №4,647,576; SC-45355 дихлорацетат фирмы Searl (3-замещенное производное пентандиоевой кислоты); имидазоловые аналоги производных мевалонолактона, описанные в заявке РСТ WO 86/07 054; производные 3-карбокси-2-гидрокси-пропан-фосфоновой кислоты, описанные в патенте Франции №2,596,393; производные 2,3-двузамещенного пиррола, фурана и тиофена, описанные в заявке на европейский патент №0,221,025; нафтиловые аналоги мевалонолактона, описанные в патенте США №4,686,237; октагидрогнафталены, описанные в патенте США №4,499,289, кето-аналоги мевинолина (ловастатин), описанные в заявке на европейский патент №0,142,146 А2, и хинолиновые и пиридиновые производные, описанные в патентах США №5,506,219 и 5,691,322.
Кроме того, соединения фосфиновой кислоты, обладающие способностью ингибировать ГМГ-КоА редуктазу и подходящие для использования в соответствии с настоящим изобретением, описаны в GB 2205837.
К ингибиторам сквален-синтетазы, подходящим для использования в соответствии с настоящим изобретением, относятся (без ограничений указанным) α-фосфоно-сульфонаты, описанные в патенте США №5,712,396; ингибиторы, описанные у Biller et al, J. Med. Chem., 1988, Vol.31, No. 10, стр. 1869-1871, включая изопреноид (фосфинил-метил) фосфонаты, а также другие известные ингибиторы сквален-синтетазы, например, описанные в патентах США №4,871,721 и 4,924,024 и Biller, S.A., Neuenschwander, K., Ponpipom, М.М., и у Poulter, CD., Current Pharmaceutical Design, 2, 1-40 (1996).
Кроме того, дополнительные ингибиторы сквален-синтетазы, подходящие для использования в соответствии с настоящим изобретением, включают терпеноид пирофосфаты, описанные Р. Otiz de Mintelliano et al, J. Med. Chem., 1977, 20, 243-249; аналоги фарнесил дифосфата А и аналоги пресквален пирофосфата (PSQ-PP), описанные Corey и Volante, J. Am. Chem. Soc, 1976, 98, 1291-1293; фосфинилфосфонаты, описанные McClard, R.W. et al, J.A.C.S., 1987, 109, 5544 и циклопропаны, описанные в диссертации Capson, T.L., июнь, 1987, Dept. Med. Chem. U of Utach, реферат, таблица, стр. 16,17, 40-43, 48-51, резюме.
Другими гиполипидемическими агентами, подходящими для использования в соответствии с настоящим изобретением, являются (без ограничений указанным) производные фибровой кислоты, такие как фенофибрат, гемфиброзил, клофибрат, безафибрат, ципрофибрат, клинофибрат и им подобные средства; пробукол и родственные соединения, описанные в патенте США №3,674,836; (робукол и гемфиброзил являются предпочтительными); секвестранты желчных кислот, такие как холестирамин, колестипол и DEAE-Сефадекс (Secholex®, Policexide®), а также липостабил (Phone-Poulenc); Eisai Е-5050 (N-замещенное производное этаноламина); иманиксил (НОЕ-402); тетрагидролипстатин (THL); истигмастанилфосфорилхолин (SPC, Roche); аминоциклодекстрин (Tanabe Seiyoki); Ajinomoto AJ-814 (производное азулена); мелинамид (Sumitomo); Sandoz 58-035, American Cyanamid CL-277,082 и CL-283, 546 (двузамещенные производные мочевины); никотиновая кислота; аципимокс; ацифран; неомицин; р-аминосалициловая кислота; аспирин, поли(диаллилметиламин) производные, описанные в патенте США №4,759,923; четвертичный амин поли(диаллилдиметиламмонийхлорид) и ионы, такие, как описанные в патенте США №4,027,009, а также другие известные средства, снижающие уровень холестерина в сыворотке крови.
Другим гиполипидемическим средством может быть ингибитор АСАТ, такой как описан, например, в публикациях "Лекарственные средства будущего 24", 9-15 (1999), (Avasimibe); "Ингибитор АСАТ, С1-1011 - эффективное средство для профилактики и замедления развития жировых полосок в аорте у хомяков", Nicolosi et al, Атеросклероз (Shannon, Irel). (1998), 137 (1), 77-85; "Фармакологический профиль FCE 27677: новый ингибитор АСАТ с потенциальной гиполипидемической активностью, обусловленной селективной супрессией печеночной секреции ApoB100-несущего липопротеина", Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16(1), 16-30; "RP 73163: биодоступный алкилсульфинил-дифенилимидазоловый АСАТ ингибитор", Smith, С, et al, Bioorg. Med. Chem. Lett. (1996), 6(1), 47-50; "Ингибиторы АСАТ: физиологические механизмы гиполипидемической и антиатеросклеротической активности у экспериментальных животных", Krause et al, Editor(s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred А., Воспаление: пути медиаторов (1995), 173-98, Издатель: CRC, Boca Raton, Fla.; "Ингибиторы АСАТ: потенциальные антиатеросклеротические агенты", Sliskovic et al, Curr. Med. Chem. (1994), 1(3), 204-25; "Ингибиторы ацил-КоА: холестерин О-ацил трансферазы (АСАТ) как гипохолестеринемические средства. б. Первый водорастворимый ингибитор АСАТ с липид-регулирующей активностью". Ингибиторы ацил-КоА: холестерин ацилтрансферазы (АСАТ)7. Разработка серии замещенных производных N-фенил-N'-[(1-фенилциклопентил)метил]мочевины, обладающих высокой гипохолестеринемической активностью", Stout et al, Chemtracts: Org. Chem. (1995), 8(6), 395-62, или TS-962 (Taisho Pharmaceutical Co. Ltd).
Гиполипидемическим средством также может быть регулятор повышающей активности рецептора LD2, такой как, MD-700 (Taisho Pharmaceutical Co. Ltd) или LY295427 (Eli Lilly).
Гиполипидемическим средством также может быть ингибитор абсорбции холестерина, предпочтительно, SCH48461 фирмы Shering-Plough, а также средства, описанные в публикациях "Атеросклероз" 115, 45-63 (1995) и J. Med. Chem. 41, 973 (1998).
Гиполипидемическим средством также может быть ингибитор кишечного Na+/желчная кислота совместного переносчика, такой как описан в публикации "Лекарственные средства будущего", 24, 425-430 (1999).
Предпочтительными гиполипидемическими средствами являются правастатин, ловастатин, симвастатин, аторвастатин, флувастатин, церивастатин, атавастатин и розувастатин.
Упомянутые выше патенты США приведены здесь в качестве ссылок. Дозировка применяемых лекарственных средств соответствует дозировке, приведенной в настольном справочнике врача или в вышеуказанных патентах.
Соединения формулы I, заявленные в соответствии с настоящим изобретением, будут применяться в весовом соотношении к гиполипидемическим средствам (в случае их использования), равном 500:1-1:100.
Используемая дозировка должна тщательным образом подбираться в соответствии с возрастом, весом и соматическим состоянием пациента, так же как и путь введения, лекарственная форма и режим введения и желаемый результат.
Дозировки и рецептуры гиполипидемических средств соответствуют изложенным в различных патентах и заявках, приведенных выше.
Дозировки и рецептуры других гиполипидемических средств, в случае их использования, соответствуют изложенным в последнем издании настольного справочника врача.
При пероральном введении удовлетворительный результат может быть получен при использовании ингибитора МТР в количестве, находящемся в пределах от 0.01 мг/кг до 500 мг, предпочтительно, от 0.1 мг до 100 мг, вводимом от одного до четырех раз в день.
Предпочтительные лекарственные формы для перорального введения, такие, как таблетки или капсулы, содержат МТР ингибитор в количестве, которое варьирует от 1 до приблизительно 500 мг, предпочтительно от 2 до приблизительно 400 мг, более предпочтительно от 5 до приблизительно 250 мг, от одного до четырех раз в день.
При пероральном введении удовлетворительный результат может быть получен при использовании ингибиторов ГМГ-Коа редуктазы, например, правастатина, ловастатина, симвастатина, аторвастатина, флувастатина или церивастатина в дозировках, указанных в настольном справочнике врача, например в количестве, находящемся в пределах от 1 до 2000 мг, предпочтительно от 4 до 2 00 мг.
Ингибитор сквален-синтетазы может вводиться в количестве, находящемся в пределах от 10 мг до 2000 мг и, предпочтительно, от 25 мг до 200 мг.
Предпочтительные лекарственные формы для перорального применения, такие как таблетки или капсулы, содержат ингибитор ГМГ-КоА редуктазы в количестве, которое варьирует от 0.1 до 100 мг, предпочтительно от 5 до 80 мг, и, более предпочтительно, от 10 мг до 4 0 мг.
Предпочтительная лекарственная форма для перорального применения, такая как таблетки или капсулы, содержат ингибитор сквален-синтетазы в количестве, которое варьирует от 10 до 500 мг, предпочтительно, от 25 до 200 мг.
Другим гиполипидемическим средством может также быть ингибитор липооксигеназы, включая ингибитор 15-липооксигеназы (15-LO), относящийся к классу бензимидазол производных, такой, как описан в WO 97/12615; LO-15 ингибиторы, описанные в WO 97/12613; изотиазолоны, описанные в WO 96/38144; и 15-LO ингибиторы, описанные Sendobry et al в публикациях "Ослабление диет-индуцированного атеросклероза у кроликов с помощью высокоселективного ингибитора 15-липоксигензы, имеющего значительную антиоксидантную активность", Brit. J. Pharmacology (1997) 120, 1199-1206, и Cornicelli et al, "15-липоксигеназа и ее ингибирование: новая терапевтическая возможность в лечении сосудистых заболеваний", Current Pharmaceutical Design, 1999, 5, 11-20.
Соединения формулы I и гиполипидемические средства могут применяться вместе как в одной лекарственной форме для перорального применения, так и в виде отдельных лекарственных форм, принимаемых одновременно.
Композиции, описанные выше, могут вводиться в лекарственных формах, описанных выше, в единичных или разделенных дозах от одного до четырех раз в день. Предпочтительно начинать лечение с комбинации низких дозировок в с постепенным повышением дозировки до высокой.
Предпочтительными гиполипидемическими средствами являются правастатин, симвастатин, ловастатин, аторвастатин, флувастатин, церивастатин, атавастатин и розувастатин.
Кроме того, другими лекарственными средствами, которые могут применяться совместно с соединениями формулы I, являются 1,2,3 или более лекарственных средств для лечения ожирения, в том числе агонисты бета-3-адренергических рецепторов, ингибиторы липазы, ингибиторы обратного захвата серотонина (и допамина), препараты для воздействия на бета-рецепторов щитовидной железы, аноректические средства, NPY антагонисты, аналоги лептина и/или агонисты МС4.
Агонистом бета-3-адренергических рецепторов, который может применяться совместно с соединением формулы I, может быть AJ9677 (Takeda/Dainippon), L750355 (Merck) или СР331648 (Pfizer) или любые другие известные агонисты бета-3-адренергических рецепторов, например, описанные в патентах США №5,541,204, 5,770,615, 5,491,134, 5,776,983 и 5,488,064; предпочтительными являются AJ9677, L750,335 и СР331648.
Ингибиторами липазы, которые могут применяться совместно с соединениями формулы I, являются орлистат и ATL-962 (Alizyme); орлистат является предпочтительным.
Ингибиторами обратного захвата серотонина (и допамина), которые могут применяться совместно с соединениями формулы I, являются сибутрамин, топирамат (Johnson & Johnson) или аксокин (Regeneron); сибутрамин и топирамат являются предпочтительными.
Препаратом для воздействия на бета-рецепторы щитовидной железы, который может применяться совместно с соединениями формулы I, может быть лиганд рецептора щитовидной железы, такой, как описан в WO 97/21993 (U. Cal SF), WO 99/00353 (KaroBio) и GB 98/284425 (KaroBio); соединения, раскрытые в заявках KaroBio, являются предпочтительными.
Аноректическими средствами, которые могут применяться совместно с соединениями формулы I, являются дексамфетамин, фентермин, фенилпропаноламин или мазиндол; дексамфетамин является предпочтительным.
Соединения формулы I и средства для лечения ожирения могут применяться вместе как в одной лекарственной форме, так и в виде отдельных лекарственных форм, принимаемых одновременно, дозировка и режим введения общеизвестен специалистам, а также указан в настольном справочнике врача.
Примерами антитромбоцитарных лекарственных средств, которые могут применяться совместно с соединениями, заявленными в соответствии с настоящим изобретением, являются абциксимаб, тиклопидин, эптифибатид, дипиридамол, аспирин, анагрелид, тирофибан и/или клопидогрел.
К антигипертензивным лекарственным средствам, которые могут применяться совместно с соединениями, заявленными в соответствии с настоящим изобретением, относятся ингибиторы АПФ, антагонисты кальция, альфа-блокаторы, диуретики, лекарственные средства центрального действия, антагонисты ангиотензина-II, бета-блокаторы и ингибиторы вазопептидазы.
Примерами ингибиторов АПФ являются лизиноприл, эналаприл, хинаприл, беназеприл, фозиноприл, рамиприл, каптоприл, эналаприлат, моексиприл, тандолаприл и периндоприл; примерами антагонистов кальция являются амлодипин, дилтиазем, нифедипин, верапамил, фелодипин, низолдипин, израдипин и никардипин; примерами альфа-блокаторов являются теразозин, доксазозин и празозин; примерами диуретиков являются гидрохлортиазид, торасемид, фуросемид, спиронолактон и индапамид; примерами лекарственных средств центрального действия являются клонидин и гуанфацин; примерами ингибиторов ангиотензина-II являются лозартан, валсартан, ирбесартан, кандесартан и телмисартан; примерами бета-блокаторов являются метопролол, пропанолол, атенолол, карведилол и соталол; и примерами ингибиторов вазопептидазы являются омапатрилат и гемопатрилат.
Способ, заявленный в соответствии с настоящим изобретением, подразумевает применение фармацевтической композиции, включающей соединение формулы I как совместно с другим антидиабетическим, гиполипидемическим или другим лекарственным средством, так и без него, а также фармацевтически приемлемый переносчик или разбавитель. Фармацевтическая композиция может быть получена при использовании обычных твердых или жидких переносчиков или разбавителей и фармацевтических добавок, соответствующих желаемому способу введения. Соединения могут вводиться млекопитающим, включая человека, обезьян, собак и т.д., как перорально, например, в форме таблеток, капсул, гранул или порошков, так и парентерально, в форме инъекционных препаратов, а также интраназально или с помощью трансдермальных пластырей. Доза для взрослой особи находится, предпочтительно, в пределах от 10 до 2000 мг в день, и может вводиться как однократно, так и быть разделена на несколько приемов от 1 до 4 раз в день.
Типичную лекарственную форму для инъекционного введения приготавливают путем помещения соединения формулы I в асептических условиях в ампулу, которую потом асептически высушивают сублимацией и герметизируют. Перед введением содержимое ампулы растворяют в 2 мл физиологического раствора для получения инъекционного препарата.
Ингибирующая активность соединения формулы I может быть определена с помощью исследований, приведенных ниже.
Исследование SGLT2 активности
Последовательность мРНК человеческого SGLT2 (GenBank #М95549) была клонирована с помощью обратной транскрипции и амплификации мРНК, взятой из почки человека, с использованием стандартных технологий молекулярной биологии. Последовательность кДНК была устойчиво трансфицирована в СНО клетки и клоны исследовали на SGLT2 активность, как описано у Ryan et al. (1994). Оценка активности ингибирования SGLT2 в клеточных линиях, отобранных с помощью клональной селекции, проводилась, как описано у Ryan et al., со следующими модификациями. Клетки выращивали в 96-луночных планшетах в течение 2-4 дней до количества 75,000 или 30,000 клеток на лунку в F-12 питательной смеси (Ham's F-12)с добавлением 10% эмбриональной бычей сыворотки, 300 мкг/мл генетицина и пенициллин-стрептомицина. В состоянии монослоя клетки отмыли дважды 10 мМоль раствором Hepes/Tris, рН 7.4, 137 мМоль N-метил-D-глюкамина, 5.4 мМоль KCl, 2.8 мМоль CaCl г, 1.2 мМоль MgSO4. Затем клетки инкубировали с 10 мкМоль [14C]AMG и 10 мкМоль ингибитора (конечный DMSO=0.5%) в 10 мМоль раствора Herpes/Tris, рН 7.4, 137 мМоль NaCl, 5.4 мМоль KCl, 2.8 мМоль CaCl г, 1.2 мМоль MgSO4 при температуре 37°С в течение 1.5 ч. Исследования по захвату метки были проведены в ледяном IX PBS, содержащем 0.5 ммоль флоризина, и клетки лизировали в 0.1% NaOH. После добавления сцинцилляционной жидкости MicroScint, клетки оставили перемешиваться на 1 час, и затем [14C]AMG подсчитали на счетчике TopCount. Контроль осуществлялся как в присутствии NaCl, так и при его отсутствии. Для определения показателя ЕС50, использовались 10-кратные концентрации ингибитора с интервалами 2 log в подходящей зоне ответа в трехкратном повторении на планшетах. Ryan MJ, Johnson G, Kirk J, Fuerstenberg SM, Zager RA и Torok-Storb B. 1994. HK-2: бессмертная клеточная линия эпителия проксимальных почечных канальцев здоровой почки взрослого человека. Kidney International 45: 48-57.
Следующие Примеры иллюстрируют настоящее изобретение. Температура, если дополнительно не оговаривается иное, выражена в значениях стоградусной шкалы.
Пример
А. 5-бром-2-хлор-4'-этоксибензофенон
К перемешанной суспензии коммерческой 5-бром-2-хлорбензойной кислоты (250 г, 1.06 моль) в 450 мл CH2Cl2, содержащей оксалил хлорид (1.1 моль) добавили 1.5 мл DMF. После прекращения интенсивного выделение газа, реакционную смесь перемешивали в течение ночи, после чего удалили летучие соединения в вакууме с помощью вращающегося испарителя. После растворения неочищенного 5-бром-2-хлорбензоилхлорида в 200 мл CH2Cl2, желтый раствор поместили в двухлитровую колбу с тремя горлышками, оснащенную наружным миксером и внутренним термометром. Данную перемешанную смесь охладили до - 3° после чего добавили фенетол (130 г, 1.08 моль).
Далее с помощью твердой воронки в течение 30 минут добавляли AlCl3 (140 г, 1.07 моль) для того, чтобы не произошло повышения температуры выше 4°. Значительное количество газообразной HCl, которая начала выделяться после добавления 60% AlCl3, улавливали путем пропускания газа через перемешанный концентрированный раствор NaOH. Жидкостная хроматография высокого разрешения (HPLC) выявила завершение реакции на 95% в течение 10 мин. после окончания добавления AlCl3. Далее смесь перемешивали при температуре 4° в течение 1 ч, после чего реакционную смесь охладили с помощью льда. Впоследствии суспензию развели Н2О (1 л) и экстрагировали трехкратно с помощью CH2Cl2. Комбинированные органические экстракты отмыли два раза 1Н. HCl, один раз Н2О, два раза 1М NaOH и два раза солевым раствором, после чего высушили над Na2SO4. После удаления летучих веществ, при использовании HPLC получили осадок, представляющий собой смесь орто/пара изомеров в соотношении 1:7.
Двухкратная рекристаллизация с помощью 400 мл абсолютного этилового спирта привела к получению 230 г (64%) 5-бром-2-хлор-4'-этоксибензофенона.
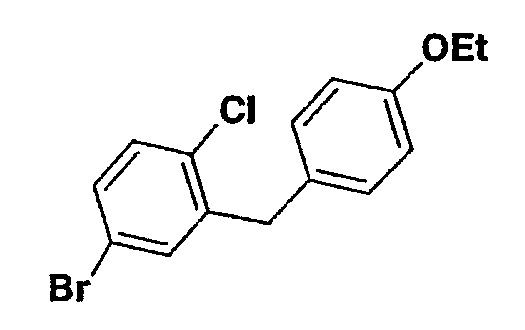
В. 5-бром-2-хлор-4'-этоксидифенилметан
К перемешанному раствору Et3SiH (400 мл, 2.51 моль) и 5-бром-2-хлор-4'-этоксибензофенона (390 г, 1.15 моль) в 900 мл смеси 1,2-дихлорэтан/MeCN в соотношении 1:2 при температуре 10° добавили BF3⋅Et2O (150 мл, 1.58 моль) при условии, что температура не превышала 20°С. В процессе добавления происходило умеренное выделение тепла.
Смесь перемешивали в течение ночи при температуре 20°С, после чего HPLC выявила завершение реакции на 90%. После добавления 40 мл Et3SiH и 15 мл BF3⋅Et2O реакционную смесь нагревали до 50°С в течение 3 ч. (замечено, высокие температуры усиливают образование продукта реакции Риттера - N-ацетил 5-бром-2-хлор-4'-этоксидифенилметиламина). Для охлаждения в реакционную смесь добавили 120 г KOH в 300 мл Н2О. После перемешивания в течение 2 ч, слои разделили. Водный слой экстрагировали дважды с помощью CH2Cl2; комбинированные органические слои отмыли однократно с помощью 300 мл 2М KOH, дважды с помощью Н2О, содержащей 10% солевого раствора, для усиления сепарации слоев, и дважды с помощью солевого раствора непосредственно перед высушиванием над Na2SO4. После удаления летучих веществ, осадок рекристаллизировали из абсолютного этилового спирта для получения 230 г 5-бром-2-хлор-4'-этоксидифенилметана в виде белого твердого вещества.
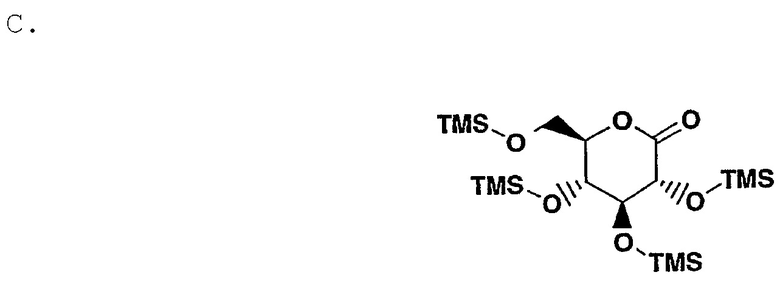
С. 2,3,4,6-тетра-О-Триметилсилил-β-D-глюколактон
К перемешанному при температуре -5°С раствору глюконолактона (239 г, 1.34 моль) и N-метилморфолина (1180 мл, 10.73 моль) в 2.4 л THF под Ar через капельную воронку добавили хлорид триметилсилила (1022 мл, 8.05 моль) таким образом, чтобы температура не превышала 5°С. Затем реакционную смесь перемешивали в течение 1 ч, после чего нагревали ее до 35°С в течение 5 часов и перемешивали в течение ночи, в результате чего произошло снижение температуры смеси до 20°С. Затем смесь развели 3.6 л толуола и охладили до 0-5°С перед аккуратным добавлением 7 л Н2О таким образом, чтобы температура не превышала 10°С. Следует обратить внимание на интенсивное выделение тепла во время добавления первой порции Н2О. После перемешивания фазы сепарировали и разделили. Органические фазы отмыли с помощью водн. NaH2PO4 (2 л), Н2О (1 л) и солевого раствора (1 л). Органический слой затем концентрировали над вакуумом, используя вращающийся испаритель; образовавшееся светло-желтое масло дважды обработали 250 мл толуола и повторно сконцентрировали до получения 616 г указанного соединения.
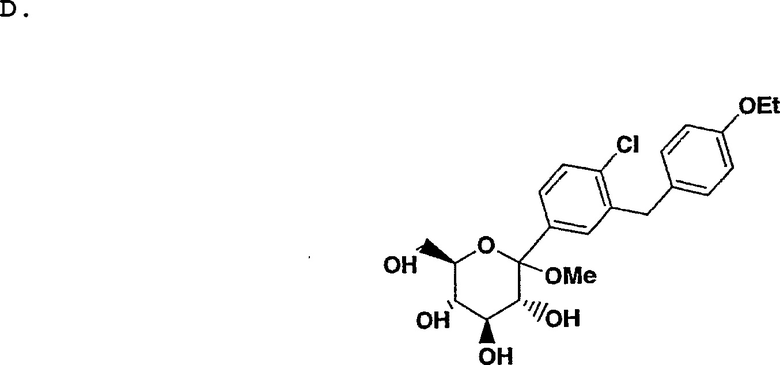
К перемешанному при температуре -78°С раствору части В 5-бром-2-хлор-4'-этоксидифенилметана (150 г, 0.4 6 моль) в 1.15 л смеси сухой THF/толулол в соотношении 1:2 под Ar добавили 184 мл 2.5 М n-BuLi в гексане по каплям, таким образом, чтобы температура не поднималась выше -70°С. После перемешивания в течение 30 мин., раствор с помощью канюли поместили в перемешанный при -78°С раствор части С 2,3,4,6-тетра-О-триметилсилил-β-D-глюколактона (236 г, 0.51 моль) в 1.1 л толуола, таким образом, чтобы температура оставалась ниже -70°С. После перемешивания раствора в течение 30 мин. при температуре -78°С его охладили путем добавления 1 л МеОН, содержащего метансульфоновую кислоту (41.8 мл, 0.64 моль). Реакционную смесь перемешивали в течение ночи и температура смеси поднялась до 20°С. HPLC выявила два новых пика, соответствующих массе ожидаемого О-метилглюкозида; соотношение варьировало в пределах от 95:5 до 80:20. Желаемый продукт соответствует главному пику с более коротким временем удержания. Следует обратить внимание на то, что более длительное протекание реакции или добавление более 50% метансульфоновой кислоты способствует превращению всего изомерного продукта в желаемый О-метилглюкозид. После завершения реакции смесь охладили с помощью добавления NaHCO3 (37 г, 0.37 моль) в 200 мл Н2О. Если рН не является слабоосновным, добавляют еще NaHCO3 перед двукратным разведением слоев H2O и трехкратном экстрагированием с помощью EtOAc. Комбинированные EtOAc фракции отмыли солевым раствором и высушили над Na2SO4. После концентрирования с помощью вращающегося испарителя, осадок растворили в горячем толуоле (150 мл). Полученный раствор влили в литр перемешанного гексана. Преципитат собрали с помощью вакуумной фильтрации; полученный осадок на фильтре отмыли два раза с помощью 500 мл гексана и затем высушили на воздухе для получения 171 г желаемого соединения в виде белого твердого вещества.
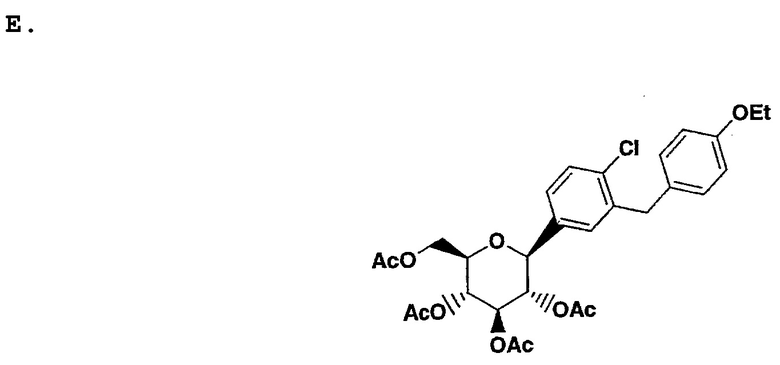
К перемешанному при -10°С раствору части D O-метилглюкозида (123 г, 0.28 моль) в 1.2 л смеси CH2Cl2/MeCN в соотношении 1:1 добавили сначала Et3SiH (65.27 г, 0.56 моль), а затем BF3⋅Et2O (59.75 г, 0.42 моль) таким образом, чтобы температура оставалась в пределах от -5° до -10°С. Температура перемешанного раствора поднималась до 0°С в течение 5 ч. Когда жидкостная хроматография высокого разрешения (HPLC) выявила завершение реакции, реакционную смесь охладили путем добавления насыщенного водного NaHCO3 (310 мл). Органические летучие вещества удалили над вакуумом с помощью вращающегося испарителя. Осадок разделили между EtOAc и Н2О, используя по 2 л каждого вещества. После разделения фаз, водный слой экстрагировали двукратно с помощью 2 л EtOAc. Комбинированные органические фазы отмыли Н2О (2 л) и солевым раствором (2 л), затем высушили над MgSO4 и сконцентрировали с помощью вращающегося испарителя до получения 104.6 г желтой застывшей пены. После растворения полученного осадка в CH2Cl2 (750 мл) добавили пиридин (200 г, 2.53 моль) и Ас2О (261.1 г, 2.56 моль) в одной порции. После того, как завершилось выделение тепла, повысившее температуру смеси с 28°С до 47°С, добавили DMAP (1.56 г, 13 мМоль). Через 1.5 ч реакционную смесь охладили путем добавления Н2О (1.8 л), HPLC выявила завершение реакции. Смесь экстрагировали двукратно с помощью CH2Cl2 (общий объем 2.7 л); комбинированные органические слои отмыли дважды 1н. HCl (1.8 л), дважды солевым раствором (1.8 л), после чего высушили над MgSO4. После концентрирования с помощью вращающегося испарителя, осадок рекристаллизировали из абсолютного этилового спирта (750 мл) для получения 8 9.5 г желаемого тетраацетилированного β-С-глюкозида в виде белого твердого вещества. Исходная жидкость содержала соответствующий α-С-глюкозид и более полярный изомер фуранозы.
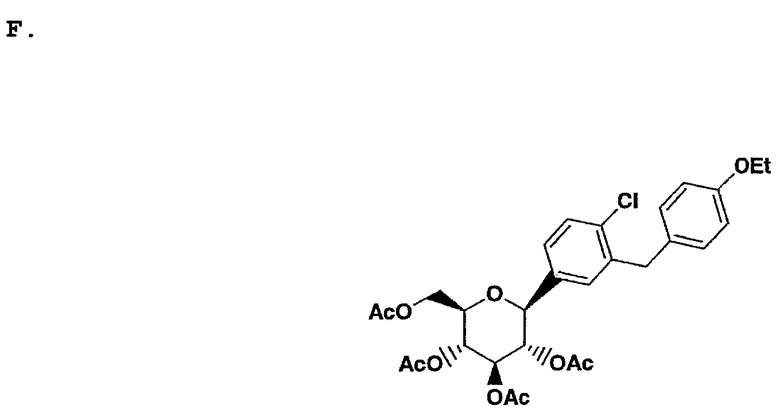
Альтернативно, часть D О-метилглюкозида может быть сначала ацетилирована, а затем восстановлена для получения желаемого тетраацетилированного С-арилгликоида, в соответствии со следующей процедурой.
Раствор части D О-метилглюкозида (3.0 г, 6.8 мМоль) в толуоле (45 мл), содержащий диизопропилэтиламин (6.9 мл, 40 мМоль)охладили до 0°С, после чего к нему добавили уксусный ангидрид (3.35 мл, 35.5 мМоль) и DMAP (84 мг, 0.6 8 мМоль). Температура раствора поднялась до 20°С в течение 6 ч, после чего тонкослойная хроматография выявила завершение образования тетраацетата. Реакционную смесь охладили путем добавления 50 мл 20% Н3РО4. После разделения слоев, водную фазу двукратно экстрагировали с помощью толуола. Комбинированные органические фазы отмыли один раз 50 мл Н2О, после чего сконцентрировали над вакуумом. Полученное масло растворили в 20 мл толуола и сконцентрировали для получения густого масла (4.15 г), которое использовалось далее без дополнительного очищения.
Раствор указанного выше неочищенного масла (4.15 г, 6.8 моль) в MeCN (60 мл), содержащий один эквивалент Н2О (123 мг, 6.8 моль) охладили до 0°С, после чего добавили сначала Et3SiH (3.27 мл, 20.5 мМоль) и затем BF3⋅Et2O (1.73 мл, 13.7 мМоль). После перемешивания в течении 1 ч. температура раствора поднялась до 20°С. Через 4 ч. осуществили однократный анализ с помощью жидкостной хроматографии высокого разрешения (HPLC), который выявил отсутствие прогрессивного течения реакции свыше 60%, после чего добавили 2 мл Et3SiH и 1 мл BF3⋅Et2O. Два часа спустя, HPLC выявила отсутствие исходного материала. После добавления водн. NaHCO3 для охлаждения реакционной смеси последнюю перемешивали в течение 30 мин., после чего трехкратно экстрагировали с помощью EtOAc. Комбинированные органические слои отмыли один раз водн. NaHCO3 и солевым раствором, после чего высушили над Na2SO4. Полученное после концентрирования над вакуумом масло растворили в 70 мл горячей 25% смеси EtOAc/гексан. В процессе охлаждения кристаллизовалось 2.45 г желаемого тетраацетилированного β-С-арилглюкозида, который последовательно выделили с помощью фильтрации.
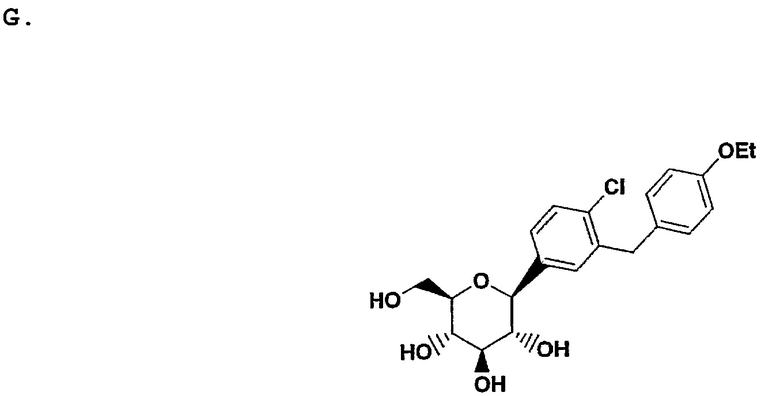
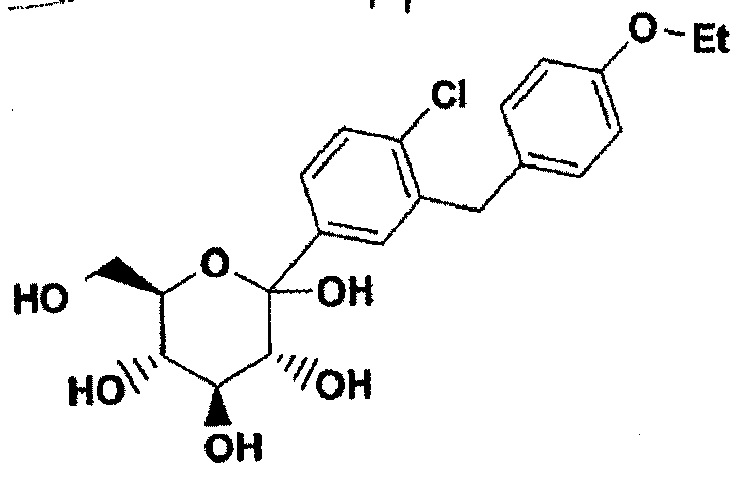
К перемешанному при температуре 20°С раствору тетраацетилированного β-С-глюкозида (27.2 г., 49 мМоль) полученного в соответствии с пунктом Е) в 480 мл смеси THF/MeOH/H2O в соотношении 2:3:1 добавили LiOH⋅H2O (2.3 г., 57 мМоль). После перемешивания в течение ночи летучие вещества удалили с помощью вращающегося испарителя. Осадок после растворения в EtOAc (300 мл) отмыли один раз солевым раствором (150 мл), один раз солевым раствором (50 мл), содержащим 10 мл 5% водн. KHSO4, и снова солевым раствором (50 мл), после чего высушили над Na2SO4. Летучие вещества удалили с помощью вращающегося испарителя и полученное масло в минимальном количестве CH2Cl2 вспенили над вакуумом для получения 2 0.4 г. желаемого С-арилглюкозида в виде стекловидного вещества белого цвета, содержащего 0.11 моль % EtOAc.
Время удержания HPLC: 7.08 мин., 94%-ная чистота, YMC S5 С-18 4.6×50 мм колонка, 2.5 мл/мин, определение при 220 нм; 8 мин - градиент 0-100% В поддерживается 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0.2% H3PO4. Растворитель В: 90% МеОН/H2O + 0.2% Н3РО4.
1Н ЯМР (500 МГц, CD3OD) δ 7.33 (d, 1Н, J=6 Гц), 7.31 (d, 1Н, J=2.2 Гц), 7.31 (dd, 1H, J=6 Гц, J=2.2 Гц), 7.07 (d, 2H, J=8.8 Гц), 6.78 (d, 2H, J=8.8 Гц), 4.07-3.90 (m, 7H), 3.85 (d, 1H, J=10.6 Гц), 3.69 (dd, 1H, J=5.3, 10.6 Гц), 3.42-3.25 (m, 4Н)Гц), 1.34 (t, 3H, J=7 Гц).
13С ЯМР (125 МГц, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2.
Anal Calcd для C21H25ClO6 LC-MS [M+Na+] 431; найден 431.
Ниже описано получение промежуточных соединений, а также их спектральные характеристики.
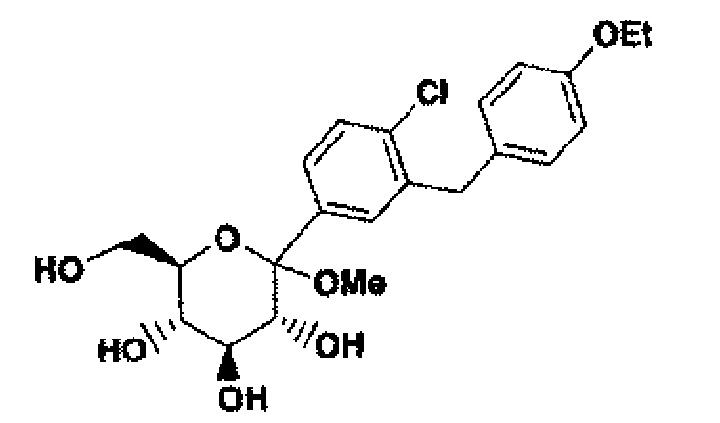
Белое твердое вещество. ЖХВР: колонка Zorbax SB С18, 4,6×75 мм, 0-100% В в течение 8 мин, выдержка 4 мин, А=10% МеОН / 90% H2O / 0,2% Н3РО4, В=10% H2O / 90% МеОН / 0,2% H3PO4, комн. темп. = 7,60 мин, степень чистоты 100%.
LC-MS: колонка Phenomex Luna 5u, 4,6×5 0 мм, 0-100% В в течение 4 мин, выдержка 1 мин, А=10% CH3CN / 90% H2O / 10 мМ NH4Ac, В=10% H2O / 90% CH3CN / 10 мМ NH4Ac, комн. темп. = 2,00 мин, рассчитано для C22H31ClNO7 (М+NH4)+ 456,2, найдено 456,3.
1H ЯМР (400 МГц, CD3OD) δ 7,77 (s, 1Н), 7,22-7,27 (m, 2Н), 7,02 (d, J=8,8, 2Н), 6,74 (d, J=9,3, 2Н), 5,01 (s, 1Н), 4,84 (s, 1H), 3,75-4, 06 (m, 8H), 3, 47-3, 60 (m, 2H), 3,17 (t, J=8,3 Гц, 1H), 3,00 (m, 1H), 2,89 (s, 3H), 1,33 (t, J=6,6, 1H). 13C ЯМР (100 МГц, CD3OD) δ 157.3, 138.6, 136.6, 134.4, 131.4, 130.0, 129.7, 129.3, 126.7, 114.4, 101.0, 74.7, 72.5, 70.1, 63.4, 62.0, 49.2, 38.5, 14.9.
Белое твердое вещество, tпл = 120°С (не корректировалась). ЖХВР tR=3,9 мин, степень чистоты 100%. Время удерживания для ЖХВР на колонке Phenomex Luna С-18, 4,6 мм × 50 мм, элюент с градиентом 4 мин 0-100% растворителя В; при этом растворитель А: 10: 90: 0,1 CH2OH = H2O - ТФК и растворитель В: 90:10:0,1 СН3ОН = H2O - ТФК.
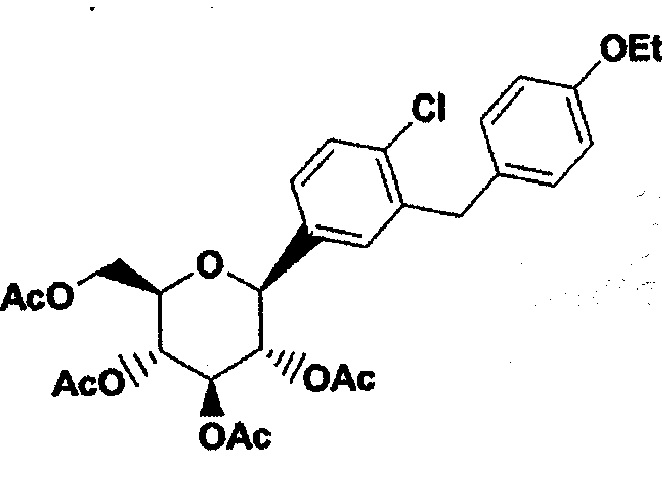
1H ЯМР (400 МГц, CDCl3) δ 7, 35 (d, J=8,4, 1Н), 7,19 (dd, J=1,8, 8,4, 1H), 7,07 (d, J=1,8, 1H), 7,05 (d, J=8,8, 2H), 6,82 (d, J=8,8, 2H), 5,28 (t, J=9,2, 1H), 5,20 (t, J=9,2, 1H), 5,05 (t, J=9,2, 1H), 4,31 (d, J=9,7, 1H), 4,26 (dd, J=4,8, 12,8, 1H), 4,14 (dd, J=2,2, 12,4, 1H), 3,95-4,07 (m, 4H), 3,80 (m, 1H), 2,08 (s, 3H), 2,04 (s, 3H), 1,99 (s, 3H), 1,71 (s, 3H), 1,40 (t, J=7,0, 3H). 13C ЯМР (100 МГц, CDCl3) δ 170.7, 170.3, 169.45, 168.7, 157.5, 139.1, 135.1, 134.6, 131.0, 129.79, 126.0, 114.5, 79.5, 76.1, 74.1, 72.5, 68.5, 63.4, 62.3, 38.2, 20.7, 20.6, 20.3, 14.8. HRMS вычислено для C29H33ClNaO10 (M+Na)+: 599, 1660; найдено: 599, 1649. Рассчитано для C29H33ClO10: С, 60, 36; Н, 5,76. Найдено: С, 60,43; Н, 5,56.
Для основного аномера: ЖХВР tR=3,45 мин, степень чистоты 100%. Время удерживания для ЖХВР на колонке Phenomex Luna С-18, 4,6 мм × 5 0 мм, элюент с градиентом 4 мин 0-100% растворителя В; при этом растворитель А: 10: 90: 0,1 СН3ОН - H2O - ТФК; растворитель В: 90: 10: 0,1 СН3ОН - H2O - ТФК.
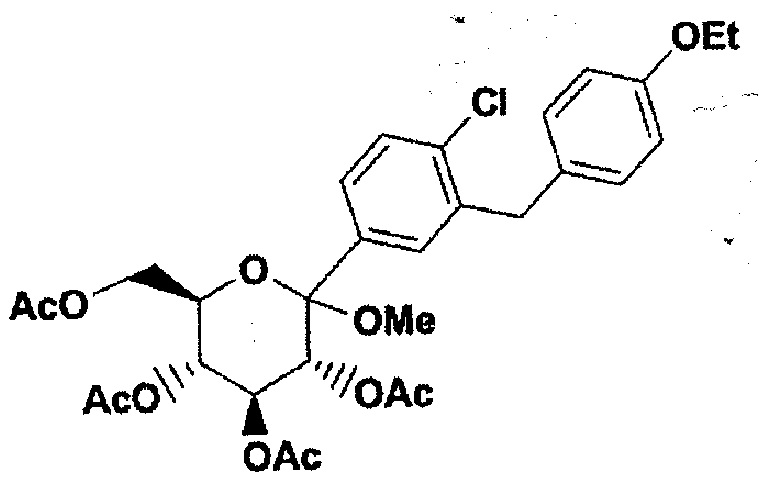
1H ЯМР (400 МГц, CD3OD) δ 7, 54 (d, J=2,2, 1Н), 7,45 (dd, J=2,2, 8,4, 1H), 7,35 (d, J=8,4, 1H), 7,08 (d, J=8,8, 2H), 6,79 (d, J=8,8, 2H), 4,08 (d, J=15,0, 1H), 3,99 (d, J=15,0, 1H), 3,98 (q, J=7,0, 2H), 3,92 (dd, J=2,2, 11,8, 1H), 3,80 (dd, J=5,3, 11,9, 1H), 3,74 (t, J=9,2, 1H), 3,57 (m, 1H), 3,41 (d, J=8,8, 1H), 3,08 (d, J=9,7, 1H), 3,06 (s, 3H), 1,35 (t, J=7,0, 3H). 13C ЯМР (100 МГц, CDCl3) δ 158.2, 139.0, 138.5, 134.2, 132.4, 131.2, 130.1, 129.2, 127.6, 114.8, 101.8, 78.0, 75.3, 74.5, 71.1, 63.8, 62.1, 49.0, 38.7, 14.5. Рассчитано для C22H27ClO7: С, 60, 20; Н, 6,20; Cl, 8,07. Найдено: С, 60, 05; Н, 6,21; Cl, 8,01.
Прозрачное масло. ЖХВР: колонка Zorbax SB С 18, 4,6 × 75 мм, 0-100% В в течение 8 мин, выдержка 4 мин, А=10% МеОН / 90% H2O / 0,2% Н3РО4, В=10% H2O / 90% МеОН / 0,2% H3PO4, комн. темп. = 8,85 мин, степень чистоты 98,1%.
LC-MS: колонка Phenomex Luna 5u, 4,6 × 50 мм, 0-100% В в течение 4 мин, выдержка 1 мин, А=10% CH3CN / 90% Н20 / 10 мМ NH4Ac, В=10% H2O / 90% CH3CN / 10 мМ NH4Ac, комн. темп. = 3,55 мин. Рассчитано для C30H39ClNO11 (М+NH4) + 624, 2, найдено 624, 3. 1H ЯМР (400 МГц, CD3OD) δ 7,38 (d, J=8,3, 1Н), 7,27 (dd, J=1,7, 8,8, 1H), 7,21 (d, J=2,2, 1H), 7,03 (d, J=8,8, 2H), 6,81 (d, J=8,2, 2H), 5,55 (t, J=9,9, 1H), 5,21 (t, J=9, 6, 1H), 4,92 (d, J=10,5, 1H), 4,34 (dd, J=5,0, 12,0, 1H), 4,20 (dd, J=2,8, 12,1, 1H), 3,97-4,08 (m, 5H), 3,09 (s, 3H), 2,10 (s, 3Н), 2,09 (s, 3Н), 1,94 (s, 3Н), 1,76 (s, 3Н), 1,39 (t, J=7,2, 3Н). 13C ЯМР (100 МГц, CD3OD) δ 170.7, 170.2, 170.1, 169.6, 169.0, 157.5, 139.0, 135.1, 134.7, 131.1, 129.8, 129.7, 129.3, 126.0, 114.6, 100.1 80.8, 73.9, 71.3, 68.9, 68.8, 63.4, 62.1, 52.1, 49.5, 38.4, 20.7, 20.6, 20.3, 14.9.
5-бром-2-хлор-4'-этоксидифенилметан
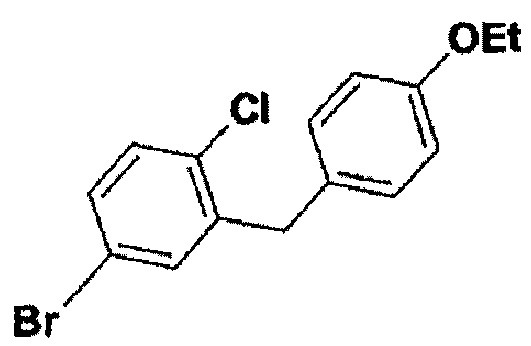
Белое твердое вещество; т.пл. 37°С (не корректировалась). ЖХВР tR=4,43 мин, степень чистоты 100%. Время удерживания для ЖХВР на колонке Phenomex Luna С-18, 4,6 мм × 50 мм, элюент с градиентом 4 мин, 0-100% растворителя В; растворитель А: 10: 90: 0,1 СН3ОН - H2O - ТФК; растворитель В: 90: 10: 0,1 СН3ОН -H2O - ТФК.
1Н ЯМР (400 МГц, CDCl3) δ 7,20-7,28 (m, 3Н), 7,08 (d, J=8,8, 2Н), 4,00 (q, J=7,0, 2H), 3,96 (s, 2H), 1,40 (t, J=7,0, 3Н). 13C ЯМР (125 МГц, CDCl3) δ 157.6, 141.3, 133.5, 133.1, 130.9, 130.5, 130.4, 130.0, 120.4, 114.6, 63.4, 38.2, 14.9. Вычислено для C15H14BrClO: С, 55, 32; Н, 4,33. Найдено: С, 55,22; Н, 4,25.
5-литий-2-хлор-4'-этоксидифенилметан
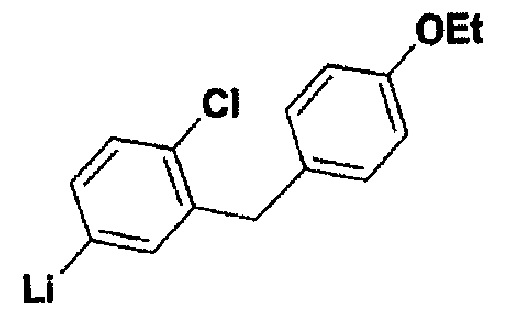
Данные не удалось получить, продукт термически неустойчив выше -75°С, происходит быстрое разложение,
или
5-бром-2-хлор-4'-этоксибензофенон
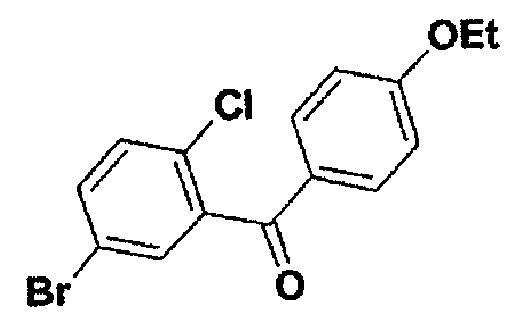
Белое твердое вещество; т.пл. = 60°С (не корректировалась). ЖХВР tR=3,9 мин, степень чистоты 100%. Время удерживания при ЖХВР с применением колонки Phenomex Luna С-18, 4,6 мм × 50 мм, элюент с градиентом 4 мин 0-100% растворителя В; растворитель А: 10: 90: 0,1 СН3ОН - Н2О - ТФК; растворитель В: 90: 10: 0,1 СН3ОН - H2O - ТФК.
1Н ЯМР (400 МГц, CDCl3) δ 7,76 (d, J=8,8, 2Н), 7,53 (dd, J=2,2, 8,4, 1H), 7,48 (d, J=2,2, 1H), 7,32 (d, J=8,8, 1H), 6,93 (d, J=8,8, 2H), 4,11 (q, J=7,0, 2H), 1,45 (t, J=7,0, 3Н). 13C ЯМР (100 МГц, CDCl3) δ 192.0, 163.9, 140.7, 133.7, 132.6, 131.4, 130.1, 128.6, 120.5, 114.5, 63.9, 14.6.
HRMS вычислено для C15H13BrClO2 (M+H)+: 338, 9787; найдено 338, 9785. Вычислено для C15H12BrClO2: С, 53, 05; Н, 3,56. Найдено: С, 53,11; Н, 3,28.
Спектральные приборы и условия
Спектры ЯМР (1Н, 13С) снимали на спектрометрах JEOL JNM-EPC-500, JEOL GSX400 и Bruker 400. Химические сдвиги указаны в ч/млн. (ppm), в области слабых полей относительно внутреннего эталона, тетраметилсилана; константы сочетания (величины J) приведены в Гц (Hz). Элементный анализ проводили в Discovery Analytical Science Department. Температуры плавления определяли на приборе Hoover Uni-melt, они не корректировались. LC/MS записывали на приборе Shimadzu LC-10AT, снабженном инжектором SIL-10A, детектор SDD-10AV обычно работал при длине волны 200 нм и с помощью масс-спектрометра Micromass ZMD. Если иное не указано, время удерживания при LC/MS или ЖХВР определяли на колонке Phenomex Luna С-18, 4,6 мм × 50 мм, элюент с градиентом 4 мин 0-100% растворителя В; при этом растворитель А: 10: 90: 0,1 СН3ОН - H2O - ТФК и растворитель В: 90: 10: 0,1 СН3ОН - H2O - ТФК.
Биологические данные
Соединение формулы I (Пример)
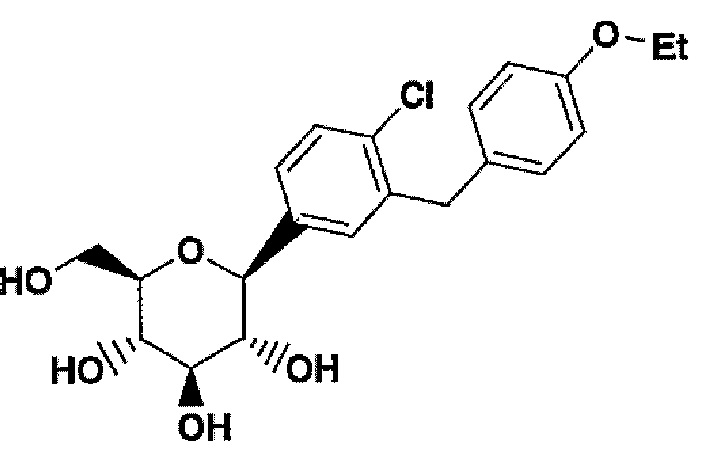
является активным и селективным ингибитором основного люминального переносчика глюкозы в почечном проксимальном канальце (SGLT2).
- При таких низких дозах, как 0,1 мг/кг, соединение формулы I стимулировал значительное выделение глюкозы в моче у обычных крыс, а при таких низких дозах, как 0,01 мг/кг, соединение формулы I стимулировало выделение глюкозы в моче у крыс ZDF, страдающих от глюкозурии вследствие диабета. Эти данные позволяют предположить, что соединение формулы I действует на диабетическую почку со снижением порогового содержания глюкозы в почке. Свойство соединения формулы I стимулировать выделение глюкозы у обычных крыс оказывается более сильным, чем в случае серглифлозина, 9Т-1095, или флоризина, введенных подкожно.
- У самцов крыс ZDF введение соединения формулы I в течение 15 дней привело к значительно меньшему ускорению и уровню глюкозы в плазме по сравнению с крысами, которым вводили носитель.
- При проведении исследования гиперинсулинемии-нормогликемии с применением клэмпов значительное увеличение инфузии глюкозы и скорости утилизации глюкозы сопровождались значительным уменьшением HGP у крыс, получавших соединение формулы I, по сравнению с крысами, которым вводили носитель.
- Доклиническая фармакология соединения формулы I позволяет предположить, что этот агент может корректировать гипергликемию в условиях окружающей среды и улучшать метаболический статус у крыс ZDF с развившимся диабетом. С учетом отсутствия побочных эффектов в желудочно-кишечном тракте или гипергликемии эти данные позволяют предположить, что соединение формулы I является возможным терапевтическим кандидатом для лечения диабета.
Повышенная селективность к SGLT2 по сравнению с SGLT1
Эффективность ингибирования in vitro SGLT (ЕС50) соединения формулы I и, в отдельных экспериментах, Примера 12 публикации W0 01/27128, оценивали путем мониторинга ингибирования накопления радиоактивно меченого α-метил-D-глюкопиранозида (AMG) клетками яичника китайского хомячка, стабильно экспрессирующими SGLT2 и SGLT1 человека.
Средние значения ЕС50 для SGLT2 и SGLT1 для каждого соединения представлены в Таблице 1 ниже.
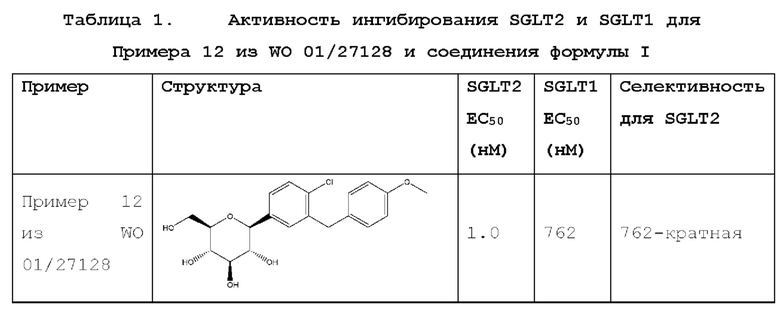
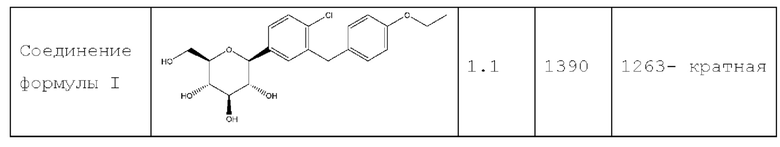
Как показано в Таблице 1, соединение формулы I демонстрирует среднее значение ЕС50 против SGLT2, равное 1.1 нМ, по сравнению со средним значением ЕС50 для Примера 12 из W0 '128, равным 1.0 нМ. По отношению к SGLT1 соединение формулы I и Пример 12 из WO '128 показали средние значения ЕС50 1390 нМ и 762 нМ, соответственно.
Эти результаты показывают, что соединение формулы I является высокоселективным (в 1,263 раза) в отношении SGLT2 по сравнению с SGLT1 и примерно в 1.7 раза более селективен в отношении SGLT2 по сравнению с SGLT1 относительно Примера 12 из WO 01/27128.
Повышенная способность снижать уровень глюкозы в крови через 5 часов после перорального введения у крыс с диабетом, индуцированным STZ (краткосрочная модель исследования на животных)
Была проведена оценка in vivo эффективности соединения формулы I и Примера 12 из WO 01/27128 с применением модели STZ-индуцированного диабета у крыс. После перорального введения соединения формулы I в виде однократной дозы 0.03 или 0.1 мг/кг уровни глюкозы в крови измеряли каждый час в течение 5 часов, в течение которых крыс лишали пищи. В отдельных исследованиях вводили однократную дозу 0.03 или 0.1 мг/кг Примера 12 из WO 01/27128.
Результаты представлены в Таблице 2 ниже. Значения за 5 часов в Таблице 2 представляют собой средний процент снижения уровней глюкозы в крови относительно исходных значений для этой когорты крыс. Поскольку крысы не имели доступа к пище в течение этого 5-часового периода, показатели глюкозы в крови как обработанных, так и необработанных крыс снизились. Чтобы скорректировать этот эффект, процентное изменение уровня глюкозы в час 5 для контрольной группы, получавшей носитель, вычитали из процентного изменения уровня глюкозы через 5 часов после введения дозы.
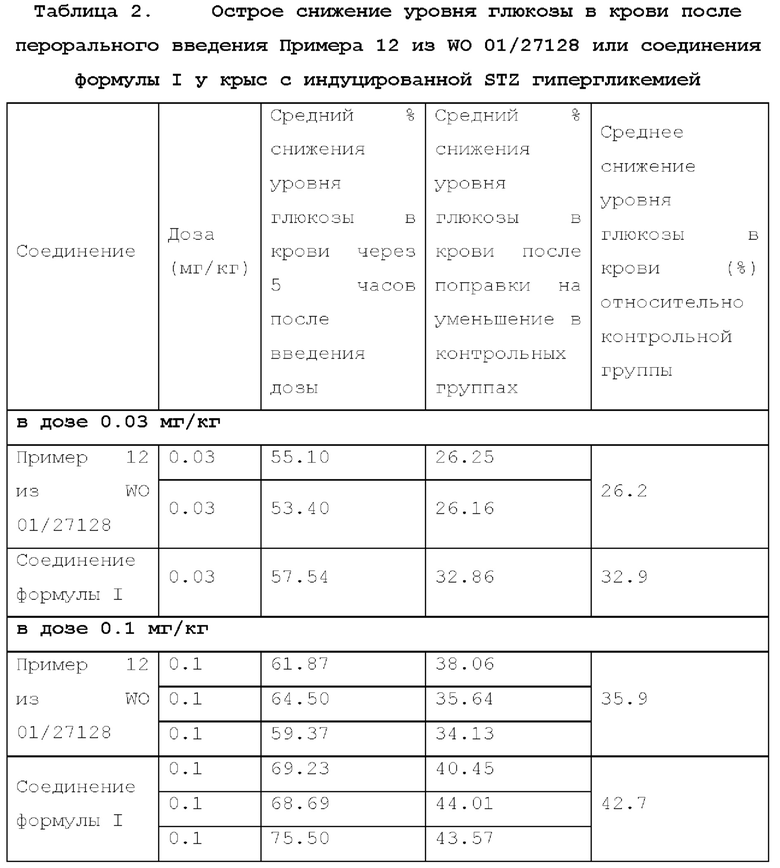
Средние значения процентных изменений через 5 часов в группах, получавших лекарственное средство, сравнивали со средними значениями процентных изменений через 5 часов, полученными в группе, получающей носитель, с использованием Т-теста. Все значения р были меньше или равны 0.0006, что считалось статистически значимым.
Как показано в Таблице 2, среднее снижение уровней глюкозы в крови по сравнению с контролем было выше для соединения формулы I, чем для Примера 12 из WO 01/27128 (32.9% против 26.2% при дозе 0.03 мг/кг; 42.7% против 35.9% при дозе 0.1 мг/кг). Это соответствует увеличению снижения уровней глюкозы в крови под действием соединения формулы I по сравнению с Примером 12 на 25% ((32.9-26.2)/26.2) и 19% ((42.7-35.9)/35.9) при 0.03 и 0.1 мг/кг, соответственно.
Повышенная способность снижать уровень глюкозы в плазме в течение 15-дневного периода после перорального введения у крыс с диабетом, индуцированным STZ (долгосрочная модель исследования на животных)
Эффективность in vivo соединения формулы I и Примера 12 из WO 01/27128 также была изучена в прямом сравнительном исследовании на модели крыс с диабетом, индуцированным STZ (диабетические тучные крысы Цукера, модель, генетически предрасположенная к развитию диабета) в течение двухнедельного субхронического исследования.
10. В этом прямом сравнительном исследовании соединение формулы I и Пример 12 вводили ежедневно в дозе 0.5 мг/кг. Концентрацию глюкозы в плазме измеряли на исходном уровне (то есть, исходную глюкозу в плазме до 1-й дозы лекарственного средства) и после 8-го и 15-го дня лечения после 18-часового голодания. Результаты приведены в Таблице 3 ниже.
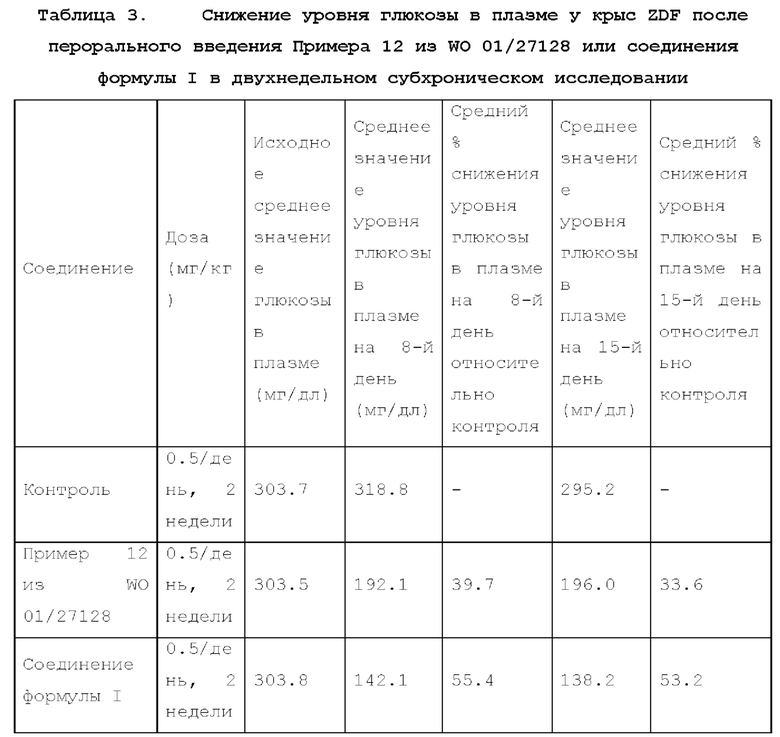
Средние значения глюкозы в плазме натощак сравнивали у крыс, получавших носитель и лекарственное средство, на 8-й и 15-й день. В проведенных Т-тестах сравнивали значения глюкозы в плазме натощак в группах, получавших лекарственное средство и получавших носитель. Значения р были меньше или равны 0.002, что считалось статистически значимым.
Как показано в Таблице 3, Пример 12 из WO 01/27128 снижал уровни глюкозы натощак примерно с 304 до 196 мг/дл (снижение на 35.4%; снижение на 33.6% по сравнению с контролем) на 15 день, тогда как соединение формулы I снижало уровни глюкозы натощак примерно от около 304 до 138 мг/дл (снижение на 54.5%; снижение на 53.2% по сравнению с контролем) за тот же период.
13. Эти данные демонстрируют, что соединение формулы I на 58% ((53.2-33.6)/33.5)) более эффективен, чем Пример 12, в снижении уровня глюкозы в плазме по сравнению с контролем в течение 15-дневного периода.
| название | год | авторы | номер документа |
|---|---|---|---|
| С-АРИЛ ГЛЮКОЗИДНЫЕ SGLT2 ИНГИБИТОРЫ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2800510C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР SGLT2 | 2008 |
|
RU2489151C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЯ ИНГИБИТОРА SGLT2 | 2013 |
|
RU2643764C2 |
| C-АРИЛ ГЛЮКОЗИДНЫЕ SGLT2 ИНГИБИТОРЫ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2337916C2 |
| О-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2269540C2 |
| С-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 | 2000 |
|
RU2262507C2 |
| С-ФЕНИЛ-1-ТИОГЛЮЦИТОЛЫ | 2007 |
|
RU2434862C2 |
| СОЕДИНЕНИЕ С-ФЕНИЛГЛИЦИТОЛА ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2007 |
|
RU2437876C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОРЫ КОТРАНСПОРТЕРОВ НАТРИЯ-ГЛЮКОЗЫ 1 И 2, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2669921C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ИХ МЕДИЦИНСКИЕ КОМПОЗИЦИИ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2005 |
|
RU2387663C2 |
Настоящее изобретение относится к соединению формулы:
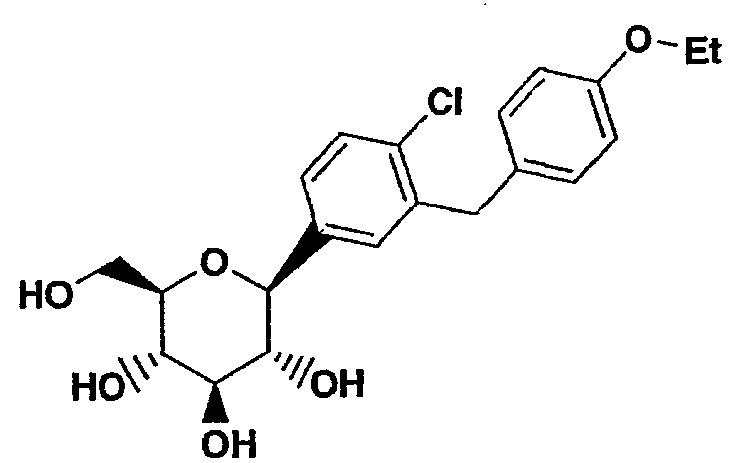
или его фармацевтически приемлемой соли или его сложному эфиру, обладающим ингибирующим действием в отношении SGLT2, а также к фармацевтической композиции. 3 н.п. ф-лы, 3 табл., 1 пр.
1. Соединение формулы:
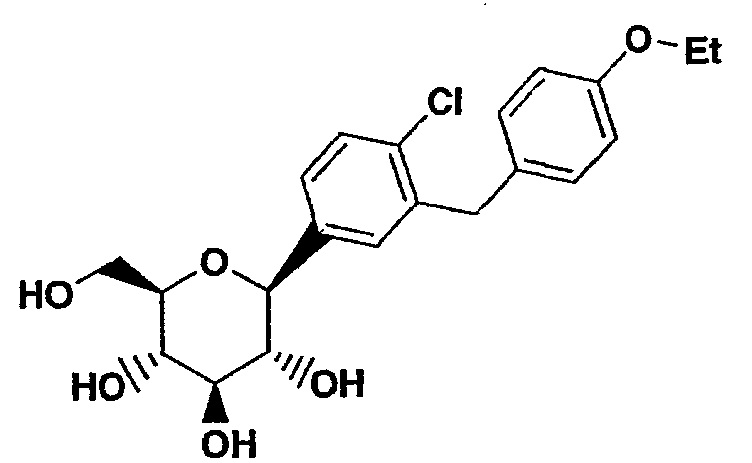
или его фармацевтически приемлемая соль или его сложный эфир, представляющий собой пролекарство.
2. Соединение формулы:
3. Фармацевтическая композиция, обладающая ингибирующим действием в отношении SGLT2, включающая соединение по п. 1 или 2 и фармацевтически приемлемый носитель.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Экономайзер | 0 |
|
SU94A1 |
| US 5965540 A, 12.10.1999 | |||
| Способ получения производных @ -D-фенилтиоксилозидов | 1988 |
|
SU1567124A3 |

